
cDNA/mRNA−タンパク質連結体の効率的合成法

【課題】 本発明はバイオテクノロジーにおけるディスプレイ技術の一つであるIVV(インビトロウィルス)法(mRNAディスプレイ法,cDNAディスプレイ法)におけるタンパク質の合成効率を向上させ、さらに作業工程の短縮をはかることを課題とする。
【解決手段】連結反応工程の条件を最適化することで、従来技術(選択図)において必須であった精製及び濃縮の工程を省略し、ディスプレイ法全体の工程短縮を図るものである。本発明の方法によれば、ディスプレイ技術のコストと時間の低減を実現し、もって進化分子工学的手法による分子探索のハイスループット化を達成する。
【解決手段】連結反応工程の条件を最適化することで、従来技術(選択図)において必須であった精製及び濃縮の工程を省略し、ディスプレイ法全体の工程短縮を図るものである。本発明の方法によれば、ディスプレイ技術のコストと時間の低減を実現し、もって進化分子工学的手法による分子探索のハイスループット化を達成する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は進化工学的ディスプレイ技術における、mRNA(メッセンジャーRNA)−リンカー−タンパク質連結体を、効率よく合成する方法に関する。
【背景技術】
【0002】
遺伝子工学の発展により、遺伝子配列を明らかにするという「ゲノム解析」の時代から遺伝子の発現産物による「機能解析」へと、開発研究のテーマは移行している。これは、遺伝子に依拠する遺伝形質は、遺伝子発現産物を介して、組織、器官及び人体等の機能として表れるものであるためである。こうした遺伝子発現産物の中でもタンパク質及びペプチドは、特に基礎的で重要な機能解析の対象である。
ポストゲノム時代においては、ゲノム解析により特定された遺伝子とその機能解明に基づき遺伝子標的をより明確にし、その作用機序を明らかにすることにより合理的な分子標的バイオ医薬(抗体医薬、ペプチド医薬)の開発が可能である。そして、近年注目されているのが核酸医薬の合成においても有効性が実証されている進化分子工学的手法である。
ペプチド医薬は、現在、広く使われるようになりつつある抗体医薬と同じ働きをしながら、抗体医薬よりも小さなタンパク質分子で同一の機能を発揮する医薬である。今後はその生産性の高さから、抗体医薬を置き換わっていくものと考えられている。
【0003】
高機能分子の新規創出を目的とする進化工学は、ランダムな配列を有する莫大なペプチドあるいはタンパク質のライブラリー中から、タンパク質に限られない特定の分子に特異的に結合するペプチド等を選択することを目的とする。
とりわけ、配列のランダム化の元となる遺伝子とその発現産物であるタンパク質とを分子的に対応付ける、又はディスプレイするIn vitro virus(IVV)法は、その中核をなす技術の一つであると言える。
IVVはタンパク質等の遺伝子発現産物の機能や表現型と、これに対応する遺伝子がコードされるmRNA等を当該分子上で1:1に対応付けることのできる分子である。IVV法においては、それぞれランダムなタンパク質を有するいくつかのIVVから、最も機能の高いタンパク質を選んだのちに、そこに連結されたmRNAを逆転写しPCR増幅等をすることでその配列を、最も機能の高い遺伝子の情報として検出することができる。
【0004】
進化工学的手法の一つであるcDNAディスプレイ法ではDNAライブラリから始まり、転写、リンカーの連結、翻訳とタンパク質の連結、固相化、逆転写、固相からの切り離し、精製、スクリーニング、cDNAの増幅と変異導入(ライブラリ化)、プロモーターの連結を、一のサイクルとし、複数回のサイクルを繰り返すのが標準とされている。一方、mRNAディスプレイ法では逆転写工程がスクリーニング工程の後に実行される。
このディスプレイ(対応付け)とスクリーニングのサイクルを繰り返すことで、前記の「スクリーニング」と分子への「変異導入」の回数を増やすことが出来るので、より優れた分子を創出し選び出すことが出来る。
進化工学的手法、及びIVV法ついては非特許文献1,2,3,7,8及び特許文献1,4,5,6に詳細が解説されている。
前記文献においてはmRNAディスプレイ法と呼ばれる方法が採られてきたが、近年のIVV法の進展の中で、In vitro virus合成後の分子の安定性向上のためcDNAディスプレイ法が開発された。これについては非特許文献4,5,6,9及び特許文献2,3,7に詳細が解説されている。
【0005】
またIVVに関連して、そのタンパク質の機能解析は、例えば、タンパク質−タンパク質相互作用やタンパク質−核酸相互作用等の試験による生化学的機能分析を通して行われている。
このようなタンパク質−タンパク質相互作用の解析法としては、イーストツーハイブリッド法、ファージディスプレイ法、GST−融合タンパク質プルダウン法、免疫共沈法等が古くから知られている。一方、タンパク質−核酸相互作用の解析法としては、電気泳動移動度シフトアッセイ法、DNaseIフットプリント法、メチル化緩衝法等が知られている。
合成されるIVVはこれらの試験に要求されるタンパク質と同等の品質を満たすものでなくてはならない。
【0006】
前記IVV法においては、遺伝子をコードするmRNAに対してタンパク質結合部位を有するリンカー(分子間連結のための分子)を連結することで、当該mRNAから合成されたタンパク質がリンカーに結合し、捕捉される。このため、1分子ごとに遺伝子と、遺伝子発現産物とを対応させることができることが知られている。
タンパク質をリンカーにトラップするには、翻訳合成系において、アミノアシルtRNAアナログを有するリンカーを伸長タンパク質鎖中に取り込ませる手法等が使われる。こうしたアナログとしてはピューロマイシン(Puromycin)等があり、これらにより修飾された核酸構築物を利用することでIVV法を実施することが可能となる。
【0007】
従来、mRNAとピューロマイシン・リンカーとを連結する工程では、反応効率の点から、微量のmRNAに対してピューロマイシン・リンカーを過剰に投入し、精製の際にこれを除去するという作業を行っていた。これらは、その後の翻訳反応において、タンパク質合成、及び連結体形成の阻害要因となると考えられていたためである(非特許文献1〜10,特許文献1〜8参照)。これらの除去は、フェノール・クロロホルム抽出や、ゲルろ過、イオン交換、ODS逆相カラム、HPLCその他のカラムを用いた精製、シリカメンブレンや限外濾過等の濾過、アクリルアミドゲル切出抽出、透析その他の種々の手法を用いて行われてきている。
また、リンカーとmRNAとを紫外光照射によるクロスリンクで連結させ、ライゲーション反応を回避し、後の精製工程を省略する方法が提案されている(非特許文献3)
一方で、あらかじめリンカーを固相に固定しておき、バッファー交換によって反応液を除去して精製を回避する方法も開発されている(特許文献3,非特許文献9)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開WO01/016600
【特許文献2】国際公開WO06/041194
【特許文献3】国際公開WO07/046520
【特許文献4】特開2002-291491
【特許文献5】特開2003-299489
【特許文献6】特開2004-097213
【特許文献7】特開2008-253176
【特許文献8】特開2005-245223
【非特許文献】
【0009】
【非特許文献1】Nemoto et al., FEBS Lett. 414, 405 (1997)
【非特許文献2】Roberts et al., Proc. Natl. Acad. Sci. USA 94, 12297 (1997)
【非特許文献3】Kurz et al., Nucleic Acids Res. 28, e83 (2000)
【非特許文献4】Kurz et al., CHEMBIOCHEM 2, 666 (2001)
【0010】
【非特許文献5】Tabuchi et al., FEBS Lett. 508, 309 (2001)
【非特許文献6】Tabuchi et al., Biol. Proced. Online 4, 49 (2002)
【非特許文献7】Miyamoto-Sato et al., Nucleic Acids Reserch 31, e78 (2003)
【非特許文献8】Ja & Roberts, Biochemistry 43, 9265 (2004)
【非特許文献9】Biyani et al., Nucleic Acids Res 34, e140 (2006)
【非特許文献10】Retic Lysate IVT Kit (Part Number AM1200) Protocol (Ambion Inc.)
【非特許文献11】Abdella et al., Biochem. Biophys. Res. Commun., 87, 734 (1979)
【発明の概要】
【発明が解決しようとする課題】
【0011】
従来の方法は、1分子ごとに遺伝子と、遺伝子発現産物とを対応させることができるという点では優れたものである。しかし、以下のような問題点があった。
すなわち、系に投入するリンカー濃度を上げるとm−RNAとリンカーとの連結体の収量は増加するが、リンカー同士やリンカーとmRNAとの凝集を招くため、投入できる濃度には限界がある。また連結効率を考えると、mRNAとリンカーの投入量とは、最大でmRNA:リンカー比=1:200程度とする必要があり、未濃縮の連結体を得るのに十分な濃度のmRNAが投入できなかった。
さらに、反応生成物であるmRNA−リンカー連結体の濃縮が必須であり、上記各カラム精製後の抽出、エタノール沈殿、限外濾過、エバポレーション等が行われてきた。
こうした精製は操作が煩雑で手間がかかるだけでなく、IVV法で得られた多種多様なペプチドのスクリーニングを自動化するには適していない。
【0012】
一方、タンパク質の収率に影響し得る未反応mRNAについても、リンカーと同様に、除去することが望ましい。あらかじめリンカーを固相に固定しておく従来の方法では、未反応mRNAを除去することはできるが、収量が著しく減少するという問題を有しており、多様なペプチドスクリーニングの需要に必ずしも応えられるものとは言えない。このように、液相中にて未反応mRNAのみを除去する有効な手段は現在見出されていない。
これに対し、リンカーとmRNAの連結にライゲーション反応ではなく、紫外光照射によるクロスリンクを用いる方法が提案されている。この方法は精製工程を省略することができる点で特に優れたものである。しかし、この方法では、リンカーの構造がさらに複雑になるため、製造コストを上昇させるほか、紫外光照射によってmRNAが損傷されることもあるという問題がある。
さらに、上述したように、進化工学的手法ではディスプレイとスクリーニングの回数を増やすほど優れた分子種を得られるため、上述した一連のサイクルは出来るだけ多いことが必要とされる。そしてサイクル中の作業工程数は少なければ少ないほどよく、特に人が関与する作業は少ないほど良い。
しかしながら、上記進化工学的手法には省略できない化学反応工程が数多く含まれており、サイクル全体の作業工程の短縮及び省力化は困難であった。
【0013】
以上のような観点から、mRNAとリンカーとの連結後の精製及び濃縮に要する手間と時間及びコストを大幅に削減することで、実用的なハイスループットスクリーニングの開発を促す、前記連結体の生成方法等に対する社会的な要請があった。
さらに、上記ペプチド医薬等の開発促進のニーズに応えるためにペプチドスクリーニング技術の発展と普及が必要とされている。このため、次世代医薬としてのリードペプチドの探索手法(ペプチドスクリーニング)においては、自動化及び省力化に対する強い要請があった。
【課題を解決するための手段】
【0014】
すなわち本発明の第1の態様は、液相中における個々の分子の反応において、タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合する工程と;を備える、mRNA−リンカー−タンパク質連結体の生成方法である。
【0015】
前記所定の割合は、前記リンカー:前記mRNA=3:1〜1:6であることが好ましく、前記リンカー:前記mRNA=1:1〜1:1.5であることが特に好ましい。前記主鎖の鎖長は16〜60塩基であることが好ましく、21〜38塩基であることが特に好ましい。
前記リンカーの主鎖は、5'位がリン酸化された主鎖5'末端部と、第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、を備えることが好ましい。
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、前記主鎖は主鎖3'末端側領域部に側鎖連結部位をさらに備え、かつ前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列をさらに有し;前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することが特に好ましい。
前記切断部位は、制限酵素認識配列を有することが好ましい。前記mRNA−リンカー−タンパク質連結体の生成方法は、前記固相結合部位を介して固相と結合させる工程と、前記第1及び第2の切断部位を制限酵素で切断する工程とをさらに備えることが好ましい。前記制限酵素はPvuIIであることが好ましい。
前記切断部位はまた、グアノシンを有することが好ましい。前記mRNA−リンカー−タンパク質連結体の生成方法は、前記固相結合部位を介して固相と結合させる工程と、前記第1及び第2の切断部位をRNA切断酵素で切断する工程とをさらに備えることが好ましい。前記RNA切断酵素はリボヌクレアーゼT1であることが特に好ましい。
【0016】
本発明の第2の態様は、液相中における個々の分子の反応において、タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合し、mRNA−リンカー−タンパク質連結体を生成する工程と;mRNA−リンカー−タンパク質連結体を前記リンカーの備える固相結合部位を介して固相と結合させる工程と;前記主鎖の3'末端を反応開始点とし、前記mRNAを鋳型として、固相上で逆転写反応を行ってcDNA鎖を形成させる工程と;前記主鎖の備える第1及び第2の切断部位を、制限酵素又はRNA切断酵素で切断する工程と;を備えるmRNA−リンカー−タンパク質−cDNA連結体の生成方法である。
【0017】
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、前記リンカーの主鎖は、5'位がリン酸化された主鎖5'末端部と、前記第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される前記固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、側鎖連結部位を備え、前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、を備えることが好ましい。また、前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することが好ましい。
【発明の効果】
【0018】
本発明がなされるまでは、ライゲーション反応後の精製濃縮工程は省略することが出来ないとするのが、cDNA/mRNAディスプレイ技術における技術常識であった。本発明の方法に拠ればこれを省略することが可能になる。
本発明の効果は、IVVすなわちmRNA−リンカー−タンパク質連結体の効率的な生成を可能とすることであり、さらにmRNAディスプレイ法、cDNAディスプレイ法、分子進化学的手法全体にとっての工程の簡略化を可能にすることである。
【図面の簡単な説明】
【0019】
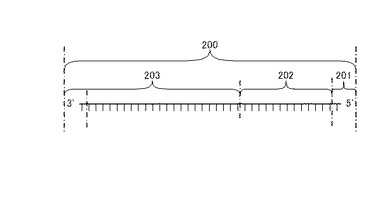
【図1】図1は、リンカー主鎖の構造図である。
【図2】図2は、リンカーの模式図である。
【図3】図3は、旧来のIVV合成法を示す模式図である。
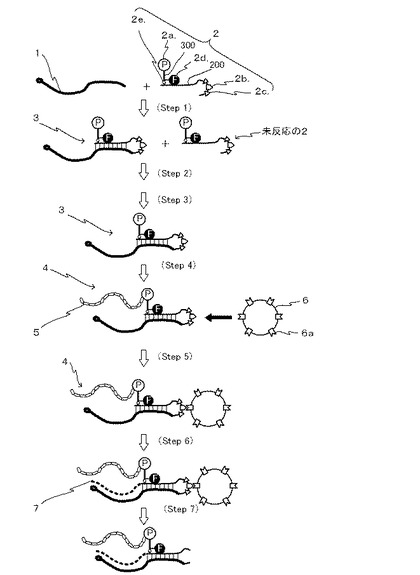
【図4】図4は、本発明のIVV合成法を示す模式図である。
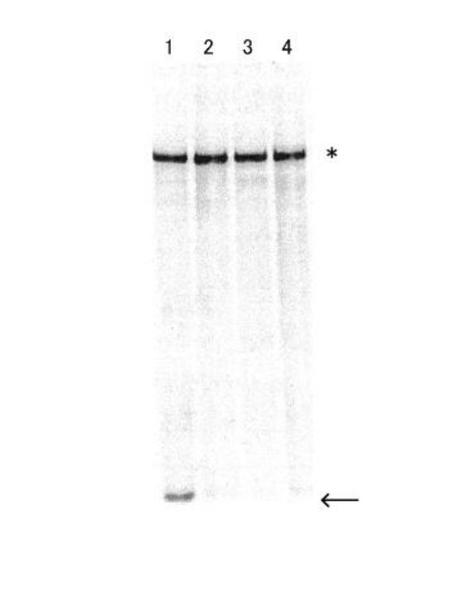
【図5】図5は、ライゲーション反応産物の電気泳動像(FITC蛍光)である。
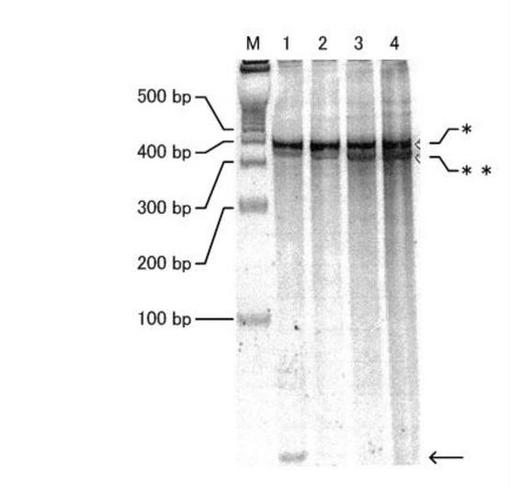
【図6】図6は、ライゲーション反応産物の電気泳動像(SYBR(登録商標) Gold)である。
【図7】図7は、翻訳連結反応産物の電気泳動像(FITC蛍光)である。
【発明を実施するための形態】
【0020】
以下に、本発明をさらに詳細に説明する。
1.連結体生成用リンカー
本発明において、「リンカー」とは、IVV法において用いられる、mRNA−リンカー連結体(連結体A)、mRNA−リンカー−タンパク質連結体(連結体B)及びmRNA−リンカー−タンパク質−cDNA連結体からなる群から選ばれるいずれかの連結体を生成する際に使用するリンカーのことをいう。
本発明において使用するリンカーは、DNA(すなわちデオキシリボ核酸単量体)、RNA(すなわちリボ核酸単量体、例えばrG:グアノシン)、及びDNAアナログからなる群から選ばれる少なくとも1以上の物質によって形成される主鎖を含み、この主鎖中に上記連結体を固相部位に結合するための固相結合部位と、前記固相結合部位を挟む位置に設けられた一対の切断部位を有することを特徴とする。ここで、DNAアナログはBiotin-dT、Fluorescein-dT等を含むものとする。
本発明において使用するリンカーは、全体として、柔軟性があり、親水性であり、かつ特異構造を形成する可能性の少ない配列を有するように、設計することが好ましい。
【0021】
本発明の詳細な説明において「液相反応」とは、通常の試験管、微量サンプルチューブ、マイクロウェルプレート等の反応容器内の均一な反応液中での反応を指すものとする。また、「固相反応」とは前記同様の反応容器内における反応ではあるが、固相ビーズや容器底面等な固相表面に反応させるべき分子を結合し、固相表面上において集中的に反応を行うことを指すものとする。
ここで「mRNAの有する所定の配列」には、遺伝子をコードする配列、連結体形成、翻訳反応促進に必要な配列その他の配列が含まれ、特に限定されない。
また、本発明の詳細な説明において「アミノアシルtRNA3’末端アナログ」とは、その3’末端がアミノアシルtRNAに化学構造骨格が類似し、翻訳系でタンパク質の合成が行われた際に、伸長中のペプチド鎖のC末端に結合する能力を有する化合物をいう。
【0022】
また、本発明の詳細な説明において「Xを有するY部位」の文言は、以下のように定義される。ここで、Xは、FITCその他の蛍光色素、RnaseT1その他の酵素の基質等の機能性分子を表わし、Yは機能性分子Xが発揮する機能を表わす。「Xを有するY部位」は、Xが結合しているか、Xを含んでいることで、機能Yを発揮する一塩基〜数十塩基からなるリンカー中の特定の配列を指す。前記特定の配列は、機能性分子X、Xに結合する修飾基のみで構成されるものであってもよく、前記特定の配列に含まれる塩基には、核酸アナログ等も含まれる。
一例を挙げると、「rGを有する切断部位」とは、グアノシン(rG)を含むことにより、リボヌクレアーゼT1がrGを特異的に認識してホスホジエステル結合を加水分解することができる部位を指す。
【0023】
また、本発明中で、リンカー主鎖はRNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるものである。本発明の方法ではリンカーの主鎖が、その全長に対して、少なくとも60%以上が、1本鎖DNAと、RNA及び/又はDNAアナログとで構成されていることが好ましい。より好ましくはリンカー主鎖の70%以上、さらに好ましくは80%以上、最も好ましくは90%以上、1本鎖DNAと、RNA及び/又はDNAアナログとで構成されている。
本発明において使用するリンカーでは、種々の反応工程での反応性、上述した連結体の生成効率などを考慮して主鎖の長さを決定するが、好ましくは16〜60merであることが好ましく、より好ましくは21〜38merである。なお、本発明において使用するリンカーは、後述するような公知の化学合成の手法を用いて合成することができる。
図1を用いて本発明に用いるリンカーの主鎖の構造を説明する。主鎖200には主鎖5'末端部201が存在する。主鎖5'末端近傍部202は主鎖5'末端部201を含まない領域であり、主鎖3'末端側領域部203は主鎖3'末端部を含まない領域である。なお、図1は上記各部の塩基長を限定したものではない。
【0024】
2.所定の配列を有するmRNA
本発明で用いるmRNAは、配列未知のもの、配列既知のものの双方を含む。前記配列は、ゲノム情報として自然界に存在する配列、又は人為的に設計した配列のいずれであってもよく、その構造は特に限定されない。
上記連結体を用いて、配列既知のタンパク質に結合する物質を探索又は定量する場合には、当該タンパク質の遺伝子をコードするmRNAを用いることができる。逆に、上記連結体を用いて、配列未知のタンパク質の配列を特定の機能に関連づけて解析する場合には、配列未知の遺伝子をコードするmRNAを用いることができる。
【0025】
こうしたmRNAとしては、例えば、配列既知の各種レセプタータンパク質をコードするmRNA、各種抗体又はその断片をコードするmRNA、各種酵素をコードするmRNA、各種遺伝子ライブラリー中のDNAから転写される配列未知のmRNA、有機合成によってランダムに合成された配列を有するDNAから転写されたランダムな配列を有するmRNA、PCR法を利用したランダム変異の挿入により配列未知のタンパク質をコードするようになったDNAから転写されるmRNAその他のmRNA等を挙げることができ、これらの中から選択される。
本発明で用いるmRNAは、自然界に存在するmRNAや、mRNA特有の構造に限定されるものではないが、タンパク質の合成効率上、5’末端に7−メチル化グアノシンキャップ構造又は3’末端にポリA尾部構造の少なくとも一方を持っていることが好ましい。翻訳の開始を促進するKozak配列や、Shine-Dlgarno配列を有することが、さらに好ましい。
mRNAはin vitro転写反応、化学合成、生体・細胞・微生物からの抽出などの所与の方法を用いて得ることができる。リンカーとの連結及び無細胞翻訳の反応効率の点から、in vitro転写反応を用いて生成することが好ましい。
本発明で用いるmRNAの長さは、反応効率の面から50〜1,000塩基であることが好ましく、200〜500塩基であることが反応効率が最も高いことから、さらに好ましい。
【0026】
3.ハイブリダイゼーション及び連結方法
mRNAとリンカーとの連結は、公知の手法を用いて、直接的又は間接的に、化学的又は物理的に行うことができる。例えば、mRNAの3’末端にリンカーの末端と相補的な配列を設けておくと、両者をハイブリダイゼーションによって連結することができる。また、末端でのライゲーション反応、又は末端を含む各部分でのソラレンを用いた光架橋反応により化学結合によって連結することもできる。
本発明をmRNAディスプレイ法の中で実施する場合、リンカーと前記mRNAとの連結は、ライゲーション反応により行われることが好ましい。mRNAとリンカーとの化学結合反応を促進するため、連結予定部位同士がハイブリダイゼーションしたことに起因して、近接していることが好ましい(アニーリング)。
アニーリングでは、第三の核酸分子がmRNAの3’末端近傍及びリンカーの5’末端近傍の両者に相補鎖を形成してもよい(Splint DNA法)が、mRNAの3’末端近傍とリンカーの5’末端近傍とが相補鎖を形成していることがより好ましい(Y-ligation法)(非特許文献6)。より具体的には、mRNAの3’末端近傍とリンカーの5’末端近傍とが、数塩基〜数十塩基に渡って相補鎖を形成することが好ましい。また、リンカーの5’末端とmRNA中途配列とが相補鎖を形成することも、ライゲーションによる連結を安定させる上でさらに好ましい。ただし、上記要件は、末端塩基に連なる数塩基が必ず相補鎖を形成していることや、相補鎖を形成する領域が必ず連続していることを要求するものではない。cDNAディスプレイ法の中で好適に使用できるリンカーの条件は後述する。
【0027】
本発明において、「ライゲーション」又は「ライゲーション反応」は、酵素により促進される反応をいう。こうした酵素は、酵素番号の6群にあたるリガーゼであり、DNAやRNAのライゲーション反応については、EC6.5.1.1(ATP依存性DNAリガーゼ)、EC6.5.1.2(NAD+依存性DNAリガーゼ)、EC6.5.1.3(RNAリガーゼ)等を利用することができる。RNA/DNA間ライゲーションを行うRNAリガーゼのほか、RNA間及びRNA/DNA間のライゲーションを行うDNAリガーゼを利用することもできる。
具体的には、T4 RNAリガーゼ又はTS2126耐熱性ファージ由来DNAリガーゼを好適に利用することができ、T4 RNAリガーゼを利用することが、連結効率の理由からさらに好ましい。
【0028】
ライゲーション反応前のアニーリングは、60〜100℃にて2〜60分間、ヒートブロック、アルミブロック、ウォーターバスその他の加温用器具にて温めた後、室温で2〜60分間放置して液温を穏やかに低下させ、さらに-5〜10℃に冷却して行うのが好ましい。例えば、90 ℃で5分間アルミブロック上にて温め、次に70 ℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した後、氷上に置くようにすることが好ましい。
ライゲーション反応液の組成は、0.1〜2.5 U/μlのT4ポリヌクレオチドキナーゼ; 0.4〜5 U/μlのT4 RNAリガーゼ; 10〜250 mMのTris-塩酸(pH 7.0〜8.0); 2.0〜50 mMの塩化マグネシウム; 2.0〜50 mMのジチオスレイトール(DTT); 0.2〜5.0のmM ATPを含むものであることが好ましい。反応効率の点から、0.5 U/μlのT4ポリヌクレオチドキナーゼ; 2.0 U/μlのT4 RNAリガーゼ; 50 mMのTris-塩酸(pH 7.5); 10 mMの塩化マグネシウム; 10 mMのDTT; 1.0 mMのATPを含有するものであることが好ましい。この反応液には、必要に応じて、SUPERase RNase inhibitor(Ambion社)などのRNA分解酵素阻害剤を添加してもよい。
【0029】
ライゲーション反応を行う反応系の体積は、4.0〜100 μlであることが好ましい。反応効率の点から20〜40μlとすることが好ましく、20μlとすると最も反応効率が高い。
この反応系中でのRNAとリンカーとの量比は、リンカー 5.0〜100 pmolに対し、mRNA5.0〜100 pmolとすることが好ましい。反応効率を上げ、残余を最少化するために、リンカー 10〜20 pmolに対し1/3〜6倍量のmRNAを反応させるのが好ましく、リンカー10 pmolに対し10〜15 pmolのmRNAを反応させるのが最も好ましい。
アニーリング反応は、T4ポリヌクレオチドキナーゼ及びT4 RNAリガーゼを除いたライゲーション反応液中で行うことができる。そして、アニーリングの終わった反応液中に、これらの酵素を必要量投入することで、ライゲーション反応を開始させることができる。
ライゲーション反応は、10〜40 ℃で1分間〜16時間行うのが好ましい。作業効率及び反応効率の面から、20〜30 ℃で5〜180分間とすることが好ましく、25℃で15分間とすることが最も好ましい。
【0030】
4.無細胞系タンパク質合成及び連結体の形成
連結体A(mRNAとリンカーとの連結体)を無細胞翻訳系と接触させることによって、タンパク質の合成を行う。ここで、タンパク質を合成するための無細胞翻訳系は、動物由来細胞、植物由来細胞、菌類及び細菌類からなる群から選択したものから得てもよく、また部分的にあるいは全体として人工的に合成したものでもよい。具体的には、大腸菌、ウサギ網状赤血球、小麦胚芽抽出物などを使用することができる(Lamfrom H., Grunberg-Manago M., Biochem. Biophys. Res. Commun., 27, 1 (1967))。
生物種により翻訳に利用されるコドンの種類が異なるので、対象となる遺伝子や遺伝子の由来に合わせて無細胞翻訳系を選択することが好ましい。または、利用したい翻訳系に合て、mRNAの配列や構造を再設計するのが好ましい。
【0031】
本発明の実施形態においては、前記無細胞翻訳系として哺乳類の網状赤血球細胞のライセートを利用することが好ましく、ウサギの血液から得られた網状赤血球細胞のライセートを利用することがさらに好ましい。また、血中の網状赤血球の割合を高めるため、前記哺乳動物に予めアセチルフェニルヒドラジンを投与して溶血性貧血等を誘導し、その後数日間飼育して採血をすることが好ましい。前記ライセートは、マイクロコッカルヌクレアーゼによって細胞由来のmRNAを分解し、グリコールエーテルジアミン四酢酸(EGTA)を加えてカルシウムをキレートし、前記ヌクレアーゼを不活化処理したもの(以下、「マイクロコッカルヌクレアーゼ処理済」という。)とすることが、より好ましい。
【0032】
翻訳反応液の組成は、3.4〜85 μlのウサギ網状赤血球ライセート(マイクロコッカルヌクレアーゼ処理済)と0.24〜10 pmolの上記連結体を含む反応バッファー(最終濃度:16〜400 mMの酢酸カリウム;0.1〜2.5 mMの酢酸マグネシウム;0.2〜50 mMのクレアチンリン酸;各0.00〜0.25 mMのアミノ酸を含む)を10〜100 μl用いるのが好ましい。反応効率の点から、17 μlのウサギ網状赤血球ライセート(マイクロコッカルヌクレアーゼ処理済)と1.2〜2.0 pmolの上記連結体とを含む反応バッファー(最終濃度:80 mMの酢酸カリウム;0.5 mMの酢酸マグネシウム;10 mMのクレアチンリン酸;各0.05 mMのアミノ酸(メチオニン及びロイシンは各々0.025 mM)を25 μl用いるのが、さらに好ましい。
また必要に応じて、この反応液に、SUPERase RNase inhibitor(Ambion社)などのRNA分解酵素阻害剤を添加してもよい。
【0033】
翻訳反応は20〜40 ℃で10〜90分間行うことが好ましく、生成効率と作業効率の点から、30℃で20分間行うことが特に好ましい。
翻訳反応後、連結体Aと翻訳産物(タンパク質)との連結(連結体Bの形成)を、高塩濃度条件下にて促進することができる。連結体Bの形成促進は、終濃度0.3〜1.6 Mの塩化カリウム及び終濃度40〜170 mMの塩化マグネシウム存在下で行うのが好ましい。生成効率の点から、25μlの上記反応液に7〜10μlの3Mの塩化カリウム、及び3μlの1Mの塩化マグネシウム(終濃度:0.60〜0.79 mMの塩化カリウム;79〜86 mMの塩化マグネシウム)を添加することが、さらに好ましい。
連結体Bの形成促進は27〜47 ℃で30分〜3時間という反応条件下で行われることが好ましい。生成効率と作業効率の点から、37 ℃で90〜120分間反応させることが特に好ましい。
【0034】
5.連結体Aとタンパク質の連結(IVV形成)
連結体Aを翻訳系に投入してタンパク質またはポリペプチド鎖を液相合成する際に、そのC末端に、前記mRNAと翻訳されたタンパク質とがリンカー及びリンカーの有するタンパク質結合部位を介して個々の分子ごとに連結される。ここで、前記タンパク質結合部位は、アミノアシルtRNA3’末端アナログを含むことが好ましく、前記アミノアシルtRNA3’末端アナログはピューロマイシンであることが好ましい。
すなわち、mRNAにピューロマイシン等を有するリンカーを結合させた連結体Aと翻訳系とを接触させると、そのmRNAがピューロマイシンを介して翻訳されたタンパク質と結合したIVVが生成する。(非特許文献1参照)。
【0035】
翻訳されたタンパク質はさらに、N末端側のペプチドの除去、糖鎖や脂質などの非アミノ酸成分の付加、アミノ酸の翻訳後修飾、又は立体構造や配列構造に基づく前記mRNAに対する親和性結合や、タンパク質の酵素活性によるmRNAの化学修飾その他の翻訳後修飾がなされてもよい。
こうした翻訳後修飾は、前記mRNAにコードされた遺伝情報に基づいて行われてもよく、前記遺伝情報ではなく外的要因に基づいて行われてもよい。また、これらの組み合わせに基づくものであってもよい。遺伝子情報に基づく反応としては、二量体及び多量体形成、自己フォールディング、分子内分解反応、元のmRNAとの複合体形成等が挙げられる。外的要因による反応としては、修飾基、脂質、又は糖鎖等の付加、分子シャペロンによるフォールディング又は複合体形成、その他の巨大構造物(ウィルス・細胞)との親和性結合等が挙げられる。遺伝子情報及び外的要因の組み合わせによる反応としては、当該タンパク質の特定配列部位における化学修飾や分解などが挙げられる。
ただし、前記反応はこれらに限定されるものではない。また、これらの反応は、解析やスクリーニング等の補助のために行うこともでき、それ自体を解析やスクリーニングの対象とすることもできる。
【0036】
アミノアシルtRNA3’末端アナログの1つであるピューロマイシンは、下記式Iで表される化合物であり、翻訳系でタンパク質の合成が行われた際に、合成されたタンパク質のC末端に結合する能力を有する。
【0037】
【化1】

【0038】
ピューロマイシンは翻訳系でタンパク質の合成が行われた際に、合成されたタンパク質のC末端に結合する能力を有する。その他のアミノアシルtRNA3’末端アナログとしては、例えば、3'-N-アミノアシルピューロマイシンアミノヌクレオシド(3'-N-Aminoacylpuromycin aminonucleoside, PANS-アミノ酸)を挙げることができる。PANS-アミノ酸としては、アミノ酸部がグリシンのPANS-Gly、バリンのPANS-Val、アラニンのPANS-Ala、又はアミノ酸部が全ての各アミノ酸に対応するPANS-アミノ酸混合物を挙げることができる。
その他には、3'-アミノアデノシンのアミノ基とアミノ酸のカルボキシル基が脱水縮合して形成される、アミド結合で連結した3'-N-アミノアシルアデノシンアミノヌクレオシド(3'-Aminoacyladenosine aminonucleoside, AANS-アミノ酸)を挙げることができる。AANS-アミノ酸としては、アミノ酸部がグリシンのAANS-Gly、バリンのAANS-Val、アラニンのAANS-Ala、又はアミノ酸部が全アミノ酸の各アミノ酸に対応するAANS-アミノ酸混合物を挙げることができる。
また、ヌクレオシドあるいはヌクレオシドとアミノ酸のエステル結合したものなども使用できる。
ピューロマイシン以外に好適に使用できるアミノアシルtRNA3'末端アナログとしては、リボシチジルピューロマイシン(rCpPur)、デオキシジルピューロマイシン(dCpPur)、デオキシウリジルピューロマイシン(dUpPur)などがあり、これらを下記式(II)〜(VI)に示す。
【0039】
【化2】
【0040】
【化3】
【0041】
【化4】
【0042】
【化5】
【0043】
【化6】
【0044】
また、アミノアシルtRNAアナログは、公知の手法によってリンカーに付加することができる。mRNAディスプレイ法では、アミノアシルtRNAアナログを有するタンパク質連結部位をリンカーの主鎖の5’末端に形成してもよい。一方、cDNAディスプレイ法において、cDNA型IVVを形成するために、リンカーの主鎖の5’末端をmRNAとの相補鎖形成に用いる場合等には、上記のタンパク質結合部位をリンカーの側鎖中に形成することが好ましい。
リンカーの側鎖が短いと、アミノアシルtRNAアナログが取り込まれるときに翻訳複合体の立体障害を受けることから、リンカーの側鎖は十分な長さを持つものであることが好ましい。リンカーの側鎖は、例えば、下記式(VII)のSpacer 18 Phosphoramiditeのようなスペーサー分子を1〜8個程度含むものであることが好ましく、リンカー合成効率と連結体形成効率のバランスとから、こうしたスペーサー分子を4個含むものであることがさらに好ましい。
【0045】
【化7】
【0046】
リンカーの主鎖と側鎖との連結は公知の方法に従って行うことができる。例えば、リンカーの主鎖が下記式(VIII)のAmino-Modifier C6 dTを含み、側鎖が下記式(IX)の5'-Thiol-Modifier C6から合成された5'末端を含む場合には、架橋剤として下記式(X)で表わされるEMCSを利用することができる。
【0047】
【化8】
【0048】
【化9】
【0049】
【化10】
【0050】
本発明で使用するリンカーは、必要に応じて、タンパク質を連結体Bから遊離させるために、切断部位(以下、「タンパク質遊離部位」ということがある。)を設けることができる。このようなタンパク質遊離部位として、例えば、光切断部位等を挙げることができる。
ここで、「光切断性部位」とは,紫外線等の光を照射すると分解される性質を持つ化合物を有する部位いい,市販の化合物を用いることもできる。例えば、下記式(XI)で表されるPC Linker Phosphoramidite(Glen research社)、フラーレンを含有してなる核酸の光切断用組成物に係る化合物(特許文献8)、光分解(SBIP)手法による鎖切断に係る化合物などが好ましい。切断効率の点から、PC Linker Phosphoramiditeを利用することが好ましい。
【0051】
【化11】
【0052】
6.IVVの検出
上記連結体A又は連結体Bは、あらかじめリンカーに標識物質を結合させておくことによって、容易に検出することができる。そのような標識物質としては、蛍光性化合物、抗原のエピトープペプチド、放射性同位体等を挙げることができる。放射性同位体としては、3H、14C、32P、125I若しくは131I等の放射性同位体を使用することが好ましい。
放射性同位体の利用が困難な場合には、蛍光性化合物を利用することが好ましい。蛍光性化合物としては、フリーの官能基(例えば、活性エステルに変換可能なカルボキシル基、ホスホアミダイドに変換可能な水酸基、又はアミノ基など)を持ち、標識された塩基としてリンカーに連結可能な種々の蛍光色素を用いることが好ましい。このような蛍光色素としては、例えば、フルオレセインイソチオシアネート(FITC)、フィコビリタンパク質、希土類金属キレート、ダンシルクロライド又はテトラメチルローダミンイソチオシアネート等を挙げることができる。蛍光標識分子としては、FITC-dTや下記式(XII)で表わされるFluorescein-dTを使用することがより好ましい。
【0053】
【化12】
【0054】
7.連結体Bの固相固定
本発明で使用するリンカーは、その主鎖中に上記連結体Bを固相部位に結合するための固相結合部位を有することが好ましい。
連結体Bが固定される固相は特に限定されず、その連結体が使用される目的に応じて適宜選択される。こうした固相としては、生体分子を固定する担体となる各種の形状のものを用いることができる。例えば、スチレンビーズ、ガラスビーズ、アガロースビーズ、セファロースビーズ、磁性体ビーズ等のビーズ;ガラス基板、シリコン(石英)基板、プラスチック基板、金属基板(例えば、金箔基板)等の基板;ガラス容器、プラスチック容器等の容器;ニトロセルロース、ポリビニリデンフロリド(PVDF)等の材料からなるメンブレンなどが挙げられる。
【0055】
上記連結体Bでは、リンカーに固相結合部位を設けることで、その固相結合部位と、固相に結合させた「固相結合部位認識部位」とを介して、連結体Bを固相に固定することができる。「固相結合部位認識部位」とは、固相に設けられた部位であって、固相結合部位の有する物質に応じて、固相結合部位に結合する部位である。上記固相結合部位は、連結体Bを所望の固相に結合し得るものであればよく、特に限定されない。例えば、リガンド、エピトープペプチドその他の特定のポリペプチドに特異的に結合する構造単位を用いる場合には、固相表面には、これらと特定に結合する特定のポリペプチド等を結合させておくことになる。
こうした固相結合部位認識部位/固相結合部位の各々の有する物質の組合せの例としては、例えば、アビジン及びストレプトアビジンその他のビオチン結合タンパク質/ビオチン、マルトース結合タンパク質/マルトース、Gタンパク質/グアニンヌクレオチド、ポリヒスチジンペプチド/ニッケルあるいはコバルト等の金属イオン、グルタチオン−S−トランスフェラーゼ/グルタチオン、配列特異的なDNA又はRNA結合タンパク質/特異配列を有するDNA又はRNA、抗体又はアプタマー/抗体又はエピトープペプチド、カルモジュリン/カルモジュリン結合ペプチド、ATP結合タンパク質/ATP、エストラジオール受容体タンパク質/エストラジオール等の各種受容体タンパク質/そのリガンド等が挙げられる。
これらのうち、上記ビオチン結合タンパク質/ビオチン、マルトース結合タンパク質/マルトース、ポリヒスチジンペプチド/上記金属イオン、グルタチオン−S−トランスフェラーゼ/グルタチオン、抗体/抗原分子(エピトープペプチド)等を使用することが好ましく、リンカー合成の容易さの面から、ストレプトアビジン/ビオチンの組合せを使用することが、さらに好ましい。
【0056】
上記固相結合部位は、例えば、連結体Bをリンカーを介して固相に結合させるための部位であり、具体的には、こうした結合を可能にする化合物、結合基などを結合し得る塩基で構成される。より具体的には、ビオチンを結合し得る塩基(例えば、デオキシチミン(dT))もしくはビオチンが結合した塩基(例えば、ビオチン−デオキシチミン(Biotin-dT))、アミノ基によって修飾された塩基(例えば、アミノ修飾デオキシチミン(例、Amino-Modifier C6-dT:Glen Research Search社製)、カルボキシ基によって修飾された塩基(例えば、カルボキシ修飾デオキシチミン(Carboxy-dT))、チオール基によって修飾された塩基(例えば、チオール修飾デオキシチミン(4-Thio-dT))等で構成される。
上記リンカーでは、固相結合部位として下記式(XIII)のビオチン−デオキシチミンを好適に使用することができる。そして、このような固相結合部位を介し、ビオチンとアビジンの親和性を利用して、ビオチンが連結された連結体Bをアビジンが固定された固相に結合させることができる。なお、上記固相結合部位として、カルボキル基やアミノ基が結合した塩基が選択された場合は、固相と連結体Bとをエステル結合やアミド結合で結合させることができる。
【0057】
【化13】
【0058】
上記の方法で固相に固定された連結体Bを用いて、洗浄後公知の方法で逆転写反応を行う。逆転写反応後、再び洗浄し、新たに加えた所定のバッファー中で固相より遊離する。
本発明で使用するリンカーでは、必要に応じて、連結体Bを固相から遊離させるために、第1及び第2切断部位(以下、「固相遊離部位」という。)を設ける。上記固相遊離部位は、前記タンパク質遊離部位と同様の条件で切断又は分解されるようにすることもできる。スクリーニング等の工程をスムーズに進める上では、当該固相遊離部位は、前記タンパク質遊離部位と同様の条件で切断又は分解されない構成とすることが好ましい。
上記リンカーでは、例えば、リボG(グアノシン;rG;Guanosine)等の特定の酵素が認識可能な塩基を含む配列として前記固相遊離部位を構成し、こうした酵素を用いて、固相に結合された連結体Bを固相から遊離させることが好ましい。
【0059】
ここで用いられる酵素は、リンカーの設計に応じて決定することができ、例えば、主鎖の鎖長に余裕があれば制限酵素(旧EC番号3.1.23, 3.1.24, 3.1.25)を利用することができる。また、リンカーがDNAを主体に形成されている場合には、RNAを切断部位として、RNA切断酵素(EC番号3.1.26, 3.1.27)を利用することができる。また、酵素切断部位を、使用する酵素の種類に応じて適宜選択し、設計することもできる。
【0060】
RNA切断酵素としては、RNaseT1、RNaseA、RNaseI、膵臓RNase、S1ヌクレアーゼ、蛇毒ヌクレアーゼ、脾臓ホスホジエステラーゼ、スタフィロコッカスヌクレアーゼ、マングビーンヌクレアーゼ、アカパンカビヌクレアーゼ等を用いることができる。上記切断部位としては、これらの酵素のうち、RNaseT1、RNaseA、RNaseI、膵臓RNaseによって切断される部位であることが好ましく、特に、RNaseT1によって認識され、切断されるリボG(rG;グアノシン)を含む配列であることが好ましい。
RNaseT1は、rGを1塩基で認識してリンカーを切断するため、rGを使用するとリンカー主鎖長を短くすることが出来る。その結果、上述したライゲーションの際に、リンカー濃度を上げても凝集しにくいという利点がある。
【0061】
リボヌクレアーゼT1(RNaseT1)等の一本鎖特異的RNA切断酵素により切断されることを予定するリンカーでは、図2に示すように、リンカー2中で固相結合部位2bが第1切断部位2c1と第2切断部位2c2との間に位置し、これらは、リンカーの主鎖の3’末端近傍の一本鎖ループに位置していなければならない。なお、図2にはリンカーと相補鎖を形成するmRNA1、リンカーの主鎖200、リンカーの側鎖300等も表されている。
【0062】
上記の特定のポリペプチド等と固相結合部位との結合は、IVVの変性を避けるために分子間の親和性のみを利用して行うことが好ましいが、その他の公知の方法を用いることもできる。そのような公知の方法としては、例えば、タンニン酸、ホルマリン、グルタルアルデヒド、ピルビックアルデヒド、ビス−ジアゾ化ベンジゾン、トルエン−2,4−ジイソシアネート等を利用する方法を挙げることができる。また、上記の特定のポリペプチド等及び固相結合部位の有する分子としては、アミノ基、カルボキシル基、又は水酸基などの官能基を構成する分子が好ましい。
上記の特定のポリペプチド等と固相を構成する要素との結合は、公知の技術によって行うことができ、上述した市販の前記ビーズや前記基板等を利用してもよい。また上記固相遊離部位は固相側に設けることもできる。上記の固定化手段は、2つの相互に親和性を有する物質を利用した固定化方法であるが、固相がスチレンビーズ、スチレン基板などのプラスチック材料で構成されているのであれば、必要に応じて、公知の手法を用いてリンカーの一部を直接それらの固相に共有結合させることもできる(Qiagen社、LiquiChip Applications Handbook等参照)。なお、本発明においては、固定化手段については、上記の方法に限定されることなく、当業者に公知であれば如何なる手段をも利用することができる。
【0063】
8.本発明におけるIVV合成法と従来法との比較
ここで、図3及び図4を参照しつつ、上記連結体A又は連結体Bを用いたIVV法における従来法と、本発明による方法と対比しつつ説明する。図中、同じ要素には同じ番号を付し、説明を省略する。
従来法を示す図3では、遺伝子情報を有するmRNA1とリンカー2とは所定の方法によって連結される。ここでは、第1切断部位2c1及び第2切断部位2c2を合わせて固相遊離部位2cと表す。図3では、ハイブリダイゼーション反応につづくライゲーション反応により、mRNA1とリンカー2とが連結され連結体Aが形成される(Step 1)。連結体の合成効率を高めるために、mRNA1とリンカー2とは1:1.5〜1:3の範囲で混合され、結果として過剰なリンカー2が未反応のまま残される。
【0064】
未反応のリンカー2を除去するためにカラム精製を行い(Step 2)、次に、精製した連結体3(連結体A)を濃縮するためエタノール沈殿を行う(Step 3)。次いで、連結体Aを、例えば、無細胞翻訳系に供することによって(Step 4)、mRNA1に対応するタンパク質5が、タンパク質結合部位2aに結合された状態で、連結体A上で合成される(Step 5)。この結果、mRNA1、リンカー2及びタンパク質5からなる連結体4(連結体B)が生成する。連結体Bは、いわゆるIn Vitro Virus (IVV)である。
固相結合部位認識部位6aを有する固相ビーズ6に、得られた連結体Bが固定され(Step 5)、これを逆転写反応に供することによって、mRNAに対応するcDNA7が生成される(Step 6)。ここで、RNaseT1などの酵素を用いて、固相遊離部位2cでリンカーを切断すると、固相ビーズ6から遊離させることができる(Step7)。さらに、標識2dを利用することで、各工程におけるタンパク質5、連結体3又は連結体4などの合成効率を定量することができる。
【0065】
また図3では省略したが、RNaseHでmRNAを分解すると、タンパク質とそれをコードするDNAが連結したDNA−タンパク質連結体(DNAのみに遺伝子を有するIVV)が生成される。タンパク質の機能や構造に基づいてスクリーニングを行いその後、光切断などの手法でタンパク質遊離部位2eにてリンカーを切断すると、タンパク質5を遊離することができる。
本発明による方法を示す図4中、図3と同一の符号を付したものは、同一の要素を表す。Step 1で合成された連結体Aは精製及び濃縮の工程を経ることなくStep 4に供され、タンパク質が合成される。その後の固相化工程(Step 5)以降は従来法と同様である。
本発明の方法を用いることにより、IVV形成またはmRNA/cDNAディスプレイ法において、二工程の短縮と、カラム精製を省略することができる。さらに、精製工程がないことから、mRNA/cDNAディスプレイ法によって、ペプチドスクリーニングも効率的に行うことができる。
【0066】
9.ランダムな遺伝子ライブラリーから目的のタンパク質をコードする遺伝子をスクリーニングする方法
本発明の別の態様によれば、上記IVVを用いたタンパク質と遺伝子との対応関係を明らかにする、効率的な方法を利用したペプチドスクリーニング法が提供される。この解析方法は、(a)一種以上の遺伝子を含むmRNAのライブラリーを、IVV合成系に投入し、連結体Bを形成する工程;(b)工程(a)において合成されたタンパク質と一以上の標的物質とを接触させる工程;及び(c)該タンパク質と該標的物質とが相互作用しているか否かを測定する工程、とを含む。
この解析方法は、例えば、(i)配列既知のタンパク質に作用する物質をスクリーニングする場合、(ii)ある特定の物質(例えば、リガンド)が結合する配列未知のタンパク質をスクリーニングする場合等に用いることができる。例えば、(i)の場合は、オーファンレセプタータンパク質をコードする核酸配列を有するmRNAと、ピューロマイシンとの連結体を複数用意しておき(すなわち、複数のオーファンレセプタータンパク質に対応するmRNAをそれぞれ有する連結体A又は連結体Bを複数用意する。)、これを翻訳系に投入する。すると、各連結体A又は連結体BのmRNAから複数のオーファンレセプタータンパク質が合成される。
【0067】
各オーファンレセプタータンパク質は、固相に固定された連結体A又は連結体Bのピューロマイシンに、C末端が結合することによって固定される。必要に応じて、不要な成分を洗浄して除去し、これに標的物質及びバッファー等を加えて、標的物質をオーファンレセプタータンパク質に結合させる試験を行う。(ii)の場合は、例えば、ある遺伝子ライブラリーから複数のmRNAを取得し、複数のmRNAとピューロマイシンとの連結体を作成し、固相に固定する。以下、同様にタンパク質の合成を行い、標的物質をそのタンパク質に接触させて結合実験を行う。
上記(b)工程では、(a)工程で合成されたタンパク質と、一以上の標的物質とを接触させる。ここで用いられる「標的物質」とは、本発明において合成されるタンパク質と相互作用するか否か調べるための物質を意味し、具体的には、タンパク質、核酸、糖鎖、低分子化合物などが挙げられる。
【0068】
前記標的物質としてのタンパク質は、アミノ酸配列及びその機能が既知のタンパク質でも、未知のタンパク質でもよく、タンパク質の全長であっても結合活性部位を含む部分ペプチドでもよい。これらは、合成されたペプチド鎖、生体より精製されたタンパク質、又はcDNAライブラリー等から適当な翻訳系を用いて翻訳し、精製したタンパク質等であってもよい。合成されたペプチド鎖には、糖鎖が結合していてもよい。これらのうち、アミノ酸配列が既知の精製されたタンパク質か、又はcDNAライブラリー等から適当な方法を用いて翻訳、精製されたタンパク質を好適に標的とすることができる。
前記標的物質としての核酸も特に制限されることはなく、DNA、RNAのいずれをも標的とすることができる。また、塩基配列又は機能が未知の核酸のみならず、既知の核酸も用いることができる。タンパク質に結合能力を有する核酸としての機能、及び塩基配列が既知のものか、又はゲノムライブラリー等から制限酵素等を用いて切断単離してきたものに対し、本発明の方法を好適に使用することができる。
【0069】
前記標的物質としての糖鎖も特に制限はなく、その糖配列又は機能が、未知の糖鎖のみならず既知の糖鎖を標的とすることができる。好ましくは、既に分離・解析され、糖配列又は機能が既知の糖鎖が用いられる。
前記標的物質としての低分子化合物についても特に制限はなく、機能が未知のもの、又はタンパク質に結合する能力が既に知られているもの等を用いることができる。
なお、これら標的物質と、リンカーに連結されたタンパク質との「相互作用」とは、通常は、タンパク質と標的分子との間の共有結合、疎水結合、水素結合、ファンデルワールス結合、及び静電力による結合のうち少なくとも1つから生じる分子間に働く力による作用を示す。ここでいう共有結合には、配位結合、双極子結合が含まれる。また、静電力による結合とは、静電結合の他、電気的反発による立体構造に起因した結合が含まれる。また、上記作用の結果生じる結合反応、合成反応、分解反応も相互作用に含有される。相互作用の具体例としては、抗原と抗体との結合及び解離、タンパク質レセプターとリガンドとの間の結合及び解離、接着分子と相手方分子との結合及び解離、酵素と基質の間の結合及び解離、核酸とそれに結合するタンパク質の間の結合及び解離、情報伝達系におけるタンパク質同士の間の結合と解離、糖タンパク質とタンパク質との間の結合及び解離、あるいは糖鎖とタンパク質との間の結合及び解離が挙げられる。
【0070】
ここで用いられる標的物質は、必要に応じて、上述した標識物質により標識して用いることができる。これらの標識物質は、標的物質と固定化タンパク質との間の相互作用に基づいて発生する信号の変化の測定又は解析方法に適したものを選択すればよい。上記標識物質の標的物質への結合は、公知の手法に基づいて行うことができる。
次いで、本解析方法によれば、工程(c)において、タンパク質と標的物質とが相互作用しているか否かを測定する。タンパク質と標的物質とが相互作用しているか否かの測定は、両分子間の相互作用に基づいて発生される信号の変化を測定、検出することにより行う。
そのような測定手法としては、例えば、表面プラズモン共鳴法(Cullen D.C., et al., Biosensors, 3(4), 211-225 (1987-88))、エバネッセント場分子イメージング法(Funatsu T., et al., Nature, 374, 555-559 (1995))、蛍光イメージングアナライズ法、固相酵素免疫検定法(Enzyme Linked Immunosorbent Assay (ELISA):Crowther J.R., Methods in Molecular Biology, 42 (1995))、蛍光偏光解消法(Perran J., et al., J. Phys. Rad., 1, 390-401 (1926))、及び蛍光相関分光法(Fluorescence Correlation Spectroscopy (FCS):Eigen M., et al., Proc. Natl. Acad. Sci. USA, 91, 5740-5747 (1994))等が挙げられる。
【0071】
本発明の解析方法においては、必要に応じて、さらに、(c)工程において相互作用していると判断されたタンパク質−標的物質結合体中の、タンパク質及び/又は標的物質を同定する。タンパク質の同定は、通常のアミノ酸配列シークエンサーで行うこともでき、こうしたタンパク質に結合しているRNAからDNAを逆転写し、得られたDNAの塩基配列を解析することによって行うこともできる。標的物質の同定は、NMR、IR、各種質量分析等を用いて行うことができる。
なお、本発明のRNAチップ及びプロテインチップを用いて、タンパク質−タンパク質間相互作用を解析する場合は、通常のプロテインチップ上のサンプル解析と同様に、飛行時間型質量分析計(MALDI−TOF MS)を用いることができる。
【実施例1】
【0072】
(rGを切断部位とするリンカーを利用したIVV合成)
以下、本発明を実施例に基づいてより具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
1.リンカーの合成
本発明で使用するShort-Biotin-ピューロマイシン・リンカー(SBPリンカー)を合成した。まず、以下の特殊DNA合成をジーンワールド(株)(東京)及び(株)BEX(東京)に委託した。
【0073】
(A)Puro-F-S[配列;5'-(S)-(PL)C(F)-(Spacer18)-(Spacer18)-(Spacer18)-(Spacer18)-CC-(Puro)-3']
ここで、(S)は5'-Thiol-Modifier C6で化合物名はS-Trityl-6-mercaptohexyl-1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite;(PL)はPC Linker Phosphoramiditeで3-(4,4'-Dimethoxytrityl)-1-(2-nitrophenyl)-1-propanyl-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite;(F)はFluorescein-dTで(5'-Dimethoxytrityloxy-5-[N-((3',6'-dipivaloylfluoresceinyl)-aminohexyl)-3-acryimido]-2'-deoxyUridine-3'-succinoyl-long chain alkylamino);(Puro)はピューロマイシン;(Spacer18)はSpacer Phosphoramidite 18(18-0-Dimethoxytritylhexaethyleneglycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)である(すべてGlen Research社製)。
(B)Hybri[(26mer) 配列番号1:5'-CC(rG)C(T-B) C(rG)CCC CGCCG CCCCC CG(T)CC T-3']
ここで、(rG)はリボG;(T)はAmino-Modifier C6 dT(5'-Dimethoxytrityl-5-[N-(trifluoroacetylaminohexyl)-3-acrylimido]-2'-deoxyUridine,3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite);(T-B)はBiotin-dT(5'-Dimethoxytrityloxy-5-[N-((4-t-butylbenzoyl)-biotinyl)-aminohexyl)-3-acrylimido]-2'-deoxyUridine-3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)である(すべてGlen Research Search社製)。
【0074】
上記SBPリンカーは、(A)Puro-F-Sと(B)Hybriを以下の方法に従って架橋し精製して得た。
Puro-F-S 10 nmolを100 μlの50 mMリン酸バッファー(pH 7.0)に溶かし、100 mM Tris[2-carboxyethyl]phosphine(TCEP、Pierce社製)を1μl加え(最終濃度 1mM)、室温で6時間放置し、Puro-F-Sのトリチル化メルカプト基をチオール基に還元した。架橋反応を行う直前に、50 mMリン酸バッファー(pH 7.0)で平衡化したNAP-5 Columns(GEヘルスケア・ジャパン(株)製)を用いてTCEPの除去及び脱塩を行った。
0.2 Mリン酸バッファー(pH 7.0)100 μlに、500 pmol/μlのHybriを20 μl、100 mMの架橋剤EMCS(6-Maleimidohexanoic acid N-hydroxysuccinide ester)、(株)同仁化学研究所製)20 μl、を加えて良く攪拌し、37 ℃で30分反応させた。その後、4℃でエタノール沈殿を行って反応産物を沈殿させ、未反応のEMCSを除去した。沈殿を500μlの冷70%エタノールにて洗浄し、減圧下で乾燥させた後、0.2 Mリン酸バッファー(pH 7.0)10 μlに溶解した。
この反応産物に、上記還元したPuro-F-S(〜10 nmol)を速やかに加えて、4℃で一晩放置した。サンプルに終濃度が4mMになるようにTCEPを加え、37 ℃で15分間放置し、チオール基の架橋反応を停止させた。室温にてエタノール沈殿を行い、合成したリンカーを沈殿させ、未反応のPuro-F-Sを取り除き、さらに未反応のHybri、及びそれらのEMCS架橋物を取り除くために、以下の条件でHPLC精製を行った。
【0075】
(HPLC条件)
カラム:COSMOSIL(登録商標)10 x 250 mm C18-AR-300(ナカライテスク(株)製)
バッファーA:0.1 M TEAA(triethylammonium acetate)
バッファーB:80 %アセトニトリル(超純水で希釈したもの)
流速:0.5 ml/分
濃度勾配:A及びBの混合バッファー濃度は85:15→65:15(A:B)で変化させる(33分間)。
HPLCにより分画された生成物を18%アクリルアミドゲル(8M 尿素、62℃)の電気泳動にて検出し、目的の画分を減圧下で乾燥させた。この後、DEPC(Diethylpyrocarbonate)処理水で溶かして、10 pmol/μlとした。
【0076】
2.mRNAの合成
モデルmRNAとしてBDA(B domain of Protein A)を用いることとした。BDA遺伝子(BDA gene;配列番号2;192塩基長)の5'側上流にT7プロモーター配列と翻訳促進配列、3'側下流にスペーサー領域、ヒスチジンタグ(Hisタグ)及びピューロマイシン・リンカーとの相補鎖領域を有する配列を付加したDNA(BDA whole;配列番号3;367塩基長)をPCRによって合成、精製した後、T7 RiboMAX Express Large Scale RNA Production System(プロメガ(株)製))を用いて、添付のプロトコールに従って5〜30 pmol/μlのmRNA(BDA mRNA;配列番号4;337塩基長)を合成した。
【0077】
3.連結体の形成
3.1 T4 RNAリガーゼを用いたライゲーション酵素反応
ライゲーション反応は、リンカー1に対して1〜6倍のmRNAを加え、20 μlのT4 RNAリガーゼバッファー(50 mM Tris-HCl, pH 7.5; 10 mM 塩化マグネシウム; 10 mM DTT; 1mM ATP)中にて行なった。酵素を加える前にアニーリングするため、90 ℃で5分間アルミブッロク上にて温めた、次に70 ℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した後、氷上に置いた。ここに、1μlのT4ポリヌクレオチドキナーゼ(10 U/μl)及び1μlのT4 RNAリガーゼ(40 U/μl)(いずれもタカラバイオ(株)製)を加え、25 ℃で15分間、保持した。
生成反応液を半分に分割して、後の実験工程にて比較するため、リンカー10 pmolに対しmRNA 10 pmolを反応させたサンプルについては、全ての物質を倍に増量して40 μlの系にて反応させた(反応系R1)。
【0078】
3.2 ライゲーション反応の結果
以下に示す混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、65℃、8M 尿素、5%変性アクリルアミドゲルを用い、200Vにて25分間の電気泳動にて行った。サンプルを各々1μlずつ分注し、3μlのローディングバッファーと2μlのDEPC水と混合して、ゲルにロードした。結果を図5に示す。バンドの検出はリンカーに結合したFITCの蛍光により行い、未反応のリンカー又は上記連結体Aを検出した。
図5は、10 pmolのmRNAに対して、10 pmolのリンカーを加え(前記40 μlの反応系にて反応させたもの)(レーン1、反応系R1)、6.6 pmol(レーン2、反応系R2)、3.3 pmol(レーン3、反応系R3)、1.66 pmol(レーン4、反応系R4)、上記の通りライゲーション反応させたものの電気泳動像を表す。なお、図5中、矢印は未反応のリンカー、*はリンカーが連結された反応産物(連結体A)を示す。
図5から分かるとおり、レーン1においてリンカーの残余が見られたが、その他のレーンではほとんど見られなかった。また混合比1:1の場合を除き、連結体Aの合成量に差はなく、いずれにおいてもほぼ等量のライゲーション反応の起きたことが観察された。
【0079】
図6には、上記のサンプルについてSYBR Goldにて染色した電気泳動像を示した。電気泳動の条件は上記と同様とした。なお、図6中、矢印は未反応のリンカー、*はリンカーが連結された合成物(連結体A)、**は連結体を形成せずに残存するmRNAをそれぞれ示す。レーンMは分子量マーカーを、その左側の数字は各バンドの分子量を表す。
図6から分かるとおり、レーン1においてリンカーの残余が見られたが、その他のレーンではほとんど見られなかった。またレーン1を除き、連結体Aの合成量に差はなく、mRNAの投入量が増えるにつれてmRNAの残余量の増加していることが観察された。
以上より、リンカーに対して1.0〜1.5倍量のmRNAがあれば連結体Aの形成には十分なことが示された。
【0080】
4.連結体Aの精製と濃縮
ライゲーション後の反応生成物の精製及び濃縮の要否を確認するために、精製及び濃縮についての実験結果を示す。
前記R1より20 μlを分取し、以下の精製濃縮工程に供した(反応系R1’)。R1の残りとR2〜R4については、ライゲーション反応後、そのままの状態で冷却して保持した。
未連結のリンカーと反応液とを除くために、RNeasy(登録商標) Kit((株)キアゲン)を用いて、添付のプロトコールに従い精製した。DEPC水により溶出した連結体A(30-50μl)は、共沈剤Quick-Precip Plus solution(EdgeBio社)を用いて添付のプロトコールに従い、エタノール沈殿させた。これを12μlのDEPC水に溶解し、翻訳用テンプレートとした。
【0081】
5.IVV形成(翻訳と並行する連結体A及びタンパク質の連結)
5.1 IVV(連結体B)合成
無細胞翻訳系にはRetic Lysate IVT Kit(Ambion社製)を利用した。混合の方法等は、キットのProtocolを参照して行った。無細胞翻訳反応に使用される全ての試薬を穏やかに撹拌して遠心した後、氷上に置いた。25μlスケールの反応液は次のような順番で混合して調製し、反応させた。
0.625μlの20X Translation Mix minus-Met、0.625μlの20X Translation Mix minus-Leu、及び17μのRetic lysateを泡立たないよう注意深くピペッテイングし、混合した。
この混合液を2.4μlのR1'及び4.0μlのR1〜4に添加した。さらに反応液が25μlになるようにDEPC水を加えた。そして再び泡がでないように丁寧に混合した。混合の後30 ℃で20分間、翻訳反応をさせた。
上記翻訳反応後に、連結体Aと合成タンパク質の連結を促進するため、上記反応液に10μlの3M 塩化カリウム、及び3μlの1M 塩化マグネシウムを加え37 ℃で90分間反応させた。
【0082】
5.2 IVV(連結体B)合成効率の比較
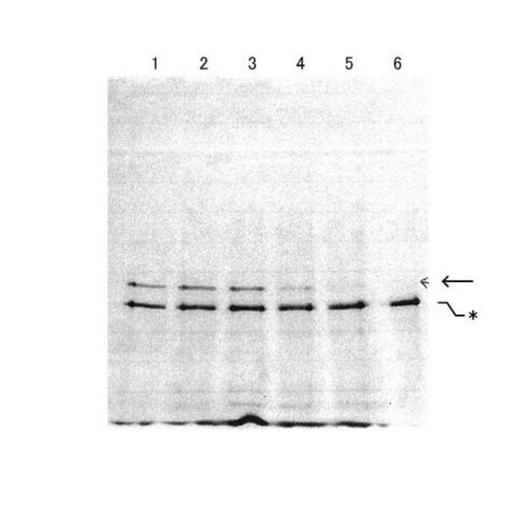
上記の通り合成した連結体Bについて、上記同様の混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、8M 尿素の6%SDSアクリルアミドゲを用いて、0.01Aで60分間、その後、さらに0.02Aで60分間、電気泳動を行った。その結果を図7に示す。バンドの検出はリンカーに結合したFITCの蛍光により行った。これにより、連結体A又はBを観察することができる。
図7は、レーン1:R1’、レーン2:R1、レーン3:R2、レーン4:R3、レーン5:R4、レーン6:連結体Aを表す。なお、図7中、*は連結体A、矢印は連結体Bのそれぞれの分子量を示す。
図7から分かるとおり、R1とR2においてはレーン1のR1'と遜色の無い連結体Bの生成が見られた。その一方で、R3とR4においては生成量が少なく、mRNAの量比が増えるにつれて連結体の形成効率が低下することが観察された。
【0083】
以上より、ライゲーション反応後のシリカゲルメンブレンカラム精製及びエタノール沈殿による濃縮を行わずとも、IVVを形成できることが示された。また、リンカーとmRNAの混合量比を1.0:1.0〜1.5に設定することで、上記精製及び濃縮を行った場合と遜色の無いIVV形成効率を得られることが示された。
本発明によれば、低コストで実施可能なIVV形成法を提供できる。また、本発明の好ましい態様における利用は、従来の方法と比較して遜色の無いIVV合成効率を有するため、mRNA/cDNAディスプレイ法の作動効率を低下させることなく、工程を簡略化できる。このことにより、本発明が、DNA及びタンパク質の機能解析及び機能タンパク質の取得を目指すハイスループットな進化分子工学的手法に応用されることで、上記新規医薬創出の要請に応えるものである。
【産業上の利用可能性】
【0084】
また、本発明は上記創薬を始めとして、医療診断、環境分析、食品分析、研究用バイオイメージングの各分野で有用である。
【符号の説明】
【0085】
1 mRNA
2 リンカー
2a タンパク質結合部位
2b 固相結合部位
2c 固相遊離部位
2c1 第1切断部位
2c2 第2切断部位
2d 標識
2e タンパク質遊離部位
3 連結体A
4 連結体B
5 タンパク質
6 固相ビーズ
6a 固相結合部位認識部位
7 cDNA
200 主鎖
201 主鎖5’末端
202 主鎖5’末端近傍部
203 主鎖3’末端側領域部
300 側鎖
【配列表フリーテキスト】
【0086】
Hybri
Guanosine
Biotin-dT
Amino-Modifier C6 dT
BDA whole
BDA mRNA
【技術分野】
【0001】
本発明は進化工学的ディスプレイ技術における、mRNA(メッセンジャーRNA)−リンカー−タンパク質連結体を、効率よく合成する方法に関する。
【背景技術】
【0002】
遺伝子工学の発展により、遺伝子配列を明らかにするという「ゲノム解析」の時代から遺伝子の発現産物による「機能解析」へと、開発研究のテーマは移行している。これは、遺伝子に依拠する遺伝形質は、遺伝子発現産物を介して、組織、器官及び人体等の機能として表れるものであるためである。こうした遺伝子発現産物の中でもタンパク質及びペプチドは、特に基礎的で重要な機能解析の対象である。
ポストゲノム時代においては、ゲノム解析により特定された遺伝子とその機能解明に基づき遺伝子標的をより明確にし、その作用機序を明らかにすることにより合理的な分子標的バイオ医薬(抗体医薬、ペプチド医薬)の開発が可能である。そして、近年注目されているのが核酸医薬の合成においても有効性が実証されている進化分子工学的手法である。
ペプチド医薬は、現在、広く使われるようになりつつある抗体医薬と同じ働きをしながら、抗体医薬よりも小さなタンパク質分子で同一の機能を発揮する医薬である。今後はその生産性の高さから、抗体医薬を置き換わっていくものと考えられている。
【0003】
高機能分子の新規創出を目的とする進化工学は、ランダムな配列を有する莫大なペプチドあるいはタンパク質のライブラリー中から、タンパク質に限られない特定の分子に特異的に結合するペプチド等を選択することを目的とする。
とりわけ、配列のランダム化の元となる遺伝子とその発現産物であるタンパク質とを分子的に対応付ける、又はディスプレイするIn vitro virus(IVV)法は、その中核をなす技術の一つであると言える。
IVVはタンパク質等の遺伝子発現産物の機能や表現型と、これに対応する遺伝子がコードされるmRNA等を当該分子上で1:1に対応付けることのできる分子である。IVV法においては、それぞれランダムなタンパク質を有するいくつかのIVVから、最も機能の高いタンパク質を選んだのちに、そこに連結されたmRNAを逆転写しPCR増幅等をすることでその配列を、最も機能の高い遺伝子の情報として検出することができる。
【0004】
進化工学的手法の一つであるcDNAディスプレイ法ではDNAライブラリから始まり、転写、リンカーの連結、翻訳とタンパク質の連結、固相化、逆転写、固相からの切り離し、精製、スクリーニング、cDNAの増幅と変異導入(ライブラリ化)、プロモーターの連結を、一のサイクルとし、複数回のサイクルを繰り返すのが標準とされている。一方、mRNAディスプレイ法では逆転写工程がスクリーニング工程の後に実行される。
このディスプレイ(対応付け)とスクリーニングのサイクルを繰り返すことで、前記の「スクリーニング」と分子への「変異導入」の回数を増やすことが出来るので、より優れた分子を創出し選び出すことが出来る。
進化工学的手法、及びIVV法ついては非特許文献1,2,3,7,8及び特許文献1,4,5,6に詳細が解説されている。
前記文献においてはmRNAディスプレイ法と呼ばれる方法が採られてきたが、近年のIVV法の進展の中で、In vitro virus合成後の分子の安定性向上のためcDNAディスプレイ法が開発された。これについては非特許文献4,5,6,9及び特許文献2,3,7に詳細が解説されている。
【0005】
またIVVに関連して、そのタンパク質の機能解析は、例えば、タンパク質−タンパク質相互作用やタンパク質−核酸相互作用等の試験による生化学的機能分析を通して行われている。
このようなタンパク質−タンパク質相互作用の解析法としては、イーストツーハイブリッド法、ファージディスプレイ法、GST−融合タンパク質プルダウン法、免疫共沈法等が古くから知られている。一方、タンパク質−核酸相互作用の解析法としては、電気泳動移動度シフトアッセイ法、DNaseIフットプリント法、メチル化緩衝法等が知られている。
合成されるIVVはこれらの試験に要求されるタンパク質と同等の品質を満たすものでなくてはならない。
【0006】
前記IVV法においては、遺伝子をコードするmRNAに対してタンパク質結合部位を有するリンカー(分子間連結のための分子)を連結することで、当該mRNAから合成されたタンパク質がリンカーに結合し、捕捉される。このため、1分子ごとに遺伝子と、遺伝子発現産物とを対応させることができることが知られている。
タンパク質をリンカーにトラップするには、翻訳合成系において、アミノアシルtRNAアナログを有するリンカーを伸長タンパク質鎖中に取り込ませる手法等が使われる。こうしたアナログとしてはピューロマイシン(Puromycin)等があり、これらにより修飾された核酸構築物を利用することでIVV法を実施することが可能となる。
【0007】
従来、mRNAとピューロマイシン・リンカーとを連結する工程では、反応効率の点から、微量のmRNAに対してピューロマイシン・リンカーを過剰に投入し、精製の際にこれを除去するという作業を行っていた。これらは、その後の翻訳反応において、タンパク質合成、及び連結体形成の阻害要因となると考えられていたためである(非特許文献1〜10,特許文献1〜8参照)。これらの除去は、フェノール・クロロホルム抽出や、ゲルろ過、イオン交換、ODS逆相カラム、HPLCその他のカラムを用いた精製、シリカメンブレンや限外濾過等の濾過、アクリルアミドゲル切出抽出、透析その他の種々の手法を用いて行われてきている。
また、リンカーとmRNAとを紫外光照射によるクロスリンクで連結させ、ライゲーション反応を回避し、後の精製工程を省略する方法が提案されている(非特許文献3)
一方で、あらかじめリンカーを固相に固定しておき、バッファー交換によって反応液を除去して精製を回避する方法も開発されている(特許文献3,非特許文献9)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開WO01/016600
【特許文献2】国際公開WO06/041194
【特許文献3】国際公開WO07/046520
【特許文献4】特開2002-291491
【特許文献5】特開2003-299489
【特許文献6】特開2004-097213
【特許文献7】特開2008-253176
【特許文献8】特開2005-245223
【非特許文献】
【0009】
【非特許文献1】Nemoto et al., FEBS Lett. 414, 405 (1997)
【非特許文献2】Roberts et al., Proc. Natl. Acad. Sci. USA 94, 12297 (1997)
【非特許文献3】Kurz et al., Nucleic Acids Res. 28, e83 (2000)
【非特許文献4】Kurz et al., CHEMBIOCHEM 2, 666 (2001)
【0010】
【非特許文献5】Tabuchi et al., FEBS Lett. 508, 309 (2001)
【非特許文献6】Tabuchi et al., Biol. Proced. Online 4, 49 (2002)
【非特許文献7】Miyamoto-Sato et al., Nucleic Acids Reserch 31, e78 (2003)
【非特許文献8】Ja & Roberts, Biochemistry 43, 9265 (2004)
【非特許文献9】Biyani et al., Nucleic Acids Res 34, e140 (2006)
【非特許文献10】Retic Lysate IVT Kit (Part Number AM1200) Protocol (Ambion Inc.)
【非特許文献11】Abdella et al., Biochem. Biophys. Res. Commun., 87, 734 (1979)
【発明の概要】
【発明が解決しようとする課題】
【0011】
従来の方法は、1分子ごとに遺伝子と、遺伝子発現産物とを対応させることができるという点では優れたものである。しかし、以下のような問題点があった。
すなわち、系に投入するリンカー濃度を上げるとm−RNAとリンカーとの連結体の収量は増加するが、リンカー同士やリンカーとmRNAとの凝集を招くため、投入できる濃度には限界がある。また連結効率を考えると、mRNAとリンカーの投入量とは、最大でmRNA:リンカー比=1:200程度とする必要があり、未濃縮の連結体を得るのに十分な濃度のmRNAが投入できなかった。
さらに、反応生成物であるmRNA−リンカー連結体の濃縮が必須であり、上記各カラム精製後の抽出、エタノール沈殿、限外濾過、エバポレーション等が行われてきた。
こうした精製は操作が煩雑で手間がかかるだけでなく、IVV法で得られた多種多様なペプチドのスクリーニングを自動化するには適していない。
【0012】
一方、タンパク質の収率に影響し得る未反応mRNAについても、リンカーと同様に、除去することが望ましい。あらかじめリンカーを固相に固定しておく従来の方法では、未反応mRNAを除去することはできるが、収量が著しく減少するという問題を有しており、多様なペプチドスクリーニングの需要に必ずしも応えられるものとは言えない。このように、液相中にて未反応mRNAのみを除去する有効な手段は現在見出されていない。
これに対し、リンカーとmRNAの連結にライゲーション反応ではなく、紫外光照射によるクロスリンクを用いる方法が提案されている。この方法は精製工程を省略することができる点で特に優れたものである。しかし、この方法では、リンカーの構造がさらに複雑になるため、製造コストを上昇させるほか、紫外光照射によってmRNAが損傷されることもあるという問題がある。
さらに、上述したように、進化工学的手法ではディスプレイとスクリーニングの回数を増やすほど優れた分子種を得られるため、上述した一連のサイクルは出来るだけ多いことが必要とされる。そしてサイクル中の作業工程数は少なければ少ないほどよく、特に人が関与する作業は少ないほど良い。
しかしながら、上記進化工学的手法には省略できない化学反応工程が数多く含まれており、サイクル全体の作業工程の短縮及び省力化は困難であった。
【0013】
以上のような観点から、mRNAとリンカーとの連結後の精製及び濃縮に要する手間と時間及びコストを大幅に削減することで、実用的なハイスループットスクリーニングの開発を促す、前記連結体の生成方法等に対する社会的な要請があった。
さらに、上記ペプチド医薬等の開発促進のニーズに応えるためにペプチドスクリーニング技術の発展と普及が必要とされている。このため、次世代医薬としてのリードペプチドの探索手法(ペプチドスクリーニング)においては、自動化及び省力化に対する強い要請があった。
【課題を解決するための手段】
【0014】
すなわち本発明の第1の態様は、液相中における個々の分子の反応において、タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合する工程と;を備える、mRNA−リンカー−タンパク質連結体の生成方法である。
【0015】
前記所定の割合は、前記リンカー:前記mRNA=3:1〜1:6であることが好ましく、前記リンカー:前記mRNA=1:1〜1:1.5であることが特に好ましい。前記主鎖の鎖長は16〜60塩基であることが好ましく、21〜38塩基であることが特に好ましい。
前記リンカーの主鎖は、5'位がリン酸化された主鎖5'末端部と、第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、を備えることが好ましい。
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、前記主鎖は主鎖3'末端側領域部に側鎖連結部位をさらに備え、かつ前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列をさらに有し;前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することが特に好ましい。
前記切断部位は、制限酵素認識配列を有することが好ましい。前記mRNA−リンカー−タンパク質連結体の生成方法は、前記固相結合部位を介して固相と結合させる工程と、前記第1及び第2の切断部位を制限酵素で切断する工程とをさらに備えることが好ましい。前記制限酵素はPvuIIであることが好ましい。
前記切断部位はまた、グアノシンを有することが好ましい。前記mRNA−リンカー−タンパク質連結体の生成方法は、前記固相結合部位を介して固相と結合させる工程と、前記第1及び第2の切断部位をRNA切断酵素で切断する工程とをさらに備えることが好ましい。前記RNA切断酵素はリボヌクレアーゼT1であることが特に好ましい。
【0016】
本発明の第2の態様は、液相中における個々の分子の反応において、タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合し、mRNA−リンカー−タンパク質連結体を生成する工程と;mRNA−リンカー−タンパク質連結体を前記リンカーの備える固相結合部位を介して固相と結合させる工程と;前記主鎖の3'末端を反応開始点とし、前記mRNAを鋳型として、固相上で逆転写反応を行ってcDNA鎖を形成させる工程と;前記主鎖の備える第1及び第2の切断部位を、制限酵素又はRNA切断酵素で切断する工程と;を備えるmRNA−リンカー−タンパク質−cDNA連結体の生成方法である。
【0017】
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、前記リンカーの主鎖は、5'位がリン酸化された主鎖5'末端部と、前記第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される前記固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、側鎖連結部位を備え、前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、を備えることが好ましい。また、前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することが好ましい。
【発明の効果】
【0018】
本発明がなされるまでは、ライゲーション反応後の精製濃縮工程は省略することが出来ないとするのが、cDNA/mRNAディスプレイ技術における技術常識であった。本発明の方法に拠ればこれを省略することが可能になる。
本発明の効果は、IVVすなわちmRNA−リンカー−タンパク質連結体の効率的な生成を可能とすることであり、さらにmRNAディスプレイ法、cDNAディスプレイ法、分子進化学的手法全体にとっての工程の簡略化を可能にすることである。
【図面の簡単な説明】
【0019】
【図1】図1は、リンカー主鎖の構造図である。
【図2】図2は、リンカーの模式図である。
【図3】図3は、旧来のIVV合成法を示す模式図である。
【図4】図4は、本発明のIVV合成法を示す模式図である。
【図5】図5は、ライゲーション反応産物の電気泳動像(FITC蛍光)である。
【図6】図6は、ライゲーション反応産物の電気泳動像(SYBR(登録商標) Gold)である。
【図7】図7は、翻訳連結反応産物の電気泳動像(FITC蛍光)である。
【発明を実施するための形態】
【0020】
以下に、本発明をさらに詳細に説明する。
1.連結体生成用リンカー
本発明において、「リンカー」とは、IVV法において用いられる、mRNA−リンカー連結体(連結体A)、mRNA−リンカー−タンパク質連結体(連結体B)及びmRNA−リンカー−タンパク質−cDNA連結体からなる群から選ばれるいずれかの連結体を生成する際に使用するリンカーのことをいう。
本発明において使用するリンカーは、DNA(すなわちデオキシリボ核酸単量体)、RNA(すなわちリボ核酸単量体、例えばrG:グアノシン)、及びDNAアナログからなる群から選ばれる少なくとも1以上の物質によって形成される主鎖を含み、この主鎖中に上記連結体を固相部位に結合するための固相結合部位と、前記固相結合部位を挟む位置に設けられた一対の切断部位を有することを特徴とする。ここで、DNAアナログはBiotin-dT、Fluorescein-dT等を含むものとする。
本発明において使用するリンカーは、全体として、柔軟性があり、親水性であり、かつ特異構造を形成する可能性の少ない配列を有するように、設計することが好ましい。
【0021】
本発明の詳細な説明において「液相反応」とは、通常の試験管、微量サンプルチューブ、マイクロウェルプレート等の反応容器内の均一な反応液中での反応を指すものとする。また、「固相反応」とは前記同様の反応容器内における反応ではあるが、固相ビーズや容器底面等な固相表面に反応させるべき分子を結合し、固相表面上において集中的に反応を行うことを指すものとする。
ここで「mRNAの有する所定の配列」には、遺伝子をコードする配列、連結体形成、翻訳反応促進に必要な配列その他の配列が含まれ、特に限定されない。
また、本発明の詳細な説明において「アミノアシルtRNA3’末端アナログ」とは、その3’末端がアミノアシルtRNAに化学構造骨格が類似し、翻訳系でタンパク質の合成が行われた際に、伸長中のペプチド鎖のC末端に結合する能力を有する化合物をいう。
【0022】
また、本発明の詳細な説明において「Xを有するY部位」の文言は、以下のように定義される。ここで、Xは、FITCその他の蛍光色素、RnaseT1その他の酵素の基質等の機能性分子を表わし、Yは機能性分子Xが発揮する機能を表わす。「Xを有するY部位」は、Xが結合しているか、Xを含んでいることで、機能Yを発揮する一塩基〜数十塩基からなるリンカー中の特定の配列を指す。前記特定の配列は、機能性分子X、Xに結合する修飾基のみで構成されるものであってもよく、前記特定の配列に含まれる塩基には、核酸アナログ等も含まれる。
一例を挙げると、「rGを有する切断部位」とは、グアノシン(rG)を含むことにより、リボヌクレアーゼT1がrGを特異的に認識してホスホジエステル結合を加水分解することができる部位を指す。
【0023】
また、本発明中で、リンカー主鎖はRNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるものである。本発明の方法ではリンカーの主鎖が、その全長に対して、少なくとも60%以上が、1本鎖DNAと、RNA及び/又はDNAアナログとで構成されていることが好ましい。より好ましくはリンカー主鎖の70%以上、さらに好ましくは80%以上、最も好ましくは90%以上、1本鎖DNAと、RNA及び/又はDNAアナログとで構成されている。
本発明において使用するリンカーでは、種々の反応工程での反応性、上述した連結体の生成効率などを考慮して主鎖の長さを決定するが、好ましくは16〜60merであることが好ましく、より好ましくは21〜38merである。なお、本発明において使用するリンカーは、後述するような公知の化学合成の手法を用いて合成することができる。
図1を用いて本発明に用いるリンカーの主鎖の構造を説明する。主鎖200には主鎖5'末端部201が存在する。主鎖5'末端近傍部202は主鎖5'末端部201を含まない領域であり、主鎖3'末端側領域部203は主鎖3'末端部を含まない領域である。なお、図1は上記各部の塩基長を限定したものではない。
【0024】
2.所定の配列を有するmRNA
本発明で用いるmRNAは、配列未知のもの、配列既知のものの双方を含む。前記配列は、ゲノム情報として自然界に存在する配列、又は人為的に設計した配列のいずれであってもよく、その構造は特に限定されない。
上記連結体を用いて、配列既知のタンパク質に結合する物質を探索又は定量する場合には、当該タンパク質の遺伝子をコードするmRNAを用いることができる。逆に、上記連結体を用いて、配列未知のタンパク質の配列を特定の機能に関連づけて解析する場合には、配列未知の遺伝子をコードするmRNAを用いることができる。
【0025】
こうしたmRNAとしては、例えば、配列既知の各種レセプタータンパク質をコードするmRNA、各種抗体又はその断片をコードするmRNA、各種酵素をコードするmRNA、各種遺伝子ライブラリー中のDNAから転写される配列未知のmRNA、有機合成によってランダムに合成された配列を有するDNAから転写されたランダムな配列を有するmRNA、PCR法を利用したランダム変異の挿入により配列未知のタンパク質をコードするようになったDNAから転写されるmRNAその他のmRNA等を挙げることができ、これらの中から選択される。
本発明で用いるmRNAは、自然界に存在するmRNAや、mRNA特有の構造に限定されるものではないが、タンパク質の合成効率上、5’末端に7−メチル化グアノシンキャップ構造又は3’末端にポリA尾部構造の少なくとも一方を持っていることが好ましい。翻訳の開始を促進するKozak配列や、Shine-Dlgarno配列を有することが、さらに好ましい。
mRNAはin vitro転写反応、化学合成、生体・細胞・微生物からの抽出などの所与の方法を用いて得ることができる。リンカーとの連結及び無細胞翻訳の反応効率の点から、in vitro転写反応を用いて生成することが好ましい。
本発明で用いるmRNAの長さは、反応効率の面から50〜1,000塩基であることが好ましく、200〜500塩基であることが反応効率が最も高いことから、さらに好ましい。
【0026】
3.ハイブリダイゼーション及び連結方法
mRNAとリンカーとの連結は、公知の手法を用いて、直接的又は間接的に、化学的又は物理的に行うことができる。例えば、mRNAの3’末端にリンカーの末端と相補的な配列を設けておくと、両者をハイブリダイゼーションによって連結することができる。また、末端でのライゲーション反応、又は末端を含む各部分でのソラレンを用いた光架橋反応により化学結合によって連結することもできる。
本発明をmRNAディスプレイ法の中で実施する場合、リンカーと前記mRNAとの連結は、ライゲーション反応により行われることが好ましい。mRNAとリンカーとの化学結合反応を促進するため、連結予定部位同士がハイブリダイゼーションしたことに起因して、近接していることが好ましい(アニーリング)。
アニーリングでは、第三の核酸分子がmRNAの3’末端近傍及びリンカーの5’末端近傍の両者に相補鎖を形成してもよい(Splint DNA法)が、mRNAの3’末端近傍とリンカーの5’末端近傍とが相補鎖を形成していることがより好ましい(Y-ligation法)(非特許文献6)。より具体的には、mRNAの3’末端近傍とリンカーの5’末端近傍とが、数塩基〜数十塩基に渡って相補鎖を形成することが好ましい。また、リンカーの5’末端とmRNA中途配列とが相補鎖を形成することも、ライゲーションによる連結を安定させる上でさらに好ましい。ただし、上記要件は、末端塩基に連なる数塩基が必ず相補鎖を形成していることや、相補鎖を形成する領域が必ず連続していることを要求するものではない。cDNAディスプレイ法の中で好適に使用できるリンカーの条件は後述する。
【0027】
本発明において、「ライゲーション」又は「ライゲーション反応」は、酵素により促進される反応をいう。こうした酵素は、酵素番号の6群にあたるリガーゼであり、DNAやRNAのライゲーション反応については、EC6.5.1.1(ATP依存性DNAリガーゼ)、EC6.5.1.2(NAD+依存性DNAリガーゼ)、EC6.5.1.3(RNAリガーゼ)等を利用することができる。RNA/DNA間ライゲーションを行うRNAリガーゼのほか、RNA間及びRNA/DNA間のライゲーションを行うDNAリガーゼを利用することもできる。
具体的には、T4 RNAリガーゼ又はTS2126耐熱性ファージ由来DNAリガーゼを好適に利用することができ、T4 RNAリガーゼを利用することが、連結効率の理由からさらに好ましい。
【0028】
ライゲーション反応前のアニーリングは、60〜100℃にて2〜60分間、ヒートブロック、アルミブロック、ウォーターバスその他の加温用器具にて温めた後、室温で2〜60分間放置して液温を穏やかに低下させ、さらに-5〜10℃に冷却して行うのが好ましい。例えば、90 ℃で5分間アルミブロック上にて温め、次に70 ℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した後、氷上に置くようにすることが好ましい。
ライゲーション反応液の組成は、0.1〜2.5 U/μlのT4ポリヌクレオチドキナーゼ; 0.4〜5 U/μlのT4 RNAリガーゼ; 10〜250 mMのTris-塩酸(pH 7.0〜8.0); 2.0〜50 mMの塩化マグネシウム; 2.0〜50 mMのジチオスレイトール(DTT); 0.2〜5.0のmM ATPを含むものであることが好ましい。反応効率の点から、0.5 U/μlのT4ポリヌクレオチドキナーゼ; 2.0 U/μlのT4 RNAリガーゼ; 50 mMのTris-塩酸(pH 7.5); 10 mMの塩化マグネシウム; 10 mMのDTT; 1.0 mMのATPを含有するものであることが好ましい。この反応液には、必要に応じて、SUPERase RNase inhibitor(Ambion社)などのRNA分解酵素阻害剤を添加してもよい。
【0029】
ライゲーション反応を行う反応系の体積は、4.0〜100 μlであることが好ましい。反応効率の点から20〜40μlとすることが好ましく、20μlとすると最も反応効率が高い。
この反応系中でのRNAとリンカーとの量比は、リンカー 5.0〜100 pmolに対し、mRNA5.0〜100 pmolとすることが好ましい。反応効率を上げ、残余を最少化するために、リンカー 10〜20 pmolに対し1/3〜6倍量のmRNAを反応させるのが好ましく、リンカー10 pmolに対し10〜15 pmolのmRNAを反応させるのが最も好ましい。
アニーリング反応は、T4ポリヌクレオチドキナーゼ及びT4 RNAリガーゼを除いたライゲーション反応液中で行うことができる。そして、アニーリングの終わった反応液中に、これらの酵素を必要量投入することで、ライゲーション反応を開始させることができる。
ライゲーション反応は、10〜40 ℃で1分間〜16時間行うのが好ましい。作業効率及び反応効率の面から、20〜30 ℃で5〜180分間とすることが好ましく、25℃で15分間とすることが最も好ましい。
【0030】
4.無細胞系タンパク質合成及び連結体の形成
連結体A(mRNAとリンカーとの連結体)を無細胞翻訳系と接触させることによって、タンパク質の合成を行う。ここで、タンパク質を合成するための無細胞翻訳系は、動物由来細胞、植物由来細胞、菌類及び細菌類からなる群から選択したものから得てもよく、また部分的にあるいは全体として人工的に合成したものでもよい。具体的には、大腸菌、ウサギ網状赤血球、小麦胚芽抽出物などを使用することができる(Lamfrom H., Grunberg-Manago M., Biochem. Biophys. Res. Commun., 27, 1 (1967))。
生物種により翻訳に利用されるコドンの種類が異なるので、対象となる遺伝子や遺伝子の由来に合わせて無細胞翻訳系を選択することが好ましい。または、利用したい翻訳系に合て、mRNAの配列や構造を再設計するのが好ましい。
【0031】
本発明の実施形態においては、前記無細胞翻訳系として哺乳類の網状赤血球細胞のライセートを利用することが好ましく、ウサギの血液から得られた網状赤血球細胞のライセートを利用することがさらに好ましい。また、血中の網状赤血球の割合を高めるため、前記哺乳動物に予めアセチルフェニルヒドラジンを投与して溶血性貧血等を誘導し、その後数日間飼育して採血をすることが好ましい。前記ライセートは、マイクロコッカルヌクレアーゼによって細胞由来のmRNAを分解し、グリコールエーテルジアミン四酢酸(EGTA)を加えてカルシウムをキレートし、前記ヌクレアーゼを不活化処理したもの(以下、「マイクロコッカルヌクレアーゼ処理済」という。)とすることが、より好ましい。
【0032】
翻訳反応液の組成は、3.4〜85 μlのウサギ網状赤血球ライセート(マイクロコッカルヌクレアーゼ処理済)と0.24〜10 pmolの上記連結体を含む反応バッファー(最終濃度:16〜400 mMの酢酸カリウム;0.1〜2.5 mMの酢酸マグネシウム;0.2〜50 mMのクレアチンリン酸;各0.00〜0.25 mMのアミノ酸を含む)を10〜100 μl用いるのが好ましい。反応効率の点から、17 μlのウサギ網状赤血球ライセート(マイクロコッカルヌクレアーゼ処理済)と1.2〜2.0 pmolの上記連結体とを含む反応バッファー(最終濃度:80 mMの酢酸カリウム;0.5 mMの酢酸マグネシウム;10 mMのクレアチンリン酸;各0.05 mMのアミノ酸(メチオニン及びロイシンは各々0.025 mM)を25 μl用いるのが、さらに好ましい。
また必要に応じて、この反応液に、SUPERase RNase inhibitor(Ambion社)などのRNA分解酵素阻害剤を添加してもよい。
【0033】
翻訳反応は20〜40 ℃で10〜90分間行うことが好ましく、生成効率と作業効率の点から、30℃で20分間行うことが特に好ましい。
翻訳反応後、連結体Aと翻訳産物(タンパク質)との連結(連結体Bの形成)を、高塩濃度条件下にて促進することができる。連結体Bの形成促進は、終濃度0.3〜1.6 Mの塩化カリウム及び終濃度40〜170 mMの塩化マグネシウム存在下で行うのが好ましい。生成効率の点から、25μlの上記反応液に7〜10μlの3Mの塩化カリウム、及び3μlの1Mの塩化マグネシウム(終濃度:0.60〜0.79 mMの塩化カリウム;79〜86 mMの塩化マグネシウム)を添加することが、さらに好ましい。
連結体Bの形成促進は27〜47 ℃で30分〜3時間という反応条件下で行われることが好ましい。生成効率と作業効率の点から、37 ℃で90〜120分間反応させることが特に好ましい。
【0034】
5.連結体Aとタンパク質の連結(IVV形成)
連結体Aを翻訳系に投入してタンパク質またはポリペプチド鎖を液相合成する際に、そのC末端に、前記mRNAと翻訳されたタンパク質とがリンカー及びリンカーの有するタンパク質結合部位を介して個々の分子ごとに連結される。ここで、前記タンパク質結合部位は、アミノアシルtRNA3’末端アナログを含むことが好ましく、前記アミノアシルtRNA3’末端アナログはピューロマイシンであることが好ましい。
すなわち、mRNAにピューロマイシン等を有するリンカーを結合させた連結体Aと翻訳系とを接触させると、そのmRNAがピューロマイシンを介して翻訳されたタンパク質と結合したIVVが生成する。(非特許文献1参照)。
【0035】
翻訳されたタンパク質はさらに、N末端側のペプチドの除去、糖鎖や脂質などの非アミノ酸成分の付加、アミノ酸の翻訳後修飾、又は立体構造や配列構造に基づく前記mRNAに対する親和性結合や、タンパク質の酵素活性によるmRNAの化学修飾その他の翻訳後修飾がなされてもよい。
こうした翻訳後修飾は、前記mRNAにコードされた遺伝情報に基づいて行われてもよく、前記遺伝情報ではなく外的要因に基づいて行われてもよい。また、これらの組み合わせに基づくものであってもよい。遺伝子情報に基づく反応としては、二量体及び多量体形成、自己フォールディング、分子内分解反応、元のmRNAとの複合体形成等が挙げられる。外的要因による反応としては、修飾基、脂質、又は糖鎖等の付加、分子シャペロンによるフォールディング又は複合体形成、その他の巨大構造物(ウィルス・細胞)との親和性結合等が挙げられる。遺伝子情報及び外的要因の組み合わせによる反応としては、当該タンパク質の特定配列部位における化学修飾や分解などが挙げられる。
ただし、前記反応はこれらに限定されるものではない。また、これらの反応は、解析やスクリーニング等の補助のために行うこともでき、それ自体を解析やスクリーニングの対象とすることもできる。
【0036】
アミノアシルtRNA3’末端アナログの1つであるピューロマイシンは、下記式Iで表される化合物であり、翻訳系でタンパク質の合成が行われた際に、合成されたタンパク質のC末端に結合する能力を有する。
【0037】
【化1】
【0038】
ピューロマイシンは翻訳系でタンパク質の合成が行われた際に、合成されたタンパク質のC末端に結合する能力を有する。その他のアミノアシルtRNA3’末端アナログとしては、例えば、3'-N-アミノアシルピューロマイシンアミノヌクレオシド(3'-N-Aminoacylpuromycin aminonucleoside, PANS-アミノ酸)を挙げることができる。PANS-アミノ酸としては、アミノ酸部がグリシンのPANS-Gly、バリンのPANS-Val、アラニンのPANS-Ala、又はアミノ酸部が全ての各アミノ酸に対応するPANS-アミノ酸混合物を挙げることができる。
その他には、3'-アミノアデノシンのアミノ基とアミノ酸のカルボキシル基が脱水縮合して形成される、アミド結合で連結した3'-N-アミノアシルアデノシンアミノヌクレオシド(3'-Aminoacyladenosine aminonucleoside, AANS-アミノ酸)を挙げることができる。AANS-アミノ酸としては、アミノ酸部がグリシンのAANS-Gly、バリンのAANS-Val、アラニンのAANS-Ala、又はアミノ酸部が全アミノ酸の各アミノ酸に対応するAANS-アミノ酸混合物を挙げることができる。
また、ヌクレオシドあるいはヌクレオシドとアミノ酸のエステル結合したものなども使用できる。
ピューロマイシン以外に好適に使用できるアミノアシルtRNA3'末端アナログとしては、リボシチジルピューロマイシン(rCpPur)、デオキシジルピューロマイシン(dCpPur)、デオキシウリジルピューロマイシン(dUpPur)などがあり、これらを下記式(II)〜(VI)に示す。
【0039】
【化2】
【0040】
【化3】
【0041】
【化4】
【0042】
【化5】
【0043】
【化6】
【0044】
また、アミノアシルtRNAアナログは、公知の手法によってリンカーに付加することができる。mRNAディスプレイ法では、アミノアシルtRNAアナログを有するタンパク質連結部位をリンカーの主鎖の5’末端に形成してもよい。一方、cDNAディスプレイ法において、cDNA型IVVを形成するために、リンカーの主鎖の5’末端をmRNAとの相補鎖形成に用いる場合等には、上記のタンパク質結合部位をリンカーの側鎖中に形成することが好ましい。
リンカーの側鎖が短いと、アミノアシルtRNAアナログが取り込まれるときに翻訳複合体の立体障害を受けることから、リンカーの側鎖は十分な長さを持つものであることが好ましい。リンカーの側鎖は、例えば、下記式(VII)のSpacer 18 Phosphoramiditeのようなスペーサー分子を1〜8個程度含むものであることが好ましく、リンカー合成効率と連結体形成効率のバランスとから、こうしたスペーサー分子を4個含むものであることがさらに好ましい。
【0045】
【化7】
【0046】
リンカーの主鎖と側鎖との連結は公知の方法に従って行うことができる。例えば、リンカーの主鎖が下記式(VIII)のAmino-Modifier C6 dTを含み、側鎖が下記式(IX)の5'-Thiol-Modifier C6から合成された5'末端を含む場合には、架橋剤として下記式(X)で表わされるEMCSを利用することができる。
【0047】
【化8】
【0048】
【化9】
【0049】
【化10】
【0050】
本発明で使用するリンカーは、必要に応じて、タンパク質を連結体Bから遊離させるために、切断部位(以下、「タンパク質遊離部位」ということがある。)を設けることができる。このようなタンパク質遊離部位として、例えば、光切断部位等を挙げることができる。
ここで、「光切断性部位」とは,紫外線等の光を照射すると分解される性質を持つ化合物を有する部位いい,市販の化合物を用いることもできる。例えば、下記式(XI)で表されるPC Linker Phosphoramidite(Glen research社)、フラーレンを含有してなる核酸の光切断用組成物に係る化合物(特許文献8)、光分解(SBIP)手法による鎖切断に係る化合物などが好ましい。切断効率の点から、PC Linker Phosphoramiditeを利用することが好ましい。
【0051】
【化11】
【0052】
6.IVVの検出
上記連結体A又は連結体Bは、あらかじめリンカーに標識物質を結合させておくことによって、容易に検出することができる。そのような標識物質としては、蛍光性化合物、抗原のエピトープペプチド、放射性同位体等を挙げることができる。放射性同位体としては、3H、14C、32P、125I若しくは131I等の放射性同位体を使用することが好ましい。
放射性同位体の利用が困難な場合には、蛍光性化合物を利用することが好ましい。蛍光性化合物としては、フリーの官能基(例えば、活性エステルに変換可能なカルボキシル基、ホスホアミダイドに変換可能な水酸基、又はアミノ基など)を持ち、標識された塩基としてリンカーに連結可能な種々の蛍光色素を用いることが好ましい。このような蛍光色素としては、例えば、フルオレセインイソチオシアネート(FITC)、フィコビリタンパク質、希土類金属キレート、ダンシルクロライド又はテトラメチルローダミンイソチオシアネート等を挙げることができる。蛍光標識分子としては、FITC-dTや下記式(XII)で表わされるFluorescein-dTを使用することがより好ましい。
【0053】
【化12】
【0054】
7.連結体Bの固相固定
本発明で使用するリンカーは、その主鎖中に上記連結体Bを固相部位に結合するための固相結合部位を有することが好ましい。
連結体Bが固定される固相は特に限定されず、その連結体が使用される目的に応じて適宜選択される。こうした固相としては、生体分子を固定する担体となる各種の形状のものを用いることができる。例えば、スチレンビーズ、ガラスビーズ、アガロースビーズ、セファロースビーズ、磁性体ビーズ等のビーズ;ガラス基板、シリコン(石英)基板、プラスチック基板、金属基板(例えば、金箔基板)等の基板;ガラス容器、プラスチック容器等の容器;ニトロセルロース、ポリビニリデンフロリド(PVDF)等の材料からなるメンブレンなどが挙げられる。
【0055】
上記連結体Bでは、リンカーに固相結合部位を設けることで、その固相結合部位と、固相に結合させた「固相結合部位認識部位」とを介して、連結体Bを固相に固定することができる。「固相結合部位認識部位」とは、固相に設けられた部位であって、固相結合部位の有する物質に応じて、固相結合部位に結合する部位である。上記固相結合部位は、連結体Bを所望の固相に結合し得るものであればよく、特に限定されない。例えば、リガンド、エピトープペプチドその他の特定のポリペプチドに特異的に結合する構造単位を用いる場合には、固相表面には、これらと特定に結合する特定のポリペプチド等を結合させておくことになる。
こうした固相結合部位認識部位/固相結合部位の各々の有する物質の組合せの例としては、例えば、アビジン及びストレプトアビジンその他のビオチン結合タンパク質/ビオチン、マルトース結合タンパク質/マルトース、Gタンパク質/グアニンヌクレオチド、ポリヒスチジンペプチド/ニッケルあるいはコバルト等の金属イオン、グルタチオン−S−トランスフェラーゼ/グルタチオン、配列特異的なDNA又はRNA結合タンパク質/特異配列を有するDNA又はRNA、抗体又はアプタマー/抗体又はエピトープペプチド、カルモジュリン/カルモジュリン結合ペプチド、ATP結合タンパク質/ATP、エストラジオール受容体タンパク質/エストラジオール等の各種受容体タンパク質/そのリガンド等が挙げられる。
これらのうち、上記ビオチン結合タンパク質/ビオチン、マルトース結合タンパク質/マルトース、ポリヒスチジンペプチド/上記金属イオン、グルタチオン−S−トランスフェラーゼ/グルタチオン、抗体/抗原分子(エピトープペプチド)等を使用することが好ましく、リンカー合成の容易さの面から、ストレプトアビジン/ビオチンの組合せを使用することが、さらに好ましい。
【0056】
上記固相結合部位は、例えば、連結体Bをリンカーを介して固相に結合させるための部位であり、具体的には、こうした結合を可能にする化合物、結合基などを結合し得る塩基で構成される。より具体的には、ビオチンを結合し得る塩基(例えば、デオキシチミン(dT))もしくはビオチンが結合した塩基(例えば、ビオチン−デオキシチミン(Biotin-dT))、アミノ基によって修飾された塩基(例えば、アミノ修飾デオキシチミン(例、Amino-Modifier C6-dT:Glen Research Search社製)、カルボキシ基によって修飾された塩基(例えば、カルボキシ修飾デオキシチミン(Carboxy-dT))、チオール基によって修飾された塩基(例えば、チオール修飾デオキシチミン(4-Thio-dT))等で構成される。
上記リンカーでは、固相結合部位として下記式(XIII)のビオチン−デオキシチミンを好適に使用することができる。そして、このような固相結合部位を介し、ビオチンとアビジンの親和性を利用して、ビオチンが連結された連結体Bをアビジンが固定された固相に結合させることができる。なお、上記固相結合部位として、カルボキル基やアミノ基が結合した塩基が選択された場合は、固相と連結体Bとをエステル結合やアミド結合で結合させることができる。
【0057】
【化13】
【0058】
上記の方法で固相に固定された連結体Bを用いて、洗浄後公知の方法で逆転写反応を行う。逆転写反応後、再び洗浄し、新たに加えた所定のバッファー中で固相より遊離する。
本発明で使用するリンカーでは、必要に応じて、連結体Bを固相から遊離させるために、第1及び第2切断部位(以下、「固相遊離部位」という。)を設ける。上記固相遊離部位は、前記タンパク質遊離部位と同様の条件で切断又は分解されるようにすることもできる。スクリーニング等の工程をスムーズに進める上では、当該固相遊離部位は、前記タンパク質遊離部位と同様の条件で切断又は分解されない構成とすることが好ましい。
上記リンカーでは、例えば、リボG(グアノシン;rG;Guanosine)等の特定の酵素が認識可能な塩基を含む配列として前記固相遊離部位を構成し、こうした酵素を用いて、固相に結合された連結体Bを固相から遊離させることが好ましい。
【0059】
ここで用いられる酵素は、リンカーの設計に応じて決定することができ、例えば、主鎖の鎖長に余裕があれば制限酵素(旧EC番号3.1.23, 3.1.24, 3.1.25)を利用することができる。また、リンカーがDNAを主体に形成されている場合には、RNAを切断部位として、RNA切断酵素(EC番号3.1.26, 3.1.27)を利用することができる。また、酵素切断部位を、使用する酵素の種類に応じて適宜選択し、設計することもできる。
【0060】
RNA切断酵素としては、RNaseT1、RNaseA、RNaseI、膵臓RNase、S1ヌクレアーゼ、蛇毒ヌクレアーゼ、脾臓ホスホジエステラーゼ、スタフィロコッカスヌクレアーゼ、マングビーンヌクレアーゼ、アカパンカビヌクレアーゼ等を用いることができる。上記切断部位としては、これらの酵素のうち、RNaseT1、RNaseA、RNaseI、膵臓RNaseによって切断される部位であることが好ましく、特に、RNaseT1によって認識され、切断されるリボG(rG;グアノシン)を含む配列であることが好ましい。
RNaseT1は、rGを1塩基で認識してリンカーを切断するため、rGを使用するとリンカー主鎖長を短くすることが出来る。その結果、上述したライゲーションの際に、リンカー濃度を上げても凝集しにくいという利点がある。
【0061】
リボヌクレアーゼT1(RNaseT1)等の一本鎖特異的RNA切断酵素により切断されることを予定するリンカーでは、図2に示すように、リンカー2中で固相結合部位2bが第1切断部位2c1と第2切断部位2c2との間に位置し、これらは、リンカーの主鎖の3’末端近傍の一本鎖ループに位置していなければならない。なお、図2にはリンカーと相補鎖を形成するmRNA1、リンカーの主鎖200、リンカーの側鎖300等も表されている。
【0062】
上記の特定のポリペプチド等と固相結合部位との結合は、IVVの変性を避けるために分子間の親和性のみを利用して行うことが好ましいが、その他の公知の方法を用いることもできる。そのような公知の方法としては、例えば、タンニン酸、ホルマリン、グルタルアルデヒド、ピルビックアルデヒド、ビス−ジアゾ化ベンジゾン、トルエン−2,4−ジイソシアネート等を利用する方法を挙げることができる。また、上記の特定のポリペプチド等及び固相結合部位の有する分子としては、アミノ基、カルボキシル基、又は水酸基などの官能基を構成する分子が好ましい。
上記の特定のポリペプチド等と固相を構成する要素との結合は、公知の技術によって行うことができ、上述した市販の前記ビーズや前記基板等を利用してもよい。また上記固相遊離部位は固相側に設けることもできる。上記の固定化手段は、2つの相互に親和性を有する物質を利用した固定化方法であるが、固相がスチレンビーズ、スチレン基板などのプラスチック材料で構成されているのであれば、必要に応じて、公知の手法を用いてリンカーの一部を直接それらの固相に共有結合させることもできる(Qiagen社、LiquiChip Applications Handbook等参照)。なお、本発明においては、固定化手段については、上記の方法に限定されることなく、当業者に公知であれば如何なる手段をも利用することができる。
【0063】
8.本発明におけるIVV合成法と従来法との比較
ここで、図3及び図4を参照しつつ、上記連結体A又は連結体Bを用いたIVV法における従来法と、本発明による方法と対比しつつ説明する。図中、同じ要素には同じ番号を付し、説明を省略する。
従来法を示す図3では、遺伝子情報を有するmRNA1とリンカー2とは所定の方法によって連結される。ここでは、第1切断部位2c1及び第2切断部位2c2を合わせて固相遊離部位2cと表す。図3では、ハイブリダイゼーション反応につづくライゲーション反応により、mRNA1とリンカー2とが連結され連結体Aが形成される(Step 1)。連結体の合成効率を高めるために、mRNA1とリンカー2とは1:1.5〜1:3の範囲で混合され、結果として過剰なリンカー2が未反応のまま残される。
【0064】
未反応のリンカー2を除去するためにカラム精製を行い(Step 2)、次に、精製した連結体3(連結体A)を濃縮するためエタノール沈殿を行う(Step 3)。次いで、連結体Aを、例えば、無細胞翻訳系に供することによって(Step 4)、mRNA1に対応するタンパク質5が、タンパク質結合部位2aに結合された状態で、連結体A上で合成される(Step 5)。この結果、mRNA1、リンカー2及びタンパク質5からなる連結体4(連結体B)が生成する。連結体Bは、いわゆるIn Vitro Virus (IVV)である。
固相結合部位認識部位6aを有する固相ビーズ6に、得られた連結体Bが固定され(Step 5)、これを逆転写反応に供することによって、mRNAに対応するcDNA7が生成される(Step 6)。ここで、RNaseT1などの酵素を用いて、固相遊離部位2cでリンカーを切断すると、固相ビーズ6から遊離させることができる(Step7)。さらに、標識2dを利用することで、各工程におけるタンパク質5、連結体3又は連結体4などの合成効率を定量することができる。
【0065】
また図3では省略したが、RNaseHでmRNAを分解すると、タンパク質とそれをコードするDNAが連結したDNA−タンパク質連結体(DNAのみに遺伝子を有するIVV)が生成される。タンパク質の機能や構造に基づいてスクリーニングを行いその後、光切断などの手法でタンパク質遊離部位2eにてリンカーを切断すると、タンパク質5を遊離することができる。
本発明による方法を示す図4中、図3と同一の符号を付したものは、同一の要素を表す。Step 1で合成された連結体Aは精製及び濃縮の工程を経ることなくStep 4に供され、タンパク質が合成される。その後の固相化工程(Step 5)以降は従来法と同様である。
本発明の方法を用いることにより、IVV形成またはmRNA/cDNAディスプレイ法において、二工程の短縮と、カラム精製を省略することができる。さらに、精製工程がないことから、mRNA/cDNAディスプレイ法によって、ペプチドスクリーニングも効率的に行うことができる。
【0066】
9.ランダムな遺伝子ライブラリーから目的のタンパク質をコードする遺伝子をスクリーニングする方法
本発明の別の態様によれば、上記IVVを用いたタンパク質と遺伝子との対応関係を明らかにする、効率的な方法を利用したペプチドスクリーニング法が提供される。この解析方法は、(a)一種以上の遺伝子を含むmRNAのライブラリーを、IVV合成系に投入し、連結体Bを形成する工程;(b)工程(a)において合成されたタンパク質と一以上の標的物質とを接触させる工程;及び(c)該タンパク質と該標的物質とが相互作用しているか否かを測定する工程、とを含む。
この解析方法は、例えば、(i)配列既知のタンパク質に作用する物質をスクリーニングする場合、(ii)ある特定の物質(例えば、リガンド)が結合する配列未知のタンパク質をスクリーニングする場合等に用いることができる。例えば、(i)の場合は、オーファンレセプタータンパク質をコードする核酸配列を有するmRNAと、ピューロマイシンとの連結体を複数用意しておき(すなわち、複数のオーファンレセプタータンパク質に対応するmRNAをそれぞれ有する連結体A又は連結体Bを複数用意する。)、これを翻訳系に投入する。すると、各連結体A又は連結体BのmRNAから複数のオーファンレセプタータンパク質が合成される。
【0067】
各オーファンレセプタータンパク質は、固相に固定された連結体A又は連結体Bのピューロマイシンに、C末端が結合することによって固定される。必要に応じて、不要な成分を洗浄して除去し、これに標的物質及びバッファー等を加えて、標的物質をオーファンレセプタータンパク質に結合させる試験を行う。(ii)の場合は、例えば、ある遺伝子ライブラリーから複数のmRNAを取得し、複数のmRNAとピューロマイシンとの連結体を作成し、固相に固定する。以下、同様にタンパク質の合成を行い、標的物質をそのタンパク質に接触させて結合実験を行う。
上記(b)工程では、(a)工程で合成されたタンパク質と、一以上の標的物質とを接触させる。ここで用いられる「標的物質」とは、本発明において合成されるタンパク質と相互作用するか否か調べるための物質を意味し、具体的には、タンパク質、核酸、糖鎖、低分子化合物などが挙げられる。
【0068】
前記標的物質としてのタンパク質は、アミノ酸配列及びその機能が既知のタンパク質でも、未知のタンパク質でもよく、タンパク質の全長であっても結合活性部位を含む部分ペプチドでもよい。これらは、合成されたペプチド鎖、生体より精製されたタンパク質、又はcDNAライブラリー等から適当な翻訳系を用いて翻訳し、精製したタンパク質等であってもよい。合成されたペプチド鎖には、糖鎖が結合していてもよい。これらのうち、アミノ酸配列が既知の精製されたタンパク質か、又はcDNAライブラリー等から適当な方法を用いて翻訳、精製されたタンパク質を好適に標的とすることができる。
前記標的物質としての核酸も特に制限されることはなく、DNA、RNAのいずれをも標的とすることができる。また、塩基配列又は機能が未知の核酸のみならず、既知の核酸も用いることができる。タンパク質に結合能力を有する核酸としての機能、及び塩基配列が既知のものか、又はゲノムライブラリー等から制限酵素等を用いて切断単離してきたものに対し、本発明の方法を好適に使用することができる。
【0069】
前記標的物質としての糖鎖も特に制限はなく、その糖配列又は機能が、未知の糖鎖のみならず既知の糖鎖を標的とすることができる。好ましくは、既に分離・解析され、糖配列又は機能が既知の糖鎖が用いられる。
前記標的物質としての低分子化合物についても特に制限はなく、機能が未知のもの、又はタンパク質に結合する能力が既に知られているもの等を用いることができる。
なお、これら標的物質と、リンカーに連結されたタンパク質との「相互作用」とは、通常は、タンパク質と標的分子との間の共有結合、疎水結合、水素結合、ファンデルワールス結合、及び静電力による結合のうち少なくとも1つから生じる分子間に働く力による作用を示す。ここでいう共有結合には、配位結合、双極子結合が含まれる。また、静電力による結合とは、静電結合の他、電気的反発による立体構造に起因した結合が含まれる。また、上記作用の結果生じる結合反応、合成反応、分解反応も相互作用に含有される。相互作用の具体例としては、抗原と抗体との結合及び解離、タンパク質レセプターとリガンドとの間の結合及び解離、接着分子と相手方分子との結合及び解離、酵素と基質の間の結合及び解離、核酸とそれに結合するタンパク質の間の結合及び解離、情報伝達系におけるタンパク質同士の間の結合と解離、糖タンパク質とタンパク質との間の結合及び解離、あるいは糖鎖とタンパク質との間の結合及び解離が挙げられる。
【0070】
ここで用いられる標的物質は、必要に応じて、上述した標識物質により標識して用いることができる。これらの標識物質は、標的物質と固定化タンパク質との間の相互作用に基づいて発生する信号の変化の測定又は解析方法に適したものを選択すればよい。上記標識物質の標的物質への結合は、公知の手法に基づいて行うことができる。
次いで、本解析方法によれば、工程(c)において、タンパク質と標的物質とが相互作用しているか否かを測定する。タンパク質と標的物質とが相互作用しているか否かの測定は、両分子間の相互作用に基づいて発生される信号の変化を測定、検出することにより行う。
そのような測定手法としては、例えば、表面プラズモン共鳴法(Cullen D.C., et al., Biosensors, 3(4), 211-225 (1987-88))、エバネッセント場分子イメージング法(Funatsu T., et al., Nature, 374, 555-559 (1995))、蛍光イメージングアナライズ法、固相酵素免疫検定法(Enzyme Linked Immunosorbent Assay (ELISA):Crowther J.R., Methods in Molecular Biology, 42 (1995))、蛍光偏光解消法(Perran J., et al., J. Phys. Rad., 1, 390-401 (1926))、及び蛍光相関分光法(Fluorescence Correlation Spectroscopy (FCS):Eigen M., et al., Proc. Natl. Acad. Sci. USA, 91, 5740-5747 (1994))等が挙げられる。
【0071】
本発明の解析方法においては、必要に応じて、さらに、(c)工程において相互作用していると判断されたタンパク質−標的物質結合体中の、タンパク質及び/又は標的物質を同定する。タンパク質の同定は、通常のアミノ酸配列シークエンサーで行うこともでき、こうしたタンパク質に結合しているRNAからDNAを逆転写し、得られたDNAの塩基配列を解析することによって行うこともできる。標的物質の同定は、NMR、IR、各種質量分析等を用いて行うことができる。
なお、本発明のRNAチップ及びプロテインチップを用いて、タンパク質−タンパク質間相互作用を解析する場合は、通常のプロテインチップ上のサンプル解析と同様に、飛行時間型質量分析計(MALDI−TOF MS)を用いることができる。
【実施例1】
【0072】
(rGを切断部位とするリンカーを利用したIVV合成)
以下、本発明を実施例に基づいてより具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
1.リンカーの合成
本発明で使用するShort-Biotin-ピューロマイシン・リンカー(SBPリンカー)を合成した。まず、以下の特殊DNA合成をジーンワールド(株)(東京)及び(株)BEX(東京)に委託した。
【0073】
(A)Puro-F-S[配列;5'-(S)-(PL)C(F)-(Spacer18)-(Spacer18)-(Spacer18)-(Spacer18)-CC-(Puro)-3']
ここで、(S)は5'-Thiol-Modifier C6で化合物名はS-Trityl-6-mercaptohexyl-1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite;(PL)はPC Linker Phosphoramiditeで3-(4,4'-Dimethoxytrityl)-1-(2-nitrophenyl)-1-propanyl-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite;(F)はFluorescein-dTで(5'-Dimethoxytrityloxy-5-[N-((3',6'-dipivaloylfluoresceinyl)-aminohexyl)-3-acryimido]-2'-deoxyUridine-3'-succinoyl-long chain alkylamino);(Puro)はピューロマイシン;(Spacer18)はSpacer Phosphoramidite 18(18-0-Dimethoxytritylhexaethyleneglycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)である(すべてGlen Research社製)。
(B)Hybri[(26mer) 配列番号1:5'-CC(rG)C(T-B) C(rG)CCC CGCCG CCCCC CG(T)CC T-3']
ここで、(rG)はリボG;(T)はAmino-Modifier C6 dT(5'-Dimethoxytrityl-5-[N-(trifluoroacetylaminohexyl)-3-acrylimido]-2'-deoxyUridine,3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite);(T-B)はBiotin-dT(5'-Dimethoxytrityloxy-5-[N-((4-t-butylbenzoyl)-biotinyl)-aminohexyl)-3-acrylimido]-2'-deoxyUridine-3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)である(すべてGlen Research Search社製)。
【0074】
上記SBPリンカーは、(A)Puro-F-Sと(B)Hybriを以下の方法に従って架橋し精製して得た。
Puro-F-S 10 nmolを100 μlの50 mMリン酸バッファー(pH 7.0)に溶かし、100 mM Tris[2-carboxyethyl]phosphine(TCEP、Pierce社製)を1μl加え(最終濃度 1mM)、室温で6時間放置し、Puro-F-Sのトリチル化メルカプト基をチオール基に還元した。架橋反応を行う直前に、50 mMリン酸バッファー(pH 7.0)で平衡化したNAP-5 Columns(GEヘルスケア・ジャパン(株)製)を用いてTCEPの除去及び脱塩を行った。
0.2 Mリン酸バッファー(pH 7.0)100 μlに、500 pmol/μlのHybriを20 μl、100 mMの架橋剤EMCS(6-Maleimidohexanoic acid N-hydroxysuccinide ester)、(株)同仁化学研究所製)20 μl、を加えて良く攪拌し、37 ℃で30分反応させた。その後、4℃でエタノール沈殿を行って反応産物を沈殿させ、未反応のEMCSを除去した。沈殿を500μlの冷70%エタノールにて洗浄し、減圧下で乾燥させた後、0.2 Mリン酸バッファー(pH 7.0)10 μlに溶解した。
この反応産物に、上記還元したPuro-F-S(〜10 nmol)を速やかに加えて、4℃で一晩放置した。サンプルに終濃度が4mMになるようにTCEPを加え、37 ℃で15分間放置し、チオール基の架橋反応を停止させた。室温にてエタノール沈殿を行い、合成したリンカーを沈殿させ、未反応のPuro-F-Sを取り除き、さらに未反応のHybri、及びそれらのEMCS架橋物を取り除くために、以下の条件でHPLC精製を行った。
【0075】
(HPLC条件)
カラム:COSMOSIL(登録商標)10 x 250 mm C18-AR-300(ナカライテスク(株)製)
バッファーA:0.1 M TEAA(triethylammonium acetate)
バッファーB:80 %アセトニトリル(超純水で希釈したもの)
流速:0.5 ml/分
濃度勾配:A及びBの混合バッファー濃度は85:15→65:15(A:B)で変化させる(33分間)。
HPLCにより分画された生成物を18%アクリルアミドゲル(8M 尿素、62℃)の電気泳動にて検出し、目的の画分を減圧下で乾燥させた。この後、DEPC(Diethylpyrocarbonate)処理水で溶かして、10 pmol/μlとした。
【0076】
2.mRNAの合成
モデルmRNAとしてBDA(B domain of Protein A)を用いることとした。BDA遺伝子(BDA gene;配列番号2;192塩基長)の5'側上流にT7プロモーター配列と翻訳促進配列、3'側下流にスペーサー領域、ヒスチジンタグ(Hisタグ)及びピューロマイシン・リンカーとの相補鎖領域を有する配列を付加したDNA(BDA whole;配列番号3;367塩基長)をPCRによって合成、精製した後、T7 RiboMAX Express Large Scale RNA Production System(プロメガ(株)製))を用いて、添付のプロトコールに従って5〜30 pmol/μlのmRNA(BDA mRNA;配列番号4;337塩基長)を合成した。
【0077】
3.連結体の形成
3.1 T4 RNAリガーゼを用いたライゲーション酵素反応
ライゲーション反応は、リンカー1に対して1〜6倍のmRNAを加え、20 μlのT4 RNAリガーゼバッファー(50 mM Tris-HCl, pH 7.5; 10 mM 塩化マグネシウム; 10 mM DTT; 1mM ATP)中にて行なった。酵素を加える前にアニーリングするため、90 ℃で5分間アルミブッロク上にて温めた、次に70 ℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した後、氷上に置いた。ここに、1μlのT4ポリヌクレオチドキナーゼ(10 U/μl)及び1μlのT4 RNAリガーゼ(40 U/μl)(いずれもタカラバイオ(株)製)を加え、25 ℃で15分間、保持した。
生成反応液を半分に分割して、後の実験工程にて比較するため、リンカー10 pmolに対しmRNA 10 pmolを反応させたサンプルについては、全ての物質を倍に増量して40 μlの系にて反応させた(反応系R1)。
【0078】
3.2 ライゲーション反応の結果
以下に示す混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、65℃、8M 尿素、5%変性アクリルアミドゲルを用い、200Vにて25分間の電気泳動にて行った。サンプルを各々1μlずつ分注し、3μlのローディングバッファーと2μlのDEPC水と混合して、ゲルにロードした。結果を図5に示す。バンドの検出はリンカーに結合したFITCの蛍光により行い、未反応のリンカー又は上記連結体Aを検出した。
図5は、10 pmolのmRNAに対して、10 pmolのリンカーを加え(前記40 μlの反応系にて反応させたもの)(レーン1、反応系R1)、6.6 pmol(レーン2、反応系R2)、3.3 pmol(レーン3、反応系R3)、1.66 pmol(レーン4、反応系R4)、上記の通りライゲーション反応させたものの電気泳動像を表す。なお、図5中、矢印は未反応のリンカー、*はリンカーが連結された反応産物(連結体A)を示す。
図5から分かるとおり、レーン1においてリンカーの残余が見られたが、その他のレーンではほとんど見られなかった。また混合比1:1の場合を除き、連結体Aの合成量に差はなく、いずれにおいてもほぼ等量のライゲーション反応の起きたことが観察された。
【0079】
図6には、上記のサンプルについてSYBR Goldにて染色した電気泳動像を示した。電気泳動の条件は上記と同様とした。なお、図6中、矢印は未反応のリンカー、*はリンカーが連結された合成物(連結体A)、**は連結体を形成せずに残存するmRNAをそれぞれ示す。レーンMは分子量マーカーを、その左側の数字は各バンドの分子量を表す。
図6から分かるとおり、レーン1においてリンカーの残余が見られたが、その他のレーンではほとんど見られなかった。またレーン1を除き、連結体Aの合成量に差はなく、mRNAの投入量が増えるにつれてmRNAの残余量の増加していることが観察された。
以上より、リンカーに対して1.0〜1.5倍量のmRNAがあれば連結体Aの形成には十分なことが示された。
【0080】
4.連結体Aの精製と濃縮
ライゲーション後の反応生成物の精製及び濃縮の要否を確認するために、精製及び濃縮についての実験結果を示す。
前記R1より20 μlを分取し、以下の精製濃縮工程に供した(反応系R1’)。R1の残りとR2〜R4については、ライゲーション反応後、そのままの状態で冷却して保持した。
未連結のリンカーと反応液とを除くために、RNeasy(登録商標) Kit((株)キアゲン)を用いて、添付のプロトコールに従い精製した。DEPC水により溶出した連結体A(30-50μl)は、共沈剤Quick-Precip Plus solution(EdgeBio社)を用いて添付のプロトコールに従い、エタノール沈殿させた。これを12μlのDEPC水に溶解し、翻訳用テンプレートとした。
【0081】
5.IVV形成(翻訳と並行する連結体A及びタンパク質の連結)
5.1 IVV(連結体B)合成
無細胞翻訳系にはRetic Lysate IVT Kit(Ambion社製)を利用した。混合の方法等は、キットのProtocolを参照して行った。無細胞翻訳反応に使用される全ての試薬を穏やかに撹拌して遠心した後、氷上に置いた。25μlスケールの反応液は次のような順番で混合して調製し、反応させた。
0.625μlの20X Translation Mix minus-Met、0.625μlの20X Translation Mix minus-Leu、及び17μのRetic lysateを泡立たないよう注意深くピペッテイングし、混合した。
この混合液を2.4μlのR1'及び4.0μlのR1〜4に添加した。さらに反応液が25μlになるようにDEPC水を加えた。そして再び泡がでないように丁寧に混合した。混合の後30 ℃で20分間、翻訳反応をさせた。
上記翻訳反応後に、連結体Aと合成タンパク質の連結を促進するため、上記反応液に10μlの3M 塩化カリウム、及び3μlの1M 塩化マグネシウムを加え37 ℃で90分間反応させた。
【0082】
5.2 IVV(連結体B)合成効率の比較
上記の通り合成した連結体Bについて、上記同様の混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、8M 尿素の6%SDSアクリルアミドゲを用いて、0.01Aで60分間、その後、さらに0.02Aで60分間、電気泳動を行った。その結果を図7に示す。バンドの検出はリンカーに結合したFITCの蛍光により行った。これにより、連結体A又はBを観察することができる。
図7は、レーン1:R1’、レーン2:R1、レーン3:R2、レーン4:R3、レーン5:R4、レーン6:連結体Aを表す。なお、図7中、*は連結体A、矢印は連結体Bのそれぞれの分子量を示す。
図7から分かるとおり、R1とR2においてはレーン1のR1'と遜色の無い連結体Bの生成が見られた。その一方で、R3とR4においては生成量が少なく、mRNAの量比が増えるにつれて連結体の形成効率が低下することが観察された。
【0083】
以上より、ライゲーション反応後のシリカゲルメンブレンカラム精製及びエタノール沈殿による濃縮を行わずとも、IVVを形成できることが示された。また、リンカーとmRNAの混合量比を1.0:1.0〜1.5に設定することで、上記精製及び濃縮を行った場合と遜色の無いIVV形成効率を得られることが示された。
本発明によれば、低コストで実施可能なIVV形成法を提供できる。また、本発明の好ましい態様における利用は、従来の方法と比較して遜色の無いIVV合成効率を有するため、mRNA/cDNAディスプレイ法の作動効率を低下させることなく、工程を簡略化できる。このことにより、本発明が、DNA及びタンパク質の機能解析及び機能タンパク質の取得を目指すハイスループットな進化分子工学的手法に応用されることで、上記新規医薬創出の要請に応えるものである。
【産業上の利用可能性】
【0084】
また、本発明は上記創薬を始めとして、医療診断、環境分析、食品分析、研究用バイオイメージングの各分野で有用である。
【符号の説明】
【0085】
1 mRNA
2 リンカー
2a タンパク質結合部位
2b 固相結合部位
2c 固相遊離部位
2c1 第1切断部位
2c2 第2切断部位
2d 標識
2e タンパク質遊離部位
3 連結体A
4 連結体B
5 タンパク質
6 固相ビーズ
6a 固相結合部位認識部位
7 cDNA
200 主鎖
201 主鎖5’末端
202 主鎖5’末端近傍部
203 主鎖3’末端側領域部
300 側鎖
【配列表フリーテキスト】
【0086】
Hybri
Guanosine
Biotin-dT
Amino-Modifier C6 dT
BDA whole
BDA mRNA
【特許請求の範囲】
【請求項1】
液相中における個々の分子の反応において、
タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;
無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;
前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合する工程と;を備える、mRNA−リンカー−タンパク質連結体の生成方法。
【請求項2】
前記所定の割合は、前記リンカー:前記mRNA=3:1〜1:6であることを特徴とする、請求項1に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項3】
前記所定の割合は、前記リンカー:前記mRNA=1:1〜1:1.5であることを特徴とする、請求項1又は2に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項4】
前記主鎖の鎖長は16〜60塩基である、請求項1〜3のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項5】
前記主鎖の鎖長は21〜38塩基である、請求項1〜4のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項6】
前記リンカーの主鎖は、
5'位がリン酸化された主鎖5'末端部と、
第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、
前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、
を備えることを特徴とする、請求項1〜5のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項7】
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、
前記主鎖は主鎖3'末端側領域部に側鎖連結部位をさらに備え、かつ前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列をさらに有し;
前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することを特徴とする、請求項6に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項8】
前記切断部位は、制限酵素認識配列を有することを特徴とする、請求項6又は7に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項9】
前記固相結合部位を介して固相と結合させる工程と、
前記第1及び第2の切断部位を制限酵素で切断する工程とをさらに備えることを特徴とする、請求項8に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項10】
前記制限酵素はPvuIIであることを特徴とする請求項9に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項11】
前記切断部位は、グアノシンを有することを特徴とする、請求項7に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項12】
前記固相結合部位を介して固相と結合させる工程と、
前記第1及び第2の切断部位をRNA切断酵素で切断する工程とをさらに備えることを特徴とする、請求項7又は11のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項13】
前記RNA切断酵素はリボヌクレアーゼT1であることを特徴とする、請求項12に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項14】
液相中における個々の分子の反応において、
タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;
無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;
前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合し、mRNA−リンカー−タンパク質連結体を生成する工程と;
mRNA−リンカー−タンパク質連結体を前記リンカーの備える固相結合部位を介して固相と結合させる工程と;
前記主鎖の3'末端を反応開始点とし、前記mRNAを鋳型として、固相上で逆転写反応を行ってcDNA鎖を形成させる工程と;
前記主鎖の備える第1及び第2の切断部位を、制限酵素またはRNA切断酵素で切断する工程と;を備えるmRNA−リンカー−タンパク質−cDNA連結体の生成方法であって、
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、
前記リンカーの主鎖は、
5'位がリン酸化された主鎖5'末端部と、
前記第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される前記固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、
側鎖連結部位を含み、前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、
を備えることを特徴とし;
前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することを特徴とする方法。
【請求項1】
液相中における個々の分子の反応において、
タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;
無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;
前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合する工程と;を備える、mRNA−リンカー−タンパク質連結体の生成方法。
【請求項2】
前記所定の割合は、前記リンカー:前記mRNA=3:1〜1:6であることを特徴とする、請求項1に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項3】
前記所定の割合は、前記リンカー:前記mRNA=1:1〜1:1.5であることを特徴とする、請求項1又は2に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項4】
前記主鎖の鎖長は16〜60塩基である、請求項1〜3のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項5】
前記主鎖の鎖長は21〜38塩基である、請求項1〜4のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項6】
前記リンカーの主鎖は、
5'位がリン酸化された主鎖5'末端部と、
第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、
前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、
を備えることを特徴とする、請求項1〜5のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項7】
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、
前記主鎖は主鎖3'末端側領域部に側鎖連結部位をさらに備え、かつ前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列をさらに有し;
前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することを特徴とする、請求項6に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項8】
前記切断部位は、制限酵素認識配列を有することを特徴とする、請求項6又は7に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項9】
前記固相結合部位を介して固相と結合させる工程と、
前記第1及び第2の切断部位を制限酵素で切断する工程とをさらに備えることを特徴とする、請求項8に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項10】
前記制限酵素はPvuIIであることを特徴とする請求項9に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項11】
前記切断部位は、グアノシンを有することを特徴とする、請求項7に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項12】
前記固相結合部位を介して固相と結合させる工程と、
前記第1及び第2の切断部位をRNA切断酵素で切断する工程とをさらに備えることを特徴とする、請求項7又は11のいずれかに記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項13】
前記RNA切断酵素はリボヌクレアーゼT1であることを特徴とする、請求項12に記載のmRNA−リンカー−タンパク質連結体の生成方法。
【請求項14】
液相中における個々の分子の反応において、
タンパク質結合部位を有し、RNA及び/又はDNAアナログならびにDNAを含む主鎖を備えるリンカーと、所定の3'末端近傍配列を有するmRNAとを、所定の割合で混合し、その後ライゲーション反応により結合させてmRNA−リンカー連結体を含むライゲーション溶液を得る工程と;
無細胞翻訳系に前記ライゲーション溶液をそのまま投入して、前記mRNA−リンカー連結体中の前記所定の配列を有するmRNAを翻訳してタンパク質を合成する工程と;
前記タンパク質が、前記mRNA−リンカー連結体中の前記タンパク質結合部位に結合し、mRNA−リンカー−タンパク質連結体を生成する工程と;
mRNA−リンカー−タンパク質連結体を前記リンカーの備える固相結合部位を介して固相と結合させる工程と;
前記主鎖の3'末端を反応開始点とし、前記mRNAを鋳型として、固相上で逆転写反応を行ってcDNA鎖を形成させる工程と;
前記主鎖の備える第1及び第2の切断部位を、制限酵素またはRNA切断酵素で切断する工程と;を備えるmRNA−リンカー−タンパク質−cDNA連結体の生成方法であって、
前記リンカーは核酸アナログとDNAとを含む側鎖をさらに備え、
前記リンカーの主鎖は、
5'位がリン酸化された主鎖5'末端部と、
前記第1及び第2の切断部位と、前記第1及び第2の切断部位の間に位置し、固相との間の結合形成に使用される前記固相結合部位とを備え、前記リンカーの主鎖、側鎖及び前記mRNAからなる群から選ばれるいずれとも非相補的な配列を有する、主鎖5'末端近傍部と、
側鎖連結部位を含み、前記主鎖の3'末端まで前記mRNAの3'末端近傍配列と相補的な配列を有する主鎖3'末端側領域部と、
を備えることを特徴とし;
前記側鎖は、5'末端に位置する前記主鎖連結部位と、3'末端に位置する前記タンパク質結合部位とを有することを特徴とする方法。
【図1】

【図2】

【図3】
【図4】

【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−87573(P2011−87573A)
【公開日】平成23年5月6日(2011.5.6)
【国際特許分類】
【出願番号】特願2010−212870(P2010−212870)
【出願日】平成22年9月22日(2010.9.22)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成21年6月15日 インターネットアドレス「http://nar.oxfordjournals.org/cgi/content/full/gkp514」に発表
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、文部科学省、地域科学技術振興事業委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504190548)国立大学法人埼玉大学 (292)
【Fターム(参考)】
【公開日】平成23年5月6日(2011.5.6)
【国際特許分類】
【出願日】平成22年9月22日(2010.9.22)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成21年6月15日 インターネットアドレス「http://nar.oxfordjournals.org/cgi/content/full/gkp514」に発表
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、文部科学省、地域科学技術振興事業委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504190548)国立大学法人埼玉大学 (292)
【Fターム(参考)】
[ Back to top ]