
制癌剤
【課題】悪性腫瘍、特に卵巣癌の治療に有効な制癌剤の提供。
【解決手段】ジフテリア毒素の変異体であってHB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質、或いは、ジフテリア毒素の一部からなり該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質又は該タンパク質を含む複合タンパク質等であってHB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を、有効成分として含む制癌剤。
【解決手段】ジフテリア毒素の変異体であってHB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質、或いは、ジフテリア毒素の一部からなり該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質又は該タンパク質を含む複合タンパク質等であってHB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を、有効成分として含む制癌剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、卵巣癌をはじめとする各種癌の治療に利用できる制癌剤に関する。
【背景技術】
【0002】
悪性腫瘍に対しては、各種の治療法及び治療薬が開発されているが、未だ充分な治療効果をあげることが出来ない場合が多い。特に、卵巣癌は婦人科領域の悪性腫瘍の中でも進行が早く、とりわけ予後の悪い悪性腫瘍である。
卵巣癌の治療法としては、現在タキソールを中心とした化学療法しか手立てが無いが、これらの化学療法も一時的効果はあるものの、後には再発を許すことが多く、新たな治療法の開発が求められている。
【0003】
HB−EGFはEGFファミリーの細胞増殖因子で、身体の形成や再生過程に必須であると同時に、血管狭窄、動脈硬化症などの発症に関係する分子として知られているが(例えば、非特許文献1参照)、この因子を悪性腫瘍の治療のターゲットとすることは現在まで全く行われていなかった。この分子は膜結合型前駆体(proHB−EGF)として合成され、細胞表面でプロテアーゼにより切断されて分泌型HB−EGFを生じる。分泌型には、増殖促進作用があるが、膜型には増殖抑制作用が観察され、分泌型と膜型は適宜使い分けられることで、組織の形成と維持に働いているものと考えられている。
【0004】
HB−EGFは、EGFリセプター(Her1)及びEGFリセプターファミリーのHer4(ErbB−4)に結合し、活性化する。しかし、EGFリセプターファミリー(Her1、Her2、Her3、Her4)はホモダイマーを形成するほか、全ての組み合わせのヘテロダイマーを形成し得るので、結果的にHB−EGFは全てのEGFリセプターファミリー分子を活性化しうる。HB−EGFは種々の組織で発現されており、広範な細胞や組織で働いていると考えられているが、線維芽細胞、平滑筋細胞あるいはケラチノサイトの増殖をよく促進することが報告されている(例えば、非特許文献2参照)。
【0005】
HB−EGFは前記のように膜結合型前駆体(proHB−EGF)として合成されるが、proHB−EGFは、N末端から、シグナル配列、プロ配列、ヘパリン結合ドメイン、EGF様ドメイン、ジャクスタメンブレンドメイン、トランスメンブレンドメイン、細胞質ドメインからなる(図1)。このproHB−EGFは、図中矢印の部分でプロテアーゼによる切断(エクトドメインシェディング)を受けて分泌型となる。proHB−EGFのエクトドメインシェディングは、リゾホスファチジン酸(LPA)等がG蛋白共役型リセプターを介して、Ras−Raf−MEK経路を活性化する経路や、フォルボールエステルがPKCを活性化する経路により刺激されることが提唱されている(例えば、非特許文献3参照)。
【0006】
分泌型HB−EGFがEGFリセプターに結合し、EGFリセプターのリン酸化を促す作用はEGF様ドメインに存在する(例えば、非特許文献1参照)。
【0007】
一方ジフテリア毒素は、ジフテリア菌が産生する分子量約59000のタンパク質であるが、HB−EGFの膜結合型前駆体(proHB−EGF)を受容体とすることが知られている(例えば、非特許文献4参照)。また、CRM197のようなジフテリア毒素の変異体は、分泌型HB−EGFの阻害剤として知られている(例えば、非特許文献5参照)。ジフテリア毒素のデータベース情報は、遺伝子についてはEMBL;K01722、アミノ酸配列はSWISS−PROT;P00588、三次元構造はPDB;1MDTあるいは1XDTで見ることができる。ジフテリア毒素の遺伝子は菌体内に溶原化しているファージがコードしている。
【0008】
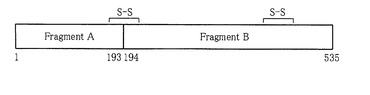
ジフテリア毒素は、アミノ酸535個からなる単純タンパク質であり(ジフテリア毒素のアミノ酸配列(配列番号1)とそれをコードする遺伝子の塩基配列(配列番号2)を図2及び図3に表し、イタリックはシグナル配列を表す)、還元剤で処理することにより分離されるフラグメントA部分とフラグメントB部分に分けられるが(図4)、立体構造解析等によれば、フラグメントB部分はさらに2つのドメインに分かれている。それぞれのドメインの機能は、フラグメントA部分に相当する触媒作用(catalytic)ドメイン(シグナル配列を除いたアミノ酸番号で1〜193)はADPリボシル化活性を、フラグメントB部分のN末端半分に相当する膜貫通(Transmembrane)ドメイン(シグナル配列を除いたアミノ酸番号で194〜378)はエンドソーム膜にチャネルを形成する性質を、フラグメントB部分のC末端半分に相当するレセプター結合(receptor−binding)ドメイン(シグナル配列を除いたアミノ酸番号で386〜535)は細胞表面のジフテリア毒素リセプターと結合する活性を有する。
【0009】
ジフテリア毒素のフラグメントA(触媒作用ドメイン部分)は、NAD存在下でEF−2(ペプチド伸長因子2)をADPリボシル化する作用を有しており、これによりタンパク質合成を阻害する。したがって、ジフテリア毒素が毒性を発揮するためには、フラグメントAが細胞質内に入らなければならない。
フラグメントAが細胞質内に入る機構は、フラグメントBにあるレセプター結合ドメインが細胞表面のリセプターであるproHB−EGFに結合することにより、ジフテリア毒素はエンドサイトーシスによりエンドソームに取り込まれ、エンドソーム内で膜貫通ドメインがエンドソーム膜に挿入され、最終的にはフラグメントAがエンドソーム膜を通過して細胞質中に遊離され、そこでEF−2を失活させる(例えば、非特許文献6参照)。
【0010】
ジフテリア毒素が毒性を発揮するためには、フラグメントAとフラグメントBの両方が必要である。したがってどちらのフラグメントに変異があっても、ジフテリア毒素の毒性を有さないタンパク質ができる。
【0011】
ジフテリア毒素には触媒作用ドメインに変異を有する無毒化された変異体、例えばCRM197が分離されている。
【0012】
一方、ジフテリア毒素の変異体が、HB−EGFとEGFレセプターとの結合を阻害する活性を有するのは、ジフテリア毒素が、分泌型HB−EGFのEGF様ドメインと結合するためである。また、この結合に関与するのはジフテリア毒素のレセプター結合ドメインである。ジフテリア毒素の516番目のLys、530番目のPheがHB―EGFの結合に重要であることが報告されている(例えば、非特許文献7参照)。また、ジフテリア毒素とHB―EGFのEGFドメインからなる複合体の結晶構造が解析されており、381−535までの間のHB―EGFとの結合に重要なアミノ酸が報告されている(例えば、非特許文献8参照)。
【0013】
このように、ジフテリア毒素変異体がHB−EGFに結合し、HB−EGFの活性を阻害することは認められていたが、HB−EGFを癌治療のターゲットとすること自体が知られていなかったこと、及び、HB−EGFを含む制御機構の複雑さから、ジフテリア毒素変異体を癌治療薬に使用しようとする試みは全くなされていなかった。
【非特許文献1】目加田英輔等著「遺伝子医学」株式会社メディカルドゥ社、vol.5 No.2 2001,p.131−134
【非特許文献2】Higashiyama, S. et al著 J.Cell Biol.(1993)122,p.933−940,
【非特許文献3】Prenzel,N.et al著 Nature (1999) 402,p.884−888
【非特許文献4】J.G.Naglich et al著 Cell (1992) 69,p.1051−1061
【非特許文献5】T.Mitamura et al J.Biol.Chem.(1995) 270,p.1015
【非特許文献6】T.Umata et al著 J.Biol.Chem.(1998) 273,p.8351
【非特許文献7】Shen, HS et al著 J. Biol. Chem. (1994) 269,p.29077−29084
【非特許文献8】Gordon VL et al著 Molecular Cell (1997) 1, p.67−78
【発明の開示】
【発明が解決しようとする課題】
【0014】
本発明は、従来における前記諸問題を解決し、以下の目的を達成することを課題とする。即ち、悪性腫瘍、特に卵巣癌の治療に有効な制癌剤を提供することを目的とする。
【課題を解決するための手段】
【0015】
発明者らは、これまで身体の形成や再生過程に必須であると同時に、血管狭窄、動脈硬化症などの発症に関係するタンパク質として知られていた分泌型HB−EGFの機能を再検討し、癌細胞の増殖、転移過程にHB−EGFが関与しているとの知見を得た。これに基づき、新たな治療法の開発を進め、その結果HB−EGF中和能を有するCRM197が、ヌードマウスに接種したヒト卵巣癌由来細胞の増殖を有意に抑制することを明らかにし、本発明に至った。
【0016】
すなわち、本発明の前記課題を解決するための手段は以下の通りである。
<1> 以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする制癌剤である。
(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質
(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質
(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質
<2> (a)、(b)及び(c)のいずれかのタンパク質が、ジフテリア毒素の触媒作用ドメインを有しない前記<1>に記載の制癌剤である。
<3> (c)のタンパク質が、GST−DTである前記<2>に記載の制癌剤である。
【発明の効果】
【0017】
本発明によると、悪性腫瘍、特に卵巣癌の治療に有効な制癌剤を提供することができる。
【発明を実施するための最良の形態】
【0018】
発明者らは、HB−EGFの機能を検討する中で、HB−EGFには造腫瘍性があるとの知見を得たことに基づき、HB−EGFのEGFレセプターへの結合阻害活性が知られているジフテリア毒素の無毒化変異体の制癌作用を実証した。
【0019】
本発明の第一の態様の制癌剤は、ジフテリア毒素の変異体であってHB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を、有効成分として含むことを特徴とする。
前記ジフテリア毒素の変異体とは、ジフテリア毒素のアミノ酸配列において1又は複数のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質を表し、例えば1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質である。また、ジフテリア毒素の25個のシグナル配列は付いていても付いていなくてもよく、いずれの配列も本発明の範囲に含まれる。
【0020】
本発明の第二の態様の制癌剤は、以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする。(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質。(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質。(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質。
【0021】
前記タンパク質は、ジフテリア毒素の一部若しくはその変異体又はそれらのタンパク質を含む複合タンパク質であって、レセプター結合ドメインを保持するものをいう。
【0022】
ここで、ジフテリア毒素の毒性とは、ジフテリア毒素が細胞表面の受容体に結合し、細胞内に侵入し、フラグメントAが保持するEF−2(ペプチド伸長因子2)をADPリボシル化する活性(ADPリボシル化活性)でもって、細胞の蛋白質合成機能を阻害することを意味し、これはタンパク質合成阻害の程度により容易に測定することができる。すなわち、培養細胞に一定量のジフテリア毒素を加えて2〜8時間程度培養し、続いて、放射性のアミノ酸存在下で短時間培養し、タンパク質に取り込まれた放射性アミノ酸量を測定する方法である。
【0023】
具体的には、Vero細胞(1×105細胞)を24ウェルプレートに接種して、CO2インキュベーターで16時間培養する。細胞が十分にプレートに接着していることを確認してから、各ウェルに冷PBS(150mM、NaCl−2.7mM KCl−10mMリン酸緩衝液、pH7.2)で1回洗う。この時の液の出し入れは細胞をはがさないように慎重に行う。血清を含むアッセイ用培養液0.5mlを加える。アッセイ用培養液は、通常の培地からロイシンの濃度を1/10程度に減らしたものを使用する。これは後で加える[3H]ロイシンの取り込み効率を高めるためである。但し、Ham’s F12培地は、ロイシン含量が少ないのでこれをアッセイ用培養液とすることもできる。血清は通常用いている濃度で加える。
次に、種々の濃度のジフテリア毒素を加えて、CO2インキュベーターで2〜5時間培養する。3.7MBq/mlの[3H]ロイシンを10μl加えて更に1時間培養する。
【0024】
培養液を捨て、ウェルをPBSで1度洗った後、細胞を0.5mlの0.1M NaOHで溶解し、その溶液をチューブに集める。0.5mlの0.1N NaOHで再度ウェルを洗浄し、その溶液を同じチューブに集める。
【0025】
これに20%のトリクロロ酢酸溶液0.5mlを加えて、Vortexミキサーでよく攪拌する。生じた沈殿をグラスフィルターでトラップし、フィルターをさらに5%トリクロロ酢酸溶液で洗浄する。
【0026】
最後にフィルターを100%エタノールで洗浄し、乾燥させる。
【0027】
フィルターをトルエン−PPOのシンチレーターにつけて、フィルターにトラップされた放射能を液体シンチレーションカウンターで測定する。ジフテリア毒素を加えなかった試料の値を測定し、これを100%として毒素を加えた時の値を%で求める。
【0028】
ジフテリア毒素の毒性を実質的に有さないタンパク質とは、ジフテリア毒素の毒性を無毒化または極めて低レベルに弱毒化されたタンパク質をいい、本発明においては、ジフテリア毒素の毒性を1ng/mlの濃度で上記Vero細胞の系で測定した場合、ジフテリア毒素を加えなかった試料または触媒作用ドメインを有しないジフテリア毒素変異体を加えた試料の値と有意差がないものをいう。前記有意差は、t検定において有意水準5%として有意差が無いことが好ましく、有意水準1%として有意差が無いことがより好ましく、有意水準0.1%として有意差が無いことがさらに好ましい。
【0029】
ただし、本発明のなかで、CRM197やDT52E148Kのようなこれまでジフテリア毒素の毒性が全くないとされていた変異体であっても、きわめて微弱な毒性(例えばCRM197では野生型のジフテリア毒素の1010分の1程度)が残存していることが証明され、このような微弱な毒性を有している変異体が本発明から排除されるものではない。ジフテリア毒素の毒性レベルとしては、CRM197と同じかそれ以下であることがジフテリア毒素の毒性による副作用を排除し、安全性を高める観点から好ましい。しかし、一方では該毒性が、制癌剤の効果に寄与することが本発明により示唆されるため、CRM197と同程度の極めて低レベルの毒性を有することも、制癌効果を高める観点から好ましい。したがって、製剤処方にあわせて、適宜ジフテリア毒素の毒性レベルを選択することができる。
【0030】
ジフテリア毒素の毒性は、ペプチド伸長因子2をADPリボシル化するのに必須である触媒作用ドメインに変異を持たせること、または、触媒作用ドメインの一部または全部を欠損させることにより調整することができる。
【0031】
変異させた触媒作用ドメインの機能は、ADPリボシル化活性を直接測定することで、正確に調べることができる。ADPリボシル化活性は、分離精製したEF−2に、フラグメントAあるいはADPリボシル化活性を測定したい蛋白質(変異させた触媒作用ドメイン等)とラジオアイソトープで標識したNADを加えて、試験管内でEF−2をADPリボシル化させ、EF−2に取り込まれた放射能を測定することで、直接測定することができる。
【0032】
具体的には、以下の文献(Moynihan, M.R. and Pappenheimer, A.M. Jr. Infect. Immun.(1981) 32, 575−582)で記載された方法で得たウサギ網状赤血球EF−2分画に、最終濃度20mMのトリス緩衝液(pH7.8)、1mMDTT(ジチオスレイトール)、0.1〜1μg/mlのフラグメントAあるいは0.1−100μg/mlのADPリボシル化活性を測定したい蛋白質を加え、さらに最終的に370KBq/mlになるように[32P]NAD(約740 GBq/mmol)を加えて混和し、37℃で10分間反応させる。
【0033】
その反応液に同容量の10%−トリクロロ酢酸溶液を加えて蛋白質を沈殿させ、生じた沈殿をグラスフィルターでトラップし、フィルターをさらに5%トリクロロ酢酸溶液で洗浄する。
【0034】
最後にフィルターを100%エタノールで洗浄し、乾燥させる。
【0035】
フィルターをトルエン−PPOのシンチレーターにつけて、フィルターにトラップされた放射能を液体シンチレーションカウンターで測定する。
【0036】
測定された放射能がADPリボシル化活性の程度を示しており、変異を加えないフラグメントAを用いたときの放射能を基準に、変異を加えた蛋白質のADPリボシル化活性の相対活性を求めることができる。
【0037】
また、分泌型HB−EGFとEGFレセプターとの結合を阻害する活性を有するという特徴は、発明者らが、ドメイン情報をもとに、より詳細に検討したところによると、レセプター結合ドメインを含む部分である378番目から535番目のアミノ酸配列を含んでいればよいことがわかった。すなわち、GST(Gluthathione−S−transferase)にジフテリア毒素の378番目から535番目までの配列を融合した遺伝子を作成し、これを大腸菌に発現させて、上記の構造を持つ融合タンパク質(GST−DT)を作成した。GST−DTは、125Iで標識したジフテリア毒素のHB―EGFへの結合を、濃度依存的に阻害した。阻害の程度から、GST−DTはジフテリア毒素と同程度の結合の強さで、HB―EGFに結合することがわかった。したがって、結合に必要なのは、378番目から535番目、すなわち、レセプター結合ドメインを含む部分であることがわかった。
【0038】
ここで、HB−EGFとEGFリセプターの結合を阻害する活性を有するかどうかは、前記のような、125Iで標識したジフテリア毒素のHB―EGFへの結合に対する阻害実験により測定することができる。
【0039】
そこで、HB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質は、レセプター結合ドメインを保持しつつ、触媒作用ドメインに変異を持たせたジフテリア毒素変異体タンパク質や、ジフテリア毒素のレセプター結合ドメインを保持しつつ触媒作用ドメインの一部または全部を欠損させたジフテリア毒素の一部であるタンパク質を作成することにより得ることができる。
【0040】
このような変異体の例としては、CRM197、DT52E148K及びGST−DTが挙げられる。これらはジフテリア毒素の毒性を実質的に持たず、HB−EGFのEGFレセプターへの結合を阻害する。CRM197は、25個のシグナル配列を除いてカウントした場合の52番目のGlyがGluに変異した変異体であり、DT52E148Kは、前記変異に加えてシグナル配列を除いてカウントした場合の148番目のGluがLysに変異した変異体である。また、GST−DTはGSTにジフテリア毒素のシグナル配列を除いてカウントした場合の378番目から535番目を含む蛋白質である。CRM197に関するアミノ酸配列(最初の25個の配列がシグナル配列を表す)を配列番号3に、それをコードする遺伝子の塩基配列を配列番号4に表す。また、GST−DTのアミノ酸配列(配列番号5)及びそれをコードする遺伝子の塩基配列(配列番号6)を図5及び図6に表す。
【0041】
ここで、CRM197については、ジフテリア毒素の毒性を有さない、即ちADPリボシル化活性を有さないとの報告が既になされている(T. Uchida and A.M. Pappenheimer Jr. (1972) Science 175, 901−903)。また、148Eに変異を有する148K変異体が極めて弱い活性しか持たないことは知られており(J.T.Barbieri and R.J. Collier (1987) Infect. Immun. 55, 1647−1651)、52E変異体であるCRM197にさらに148Kの変異を入れたダブルミュータントであるDT52E148Kは、さらに安全な変異体として好ましい。また、これらの変異体の毒性は、前記タンパク質合成阻害実験によってもジフテリア毒素を加えなかった試料の値と有意差がないことが確かめられた。ただし、前述のように、これらの変異体であっても、極めて微弱なジフテリア毒素の毒性が残存していることが、本発明の、より詳細な解析により明らかになっている。
【0042】
なお、GST−DTがジフテリア毒素の毒性を有しないことは、触媒作用ドメインを完全に欠如することから明らかである。
(配列番号3)
MSRKLFASILIGALLGIGAPPSAHAGADDVVDSSKSFVMENFSSYHGTKPGYVDSIQKGIQKPKSGTQGNYDDDWKEFYSTDNKYDAAGYSVDNENPLSGKAGGVVKVTYPGLTKVLALKVDNAETIKKELGLSLTEPLMEQVGTEEFIKRFGDGASRVVLSLPFAEGSSSVEYINNWEQAKALSVELEINFETRGKRGQDAMYEYMAQACAGNRVRRSVGSSLSCINLDWDVIRDKTKTKIESLKEHGPIKNKMSESPNKTVSEEKAKQYLEEFHQTALEHPELSELKTVTGTNPVFAGANYAAWAVNVAQVIDSETADNLEKTTAALSILPGIGSVMGIADGAVHHNTEEIVAQSIALSSLMVAQAIPLVGELVDIGFAAYNFVESIINLFQVVHNSYNRPAYSPGHKTQPFLHDGYAVSWNTVEDSIIRTGFQGESGHDIKITAENTPLPIAGVLLPTIPGKLDVNKSKTHISVNGRKIRMRCRAIDGDVTFCRPKSPVYVGNGVHANLHVAFHRSSSEKIHSNEISSDSIGVLGYQKTVDHTKVNSKLSLFFEIKS
(配列番号4)
GTGAGCAGAAAACTGTTTGCGTCAATCTTAATAGGGGCGCTACTGGGGATAGGGGCCCCACCTTCAGCCCATGCAGGCGCTGATGATGTTGTTGATTCTTCTAAATCTTTTGTGATGGAAAACTTTTCTTCGTACCACGGGACTAAACCTGGTTATGTAGATTCCATTCAAAAAGGTATACAAAAGCCAAAATCTGGTACACAAGGAAATTATGACGATGATTGGAAAGAGTTTTATAGTACCGACAATAAATACGACGCTGCGGGATACTCTGTAGATAATGAAAACCCGCTCTCTGGAAAAGCTGGAGGCGTGGTCAAAGTGACGTATCCAGGACTGACGAAGGTTCTCGCACTAAAAGTGGATAATGCCGAAACTATTAAGAAAGAGTTAGGTTTAAGTCTCACTGAACCGTTGATGGAGCAAGTCGGAACGGAAGAGTTTATCAAAAGGTTCGGTGATGGTGCTTCGCGTGTAGTGCTCAGCCTTCCCTTCGCTGAGGGGAGTTCTAGCGTTGAATATATTAATAACTGGGAACAGGCGAAAGCGTTAAGCGTAGAACTTGAGATTAATTTTGAAACCCGTGGAAAACGTGGCCAAGATGCGATGTATGAGTATATGGCTCAAGCCTGTGCAGGAAATCGTGTCAGGCGATCAGTAGGTAGCTCATTGTCATGCATAAATCTTGATTGGGATGTCATAAGGGATAAAACTAAGACAAAGATAGAGTCTTTGAAAGAGCATGGCCCTATCAAAAATAAAATGAGCGAAAGTCCCAATAAAACAGTATCTGAGGAAAAAGCTAAACAATACCTAGAAGAATTTCATCAAACGGCATTAGAGCATCCTGAATTGTCAGAACTTAAAACCGTTACTGGGACCAATCCTGTATTCGCTGGGGCTAACTATGCGGCGTGGGCAGTAAACGTTGCGCAAGTTATCGATAGCGAAACAGCTGATAATTTGGAAAAGACAACTGCTGCTCTTTCGATACTTCCTGGTATCGGTAGCGTAATGGGCATTGCAGACGGTGCCGTTCACCACAATACAGAAGAGATAGTGGCACAATCAATAGCTTTATCGTCTTTAATGGTTGCTCAAGCTATTCCATTGGTAGGAGAGCTAGTTGATATTGGTTTCGCTGCATATAATTTTGTAGAGAGTATTATCAATTTATTTCAAGTAGTTCATAATTCGTATAATCGTCCCGCGTATTCTCCGGGGCATAAAACGCAACCATTTCTTCATGACGGGTATGCTGTCAGTTGGAACACTGTTGAAGATTCGATAATCCGAACTGGTTTTCAAGGGGAGAGTGGGCACGACATAAAAATTACTGCTGAAAATACCCCGCTTCCAATCGCGGGTGTCCTACTACCGACTATTCCTGGAAAGCTGGACGTTAATAAGTCCAAGACTCATATTTCCGTAAATGGTCGGAAAATAAGGATGCGTTGCAGAGCTATAGACGGTGATGTAACTTTTTGTCGCCCTAAATCTCCTGTTTATGTTGGTAATGGTGTGCATGCGAATCTTCACGTGGCATTTCACAGAAGCAGCTCGGAGAAAATTCATTCTAATGAAATTTCGTCGGATTCCATAGGCGTTCTTGGGTACCAGAAAACAGTAGATCACACCAAGGTTAATTCTAAGCTATCGCTATTTTTTGAAATCAAAAGC
TGA
【0043】
なお、レセプター結合ドメインを含む断片は、プラスミッドに組み込まれたCRM197をコードする遺伝子(Pβ197)を鋳型にして、PCR法にてレセプター結合ドメイン部分のDNA配列を合成して、これをGST融合タンパク質やヒスチジンタグを合成させるための発現ベクター(pGEX−3X、pQE−30)のマルチクローニングサイトに挿入し、得られたプラスミッドを大腸菌に組み込み、プラスミッドがコードする遺伝子を大腸菌で合成させることで作成できる。
【0044】
また、触媒作用ドメインに変異を有する変異体は、以下のようにして作成することができる。プラスミッドに組み込まれたCRM197をコードする遺伝子(Pβ197)を鋳型にして、変異を持たせたい部位をプライマーとして、PCR法にてCRM197領域を合成する。プライマーは、変異を持つように点突然変異を導入したものを合成し、使用する。合成したDNAを、大腸菌用の遺伝子発現ベクター(pET−22b)に組み込み、大腸菌に形質導入を行い、変異体を大腸菌で発現させて、作成することができる。
【0045】
本発明の制癌剤は、乳癌、前立腺癌、甲状腺癌、卵巣癌等、悪性腫瘍全般の治療に用いることができるが、好ましくはHB−EGFを発現している悪性腫瘍に用いることができ、特に卵巣癌に好適に用いることができる。
本発明の制癌剤は、前記ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、かつ、前記ジフテリア毒素の変異体の細胞への結合を抑制しない前記ジフテリア毒素の変異体のモノクローナル抗体をさらに含み、
前記ジフテリア毒素の変異体のモノクローナル抗体と、ジフテリア毒素の変異体とが複合体を形成することも好ましい。
【0046】
前記ジフテリア毒素の変異体は、変異を導入することによりジフテリア毒素の毒性が弱毒化されており、実質的にジフテリア毒素の毒性を有しないものであるが、極めて厳格なレベルで見ればジフテリア毒素の毒性が残存している場合を含むことは前述のとおりである。そこで、ジフテリア毒素の変異体は、該ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、かつ、該ジフテリア毒素の変異体の細胞への結合を抑制しないモノクローナル抗体と複合体を形成することによってさらにジフテリア毒素の毒性を低下させることができる。
【0047】
前記ジフテリア毒素の変異体のモノクローナル抗体であって、前記ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、ジフテリア毒素の変異体の細胞への結合を抑制しない(即ち、proHB−EGFとの結合活性を阻害しない)モノクローナル抗体は、以下の文献(Hayakawa S, J. Biol. Chem. 258, 4311−4317, 1983) によって示されるような常法に従い作成することができる。すなわち、ジフテリア毒素の変異体に対する抗体を作成しているクローンの中で、最終的にジフテリア毒素の毒性を中和する活性は持つが、ジフテリア毒素の変異体の細胞への結合を抑制しない抗体を作成しているクローンを分離して作成できる。また、公知のジフテリア毒素モノクローナル抗体の中から、使用するジフテリア毒素変異体に対して前記性質を有するものを選択することもできる。
【0048】
ジフテリア毒素の毒性を中和する活性は、ジフテリア毒素の変異体と該ジフテリア毒素モノクローナル抗体との複合体をVero−H細胞に加えて、ジフテリア毒素の変異体を単体で加えたときに起きるコロニー形成の抑制が、阻害されるかどうかを見ることにより容易に測定できる。
【0049】
一方、該モノクローナル抗体が、ジフテリア毒素の変異体の細胞への結合を抑制しないことは、前記125Iで標識したジフテリア毒素のHB―EGFへの結合の阻害や、後述するIL−3依存的に増殖能を示す32D cells(ATCCより入手)に上皮細胞増殖因子受容体遺伝子を発現させた、DER cellにおいて、HB−EGFの増殖活性阻害作用を測定すること等により調べることができる。
【0050】
ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、ジフテリア毒素の変異体の細胞への結合を抑制しないジフテリア毒素の変異体のモノクローナル抗体としては、例えば#2 anti−DT mAb(特許生物寄託センター 寄託番号:FERM P−19551のハイブリドーマより産生されるモノクローナル抗体)が挙げられる。
【0051】
ジフテリア毒素の変異体との複合体は、ジフテリア毒素の変異体とモノクローナル抗体とを適当な比により混合することにより作成できる。
【0052】
本発明の制癌剤は、上記有効成分をそのまま、または、薬学的に許容される医薬用担体と組合せて製剤化することができる。
【0053】
前記制癌剤は、経口的又は非経口的(例えば、静脈内、筋肉内、腹腔内、皮下又は皮内等への注射、直腸内投与、経粘膜投与、経気道投与など)に投与することができる。卵巣癌等腹腔内に播種する悪性腫瘍に適用する場合には、腹腔内に注射により投与することが、癌細胞に直接運搬される点で好ましい。
【0054】
経口投与に適する医薬組成物としては、例えば、錠剤、顆粒剤、カプセル剤、散剤、溶液剤、懸濁剤、シロップ剤などを挙げることができ、非経口投与に適する医薬組成物としては、例えば、注射剤、点滴剤、坐剤、経皮吸収剤などを挙げることができるが、剤形はこれらに限定されることはない。
【0055】
前記制癌剤の製造に用いられる製剤用添加物の種類は特に限定されず、当業者が適宜選択可能である。例えば、賦形剤、崩壊剤又は崩壊補助剤、結合剤、滑沢剤、コーティング剤、基剤、溶解剤又は溶解補助剤、分散剤、懸濁剤、乳化剤、緩衝剤、抗酸化剤、防腐剤、等張化剤、pH調節剤、溶解剤、安定化剤などを用いることができ、これらの目的で使用される個々の具体的成分は当業者に周知されている。
【0056】
経口投与用の製剤の調製に用いることができる製剤用添加物として、例えば、ブドウ糖、乳糖、D−マンニトール、デンプン、又は結晶セルロース等の賦形剤;カルボキシメチルセルロース、デンプン、又はカルボキシメチルセルロースカルシウム等の崩壊剤又は崩壊補助剤;ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、又はゼラチン等の結合剤;ステアリン酸マグネシウム又はタルク等の滑沢剤;ヒドロキシプロピルメチルセルロース、白糖、ポリエチレングリコール又は酸化チタン等のコーティング剤;ワセリン、流動パラフィン、ポリエチレングリコール、ゼラチン、カオリン、グリセリン、精製水、又はハードファット等の基剤を用いることができる。
【0057】
注射あるいは点滴用の製剤の調製に用いることができる製剤用添加物としては、注射用蒸留水、生理食塩水、プロピレングリコール等の水性あるいは用時溶解型注射剤を構成しうる溶解剤又は溶解補助剤;ブドウ糖、塩化ナトリウム、D−マンニトール、グリセリン等の等張化剤;無機酸、有機酸、無機塩基又は有機塩基等のpH調節剤等の製剤用添加物を用いることができる。
前記本発明の制癌剤に含まれる有効成分の量は、制癌剤の製剤形態または投与経路によって異なり、一概に規定することはできないが、通常、最終製剤中に約0.0001%から70%の範囲から適宜選択して決定することができる。
【0058】
本発明の制癌剤は、ヒトを含む哺乳動物に投与することができる。
【0059】
本発明の制癌剤の投与量は、患者の年齢、性別、体重、症状、及び投与経路などの条件に応じて適宜増減されるべきであるが、成人一日あたりの有効成分の量として、体重1kgあたり1μgから30mg程度の範囲であることが好ましい。上記投与量の医薬は一日一回に投与してもよいし、数回に分けて投与してもよい。また、数日から数週間に1度又は単発的に投与してもよい。また、ステロイド等、副作用を抑える成分とともに投与することもできる。
【実施例】
【0060】
以下、本発明の実施例について説明するが、本発明はこの実施例に何ら限定されるものではない。
(実施例1)
CRM197タンパク質の生成
C7(β197)の溶原菌のストック(C7(β197)ファージを溶原化したジフテリア菌C7(beta197)M1としてATCC(American Type Culture Collection)Bacteria collection(No.39255)から入手可能)を培養し、対数増殖期の後期の菌液を、2%の濾過したマルトースを加えたC−Y培地に、最初のOD590が約0.05になるように加える。このODは、約5×107菌体/mlにあたる。フラスコは毎分200回転のロータリーシェーカーに載せ、35℃で16時間から17時間培養する。ODが10から15になった時点で、培養を終了する。
【0061】
前記C−Y培地は、以下のように調製する。即ち、10gのカサミノ酸、20gの酵母抽出液、5gのKH2PO4を1lの蒸留水に溶かす。2mlの50%CaCl2.2H2Oを加えた後、pHを7.4に調整する。沸騰させた後、濾過する。2mlのMuellerとMillerの溶液II(22.5g MgSO4、0.115g βアラニン、0.115g ニコチン酸、7.5mg ピメリン酸、1g CuSO4・5H2O、1g ZnSO4・5H2O、1g MnCl2・4H2O/100ml H2O)と1mlのMuellerとMillerの溶液III(20g L−シスチン、20ml 濃塩酸/100ml H2O)を加える。100mLずつ分注し、オートクレーブをかけることによりC−Y培地を得た。
【0062】
CRMタンパクの精製は、以下のようにして行う。
【0063】
培養液は、10000gで15分間遠心する。培養上清に硫安を65%の飽和状態になるように加える。氷室で24から48時間放置する。沈殿物を集めて0.02MのpH7.2のトリス−塩酸バッファーに溶かし、同液にて透析する。
【0064】
遠心して、不溶物を除き、DE52カラムに添加し、0.02MのpH7.2のトリス−塩酸バッファー中のNaClの濃度勾配により溶出する。CRM197は、NaClが0.08Mのところで溶出する。溶出した液を硫安で65%飽和状態にする。沈殿物を0.01Mのトリス−塩酸バッファーに溶かし、再び平衡化する。そして、DE52カラムに再び掛け、再度硫安沈殿する。続いて、SephacrylS−200のカラムにかけ、HEPES−NaOH、pH7.2、0.15M NaClの溶液で溶出する。溶出されたCRM197をDeToxiゲルにアプライして、CRM197試料に含まれるLPS類似物質を取り除き、実験に使用する。CRM197の280nmにおける吸収は、1ODが約0.67 mg/mlに相当する。
【0065】
(実施例2)
ヌードマウスを用いた造腫瘍性実験を行った。
【0066】
RPMI+10%FBSで培養した卵巣癌細胞株SKOV−3(ATCCから入手可)を、EDTA/PBS(−)で洗浄し、0.25%トリプシンで回収した。RPMI+10%FBSで2回、RPMI(血清なし)で2回洗浄し、5×106細胞ずつRPMI(血清あり)250μLへ添加し、これをヌードマウスの背部に皮下注射により接種した。
【0067】
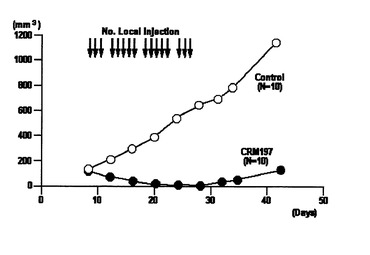
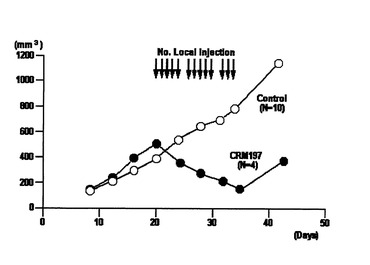
一群のヌードマウスについては、SKOV−3細胞接種1週間後よりCRM197の投与を開始し、腹腔内に200μg/日で週5回を4週に渡って投与した(図7)。また、別の群のヌードマウスについては、SKOV−3細胞接種1週間後よりCRM197の投与を開始し、接種した腫瘍に直接100μg/日で週5回を4週に渡って投与した(図8)。さらに、別の群のヌードマウスについては、SKOV−3細胞接種20日後よりCRM197の投与を開始し、接種した腫瘍に直接100μg/日で週5回を3週に渡って投与した(図9)。いずれの場合もCRM197を投与しないヌードマウスを対照実験とした。投与時期と腫瘍体積の関係を図7から図9に示す。ここで、腫瘍体積は、できた腫瘍の長径、短径を3〜4日ごとに測定し、長径×短径×短径×1/2で求めた。
【0068】
これらの結果から、いずれの場合も、CRM197の投与により、腫瘍の成長が抑えられることが分かった。
【0069】
(実施例3)
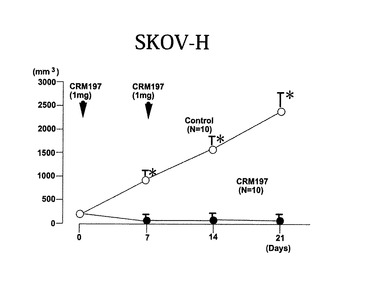
卵巣癌細胞株RMG−1(Japanese Collection of Research Bioresources Cell Bankに登録)、卵巣癌細胞株OV47、及び、卵巣癌細胞株SKOV−3細胞にHB−EGF遺伝子をトランスフェクションして作製した細胞であるSKOV−Hを、前記実施例2におけるSKOV−3細胞と同様に回収し、SKOV−3細胞の代わりにヌードマウス背部に皮下注射により接種した。
【0070】
いずれの細胞を接種した場合にも、細胞接種直後と一週間後にCRM197を腹腔内に1mg投与した。また、それぞれCRM197を投与しないヌードマウスを対照実験とした。
【0071】
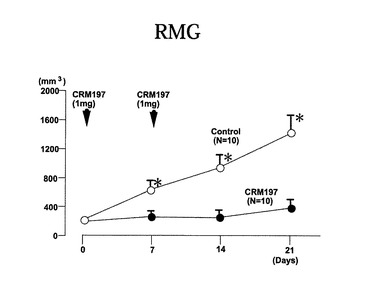
投与時期と腫瘍体積の関係を図10から図12に示す。図10はRMG−1細胞、図11はOV47細胞、図12はSKOV−H細胞を接種したヌードマウスの場合の結果を示す。
【0072】
これらの結果からも、いずれの場合も、CRM197の投与により、腫瘍の成長が抑えられることが分かった。
【0073】
(実施例4)
CRM197の細胞毒性試験
これまでの実験では、CRM197のフラグメントAにはADPリボース転移酵素活性が認められず、全く無毒の分子であると考えられてきたが、全く毒性がないか、わずかながらもジフテリア毒素としての毒性を有しているかを明らかにすることは、本発明において極めて重大であるので、CRM197のADPリボース転移酵素活性について、さらに詳しく解析した。
【0074】
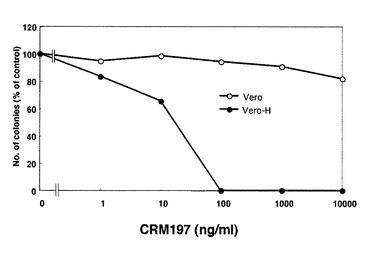
まず、CRM197の細胞毒性を調べるためにVero細胞(ATCCから入手可能)、及びVero細胞にHB−EGFを高発現させたVero−H細胞(OriGene Technologies社製ヒトHB−EGFcDNAをpCDNA3.1プラスミド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成した細胞)に種々の濃度のCRM197を加えて1週間培養し、コロニー形成率を調べた。
【0075】
図13はVero細胞及びVero−H細胞を6wellプレートに300cell/wellの密度で蒔き、10時間培養し、これにCRM197を加えて24時間培養し、その後CRM197を含まない培養液で6日間培養してコロニー数をカウントした場合のコロニー形成率を示している。図14はVero細胞及びVero−H細胞にCRM197を加えて一週間培養した場合のコロニー形成率を示している。その結果、Vero−H細胞は、CRM197を加えて24時間培養した場合には1μg/mlのCRM197で、CRM197存在下で一週間培養した場合には100ng/mlのCRM197で、コロニー数の減少が確認された。Vero細胞ではどちらの条件においても有意なコロニー数の減少は認められなかった。CRM197がVero細胞よりもVero−H細胞に対して強い毒性を示したことから、この毒性がジフテリア毒素リセプター(proHB−EGF)に関係したものであることが示唆された。なお、CRM197の代わりにジフテリア毒素を24時間作用させ、その後ジフテリア毒素を含まない培養液で6日間培養した場合、ジフテリア毒素は約1fg/mlの濃度でコロニーの出現をほとんど見なかった(毒素を加えない場合に得られるコロニー数の約3%)。したがって、ここで用いたコロニー形成法で測定されるCRM197の細胞毒性はジフテリア毒素の1010以下であり、CRM197の細胞毒性は極めて微弱なものであることがわかった。
【0076】
(実施例5)
CRM197によるタンパク合成阻害試験
ジフテリア毒素の毒素作用は、EF2のADPリボシル化に基づく、タンパク合成阻害作用である。そこで、CRM197の毒性がタンパク質合成阻害によるものかどうかを検討するために、CRM197によるタンパク合成阻害作用を調べた。実施例4で使用したのと同様のVero細胞、及びVero−H細胞にCRM197を36時間暴露させ、そのタンパク質合成阻害能を[3H]Leuの蛋白への取り込み率から調べた。具体的には、24wellプレートにVero細胞、Vero−H細胞を1×105cell/mlの密度で蒔き、16時間培養後、CRM197を加えて、さらに36時間培養した。その後、[3H]Leuを添加し、1時間インキュベートしたのち、タンパク質に取り込まれた[3H]Leuの放射能量を液体シンチレーションカウンターにて測定し、タンパク質の合成阻害能を求めた。
【0077】
その結果、Vero−H細胞では100 ng/ml以上の濃度のCRM197でタンパク質合成の阻害が観察された。この条件でVero細胞ではタンパク合成阻害作用はほとんど観察されなかった(図15)。
【0078】
(実施例6)
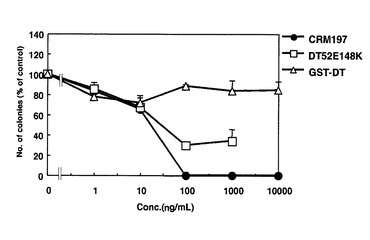
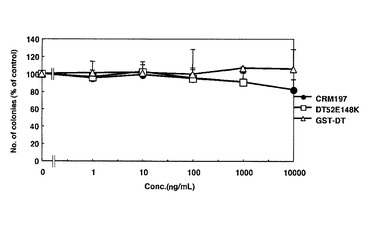
DT52E148Kの細胞毒性試験
ジフテリアトキシンに2カ所の変異を持つ変異体であるDT52E148K及びリコンビナント蛋白GST−DTについても実施例4と同様にその毒性をコロニー形成法で測定した。その結果、CRM197よりもさらに微弱ではあるが、DT52E148KはVero−H細胞対して毒性を示した(図16)。一方、Vero細胞には細胞毒性は認められなかった(図17)。したがって、完全に毒性を消失させるためには、この2カ所の変異だけでは不十分であることが示された。GST−DTはVero細胞に対してもVero−H細胞に対しても、全く毒性を示さなかった。
【0079】
(実施例7)
Vdtr細胞のCRM197及びDT52E148Kに対する耐性
これまで示されたCRM197及びDT52E148Kの細胞毒性、タンパク質合成阻害作用がジフテリア毒素の持つADPリボシル化活性によるEF−2の不活性化であるのかどうかを検討するために、ジフテリア毒素耐性株Vdtr細胞を作成した。具体的には以下の文献(Moehring JM and Moehring TJ, Somat. Cell Genet. 5, 453−468, 1979;
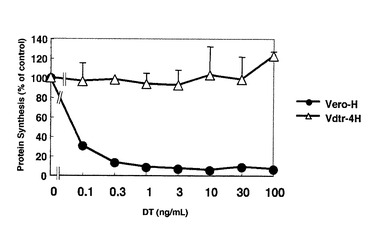
Kohno K et al., Somat. Cell Genet. 11, 421−423, 1985)に従って、Vero細胞をEMS処理した後、ジフテリア毒素を加えて培養し、生存する細胞を得た。これらの中から、高濃度のジフテリア毒素を加えても耐性である細胞を選別し、Vdtr細胞を得た。次ぎに、このVdtr細胞にHB−EGFを高発現させ、Vdtr−4H細胞を得た(OriGene Technologies社製ヒト由来HB−EGF遺伝子をpCDNA3.1プラスミッド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成した細胞)。Vdtr−4H細胞にジフテリア毒素を加えたときの蛋白合成阻害率を図18に、この細胞より得た細胞破砕液にフラグメントAを加えたcell−freeのADPリボシル化アッセイの結果を図19に示す。この細胞ではcell−freeのADPリボシル化アッセイにおいてもEF2のADPリボシル化が全く認められないことから、この細胞ではジフテリア毒素耐性はEF2に原因があることが示された。
【0080】
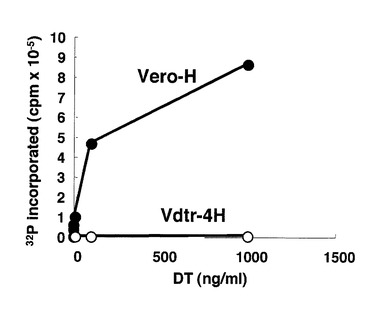
CRM197の持つ細胞毒性がADPリボシル化活性によるものかどうかを確認するために、このVdtr細胞にHB−EGFを高発現させたVdtr−4H細胞に対する、CRM197の毒性を検討した。Vdtr−4H細胞にCRM197を加え、実施例4と同様にコロニー形成法によってその毒性を検討した。その結果、100μg/mlのCRM197を加えてもVdtr−4H細胞に全く毒性を示さなかった(図20)。また、DT52E148Kについても同様に全く毒性を示さなかった(図20)。この結果からCRM197の示す細胞毒性、及びタンパク合成阻害はフラグメントAのADPリボシルトランスフェラーゼ活性によるものであることが明らかとなった。
【0081】
(実施例8)
無細胞系でのEF−2のADPリボシル化実験
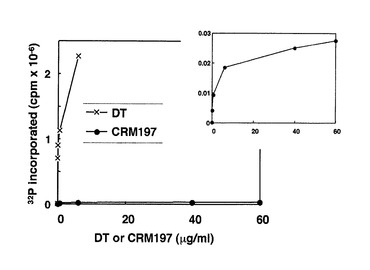
実施例7のVdtr細胞を用いた実験結果は、CRM197及びDT52E148Kに存在する細胞毒性が、残存するADPリボシルトランスフェラーゼ活性によるものであることを示している。これをさらに確認するために、Cell−freeの条件下でADPリボシル化実験を行った。以下の文献Gill, DM and Pappenheimer, AM Jr. J. Biol. Chem. 246, 1492−1495,1971) に示された方法を用いて、ウサギ肝より抽出したEF−2にCRM197又はDT52E148Kを加え、これに[32P]NADを加えて、37℃で10分間インキュベートし、cell−freeでADPリボシル化反応を行った。その後、液体シンチレーションカウンターにて放射能量を測定した。その結果、極めて弱い活性であるが、CRM197及びDT52E148KにEF−2をADPリボシル化する活性が認められた(図21及び図22)。なお、図21におけるの右上の図は、CRM197のADPリボシル化活性を縦軸の尺度を拡大して示したものである。この結果から、CRM197及びDT52E148Kの両者にEF2をADPリボシル化する活性がわずかではあるが残存することが結論づけられた。
【0082】
(実施例9)
ジフテリア毒素モノクローナル抗体#2 anti−DT mAb(diphtheria toxin monoclonal antibodies ♯2)によるCRM197の細胞毒性の中和
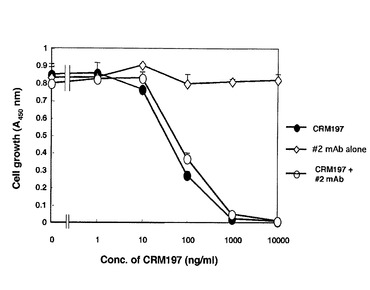
CRM197をHB−EGF増殖活性阻害物質として利用する場合、CRM197に微弱ではあるが細胞毒性があることは、場合によっては望ましくない。そこで、次にCRM197に残る毒性を抑制する条件を検討した。ジフテリア毒素に対するモノクローナル抗体は、多数分離されている(Hayakawa S, J. Biol. Chem. 258, 4311−4317, 1983)。これらの中には、ジフテリア毒素の細胞毒性は抑制するが、ジフテリア毒素の受容体への結合は抑制しない抗体がある。これらの中で、#2 anti−DT mAbが、ジフテリア毒素の場合と同様にCRM197の毒性は中和するが、CRM197のHB−EGFへの結合は抑制しないことを見出した。
【0083】
CRM197と同時に#2 anti−DT mAb(特許生物寄託センター 受託番号:FERM P−19551のハイブリドーマより産生されるモノクローナル抗体)を加え、CRM197の毒性が中和されるかを検討した。
【0084】
ジフテリア毒素に対するモノクローナル抗体の作成は、以下の文献(Hayakawa S,J. Biol. Chem. 258, 4311−4317, 1983) によって示されるが、簡単に記述すると以下のようになる。BALB/cマウスの腹腔に、ホルマリン処理したジフテリア毒素0.1 mgを、フロインドアジュバントと共に接種し、これを1週間ごとに合計3回行った。最後の接種から数日後に、このマウスの脾臓細胞を取り出し、これをマウスミエローマ細胞SP2/0細胞と融合させた。融合反応後の細胞をHAT選択培地で培養し、増殖細胞の中から、ジフテリア毒素に対する抗体を作成しているクローンを分離した。ジフテリア毒素に対する抗体を作成しているクローンの中で、最終的にジフテリア毒素の毒性を中和する活性は持つが、ジフテリア毒素の細胞への結合を抑制しない抗体を作成しているクローンを分離した。
【0085】
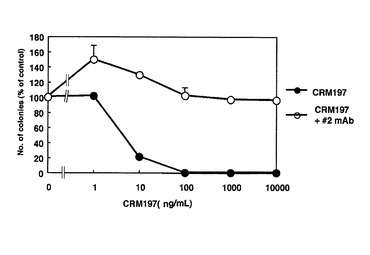
CRM197/#2 anti−DT mAb 複合体は、まず、CRM197(1mg)と#2 anti−DT mAb(10mg)を混ぜ、37℃で1時間インキュベートして、作成した。Vero−H細胞にこの複合体を種々の濃度で加えて1週間培養し、コロニー形成率を調べた。その結果、たとえば、CRM197単独の場合は、100ng/mlのCRM197で死滅するが、CRM197/#2 anti−DT mAb 複合体の場合、最大量10μg/mLでもコロニーの形成は全く抑制されず、#2 anti−DT mAbがCRM197の細胞毒性を完全に阻害することがわかった(図23)。
【0086】
(実施例10)
CRM197/#2 anti−DT mAb 複合体のHB−EGF増殖活性阻害作用
CRM197/#2 anti−DT mAb 複合体のHB−EGF増殖活性阻害作用(HB−EGFの増殖活性を阻害する作用)について検討した。IL−3依存的に増殖能を示す32D cells(ATCCより入手)に上皮細胞増殖因子受容体遺伝子(EGFR遺伝子、OriGene Technologies社製)を発現させて、DER cellを作成した(EGFR遺伝子を、pCDNA3.1プラスミド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成)。この細胞は、IL−3非存在下では、HB−EGFの増殖活性により増殖する。この細胞に対し、HB−EGF存在下で、CRM197及びCRM197/#2 anti−DT mAb 複合体を添加した。細胞を48時間培養し、増殖した細胞数をMTT assayによって測定した。CRM197及びCRM197/#2 anti−DT mAb 複合体が存在しない条件ではDER cellは増殖したが、CRM197やCRM197/#2 anti−DT mAb 複合体が存在する条件では、DER cellの増殖は抑制された(図24)。CRM197とCRM197/#2 anti−DT mAb複合体の増殖活性阻害作用を比較したところ、両者にはほとんど違いはなく、同様の阻害効果を示した。すなわち、CRM197/#2 anti−DT mAb 複合体は、CRM197が持つ細胞毒性は抑制されているが、HB−EGFの増殖活性阻害作用についてはCRM197単独と同様の阻害活性を保持していることがわかった。
【0087】
(実施例11)
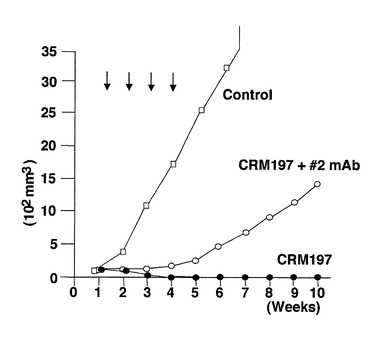
CRM197/#2 anti−DT mAb 複合体による腫瘍抑制効果
一群のヌードマウス(3個体づつ)について、SKOV−H細胞をそれぞれ接種し、接種から1週間後よりCRM197(1mg/1個体/week)、あるいはCRM197/#2 anti−DT mAb 複合体(CRM197 1mgを含む/week)の投与を開始し、腹腔内に週1回を4週に渡って投与した)。CRM197/#2 anti−DT mAb 複合体は1mgのCRM197と8 mgの#2 anti−DT mAbを1時間、室温でインキュベーションしたものを使用した。また、CRM197を投与しないヌードマウスを対照実験とした。CRM197、あるいはCRM197/#2 anti−DT mAb 複合体投与と腫瘍体積の関係を図25に示す。腫瘍体積の測定は実施例2と同様に行った。この実験から、CRM197/#2 anti−DT mAb 複合体は腫瘍の増殖を抑制することが明らかになったが、その効果はCRM197単独よりも弱いことが示された。
【0088】
CRM197/#2 anti−DT mAb 複合体は、すでに示した図から、CRM197の持つ微弱な細胞毒性を完全に抑えることから、CRM197単独使用よりもより高い安全性が期待できる。一方、CRM197を単独で用いると腫瘍の増殖を抑制する効果がより強い。これは、CRM197がもつHB−EGF増殖活性阻害作用に加えて、CRM197が持つ微弱な細胞毒性が加わった結果であると考えられる。したがって、CRM197の使用にあたっては、安全性をより重視する場合には#2 anti−DT mAbと複合体として投与することも可能であり、効果をより重視する場合にはCRM197単独投与も有効であることが考えられる。
【0089】
(実施例12)
DT52E148KとCRM197について、投与が週1回を3週にわたっての投与であった以外は、実施例11と同様に腫瘍抑制効果を調べた。DT52E148K又はCRM197と、腫瘍体積との関係を図26に示す。この実験から、DT52E148Kは腫瘍の増殖を抑制することが明らかになったが、その効果はCRM197よりも弱いことが示された。ADPリボース転移酵素活性を有するAフラグメントに2箇所の変異があり、細胞毒性が低い(図16)DT52E148Kに関するこの結果は、前記、CRM197においてはCRM197がもつHB−EGF増殖活性阻害作用に加えて、CRM197が持つ微弱な細胞毒性が腫瘍の増殖の抑制に効果を有しているという推定を裏付けるものである。
【0090】
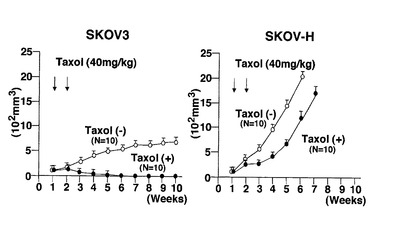
(参考例)
一群のヌードマウス(10個体づつ)について、SKOV−3細胞又はSKOV−H細胞をそれぞれ接種し、接種から1週間後及び2週間後に、タキソール(Taxol)40mg/kg/weekを腹腔内に投与した。結果を図27に示す。タキソールは、SKOV−3細胞には効果があるが、SKOV−H細胞には効果が少ないことが分かった。SKOV−H細胞は、HB−EGFを多く発現する性質があることから、タキソールは本発明の制癌剤とは作用機序が異なることが分かった。したがって、本発明の制癌剤は、タキソールがあまり有効でない症例にも効果があることが示唆される。
【産業上の利用可能性】
【0091】
本発明は、卵巣癌をはじめとする各種癌の治療に有効な制癌剤の製造に利用できる。
【図面の簡単な説明】
【0092】
【図1】図1は、proHB−EGFの構造を表す模式図である。
【図2】図2は、ジフテリア毒素のアミノ酸配列及び塩基配列を表す図である。
【図3】図3は、ジフテリア毒素のアミノ酸配列及び塩基配列を表す図である(図2の続き)。
【図4】図4は、ジフテリア毒素のドメイン構造を表す模式図である。
【図5】図5は、GST−DTのアミノ酸配列及び塩基配列を表す図である。
【図6】図6は、GST−DTのアミノ酸配列及び塩基配列を表す図である(図5の続き)。
【図7】図7は、SKOV−3細胞注射ヌードマウスにおけるCRM197腹腔内注射による腫瘍増殖に対する効果を表す図である。
【図8】図8は、SKOV−3細胞注射ヌードマウスにおけるCRM197局所注射による腫瘍増殖に対する効果を表す図である。
【図9】図9は、SKOV−3細胞注射ヌードマウスにおけるCRM197局所注射による腫瘍増殖に対する効果を表す図である。
【図10】図10は、RMG−1細胞注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図11】図11は、OV47細胞注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図12】図12は、SKOV−H注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図13】図13は、CRM197を24時間作用させた場合のVero細胞及びVero−H細胞に対する毒性を表す図である。
【図14】図14は、CRM197を1週間作用させた場合のVero細胞及びVero−H細胞に対する毒性を表す図である。
【図15】図15は、CRM197によるタンパク合成阻害作用を表す図である。
【図16】図16は、CRM197、DT52E148K及びGST−DTのVero−H細胞に対する毒性を表す図である。
【図17】図17は、CRM197、DT52E148K及びGST−DTのVero細胞に対する毒性を表す図である。
【図18】図18は、Vdtr−4H細胞のDT耐性を表す図である。
【図19】図19は、無細胞条件におけるVdtr−4H細胞及びVero−H細胞破砕液のEF−2のADP−リボシル化反応を表す図である。
【図20】図20は、Vdtr−4H細胞のCRM197及びDT52E148Kに対する耐性を表す図である。
【図21】図21は、CRM197及びDTによるEF−2のADP−リボシル化を表す図である。
【図22】図22は、DT52E148KによるEF−2のADP−リボシル化を表す図である。
【図23】図23は、#2 anti−DT mAbによるCRM197の毒性の中和を表す図である。
【図24】図24は、CRM197/#2 anti−DT mAb複合体による、HB−EGF細胞増殖活性の阻害を表す図である。
【図25】図25は、CRM197及びCRM197/#2 anti−DT mAb複合体の腹腔内投与によるヌードマウスの癌細胞の増殖を表す図である。
【図26】図26は、CRM197及びDT52E148Kの腹腔内投与によるヌードマウスの癌細胞の増殖を表す図である。
【図27】図27は、タキソール投与の投与によるXenograftマウスの癌細胞の増殖を表す図である。
【技術分野】
【0001】
本発明は、卵巣癌をはじめとする各種癌の治療に利用できる制癌剤に関する。
【背景技術】
【0002】
悪性腫瘍に対しては、各種の治療法及び治療薬が開発されているが、未だ充分な治療効果をあげることが出来ない場合が多い。特に、卵巣癌は婦人科領域の悪性腫瘍の中でも進行が早く、とりわけ予後の悪い悪性腫瘍である。
卵巣癌の治療法としては、現在タキソールを中心とした化学療法しか手立てが無いが、これらの化学療法も一時的効果はあるものの、後には再発を許すことが多く、新たな治療法の開発が求められている。
【0003】
HB−EGFはEGFファミリーの細胞増殖因子で、身体の形成や再生過程に必須であると同時に、血管狭窄、動脈硬化症などの発症に関係する分子として知られているが(例えば、非特許文献1参照)、この因子を悪性腫瘍の治療のターゲットとすることは現在まで全く行われていなかった。この分子は膜結合型前駆体(proHB−EGF)として合成され、細胞表面でプロテアーゼにより切断されて分泌型HB−EGFを生じる。分泌型には、増殖促進作用があるが、膜型には増殖抑制作用が観察され、分泌型と膜型は適宜使い分けられることで、組織の形成と維持に働いているものと考えられている。
【0004】
HB−EGFは、EGFリセプター(Her1)及びEGFリセプターファミリーのHer4(ErbB−4)に結合し、活性化する。しかし、EGFリセプターファミリー(Her1、Her2、Her3、Her4)はホモダイマーを形成するほか、全ての組み合わせのヘテロダイマーを形成し得るので、結果的にHB−EGFは全てのEGFリセプターファミリー分子を活性化しうる。HB−EGFは種々の組織で発現されており、広範な細胞や組織で働いていると考えられているが、線維芽細胞、平滑筋細胞あるいはケラチノサイトの増殖をよく促進することが報告されている(例えば、非特許文献2参照)。
【0005】
HB−EGFは前記のように膜結合型前駆体(proHB−EGF)として合成されるが、proHB−EGFは、N末端から、シグナル配列、プロ配列、ヘパリン結合ドメイン、EGF様ドメイン、ジャクスタメンブレンドメイン、トランスメンブレンドメイン、細胞質ドメインからなる(図1)。このproHB−EGFは、図中矢印の部分でプロテアーゼによる切断(エクトドメインシェディング)を受けて分泌型となる。proHB−EGFのエクトドメインシェディングは、リゾホスファチジン酸(LPA)等がG蛋白共役型リセプターを介して、Ras−Raf−MEK経路を活性化する経路や、フォルボールエステルがPKCを活性化する経路により刺激されることが提唱されている(例えば、非特許文献3参照)。
【0006】
分泌型HB−EGFがEGFリセプターに結合し、EGFリセプターのリン酸化を促す作用はEGF様ドメインに存在する(例えば、非特許文献1参照)。
【0007】
一方ジフテリア毒素は、ジフテリア菌が産生する分子量約59000のタンパク質であるが、HB−EGFの膜結合型前駆体(proHB−EGF)を受容体とすることが知られている(例えば、非特許文献4参照)。また、CRM197のようなジフテリア毒素の変異体は、分泌型HB−EGFの阻害剤として知られている(例えば、非特許文献5参照)。ジフテリア毒素のデータベース情報は、遺伝子についてはEMBL;K01722、アミノ酸配列はSWISS−PROT;P00588、三次元構造はPDB;1MDTあるいは1XDTで見ることができる。ジフテリア毒素の遺伝子は菌体内に溶原化しているファージがコードしている。
【0008】
ジフテリア毒素は、アミノ酸535個からなる単純タンパク質であり(ジフテリア毒素のアミノ酸配列(配列番号1)とそれをコードする遺伝子の塩基配列(配列番号2)を図2及び図3に表し、イタリックはシグナル配列を表す)、還元剤で処理することにより分離されるフラグメントA部分とフラグメントB部分に分けられるが(図4)、立体構造解析等によれば、フラグメントB部分はさらに2つのドメインに分かれている。それぞれのドメインの機能は、フラグメントA部分に相当する触媒作用(catalytic)ドメイン(シグナル配列を除いたアミノ酸番号で1〜193)はADPリボシル化活性を、フラグメントB部分のN末端半分に相当する膜貫通(Transmembrane)ドメイン(シグナル配列を除いたアミノ酸番号で194〜378)はエンドソーム膜にチャネルを形成する性質を、フラグメントB部分のC末端半分に相当するレセプター結合(receptor−binding)ドメイン(シグナル配列を除いたアミノ酸番号で386〜535)は細胞表面のジフテリア毒素リセプターと結合する活性を有する。
【0009】
ジフテリア毒素のフラグメントA(触媒作用ドメイン部分)は、NAD存在下でEF−2(ペプチド伸長因子2)をADPリボシル化する作用を有しており、これによりタンパク質合成を阻害する。したがって、ジフテリア毒素が毒性を発揮するためには、フラグメントAが細胞質内に入らなければならない。
フラグメントAが細胞質内に入る機構は、フラグメントBにあるレセプター結合ドメインが細胞表面のリセプターであるproHB−EGFに結合することにより、ジフテリア毒素はエンドサイトーシスによりエンドソームに取り込まれ、エンドソーム内で膜貫通ドメインがエンドソーム膜に挿入され、最終的にはフラグメントAがエンドソーム膜を通過して細胞質中に遊離され、そこでEF−2を失活させる(例えば、非特許文献6参照)。
【0010】
ジフテリア毒素が毒性を発揮するためには、フラグメントAとフラグメントBの両方が必要である。したがってどちらのフラグメントに変異があっても、ジフテリア毒素の毒性を有さないタンパク質ができる。
【0011】
ジフテリア毒素には触媒作用ドメインに変異を有する無毒化された変異体、例えばCRM197が分離されている。
【0012】
一方、ジフテリア毒素の変異体が、HB−EGFとEGFレセプターとの結合を阻害する活性を有するのは、ジフテリア毒素が、分泌型HB−EGFのEGF様ドメインと結合するためである。また、この結合に関与するのはジフテリア毒素のレセプター結合ドメインである。ジフテリア毒素の516番目のLys、530番目のPheがHB―EGFの結合に重要であることが報告されている(例えば、非特許文献7参照)。また、ジフテリア毒素とHB―EGFのEGFドメインからなる複合体の結晶構造が解析されており、381−535までの間のHB―EGFとの結合に重要なアミノ酸が報告されている(例えば、非特許文献8参照)。
【0013】
このように、ジフテリア毒素変異体がHB−EGFに結合し、HB−EGFの活性を阻害することは認められていたが、HB−EGFを癌治療のターゲットとすること自体が知られていなかったこと、及び、HB−EGFを含む制御機構の複雑さから、ジフテリア毒素変異体を癌治療薬に使用しようとする試みは全くなされていなかった。
【非特許文献1】目加田英輔等著「遺伝子医学」株式会社メディカルドゥ社、vol.5 No.2 2001,p.131−134
【非特許文献2】Higashiyama, S. et al著 J.Cell Biol.(1993)122,p.933−940,
【非特許文献3】Prenzel,N.et al著 Nature (1999) 402,p.884−888
【非特許文献4】J.G.Naglich et al著 Cell (1992) 69,p.1051−1061
【非特許文献5】T.Mitamura et al J.Biol.Chem.(1995) 270,p.1015
【非特許文献6】T.Umata et al著 J.Biol.Chem.(1998) 273,p.8351
【非特許文献7】Shen, HS et al著 J. Biol. Chem. (1994) 269,p.29077−29084
【非特許文献8】Gordon VL et al著 Molecular Cell (1997) 1, p.67−78
【発明の開示】
【発明が解決しようとする課題】
【0014】
本発明は、従来における前記諸問題を解決し、以下の目的を達成することを課題とする。即ち、悪性腫瘍、特に卵巣癌の治療に有効な制癌剤を提供することを目的とする。
【課題を解決するための手段】
【0015】
発明者らは、これまで身体の形成や再生過程に必須であると同時に、血管狭窄、動脈硬化症などの発症に関係するタンパク質として知られていた分泌型HB−EGFの機能を再検討し、癌細胞の増殖、転移過程にHB−EGFが関与しているとの知見を得た。これに基づき、新たな治療法の開発を進め、その結果HB−EGF中和能を有するCRM197が、ヌードマウスに接種したヒト卵巣癌由来細胞の増殖を有意に抑制することを明らかにし、本発明に至った。
【0016】
すなわち、本発明の前記課題を解決するための手段は以下の通りである。
<1> 以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする制癌剤である。
(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質
(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質
(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質
<2> (a)、(b)及び(c)のいずれかのタンパク質が、ジフテリア毒素の触媒作用ドメインを有しない前記<1>に記載の制癌剤である。
<3> (c)のタンパク質が、GST−DTである前記<2>に記載の制癌剤である。
【発明の効果】
【0017】
本発明によると、悪性腫瘍、特に卵巣癌の治療に有効な制癌剤を提供することができる。
【発明を実施するための最良の形態】
【0018】
発明者らは、HB−EGFの機能を検討する中で、HB−EGFには造腫瘍性があるとの知見を得たことに基づき、HB−EGFのEGFレセプターへの結合阻害活性が知られているジフテリア毒素の無毒化変異体の制癌作用を実証した。
【0019】
本発明の第一の態様の制癌剤は、ジフテリア毒素の変異体であってHB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を、有効成分として含むことを特徴とする。
前記ジフテリア毒素の変異体とは、ジフテリア毒素のアミノ酸配列において1又は複数のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質を表し、例えば1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質である。また、ジフテリア毒素の25個のシグナル配列は付いていても付いていなくてもよく、いずれの配列も本発明の範囲に含まれる。
【0020】
本発明の第二の態様の制癌剤は、以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする。(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質。(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質。(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質。
【0021】
前記タンパク質は、ジフテリア毒素の一部若しくはその変異体又はそれらのタンパク質を含む複合タンパク質であって、レセプター結合ドメインを保持するものをいう。
【0022】
ここで、ジフテリア毒素の毒性とは、ジフテリア毒素が細胞表面の受容体に結合し、細胞内に侵入し、フラグメントAが保持するEF−2(ペプチド伸長因子2)をADPリボシル化する活性(ADPリボシル化活性)でもって、細胞の蛋白質合成機能を阻害することを意味し、これはタンパク質合成阻害の程度により容易に測定することができる。すなわち、培養細胞に一定量のジフテリア毒素を加えて2〜8時間程度培養し、続いて、放射性のアミノ酸存在下で短時間培養し、タンパク質に取り込まれた放射性アミノ酸量を測定する方法である。
【0023】
具体的には、Vero細胞(1×105細胞)を24ウェルプレートに接種して、CO2インキュベーターで16時間培養する。細胞が十分にプレートに接着していることを確認してから、各ウェルに冷PBS(150mM、NaCl−2.7mM KCl−10mMリン酸緩衝液、pH7.2)で1回洗う。この時の液の出し入れは細胞をはがさないように慎重に行う。血清を含むアッセイ用培養液0.5mlを加える。アッセイ用培養液は、通常の培地からロイシンの濃度を1/10程度に減らしたものを使用する。これは後で加える[3H]ロイシンの取り込み効率を高めるためである。但し、Ham’s F12培地は、ロイシン含量が少ないのでこれをアッセイ用培養液とすることもできる。血清は通常用いている濃度で加える。
次に、種々の濃度のジフテリア毒素を加えて、CO2インキュベーターで2〜5時間培養する。3.7MBq/mlの[3H]ロイシンを10μl加えて更に1時間培養する。
【0024】
培養液を捨て、ウェルをPBSで1度洗った後、細胞を0.5mlの0.1M NaOHで溶解し、その溶液をチューブに集める。0.5mlの0.1N NaOHで再度ウェルを洗浄し、その溶液を同じチューブに集める。
【0025】
これに20%のトリクロロ酢酸溶液0.5mlを加えて、Vortexミキサーでよく攪拌する。生じた沈殿をグラスフィルターでトラップし、フィルターをさらに5%トリクロロ酢酸溶液で洗浄する。
【0026】
最後にフィルターを100%エタノールで洗浄し、乾燥させる。
【0027】
フィルターをトルエン−PPOのシンチレーターにつけて、フィルターにトラップされた放射能を液体シンチレーションカウンターで測定する。ジフテリア毒素を加えなかった試料の値を測定し、これを100%として毒素を加えた時の値を%で求める。
【0028】
ジフテリア毒素の毒性を実質的に有さないタンパク質とは、ジフテリア毒素の毒性を無毒化または極めて低レベルに弱毒化されたタンパク質をいい、本発明においては、ジフテリア毒素の毒性を1ng/mlの濃度で上記Vero細胞の系で測定した場合、ジフテリア毒素を加えなかった試料または触媒作用ドメインを有しないジフテリア毒素変異体を加えた試料の値と有意差がないものをいう。前記有意差は、t検定において有意水準5%として有意差が無いことが好ましく、有意水準1%として有意差が無いことがより好ましく、有意水準0.1%として有意差が無いことがさらに好ましい。
【0029】
ただし、本発明のなかで、CRM197やDT52E148Kのようなこれまでジフテリア毒素の毒性が全くないとされていた変異体であっても、きわめて微弱な毒性(例えばCRM197では野生型のジフテリア毒素の1010分の1程度)が残存していることが証明され、このような微弱な毒性を有している変異体が本発明から排除されるものではない。ジフテリア毒素の毒性レベルとしては、CRM197と同じかそれ以下であることがジフテリア毒素の毒性による副作用を排除し、安全性を高める観点から好ましい。しかし、一方では該毒性が、制癌剤の効果に寄与することが本発明により示唆されるため、CRM197と同程度の極めて低レベルの毒性を有することも、制癌効果を高める観点から好ましい。したがって、製剤処方にあわせて、適宜ジフテリア毒素の毒性レベルを選択することができる。
【0030】
ジフテリア毒素の毒性は、ペプチド伸長因子2をADPリボシル化するのに必須である触媒作用ドメインに変異を持たせること、または、触媒作用ドメインの一部または全部を欠損させることにより調整することができる。
【0031】
変異させた触媒作用ドメインの機能は、ADPリボシル化活性を直接測定することで、正確に調べることができる。ADPリボシル化活性は、分離精製したEF−2に、フラグメントAあるいはADPリボシル化活性を測定したい蛋白質(変異させた触媒作用ドメイン等)とラジオアイソトープで標識したNADを加えて、試験管内でEF−2をADPリボシル化させ、EF−2に取り込まれた放射能を測定することで、直接測定することができる。
【0032】
具体的には、以下の文献(Moynihan, M.R. and Pappenheimer, A.M. Jr. Infect. Immun.(1981) 32, 575−582)で記載された方法で得たウサギ網状赤血球EF−2分画に、最終濃度20mMのトリス緩衝液(pH7.8)、1mMDTT(ジチオスレイトール)、0.1〜1μg/mlのフラグメントAあるいは0.1−100μg/mlのADPリボシル化活性を測定したい蛋白質を加え、さらに最終的に370KBq/mlになるように[32P]NAD(約740 GBq/mmol)を加えて混和し、37℃で10分間反応させる。
【0033】
その反応液に同容量の10%−トリクロロ酢酸溶液を加えて蛋白質を沈殿させ、生じた沈殿をグラスフィルターでトラップし、フィルターをさらに5%トリクロロ酢酸溶液で洗浄する。
【0034】
最後にフィルターを100%エタノールで洗浄し、乾燥させる。
【0035】
フィルターをトルエン−PPOのシンチレーターにつけて、フィルターにトラップされた放射能を液体シンチレーションカウンターで測定する。
【0036】
測定された放射能がADPリボシル化活性の程度を示しており、変異を加えないフラグメントAを用いたときの放射能を基準に、変異を加えた蛋白質のADPリボシル化活性の相対活性を求めることができる。
【0037】
また、分泌型HB−EGFとEGFレセプターとの結合を阻害する活性を有するという特徴は、発明者らが、ドメイン情報をもとに、より詳細に検討したところによると、レセプター結合ドメインを含む部分である378番目から535番目のアミノ酸配列を含んでいればよいことがわかった。すなわち、GST(Gluthathione−S−transferase)にジフテリア毒素の378番目から535番目までの配列を融合した遺伝子を作成し、これを大腸菌に発現させて、上記の構造を持つ融合タンパク質(GST−DT)を作成した。GST−DTは、125Iで標識したジフテリア毒素のHB―EGFへの結合を、濃度依存的に阻害した。阻害の程度から、GST−DTはジフテリア毒素と同程度の結合の強さで、HB―EGFに結合することがわかった。したがって、結合に必要なのは、378番目から535番目、すなわち、レセプター結合ドメインを含む部分であることがわかった。
【0038】
ここで、HB−EGFとEGFリセプターの結合を阻害する活性を有するかどうかは、前記のような、125Iで標識したジフテリア毒素のHB―EGFへの結合に対する阻害実験により測定することができる。
【0039】
そこで、HB−EGFとEGFレセプターとの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質は、レセプター結合ドメインを保持しつつ、触媒作用ドメインに変異を持たせたジフテリア毒素変異体タンパク質や、ジフテリア毒素のレセプター結合ドメインを保持しつつ触媒作用ドメインの一部または全部を欠損させたジフテリア毒素の一部であるタンパク質を作成することにより得ることができる。
【0040】
このような変異体の例としては、CRM197、DT52E148K及びGST−DTが挙げられる。これらはジフテリア毒素の毒性を実質的に持たず、HB−EGFのEGFレセプターへの結合を阻害する。CRM197は、25個のシグナル配列を除いてカウントした場合の52番目のGlyがGluに変異した変異体であり、DT52E148Kは、前記変異に加えてシグナル配列を除いてカウントした場合の148番目のGluがLysに変異した変異体である。また、GST−DTはGSTにジフテリア毒素のシグナル配列を除いてカウントした場合の378番目から535番目を含む蛋白質である。CRM197に関するアミノ酸配列(最初の25個の配列がシグナル配列を表す)を配列番号3に、それをコードする遺伝子の塩基配列を配列番号4に表す。また、GST−DTのアミノ酸配列(配列番号5)及びそれをコードする遺伝子の塩基配列(配列番号6)を図5及び図6に表す。
【0041】
ここで、CRM197については、ジフテリア毒素の毒性を有さない、即ちADPリボシル化活性を有さないとの報告が既になされている(T. Uchida and A.M. Pappenheimer Jr. (1972) Science 175, 901−903)。また、148Eに変異を有する148K変異体が極めて弱い活性しか持たないことは知られており(J.T.Barbieri and R.J. Collier (1987) Infect. Immun. 55, 1647−1651)、52E変異体であるCRM197にさらに148Kの変異を入れたダブルミュータントであるDT52E148Kは、さらに安全な変異体として好ましい。また、これらの変異体の毒性は、前記タンパク質合成阻害実験によってもジフテリア毒素を加えなかった試料の値と有意差がないことが確かめられた。ただし、前述のように、これらの変異体であっても、極めて微弱なジフテリア毒素の毒性が残存していることが、本発明の、より詳細な解析により明らかになっている。
【0042】
なお、GST−DTがジフテリア毒素の毒性を有しないことは、触媒作用ドメインを完全に欠如することから明らかである。
(配列番号3)
MSRKLFASILIGALLGIGAPPSAHAGADDVVDSSKSFVMENFSSYHGTKPGYVDSIQKGIQKPKSGTQGNYDDDWKEFYSTDNKYDAAGYSVDNENPLSGKAGGVVKVTYPGLTKVLALKVDNAETIKKELGLSLTEPLMEQVGTEEFIKRFGDGASRVVLSLPFAEGSSSVEYINNWEQAKALSVELEINFETRGKRGQDAMYEYMAQACAGNRVRRSVGSSLSCINLDWDVIRDKTKTKIESLKEHGPIKNKMSESPNKTVSEEKAKQYLEEFHQTALEHPELSELKTVTGTNPVFAGANYAAWAVNVAQVIDSETADNLEKTTAALSILPGIGSVMGIADGAVHHNTEEIVAQSIALSSLMVAQAIPLVGELVDIGFAAYNFVESIINLFQVVHNSYNRPAYSPGHKTQPFLHDGYAVSWNTVEDSIIRTGFQGESGHDIKITAENTPLPIAGVLLPTIPGKLDVNKSKTHISVNGRKIRMRCRAIDGDVTFCRPKSPVYVGNGVHANLHVAFHRSSSEKIHSNEISSDSIGVLGYQKTVDHTKVNSKLSLFFEIKS
(配列番号4)
GTGAGCAGAAAACTGTTTGCGTCAATCTTAATAGGGGCGCTACTGGGGATAGGGGCCCCACCTTCAGCCCATGCAGGCGCTGATGATGTTGTTGATTCTTCTAAATCTTTTGTGATGGAAAACTTTTCTTCGTACCACGGGACTAAACCTGGTTATGTAGATTCCATTCAAAAAGGTATACAAAAGCCAAAATCTGGTACACAAGGAAATTATGACGATGATTGGAAAGAGTTTTATAGTACCGACAATAAATACGACGCTGCGGGATACTCTGTAGATAATGAAAACCCGCTCTCTGGAAAAGCTGGAGGCGTGGTCAAAGTGACGTATCCAGGACTGACGAAGGTTCTCGCACTAAAAGTGGATAATGCCGAAACTATTAAGAAAGAGTTAGGTTTAAGTCTCACTGAACCGTTGATGGAGCAAGTCGGAACGGAAGAGTTTATCAAAAGGTTCGGTGATGGTGCTTCGCGTGTAGTGCTCAGCCTTCCCTTCGCTGAGGGGAGTTCTAGCGTTGAATATATTAATAACTGGGAACAGGCGAAAGCGTTAAGCGTAGAACTTGAGATTAATTTTGAAACCCGTGGAAAACGTGGCCAAGATGCGATGTATGAGTATATGGCTCAAGCCTGTGCAGGAAATCGTGTCAGGCGATCAGTAGGTAGCTCATTGTCATGCATAAATCTTGATTGGGATGTCATAAGGGATAAAACTAAGACAAAGATAGAGTCTTTGAAAGAGCATGGCCCTATCAAAAATAAAATGAGCGAAAGTCCCAATAAAACAGTATCTGAGGAAAAAGCTAAACAATACCTAGAAGAATTTCATCAAACGGCATTAGAGCATCCTGAATTGTCAGAACTTAAAACCGTTACTGGGACCAATCCTGTATTCGCTGGGGCTAACTATGCGGCGTGGGCAGTAAACGTTGCGCAAGTTATCGATAGCGAAACAGCTGATAATTTGGAAAAGACAACTGCTGCTCTTTCGATACTTCCTGGTATCGGTAGCGTAATGGGCATTGCAGACGGTGCCGTTCACCACAATACAGAAGAGATAGTGGCACAATCAATAGCTTTATCGTCTTTAATGGTTGCTCAAGCTATTCCATTGGTAGGAGAGCTAGTTGATATTGGTTTCGCTGCATATAATTTTGTAGAGAGTATTATCAATTTATTTCAAGTAGTTCATAATTCGTATAATCGTCCCGCGTATTCTCCGGGGCATAAAACGCAACCATTTCTTCATGACGGGTATGCTGTCAGTTGGAACACTGTTGAAGATTCGATAATCCGAACTGGTTTTCAAGGGGAGAGTGGGCACGACATAAAAATTACTGCTGAAAATACCCCGCTTCCAATCGCGGGTGTCCTACTACCGACTATTCCTGGAAAGCTGGACGTTAATAAGTCCAAGACTCATATTTCCGTAAATGGTCGGAAAATAAGGATGCGTTGCAGAGCTATAGACGGTGATGTAACTTTTTGTCGCCCTAAATCTCCTGTTTATGTTGGTAATGGTGTGCATGCGAATCTTCACGTGGCATTTCACAGAAGCAGCTCGGAGAAAATTCATTCTAATGAAATTTCGTCGGATTCCATAGGCGTTCTTGGGTACCAGAAAACAGTAGATCACACCAAGGTTAATTCTAAGCTATCGCTATTTTTTGAAATCAAAAGC
TGA
【0043】
なお、レセプター結合ドメインを含む断片は、プラスミッドに組み込まれたCRM197をコードする遺伝子(Pβ197)を鋳型にして、PCR法にてレセプター結合ドメイン部分のDNA配列を合成して、これをGST融合タンパク質やヒスチジンタグを合成させるための発現ベクター(pGEX−3X、pQE−30)のマルチクローニングサイトに挿入し、得られたプラスミッドを大腸菌に組み込み、プラスミッドがコードする遺伝子を大腸菌で合成させることで作成できる。
【0044】
また、触媒作用ドメインに変異を有する変異体は、以下のようにして作成することができる。プラスミッドに組み込まれたCRM197をコードする遺伝子(Pβ197)を鋳型にして、変異を持たせたい部位をプライマーとして、PCR法にてCRM197領域を合成する。プライマーは、変異を持つように点突然変異を導入したものを合成し、使用する。合成したDNAを、大腸菌用の遺伝子発現ベクター(pET−22b)に組み込み、大腸菌に形質導入を行い、変異体を大腸菌で発現させて、作成することができる。
【0045】
本発明の制癌剤は、乳癌、前立腺癌、甲状腺癌、卵巣癌等、悪性腫瘍全般の治療に用いることができるが、好ましくはHB−EGFを発現している悪性腫瘍に用いることができ、特に卵巣癌に好適に用いることができる。
本発明の制癌剤は、前記ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、かつ、前記ジフテリア毒素の変異体の細胞への結合を抑制しない前記ジフテリア毒素の変異体のモノクローナル抗体をさらに含み、
前記ジフテリア毒素の変異体のモノクローナル抗体と、ジフテリア毒素の変異体とが複合体を形成することも好ましい。
【0046】
前記ジフテリア毒素の変異体は、変異を導入することによりジフテリア毒素の毒性が弱毒化されており、実質的にジフテリア毒素の毒性を有しないものであるが、極めて厳格なレベルで見ればジフテリア毒素の毒性が残存している場合を含むことは前述のとおりである。そこで、ジフテリア毒素の変異体は、該ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、かつ、該ジフテリア毒素の変異体の細胞への結合を抑制しないモノクローナル抗体と複合体を形成することによってさらにジフテリア毒素の毒性を低下させることができる。
【0047】
前記ジフテリア毒素の変異体のモノクローナル抗体であって、前記ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、ジフテリア毒素の変異体の細胞への結合を抑制しない(即ち、proHB−EGFとの結合活性を阻害しない)モノクローナル抗体は、以下の文献(Hayakawa S, J. Biol. Chem. 258, 4311−4317, 1983) によって示されるような常法に従い作成することができる。すなわち、ジフテリア毒素の変異体に対する抗体を作成しているクローンの中で、最終的にジフテリア毒素の毒性を中和する活性は持つが、ジフテリア毒素の変異体の細胞への結合を抑制しない抗体を作成しているクローンを分離して作成できる。また、公知のジフテリア毒素モノクローナル抗体の中から、使用するジフテリア毒素変異体に対して前記性質を有するものを選択することもできる。
【0048】
ジフテリア毒素の毒性を中和する活性は、ジフテリア毒素の変異体と該ジフテリア毒素モノクローナル抗体との複合体をVero−H細胞に加えて、ジフテリア毒素の変異体を単体で加えたときに起きるコロニー形成の抑制が、阻害されるかどうかを見ることにより容易に測定できる。
【0049】
一方、該モノクローナル抗体が、ジフテリア毒素の変異体の細胞への結合を抑制しないことは、前記125Iで標識したジフテリア毒素のHB―EGFへの結合の阻害や、後述するIL−3依存的に増殖能を示す32D cells(ATCCより入手)に上皮細胞増殖因子受容体遺伝子を発現させた、DER cellにおいて、HB−EGFの増殖活性阻害作用を測定すること等により調べることができる。
【0050】
ジフテリア毒素の変異体に残存するジフテリア毒素の毒性を中和する活性を有し、ジフテリア毒素の変異体の細胞への結合を抑制しないジフテリア毒素の変異体のモノクローナル抗体としては、例えば#2 anti−DT mAb(特許生物寄託センター 寄託番号:FERM P−19551のハイブリドーマより産生されるモノクローナル抗体)が挙げられる。
【0051】
ジフテリア毒素の変異体との複合体は、ジフテリア毒素の変異体とモノクローナル抗体とを適当な比により混合することにより作成できる。
【0052】
本発明の制癌剤は、上記有効成分をそのまま、または、薬学的に許容される医薬用担体と組合せて製剤化することができる。
【0053】
前記制癌剤は、経口的又は非経口的(例えば、静脈内、筋肉内、腹腔内、皮下又は皮内等への注射、直腸内投与、経粘膜投与、経気道投与など)に投与することができる。卵巣癌等腹腔内に播種する悪性腫瘍に適用する場合には、腹腔内に注射により投与することが、癌細胞に直接運搬される点で好ましい。
【0054】
経口投与に適する医薬組成物としては、例えば、錠剤、顆粒剤、カプセル剤、散剤、溶液剤、懸濁剤、シロップ剤などを挙げることができ、非経口投与に適する医薬組成物としては、例えば、注射剤、点滴剤、坐剤、経皮吸収剤などを挙げることができるが、剤形はこれらに限定されることはない。
【0055】
前記制癌剤の製造に用いられる製剤用添加物の種類は特に限定されず、当業者が適宜選択可能である。例えば、賦形剤、崩壊剤又は崩壊補助剤、結合剤、滑沢剤、コーティング剤、基剤、溶解剤又は溶解補助剤、分散剤、懸濁剤、乳化剤、緩衝剤、抗酸化剤、防腐剤、等張化剤、pH調節剤、溶解剤、安定化剤などを用いることができ、これらの目的で使用される個々の具体的成分は当業者に周知されている。
【0056】
経口投与用の製剤の調製に用いることができる製剤用添加物として、例えば、ブドウ糖、乳糖、D−マンニトール、デンプン、又は結晶セルロース等の賦形剤;カルボキシメチルセルロース、デンプン、又はカルボキシメチルセルロースカルシウム等の崩壊剤又は崩壊補助剤;ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、又はゼラチン等の結合剤;ステアリン酸マグネシウム又はタルク等の滑沢剤;ヒドロキシプロピルメチルセルロース、白糖、ポリエチレングリコール又は酸化チタン等のコーティング剤;ワセリン、流動パラフィン、ポリエチレングリコール、ゼラチン、カオリン、グリセリン、精製水、又はハードファット等の基剤を用いることができる。
【0057】
注射あるいは点滴用の製剤の調製に用いることができる製剤用添加物としては、注射用蒸留水、生理食塩水、プロピレングリコール等の水性あるいは用時溶解型注射剤を構成しうる溶解剤又は溶解補助剤;ブドウ糖、塩化ナトリウム、D−マンニトール、グリセリン等の等張化剤;無機酸、有機酸、無機塩基又は有機塩基等のpH調節剤等の製剤用添加物を用いることができる。
前記本発明の制癌剤に含まれる有効成分の量は、制癌剤の製剤形態または投与経路によって異なり、一概に規定することはできないが、通常、最終製剤中に約0.0001%から70%の範囲から適宜選択して決定することができる。
【0058】
本発明の制癌剤は、ヒトを含む哺乳動物に投与することができる。
【0059】
本発明の制癌剤の投与量は、患者の年齢、性別、体重、症状、及び投与経路などの条件に応じて適宜増減されるべきであるが、成人一日あたりの有効成分の量として、体重1kgあたり1μgから30mg程度の範囲であることが好ましい。上記投与量の医薬は一日一回に投与してもよいし、数回に分けて投与してもよい。また、数日から数週間に1度又は単発的に投与してもよい。また、ステロイド等、副作用を抑える成分とともに投与することもできる。
【実施例】
【0060】
以下、本発明の実施例について説明するが、本発明はこの実施例に何ら限定されるものではない。
(実施例1)
CRM197タンパク質の生成
C7(β197)の溶原菌のストック(C7(β197)ファージを溶原化したジフテリア菌C7(beta197)M1としてATCC(American Type Culture Collection)Bacteria collection(No.39255)から入手可能)を培養し、対数増殖期の後期の菌液を、2%の濾過したマルトースを加えたC−Y培地に、最初のOD590が約0.05になるように加える。このODは、約5×107菌体/mlにあたる。フラスコは毎分200回転のロータリーシェーカーに載せ、35℃で16時間から17時間培養する。ODが10から15になった時点で、培養を終了する。
【0061】
前記C−Y培地は、以下のように調製する。即ち、10gのカサミノ酸、20gの酵母抽出液、5gのKH2PO4を1lの蒸留水に溶かす。2mlの50%CaCl2.2H2Oを加えた後、pHを7.4に調整する。沸騰させた後、濾過する。2mlのMuellerとMillerの溶液II(22.5g MgSO4、0.115g βアラニン、0.115g ニコチン酸、7.5mg ピメリン酸、1g CuSO4・5H2O、1g ZnSO4・5H2O、1g MnCl2・4H2O/100ml H2O)と1mlのMuellerとMillerの溶液III(20g L−シスチン、20ml 濃塩酸/100ml H2O)を加える。100mLずつ分注し、オートクレーブをかけることによりC−Y培地を得た。
【0062】
CRMタンパクの精製は、以下のようにして行う。
【0063】
培養液は、10000gで15分間遠心する。培養上清に硫安を65%の飽和状態になるように加える。氷室で24から48時間放置する。沈殿物を集めて0.02MのpH7.2のトリス−塩酸バッファーに溶かし、同液にて透析する。
【0064】
遠心して、不溶物を除き、DE52カラムに添加し、0.02MのpH7.2のトリス−塩酸バッファー中のNaClの濃度勾配により溶出する。CRM197は、NaClが0.08Mのところで溶出する。溶出した液を硫安で65%飽和状態にする。沈殿物を0.01Mのトリス−塩酸バッファーに溶かし、再び平衡化する。そして、DE52カラムに再び掛け、再度硫安沈殿する。続いて、SephacrylS−200のカラムにかけ、HEPES−NaOH、pH7.2、0.15M NaClの溶液で溶出する。溶出されたCRM197をDeToxiゲルにアプライして、CRM197試料に含まれるLPS類似物質を取り除き、実験に使用する。CRM197の280nmにおける吸収は、1ODが約0.67 mg/mlに相当する。
【0065】
(実施例2)
ヌードマウスを用いた造腫瘍性実験を行った。
【0066】
RPMI+10%FBSで培養した卵巣癌細胞株SKOV−3(ATCCから入手可)を、EDTA/PBS(−)で洗浄し、0.25%トリプシンで回収した。RPMI+10%FBSで2回、RPMI(血清なし)で2回洗浄し、5×106細胞ずつRPMI(血清あり)250μLへ添加し、これをヌードマウスの背部に皮下注射により接種した。
【0067】
一群のヌードマウスについては、SKOV−3細胞接種1週間後よりCRM197の投与を開始し、腹腔内に200μg/日で週5回を4週に渡って投与した(図7)。また、別の群のヌードマウスについては、SKOV−3細胞接種1週間後よりCRM197の投与を開始し、接種した腫瘍に直接100μg/日で週5回を4週に渡って投与した(図8)。さらに、別の群のヌードマウスについては、SKOV−3細胞接種20日後よりCRM197の投与を開始し、接種した腫瘍に直接100μg/日で週5回を3週に渡って投与した(図9)。いずれの場合もCRM197を投与しないヌードマウスを対照実験とした。投与時期と腫瘍体積の関係を図7から図9に示す。ここで、腫瘍体積は、できた腫瘍の長径、短径を3〜4日ごとに測定し、長径×短径×短径×1/2で求めた。
【0068】
これらの結果から、いずれの場合も、CRM197の投与により、腫瘍の成長が抑えられることが分かった。
【0069】
(実施例3)
卵巣癌細胞株RMG−1(Japanese Collection of Research Bioresources Cell Bankに登録)、卵巣癌細胞株OV47、及び、卵巣癌細胞株SKOV−3細胞にHB−EGF遺伝子をトランスフェクションして作製した細胞であるSKOV−Hを、前記実施例2におけるSKOV−3細胞と同様に回収し、SKOV−3細胞の代わりにヌードマウス背部に皮下注射により接種した。
【0070】
いずれの細胞を接種した場合にも、細胞接種直後と一週間後にCRM197を腹腔内に1mg投与した。また、それぞれCRM197を投与しないヌードマウスを対照実験とした。
【0071】
投与時期と腫瘍体積の関係を図10から図12に示す。図10はRMG−1細胞、図11はOV47細胞、図12はSKOV−H細胞を接種したヌードマウスの場合の結果を示す。
【0072】
これらの結果からも、いずれの場合も、CRM197の投与により、腫瘍の成長が抑えられることが分かった。
【0073】
(実施例4)
CRM197の細胞毒性試験
これまでの実験では、CRM197のフラグメントAにはADPリボース転移酵素活性が認められず、全く無毒の分子であると考えられてきたが、全く毒性がないか、わずかながらもジフテリア毒素としての毒性を有しているかを明らかにすることは、本発明において極めて重大であるので、CRM197のADPリボース転移酵素活性について、さらに詳しく解析した。
【0074】
まず、CRM197の細胞毒性を調べるためにVero細胞(ATCCから入手可能)、及びVero細胞にHB−EGFを高発現させたVero−H細胞(OriGene Technologies社製ヒトHB−EGFcDNAをpCDNA3.1プラスミド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成した細胞)に種々の濃度のCRM197を加えて1週間培養し、コロニー形成率を調べた。
【0075】
図13はVero細胞及びVero−H細胞を6wellプレートに300cell/wellの密度で蒔き、10時間培養し、これにCRM197を加えて24時間培養し、その後CRM197を含まない培養液で6日間培養してコロニー数をカウントした場合のコロニー形成率を示している。図14はVero細胞及びVero−H細胞にCRM197を加えて一週間培養した場合のコロニー形成率を示している。その結果、Vero−H細胞は、CRM197を加えて24時間培養した場合には1μg/mlのCRM197で、CRM197存在下で一週間培養した場合には100ng/mlのCRM197で、コロニー数の減少が確認された。Vero細胞ではどちらの条件においても有意なコロニー数の減少は認められなかった。CRM197がVero細胞よりもVero−H細胞に対して強い毒性を示したことから、この毒性がジフテリア毒素リセプター(proHB−EGF)に関係したものであることが示唆された。なお、CRM197の代わりにジフテリア毒素を24時間作用させ、その後ジフテリア毒素を含まない培養液で6日間培養した場合、ジフテリア毒素は約1fg/mlの濃度でコロニーの出現をほとんど見なかった(毒素を加えない場合に得られるコロニー数の約3%)。したがって、ここで用いたコロニー形成法で測定されるCRM197の細胞毒性はジフテリア毒素の1010以下であり、CRM197の細胞毒性は極めて微弱なものであることがわかった。
【0076】
(実施例5)
CRM197によるタンパク合成阻害試験
ジフテリア毒素の毒素作用は、EF2のADPリボシル化に基づく、タンパク合成阻害作用である。そこで、CRM197の毒性がタンパク質合成阻害によるものかどうかを検討するために、CRM197によるタンパク合成阻害作用を調べた。実施例4で使用したのと同様のVero細胞、及びVero−H細胞にCRM197を36時間暴露させ、そのタンパク質合成阻害能を[3H]Leuの蛋白への取り込み率から調べた。具体的には、24wellプレートにVero細胞、Vero−H細胞を1×105cell/mlの密度で蒔き、16時間培養後、CRM197を加えて、さらに36時間培養した。その後、[3H]Leuを添加し、1時間インキュベートしたのち、タンパク質に取り込まれた[3H]Leuの放射能量を液体シンチレーションカウンターにて測定し、タンパク質の合成阻害能を求めた。
【0077】
その結果、Vero−H細胞では100 ng/ml以上の濃度のCRM197でタンパク質合成の阻害が観察された。この条件でVero細胞ではタンパク合成阻害作用はほとんど観察されなかった(図15)。
【0078】
(実施例6)
DT52E148Kの細胞毒性試験
ジフテリアトキシンに2カ所の変異を持つ変異体であるDT52E148K及びリコンビナント蛋白GST−DTについても実施例4と同様にその毒性をコロニー形成法で測定した。その結果、CRM197よりもさらに微弱ではあるが、DT52E148KはVero−H細胞対して毒性を示した(図16)。一方、Vero細胞には細胞毒性は認められなかった(図17)。したがって、完全に毒性を消失させるためには、この2カ所の変異だけでは不十分であることが示された。GST−DTはVero細胞に対してもVero−H細胞に対しても、全く毒性を示さなかった。
【0079】
(実施例7)
Vdtr細胞のCRM197及びDT52E148Kに対する耐性
これまで示されたCRM197及びDT52E148Kの細胞毒性、タンパク質合成阻害作用がジフテリア毒素の持つADPリボシル化活性によるEF−2の不活性化であるのかどうかを検討するために、ジフテリア毒素耐性株Vdtr細胞を作成した。具体的には以下の文献(Moehring JM and Moehring TJ, Somat. Cell Genet. 5, 453−468, 1979;
Kohno K et al., Somat. Cell Genet. 11, 421−423, 1985)に従って、Vero細胞をEMS処理した後、ジフテリア毒素を加えて培養し、生存する細胞を得た。これらの中から、高濃度のジフテリア毒素を加えても耐性である細胞を選別し、Vdtr細胞を得た。次ぎに、このVdtr細胞にHB−EGFを高発現させ、Vdtr−4H細胞を得た(OriGene Technologies社製ヒト由来HB−EGF遺伝子をpCDNA3.1プラスミッド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成した細胞)。Vdtr−4H細胞にジフテリア毒素を加えたときの蛋白合成阻害率を図18に、この細胞より得た細胞破砕液にフラグメントAを加えたcell−freeのADPリボシル化アッセイの結果を図19に示す。この細胞ではcell−freeのADPリボシル化アッセイにおいてもEF2のADPリボシル化が全く認められないことから、この細胞ではジフテリア毒素耐性はEF2に原因があることが示された。
【0080】
CRM197の持つ細胞毒性がADPリボシル化活性によるものかどうかを確認するために、このVdtr細胞にHB−EGFを高発現させたVdtr−4H細胞に対する、CRM197の毒性を検討した。Vdtr−4H細胞にCRM197を加え、実施例4と同様にコロニー形成法によってその毒性を検討した。その結果、100μg/mlのCRM197を加えてもVdtr−4H細胞に全く毒性を示さなかった(図20)。また、DT52E148Kについても同様に全く毒性を示さなかった(図20)。この結果からCRM197の示す細胞毒性、及びタンパク合成阻害はフラグメントAのADPリボシルトランスフェラーゼ活性によるものであることが明らかとなった。
【0081】
(実施例8)
無細胞系でのEF−2のADPリボシル化実験
実施例7のVdtr細胞を用いた実験結果は、CRM197及びDT52E148Kに存在する細胞毒性が、残存するADPリボシルトランスフェラーゼ活性によるものであることを示している。これをさらに確認するために、Cell−freeの条件下でADPリボシル化実験を行った。以下の文献Gill, DM and Pappenheimer, AM Jr. J. Biol. Chem. 246, 1492−1495,1971) に示された方法を用いて、ウサギ肝より抽出したEF−2にCRM197又はDT52E148Kを加え、これに[32P]NADを加えて、37℃で10分間インキュベートし、cell−freeでADPリボシル化反応を行った。その後、液体シンチレーションカウンターにて放射能量を測定した。その結果、極めて弱い活性であるが、CRM197及びDT52E148KにEF−2をADPリボシル化する活性が認められた(図21及び図22)。なお、図21におけるの右上の図は、CRM197のADPリボシル化活性を縦軸の尺度を拡大して示したものである。この結果から、CRM197及びDT52E148Kの両者にEF2をADPリボシル化する活性がわずかではあるが残存することが結論づけられた。
【0082】
(実施例9)
ジフテリア毒素モノクローナル抗体#2 anti−DT mAb(diphtheria toxin monoclonal antibodies ♯2)によるCRM197の細胞毒性の中和
CRM197をHB−EGF増殖活性阻害物質として利用する場合、CRM197に微弱ではあるが細胞毒性があることは、場合によっては望ましくない。そこで、次にCRM197に残る毒性を抑制する条件を検討した。ジフテリア毒素に対するモノクローナル抗体は、多数分離されている(Hayakawa S, J. Biol. Chem. 258, 4311−4317, 1983)。これらの中には、ジフテリア毒素の細胞毒性は抑制するが、ジフテリア毒素の受容体への結合は抑制しない抗体がある。これらの中で、#2 anti−DT mAbが、ジフテリア毒素の場合と同様にCRM197の毒性は中和するが、CRM197のHB−EGFへの結合は抑制しないことを見出した。
【0083】
CRM197と同時に#2 anti−DT mAb(特許生物寄託センター 受託番号:FERM P−19551のハイブリドーマより産生されるモノクローナル抗体)を加え、CRM197の毒性が中和されるかを検討した。
【0084】
ジフテリア毒素に対するモノクローナル抗体の作成は、以下の文献(Hayakawa S,J. Biol. Chem. 258, 4311−4317, 1983) によって示されるが、簡単に記述すると以下のようになる。BALB/cマウスの腹腔に、ホルマリン処理したジフテリア毒素0.1 mgを、フロインドアジュバントと共に接種し、これを1週間ごとに合計3回行った。最後の接種から数日後に、このマウスの脾臓細胞を取り出し、これをマウスミエローマ細胞SP2/0細胞と融合させた。融合反応後の細胞をHAT選択培地で培養し、増殖細胞の中から、ジフテリア毒素に対する抗体を作成しているクローンを分離した。ジフテリア毒素に対する抗体を作成しているクローンの中で、最終的にジフテリア毒素の毒性を中和する活性は持つが、ジフテリア毒素の細胞への結合を抑制しない抗体を作成しているクローンを分離した。
【0085】
CRM197/#2 anti−DT mAb 複合体は、まず、CRM197(1mg)と#2 anti−DT mAb(10mg)を混ぜ、37℃で1時間インキュベートして、作成した。Vero−H細胞にこの複合体を種々の濃度で加えて1週間培養し、コロニー形成率を調べた。その結果、たとえば、CRM197単独の場合は、100ng/mlのCRM197で死滅するが、CRM197/#2 anti−DT mAb 複合体の場合、最大量10μg/mLでもコロニーの形成は全く抑制されず、#2 anti−DT mAbがCRM197の細胞毒性を完全に阻害することがわかった(図23)。
【0086】
(実施例10)
CRM197/#2 anti−DT mAb 複合体のHB−EGF増殖活性阻害作用
CRM197/#2 anti−DT mAb 複合体のHB−EGF増殖活性阻害作用(HB−EGFの増殖活性を阻害する作用)について検討した。IL−3依存的に増殖能を示す32D cells(ATCCより入手)に上皮細胞増殖因子受容体遺伝子(EGFR遺伝子、OriGene Technologies社製)を発現させて、DER cellを作成した(EGFR遺伝子を、pCDNA3.1プラスミド(インビトロゲン社製)にクローニングしたものを、トランスフェクションして作成)。この細胞は、IL−3非存在下では、HB−EGFの増殖活性により増殖する。この細胞に対し、HB−EGF存在下で、CRM197及びCRM197/#2 anti−DT mAb 複合体を添加した。細胞を48時間培養し、増殖した細胞数をMTT assayによって測定した。CRM197及びCRM197/#2 anti−DT mAb 複合体が存在しない条件ではDER cellは増殖したが、CRM197やCRM197/#2 anti−DT mAb 複合体が存在する条件では、DER cellの増殖は抑制された(図24)。CRM197とCRM197/#2 anti−DT mAb複合体の増殖活性阻害作用を比較したところ、両者にはほとんど違いはなく、同様の阻害効果を示した。すなわち、CRM197/#2 anti−DT mAb 複合体は、CRM197が持つ細胞毒性は抑制されているが、HB−EGFの増殖活性阻害作用についてはCRM197単独と同様の阻害活性を保持していることがわかった。
【0087】
(実施例11)
CRM197/#2 anti−DT mAb 複合体による腫瘍抑制効果
一群のヌードマウス(3個体づつ)について、SKOV−H細胞をそれぞれ接種し、接種から1週間後よりCRM197(1mg/1個体/week)、あるいはCRM197/#2 anti−DT mAb 複合体(CRM197 1mgを含む/week)の投与を開始し、腹腔内に週1回を4週に渡って投与した)。CRM197/#2 anti−DT mAb 複合体は1mgのCRM197と8 mgの#2 anti−DT mAbを1時間、室温でインキュベーションしたものを使用した。また、CRM197を投与しないヌードマウスを対照実験とした。CRM197、あるいはCRM197/#2 anti−DT mAb 複合体投与と腫瘍体積の関係を図25に示す。腫瘍体積の測定は実施例2と同様に行った。この実験から、CRM197/#2 anti−DT mAb 複合体は腫瘍の増殖を抑制することが明らかになったが、その効果はCRM197単独よりも弱いことが示された。
【0088】
CRM197/#2 anti−DT mAb 複合体は、すでに示した図から、CRM197の持つ微弱な細胞毒性を完全に抑えることから、CRM197単独使用よりもより高い安全性が期待できる。一方、CRM197を単独で用いると腫瘍の増殖を抑制する効果がより強い。これは、CRM197がもつHB−EGF増殖活性阻害作用に加えて、CRM197が持つ微弱な細胞毒性が加わった結果であると考えられる。したがって、CRM197の使用にあたっては、安全性をより重視する場合には#2 anti−DT mAbと複合体として投与することも可能であり、効果をより重視する場合にはCRM197単独投与も有効であることが考えられる。
【0089】
(実施例12)
DT52E148KとCRM197について、投与が週1回を3週にわたっての投与であった以外は、実施例11と同様に腫瘍抑制効果を調べた。DT52E148K又はCRM197と、腫瘍体積との関係を図26に示す。この実験から、DT52E148Kは腫瘍の増殖を抑制することが明らかになったが、その効果はCRM197よりも弱いことが示された。ADPリボース転移酵素活性を有するAフラグメントに2箇所の変異があり、細胞毒性が低い(図16)DT52E148Kに関するこの結果は、前記、CRM197においてはCRM197がもつHB−EGF増殖活性阻害作用に加えて、CRM197が持つ微弱な細胞毒性が腫瘍の増殖の抑制に効果を有しているという推定を裏付けるものである。
【0090】
(参考例)
一群のヌードマウス(10個体づつ)について、SKOV−3細胞又はSKOV−H細胞をそれぞれ接種し、接種から1週間後及び2週間後に、タキソール(Taxol)40mg/kg/weekを腹腔内に投与した。結果を図27に示す。タキソールは、SKOV−3細胞には効果があるが、SKOV−H細胞には効果が少ないことが分かった。SKOV−H細胞は、HB−EGFを多く発現する性質があることから、タキソールは本発明の制癌剤とは作用機序が異なることが分かった。したがって、本発明の制癌剤は、タキソールがあまり有効でない症例にも効果があることが示唆される。
【産業上の利用可能性】
【0091】
本発明は、卵巣癌をはじめとする各種癌の治療に有効な制癌剤の製造に利用できる。
【図面の簡単な説明】
【0092】
【図1】図1は、proHB−EGFの構造を表す模式図である。
【図2】図2は、ジフテリア毒素のアミノ酸配列及び塩基配列を表す図である。
【図3】図3は、ジフテリア毒素のアミノ酸配列及び塩基配列を表す図である(図2の続き)。
【図4】図4は、ジフテリア毒素のドメイン構造を表す模式図である。
【図5】図5は、GST−DTのアミノ酸配列及び塩基配列を表す図である。
【図6】図6は、GST−DTのアミノ酸配列及び塩基配列を表す図である(図5の続き)。
【図7】図7は、SKOV−3細胞注射ヌードマウスにおけるCRM197腹腔内注射による腫瘍増殖に対する効果を表す図である。
【図8】図8は、SKOV−3細胞注射ヌードマウスにおけるCRM197局所注射による腫瘍増殖に対する効果を表す図である。
【図9】図9は、SKOV−3細胞注射ヌードマウスにおけるCRM197局所注射による腫瘍増殖に対する効果を表す図である。
【図10】図10は、RMG−1細胞注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図11】図11は、OV47細胞注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図12】図12は、SKOV−H注射ヌードマウスにおけるCRM197注射後の癌増殖率に対する効果を表す図である。
【図13】図13は、CRM197を24時間作用させた場合のVero細胞及びVero−H細胞に対する毒性を表す図である。
【図14】図14は、CRM197を1週間作用させた場合のVero細胞及びVero−H細胞に対する毒性を表す図である。
【図15】図15は、CRM197によるタンパク合成阻害作用を表す図である。
【図16】図16は、CRM197、DT52E148K及びGST−DTのVero−H細胞に対する毒性を表す図である。
【図17】図17は、CRM197、DT52E148K及びGST−DTのVero細胞に対する毒性を表す図である。
【図18】図18は、Vdtr−4H細胞のDT耐性を表す図である。
【図19】図19は、無細胞条件におけるVdtr−4H細胞及びVero−H細胞破砕液のEF−2のADP−リボシル化反応を表す図である。
【図20】図20は、Vdtr−4H細胞のCRM197及びDT52E148Kに対する耐性を表す図である。
【図21】図21は、CRM197及びDTによるEF−2のADP−リボシル化を表す図である。
【図22】図22は、DT52E148KによるEF−2のADP−リボシル化を表す図である。
【図23】図23は、#2 anti−DT mAbによるCRM197の毒性の中和を表す図である。
【図24】図24は、CRM197/#2 anti−DT mAb複合体による、HB−EGF細胞増殖活性の阻害を表す図である。
【図25】図25は、CRM197及びCRM197/#2 anti−DT mAb複合体の腹腔内投与によるヌードマウスの癌細胞の増殖を表す図である。
【図26】図26は、CRM197及びDT52E148Kの腹腔内投与によるヌードマウスの癌細胞の増殖を表す図である。
【図27】図27は、タキソール投与の投与によるXenograftマウスの癌細胞の増殖を表す図である。
【特許請求の範囲】
【請求項1】
以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする制癌剤。
(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質
(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質
(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質
【請求項2】
(a)、(b)及び(c)のいずれかのタンパク質が、ジフテリア毒素の触媒作用ドメインを有しない請求項1に記載の制癌剤。
【請求項3】
(c)のタンパク質が、GST−DTである請求項2に記載の制癌剤。
【請求項1】
以下の(a)、(b)及び(c)のいずれかのタンパク質であって、HB−EGFのEGFレセプターへの結合を阻害する活性を有しかつジフテリア毒素の毒性を実質的に有さないタンパク質を有効成分として含むことを特徴とする制癌剤。
(a) ジフテリア毒素の一部からなり、該ジフテリア毒素のレセプター結合ドメインを少なくとも含むタンパク質
(b) (a)のタンパク質のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質
(c) (a)及び(b)のいずれかのタンパク質を含む複合タンパク質
【請求項2】
(a)、(b)及び(c)のいずれかのタンパク質が、ジフテリア毒素の触媒作用ドメインを有しない請求項1に記載の制癌剤。
【請求項3】
(c)のタンパク質が、GST−DTである請求項2に記載の制癌剤。
【図1】



【図2】

【図3】

【図4】
【図5】

【図6】

【図7】

【図8】
【図9】
【図10】
【図11】

【図12】
【図13】

【図14】
【図15】

【図16】
【図17】
【図18】
【図19】
【図20】

【図21】
【図22】

【図23】
【図24】
【図25】
【図26】

【図27】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2008−285491(P2008−285491A)
【公開日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願番号】特願2008−143242(P2008−143242)
【出願日】平成20年5月30日(2008.5.30)
【分割の表示】特願2003−355731(P2003−355731)の分割
【原出願日】平成15年10月15日(2003.10.15)
【出願人】(000173692)財団法人阪大微生物病研究会 (23)
【Fターム(参考)】
【公開日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願日】平成20年5月30日(2008.5.30)
【分割の表示】特願2003−355731(P2003−355731)の分割
【原出願日】平成15年10月15日(2003.10.15)
【出願人】(000173692)財団法人阪大微生物病研究会 (23)
【Fターム(参考)】
[ Back to top ]