
膵臓腫瘍形成の根底にある経路および遺伝性の膵癌遺伝子
本発明は、24例の膵臓腫瘍において改変された遺伝子を解析することによる、膵癌の病因に対する新しい洞察に関する。第1に、本発明者らは、これらの腫瘍に由来するDNA中の20,583個のタンパク質コード遺伝子に相当する、23,781個の転写物の配列を決定した。第2に、本発明者らは、各試料において100万個の一塩基多型を検索するマイクロアレイを用いて、ホモ接合性欠失および増幅を探索した。第3に、本発明者らは、SAGE技術および次世代の合成による配列決定技術を用いて、同じ試料のトランスクリプトームを解析した。本発明者らは、膵癌が平均63個の遺伝的改変を含み、その内の49個が点変異であり、8個がホモ接合性欠失であり、6個が増幅であることを発見した。さらなる解析により、試料の70%〜100%においてそれぞれ遺伝的に改変されている12個の調節プロセスまたは経路からなる中心的セットが明らかになった。このデータから、経路のこの中心的セットの調節不全が、膵臓腫瘍形成の主要な特徴の原因であることが示唆される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、米国政府からの資金を用いて行われた。米国政府は、NIH助成金CA 43460、CA 57345、CA 62924、CA123483、RO1CA97075、およびCA 121113の規定に従い、本発明において一定の権利を保有する。
【0002】
発明の技術分野
本発明は、膵癌の分野に関する。特に、膵癌の診断、治療、特徴付け、モニタリング、検出、および層別化に関する。
【背景技術】
【0003】
発明の背景
世界中で213,000名の患者が2008年に膵癌を発症し、ほぼ全員がこの疾患で死亡する(1)。死亡率は非常に高く、その理由の一つは、この疾患が、既に局部的に広がっているか、または肝臓、腹膜、もしくは他の器官に転移するまで一般に検出されないことにある。この腫瘍は男性および女性に比較的等しく発症し、西洋社会で使用されている積極的治療を用いた場合でさえ、全生存率は5%未満である(2、3)。タバコの喫煙、長期に渡る慢性膵炎、およびある種の食生活にいくらか関連はあるものの、環境因子が膵臓新形成をもたらすメカニズムについてはほとんど知られていない。同様に、膵癌患者の約10%は、この疾患に対する家族性素因を有すると思われる。これらの患者のごく一部は、BRCA2、CDKN2A、LKB1、PRSS1、STK11、またはMSH2の生殖系列変異を含むが、膵癌に対する家族性素因を有する患者の圧倒的多数に関与する遺伝子は、まだ発見されていない(4)。
【0004】
膵臓腫瘍は、結腸直腸腫瘍のものとよく似た、いくつかの中間段階を経て進行すると思われる。浸潤癌に先行する非浸潤性段階は、膵臓上皮内新形成(PanIN)と呼ばれ、組織病理学的検査の際に明らかな進行性異形成を伴う(5)。これらの病変、ならびにそれらから最終的に発達する完全に浸潤性の癌腫において、いくつかの遺伝的改変が同定されている(6〜10)。改変された遺伝子には、腫瘍抑制遺伝子CDKN2A、SMAD4、およびTP53、ならびにKRAS癌遺伝子が含まれ、これらの各遺伝子は、かなりの割合の後期癌、および様々な割合の前浸潤新生物において変異していることが見出されている。これらの遺伝子の発見は、この疾患の自然史に対する独自の洞察をもたらし、より優れた診断用物質および治療物質を開発する試みにはずみをつけた(11)。
【0005】
膵癌の遺伝的要因を詳細に理解することが、当技術分野において引き続き必要とされている。膵癌に関連し、かつ重要である、その他の遺伝子および経路が引き続き必要とされている。
【発明の概要】
【0006】
本発明の1つの局面は、ある疾患に対する素因を有する個体を同定する方法である。その個体の組織に由来する鋳型核酸のタンパク質コード遺伝子の複数のエクソンにおいて配列決定反応を実施する。個体の複数のエクソンの配列を疾患に罹患していない個体の配列と比較して、その疾患に罹患していない個体には存在しない、前記個体のタンパク質コード遺伝子中の変異対立遺伝子を同定する。変異対立遺伝子の存在により、その個体がその疾患に罹患しやすいことが示唆される。
【0007】
本発明の別の局面は、遺伝性癌に関与している遺伝子を同定する方法である。少なくとも、タンパク質コード遺伝子のエクソンの鋳型核酸に基づいて配列決定反応を実施する。鋳型核酸は、家族性癌に罹患した第1のヒト個体の腫瘍に由来する。野生型対立遺伝子が存在していない、腫瘍中のタンパク質コード遺伝子を同定する。第1のヒト個体と同じ器官の家族性癌に罹患している複数のヒト個体のタンパク質コード遺伝子の鋳型核酸に基づいて配列決定反応を実施する。第1のヒト個体中の対立遺伝子と異なる、複数のヒト個体(plurality)のタンパク質コード遺伝子中の1つまたは複数の変異対立遺伝子を同定し、それによって、そのタンパク質コード遺伝子が家族性癌に対する易罹患性を与えることを確認する。
【0008】
本発明のさらに別の局面は、膵癌に対する易罹患性を判定する方法である。ある個体を、その個体の家族において見出されるPALB2遺伝子中の変異の存在に関して試験する。変異が存在する場合、その個体は膵癌を発症するリスクが高いと判定し、変異が存在しない場合、その個体のリスクは通常であると判定する。
【0009】
本発明のさらに別の局面は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブである。
【0010】
本発明のさらなる局面は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列をそれぞれが含むプローブまたはプライマーを含む、プライマーまたはプローブのキットである。
【0011】
本発明のある局面は、ある個体の膵癌に対する易罹患性を判定する方法である。その個体に由来する鋳型核酸に基づいてPALB2遺伝子配列の配列決定反応を実施する。PALB2配列中の変異を同定し、それによって、その個体は膵癌に対する易罹患性が高いと判定する。
【0012】
本発明の別の局面は、ある個体の膵癌に対する易罹患性を判定する方法である。TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む核酸プライマーまたは核酸プローブを、その個体に由来する核酸中のPALB2遺伝子配列にハイブリダイズさせる。その個体のPALB2配列中の該変異の内の1つを同定し、それによって、その個体は膵癌に対する易罹患性が高いと判定する。
【0013】
本発明の1つの態様によれば、ヒトにおいて膵癌もしくは微小残存病変または分子再発を検出または診断するための方法が提供される。試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異は、前記ヒトの正常試料と比べて確認される。この遺伝子は、表S7または表S3に列挙したものからなる群より選択される;しかし、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。体細胞変異が確認される場合、その個体は、膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定される。
【0014】
また、ヒトの膵癌を特徴付ける方法も提供される。試験試料中のCAN遺伝子変異シグネチャー(signature)は、ある遺伝子またはそのコードされたcDNAもしくはタンパク質における少なくとも1つの体細胞変異を確認することにより、前記ヒトの正常試料と比べて確認される。この遺伝子は、表S7または表S3に列挙したものからなる群より選択される;しかし、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。
【0015】
本発明の別の局面は、ヒトの膵臓腫瘍を特徴付ける方法である。表S8に示すものからなる群より選択される変異した経路は、試験試料における少なくとも1つの体細胞変異を前記ヒトの正常試料と比べて確認することにより、膵臓腫瘍において確認される。この少なくとも1つの体細胞変異は、その経路における1つまたは複数の遺伝子中にある。膵臓腫瘍は、経路中に変異を有する第1群の膵臓腫瘍に割り当てられる;この第1群は、変異を有する経路における遺伝子に対して異種性であるが、その経路に対しては同種である。
【0016】
本発明のさらなる局面は、個体において早期癌もしくは微小残存病変または分子再発を検出する方法である。表S6または表S12に示すもの(SAGEによる膵臓で過剰発現される遺伝子)より選択される遺伝子からのmRNAまたはタンパク質の発現の増大が、その個体から採取された臨床試料において検出される。この増大は、健常個体の集団と比べるか、または異なる時点に採取した同じ個体の臨床試料と比べる。臨床試料が対照と比べて高い発現を示す場合、その個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定される。
【0017】
本発明のさらに別の局面は、膵癌量をモニターするための方法である。表S6または表S12に列挙する1つまたは複数の遺伝子(SAGEによる膵臓で過剰発現される遺伝子)の臨床試料における発現が測定される。発現を測定する段階は、前記のように1回または複数回繰り返される。経時的な発現の増大、減少、または安定なレベルが確認される。
【0018】
本発明のさらに別の局面は、膵癌を検出または診断するための方法である。表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料における発現が測定される。臨床試料中の1つまたは複数の遺伝子の発現は、対照ヒトもしくは対照ヒト群またはその患者の正常組織といった対応する試料におけるそれら1つまたは複数の遺伝子の発現と比較される。対照と比べて低い発現を示す臨床試料は、膵癌を有する可能性が高いと判定される。
【0019】
さらに、膵癌量をモニターするための方法も提供される。発現は、表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料において測定される。測定する段階は、1回または複数回繰り返される。経時的な発現の増大、減少、または安定なレベルが確認される。
【0020】
また、試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異が、前記ヒトの正常試料と比べて確認される、膵癌量をモニターするための方法も、本発明によって提供される。この遺伝子は、表S7に列挙したものからなる群より選択されるが、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。確認する段階は、1回または複数回繰り返される。試験試料における前記変異の経時的な増加、減少、または安定なレベルが確認される。
【0021】
本明細書を読めば当業者に明らかになると考えられるこれらおよび他の態様は、膵癌を特徴付け、治療、予後判定、診断、および層別化するための手段(tool)および方法を当技術分野に提供する。
【図面の簡単な説明】
【0022】
【図1】図1A〜1Dは、変異の構造モデルの例を示す。(図1A)プロテインキナーゼC(PKC)γ(PDBID:2UZP)のC2ドメインのX線結晶構造。Arg252は、大きな空間充填物(space-fill)として示され、Ca2+イオンは小型球体として示されている。リガンドである1,2-エタンジオールおよびピリドキサール-5’-ホスファートは、玉および棒の図として示されている。R252H変異は、PRKCGのC2ドメインの膜結合を減少させ、それによって機能に影響を及ぼし得る。(図1B)ヒトクルッペル様因子5(KLF5)(PDBID:2EBT)に由来するzf-C2H2ドメインの3つのタンデムリピートのNMR溶液構造。His389は、空間充填物として示され、Zn2+イオンは小型球体として示されている。C2H2基を含み近隣のZn2+イオンに配位結合する残基であるH393およびH397は、玉および棒の図として示され、Cys380およびCys375は玉および棒の図として示されている。389位での変異(H389N)は、亜鉛フィンガーまたは近隣の亜鉛配位部位の構造を混乱させる可能性がある。(図1C)SMAD3(ほぼ垂直方向のリボンとして示されている2つのサブユニット)およびSMAD4(水平方向のリボンとして示されている1つのサブユニット)(PDBID:1U7F)のヘテロ三量体のX線結晶構造。変異位置の内の2つに対応する残基(F260SおよびS422F、鎖A中の空間充填物として示されている)は、境界面に位置しており、Smad3-Smad3相互作用またはSmad3-Smad4相互作用を乱し得る。鎖Bにおいて、F260およびS422は、空間充填物として示されている。(図1D)ホモ二量体としてのヒトDPP6(PDBID:1XFD)の細胞外ドメインのX線結晶構造。この研究で発見された変異残基の内の2つ、すなわちT409IおよびD475N(空間充填物として示されている)は、空間的に近接しており、グリコシル化部位の内の1つであるAsn471(空間充填物として示されている)に近接している。これらの変異は、タンパク質同士の相互作用に関与していると考えられているタンパク質のβ-プロペラドメイン(残基142〜322および351〜581)中にある。A778T変異(空間充填物として示されている)は、α/βヒドロラーゼドメイン(残基127〜142および581〜849)中にあり、このタンパク質のホモ二量体領域に近接し、ホモ二量体会合を乱し得る。グリコシル化部位を有する炭水化物は、棒の図として示されている。
【図2】配列決定を通じて検出された遺伝的改変の数および24例の各癌のコピー数解析を示す。棒の下部は変異を表し、棒の中央は増幅を表し、棒の上部は欠失を表す。
【図3】図3A〜3Cは、経路および調節プロセスを示す。(図3A)大半の膵癌において構成要素遺伝子が遺伝的に改変された12個の経路およびプロセス。(図3B、図3C)2種の膵癌(Pa14CおよびPa10X)ならびにそれらにおいて変異している特定の遺伝子。(図3B)および(図3C)の円の周りの位置は、(図3A)の経路およびプロセスに対応する。Pa10XにおいてSMAD4シグナル伝達経路およびヘッジホッグ(Hedgehog)シグナル伝達経路の両方を混乱させる可能性が高いBMPR2変異によって例示されるように、いくつかの経路構成要素が重複していた。さらに、DNA損傷制御(damage control)に影響を及ぼすことが公知である変異がPa10Xにおいて観察されない(N.O.、観察されず)ことによって例示されるように、すべての膵癌において12個のプロセスおよび経路のすべてが改変されているわけではなかった。
【図4】PALB2遺伝子中の変異の位置を示す。エクソンは四角形として表し、イントロンは黒い線として表している(原寸に比例しない)。家族性乳癌患者またはファンコニー貧血患者において以前に同定された変異を遺伝子の下に示している。家族性膵癌患者において同定された生殖系列変異を遺伝子の上に示している。
【図5】補足的な表S3(Discovery Screen(発見スクリーニング)における変異)、S4(Mutation Prevalence(変異保有率スクリーニング)、S5(ホモ接合性欠失)、S6(増幅された遺伝子)、S7(CAN遺伝子)、S8(頻繁に変異している経路)、S12(SAGEによる過剰発現遺伝子)、およびS13(過剰発現された細胞外遺伝子)を示す。
【発明を実施するための形態】
【0023】
発明の詳細な説明
本発明者らは、膵臓腫瘍を徹底的に解析し、得られた解析に基づいて、新規な療法、予後因子(prognosticator)、手段、およびストラティフィアー(stratifier)を開発した。いくつかの独特なアプローチ、例えば、変異、増幅、および欠失を検出するための配列決定、ならびに発現定量化を用いて、本発明者らは、重要な遺伝子、経路、および変異を同定した。個々の膵臓腫瘍間の遺伝的異質性は大きいものの、しばしば変異している遺伝子および経路のパターンが検出された。
【0024】
体細胞変異は、個々の生物の一生の間に体細胞の特定のクローンにおいて生じる変異である。したがって、この変異は、親から受け継がれることもなく、子孫に受け渡されることもない。この変異は、他の細胞、組織、器官との差異として現れる。癌性と推測される膵臓組織における体細胞変異について試験する場合、非新生物性と思われる正常膵臓組織、もしくは血液細胞のような非膵臓試料、または罹患していない個体に由来する試料と比較することができる。
【0025】
膵臓腫瘍中で見出されている変異を表S7または表2に示している。これらの変異は、試験試料、例えば、推測される腫瘍組織試料、血液、膵管液、尿、唾液、リンパなどにおいて検出され得る。典型的には、体細胞変異は、試験試料中の配列を正常対照試料中の配列、例えば健常な膵臓組織に由来するものと比較することによって確認される。1つまたは複数の変異が、この目的のために使用され得る。患者が手術を受けた場合には、腫瘍辺縁部または残存する隣接組織中の変異の検出を用いて、微小残存病変または分子再発を検出することができる。それまでに膵癌が診断未確定であった場合、この変異は、例えば、生化学的マーカーおよび放射線医学的所見を含む研究室結果の他の物理的所見と組み合わせて、診断を助けるのに役立つ可能性がある。変異を利用して患者を層別化して、薬物または他の治療に感受性または抵抗性である患者または患者群を同定することができる。
【0026】
CAN遺伝子シグネチャーは、膵臓腫瘍を特徴付けるために確認され得る。シグネチャーは、CAN遺伝子における1つまたは複数の体細胞変異のセットである。膵臓に関するCAN遺伝子は、表S7および表2に列挙する。このようなシグネチャーを確認した後で、膵臓腫瘍を、そのシグネチャーを共有する膵臓腫瘍群に割り付けることができる。この群を用いて、予後を割り付けること、臨床試験群に割り付けること、治療計画に割り付けること、ならびに/またはさらなる特徴付けおよび研究のために割り付けることができる。臨床試験群において、そのシグネチャーを有する膵臓腫瘍および有さない膵臓腫瘍に差次的に影響を及ぼす能力について薬物を評価することができる。差次的な効果が測定されたら、そのシグネチャーを利用して、患者を投薬計画に割り付けるか、またはその薬物が有益な効果を持たないであろう患者を不必要に治療するのを回避することができる。臨床試験における薬物は、別の目的のためにこれまで公知であるか、膵癌を治療するためにこれまで公知であるか、または治療物質としてこれまで未知であるものでよい。CAN遺伝子シグネチャーは、少なくとも1個、少なくとも2個、少なくとも3個、少なくとも4個、少なくとも5個、少なくとも6個、少なくとも7個、少なくとも8個、少なくとも9個、少なくとも10個の遺伝子を含んでよい。個々のシグネチャー中の遺伝子または変異の数は、そのシグネチャー中のCAN遺伝子のアイデンティティに応じて変動し得る。
【0027】
解析された膵臓腫瘍中の変異遺伝子の解析により、経路の興味深い関与が明らかになった。いくつかの経路は、頻繁に、膵臓腫瘍において変異を有する。しばしば、単一遺伝子変異は、個々の腫瘍のその経路における別の遺伝子における変異の存在を排除する。膵臓腫瘍中の頻繁に変異している経路を、表S8および表2に列挙する。経路は、MetaCore Gene Ontology(GO)データベース、MetaCore古典的遺伝子経路マップ(MA)データベース、MetaCore GeneGo (GG)データベース、Pantherデータベース、TRMPデータベース、KEGGデータベース、およびSPADデータベースなどの標準的な参照データベースのいずれかを用いて定義することができる。群は、特定の経路における変異の存在または不在に基づいて形成させることができる。このような群は、変異遺伝子に関して異種性であるが、変異経路に関しては同種性である。CAN遺伝子シグネチャーと同様に、これらの群を利用して膵臓を特徴付けることができる。ある経路における変異を確認した後で、膵臓を、その変異経路を共有する膵臓腫瘍群に割り付けることができる。この群を用いて、予後を割り付けること、臨床試験群に割り付けること、治療計画に割り付けること、ならびに/またはさらなる特徴付けおよび研究のために割り付けることができる。臨床試験群において、その変異経路を有する膵臓腫瘍および有さない膵臓腫瘍に差次的に影響を及ぼす能力について薬物を評価することができる。差次的な効果が測定されたら、その経路を利用して、患者を投薬計画に割り付けるか、またはその薬物が有益な効果を持たないであろう患者を不必要に治療するのを回避することができる。臨床試験における薬物は、別の目的のためにこれまで公知であるか、膵臓を治療するためにこれまで公知であるか、または治療物質としてこれまで未知であるものでよい。
【0028】
発現レベルを測定することができ、過剰発現は、新たな膵臓腫瘍、膵臓の分子再発、または微小残存病変を示す可能性がある。膵臓腫瘍中で見出される著しく増大した発現を表S6および表S12に示している。これらの過剰発現された遺伝子は、試験試料、例えば、推測される腫瘍組織試料、血液、膵管液、尿、唾液、リンパなどにおいて検出され得る。典型的には、高発現は、試験試料中の遺伝子の発現を正常試料中の遺伝子、例えば健常な膵臓組織に由来するものの発現と比較することによって確認される。1つまたは複数の遺伝子の高発現が、この目的のために使用され得る。患者が手術を受けた場合には、腫瘍辺縁部または残存する隣接組織中の高発現の検出を用いて、微小残存病変または分子再発を検出することができる。それまでに膵臓が診断未確定であった場合、この高発現は、例えば、生化学的マーカーおよび放射線医学的所見を含む研究室結果の他の物理的所見と組み合わせて、診断を助けるのに役立つ可能性がある。増加したmRNAを検出するためのSAGEまたはマイクロアレイ、およびタンパク質高発現を検出するために様々なアッセイ形式で使用される抗体を含む、当技術分野において公知である発現を定量するための任意の手段が、これらの目的のために使用され得る。タンパク質発現を検出するためには、表S13に列挙した遺伝子が特に有用である。
【0029】
腫瘍量は、表S7に列挙した変異を利用してモニターすることができる。慎重な経過観察の様式で、または療法期間中にこれを利用して、例えば有効性をモニターすることができる。マーカーとしての体細胞変異の使用および検出可能なDNA、mRNA、またはタンパク質のレベルの経時的な分析により、腫瘍量を示すことができる。試料中の変異のレベルは、解析期間を通して、上昇するか、低下するか、または一定のままであり得る。治療的処置およびタイミングは、このようなモニタリングに基づいて管理することができる。
【0030】
膵臓腫瘍の解析により、ホモ接合性に欠失している遺伝子が明らかになった。これらを表S5に列挙する。1つまたは複数のこれらの遺伝子の発現喪失の測定は、膵癌のマーカーとして利用され得る。これは、血液もしくはリンパ節の試料において、または膵臓組織試料において実施され得る。1つまたは複数のこれらの遺伝子の発現を試験してよい。ELISAまたはIHCなどの技術を用いて、試料中のタンパク質発現の減少または喪失を検出することができる。同様に、表S5に列挙したホモ接合性に欠失した遺伝子(および表S6の増幅された遺伝子)を用いて、腫瘍量を経時的にモニターすることもできる。発現の増大、減少、または安定なレベルを確認できるように、発現を繰り返しモニターしてよい。
【0031】
変異およびコピー数改変のこの統合された解析の結果得られるデータは、膵臓腫瘍の遺伝的景観の異なる観点を与えた。点変異、増幅、および欠失を含む様々なタイプの遺伝的データを組み合わせることにより、個々のCAN遺伝子ならびに膵臓腫瘍の複雑な細胞経路およびプロセスにおいて優先的に影響を受ける可能性がある遺伝子群の同定が可能になる。膵臓腫瘍において変異、増幅、または欠失による影響を受けることが以前に示されているほぼすべての遺伝子の同定により、本発明者らが使用した包括的ゲノムアプローチが有効になる。
【0032】
本明細書において説明する広範囲の遺伝的研究から、膵癌を理解するための鍵は、調節プロセスおよび経路の中心的(core)セットを理解することにあることが示唆される。本発明者らは、膵癌の大多数において遺伝的に改変されている12個のこのようなプロセスを同定した(図3A)。しかしながら、任意の個々の腫瘍において改変されている経路構成要素は、多種多様である(図3B、3C)。例えば、図3Bおよび3Cに示す2種の腫瘍はそれぞれ、TGF-β経路に関与している遺伝子の変異を含む(一方はSMAD4、他方はBMPR2)。同様に、これら2種の腫瘍は両方とも、他の11個の中心的プロセス/経路の大半に関与している遺伝子の変異を含むが、各腫瘍中の改変されたこれらの遺伝子は大いに異なる。本発明者らは、同定された変異のすべてが、関係があるとされている経路またはプロセスにおいて機能的役割を果たしていることに確信は持てないが、現在公開されている遺伝的データおよび以前に公開された遺伝的データの両方から、ならびに過去の機能的研究から、それらの内の多くがこれらの経路に影響を与えている可能性が高いことは明らかである。
【0033】
この見込みは、全部ではないが大半の上皮性腫瘍に当てはまる可能性が高い。これは、遺伝的改変を山(高頻度の変異)または丘(低頻度の変異)に分類することができ、丘の方が、関与している改変の総数の点で優勢であるという考えと完全に一致している(16)。経路構成要素間の異種性および個々の遺伝子内の変異の異なる性質は、すべての固形腫瘍の基本的な側面である腫瘍異質性の説明となり得る(39)。
【0034】
知的観点から、この経路という見方は、非常に複雑な疾患に対して秩序および初歩的理解をもたらすのを助ける(40〜42)。一般的な新形成を理解する上での調節プロセスおよび経路の重要性は認識されているが(43、44)、本研究で実施されるもののようなゲノム全域に渡る遺伝的解析により、各患者の腫瘍におけるそれらの調節不全に関与する厳密な遺伝的改変を同定することができる。腫瘍病因に対する洞察をもたらすことに加えて、このような研究は、個別化された癌医薬品に基づくアプローチのために必要とされるデータを提供する。単一の標的指向性癌遺伝子によって腫瘍形成が推進されると思われる特定の型の白血病とは違って、膵癌は、比較的少数の経路およびプロセスを介して機能する多数の遺伝子の遺伝的改変に起因する。KRAS癌遺伝子はこれまでのところ、ターゲティングに成功しておらず、遍在的に改変された同様の新規な標的は明らかでないため、本発明者らの研究により、治療物質開発の最も優れた希望は、それらの個々の遺伝子構成要素ではなく、改変された経路および調節プロセスの生理的効果を標的とする作用物質の発見にあることが示唆される。これらの効果には、代謝妨害、血管新生、細胞表面タンパク質の異所性発現、細胞周期の改変、細胞骨格異常、およびゲノム損傷を修復する能力の障害が含まれる(表S8)。
【0035】
膵癌のために使用されている方法は、より広範囲の用途を有する。遺伝性疾患に関与している遺伝子を同定する方法は、他の癌および他の疾患のために使用され得る。
【0036】
膵癌に対する易罹患性に関与していると判定された1つの遺伝子は、PALB2である。PALB2中の変異は、患者の膵癌において同定される。ついで、家族を試験して、彼らもまたその変異を保有するかどうか確認することができる。家族がその変異を有する場合、その人は、膵癌を発症するリスクが高い。患者のPALB2変異が家族には存在しない場合、その人は、一般集団と同じリスクを有する。試験は、当業者に公知の任意の方法によって実施することができる。核酸プローブまたはプライマーへの家族の鋳型核酸のハイブリダイゼーションを用いて、変異を分析することができる。鋳型核酸は、例として、ゲノムまたはmRNAもしくはcDNAでよい。プローブまたはプライマーは、少なくとも14個、16個、18個、20個、22個、24個、26個、または30個の核酸塩基を含んでよい。プローブまたはプライマーは、膵臓腫瘍中に存在する変異を含む、PALB2の一部分を含んでよい。プライマーは、変異部位に隣接し、アンプリコン中の変異の増幅および解析を可能にし得る。確認され得る具体的な変異は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tである。
【0037】
変異特異的なPALB2プローブまたはPALB2プライマーをキット中で組み合わせてもよい。これらのキットは、分割された容器または分割されていない容器を含んでよい。キットの構成要素は、別々でもよく、または混合されてもよい。容器に加えて、キットの他の要素には、取扱い説明書、緩衝剤のような試薬、およびポリメラーゼのような酵素が含まれ得る。固体支持体、反応チューブ、ビーズなどが、キットに含まれてよい。これらのキットは、少なくとも2個、3個、または4個の異なる変異特異的試薬を含んでよい。
【0038】
上記の開示は、本発明を全体として説明する。本明細書において開示される参考文献はすべて、参照により明確に組み入れられる。より完全な理解は、以下の具体的な実施例を参照することにより得ることができ、これらの実施例は、例証のために本明細書において提供されるにすぎず、本発明の範囲を限定することを意図しない。
【実施例】
【0039】
実施例1
試料選択
任意の癌ゲノム研究と同様に、試料の選択は重大な意味を持つ。この研究のために、本発明者らは、24例の進行した腺癌を選択した。これらはそれぞれ、異なる無関係の患者に由来した(表S1)。初期の癌は1つのサブセットしか含まない可能性があるのに対し、進行した膵癌は、腫瘍の開始および進行の原因である遺伝的改変のすべてを含むと予想できるため、進行した膵癌を選択した。これら24例の癌をインビトロで細胞株として、またはヌードマウス中で異種移植片として継代して、変異の検出を容易にした。このような継代は、それらの腫瘍中に元々存在する混入非新生細胞を除去するため、サンガー配列決定またはコピー数解析のために原発腫瘍よりも優れたDNA鋳型を提供することが示されている(12)。また、細胞株および異種移植片中に存在するクローン変異は、あるとしても、エクスビボでの培養中にめったに起こらないことも実証されている(12〜14)。
【0040】
実施例2
配列決定戦略
Consensus Coding Sequence Database(リリース1)、Reference Sequence Database(リリース16)、およびEnsembl Database(リリース31)において見出されるタンパク質をコードするエクソンの配列を取り出し、ゲノムDNAを増幅するためのプライマーを設計するために使用した(図S1)。乳癌および結腸直腸癌に関する本発明者らの過去の研究において以前に設計したプライマーが好結果であると判明した場合は(15、16)、同じプライマーを使用した。以前に研究されていないエクソン11,579個ならびに以前に設計されたプライマーが不十分であると判明したエクソンに対して、新しいプライマーセットを設計した(下記を参照されたい)(17)。次いで、これらの結果として得られた各エクソンの配列を、色素ターミネーター配列決定および表S2に列挙したプライマー416,622個を用いて、24例の膵癌において決定した。変異配列を含むエクソンを、腫瘍DNAから再増幅および再配列決定して、観察された改変を確認した。すべての場合において、変異を有する患者の正常組織に由来するDNAをさらに検査した。このアプローチにより、その改変が正常細胞中に存在するか(したがって、生殖系列変異体であるか)、またはその個体の癌細胞に特異的な体細胞変異に相当するかを判定した。
【0041】
将来の医学的再配列決定プロジェクトは、次世代の合成化学反応による配列決定を使用し得るため、本研究で使用する従来の色素ターミネーター配列決定法を用いて得られる有効範囲を決定することは関心対象であった。本発明者らは、遺伝子20,735個に相当する、転写物23,962個のタンパク質コードエクソンの配列を評価することを試みた。標的配列は、各エクソンのタンパク質コード部分全て、ならびに上流の塩基4個および下流の塩基4個を含んだ。これらの領域を網羅するために、本発明者らは、アンプリコン219,229個に対するプライマーを設計し、その内の208,311個(95%)から得られたPCR産物は、首尾よく配列決定され、さらなる変異解析のための本発明者らの品質管理条件を満たした(17)。これらの品質管理されたアンプリコンは、標的とされたコード領域の94.5%を網羅し、それらのアンプリコン内部の標的塩基の98.5%に関する高品質の配列決定データをもたらした。全体で、本発明者らは、これら24名の患者において、標的とされた転写物のコード領域中の塩基の93.1%に相当する752,843,968bpを成功裡に配列決定することができた。これにより、遺伝子20,661個に相当する転写物23,219個に関する変異データが得られた。増幅のために使用されるプライマーは最低でも「第2世代」プライマーであり、失敗したプライマーは、本発明者らの研究室で以前に実施された各大規模配列決定プロジェクトの間に、新しいプライマーで置き換えられ、改善されたことに留意されたい。したがって、この93.1%という値は、色素ターミネーター技術を用いて達成できる最大値に近いことになる。さらに、配列決定できない領域の圧倒的多数は、配列決定の失敗それ自体よりはむしろ、反復されたエレメントを意味した。反復された領域は、短い読取り長(short read length)を生じる方法の場合はなおさら問題があるため、この配列包括度が次世代技術によって増加する可能性は低い。
【0042】
実施例3
体細胞変異
体細胞変異1562個の内で、25.5%は同義であり、62.4%はミスセンスであり、3.8%はナンセンスであり、5.0%は小規模の挿入および欠失であり、3.3%は、スプライス部位にあるか、またはUTR内部に存在した(表1)。体細胞変異のスペクトルは、潜在的な発癌物質および他の環境曝露への洞察をもたらし得る。表1では、タンパク質コード遺伝子の大多数の大規模配列決定解析に供された4例の腫瘍において観察されたスペクトルを記載している。乳房腫瘍は独特な体細胞変異スペクトルを有しており、5'-TpC部位における変異が優勢であり、5'-CpG部位における変異は比較的少数であることが明らかである。しかしながら、結腸直腸腫瘍、脳腫瘍(18)、および膵臓腫瘍のスペクトルは類似していることから、乳房上皮細胞が、異なるレベルもしくはタイプの発癌物質に曝露されるか、または他の腫瘍を生じる細胞とは異なる修復システムを使用することが示唆される(19、20)。膵臓および結腸のものなど胃腸管中の細胞は、乳房細胞または脳細胞よりも食事性発癌物質に多く曝露されると予想されることから、これらの結果の1つの解釈は、食事性成分は、ヒト癌中に存在する変異の大半の直接的原因ではないというものである。
【0043】
(表1)4種の腫瘍型における体細胞変異の要約

*本研究において解析した24例の腫瘍に基づく
†Parsons et al., Science, 2008 (印刷中)において解析された21例の非高変異性(nonhypermutable)腫瘍に基づく。
‡Wood et al., Science 20:1108-13 2007において解析された11例の乳房腫瘍および11例の結腸直腸腫瘍。
§括弧内の数字は、非同義変異の総数の比率(%)を示す。
**指定の研究において同定された同義変異ならびに非同義変異を含む。
††括弧内の数字は、置換の総数の比率(%)を示す。
【0044】
調査した24例の癌において、配列決定によって解析した遺伝子20,661個の内で、1327個は少なくとも1つの変異を有し、148個は2個またはそれ以上の変異を有していた(表S3)。ある遺伝子における変異出現率に加えて、変異のタイプは、疾患におけるその潜在的役割を評価するために有用な情報を提供し得る(21)。ナンセンス変異、アウトオブフレームな挿入または欠失、およびスプライス部位の変化は、一般に、タンパク質産生物の不活性化を招く。ミスセンス変異が起こしそうな影響は、進化的手段または構造的手段によって変異残基を評価することによって評価することができる。ミスセンス変異を評価するために、本発明者らは、置換に関与しているアミノ酸の物理的-化学的特性および保存されているタンパク質の等価な位置におけるそれらの進化的保存に基づく58個の予測的特徴の機械学習を使用する新規なアルゴリズムを開発した(17)。このアルゴリズムを用いて採点できるミスセンス変異926個の内で、160個(17.3%)は、この方法によって評価した場合、腫瘍形成に寄与していると予測された(表S3)。
【0045】
本発明者らはまた、本研究で同定したミスセンス変異の内の404個の構造モデルを作ることに成功した((22)で入手可能な構造モデルに関連する)。各場合において、モデルは、正常タンパク質または近縁ホモログのX線結晶解析または核磁気共鳴分光法に基づいた。この解析により、変異244個の内の55個が、ドメイン境界面またはリガンド結合部位の近くに位置しており、機能に強い影響を与える可能性が高いことが示された(図1の例)。
【0046】
タンパク質コード遺伝子すべてに関する本発明者らの解析により、個々の腫瘍中の遺伝的改変の概要の詳細な図が提供される。図2に示したように、膵癌は、腫瘍1個当たりタンパク質コード遺伝子において平均48個の体細胞変異を有していた。この数の変動は、腫瘍形成プロセスの複雑さおよび患者の様々な年齢を考慮すれば、著しく小さかった(表S1)。膵癌における体細胞変異の平均数は、乳癌または結腸直腸癌の場合よりもかなり少なく(p<0.001)、後者の2種の腫瘍型の方が少ない遺伝子を配列決定したにも関わらず、この結果であった(16)。この低い割合に関して妥当と思われる1つの説明は、膵臓腫瘍形成を開始する細胞の方が、結腸直腸癌細胞または乳癌細胞よりも少ない分裂を経たというものである。結腸直腸癌において観察される変異の大多数は、最初の(initiating)新生細胞を生じた正常幹細胞において起こった可能性が高いことが以前に示されている(14)。したがって、本発明者らのデータは、膵臓上皮細胞がまれに分裂するのに対し(23、24)、乳房上皮細胞および結腸直腸上皮細胞は頻繁に分裂する(前者はホルモン刺激の期間中、後者は一生を通じて)ことを示す観察結果と一致している。
【0047】
本発明者らはさらに、膵癌90個からなるPrevalenceスクリーニングにおいて、24例のDiscovery Screen癌の内の複数において変異してる遺伝子39個を評価した。このスクリーニングにおいて、本発明者らは、遺伝子23個において非サイレント体細胞変異255個を検出した(表S4)。Prevalenceスクリーニングにおけるこれらの遺伝子(KRAS、TP53、CDK2NA、およびSMAD4を除く)の非サイレント変異の比率は、Discovery Screenにおけるものよりも高かった(3.6 対 1.47の非サイレント変異/Mb、p<0.0001)。これら19個の遺伝子において観察された非サイレント変異の割合もまた、Discovery Screenにおいて観察されたものよりも高かった(p<0.052)。これらのデータは、Prevalenceスクリーニングにおいて試験した遺伝子の大部分(greater fraction)が腫瘍形成の間に陽性選択されたという仮説と一致している。
【0048】
実施例4
欠失
本研究の設計の重要な局面は、細胞株または異種移植片に由来するDNAの使用であった。このDNAは、真のホモ接合性欠失の確信的な検出を可能にする。これは、一番最初の原発腫瘍検査材料に由来するDNAを用いた場合は、非新生物性の間質細胞および炎症細胞の混入が原因で、非常に困難な任務である。SNPアレイデータ、デジタル核型解析(Digital Karyotyping)、およびリアルタイムPCR解析の比較を通して、本発明者らは、SNPアレイデータからこのような試料における欠失事象を確信的に同定するための確固としたアルゴリズムを以前に開発していた(25)。これらのアルゴリズムを用いて、1,069,688個のSNPに対するプローブを含むIllumina社製オリゴヌクレオチドアレイから得たデータを解析したところ、本発明者らは、変異解析のために使用された膵癌24個において198個の別個のホモ接合性欠失を検出した(表S5)。これらの欠失の平均サイズは335,000bpであった。ホモ接合性欠失のほかに、本発明者らは、染色体全体または染色体腕全体の喪失を含む、ヘテロ接合性の喪失としてしばしば明らかである単一コピー喪失を経験した多くの領域を観察した。欠失していない染色体上の遺伝子の残存コピーが変異している場合を除いては、このような大きな領域から標的遺伝子を確実に同定することは困難であるため、本発明者らは、これらの変化を追究しなかった。このような標的遺伝子は、Discovery配列決定スクリーニングの結果によって本発明者らの注目に既になっており、ホモ接合性変化として記録されていた(表S3)。
【0049】
対立遺伝子のツーヒット仮説によれば、ホモ接合性欠失の存在から、欠失した領域内に腫瘍抑制遺伝子が存在することが示唆される(26)。これらの欠失内の最も可能性が高い標的を決定するために、本発明者らは、本発明者らの新しい変異解析および発現解析の結果ならびに過去の研究からのデータを使用した。ある遺伝子が候補標的とみなされるためには、そのコード領域の一部分が、ホモ接合性欠失による影響を受けていなければならず、かつ、(i)その遺伝子は、Discovery Screenにより、異なる腫瘍中で非サイレント配列改変を含まなければならないか、または(ii)十分に考証された腫瘍抑制遺伝子でなければならないか、または(iii)裏付けとなる発現データを有していなければならなかった(下記の遺伝子発現セクションを参照されたい)。これらの基準を満たした、各ホモ接合性欠失に対する推定上の標的遺伝子を表S5に列挙する。このリストは、古典的な腫瘍抑制遺伝子であるCDKN2A(p16)、SMAD4、およびTP53、ならびにこれまでは膵臓腫瘍形成に関係があるとされていなかった様々な他の遺伝子を含む。
【0050】
SNPアレイを通じて発見されたホモ接合性欠失を確認するために、本発明者らは配列決定データを再解析した。ある遺伝子のエクソンが、腫瘍中で本当に欠失している場合、そのエクソンの増幅を試みても、配列決定情報は得られないはずである。例外無く、配列決定データによって、その結果、マイクロアレイハイブリダイゼーションを通じて同定された欠失が確認された。さらに、マイクロアレイハイブリダイゼーションにおいては明らかではなく、配列決定によって明らかにされたホモ接合性欠失が1つだけ存在した(1つの腫瘍におけるSMAD4の4つのエクソンの欠失)。
【0051】
ある腫瘍中の欠失の数は、体細胞変異の数よりも多様であり、腫瘍1個当たり平均8.2個で、2〜20個の間の範囲であった(図2)。しかしながら、各ホモ接合性欠失は、標的遺伝子ならびに欠失領域内の他のすべての遺伝子の機能を完全に抑制したのに対し、体細胞変異のごく一部しか、遺伝子の機能を改変することが予測されなかったことに、留意すべきである。平均的な膵癌において、合計約10個の遺伝子(標的および欠失内の近隣遺伝子を含む)が、ホモ接合性欠失によって腫瘍ゲノムから失われることは、このような喪失を標的とする治療戦略に想像力豊かな根拠を与える(27、28)。
【0052】
実施例5
増幅
欠失と同様に、本発明者らは、SNPアレイデータから増幅を確信的に同定するためのアルゴリズムを開発した(25)。Illumina社製アレイから得た個々の蛍光強度比の測定値、ならびにコピー数が変化する連続した領域における最小強度比、最大強度比、および平均強度比の組合せを用いて、本発明者らは、染色体全体、染色体腕、または他の大きなゲノム領域の様々な少しの(low)コピー数増加(gain)を確認した。このような大きな染色体領域から候補癌遺伝子を確実に同定することは困難であるため、本発明者らは、これらのコピー数変化をそれ以上追究しなかった。さらに、腫瘍増殖または薬物耐性を促進する十分に考証されているほぼすべての増幅は、比較的小さな増幅領域を使用する(29)。したがって、本発明者らは、異数性ではなく、真の増幅の結果であることが明らかな局所的増幅に焦点を合わせた。
【0053】
核1つにつき増幅領域の12個超のコピーが存在することを含む局所的増幅の厳密な基準を用いて(17)、本発明者らは、24例の膵癌において144個の増幅を同定した(表S6)。これらの増幅の最も可能性が高い標的を決定するために、本発明者らは、本発明者らの変異解析および発現解析の結果ならびに以前に公表したデータを再び使用した。ある遺伝子が増幅の標的とみなされるためには、そのコード領域全体が、増幅された領域中に含まれなければならず、かつ、それは、(i)Discovery Screenにより、異なる腫瘍において変異していなければならないか、または(ii)十分に考証された癌遺伝子でなければならないか、または(iii)裏付けとなる発現データを有していなければならなかった(下記の遺伝子発現セクションを参照されたい)。これらの基準を満たした、各増幅に対する推定上の標的遺伝子を表S6に列挙する。大半の膵臓腫瘍において、増幅は、ホモ接合性欠失または点変異よりも少なかった(図2)。
【0054】
実施例6
パッセンジャー変異率
癌ゲノム研究の最も重要な目標は、新生物プロセスにおいて原因となる役割を果たす遺伝子(ドライバー)の同定である。しかしながら、多くの遺伝子は、数十年の長さのこのプロセスの間に、比較的無害な変異(パッセンジャー)を蓄積する。したがって、大半の変異遺伝子において、その変異のみに基づいて、原因となる役割をその遺伝子と決定的に結びつけることは困難である(12、15、30)。しかしながら、それらの変異の出現率およびタイプに基づいて、最も優れた候補癌遺伝子(CAN遺伝子)を分類することはできる。どの遺伝子が腫瘍形成を推進する可能性が高いかを判定するために、パッセンジャー変異率の推定が必要とされる(16、30)。
【0055】
パッセンジャー変異とドライバー変異を先験的に区別することは不可能であるため、パッセンジャー変異率を変異データから直接決定することはできない。しかしながら、大半のサイレント(同義またはS)変異が、細胞増殖に対して正の効果も負の効果ももたらさないと推測することは妥当である。本研究において観察された同義変異から、24例の癌における非同義(NS)変異のパッセンジャー比率の下界を推定することが可能である(17)。下界は、ヒト多型のHapMapデータベースにおいて観察された同義変異率とNS:S比(1.02)の積と定義した。ある種の非同義変異に対する選択は、体細胞よりも生殖系列において、よりストリンジェントであり得るため、これは過小評価である可能性が高い。上界は、(SMAD4、CDK2NA、TP53、およびKRASにおける変異を除いた後に)観察された変異の総数に基づいて決定した。これまでに公知の遺伝子中の変異以外の変異のどれもドライバーではなかったと推測されるため、これは過大評価である可能性が高い。
【0056】
体細胞変異を含む各遺伝子について、変異率低限界値および変異率高限界値、ならびにこれら2つの平均である中間率(mid-rate)を用いてパッセンジャー確率を決定した。これらのパッセンジャー確率は、その遺伝子のサイズ、そのヌクレオチド(nt)組成、ならびに膵癌中の個々のヌクレオチドおよびジヌクレオチドにおける変異の相対的出現率を考慮に入れた(表1および(17))。所与の遺伝子が増幅または欠失に関与していると思われる確率を解析するために、本発明者らは、観察したすべての増幅および欠失の全頻度が、パッセンジャー変異率に相当するという控えめな仮説を立てた。次いで、全腫瘍中の各遺伝子に影響を及ぼす実際のコピー数変化の数を、遺伝子サイズおよびSNP位置の分布を考慮に入れてシミュレートした、予想されるパッセンジャーコピー数変化の数と比較した。
【0057】
次いで、点変異、小規模の欠失もしくは挿入、ホモ接合性欠失、または増幅の合計パッセンジャー確率が低いことに基づいて、変異遺伝子のリストからCAN遺伝子を選択することができる。トップクラスのCAN遺伝子を表S7に列挙しており、これには、膵癌において重要な役割を果たすことがかねてより公知である遺伝子すべて(例えば、RAS、SMAD4、CDKN2A、およびTP53)が含まれている。これらの遺伝子における変異およびコピー数変化の同定により、本発明者らの一般的アプローチの明白な実験的確認が得られた。重要なことには、これらのCAN遺伝子には、潜在的に生物学的関心対象である他の多数の遺伝子が含まれており、その多くは、この腫瘍型において役割を果たすことがこれまで確認されていなかった。例には、転写活性化因子MLL3、TGF-β受容体TGBBR2、カドヘリンホモログCDH10、PCDH15、およびPCDH18、α-カテニンCTNNA2、ジペプチジルペプチダーゼDPP6、血管新生阻害物質BAI3、Gタンパク質共役型受容体GPR133、グアニル酸シクラーゼGUCY1A2、プロテインキナーゼPRKCG、ならびに機能が未知の遺伝子であるQ9H5F0が含まれる。これらの遺伝子は一般に、膵癌において変異していることが以前に確認されているものよりもはるかに低い頻度で変異していた。これは、従来の戦略では、頻繁に変異した遺伝子を同定できるが、膵癌において遺伝的に改変される遺伝子の大多数を同定することはできなかったという考えと矛盾しない。
【0058】
実施例7
膵臓腫瘍形成を促進する候補経路
ヒトゲノム中の全タンパク質コード遺伝子を本研究において評価したため、このデータは、遺伝的に改変された経路およびプロセスをゲノム全域レベルで調査する独特な機会を与える。本発明者らは、本研究で評価したすべてのタイプの遺伝的改変を考慮に入れて、ある経路またはプロセスがドライバー改変を含む合計確率を与える統計学的アプローチを開発した(22)。次いで、本発明者らは、十分にアノテーションされた3つのGeneGo MetaCoreデータベース:遺伝子オントロジー(GO)、古典的遺伝子経路マップ(MA)、ならびに所定の細胞プロセスおよび細胞ネットワークに関与している遺伝子(GG)を通じて定義された細胞経路またはプロセスに関与している遺伝子群にこのアプローチを適用した(31)。各遺伝子群に関して、本発明者らは、その構成要素遺伝子が、パッセンジャー比率から予測されるよりも、遺伝的改変の影響を受ける可能性が高いかどうか考察した。これらの解析は、個々の遺伝子群内での変異総数ではなく、各群内の改変遺伝子の順位付けの解析に基づいた。
【0059】
これらの解析により、統計学的に有意なだけではなく、検査した24例の癌の大多数においても改変していた経路および調節プロセスが同定された(表2および表S8)。これらには、単一の頻繁に改変された遺伝子が優勢である経路、例えば、KRASシグナル伝達およびG1/S移行の調節の経路;少数の改変遺伝子が優勢である経路、例えば、TGF-βシグナル伝達の経路;ならびに多数の異なる遺伝子が改変されている経路、例えば、インテグリンシグナル伝達、浸潤の調節、同種親和性細胞接着、および低分子量GTPアーゼ依存性シグナル伝達の経路が含まれた。
【0060】
(表2)大半の膵癌において遺伝的に改変されている中心的なシグナル伝達経路およびプロセス
*これらのシグナル伝達経路およびプロセスを規定する遺伝子セットの完全なリストならびに各遺伝子セットの統計的有意性は、表S8において提供される。
【0061】
実施例8
遺伝子発現の解析
遺伝子発現パターンは、配列決定またはコピー数解析によって検出できない後成的改変を反映し得るため、経路の解析に情報を与え得る。それらはまた、前述の改変された経路に起因する、遺伝子発現への下流の影響を指摘することもできる。膵癌のトランスクリプトームを解析するために、本発明者らは、変異解析のために使用したのと同じ24例の癌に由来するRNAに対してSAGE(serial analysis of gene expression、(32))を実施した。合成による超並列配列決定と組み合わせた場合、SAGEは、定量性および感受性が高い遺伝子発現の手段を提供する。この解析を実施するために使用される合成による配列決定のアプローチは、最近のRNA-Seq研究において使用したものと類似していた(33〜36)が、SAGEは、定量が転写物の長さに依存せず、その結果、所与の数のタグの配列から得られる情報を最大にするという利点を有する。
【0062】
本研究用の対照として、本発明者らは、組織学的に正常な膵管上皮細胞を顕微解剖した。この顕微解剖は技術的に難易度が高いが、これらの細胞は、膵癌の推定上の前駆体である。その他の対照として、本発明者らは、正常な管上皮細胞と共通な多くの特性を有することが示されている、HPVによって不死化した膵管上皮細胞(HPDE)を使用した(37、38)。これらの細胞ならびに24例の膵癌からSAGEライブラリーを調製した;平均5,737,000個のタグが各ライブラリーから得られ、ライブラリー1つ当たり平均2,268,000個のタグが、公知の転写物の配列と一致した。
【0063】
本研究で同定された、増幅された領域およびホモ接合性に欠失した領域から標的遺伝子を同定するのを助けるために、転写物解析が最初に使用された。これらの領域のごく一部しか、公知の腫瘍抑制遺伝子または癌遺伝子を含まなかったが、多くは、癌にこれまで関係があるとされていなかった複数の遺伝子を含んだ。表S5およびS6では、変異データならびに転写データを用いて、推定上の標的遺伝子がこれらの領域内で同定された。例えば、本発明者らは、増幅を含む腫瘍においてある遺伝子が発現されない場合、その遺伝子は増幅事象の標的であり得なかったと想定した。同様に、本発明者らは、欠失内の真の腫瘍抑制遺伝子は、正常膵管上皮では発現されるが、対応する癌では発現されないはずであると想定した。
【0064】
二番目に、本発明者らは、前述した中心的なシグナル伝達経路およびプロセス中の遺伝子が差次的に発現されるかどうかを判定した。遺伝的改変を含む経路およびプロセスが、実際に腫瘍形成の原因であった場合、これらの経路内の遺伝子の多くが異常に発現されることが予想され得る。この仮説を検証するために、本発明者らは、12個の中心的なシグナル伝達経路およびプロセスを構成する遺伝子セットの発現を検査した(表2および表S8)。これらの経路を構成する31個の遺伝子セットの方が、3041個の残りの遺伝子セットよりも、差次的に発現された遺伝子が著しく多かった(p<0.001)。したがって、これらの発現データは、これらのシグナル伝達経路およびプロセスが膵臓腫瘍形成に寄与することを単独で裏付ける。
【0065】
最後に、本発明者らは、癌において差次的に発現される経路ではなく、個々の遺伝子の同定を試みた。集められたデータは、これまでに任意の腫瘍型に関して得られたデジタル発現データの最大の一覧表に相当する。(正常な膵管細胞またはHPDEと比べて)24例の癌の90%超において少なくとも10倍過剰発現された遺伝子が目立って多かった(541)。これらの遺伝子が、細胞株を作製した元の原発腫瘍においても過剰発現されたかどうか判定するために、本発明者らは、5例のこのような原発腫瘍においてSAGEを実施した。これらの結果から、インサイチューでのこれら541個の遺伝子の過剰発現が確認された:これらの遺伝子は、正常管上皮細胞と比べて、平均で、細胞株においては75倍高いレベルで発現され、原発腫瘍においては88倍高いレベルで発現された。過剰発現された遺伝子の内の54個が、細胞表面で分泌または発現されると予測されるタンパク質をコードすることは注目に値した。これらの過剰発現された遺伝子は、様々な診断的アプローチおよび治療的アプローチのためのリードを提供する。
【0066】
参考文献
引用される各参考文献の開示は、本明細書に明確に組み入れられる。
参考文献および注釈
【0067】
実施例9
材料および方法
遺伝子選択
20,735個の独特な遺伝子に対応する23,781個の転写物に由来するタンパク質コードエクソンを、配列決定のための標的とした。このセットは、高度にキュレーションされたConsensus Coding Sequence(CCDS)データベース(http://www.ncbi.nlm.nih.gov/CCDS/)に由来する14,554個の転写物、Reference Sequence(RefSeq)データベース(http://www.ncbi.nlm.nih.gov/projects/RefSeq/)に由来するさらに6,019個の転写物、およびEnsemblデータベース(http://www.ensembl.org/)に由来し、完全なオープンリーディングフレームを有するさらに3,208個の転写物を含んだ。本発明者らは、Y染色体上に位置しているか、またはゲノム内で完全に(precisely)重複している遺伝子からの転写物を除外した。下記に詳述するように、20,661個の遺伝子に対応する23,219個の転写物の配列決定に成功した。
【0068】
バイオインフォマティクス情報源
Consensus Coding Sequence(リリース1)RefSeq(リリース16,2006年3月)およびEnsembl(リリース31)の遺伝子座標および配列は、UCSC Santa Cruz Genome Bioinformatics Site(http://genome.ucsc.edu)から獲得した。補足的表(Supplementary Tables)に挙げた位置は、UCSC Santa Cruz hg17、build 35.1に対応する。公知のSNPをふるい落とすために使用される一塩基多型は、HapMapプロジェクトによって確認されていたdbSNP(リリース125)に存在するものであった。BLATおよびインシリコ(In Silico)PCR(http://genome.ucsc.edu/cgi-bin/hgPcr)を用いて、ヒトゲノムおよびマウスゲノムにおいてホモロジー検索を実施した。
【0069】
プライマー設計
プライマー3ソフトウェア(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)を用いて、標的とする境界面から50bp以上離れたプライマーを作製して、300〜600bpの産生物を得た。350bpを超えるエクソンは、共通の部分があるいくつかのアンプリコンに分割した。インシリコPCRおよびBLATを用いて、独特なゲノム位置から単一のPCR産物を生じるプライマー対を選択した。複数のインシリコPCRまたはBLATヒットを与える重複領域のためのプライマー対は、標的配列と重複配列とでは最大限に異なる位置で再設計した。ユニバーサールプライマー
を、それ自体と標的領域の間に最も少ない数のモノヌクレオチドまたはジヌクレオチドの繰り返しを有するプライマーの5’末端に付加した。この研究で使用したプライマー配列は表S2に列挙する。
【0070】
腫瘍試料
浸潤性の管腺癌の異種移植片および細胞株、ならびにマッチされた正常組織または末梢血に由来するDNA試料を、以前に説明されているようにして得た(1)。Discovery Screenのために使用した24個の試料には、14個の細胞株および10個の異種移植片が含まれた。これらは、外科的に切除された17例の癌腫および本発明者らのGastrointestinal Cancer Rapid Medical Donation Program(GICRMDP)の一環として迅速な剖検を受けた7名の患者に由来した。これらの癌腫の内の22例は、膵臓の原発性管腺癌であり、2つは、膵臓内の胆管を中心とする浸潤性腺癌であった。本発明者らは以前に、これら後者の新生物が膵臓腺癌に遺伝的に類似していることを示していた。進行期癌ならびに公的に入手可能な癌腫を含むように、Discovery Screen用の癌を選択した。具体的には、Discovery Screenは、7例の転移性癌および外科的に切除された15例の後期(IIb期またはIV期)癌、ならびにATCCを通じて入手可能な3個の細胞株(Pa14CはPanc8.13であり、Pa16CはPanc10.05であり、Pa18CはPanc5.04である)を含んだ。Prevalence Screenで使用した90個の試料には、79個の異種移植片および11個の細胞株が含まれた。Prevalence Screenのための症例は、均一性を向上させるように選択した。したがって、膵臓の浸潤性管腺癌のみを含めた。浸潤性管腺癌の変種(例えば、膠様癌)および膵管内乳頭粘液性腫瘍に付随して発生する浸潤性管腺癌は除外した。試料はすべて、医療保険の相互運用性と説明責任に関する法律(Health Insurance Portability and Accountability Act)(HIPAA)に従って取得した。以前に説明されているように、腫瘍と正常物のペアがマッチしているかは、PowerPlex 2.1 System (Promega, Madison, WI)を用いて9個のSTR座位を型解析することにより確認し、試料のアイデンティティは、DiscoveryスクリーニングおよびPrevalenceスクリーニングの間を通して、HLA-A遺伝子のエクソン3を配列決定することにより検査した。PCRおよび配列決定は、(1)で説明されているようにして実施した。
【0071】
変異発見スクリーニング(Discovery Screen)
CCDS遺伝子、RefSeq遺伝子、およびEnsembl遺伝子を、24個の膵癌試料および無関係な患者の正常組織に由来する1個の対照試料において増幅させた。すべてのコード配列および隣接する4bpを、関係データベース(Microsoft SQL Server)と連結されたMutations Surveyor(Softgenetics, State College, PA)を用いて解析した。アンプリコンをさらに解析するためには、これらの腫瘍の少なくとも4分の3が、関心対照の領域中にPhredクオリティスコアが20以上である塩基を90%またはそれ以上有することを必要とした。この品質管理に合格したアンプリコンにおいて、正常試料において観察されたもの、ならびに公知の一塩基多型と同一の変異は除去した。次いで、検出された各変異の配列決定クロマトグラムを視覚的に点検して、偽陽性コール(call)をソフトウェアにより除去した。腫瘍DNAにおいてすべての推定上の変異を再増幅させ配列決定して、人為的結果(artifact)を排除した。変異が同定された同じ患者の正常組織に由来するDNAを増幅させ配列決定して、それらの変異が体細胞性であるかどうか判定した。変異が判明した場合、BLATを用いて、関連するエクソンを求めてヒトゲノムおよびマウスゲノムを検索して、推定上の変異が相同配列の増幅の結果であることを確認した。標的領域の90%に渡って同一性が90%である類似配列があった場合、追加の段階を実施した。これら2つの配列を区別するために設計されたプライマーを用いて、ヒト重複に潜在的に起因する変異を再増幅させた。新しいプライマー対を用いて観察されない変異は除外した。残りは、BLATによって同定された相同配列中に変異塩基が存在しない限りにおいて、含めた。マウス異種移植片において最初に観察された変異は、原発腫瘍由来のDNAにおいて再増幅させ、原発腫瘍中にその変異が存在する場合、またはBLATによって同定されたマウス相同配列中ではその変異が同定されない場合は、含めた。膵癌で同定された体細胞変異の数を乳癌または結腸直腸癌において同定された数と比較するために、独立群の平均値のt検定を用いた。
【0072】
変異保有率スクリーニング(Prevalence Screen)
Discovery Screenにおいて2つまたはそれ以上の腫瘍で変異している遺伝子39個のサブセットを、Prevalenceスクリーニングでの解析のために選択した。表 S2に記載するプライマーを用いて、さらに90例の膵癌のこれらの遺伝子を増幅させ、配列決定した。同じ患者90名に由来するマッチさせた正常組織を用い、Discoveryスクリーニングにおいて説明したようにして、変異解析、体細胞状態の確認および判定を実施した。
【0073】
コピー数の解析
BeadChipプラットホームを使用するIllumina社製Infinium II Whole Genome Genotyping Assayを用いて、1,072,820個の(1M)SNP座位において腫瘍試料を解析した。SNP位置はすべて、ヒトゲノム参照配列のhg18(NCBI Build 36、2006年3月)版に基づいた。遺伝子型解析アッセイ法は、50ヌクレオチドのオリゴへのハイブリダイゼーションとそれに続く2色蛍光一塩基伸長で開始する。Illumina社製BeadStationソフトウェアを用いて蛍光強度画像ファイルを処理して、各SNP位置に関して正規化した強度値(R)を求めた。各SNPについて、正規化した実験的強度値(R)を、正常試料の学習用セットに由来するそのSNPに対する強度値と比較し、log2(R(実験)/R(練習用セット))という比(「Log R Ratio(R比の対数)」と呼ぶ)として表した。
【0074】
以前に説明されている方法の改良法を用いて、SNPアレイデータを解析した(2)。ホモ接合性欠失(HD)は、Log R Ratioの値が-2以下である3つまたはそれ以上の連続的SNPと定義した。HD領域の最初のSNPおよび最後のSNPは、その後の解析のための改変の境界であるとみなした。チップ人為的結果(artifact)および潜在的なコピー数多型を排除するために、本発明者らは、コピー数多型データベースに含まれるHDすべてを除去した。3つまたはそれ以下のSNPで隔てられた隣接するホモ接合性欠失は、HDが互いから100,000 bp以内であったため、同じ欠失の一部分であるとみなした。HDの影響を受ける標的遺伝子を同定するために、本発明者らは、RefSeqデータベース、CCDSデータベース、およびEnsemblデータベース中のコードエクソンの場所を、観察したHDのゲノム座標と比較した。そのコード領域の一部分がホモ接合性欠失内に含まれる任意の遺伝子は、その欠失の影響を受けるとみなした。
【0075】
(2)で概説したように、平均Log R比が0.9以上である3つ以上のSNPを含み、少なくとも1つのSNPが1.4以上のLog R比を有する領域に基づいて、増幅物を定義した。HDと同様に、本発明者らは、複数の試料において同一の境界を有する推定上の増幅物はすべて除外した。局所的増幅物の方が、特定の標的遺伝子を同定する際に有用である可能性が高いため、第2の判定基準セットを用いて、複雑な増幅物、大きな染色体領域、またはコピー数増加を示した染色体全体を除去した。サイズが3Mbを超える増幅物および同様にサイズが3Mbを超える近隣増幅物(1Mb以内)群を複雑とみなした。10Mb領域中に4回以上の別個の増幅、または染色体1個当たり5回以上の増幅の頻度で発生した増幅物または増幅物群は、複雑であるとみなした。これらのふるい分け段階の後に残る増幅物は、局所的増幅物であるとみなされ、その後の統計学的解析に含まれる唯一の増幅物であった。増幅の影響を受けるタンパク質コード遺伝子を同定するために、本発明者らは、RefSeqデータベース、CCDSデータベース、およびEnsmblデータベース内の各遺伝子の開始位置および停止位置の場所を、観察した増幅のゲノム座標と比較した。ある遺伝子の一部分のみを含む増幅物は、機能的意義を有する可能性が低いため、本発明者らは、観察した増幅物中にそのコード領域全体が含まれた遺伝子のみを考察した。
【0076】
パッセンジャー変異率の概算
Discovery Screenで観察された同義変異から、本発明者らは、パッセンジャー率の下界を概算した。下界は、ヒト多型性のHapMapデータベースにおいて観察された同義変異率とNS:S比(1.02)の積と定義した。非同義変異に対する選択は、体細胞よりも生殖系列において、よりストリンジェントであり得るため、変異0.54個/配列決定に成功したMbと算出された比率は、過小評価である可能性が高い。以前の研究からドライバーであることが公知である最も著しく変異した遺伝子(SMAD4、CDK2NA、TP53、およびKRAS)を除外した後に、観察された非同義変異の総数/Mbから、上界を算出した。SMAD4、CDK2NA、TP53、およびKRAS以外の遺伝子における変異のいくつかはドライバーである可能性が高かったため、非同義変異1.38個/Mbという結果として得られるパッセンジャー変異率は、バックグラウンド比率の過大評価に相当する。変異0.96個/Mbという「中央」測定値は、下界比率と上界比率の平均から得た。Discovery ScreenおよびPrevalence Screenにおいて同定された体細胞変異の数およびタイプを比較するために、本発明者らは、R統計パッケージ中の関数prop.testによって実行される、2つの比率を比較するための二項検定を用いた。
【0077】
発現解析
Digital Gene Expression-Tag Profiling調製キット(Illumina, San Diego, CA)を製造業者の推奨に従って用いて、SAGEタグを作製した。手短に言えば、グアニジン(guianidine)イソチオシアナートを用いてRNAを精製し、各試料に由来する全RNA約1ugに対して、オリゴdT磁性ビーズを用いた逆転写を実施した。RNAse HニッキングおよびDNAポリメラーゼI伸張によって、第2の鎖合成を遂行した。二本鎖cDNAを制限エンドヌクレアーゼ(enonuclease)Nla IIIで消化し、Mme I制限部位を含むアダプターに連結した。Mme I消化後、第2のアダプターを連結し、アダプターを連結されたcDNA構築物を18サイクルのPCRによって増量し、85bpの断片をポリアクリルアミドゲルから精製した。リアルタイムPCRおよびGenome Analyzer System(Illumina, San Diego, CA)において配列決定されたタグを用いてライブラリーサイズを推定した。
【0078】
統計学的解析
統計学的解析の概要
統計学的解析は、ある遺伝子または生物学的に定められた遺伝子セットにおける変異が、パッセンジャー比率よりも高い潜在的な変異率を反映するという証拠を定量することに焦点を合わせた。両方の場合において、解析では、点変異に関するデータとコピー数改変(CNA)に関するデータを統合する。点変異を解析するための方法論は、(3)に記載されているものに基づくが、点変異およびCNAを統合するための方法論は(2)に基づく。以前に説明されている方法にはいくつかの改良が必要とされたため、本発明者らは、本明細書においてそれを読めば理解できる(self-contained)要約を提供する。
【0079】
CAN遺伝子の統計学的解析
ある遺伝子の変異プロファイルとは、以前に定義された25種の状況特異型の各変異の数を指す(3)。変異プロファイルに関する証拠は、実験結果をパッセンジャー遺伝子のみから構成されるゲノムに相当する参照分布と比較するEmpirical Bayes解析(4)を用いて評価する。これは、実験計画を正確に再現するように、パッセンジャー比率の変異をシミュレートすることによって得られる。具体的には、本発明者らは、各遺伝子を順に考察し、状況特異的パッセンジャー比率に等しい成功確率を有する二項分布から各型の変異の数をシミュレートする。各状況において利用可能なヌクレオチドの数は、その特定の状況および研究した試料中の遺伝子に関して成功裡に配列決定されたヌクレオチドの数である。挿入欠失(indel)以外の非同義変異を考慮する場合、本発明者らは、以前に明確にされているように、リスクがあるヌクレオチドに焦点を合わせる(3)。
【0080】
これらのシミュレートされたデータセットを用いて、本発明者らは、この研究で解析した各遺伝子のパッセンジャー確率を評価した。これらのパッセンジャー確率は、遺伝子群に関するよりはむしろ、個々の遺伝子に関する記載に相当する。各パッセンジャー確率は、尤度比の論理に関連した論理を用いて得られる:ある遺伝子がパッセンジャーである場合にその遺伝子において特定のスコアを観察する尤度を、実際のデータでそれを観察する尤度と比較する。本発明者らの解析において使用する遺伝子特異的スコアは、考察中の遺伝子に関して、変異率がパッセンジャー変異率と同じであるという帰無仮説に対するLikelihood Ratio Test(LRT)に基づいている。スコアを得るために、本発明者らは単純に、LRTをs=log(LRT)に変換する。より高いスコアが、パッセンジャー比率を上回る変異率の証拠を示す。パッセンジャー確率を評価するためのこの一般的アプローチは、EfronおよびTibshiraniによって説明されているものに従う(4)。具体的には、所与の任意のスコアsに関して、F(s)は、実験データ中のsより高いスコアを有するシミュレート遺伝子の比率を表し、F0は、シミュレートデータ中の対応する比率であり、p0は、パッセンジャー遺伝子の推定される合計比率である(下記に考察する)。シミュレーション間の変動は小さいが、それでもなお、本発明者らは、100個のデータセットを作成し照合してF0を推定した。次に、本発明者らは、FおよびF0に対応する密度関数fおよびf0を数値的に推定し、各スコアsに関して、「局所的(local)誤発見率」としても公知の比率p0・f0(s)/f(s)を算出した(4)。デフォルト設定の統計用プログラミング言語Rにおいて「密度」関数を用いて、密度推定を実施した。パッセンジャー確率の計算は、p0、すなわち真のパッセンジャーの比率の推定値に依存する。本発明者らの実施は、p0に上界を与え、したがって、パッセンジャー確率の控えめに高い推定値を提供しようとするものである。このために、本発明者らはp0=1に設定した。本発明者らははまた、最も低い値で開始し、減少する値を右側の次の値に繰り返し設定することによって、スコアと共に単調に変化することをパッセンジャー確率に強いた。本発明者らは同様に、パッセンジャー比率と共に単調に変化することもパッセンジャー確率に強いる。
【0081】
R統計学的環境においてこれらの計算を実施するためのオープンソースパッケージは、CancerMutationAnalysisと名付けられており、http://astor.som.jhmi.edu/~gp/software/CancerMutationAnalysis/cma.htmで入手可能である。本発明者らの具体的な実施の詳細な数学的説明は、(5)に提供されており、一般的な解析論点は(6)で考察されている。
【0082】
CNAの統計学的解析
増幅または欠失に関与している各遺伝子について、本発明者らはさらに、それらのパッセンジャー確率の推定を通して、それらが腫瘍形成を推進するという証拠の強さを数量化した。各場合において、本発明者らは、(3)の体細胞変異解析から得た情報を本論文中て提示するデータと統合する帰納的確率としてパッセンジャー確率を得る。点変異解析から得られたパッセンジャー確率は、演繹的確率となる。これらは、パッセンジャー変異率の3つの異なる筋書きのために利用可能であり、結果はそれぞれ別に表S3に提示する。次いで、「ドライバー」対「パッセンジャー」の尤度比を、ある遺伝子が増幅(または欠失)されていることが判明した試料の数を根拠として用いて評価した。パッセンジャー項(term)は、問題の遺伝子が、観察された頻度で増幅(または欠失)されている確率である。各試料について、本発明者らは、観察された増幅(および欠失)が問題の遺伝子を偶然に含むと考えられる確率を計算することによって開始する。増幅の場合は、入手可能な全SNPを含めることが必要であるのに対し、欠失の場合は、SNPの任意の重複で十分である。具体的には、ある特定の試料において、N SNPが型決定され、かつ、K増幅が見出され、関与するSNPの観点からそのサイズがA1〜AKである場合、G SNPを有する遺伝子がランダムに含まれると考えられ、増幅の確率は(A1-G+1)/N+....+(AK-G+1)/Nであり、欠失の確率は(A1+G-1)/N+....+(AK+G-1)/Nである。次いで、本発明者らは、それらの試料は独立しているが、同一に分布したベルヌーイ(Bernoulli)確率変数ではないと想定して、ThomasおよびTraubのアルゴリズムを用いて(7)、観察された増幅(または欠失)数の確率を計算する。帰無仮説のもとでの尤度評価への本発明者らのアプローチは、観察された欠失および増幅のすべてのみがパッセンジャーを含むと想定するため、極めて控えめである。尤度比のドライバー項(term)は、関心対照の増加(対立仮説)を反映する遺伝子特異的係数(factor)を上記の試料特異的なパッセンジャー比率に掛けた後、パッセンジャー項と同様に概算した。この増加は、その遺伝子の経験的な欠失比率と合計欠失比率との比に基づいて推定する。
【0083】
この組合せアプローチにより、増幅および欠失の独立性がおおよそ想定される。実際は、増幅された遺伝子は欠失されることができず、したがって、独立性は技術的に侵害される。しかしながら、増幅事象および欠失事象は比較的少数であるため、この想定は、本発明者らの解析の目的のために支持できる。対数目盛での尤度の調査により、事象の総数において尤度がほぼ直線状であることが示されることから、スコア付け方式としてこの想定が妥当であることが裏付けられる。
【0084】
変異遺伝子経路および変異遺伝子群の解析
次の4つのタイプのデータをMetaCoreデータベース(GeneGo, Inc., St. Joseph, MI)から入手した:経路マップ、Gene Ontology (GO)プロセス、GeneGoプロセスネットワーク、およびタンパク質間相互作用。これらのカテゴリーの23,781個の各転写物のメンバーシップ(membership)を、RefSeq識別子を用いてデータベースから取得した。GeneGo経路マップにおいて、4,175個の転写物および509個の経路に関わる22,622個の関係が確認された。Gene Ontologyプロセスの場合、12,373個の転写物および4,426個のGO群に関わる合計66,397個の対関係が確認された。GeneGoプロセスネットワークの場合、6,158個の転写物および127個のプロセスに関わる合計23,356個の対関係が確認された。各変異遺伝子の予測されるタンパク質産物もまた、MetaCoreデータベースから推測されるように、他の変異遺伝子によってコードされるタンパク質との物理的相互作用に関して評価した。
【0085】
考察する各遺伝子セットに関して、本発明者らは、セットの大きさを考慮した後に、平均より高い比率の発癌ドライバーをそれらが含むという証拠の強さを数量化した。この目的のために、本発明者らは、(変異、ホモ接合性欠失、および増幅を考慮に入れた)前述の組み合わせたパッセンジャー確率に基づいたスコアを基準として、これらの遺伝子を分類した。本発明者らは、BioconductorのLimmaパッケージを用いて実行されるように(8)、ウィルコクソン検定を用いて、そのセット中に含まれる遺伝子の順位をセット外のものの順位と比較し、次いで、αの値を0.2としたq値法により、多重性を補正した(9)。本発明者らは同様に、SAGEデータから、遺伝子セットが正常膵管細胞と比べて、差次的に発現される遺伝子を平均より高い比率で含むという証拠の強さを数量化した。組み合わされた遺伝子改変が豊富な遺伝子セットと他の遺伝子セットの発現q値を比較するために、本発明者らは、独立群の平均値のt検定を用いた。
【0086】
バイオインフォマティクス解析
バイオインフォマティクス解析の概要
本発明者らは、体細胞性ミスセンス変異をそれらがパッセンジャーである尤度に基づいて順位付けするためのスコア(LS-Mut)を計算するために、新規なバイオインフォマティクスソフトウェアパイプライン(下記に示す)を開発した。これらのスコアは、タンパク質配列、タンパク質内でのアミノ酸残基の変化および位置に由来する特性に基づいている。このパイプラインの一環として、本発明者らはまた、タンパク質構造相同性モデルに基づいた各変異の定性的アノテーションも開発した。
【0087】
変異スコア
本発明者らは、いくつかの教師付き機械学習アルゴリズムを試験して、おそらく何の変化ももたらさない多型と癌関連変異とを確実に区別すると思われるものを特定した。最も優れたアルゴリズムはランダムフォレスト(Random Forest)(11)であった。本発明者らは、並行ランダムフォレストソフトウェア(PARF)[http://www.irb.hr/en/cir/projects/info/parf]を用いて、SwissProt Variant Pages(12)から得た2,840個の癌関連変異および19,503個の多型においてこのアルゴリズムを学習させた。癌関連変異は、「癌」、「癌腫」、「肉腫」、「芽腫」、「黒色腫」、「リンパ腫」、「腺腫」、および「神経膠腫」というキーワードに関して解析することによって同定した。各変異または多型に対して、本発明者らは、58個の数値的およびカテゴリー的な特徴を計算した(下記の表を参照されたい)。学習用セットは、癌関連変異よりも約7倍多い多型を含んだため、本発明者らは、少数派クラスのウェイトを上げる(up-weight)ためにクラス別ウェイト(class weight)を使用した(癌関連変異のウェイトは5.0であり、多型のウェイトは1.0であった)。mtryパラメーターは8に設定し、フォレストサイズは500個のツリーに設定した。ランダムフォレストの近接性に基づく(proximity-based)補完アルゴリズム(13)を6回繰り返して使用して、欠けている特徴の値を埋めた。RandomForestを作るために使用した全体のパラメーター設定および全データは、要求に応じて入手可能である。
【0088】
次いで、本発明者らは、906個の異なる膵臓ミスセンス変異、および11例の結腸直腸癌において変異していないことが判明している78個の遺伝子の転写物においてランダムに作製したミスセンス変異142個からなる対照セットに、学習させたフォレストを適用した(2)。各変異に関して、58個の予測的特徴を前述したようにして計算し、学習させたフォレストを用いて、これらの変異を順位付けするための予測的スコアを計算した。具体的には、使用したスコアは、各変異に関して「多型」クラスの支持を示した(voted in favor of)ツリーの割合である。
【0089】
上位に順位付けされたCAN遺伝子の膵癌中のミスセンス変異のスコアの分布は、ランダムなミスセンス変異とは異なっていたという仮説を検証するために、本発明者らは、改良型コルモゴロフ・スミルノフ(KS)検定を適用した。この検定では、各スコアに非常に小さな乱数を加えることによって、均衡を破る。上位32個の膵臓CAN遺伝子のミスセンス変異のスコアは、対照セットの変異とは有意に異なっていることが判明した(P<0.001)。
【0090】
これらの比較に基づいて、本発明者らは、スコア£0.7を有する変異(膵癌のミスセンス変異の約17%)がパッセンジャーである見込みは少ないと推定する。この閾値は、SwissProt Variantセット(その約2%しかスコア£0.7を有さない)の中立的な多型に対するパッセンジャーの推定上の類似性に基づいている。膵癌変異スコアの閾値を定める(threshold)ために使用され得るSwissProt 変異体の不偏性スコアを計算するために、本発明者らは、22,343個の変異体を2つの群(fold)に無作為に分け、(前述したように)それぞれにおいてRandomForestを学習させた。次いで、各群の変異体を、他方の群において学習させたRandomForestを用いて採点した。
【0091】
相同性モデル
体細胞性ミスセンス変異を有することが判明したmRNA転写物のタンパク質翻訳物を、ModPipe 1.0/MODELLER 9.1相同性モデル作製ソフトウェアに入力した(13)。各変異について、本発明者らは、変異位置を含むモデルすべてを同定した。1つの変異に対して複数のモデルが作製された場合、本発明者らは、その鋳型構造物との配列同一性が最も高いモデルを選択した。結果として得られたモデルを用いて、変異位置における野生型残基の溶媒接触性をDSSPソフトウェアによって計算した(14)。トリペプチドGly-X-Gly中の各側鎖タイプの最大残基溶媒接触性で割ることによって、接触性の値を正規化した(15)。36%より大きな溶媒接触性は「露出」とみなし、9%〜35%の溶媒接触性は「中間」とみなし、9%未満のものは「埋もれている」とみなした。また、DSSPを用いて、変異位置の二次構造を計算した。本発明者らは、LigBaseデータベース(15)およびPiBaseデータベース(16)を用いて、鋳型構造体の対応する位置ではリガンドまたはドメイン境界面に近い、相同性モデル中の変異残基位置を同定した。最後に、各変異について、本発明者らは、その相同性モデル上にマッピングした変異のイメージをUCSF Chimeraによって作製した(17)。各変異に関するイメージおよび関連する情報は、http://karchinlab.org/Mutants/CAN-genes/pancreatic/Pancreatic_cancer.htmlで入手可能である。モデルの座標は、要求に応じて入手可能である。
【0092】
ランダムフォレストを学習させるために使用した56個の数値的およびカテゴリー的な特徴
【0093】
実施例9のみの参考文献
【0094】
実施例10
パーソナルゲノム配列決定の価値についてはかなりの論議がなされている(1)。ゲノム全体が配列決定された5名の個体に加えて、68名の患者のタンパク質コード遺伝子の全エクソンにおける腫瘍特異的変異が評価された(エキソミック配列決定)。これは同時に、これらの個体における生殖系列配列変異に関する情報ももたらした(2〜4)。このような情報の有用性を調査するために、本発明者らは、腫瘍DNAが(4)において配列決定されていた膵癌患者(Pa10)を評価した。この患者は、その女兄弟もこの疾患を発症していたことから明確にされるように、家族性膵癌に罹患していた。
【0095】
解析した20,661個のコード遺伝子の内で、本発明者らは、参照ヒトゲノム中に存在しない15,461個の生殖系列変異体をPa10において同定した。これらの内で、7318個は同義であり、7721個はミスセンスであり、64個はナンセンスであり、108個はスプライス部位にあり、250個は小規模な欠失または挿入(54%インフレーム)であった。過去の研究により、遺伝性素因を有する患者において発生する腫瘍は、原因である遺伝子の正常な対立遺伝子を含まないことが示されている:一方の対立遺伝子は、変異型で遺伝されており、しばしば停止コドンをもたらし、他方(野生型)の対立遺伝子は、腫瘍形成の間に体細胞変異によって不活化される。Pa10において、3つの遺伝子だけが、これらの基準を満たした:SERPINB12、RAGE、およびPALB2。これらの内で、本発明者らは、PALB2が最も優れた候補であるとみなした。その理由は、SERPINB12およびRAGE中の生殖系列停止コドンは健常個体で比較的一般的であるがPALB2中のものはそうではないこと、および生殖系列PALB2変異は以前に乳癌素因およびファンコニー貧血に関連付けられているが(5)、その機能は十分に理解されていないことであった。Pa10は、コドン58においてフレームシフトを生じる4bp(c.172〜175のTTGT)の生殖系列欠失を含んだ:Pa10において発症した膵癌はまた、エクソン10に対する古典的スプライス部位(IVS10+2)においてトランジション変異(CからT)を体細胞的に獲得していた。
【0096】
PALB2変異が他の家族性膵癌患者において発生するかどうかを判定するために、本発明者らは、家族性膵癌患者96名(その内90名が白人家系であった)のコホートにおいてこの遺伝子を配列決定した。これらの患者の内の16名が、膵癌に罹患した1名の第一度近親者を有し、80名が、この疾患に罹患した少なくとも2名のその他の親戚を有し、その内の少なくとも1名が第一度近親者であった。これらの患者96名の内の3名において切断型変異が同定され、それぞれ、異なる停止コドンを生じた(図1)。これらの家族における膵癌の平均発症年齢は66.7歳であり、PALB2変異無しの家族における平均発症年齢65.3歳と類似していた。本発明者らが、これらの家系の内の1つにおいて罹患した男兄弟の生殖系列配列を決定したところ、彼は同じ停止コドンを有していた。PALB2中の切断型変異は、癌に罹患していない個体においてはまれである;本発明者らのものと類似した民族性のコホートを用いた以前の研究において、1,084名の正常個体において一つも報告されていない(6)。PALB2停止変異を有すると本発明者らが判定したいくつかの家族は、乳癌および膵癌の両方の病歴を有していたが、乳癌は全家族で観察されたわけではなかった。これらのデータから、PALB2は、遺伝性膵癌において2番目によく変異する遺伝子であると思われる。興味深いことに、最もよく変異する遺伝子はBRCA2であり(7)、そのタンパク質産物はPALB2タンパク質の結合相手である(8)。
【0097】
要約すれば、タンパク質コード遺伝子の全面的な先入観の無い配列決定によって、本発明者らは、遺伝性疾患に関与する遺伝子を発見した。本発明者らは、このアプローチが、単一遺伝子疾患に罹患した大家族を欠く場合は困難となり得る連鎖解析のような従来の遺伝子発見法に依存しないことに注目している。本発明者らは、本明細書において説明するアプローチの変形が、疾患関連遺伝子を発見するための標準手段にすぐになるであろうと予測している。
【0098】
参考文献(実施例10のみのため)
【技術分野】
【0001】
本発明は、米国政府からの資金を用いて行われた。米国政府は、NIH助成金CA 43460、CA 57345、CA 62924、CA123483、RO1CA97075、およびCA 121113の規定に従い、本発明において一定の権利を保有する。
【0002】
発明の技術分野
本発明は、膵癌の分野に関する。特に、膵癌の診断、治療、特徴付け、モニタリング、検出、および層別化に関する。
【背景技術】
【0003】
発明の背景
世界中で213,000名の患者が2008年に膵癌を発症し、ほぼ全員がこの疾患で死亡する(1)。死亡率は非常に高く、その理由の一つは、この疾患が、既に局部的に広がっているか、または肝臓、腹膜、もしくは他の器官に転移するまで一般に検出されないことにある。この腫瘍は男性および女性に比較的等しく発症し、西洋社会で使用されている積極的治療を用いた場合でさえ、全生存率は5%未満である(2、3)。タバコの喫煙、長期に渡る慢性膵炎、およびある種の食生活にいくらか関連はあるものの、環境因子が膵臓新形成をもたらすメカニズムについてはほとんど知られていない。同様に、膵癌患者の約10%は、この疾患に対する家族性素因を有すると思われる。これらの患者のごく一部は、BRCA2、CDKN2A、LKB1、PRSS1、STK11、またはMSH2の生殖系列変異を含むが、膵癌に対する家族性素因を有する患者の圧倒的多数に関与する遺伝子は、まだ発見されていない(4)。
【0004】
膵臓腫瘍は、結腸直腸腫瘍のものとよく似た、いくつかの中間段階を経て進行すると思われる。浸潤癌に先行する非浸潤性段階は、膵臓上皮内新形成(PanIN)と呼ばれ、組織病理学的検査の際に明らかな進行性異形成を伴う(5)。これらの病変、ならびにそれらから最終的に発達する完全に浸潤性の癌腫において、いくつかの遺伝的改変が同定されている(6〜10)。改変された遺伝子には、腫瘍抑制遺伝子CDKN2A、SMAD4、およびTP53、ならびにKRAS癌遺伝子が含まれ、これらの各遺伝子は、かなりの割合の後期癌、および様々な割合の前浸潤新生物において変異していることが見出されている。これらの遺伝子の発見は、この疾患の自然史に対する独自の洞察をもたらし、より優れた診断用物質および治療物質を開発する試みにはずみをつけた(11)。
【0005】
膵癌の遺伝的要因を詳細に理解することが、当技術分野において引き続き必要とされている。膵癌に関連し、かつ重要である、その他の遺伝子および経路が引き続き必要とされている。
【発明の概要】
【0006】
本発明の1つの局面は、ある疾患に対する素因を有する個体を同定する方法である。その個体の組織に由来する鋳型核酸のタンパク質コード遺伝子の複数のエクソンにおいて配列決定反応を実施する。個体の複数のエクソンの配列を疾患に罹患していない個体の配列と比較して、その疾患に罹患していない個体には存在しない、前記個体のタンパク質コード遺伝子中の変異対立遺伝子を同定する。変異対立遺伝子の存在により、その個体がその疾患に罹患しやすいことが示唆される。
【0007】
本発明の別の局面は、遺伝性癌に関与している遺伝子を同定する方法である。少なくとも、タンパク質コード遺伝子のエクソンの鋳型核酸に基づいて配列決定反応を実施する。鋳型核酸は、家族性癌に罹患した第1のヒト個体の腫瘍に由来する。野生型対立遺伝子が存在していない、腫瘍中のタンパク質コード遺伝子を同定する。第1のヒト個体と同じ器官の家族性癌に罹患している複数のヒト個体のタンパク質コード遺伝子の鋳型核酸に基づいて配列決定反応を実施する。第1のヒト個体中の対立遺伝子と異なる、複数のヒト個体(plurality)のタンパク質コード遺伝子中の1つまたは複数の変異対立遺伝子を同定し、それによって、そのタンパク質コード遺伝子が家族性癌に対する易罹患性を与えることを確認する。
【0008】
本発明のさらに別の局面は、膵癌に対する易罹患性を判定する方法である。ある個体を、その個体の家族において見出されるPALB2遺伝子中の変異の存在に関して試験する。変異が存在する場合、その個体は膵癌を発症するリスクが高いと判定し、変異が存在しない場合、その個体のリスクは通常であると判定する。
【0009】
本発明のさらに別の局面は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブである。
【0010】
本発明のさらなる局面は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列をそれぞれが含むプローブまたはプライマーを含む、プライマーまたはプローブのキットである。
【0011】
本発明のある局面は、ある個体の膵癌に対する易罹患性を判定する方法である。その個体に由来する鋳型核酸に基づいてPALB2遺伝子配列の配列決定反応を実施する。PALB2配列中の変異を同定し、それによって、その個体は膵癌に対する易罹患性が高いと判定する。
【0012】
本発明の別の局面は、ある個体の膵癌に対する易罹患性を判定する方法である。TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む核酸プライマーまたは核酸プローブを、その個体に由来する核酸中のPALB2遺伝子配列にハイブリダイズさせる。その個体のPALB2配列中の該変異の内の1つを同定し、それによって、その個体は膵癌に対する易罹患性が高いと判定する。
【0013】
本発明の1つの態様によれば、ヒトにおいて膵癌もしくは微小残存病変または分子再発を検出または診断するための方法が提供される。試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異は、前記ヒトの正常試料と比べて確認される。この遺伝子は、表S7または表S3に列挙したものからなる群より選択される;しかし、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。体細胞変異が確認される場合、その個体は、膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定される。
【0014】
また、ヒトの膵癌を特徴付ける方法も提供される。試験試料中のCAN遺伝子変異シグネチャー(signature)は、ある遺伝子またはそのコードされたcDNAもしくはタンパク質における少なくとも1つの体細胞変異を確認することにより、前記ヒトの正常試料と比べて確認される。この遺伝子は、表S7または表S3に列挙したものからなる群より選択される;しかし、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。
【0015】
本発明の別の局面は、ヒトの膵臓腫瘍を特徴付ける方法である。表S8に示すものからなる群より選択される変異した経路は、試験試料における少なくとも1つの体細胞変異を前記ヒトの正常試料と比べて確認することにより、膵臓腫瘍において確認される。この少なくとも1つの体細胞変異は、その経路における1つまたは複数の遺伝子中にある。膵臓腫瘍は、経路中に変異を有する第1群の膵臓腫瘍に割り当てられる;この第1群は、変異を有する経路における遺伝子に対して異種性であるが、その経路に対しては同種である。
【0016】
本発明のさらなる局面は、個体において早期癌もしくは微小残存病変または分子再発を検出する方法である。表S6または表S12に示すもの(SAGEによる膵臓で過剰発現される遺伝子)より選択される遺伝子からのmRNAまたはタンパク質の発現の増大が、その個体から採取された臨床試料において検出される。この増大は、健常個体の集団と比べるか、または異なる時点に採取した同じ個体の臨床試料と比べる。臨床試料が対照と比べて高い発現を示す場合、その個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定される。
【0017】
本発明のさらに別の局面は、膵癌量をモニターするための方法である。表S6または表S12に列挙する1つまたは複数の遺伝子(SAGEによる膵臓で過剰発現される遺伝子)の臨床試料における発現が測定される。発現を測定する段階は、前記のように1回または複数回繰り返される。経時的な発現の増大、減少、または安定なレベルが確認される。
【0018】
本発明のさらに別の局面は、膵癌を検出または診断するための方法である。表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料における発現が測定される。臨床試料中の1つまたは複数の遺伝子の発現は、対照ヒトもしくは対照ヒト群またはその患者の正常組織といった対応する試料におけるそれら1つまたは複数の遺伝子の発現と比較される。対照と比べて低い発現を示す臨床試料は、膵癌を有する可能性が高いと判定される。
【0019】
さらに、膵癌量をモニターするための方法も提供される。発現は、表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料において測定される。測定する段階は、1回または複数回繰り返される。経時的な発現の増大、減少、または安定なレベルが確認される。
【0020】
また、試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異が、前記ヒトの正常試料と比べて確認される、膵癌量をモニターするための方法も、本発明によって提供される。この遺伝子は、表S7に列挙したものからなる群より選択されるが、この遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない。確認する段階は、1回または複数回繰り返される。試験試料における前記変異の経時的な増加、減少、または安定なレベルが確認される。
【0021】
本明細書を読めば当業者に明らかになると考えられるこれらおよび他の態様は、膵癌を特徴付け、治療、予後判定、診断、および層別化するための手段(tool)および方法を当技術分野に提供する。
【図面の簡単な説明】
【0022】
【図1】図1A〜1Dは、変異の構造モデルの例を示す。(図1A)プロテインキナーゼC(PKC)γ(PDBID:2UZP)のC2ドメインのX線結晶構造。Arg252は、大きな空間充填物(space-fill)として示され、Ca2+イオンは小型球体として示されている。リガンドである1,2-エタンジオールおよびピリドキサール-5’-ホスファートは、玉および棒の図として示されている。R252H変異は、PRKCGのC2ドメインの膜結合を減少させ、それによって機能に影響を及ぼし得る。(図1B)ヒトクルッペル様因子5(KLF5)(PDBID:2EBT)に由来するzf-C2H2ドメインの3つのタンデムリピートのNMR溶液構造。His389は、空間充填物として示され、Zn2+イオンは小型球体として示されている。C2H2基を含み近隣のZn2+イオンに配位結合する残基であるH393およびH397は、玉および棒の図として示され、Cys380およびCys375は玉および棒の図として示されている。389位での変異(H389N)は、亜鉛フィンガーまたは近隣の亜鉛配位部位の構造を混乱させる可能性がある。(図1C)SMAD3(ほぼ垂直方向のリボンとして示されている2つのサブユニット)およびSMAD4(水平方向のリボンとして示されている1つのサブユニット)(PDBID:1U7F)のヘテロ三量体のX線結晶構造。変異位置の内の2つに対応する残基(F260SおよびS422F、鎖A中の空間充填物として示されている)は、境界面に位置しており、Smad3-Smad3相互作用またはSmad3-Smad4相互作用を乱し得る。鎖Bにおいて、F260およびS422は、空間充填物として示されている。(図1D)ホモ二量体としてのヒトDPP6(PDBID:1XFD)の細胞外ドメインのX線結晶構造。この研究で発見された変異残基の内の2つ、すなわちT409IおよびD475N(空間充填物として示されている)は、空間的に近接しており、グリコシル化部位の内の1つであるAsn471(空間充填物として示されている)に近接している。これらの変異は、タンパク質同士の相互作用に関与していると考えられているタンパク質のβ-プロペラドメイン(残基142〜322および351〜581)中にある。A778T変異(空間充填物として示されている)は、α/βヒドロラーゼドメイン(残基127〜142および581〜849)中にあり、このタンパク質のホモ二量体領域に近接し、ホモ二量体会合を乱し得る。グリコシル化部位を有する炭水化物は、棒の図として示されている。
【図2】配列決定を通じて検出された遺伝的改変の数および24例の各癌のコピー数解析を示す。棒の下部は変異を表し、棒の中央は増幅を表し、棒の上部は欠失を表す。
【図3】図3A〜3Cは、経路および調節プロセスを示す。(図3A)大半の膵癌において構成要素遺伝子が遺伝的に改変された12個の経路およびプロセス。(図3B、図3C)2種の膵癌(Pa14CおよびPa10X)ならびにそれらにおいて変異している特定の遺伝子。(図3B)および(図3C)の円の周りの位置は、(図3A)の経路およびプロセスに対応する。Pa10XにおいてSMAD4シグナル伝達経路およびヘッジホッグ(Hedgehog)シグナル伝達経路の両方を混乱させる可能性が高いBMPR2変異によって例示されるように、いくつかの経路構成要素が重複していた。さらに、DNA損傷制御(damage control)に影響を及ぼすことが公知である変異がPa10Xにおいて観察されない(N.O.、観察されず)ことによって例示されるように、すべての膵癌において12個のプロセスおよび経路のすべてが改変されているわけではなかった。
【図4】PALB2遺伝子中の変異の位置を示す。エクソンは四角形として表し、イントロンは黒い線として表している(原寸に比例しない)。家族性乳癌患者またはファンコニー貧血患者において以前に同定された変異を遺伝子の下に示している。家族性膵癌患者において同定された生殖系列変異を遺伝子の上に示している。
【図5】補足的な表S3(Discovery Screen(発見スクリーニング)における変異)、S4(Mutation Prevalence(変異保有率スクリーニング)、S5(ホモ接合性欠失)、S6(増幅された遺伝子)、S7(CAN遺伝子)、S8(頻繁に変異している経路)、S12(SAGEによる過剰発現遺伝子)、およびS13(過剰発現された細胞外遺伝子)を示す。
【発明を実施するための形態】
【0023】
発明の詳細な説明
本発明者らは、膵臓腫瘍を徹底的に解析し、得られた解析に基づいて、新規な療法、予後因子(prognosticator)、手段、およびストラティフィアー(stratifier)を開発した。いくつかの独特なアプローチ、例えば、変異、増幅、および欠失を検出するための配列決定、ならびに発現定量化を用いて、本発明者らは、重要な遺伝子、経路、および変異を同定した。個々の膵臓腫瘍間の遺伝的異質性は大きいものの、しばしば変異している遺伝子および経路のパターンが検出された。
【0024】
体細胞変異は、個々の生物の一生の間に体細胞の特定のクローンにおいて生じる変異である。したがって、この変異は、親から受け継がれることもなく、子孫に受け渡されることもない。この変異は、他の細胞、組織、器官との差異として現れる。癌性と推測される膵臓組織における体細胞変異について試験する場合、非新生物性と思われる正常膵臓組織、もしくは血液細胞のような非膵臓試料、または罹患していない個体に由来する試料と比較することができる。
【0025】
膵臓腫瘍中で見出されている変異を表S7または表2に示している。これらの変異は、試験試料、例えば、推測される腫瘍組織試料、血液、膵管液、尿、唾液、リンパなどにおいて検出され得る。典型的には、体細胞変異は、試験試料中の配列を正常対照試料中の配列、例えば健常な膵臓組織に由来するものと比較することによって確認される。1つまたは複数の変異が、この目的のために使用され得る。患者が手術を受けた場合には、腫瘍辺縁部または残存する隣接組織中の変異の検出を用いて、微小残存病変または分子再発を検出することができる。それまでに膵癌が診断未確定であった場合、この変異は、例えば、生化学的マーカーおよび放射線医学的所見を含む研究室結果の他の物理的所見と組み合わせて、診断を助けるのに役立つ可能性がある。変異を利用して患者を層別化して、薬物または他の治療に感受性または抵抗性である患者または患者群を同定することができる。
【0026】
CAN遺伝子シグネチャーは、膵臓腫瘍を特徴付けるために確認され得る。シグネチャーは、CAN遺伝子における1つまたは複数の体細胞変異のセットである。膵臓に関するCAN遺伝子は、表S7および表2に列挙する。このようなシグネチャーを確認した後で、膵臓腫瘍を、そのシグネチャーを共有する膵臓腫瘍群に割り付けることができる。この群を用いて、予後を割り付けること、臨床試験群に割り付けること、治療計画に割り付けること、ならびに/またはさらなる特徴付けおよび研究のために割り付けることができる。臨床試験群において、そのシグネチャーを有する膵臓腫瘍および有さない膵臓腫瘍に差次的に影響を及ぼす能力について薬物を評価することができる。差次的な効果が測定されたら、そのシグネチャーを利用して、患者を投薬計画に割り付けるか、またはその薬物が有益な効果を持たないであろう患者を不必要に治療するのを回避することができる。臨床試験における薬物は、別の目的のためにこれまで公知であるか、膵癌を治療するためにこれまで公知であるか、または治療物質としてこれまで未知であるものでよい。CAN遺伝子シグネチャーは、少なくとも1個、少なくとも2個、少なくとも3個、少なくとも4個、少なくとも5個、少なくとも6個、少なくとも7個、少なくとも8個、少なくとも9個、少なくとも10個の遺伝子を含んでよい。個々のシグネチャー中の遺伝子または変異の数は、そのシグネチャー中のCAN遺伝子のアイデンティティに応じて変動し得る。
【0027】
解析された膵臓腫瘍中の変異遺伝子の解析により、経路の興味深い関与が明らかになった。いくつかの経路は、頻繁に、膵臓腫瘍において変異を有する。しばしば、単一遺伝子変異は、個々の腫瘍のその経路における別の遺伝子における変異の存在を排除する。膵臓腫瘍中の頻繁に変異している経路を、表S8および表2に列挙する。経路は、MetaCore Gene Ontology(GO)データベース、MetaCore古典的遺伝子経路マップ(MA)データベース、MetaCore GeneGo (GG)データベース、Pantherデータベース、TRMPデータベース、KEGGデータベース、およびSPADデータベースなどの標準的な参照データベースのいずれかを用いて定義することができる。群は、特定の経路における変異の存在または不在に基づいて形成させることができる。このような群は、変異遺伝子に関して異種性であるが、変異経路に関しては同種性である。CAN遺伝子シグネチャーと同様に、これらの群を利用して膵臓を特徴付けることができる。ある経路における変異を確認した後で、膵臓を、その変異経路を共有する膵臓腫瘍群に割り付けることができる。この群を用いて、予後を割り付けること、臨床試験群に割り付けること、治療計画に割り付けること、ならびに/またはさらなる特徴付けおよび研究のために割り付けることができる。臨床試験群において、その変異経路を有する膵臓腫瘍および有さない膵臓腫瘍に差次的に影響を及ぼす能力について薬物を評価することができる。差次的な効果が測定されたら、その経路を利用して、患者を投薬計画に割り付けるか、またはその薬物が有益な効果を持たないであろう患者を不必要に治療するのを回避することができる。臨床試験における薬物は、別の目的のためにこれまで公知であるか、膵臓を治療するためにこれまで公知であるか、または治療物質としてこれまで未知であるものでよい。
【0028】
発現レベルを測定することができ、過剰発現は、新たな膵臓腫瘍、膵臓の分子再発、または微小残存病変を示す可能性がある。膵臓腫瘍中で見出される著しく増大した発現を表S6および表S12に示している。これらの過剰発現された遺伝子は、試験試料、例えば、推測される腫瘍組織試料、血液、膵管液、尿、唾液、リンパなどにおいて検出され得る。典型的には、高発現は、試験試料中の遺伝子の発現を正常試料中の遺伝子、例えば健常な膵臓組織に由来するものの発現と比較することによって確認される。1つまたは複数の遺伝子の高発現が、この目的のために使用され得る。患者が手術を受けた場合には、腫瘍辺縁部または残存する隣接組織中の高発現の検出を用いて、微小残存病変または分子再発を検出することができる。それまでに膵臓が診断未確定であった場合、この高発現は、例えば、生化学的マーカーおよび放射線医学的所見を含む研究室結果の他の物理的所見と組み合わせて、診断を助けるのに役立つ可能性がある。増加したmRNAを検出するためのSAGEまたはマイクロアレイ、およびタンパク質高発現を検出するために様々なアッセイ形式で使用される抗体を含む、当技術分野において公知である発現を定量するための任意の手段が、これらの目的のために使用され得る。タンパク質発現を検出するためには、表S13に列挙した遺伝子が特に有用である。
【0029】
腫瘍量は、表S7に列挙した変異を利用してモニターすることができる。慎重な経過観察の様式で、または療法期間中にこれを利用して、例えば有効性をモニターすることができる。マーカーとしての体細胞変異の使用および検出可能なDNA、mRNA、またはタンパク質のレベルの経時的な分析により、腫瘍量を示すことができる。試料中の変異のレベルは、解析期間を通して、上昇するか、低下するか、または一定のままであり得る。治療的処置およびタイミングは、このようなモニタリングに基づいて管理することができる。
【0030】
膵臓腫瘍の解析により、ホモ接合性に欠失している遺伝子が明らかになった。これらを表S5に列挙する。1つまたは複数のこれらの遺伝子の発現喪失の測定は、膵癌のマーカーとして利用され得る。これは、血液もしくはリンパ節の試料において、または膵臓組織試料において実施され得る。1つまたは複数のこれらの遺伝子の発現を試験してよい。ELISAまたはIHCなどの技術を用いて、試料中のタンパク質発現の減少または喪失を検出することができる。同様に、表S5に列挙したホモ接合性に欠失した遺伝子(および表S6の増幅された遺伝子)を用いて、腫瘍量を経時的にモニターすることもできる。発現の増大、減少、または安定なレベルを確認できるように、発現を繰り返しモニターしてよい。
【0031】
変異およびコピー数改変のこの統合された解析の結果得られるデータは、膵臓腫瘍の遺伝的景観の異なる観点を与えた。点変異、増幅、および欠失を含む様々なタイプの遺伝的データを組み合わせることにより、個々のCAN遺伝子ならびに膵臓腫瘍の複雑な細胞経路およびプロセスにおいて優先的に影響を受ける可能性がある遺伝子群の同定が可能になる。膵臓腫瘍において変異、増幅、または欠失による影響を受けることが以前に示されているほぼすべての遺伝子の同定により、本発明者らが使用した包括的ゲノムアプローチが有効になる。
【0032】
本明細書において説明する広範囲の遺伝的研究から、膵癌を理解するための鍵は、調節プロセスおよび経路の中心的(core)セットを理解することにあることが示唆される。本発明者らは、膵癌の大多数において遺伝的に改変されている12個のこのようなプロセスを同定した(図3A)。しかしながら、任意の個々の腫瘍において改変されている経路構成要素は、多種多様である(図3B、3C)。例えば、図3Bおよび3Cに示す2種の腫瘍はそれぞれ、TGF-β経路に関与している遺伝子の変異を含む(一方はSMAD4、他方はBMPR2)。同様に、これら2種の腫瘍は両方とも、他の11個の中心的プロセス/経路の大半に関与している遺伝子の変異を含むが、各腫瘍中の改変されたこれらの遺伝子は大いに異なる。本発明者らは、同定された変異のすべてが、関係があるとされている経路またはプロセスにおいて機能的役割を果たしていることに確信は持てないが、現在公開されている遺伝的データおよび以前に公開された遺伝的データの両方から、ならびに過去の機能的研究から、それらの内の多くがこれらの経路に影響を与えている可能性が高いことは明らかである。
【0033】
この見込みは、全部ではないが大半の上皮性腫瘍に当てはまる可能性が高い。これは、遺伝的改変を山(高頻度の変異)または丘(低頻度の変異)に分類することができ、丘の方が、関与している改変の総数の点で優勢であるという考えと完全に一致している(16)。経路構成要素間の異種性および個々の遺伝子内の変異の異なる性質は、すべての固形腫瘍の基本的な側面である腫瘍異質性の説明となり得る(39)。
【0034】
知的観点から、この経路という見方は、非常に複雑な疾患に対して秩序および初歩的理解をもたらすのを助ける(40〜42)。一般的な新形成を理解する上での調節プロセスおよび経路の重要性は認識されているが(43、44)、本研究で実施されるもののようなゲノム全域に渡る遺伝的解析により、各患者の腫瘍におけるそれらの調節不全に関与する厳密な遺伝的改変を同定することができる。腫瘍病因に対する洞察をもたらすことに加えて、このような研究は、個別化された癌医薬品に基づくアプローチのために必要とされるデータを提供する。単一の標的指向性癌遺伝子によって腫瘍形成が推進されると思われる特定の型の白血病とは違って、膵癌は、比較的少数の経路およびプロセスを介して機能する多数の遺伝子の遺伝的改変に起因する。KRAS癌遺伝子はこれまでのところ、ターゲティングに成功しておらず、遍在的に改変された同様の新規な標的は明らかでないため、本発明者らの研究により、治療物質開発の最も優れた希望は、それらの個々の遺伝子構成要素ではなく、改変された経路および調節プロセスの生理的効果を標的とする作用物質の発見にあることが示唆される。これらの効果には、代謝妨害、血管新生、細胞表面タンパク質の異所性発現、細胞周期の改変、細胞骨格異常、およびゲノム損傷を修復する能力の障害が含まれる(表S8)。
【0035】
膵癌のために使用されている方法は、より広範囲の用途を有する。遺伝性疾患に関与している遺伝子を同定する方法は、他の癌および他の疾患のために使用され得る。
【0036】
膵癌に対する易罹患性に関与していると判定された1つの遺伝子は、PALB2である。PALB2中の変異は、患者の膵癌において同定される。ついで、家族を試験して、彼らもまたその変異を保有するかどうか確認することができる。家族がその変異を有する場合、その人は、膵癌を発症するリスクが高い。患者のPALB2変異が家族には存在しない場合、その人は、一般集団と同じリスクを有する。試験は、当業者に公知の任意の方法によって実施することができる。核酸プローブまたはプライマーへの家族の鋳型核酸のハイブリダイゼーションを用いて、変異を分析することができる。鋳型核酸は、例として、ゲノムまたはmRNAもしくはcDNAでよい。プローブまたはプライマーは、少なくとも14個、16個、18個、20個、22個、24個、26個、または30個の核酸塩基を含んでよい。プローブまたはプライマーは、膵臓腫瘍中に存在する変異を含む、PALB2の一部分を含んでよい。プライマーは、変異部位に隣接し、アンプリコン中の変異の増幅および解析を可能にし得る。確認され得る具体的な変異は、TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tである。
【0037】
変異特異的なPALB2プローブまたはPALB2プライマーをキット中で組み合わせてもよい。これらのキットは、分割された容器または分割されていない容器を含んでよい。キットの構成要素は、別々でもよく、または混合されてもよい。容器に加えて、キットの他の要素には、取扱い説明書、緩衝剤のような試薬、およびポリメラーゼのような酵素が含まれ得る。固体支持体、反応チューブ、ビーズなどが、キットに含まれてよい。これらのキットは、少なくとも2個、3個、または4個の異なる変異特異的試薬を含んでよい。
【0038】
上記の開示は、本発明を全体として説明する。本明細書において開示される参考文献はすべて、参照により明確に組み入れられる。より完全な理解は、以下の具体的な実施例を参照することにより得ることができ、これらの実施例は、例証のために本明細書において提供されるにすぎず、本発明の範囲を限定することを意図しない。
【実施例】
【0039】
実施例1
試料選択
任意の癌ゲノム研究と同様に、試料の選択は重大な意味を持つ。この研究のために、本発明者らは、24例の進行した腺癌を選択した。これらはそれぞれ、異なる無関係の患者に由来した(表S1)。初期の癌は1つのサブセットしか含まない可能性があるのに対し、進行した膵癌は、腫瘍の開始および進行の原因である遺伝的改変のすべてを含むと予想できるため、進行した膵癌を選択した。これら24例の癌をインビトロで細胞株として、またはヌードマウス中で異種移植片として継代して、変異の検出を容易にした。このような継代は、それらの腫瘍中に元々存在する混入非新生細胞を除去するため、サンガー配列決定またはコピー数解析のために原発腫瘍よりも優れたDNA鋳型を提供することが示されている(12)。また、細胞株および異種移植片中に存在するクローン変異は、あるとしても、エクスビボでの培養中にめったに起こらないことも実証されている(12〜14)。
【0040】
実施例2
配列決定戦略
Consensus Coding Sequence Database(リリース1)、Reference Sequence Database(リリース16)、およびEnsembl Database(リリース31)において見出されるタンパク質をコードするエクソンの配列を取り出し、ゲノムDNAを増幅するためのプライマーを設計するために使用した(図S1)。乳癌および結腸直腸癌に関する本発明者らの過去の研究において以前に設計したプライマーが好結果であると判明した場合は(15、16)、同じプライマーを使用した。以前に研究されていないエクソン11,579個ならびに以前に設計されたプライマーが不十分であると判明したエクソンに対して、新しいプライマーセットを設計した(下記を参照されたい)(17)。次いで、これらの結果として得られた各エクソンの配列を、色素ターミネーター配列決定および表S2に列挙したプライマー416,622個を用いて、24例の膵癌において決定した。変異配列を含むエクソンを、腫瘍DNAから再増幅および再配列決定して、観察された改変を確認した。すべての場合において、変異を有する患者の正常組織に由来するDNAをさらに検査した。このアプローチにより、その改変が正常細胞中に存在するか(したがって、生殖系列変異体であるか)、またはその個体の癌細胞に特異的な体細胞変異に相当するかを判定した。
【0041】
将来の医学的再配列決定プロジェクトは、次世代の合成化学反応による配列決定を使用し得るため、本研究で使用する従来の色素ターミネーター配列決定法を用いて得られる有効範囲を決定することは関心対象であった。本発明者らは、遺伝子20,735個に相当する、転写物23,962個のタンパク質コードエクソンの配列を評価することを試みた。標的配列は、各エクソンのタンパク質コード部分全て、ならびに上流の塩基4個および下流の塩基4個を含んだ。これらの領域を網羅するために、本発明者らは、アンプリコン219,229個に対するプライマーを設計し、その内の208,311個(95%)から得られたPCR産物は、首尾よく配列決定され、さらなる変異解析のための本発明者らの品質管理条件を満たした(17)。これらの品質管理されたアンプリコンは、標的とされたコード領域の94.5%を網羅し、それらのアンプリコン内部の標的塩基の98.5%に関する高品質の配列決定データをもたらした。全体で、本発明者らは、これら24名の患者において、標的とされた転写物のコード領域中の塩基の93.1%に相当する752,843,968bpを成功裡に配列決定することができた。これにより、遺伝子20,661個に相当する転写物23,219個に関する変異データが得られた。増幅のために使用されるプライマーは最低でも「第2世代」プライマーであり、失敗したプライマーは、本発明者らの研究室で以前に実施された各大規模配列決定プロジェクトの間に、新しいプライマーで置き換えられ、改善されたことに留意されたい。したがって、この93.1%という値は、色素ターミネーター技術を用いて達成できる最大値に近いことになる。さらに、配列決定できない領域の圧倒的多数は、配列決定の失敗それ自体よりはむしろ、反復されたエレメントを意味した。反復された領域は、短い読取り長(short read length)を生じる方法の場合はなおさら問題があるため、この配列包括度が次世代技術によって増加する可能性は低い。
【0042】
実施例3
体細胞変異
体細胞変異1562個の内で、25.5%は同義であり、62.4%はミスセンスであり、3.8%はナンセンスであり、5.0%は小規模の挿入および欠失であり、3.3%は、スプライス部位にあるか、またはUTR内部に存在した(表1)。体細胞変異のスペクトルは、潜在的な発癌物質および他の環境曝露への洞察をもたらし得る。表1では、タンパク質コード遺伝子の大多数の大規模配列決定解析に供された4例の腫瘍において観察されたスペクトルを記載している。乳房腫瘍は独特な体細胞変異スペクトルを有しており、5'-TpC部位における変異が優勢であり、5'-CpG部位における変異は比較的少数であることが明らかである。しかしながら、結腸直腸腫瘍、脳腫瘍(18)、および膵臓腫瘍のスペクトルは類似していることから、乳房上皮細胞が、異なるレベルもしくはタイプの発癌物質に曝露されるか、または他の腫瘍を生じる細胞とは異なる修復システムを使用することが示唆される(19、20)。膵臓および結腸のものなど胃腸管中の細胞は、乳房細胞または脳細胞よりも食事性発癌物質に多く曝露されると予想されることから、これらの結果の1つの解釈は、食事性成分は、ヒト癌中に存在する変異の大半の直接的原因ではないというものである。
【0043】
(表1)4種の腫瘍型における体細胞変異の要約
*本研究において解析した24例の腫瘍に基づく
†Parsons et al., Science, 2008 (印刷中)において解析された21例の非高変異性(nonhypermutable)腫瘍に基づく。
‡Wood et al., Science 20:1108-13 2007において解析された11例の乳房腫瘍および11例の結腸直腸腫瘍。
§括弧内の数字は、非同義変異の総数の比率(%)を示す。
**指定の研究において同定された同義変異ならびに非同義変異を含む。
††括弧内の数字は、置換の総数の比率(%)を示す。
【0044】
調査した24例の癌において、配列決定によって解析した遺伝子20,661個の内で、1327個は少なくとも1つの変異を有し、148個は2個またはそれ以上の変異を有していた(表S3)。ある遺伝子における変異出現率に加えて、変異のタイプは、疾患におけるその潜在的役割を評価するために有用な情報を提供し得る(21)。ナンセンス変異、アウトオブフレームな挿入または欠失、およびスプライス部位の変化は、一般に、タンパク質産生物の不活性化を招く。ミスセンス変異が起こしそうな影響は、進化的手段または構造的手段によって変異残基を評価することによって評価することができる。ミスセンス変異を評価するために、本発明者らは、置換に関与しているアミノ酸の物理的-化学的特性および保存されているタンパク質の等価な位置におけるそれらの進化的保存に基づく58個の予測的特徴の機械学習を使用する新規なアルゴリズムを開発した(17)。このアルゴリズムを用いて採点できるミスセンス変異926個の内で、160個(17.3%)は、この方法によって評価した場合、腫瘍形成に寄与していると予測された(表S3)。
【0045】
本発明者らはまた、本研究で同定したミスセンス変異の内の404個の構造モデルを作ることに成功した((22)で入手可能な構造モデルに関連する)。各場合において、モデルは、正常タンパク質または近縁ホモログのX線結晶解析または核磁気共鳴分光法に基づいた。この解析により、変異244個の内の55個が、ドメイン境界面またはリガンド結合部位の近くに位置しており、機能に強い影響を与える可能性が高いことが示された(図1の例)。
【0046】
タンパク質コード遺伝子すべてに関する本発明者らの解析により、個々の腫瘍中の遺伝的改変の概要の詳細な図が提供される。図2に示したように、膵癌は、腫瘍1個当たりタンパク質コード遺伝子において平均48個の体細胞変異を有していた。この数の変動は、腫瘍形成プロセスの複雑さおよび患者の様々な年齢を考慮すれば、著しく小さかった(表S1)。膵癌における体細胞変異の平均数は、乳癌または結腸直腸癌の場合よりもかなり少なく(p<0.001)、後者の2種の腫瘍型の方が少ない遺伝子を配列決定したにも関わらず、この結果であった(16)。この低い割合に関して妥当と思われる1つの説明は、膵臓腫瘍形成を開始する細胞の方が、結腸直腸癌細胞または乳癌細胞よりも少ない分裂を経たというものである。結腸直腸癌において観察される変異の大多数は、最初の(initiating)新生細胞を生じた正常幹細胞において起こった可能性が高いことが以前に示されている(14)。したがって、本発明者らのデータは、膵臓上皮細胞がまれに分裂するのに対し(23、24)、乳房上皮細胞および結腸直腸上皮細胞は頻繁に分裂する(前者はホルモン刺激の期間中、後者は一生を通じて)ことを示す観察結果と一致している。
【0047】
本発明者らはさらに、膵癌90個からなるPrevalenceスクリーニングにおいて、24例のDiscovery Screen癌の内の複数において変異してる遺伝子39個を評価した。このスクリーニングにおいて、本発明者らは、遺伝子23個において非サイレント体細胞変異255個を検出した(表S4)。Prevalenceスクリーニングにおけるこれらの遺伝子(KRAS、TP53、CDK2NA、およびSMAD4を除く)の非サイレント変異の比率は、Discovery Screenにおけるものよりも高かった(3.6 対 1.47の非サイレント変異/Mb、p<0.0001)。これら19個の遺伝子において観察された非サイレント変異の割合もまた、Discovery Screenにおいて観察されたものよりも高かった(p<0.052)。これらのデータは、Prevalenceスクリーニングにおいて試験した遺伝子の大部分(greater fraction)が腫瘍形成の間に陽性選択されたという仮説と一致している。
【0048】
実施例4
欠失
本研究の設計の重要な局面は、細胞株または異種移植片に由来するDNAの使用であった。このDNAは、真のホモ接合性欠失の確信的な検出を可能にする。これは、一番最初の原発腫瘍検査材料に由来するDNAを用いた場合は、非新生物性の間質細胞および炎症細胞の混入が原因で、非常に困難な任務である。SNPアレイデータ、デジタル核型解析(Digital Karyotyping)、およびリアルタイムPCR解析の比較を通して、本発明者らは、SNPアレイデータからこのような試料における欠失事象を確信的に同定するための確固としたアルゴリズムを以前に開発していた(25)。これらのアルゴリズムを用いて、1,069,688個のSNPに対するプローブを含むIllumina社製オリゴヌクレオチドアレイから得たデータを解析したところ、本発明者らは、変異解析のために使用された膵癌24個において198個の別個のホモ接合性欠失を検出した(表S5)。これらの欠失の平均サイズは335,000bpであった。ホモ接合性欠失のほかに、本発明者らは、染色体全体または染色体腕全体の喪失を含む、ヘテロ接合性の喪失としてしばしば明らかである単一コピー喪失を経験した多くの領域を観察した。欠失していない染色体上の遺伝子の残存コピーが変異している場合を除いては、このような大きな領域から標的遺伝子を確実に同定することは困難であるため、本発明者らは、これらの変化を追究しなかった。このような標的遺伝子は、Discovery配列決定スクリーニングの結果によって本発明者らの注目に既になっており、ホモ接合性変化として記録されていた(表S3)。
【0049】
対立遺伝子のツーヒット仮説によれば、ホモ接合性欠失の存在から、欠失した領域内に腫瘍抑制遺伝子が存在することが示唆される(26)。これらの欠失内の最も可能性が高い標的を決定するために、本発明者らは、本発明者らの新しい変異解析および発現解析の結果ならびに過去の研究からのデータを使用した。ある遺伝子が候補標的とみなされるためには、そのコード領域の一部分が、ホモ接合性欠失による影響を受けていなければならず、かつ、(i)その遺伝子は、Discovery Screenにより、異なる腫瘍中で非サイレント配列改変を含まなければならないか、または(ii)十分に考証された腫瘍抑制遺伝子でなければならないか、または(iii)裏付けとなる発現データを有していなければならなかった(下記の遺伝子発現セクションを参照されたい)。これらの基準を満たした、各ホモ接合性欠失に対する推定上の標的遺伝子を表S5に列挙する。このリストは、古典的な腫瘍抑制遺伝子であるCDKN2A(p16)、SMAD4、およびTP53、ならびにこれまでは膵臓腫瘍形成に関係があるとされていなかった様々な他の遺伝子を含む。
【0050】
SNPアレイを通じて発見されたホモ接合性欠失を確認するために、本発明者らは配列決定データを再解析した。ある遺伝子のエクソンが、腫瘍中で本当に欠失している場合、そのエクソンの増幅を試みても、配列決定情報は得られないはずである。例外無く、配列決定データによって、その結果、マイクロアレイハイブリダイゼーションを通じて同定された欠失が確認された。さらに、マイクロアレイハイブリダイゼーションにおいては明らかではなく、配列決定によって明らかにされたホモ接合性欠失が1つだけ存在した(1つの腫瘍におけるSMAD4の4つのエクソンの欠失)。
【0051】
ある腫瘍中の欠失の数は、体細胞変異の数よりも多様であり、腫瘍1個当たり平均8.2個で、2〜20個の間の範囲であった(図2)。しかしながら、各ホモ接合性欠失は、標的遺伝子ならびに欠失領域内の他のすべての遺伝子の機能を完全に抑制したのに対し、体細胞変異のごく一部しか、遺伝子の機能を改変することが予測されなかったことに、留意すべきである。平均的な膵癌において、合計約10個の遺伝子(標的および欠失内の近隣遺伝子を含む)が、ホモ接合性欠失によって腫瘍ゲノムから失われることは、このような喪失を標的とする治療戦略に想像力豊かな根拠を与える(27、28)。
【0052】
実施例5
増幅
欠失と同様に、本発明者らは、SNPアレイデータから増幅を確信的に同定するためのアルゴリズムを開発した(25)。Illumina社製アレイから得た個々の蛍光強度比の測定値、ならびにコピー数が変化する連続した領域における最小強度比、最大強度比、および平均強度比の組合せを用いて、本発明者らは、染色体全体、染色体腕、または他の大きなゲノム領域の様々な少しの(low)コピー数増加(gain)を確認した。このような大きな染色体領域から候補癌遺伝子を確実に同定することは困難であるため、本発明者らは、これらのコピー数変化をそれ以上追究しなかった。さらに、腫瘍増殖または薬物耐性を促進する十分に考証されているほぼすべての増幅は、比較的小さな増幅領域を使用する(29)。したがって、本発明者らは、異数性ではなく、真の増幅の結果であることが明らかな局所的増幅に焦点を合わせた。
【0053】
核1つにつき増幅領域の12個超のコピーが存在することを含む局所的増幅の厳密な基準を用いて(17)、本発明者らは、24例の膵癌において144個の増幅を同定した(表S6)。これらの増幅の最も可能性が高い標的を決定するために、本発明者らは、本発明者らの変異解析および発現解析の結果ならびに以前に公表したデータを再び使用した。ある遺伝子が増幅の標的とみなされるためには、そのコード領域全体が、増幅された領域中に含まれなければならず、かつ、それは、(i)Discovery Screenにより、異なる腫瘍において変異していなければならないか、または(ii)十分に考証された癌遺伝子でなければならないか、または(iii)裏付けとなる発現データを有していなければならなかった(下記の遺伝子発現セクションを参照されたい)。これらの基準を満たした、各増幅に対する推定上の標的遺伝子を表S6に列挙する。大半の膵臓腫瘍において、増幅は、ホモ接合性欠失または点変異よりも少なかった(図2)。
【0054】
実施例6
パッセンジャー変異率
癌ゲノム研究の最も重要な目標は、新生物プロセスにおいて原因となる役割を果たす遺伝子(ドライバー)の同定である。しかしながら、多くの遺伝子は、数十年の長さのこのプロセスの間に、比較的無害な変異(パッセンジャー)を蓄積する。したがって、大半の変異遺伝子において、その変異のみに基づいて、原因となる役割をその遺伝子と決定的に結びつけることは困難である(12、15、30)。しかしながら、それらの変異の出現率およびタイプに基づいて、最も優れた候補癌遺伝子(CAN遺伝子)を分類することはできる。どの遺伝子が腫瘍形成を推進する可能性が高いかを判定するために、パッセンジャー変異率の推定が必要とされる(16、30)。
【0055】
パッセンジャー変異とドライバー変異を先験的に区別することは不可能であるため、パッセンジャー変異率を変異データから直接決定することはできない。しかしながら、大半のサイレント(同義またはS)変異が、細胞増殖に対して正の効果も負の効果ももたらさないと推測することは妥当である。本研究において観察された同義変異から、24例の癌における非同義(NS)変異のパッセンジャー比率の下界を推定することが可能である(17)。下界は、ヒト多型のHapMapデータベースにおいて観察された同義変異率とNS:S比(1.02)の積と定義した。ある種の非同義変異に対する選択は、体細胞よりも生殖系列において、よりストリンジェントであり得るため、これは過小評価である可能性が高い。上界は、(SMAD4、CDK2NA、TP53、およびKRASにおける変異を除いた後に)観察された変異の総数に基づいて決定した。これまでに公知の遺伝子中の変異以外の変異のどれもドライバーではなかったと推測されるため、これは過大評価である可能性が高い。
【0056】
体細胞変異を含む各遺伝子について、変異率低限界値および変異率高限界値、ならびにこれら2つの平均である中間率(mid-rate)を用いてパッセンジャー確率を決定した。これらのパッセンジャー確率は、その遺伝子のサイズ、そのヌクレオチド(nt)組成、ならびに膵癌中の個々のヌクレオチドおよびジヌクレオチドにおける変異の相対的出現率を考慮に入れた(表1および(17))。所与の遺伝子が増幅または欠失に関与していると思われる確率を解析するために、本発明者らは、観察したすべての増幅および欠失の全頻度が、パッセンジャー変異率に相当するという控えめな仮説を立てた。次いで、全腫瘍中の各遺伝子に影響を及ぼす実際のコピー数変化の数を、遺伝子サイズおよびSNP位置の分布を考慮に入れてシミュレートした、予想されるパッセンジャーコピー数変化の数と比較した。
【0057】
次いで、点変異、小規模の欠失もしくは挿入、ホモ接合性欠失、または増幅の合計パッセンジャー確率が低いことに基づいて、変異遺伝子のリストからCAN遺伝子を選択することができる。トップクラスのCAN遺伝子を表S7に列挙しており、これには、膵癌において重要な役割を果たすことがかねてより公知である遺伝子すべて(例えば、RAS、SMAD4、CDKN2A、およびTP53)が含まれている。これらの遺伝子における変異およびコピー数変化の同定により、本発明者らの一般的アプローチの明白な実験的確認が得られた。重要なことには、これらのCAN遺伝子には、潜在的に生物学的関心対象である他の多数の遺伝子が含まれており、その多くは、この腫瘍型において役割を果たすことがこれまで確認されていなかった。例には、転写活性化因子MLL3、TGF-β受容体TGBBR2、カドヘリンホモログCDH10、PCDH15、およびPCDH18、α-カテニンCTNNA2、ジペプチジルペプチダーゼDPP6、血管新生阻害物質BAI3、Gタンパク質共役型受容体GPR133、グアニル酸シクラーゼGUCY1A2、プロテインキナーゼPRKCG、ならびに機能が未知の遺伝子であるQ9H5F0が含まれる。これらの遺伝子は一般に、膵癌において変異していることが以前に確認されているものよりもはるかに低い頻度で変異していた。これは、従来の戦略では、頻繁に変異した遺伝子を同定できるが、膵癌において遺伝的に改変される遺伝子の大多数を同定することはできなかったという考えと矛盾しない。
【0058】
実施例7
膵臓腫瘍形成を促進する候補経路
ヒトゲノム中の全タンパク質コード遺伝子を本研究において評価したため、このデータは、遺伝的に改変された経路およびプロセスをゲノム全域レベルで調査する独特な機会を与える。本発明者らは、本研究で評価したすべてのタイプの遺伝的改変を考慮に入れて、ある経路またはプロセスがドライバー改変を含む合計確率を与える統計学的アプローチを開発した(22)。次いで、本発明者らは、十分にアノテーションされた3つのGeneGo MetaCoreデータベース:遺伝子オントロジー(GO)、古典的遺伝子経路マップ(MA)、ならびに所定の細胞プロセスおよび細胞ネットワークに関与している遺伝子(GG)を通じて定義された細胞経路またはプロセスに関与している遺伝子群にこのアプローチを適用した(31)。各遺伝子群に関して、本発明者らは、その構成要素遺伝子が、パッセンジャー比率から予測されるよりも、遺伝的改変の影響を受ける可能性が高いかどうか考察した。これらの解析は、個々の遺伝子群内での変異総数ではなく、各群内の改変遺伝子の順位付けの解析に基づいた。
【0059】
これらの解析により、統計学的に有意なだけではなく、検査した24例の癌の大多数においても改変していた経路および調節プロセスが同定された(表2および表S8)。これらには、単一の頻繁に改変された遺伝子が優勢である経路、例えば、KRASシグナル伝達およびG1/S移行の調節の経路;少数の改変遺伝子が優勢である経路、例えば、TGF-βシグナル伝達の経路;ならびに多数の異なる遺伝子が改変されている経路、例えば、インテグリンシグナル伝達、浸潤の調節、同種親和性細胞接着、および低分子量GTPアーゼ依存性シグナル伝達の経路が含まれた。
【0060】
(表2)大半の膵癌において遺伝的に改変されている中心的なシグナル伝達経路およびプロセス
*これらのシグナル伝達経路およびプロセスを規定する遺伝子セットの完全なリストならびに各遺伝子セットの統計的有意性は、表S8において提供される。
【0061】
実施例8
遺伝子発現の解析
遺伝子発現パターンは、配列決定またはコピー数解析によって検出できない後成的改変を反映し得るため、経路の解析に情報を与え得る。それらはまた、前述の改変された経路に起因する、遺伝子発現への下流の影響を指摘することもできる。膵癌のトランスクリプトームを解析するために、本発明者らは、変異解析のために使用したのと同じ24例の癌に由来するRNAに対してSAGE(serial analysis of gene expression、(32))を実施した。合成による超並列配列決定と組み合わせた場合、SAGEは、定量性および感受性が高い遺伝子発現の手段を提供する。この解析を実施するために使用される合成による配列決定のアプローチは、最近のRNA-Seq研究において使用したものと類似していた(33〜36)が、SAGEは、定量が転写物の長さに依存せず、その結果、所与の数のタグの配列から得られる情報を最大にするという利点を有する。
【0062】
本研究用の対照として、本発明者らは、組織学的に正常な膵管上皮細胞を顕微解剖した。この顕微解剖は技術的に難易度が高いが、これらの細胞は、膵癌の推定上の前駆体である。その他の対照として、本発明者らは、正常な管上皮細胞と共通な多くの特性を有することが示されている、HPVによって不死化した膵管上皮細胞(HPDE)を使用した(37、38)。これらの細胞ならびに24例の膵癌からSAGEライブラリーを調製した;平均5,737,000個のタグが各ライブラリーから得られ、ライブラリー1つ当たり平均2,268,000個のタグが、公知の転写物の配列と一致した。
【0063】
本研究で同定された、増幅された領域およびホモ接合性に欠失した領域から標的遺伝子を同定するのを助けるために、転写物解析が最初に使用された。これらの領域のごく一部しか、公知の腫瘍抑制遺伝子または癌遺伝子を含まなかったが、多くは、癌にこれまで関係があるとされていなかった複数の遺伝子を含んだ。表S5およびS6では、変異データならびに転写データを用いて、推定上の標的遺伝子がこれらの領域内で同定された。例えば、本発明者らは、増幅を含む腫瘍においてある遺伝子が発現されない場合、その遺伝子は増幅事象の標的であり得なかったと想定した。同様に、本発明者らは、欠失内の真の腫瘍抑制遺伝子は、正常膵管上皮では発現されるが、対応する癌では発現されないはずであると想定した。
【0064】
二番目に、本発明者らは、前述した中心的なシグナル伝達経路およびプロセス中の遺伝子が差次的に発現されるかどうかを判定した。遺伝的改変を含む経路およびプロセスが、実際に腫瘍形成の原因であった場合、これらの経路内の遺伝子の多くが異常に発現されることが予想され得る。この仮説を検証するために、本発明者らは、12個の中心的なシグナル伝達経路およびプロセスを構成する遺伝子セットの発現を検査した(表2および表S8)。これらの経路を構成する31個の遺伝子セットの方が、3041個の残りの遺伝子セットよりも、差次的に発現された遺伝子が著しく多かった(p<0.001)。したがって、これらの発現データは、これらのシグナル伝達経路およびプロセスが膵臓腫瘍形成に寄与することを単独で裏付ける。
【0065】
最後に、本発明者らは、癌において差次的に発現される経路ではなく、個々の遺伝子の同定を試みた。集められたデータは、これまでに任意の腫瘍型に関して得られたデジタル発現データの最大の一覧表に相当する。(正常な膵管細胞またはHPDEと比べて)24例の癌の90%超において少なくとも10倍過剰発現された遺伝子が目立って多かった(541)。これらの遺伝子が、細胞株を作製した元の原発腫瘍においても過剰発現されたかどうか判定するために、本発明者らは、5例のこのような原発腫瘍においてSAGEを実施した。これらの結果から、インサイチューでのこれら541個の遺伝子の過剰発現が確認された:これらの遺伝子は、正常管上皮細胞と比べて、平均で、細胞株においては75倍高いレベルで発現され、原発腫瘍においては88倍高いレベルで発現された。過剰発現された遺伝子の内の54個が、細胞表面で分泌または発現されると予測されるタンパク質をコードすることは注目に値した。これらの過剰発現された遺伝子は、様々な診断的アプローチおよび治療的アプローチのためのリードを提供する。
【0066】
参考文献
引用される各参考文献の開示は、本明細書に明確に組み入れられる。
参考文献および注釈
【0067】
実施例9
材料および方法
遺伝子選択
20,735個の独特な遺伝子に対応する23,781個の転写物に由来するタンパク質コードエクソンを、配列決定のための標的とした。このセットは、高度にキュレーションされたConsensus Coding Sequence(CCDS)データベース(http://www.ncbi.nlm.nih.gov/CCDS/)に由来する14,554個の転写物、Reference Sequence(RefSeq)データベース(http://www.ncbi.nlm.nih.gov/projects/RefSeq/)に由来するさらに6,019個の転写物、およびEnsemblデータベース(http://www.ensembl.org/)に由来し、完全なオープンリーディングフレームを有するさらに3,208個の転写物を含んだ。本発明者らは、Y染色体上に位置しているか、またはゲノム内で完全に(precisely)重複している遺伝子からの転写物を除外した。下記に詳述するように、20,661個の遺伝子に対応する23,219個の転写物の配列決定に成功した。
【0068】
バイオインフォマティクス情報源
Consensus Coding Sequence(リリース1)RefSeq(リリース16,2006年3月)およびEnsembl(リリース31)の遺伝子座標および配列は、UCSC Santa Cruz Genome Bioinformatics Site(http://genome.ucsc.edu)から獲得した。補足的表(Supplementary Tables)に挙げた位置は、UCSC Santa Cruz hg17、build 35.1に対応する。公知のSNPをふるい落とすために使用される一塩基多型は、HapMapプロジェクトによって確認されていたdbSNP(リリース125)に存在するものであった。BLATおよびインシリコ(In Silico)PCR(http://genome.ucsc.edu/cgi-bin/hgPcr)を用いて、ヒトゲノムおよびマウスゲノムにおいてホモロジー検索を実施した。
【0069】
プライマー設計
プライマー3ソフトウェア(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)を用いて、標的とする境界面から50bp以上離れたプライマーを作製して、300〜600bpの産生物を得た。350bpを超えるエクソンは、共通の部分があるいくつかのアンプリコンに分割した。インシリコPCRおよびBLATを用いて、独特なゲノム位置から単一のPCR産物を生じるプライマー対を選択した。複数のインシリコPCRまたはBLATヒットを与える重複領域のためのプライマー対は、標的配列と重複配列とでは最大限に異なる位置で再設計した。ユニバーサールプライマー
を、それ自体と標的領域の間に最も少ない数のモノヌクレオチドまたはジヌクレオチドの繰り返しを有するプライマーの5’末端に付加した。この研究で使用したプライマー配列は表S2に列挙する。
【0070】
腫瘍試料
浸潤性の管腺癌の異種移植片および細胞株、ならびにマッチされた正常組織または末梢血に由来するDNA試料を、以前に説明されているようにして得た(1)。Discovery Screenのために使用した24個の試料には、14個の細胞株および10個の異種移植片が含まれた。これらは、外科的に切除された17例の癌腫および本発明者らのGastrointestinal Cancer Rapid Medical Donation Program(GICRMDP)の一環として迅速な剖検を受けた7名の患者に由来した。これらの癌腫の内の22例は、膵臓の原発性管腺癌であり、2つは、膵臓内の胆管を中心とする浸潤性腺癌であった。本発明者らは以前に、これら後者の新生物が膵臓腺癌に遺伝的に類似していることを示していた。進行期癌ならびに公的に入手可能な癌腫を含むように、Discovery Screen用の癌を選択した。具体的には、Discovery Screenは、7例の転移性癌および外科的に切除された15例の後期(IIb期またはIV期)癌、ならびにATCCを通じて入手可能な3個の細胞株(Pa14CはPanc8.13であり、Pa16CはPanc10.05であり、Pa18CはPanc5.04である)を含んだ。Prevalence Screenで使用した90個の試料には、79個の異種移植片および11個の細胞株が含まれた。Prevalence Screenのための症例は、均一性を向上させるように選択した。したがって、膵臓の浸潤性管腺癌のみを含めた。浸潤性管腺癌の変種(例えば、膠様癌)および膵管内乳頭粘液性腫瘍に付随して発生する浸潤性管腺癌は除外した。試料はすべて、医療保険の相互運用性と説明責任に関する法律(Health Insurance Portability and Accountability Act)(HIPAA)に従って取得した。以前に説明されているように、腫瘍と正常物のペアがマッチしているかは、PowerPlex 2.1 System (Promega, Madison, WI)を用いて9個のSTR座位を型解析することにより確認し、試料のアイデンティティは、DiscoveryスクリーニングおよびPrevalenceスクリーニングの間を通して、HLA-A遺伝子のエクソン3を配列決定することにより検査した。PCRおよび配列決定は、(1)で説明されているようにして実施した。
【0071】
変異発見スクリーニング(Discovery Screen)
CCDS遺伝子、RefSeq遺伝子、およびEnsembl遺伝子を、24個の膵癌試料および無関係な患者の正常組織に由来する1個の対照試料において増幅させた。すべてのコード配列および隣接する4bpを、関係データベース(Microsoft SQL Server)と連結されたMutations Surveyor(Softgenetics, State College, PA)を用いて解析した。アンプリコンをさらに解析するためには、これらの腫瘍の少なくとも4分の3が、関心対照の領域中にPhredクオリティスコアが20以上である塩基を90%またはそれ以上有することを必要とした。この品質管理に合格したアンプリコンにおいて、正常試料において観察されたもの、ならびに公知の一塩基多型と同一の変異は除去した。次いで、検出された各変異の配列決定クロマトグラムを視覚的に点検して、偽陽性コール(call)をソフトウェアにより除去した。腫瘍DNAにおいてすべての推定上の変異を再増幅させ配列決定して、人為的結果(artifact)を排除した。変異が同定された同じ患者の正常組織に由来するDNAを増幅させ配列決定して、それらの変異が体細胞性であるかどうか判定した。変異が判明した場合、BLATを用いて、関連するエクソンを求めてヒトゲノムおよびマウスゲノムを検索して、推定上の変異が相同配列の増幅の結果であることを確認した。標的領域の90%に渡って同一性が90%である類似配列があった場合、追加の段階を実施した。これら2つの配列を区別するために設計されたプライマーを用いて、ヒト重複に潜在的に起因する変異を再増幅させた。新しいプライマー対を用いて観察されない変異は除外した。残りは、BLATによって同定された相同配列中に変異塩基が存在しない限りにおいて、含めた。マウス異種移植片において最初に観察された変異は、原発腫瘍由来のDNAにおいて再増幅させ、原発腫瘍中にその変異が存在する場合、またはBLATによって同定されたマウス相同配列中ではその変異が同定されない場合は、含めた。膵癌で同定された体細胞変異の数を乳癌または結腸直腸癌において同定された数と比較するために、独立群の平均値のt検定を用いた。
【0072】
変異保有率スクリーニング(Prevalence Screen)
Discovery Screenにおいて2つまたはそれ以上の腫瘍で変異している遺伝子39個のサブセットを、Prevalenceスクリーニングでの解析のために選択した。表 S2に記載するプライマーを用いて、さらに90例の膵癌のこれらの遺伝子を増幅させ、配列決定した。同じ患者90名に由来するマッチさせた正常組織を用い、Discoveryスクリーニングにおいて説明したようにして、変異解析、体細胞状態の確認および判定を実施した。
【0073】
コピー数の解析
BeadChipプラットホームを使用するIllumina社製Infinium II Whole Genome Genotyping Assayを用いて、1,072,820個の(1M)SNP座位において腫瘍試料を解析した。SNP位置はすべて、ヒトゲノム参照配列のhg18(NCBI Build 36、2006年3月)版に基づいた。遺伝子型解析アッセイ法は、50ヌクレオチドのオリゴへのハイブリダイゼーションとそれに続く2色蛍光一塩基伸長で開始する。Illumina社製BeadStationソフトウェアを用いて蛍光強度画像ファイルを処理して、各SNP位置に関して正規化した強度値(R)を求めた。各SNPについて、正規化した実験的強度値(R)を、正常試料の学習用セットに由来するそのSNPに対する強度値と比較し、log2(R(実験)/R(練習用セット))という比(「Log R Ratio(R比の対数)」と呼ぶ)として表した。
【0074】
以前に説明されている方法の改良法を用いて、SNPアレイデータを解析した(2)。ホモ接合性欠失(HD)は、Log R Ratioの値が-2以下である3つまたはそれ以上の連続的SNPと定義した。HD領域の最初のSNPおよび最後のSNPは、その後の解析のための改変の境界であるとみなした。チップ人為的結果(artifact)および潜在的なコピー数多型を排除するために、本発明者らは、コピー数多型データベースに含まれるHDすべてを除去した。3つまたはそれ以下のSNPで隔てられた隣接するホモ接合性欠失は、HDが互いから100,000 bp以内であったため、同じ欠失の一部分であるとみなした。HDの影響を受ける標的遺伝子を同定するために、本発明者らは、RefSeqデータベース、CCDSデータベース、およびEnsemblデータベース中のコードエクソンの場所を、観察したHDのゲノム座標と比較した。そのコード領域の一部分がホモ接合性欠失内に含まれる任意の遺伝子は、その欠失の影響を受けるとみなした。
【0075】
(2)で概説したように、平均Log R比が0.9以上である3つ以上のSNPを含み、少なくとも1つのSNPが1.4以上のLog R比を有する領域に基づいて、増幅物を定義した。HDと同様に、本発明者らは、複数の試料において同一の境界を有する推定上の増幅物はすべて除外した。局所的増幅物の方が、特定の標的遺伝子を同定する際に有用である可能性が高いため、第2の判定基準セットを用いて、複雑な増幅物、大きな染色体領域、またはコピー数増加を示した染色体全体を除去した。サイズが3Mbを超える増幅物および同様にサイズが3Mbを超える近隣増幅物(1Mb以内)群を複雑とみなした。10Mb領域中に4回以上の別個の増幅、または染色体1個当たり5回以上の増幅の頻度で発生した増幅物または増幅物群は、複雑であるとみなした。これらのふるい分け段階の後に残る増幅物は、局所的増幅物であるとみなされ、その後の統計学的解析に含まれる唯一の増幅物であった。増幅の影響を受けるタンパク質コード遺伝子を同定するために、本発明者らは、RefSeqデータベース、CCDSデータベース、およびEnsmblデータベース内の各遺伝子の開始位置および停止位置の場所を、観察した増幅のゲノム座標と比較した。ある遺伝子の一部分のみを含む増幅物は、機能的意義を有する可能性が低いため、本発明者らは、観察した増幅物中にそのコード領域全体が含まれた遺伝子のみを考察した。
【0076】
パッセンジャー変異率の概算
Discovery Screenで観察された同義変異から、本発明者らは、パッセンジャー率の下界を概算した。下界は、ヒト多型性のHapMapデータベースにおいて観察された同義変異率とNS:S比(1.02)の積と定義した。非同義変異に対する選択は、体細胞よりも生殖系列において、よりストリンジェントであり得るため、変異0.54個/配列決定に成功したMbと算出された比率は、過小評価である可能性が高い。以前の研究からドライバーであることが公知である最も著しく変異した遺伝子(SMAD4、CDK2NA、TP53、およびKRAS)を除外した後に、観察された非同義変異の総数/Mbから、上界を算出した。SMAD4、CDK2NA、TP53、およびKRAS以外の遺伝子における変異のいくつかはドライバーである可能性が高かったため、非同義変異1.38個/Mbという結果として得られるパッセンジャー変異率は、バックグラウンド比率の過大評価に相当する。変異0.96個/Mbという「中央」測定値は、下界比率と上界比率の平均から得た。Discovery ScreenおよびPrevalence Screenにおいて同定された体細胞変異の数およびタイプを比較するために、本発明者らは、R統計パッケージ中の関数prop.testによって実行される、2つの比率を比較するための二項検定を用いた。
【0077】
発現解析
Digital Gene Expression-Tag Profiling調製キット(Illumina, San Diego, CA)を製造業者の推奨に従って用いて、SAGEタグを作製した。手短に言えば、グアニジン(guianidine)イソチオシアナートを用いてRNAを精製し、各試料に由来する全RNA約1ugに対して、オリゴdT磁性ビーズを用いた逆転写を実施した。RNAse HニッキングおよびDNAポリメラーゼI伸張によって、第2の鎖合成を遂行した。二本鎖cDNAを制限エンドヌクレアーゼ(enonuclease)Nla IIIで消化し、Mme I制限部位を含むアダプターに連結した。Mme I消化後、第2のアダプターを連結し、アダプターを連結されたcDNA構築物を18サイクルのPCRによって増量し、85bpの断片をポリアクリルアミドゲルから精製した。リアルタイムPCRおよびGenome Analyzer System(Illumina, San Diego, CA)において配列決定されたタグを用いてライブラリーサイズを推定した。
【0078】
統計学的解析
統計学的解析の概要
統計学的解析は、ある遺伝子または生物学的に定められた遺伝子セットにおける変異が、パッセンジャー比率よりも高い潜在的な変異率を反映するという証拠を定量することに焦点を合わせた。両方の場合において、解析では、点変異に関するデータとコピー数改変(CNA)に関するデータを統合する。点変異を解析するための方法論は、(3)に記載されているものに基づくが、点変異およびCNAを統合するための方法論は(2)に基づく。以前に説明されている方法にはいくつかの改良が必要とされたため、本発明者らは、本明細書においてそれを読めば理解できる(self-contained)要約を提供する。
【0079】
CAN遺伝子の統計学的解析
ある遺伝子の変異プロファイルとは、以前に定義された25種の状況特異型の各変異の数を指す(3)。変異プロファイルに関する証拠は、実験結果をパッセンジャー遺伝子のみから構成されるゲノムに相当する参照分布と比較するEmpirical Bayes解析(4)を用いて評価する。これは、実験計画を正確に再現するように、パッセンジャー比率の変異をシミュレートすることによって得られる。具体的には、本発明者らは、各遺伝子を順に考察し、状況特異的パッセンジャー比率に等しい成功確率を有する二項分布から各型の変異の数をシミュレートする。各状況において利用可能なヌクレオチドの数は、その特定の状況および研究した試料中の遺伝子に関して成功裡に配列決定されたヌクレオチドの数である。挿入欠失(indel)以外の非同義変異を考慮する場合、本発明者らは、以前に明確にされているように、リスクがあるヌクレオチドに焦点を合わせる(3)。
【0080】
これらのシミュレートされたデータセットを用いて、本発明者らは、この研究で解析した各遺伝子のパッセンジャー確率を評価した。これらのパッセンジャー確率は、遺伝子群に関するよりはむしろ、個々の遺伝子に関する記載に相当する。各パッセンジャー確率は、尤度比の論理に関連した論理を用いて得られる:ある遺伝子がパッセンジャーである場合にその遺伝子において特定のスコアを観察する尤度を、実際のデータでそれを観察する尤度と比較する。本発明者らの解析において使用する遺伝子特異的スコアは、考察中の遺伝子に関して、変異率がパッセンジャー変異率と同じであるという帰無仮説に対するLikelihood Ratio Test(LRT)に基づいている。スコアを得るために、本発明者らは単純に、LRTをs=log(LRT)に変換する。より高いスコアが、パッセンジャー比率を上回る変異率の証拠を示す。パッセンジャー確率を評価するためのこの一般的アプローチは、EfronおよびTibshiraniによって説明されているものに従う(4)。具体的には、所与の任意のスコアsに関して、F(s)は、実験データ中のsより高いスコアを有するシミュレート遺伝子の比率を表し、F0は、シミュレートデータ中の対応する比率であり、p0は、パッセンジャー遺伝子の推定される合計比率である(下記に考察する)。シミュレーション間の変動は小さいが、それでもなお、本発明者らは、100個のデータセットを作成し照合してF0を推定した。次に、本発明者らは、FおよびF0に対応する密度関数fおよびf0を数値的に推定し、各スコアsに関して、「局所的(local)誤発見率」としても公知の比率p0・f0(s)/f(s)を算出した(4)。デフォルト設定の統計用プログラミング言語Rにおいて「密度」関数を用いて、密度推定を実施した。パッセンジャー確率の計算は、p0、すなわち真のパッセンジャーの比率の推定値に依存する。本発明者らの実施は、p0に上界を与え、したがって、パッセンジャー確率の控えめに高い推定値を提供しようとするものである。このために、本発明者らはp0=1に設定した。本発明者らははまた、最も低い値で開始し、減少する値を右側の次の値に繰り返し設定することによって、スコアと共に単調に変化することをパッセンジャー確率に強いた。本発明者らは同様に、パッセンジャー比率と共に単調に変化することもパッセンジャー確率に強いる。
【0081】
R統計学的環境においてこれらの計算を実施するためのオープンソースパッケージは、CancerMutationAnalysisと名付けられており、http://astor.som.jhmi.edu/~gp/software/CancerMutationAnalysis/cma.htmで入手可能である。本発明者らの具体的な実施の詳細な数学的説明は、(5)に提供されており、一般的な解析論点は(6)で考察されている。
【0082】
CNAの統計学的解析
増幅または欠失に関与している各遺伝子について、本発明者らはさらに、それらのパッセンジャー確率の推定を通して、それらが腫瘍形成を推進するという証拠の強さを数量化した。各場合において、本発明者らは、(3)の体細胞変異解析から得た情報を本論文中て提示するデータと統合する帰納的確率としてパッセンジャー確率を得る。点変異解析から得られたパッセンジャー確率は、演繹的確率となる。これらは、パッセンジャー変異率の3つの異なる筋書きのために利用可能であり、結果はそれぞれ別に表S3に提示する。次いで、「ドライバー」対「パッセンジャー」の尤度比を、ある遺伝子が増幅(または欠失)されていることが判明した試料の数を根拠として用いて評価した。パッセンジャー項(term)は、問題の遺伝子が、観察された頻度で増幅(または欠失)されている確率である。各試料について、本発明者らは、観察された増幅(および欠失)が問題の遺伝子を偶然に含むと考えられる確率を計算することによって開始する。増幅の場合は、入手可能な全SNPを含めることが必要であるのに対し、欠失の場合は、SNPの任意の重複で十分である。具体的には、ある特定の試料において、N SNPが型決定され、かつ、K増幅が見出され、関与するSNPの観点からそのサイズがA1〜AKである場合、G SNPを有する遺伝子がランダムに含まれると考えられ、増幅の確率は(A1-G+1)/N+....+(AK-G+1)/Nであり、欠失の確率は(A1+G-1)/N+....+(AK+G-1)/Nである。次いで、本発明者らは、それらの試料は独立しているが、同一に分布したベルヌーイ(Bernoulli)確率変数ではないと想定して、ThomasおよびTraubのアルゴリズムを用いて(7)、観察された増幅(または欠失)数の確率を計算する。帰無仮説のもとでの尤度評価への本発明者らのアプローチは、観察された欠失および増幅のすべてのみがパッセンジャーを含むと想定するため、極めて控えめである。尤度比のドライバー項(term)は、関心対照の増加(対立仮説)を反映する遺伝子特異的係数(factor)を上記の試料特異的なパッセンジャー比率に掛けた後、パッセンジャー項と同様に概算した。この増加は、その遺伝子の経験的な欠失比率と合計欠失比率との比に基づいて推定する。
【0083】
この組合せアプローチにより、増幅および欠失の独立性がおおよそ想定される。実際は、増幅された遺伝子は欠失されることができず、したがって、独立性は技術的に侵害される。しかしながら、増幅事象および欠失事象は比較的少数であるため、この想定は、本発明者らの解析の目的のために支持できる。対数目盛での尤度の調査により、事象の総数において尤度がほぼ直線状であることが示されることから、スコア付け方式としてこの想定が妥当であることが裏付けられる。
【0084】
変異遺伝子経路および変異遺伝子群の解析
次の4つのタイプのデータをMetaCoreデータベース(GeneGo, Inc., St. Joseph, MI)から入手した:経路マップ、Gene Ontology (GO)プロセス、GeneGoプロセスネットワーク、およびタンパク質間相互作用。これらのカテゴリーの23,781個の各転写物のメンバーシップ(membership)を、RefSeq識別子を用いてデータベースから取得した。GeneGo経路マップにおいて、4,175個の転写物および509個の経路に関わる22,622個の関係が確認された。Gene Ontologyプロセスの場合、12,373個の転写物および4,426個のGO群に関わる合計66,397個の対関係が確認された。GeneGoプロセスネットワークの場合、6,158個の転写物および127個のプロセスに関わる合計23,356個の対関係が確認された。各変異遺伝子の予測されるタンパク質産物もまた、MetaCoreデータベースから推測されるように、他の変異遺伝子によってコードされるタンパク質との物理的相互作用に関して評価した。
【0085】
考察する各遺伝子セットに関して、本発明者らは、セットの大きさを考慮した後に、平均より高い比率の発癌ドライバーをそれらが含むという証拠の強さを数量化した。この目的のために、本発明者らは、(変異、ホモ接合性欠失、および増幅を考慮に入れた)前述の組み合わせたパッセンジャー確率に基づいたスコアを基準として、これらの遺伝子を分類した。本発明者らは、BioconductorのLimmaパッケージを用いて実行されるように(8)、ウィルコクソン検定を用いて、そのセット中に含まれる遺伝子の順位をセット外のものの順位と比較し、次いで、αの値を0.2としたq値法により、多重性を補正した(9)。本発明者らは同様に、SAGEデータから、遺伝子セットが正常膵管細胞と比べて、差次的に発現される遺伝子を平均より高い比率で含むという証拠の強さを数量化した。組み合わされた遺伝子改変が豊富な遺伝子セットと他の遺伝子セットの発現q値を比較するために、本発明者らは、独立群の平均値のt検定を用いた。
【0086】
バイオインフォマティクス解析
バイオインフォマティクス解析の概要
本発明者らは、体細胞性ミスセンス変異をそれらがパッセンジャーである尤度に基づいて順位付けするためのスコア(LS-Mut)を計算するために、新規なバイオインフォマティクスソフトウェアパイプライン(下記に示す)を開発した。これらのスコアは、タンパク質配列、タンパク質内でのアミノ酸残基の変化および位置に由来する特性に基づいている。このパイプラインの一環として、本発明者らはまた、タンパク質構造相同性モデルに基づいた各変異の定性的アノテーションも開発した。
【0087】
変異スコア
本発明者らは、いくつかの教師付き機械学習アルゴリズムを試験して、おそらく何の変化ももたらさない多型と癌関連変異とを確実に区別すると思われるものを特定した。最も優れたアルゴリズムはランダムフォレスト(Random Forest)(11)であった。本発明者らは、並行ランダムフォレストソフトウェア(PARF)[http://www.irb.hr/en/cir/projects/info/parf]を用いて、SwissProt Variant Pages(12)から得た2,840個の癌関連変異および19,503個の多型においてこのアルゴリズムを学習させた。癌関連変異は、「癌」、「癌腫」、「肉腫」、「芽腫」、「黒色腫」、「リンパ腫」、「腺腫」、および「神経膠腫」というキーワードに関して解析することによって同定した。各変異または多型に対して、本発明者らは、58個の数値的およびカテゴリー的な特徴を計算した(下記の表を参照されたい)。学習用セットは、癌関連変異よりも約7倍多い多型を含んだため、本発明者らは、少数派クラスのウェイトを上げる(up-weight)ためにクラス別ウェイト(class weight)を使用した(癌関連変異のウェイトは5.0であり、多型のウェイトは1.0であった)。mtryパラメーターは8に設定し、フォレストサイズは500個のツリーに設定した。ランダムフォレストの近接性に基づく(proximity-based)補完アルゴリズム(13)を6回繰り返して使用して、欠けている特徴の値を埋めた。RandomForestを作るために使用した全体のパラメーター設定および全データは、要求に応じて入手可能である。
【0088】
次いで、本発明者らは、906個の異なる膵臓ミスセンス変異、および11例の結腸直腸癌において変異していないことが判明している78個の遺伝子の転写物においてランダムに作製したミスセンス変異142個からなる対照セットに、学習させたフォレストを適用した(2)。各変異に関して、58個の予測的特徴を前述したようにして計算し、学習させたフォレストを用いて、これらの変異を順位付けするための予測的スコアを計算した。具体的には、使用したスコアは、各変異に関して「多型」クラスの支持を示した(voted in favor of)ツリーの割合である。
【0089】
上位に順位付けされたCAN遺伝子の膵癌中のミスセンス変異のスコアの分布は、ランダムなミスセンス変異とは異なっていたという仮説を検証するために、本発明者らは、改良型コルモゴロフ・スミルノフ(KS)検定を適用した。この検定では、各スコアに非常に小さな乱数を加えることによって、均衡を破る。上位32個の膵臓CAN遺伝子のミスセンス変異のスコアは、対照セットの変異とは有意に異なっていることが判明した(P<0.001)。
【0090】
これらの比較に基づいて、本発明者らは、スコア£0.7を有する変異(膵癌のミスセンス変異の約17%)がパッセンジャーである見込みは少ないと推定する。この閾値は、SwissProt Variantセット(その約2%しかスコア£0.7を有さない)の中立的な多型に対するパッセンジャーの推定上の類似性に基づいている。膵癌変異スコアの閾値を定める(threshold)ために使用され得るSwissProt 変異体の不偏性スコアを計算するために、本発明者らは、22,343個の変異体を2つの群(fold)に無作為に分け、(前述したように)それぞれにおいてRandomForestを学習させた。次いで、各群の変異体を、他方の群において学習させたRandomForestを用いて採点した。
【0091】
相同性モデル
体細胞性ミスセンス変異を有することが判明したmRNA転写物のタンパク質翻訳物を、ModPipe 1.0/MODELLER 9.1相同性モデル作製ソフトウェアに入力した(13)。各変異について、本発明者らは、変異位置を含むモデルすべてを同定した。1つの変異に対して複数のモデルが作製された場合、本発明者らは、その鋳型構造物との配列同一性が最も高いモデルを選択した。結果として得られたモデルを用いて、変異位置における野生型残基の溶媒接触性をDSSPソフトウェアによって計算した(14)。トリペプチドGly-X-Gly中の各側鎖タイプの最大残基溶媒接触性で割ることによって、接触性の値を正規化した(15)。36%より大きな溶媒接触性は「露出」とみなし、9%〜35%の溶媒接触性は「中間」とみなし、9%未満のものは「埋もれている」とみなした。また、DSSPを用いて、変異位置の二次構造を計算した。本発明者らは、LigBaseデータベース(15)およびPiBaseデータベース(16)を用いて、鋳型構造体の対応する位置ではリガンドまたはドメイン境界面に近い、相同性モデル中の変異残基位置を同定した。最後に、各変異について、本発明者らは、その相同性モデル上にマッピングした変異のイメージをUCSF Chimeraによって作製した(17)。各変異に関するイメージおよび関連する情報は、http://karchinlab.org/Mutants/CAN-genes/pancreatic/Pancreatic_cancer.htmlで入手可能である。モデルの座標は、要求に応じて入手可能である。
【0092】
ランダムフォレストを学習させるために使用した56個の数値的およびカテゴリー的な特徴
【0093】
実施例9のみの参考文献
【0094】
実施例10
パーソナルゲノム配列決定の価値についてはかなりの論議がなされている(1)。ゲノム全体が配列決定された5名の個体に加えて、68名の患者のタンパク質コード遺伝子の全エクソンにおける腫瘍特異的変異が評価された(エキソミック配列決定)。これは同時に、これらの個体における生殖系列配列変異に関する情報ももたらした(2〜4)。このような情報の有用性を調査するために、本発明者らは、腫瘍DNAが(4)において配列決定されていた膵癌患者(Pa10)を評価した。この患者は、その女兄弟もこの疾患を発症していたことから明確にされるように、家族性膵癌に罹患していた。
【0095】
解析した20,661個のコード遺伝子の内で、本発明者らは、参照ヒトゲノム中に存在しない15,461個の生殖系列変異体をPa10において同定した。これらの内で、7318個は同義であり、7721個はミスセンスであり、64個はナンセンスであり、108個はスプライス部位にあり、250個は小規模な欠失または挿入(54%インフレーム)であった。過去の研究により、遺伝性素因を有する患者において発生する腫瘍は、原因である遺伝子の正常な対立遺伝子を含まないことが示されている:一方の対立遺伝子は、変異型で遺伝されており、しばしば停止コドンをもたらし、他方(野生型)の対立遺伝子は、腫瘍形成の間に体細胞変異によって不活化される。Pa10において、3つの遺伝子だけが、これらの基準を満たした:SERPINB12、RAGE、およびPALB2。これらの内で、本発明者らは、PALB2が最も優れた候補であるとみなした。その理由は、SERPINB12およびRAGE中の生殖系列停止コドンは健常個体で比較的一般的であるがPALB2中のものはそうではないこと、および生殖系列PALB2変異は以前に乳癌素因およびファンコニー貧血に関連付けられているが(5)、その機能は十分に理解されていないことであった。Pa10は、コドン58においてフレームシフトを生じる4bp(c.172〜175のTTGT)の生殖系列欠失を含んだ:Pa10において発症した膵癌はまた、エクソン10に対する古典的スプライス部位(IVS10+2)においてトランジション変異(CからT)を体細胞的に獲得していた。
【0096】
PALB2変異が他の家族性膵癌患者において発生するかどうかを判定するために、本発明者らは、家族性膵癌患者96名(その内90名が白人家系であった)のコホートにおいてこの遺伝子を配列決定した。これらの患者の内の16名が、膵癌に罹患した1名の第一度近親者を有し、80名が、この疾患に罹患した少なくとも2名のその他の親戚を有し、その内の少なくとも1名が第一度近親者であった。これらの患者96名の内の3名において切断型変異が同定され、それぞれ、異なる停止コドンを生じた(図1)。これらの家族における膵癌の平均発症年齢は66.7歳であり、PALB2変異無しの家族における平均発症年齢65.3歳と類似していた。本発明者らが、これらの家系の内の1つにおいて罹患した男兄弟の生殖系列配列を決定したところ、彼は同じ停止コドンを有していた。PALB2中の切断型変異は、癌に罹患していない個体においてはまれである;本発明者らのものと類似した民族性のコホートを用いた以前の研究において、1,084名の正常個体において一つも報告されていない(6)。PALB2停止変異を有すると本発明者らが判定したいくつかの家族は、乳癌および膵癌の両方の病歴を有していたが、乳癌は全家族で観察されたわけではなかった。これらのデータから、PALB2は、遺伝性膵癌において2番目によく変異する遺伝子であると思われる。興味深いことに、最もよく変異する遺伝子はBRCA2であり(7)、そのタンパク質産物はPALB2タンパク質の結合相手である(8)。
【0097】
要約すれば、タンパク質コード遺伝子の全面的な先入観の無い配列決定によって、本発明者らは、遺伝性疾患に関与する遺伝子を発見した。本発明者らは、このアプローチが、単一遺伝子疾患に罹患した大家族を欠く場合は困難となり得る連鎖解析のような従来の遺伝子発見法に依存しないことに注目している。本発明者らは、本明細書において説明するアプローチの変形が、疾患関連遺伝子を発見するための標準手段にすぐになるであろうと予測している。
【0098】
参考文献(実施例10のみのため)
【特許請求の範囲】
【請求項1】
個体の組織に由来する鋳型核酸に基づいて、タンパク質コード遺伝子の複数のエクソンにおいて配列決定反応を実施する段階;
個体の前記複数のエクソンの配列を疾患に罹患していない個体の配列と比較して、該疾患に罹患していない個体には存在しない、前記個体のタンパク質コード遺伝子中の変異対立遺伝子を同定する段階であって、該変異対立遺伝子の存在により、該個体が該疾患に罹患しやすいことが示唆される、段階
により、ある疾患に対する素因を有する個体を同定する方法。
【請求項2】
生殖系列中に存在しかつ切断型変異を含む個体中の遺伝子の対立遺伝子を同定する段階をさらに含む、請求項1記載の方法。
【請求項3】
切断型変異がナンセンス変異である、請求項2記載の方法。
【請求項4】
切断型変異がフレームシフト変異である、請求項2記載の方法。
【請求項5】
切断型変異がスプライス部位中にある、請求項2記載の方法。
【請求項6】
1つまたは複数の変異対立遺伝子が切断型変異を含む、請求項1記載の方法。
【請求項7】
鋳型核酸が腫瘍DNAである、請求項1記載の方法。
【請求項8】
タンパク質コード遺伝子の全エクソンにおいて配列決定反応を実施する、請求項1記載の方法。
【請求項9】
タンパク質コード遺伝子の不偏性の試料(unbiased sample)において配列決定反応を実施する、請求項1記載の方法。
【請求項10】
連鎖解析無しで配列決定反応を実施する、請求項1記載の方法。
【請求項11】
野生型対立遺伝子が存在しないかどうか、およびタンパク質コード遺伝子の残りの対立遺伝子の内の少なくとも1つが切断型変異を含むかどうか、タンパク質コード遺伝子を確認する、請求項1記載の方法。
【請求項12】
疾患素因が癌である、請求項1記載の方法。
【請求項13】
疾患素因が、少なくとも1名の罹患した第一度近親者を有する罹患個体によって定義される家族性癌である、請求項1記載の方法。
【請求項14】
以下の段階を含む、遺伝性癌に関与している遺伝子を同定する方法:
少なくともタンパク質コード遺伝子のエクソンの鋳型核酸に基づいて配列決定反応を実施する段階であって、該鋳型核酸が、家族性癌に罹患した第1のヒト個体の腫瘍に由来する、段階;
野生型対立遺伝子が存在していない、腫瘍中のタンパク質コード遺伝子を同定する段階;
前記第1のヒト個体と同じ器官の家族性癌に罹患している複数のヒト個体のタンパク質コード遺伝子の鋳型核酸に基づいて配列決定反応を実施する段階;
前記第1のヒト個体中の対立遺伝子と異なる、前記複数のヒト個体のタンパク質コード遺伝子中の1つまたは複数の変異対立遺伝子を同定し、それによって、該タンパク質コード遺伝子が前記家族性癌に対する易罹患性を与えることを確認する段階。
【請求項15】
以下の段階を含む、膵癌に対する易罹患性を判定する方法:
ある個体を、前記個体の家族において見出されるPALB2遺伝子中の変異の存在に関して試験する段階;
前記変異が存在する場合に、前記個体は膵癌を発症するリスクが高いと判定し、前記変異が存在しない場合に、前記個体のリスクは通常であると判定する段階。
【請求項16】
前記試験する段階が、PALB2遺伝子またはPALB2転写物への核酸プローブまたは核酸プライマーのハイブリダイゼーションの段階を含む、請求項15記載の方法。
【請求項17】
TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブ。
【請求項18】
請求項17記載の4種類のプローブまたはプライマーを含む、プライマーまたはプローブのキット。
【請求項19】
以下の段階を含む、ある個体の膵癌に対する易罹患性を判定する方法:
前記個体に由来する鋳型核酸に基づいてPALB2遺伝子配列の配列決定反応を実施する段階;
前記PALB2配列中の変異を同定し、それによって、前記個体は膵癌に対する易罹患性が高いと判定する段階。
【請求項20】
変異が切断型変異である、請求項19記載の方法。
【請求項21】
以下の段階を含む、ある個体の膵癌に対する易罹患性を判定する方法:
TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブを、前記個体に由来する核酸上のPALB2遺伝子配列にハイブリダイズさせる段階;
前記PALB2配列中の前記変異の内の1つを同定し、それによって、前記個体は膵癌に対する易罹患性が高いと判定する段階。
【請求項22】
以下の段階を含む、ヒトにおいて膵癌もしくは微小残存病変または分子再発を検出または診断する方法:
試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異を、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7または表S3に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階;
体細胞変異が確認される場合に、前記個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定する段階。
【請求項23】
試験試料が、膵臓組織試料、推測される膵癌転移、血漿、血清、全血、膵管液、便、および息である、請求項22記載の方法。
【請求項24】
正常試料が膵臓組織試料である、請求項22記載の方法。
【請求項25】
以下の段階を含む、ヒトの膵癌を特徴付ける方法:
試験試料中のCAN遺伝子変異シグネチャーを、ある遺伝子またはそのコードされたcDNAもしくはタンパク質における少なくとも1つの体細胞変異を確認することによって、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7または表S3に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階。
【請求項26】
以下の段階をさらに含む、請求項4記載の方法:
CAN遺伝子変異シグネチャーを有する第1の膵癌群を形成する段階;
前記第1の群に対する候補または公知の抗癌治療物質の有効性を、異なるCAN遺伝子変異シグネチャーを有する第2の膵癌群に対する有効性と比較する段階;
前記候補または公知の抗癌治療物質の、他の群と比べて高いまたは低い有効性と相互関係があるCAN遺伝子変異シグネチャーを同定する段階。
【請求項27】
試験試料が膵臓組織試料である、請求項25記載の方法。
【請求項28】
正常試料が膵臓組織試料である、請求項25記載の方法。
【請求項29】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも2個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項30】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも3個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項31】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも4個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項32】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも5個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項33】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも6個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項34】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも7個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項35】
以下の段階を含む、ヒトの膵臓腫瘍を特徴付ける方法:
試験試料における少なくとも1つの体細胞変異を前記ヒトの正常試料と比べて確認することにより、膵臓腫瘍における表S8に示すものからなる群より選択される変異した経路を確認する段階であって、該少なくとも1つの体細胞変異が、該経路における1つまたは複数の遺伝子中にある、段階;
前記膵臓腫瘍を、前記経路において変異を有する第1群の膵臓腫瘍に割り当てる段階であって、該第1群が、変異を有する該経路における遺伝子に対して異種性である、段階。
【請求項36】
経路が、表2に示すものより選択される、請求項35記載の方法。
【請求項37】
以下の段階をさらに含む、請求項35記載の方法:
前記第1の群に対する候補または公知の抗癌治療物質の有効性を、第1の経路において変異を有さない第2の膵臓腫瘍群に対する有効性と比較する段階;
前記第1の経路が、前記候補または公知の抗癌治療物質の、他の群と比べて高いまたは低い有効性との相互関係を有する場合に、該第1の経路は層別化に有用であると判定する段階。
【請求項38】
経路が、表2に示すものより選択される、請求項37記載の方法。
【請求項39】
以下の段階を含む、個体において早期癌もしくは微小残存病変または分子再発を検出する方法:
表S6または表S12に示すもの(SAGEによる膵臓で過剰発現される遺伝子)より選択される遺伝子からのmRNAまたはタンパク質の発現の増大を、前記個体から採取された臨床試料において検出する段階であって、該増大が、健常個体の集団と比べられるか、または異なる時点に採取した同じ個体の臨床試料と比べられる、段階;
前記臨床試料が対照と比べて高い発現を示す場合に、前記個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定する段階。
【請求項40】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項39記載の方法。
【請求項41】
1つまたは複数の遺伝子のmRNA発現を測定する、請求項39記載の方法。
【請求項42】
1つまたは複数の遺伝子が、表S13に列挙したもの(SAGEによって発見された細胞外タンパク質)より選択される、請求項41記載の方法。
【請求項43】
以下の段階を含む、膵癌量をモニターするための方法:
表S6または表S12に列挙する1つまたは複数の遺伝子(SAGEによる膵臓で過剰発現される遺伝子)の臨床試料における発現を測定する段階;
前記測定する段階を1回または複数回繰り返す段階;および
経時的な発現の増大、減少、または安定なレベルを確認する段階。
【請求項44】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項43記載の方法。
【請求項45】
1つまたは複数の遺伝子のmRNA発現を測定する、請求項43記載の方法。
【請求項46】
1つまたは複数の遺伝子が、表S13に列挙したもの(SAGEによって発見された細胞外タンパク質)より選択される、請求項43記載の方法。
【請求項47】
検査材料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項39または43記載の方法。
【請求項48】
以下の段階を含む、ヒトの膵癌を検出または診断するための方法:
表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)のヒト臨床試料における発現を測定する段階;
前記臨床試料中の前記1つまたは複数の遺伝子の発現を、対照ヒトもしくは対照ヒト群または該ヒトの対照組織の対応する試料における前記1つまたは複数の遺伝子の発現と比較する段階;
対照と比べて低い発現を示す臨床試料は膵癌を有する可能性が高いと判定する段階。
【請求項49】
臨床試料が血液試料またはリンパ節試料である、請求項48記載の方法。
【請求項50】
臨床試料が膵臓組織試料である、請求項48記載の方法。
【請求項51】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項48記載の方法。
【請求項52】
以下の段階を含む、膵癌量をモニターするための方法:
表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料における発現を測定する段階;
前記測定する段階を1回または複数回繰り返す段階;および
経時的な発現の増大、減少、または安定なレベルを確認する段階。
【請求項53】
臨床試料が血液試料またはリンパ節試料である、請求項52記載の方法。
【請求項54】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項52記載の方法。
【請求項55】
検査材料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項48または52記載の方法。
【請求項56】
以下の段階を含む、膵癌量をモニターするための方法:
試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異を、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階;
前記確認する段階を1回または複数回繰り返す段階;および
前記試験試料における前記変異の経時的な増加、減少、または安定なレベルを確認する段階。
【請求項57】
複数の遺伝子の体細胞変異を確認する、請求項56記載の方法。
【請求項58】
試験試料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項56記載の方法。
【請求項59】
膵癌と診断されるか、または膵癌の家族歴を有する患者から得られた試料において、PALB2遺伝子の変異を検出する段階を含む、方法。
【請求項60】
前記検出する段階が、試料中のゲノムDNAを解析する段階を含む、請求項59記載の方法。
【請求項61】
前記検出する段階が、試料中のPALB2タンパク質を解析する段階を含む、請求項59記載の方法。
【請求項62】
以下の段階を含む、患者の膵癌に対する素因を診断する方法:
ある個体から得られた非腫瘍試料において、PALB2遺伝子の生殖系列変異を検出する段階であって、該生殖系列変異の存在により、膵癌に対する素因が示唆される、段階。
【請求項63】
前記検出する段階が、試料中のゲノムDNAを解析する段階を含む、請求項62記載の方法。
【請求項64】
前記検出する段階が、試料中のPALB2タンパク質を解析する段階を含む、請求項62記載の方法。
【請求項65】
個体が、膵癌に罹患していると診断されるか、もしくは推測されるか、または膵癌の家族歴を有する、請求項62記載の方法。
【請求項1】
個体の組織に由来する鋳型核酸に基づいて、タンパク質コード遺伝子の複数のエクソンにおいて配列決定反応を実施する段階;
個体の前記複数のエクソンの配列を疾患に罹患していない個体の配列と比較して、該疾患に罹患していない個体には存在しない、前記個体のタンパク質コード遺伝子中の変異対立遺伝子を同定する段階であって、該変異対立遺伝子の存在により、該個体が該疾患に罹患しやすいことが示唆される、段階
により、ある疾患に対する素因を有する個体を同定する方法。
【請求項2】
生殖系列中に存在しかつ切断型変異を含む個体中の遺伝子の対立遺伝子を同定する段階をさらに含む、請求項1記載の方法。
【請求項3】
切断型変異がナンセンス変異である、請求項2記載の方法。
【請求項4】
切断型変異がフレームシフト変異である、請求項2記載の方法。
【請求項5】
切断型変異がスプライス部位中にある、請求項2記載の方法。
【請求項6】
1つまたは複数の変異対立遺伝子が切断型変異を含む、請求項1記載の方法。
【請求項7】
鋳型核酸が腫瘍DNAである、請求項1記載の方法。
【請求項8】
タンパク質コード遺伝子の全エクソンにおいて配列決定反応を実施する、請求項1記載の方法。
【請求項9】
タンパク質コード遺伝子の不偏性の試料(unbiased sample)において配列決定反応を実施する、請求項1記載の方法。
【請求項10】
連鎖解析無しで配列決定反応を実施する、請求項1記載の方法。
【請求項11】
野生型対立遺伝子が存在しないかどうか、およびタンパク質コード遺伝子の残りの対立遺伝子の内の少なくとも1つが切断型変異を含むかどうか、タンパク質コード遺伝子を確認する、請求項1記載の方法。
【請求項12】
疾患素因が癌である、請求項1記載の方法。
【請求項13】
疾患素因が、少なくとも1名の罹患した第一度近親者を有する罹患個体によって定義される家族性癌である、請求項1記載の方法。
【請求項14】
以下の段階を含む、遺伝性癌に関与している遺伝子を同定する方法:
少なくともタンパク質コード遺伝子のエクソンの鋳型核酸に基づいて配列決定反応を実施する段階であって、該鋳型核酸が、家族性癌に罹患した第1のヒト個体の腫瘍に由来する、段階;
野生型対立遺伝子が存在していない、腫瘍中のタンパク質コード遺伝子を同定する段階;
前記第1のヒト個体と同じ器官の家族性癌に罹患している複数のヒト個体のタンパク質コード遺伝子の鋳型核酸に基づいて配列決定反応を実施する段階;
前記第1のヒト個体中の対立遺伝子と異なる、前記複数のヒト個体のタンパク質コード遺伝子中の1つまたは複数の変異対立遺伝子を同定し、それによって、該タンパク質コード遺伝子が前記家族性癌に対する易罹患性を与えることを確認する段階。
【請求項15】
以下の段階を含む、膵癌に対する易罹患性を判定する方法:
ある個体を、前記個体の家族において見出されるPALB2遺伝子中の変異の存在に関して試験する段階;
前記変異が存在する場合に、前記個体は膵癌を発症するリスクが高いと判定し、前記変異が存在しない場合に、前記個体のリスクは通常であると判定する段階。
【請求項16】
前記試験する段階が、PALB2遺伝子またはPALB2転写物への核酸プローブまたは核酸プライマーのハイブリダイゼーションの段階を含む、請求項15記載の方法。
【請求項17】
TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブ。
【請求項18】
請求項17記載の4種類のプローブまたはプライマーを含む、プライマーまたはプローブのキット。
【請求項19】
以下の段階を含む、ある個体の膵癌に対する易罹患性を判定する方法:
前記個体に由来する鋳型核酸に基づいてPALB2遺伝子配列の配列決定反応を実施する段階;
前記PALB2配列中の変異を同定し、それによって、前記個体は膵癌に対する易罹患性が高いと判定する段階。
【請求項20】
変異が切断型変異である、請求項19記載の方法。
【請求項21】
以下の段階を含む、ある個体の膵癌に対する易罹患性を判定する方法:
TTGT 172〜175の欠失、IVS5-1におけるG>T、3116位におけるAの欠失、および3256位におけるC>Tからなる群より選択される変異を含む少なくとも18ヌクレオチドのPALB2配列を含む、核酸プライマーまたは核酸プローブを、前記個体に由来する核酸上のPALB2遺伝子配列にハイブリダイズさせる段階;
前記PALB2配列中の前記変異の内の1つを同定し、それによって、前記個体は膵癌に対する易罹患性が高いと判定する段階。
【請求項22】
以下の段階を含む、ヒトにおいて膵癌もしくは微小残存病変または分子再発を検出または診断する方法:
試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異を、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7または表S3に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階;
体細胞変異が確認される場合に、前記個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定する段階。
【請求項23】
試験試料が、膵臓組織試料、推測される膵癌転移、血漿、血清、全血、膵管液、便、および息である、請求項22記載の方法。
【請求項24】
正常試料が膵臓組織試料である、請求項22記載の方法。
【請求項25】
以下の段階を含む、ヒトの膵癌を特徴付ける方法:
試験試料中のCAN遺伝子変異シグネチャーを、ある遺伝子またはそのコードされたcDNAもしくはタンパク質における少なくとも1つの体細胞変異を確認することによって、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7または表S3に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階。
【請求項26】
以下の段階をさらに含む、請求項4記載の方法:
CAN遺伝子変異シグネチャーを有する第1の膵癌群を形成する段階;
前記第1の群に対する候補または公知の抗癌治療物質の有効性を、異なるCAN遺伝子変異シグネチャーを有する第2の膵癌群に対する有効性と比較する段階;
前記候補または公知の抗癌治療物質の、他の群と比べて高いまたは低い有効性と相互関係があるCAN遺伝子変異シグネチャーを同定する段階。
【請求項27】
試験試料が膵臓組織試料である、請求項25記載の方法。
【請求項28】
正常試料が膵臓組織試料である、請求項25記載の方法。
【請求項29】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも2個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項30】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも3個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項31】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも4個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項32】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも5個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項33】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも6個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項34】
CAN遺伝子変異シグネチャーが、表S7または表S3より選択される少なくとも7個の遺伝子を含み、該遺伝子が、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、請求項25記載の方法。
【請求項35】
以下の段階を含む、ヒトの膵臓腫瘍を特徴付ける方法:
試験試料における少なくとも1つの体細胞変異を前記ヒトの正常試料と比べて確認することにより、膵臓腫瘍における表S8に示すものからなる群より選択される変異した経路を確認する段階であって、該少なくとも1つの体細胞変異が、該経路における1つまたは複数の遺伝子中にある、段階;
前記膵臓腫瘍を、前記経路において変異を有する第1群の膵臓腫瘍に割り当てる段階であって、該第1群が、変異を有する該経路における遺伝子に対して異種性である、段階。
【請求項36】
経路が、表2に示すものより選択される、請求項35記載の方法。
【請求項37】
以下の段階をさらに含む、請求項35記載の方法:
前記第1の群に対する候補または公知の抗癌治療物質の有効性を、第1の経路において変異を有さない第2の膵臓腫瘍群に対する有効性と比較する段階;
前記第1の経路が、前記候補または公知の抗癌治療物質の、他の群と比べて高いまたは低い有効性との相互関係を有する場合に、該第1の経路は層別化に有用であると判定する段階。
【請求項38】
経路が、表2に示すものより選択される、請求項37記載の方法。
【請求項39】
以下の段階を含む、個体において早期癌もしくは微小残存病変または分子再発を検出する方法:
表S6または表S12に示すもの(SAGEによる膵臓で過剰発現される遺伝子)より選択される遺伝子からのmRNAまたはタンパク質の発現の増大を、前記個体から採取された臨床試料において検出する段階であって、該増大が、健常個体の集団と比べられるか、または異なる時点に採取した同じ個体の臨床試料と比べられる、段階;
前記臨床試料が対照と比べて高い発現を示す場合に、前記個体は膵癌、微小残存病変、または膵癌の分子再発を有する可能性が高いと判定する段階。
【請求項40】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項39記載の方法。
【請求項41】
1つまたは複数の遺伝子のmRNA発現を測定する、請求項39記載の方法。
【請求項42】
1つまたは複数の遺伝子が、表S13に列挙したもの(SAGEによって発見された細胞外タンパク質)より選択される、請求項41記載の方法。
【請求項43】
以下の段階を含む、膵癌量をモニターするための方法:
表S6または表S12に列挙する1つまたは複数の遺伝子(SAGEによる膵臓で過剰発現される遺伝子)の臨床試料における発現を測定する段階;
前記測定する段階を1回または複数回繰り返す段階;および
経時的な発現の増大、減少、または安定なレベルを確認する段階。
【請求項44】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項43記載の方法。
【請求項45】
1つまたは複数の遺伝子のmRNA発現を測定する、請求項43記載の方法。
【請求項46】
1つまたは複数の遺伝子が、表S13に列挙したもの(SAGEによって発見された細胞外タンパク質)より選択される、請求項43記載の方法。
【請求項47】
検査材料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項39または43記載の方法。
【請求項48】
以下の段階を含む、ヒトの膵癌を検出または診断するための方法:
表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)のヒト臨床試料における発現を測定する段階;
前記臨床試料中の前記1つまたは複数の遺伝子の発現を、対照ヒトもしくは対照ヒト群または該ヒトの対照組織の対応する試料における前記1つまたは複数の遺伝子の発現と比較する段階;
対照と比べて低い発現を示す臨床試料は膵癌を有する可能性が高いと判定する段階。
【請求項49】
臨床試料が血液試料またはリンパ節試料である、請求項48記載の方法。
【請求項50】
臨床試料が膵臓組織試料である、請求項48記載の方法。
【請求項51】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項48記載の方法。
【請求項52】
以下の段階を含む、膵癌量をモニターするための方法:
表S5に列挙する1つまたは複数の遺伝子(ホモ接合性欠失)の臨床試料における発現を測定する段階;
前記測定する段階を1回または複数回繰り返す段階;および
経時的な発現の増大、減少、または安定なレベルを確認する段階。
【請求項53】
臨床試料が血液試料またはリンパ節試料である、請求項52記載の方法。
【請求項54】
1つまたは複数の遺伝子のタンパク質発現を測定する、請求項52記載の方法。
【請求項55】
検査材料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項48または52記載の方法。
【請求項56】
以下の段階を含む、膵癌量をモニターするための方法:
試験試料中のある遺伝子またはそのコードされたcDNAもしくはタンパク質における体細胞変異を、前記ヒトの正常試料と比べて確認する段階であって、該遺伝子は表S7に列挙したものからなる群より選択され、該遺伝子は、RAS、SMAD4、CDKN2A、およびTP53のいずれでもない、段階;
前記確認する段階を1回または複数回繰り返す段階;および
前記試験試料における前記変異の経時的な増加、減少、または安定なレベルを確認する段階。
【請求項57】
複数の遺伝子の体細胞変異を確認する、請求項56記載の方法。
【請求項58】
試験試料が、血漿、血清、全血、膵管液、便、および息からなる群より選択される、請求項56記載の方法。
【請求項59】
膵癌と診断されるか、または膵癌の家族歴を有する患者から得られた試料において、PALB2遺伝子の変異を検出する段階を含む、方法。
【請求項60】
前記検出する段階が、試料中のゲノムDNAを解析する段階を含む、請求項59記載の方法。
【請求項61】
前記検出する段階が、試料中のPALB2タンパク質を解析する段階を含む、請求項59記載の方法。
【請求項62】
以下の段階を含む、患者の膵癌に対する素因を診断する方法:
ある個体から得られた非腫瘍試料において、PALB2遺伝子の生殖系列変異を検出する段階であって、該生殖系列変異の存在により、膵癌に対する素因が示唆される、段階。
【請求項63】
前記検出する段階が、試料中のゲノムDNAを解析する段階を含む、請求項62記載の方法。
【請求項64】
前記検出する段階が、試料中のPALB2タンパク質を解析する段階を含む、請求項62記載の方法。
【請求項65】
個体が、膵癌に罹患していると診断されるか、もしくは推測されるか、または膵癌の家族歴を有する、請求項62記載の方法。
【図1】

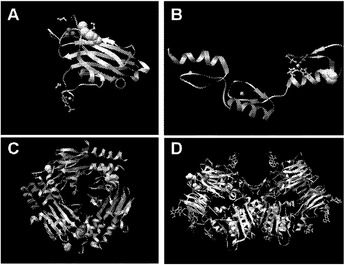
【図2】
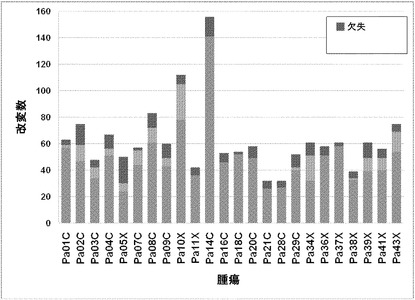
【図3】
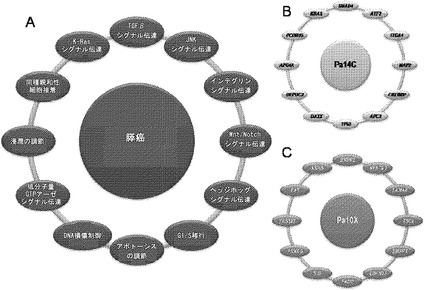
【図4】
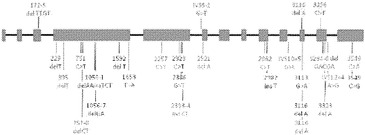
【図5−001】
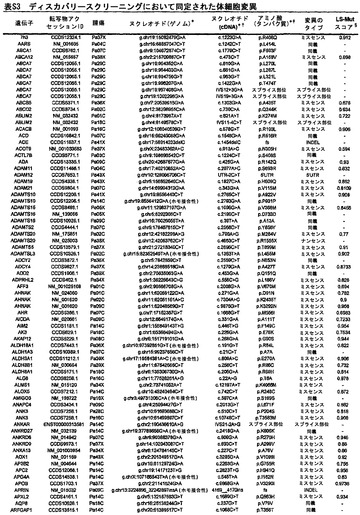
【図5−002】
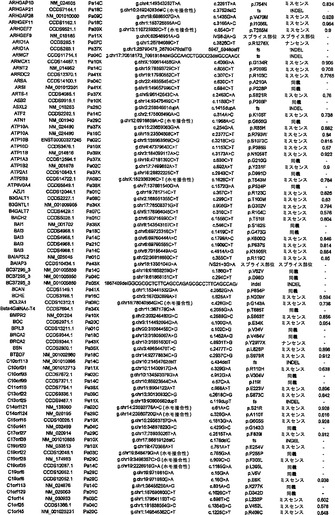
【図5−003】
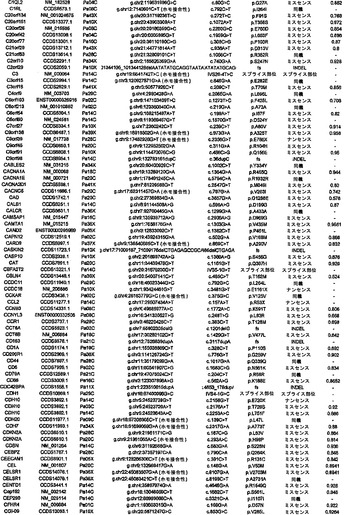
【図5−004】
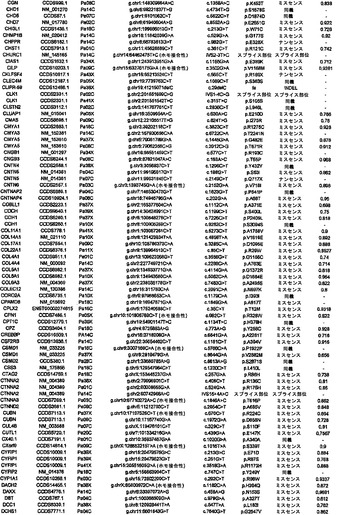
【図5−005】
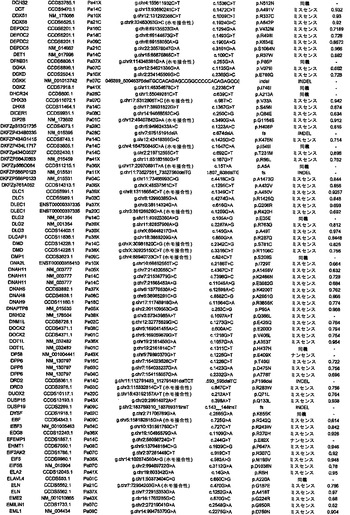
【図5−006】
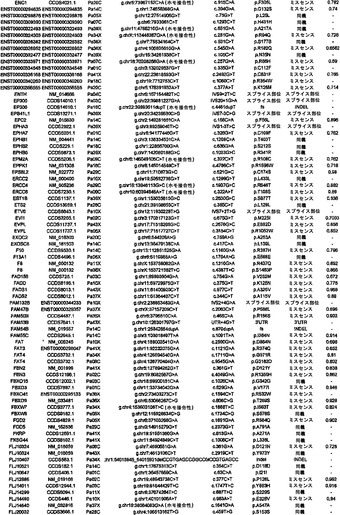
【図5−007】
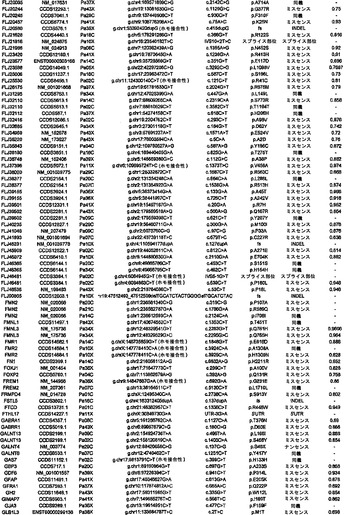
【図5−008】
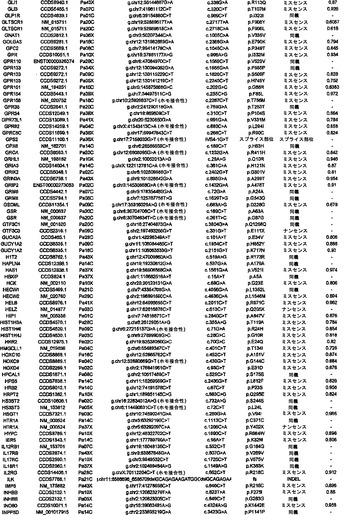
【図5−009】
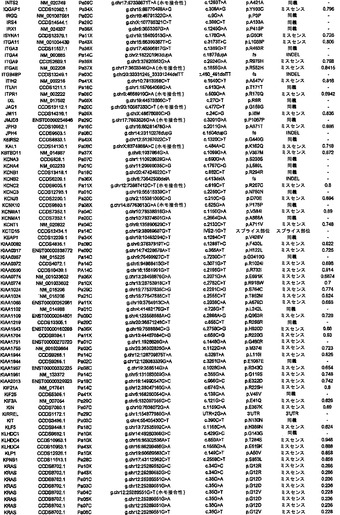
【図5−010】
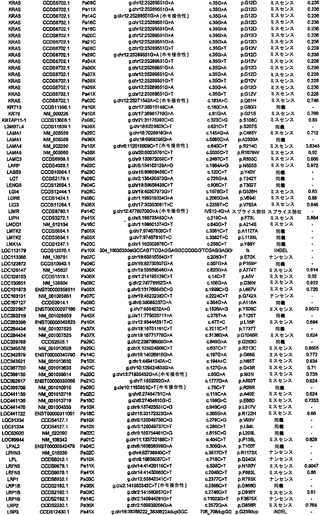
【図5−011】
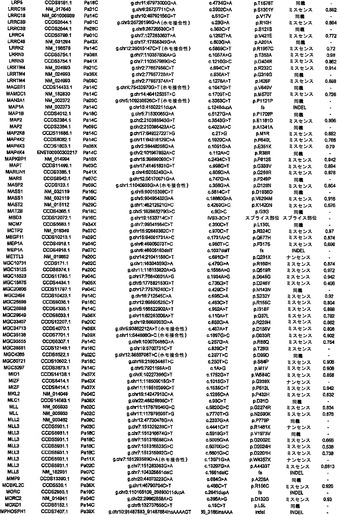
【図5−012】
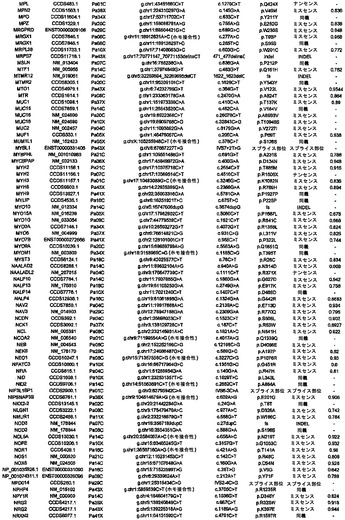
【図5−013】
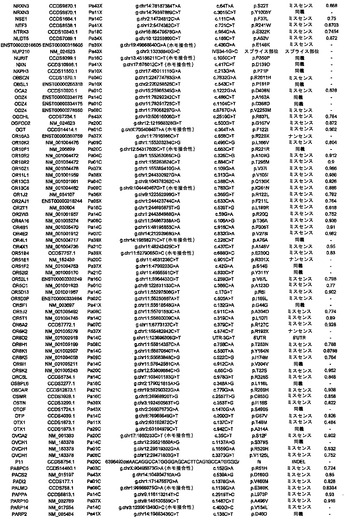
【図5−014】
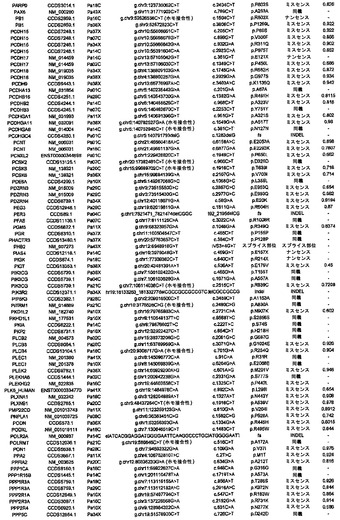
【図5−015】
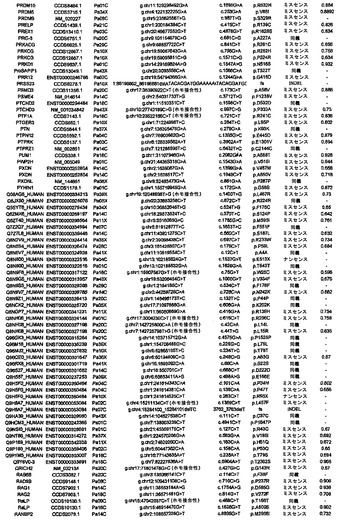
【図5−016】
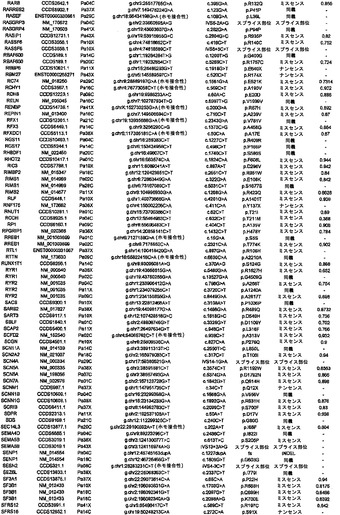
【図5−017】
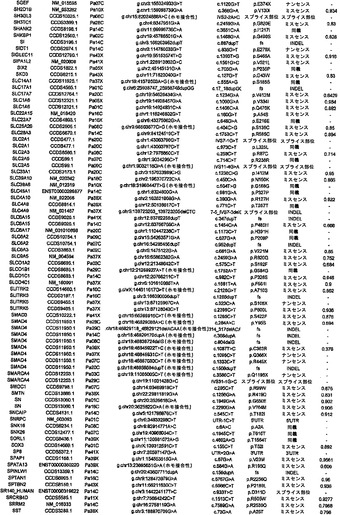
【図5−018】
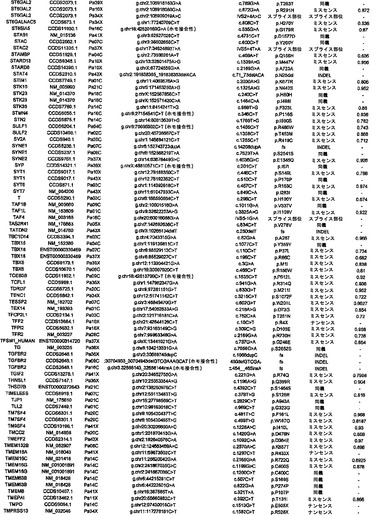
【図5−019】
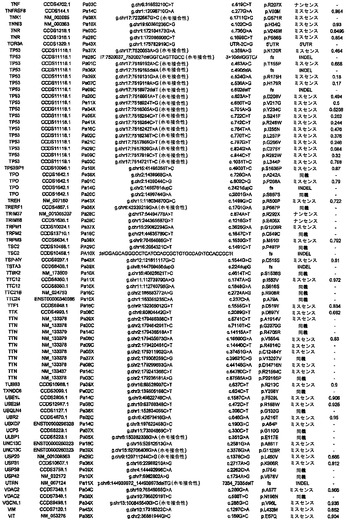
【図5−020】
【図5−021】

【図5−022】
【図5−023】

【図5−024】

【図5−025】
【図5−026】

【図5−027】
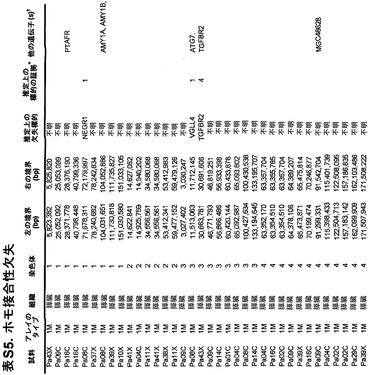
【図5−028】
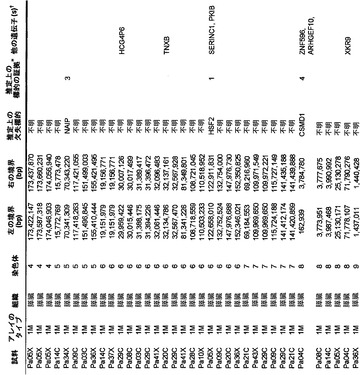
【図5−029】
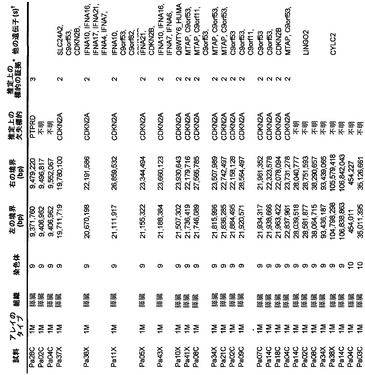
【図5−030】
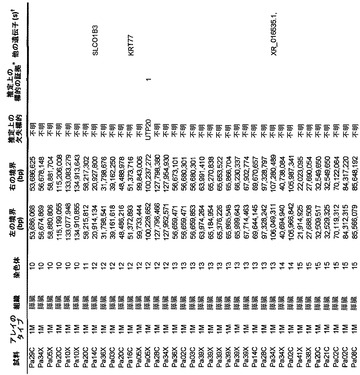
【図5−031】
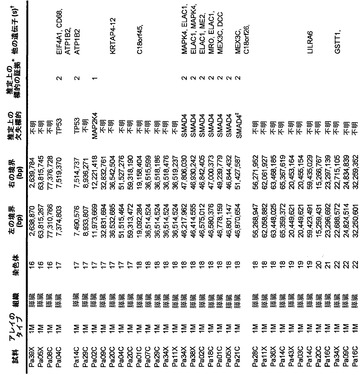
【図5−032】
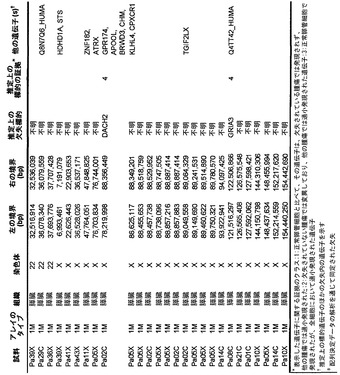
【図5−033】
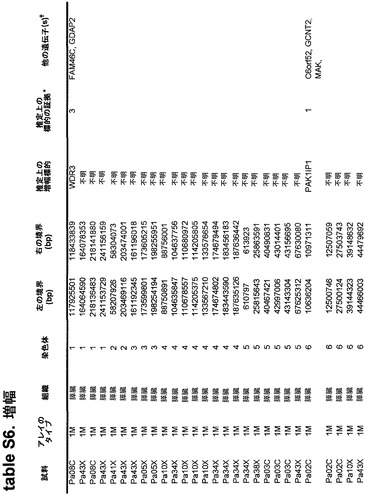
【図5−034】

【図5−035】

【図5−036】
【図5−037】

【図5−038】
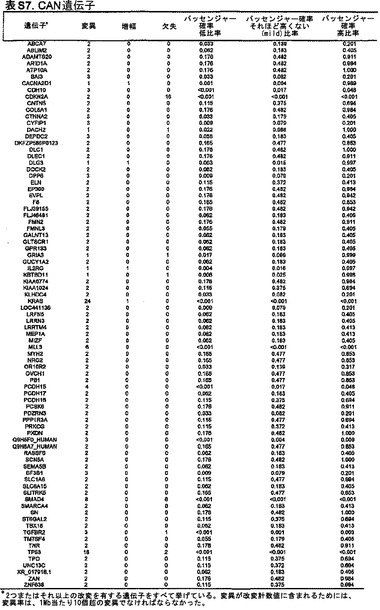
【図5−039】
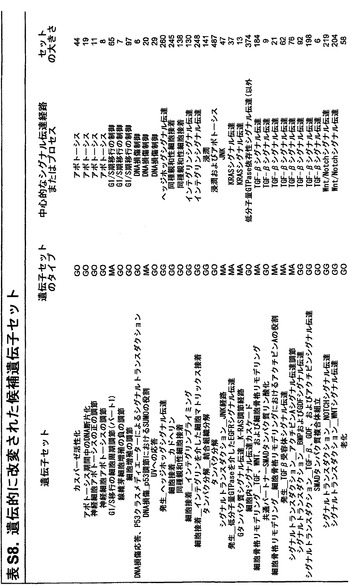
【図5−040】
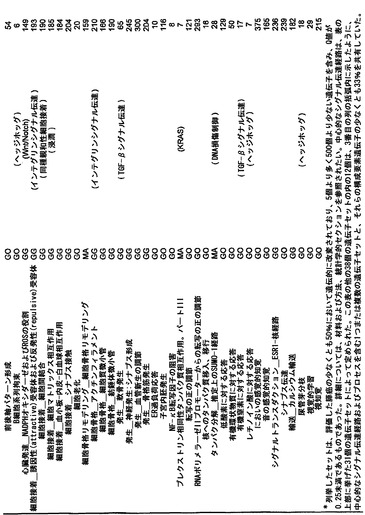
【図5−041】

【図5−042】

【図5−043】
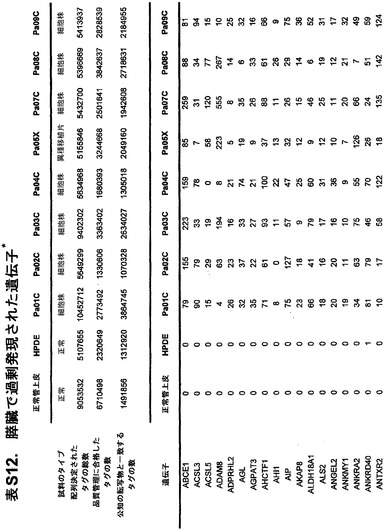
【図5−044】

【図5−045】

【図5−046】

【図5−047】

【図5−048】

【図5−049】

【図5−050】

【図5−051】

【図5−052】

【図5−053】

【図5−054】

【図5−055】

【図5−056】

【図5−057】

【図5−058】

【図5−059】

【図5−060】
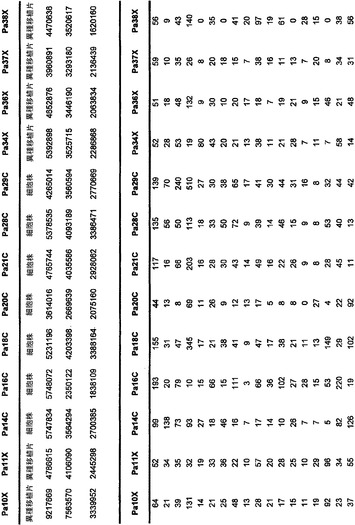
【図5−061】

【図5−062】

【図5−063】

【図5−064】

【図5−065】

【図5−066】

【図5−067】

【図5−068】

【図5−069】

【図5−070】

【図5−071】

【図5−072】

【図5−073】

【図5−074】

【図5−075】

【図5−076】

【図5−077】
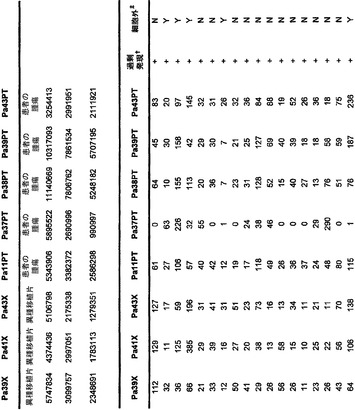
【図5−078】

【図5−079】

【図5−080】

【図5−081】

【図5−082】

【図5−083】

【図5−084】

【図5−085】

【図5−086】

【図5−087】

【図5−088】

【図5−089】

【図5−090】

【図5−091】

【図5−092】

【図5−093】
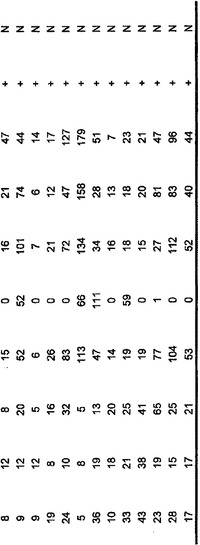
【図5−094】
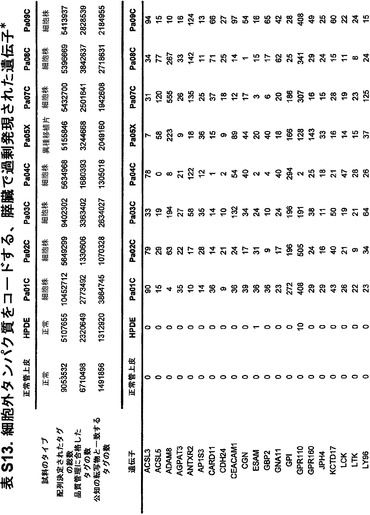
【図5−095】
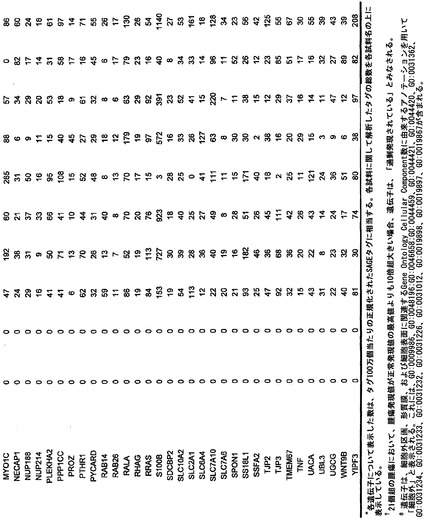
【図5−096】
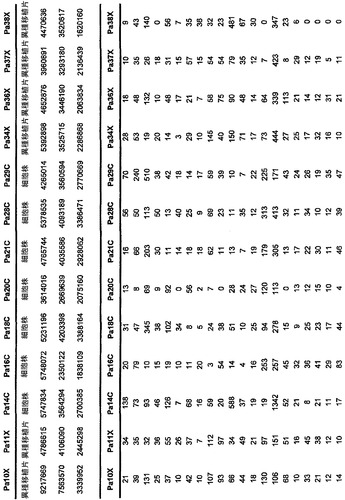
【図5−097】

【図5−098】
【図5−099】

【図2】
【図3】
【図4】
【図5−001】
【図5−002】
【図5−003】
【図5−004】
【図5−005】
【図5−006】
【図5−007】
【図5−008】
【図5−009】
【図5−010】
【図5−011】
【図5−012】
【図5−013】
【図5−014】
【図5−015】
【図5−016】
【図5−017】
【図5−018】
【図5−019】
【図5−020】
【図5−021】
【図5−022】
【図5−023】
【図5−024】
【図5−025】
【図5−026】
【図5−027】
【図5−028】
【図5−029】
【図5−030】
【図5−031】
【図5−032】
【図5−033】
【図5−034】
【図5−035】
【図5−036】
【図5−037】
【図5−038】
【図5−039】
【図5−040】
【図5−041】
【図5−042】
【図5−043】
【図5−044】
【図5−045】
【図5−046】
【図5−047】
【図5−048】
【図5−049】
【図5−050】
【図5−051】
【図5−052】
【図5−053】
【図5−054】
【図5−055】
【図5−056】
【図5−057】
【図5−058】
【図5−059】
【図5−060】
【図5−061】
【図5−062】
【図5−063】
【図5−064】
【図5−065】
【図5−066】
【図5−067】
【図5−068】
【図5−069】
【図5−070】
【図5−071】
【図5−072】
【図5−073】
【図5−074】
【図5−075】
【図5−076】
【図5−077】
【図5−078】
【図5−079】
【図5−080】
【図5−081】
【図5−082】
【図5−083】
【図5−084】
【図5−085】
【図5−086】
【図5−087】
【図5−088】
【図5−089】
【図5−090】
【図5−091】
【図5−092】
【図5−093】
【図5−094】
【図5−095】
【図5−096】
【図5−097】
【図5−098】
【図5−099】
【公表番号】特表2012−501651(P2012−501651A)
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願番号】特願2011−526179(P2011−526179)
【出願日】平成21年9月3日(2009.9.3)
【国際出願番号】PCT/US2009/055802
【国際公開番号】WO2010/028098
【国際公開日】平成22年3月11日(2010.3.11)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願日】平成21年9月3日(2009.9.3)
【国際出願番号】PCT/US2009/055802
【国際公開番号】WO2010/028098
【国際公開日】平成22年3月11日(2010.3.11)
【出願人】(398076227)ザ・ジョンズ・ホプキンス・ユニバーシティー (35)
【Fターム(参考)】
[ Back to top ]