
変異体マルトース結合タンパク質に融合された標的タンパク質の可溶化及び精製
(i)マルトデキストリン基質に対する標的タンパク質の結合親和性及び/又は(ii)標的タンパク質の溶解度の少なくとも1つを増加させるための方法及び組成物が提供される。本方法及び組成物は、修飾されたマルトース結合タンパク質に関する。

【発明の詳細な説明】
【背景技術】
【0001】
純粋なタンパク質の大量が必要とされる場合には常に、組み換えタンパク質は、バイオテクノロジーにおいて多くの用途を有している。エシェリヒア・コリ(E.coli)及び酵母などの微生物発現系は、しばしば、低いコストと高い収率のために、第一の選択肢となる。イー・コリ中で外来タンパク質を発現させる場合には、タンパク質の発現の低いレベル及び/又は不溶性という問題に遭遇することが珍しくない。タンパク質が十分に発現され、可溶性を保ったとしても、細胞抽出液中の他のタンパク質の多数から精製しなければならない。標的タンパク質の発現及び精製を容易にするために、一般的に使用されている1つの方法は、タンパク質に親和性タグを融合することである。優れた親和性タグは、標的タンパク質のN末端に融合された場合に、高いレベルの発現を促進するとともに、発現環境から標的タンパク質を精製可能とする単純な一工程のアフィニティー精製を提供する特性を有する。
【0002】
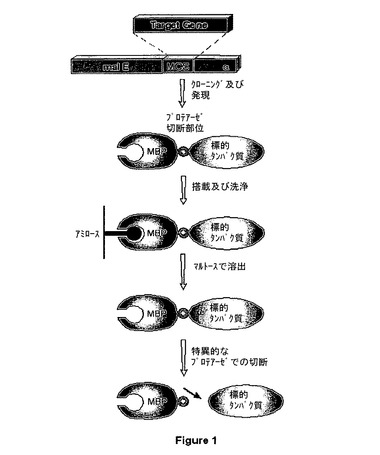
イー・コリのマルトース結合タンパク質(MBP)は、イー・コリ中で産生された外来タンパク質の発現及び精製用のアフィニティータグとして一般に使用される。MBPの本来の役割は、外膜ポリンのマルトデキストリンを結合し、内膜中のMalEFK輸送装置にマルトデキストリンを放出することである。標的タンパク質のN末端にMBPのC末端を融合することによって、イー・コリ中での融合タンパク質の発現が可能となる(図1)。MBP及びMBP融合物は、アミロース、デンプン又は他のマルトデキストリンなどの多数のグルコース−α−>4−グルコース多糖の何れをも含有するクロマトグラフィーマトリックスへの結合によって、単一の工程で精製することができる(米国特許第5,643,758号)。本来の宿主中において可溶性である多くのタンパク質は、組み換えタンパク質として発現された場合には不溶性である。これらのタンパク質の多くは、MBPへの融合によって、可溶性となる(Kapust & Waugh,Protein Sci.8:1668−74(1999))。
【0003】
親和性タグとしてのMBPの有用性は、MBP−標的タンパク質精製におけるタンパク質に応じて、他の融合程うまくアフィニティーマトリックスに結合しない融合もあるという事実によって制限される。さらに、TritonX100及びTween20などの非イオン性界面活性剤の存在は、特に、本質的により低い親和性を有するMBP−標的タンパク質融合物に対する結合を妨害し得る。
【0004】
研究者は、MBPの結合特性を変化させるために、MBPの構造を使用し指定された変異を作製してきた。MBPのX線結晶構造は、「Spurlino et al.,J.Biol.Chem.266:5202−5219(1991)」によって報告されてきた。MBPは2つのドメインからなり、ドメイン間に間隙を有し、ここに多糖が結合する。N末端を含有するドメインはNドメインと名付けられ、C末端を含有するドメインはCドメインと名付けられている。3つのループは、2つのドメイン間を横断して、ヒンジを形成する。2つのグループは、マルトース及びマルトトリオースに対するMBPの親和性を増加させるヒンジ後方の領域に対して指定された変異を施すために、本構造を使用してきた(Marvin et al., Nature Structural Biology 8:795−798(2001);Telmer & Shilton, Journal of Biol.Chem.278:34555−34567 (2003))。しかしながら、MBPへの修飾はタンパク質の表面の疎水性を増加させ、従って、その溶解度を減少させるので、このアプローチは固有の欠点を有している。このため、融合タンパク質を不溶性にするその傾向を増加させることによって、親和性タグとしてのその有用性が低下する。
【発明の開示】
【0005】
本発明の一実施形態において、修飾されたマルトース結合タンパク質(MBP)であり、修飾されたMBP融合タンパク質が(i)マルトデキストリン基質に対する増加した親和性及び(ii)限定された溶解度を有する標的タンパク質に融合された際の増加した溶解度から選択される少なくとも1つの特性を有するようにする変異を有するMBPアミノ酸配列によって特徴付けられる修飾されたMBPが提供される。本発明のさらなる実施形態において、修飾されたMBPは、へリックスXIとXIIの間のヒンジ領域中に、又はタンパク質のA313の10オングストローム内の領域中に位置する変異を有する。例えば、変異は、Cドメイン中に、又はMBPのβシートFの先頭のS146の10オングストローム以内の領域中に位置し得る。修飾は、具体的には、A313V又はS146Tであり得る。
【0006】
本発明のさらなる実施形態において、修飾されたMBPは、融合タンパク質を形成するために標的タンパク質に融合され得、例えば、標的タンパク質に融合された修飾されていないMBPタンパク質の溶解度を上回る溶解度を有し得る。
【0007】
本発明の一実施形態において、修飾されたMBPは、配列番号4若しくは配列番号6から選択されるアミノ酸配列又は配列番号3若しくは配列番号5から選択されるタンパク質をコードするDNA配列を有し得る。DNAは、クルイベロミセス又はイー・コリなどの宿主細胞中で発現用pKLAC1ベクターなどのベクター中に取り込まれ得る。
【0008】
本発明の一実施形態において、クルイベロミセス又はイー・コリなどの宿主細胞中で、上記修飾されたMBP及び標的タンパク質の何れかを含む融合タンパク質を発現させることを含む、タンパク質を精製するための方法が提供される。修飾されたMBP融合タンパク質は、マルトデキストリンを例とする多糖などのマトリックスに結合することが可能である。次いで、精製されたタンパク質を取得するために、融合タンパク質は、選択された緩衝液中のマトリックスから溶出することが可能である。
【0009】
本発明のさらなる実施形態において、融合タンパク質が、変異体MBPの不存在下で観察できるより大きな程度まで、及び変異されていないMBPを用いて観察されるより大きな程度まで可溶化されるように、標的タンパク質に融合されたMBP変異体を発現させることを含む、標的タンパク質を可溶化する方法が提供される。変異の例には、A313V変異及び/又はS146T変異が含まれる。
【発明を実施するための最良の形態】
【0010】
本明細書において使用される用語が、以下に論述されている。
【0011】
「野生型」MBPは、ポリリンカー中に停止コドンを有するpMAL−2プラスミドの1つ、例えば、pKO1483の誘導体からの発現によって産生されたMBPタンパク質が含まれる。
【0012】
「変異体MBPに融合されたタンパク質の増強された溶解度」とは、野生型MBPに融合された同じタンパク質と比べた場合における可溶性タンパク質の量の増加である。溶解度は、例えば、不溶性材料が、例えば、遠心によって不溶性物質が除去される前に存在する当該タンパク質の全量に対する可溶性タンパク質の比として表すことができる。
【0013】
「変異体MBPの増加された親和性」又は「変異体MBP融合タンパク質」には、一連の所定の条件下における、マルトデキストリンなどの固体基質に結合するタンパク質の量の増加が含まれる。アフィニティー精製の効率は、カラムに適用されたタンパク質の総量に対する、指定の条件下でのマルトデキストリンに結合し、次いで、指定の緩衝液で溶出されるタンパク質の比として表すことができる。
【0014】
本発明の本実施形態は、標的タンパク質に融合された場合に、インビボでの発現の間に、融合タンパク質の溶解度を増強し、精製の間の融合タンパク質の親和性を改善することもできるMBP変異体を提供する。本実施形態には、野生型MBPが部分的な結合を示す条件下で培地に適用された場合に、多糖溶媒(マルトデキストリンを含むものなど)への増加した結合を示す変異体MBPが含まれる。次いで、例えば、可溶性マルトデキストリンの溶液を用いて、修飾されたMBPを培地から溶出し、野生型MBPと比較した場合に、少なくとも1.5から10倍多いタンパク質を与える。
【0015】
MBPのこれらの改良された変異体を発見するために、多数の試料を取り扱えるようにする技術を開発することなどの技術的な障壁を乗り越えなければならない。大規模な精製に対して音波処理が実用的でなく、溶解緩衝液がMBPのアフィニティー結合を妨害し得る場合に、これには、可溶化された融合タンパク質を放出するために宿主細胞を切断するための改善された方法が必要とされる。界面活性剤及びリゾチームを滴定することによって、結合親和性に対して悪影響を与えずに、宿主細胞を効果的に切断するためのこれらの溶解試薬の適切な濃度及び比を同定することが可能であることが発見された。
【0016】
所望の結合親和性特性を有する変異体に対してスクリーニングするために、各ウェルが融合タンパク質を結合するためのミクロマトリックスを含有し、フィルター装置がろ液中のきょう雑物を除去する96ウェルミクロプレートが使用された。これにより、多数の試料の迅速なスクリーニングが可能となった。
【0017】
タンパク質を精製するための改善されたタグとして、修飾された変異体MBPタンパク質を取得及び検査するためのスクリーニング法は、実施例に記載されている。マトリックスに対してより高い親和性を有するこのような修飾されたMBPは、マトリックスへの結合が乏しい、又は非イオン性界面活性剤の存在によって、結合が破壊されるMBP融合タンパク質の野生型MBPに付随する問題を解決する。
【0018】
実施例において、スクリーニング操作を用いて単離された、改善された溶解度及び多糖マトリックスへの改善された結合親和性という所望の特性を有する2つの変異(S146T及びA313V)が記載されている。S146T変異は、βシートFの先頭のMBPのCドメイン中に存在する。不溶性である標的タンパク質がこの変異を含有するMBPに融合されると、融合タンパク質の溶解度が増強される。本明細書中に記載されているA313V変異は、2つのドメイン間を横切る第三のヒンジ領域中、特に、ヘリックスXIとXIIの間のループ中に位置する。この変異は、融合タンパク質の溶解度と親和性の両者を増強する。イー・コリ中の外来タンパク質を発現させると、タンパク質は、不溶性凝集物の形態で部分的に又は完全に発現され得る。特定の実施例では、溶解度は、総溶解度の上限を伴って、1.1又はそれ以上、上方向に増加され得る。
【0019】
本明細書に引用される全ての参考文献及び2006年4月14日に出願された米国仮出願第60/792,133号は、参照により、組み込まれる。
【0020】
実施例
材料
制限酵素、β−アガラーゼ、DNAポリメラーゼ、T4リガーゼ、南極ホスファターゼ、Litmus38、pMAL−c2X及びpMAL−c2Gを含むpMALタンパク質融合及び精製系、アミロース樹脂(#E8021)、西洋ワサビペルオキシダーゼに連結された抗MBPモノクローナル抗体(#E8038)、USERFriendly Cloning キット、ベクターpKLAC1、宿主株TB1、ER1992、ER2502、ER2984、NEB5−α及びNEBTurboを含むK.ラクティスタンパク質発現キット並びに合成オリゴヌクレオチドは、New England Biolabs、 Inc.(NEB)、Ipswich、MAから取得した。フィルター底付きのUnifilter800マイクロタイターマイクロプレートは、Whatman、Brentford、Englandから購入した。Minelute DNA Extraction and Qiaprep Spinキットは、Qiagen、Valencia、CAから購入した。Mega10は、Dojindo、Gaithersburg、MDから購入した。ニワトリ卵白リゾチーム、クマシーブリリアントブルーR及び酸洗浄されたガラスビーズ(425から600ミクロン)は、Sigma−Aldrich、St.Louis、MOから購入した。Sea Plaque GTG低融点アガロースは、Cambrex、 E. Rutherford、 NJから購入した。使い捨てポリプロピレンカラム(#732−6008)は、BioRad、Hercules、CAから購入した。10から20%のグラジエントゲルは、Daiichi、Tokyo、Japan又はInVitrogen/Novex、Carlsbad、CAの何れかから購入した。CompleteTMプロテアーゼ阻害剤カクテルは、Roche、Basel、Switzerlandから購入した。SimplyBlue Safestainは、Invitrogen、Carlsbad、CAから購入した。ヒトジヒドロ葉酸還元酵素(DHFR)cDNAクローンpOTB7−DHFRは、Invitrogen(MGC:857)から購入した。GAPDH遺伝子は、pJF931から入手した(Fox et al.FEBS Lett.537:53−57(2003))。
【0021】
技術
セラチア・マルセッセンスヌクレアーゼは、PCT/US05/28739に記載されているとおりに取得した。プラスミドDNAのMiniprepは、Qiaprep Spinキットを用いて調製した。ランダムPCR突然変異導入は、「Fromant et al.Analytical Biochemistry 224, 347−353 (1995)」に記載されているとおりに実施した。PCRは、注記されていることを除き、Vent(R)DNAポリメラーゼを用いて行った。1%SeaPlaqueGTG低融点アガロース上で電気泳動を行い、バンドを切り出し、MineluteDNAExtractionキットを用いてDNAを精製し、又は75℃で5分間、DNAを融解させ、37℃まで冷却し、βアガロースで1から2時間消化することによって、DNA断片をゲル精製した。DNA配列決定は、BigDye標識された色素ターミネーター化学(ABI, Foster City, CA)を使用し、Applied Biosystemsの(ABIの)自動化されたDNASequencerモデル3100ABI上で行った。アクリルアミドゲルの供給者の指示書に従って、SDS−PAGEを実施し、別段の記載がある場合を除いて、クマシーブリリアントブルーRでの染色によって、タンパク質を可視化した。
【0022】
pMal−c2X又はpMal−c2G又はpMal−c2Gの誘導体の何れかからMBPを発現させた。malE中の変異を特定するための塩基の付番は、pMAL−c2X配列中の塩基番号を表す(図2A−1、2A−2、2B−1、2B−2、2C−1及び2C−2(配列番号1から6))。pMAL−c2G誘導体pSN1578は、BsmI及びBsiWIでプラスミドを切断し、4つのdNTPを全て加えて、DNAポリメラーゼKlenow断片で産物を処理した後、malE遺伝子内に欠失を作出するために連結することによって作製した。
【0023】
「Guan et al.Nucleic Acid Research, 33:6225−6234(2005)」に記載されているように、位置指定突然変異導入は、4プライマーのPCR突然変異導入を用いて実施した。MBP及びMBP融合タンパク質は、幾つかの事例において、音波処理の代わりにリゾチーム/界面活性剤溶液で細胞を溶解したことを除き、pMALProtein Fusion and Purification Systemの指示書に記載されているとおりに精製した。
【0024】
培養の500ないし1000mLから調製された未精製細胞抽出を用いて、大規模な精製を実施し、アミロース樹脂15mLを含有する2.5cm直径のカラム上に搭載した(NEB #E8021, Ipswich, MA)。培養の67mLから調製された未精製抽出物を用いて、小規模な精製を実施し、アミロース樹脂1mLを含有する使い捨てポリプロピレンカラム上に搭載した。10から20%グラジエントゲルを用いて、SDS−ポリアクリルアミドゲル電気泳動を実施した。ゲルバンドの定量のために、セロファンシートの間でゲルを乾燥させ、Microtek III scanner Microtek, Carson, CAを用いて走査し、ImageJ(NIH)を使用して濃度測定行った。
【実施例1】
【0025】
改善された特性を有するMBP中の変異体の単離
精製後の改善された収率に対するスクリーニング:
pMAL−c2xから得られるmalE遺伝子のランダムな突然変異導入は、プライマー:オリゴ1:5’GGAGACAUGAATTCAATGAAAATCGAAGAA(配列番号9)及びオリゴ2:5’GGGAAAGUAAGCTTAATCCTTCCCTCGATC(配列番号10)を使用する変異性のPCRによって達成した。
【0026】
製造業者の指示書に従い、USERFriendlyCloningKitを用いて、直鎖化されたpNEB208A中にPCR断片をクローニングした。1mLLB+1mMIPTG及び100μg/mLアンピシリン中で形質転換体を一晩増殖させた後、0.3mg/mLリゾチーム及びS.マルッセンスヌクレアーゼ20単位を添加によって溶解させ、10分間温置し、次いで、2%Tween20の0.1mLを添加することによって溶解した。
【0027】
Unifilter800マイクロプレート中のアミロース樹脂カラム(NEB#E8021,Ipswich,MA)50μLに、未精製抽出物を適用し、20mMTris−Cl、0.2MNaCl、1mMEDTA、pH7.4の0.7mL(カラム緩衝液)で、次いで、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.7mLで各ウェルを洗浄した。次いで、10mMマルトース、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.2mLで、アミロース樹脂に結合されたタンパク質を溶出した。Immulon2HBミクロタイタープレート(ThermoFisherScientific,Waltham,MA)に溶出液を移し、4℃で一晩温置した。次いで、マイクロタイターウェルを空にし、20mMTris−Cl、150mMNaCl、pH7.5(TBST)で二回洗浄し、次いで、37℃で1時間、0.36mLTBST+3%ウシ血清アルブミンでブロッキングを行った。
【0028】
TBSTで、ウェルを二回洗浄し、次いで、TBST+3%ウシ血清アルブミン中の、西洋ワサビペルオキシダーゼに連結された抗MBPモノクローナル抗体の1:2000希釈物の0.1mLを各ウェルに添加し、37℃で1時間、プレートを温置した。ウェルを空にし、次いで、TBSTで二回洗浄した。0.01%o−フェニレンジアミン、0.003%過酸化水素水で、ウェルを展開した。4MH2SO40.025mLを添加することによって検出反応を停止させ、490nmで、ウェルを分光学的にアッセイした。野生型MBPと比べて、より高い結合及び溶出を示した試料に対応する可溶化液から細胞を回収した。より高い結合及び溶出を確認するために、これらの候補を増殖し、再度検査した。
【0029】
ランダム突然変異誘発後に得られた変異の性質決定及び分離
増加した結合及び溶出特性を有するUSER中のライブラリーから得た2つの単離株を配列決定した(図2)。1つの単離株G8−1は、サイレント変異とともに、単一のミスセンス変異G1964Cを有することが見出された。G1964C変異は、MBP中のアミノ酸変化S146Tに対応する。他の単離株A9は、サイレント変異とともに、3つのミスセンス変異A1583G、A2419G及びC2465Tを有することが見出された。A1583G、A2419G及びC2465T変異は、それぞれ、アミノ酸変化N19S、K298E及びA313Vに対応する。
【0030】
pMal−C2X又はpSN1578中へのサブクローニング
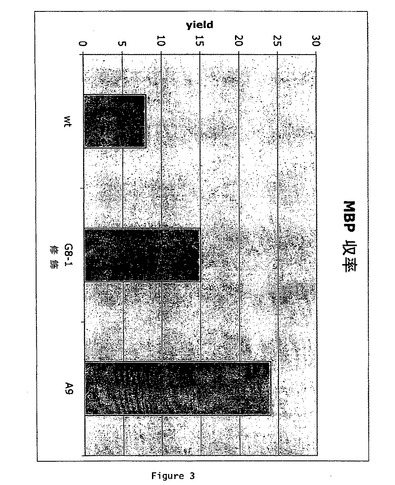
以下のプライマー:オリゴ3:5’GACTCATATGAAAATCGAAGAAGGTAAACTGGTAATCTGGATTAACGGC(配列番号11)及びオリゴ4:5’ATATAAGCTTTCACCTTCCCTCGATCCCGAGGT(配列番号12)を用いて、各単離株をPCRによって増幅した。増幅されたDNAをエタノール沈殿し、NEBuffer4(NEB,Ipswich,MA)中のNdeI及びHindIIIで切断し、ゲル精製した。NdeI及びHindIIIでpSN1578を切断し、ベクター骨格をゲル精製した。G8−1及びA9断片をpSN1578断片と混合し、連結し、TB1を形質転換するために、この連結を使用した。各形質転換体から得たプラスミド調製物の配列を決定し、G8−1に関してはpIH1596と命名し、A9に関してはpIH1593と命名した。本実験における3’プライマーは、malE翻訳がpMALのlacZα断片中に進行するのを防ぐために、正しいリーディングフレーム中に停止コドンを有する。従って、これらのサブクローンは、ポリリンカーによってコードされるアミノ酸配列...IEGRの後で終了する修飾されたMBPを産生する。野生型malE遺伝子後に停止コドンを含有する対照プラスミドは、malEとlacZaの間のポリリンカー中のpMAL−c2XをXbaIで切断することによって構築された。DNAポリメラーゼI、巨大断片(Klenow)及び4つの全てのdNTPを用いて、XbaI突出部を充填し、次いで、T4リガーゼでの処理によって、プラスミドを再度環状化した。これは、malEと同じ読み枠中に停止コドンを導入し、この誘導体は、ポリリンカーによってコードされる8残基の伸長を除き、G8−1及びA9によって産生されるものと同等のMBPを産生する。この対照プラスミドは、pKO1483と名付けられた。LB+0.1%グルコース及び100μg/mLアンピシリンの500mL培養物中で、2×108細胞/mLになるまで、pKO1483、pIH1596及びpIH1593を含有するイー・コリTB1を増殖させ、0.3mMIPTGで誘導し、37℃で2時間増殖させ、次いで、採集した。カラム緩衝液25mL(10mMマルトース、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.2mL)+10mMβ−メルカプトエタノール中に細胞を再懸濁し、次いで、音波処理によって溶解された。9000×gで30分間遠心することによって、抽出物を清澄化し、次いで、カラム緩衝液を用いて1:4希釈し、アミロース樹脂の15mLカラム上に搭載した。約125mLカラム緩衝液でカラムを洗浄し、カラム緩衝液+10mMマルトースで溶出した。3つの株の間で、MBPの収率を比較した(図3)。結果は、修飾されたMBPがアミロースへの増加した結合及び適切な緩衝液中への溶出を示したことを確認する。
【0031】
3つの変異のうち何れがA9バリアントの増加した結合に必要であるかを確認するために、pSN1578(malE遺伝子の内部に欠失(これは、挿入物を受容したクローンの簡単な同定を可能とする。)を有するpMAL−c2G誘導体)中に、3つの変異を別個にサブクローニングした。A1583G及びA2419変異は何れも、アフィニティー精製におけるMBPの収率にまったく影響がなく、又は収率を低下させ、廃棄された。N末端PCR断片を作製するためにプライマーオリゴ5:5’CTTCAAGGGTCAACCATCCAAACC(配列番号13)及びオリゴ6:5’AATACGCGGATCTTTCACCAACTCTTC(配列番号14)を用い、並びにC末端PCR断片を作製するためにプライマーオリゴ7:5’GAAGAGTTGGTGAAAGATCCGCGTATT(配列番号15)及びオリゴ8:5’CTGAGAATTCTGAAATCCTTCCCTCGAT(配列番号16)を用いる、第一のテンプレートとしてpMAL−c2Xを使用する4プライマーの位置指定PCR突然変異誘発によって、C2465T変異を分離して再現した。テンプレートとしてのゲル精製されたN末端及びC末端断片並びにプライマーオリゴ5及びオリゴ8を用いて、集合工程を実施した。最終PCR断片をBlpI及びAvaIを用いて切断し、ゲル精製し、BlpI及びAvaIを用いて切断されたpMAL−c2Xに連結し、ゲル精製した。TB1を形質転換するために連結を使用し、形質転換体からプラスミドを精製し、C2465T変異を確認するために配列を決定した。さらなる研究のために単離株を選択し、pIH1606と名付けた。
【0032】
pIH1606の構築において、MBPの末端の停止コドンは保存されていなかった。この構築物は、LacZaa断片に融合されたMBPを発現する。C2465T変異の効果をその親A9に対して比較するために、pIH1606中のmalE遺伝子の後に、停止コドンを導入した。XbaIで、本プラスミドを切断し、Klenow+dNTPで充填し、pKO1483に関して上述されているように、再連結した。停止コドンを有するC2465T誘導体は、pPR1610と称された。pKO1483及びA9と平行して行われた、このプラスミドを有するTB1の大規模MBP精製は、A9において見出されたMBPの収率の増加が全て、C2465T変異によって説明され得ることを示した。この変異は、MBPのアラニン313をバリン(A313V)に変化させる。
【0033】
MBP(S146T)を野生型MBP及び正確に同性質をもつ構築物を有する誘導体中のMBP(A313V)と比較できるようにするために、pKO1483と同じ停止コドンを有するMBP(S146T)のバージョンを構築した。pIH1596からのNdeI、BlpI断片を精製し、NdeI及びBlpIを用いて切断されたpKO1493中にサブクローニングし、pIH1619を作製した。
【実施例2】
【0034】
MBP融合タンパク質の増加した収率
A:MBP−Klenow
修飾されたMBPが、精製後に、融合タンパク質の収率を増加させることができるかどうかを検査するために、イー・コリDNAポリメラーゼIのKlenow断片をコードする遺伝子を、pMAL−c2X、pIH1619(S146T)及びpPR1610(A313V)中にクローニングした。アミロースカラムに対して本質的に低い親和性を有し、親和性精製の間に、MBP−Klenowタンパク質の幾つかは、結合せずに、アミロースカラムを流出するので、MBP−Klenow融合物を選択した。BglII及びHindIII部位間において、pBR322中にクローニングされたpolA遺伝子(GenbankecopolA:206−4127)のBglII−HindIII断片を含有するプラスミドpPolAから、DNAポリメラーゼIのKlenow部分にPCR反応を行った。プライマー:オリゴ9:5’CCAGAAGTGACGGCAACGGTGATT(配列番号17)及びオリゴ10:5’AAGTGCGGCGACGATAGTCATGCCCCGCGC(配列番号18)を用いて、PCRを実施した。
【0035】
HindIIIを用いてPCR断片を切断し、pMAL−c2に連結し、XmnI及びHindIIIでこれを切断した。得られた構築物は、pIH1040と名付けた。挿入物中でのPCRの誤りの可能性を減らすために、XhoIからHindIIIまでの領域を、pPolA由来の対応する断片で置換した。この構築物を配列決定によってチェックし、pIH1062と名付けた。pPolAをテンプレートとして使用し、プライマー:オリゴ11:5’GTGATTTCTTATGACAACTACGTCACCATCCTTGATG(配列番号19)及びオリゴ12:5’TTAAGGATCCTTAGTGCGCCTGATCCCAGT(配列番号20)を使用するPCRによって、Klenow断片をコードする遺伝子をpPR1610及びpIH1619中にクローニングした。
【0036】
BamHIを用いて、PCR断片を切断し、XmnI及びBamHIを用いて、ベクターを切断し、精製し、PCR断片と混合し、連結した。NEBTurbo(Ipswich,MA)を形質転換するために、この連結物を使用し、制限分析によって、並びにMBP−Klenow融合物の発現及びSDS−PAGEによる分析によって、正しい構造に関して形質転換体をチェックした。pIH1610−Klenow構築物をpIH1643と名付け、pIH1619−Klenow構築物をpIH1644と名付けた。
【0037】
pIH1062、pIH1643及びpIH1644を含有する細胞を用いて、MBP−Klenowのアフィニティー精製を行った。各株から得られた未精製抽出物、カラム流出物及び溶出液画分を、SDS−PAGEによって分析した(図4)。A280を測定することによって、溶出されたタンパク質を定量し、MBP−Klenowタンパク質の予想吸光係数を用いて定量した(表1)。pIH1643を有し、MBP−A313V修飾を担持する細胞は、野生型MBP又はS146T修飾されたMBP融合プラスミドを含有する細胞より2倍多くの融合タンパク質を生成した。
【0038】
B.MBP−キチン結合ドメイン:
変異体MBPが親和性精製からの融合タンパク質の収率を増加させる能力が一般的な特性であるかどうかを検査するために、アミロースに対して本質的に低い親和性を有する別の融合タンパク質を検査した。バチラス・サーキュランス(Bacillus circulans)のキチン結合ドメインに融合されたMBP(MBP−CBD)は、プラスミドpMB50によってコードされる(図5)。MBP−Klenowと同様に、このMBP−CBD融合タンパク質のかなりの割合が、アフィニティー精製の間に、アミロースカラムから流出する傾向がある。MBP(A313V)をコードするpPR1610の一部を、HapI及びSacIを用いて、プラスミドから切断し、断片を精製した。同じ酵素でpMB50を切断し、骨格も精製し、2つの断片を連結し、ER2523中に形質転換した。得られたプラスミドは、pIH1660と名付けられた。
【0039】
500mL培養物から採集されたpMB50及びpIH1660を含有する細胞を用いて、MBP−CBDのアフィニティー精製を行った。A280を測定し、MBP−CBDタンパク質の予想吸光係数を使用することによって、溶出されたタンパク質を定量した(表2)。pIH1660を有し、MBP−A313V修飾を担持する細胞は、野生型MBP融合プラスミドを含有する細胞よりほぼ2倍多くの融合タンパク質を生成した。
【実施例3】
【0040】
MBP融合タンパク質の溶解度の増強
MBP融合物の構築
修飾されたMBPがMBPに融合されたタンパク質の溶解度を増強する能力を保持するかどうかを調べるために、イー・コリ中で不溶性である傾向がある2つのタンパク質であるジヒドロ葉酸還元酵素(DHFR)及びグリセルアルデヒド−3−リン酸脱水素酵素(GAPDH)を、pIH1619(S146T)及びpIH1610(A313V)中にクローニングした。対照として、「MBP−DM」と名付けられた、Telmerらの修飾されたMBP中に変異を含有するpMAL−c2X及びpMAL−c2G誘導体中に、同じ2つのタンパク質をクローニングした。MBP−DMをコードするベクターを、以下のように構築した。まず、4プライマー位置指定PCRによって、ヌクレオチド2030に翻訳的にサイレントなNsiI部位を有するpMAL−c2G誘導体を構築した。N末端及びC末端断片の両方に対するテンプレートは、pMAL−c2Xであった。N末端断片を作製するために、プライマーオリゴ13:5’CCATAGCATATGAAAATCGAAGAAG(配列番号21)及びオリゴ14:5’CTTGAATGCATAACCCCCGTCAGCAGC(配列番号22)を使用し、C末端断片を作製するために、プライマーオリゴ15:5’GGTTATGCATTCAAGTATGAAAACGGCAAG(配列番号23)及びオリゴ8を使用した。PCR断片をゲル精製し、次いで、プライマーオリゴ13及びオリゴ8を用いた集合工程中でのテンプレートとして使用した。最終PCR断片をエタノール沈殿し、NdeI及びEcoRIで切断し、ゲル精製した。NdeI及びEcoRIでpMAL−c2Xを切断し、ゲル精製し、2つの断片を混合し、連結した。ER2502を形質転換するために、連結物を使用し、培養物を一晩増殖させ、ミニプレプDNAを調製した。NsiI部位を獲得した、正しいサイズの幾つかの形質転換体を取得したが、予想に反して、それらの全てはEcoRI部位を欠如していた。この組の代表(pPR1629と名付けられた。)をNdeI及びAvaIで切断し、malE断片をゲル精製した。同じ2つの酵素でpSN1578を切断し、プラスミド骨格をゲル精製した。これら2つのDNA断片を連結し、ER2502を形質転換するためにこの連結物を使用し、1つの形質転換体から得られるプラスミドDNAを配列決定し、pPR1633と名付けた。次に、Telmer及びShiltonによってDMと称される修飾されたMBPに対する遺伝子を、以下のように、4プライマーのPCR部位指定突然変異生成によって構築した。両N及びC末端断片に対するテンプレートは、pMAL−c2Xであった。N末端断片に対して使用されたプライマーは、オリゴ16:5’TATGCATTCAAATACGGTGACATTAAAGACGTGGGCGTGGAT(配列番号24)及びオリゴ17:5’GCGGCGTTTTCCGCAGTGGCGGCAATACGTGGATCTTTC(配列番号25)であった。C末端断片は、プライマーオリゴ18:5’GCGGAAAACGCCGCGAAAGGTGAAATCATGCCGAACATC(配列番号26)及びオリゴ8を用いて作製された。オリゴ16及びオリゴ8をプライマーとして使用し、精製されたN末端及びC末端断片を用いて集合PCRを実行した。PCR断片をエタノール沈殿し、EcoRVで直鎖化されたLitmus38に連結し、南極ホスファターゼで処理した。NEB5−α(Ipswich,MA)を形質転換するために連結物を使用し、構築物を確認するために1つの形質転換体から得られたプラスミドDNAを配列決定し、pPR1638と名付けた。プラスミドDNApPR1633及びpPR1638をNsiI及びAvaIで切断し、pPR1633由来のプラスミド骨格及びpPR1638由来のmalE断片をゲル精製し、連結し、NEBTurboを形質転換するために連結物を使用した。配列決定のために、1つの形質転換体由来のプラスミドDNAを選択し、pPR1639と名付けた。
【0041】
pOTB7−DHFRをテンプレートとして使用し、プライマーオリゴ19:5’GGATGGTTGGTTCGCTAAACTGCATCGTC(配列番号27)及びオリゴ20:5’TATTAATCATTCTTCTCATATACTTCAAA(配列番号28)を使用して、DHFR遺伝子にPCRを行い、次いで、室温で15分間、T4ポリメラーゼ及びdTでPCR断片を処理した。この処理によって、断片の各末端上に短い3‘突出(DHFR遺伝子の上流末端上にGG及び下流末端上にA)が生じる。XmnIで切断し、次いで、T4ポリメラーゼ及びdAで処理することによって、ベクターpMAL−c2X及びpPR1610を調製し、同様に、malE領域の末端に3’CC突出部及びlacZa末端上にTを生成する。ベクターDNAをDHFR断片に連結し、ER1992を形質転換するためにこの連結物を使用した。各ベクターに関して、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって形質転換体を確認した。pMAL−c2X−DHFR単離株をpIH1616と名付け、pIH1610−DHFR単離株をpIH1617と名付けた。次いで、以下のように、DHFR挿入物をpIH1596中にサブクローニングした。NdeI及びBlpIでpIH1616を切断し、消化物をゲル上で走行させ、(DHFR遺伝子を含む)骨格を切除し、ゲル精製した。NdeI及びBlpIでpIH1596を切断し、malE’をゲル精製した。malE’断片にベクター骨格を連結し、ER1992を形質転換するためにこの連結物を使用した。制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって、単離株を確認し、pIH1618と名付けた。pPR1639及びpIH1616の両者をAvaI及びSalIで切断し、pPR1639由来のベクター骨格及びpIH1616由来のDFHR断片をゲル精製し、2つの断片を連結することによって、DHFR挿入物をpPR1639中にサブクローニングした。NEBTurboを形質転換するために、この連結物を使用し、配列決定によって形質転換体を確認し、pIH1646と名付けた。
【0042】
pJF931をテンプレートとして使用し、プライマーオリゴ21:5’GGATGGTGAAGGTCGGTGTGAACGG(配列番号29)及びオリゴ22:5’TATTACTCCTTGGAGGCCATGTAGGCCA(配列番号30)を使用して、GAPDH遺伝子にPCRに供し、次いで、T4ポリメラーゼ及びdTでPCR断片を処理した。
【0043】
XmnIで切断した後、上述のようにT4ポリメラーゼ及びdAで処理することによって、ベクターpMAL−c2X、pPR1610及びpIH1619を調製した。ベクターDNAをGAPDH断片に連結し、ER1992を形質転換するためにこの連結物を使用した。各ベクターに関して、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって形質転換体を確認した。pMAL−c2X−GAPDH単離株をpIH1625と名付け、pPR1610−GAPDH単離株をpIH1626と名付け、pIH1619−GAPDH単離株をpIH1627と名付けた。次いで、pPR1639及びpIH1625の両者をAvaI及びSalIで切断し、pPR1639由来のベクター骨格及びpIH1625由来のGAPDH断片をゲル精製し、2つの断片を連結することによって、GAPDH挿入物をpPR1639中にサブクローニングした。NEBTurboを形質転換するために、この連結物を使用し、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって、形質転換体を確認し、pIH1645と名付けた。
【0044】
溶解度特性
8つのpMALプラスミド(pMAL−c2X、pIH1619(S146Tを担持する。)及びpPR1610(A313Vを担持する。)、各プラスミドは、DHFR又はGApDHをコードするDNJAをさらに含有する。)を含有するTB1の20mL培養物を、LBamp中で、2×108/mlまで増殖させ、0.3mMIPTGを用いて誘導し、さらに2時間温置し、次いで、微量遠心管中にて、3000×gでの遠心によって、細胞を採集した。50mMTris−Cl、pH7.9、50mMNaCl、0.75mMEDTA、0.6%Mega10、150μg/mLリゾチーム及び20Kunitz単位/mLセラチア・マルセッセンスヌクレアーゼ2mL中に各ペレットを再懸濁し、室温で10分間温置した。再懸濁されたペレットは、全細胞抽出物と表記した。125μLの試料を取り出し、14,000×gで、2分間遠心した。上清を取り出し、可溶性画分と表記した。同じ緩衝液125μL中にペレットを再懸濁し、不溶性画分と表記した。各株に対して、SDS−PAGE上に各画分の試料(5μL)を走行させた(図5)。ゲルを乾燥させ、走査し、遠心前の細胞可溶化液中に存在するタンパク質に対する可溶性タンパク質の比として、各レーン中のMBP融合タンパク質の量を定量した(表2)。DHFR及びGAPDH融合物の両者に関して、A313V及びS146T修飾されたMBPは、野生型MBPに比べて、融合タンパク質の溶解度を増強した。DMによって修飾されたMBPを用いて作製された融合物は、予想通り、野生型MBPと比べて、低下した溶解度を示した。
【実施例4】
【0045】
K.ラクティス中でのMBP変異体の使用
K.ラクティスMBP(A313V)−融合発現ベクターの構築
それぞれ、フォワード及びリバースプライマー:
【0046】
【化1】
を使用するPCRによって、変異体マルトース結合タンパク質MBP(A313V)をコードする遺伝子を増幅した。フォワードプライマーは、HindIII制限酵素部位(太字)を含有し、直前にmalE遺伝子開始コドン(下線)が先行するKozakコンセンサス配列(斜字体)が続くように構築された。リバースプライマーは、XhoI制限酵素部位(太字)を含有し、プロテアーゼエンテロキナーゼのタンパク分解認識部位(斜字及び下線)をコードするDNAが直後に続くように構築された。フレーム内C末端MBP(A313V)融合発現カセットの構築を可能とするために、リバースプライマー上に停止コドンは取り込まれなかった。Phusionポリメラーゼを用いて、完全長遺伝子を含有するプラスミドpPR1610からmalE遺伝子を増幅した。K.ラクティスMBP(A313V)融合発現ベクター(pKIMF2)を作製するために、K.ラクティス発現ベクターpkLAC1のHindIII及びXhoI制限部位中に、増幅された遺伝子がクローニングされた。このクローニング戦略は、malE遺伝子による、pKLAC1中のK.ラクティスα接合因子プレプロシグナル配列の置換をもたらす。従って、MBP及びMBP(A313V)融合タンパク質は、分泌経路に誘導されず、代わりに、酵母サイトゾル中に保持される。
【0047】
K.ラクティス中のMBP(A313V)融合タンパク質の発現及び精製
以下のプライマーを用いて、パラミオシンの切断型をコードする遺伝子(Steel et al., J. Immunol.145:3917−3923(1990))をPCRによって増幅した。
【0048】
【化2】
フォワードプライマーは、XhoI制限部位(太字)を含有するように構築された。リバースプライマーは、PMデルタSal停止コドン(斜字体)のすぐ上流にNotI制限部位(太字)を含有するように構築された。
【0049】
クローニング戦略は、以下のとおりであった。約750bpパラミオシン遺伝子を増幅し、精製した。XhoI及びNotIで、断片を二重消化し、発現ベクターpKIMF2のXhoI及びNotI制限部位中にクローニングし、PMデルタSalのC末端及びN末端の間にフレーム内融合物を作出する。このようにして構築されたプラスミドは、pKLMF2−PMデルタSalと名付けられた(図7)。
【0050】
SacII制限消化によって、MBP(A313V)−PMデルタ融合ベクター(pKIMF2−PMデルタSal)を直鎖化し、その指示書に従って、K.ラクティスProtein Expression Kitを用いて、化学的に形質転換受容性のK.ラクティス細胞中に精製された産物を形質転換した。アセトアミドプレート上で、形質転換体コロニーを選択し、上記キットに記載されているように、細胞全体のPCRによって、正しく組み込まれたMBP(A313V)−PMデルタSal発現カセットを含有するクローンを同定した。
【0051】
YPGalactose培地(1%酵母抽出物、2%ペプトン、2%ガラクトース)20mL培養物に、複数コピーの組み込まれたMBP(A313V)−PMデルタSal発現カセットを含有するK.ラクティス細胞の株を接種し、振盪プラットフォームを有する温置装置中において、4日間、30℃で増殖させた。増殖後の培養物の合計細胞含量は、約8.2×1010細胞(すなわち、約4.1×108細胞/mL)であると測定された。4℃で10分間、8,000rpmの遠心によって、細胞を採集した。過剰な培地を除去するために、蒸留水中で、細胞を一回洗浄した。
【0052】
可溶化液の調製のために、1Mソルビトール中の10mg/mLリチカーゼの2mL溶液中に細胞を再懸濁し、37℃で1時間温置した。微量遠心管中での2分間の穏やかな遠心(7,000rpm)によって、細胞を採集した。プロテアーゼ阻害剤カクテル(CompleteTMプロテアーゼ阻害剤カクテル、Roche)を含有する氷冷アミロースカラム緩衝液(20mMTris−ClpH7.4、0.2MNaCl、1mMEDTA)3mLの総容量中に、細胞ペレットを再懸濁した。細胞スラリーをガラスチューブに移し、氷の上に配置した。酸洗浄されたガラスビーズ(425から600ミクロン、Sigma)の等量を細胞スラリーに添加し、それぞれ、1分間10回のボルテックスによって、細胞を破壊した。細胞可溶化液を新しいチューブに移し、カラム緩衝液1mLで、ガラスビーズを4回洗浄した。細胞可溶化液及び洗浄液をプールし、遠心によって、細胞破砕物を除去した。清澄化された細胞可溶化液を、カラム緩衝液で、8mLになるように希釈した。
【0053】
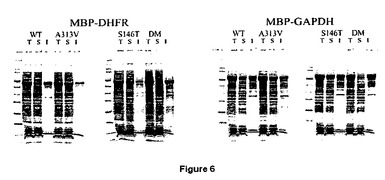
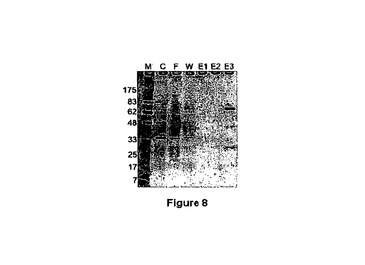
融合タンパク質精製のために、カラム緩衝液15mLで予め平衡化された1.6mLアミロース樹脂カラム上に、細胞可溶化液を通過させた。カラム緩衝液24mLでカラムを洗浄し、10mMマルトースを含有するカラム緩衝液で、0.4mL画分中に、結合されたタンパク質を溶出した。10から20%Tris−グリシン勾配ゲル上でのSDS−PAGEによって、細胞可溶化液及び精製されたタンパク質を分離した。SimplyBlue Safestainによって、タンパク質を同定した。図6は、主要な溶出されたタンパク質が、MBP(A313V)−PMデルタSal融合タンパク質に対して予想されたサイズ(68.5kDa)に対応することを示している。
【0054】
【表1】
【0055】
【表2】
【0056】
【表3】
【図面の簡単な説明】
【0057】
【図1】図1は、標的タンパク質に融合されたMBPをコードするDNAを発現し、融合タンパク質をアミロースに選択的に結合させ、マルトース含有緩衝液中に標的タンパク質を溶出し、次いで、プロテアーゼ切断によって、精製された融合タンパク質から標的タンパク質を回収することによる、標的タンパク質のクローニング及び精製を記載する模式図を示している。
【図2A−1】野生型MBPを修飾されたMBPと比較する配列を提供する。pMAL−c2Xから得られる、野生型MBP(配列番号2)をコードするDNA配列(配列番号1)。
【図2A−2】野生型MBPを修飾されたMBPと比較する配列を提供する。pMAL−c2Xから得られる、野生型MBP(配列番号2)をコードするDNA配列(配列番号1)。
【図2B−1】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体A9(配列番号4)をコードするDNA配列(配列番号3)。修飾されたMBP配列中の変化は、太字で示されている。
【図2B−2】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体A9(配列番号4)をコードするDNA配列(配列番号3)。修飾されたMBP配列中の変化は、太字で示されている。
【図2C−1】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体G8−1(配列番号6)をコードするDNA配列(配列番号5)。修飾されたMBPDNA及びアミノ酸配列中の変化は、太字で示されている。
【図2C−2】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体G8−1(配列番号6)をコードするDNA配列(配列番号5)。修飾されたMBPDNA及びアミノ酸配列中の変化は、太字で示されている。
【図3】野生型MBP(pKO1483)又は修飾されたMBP(A9又はG8−1)を用いたタンパク質の収率を比較するヒストグラムを示している。MBPの収率は、mg/500ml培養で記載されている。
【図4】野生型MBP、MBPA313V及びS146T修飾を含有するpMALからのMBP−Klenowの精製された産物をSDS−PAGEゲル上に示している。プラスミド:WT=pIH1062、A313V=pIH1643、S136T=pIH1644。レーン:CE=未精製抽出物、FT=流出物及びE=溶出液。表記画分の等しい一部を、各レーンに搭載した。MBP融合タンパク質の有意により高い収率が、野生型MBPからより、A313V変異体MBPから得られた(表1も参照)。
【図5−1】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−2】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−3】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−4】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−5】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図6】溶解度に対する、DHFR及びGAPDHへの野生型及び修飾されたMBPの融合の効果を示すSDS−PAGEゲル。DHFR融合プラスミド:WT=pIH1616;A313V=pIH1617;S146T=pIH1618;DM=pIH1646。GAPDH融合プラスミド:WT=pIH1625;A313V=pIH1626;S146T=pIH1627;DM=pIH1645。レーン:T=総細胞抽出物;S=可溶性抽出物;I=不溶性材料の再懸濁。表記画分の等しい一部を、各レーンに搭載した。不溶性タンパク質に対する可溶性タンパク質の比は、野生型MBPと比較して、両変異体に関してより大きかった。
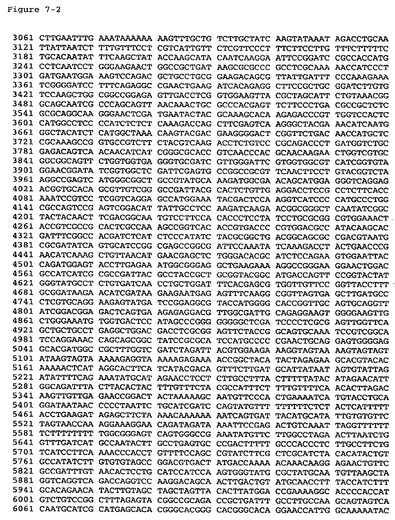
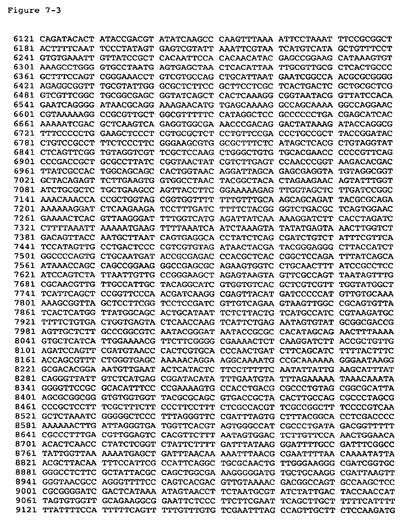
【図7−1】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−2】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−3】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−4】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図8】アミロース樹脂カラムを用いて、SDS−PAGEゲル上の融合タンパク質のアフィニティー精製から得られた画分を示している。MBP(A313V)−PMデルタSal発現カセットを担持するクルイベロミセス・ラクティス(Kluyveromyces lactis)細胞中のMBP(A313V)PMデルタSalを発現させることによって、修飾されたMBP融合タンパク質の増強された収率が産生された。 レーン1:NEB広範囲マーカー、Ipswich、MA; レーン2−未精製抽出搭載物; レーン3−カラム流出物; レーン4−カラム洗浄物; レーン5,6及び7、カラム緩衝液+マルトースで溶出。
【背景技術】
【0001】
純粋なタンパク質の大量が必要とされる場合には常に、組み換えタンパク質は、バイオテクノロジーにおいて多くの用途を有している。エシェリヒア・コリ(E.coli)及び酵母などの微生物発現系は、しばしば、低いコストと高い収率のために、第一の選択肢となる。イー・コリ中で外来タンパク質を発現させる場合には、タンパク質の発現の低いレベル及び/又は不溶性という問題に遭遇することが珍しくない。タンパク質が十分に発現され、可溶性を保ったとしても、細胞抽出液中の他のタンパク質の多数から精製しなければならない。標的タンパク質の発現及び精製を容易にするために、一般的に使用されている1つの方法は、タンパク質に親和性タグを融合することである。優れた親和性タグは、標的タンパク質のN末端に融合された場合に、高いレベルの発現を促進するとともに、発現環境から標的タンパク質を精製可能とする単純な一工程のアフィニティー精製を提供する特性を有する。
【0002】
イー・コリのマルトース結合タンパク質(MBP)は、イー・コリ中で産生された外来タンパク質の発現及び精製用のアフィニティータグとして一般に使用される。MBPの本来の役割は、外膜ポリンのマルトデキストリンを結合し、内膜中のMalEFK輸送装置にマルトデキストリンを放出することである。標的タンパク質のN末端にMBPのC末端を融合することによって、イー・コリ中での融合タンパク質の発現が可能となる(図1)。MBP及びMBP融合物は、アミロース、デンプン又は他のマルトデキストリンなどの多数のグルコース−α−>4−グルコース多糖の何れをも含有するクロマトグラフィーマトリックスへの結合によって、単一の工程で精製することができる(米国特許第5,643,758号)。本来の宿主中において可溶性である多くのタンパク質は、組み換えタンパク質として発現された場合には不溶性である。これらのタンパク質の多くは、MBPへの融合によって、可溶性となる(Kapust & Waugh,Protein Sci.8:1668−74(1999))。
【0003】
親和性タグとしてのMBPの有用性は、MBP−標的タンパク質精製におけるタンパク質に応じて、他の融合程うまくアフィニティーマトリックスに結合しない融合もあるという事実によって制限される。さらに、TritonX100及びTween20などの非イオン性界面活性剤の存在は、特に、本質的により低い親和性を有するMBP−標的タンパク質融合物に対する結合を妨害し得る。
【0004】
研究者は、MBPの結合特性を変化させるために、MBPの構造を使用し指定された変異を作製してきた。MBPのX線結晶構造は、「Spurlino et al.,J.Biol.Chem.266:5202−5219(1991)」によって報告されてきた。MBPは2つのドメインからなり、ドメイン間に間隙を有し、ここに多糖が結合する。N末端を含有するドメインはNドメインと名付けられ、C末端を含有するドメインはCドメインと名付けられている。3つのループは、2つのドメイン間を横断して、ヒンジを形成する。2つのグループは、マルトース及びマルトトリオースに対するMBPの親和性を増加させるヒンジ後方の領域に対して指定された変異を施すために、本構造を使用してきた(Marvin et al., Nature Structural Biology 8:795−798(2001);Telmer & Shilton, Journal of Biol.Chem.278:34555−34567 (2003))。しかしながら、MBPへの修飾はタンパク質の表面の疎水性を増加させ、従って、その溶解度を減少させるので、このアプローチは固有の欠点を有している。このため、融合タンパク質を不溶性にするその傾向を増加させることによって、親和性タグとしてのその有用性が低下する。
【発明の開示】
【0005】
本発明の一実施形態において、修飾されたマルトース結合タンパク質(MBP)であり、修飾されたMBP融合タンパク質が(i)マルトデキストリン基質に対する増加した親和性及び(ii)限定された溶解度を有する標的タンパク質に融合された際の増加した溶解度から選択される少なくとも1つの特性を有するようにする変異を有するMBPアミノ酸配列によって特徴付けられる修飾されたMBPが提供される。本発明のさらなる実施形態において、修飾されたMBPは、へリックスXIとXIIの間のヒンジ領域中に、又はタンパク質のA313の10オングストローム内の領域中に位置する変異を有する。例えば、変異は、Cドメイン中に、又はMBPのβシートFの先頭のS146の10オングストローム以内の領域中に位置し得る。修飾は、具体的には、A313V又はS146Tであり得る。
【0006】
本発明のさらなる実施形態において、修飾されたMBPは、融合タンパク質を形成するために標的タンパク質に融合され得、例えば、標的タンパク質に融合された修飾されていないMBPタンパク質の溶解度を上回る溶解度を有し得る。
【0007】
本発明の一実施形態において、修飾されたMBPは、配列番号4若しくは配列番号6から選択されるアミノ酸配列又は配列番号3若しくは配列番号5から選択されるタンパク質をコードするDNA配列を有し得る。DNAは、クルイベロミセス又はイー・コリなどの宿主細胞中で発現用pKLAC1ベクターなどのベクター中に取り込まれ得る。
【0008】
本発明の一実施形態において、クルイベロミセス又はイー・コリなどの宿主細胞中で、上記修飾されたMBP及び標的タンパク質の何れかを含む融合タンパク質を発現させることを含む、タンパク質を精製するための方法が提供される。修飾されたMBP融合タンパク質は、マルトデキストリンを例とする多糖などのマトリックスに結合することが可能である。次いで、精製されたタンパク質を取得するために、融合タンパク質は、選択された緩衝液中のマトリックスから溶出することが可能である。
【0009】
本発明のさらなる実施形態において、融合タンパク質が、変異体MBPの不存在下で観察できるより大きな程度まで、及び変異されていないMBPを用いて観察されるより大きな程度まで可溶化されるように、標的タンパク質に融合されたMBP変異体を発現させることを含む、標的タンパク質を可溶化する方法が提供される。変異の例には、A313V変異及び/又はS146T変異が含まれる。
【発明を実施するための最良の形態】
【0010】
本明細書において使用される用語が、以下に論述されている。
【0011】
「野生型」MBPは、ポリリンカー中に停止コドンを有するpMAL−2プラスミドの1つ、例えば、pKO1483の誘導体からの発現によって産生されたMBPタンパク質が含まれる。
【0012】
「変異体MBPに融合されたタンパク質の増強された溶解度」とは、野生型MBPに融合された同じタンパク質と比べた場合における可溶性タンパク質の量の増加である。溶解度は、例えば、不溶性材料が、例えば、遠心によって不溶性物質が除去される前に存在する当該タンパク質の全量に対する可溶性タンパク質の比として表すことができる。
【0013】
「変異体MBPの増加された親和性」又は「変異体MBP融合タンパク質」には、一連の所定の条件下における、マルトデキストリンなどの固体基質に結合するタンパク質の量の増加が含まれる。アフィニティー精製の効率は、カラムに適用されたタンパク質の総量に対する、指定の条件下でのマルトデキストリンに結合し、次いで、指定の緩衝液で溶出されるタンパク質の比として表すことができる。
【0014】
本発明の本実施形態は、標的タンパク質に融合された場合に、インビボでの発現の間に、融合タンパク質の溶解度を増強し、精製の間の融合タンパク質の親和性を改善することもできるMBP変異体を提供する。本実施形態には、野生型MBPが部分的な結合を示す条件下で培地に適用された場合に、多糖溶媒(マルトデキストリンを含むものなど)への増加した結合を示す変異体MBPが含まれる。次いで、例えば、可溶性マルトデキストリンの溶液を用いて、修飾されたMBPを培地から溶出し、野生型MBPと比較した場合に、少なくとも1.5から10倍多いタンパク質を与える。
【0015】
MBPのこれらの改良された変異体を発見するために、多数の試料を取り扱えるようにする技術を開発することなどの技術的な障壁を乗り越えなければならない。大規模な精製に対して音波処理が実用的でなく、溶解緩衝液がMBPのアフィニティー結合を妨害し得る場合に、これには、可溶化された融合タンパク質を放出するために宿主細胞を切断するための改善された方法が必要とされる。界面活性剤及びリゾチームを滴定することによって、結合親和性に対して悪影響を与えずに、宿主細胞を効果的に切断するためのこれらの溶解試薬の適切な濃度及び比を同定することが可能であることが発見された。
【0016】
所望の結合親和性特性を有する変異体に対してスクリーニングするために、各ウェルが融合タンパク質を結合するためのミクロマトリックスを含有し、フィルター装置がろ液中のきょう雑物を除去する96ウェルミクロプレートが使用された。これにより、多数の試料の迅速なスクリーニングが可能となった。
【0017】
タンパク質を精製するための改善されたタグとして、修飾された変異体MBPタンパク質を取得及び検査するためのスクリーニング法は、実施例に記載されている。マトリックスに対してより高い親和性を有するこのような修飾されたMBPは、マトリックスへの結合が乏しい、又は非イオン性界面活性剤の存在によって、結合が破壊されるMBP融合タンパク質の野生型MBPに付随する問題を解決する。
【0018】
実施例において、スクリーニング操作を用いて単離された、改善された溶解度及び多糖マトリックスへの改善された結合親和性という所望の特性を有する2つの変異(S146T及びA313V)が記載されている。S146T変異は、βシートFの先頭のMBPのCドメイン中に存在する。不溶性である標的タンパク質がこの変異を含有するMBPに融合されると、融合タンパク質の溶解度が増強される。本明細書中に記載されているA313V変異は、2つのドメイン間を横切る第三のヒンジ領域中、特に、ヘリックスXIとXIIの間のループ中に位置する。この変異は、融合タンパク質の溶解度と親和性の両者を増強する。イー・コリ中の外来タンパク質を発現させると、タンパク質は、不溶性凝集物の形態で部分的に又は完全に発現され得る。特定の実施例では、溶解度は、総溶解度の上限を伴って、1.1又はそれ以上、上方向に増加され得る。
【0019】
本明細書に引用される全ての参考文献及び2006年4月14日に出願された米国仮出願第60/792,133号は、参照により、組み込まれる。
【0020】
実施例
材料
制限酵素、β−アガラーゼ、DNAポリメラーゼ、T4リガーゼ、南極ホスファターゼ、Litmus38、pMAL−c2X及びpMAL−c2Gを含むpMALタンパク質融合及び精製系、アミロース樹脂(#E8021)、西洋ワサビペルオキシダーゼに連結された抗MBPモノクローナル抗体(#E8038)、USERFriendly Cloning キット、ベクターpKLAC1、宿主株TB1、ER1992、ER2502、ER2984、NEB5−α及びNEBTurboを含むK.ラクティスタンパク質発現キット並びに合成オリゴヌクレオチドは、New England Biolabs、 Inc.(NEB)、Ipswich、MAから取得した。フィルター底付きのUnifilter800マイクロタイターマイクロプレートは、Whatman、Brentford、Englandから購入した。Minelute DNA Extraction and Qiaprep Spinキットは、Qiagen、Valencia、CAから購入した。Mega10は、Dojindo、Gaithersburg、MDから購入した。ニワトリ卵白リゾチーム、クマシーブリリアントブルーR及び酸洗浄されたガラスビーズ(425から600ミクロン)は、Sigma−Aldrich、St.Louis、MOから購入した。Sea Plaque GTG低融点アガロースは、Cambrex、 E. Rutherford、 NJから購入した。使い捨てポリプロピレンカラム(#732−6008)は、BioRad、Hercules、CAから購入した。10から20%のグラジエントゲルは、Daiichi、Tokyo、Japan又はInVitrogen/Novex、Carlsbad、CAの何れかから購入した。CompleteTMプロテアーゼ阻害剤カクテルは、Roche、Basel、Switzerlandから購入した。SimplyBlue Safestainは、Invitrogen、Carlsbad、CAから購入した。ヒトジヒドロ葉酸還元酵素(DHFR)cDNAクローンpOTB7−DHFRは、Invitrogen(MGC:857)から購入した。GAPDH遺伝子は、pJF931から入手した(Fox et al.FEBS Lett.537:53−57(2003))。
【0021】
技術
セラチア・マルセッセンスヌクレアーゼは、PCT/US05/28739に記載されているとおりに取得した。プラスミドDNAのMiniprepは、Qiaprep Spinキットを用いて調製した。ランダムPCR突然変異導入は、「Fromant et al.Analytical Biochemistry 224, 347−353 (1995)」に記載されているとおりに実施した。PCRは、注記されていることを除き、Vent(R)DNAポリメラーゼを用いて行った。1%SeaPlaqueGTG低融点アガロース上で電気泳動を行い、バンドを切り出し、MineluteDNAExtractionキットを用いてDNAを精製し、又は75℃で5分間、DNAを融解させ、37℃まで冷却し、βアガロースで1から2時間消化することによって、DNA断片をゲル精製した。DNA配列決定は、BigDye標識された色素ターミネーター化学(ABI, Foster City, CA)を使用し、Applied Biosystemsの(ABIの)自動化されたDNASequencerモデル3100ABI上で行った。アクリルアミドゲルの供給者の指示書に従って、SDS−PAGEを実施し、別段の記載がある場合を除いて、クマシーブリリアントブルーRでの染色によって、タンパク質を可視化した。
【0022】
pMal−c2X又はpMal−c2G又はpMal−c2Gの誘導体の何れかからMBPを発現させた。malE中の変異を特定するための塩基の付番は、pMAL−c2X配列中の塩基番号を表す(図2A−1、2A−2、2B−1、2B−2、2C−1及び2C−2(配列番号1から6))。pMAL−c2G誘導体pSN1578は、BsmI及びBsiWIでプラスミドを切断し、4つのdNTPを全て加えて、DNAポリメラーゼKlenow断片で産物を処理した後、malE遺伝子内に欠失を作出するために連結することによって作製した。
【0023】
「Guan et al.Nucleic Acid Research, 33:6225−6234(2005)」に記載されているように、位置指定突然変異導入は、4プライマーのPCR突然変異導入を用いて実施した。MBP及びMBP融合タンパク質は、幾つかの事例において、音波処理の代わりにリゾチーム/界面活性剤溶液で細胞を溶解したことを除き、pMALProtein Fusion and Purification Systemの指示書に記載されているとおりに精製した。
【0024】
培養の500ないし1000mLから調製された未精製細胞抽出を用いて、大規模な精製を実施し、アミロース樹脂15mLを含有する2.5cm直径のカラム上に搭載した(NEB #E8021, Ipswich, MA)。培養の67mLから調製された未精製抽出物を用いて、小規模な精製を実施し、アミロース樹脂1mLを含有する使い捨てポリプロピレンカラム上に搭載した。10から20%グラジエントゲルを用いて、SDS−ポリアクリルアミドゲル電気泳動を実施した。ゲルバンドの定量のために、セロファンシートの間でゲルを乾燥させ、Microtek III scanner Microtek, Carson, CAを用いて走査し、ImageJ(NIH)を使用して濃度測定行った。
【実施例1】
【0025】
改善された特性を有するMBP中の変異体の単離
精製後の改善された収率に対するスクリーニング:
pMAL−c2xから得られるmalE遺伝子のランダムな突然変異導入は、プライマー:オリゴ1:5’GGAGACAUGAATTCAATGAAAATCGAAGAA(配列番号9)及びオリゴ2:5’GGGAAAGUAAGCTTAATCCTTCCCTCGATC(配列番号10)を使用する変異性のPCRによって達成した。
【0026】
製造業者の指示書に従い、USERFriendlyCloningKitを用いて、直鎖化されたpNEB208A中にPCR断片をクローニングした。1mLLB+1mMIPTG及び100μg/mLアンピシリン中で形質転換体を一晩増殖させた後、0.3mg/mLリゾチーム及びS.マルッセンスヌクレアーゼ20単位を添加によって溶解させ、10分間温置し、次いで、2%Tween20の0.1mLを添加することによって溶解した。
【0027】
Unifilter800マイクロプレート中のアミロース樹脂カラム(NEB#E8021,Ipswich,MA)50μLに、未精製抽出物を適用し、20mMTris−Cl、0.2MNaCl、1mMEDTA、pH7.4の0.7mL(カラム緩衝液)で、次いで、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.7mLで各ウェルを洗浄した。次いで、10mMマルトース、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.2mLで、アミロース樹脂に結合されたタンパク質を溶出した。Immulon2HBミクロタイタープレート(ThermoFisherScientific,Waltham,MA)に溶出液を移し、4℃で一晩温置した。次いで、マイクロタイターウェルを空にし、20mMTris−Cl、150mMNaCl、pH7.5(TBST)で二回洗浄し、次いで、37℃で1時間、0.36mLTBST+3%ウシ血清アルブミンでブロッキングを行った。
【0028】
TBSTで、ウェルを二回洗浄し、次いで、TBST+3%ウシ血清アルブミン中の、西洋ワサビペルオキシダーゼに連結された抗MBPモノクローナル抗体の1:2000希釈物の0.1mLを各ウェルに添加し、37℃で1時間、プレートを温置した。ウェルを空にし、次いで、TBSTで二回洗浄した。0.01%o−フェニレンジアミン、0.003%過酸化水素水で、ウェルを展開した。4MH2SO40.025mLを添加することによって検出反応を停止させ、490nmで、ウェルを分光学的にアッセイした。野生型MBPと比べて、より高い結合及び溶出を示した試料に対応する可溶化液から細胞を回収した。より高い結合及び溶出を確認するために、これらの候補を増殖し、再度検査した。
【0029】
ランダム突然変異誘発後に得られた変異の性質決定及び分離
増加した結合及び溶出特性を有するUSER中のライブラリーから得た2つの単離株を配列決定した(図2)。1つの単離株G8−1は、サイレント変異とともに、単一のミスセンス変異G1964Cを有することが見出された。G1964C変異は、MBP中のアミノ酸変化S146Tに対応する。他の単離株A9は、サイレント変異とともに、3つのミスセンス変異A1583G、A2419G及びC2465Tを有することが見出された。A1583G、A2419G及びC2465T変異は、それぞれ、アミノ酸変化N19S、K298E及びA313Vに対応する。
【0030】
pMal−C2X又はpSN1578中へのサブクローニング
以下のプライマー:オリゴ3:5’GACTCATATGAAAATCGAAGAAGGTAAACTGGTAATCTGGATTAACGGC(配列番号11)及びオリゴ4:5’ATATAAGCTTTCACCTTCCCTCGATCCCGAGGT(配列番号12)を用いて、各単離株をPCRによって増幅した。増幅されたDNAをエタノール沈殿し、NEBuffer4(NEB,Ipswich,MA)中のNdeI及びHindIIIで切断し、ゲル精製した。NdeI及びHindIIIでpSN1578を切断し、ベクター骨格をゲル精製した。G8−1及びA9断片をpSN1578断片と混合し、連結し、TB1を形質転換するために、この連結を使用した。各形質転換体から得たプラスミド調製物の配列を決定し、G8−1に関してはpIH1596と命名し、A9に関してはpIH1593と命名した。本実験における3’プライマーは、malE翻訳がpMALのlacZα断片中に進行するのを防ぐために、正しいリーディングフレーム中に停止コドンを有する。従って、これらのサブクローンは、ポリリンカーによってコードされるアミノ酸配列...IEGRの後で終了する修飾されたMBPを産生する。野生型malE遺伝子後に停止コドンを含有する対照プラスミドは、malEとlacZaの間のポリリンカー中のpMAL−c2XをXbaIで切断することによって構築された。DNAポリメラーゼI、巨大断片(Klenow)及び4つの全てのdNTPを用いて、XbaI突出部を充填し、次いで、T4リガーゼでの処理によって、プラスミドを再度環状化した。これは、malEと同じ読み枠中に停止コドンを導入し、この誘導体は、ポリリンカーによってコードされる8残基の伸長を除き、G8−1及びA9によって産生されるものと同等のMBPを産生する。この対照プラスミドは、pKO1483と名付けられた。LB+0.1%グルコース及び100μg/mLアンピシリンの500mL培養物中で、2×108細胞/mLになるまで、pKO1483、pIH1596及びpIH1593を含有するイー・コリTB1を増殖させ、0.3mMIPTGで誘導し、37℃で2時間増殖させ、次いで、採集した。カラム緩衝液25mL(10mMマルトース、10mMリン酸ナトリウム、0.2MNaCl、1mMEDTA、pH7.2の0.2mL)+10mMβ−メルカプトエタノール中に細胞を再懸濁し、次いで、音波処理によって溶解された。9000×gで30分間遠心することによって、抽出物を清澄化し、次いで、カラム緩衝液を用いて1:4希釈し、アミロース樹脂の15mLカラム上に搭載した。約125mLカラム緩衝液でカラムを洗浄し、カラム緩衝液+10mMマルトースで溶出した。3つの株の間で、MBPの収率を比較した(図3)。結果は、修飾されたMBPがアミロースへの増加した結合及び適切な緩衝液中への溶出を示したことを確認する。
【0031】
3つの変異のうち何れがA9バリアントの増加した結合に必要であるかを確認するために、pSN1578(malE遺伝子の内部に欠失(これは、挿入物を受容したクローンの簡単な同定を可能とする。)を有するpMAL−c2G誘導体)中に、3つの変異を別個にサブクローニングした。A1583G及びA2419変異は何れも、アフィニティー精製におけるMBPの収率にまったく影響がなく、又は収率を低下させ、廃棄された。N末端PCR断片を作製するためにプライマーオリゴ5:5’CTTCAAGGGTCAACCATCCAAACC(配列番号13)及びオリゴ6:5’AATACGCGGATCTTTCACCAACTCTTC(配列番号14)を用い、並びにC末端PCR断片を作製するためにプライマーオリゴ7:5’GAAGAGTTGGTGAAAGATCCGCGTATT(配列番号15)及びオリゴ8:5’CTGAGAATTCTGAAATCCTTCCCTCGAT(配列番号16)を用いる、第一のテンプレートとしてpMAL−c2Xを使用する4プライマーの位置指定PCR突然変異誘発によって、C2465T変異を分離して再現した。テンプレートとしてのゲル精製されたN末端及びC末端断片並びにプライマーオリゴ5及びオリゴ8を用いて、集合工程を実施した。最終PCR断片をBlpI及びAvaIを用いて切断し、ゲル精製し、BlpI及びAvaIを用いて切断されたpMAL−c2Xに連結し、ゲル精製した。TB1を形質転換するために連結を使用し、形質転換体からプラスミドを精製し、C2465T変異を確認するために配列を決定した。さらなる研究のために単離株を選択し、pIH1606と名付けた。
【0032】
pIH1606の構築において、MBPの末端の停止コドンは保存されていなかった。この構築物は、LacZaa断片に融合されたMBPを発現する。C2465T変異の効果をその親A9に対して比較するために、pIH1606中のmalE遺伝子の後に、停止コドンを導入した。XbaIで、本プラスミドを切断し、Klenow+dNTPで充填し、pKO1483に関して上述されているように、再連結した。停止コドンを有するC2465T誘導体は、pPR1610と称された。pKO1483及びA9と平行して行われた、このプラスミドを有するTB1の大規模MBP精製は、A9において見出されたMBPの収率の増加が全て、C2465T変異によって説明され得ることを示した。この変異は、MBPのアラニン313をバリン(A313V)に変化させる。
【0033】
MBP(S146T)を野生型MBP及び正確に同性質をもつ構築物を有する誘導体中のMBP(A313V)と比較できるようにするために、pKO1483と同じ停止コドンを有するMBP(S146T)のバージョンを構築した。pIH1596からのNdeI、BlpI断片を精製し、NdeI及びBlpIを用いて切断されたpKO1493中にサブクローニングし、pIH1619を作製した。
【実施例2】
【0034】
MBP融合タンパク質の増加した収率
A:MBP−Klenow
修飾されたMBPが、精製後に、融合タンパク質の収率を増加させることができるかどうかを検査するために、イー・コリDNAポリメラーゼIのKlenow断片をコードする遺伝子を、pMAL−c2X、pIH1619(S146T)及びpPR1610(A313V)中にクローニングした。アミロースカラムに対して本質的に低い親和性を有し、親和性精製の間に、MBP−Klenowタンパク質の幾つかは、結合せずに、アミロースカラムを流出するので、MBP−Klenow融合物を選択した。BglII及びHindIII部位間において、pBR322中にクローニングされたpolA遺伝子(GenbankecopolA:206−4127)のBglII−HindIII断片を含有するプラスミドpPolAから、DNAポリメラーゼIのKlenow部分にPCR反応を行った。プライマー:オリゴ9:5’CCAGAAGTGACGGCAACGGTGATT(配列番号17)及びオリゴ10:5’AAGTGCGGCGACGATAGTCATGCCCCGCGC(配列番号18)を用いて、PCRを実施した。
【0035】
HindIIIを用いてPCR断片を切断し、pMAL−c2に連結し、XmnI及びHindIIIでこれを切断した。得られた構築物は、pIH1040と名付けた。挿入物中でのPCRの誤りの可能性を減らすために、XhoIからHindIIIまでの領域を、pPolA由来の対応する断片で置換した。この構築物を配列決定によってチェックし、pIH1062と名付けた。pPolAをテンプレートとして使用し、プライマー:オリゴ11:5’GTGATTTCTTATGACAACTACGTCACCATCCTTGATG(配列番号19)及びオリゴ12:5’TTAAGGATCCTTAGTGCGCCTGATCCCAGT(配列番号20)を使用するPCRによって、Klenow断片をコードする遺伝子をpPR1610及びpIH1619中にクローニングした。
【0036】
BamHIを用いて、PCR断片を切断し、XmnI及びBamHIを用いて、ベクターを切断し、精製し、PCR断片と混合し、連結した。NEBTurbo(Ipswich,MA)を形質転換するために、この連結物を使用し、制限分析によって、並びにMBP−Klenow融合物の発現及びSDS−PAGEによる分析によって、正しい構造に関して形質転換体をチェックした。pIH1610−Klenow構築物をpIH1643と名付け、pIH1619−Klenow構築物をpIH1644と名付けた。
【0037】
pIH1062、pIH1643及びpIH1644を含有する細胞を用いて、MBP−Klenowのアフィニティー精製を行った。各株から得られた未精製抽出物、カラム流出物及び溶出液画分を、SDS−PAGEによって分析した(図4)。A280を測定することによって、溶出されたタンパク質を定量し、MBP−Klenowタンパク質の予想吸光係数を用いて定量した(表1)。pIH1643を有し、MBP−A313V修飾を担持する細胞は、野生型MBP又はS146T修飾されたMBP融合プラスミドを含有する細胞より2倍多くの融合タンパク質を生成した。
【0038】
B.MBP−キチン結合ドメイン:
変異体MBPが親和性精製からの融合タンパク質の収率を増加させる能力が一般的な特性であるかどうかを検査するために、アミロースに対して本質的に低い親和性を有する別の融合タンパク質を検査した。バチラス・サーキュランス(Bacillus circulans)のキチン結合ドメインに融合されたMBP(MBP−CBD)は、プラスミドpMB50によってコードされる(図5)。MBP−Klenowと同様に、このMBP−CBD融合タンパク質のかなりの割合が、アフィニティー精製の間に、アミロースカラムから流出する傾向がある。MBP(A313V)をコードするpPR1610の一部を、HapI及びSacIを用いて、プラスミドから切断し、断片を精製した。同じ酵素でpMB50を切断し、骨格も精製し、2つの断片を連結し、ER2523中に形質転換した。得られたプラスミドは、pIH1660と名付けられた。
【0039】
500mL培養物から採集されたpMB50及びpIH1660を含有する細胞を用いて、MBP−CBDのアフィニティー精製を行った。A280を測定し、MBP−CBDタンパク質の予想吸光係数を使用することによって、溶出されたタンパク質を定量した(表2)。pIH1660を有し、MBP−A313V修飾を担持する細胞は、野生型MBP融合プラスミドを含有する細胞よりほぼ2倍多くの融合タンパク質を生成した。
【実施例3】
【0040】
MBP融合タンパク質の溶解度の増強
MBP融合物の構築
修飾されたMBPがMBPに融合されたタンパク質の溶解度を増強する能力を保持するかどうかを調べるために、イー・コリ中で不溶性である傾向がある2つのタンパク質であるジヒドロ葉酸還元酵素(DHFR)及びグリセルアルデヒド−3−リン酸脱水素酵素(GAPDH)を、pIH1619(S146T)及びpIH1610(A313V)中にクローニングした。対照として、「MBP−DM」と名付けられた、Telmerらの修飾されたMBP中に変異を含有するpMAL−c2X及びpMAL−c2G誘導体中に、同じ2つのタンパク質をクローニングした。MBP−DMをコードするベクターを、以下のように構築した。まず、4プライマー位置指定PCRによって、ヌクレオチド2030に翻訳的にサイレントなNsiI部位を有するpMAL−c2G誘導体を構築した。N末端及びC末端断片の両方に対するテンプレートは、pMAL−c2Xであった。N末端断片を作製するために、プライマーオリゴ13:5’CCATAGCATATGAAAATCGAAGAAG(配列番号21)及びオリゴ14:5’CTTGAATGCATAACCCCCGTCAGCAGC(配列番号22)を使用し、C末端断片を作製するために、プライマーオリゴ15:5’GGTTATGCATTCAAGTATGAAAACGGCAAG(配列番号23)及びオリゴ8を使用した。PCR断片をゲル精製し、次いで、プライマーオリゴ13及びオリゴ8を用いた集合工程中でのテンプレートとして使用した。最終PCR断片をエタノール沈殿し、NdeI及びEcoRIで切断し、ゲル精製した。NdeI及びEcoRIでpMAL−c2Xを切断し、ゲル精製し、2つの断片を混合し、連結した。ER2502を形質転換するために、連結物を使用し、培養物を一晩増殖させ、ミニプレプDNAを調製した。NsiI部位を獲得した、正しいサイズの幾つかの形質転換体を取得したが、予想に反して、それらの全てはEcoRI部位を欠如していた。この組の代表(pPR1629と名付けられた。)をNdeI及びAvaIで切断し、malE断片をゲル精製した。同じ2つの酵素でpSN1578を切断し、プラスミド骨格をゲル精製した。これら2つのDNA断片を連結し、ER2502を形質転換するためにこの連結物を使用し、1つの形質転換体から得られるプラスミドDNAを配列決定し、pPR1633と名付けた。次に、Telmer及びShiltonによってDMと称される修飾されたMBPに対する遺伝子を、以下のように、4プライマーのPCR部位指定突然変異生成によって構築した。両N及びC末端断片に対するテンプレートは、pMAL−c2Xであった。N末端断片に対して使用されたプライマーは、オリゴ16:5’TATGCATTCAAATACGGTGACATTAAAGACGTGGGCGTGGAT(配列番号24)及びオリゴ17:5’GCGGCGTTTTCCGCAGTGGCGGCAATACGTGGATCTTTC(配列番号25)であった。C末端断片は、プライマーオリゴ18:5’GCGGAAAACGCCGCGAAAGGTGAAATCATGCCGAACATC(配列番号26)及びオリゴ8を用いて作製された。オリゴ16及びオリゴ8をプライマーとして使用し、精製されたN末端及びC末端断片を用いて集合PCRを実行した。PCR断片をエタノール沈殿し、EcoRVで直鎖化されたLitmus38に連結し、南極ホスファターゼで処理した。NEB5−α(Ipswich,MA)を形質転換するために連結物を使用し、構築物を確認するために1つの形質転換体から得られたプラスミドDNAを配列決定し、pPR1638と名付けた。プラスミドDNApPR1633及びpPR1638をNsiI及びAvaIで切断し、pPR1633由来のプラスミド骨格及びpPR1638由来のmalE断片をゲル精製し、連結し、NEBTurboを形質転換するために連結物を使用した。配列決定のために、1つの形質転換体由来のプラスミドDNAを選択し、pPR1639と名付けた。
【0041】
pOTB7−DHFRをテンプレートとして使用し、プライマーオリゴ19:5’GGATGGTTGGTTCGCTAAACTGCATCGTC(配列番号27)及びオリゴ20:5’TATTAATCATTCTTCTCATATACTTCAAA(配列番号28)を使用して、DHFR遺伝子にPCRを行い、次いで、室温で15分間、T4ポリメラーゼ及びdTでPCR断片を処理した。この処理によって、断片の各末端上に短い3‘突出(DHFR遺伝子の上流末端上にGG及び下流末端上にA)が生じる。XmnIで切断し、次いで、T4ポリメラーゼ及びdAで処理することによって、ベクターpMAL−c2X及びpPR1610を調製し、同様に、malE領域の末端に3’CC突出部及びlacZa末端上にTを生成する。ベクターDNAをDHFR断片に連結し、ER1992を形質転換するためにこの連結物を使用した。各ベクターに関して、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって形質転換体を確認した。pMAL−c2X−DHFR単離株をpIH1616と名付け、pIH1610−DHFR単離株をpIH1617と名付けた。次いで、以下のように、DHFR挿入物をpIH1596中にサブクローニングした。NdeI及びBlpIでpIH1616を切断し、消化物をゲル上で走行させ、(DHFR遺伝子を含む)骨格を切除し、ゲル精製した。NdeI及びBlpIでpIH1596を切断し、malE’をゲル精製した。malE’断片にベクター骨格を連結し、ER1992を形質転換するためにこの連結物を使用した。制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって、単離株を確認し、pIH1618と名付けた。pPR1639及びpIH1616の両者をAvaI及びSalIで切断し、pPR1639由来のベクター骨格及びpIH1616由来のDFHR断片をゲル精製し、2つの断片を連結することによって、DHFR挿入物をpPR1639中にサブクローニングした。NEBTurboを形質転換するために、この連結物を使用し、配列決定によって形質転換体を確認し、pIH1646と名付けた。
【0042】
pJF931をテンプレートとして使用し、プライマーオリゴ21:5’GGATGGTGAAGGTCGGTGTGAACGG(配列番号29)及びオリゴ22:5’TATTACTCCTTGGAGGCCATGTAGGCCA(配列番号30)を使用して、GAPDH遺伝子にPCRに供し、次いで、T4ポリメラーゼ及びdTでPCR断片を処理した。
【0043】
XmnIで切断した後、上述のようにT4ポリメラーゼ及びdAで処理することによって、ベクターpMAL−c2X、pPR1610及びpIH1619を調製した。ベクターDNAをGAPDH断片に連結し、ER1992を形質転換するためにこの連結物を使用した。各ベクターに関して、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって形質転換体を確認した。pMAL−c2X−GAPDH単離株をpIH1625と名付け、pPR1610−GAPDH単離株をpIH1626と名付け、pIH1619−GAPDH単離株をpIH1627と名付けた。次いで、pPR1639及びpIH1625の両者をAvaI及びSalIで切断し、pPR1639由来のベクター骨格及びpIH1625由来のGAPDH断片をゲル精製し、2つの断片を連結することによって、GAPDH挿入物をpPR1639中にサブクローニングした。NEBTurboを形質転換するために、この連結物を使用し、制限分析及びSDS−PAGEによって分析された予想サイズの融合タンパク質の発現によって、形質転換体を確認し、pIH1645と名付けた。
【0044】
溶解度特性
8つのpMALプラスミド(pMAL−c2X、pIH1619(S146Tを担持する。)及びpPR1610(A313Vを担持する。)、各プラスミドは、DHFR又はGApDHをコードするDNJAをさらに含有する。)を含有するTB1の20mL培養物を、LBamp中で、2×108/mlまで増殖させ、0.3mMIPTGを用いて誘導し、さらに2時間温置し、次いで、微量遠心管中にて、3000×gでの遠心によって、細胞を採集した。50mMTris−Cl、pH7.9、50mMNaCl、0.75mMEDTA、0.6%Mega10、150μg/mLリゾチーム及び20Kunitz単位/mLセラチア・マルセッセンスヌクレアーゼ2mL中に各ペレットを再懸濁し、室温で10分間温置した。再懸濁されたペレットは、全細胞抽出物と表記した。125μLの試料を取り出し、14,000×gで、2分間遠心した。上清を取り出し、可溶性画分と表記した。同じ緩衝液125μL中にペレットを再懸濁し、不溶性画分と表記した。各株に対して、SDS−PAGE上に各画分の試料(5μL)を走行させた(図5)。ゲルを乾燥させ、走査し、遠心前の細胞可溶化液中に存在するタンパク質に対する可溶性タンパク質の比として、各レーン中のMBP融合タンパク質の量を定量した(表2)。DHFR及びGAPDH融合物の両者に関して、A313V及びS146T修飾されたMBPは、野生型MBPに比べて、融合タンパク質の溶解度を増強した。DMによって修飾されたMBPを用いて作製された融合物は、予想通り、野生型MBPと比べて、低下した溶解度を示した。
【実施例4】
【0045】
K.ラクティス中でのMBP変異体の使用
K.ラクティスMBP(A313V)−融合発現ベクターの構築
それぞれ、フォワード及びリバースプライマー:
【0046】
【化1】
を使用するPCRによって、変異体マルトース結合タンパク質MBP(A313V)をコードする遺伝子を増幅した。フォワードプライマーは、HindIII制限酵素部位(太字)を含有し、直前にmalE遺伝子開始コドン(下線)が先行するKozakコンセンサス配列(斜字体)が続くように構築された。リバースプライマーは、XhoI制限酵素部位(太字)を含有し、プロテアーゼエンテロキナーゼのタンパク分解認識部位(斜字及び下線)をコードするDNAが直後に続くように構築された。フレーム内C末端MBP(A313V)融合発現カセットの構築を可能とするために、リバースプライマー上に停止コドンは取り込まれなかった。Phusionポリメラーゼを用いて、完全長遺伝子を含有するプラスミドpPR1610からmalE遺伝子を増幅した。K.ラクティスMBP(A313V)融合発現ベクター(pKIMF2)を作製するために、K.ラクティス発現ベクターpkLAC1のHindIII及びXhoI制限部位中に、増幅された遺伝子がクローニングされた。このクローニング戦略は、malE遺伝子による、pKLAC1中のK.ラクティスα接合因子プレプロシグナル配列の置換をもたらす。従って、MBP及びMBP(A313V)融合タンパク質は、分泌経路に誘導されず、代わりに、酵母サイトゾル中に保持される。
【0047】
K.ラクティス中のMBP(A313V)融合タンパク質の発現及び精製
以下のプライマーを用いて、パラミオシンの切断型をコードする遺伝子(Steel et al., J. Immunol.145:3917−3923(1990))をPCRによって増幅した。
【0048】
【化2】
フォワードプライマーは、XhoI制限部位(太字)を含有するように構築された。リバースプライマーは、PMデルタSal停止コドン(斜字体)のすぐ上流にNotI制限部位(太字)を含有するように構築された。
【0049】
クローニング戦略は、以下のとおりであった。約750bpパラミオシン遺伝子を増幅し、精製した。XhoI及びNotIで、断片を二重消化し、発現ベクターpKIMF2のXhoI及びNotI制限部位中にクローニングし、PMデルタSalのC末端及びN末端の間にフレーム内融合物を作出する。このようにして構築されたプラスミドは、pKLMF2−PMデルタSalと名付けられた(図7)。
【0050】
SacII制限消化によって、MBP(A313V)−PMデルタ融合ベクター(pKIMF2−PMデルタSal)を直鎖化し、その指示書に従って、K.ラクティスProtein Expression Kitを用いて、化学的に形質転換受容性のK.ラクティス細胞中に精製された産物を形質転換した。アセトアミドプレート上で、形質転換体コロニーを選択し、上記キットに記載されているように、細胞全体のPCRによって、正しく組み込まれたMBP(A313V)−PMデルタSal発現カセットを含有するクローンを同定した。
【0051】
YPGalactose培地(1%酵母抽出物、2%ペプトン、2%ガラクトース)20mL培養物に、複数コピーの組み込まれたMBP(A313V)−PMデルタSal発現カセットを含有するK.ラクティス細胞の株を接種し、振盪プラットフォームを有する温置装置中において、4日間、30℃で増殖させた。増殖後の培養物の合計細胞含量は、約8.2×1010細胞(すなわち、約4.1×108細胞/mL)であると測定された。4℃で10分間、8,000rpmの遠心によって、細胞を採集した。過剰な培地を除去するために、蒸留水中で、細胞を一回洗浄した。
【0052】
可溶化液の調製のために、1Mソルビトール中の10mg/mLリチカーゼの2mL溶液中に細胞を再懸濁し、37℃で1時間温置した。微量遠心管中での2分間の穏やかな遠心(7,000rpm)によって、細胞を採集した。プロテアーゼ阻害剤カクテル(CompleteTMプロテアーゼ阻害剤カクテル、Roche)を含有する氷冷アミロースカラム緩衝液(20mMTris−ClpH7.4、0.2MNaCl、1mMEDTA)3mLの総容量中に、細胞ペレットを再懸濁した。細胞スラリーをガラスチューブに移し、氷の上に配置した。酸洗浄されたガラスビーズ(425から600ミクロン、Sigma)の等量を細胞スラリーに添加し、それぞれ、1分間10回のボルテックスによって、細胞を破壊した。細胞可溶化液を新しいチューブに移し、カラム緩衝液1mLで、ガラスビーズを4回洗浄した。細胞可溶化液及び洗浄液をプールし、遠心によって、細胞破砕物を除去した。清澄化された細胞可溶化液を、カラム緩衝液で、8mLになるように希釈した。
【0053】
融合タンパク質精製のために、カラム緩衝液15mLで予め平衡化された1.6mLアミロース樹脂カラム上に、細胞可溶化液を通過させた。カラム緩衝液24mLでカラムを洗浄し、10mMマルトースを含有するカラム緩衝液で、0.4mL画分中に、結合されたタンパク質を溶出した。10から20%Tris−グリシン勾配ゲル上でのSDS−PAGEによって、細胞可溶化液及び精製されたタンパク質を分離した。SimplyBlue Safestainによって、タンパク質を同定した。図6は、主要な溶出されたタンパク質が、MBP(A313V)−PMデルタSal融合タンパク質に対して予想されたサイズ(68.5kDa)に対応することを示している。
【0054】
【表1】
【0055】
【表2】
【0056】
【表3】
【図面の簡単な説明】
【0057】
【図1】図1は、標的タンパク質に融合されたMBPをコードするDNAを発現し、融合タンパク質をアミロースに選択的に結合させ、マルトース含有緩衝液中に標的タンパク質を溶出し、次いで、プロテアーゼ切断によって、精製された融合タンパク質から標的タンパク質を回収することによる、標的タンパク質のクローニング及び精製を記載する模式図を示している。
【図2A−1】野生型MBPを修飾されたMBPと比較する配列を提供する。pMAL−c2Xから得られる、野生型MBP(配列番号2)をコードするDNA配列(配列番号1)。
【図2A−2】野生型MBPを修飾されたMBPと比較する配列を提供する。pMAL−c2Xから得られる、野生型MBP(配列番号2)をコードするDNA配列(配列番号1)。
【図2B−1】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体A9(配列番号4)をコードするDNA配列(配列番号3)。修飾されたMBP配列中の変化は、太字で示されている。
【図2B−2】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体A9(配列番号4)をコードするDNA配列(配列番号3)。修飾されたMBP配列中の変化は、太字で示されている。
【図2C−1】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体G8−1(配列番号6)をコードするDNA配列(配列番号5)。修飾されたMBPDNA及びアミノ酸配列中の変化は、太字で示されている。
【図2C−2】野生型MBPを修飾されたMBPと比較する配列を提供する。MBP変異体G8−1(配列番号6)をコードするDNA配列(配列番号5)。修飾されたMBPDNA及びアミノ酸配列中の変化は、太字で示されている。
【図3】野生型MBP(pKO1483)又は修飾されたMBP(A9又はG8−1)を用いたタンパク質の収率を比較するヒストグラムを示している。MBPの収率は、mg/500ml培養で記載されている。
【図4】野生型MBP、MBPA313V及びS146T修飾を含有するpMALからのMBP−Klenowの精製された産物をSDS−PAGEゲル上に示している。プラスミド:WT=pIH1062、A313V=pIH1643、S136T=pIH1644。レーン:CE=未精製抽出物、FT=流出物及びE=溶出液。表記画分の等しい一部を、各レーンに搭載した。MBP融合タンパク質の有意により高い収率が、野生型MBPからより、A313V変異体MBPから得られた(表1も参照)。
【図5−1】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−2】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−3】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−4】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図5−5】MBP−CBD融合タンパク質を発現させるために使用されるpMB50(配列番号7)の配列(表2参照)。
【図6】溶解度に対する、DHFR及びGAPDHへの野生型及び修飾されたMBPの融合の効果を示すSDS−PAGEゲル。DHFR融合プラスミド:WT=pIH1616;A313V=pIH1617;S146T=pIH1618;DM=pIH1646。GAPDH融合プラスミド:WT=pIH1625;A313V=pIH1626;S146T=pIH1627;DM=pIH1645。レーン:T=総細胞抽出物;S=可溶性抽出物;I=不溶性材料の再懸濁。表記画分の等しい一部を、各レーンに搭載した。不溶性タンパク質に対する可溶性タンパク質の比は、野生型MBPと比較して、両変異体に関してより大きかった。
【図7−1】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−2】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−3】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図7−4】pKLMF2−PMデルタSal(配列番号8)の配列を提供する。
【図8】アミロース樹脂カラムを用いて、SDS−PAGEゲル上の融合タンパク質のアフィニティー精製から得られた画分を示している。MBP(A313V)−PMデルタSal発現カセットを担持するクルイベロミセス・ラクティス(Kluyveromyces lactis)細胞中のMBP(A313V)PMデルタSalを発現させることによって、修飾されたMBP融合タンパク質の増強された収率が産生された。 レーン1:NEB広範囲マーカー、Ipswich、MA; レーン2−未精製抽出搭載物; レーン3−カラム流出物; レーン4−カラム洗浄物; レーン5,6及び7、カラム緩衝液+マルトースで溶出。
【特許請求の範囲】
【請求項1】
修飾されたマルトース結合タンパク質(MBP)であり、タンパク質に融合された場合に、前記修飾されたMBPが(i)マルトデキストリン基質に対する結合親和性及び(ii)前記タンパク質が限定された溶解度を有する場合における溶解度のうち少なくとも1つの増加を有するようにする変異を有するMBPアミノ酸配列を含む、前記修飾されたマルトース結合タンパク質。
【請求項2】
変異が、タンパク質のへリックスXIとXIIの間のヒンジ領域中に位置し、又はA313の10オングストローム内の領域中に位置する、請求項1に記載の修飾されたMBP。
【請求項3】
変異が、タンパク質のCドメイン中に位置し、又はβシートFの先頭のS146の10オングストローム以内の領域中に位置する、請求項1に記載の修飾されたMBP。
【請求項4】
変異がA313Vである、請求項1に記載の修飾されたMBP。
【請求項5】
変異がS146Tである、請求項1に記載の修飾されたMBP。
【請求項6】
融合タンパク質を形成するために標的タンパク質に融合された、請求項1に記載の修飾されたMBP。
【請求項7】
融合タンパク質が、標的タンパク質に融合された修飾されていないMBPタンパク質の溶解度より大きな溶解度を有する、請求項6に記載の修飾されたMBP。
【請求項8】
配列番号4又は配列番号6を含む、請求項1に記載された修飾されたMBP。
【請求項9】
配列番号3又は配列番号5を含む、修飾されたMBPをコードする単離されたDNA。
【請求項10】
請求項9に記載のDNAを含むベクター。
【請求項11】
請求項10に記載のベクターで形質転換された宿主細胞。
【請求項12】
形質転換された宿主細胞が形質転換されたイー・コリ宿主細胞である、請求項11に記載の宿主細胞。
【請求項13】
形質転換された宿主細胞が形質転換されたクルイベロミセス宿主細胞である、請求項11に記載の宿主細胞。
【請求項14】
ベクターがpKIMF2である、請求項11に記載の宿主細胞。
【請求項15】
請求項1に記載の修飾されたMBPと標的タンパク質を含む融合タンパク質を宿主細胞中で発現させること、
修飾されたMBP融合タンパク質をマトリックスに結合させること、及び
精製されたタンパク質を取得するために、選択された緩衝液中のマトリックスから融合タンパク質を溶出することを含む、
タンパク質を精製する方法。
【請求項16】
マトリックスが多糖である、請求項15に記載の方法。
【請求項17】
炭水化物がマルトデキストリンである、請求項15に記載の方法。
【請求項18】
修飾されたMBPがA313V及びS146Tから選択される変異を有する、請求項15に記載の方法。
【請求項19】
修飾されたMBPが配列番号4を含む、請求項15に記載の方法。
【請求項20】
修飾されたMBPが配列番号6を含む、請求項15に記載の方法。
【請求項21】
修飾されたMBPが配列番号3又は配列番号5によってコードされる、請求項15に記載の方法。
【請求項22】
宿主細胞がクルイベロミセス又はイー・コリである、請求項15に記載の方法。
【請求項23】
インビボにおいて、融合タンパク質が、変異体MBPの不存在下で観察できるより大きな程度まで、及び変異されていないMBPを用いて観察されるより大きな程度まで可溶化されるように、標的タンパク質に融合された修飾されたMBPを発現させることを含む、標的タンパク質を可溶化する方法。
【請求項24】
修飾されたMBPがA313V変異又はS146T変異を有する、請求項23に記載の方法。
【請求項25】
修飾されたMBPがA313V変異及びS146T変異を有する、請求項24に記載の方法。
【請求項1】
修飾されたマルトース結合タンパク質(MBP)であり、タンパク質に融合された場合に、前記修飾されたMBPが(i)マルトデキストリン基質に対する結合親和性及び(ii)前記タンパク質が限定された溶解度を有する場合における溶解度のうち少なくとも1つの増加を有するようにする変異を有するMBPアミノ酸配列を含む、前記修飾されたマルトース結合タンパク質。
【請求項2】
変異が、タンパク質のへリックスXIとXIIの間のヒンジ領域中に位置し、又はA313の10オングストローム内の領域中に位置する、請求項1に記載の修飾されたMBP。
【請求項3】
変異が、タンパク質のCドメイン中に位置し、又はβシートFの先頭のS146の10オングストローム以内の領域中に位置する、請求項1に記載の修飾されたMBP。
【請求項4】
変異がA313Vである、請求項1に記載の修飾されたMBP。
【請求項5】
変異がS146Tである、請求項1に記載の修飾されたMBP。
【請求項6】
融合タンパク質を形成するために標的タンパク質に融合された、請求項1に記載の修飾されたMBP。
【請求項7】
融合タンパク質が、標的タンパク質に融合された修飾されていないMBPタンパク質の溶解度より大きな溶解度を有する、請求項6に記載の修飾されたMBP。
【請求項8】
配列番号4又は配列番号6を含む、請求項1に記載された修飾されたMBP。
【請求項9】
配列番号3又は配列番号5を含む、修飾されたMBPをコードする単離されたDNA。
【請求項10】
請求項9に記載のDNAを含むベクター。
【請求項11】
請求項10に記載のベクターで形質転換された宿主細胞。
【請求項12】
形質転換された宿主細胞が形質転換されたイー・コリ宿主細胞である、請求項11に記載の宿主細胞。
【請求項13】
形質転換された宿主細胞が形質転換されたクルイベロミセス宿主細胞である、請求項11に記載の宿主細胞。
【請求項14】
ベクターがpKIMF2である、請求項11に記載の宿主細胞。
【請求項15】
請求項1に記載の修飾されたMBPと標的タンパク質を含む融合タンパク質を宿主細胞中で発現させること、
修飾されたMBP融合タンパク質をマトリックスに結合させること、及び
精製されたタンパク質を取得するために、選択された緩衝液中のマトリックスから融合タンパク質を溶出することを含む、
タンパク質を精製する方法。
【請求項16】
マトリックスが多糖である、請求項15に記載の方法。
【請求項17】
炭水化物がマルトデキストリンである、請求項15に記載の方法。
【請求項18】
修飾されたMBPがA313V及びS146Tから選択される変異を有する、請求項15に記載の方法。
【請求項19】
修飾されたMBPが配列番号4を含む、請求項15に記載の方法。
【請求項20】
修飾されたMBPが配列番号6を含む、請求項15に記載の方法。
【請求項21】
修飾されたMBPが配列番号3又は配列番号5によってコードされる、請求項15に記載の方法。
【請求項22】
宿主細胞がクルイベロミセス又はイー・コリである、請求項15に記載の方法。
【請求項23】
インビボにおいて、融合タンパク質が、変異体MBPの不存在下で観察できるより大きな程度まで、及び変異されていないMBPを用いて観察されるより大きな程度まで可溶化されるように、標的タンパク質に融合された修飾されたMBPを発現させることを含む、標的タンパク質を可溶化する方法。
【請求項24】
修飾されたMBPがA313V変異又はS146T変異を有する、請求項23に記載の方法。
【請求項25】
修飾されたMBPがA313V変異及びS146T変異を有する、請求項24に記載の方法。
【図1】

【図2A−1】

【図2A−2】

【図2B−1】

【図2B−2】

【図2C−1】

【図2C−2】

【図3】
【図4】

【図5−1】

【図5−2】

【図5−3】

【図5−4】

【図5−5】

【図6】
【図7−1】
【図7−2】
【図7−3】
【図7−4】

【図8】
【図2A−1】
【図2A−2】
【図2B−1】
【図2B−2】
【図2C−1】
【図2C−2】
【図3】
【図4】
【図5−1】
【図5−2】
【図5−3】
【図5−4】
【図5−5】
【図6】
【図7−1】
【図7−2】
【図7−3】
【図7−4】
【図8】
【公表番号】特表2009−533058(P2009−533058A)
【公表日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願番号】特願2009−505493(P2009−505493)
【出願日】平成19年4月14日(2007.4.14)
【国際出願番号】PCT/US2007/009100
【国際公開番号】WO2007/120809
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(591021970)ニユー・イングランド・バイオレイブス・インコーポレイテツド (18)
【Fターム(参考)】
【公表日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願日】平成19年4月14日(2007.4.14)
【国際出願番号】PCT/US2007/009100
【国際公開番号】WO2007/120809
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(591021970)ニユー・イングランド・バイオレイブス・インコーポレイテツド (18)
【Fターム(参考)】
[ Back to top ]