
タンパク質を核内に導入する方法、およびその利用

【課題】部位特異的遺伝子除去方法または部位特異的遺伝子組換え方法において、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための工程、部位特異的組換え酵素発現プラスミドを脱落する工程等の煩雑な工程を必要としない、簡便、且つ迅速な方法を実現することにある。
【解決手段】本発明にかかるタンパク質を核内へ導入する方法は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む。
【解決手段】本発明にかかるタンパク質を核内へ導入する方法は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質を核内に導入する方法、およびその利用に関する。具体的には、タンパク質をコードする遺伝子を細胞に導入してタンパク質を発現させる工程を必要とせず、タンパク質を核内に簡便に導入する方法およびその利用に関する。
【背景技術】
【0002】
従来、外来タンパク質を細胞内で機能させる場合は、目的のタンパク質をコードする遺伝子のcDNAを含むプラスミドを細胞内に導入し、細胞内でタンパク質を発現させる方法(遺伝子導入法)が用いられてきた。
【0003】
例えば、遺伝子の機能解明を行う場合、一般に、Cre−loxPシステムを用いた目的遺伝子の除去または挿入が行われている。図1(A)は、上記Cre−loxPシステムの概要を示す図である。大腸菌を宿主とするP1バクテリオファージの部位特異的組換え酵素であるCreリコンビナーゼは、38kDaのタンパク質であり、34塩基からなるloxP配列を特異的に認識することにより、2つのloxP配列間において特異的組換えを効率よく引き起こす(非特許文献1)。図1(A)に示すように、特に、同一DNA鎖上に、2つのloxP配列が同方向にある場合、loxPで挟まれたDNA領域を、効率よく環状に切り出して除去する。一方、図2(A)に示すように、DNA鎖上に1つのloxP配列がある場合は、上記DNA鎖上のloxP配列を標的として、分子内にloxP配列を含む環状DNAを挿入することができる。
【0004】
糸状菌を例に挙げると、Aspergillus fumigatus菌体内(非特許文献2)、または、A. nidulans菌体内(非特許文献3)において、Creリコンビナーゼを発現させ、染色体上にある除去対象遺伝子の上流および下流に連結されたloxP配列との組換えを行う事で、染色体上のloxP配列の間にある除去対象遺伝子を除去することに成功している。また、Cre−loxPシステムを用いた目的遺伝子の除去方法または挿入方法は、酵母等でも一般に行われている確立された方法である(非特許文献4)。
【非特許文献1】N. Sternberg and D. Hamilton, J Mol Biol 150 (1981) 467-86.
【非特許文献2】S. Krappmann, O. Bayram and G.H. Braus, Eukaryot Cell 4 (2005) 1298-307.
【非特許文献3】J.V. Forment, D. Ramon and A.P. MacCabe, Curr Genet 50 (2006) 217-24.
【非特許文献4】B. Sauer, Mol Cell Biol 7 (1987) 2087-96.
【非特許文献5】A. Kuspa and W. F. Loomis, Proc. Natl. Acad. Sci. USA 89 (1992) 8803-8807.
【非特許文献6】X. Jin, M. Ming-He, Z. Wei, H. Xiao-Wei and Z. Ke-Qin, The Journal of Microbiology, Vol. 43, No. 5 (2005), 417-423.
【発明の開示】
【発明が解決しようとする課題】
【0005】
しかしながら、上述のように目的遺伝子を染色体上の特定の部位に挿入する方法は確立されてはいるものの、上記従来の構成では、多くの煩雑なステップと時間が必要であった。図2を参照しながら具体的に説明すると、まず、Creリコンビナーゼを発現するプラスミドを細胞内に導入し、プラスミド保持細胞を選抜する。次いで、Creリコンビナーゼの発現を誘導し、loxP配列に挟まれた第一遺伝子を除去する。その後、loxP配列をターゲットに第二遺伝子を挿入するためには、図2に示すような、特異的部位を含む環状目的配列を細胞内に導入し、誘導をかけて部位特異的組換え酵素を発現させるという煩雑なステップが必要であり、さらに目的遺伝子の脱落を防ぐためには、部位特異的組換え酵素発現カセットプラスミドを脱落させ、細胞内で部位特異的組換え酵素の存在をなくす必要があり、時間と手間がかかる作業であった(図2(A))。
【0006】
本発明は、上記の問題点に鑑みてなされたものであり、その主たる目的は、上述したような、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための工程、部位特異的組換え酵素発現プラスミドを脱落する工程等の煩雑な工程を必要としない、簡便、且つ迅速な部位特異的遺伝子除去方法および部位特異的遺伝子組換え方法を実現することにある。
【課題を解決するための手段】
【0007】
本発明者は、上記課題に鑑み、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるのではなく、部位特異的組換え酵素タンパク質そのものを細胞の外部から細胞の内部に導入することができれば、極めて効率的になると考え、タンパク質の核内導入方法について鋭意検討した。その結果、核酸をキャリアとして部位特異的組換え酵素タンパク質を細胞へ導入する事で、部位特異的組換え酵素タンパク質を効率良く核内に導入しうることを見出し、本発明を完成するに至った。
【0008】
すなわち、本発明にかかるタンパク質を核内へ導入する方法(以下、「タンパク質核内導入方法」という)は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴としている。
【0009】
本発明にかかるタンパク質核内導入方法によれば、従来の遺伝子導入方法のように目的のタンパク質をコードする遺伝子を細胞に導入して目的タンパク質を発現させる工程を必要としないため、タンパク質を核内に簡便に導入することができる。また、従来の遺伝子導入方法では、タンパク質が細胞内で機能する時期を調節するためには、タンパク質の発現を調節可能な別のシステムをさらに導入するか、目的のタンパク質をコードする遺伝子を脱落させる必要がある。一方、本発明にかかるタンパク質核内導入方法によれば、タンパク質を細胞内に導入するため、導入されたタンパク質が分解された後には、タンパク質が機能することはなく、細胞内でタンパク質を機能させる時期を容易に調節することができる。
【0010】
ところで、非特許文献5および6には、形質転向率や相同組み換え効率を上昇させるために、制限酵素と核酸とを細胞内に導入する方法(REMI法)について記載されている。上記REMI法において細胞内に導入される制限酵素を制限酵素Aとすると、細胞内に導入される核酸としては、両端が制限酵素Aによって消化された直鎖状の核酸が用いられる。上記REMI法を用いれば、細胞の染色体上に存在する制限酵素Aの特異的認識配列を制限酵素Aを用いて切断するため、切断された部位に目的の核酸を効率よく導入することができる。非特許文献5または6に記載のREMI法は、核酸を染色体内の所望の部位に導入することを目的として制限酵素と核酸とを細胞内に導入し、これにより形質転換効率や相同組換え効率を上昇させる方法である。よって、本発明のタンパク質核内導入方法とは目的も効果も全く違う方法である。
【0011】
本発明にかかるタンパク質核内導入方法においては、上記タンパク質は、部位特異的組換え酵素であることが好ましい。上記タンパク質が部位特異的組換え酵素であれば、後述する部位特異的遺伝子除去方法および部位特異的遺伝子組換え方法を簡便、且つ迅速に行うことができる。
【0012】
本発明にかかるタンパク質核内導入方法においては、上記核酸は、全長が50塩基〜300,000塩基の範囲内であることが好ましい。核酸の全長が上記範囲内であれは、より効率よくタンパク質を核内に導入することができる。
【0013】
また、本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴としている。
【0014】
図1(A)に示すように、従来の部位特異的遺伝子除去方法では、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるために複数の工程を行う必要があったが、本発明にかかる部位特異的遺伝子除去方法を用いれば、これらの工程を省略することができる。従って、除去対象遺伝子の除去を簡便、且つ迅速に行うことができる。
【0015】
また、本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含むことを特徴としている。
【0016】
図2(A)に示すように、従来の部位特異的遺伝子組換え方法では、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程に加えて、目的遺伝子が挿入された細胞において部位特異的組換え酵素が発現し続けることにより、挿入部位から当該目的遺伝子が除去されることを防ぐために、上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を行う必要があった。本発明にかかる部位特異的遺伝子組換え方法を用いれば、部位特異的組換え酵素のタンパク質を細胞内に導入するため、導入されたタンパク質が分解された後には、上記部位特異的組換え酵素は機能しない。よって、部位特異的組換え酵素を細胞内で発現させるための複数の工程および上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を省略することができ、部位特異的遺伝子組換えをより簡便に行うことができる。
【0017】
本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであることを特徴としている。
【0018】
核酸が、目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであれば、部位特異的組換え酵素と目的遺伝子とを1つの工程で同時に細胞内に導入することができるため、本発明にかかる部位特異的遺伝子組換え方法をより簡便に行うことができる。
【0019】
また、本発明にかかる部位特異的遺伝子除去キットは、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴としている。
【0020】
上記構成によれば、少なくとも部位特異的組換え酵素と核酸とをキット内に含むため、部位特異的組換え酵素を簡便に核内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができる。
【0021】
また、本発明にかかる部位特異的遺伝子組換えキットは、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴としている。
【0022】
上記構成によれば、少なくとも部位特異的組換え酵素と核酸とをキット内に含むため、部位特異的組換え酵素を簡便に核内に導入することができ、目的遺伝子の挿入を簡便、且つ迅速に行うことができる。
【発明の効果】
【0023】
本発明にかかるタンパク質核内導入方法は、以上のように、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含んでいるので、タンパク質をコードする遺伝子を細胞に導入し、発現させる工程を必要とせず、簡便且つ容易にタンパク質を核内に導入することができるという効果を奏する。また、タンパク質を細胞内に導入するため、細胞内でタンパク質を機能させる時期を容易に調節することができるというさらなる効果を奏する。
【0024】
本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むため、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程を行う必要が無く、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0025】
本発明にかかる部位特異的遺伝子組換え方法は、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含むため、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程、および遺伝子挿入後に上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を行う必要が無く、部位特異的遺伝子組換えをより簡便に行うことができるという効果を奏する。
【0026】
本発明にかかる部位特異的遺伝子組換え方法は、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであるため、部位特異的組換え酵素と目的遺伝子とを1つの工程で同時に細胞内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0027】
本発明にかかる部位特異的遺伝子除去キットは、以上のように除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むため、部位特異的組換え酵素を簡便に核内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0028】
本発明にかかる部位特異的遺伝子組換えキットは、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むため、部位特異的組換え酵素を簡便に核内に導入することができ、目的遺伝子の挿入を簡便、且つ迅速に行うことができるという効果を奏する。
【発明を実施するための最良の形態】
【0029】
本発明の実施の形態について説明すれば以下のとおりであるが、本発明はこれに限定されるものではない。
【0030】
なお、本明細書中において範囲を示す「A〜B」は、A以上B以下であることを示す。本発明の一実施形態について説明すると以下の通りである。
【0031】
〔1.タンパク質核内導入方法〕
一実施形態において、本発明にかかるタンパク質核内導入方法は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む。上記タンパク質としては、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、部位特異的組換え酵素、DNA修復に関与するDNA結合タンパク質やリガーゼといった酵素、抗体等を用いることができる。上記部位特異的組換え酵素については、後述する。
【0032】
上記DNA修復に関与するタンパク質としては、特に限定されるものではないが、例えば、Rad51、Rad52、Rad54等を挙げることができる。上記抗体としては、特に限定されるものではないが、Kuタンパク質、DNAリガーゼ等に対する抗体を挙げることができる。
【0033】
Rad51、Rad52、Rad54等の相同組換えに関与するタンパク質群を細胞核内に導入することにより、相同組換え効率を上昇させることができる。または、非相同末端結合に関与するタンパク質に対する抗体、例えば、Kuタンパク質、DNAリガーゼ等に対する抗体を細胞核内に導入し、抗原抗体反応により、核内で正常に機能する非相同末端結合に関与するタンパク質を減少させる事で、ターゲティング効率を上昇させることができる。
【0034】
また、上記タンパク質は、天然の細胞由来であっても、遺伝子組換えによって製造されたものでもよい。また、上記タンパク質は、一種類を単独で用いてもよいし、二種類以上を併用してもよい。
【0035】
本発明で用いられる「核酸」とは、本発明に係るタンパク質核内導入方法においてタンパク質とともに細胞に導入される核酸を意味する。また、「核酸をキャリアとする」とは、特に限定されるものではないが、例えば、タンパク質と核酸とをともに細胞に導入することを意味する。このとき核酸は対象タンパク質を核内に運ぶキャリアとしての役割を果たす。発明者らの検討によれば、上記核酸としては、リン酸、ペントース、および塩基からなるヌクレオチドが重合したポリヌクレオチドである核酸であれば塩基配列や長さは特に限定されるものではないことが分かっている。また上記核酸としては、デオキシリボ核酸(DNA)であってもよいし、リボ核酸(RNA)であってもよい。なお、RNAと比較して、DNAは非常に安定性が高く、取り扱い性に優れるため、上記核酸としては、DNAを用いることが好ましい。
【0036】
また、本発明で用いられる核酸は、一本鎖構造を有するものであってもよいし二本鎖構造を有するものであってもよい。一本鎖構造を有する核酸と比較して、二本鎖構造を有する核酸の方が、一般に安定性が高く、取り扱い性に優れるため、本発明で用いられる核酸としては、二本鎖構造を有するものを用いることが好ましい。
【0037】
また、本発明で用いられる核酸は、直鎖状であってもよいし、環状であってもよい。本発明で用いられる核酸の全長として特に限定されるものではないが、50塩基〜300,000塩基であることが好ましく、300〜100,000塩基であることがより好ましい。核酸の全長が50塩基以上であれば、対象タンパク質を核内に運ぶキャリアとしてより好ましく機能する。また、300,000塩基以下であれば、細胞内への核酸の導入効率が高くなる。
【0038】
本発明で用いられる核酸としては、例えば、細胞由来の核酸、タンパク質が導入される細胞が由来する生物以外の生物由来の核酸、プラスミド、PCR増幅断片、合成核酸等を利用することができる。
【0039】
本発明で用いられる核酸として、細胞由来の核酸を用いる場合、当該細胞は、微生物(例えば、バクテリア、酵母、真菌等)由来であってもよいし、植物由来であってもよいし、動物(例えば、マウス、ヒト等)由来であってもよく、適宜選択することができる。さらに、上記細胞由来の核酸の種類としては、例えば、染色体DNAであってもよく、totalRNAであってもよく、mRNAであってもよい。安定性が高く取り扱い性に優れることから、染色体DNAであることがより好ましい。また、細胞由来の核酸は、従来公知の核酸分離法を用いることにより核酸のみを単離することができる。例えば、染色体DNAを単離したい場合、従来公知のフェノール・クロロホルム溶解及び抽出/エタノール析出を用いることができる。また、totalRNAまたはmRNAを単離したい場合は、従来公知のホットフェノール法を用いることができる。
【0040】
上記核酸が有する塩基配列は、特に限定されるものではなく、どのような塩基配列の核酸も好適に用いることができる。例えば、塩基をランダムに選択して合成した合成核酸を用いることもできる。
【0041】
上記核酸は、一種類を単独で用いてもよいし、二種類以上を併用してもよい。
【0042】
また、タンパク質が導入される細胞としては、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物(例えば、バクテリア、酵母、真菌等)、植物、動物(例えば、マウス、ヒト等)の細胞を挙げることができる。一例として、微生物細胞を用いる場合は、特に限定されるものではないが、例えば、麹菌、クリプトコッカス等の細胞を好適に用いることができる。また、一例として、動物細胞を用いる場合は、細胞種としては、特に限定されるものではないが、例えば、線維芽細胞、臍帯静脈内皮細胞等の細胞を好適に用いることができる。
【0043】
上記「タンパク質を核内に導入する」方法としては、タンパク質と核酸とを細胞内に導入できる限り特に限定されるものではなく、従来公知の形質転換法を用いて行うことができる。上記形質転換法としては、例えば、プロトプラスト−PEG法(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)、エレクトロポレーション法(BN Chakrabory and M Kapoor, Nucleic Acids Res 25 (1990) 6737を参照)等を用いることができ、タンパク質を導入される生物種および細胞種によって最適な形質転換法を選択することが好ましい。
【0044】
例えば、上記プロトプラスト−PEGを用いる場合、まず、部位特異的組換え酵素を導入するために、プロトプラスト細胞を作製する。当該プロトプラストの作製は、当業者に公知の任意の方法・手段を用いて作製することが可能である(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)。例えば、市販されているヤタラーゼ(TO17、TaKaRa製)、セルラーゼ(201093、Yakult Pharmaceutical製)、ライジングエンザイム(L1412、Sigma Chemical製)等を単独、または混合して用いることができる。次いで、部位特異的組換え酵素、核酸、プロトプラスト、およびPEGを含むプロトプラスト等張液を混合し、氷上にて20〜30分間インキュベートを行う。以後の操作は、当業者に公知のプロトプラスト−PEG法による形質転換法と同様に行う事ができる。
【0045】
また、上記「タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程」において、タンパク質と核酸とを別々に導入してもよいし、同時に導入してもよい。タンパク質と核酸とを別々に導入する場合、導入する順序については特に限定されるものではなく、タンパク質を導入した後に核酸を導入してもよいし、核酸を導入した後にタンパク質を導入してもよい。
【0046】
導入の際に細胞に与えるダメージを考慮すると、タンパク質と核酸とを同時に細胞内に導入することが好ましい。タンパク質と核酸とを同時に細胞内に導入することにより、導入操作を1回行えばよく、細胞に与えるダメージも少ないため好ましい。
【0047】
〔2.部位特異的遺伝子除去方法〕
一実施形態において、本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記除去対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含む。
【0048】
上記「部位特異的組換え酵素」は、DNAの分子内あるいは分子間の特定部位で起こる組換えの過程を触媒する酵素のことを指す。また、上記「特異的認識配列」は、上記部位特異的組換え酵素によって特異的に認識される配列のことを指す。当該部位特異的組換え酵素は、上記特異的認識配列に挟まれたDNA断片を切り出し、環状化させる機能を有する。さらに、上記部位特異的組換え酵素は、分子内に上記特異的認識配列を有する環状DNAを、上記特異的認識配列を有する別のDNA分子内に、当該特異的認識配列を介して挿入する反応も行うこともできる。上記「部位特異的組換え酵素」としては、例えば、Creリコンビナーゼ、Flpリコンビナーゼ等を挙げることができる。これらの部位特異的組換え酵素は、天然の細胞由来であっても、遺伝子組換えによって製造されたものであってもよい。
【0049】
例えば、上記部位特異的組換え酵素としてCreリコンビナーゼを用いる場合、上記「特異的認識配列」としては、loxP配列が用いられる。当該loxP配列は、Creリコンビナーゼによって特異的に認識される塩基配列である(K Abremski, R Hoess and N Sternberg, Cell 32 (1983) 1301-11を参照)。他に上記部位特異的組換え酵素としてFlpリコンビナーゼを用いる場合、上記「特異的認識配列」としては、FRT配列が用いられる(D Babineau, D Vetter, BJ Andrews, RM Gronostajski, GA Proteau, LG Beatty, and PD Sadowski, J Biol Chem 260 (1985) 12313-9を参照)。
【0050】
上記除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖(例えば、「loxP:: 除去対象遺伝子」で表すことができる)は、細胞に1つ以上備えられていればよい。
【0051】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0052】
本発明にかかる部位特異的遺伝子除去方法で用いられる細胞としては、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。
【0053】
ここで「除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞」とは、染色体上に除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を有する細胞、または染色体外に除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖(例えば、プラスミド等)を保持している細胞を意味する。
【0054】
本発明にかかる部位特異的遺伝子除去方法で用いられる、細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0055】
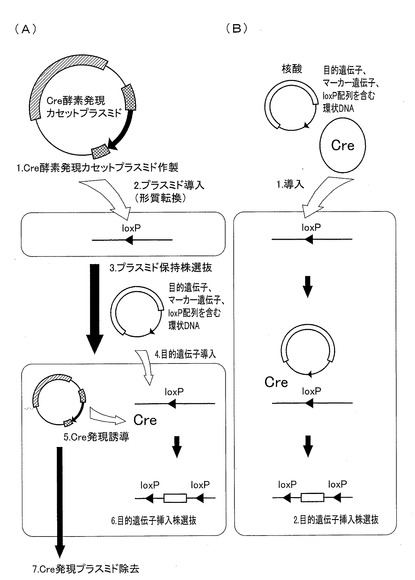
ここで、Cre−loxPシステムを用いた部位特異的遺伝子除去方法について、図1を参照しながら説明する。図1において、(A)は従来の部位特異的遺伝子除去方法を示し、(B)は本発明にかかる部位特異的遺伝子除去方法を示す。図1(A)に示されるように、従来の部位特異的遺伝子除去方法を用いた場合、以下の工程が必要である。具体的には、(1)除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞において機能するプロモーター配列の下流に、Cre遺伝子を連結したCre発現カセットを含むCre発現プラスミドを作製する。(2)作製したCre発現プラスミドを目的の細胞に形質転換する。(3)上記Cre発現プラスミドが形質転換された細胞を選抜する。(4)上記選抜された細胞内においてCreリコンビナーゼタンパク質の発現を誘導し、除去対象遺伝子を除去する。(5)除去対象遺伝子が除去された細胞を選抜する。
【0056】
一方、本発明にかかる部位特異的遺伝子除去方法によれば、図1(B)に示されるように、少なくともCreリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含めば、除去対象遺伝子を簡便に除去することができる。従来の部位特異的遺伝子除去方法を用いた場合に必要であった上記(1)〜(4)の工程の代わりに、Creリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含めばよく、非常に簡便に部位特異的遺伝子除去を行うことができる。
【0057】
〔3.部位特異的遺伝子組換え方法〕
一実施形態において、本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含む。
【0058】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。核酸としては、核酸であれば特に限定されるものではなく、上記〔1.タンパク質核内導入方法〕で説明したものを好適に用いることができる。
【0059】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞としては、少なくとも1つ以上の特異的認識配列を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。なお、〔2.部位特異的遺伝子除去方法〕によって除去対象遺伝子が除去された細胞は、当該細胞内に特異的認識配列を備えるため、この細胞を用いれば、特異的認識配列を細胞に導入する工程を省略することができるため、好適に用いることができる。
【0060】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0061】
上記部位特異的組換え酵素が、細胞に備えられた1つ以上の特異的認識配列に、当該特異的認識配列を介して目的遺伝子を挿入するためには、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する必要がある。よって、本発明にかかる部位特異的遺伝子組換え方法は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程に加えて、さらに上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程(以下、環状二本鎖DNA導入工程という)を含む。当該環状二本鎖DNA導入工程は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入することができる限り、特に限定されるものではなく、従来公知の形質転換法を用いて上記環状二本鎖DNAを細胞内に導入することが出来る。例えば、プロトプラスト−PEG法、エレクトロポレーション法を用いることができる。
【0062】
当該環状二本鎖DNA導入工程は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程の前に行ってもよいし、後で行ってもよい。細胞内に導入された部位特異的組換えタンパク質の細胞内での安定性を考慮に入れると、上記遺伝子導入工程を行った後に、部位特異的組換え酵素タンパク質の核内導入を行うことが好ましい。
【0063】
一実施形態において、本発明にかかる部位特異的遺伝子組換え方法は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAである。
【0064】
本発明にかかる部位特異的遺伝子組換え方法において、上記部位特異的組換え酵素は、環状分子を、特異的認識配列を介して挿入するため、本発明で用いられる核酸として目的遺伝子を含む核酸を用いる場合は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAである必要がある。
【0065】
本発明において核酸として用いられる環状二本鎖DNAの全長は、導入したい目的遺伝子の全長によるが、50塩基〜300,000塩基対であることが好ましく、2,000〜200,000塩基対であることがより好ましい。核酸として用いられる上記環状二本鎖DNAの全長が300,000塩基対以下であれば細胞内への核酸の導入効率が高くなる。
【0066】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。本発明で用いられる核酸としては、核酸であれば特に限定されるものではなく、上記〔1.タンパク質核内導入方法〕で説明したものを好適に用いることができる。
【0067】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞としては、少なくとも1つ以上の特異的認識配列を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。なお、〔2.部位特異的遺伝子除去方法〕によって除去対象遺伝子が除去された細胞は、当該細胞内上に特異的認識配列を備えるため、この細胞を用いれば、特異的認識配列を細胞に導入する工程を省略することができるため、好適に用いることができる。
【0068】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0069】
ここで、Cre−loxPシステムを用いた部位特異的遺伝子組換え方法について、図2を参照しながら説明する。図2において、(A)は従来の部位特異的遺伝子組換え方法、(B)は本発明にかかる部位特異的遺伝子組換え方法を示す。図2(A)に示されるように、従来の部位特異的遺伝子組換え方法を用いた場合、以下の工程が必要である。具体的には、(1)目的遺伝子を導入したい細胞において機能するプロモーターの下流にCre遺伝子をつないだCre発現カセットを含むプラスミドを作製する。(2)作製したCre発現プラスミドを目的の細胞に形質転換する。(3)上記Cre発現プラスミドが形質転換された細胞を選抜する。(4)目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する。(5)上記細胞内においてCreリコンビナーゼタンパク質の発現を誘導する。(6)目的遺伝子が導入された細胞を選抜する。(7)導入された目的遺伝子が除去されることを防ぐために、Creリコンビナーゼ発現カセットを含むプラスミドを脱落させる。尚、図2(A)の工程(4)では、一例として目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNAを用いているが、目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを用いてもよい。
【0070】
本発明にかかる部位特異的遺伝子組換えは、図2(A)に示されるような従来の部位特異的遺伝子組換え方法を用いた場合に必要であった上記(1)〜(4)の工程の代わりに、Creリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入すればよく、非常に簡便に部位特異的遺伝子組換えを行うことができる。一実施形態において、本発明で用いられる核酸として目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを利用すれば、Creリコンビナーゼタンパク質と目的遺伝子とを細胞内に導入することができるため、さらに簡便に部位特異的遺伝子組換えを行うことができる。
【0071】
また、本発明にかかる部位特異的遺伝子組換え方法を用いれば、細胞内に導入されたCreリコンビナーゼタンパク質は細胞分裂とともに細胞内で希釈され、時間とともに細胞内のタンパク質分解酵素により分解されるため、従来の部位特異的遺伝子組換え方法のようにプラスミドを脱落させる必要が無い。
【0072】
〔4.部位特異的遺伝子除去キット〕
本発明にかかる部位特異的遺伝子除去キットは、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含む。
【0073】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。
【0074】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0075】
本発明にかかる部位特異的遺伝子除去キットは、少なくとも上記部位特異的組換え酵素と上記核酸とを含んでいればよく、これらは別々の容器に充填されていてもよいし、同じ容器に充填されていてもよい。タンパク質と核酸とでは、安定性が異なるため、取り扱い性を考慮に入れると、別々の容器に充填されていることが好ましい。
【0076】
また、本発明にかかる部位特異的遺伝子除去キットは、上記部位特異的組換え酵素および上記核酸以外の成分を含んでいてもよい。例えば、形質転換に必要な試薬、2つの特異的認識配列の間に、除去対象遺伝子を連結することができるマルチクローニングサイトを備えるプラスミド、プラスミド作成用オリゴプライマー、除去株確認用オリゴプライマー等を挙げることができる。なお、上記除去株確認用オリゴプライマーは、除去対象遺伝子が除去された株を選択するPCRに用いるためのオリゴプライマーを指す。本発明にかかる部位特異的遺伝子除去キットを用いて部位特異的遺伝子除去を行うためには、予め、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞を準備する必要がある。よって、上記2つの特異的認識配列の間に、除去対象遺伝子を連結することができるマルチクローニングサイトを備えるプラスミドは、上記細胞を得るために用いることができる。
【0077】
〔5.部位特異的遺伝子組換えキット〕
本発明にかかる部位特異的遺伝子組換えキットは、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含む。
【0078】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。
【0079】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0080】
本発明にかかる部位特異的遺伝子組換えキットは、少なくとも上記部位特異的組換え酵素と上記核酸とを含んでいればよく、これらは別々の容器に充填されていてもよいし、同じ容器に充填されていてもよい。タンパク質と核酸とでは、安定性が異なるため、取り扱い性を考慮に入れると、別々の容器に充填されていることが好ましい。
【0081】
また、本発明にかかる部位特異的遺伝子組換えキットは、上記部位特異的組換え酵素および上記核酸以外の成分を含んでいてもよい。例えば、形質転換に必要な試薬、1つ以上の特異的認識配列を含むプラスミド、1つの特異的認識配列と目的遺伝子を連結することができるマルチクローニングサイトとを備えるプラスミド、2つの特異的認識配列の間に、目的遺伝子を連結することができるマルチクローニングサイトを備えるプラスミド、プラスミド作成用オリゴプライマー、遺伝子組換え株確認用オリゴプライマー等を挙げることができる。なお、上記遺伝子組換え株確認用オリゴプライマーは、目的遺伝子が挿入された株を選択するPCRに用いるためのオリゴプライマーを指す。本発明にかかる部位特異的遺伝子組換えキットを用いて部位特異的遺伝子組換えを行うためには、予め、少なくとも1つ以上の特異的認識配列を備える細胞を準備する必要がある。よって、上記1つ以上の特異的認識配列を含むプラスミドは、上記細胞を得るために用いることができる。
【0082】
また、本発明にかかる部位特異的遺伝子組換えキットを用いて部位特異的遺伝子組換えを行うためには、目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する必要がある。上記目的遺伝子は、必要に応じて任意に変更可能であることが好ましい。上記1つの特異的認識配列と目的遺伝子を連結することができるマルチクローニングサイトとを備えるプラスミド、または上記2つの特異的認識配列の間に、目的遺伝子を連結することができるマルチクローニングサイトを備えるプラスミドを用いれば、上記環状二本鎖DNAを構築する際に、目的遺伝子を容易に導入することができる。また、一実施形態において、本発明で用いられる核酸として上記環状二本鎖DNAを用いることも可能である。
【0083】
なお、本発明は上記の各実施形態に限定されるものではなく、請求項に示した範囲で種種の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0084】
以下に、実施例を挙げて本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。
【0085】
本発明の実施例では、タンパク質が核に移行し、正常に機能することを確認するためのモデル細胞として真菌を用いた。当該真菌としては麹菌NS4株(niaD−,sC−)(以下、NS4株という)を用いた。当該NS4株は、独立行政法人酒類総合研究所にて保存される麹菌株であり、硝酸塩および硫酸塩を資化することができない表現型を有する麹菌株である。
【0086】
また、本発明の実施例では、タンパク質が核に移行し、正常に機能することを確認するための手段として、従来公知のCre−loxPシステムを用いた。当該システムを用いれば、loxP配列が上流および下流に連結された除去対象遺伝子がCreリコンビナーゼによって除去されたことを指標として、タンパク質が核に移行し、且つ正常に機能したことを確認することができる。
【0087】
さらに、本発明の実施例では、A. nidulansのsC遺伝子(以下、sCマーカー遺伝子という)を除去対象遺伝子として用いた。上記sCマーカー遺伝子は、硫酸塩資化性に関与することが知られており、また、当該遺伝子の欠損株はセレン酸塩耐性を示す。従って当該遺伝子をマーカーとして上記NS4株に導入することにより、表現型として硫酸塩資化性およびセレン酸塩感受性を示すNS4株(以下、loxP::sC/NS4株という)を得ることができる。そのため、Creリコンビナーゼタンパク質の導入後に細胞がセレン酸塩耐性を獲得し、且つ硫酸塩資化性が消失したことを指標として、Cre−loxPシステムが正常に機能し、除去対象遺伝子が除去できたことを容易に確認することができる。
【0088】
本発明の実施例で用いたプラスミドを構築するために用いたloxP配列を含むプラスミド(以下、pUG6プラスミドという)、sCマーカー遺伝子が搭載されたプラスミド(以下、pUsCプラスミドという)、およびniaDマーカー遺伝子が搭載されたプラスミド(以下、pNGA142プラスミドという)は、全て東北大学の五味博士より供与して頂いた。なお、pUG6プラスミドについてはU Guldener, S Heck, T Fielder, J Beinhauer, JH Hegemann, Nucleic Acids Res 24 (1996) 2519-24を、pUsCプラスミドについてはO Yamada, BR Lee and K Gomi, Biosci. Biotech. Biochem. 61 (1997) 1367-69を、pNGA142プラスミドについてはS Tamalampudi, MMT Rahaman, S Hama, Y Suzuki, A Kondo and H Fukuda, Appl. Microbiol. Biotechnol 75 (2007) 387-95を参照のこと。
【0089】
〔実施例1:麹菌におけるsCマーカー遺伝子の除去〕
(1)loxP::sC搭載プラスミドの構築
pUG6のプラスミド地図を図7に示す。pUG6プラスミドは、2つのloxP配列(loxP1および2)に挟まれたカナマイシン耐性遺伝子(KanMX)を有し、図には示さないが、さらにアンピシリン耐性遺伝子及び大腸菌のOri領域を有するプラスミドである。
【0090】
上記pUG6プラスミド内にsCマーカー遺伝子が導入できるよう、図7中に示すSacIサイトから5’側22bp上流の部分に、QuikChange site-directed mutagenesis法により制限酵素サイトであるSphIサイトを導入した。
【0091】
具体的には、まず、SphIサイトを含む、配列番号1および配列番号2のプライマーを用い、pUG6プラスミドをテンプレートとして用いてPCRを行った。PCRの反応条件を表1に示す。
【0092】
【表1】

【0093】
反応終了後、制限酵素DpnIを1μl加え、37℃で2時間インキュベートし、テンプレートDNAを消化した。この溶液を用いて大腸菌の形質転換を行い、SphIサイトが新たに導入されたプラスミド(以下、pUG6SphIという)を得た。
【0094】
sCマーカー遺伝子が搭載されたpUsCプラスミドを制限酵素XbaIおよびSphIで切断し、sCマーカー遺伝子の全長cDNAを含むDNA断片を得た。得られたDNA断片を、予め制限酵素XbaIおよびSphIで切断したpUG6SphIプラスミドにライゲーションし、sCマーカー遺伝子が新たに導入されたプラスミド(以下、pUG6sCという)を得た。
【0095】
続いて、niaDマーカー遺伝子が搭載されたpNGA142プラスミドを制限酵素AvrIIおよびSpeIで切断し、硝酸塩資化性マーカーであるniaD遺伝子の全長cDNAを含むDNA断片を得た。得られたDNA断片を、予めSpeIで切断し、且つBAP処理を行ったpUG6sCプラスミドにライゲーションし、loxP::sC搭載プラスミド(以下、pUGsCniaDという)を得た。
【0096】
(2)NS4株へのloxP::sCプラスミドの導入
上記(1)で得られたloxP::sC搭載プラスミドであるpUGsCniaDを制限酵素BamHIで消化して直鎖状にしたものを用いて、NS4株の形質転換を行った。
【0097】
loxP::sCマーカーが導入された株の選抜は、配列番号3および配列番号4のプライマーを用いて、形質転換体のゲノムDNAに対してPCRを行い、約3.7kbの増幅断片のみが確認されたものを候補株とした。最終的に、sCマーカー遺伝子の部分配列をプローブとして用いたサザンハイブリダイゼーションにより、NS4株の染色体DNA中にloxP::sCマーカーが導入されたことを確認した。得られた形質転換体は、loxP::sC/NS4株と命名した。図3(A)に、loxP::sC/NS4株が有する遺伝子を表す。Cre−loxPシステムが機能すれば、得られるloxP/NS4株は、sC遺伝子が除去された図3(B)に示すような遺伝子を有すると予測された。
【0098】
(3)loxP::sC/NS4株へのCre酵素タンパク質の導入
まず、loxP::sC/NS4株のプロトプラスト化を行った。loxP::sC/NS4株の分生子懸濁液をYPD液体培地に添加し、30℃において20時間振盪培養した後に、滅菌したガラスフィルターを用いて菌体を回収した。回収した菌体を50ml容の遠心チューブに移し、10mlのプロトプラスト化溶液(1.2M ソルビトール、10mM リン酸二水素ナトリウム(pH6.0)、5mg/ml Lysing enzyme(Sigma Chemical Co.)、10mg/ml Cellulase Onozuka R−10(Yakult Pharmaceutical Ind.Co.,Ltd.)、10mg/ml Yatalase(TaKaRa))を加えて菌体を懸濁し、30℃において、90rpm、3時間振盪し、プロトプラスト化反応を行った。
【0099】
プロトプラスト化反応後に、セルストレイナー(352350、BD Falcon製)にて濾過し、濾液中のプロトプラストを、4℃において、3,000×gで5分間、遠心分離することにより、菌体を沈殿として得た。得られた菌体を、洗浄液(1.2M ソルビトール、10mM リン酸二水素ナトリウム(pH6.0))を用いて3回洗浄した。4℃において、3,000×gで5分間、遠心分離することにより、沈殿としてプロトプラストを得た。
【0100】
Creリコンビナーゼタンパク質をloxP::sC/NS4株のプロトプラストに導入するために、核酸として、Creリコンビナーゼのターゲット配列であるloxP配列を搭載したpUG6プラスミドを用いた。
【0101】
以下の2つの条件により、Creリコンビナーゼタンパク質溶液と沈殿として得られたプロトプラストとを混合し、氷上にて30分間インキュベートした。以後の操作は、通常のプロトプラスト−PEG法による真菌の形質転換法(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)に従って行った。
【0102】
<条件1:Cre+pUG6>
溶液Bに懸濁したプロトプラスト 100μl(細胞数:1×108個)
溶液C 12.5μl
Creリコンビナーゼタンパク質(631614、Clontech製) 10μl
pUG6プラスミド 3μg
<条件2:Creのみ>
溶液Bに懸濁したプロトプラスト 100μl(細胞数:1×108個)
溶液C 12.5μl
Creリコンビナーゼタンパク質 10μl
以下に溶液B及び溶液Cの組成を示す。
(溶液B)
1.2M ソルビトール、50mM CaCl2、10mM Tris−HCl(pH7.5)
(溶液C)
50% PEG4000、50mM CaCl2、10mM Tris−HCl(pH7.5)
Cre−loxPシステムが機能し、sCマーカー遺伝子が除去されたことを確認するために、最終濃度0.1mMのセレン酸を含有するプレートに、最終濃度0.8MとなるようにNaClを加えたものを用いた。結果は図には示さないが、Cre酵素タンパク質と核酸(pUG6プラスミド)とを用いた方法で、Cre−loxPシステムが正常に機能し、Cre酵素タンパク質のみを用いた場合と比較して、得られる形質転換体の数が有意に増加した。
【0103】
(4)Cre−loxPシステムの機能確認
sCマーカー遺伝子が除去されていることを確認するために、得られた候補株に対して、コロニーPCRを行った。図3は、sCマーカー遺伝子除去方法の概念を説明するための図を表す。図3(A)は、loxP::sC/NS4株が有する遺伝子を表し、図3(B)は、sCマーカー遺伝子が除去された株(loxP/NS4株)が有する遺伝子を表す。図3中の矢印に挟まれた部分をPCRによって増幅することができるような配列番号5および配列番号6のプライマーを作製した。これらのプライマーセットを用いてPCRを行えば、sCマーカー遺伝子がゲノム上に存在する場合には全長4.1kbのDNA断片が増幅されるが、sCマーカー遺伝子が除去され、ゲノム上に存在しない場合には、全長0.8kbのDNA断片が増幅される。
【0104】
上記PCR反応は酵素としてEx taq(RR001A、TaKaRa製)を用いた。反応条件を表2に示す。
【0105】
【表2】
【0106】
コロニーPCRの結果を図4に示す。図4中、レーン1〜4は、Creのみを用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン5〜9は、Cre酵素をpUG6プラスミドをキャリアとして用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン10は、loxP::sC/NS4株のゲノムをテンプレートにしてコロニーPCRを行った結果を表す。Creリコンビナーゼタンパク質と核酸(pUG6プラスミド)とを導入した場合にのみ0.8kbのバンドの増幅が確認された。PCRにて目的の長さのバンドが確認された形質転換体について、さらに、sCマーカー遺伝子の部分配列をプローブとしてサザンハイブリダイゼーションを行うことにより、sCマーカー遺伝子が除去されたことを確認した。得られた株をloxP/NS4株と命名した。
【0107】
loxP/NS4株、及びその親株であるloxP::sC/NS4株、さらにその親株であるNS4株について、栄養要求性による表現型観察を行い、マーカー除去の確認を行った。結果を図5に示す。図5は、sCマーカー遺伝子除去株の栄養要求性を示す図である。図5中のCDは、Czapek−DoxにN源として硝酸塩(NO3)を加え、S源として硫酸塩(SO4)を加えた最小培地(以下、CD培地という)を表す。図5中のCDMは、CD培地において、S源としての硫酸塩の代わりにメチオニンを最終濃度が30mg/mlとなるように加えた培地を表す(以下、CDM培地という)。図5中のCDMEは、CDM培地において、N源としての硝酸塩の代わりにグルタミン酸塩を最終濃度が70mMとなるように加えた培地を表す。
【0108】
それぞれの株の分生子を各培地上にそれぞれ約103個となるようにスポット植菌し、30℃で4日間培養した。NS4株は、NO3およびSO4を資化できないため、CD培地およびCDM培地では生育できなかった。また、loxP::sC/NS4株は、niaD遺伝子およびsC遺伝子が導入されているため、NO3およびSO4をともに資化することができ、最小培地であるCD培地においても生育が可能であった。一方、loxP/NS4株は、CD培地のみで生育することができず、sCマーカー遺伝子が除去された事によりSO4を資化することができない硫酸塩非資化性株となったことが明らかになった。
【0109】
以上の結果からCreリコンビナーゼタンパク質と核酸とを用いた方法でCre−loxPシステムが機能することが明らかとなった。
【0110】
〔実施例2〕
核酸として、一本鎖DNA(サケの精子由来)(201190、STRATGENE製)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0111】
〔実施例3〕
核酸として、pUC18プラスミド(3218、TaKaRa製)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0112】
〔実施例4〕
核酸として、pUC18断片(実施例3で用いたpUC18プラスミドを制限酵素XbaIで切断し断片化したもの)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0113】
実施例2で得られた形質転換体のコロニーPCRの結果を図6に示す。図6中、レーン1〜14は、Cre酵素と一本鎖DNAとを用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン15は、loxP::sC/NS4株のゲノムをテンプレートにしてコロニーPCRを行った結果を表す。図6からわかるように、核酸として一本鎖DNAを用いた場合であっても、0.8kbのバンドが増幅され、Cre−loxPシステムが正常に機能し、sCマーカー遺伝子が除去されたことが明らかになった。図には示さないが、実施例3および4で得られた形質転換体においても、コロニーPCRの結果からCre−loxPシステムが正常に機能し、sCマーカー遺伝子が除去されたことが明らかになった。以上の結果は、核酸であればキャリアとして機能する事を示すものである。
【0114】
本発明にかかる核内へタンパク質を導入する方法は、キャリアとしての核酸の形態を選ばないため、上述したCre−loxPシステムを用いた部位特異的遺伝子除去のみならず、Cre−loxPシステムを用いた従来公知の部位特異的遺伝子組換えにも適用することができることは言うまでもない。
【産業上の利用可能性】
【0115】
本発明にかかるタンパク質核内導入方法によれば、タンパク質を機能させたい細胞において、目的のタンパク質をコードする遺伝子が導入された形質転換体を作製する手間を省くことができる。また、タンパク質を細胞内に導入するため、遺伝子発現カセットプラスミドを脱落させることなく、必要な時期に一過的にタンパク質を機能させることができる。よって、部位特異的遺伝子除去方法又は部位特異的遺伝子組換え方法を簡便、且つ迅速に行うことができるため、上記方法を用いる医薬品産業等の産業において好適に利用することができる。
【図面の簡単な説明】
【0116】
【図1】Cre−loxPシステムを用いた従来の部位特異的遺伝子除去方法と本発明にかかる部位特異的遺伝子除去方法とを比較した図である。(A)は、従来の部位特異的遺伝子除去方法を表し、(B)は、本発明にかかる部位特異的遺伝子除去方法を表す。
【図2】Cre−loxPシステムを用いた従来の部位特異的遺伝子組換え方法と本発明にかかる部位特異的遺伝子組換え方法とを比較した図である。(A)は、従来の部位特異的遺伝子組換え方法を表し、(B)は、本発明にかかる部位特異的遺伝子組換え方法を表す。
【図3】sCマーカー遺伝子除去方法の概念を説明するための図であり、(A)は、loxP::sC/NS4株が有する遺伝子を表し、(B)は、loxP/NS4株が有する遺伝子を表す。
【図4】Creリコンビナーゼタンパク質のみを形質転換した場合のコロニーPCRの結果と、Creリコンビナーゼタンパク質と核酸とを形質転換した場合のコロニーPCRの結果とを示す図である。
【図5】sCマーカー遺伝子除去株の栄養要求性を検討した結果を示す図である。
【図6】一本鎖DNAをキャリアとして用いた際のコロニーPCRの結果を示す図である。
【図7】pUG6のプラスミド地図である。
【技術分野】
【0001】
本発明は、タンパク質を核内に導入する方法、およびその利用に関する。具体的には、タンパク質をコードする遺伝子を細胞に導入してタンパク質を発現させる工程を必要とせず、タンパク質を核内に簡便に導入する方法およびその利用に関する。
【背景技術】
【0002】
従来、外来タンパク質を細胞内で機能させる場合は、目的のタンパク質をコードする遺伝子のcDNAを含むプラスミドを細胞内に導入し、細胞内でタンパク質を発現させる方法(遺伝子導入法)が用いられてきた。
【0003】
例えば、遺伝子の機能解明を行う場合、一般に、Cre−loxPシステムを用いた目的遺伝子の除去または挿入が行われている。図1(A)は、上記Cre−loxPシステムの概要を示す図である。大腸菌を宿主とするP1バクテリオファージの部位特異的組換え酵素であるCreリコンビナーゼは、38kDaのタンパク質であり、34塩基からなるloxP配列を特異的に認識することにより、2つのloxP配列間において特異的組換えを効率よく引き起こす(非特許文献1)。図1(A)に示すように、特に、同一DNA鎖上に、2つのloxP配列が同方向にある場合、loxPで挟まれたDNA領域を、効率よく環状に切り出して除去する。一方、図2(A)に示すように、DNA鎖上に1つのloxP配列がある場合は、上記DNA鎖上のloxP配列を標的として、分子内にloxP配列を含む環状DNAを挿入することができる。
【0004】
糸状菌を例に挙げると、Aspergillus fumigatus菌体内(非特許文献2)、または、A. nidulans菌体内(非特許文献3)において、Creリコンビナーゼを発現させ、染色体上にある除去対象遺伝子の上流および下流に連結されたloxP配列との組換えを行う事で、染色体上のloxP配列の間にある除去対象遺伝子を除去することに成功している。また、Cre−loxPシステムを用いた目的遺伝子の除去方法または挿入方法は、酵母等でも一般に行われている確立された方法である(非特許文献4)。
【非特許文献1】N. Sternberg and D. Hamilton, J Mol Biol 150 (1981) 467-86.
【非特許文献2】S. Krappmann, O. Bayram and G.H. Braus, Eukaryot Cell 4 (2005) 1298-307.
【非特許文献3】J.V. Forment, D. Ramon and A.P. MacCabe, Curr Genet 50 (2006) 217-24.
【非特許文献4】B. Sauer, Mol Cell Biol 7 (1987) 2087-96.
【非特許文献5】A. Kuspa and W. F. Loomis, Proc. Natl. Acad. Sci. USA 89 (1992) 8803-8807.
【非特許文献6】X. Jin, M. Ming-He, Z. Wei, H. Xiao-Wei and Z. Ke-Qin, The Journal of Microbiology, Vol. 43, No. 5 (2005), 417-423.
【発明の開示】
【発明が解決しようとする課題】
【0005】
しかしながら、上述のように目的遺伝子を染色体上の特定の部位に挿入する方法は確立されてはいるものの、上記従来の構成では、多くの煩雑なステップと時間が必要であった。図2を参照しながら具体的に説明すると、まず、Creリコンビナーゼを発現するプラスミドを細胞内に導入し、プラスミド保持細胞を選抜する。次いで、Creリコンビナーゼの発現を誘導し、loxP配列に挟まれた第一遺伝子を除去する。その後、loxP配列をターゲットに第二遺伝子を挿入するためには、図2に示すような、特異的部位を含む環状目的配列を細胞内に導入し、誘導をかけて部位特異的組換え酵素を発現させるという煩雑なステップが必要であり、さらに目的遺伝子の脱落を防ぐためには、部位特異的組換え酵素発現カセットプラスミドを脱落させ、細胞内で部位特異的組換え酵素の存在をなくす必要があり、時間と手間がかかる作業であった(図2(A))。
【0006】
本発明は、上記の問題点に鑑みてなされたものであり、その主たる目的は、上述したような、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための工程、部位特異的組換え酵素発現プラスミドを脱落する工程等の煩雑な工程を必要としない、簡便、且つ迅速な部位特異的遺伝子除去方法および部位特異的遺伝子組換え方法を実現することにある。
【課題を解決するための手段】
【0007】
本発明者は、上記課題に鑑み、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるのではなく、部位特異的組換え酵素タンパク質そのものを細胞の外部から細胞の内部に導入することができれば、極めて効率的になると考え、タンパク質の核内導入方法について鋭意検討した。その結果、核酸をキャリアとして部位特異的組換え酵素タンパク質を細胞へ導入する事で、部位特異的組換え酵素タンパク質を効率良く核内に導入しうることを見出し、本発明を完成するに至った。
【0008】
すなわち、本発明にかかるタンパク質を核内へ導入する方法(以下、「タンパク質核内導入方法」という)は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴としている。
【0009】
本発明にかかるタンパク質核内導入方法によれば、従来の遺伝子導入方法のように目的のタンパク質をコードする遺伝子を細胞に導入して目的タンパク質を発現させる工程を必要としないため、タンパク質を核内に簡便に導入することができる。また、従来の遺伝子導入方法では、タンパク質が細胞内で機能する時期を調節するためには、タンパク質の発現を調節可能な別のシステムをさらに導入するか、目的のタンパク質をコードする遺伝子を脱落させる必要がある。一方、本発明にかかるタンパク質核内導入方法によれば、タンパク質を細胞内に導入するため、導入されたタンパク質が分解された後には、タンパク質が機能することはなく、細胞内でタンパク質を機能させる時期を容易に調節することができる。
【0010】
ところで、非特許文献5および6には、形質転向率や相同組み換え効率を上昇させるために、制限酵素と核酸とを細胞内に導入する方法(REMI法)について記載されている。上記REMI法において細胞内に導入される制限酵素を制限酵素Aとすると、細胞内に導入される核酸としては、両端が制限酵素Aによって消化された直鎖状の核酸が用いられる。上記REMI法を用いれば、細胞の染色体上に存在する制限酵素Aの特異的認識配列を制限酵素Aを用いて切断するため、切断された部位に目的の核酸を効率よく導入することができる。非特許文献5または6に記載のREMI法は、核酸を染色体内の所望の部位に導入することを目的として制限酵素と核酸とを細胞内に導入し、これにより形質転換効率や相同組換え効率を上昇させる方法である。よって、本発明のタンパク質核内導入方法とは目的も効果も全く違う方法である。
【0011】
本発明にかかるタンパク質核内導入方法においては、上記タンパク質は、部位特異的組換え酵素であることが好ましい。上記タンパク質が部位特異的組換え酵素であれば、後述する部位特異的遺伝子除去方法および部位特異的遺伝子組換え方法を簡便、且つ迅速に行うことができる。
【0012】
本発明にかかるタンパク質核内導入方法においては、上記核酸は、全長が50塩基〜300,000塩基の範囲内であることが好ましい。核酸の全長が上記範囲内であれは、より効率よくタンパク質を核内に導入することができる。
【0013】
また、本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴としている。
【0014】
図1(A)に示すように、従来の部位特異的遺伝子除去方法では、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるために複数の工程を行う必要があったが、本発明にかかる部位特異的遺伝子除去方法を用いれば、これらの工程を省略することができる。従って、除去対象遺伝子の除去を簡便、且つ迅速に行うことができる。
【0015】
また、本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含むことを特徴としている。
【0016】
図2(A)に示すように、従来の部位特異的遺伝子組換え方法では、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程に加えて、目的遺伝子が挿入された細胞において部位特異的組換え酵素が発現し続けることにより、挿入部位から当該目的遺伝子が除去されることを防ぐために、上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を行う必要があった。本発明にかかる部位特異的遺伝子組換え方法を用いれば、部位特異的組換え酵素のタンパク質を細胞内に導入するため、導入されたタンパク質が分解された後には、上記部位特異的組換え酵素は機能しない。よって、部位特異的組換え酵素を細胞内で発現させるための複数の工程および上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を省略することができ、部位特異的遺伝子組換えをより簡便に行うことができる。
【0017】
本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであることを特徴としている。
【0018】
核酸が、目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであれば、部位特異的組換え酵素と目的遺伝子とを1つの工程で同時に細胞内に導入することができるため、本発明にかかる部位特異的遺伝子組換え方法をより簡便に行うことができる。
【0019】
また、本発明にかかる部位特異的遺伝子除去キットは、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴としている。
【0020】
上記構成によれば、少なくとも部位特異的組換え酵素と核酸とをキット内に含むため、部位特異的組換え酵素を簡便に核内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができる。
【0021】
また、本発明にかかる部位特異的遺伝子組換えキットは、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴としている。
【0022】
上記構成によれば、少なくとも部位特異的組換え酵素と核酸とをキット内に含むため、部位特異的組換え酵素を簡便に核内に導入することができ、目的遺伝子の挿入を簡便、且つ迅速に行うことができる。
【発明の効果】
【0023】
本発明にかかるタンパク質核内導入方法は、以上のように、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含んでいるので、タンパク質をコードする遺伝子を細胞に導入し、発現させる工程を必要とせず、簡便且つ容易にタンパク質を核内に導入することができるという効果を奏する。また、タンパク質を細胞内に導入するため、細胞内でタンパク質を機能させる時期を容易に調節することができるというさらなる効果を奏する。
【0024】
本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むため、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程を行う必要が無く、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0025】
本発明にかかる部位特異的遺伝子組換え方法は、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含むため、部位特異的組換え酵素をコードする遺伝子を細胞内で発現させるための複数の工程、および遺伝子挿入後に上記部位特異的組換え酵素を発現するプラスミドを細胞内から脱落させる工程を行う必要が無く、部位特異的遺伝子組換えをより簡便に行うことができるという効果を奏する。
【0026】
本発明にかかる部位特異的遺伝子組換え方法は、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであるため、部位特異的組換え酵素と目的遺伝子とを1つの工程で同時に細胞内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0027】
本発明にかかる部位特異的遺伝子除去キットは、以上のように除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むため、部位特異的組換え酵素を簡便に核内に導入することができ、除去対象遺伝子の除去を簡便、且つ迅速に行うことができるという効果を奏する。
【0028】
本発明にかかる部位特異的遺伝子組換えキットは、以上のように少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むため、部位特異的組換え酵素を簡便に核内に導入することができ、目的遺伝子の挿入を簡便、且つ迅速に行うことができるという効果を奏する。
【発明を実施するための最良の形態】
【0029】
本発明の実施の形態について説明すれば以下のとおりであるが、本発明はこれに限定されるものではない。
【0030】
なお、本明細書中において範囲を示す「A〜B」は、A以上B以下であることを示す。本発明の一実施形態について説明すると以下の通りである。
【0031】
〔1.タンパク質核内導入方法〕
一実施形態において、本発明にかかるタンパク質核内導入方法は、タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む。上記タンパク質としては、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、部位特異的組換え酵素、DNA修復に関与するDNA結合タンパク質やリガーゼといった酵素、抗体等を用いることができる。上記部位特異的組換え酵素については、後述する。
【0032】
上記DNA修復に関与するタンパク質としては、特に限定されるものではないが、例えば、Rad51、Rad52、Rad54等を挙げることができる。上記抗体としては、特に限定されるものではないが、Kuタンパク質、DNAリガーゼ等に対する抗体を挙げることができる。
【0033】
Rad51、Rad52、Rad54等の相同組換えに関与するタンパク質群を細胞核内に導入することにより、相同組換え効率を上昇させることができる。または、非相同末端結合に関与するタンパク質に対する抗体、例えば、Kuタンパク質、DNAリガーゼ等に対する抗体を細胞核内に導入し、抗原抗体反応により、核内で正常に機能する非相同末端結合に関与するタンパク質を減少させる事で、ターゲティング効率を上昇させることができる。
【0034】
また、上記タンパク質は、天然の細胞由来であっても、遺伝子組換えによって製造されたものでもよい。また、上記タンパク質は、一種類を単独で用いてもよいし、二種類以上を併用してもよい。
【0035】
本発明で用いられる「核酸」とは、本発明に係るタンパク質核内導入方法においてタンパク質とともに細胞に導入される核酸を意味する。また、「核酸をキャリアとする」とは、特に限定されるものではないが、例えば、タンパク質と核酸とをともに細胞に導入することを意味する。このとき核酸は対象タンパク質を核内に運ぶキャリアとしての役割を果たす。発明者らの検討によれば、上記核酸としては、リン酸、ペントース、および塩基からなるヌクレオチドが重合したポリヌクレオチドである核酸であれば塩基配列や長さは特に限定されるものではないことが分かっている。また上記核酸としては、デオキシリボ核酸(DNA)であってもよいし、リボ核酸(RNA)であってもよい。なお、RNAと比較して、DNAは非常に安定性が高く、取り扱い性に優れるため、上記核酸としては、DNAを用いることが好ましい。
【0036】
また、本発明で用いられる核酸は、一本鎖構造を有するものであってもよいし二本鎖構造を有するものであってもよい。一本鎖構造を有する核酸と比較して、二本鎖構造を有する核酸の方が、一般に安定性が高く、取り扱い性に優れるため、本発明で用いられる核酸としては、二本鎖構造を有するものを用いることが好ましい。
【0037】
また、本発明で用いられる核酸は、直鎖状であってもよいし、環状であってもよい。本発明で用いられる核酸の全長として特に限定されるものではないが、50塩基〜300,000塩基であることが好ましく、300〜100,000塩基であることがより好ましい。核酸の全長が50塩基以上であれば、対象タンパク質を核内に運ぶキャリアとしてより好ましく機能する。また、300,000塩基以下であれば、細胞内への核酸の導入効率が高くなる。
【0038】
本発明で用いられる核酸としては、例えば、細胞由来の核酸、タンパク質が導入される細胞が由来する生物以外の生物由来の核酸、プラスミド、PCR増幅断片、合成核酸等を利用することができる。
【0039】
本発明で用いられる核酸として、細胞由来の核酸を用いる場合、当該細胞は、微生物(例えば、バクテリア、酵母、真菌等)由来であってもよいし、植物由来であってもよいし、動物(例えば、マウス、ヒト等)由来であってもよく、適宜選択することができる。さらに、上記細胞由来の核酸の種類としては、例えば、染色体DNAであってもよく、totalRNAであってもよく、mRNAであってもよい。安定性が高く取り扱い性に優れることから、染色体DNAであることがより好ましい。また、細胞由来の核酸は、従来公知の核酸分離法を用いることにより核酸のみを単離することができる。例えば、染色体DNAを単離したい場合、従来公知のフェノール・クロロホルム溶解及び抽出/エタノール析出を用いることができる。また、totalRNAまたはmRNAを単離したい場合は、従来公知のホットフェノール法を用いることができる。
【0040】
上記核酸が有する塩基配列は、特に限定されるものではなく、どのような塩基配列の核酸も好適に用いることができる。例えば、塩基をランダムに選択して合成した合成核酸を用いることもできる。
【0041】
上記核酸は、一種類を単独で用いてもよいし、二種類以上を併用してもよい。
【0042】
また、タンパク質が導入される細胞としては、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物(例えば、バクテリア、酵母、真菌等)、植物、動物(例えば、マウス、ヒト等)の細胞を挙げることができる。一例として、微生物細胞を用いる場合は、特に限定されるものではないが、例えば、麹菌、クリプトコッカス等の細胞を好適に用いることができる。また、一例として、動物細胞を用いる場合は、細胞種としては、特に限定されるものではないが、例えば、線維芽細胞、臍帯静脈内皮細胞等の細胞を好適に用いることができる。
【0043】
上記「タンパク質を核内に導入する」方法としては、タンパク質と核酸とを細胞内に導入できる限り特に限定されるものではなく、従来公知の形質転換法を用いて行うことができる。上記形質転換法としては、例えば、プロトプラスト−PEG法(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)、エレクトロポレーション法(BN Chakrabory and M Kapoor, Nucleic Acids Res 25 (1990) 6737を参照)等を用いることができ、タンパク質を導入される生物種および細胞種によって最適な形質転換法を選択することが好ましい。
【0044】
例えば、上記プロトプラスト−PEGを用いる場合、まず、部位特異的組換え酵素を導入するために、プロトプラスト細胞を作製する。当該プロトプラストの作製は、当業者に公知の任意の方法・手段を用いて作製することが可能である(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)。例えば、市販されているヤタラーゼ(TO17、TaKaRa製)、セルラーゼ(201093、Yakult Pharmaceutical製)、ライジングエンザイム(L1412、Sigma Chemical製)等を単独、または混合して用いることができる。次いで、部位特異的組換え酵素、核酸、プロトプラスト、およびPEGを含むプロトプラスト等張液を混合し、氷上にて20〜30分間インキュベートを行う。以後の操作は、当業者に公知のプロトプラスト−PEG法による形質転換法と同様に行う事ができる。
【0045】
また、上記「タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程」において、タンパク質と核酸とを別々に導入してもよいし、同時に導入してもよい。タンパク質と核酸とを別々に導入する場合、導入する順序については特に限定されるものではなく、タンパク質を導入した後に核酸を導入してもよいし、核酸を導入した後にタンパク質を導入してもよい。
【0046】
導入の際に細胞に与えるダメージを考慮すると、タンパク質と核酸とを同時に細胞内に導入することが好ましい。タンパク質と核酸とを同時に細胞内に導入することにより、導入操作を1回行えばよく、細胞に与えるダメージも少ないため好ましい。
【0047】
〔2.部位特異的遺伝子除去方法〕
一実施形態において、本発明にかかる部位特異的遺伝子除去方法は、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記除去対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含む。
【0048】
上記「部位特異的組換え酵素」は、DNAの分子内あるいは分子間の特定部位で起こる組換えの過程を触媒する酵素のことを指す。また、上記「特異的認識配列」は、上記部位特異的組換え酵素によって特異的に認識される配列のことを指す。当該部位特異的組換え酵素は、上記特異的認識配列に挟まれたDNA断片を切り出し、環状化させる機能を有する。さらに、上記部位特異的組換え酵素は、分子内に上記特異的認識配列を有する環状DNAを、上記特異的認識配列を有する別のDNA分子内に、当該特異的認識配列を介して挿入する反応も行うこともできる。上記「部位特異的組換え酵素」としては、例えば、Creリコンビナーゼ、Flpリコンビナーゼ等を挙げることができる。これらの部位特異的組換え酵素は、天然の細胞由来であっても、遺伝子組換えによって製造されたものであってもよい。
【0049】
例えば、上記部位特異的組換え酵素としてCreリコンビナーゼを用いる場合、上記「特異的認識配列」としては、loxP配列が用いられる。当該loxP配列は、Creリコンビナーゼによって特異的に認識される塩基配列である(K Abremski, R Hoess and N Sternberg, Cell 32 (1983) 1301-11を参照)。他に上記部位特異的組換え酵素としてFlpリコンビナーゼを用いる場合、上記「特異的認識配列」としては、FRT配列が用いられる(D Babineau, D Vetter, BJ Andrews, RM Gronostajski, GA Proteau, LG Beatty, and PD Sadowski, J Biol Chem 260 (1985) 12313-9を参照)。
【0050】
上記除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖(例えば、「loxP:: 除去対象遺伝子」で表すことができる)は、細胞に1つ以上備えられていればよい。
【0051】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0052】
本発明にかかる部位特異的遺伝子除去方法で用いられる細胞としては、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。
【0053】
ここで「除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞」とは、染色体上に除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を有する細胞、または染色体外に除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖(例えば、プラスミド等)を保持している細胞を意味する。
【0054】
本発明にかかる部位特異的遺伝子除去方法で用いられる、細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0055】
ここで、Cre−loxPシステムを用いた部位特異的遺伝子除去方法について、図1を参照しながら説明する。図1において、(A)は従来の部位特異的遺伝子除去方法を示し、(B)は本発明にかかる部位特異的遺伝子除去方法を示す。図1(A)に示されるように、従来の部位特異的遺伝子除去方法を用いた場合、以下の工程が必要である。具体的には、(1)除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞において機能するプロモーター配列の下流に、Cre遺伝子を連結したCre発現カセットを含むCre発現プラスミドを作製する。(2)作製したCre発現プラスミドを目的の細胞に形質転換する。(3)上記Cre発現プラスミドが形質転換された細胞を選抜する。(4)上記選抜された細胞内においてCreリコンビナーゼタンパク質の発現を誘導し、除去対象遺伝子を除去する。(5)除去対象遺伝子が除去された細胞を選抜する。
【0056】
一方、本発明にかかる部位特異的遺伝子除去方法によれば、図1(B)に示されるように、少なくともCreリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含めば、除去対象遺伝子を簡便に除去することができる。従来の部位特異的遺伝子除去方法を用いた場合に必要であった上記(1)〜(4)の工程の代わりに、Creリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含めばよく、非常に簡便に部位特異的遺伝子除去を行うことができる。
【0057】
〔3.部位特異的遺伝子組換え方法〕
一実施形態において、本発明にかかる部位特異的遺伝子組換え方法は、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程と、を含む。
【0058】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。核酸としては、核酸であれば特に限定されるものではなく、上記〔1.タンパク質核内導入方法〕で説明したものを好適に用いることができる。
【0059】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞としては、少なくとも1つ以上の特異的認識配列を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。なお、〔2.部位特異的遺伝子除去方法〕によって除去対象遺伝子が除去された細胞は、当該細胞内に特異的認識配列を備えるため、この細胞を用いれば、特異的認識配列を細胞に導入する工程を省略することができるため、好適に用いることができる。
【0060】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0061】
上記部位特異的組換え酵素が、細胞に備えられた1つ以上の特異的認識配列に、当該特異的認識配列を介して目的遺伝子を挿入するためには、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する必要がある。よって、本発明にかかる部位特異的遺伝子組換え方法は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程に加えて、さらに上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程(以下、環状二本鎖DNA導入工程という)を含む。当該環状二本鎖DNA導入工程は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入することができる限り、特に限定されるものではなく、従来公知の形質転換法を用いて上記環状二本鎖DNAを細胞内に導入することが出来る。例えば、プロトプラスト−PEG法、エレクトロポレーション法を用いることができる。
【0062】
当該環状二本鎖DNA導入工程は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程の前に行ってもよいし、後で行ってもよい。細胞内に導入された部位特異的組換えタンパク質の細胞内での安定性を考慮に入れると、上記遺伝子導入工程を行った後に、部位特異的組換え酵素タンパク質の核内導入を行うことが好ましい。
【0063】
一実施形態において、本発明にかかる部位特異的遺伝子組換え方法は、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAである。
【0064】
本発明にかかる部位特異的遺伝子組換え方法において、上記部位特異的組換え酵素は、環状分子を、特異的認識配列を介して挿入するため、本発明で用いられる核酸として目的遺伝子を含む核酸を用いる場合は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAである必要がある。
【0065】
本発明において核酸として用いられる環状二本鎖DNAの全長は、導入したい目的遺伝子の全長によるが、50塩基〜300,000塩基対であることが好ましく、2,000〜200,000塩基対であることがより好ましい。核酸として用いられる上記環状二本鎖DNAの全長が300,000塩基対以下であれば細胞内への核酸の導入効率が高くなる。
【0066】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。本発明で用いられる核酸としては、核酸であれば特に限定されるものではなく、上記〔1.タンパク質核内導入方法〕で説明したものを好適に用いることができる。
【0067】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞としては、少なくとも1つ以上の特異的認識配列を備える細胞である限り、特に限定されるものではなく、目的に応じて適宜選択することができる。例えば、微生物由来であってもよく、植物由来であってもよく、動物由来であってもよい。なお、〔2.部位特異的遺伝子除去方法〕によって除去対象遺伝子が除去された細胞は、当該細胞内上に特異的認識配列を備えるため、この細胞を用いれば、特異的認識配列を細胞に導入する工程を省略することができるため、好適に用いることができる。
【0068】
本発明にかかる部位特異的遺伝子組換え方法で用いられる細胞内への部位特異的組換え酵素および核酸の導入方法としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0069】
ここで、Cre−loxPシステムを用いた部位特異的遺伝子組換え方法について、図2を参照しながら説明する。図2において、(A)は従来の部位特異的遺伝子組換え方法、(B)は本発明にかかる部位特異的遺伝子組換え方法を示す。図2(A)に示されるように、従来の部位特異的遺伝子組換え方法を用いた場合、以下の工程が必要である。具体的には、(1)目的遺伝子を導入したい細胞において機能するプロモーターの下流にCre遺伝子をつないだCre発現カセットを含むプラスミドを作製する。(2)作製したCre発現プラスミドを目的の細胞に形質転換する。(3)上記Cre発現プラスミドが形質転換された細胞を選抜する。(4)目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する。(5)上記細胞内においてCreリコンビナーゼタンパク質の発現を誘導する。(6)目的遺伝子が導入された細胞を選抜する。(7)導入された目的遺伝子が除去されることを防ぐために、Creリコンビナーゼ発現カセットを含むプラスミドを脱落させる。尚、図2(A)の工程(4)では、一例として目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNAを用いているが、目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを用いてもよい。
【0070】
本発明にかかる部位特異的遺伝子組換えは、図2(A)に示されるような従来の部位特異的遺伝子組換え方法を用いた場合に必要であった上記(1)〜(4)の工程の代わりに、Creリコンビナーゼタンパク質を、核酸をキャリアとすることによって細胞内に導入すればよく、非常に簡便に部位特異的遺伝子組換えを行うことができる。一実施形態において、本発明で用いられる核酸として目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを利用すれば、Creリコンビナーゼタンパク質と目的遺伝子とを細胞内に導入することができるため、さらに簡便に部位特異的遺伝子組換えを行うことができる。
【0071】
また、本発明にかかる部位特異的遺伝子組換え方法を用いれば、細胞内に導入されたCreリコンビナーゼタンパク質は細胞分裂とともに細胞内で希釈され、時間とともに細胞内のタンパク質分解酵素により分解されるため、従来の部位特異的遺伝子組換え方法のようにプラスミドを脱落させる必要が無い。
【0072】
〔4.部位特異的遺伝子除去キット〕
本発明にかかる部位特異的遺伝子除去キットは、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含む。
【0073】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。
【0074】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0075】
本発明にかかる部位特異的遺伝子除去キットは、少なくとも上記部位特異的組換え酵素と上記核酸とを含んでいればよく、これらは別々の容器に充填されていてもよいし、同じ容器に充填されていてもよい。タンパク質と核酸とでは、安定性が異なるため、取り扱い性を考慮に入れると、別々の容器に充填されていることが好ましい。
【0076】
また、本発明にかかる部位特異的遺伝子除去キットは、上記部位特異的組換え酵素および上記核酸以外の成分を含んでいてもよい。例えば、形質転換に必要な試薬、2つの特異的認識配列の間に、除去対象遺伝子を連結することができるマルチクローニングサイトを備えるプラスミド、プラスミド作成用オリゴプライマー、除去株確認用オリゴプライマー等を挙げることができる。なお、上記除去株確認用オリゴプライマーは、除去対象遺伝子が除去された株を選択するPCRに用いるためのオリゴプライマーを指す。本発明にかかる部位特異的遺伝子除去キットを用いて部位特異的遺伝子除去を行うためには、予め、除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞を準備する必要がある。よって、上記2つの特異的認識配列の間に、除去対象遺伝子を連結することができるマルチクローニングサイトを備えるプラスミドは、上記細胞を得るために用いることができる。
【0077】
〔5.部位特異的遺伝子組換えキット〕
本発明にかかる部位特異的遺伝子組換えキットは、少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含む。
【0078】
上記部位特異的組換え酵素としては、上記〔2.部位特異的遺伝子除去方法〕で説明したとおりである。
【0079】
上記核酸としては、上記〔1.タンパク質核内導入方法〕で説明したとおりである。
【0080】
本発明にかかる部位特異的遺伝子組換えキットは、少なくとも上記部位特異的組換え酵素と上記核酸とを含んでいればよく、これらは別々の容器に充填されていてもよいし、同じ容器に充填されていてもよい。タンパク質と核酸とでは、安定性が異なるため、取り扱い性を考慮に入れると、別々の容器に充填されていることが好ましい。
【0081】
また、本発明にかかる部位特異的遺伝子組換えキットは、上記部位特異的組換え酵素および上記核酸以外の成分を含んでいてもよい。例えば、形質転換に必要な試薬、1つ以上の特異的認識配列を含むプラスミド、1つの特異的認識配列と目的遺伝子を連結することができるマルチクローニングサイトとを備えるプラスミド、2つの特異的認識配列の間に、目的遺伝子を連結することができるマルチクローニングサイトを備えるプラスミド、プラスミド作成用オリゴプライマー、遺伝子組換え株確認用オリゴプライマー等を挙げることができる。なお、上記遺伝子組換え株確認用オリゴプライマーは、目的遺伝子が挿入された株を選択するPCRに用いるためのオリゴプライマーを指す。本発明にかかる部位特異的遺伝子組換えキットを用いて部位特異的遺伝子組換えを行うためには、予め、少なくとも1つ以上の特異的認識配列を備える細胞を準備する必要がある。よって、上記1つ以上の特異的認識配列を含むプラスミドは、上記細胞を得るために用いることができる。
【0082】
また、本発明にかかる部位特異的遺伝子組換えキットを用いて部位特異的遺伝子組換えを行うためには、目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを細胞内に導入する必要がある。上記目的遺伝子は、必要に応じて任意に変更可能であることが好ましい。上記1つの特異的認識配列と目的遺伝子を連結することができるマルチクローニングサイトとを備えるプラスミド、または上記2つの特異的認識配列の間に、目的遺伝子を連結することができるマルチクローニングサイトを備えるプラスミドを用いれば、上記環状二本鎖DNAを構築する際に、目的遺伝子を容易に導入することができる。また、一実施形態において、本発明で用いられる核酸として上記環状二本鎖DNAを用いることも可能である。
【0083】
なお、本発明は上記の各実施形態に限定されるものではなく、請求項に示した範囲で種種の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0084】
以下に、実施例を挙げて本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。
【0085】
本発明の実施例では、タンパク質が核に移行し、正常に機能することを確認するためのモデル細胞として真菌を用いた。当該真菌としては麹菌NS4株(niaD−,sC−)(以下、NS4株という)を用いた。当該NS4株は、独立行政法人酒類総合研究所にて保存される麹菌株であり、硝酸塩および硫酸塩を資化することができない表現型を有する麹菌株である。
【0086】
また、本発明の実施例では、タンパク質が核に移行し、正常に機能することを確認するための手段として、従来公知のCre−loxPシステムを用いた。当該システムを用いれば、loxP配列が上流および下流に連結された除去対象遺伝子がCreリコンビナーゼによって除去されたことを指標として、タンパク質が核に移行し、且つ正常に機能したことを確認することができる。
【0087】
さらに、本発明の実施例では、A. nidulansのsC遺伝子(以下、sCマーカー遺伝子という)を除去対象遺伝子として用いた。上記sCマーカー遺伝子は、硫酸塩資化性に関与することが知られており、また、当該遺伝子の欠損株はセレン酸塩耐性を示す。従って当該遺伝子をマーカーとして上記NS4株に導入することにより、表現型として硫酸塩資化性およびセレン酸塩感受性を示すNS4株(以下、loxP::sC/NS4株という)を得ることができる。そのため、Creリコンビナーゼタンパク質の導入後に細胞がセレン酸塩耐性を獲得し、且つ硫酸塩資化性が消失したことを指標として、Cre−loxPシステムが正常に機能し、除去対象遺伝子が除去できたことを容易に確認することができる。
【0088】
本発明の実施例で用いたプラスミドを構築するために用いたloxP配列を含むプラスミド(以下、pUG6プラスミドという)、sCマーカー遺伝子が搭載されたプラスミド(以下、pUsCプラスミドという)、およびniaDマーカー遺伝子が搭載されたプラスミド(以下、pNGA142プラスミドという)は、全て東北大学の五味博士より供与して頂いた。なお、pUG6プラスミドについてはU Guldener, S Heck, T Fielder, J Beinhauer, JH Hegemann, Nucleic Acids Res 24 (1996) 2519-24を、pUsCプラスミドについてはO Yamada, BR Lee and K Gomi, Biosci. Biotech. Biochem. 61 (1997) 1367-69を、pNGA142プラスミドについてはS Tamalampudi, MMT Rahaman, S Hama, Y Suzuki, A Kondo and H Fukuda, Appl. Microbiol. Biotechnol 75 (2007) 387-95を参照のこと。
【0089】
〔実施例1:麹菌におけるsCマーカー遺伝子の除去〕
(1)loxP::sC搭載プラスミドの構築
pUG6のプラスミド地図を図7に示す。pUG6プラスミドは、2つのloxP配列(loxP1および2)に挟まれたカナマイシン耐性遺伝子(KanMX)を有し、図には示さないが、さらにアンピシリン耐性遺伝子及び大腸菌のOri領域を有するプラスミドである。
【0090】
上記pUG6プラスミド内にsCマーカー遺伝子が導入できるよう、図7中に示すSacIサイトから5’側22bp上流の部分に、QuikChange site-directed mutagenesis法により制限酵素サイトであるSphIサイトを導入した。
【0091】
具体的には、まず、SphIサイトを含む、配列番号1および配列番号2のプライマーを用い、pUG6プラスミドをテンプレートとして用いてPCRを行った。PCRの反応条件を表1に示す。
【0092】
【表1】
【0093】
反応終了後、制限酵素DpnIを1μl加え、37℃で2時間インキュベートし、テンプレートDNAを消化した。この溶液を用いて大腸菌の形質転換を行い、SphIサイトが新たに導入されたプラスミド(以下、pUG6SphIという)を得た。
【0094】
sCマーカー遺伝子が搭載されたpUsCプラスミドを制限酵素XbaIおよびSphIで切断し、sCマーカー遺伝子の全長cDNAを含むDNA断片を得た。得られたDNA断片を、予め制限酵素XbaIおよびSphIで切断したpUG6SphIプラスミドにライゲーションし、sCマーカー遺伝子が新たに導入されたプラスミド(以下、pUG6sCという)を得た。
【0095】
続いて、niaDマーカー遺伝子が搭載されたpNGA142プラスミドを制限酵素AvrIIおよびSpeIで切断し、硝酸塩資化性マーカーであるniaD遺伝子の全長cDNAを含むDNA断片を得た。得られたDNA断片を、予めSpeIで切断し、且つBAP処理を行ったpUG6sCプラスミドにライゲーションし、loxP::sC搭載プラスミド(以下、pUGsCniaDという)を得た。
【0096】
(2)NS4株へのloxP::sCプラスミドの導入
上記(1)で得られたloxP::sC搭載プラスミドであるpUGsCniaDを制限酵素BamHIで消化して直鎖状にしたものを用いて、NS4株の形質転換を行った。
【0097】
loxP::sCマーカーが導入された株の選抜は、配列番号3および配列番号4のプライマーを用いて、形質転換体のゲノムDNAに対してPCRを行い、約3.7kbの増幅断片のみが確認されたものを候補株とした。最終的に、sCマーカー遺伝子の部分配列をプローブとして用いたサザンハイブリダイゼーションにより、NS4株の染色体DNA中にloxP::sCマーカーが導入されたことを確認した。得られた形質転換体は、loxP::sC/NS4株と命名した。図3(A)に、loxP::sC/NS4株が有する遺伝子を表す。Cre−loxPシステムが機能すれば、得られるloxP/NS4株は、sC遺伝子が除去された図3(B)に示すような遺伝子を有すると予測された。
【0098】
(3)loxP::sC/NS4株へのCre酵素タンパク質の導入
まず、loxP::sC/NS4株のプロトプラスト化を行った。loxP::sC/NS4株の分生子懸濁液をYPD液体培地に添加し、30℃において20時間振盪培養した後に、滅菌したガラスフィルターを用いて菌体を回収した。回収した菌体を50ml容の遠心チューブに移し、10mlのプロトプラスト化溶液(1.2M ソルビトール、10mM リン酸二水素ナトリウム(pH6.0)、5mg/ml Lysing enzyme(Sigma Chemical Co.)、10mg/ml Cellulase Onozuka R−10(Yakult Pharmaceutical Ind.Co.,Ltd.)、10mg/ml Yatalase(TaKaRa))を加えて菌体を懸濁し、30℃において、90rpm、3時間振盪し、プロトプラスト化反応を行った。
【0099】
プロトプラスト化反応後に、セルストレイナー(352350、BD Falcon製)にて濾過し、濾液中のプロトプラストを、4℃において、3,000×gで5分間、遠心分離することにより、菌体を沈殿として得た。得られた菌体を、洗浄液(1.2M ソルビトール、10mM リン酸二水素ナトリウム(pH6.0))を用いて3回洗浄した。4℃において、3,000×gで5分間、遠心分離することにより、沈殿としてプロトプラストを得た。
【0100】
Creリコンビナーゼタンパク質をloxP::sC/NS4株のプロトプラストに導入するために、核酸として、Creリコンビナーゼのターゲット配列であるloxP配列を搭載したpUG6プラスミドを用いた。
【0101】
以下の2つの条件により、Creリコンビナーゼタンパク質溶液と沈殿として得られたプロトプラストとを混合し、氷上にて30分間インキュベートした。以後の操作は、通常のプロトプラスト−PEG法による真菌の形質転換法(K Gomi, Y Iimura and S Hara, Agric Biol Chem 51 (1987) 2549-55を参照)に従って行った。
【0102】
<条件1:Cre+pUG6>
溶液Bに懸濁したプロトプラスト 100μl(細胞数:1×108個)
溶液C 12.5μl
Creリコンビナーゼタンパク質(631614、Clontech製) 10μl
pUG6プラスミド 3μg
<条件2:Creのみ>
溶液Bに懸濁したプロトプラスト 100μl(細胞数:1×108個)
溶液C 12.5μl
Creリコンビナーゼタンパク質 10μl
以下に溶液B及び溶液Cの組成を示す。
(溶液B)
1.2M ソルビトール、50mM CaCl2、10mM Tris−HCl(pH7.5)
(溶液C)
50% PEG4000、50mM CaCl2、10mM Tris−HCl(pH7.5)
Cre−loxPシステムが機能し、sCマーカー遺伝子が除去されたことを確認するために、最終濃度0.1mMのセレン酸を含有するプレートに、最終濃度0.8MとなるようにNaClを加えたものを用いた。結果は図には示さないが、Cre酵素タンパク質と核酸(pUG6プラスミド)とを用いた方法で、Cre−loxPシステムが正常に機能し、Cre酵素タンパク質のみを用いた場合と比較して、得られる形質転換体の数が有意に増加した。
【0103】
(4)Cre−loxPシステムの機能確認
sCマーカー遺伝子が除去されていることを確認するために、得られた候補株に対して、コロニーPCRを行った。図3は、sCマーカー遺伝子除去方法の概念を説明するための図を表す。図3(A)は、loxP::sC/NS4株が有する遺伝子を表し、図3(B)は、sCマーカー遺伝子が除去された株(loxP/NS4株)が有する遺伝子を表す。図3中の矢印に挟まれた部分をPCRによって増幅することができるような配列番号5および配列番号6のプライマーを作製した。これらのプライマーセットを用いてPCRを行えば、sCマーカー遺伝子がゲノム上に存在する場合には全長4.1kbのDNA断片が増幅されるが、sCマーカー遺伝子が除去され、ゲノム上に存在しない場合には、全長0.8kbのDNA断片が増幅される。
【0104】
上記PCR反応は酵素としてEx taq(RR001A、TaKaRa製)を用いた。反応条件を表2に示す。
【0105】
【表2】
【0106】
コロニーPCRの結果を図4に示す。図4中、レーン1〜4は、Creのみを用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン5〜9は、Cre酵素をpUG6プラスミドをキャリアとして用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン10は、loxP::sC/NS4株のゲノムをテンプレートにしてコロニーPCRを行った結果を表す。Creリコンビナーゼタンパク質と核酸(pUG6プラスミド)とを導入した場合にのみ0.8kbのバンドの増幅が確認された。PCRにて目的の長さのバンドが確認された形質転換体について、さらに、sCマーカー遺伝子の部分配列をプローブとしてサザンハイブリダイゼーションを行うことにより、sCマーカー遺伝子が除去されたことを確認した。得られた株をloxP/NS4株と命名した。
【0107】
loxP/NS4株、及びその親株であるloxP::sC/NS4株、さらにその親株であるNS4株について、栄養要求性による表現型観察を行い、マーカー除去の確認を行った。結果を図5に示す。図5は、sCマーカー遺伝子除去株の栄養要求性を示す図である。図5中のCDは、Czapek−DoxにN源として硝酸塩(NO3)を加え、S源として硫酸塩(SO4)を加えた最小培地(以下、CD培地という)を表す。図5中のCDMは、CD培地において、S源としての硫酸塩の代わりにメチオニンを最終濃度が30mg/mlとなるように加えた培地を表す(以下、CDM培地という)。図5中のCDMEは、CDM培地において、N源としての硝酸塩の代わりにグルタミン酸塩を最終濃度が70mMとなるように加えた培地を表す。
【0108】
それぞれの株の分生子を各培地上にそれぞれ約103個となるようにスポット植菌し、30℃で4日間培養した。NS4株は、NO3およびSO4を資化できないため、CD培地およびCDM培地では生育できなかった。また、loxP::sC/NS4株は、niaD遺伝子およびsC遺伝子が導入されているため、NO3およびSO4をともに資化することができ、最小培地であるCD培地においても生育が可能であった。一方、loxP/NS4株は、CD培地のみで生育することができず、sCマーカー遺伝子が除去された事によりSO4を資化することができない硫酸塩非資化性株となったことが明らかになった。
【0109】
以上の結果からCreリコンビナーゼタンパク質と核酸とを用いた方法でCre−loxPシステムが機能することが明らかとなった。
【0110】
〔実施例2〕
核酸として、一本鎖DNA(サケの精子由来)(201190、STRATGENE製)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0111】
〔実施例3〕
核酸として、pUC18プラスミド(3218、TaKaRa製)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0112】
〔実施例4〕
核酸として、pUC18断片(実施例3で用いたpUC18プラスミドを制限酵素XbaIで切断し断片化したもの)を用いた以外は、実施例1と同様の方法により、Cre酵素タンパク質を導入した。
【0113】
実施例2で得られた形質転換体のコロニーPCRの結果を図6に示す。図6中、レーン1〜14は、Cre酵素と一本鎖DNAとを用いて形質転換を行った形質転換体のゲノムをテンプレートにしてコロニーPCRを行った結果を表し、レーン15は、loxP::sC/NS4株のゲノムをテンプレートにしてコロニーPCRを行った結果を表す。図6からわかるように、核酸として一本鎖DNAを用いた場合であっても、0.8kbのバンドが増幅され、Cre−loxPシステムが正常に機能し、sCマーカー遺伝子が除去されたことが明らかになった。図には示さないが、実施例3および4で得られた形質転換体においても、コロニーPCRの結果からCre−loxPシステムが正常に機能し、sCマーカー遺伝子が除去されたことが明らかになった。以上の結果は、核酸であればキャリアとして機能する事を示すものである。
【0114】
本発明にかかる核内へタンパク質を導入する方法は、キャリアとしての核酸の形態を選ばないため、上述したCre−loxPシステムを用いた部位特異的遺伝子除去のみならず、Cre−loxPシステムを用いた従来公知の部位特異的遺伝子組換えにも適用することができることは言うまでもない。
【産業上の利用可能性】
【0115】
本発明にかかるタンパク質核内導入方法によれば、タンパク質を機能させたい細胞において、目的のタンパク質をコードする遺伝子が導入された形質転換体を作製する手間を省くことができる。また、タンパク質を細胞内に導入するため、遺伝子発現カセットプラスミドを脱落させることなく、必要な時期に一過的にタンパク質を機能させることができる。よって、部位特異的遺伝子除去方法又は部位特異的遺伝子組換え方法を簡便、且つ迅速に行うことができるため、上記方法を用いる医薬品産業等の産業において好適に利用することができる。
【図面の簡単な説明】
【0116】
【図1】Cre−loxPシステムを用いた従来の部位特異的遺伝子除去方法と本発明にかかる部位特異的遺伝子除去方法とを比較した図である。(A)は、従来の部位特異的遺伝子除去方法を表し、(B)は、本発明にかかる部位特異的遺伝子除去方法を表す。
【図2】Cre−loxPシステムを用いた従来の部位特異的遺伝子組換え方法と本発明にかかる部位特異的遺伝子組換え方法とを比較した図である。(A)は、従来の部位特異的遺伝子組換え方法を表し、(B)は、本発明にかかる部位特異的遺伝子組換え方法を表す。
【図3】sCマーカー遺伝子除去方法の概念を説明するための図であり、(A)は、loxP::sC/NS4株が有する遺伝子を表し、(B)は、loxP/NS4株が有する遺伝子を表す。
【図4】Creリコンビナーゼタンパク質のみを形質転換した場合のコロニーPCRの結果と、Creリコンビナーゼタンパク質と核酸とを形質転換した場合のコロニーPCRの結果とを示す図である。
【図5】sCマーカー遺伝子除去株の栄養要求性を検討した結果を示す図である。
【図6】一本鎖DNAをキャリアとして用いた際のコロニーPCRの結果を示す図である。
【図7】pUG6のプラスミド地図である。
【特許請求の範囲】
【請求項1】
タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む、タンパク質を核内に導入する方法。
【請求項2】
上記タンパク質は、部位特異的組換え酵素であることを特徴とする、請求項1に記載の方法。
【請求項3】
上記核酸は、全長が50塩基〜300,000塩基の範囲内であることを特徴とする、請求項1または2に記載の方法。
【請求項4】
除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴とする上記方法。
【請求項5】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程とを含むことを特徴とする上記方法。
【請求項6】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであることを特徴とする上記方法。
【請求項7】
除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴とする上記キット。
【請求項8】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴とする上記キット。
【請求項1】
タンパク質を、核酸をキャリアとすることによって細胞内に導入する工程を含む、タンパク質を核内に導入する方法。
【請求項2】
上記タンパク質は、部位特異的組換え酵素であることを特徴とする、請求項1に記載の方法。
【請求項3】
上記核酸は、全長が50塩基〜300,000塩基の範囲内であることを特徴とする、請求項1または2に記載の方法。
【請求項4】
除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含むことを特徴とする上記方法。
【請求項5】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、当該目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは当該目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAを上記細胞内に導入する工程と、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程とを含むことを特徴とする上記方法。
【請求項6】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入する方法であって、部位特異的組換え酵素を、核酸をキャリアとすることによって細胞内に導入する工程を含み、上記核酸は、上記目的遺伝子の上流または下流に特異的認識配列が連結された環状二本鎖DNA、或いは上記目的遺伝子の上流および下流に特異的認識配列が連結された環状二本鎖DNAであることを特徴とする上記方法。
【請求項7】
除去対象遺伝子の上流および下流に特異的認識配列が連結されたヌクレオチド鎖を備える細胞から上記対象遺伝子を除去するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴とする上記キット。
【請求項8】
少なくとも1つ以上の特異的認識配列を備える細胞において、当該特異的認識配列に目的遺伝子を挿入するためのキットであって、部位特異的組換え酵素と核酸とを含むことを特徴とする上記キット。
【図1】


【図2】
【図3】

【図7】

【図4】

【図5】

【図6】

【図2】
【図3】
【図7】
【図4】
【図5】
【図6】
【公開番号】特開2010−148359(P2010−148359A)
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2008−326681(P2008−326681)
【出願日】平成20年12月23日(2008.12.23)
【出願人】(301025634)独立行政法人酒類総合研究所 (55)
【Fターム(参考)】
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願日】平成20年12月23日(2008.12.23)
【出願人】(301025634)独立行政法人酒類総合研究所 (55)
【Fターム(参考)】
[ Back to top ]