
ウイルス及び細胞結合部位を持つ分子を用いたウイルス媒介性DNA導入を増強する方法
【課題】細胞へのウイルス媒介性遺伝子導入の増強方法を提供する。
【解決手段】フィブロネクチンまたはフィブロネクチンフラグメントの存在下で、細胞を感染させることを含むレトロウイルスによる造血細胞およびその他の細胞の形質転換効率を増加させる方法、および遺伝子トランスファーの強化、造血細胞集団、および細胞内へのレトロウイスル仲介DNAトランスファーを強化する構築物、を利用する体細胞遺伝子療法の改良された方法ならびにそれらの使用。フィブロネクチンおよびフィブロネクチンフラグメントは、細胞、特に、前駆細胞および初期造血幹細胞を含む造血細胞、へのレトロウイスル仲介遺伝子トランスファーを強化する。
【解決手段】フィブロネクチンまたはフィブロネクチンフラグメントの存在下で、細胞を感染させることを含むレトロウイルスによる造血細胞およびその他の細胞の形質転換効率を増加させる方法、および遺伝子トランスファーの強化、造血細胞集団、および細胞内へのレトロウイスル仲介DNAトランスファーを強化する構築物、を利用する体細胞遺伝子療法の改良された方法ならびにそれらの使用。フィブロネクチンおよびフィブロネクチンフラグメントは、細胞、特に、前駆細胞および初期造血幹細胞を含む造血細胞、へのレトロウイスル仲介遺伝子トランスファーを強化する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、一般的にはウイルスによる細胞の形質導入の効率を増加させること、より具体的には、ウイルスに結合する領域と、細胞に結合する領域を含むポリペプチド類のような分子を用いて、細胞へのウイルス媒介性遺伝子導入を増強させる方法に関連する。
【背景技術】
【0002】
発明の背景
遺伝子導入技術の進歩と同様に、多くのヒト疾患の分子的基礎の理解の進歩により、重度の遺伝病に対する体細胞遺伝子治療のプロトコールを開発するための最近の試みがもたらされた。最近、ヒト遺伝子治療に対する、見込みのある疾患候補は、酵素、もしくは他のタンパク質が不完全または見られないものであって、酵素もしくはタンパク質のレベルが厳密に調節される必要がないもの、特に本質的には調節されており、そしてその欠陥が骨髄で発見されたものを含む。
【0003】
例えば、遺伝子治療の候補の一つの疾患は、重症複合免疫不全症(SCID)を引き起こすアデノシンデアミナーゼ(ADA)欠損症である。ADA欠損患者は、骨髄細胞で検出できる酵素はわずか、もしくは存在しない。しかしながら、ADA欠損は適合した骨髄移植によって治療された。ADA正常細胞は、ADA欠損細胞に比べて、選択的優位であり、正常に患者の骨髄に再配置(repopulate)される。
【0004】
骨髄細胞は、骨髄組織が簡単にin vitroで扱うことができ、再配置する細胞を含むことから、体細胞遺伝子治療のよい標的である。別の方法としては、ヒト臍帯血も多数の原始前駆細胞を含んでいることが以前に明らかにされている。長期にわたり再配置する細胞である造血幹細胞への遺伝子導入の成功は、これらの細胞の子孫において明示された様々な疾患に対して修正の治癒を導く可能性がある。
【0005】
再配置する幹細胞への遺伝子導入、及び長期遺伝子発現は、若干の研究者によってネズミモデルで成し遂げられた。しかしながら、イヌ及び霊長類の様なより大きい動物での in vivo 実験は限られた成功しかなく、これはほとんど原始造血幹細胞の感染の低効率による。現在の遺伝子導入技術の限界は、成人骨髄(ABM)に存在する幹細胞の数の少なさ、これらの細胞を精製する適当な方法がないこと、及び細胞周期でのこれらの原始細胞の画分が低いことを含む、いくつかの因子によって、ヒトプロトコールに利用する際、さらに複雑になる。
【0006】
骨髄細胞を伴うネズミの実験、及び大動物の実験双方において、最も有効なプロトコールが、レトロウイルス産生細胞株と標的細胞の共培養を利用していることが注目された。また、ヒトでのFDA−立証済み遺伝子導入試験のほとんどは、遺伝子形質導入のために、組換えレトロウイルスに依存している。組換えレトロウイルスベクターは、導入に効果的であり、細胞DNAへ外来DNAを正確に、及び安定に組み込むことから、遺伝子治療に好ましい。これらのベクターは、遺伝子導入のための外来DNAを含み、ウイルスの病原性を除去するために、さらに修飾される。これらの修飾のために、ウイルスの産生は一般的にレトロウイルスパッケージング細胞を用いることによって行われる。しかしながら、臨床遺伝子治療のために、無細胞の形質導入が、生物学的に安全で、品質管理についての関心のためにさらに好ましい。不幸なことに、幹細胞などの造血細胞への効率のよい遺伝子導入は、一般的にウイルス産生細胞との共培養なしには不可能であった。
【0007】
最近、遺伝子導入効率は、感染中に標的細胞を間質細胞へ暴露することで増加されうることが示された。間質細胞は、造血微環境(HM)の主要な構成物である。HMは、マクロファージ、間質細胞、内皮細胞、含脂肪細胞、及び様々な特定の接着分子によって構成されている複合細胞外マトリックス(ECM)の組織化されたネットワークによって成り立っている。ラミニン、コラーゲン、トロンボスポンジン、プロテオグリカン類、グリコサミノグリカン類、及びフィブロネクチンなどのECM分子は、造血細胞及び成長因子双方のための固定部分を提供する。このレトロウイルス感染における間質の促進効果の基礎をなす機構は明らかではないが、造血細胞の増殖及び分化の生理学的調節が、これらの細胞がHMの細胞と直接接触されたときに起こるということが以前から知られていた。
【0008】
長期にわたり再配置する造血幹細胞及び他の細胞への効率的な遺伝子導入は問題を残しており、現在の造血性の疾患及び他の疾患の治癒を目指す療法への遺伝子導入プロトコールの広範囲での適用を阻害する。求められていることは、過去の方法の危険および限界を排除したほ乳類細胞への遺伝的物質の効果的な導入の方法を求めることであり続けている。本発明は、この必要性に取り組んだものである。
【発明の概要】
【発明が解決しようとする課題】
【0009】
発明の概要
簡単に、本発明の一つの好ましい様態は、レトロウイルスベクターによる造血細胞の形質導入の効率を増加させる方法を提供する。この方法は、レトロウイルスによる細胞の形質導入の効率を増加させるのに効果的な、十分に純粋なフィブロネクチンおよび/またはフィブロネクチン断片の存在下で、複製欠損組換えレトロウイルスベクターを、生存している造血細胞に感染することを含む。フィブロネクチンおよび/またはフィブロネクチン断片は、天然に生じる物質から由来しうるか、また合成的に由来することができ(例えば、遺伝的に組換え、もしくは化学的合成技術によって設計される)、もしくは、天然に生じるもの及び合成物質の組み合わせにより由来しうる。加えて、本発明で用いたフィブロネクチンポリペプチド、もしくはペプチド類は、本発明に従って、形質導入を高めることができるのに必要な接着特性を持っている、機能的ポリペプチドをそれでもなお提供する、自然に生ずるフィブロネクチンのアミノ酸配列の変異を含む可能性があることが理解されるであろう。
【0010】
他の本発明の好ましい様態は、レトロウイルスによる細胞形質導入の効率が増加するのに効果的な量の、固定化されたフィブロネクチン、固定化されたフィブロネクチン断片、もしくは固定化されたそれらの混合物存在下で、生存している造血細胞へ外来DNAを運ぶ複製欠損組換えレトロウイルスを感染することを含む、形質導入された造血細胞を産生する方法を提供する。
【0011】
本発明の他の好ましい様態は、細胞移植の改善された方法を提供する。この方法は、ドナー動物からの生存している造血細胞を得る工程、形質導入された生存している造血細胞産生のために、固定化された形で、形質導入の効率を増加させるのに効果的なフィブロネクチンおよび/またはその断片の存在下で、造血細胞を組換えレトロウイルスベクターにて感染させる工程、及び細胞移植片として、この形質導入された生存している造血細胞をレシピエント動物に導入する工程を含む。一つの好ましい方法において、感染細胞は自己提供体に導入することができる。
【0012】
本発明の他の好ましい様態は、細胞移植法に適した、形質導入された臍帯血細胞を得る方法を提供する。この方法は、レトロウイルスベクターによる造血細胞の形質導入効率を増加するために効果的な量の固定化されたフィブロネクチンおよび/またはフィブロネクチン断片の存在下で、臍帯血由来の造血細胞に、複製欠損組換えレトロウイルスベクターを感染させることを含む。本発明はまた、これらの方法で得ることのできる臍帯血由来の生存している形質導入された細胞集団を含み、そして細胞移植片として動物体内へ形質導入された細胞集団を導入することを含む、細胞移植方法を含む。
【0013】
本発明の上記言及の様態のさらに固有の局面に従って、使用されたフィブロネクチンまたはフィブロネクチン断片は、フィブロネクチンのヘパリン−II結合部位のレトロウイルス結合活性を提供する第一のアミノ酸配列、及び、フィブロネクチンのCS−1部位の細胞結合活性を提供する第二のアミノ酸配列を含みうる。これらの二つのフィブロネクチンの結合部位を一緒に用いることは、レトロウイルスによる標的細胞の形質導入効率を非常に顕著に高めることが証明された。
【0014】
本発明の他の好ましい様態は、レトロウイルスベクターによる予め決定した標的細胞の形質導入の効率を高めるための構築物を産生する方法を提供する。この方法は、前記標的細胞と結合するリガンドと、フィブロネクチンのヘパリン−II結合部位のレトロウイルス結合活性を提供するアミノ酸配列を含むポリペプチドとを、共有的に結合する工程を含む。本発明はまた、レトロウイルスによる、予め決定した標的細胞の形質導入の効率を増加させるために、そしてこの形質導入された細胞を用いて細胞移植処置法のために、これらの構築物を利用することを含む方法を含む。
【0015】
本発明の他の好ましい様態は、多量のウイルスを局在化するために、フィブロネクチンのヘパリン−II結合部位のウイルス結合活性を含む、固定化された効果的な量のフィブロネクチンまたはフィブロネクチン断片の存在下で、ウイルスを含む培養液をインキュベートすることを含む、多量のウイルスを局在化するための方法を提供する。
【0016】
さらに、本発明の他の好ましい様態は、一般的に、以下の節でさらに示すように、十分に純粋なおよび/または固定されたフィブロネクチン、またはそのフラグメントを含む、形質導入された生存している造血細胞、及び細胞培養を提供すると同時に、細胞へのレトロウイルス媒介性DNA導入を行うためのキットを提供する。
【0017】
さらなる本発明の好ましい様態は、レトロウイルスと細胞を共局在化させ、細胞の形質導入効率を増加させるために、細胞と結合するリガンドと、レトロウイルスと結合するリガンドとを含む、特定の量の固定化物質である、効果的な固定された量のポリペプチドのような物質の存在下で、レトロウイルスを細胞に感染させることを含む、レトロウイルスによる生存している細胞の形質導入された群を得るための方法を提供する。さらに驚いたことに、それらの工程は、さらに都合よいことに、今まで、形質導入効率を増加させることを望むために、遺伝子導入プロトコールにおいて使用されていたものである、ヘキサジメトリンブロミド(1,5−ジメチル−1,5−ジアゾウンデカメチレンポリメトブロミドとしても同定されている)の非存在下で、もしくは少なくとも実質的に存在しない状態で行われる。しかしながら、驚くべきことに、ヘキサジメトリンブロミドの存在は、本発明の共局在化媒介性遺伝子導入工程における形質導入効率を増大させるのではなく、むしろ減少させることが発見された。従って、さらに好ましい本発明の様態は、ヘキサジメトリンブロミド、もしくは共局在化のための物質の非存在下で行われた同じ様なプロトコール、例えば類似の共培養プロトコールにおける、形質導入効率を増加させる他の物質の非存在下で行われる。結果として生ずる改善した細胞集団、及び細胞移植方法はまた、本発明の一部分を形成する。
【0018】
本発明の他の好ましい様態は、細胞に結合するアミノ酸配列と、レトロウイルスに結合する、V型コラーゲン、または繊維芽細胞増殖因子由来のアミノ酸配列を含むポリペプチドの存在下での、レトロウイルスと細胞の感染の過程を含む、レトロウイルスによる生存している細胞の形質導入群を得るための方法を提供する。
【0019】
本発明の他の好ましい様態は、レトロウイルスと細胞を共局在化し、細胞の形質導入効率を増加させるために、T細胞に結合するリガンドとレトロウイルスに結合するリガンドとを含む物質の存在下で、レトロウイルスを細胞に感染させることを含む、レトロウイルスによるT細胞の形質導入の方法を提供する。好ましい形において、本方法で用いた物質は、T細胞に結合する第一のアミノ酸配列と、レトロウイルスに結合する第二のアミノ酸配列を含むポリペプチドである。
【0020】
本発明の他の好ましい様態は、レトロウイルスを局在化させる方法を提供する。レトロウイルスに結合するV型コラーゲンまたは繊維芽細胞増殖因子由来のアミノ酸配列を持つ、効果的な量の分離されたポリペプチドに、レトロウイルスを接触させることを含む。
【0021】
ほ乳類細胞の効果的なレトロウイルス感染の方法を提供することは本発明の目的である。
共培養の必要性を取りのぞいた、レトロウイルスベクターによる遺伝子導入の方法を提供することは、本発明のさらなる目的である。
【0022】
自己および/または同種の細胞移植のための、改善した方法および細胞培養を提供することは、本発明のさらなる目的である。
本発明のこれらのそして他の目的、利点、及び特徴は、すぐに以下の記載によって、当業者に対しては容易に明らかになるであろう。
【課題を解決するための手段】
【0023】
好ましい様態の記述
本発明の原理の理解を促進する目的のために、参考文献は現在、それらの特定の態様をなし、固有言語は同様に記載するために使用しうる。それにも関わらず、本発明の範囲を制限しないことがそれによって、このような変化、さらには改良、そして本発明が関係する技術分野での当業者が想到したであろう様に企画され、本明細書に示されたように、このような本発明の原理の応用を意図していると理解されるであろう。
【0024】
以上に指摘したように、本発明は、レトロウイルスなどのウイルスによる、生存している細胞の形質導入の頻度を増加させる方法を提供する。本発明はまた、組換えレトロウイルスベクターを用いて、生存している細胞への効果的な遺伝子導入の方法、形質導入された細胞を得る方法、そして自己及び他の細胞移植を行うための方法と物質も提供する。
【0025】
本発明の一つの特徴は、フィブロネクチン(FN)及びFNの細胞結合部位CS−1を含むフィブロネクチン断片が、フィブロネクチン受容体を持ち、それによって、フィブロネクチンもしくはその断片への結合能力を示す、たとえば投入された前駆細胞及び原始造血幹細胞、もしくは長期培養始原細胞(LTC−IC)といった造血細胞などの細胞へのレトロウイルス媒介性遺伝子導入を顕著に増強するという発見である。都合よいことに、この上昇した効率は、ウイルス産生細胞との共培養を必要としない。本発明の他の特徴は、ヘパリン−II結合部位の中に位置するフィブロネクチンのウイルス結合部位の発見を利用する。本発明の別の特徴は、ヘパリン−II結合部位に局在するフィブロネクチンのウィルス結合部位の発見を利用する。このウイルス結合部位は、例えば、ウイルスを標的細胞に送達するための広い範囲の構造を含む多くの応用において、ウイルス粒子を局在化させるために使うことができる。
【0026】
本発明のある好ましい観点に従った組換えウイルスベクターは、外来DNAを含み、そして非病原性、すなわち複製欠損である。これらのベクターは、外来DNAを動物細胞、特にほ乳類細胞のような宿主細胞の細胞DNAの中に効果的に導入し、正確にそして安定に組み込む。例えば、本発明において、関心の遺伝子のコード配列からの塩基の列を含むヌクレオチド配列を、遺伝子を駆動するための適当なプロモーター、一般的には外来のプロモーターの調節下で、組換えレトロウイルスベクターの中へ組み込むことができる。これに関連して、外来DNAは、天然、もしくは人工的どちらかで産生したDNAを含むことができ、異質組織源から得られた部分から由来することができる。この異質組織源の部分は、天然に生じたもの、もしくは化学的に合成された分子である可能性があり、そしてこれらの部分は、ライゲーションもしくはこの分野で既知の他の方法で連結する。
【0027】
ウイルスに組み込まれた外来DNAは、細胞への導入のための関心のいずれかのDNAであり得る。例えば、外来DNAは、知られている異常と関係するADAなどのタンパク質、もしくはアンチセンスRNA、リボザイム、もしくは疑似プライマー(例えば、1990年11月15日発行のWO 90/13641 を参照)、細胞内抗体(例えば、1994年2月3日発行の WO 94/02610 を参照)、成長因子、もしくは類似のものをコードすることができる。
【0028】
示したように、導入されたヌクレオチド配列は、プロモーター調節下にあり、従って一般的にプロモーターの下流にあるであろう。別の言い方をすれば、プロモーター配列は、一般的にコード配列の上流(すなわち5’末端)にあるであろう。このような傾向において、プロモーターと共同する他の調節因子(例えばエンハンサー配列)や、外来コード配列の転写を行うための転写開始コドンが存在しうるか否かについてはよく知られている。「調節下」の語は、導入された遺伝子の転写を達成するために必要であるような、他の成分の存在を企図する。同様に組換えDNAは好ましくは導入されたコード配列より下流に終止配列を含みうる。
【0029】
選択マーカー、もしくは他の選別できる好要因を提供する外来DNAを含むレトロウイルスベクターを使用することができる。例えば、ベクターはネオマイシンなどの抗生物質を含む、様々な選択薬剤に対する抵抗性を提供する一つもしくはそれ以上の外来性遺伝子を含むことができる。本発明で用いることのできた典型的なベクターは、例えば、すべてすでに Moritz et al. (1993) J. Exp. Med. 178:529 によって報告されている、 N2/Zip TKNEOベクター(TKNEO)(NIH/3T3細胞上の力価:1×105 G418r cfu/ml)、ZipPGK−hADAベクター、及びZipPGK−mADAベクターである。TKNEOベクターでは、ネオホスホタランスフェラーゼ配列がヘルペス・シンプレックスのチミジンキナーゼプロモーターを介して、センス方向に発現している(5’長末端繰り返しLTRに関係する)。このベクターは形質導入された細胞の同定を促進するネオマイシン耐性を提供する選択マーカー遺伝子を含む。ZipPGK−hADAベクターにおいて、ヒトADA(”hADA”)cDNAは、ヒトホスフォグリセレートキナーゼ(PGK)プロモーターを介して、5’LTRと関連するセンス方向に発現している。これは、ただ一つの発現しうる遺伝子配列を含み、優性選択マーカーを欠く。ZipPGK−mADA(PGK−mADA)ベクターは、ヒトADA cDNAがネズミADA(”mADA”)DNAに置き換えられたこと以外は、ZipPGK−hADAと同じものである。これら、及び他のウイルスベクター、及びこれらの産生手法はよく知られており、これらの本発明における実行、及び使用は、本明細書中の開示を与えられた当業者の間ではよくあることである。
【0030】
本発明中で使用したウイルスベクターは、ヒトフィブロネクチンのアミノ酸配列を含む、フィブロネクチンのヘパリン−II結合部位のアミノ酸配列に結合する能力を示す。以下で議論するように、本発明はいずれかの説に限られるわけではないが、それぞれの機能部位に対する、ウイルスと細胞の結合を介して、ウイルスと標的細胞とが共局在化することは、ウイルスによる細胞の形質導入の増大を促進すると信じられている。この考えでは、ヘパリン−II結合部位のアミノ酸配列に対するウィルスの結合能力、そして従って、本発明で効果的に役に立つためのウイルスの能力は、以下の実施例8、及び9で記載したような決まった操作を用いてすぐに確かめられる。一般的にいえば、これらのアッセイは、固定化ポリペプチドマトリックスからの洗浄に耐えるために、ウイルス粒子が、ヘパリン−II結合部位を含む固定化ペプチドに結合する範囲を決定する。簡単に言えば、例えば、フィブロネクチンヘパリン−II結合部位を含む、固定化ポリペプチドを含むウェルの中で、ウイルス含有上清をインキュベートできる。次いでこのウェルをさらに生理食塩水緩衝液で洗い、その後、ウイルスに対する標的細胞を、ウェルに残っている感染活性のレベルを決定するために、ウェルの中でインキュベートする。初期ウイルス上清と比べた感染活性、もしくは力価の減少が測定され、同じ対照実行(例えばBSA−コートウェルの使用)と比較した。対照ウェルに比べてヘパリン−II部分を含むウェルでの顕著に高い力価は、本発明の観点における使用に、対象ウイルスが適していることを明らかにしている。このスクリーニング手法を促進するために、ウイルスベクターは、以上で議論したように、選択マーカー遺伝子を含んでもよい。
【0031】
本発明で使用するためのフィブロネクチン断片は、天然、もしくは合成起源のものでよく、例えば以前に、 Ruoslahti et al. (1981) J. Biol. Chem. 256:7277 ; Patel and Lodish (1986) J. Cell. Biol. 102:449 ; 及び Bernardi et al. (1987) J. Cell. Biol. 105:489 によって記載されたような天然に生ずる物質からの十分な純度で調整されうる。この考えにおいて、本明細書の、十分に純粋なフィブロネクチンもしくはフィブロネクチン断片についての言及は、それらがフィブロネクチンが天然で生じる他のタンパク質から、本質的に遊離していることを意味するつもりである。本発明で使用するための、十分に純粋なフィブロネクチンもしくはフィブロネクチン断片は、例えば、一般的にはTaguchiら、そして日本、京都の宝酒造株式会社に与えられた1993年3月30日の米国特許 No. 5,198,423で記載されたように、組換えにより産生することができる。特に、以下の実施例でH−271、H−296、CH271、CH296及びC−CS−1として同定された組換え断片、及びそれらを入手する方法は、この ’423特許で詳細に記載されている。以下の実施例で用いたC274断片は、米国特許 No. 5,102,983で記載されているようにして入手した。これらの断片、もしくは断片からの日常的に引き出すことのできるものは、これも米国特許 No. 5,198,423 に記載されているようにFERM P−10721(H−296)、FERM BP−2799(メチオニンを介してH−271に結合するC−277)、FERM BP−2800(メチオニンを介してH−296に結合するC−277)、及びFERM BP−2264(H−271)として工業技術院(日本)の機関である発酵研究所(Fermentation Research Institute)に寄託されている、E. coliの培養によって入手できる。加えて、本明細書中の使用可能なフィブロネクチン断片に関する、もしくはこのような断片のための開始の物質に関する役に立つ情報は、上記記載の組換え断片についてさらに報告されているKimizukaら(J. Biochem., 110, 284−291 (1991))、ヒトフィブロネクチン遺伝子の構造について報告しているもの(EMBO J., 4, 1755−1759 (1985))、ヒトフィブロネクチンのヘパリン−II結合部位について報告しているもの(Biochemistry, 25, 4936−4941 (1986))において見いだしうる。CS−1細胞接着部位とヘパリン−II結合部位双方を含むフィブロネクチン断片、例えば、約30もしくは35kD断片(30/35FN)、そして以下の実施例で報告する様々な組換え断片を含むものは、このようなさらなる作業の中で造血細胞への遺伝子導入の効率を顕著に高めることが発見されており、そして本発明での使用に好ましい。従って、大きくいえば、本発明で用いたフィブロネクチン関連ポリペプチド、もしくはポリペプチド類は、ウイルスに結合するフィブロネクチンのヘパリン−II結合部位のアミノ酸配列と同様に、フィブロネクチンのCS−1細胞接着の細胞結合活性を与えるアミノ酸配列を提供すると理解されるだろう。当業者は、細胞及びウイルス結合活性の必要性は、これらの機能的フィブロネクチン部位の天然アミノ酸配列、及び天然アミノ酸配列と違うが、十分に細胞結合及びウイルス結合活性を示す点では似ているアミノ酸配列両方により提供されうるということを理解するであろう。これらの類似アミノ酸配列は、これらの対応する天然配列との実質的な配列相同性を示し、望ましい細胞結合特性もしくは、ウイルス結合特性を持つアミノ酸配列を提供する一方で、アミノ酸が削除されたもの、置換されたものおよび/または修飾されたものを含むことができる。
【0032】
この考えでは、適切な生物技術分野は、対象の機能的部位内でアミノ酸の削除、置換、付加、もしくはその他の修飾を日常的に起こしうるような状態に進歩した。次いで結果としてのアミノ酸配列は、望ましい細胞結合活性、もしくはウイルス結合活性を求めて、日常的にスクリーニングされうる。例えば、フィブロネクチンのヘパリン−II結合部位の変異体、もしくは修飾型のウイルス結合活性は、一般的に上記で議論し、さらに具体的には、ウイルスインキュベート、洗浄、対照と比較した感染維持力を決定するためのウイルス力価アッセイを用いた、以下の実施例8及び9のようにしてスクリーニングすることができる。本明細書中で提供した技術を得れば、これらの結合アッセイは代表的で、しかし当業者に対しては日常的な実験であり得る。
【0033】
フィブロネクチンのCS−1細胞接着部位に対する修飾、もしくは変異型に対する細胞の結合、もしくは他の細胞結合ポリペプチド類の細胞結合は、同じく慣例の手法を用いてアッセイされうる。例えば、このような手法は、 Nature, 352:438−441 (1991)に記載のものを含む。簡単にいえば、細胞結合ポリペプチドをプラスチックディッシュにコートし、分析すべき細胞集団を培養液中で30分〜2時間重層する。このインキュベート期間後、タンパク質への非接着の細胞を回収し、計数し、生存数をアッセイする。ポリペプチドへ接着した細胞も、トリプシン、もしくは細胞解離緩衝液(例えば Gibco)を用いて回収し、計数し生存数を調べる。いくつかの場合、例えば、造血コロニー形成細胞のために、この細胞のコロニー形成特性を確かめるために、細胞をさらに12〜14日間培養する。次いで付着細胞の割合を測定し、プラスチックディッシュにコートしたウシ血清アルブミン(BSA)などの標準対照と比較する。アッセイしたポリペプチドに対する標的細胞の実質的な結合は、ポリペプチド/細胞の組み合わせが本発明に適合するという指摘を提供し、このポリペプチドは、ウイルスベクターによる標的細胞の感染を高めるための、本発明の構築物を作製するために、フィブロネクチン由来のレトロウイルス結合断片に結合しうる。
【0034】
本発明の、さらに固有の観点により、レトロウイルスベクターによる形質導入を高めるために使用した、ウイルス結合ポリペプチドは、(i)ヒトフィブロネクチンのヘパリン−II結合部位のAla1690−Thr1960に対応する第一のアミノ酸配列を含み、これは式により示す(配列番号1)
【0035】
【化1】

【0036】
もしくは、レトロウイルスの結合能力を示すための、それに対して十分に類似のアミノ酸配列;
及び(ii)式により示される、ヒトフィブロネクチンのIIICS結合部位(CS−1細胞結合部位)の一つの部分に対応する、第二のアミノ酸配列(配列番号2)、
Asp Glu Leu Pro Gln Leu Val Thr Leu Pro His Pro Asn Leu His Gly Pro Glu Ile Leu Asp Val Pro Ser Thr ;
もしくは、原始前駆細胞および/または長期にわたり再配置する(幹)細胞などの造血細胞結合能を示すための、それに対して十分に類似のアミノ酸配列を含みうる。
【0037】
以前に言及したように、結果としてのアミノ酸配列が、ウイルス結合能(ヘパリン−II結合部位の場合)、及び標的細胞結合能(CS−1部位の場合)を示すために、これらの天然配列のある修飾および/または変異は、本発明の実施内で可能であることが理解されるであろう。
【0038】
例えば、ヘパリンと結合し、フィブロネクチンのヘパリン−II結合部位との本質的に配列同一性を示す配列を持つ既知のポリペプチド類は、例えば、V型コラーゲン及び繊維芽細胞増殖因子を含む。以下の特異的な実施例16で記載のように、VLA−4および/またはVLA−5のための結合部位などのような細胞結合配列と連結する、これらの配列を含むポリペプチド類は、細胞結合配列に結合する細胞への、レトロウイルス媒介性DNA導入を高めるために使用できる。
【0039】
本発明の一つの観点は、in vitroの細胞治療、及び続いて宿主への標的細胞の移植を伴う、すなわち形質導入された標的細胞による宿主への「移植」として知られる、宿主への標的細胞の形質導入の体細胞の遺伝子治療の方法を提供する。造血、または他の細胞、例えば骨髄もしくは末梢血から単離された幹細胞、胚性幹細胞、もしくはそれ以外のCD34+、および/もしくはC−kit+として特性化された細胞は、ヒトもしくはその他の哺乳動物源より標準プロトコールを用いて集められる。例えば、造血細胞は、ヒトドナーの骨髄または末梢血、もしくはヒト胎児臍帯血から集めることができる。いったん回収した後、造血細胞は所望により幹細胞および/または始原前駆細胞内で、それらを濃縮するために、標準手技によって処理する。次いで造血細胞は、例えば組織培養プレート上で適当にインキュベートされうる。所望により、この期間中、接着陰性低密度単核細胞は、レトロウイルス感染の前に前刺激されうる。当該技術分野で既知の、そして本明細書で用いられている前刺激は、レトロウイルスに暴露する前に、成長刺激因子に細胞を暴露する過程と関連する。このような前刺激は、レトロウイルスによる造血細胞の形質導入を改善することが証明された。
【0040】
前刺激に続いて、細胞を採集し、レトロウイルスによる細胞形質導入の効率を高める、本明細書に記載される、フィブロネクチンまたはその断片とともにインキュベートする。好ましくは、細胞は精製されたおよび/または不溶性、例えば、固定化フィブロネクチンもしくはフィブロネクチン断片とともにインキュベートする。次いで細胞は、ウイルスによる細胞の形質導入の効率を増加させるのに効果的な量のフィブロネクチンもしくはフィブロネクチン断片の存在下で、組換えウイルス、例えば細胞中の酵素もしくは他のタンパク質の欠損、もしくは異常を修正するための遺伝子を含んだレトロウイルスに感染させることができる。次いで結果としての形質導入造血細胞は、慣習的に、例えば、静脈注射で動物細胞移植レシピエントへ導入し、自己ドナーが望ましいが、異種遺伝子移植も含まれ、後者は特に、以下で議論するように、移植に対して臍帯血細胞が使用される。
【0041】
本発明の方法は、例えば、癌、白血病、タンパク質欠損もしくは異常に関連する疾患を含む、骨髄疾患を含む様々な疾患に対する遺伝子標識プロトコル、もしくは遺伝子治療プロトコール、及び、化学療法などの他の治療プロトコールへの抵抗性を改善するために、造血細胞を修飾する治療の中で使用されうる。本発明が使用される可能性のある代表的な疾患は、従って、ADA欠損、例えばADA−欠損SCID、小児急性骨髄性白血病(AML)、神経芽細胞腫、及び成人AML、及び急性リンパ球性白血病(ALL)を含む。
【0042】
本発明の一つの特に好ましい様態において、細胞移植のために用いられた細胞は、ヒトの臍帯血から入手する。従って、ヒト臍帯血は収集され、例えば接着陰性低密度単核細胞集団を入手することにより、生存している始原造血前駆細胞および/または幹細胞に濃縮される。その後この集団は、そこで所望により、前刺激され、そしてレトロウイルスベクターと、ベクターによる細胞の形質導入の効率を高めるための、固定化および/または精製フィブロネクチンもしくはフィブロネクチン断片の存在下で、インキュベートする。この考えにおいて、臍帯血由来の始原造血細胞および/または幹細胞は、たとえフィブロネクチンが臍帯血中のECMの構成成分ではなく、たとえ臍帯血由来の始原前駆細胞及び幹細胞が、骨髄からのものと違う特徴を有する場合でも、フィブロネクチン、もしくはフィブロネクチン断片の存在下で、かなり高められる。特に、臍帯血幹細胞は、CD34+、HLA−DR+として特徴づけられており、一方、骨髄からの幹細胞は、CD34+、HLA−DR−として特徴づけられた。臍帯血由来の始原前駆細胞が、フィブロネクチンもしくはフィブロネクチン断片の存在下で高められた様式で、効率的に形質導入されることの発見により、造血細胞の、簡単で高い幹細胞濃縮源の使用を可能になった。さらに、始原前駆細胞、及び幹細胞を濃縮した臍帯血由来の異種遺伝子移植による、多くの患者の移植の成功についての証拠により、臍帯血を造血細胞のとても好ましい源とすることができる(Kohli−Kummer et al., Brit. Heaematol. 85:419−422 (1993) ; Broxmeyer et al., Blood Cell 17:313−329 (1991) ; Gluckman et al., Br. J. Heaematol. 45:557 (1980) ; Heidelberg:Springer−Verlag pp. 60−68 (1989) ; Wagner et al., Blood 79:1874−1881 (1992) ; Wagner et al., Blood 82−86a (要約)を参照)。
【0043】
もし望めば、回収した、形質導入した造血もしくは他の細胞を、形質導入の効率及び遺伝子発現について試験することができる。例えば、本発明によって提供される、レトロウイルス媒介性遺伝子導入での顕著な改善は、以下の特定の実施例によって示されており、フィブロネクチンもしくは効果的なフィブロネクチン断片の存在下で、レトロウイルスによる高感染及び遺伝子導入効率を示す、いくつかの試験を記載している。特に、PGK−hADAレトロウイルスに感染されたネズミ造血細胞は、高いレベルの形質導入ADA cDNAを発現した。同様に、ヒト前駆細胞コロニーに感染させた個々のPGK−mADAウイルスは、ネズミADAのレベルが、内在性ヒトADAタンパク質より10倍も上昇して発現した。従って、導入効率の説得力のある解析をするために、前駆細胞コロニーは、もし形質導入されたmADAの発現が、内在性ヒトADAレベルと同等か、それ以上ある場合にのみ、形質導入されたとみなされた。TKNEOベクター由来のネオの高レベルの発現は、ネオホスフォトランスフェラーゼ(ネオ遺伝子産物)活性に対するアッセイとしてG418薬剤耐性によって検出した。
【0044】
上で示したように、本発明の方法は、レトロウイルス産生細胞の存在下での共培養を必要とすることなく、都合よく行われる。従って、本発明の一つの観点によると、レトロウイルス媒介性遺伝子導入は、標的造血細胞、もしくは他の細胞以外の細胞が実質上非存在下において実行しうる。例えば、レトロウイルスベクタープラスミドを含む産生細胞を培養することができ、上清が回収される。次いでレトロウイルス含有上清は、好ましくは固定型の、すなわち、感染が行われる基質上、さもなければ、感染用の培養液と接触する場合のフィブロネクチンおよび/またはフィブロネクチン断片存在下で、造血細胞の感染に用いることができる。この考えにおいては、高力価、ヘルパーなしのレトロウイルスを産生する産生細胞も、本発明での使用に適合することが企図される。これらは、例えば、本分野で既知の、多くの他のパッケージング細胞株と同様に、Psi−2、C2、PA12、PA317、GP+envAM12などのパッケージング細胞を含む。
【0045】
本発明の他の特徴によると、フィブロネクチンのヘパリン−II結合部位内のアミノ酸に対する、強いウイルスの結合は、広い範囲の細胞型にわたるウィルス療法に対する送達システムを構築するために使用しうる。この目的のために、フィブロネクチン由来のレトロウイルス結合部位を含むポリペプチドは、標的細胞に特異的な、この構造を与える、いずれかのリガンドとも共有的に結合しうる。このアプローチは、それぞれの標的細胞に対する特異的レトロウイルス細胞株の構築の、前もった必要性を回避しうる(Kasahara, N., A. M. Dozy, and Y.W. Kan., Science, Vol. 266, pp. 1373−1376 (1994) ;そしてValsesia−Wittmann, S., A. Drynda, G. Deleage, M. Aumailley, J. M. Heard, O. Danos, G. Verdier, and F. L. Cosset, J. Virol., Vol. 68, pp 4609−4619 (1994))。標的構造の特異性は、例えば、1)細胞接着タンパク質、2)ホルモン、もしくはサイトカイン、3)標的細胞に対するモノクローナル抗体、4)標的細胞に結合する糖鎖(G.Ashwell, et al., Annu. Rev. Biochem., Vol 51, pp. 531−554 (1982))、5)標的細胞代謝物、もしくは6)標的細胞に結合する機能的ポリペプチド類を含む、リガンドを使用することにより提供されうる。遺伝子送達のための構築物の効率は、いくつかのヘパリン−IIウイルス結合部位を含むこと、そして従って、標的細胞に対して送達されるウイルス粒子の量を増加させることにより改善されうる。例えば、米国特許 No.5,198,423に記載のような、Pro1239−Ser1515に対応しているヒトフィブロネクチンの細胞結合部位は、BHK及びB16−F10細胞(Kimizuka et al., J. Biochem. Vol. 110, pp. 285−291 (1991))を含む細胞に結合することが示された。加えて、ヘパリン−II部位、それ自身は、繊維芽細胞、内皮細胞、及び腫瘍細胞に結合することが示された。これらのポリペプチド配列は、レトロウイルスによる感染のために、予め決定した細胞を標的にするために、フィブロネクチン由来の、レトロウイルス結合部位と結合しうる。
【0046】
造血系における、典型的な応用例は、それぞれ、高度特異的な赤血球、もしくは顆粒球前駆細胞を標的にするための、フィブロネクチンのレトロウイルス結合部位と結合する、エリスロポエチンもしくはG−CSFの構造をも含む。他の本発明に従った共通の応用は、特異的もしくは優位的に悪性細胞と結合するリガンドと、レトロウイルス結合部位、または部位群を組み合わせることでありうる。例えば、in vitro、及びin vivoでも、乳癌細胞の成長は、フィブロネクチンからの一つもしくはそれ以上のウイルス結合部位を含んでいる本発明の構造の中のリガンドとして、供給されるであろう、黄体形成ホルモン放出誘導体(Emons, G. et al., Hum. Reprod. 9:1364−1379 (1994))、エストロジェン(Tolcher, A. W., Oncol. 8:39−43 (1994))、もしくは抗エストロジェン(Howell, A. et al., Lancet 345:29−30 (1995))、プロゲステロジェン、または抗プロゲステロジェン(Klijn. F. G et al., Hum. Reprod. 9 Suppl. 1:181−189 (1994) ; Griffiths, K. et al., Semin. Oncol. 21:672−687 (1994))のような標的細胞上の受容体に結合する物質を使用することで、影響されうることが示された。さらなる例として、甲状腺(癌)細胞はJodidの構築物を用いることで高度に、特異的に標的化されうる。肝(癌)細胞は、HDL、もしくはその一部を含む構築物により標的化されうる。最後に、モノクローナル抗体とフィブロネクチンのレトロウイルス結合部位の構造は、抗体が入手できるどんな細胞及び臓器を標的化することもできる。従って広い範囲のほ乳類細胞型が、ウイルスベクターに結合し局在化するために、フィブロネクチンのレトロウイルス結合部位の能力を利用することで、標的になりうる。
【0047】
従って、本発明の他の好ましい様態は、標的細胞のウイルス形質導入を高めるために用いることのできる構築物を準備することを含む。フィブロネクチンのヘパリン−II結合部位の、ウイルス結合アミノ酸配列は、標的細胞が結合するリガンドと結合する。以上で議論したように、リガンドは、例えば、フィブロネクチンもしくは他のタンパク質(細胞接着タンパク質、例えば、ラミニン、コラーゲン、ビトロネクチン、オステオポンチン、もしくはトロンボスポンジンを含む)由来ののポリペプチド、ホルモン、代謝物、抗体(モノクローナル抗体を含む)、もしくは標的細胞に結合する、好ましくは特異的に結合する能力のあることを示しているいずれかの他のリガンドである可能性がある。結果のすべての構築物は、以下の実施例にて特異的に例示した、フィブロネクチンポリペプチド鎖として使用する場合と同様の様式にて、固定型として用いることができる。
【0048】
このような構築物と、細胞標的アプローチは、上記に議論したようにin vitroで用いられる可能性があり、そして同様に生理学的条件下での構造の安定化、及び特異性と、レトロウイルス構造の相互作用などの、様々な因子を考慮することで、レトロウイルスのin vivoでの標的化にも使用される可能性がある。特異性は、例えば、標的肝細胞への門脈静脈のカテーテル挿入などの、標的細胞への構築物の送達の局在化のための、送達系の改良によって改善される可能性もある。
【0049】
他の本発明の観点は、ウイルスと細胞の共局在化に関与する、本発明の形質導入過程が、ヘキサジメトリンブロミドの非存在下で、もしくは実質的に非存在の状況で行ったときに、より優位であるという発見に関連する。ヘキサジメトリンブロミド(商業的にはポリブレンRの名前で入手できる)は、レトロウイルスによる形質導入の効率を改善する目的のために、レトロウイルス媒介性遺伝子導入プロトコールで広く用いられていたポリカチオン性の物質である。それにも関わらず、本明細書で記載したような遺伝子導入プロトコールを高めるような共局在化した際の、ヘキサジメトリンブロミドの存在は、形質導入効率を減少させることが発見された。従って、本発明の改善された過程は、少なくとも実質的にヘキサジメトリンブロミドの非存在下で(すなわち、約1μg/ml未満のヘキサジメトリンブロミドのみ含む)、及びさらに好ましくはヘキサジメトリンブロミドの非存在下の培養液中で行われる。このような過程は、レトロウイルス産生細胞及びヘキサジメトリンブロミド双方とも実質的に存在しない、実質的なレトロウイルス形質導入生存細胞集団を含む、本発明の好ましい細胞構成物を提供する。この考えにおいては、本明細書中で用いたような、実質的に形質導入された生存している細胞群は、少なくとも約20%のこの集団の細胞が、レトロウイルスによって形質導入されたことを意味することを意図する。さらに好ましい集団は、少なくとも約50%の形質導入細胞を持ち、最も好ましいものは、少なくとも約75%の形質導入細胞を持ちうる。本発明のこの観点による、好ましい細胞構成物は、造血細胞を含み、さらに好ましくは、始原前駆細胞及び幹細胞を濃縮した、造血細胞集団を含みうる。従って一般的にいえば、本発明の都合のよい過程は、ポリカチオン性の薬剤、もしくは、フィブロネクチン断片、もしくは共局在化のための他の物質の存在しない、類似のレトロウイルス感染プロトコール(例えば共培養)に対応して、形質導入効率の増加を導くような、しかし共局在化のための物質の存在下での形質導入効率を減少させるようなその他の薬剤が存在しない状態で行うことができる。
【0050】
非常に簡単な、レトロウイルス媒介性DNA導入が、本発明の方法を実施するように特にデザインされたキットを用いて実行しうることが企画される。従って、本発明の他の側面は、レトロウイルス感染を行うことができる人工基質とともに、レトロウイルスによる標的細胞の形質導入を高めるよう上記で議論した一定量の実質的に純粋なポリペプチド、もしくは構築物を含むキットを提供する。ポリペプチド、もしくは他の構築物は、別々に提供され、もしくは人工基質上にコートされうる。ヒト造血細胞のための感染プロトコールの場合、キットは、細胞前刺激のための造血細胞増殖因子をも含みうる。加えて、このキットは、形質導入のために、上記で議論したように、組換えレトロウイルスベクターを含みうる。一般的にいえば、このキットは、このキットを使用している間、構成物の破損を防ぐのにもう一つの十分な空間的な関連において、これらの様々なキット構成物を収納する、無菌包装を含みうる。例えば、複合構成物、もしくは空間的な関連におけるキット構成物を保持するための部分を有する成形プラスチック商品を用いることが共通の慣例である。
【図面の簡単な説明】
【0051】
【図1】図1は、キモトリプシン断片を含むフィブロネクチン分子の概略図表示を提供する。
【図2】図2は、以下の実施例1でさらに記載したように、TKNEOベクターを用いた、フィブロネクチン断片の存在下での、投入された(comitted)ヒト前駆細胞の感染効率を表す。
【図3】図3は、以下の実施例1でさらに記載したように、TKNEOベクターを用いた、フィブロネクチン及びその断片の存在下での、様々な投入されたヒト造血前駆細胞の感染効率を比較する。
【図4】図4は、以下の実施例7でさらに記載したように、(i)共培養方法(レーン2−4)、(ii)固定化フィブロネクチン断片存在下での上清感染(レーン5−7)、及び(iii)BSA上での上清感染(レーン8−10)によって形質導入された骨髄細胞を移植されたマウスの、hADAの存在を比較する。hADAの対照はレーン1及び12に示され、ネズミADAに対する対照はレーン11に示される。
【図5】図5は、以下の実施例8でさらに記載したように、フィブロネクチン断片へのウイルス結合を示す。
【図6】図6は、以下の実施例8でさらに記載したように、フィブロネクチン断片への、レトロウイルスの結合が、濃度依存的であることを示す。
【図7】図7は、以下の実施例9−11で用いた、様々な組換えフィブロネクチン断片を図示している略図を提供する。
【図8】図8は、以下の実施例9で記載したように、いくつかの組換え断片を含む、様々なフィブロネクチン断片に結合するレトロウイルスを示す。
【図9】図9は、以下の実施例9で記載したように、フィブロネクチン断片と結合するレトロウイルスを、ヘパリンが阻止することを示す。
【図10】図10は、以下の実施例10でさらに報告したように、様々なフィブロネクチン断片の存在下での、ネズミ造血細胞のレトロウイルス感染効率を示す。
【図11】図11及び12は、以下の実施例11で記載するように、(i)共培養法、(ii)様々なフィブロネクチン断片上での上清感染、及び(iii)BSA上での上清感染によって形質導入された骨髄細胞を移植されたマウスにおける、hADAの存在を比較している。
【図12】図11及び12は、以下の実施例11で記載するように、(i)共培養法、(ii)様々なフィブロネクチン断片上での上清感染、及び(iii)BSA上での上清感染によって形質導入された骨髄細胞を移植されたマウスにおける、hADAの存在を比較している。
【図13】図13は、フィブロネクチンのα−鎖の構造と、実施例で用いた組換え断片とのその関連とを示している。フィブロネクチンI型、II型、及びIII型リピートが示され、III型リピートは1〜14まで番号付けした。細胞への3つの結合部位は、細胞結合部位(CBD)に対してはCELL、ヘパリン結合部位(HBD)に対してはHEPARIN、そしてオルタナティブにスプライシングされたIIICS部位のはじめの25アミノ酸によって形成された、VLA−4結合部位CS−1に対してはCS−1として表示する。
【図14】図14は、以下の実施例12でさらに報告したように、様々なフィブロネクチン断片の存在下での、NIH/3T3細胞のレトロウイルス感染の効率を示す。
【図15】図15は、以下の実施例12でさらに報告したように、様々なフィブロネクチン断片の存在下で、非接着性HL60細胞のレトロウイルス感染の効率を示す。
【図16】図16は、フィブロネクチンへ結合するレトロウイルスに対する、低分子量及び高分子量ヘパリンの影響を示す。
【図17】図17は以下の実施例13で議論したように、組換えフィブロネクチン断片の存在下で、CD34+細胞集団内の様々な型の前駆細胞のレトロウイルス感染の効率を示す。
【図18】図18は、以下の実施例14で議論したように、組換えフィブロネクチンの存在下で、C−kit+細胞集団中の、HPP−CFC細胞のレトロウイルス感染の効率を示す。
【図19】図19は、以下の実施例15で議論したように、ヘキサジメトリンブロミドの濃度を増加させるに従って、NIH/3T3細胞のレトロウイルス感染効率が減少することを示す。
【図20】図20は、以下の実施例15で議論したように、ヘキサジメトリンブロミドの濃度を増加させるに従って、クローン原性骨髄細胞のレトロウイルス感染効率が減少することを示す。
【図21】図21は、T細胞がフィブロネクチン受容体を発現することを示す、フローサイトメトリー試験の結果を示す。
【図22】図22は、ヒトT細胞の前刺激を示すフローサイトメトリー試験の結果を示す。
【図23】図23は、ADAイソ酵素アッセイによる、ヒトT細胞への遺伝子導入の効率の解析の結果を示す。
【図24】図24は、さらに実施例16で記載する、感染プロトコールで用いたポリペプチド類を示す略図を提供する。
【図25】図25は、図24で同定した、ペプチド類C−FGF、C−COL、及びbFGFの存在下で、NIH/3T3細胞の形質導入の効率を示す図である。
【図26】図26は、フローサイトメトリー解析でアッセイしたように、図24で同定したペプチドの存在下で、HEL細胞へのレトロウイルス遺伝子導入の効率を示す。
【図27】図27は、図24で同定したポリペプチドの存在下で、CD34+骨髄細胞の形質導入の効率を示す図である。
【発明を実施するための形態】
【0052】
本発明は好ましくは以下の形態を含む。
1. レトロウイルスによって形質転換された生存可能な細胞集団を得る方法であって:
細胞に結合するリガンドおよびレトロウイルスに結合するリガンドを含む、有効に固定化される量の物質の存在下で、細胞をレトロウイルスに感染させ、よってレトロウイルスおよび細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ここにおいて前記感染は本質的にヘキサジメチリンブロミドを含まない培地で行われる:
ことを含む、前記の方法。
【0053】
2. 細胞が造血幹細胞を含む、態様1記載の方法。
3. 態様1記載の方法によって製造される生存可能な細胞集団。
4. 造血幹細胞を含む、態様3記載の生存可能な細胞集団。
【0054】
5. 態様1記載の方法によって製造される生存可能な細胞集団を哺乳動物に移植する:ことを含む、細胞移植方法。
6. 実質上レトロウイルスで形質転換された生存可能な細胞集団を含み、レトロウイルス産生細胞およびヘキサジメチリンブロミドの両方を本質的に含まない:細胞組成物。
【0055】
7. 前記の生存可能な細胞が造血幹細胞を含む、態様6記載の細胞組成物。
8. 態様6記載の細胞組成物を哺乳動物に移植する;ことを含む、細胞移植方法。
9. 細胞集団が造血幹細胞を含む、態様8記載の方法。
10. レトロウイルスによって形質転換された生存可能な細胞集団を得る方法であって:
細胞と結合するリガンドおよびレトロウイルスと結合するリガンドを含む、有効に固定化される量の物質の存在下で、細胞をレトロウイルスに感染させ、よって、レトロウイルスと細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ここにおいて前記感染は、共培養中ではレトロウイルスによる細胞を形質転換する効率を上昇させるが、前記物質存在下ではレトロウイルスによる細胞の形質転換の効率を減少させる薬剤を実質上含まない培地で行われる:
ことを含む、前記の方法。
11. レトロウイルスによって形質転換された生存可能な細胞の集団を得る方法であって:
細胞を結合するアミノ酸配列、およびレトロウイルスと結合するコラーゲンVまたは繊維芽細胞増殖因子からのアミノ酸配列を含むポリペプチド存在下で、細胞をレトロウイルスに感染させる:
ことを含む、前記の方法。
12. 前記の感染が、レトロウイルス−プロデューサー細胞の非存在下で、レトロウイルスと細胞を接触させることを含む、態様11記載の方法。
13. T細胞をレトロウイルスで形質転換する方法であって、T細胞と結合するリガンド、およびレトロウイルスと結合するリガンドを含む物質存在下で、細胞をレトロウイルスに感染させ、よって、レトロウイルスと細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ことを含む、前記の方法。
14. 物質が、T細胞と結合する第一のアミノ酸配列、およびレトロウイルスと結合する第二のアミノ酸配列を含むポリペプチドであって、第二のアミノ酸配列が、配列:
【0056】
【化2】
【0057】
またはそれと充分に類似するアミノ酸配列を持ち、レトロウイスルと結合する能力を示すものである;
態様13記載の方法。
15. レトロウイスルを結合するコラーゲンVまたは繊維芽細胞増殖因子からのアミノ酸配列を持つ、有効量の単離されたポリペプチドとレトロウイルスを接触させることを含む、レトロウイルスを配置する方法。
【実施例】
【0058】
本発明のさらなる理解、及び評価を促進するために、以下に続く固有の実施例を提供する。これらの実施例は、説示的なものであり、その本質を限定しているものではないことが理解されよう。
【0059】
実施例1 TKNEOを用いた骨髄細胞への遺伝子導入
1.1.ウイルス上清の調整
レトロウイルスプラスミドTKNEOベクターを含むGP+EnvAM12産生細胞(Markowitz et al. (1988) Virology 167:400 参照)を、10%胎児子牛血清(FCS、Hyclone, Logan UT)、及び100 units/ml ペニシリン、及び100 microgram/ml ストレプトマイシン(P/S、双方 Gibco)を含むIscove’s Modified Dulbeccos Medium (IMDM、Gibco, Gaithersburg,MD)中で培養した。ウイルスを含む上清を、20%FCSを含むIMDM10 ml を加えて一晩プレートをコンフルエンスにすることによって回収する。回収した培養液を、0.45ミクロンフィルター(Gelman Science, Ann Arbor, MI)を通して濾過し、使用まで−80℃にて保存した。
【0060】
1.2.フィブロネクチン断片の用意
FNを、FNを4M尿素で溶出する前に、ゼラチン−アガロースカラムを1M尿素で洗浄した以外は、すでに、 Ruoslahti et al., Methods. Enzymol. 82:803−831 (1982) で記載されているように、ヒト血漿(Lifesource, Glenview,IL)から精製した。精製したFNは、4℃にて,10mM 3−(シクロヘキシルアミノ)−1−プロパン−スルホン酸(CAPS)、150mM NaCl、2mM CaCl2 pH 11.0に対して透析し、−80℃にて等分して保存した。FNのキモトリプシン様細胞結合部位(CBD)(CS−1)、及び、ヘパリン−II結合断片は、すでに記載されているように(Ruoslahti et al. (1982), supra, Patel and Lodish, J. Cell. Biol. 102, pp. 449−456 (1986), and Bernardi et al., J. Cell. Biol. 105, pp. 489−498 (1987))精製した。三つの主なヘパリン断片(30kD、35kD、42kD)が、ヘパリン−アガロースカラムからの1M NaCl中の溶出で得られた。これらのヘパリン結合断片を、さらに精製するために、1M NaCl溶出物を4℃にて1晩、10mM Tris−HCl pH7.0に対して透析し、10mM Tris−HCl pH7.0で平衡化された陽イオン交換カラム(2ml DEAE セファロース fast flow, (Pharmacia Fine Chemicals, Uppsala, Sweden) /mg タンパク質)に通した。30/35kD断片を、非結合フラクションで回収し、一方42kD断片はカラムより100mM NaClにて溶出した。FN 500mg から、おおよそ26mgの30/35kD断片、及び4mgの42kD断片を得た。30/35kD断片でなく、42kD断片は、ウエスタンブロッティング手法によって決定したように、フィブリン結合部位に対する抗体によって認識された。また、42kD断片は、フィブリン−セルロースアフィニティーカラムへ結合する。
【0061】
感染プロトコールで使用するために、フィブロネクチン断片を Patel and Lodish (1986), supra によって記載されたように、75pmol/cm2の濃度にて、35、もしくは100mmペトリディシュ(Falcon,Lincoln Park,NJ)に固定化した。コントロールのプレートは、2%(FNなし)ウシ血清アルブミン(BSA,Boehringer Mannheim, Mannheim,Germany)を用いて類似の方法でコートした。
【0062】
1.3.レトロウイルス感染プロトコール
健康な大人提供者からの、骨髄サンプルを、the Institutional Review Board of Indiana University School of Medicine によって立証されたプロトコールに従った、無菌保存剤なしの培養液硫酸ヘパリンを含むチューブに集めた。低密度単核細胞を、フィコール−ハイパック(密度 1.077 g/ml, Pharmacia, Piscataway, NJ)において、25℃にて45分間遠心することで用意した。プラスチック接着細胞を、低密度骨髄細胞から2%FCS入りIMDM中、5%CO2中で37℃にて4〜16時間さらに組織培養プレートをインキュベートすることで取り除いた。
【0063】
接着陰性低密度単核細胞は、すでに、Luskey er al. (1992) Blood 80:396 に記載されたように、レトロウイルス感染前にペトリディシュ上1×106 cells/ml の細胞密度で、20%FCS、100 U/ml rhIL−6、100 ng/ml rhSCF(両方 Amgen,Thousand Oaks, CA)、及びP/Sを含むIMDM中、5%CO2、37℃で48時間前刺激した。前刺激した細胞を、プラスチックへ弱い接着をする細胞を取り除くために、勢いよくピペッティングすることで回収した。
【0064】
前刺激した細胞(5×105 cells/ml)を、BSA(コントロールプレート)、もしくはフィブロネクチンもしくはその断片(プラスチックプレートへタンパク質がより接着するようにUV照射した。)でコートされたプレート上で6時間インキュベートし、成長因子(上記の通り)、及び7.5 microgram/ml ポリブレン(Aldrich Chemical, Milwaukee, WL)存在下、ウイルス上清にて感染させた。ウイルス上清は、2時間後置き換え、(成長因子、及び5.0 microgram/ml ポリブレンを含むもの)、細胞をさらに12〜24時間インキュベートした。非接着細胞はそれぞれの培養液交換で再び加えた。
【0065】
感染プロトコールに従って、非接着細胞をデカントし、接着造血細胞は製品の使用説明書に従って、細胞解離緩衝液(CDB)(酵素なし/PBSベース、Gibco)を用いて回収した。接着細胞を、非接着細胞フラクシャンに加え、2回洗浄し、計数した。回収した細胞は、クローン原性メチルセルロース前駆体分析を行うか、あるいは長期骨髄培養を行った。
【0066】
1.4.長期骨髄培養
LTC−IC(ヒト幹細胞)分析を、すでに記載された方法に従って、若干の改良をして行った。Sutherland et al. Blood 74:1563 (1989)。簡単には10%FCS、10%ウマ血清(Sigma)及びP/S、1×10−5Mヒドロコルチゾン(Upjohn, Kalamazoo,MI)、及び、6ウェル組織培養プレート上に、320モスモル塩化ナトリウム(Costar, Cambridge,MA)を含む5 ml IMDM中でのコンフレントな(上記のような)前照射を受けた同種ヒト骨髄繊維芽細胞(BMF)において、0.5−1×106の感染細胞を播種した。LTMCは、33℃で5%CO2、及び毎週培養液及び非接着細胞の50%を取り去ることで栄養を与えてインキュベートした。5週間後、LTC−IC培養は、BMFから、接着造血細胞を取り除くために、CDBを用いて回収した。非接着、及び接着造血細胞双方を一緒にし、LTC−ICから由来するコロニーを得るために、メチルセルロースにプレートした。
【0067】
1.5.クローン原性メチルセルロース分析
メチルセルロース分析は、すでに Toksoz et al., Prc. Natl.Acad. Sci., USA, Vol. 89, p7350 (1992) で記載されたものに、若干の改良を加えて行った。2.5×104感染大人骨髄細胞を、25%FCS、10%ヒト血漿、10−5Mベータ−メルカプトエタノール(Sigma)、及びP/Sを含む1 mlの2.4%IMDMメチルセルロース(Fluka, Ronkonkoma, NY)中で、5 units/ml エリスロポエチン(Epo, Amgen)、100 ng/ml rhSCF、10 ng/ml rhIL−3(Genzyme, Cambridge, MA)とともにプレートした。培養は、37℃にて、5%CO2/95%空気でインキュベートし、コロニー(>50細胞数)を13日目に反転顕微鏡で見ることで、CFU−GM(顆粒球及びマクロファージを含む)、CFU−混合(骨髄及び赤血球成分を含む)、BFU−E(赤血球成分のみを含む)として計数した。
【0068】
1.6.レトロウイルス感染分析
TKNEOウイルスによる感染効率は13日目の1.5 mg/ml(乾燥粉、Gibco)G418抵抗性メチルセルロ−スコロニ−の割合を決めることで解析した。擬感染は、骨髄を組換えウイルスをつくらないGP+EnvAM12パッケージング株上でインキュベートすることでそれぞれの実験で行った。1.5 mg/ml G418とのこれらの擬感染細胞の培養は一貫して、<1%バックグラウンドコロニーを示す。
【0069】
1.7.前駆細胞への遺伝子導入効率
形質導入効率は、30/35 FN−、もしくはBSAコートディッシュにまき、骨髄細胞に感染することで比較した。これらの状態間では、選択なしの感染の後に得られたコロニー数においては、違いが観察されなかった。図2は感染後のG418rコロニーの割合を示す。多系統(CFU−混合)前駆細胞と同様に、系統制限(BFU−E、及びCFU−GM)から由来するものを含む、すべての前駆体型からの、30/35 FN上で高い割合のG418rコロニーが確認された。すべての前駆体への感染はBSAに比べ、30/35 FNで9倍増加した。
【0070】
1.8.長期培養イニシエーション細胞への遺伝子導入
LTC−ICへの遺伝子導入はTKNEOベクターを用いて行った。コロニー由来LTC−ICへの遺伝子導入は、30/35 FN上での感染後、検出されたのみである。(30/35 FN対BSA 16%G418r対0%G418rコロニー)。
【0071】
1.9.造血細胞の感染効率への30/35FN効果の特異性
30/35 FNにおいて見られた遺伝子導入効率の増加の特異性を決めるためにTKNEOによる感染を、BSA、30/35 FN、無傷の(intact)フィブロネクチン、RGDSテトラペプチド配列を含む中心細胞結合部位(CBD)を含む115kD FN断片、CS−1配列は欠損しているが、ヘパリン−II結合部位を持つことによって特徴づけられる42kD C−末端FN断片(42FN)上で行った(図1)。BSA上での感染は3±1%G418r BFU−E、1±1% G418r CFU−GM、及び0±0% G418r CFU−混合を示した。CBD上で観察されたG418rコロニー群は明らかな増加が見られず、一方42 FN上ではBFU−Eのわずかに高い感染(6.0±−1%)が見られた(図3)。しかしながら、無傷のFNはすべての前駆体への遺伝子導入の増加を促進した。無傷のFN上での感染後のG418rコロニーの割合は30/35FN上のものより少なかったが、それぞれBFU−E(16±−2対24±4%)、CFU−GM(5±2対20±4%)、及びCFU−混合(6±1対9±1%)(無傷FN対30/35FN)であった。
【0072】
実施例2 PGK−mADAを用いた骨髄細胞への遺伝子導入
2.1.全体的な手順
PGK−mADAウイルス上清は実施例1のTKNEOのための記載と同様に準備した。フィブロネクチンのキモトリプシン様断片は(図1)、すでに実施例1で記載のように準備し、実施例1のレトロウイルス感染プロトコールに従った。LTC−IC(ヒト幹細胞)分析、及びメチルセルロース分析は実施例1に従って行った。
【0073】
2.2. レトロウイルス感染の分析
ADAイソ酵素電気泳動によるタンパク質分析によって、PGK−mADAベクターの感染効率を定量した。個々の前駆(progenitor)細胞コロニーの分析は、Moritz(1993)およびLimら(1989)、Proc.Natl.Acad.Sci.USA,86巻、8892頁に前記した方法に従って、行なわれた。トランスファー効率を厳密に分析するために、内在性ヒトADAと同じか、またはより高レベルのmADAを発現するコロニーのみを形質転換されたと考えた。プールしたコロニーを分析するために、メチルセルロース培地より選択(pick out)したコロニーを、1.5mlのミクロチューブ(Rainin,Woburn,MA)内に集め、暖めた培地およびリン酸緩衝塩類溶液(PBS)で洗浄し、遠心分離し、そして−20℃に保存した。ADA分析のため、凍結−融解サイクルを繰り返すことによって、5μlの溶解バッファーに細胞を溶解し、そしてイソ酵素電気泳動を、前記の方法に従って行った。
【0074】
2.3. 前駆細胞内への遺伝子トランスファー効率
30/35FN−またはBSA−コート皿上にプレートした間、骨髄細胞に感染させることによって、形質転換効率を比較した。選別しなかった場合、感染後得られたコロニー数の差は、これらの条件間では認められなかった。表1に示したように、すべての前駆細胞内への感染効率は、実質上、BSAに対して30/35FN上で増加した。高力価(〜1x107ビリオン(virons)/ml)ベクターで予想されるように、前駆細胞の形質転換効率は、非常に高かった。表1に示すように、30/35FN上での骨髄へのPGK−mADAの感染は、2回の別々に行った実験で、前駆細胞の100%近い形質転換が得られた。
【0075】
【表1】
【0076】
2.4. 長期培養−始原細胞(initiating cell)内への遺伝子トランスファー効率
PGK−mADAで行った4回の独立した実験では、週齢5週のLTMCから誘導された前駆細胞コロニー(即ち、LTC−IC誘導コロニー)の有意な割合が、トランスファーされたマウスADA遺伝子を発現していた。発現は、分析したコロニーの2/12(17%)から6/6(100%)までの範囲であった(表2)。導入されたmADA遺伝子の発現は、陽性と考えられるすべてのコロニー内で、内在性ヒトADA活性量を越えるか、または少なくとも等しかった。PGK−mADAの感染効率は、TKNEOより高かった。表2に示すように、30/35FN上の骨髄へのPGK−mADAの感染では、2回の別々の実験で、前駆細胞の100%近い形質転換が得られた。
【0077】
【表2】
【0078】
2.5. 造血細胞の感染効率への30/35FN効果の特異性
LTC−IC内への遺伝子トランスファー効率は、30/35FN上で増加した。これらの二次のLTC−IC誘導コロニーは比較的小サイズであるため、単一コロニーでタンパク質分析を成し遂げる能力には限界がある。BSA上でのPGK−mADAでの感染後、完全な(intact)フィブロネクチンならびに42FN 0/6、0/4および0/3 LTC−IC誘導コロニーは、それぞれ、マウスADAの発現を示したが、30/35FN上で感染させた3/5LTC−IC−誘導コロニーは、mADAを発現した。さらに、複数のLTC−IC−誘導コロニーを2回の追加実験で分析する前にプールしたところ、mADA発現は、30/35FN上で感染後のみに検出され、完全なFN上ではより少ない程度であり、42FNまたはBSA上では検出されなかった。
【0079】
実施例3 PGK−hADAを用いた骨髄細胞内への遺伝子トランスファー
3.1. 一般的方法
実施例1のTKNEOで記載した方法に従って、PGK−hADAウイルス上清を調製した。実施例1に前述した方法に従って、フィブロネクチンのキモトリプシンフラグメント(図1)を調製し、次に実施例1のレトロウイルス感染プロトコールを行った。LTC−ICおよびメチルセルロースアッセイを、実施例1記載の方法に従って行った。
【0080】
3.2. レトロウイスル感染の分析
プールしたコロニーを分析するために、メチルセルロース培地より選択したコロニーを1.5mlのミクロチューブ(Rainin,Woburn,MA)に集め、暖めた培地およびPBSで洗浄し、遠心分離し、そして−20℃で保存した。ADA分析のために、細胞を、凍結−解凍サイクルを繰り返すことによって5mlの溶解バッファーに溶解し、そして前述の方法に従って、イソ酵素を電気泳動した。
【0081】
実施例4 TKNEOを用いた臍帯血細胞内への遺伝子トランスファー
4.1. 一般的方法
TKNEOウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(図1)は、実施例1に前述した方法に従って、調製された。次いで、実施例1に記載のレトロウイルス感染プロトコールを行ったが、骨髄細胞の代わりに、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineで是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−IC(ヒト幹細胞)およびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0082】
4.2. 前駆細胞の遺伝子トランスファー効率
FN30/35上での感染は、3回の別々の実験において、BSAと比較して4倍以上増加していた(表3)
【0083】
【表3】
【0084】
実施例5 PGK−mADAを用いた臍帯血細胞内への遺伝子トランスファー
5.1. 一般的方法
PGK−mADAウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(図1)は、実施例1に前記された方法に従って、調製された。次いで、実施例1記載のレトロウイルス感染プロトコールを行ったが、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineに是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−ICおよびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0085】
5.2. 長期培養した始原細胞内への遺伝子トランスファー効率
より力価の高いPGK−mADAを用いると、LTC−IC−誘導コロニーの分析は、FN30/35上で上清を用いて感染させた臍帯血から確立された培地からのみ、導入したmADA cDNAの高レベル発現を示した。BSA対照プレート内で感染させたLTC−IC誘導コロニー内では、mADAの発現はほとんど検出されなかった。
【0086】
実施例4および5に示した結果は、FN30/35を用いた感染効率の改善が、臍帯血前駆細胞および幹細胞を用いた場合にも達成できることを、証明している。
実施例6 PGK−hADAを用いての臍帯血細胞内への遺伝子トランスファー
PGK−hADAウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(表1)は、実施例1のTKNEOに記載の方法に従って、調製された。次に、実施例1のレトロウイルス感染プロトコールを行ったが、骨髄細胞の代わりに、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineに是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−ICおよびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0087】
実施例7−11 実施例7−11のためのレトロウイルスベクターおよび産生細胞(producer cell)系
実施例7−11に関して、2つのレトロウイルス産生パッケージング細胞系(retrovirus−producing packaging cell line)を用いた:それぞれ、エコトロピック(ecotropic)GP+E86(Markowitz,D.,S.GoffおよびA.Bank,J.Virol.,62,1120−1124、1988)ならびにアンホトロピック(amphotropic)GP+envAM12細胞系(Markowitz,D.,S.GoffおよびA.Bank,Virology,167,400−406,1988):ここに記載された研究に用いられたレトロウイルスベクターおよび産生細胞クローンを、表4に列挙した。
【0088】
【表4】
【0089】
すべての細胞系は、EAL2a細胞を除き、10%のウシ胎児血清(FCS,Hyclone,Logan,UT)および100ユニット/mlのペニシリンおよび100μg/mlのストレプトマイシン(P/S、両方共Gibco)を含むダルベッコの修飾Eagle培地(DME,Gibco,GrandIsland,NY)内で培養され、EAL2a細胞は、10% FCS+P/Sを含むDME−F12(Gibco)内で増殖させた。ウイルスを含む上清は、マウス細胞には10mlのα−最少必須培地(αMEM,Gibco)、またはヒト細胞にはイスコフのダルベッコ培地(IMDM,Gibco)(それぞれ10%のFCS+P/Sを含む)を10cmの集密プレートに一晩加えることによって、集められた。収集した培地を、0.45ミクロンフィルター(Gelman Sciences,Ann Arbor,MI)でろ過し、使用するまで−80℃に保存した。
【0090】
実施例7 マウスの一次造血細胞の形質転換
7.1. 実験方法
マウスの細胞で研究するために、150mg/kgの5−フルオロウラシルの投与後、2日の週齢6から8週のC3H/HeJマウスの大腿骨および頸骨から、骨髄を収集した(SoloPack Laboratories,Franklin Park,IL)(Lim,B.,J.F.Apperley,S.H.OrkinおよびD.A.Williams,Proc.Natl.Acad.Sci.USA,86、8892−8896、1989)。細胞は、IMDM/20%FCS+P/S中の濃度5x106細胞/ml、並びに100ng/mlのラット組換え幹細胞因子(rrSCF;Amgen.Thousands Oaks,CA)および100ユニット/mlのヒト組換えインターロイキン−6(rhIL−6;Pepro Tech Inc.,Rock Hill,NJ)で、48時間、前刺激(prestimulate)した(Luskey,B.D.,M.Rosenblatt,K.ZseboおよびD.A.Williams,Blood,Vol.80,pp.396−402、1992)。次に、EPHA−5産生細胞によって作り出されたPGK−hADAベクターでの遺伝子トランスファー効率を、3種類の異なる感染プロトコールを用いて比較した:1)上清感染;2)FN30/35上での上清感染;3)EPHA−5産生細胞上での同時培養。そのために、100mmの細菌皿を、5mlのリン酸緩衝溶液(PBS;Gibco)中に溶解した2.5μg/cm2のFN30/35(75pmol/cm2に等しい)で、室温、UV光下で、1時間、皿のフタを開けたままで、さらに1時間、皿のふたを閉めてコートした。2%のウシ血清アルブミン(BSA,Fraction V:Boehringer Mannheim,Indianapolis,IN)で、室温で30分間、ブロックした後、皿を、2.5%(v/v) 1M Hepesを添加したハンクス液(Hank’s Balanced Salt Solution)(HBSS)(両方共Gibco)で、一度洗浄した。上清感染では、皿をBSAのみでコートした。5x106の前刺激したドナー細胞を、EPHA−5細胞より上記の方法に従って得られた10mlのウイルス上清に、100U/mlのrhIL−6、100ng/mlのhrSCFおよび7.5μg/mlのポリブレン(polybrene)を添加したものと共に、インキュベートした。非粘着性細胞を集め、新たにウイルス上清を再付加した。共培養のために、4ml培地中のEPHA−5細胞を、10μg/mlのマイトマイシンCと共に、2時間、37℃でインキュベートし、洗浄し、トリプシン処理し、そして10mlのαMEM/20%FCS+P/S中、3x106の細胞濃度で、100mmの組織培養皿上に接種した。翌日、5x106の前刺激した骨髄細胞を、100U/mlのrhIL−6、100ng/mlのrrSCFおよび4μg/mlのポリブレンと共に、48時間加えた。感染プロトコールに次いで、非粘着性細胞をデカントし、そして、粘着性造血細胞を、製造者の指示書に従って、Cell Dissociation Buffer(CDB)(酵素非含有/PBSをベースとする、Gibco)を用いて、培養液から収集した。粘着性細胞を非粘着性画分に加え、二度洗浄し、そして、おおよそ1mlのHBSS/Hepes中に懸濁した。5x106の前刺激した細胞から得られた全細胞を、致死量の全身照射(700+400cGy、137Cs−源)(Luskey,B.D.,M.Rosenblatt,K.ZseboおよびD.A.Williams,Blood,Vol.80,396−402、1992)を受けた3匹のレシピエントマウスの尾静脈内に注射した。造血幹細胞の形質転換は、導入されたヒトADA cDNAの発現用に再構築されたマウスを試験することによって、分析された。このADAイソ酵素分析は、原位置での酢酸セルロースのin vivo酵素分析によって、hADAタンパク質の存在について、末梢血液細胞を試験することによって、移植されたマウス内で成し遂げられた(Lim,B.,D.A.WilliamsおよびS.H.Orkin,Mol.Cell.Biol.,7,3459−3465、1987)。試験は、最初4ヶ月間、追加移植を行い、月に一度繰り返した。
【0091】
7.2. 結果
遺伝子操作された造血幹細胞でのマウスの骨髄再構築が長期に及ぶことは、一般的に、4ヶ月の追加移植期間後の幹細胞の形質転換効率を定量するために、適当であるとして受け入れられる。形質転換骨髄の7ヶ月後のレシピエントのイソ酵素分析による分析は、1)ヒトADA cDNA発現は、同時培養またはFN30/35上での上清感染のいずれかを用いると存在するが、FN30/35なしでの上清感染後移植したグループでは、存在せず、そして2)発現のレベルは、共培養とFN30/35グループで同等である:ことを示した。図4、列2−4に示したように、EPHA−5細胞上での同時培養によって形質転換された骨髄を移植した3匹のマウスでは、ヒトADAが容易に検出できることを証明した。同様のレベルのヒトADAは、FN20/35の上清感染によって形質転換された造血細胞を移植した3匹のマウスでも検出された(図4、列5−7)。これに対して、BSA上での上清感染によって形質転換された造血細胞を移植した3匹のマウスでは、ヒトADAは検出されなかった(図4、列8−10)。ヒトADAの位置コントロールを図4の列1および12に、ならびにマウスのADAを列11に示す。列2−10のマウスのバンドは、同量のタンパク質が負荷されたことを示す。これらのデータは、FN30/35上での上清感染によって長期にわたって再構築される造血幹細胞の形質転換が 同時培養と等価であり、FN30/35なしでの上清感染より遥かに優れていることを証明している。
【0092】
実施例8 TN30/35に結合したレトロウイルスベクターによる改良された形質転換メカニズム
8.1. 実験方法
形質転換の増加が、ウイルスと造血細胞を共に配置する(co−localization)結果であるか否かを試験するため、我々は、組換えレトロウイルス粒子がFN30/35との結合を示すか否かについて分析した。そのため、FN30/35−コートプレートを、TKNeoウイルスを含む上清と共に、30分間プレインキュベートし、その後、広範囲にわたって洗浄した。上清のウイルス力価をNIH/3T3を用いて、標準的方法に従って、定量した(Markowitz,D.,S.GoffおよびA.Bank,J.Virol.,62,1120−1124、1988)。簡単に言えば、3T3細胞を、6穴の組織培養プレートに、濃度1000細胞/穴でプレートし、一晩増殖させた。ウイルス上清の一連の希釈物を、7.5μg/mlのポリブレンと共に、それぞれの穴に加え、そして2mlの培地を加えた後、37℃で2.5時間インキュベートした。24時間後、培地は、G418(0.75mg/ml、乾燥粉末、Gibco)を含む培地と置き換え、そしてプレートを10−12日間インキュベートした。G418−耐性コロニー(G418r)を10−12日後に染色し、そして数を記入した。ウイルス上清の希釈倍率を掛けたコロニー/穴の数を、上清の感染粒子(cfu)/mlとして用いた。我々は、ウイルス上清とプレインキュベーションし、そして1000NIH/3T3/35mm細菌皿細胞+ポリブレンを加えることによって集中的に洗浄した後、FN30/35−コートしたまたはBSA−コートした35mmプレート上に残っているレトロウイルス粒子の量を、評価/”滴定”した。24時間後、培養液に、0.75mg/mlのG418(乾燥粉末)を含む培地を供給し、そしてさらに細胞を10−12日間インキュベートした。このインキュベーションの後、G418−耐性NIH/3T3コロニーを数えることによって、粘着性ウイルスの存在を定量した。
【0093】
FN30/35へのウイルスの結合が、服用量依存様式で起こるか否かを評価するために、皿をコートするFN30/35の濃度を上昇させて、上記の実験を繰り返した。そのため、上記の方法に従って、35mmの細胞皿を、1、4、10および20μg/cm2のFN30/35でコートした。前もって1x104感染粒子/mlと滴定されたTKNeoウイルスストックの1:50希釈物を、FN30/35−コートプレート上で30分間インキュベートした。集中的に洗浄した後、2000NIH/3T3細胞をそれぞれの穴に加えた。選別を上記の通り行い、そしてG418−耐性NIH/3T3コロニーを、選別の10日後、計数した。
【0094】
8.2. 結果
図5は、3回の実験の内の典型的な一つの結果を示す。TKNeo上清を用いて、NIH/3T3細胞内のG418rコロニーによって測定されたレトロウイルスの力価は、BSA−コートプレート上では、3logより多く(4x103から0)減少したが、これに対して、30/35FNでコートしたプレート上では、力価の減少は、1logのみであった。これらのデータは、レトロウイルスは、FN30/35と定量的に結合するが、しかしBSAでコートした皿(対照皿)とは結合しないことを、証明している。図6は、ウイルス−含有上清を、FN30/35の濃度を増加させてコートしたプレート上でインキュベートした場合、G418−耐性コロニーの数の増加が検出されたことを示している。このように、FN30/35へのウイルスの結合は、服用量依存様式で起こっている。
【0095】
実施例9 組換えフィブロネクチンフラグメントへのウイルスの結合
9.1. 実験方法
Kimizukaらは、以前、大腸菌内にクローン化したFN DNA配列の発現について報告している(Kimizuka,F.,Y.Tatuchi,Y.Ohdate,Y.Kawase,T.Shimojo,D.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)。クローン化したキメラペプチドは、細胞粘着性に関係することが知られているフィブロネクチン内の様々な重要配列(RGDS、CS−1およびヘパリン結合部位)の内の一つまたはその組み合わせを含む(図7参照のこと)。レトロウイルスがこれらの組み換えFNフラグメントと結合することができるか否かを分析するために、3T3細胞コロニー形成アッセイを、1x104感染粒子/mlの凍結レトロウイルスTKNeoストックの2つの異なる希釈物(1:10および1:100)を用いて、組換えフラグメントC−274、H−271、H−296、CH−271、CH−296およびC−CS1ならびに陽性対照としてFN30/35でコートしたプレート上で繰り返した。FNフラグメントは、120−130pmol/cm2の濃度(C−274、H−271、H−296、C−CS1、FN30/35では4μg/cm2に等しく、CH−271およびCH296では8μg/cm2に等しい)で用いられた。簡単に言えば、プレートをコートし、ウイスルを加え、プレートを広範囲に洗浄し、NIH/3T3細胞を24時間加え、そして10日間選択培地内で増殖させ、その後、コロニーを染色し、数を数えた。
【0096】
9.2. 結果
図8は、G418−耐性コロニーの数(およびそのウイルス粘着性)が、フラグメントH−271、H−296、CH−271およびCH−296で増加したことを示している。さらに、図8は、ウイルス結合量が、これら組み換えフラグメントとFN30/35の間では概略的に比較できるが、この仕事中では、CH−271フラグメントが最も高いレベルのウイルス結合を示したことを、示している。これらの5FNフラグメントのすべてに共通なのは、高アフィニティーのヘパリン結合部位を含むタイプIIIリピート12−14であり(Ruoslahti,E.,Ann.Rev.Biochem.,57,375−413、1988、およびKimizuka,F.,Y.Taguchi,Y.Ohdate,Y.Kawase,T.Shimojo.K.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)、多分、リピート13に位置する(Kimizuka,F.,Y.Taguchi,Y.Ohdate,Y.Kawase,T.Shimoko,K.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)。このことは、ウイルス結合が、この既知の粘着部位を通して起こることを示唆している。このことは、4μg/cm2のCH−271でコートした皿を、ヘパリン結合部位との細胞結合を阻害することが知られている高荷電分子である、硫酸ヘパリンの濃度を上昇(10−1000μg/ml)させて、プレインキュベーションすることによって、証明された。図9に示したように、G418−耐性コロニーの数は、高濃度の硫酸ヘパリンと共にCH−271をプレインキュベーションした後には、減少している。これらのデータは、FNへのウイルス結合が、FNのカルボキシ末端ドメイン内のCS−1部位のすぐ隣りに位置する高アフィニティーのヘパリン結合部位を通して仲介されることを、示唆している。
【0097】
実施例10 組換えフィブロネクチンフラグメント上での造血細胞の形質転換
10.1. 実験方法
前記したように、FN30/35上での造血細胞の形質転換の上昇が、組換えFNフラグメントでもまた認めることができるか否かを分析するために、我々は、高増殖潜在性−コロニー形成細胞(high proliferative potential− colony forming cell)(HPP−CFC)アッセイを用いて、インビトロでの上清感染の形質転換効率を評価した。EAL2aベクターを用いることによって、我々は、BSA上での様々な組換えFNフラグメント対FN30/35の上清感染の、造血細胞の形質転換への影響を、遺伝子トランスファーのインジケーターとしてG418−耐性コロニーの増殖を利用して、比較した。さらに、我々は、すでにFNフラグメントに粘着したウイルス粒子(対上清ウイルス)が造血細胞を形質転換する能力を比較した。上記の方法に従って、0.5から1x106の前刺激した骨髄細胞を、35mmのFN−コートペトリ皿上、1−2mlのEAL2aウイルス含有上清中、増殖因子および5μg/mlのポリブレンと共に、インキュベートした。FNフラグメントに結合したウイルスによる造血細胞の形質転換を評価するために、ウイルス含有上清と共にインキュベートした35mmのFN−コート皿を各2mlのPBSで3回洗浄した。次に、0.5から1x106の前刺激した骨髄細胞を、増殖因子およびポリブレンを添加した2mlの培地内に加えた。22時間後、細胞を収集し、そして、Bradley,T.R.およびD.Metcalf,Aust,J.Exp.Biol.Med.Sci.、44、287ー293,1966に記載の方法に従って、1.5mg/mlのG418を加えておよび加えずに、HPP−CFCアッセイに塗布した。培養液を、14日間、7%CO2中、33℃でインキュベートし、遺伝子トランスファー効率を、G418耐性コロニーの%として計算した。
【0098】
10.2. 結果
上清感染を通しての初期造血細胞(primitive hematopoietic cells)の形質転換(図10)は、ヘパリン−結合部位(heparin−binding site)(HBS)および少なくとも一つより多くの活性細胞粘着部位(立体棒)を含むすべてのフラグメントで、BSA上での上清感染より有意に高い。組み換えフラグメントCH−271、H−296、CH−296およびC−CS1(3つのフラグメントはすべて、ヘパリン−結合部位およびCS−1部位の両方を含む)の形質転換効率は、FN30/35の効果と同等であった。その他のすべての場合において、形質転換は、劇的に減少した。これらのデータは、以前にFN30/35上で示された初期造血細胞の形質転換の増加を、組み換えFNフラグメント上でも複製できることを、証明している。さらに、フィブロネクチンに直接結合したウイルスは、造血細胞を遺伝的に形質転換する能力を持つことを、証明している。最終的に、初期造血前駆体(幹)細胞を形質転換するために、CS−1およびヘパリン結合部位の両方の存在が有用であることが、確認された。
【0099】
実施例11 組換えフィブロネクチンフラグメント上でのマウスのドナー細胞の形質転換を用いる、マウスの長期にわたる骨髄の再構築
11.1. 実験方法
造血幹細胞の再構築への効果を分析するために、初期造血前駆細胞について、BS対FN30/35対組み換えFNフラグメント上での上清感染、対同時培養を、骨髄移植を用いて比較する、上記のインビトロでの研究(実施例10から)を繰り返した。簡単に言えば、致死照射マウスに、ヒトADA cDNAを含むEPHA−5ベクターで形質転換したドナー細胞を注射した。1ヶ月後およびおおよそ6ヶ月後、遺伝子トランスファー効果を、ADAイソ酵素アッセイで末梢血液から分析した。
【0100】
11.2. 結果
図11は、1ヶ月後の結果を示し、ヘパリン結合部位およびCS−1の両方を含むフィブロネクチンフラグメントH−296では、FN30/35および共培養と同様の結果が得られることを、はっきりと示している。これら部位を両方共含まないフラグメントは、移植可能な造血細胞の形質転換に、あまり有効ではない。これらのデータは、初期造血/幹細胞ならびにCS−1およびヘパリン結合部位を両方共含む組換えFNフラグメントに結合したレトロウイルスが共に配置されることが、移植可能な造血細胞を効果的に形質転換することを証明している。
【0101】
図12は、移植後4ヶ月および6ヶ月の結果を示している。この時点で、造血幹細胞を再集団化する遺伝的形質転換は、対照プレート上またはCBD(C−274)あるいはIII12−14(H−271)のみを含むフラグメント上で形質転換した細胞を移植した動物内では、示すことができなかった。HSCsの遺伝的形質転換は、C−CS1フラグメント上ではより少ない頻度で認められ、この場合、1/3の動物がヒトのタンパク質陽性であった。インビボで再集団化した幹細胞内への遺伝子トランスファーは、CBD(CH−271)またはCS1(H−296)のいずれかと組み合わされたHBDを含むフラグメント上で、一律に認められた。3つの細胞結合部位をすべて含むフラグメント(CH−296)上での形質転換は、標的細胞、産生細胞系、の同時培養で最も効果的に比較できた。これらのデータは、VLA−4および/またはVLA−5の結合部位と組み合わせてIII12−14を含むフィブロネクチンフラグメントがマウスの造血前駆細胞および幹細胞内へのレトロウイルスが仲介する遺伝子トランスファーを増加させる能力を有するを示している。
【0102】
実施例12
フィブロネクチンは、少なくとも3つの部位(図13):インテグリンVLA−5を通してリピート10内にテトラペプチドRGDSを含む細胞結合ドメイン(cell binding domain)(CBD);細胞表面のプロテオグリカン分子を通してタイプIIIリピート12−14(III12−14)内に含まれるヘパリン結合ドメイン;および、インテグリンVLA−4を通して変化するスプライス領域IIICS内のCS1配列:を通して細胞粘着性を支配する。これらの研究では、これらの単一の細胞粘着部位を単独で含むか、あるいはペプチド内に組み合わせて含む、図13に示す6つのキメラ組換えFNフラグメントを用いた。
【0103】
FN−コートした細菌皿を、アンホトロピック(amphotropic)パッケージング細胞系TKNeoからの2mlの上清(SN)内の200cfuと共に、インキュベートした。37℃に30分間置いた後、皿をPBSで3回洗浄し、次いで、2000NIH/3T3細胞(繊維芽細胞)を、10%のウシ血清(CS;Sigma,U.S.A.)および2%のP/Sを添加した1mlのDMEM内に加えた。翌日、細胞は、0.75mg/mlのG418を含む選択培地内に置かれた。8−10日後、皿を、Stat Stain(Volu−Sol,U.S.A.)で染色し、そしてG418rコロニーをカウントした。このアッセイでは、レトロウイルスは、コートしていないかまたはBSA−コートしたプレートとは結合しない。図14に示したように、NIH/3T3細胞内への遺伝子トランスファーは、ここでは”III12−14”と名付けられたヘパリン−II結合ドメインを含むフラグメント(H−271、CH−271、H−296、CH−296)上でのみ起こった。これらのデータは、レトロウイルス粒子がフィブロネクチンのIII12−14リピート内の配列に直接的に結合することを証明している。NIH/3T3細胞の感染は、III12−14のみの場合と比較して、III12−14およびCBDの両方を含むFNフラグメント(H−271対CH−271を比較する)で有意により高かった。注目すべきことに、200のNEOcfu/プレートのインプットから最大で80のG418rNIH/3T3コロニー/プレートが生成した。これとは対照的に、CS1を加えても、NIH/3T3形質転換にいかなる効果も示さなかった(H−271対H−296、CH−271対CH−296)。
【0104】
FNがレトロウイルス−仲介の遺伝子トランスファーを増加させるメカニズムがフラグメントへのレトロウイルスおよび標的細胞の同時結合によるものであることを証拠付けるため、非粘着細胞系(HL60、既知の前骨髄細胞白血病細胞系)を用いて、実験を行った。HL60細胞を、直接フルオロクロム−結合のVLA−4に対するモノクローナル抗体(FITC:Immunotech,U.S.A.)およびVLA−5に対するモノクローナル抗体(PE;Antigenix America,U.S.A.)で、またはIgG1およびIgG2a(Becton Dickinson)をイソ型対照として、染色した。死滅細胞を、プロピフィウムヨード(propifium iodine)染色によって、排除した。次いで、細胞をFACScan(Becton Dickinson)上で分析した。遺伝子トランスファーを研究するために、104のHL60細胞を、神経増殖因子レセプター(NGF−R)(力価105cfu/ml)を含むアンホトロピックパッケージング細胞系DAG(St.Jude’s Children Research Hospital,Memphis,TN.U.S.A.から得られた)からのウイルス上清と共に、懸濁し、そして、濃度2μg/cm2 の6つのFNフラグメントでコートした細胞6穴プレートに加えた。4時間後、培養中のHL60培養液から2mlの順化培地を加えた。4mlの新たな培地(5%のFCSおよび2%のP/Sを含むRPMI, Gibco)を、4−5日後に加えた。細胞を全体で8日間増殖させ、次いで、NGF−R(Boehringer,U.S.A.)に対するモノクローナル抗体8211で、または対照としてイソ型IgG1(Dako,U.S.A.)で染色し、次いで、FITC−複合 ヤギ−抗−マウスの二次血清(Boehringer)と反応させた。細胞デブリスを排除するために、サンプルをプロピジウムヨード(propidium iodine)(PI)と共にインキュベートし、次に、FACScanで測定した。遺伝子トランスファーは、NGF−R発現の分析によって、生きた細胞の通門(gating)の後に、証明された。遺伝子トランスファーの研究はすべて、ポリブレンまたはプロタミンなしで、行われた。図15に示すように、HL60細胞は、フローサイトメトリー分析(A+B)では、VLA−4を発現するが、VLA−5を発現しない。発現データと一致して、HL60細胞は、CS1を含むフラグメント(H−296、CH−296、C−CS1)でコートされたプレートのみに粘着した。図15に示すように、HL−60細胞の遺伝子形質転換は、CS1と組み合わせてIII12−14を含むフラグメント(H−296、CH−296)上でのみ起こった。HL−60細胞は、それらのVLA−4インテグリンを通してC−CS1フラグメントと粘着するが、レトロウイルス結合が劇的に起こるIII12−14 が存在しないと、HL60細胞の形質転換は減少する。
【0105】
実験のもう一つのセットでは、濃度を増加させた高分子量(HMW) のヘパリン(Sigma,U.S.A.、MW約7000−25000)は、2.5%(v/v)の1M Hepes(HBSS/Hepes;すべてGibco)を添加した2mlのハンクス液(Hanks Balanced Salt Solution)に溶解させ、そして濃度4μg/cm2 の異なるFNフラグメントでコートした細菌の6穴プレートに加えた。30分後、プレートを、PBSで3回洗浄し、次に400cfu/2mlのアンホトロピックTKNeoベクターを加えた。37℃で30分後、プレートを再びPBSで洗浄し、次いで10%のCSおよび2%のP/Sと共に、DMEM中の2000NIH/3T3細胞を加えた。上記の方法に従って、選別を行い、遺伝子トランスファー効率を、8−10日の培養の後のG418r コロニーの数として計数した。同様の実験セットでは、分画した低分子量(LMW)ヘパリン(Sigma,U.S.A.、MW約3000)は、濃度を増加させた2mlのHBSS/Hepes中に溶解させた。次いで、HMW ヘパリンに関して上記した方法に従って、実験をデザインした。遺伝子トランスファー効率は、8−10日後のG418r の数として計数した。これらの遺伝子トランスファー研究のすべては、ポリブレンまたはプロタミンなしで行った。これらの実験の結果を図16に示す。示したように、FNへのウイルスの結合は、高分子量ヘパリンでは服用量依存様式で競合できる(16A)が、低分子量ヘパリンでは競合しない(16B)。これらは、結果として、レトロウイルス粒子がIII12−14内の配列に結合すること証拠となる。
【0106】
実施例13 CD34+ 細胞集団内でのBFU−E細胞の選択的形質転換
この実施例では、混合した細胞集団内で選別された細胞を、ウイルス結合ドメインおよび細胞結合ドメインを持ち、標的細胞に特異的であるポリペプチドを用いることによって、形質転換の標的とすることができることを証明した。一般的TKNEO感染および実施例1のアッセイプロトコールを繰り返したが、BFU−E、CFU−GMおよびCFU−Mix細胞[臍帯血(CB)細胞よりアフィニティクロマトグラフィーによって得られた]を含むCD34+細胞は、実質的に均一な集団を用い、組換えFNフラグメントCH−271の濃度は1、2、4、8、16および20μg/cm2 に変化させて用いた。結果を図17に示す。示した通り、BFU−E細胞は、VLA−5結合部位に結合し、高い効率で形質転換された(G418耐性コロニーの25%より多い)が、これに対してCFU−GMおよびCFU−Mix集団は、有意なVLA−5への結合を示さず、実質的により低い効率で形質転換された(G418耐性コロニーの約5%より少ない)。
【0107】
実施例14 c−KIT+細胞の形質転換
本実施例では、c−KIT+として実質的に均一な細胞集団を、本発明の方法に従って、形質転換した。このため、c−KIT+細胞は、フローサイトメトリー選択技術を用いて、マウスの骨髄から単離された。細胞は、一般的には実施例1記載の方法に従って、TKNEOベクターを用いた感染プロトコールに委ねたが、FNフラグメントは、図18に示すように変化させ、感染した細胞は、HPP−CFCでアッセイした。図18に示すように、III12−14およびVLA−4結合部位を含むFNフラグメント(FN30/35、H296およびCH−296)は、高い形質転換効率を示した(G418耐性コロニーの80%より高い)が、これらの2つのドメインを欠くFNフラグメントは、結果として有意な形質転換を生じない(C274およびC−CS1)か、または実質的に低い効率を示す(H271、CH271、G418耐性コロニーの20%より少ない)。
【0108】
実施例15 ポリブレンの濃度を変化させて用いた場合のNIH/3T3およびクローン原性(clonogenic)BM細胞の形質転換
この実験セットでは、本発明の有用な形質転換法が、ヘキサジメチリンブロミド非存在下で行われることを証明した。NIH/3T3(繊維芽細胞)細胞およびクロ−ン原性骨髄細胞は、一般的には、それぞれ実施例12および1に記載した通りのTKNEO感染プロトコールに委ねたが、ベクター、細胞および様々な濃度のポリブレンを最初に一緒に懸濁し、次いで、FNフラグメントCH−296(8μg/ml)でコートした基質に適用した。アッセイは、これらの細胞型に関して前記したとおりである。結果を図19および20に示す。図19に示すように、G418耐性NIH/3T3コロニーの数は、ポリブレン濃度を増加させると、劇的に減少する(その範囲は、ポリブレンを使わなかった場合約14から、12.5μg/mlのポリブレンを用いた場合約4に低下する)。同様に、図20は、プロトコールがポリブレンの非存在下に委ねられた場合に40近いコロニーが得られたことを、表しているが、これに対して10μg/mlのポリブレンを用いた場合の関連値は、15より少なかった。
【0109】
実施例16 T細胞の形質転換、ならびにコラーゲンVおよびFGFウイルス結合配列を持つポリペプチドの使用
この実施例では、ヒトT細胞内へのDNAトランスファーの強化、およびコラーゲンVおよび繊維芽細胞増殖因子(FGF)からのレトロウイルス結合配列(フィブロネクチンのヘパリンII結合部位と実質的に相同である)の発明への使用について、説明している。このために、全血を、モノクローナル抗体(CD3 perCPに対するMoAbs;Becton Dickinson)、CD49e(PE;Anitenix America)、CD49d(FITC,PE,それぞれ:Becton Dickinson)で、15分間、室温で染色し、次いで、製造者らの勧告に従って、FACS溶解溶液(BECTIN DICKINSON)と共にFACScan(BECTIN DICKINSON)での分析用に、調製した。末梢血液−誘導単核細胞(peripheral blood−derived mononuclear cells)(PB−MNCs)は、1x106細胞/mlの濃度で、1μg/mlのOKT−3(Ortho)および50U/mlのIL−2(Cetus)で、前刺激した。2−3日後、細胞を収集し、洗浄し、次いで、7.5μg/mlのポリブレン(Aldrich)の連続存在下、8μg/cm2 のCH296で又は2%のウシ血清アルブミン(BSA,Boehringer)でコートした組織培養処理しなかったプレート上、PGKプロモーターの調節下で、マウスのアデノシンデアミナーゼ(ADA)cDNAを含むアンホトロピックレトロウイスル55/6で、1または2日間形質転換した。一日おきに、50%の培地を、50U/mlのIL−2を含む培地に新しく置き換える。感染から4−6日後、細胞を収集し、CD3、CD56、TCR−αβおよびHLA−DRに対するMoAbs(すべてBectin Dickinson)を用いて、形態学的に分析した。遺伝子トランスファー効率の分析は、ADAイソ酵素アッセイ(Bodine,D.M.ら、Blood,82,1975−80,1993;Moritz,T.ら、J.Exp.Med.178,529−36、1993)でヒトの内在性のADAタンパク質と比較して、形質転換されたマウスのADA酵素の活性を測定することによって、非選択集団上で、行われた。
【0110】
結果:
A. T細胞
最初に、ヒトT細胞を、FNレセプターVLA−4およびVLA−5の発現について分析した。このために、末梢血液(PB)−誘導単核細胞(MNCs)を、CD3、CD49dおよびCD49eに対するモノクローナル抗体(MoAbs)で染色し、フローサイトメトリーで分析した。図21に示すように、細胞を、すべての末梢血液T細胞を表すCD3陽性リンパ球(R1およびR4)について通門した。PB T細胞の94%は、CD49d陽性であり、96%はCD49e陽性である。T細胞の90%は、VLA−4およびVLA−5の両方を発現している。
【0111】
次に、PB−誘導PB−MNCsを、2−3日間、CD3MoAb OKT−3および組換えIL−2で前刺激し、次いで、1−2日間、組換えFNフラグメントCH−296かまたはBSAでコートした非組織培養処理したプレート上、PGKプロモーター調節下で、形質転換した;対照には、マウスのアデノシンデアミナーゼ(ADA)cDNAを含むアンホトロピック産生細胞系AMmA55/6(Bodine,D.M.ら、Blood,82,1975−80,1993;Moritz,T.ら、J.Exp.Med.,178,529−36,1993)からのレトロウイルス上清でコートしたプレートを用いた。
【0112】
感染4−6日後のフロー分析は、細胞培養液が>90%のT細胞を含み、その大部分は、それらのHLA−DRおよびCD56発現によって示されるように強く活性化された(図22)。
【0113】
ADAイソ酵素アッセイによる遺伝子トランスファー効率の分析(図23)は、誘導されたマウスADAタンパク質の活性が、形質転換プロトコールが組み換えペプチドCH−296を含む場合には、ヒト内因性タンパク質の活性と等しいことを証明した。BSA上での感染では、アッセイの感受性以上のレベルの遺伝子トランスファーが得られた。
【0114】
これらのデータは、ヒト一次T細胞(primary T cells)が、ポリブレンの様なポリカチオンの非存在下で、組換えFNフラグメントCH−296上の無細胞レトロウイルス−含有上清で効率よく形質転換されうることを、証明している。遺伝子配達効率は、上清+ポリブレンのみを用いた標準的方法と比較して、非常に高い。
【0115】
B. 新しい組換えペプチド
コラーゲンV(COL)および塩基性繊維芽細胞増殖因子(bFGF)の両方が、フィブロネクチンのHBDと配列相同性を示し、ヘパリン結合能力もまた有するため、図24に示す7つの組み換えタンパク質を、レトロウイルス結合活性について分析した。
【0116】
最初に、COLおよびFGFを分析して、それらが機能的レトロウイルス結合配列を持つか否かについて決定した。最後に、細菌プレートを組換えペプチドでコートし、次いで、アンホトロピックNEOレトロウイルスを含む上清を加えた。30分後、プレートを徹底的に洗浄してすべての非結合レトロウイルス粒子を除去し、次いで、2000NIH/3T3細胞をそれぞれのプレートに加えた。遺伝子トランスファー効率は、8−10日後、G418耐性(G418r )NIH/3T3細胞コロニーの数で、評価した。
【0117】
図25に示すように、NIH/3T3細胞内への充分な遺伝子配達は、HBD(フラグメントCH−271)と又はコラーゲンV(C−COL)あるいはFGF(C−FGF)の配列と組み合わせてFNの細胞結合ドメイン(CBD)を含む組換えペプチド上で起こるG418rコロニーの増殖で、評価した。これらの結果は、レトロウイルス粒子がコラーゲンVまたはbFGF内の配列と直接的に結合することをも証明し、これらの配列を含む組換えペプチドを、造血細胞の標的に用いることができることの証拠となる。
【0118】
VLA−4およびVLA−5を発現するHEL細胞は、このように、組換えフラグメントC−274(CBD)、H−271(HBD)、CH−271(CBD−HBD)、COL、C−COL(CBD+COL)、C−FGF、C−FGF−CS1上で、および対照としてBSA上で、切除型(truncated)神経増殖因子レセプターp75鎖(NGF−R)cDNAを含むアンホトロピックレトロウイルスで形質転換された。遺伝子トランスファ−効率を、8日後、NGF−R発現のフローサイトメトリー分析によって分析した。
【0119】
図26に示したように、低レベルの遺伝子トランスファーは、C−274でコートしたプレートまたはBSAでコートしたプレート上で起こった。H−271(HBD)およびCOL上での形質転換は、結果として33−36%のNGF−R陽性細胞を生じた。HBD(CH−271)へのCBDの付加は、切除型細胞の百分率を、この実験で最も高いレベルに上昇させた。興味深いことに、C−COLフラグメント中のCOLへのCBD(VAL−5結合部位)付加も、また、フラグメントC−FGF−CS1内のCS−1(=VAL−4結合部位)の付加も、HEL細胞の遺伝子形質転換を有意には上昇させなかった。
【0120】
最後に、1日IL−6およびSCFで前刺激したヒトCD34+骨髄(BM)細胞を、アンホトロピックNEOレトロウイルスと共にキメラペプチド上で、1日間形質転換し、次いで、1.5mg/mlのG418の存在下または非存在下で前駆細胞アッセイのため、塗布した。
【0121】
G418rコロニーの%として培養14日後に評価した効率良い遺伝子トランスファーは、細胞結合レセプターと組み合わせてH−271又はFGF内にレトロウイルス結合配列を含むフラグメント上で起こっていた。特記すべきことに、NGF−RレトロウイルスでHEL細胞を遺伝的に修飾することによって得られる結果(図27)に対して、C−FGFにCS1部位を付加すると、クローン原性CD34+BM細胞内への遺伝子トランスファー効率が著しく促進されて、フラグメントCH−296(CBD+HBD+CD1)で得られたレベルに近づく。
【0122】
このように、一次T細胞は、それらのVLA−4又はVLA−5インテグリンを通してフィブロネクチンと結合するT細胞の能力を用いるフィブロネクチンの粘着性ドメイン上のレトロウイルスベクターで効率よく形質転換できることが、証明された。また、レトロウイルス粒子は、フィブロネクチンのヘパリンII結合ドメインと相同性を示すコラーゲンVまたはbFGF内の配列に粘着すること、ならびに造血細胞および細胞系は、標的細胞のための結合部位と組み合わせてコラーゲンVまたはbFGFのレトロウイルス結合配列を含む組み換えペプチド上で、効率よく遺伝的に修飾されうること、もまた示した。
【0123】
本発明は、前の記述を詳細に説明し記載するが、本発明は、その特徴を説明するものとして考えられるべきであって、特徴を制限するものとして考えるべきではなく、望ましい実施態様のみを記載すること、および本発明の思想の中に入る変化および修飾のすべてが保護されることを所望することを、理解するであろう。
【0124】
ここに引用されたすべての文献は、そのそれぞれが独立的に参照として採用される様に、ここにその全体を参照として採用される。さらに、同時係属の米国特許出願08/218,355、1994年3月25日出願、および同時係属の国際特許出願PCT/US95/03817、1995年3月27日に出願され合衆国指定、はその全体を参照としてここに採用する。
【技術分野】
【0001】
発明の分野
本発明は、一般的にはウイルスによる細胞の形質導入の効率を増加させること、より具体的には、ウイルスに結合する領域と、細胞に結合する領域を含むポリペプチド類のような分子を用いて、細胞へのウイルス媒介性遺伝子導入を増強させる方法に関連する。
【背景技術】
【0002】
発明の背景
遺伝子導入技術の進歩と同様に、多くのヒト疾患の分子的基礎の理解の進歩により、重度の遺伝病に対する体細胞遺伝子治療のプロトコールを開発するための最近の試みがもたらされた。最近、ヒト遺伝子治療に対する、見込みのある疾患候補は、酵素、もしくは他のタンパク質が不完全または見られないものであって、酵素もしくはタンパク質のレベルが厳密に調節される必要がないもの、特に本質的には調節されており、そしてその欠陥が骨髄で発見されたものを含む。
【0003】
例えば、遺伝子治療の候補の一つの疾患は、重症複合免疫不全症(SCID)を引き起こすアデノシンデアミナーゼ(ADA)欠損症である。ADA欠損患者は、骨髄細胞で検出できる酵素はわずか、もしくは存在しない。しかしながら、ADA欠損は適合した骨髄移植によって治療された。ADA正常細胞は、ADA欠損細胞に比べて、選択的優位であり、正常に患者の骨髄に再配置(repopulate)される。
【0004】
骨髄細胞は、骨髄組織が簡単にin vitroで扱うことができ、再配置する細胞を含むことから、体細胞遺伝子治療のよい標的である。別の方法としては、ヒト臍帯血も多数の原始前駆細胞を含んでいることが以前に明らかにされている。長期にわたり再配置する細胞である造血幹細胞への遺伝子導入の成功は、これらの細胞の子孫において明示された様々な疾患に対して修正の治癒を導く可能性がある。
【0005】
再配置する幹細胞への遺伝子導入、及び長期遺伝子発現は、若干の研究者によってネズミモデルで成し遂げられた。しかしながら、イヌ及び霊長類の様なより大きい動物での in vivo 実験は限られた成功しかなく、これはほとんど原始造血幹細胞の感染の低効率による。現在の遺伝子導入技術の限界は、成人骨髄(ABM)に存在する幹細胞の数の少なさ、これらの細胞を精製する適当な方法がないこと、及び細胞周期でのこれらの原始細胞の画分が低いことを含む、いくつかの因子によって、ヒトプロトコールに利用する際、さらに複雑になる。
【0006】
骨髄細胞を伴うネズミの実験、及び大動物の実験双方において、最も有効なプロトコールが、レトロウイルス産生細胞株と標的細胞の共培養を利用していることが注目された。また、ヒトでのFDA−立証済み遺伝子導入試験のほとんどは、遺伝子形質導入のために、組換えレトロウイルスに依存している。組換えレトロウイルスベクターは、導入に効果的であり、細胞DNAへ外来DNAを正確に、及び安定に組み込むことから、遺伝子治療に好ましい。これらのベクターは、遺伝子導入のための外来DNAを含み、ウイルスの病原性を除去するために、さらに修飾される。これらの修飾のために、ウイルスの産生は一般的にレトロウイルスパッケージング細胞を用いることによって行われる。しかしながら、臨床遺伝子治療のために、無細胞の形質導入が、生物学的に安全で、品質管理についての関心のためにさらに好ましい。不幸なことに、幹細胞などの造血細胞への効率のよい遺伝子導入は、一般的にウイルス産生細胞との共培養なしには不可能であった。
【0007】
最近、遺伝子導入効率は、感染中に標的細胞を間質細胞へ暴露することで増加されうることが示された。間質細胞は、造血微環境(HM)の主要な構成物である。HMは、マクロファージ、間質細胞、内皮細胞、含脂肪細胞、及び様々な特定の接着分子によって構成されている複合細胞外マトリックス(ECM)の組織化されたネットワークによって成り立っている。ラミニン、コラーゲン、トロンボスポンジン、プロテオグリカン類、グリコサミノグリカン類、及びフィブロネクチンなどのECM分子は、造血細胞及び成長因子双方のための固定部分を提供する。このレトロウイルス感染における間質の促進効果の基礎をなす機構は明らかではないが、造血細胞の増殖及び分化の生理学的調節が、これらの細胞がHMの細胞と直接接触されたときに起こるということが以前から知られていた。
【0008】
長期にわたり再配置する造血幹細胞及び他の細胞への効率的な遺伝子導入は問題を残しており、現在の造血性の疾患及び他の疾患の治癒を目指す療法への遺伝子導入プロトコールの広範囲での適用を阻害する。求められていることは、過去の方法の危険および限界を排除したほ乳類細胞への遺伝的物質の効果的な導入の方法を求めることであり続けている。本発明は、この必要性に取り組んだものである。
【発明の概要】
【発明が解決しようとする課題】
【0009】
発明の概要
簡単に、本発明の一つの好ましい様態は、レトロウイルスベクターによる造血細胞の形質導入の効率を増加させる方法を提供する。この方法は、レトロウイルスによる細胞の形質導入の効率を増加させるのに効果的な、十分に純粋なフィブロネクチンおよび/またはフィブロネクチン断片の存在下で、複製欠損組換えレトロウイルスベクターを、生存している造血細胞に感染することを含む。フィブロネクチンおよび/またはフィブロネクチン断片は、天然に生じる物質から由来しうるか、また合成的に由来することができ(例えば、遺伝的に組換え、もしくは化学的合成技術によって設計される)、もしくは、天然に生じるもの及び合成物質の組み合わせにより由来しうる。加えて、本発明で用いたフィブロネクチンポリペプチド、もしくはペプチド類は、本発明に従って、形質導入を高めることができるのに必要な接着特性を持っている、機能的ポリペプチドをそれでもなお提供する、自然に生ずるフィブロネクチンのアミノ酸配列の変異を含む可能性があることが理解されるであろう。
【0010】
他の本発明の好ましい様態は、レトロウイルスによる細胞形質導入の効率が増加するのに効果的な量の、固定化されたフィブロネクチン、固定化されたフィブロネクチン断片、もしくは固定化されたそれらの混合物存在下で、生存している造血細胞へ外来DNAを運ぶ複製欠損組換えレトロウイルスを感染することを含む、形質導入された造血細胞を産生する方法を提供する。
【0011】
本発明の他の好ましい様態は、細胞移植の改善された方法を提供する。この方法は、ドナー動物からの生存している造血細胞を得る工程、形質導入された生存している造血細胞産生のために、固定化された形で、形質導入の効率を増加させるのに効果的なフィブロネクチンおよび/またはその断片の存在下で、造血細胞を組換えレトロウイルスベクターにて感染させる工程、及び細胞移植片として、この形質導入された生存している造血細胞をレシピエント動物に導入する工程を含む。一つの好ましい方法において、感染細胞は自己提供体に導入することができる。
【0012】
本発明の他の好ましい様態は、細胞移植法に適した、形質導入された臍帯血細胞を得る方法を提供する。この方法は、レトロウイルスベクターによる造血細胞の形質導入効率を増加するために効果的な量の固定化されたフィブロネクチンおよび/またはフィブロネクチン断片の存在下で、臍帯血由来の造血細胞に、複製欠損組換えレトロウイルスベクターを感染させることを含む。本発明はまた、これらの方法で得ることのできる臍帯血由来の生存している形質導入された細胞集団を含み、そして細胞移植片として動物体内へ形質導入された細胞集団を導入することを含む、細胞移植方法を含む。
【0013】
本発明の上記言及の様態のさらに固有の局面に従って、使用されたフィブロネクチンまたはフィブロネクチン断片は、フィブロネクチンのヘパリン−II結合部位のレトロウイルス結合活性を提供する第一のアミノ酸配列、及び、フィブロネクチンのCS−1部位の細胞結合活性を提供する第二のアミノ酸配列を含みうる。これらの二つのフィブロネクチンの結合部位を一緒に用いることは、レトロウイルスによる標的細胞の形質導入効率を非常に顕著に高めることが証明された。
【0014】
本発明の他の好ましい様態は、レトロウイルスベクターによる予め決定した標的細胞の形質導入の効率を高めるための構築物を産生する方法を提供する。この方法は、前記標的細胞と結合するリガンドと、フィブロネクチンのヘパリン−II結合部位のレトロウイルス結合活性を提供するアミノ酸配列を含むポリペプチドとを、共有的に結合する工程を含む。本発明はまた、レトロウイルスによる、予め決定した標的細胞の形質導入の効率を増加させるために、そしてこの形質導入された細胞を用いて細胞移植処置法のために、これらの構築物を利用することを含む方法を含む。
【0015】
本発明の他の好ましい様態は、多量のウイルスを局在化するために、フィブロネクチンのヘパリン−II結合部位のウイルス結合活性を含む、固定化された効果的な量のフィブロネクチンまたはフィブロネクチン断片の存在下で、ウイルスを含む培養液をインキュベートすることを含む、多量のウイルスを局在化するための方法を提供する。
【0016】
さらに、本発明の他の好ましい様態は、一般的に、以下の節でさらに示すように、十分に純粋なおよび/または固定されたフィブロネクチン、またはそのフラグメントを含む、形質導入された生存している造血細胞、及び細胞培養を提供すると同時に、細胞へのレトロウイルス媒介性DNA導入を行うためのキットを提供する。
【0017】
さらなる本発明の好ましい様態は、レトロウイルスと細胞を共局在化させ、細胞の形質導入効率を増加させるために、細胞と結合するリガンドと、レトロウイルスと結合するリガンドとを含む、特定の量の固定化物質である、効果的な固定された量のポリペプチドのような物質の存在下で、レトロウイルスを細胞に感染させることを含む、レトロウイルスによる生存している細胞の形質導入された群を得るための方法を提供する。さらに驚いたことに、それらの工程は、さらに都合よいことに、今まで、形質導入効率を増加させることを望むために、遺伝子導入プロトコールにおいて使用されていたものである、ヘキサジメトリンブロミド(1,5−ジメチル−1,5−ジアゾウンデカメチレンポリメトブロミドとしても同定されている)の非存在下で、もしくは少なくとも実質的に存在しない状態で行われる。しかしながら、驚くべきことに、ヘキサジメトリンブロミドの存在は、本発明の共局在化媒介性遺伝子導入工程における形質導入効率を増大させるのではなく、むしろ減少させることが発見された。従って、さらに好ましい本発明の様態は、ヘキサジメトリンブロミド、もしくは共局在化のための物質の非存在下で行われた同じ様なプロトコール、例えば類似の共培養プロトコールにおける、形質導入効率を増加させる他の物質の非存在下で行われる。結果として生ずる改善した細胞集団、及び細胞移植方法はまた、本発明の一部分を形成する。
【0018】
本発明の他の好ましい様態は、細胞に結合するアミノ酸配列と、レトロウイルスに結合する、V型コラーゲン、または繊維芽細胞増殖因子由来のアミノ酸配列を含むポリペプチドの存在下での、レトロウイルスと細胞の感染の過程を含む、レトロウイルスによる生存している細胞の形質導入群を得るための方法を提供する。
【0019】
本発明の他の好ましい様態は、レトロウイルスと細胞を共局在化し、細胞の形質導入効率を増加させるために、T細胞に結合するリガンドとレトロウイルスに結合するリガンドとを含む物質の存在下で、レトロウイルスを細胞に感染させることを含む、レトロウイルスによるT細胞の形質導入の方法を提供する。好ましい形において、本方法で用いた物質は、T細胞に結合する第一のアミノ酸配列と、レトロウイルスに結合する第二のアミノ酸配列を含むポリペプチドである。
【0020】
本発明の他の好ましい様態は、レトロウイルスを局在化させる方法を提供する。レトロウイルスに結合するV型コラーゲンまたは繊維芽細胞増殖因子由来のアミノ酸配列を持つ、効果的な量の分離されたポリペプチドに、レトロウイルスを接触させることを含む。
【0021】
ほ乳類細胞の効果的なレトロウイルス感染の方法を提供することは本発明の目的である。
共培養の必要性を取りのぞいた、レトロウイルスベクターによる遺伝子導入の方法を提供することは、本発明のさらなる目的である。
【0022】
自己および/または同種の細胞移植のための、改善した方法および細胞培養を提供することは、本発明のさらなる目的である。
本発明のこれらのそして他の目的、利点、及び特徴は、すぐに以下の記載によって、当業者に対しては容易に明らかになるであろう。
【課題を解決するための手段】
【0023】
好ましい様態の記述
本発明の原理の理解を促進する目的のために、参考文献は現在、それらの特定の態様をなし、固有言語は同様に記載するために使用しうる。それにも関わらず、本発明の範囲を制限しないことがそれによって、このような変化、さらには改良、そして本発明が関係する技術分野での当業者が想到したであろう様に企画され、本明細書に示されたように、このような本発明の原理の応用を意図していると理解されるであろう。
【0024】
以上に指摘したように、本発明は、レトロウイルスなどのウイルスによる、生存している細胞の形質導入の頻度を増加させる方法を提供する。本発明はまた、組換えレトロウイルスベクターを用いて、生存している細胞への効果的な遺伝子導入の方法、形質導入された細胞を得る方法、そして自己及び他の細胞移植を行うための方法と物質も提供する。
【0025】
本発明の一つの特徴は、フィブロネクチン(FN)及びFNの細胞結合部位CS−1を含むフィブロネクチン断片が、フィブロネクチン受容体を持ち、それによって、フィブロネクチンもしくはその断片への結合能力を示す、たとえば投入された前駆細胞及び原始造血幹細胞、もしくは長期培養始原細胞(LTC−IC)といった造血細胞などの細胞へのレトロウイルス媒介性遺伝子導入を顕著に増強するという発見である。都合よいことに、この上昇した効率は、ウイルス産生細胞との共培養を必要としない。本発明の他の特徴は、ヘパリン−II結合部位の中に位置するフィブロネクチンのウイルス結合部位の発見を利用する。本発明の別の特徴は、ヘパリン−II結合部位に局在するフィブロネクチンのウィルス結合部位の発見を利用する。このウイルス結合部位は、例えば、ウイルスを標的細胞に送達するための広い範囲の構造を含む多くの応用において、ウイルス粒子を局在化させるために使うことができる。
【0026】
本発明のある好ましい観点に従った組換えウイルスベクターは、外来DNAを含み、そして非病原性、すなわち複製欠損である。これらのベクターは、外来DNAを動物細胞、特にほ乳類細胞のような宿主細胞の細胞DNAの中に効果的に導入し、正確にそして安定に組み込む。例えば、本発明において、関心の遺伝子のコード配列からの塩基の列を含むヌクレオチド配列を、遺伝子を駆動するための適当なプロモーター、一般的には外来のプロモーターの調節下で、組換えレトロウイルスベクターの中へ組み込むことができる。これに関連して、外来DNAは、天然、もしくは人工的どちらかで産生したDNAを含むことができ、異質組織源から得られた部分から由来することができる。この異質組織源の部分は、天然に生じたもの、もしくは化学的に合成された分子である可能性があり、そしてこれらの部分は、ライゲーションもしくはこの分野で既知の他の方法で連結する。
【0027】
ウイルスに組み込まれた外来DNAは、細胞への導入のための関心のいずれかのDNAであり得る。例えば、外来DNAは、知られている異常と関係するADAなどのタンパク質、もしくはアンチセンスRNA、リボザイム、もしくは疑似プライマー(例えば、1990年11月15日発行のWO 90/13641 を参照)、細胞内抗体(例えば、1994年2月3日発行の WO 94/02610 を参照)、成長因子、もしくは類似のものをコードすることができる。
【0028】
示したように、導入されたヌクレオチド配列は、プロモーター調節下にあり、従って一般的にプロモーターの下流にあるであろう。別の言い方をすれば、プロモーター配列は、一般的にコード配列の上流(すなわち5’末端)にあるであろう。このような傾向において、プロモーターと共同する他の調節因子(例えばエンハンサー配列)や、外来コード配列の転写を行うための転写開始コドンが存在しうるか否かについてはよく知られている。「調節下」の語は、導入された遺伝子の転写を達成するために必要であるような、他の成分の存在を企図する。同様に組換えDNAは好ましくは導入されたコード配列より下流に終止配列を含みうる。
【0029】
選択マーカー、もしくは他の選別できる好要因を提供する外来DNAを含むレトロウイルスベクターを使用することができる。例えば、ベクターはネオマイシンなどの抗生物質を含む、様々な選択薬剤に対する抵抗性を提供する一つもしくはそれ以上の外来性遺伝子を含むことができる。本発明で用いることのできた典型的なベクターは、例えば、すべてすでに Moritz et al. (1993) J. Exp. Med. 178:529 によって報告されている、 N2/Zip TKNEOベクター(TKNEO)(NIH/3T3細胞上の力価:1×105 G418r cfu/ml)、ZipPGK−hADAベクター、及びZipPGK−mADAベクターである。TKNEOベクターでは、ネオホスホタランスフェラーゼ配列がヘルペス・シンプレックスのチミジンキナーゼプロモーターを介して、センス方向に発現している(5’長末端繰り返しLTRに関係する)。このベクターは形質導入された細胞の同定を促進するネオマイシン耐性を提供する選択マーカー遺伝子を含む。ZipPGK−hADAベクターにおいて、ヒトADA(”hADA”)cDNAは、ヒトホスフォグリセレートキナーゼ(PGK)プロモーターを介して、5’LTRと関連するセンス方向に発現している。これは、ただ一つの発現しうる遺伝子配列を含み、優性選択マーカーを欠く。ZipPGK−mADA(PGK−mADA)ベクターは、ヒトADA cDNAがネズミADA(”mADA”)DNAに置き換えられたこと以外は、ZipPGK−hADAと同じものである。これら、及び他のウイルスベクター、及びこれらの産生手法はよく知られており、これらの本発明における実行、及び使用は、本明細書中の開示を与えられた当業者の間ではよくあることである。
【0030】
本発明中で使用したウイルスベクターは、ヒトフィブロネクチンのアミノ酸配列を含む、フィブロネクチンのヘパリン−II結合部位のアミノ酸配列に結合する能力を示す。以下で議論するように、本発明はいずれかの説に限られるわけではないが、それぞれの機能部位に対する、ウイルスと細胞の結合を介して、ウイルスと標的細胞とが共局在化することは、ウイルスによる細胞の形質導入の増大を促進すると信じられている。この考えでは、ヘパリン−II結合部位のアミノ酸配列に対するウィルスの結合能力、そして従って、本発明で効果的に役に立つためのウイルスの能力は、以下の実施例8、及び9で記載したような決まった操作を用いてすぐに確かめられる。一般的にいえば、これらのアッセイは、固定化ポリペプチドマトリックスからの洗浄に耐えるために、ウイルス粒子が、ヘパリン−II結合部位を含む固定化ペプチドに結合する範囲を決定する。簡単に言えば、例えば、フィブロネクチンヘパリン−II結合部位を含む、固定化ポリペプチドを含むウェルの中で、ウイルス含有上清をインキュベートできる。次いでこのウェルをさらに生理食塩水緩衝液で洗い、その後、ウイルスに対する標的細胞を、ウェルに残っている感染活性のレベルを決定するために、ウェルの中でインキュベートする。初期ウイルス上清と比べた感染活性、もしくは力価の減少が測定され、同じ対照実行(例えばBSA−コートウェルの使用)と比較した。対照ウェルに比べてヘパリン−II部分を含むウェルでの顕著に高い力価は、本発明の観点における使用に、対象ウイルスが適していることを明らかにしている。このスクリーニング手法を促進するために、ウイルスベクターは、以上で議論したように、選択マーカー遺伝子を含んでもよい。
【0031】
本発明で使用するためのフィブロネクチン断片は、天然、もしくは合成起源のものでよく、例えば以前に、 Ruoslahti et al. (1981) J. Biol. Chem. 256:7277 ; Patel and Lodish (1986) J. Cell. Biol. 102:449 ; 及び Bernardi et al. (1987) J. Cell. Biol. 105:489 によって記載されたような天然に生ずる物質からの十分な純度で調整されうる。この考えにおいて、本明細書の、十分に純粋なフィブロネクチンもしくはフィブロネクチン断片についての言及は、それらがフィブロネクチンが天然で生じる他のタンパク質から、本質的に遊離していることを意味するつもりである。本発明で使用するための、十分に純粋なフィブロネクチンもしくはフィブロネクチン断片は、例えば、一般的にはTaguchiら、そして日本、京都の宝酒造株式会社に与えられた1993年3月30日の米国特許 No. 5,198,423で記載されたように、組換えにより産生することができる。特に、以下の実施例でH−271、H−296、CH271、CH296及びC−CS−1として同定された組換え断片、及びそれらを入手する方法は、この ’423特許で詳細に記載されている。以下の実施例で用いたC274断片は、米国特許 No. 5,102,983で記載されているようにして入手した。これらの断片、もしくは断片からの日常的に引き出すことのできるものは、これも米国特許 No. 5,198,423 に記載されているようにFERM P−10721(H−296)、FERM BP−2799(メチオニンを介してH−271に結合するC−277)、FERM BP−2800(メチオニンを介してH−296に結合するC−277)、及びFERM BP−2264(H−271)として工業技術院(日本)の機関である発酵研究所(Fermentation Research Institute)に寄託されている、E. coliの培養によって入手できる。加えて、本明細書中の使用可能なフィブロネクチン断片に関する、もしくはこのような断片のための開始の物質に関する役に立つ情報は、上記記載の組換え断片についてさらに報告されているKimizukaら(J. Biochem., 110, 284−291 (1991))、ヒトフィブロネクチン遺伝子の構造について報告しているもの(EMBO J., 4, 1755−1759 (1985))、ヒトフィブロネクチンのヘパリン−II結合部位について報告しているもの(Biochemistry, 25, 4936−4941 (1986))において見いだしうる。CS−1細胞接着部位とヘパリン−II結合部位双方を含むフィブロネクチン断片、例えば、約30もしくは35kD断片(30/35FN)、そして以下の実施例で報告する様々な組換え断片を含むものは、このようなさらなる作業の中で造血細胞への遺伝子導入の効率を顕著に高めることが発見されており、そして本発明での使用に好ましい。従って、大きくいえば、本発明で用いたフィブロネクチン関連ポリペプチド、もしくはポリペプチド類は、ウイルスに結合するフィブロネクチンのヘパリン−II結合部位のアミノ酸配列と同様に、フィブロネクチンのCS−1細胞接着の細胞結合活性を与えるアミノ酸配列を提供すると理解されるだろう。当業者は、細胞及びウイルス結合活性の必要性は、これらの機能的フィブロネクチン部位の天然アミノ酸配列、及び天然アミノ酸配列と違うが、十分に細胞結合及びウイルス結合活性を示す点では似ているアミノ酸配列両方により提供されうるということを理解するであろう。これらの類似アミノ酸配列は、これらの対応する天然配列との実質的な配列相同性を示し、望ましい細胞結合特性もしくは、ウイルス結合特性を持つアミノ酸配列を提供する一方で、アミノ酸が削除されたもの、置換されたものおよび/または修飾されたものを含むことができる。
【0032】
この考えでは、適切な生物技術分野は、対象の機能的部位内でアミノ酸の削除、置換、付加、もしくはその他の修飾を日常的に起こしうるような状態に進歩した。次いで結果としてのアミノ酸配列は、望ましい細胞結合活性、もしくはウイルス結合活性を求めて、日常的にスクリーニングされうる。例えば、フィブロネクチンのヘパリン−II結合部位の変異体、もしくは修飾型のウイルス結合活性は、一般的に上記で議論し、さらに具体的には、ウイルスインキュベート、洗浄、対照と比較した感染維持力を決定するためのウイルス力価アッセイを用いた、以下の実施例8及び9のようにしてスクリーニングすることができる。本明細書中で提供した技術を得れば、これらの結合アッセイは代表的で、しかし当業者に対しては日常的な実験であり得る。
【0033】
フィブロネクチンのCS−1細胞接着部位に対する修飾、もしくは変異型に対する細胞の結合、もしくは他の細胞結合ポリペプチド類の細胞結合は、同じく慣例の手法を用いてアッセイされうる。例えば、このような手法は、 Nature, 352:438−441 (1991)に記載のものを含む。簡単にいえば、細胞結合ポリペプチドをプラスチックディッシュにコートし、分析すべき細胞集団を培養液中で30分〜2時間重層する。このインキュベート期間後、タンパク質への非接着の細胞を回収し、計数し、生存数をアッセイする。ポリペプチドへ接着した細胞も、トリプシン、もしくは細胞解離緩衝液(例えば Gibco)を用いて回収し、計数し生存数を調べる。いくつかの場合、例えば、造血コロニー形成細胞のために、この細胞のコロニー形成特性を確かめるために、細胞をさらに12〜14日間培養する。次いで付着細胞の割合を測定し、プラスチックディッシュにコートしたウシ血清アルブミン(BSA)などの標準対照と比較する。アッセイしたポリペプチドに対する標的細胞の実質的な結合は、ポリペプチド/細胞の組み合わせが本発明に適合するという指摘を提供し、このポリペプチドは、ウイルスベクターによる標的細胞の感染を高めるための、本発明の構築物を作製するために、フィブロネクチン由来のレトロウイルス結合断片に結合しうる。
【0034】
本発明の、さらに固有の観点により、レトロウイルスベクターによる形質導入を高めるために使用した、ウイルス結合ポリペプチドは、(i)ヒトフィブロネクチンのヘパリン−II結合部位のAla1690−Thr1960に対応する第一のアミノ酸配列を含み、これは式により示す(配列番号1)
【0035】
【化1】
【0036】
もしくは、レトロウイルスの結合能力を示すための、それに対して十分に類似のアミノ酸配列;
及び(ii)式により示される、ヒトフィブロネクチンのIIICS結合部位(CS−1細胞結合部位)の一つの部分に対応する、第二のアミノ酸配列(配列番号2)、
Asp Glu Leu Pro Gln Leu Val Thr Leu Pro His Pro Asn Leu His Gly Pro Glu Ile Leu Asp Val Pro Ser Thr ;
もしくは、原始前駆細胞および/または長期にわたり再配置する(幹)細胞などの造血細胞結合能を示すための、それに対して十分に類似のアミノ酸配列を含みうる。
【0037】
以前に言及したように、結果としてのアミノ酸配列が、ウイルス結合能(ヘパリン−II結合部位の場合)、及び標的細胞結合能(CS−1部位の場合)を示すために、これらの天然配列のある修飾および/または変異は、本発明の実施内で可能であることが理解されるであろう。
【0038】
例えば、ヘパリンと結合し、フィブロネクチンのヘパリン−II結合部位との本質的に配列同一性を示す配列を持つ既知のポリペプチド類は、例えば、V型コラーゲン及び繊維芽細胞増殖因子を含む。以下の特異的な実施例16で記載のように、VLA−4および/またはVLA−5のための結合部位などのような細胞結合配列と連結する、これらの配列を含むポリペプチド類は、細胞結合配列に結合する細胞への、レトロウイルス媒介性DNA導入を高めるために使用できる。
【0039】
本発明の一つの観点は、in vitroの細胞治療、及び続いて宿主への標的細胞の移植を伴う、すなわち形質導入された標的細胞による宿主への「移植」として知られる、宿主への標的細胞の形質導入の体細胞の遺伝子治療の方法を提供する。造血、または他の細胞、例えば骨髄もしくは末梢血から単離された幹細胞、胚性幹細胞、もしくはそれ以外のCD34+、および/もしくはC−kit+として特性化された細胞は、ヒトもしくはその他の哺乳動物源より標準プロトコールを用いて集められる。例えば、造血細胞は、ヒトドナーの骨髄または末梢血、もしくはヒト胎児臍帯血から集めることができる。いったん回収した後、造血細胞は所望により幹細胞および/または始原前駆細胞内で、それらを濃縮するために、標準手技によって処理する。次いで造血細胞は、例えば組織培養プレート上で適当にインキュベートされうる。所望により、この期間中、接着陰性低密度単核細胞は、レトロウイルス感染の前に前刺激されうる。当該技術分野で既知の、そして本明細書で用いられている前刺激は、レトロウイルスに暴露する前に、成長刺激因子に細胞を暴露する過程と関連する。このような前刺激は、レトロウイルスによる造血細胞の形質導入を改善することが証明された。
【0040】
前刺激に続いて、細胞を採集し、レトロウイルスによる細胞形質導入の効率を高める、本明細書に記載される、フィブロネクチンまたはその断片とともにインキュベートする。好ましくは、細胞は精製されたおよび/または不溶性、例えば、固定化フィブロネクチンもしくはフィブロネクチン断片とともにインキュベートする。次いで細胞は、ウイルスによる細胞の形質導入の効率を増加させるのに効果的な量のフィブロネクチンもしくはフィブロネクチン断片の存在下で、組換えウイルス、例えば細胞中の酵素もしくは他のタンパク質の欠損、もしくは異常を修正するための遺伝子を含んだレトロウイルスに感染させることができる。次いで結果としての形質導入造血細胞は、慣習的に、例えば、静脈注射で動物細胞移植レシピエントへ導入し、自己ドナーが望ましいが、異種遺伝子移植も含まれ、後者は特に、以下で議論するように、移植に対して臍帯血細胞が使用される。
【0041】
本発明の方法は、例えば、癌、白血病、タンパク質欠損もしくは異常に関連する疾患を含む、骨髄疾患を含む様々な疾患に対する遺伝子標識プロトコル、もしくは遺伝子治療プロトコール、及び、化学療法などの他の治療プロトコールへの抵抗性を改善するために、造血細胞を修飾する治療の中で使用されうる。本発明が使用される可能性のある代表的な疾患は、従って、ADA欠損、例えばADA−欠損SCID、小児急性骨髄性白血病(AML)、神経芽細胞腫、及び成人AML、及び急性リンパ球性白血病(ALL)を含む。
【0042】
本発明の一つの特に好ましい様態において、細胞移植のために用いられた細胞は、ヒトの臍帯血から入手する。従って、ヒト臍帯血は収集され、例えば接着陰性低密度単核細胞集団を入手することにより、生存している始原造血前駆細胞および/または幹細胞に濃縮される。その後この集団は、そこで所望により、前刺激され、そしてレトロウイルスベクターと、ベクターによる細胞の形質導入の効率を高めるための、固定化および/または精製フィブロネクチンもしくはフィブロネクチン断片の存在下で、インキュベートする。この考えにおいて、臍帯血由来の始原造血細胞および/または幹細胞は、たとえフィブロネクチンが臍帯血中のECMの構成成分ではなく、たとえ臍帯血由来の始原前駆細胞及び幹細胞が、骨髄からのものと違う特徴を有する場合でも、フィブロネクチン、もしくはフィブロネクチン断片の存在下で、かなり高められる。特に、臍帯血幹細胞は、CD34+、HLA−DR+として特徴づけられており、一方、骨髄からの幹細胞は、CD34+、HLA−DR−として特徴づけられた。臍帯血由来の始原前駆細胞が、フィブロネクチンもしくはフィブロネクチン断片の存在下で高められた様式で、効率的に形質導入されることの発見により、造血細胞の、簡単で高い幹細胞濃縮源の使用を可能になった。さらに、始原前駆細胞、及び幹細胞を濃縮した臍帯血由来の異種遺伝子移植による、多くの患者の移植の成功についての証拠により、臍帯血を造血細胞のとても好ましい源とすることができる(Kohli−Kummer et al., Brit. Heaematol. 85:419−422 (1993) ; Broxmeyer et al., Blood Cell 17:313−329 (1991) ; Gluckman et al., Br. J. Heaematol. 45:557 (1980) ; Heidelberg:Springer−Verlag pp. 60−68 (1989) ; Wagner et al., Blood 79:1874−1881 (1992) ; Wagner et al., Blood 82−86a (要約)を参照)。
【0043】
もし望めば、回収した、形質導入した造血もしくは他の細胞を、形質導入の効率及び遺伝子発現について試験することができる。例えば、本発明によって提供される、レトロウイルス媒介性遺伝子導入での顕著な改善は、以下の特定の実施例によって示されており、フィブロネクチンもしくは効果的なフィブロネクチン断片の存在下で、レトロウイルスによる高感染及び遺伝子導入効率を示す、いくつかの試験を記載している。特に、PGK−hADAレトロウイルスに感染されたネズミ造血細胞は、高いレベルの形質導入ADA cDNAを発現した。同様に、ヒト前駆細胞コロニーに感染させた個々のPGK−mADAウイルスは、ネズミADAのレベルが、内在性ヒトADAタンパク質より10倍も上昇して発現した。従って、導入効率の説得力のある解析をするために、前駆細胞コロニーは、もし形質導入されたmADAの発現が、内在性ヒトADAレベルと同等か、それ以上ある場合にのみ、形質導入されたとみなされた。TKNEOベクター由来のネオの高レベルの発現は、ネオホスフォトランスフェラーゼ(ネオ遺伝子産物)活性に対するアッセイとしてG418薬剤耐性によって検出した。
【0044】
上で示したように、本発明の方法は、レトロウイルス産生細胞の存在下での共培養を必要とすることなく、都合よく行われる。従って、本発明の一つの観点によると、レトロウイルス媒介性遺伝子導入は、標的造血細胞、もしくは他の細胞以外の細胞が実質上非存在下において実行しうる。例えば、レトロウイルスベクタープラスミドを含む産生細胞を培養することができ、上清が回収される。次いでレトロウイルス含有上清は、好ましくは固定型の、すなわち、感染が行われる基質上、さもなければ、感染用の培養液と接触する場合のフィブロネクチンおよび/またはフィブロネクチン断片存在下で、造血細胞の感染に用いることができる。この考えにおいては、高力価、ヘルパーなしのレトロウイルスを産生する産生細胞も、本発明での使用に適合することが企図される。これらは、例えば、本分野で既知の、多くの他のパッケージング細胞株と同様に、Psi−2、C2、PA12、PA317、GP+envAM12などのパッケージング細胞を含む。
【0045】
本発明の他の特徴によると、フィブロネクチンのヘパリン−II結合部位内のアミノ酸に対する、強いウイルスの結合は、広い範囲の細胞型にわたるウィルス療法に対する送達システムを構築するために使用しうる。この目的のために、フィブロネクチン由来のレトロウイルス結合部位を含むポリペプチドは、標的細胞に特異的な、この構造を与える、いずれかのリガンドとも共有的に結合しうる。このアプローチは、それぞれの標的細胞に対する特異的レトロウイルス細胞株の構築の、前もった必要性を回避しうる(Kasahara, N., A. M. Dozy, and Y.W. Kan., Science, Vol. 266, pp. 1373−1376 (1994) ;そしてValsesia−Wittmann, S., A. Drynda, G. Deleage, M. Aumailley, J. M. Heard, O. Danos, G. Verdier, and F. L. Cosset, J. Virol., Vol. 68, pp 4609−4619 (1994))。標的構造の特異性は、例えば、1)細胞接着タンパク質、2)ホルモン、もしくはサイトカイン、3)標的細胞に対するモノクローナル抗体、4)標的細胞に結合する糖鎖(G.Ashwell, et al., Annu. Rev. Biochem., Vol 51, pp. 531−554 (1982))、5)標的細胞代謝物、もしくは6)標的細胞に結合する機能的ポリペプチド類を含む、リガンドを使用することにより提供されうる。遺伝子送達のための構築物の効率は、いくつかのヘパリン−IIウイルス結合部位を含むこと、そして従って、標的細胞に対して送達されるウイルス粒子の量を増加させることにより改善されうる。例えば、米国特許 No.5,198,423に記載のような、Pro1239−Ser1515に対応しているヒトフィブロネクチンの細胞結合部位は、BHK及びB16−F10細胞(Kimizuka et al., J. Biochem. Vol. 110, pp. 285−291 (1991))を含む細胞に結合することが示された。加えて、ヘパリン−II部位、それ自身は、繊維芽細胞、内皮細胞、及び腫瘍細胞に結合することが示された。これらのポリペプチド配列は、レトロウイルスによる感染のために、予め決定した細胞を標的にするために、フィブロネクチン由来の、レトロウイルス結合部位と結合しうる。
【0046】
造血系における、典型的な応用例は、それぞれ、高度特異的な赤血球、もしくは顆粒球前駆細胞を標的にするための、フィブロネクチンのレトロウイルス結合部位と結合する、エリスロポエチンもしくはG−CSFの構造をも含む。他の本発明に従った共通の応用は、特異的もしくは優位的に悪性細胞と結合するリガンドと、レトロウイルス結合部位、または部位群を組み合わせることでありうる。例えば、in vitro、及びin vivoでも、乳癌細胞の成長は、フィブロネクチンからの一つもしくはそれ以上のウイルス結合部位を含んでいる本発明の構造の中のリガンドとして、供給されるであろう、黄体形成ホルモン放出誘導体(Emons, G. et al., Hum. Reprod. 9:1364−1379 (1994))、エストロジェン(Tolcher, A. W., Oncol. 8:39−43 (1994))、もしくは抗エストロジェン(Howell, A. et al., Lancet 345:29−30 (1995))、プロゲステロジェン、または抗プロゲステロジェン(Klijn. F. G et al., Hum. Reprod. 9 Suppl. 1:181−189 (1994) ; Griffiths, K. et al., Semin. Oncol. 21:672−687 (1994))のような標的細胞上の受容体に結合する物質を使用することで、影響されうることが示された。さらなる例として、甲状腺(癌)細胞はJodidの構築物を用いることで高度に、特異的に標的化されうる。肝(癌)細胞は、HDL、もしくはその一部を含む構築物により標的化されうる。最後に、モノクローナル抗体とフィブロネクチンのレトロウイルス結合部位の構造は、抗体が入手できるどんな細胞及び臓器を標的化することもできる。従って広い範囲のほ乳類細胞型が、ウイルスベクターに結合し局在化するために、フィブロネクチンのレトロウイルス結合部位の能力を利用することで、標的になりうる。
【0047】
従って、本発明の他の好ましい様態は、標的細胞のウイルス形質導入を高めるために用いることのできる構築物を準備することを含む。フィブロネクチンのヘパリン−II結合部位の、ウイルス結合アミノ酸配列は、標的細胞が結合するリガンドと結合する。以上で議論したように、リガンドは、例えば、フィブロネクチンもしくは他のタンパク質(細胞接着タンパク質、例えば、ラミニン、コラーゲン、ビトロネクチン、オステオポンチン、もしくはトロンボスポンジンを含む)由来ののポリペプチド、ホルモン、代謝物、抗体(モノクローナル抗体を含む)、もしくは標的細胞に結合する、好ましくは特異的に結合する能力のあることを示しているいずれかの他のリガンドである可能性がある。結果のすべての構築物は、以下の実施例にて特異的に例示した、フィブロネクチンポリペプチド鎖として使用する場合と同様の様式にて、固定型として用いることができる。
【0048】
このような構築物と、細胞標的アプローチは、上記に議論したようにin vitroで用いられる可能性があり、そして同様に生理学的条件下での構造の安定化、及び特異性と、レトロウイルス構造の相互作用などの、様々な因子を考慮することで、レトロウイルスのin vivoでの標的化にも使用される可能性がある。特異性は、例えば、標的肝細胞への門脈静脈のカテーテル挿入などの、標的細胞への構築物の送達の局在化のための、送達系の改良によって改善される可能性もある。
【0049】
他の本発明の観点は、ウイルスと細胞の共局在化に関与する、本発明の形質導入過程が、ヘキサジメトリンブロミドの非存在下で、もしくは実質的に非存在の状況で行ったときに、より優位であるという発見に関連する。ヘキサジメトリンブロミド(商業的にはポリブレンRの名前で入手できる)は、レトロウイルスによる形質導入の効率を改善する目的のために、レトロウイルス媒介性遺伝子導入プロトコールで広く用いられていたポリカチオン性の物質である。それにも関わらず、本明細書で記載したような遺伝子導入プロトコールを高めるような共局在化した際の、ヘキサジメトリンブロミドの存在は、形質導入効率を減少させることが発見された。従って、本発明の改善された過程は、少なくとも実質的にヘキサジメトリンブロミドの非存在下で(すなわち、約1μg/ml未満のヘキサジメトリンブロミドのみ含む)、及びさらに好ましくはヘキサジメトリンブロミドの非存在下の培養液中で行われる。このような過程は、レトロウイルス産生細胞及びヘキサジメトリンブロミド双方とも実質的に存在しない、実質的なレトロウイルス形質導入生存細胞集団を含む、本発明の好ましい細胞構成物を提供する。この考えにおいては、本明細書中で用いたような、実質的に形質導入された生存している細胞群は、少なくとも約20%のこの集団の細胞が、レトロウイルスによって形質導入されたことを意味することを意図する。さらに好ましい集団は、少なくとも約50%の形質導入細胞を持ち、最も好ましいものは、少なくとも約75%の形質導入細胞を持ちうる。本発明のこの観点による、好ましい細胞構成物は、造血細胞を含み、さらに好ましくは、始原前駆細胞及び幹細胞を濃縮した、造血細胞集団を含みうる。従って一般的にいえば、本発明の都合のよい過程は、ポリカチオン性の薬剤、もしくは、フィブロネクチン断片、もしくは共局在化のための他の物質の存在しない、類似のレトロウイルス感染プロトコール(例えば共培養)に対応して、形質導入効率の増加を導くような、しかし共局在化のための物質の存在下での形質導入効率を減少させるようなその他の薬剤が存在しない状態で行うことができる。
【0050】
非常に簡単な、レトロウイルス媒介性DNA導入が、本発明の方法を実施するように特にデザインされたキットを用いて実行しうることが企画される。従って、本発明の他の側面は、レトロウイルス感染を行うことができる人工基質とともに、レトロウイルスによる標的細胞の形質導入を高めるよう上記で議論した一定量の実質的に純粋なポリペプチド、もしくは構築物を含むキットを提供する。ポリペプチド、もしくは他の構築物は、別々に提供され、もしくは人工基質上にコートされうる。ヒト造血細胞のための感染プロトコールの場合、キットは、細胞前刺激のための造血細胞増殖因子をも含みうる。加えて、このキットは、形質導入のために、上記で議論したように、組換えレトロウイルスベクターを含みうる。一般的にいえば、このキットは、このキットを使用している間、構成物の破損を防ぐのにもう一つの十分な空間的な関連において、これらの様々なキット構成物を収納する、無菌包装を含みうる。例えば、複合構成物、もしくは空間的な関連におけるキット構成物を保持するための部分を有する成形プラスチック商品を用いることが共通の慣例である。
【図面の簡単な説明】
【0051】
【図1】図1は、キモトリプシン断片を含むフィブロネクチン分子の概略図表示を提供する。
【図2】図2は、以下の実施例1でさらに記載したように、TKNEOベクターを用いた、フィブロネクチン断片の存在下での、投入された(comitted)ヒト前駆細胞の感染効率を表す。
【図3】図3は、以下の実施例1でさらに記載したように、TKNEOベクターを用いた、フィブロネクチン及びその断片の存在下での、様々な投入されたヒト造血前駆細胞の感染効率を比較する。
【図4】図4は、以下の実施例7でさらに記載したように、(i)共培養方法(レーン2−4)、(ii)固定化フィブロネクチン断片存在下での上清感染(レーン5−7)、及び(iii)BSA上での上清感染(レーン8−10)によって形質導入された骨髄細胞を移植されたマウスの、hADAの存在を比較する。hADAの対照はレーン1及び12に示され、ネズミADAに対する対照はレーン11に示される。
【図5】図5は、以下の実施例8でさらに記載したように、フィブロネクチン断片へのウイルス結合を示す。
【図6】図6は、以下の実施例8でさらに記載したように、フィブロネクチン断片への、レトロウイルスの結合が、濃度依存的であることを示す。
【図7】図7は、以下の実施例9−11で用いた、様々な組換えフィブロネクチン断片を図示している略図を提供する。
【図8】図8は、以下の実施例9で記載したように、いくつかの組換え断片を含む、様々なフィブロネクチン断片に結合するレトロウイルスを示す。
【図9】図9は、以下の実施例9で記載したように、フィブロネクチン断片と結合するレトロウイルスを、ヘパリンが阻止することを示す。
【図10】図10は、以下の実施例10でさらに報告したように、様々なフィブロネクチン断片の存在下での、ネズミ造血細胞のレトロウイルス感染効率を示す。
【図11】図11及び12は、以下の実施例11で記載するように、(i)共培養法、(ii)様々なフィブロネクチン断片上での上清感染、及び(iii)BSA上での上清感染によって形質導入された骨髄細胞を移植されたマウスにおける、hADAの存在を比較している。
【図12】図11及び12は、以下の実施例11で記載するように、(i)共培養法、(ii)様々なフィブロネクチン断片上での上清感染、及び(iii)BSA上での上清感染によって形質導入された骨髄細胞を移植されたマウスにおける、hADAの存在を比較している。
【図13】図13は、フィブロネクチンのα−鎖の構造と、実施例で用いた組換え断片とのその関連とを示している。フィブロネクチンI型、II型、及びIII型リピートが示され、III型リピートは1〜14まで番号付けした。細胞への3つの結合部位は、細胞結合部位(CBD)に対してはCELL、ヘパリン結合部位(HBD)に対してはHEPARIN、そしてオルタナティブにスプライシングされたIIICS部位のはじめの25アミノ酸によって形成された、VLA−4結合部位CS−1に対してはCS−1として表示する。
【図14】図14は、以下の実施例12でさらに報告したように、様々なフィブロネクチン断片の存在下での、NIH/3T3細胞のレトロウイルス感染の効率を示す。
【図15】図15は、以下の実施例12でさらに報告したように、様々なフィブロネクチン断片の存在下で、非接着性HL60細胞のレトロウイルス感染の効率を示す。
【図16】図16は、フィブロネクチンへ結合するレトロウイルスに対する、低分子量及び高分子量ヘパリンの影響を示す。
【図17】図17は以下の実施例13で議論したように、組換えフィブロネクチン断片の存在下で、CD34+細胞集団内の様々な型の前駆細胞のレトロウイルス感染の効率を示す。
【図18】図18は、以下の実施例14で議論したように、組換えフィブロネクチンの存在下で、C−kit+細胞集団中の、HPP−CFC細胞のレトロウイルス感染の効率を示す。
【図19】図19は、以下の実施例15で議論したように、ヘキサジメトリンブロミドの濃度を増加させるに従って、NIH/3T3細胞のレトロウイルス感染効率が減少することを示す。
【図20】図20は、以下の実施例15で議論したように、ヘキサジメトリンブロミドの濃度を増加させるに従って、クローン原性骨髄細胞のレトロウイルス感染効率が減少することを示す。
【図21】図21は、T細胞がフィブロネクチン受容体を発現することを示す、フローサイトメトリー試験の結果を示す。
【図22】図22は、ヒトT細胞の前刺激を示すフローサイトメトリー試験の結果を示す。
【図23】図23は、ADAイソ酵素アッセイによる、ヒトT細胞への遺伝子導入の効率の解析の結果を示す。
【図24】図24は、さらに実施例16で記載する、感染プロトコールで用いたポリペプチド類を示す略図を提供する。
【図25】図25は、図24で同定した、ペプチド類C−FGF、C−COL、及びbFGFの存在下で、NIH/3T3細胞の形質導入の効率を示す図である。
【図26】図26は、フローサイトメトリー解析でアッセイしたように、図24で同定したペプチドの存在下で、HEL細胞へのレトロウイルス遺伝子導入の効率を示す。
【図27】図27は、図24で同定したポリペプチドの存在下で、CD34+骨髄細胞の形質導入の効率を示す図である。
【発明を実施するための形態】
【0052】
本発明は好ましくは以下の形態を含む。
1. レトロウイルスによって形質転換された生存可能な細胞集団を得る方法であって:
細胞に結合するリガンドおよびレトロウイルスに結合するリガンドを含む、有効に固定化される量の物質の存在下で、細胞をレトロウイルスに感染させ、よってレトロウイルスおよび細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ここにおいて前記感染は本質的にヘキサジメチリンブロミドを含まない培地で行われる:
ことを含む、前記の方法。
【0053】
2. 細胞が造血幹細胞を含む、態様1記載の方法。
3. 態様1記載の方法によって製造される生存可能な細胞集団。
4. 造血幹細胞を含む、態様3記載の生存可能な細胞集団。
【0054】
5. 態様1記載の方法によって製造される生存可能な細胞集団を哺乳動物に移植する:ことを含む、細胞移植方法。
6. 実質上レトロウイルスで形質転換された生存可能な細胞集団を含み、レトロウイルス産生細胞およびヘキサジメチリンブロミドの両方を本質的に含まない:細胞組成物。
【0055】
7. 前記の生存可能な細胞が造血幹細胞を含む、態様6記載の細胞組成物。
8. 態様6記載の細胞組成物を哺乳動物に移植する;ことを含む、細胞移植方法。
9. 細胞集団が造血幹細胞を含む、態様8記載の方法。
10. レトロウイルスによって形質転換された生存可能な細胞集団を得る方法であって:
細胞と結合するリガンドおよびレトロウイルスと結合するリガンドを含む、有効に固定化される量の物質の存在下で、細胞をレトロウイルスに感染させ、よって、レトロウイルスと細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ここにおいて前記感染は、共培養中ではレトロウイルスによる細胞を形質転換する効率を上昇させるが、前記物質存在下ではレトロウイルスによる細胞の形質転換の効率を減少させる薬剤を実質上含まない培地で行われる:
ことを含む、前記の方法。
11. レトロウイルスによって形質転換された生存可能な細胞の集団を得る方法であって:
細胞を結合するアミノ酸配列、およびレトロウイルスと結合するコラーゲンVまたは繊維芽細胞増殖因子からのアミノ酸配列を含むポリペプチド存在下で、細胞をレトロウイルスに感染させる:
ことを含む、前記の方法。
12. 前記の感染が、レトロウイルス−プロデューサー細胞の非存在下で、レトロウイルスと細胞を接触させることを含む、態様11記載の方法。
13. T細胞をレトロウイルスで形質転換する方法であって、T細胞と結合するリガンド、およびレトロウイルスと結合するリガンドを含む物質存在下で、細胞をレトロウイルスに感染させ、よって、レトロウイルスと細胞を共に配置し、そして細胞の形質転換効率を上昇させる、ことを含む、前記の方法。
14. 物質が、T細胞と結合する第一のアミノ酸配列、およびレトロウイルスと結合する第二のアミノ酸配列を含むポリペプチドであって、第二のアミノ酸配列が、配列:
【0056】
【化2】
【0057】
またはそれと充分に類似するアミノ酸配列を持ち、レトロウイスルと結合する能力を示すものである;
態様13記載の方法。
15. レトロウイスルを結合するコラーゲンVまたは繊維芽細胞増殖因子からのアミノ酸配列を持つ、有効量の単離されたポリペプチドとレトロウイルスを接触させることを含む、レトロウイルスを配置する方法。
【実施例】
【0058】
本発明のさらなる理解、及び評価を促進するために、以下に続く固有の実施例を提供する。これらの実施例は、説示的なものであり、その本質を限定しているものではないことが理解されよう。
【0059】
実施例1 TKNEOを用いた骨髄細胞への遺伝子導入
1.1.ウイルス上清の調整
レトロウイルスプラスミドTKNEOベクターを含むGP+EnvAM12産生細胞(Markowitz et al. (1988) Virology 167:400 参照)を、10%胎児子牛血清(FCS、Hyclone, Logan UT)、及び100 units/ml ペニシリン、及び100 microgram/ml ストレプトマイシン(P/S、双方 Gibco)を含むIscove’s Modified Dulbeccos Medium (IMDM、Gibco, Gaithersburg,MD)中で培養した。ウイルスを含む上清を、20%FCSを含むIMDM10 ml を加えて一晩プレートをコンフルエンスにすることによって回収する。回収した培養液を、0.45ミクロンフィルター(Gelman Science, Ann Arbor, MI)を通して濾過し、使用まで−80℃にて保存した。
【0060】
1.2.フィブロネクチン断片の用意
FNを、FNを4M尿素で溶出する前に、ゼラチン−アガロースカラムを1M尿素で洗浄した以外は、すでに、 Ruoslahti et al., Methods. Enzymol. 82:803−831 (1982) で記載されているように、ヒト血漿(Lifesource, Glenview,IL)から精製した。精製したFNは、4℃にて,10mM 3−(シクロヘキシルアミノ)−1−プロパン−スルホン酸(CAPS)、150mM NaCl、2mM CaCl2 pH 11.0に対して透析し、−80℃にて等分して保存した。FNのキモトリプシン様細胞結合部位(CBD)(CS−1)、及び、ヘパリン−II結合断片は、すでに記載されているように(Ruoslahti et al. (1982), supra, Patel and Lodish, J. Cell. Biol. 102, pp. 449−456 (1986), and Bernardi et al., J. Cell. Biol. 105, pp. 489−498 (1987))精製した。三つの主なヘパリン断片(30kD、35kD、42kD)が、ヘパリン−アガロースカラムからの1M NaCl中の溶出で得られた。これらのヘパリン結合断片を、さらに精製するために、1M NaCl溶出物を4℃にて1晩、10mM Tris−HCl pH7.0に対して透析し、10mM Tris−HCl pH7.0で平衡化された陽イオン交換カラム(2ml DEAE セファロース fast flow, (Pharmacia Fine Chemicals, Uppsala, Sweden) /mg タンパク質)に通した。30/35kD断片を、非結合フラクションで回収し、一方42kD断片はカラムより100mM NaClにて溶出した。FN 500mg から、おおよそ26mgの30/35kD断片、及び4mgの42kD断片を得た。30/35kD断片でなく、42kD断片は、ウエスタンブロッティング手法によって決定したように、フィブリン結合部位に対する抗体によって認識された。また、42kD断片は、フィブリン−セルロースアフィニティーカラムへ結合する。
【0061】
感染プロトコールで使用するために、フィブロネクチン断片を Patel and Lodish (1986), supra によって記載されたように、75pmol/cm2の濃度にて、35、もしくは100mmペトリディシュ(Falcon,Lincoln Park,NJ)に固定化した。コントロールのプレートは、2%(FNなし)ウシ血清アルブミン(BSA,Boehringer Mannheim, Mannheim,Germany)を用いて類似の方法でコートした。
【0062】
1.3.レトロウイルス感染プロトコール
健康な大人提供者からの、骨髄サンプルを、the Institutional Review Board of Indiana University School of Medicine によって立証されたプロトコールに従った、無菌保存剤なしの培養液硫酸ヘパリンを含むチューブに集めた。低密度単核細胞を、フィコール−ハイパック(密度 1.077 g/ml, Pharmacia, Piscataway, NJ)において、25℃にて45分間遠心することで用意した。プラスチック接着細胞を、低密度骨髄細胞から2%FCS入りIMDM中、5%CO2中で37℃にて4〜16時間さらに組織培養プレートをインキュベートすることで取り除いた。
【0063】
接着陰性低密度単核細胞は、すでに、Luskey er al. (1992) Blood 80:396 に記載されたように、レトロウイルス感染前にペトリディシュ上1×106 cells/ml の細胞密度で、20%FCS、100 U/ml rhIL−6、100 ng/ml rhSCF(両方 Amgen,Thousand Oaks, CA)、及びP/Sを含むIMDM中、5%CO2、37℃で48時間前刺激した。前刺激した細胞を、プラスチックへ弱い接着をする細胞を取り除くために、勢いよくピペッティングすることで回収した。
【0064】
前刺激した細胞(5×105 cells/ml)を、BSA(コントロールプレート)、もしくはフィブロネクチンもしくはその断片(プラスチックプレートへタンパク質がより接着するようにUV照射した。)でコートされたプレート上で6時間インキュベートし、成長因子(上記の通り)、及び7.5 microgram/ml ポリブレン(Aldrich Chemical, Milwaukee, WL)存在下、ウイルス上清にて感染させた。ウイルス上清は、2時間後置き換え、(成長因子、及び5.0 microgram/ml ポリブレンを含むもの)、細胞をさらに12〜24時間インキュベートした。非接着細胞はそれぞれの培養液交換で再び加えた。
【0065】
感染プロトコールに従って、非接着細胞をデカントし、接着造血細胞は製品の使用説明書に従って、細胞解離緩衝液(CDB)(酵素なし/PBSベース、Gibco)を用いて回収した。接着細胞を、非接着細胞フラクシャンに加え、2回洗浄し、計数した。回収した細胞は、クローン原性メチルセルロース前駆体分析を行うか、あるいは長期骨髄培養を行った。
【0066】
1.4.長期骨髄培養
LTC−IC(ヒト幹細胞)分析を、すでに記載された方法に従って、若干の改良をして行った。Sutherland et al. Blood 74:1563 (1989)。簡単には10%FCS、10%ウマ血清(Sigma)及びP/S、1×10−5Mヒドロコルチゾン(Upjohn, Kalamazoo,MI)、及び、6ウェル組織培養プレート上に、320モスモル塩化ナトリウム(Costar, Cambridge,MA)を含む5 ml IMDM中でのコンフレントな(上記のような)前照射を受けた同種ヒト骨髄繊維芽細胞(BMF)において、0.5−1×106の感染細胞を播種した。LTMCは、33℃で5%CO2、及び毎週培養液及び非接着細胞の50%を取り去ることで栄養を与えてインキュベートした。5週間後、LTC−IC培養は、BMFから、接着造血細胞を取り除くために、CDBを用いて回収した。非接着、及び接着造血細胞双方を一緒にし、LTC−ICから由来するコロニーを得るために、メチルセルロースにプレートした。
【0067】
1.5.クローン原性メチルセルロース分析
メチルセルロース分析は、すでに Toksoz et al., Prc. Natl.Acad. Sci., USA, Vol. 89, p7350 (1992) で記載されたものに、若干の改良を加えて行った。2.5×104感染大人骨髄細胞を、25%FCS、10%ヒト血漿、10−5Mベータ−メルカプトエタノール(Sigma)、及びP/Sを含む1 mlの2.4%IMDMメチルセルロース(Fluka, Ronkonkoma, NY)中で、5 units/ml エリスロポエチン(Epo, Amgen)、100 ng/ml rhSCF、10 ng/ml rhIL−3(Genzyme, Cambridge, MA)とともにプレートした。培養は、37℃にて、5%CO2/95%空気でインキュベートし、コロニー(>50細胞数)を13日目に反転顕微鏡で見ることで、CFU−GM(顆粒球及びマクロファージを含む)、CFU−混合(骨髄及び赤血球成分を含む)、BFU−E(赤血球成分のみを含む)として計数した。
【0068】
1.6.レトロウイルス感染分析
TKNEOウイルスによる感染効率は13日目の1.5 mg/ml(乾燥粉、Gibco)G418抵抗性メチルセルロ−スコロニ−の割合を決めることで解析した。擬感染は、骨髄を組換えウイルスをつくらないGP+EnvAM12パッケージング株上でインキュベートすることでそれぞれの実験で行った。1.5 mg/ml G418とのこれらの擬感染細胞の培養は一貫して、<1%バックグラウンドコロニーを示す。
【0069】
1.7.前駆細胞への遺伝子導入効率
形質導入効率は、30/35 FN−、もしくはBSAコートディッシュにまき、骨髄細胞に感染することで比較した。これらの状態間では、選択なしの感染の後に得られたコロニー数においては、違いが観察されなかった。図2は感染後のG418rコロニーの割合を示す。多系統(CFU−混合)前駆細胞と同様に、系統制限(BFU−E、及びCFU−GM)から由来するものを含む、すべての前駆体型からの、30/35 FN上で高い割合のG418rコロニーが確認された。すべての前駆体への感染はBSAに比べ、30/35 FNで9倍増加した。
【0070】
1.8.長期培養イニシエーション細胞への遺伝子導入
LTC−ICへの遺伝子導入はTKNEOベクターを用いて行った。コロニー由来LTC−ICへの遺伝子導入は、30/35 FN上での感染後、検出されたのみである。(30/35 FN対BSA 16%G418r対0%G418rコロニー)。
【0071】
1.9.造血細胞の感染効率への30/35FN効果の特異性
30/35 FNにおいて見られた遺伝子導入効率の増加の特異性を決めるためにTKNEOによる感染を、BSA、30/35 FN、無傷の(intact)フィブロネクチン、RGDSテトラペプチド配列を含む中心細胞結合部位(CBD)を含む115kD FN断片、CS−1配列は欠損しているが、ヘパリン−II結合部位を持つことによって特徴づけられる42kD C−末端FN断片(42FN)上で行った(図1)。BSA上での感染は3±1%G418r BFU−E、1±1% G418r CFU−GM、及び0±0% G418r CFU−混合を示した。CBD上で観察されたG418rコロニー群は明らかな増加が見られず、一方42 FN上ではBFU−Eのわずかに高い感染(6.0±−1%)が見られた(図3)。しかしながら、無傷のFNはすべての前駆体への遺伝子導入の増加を促進した。無傷のFN上での感染後のG418rコロニーの割合は30/35FN上のものより少なかったが、それぞれBFU−E(16±−2対24±4%)、CFU−GM(5±2対20±4%)、及びCFU−混合(6±1対9±1%)(無傷FN対30/35FN)であった。
【0072】
実施例2 PGK−mADAを用いた骨髄細胞への遺伝子導入
2.1.全体的な手順
PGK−mADAウイルス上清は実施例1のTKNEOのための記載と同様に準備した。フィブロネクチンのキモトリプシン様断片は(図1)、すでに実施例1で記載のように準備し、実施例1のレトロウイルス感染プロトコールに従った。LTC−IC(ヒト幹細胞)分析、及びメチルセルロース分析は実施例1に従って行った。
【0073】
2.2. レトロウイルス感染の分析
ADAイソ酵素電気泳動によるタンパク質分析によって、PGK−mADAベクターの感染効率を定量した。個々の前駆(progenitor)細胞コロニーの分析は、Moritz(1993)およびLimら(1989)、Proc.Natl.Acad.Sci.USA,86巻、8892頁に前記した方法に従って、行なわれた。トランスファー効率を厳密に分析するために、内在性ヒトADAと同じか、またはより高レベルのmADAを発現するコロニーのみを形質転換されたと考えた。プールしたコロニーを分析するために、メチルセルロース培地より選択(pick out)したコロニーを、1.5mlのミクロチューブ(Rainin,Woburn,MA)内に集め、暖めた培地およびリン酸緩衝塩類溶液(PBS)で洗浄し、遠心分離し、そして−20℃に保存した。ADA分析のため、凍結−融解サイクルを繰り返すことによって、5μlの溶解バッファーに細胞を溶解し、そしてイソ酵素電気泳動を、前記の方法に従って行った。
【0074】
2.3. 前駆細胞内への遺伝子トランスファー効率
30/35FN−またはBSA−コート皿上にプレートした間、骨髄細胞に感染させることによって、形質転換効率を比較した。選別しなかった場合、感染後得られたコロニー数の差は、これらの条件間では認められなかった。表1に示したように、すべての前駆細胞内への感染効率は、実質上、BSAに対して30/35FN上で増加した。高力価(〜1x107ビリオン(virons)/ml)ベクターで予想されるように、前駆細胞の形質転換効率は、非常に高かった。表1に示すように、30/35FN上での骨髄へのPGK−mADAの感染は、2回の別々に行った実験で、前駆細胞の100%近い形質転換が得られた。
【0075】
【表1】
【0076】
2.4. 長期培養−始原細胞(initiating cell)内への遺伝子トランスファー効率
PGK−mADAで行った4回の独立した実験では、週齢5週のLTMCから誘導された前駆細胞コロニー(即ち、LTC−IC誘導コロニー)の有意な割合が、トランスファーされたマウスADA遺伝子を発現していた。発現は、分析したコロニーの2/12(17%)から6/6(100%)までの範囲であった(表2)。導入されたmADA遺伝子の発現は、陽性と考えられるすべてのコロニー内で、内在性ヒトADA活性量を越えるか、または少なくとも等しかった。PGK−mADAの感染効率は、TKNEOより高かった。表2に示すように、30/35FN上の骨髄へのPGK−mADAの感染では、2回の別々の実験で、前駆細胞の100%近い形質転換が得られた。
【0077】
【表2】
【0078】
2.5. 造血細胞の感染効率への30/35FN効果の特異性
LTC−IC内への遺伝子トランスファー効率は、30/35FN上で増加した。これらの二次のLTC−IC誘導コロニーは比較的小サイズであるため、単一コロニーでタンパク質分析を成し遂げる能力には限界がある。BSA上でのPGK−mADAでの感染後、完全な(intact)フィブロネクチンならびに42FN 0/6、0/4および0/3 LTC−IC誘導コロニーは、それぞれ、マウスADAの発現を示したが、30/35FN上で感染させた3/5LTC−IC−誘導コロニーは、mADAを発現した。さらに、複数のLTC−IC−誘導コロニーを2回の追加実験で分析する前にプールしたところ、mADA発現は、30/35FN上で感染後のみに検出され、完全なFN上ではより少ない程度であり、42FNまたはBSA上では検出されなかった。
【0079】
実施例3 PGK−hADAを用いた骨髄細胞内への遺伝子トランスファー
3.1. 一般的方法
実施例1のTKNEOで記載した方法に従って、PGK−hADAウイルス上清を調製した。実施例1に前述した方法に従って、フィブロネクチンのキモトリプシンフラグメント(図1)を調製し、次に実施例1のレトロウイルス感染プロトコールを行った。LTC−ICおよびメチルセルロースアッセイを、実施例1記載の方法に従って行った。
【0080】
3.2. レトロウイスル感染の分析
プールしたコロニーを分析するために、メチルセルロース培地より選択したコロニーを1.5mlのミクロチューブ(Rainin,Woburn,MA)に集め、暖めた培地およびPBSで洗浄し、遠心分離し、そして−20℃で保存した。ADA分析のために、細胞を、凍結−解凍サイクルを繰り返すことによって5mlの溶解バッファーに溶解し、そして前述の方法に従って、イソ酵素を電気泳動した。
【0081】
実施例4 TKNEOを用いた臍帯血細胞内への遺伝子トランスファー
4.1. 一般的方法
TKNEOウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(図1)は、実施例1に前述した方法に従って、調製された。次いで、実施例1に記載のレトロウイルス感染プロトコールを行ったが、骨髄細胞の代わりに、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineで是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−IC(ヒト幹細胞)およびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0082】
4.2. 前駆細胞の遺伝子トランスファー効率
FN30/35上での感染は、3回の別々の実験において、BSAと比較して4倍以上増加していた(表3)
【0083】
【表3】
【0084】
実施例5 PGK−mADAを用いた臍帯血細胞内への遺伝子トランスファー
5.1. 一般的方法
PGK−mADAウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(図1)は、実施例1に前記された方法に従って、調製された。次いで、実施例1記載のレトロウイルス感染プロトコールを行ったが、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineに是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−ICおよびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0085】
5.2. 長期培養した始原細胞内への遺伝子トランスファー効率
より力価の高いPGK−mADAを用いると、LTC−IC−誘導コロニーの分析は、FN30/35上で上清を用いて感染させた臍帯血から確立された培地からのみ、導入したmADA cDNAの高レベル発現を示した。BSA対照プレート内で感染させたLTC−IC誘導コロニー内では、mADAの発現はほとんど検出されなかった。
【0086】
実施例4および5に示した結果は、FN30/35を用いた感染効率の改善が、臍帯血前駆細胞および幹細胞を用いた場合にも達成できることを、証明している。
実施例6 PGK−hADAを用いての臍帯血細胞内への遺伝子トランスファー
PGK−hADAウイルス上清およびフィブロネクチンのキモトリプシンフラグメント(表1)は、実施例1のTKNEOに記載の方法に従って、調製された。次に、実施例1のレトロウイルス感染プロトコールを行ったが、骨髄細胞の代わりに、正常な満期産新生児からの臍帯血を、the Institutional Review Board of Indiana University School of Medicineに是認されたプロトコールに従って、ヘパリン含有チューブに集めて用いた。LTC−ICおよびメチルセルロースアッセイは、実施例1に記載の方法に従って、行われた。
【0087】
実施例7−11 実施例7−11のためのレトロウイルスベクターおよび産生細胞(producer cell)系
実施例7−11に関して、2つのレトロウイルス産生パッケージング細胞系(retrovirus−producing packaging cell line)を用いた:それぞれ、エコトロピック(ecotropic)GP+E86(Markowitz,D.,S.GoffおよびA.Bank,J.Virol.,62,1120−1124、1988)ならびにアンホトロピック(amphotropic)GP+envAM12細胞系(Markowitz,D.,S.GoffおよびA.Bank,Virology,167,400−406,1988):ここに記載された研究に用いられたレトロウイルスベクターおよび産生細胞クローンを、表4に列挙した。
【0088】
【表4】
【0089】
すべての細胞系は、EAL2a細胞を除き、10%のウシ胎児血清(FCS,Hyclone,Logan,UT)および100ユニット/mlのペニシリンおよび100μg/mlのストレプトマイシン(P/S、両方共Gibco)を含むダルベッコの修飾Eagle培地(DME,Gibco,GrandIsland,NY)内で培養され、EAL2a細胞は、10% FCS+P/Sを含むDME−F12(Gibco)内で増殖させた。ウイルスを含む上清は、マウス細胞には10mlのα−最少必須培地(αMEM,Gibco)、またはヒト細胞にはイスコフのダルベッコ培地(IMDM,Gibco)(それぞれ10%のFCS+P/Sを含む)を10cmの集密プレートに一晩加えることによって、集められた。収集した培地を、0.45ミクロンフィルター(Gelman Sciences,Ann Arbor,MI)でろ過し、使用するまで−80℃に保存した。
【0090】
実施例7 マウスの一次造血細胞の形質転換
7.1. 実験方法
マウスの細胞で研究するために、150mg/kgの5−フルオロウラシルの投与後、2日の週齢6から8週のC3H/HeJマウスの大腿骨および頸骨から、骨髄を収集した(SoloPack Laboratories,Franklin Park,IL)(Lim,B.,J.F.Apperley,S.H.OrkinおよびD.A.Williams,Proc.Natl.Acad.Sci.USA,86、8892−8896、1989)。細胞は、IMDM/20%FCS+P/S中の濃度5x106細胞/ml、並びに100ng/mlのラット組換え幹細胞因子(rrSCF;Amgen.Thousands Oaks,CA)および100ユニット/mlのヒト組換えインターロイキン−6(rhIL−6;Pepro Tech Inc.,Rock Hill,NJ)で、48時間、前刺激(prestimulate)した(Luskey,B.D.,M.Rosenblatt,K.ZseboおよびD.A.Williams,Blood,Vol.80,pp.396−402、1992)。次に、EPHA−5産生細胞によって作り出されたPGK−hADAベクターでの遺伝子トランスファー効率を、3種類の異なる感染プロトコールを用いて比較した:1)上清感染;2)FN30/35上での上清感染;3)EPHA−5産生細胞上での同時培養。そのために、100mmの細菌皿を、5mlのリン酸緩衝溶液(PBS;Gibco)中に溶解した2.5μg/cm2のFN30/35(75pmol/cm2に等しい)で、室温、UV光下で、1時間、皿のフタを開けたままで、さらに1時間、皿のふたを閉めてコートした。2%のウシ血清アルブミン(BSA,Fraction V:Boehringer Mannheim,Indianapolis,IN)で、室温で30分間、ブロックした後、皿を、2.5%(v/v) 1M Hepesを添加したハンクス液(Hank’s Balanced Salt Solution)(HBSS)(両方共Gibco)で、一度洗浄した。上清感染では、皿をBSAのみでコートした。5x106の前刺激したドナー細胞を、EPHA−5細胞より上記の方法に従って得られた10mlのウイルス上清に、100U/mlのrhIL−6、100ng/mlのhrSCFおよび7.5μg/mlのポリブレン(polybrene)を添加したものと共に、インキュベートした。非粘着性細胞を集め、新たにウイルス上清を再付加した。共培養のために、4ml培地中のEPHA−5細胞を、10μg/mlのマイトマイシンCと共に、2時間、37℃でインキュベートし、洗浄し、トリプシン処理し、そして10mlのαMEM/20%FCS+P/S中、3x106の細胞濃度で、100mmの組織培養皿上に接種した。翌日、5x106の前刺激した骨髄細胞を、100U/mlのrhIL−6、100ng/mlのrrSCFおよび4μg/mlのポリブレンと共に、48時間加えた。感染プロトコールに次いで、非粘着性細胞をデカントし、そして、粘着性造血細胞を、製造者の指示書に従って、Cell Dissociation Buffer(CDB)(酵素非含有/PBSをベースとする、Gibco)を用いて、培養液から収集した。粘着性細胞を非粘着性画分に加え、二度洗浄し、そして、おおよそ1mlのHBSS/Hepes中に懸濁した。5x106の前刺激した細胞から得られた全細胞を、致死量の全身照射(700+400cGy、137Cs−源)(Luskey,B.D.,M.Rosenblatt,K.ZseboおよびD.A.Williams,Blood,Vol.80,396−402、1992)を受けた3匹のレシピエントマウスの尾静脈内に注射した。造血幹細胞の形質転換は、導入されたヒトADA cDNAの発現用に再構築されたマウスを試験することによって、分析された。このADAイソ酵素分析は、原位置での酢酸セルロースのin vivo酵素分析によって、hADAタンパク質の存在について、末梢血液細胞を試験することによって、移植されたマウス内で成し遂げられた(Lim,B.,D.A.WilliamsおよびS.H.Orkin,Mol.Cell.Biol.,7,3459−3465、1987)。試験は、最初4ヶ月間、追加移植を行い、月に一度繰り返した。
【0091】
7.2. 結果
遺伝子操作された造血幹細胞でのマウスの骨髄再構築が長期に及ぶことは、一般的に、4ヶ月の追加移植期間後の幹細胞の形質転換効率を定量するために、適当であるとして受け入れられる。形質転換骨髄の7ヶ月後のレシピエントのイソ酵素分析による分析は、1)ヒトADA cDNA発現は、同時培養またはFN30/35上での上清感染のいずれかを用いると存在するが、FN30/35なしでの上清感染後移植したグループでは、存在せず、そして2)発現のレベルは、共培養とFN30/35グループで同等である:ことを示した。図4、列2−4に示したように、EPHA−5細胞上での同時培養によって形質転換された骨髄を移植した3匹のマウスでは、ヒトADAが容易に検出できることを証明した。同様のレベルのヒトADAは、FN20/35の上清感染によって形質転換された造血細胞を移植した3匹のマウスでも検出された(図4、列5−7)。これに対して、BSA上での上清感染によって形質転換された造血細胞を移植した3匹のマウスでは、ヒトADAは検出されなかった(図4、列8−10)。ヒトADAの位置コントロールを図4の列1および12に、ならびにマウスのADAを列11に示す。列2−10のマウスのバンドは、同量のタンパク質が負荷されたことを示す。これらのデータは、FN30/35上での上清感染によって長期にわたって再構築される造血幹細胞の形質転換が 同時培養と等価であり、FN30/35なしでの上清感染より遥かに優れていることを証明している。
【0092】
実施例8 TN30/35に結合したレトロウイルスベクターによる改良された形質転換メカニズム
8.1. 実験方法
形質転換の増加が、ウイルスと造血細胞を共に配置する(co−localization)結果であるか否かを試験するため、我々は、組換えレトロウイルス粒子がFN30/35との結合を示すか否かについて分析した。そのため、FN30/35−コートプレートを、TKNeoウイルスを含む上清と共に、30分間プレインキュベートし、その後、広範囲にわたって洗浄した。上清のウイルス力価をNIH/3T3を用いて、標準的方法に従って、定量した(Markowitz,D.,S.GoffおよびA.Bank,J.Virol.,62,1120−1124、1988)。簡単に言えば、3T3細胞を、6穴の組織培養プレートに、濃度1000細胞/穴でプレートし、一晩増殖させた。ウイルス上清の一連の希釈物を、7.5μg/mlのポリブレンと共に、それぞれの穴に加え、そして2mlの培地を加えた後、37℃で2.5時間インキュベートした。24時間後、培地は、G418(0.75mg/ml、乾燥粉末、Gibco)を含む培地と置き換え、そしてプレートを10−12日間インキュベートした。G418−耐性コロニー(G418r)を10−12日後に染色し、そして数を記入した。ウイルス上清の希釈倍率を掛けたコロニー/穴の数を、上清の感染粒子(cfu)/mlとして用いた。我々は、ウイルス上清とプレインキュベーションし、そして1000NIH/3T3/35mm細菌皿細胞+ポリブレンを加えることによって集中的に洗浄した後、FN30/35−コートしたまたはBSA−コートした35mmプレート上に残っているレトロウイルス粒子の量を、評価/”滴定”した。24時間後、培養液に、0.75mg/mlのG418(乾燥粉末)を含む培地を供給し、そしてさらに細胞を10−12日間インキュベートした。このインキュベーションの後、G418−耐性NIH/3T3コロニーを数えることによって、粘着性ウイルスの存在を定量した。
【0093】
FN30/35へのウイルスの結合が、服用量依存様式で起こるか否かを評価するために、皿をコートするFN30/35の濃度を上昇させて、上記の実験を繰り返した。そのため、上記の方法に従って、35mmの細胞皿を、1、4、10および20μg/cm2のFN30/35でコートした。前もって1x104感染粒子/mlと滴定されたTKNeoウイルスストックの1:50希釈物を、FN30/35−コートプレート上で30分間インキュベートした。集中的に洗浄した後、2000NIH/3T3細胞をそれぞれの穴に加えた。選別を上記の通り行い、そしてG418−耐性NIH/3T3コロニーを、選別の10日後、計数した。
【0094】
8.2. 結果
図5は、3回の実験の内の典型的な一つの結果を示す。TKNeo上清を用いて、NIH/3T3細胞内のG418rコロニーによって測定されたレトロウイルスの力価は、BSA−コートプレート上では、3logより多く(4x103から0)減少したが、これに対して、30/35FNでコートしたプレート上では、力価の減少は、1logのみであった。これらのデータは、レトロウイルスは、FN30/35と定量的に結合するが、しかしBSAでコートした皿(対照皿)とは結合しないことを、証明している。図6は、ウイルス−含有上清を、FN30/35の濃度を増加させてコートしたプレート上でインキュベートした場合、G418−耐性コロニーの数の増加が検出されたことを示している。このように、FN30/35へのウイルスの結合は、服用量依存様式で起こっている。
【0095】
実施例9 組換えフィブロネクチンフラグメントへのウイルスの結合
9.1. 実験方法
Kimizukaらは、以前、大腸菌内にクローン化したFN DNA配列の発現について報告している(Kimizuka,F.,Y.Tatuchi,Y.Ohdate,Y.Kawase,T.Shimojo,D.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)。クローン化したキメラペプチドは、細胞粘着性に関係することが知られているフィブロネクチン内の様々な重要配列(RGDS、CS−1およびヘパリン結合部位)の内の一つまたはその組み合わせを含む(図7参照のこと)。レトロウイルスがこれらの組み換えFNフラグメントと結合することができるか否かを分析するために、3T3細胞コロニー形成アッセイを、1x104感染粒子/mlの凍結レトロウイルスTKNeoストックの2つの異なる希釈物(1:10および1:100)を用いて、組換えフラグメントC−274、H−271、H−296、CH−271、CH−296およびC−CS1ならびに陽性対照としてFN30/35でコートしたプレート上で繰り返した。FNフラグメントは、120−130pmol/cm2の濃度(C−274、H−271、H−296、C−CS1、FN30/35では4μg/cm2に等しく、CH−271およびCH296では8μg/cm2に等しい)で用いられた。簡単に言えば、プレートをコートし、ウイスルを加え、プレートを広範囲に洗浄し、NIH/3T3細胞を24時間加え、そして10日間選択培地内で増殖させ、その後、コロニーを染色し、数を数えた。
【0096】
9.2. 結果
図8は、G418−耐性コロニーの数(およびそのウイルス粘着性)が、フラグメントH−271、H−296、CH−271およびCH−296で増加したことを示している。さらに、図8は、ウイルス結合量が、これら組み換えフラグメントとFN30/35の間では概略的に比較できるが、この仕事中では、CH−271フラグメントが最も高いレベルのウイルス結合を示したことを、示している。これらの5FNフラグメントのすべてに共通なのは、高アフィニティーのヘパリン結合部位を含むタイプIIIリピート12−14であり(Ruoslahti,E.,Ann.Rev.Biochem.,57,375−413、1988、およびKimizuka,F.,Y.Taguchi,Y.Ohdate,Y.Kawase,T.Shimojo.K.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)、多分、リピート13に位置する(Kimizuka,F.,Y.Taguchi,Y.Ohdate,Y.Kawase,T.Shimoko,K.Hashino,I.Kato,K.SekiguchiおよびK.Titani,J.Biochem.,110,284−291、1991)。このことは、ウイルス結合が、この既知の粘着部位を通して起こることを示唆している。このことは、4μg/cm2のCH−271でコートした皿を、ヘパリン結合部位との細胞結合を阻害することが知られている高荷電分子である、硫酸ヘパリンの濃度を上昇(10−1000μg/ml)させて、プレインキュベーションすることによって、証明された。図9に示したように、G418−耐性コロニーの数は、高濃度の硫酸ヘパリンと共にCH−271をプレインキュベーションした後には、減少している。これらのデータは、FNへのウイルス結合が、FNのカルボキシ末端ドメイン内のCS−1部位のすぐ隣りに位置する高アフィニティーのヘパリン結合部位を通して仲介されることを、示唆している。
【0097】
実施例10 組換えフィブロネクチンフラグメント上での造血細胞の形質転換
10.1. 実験方法
前記したように、FN30/35上での造血細胞の形質転換の上昇が、組換えFNフラグメントでもまた認めることができるか否かを分析するために、我々は、高増殖潜在性−コロニー形成細胞(high proliferative potential− colony forming cell)(HPP−CFC)アッセイを用いて、インビトロでの上清感染の形質転換効率を評価した。EAL2aベクターを用いることによって、我々は、BSA上での様々な組換えFNフラグメント対FN30/35の上清感染の、造血細胞の形質転換への影響を、遺伝子トランスファーのインジケーターとしてG418−耐性コロニーの増殖を利用して、比較した。さらに、我々は、すでにFNフラグメントに粘着したウイルス粒子(対上清ウイルス)が造血細胞を形質転換する能力を比較した。上記の方法に従って、0.5から1x106の前刺激した骨髄細胞を、35mmのFN−コートペトリ皿上、1−2mlのEAL2aウイルス含有上清中、増殖因子および5μg/mlのポリブレンと共に、インキュベートした。FNフラグメントに結合したウイルスによる造血細胞の形質転換を評価するために、ウイルス含有上清と共にインキュベートした35mmのFN−コート皿を各2mlのPBSで3回洗浄した。次に、0.5から1x106の前刺激した骨髄細胞を、増殖因子およびポリブレンを添加した2mlの培地内に加えた。22時間後、細胞を収集し、そして、Bradley,T.R.およびD.Metcalf,Aust,J.Exp.Biol.Med.Sci.、44、287ー293,1966に記載の方法に従って、1.5mg/mlのG418を加えておよび加えずに、HPP−CFCアッセイに塗布した。培養液を、14日間、7%CO2中、33℃でインキュベートし、遺伝子トランスファー効率を、G418耐性コロニーの%として計算した。
【0098】
10.2. 結果
上清感染を通しての初期造血細胞(primitive hematopoietic cells)の形質転換(図10)は、ヘパリン−結合部位(heparin−binding site)(HBS)および少なくとも一つより多くの活性細胞粘着部位(立体棒)を含むすべてのフラグメントで、BSA上での上清感染より有意に高い。組み換えフラグメントCH−271、H−296、CH−296およびC−CS1(3つのフラグメントはすべて、ヘパリン−結合部位およびCS−1部位の両方を含む)の形質転換効率は、FN30/35の効果と同等であった。その他のすべての場合において、形質転換は、劇的に減少した。これらのデータは、以前にFN30/35上で示された初期造血細胞の形質転換の増加を、組み換えFNフラグメント上でも複製できることを、証明している。さらに、フィブロネクチンに直接結合したウイルスは、造血細胞を遺伝的に形質転換する能力を持つことを、証明している。最終的に、初期造血前駆体(幹)細胞を形質転換するために、CS−1およびヘパリン結合部位の両方の存在が有用であることが、確認された。
【0099】
実施例11 組換えフィブロネクチンフラグメント上でのマウスのドナー細胞の形質転換を用いる、マウスの長期にわたる骨髄の再構築
11.1. 実験方法
造血幹細胞の再構築への効果を分析するために、初期造血前駆細胞について、BS対FN30/35対組み換えFNフラグメント上での上清感染、対同時培養を、骨髄移植を用いて比較する、上記のインビトロでの研究(実施例10から)を繰り返した。簡単に言えば、致死照射マウスに、ヒトADA cDNAを含むEPHA−5ベクターで形質転換したドナー細胞を注射した。1ヶ月後およびおおよそ6ヶ月後、遺伝子トランスファー効果を、ADAイソ酵素アッセイで末梢血液から分析した。
【0100】
11.2. 結果
図11は、1ヶ月後の結果を示し、ヘパリン結合部位およびCS−1の両方を含むフィブロネクチンフラグメントH−296では、FN30/35および共培養と同様の結果が得られることを、はっきりと示している。これら部位を両方共含まないフラグメントは、移植可能な造血細胞の形質転換に、あまり有効ではない。これらのデータは、初期造血/幹細胞ならびにCS−1およびヘパリン結合部位を両方共含む組換えFNフラグメントに結合したレトロウイルスが共に配置されることが、移植可能な造血細胞を効果的に形質転換することを証明している。
【0101】
図12は、移植後4ヶ月および6ヶ月の結果を示している。この時点で、造血幹細胞を再集団化する遺伝的形質転換は、対照プレート上またはCBD(C−274)あるいはIII12−14(H−271)のみを含むフラグメント上で形質転換した細胞を移植した動物内では、示すことができなかった。HSCsの遺伝的形質転換は、C−CS1フラグメント上ではより少ない頻度で認められ、この場合、1/3の動物がヒトのタンパク質陽性であった。インビボで再集団化した幹細胞内への遺伝子トランスファーは、CBD(CH−271)またはCS1(H−296)のいずれかと組み合わされたHBDを含むフラグメント上で、一律に認められた。3つの細胞結合部位をすべて含むフラグメント(CH−296)上での形質転換は、標的細胞、産生細胞系、の同時培養で最も効果的に比較できた。これらのデータは、VLA−4および/またはVLA−5の結合部位と組み合わせてIII12−14を含むフィブロネクチンフラグメントがマウスの造血前駆細胞および幹細胞内へのレトロウイルスが仲介する遺伝子トランスファーを増加させる能力を有するを示している。
【0102】
実施例12
フィブロネクチンは、少なくとも3つの部位(図13):インテグリンVLA−5を通してリピート10内にテトラペプチドRGDSを含む細胞結合ドメイン(cell binding domain)(CBD);細胞表面のプロテオグリカン分子を通してタイプIIIリピート12−14(III12−14)内に含まれるヘパリン結合ドメイン;および、インテグリンVLA−4を通して変化するスプライス領域IIICS内のCS1配列:を通して細胞粘着性を支配する。これらの研究では、これらの単一の細胞粘着部位を単独で含むか、あるいはペプチド内に組み合わせて含む、図13に示す6つのキメラ組換えFNフラグメントを用いた。
【0103】
FN−コートした細菌皿を、アンホトロピック(amphotropic)パッケージング細胞系TKNeoからの2mlの上清(SN)内の200cfuと共に、インキュベートした。37℃に30分間置いた後、皿をPBSで3回洗浄し、次いで、2000NIH/3T3細胞(繊維芽細胞)を、10%のウシ血清(CS;Sigma,U.S.A.)および2%のP/Sを添加した1mlのDMEM内に加えた。翌日、細胞は、0.75mg/mlのG418を含む選択培地内に置かれた。8−10日後、皿を、Stat Stain(Volu−Sol,U.S.A.)で染色し、そしてG418rコロニーをカウントした。このアッセイでは、レトロウイルスは、コートしていないかまたはBSA−コートしたプレートとは結合しない。図14に示したように、NIH/3T3細胞内への遺伝子トランスファーは、ここでは”III12−14”と名付けられたヘパリン−II結合ドメインを含むフラグメント(H−271、CH−271、H−296、CH−296)上でのみ起こった。これらのデータは、レトロウイルス粒子がフィブロネクチンのIII12−14リピート内の配列に直接的に結合することを証明している。NIH/3T3細胞の感染は、III12−14のみの場合と比較して、III12−14およびCBDの両方を含むFNフラグメント(H−271対CH−271を比較する)で有意により高かった。注目すべきことに、200のNEOcfu/プレートのインプットから最大で80のG418rNIH/3T3コロニー/プレートが生成した。これとは対照的に、CS1を加えても、NIH/3T3形質転換にいかなる効果も示さなかった(H−271対H−296、CH−271対CH−296)。
【0104】
FNがレトロウイルス−仲介の遺伝子トランスファーを増加させるメカニズムがフラグメントへのレトロウイルスおよび標的細胞の同時結合によるものであることを証拠付けるため、非粘着細胞系(HL60、既知の前骨髄細胞白血病細胞系)を用いて、実験を行った。HL60細胞を、直接フルオロクロム−結合のVLA−4に対するモノクローナル抗体(FITC:Immunotech,U.S.A.)およびVLA−5に対するモノクローナル抗体(PE;Antigenix America,U.S.A.)で、またはIgG1およびIgG2a(Becton Dickinson)をイソ型対照として、染色した。死滅細胞を、プロピフィウムヨード(propifium iodine)染色によって、排除した。次いで、細胞をFACScan(Becton Dickinson)上で分析した。遺伝子トランスファーを研究するために、104のHL60細胞を、神経増殖因子レセプター(NGF−R)(力価105cfu/ml)を含むアンホトロピックパッケージング細胞系DAG(St.Jude’s Children Research Hospital,Memphis,TN.U.S.A.から得られた)からのウイルス上清と共に、懸濁し、そして、濃度2μg/cm2 の6つのFNフラグメントでコートした細胞6穴プレートに加えた。4時間後、培養中のHL60培養液から2mlの順化培地を加えた。4mlの新たな培地(5%のFCSおよび2%のP/Sを含むRPMI, Gibco)を、4−5日後に加えた。細胞を全体で8日間増殖させ、次いで、NGF−R(Boehringer,U.S.A.)に対するモノクローナル抗体8211で、または対照としてイソ型IgG1(Dako,U.S.A.)で染色し、次いで、FITC−複合 ヤギ−抗−マウスの二次血清(Boehringer)と反応させた。細胞デブリスを排除するために、サンプルをプロピジウムヨード(propidium iodine)(PI)と共にインキュベートし、次に、FACScanで測定した。遺伝子トランスファーは、NGF−R発現の分析によって、生きた細胞の通門(gating)の後に、証明された。遺伝子トランスファーの研究はすべて、ポリブレンまたはプロタミンなしで、行われた。図15に示すように、HL60細胞は、フローサイトメトリー分析(A+B)では、VLA−4を発現するが、VLA−5を発現しない。発現データと一致して、HL60細胞は、CS1を含むフラグメント(H−296、CH−296、C−CS1)でコートされたプレートのみに粘着した。図15に示すように、HL−60細胞の遺伝子形質転換は、CS1と組み合わせてIII12−14を含むフラグメント(H−296、CH−296)上でのみ起こった。HL−60細胞は、それらのVLA−4インテグリンを通してC−CS1フラグメントと粘着するが、レトロウイルス結合が劇的に起こるIII12−14 が存在しないと、HL60細胞の形質転換は減少する。
【0105】
実験のもう一つのセットでは、濃度を増加させた高分子量(HMW) のヘパリン(Sigma,U.S.A.、MW約7000−25000)は、2.5%(v/v)の1M Hepes(HBSS/Hepes;すべてGibco)を添加した2mlのハンクス液(Hanks Balanced Salt Solution)に溶解させ、そして濃度4μg/cm2 の異なるFNフラグメントでコートした細菌の6穴プレートに加えた。30分後、プレートを、PBSで3回洗浄し、次に400cfu/2mlのアンホトロピックTKNeoベクターを加えた。37℃で30分後、プレートを再びPBSで洗浄し、次いで10%のCSおよび2%のP/Sと共に、DMEM中の2000NIH/3T3細胞を加えた。上記の方法に従って、選別を行い、遺伝子トランスファー効率を、8−10日の培養の後のG418r コロニーの数として計数した。同様の実験セットでは、分画した低分子量(LMW)ヘパリン(Sigma,U.S.A.、MW約3000)は、濃度を増加させた2mlのHBSS/Hepes中に溶解させた。次いで、HMW ヘパリンに関して上記した方法に従って、実験をデザインした。遺伝子トランスファー効率は、8−10日後のG418r の数として計数した。これらの遺伝子トランスファー研究のすべては、ポリブレンまたはプロタミンなしで行った。これらの実験の結果を図16に示す。示したように、FNへのウイルスの結合は、高分子量ヘパリンでは服用量依存様式で競合できる(16A)が、低分子量ヘパリンでは競合しない(16B)。これらは、結果として、レトロウイルス粒子がIII12−14内の配列に結合すること証拠となる。
【0106】
実施例13 CD34+ 細胞集団内でのBFU−E細胞の選択的形質転換
この実施例では、混合した細胞集団内で選別された細胞を、ウイルス結合ドメインおよび細胞結合ドメインを持ち、標的細胞に特異的であるポリペプチドを用いることによって、形質転換の標的とすることができることを証明した。一般的TKNEO感染および実施例1のアッセイプロトコールを繰り返したが、BFU−E、CFU−GMおよびCFU−Mix細胞[臍帯血(CB)細胞よりアフィニティクロマトグラフィーによって得られた]を含むCD34+細胞は、実質的に均一な集団を用い、組換えFNフラグメントCH−271の濃度は1、2、4、8、16および20μg/cm2 に変化させて用いた。結果を図17に示す。示した通り、BFU−E細胞は、VLA−5結合部位に結合し、高い効率で形質転換された(G418耐性コロニーの25%より多い)が、これに対してCFU−GMおよびCFU−Mix集団は、有意なVLA−5への結合を示さず、実質的により低い効率で形質転換された(G418耐性コロニーの約5%より少ない)。
【0107】
実施例14 c−KIT+細胞の形質転換
本実施例では、c−KIT+として実質的に均一な細胞集団を、本発明の方法に従って、形質転換した。このため、c−KIT+細胞は、フローサイトメトリー選択技術を用いて、マウスの骨髄から単離された。細胞は、一般的には実施例1記載の方法に従って、TKNEOベクターを用いた感染プロトコールに委ねたが、FNフラグメントは、図18に示すように変化させ、感染した細胞は、HPP−CFCでアッセイした。図18に示すように、III12−14およびVLA−4結合部位を含むFNフラグメント(FN30/35、H296およびCH−296)は、高い形質転換効率を示した(G418耐性コロニーの80%より高い)が、これらの2つのドメインを欠くFNフラグメントは、結果として有意な形質転換を生じない(C274およびC−CS1)か、または実質的に低い効率を示す(H271、CH271、G418耐性コロニーの20%より少ない)。
【0108】
実施例15 ポリブレンの濃度を変化させて用いた場合のNIH/3T3およびクローン原性(clonogenic)BM細胞の形質転換
この実験セットでは、本発明の有用な形質転換法が、ヘキサジメチリンブロミド非存在下で行われることを証明した。NIH/3T3(繊維芽細胞)細胞およびクロ−ン原性骨髄細胞は、一般的には、それぞれ実施例12および1に記載した通りのTKNEO感染プロトコールに委ねたが、ベクター、細胞および様々な濃度のポリブレンを最初に一緒に懸濁し、次いで、FNフラグメントCH−296(8μg/ml)でコートした基質に適用した。アッセイは、これらの細胞型に関して前記したとおりである。結果を図19および20に示す。図19に示すように、G418耐性NIH/3T3コロニーの数は、ポリブレン濃度を増加させると、劇的に減少する(その範囲は、ポリブレンを使わなかった場合約14から、12.5μg/mlのポリブレンを用いた場合約4に低下する)。同様に、図20は、プロトコールがポリブレンの非存在下に委ねられた場合に40近いコロニーが得られたことを、表しているが、これに対して10μg/mlのポリブレンを用いた場合の関連値は、15より少なかった。
【0109】
実施例16 T細胞の形質転換、ならびにコラーゲンVおよびFGFウイルス結合配列を持つポリペプチドの使用
この実施例では、ヒトT細胞内へのDNAトランスファーの強化、およびコラーゲンVおよび繊維芽細胞増殖因子(FGF)からのレトロウイルス結合配列(フィブロネクチンのヘパリンII結合部位と実質的に相同である)の発明への使用について、説明している。このために、全血を、モノクローナル抗体(CD3 perCPに対するMoAbs;Becton Dickinson)、CD49e(PE;Anitenix America)、CD49d(FITC,PE,それぞれ:Becton Dickinson)で、15分間、室温で染色し、次いで、製造者らの勧告に従って、FACS溶解溶液(BECTIN DICKINSON)と共にFACScan(BECTIN DICKINSON)での分析用に、調製した。末梢血液−誘導単核細胞(peripheral blood−derived mononuclear cells)(PB−MNCs)は、1x106細胞/mlの濃度で、1μg/mlのOKT−3(Ortho)および50U/mlのIL−2(Cetus)で、前刺激した。2−3日後、細胞を収集し、洗浄し、次いで、7.5μg/mlのポリブレン(Aldrich)の連続存在下、8μg/cm2 のCH296で又は2%のウシ血清アルブミン(BSA,Boehringer)でコートした組織培養処理しなかったプレート上、PGKプロモーターの調節下で、マウスのアデノシンデアミナーゼ(ADA)cDNAを含むアンホトロピックレトロウイスル55/6で、1または2日間形質転換した。一日おきに、50%の培地を、50U/mlのIL−2を含む培地に新しく置き換える。感染から4−6日後、細胞を収集し、CD3、CD56、TCR−αβおよびHLA−DRに対するMoAbs(すべてBectin Dickinson)を用いて、形態学的に分析した。遺伝子トランスファー効率の分析は、ADAイソ酵素アッセイ(Bodine,D.M.ら、Blood,82,1975−80,1993;Moritz,T.ら、J.Exp.Med.178,529−36、1993)でヒトの内在性のADAタンパク質と比較して、形質転換されたマウスのADA酵素の活性を測定することによって、非選択集団上で、行われた。
【0110】
結果:
A. T細胞
最初に、ヒトT細胞を、FNレセプターVLA−4およびVLA−5の発現について分析した。このために、末梢血液(PB)−誘導単核細胞(MNCs)を、CD3、CD49dおよびCD49eに対するモノクローナル抗体(MoAbs)で染色し、フローサイトメトリーで分析した。図21に示すように、細胞を、すべての末梢血液T細胞を表すCD3陽性リンパ球(R1およびR4)について通門した。PB T細胞の94%は、CD49d陽性であり、96%はCD49e陽性である。T細胞の90%は、VLA−4およびVLA−5の両方を発現している。
【0111】
次に、PB−誘導PB−MNCsを、2−3日間、CD3MoAb OKT−3および組換えIL−2で前刺激し、次いで、1−2日間、組換えFNフラグメントCH−296かまたはBSAでコートした非組織培養処理したプレート上、PGKプロモーター調節下で、形質転換した;対照には、マウスのアデノシンデアミナーゼ(ADA)cDNAを含むアンホトロピック産生細胞系AMmA55/6(Bodine,D.M.ら、Blood,82,1975−80,1993;Moritz,T.ら、J.Exp.Med.,178,529−36,1993)からのレトロウイルス上清でコートしたプレートを用いた。
【0112】
感染4−6日後のフロー分析は、細胞培養液が>90%のT細胞を含み、その大部分は、それらのHLA−DRおよびCD56発現によって示されるように強く活性化された(図22)。
【0113】
ADAイソ酵素アッセイによる遺伝子トランスファー効率の分析(図23)は、誘導されたマウスADAタンパク質の活性が、形質転換プロトコールが組み換えペプチドCH−296を含む場合には、ヒト内因性タンパク質の活性と等しいことを証明した。BSA上での感染では、アッセイの感受性以上のレベルの遺伝子トランスファーが得られた。
【0114】
これらのデータは、ヒト一次T細胞(primary T cells)が、ポリブレンの様なポリカチオンの非存在下で、組換えFNフラグメントCH−296上の無細胞レトロウイルス−含有上清で効率よく形質転換されうることを、証明している。遺伝子配達効率は、上清+ポリブレンのみを用いた標準的方法と比較して、非常に高い。
【0115】
B. 新しい組換えペプチド
コラーゲンV(COL)および塩基性繊維芽細胞増殖因子(bFGF)の両方が、フィブロネクチンのHBDと配列相同性を示し、ヘパリン結合能力もまた有するため、図24に示す7つの組み換えタンパク質を、レトロウイルス結合活性について分析した。
【0116】
最初に、COLおよびFGFを分析して、それらが機能的レトロウイルス結合配列を持つか否かについて決定した。最後に、細菌プレートを組換えペプチドでコートし、次いで、アンホトロピックNEOレトロウイルスを含む上清を加えた。30分後、プレートを徹底的に洗浄してすべての非結合レトロウイルス粒子を除去し、次いで、2000NIH/3T3細胞をそれぞれのプレートに加えた。遺伝子トランスファー効率は、8−10日後、G418耐性(G418r )NIH/3T3細胞コロニーの数で、評価した。
【0117】
図25に示すように、NIH/3T3細胞内への充分な遺伝子配達は、HBD(フラグメントCH−271)と又はコラーゲンV(C−COL)あるいはFGF(C−FGF)の配列と組み合わせてFNの細胞結合ドメイン(CBD)を含む組換えペプチド上で起こるG418rコロニーの増殖で、評価した。これらの結果は、レトロウイルス粒子がコラーゲンVまたはbFGF内の配列と直接的に結合することをも証明し、これらの配列を含む組換えペプチドを、造血細胞の標的に用いることができることの証拠となる。
【0118】
VLA−4およびVLA−5を発現するHEL細胞は、このように、組換えフラグメントC−274(CBD)、H−271(HBD)、CH−271(CBD−HBD)、COL、C−COL(CBD+COL)、C−FGF、C−FGF−CS1上で、および対照としてBSA上で、切除型(truncated)神経増殖因子レセプターp75鎖(NGF−R)cDNAを含むアンホトロピックレトロウイルスで形質転換された。遺伝子トランスファ−効率を、8日後、NGF−R発現のフローサイトメトリー分析によって分析した。
【0119】
図26に示したように、低レベルの遺伝子トランスファーは、C−274でコートしたプレートまたはBSAでコートしたプレート上で起こった。H−271(HBD)およびCOL上での形質転換は、結果として33−36%のNGF−R陽性細胞を生じた。HBD(CH−271)へのCBDの付加は、切除型細胞の百分率を、この実験で最も高いレベルに上昇させた。興味深いことに、C−COLフラグメント中のCOLへのCBD(VAL−5結合部位)付加も、また、フラグメントC−FGF−CS1内のCS−1(=VAL−4結合部位)の付加も、HEL細胞の遺伝子形質転換を有意には上昇させなかった。
【0120】
最後に、1日IL−6およびSCFで前刺激したヒトCD34+骨髄(BM)細胞を、アンホトロピックNEOレトロウイルスと共にキメラペプチド上で、1日間形質転換し、次いで、1.5mg/mlのG418の存在下または非存在下で前駆細胞アッセイのため、塗布した。
【0121】
G418rコロニーの%として培養14日後に評価した効率良い遺伝子トランスファーは、細胞結合レセプターと組み合わせてH−271又はFGF内にレトロウイルス結合配列を含むフラグメント上で起こっていた。特記すべきことに、NGF−RレトロウイルスでHEL細胞を遺伝的に修飾することによって得られる結果(図27)に対して、C−FGFにCS1部位を付加すると、クローン原性CD34+BM細胞内への遺伝子トランスファー効率が著しく促進されて、フラグメントCH−296(CBD+HBD+CD1)で得られたレベルに近づく。
【0122】
このように、一次T細胞は、それらのVLA−4又はVLA−5インテグリンを通してフィブロネクチンと結合するT細胞の能力を用いるフィブロネクチンの粘着性ドメイン上のレトロウイルスベクターで効率よく形質転換できることが、証明された。また、レトロウイルス粒子は、フィブロネクチンのヘパリンII結合ドメインと相同性を示すコラーゲンVまたはbFGF内の配列に粘着すること、ならびに造血細胞および細胞系は、標的細胞のための結合部位と組み合わせてコラーゲンVまたはbFGFのレトロウイルス結合配列を含む組み換えペプチド上で、効率よく遺伝的に修飾されうること、もまた示した。
【0123】
本発明は、前の記述を詳細に説明し記載するが、本発明は、その特徴を説明するものとして考えられるべきであって、特徴を制限するものとして考えるべきではなく、望ましい実施態様のみを記載すること、および本発明の思想の中に入る変化および修飾のすべてが保護されることを所望することを、理解するであろう。
【0124】
ここに引用されたすべての文献は、そのそれぞれが独立的に参照として採用される様に、ここにその全体を参照として採用される。さらに、同時係属の米国特許出願08/218,355、1994年3月25日出願、および同時係属の国際特許出願PCT/US95/03817、1995年3月27日に出願され合衆国指定、はその全体を参照としてここに採用する。
【特許請求の範囲】
【請求項1】
ポリカチオン性剤を含まず、そして組換えフィブロネクチンフラグメントCH−296を含む、生存可能な哺乳類の細胞集団であって、
当該細胞集団は、レトロウイルスによって形質導入された生存可能な細胞集団を得る方法であって、
レトロウイルスによる細胞の形質導入の効率を増加させるために、有効な固定化された量の組換えフィブロネクチンフラグメントCH−296の存在下で、細胞をレトロウイルスに感染させる、
ここにおいて、前記感染は、共培養中レトロウイルスによる細胞の形質導入の効率を増加させるが、前記組換えフィブロネクチンフラグメントCH−296の存在下でレトロウイルスによる細胞の形質導入の効率を減少させる、ポリカチオン性剤を含まない培地で行われる、
ことを含む方法によって製造される、
前記前記哺乳類の細胞集団。
【請求項2】
造血幹細胞を含む、請求項1記載の生存可能な哺乳類の細胞集団。
【請求項3】
請求項1記載の生存可能な哺乳類の細胞集団を哺乳動物に移植することを含む、細胞移植方法のために使用される、請求項1記載の生存可能な哺乳類の細胞集団。
【請求項4】
レトロウイルスで形質導入されたin vitroの生存可能な哺乳類の細胞集団を含む哺乳類の細胞組成物であって、レトロウイルス産生細胞および共培養中レトロウイルスによる細胞の形質導入の効率を増加させるポリカチオン性剤を含まず、そして、組換えフィブロネクチンフラグメントCH−296をさらに含む、前記哺乳類の細胞組成物。
【請求項5】
前記の生存可能な細胞が造血幹細胞を含む、請求項4記載の哺乳類の細胞組成物。
【請求項6】
請求項4記載の組成物から得られた生存可能な哺乳類の細胞集団を哺乳動物に移植することを含む、細胞移植方法のために使用される、請求項4記載の哺乳類の細胞組成物。
【請求項7】
細胞が造血幹細胞を含む、請求項6記載の哺乳類の細胞組成物。
【請求項8】
前記哺乳類の細胞集団が、造血幹細胞を含むヒト造血細胞集団である、請求項2記載の生存可能な哺乳類の細胞集団。
【請求項9】
生存可能な哺乳類の細胞集団が、哺乳類の同じ種に由来する、請求項6記載の哺乳類の細胞組成物。
【請求項10】
哺乳類がヒトであり、そして生存可能な哺乳類の細胞集団がヒトの細胞集団である、請求項9記載の哺乳類の細胞組成物。
【請求項11】
細胞がヒト造血細胞である、請求項4に記載の哺乳類の細胞組成物。
【請求項1】
ポリカチオン性剤を含まず、そして組換えフィブロネクチンフラグメントCH−296を含む、生存可能な哺乳類の細胞集団であって、
当該細胞集団は、レトロウイルスによって形質導入された生存可能な細胞集団を得る方法であって、
レトロウイルスによる細胞の形質導入の効率を増加させるために、有効な固定化された量の組換えフィブロネクチンフラグメントCH−296の存在下で、細胞をレトロウイルスに感染させる、
ここにおいて、前記感染は、共培養中レトロウイルスによる細胞の形質導入の効率を増加させるが、前記組換えフィブロネクチンフラグメントCH−296の存在下でレトロウイルスによる細胞の形質導入の効率を減少させる、ポリカチオン性剤を含まない培地で行われる、
ことを含む方法によって製造される、
前記前記哺乳類の細胞集団。
【請求項2】
造血幹細胞を含む、請求項1記載の生存可能な哺乳類の細胞集団。
【請求項3】
請求項1記載の生存可能な哺乳類の細胞集団を哺乳動物に移植することを含む、細胞移植方法のために使用される、請求項1記載の生存可能な哺乳類の細胞集団。
【請求項4】
レトロウイルスで形質導入されたin vitroの生存可能な哺乳類の細胞集団を含む哺乳類の細胞組成物であって、レトロウイルス産生細胞および共培養中レトロウイルスによる細胞の形質導入の効率を増加させるポリカチオン性剤を含まず、そして、組換えフィブロネクチンフラグメントCH−296をさらに含む、前記哺乳類の細胞組成物。
【請求項5】
前記の生存可能な細胞が造血幹細胞を含む、請求項4記載の哺乳類の細胞組成物。
【請求項6】
請求項4記載の組成物から得られた生存可能な哺乳類の細胞集団を哺乳動物に移植することを含む、細胞移植方法のために使用される、請求項4記載の哺乳類の細胞組成物。
【請求項7】
細胞が造血幹細胞を含む、請求項6記載の哺乳類の細胞組成物。
【請求項8】
前記哺乳類の細胞集団が、造血幹細胞を含むヒト造血細胞集団である、請求項2記載の生存可能な哺乳類の細胞集団。
【請求項9】
生存可能な哺乳類の細胞集団が、哺乳類の同じ種に由来する、請求項6記載の哺乳類の細胞組成物。
【請求項10】
哺乳類がヒトであり、そして生存可能な哺乳類の細胞集団がヒトの細胞集団である、請求項9記載の哺乳類の細胞組成物。
【請求項11】
細胞がヒト造血細胞である、請求項4に記載の哺乳類の細胞組成物。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】
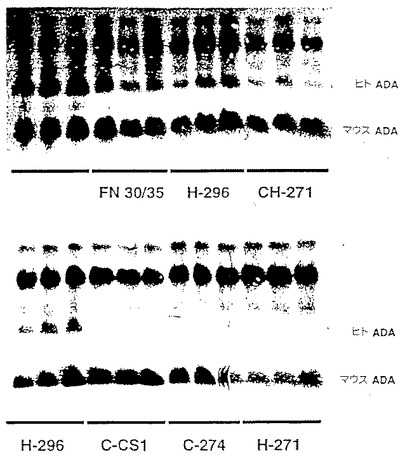
【図12】
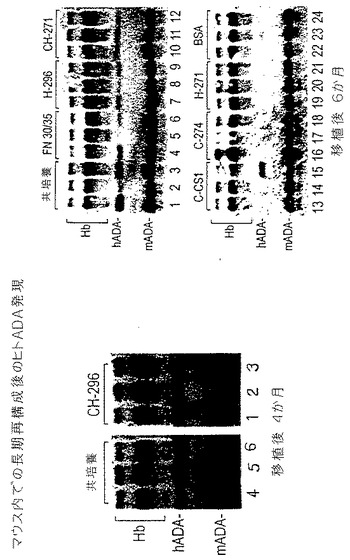
【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図24】

【図25】

【図26】

【図27】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2009−261407(P2009−261407A)
【公開日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願番号】特願2009−155855(P2009−155855)
【出願日】平成21年6月30日(2009.6.30)
【分割の表示】特願平9−513758の分割
【原出願日】平成8年9月30日(1996.9.30)
【出願人】(301046787)インディアナ・ユニバーシティ・リサーチ・アンド・テクノロジー・コーポレーション (24)
【Fターム(参考)】
【公開日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願日】平成21年6月30日(2009.6.30)
【分割の表示】特願平9−513758の分割
【原出願日】平成8年9月30日(1996.9.30)
【出願人】(301046787)インディアナ・ユニバーシティ・リサーチ・アンド・テクノロジー・コーポレーション (24)
【Fターム(参考)】
[ Back to top ]