
医薬組成物
少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットの構造、発現、および/または活性の制御または撹乱のための薬剤的に許容される調節因子、例えばヒトを含む温血動物のピルビン酸脱水素酵素(PDH)複合体のミトコンドリアエネルギー代謝調節の変化を通したもの、およびその使用方法は、複合体のリン酸化状態に影響を与えるための、少なくとも1の有効量のリポ酸誘導体および少なくとも1のその薬剤的に許容される担体を含む。PDHキナーゼ活性の増大および/またはPDHホスファターゼ活性の減少により、この調節因子は、PDH複合体のElαサブユニット活性の阻害を通して嫌気性解糖作用の有毒代謝産物の解毒を阻止し、ミトコンドリアの酸化的なリン酸化活性を強制的に増大させる。腫瘍細胞のような過剰増殖により特徴付けられる細胞は、PDH複合体のE2サブユニット活性の阻害における調節因子のさらなる作用のためにアセチル−CoAおよびNADHもまた産生できず、ミトコンドリアの膜分極は失われ、細胞死が促進される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、治療用および診断用の組成物に関し、さらに詳細には、癌細胞を含む過剰増殖により特徴付けられる細胞内への選択的な取り込みを示し、かつ酵素の構造、発現、および/または活性の制御または撹乱を調節し、これによってそれらの細胞の検出および処置または破壊を促進する医薬組成物、およびそれらの使用方法に関する。さらに特異的には、これらの薬剤は、大部分の癌に付随する修飾されたピルビン酸脱水素酵素(PDH)複合体などとして観察される、変化したミトコンドリアエネルギー代謝の活性またはその制御を標的とし、撹乱する。
【背景技術】
【0002】
ミトコンドリアは、真核細胞におけるエネルギー産生および生と死の工程に対する主要なコントロールセンターである。残りの細胞との様々な連絡機構が存在するが、可逆的リン酸化はミトコンドリアの機能を制御するための重要な手段である(Pagliarini D.J.and Dixon J.E.(2006).Mitochondrial modulation: reversible phosphorylation takes center stage? TRENDS in Biochem.Sci.31:26−34、本明細書の各所に参照として取り込まれる)。報告されたミトコンドリアキナーゼ、ホスファターゼ、およびリン酸化タンパク質の数の着実な増加は、リン酸化がミトコンドリアでの工程の制御における共通のテーマとして現れているであろうことを示唆する。ミトコンドリア酵素の構造、機能および活性制御に付随する病理的または遺伝的な変化は、疾病の治療に貢献し、かつその重要な標的であり得る。
【0003】
様々な細胞機能の収束点および制御因子としてのそれらの役割に一致して、ミトコンドリアはアポトーシス、活性酸素種(ROS)の産生、および細胞ATPの90%を超える産生を含む多数の代謝工程において決定的な役割を持つ。さらに、細胞の増殖および分裂に従って新たなミトコンドリアが作られる必要があり、この工程それ自体が、核およびミトコンドリアDNAの転写および翻訳の注意深い協調を要求する。最終的に、細胞エネルギーが変化を必要とするとき、ミトコンドリアはそのATP産出を回転させることによって迅速に応答しなければならない。それ故に、ミトコンドリアは細胞機能と連絡する複雑な系を要求する。図1から明らかなように、ミトコンドリアへ出入りするシグナル分子はイオン、気体、代謝産物、ホルモン、転写因子およびタンパク質を含む。結果的に、細胞シグナルを受け取り、統合し、伝達するための中心としてミトコンドリアを認識することは、医薬品の設計および試験における重要な進歩である。
【0004】
ミトコンドリア制御におけるシグナル伝達の礎石は可逆的リン酸化である。しかしながら、タンパク質キナーゼ現象の最初の実証が1954年に報告され、40年近く前に可逆的リン酸化によってミトコンドリアの中心的機能が制御できるという事実が発見されたにもかかわらず、ミトコンドリアのリン酸化現象についての報告は少ない。このことは、1980年代および1990年代において、細胞膜を横断し、サイトゾルを通って核へと伸展する多数のシグナル伝達リン酸化カスケードが認識されたことにも全くかかわりなくである。このような知識の不足は、ミトコンドリアタンパク質機構の大部分が二つの脂質二重層の後ろに位置する事実により、サイトゾルシグナル伝達カスケードはミトコンドリアタンパク質に到達できないと考えられていることに部分的な原因があるかもしれない。いずれの現象でも、疾病管理の鍵となる方法において、ミトコンドリアが可逆的リン酸化によりシグナル伝達を制御することは広く受け入れられてはいなかった。それにもかかわらず、表1に示すように、2006年までに全てのミトコンドリアの区画(すなわち、マトリックス、内膜、膜間腔、および細胞質に面する外表面を含む外膜)内の60を超えるタンパク質が、ミトコンドリアの機能の広いスペクトルに関係付けられるリン酸化タンパク質として同定された。増加するデータはさらに、ミトコンドリアの標的の可逆的リン酸化の重要性、およびそれを対象とする、改善された癌治療のための組成物の使用について実証している。
【0005】
表1 ミトコンドリアのリン酸化タンパク質

a略語:AA,アミノ酸(S,セリン;T,スレオニン;Y,チロシン);ANT,アデニンヌクレオチド輸送体;Axmito,ミトコンドリアアネキシン;β−OX,β−酸化:BCKAD,分岐鎖ケト酸脱水素酵素;CI−CV,呼吸鎖複合体1−5;CPT,カルニチンパルミトイルトランスフェラーゼ;CREB,cAMP反応性エレメント(Cre)結合タンパク質:CYP,チトクロムP450;DBP,十二量体結合タンパク質;EF,伸長因子;GST,グルタチオンS−トランスフェラーゼ;HSP,熱ショックタンパク質;IM,内膜;IMS,膜間腔;M,マトリックス;MnSOD,マンガンスーパーオキシドジスムターゼ;mTERF,ミトコンドリア転写終結因子;mtGAT,ミトコンドリアグリセロール−3−ホスファターゼアセチルトランスフェラーゼ;mthsp,ミトコンドリアHSP;mtTBP,ミトコンドリアテロメア結合タンパク質;NDK,ヌクレオシド二リン酸キナーゼ;OM,外膜;OXPHOS,酸化的リン酸化;P部位,リン酸化部位;PDC E1/3,ピルビン酸脱水素酵素複合体E1/3サブユニット;PDK,ピルビン酸脱水素酵素キナーゼ;PLA,ホスホリパーゼA;ScIRP,サブユニットc−免疫反応性ペプチド;SSAT,スペルミジン/スペルミンアセチルトランスフェラーゼ;StAR,ステロイド産生急性調節タンパク質;TCA,トリカルボン酸サイクル;TRAP−1,腫瘍壊死因子タイプ1受容体付随タンパク質;VDAC,電位依存性陰イオンチャネル。
b組み換えタンパク質において、リン酸化はin vitroでのみ観察される。
cタンパク質はミトコンドリアへ移動する(すなわちタンパク質はミトコンドリアの常在タンパク質ではない)。
【0006】
ヒトゲノムにおける最も大きなキナーゼおよびホスファターゼファミリーであるタンパク質キナーゼ(PK)およびタンパク質チロシンホスファターゼ(PTP)は、それぞれ500を超える、および100を超えるメンバーを有する。キナーゼおよびホスファターゼの比較的小さなファミリーを合わせると、これらのシグナル伝達分子はヒトゲノムによりコードされる全てのタンパク質の3パーセント近くを構成する。前述のリン酸化タンパク質と同様に、驚くべき数の研究によってキナーゼおよびホスファターゼがミトコンドリアの機能に関係付けられており、図2に示すように、これまでに少なくとも25のキナーゼおよび8のホスファターゼがミトコンドリアに局在していることが報告されている。これらのキナーゼおよびホスファターゼが一つのグループまたはファミリーに限定されていないことは明らかで、むしろそれらはほとんど全ての既知の哺乳類キナーゼおよびホスファターゼのサブグループに相当しており、これはミトコンドリアに影響を及ぼすと考えられるシグナル伝達経路の範囲を反映している。これらのシグナル伝達分子は基質の特異性が変化するキナーゼおよびホスファターゼ(例えばチロシンキナーゼ、古典的PTPサブグループ、セリン/スレオニンキナーゼ、および二重特異性PTP)を含み、これらの変化は触媒的な機構(例えばシステインベースのPTP、アスパラギン酸ベースのPTP、および金属依存性ホスファターゼ)および進化的保存性(例えば最近関連ピルビン酸脱水素酵素キナーゼ(PDK)およびホスファターゼ(PDP)、分岐鎖ケト酸脱水素酵素キナーゼ(BCKDK)およびホスファターゼ(BCKDP)、および多数の哺乳類特異的酵素)による。
【0007】
これらのシグナル伝達分子の大部分は、細胞内でミトコンドリアに関係しないその他の役割を有し、主にミトコンドリアの外に存在することが見出された。大部分のタンパク質について、それらのミトコンドリアへの移行の起動力または機構はよく知られていない。しかしながら、図3から明らかなように、先に記載したリン酸化タンパク質のようなキナーゼおよびホスファターゼがミトコンドリアの全ての区画に存在し、かつそれらの活性が様々なミトコンドリアの機能に影響を与えることははっきりしている。
【0008】
しかしながら少数のシグナル伝達分子は主にミトコンドリアに局在すると考えられる。PDKおよびPDPに加え、このグループはPTEN誘導性キナーゼであるPINK1、ミトコンドリアPTPMT1を標的とする二重特異性PTP、およびアスパラギン酸ベースのホスファターゼ/ATPアーゼであるTim50を含む。現在これらのタンパク質の多くに対する基質は未知であるが、生物学的および遺伝的データからそれらがミトコンドリアにおいて決定的な機能を有することは明らかである。例えば、そのミトコンドリア下の局在はまだ決定されていないが、PINK1はN末端シグナル配列によってミトコンドリアを標的とする。Ca2+/カルモジュリン調節性キナーゼファミリーと高い配列相同性を共有するこのキナーゼは、生存促進性の活性に関与すると考えられる。同様にPTPMT1は、主にミトコンドリア内部に局在する最初のPTPとして最近同定され、PINK1のようにN末端シグナルペプチドによってミトコンドリアを標的とし、かつミトコンドリアの内膜のマトリックス面に密接に結合することが見出された。PTPMT1は、そのミトコンドリアが糖代謝とインスリン分泌の連関に重要な機能を持つ膵臓β細胞に高度に発現する。最後に、TIM(内膜の交換輸送体)複合体の鍵となる成分であるTim50は、アスパラギン酸ベースのホスファターゼ/ATPアーゼのCTDファミリーに対する配列相同性を有する。このファミリーの他のメンバーと同様に、Tim50はATPアーゼとして機能し得るが、in vitroにおいて、ホスホチロシン類似体であるパラニトロフェニルホスフェートに対するホスファターゼ活性を有することもまた示されてきた。上記のことから、細胞内の他の場所からミトコンドリアへ動員されるキナーゼおよびホスファターゼのみが見られるのではなく、ミトコンドリア自体が常在するシグナル伝達分子のグループを有すると考えられる。
【0009】
時おり原因となるキナーゼおよびホスファターゼが同定されていないことから、最もよく知られたミトコンドリアリン酸化タンパク質におけるリン酸化の効果は明らかではないが、幾つかのリン酸化現象が部分的に特徴付けられている。これらの例は、先に考察したリン酸化タンパク質およびシグナル伝達分子のように、ミトコンドリアの一領域に限定されてはいない。
【0010】
ミトコンドリア外膜におけるリン酸化は、アポトーシスの制御に決定的な役割を持つ。特によく定義されている現象は、BCL−2ファミリーのアポトーシス促進性メンバーであるBADのリン酸化である。生存促進性サイトカインであるインターロイキン−3で処理した後、PKAが外膜へ移動することが示されている。一旦、外膜のA−キナーゼアンカータンパク質(AKAP)に固定された後、PKAはBADをSer112においてリン酸化して、図3Aに示す工程であるBADの不活性化およびミトコンドリアからの分離を導く。p70S6キナーゼによるSer136、および未同定のキナーゼによるSer155におけるBADのリン酸化もまた、BADの不活性化に関係付けられている。
【0011】
健常な細胞ミトコンドリアにおける制御機構として作用する可逆的リン酸化の最もよく確立された例は、マトリックス内のPDH複合体であり、その単純化された下絵を図3Bに示す。この複合体は、解糖系由来のピルビン酸塩の、トリカルボン酸(TCA)サイクルの主要な前駆体であるアセチルコエンザイムA(CoA)への変換を触媒する。これら二つの主要なエネルギー産生経路をつなぐものとして、PDH複合体は細胞内グルコースの恒常性維持のために適切に制御されなければならない。
【0012】
ミトコンドリアリン酸化タンパク質として最初に同定されたことから、PDH複合体および可逆的リン酸化によるその制御は徹底的に研究されてきた。PDH複合体のリン酸化および脱リン酸化は、それぞれPDKおよびPDPにより行われる。少なくとも4のPDKアイソフォームおよび2のPDPアイソフォームが知られており、それら全てはPDH複合体のE2サブユニットに結合する。リン酸化現象はE1サブユニットの別個の三つのセリン残基に起こり、それぞれがこの複合体の著しい不活性化を導く。特に、PDKそれ自体のリン酸化については現在少なくとも一報告がある。PKCにより行われるこのリン酸化はPDKを不活性化することが示されており、可逆的リン酸化によるPDH複合体制御のさらなるレベルを潜在的に証明している。従ってPDH複合体は、他の保存された工程に制御の段階を加えるためにリン酸化を用いるミトコンドリアの最も重要な例である。
【0013】
先に記載したミトコンドリアのチロシンキナーゼおよびホスファターゼによって示されるように、この細胞小器官内でのリン酸化はセリンおよびスレオニン残基に限定されない。ミトコンドリアのエネルギーに影響を与えるチロシンリン酸化の例は、図3Cに示す内膜でのチトクロムCオキシダーゼ(COX)の制御に見られる。呼吸鎖における終末酵素としてのCOXは、ポンプによってプロトンが内膜を横切る間、協調的に酸素を水に還元する。PDH複合体と同じく、COXはATPおよびADP、同様に甲状腺ホルモンT2およびおそらくはCa2+イオンによりアロステリックに制御される。これらの制御形式に加え、COXはin vitroおよびHepG2細胞におけるin vivoの両方で、cAMP依存性の様式によりリン酸化されることが示されている。COXは13のサブユニットを含み、二量体として結晶化される。リン酸化部位はサブユニット1のTyr304であると同定され、これは膜間の二量体インターフェースに位置する。リン酸化現象は、おそらく二量体形成の中断によってCOX活性を顕著に阻害する。
【0014】
COXのチロシンリン酸化の第二の例である非受容体型チロシンキナーゼc−Srcの一部は、Lynチロシンキナーゼと同様に、破骨細胞においてミトコンドリア内部に局在してCOXのサブユニットIIの未同定部位にチロシンリン酸化を導く。リン酸化現象の結果は、COX活性の増大を導くサブユニットIに見られる結果とは逆である。
【0015】
ミトコンドリアシグナル伝達の重要な態様は、キナーゼおよびホスファターゼがそれら自体でどのように制御されるかである。Abl、Akt、GSK313およびPKCδのような、主に細胞内の他の場所に存在しているがミトコンドリアを標的とするようになる多数のキナーゼは、それらが活性化された状態においてのみそうするようになると考えられる。従ってミトコンドリア内での幾つかのキナーゼ活性の範囲は、単純に細胞小器官内へ移入される酵素の数によって決定されるであろう。
【0016】
しかしながら常在するシグナル伝達分子には、異なる制御手段が実施されなければならない。これらの工程は未だ確定されていないものの、セカンドメッセンジャーの役割が鍵となるように思われる。PDKおよびPDPの活性は、Mg2+、Ca2+、K+およびADPのようなイオンおよび小分子によって制御されることが知られている。ミトコンドリア一酸化窒素合成酵素の性質決定および最近のミトコンドリアにおける可溶性アデニル酸シクラーゼの発見は、ミトコンドリアシグナル伝達分子の制御に寄与するセカンドメッセンジャーに対してさらなる機会を提供する。最後に、細胞内の他の場所でシグナル伝達分子を制御する手段として確立されているROSは、大量の活性酸素種が産生されるミトコンドリアにおけるキナーゼおよびホスファターゼの制御にほとんど確実に関与するであろう。キナーゼおよびホスファターゼのアイソフォームの相対的な発現レベルは、病理学における重要な役割を果たし、かつ疾病に付随するその他のシグナル伝達現象に関連付けられる可能性がある。遺伝子および発現レベルでの変化もまた、これらの変化に関連し得る。
【0017】
幾つかのプロテオミクス調査を通した後であっても、哺乳類ミトコンドリアプロテオームの3分の2のみが知られていると推定される。残りの3分の1の多くは、これらのマススペクトロメトリー分析の検出レベル未満である、シグナル伝達タンパク質のような少量のタンパク質により構成されると思われる。これらの研究から、異なる組織由来のミトコンドリアの間で、含まれるタンパク質に高い可変性があることも明らかになった。例えばこれらのプロテオミクスの試みにおいて、〜50%のタンパク質のみが、調査された四つの組織(すなわち脳、心臓、肝臓および骨格筋)にわたって保存されることが見出された。異なるミトコンドリアシグナル伝達経路は、組織から組織において同様の方向で変動し得るのみではなく、この観察されるミトコンドリアの多様性に非常によく寄与していると考えられる。
【0018】
それにもかかわらず、可逆的リン酸化がミトコンドリアでの工程の制御に関与すると結論付けるための十分すぎる証拠が存在する。報告された60を超えるリン酸化タンパク質、30のキナーゼおよびホスファターゼ、および様々な補助的シグナル伝達タンパク質によれば、ミトコンドリアは確かに可逆的リン酸化によるシグナル伝達のための場として正当に評価されておらず、事実としてこのような制御は癌のような過剰増殖疾患に有用となるであろう。
【0019】
増殖の速い腫瘍細胞の大多数は、形質転換されていない細胞に対する著明な遺伝的、生化学的、および組織学的な相違を示す。これらの多くは、元の組織に比較して変化したエネルギー代謝に関連する。腫瘍細胞において最も悪名高く、かつよく知られたエネルギー代謝の変化は、ワールブルク効果として知られている、高濃度のO2存在下であっても解糖能が亢進する現象である。
【0020】
ワールブルクは最初、腫瘍細胞における亢進した解糖の推進力は、ミトコンドリアの機能の非可逆的損傷によって起こるエネルギー欠乏であると提唱し、この場合嫌気性の筋肉と同様に、解糖を通してグルコースが後に分泌される乳酸に変換される。腫瘍細胞におけるこの解糖の流れの増大は、酸素の供給が不十分な固形腫瘍に見られる部分的な低酸素状態のような、O2濃度の低い環境での生存および増殖を保証するための代謝の戦略であると提唱されてきた。特に、多くのヒト低酸素性腫瘍におけるO2濃度は20μMよりも低いことから、そこでの酸化的リン酸化は限定される。結果的に、解糖は固形腫瘍(例えば増殖の遅いメラノーマおよび乳腺腺癌)における主要なエネルギー経路であると考えられる。
【0021】
細胞増殖の比率とATP供給の比率との間の比例関係は、増殖の速い腫瘍細胞について確立されてきた。何人かの著者は、高度に脱分化した増殖の速い腫瘍における解糖の比率が、増殖の遅い腫瘍および正常細胞における解糖の比率よりも高くなるように、解糖活性は腫瘍の悪性度と相関することを提唱した。実際に高レベルの乳酸は悪性腫瘍の前兆であると提唱されてきた。これらの現象がさらなるシグナル伝達現象および遺伝的変化に関連すると考えられ、例として低酸素誘導因子および血管新生因子の産生および放出が含まれる。
【0022】
図4に示すように、何年もの間トリカルボン酸(TCA)サイクルは、生命体のエネルギー源としてのATPの産生におけるその役割のみに対して、生物学的に重要であるとみなされるだけであった。しかしながら最近の研究により、TCAサイクルの活性は、細胞増殖およびアポトーシスの決定を含むシグナル伝達経路の機能にもまた影響を与え、関係する解糖およびTCAサイクル酵素の増加または減少を制御できることが示された。腫瘍の進行と、解糖酵素であるヘキソキナーゼおよびホスホフルクトキナーゼ(PFK)1との間には直接の相関もあり、これらは増殖の速い腫瘍細胞で大きく増加している。従って、その酸化能に不足を示す腫瘍細胞は、活発な酸化的リン酸化を有する腫瘍細胞よりもさらに悪性であると想定されている。低酸素性または好気性の状態にあるか否かに関わらず、癌組織での解糖への依存は悪性度の増大に関連する。
【0023】
健常な細胞のPDH複合体におけるリポ酸の役割については、よく研究されてきた。PDH複合体は三つの中心的なサブユニット、E1、E2、およびE3(それぞれピルビン酸脱水素酵素、ジヒドロリポイルトランスアセチラーゼ、およびジヒドロリポアミド脱水素酵素)を有する。これらの複合体は中心にE2コアを持ち、このコアをその他のサブユニットが取り囲んで複合体を形成する。これら二つのサブユニット間のギャップ内で、リポイルドメインが活性部位間での中間体の輸送を行う。リポイルドメイン自体は、可動性のリンカーによってE2コアに付着する。ピルビン酸塩とチアミンピロリン酸の反応によるヘミチオアセタールの形成によって、この陰イオンは、リジン残基に付着する酸化されたリポ酸塩種のS1を攻撃する。結果的に、リポ酸塩S2は硫化物またはスルフヒドリル成分として置換され、続く四面体ヘミチオアセタールの崩壊によりチアゾールが放出され、これによりTPP補助因子が放出され、かつリポ酸塩のS1にチオアセテートが産生される。この時点でリポ酸塩−チオエステルの機能性はE2活性部位内へ移動され、アシル基転移反応がアセチル基をリポ酸塩の「自在アーム」からコエンザイムAのチオール基へ転移させる。これによりアセチルCoAが産生され、酵素複合体から放出され、続いてTCAサイクルに入る。ジヒドロリポ酸塩は複合体のリジン残基にまだ結合しているが、その後E3活性部位へ移動し、フラビンにより仲介される酸化によってそのリポ酸塩静止状態へ戻り、FADH2(および最終的にはNADH)を産生し、かつリポ酸塩を再生して、コンピテントなアシルアクセプターへ戻る。このリポ酸塩種が遮断された場合、FADH2への電子の流れまたはアセチルCoAの産生がなくなり得る結果として細胞内にピルビン酸の毒性が蓄積する。癌細胞では、細胞の解毒および生存のためにアセトインの産生が必要なことが示唆されている。
【0024】
以前に述べたように、ミトコンドリア内でのPDH複合体の活性は様々なアロステリックエフェクターおよび共有結合性の修飾によって高度に制御される。PDH活性はそのリン酸化状態によって制御され、これは脱リン酸化状態で最も活性である。PDHのリン酸化はPDKにより触媒される。PDK活性はATP、NADH、およびアセチルCoAレベルの増加により増大する。PDKの負のエフェクターは、ATPレベルが低下した時に増加するADP、NAD+、CoA−SH、およびピルビン酸である。脱リン酸化を通してPDHを活性化する酵素であるPDPの制御については完全に理解されていないが、Mg2+およびCa2+がPDPを活性化することが知られている。
【0025】
複合体の二つの産物であるNADHおよびアセチルCoAは、PDHの脱リン酸化活性型であるPDH−aに対する負のアロステリックエフェクターである。これらのエフェクターは、ピルビン酸に対する酵素のアフィニティーを減少させることによって、PDH複合体を通した炭素の流れを限定する。加えてNADHおよびアセチルCoAは、PDHをリン酸化PDH−b型に変換することにより不活性化する酵素であるPDKの、強力な正のエフェクターである。細胞のエネルギーチャージが高い際にNADHおよびアセチルCoAが蓄積されることから、エネルギーの豊富な細胞内で高レベルのATPがPDKの活性もまた上昇させ、PDHの活性を減少させることは不思議ではない。しかしながらピルビン酸がPDKの強力な負のエフェクターであることから、ピルビン酸のレベルが増加するときには、高レベルのNADHおよびアセチルCoA存在下であってもPDH−aになりやすいであろう。
【0026】
ATPの豊富な細胞内ではPDH−aを維持するピルビン酸の濃度が十分に高く、アロステリックに減少したPDHの高Km型はそれにもかかわらずピルビン酸をアセチルCoAに変換できる。高いATPおよびNADHレベルを有する細胞内での大量のピルビン酸により、ピルビン酸の炭素は炭素の二つの主な貯蔵型(糖新生を通したグリコーゲンおよび脂肪酸合成を通した脂肪の産生)に方向付けられ得り、ここではアセチルCoAが主要な炭素ドナーである。PDP−bの制御はよく理解されていないが、減少したATP濃度下でピルビン酸の酸化を最大化、および高いATP濃度下でPDH活性を最小化するための制御である可能性がかなり高い。
【0027】
腫瘍細胞は、ピルビン酸および付随するアルデヒドおよびラジカル、例えばアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを、これらの分子が高濃度において細胞内pHを大幅に低下させるという機構により細胞傷害性であることから、無制限に蓄積できない。従ってAS−30Dおよびエールリッヒヘパトーマについては、ミトコンドリアピルビン酸の有意な画分が、PDH複合体のE1成分により、結合したβ−ヒドロキシエチルチアミンピロリン酸を通して、活性アセトアルデヒドに脱炭酸されることが記載されている。この活性アセトアルデヒドは第二のアセトアルデヒドと共に次々に縮合し、最終的には元のピルビン酸をアミノ酸のグルタミンまたはリポ酸のいずれかを用いて脱酸または還元して、競合的にPDHを阻害しかつそのピルビン酸前駆体よりも細胞に対する毒性の少ない(例えば細胞内でのpH恒常性を維持することにより)化合物であるアセトイン(3−ヒドロキシブタノン)を産生する。しかしながら、ATP産生の源としての解糖への依存によるピルビン酸蓄積の結果としての腫瘍細胞の解毒経路におけるアセトインの重要性にもかかわらず、先行技術には腫瘍細胞生存率に関して、アセトイン産生を遮断する効果についての参考文献は少ない。
【0028】
最近の研究は、癌細胞にさらなる好気性の代謝を強制することによって腫瘍の増殖が抑制されることを示唆している。従って、ワールブルク代謝への移行はPDH複合体の遮断を必要とする。図5に示すように、この移行において癌細胞での低酸素誘導因子(HIF)によるシグナル伝達の増大が見られることは、グルコースの代謝におけるHIFの重要な役割を考えれば不思議ではない。直接または間接にHIFのシグナル伝達を引き起こす変異は、実際に癌の発生における一般的な機構であると考えられる。HIFはPDK1の過剰発現を誘導し、これが次にPDH複合体の減少に働く。PDK1によるリン酸化は、このアイソフォームがPDH複合体の第1のサブユニットであるE1のアルファサブユニットの三つのセリン残基を独自にリン酸化することから、不活性なPDH複合体の維持に特に有効となる。E1の再活性化は、三つのリン酸基全ての除去を必要とする。さらに、PDH複合体の活性化はROS産生の増大を導き得り、これが順にアポトーシスを導き得る。しかしながら、癌に見られるPDK1の変化は、その濃度の変化によるのみではなく、その活性の変化、およびおそらくは、実に一腫瘍型または一患者と他方との間でのそのアミノ酸配列の変化にもよる可能性がある。加えてPDK1は、腫瘍の型に依存して、腫瘍に付随する様々な分子と共に異なる複合体を形成し得る。従ってPDK1の阻害は、腫瘍にアポトーシスを起こすための潜在的な標的であり得る。しかしながら今日までに、既知のPDK1阻害剤は、このアイソザイムを最大60%のみ阻害することが示されている。
【0029】
伝統的な化学療法の標的は細胞の分裂、増殖であるが、全ての臨床的に許容される化学療法的な治療には大量の薬物が用いられ、これがホスト細胞の正常な増殖に深刻な損傷もまた誘導する。従って癌の治療には、さらに選択的な標的指向化が必要とされる。化学療法に付随する別の問題は、多くの腫瘍型において、抗新生物剤に対する固有であるかまたは獲得された抵抗性が存在することである。概して伝統的な化学療法は、現在ほとんどの悪性腫瘍に対する長期的な利点が少なく、かつしばしば生存期間またはその質を減少させる有害な副作用に関連する。従って、生命の適正な質を可能にしながら腫瘍の長期的な管理を提供できる、徹底的な新しいアプローチが必要とされている。
【0030】
新しい化学療法の開発において、薬物の有効性、送達、および副作用は確実に解決されなければならない問題である。固形腫瘍における低酸素性の領域への送達は、薬物が異なる細胞層を容易に透過できないときに困難であり得る。このような不確実性を避けるため、少なくともマイクロモル以下の範囲で一定に代謝阻害する抗癌剤の設計に今日的な意味があると考えられる。癌細胞が系統から系統にわたって遺伝的および表現型的に異質であることが立証され得るが、全ての腫瘍細胞株はATP供給のための解糖および酸化的リン酸化に依存する。ワールブルク効果に集中することは、健常ホスト組織および臓器の機能性に影響を与えることなく単に腫瘍の代謝および増殖に選択的に影響を与える送達および治療的戦略の設計を促進する、腫瘍および正常細胞の間での物理化学的および生化学的エネルギーの相違に基づく薬物設計を可能にする。
【0031】
Binghamらの米国特許第6,331,559号および6,951,887号、同様にBinghamらの米国仮出願第60/912,598号は全て参照としてここに取り込まれ、腫瘍細胞およびその他の特定の型の病的な細胞の両方を選択的に標的化しかつ死滅させる、新しい種類のリポ酸誘導体治療薬を開示する。これらの特許はさらに、有効量のこれらのリポ酸誘導体を薬剤的に許容される担体と共に含む医薬組成物、およびその使用法について開示する。しかしながら、これらの特許にはこれらのリポ酸誘導体の構造および一般的使用についてさらに記載されているが、いずれの特許にも、これらの誘導体がPDH複合体の構造および/または発現レベル、および/または活性の制御における調節に有用であることは指摘されていない。
【0032】
PDH複合体の構造および/または活性が腫瘍活性の重大な決定要因であることが示されていることから、PDH複合体の構造および/または活性、またはさらに発現レベルの、薬剤的に許容される調節因子、およびその使用法を提供することは有益であろう。
【0033】
発明の目的及び産業上の利用可能性
結果的に本発明の目的は、癌のような細胞の過剰増殖によって特徴付けられる疾病、状態、または症候群の治療または診断に使用され、腫瘍細胞に選択的活性を示す医薬組成物の提供である。
【0034】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、投与による副作用が最少である医薬組成物の提供である。
【0035】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、可能な最少の費用で容易に製造され、かつ可能な最長期間保存できる医薬組成物の提供である。
【0036】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、特に腫瘍細胞のミトコンドリア内のPDH複合体の構造、活性、および/または発現レベルを通してミトコンドリアのエネルギー代謝を調節する医薬組成物の提供である。
【発明の概要】
【0037】
前記の目的を達成するため、本発明は、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化により特徴付けられる、ヒトを含む温血動物の癌またはその症候群のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、または症候群の治療、診断、または予防に有用な医薬組成物を広く提供し、ここで該医薬組成物は、その全てが参照としてここに取り込まれる米国特許第6,331,559号および6,951,887号、ならびに米国仮出願第60/912,598号に記載されるものを含む、少なくとも1の有効量のリポ酸誘導体、および少なくとも1のその薬剤的に許容される担体を含む。
【0038】
ミトコンドリアのエネルギー代謝を阻害することにより、本発明のリポ酸誘導体は病的な細胞におけるミトコンドリア膜電位およびその他のミトコンドリア事象両方の損失の原因となり、結果として非可逆的な細胞死が開始される。本発明のリポ酸誘導体はまた、PDKの活性化および/またはPDPの阻害によるか、またはPDH複合体のE1サブユニットの活性阻害を通した、毒性の少ない分子であるアセトインへのピルビン酸の変換の阻害により、ミトコンドリアのエネルギー代謝も阻害し得る。アセトイン合成の阻害は、酸化還元バランスを含むその他の工程を歪め、かつアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを含む有毒な副生成物産生の原因となる可能性もあり、結果としてこれらの副生成物自体は病的な細胞のミトコンドリアに非可逆的な損傷を引き起こす。
【0039】
本発明の医薬組成物は、PDK1、PDK2、PDK3、PDK4、およびそれらの各変異体またはアイソフォームの効果を調節し得る。該医薬化合物はまた、PDP1、PDP2,およびそれらの各アイソフォームの効果も調節し得る。
【0040】
本発明の医薬組成物はまた、PDH複合体中に見出されるホスホリラーゼ、キナーゼ、および脱水素酵素の酵素構成物質の発現レベルも調節し得る。この調節は、転写、翻訳、または適切な遺伝子のエピジェネティックなサイレンシングを含む後翻訳段階において起こり得る。
【0041】
TCAサイクルに基本的に付随する分子由来の化合物として、かつ解糖の伸展により、本発明の医薬組成物は腫瘍細胞内への選択的な取り込みを示す。さらにこのような選択的な腫瘍細胞への取り込みは、この医薬組成物の投与が健常な非形質転換細胞および組織に起こし得る副作用を最小化する。
【0042】
本発明の第1の実施形態によれば、リポ酸誘導体は一般式1
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す);芳香族;R3C(O)−として定義されるアシル;ヘテロアリール;R4C(=NH)−として定義されるイミドイル;有機金属アリール;アルキル−有機金属アリール;およびセミアセタール R5CH(OH)−S−から成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R4は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0043】
本発明の第2の実施形態によれば、リポ酸誘導体は一般式2
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R3C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R3C(=NH)−として定義されるイミドイル、およびR4CH(OH)−S−として定義されるヘミアセタールから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3およびR4は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0044】
本発明の第3の実施形態によれば、リポ酸誘導体は一般式3
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0045】
本発明の第4の実施形態によれば、リポ酸誘導体は一般式4
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0046】
さらに、これらの一般構造のいずれかもしくは全てが細胞またはミトコンドリア内で代謝され得ることから、上に言及された構造の代謝産物は本発明の範囲内にあることが明確に意図される。
【0047】
それぞれの一般式内で、特定のリポ酸誘導体それぞれの(R)−異性体は、(S)−異性体よりも大きな生理活性を有する。結果的にリポ酸誘導体は、単にその(R)−異性体型かまたは(R)−および(S)−異性体の混合物として存在するべきである。
【0048】
本発明のさらなる態様によれば、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または機能の制御または撹乱の変化を含む、ヒトを含む温血動物の癌のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、症候群、または症状を診断、治療、または予防するための方法が提供され、ここで該方法は、有効量のここに開示される医薬組成物をこのような動物へ投与することを含む。
【0049】
本発明のさらなる態様によれば、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化を含む、癌のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、もしくは症候群、またはそれらの症状を呈する患者の診断および利益を予想するための方法が提供され、該方法は、患者から細胞サンプルを得ること、有効量の本発明の医薬組成物をin vitroで細胞へ投与すること、およびそこから結果を得ることを含む。
【図面の簡単な説明】
【0050】
以下の図面は本発明の実施形態の図解であり、明細書および請求項の全体により包含される本出願の範囲を限定しようとするものではない。
【図1】ミトコンドリアへの出入りの両方を標的とする一般的なシグナル伝達分子およびそれらの効果を示した図である。
【図2】ミトコンドリアキナーゼおよびホスファターゼの一覧、およびミトコンドリア内でのそれらの位置を示した図である。
【図3】図3Aは、ミトコンドリア外膜でのPKAによるBADのリン酸化、およびそこにおける可逆的リン酸化の効果を示した図である。図3Bは、PDH複合体の作用による、ミトコンドリアマトリックスでのピルビン酸のアセチルCoAへの変換、およびそこにおける可逆的リン酸化の効果を示した図である。図3Cは、酸素の水への還元およびポンプによるプロトンのミトコンドリア内膜の横断におけるCOXの作用、およびそこにおける可逆的リン酸化の効果を示した図である。
【図4】ピルビン酸の解糖による産生における基質および産物の構造を図示し、ATPおよびNADHの産生ならびに付随する酵素もまた示した図である。
【図5】HIF−1によるグルコース代謝の制御を示した図である。
【図6】図6Aは、in vivoにおける、正常組織と癌組織の間でのエネルギー代謝の相違を示した図である。図6Bは、PDH複合体中のリポ酸の生体型と、本発明の医薬組成物の一部を形成するリポ酸誘導体との間の相違を示した図である。図6Cは、PDKのリポイル残基効果による、PDH複合体の制御を示した図である。
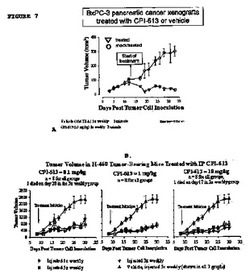
【図7】異種移植腫瘍の増殖における、本発明の医薬組成物の効果を示した図である。
【図8】三つの腫瘍細胞型および非形質転換細胞における、本発明の医薬組成物を用いた処理の効果を示した図である。
【図9】図9Aは、致死閾値またはそれを超える量の本発明の医薬組成物により処理した肺癌細胞におけるATPレベルを示した図である。図9Bは、ピルビン酸を含む培地とグルコースを含む培地中での、本発明の医薬組成物によるATP合成阻害の比較を示した図である。図9Cは、乳癌細胞と正常乳房細胞における、本発明の医薬組成物によるATP合成の阻害を比較した図である。図9Dは、肺癌細胞における、本発明の医薬組成物によるATP合成の阻害と、リポ酸および本発明の不活性型によるATP合成の阻害との比較を示した図である。
【図10】PDH複合体およびアルファケトグルタル酸(αKDH)脱水素酵素の酵素活性の腫瘍細胞ミトコンドリアでのレベルにおける、本発明の医薬組成物の効果を示した図である。
【図11】図11Aは、本発明の医薬組成物による処理またはモック処理した肺癌細胞抽出物の、二次元ゲルのウェスタン分析を示した図である。図11Bは、本発明の医薬組成物による処理またはモック処理した対の二次元ゲルサンプルを、拡大して示した図である。
【図12】図12Aは、PDH複合体E2サブユニットに共有結合した内在性リポ酸塩により調節される、PDKの制御的役割を示した図である。図12Bは、本発明の医薬組成物による腫瘍細胞PDH複合体の差次的な不活性化の、考えられる機構を示した図である。
【図13】H460肺癌細胞のミトコンドリア膜電位における、本発明の医薬組成物の効果を示した図である。
【図14】多様な腫瘍細胞型における本発明の医薬組成物による細胞死経路の、ウェスタンブロット分析の結果を示した図である。
【0051】
発明の詳細な説明
本発明は一般的に、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化を含む、温血動物の癌のような細胞の過剰増殖によって特徴付けられるもの、またはそれらの症状を含む疾病、状態、もしくは症候群、またはそれらの症状を治療、診断、または予防するための医薬組成物に方向付けられる。このような動物は、癌を含む細胞の過剰増殖によって特徴付けられる疾病ならびにその他の病態および症候群にかかりやすい、ヒト、ウマ、ウシ、イヌおよびネコを含む家畜などのような哺乳類を含む。本発明の医薬組成物は、チオクタンとしても知られる、米国特許第6,331,559号および6,951,887号、ならびに米国仮出願第60/912,598号に記載されるものを含む、少なくとも1の有効量のリポ酸誘導体、およびそのための薬剤的に許容される担体または賦形剤を含む。ミトコンドリア内に通常見出されるのみではなく、ワールブルク効果に見られるような腫瘍細胞の増大した解糖活性にも役立つ誘導体分子として、本発明のリポ酸誘導体は、腫瘍のような過剰増殖により特徴付けられる細胞および組織のミトコンドリア内への選択的送達ならびに有効な濃度について特によく適しており、これにより正常な細胞および組織は該組成物の効果から逃れる。
【0052】
本発明の医薬組成物は、PDK1、PDK2、PDK3、PDK4、およびそれらの各アイソフォームの効果を可逆的リン酸化を通して調節し得る。該医薬組成物はまた、PDP1、PDP2,およびそれらの各アイソフォームおよび/または変異体の効果も、可逆的リン酸化を通して調節し得る。これらの調節は、キナーゼもしくはホスファターゼ活性の促進または阻害のいずれかを通して起こり得る。
【0053】
ミトコンドリアのエネルギー代謝を阻害することにより、本発明のリポ酸誘導体は病的な細胞におけるミトコンドリア膜電位およびその他のミトコンドリア事象両方の損失の原因となり、結果として非可逆的な細胞死が開始される。本発明のリポ酸誘導体はまた、PDKの活性化および/またはPDPの阻害によるか、またはPDH複合体のE1サブユニットの活性阻害を通した、毒性の少ない分子であるアセトインへのピルビン酸の変換の阻害により、ミトコンドリアのエネルギー代謝も阻害し得る。アセトイン合成の阻害は、酸化還元バランスを含むその他の工程を歪め、かつアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを含む有毒な副生成物産生の原因となる可能性もあり、結果としてこれらの副生成物自体は病的な細胞のミトコンドリアに非可逆的な損傷を引き起こす。
【0054】
本発明の第1の実施形態によれば、リポ酸誘導体は一般式1
[式中、xは0〜16を表し、R1およびR2は独立して
(1)アシル基 R3C(O)−であって、R3がアルキル、アリール、または有機金属アリール基を表し、チオエステル結合を通して連結され、限定はされないがアセチルおよびブタリル(butaryl)を含み、特定の例としてビスアセチルリポ酸塩となるアシル基;
(2)チオエステル結合を通して連結される芳香族基であって、限定はされないがベンゾイルまたはベンゾイル誘導体を含み、特定の例としてビスベンゾイルリポ酸塩となる芳香族基;
(3)アルキル基 CnH2n+1であって、nが1〜10を表し、他の成分、例えば−OH、−Cl、または−NH2により置換されたこのようなアルキル基とチオエーテル結合を通して連結され、限定はされないがメチル、エチル、ブチル、デカニル、および6,8−ビスカルボモイル(carbomoyl)メチルリポ酸塩を含むアルキル基;
(4)アルケニル基 CnH2n−1であって、nが2〜10を表し、チオエーテル結合を通して連結され、限定はされないがプロピレン、2,3ジメチル−2−ブテン、およびヘプテンを含むアルケニル基;
(5)アルキニル基 CnH2n−3であって、nが2〜10を表し、チオエーテル結合を通して連結され、限定はされないがアセチレン、プロピン、およびオクチンを含むアルキニル基;
(6)鎖式、または複素環を形成するいずれの炭素の付加もしくは置換を有する脂環基による脂環式のいずれかであってよいアルキル、アルケニル、およびアルキニル基であって、限定はされないがシクロプロパン、シクロペンテン、および6,8メチル−スクシンイミドリポ酸塩を含むアルキル、アルケニル、およびアルキニル基;
(7)それらのいずれの炭素にも付加を有してよいアルキル、アルケニル、およびアルキニル基であって、限定はされないがヒドロキシルおよびアミンを含むアルキル、アルケニル、およびアルキニル基;
(8)チオエーテル結合を通して連結され、ベンゼンまたはベンゼン誘導体であってよい芳香族またはアリール基であって、限定はされないがトルエンおよびアニリンを含む芳香族またはアリール基;
(9)硫化アルキル基 CH3(CH2)n−S−であって、限定はされないがnは0〜9を表してよく、ジスルフィド結合を通して連結される硫化アルキル基;
(10)イミドイル基 CHR4C(=NH)−であって、限定はされないがnは1〜10を表してよく、チオアミド結合を通して連結されるイミドイル基;および
(11)セミアセタール基 R5CH(OH)−S−であって、R5が強力な電子求引性の置換基をもつ化合物に限定され、限定はされないがトリクロロアセトアルデヒドおよびピルビン酸を含むセミアセタール基であってよい]
またはその塩により定義される。
【0055】
上に定義されるR1およびR2は非置換または置換であってよく、スルホキシドまたはスルホン産生のために酸化できるチオエステル、例えばC−S(O)−RおよびC−S(O)2−Rもまた含んでよい。
【0056】
R1およびR2はさらに、チオスルフィン酸またはチオスルホン酸へと酸化できるジスルフィド、例えばそれぞれC−S(O)−S−RおよびC−S(O)2−S−Rを含んでよい。
【0057】
R3は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよい。同様に、R4は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよい。
【0058】
R5は−CCl3、−CF3、または−COOHを表す。
【0059】
本発明の第2の実施形態によれば、リポ酸誘導体は一般式2
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R3C(O)−、アルキル CnH2n+1、CnH2n−1として定義されるアルケニル、CnH2n−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R3C(=NH)−として定義されるイミドイル、およびR4CH(OH)−S−として定義されるヘミアセタールから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3およびR4は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5は−CCl3、−CF3、または−COOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、およびnは0〜10を表す)
またはその塩により定義される。
【0060】
本発明の第3の実施形態によれば、リポ酸誘導体は一般式3
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0061】
本発明の第4の実施形態によれば、リポ酸誘導体は一般式4
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0062】
さらに、これらの一般構造のいずれかもしくは全てが細胞またはミトコンドリア内で代謝され得ることから、上に言及された構造の代謝産物は本発明の範囲内にあることが明確に意図される。
【0063】
第1および第2両方の一般式内で、特定のリポ酸誘導体それぞれの(R)−異性体は、(S)−異性体よりも大きな生理活性を有することが観察されている。従って、本発明における使用のためには両方の異性体が意図されるが、特に好ましい実施形態では(R)−異性体を優先的に用いるか、または(S)−異性体との混合物中に(R)−異性体が存在することが特に意図される。
【0064】
本発明の医薬組成物はまた、PDH複合体中に見出されるホスファターゼ、キナーゼ、および脱水素酵素の酵素構成物質の発現レベルも調節し得る。この調節は、転写、翻訳、または適切な遺伝子のエピジェネティックなサイレンシングを含む後翻訳段階において起こり得る。
【0065】
本発明の組成物は、薬剤的に許容される担体または賦形剤をさらに含んでよい。薬剤的に許容される担体の例は当該技術分野において公知であり、医薬組成物中で通常用いられるもの、例えば限定はされないが、抗酸化剤、緩衝剤、キレート剤、香味料(flavorants)、着色剤、保存料、生物学的利用能を増大させるための吸収促進剤、抗菌剤、およびそれらの組み合わせを含む。このような添加物の量は望まれる性質に依存し、当業者によって容易に決定できる。
【0066】
通常本発明の医薬組成物は、塩、緩衝剤、保存料、および適合する担体を、任意にその他の治療的成分と組み合わせて含んでよい。医薬に用いられる場合、塩は薬剤的に許容されなければならないが、薬剤的に許容されない塩が、その薬剤的に許容される塩の調製のために便利に用いられてよく、本発明の範囲から除外されるものではない。このような薬理学的および薬剤的に許容される塩は、限定はされないが、以下の酸:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、p−トルエンスルホン酸、酒石酸、クエン酸、メタンスルホン酸、マロン酸、ギ酸、コハク酸、ナフタレン−2−スルホン酸、およびベンゼンスルホン酸から調製されるものを含む。また、薬剤的に許容される塩は、カルボン酸基のナトリウム、カリウム、またはカルシウム塩のような、アルカリ金属またはアルカリ土類塩として調製できる。
【0067】
本発明はさらに、細胞に有効量の少なくとも1の治療薬または診断薬を送達することによる、患者を治療薬または診断薬によって治療または診断するための、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットの構造、発現、および/または活性の制御または撹乱の変化を含む方法を、細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、もしくは症候群、またはそれらの症状を予防、診断、または治療するために提供する。癌の改良された治療としてのPDH複合体の調節が特に意図され、これは腫瘍細胞増殖、血管新生、転移性増殖、アポトーシスの制御による原発腫瘍の治療、および外科的な除去の後または同時に起こる微小転移の発生の治療;および原発腫瘍の放射線またはその他の化学療法による治療を含む。本発明の医薬組成物は、原発性または転移性のメラノーマ、リンパ腫、肉腫、肺癌、肝臓癌、ホジキンおよび非ホジキンリンパ腫、白血病、子宮癌、子宮頸癌、膀胱癌、腎癌、大腸癌、ならびに腺癌、例えば乳癌、前立腺癌、卵巣癌、および膵臓癌のような癌の型に有用である。
【0068】
治療および診断への応用のため、該医薬組成物は、薬剤的に許容される担体と組み合わせた場合に患者へ直接投与できる。この方法は、治療薬もしくは診断薬を単独、または有効量のその他の治療薬もしくは診断薬、限定はされないが解糖阻害剤、微小管相互作用剤、細胞増殖抑制剤、葉酸阻害剤、アルキル化剤、トポイソメラーゼ阻害剤、チロシンキナーゼ阻害剤、ポドフィロトキシンまたはその誘導体、抗腫瘍剤、抗生剤、化学療法剤、アポトーシス誘導剤、抗血管新生剤、ナイトロジェンマスタード、核酸挿入剤、およびそれらの組み合わせ、と組み合わせて投与することにより実行されてよい。このような治療薬は、その他の代謝阻害剤をさらに含んでよい。このような治療薬の多くは公知である。組み合わせによる治療法は、治療法に対する患者の反応を増幅または確実にする必要に応じたこのような状態の治療において、同時、連続的、または別々の使用を提供する。
【0069】
本発明の方法は、医学的に許容される投与のいずれの様式を用いて実行されてもよく、臨床的に許容されない有害作用を起こすことなく、有効濃度の活性化合物を産生する。製剤は特に非経口投与に適することが好ましいが、本発明の組成物はエアロゾル、スプレー、散剤、ゲル、ローション、クリーム、坐薬、軟膏などの形態において、吸入、経口、外用、経皮、経鼻、眼内、肺内、直腸内、経粘膜、静脈内、筋肉内、皮下、腹腔内、胸腔内、胸膜内、子宮内、腫瘍内、または点滴の方法論または投与のためにもまた処方できる。このような製剤が望まれる場合、望まれる一貫性およびその他の性質を製剤に与えるため、当該技術分野において公知のその他の添加物が含まれてよい。
【0070】
当業者は、治療薬または診断薬の特定の投与様式は、投与が疾病、状態、症候群、またはそれらの症状の治療、診断、または予防;治療または診断される医学疾患の重症度;および治療的有効性に要求される投薬量のためであるか否かに関わらず、選択された特定の薬剤に依存することを認識するであろう。例えば、白血病治療のための抗癌剤投与の好ましい様式は静脈内投与を含み得るが、皮膚癌治療の好ましい方法は、局所または皮内投与を含んでよい。
【0071】
ここで用いられる「有効量」は、望まれる治療効果または診断効果が達成される、治療薬もしくは診断薬の投薬量または複数の投薬量を指す。一般に治療薬または診断薬の有効量は、用いられる特定の薬剤の活性;その薬剤の代謝の安定性および作用の長さ;種、年齢、体重、一般的健康、食事状態、性別および被験体の食事制限;投与の様式および時間;排泄速度;場合により薬物の組み合わせ;および治療される特定の状態の症状および/または重症度の程度によって変動し得る。望まれる結果を得るための1日当たりの1回または数回の投与における正確な投薬量は、当業者によって過度の実験なしに決定でき、かつ該投薬量は望まれる治療効果を達成するためか、またはいずれの合併症の現象において、個々の実行者により調整されてよい。重要なことは、癌治療に用いる際、使用される治療薬の投薬量は正常細胞には実質的に無害であるものの、腫瘍細胞の死滅には十分でなければならないことである。
【0072】
本発明の医薬組成物に含まれる治療薬または診断薬は、患者へ安全に投与できる最大量までの、望まれるいずれの量においても調製できる。治療薬または診断薬の量は、0.01mg/mL未満から1000mg/mLを超えるまで、好ましくは約50mg/mLに変動してよい。
【0073】
一般に本発明の医薬組成物は、PDH複合体の構造および/または活性の調節に有効な量を患者へ投与するために十分な様式で送達され得る。従って投薬量は、約0.3mg/m2から2000mg/m2、好ましくは約60mg/m2に変動してよい。投薬量は単一用量において、または個別に分けられた用量の形、例えば1日当たり1から4以上において投与されてよい。被験体の反応がある用量において不十分な場合は、より高い用量(または異なる、さらに限局性の送達経路による有効なより高い用量)が、患者の耐性の範囲まで用いられてよい。1日当たりの複数の用量は、治療薬もしくは診断薬の適切な全身性または標的濃度を達成するために意図される。
【0074】
さらに本発明の別の実施形態によれば、本発明のリポ酸誘導体はin vitroでの診断薬および予測薬(predictive agents)として用いられてよい。先に述べたように、問題となる特定の腫瘍細胞または細胞型に依存して、様々なリポ酸誘導体は、PDH複合体の調節を通した異なる腫瘍の種類の阻害について多かれ少なかれ有効であり得る。従って、例えば診断または適切な化学療法的戦略の選択が困難であり得る場合、in vitroでの腫瘍細胞の培養を、特定の腫瘍細胞型を標的にすることが知られているリポ酸誘導体と共に試験することにより、腫瘍型および有効な治療を特定するための別のアプローチが提供される。
【0075】
図面に戻ると、図6Aは、正常組織および腫瘍細胞のin vivoでのエネルギー代謝において考えられる多数の相違を図示する。相当する条件下で正常細胞に比べると、腫瘍細胞はしばしばATP産生のためにミトコンドリアのエネルギー代謝よりも細胞質の解糖に強く依存する。PDH複合体の発現および制御の変化は、明らかにこの腫瘍特異的な適応の一部である。これらの効果を生み出す、PDHの触媒成分のレベルの減少および/または抑制性PDKのレベルの増加により、腫瘍細胞は、PDH複合体を攻撃する薬剤に対して正常細胞よりもさらに脆弱となるであろう。
【0076】
図6Bは、リポ酸がPDH複合体中でピルビン酸からのアセチルCoA合成に関わる正常の反応を触媒することから、この構造を示す。リポ酸はin vivoでそのカルボキシル末端を通し、非ペプチドアミド結合によって、E2リポイルドメイン活性部位におけるリジンのイプシロンアミノ基へ連結する。PDH E2結合リポ酸塩の酸化/還元/アセチル化状態は、E1αPDHサブユニットのリン酸化の不活性化を制御することによってPDH活性を制御する、キナーゼおよびホスファターゼによりモニターされることも認められる。この図面はまた、本発明において使用されてよい三つの代表的なリポ酸誘導体の構造も示す。細胞培養において、CPI−613およびCPI−045は高い抗癌効力を持つが、CPI−157は活性が少ないかまたは無であり、幾つかの実験ではコントロールとして有用である。
【0077】
図6Cは、E2をその結合リポ酸塩であるE1と共に含むPDH複合体成分と、調節性PDKとの間の関連性を示す。高レベルのアセチルリポ酸塩またはジヒドロリポ酸塩(図示していない)はPDKを活性化し、次にこれがPDH複合体触媒作用の第1段階を触媒するサブユニットであるE1αを不活性化することによって、PDH複合体を通したさらなる流れを抑制する。図3Aに見られるように、この工程はPDH複合体を通した炭素/エネルギーの流れの調速機として作用し、かつこの調節工程は明らかに、様々な腫瘍細胞のエネルギー代謝を支持するために実質的に変更される。
【0078】
図7は、異種移植腫瘍の増殖における本発明の医薬組成物の効果を示す。実施例2に記載するように、脇腹の背側に細胞を皮下移植した。次に、図に示す日にマウスへの薬物(またはビヒクルのみ;「モック」)の腹腔内注射を開始した。左のパネルに、週に3回1mg/kgの本発明またはビヒクルコントロールと共に注射した膵臓腫瘍モデルを示す。この実験は、BxPC−3細胞により行った2実験、およびAsPC−1細胞により行った2実験の代表である。右の三つのパネルは、週に1回(丸)、週に3回(逆三角形)、または週に5回(ビヒクル処置は三角形、薬物処置は四角形)のいずれかにより示される濃度と共に注射したH460肺腫瘍モデルを示す。この実験は、H460細胞により行った4実験の代表である。
【0079】
図8は、三つの腫瘍細胞型および非形質転換細胞型(MDCK)を、200〜300μM(「処理」)またはモック処理(「モック処理」)のいずれかで、本発明の医薬組成物により処理した効果を示す。細胞は10%血清を含む適切な組織培地により、または48時間処理した。実施例2に記載の方法論を通し、三つの癌細胞株にアポトーシスまたはアポトーシス様経路(図11も参照)による広範な細胞死が観察される。対照的に、非形質転換MDCK細胞は、明らかにこの用量での薬物処理によって影響されない。
【0080】
図9Aは、致死閾値(10%血清中、200μM)またはそれを超える量の本発明の医薬組成物により処理したH460肺癌細胞におけるATPレベルを示す。破線は表示される濃度での処理を示す。対応するテクスチャの実線は、表示される時間後に薬物を除去し、薬物を含まない培地中で60分間回復させる処理を示す。一組の矢印(Block arrows)は、ATP回復の間隔を示す。
【0081】
図9Bは、ピルビン酸(ピルビン酸メチルの形)が主要な炭素原である培地(破線)とグルコースが主要な炭素原である培地(実線)中での、ATP合成の阻害を比較する。本発明の医薬組成物は、両方の培地中で、同じ閾値濃度において最終的に細胞死を引き起こすが、細胞の全ATPレベルの早い枯渇はピルビン酸を含む培地中で高度であり、グルコースを含む培地中では見られないことが認められる。また、細胞死の開始は200μMよりも300μMの薬物濃度でより迅速である。
【0082】
図9Cは、SK−Br−3乳癌細胞とHMEC正常乳房細胞における、本発明の医薬組成物によるATP合成の阻害を比較する。図6Aおよび図6Bにその結果が示される実験とは対照的に、これらの実験は無血清培地(MEBM)中で行った。結果として薬物の致死閾値は、約50μMと低めであった。正常細胞サンプルにおける22時間目のATPレベルの小さな低下は薬物用量には関連せず、普通の実験的変動を反映していることに留意されたい。
【0083】
図9Dは、本発明の医薬組成物(左のグラフ)、リポ酸(中央のグラフ)、および本発明の不活性型(右のグラフ)による、H460肺癌細胞におけるATP合成の阻害を比較する。図9Cに示すように、これらの実験は、薬物の致死閾値が約50μMとなるように無血清培地中で行った。
【0084】
図10は、PDH(PDC)および(αKDH)の酵素活性の、腫瘍細胞ミトコンドリアでのレベルにおける、本発明の医薬組成物(10%血清を含むDMEM中で400μM)の効果を図示する。PDHは強力に阻害されるが、αKDHはそうでないことが認められる。実施例2に記載するように、精製したミトコンドリア抽出物中の酵素活性レベルは、添加炭素源に反応するレサズリン還元を用いて測定する。バックグラウンドラインは、添加炭素源なしのレサズリン還元に相当する。
【0085】
次に図11Aでは、本発明の医薬組成物により処理(+)またはモック処理(−)した(10%血清を添加したRPMI培地中で、400μMで120分間)H460肺癌細胞抽出物の、二次元ゲルのウェスタン分析を行った。ウェスタン転写は、PDH複合体のE1αおよびE2サブユニットに対するモノクローナル抗体のカクテルを用いて探査した。ウェスタン転写はE2に合わせた。薬物処理サンプル中のE1には、高リン酸化型の実質的に高いレベルおよび低リン酸化型の減少したレベルが認められる。左の垂直な白線は、ゲル整列の基準の一つである、E2サブユニットの移動度を示す。右の垂直な白線は、推定的、酵素的に活性のある成分である、リン酸化の少ないE1α型を通る。
【0086】
図11Bは、本発明の医薬組成物による処理またはモック処理した対の二次元ゲルサンプルを、拡大して示す。エレメントAは、図8Aの一部の拡大である。エレメントBは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で180分間処理(+)されたSK−Br−3乳癌細胞である。エレメントCは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で240分間処理(+)されたHMEC正常乳房上皮細胞である。エレメントDは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で240分間処理(+)されたSK−Br−3乳癌細胞である。垂直な白線は、推定的、酵素的に活性のある成分である、リン酸化の少ないE1α型を通る。
【0087】
図12Aおよび12Bは、本発明の医薬組成物のin vivoでの強力で選択的な抗癌効果に対する作業仮説を示す。例えば図12Aは、PDC E2サブユニットに共有結合した内在性リポ酸塩により調節される、PDKの制御的役割を示す。PDKは通常、高レベルの還元および/またはアセチル化リポ酸塩に反応してPDCを不活性化し、これは腫瘍細胞内で明らかに実質的に修飾される工程である。
【0088】
同時に図12Bは、PDC内における、PDKのその基質であるPDC−E1に対する比の大きな量的相違を示し、このことはin vivoで正常および腫瘍細胞を区別すると考えられる。正常細胞では、低レベルのPDKがPDH複合体の周囲をhand−over−handに「歩く」(その二つの二量体サブユニットを通して)と考えられており、徐々にE1をリン酸化する。このリン酸化は、PDPの脱リン酸化と定常的に平衡状態である(図示していない)。ここに図示される作業仮説では、チオクタンが、通常アセチルリポ酸塩および/またはジヒドロリポ酸塩が結合するのと同じ部位を通じてPDKを刺激し、これにより1以上のPDKアイソフォームを不活性なE1αへと人工的に刺激する。癌細胞では、さらに高レベルのPDKが、チオクタンによりさらに効果的にこの刺激を作り出し、PDCの酵素活性およびミトコンドリアエネルギー代謝を停止させるであろう。
【発明を実施するための形態】
【0089】
以下の限定されない実施例は、本発明の医薬組成物についての理解を促進するために提供される。
【実施例1】
【0090】
チオクタンの化学合成
リポ酸誘導体(すなわちチオクタン)CPI−613およびCPI−157は、米国特許第6,331,559 B1号および米国特許第6,951,887 B2号に記載の方法の改変により、6,8−ビスメルカプトオクタン酸を出発物質として用いて合成した。チオクタンCPI−045は、米国特許第6,331,559 B1号に記載の通りに合成した。
【0091】
三つのチオクタンの構造解析について以下に述べる。CPI−045とCPI−613の独立した複数の合成は、これらの抗癌特性について区別できなかった。異種移植(図7)およびATP測定(図9)に用いたCPI−613の純度は99%を超えていた。その他の全ての調製品は98%を超える純度であった。
【0092】
CPI−613:6,8−ビスベンジルスルファニルオクタン酸:白色の結晶性固体、m.p.65〜66℃(lit.167.5〜69°);1H−NMR(250MHz,CDCl3):δ7.15−7.4(m,10H)、3.66(s,2H)、3.64(s,2H)、2.52−2.62(m,1H)、2.50(t,J=7.6Hz,2H)、2.29(t,J=7.6Hz,2H)、1.2−1.8(m,8H);13C−NMR(62.9MHz,CDC13):δ179.6、138.6、138.5、128.9、128.8、128.5、128.4、126.9、44.1、36.4、35.1、34.4、33.8、28.7、26.0、24.4。
【0093】
CPI−157:6,8−ビスエチルスルファニルオクタン酸:無色の油;TLC
(EtAc:ヘキサン:HAc,200:200:1v/v):Rf=0.60;1H−NMR(300MHz,CDCl3):δ2.64−2.76(m,1H)、2.65(t,J=7.5Hz,2H)、2.52(q,J=7.5Hz,2H)、2.49(q,J=7.2Hz,2H)、2.36(t,J=7.4Hz,2H)、1.40−1.85(m,8H)、1.25(t,J=7.2Hz,3H)、1.22(t,J=7.5Hz,3H);13C−NMR(75MHz,CDCl3):δ180.0、44.3、34.6、33.9、28.9、26.2、25.9、24.5、24.2、14.9、14.7;IR(フィルム):2963、1708、1449、1423、1283、1263cm−1。
【0094】
CPI−045:6,8−ビスベンゾイルスルファニルオクタン酸:無色の粘性油;TLC(ヘキサン:EtAc:HAc,100:50:1v/v):Rf=0.30;1H−NMR(250MHz,CDCl3):δ7.9−8.1(m,4H)、7.38−7.60(m,6H)、3.8−4.0(m,1H)、3.0−3.3(m,2H)、2.34(t,J=7.1Hz,2H)、1.4−2.2(m,8H);13C−NMR(62.9MHz,CDCl3):δ191.7、191.5、179.7、137.0、136.9、133.3、128.5、127.3、127.1、43.6、35.0、34.6、33.8、26.4、26.2、24.3;IR(フィルム):2973、1710、1704、1667、1665、1662、1448、1207、1175、911、773、757、733、688、648cm−1。
【実施例2】
【0095】
チオクタンの抗癌効果決定のために用いた方法
細胞:ヒト腫瘍細胞株はATCCから入手し、ATCCの勧告に従って増殖させた。ヒト乳房上皮細胞(HMEC)、末梢気道上皮細胞(SAEC)、および正常ヒト表皮角化細胞(NHEK)の初代細胞は、LONZA Walkersville, Inc(ウォーカーズビル、メリーランド州)から得た。各細胞株は、開発した供給業者から購入した適切な培地中で、供給者の指示に従って維持および増殖させた。ここに報告する実験では、3ないし6回継代した正常細胞を使用した。
【0096】
腫瘍増殖阻害の研究:CD1−Nu/Nu雌マウスに、ヒトBxPC−3もしくはAsPC−1膵臓腫瘍細胞、またはH460 NSCLCを皮下(SC)注射によって移植した。約8〜12日後、図の説明文に示される用量およびスケジュールによりマウスに腹腔内(IP)注射した。薬物またはビヒクルは、体重25gあたり約2ml注射した。薬物濃度は1.25mg/ml(約3.1mM)以下であった。ビヒクル/溶媒は、25mM以下のトリエタノールアミン水溶液により構成された。モック処置動物に注射されたビヒクルは、この実験で最も高い薬物用量が注射された溶媒と常に同一であった。マウスは、健康状態および死亡率について毎日モニターされた。体重および腫瘍の体積を、処置前には毎日、処置中および処置後には週に約3回評価した。マウスはStony Brook大学動物施設において、12時間の明/暗サイクルに置かれ、自由に摂食し、施設の指針に従って飼育された。
【0097】
細胞死アッセイ:細胞生存率評価のために、ほとんどの場合CellTiter−Gloアッセイ(Promega)を、チオクタンによるATP合成の初期の阻害によって混乱させないための十分に長い時間使用した(図9)。典型的な実験では、黒く透明な底の96ウェルプレートに、細胞をウェルあたり5,000細胞まいた。18〜25時間後に培地を、薬物溶媒(血清を含む培地中に2.8mM、無血清培地中に0.7mMの、トリエタノールアミン水溶液)または同じ溶媒中のチオクタンCPI−613を含む新鮮な培地に交換した。製造者の指示に従い、薬物用量に応じて、薬物添加後24または48時間後にアッセイを行った。
【0098】
幾つかの場合には、細胞は48ウェルプレートにウェルあたり10,000細胞まき、18〜25時間後に培地を、薬物溶媒(最終濃度が1%のEtOH)または同じ溶媒中の様々な濃度のチオクタンCPI−045を含む新鮮な培地に交換した。残りの実験の間、細胞を溶媒または薬物を含む培地中にとどめた。薬物添加後24、48、および72時間後にプレートを点検し、コンフルエンスのパーセンテージから細胞数を推定した。これらの条件下でチオクタンにより誘導された細胞死は、閾値に近い用量で高度にアポトーシス性であり、細胞数の推定は非常に信頼できる死の指標である(図10)。72時間後にも完全な細胞が残っていた場合には、トリパンブルー排除試験を行った。
【0099】
表2に、腫瘍細胞に対するチオクタンのin vitroでの作用についてのデータを示す。CPI−613および/またはCPI−045による殺傷に対する感受性について本発明者らが調査した、ヒト腫瘍および初代ヒト細胞を収載する。「+」は、約200〜300μM(10%血清存在下で)および無血清培地中で約50μMの用量でアポトーシスまたはネクローシス様の細胞死を起こした細胞を示す(図8および図9)。「−−」は、相当する培地中で細胞死誘導のために5倍高い薬物用量を必要とした細胞を示す。「nt」は、未試験の組み合わせを示す。図8でのMDCK正常細胞と同様に、全ての腫瘍細胞株は10%血清を含む適切な培地中で分析された。加えて、HMEC、SAEC、NHKC初代ヒト細胞、およびSK−BR−3、A549およびH460腫瘍株もまた、適切な無血清培地中で分析された。初代細胞は接触阻止され、形質転換細胞は同程度の濃度であった。
【0100】
ATPアッセイ:黒く透明な底の96ウェルプレートに、細胞をウェルあたり5,000細胞まいた。18〜25時間後に培地を、薬物溶媒(トリエタノールアミン)またはチオクタン(CPI−613またはCPI−157)またはリポ酸を含む新鮮な培地に、示される時間間隔および薬物濃度において交換した。トリパンブルー排除を用い、細胞の生存率および完全性を、薬物を除去した後の回収率によって評価した。ATPは、CellTiter−Glo luminescence assay(Promega)を用い、製造者の指示に従って測定した。全ての測定は二重に行い、高い一貫性が示された。平均の標準誤差は、測定値の0.1〜2%の範囲であった。その結果、図9ではエラーバーを省略した。図9のピルビン酸メチル培地は、10%透析ウシ胎仔血清、5mM HEPES(pH7.4)、および10mMピルビン酸メチル(Sigma−Aldrich)を補充した、グルコースを含まないRPMI(Invitrogen)から成り、対応するグルコース培地は従来のRPMI(Invitrogen)であった。
【0101】
PDHおよびαKDH酵素アッセイ:腫瘍細胞を15cmプレート内で80%コンフルエンスに増殖させ、示されているようにCPI−613によって処理した。MoreadithおよびFiskum1の方法に従ってミトコンドリアを分離した。ミトコンドリアを0.4%ラウリルマルトシド中で溶解した。50μlのミトコンドリアライセートを96ウェルプレートへ加えた。50μlの反応混合物(50mMトリス、pH7.5、2mM β−NAD+、225μMv TPP、2mMピルビン酸またはα−ケトグルタル酸、150μMコエンザイムA、2.6mMシステイン、1mM MgCl2)をミトコンドリアライセートに加え、混合物を37℃で45分間インキュベートした。この時点で、混合物に15μMのレサズリンおよび0.5U/mlのジアホラーゼを加え、さらに5分間インキュベートした。マイクロプレートリーダー(Fluorostar)内での530nmの励起波長および590nmの発光波長を用いた蛍光測定により、NADHの産生をモニターした。全ての測定は二重に行い、高い一貫性が示された。平均の標準誤差は、測定値の0.3〜4%の範囲であった。その結果、図10ではエラーバーを省略した。
【0102】
E1αのリン酸化:
2−Dゲルのための細胞ライセート:細胞を60mmディッシュ内で95%コンフルエンスに増殖させ、示されているように薬物または溶媒によって処理した。細胞をin situで450μlの溶解緩衝液A[455μl Zoom 2D protein solubilizer 1(Invitrogen)、2.5μl 1Mトリスベース、5μl 100Xプロテアーゼ阻害剤カクテル(EDTAを含まないComplete mini、Roche);5μl 2M DTT]により溶解した。細胞ライセートを1.5ml微量遠心チューブへ移し、氷上で50%出力にて15段階の超音波処理を行った。室温での10分間のインキュベーション後に2.5μlのジメチルアクリルアミド(DMA、Sigma−Aldrich)を加え、ライセートをさらに10分間インキュベートした。5μlの2M DTTを加えて、過剰のDMAを中和した。ライセートを16,000xgで15分間遠心した。
【0103】
2−Dゲル:Zoom Benchtop proteomics system(Invitrogen)を、製造者の指示に従って使用した。簡単に述べると、30〜50μlのライセートを0.8μl pH3〜10両性電解質、0.75μl 2M DTTと混合し、Zoom 2D protein solubilizer 1にて150μlにした。150μlのサンプルをIPG runner内に装填し、pH3〜10のNL IPGストリップを加えた。ストリップを室温で一晩浸漬した。等電点(isolectric)電気泳動には段階的プロトコルを使用した(250V、20分;450V、15分;750V、15分 2000V、30分)。ストリップを1Xローディング緩衝液で15分間、続いて1Xローディング緩衝液および160mMヨード酢酸で15分間処理した。ストリップはNuPAGE 4−12% Bis Tris ZOOMゲル(Invitrogen)で電気泳動した。
【0104】
表2 腫瘍および初代細胞に対するチオクタンのin vitroでの効果
【0105】
ウェスタン:タンパク質はPVDF 4.5μmメンブレンにブロットした。mAb(Invitrogen)を用いて、PDH E1αおよびE2を検出した。
【0106】
カスパーゼ−3およびPARPの切断:Roy and Nicholson.2に従い、切断されたカスパーゼ−3をウェスタンブロットにて検出した。簡単に述べると、薬物または溶媒処理後に細胞をかき取り、培地/細胞/アポトーシス小体の混合物を6000xgにて遠心した。ペレットを溶解緩衝液C(4M尿素、10%グリセロール、2%SDS、0.003%BPB;5% 2−メルカプトエタノール)により溶解した。ウェルあたり30μgの全細胞ライセートタンパク質を、12%ビス−トリスゲルにロードした。タンパク質はPVDF 4.5μmメンブレンにブロットした。抗カスパーゼ3mAb(マウスモノクローナル[31A1067];abcam)により、プロ−および活性化カスパーゼ3を検出した。モノクローナル抗ポリ(ADPリボース)ポリメラーゼ抗体、クローンC−2−10(Sigma−Aldrich)を用いて、PARP切断を検出した。
【0107】
ミトコンドリアCa2+の検出:細胞を35mmガラス底プレート(BD Biosciences)に3x105まき、一晩増殖させて、示されているように薬物または溶媒によって処理した。次に細胞をカルシウム色素Fluo−4、X−Rhod−1またはRhod−2(4μM、Invitrogen)と共にフェノールレッドを含まない培地中にロードし、37℃で10分間インキュベートした。細胞をPBSで1回洗浄し、Axiovert 200M(Zeiss)デコンボリューション顕微鏡を用い、固定した露光時間で、FITCフィルターを用いて画像を得た。蛍光の定量化は、製造者より提供されたソフトウェアを用いて行った。X−Rhod−1およびRhod−2からは同様の結果が得られ(図13)、これらの色素はミトコンドリアCa2+シグナルを測定していることが示された3−4。
【0108】
参考文献
1.Moreadith RW and Fiskum G. Isolation of Mitochondria from Ascites Tumour−Cells Permeabilized with Digitonin. Analytical Biochemistry 137,360−367(1984).
2.Roy S and Nicholson DW.Criteria for identifying authentic caspase substrates during apoptosis.Apoptosis 322,110−125(2000).
3.Gerencser AA and Adam−Vizi V. Selective,high−resolution fluorescence imaging of mitochondrial Ca2+ concentration.Cell Calcium 30,311−321(2001).
4.Gyorgy H,Gyorgy C,Das S,Garcia−Perez C,Saotome M,Roy SS,and Yi MQ. Mitochondrial calcium signalling and cell death:Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis.Cell Calcium 40,553−560(2006).
【実施例3】
【0109】
チオクタンによるミトコンドリア膜電位およびCa2+取り込みの撹乱
チオクタンによるATP合成の阻害における基質効果(図9)は、該薬物がミトコンドリアマトリックス内でTCAサイクルに干渉することを示す。これが事実であれば、ミトコンドリア膜電位は致死閾値用量以上で損傷し得ると本発明者らは予測する1。電位感受性の色素であるTMREを用い、本発明者らは予想された効果を観察する(図13)。ミトコンドリア膜電位は、薬物処理の開始によって迅速に低下する。膜電位低下の動態は、ミトコンドリア基質存在下でのATP合成の損失によく似ている(図9)。
【0110】
ミトコンドリアでのATPの枯渇は、小胞体を含む細胞質貯蔵から放出されたCa2+の取り込みを含む恒常性反応を誘発することが知られている2。さらに、このCa2+のミトコンドリアマトリックス内への移入には、ミトコンドリア膜電位が必要であると考えられている。従って、進行的に損なわれる膜電位を考えると、本発明者らは、致死閾値以上でのチオクタン処理が持続性のCa2+の細胞質放出と共にイオンの一過性のミトコンドリアへの取り込みを起こし得ると予測する。本発明者らはミトコンドリアCa2+の測定にX−Rhod−1およびRhod−2、および細胞質Ca2+の測定にFluo−4を用いて、これらの予想される効果を観察する(図13)。
【0111】
致死閾値かまたはそれよりもわずかに高いCPIの用量において、約2時間までに(図9および13を比較されたい)ミトコンドリア膜電位が低下するに従い、この最初のミトコンドリアの一過性Ca2+の取り込みは減衰する。この4〜6時間後、カルシウム依存性細胞死経路の開始に付随すると推定される、第2の大きなミトコンドリアCa2+ピークが続く3。
【0112】
参考文献
1.Garrett R and Grisham CM. Biochemistry.Thomson Brooks/Cole,Southbank,Vic,Australia; Belmont,CA.(2007).
2.Graier WF,Frieden M,and Malli R.Mitochondria and Ca2+ signaling:old guests,new functions.Pflugers Archiv−European Journal of Physiology 455,375−396(2007).
3.Gyorgy H,Gyorgy C,Das S,Garcia−Perez C,Saotome M,Roy SS,and Yi MQ. Mitochondrial calcium signalling and cell death:Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis.Cell Calcium 40,553−560(2006).
【実施例4】
【0113】
チオクタンにより誘導される多様な細胞死プログラム
詳しい機構は完全には理解されていないが、ある状況下では、ミトコンドリアエネルギー代謝の減少は細胞死経路へ入るための決定に関連することが知られている1−3。閾値を超えるが、この最小の殺傷用量の2倍以内であるチオクタン用量においては、試験された全ての癌細胞型が形態的には主にアポトーシス様である細胞死を起こす(図8)。この条件下でのアポトーシス様の死は、従来のアネキシン免疫染色およびTUNEL DNA末端標識アッセイによって確認した(結果は提示していない)。
【0114】
さらに高い薬物用量(閾値の2倍を超える値よりも多い)において、活性のあるチオクタンは、形態的なアポトーシスへの関連を示さずに細胞死を誘導し(再プレートによる生存率アッセイ、およびトリパンブルーまたはプロピジウム排除による評価として)、これはネクローシス様経路を示唆している(結果は提示していない)。
【0115】
これらのデータは、チオクタンCPI−613によるミトコンドリアエネルギー代謝の阻害が、細胞死誘導に正確に関連することを確証する。
【0116】
異なる多様な細胞死経路についての不活性化変異を含むと知られているかまたは推定される多様な腫瘍細胞4は全て、ほとんど同様のチオクタン用量において殺傷される(図8および表2)という知見が著しい。この知見はこれらの薬物が、複数の、潜在的に重複性である遠位細胞死遂行経路に関与できるマスターシグナルを誘導することを示唆する5。
【0117】
この可能性に一致して、本発明者らは一般的なカスパーゼ阻害剤であるZ−VAD−FMKが、チオクタンで処理された細胞における細胞死の形態をわずかに変化させるが、該薬剤の致死閾値用量では識別可能な効果をもたないことを見出している。
【0118】
チオクタン誘導性の細胞死が複数の末端遂行機構を通して進行できる可能性をさらに試験するため、本発明者らは異なる細胞死経路の指標であるカスパーゼ−3およびPARP−Iによる切断5について検討した。本発明者らは、チオクタンCPI−613およびCPI−045の両方が、異なる細胞において非常に変化しやすいレベルのこれらの二つの切断現象を誘導することを見出している(図14)。
【0119】
まとめるとこれらの結果は、チオクタンが、薬物用量および細胞型に依存して死への戦略的なコミットメントを誘導することができ、その決定の末端での戦術的な遂行について作動性であることを示す。
【0120】
参考文献
1.Watabe M and Nakaki T. ATP depletion does not account for apoptosis induced by inhibition of mitochondrial electron transport chain in human dopaminergic cells.Neuropharmacology 52,536−541(2007).
2.Yuneva M,Zamboni N,Oefher P,Sachidanandam R,and Lazebnik Y. Deficiency in glutamine but not glucose induces MYC−dependent apoptosis in human cells.Journal of Cell Biology 178,93−105(2007).
3.Skulachev VP. Bioenergetic aspects of apoptosis,necrosis and mitoptosis.Apoptosis 11,473−485 (2006).
4.Johnstone RW,Ruefli AA,and Lowe SW. Apoptosis:A link between cancer genetics and chemotherapy. Cell 108,153−164(2002).
5.Cregan SP,Dawson VL,and Slack RS. Role of AIF in caspase−dependent and caspase−independent cell death.Oncogene 23,2785−2796(2004).
【実施例5】
【0121】
リポ酸誘導体類似体の構造
以下に提示する様々なリポ酸誘導体類似体の限定されない例は、製造され、ここに開示される。
【0122】
前述の考察は、本発明の単なる典型的な実施形態を開示および記述するものである。当業者はこれらの考察および添付の請求項から、以下の請求項に定義される本発明の精神および範囲から逸脱することなく、ここに様々な変更、改変、および変動があってよいことを容易に認識するであろう。さらに、ここには典型的な実施形態が表されているが、その他の当業者は本発明のその他の設計または使用について気付くであろう。従って、本発明はその典型的な実施形態と関連して記述されているが、設計および使用の両方に対する多数の改変が当業者に明らかとなるであろうこと、およびこの応用がそのいずれの適応または変動も網羅するように意図されることが理解されるであろう。それ故本発明は、請求項およびその均等物のみによって限定されることが明白に意図される。
【技術分野】
【0001】
本発明は、治療用および診断用の組成物に関し、さらに詳細には、癌細胞を含む過剰増殖により特徴付けられる細胞内への選択的な取り込みを示し、かつ酵素の構造、発現、および/または活性の制御または撹乱を調節し、これによってそれらの細胞の検出および処置または破壊を促進する医薬組成物、およびそれらの使用方法に関する。さらに特異的には、これらの薬剤は、大部分の癌に付随する修飾されたピルビン酸脱水素酵素(PDH)複合体などとして観察される、変化したミトコンドリアエネルギー代謝の活性またはその制御を標的とし、撹乱する。
【背景技術】
【0002】
ミトコンドリアは、真核細胞におけるエネルギー産生および生と死の工程に対する主要なコントロールセンターである。残りの細胞との様々な連絡機構が存在するが、可逆的リン酸化はミトコンドリアの機能を制御するための重要な手段である(Pagliarini D.J.and Dixon J.E.(2006).Mitochondrial modulation: reversible phosphorylation takes center stage? TRENDS in Biochem.Sci.31:26−34、本明細書の各所に参照として取り込まれる)。報告されたミトコンドリアキナーゼ、ホスファターゼ、およびリン酸化タンパク質の数の着実な増加は、リン酸化がミトコンドリアでの工程の制御における共通のテーマとして現れているであろうことを示唆する。ミトコンドリア酵素の構造、機能および活性制御に付随する病理的または遺伝的な変化は、疾病の治療に貢献し、かつその重要な標的であり得る。
【0003】
様々な細胞機能の収束点および制御因子としてのそれらの役割に一致して、ミトコンドリアはアポトーシス、活性酸素種(ROS)の産生、および細胞ATPの90%を超える産生を含む多数の代謝工程において決定的な役割を持つ。さらに、細胞の増殖および分裂に従って新たなミトコンドリアが作られる必要があり、この工程それ自体が、核およびミトコンドリアDNAの転写および翻訳の注意深い協調を要求する。最終的に、細胞エネルギーが変化を必要とするとき、ミトコンドリアはそのATP産出を回転させることによって迅速に応答しなければならない。それ故に、ミトコンドリアは細胞機能と連絡する複雑な系を要求する。図1から明らかなように、ミトコンドリアへ出入りするシグナル分子はイオン、気体、代謝産物、ホルモン、転写因子およびタンパク質を含む。結果的に、細胞シグナルを受け取り、統合し、伝達するための中心としてミトコンドリアを認識することは、医薬品の設計および試験における重要な進歩である。
【0004】
ミトコンドリア制御におけるシグナル伝達の礎石は可逆的リン酸化である。しかしながら、タンパク質キナーゼ現象の最初の実証が1954年に報告され、40年近く前に可逆的リン酸化によってミトコンドリアの中心的機能が制御できるという事実が発見されたにもかかわらず、ミトコンドリアのリン酸化現象についての報告は少ない。このことは、1980年代および1990年代において、細胞膜を横断し、サイトゾルを通って核へと伸展する多数のシグナル伝達リン酸化カスケードが認識されたことにも全くかかわりなくである。このような知識の不足は、ミトコンドリアタンパク質機構の大部分が二つの脂質二重層の後ろに位置する事実により、サイトゾルシグナル伝達カスケードはミトコンドリアタンパク質に到達できないと考えられていることに部分的な原因があるかもしれない。いずれの現象でも、疾病管理の鍵となる方法において、ミトコンドリアが可逆的リン酸化によりシグナル伝達を制御することは広く受け入れられてはいなかった。それにもかかわらず、表1に示すように、2006年までに全てのミトコンドリアの区画(すなわち、マトリックス、内膜、膜間腔、および細胞質に面する外表面を含む外膜)内の60を超えるタンパク質が、ミトコンドリアの機能の広いスペクトルに関係付けられるリン酸化タンパク質として同定された。増加するデータはさらに、ミトコンドリアの標的の可逆的リン酸化の重要性、およびそれを対象とする、改善された癌治療のための組成物の使用について実証している。
【0005】
表1 ミトコンドリアのリン酸化タンパク質
a略語:AA,アミノ酸(S,セリン;T,スレオニン;Y,チロシン);ANT,アデニンヌクレオチド輸送体;Axmito,ミトコンドリアアネキシン;β−OX,β−酸化:BCKAD,分岐鎖ケト酸脱水素酵素;CI−CV,呼吸鎖複合体1−5;CPT,カルニチンパルミトイルトランスフェラーゼ;CREB,cAMP反応性エレメント(Cre)結合タンパク質:CYP,チトクロムP450;DBP,十二量体結合タンパク質;EF,伸長因子;GST,グルタチオンS−トランスフェラーゼ;HSP,熱ショックタンパク質;IM,内膜;IMS,膜間腔;M,マトリックス;MnSOD,マンガンスーパーオキシドジスムターゼ;mTERF,ミトコンドリア転写終結因子;mtGAT,ミトコンドリアグリセロール−3−ホスファターゼアセチルトランスフェラーゼ;mthsp,ミトコンドリアHSP;mtTBP,ミトコンドリアテロメア結合タンパク質;NDK,ヌクレオシド二リン酸キナーゼ;OM,外膜;OXPHOS,酸化的リン酸化;P部位,リン酸化部位;PDC E1/3,ピルビン酸脱水素酵素複合体E1/3サブユニット;PDK,ピルビン酸脱水素酵素キナーゼ;PLA,ホスホリパーゼA;ScIRP,サブユニットc−免疫反応性ペプチド;SSAT,スペルミジン/スペルミンアセチルトランスフェラーゼ;StAR,ステロイド産生急性調節タンパク質;TCA,トリカルボン酸サイクル;TRAP−1,腫瘍壊死因子タイプ1受容体付随タンパク質;VDAC,電位依存性陰イオンチャネル。
b組み換えタンパク質において、リン酸化はin vitroでのみ観察される。
cタンパク質はミトコンドリアへ移動する(すなわちタンパク質はミトコンドリアの常在タンパク質ではない)。
【0006】
ヒトゲノムにおける最も大きなキナーゼおよびホスファターゼファミリーであるタンパク質キナーゼ(PK)およびタンパク質チロシンホスファターゼ(PTP)は、それぞれ500を超える、および100を超えるメンバーを有する。キナーゼおよびホスファターゼの比較的小さなファミリーを合わせると、これらのシグナル伝達分子はヒトゲノムによりコードされる全てのタンパク質の3パーセント近くを構成する。前述のリン酸化タンパク質と同様に、驚くべき数の研究によってキナーゼおよびホスファターゼがミトコンドリアの機能に関係付けられており、図2に示すように、これまでに少なくとも25のキナーゼおよび8のホスファターゼがミトコンドリアに局在していることが報告されている。これらのキナーゼおよびホスファターゼが一つのグループまたはファミリーに限定されていないことは明らかで、むしろそれらはほとんど全ての既知の哺乳類キナーゼおよびホスファターゼのサブグループに相当しており、これはミトコンドリアに影響を及ぼすと考えられるシグナル伝達経路の範囲を反映している。これらのシグナル伝達分子は基質の特異性が変化するキナーゼおよびホスファターゼ(例えばチロシンキナーゼ、古典的PTPサブグループ、セリン/スレオニンキナーゼ、および二重特異性PTP)を含み、これらの変化は触媒的な機構(例えばシステインベースのPTP、アスパラギン酸ベースのPTP、および金属依存性ホスファターゼ)および進化的保存性(例えば最近関連ピルビン酸脱水素酵素キナーゼ(PDK)およびホスファターゼ(PDP)、分岐鎖ケト酸脱水素酵素キナーゼ(BCKDK)およびホスファターゼ(BCKDP)、および多数の哺乳類特異的酵素)による。
【0007】
これらのシグナル伝達分子の大部分は、細胞内でミトコンドリアに関係しないその他の役割を有し、主にミトコンドリアの外に存在することが見出された。大部分のタンパク質について、それらのミトコンドリアへの移行の起動力または機構はよく知られていない。しかしながら、図3から明らかなように、先に記載したリン酸化タンパク質のようなキナーゼおよびホスファターゼがミトコンドリアの全ての区画に存在し、かつそれらの活性が様々なミトコンドリアの機能に影響を与えることははっきりしている。
【0008】
しかしながら少数のシグナル伝達分子は主にミトコンドリアに局在すると考えられる。PDKおよびPDPに加え、このグループはPTEN誘導性キナーゼであるPINK1、ミトコンドリアPTPMT1を標的とする二重特異性PTP、およびアスパラギン酸ベースのホスファターゼ/ATPアーゼであるTim50を含む。現在これらのタンパク質の多くに対する基質は未知であるが、生物学的および遺伝的データからそれらがミトコンドリアにおいて決定的な機能を有することは明らかである。例えば、そのミトコンドリア下の局在はまだ決定されていないが、PINK1はN末端シグナル配列によってミトコンドリアを標的とする。Ca2+/カルモジュリン調節性キナーゼファミリーと高い配列相同性を共有するこのキナーゼは、生存促進性の活性に関与すると考えられる。同様にPTPMT1は、主にミトコンドリア内部に局在する最初のPTPとして最近同定され、PINK1のようにN末端シグナルペプチドによってミトコンドリアを標的とし、かつミトコンドリアの内膜のマトリックス面に密接に結合することが見出された。PTPMT1は、そのミトコンドリアが糖代謝とインスリン分泌の連関に重要な機能を持つ膵臓β細胞に高度に発現する。最後に、TIM(内膜の交換輸送体)複合体の鍵となる成分であるTim50は、アスパラギン酸ベースのホスファターゼ/ATPアーゼのCTDファミリーに対する配列相同性を有する。このファミリーの他のメンバーと同様に、Tim50はATPアーゼとして機能し得るが、in vitroにおいて、ホスホチロシン類似体であるパラニトロフェニルホスフェートに対するホスファターゼ活性を有することもまた示されてきた。上記のことから、細胞内の他の場所からミトコンドリアへ動員されるキナーゼおよびホスファターゼのみが見られるのではなく、ミトコンドリア自体が常在するシグナル伝達分子のグループを有すると考えられる。
【0009】
時おり原因となるキナーゼおよびホスファターゼが同定されていないことから、最もよく知られたミトコンドリアリン酸化タンパク質におけるリン酸化の効果は明らかではないが、幾つかのリン酸化現象が部分的に特徴付けられている。これらの例は、先に考察したリン酸化タンパク質およびシグナル伝達分子のように、ミトコンドリアの一領域に限定されてはいない。
【0010】
ミトコンドリア外膜におけるリン酸化は、アポトーシスの制御に決定的な役割を持つ。特によく定義されている現象は、BCL−2ファミリーのアポトーシス促進性メンバーであるBADのリン酸化である。生存促進性サイトカインであるインターロイキン−3で処理した後、PKAが外膜へ移動することが示されている。一旦、外膜のA−キナーゼアンカータンパク質(AKAP)に固定された後、PKAはBADをSer112においてリン酸化して、図3Aに示す工程であるBADの不活性化およびミトコンドリアからの分離を導く。p70S6キナーゼによるSer136、および未同定のキナーゼによるSer155におけるBADのリン酸化もまた、BADの不活性化に関係付けられている。
【0011】
健常な細胞ミトコンドリアにおける制御機構として作用する可逆的リン酸化の最もよく確立された例は、マトリックス内のPDH複合体であり、その単純化された下絵を図3Bに示す。この複合体は、解糖系由来のピルビン酸塩の、トリカルボン酸(TCA)サイクルの主要な前駆体であるアセチルコエンザイムA(CoA)への変換を触媒する。これら二つの主要なエネルギー産生経路をつなぐものとして、PDH複合体は細胞内グルコースの恒常性維持のために適切に制御されなければならない。
【0012】
ミトコンドリアリン酸化タンパク質として最初に同定されたことから、PDH複合体および可逆的リン酸化によるその制御は徹底的に研究されてきた。PDH複合体のリン酸化および脱リン酸化は、それぞれPDKおよびPDPにより行われる。少なくとも4のPDKアイソフォームおよび2のPDPアイソフォームが知られており、それら全てはPDH複合体のE2サブユニットに結合する。リン酸化現象はE1サブユニットの別個の三つのセリン残基に起こり、それぞれがこの複合体の著しい不活性化を導く。特に、PDKそれ自体のリン酸化については現在少なくとも一報告がある。PKCにより行われるこのリン酸化はPDKを不活性化することが示されており、可逆的リン酸化によるPDH複合体制御のさらなるレベルを潜在的に証明している。従ってPDH複合体は、他の保存された工程に制御の段階を加えるためにリン酸化を用いるミトコンドリアの最も重要な例である。
【0013】
先に記載したミトコンドリアのチロシンキナーゼおよびホスファターゼによって示されるように、この細胞小器官内でのリン酸化はセリンおよびスレオニン残基に限定されない。ミトコンドリアのエネルギーに影響を与えるチロシンリン酸化の例は、図3Cに示す内膜でのチトクロムCオキシダーゼ(COX)の制御に見られる。呼吸鎖における終末酵素としてのCOXは、ポンプによってプロトンが内膜を横切る間、協調的に酸素を水に還元する。PDH複合体と同じく、COXはATPおよびADP、同様に甲状腺ホルモンT2およびおそらくはCa2+イオンによりアロステリックに制御される。これらの制御形式に加え、COXはin vitroおよびHepG2細胞におけるin vivoの両方で、cAMP依存性の様式によりリン酸化されることが示されている。COXは13のサブユニットを含み、二量体として結晶化される。リン酸化部位はサブユニット1のTyr304であると同定され、これは膜間の二量体インターフェースに位置する。リン酸化現象は、おそらく二量体形成の中断によってCOX活性を顕著に阻害する。
【0014】
COXのチロシンリン酸化の第二の例である非受容体型チロシンキナーゼc−Srcの一部は、Lynチロシンキナーゼと同様に、破骨細胞においてミトコンドリア内部に局在してCOXのサブユニットIIの未同定部位にチロシンリン酸化を導く。リン酸化現象の結果は、COX活性の増大を導くサブユニットIに見られる結果とは逆である。
【0015】
ミトコンドリアシグナル伝達の重要な態様は、キナーゼおよびホスファターゼがそれら自体でどのように制御されるかである。Abl、Akt、GSK313およびPKCδのような、主に細胞内の他の場所に存在しているがミトコンドリアを標的とするようになる多数のキナーゼは、それらが活性化された状態においてのみそうするようになると考えられる。従ってミトコンドリア内での幾つかのキナーゼ活性の範囲は、単純に細胞小器官内へ移入される酵素の数によって決定されるであろう。
【0016】
しかしながら常在するシグナル伝達分子には、異なる制御手段が実施されなければならない。これらの工程は未だ確定されていないものの、セカンドメッセンジャーの役割が鍵となるように思われる。PDKおよびPDPの活性は、Mg2+、Ca2+、K+およびADPのようなイオンおよび小分子によって制御されることが知られている。ミトコンドリア一酸化窒素合成酵素の性質決定および最近のミトコンドリアにおける可溶性アデニル酸シクラーゼの発見は、ミトコンドリアシグナル伝達分子の制御に寄与するセカンドメッセンジャーに対してさらなる機会を提供する。最後に、細胞内の他の場所でシグナル伝達分子を制御する手段として確立されているROSは、大量の活性酸素種が産生されるミトコンドリアにおけるキナーゼおよびホスファターゼの制御にほとんど確実に関与するであろう。キナーゼおよびホスファターゼのアイソフォームの相対的な発現レベルは、病理学における重要な役割を果たし、かつ疾病に付随するその他のシグナル伝達現象に関連付けられる可能性がある。遺伝子および発現レベルでの変化もまた、これらの変化に関連し得る。
【0017】
幾つかのプロテオミクス調査を通した後であっても、哺乳類ミトコンドリアプロテオームの3分の2のみが知られていると推定される。残りの3分の1の多くは、これらのマススペクトロメトリー分析の検出レベル未満である、シグナル伝達タンパク質のような少量のタンパク質により構成されると思われる。これらの研究から、異なる組織由来のミトコンドリアの間で、含まれるタンパク質に高い可変性があることも明らかになった。例えばこれらのプロテオミクスの試みにおいて、〜50%のタンパク質のみが、調査された四つの組織(すなわち脳、心臓、肝臓および骨格筋)にわたって保存されることが見出された。異なるミトコンドリアシグナル伝達経路は、組織から組織において同様の方向で変動し得るのみではなく、この観察されるミトコンドリアの多様性に非常によく寄与していると考えられる。
【0018】
それにもかかわらず、可逆的リン酸化がミトコンドリアでの工程の制御に関与すると結論付けるための十分すぎる証拠が存在する。報告された60を超えるリン酸化タンパク質、30のキナーゼおよびホスファターゼ、および様々な補助的シグナル伝達タンパク質によれば、ミトコンドリアは確かに可逆的リン酸化によるシグナル伝達のための場として正当に評価されておらず、事実としてこのような制御は癌のような過剰増殖疾患に有用となるであろう。
【0019】
増殖の速い腫瘍細胞の大多数は、形質転換されていない細胞に対する著明な遺伝的、生化学的、および組織学的な相違を示す。これらの多くは、元の組織に比較して変化したエネルギー代謝に関連する。腫瘍細胞において最も悪名高く、かつよく知られたエネルギー代謝の変化は、ワールブルク効果として知られている、高濃度のO2存在下であっても解糖能が亢進する現象である。
【0020】
ワールブルクは最初、腫瘍細胞における亢進した解糖の推進力は、ミトコンドリアの機能の非可逆的損傷によって起こるエネルギー欠乏であると提唱し、この場合嫌気性の筋肉と同様に、解糖を通してグルコースが後に分泌される乳酸に変換される。腫瘍細胞におけるこの解糖の流れの増大は、酸素の供給が不十分な固形腫瘍に見られる部分的な低酸素状態のような、O2濃度の低い環境での生存および増殖を保証するための代謝の戦略であると提唱されてきた。特に、多くのヒト低酸素性腫瘍におけるO2濃度は20μMよりも低いことから、そこでの酸化的リン酸化は限定される。結果的に、解糖は固形腫瘍(例えば増殖の遅いメラノーマおよび乳腺腺癌)における主要なエネルギー経路であると考えられる。
【0021】
細胞増殖の比率とATP供給の比率との間の比例関係は、増殖の速い腫瘍細胞について確立されてきた。何人かの著者は、高度に脱分化した増殖の速い腫瘍における解糖の比率が、増殖の遅い腫瘍および正常細胞における解糖の比率よりも高くなるように、解糖活性は腫瘍の悪性度と相関することを提唱した。実際に高レベルの乳酸は悪性腫瘍の前兆であると提唱されてきた。これらの現象がさらなるシグナル伝達現象および遺伝的変化に関連すると考えられ、例として低酸素誘導因子および血管新生因子の産生および放出が含まれる。
【0022】
図4に示すように、何年もの間トリカルボン酸(TCA)サイクルは、生命体のエネルギー源としてのATPの産生におけるその役割のみに対して、生物学的に重要であるとみなされるだけであった。しかしながら最近の研究により、TCAサイクルの活性は、細胞増殖およびアポトーシスの決定を含むシグナル伝達経路の機能にもまた影響を与え、関係する解糖およびTCAサイクル酵素の増加または減少を制御できることが示された。腫瘍の進行と、解糖酵素であるヘキソキナーゼおよびホスホフルクトキナーゼ(PFK)1との間には直接の相関もあり、これらは増殖の速い腫瘍細胞で大きく増加している。従って、その酸化能に不足を示す腫瘍細胞は、活発な酸化的リン酸化を有する腫瘍細胞よりもさらに悪性であると想定されている。低酸素性または好気性の状態にあるか否かに関わらず、癌組織での解糖への依存は悪性度の増大に関連する。
【0023】
健常な細胞のPDH複合体におけるリポ酸の役割については、よく研究されてきた。PDH複合体は三つの中心的なサブユニット、E1、E2、およびE3(それぞれピルビン酸脱水素酵素、ジヒドロリポイルトランスアセチラーゼ、およびジヒドロリポアミド脱水素酵素)を有する。これらの複合体は中心にE2コアを持ち、このコアをその他のサブユニットが取り囲んで複合体を形成する。これら二つのサブユニット間のギャップ内で、リポイルドメインが活性部位間での中間体の輸送を行う。リポイルドメイン自体は、可動性のリンカーによってE2コアに付着する。ピルビン酸塩とチアミンピロリン酸の反応によるヘミチオアセタールの形成によって、この陰イオンは、リジン残基に付着する酸化されたリポ酸塩種のS1を攻撃する。結果的に、リポ酸塩S2は硫化物またはスルフヒドリル成分として置換され、続く四面体ヘミチオアセタールの崩壊によりチアゾールが放出され、これによりTPP補助因子が放出され、かつリポ酸塩のS1にチオアセテートが産生される。この時点でリポ酸塩−チオエステルの機能性はE2活性部位内へ移動され、アシル基転移反応がアセチル基をリポ酸塩の「自在アーム」からコエンザイムAのチオール基へ転移させる。これによりアセチルCoAが産生され、酵素複合体から放出され、続いてTCAサイクルに入る。ジヒドロリポ酸塩は複合体のリジン残基にまだ結合しているが、その後E3活性部位へ移動し、フラビンにより仲介される酸化によってそのリポ酸塩静止状態へ戻り、FADH2(および最終的にはNADH)を産生し、かつリポ酸塩を再生して、コンピテントなアシルアクセプターへ戻る。このリポ酸塩種が遮断された場合、FADH2への電子の流れまたはアセチルCoAの産生がなくなり得る結果として細胞内にピルビン酸の毒性が蓄積する。癌細胞では、細胞の解毒および生存のためにアセトインの産生が必要なことが示唆されている。
【0024】
以前に述べたように、ミトコンドリア内でのPDH複合体の活性は様々なアロステリックエフェクターおよび共有結合性の修飾によって高度に制御される。PDH活性はそのリン酸化状態によって制御され、これは脱リン酸化状態で最も活性である。PDHのリン酸化はPDKにより触媒される。PDK活性はATP、NADH、およびアセチルCoAレベルの増加により増大する。PDKの負のエフェクターは、ATPレベルが低下した時に増加するADP、NAD+、CoA−SH、およびピルビン酸である。脱リン酸化を通してPDHを活性化する酵素であるPDPの制御については完全に理解されていないが、Mg2+およびCa2+がPDPを活性化することが知られている。
【0025】
複合体の二つの産物であるNADHおよびアセチルCoAは、PDHの脱リン酸化活性型であるPDH−aに対する負のアロステリックエフェクターである。これらのエフェクターは、ピルビン酸に対する酵素のアフィニティーを減少させることによって、PDH複合体を通した炭素の流れを限定する。加えてNADHおよびアセチルCoAは、PDHをリン酸化PDH−b型に変換することにより不活性化する酵素であるPDKの、強力な正のエフェクターである。細胞のエネルギーチャージが高い際にNADHおよびアセチルCoAが蓄積されることから、エネルギーの豊富な細胞内で高レベルのATPがPDKの活性もまた上昇させ、PDHの活性を減少させることは不思議ではない。しかしながらピルビン酸がPDKの強力な負のエフェクターであることから、ピルビン酸のレベルが増加するときには、高レベルのNADHおよびアセチルCoA存在下であってもPDH−aになりやすいであろう。
【0026】
ATPの豊富な細胞内ではPDH−aを維持するピルビン酸の濃度が十分に高く、アロステリックに減少したPDHの高Km型はそれにもかかわらずピルビン酸をアセチルCoAに変換できる。高いATPおよびNADHレベルを有する細胞内での大量のピルビン酸により、ピルビン酸の炭素は炭素の二つの主な貯蔵型(糖新生を通したグリコーゲンおよび脂肪酸合成を通した脂肪の産生)に方向付けられ得り、ここではアセチルCoAが主要な炭素ドナーである。PDP−bの制御はよく理解されていないが、減少したATP濃度下でピルビン酸の酸化を最大化、および高いATP濃度下でPDH活性を最小化するための制御である可能性がかなり高い。
【0027】
腫瘍細胞は、ピルビン酸および付随するアルデヒドおよびラジカル、例えばアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを、これらの分子が高濃度において細胞内pHを大幅に低下させるという機構により細胞傷害性であることから、無制限に蓄積できない。従ってAS−30Dおよびエールリッヒヘパトーマについては、ミトコンドリアピルビン酸の有意な画分が、PDH複合体のE1成分により、結合したβ−ヒドロキシエチルチアミンピロリン酸を通して、活性アセトアルデヒドに脱炭酸されることが記載されている。この活性アセトアルデヒドは第二のアセトアルデヒドと共に次々に縮合し、最終的には元のピルビン酸をアミノ酸のグルタミンまたはリポ酸のいずれかを用いて脱酸または還元して、競合的にPDHを阻害しかつそのピルビン酸前駆体よりも細胞に対する毒性の少ない(例えば細胞内でのpH恒常性を維持することにより)化合物であるアセトイン(3−ヒドロキシブタノン)を産生する。しかしながら、ATP産生の源としての解糖への依存によるピルビン酸蓄積の結果としての腫瘍細胞の解毒経路におけるアセトインの重要性にもかかわらず、先行技術には腫瘍細胞生存率に関して、アセトイン産生を遮断する効果についての参考文献は少ない。
【0028】
最近の研究は、癌細胞にさらなる好気性の代謝を強制することによって腫瘍の増殖が抑制されることを示唆している。従って、ワールブルク代謝への移行はPDH複合体の遮断を必要とする。図5に示すように、この移行において癌細胞での低酸素誘導因子(HIF)によるシグナル伝達の増大が見られることは、グルコースの代謝におけるHIFの重要な役割を考えれば不思議ではない。直接または間接にHIFのシグナル伝達を引き起こす変異は、実際に癌の発生における一般的な機構であると考えられる。HIFはPDK1の過剰発現を誘導し、これが次にPDH複合体の減少に働く。PDK1によるリン酸化は、このアイソフォームがPDH複合体の第1のサブユニットであるE1のアルファサブユニットの三つのセリン残基を独自にリン酸化することから、不活性なPDH複合体の維持に特に有効となる。E1の再活性化は、三つのリン酸基全ての除去を必要とする。さらに、PDH複合体の活性化はROS産生の増大を導き得り、これが順にアポトーシスを導き得る。しかしながら、癌に見られるPDK1の変化は、その濃度の変化によるのみではなく、その活性の変化、およびおそらくは、実に一腫瘍型または一患者と他方との間でのそのアミノ酸配列の変化にもよる可能性がある。加えてPDK1は、腫瘍の型に依存して、腫瘍に付随する様々な分子と共に異なる複合体を形成し得る。従ってPDK1の阻害は、腫瘍にアポトーシスを起こすための潜在的な標的であり得る。しかしながら今日までに、既知のPDK1阻害剤は、このアイソザイムを最大60%のみ阻害することが示されている。
【0029】
伝統的な化学療法の標的は細胞の分裂、増殖であるが、全ての臨床的に許容される化学療法的な治療には大量の薬物が用いられ、これがホスト細胞の正常な増殖に深刻な損傷もまた誘導する。従って癌の治療には、さらに選択的な標的指向化が必要とされる。化学療法に付随する別の問題は、多くの腫瘍型において、抗新生物剤に対する固有であるかまたは獲得された抵抗性が存在することである。概して伝統的な化学療法は、現在ほとんどの悪性腫瘍に対する長期的な利点が少なく、かつしばしば生存期間またはその質を減少させる有害な副作用に関連する。従って、生命の適正な質を可能にしながら腫瘍の長期的な管理を提供できる、徹底的な新しいアプローチが必要とされている。
【0030】
新しい化学療法の開発において、薬物の有効性、送達、および副作用は確実に解決されなければならない問題である。固形腫瘍における低酸素性の領域への送達は、薬物が異なる細胞層を容易に透過できないときに困難であり得る。このような不確実性を避けるため、少なくともマイクロモル以下の範囲で一定に代謝阻害する抗癌剤の設計に今日的な意味があると考えられる。癌細胞が系統から系統にわたって遺伝的および表現型的に異質であることが立証され得るが、全ての腫瘍細胞株はATP供給のための解糖および酸化的リン酸化に依存する。ワールブルク効果に集中することは、健常ホスト組織および臓器の機能性に影響を与えることなく単に腫瘍の代謝および増殖に選択的に影響を与える送達および治療的戦略の設計を促進する、腫瘍および正常細胞の間での物理化学的および生化学的エネルギーの相違に基づく薬物設計を可能にする。
【0031】
Binghamらの米国特許第6,331,559号および6,951,887号、同様にBinghamらの米国仮出願第60/912,598号は全て参照としてここに取り込まれ、腫瘍細胞およびその他の特定の型の病的な細胞の両方を選択的に標的化しかつ死滅させる、新しい種類のリポ酸誘導体治療薬を開示する。これらの特許はさらに、有効量のこれらのリポ酸誘導体を薬剤的に許容される担体と共に含む医薬組成物、およびその使用法について開示する。しかしながら、これらの特許にはこれらのリポ酸誘導体の構造および一般的使用についてさらに記載されているが、いずれの特許にも、これらの誘導体がPDH複合体の構造および/または発現レベル、および/または活性の制御における調節に有用であることは指摘されていない。
【0032】
PDH複合体の構造および/または活性が腫瘍活性の重大な決定要因であることが示されていることから、PDH複合体の構造および/または活性、またはさらに発現レベルの、薬剤的に許容される調節因子、およびその使用法を提供することは有益であろう。
【0033】
発明の目的及び産業上の利用可能性
結果的に本発明の目的は、癌のような細胞の過剰増殖によって特徴付けられる疾病、状態、または症候群の治療または診断に使用され、腫瘍細胞に選択的活性を示す医薬組成物の提供である。
【0034】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、投与による副作用が最少である医薬組成物の提供である。
【0035】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、可能な最少の費用で容易に製造され、かつ可能な最長期間保存できる医薬組成物の提供である。
【0036】
本発明のさらなる目的は、前記の疾病、状態、または症候群の治療または診断に使用され、特に腫瘍細胞のミトコンドリア内のPDH複合体の構造、活性、および/または発現レベルを通してミトコンドリアのエネルギー代謝を調節する医薬組成物の提供である。
【発明の概要】
【0037】
前記の目的を達成するため、本発明は、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化により特徴付けられる、ヒトを含む温血動物の癌またはその症候群のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、または症候群の治療、診断、または予防に有用な医薬組成物を広く提供し、ここで該医薬組成物は、その全てが参照としてここに取り込まれる米国特許第6,331,559号および6,951,887号、ならびに米国仮出願第60/912,598号に記載されるものを含む、少なくとも1の有効量のリポ酸誘導体、および少なくとも1のその薬剤的に許容される担体を含む。
【0038】
ミトコンドリアのエネルギー代謝を阻害することにより、本発明のリポ酸誘導体は病的な細胞におけるミトコンドリア膜電位およびその他のミトコンドリア事象両方の損失の原因となり、結果として非可逆的な細胞死が開始される。本発明のリポ酸誘導体はまた、PDKの活性化および/またはPDPの阻害によるか、またはPDH複合体のE1サブユニットの活性阻害を通した、毒性の少ない分子であるアセトインへのピルビン酸の変換の阻害により、ミトコンドリアのエネルギー代謝も阻害し得る。アセトイン合成の阻害は、酸化還元バランスを含むその他の工程を歪め、かつアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを含む有毒な副生成物産生の原因となる可能性もあり、結果としてこれらの副生成物自体は病的な細胞のミトコンドリアに非可逆的な損傷を引き起こす。
【0039】
本発明の医薬組成物は、PDK1、PDK2、PDK3、PDK4、およびそれらの各変異体またはアイソフォームの効果を調節し得る。該医薬化合物はまた、PDP1、PDP2,およびそれらの各アイソフォームの効果も調節し得る。
【0040】
本発明の医薬組成物はまた、PDH複合体中に見出されるホスホリラーゼ、キナーゼ、および脱水素酵素の酵素構成物質の発現レベルも調節し得る。この調節は、転写、翻訳、または適切な遺伝子のエピジェネティックなサイレンシングを含む後翻訳段階において起こり得る。
【0041】
TCAサイクルに基本的に付随する分子由来の化合物として、かつ解糖の伸展により、本発明の医薬組成物は腫瘍細胞内への選択的な取り込みを示す。さらにこのような選択的な腫瘍細胞への取り込みは、この医薬組成物の投与が健常な非形質転換細胞および組織に起こし得る副作用を最小化する。
【0042】
本発明の第1の実施形態によれば、リポ酸誘導体は一般式1
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す);芳香族;R3C(O)−として定義されるアシル;ヘテロアリール;R4C(=NH)−として定義されるイミドイル;有機金属アリール;アルキル−有機金属アリール;およびセミアセタール R5CH(OH)−S−から成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R4は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0043】
本発明の第2の実施形態によれば、リポ酸誘導体は一般式2
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R3C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R3C(=NH)−として定義されるイミドイル、およびR4CH(OH)−S−として定義されるヘミアセタールから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3およびR4は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0044】
本発明の第3の実施形態によれば、リポ酸誘導体は一般式3
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0045】
本発明の第4の実施形態によれば、リポ酸誘導体は一般式4
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0046】
さらに、これらの一般構造のいずれかもしくは全てが細胞またはミトコンドリア内で代謝され得ることから、上に言及された構造の代謝産物は本発明の範囲内にあることが明確に意図される。
【0047】
それぞれの一般式内で、特定のリポ酸誘導体それぞれの(R)−異性体は、(S)−異性体よりも大きな生理活性を有する。結果的にリポ酸誘導体は、単にその(R)−異性体型かまたは(R)−および(S)−異性体の混合物として存在するべきである。
【0048】
本発明のさらなる態様によれば、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または機能の制御または撹乱の変化を含む、ヒトを含む温血動物の癌のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、症候群、または症状を診断、治療、または予防するための方法が提供され、ここで該方法は、有効量のここに開示される医薬組成物をこのような動物へ投与することを含む。
【0049】
本発明のさらなる態様によれば、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化を含む、癌のような細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、もしくは症候群、またはそれらの症状を呈する患者の診断および利益を予想するための方法が提供され、該方法は、患者から細胞サンプルを得ること、有効量の本発明の医薬組成物をin vitroで細胞へ投与すること、およびそこから結果を得ることを含む。
【図面の簡単な説明】
【0050】
以下の図面は本発明の実施形態の図解であり、明細書および請求項の全体により包含される本出願の範囲を限定しようとするものではない。
【図1】ミトコンドリアへの出入りの両方を標的とする一般的なシグナル伝達分子およびそれらの効果を示した図である。
【図2】ミトコンドリアキナーゼおよびホスファターゼの一覧、およびミトコンドリア内でのそれらの位置を示した図である。
【図3】図3Aは、ミトコンドリア外膜でのPKAによるBADのリン酸化、およびそこにおける可逆的リン酸化の効果を示した図である。図3Bは、PDH複合体の作用による、ミトコンドリアマトリックスでのピルビン酸のアセチルCoAへの変換、およびそこにおける可逆的リン酸化の効果を示した図である。図3Cは、酸素の水への還元およびポンプによるプロトンのミトコンドリア内膜の横断におけるCOXの作用、およびそこにおける可逆的リン酸化の効果を示した図である。
【図4】ピルビン酸の解糖による産生における基質および産物の構造を図示し、ATPおよびNADHの産生ならびに付随する酵素もまた示した図である。
【図5】HIF−1によるグルコース代謝の制御を示した図である。
【図6】図6Aは、in vivoにおける、正常組織と癌組織の間でのエネルギー代謝の相違を示した図である。図6Bは、PDH複合体中のリポ酸の生体型と、本発明の医薬組成物の一部を形成するリポ酸誘導体との間の相違を示した図である。図6Cは、PDKのリポイル残基効果による、PDH複合体の制御を示した図である。
【図7】異種移植腫瘍の増殖における、本発明の医薬組成物の効果を示した図である。
【図8】三つの腫瘍細胞型および非形質転換細胞における、本発明の医薬組成物を用いた処理の効果を示した図である。
【図9】図9Aは、致死閾値またはそれを超える量の本発明の医薬組成物により処理した肺癌細胞におけるATPレベルを示した図である。図9Bは、ピルビン酸を含む培地とグルコースを含む培地中での、本発明の医薬組成物によるATP合成阻害の比較を示した図である。図9Cは、乳癌細胞と正常乳房細胞における、本発明の医薬組成物によるATP合成の阻害を比較した図である。図9Dは、肺癌細胞における、本発明の医薬組成物によるATP合成の阻害と、リポ酸および本発明の不活性型によるATP合成の阻害との比較を示した図である。
【図10】PDH複合体およびアルファケトグルタル酸(αKDH)脱水素酵素の酵素活性の腫瘍細胞ミトコンドリアでのレベルにおける、本発明の医薬組成物の効果を示した図である。
【図11】図11Aは、本発明の医薬組成物による処理またはモック処理した肺癌細胞抽出物の、二次元ゲルのウェスタン分析を示した図である。図11Bは、本発明の医薬組成物による処理またはモック処理した対の二次元ゲルサンプルを、拡大して示した図である。
【図12】図12Aは、PDH複合体E2サブユニットに共有結合した内在性リポ酸塩により調節される、PDKの制御的役割を示した図である。図12Bは、本発明の医薬組成物による腫瘍細胞PDH複合体の差次的な不活性化の、考えられる機構を示した図である。
【図13】H460肺癌細胞のミトコンドリア膜電位における、本発明の医薬組成物の効果を示した図である。
【図14】多様な腫瘍細胞型における本発明の医薬組成物による細胞死経路の、ウェスタンブロット分析の結果を示した図である。
【0051】
発明の詳細な説明
本発明は一般的に、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニット、例えばPDH複合体の構造、発現、および/または活性の制御または撹乱の変化を含む、温血動物の癌のような細胞の過剰増殖によって特徴付けられるもの、またはそれらの症状を含む疾病、状態、もしくは症候群、またはそれらの症状を治療、診断、または予防するための医薬組成物に方向付けられる。このような動物は、癌を含む細胞の過剰増殖によって特徴付けられる疾病ならびにその他の病態および症候群にかかりやすい、ヒト、ウマ、ウシ、イヌおよびネコを含む家畜などのような哺乳類を含む。本発明の医薬組成物は、チオクタンとしても知られる、米国特許第6,331,559号および6,951,887号、ならびに米国仮出願第60/912,598号に記載されるものを含む、少なくとも1の有効量のリポ酸誘導体、およびそのための薬剤的に許容される担体または賦形剤を含む。ミトコンドリア内に通常見出されるのみではなく、ワールブルク効果に見られるような腫瘍細胞の増大した解糖活性にも役立つ誘導体分子として、本発明のリポ酸誘導体は、腫瘍のような過剰増殖により特徴付けられる細胞および組織のミトコンドリア内への選択的送達ならびに有効な濃度について特によく適しており、これにより正常な細胞および組織は該組成物の効果から逃れる。
【0052】
本発明の医薬組成物は、PDK1、PDK2、PDK3、PDK4、およびそれらの各アイソフォームの効果を可逆的リン酸化を通して調節し得る。該医薬組成物はまた、PDP1、PDP2,およびそれらの各アイソフォームおよび/または変異体の効果も、可逆的リン酸化を通して調節し得る。これらの調節は、キナーゼもしくはホスファターゼ活性の促進または阻害のいずれかを通して起こり得る。
【0053】
ミトコンドリアのエネルギー代謝を阻害することにより、本発明のリポ酸誘導体は病的な細胞におけるミトコンドリア膜電位およびその他のミトコンドリア事象両方の損失の原因となり、結果として非可逆的な細胞死が開始される。本発明のリポ酸誘導体はまた、PDKの活性化および/またはPDPの阻害によるか、またはPDH複合体のE1サブユニットの活性阻害を通した、毒性の少ない分子であるアセトインへのピルビン酸の変換の阻害により、ミトコンドリアのエネルギー代謝も阻害し得る。アセトイン合成の阻害は、酸化還元バランスを含むその他の工程を歪め、かつアセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルを含む有毒な副生成物産生の原因となる可能性もあり、結果としてこれらの副生成物自体は病的な細胞のミトコンドリアに非可逆的な損傷を引き起こす。
【0054】
本発明の第1の実施形態によれば、リポ酸誘導体は一般式1
[式中、xは0〜16を表し、R1およびR2は独立して
(1)アシル基 R3C(O)−であって、R3がアルキル、アリール、または有機金属アリール基を表し、チオエステル結合を通して連結され、限定はされないがアセチルおよびブタリル(butaryl)を含み、特定の例としてビスアセチルリポ酸塩となるアシル基;
(2)チオエステル結合を通して連結される芳香族基であって、限定はされないがベンゾイルまたはベンゾイル誘導体を含み、特定の例としてビスベンゾイルリポ酸塩となる芳香族基;
(3)アルキル基 CnH2n+1であって、nが1〜10を表し、他の成分、例えば−OH、−Cl、または−NH2により置換されたこのようなアルキル基とチオエーテル結合を通して連結され、限定はされないがメチル、エチル、ブチル、デカニル、および6,8−ビスカルボモイル(carbomoyl)メチルリポ酸塩を含むアルキル基;
(4)アルケニル基 CnH2n−1であって、nが2〜10を表し、チオエーテル結合を通して連結され、限定はされないがプロピレン、2,3ジメチル−2−ブテン、およびヘプテンを含むアルケニル基;
(5)アルキニル基 CnH2n−3であって、nが2〜10を表し、チオエーテル結合を通して連結され、限定はされないがアセチレン、プロピン、およびオクチンを含むアルキニル基;
(6)鎖式、または複素環を形成するいずれの炭素の付加もしくは置換を有する脂環基による脂環式のいずれかであってよいアルキル、アルケニル、およびアルキニル基であって、限定はされないがシクロプロパン、シクロペンテン、および6,8メチル−スクシンイミドリポ酸塩を含むアルキル、アルケニル、およびアルキニル基;
(7)それらのいずれの炭素にも付加を有してよいアルキル、アルケニル、およびアルキニル基であって、限定はされないがヒドロキシルおよびアミンを含むアルキル、アルケニル、およびアルキニル基;
(8)チオエーテル結合を通して連結され、ベンゼンまたはベンゼン誘導体であってよい芳香族またはアリール基であって、限定はされないがトルエンおよびアニリンを含む芳香族またはアリール基;
(9)硫化アルキル基 CH3(CH2)n−S−であって、限定はされないがnは0〜9を表してよく、ジスルフィド結合を通して連結される硫化アルキル基;
(10)イミドイル基 CHR4C(=NH)−であって、限定はされないがnは1〜10を表してよく、チオアミド結合を通して連結されるイミドイル基;および
(11)セミアセタール基 R5CH(OH)−S−であって、R5が強力な電子求引性の置換基をもつ化合物に限定され、限定はされないがトリクロロアセトアルデヒドおよびピルビン酸を含むセミアセタール基であってよい]
またはその塩により定義される。
【0055】
上に定義されるR1およびR2は非置換または置換であってよく、スルホキシドまたはスルホン産生のために酸化できるチオエステル、例えばC−S(O)−RおよびC−S(O)2−Rもまた含んでよい。
【0056】
R1およびR2はさらに、チオスルフィン酸またはチオスルホン酸へと酸化できるジスルフィド、例えばそれぞれC−S(O)−S−RおよびC−S(O)2−S−Rを含んでよい。
【0057】
R3は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよい。同様に、R4は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよい。
【0058】
R5は−CCl3、−CF3、または−COOHを表す。
【0059】
本発明の第2の実施形態によれば、リポ酸誘導体は一般式2
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R3C(O)−、アルキル CnH2n+1、CnH2n−1として定義されるアルケニル、CnH2n−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R3C(=NH)−として定義されるイミドイル、およびR4CH(OH)−S−として定義されるヘミアセタールから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3およびR4は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5は−CCl3、−CF3、または−COOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、およびnは0〜10を表す)
またはその塩により定義される。
【0060】
本発明の第3の実施形態によれば、リポ酸誘導体は一般式3
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
またはその塩である。
【0061】
本発明の第4の実施形態によれば、リポ酸誘導体は一般式4
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、上に定義されるR1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
またはその塩により定義される。
【0062】
さらに、これらの一般構造のいずれかもしくは全てが細胞またはミトコンドリア内で代謝され得ることから、上に言及された構造の代謝産物は本発明の範囲内にあることが明確に意図される。
【0063】
第1および第2両方の一般式内で、特定のリポ酸誘導体それぞれの(R)−異性体は、(S)−異性体よりも大きな生理活性を有することが観察されている。従って、本発明における使用のためには両方の異性体が意図されるが、特に好ましい実施形態では(R)−異性体を優先的に用いるか、または(S)−異性体との混合物中に(R)−異性体が存在することが特に意図される。
【0064】
本発明の医薬組成物はまた、PDH複合体中に見出されるホスファターゼ、キナーゼ、および脱水素酵素の酵素構成物質の発現レベルも調節し得る。この調節は、転写、翻訳、または適切な遺伝子のエピジェネティックなサイレンシングを含む後翻訳段階において起こり得る。
【0065】
本発明の組成物は、薬剤的に許容される担体または賦形剤をさらに含んでよい。薬剤的に許容される担体の例は当該技術分野において公知であり、医薬組成物中で通常用いられるもの、例えば限定はされないが、抗酸化剤、緩衝剤、キレート剤、香味料(flavorants)、着色剤、保存料、生物学的利用能を増大させるための吸収促進剤、抗菌剤、およびそれらの組み合わせを含む。このような添加物の量は望まれる性質に依存し、当業者によって容易に決定できる。
【0066】
通常本発明の医薬組成物は、塩、緩衝剤、保存料、および適合する担体を、任意にその他の治療的成分と組み合わせて含んでよい。医薬に用いられる場合、塩は薬剤的に許容されなければならないが、薬剤的に許容されない塩が、その薬剤的に許容される塩の調製のために便利に用いられてよく、本発明の範囲から除外されるものではない。このような薬理学的および薬剤的に許容される塩は、限定はされないが、以下の酸:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、p−トルエンスルホン酸、酒石酸、クエン酸、メタンスルホン酸、マロン酸、ギ酸、コハク酸、ナフタレン−2−スルホン酸、およびベンゼンスルホン酸から調製されるものを含む。また、薬剤的に許容される塩は、カルボン酸基のナトリウム、カリウム、またはカルシウム塩のような、アルカリ金属またはアルカリ土類塩として調製できる。
【0067】
本発明はさらに、細胞に有効量の少なくとも1の治療薬または診断薬を送達することによる、患者を治療薬または診断薬によって治療または診断するための、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットの構造、発現、および/または活性の制御または撹乱の変化を含む方法を、細胞の過剰増殖によって特徴付けられるものを含む疾病、状態、もしくは症候群、またはそれらの症状を予防、診断、または治療するために提供する。癌の改良された治療としてのPDH複合体の調節が特に意図され、これは腫瘍細胞増殖、血管新生、転移性増殖、アポトーシスの制御による原発腫瘍の治療、および外科的な除去の後または同時に起こる微小転移の発生の治療;および原発腫瘍の放射線またはその他の化学療法による治療を含む。本発明の医薬組成物は、原発性または転移性のメラノーマ、リンパ腫、肉腫、肺癌、肝臓癌、ホジキンおよび非ホジキンリンパ腫、白血病、子宮癌、子宮頸癌、膀胱癌、腎癌、大腸癌、ならびに腺癌、例えば乳癌、前立腺癌、卵巣癌、および膵臓癌のような癌の型に有用である。
【0068】
治療および診断への応用のため、該医薬組成物は、薬剤的に許容される担体と組み合わせた場合に患者へ直接投与できる。この方法は、治療薬もしくは診断薬を単独、または有効量のその他の治療薬もしくは診断薬、限定はされないが解糖阻害剤、微小管相互作用剤、細胞増殖抑制剤、葉酸阻害剤、アルキル化剤、トポイソメラーゼ阻害剤、チロシンキナーゼ阻害剤、ポドフィロトキシンまたはその誘導体、抗腫瘍剤、抗生剤、化学療法剤、アポトーシス誘導剤、抗血管新生剤、ナイトロジェンマスタード、核酸挿入剤、およびそれらの組み合わせ、と組み合わせて投与することにより実行されてよい。このような治療薬は、その他の代謝阻害剤をさらに含んでよい。このような治療薬の多くは公知である。組み合わせによる治療法は、治療法に対する患者の反応を増幅または確実にする必要に応じたこのような状態の治療において、同時、連続的、または別々の使用を提供する。
【0069】
本発明の方法は、医学的に許容される投与のいずれの様式を用いて実行されてもよく、臨床的に許容されない有害作用を起こすことなく、有効濃度の活性化合物を産生する。製剤は特に非経口投与に適することが好ましいが、本発明の組成物はエアロゾル、スプレー、散剤、ゲル、ローション、クリーム、坐薬、軟膏などの形態において、吸入、経口、外用、経皮、経鼻、眼内、肺内、直腸内、経粘膜、静脈内、筋肉内、皮下、腹腔内、胸腔内、胸膜内、子宮内、腫瘍内、または点滴の方法論または投与のためにもまた処方できる。このような製剤が望まれる場合、望まれる一貫性およびその他の性質を製剤に与えるため、当該技術分野において公知のその他の添加物が含まれてよい。
【0070】
当業者は、治療薬または診断薬の特定の投与様式は、投与が疾病、状態、症候群、またはそれらの症状の治療、診断、または予防;治療または診断される医学疾患の重症度;および治療的有効性に要求される投薬量のためであるか否かに関わらず、選択された特定の薬剤に依存することを認識するであろう。例えば、白血病治療のための抗癌剤投与の好ましい様式は静脈内投与を含み得るが、皮膚癌治療の好ましい方法は、局所または皮内投与を含んでよい。
【0071】
ここで用いられる「有効量」は、望まれる治療効果または診断効果が達成される、治療薬もしくは診断薬の投薬量または複数の投薬量を指す。一般に治療薬または診断薬の有効量は、用いられる特定の薬剤の活性;その薬剤の代謝の安定性および作用の長さ;種、年齢、体重、一般的健康、食事状態、性別および被験体の食事制限;投与の様式および時間;排泄速度;場合により薬物の組み合わせ;および治療される特定の状態の症状および/または重症度の程度によって変動し得る。望まれる結果を得るための1日当たりの1回または数回の投与における正確な投薬量は、当業者によって過度の実験なしに決定でき、かつ該投薬量は望まれる治療効果を達成するためか、またはいずれの合併症の現象において、個々の実行者により調整されてよい。重要なことは、癌治療に用いる際、使用される治療薬の投薬量は正常細胞には実質的に無害であるものの、腫瘍細胞の死滅には十分でなければならないことである。
【0072】
本発明の医薬組成物に含まれる治療薬または診断薬は、患者へ安全に投与できる最大量までの、望まれるいずれの量においても調製できる。治療薬または診断薬の量は、0.01mg/mL未満から1000mg/mLを超えるまで、好ましくは約50mg/mLに変動してよい。
【0073】
一般に本発明の医薬組成物は、PDH複合体の構造および/または活性の調節に有効な量を患者へ投与するために十分な様式で送達され得る。従って投薬量は、約0.3mg/m2から2000mg/m2、好ましくは約60mg/m2に変動してよい。投薬量は単一用量において、または個別に分けられた用量の形、例えば1日当たり1から4以上において投与されてよい。被験体の反応がある用量において不十分な場合は、より高い用量(または異なる、さらに限局性の送達経路による有効なより高い用量)が、患者の耐性の範囲まで用いられてよい。1日当たりの複数の用量は、治療薬もしくは診断薬の適切な全身性または標的濃度を達成するために意図される。
【0074】
さらに本発明の別の実施形態によれば、本発明のリポ酸誘導体はin vitroでの診断薬および予測薬(predictive agents)として用いられてよい。先に述べたように、問題となる特定の腫瘍細胞または細胞型に依存して、様々なリポ酸誘導体は、PDH複合体の調節を通した異なる腫瘍の種類の阻害について多かれ少なかれ有効であり得る。従って、例えば診断または適切な化学療法的戦略の選択が困難であり得る場合、in vitroでの腫瘍細胞の培養を、特定の腫瘍細胞型を標的にすることが知られているリポ酸誘導体と共に試験することにより、腫瘍型および有効な治療を特定するための別のアプローチが提供される。
【0075】
図面に戻ると、図6Aは、正常組織および腫瘍細胞のin vivoでのエネルギー代謝において考えられる多数の相違を図示する。相当する条件下で正常細胞に比べると、腫瘍細胞はしばしばATP産生のためにミトコンドリアのエネルギー代謝よりも細胞質の解糖に強く依存する。PDH複合体の発現および制御の変化は、明らかにこの腫瘍特異的な適応の一部である。これらの効果を生み出す、PDHの触媒成分のレベルの減少および/または抑制性PDKのレベルの増加により、腫瘍細胞は、PDH複合体を攻撃する薬剤に対して正常細胞よりもさらに脆弱となるであろう。
【0076】
図6Bは、リポ酸がPDH複合体中でピルビン酸からのアセチルCoA合成に関わる正常の反応を触媒することから、この構造を示す。リポ酸はin vivoでそのカルボキシル末端を通し、非ペプチドアミド結合によって、E2リポイルドメイン活性部位におけるリジンのイプシロンアミノ基へ連結する。PDH E2結合リポ酸塩の酸化/還元/アセチル化状態は、E1αPDHサブユニットのリン酸化の不活性化を制御することによってPDH活性を制御する、キナーゼおよびホスファターゼによりモニターされることも認められる。この図面はまた、本発明において使用されてよい三つの代表的なリポ酸誘導体の構造も示す。細胞培養において、CPI−613およびCPI−045は高い抗癌効力を持つが、CPI−157は活性が少ないかまたは無であり、幾つかの実験ではコントロールとして有用である。
【0077】
図6Cは、E2をその結合リポ酸塩であるE1と共に含むPDH複合体成分と、調節性PDKとの間の関連性を示す。高レベルのアセチルリポ酸塩またはジヒドロリポ酸塩(図示していない)はPDKを活性化し、次にこれがPDH複合体触媒作用の第1段階を触媒するサブユニットであるE1αを不活性化することによって、PDH複合体を通したさらなる流れを抑制する。図3Aに見られるように、この工程はPDH複合体を通した炭素/エネルギーの流れの調速機として作用し、かつこの調節工程は明らかに、様々な腫瘍細胞のエネルギー代謝を支持するために実質的に変更される。
【0078】
図7は、異種移植腫瘍の増殖における本発明の医薬組成物の効果を示す。実施例2に記載するように、脇腹の背側に細胞を皮下移植した。次に、図に示す日にマウスへの薬物(またはビヒクルのみ;「モック」)の腹腔内注射を開始した。左のパネルに、週に3回1mg/kgの本発明またはビヒクルコントロールと共に注射した膵臓腫瘍モデルを示す。この実験は、BxPC−3細胞により行った2実験、およびAsPC−1細胞により行った2実験の代表である。右の三つのパネルは、週に1回(丸)、週に3回(逆三角形)、または週に5回(ビヒクル処置は三角形、薬物処置は四角形)のいずれかにより示される濃度と共に注射したH460肺腫瘍モデルを示す。この実験は、H460細胞により行った4実験の代表である。
【0079】
図8は、三つの腫瘍細胞型および非形質転換細胞型(MDCK)を、200〜300μM(「処理」)またはモック処理(「モック処理」)のいずれかで、本発明の医薬組成物により処理した効果を示す。細胞は10%血清を含む適切な組織培地により、または48時間処理した。実施例2に記載の方法論を通し、三つの癌細胞株にアポトーシスまたはアポトーシス様経路(図11も参照)による広範な細胞死が観察される。対照的に、非形質転換MDCK細胞は、明らかにこの用量での薬物処理によって影響されない。
【0080】
図9Aは、致死閾値(10%血清中、200μM)またはそれを超える量の本発明の医薬組成物により処理したH460肺癌細胞におけるATPレベルを示す。破線は表示される濃度での処理を示す。対応するテクスチャの実線は、表示される時間後に薬物を除去し、薬物を含まない培地中で60分間回復させる処理を示す。一組の矢印(Block arrows)は、ATP回復の間隔を示す。
【0081】
図9Bは、ピルビン酸(ピルビン酸メチルの形)が主要な炭素原である培地(破線)とグルコースが主要な炭素原である培地(実線)中での、ATP合成の阻害を比較する。本発明の医薬組成物は、両方の培地中で、同じ閾値濃度において最終的に細胞死を引き起こすが、細胞の全ATPレベルの早い枯渇はピルビン酸を含む培地中で高度であり、グルコースを含む培地中では見られないことが認められる。また、細胞死の開始は200μMよりも300μMの薬物濃度でより迅速である。
【0082】
図9Cは、SK−Br−3乳癌細胞とHMEC正常乳房細胞における、本発明の医薬組成物によるATP合成の阻害を比較する。図6Aおよび図6Bにその結果が示される実験とは対照的に、これらの実験は無血清培地(MEBM)中で行った。結果として薬物の致死閾値は、約50μMと低めであった。正常細胞サンプルにおける22時間目のATPレベルの小さな低下は薬物用量には関連せず、普通の実験的変動を反映していることに留意されたい。
【0083】
図9Dは、本発明の医薬組成物(左のグラフ)、リポ酸(中央のグラフ)、および本発明の不活性型(右のグラフ)による、H460肺癌細胞におけるATP合成の阻害を比較する。図9Cに示すように、これらの実験は、薬物の致死閾値が約50μMとなるように無血清培地中で行った。
【0084】
図10は、PDH(PDC)および(αKDH)の酵素活性の、腫瘍細胞ミトコンドリアでのレベルにおける、本発明の医薬組成物(10%血清を含むDMEM中で400μM)の効果を図示する。PDHは強力に阻害されるが、αKDHはそうでないことが認められる。実施例2に記載するように、精製したミトコンドリア抽出物中の酵素活性レベルは、添加炭素源に反応するレサズリン還元を用いて測定する。バックグラウンドラインは、添加炭素源なしのレサズリン還元に相当する。
【0085】
次に図11Aでは、本発明の医薬組成物により処理(+)またはモック処理(−)した(10%血清を添加したRPMI培地中で、400μMで120分間)H460肺癌細胞抽出物の、二次元ゲルのウェスタン分析を行った。ウェスタン転写は、PDH複合体のE1αおよびE2サブユニットに対するモノクローナル抗体のカクテルを用いて探査した。ウェスタン転写はE2に合わせた。薬物処理サンプル中のE1には、高リン酸化型の実質的に高いレベルおよび低リン酸化型の減少したレベルが認められる。左の垂直な白線は、ゲル整列の基準の一つである、E2サブユニットの移動度を示す。右の垂直な白線は、推定的、酵素的に活性のある成分である、リン酸化の少ないE1α型を通る。
【0086】
図11Bは、本発明の医薬組成物による処理またはモック処理した対の二次元ゲルサンプルを、拡大して示す。エレメントAは、図8Aの一部の拡大である。エレメントBは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で180分間処理(+)されたSK−Br−3乳癌細胞である。エレメントCは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で240分間処理(+)されたHMEC正常乳房上皮細胞である。エレメントDは、MEBM無血清乳房上皮細胞培地中で、モック処理(−)、および80μMの組成物で240分間処理(+)されたSK−Br−3乳癌細胞である。垂直な白線は、推定的、酵素的に活性のある成分である、リン酸化の少ないE1α型を通る。
【0087】
図12Aおよび12Bは、本発明の医薬組成物のin vivoでの強力で選択的な抗癌効果に対する作業仮説を示す。例えば図12Aは、PDC E2サブユニットに共有結合した内在性リポ酸塩により調節される、PDKの制御的役割を示す。PDKは通常、高レベルの還元および/またはアセチル化リポ酸塩に反応してPDCを不活性化し、これは腫瘍細胞内で明らかに実質的に修飾される工程である。
【0088】
同時に図12Bは、PDC内における、PDKのその基質であるPDC−E1に対する比の大きな量的相違を示し、このことはin vivoで正常および腫瘍細胞を区別すると考えられる。正常細胞では、低レベルのPDKがPDH複合体の周囲をhand−over−handに「歩く」(その二つの二量体サブユニットを通して)と考えられており、徐々にE1をリン酸化する。このリン酸化は、PDPの脱リン酸化と定常的に平衡状態である(図示していない)。ここに図示される作業仮説では、チオクタンが、通常アセチルリポ酸塩および/またはジヒドロリポ酸塩が結合するのと同じ部位を通じてPDKを刺激し、これにより1以上のPDKアイソフォームを不活性なE1αへと人工的に刺激する。癌細胞では、さらに高レベルのPDKが、チオクタンによりさらに効果的にこの刺激を作り出し、PDCの酵素活性およびミトコンドリアエネルギー代謝を停止させるであろう。
【発明を実施するための形態】
【0089】
以下の限定されない実施例は、本発明の医薬組成物についての理解を促進するために提供される。
【実施例1】
【0090】
チオクタンの化学合成
リポ酸誘導体(すなわちチオクタン)CPI−613およびCPI−157は、米国特許第6,331,559 B1号および米国特許第6,951,887 B2号に記載の方法の改変により、6,8−ビスメルカプトオクタン酸を出発物質として用いて合成した。チオクタンCPI−045は、米国特許第6,331,559 B1号に記載の通りに合成した。
【0091】
三つのチオクタンの構造解析について以下に述べる。CPI−045とCPI−613の独立した複数の合成は、これらの抗癌特性について区別できなかった。異種移植(図7)およびATP測定(図9)に用いたCPI−613の純度は99%を超えていた。その他の全ての調製品は98%を超える純度であった。
【0092】
CPI−613:6,8−ビスベンジルスルファニルオクタン酸:白色の結晶性固体、m.p.65〜66℃(lit.167.5〜69°);1H−NMR(250MHz,CDCl3):δ7.15−7.4(m,10H)、3.66(s,2H)、3.64(s,2H)、2.52−2.62(m,1H)、2.50(t,J=7.6Hz,2H)、2.29(t,J=7.6Hz,2H)、1.2−1.8(m,8H);13C−NMR(62.9MHz,CDC13):δ179.6、138.6、138.5、128.9、128.8、128.5、128.4、126.9、44.1、36.4、35.1、34.4、33.8、28.7、26.0、24.4。
【0093】
CPI−157:6,8−ビスエチルスルファニルオクタン酸:無色の油;TLC
(EtAc:ヘキサン:HAc,200:200:1v/v):Rf=0.60;1H−NMR(300MHz,CDCl3):δ2.64−2.76(m,1H)、2.65(t,J=7.5Hz,2H)、2.52(q,J=7.5Hz,2H)、2.49(q,J=7.2Hz,2H)、2.36(t,J=7.4Hz,2H)、1.40−1.85(m,8H)、1.25(t,J=7.2Hz,3H)、1.22(t,J=7.5Hz,3H);13C−NMR(75MHz,CDCl3):δ180.0、44.3、34.6、33.9、28.9、26.2、25.9、24.5、24.2、14.9、14.7;IR(フィルム):2963、1708、1449、1423、1283、1263cm−1。
【0094】
CPI−045:6,8−ビスベンゾイルスルファニルオクタン酸:無色の粘性油;TLC(ヘキサン:EtAc:HAc,100:50:1v/v):Rf=0.30;1H−NMR(250MHz,CDCl3):δ7.9−8.1(m,4H)、7.38−7.60(m,6H)、3.8−4.0(m,1H)、3.0−3.3(m,2H)、2.34(t,J=7.1Hz,2H)、1.4−2.2(m,8H);13C−NMR(62.9MHz,CDCl3):δ191.7、191.5、179.7、137.0、136.9、133.3、128.5、127.3、127.1、43.6、35.0、34.6、33.8、26.4、26.2、24.3;IR(フィルム):2973、1710、1704、1667、1665、1662、1448、1207、1175、911、773、757、733、688、648cm−1。
【実施例2】
【0095】
チオクタンの抗癌効果決定のために用いた方法
細胞:ヒト腫瘍細胞株はATCCから入手し、ATCCの勧告に従って増殖させた。ヒト乳房上皮細胞(HMEC)、末梢気道上皮細胞(SAEC)、および正常ヒト表皮角化細胞(NHEK)の初代細胞は、LONZA Walkersville, Inc(ウォーカーズビル、メリーランド州)から得た。各細胞株は、開発した供給業者から購入した適切な培地中で、供給者の指示に従って維持および増殖させた。ここに報告する実験では、3ないし6回継代した正常細胞を使用した。
【0096】
腫瘍増殖阻害の研究:CD1−Nu/Nu雌マウスに、ヒトBxPC−3もしくはAsPC−1膵臓腫瘍細胞、またはH460 NSCLCを皮下(SC)注射によって移植した。約8〜12日後、図の説明文に示される用量およびスケジュールによりマウスに腹腔内(IP)注射した。薬物またはビヒクルは、体重25gあたり約2ml注射した。薬物濃度は1.25mg/ml(約3.1mM)以下であった。ビヒクル/溶媒は、25mM以下のトリエタノールアミン水溶液により構成された。モック処置動物に注射されたビヒクルは、この実験で最も高い薬物用量が注射された溶媒と常に同一であった。マウスは、健康状態および死亡率について毎日モニターされた。体重および腫瘍の体積を、処置前には毎日、処置中および処置後には週に約3回評価した。マウスはStony Brook大学動物施設において、12時間の明/暗サイクルに置かれ、自由に摂食し、施設の指針に従って飼育された。
【0097】
細胞死アッセイ:細胞生存率評価のために、ほとんどの場合CellTiter−Gloアッセイ(Promega)を、チオクタンによるATP合成の初期の阻害によって混乱させないための十分に長い時間使用した(図9)。典型的な実験では、黒く透明な底の96ウェルプレートに、細胞をウェルあたり5,000細胞まいた。18〜25時間後に培地を、薬物溶媒(血清を含む培地中に2.8mM、無血清培地中に0.7mMの、トリエタノールアミン水溶液)または同じ溶媒中のチオクタンCPI−613を含む新鮮な培地に交換した。製造者の指示に従い、薬物用量に応じて、薬物添加後24または48時間後にアッセイを行った。
【0098】
幾つかの場合には、細胞は48ウェルプレートにウェルあたり10,000細胞まき、18〜25時間後に培地を、薬物溶媒(最終濃度が1%のEtOH)または同じ溶媒中の様々な濃度のチオクタンCPI−045を含む新鮮な培地に交換した。残りの実験の間、細胞を溶媒または薬物を含む培地中にとどめた。薬物添加後24、48、および72時間後にプレートを点検し、コンフルエンスのパーセンテージから細胞数を推定した。これらの条件下でチオクタンにより誘導された細胞死は、閾値に近い用量で高度にアポトーシス性であり、細胞数の推定は非常に信頼できる死の指標である(図10)。72時間後にも完全な細胞が残っていた場合には、トリパンブルー排除試験を行った。
【0099】
表2に、腫瘍細胞に対するチオクタンのin vitroでの作用についてのデータを示す。CPI−613および/またはCPI−045による殺傷に対する感受性について本発明者らが調査した、ヒト腫瘍および初代ヒト細胞を収載する。「+」は、約200〜300μM(10%血清存在下で)および無血清培地中で約50μMの用量でアポトーシスまたはネクローシス様の細胞死を起こした細胞を示す(図8および図9)。「−−」は、相当する培地中で細胞死誘導のために5倍高い薬物用量を必要とした細胞を示す。「nt」は、未試験の組み合わせを示す。図8でのMDCK正常細胞と同様に、全ての腫瘍細胞株は10%血清を含む適切な培地中で分析された。加えて、HMEC、SAEC、NHKC初代ヒト細胞、およびSK−BR−3、A549およびH460腫瘍株もまた、適切な無血清培地中で分析された。初代細胞は接触阻止され、形質転換細胞は同程度の濃度であった。
【0100】
ATPアッセイ:黒く透明な底の96ウェルプレートに、細胞をウェルあたり5,000細胞まいた。18〜25時間後に培地を、薬物溶媒(トリエタノールアミン)またはチオクタン(CPI−613またはCPI−157)またはリポ酸を含む新鮮な培地に、示される時間間隔および薬物濃度において交換した。トリパンブルー排除を用い、細胞の生存率および完全性を、薬物を除去した後の回収率によって評価した。ATPは、CellTiter−Glo luminescence assay(Promega)を用い、製造者の指示に従って測定した。全ての測定は二重に行い、高い一貫性が示された。平均の標準誤差は、測定値の0.1〜2%の範囲であった。その結果、図9ではエラーバーを省略した。図9のピルビン酸メチル培地は、10%透析ウシ胎仔血清、5mM HEPES(pH7.4)、および10mMピルビン酸メチル(Sigma−Aldrich)を補充した、グルコースを含まないRPMI(Invitrogen)から成り、対応するグルコース培地は従来のRPMI(Invitrogen)であった。
【0101】
PDHおよびαKDH酵素アッセイ:腫瘍細胞を15cmプレート内で80%コンフルエンスに増殖させ、示されているようにCPI−613によって処理した。MoreadithおよびFiskum1の方法に従ってミトコンドリアを分離した。ミトコンドリアを0.4%ラウリルマルトシド中で溶解した。50μlのミトコンドリアライセートを96ウェルプレートへ加えた。50μlの反応混合物(50mMトリス、pH7.5、2mM β−NAD+、225μMv TPP、2mMピルビン酸またはα−ケトグルタル酸、150μMコエンザイムA、2.6mMシステイン、1mM MgCl2)をミトコンドリアライセートに加え、混合物を37℃で45分間インキュベートした。この時点で、混合物に15μMのレサズリンおよび0.5U/mlのジアホラーゼを加え、さらに5分間インキュベートした。マイクロプレートリーダー(Fluorostar)内での530nmの励起波長および590nmの発光波長を用いた蛍光測定により、NADHの産生をモニターした。全ての測定は二重に行い、高い一貫性が示された。平均の標準誤差は、測定値の0.3〜4%の範囲であった。その結果、図10ではエラーバーを省略した。
【0102】
E1αのリン酸化:
2−Dゲルのための細胞ライセート:細胞を60mmディッシュ内で95%コンフルエンスに増殖させ、示されているように薬物または溶媒によって処理した。細胞をin situで450μlの溶解緩衝液A[455μl Zoom 2D protein solubilizer 1(Invitrogen)、2.5μl 1Mトリスベース、5μl 100Xプロテアーゼ阻害剤カクテル(EDTAを含まないComplete mini、Roche);5μl 2M DTT]により溶解した。細胞ライセートを1.5ml微量遠心チューブへ移し、氷上で50%出力にて15段階の超音波処理を行った。室温での10分間のインキュベーション後に2.5μlのジメチルアクリルアミド(DMA、Sigma−Aldrich)を加え、ライセートをさらに10分間インキュベートした。5μlの2M DTTを加えて、過剰のDMAを中和した。ライセートを16,000xgで15分間遠心した。
【0103】
2−Dゲル:Zoom Benchtop proteomics system(Invitrogen)を、製造者の指示に従って使用した。簡単に述べると、30〜50μlのライセートを0.8μl pH3〜10両性電解質、0.75μl 2M DTTと混合し、Zoom 2D protein solubilizer 1にて150μlにした。150μlのサンプルをIPG runner内に装填し、pH3〜10のNL IPGストリップを加えた。ストリップを室温で一晩浸漬した。等電点(isolectric)電気泳動には段階的プロトコルを使用した(250V、20分;450V、15分;750V、15分 2000V、30分)。ストリップを1Xローディング緩衝液で15分間、続いて1Xローディング緩衝液および160mMヨード酢酸で15分間処理した。ストリップはNuPAGE 4−12% Bis Tris ZOOMゲル(Invitrogen)で電気泳動した。
【0104】
表2 腫瘍および初代細胞に対するチオクタンのin vitroでの効果
【0105】
ウェスタン:タンパク質はPVDF 4.5μmメンブレンにブロットした。mAb(Invitrogen)を用いて、PDH E1αおよびE2を検出した。
【0106】
カスパーゼ−3およびPARPの切断:Roy and Nicholson.2に従い、切断されたカスパーゼ−3をウェスタンブロットにて検出した。簡単に述べると、薬物または溶媒処理後に細胞をかき取り、培地/細胞/アポトーシス小体の混合物を6000xgにて遠心した。ペレットを溶解緩衝液C(4M尿素、10%グリセロール、2%SDS、0.003%BPB;5% 2−メルカプトエタノール)により溶解した。ウェルあたり30μgの全細胞ライセートタンパク質を、12%ビス−トリスゲルにロードした。タンパク質はPVDF 4.5μmメンブレンにブロットした。抗カスパーゼ3mAb(マウスモノクローナル[31A1067];abcam)により、プロ−および活性化カスパーゼ3を検出した。モノクローナル抗ポリ(ADPリボース)ポリメラーゼ抗体、クローンC−2−10(Sigma−Aldrich)を用いて、PARP切断を検出した。
【0107】
ミトコンドリアCa2+の検出:細胞を35mmガラス底プレート(BD Biosciences)に3x105まき、一晩増殖させて、示されているように薬物または溶媒によって処理した。次に細胞をカルシウム色素Fluo−4、X−Rhod−1またはRhod−2(4μM、Invitrogen)と共にフェノールレッドを含まない培地中にロードし、37℃で10分間インキュベートした。細胞をPBSで1回洗浄し、Axiovert 200M(Zeiss)デコンボリューション顕微鏡を用い、固定した露光時間で、FITCフィルターを用いて画像を得た。蛍光の定量化は、製造者より提供されたソフトウェアを用いて行った。X−Rhod−1およびRhod−2からは同様の結果が得られ(図13)、これらの色素はミトコンドリアCa2+シグナルを測定していることが示された3−4。
【0108】
参考文献
1.Moreadith RW and Fiskum G. Isolation of Mitochondria from Ascites Tumour−Cells Permeabilized with Digitonin. Analytical Biochemistry 137,360−367(1984).
2.Roy S and Nicholson DW.Criteria for identifying authentic caspase substrates during apoptosis.Apoptosis 322,110−125(2000).
3.Gerencser AA and Adam−Vizi V. Selective,high−resolution fluorescence imaging of mitochondrial Ca2+ concentration.Cell Calcium 30,311−321(2001).
4.Gyorgy H,Gyorgy C,Das S,Garcia−Perez C,Saotome M,Roy SS,and Yi MQ. Mitochondrial calcium signalling and cell death:Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis.Cell Calcium 40,553−560(2006).
【実施例3】
【0109】
チオクタンによるミトコンドリア膜電位およびCa2+取り込みの撹乱
チオクタンによるATP合成の阻害における基質効果(図9)は、該薬物がミトコンドリアマトリックス内でTCAサイクルに干渉することを示す。これが事実であれば、ミトコンドリア膜電位は致死閾値用量以上で損傷し得ると本発明者らは予測する1。電位感受性の色素であるTMREを用い、本発明者らは予想された効果を観察する(図13)。ミトコンドリア膜電位は、薬物処理の開始によって迅速に低下する。膜電位低下の動態は、ミトコンドリア基質存在下でのATP合成の損失によく似ている(図9)。
【0110】
ミトコンドリアでのATPの枯渇は、小胞体を含む細胞質貯蔵から放出されたCa2+の取り込みを含む恒常性反応を誘発することが知られている2。さらに、このCa2+のミトコンドリアマトリックス内への移入には、ミトコンドリア膜電位が必要であると考えられている。従って、進行的に損なわれる膜電位を考えると、本発明者らは、致死閾値以上でのチオクタン処理が持続性のCa2+の細胞質放出と共にイオンの一過性のミトコンドリアへの取り込みを起こし得ると予測する。本発明者らはミトコンドリアCa2+の測定にX−Rhod−1およびRhod−2、および細胞質Ca2+の測定にFluo−4を用いて、これらの予想される効果を観察する(図13)。
【0111】
致死閾値かまたはそれよりもわずかに高いCPIの用量において、約2時間までに(図9および13を比較されたい)ミトコンドリア膜電位が低下するに従い、この最初のミトコンドリアの一過性Ca2+の取り込みは減衰する。この4〜6時間後、カルシウム依存性細胞死経路の開始に付随すると推定される、第2の大きなミトコンドリアCa2+ピークが続く3。
【0112】
参考文献
1.Garrett R and Grisham CM. Biochemistry.Thomson Brooks/Cole,Southbank,Vic,Australia; Belmont,CA.(2007).
2.Graier WF,Frieden M,and Malli R.Mitochondria and Ca2+ signaling:old guests,new functions.Pflugers Archiv−European Journal of Physiology 455,375−396(2007).
3.Gyorgy H,Gyorgy C,Das S,Garcia−Perez C,Saotome M,Roy SS,and Yi MQ. Mitochondrial calcium signalling and cell death:Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis.Cell Calcium 40,553−560(2006).
【実施例4】
【0113】
チオクタンにより誘導される多様な細胞死プログラム
詳しい機構は完全には理解されていないが、ある状況下では、ミトコンドリアエネルギー代謝の減少は細胞死経路へ入るための決定に関連することが知られている1−3。閾値を超えるが、この最小の殺傷用量の2倍以内であるチオクタン用量においては、試験された全ての癌細胞型が形態的には主にアポトーシス様である細胞死を起こす(図8)。この条件下でのアポトーシス様の死は、従来のアネキシン免疫染色およびTUNEL DNA末端標識アッセイによって確認した(結果は提示していない)。
【0114】
さらに高い薬物用量(閾値の2倍を超える値よりも多い)において、活性のあるチオクタンは、形態的なアポトーシスへの関連を示さずに細胞死を誘導し(再プレートによる生存率アッセイ、およびトリパンブルーまたはプロピジウム排除による評価として)、これはネクローシス様経路を示唆している(結果は提示していない)。
【0115】
これらのデータは、チオクタンCPI−613によるミトコンドリアエネルギー代謝の阻害が、細胞死誘導に正確に関連することを確証する。
【0116】
異なる多様な細胞死経路についての不活性化変異を含むと知られているかまたは推定される多様な腫瘍細胞4は全て、ほとんど同様のチオクタン用量において殺傷される(図8および表2)という知見が著しい。この知見はこれらの薬物が、複数の、潜在的に重複性である遠位細胞死遂行経路に関与できるマスターシグナルを誘導することを示唆する5。
【0117】
この可能性に一致して、本発明者らは一般的なカスパーゼ阻害剤であるZ−VAD−FMKが、チオクタンで処理された細胞における細胞死の形態をわずかに変化させるが、該薬剤の致死閾値用量では識別可能な効果をもたないことを見出している。
【0118】
チオクタン誘導性の細胞死が複数の末端遂行機構を通して進行できる可能性をさらに試験するため、本発明者らは異なる細胞死経路の指標であるカスパーゼ−3およびPARP−Iによる切断5について検討した。本発明者らは、チオクタンCPI−613およびCPI−045の両方が、異なる細胞において非常に変化しやすいレベルのこれらの二つの切断現象を誘導することを見出している(図14)。
【0119】
まとめるとこれらの結果は、チオクタンが、薬物用量および細胞型に依存して死への戦略的なコミットメントを誘導することができ、その決定の末端での戦術的な遂行について作動性であることを示す。
【0120】
参考文献
1.Watabe M and Nakaki T. ATP depletion does not account for apoptosis induced by inhibition of mitochondrial electron transport chain in human dopaminergic cells.Neuropharmacology 52,536−541(2007).
2.Yuneva M,Zamboni N,Oefher P,Sachidanandam R,and Lazebnik Y. Deficiency in glutamine but not glucose induces MYC−dependent apoptosis in human cells.Journal of Cell Biology 178,93−105(2007).
3.Skulachev VP. Bioenergetic aspects of apoptosis,necrosis and mitoptosis.Apoptosis 11,473−485 (2006).
4.Johnstone RW,Ruefli AA,and Lowe SW. Apoptosis:A link between cancer genetics and chemotherapy. Cell 108,153−164(2002).
5.Cregan SP,Dawson VL,and Slack RS. Role of AIF in caspase−dependent and caspase−independent cell death.Oncogene 23,2785−2796(2004).
【実施例5】
【0121】
リポ酸誘導体類似体の構造
以下に提示する様々なリポ酸誘導体類似体の限定されない例は、製造され、ここに開示される。
【0122】
前述の考察は、本発明の単なる典型的な実施形態を開示および記述するものである。当業者はこれらの考察および添付の請求項から、以下の請求項に定義される本発明の精神および範囲から逸脱することなく、ここに様々な変更、改変、および変動があってよいことを容易に認識するであろう。さらに、ここには典型的な実施形態が表されているが、その他の当業者は本発明のその他の設計または使用について気付くであろう。従って、本発明はその典型的な実施形態と関連して記述されているが、設計および使用の両方に対する多数の改変が当業者に明らかとなるであろうこと、およびこの応用がそのいずれの適応または変動も網羅するように意図されることが理解されるであろう。それ故本発明は、請求項およびその均等物のみによって限定されることが明白に意図される。
【特許請求の範囲】
【請求項1】
ヒトを含む温血動物の病的な細胞のミトコンドリア内の、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態のための薬剤的に許容される調節因子であって、該調節因子が、
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、R1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
その代謝産物;
またはその塩;
および
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、R1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
その代謝産物;
またはその塩から成る群より選択される調節因子。
【請求項2】
調節が可逆的リン酸化もしくは脱リン酸化を含む、請求項1に記載の調節因子。
【請求項3】
可逆的リン酸化または脱リン酸化が、酵素もしくは酵素複合体、またはそれらのサブユニットのキナーゼ、ホスファターゼ、および/または脱水素酵素に起こる、請求項2に記載の調節因子。
【請求項4】
調節因子がキナーゼ活性を促進または阻害する、請求項3に記載の調節因子。
【請求項5】
キナーゼがピルビン酸脱水素酵素キナーゼ(PDK)1、PDK2、PDK3、PDK4、およびそれらの各アイソフォームを含む群より選択される、請求項4に記載の調節因子。
【請求項6】
調節因子がホスファターゼ活性を促進または阻害する、請求項3に記載の調節因子。
【請求項7】
ホスファターゼがピルビン酸脱水素酵素ホスファターゼ(PDP)1、PDP2、およびそれらの各アイソフォームを含む群より選択される、請求項6に記載の調節因子。
【請求項8】
調節因子が脱水素酵素活性を促進または阻害する、請求項3に記載の調節因子。
【請求項9】
可逆的リン酸化または脱リン酸化がピルビン酸脱水素酵素(PDH)複合体に起こる、請求項2に記載の調節因子。
【請求項10】
調節がPDH複合体のE1αサブユニットに起こる、請求項9に記載の調節因子。
【請求項11】
調節がPDPならびにそのアイソフォームおよび変異型の不活性化によって起こる、請求項10に記載の調節因子。
【請求項12】
PDPの不活性がPDP発現の抑制によって起こる、請求項11に記載の調節因子。
【請求項13】
調節がPDKならびにそのアイソフォームおよび変異型の活性化によって起こる、請求項10に記載の調節因子。
【請求項14】
病的な細胞が請求項1に記載の調節因子による処理に感受性または非感受性を示す、請求項1に記載の調節因子。
【請求項15】
、処理に非感受性の病的な細胞が、それらが処理感受性となるように、少なくとも1の修飾された酵素もしくは酵素複合体、またはそれらのサブユニットの発現を誘導されてよい、請求項14に記載の調節因子。
【請求項16】
発現が遺伝操作によって誘導される、請求項15に記載の調節因子。
【請求項17】
誘導が転写操作によって達成される、請求項16に記載の調節因子。
【請求項18】
誘導が翻訳操作によって達成される、請求項16に記載の調節因子。
【請求項19】
誘導が翻訳後操作によって達成される、請求項16に記載の調節因子。
【請求項20】
発現がエピジェネティック操作によって誘導される、請求項15に記載の調節因子。
【請求項21】
発現が表現型操作によって誘導される、請求項15に記載の調節因子。
【請求項22】
病的な細胞が、請求項1に記載の調節因子による処理で少なくとも1の修飾された酵素または酵素複合体を発現する、請求項14に記載の調節因子。
【請求項23】
調節因子がPDKおよびそのアイソフォームおよび変異型の発現レベルに影響する、請求項1に記載の調節因子。
【請求項24】
調節因子がPDPおよびそのアイソフォームおよび変異型の発現レベルに影響する、請求項1に記載の調節因子。
【請求項25】
発現レベルが転写、翻訳、または翻訳後レベルにおいて変化する、請求項23または24に記載の調節因子。
【請求項26】
変化がエピジェネティックである、請求項25に記載の調節因子。
【請求項27】
調節因子が有毒代謝産物の生成を阻害する、請求項9に記載の調節因子。
【請求項28】
調節因子が有毒代謝産物の解毒を促進する、請求項9に記載の調節因子。
【請求項29】
代謝産物が、アセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルから成る群より選択される、請求項27または28に記載の調節因子。
【請求項30】
調節効果がアセトイン産生の減少により観察される、請求項28に記載の調節因子。
【請求項31】
可逆的リン酸化または脱リン酸化が非可逆的となる、請求項9に記載の調節因子。
【請求項32】
リン酸化または脱リン酸化の効果が細胞死をもたらす、請求項31に記載の調節因子。
【請求項33】
効果がアポトーシスである、請求項32に記載の調節因子。
【請求項34】
効果がネクローシスである、請求項32に記載の調節因子。
【請求項35】
調節因子が、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、もしくは症候群、またはそれらの症状の治療および予防に有用である、請求項1に記載の調節因子。
【請求項36】
少なくとも1の酵素複合体がPDH複合体である、請求項35に記載の調節因子。
【請求項37】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項35に記載の調節因子。
【請求項38】
疾病、状態、または症候群が癌である、請求項37に記載の調節因子。
【請求項39】
有効量の請求項1に記載の調節因子の投与を含む、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、もしくは症候群、またはそれらの症状を呈する患者において、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットを調節する方法。
【請求項40】
少なくとも1の酵素複合体がPDH複合体である、請求項39に記載の方法。
【請求項41】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項39に記載の方法。
【請求項42】
疾病、状態、または症候群が癌である、請求項41に記載の方法。
【請求項43】
患者から細胞サンプルを得ること、有効量の請求項1に記載の調節因子をin vitroで細胞へ投与すること、およびそこから結果を得ることを含む、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、または症候群の症状を呈する患者の診断および利益を予想するための方法。
【請求項44】
少なくとも1の酵素複合体がPDH複合体である、請求項43に記載の方法。
【請求項45】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項43に記載の方法。
【請求項46】
疾病、状態、または症候群が癌である、請求項45に記載の方法。
【請求項1】
ヒトを含む温血動物の病的な細胞のミトコンドリア内の、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態のための薬剤的に許容される調節因子であって、該調節因子が、
[式中、R1およびR2は水素、アルキル CnH2n+1、アルケン CnH2n、アルケニル CnH2n−1、アルキン CnH2n−2、アルキニル CnH2n−3、硫化アルキル CH3(CH2)n−S−、ジスルフィドアルキル CH3CHt−S−S−、チオカルバミン酸エステル (CH2)nC=NH−、およびセミチオアセタール CH3CH(OH)−S−(ここでnは1〜10、およびtは0〜9を表す)、芳香族、R4C(O)−として定義されるアシル、ヘテロアリール、R5C(=NH)−として定義されるイミドイル、有機金属アリール、アルキル−有機金属アリール、セミアセタール R6CH(OH)−S−、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され、;
式中、R1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4は水素、アルケニル、アルキニル、アルキルアリール、ヘテロアリール、アルキルヘテロアリール、および有機金属アリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R5は水素、アルケニル、アルキニル、アリール、アルキルアリール、ヘテロアリール、およびアルキルヘテロアリールから成る群より選択され、そのいずれもが置換または非置換であってよく;
式中、R6はCCl3、CF3、またはCOOHを表し;かつ
式中、xは0〜16を表す]
その代謝産物;
またはその塩;
および
(式中、Mは共有結合、−[C(R1)(R2)]z−、または金属がパラジウムではない金属キレートもしくはその他の金属複合体を表し;
式中、R1およびR2は水素、アシル R4C(O)−、アルキル CnH2n+1、CmH2m−1として定義されるアルケニル、CmH2m−3として定義されるアルキニル、アリール、ヘテロアリール、硫化アルキル CH3(CH2)n−S−、R4C(=NH)−として定義されるイミドイル、R6CH(OH)−S−として定義されるヘミアセタール、アミノ酸、炭水化物、核酸、脂質、ならびにそれらの多量体および組み合わせから成る群より独立して選択され;
式中、R1およびR2は非置換または置換であってよく;
式中、R3はアミノ酸、炭水化物、核酸、脂質、およびそれらの多量体から成る群より選択され;
式中、R4およびR5は水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルアリール、ヘテロアリール、およびヘテロシクリルから成る群より独立して選択され、そのいずれもが置換または非置換であってよく;
式中、R5はCCl3、CF3、またはCOOHから成る群より選択され;かつ
式中、xは0〜16、zは0〜5、nは0〜10、およびmは2〜10を表す)
その代謝産物;
またはその塩から成る群より選択される調節因子。
【請求項2】
調節が可逆的リン酸化もしくは脱リン酸化を含む、請求項1に記載の調節因子。
【請求項3】
可逆的リン酸化または脱リン酸化が、酵素もしくは酵素複合体、またはそれらのサブユニットのキナーゼ、ホスファターゼ、および/または脱水素酵素に起こる、請求項2に記載の調節因子。
【請求項4】
調節因子がキナーゼ活性を促進または阻害する、請求項3に記載の調節因子。
【請求項5】
キナーゼがピルビン酸脱水素酵素キナーゼ(PDK)1、PDK2、PDK3、PDK4、およびそれらの各アイソフォームを含む群より選択される、請求項4に記載の調節因子。
【請求項6】
調節因子がホスファターゼ活性を促進または阻害する、請求項3に記載の調節因子。
【請求項7】
ホスファターゼがピルビン酸脱水素酵素ホスファターゼ(PDP)1、PDP2、およびそれらの各アイソフォームを含む群より選択される、請求項6に記載の調節因子。
【請求項8】
調節因子が脱水素酵素活性を促進または阻害する、請求項3に記載の調節因子。
【請求項9】
可逆的リン酸化または脱リン酸化がピルビン酸脱水素酵素(PDH)複合体に起こる、請求項2に記載の調節因子。
【請求項10】
調節がPDH複合体のE1αサブユニットに起こる、請求項9に記載の調節因子。
【請求項11】
調節がPDPならびにそのアイソフォームおよび変異型の不活性化によって起こる、請求項10に記載の調節因子。
【請求項12】
PDPの不活性がPDP発現の抑制によって起こる、請求項11に記載の調節因子。
【請求項13】
調節がPDKならびにそのアイソフォームおよび変異型の活性化によって起こる、請求項10に記載の調節因子。
【請求項14】
病的な細胞が請求項1に記載の調節因子による処理に感受性または非感受性を示す、請求項1に記載の調節因子。
【請求項15】
、処理に非感受性の病的な細胞が、それらが処理感受性となるように、少なくとも1の修飾された酵素もしくは酵素複合体、またはそれらのサブユニットの発現を誘導されてよい、請求項14に記載の調節因子。
【請求項16】
発現が遺伝操作によって誘導される、請求項15に記載の調節因子。
【請求項17】
誘導が転写操作によって達成される、請求項16に記載の調節因子。
【請求項18】
誘導が翻訳操作によって達成される、請求項16に記載の調節因子。
【請求項19】
誘導が翻訳後操作によって達成される、請求項16に記載の調節因子。
【請求項20】
発現がエピジェネティック操作によって誘導される、請求項15に記載の調節因子。
【請求項21】
発現が表現型操作によって誘導される、請求項15に記載の調節因子。
【請求項22】
病的な細胞が、請求項1に記載の調節因子による処理で少なくとも1の修飾された酵素または酵素複合体を発現する、請求項14に記載の調節因子。
【請求項23】
調節因子がPDKおよびそのアイソフォームおよび変異型の発現レベルに影響する、請求項1に記載の調節因子。
【請求項24】
調節因子がPDPおよびそのアイソフォームおよび変異型の発現レベルに影響する、請求項1に記載の調節因子。
【請求項25】
発現レベルが転写、翻訳、または翻訳後レベルにおいて変化する、請求項23または24に記載の調節因子。
【請求項26】
変化がエピジェネティックである、請求項25に記載の調節因子。
【請求項27】
調節因子が有毒代謝産物の生成を阻害する、請求項9に記載の調節因子。
【請求項28】
調節因子が有毒代謝産物の解毒を促進する、請求項9に記載の調節因子。
【請求項29】
代謝産物が、アセトアルデヒド、スーパーオキシド、過酸化水素、およびヒドロキシルラジカルから成る群より選択される、請求項27または28に記載の調節因子。
【請求項30】
調節効果がアセトイン産生の減少により観察される、請求項28に記載の調節因子。
【請求項31】
可逆的リン酸化または脱リン酸化が非可逆的となる、請求項9に記載の調節因子。
【請求項32】
リン酸化または脱リン酸化の効果が細胞死をもたらす、請求項31に記載の調節因子。
【請求項33】
効果がアポトーシスである、請求項32に記載の調節因子。
【請求項34】
効果がネクローシスである、請求項32に記載の調節因子。
【請求項35】
調節因子が、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、もしくは症候群、またはそれらの症状の治療および予防に有用である、請求項1に記載の調節因子。
【請求項36】
少なくとも1の酵素複合体がPDH複合体である、請求項35に記載の調節因子。
【請求項37】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項35に記載の調節因子。
【請求項38】
疾病、状態、または症候群が癌である、請求項37に記載の調節因子。
【請求項39】
有効量の請求項1に記載の調節因子の投与を含む、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、もしくは症候群、またはそれらの症状を呈する患者において、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットを調節する方法。
【請求項40】
少なくとも1の酵素複合体がPDH複合体である、請求項39に記載の方法。
【請求項41】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項39に記載の方法。
【請求項42】
疾病、状態、または症候群が癌である、請求項41に記載の方法。
【請求項43】
患者から細胞サンプルを得ること、有効量の請求項1に記載の調節因子をin vitroで細胞へ投与すること、およびそこから結果を得ることを含む、少なくとも1の酵素および/または酵素複合体、またはそれらのサブユニットのリン酸化状態の変化を含む、疾病、状態、または症候群の症状を呈する患者の診断および利益を予想するための方法。
【請求項44】
少なくとも1の酵素複合体がPDH複合体である、請求項43に記載の方法。
【請求項45】
疾病、状態、または症候群が、細胞の過剰増殖によってさらに特徴付けられる、請求項43に記載の方法。
【請求項46】
疾病、状態、または症候群が癌である、請求項45に記載の方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2011−516473(P2011−516473A)
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願番号】特願2011−502908(P2011−502908)
【出願日】平成20年4月4日(2008.4.4)
【国際出願番号】PCT/US2008/004410
【国際公開番号】WO2009/123597
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(510237262)
【氏名又は名称原語表記】SHORR,Robert
【出願人】(510237273)
【氏名又は名称原語表記】RODRIGUEZ,Robert
【Fターム(参考)】
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願日】平成20年4月4日(2008.4.4)
【国際出願番号】PCT/US2008/004410
【国際公開番号】WO2009/123597
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(510237262)
【氏名又は名称原語表記】SHORR,Robert
【出願人】(510237273)
【氏名又は名称原語表記】RODRIGUEZ,Robert
【Fターム(参考)】
[ Back to top ]