
遺伝子断片のクローニング方法
【課題】 遺伝子の全長配列又は近接する領域をクローニングするための簡便かつ高感度な方法の提供。
【解決手段】(a)DNAを断片化するステップ、(b)断片化DNAを環状化するステップ、(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、(d)任意により、得られる増幅産物を回収するステップを含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法。
【解決手段】(a)DNAを断片化するステップ、(b)断片化DNAを環状化するステップ、(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、(d)任意により、得られる増幅産物を回収するステップを含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、既知の塩基配列に基づいて、DNA(例えば環境メタゲノム)から、既知の塩基配列と同一の若しくは相同性を有する塩基配列、又は該既知の塩基配列に近接する領域をクローニングする方法に関する。
【背景技術】
【0002】
近年、微生物等の有する代謝系酵素を利用したバイオプロセスが特定の化学物質の生産に利用されるようになってきている。これに伴い、微生物等から新規代謝系酵素の遺伝子を獲得する試みが盛んに行われるようになった。特定の代謝系酵素の遺伝子を獲得する際には、通常、まず目的の活性を持った生物を環境サンプルから単離し、次にその単離された生物から代謝系遺伝子のクローニングを試みる。しかし近年、分子生態解析の手法が確立されてくると、環境中の大多数の微生物は未だに培養・単離されておらず、またそれらの多くが難培養であることがわかってきた(非特許文献1参照)。これは、微生物等の培養・単離を介したのでは、環境中の多様な遺伝子の大多数を獲得できないことを意味している。そこで最近、微生物等を単離培養することなく直接メタゲノム(環境サンプル等から抽出されたDNA)を抽出し、そこから作製されたメタゲノムライブラリーを、特定機能を有する遺伝子のスクリーニングに利用する試みがなされるようになってきている(非特許文献2参照)。
【0003】
単離された生物のゲノムや環境メタゲノムからの遺伝子のスクリーニング法として、遺伝子配列に依存したスクリーニング法がある。遺伝子配列に依存したスクリーニングとは、既知の遺伝子配列から設計されたプローブやプライマーを用いて類似遺伝子を獲得しようとするものである(特許文献1及び非特許文献3参照)。しかしながら、この方法においてクローニング可能な塩基配列は、設計したプライマーの内部配列のみであるため、環境中で実際に機能している酵素をコードする完全な遺伝子配列を決定することが出来ない。通常、酵素はいくつかのドメインからなり、活性中心を持つドメイン以外にもその酵素の折り畳みを助けるようなドメインや、酵素活性を高めるために重要なドメインなどから形成される。通常遺伝子配列に依存したクローニング法においては、高度に保存された活性中心の遺伝子配列からプライマーを設計するため、周辺ドメインの遺伝子配列はクローニングされてはこない。
【0004】
そのような問題を解決するため、環境メタゲノムを鋳型とし、遺伝子配列に依存したクローニング法で得られた遺伝子配列から、さらにその前後の未知の遺伝子配列をクローニングし、最終的に酵素遺伝子全体をクローニングするような試みがおこなわれはじめている(非特許文献4及び5参照)。これらの方法においては、(1)始めに遺伝子配列に依存したクローニングで、遺伝子配列を決定し、(2)決定した遺伝子配列を元にプライマーを作製し、(3)環境から抽出した染色体DNAを適当な制限酵素で処理し、リンカーをつなげ、(4)リンカー特異的なプライマーと、決定した遺伝子配列を元にしたプライマーを用いてPCRを行い、(5)増幅断片をクローニングし、始めに決定した遺伝子配列の外側に位置する未知塩基配列を決定する、というスキームをとっている。しかしながらこれらの方法においては、わずか1〜2種類程度のクローンしか得られていない。これはおそらく、メタゲノム中に混在する大量のDNAがPCR反応を阻害し、低コピー数でしか存在しない遺伝子からの増幅を困難にしていることが原因と考えられる。
【0005】
そのため、このような問題を解決する新たなクローニング法を開発し、低コピー数でしか存在しない多様な未知遺伝子及び未知ドメインのクローニングを行うことが望まれている。
【0006】
【特許文献1】特表2005−525793号公報
【非特許文献1】Amann RI, Ludwig W, 及びSchleifer KH,"Phylogenetic identification and in situ detection of individual microbial cells without cultivation",Microbiol Rev.,第59巻第143〜169頁,1995年
【非特許文献2】Lorenz P, Liebeton K, Niehaus F, 及びEck J,"Screening for novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequence space",Curr Opin Biotechnol.,第13巻第572〜577頁,2002年
【非特許文献3】Stokes HW, Holmes AJ, Nield BS, Holley MP, Nevalainen KM, Mabbutt BC, 及びGillings MR,"Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA",Appl Environ Microbiol.,第67巻第5240〜5246頁,2001年
【非特許文献4】Eschenfeldt WH, Stols L, Rosenbaum H, Khambatta ZS, Quaite-Randall E, Wu S, Kilgore DC, Trent JD, 及びDonnelly MI,"DNA from uncultured organisms as a source of 2,5-diketo-D-gluconic acid reductases",Appl Environ Microbiol.,第67巻第4206〜4214頁,2001年
【非特許文献5】Bell PJL, Sunna A, Gibbs MD, Curach NC, Nevalainen H, 及びBergquist PL,"Prospecting for novel lipase genes using PCR",Microbiol.,第148巻第2283〜2291頁,2002年
【発明の開示】
【発明が解決しようとする課題】
【0007】
上に述べたように、従来法では環境メタゲノムから得られた遺伝子断片の塩基配列情報を元にして、環境メタゲノム中に存在する周辺領域を増幅することは非常に困難であった。
【0008】
そこで本発明は、今までクローニングできなかった遺伝子の全長配列又は近接する領域をクローニングするための簡便かつ高感度な方法を提供するとともに、巨大な環境メタゲノムから低コピー数にしか存在しない遺伝子断片をクローニングするためにも有用な方法を提供する。
【課題を解決するための手段】
【0009】
本発明者は、上記課題を解決するために鋭意検討を重ねた結果、一回目の増幅反応を行った後に二回目の増幅反応を行うことにより高効率に遺伝子断片の回収ができ、さらに一回目の増幅反応の後に増幅産物を回収して、二回目の増幅反応を行うことにより高感度かつ高効率に遺伝子断片を増幅することができることを見出し、本発明を完成させた。また、本発明の方法において、一度目の増幅反応としてインバースPCRを行うことにより、遺伝子の周辺領域のクローニングが可能となる(ゲノムウォーキング)ことも見出した。
【0010】
すなわち、本発明は、以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)任意により、得られる増幅産物を回収するステップ、
(e)回収した増幅産物を増幅可能なプライマーを用いて該増幅産物を鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法に関する。
【0011】
また本発明は、以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)得られた増幅産物中の上記既知の塩基配列以外の領域に基づいて設計した1つのプライマーと上記既知の塩基配列に基づいて設計した1つのプライマーを用いて、上記DNAを鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域をクローニングする方法に関する。
【0012】
上記方法において、DNAの断片化は、例えば制限酵素処理により行うことができる。制限酵素としては、付着末端を生じる制限酵素を使用することが好ましく、例えば限定されるものではないが、EcoR I、Xho I又はApaL Iを用いることができる。
【0013】
また上記方法において、DNAとしては、ゲノムDNA、プラスミドDNA、及び/又は、ファージDNAを用いることができる。ゲノムDNAは、単一の生物に由来するものであってもよいし、又は複数の生物に由来するものであってもよい。DNAは、例えば環境サンプル、発酵槽サンプル及び集積培養サンプルから抽出されたDNA(メタゲノム)とすることができる。
【0014】
上記方法において、断片化DNAの環状化は、例えばセルフライゲーションにより行われる。
【0015】
上記方法において、既知の塩基配列としては、例えば限定されるものではないが、代謝系遺伝子、転写制御系遺伝子、又はトランスポゾンをコードするものが挙げられる。また、既知の塩基配列に近接する領域は、未知の配列を含んでもよい。
【0016】
上記方法において、使用するプライマーのうち少なくとも1つはランダムプライマーであってもよい。
【0017】
また上記方法において、ステップ(c)において使用するプライマーの一方又は両方は、標識されていることが好ましい。標識としては、限定されるものではないが、ビオチンが挙げられる。プライマーに標識を付加した場合、ステップ(d)において、標識を利用して増幅産物を回収することができる。例えば、プライマーにビオチン標識を付加した場合、ステップ(d)においては、例えばストレプトアビジンを固定化した磁気ビーズを用いて、ビオチン標識を有する増幅産物を回収することができる。
【発明の効果】
【0018】
本発明により、目的の遺伝子のクローニング、さらには目的の遺伝子と相同性を有する遺伝子や目的の遺伝子に近接する領域のクローニングを簡便、高効率、かつ高感度に行うことが可能となる。そのため、クローニング対象の配列が低コピー数にしか存在しない場合であっても、確実にその全長配列をクローニングすることができ、有用である。
【発明を実施するための最良の形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
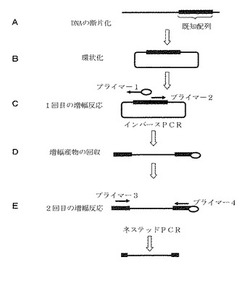
本発明は、既知の塩基配列に基づいて、DNAから、既知の塩基配列と同一の若しくは相同性を有する塩基配列、又は該既知の塩基配列に近接する領域をクローニングする方法に関する。本発明の方法の概要を図1に示す。図1において、太線は既知の塩基配列で、細線は既知の塩基配列に近接する領域を表す。矢印を有する線はプライマーを表し、矢印の向きはプライマーから伸長される方向を示す。白抜きの丸は標識を表す。本発明の方法においては、2回の増幅反応(図1におけるCとE)を行うことによって、わずかにしか存在しないDNAに基づいて、既知の塩基配列、及びそれに近接する未知の配列を含む領域を、簡便かつ高感度にクローニングすることが可能である。
【0021】
最初に、目的の塩基配列又はそれに近接する領域のクローニングの鋳型となるDNAを準備する。鋳型とするDNAは、特に限定されるものではなく、例えばゲノムDNA、ファージDNA、プラスミドDNAなどである。また例えば、ある単一の生物に由来するDNA(例えばゲノムDNA、染色体DNA)であってもよいし、複数の生物に由来するDNAであってもよい。また、生物は、単離された生物(例えば微生物、微生物混合物など)であってもよいし、単離されていなくてもよい。例えば、環境サンプル(例えば、土壌、湖沼水、海水、河川水など)、微生物の培養物、発酵槽サンプル、集積培養サンプルなどに由来するDNA(メタゲノム)を用いることが可能である。さらに、DNAは、ゲノムDNAライブラリであってもよい。DNAの調製は当技術分野で公知であり、任意の方法(例えばフェノール・クロロホルム法等)又は任意の市販キットを用いてDNAを調製することができる。また例えば、土壌中からメタゲノムをクローニングする方法が記載されており、この文献に記載の手法も本発明において用いることができる(Rondon, MR. et al., Appl Environ Microbiol. 2000, 66:2541-2547)。
【0022】
続いて、DNAを断片化する(図1のA)。DNAの断片化は、制限酵素処理により行うことができる。使用する制限酵素の種類は特に限定されるものではないが、後述するDNAの環状化を考慮すると、付着末端を生じる制限酵素を使用することが好ましい。好ましくは、制限酵素としてEcoR I、Xho I、ApaL Iなどを使用する。
【0023】
断片化されたDNAは、当技術分野で公知の方法により環状化する(図1のB)。環状化は、例えばセルフライゲーションにより行うことができる。セルフライゲーションとは、断片化DNAの両端に存在する制限酵素切断部位がリガーゼにより連結されることをいう。従って、断片化DNAは、適当なリガーゼ(例えばT4 DNAリガーゼ等)を用いて環状化することができる。
【0024】
次に、環状化DNAを鋳型とした増幅反応を行う(図1のC)。プライマーは、既知の塩基配列に基づいて設計する。選択する「既知の塩基配列」は、塩基配列が既知であれば限定されるものではない。例えば、遺伝子(代謝系遺伝子、転写制御系遺伝子等)、トランスポゾン、トランスポーター、ハウスキーピングタンパク質(例えばDNAポリメラーゼ、DNAリガーゼ)などをコードする塩基配列を選択することができる。また、遺伝子全体が既知の塩基配列である場合に加えて、一部のみが配列決定されている場合であっても、遺伝子全体又は遺伝子の周辺領域(近接する領域)をクローニングするため、その一部の既知配列を選択することができる。
【0025】
メタゲノムに存在する有用な遺伝子の例としては、限定されるものではないが、代謝系遺伝子、すなわち特定の化合物又は生体分子の化学的構造の変換を触媒する酵素をコードする遺伝子が挙げられる。例えば、フェノールの芳香環に酸素分子由来の酸素原子を導入しカテコールを生産する酵素であるフェノール水酸化酵素(phenol hydroxylase)EC 1.14.13.7(Nordlund I, Powlowski J, Shingler V. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 1990 172:6826-6833)などが挙げられる。また、限定されるものではないが、ハイドラーゼ、エステラーゼ、プロテアーゼ、リパーゼ、ベータガラクトシダーゼ、ラクターゼ、ポリガラクツロナーゼ、ベータグルコアミラーゼ、エステラーゼ、ヘミセルラーゼ、ペルオキシダーゼ、オキシダーゼ、オキシゲナーゼ、ラッカーゼ、グルコースオキシダーゼ、ペクチナーゼ、アミラーゼ、ガラクタナーゼ、アラビナーゼ、キチナーゼ、キシラナーゼ、DNAポリメラーゼ、DNAリガーゼ又はセルラーゼなどの酵素をコードする遺伝子の塩基配列も、本発明の方法において既知の塩基配列として選択することができる。
【0026】
さらに、上記の代謝系酵素をコードする遺伝子に加え、「代謝系遺伝子」には、代謝反応において基質となる化学物質の細胞内外への輸送に関与するタンパク質をコードする遺伝子も含まれる。例えば、トルエンなどの化学物質の取り込みに関与するタンパク質XylN(Kasai Y, Inoue J, Harayama S. The TOL plasmid pWW0 xylN gene product from Pseudomonas putida is involved in m-xylene uptake. J Bacteriol. 2001 183:6662-6)などである。または、トルエンなどの溶媒分子の細胞外への排出に関与するタンパク質である排出ポンプ(Duque E, Segura A, Mosqueda G, Ramos JL. Global and cognate regulators control the expression of the organic solvent efflux pumps TtgABC and TtgDEF of Pseudomonas putida. Mol Microbiol. 2001 39:1100-6)なども含まれる。
【0027】
またさらに、代謝系オペロンを制御するような転写制御因子をコードする転写制御系遺伝子の塩基配列を選択することもできる。「発現制御因子」とは、特定の基質の存在下にて代謝系遺伝子が誘導的に発現される場合にその発現を制御する遺伝的要素を指す。例えば、ナフタレンやサリチル酸の代謝系オペロンの発現を制御しているタンパク質NahR(Schell MA, Wender PE. Identification of the nahR gene product and nucleotide sequences required for its activation of the sal operon. J. Bacteriol. 1986 166:9-14)などである。他の発現制御因子としては、例えば、XylR、XylS、DmpR、PhcRなどが挙げられる。
【0028】
また、遺伝子の水平伝播に関わるとされるトランスポゾン(Shintani M, Yoshida T, Habe H, Omori T, Nojiri H. Large plasmid pCAR2 and class II transposon Tn4676 are functional mobile genetic elements to distribute the carbazole/dioxin-degradative car gene cluster in different bacteria. Appl. Microbiol. Biotechnol. 2005 67:370-82)をコードする塩基配列を選択してもよい。
【0029】
プライマーを設計する領域は、当業者であれば容易に選択することができる。上記増幅反応において、既知の塩基配列に近接する領域をクローニングする場合には、設計する2つのプライマーのうち、3’側をフォワードプライマーとして、5’側をリバースプライマーとして用いる(図1のCの左側におけるプライマー1及び2)。このようにプライマーを設計することによって、図1のCの左側に示すように環状化DNAを鋳型とした増幅反応を行い、既知の塩基配列の一部と同一の又は相同性を有する領域とそれに近接する領域を含む断片を増幅することができる(図1のDの左側)。
【0030】
さらに、既知の塩基配列と相同性を有する領域をクローニングする場合、プライマーをランダムプライマーとして設計してもよい。それにより、既知の塩基配列とは完全に同一ではないが、プライマーがアニーリングできる程度に相同性を有する塩基配列の領域が増幅されることになる。
【0031】
プライマーの設計は、当業者に公知の方法に従って行うことができる。プライマーとして実質的な機能を有する長さとしては、10塩基以上が好ましく、さらに好ましくは16〜50塩基であり、さらに好ましくは20〜30塩基である。また設計の際には、公知のプライマー又はプローブ設計用ソフトウエアを利用することができ、本発明で利用可能なソフトウエアとしては、例えばOligoTM[National Bioscience Inc.(米国)製]、GENETYX[ソフトウェア開発(株)(日本)製]等などが挙げられる。上述のように設計したプライマーは、当業者に公知の方法に従って、例えばオリゴヌクレオチド合成機を用いることにより、調製することができる。
【0032】
プライマーには、後述する増幅産物の回収のために、回収を容易にする標識を付加してもよい。例えば、物質間の相互作用性質を利用して増幅産物の回収を行う場合には、その物質の一方を用いてプライマーを標識することができる。このような標識の種類や標識の導入方法等は、特に限定されるものではなく、従来公知の各種手段を用いることができる。例えば、ビオチン−アビジン系を利用してプライマーにビオチンを標識することや、抗原−抗体反応を利用してプライマーに抗原を標識することができる。標識は、フォワードプライマー若しくはリバースプライマーの一方、又は両方に付加することができる。
【0033】
上述の通り作製したプライマーを用いて、上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列(既知の塩基配列と相同性を有する領域を含む)、及び/又は上記既知の塩基配列に近接する領域を増幅する(図1のC)。ここで、「近接する領域」とは、既知の塩基配列に隣接する領域及び既知の塩基配列の周囲領域を意味し、具体的には、既知の塩基配列から約6000bp、好ましくは約3000bpまでの領域である。
【0034】
増幅反応は、上述のように設計したプライマーを用いて、環状化DNAを鋳型とした増幅を行うことができる方法であれば、特に限定されるものではない。ゲノムDNAから任意の塩基配列をクローニングするため、通常はポリメラーゼ連鎖反応(PCR)を応用した方法を利用する。例えば、インバースPCR法(Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics 1988 120:621-623)、ランダムプライマーPCR法(Liu Y, Whittier RF. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995 25:674-681)、アダプターライゲーションPCR法(Ochman H. et al. Use of polymerase chain reaction to amplify segments outside boundaries of known sequences. Methods Enzymol. 1993 218:309-321)、ユニバーサルファーストウォーキングPCR法(Myrick, K.V. and Gelbart, W.M. 2002 Universal fast walking for direct and versatile determination of flanking sequence. Gene 284:125-131)、パンハンドル PCR法(Megonigal, M.D. et al. 2000 Panhandle PCR for cDNA: a rapid method for isolation of MLL fusion transcripts involving unknown partner genes. PNAS 97:9597-9602)などが挙げられる。
【0035】
増幅反応後、任意により増幅産物を回収してもよい(図1のD)。この回収ステップを行うことにより、2回目の増幅反応において鋳型として使用される増幅産物が濃縮され、目的とする配列のクローニングが高感度かつ高効率となる。増幅産物の回収は、当技術分野で公知の任意の方法により行うことができる。例えば増幅反応において標識プライマーを用いた場合には、その標識を利用して増幅産物を回収することができる。具体的には、標識と特異的に結合することができる物質を利用して、その物質とプライマーの標識とを結合させ、未結合のDNAを除去することによって、増幅産物を回収する。好ましくは、ビオチンで標識したプライマーを用いて増幅反応を行い、増幅反応溶液をストレプトアビジンを表面にコートした磁気ビーズ(例えば、Dynabeads kilobaseBINDER Kit(DYNAL)として市販されている)と接触させ、磁気ビーズに結合していない反応液を洗浄することにより、増幅産物を回収することができる。回収ステップは、例えばメタゲノムなどの多様なDNAを含むサンプル(このようなサンプルは増幅反応を阻害することがある)から目的の配列をクローニングする場合に特に有用である。
【0036】
続いて、得られた増幅産物を鋳型とした2回目の増幅反応を行う(図1のE)。この増幅反応においては、得られた増幅産物を増幅可能なプライマーを用いる。そのようなプライマーは、既知の塩基配列に基づいて設計することができ、具体的には、既知の塩基配列に基づいて、1回目の増幅反応において用いたプライマー(図1におけるプライマー1及び2)の領域よりも内側の領域にプライマー(図1におけるプライマー3及び4)を設計する。プライマーの設計は、上述と同様に、当技術分野で公知の方法に従って行うことができる。なお、2回目の増幅反応においても、プライマーとしてランダムプライマーを用いることができる。
【0037】
2回目の増幅反応は、DNAを鋳型とした増幅反応を行うことができれば、当技術分野で公知の任意の方法により行うことができる。このような増幅反応によって、既知の塩基配列、既知の塩基配列と相同性を有する領域、及び/又は既知の塩基配列に近接する領域を増幅し、クローニングすることができる。
【0038】
また、1回目の増幅反応又は2回目の増幅反応において得られた増幅産物の塩基配列を決定することによって、既知の塩基配列に近接する領域の配列情報を明らかにすることができる。また、その決定された塩基配列に基づいて設計した1つのプライマーと、既知の塩基配列に基づいて設計した1つのプライマーとを用いて、DNAを鋳型とした増幅反応を行うことにより、既知の塩基配列の一部とそれに近接する領域をクローニングすることができる。
【0039】
さらに、決定した塩基配列に基づいて設計されたプローブを用いたハイブリダイゼーションを行うことによっても、既知の塩基配列に近接する領域をクローニングすることが可能である。
上述のように、本発明の方法により、既知の塩基配列の一部若しくは全体、又はそれに近接する領域を、効率的にクローニングすることができるため、例えば代謝系酵素遺伝子オペロンの全体構造の解明に有用である。
【0040】
以下の実施例は、本発明をさらに具体的に説明するためのものであり、本発明を限定するものではない。
【実施例1】
【0041】
本実施例においては、本発明の方法の有効性を実証するために、次のような実験を行った。
【0042】
始めにRalstonia eutropha E2株とEscherichia coliJM109株の2種の染色体DNAを用意し、それぞれを混合した。染色体DNAの抽出は、Silhavyらの方法(Silhavy TJ, Berman ML, and Enquist LW. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y. 1984.)によりおこなった。E. coli JM109株のDNAは一定量、R. eutropha E2株のDNAはE. coliJM109株のDNA濃度に対して段階希釈(10-1〜10-6)し、混合した。次にDNAの混合物をXhoIで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。
【0043】
次に、R. eutropha E2株のフェノール代謝系遺伝子オペロン(poxオペロン)(Hino S, Watanabe K, Takahashi N. Phenol hydroxylase cloned from Ralstoniaeutropha strain E2 exhibits novel kinetic properties. Microbiology 1998 144:1765-72)に近接する領域をインバースPCRで特異的に増幅させうるような、以下のプライマー1及び2(それぞれ配列番号1及び2)を用いてPCRを行い(図1のC)、図2に示すようにR. eutropha E2株のpoxオペロンの一部、すなわちXho I切断部位で囲まれた3.8 kbの増幅産物の増幅を試みた。
【0044】
プライマー1:5’Biotinirated-gaacaactcggactcgatcagttcgtcg-3’(配列番号1)
プライマー2:5’-ttcgtgctgtttgccttcagcgtcg-3’(配列番号2)
PCRの反応条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃4分からなるサイクルを32サイクルとした。
【0045】
インバースPCRの後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物を回収した(図1のD)。また、回収を行った場合及び回収を行わなかった場合の増幅産物に対して以下のプライマー3及び4(配列番号3及び4)を用いたネステッドPCRを行った(図1のE):
プライマー3:5’-atcaagacgcagcagatcc-3’(配列番号3)
プライマー4:5’-caaggccaggatggtatcg-3’(配列番号4)
ネステッドPCRの条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃4分からなるサイクルを32サイクルとした。
【0046】
インバースPCRのみを行った場合(条件1)、インバースPCR後、増幅産物をネステッドPCRでさらに増幅させた場合(条件2)、インバースPCR後、増幅産物をDynabeads kilobaseBINDER Kitを用いて濃縮し、これらをネステッドPCRでさらに増幅させた場合(条件3)の3種の条件で、目的の遺伝子断片の増幅検出限界を0.7%アガロースゲル電気泳動により確認した。
【0047】
実験の結果を図3に示す。実験の結果、(条件1)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 1 ngを混合した場合(10-1)が、増幅産物の検出限界であった。(条件2)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 10 pgを混合した場合(10-3)が、増幅産物の検出限界であった。(条件3)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 100 fgを混合した場合(10-5)が、増幅産物の検出限界であった。実験の結果、インバースPCRとネステッドPCRとを組み合わせた本方法(条件2及び3)を用いることにより検出限界が高まることが確認された。特に、インバースPCRを行った後に増幅産物を回収した条件3によって、もっとも高い検出限界を達成することに成功した。従って、本発明の方法は、低コピー数のDNAしか存在しない場合であっても、効率的かつ高感度に目的遺伝子の近接領域を増幅することができることがわかった。
【実施例2】
【0048】
本発明の方法の有効性を実証するために、本方法を用いて実際にメタゲノムからP450様モノオキシゲナーゼを含むオペロンの全長のクローニングを試みた。
【0049】
環境メタゲノムは、地下石油備蓄基地の湧水(Watanabe K, Kodama Y, Syutsubo K, Harayama S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude-oil-storage cavities. Appl. Environ. Microbiol. 2000 66:4803-4809)から抽出した染色体DNAを使用し、P450様モノオキシゲナーゼ(Uchiyama T, Abe T, Ikemura T, Watanabe K. Substrate-induced gene-expression screening of environmental metagnome libraries for isolation of catabolic genes. Nat. Biotechnol. 2005 1:88-93)を含むオペロンの全長クローニングを試みた。
【0050】
始めにメタゲノムをEcoR Iで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。次に、P450様モノオキシゲナーゼを含むオペロンの領域を特異的に増幅させうるような、以下のプライマー71A及び71B(それぞれ配列番号5及び6)を用いてインバースPCRを行い(図1のC)、図4に示すようにオペロンの一部の増幅を試みた。
【0051】
プライマー71A:5’Biotinirated-tgcgtagattacgaaacggccaattccagc -3’(配列番号5)
プライマー71B:5’-agcaccacagtcagcctgaatgacg -3’(配列番号6)
インバースPCRの反応条件は、94℃にて1分の後、98℃20秒、58℃20秒及び72℃8分からなるサイクルを32サイクルとした。
【0052】
インバースPCRの後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物を回収し(図1のD)、その増幅産物に対して以下のプライマー71C及び71D(それぞれ配列番号7及び8)を用いたネステッドPCRを行った(図1のE)。
【0053】
プライマー71C:5’-gccaacggcatcgacggtattctgg -3’(配列番号7)
プライマー71D:5’-tgatggcatccacggcaattggtcg -3’(配列番号8)
【0054】
プライマー71C及び71Dは、インバースPCRに使用したプライマー71A及び71Dに近接するように設計した(図4)。ネステッドPCRの条件は、94℃にて1分の後、98℃20秒、58℃20秒及び72℃8分からなるサイクルを32サイクルとした。図4において、白抜きのバーは本実験によって増幅された既知塩基配列と未知塩基配列の遺伝子断片を示す。
【0055】
また、回収された増幅産物に対して、以下のプライマー71OperonF及び71OperonR(それぞれ配列番号9及び10)を用いたPCRを行った:
プライマー71OperonF:5’-aggccatggcggtgaagtttgttcc -3’(配列番号9)
プライマー71OperonR:5’-gaattcacccgaccggtgctcgacg -3’(配列番号10)
【0056】
プライマー71OperonFは既知の塩基配列に基づいて設計し、プライマー71OperonRはクローニングされた未知の配列に基づいて設計した(図4)。PCRの条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃5分からなるサイクルを32サイクルとした。図4に示す灰色のバーは、既知の塩基配列と今回決定された未知の塩基配列を含む遺伝子断片を示す。
【0057】
実験は、プライマー71A及び71BでインバースPCRを行った際の増幅産物を泳動した条件(条件1)、その増幅産物を濃縮し、プライマー71C及び71DでネステッドPCRを行った後の増幅産物を泳動した条件(条件2)、及びプライマー71OperonF及びプライマー71OperonRでPCRを行った後の増幅産物を泳動した条件(条件3)で行った。目的の遺伝子断片の増幅は、0.7%アガロースゲル電気泳動により確認した。
【0058】
結果を図5に示す。図5において、レーン1〜3は、それぞれ条件1〜3で得られた増幅産物の泳動を示す。実験の結果、P450様モノオキシゲナーゼに近接する領域の未知塩基配列の決定に成功した。レーン2に示されるように、ネステッドPCRを行うことにより、オペロン全長を取得することができたが、これは、P450様モノオキシゲナーゼオペロンから設計したプライマーと、新たに決定した塩基配列から設計したプライマーを用いてPCRを行って、オペロン全長が増幅されていることを確認できた(図5のレーン3)。以上より、本発明の方法を行うことによりメタゲノムから任意の塩基配列とそれに近接する領域(未知の塩基配列を含む)をクローニングできることが確認された。
【実施例3】
【0059】
本発明の方法の有効性を実証するため、メタゲノムを鋳型とし、酵素のアミノ酸相同性を基に設計したプライマーを用いて遺伝子の一部をクローニングし、その塩基配列を基に本発明の方法によって遺伝子全体の塩基配列を決定するような実験を試みた。
【0060】
(1)キチナーゼ遺伝子の単離
環境メタゲノムは地下石油備蓄基地の湧水(Watanabe K, Kodama Y, Syutsubo K, Harayama S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude-oil-storage cavities. Appl. Environ. Microbiol. 2000 66:4803-9)から抽出した染色体DNAを使用した。酵素のアミノ酸相同性を基に設計したプライマーは論文(Hobel CFV, Marteinsson VT, Hreggvidsson GO, Kristjansson JK. Investigation of the microbial ecology of intertidal hot springs by using diversity analysis of 16S rRNA and chitinase genes. 2005 71:2771-6)で使用された、キチナーゼ遺伝子の活性ドメインをコードする塩基配列の一部を増幅させる以下のプライマーを使用した。
【0061】
プライマーChiA_F1, 5'-ACG GCG TGG ACA TCG AYT GGG ART-3'(配列番号11)
プライマーChiA_F2, 5'-CGT GGA CAT CGA CTG GGA RTW YCC-3'(配列番号12)
プライマーChiA_R1, 5'-CCC AGG CGC CGT AGA RRT CRTAYS-3'(配列番号13)
プライマーChiA_R2, 5'-CCC AGG CGC CGT AGA RRT CRT ARS WCA-5'(配列番号14)
【0062】
なお、配列番号11〜14における「Y」はT又はCを表し、「R」はG又はAを表し、「W」はA又はTを表し、「S」はG又はCを表す。
【0063】
始めにメタゲノムを鋳型に、キチナーゼ遺伝子の活性ドメインを増幅させる上記プライマーを用いて、増幅産物のランダムクローニングを行った。得られた増幅産物を2.0%アガロースゲル中で電気泳動し、300 bp前後の大きさの増幅産物をQIAquick gel extraction kit(Qiagen)によりゲル中から抽出した。抽出した増幅産物は、TA cloning vector(タカラ酒造)とライゲーションさせた。これを用いエレクトロポレーションによりEscherichia coliJM109(タカラ酒造)を形質転換させ、IPTGおよびX-Galを含んだLB培地プレートに塗布した。形質転換体を100個からプラスミドを抽出し、クローニングされた塩基配列を決定した。決定した塩基配列をアミノ酸配列に変換し、既知のキチナーゼ配列と併せてClustalWプログラムにより解析を行い、系統樹を描かせた(図6)。
【0064】
その結果、多様なキチナーゼ遺伝子を得ることに成功した。図6において、本実験でクローニングしたクローンはCHIと表記し、クローンナンバーと得られた陽性クローンの数を表記した。
【0065】
(2)キチナーゼ遺伝子全体のクローニング
(1)の実験で単離されたキチナーゼ遺伝子の中から10クローンを適当に選び出し(図6中、黒丸でマークされたもの)、本発明の方法を用いてキチナーゼ遺伝子全体をクローニングすることを試みた。
【0066】
始めにメタゲノムをApaL I又はEcoR Iで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。次に、キチナーゼ遺伝子に近接する未知配列を増幅させうるような、以下のプライマー(配列番号15〜34)を用いてインバースPCRを行った。なお、インバースPCRの反応条件は、94℃にて1分の後、98℃20秒、56℃20秒及び72℃8分からなるサイクルを32サイクルとした。
【0067】
CHI01用プライマー
CHI01-biotin:5’biotinirated-ccgacaggctgtccagggccttgcgcaagg-3’(配列番号15)
CHI01-B:5’-tacctggtgaccagtgcgatcaatg-3’(配列番号16)
CHI03用プライマー
CHI03-biotin:5’biotinirated-cattcaaacagtacctgaccaggaccgagc-3’(配列番号17)
CHI03-B:5’-cccggagcagcagtgtgaagtttgc-3’(配列番号18)
CHI05用プライマー
CHI05-biotin:5’biotinirated-aggcgtcgacaagatccgggttaccgatcc-3’(配列番号19)
CHI05-B:5’-ggcatcgagttgccgcctgaactcc-3’(配列番号20)
CHI08用プライマー
CHI08-biotin:5’biotinirated-ggcaccaaatggggatgccgcttcaccgac-3’(配列番号21)
CHI08-B:5’-cgccggaaaccgatcgccatcgagg-3’(配列番号22)
CHI12用プライマー
CHI12-biotin:5’biotinirated-gacctggcagcatccgcaatctggatctgc-3’(配列番号23)
CHI12-B:5’-cagcgtcaaattggcagtgtcatcg-3’(配列番号24)
CHI14用プライマー
CHI14-biotin:5’biotinirated-tcgtttcgtgcgcttgctacgctcatcgag-3’(配列番号25)
CHI14-B:5’-cctgctgccattctatgaaatcgag-3’(配列番号26)
CHI19用プライマー
CHI19-biotin:5’biotinirated-cgttgatccagttgatgacgaagttcttgc-3’(配列番号27)
CHI19-B:5’-gtagacaagattgttaagtcggagc-3’(配列番号28)
CHI23用プライマー
CHI23-biotin:5’biotinirated-gcagttcctcgaccatcgtggcatggtctg-3’(配列番号29)
CHI23-B:5’-gactcggactccgtgttcgatcacc-3’(配列番号30)
CHI31用プライマー
CHI31-biotin:5’biotinirated-aactcctgagttcctgcttcagttgcgacc-3’(配列番号31)
CHI31-B:5’-cagcgacgtgaagttgtggaaatcg-3’(配列番号32)
CHI32用プライマー
CHI32-biotin:5’biotinirated-gcatcgagccgcgagcgaagctcttgcagc-3’(配列番号33)
CHI32-B:5’-ccttcgacgatcgcgaacatgccag-3’(配列番号34)
【0068】
インバースPCR後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物の濃縮を行い、以下のプライマー(配列番号35〜54)を用いてネステッドPCRを行い、最終的にキチナーゼ遺伝子の全長配列の決定を試みた(図7)。ネステッドPCRの反応条件は、94℃にて1分の後、98℃20秒、56℃20秒及び72℃8分からなるサイクルを32サイクルとした。また、実験の際にはインバースPCR後の増幅産物の濃縮を行なわずにネステッドPCRを行う条件も試した。
【0069】
CHI01用プライマー
CHI01-C:5’-gtgcatcagatccgcataaccttgc-3’(配列番号35)
CHI01-D:5’-tctctggtgggcaagatcaactacc-3’(配列番号36)
CHI03用プライマー
CHI03-C:5’-ttgtcctccggacggaagacattcc-3’(配列番号37)
CHI03-D:5’-attcgcgcagtacgtcaattacgtc-3’(配列番号38)
CHI05用プライマー
CHI05-C:5’-cggtgaagttctcgcggtcttcagg-3’(配列番号39)
CHI05-D:5’-ccagtacctcgacgccatcctggtc-3’(配列番号40)
CHI08用プライマー
CHI08-C:5’-aggccgtcgcgctcctgcatgttcg-3’(配列番号41)
CHI08-D:5’-ctcccgcttctcgatttcatcaacg-3’(配列番号42)
CHI12用プライマー
CHI12-C:5’-aaccgttggtcaatccaccgctgac-3’(配列番号43)
CHI12-D:5’-cgtcgctgtggactggatcaacctg-3’(配列番号44)
CHI14用プライマー
CHI14-C:5’-cttgtcttcaggtcgcgaggcatcc-3’(配列番号45)
CHI14-D:5’-gcaggttacggactggttcaacctg-3’(配列番号46)
CHI19用プライマー
CHI19-C:5’-gtcttgagggcgaacggtattttcg-3’(配列番号47)
CHI19-D:5’-catgtctgtcagcctggactggatg-3’(配列番号48)
CHI23用プライマー
CHI23-C:5’-gtctgagcgctctttggcgttcttc-3’(配列番号49)
CHI23-D:5’-ccgtctgaacaagtcgctcgactgg-3’(配列番号50)
CHI31用プライマー
CHI31-C:5’-tggtcggaaatcgacaagtatctcg-3’(配列番号51)
CHI31-D:5’-cgttccctgataattcatgccctcg-3’(配列番号52)
CHI32用プライマー
CHI32-C:5’-taacgtgtagttctgcttgtcctcg-3’(配列番号53)
CHI32-D:5’-catgcacgcgcacctcgactggatc-3’(配列番号54)
【0070】
上記方法によりメタゲノムからのキチナーゼ遺伝子を回収した結果を図7に示す。図7においては、左側にインバースPCRとネステッドPCRのみを行った条件、右側にインバースPCRの後、増幅産物を濃縮してからネステッドPCRを行った条件を示す。また、図7の(A)は、ApaLI処理ライブラリーを鋳型にしたサンプル群であり、レーン1:CHI32、レーン2:CHI23、レーン3:CHI19、レーン4:CHI14、レーン5:CHI08、レーン6:CHI01、レーン7:CHI32、レーン8:CHI23、レーン9:CHI19、レーン10:CHI14、レーン11:CHI08、レーン12:CHI01である。図7の(B)は、EcoRI処理ライブラリーを鋳型にしたサンプル群であり、レーン1:CHI31;レーン2:CHI12;レーン3:CHI05;レーン4:CHI03;レーン5:CHI31;レーン6:CHI12;レーン7:CHI05;レーン8:CHI03である。なお、MはλHindIIIマーカーである。
【0071】
実験の結果、未知配列を含む領域を増幅(クローニング)することに成功した。インバースPCR後の増幅産物の濃縮を行わなかった場合は、5クローンの未知遺伝子配列を増幅することができ、また増幅産物の濃縮を行った場合には、10クローンすべての未知遺伝子配列を増幅することができた。本発明の方法により増幅された遺伝子断片の塩基配列を決定し、Swiss Prot等のアミノ酸配列データベース上のアミノ酸データとBlastX検索プログラムで比較したところ、7クローンについては、全キチナーゼ遺伝子配列の決定に成功していることが明らかとなった。さらにそのうちの5クローンについては、既知のキチナーゼとのアミノ酸相同性が40%以下の新規性の高いものであることがあきらかとなった(表1)。
【0072】
【表1】

【0073】
以上の実験結果から、本発明の方法によりメタゲノムから目的の遺伝子、特に目的の遺伝子の全長配列が高効率に得られることが確認された。
【産業上の利用可能性】
【0074】
本発明により、目的の遺伝子のクローニング、さらには目的の遺伝子と相同性を有する遺伝子や目的の遺伝子に近接する領域のクローニングを簡便、高効率、かつ高感度に行うことが可能となる。そのため、クローニング対象の配列が低コピー数にしか存在しない場合であっても、確実にその全長配列をクローニングすることができ、有用である。
【図面の簡単な説明】
【0075】
【図1】本発明の方法の概要を示す。
【図2】本発明の方法のクローニング効率試験に用いたR. eutropha 由来のpoxオペロン遺伝子断片を示す。
【図3】本発明の方法のクローニング効率試験の結果を示す。レーンMはλHind IIIマーカーである。
【図4】本発明の方法を用いたメタゲノムからのP450様モノオキシゲナーゼを含むオペロンの全長クローニングにおいて、決定されたP450様モノオキシゲナーゼを含むオペロンの全体図を示す。
【図5】本発明の方法を用いたメタゲノムからのP450様モノオキシゲナーゼを含むオペロンの全長クローニングの結果を示す。レーンMはλHind IIIマーカーである。
【図6】メタゲノムからクローニングされたキチナーゼ遺伝子の一部と、既知のキチナーゼ遺伝子のアミノ酸配列を基に作成した無根系統樹を示す。
【図7】本発明の方法によるメタゲノムからのキチナーゼ遺伝子の回収を示す。(A)ApaLI処理ライブラリーを鋳型にしたサンプル群。(B)EcoRI処理ライブラリーを鋳型にしたサンプル群。レーンMはλHind IIIマーカーである。
【配列表フリーテキスト】
【0076】
配列番号1〜54:合成オリゴヌクレオチド
配列中、「Y」はT又はCを表し、「R」はG又はAを表し、「W」はA又はTを表し、「S」はG又はCを表す。
【技術分野】
【0001】
本発明は、既知の塩基配列に基づいて、DNA(例えば環境メタゲノム)から、既知の塩基配列と同一の若しくは相同性を有する塩基配列、又は該既知の塩基配列に近接する領域をクローニングする方法に関する。
【背景技術】
【0002】
近年、微生物等の有する代謝系酵素を利用したバイオプロセスが特定の化学物質の生産に利用されるようになってきている。これに伴い、微生物等から新規代謝系酵素の遺伝子を獲得する試みが盛んに行われるようになった。特定の代謝系酵素の遺伝子を獲得する際には、通常、まず目的の活性を持った生物を環境サンプルから単離し、次にその単離された生物から代謝系遺伝子のクローニングを試みる。しかし近年、分子生態解析の手法が確立されてくると、環境中の大多数の微生物は未だに培養・単離されておらず、またそれらの多くが難培養であることがわかってきた(非特許文献1参照)。これは、微生物等の培養・単離を介したのでは、環境中の多様な遺伝子の大多数を獲得できないことを意味している。そこで最近、微生物等を単離培養することなく直接メタゲノム(環境サンプル等から抽出されたDNA)を抽出し、そこから作製されたメタゲノムライブラリーを、特定機能を有する遺伝子のスクリーニングに利用する試みがなされるようになってきている(非特許文献2参照)。
【0003】
単離された生物のゲノムや環境メタゲノムからの遺伝子のスクリーニング法として、遺伝子配列に依存したスクリーニング法がある。遺伝子配列に依存したスクリーニングとは、既知の遺伝子配列から設計されたプローブやプライマーを用いて類似遺伝子を獲得しようとするものである(特許文献1及び非特許文献3参照)。しかしながら、この方法においてクローニング可能な塩基配列は、設計したプライマーの内部配列のみであるため、環境中で実際に機能している酵素をコードする完全な遺伝子配列を決定することが出来ない。通常、酵素はいくつかのドメインからなり、活性中心を持つドメイン以外にもその酵素の折り畳みを助けるようなドメインや、酵素活性を高めるために重要なドメインなどから形成される。通常遺伝子配列に依存したクローニング法においては、高度に保存された活性中心の遺伝子配列からプライマーを設計するため、周辺ドメインの遺伝子配列はクローニングされてはこない。
【0004】
そのような問題を解決するため、環境メタゲノムを鋳型とし、遺伝子配列に依存したクローニング法で得られた遺伝子配列から、さらにその前後の未知の遺伝子配列をクローニングし、最終的に酵素遺伝子全体をクローニングするような試みがおこなわれはじめている(非特許文献4及び5参照)。これらの方法においては、(1)始めに遺伝子配列に依存したクローニングで、遺伝子配列を決定し、(2)決定した遺伝子配列を元にプライマーを作製し、(3)環境から抽出した染色体DNAを適当な制限酵素で処理し、リンカーをつなげ、(4)リンカー特異的なプライマーと、決定した遺伝子配列を元にしたプライマーを用いてPCRを行い、(5)増幅断片をクローニングし、始めに決定した遺伝子配列の外側に位置する未知塩基配列を決定する、というスキームをとっている。しかしながらこれらの方法においては、わずか1〜2種類程度のクローンしか得られていない。これはおそらく、メタゲノム中に混在する大量のDNAがPCR反応を阻害し、低コピー数でしか存在しない遺伝子からの増幅を困難にしていることが原因と考えられる。
【0005】
そのため、このような問題を解決する新たなクローニング法を開発し、低コピー数でしか存在しない多様な未知遺伝子及び未知ドメインのクローニングを行うことが望まれている。
【0006】
【特許文献1】特表2005−525793号公報
【非特許文献1】Amann RI, Ludwig W, 及びSchleifer KH,"Phylogenetic identification and in situ detection of individual microbial cells without cultivation",Microbiol Rev.,第59巻第143〜169頁,1995年
【非特許文献2】Lorenz P, Liebeton K, Niehaus F, 及びEck J,"Screening for novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequence space",Curr Opin Biotechnol.,第13巻第572〜577頁,2002年
【非特許文献3】Stokes HW, Holmes AJ, Nield BS, Holley MP, Nevalainen KM, Mabbutt BC, 及びGillings MR,"Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA",Appl Environ Microbiol.,第67巻第5240〜5246頁,2001年
【非特許文献4】Eschenfeldt WH, Stols L, Rosenbaum H, Khambatta ZS, Quaite-Randall E, Wu S, Kilgore DC, Trent JD, 及びDonnelly MI,"DNA from uncultured organisms as a source of 2,5-diketo-D-gluconic acid reductases",Appl Environ Microbiol.,第67巻第4206〜4214頁,2001年
【非特許文献5】Bell PJL, Sunna A, Gibbs MD, Curach NC, Nevalainen H, 及びBergquist PL,"Prospecting for novel lipase genes using PCR",Microbiol.,第148巻第2283〜2291頁,2002年
【発明の開示】
【発明が解決しようとする課題】
【0007】
上に述べたように、従来法では環境メタゲノムから得られた遺伝子断片の塩基配列情報を元にして、環境メタゲノム中に存在する周辺領域を増幅することは非常に困難であった。
【0008】
そこで本発明は、今までクローニングできなかった遺伝子の全長配列又は近接する領域をクローニングするための簡便かつ高感度な方法を提供するとともに、巨大な環境メタゲノムから低コピー数にしか存在しない遺伝子断片をクローニングするためにも有用な方法を提供する。
【課題を解決するための手段】
【0009】
本発明者は、上記課題を解決するために鋭意検討を重ねた結果、一回目の増幅反応を行った後に二回目の増幅反応を行うことにより高効率に遺伝子断片の回収ができ、さらに一回目の増幅反応の後に増幅産物を回収して、二回目の増幅反応を行うことにより高感度かつ高効率に遺伝子断片を増幅することができることを見出し、本発明を完成させた。また、本発明の方法において、一度目の増幅反応としてインバースPCRを行うことにより、遺伝子の周辺領域のクローニングが可能となる(ゲノムウォーキング)ことも見出した。
【0010】
すなわち、本発明は、以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)任意により、得られる増幅産物を回収するステップ、
(e)回収した増幅産物を増幅可能なプライマーを用いて該増幅産物を鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法に関する。
【0011】
また本発明は、以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)得られた増幅産物中の上記既知の塩基配列以外の領域に基づいて設計した1つのプライマーと上記既知の塩基配列に基づいて設計した1つのプライマーを用いて、上記DNAを鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域をクローニングする方法に関する。
【0012】
上記方法において、DNAの断片化は、例えば制限酵素処理により行うことができる。制限酵素としては、付着末端を生じる制限酵素を使用することが好ましく、例えば限定されるものではないが、EcoR I、Xho I又はApaL Iを用いることができる。
【0013】
また上記方法において、DNAとしては、ゲノムDNA、プラスミドDNA、及び/又は、ファージDNAを用いることができる。ゲノムDNAは、単一の生物に由来するものであってもよいし、又は複数の生物に由来するものであってもよい。DNAは、例えば環境サンプル、発酵槽サンプル及び集積培養サンプルから抽出されたDNA(メタゲノム)とすることができる。
【0014】
上記方法において、断片化DNAの環状化は、例えばセルフライゲーションにより行われる。
【0015】
上記方法において、既知の塩基配列としては、例えば限定されるものではないが、代謝系遺伝子、転写制御系遺伝子、又はトランスポゾンをコードするものが挙げられる。また、既知の塩基配列に近接する領域は、未知の配列を含んでもよい。
【0016】
上記方法において、使用するプライマーのうち少なくとも1つはランダムプライマーであってもよい。
【0017】
また上記方法において、ステップ(c)において使用するプライマーの一方又は両方は、標識されていることが好ましい。標識としては、限定されるものではないが、ビオチンが挙げられる。プライマーに標識を付加した場合、ステップ(d)において、標識を利用して増幅産物を回収することができる。例えば、プライマーにビオチン標識を付加した場合、ステップ(d)においては、例えばストレプトアビジンを固定化した磁気ビーズを用いて、ビオチン標識を有する増幅産物を回収することができる。
【発明の効果】
【0018】
本発明により、目的の遺伝子のクローニング、さらには目的の遺伝子と相同性を有する遺伝子や目的の遺伝子に近接する領域のクローニングを簡便、高効率、かつ高感度に行うことが可能となる。そのため、クローニング対象の配列が低コピー数にしか存在しない場合であっても、確実にその全長配列をクローニングすることができ、有用である。
【発明を実施するための最良の形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
本発明は、既知の塩基配列に基づいて、DNAから、既知の塩基配列と同一の若しくは相同性を有する塩基配列、又は該既知の塩基配列に近接する領域をクローニングする方法に関する。本発明の方法の概要を図1に示す。図1において、太線は既知の塩基配列で、細線は既知の塩基配列に近接する領域を表す。矢印を有する線はプライマーを表し、矢印の向きはプライマーから伸長される方向を示す。白抜きの丸は標識を表す。本発明の方法においては、2回の増幅反応(図1におけるCとE)を行うことによって、わずかにしか存在しないDNAに基づいて、既知の塩基配列、及びそれに近接する未知の配列を含む領域を、簡便かつ高感度にクローニングすることが可能である。
【0021】
最初に、目的の塩基配列又はそれに近接する領域のクローニングの鋳型となるDNAを準備する。鋳型とするDNAは、特に限定されるものではなく、例えばゲノムDNA、ファージDNA、プラスミドDNAなどである。また例えば、ある単一の生物に由来するDNA(例えばゲノムDNA、染色体DNA)であってもよいし、複数の生物に由来するDNAであってもよい。また、生物は、単離された生物(例えば微生物、微生物混合物など)であってもよいし、単離されていなくてもよい。例えば、環境サンプル(例えば、土壌、湖沼水、海水、河川水など)、微生物の培養物、発酵槽サンプル、集積培養サンプルなどに由来するDNA(メタゲノム)を用いることが可能である。さらに、DNAは、ゲノムDNAライブラリであってもよい。DNAの調製は当技術分野で公知であり、任意の方法(例えばフェノール・クロロホルム法等)又は任意の市販キットを用いてDNAを調製することができる。また例えば、土壌中からメタゲノムをクローニングする方法が記載されており、この文献に記載の手法も本発明において用いることができる(Rondon, MR. et al., Appl Environ Microbiol. 2000, 66:2541-2547)。
【0022】
続いて、DNAを断片化する(図1のA)。DNAの断片化は、制限酵素処理により行うことができる。使用する制限酵素の種類は特に限定されるものではないが、後述するDNAの環状化を考慮すると、付着末端を生じる制限酵素を使用することが好ましい。好ましくは、制限酵素としてEcoR I、Xho I、ApaL Iなどを使用する。
【0023】
断片化されたDNAは、当技術分野で公知の方法により環状化する(図1のB)。環状化は、例えばセルフライゲーションにより行うことができる。セルフライゲーションとは、断片化DNAの両端に存在する制限酵素切断部位がリガーゼにより連結されることをいう。従って、断片化DNAは、適当なリガーゼ(例えばT4 DNAリガーゼ等)を用いて環状化することができる。
【0024】
次に、環状化DNAを鋳型とした増幅反応を行う(図1のC)。プライマーは、既知の塩基配列に基づいて設計する。選択する「既知の塩基配列」は、塩基配列が既知であれば限定されるものではない。例えば、遺伝子(代謝系遺伝子、転写制御系遺伝子等)、トランスポゾン、トランスポーター、ハウスキーピングタンパク質(例えばDNAポリメラーゼ、DNAリガーゼ)などをコードする塩基配列を選択することができる。また、遺伝子全体が既知の塩基配列である場合に加えて、一部のみが配列決定されている場合であっても、遺伝子全体又は遺伝子の周辺領域(近接する領域)をクローニングするため、その一部の既知配列を選択することができる。
【0025】
メタゲノムに存在する有用な遺伝子の例としては、限定されるものではないが、代謝系遺伝子、すなわち特定の化合物又は生体分子の化学的構造の変換を触媒する酵素をコードする遺伝子が挙げられる。例えば、フェノールの芳香環に酸素分子由来の酸素原子を導入しカテコールを生産する酵素であるフェノール水酸化酵素(phenol hydroxylase)EC 1.14.13.7(Nordlund I, Powlowski J, Shingler V. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 1990 172:6826-6833)などが挙げられる。また、限定されるものではないが、ハイドラーゼ、エステラーゼ、プロテアーゼ、リパーゼ、ベータガラクトシダーゼ、ラクターゼ、ポリガラクツロナーゼ、ベータグルコアミラーゼ、エステラーゼ、ヘミセルラーゼ、ペルオキシダーゼ、オキシダーゼ、オキシゲナーゼ、ラッカーゼ、グルコースオキシダーゼ、ペクチナーゼ、アミラーゼ、ガラクタナーゼ、アラビナーゼ、キチナーゼ、キシラナーゼ、DNAポリメラーゼ、DNAリガーゼ又はセルラーゼなどの酵素をコードする遺伝子の塩基配列も、本発明の方法において既知の塩基配列として選択することができる。
【0026】
さらに、上記の代謝系酵素をコードする遺伝子に加え、「代謝系遺伝子」には、代謝反応において基質となる化学物質の細胞内外への輸送に関与するタンパク質をコードする遺伝子も含まれる。例えば、トルエンなどの化学物質の取り込みに関与するタンパク質XylN(Kasai Y, Inoue J, Harayama S. The TOL plasmid pWW0 xylN gene product from Pseudomonas putida is involved in m-xylene uptake. J Bacteriol. 2001 183:6662-6)などである。または、トルエンなどの溶媒分子の細胞外への排出に関与するタンパク質である排出ポンプ(Duque E, Segura A, Mosqueda G, Ramos JL. Global and cognate regulators control the expression of the organic solvent efflux pumps TtgABC and TtgDEF of Pseudomonas putida. Mol Microbiol. 2001 39:1100-6)なども含まれる。
【0027】
またさらに、代謝系オペロンを制御するような転写制御因子をコードする転写制御系遺伝子の塩基配列を選択することもできる。「発現制御因子」とは、特定の基質の存在下にて代謝系遺伝子が誘導的に発現される場合にその発現を制御する遺伝的要素を指す。例えば、ナフタレンやサリチル酸の代謝系オペロンの発現を制御しているタンパク質NahR(Schell MA, Wender PE. Identification of the nahR gene product and nucleotide sequences required for its activation of the sal operon. J. Bacteriol. 1986 166:9-14)などである。他の発現制御因子としては、例えば、XylR、XylS、DmpR、PhcRなどが挙げられる。
【0028】
また、遺伝子の水平伝播に関わるとされるトランスポゾン(Shintani M, Yoshida T, Habe H, Omori T, Nojiri H. Large plasmid pCAR2 and class II transposon Tn4676 are functional mobile genetic elements to distribute the carbazole/dioxin-degradative car gene cluster in different bacteria. Appl. Microbiol. Biotechnol. 2005 67:370-82)をコードする塩基配列を選択してもよい。
【0029】
プライマーを設計する領域は、当業者であれば容易に選択することができる。上記増幅反応において、既知の塩基配列に近接する領域をクローニングする場合には、設計する2つのプライマーのうち、3’側をフォワードプライマーとして、5’側をリバースプライマーとして用いる(図1のCの左側におけるプライマー1及び2)。このようにプライマーを設計することによって、図1のCの左側に示すように環状化DNAを鋳型とした増幅反応を行い、既知の塩基配列の一部と同一の又は相同性を有する領域とそれに近接する領域を含む断片を増幅することができる(図1のDの左側)。
【0030】
さらに、既知の塩基配列と相同性を有する領域をクローニングする場合、プライマーをランダムプライマーとして設計してもよい。それにより、既知の塩基配列とは完全に同一ではないが、プライマーがアニーリングできる程度に相同性を有する塩基配列の領域が増幅されることになる。
【0031】
プライマーの設計は、当業者に公知の方法に従って行うことができる。プライマーとして実質的な機能を有する長さとしては、10塩基以上が好ましく、さらに好ましくは16〜50塩基であり、さらに好ましくは20〜30塩基である。また設計の際には、公知のプライマー又はプローブ設計用ソフトウエアを利用することができ、本発明で利用可能なソフトウエアとしては、例えばOligoTM[National Bioscience Inc.(米国)製]、GENETYX[ソフトウェア開発(株)(日本)製]等などが挙げられる。上述のように設計したプライマーは、当業者に公知の方法に従って、例えばオリゴヌクレオチド合成機を用いることにより、調製することができる。
【0032】
プライマーには、後述する増幅産物の回収のために、回収を容易にする標識を付加してもよい。例えば、物質間の相互作用性質を利用して増幅産物の回収を行う場合には、その物質の一方を用いてプライマーを標識することができる。このような標識の種類や標識の導入方法等は、特に限定されるものではなく、従来公知の各種手段を用いることができる。例えば、ビオチン−アビジン系を利用してプライマーにビオチンを標識することや、抗原−抗体反応を利用してプライマーに抗原を標識することができる。標識は、フォワードプライマー若しくはリバースプライマーの一方、又は両方に付加することができる。
【0033】
上述の通り作製したプライマーを用いて、上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列(既知の塩基配列と相同性を有する領域を含む)、及び/又は上記既知の塩基配列に近接する領域を増幅する(図1のC)。ここで、「近接する領域」とは、既知の塩基配列に隣接する領域及び既知の塩基配列の周囲領域を意味し、具体的には、既知の塩基配列から約6000bp、好ましくは約3000bpまでの領域である。
【0034】
増幅反応は、上述のように設計したプライマーを用いて、環状化DNAを鋳型とした増幅を行うことができる方法であれば、特に限定されるものではない。ゲノムDNAから任意の塩基配列をクローニングするため、通常はポリメラーゼ連鎖反応(PCR)を応用した方法を利用する。例えば、インバースPCR法(Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics 1988 120:621-623)、ランダムプライマーPCR法(Liu Y, Whittier RF. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995 25:674-681)、アダプターライゲーションPCR法(Ochman H. et al. Use of polymerase chain reaction to amplify segments outside boundaries of known sequences. Methods Enzymol. 1993 218:309-321)、ユニバーサルファーストウォーキングPCR法(Myrick, K.V. and Gelbart, W.M. 2002 Universal fast walking for direct and versatile determination of flanking sequence. Gene 284:125-131)、パンハンドル PCR法(Megonigal, M.D. et al. 2000 Panhandle PCR for cDNA: a rapid method for isolation of MLL fusion transcripts involving unknown partner genes. PNAS 97:9597-9602)などが挙げられる。
【0035】
増幅反応後、任意により増幅産物を回収してもよい(図1のD)。この回収ステップを行うことにより、2回目の増幅反応において鋳型として使用される増幅産物が濃縮され、目的とする配列のクローニングが高感度かつ高効率となる。増幅産物の回収は、当技術分野で公知の任意の方法により行うことができる。例えば増幅反応において標識プライマーを用いた場合には、その標識を利用して増幅産物を回収することができる。具体的には、標識と特異的に結合することができる物質を利用して、その物質とプライマーの標識とを結合させ、未結合のDNAを除去することによって、増幅産物を回収する。好ましくは、ビオチンで標識したプライマーを用いて増幅反応を行い、増幅反応溶液をストレプトアビジンを表面にコートした磁気ビーズ(例えば、Dynabeads kilobaseBINDER Kit(DYNAL)として市販されている)と接触させ、磁気ビーズに結合していない反応液を洗浄することにより、増幅産物を回収することができる。回収ステップは、例えばメタゲノムなどの多様なDNAを含むサンプル(このようなサンプルは増幅反応を阻害することがある)から目的の配列をクローニングする場合に特に有用である。
【0036】
続いて、得られた増幅産物を鋳型とした2回目の増幅反応を行う(図1のE)。この増幅反応においては、得られた増幅産物を増幅可能なプライマーを用いる。そのようなプライマーは、既知の塩基配列に基づいて設計することができ、具体的には、既知の塩基配列に基づいて、1回目の増幅反応において用いたプライマー(図1におけるプライマー1及び2)の領域よりも内側の領域にプライマー(図1におけるプライマー3及び4)を設計する。プライマーの設計は、上述と同様に、当技術分野で公知の方法に従って行うことができる。なお、2回目の増幅反応においても、プライマーとしてランダムプライマーを用いることができる。
【0037】
2回目の増幅反応は、DNAを鋳型とした増幅反応を行うことができれば、当技術分野で公知の任意の方法により行うことができる。このような増幅反応によって、既知の塩基配列、既知の塩基配列と相同性を有する領域、及び/又は既知の塩基配列に近接する領域を増幅し、クローニングすることができる。
【0038】
また、1回目の増幅反応又は2回目の増幅反応において得られた増幅産物の塩基配列を決定することによって、既知の塩基配列に近接する領域の配列情報を明らかにすることができる。また、その決定された塩基配列に基づいて設計した1つのプライマーと、既知の塩基配列に基づいて設計した1つのプライマーとを用いて、DNAを鋳型とした増幅反応を行うことにより、既知の塩基配列の一部とそれに近接する領域をクローニングすることができる。
【0039】
さらに、決定した塩基配列に基づいて設計されたプローブを用いたハイブリダイゼーションを行うことによっても、既知の塩基配列に近接する領域をクローニングすることが可能である。
上述のように、本発明の方法により、既知の塩基配列の一部若しくは全体、又はそれに近接する領域を、効率的にクローニングすることができるため、例えば代謝系酵素遺伝子オペロンの全体構造の解明に有用である。
【0040】
以下の実施例は、本発明をさらに具体的に説明するためのものであり、本発明を限定するものではない。
【実施例1】
【0041】
本実施例においては、本発明の方法の有効性を実証するために、次のような実験を行った。
【0042】
始めにRalstonia eutropha E2株とEscherichia coliJM109株の2種の染色体DNAを用意し、それぞれを混合した。染色体DNAの抽出は、Silhavyらの方法(Silhavy TJ, Berman ML, and Enquist LW. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y. 1984.)によりおこなった。E. coli JM109株のDNAは一定量、R. eutropha E2株のDNAはE. coliJM109株のDNA濃度に対して段階希釈(10-1〜10-6)し、混合した。次にDNAの混合物をXhoIで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。
【0043】
次に、R. eutropha E2株のフェノール代謝系遺伝子オペロン(poxオペロン)(Hino S, Watanabe K, Takahashi N. Phenol hydroxylase cloned from Ralstoniaeutropha strain E2 exhibits novel kinetic properties. Microbiology 1998 144:1765-72)に近接する領域をインバースPCRで特異的に増幅させうるような、以下のプライマー1及び2(それぞれ配列番号1及び2)を用いてPCRを行い(図1のC)、図2に示すようにR. eutropha E2株のpoxオペロンの一部、すなわちXho I切断部位で囲まれた3.8 kbの増幅産物の増幅を試みた。
【0044】
プライマー1:5’Biotinirated-gaacaactcggactcgatcagttcgtcg-3’(配列番号1)
プライマー2:5’-ttcgtgctgtttgccttcagcgtcg-3’(配列番号2)
PCRの反応条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃4分からなるサイクルを32サイクルとした。
【0045】
インバースPCRの後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物を回収した(図1のD)。また、回収を行った場合及び回収を行わなかった場合の増幅産物に対して以下のプライマー3及び4(配列番号3及び4)を用いたネステッドPCRを行った(図1のE):
プライマー3:5’-atcaagacgcagcagatcc-3’(配列番号3)
プライマー4:5’-caaggccaggatggtatcg-3’(配列番号4)
ネステッドPCRの条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃4分からなるサイクルを32サイクルとした。
【0046】
インバースPCRのみを行った場合(条件1)、インバースPCR後、増幅産物をネステッドPCRでさらに増幅させた場合(条件2)、インバースPCR後、増幅産物をDynabeads kilobaseBINDER Kitを用いて濃縮し、これらをネステッドPCRでさらに増幅させた場合(条件3)の3種の条件で、目的の遺伝子断片の増幅検出限界を0.7%アガロースゲル電気泳動により確認した。
【0047】
実験の結果を図3に示す。実験の結果、(条件1)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 1 ngを混合した場合(10-1)が、増幅産物の検出限界であった。(条件2)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 10 pgを混合した場合(10-3)が、増幅産物の検出限界であった。(条件3)においては鋳型としてE. coliJM109株のDNA 10 ngに対し、R. eutrophaE2株のDNA 100 fgを混合した場合(10-5)が、増幅産物の検出限界であった。実験の結果、インバースPCRとネステッドPCRとを組み合わせた本方法(条件2及び3)を用いることにより検出限界が高まることが確認された。特に、インバースPCRを行った後に増幅産物を回収した条件3によって、もっとも高い検出限界を達成することに成功した。従って、本発明の方法は、低コピー数のDNAしか存在しない場合であっても、効率的かつ高感度に目的遺伝子の近接領域を増幅することができることがわかった。
【実施例2】
【0048】
本発明の方法の有効性を実証するために、本方法を用いて実際にメタゲノムからP450様モノオキシゲナーゼを含むオペロンの全長のクローニングを試みた。
【0049】
環境メタゲノムは、地下石油備蓄基地の湧水(Watanabe K, Kodama Y, Syutsubo K, Harayama S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude-oil-storage cavities. Appl. Environ. Microbiol. 2000 66:4803-4809)から抽出した染色体DNAを使用し、P450様モノオキシゲナーゼ(Uchiyama T, Abe T, Ikemura T, Watanabe K. Substrate-induced gene-expression screening of environmental metagnome libraries for isolation of catabolic genes. Nat. Biotechnol. 2005 1:88-93)を含むオペロンの全長クローニングを試みた。
【0050】
始めにメタゲノムをEcoR Iで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。次に、P450様モノオキシゲナーゼを含むオペロンの領域を特異的に増幅させうるような、以下のプライマー71A及び71B(それぞれ配列番号5及び6)を用いてインバースPCRを行い(図1のC)、図4に示すようにオペロンの一部の増幅を試みた。
【0051】
プライマー71A:5’Biotinirated-tgcgtagattacgaaacggccaattccagc -3’(配列番号5)
プライマー71B:5’-agcaccacagtcagcctgaatgacg -3’(配列番号6)
インバースPCRの反応条件は、94℃にて1分の後、98℃20秒、58℃20秒及び72℃8分からなるサイクルを32サイクルとした。
【0052】
インバースPCRの後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物を回収し(図1のD)、その増幅産物に対して以下のプライマー71C及び71D(それぞれ配列番号7及び8)を用いたネステッドPCRを行った(図1のE)。
【0053】
プライマー71C:5’-gccaacggcatcgacggtattctgg -3’(配列番号7)
プライマー71D:5’-tgatggcatccacggcaattggtcg -3’(配列番号8)
【0054】
プライマー71C及び71Dは、インバースPCRに使用したプライマー71A及び71Dに近接するように設計した(図4)。ネステッドPCRの条件は、94℃にて1分の後、98℃20秒、58℃20秒及び72℃8分からなるサイクルを32サイクルとした。図4において、白抜きのバーは本実験によって増幅された既知塩基配列と未知塩基配列の遺伝子断片を示す。
【0055】
また、回収された増幅産物に対して、以下のプライマー71OperonF及び71OperonR(それぞれ配列番号9及び10)を用いたPCRを行った:
プライマー71OperonF:5’-aggccatggcggtgaagtttgttcc -3’(配列番号9)
プライマー71OperonR:5’-gaattcacccgaccggtgctcgacg -3’(配列番号10)
【0056】
プライマー71OperonFは既知の塩基配列に基づいて設計し、プライマー71OperonRはクローニングされた未知の配列に基づいて設計した(図4)。PCRの条件は、94℃にて1分の後、98℃20秒、62℃20秒及び72℃5分からなるサイクルを32サイクルとした。図4に示す灰色のバーは、既知の塩基配列と今回決定された未知の塩基配列を含む遺伝子断片を示す。
【0057】
実験は、プライマー71A及び71BでインバースPCRを行った際の増幅産物を泳動した条件(条件1)、その増幅産物を濃縮し、プライマー71C及び71DでネステッドPCRを行った後の増幅産物を泳動した条件(条件2)、及びプライマー71OperonF及びプライマー71OperonRでPCRを行った後の増幅産物を泳動した条件(条件3)で行った。目的の遺伝子断片の増幅は、0.7%アガロースゲル電気泳動により確認した。
【0058】
結果を図5に示す。図5において、レーン1〜3は、それぞれ条件1〜3で得られた増幅産物の泳動を示す。実験の結果、P450様モノオキシゲナーゼに近接する領域の未知塩基配列の決定に成功した。レーン2に示されるように、ネステッドPCRを行うことにより、オペロン全長を取得することができたが、これは、P450様モノオキシゲナーゼオペロンから設計したプライマーと、新たに決定した塩基配列から設計したプライマーを用いてPCRを行って、オペロン全長が増幅されていることを確認できた(図5のレーン3)。以上より、本発明の方法を行うことによりメタゲノムから任意の塩基配列とそれに近接する領域(未知の塩基配列を含む)をクローニングできることが確認された。
【実施例3】
【0059】
本発明の方法の有効性を実証するため、メタゲノムを鋳型とし、酵素のアミノ酸相同性を基に設計したプライマーを用いて遺伝子の一部をクローニングし、その塩基配列を基に本発明の方法によって遺伝子全体の塩基配列を決定するような実験を試みた。
【0060】
(1)キチナーゼ遺伝子の単離
環境メタゲノムは地下石油備蓄基地の湧水(Watanabe K, Kodama Y, Syutsubo K, Harayama S. Molecular characterization of bacterial populations in petroleum-contaminated groundwater discharged from underground crude-oil-storage cavities. Appl. Environ. Microbiol. 2000 66:4803-9)から抽出した染色体DNAを使用した。酵素のアミノ酸相同性を基に設計したプライマーは論文(Hobel CFV, Marteinsson VT, Hreggvidsson GO, Kristjansson JK. Investigation of the microbial ecology of intertidal hot springs by using diversity analysis of 16S rRNA and chitinase genes. 2005 71:2771-6)で使用された、キチナーゼ遺伝子の活性ドメインをコードする塩基配列の一部を増幅させる以下のプライマーを使用した。
【0061】
プライマーChiA_F1, 5'-ACG GCG TGG ACA TCG AYT GGG ART-3'(配列番号11)
プライマーChiA_F2, 5'-CGT GGA CAT CGA CTG GGA RTW YCC-3'(配列番号12)
プライマーChiA_R1, 5'-CCC AGG CGC CGT AGA RRT CRTAYS-3'(配列番号13)
プライマーChiA_R2, 5'-CCC AGG CGC CGT AGA RRT CRT ARS WCA-5'(配列番号14)
【0062】
なお、配列番号11〜14における「Y」はT又はCを表し、「R」はG又はAを表し、「W」はA又はTを表し、「S」はG又はCを表す。
【0063】
始めにメタゲノムを鋳型に、キチナーゼ遺伝子の活性ドメインを増幅させる上記プライマーを用いて、増幅産物のランダムクローニングを行った。得られた増幅産物を2.0%アガロースゲル中で電気泳動し、300 bp前後の大きさの増幅産物をQIAquick gel extraction kit(Qiagen)によりゲル中から抽出した。抽出した増幅産物は、TA cloning vector(タカラ酒造)とライゲーションさせた。これを用いエレクトロポレーションによりEscherichia coliJM109(タカラ酒造)を形質転換させ、IPTGおよびX-Galを含んだLB培地プレートに塗布した。形質転換体を100個からプラスミドを抽出し、クローニングされた塩基配列を決定した。決定した塩基配列をアミノ酸配列に変換し、既知のキチナーゼ配列と併せてClustalWプログラムにより解析を行い、系統樹を描かせた(図6)。
【0064】
その結果、多様なキチナーゼ遺伝子を得ることに成功した。図6において、本実験でクローニングしたクローンはCHIと表記し、クローンナンバーと得られた陽性クローンの数を表記した。
【0065】
(2)キチナーゼ遺伝子全体のクローニング
(1)の実験で単離されたキチナーゼ遺伝子の中から10クローンを適当に選び出し(図6中、黒丸でマークされたもの)、本発明の方法を用いてキチナーゼ遺伝子全体をクローニングすることを試みた。
【0066】
始めにメタゲノムをApaL I又はEcoR Iで完全分解した(図1のA)。QIAquick PCR Purification Kit(QIAGEN)を用いて分解物を精製し、T4 DNA ligaseを用いてセルフライゲーションさせ、環状化DNAを作製した(図1のB)。次に、キチナーゼ遺伝子に近接する未知配列を増幅させうるような、以下のプライマー(配列番号15〜34)を用いてインバースPCRを行った。なお、インバースPCRの反応条件は、94℃にて1分の後、98℃20秒、56℃20秒及び72℃8分からなるサイクルを32サイクルとした。
【0067】
CHI01用プライマー
CHI01-biotin:5’biotinirated-ccgacaggctgtccagggccttgcgcaagg-3’(配列番号15)
CHI01-B:5’-tacctggtgaccagtgcgatcaatg-3’(配列番号16)
CHI03用プライマー
CHI03-biotin:5’biotinirated-cattcaaacagtacctgaccaggaccgagc-3’(配列番号17)
CHI03-B:5’-cccggagcagcagtgtgaagtttgc-3’(配列番号18)
CHI05用プライマー
CHI05-biotin:5’biotinirated-aggcgtcgacaagatccgggttaccgatcc-3’(配列番号19)
CHI05-B:5’-ggcatcgagttgccgcctgaactcc-3’(配列番号20)
CHI08用プライマー
CHI08-biotin:5’biotinirated-ggcaccaaatggggatgccgcttcaccgac-3’(配列番号21)
CHI08-B:5’-cgccggaaaccgatcgccatcgagg-3’(配列番号22)
CHI12用プライマー
CHI12-biotin:5’biotinirated-gacctggcagcatccgcaatctggatctgc-3’(配列番号23)
CHI12-B:5’-cagcgtcaaattggcagtgtcatcg-3’(配列番号24)
CHI14用プライマー
CHI14-biotin:5’biotinirated-tcgtttcgtgcgcttgctacgctcatcgag-3’(配列番号25)
CHI14-B:5’-cctgctgccattctatgaaatcgag-3’(配列番号26)
CHI19用プライマー
CHI19-biotin:5’biotinirated-cgttgatccagttgatgacgaagttcttgc-3’(配列番号27)
CHI19-B:5’-gtagacaagattgttaagtcggagc-3’(配列番号28)
CHI23用プライマー
CHI23-biotin:5’biotinirated-gcagttcctcgaccatcgtggcatggtctg-3’(配列番号29)
CHI23-B:5’-gactcggactccgtgttcgatcacc-3’(配列番号30)
CHI31用プライマー
CHI31-biotin:5’biotinirated-aactcctgagttcctgcttcagttgcgacc-3’(配列番号31)
CHI31-B:5’-cagcgacgtgaagttgtggaaatcg-3’(配列番号32)
CHI32用プライマー
CHI32-biotin:5’biotinirated-gcatcgagccgcgagcgaagctcttgcagc-3’(配列番号33)
CHI32-B:5’-ccttcgacgatcgcgaacatgccag-3’(配列番号34)
【0068】
インバースPCR後、Dynabeads(登録商標)kilobaseBINDER Kit(Dynal Biotech製)を用いて増幅産物の濃縮を行い、以下のプライマー(配列番号35〜54)を用いてネステッドPCRを行い、最終的にキチナーゼ遺伝子の全長配列の決定を試みた(図7)。ネステッドPCRの反応条件は、94℃にて1分の後、98℃20秒、56℃20秒及び72℃8分からなるサイクルを32サイクルとした。また、実験の際にはインバースPCR後の増幅産物の濃縮を行なわずにネステッドPCRを行う条件も試した。
【0069】
CHI01用プライマー
CHI01-C:5’-gtgcatcagatccgcataaccttgc-3’(配列番号35)
CHI01-D:5’-tctctggtgggcaagatcaactacc-3’(配列番号36)
CHI03用プライマー
CHI03-C:5’-ttgtcctccggacggaagacattcc-3’(配列番号37)
CHI03-D:5’-attcgcgcagtacgtcaattacgtc-3’(配列番号38)
CHI05用プライマー
CHI05-C:5’-cggtgaagttctcgcggtcttcagg-3’(配列番号39)
CHI05-D:5’-ccagtacctcgacgccatcctggtc-3’(配列番号40)
CHI08用プライマー
CHI08-C:5’-aggccgtcgcgctcctgcatgttcg-3’(配列番号41)
CHI08-D:5’-ctcccgcttctcgatttcatcaacg-3’(配列番号42)
CHI12用プライマー
CHI12-C:5’-aaccgttggtcaatccaccgctgac-3’(配列番号43)
CHI12-D:5’-cgtcgctgtggactggatcaacctg-3’(配列番号44)
CHI14用プライマー
CHI14-C:5’-cttgtcttcaggtcgcgaggcatcc-3’(配列番号45)
CHI14-D:5’-gcaggttacggactggttcaacctg-3’(配列番号46)
CHI19用プライマー
CHI19-C:5’-gtcttgagggcgaacggtattttcg-3’(配列番号47)
CHI19-D:5’-catgtctgtcagcctggactggatg-3’(配列番号48)
CHI23用プライマー
CHI23-C:5’-gtctgagcgctctttggcgttcttc-3’(配列番号49)
CHI23-D:5’-ccgtctgaacaagtcgctcgactgg-3’(配列番号50)
CHI31用プライマー
CHI31-C:5’-tggtcggaaatcgacaagtatctcg-3’(配列番号51)
CHI31-D:5’-cgttccctgataattcatgccctcg-3’(配列番号52)
CHI32用プライマー
CHI32-C:5’-taacgtgtagttctgcttgtcctcg-3’(配列番号53)
CHI32-D:5’-catgcacgcgcacctcgactggatc-3’(配列番号54)
【0070】
上記方法によりメタゲノムからのキチナーゼ遺伝子を回収した結果を図7に示す。図7においては、左側にインバースPCRとネステッドPCRのみを行った条件、右側にインバースPCRの後、増幅産物を濃縮してからネステッドPCRを行った条件を示す。また、図7の(A)は、ApaLI処理ライブラリーを鋳型にしたサンプル群であり、レーン1:CHI32、レーン2:CHI23、レーン3:CHI19、レーン4:CHI14、レーン5:CHI08、レーン6:CHI01、レーン7:CHI32、レーン8:CHI23、レーン9:CHI19、レーン10:CHI14、レーン11:CHI08、レーン12:CHI01である。図7の(B)は、EcoRI処理ライブラリーを鋳型にしたサンプル群であり、レーン1:CHI31;レーン2:CHI12;レーン3:CHI05;レーン4:CHI03;レーン5:CHI31;レーン6:CHI12;レーン7:CHI05;レーン8:CHI03である。なお、MはλHindIIIマーカーである。
【0071】
実験の結果、未知配列を含む領域を増幅(クローニング)することに成功した。インバースPCR後の増幅産物の濃縮を行わなかった場合は、5クローンの未知遺伝子配列を増幅することができ、また増幅産物の濃縮を行った場合には、10クローンすべての未知遺伝子配列を増幅することができた。本発明の方法により増幅された遺伝子断片の塩基配列を決定し、Swiss Prot等のアミノ酸配列データベース上のアミノ酸データとBlastX検索プログラムで比較したところ、7クローンについては、全キチナーゼ遺伝子配列の決定に成功していることが明らかとなった。さらにそのうちの5クローンについては、既知のキチナーゼとのアミノ酸相同性が40%以下の新規性の高いものであることがあきらかとなった(表1)。
【0072】
【表1】
【0073】
以上の実験結果から、本発明の方法によりメタゲノムから目的の遺伝子、特に目的の遺伝子の全長配列が高効率に得られることが確認された。
【産業上の利用可能性】
【0074】
本発明により、目的の遺伝子のクローニング、さらには目的の遺伝子と相同性を有する遺伝子や目的の遺伝子に近接する領域のクローニングを簡便、高効率、かつ高感度に行うことが可能となる。そのため、クローニング対象の配列が低コピー数にしか存在しない場合であっても、確実にその全長配列をクローニングすることができ、有用である。
【図面の簡単な説明】
【0075】
【図1】本発明の方法の概要を示す。
【図2】本発明の方法のクローニング効率試験に用いたR. eutropha 由来のpoxオペロン遺伝子断片を示す。
【図3】本発明の方法のクローニング効率試験の結果を示す。レーンMはλHind IIIマーカーである。
【図4】本発明の方法を用いたメタゲノムからのP450様モノオキシゲナーゼを含むオペロンの全長クローニングにおいて、決定されたP450様モノオキシゲナーゼを含むオペロンの全体図を示す。
【図5】本発明の方法を用いたメタゲノムからのP450様モノオキシゲナーゼを含むオペロンの全長クローニングの結果を示す。レーンMはλHind IIIマーカーである。
【図6】メタゲノムからクローニングされたキチナーゼ遺伝子の一部と、既知のキチナーゼ遺伝子のアミノ酸配列を基に作成した無根系統樹を示す。
【図7】本発明の方法によるメタゲノムからのキチナーゼ遺伝子の回収を示す。(A)ApaLI処理ライブラリーを鋳型にしたサンプル群。(B)EcoRI処理ライブラリーを鋳型にしたサンプル群。レーンMはλHind IIIマーカーである。
【配列表フリーテキスト】
【0076】
配列番号1〜54:合成オリゴヌクレオチド
配列中、「Y」はT又はCを表し、「R」はG又はAを表し、「W」はA又はTを表し、「S」はG又はCを表す。
【特許請求の範囲】
【請求項1】
以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)任意により、得られる増幅産物を回収するステップ、
(e)回収した増幅産物を増幅可能なプライマーを用いて該増幅産物を鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法。
【請求項2】
以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)得られた増幅産物中の上記既知の塩基配列以外の領域に基づいて設計した1つのプライマーと上記既知の塩基配列に基づいて設計した1つのプライマーを用いて、上記DNAを鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域をクローニングする方法。
【請求項3】
DNAの断片化を制限酵素処理により行う、請求項1又は2記載の方法。
【請求項4】
制限酵素が付着末端を生じる制限酵素である、請求項3記載の方法。
【請求項5】
制限酵素がEcoR I、Xho I及びApaL Iからなる群より選択される、請求項3又は4記載の方法。
【請求項6】
DNAがゲノムDNA、プラスミドDNA、及びファージDNAからなる群より選択される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
DNAが単一の生物に由来するものである、請求項6記載の方法。
【請求項8】
DNAが複数の生物に由来するものである、請求項6記載の方法。
【請求項9】
DNAが、環境サンプル、発酵槽サンプル及び集積培養サンプルから抽出されたDNAである、請求項6記載の方法。
【請求項10】
断片化DNAの環状化がセルフライゲーションにより行われる、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
既知の塩基配列が、代謝系遺伝子、転写制御系遺伝子、又はトランスポゾンをコードするものである、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
既知の塩基配列に近接する領域が未知の配列を含む、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
プライマーのうち少なくとも1つがランダムプライマーである、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
ステップ(c)において使用するプライマーの一方又は両方が標識されたものである、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
標識がビオチンである、請求項14記載の方法。
【請求項16】
ステップ(d)において、標識を利用して増幅産物を回収する、請求項14又は15記載の方法。
【請求項17】
ステップ(d)において、ストレプトアビジンを固定化した磁気ビーズを用いて、ビオチン標識を有する増幅産物を回収する、請求項15又は16記載の方法。
【請求項1】
以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)任意により、得られる増幅産物を回収するステップ、
(e)回収した増幅産物を増幅可能なプライマーを用いて該増幅産物を鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域、又は既知の塩基配列と同一の若しくは相同性を有する領域をクローニングする方法。
【請求項2】
以下のステップ:
(a)DNAを断片化するステップ、
(b)断片化DNAを環状化するステップ、
(c)既知の塩基配列に基づいて設計した2つのプライマーのうち、3’側をフォワードプライマーとしてかつ5’側をリバースプライマーとして用いて上記環状化DNAを鋳型とした増幅反応を行い、上記既知の塩基配列に近接する領域を増幅するステップ、
(d)得られた増幅産物中の上記既知の塩基配列以外の領域に基づいて設計した1つのプライマーと上記既知の塩基配列に基づいて設計した1つのプライマーを用いて、上記DNAを鋳型とした増幅反応を行うステップ、
を含む、既知の塩基配列に近接する領域をクローニングする方法。
【請求項3】
DNAの断片化を制限酵素処理により行う、請求項1又は2記載の方法。
【請求項4】
制限酵素が付着末端を生じる制限酵素である、請求項3記載の方法。
【請求項5】
制限酵素がEcoR I、Xho I及びApaL Iからなる群より選択される、請求項3又は4記載の方法。
【請求項6】
DNAがゲノムDNA、プラスミドDNA、及びファージDNAからなる群より選択される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
DNAが単一の生物に由来するものである、請求項6記載の方法。
【請求項8】
DNAが複数の生物に由来するものである、請求項6記載の方法。
【請求項9】
DNAが、環境サンプル、発酵槽サンプル及び集積培養サンプルから抽出されたDNAである、請求項6記載の方法。
【請求項10】
断片化DNAの環状化がセルフライゲーションにより行われる、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
既知の塩基配列が、代謝系遺伝子、転写制御系遺伝子、又はトランスポゾンをコードするものである、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
既知の塩基配列に近接する領域が未知の配列を含む、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
プライマーのうち少なくとも1つがランダムプライマーである、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
ステップ(c)において使用するプライマーの一方又は両方が標識されたものである、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
標識がビオチンである、請求項14記載の方法。
【請求項16】
ステップ(d)において、標識を利用して増幅産物を回収する、請求項14又は15記載の方法。
【請求項17】
ステップ(d)において、ストレプトアビジンを固定化した磁気ビーズを用いて、ビオチン標識を有する増幅産物を回収する、請求項15又は16記載の方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2007−228850(P2007−228850A)
【公開日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願番号】特願2006−52921(P2006−52921)
【出願日】平成18年2月28日(2006.2.28)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成17年度、独立行政法人新エネルギー・産業技術総合開発機構「生分解・処理メカニズムの解析と制御技術の開発 土壌中難分解性物質等の生分解・処理技術の開発」委託研究、産業再生法第30条の適用を受ける特許出願
【出願人】(591001949)株式会社海洋バイオテクノロジー研究所 (33)
【Fターム(参考)】
【公開日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願日】平成18年2月28日(2006.2.28)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成17年度、独立行政法人新エネルギー・産業技術総合開発機構「生分解・処理メカニズムの解析と制御技術の開発 土壌中難分解性物質等の生分解・処理技術の開発」委託研究、産業再生法第30条の適用を受ける特許出願
【出願人】(591001949)株式会社海洋バイオテクノロジー研究所 (33)
【Fターム(参考)】
[ Back to top ]