
樹脂複合材料及び樹脂複合材料の製造方法
【課題】グラフェン構造を有する炭素材料が合成樹脂中に分散されている樹脂複合材料であって、機械的強度が高く、線膨張率が低い樹脂複合材料及び上記樹脂複合材料を製造する方法を提供する。
【解決手段】合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である樹脂複合材料、及び合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、上記工程と同時にまたは上記工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程とを備える樹脂複合材料の製造方法。
【解決手段】合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である樹脂複合材料、及び合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、上記工程と同時にまたは上記工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程とを備える樹脂複合材料の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、炭素材料が合成樹脂中に分散されてなる樹脂複合材料の製造方法及び樹脂複合材料に関し、特に、炭素材料がグラフェン構造を有する炭素材料である、樹脂複合材料の製造方法及び樹脂複合材料に関する。
【背景技術】
【0002】
高い弾性率や高い導電性を有することから、グラフェンシート構造を有する炭素材料が注目されてきている。このようなグラフェン構造を有する炭素材料を、合成樹脂に複合することにより、合成樹脂からなる製品を補強したり、導電性を付与したりすることができる。特に、グラフェンシート、カーボンナノチューブまたは薄膜化グラファイトなどは、ナノサイズの寸法を有し、かつ比表面積が大きい。そのため、樹脂に複合させた場合の補強効果が高いと考えられている。
【0003】
一般に、複合材料としての効果を高めるには、上記炭素材料をマトリクス樹脂に均一に分散させることが好ましい。そこで、下記の特許文献1には、熱可塑性樹脂と炭素材料との良溶媒を用いて、均一な分散状態を得る方法が開示されている。この方法によれば、熱可塑性樹脂と炭素材料とに共通溶媒が存在する限りにおいて、均一な分散状態の樹脂複合材料を得ることが可能である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−264059号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、特許文献1に記載の樹脂複合材料では、溶媒を揮発させた後に形成される熱可塑性樹脂と炭素材料との界面の密着強度が低かった。そのため、得られた樹脂複合材料に外力が作用した場合、上記界面で剥離が生じることがあった。加えて、上記のような炭素材料は、πスタッキング力による凝集力が大きいため、溶媒中の分散性が極めて悪かった。従って、充分な機械的強度を発現しないという問題があった。
【0006】
本発明の目的は、グラフェン構造を有する炭素材料が合成樹脂中に分散されている樹脂複合材料であって、機械的強度が高く、線膨張率が低い樹脂複合材料及び上記樹脂複合材料を製造する方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明の樹脂複合材料は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である。
【0008】
本発明の樹脂複合材料のある特定の局面では、上記樹脂複合材料が、上記合成樹脂と異なる種類の樹脂をさらに含んでいる。その場合には、様々な物性の樹脂複合材料を容易に提供することができる。
【0009】
本発明の樹脂複合材料の他の特定の局面では、上記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである。その場合には、上記炭素材料はナノサイズを有し、かつ比表面積が大きい。そのため、樹脂複合材料の機械的強度をさらに高めること及び線膨張率をさらに低めることができる。
【0010】
本発明の樹脂複合材料の別の特定の局面では、上記合成樹脂が熱可塑性樹脂である。その場合には、樹脂複合材料の成形が容易であるため、様々な形状の樹脂複合材料製品を容易に提供することができる。より好ましくは、熱可塑性樹脂としてポリオレフィンが用いられる。この場合には、汎用されているポリオレフィンを用いているので、樹脂複合材料のコストを低減することができる。
【0011】
本発明の樹脂複合材料のさらに他の特定の局面では、上記熱可塑性樹脂が非晶性樹脂である。その場合には、上記炭素材料が非晶性樹脂中に均一に分散されているので、非晶性樹脂の流動性を効果的に抑制することができる。従って、樹脂複合材料の機械的強度を効果的に高めることができ、かつ線膨張率が効果的に低めることができる。
【0012】
本発明の樹脂複合材料の製造方法は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、上記工程と同時にまたは上記工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程とを備える。
【0013】
本発明の樹脂複合材料の製造方法のある特定の局面では、上記グラフト化させる工程が、上記樹脂組成物に電子線を照射することにより行われる。その場合には、上記樹脂組成物に電子線を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0014】
本発明の樹脂複合材料の製造方法の他の特定の局面では、上記グラフト化させる工程が、上記合成樹脂及び上記炭素材料に、ラジカル開始剤を混合することにより行われる。その場合には、上記合成樹脂及び上記炭素材料にラジカル開始剤を混合することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0015】
本発明の樹脂複合材料の製造方法の別の特定の局面では、上記グラフト化させる工程が、上記合成樹脂及び上記炭素材料を、混練用スクリューを備えるせん断混練装置により加熱混練することにより行われる。さらに、上記加熱混練の際に、上記スクリューを500rpm〜5000rpmの回転速度で回転させる。その場合には、グラフェン構造を有する炭素材料を加熱混練する過程で、上記合成樹脂の分子鎖が切断され、上記合成樹脂内にフリーラジカルが生じる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0016】
好ましくは、上記加熱混練は動的な加熱混練により行われる。その場合には、炭素材料と合成樹脂との間の親和性がより高められるため、上記炭素材料を上記合成樹脂中に容易に均一分散させることができる。
【0017】
本発明の樹脂複合材料の製造方法のさらに他の特定の局面では、上記グラフト化させる工程が、上記樹脂組成物にマイクロ波を照射することにより行われる。上記樹脂組成物にマイクロ波を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0018】
上記のようなマイクロ波を照射することによるグラフト化は、フリーラジカルを発生させる工程が簡便である。さらに、上記炭素材料との界面のみにおいて上記合成樹脂の分子が切断されるため、上記合成樹脂の分子が過度に切断されない。そのため、得られる樹脂複合材料の機械的強度を効率的に高めること及び線膨張率を効率的に低めることができる。
【0019】
本発明の樹脂複合材料の製造方法のさらに別の特定の局面では、上記グラフト化させる工程において、上記炭素材料のグラフト化率が5重量%〜3300重量%の範囲となるように上記合成樹脂を上記炭素材料にグラフト化させる。その場合には、上記合成樹脂と上記炭素材料との密着性がより一層高められる。そのため、上記炭素材料を上記合成樹脂中により一層均一に分散することができる。従って、機械的強度がさらに高められ、線膨張率がさらに低められた樹脂複合材料を得ることができる。
【0020】
本発明の樹脂複合材料の製造方法のまた他の特定の局面では、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を上記樹脂組成物中より分離する分離工程と、上記工程の後に、上記合成樹脂と同種または異なる新たな合成樹脂と、上記合成樹脂がグラフト化している上記炭素材料とを混合する工程とをさらに含む。その場合には、上記新たな合成樹脂を混合することにより、様々な物性の樹脂複合材料を容易に製造することができる。
【0021】
本発明の樹脂複合材料の製造方法のまた他の特定の局面では、上記グラフト化させる工程の後に、上記合成樹脂と同種または異なる樹脂を混合する工程をさらに備える。その場合には、様々な物性の樹脂複合材料を容易に製造することができる。
【0022】
本発明の樹脂複合材料の製造方法のまた別の特定の局面では、上記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである。その場合には、上記炭素材料はナノサイズを有し、かつ比表面積が大きい。そのため、機械的強度がさらに高められた樹脂複合材料を容易に製造することができる。
【0023】
本発明の樹脂複合材料の製造方法のさらに他の特定の局面では、上記合成樹脂が熱可塑性樹脂である。その場合には、樹脂複合材料の成形が容易であるため、様々な形状の樹脂複合材料製品を容易に製造することができる。より好ましくは、熱可塑性樹脂としてポリオレフィンが用いられる。その場合には、汎用されているポリオレフィンを用いているので、樹脂複合材料の製造コストを低減することができる。
【0024】
本発明の樹脂複合材料の製造方法のさらに別の特定の局面では、上記熱可塑性樹脂が非晶性樹脂である。その場合には、上記炭素材料が非晶性樹脂中に均一に分散されているので、非晶性樹脂の流動性を効果的に抑制することができる。従って、機械的強度が効果的に高められ、線膨張率が効果的に低められた樹脂複合材料を得ることができる。
【発明の効果】
【0025】
本発明に係る樹脂複合材料によれば、グラフェン構造を有する炭素材料に合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%であるため、樹脂複合材料中において上記炭素材料と上記合成樹脂との密着性が高められている。さらに、上記合成樹脂によりグラフト化された上記炭素材料は、上記合成樹脂との親和性が高められているため、上記合成樹脂中に均一に分散されている。そのため、上記炭素材料は、上記合成樹脂中に均一に分散されている。従って、機械的強度が高められ、線膨張率が低められた樹脂複合材料を提供することができる。
【0026】
また、本発明に係る樹脂複合材料の製造方法では、グラフェン構造を有する炭素材料に合成樹脂をグラフト化させるため、上記炭素材料と上記合成樹脂との間に化学結合が形成される。それによって、上記炭素材料と上記合成樹脂との密着性が高められる。加えて、上記合成樹脂によりグラフト化された上記炭素材料は、上記合成樹脂との親和性が高められているため、上記炭素材料を上記合成樹脂中に均一に分散することができる。従って、機械的強度が高められ、線膨張率が低められた樹脂複合材料を得ることができる。
【0027】
さらに、本発明に係る樹脂複合材料の製造方法においては、グラフェン構造を有する炭素材料を用いているため、合成樹脂をグラフト化させる際に生じるフリーラジカルが、上記炭素材料のグラフェン構造により非局在化される。そのため、上記炭素材料と上記合成樹脂との間において選択的な反応が進行する。従って、炭素材料に合成樹脂を効率的にグラフト化することができる。よって、本発明の製造方法によれば、機械的強度が高められ、線膨張率が低められた樹脂複合材料を効率的に得ることができる。
【図面の簡単な説明】
【0028】
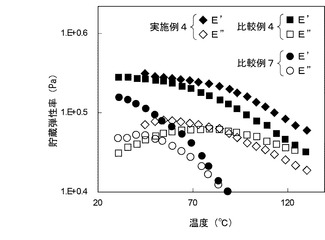
【図1】図1は、実施例4並びに比較例4及び7により得られた樹脂複合材料シートの粘弾性測定において、樹脂複合材料シートの温度と貯蔵弾性率(E’)及び損失弾性率(E’’)を示す図である。
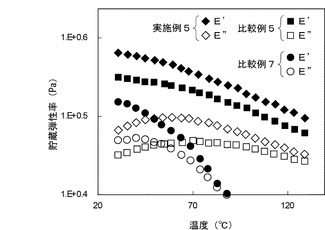
【図2】図2は、実施例5並びに比較例5及び7により得られた樹脂複合材料シートの粘弾性測定において、樹脂複合材料シートの温度と貯蔵弾性率(E’)及び損失弾性率(E’’)を示す図である。
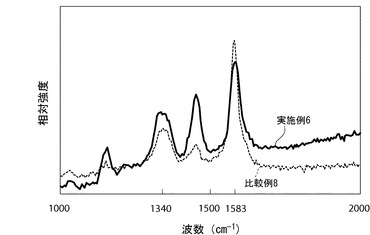
【図3】図3は、実施例6、比較例8によって得られた樹脂複合材料シートの顕微ラマン測定によるラマンスペクトルを示す図である。
【発明を実施するための形態】
【0029】
以下、本発明の詳細を説明する。
【0030】
<樹脂複合材料>
本発明の樹脂複合材料は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む。本発明の樹脂複合材料においては、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である。
【0031】
(合成樹脂)
本発明の樹脂複合材料に含まれる上記合成樹脂は特に限定されず、様々な公知の合成樹脂を用いることができる。好ましくは、上記合成樹脂として熱可塑性樹脂が用いられる。 熱可塑性樹脂を用いた樹脂複合材料では、加熱下により様々な成形方法を用いて、様々な成形品を容易に得ることができる。上記熱可塑性樹脂としては、高密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレンなどのポリエチレン類や、ホモポリプロピレン、ブロックポリプロピレン、ランダムポリプロピレンなどのポリプロピレン類に代表されるポリオレフィン、ノルボルネン樹脂等の環状ポリオレフィン類、ポリ酢酸ビニル、エチレン酢酸ビニル等の酢酸ビニル共重合体類や、ポリビニルアルコール、ポリビニルブチラール等のポリ酢酸ビニル誘導体類、PET、ポリカーボネート、ポリ乳酸等のポリエステル類、ポリエチレンオキサイド、ポリフェニレンエーテル、ポリエーテルエーテルケトン等のポリエーテル樹脂類、PMMA等のアクリル系樹脂類、ポリスルホン、ポリエーテルスルホン等のスルホン系樹脂類、PTFE、PVDF等のフッ素化樹脂類、ナイロン等のポリアミド樹脂類、ポリ塩化ビニル、塩化ビニリデン等のハロゲン化樹脂類、ポリスチレン、ポリアクリロニトリルやそれらの共重合樹脂類などを挙げることができる。上記合成樹脂は1種のみが用いられてもよく、2種以上が併用されてもよい。特に好ましくは、安価であり、加熱下の成形が容易であるポリオレフィンが望ましい。
【0032】
さらに、上記合成樹脂としては、結晶性樹脂を用いてもよく、非晶性樹脂を用いてもよい。上記合成樹脂として非晶性樹脂を用いた場合には、上記炭素材料を上記非晶性樹脂に分散させることにより、上記非晶性樹脂の流動性を効果的に抑制することができる。上記非晶性樹脂は特に限定されず、適宜の非晶性樹脂を用いることができる。上記非晶性樹脂としては、例えば、アタクチックポリプロピレン、非晶性ノルボルネン、非晶性PET、非晶性ポリカーボネート、ポリフェニレンエーテル、ポリエーテルエーテルケトン、アタクチックPMMA、ポリスルホン、ポリエーテルスルホン、及びアタクチックポリスチレン等が挙げられる。より好ましくは、安価なアタクチックポリプロピレンを用いることができる。
【0033】
(グラフェン構造を有する炭素材料)
本発明の樹脂複合材料では、上記グラフェン構造を有する炭素材料が、上記合成樹脂に分散されている。それによって、本発明の樹脂複合材料の機械的強度を高めること及び線膨張係数を低めることができる。さらに、場合によっては、本発明の樹脂複合材料に導電性を発現させることもできる。そのため、本発明の樹脂複合材料は、導電性を発現する材料としても用いることができる可能性を有する。
【0034】
加えて、上記炭素材料には、上記合成樹脂がグラフト化している。そのため、本発明の樹脂複合材料では、上記合成樹脂と上記炭素材料との密着性がより一層高められている。さらに、上記合成樹脂がグラフト化した上記炭素材料は、上記合成樹脂との親和性が高められている。そのため、本発明の樹脂複合材料では、上記グラフト化された炭素材料が上記合成樹脂中に均一に分散されている。従って、本発明の樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0035】
上記グラフェン構造を有する炭素材料としては特に限定されないが、好ましくは、薄片化黒鉛またはカーボンナノチューブを用いることができる。より好ましくは、上記炭素材料としては、複数のグラフェンシートの積層体、すなわち薄片化黒鉛が用いられる。本発明において、薄片化黒鉛とは、元の黒鉛を剥離処理して得られるものであり、元の黒鉛よりも薄いグラフェンシート積層体をいう。薄片化黒鉛におけるグラフェンシート積層数は、元の黒鉛より少なければよいが、通常数層〜200層程度である。
【0036】
上記薄片化黒鉛は、比表面積が比較的大きい形状を有する。従って、本発明の樹脂複合材料では、上記薄片化黒鉛が分散されているため、上記薄片化黒鉛のグラフェンシート積層面に交差する方向に加わる外力に対する機械的強度を効果的に高めることができる。なお、本発明において比表面積とは、BET3点法により測定されたBET比表面積をいう。
【0037】
上記薄片化黒鉛のBET比表面積の好ましい下限は15m2/gであり、好ましい上限は2700m2/gである。上記薄片化黒鉛の比表面積が15m2/gより低いと、上記積層面に交差する方向に加わった外力に対する機械的強度が充分に高められないことがある。一方で、単層グラフェンシートの理論BET比表面積は2700m2/gであり、限界値である。
【0038】
上記炭素材料と上記合成樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記炭素材料の配合量を1〜50重量部の範囲とすることが好ましい。上記炭素材料の配合量が1重量部未満では、機械的強度が充分に高められないこと及び線膨張係数が充分に高められないことがある。上記炭素材料の配合量が50重量部を超えると、樹脂複合材料の剛性が高められるものの、樹脂複合材料が脆くなり、割れやすくなることがある。
【0039】
本発明の樹脂複合材料では、上記炭素材料のグラフト化率が5重量%〜3300重量%の範囲である。本発明において炭素材料のグラフト化率とは、樹脂複合材料内に含まれる上記炭素材料の重量に対する、樹脂複合材料内において上記炭素材料にグラフト化により直接化学結合を形成している合成樹脂の重量の比率をいう。上記炭素材料のグラフト化率を上記範囲とすることにより、本発明の樹脂複合材料の機械的強度を効果的に高めることができ、線膨張係数を効果的に低めることができる。
【0040】
上記炭素材料のグラフト化率が5重量%より低いと、上記合成樹脂と上記炭素材料との密着性が充分に高められないことがある。そのため、樹脂複合材料の機械的強度を充分に高めること及び線膨張係数を充分に低めることができないことがある。上記炭素材料のグラフト化率が3300重量%より高いと、効果が飽和してそれ以上機械的強度が高められないこと及び線膨張係数が低められないことがある。好ましくは、上記炭素材料のグラフト化率は10重量%〜2000重量%の範囲であり、さらに好ましくは30重量%〜1000重量%の範囲である。
【0041】
なお、樹脂複合材料に含まれる炭素材料のグラフト化率は、以下の方法により測定することができる。例えば、樹脂複合材料に含まれるグラフト化していない合成樹脂を溶媒により溶解除去して、グラフト化された炭素材料を単離する。その後、空気雰囲気下、30〜600℃の温度範囲において、10℃/分の昇温速度により上記グラフト化された炭素材料を熱重量測定(TGA測定)を行う。このとき、500℃に昇温されるまでに分解した分解物の量をA重量%、500℃まで昇温しても分解しなかった未分解物の量をB重量%として、下記の式により上記炭素材料のグラフト化率を求めることができる。
【0042】
グラフト化率(重量%)=A/B×100
【0043】
上記溶媒としては、上記グラフト化していない合成樹脂を溶解し、上記グラフト化された炭素材料をほとんど溶解しない限り特に限定されず、適宜の溶媒を用いることができる。例えば、上記合成樹脂がオレフィン系樹脂の場合には130℃熱キシレンなど、PMMA等のアクリル系樹脂の場合にはアセトンまたはジクロロベンゼンなど、ナイロン等のポリアミド系樹脂の場合には200℃熱ベンジルアルコールまたは200℃熱ニトロベンゼンなど、ポリスチレン系樹脂の場合にはTHFまたはジクロロベンゼンなど、ポリカーボネート系樹脂の場合にはTHFまたはジクロロメタンなどを用いることができる。
【0044】
(追加の樹脂)
本発明の樹脂複合材料では、上記合成樹脂と異なる種類の追加の樹脂をさらに含んでいてもよい。その場合には、樹脂複合材料が上記追加の樹脂を含むことにより、様々な物性の樹脂複合材料を容易に提供することができる。上記追加の樹脂は、上記炭素材料にグラフト化していてもよく、グラフト化していなくてもよい。
【0045】
上記追加の樹脂は特に限定されず、様々な熱可塑性樹脂や熱硬化性樹脂を用いることができる。熱可塑性樹脂としては、例えば、先に述べた上記合成樹脂として用いることのできる種々の熱可塑性樹脂などが挙げられる。熱硬化性樹脂としては、例えば、エポキシ樹脂、ポリウレタン樹脂などが挙げられる。また、上記追加の樹脂は結晶性樹脂であってもよく、先に述べたような非晶性樹脂であってもよい。もっとも、一般に結晶性樹脂は非晶性樹脂に比べて弾性率等の機械物性や成形加工性に優れるため、上記追加の樹脂は結晶性樹脂であることが好ましい。
【0046】
上記合成樹脂と上記追加の樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記追加の樹脂の配合量を1000重量部以下とすることが好ましい。上記追加の樹脂の配合量が1000重量部を超えると、樹脂複合材料に含まれる上記炭素材料による機械的強度の向上及び線膨張率の低下効果を充分に発揮できないことがある。
【0047】
(他の成分)
本発明の樹脂複合材料においては、本発明の目的を阻害しない範囲で、様々な添加剤を含んでいてもよい。このような添加剤としては、フェノール系、リン系、アミン系もしくはイオウ系等の酸化防止剤;金属害防止剤;ヘキサブロモビフェニルエーテルもしくはデカブロモジフェニルエーテル等のハロゲン化難燃剤;ポリリン酸アンモニウムもしくはトリメチルフォスフェート等の難燃剤;各種充填剤;帯電防止剤;安定剤;顔料等を挙げることができる。
【0048】
また、本発明の樹脂複合材料においては、ラジカル反応を促進するのに一般的に用いられている適宜の反応助剤を含んでいてもよい。このような反応助剤は、本発明の樹脂複合材料を製造する際において、上記合成樹脂の上記炭素材料へのグラフト化反応を促進するために用いられることがある。上記反応助剤としては、例えば、ジビニルベンゼン、トリメチロールプロパントリメタクリレート、1,9−ノナンジオールジメタクリレート、1,10−デカンジオールジメタクリレート、トリメリット酸トリアリルエステル、トリアリルイソシアヌレート、エチルビニルベンゼン、ネオペンチルグリコールジメタクリレート、1,6−ヘキサンジオールジメタクリレート、ラウリルメタクリレート、ステアリルメタクリレート、フタル酸ジアリル、テレフタル酸ジアリル、イソフタル酸ジアリル等を挙げることができる。
【0049】
<樹脂複合材料の製造方法>
次に、本発明の樹脂複合材料の製造方法について説明する。
まず、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程を行う。
【0050】
上記合成樹脂及び上記グラフェン構造を有する炭素材料は、本発明の樹脂複合材料の説明において先に述べた合成樹脂及びグラフェン構造を有する炭素材料を使用することができる。もっとも、本発明の樹脂複合材料の製造方法では、後述する上記炭素材料に上記合成樹脂をグラフト化させる工程において、上記合成樹脂にフリーラジカルが生じる。それによって、フリーラジカルを有する上記合成樹脂を上記炭素材料にグラフト化させる。そのため、上記合成樹脂としてはラジカルが生じやすいものがより好適であり、特にポリオレフィンが望ましい。
【0051】
上記樹脂組成物を用意する方法としては、例えば、合成樹脂とグラフェン構造を有する炭素材料とを混合し、上記炭素材料を上記合成樹脂中に分散させる方法が挙げられる。上記混合方法は特に限定されないが、例えば、上記合成樹脂と上記炭素材料とを溶融混練する方法や、上記合成樹脂と上記炭素材料とを溶媒中に溶解または分散させる方法等が挙げられる。
【0052】
上記混合方法が上記合成樹脂と上記炭素材料とを溶融混練する方法である場合には、上記溶融混練は、例えば、プラストミル、単軸押出機、二軸押出機、バンバリーミキサー、ロールなどの適宜の混練装置を用いて行うことができる。
【0053】
上記混合方法が上記合成樹脂と上記炭素材料とを溶媒中に溶解または分散する方法である場合には、上記溶媒は、上記合成樹脂と上記炭素材料を溶解または分散させることができる限り、特に限定されない。上記溶媒としては、例えば、ジクロロベンゼン、N−メチル−2−ピロリドン、DMF及び高級アルコール類等が挙げられる。
【0054】
上記樹脂組成物中における上記炭素材料と上記合成樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記炭素材料の配合量を1〜50重量部の範囲とすることが好ましい。上記炭素材料の配合量が1重量部未満では、機械的強度が充分に高められないこと及び線膨張係数が充分に高められないことがある。上記炭素材料の配合量が50重量部を超えると、樹脂複合材料の剛性が高められるものの、樹脂複合材料が脆くなり、割れやすくなることがある。
【0055】
上記樹脂組成物は、先に述べたラジカル反応を促進するのに一般的に用いられている適宜の反応助剤を、必要に応じてさらに含んでいてもよい。反応助剤の添加により、後述する上記炭素材料に上記合成樹脂をグラフト化させる工程において、効率的にグラフト反応を引き起こさせることができる。また、グラフト化反応にともなう問題、すなわち分子鎖の過度の切断等による樹脂劣化を抑制することもできる。
【0056】
上記反応助剤としては、好ましくは、多官能の化合物を用いることができる。また、上記反応助剤は1種のみが用いられてもよく、2種以上の反応助剤が併用されてもよい。
【0057】
上記反応助剤の添加量が少ないと、上記フリーラジカルが充分に発生せず、上記合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。従って、上記反応助剤は、上記合成樹脂100重量部に対し、0.1重量部以上配合されることが好ましく、より好ましくは0.2重量部以上である。
【0058】
上記反応助剤の添加量が多すぎると、上記反応助剤の重合物が多く生成してしまうことがある、そのため、得られる樹脂複合材料の外観性が低下することがある。従って、上記反応助剤は、上記合成樹脂100重量部に対し、10重量部以下配合されることが好ましく、より好ましくは8重量部以下である。
【0059】
また、上記樹脂組成物は、先に述べた様々な添加剤を含んでいてもよい。それによって、得られる樹脂複合材料に様々な性質を付与することができる。
【0060】
上記反応助剤及び/または上記添加剤を含む上記樹脂組成物を用意する方法としては、先に述べたような溶融混練する方法や、溶媒中に溶解または分散する方法などの混合方法が挙げられる。上記反応助剤及び/または上記添加剤は、上記炭素材料と上記合成樹脂とを混合させる際において添加してもよく、別の時に添加してもよい。
【0061】
次に、上記樹脂組成物を用意する工程と同時にまたは上記樹脂組成物を用意する工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程を行う。上記工程では、合成樹脂にラジカルを発生させる目的で一般的に用いられる適宜の方法により、上記合成樹脂にフリーラジカルを発生させる。一方、グラフェン構造を有する炭素材料は、フリーラジカルを非局在化することにより、フリーラジカルを吸着しやすいという性質を有する。このため、上記フリーラジカルは、上記炭素材料に吸着され、上記炭素材料と上記合成樹脂とがラジカルグラフト反応により強固に結合する。従って、上記樹脂混合物中において、上記合成樹脂が上記炭素材料の表面にグラフト化した炭素材料が生成される。
【0062】
上記工程においては、上記炭素材料に上記合成樹脂をグラフト化させることにより、上記炭素材料のグラフト化率を5重量%〜3300重量%の範囲とすることが好ましい。本発明において炭素材料のグラフト化率とは、樹脂複合材料内に含まれる上記炭素材料の重量に対する、樹脂複合材料内において上記炭素材料にグラフト化により直接化学結合を形成している合成樹脂の重量の比率をいう。上記炭素材料のグラフト化率を上記範囲とすることにより、本発明の製造方法により得られる樹脂複合材料の機械的強度を効果的に高めることができ、線膨張係数を効果的に低めることができる。
【0063】
上記炭素材料のグラフト化率が5重量%より低いと、上記合成樹脂と上記炭素材料との密着性が充分に高められないことがある。そのため、得られる樹脂複合材料の機械的強度を充分に高めること及び線膨張係数を充分に低めることができないことがある。上記炭素材料のグラフト化率が3300重量%より高いと、効果が飽和してそれ以上機械的強度が高められないこと及び線膨張係数が低められないことがある。好ましくは、上記炭素材料のグラフト化率は10重量%〜2000重量%の範囲であり、さらに好ましくは30重量%〜1000重量%の範囲である。
【0064】
上記炭素材料に上記合成樹脂をグラフト化させる方法としては、例えば、上記樹脂組成物に電子線を照射する方法が挙げられる。電子線を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0065】
上記電子線としては、ラジカルを生じさせる目的で一般的に用いられている種類のものであれば特に限定されず、例えば、α線、β線、γ線などの電離性放射線の他、様々な電子線を用いることができる。電子線の照射方法については特に限定されず、公知の電子線照射装置を用いる方法により行うことができる。
【0066】
上記電子線の照射量は特に限定されないが、0.01〜10Mradの範囲とすることが好ましい。0.01Mrad未満だと、上記フリーラジカルが充分に発生せず、上記合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。10Mradを超えると、上記フリーラジカルによる上記合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。より好ましくは、上記電子線の照射量は0.02〜5Mradの範囲である。
【0067】
また、上記炭素材料に上記合成樹脂をグラフト化させる他の方法としては、例えば、ラジカル開始剤をさらに混合する方法が挙げられる。ラジカル開始剤を混合することにより、上記合成樹脂内にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0068】
上記ラジカル開始剤の混合では、上記樹脂組成物を用意する工程と同時に、例えば、上記合成樹脂と上記炭素材料とを混合する際において上記ラジカル開始剤を添加してもよい。また、上記樹脂組成物を用意する工程の後に、上記樹脂組成物と上記ラジカル開始剤とを混合してもよい。上記混合方法は、先に述べた混合方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0069】
上記ラジカル開始剤としては、ラジカルを生じさせる目的で一般的に用いられる適宜の開始剤を用いることができる。このようなラジカル開始剤としてはベンゾイルパーオキサイド、ジクミルパーオキサイド等の過酸化物、過酸化物化合物、アゾ系化合物、ジハロゲン系化合物等が挙げられる。上記ラジカル開始剤は1種のみが用いられてもよく、2種以上が併用されてもよい。
【0070】
上記ラジカル開始剤の配合割合は特に限定されないが、上記合成樹脂100重量部に対し、0.1重量部以上とすることが好ましい。上記ラジカル開始剤の配合割合が0.1重量部未満だと、上記フリーラジカルが充分に発生せず、十分なグラフト化反応を引き起こさせることができないことがある。より好ましくは、上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、0.5重量部以上である。
【0071】
上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、10重量部以下とすることが好ましい。上記ラジカル開始剤の配合割合が10重量部を超えると、上記フリーラジカルによる上記合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。また、反応時の発熱による爆発等の危険性が増大することがある。より好ましくは、上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、8重量部以下である。
【0072】
また、上記炭素材料に上記合成樹脂をグラフト化させる別の方法としては、例えば、上記樹脂組成物を用意する工程において、上記合成樹脂と上記炭素材料とを、高せん断により混練する方法が挙げられる。上記方法では、高せん断混練によって上記合成樹脂の分子が切断される。それによって、切断された上記合成樹脂にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0073】
上記高せん断混練は、高速回転スクリューを備える高せん断混練装置により行うことができる。上記高せん断装置のスクリュー回転数は、好ましくは500〜5000rpmの範囲である。500rpm未満であると、上記フリーラジカルが充分に発生せず、上記非晶性合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。5000rpmを超えると、上記非晶性合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。
【0074】
上記高せん断混練は、好ましくは、動的な加熱混練により行われる。それによって、炭素材料と合成樹脂との間の親和性がより高められる。従って、上記炭素材料を上記合成樹脂中に容易に均一分散させることができる。
【0075】
上記高せん断装置としては特に限定されないが、好ましくは、回転によるせん断効率の優れた二軸押出機が用いられる。また、上記高せん断混練装置の高速回転スクリューによるせん断では、スクリュー回転数に反比例して混練時間が減少する。このため、上記高速回転スクリューとしては、充分な混練時間を担保することができる長尺のスクリュー長をもつ押出装置や、スクリュー先端に達した試料を再びスクリュー後部に戻すことで任意の混練時間を設定できる内部還流式スクリュー等を用いることが好ましい。
【0076】
また、上記炭素材料に上記合成樹脂をグラフト化させるさらに他の方法としては、例えば、上記樹脂組成物にマイクロ波を照射する方法が挙げられる。上記マイクロ波の照射方法については特に限定されず、電子レンジ等の公知のマイクロ波照射装置を用いる方法により行うことができる。
【0077】
この方法により上記合成樹脂が上記炭素材料にグラフト化する理由については定かではないが、以下の理由が考えられる。上記グラフェン構造を有する炭素材料は導電性を有する。そのため、上記炭素材料を含む上記樹脂組成物にマイクロ波を照射すると、上記炭素材料内部の電子が励起され、上記樹脂組成物内部に電位差が生じる。それによって、上記樹脂組成物内において上記炭素材料が放電し、電子を放出する。このとき放出される電子は、大型加速器により放出される電子線と同様に、上記合成樹脂に照射されることによってフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0078】
あるいは、上記理由としては以下のようにも考えられる。マイクロ波がもたらす電磁誘導により、上記炭素材料が非常に高温にまで加熱される。それによって、上記樹脂組成物内において上記炭素材料との界面に存在する上記合成樹脂の分子が切断されることにより、フリーラジカルが発生する。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0079】
上記方法では、マイクロ波を照射するだけでよいため、フリーラジカルを発生させる工程が簡便である。さらに、上記炭素材料との界面のみにおいて上記合成樹脂の分子が切断されるため、上記合成樹脂の分子が過度に切断されない。そのため、得られる樹脂複合材料の機械的強度を効率的に高めること及び線膨張率を効率的に低めることができる。
【0080】
上記マイクロ波を照射する際の照射電力は特に限定されないが、10W〜1000Wの範囲とすることが好ましい。10W未満だと、上記フリーラジカルが充分に発生せず、上記非晶性合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。1000Wを超えると、上記フリーラジカルによる上記合成樹脂の分子が過剰に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。より好ましくは、上記照射電力は50W〜700Wの範囲である。
【0081】
上記炭素材料に上記合成樹脂をグラフト化させる工程により、上記炭素材料に上記合成樹脂がグラフト化した炭素材料と、上記炭素材料にグラフト化していない未反応の上記合成樹脂とを含む上記樹脂組成物を得ることができる。このようにして得られた上記樹脂組成物を、本発明の製造方法により得られる、上記未反応の合成樹脂をマトリクス樹脂とした樹脂複合材料とすることができる。
【0082】
上記樹脂複合材料は、上記グラフェン構造を有する炭素材料の表面に上記合成樹脂がグラフト化している。そのため、本発明の樹脂複合材料では、上記合成樹脂と上記炭素材料との密着性がより一層高められている。さらに、上記グラフト化された炭素材料は、上記合成樹脂との親和性が高められている。そのため、上記合成樹脂を備える上記樹脂複合材料中では、上記合成樹脂中に上記グラフト化された炭素材料が均一に分散されている。従って、上記樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0083】
さらに、本発明の樹脂複合材料の製造方法においては、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物から、上記グラフト化された炭素材料を分離した後、分離したグラフト化された炭素材料と新たな合成樹脂とを混合することにより、上記新たな合成樹脂をマトリクス樹脂とする新たな樹脂複合材料を得ることもできる。上記グラフト化させる工程に用いられる上記合成樹脂に代えて、上記新たな合成樹脂をマトリクス樹脂として用いることにより、様々な物性の樹脂複合材料を容易に製造することができる。
【0084】
上記樹脂組成物から上記グラフト化された炭素材料を分離する方法は、特に限定されない。上記分離方法としては、例えば、上記樹脂組成物に含まれるグラフト化していない合成樹脂を溶媒により溶解除去して、グラフト化された炭素材料を単離する方法等が挙げられる。上記溶媒としては、上記グラフト化していない合成樹脂を溶解し、上記グラフト化された炭素材料をほとんど溶解しない限り特に限定されず、適宜の溶媒を用いることができる。例えば、上記合成樹脂がオレフィン系樹脂の場合には130℃熱キシレンなど、PMMA等のアクリル系樹脂の場合にはアセトンまたはジクロロベンゼンなど、ナイロン等のポリアミド系樹脂の場合には200℃熱ベンジルアルコールまたは200℃熱ニトロベンゼンなど、ポリスチレン系樹脂の場合にはTHFまたはジクロロベンゼンなど、ポリカーボネート系樹脂の場合にはTHFまたはジクロロメタンなどを用いることができる。
【0085】
上記グラフト化された炭素材料は、グラフェン構造を有する炭素材料が合成樹脂により表面修飾されているため、上記新たな合成樹脂中においても親和性が高められている。そのため、上記新たな合成樹脂をマトリクス樹脂とする上記新たな樹脂複合材料においても、上記新たな合成樹脂中に上記グラフト化された炭素材料が均一に分散されている。従って、樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0086】
上記新たな合成樹脂としては特に限定されず、先に述べた熱可塑性樹脂や熱硬化性樹脂などを用いることができる。好ましくは、上記新たな合成樹脂としては熱可塑性樹脂が用いられ、さらに好ましくは安価かつ成型性に優れたポリオレフィンが用いられる。また、上記新たな合成樹脂は結晶性樹脂であってもよく、先に述べたような非晶性樹脂であってもよい。もっとも、一般に結晶性樹脂は非晶性樹脂に比べて弾性率等の機械物性や成形加工性に優れるため、上記追加の樹脂は結晶性樹脂であることが好ましい。
【0087】
上記新たな合成樹脂と、上記グラフト化させる工程に用いられる上記合成樹脂とは、同一の種類の樹脂であってもよく、異なる種類の樹脂であってもよい。また、上記新たな合成樹脂は、1種のみが用いられてもよく、2種以上が併用されてもよい。
【0088】
上記グラフト化された炭素材料と上記新たな合成樹脂との混合は、先に述べた上記炭素材料と上記合成樹脂とを混合する方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0089】
さらに、本発明の樹脂複合材料の製造方法においては、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物に、先に述べた上記追加の樹脂をさらに混合することにより、上記合成樹脂と上記追加の樹脂との両方をマトリクス樹脂とする新たな樹脂合成材料を得ることもできる。それによって、様々な物性の樹脂複合材料を容易に製造することができる。
【0090】
上記合成樹脂と上記追加の樹脂との混合方法は、先に述べた上記炭素材料と上記合成樹脂とを混合する方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0091】
上記追加の樹脂と上記合成樹脂とは、同一の種類の樹脂であってもよく、異なる種類の樹脂であってもよい。また、追加の樹脂は、1種のみが用いられてもよく、2種以上が併用されてもよい。
【0092】
上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物と上記追加の樹脂との配合割合は特に限定されないが、上記樹脂組成物100重量部に対し、上記追加の樹脂の配合量を1000重量部以下とすることが好ましい。上記追加の樹脂の配合量が1000重量部を超えると、樹脂複合材料に含まれる上記炭素材料による機械的強度の向上及び線膨張率の低下効果を充分に発揮できないことがある。
【0093】
本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料は、例えば、シート状にプレス成形することにより、引張弾性率や曲げ弾性率等の機械的強度が高く、線膨張率が低い樹脂複合材料シートを製造することができる。もっとも、本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料により製造される製品は、特に限定されない。本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料を用いることにより、様々な機械的強度が高く、線膨張率が低い製品を製造することができる。
【0094】
また、本発明の樹脂複合材料の製造方法において、上記樹脂組成物を用意する工程と上記グラフト化させる工程とを同時に行わない場合には、上記グラフト化の前に上記樹脂組成物を成形することができることがある。例えば、上述の電子線またはマイクロ波の照射によりグラフト化を行う場合には、上記グラフト化の前に上記樹脂組成物をプレス成形等によって成形した後に、電子線またはマイクロ波を照射して上記樹脂組成物のグラフト化をすることができる。
【0095】
〔実施例及び比較例〕
以下、本発明の具体的な実施例及び比較例を挙げることにより、本発明を明らかにする。なお、本発明は以下の実施例に限定されるものではない。
本発明の実施例及び比較例において使用した材料は、以下の通りである。
【0096】
(薄片化黒鉛A)
XG SCIENCE社製、商品名「XGnP−5」の薄片化黒鉛を、薄片化黒鉛Aとした。この薄片化黒鉛Aを、後述の実施例1〜3,6,13及び16〜22並びに比較例1〜3,8,15及び18〜24において用いた。
【0097】
薄片化黒鉛Aにおいては、使用前にSEMを用いて観察した層面の面方向における最大寸法が約5.0μm、層厚みが約60nm、グラフェンの積層数が約180層、BET比表面積が75m2/gであった。
【0098】
(薄片化黒鉛B)
実施例及び比較例に用いる薄片化黒鉛Bを、以下の方法で製造した。
【0099】
黒鉛単結晶粉末0.25gを65重量%の濃硫酸11.5mlに供給して、得られた混合物を10℃の水浴により冷却しながら撹拌した。次に、黒鉛単結晶粉末と濃硫酸との撹拌によって得られた混合物に、過マンガン酸カリウム1.5gを徐々に加えながら混合物を撹拌し、混合物を35℃で30分にわたって反応させた。
【0100】
次に、反応混合物に水23gを徐々に加えて、混合物を98℃で15分にわたって反応させた。しかる後、反応混合物に水70gと30重量%の過酸化水素水4.5gを加えて反応を停止させた。続いて、混合物を14000rpmの回転速度にて30分にわたって遠心分離した。その後、得られた酸化黒鉛を5重量%の希塩酸及び水により十分に洗浄して、しかる後に乾燥させた。得られた酸化黒鉛を0.2mg/mlの量にて水に分散させた後、超音波洗浄機を45kHz、100Wの条件下にて用いて、酸化黒鉛に超音波を60分にわたって照射することにより、酸化黒鉛をその層界面間において剥離断片化して、層面が酸化されてなる薄片化黒鉛を得た。得られた層面が酸化されてなる薄片化黒鉛にヒドラジンを添加して、3分間にわたって還元処理を行って、薄片化黒鉛Bの混合液を得た。
【0101】
その後、得られた薄片化黒鉛Bの混合液を濾過し、濾別された薄片化黒鉛Bを乾燥させて、薄片化黒鉛Bを得た。得られた薄片化黒鉛Bを、後述の実施例7及び比較例9において用いた。
【0102】
薄片化黒鉛Bにおいては、使用前にAFM(原子間力顕微鏡)を用いて観察した層面の面方向における最大寸法が約5.0μm、層厚みが約2nm、グラフェンの積層数が約6層、BET比表面積が450m2/gであった。
【0103】
(薄片化黒鉛Bのo−ジクロロベンゼン溶液)
上記方法と同様にして、薄片化黒鉛Bの混合液を得た。この薄片化黒鉛Bの混合液には、薄片化黒鉛Bが0.25g含まれていた。
【0104】
次に、得られた薄片化黒鉛Bの混合液に200gの水を加えた後、500gのo−ジクロロベンゼンを加え、混合溶液を得た。上記混合溶液を充分に撹拌して、薄片化黒鉛を上記混合溶液の有機層に抽出した。その後、上記混合溶液を静置することにより、上記混合溶液を黒色のo−ジクロロベンゼン層からなる下層と、無色透明の水溶液層からなる上層とに分離させた。続いて、上記o−ジクロロベンゼン層からなる下層を回収した。その後、抽出されたo−ジクロロベンゼン層に200gの水を加えて、同様の操作により再び撹拌し、o−ジクロロベンゼン層を回収することによって、上記o−ジクロロベンゼン層から不純物を除去した。このようにして、上記o−ジクロロベンゼン層からなる薄片化黒鉛Bのo−ジクロロベンゼン溶液を500g得た。得られた薄片化黒鉛Bのo−ジクロロベンゼン溶液500g中には、薄片化黒鉛Bが0.25g含まれていた。
【0105】
このようにして得られた薄片化黒鉛Bのo−ジクロロベンゼン溶液のうち250gを、後述の実施例4,8〜10,12,14,15,23及び24並びに比較例4,10〜12,14,16,17,25及び26において用いた。上記薄片化黒鉛Bのo−ジクロロベンゼン溶液250g中には、薄片化黒鉛Bが0.125g含まれていた。
(カーボンナノチューブ(CNT))
【0106】
Nanocyl社製、商標名「Nanocyl−3100」の多層カーボンナノチューブを、実施例5,11及び25並びに比較例5,13及び27において用いた。
上記カーボンナノチューブの平均長は約1.5μm、平均径は約9.5nmであった。
【0107】
(合成樹脂)
本発明の実施例及び比較例において使用した合成樹脂は、以下の通りである。
【0108】
(1)ポリプロピレン系樹脂(PP)…プライムポリマー社製、商品名「J−721GR」、23℃における引張弾性率:1.2GPa、線膨張係数:11×10−5/K
【0109】
(2)高密度ポリエチレン系樹脂(PE)…日本ポリエチレン社製、商品名「HF560」、23℃における引張弾性率:1.0GPa
【0110】
(3)シラン変性ポリプロピレン系樹脂(Silane−PP)…三井化学社製、商品名「リンクロンXPM800HM」、23℃における引張弾性率:1.1GPa、線膨張係数:14×10−5/K
【0111】
(4)ポリメチルメタクリレート系樹脂(PMMA)…住友化学社製 商品名「スミペックスES」、23℃における曲げ弾性率:2.9GPa、線膨張係数:7×10−5/K
【0112】
(5)ポリアミド系樹脂(PA)…ユニチカ社製 商品名「A−125J」、23℃における引張弾性率:1.0GPa、線膨張係数:9×10−5/K
【0113】
(6)ポリアクリロニトリル系樹脂(PAN)…三井化学社製 商品名「バレックス#1000」、23℃における引張弾性率:3.3GPa、線膨張係数:8×10−5/K
【0114】
(7)ポリスチレン系樹脂(PSt)…東洋スチレン社製「トーヨースチロールGP RMS26」、23℃における曲げ弾性率:3.2GPa
【0115】
(8)ポリカーボネート系樹脂(PC)…住化スタイロン ポリカーボネート社製 商品名「ガリバー301−15」、23℃における引張弾性率:2.3GPa、線膨張係数:7×10−5/K
【0116】
(9)アタクチックポリプロピレン系樹脂(aPP)…三菱化学社製、商品名「タフセレンT3712」、23℃における引張弾性率:40MPa
【0117】
(ラジカル開始剤)
後述の実施例6〜17において使用するラジカル開始剤としては、Sigma−Aldrich社製のベンゾイルパーオキサイドを用いた。
【0118】
<電子線照射を用いた製造方法により得られた樹脂複合材料シート>
(実施例1)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部と、反応助剤としてジビニルベンゼン3重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0119】
(実施例2)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを160℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0120】
(実施例3)
ポリカーボネート系樹脂100重量部と、薄片化黒鉛A20重量部とを290℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を3.0Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0121】
(実施例4)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にアタクチックポリプロピレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記アタクチックポリプロピレン系樹脂を溶解させて、混合溶液を得た。得られた混合溶液を室温において放冷し、濾過して、続いて真空乾燥させた。
【0122】
その後、上記混合溶液を180℃の温度でプレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に収縮を防止する目的により離型PETフィルムを両面に貼り合わせた。その後、上記シート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0123】
(実施例5)
アタクチックポリプロピレン系樹脂100重量部と、多層カーボンナノチューブ40重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に収縮を防止する目的により離型PETフィルムを両面に貼り合わせた。その後、上記シート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0124】
(比較例1〜5)
電離性放射線を照射しなかったこと以外は実施例1〜5と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0125】
(比較例6)
薄片化黒鉛Aを添加しなかったこと以外は実施例1と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。なお、電離性放射線を照射する前のシート状成型体においては、23℃における引張弾性率は1.7GPaであった。
【0126】
(比較例7)
多層カーボンナノチューブを添加しなかったこと以外は実施例5と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0127】
<ラジカル開始剤を用いた製造方法により得られた樹脂複合材料シート>
(実施例6)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して溶融混練し、ポリオレフィン系樹脂組成物を得た。
【0128】
上記ポリオレフィン樹脂組成物120重量部と、新たなポリプロピレン系樹脂100重量部とを押出機に供給して、190℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0129】
(実施例7)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛B10重量部と、ラジカル開始剤としてベンゾイルパーオキサイド3重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚みが0.5mmの樹脂複合材料シートを得た。
【0130】
(実施例8)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にポリプロピレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記ポリプロピレン系樹脂を溶解させて、混合溶液を得た。次に、上記混合溶液に、ラジカル開始剤として0.1重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを0.4重量部含む)を、窒素雰囲気下において滴下した。続いて、2時間にわたって撹拌を続けて、グラフト化反応物を得た。その後、上記反応物を室温において放冷し、濾過して、続いて真空乾燥させた。
【0131】
その後、真空乾燥させた上記反応物を180℃の温度でプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0132】
(実施例9)
0.5重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを2重量部含む)を用いたこと以外は実施例8と同様にして、樹脂複合材料シートを得た。
【0133】
上記樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、130℃に加熱された過剰量の熱キシレンに12時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していないポリプロピレン系樹脂を溶解除去した。その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、精製されたグラフト化薄片化黒鉛を回収した。
【0134】
その後、ポリプロピレン樹脂100重量部と、得られたグラフト化薄片化黒鉛5重量部とを180℃のプラストミルに供給して溶融混練し、180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。このようにして得られた樹脂複合材料シートを、実施例9の樹脂複合材料シートとした。
【0135】
(実施例10)
薄片化黒鉛Bのo−ジクロロベンゼン溶液に加えたポリプロピレン系樹脂の代わりに、アタクチックポリプロピレン系樹脂用いたこと以外は実施例9と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0136】
(実施例11)
最初に使用したポリプロピレン系樹脂の代わりにアタクチックポリプロピレン系樹脂を用いたこと、及び薄片化黒鉛A20重量部の代わりにカーボンナノチューブ40重量部を用いたこと以外は実施例6と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0137】
(実施例12)
グラフト化に用いたポリプロピレン系樹脂2.5gの代わりに高密度ポリエチレン樹脂2.5g(100重量部)を用いたこと、及び0.7重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを2.8重量部含む)を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0138】
(実施例13)
シラン変性ポリプロピレン系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド4重量部とを押出機に供給して、190℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0139】
(実施例14)
ポリプロピレン系樹脂の代わりにポリメチルメタクリレート系樹脂を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0140】
(実施例15)
ポリプロピレン系樹脂の代わりにポリスチレン系樹脂を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0141】
(実施例16)
ポリアミド系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して、270℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0142】
(実施例17)
ポリアクリロニトリル系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して、200℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0143】
(比較例8〜19)
ベンゾイルパーオキサイドを用いなかったこと以外は実施例6〜17と同様にして、厚み0.5mmの樹脂複合材料シートを得た。
【0144】
<高せん断混練を用いた製造方法により得られた樹脂複合材料シート>
(実施例18)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて200〜250℃に加熱溶融して混練(スクリュー回転数=2500rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0145】
(実施例19)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて200〜280℃に加熱溶融して混練(スクリュー回転数=1000rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0146】
(実施例20)
ポリスチレン系樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて120〜200℃に加熱溶融して混練(スクリュー回転数=4500rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0147】
(比較例20〜22)
スクリュー回転数を200rpmとしたこと以外は実施例18〜20と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0148】
<マイクロ波照射を用いた製造方法により得られた樹脂複合材料シート>
(実施例21)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部とを180℃プラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、300W電子レンジを用いてマイクロ波を60秒間照射した。続いて、上記シート状成形物を室温まで放冷し、再び300W電子レンジを用いてマイクロ波を60秒間照射した。その後、上記シート状成形物を180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0149】
(実施例22)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを180℃プラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、100W電子レンジを用いてマイクロ波を60秒間照射した。続いて、上記シート状成形物を室温まで放冷し、再び100W電子レンジを用いてマイクロ波を60秒間照射した。その後、上記シート状成形物を180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0150】
(実施例23)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にポリスチレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記ポリスチレン系樹脂系樹脂を溶解させて、混合溶液を得た。得られた混合溶液を室温において放冷し、濾過して、続いて真空乾燥させた。
【0151】
その後、上記混合溶液を160℃の温度でプレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、500W電子レンジを用いてマイクロ波を30秒間照射した。続いて上記シート状成形物を室温まで放冷した後、上記マイクロ波照射及び放冷をさらに2度繰り返した。その後、上記シート状成形物を160℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0152】
(実施例24)
実施例23と同様にして、樹脂複合材料シートを得た。この樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、過剰量のTHFに24時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していないポリスチレン系樹脂を溶解除去した。その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、精製されたグラフト化薄片化黒鉛を回収した。
【0153】
その後、ポリスチレン樹脂100重量部と、得られたグラフト化薄片化黒鉛5重量部とを160℃のプラストミルに供給して溶融混練し、プレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。このようにして得られた樹脂複合材料シートを、実施例24の樹脂複合材料シートとした。
【0154】
(実施例25)
薄片化黒鉛A20重量部の代わりに多層カーボンナノチューブ40重量部を用いたこと以外は実施例21と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0155】
(比較例23〜27)
マイクロ波を照射しなかったこと以外は実施例21〜25と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0156】
(実施例及び比較例の評価)
得られた樹脂複合材料のグラフト化率、引張弾性率、曲げ弾性率、線膨張係数、及びラマンスペクトルを、下記の方法により測定した。
【0157】
(1)グラフト化率測定
実施例及び比較例により得られた樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、過剰量の溶媒に12時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していない合成樹脂を溶解除去した。
【0158】
上記溶媒としては、用いた合成樹脂がポリプロピレン系樹脂、高密度ポリエチレン系樹脂、シラン変性ポリプロピレン系樹脂またはアタクチックポリプロピレン系樹脂の場合には、130℃熱キシレンを使用した。用いた合成樹脂がポリメチルメタクリレート系樹脂の場合にはジクロロベンゼンを使用した。用いた合成樹脂がポリアミド系樹脂の場合には200℃熱ニトロベンゼンを使用した。用いた合成樹脂がポリスチレン系樹脂の場合にはジクロロベンゼンを使用した。用いた合成樹脂がポリカーボネート系樹脂の場合にはTHFを使用した。
【0159】
その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、上記グラフト化薄片化黒鉛を単離した。
【0160】
このようにして単離されたグラフト化薄片化黒鉛を、空気雰囲気下、30〜600℃の温度範囲において、10℃/分の昇温速度により熱重量測定(TGA測定)を行った。このとき、上記グラフト化薄片化黒鉛において、500℃に昇温されるまでに分解した分解物の量をA重量%、500℃まで昇温しても分解しなかった未分解物の量をB重量%として、下記の式によりグラフト化率を求めた。結果を下記の表1〜表5に示す。
【0161】
グラフト化率(重量%)=A/B×100
【0162】
(2)引張弾性率測定
実施例及び比較例により得られた樹脂複合材料シートの23℃における引張り弾性率を、JIS K6767に準拠して測定した。結果を下記の表1〜表5に示す。
【0163】
(3)曲げ弾性率測定
実施例及び比較例により得られた樹脂複合材料シートの23℃における見かけ曲げ弾性率を、JIS K7161に準拠して測定した。結果を下記の表1〜表5に示す。
【0164】
(4)線膨脹係数測定
実施例及び比較例により得られた樹脂複合シートの−30〜100℃における線膨張係数を、JIS K7197に準拠して測定した。結果を下記の表1〜表5に示す。
【0165】
【表1】

【0166】
【表2】
【0167】
【表3】
【0168】
【表4】
【0169】
【表5】
【0170】
表1〜表5から明らかなように、各実施例の樹脂複合材料シートでは、薄片化黒鉛やカーボンナノチューブ等の炭素材料に合成樹脂がグラフト化しており、そのグラフト化率が5重量%〜3300重量%の範囲にある。さらに、各実施例の樹脂複合材料シートは、対応する各比較例の樹脂複合材料シートと比較して、引張弾性率及び/または曲げ弾性率が大幅に高められており、かつ線膨張係数が大幅に低められている。これは、合成樹脂が炭素材料にグラフト化し、樹脂と薄片化黒鉛との界面における密着性が高められていることによると考えられる。
【0171】
(5)粘弾性測定
実施例4及び5並びに比較例4,5及び7により得られた樹脂複合材料シートについて、以下のようにして粘弾性を測定した。
【0172】
動的粘弾性測定装置(TA Instruments社製、「Ares」)を用いて、昇温速度5℃/分で30℃から130℃まで昇温しながら周波数10rad/秒の条件により、得られた樹脂複合材料シートに1.0%のひずみ量を繰り返し加えた。その時の樹脂複合材料の貯蔵弾性率E’及び損失弾性率E’’を測定した。結果を図1及び図2に示した。
【0173】
図1から明らかなように、実施例4により得られた樹脂複合材料シートは、比較例4により得られた樹脂複合材料シートと比較して、高温での貯蔵弾性率(E’)が高められている。加えて、実施例4により得られた樹脂複合材料シートでは、貯蔵弾性率(E’)が損失弾性率(E”)を下回る領域が存在しない。このことは、実施例4により得られた樹脂複合材料シートでは、アタクチックポリプロピレン系樹脂が薄片化黒鉛Bにラジカルグラフト化することにより、上記樹脂複合材料シートから樹脂の流動域が消失していることによると考えられる。
【0174】
同様に、図2から明らかなように、実施例5により得られた樹脂複合材料シートは、比較例5により得られた樹脂複合材料シートと比較して、貯蔵弾性率(E’)が高められており、貯蔵弾性率(E’)が損失弾性率(E”)を下回る領域が存在しない。このことは、上記と同様に、アタクチックポリプロピレン系樹脂が多層カーボンナノチューブにラジカルグラフト化することによると考えられる。
【0175】
さらに、図1及び図2によると、比較例7により得られた樹脂複合材料シートは、比較例4及び5により得られた樹脂複合材料シートと比較して、貯蔵弾性率(E’)及び損失弾性率(E”)が大幅に低下している。これは、パーオキサイドまたは電離性放射線によって生じたフリーラジカルにより、アタクチックポリプロピレン系樹脂が劣化したことによると考えられる。一方、実施例4及び5により得られた樹脂複合材料シートでは、上記と同様の樹脂劣化が起きているにもかかわらず、高い貯蔵弾性率(E’)及び損失弾性率(E”)を示している。これは、薄片化黒鉛B及び多層カーボンナノチューブの上記ラジカルグラフト化による貯蔵弾性率(E’)及び損失弾性率(E”)の向上効果が、樹脂劣化による貯蔵弾性率(E’)及び損失弾性率(E”)の低下を上回ったことによると考えられる。
【0176】
(6)ラマンスペクトル測定
実施例6及び比較例8によって得られた樹脂複合材料シートのラマンスペクトルを、波長532nmのレーザーを用いて、露光時間1秒、スキャン回数32回により顕微ラマン測定を行った。結果を図3に示した。
【0177】
ラマンスペクトルにおける1583cm−1付近のピークは炭素材料のグラフェン構造に由来し、1340cm−1付近のピークはグラフト化反応等によるグラフェン構造の乱れに由来する。実施例6の樹脂複合材料シートのラマンスペクトルに見られる1583cm−1付近のピークの低下と、1340cm−1付近のピークの上昇は、薄片化黒鉛Aに新たな化学結合が導入されたことを示唆する結果であると考えられる。
【技術分野】
【0001】
本発明は、炭素材料が合成樹脂中に分散されてなる樹脂複合材料の製造方法及び樹脂複合材料に関し、特に、炭素材料がグラフェン構造を有する炭素材料である、樹脂複合材料の製造方法及び樹脂複合材料に関する。
【背景技術】
【0002】
高い弾性率や高い導電性を有することから、グラフェンシート構造を有する炭素材料が注目されてきている。このようなグラフェン構造を有する炭素材料を、合成樹脂に複合することにより、合成樹脂からなる製品を補強したり、導電性を付与したりすることができる。特に、グラフェンシート、カーボンナノチューブまたは薄膜化グラファイトなどは、ナノサイズの寸法を有し、かつ比表面積が大きい。そのため、樹脂に複合させた場合の補強効果が高いと考えられている。
【0003】
一般に、複合材料としての効果を高めるには、上記炭素材料をマトリクス樹脂に均一に分散させることが好ましい。そこで、下記の特許文献1には、熱可塑性樹脂と炭素材料との良溶媒を用いて、均一な分散状態を得る方法が開示されている。この方法によれば、熱可塑性樹脂と炭素材料とに共通溶媒が存在する限りにおいて、均一な分散状態の樹脂複合材料を得ることが可能である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−264059号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、特許文献1に記載の樹脂複合材料では、溶媒を揮発させた後に形成される熱可塑性樹脂と炭素材料との界面の密着強度が低かった。そのため、得られた樹脂複合材料に外力が作用した場合、上記界面で剥離が生じることがあった。加えて、上記のような炭素材料は、πスタッキング力による凝集力が大きいため、溶媒中の分散性が極めて悪かった。従って、充分な機械的強度を発現しないという問題があった。
【0006】
本発明の目的は、グラフェン構造を有する炭素材料が合成樹脂中に分散されている樹脂複合材料であって、機械的強度が高く、線膨張率が低い樹脂複合材料及び上記樹脂複合材料を製造する方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明の樹脂複合材料は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である。
【0008】
本発明の樹脂複合材料のある特定の局面では、上記樹脂複合材料が、上記合成樹脂と異なる種類の樹脂をさらに含んでいる。その場合には、様々な物性の樹脂複合材料を容易に提供することができる。
【0009】
本発明の樹脂複合材料の他の特定の局面では、上記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである。その場合には、上記炭素材料はナノサイズを有し、かつ比表面積が大きい。そのため、樹脂複合材料の機械的強度をさらに高めること及び線膨張率をさらに低めることができる。
【0010】
本発明の樹脂複合材料の別の特定の局面では、上記合成樹脂が熱可塑性樹脂である。その場合には、樹脂複合材料の成形が容易であるため、様々な形状の樹脂複合材料製品を容易に提供することができる。より好ましくは、熱可塑性樹脂としてポリオレフィンが用いられる。この場合には、汎用されているポリオレフィンを用いているので、樹脂複合材料のコストを低減することができる。
【0011】
本発明の樹脂複合材料のさらに他の特定の局面では、上記熱可塑性樹脂が非晶性樹脂である。その場合には、上記炭素材料が非晶性樹脂中に均一に分散されているので、非晶性樹脂の流動性を効果的に抑制することができる。従って、樹脂複合材料の機械的強度を効果的に高めることができ、かつ線膨張率が効果的に低めることができる。
【0012】
本発明の樹脂複合材料の製造方法は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、上記工程と同時にまたは上記工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程とを備える。
【0013】
本発明の樹脂複合材料の製造方法のある特定の局面では、上記グラフト化させる工程が、上記樹脂組成物に電子線を照射することにより行われる。その場合には、上記樹脂組成物に電子線を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0014】
本発明の樹脂複合材料の製造方法の他の特定の局面では、上記グラフト化させる工程が、上記合成樹脂及び上記炭素材料に、ラジカル開始剤を混合することにより行われる。その場合には、上記合成樹脂及び上記炭素材料にラジカル開始剤を混合することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0015】
本発明の樹脂複合材料の製造方法の別の特定の局面では、上記グラフト化させる工程が、上記合成樹脂及び上記炭素材料を、混練用スクリューを備えるせん断混練装置により加熱混練することにより行われる。さらに、上記加熱混練の際に、上記スクリューを500rpm〜5000rpmの回転速度で回転させる。その場合には、グラフェン構造を有する炭素材料を加熱混練する過程で、上記合成樹脂の分子鎖が切断され、上記合成樹脂内にフリーラジカルが生じる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0016】
好ましくは、上記加熱混練は動的な加熱混練により行われる。その場合には、炭素材料と合成樹脂との間の親和性がより高められるため、上記炭素材料を上記合成樹脂中に容易に均一分散させることができる。
【0017】
本発明の樹脂複合材料の製造方法のさらに他の特定の局面では、上記グラフト化させる工程が、上記樹脂組成物にマイクロ波を照射することにより行われる。上記樹脂組成物にマイクロ波を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。それによって、フリーラジカルを有する上記合成樹脂が上記炭素材料にグラフト化することができる。
【0018】
上記のようなマイクロ波を照射することによるグラフト化は、フリーラジカルを発生させる工程が簡便である。さらに、上記炭素材料との界面のみにおいて上記合成樹脂の分子が切断されるため、上記合成樹脂の分子が過度に切断されない。そのため、得られる樹脂複合材料の機械的強度を効率的に高めること及び線膨張率を効率的に低めることができる。
【0019】
本発明の樹脂複合材料の製造方法のさらに別の特定の局面では、上記グラフト化させる工程において、上記炭素材料のグラフト化率が5重量%〜3300重量%の範囲となるように上記合成樹脂を上記炭素材料にグラフト化させる。その場合には、上記合成樹脂と上記炭素材料との密着性がより一層高められる。そのため、上記炭素材料を上記合成樹脂中により一層均一に分散することができる。従って、機械的強度がさらに高められ、線膨張率がさらに低められた樹脂複合材料を得ることができる。
【0020】
本発明の樹脂複合材料の製造方法のまた他の特定の局面では、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を上記樹脂組成物中より分離する分離工程と、上記工程の後に、上記合成樹脂と同種または異なる新たな合成樹脂と、上記合成樹脂がグラフト化している上記炭素材料とを混合する工程とをさらに含む。その場合には、上記新たな合成樹脂を混合することにより、様々な物性の樹脂複合材料を容易に製造することができる。
【0021】
本発明の樹脂複合材料の製造方法のまた他の特定の局面では、上記グラフト化させる工程の後に、上記合成樹脂と同種または異なる樹脂を混合する工程をさらに備える。その場合には、様々な物性の樹脂複合材料を容易に製造することができる。
【0022】
本発明の樹脂複合材料の製造方法のまた別の特定の局面では、上記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである。その場合には、上記炭素材料はナノサイズを有し、かつ比表面積が大きい。そのため、機械的強度がさらに高められた樹脂複合材料を容易に製造することができる。
【0023】
本発明の樹脂複合材料の製造方法のさらに他の特定の局面では、上記合成樹脂が熱可塑性樹脂である。その場合には、樹脂複合材料の成形が容易であるため、様々な形状の樹脂複合材料製品を容易に製造することができる。より好ましくは、熱可塑性樹脂としてポリオレフィンが用いられる。その場合には、汎用されているポリオレフィンを用いているので、樹脂複合材料の製造コストを低減することができる。
【0024】
本発明の樹脂複合材料の製造方法のさらに別の特定の局面では、上記熱可塑性樹脂が非晶性樹脂である。その場合には、上記炭素材料が非晶性樹脂中に均一に分散されているので、非晶性樹脂の流動性を効果的に抑制することができる。従って、機械的強度が効果的に高められ、線膨張率が効果的に低められた樹脂複合材料を得ることができる。
【発明の効果】
【0025】
本発明に係る樹脂複合材料によれば、グラフェン構造を有する炭素材料に合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%であるため、樹脂複合材料中において上記炭素材料と上記合成樹脂との密着性が高められている。さらに、上記合成樹脂によりグラフト化された上記炭素材料は、上記合成樹脂との親和性が高められているため、上記合成樹脂中に均一に分散されている。そのため、上記炭素材料は、上記合成樹脂中に均一に分散されている。従って、機械的強度が高められ、線膨張率が低められた樹脂複合材料を提供することができる。
【0026】
また、本発明に係る樹脂複合材料の製造方法では、グラフェン構造を有する炭素材料に合成樹脂をグラフト化させるため、上記炭素材料と上記合成樹脂との間に化学結合が形成される。それによって、上記炭素材料と上記合成樹脂との密着性が高められる。加えて、上記合成樹脂によりグラフト化された上記炭素材料は、上記合成樹脂との親和性が高められているため、上記炭素材料を上記合成樹脂中に均一に分散することができる。従って、機械的強度が高められ、線膨張率が低められた樹脂複合材料を得ることができる。
【0027】
さらに、本発明に係る樹脂複合材料の製造方法においては、グラフェン構造を有する炭素材料を用いているため、合成樹脂をグラフト化させる際に生じるフリーラジカルが、上記炭素材料のグラフェン構造により非局在化される。そのため、上記炭素材料と上記合成樹脂との間において選択的な反応が進行する。従って、炭素材料に合成樹脂を効率的にグラフト化することができる。よって、本発明の製造方法によれば、機械的強度が高められ、線膨張率が低められた樹脂複合材料を効率的に得ることができる。
【図面の簡単な説明】
【0028】
【図1】図1は、実施例4並びに比較例4及び7により得られた樹脂複合材料シートの粘弾性測定において、樹脂複合材料シートの温度と貯蔵弾性率(E’)及び損失弾性率(E’’)を示す図である。
【図2】図2は、実施例5並びに比較例5及び7により得られた樹脂複合材料シートの粘弾性測定において、樹脂複合材料シートの温度と貯蔵弾性率(E’)及び損失弾性率(E’’)を示す図である。
【図3】図3は、実施例6、比較例8によって得られた樹脂複合材料シートの顕微ラマン測定によるラマンスペクトルを示す図である。
【発明を実施するための形態】
【0029】
以下、本発明の詳細を説明する。
【0030】
<樹脂複合材料>
本発明の樹脂複合材料は、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む。本発明の樹脂複合材料においては、上記炭素材料に上記合成樹脂がグラフト化しており、上記炭素材料のグラフト化率が5重量%〜3300重量%である。
【0031】
(合成樹脂)
本発明の樹脂複合材料に含まれる上記合成樹脂は特に限定されず、様々な公知の合成樹脂を用いることができる。好ましくは、上記合成樹脂として熱可塑性樹脂が用いられる。 熱可塑性樹脂を用いた樹脂複合材料では、加熱下により様々な成形方法を用いて、様々な成形品を容易に得ることができる。上記熱可塑性樹脂としては、高密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレンなどのポリエチレン類や、ホモポリプロピレン、ブロックポリプロピレン、ランダムポリプロピレンなどのポリプロピレン類に代表されるポリオレフィン、ノルボルネン樹脂等の環状ポリオレフィン類、ポリ酢酸ビニル、エチレン酢酸ビニル等の酢酸ビニル共重合体類や、ポリビニルアルコール、ポリビニルブチラール等のポリ酢酸ビニル誘導体類、PET、ポリカーボネート、ポリ乳酸等のポリエステル類、ポリエチレンオキサイド、ポリフェニレンエーテル、ポリエーテルエーテルケトン等のポリエーテル樹脂類、PMMA等のアクリル系樹脂類、ポリスルホン、ポリエーテルスルホン等のスルホン系樹脂類、PTFE、PVDF等のフッ素化樹脂類、ナイロン等のポリアミド樹脂類、ポリ塩化ビニル、塩化ビニリデン等のハロゲン化樹脂類、ポリスチレン、ポリアクリロニトリルやそれらの共重合樹脂類などを挙げることができる。上記合成樹脂は1種のみが用いられてもよく、2種以上が併用されてもよい。特に好ましくは、安価であり、加熱下の成形が容易であるポリオレフィンが望ましい。
【0032】
さらに、上記合成樹脂としては、結晶性樹脂を用いてもよく、非晶性樹脂を用いてもよい。上記合成樹脂として非晶性樹脂を用いた場合には、上記炭素材料を上記非晶性樹脂に分散させることにより、上記非晶性樹脂の流動性を効果的に抑制することができる。上記非晶性樹脂は特に限定されず、適宜の非晶性樹脂を用いることができる。上記非晶性樹脂としては、例えば、アタクチックポリプロピレン、非晶性ノルボルネン、非晶性PET、非晶性ポリカーボネート、ポリフェニレンエーテル、ポリエーテルエーテルケトン、アタクチックPMMA、ポリスルホン、ポリエーテルスルホン、及びアタクチックポリスチレン等が挙げられる。より好ましくは、安価なアタクチックポリプロピレンを用いることができる。
【0033】
(グラフェン構造を有する炭素材料)
本発明の樹脂複合材料では、上記グラフェン構造を有する炭素材料が、上記合成樹脂に分散されている。それによって、本発明の樹脂複合材料の機械的強度を高めること及び線膨張係数を低めることができる。さらに、場合によっては、本発明の樹脂複合材料に導電性を発現させることもできる。そのため、本発明の樹脂複合材料は、導電性を発現する材料としても用いることができる可能性を有する。
【0034】
加えて、上記炭素材料には、上記合成樹脂がグラフト化している。そのため、本発明の樹脂複合材料では、上記合成樹脂と上記炭素材料との密着性がより一層高められている。さらに、上記合成樹脂がグラフト化した上記炭素材料は、上記合成樹脂との親和性が高められている。そのため、本発明の樹脂複合材料では、上記グラフト化された炭素材料が上記合成樹脂中に均一に分散されている。従って、本発明の樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0035】
上記グラフェン構造を有する炭素材料としては特に限定されないが、好ましくは、薄片化黒鉛またはカーボンナノチューブを用いることができる。より好ましくは、上記炭素材料としては、複数のグラフェンシートの積層体、すなわち薄片化黒鉛が用いられる。本発明において、薄片化黒鉛とは、元の黒鉛を剥離処理して得られるものであり、元の黒鉛よりも薄いグラフェンシート積層体をいう。薄片化黒鉛におけるグラフェンシート積層数は、元の黒鉛より少なければよいが、通常数層〜200層程度である。
【0036】
上記薄片化黒鉛は、比表面積が比較的大きい形状を有する。従って、本発明の樹脂複合材料では、上記薄片化黒鉛が分散されているため、上記薄片化黒鉛のグラフェンシート積層面に交差する方向に加わる外力に対する機械的強度を効果的に高めることができる。なお、本発明において比表面積とは、BET3点法により測定されたBET比表面積をいう。
【0037】
上記薄片化黒鉛のBET比表面積の好ましい下限は15m2/gであり、好ましい上限は2700m2/gである。上記薄片化黒鉛の比表面積が15m2/gより低いと、上記積層面に交差する方向に加わった外力に対する機械的強度が充分に高められないことがある。一方で、単層グラフェンシートの理論BET比表面積は2700m2/gであり、限界値である。
【0038】
上記炭素材料と上記合成樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記炭素材料の配合量を1〜50重量部の範囲とすることが好ましい。上記炭素材料の配合量が1重量部未満では、機械的強度が充分に高められないこと及び線膨張係数が充分に高められないことがある。上記炭素材料の配合量が50重量部を超えると、樹脂複合材料の剛性が高められるものの、樹脂複合材料が脆くなり、割れやすくなることがある。
【0039】
本発明の樹脂複合材料では、上記炭素材料のグラフト化率が5重量%〜3300重量%の範囲である。本発明において炭素材料のグラフト化率とは、樹脂複合材料内に含まれる上記炭素材料の重量に対する、樹脂複合材料内において上記炭素材料にグラフト化により直接化学結合を形成している合成樹脂の重量の比率をいう。上記炭素材料のグラフト化率を上記範囲とすることにより、本発明の樹脂複合材料の機械的強度を効果的に高めることができ、線膨張係数を効果的に低めることができる。
【0040】
上記炭素材料のグラフト化率が5重量%より低いと、上記合成樹脂と上記炭素材料との密着性が充分に高められないことがある。そのため、樹脂複合材料の機械的強度を充分に高めること及び線膨張係数を充分に低めることができないことがある。上記炭素材料のグラフト化率が3300重量%より高いと、効果が飽和してそれ以上機械的強度が高められないこと及び線膨張係数が低められないことがある。好ましくは、上記炭素材料のグラフト化率は10重量%〜2000重量%の範囲であり、さらに好ましくは30重量%〜1000重量%の範囲である。
【0041】
なお、樹脂複合材料に含まれる炭素材料のグラフト化率は、以下の方法により測定することができる。例えば、樹脂複合材料に含まれるグラフト化していない合成樹脂を溶媒により溶解除去して、グラフト化された炭素材料を単離する。その後、空気雰囲気下、30〜600℃の温度範囲において、10℃/分の昇温速度により上記グラフト化された炭素材料を熱重量測定(TGA測定)を行う。このとき、500℃に昇温されるまでに分解した分解物の量をA重量%、500℃まで昇温しても分解しなかった未分解物の量をB重量%として、下記の式により上記炭素材料のグラフト化率を求めることができる。
【0042】
グラフト化率(重量%)=A/B×100
【0043】
上記溶媒としては、上記グラフト化していない合成樹脂を溶解し、上記グラフト化された炭素材料をほとんど溶解しない限り特に限定されず、適宜の溶媒を用いることができる。例えば、上記合成樹脂がオレフィン系樹脂の場合には130℃熱キシレンなど、PMMA等のアクリル系樹脂の場合にはアセトンまたはジクロロベンゼンなど、ナイロン等のポリアミド系樹脂の場合には200℃熱ベンジルアルコールまたは200℃熱ニトロベンゼンなど、ポリスチレン系樹脂の場合にはTHFまたはジクロロベンゼンなど、ポリカーボネート系樹脂の場合にはTHFまたはジクロロメタンなどを用いることができる。
【0044】
(追加の樹脂)
本発明の樹脂複合材料では、上記合成樹脂と異なる種類の追加の樹脂をさらに含んでいてもよい。その場合には、樹脂複合材料が上記追加の樹脂を含むことにより、様々な物性の樹脂複合材料を容易に提供することができる。上記追加の樹脂は、上記炭素材料にグラフト化していてもよく、グラフト化していなくてもよい。
【0045】
上記追加の樹脂は特に限定されず、様々な熱可塑性樹脂や熱硬化性樹脂を用いることができる。熱可塑性樹脂としては、例えば、先に述べた上記合成樹脂として用いることのできる種々の熱可塑性樹脂などが挙げられる。熱硬化性樹脂としては、例えば、エポキシ樹脂、ポリウレタン樹脂などが挙げられる。また、上記追加の樹脂は結晶性樹脂であってもよく、先に述べたような非晶性樹脂であってもよい。もっとも、一般に結晶性樹脂は非晶性樹脂に比べて弾性率等の機械物性や成形加工性に優れるため、上記追加の樹脂は結晶性樹脂であることが好ましい。
【0046】
上記合成樹脂と上記追加の樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記追加の樹脂の配合量を1000重量部以下とすることが好ましい。上記追加の樹脂の配合量が1000重量部を超えると、樹脂複合材料に含まれる上記炭素材料による機械的強度の向上及び線膨張率の低下効果を充分に発揮できないことがある。
【0047】
(他の成分)
本発明の樹脂複合材料においては、本発明の目的を阻害しない範囲で、様々な添加剤を含んでいてもよい。このような添加剤としては、フェノール系、リン系、アミン系もしくはイオウ系等の酸化防止剤;金属害防止剤;ヘキサブロモビフェニルエーテルもしくはデカブロモジフェニルエーテル等のハロゲン化難燃剤;ポリリン酸アンモニウムもしくはトリメチルフォスフェート等の難燃剤;各種充填剤;帯電防止剤;安定剤;顔料等を挙げることができる。
【0048】
また、本発明の樹脂複合材料においては、ラジカル反応を促進するのに一般的に用いられている適宜の反応助剤を含んでいてもよい。このような反応助剤は、本発明の樹脂複合材料を製造する際において、上記合成樹脂の上記炭素材料へのグラフト化反応を促進するために用いられることがある。上記反応助剤としては、例えば、ジビニルベンゼン、トリメチロールプロパントリメタクリレート、1,9−ノナンジオールジメタクリレート、1,10−デカンジオールジメタクリレート、トリメリット酸トリアリルエステル、トリアリルイソシアヌレート、エチルビニルベンゼン、ネオペンチルグリコールジメタクリレート、1,6−ヘキサンジオールジメタクリレート、ラウリルメタクリレート、ステアリルメタクリレート、フタル酸ジアリル、テレフタル酸ジアリル、イソフタル酸ジアリル等を挙げることができる。
【0049】
<樹脂複合材料の製造方法>
次に、本発明の樹脂複合材料の製造方法について説明する。
まず、合成樹脂と、上記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程を行う。
【0050】
上記合成樹脂及び上記グラフェン構造を有する炭素材料は、本発明の樹脂複合材料の説明において先に述べた合成樹脂及びグラフェン構造を有する炭素材料を使用することができる。もっとも、本発明の樹脂複合材料の製造方法では、後述する上記炭素材料に上記合成樹脂をグラフト化させる工程において、上記合成樹脂にフリーラジカルが生じる。それによって、フリーラジカルを有する上記合成樹脂を上記炭素材料にグラフト化させる。そのため、上記合成樹脂としてはラジカルが生じやすいものがより好適であり、特にポリオレフィンが望ましい。
【0051】
上記樹脂組成物を用意する方法としては、例えば、合成樹脂とグラフェン構造を有する炭素材料とを混合し、上記炭素材料を上記合成樹脂中に分散させる方法が挙げられる。上記混合方法は特に限定されないが、例えば、上記合成樹脂と上記炭素材料とを溶融混練する方法や、上記合成樹脂と上記炭素材料とを溶媒中に溶解または分散させる方法等が挙げられる。
【0052】
上記混合方法が上記合成樹脂と上記炭素材料とを溶融混練する方法である場合には、上記溶融混練は、例えば、プラストミル、単軸押出機、二軸押出機、バンバリーミキサー、ロールなどの適宜の混練装置を用いて行うことができる。
【0053】
上記混合方法が上記合成樹脂と上記炭素材料とを溶媒中に溶解または分散する方法である場合には、上記溶媒は、上記合成樹脂と上記炭素材料を溶解または分散させることができる限り、特に限定されない。上記溶媒としては、例えば、ジクロロベンゼン、N−メチル−2−ピロリドン、DMF及び高級アルコール類等が挙げられる。
【0054】
上記樹脂組成物中における上記炭素材料と上記合成樹脂との配合割合は特に限定されないが、上記合成樹脂100重量部に対し、上記炭素材料の配合量を1〜50重量部の範囲とすることが好ましい。上記炭素材料の配合量が1重量部未満では、機械的強度が充分に高められないこと及び線膨張係数が充分に高められないことがある。上記炭素材料の配合量が50重量部を超えると、樹脂複合材料の剛性が高められるものの、樹脂複合材料が脆くなり、割れやすくなることがある。
【0055】
上記樹脂組成物は、先に述べたラジカル反応を促進するのに一般的に用いられている適宜の反応助剤を、必要に応じてさらに含んでいてもよい。反応助剤の添加により、後述する上記炭素材料に上記合成樹脂をグラフト化させる工程において、効率的にグラフト反応を引き起こさせることができる。また、グラフト化反応にともなう問題、すなわち分子鎖の過度の切断等による樹脂劣化を抑制することもできる。
【0056】
上記反応助剤としては、好ましくは、多官能の化合物を用いることができる。また、上記反応助剤は1種のみが用いられてもよく、2種以上の反応助剤が併用されてもよい。
【0057】
上記反応助剤の添加量が少ないと、上記フリーラジカルが充分に発生せず、上記合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。従って、上記反応助剤は、上記合成樹脂100重量部に対し、0.1重量部以上配合されることが好ましく、より好ましくは0.2重量部以上である。
【0058】
上記反応助剤の添加量が多すぎると、上記反応助剤の重合物が多く生成してしまうことがある、そのため、得られる樹脂複合材料の外観性が低下することがある。従って、上記反応助剤は、上記合成樹脂100重量部に対し、10重量部以下配合されることが好ましく、より好ましくは8重量部以下である。
【0059】
また、上記樹脂組成物は、先に述べた様々な添加剤を含んでいてもよい。それによって、得られる樹脂複合材料に様々な性質を付与することができる。
【0060】
上記反応助剤及び/または上記添加剤を含む上記樹脂組成物を用意する方法としては、先に述べたような溶融混練する方法や、溶媒中に溶解または分散する方法などの混合方法が挙げられる。上記反応助剤及び/または上記添加剤は、上記炭素材料と上記合成樹脂とを混合させる際において添加してもよく、別の時に添加してもよい。
【0061】
次に、上記樹脂組成物を用意する工程と同時にまたは上記樹脂組成物を用意する工程の後に、上記炭素材料に上記合成樹脂をグラフト化させる工程を行う。上記工程では、合成樹脂にラジカルを発生させる目的で一般的に用いられる適宜の方法により、上記合成樹脂にフリーラジカルを発生させる。一方、グラフェン構造を有する炭素材料は、フリーラジカルを非局在化することにより、フリーラジカルを吸着しやすいという性質を有する。このため、上記フリーラジカルは、上記炭素材料に吸着され、上記炭素材料と上記合成樹脂とがラジカルグラフト反応により強固に結合する。従って、上記樹脂混合物中において、上記合成樹脂が上記炭素材料の表面にグラフト化した炭素材料が生成される。
【0062】
上記工程においては、上記炭素材料に上記合成樹脂をグラフト化させることにより、上記炭素材料のグラフト化率を5重量%〜3300重量%の範囲とすることが好ましい。本発明において炭素材料のグラフト化率とは、樹脂複合材料内に含まれる上記炭素材料の重量に対する、樹脂複合材料内において上記炭素材料にグラフト化により直接化学結合を形成している合成樹脂の重量の比率をいう。上記炭素材料のグラフト化率を上記範囲とすることにより、本発明の製造方法により得られる樹脂複合材料の機械的強度を効果的に高めることができ、線膨張係数を効果的に低めることができる。
【0063】
上記炭素材料のグラフト化率が5重量%より低いと、上記合成樹脂と上記炭素材料との密着性が充分に高められないことがある。そのため、得られる樹脂複合材料の機械的強度を充分に高めること及び線膨張係数を充分に低めることができないことがある。上記炭素材料のグラフト化率が3300重量%より高いと、効果が飽和してそれ以上機械的強度が高められないこと及び線膨張係数が低められないことがある。好ましくは、上記炭素材料のグラフト化率は10重量%〜2000重量%の範囲であり、さらに好ましくは30重量%〜1000重量%の範囲である。
【0064】
上記炭素材料に上記合成樹脂をグラフト化させる方法としては、例えば、上記樹脂組成物に電子線を照射する方法が挙げられる。電子線を照射することにより、上記合成樹脂内にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0065】
上記電子線としては、ラジカルを生じさせる目的で一般的に用いられている種類のものであれば特に限定されず、例えば、α線、β線、γ線などの電離性放射線の他、様々な電子線を用いることができる。電子線の照射方法については特に限定されず、公知の電子線照射装置を用いる方法により行うことができる。
【0066】
上記電子線の照射量は特に限定されないが、0.01〜10Mradの範囲とすることが好ましい。0.01Mrad未満だと、上記フリーラジカルが充分に発生せず、上記合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。10Mradを超えると、上記フリーラジカルによる上記合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。より好ましくは、上記電子線の照射量は0.02〜5Mradの範囲である。
【0067】
また、上記炭素材料に上記合成樹脂をグラフト化させる他の方法としては、例えば、ラジカル開始剤をさらに混合する方法が挙げられる。ラジカル開始剤を混合することにより、上記合成樹脂内にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0068】
上記ラジカル開始剤の混合では、上記樹脂組成物を用意する工程と同時に、例えば、上記合成樹脂と上記炭素材料とを混合する際において上記ラジカル開始剤を添加してもよい。また、上記樹脂組成物を用意する工程の後に、上記樹脂組成物と上記ラジカル開始剤とを混合してもよい。上記混合方法は、先に述べた混合方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0069】
上記ラジカル開始剤としては、ラジカルを生じさせる目的で一般的に用いられる適宜の開始剤を用いることができる。このようなラジカル開始剤としてはベンゾイルパーオキサイド、ジクミルパーオキサイド等の過酸化物、過酸化物化合物、アゾ系化合物、ジハロゲン系化合物等が挙げられる。上記ラジカル開始剤は1種のみが用いられてもよく、2種以上が併用されてもよい。
【0070】
上記ラジカル開始剤の配合割合は特に限定されないが、上記合成樹脂100重量部に対し、0.1重量部以上とすることが好ましい。上記ラジカル開始剤の配合割合が0.1重量部未満だと、上記フリーラジカルが充分に発生せず、十分なグラフト化反応を引き起こさせることができないことがある。より好ましくは、上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、0.5重量部以上である。
【0071】
上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、10重量部以下とすることが好ましい。上記ラジカル開始剤の配合割合が10重量部を超えると、上記フリーラジカルによる上記合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。また、反応時の発熱による爆発等の危険性が増大することがある。より好ましくは、上記ラジカル開始剤の配合割合は、上記合成樹脂100重量部に対し、8重量部以下である。
【0072】
また、上記炭素材料に上記合成樹脂をグラフト化させる別の方法としては、例えば、上記樹脂組成物を用意する工程において、上記合成樹脂と上記炭素材料とを、高せん断により混練する方法が挙げられる。上記方法では、高せん断混練によって上記合成樹脂の分子が切断される。それによって、切断された上記合成樹脂にフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0073】
上記高せん断混練は、高速回転スクリューを備える高せん断混練装置により行うことができる。上記高せん断装置のスクリュー回転数は、好ましくは500〜5000rpmの範囲である。500rpm未満であると、上記フリーラジカルが充分に発生せず、上記非晶性合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。5000rpmを超えると、上記非晶性合成樹脂の分子が過度に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。
【0074】
上記高せん断混練は、好ましくは、動的な加熱混練により行われる。それによって、炭素材料と合成樹脂との間の親和性がより高められる。従って、上記炭素材料を上記合成樹脂中に容易に均一分散させることができる。
【0075】
上記高せん断装置としては特に限定されないが、好ましくは、回転によるせん断効率の優れた二軸押出機が用いられる。また、上記高せん断混練装置の高速回転スクリューによるせん断では、スクリュー回転数に反比例して混練時間が減少する。このため、上記高速回転スクリューとしては、充分な混練時間を担保することができる長尺のスクリュー長をもつ押出装置や、スクリュー先端に達した試料を再びスクリュー後部に戻すことで任意の混練時間を設定できる内部還流式スクリュー等を用いることが好ましい。
【0076】
また、上記炭素材料に上記合成樹脂をグラフト化させるさらに他の方法としては、例えば、上記樹脂組成物にマイクロ波を照射する方法が挙げられる。上記マイクロ波の照射方法については特に限定されず、電子レンジ等の公知のマイクロ波照射装置を用いる方法により行うことができる。
【0077】
この方法により上記合成樹脂が上記炭素材料にグラフト化する理由については定かではないが、以下の理由が考えられる。上記グラフェン構造を有する炭素材料は導電性を有する。そのため、上記炭素材料を含む上記樹脂組成物にマイクロ波を照射すると、上記炭素材料内部の電子が励起され、上記樹脂組成物内部に電位差が生じる。それによって、上記樹脂組成物内において上記炭素材料が放電し、電子を放出する。このとき放出される電子は、大型加速器により放出される電子線と同様に、上記合成樹脂に照射されることによってフリーラジカルを発生させることができる。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0078】
あるいは、上記理由としては以下のようにも考えられる。マイクロ波がもたらす電磁誘導により、上記炭素材料が非常に高温にまで加熱される。それによって、上記樹脂組成物内において上記炭素材料との界面に存在する上記合成樹脂の分子が切断されることにより、フリーラジカルが発生する。上記フリーラジカルにより、上記合成樹脂が上記炭素材料にグラフト化することができる。
【0079】
上記方法では、マイクロ波を照射するだけでよいため、フリーラジカルを発生させる工程が簡便である。さらに、上記炭素材料との界面のみにおいて上記合成樹脂の分子が切断されるため、上記合成樹脂の分子が過度に切断されない。そのため、得られる樹脂複合材料の機械的強度を効率的に高めること及び線膨張率を効率的に低めることができる。
【0080】
上記マイクロ波を照射する際の照射電力は特に限定されないが、10W〜1000Wの範囲とすることが好ましい。10W未満だと、上記フリーラジカルが充分に発生せず、上記非晶性合成樹脂と上記炭素材料との化学結合が充分に形成されないことがある。1000Wを超えると、上記フリーラジカルによる上記合成樹脂の分子が過剰に切断されることがある。そのため、得られる樹脂複合材料の機械的強度が低下することがある。より好ましくは、上記照射電力は50W〜700Wの範囲である。
【0081】
上記炭素材料に上記合成樹脂をグラフト化させる工程により、上記炭素材料に上記合成樹脂がグラフト化した炭素材料と、上記炭素材料にグラフト化していない未反応の上記合成樹脂とを含む上記樹脂組成物を得ることができる。このようにして得られた上記樹脂組成物を、本発明の製造方法により得られる、上記未反応の合成樹脂をマトリクス樹脂とした樹脂複合材料とすることができる。
【0082】
上記樹脂複合材料は、上記グラフェン構造を有する炭素材料の表面に上記合成樹脂がグラフト化している。そのため、本発明の樹脂複合材料では、上記合成樹脂と上記炭素材料との密着性がより一層高められている。さらに、上記グラフト化された炭素材料は、上記合成樹脂との親和性が高められている。そのため、上記合成樹脂を備える上記樹脂複合材料中では、上記合成樹脂中に上記グラフト化された炭素材料が均一に分散されている。従って、上記樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0083】
さらに、本発明の樹脂複合材料の製造方法においては、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物から、上記グラフト化された炭素材料を分離した後、分離したグラフト化された炭素材料と新たな合成樹脂とを混合することにより、上記新たな合成樹脂をマトリクス樹脂とする新たな樹脂複合材料を得ることもできる。上記グラフト化させる工程に用いられる上記合成樹脂に代えて、上記新たな合成樹脂をマトリクス樹脂として用いることにより、様々な物性の樹脂複合材料を容易に製造することができる。
【0084】
上記樹脂組成物から上記グラフト化された炭素材料を分離する方法は、特に限定されない。上記分離方法としては、例えば、上記樹脂組成物に含まれるグラフト化していない合成樹脂を溶媒により溶解除去して、グラフト化された炭素材料を単離する方法等が挙げられる。上記溶媒としては、上記グラフト化していない合成樹脂を溶解し、上記グラフト化された炭素材料をほとんど溶解しない限り特に限定されず、適宜の溶媒を用いることができる。例えば、上記合成樹脂がオレフィン系樹脂の場合には130℃熱キシレンなど、PMMA等のアクリル系樹脂の場合にはアセトンまたはジクロロベンゼンなど、ナイロン等のポリアミド系樹脂の場合には200℃熱ベンジルアルコールまたは200℃熱ニトロベンゼンなど、ポリスチレン系樹脂の場合にはTHFまたはジクロロベンゼンなど、ポリカーボネート系樹脂の場合にはTHFまたはジクロロメタンなどを用いることができる。
【0085】
上記グラフト化された炭素材料は、グラフェン構造を有する炭素材料が合成樹脂により表面修飾されているため、上記新たな合成樹脂中においても親和性が高められている。そのため、上記新たな合成樹脂をマトリクス樹脂とする上記新たな樹脂複合材料においても、上記新たな合成樹脂中に上記グラフト化された炭素材料が均一に分散されている。従って、樹脂複合材料の機械的強度を効果的に高めること及び線膨張率を効果的に低めることができる。
【0086】
上記新たな合成樹脂としては特に限定されず、先に述べた熱可塑性樹脂や熱硬化性樹脂などを用いることができる。好ましくは、上記新たな合成樹脂としては熱可塑性樹脂が用いられ、さらに好ましくは安価かつ成型性に優れたポリオレフィンが用いられる。また、上記新たな合成樹脂は結晶性樹脂であってもよく、先に述べたような非晶性樹脂であってもよい。もっとも、一般に結晶性樹脂は非晶性樹脂に比べて弾性率等の機械物性や成形加工性に優れるため、上記追加の樹脂は結晶性樹脂であることが好ましい。
【0087】
上記新たな合成樹脂と、上記グラフト化させる工程に用いられる上記合成樹脂とは、同一の種類の樹脂であってもよく、異なる種類の樹脂であってもよい。また、上記新たな合成樹脂は、1種のみが用いられてもよく、2種以上が併用されてもよい。
【0088】
上記グラフト化された炭素材料と上記新たな合成樹脂との混合は、先に述べた上記炭素材料と上記合成樹脂とを混合する方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0089】
さらに、本発明の樹脂複合材料の製造方法においては、上記グラフト化させる工程の後に、上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物に、先に述べた上記追加の樹脂をさらに混合することにより、上記合成樹脂と上記追加の樹脂との両方をマトリクス樹脂とする新たな樹脂合成材料を得ることもできる。それによって、様々な物性の樹脂複合材料を容易に製造することができる。
【0090】
上記合成樹脂と上記追加の樹脂との混合方法は、先に述べた上記炭素材料と上記合成樹脂とを混合する方法と同様に、溶融混練、溶媒中への溶解または分散等の適宜の方法により行うことができる。
【0091】
上記追加の樹脂と上記合成樹脂とは、同一の種類の樹脂であってもよく、異なる種類の樹脂であってもよい。また、追加の樹脂は、1種のみが用いられてもよく、2種以上が併用されてもよい。
【0092】
上記合成樹脂がグラフト化している上記炭素材料を含む上記樹脂組成物と上記追加の樹脂との配合割合は特に限定されないが、上記樹脂組成物100重量部に対し、上記追加の樹脂の配合量を1000重量部以下とすることが好ましい。上記追加の樹脂の配合量が1000重量部を超えると、樹脂複合材料に含まれる上記炭素材料による機械的強度の向上及び線膨張率の低下効果を充分に発揮できないことがある。
【0093】
本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料は、例えば、シート状にプレス成形することにより、引張弾性率や曲げ弾性率等の機械的強度が高く、線膨張率が低い樹脂複合材料シートを製造することができる。もっとも、本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料により製造される製品は、特に限定されない。本発明の樹脂複合材料及び本発明の製造方法により得られた樹脂複合材料を用いることにより、様々な機械的強度が高く、線膨張率が低い製品を製造することができる。
【0094】
また、本発明の樹脂複合材料の製造方法において、上記樹脂組成物を用意する工程と上記グラフト化させる工程とを同時に行わない場合には、上記グラフト化の前に上記樹脂組成物を成形することができることがある。例えば、上述の電子線またはマイクロ波の照射によりグラフト化を行う場合には、上記グラフト化の前に上記樹脂組成物をプレス成形等によって成形した後に、電子線またはマイクロ波を照射して上記樹脂組成物のグラフト化をすることができる。
【0095】
〔実施例及び比較例〕
以下、本発明の具体的な実施例及び比較例を挙げることにより、本発明を明らかにする。なお、本発明は以下の実施例に限定されるものではない。
本発明の実施例及び比較例において使用した材料は、以下の通りである。
【0096】
(薄片化黒鉛A)
XG SCIENCE社製、商品名「XGnP−5」の薄片化黒鉛を、薄片化黒鉛Aとした。この薄片化黒鉛Aを、後述の実施例1〜3,6,13及び16〜22並びに比較例1〜3,8,15及び18〜24において用いた。
【0097】
薄片化黒鉛Aにおいては、使用前にSEMを用いて観察した層面の面方向における最大寸法が約5.0μm、層厚みが約60nm、グラフェンの積層数が約180層、BET比表面積が75m2/gであった。
【0098】
(薄片化黒鉛B)
実施例及び比較例に用いる薄片化黒鉛Bを、以下の方法で製造した。
【0099】
黒鉛単結晶粉末0.25gを65重量%の濃硫酸11.5mlに供給して、得られた混合物を10℃の水浴により冷却しながら撹拌した。次に、黒鉛単結晶粉末と濃硫酸との撹拌によって得られた混合物に、過マンガン酸カリウム1.5gを徐々に加えながら混合物を撹拌し、混合物を35℃で30分にわたって反応させた。
【0100】
次に、反応混合物に水23gを徐々に加えて、混合物を98℃で15分にわたって反応させた。しかる後、反応混合物に水70gと30重量%の過酸化水素水4.5gを加えて反応を停止させた。続いて、混合物を14000rpmの回転速度にて30分にわたって遠心分離した。その後、得られた酸化黒鉛を5重量%の希塩酸及び水により十分に洗浄して、しかる後に乾燥させた。得られた酸化黒鉛を0.2mg/mlの量にて水に分散させた後、超音波洗浄機を45kHz、100Wの条件下にて用いて、酸化黒鉛に超音波を60分にわたって照射することにより、酸化黒鉛をその層界面間において剥離断片化して、層面が酸化されてなる薄片化黒鉛を得た。得られた層面が酸化されてなる薄片化黒鉛にヒドラジンを添加して、3分間にわたって還元処理を行って、薄片化黒鉛Bの混合液を得た。
【0101】
その後、得られた薄片化黒鉛Bの混合液を濾過し、濾別された薄片化黒鉛Bを乾燥させて、薄片化黒鉛Bを得た。得られた薄片化黒鉛Bを、後述の実施例7及び比較例9において用いた。
【0102】
薄片化黒鉛Bにおいては、使用前にAFM(原子間力顕微鏡)を用いて観察した層面の面方向における最大寸法が約5.0μm、層厚みが約2nm、グラフェンの積層数が約6層、BET比表面積が450m2/gであった。
【0103】
(薄片化黒鉛Bのo−ジクロロベンゼン溶液)
上記方法と同様にして、薄片化黒鉛Bの混合液を得た。この薄片化黒鉛Bの混合液には、薄片化黒鉛Bが0.25g含まれていた。
【0104】
次に、得られた薄片化黒鉛Bの混合液に200gの水を加えた後、500gのo−ジクロロベンゼンを加え、混合溶液を得た。上記混合溶液を充分に撹拌して、薄片化黒鉛を上記混合溶液の有機層に抽出した。その後、上記混合溶液を静置することにより、上記混合溶液を黒色のo−ジクロロベンゼン層からなる下層と、無色透明の水溶液層からなる上層とに分離させた。続いて、上記o−ジクロロベンゼン層からなる下層を回収した。その後、抽出されたo−ジクロロベンゼン層に200gの水を加えて、同様の操作により再び撹拌し、o−ジクロロベンゼン層を回収することによって、上記o−ジクロロベンゼン層から不純物を除去した。このようにして、上記o−ジクロロベンゼン層からなる薄片化黒鉛Bのo−ジクロロベンゼン溶液を500g得た。得られた薄片化黒鉛Bのo−ジクロロベンゼン溶液500g中には、薄片化黒鉛Bが0.25g含まれていた。
【0105】
このようにして得られた薄片化黒鉛Bのo−ジクロロベンゼン溶液のうち250gを、後述の実施例4,8〜10,12,14,15,23及び24並びに比較例4,10〜12,14,16,17,25及び26において用いた。上記薄片化黒鉛Bのo−ジクロロベンゼン溶液250g中には、薄片化黒鉛Bが0.125g含まれていた。
(カーボンナノチューブ(CNT))
【0106】
Nanocyl社製、商標名「Nanocyl−3100」の多層カーボンナノチューブを、実施例5,11及び25並びに比較例5,13及び27において用いた。
上記カーボンナノチューブの平均長は約1.5μm、平均径は約9.5nmであった。
【0107】
(合成樹脂)
本発明の実施例及び比較例において使用した合成樹脂は、以下の通りである。
【0108】
(1)ポリプロピレン系樹脂(PP)…プライムポリマー社製、商品名「J−721GR」、23℃における引張弾性率:1.2GPa、線膨張係数:11×10−5/K
【0109】
(2)高密度ポリエチレン系樹脂(PE)…日本ポリエチレン社製、商品名「HF560」、23℃における引張弾性率:1.0GPa
【0110】
(3)シラン変性ポリプロピレン系樹脂(Silane−PP)…三井化学社製、商品名「リンクロンXPM800HM」、23℃における引張弾性率:1.1GPa、線膨張係数:14×10−5/K
【0111】
(4)ポリメチルメタクリレート系樹脂(PMMA)…住友化学社製 商品名「スミペックスES」、23℃における曲げ弾性率:2.9GPa、線膨張係数:7×10−5/K
【0112】
(5)ポリアミド系樹脂(PA)…ユニチカ社製 商品名「A−125J」、23℃における引張弾性率:1.0GPa、線膨張係数:9×10−5/K
【0113】
(6)ポリアクリロニトリル系樹脂(PAN)…三井化学社製 商品名「バレックス#1000」、23℃における引張弾性率:3.3GPa、線膨張係数:8×10−5/K
【0114】
(7)ポリスチレン系樹脂(PSt)…東洋スチレン社製「トーヨースチロールGP RMS26」、23℃における曲げ弾性率:3.2GPa
【0115】
(8)ポリカーボネート系樹脂(PC)…住化スタイロン ポリカーボネート社製 商品名「ガリバー301−15」、23℃における引張弾性率:2.3GPa、線膨張係数:7×10−5/K
【0116】
(9)アタクチックポリプロピレン系樹脂(aPP)…三菱化学社製、商品名「タフセレンT3712」、23℃における引張弾性率:40MPa
【0117】
(ラジカル開始剤)
後述の実施例6〜17において使用するラジカル開始剤としては、Sigma−Aldrich社製のベンゾイルパーオキサイドを用いた。
【0118】
<電子線照射を用いた製造方法により得られた樹脂複合材料シート>
(実施例1)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部と、反応助剤としてジビニルベンゼン3重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0119】
(実施例2)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを160℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0120】
(実施例3)
ポリカーボネート系樹脂100重量部と、薄片化黒鉛A20重量部とを290℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に、加速電圧700KVで電離性放射線を3.0Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0121】
(実施例4)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にアタクチックポリプロピレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記アタクチックポリプロピレン系樹脂を溶解させて、混合溶液を得た。得られた混合溶液を室温において放冷し、濾過して、続いて真空乾燥させた。
【0122】
その後、上記混合溶液を180℃の温度でプレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に収縮を防止する目的により離型PETフィルムを両面に貼り合わせた。その後、上記シート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0123】
(実施例5)
アタクチックポリプロピレン系樹脂100重量部と、多層カーボンナノチューブ40重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み1mmのシート状成形物を得た。得られたシート状成形物に収縮を防止する目的により離型PETフィルムを両面に貼り合わせた。その後、上記シート状成形物に、加速電圧700KVで電離性放射線を1.5Mrad照射することにより、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0124】
(比較例1〜5)
電離性放射線を照射しなかったこと以外は実施例1〜5と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0125】
(比較例6)
薄片化黒鉛Aを添加しなかったこと以外は実施例1と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。なお、電離性放射線を照射する前のシート状成型体においては、23℃における引張弾性率は1.7GPaであった。
【0126】
(比較例7)
多層カーボンナノチューブを添加しなかったこと以外は実施例5と同様にして、表面平滑な厚み1mmの樹脂複合材料シートを得た。
【0127】
<ラジカル開始剤を用いた製造方法により得られた樹脂複合材料シート>
(実施例6)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して溶融混練し、ポリオレフィン系樹脂組成物を得た。
【0128】
上記ポリオレフィン樹脂組成物120重量部と、新たなポリプロピレン系樹脂100重量部とを押出機に供給して、190℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0129】
(実施例7)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛B10重量部と、ラジカル開始剤としてベンゾイルパーオキサイド3重量部とを180℃のプラストミルに供給して混練し、プレス成形することによって、表面平滑な厚みが0.5mmの樹脂複合材料シートを得た。
【0130】
(実施例8)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にポリプロピレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記ポリプロピレン系樹脂を溶解させて、混合溶液を得た。次に、上記混合溶液に、ラジカル開始剤として0.1重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを0.4重量部含む)を、窒素雰囲気下において滴下した。続いて、2時間にわたって撹拌を続けて、グラフト化反応物を得た。その後、上記反応物を室温において放冷し、濾過して、続いて真空乾燥させた。
【0131】
その後、真空乾燥させた上記反応物を180℃の温度でプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0132】
(実施例9)
0.5重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを2重量部含む)を用いたこと以外は実施例8と同様にして、樹脂複合材料シートを得た。
【0133】
上記樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、130℃に加熱された過剰量の熱キシレンに12時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していないポリプロピレン系樹脂を溶解除去した。その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、精製されたグラフト化薄片化黒鉛を回収した。
【0134】
その後、ポリプロピレン樹脂100重量部と、得られたグラフト化薄片化黒鉛5重量部とを180℃のプラストミルに供給して溶融混練し、180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。このようにして得られた樹脂複合材料シートを、実施例9の樹脂複合材料シートとした。
【0135】
(実施例10)
薄片化黒鉛Bのo−ジクロロベンゼン溶液に加えたポリプロピレン系樹脂の代わりに、アタクチックポリプロピレン系樹脂用いたこと以外は実施例9と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0136】
(実施例11)
最初に使用したポリプロピレン系樹脂の代わりにアタクチックポリプロピレン系樹脂を用いたこと、及び薄片化黒鉛A20重量部の代わりにカーボンナノチューブ40重量部を用いたこと以外は実施例6と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0137】
(実施例12)
グラフト化に用いたポリプロピレン系樹脂2.5gの代わりに高密度ポリエチレン樹脂2.5g(100重量部)を用いたこと、及び0.7重量%濃度のベンゾイルパーオキサイド・o−ジクロロベンゼン溶液10g(ベンゾイルパーオキサイドを2.8重量部含む)を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0138】
(実施例13)
シラン変性ポリプロピレン系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド4重量部とを押出機に供給して、190℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0139】
(実施例14)
ポリプロピレン系樹脂の代わりにポリメチルメタクリレート系樹脂を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0140】
(実施例15)
ポリプロピレン系樹脂の代わりにポリスチレン系樹脂を用いたこと以外は実施例8と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0141】
(実施例16)
ポリアミド系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して、270℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0142】
(実施例17)
ポリアクリロニトリル系樹脂100重量部と、薄片化黒鉛A3重量部と、ラジカル開始剤としてベンゾイルパーオキサイド6重量部とを押出機に供給して、200℃において溶融混練して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0143】
(比較例8〜19)
ベンゾイルパーオキサイドを用いなかったこと以外は実施例6〜17と同様にして、厚み0.5mmの樹脂複合材料シートを得た。
【0144】
<高せん断混練を用いた製造方法により得られた樹脂複合材料シート>
(実施例18)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて200〜250℃に加熱溶融して混練(スクリュー回転数=2500rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0145】
(実施例19)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて200〜280℃に加熱溶融して混練(スクリュー回転数=1000rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0146】
(実施例20)
ポリスチレン系樹脂100重量部と、薄片化黒鉛A20重量部とを、同方向二軸押出機(スクリュー径=15mm、スクリュー有効長さ/スクリュー径=120)を用いて120〜200℃に加熱溶融して混練(スクリュー回転数=4500rpm)して、溶融混練物とした。その後、上記溶融混練物を押出機の先端に取り付けたTダイから押出し、冷却ロールにてシート成形することにより、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0147】
(比較例20〜22)
スクリュー回転数を200rpmとしたこと以外は実施例18〜20と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0148】
<マイクロ波照射を用いた製造方法により得られた樹脂複合材料シート>
(実施例21)
ポリプロピレン系樹脂100重量部と、薄片化黒鉛A20重量部とを180℃プラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、300W電子レンジを用いてマイクロ波を60秒間照射した。続いて、上記シート状成形物を室温まで放冷し、再び300W電子レンジを用いてマイクロ波を60秒間照射した。その後、上記シート状成形物を180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0149】
(実施例22)
高密度ポリエチレン樹脂100重量部と、薄片化黒鉛A20重量部とを180℃プラストミルに供給して混練し、プレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、100W電子レンジを用いてマイクロ波を60秒間照射した。続いて、上記シート状成形物を室温まで放冷し、再び100W電子レンジを用いてマイクロ波を60秒間照射した。その後、上記シート状成形物を180℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0150】
(実施例23)
130℃に加熱した薄片化黒鉛Bのo−ジクロロベンゼン溶液250g(薄片化黒鉛を0.125g(5重量部)含む)にポリスチレン系樹脂2.5g(100重量部)を加え、130℃で2時間撹拌することにより上記ポリスチレン系樹脂系樹脂を溶解させて、混合溶液を得た。得られた混合溶液を室温において放冷し、濾過して、続いて真空乾燥させた。
【0151】
その後、上記混合溶液を160℃の温度でプレス成形することによって、表面平滑な厚み0.5mmのシート状成形物を得た。得られたシート状成形物に、500W電子レンジを用いてマイクロ波を30秒間照射した。続いて上記シート状成形物を室温まで放冷した後、上記マイクロ波照射及び放冷をさらに2度繰り返した。その後、上記シート状成形物を160℃においてプレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0152】
(実施例24)
実施例23と同様にして、樹脂複合材料シートを得た。この樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、過剰量のTHFに24時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していないポリスチレン系樹脂を溶解除去した。その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、精製されたグラフト化薄片化黒鉛を回収した。
【0153】
その後、ポリスチレン樹脂100重量部と、得られたグラフト化薄片化黒鉛5重量部とを160℃のプラストミルに供給して溶融混練し、プレス成形することによって、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。このようにして得られた樹脂複合材料シートを、実施例24の樹脂複合材料シートとした。
【0154】
(実施例25)
薄片化黒鉛A20重量部の代わりに多層カーボンナノチューブ40重量部を用いたこと以外は実施例21と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0155】
(比較例23〜27)
マイクロ波を照射しなかったこと以外は実施例21〜25と同様にして、表面平滑な厚み0.5mmの樹脂複合材料シートを得た。
【0156】
(実施例及び比較例の評価)
得られた樹脂複合材料のグラフト化率、引張弾性率、曲げ弾性率、線膨張係数、及びラマンスペクトルを、下記の方法により測定した。
【0157】
(1)グラフト化率測定
実施例及び比較例により得られた樹脂複合材料シートを小さく裁断し、樹脂複合材料片とした。次に、上記樹脂複合材料片を濾紙で包んだ。上記濾紙から上記樹脂複合材料片が漏れ出ないように上記濾紙の端を折り込み、さらにその周囲を金属クリップで封止した。このようにして得られた包装体を、過剰量の溶媒に12時間浸した。それによって、樹脂複合材料シートに含まれるグラフト化していない合成樹脂を溶解除去した。
【0158】
上記溶媒としては、用いた合成樹脂がポリプロピレン系樹脂、高密度ポリエチレン系樹脂、シラン変性ポリプロピレン系樹脂またはアタクチックポリプロピレン系樹脂の場合には、130℃熱キシレンを使用した。用いた合成樹脂がポリメチルメタクリレート系樹脂の場合にはジクロロベンゼンを使用した。用いた合成樹脂がポリアミド系樹脂の場合には200℃熱ニトロベンゼンを使用した。用いた合成樹脂がポリスチレン系樹脂の場合にはジクロロベンゼンを使用した。用いた合成樹脂がポリカーボネート系樹脂の場合にはTHFを使用した。
【0159】
その後、上記包装体を溶媒から取り出し、真空乾燥させることにより、上記グラフト化薄片化黒鉛を単離した。
【0160】
このようにして単離されたグラフト化薄片化黒鉛を、空気雰囲気下、30〜600℃の温度範囲において、10℃/分の昇温速度により熱重量測定(TGA測定)を行った。このとき、上記グラフト化薄片化黒鉛において、500℃に昇温されるまでに分解した分解物の量をA重量%、500℃まで昇温しても分解しなかった未分解物の量をB重量%として、下記の式によりグラフト化率を求めた。結果を下記の表1〜表5に示す。
【0161】
グラフト化率(重量%)=A/B×100
【0162】
(2)引張弾性率測定
実施例及び比較例により得られた樹脂複合材料シートの23℃における引張り弾性率を、JIS K6767に準拠して測定した。結果を下記の表1〜表5に示す。
【0163】
(3)曲げ弾性率測定
実施例及び比較例により得られた樹脂複合材料シートの23℃における見かけ曲げ弾性率を、JIS K7161に準拠して測定した。結果を下記の表1〜表5に示す。
【0164】
(4)線膨脹係数測定
実施例及び比較例により得られた樹脂複合シートの−30〜100℃における線膨張係数を、JIS K7197に準拠して測定した。結果を下記の表1〜表5に示す。
【0165】
【表1】
【0166】
【表2】
【0167】
【表3】
【0168】
【表4】
【0169】
【表5】
【0170】
表1〜表5から明らかなように、各実施例の樹脂複合材料シートでは、薄片化黒鉛やカーボンナノチューブ等の炭素材料に合成樹脂がグラフト化しており、そのグラフト化率が5重量%〜3300重量%の範囲にある。さらに、各実施例の樹脂複合材料シートは、対応する各比較例の樹脂複合材料シートと比較して、引張弾性率及び/または曲げ弾性率が大幅に高められており、かつ線膨張係数が大幅に低められている。これは、合成樹脂が炭素材料にグラフト化し、樹脂と薄片化黒鉛との界面における密着性が高められていることによると考えられる。
【0171】
(5)粘弾性測定
実施例4及び5並びに比較例4,5及び7により得られた樹脂複合材料シートについて、以下のようにして粘弾性を測定した。
【0172】
動的粘弾性測定装置(TA Instruments社製、「Ares」)を用いて、昇温速度5℃/分で30℃から130℃まで昇温しながら周波数10rad/秒の条件により、得られた樹脂複合材料シートに1.0%のひずみ量を繰り返し加えた。その時の樹脂複合材料の貯蔵弾性率E’及び損失弾性率E’’を測定した。結果を図1及び図2に示した。
【0173】
図1から明らかなように、実施例4により得られた樹脂複合材料シートは、比較例4により得られた樹脂複合材料シートと比較して、高温での貯蔵弾性率(E’)が高められている。加えて、実施例4により得られた樹脂複合材料シートでは、貯蔵弾性率(E’)が損失弾性率(E”)を下回る領域が存在しない。このことは、実施例4により得られた樹脂複合材料シートでは、アタクチックポリプロピレン系樹脂が薄片化黒鉛Bにラジカルグラフト化することにより、上記樹脂複合材料シートから樹脂の流動域が消失していることによると考えられる。
【0174】
同様に、図2から明らかなように、実施例5により得られた樹脂複合材料シートは、比較例5により得られた樹脂複合材料シートと比較して、貯蔵弾性率(E’)が高められており、貯蔵弾性率(E’)が損失弾性率(E”)を下回る領域が存在しない。このことは、上記と同様に、アタクチックポリプロピレン系樹脂が多層カーボンナノチューブにラジカルグラフト化することによると考えられる。
【0175】
さらに、図1及び図2によると、比較例7により得られた樹脂複合材料シートは、比較例4及び5により得られた樹脂複合材料シートと比較して、貯蔵弾性率(E’)及び損失弾性率(E”)が大幅に低下している。これは、パーオキサイドまたは電離性放射線によって生じたフリーラジカルにより、アタクチックポリプロピレン系樹脂が劣化したことによると考えられる。一方、実施例4及び5により得られた樹脂複合材料シートでは、上記と同様の樹脂劣化が起きているにもかかわらず、高い貯蔵弾性率(E’)及び損失弾性率(E”)を示している。これは、薄片化黒鉛B及び多層カーボンナノチューブの上記ラジカルグラフト化による貯蔵弾性率(E’)及び損失弾性率(E”)の向上効果が、樹脂劣化による貯蔵弾性率(E’)及び損失弾性率(E”)の低下を上回ったことによると考えられる。
【0176】
(6)ラマンスペクトル測定
実施例6及び比較例8によって得られた樹脂複合材料シートのラマンスペクトルを、波長532nmのレーザーを用いて、露光時間1秒、スキャン回数32回により顕微ラマン測定を行った。結果を図3に示した。
【0177】
ラマンスペクトルにおける1583cm−1付近のピークは炭素材料のグラフェン構造に由来し、1340cm−1付近のピークはグラフト化反応等によるグラフェン構造の乱れに由来する。実施例6の樹脂複合材料シートのラマンスペクトルに見られる1583cm−1付近のピークの低下と、1340cm−1付近のピークの上昇は、薄片化黒鉛Aに新たな化学結合が導入されたことを示唆する結果であると考えられる。
【特許請求の範囲】
【請求項1】
合成樹脂と、前記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、前記炭素材料に前記合成樹脂がグラフト化しており、前記炭素材料のグラフト化率が5重量%〜3300重量%である、樹脂複合材料。
【請求項2】
前記合成樹脂と異なる種類の樹脂をさらに含んでいる、請求項1に記載の樹脂複合材料。
【請求項3】
前記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである、請求項1または2に記載の樹脂複合材料。
【請求項4】
前記合成樹脂が熱可塑性樹脂である、請求項1〜3のいずれか1項に記載の樹脂複合材料。
【請求項5】
前記熱可塑性樹脂がポリオレフィンである、請求項4に記載の樹脂複合材料。
【請求項6】
前記熱可塑性樹脂が非晶性樹脂である、請求項4に記載の樹脂複合材料。
【請求項7】
合成樹脂と、前記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、
前記樹脂組成物を用意する工程と同時にまたは前記樹脂組成物を用意する工程の後に、前記炭素材料に前記合成樹脂をグラフト化させる工程とを備える、樹脂複合材料の製造方法。
【請求項8】
前記グラフト化させる工程が、前記合成樹脂及び前記炭素材料を、混練用スクリューを備えるせん断混練装置により加熱混練することにより行われる、請求項7に記載の樹脂複合材料の製造方法であって、
前記加熱混練の際に、前記スクリューを500rpm〜5000rpmの回転速度で回転させる、樹脂複合材料の製造方法。
【請求項9】
前記加熱混練が動的な加熱混練により行われる、請求項8に記載の樹脂複合材料の製造方法。
【請求項10】
前記グラフト化させる工程において、前記炭素材料のグラフト化率が5重量%〜3300重量%の範囲となるように前記合成樹脂を前記炭素材料にグラフト化させる、請求項7〜9のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項11】
前記グラフト化させる工程の後に、前記合成樹脂と同種または異なる樹脂を混合する工程をさらに備える、請求項7〜10のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項12】
前記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである、請求項7〜11のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項13】
前記合成樹脂が熱可塑性樹脂である、請求項7〜12のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項14】
前記熱可塑性樹脂がポリオレフィンである、請求項13に記載の樹脂複合材料の製造方法。
【請求項15】
前記熱可塑性樹脂が非晶性樹脂である、請求項13に記載の樹脂複合材料の製造方法。
【請求項1】
合成樹脂と、前記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂複合材料であって、前記炭素材料に前記合成樹脂がグラフト化しており、前記炭素材料のグラフト化率が5重量%〜3300重量%である、樹脂複合材料。
【請求項2】
前記合成樹脂と異なる種類の樹脂をさらに含んでいる、請求項1に記載の樹脂複合材料。
【請求項3】
前記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである、請求項1または2に記載の樹脂複合材料。
【請求項4】
前記合成樹脂が熱可塑性樹脂である、請求項1〜3のいずれか1項に記載の樹脂複合材料。
【請求項5】
前記熱可塑性樹脂がポリオレフィンである、請求項4に記載の樹脂複合材料。
【請求項6】
前記熱可塑性樹脂が非晶性樹脂である、請求項4に記載の樹脂複合材料。
【請求項7】
合成樹脂と、前記合成樹脂中に分散されたグラフェン構造を有する炭素材料とを含む樹脂組成物を用意する工程と、
前記樹脂組成物を用意する工程と同時にまたは前記樹脂組成物を用意する工程の後に、前記炭素材料に前記合成樹脂をグラフト化させる工程とを備える、樹脂複合材料の製造方法。
【請求項8】
前記グラフト化させる工程が、前記合成樹脂及び前記炭素材料を、混練用スクリューを備えるせん断混練装置により加熱混練することにより行われる、請求項7に記載の樹脂複合材料の製造方法であって、
前記加熱混練の際に、前記スクリューを500rpm〜5000rpmの回転速度で回転させる、樹脂複合材料の製造方法。
【請求項9】
前記加熱混練が動的な加熱混練により行われる、請求項8に記載の樹脂複合材料の製造方法。
【請求項10】
前記グラフト化させる工程において、前記炭素材料のグラフト化率が5重量%〜3300重量%の範囲となるように前記合成樹脂を前記炭素材料にグラフト化させる、請求項7〜9のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項11】
前記グラフト化させる工程の後に、前記合成樹脂と同種または異なる樹脂を混合する工程をさらに備える、請求項7〜10のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項12】
前記グラフェン構造を有する炭素材料が、薄片化黒鉛またはカーボンナノチューブである、請求項7〜11のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項13】
前記合成樹脂が熱可塑性樹脂である、請求項7〜12のいずれか1項に記載の樹脂複合材料の製造方法。
【請求項14】
前記熱可塑性樹脂がポリオレフィンである、請求項13に記載の樹脂複合材料の製造方法。
【請求項15】
前記熱可塑性樹脂が非晶性樹脂である、請求項13に記載の樹脂複合材料の製造方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2012−136712(P2012−136712A)
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2012−90086(P2012−90086)
【出願日】平成24年4月11日(2012.4.11)
【分割の表示】特願2011−544517(P2011−544517)の分割
【原出願日】平成23年9月2日(2011.9.2)
【出願人】(000002174)積水化学工業株式会社 (5,781)
【Fターム(参考)】
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願日】平成24年4月11日(2012.4.11)
【分割の表示】特願2011−544517(P2011−544517)の分割
【原出願日】平成23年9月2日(2011.9.2)
【出願人】(000002174)積水化学工業株式会社 (5,781)
【Fターム(参考)】
[ Back to top ]