
トリフラート化化合物の安定化
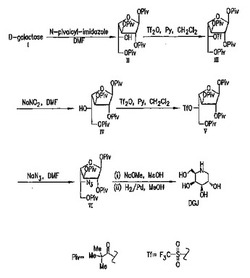
合成トリフラート化糖の新規な工程が記載されている。これらの糖は、化合物、例えば、D−1−デオキシノジリマイシン(DNJ)及びD−1−デオキシガラクトノジリマイシン(DGJ)の産生に有用である。特に、イミノ糖の合成のための数キログラム規模の安定化方法が記載されている。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本出願は、2005年6月8日に出願された米国仮特許出願第60/689,131号の優先権を主張し、その開示はその全体が参照として本明細書に組み入れられている。
【0002】
[背景]
トリフルオロメタンスルホニル、すなわちトリフラートは、ヒドロキシル基のための周知の保護基である。ヒドロキシ基は、トリフラートによって保護されると、非常に反応性の離脱基となる。この特徴は、アルコールの使用による合成目的で求核置換を実施するために幅広く使用される。炭水化物化学において、トリフラートの使用は特に一般的である。トリフラート保護されたヒドロキシル基は、SN2機構によって生じる求核置換反応における配置の完全な反転により、いずれかの求核試薬と置換されうる。トリフラートは、不飽和及び天然アルコールの両方を含む第1級及び第2級アルコールの軽度の酸化にも影響する。トリフラート化アルコールは対応するカルボニル化合物に酸化可能であり、次に離脱基はトリフラートを除去するために開裂される。
【0003】
しかしながら、トリフラート化化合物は水分に対して感受性である。低速の反応では、中間体は分解して、それにより反応収率の低下を引き起こす傾向がある。トリフラート化合物は、不飽和二重結合に対する排除を受けることがあり、本プロセスの副生成物はトリフル酸であり、トリフル酸は非常に強力な酸で、さらに加速された分解を引き起こしうる。大規模反応はミリグラム又はグラム規模の反応よりもはるかに時間がかかるので、このような問題は反応を数キログラム規模にスケールアップするときに重大となる。このような時間の延長は、少なくとも一部は、溶媒蒸発、反応容器への、及び反応容器からの生成物の移動に必要な時間の延長、並びに所望の温度に到達するのに必要なより長い加熱及び冷却時間のためである。したがってトリフラート化糖中間体を安定化する手段への要求が存在する。
【0004】
トリフラート自体は安定化されうる。1−ベンゼンスルフィニルピペリジン(BSP)及びトリフルオロメタンスルホン酸無水物の組合せは、ジクロロメタン中でグリコシルトリフラートを介してチオグリコシドを活性化できる金属を含まないチオフィルを形成して、トリフラート安定性に関連する問題を削減することが見出された(Crich D,Smith M.J Am Chem Soc.2001 Sep 19;123(37):9015−20)。
【0005】
発明者らの知る限りでは、現在、トリフラート保護糖化合物、例えば、D−ガラクトースのデオキシノジリマイシンの類似体であるD−1−デオキシガラクトノジリマイシン(DGJ)の中間体を特に産業規模で安定化するための既知の簡単な方法はない。D−1−デオキシガラクトノジリマイシン(DGJ)は、α−及びβ−D−ガラクトシダーゼの両方の強力な阻害物質である。ガラクトシダーゼは、グリコシド結合の加水分解を触媒し、複合糖質の代謝において重要である。DGJなどのガラクトシダーゼ阻害物質は、糖尿病(例えば、米国特許第4,634,765号)、癌(例えば、米国特許第5,250,545号)、ヘルペス(例えば、米国特許第4,957,926号)、HIV及びファブリ病(Fanら,Nat.Med.1999 5:1,112−5)を含む、多くの疾患及び状態の治療に使用されうる。
【0006】
文献で発表されたD−1−デオキシガラクトノジリマイシン(DGJ)の複数の製剤があるが、その大半は、予備規模手順(>100g)で産業研究所での反復には適切でない。これらの合成の一部としては、D−グルコース(Legler Gら,Carbohydr Res.1986 Nov 1;155:119−29);D−ガラクトース(Uriel,C.,Santoyo−Gonzalez,F.ら,Synlett 1999 593−595;Synthesis 1998 1787−1792);ガラクトピラノース(Bernotas RCら,Carbohydr Res.1987 Sep 15;167:305−ll);L−酒石酸(Aoyagiら,J.Org.Chem.1991,56,815);ケブラコイトール(quebrachoitol)(Chidaら,J.Chem.Soc.,Chem Commun.1994,1247);ガラクトフラノース(Paulsenら,Chem.Ber.1980,113,2601);ベンゼン(Johnsonら,Tetrahedron Lett.1995,36,653);アラビノ−ヘキソース−5−ウロース(Bariliら,tetrahedron 1997,3407);5−アジド−1,4−ラクトン(Shilvockら,Synlett,1998,554);ドキシノジリマイシン(Takahashiら,J.Carbohydr.Chem.1998,17,117);アセチルグルコサミン(Heightmanら,Helv.Chim.Acta 1995,78,514);ミオ−イノシトール(Chida N,ら,Carbohydr Res.1992 Dec 31;237:185−94);ジオキサニルピペリデン(Takahataら,Org.Lett.2003;5(14);2527−2529);及び(E)−2,4−ペンタジエノール(Martin Rら,Org Lett.2000 Jan;2(1):93−5)(Hughes ABら,Nat Prod Rep.1994 Apr;11(2):135−62)からの合成を含む。N−メチル−1−デオキシノジリマイシン含有オリゴサッカライドの合成は、Kisoによって記載されている(Bioorg Med Chem.1994 Nov;2(11):1295−308)。Kisoは、保護された1−デオキシノジリマイシン誘導体に、D−ガラクトースのメチル−1−チオグリコシド(グリコシル供与体)をグリコシルプロモータ(glycosyl promoter)として使用されたトリフレートと共に結合させた。
【0007】
Fred−Robert Heiker,Alfred Matthias Schueller,Carbohydrate Research,1986,119−129)は、イオン交換樹脂と共に撹拌することによってDGJが単離され、エタノールの添加によって結晶化される、DGJを13g規模で調製する方法を開示している。しかしながら本プロセスは、数キログラム量を産生するための産業規模にはただちに適用できない。
【0008】
DGJ産生のための別のプロセスは、Francisco Santoyo−Gonzalez及び共同研究者によって開発された手順である(Santoyo−Gonzalezら,Synlett 1999 593−595;Synthesis 1998 1787−1792)。本合成の方法は、D−ガラクトースのヒドロキシル基の保護;生じたガラクトフラノシドのトリフラート化;及びアルトロフラノシドへの変換;を含む。アルトロフラノシドを次にトリフラート化し、アジドと反応して、5−アジド化合物を産生する。本化合物を次に脱保護及び還元して、DGJを得る。Santoyo−Gonzalezによって記載されたようなDGJの合成手順は、その収率が非常に低く、例えば、約20%の全収率であるために、小規模合成により適している。本合成の問題の1つは、トリフラート化フラノシドが不安定であり、分解しやすく、低い収率を生じて、場合により反応を汚染するということである。
【0009】
したがって、糖を分解及び加水分解から保護するために、トリフラート化糖、例えば、DGJの中間体として使用されるトリフラート化糖を安定化させる方法への要求がある。例えば、そのような安定化されたトリフラート化中間体は、D−ガラクトースからのDGJの合成の全収率を改善するために使用されうる。
【0010】
[発明の概要]
本発明は、トリフラート化糖を溶媒中で第2級又は第3級アルキルアミンと結合するステップと、当該溶媒を除去するステップと、によってトリフラート化糖を安定化する方法を提供する。本発明は、第2級又は第3級アミンが使用されない場合よりも安定であるトリフラート化糖を提供する。
【0011】
一実施形態において、トリフラート化糖はテトラピバロイルフラノース又はピラノースである。別の態様において、第3級アルキルアミンは、N,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンであり、第3級アルキルアミンはトリフラート化糖と比較して約0.1〜0.3当量で供給される。
【0012】
本発明の別の態様は、トリフラート化糖を産生するために溶媒中で糖開始物質にトリフルオロメタンスルホニル試薬を反応させるステップと、第2級又は第3級アミンをトリフラート化糖に添加するステップと、溶媒を濃縮するステップと、トリフラート糖を産生するために還元するステップと、によって、糖生成物の反応収率を上昇させる方法を備える。亜硝酸ナトリウムも同様に反応に添加されうる。
【0013】
本発明の他の特徴、利点及び実施形態は、次の説明、付随するデータ及び添付請求項から当業者に明らかとなるであろう。
【0014】
[好ましい実施形態の詳細な説明]
次の図面は、本明細書の一部を形成し、本発明のある態様をさらに説明するために含まれている。本発明は、本明細書に示す具体的な実施形態の詳細な説明と併せた、これらの図面の1つ以上への参照によって、さらに良好に理解されうる。
【0015】
本明細書で使用するように、「安定化する」又は「安定化された」という用語は、安定化された化合物が、安定化なしで化合物が分解する条件下でより分解しにくいことを意味する。好ましくは、安定化トリフラートは、同じ期間、例えば、1日又は1週間では、非安定化トリフラートと比べてより少なく分解する。分解は、安定化及び非安定化トリフラートをそれぞれ亜硝酸塩又はアジドと反応させて、反応のより高い収率を与える安定化トリフラートを決定しうる、「使用試験」を用いて試験されうる。好ましい実施形態において、「安定化させる」又は「安定化された」という用語は、安定化トリフラートの分解が、標準の分析方法、例えば、MR又はTLCによって1時間、好ましくは1日、さらにより好ましくは1週間以内に検出されないことを意味する。
【0016】
本明細書で使用するように、「数キログラム」及び「予備規模」という用語は、生成物が、1回のパスで生成物1kgを超える、あるいはさらに好ましくは10kgすら超える量である合成規模を示す。
【0017】
本明細書で使用するように、「反応収率」とは、制限開始材料が定量的に生成物に変換される場合に得られうる本生成物のグラム数と比較された、単離生成物のグラム数を意味する。「反応収率を上昇させる」とは、本工程を使用すると使用しないよりも、反応収率が少なくとも10%高いことを意味する。好ましくは、反応収率は少なくとも20%、又は30%、又は40%高い。さらにより好ましくは、反応収率は少なくとも50%以上である。加えて、好ましい実施形態において、中間体の分解による反応収率のいずれの低下はごくわずかである。
【0018】
「アルキル」という用語は、炭素及び水素原子のみから成り、不飽和を含有せず、単結合によって分子の残りに結合した、直鎖又は分岐C1〜C20炭化水素基、例えば、メチル、エチル、n−プロピル、1−メチルエチル(イソプロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)を指す。本明細書で使用するアルキルは、好ましくはC1〜C8アルキルである。
【0019】
「アルケニル」という用語は、少なくとも1個の炭素間二重結合を含有し、直鎖又は分岐鎖でありうるC2〜C20脂肪族炭化水素基、例えば、エタノール(ethanol)、1−プロジェニー(progeny)、2−プロジェニー(アリル)(ally)、イソプロペニル、2−メチル−1−プロペニル、1−ブテニル、2−ブテニルを指す。
【0020】
「シクロアルキル」という用語は、不飽和、非芳香族単環又は多環式炭化水素環系、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルを示す。多環式シクロアルキル基の例としては、パーヒドロナフチル(perhydronapththyl)、アダマンチル及びノルボルニル基、架橋環式基又はスピロ2環式基、例えば、スピロ(4,4)ノン−2−イル(spiro(4,4)non−2−yl)が挙げられる。
【0021】
「シクロアルカルキル」という用語は、上記に定義したようなアルキル基に直接結合した、上記に定義したようなシクロアルキルを指し、当該シクロアルキルはシクロプロピルメチル、シクロブチルエチル、シクロペンチルエチルなどの安定した構造の生成をもたらす。
【0022】
「アルキルエーテル」という用語は、アルキル鎖内に包含された少なくとも1個の酸素を有する、上記に定義したようなアルキル基又はシクロアルキル基、例えば、メチルエチルエーテル、ジエチルエーテル、テトラヒドロフランを指す。
【0023】
「アルキルアミン」という用語は、少なくとも1個の窒素原子を有する、上記に定義したようなアルキル基又はシクロアルキル基、例えば、n−ブチルアミン及びテトラヒドロオキサジンを指す。
【0024】
「アリール」という用語は、約6〜約14個の範囲の炭素原子を有する芳香族基、例えば、フェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニルを指す。
【0025】
「アリールアルキル」という用語は、上記に定義したようなアルキル基に直接結合した、上記に定義したようなアリール基、例えば、−CH2C6H5、及び−C2H4C6H5を指す。
【0026】
「複素環式」という用語は、炭素原子及び窒素、リン、酸素及び硫黄から成る群より選択される1〜5個のヘテロ原子より成る、安定な3〜15員環ラジカルを指す。本発明の目的では、縮合、架橋又はスピロ環系を含むことがあり、複素環式環基中の窒素、リン、炭素、酸素又は硫黄原子が各種の酸化状態へ場合により酸化されうる、複素環式環基は、単環式、2環式又は3環式環系でありうる。加えて、窒素原子は場合により4級化されうる。環基は、一部又は完全に飽和されうる(すなわちヘテロ芳香族又はヘテロアリール芳香族)。そのような複素環式環基の例としては、これに限定されるわけではないが、アゼチジニル、アクリジニル、ベンゾジオキソリル、ベンゾジオキサニル、ベンゾフラニル、カルバゾリル、シンノリニル、ジオキソラニル、インドリジニル、ナフチリジニル、パーヒドロアゼピニル、フェナジニル、フェノチアジニル、フェノキサジニル、フタラジニル、ピリジル、プテリジニル、プリニル、キナゾリニル、キノキサリニル、キノリニル、イソキノリニル、テトラゾイル、イミダゾリル、テトラヒドロイソイノリル(tetrahydroisouinolyl)、ピペリジニル、ピペラジニル、2−オキソピペラジニル、2−オキソピペリジニル、2−オキソピロリジニル、2−オキソアゼピニル、アゼピニル、ピロリル、4−ピペリドニル、ピロリジニル、ピラジニル、ピリミジニル、ピリダジニル、オキサゾリル、オキサゾリニル、オキサソリジニル、トリアゾリル、インダニル、イソキサゾリル、イソキサソリジニル、モルホリニル、チアゾリル、チアゾリニル、チアゾリジニル、イソチアゾリル、キヌクリジニル、イソチアゾリジニル、インドリル、イソインドリル、インドリニル、イソインドリニル、オクタヒドロインドリル、オクタヒドロイソインドリル、キノリル、イソキノリル、デカヒドロイソキノリル、ベンズイミダゾリル、チアジアゾリル、ベンゾピラニル、ベンゾチアゾリル、ベンゾオキサゾリル、フリル、テトラヒドロフルチル(tetrahydrofurtyl)、テトラヒドロピラニル、チエニル、ベンゾチエニル、チアモルホリニル、チアモルホリニルスルホキシド、チアモルホリニル、スルホニル、ジオキサホスホラニル、オキサジアゾリル、クロマニル、イソクロマニルが挙げられる。
【0027】
複素環式環基は、主構造にいずれかのヘテロ原子又は炭素原子にて結合され、安定な構造の生成をもたらす。
【0028】
「ヘテロアリール」という用語は、環が芳香族である複素環式環を指す。
【0029】
「ヘテロアリールアルキル」という用語は、アルキル基に直接結合した、上記に定義したようなヘテロアリール環基を指す。ヘテロアリールアルキル基は、主構造にアルキル基からのいずれかの炭素原子にて結合され、安定な構造の生成をもたらす。
【0030】
「ヘテロシクリル」という用語は、上記に定義したような複素環式環基を指す。ヘテロシクリル環基は、主構造にいずれかのヘテロ原子又は炭素原子にて結合され、安定な構造の生成をもたらす。
【0031】
「ヘテロシクリルアルキル」という用語は、アルキル基に直接結合した、上記に定義したような複素環式環基を指す。ヘテロシクリルアルキル基は、主構造に、アルキル基内のいずれかの炭素原子にて結合され、安定な構造の生成をもたらす。
【0032】
「置換アルキル」、「置換アルケニル」、「置換アルキニル」、「置換シクロアルキル」、「置換シクロアルカルキル」、「置換シクロアルケニル」、「置換アリールアルキル」、「置換アリール」、「置換複素環式環」、「置換へテロアリール環」、「置換へテロアリールアルキル」、又は「置換ヘテロシクリルアルキル環」における置換基は、水素、ヒドロキシル、ハロゲン、カルボキシル、シアノ、アミノ、ニトロ、オキソ(=O)、チオ(=S)の基の群より選択される1個以上、あるいはアルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アリール、ヘテロアリール、ヘテロアリールアルキル、複素環式環、−COORx、−C(O)Rx、−C(S)Rx、−C(O)NRxRy、−C(O)ONRxRy、−NRxCONRyRz、−N(Rx)SORy、−N(Rx)SO2Ry、−(=N−N(Rx)Ry)、−NRxC(O)ORy、−NRxRy、−NRxC(O)Ry−、−NRxC(S)Ry、−NRxC(S)NRyRz、−SONRxRy−、−SO2NRxRy−、−ORx、−ORxC(O)NRyRz、−ORxC(O)ORy−、−OC(O)Rx、−OC(O)NRxRy、−RxNRyRz、−RxRyRz、−RxCF3、−RxNRyC(O)Rz、−RxORy、−RxC(O)ORy、−RxC(O)NRyRz、−RxC(O)Rx、−RxOC(O)Ry、−SRx、−SORx、−SO2Rx、−ONO2より選択される場合により置換された基と同じ又は異なっており、上の基のそれぞれにおけるRx、Ry及びRzは、水素原子、置換又は非置換アルキル、ハロアルキル、置換又は非置換アリールアルキル、置換又は非置換アリール、置換又は非置換シクロアルキル、置換又は非置換シクロアルカルキル、置換又は非置換複素環式環、置換又は非置換ヘテロシクリルアルキル、置換又は非置換へテロアリール並びに置換又は非置換へテロアリールアルキルでありうる。
【0033】
「ハロゲン」という用語は、フッ素、塩素、臭素及びヨウ素の基を指す。
【0034】
安定なトリフラート糖、例えば、ガラクトフラノシド及びアルトロフラノシドを提供する方法が、本明細書で開示される。これらの糖は、単純及び安価な糖、例えば、D−ガラクトースより作製可能であり、イミノ糖、例えば、ノジリマイシンの誘導体であるDGJ((2R,3S,4R,5S)−2−ヒドロキシメチル−3,4,5−トリヒドロキシピペリジン;1−デオキシ−ガラクトスタチン;又は1−デオキシ−ガラクトスタチンとしても記載される)の産生で有用である。本明細書に記載する安定なトリフラート化糖は、高い純度及び良好な収率での数キログラム規模の合成を可能にする。
【0035】
トリフルオロメタンスルホニル保護基を有する糖(トリフラート化糖)は、本発明の方法を使用して安定化されうる。トリフラート化部分を有する、フラノース及びピラノースを含む環式ヘキソース糖は、本明細書に記載する方法を使用して安定化されうる。フラノース及びピラノース中間体は、次の構造A及びBによってそれぞれ記載される。
【化1】

式中、少なくとも1個のRはトリフラートであり、各追加のRは独立して、トリフラート、H、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキル、OS(=O)2R2、C(=O)R2、炭水化物化学分野で理解されるような他のO−保護基である。R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである。一部の好ましいR基としては、ハロアルキル、ポリハロアルキル、クロロアセチル、ジクロロアセチル及びトリクロロアセチルが挙げられる。少なくとも1個のRはトリフラート保護基であるため、トリフラートとヒドロキシとの間の反応を防止する、糖上の遊離ヒドロキシル基は存在しない。トリフラート化糖は、トリフラート化D−マンノースでない。
【0036】
ペントース糖も本発明で考慮される。これらの5炭糖は、本明細書に記載する方法によってトリフラート化及び安定化されうる。ペントース糖は、次式
【化2】
によって定義でき、式中、Rは、ヘキソース糖で定義されたように定義される。
【0037】
ヘプトース糖も本発明で考慮される。これらの7炭糖は、本明細書に記載する方法によってトリフラート化及び安定化されうる。ヘプトース糖は、次式
【化3】
によって定義でき、式中、Rは、ヘキソース糖で定義されたように定義される。
【0038】
本発明のトリフラート化糖は、既知のプロセスによって調製されうる。本発明のトリフラート化糖は例えば、トリフラート化モノサッカライド及びオリゴサッカライド、例えば、モノ、ジ、トリ、テトラ及びペンタサッカライドでありうる。一実施形態において、トリフラート化フラノースは、D−グルコース、D−ガラクトース、D−アルトロース、D−ケトース、D−アルドース、D−プシコース、D−フルクトース、D−ソルボース又はD−タグトースからなる群より選択される。別の実施形態において、トリフラート化ピラノースは、D−リボース、D−アラビノース、D−キシロース又はD−リキソースからなる群より選択される。あるいはトリフラート化ヘキソースは、D−アロース、D−アルトロース、D−グルコース、D−グロース、D−イドース、D−ガラクトース又はD−タロースより選択され、ここで少なくとも1個のヒドロキシル基がトリフラート基によって保護される。
【0039】
トリフラート保護ジサッカライド及びトリサッカライドも、本明細書に記載した方法を使用して安定化されうる。ある実施形態において、ジサッカライドは、トレハロース、ソホロース、コージビオース、ラミナリビオース、マルトース、セロビオース、イソマルトース、ゲントビオース(gentobiose)、スクロース、ラフィノース又はラクトースであり、ここで少なくとも1個のヒドロキシル基がトリフラート基を使用して保護される。
【0040】
トリフラート化糖は、糖、例えば、テトラピバロイルフラノースをいずれかのトリフルオロメタンスルホニル化剤、例えば、トリフルオロメタンスルホン酸無水物(trifluoromethanesulfonic acid anhydride)(trifluoromethanesulfonic anhydride、triflic anhydride)、トリフルオロメタンスルホニルクロライド、N−フェニルトリフルオロメタンスルホンイミドなどと、塩基の存在下で反応させることによって生成される。本反応に好ましい塩基はピリジンであるが、しかしながら他の塩基、例えば、トリエチルアミン、n−ブチルアミン、N,N−ジメチルアミノピリジンが使用されうる。アルカリ金属塩、例えば、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、及び炭酸水素カリウムは、生成されたときにトリフラートの分解を引き起こさない限り、塩基として使用されうる(例えば、塩基は比較的弱くなければならない)。
【0041】
トリフラート糖は、特に炭水化物化学において、各種の反応で有用である。安定な反応中間体としてのトリフラート糖の使用の一例は、DGJの合成においてD−ガラクトースを開始物質として使用する、Santoyo−Gonzalezによって記載されたDGJの合成にある。Santoyo−Gonzalezによる合成は、本明細書で開示された方法によって改良され、安定なトリフラート中間体を提供して、それによりDGJの合成を数kg規模で可能にする反応スキームを提供しうる。
【0042】
加えて、安定化糖はピボイル化糖(pivoylated sugar)を含む反応において有用である。これらの糖は、結晶化によって安価で容易に単離及び精製され、アルコール部分の保護を必要とする反応で開始物質として使用されうる。
【0043】
小規模(例えば、ミリグラム量)では、特に好ましいトリフラート化糖、5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースIII(この糖は1,2,3,6−テトラ−O−ピバロイル−5−O−トリフルオロメタンスルホニル−α−D−ガラクトフラノースとも記載されうる)の調製及びそのさらなる反応は、Santoyo−Gonzalezらによって記載されているように、中程度から高い収率で進行しうる。本合成において、ピバロイル保護された糖1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースAは、CH2Cl2中でトリフルオロメタンスルホン酸無水物と、次にワークアップ(work−up)の後に、亜硝酸ナトリウムとただちに反応させて、転化1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(IV)を得る。HPLCは、D−ガラクト(A)及びL−アルトロ(IV)誘導体の顕著に異なる保持時間のために、転化アルコールへの完全な変換を証明した。
【化4】
しかしながら本転化反応は、この規模ではIVの中程度の収率(例えば、30〜50%)しか与えない。この低い収率は、少なくとも一部は、ワークアップ中に、又は亜硝酸塩と反応する間に他の生成物を与える競合的な副排除(side elimination)又は加水分解反応に入る、比較的不安定な中間体トリフラートIIIによって引き起こされる。
【0044】
本反応がより大規模で実施されるとき、溶媒の除去を必要とするトリフラートの単離は、除去される溶媒の対応するより多くの量のために、なおさらに問題となる。溶媒の濃縮中に、トリフラートの著しい分解が観察される。小規模では、トリフラートIIIは、白色又はオフホワイト固体として単離されるが、キログラム規模ではトリフラートIIIは、分解の明らかな徴候である褐色固体又は液体としてすら単離されることが多い。この分解の1つの原因は、溶媒(例えば、塩化メチレン)中に存在する微量の水である。本分解の1つの考えられる機構は、トリフラートIIIを開裂させてトリフル酸及び開始化合物を産生することを含みうる(図3を参照)。トリフル酸はまた、トリフラートIIIの分解をさらに促進して、自己触媒工程で不飽和化合物VIIを生成させる。ある例において、さらなるスケールアップとして、本工程は、濃縮末期にトリフラートIIIすべてを完全に分解させる。トリフラート開裂に加えて、フラスコ内の高温及び成分のより高い濃度は、トリフラート分解に対してさらなる寄与となった。
【0045】
不安定なトリフラートの分解は、pHを中性から約1まで非常に急激に低下させることができ、次に分解は自己加速する。最初に本分解は低速であり、小規模合成の場合は安定化は不要でありうる。例えば、転化反応(ガラクトからアルトロへ)は、第2級又は第3級アミンによる安定化なしで最大500gまで再現されうる。しかしながら対応するより長いワークアップ時間を伴う、より大型の反応では、安定化が要求される。
【0046】
今や不安定な中間体IIIを安定させるためにはもちろんのこと、他のトリフラート化糖を安定化させるためにも新たな手順が使用されうることが見出されている。濃縮ステップ中の第2級又は第3級アミン塩基の添加は、初期分解で生成されたトリフル酸が次に反応停止されて塩IVを生成し、いずれのさらなる分解も触媒することが許容されないために、生成物を安定化させる。
【0047】
同様に、不安定な中間体Vは、第2級又は第3級アミン塩基との組合せによって安定化される。本中間体は、対応するアジドVIへただちに変換される。
【化5】
【0048】
安定化のために、化合物Vは高い収率で得られうる。その上、添加されたアミン塩基は化合物VIの生成には影響しないので、高い全収率が達成されうる。
【0049】
トリフラート糖の安定化の後、トリフラート部分は、化合物を溶媒和させて、溶媒和させた化合物を硝酸塩などの化合物と反応させることによって除去され、中和されうる。該生成物は次に、溶媒系、例えば、ヘプタン/酢酸エチルによって抽出され、溶媒、例えば、ぺプタンから結晶化されうる。
【0050】
本発明によって考慮される、トリフラートのワークアップのための他の標準手順がある。例えば、トリフラートは、ピリジンを除去するためにトルエンと同時蒸発させられうる。しかしながら、大規模での好都合な産生及びトルエンによるワークアップ中にトリフラートが加熱されるときに産生される副生成物などの、副生成物の最小限の産生を可能にするワークアップを使用することが好ましい。
【0051】
本発明に従って調製された安定化トリフラートは、将来使用するためにその著しい分解を伴わずに乾燥及び貯蔵されうる。
【0052】
化合物III又はVとして定義される粗生成物は、溶液、例えば、水/DMF溶液からの結晶化によって単離されうる。本結晶化は低速であり、最大2日間を要しうる。粗生成物がいったん回収されると、粗生成物はヘプタン/酢酸エチルなどの溶液に溶解されうる。粗生成物は洗浄、乾燥、濃縮、及び例えば、ヘプタンからの再結晶化によって精製され、母液中にペンタ−ピバロイラート化合物を残す。本結晶化もまた、低速であり、最大2日間を要しうる。本ステップの代表的な収率範囲は30〜33%である。D−ガラクトースを包含する反応では、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド生成物は、高純度を有する白色結晶性粉末である。
【0053】
トリフラート化糖を安定化させるために使用されたアミン塩基は、中でトリフラート化糖が調製され、トリフラート化糖とのいずれの副反応も引き起こさない、同じ溶媒に溶解されうる有機アミンである。有機アミンは、好ましくは第2級又は第3級アルキルアミンであり、さらに好ましくは第3級アルキルアルキルアミンである。
【0054】
第2級アミンは例えば、アルキル鎖当たり3個以上の炭素を有するジアルキルアミンを含みうる。好ましいジアルキルアミンは、各アルキル鎖上に3、4、5、6、7、又は8個の炭素を有するであろう。第3級アミンとしては、アルキル鎖当たり1個以上の炭素を有するトリアルキルアミンが挙げられる。好ましいトリアルキルアミンは、2又は3個のアルキル鎖上に3、4、5、6、7、又は8個の炭素を有するであろう。ジアルキルアミン及びトリアルキルアミンの両方におけるアルキル鎖は相互に連結して、環式、2環式、又は3環式化合物を生成しうる。
【0055】
好ましくは、該塩基はヒンダード第2級アミン又は第3級アミンであろう。当該塩基は、これに限定されるわけではないが、ヒューニッヒ塩基(ジイソプロピルエチルアミン)、トリエチルアミン、トリブチルアミン、ジイソプロピルメチルアミン、ジイソプロピルブチルアミン、ジイソプロピルプロプリアミン(diisopropylproply amine)、トリプロピルアミン、トリイソプロピルアミン、トリイソブチルアミン、トリ−tert−ブチルアミン、ジイソブチルメチルアミン、ジイソブチルエチルアミン、ジイソブチルプロピルアミン、ジイソブチブチルアミン(diisobutybutyl amine)、ジイソプロピルアミン、及びジ−tert−ブチルアミンでありうる。当該有機塩基は、ピリジン、モルホリンなどの単環式環を含む第2級又は第3級環式アミン、及びウロトロピン、又はジアザビシクロウンデカンにおける2環式又は3環式環などの、2環式又は3環式環でもありうる。1つの特に好ましい有機塩基は、ヒューニッヒ塩基である。
【0056】
トリフラート化化合物を安定化させるのに有用なアミン塩基の構造は、トリフラートが位置する糖上の位置によって変わる。より反応性の糖は、より反応性でないアミン塩基の使用を必要とする。例えば、糖C6位置が最も反応性であるので、C6位置でトリフラート化された糖は短い(例えば、1〜3個の炭素)ジアルキルアミンによって安定化されない。これらの組成物では、より多くのアルキル炭素を有する塩基が好ましい(例えば、ジイソプロピルアミン)。
【0057】
アミン塩基は、トリフラート化糖の1モル当量以下、好ましくは0.5当量、さらに好ましくは0.2当量である量で使用されうる。
【実施例】
【0058】
本発明は、本発明の範囲を限定すると解釈されるべきではない次の実施例でさらに説明される。
【0059】
[実施例1]
3−トリフルオロメトキシ−3−デオキシ−1,2,1,8−テトラピバロイル−α−D−ガラクトフラノシドの調製及び安定化
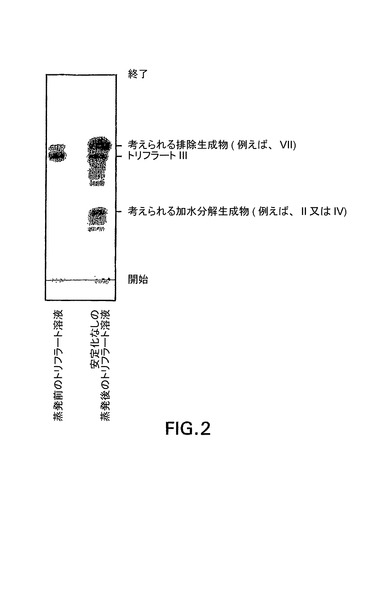
1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させた。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄した。トリフラートの塩化メチレン溶液に、ヒューニッヒ塩基0.2当量(230mL)を添加し、当該溶液を蒸発させて表題化合物を得た。本化合物の分解は、蒸発前に塩基が添加されない場合に図2で見られる。
【0060】
[実施例2]
テトラピバロイルフラノースの安定化
実施例1に記載した工程に従って、ピバロイル化ガラクトフラノシド1.5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させた。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄した。トリフラートの塩化メチレン溶液に、ヒューニッヒ塩基0.2当量(230mL)を添加し、当該溶液を蒸発させて表題化合物を得た。
【0061】
[実施例3]
3−トリフルオロメトキシ−3−デオキシ−1,2,1,8−テトラピバロイル−α−D−ガラクトフラノシドの調製及び安定化
1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド1 5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させる。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄する。トリフラートの塩化メチレン溶液に、トリエチルアミン0.2当量を添加し、当該溶液を蒸発させて表題化合物を得た。
【0062】
[実施例4]
テトラピバロイルフラノースの安定化
実施例1に記載した工程に従って、ピバロイル化ガラクトフラノシド5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させる。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄する。トリフラートの塩化メチレン溶液に、トリエチルアミン0.2当量を添加し、当該溶液を蒸発させて表題化合物を得た。
【0063】
本発明の多くの変形は、上述の説明に照らして当業者にそれ自体を示唆するであろう。そのようなすべての明らかな変形は、添付請求項の十分に意図された範囲内である。
【0064】
当業者は、本開示に照らして、本明細書で開示される具体的な実施形態で変更を実施して、本発明の精神及び範囲から逸脱せずに同じ又は同様の結果が得られることを認識すべきである。
【0065】
上記の特許、出願、試験方法、刊行物は、その全体が参照により本明細書に組み入れられている。
【図面の簡単な説明】
【0066】
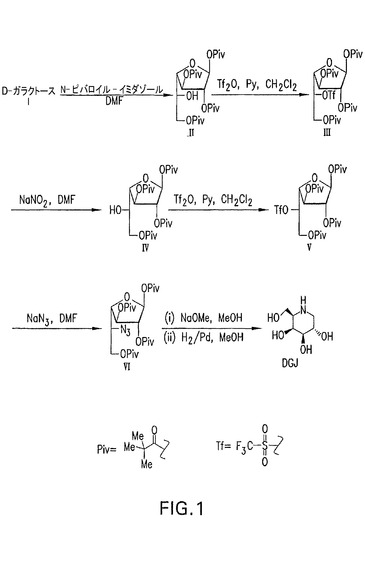
【図1】D−ガラクトースから開始して、トリフラート化中間体III及びVを有するDGJの合成を示す、合成スキーム。
【図2】トリフラートIII分解の薄層クロマトグラフィー。溶離はヘキサン:酢酸エチル(4:1)により行い、5%硫酸及び加熱により染色された。
【図3】トリフラート分解及びトリフラート安定化の経路。
【発明の詳細な説明】
【0001】
本出願は、2005年6月8日に出願された米国仮特許出願第60/689,131号の優先権を主張し、その開示はその全体が参照として本明細書に組み入れられている。
【0002】
[背景]
トリフルオロメタンスルホニル、すなわちトリフラートは、ヒドロキシル基のための周知の保護基である。ヒドロキシ基は、トリフラートによって保護されると、非常に反応性の離脱基となる。この特徴は、アルコールの使用による合成目的で求核置換を実施するために幅広く使用される。炭水化物化学において、トリフラートの使用は特に一般的である。トリフラート保護されたヒドロキシル基は、SN2機構によって生じる求核置換反応における配置の完全な反転により、いずれかの求核試薬と置換されうる。トリフラートは、不飽和及び天然アルコールの両方を含む第1級及び第2級アルコールの軽度の酸化にも影響する。トリフラート化アルコールは対応するカルボニル化合物に酸化可能であり、次に離脱基はトリフラートを除去するために開裂される。
【0003】
しかしながら、トリフラート化化合物は水分に対して感受性である。低速の反応では、中間体は分解して、それにより反応収率の低下を引き起こす傾向がある。トリフラート化合物は、不飽和二重結合に対する排除を受けることがあり、本プロセスの副生成物はトリフル酸であり、トリフル酸は非常に強力な酸で、さらに加速された分解を引き起こしうる。大規模反応はミリグラム又はグラム規模の反応よりもはるかに時間がかかるので、このような問題は反応を数キログラム規模にスケールアップするときに重大となる。このような時間の延長は、少なくとも一部は、溶媒蒸発、反応容器への、及び反応容器からの生成物の移動に必要な時間の延長、並びに所望の温度に到達するのに必要なより長い加熱及び冷却時間のためである。したがってトリフラート化糖中間体を安定化する手段への要求が存在する。
【0004】
トリフラート自体は安定化されうる。1−ベンゼンスルフィニルピペリジン(BSP)及びトリフルオロメタンスルホン酸無水物の組合せは、ジクロロメタン中でグリコシルトリフラートを介してチオグリコシドを活性化できる金属を含まないチオフィルを形成して、トリフラート安定性に関連する問題を削減することが見出された(Crich D,Smith M.J Am Chem Soc.2001 Sep 19;123(37):9015−20)。
【0005】
発明者らの知る限りでは、現在、トリフラート保護糖化合物、例えば、D−ガラクトースのデオキシノジリマイシンの類似体であるD−1−デオキシガラクトノジリマイシン(DGJ)の中間体を特に産業規模で安定化するための既知の簡単な方法はない。D−1−デオキシガラクトノジリマイシン(DGJ)は、α−及びβ−D−ガラクトシダーゼの両方の強力な阻害物質である。ガラクトシダーゼは、グリコシド結合の加水分解を触媒し、複合糖質の代謝において重要である。DGJなどのガラクトシダーゼ阻害物質は、糖尿病(例えば、米国特許第4,634,765号)、癌(例えば、米国特許第5,250,545号)、ヘルペス(例えば、米国特許第4,957,926号)、HIV及びファブリ病(Fanら,Nat.Med.1999 5:1,112−5)を含む、多くの疾患及び状態の治療に使用されうる。
【0006】
文献で発表されたD−1−デオキシガラクトノジリマイシン(DGJ)の複数の製剤があるが、その大半は、予備規模手順(>100g)で産業研究所での反復には適切でない。これらの合成の一部としては、D−グルコース(Legler Gら,Carbohydr Res.1986 Nov 1;155:119−29);D−ガラクトース(Uriel,C.,Santoyo−Gonzalez,F.ら,Synlett 1999 593−595;Synthesis 1998 1787−1792);ガラクトピラノース(Bernotas RCら,Carbohydr Res.1987 Sep 15;167:305−ll);L−酒石酸(Aoyagiら,J.Org.Chem.1991,56,815);ケブラコイトール(quebrachoitol)(Chidaら,J.Chem.Soc.,Chem Commun.1994,1247);ガラクトフラノース(Paulsenら,Chem.Ber.1980,113,2601);ベンゼン(Johnsonら,Tetrahedron Lett.1995,36,653);アラビノ−ヘキソース−5−ウロース(Bariliら,tetrahedron 1997,3407);5−アジド−1,4−ラクトン(Shilvockら,Synlett,1998,554);ドキシノジリマイシン(Takahashiら,J.Carbohydr.Chem.1998,17,117);アセチルグルコサミン(Heightmanら,Helv.Chim.Acta 1995,78,514);ミオ−イノシトール(Chida N,ら,Carbohydr Res.1992 Dec 31;237:185−94);ジオキサニルピペリデン(Takahataら,Org.Lett.2003;5(14);2527−2529);及び(E)−2,4−ペンタジエノール(Martin Rら,Org Lett.2000 Jan;2(1):93−5)(Hughes ABら,Nat Prod Rep.1994 Apr;11(2):135−62)からの合成を含む。N−メチル−1−デオキシノジリマイシン含有オリゴサッカライドの合成は、Kisoによって記載されている(Bioorg Med Chem.1994 Nov;2(11):1295−308)。Kisoは、保護された1−デオキシノジリマイシン誘導体に、D−ガラクトースのメチル−1−チオグリコシド(グリコシル供与体)をグリコシルプロモータ(glycosyl promoter)として使用されたトリフレートと共に結合させた。
【0007】
Fred−Robert Heiker,Alfred Matthias Schueller,Carbohydrate Research,1986,119−129)は、イオン交換樹脂と共に撹拌することによってDGJが単離され、エタノールの添加によって結晶化される、DGJを13g規模で調製する方法を開示している。しかしながら本プロセスは、数キログラム量を産生するための産業規模にはただちに適用できない。
【0008】
DGJ産生のための別のプロセスは、Francisco Santoyo−Gonzalez及び共同研究者によって開発された手順である(Santoyo−Gonzalezら,Synlett 1999 593−595;Synthesis 1998 1787−1792)。本合成の方法は、D−ガラクトースのヒドロキシル基の保護;生じたガラクトフラノシドのトリフラート化;及びアルトロフラノシドへの変換;を含む。アルトロフラノシドを次にトリフラート化し、アジドと反応して、5−アジド化合物を産生する。本化合物を次に脱保護及び還元して、DGJを得る。Santoyo−Gonzalezによって記載されたようなDGJの合成手順は、その収率が非常に低く、例えば、約20%の全収率であるために、小規模合成により適している。本合成の問題の1つは、トリフラート化フラノシドが不安定であり、分解しやすく、低い収率を生じて、場合により反応を汚染するということである。
【0009】
したがって、糖を分解及び加水分解から保護するために、トリフラート化糖、例えば、DGJの中間体として使用されるトリフラート化糖を安定化させる方法への要求がある。例えば、そのような安定化されたトリフラート化中間体は、D−ガラクトースからのDGJの合成の全収率を改善するために使用されうる。
【0010】
[発明の概要]
本発明は、トリフラート化糖を溶媒中で第2級又は第3級アルキルアミンと結合するステップと、当該溶媒を除去するステップと、によってトリフラート化糖を安定化する方法を提供する。本発明は、第2級又は第3級アミンが使用されない場合よりも安定であるトリフラート化糖を提供する。
【0011】
一実施形態において、トリフラート化糖はテトラピバロイルフラノース又はピラノースである。別の態様において、第3級アルキルアミンは、N,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンであり、第3級アルキルアミンはトリフラート化糖と比較して約0.1〜0.3当量で供給される。
【0012】
本発明の別の態様は、トリフラート化糖を産生するために溶媒中で糖開始物質にトリフルオロメタンスルホニル試薬を反応させるステップと、第2級又は第3級アミンをトリフラート化糖に添加するステップと、溶媒を濃縮するステップと、トリフラート糖を産生するために還元するステップと、によって、糖生成物の反応収率を上昇させる方法を備える。亜硝酸ナトリウムも同様に反応に添加されうる。
【0013】
本発明の他の特徴、利点及び実施形態は、次の説明、付随するデータ及び添付請求項から当業者に明らかとなるであろう。
【0014】
[好ましい実施形態の詳細な説明]
次の図面は、本明細書の一部を形成し、本発明のある態様をさらに説明するために含まれている。本発明は、本明細書に示す具体的な実施形態の詳細な説明と併せた、これらの図面の1つ以上への参照によって、さらに良好に理解されうる。
【0015】
本明細書で使用するように、「安定化する」又は「安定化された」という用語は、安定化された化合物が、安定化なしで化合物が分解する条件下でより分解しにくいことを意味する。好ましくは、安定化トリフラートは、同じ期間、例えば、1日又は1週間では、非安定化トリフラートと比べてより少なく分解する。分解は、安定化及び非安定化トリフラートをそれぞれ亜硝酸塩又はアジドと反応させて、反応のより高い収率を与える安定化トリフラートを決定しうる、「使用試験」を用いて試験されうる。好ましい実施形態において、「安定化させる」又は「安定化された」という用語は、安定化トリフラートの分解が、標準の分析方法、例えば、MR又はTLCによって1時間、好ましくは1日、さらにより好ましくは1週間以内に検出されないことを意味する。
【0016】
本明細書で使用するように、「数キログラム」及び「予備規模」という用語は、生成物が、1回のパスで生成物1kgを超える、あるいはさらに好ましくは10kgすら超える量である合成規模を示す。
【0017】
本明細書で使用するように、「反応収率」とは、制限開始材料が定量的に生成物に変換される場合に得られうる本生成物のグラム数と比較された、単離生成物のグラム数を意味する。「反応収率を上昇させる」とは、本工程を使用すると使用しないよりも、反応収率が少なくとも10%高いことを意味する。好ましくは、反応収率は少なくとも20%、又は30%、又は40%高い。さらにより好ましくは、反応収率は少なくとも50%以上である。加えて、好ましい実施形態において、中間体の分解による反応収率のいずれの低下はごくわずかである。
【0018】
「アルキル」という用語は、炭素及び水素原子のみから成り、不飽和を含有せず、単結合によって分子の残りに結合した、直鎖又は分岐C1〜C20炭化水素基、例えば、メチル、エチル、n−プロピル、1−メチルエチル(イソプロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)を指す。本明細書で使用するアルキルは、好ましくはC1〜C8アルキルである。
【0019】
「アルケニル」という用語は、少なくとも1個の炭素間二重結合を含有し、直鎖又は分岐鎖でありうるC2〜C20脂肪族炭化水素基、例えば、エタノール(ethanol)、1−プロジェニー(progeny)、2−プロジェニー(アリル)(ally)、イソプロペニル、2−メチル−1−プロペニル、1−ブテニル、2−ブテニルを指す。
【0020】
「シクロアルキル」という用語は、不飽和、非芳香族単環又は多環式炭化水素環系、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルを示す。多環式シクロアルキル基の例としては、パーヒドロナフチル(perhydronapththyl)、アダマンチル及びノルボルニル基、架橋環式基又はスピロ2環式基、例えば、スピロ(4,4)ノン−2−イル(spiro(4,4)non−2−yl)が挙げられる。
【0021】
「シクロアルカルキル」という用語は、上記に定義したようなアルキル基に直接結合した、上記に定義したようなシクロアルキルを指し、当該シクロアルキルはシクロプロピルメチル、シクロブチルエチル、シクロペンチルエチルなどの安定した構造の生成をもたらす。
【0022】
「アルキルエーテル」という用語は、アルキル鎖内に包含された少なくとも1個の酸素を有する、上記に定義したようなアルキル基又はシクロアルキル基、例えば、メチルエチルエーテル、ジエチルエーテル、テトラヒドロフランを指す。
【0023】
「アルキルアミン」という用語は、少なくとも1個の窒素原子を有する、上記に定義したようなアルキル基又はシクロアルキル基、例えば、n−ブチルアミン及びテトラヒドロオキサジンを指す。
【0024】
「アリール」という用語は、約6〜約14個の範囲の炭素原子を有する芳香族基、例えば、フェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニルを指す。
【0025】
「アリールアルキル」という用語は、上記に定義したようなアルキル基に直接結合した、上記に定義したようなアリール基、例えば、−CH2C6H5、及び−C2H4C6H5を指す。
【0026】
「複素環式」という用語は、炭素原子及び窒素、リン、酸素及び硫黄から成る群より選択される1〜5個のヘテロ原子より成る、安定な3〜15員環ラジカルを指す。本発明の目的では、縮合、架橋又はスピロ環系を含むことがあり、複素環式環基中の窒素、リン、炭素、酸素又は硫黄原子が各種の酸化状態へ場合により酸化されうる、複素環式環基は、単環式、2環式又は3環式環系でありうる。加えて、窒素原子は場合により4級化されうる。環基は、一部又は完全に飽和されうる(すなわちヘテロ芳香族又はヘテロアリール芳香族)。そのような複素環式環基の例としては、これに限定されるわけではないが、アゼチジニル、アクリジニル、ベンゾジオキソリル、ベンゾジオキサニル、ベンゾフラニル、カルバゾリル、シンノリニル、ジオキソラニル、インドリジニル、ナフチリジニル、パーヒドロアゼピニル、フェナジニル、フェノチアジニル、フェノキサジニル、フタラジニル、ピリジル、プテリジニル、プリニル、キナゾリニル、キノキサリニル、キノリニル、イソキノリニル、テトラゾイル、イミダゾリル、テトラヒドロイソイノリル(tetrahydroisouinolyl)、ピペリジニル、ピペラジニル、2−オキソピペラジニル、2−オキソピペリジニル、2−オキソピロリジニル、2−オキソアゼピニル、アゼピニル、ピロリル、4−ピペリドニル、ピロリジニル、ピラジニル、ピリミジニル、ピリダジニル、オキサゾリル、オキサゾリニル、オキサソリジニル、トリアゾリル、インダニル、イソキサゾリル、イソキサソリジニル、モルホリニル、チアゾリル、チアゾリニル、チアゾリジニル、イソチアゾリル、キヌクリジニル、イソチアゾリジニル、インドリル、イソインドリル、インドリニル、イソインドリニル、オクタヒドロインドリル、オクタヒドロイソインドリル、キノリル、イソキノリル、デカヒドロイソキノリル、ベンズイミダゾリル、チアジアゾリル、ベンゾピラニル、ベンゾチアゾリル、ベンゾオキサゾリル、フリル、テトラヒドロフルチル(tetrahydrofurtyl)、テトラヒドロピラニル、チエニル、ベンゾチエニル、チアモルホリニル、チアモルホリニルスルホキシド、チアモルホリニル、スルホニル、ジオキサホスホラニル、オキサジアゾリル、クロマニル、イソクロマニルが挙げられる。
【0027】
複素環式環基は、主構造にいずれかのヘテロ原子又は炭素原子にて結合され、安定な構造の生成をもたらす。
【0028】
「ヘテロアリール」という用語は、環が芳香族である複素環式環を指す。
【0029】
「ヘテロアリールアルキル」という用語は、アルキル基に直接結合した、上記に定義したようなヘテロアリール環基を指す。ヘテロアリールアルキル基は、主構造にアルキル基からのいずれかの炭素原子にて結合され、安定な構造の生成をもたらす。
【0030】
「ヘテロシクリル」という用語は、上記に定義したような複素環式環基を指す。ヘテロシクリル環基は、主構造にいずれかのヘテロ原子又は炭素原子にて結合され、安定な構造の生成をもたらす。
【0031】
「ヘテロシクリルアルキル」という用語は、アルキル基に直接結合した、上記に定義したような複素環式環基を指す。ヘテロシクリルアルキル基は、主構造に、アルキル基内のいずれかの炭素原子にて結合され、安定な構造の生成をもたらす。
【0032】
「置換アルキル」、「置換アルケニル」、「置換アルキニル」、「置換シクロアルキル」、「置換シクロアルカルキル」、「置換シクロアルケニル」、「置換アリールアルキル」、「置換アリール」、「置換複素環式環」、「置換へテロアリール環」、「置換へテロアリールアルキル」、又は「置換ヘテロシクリルアルキル環」における置換基は、水素、ヒドロキシル、ハロゲン、カルボキシル、シアノ、アミノ、ニトロ、オキソ(=O)、チオ(=S)の基の群より選択される1個以上、あるいはアルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アリール、ヘテロアリール、ヘテロアリールアルキル、複素環式環、−COORx、−C(O)Rx、−C(S)Rx、−C(O)NRxRy、−C(O)ONRxRy、−NRxCONRyRz、−N(Rx)SORy、−N(Rx)SO2Ry、−(=N−N(Rx)Ry)、−NRxC(O)ORy、−NRxRy、−NRxC(O)Ry−、−NRxC(S)Ry、−NRxC(S)NRyRz、−SONRxRy−、−SO2NRxRy−、−ORx、−ORxC(O)NRyRz、−ORxC(O)ORy−、−OC(O)Rx、−OC(O)NRxRy、−RxNRyRz、−RxRyRz、−RxCF3、−RxNRyC(O)Rz、−RxORy、−RxC(O)ORy、−RxC(O)NRyRz、−RxC(O)Rx、−RxOC(O)Ry、−SRx、−SORx、−SO2Rx、−ONO2より選択される場合により置換された基と同じ又は異なっており、上の基のそれぞれにおけるRx、Ry及びRzは、水素原子、置換又は非置換アルキル、ハロアルキル、置換又は非置換アリールアルキル、置換又は非置換アリール、置換又は非置換シクロアルキル、置換又は非置換シクロアルカルキル、置換又は非置換複素環式環、置換又は非置換ヘテロシクリルアルキル、置換又は非置換へテロアリール並びに置換又は非置換へテロアリールアルキルでありうる。
【0033】
「ハロゲン」という用語は、フッ素、塩素、臭素及びヨウ素の基を指す。
【0034】
安定なトリフラート糖、例えば、ガラクトフラノシド及びアルトロフラノシドを提供する方法が、本明細書で開示される。これらの糖は、単純及び安価な糖、例えば、D−ガラクトースより作製可能であり、イミノ糖、例えば、ノジリマイシンの誘導体であるDGJ((2R,3S,4R,5S)−2−ヒドロキシメチル−3,4,5−トリヒドロキシピペリジン;1−デオキシ−ガラクトスタチン;又は1−デオキシ−ガラクトスタチンとしても記載される)の産生で有用である。本明細書に記載する安定なトリフラート化糖は、高い純度及び良好な収率での数キログラム規模の合成を可能にする。
【0035】
トリフルオロメタンスルホニル保護基を有する糖(トリフラート化糖)は、本発明の方法を使用して安定化されうる。トリフラート化部分を有する、フラノース及びピラノースを含む環式ヘキソース糖は、本明細書に記載する方法を使用して安定化されうる。フラノース及びピラノース中間体は、次の構造A及びBによってそれぞれ記載される。
【化1】
式中、少なくとも1個のRはトリフラートであり、各追加のRは独立して、トリフラート、H、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキル、OS(=O)2R2、C(=O)R2、炭水化物化学分野で理解されるような他のO−保護基である。R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである。一部の好ましいR基としては、ハロアルキル、ポリハロアルキル、クロロアセチル、ジクロロアセチル及びトリクロロアセチルが挙げられる。少なくとも1個のRはトリフラート保護基であるため、トリフラートとヒドロキシとの間の反応を防止する、糖上の遊離ヒドロキシル基は存在しない。トリフラート化糖は、トリフラート化D−マンノースでない。
【0036】
ペントース糖も本発明で考慮される。これらの5炭糖は、本明細書に記載する方法によってトリフラート化及び安定化されうる。ペントース糖は、次式
【化2】
によって定義でき、式中、Rは、ヘキソース糖で定義されたように定義される。
【0037】
ヘプトース糖も本発明で考慮される。これらの7炭糖は、本明細書に記載する方法によってトリフラート化及び安定化されうる。ヘプトース糖は、次式
【化3】
によって定義でき、式中、Rは、ヘキソース糖で定義されたように定義される。
【0038】
本発明のトリフラート化糖は、既知のプロセスによって調製されうる。本発明のトリフラート化糖は例えば、トリフラート化モノサッカライド及びオリゴサッカライド、例えば、モノ、ジ、トリ、テトラ及びペンタサッカライドでありうる。一実施形態において、トリフラート化フラノースは、D−グルコース、D−ガラクトース、D−アルトロース、D−ケトース、D−アルドース、D−プシコース、D−フルクトース、D−ソルボース又はD−タグトースからなる群より選択される。別の実施形態において、トリフラート化ピラノースは、D−リボース、D−アラビノース、D−キシロース又はD−リキソースからなる群より選択される。あるいはトリフラート化ヘキソースは、D−アロース、D−アルトロース、D−グルコース、D−グロース、D−イドース、D−ガラクトース又はD−タロースより選択され、ここで少なくとも1個のヒドロキシル基がトリフラート基によって保護される。
【0039】
トリフラート保護ジサッカライド及びトリサッカライドも、本明細書に記載した方法を使用して安定化されうる。ある実施形態において、ジサッカライドは、トレハロース、ソホロース、コージビオース、ラミナリビオース、マルトース、セロビオース、イソマルトース、ゲントビオース(gentobiose)、スクロース、ラフィノース又はラクトースであり、ここで少なくとも1個のヒドロキシル基がトリフラート基を使用して保護される。
【0040】
トリフラート化糖は、糖、例えば、テトラピバロイルフラノースをいずれかのトリフルオロメタンスルホニル化剤、例えば、トリフルオロメタンスルホン酸無水物(trifluoromethanesulfonic acid anhydride)(trifluoromethanesulfonic anhydride、triflic anhydride)、トリフルオロメタンスルホニルクロライド、N−フェニルトリフルオロメタンスルホンイミドなどと、塩基の存在下で反応させることによって生成される。本反応に好ましい塩基はピリジンであるが、しかしながら他の塩基、例えば、トリエチルアミン、n−ブチルアミン、N,N−ジメチルアミノピリジンが使用されうる。アルカリ金属塩、例えば、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、及び炭酸水素カリウムは、生成されたときにトリフラートの分解を引き起こさない限り、塩基として使用されうる(例えば、塩基は比較的弱くなければならない)。
【0041】
トリフラート糖は、特に炭水化物化学において、各種の反応で有用である。安定な反応中間体としてのトリフラート糖の使用の一例は、DGJの合成においてD−ガラクトースを開始物質として使用する、Santoyo−Gonzalezによって記載されたDGJの合成にある。Santoyo−Gonzalezによる合成は、本明細書で開示された方法によって改良され、安定なトリフラート中間体を提供して、それによりDGJの合成を数kg規模で可能にする反応スキームを提供しうる。
【0042】
加えて、安定化糖はピボイル化糖(pivoylated sugar)を含む反応において有用である。これらの糖は、結晶化によって安価で容易に単離及び精製され、アルコール部分の保護を必要とする反応で開始物質として使用されうる。
【0043】
小規模(例えば、ミリグラム量)では、特に好ましいトリフラート化糖、5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースIII(この糖は1,2,3,6−テトラ−O−ピバロイル−5−O−トリフルオロメタンスルホニル−α−D−ガラクトフラノースとも記載されうる)の調製及びそのさらなる反応は、Santoyo−Gonzalezらによって記載されているように、中程度から高い収率で進行しうる。本合成において、ピバロイル保護された糖1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースAは、CH2Cl2中でトリフルオロメタンスルホン酸無水物と、次にワークアップ(work−up)の後に、亜硝酸ナトリウムとただちに反応させて、転化1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(IV)を得る。HPLCは、D−ガラクト(A)及びL−アルトロ(IV)誘導体の顕著に異なる保持時間のために、転化アルコールへの完全な変換を証明した。
【化4】
しかしながら本転化反応は、この規模ではIVの中程度の収率(例えば、30〜50%)しか与えない。この低い収率は、少なくとも一部は、ワークアップ中に、又は亜硝酸塩と反応する間に他の生成物を与える競合的な副排除(side elimination)又は加水分解反応に入る、比較的不安定な中間体トリフラートIIIによって引き起こされる。
【0044】
本反応がより大規模で実施されるとき、溶媒の除去を必要とするトリフラートの単離は、除去される溶媒の対応するより多くの量のために、なおさらに問題となる。溶媒の濃縮中に、トリフラートの著しい分解が観察される。小規模では、トリフラートIIIは、白色又はオフホワイト固体として単離されるが、キログラム規模ではトリフラートIIIは、分解の明らかな徴候である褐色固体又は液体としてすら単離されることが多い。この分解の1つの原因は、溶媒(例えば、塩化メチレン)中に存在する微量の水である。本分解の1つの考えられる機構は、トリフラートIIIを開裂させてトリフル酸及び開始化合物を産生することを含みうる(図3を参照)。トリフル酸はまた、トリフラートIIIの分解をさらに促進して、自己触媒工程で不飽和化合物VIIを生成させる。ある例において、さらなるスケールアップとして、本工程は、濃縮末期にトリフラートIIIすべてを完全に分解させる。トリフラート開裂に加えて、フラスコ内の高温及び成分のより高い濃度は、トリフラート分解に対してさらなる寄与となった。
【0045】
不安定なトリフラートの分解は、pHを中性から約1まで非常に急激に低下させることができ、次に分解は自己加速する。最初に本分解は低速であり、小規模合成の場合は安定化は不要でありうる。例えば、転化反応(ガラクトからアルトロへ)は、第2級又は第3級アミンによる安定化なしで最大500gまで再現されうる。しかしながら対応するより長いワークアップ時間を伴う、より大型の反応では、安定化が要求される。
【0046】
今や不安定な中間体IIIを安定させるためにはもちろんのこと、他のトリフラート化糖を安定化させるためにも新たな手順が使用されうることが見出されている。濃縮ステップ中の第2級又は第3級アミン塩基の添加は、初期分解で生成されたトリフル酸が次に反応停止されて塩IVを生成し、いずれのさらなる分解も触媒することが許容されないために、生成物を安定化させる。
【0047】
同様に、不安定な中間体Vは、第2級又は第3級アミン塩基との組合せによって安定化される。本中間体は、対応するアジドVIへただちに変換される。
【化5】
【0048】
安定化のために、化合物Vは高い収率で得られうる。その上、添加されたアミン塩基は化合物VIの生成には影響しないので、高い全収率が達成されうる。
【0049】
トリフラート糖の安定化の後、トリフラート部分は、化合物を溶媒和させて、溶媒和させた化合物を硝酸塩などの化合物と反応させることによって除去され、中和されうる。該生成物は次に、溶媒系、例えば、ヘプタン/酢酸エチルによって抽出され、溶媒、例えば、ぺプタンから結晶化されうる。
【0050】
本発明によって考慮される、トリフラートのワークアップのための他の標準手順がある。例えば、トリフラートは、ピリジンを除去するためにトルエンと同時蒸発させられうる。しかしながら、大規模での好都合な産生及びトルエンによるワークアップ中にトリフラートが加熱されるときに産生される副生成物などの、副生成物の最小限の産生を可能にするワークアップを使用することが好ましい。
【0051】
本発明に従って調製された安定化トリフラートは、将来使用するためにその著しい分解を伴わずに乾燥及び貯蔵されうる。
【0052】
化合物III又はVとして定義される粗生成物は、溶液、例えば、水/DMF溶液からの結晶化によって単離されうる。本結晶化は低速であり、最大2日間を要しうる。粗生成物がいったん回収されると、粗生成物はヘプタン/酢酸エチルなどの溶液に溶解されうる。粗生成物は洗浄、乾燥、濃縮、及び例えば、ヘプタンからの再結晶化によって精製され、母液中にペンタ−ピバロイラート化合物を残す。本結晶化もまた、低速であり、最大2日間を要しうる。本ステップの代表的な収率範囲は30〜33%である。D−ガラクトースを包含する反応では、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド生成物は、高純度を有する白色結晶性粉末である。
【0053】
トリフラート化糖を安定化させるために使用されたアミン塩基は、中でトリフラート化糖が調製され、トリフラート化糖とのいずれの副反応も引き起こさない、同じ溶媒に溶解されうる有機アミンである。有機アミンは、好ましくは第2級又は第3級アルキルアミンであり、さらに好ましくは第3級アルキルアルキルアミンである。
【0054】
第2級アミンは例えば、アルキル鎖当たり3個以上の炭素を有するジアルキルアミンを含みうる。好ましいジアルキルアミンは、各アルキル鎖上に3、4、5、6、7、又は8個の炭素を有するであろう。第3級アミンとしては、アルキル鎖当たり1個以上の炭素を有するトリアルキルアミンが挙げられる。好ましいトリアルキルアミンは、2又は3個のアルキル鎖上に3、4、5、6、7、又は8個の炭素を有するであろう。ジアルキルアミン及びトリアルキルアミンの両方におけるアルキル鎖は相互に連結して、環式、2環式、又は3環式化合物を生成しうる。
【0055】
好ましくは、該塩基はヒンダード第2級アミン又は第3級アミンであろう。当該塩基は、これに限定されるわけではないが、ヒューニッヒ塩基(ジイソプロピルエチルアミン)、トリエチルアミン、トリブチルアミン、ジイソプロピルメチルアミン、ジイソプロピルブチルアミン、ジイソプロピルプロプリアミン(diisopropylproply amine)、トリプロピルアミン、トリイソプロピルアミン、トリイソブチルアミン、トリ−tert−ブチルアミン、ジイソブチルメチルアミン、ジイソブチルエチルアミン、ジイソブチルプロピルアミン、ジイソブチブチルアミン(diisobutybutyl amine)、ジイソプロピルアミン、及びジ−tert−ブチルアミンでありうる。当該有機塩基は、ピリジン、モルホリンなどの単環式環を含む第2級又は第3級環式アミン、及びウロトロピン、又はジアザビシクロウンデカンにおける2環式又は3環式環などの、2環式又は3環式環でもありうる。1つの特に好ましい有機塩基は、ヒューニッヒ塩基である。
【0056】
トリフラート化化合物を安定化させるのに有用なアミン塩基の構造は、トリフラートが位置する糖上の位置によって変わる。より反応性の糖は、より反応性でないアミン塩基の使用を必要とする。例えば、糖C6位置が最も反応性であるので、C6位置でトリフラート化された糖は短い(例えば、1〜3個の炭素)ジアルキルアミンによって安定化されない。これらの組成物では、より多くのアルキル炭素を有する塩基が好ましい(例えば、ジイソプロピルアミン)。
【0057】
アミン塩基は、トリフラート化糖の1モル当量以下、好ましくは0.5当量、さらに好ましくは0.2当量である量で使用されうる。
【実施例】
【0058】
本発明は、本発明の範囲を限定すると解釈されるべきではない次の実施例でさらに説明される。
【0059】
[実施例1]
3−トリフルオロメトキシ−3−デオキシ−1,2,1,8−テトラピバロイル−α−D−ガラクトフラノシドの調製及び安定化
1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させた。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄した。トリフラートの塩化メチレン溶液に、ヒューニッヒ塩基0.2当量(230mL)を添加し、当該溶液を蒸発させて表題化合物を得た。本化合物の分解は、蒸発前に塩基が添加されない場合に図2で見られる。
【0060】
[実施例2]
テトラピバロイルフラノースの安定化
実施例1に記載した工程に従って、ピバロイル化ガラクトフラノシド1.5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させた。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄した。トリフラートの塩化メチレン溶液に、ヒューニッヒ塩基0.2当量(230mL)を添加し、当該溶液を蒸発させて表題化合物を得た。
【0061】
[実施例3]
3−トリフルオロメトキシ−3−デオキシ−1,2,1,8−テトラピバロイル−α−D−ガラクトフラノシドの調製及び安定化
1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド1 5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させる。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄する。トリフラートの塩化メチレン溶液に、トリエチルアミン0.2当量を添加し、当該溶液を蒸発させて表題化合物を得た。
【0062】
[実施例4]
テトラピバロイルフラノースの安定化
実施例1に記載した工程に従って、ピバロイル化ガラクトフラノシド5kgを、トリフルオロメタンスルホン酸無水物1.2当量(3.3kg)及びピリジン5当量(3.8kg)と、0℃の塩化メチレン25L中で結合させる。約2時間後、反応混合物を冷塩酸溶液で、次に重炭酸ナトリウム溶液で、当該反応物のpHが中性になるまで洗浄する。トリフラートの塩化メチレン溶液に、トリエチルアミン0.2当量を添加し、当該溶液を蒸発させて表題化合物を得た。
【0063】
本発明の多くの変形は、上述の説明に照らして当業者にそれ自体を示唆するであろう。そのようなすべての明らかな変形は、添付請求項の十分に意図された範囲内である。
【0064】
当業者は、本開示に照らして、本明細書で開示される具体的な実施形態で変更を実施して、本発明の精神及び範囲から逸脱せずに同じ又は同様の結果が得られることを認識すべきである。
【0065】
上記の特許、出願、試験方法、刊行物は、その全体が参照により本明細書に組み入れられている。
【図面の簡単な説明】
【0066】
【図1】D−ガラクトースから開始して、トリフラート化中間体III及びVを有するDGJの合成を示す、合成スキーム。
【図2】トリフラートIII分解の薄層クロマトグラフィー。溶離はヘキサン:酢酸エチル(4:1)により行い、5%硫酸及び加熱により染色された。
【図3】トリフラート分解及びトリフラート安定化の経路。
【特許請求の範囲】
【請求項1】
トリフラート化糖を安定化させる方法であって、
(a)トリフラート化糖を有機塩基と溶媒中で結合するステップと、
(b)前記溶媒を除去するステップであって、溶媒の除去時にトリフラート化糖が第2級又は第3級アルキルアミンと結合されていないトリフラート化糖よりも安定である、溶媒を除去するステップと、
を備える方法。
【請求項2】
前記トリフラート化糖が次式を有する、請求項1に記載の方法。
【化1】
[式中、少なくとも1個のRはトリフラートであり、
各追加のRは独立して、トリフラート、H、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル、C5〜C12ヘテロアリールアルキル、S(=O)2R2、C(=O)R2、又は他のO−保護基であり、
R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである。]
【請求項3】
前記トリフラート化糖がテトラピバロイルフラノースである、請求項1に記載の方法。
【請求項4】
前記有機塩基が第2級又は第3級アミンである、請求項1に記載の方法。
【請求項5】
前記第2級又は第3級アミンがN,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンである、請求項4に記載の方法。
【請求項6】
前記有機塩基がN,N−ジイソプロピルエチルアミンである、請求項1に記載の方法。
【請求項7】
N,N−ジイソプロピルエチルアミンの0.1〜0.3当量が使用される、請求項1に記載の方法。
【請求項8】
N,N−ジイソプロピルエチルアミンの量が使用された前記トリフラート化糖の約0.2当量である、請求項7に記載の方法。
【請求項9】
前記トリフラート化糖がピラノースである、請求項2に記載の方法。
【請求項10】
前記トリフラート化糖がフラノースである、請求項2に記載の方法。
【請求項11】
前記フラノースがα−D−ガラクトフラノースである、請求項10に記載の方法。
【請求項12】
前記溶媒を除去するステップが、前記溶媒を微量レベルまで蒸発させる工程を備える、請求項1に記載の方法。
【請求項13】
糖生成物の反応収率を増加させる方法であって、
(a)トリフラート化糖を産生するために、糖開始物質をトリフルオロメタンスルホニル試薬と溶媒中で反応させるステップと、
(b)前記トリフラート化糖に第2級又は第3級アミンを添加するステップと、
(c)安定化トリフラートを得るために溶媒を濃縮するステップと、
を備える方法。
【請求項14】
前記第2級又は第3級アミンの量が前記糖の約0.2当量である、請求項13に記載の方法。
【請求項15】
前記濃縮するステップが、前記溶媒を微量レベルまで蒸発させる工程を含む、請求項13に記載の方法。
【請求項16】
前記トリフラート化糖がフラノースである、請求項13に記載の方法。
【請求項17】
さらに、以下の工程:
d)フラノシド開始物質の異性体であるフラノシドを産生するために、亜硝酸ナトリウムを添加するステップ、
を備える、請求項16に記載の方法。
【請求項18】
前記糖生成物がフラノシドである、請求項16に記載の方法。
【請求項19】
前記糖生成物がピラノシドである、請求項13に記載の方法。
【請求項20】
前記糖生成物が前記糖開始物質の異性体である、請求項13に記載の方法。
【請求項21】
前記アミンがN,N−ジイソプロピルエチルアミンである、請求項13に記載の方法。
【請求項22】
前記トリフラート化糖が少なくとも500g産生される、請求項13に記載の方法。
【請求項23】
第2級又は第3級アルキルアミン及びトリフラート化糖を含む安定化トリフラート化糖組成物であって、前記糖が次式を有する、安定化トリフラート化糖組成物。
【化2】
[式中、少なくとも1個のRはトリフラートであり、
各追加のRは独立して、トリフラート、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル、C5〜C12ヘテロアリールアルキル、S(=O)2R2、C(=O)R2、又は他のO−保護基であり、
R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである]。
【請求項24】
前記トリフラート化糖がテトラピバロイルフラノースである、請求項23に記載の方法。
【請求項25】
前記第2級又は第3級アミンがN,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンである、請求項23に記載の方法。
【請求項26】
前記アミンがN,N−ジイソプロピルエチルアミンである、請求項25に記載の方法。
【請求項27】
前記N,N−ジイソプロピルエチルアミンの量が前記糖の約0.1〜0.3当量である、請求項26に記載の方法。
【請求項28】
前記量が前記糖の約0.2当量である、請求項27に記載の方法。
【請求項29】
前記トリフラート化糖がピラノースである、請求項23に記載の方法。
【請求項30】
前記トリフラート化糖がフラノースである、請求項23に記載の方法。
【請求項31】
前記フラノースがα−D−ガラクトフラノースである、請求項30に記載の方法。
【請求項1】
トリフラート化糖を安定化させる方法であって、
(a)トリフラート化糖を有機塩基と溶媒中で結合するステップと、
(b)前記溶媒を除去するステップであって、溶媒の除去時にトリフラート化糖が第2級又は第3級アルキルアミンと結合されていないトリフラート化糖よりも安定である、溶媒を除去するステップと、
を備える方法。
【請求項2】
前記トリフラート化糖が次式を有する、請求項1に記載の方法。
【化1】
[式中、少なくとも1個のRはトリフラートであり、
各追加のRは独立して、トリフラート、H、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル、C5〜C12ヘテロアリールアルキル、S(=O)2R2、C(=O)R2、又は他のO−保護基であり、
R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである。]
【請求項3】
前記トリフラート化糖がテトラピバロイルフラノースである、請求項1に記載の方法。
【請求項4】
前記有機塩基が第2級又は第3級アミンである、請求項1に記載の方法。
【請求項5】
前記第2級又は第3級アミンがN,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンである、請求項4に記載の方法。
【請求項6】
前記有機塩基がN,N−ジイソプロピルエチルアミンである、請求項1に記載の方法。
【請求項7】
N,N−ジイソプロピルエチルアミンの0.1〜0.3当量が使用される、請求項1に記載の方法。
【請求項8】
N,N−ジイソプロピルエチルアミンの量が使用された前記トリフラート化糖の約0.2当量である、請求項7に記載の方法。
【請求項9】
前記トリフラート化糖がピラノースである、請求項2に記載の方法。
【請求項10】
前記トリフラート化糖がフラノースである、請求項2に記載の方法。
【請求項11】
前記フラノースがα−D−ガラクトフラノースである、請求項10に記載の方法。
【請求項12】
前記溶媒を除去するステップが、前記溶媒を微量レベルまで蒸発させる工程を備える、請求項1に記載の方法。
【請求項13】
糖生成物の反応収率を増加させる方法であって、
(a)トリフラート化糖を産生するために、糖開始物質をトリフルオロメタンスルホニル試薬と溶媒中で反応させるステップと、
(b)前記トリフラート化糖に第2級又は第3級アミンを添加するステップと、
(c)安定化トリフラートを得るために溶媒を濃縮するステップと、
を備える方法。
【請求項14】
前記第2級又は第3級アミンの量が前記糖の約0.2当量である、請求項13に記載の方法。
【請求項15】
前記濃縮するステップが、前記溶媒を微量レベルまで蒸発させる工程を含む、請求項13に記載の方法。
【請求項16】
前記トリフラート化糖がフラノースである、請求項13に記載の方法。
【請求項17】
さらに、以下の工程:
d)フラノシド開始物質の異性体であるフラノシドを産生するために、亜硝酸ナトリウムを添加するステップ、
を備える、請求項16に記載の方法。
【請求項18】
前記糖生成物がフラノシドである、請求項16に記載の方法。
【請求項19】
前記糖生成物がピラノシドである、請求項13に記載の方法。
【請求項20】
前記糖生成物が前記糖開始物質の異性体である、請求項13に記載の方法。
【請求項21】
前記アミンがN,N−ジイソプロピルエチルアミンである、請求項13に記載の方法。
【請求項22】
前記トリフラート化糖が少なくとも500g産生される、請求項13に記載の方法。
【請求項23】
第2級又は第3級アルキルアミン及びトリフラート化糖を含む安定化トリフラート化糖組成物であって、前記糖が次式を有する、安定化トリフラート化糖組成物。
【化2】
[式中、少なくとも1個のRはトリフラートであり、
各追加のRは独立して、トリフラート、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル、C5〜C12ヘテロアリールアルキル、S(=O)2R2、C(=O)R2、又は他のO−保護基であり、
R2は、置換又は非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12ヘテロシクロアルキル又はC5〜C12ヘテロアリールアルキルである]。
【請求項24】
前記トリフラート化糖がテトラピバロイルフラノースである、請求項23に記載の方法。
【請求項25】
前記第2級又は第3級アミンがN,N−ジイソプロピルエチルアミン、N,N,N−トリブチルアミン、又はN,N,N−トリエチルアミンである、請求項23に記載の方法。
【請求項26】
前記アミンがN,N−ジイソプロピルエチルアミンである、請求項25に記載の方法。
【請求項27】
前記N,N−ジイソプロピルエチルアミンの量が前記糖の約0.1〜0.3当量である、請求項26に記載の方法。
【請求項28】
前記量が前記糖の約0.2当量である、請求項27に記載の方法。
【請求項29】
前記トリフラート化糖がピラノースである、請求項23に記載の方法。
【請求項30】
前記トリフラート化糖がフラノースである、請求項23に記載の方法。
【請求項31】
前記フラノースがα−D−ガラクトフラノースである、請求項30に記載の方法。
【図1】

【図2】
【図3】

【図2】
【図3】
【公表番号】特表2008−545800(P2008−545800A)
【公表日】平成20年12月18日(2008.12.18)
【国際特許分類】
【出願番号】特願2008−516016(P2008−516016)
【出願日】平成18年6月8日(2006.6.8)
【国際出願番号】PCT/US2006/022757
【国際公開番号】WO2006/133448
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(507170099)アミカス セラピューティックス インコーポレイテッド (21)
【Fターム(参考)】
【公表日】平成20年12月18日(2008.12.18)
【国際特許分類】
【出願日】平成18年6月8日(2006.6.8)
【国際出願番号】PCT/US2006/022757
【国際公開番号】WO2006/133448
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(507170099)アミカス セラピューティックス インコーポレイテッド (21)
【Fターム(参考)】
[ Back to top ]