
新規の種類のヒストン脱アセチル化酵素阻害剤としてのZn2+キレートモチーフ係留短鎖脂肪酸
【課題】ヒストン脱アセチル化酵素(HDAC)阻害剤としての、Zn2+キレートモチーフで係留された脂肪酸を提供すること。
【解決手段】化合物はインビトロおよびインビボテストにて良く効果を発揮した。本発明は、例えばバルプロエート、ブチレート、フェニルおよびフェニルブチレートなどの短鎖脂肪酸を含む脂肪酸の構造的な改変に基づく。本発明は一般に、脂肪酸を、芳香族ωアミノ酸リンカーを介してZn2+キレートモチーフ(ヒドロキサム酸、ο‐フェニレンジアミンが挙げられるが、これらに限定されない)と結合することを含む。
【解決手段】化合物はインビトロおよびインビボテストにて良く効果を発揮した。本発明は、例えばバルプロエート、ブチレート、フェニルおよびフェニルブチレートなどの短鎖脂肪酸を含む脂肪酸の構造的な改変に基づく。本発明は一般に、脂肪酸を、芳香族ωアミノ酸リンカーを介してZn2+キレートモチーフ(ヒドロキサム酸、ο‐フェニレンジアミンが挙げられるが、これらに限定されない)と結合することを含む。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の説明)
本発明は、Army Grant DAMD17−02−1−0117およびNational Institute of Health Grant CA94829によって支援された。政府は、本発明において一定の権利を有する。本願は、2002年12月2日に出願された米国仮特許出願60/526,348号に対する優先権を主張する。
【0002】
(発明の分野)
本発明はヒストン脱アセチル化酵素阻害剤に関係し、特にZn2+キレートモチーフを包含するヒストン脱アセチル化酵素阻害剤に関する。より詳細には、本発明は、短鎖脂肪酸を基本とする、Zn2+キレートモチーフを包含するヒストン脱アセチル化酵素阻害剤に関する。
【背景技術】
【0003】
(発明の背景)
コアヒストンのアセチル化状態は、DNAのヌクレオソーム・パッケージングの調節を介して、遺伝子の転写調節において中枢的役割を果たしている(非特許文献1;非特許文献2;非特許文献3)。低アセチル化状態では、ヌクレオソームは緊密に凝縮されており、その結果転写因子の標的DNAへの接触が制限されることに起因して、転写が抑制される。反対に、ヒストンがアセチル化されるとヌクレオソーム構造は緩み、クロマチンは転写許容状態となる。どちらも配列特異的転写活性因子との複合体として標的遺伝子へ動員される、ヒストンアセチル基転移酵素(HAT)の活性とヒストン脱アセチル化酵素(HDAC)の活性との間の動力学的なバランスが、この翻訳後修飾のレベルを維持している。この後成的なマーキング(marking)システムの調節異常が、多くの形態の癌の主要な病因である不適切な遺伝子発現を引き起こすことが示されている(非特許文献4;非特許文献5;非特許文献6)。さらに、多くの種類の腫瘍細胞でHDACを阻害すると、少数の遺伝子の転写が再活性化されることによって、成長停止、分化および/またはアポトーシスが誘引されることを例証する多くの証拠がある(非特許文献7;非特許文献8;非特許文献9;非特許文献10;非特許文献11)。異種移植片モデルでもまた、これらインビトロでの知見が確認されており、これらのことはHDAC機能の調節が、癌の予防的および/または治療的介入のための標的となることを示唆している。
【0004】
現時点で、構造的に異なる種類のいくつかのHDAC阻害剤が報告されており(非特許文献8;非特許文献9;非特許文献10;非特許文献11)、これとしては短鎖脂肪酸(例えば、ブチレート(butyrate)、バルプロエート(valproate)、フェニルアセテート(phenylacetate)およびフェニルブチレート(phenylbutyrate))(非特許文献12;非特許文献13;非特許文献14;非特許文献15)、ベンズアミド誘導体類(例えばMS−27−275)(非特許文献16;非特許文献17)、トリコスタチンA(TSA)および類似体(非特許文献18;非特許文献19;非特許文献20)、ハイブリッド極性化合物(例えばスベロイルアニリドヒドロキサム酸(SAHA))(非特許文献21;非特許文献22)、環状テトラペプチド類(例えばアピシジン(apicidin))(非特許文献23;非特許文献24;非特許文献25;非特許文献26)、並びにデプシペプチドFR901228(非特許文献26)が挙げられる。これらの薬剤の内、短鎖脂肪酸は、他の種類のHDAC阻害剤のIC50がμMまたはnM単位ですらあることと比較して、IC50がmM単位である最も弱い阻害剤である。短鎖脂肪酸を癌治療に使用することが報告されているが、低い抗増殖活性、速い代謝、および非特異的様式での作用のため、その治療効果は限られている。
【0005】
最近、細菌性のHDAC相同体であるHDLP(ヒストン脱アセチル化酵素様タンパク質)のX線結晶分析から、タンパク質−リガンド相互作用(それによりTSAおよびSAHAが酵素阻害を媒介する)の特異な様式が示唆された(非特許文献27)。HDAC触媒ドメインは、炭素数4から6の直鎖に相当する長さにわたる、細い管状ポケット構造から成ることが明白となっている。この酵素のポケット構造の底部近くにZn2+陽イオンが位置し、これが、2つのHis−Asp電荷伝達系と協同して脱アセチル化触媒を促進していると考えられている。
【0006】
当該分野の他の研究を慎重に考察し、短鎖脂肪酸のHDAC阻害効力が弱いのは、短鎖脂肪酸が、本発明者らが脱アセチル化触媒の中心的役割を果たしていると考えている、活性部位ポケット内のZn2+陽イオンに接近できないことに一部起因すると、本発明者らは理解した。この考察およびさらなる研究を基に、本発明者らは、芳香族リンカーを介してZn2+キレートモチーフに短鎖脂肪酸を係留(tethering)することによって、短鎖脂肪酸を構造的に改変した。本発明者らの発見および研究によって、本発明者らは、新しい種類のZn2+キレートモチーフに係留された短鎖脂肪酸を発明するに至った。これらのうちのいくつかはnMの単位でHDAC活性並びに癌細胞増殖の阻害を示すものもあり、これは親化合物と比較して3桁の作用効力向上である。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Kouzarides,「Histone acetylases and deacetylases in cell proliferation.」Curr Opin Genet Dev(1999)9:40−48
【非特許文献2】Gray and Ekstrom,「The human histone deacetylase family.」Exp Cell Res(2001)262:75−83
【非特許文献3】Jenuwein and Allis,「Translating the histone code.」Science(2001)293:1074−1080
【非特許文献4】Wade,「Transcriptional control at regulatory checkpoints by histone deacetylases:molecular connections between cancer and chromatin.」Hum Mol Genet(2001)10:693−698
【非特許文献5】Cress and Seto,「Histone deacetylases,treanscriptional control,and cancer.」J Cell Physiol(2000)184:1−16
【非特許文献6】Marks et al.,「Histone deacetylases and cancer: causes and thrapies.」Natl Rev Cancer(2001)1:194−202
【非特許文献7】Jung,「Inhibitors of histone deacetylases as new anticancer agents.」Curr Med Chem(2001)8 1505−1511
【非特許文献8】Grozinger and Schreiber,「Deacetylase enzymes:biological functions and the use of small−molecule inhibitors.」Chem Biol(2002)9:3−16
【非特許文献9】Johnstone,「Histone−deacetylase inhibitors:novel drugs for the treatment of cancer.」Nat Rev Drug Discov(2002)1:287−299
【非特許文献10】Kramer et al.,「Histone deacetylase as a thrapeutic target.」Trends Endocrinol Metab(2001)12:294−300
【非特許文献11】Marks et al.,「Histone deacetylase inhibitors:inducers of differentiation or apoptosis of transformed cells」J Natl Cancer Inst(2000)92:1210−1216
【非特許文献12】Lea and Tulsyan,「Discordant effects of butylate anologues on erythroleukemia cell proliferation,differentiation and histone deacetylase.」Anticancer Res(1995)15:879:883
【非特許文献13】Kruh,「Effects of sodium butyrate,a new pharmacological agent,on cells in culture.」Mol Cell Biochem(1982)42:65−82
【非特許文献14】Newmark and Young,「Butyrate and phenylacetate as differentiating agents:practical problems and opportunities.」J Cell Biochem Suppl(1995)22:247−253
【非特許文献15】Phiel et al.,「Histone deacetylase is a direct target of valproic acid,a potent anticonvulsant,mood stabilizer,and teratogen.」J Biol Chem(2001)276:36734−36741
【非特許文献16】Suzuki et al.,「Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives.」J Med Chem(1999)42:3001−3003
【非特許文献17】Saito et al.,「A synthetic inhibitor of histone deacetylase,MS−27−275,with marked in vivo antitumor activity against human tumors.」Proc Natl Acad Sci USA(1999)96:4592−4597
【非特許文献18】Tsuji et al.,「A new antifungal antibiotic,trichostatin.」J Antibiot(Tokyo)(1976)29:1−6
【非特許文献19】Jung et al.「Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation.」J Med Chem(1999)42:4669−4679
【非特許文献20】Furumai et al.「Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin.」Proc Natl Acad Sci USA(2001)98:87−92
【非特許文献21】Richon et al.,「A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases.」Proc Natl Acad Sci USA(1998)95:3003−3007
【非特許文献22】Remiszewski et al.,「Inhibitors of human histone deacetylase:synthesis and enzyme and cellular activity of straight chain hydroxamates.」J Med Chem(2002)45:753−757
【非特許文献23】Kijima et al.,「Trapoxin,an antitumor cyclic tetrapeptide,is an inrreversible inhibitor of mammalian histone deacetylase.」J Biol Chem(1993)268:22429−22435
【非特許文献24】Shute et al.,「Analogues of the cytostatic and antimitogenic agents chlamydocin and HC−toxin:synthesis and biological activity of chloromethyl ketone and diazomethyl ketone functionalized cyclic tetrapeptides.」J Med Chem(1987)30:71−78
【非特許文献25】Han et al.,「Apicidin,a histone deacetylase inhibitor,inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin.」Cancer Res(2000)60:6068−6074
【非特許文献26】Nakajima et al.「FR901228,a potent antitumor antibiotic,is a novel histone deacetylase inhibitor.」Exp Cell Res(1998)241:126−133
【非特許文献27】Finnin et al.,「Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors」Nature(1999)401:188−193
【発明の概要】
【課題を解決するための手段】
【0008】
(本発明の要旨)
本発明者らは、短鎖脂肪酸のヒストン脱アセチル化酵素(HDAC)阻害剤としての効力が弱いのは、脱アセチル化触媒に重要と考えられるHDAC活性部位ポケット内の亜鉛陽イオンに短鎖脂肪酸が接近できないことに一部起因すると理解した。本発明は、例えばバルプロエート、ブチレート、フェニルアセテートおよびフェニルブチレートなどの短鎖脂肪酸を含む脂肪酸の構造的な改変に基づく。本発明は一般に、脂肪酸を、芳香族ωアミノ酸リンカーを介してZn2+キレートモチーフ(ヒドロキサム酸、ο‐フェニレンジアミンが挙げられるが、これらに限定されない)と結合することを含む。新しい種類のZn2+キレートモチーフ係留短鎖脂肪酸を含む本発明は、このストラテジーから導かれた。
【0009】
本発明の化合物のHDAC阻害における効力は、HDLPリガンド複合体の結晶構造から提供されるフレームワークに基づいて、強力なHDAC阻害剤が設計され得ることを示している。本発明は、短鎖脂肪酸、ωアミノ酸、ヒドロキサマト(hydroxamato)などの亜鉛キレート剤の様々な組み合わせにより、化合物の大きなライブラリを創出することを可能にする係留ストラテジーに基づいている。
【0010】
本発明は、以下の式:
【0011】
【化6】

を有するヒストン脱アセチル化酵素阻害剤を含む。ここで:
Xは、HおよびCH3から選択され;
Yは、nが0〜2の(CH2)nであり;
Zは、mが0〜3である(CH2)mおよび(CH)2から選択され;
Aは、ヒドロカルビル基であり;
Bは、ο‐アミノフェニル基またはヒドロキシ基であり;
Qは、ハロゲン、水素、またはメチル基である。
【0012】
一実施例において、Aは脂肪族を含み得、この脂肪族は分岐であり得る。Aはまた、置換されていても置換されていなくてもよい芳香族も含む。この式において、Bはο‐アミノフェニル基またはヒドロキシ基である。いくつかの実施例において、Yはnが0である(CH2)nであり得、Aは芳香族基を含み、Bはヒドロキシで、かつQは水素である。
【0013】
いくつかの実施例では、mは0かつXはHである。これらの特徴を有する化合物としては以下のものが含まれるが、これらに限定されない。
【0014】
【化7】
【0015】
【化8】
いくつかの実施例では、XはHであり、かつAは以下:
【0016】
【化9】
から選択され、ここでRは分岐または非分岐の、置換または非置換の、飽和または不飽和の、脂肪族または芳香族を含む。
【0017】
本発明は、本発明の阻害剤を含む組成物を包含し、この組成物はR立体異性体と比較してS立体異性体が富化されている。
【0018】
本発明の具体的な阻害剤としては、N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;2−プロピル−ペンタン酸−{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;2−プロピル−ペンタン酸−{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;2−プロピル−ペンタン酸{4−2−[2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;2−プロピル−ペンタン酸−[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミド;N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド;N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(3−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロシキ−ベンズアミド;4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;ピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミド;N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;4−(2,2−ジメチル−4−フェニル‐ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−メトキシ−2−フェニル−アセチルアミノ)−ベンズアミド;4−ジフェニルアセチルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;N−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミド;N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;ならびにN−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミド;N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;N−[4−(2−ヒドロキシカルバモイルーエチル)−フェニル]−2−(S)−フェニル−ブチリルアミド;N−[4−(2−ヒドロキシカルバモイルーエチル)−フェニル]−2−(R)−フェニルブチルアミド;N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミドが含まれる。
【0019】
本発明はまた、これらの阻害剤と1つ以上の薬学的に受容可能な賦形剤とを含む薬学的組成物を含む。さらになお、本発明は、動物(例えば、ヒト)内で腫瘍性細胞増殖を阻害する方法を含み、、これは治療的有効量の少なくとも1種の本発明の阻害剤を投与する工程を包含する。
【0020】
本発明のさらなる特性および利点は、以下に続く説明にてその一部が記述される。その一部はこの説明から明白となるか、または本発明の実施によって習得され得る。本発明の特性および利点は、添付の特許請求の範囲において具体的に示される要素および組み合わせによって理解され、達成されることとなる。
【0021】
前述の一般的な説明および以下の詳細な説明はいずれも、例示であって説明にすぎず、特許請求の範囲のように本発明を制限するものではないことが、理解されるべきである。この明細に添付され、この明細の一部を構成する付属の図面は、本発明の実施形態を実証し、説明と一緒になって本発明の本質を説明するのに役立つものである。
例えば、本発明は以下の項目を提供する。
(項目1)
式
【化1】
を有する、ヒストン脱アセチル化酵素阻害剤であって、ここで:
Xは、HおよびCH3から選択され;
Yは、nが0〜2である、(CH2)nから選択され;
Zは、mが0〜3である(CH2)mおよび(CH)2から選択され;
Aは、ヒドロカルビル基であり;
Bは、o−アミノフェニル基またはヒドロキシル基であり;そして
Qが、ハロゲン、水素、またはメチルである、阻害剤。
(項目2)
請求項1に記載の阻害剤であって、Aが脂肪族基を含む、阻害剤。
(項目3)
請求項2に記載の阻害剤であって、前記脂肪族基が分枝である、阻害剤。
(項目4)
請求項1に記載の阻害剤であって、Aが芳香族基を含む、阻害剤。
(項目5)
請求項4に記載の阻害剤であって、Aが、置換した芳香族基を含む、阻害剤。
(項目6)
請求項1に記載の阻害剤であって、Bがo−アミノフェニルである、阻害剤。
(項目7)
請求項1に記載の阻害剤であって、Bがヒドロキシである、阻害剤。
(項目8)
請求項1に記載の阻害剤であって、nが0であり、Aが芳香族基を含み、Bがヒドロキシであり、そしてQが水素である、阻害剤。
(項目9)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;2−プロピル−ペンタン酸{4−2−(2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;および2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミドから選択される、阻害剤。
(項目10)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;およびN−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミドから選択される、阻害剤。
(項目11)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;およびN−ヒドロキシ−4−[(3−フェニルーブチリルアミノ)−メチル]−ベンズアミドから選択される、阻害剤。
(項目12)
請求項1に記載の阻害剤であって、該阻害剤が、4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミドおよびピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミドから選択される、阻害剤。
(項目13)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−メトキシ−2−フェニルーアセチルアミノ)−ベンズアミド;4−ジフェニルアセチルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;およびN−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミドから選択される、阻害剤。
(項目14)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミドから選択される、阻害剤。
(項目15)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(S)−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(R)−フェニル−ブチルアミド;N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミドから選択される、阻害剤。
(項目16)
請求項1に記載の阻害剤であって、該阻害剤が、エステルまたは塩である、阻害剤。
(項目17)
請求項1に記載の阻害剤と、少なくとも一つ薬学的に受容可能な賦形剤とを含む、薬学的組成物。
(項目18)
動物における腫瘍性細胞増殖を阻害する方法であって、治療上効果的な量の、請求項1に記載される少なくとも一つの阻害剤を投与する工程、を包含する、方法。
(項目19)
請求項18に記載の方法であって、前記動物がヒトである、方法。
(項目20)
請求項8に記載の阻害剤であって、m=0であり、そしてX=Hである、方法。
(項目21)
請求項20に記載の阻害剤であって、前記化合物が、
【化2】
である、阻害剤。
(項目22)
請求項20に記載の阻害剤であって、前記化合物が、
【化3】
である、阻害剤。
(項目23)
請求項20に記載の阻害剤であって、前記化合物が、
【化4】
である、阻害剤。
(項目24)
請求項21に記載の阻害剤を含む組成物であって、該組成物が、R−立体異性体と比べて、S−立体異性体が富化されている、組成物。
(項目25)
請求項1に記載の阻害剤であって、X=Hであり、そしてAが、
【化5】
から選択され、Rが、分枝または非分枝の、置換または非置換の、飽和または不飽和の、脂肪族基または芳香族基である、阻害剤。
【図面の簡単な説明】
【0022】
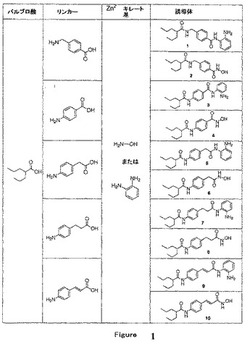
【図1】図1は、化合物1〜10を生成するための、バルプロ酸と5つの芳香族ωアミノ酸および2つのZn2+キレート部分との様々な結合体を示す。
【図2】図2は、化合物1〜10のHDAC阻害効力を示す。インビトロHDACアッセイは、本明細書中で記載されている市販の酵素アッセイキットを使用して行った。データは、平均値±S.D.(n=3)である。
【図3】図3は、化合物11〜22の構造およびHDAC阻害効力を示す。値は、平均値±S.D.(n=3)である。
【図4】図4は、DU−145細胞におけるヒストンアセチル化およびp21WAF/CIP1の発現に対する、HTPB、TSAおよびフェニルブチレートの影響を示す。10%FBS添加RPMI 1640培地中で、DU−145細胞を、HTPB、TSAおよびフェニルブチレートに、示された濃度で24時間曝露した。それぞれの溶解液からの等量のタンパク質を電気泳動し、それぞれの抗体を使用するウエスタンブロットによって試験した。内部参照タンパク質としてアクチンを使用した。
【図5】図5は、DU−145細胞に対するHTPBの増殖阻害効果を示す。(A)細胞生存率に対するHTPB用量依存的な影響の時間経過。DU−145細胞を、10%FBS含有RPMI 1640培地中の0〜2.5μMのHTPBで、示された時間、処理した。MTTアッセイによって生細胞を試験した。データは平均値±S.D.である(n=6)。(B)24時間曝露後のヌクレオソームDNAの形成に対する、HTPB用量依存的な影響。ヌクレオソームの形成は、各アッセイについて2×103の細胞に相当する溶解物を用いたCell Death Detection ELISAによって、定量的に測定した。データは、2回の独立した定量の平均である。
【図6】図6は、本発明の化合物を合成するためのスキームの例を示す。
【図7−1】図7(フレーム1)は、本発明に従う種々の化合物の例を表示する。
【図7−2】図7(フレーム2)は、本発明に従う種々の化合物の例を表示する。
【図7−3】図7(フレーム3)は、本発明に従う種々の化合物の例を表示する。
【図7−4】図7(フレーム4)は、本発明に従う種々の化合物の例を表示する。
【図7−5】図7(フレーム5)は、本発明に従う種々の化合物の例を表示する。
【図7−6】図7(フレーム6)は、本発明に従う種々の化合物の例を表示する。
【図7−7】図7(フレーム7)は、本発明に従う種々の化合物の例を表示する。
【図7−8】図7(フレーム8)は、本発明に従う種々の化合物の例を表示する。
【図7−9】図7(フレーム9)は、本発明に従う種々の化合物の例を表示する。
【図7−10】図7(フレーム10)は、本発明に従う種々の化合物の例を表示する。
【図7−11】図7(フレーム11)は、本発明に従う種々の化合物の例を表示する。
【図8−1】図8(フレーム1)は、本発明に従って使用され得る亜鉛キレートモチーフの例を表示する。
【図8−2】図8(フレーム2)は、本発明に従って使用され得る亜鉛キレートモチーフの例を表示する。
【図9】図9は、化合物19のリガンドドッキングの分子モデリング研究である。
【図10】図10は、化合物19とその結合部位との相互作用の過程で起こると考えられるプロトン移動を図示する。
【図11】図11は、化合物42のリガンドドッキングの分子モデリング研究である。
【図12】図12は、PC−3アンドロゲン非依存性前立腺癌細胞における、ヒストンH−4過アセチル化およびp21WAF/CIP1発現に対する、化合物42、SAHA、およびTSAの影響を示す。
【図13】図13は、PC−3細胞におけるAktの活性化状態に対する、化合物42、SAHA、およびTSAの影響を示す。
【図14】図14は、いくつかのリン酸化酵素に対する化合物42の影響を示す。
【図15】図15は、定着したPC−3異種移植片腫瘍の増殖に対する、経口投与した化合物42の影響を示す。
【図16】図16は、胸腺欠損マウスにおける皮下PC−3異種移植片腫瘍の増殖に対する、化合物42、化合物44、およびSAHAの影響を示す。
【図17】図17は、化合物42、SAHA、またはコントロールで処理した2つの代表的なPC−3腫瘍ホモジネートの、ヒストンH3並びにアセチル化H3のウエスタンブロットを示す。
【図18】図18は、代表的な乳癌細胞株、肺癌細胞株および甲状腺癌細胞株に対する、化合物42の用量依存的な抗増殖効果を示す。
【図19】図19は、初代CLL細胞に対する、化合物42の影響を示す。
【図20】図20は、化合物42の化学合成スキームを図示する。
【図21】図21は、本発明に従う化合物に関する一般的な化学合成スキームを図示する。
【発明を実施するための形態】
【0023】
(発明の実施形態の説明)
一般に、短鎖脂肪酸は、アシル鎖の構造に係わらず、mM範囲でHDAC阻害および抗増殖活性を示す(Grozinger and Schreiber,Chem Biol 9:3−16(2002);Johnstone,Nat Rev Drug Discov 1:287−299(2002);Kramer et al.,Trends Endocrinol Metab 12:294−300(2001))。本発明はとりわけ、これらの脂肪酸が、酵素ポケット入口に位置する表面残基および/またはこの管状ポケット内の疎水性部分との非特異的な疎水性相互作用を介して、HDAC阻害を発揮するという発見に基づく。本発明者らは、疎水性スペーサーを介してZn2+キレート部分を係留することによって、これら短鎖脂肪酸のHDAC阻害効力を増大させた。
【0024】
ヒストン脱アセチル化酵素の酵素活性の測定は、公知の方法論を用いて達成され得る。例えば、Yoshidaら(J.Biol.Chem.265:17174−17179(1990))は、トリコスタチンAで処理した細胞においてアセチル化ヒストンを検出することによる、ヒストン脱アセチル化酵素の酵素活性の評価を記載している。同様に、Tauntonら(Science 272:408−411(1996))は、内在性HDAC−1および組換えHDAC−1を使用してヒストン脱アセチル化酵素の酵素活性を測定する方法を記載している。Yoshidaら(J.Biol.Chem.265:17174−17179,1990)およびTauntonら(Science 272:408−411,1996)はいずれも、本明細書中に参考として援用される。
【0025】
この開示を通じて、本発明に従う化合物に対する参照がなされる。この明細書および特許請求の範囲においてそのような化合物に対する参照は、その化合物のエステルおよび塩を包含する。従って、たとえ明確に列挙されていなくても、その化合物自体への参照によって、そのようなエステルおよび塩は企図および包含される。
【0026】
本発明に従って使用され得る脂肪酸は、ヒドロカルビル部分およびカルボン酸部分を含む。本明細書中で使用される場合、用語「ヒドロカルビル」は、「脂肪族」、「環式脂肪族」、および「芳香族」が含まれると理解される。このヒドロカルビル基には、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アリール基、アラルキル基、およびアルカリル基が含まれると理解される。さらに、「ヒドロカルビル」には、非置換ヒドロカルビル基と置換ヒドロカルビル基とのいずれもが含まれると理解され、後者は炭素および水素以外の付加置換基を有する炭化水素部分を指す。さらに、「カルボン酸」はこの化合物を指すために使用されるが、そのような酸の塩(例えば、カルボン酸塩など)も明確に企図される。さらに、本明細書中においてカルボン酸とカルボン酸塩は交換可能なものとして使用され得る。
【0027】
脂肪酸には、特に、約3個〜約14個の炭素の長さの非分枝脂肪酸の長さに匹敵する長さの分子鎖を有するものが含まれるが、これらに限定されない。従って、この分子鎖は、例えば約3個、4個、5個、6個、7個、8個、9個、10個、11個、12個または13個の炭素の長さであり得る。この鎖は、最大で、例えば約14個、13個、12個、11個、9個、8個、7個、6個、5個または4個の炭素の長さであり得る。この脂肪酸は直鎖であっても分枝であってもよく、単結合、二重結合および/または三重結合を含み得る。脂肪酸の非限定的な例としては、バルプロエート、ブチレート、フェニルアセテートおよびフェニルブチレートが含まれる。
【0028】
本発明に従って企図されるZn2+キレートモチーフとしては、ヒドロキサム酸およびο−フェニレンジアミンが挙げられるが、これらに限定されない。他の例としては、トリフルオロメチルケトン、α−ケトアミド、α−ケトチアゾール、2−ケト1−メチル−1H−イミダゾール、α−ケト1H−テトラゾール、α−ケト1H−イミダゾール、5−ケト1メチル−1H−イミダゾール、α−ケトオキサゾール、α−ケト4,5−シヒドロ−オキサゾール、α−ケトベンゾオキサゾール、α−ケトオキサゾール[4,5−b]ピリジンおよびα−ケトピリジンが含まれる。これらのモチーフの構造は、図8に示される。
【0029】
スペーサーは任意のヒドロカルビルスペーサーであり得るが、好ましくは芳香族成分を含む。芳香族リンカーは、以下の利点を有していると考えられている:1)上記結合体の構造硬性を増大させる、および2)ポケットの管様疎水領域とのファンデルワールス接触を増大させて結合親和力を向上させる。リンカーの例としては、芳香族ω‐アミノ酸が挙げられるが、これに限定されない。
【0030】
このリンカーは、炭素数4から8の直鎖(例えば炭素数4、5、6、7、あるいは8の直鎖)と同じ長さを示す。従って、その長さは炭素数4〜7または4〜6の直鎖と同じであり得る。その長さは、結合ポケットの疎水領域の深さに基づき得る。本発明のリンカーの例としては、4−(アミノメチル)安息香酸、4−アミノ安息香酸、(4−アミノフェニル)酢酸、3−(4−アミノフェニル)プロピオン酸、および3−(4−アミノフェニル)−アクリル酸が挙げられるが、これらに限定されない。とりわけ、4−(アミノメチル)安息香酸は、MS−27−275(Saito et al.,Proc Natl
Acd Sci USA 96:4592−4597(1999))のリンカーとして使用されている。
【0031】
以下の化合物が具体的に企図される:
N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;
N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;
2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;
2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;
2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;
2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;
2−プロピル−ペンタン酸{4−[2−(2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;
2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミド;
N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;
N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;
N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;
N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド;
N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;
N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;
N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
N−ヒドロキシ−4−[(3−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;]
4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
ピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミド;
N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;
4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;
N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(2−メトキシ−2−フェニル−アセチルアミノ)−ベンズアミド;
4−ジフェニルアセチルアミノ−N−ヒドロキキシ−ベンズアミド;
N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;
N−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミド;
N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;
N−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミド;
N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(S)−フェニル−ブチルアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(R)−フェニル−ブチルアミド;
N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;および
N−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミド。
【0032】
本発明の化合物は、ラセミ化合物あるいはラセミ混合物である場合がある。本明細書中で使用される場合、用語「ラセミ」は、本発明の化合物の(R)鏡像異性体と(S)鏡像異性体との混合物、あるいは立体異性体混合物を意味し、ここで鏡像異性体もしくは立体異性体はいずれも、実質的に他方から精製されていない。
【0033】
本発明の(R)立体異性体または(S)立体異性体を記述するために本明細書中で使用される「富化」という語は、(S)立体異性体よりも(R)立体異性体の量が多いか、またはその逆の組成物を指す。例えば、この組成物は、化合物42の全重量に対して、50重量%、55重量%、あるいは60重量%以上の化合物42の(S)立体異性体を含有し得る。一実施例において、富化された(S)化合物42の量はより多いいものであり得、例えば、化合物42の全重量に対し、65重量%、70重量%、75重量%、80重量%、81重量%、82重量%、83重量%、84重量%、85重量%、86重量%、87重量%、88重量%、89重量%、90重量%、91重量%、92重量%、93重量%、94重量%、95重量%、96重量%、97重量%、98重量%、99重量%以上、もしくはこれらの任意の分数量(例えば90.1重量%、90.2重量%等)の(S)‐化合物42であり得る。特定の実施例において、富化された(S)化合物42の量は、化合物42の全重量に対し、99重量%、99.1重量%、99.2重量%、99.3重量%、99.4重量%、99.5重量%、99.6重量%、99.7重量%、99.8重量%、99.9重量%よりも多いものであってもよいし、100重量%であってもよい。これらの用語はまた、(S)化合物42の薬学的に受容可能な任意の塩の量を定義する。これらは非限定的な例であり、本発明の他のラセミ化合物に対しても、同じ富化が達成され得る。
【0034】
本発明の鏡像異性体富化した組成物を投与することにより、所望の治療効果が得られる。即ち、鏡像異性体富化した組成物は、低い合計濃度で治療効果をもたらし得るか、または所望されな鏡像異性体の存在に起因する副作用を軽減し得る。これらの利点は特に企図される。
【0035】
本発明の方法にて用いられる任意の本発明の化合物は、経口的に、非経口的に(IV、IM、デポーIMS、SQ、およびデポーSQ)、舌下に、経鼻的に(吸入)、髄腔内に、局所的に、あるいは直腸的に投与され得る。当業者に公知の投与形態は、本発明の方法において用いられる本発明の化合物の送達に適している。
【0036】
治療的有効量の、本発明の方法において用いられる本発明の化合物を含有する組成物が提供される。この化合物は、錠剤、カプセル、あるいはエリキシル等経口投与に適切な薬学的調製物に、あるいは非経口投与のための滅菌溶液もしくは滅菌懸濁液に処方することができる。本明細書中に記載される化合物は、当該分野で周知の技術および手順を用いて薬学的組成物に処方することができる。
【0037】
約0.1mgから1000mgの、本発明の方法において用いられる本発明の化合物あるいは発明の化合物の混合物、あるいは生理学的に受容可能な塩もしくはエステルは、認可された製薬業務に必要な単位投与量形態内で、生理学的に許容できるビヒクル、キャリア、賦形剤、結合剤、保存剤、安定剤、香味料等と混合される。これらの組成物あるいは調製物中の活性物質量は、記載した単位の適切な投与量が得られる量である。この組成物は単位投薬形態に処方され得、各投与形態物は、約1〜約500mg、または約10〜約100mgの活性成分を含有する。「単位用量形態」という語は、ヒト被験体および他の哺乳類のための単位投与物として適切な、物理的に別々の単位を指し、各単位物は、適切な賦形剤と共に、所望する治療効果が生じるよう計算された、前決定された量の活性物質を含有する。
【0038】
組成物を調製するため、本発明の方法で用いられる1つ以上の発明の化合物は、適切な薬学的に受容可能なキャリアと混合される。この化合物を混合あるいは添加すると、溶液、懸濁液、乳液などが生じ得る。薬学的に適切なキャリアとして、リポソーム懸濁液もまた、使用され得る。これらは、当業者に周知の方法に従って調製され得る。得られる混合物の形態は、多数の因子に依存し、この因子としては、意図する投与様式および選択したキャリアまたはビヒクルにおけるこの化合物の溶解性が挙げられる。有効濃度は、処置される疾患、障害または状態の1つ以上の症状が軽減または改善されるのに十分であり、経験的に決定され得る。
【0039】
本明細書中で提供される化合物の投与に適した薬学的キャリアまたは薬学的ビヒクルとしては、特定の投与様式に適する任意のキャリアが含まれる。さらに、活性物質もまた、所望の活性を損なわない他の活性物質と、または所望の活性を補う他の活性物質と、または他の活性を有する活性物質と、混合され得る。この化合物は、組成物内で唯一の薬学的活性成分として処方されてもよいし、他の活性成分と組み合わせられてもよい。
【0040】
この化合物が不十分な溶解性を示す場合、可溶化のための方法が使用され得る。そのような方法は公知であり、これとしてはジメチルスルホキシド(DNSO)のような共溶媒の使用、TWEENのような界面活性剤の使用、および炭酸水素ナトリウム水溶液への溶解が含まれるが、これらに限定されない。有効な薬学的組成物の処方において、塩またはプロドラッグのような、この化合物の誘導体もまた使用され得る。
【0041】
化合物の濃度は、化合物投与の対象である障害の少なくとも1つの症状を軽減または改善する投与量の送達に有効なものである。代表的には、この組成物は、単回用量投与のために処方される。
【0042】
本発明の方法において用いられる本発明の化合物は、徐放性処方物またはコーティングのような、体内からの急速な排出に対して化合物を保護するキャリアと一緒に、調製され得る。そのようなキャリアとしては、マイクロカプセル化された送達系などの徐放性処方物が挙げられるが、これに限定されない。この活性化合物は、治療を受ける患者に有害な副作用を与えずに、治療的に有用な効果を発揮するのに十分な量で、薬学的に受容可能なキャリア中に含有され得る。治療的に有効な濃度は、処置される疾患に対するインビトロモデル系およびインビボモデル系において、公知の化合物を試験することによって、経験的に決定され得る。
【0043】
本発明の化合物および組成物は、複数回投与容器あるいは1回投与容器内に収められ得る。この封入された化合物および組成物は、例えば、使用のために組み立てられ得る成分部分を含有するキットにおいて、提供され得る。例えば、凍結乾燥形態の発明の化合物と適切な稀釈剤とが、使用前に混合するために別々の成分部分として提供され得る。キットは、本発明の化合物と、共投与のための第二の治療剤とを包含し得る。本発明の化合物および第二の治療剤は、別々の成分部分として提供され得る。キットは複数の容器を備え得、各容器は、本発明の方法において用いられる本発明の化合物の1単位用量以上を含む。この容器は所望の投与様式に適合され得、これとしては、経口投与のための錠剤、ゲルカプセル、徐放性カプセルなど;非経口投与のためのデポー製品、充填済み注射器、アンプル、バイアルなど;局所投与のためのパッチ、医療用パッド、クリームなど、が挙げられるが、これらに限定されない。
【0044】
薬物組成物中の本発明の活性化合物の濃度は、その活性化合物の吸収性、不活性化性および排出速度、投与スケジュール、投与量、ならびに当業者に公知の他の要因に依存する。
【0045】
この活性成分は、一度に投与されてもよいし、時間間隔ごとに投与される、より小さないくつかの投与量に分割されてもよい。正確な用量および処置期間は、処置される疾患の関数であり、公知の試験プロトコルを用いて、あるいはインビボもしくはインビトロの試験データから外挿することによって、経験的に決定され得る。また、濃度および投与値は、緩和されるべき状態の重篤度によって変動し得ることに、注意するべきである。任意の特定の患者に関して、特定の投薬レジメンは、その患者の必要性および、この組成物を投与するかまたは組成物の投与を監督する者である専門家による判断に従って、時間経過と共に調整するべきであること、そして、この明細で記載される濃度の範囲は例示というだけであって、特許請求される組成物の範囲または実施の制限を意図したものでないことが、さらに理解されるべきである。
【0046】
経口投与が所望される場合、胃の酸性環境から化合物を保護する組成で、この化合物は提供され得る。例えば、この組成物は、胃の中でその保全性を維持し、そして腸内でこの活性化合物を放出する腸溶コーティング内に処方され得る。この組成物はまた、制酸薬あるいは他の同様の成分と組み合わせて処方され得る。
【0047】
経口組成物は、一般に、不活性の希釈剤あるいは食用のキャリアが含み、そして圧縮されて錠剤となるか、またはゼラチンカプセル内に封入され得る。経口治療投与の目的のために、その活性化合物を賦形剤と混合し、錠剤、カプセル、あるいはトローチの形態にて使用され得る。この組成物の一部として、薬学的に適合性の結合剤および補助物質が含有され得る。
【0048】
この錠剤、丸剤、カプセル、トローチなどは、任意の以下の成分あるいは同様の性質の化合物を含有し得る:結合剤(例えば、トラガカントゴム、アカシアゴム、トウモロコシ澱粉またはゼラチンが挙げられるが、これらに限定されない);賦形剤(例えば、ミクロクリスタリンセルロース、澱粉、あるいは乳糖);崩壊剤(例えば、アルギン酸およびトウモロコシ澱粉が挙げられるが、これらに限定されない);潤滑剤(lubricant)(例えば、ステアリン酸マグネシウムが挙げられるが、これに限定されない);潤滑剤(glidant)(例えば、二酸化ケイ素コロイドが挙げられるが、これに限定されない);甘味料(例えば、ショ糖またはサッカリン);香味料(例えば、ペパーミント、サルチル酸メチルまたはフルーツ香料)。
【0049】
投与単位形態がカプセルである場合、これは上記の種類の物質に加えて脂肪油などの液体キャリアを含有し得る。さらに、投与単位形態は、例えば糖衣あるいは他の腸溶剤のような、この投与単位形態の物理的形態を改変する様々な他の物質を含有し得る。この化合物はまた、エリキシル剤、懸濁液、シロップ、カシェ剤、チューイングガムなどの成分として投与され得る。シロップは、この活性成分に加えて、甘味料、およびある種の保存料としてショ糖、色素および着色剤、ならびに香味料を含有し得る。
【0050】
この活性物質はまた、所望の作用を損なわない他の活性物質と、あるいは所望の作用を補う物質と、混合され得る。本発明の化合物は、例えば抗がん剤、ホルモン、ステロイド、またはレチノイドと併用して使用され得る。抗がん剤は、アルキル化剤、代謝拮抗剤、ホルモン剤、抗生剤、コルヒチン、ビンカアルカロイド、L−アスパラギナーゼ、プロカルバジン、ヒドロキシ尿素、ミトタン、ニトロソウレア類、あるいはイミダゾールカルボキサミドのような多数の化学治療剤のうちの1つであり得る。適切な薬剤としては、チューブリンの脱分極を促進する薬剤が挙げられる。例としては、コルヒチンならびにビンカアルカロイド(例えば、ビンブラスチンおよびビンクリスチン)が挙げられる。
【0051】
経口、皮内、皮下、あるいは局所投与に使用する溶液または懸濁液は、以下の任意の成分を含有し得る:滅菌希釈剤(注射用水、生理食塩水、固定油、例えばゴマ油、ココナッツ油、ピーナッツ油、綿果油などのような天然植物油、あるいはオレイン酸エチルなどの合成脂質性ビヒクル、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒);抗菌剤(例えば、ベンジルアルコールおよびメチルパラベン);抗酸化剤(例えば、アスコルビン酸および重亜硫酸ナトリウム);キレート剤(例えば、エチレンジアミンテトラ酢酸(EDTA));緩衝液(例えば、酢酸、クエン酸、およびリン酸);浸透圧を調整するための薬剤(例えば、塩化ナトリウムおよびデキストロース)。非経口調製物は、アンプル、使い捨て注射器、あるいはガラス、プラスチック、もしくは他の適切な物質からできた複数回投与用バイアル内に、封入することができる。必要に応じて、緩衝液、保存剤、抗酸化剤などが組み入れられ得る。
【0052】
静脈経由で投与する場合、適切なキャリアとしては、生理食塩水、リン酸緩衝化生理食塩水(PBS)、ならびに増粘剤および溶解剤を含有する溶液(例えば、グルコース、ポリエチレングリコール、ポリプロピレングリコールおよびこれらの混合物)が挙げられるが、これらに限定されない。組織標的リポソームを含むリポソーム懸濁液もまた、薬学的に受容可能なキャリアであり得る。これらは、当該分野で公知な方法に従って調製され得る。
【0053】
本発明の化合物は、時間放出処方あるいはコーティングのような、体外への急速な排出から化合物を保護するキャリアと共に調製され得る。そのようなキャリアとしては、インプラントおよびマイクロカプセル送達系、ならびに生分解性生体適合ポリマー(例えば、コラーゲン、エチレンビニル酢酸、ポリ無水物、ポリグリコール酸、ポリオルトエステル、ポリ乳酸など)のような徐放性組成物が挙げられるが、これらに限定されない。そのような組成物を調製するための方法は、当業者に公知である。
【0054】
本発明の方法において用いられる化合物は、経腸的にあるいは非経口的に投与され得る。経口的に投与される場合、本発明の方法で用いられる化合物は、当業者に周知の、経口投与のための通常の投薬形態で投与され得る。これらの投薬形態としては、錠剤並びにカプセルの通常の固形単位用量形態、ならびに溶液、懸濁液、およびエリキシルなどの液体投与形態が挙げられる。固形投与形態を使用する場合、その形態は徐放性型であり得、その結果、本発明の方法で用いられる化合物は1日1、2回の投与のみが必要とされる。
【0055】
経口投与形態は、1日1回、2回、3回、または4回患者に投与され得る。本発明の方法で用いられる本発明の化合物は、1日3回以下、もしくは1日1回あるいは2回のみ投与され得る。つまり、本発明の方法で用いられる本発明の化合物は、経口投与形態にて投与される。どのような経口投与形態を用いる場合でも、本発明の方法で用いられる化合物を胃の酸性環境から保護するように、これらは設計され得る。腸溶性コーティング錠剤が、当業者に周知である。さらに、酸性である胃から保護するよう一つ一つがコーティングされた小球体が充填されたカプセルもまた、当業者に周知である。
【0056】
本発明の方法で用いられる本発明の化合物はまた、ナノ結晶分散処方物にて、有利に送達され得る。そのような処方物の調製は、例えば米国特許第5,145,684号に記載されており、その全内容が、参考として援用される。HIVプロテアーゼ阻害剤のナノ結晶分散物およびその使用法は、米国特許第6,045,829号に記載されており、その全内容が、参考として援用される。ナノ結晶処方物は、代表的には薬物化合物の生物学的有用性を増大させる。
【0057】
本発明の化合物および方法は、動物において腫瘍性細胞の増殖を抑制するために使用され得る。この方法は、体内に1つ以上の腫瘍性細胞を有する動物に、治療的有効量の1種以上の本発明の化合物を、上記のような組成物にて投与する工程を包含する。この動物は哺乳類(家畜を含む)であり得る。この動物はヒトであり得る。
【0058】
用語「腫瘍性細胞」は、異常な細胞増殖を示す細胞を指すために使用される。腫瘍性細胞の異常な増殖には、細胞増殖の増大が含まれる。例えば、腫瘍性細胞は、過増殖性細胞、またはインビトロにて増殖の接触抑制の欠如を示す細胞、インビボで転移することのできない良性腫瘍細胞、インビボで転移することができかつ除去の試み後に再発し得る癌細胞であり得る。用語「腫瘍形成」は、腫瘍性増殖の発症へと導く細胞増殖の誘導を示すために使用される。
【0059】
用語「治療的有効量」および「治療的有効期間」は、腫瘍性細胞増殖の軽減に有効な投与量で腫瘍性細胞増殖の軽減に有効な期間に渡る処置を示すために使用される。上記のように、そのような投与は、非経口的、経口的、舌下的、経皮的、局所的、経鼻的、あるいは直腸内的であり得る。全身投与する場合、本発明の化合物の血中濃度が約0.1μM〜約100mMを達成するのに十分な投与量で、その治療的組成物は投与され得る。局所的な投与については、これよりもはるかに少量が有効であり得、またはるかに高い濃度が許容される場合がある。当業者は、ヒストン脱アセチル化酵素阻害剤の有効濃度の低下をもたらすそのような治療効果は、本発明に従って処置される組織、器官、あるいは特定の動物もしくは患者によって著しく変動し得ることを認識する。また、患者がある投与量で開始しても、その患者の状態が変化するため、投与量は経時的に変化し得ることもまた、理解される。
【0060】
本発明は、動物の細胞増殖性疾患あるいは状態を治療するための組成物並びに方法を提供し、この方法は、そのような処置を必要とする動物に治療的有効量の、本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。記述したように、この動物は哺乳動物(家畜を含む)であり得る。この動物はヒトであり得る。
【0061】
用語「細胞増殖性疾患または細胞増殖性状態」は、異常な細胞増殖(好ましくは、異常に増大した細胞増殖)によって特徴づけられる任意の状態を指すことが意味される。そのような細胞増殖性疾患または状態の例としては、癌、再狭窄および乾癬が挙げられるが、これらに限定されない。いくつかの実施例において、本発明は、動物内の腫瘍性細胞増殖を阻害するための方法を提供し、この方法は、体内に1つ以上の腫瘍性細胞を有する動物に、治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。本発明に従って治療することのできる癌としては、前立腺癌、肺癌、急性白血病、多発性硬化症、膀胱癌腫、腎臓癌腫、乳癌腫、結腸直腸癌腫、神経芽腫、あるいは黒色腫が挙げられるが、これらに限定されない。
【0062】
本発明の化合物のいくつかは、原生動物由来のヒストン脱アセチル化酵素に対する阻害活性を有する。従って、本発明はまた、原生動物性疾患または原生動物性感染を処置もしくは予防するための方法を提供し、この方法は、そのような処置を必要とする動物に、治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。ここでも、動物は哺乳動物であり得、そしてヒトであり得る。いくつかの実施形態において、このヒストン脱アセチル化酵素阻害剤は、哺乳動物ヒストン脱アセチル化酵素、特にヒトヒストン脱アセチル化酵素阻害剤を阻害するよりも高い程度で、原生動物ヒストン脱アセチル化酵素を阻害する。
【0063】
本発明はさらに、真菌性疾患または真菌性感染を処置するための方法を提供し、この方法は、そのような処置を必要とする動物に治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。この動物は、ヒトを含む哺乳動物であり得る。本発明のこの実施形態に従って使用されるヒストン脱アセチル化酵素阻害剤は、哺乳類ヒストン脱アセチル化酵素、特にヒトヒストン脱アセチル化酵素阻害剤を阻害するよりも高い程度で、真菌のヒストン脱アセチル化酵素を阻害し得る。
【0064】
当業者である投与を行う医療従事者に周知なように、正確な投与量および投与の頻度が、本発明の方法で用いられる投与される特定の化合物、処置される患者の具体的な状態、処置される状態の重篤度、特定の患者の年齢、体重、全身の身体状態、およびその患者が受けている可能性のある他の医薬に依存することは、当業者には明白であるはずである。
【実施例】
【0065】
(実験)
化学試薬および有機溶媒は、他に記載のない限り、Aldrichから購入した。核磁気共鳴スペクトル(1H NMR)は、Bruker 250 MHzで測定した。化学シフト(δ)は、TMSピークに対する100万分の1(ppm)で記載する。エレクトロスプレーイオン化(ESI)質量分析は、3−Tesla Finnigan FTMS−2000 フーリエ変換質量分析器で実行した。元素分析値は、計算値の±0.4%以内であった。
【0066】
シリカゲル(230−400メッシュ)を用いて、フラッシュカラムクロマトグラフィーを行った。ωアミノ酸メチルエステルを、メタノール/TMSClを使用して市販の酸から調製し、(2−アミノ−フェニル)カルバミン酸ベンジルエステルを、標準的な手順に従ってο−フェニレンジアミンおよびギ酸ベンジルクロリドから合成した。ウサギ抗アセチルヒストンH3ポリクローナル抗体およびウサギ抗アセチルヒストンH4ポリクローナル抗体は、Upstate Biotechnology(Lake Placid,
NY)から購入し、ウサギ抗p21抗体はSanta Cruz Biotechnology(Santa Cruz,CA)から購入した。マウス抗アクチンモノクローナル抗体は、ICN Biomedicals(Irvine,CA)製であった。HRP結合対化ヤギ抗ウサギIgGおよびHRP結合体化ヤギ抗マウスIgGは、Jackson ImmunoResearch(West Grove,PA)から購入した。
【0067】
化合物1〜8および11〜22は、以下に記載する方法(図6、スキーム1A)に従って合成し、化合物9および10は、3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリル酸から、それぞれ方法fおよび方法g(図6、スキーム1B)によって調製した。これらは表題化合物の下に別々に記載している。
【0068】
方法a 塩酸[1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(EDC)カップリング]
個々の短鎖脂肪酸の無水THF(5〜10 mmol/mL)溶液に、N2下で、様々なωアミノ酸メチルエステル(1当量)を加え、続いてEDC(1.3当量)を加えた。一晩撹拌した後、減圧下でTHFを除去し、残渣を酢酸エチル(100mL)に溶解した。この混合物を、水(50mL)で2回、さらに飽和食塩水(50mL)で続けて洗浄した。有機相をNa2SO4上で乾燥し、真空下で濃縮した。生じた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0069】
方法b(エステル開裂)
方法aから得られたエステルを2M KOH/MeOH溶液に溶解した。この混合物を80℃で1時間撹拌して0℃まで冷却した。さらに2N HClでpH 3まで酸性化し、真空下で濃縮して、酢酸エチル(100mL)およびH2O(50mL)を加えた。この有機相を分離して、それぞれ50mLの水および飽和食塩水で続けて洗浄した。さらにNa2SO4上で乾燥し、真空下で濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0070】
方法c [bis(2−オキソ−3−オキザゾリジニル)ホスホルジアミド(BOP−Cl)カップリング]
方法bから得られる酸の無水THF(5〜10mmol/mL)溶液に、N2下で、トリエチルアミン(TEA、1当量)を加えた。この混合物を室温で10分間撹拌し、BOP−Cl(1.1当量)、O−ベンジルヒドロキシルアミン塩酸(1当量)、およびTEA(3当量)を加えた。室温で一晩撹拌した後、この溶液を真空下で濃縮し、酢酸エチル(100mL)を加え、次に3%NaHCO3(50mL)を加えた。この有機相を分離して、それぞれ50mLの水および飽和食塩水で続けて洗浄した。さらにNa2SO4上で乾燥し、真空下で濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0071】
方法d(EDCカップリング)
方法bから得られる個々の酸の無水THF(5〜10mmol/mL)溶液に、N2下で、(2−アミノフェニル)カルバミン酸ベンジルエステル(1当量)を加え、次にEDC(1.3当量)を加えた。一晩撹拌した後、この混合物を真空下で濃縮し、酢酸エチル(100mL)を加えた。有機相を水(50mL)で2回、次に飽和食塩水(50mL)で続けて洗浄し、さらにNa2SO4上で乾燥して濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0072】
方法e(水素化分解)
方法cまたは方法dから得られたN−ベンジルオキシもしくはN−Cbz誘導体を、メタノール/THF(5〜10mmol/mL)1:1溶液に溶解し、木炭上10%パラジウム(10% w/w)を加えた。この混合物を大気圧下で2時間、水素で処理し、ろ過した。溶媒をエバポレートし、残渣を酢酸エチルで再結晶化した。
【0073】
N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド(1)
【0074】
【化10−1】
【0075】
【化10−2】
N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド(2)
【0076】
【化10−3】
N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド(3)
【0077】
【化10−4】
N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド(4)
【0078】
【化10−5】
2−プロピル−ペンタン酸{4−[(2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド(5)
【0079】
【化10−6】
2−プロピル−ペンタン酸(4−ヒドロキシフェニルカルバモイルメチル−フェニル)−アミド(6)
【0080】
【化10−7】
【0081】
【化10−8】
2−プロピル−ペンタン酸{4−[2−(2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド(7)
【0082】
【化10−9】
2−プロピル−ペンタン酸[4−(2−ヒドロキシフェニルカルバモイル)−エチル)−フェニル]−アミド(8)
【0083】
【化10−10】
3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸
化合物9および10の前駆体であるこの化合物は、2−プロピル−ペンタン酸(0.78mL、4.9mmol)および3−(4−アミノ−フェニル)−アクリル酸メチルエステル(0.86g、4.9mmol)から、前述の方法aおよび方法bに従って合成した。全収量、1.05g(2工程で70%);
【0084】
【化10−11】
N−(2−アミノ−フェニル)−3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリルアミド(9)
3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸(200mg、0.7mmol)の無水THF溶液に、N2下で、ベンゼン−1,2−ジアミン(450mg、4.2mmol)を、次にEDC(180mg、0.9mmol)を加えた。一晩撹拌した後、この混合物を真空下で濃縮し、酢酸エチル(50mL)を加え、水(30mL)で2回、そして飽和食塩水(30mL)で続けて洗浄した。有機相をNa2SO4上で乾燥し、真空下で濃縮した。この粗生成物をフラッシュクロマトグラフィー(酢酸エチル−ヘキサン、1:1)によって精製し、白色の固体として化合物9(200mg、収量76%)を得た。
【0085】
【化10−12】
N−ヒドロキシ−3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリルアミド(10)
3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリル酸(100mg、0.35mmol)の無水DMF(3mL)溶液に、窒素下で、EDC(79mg、0.53mmol)およびヒドロキシベンゾトリアゾール水和物(HOBT)(62mg、0.46mmol)を加えた。この混合物を1時間撹拌し、塩酸ヒドロキシルアミン(27.4mg、0.39mmol)およびTEA(54μL)を加え、さらに12時間撹拌し、真空下で濃縮した。さらに、酢酸エチル(40mL)および飽和NaHCO3溶液(15mL)を加えた。有機相を分離し、それぞれ20mLの水および飽和食塩水で続けて洗浄した。この有機相をNa2SO4上で乾燥し、真空下で濃縮した。この粗生成物をフラッシュクロマトグラフィー[酢酸エチル−MeOH(9:1)]によって精製し、白色固体の化合物10(45mg、収量40%)を得た。
【0086】
【化10−13】
N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド(11)
【0087】
【化10−14】
N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド(12)
【0088】
【化10−15】
N−(2−アミノ−フェニル)−4−[(4−フェニルブチリルアミノ)−メチル]−ベンズアミド(13)
【0089】
【化10−16】
4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド(14)
【0090】
【化10−17】
N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド(15)
【0091】
【化10−18】
【0092】
【化10−19】
N−ヒドロキシ−4−[(4−フェニルブチリルアミノ)−メチル]−ベンズアミド(16)
【0093】
【化10−20】
4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド(17)
【0094】
【化10−21】
N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド(18)
【0095】
【化10−22】
N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド(19)
【0096】
【化10−23】
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド(20)
【0097】
【化10−24】
【0098】
【化10−25】
N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド(21)
【0099】
【化10−26】
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド(22)
【0100】
【化10−27】
さらに別の化合物23〜67を含む、全ての化合物の一覧を図6に示す。
【0101】
(インビトロHDACアッセイ)
ヒストン脱アセチル化酵素アッセイキット(Upstate Biotechnology,Lake Placid,NY)を使用して、製造者による使用説明書にわずかな改変を加えてこれに従い、HDAC活性を測定した。このアッセイは、ヒストン脱アセチル化酵素活性に富むDU−145核抽出物が、ストレプトアビジンアガロースビーズに結合させたビオチン化[3H]−アセチルヒストンH4ペプチドの脱アセチル化を媒介する能力に基づく。上清への遊離した[3H]−アセテートの放出を測定し、HDAC活性を計算した。ナトリウムブチレート(0.25〜1mM)を陽性コントロールとして使用した。
【0102】
(細胞生存率アッセイ)
各ウェル当たり4,000個のDU−145細胞を播種した96ウェルの平底プレートにて、細胞生存率に対するHTPBの影響を、MTT{[3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニル−2H−臭化テトラゾリウム]}アッセイによって分析した。細胞を、5%CO2下37℃で、記載した時間、10% FBS添加RPMI−1640培地に記載した濃度にて溶解したHTPBに曝露した。この培地を取り除いて、RPMI−1640培地に溶解した0.5mg/ml MTT 150μlに置き換え、細胞を、37℃で2時間、CO2インキュベーターにてインキュベートした。ウェルから上清を除去し、還元されたMTT色素を200μl/ウェルのDMSOで可溶化した。プレートリーダーで、570nmにて吸光度を測定した。各処理は6ウェルずつ繰り返した。
【0103】
(酵素結合免疫吸着測定法(ELISA)によるアポトーシスの検出)
Cell Death ELISA(Roche Diagnostics,Manheim,Germany)を製造者の使用説明書に従って使用して、アポトーシスの誘導を評価した。この試験は、アポトーシス性細胞死誘導後の、モノヌクレオソームおよびオリゴヌクレオソーム形態の細胞質性ヒストン結合DNAフラグメントの定量的測定に基づいている。簡潔に述べると、実験の24時間前に1×106のDU−145細胞をT−75フラスコ内で培養した。細胞を、10% FBS添加RPMI−1640培地に、記載した濃度のHTPBで処理した。浮遊細胞および吸着細胞の双方を回収し、2×103個の細胞に相当する細胞溶解物をELISAに使用した。
【0104】
(ウェエスタンブロット分析)
10% FBS添加RPMI−1640培地中で、記載した濃度のHTPBで24時間処理したDU−145細胞(1×106個)を回収し、超音波処理した。Bradfordタンパク質アッセイキット(Bio−Rad,Hercules,CA)を使用して、この溶解物のタンパク質濃度を測定した;各溶解物由来の等量のタンパク質を10% SDSポリアクリルアミドゲルに溶解し、次いで半乾式トランスファーセルにてイモビロン−ニトロセルロース膜(Millipore,Bellerica,MA)上へ移した。0.1%のTWEEN20含有トリス緩衝化生理食塩水(TBS)(TBST)で、このトランスブロットした膜を2回洗浄した。5%ノンファットミルク含有TBSTで40分間ブロッキングした後、この膜をTBST−1%ノンファットミルクに溶解した一次抗体(1:1000希釈)と共に4℃で一晩インキュベートした。一次抗体で処理した後、この膜を計15分間、TBSTで3回洗浄し、続いてヤギ抗ウサギ、もしくは抗マウスIgG西洋ワサビペルオキシダーゼ結合体(1:3000稀釈)を室温で1時間処理し、さらにTBSTで3回、計1時間洗浄した。この免疫ブロットを高感度化学発光によって可視化した。
【0105】
(結果)
第一のシリーズの化合物に関して、本発明者らは、スキーム1(A、化合物1〜8;B、化合物9および10)にて記載した手順に従って、Zn2+係留結合体(図1)を合成する先導化合物としてバルプロ酸を用いた。化合物1〜8に関して、EDC活性化を介して、バルプロ酸を4つの異なるωアミノ酸メチルエステルスペーサーと結合させた。得られたエステルを、アルカリ加水分解により酸に分解した。代表的なペプチドカップリング条件(BOPClまたはEDC)の下で、得られた酸を、Bn保護ヒドロキシアミンおよびCbz保護ο‐フェニレンジアミンで処理し、水素化分解後に、それぞれアニリドおよびヒドロキサム酸を形成した(スキーム1A)。化合物9および10は、3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸を、それぞれο‐フェニレンジアミンおよびヒドロキシルアミンと、代表的なEDCカップリング条件下で直接カップリングさせることによって合成した(スキーム1B)。
【0106】
(Zn2+キレートモチーフ係留バルプロエート誘導)
バルプロ酸を5種類の芳香族リンカーと、続いて2つのZn2+結合モチーフと様々に結合体化することによって、化合物1〜10を得た。これらの係留結合体は、様々な程度のHDAC阻害効力を示し(図2)、そのIC50値は5μM(化合物8)から80μM(化合物5)の範囲にわたった。この抑効力は、親分子(IC50、0.4mM)の8倍から5倍改善された。これらの結合体のうちのいずれかからバルプロイル部分もしくはZn2+キレートモチーフを除去すると、HDAC阻害活性は完全に失われ(データは示していない)、このことはタンパク質‐リガンド相互作用において、アシル基およびZn2+キレートモチーフが重要であることを示している。
【0107】
試験した10個の係留結合体の中で、化合物1、2、4および8は最適な誘導体(IC50、5〜8μM)であり、3、9、および10がこれに次ぐ(IC50、10〜20μM)。相対的に、ヒドロキサマト(化合物2、4、6、8および10)は、一般に、対応するフェニレンジアミン(化合物1、3、5、7および9)よりも作用効力が強かった。さらに、芳香族リンカーは、HDAC阻害活性に対して微小な影響を示した。試験した5つの芳香族ωアミノ酸の中で、(4−アミノフェニル)アセテートは、HDAC阻害効力が最も弱い結合体(5および6)となったが、4−(アミノメチル)ベンゾエートは最適であるようであった。さらなる構造的改変については、化合物1、2、4および8が、全てIC50<10μMを示したので、これらを先導化合物として使用した。
【0108】
(構造的改変)
バルプロイル基を除去すると、この結合体の阻害活性が完全に失われるという発見によって、活性部位ポケットと相互作用する際のアシル部分の重要性が強調された。従って、本発明者らは、化合物1、2、4および8のバルプロイル基を、ブチリル基、フェニルアセチル基またはフェニルブチリル基に置換して、HDAC阻害に対する立体電子効果を増大させた(図3)。これら全ての誘導体は、バルプロイル対応物と比較して、向上したHDAC阻害作用効力を示した。試験した様々なアシル官能基の中で、同一のスペーサーおよびZn2+キレートモチーフと結合させた場合、相対的効力は、フェニルブチリル基>フェニルアセチル基>ブチリル基>バルプロイル基の順であった。
【0109】
これらの12個の誘導体の中で、化合物19は特に注目すべきものであった。このヒドロキサマト係留フェニルブチレート(HTPB)(すなわち、化合物19)は、44nMというIC50を示し、これはフェニルブチレートに対して強度4倍の向上である。この化合物を使用して、DU−145前立腺癌細胞におけるHDAC活性に対する影響を調べた。
【0110】
(DU−145前立腺癌細胞におけるヒストンアセチル化およびp21WAF/CIP1発現に対するHTPBの影響)
ヒストンの過アセチル化(hyperacetylation)およびサイクリン依存性リン酸化酵素阻害因子であるp21WAF/CIP1の発現増加は、細胞内HDAC阻害に関する二つの特性である(Marks et al.,Nat Rev Cancer 1:194−202(2001))。従って、本発明者らは、ヒストンアセチル化およびp21WAF/CIP1発現の状態の特性を明らかにすることによって、DU−145前立腺癌細胞におけるHDAC活性に対するHTPBの影響を、TSAおよびフェニルブチレートと比較して試験した。
【0111】
DU−145細胞を、10%FBS添加RPMI 1640培地中で24時間、0.5、1、2.5μMのHTPB、0.25および0.5μMのTSA、または1、2.5、および5mMのフェニルブチレートに曝露した。細胞溶解物のウエスタンブロット分析は、これらの薬剤の処理によってアセチル化ヒストンH3およびH4、ならびにp21WAF/CIP1のレベルが上昇したことを示している(図4)。これらのバイオマーカーに対する1μM HTPBの影響は、0.25μM TSA、もしくは2.5mMフェニルブチレートの影響に近かった。DU−145細胞は、少量だが有意な量の内在性p21WAF/CIP1を示し、そのレベルは、0.5μMという低濃度のHTPBへ曝露した後に、実質的に増加した。まとめると、これらのデータによって、HTPBはDU−145細胞にてHDAC活性を標的としていることを確認した。
【0112】
(DU−145細胞の生存率に対するHTPBの影響)
10%FBS添加RPMI 1640培地中のDU−145細胞において、癌細胞生存率に対するHTPBの影響を調べた。これらの細胞は、HTPBに対して高度の感受性を示し、増殖阻害におけるIC50は、約0.5μMと1μMの間であった(図5A)。DNAフラグメント化によって証明されたように、HTPBは用量依存的様式でDU−145細胞のアポトーシスに対する感受性を増加させた(図5B)。示したように、薬物の濃度が1μMを超えたときに、24時間にて広範囲のアポトーシスが発生し、これは少なくとも部分的には、この細胞毒性効果がHDAC阻害によってアポトーシスが誘導されたことに起因することを示している。試験した他の細胞株としては、AN3CA子宮内膜癌細胞、ならびにSW−48およびHCT−15結腸直腸癌細胞が挙げられる。これらの癌細胞もまた、同程度の効力でHTPBの細胞毒性効果に対して感受性であった(データは示さず)。
【0113】
(考察)
本明細書中で、本発明者らは、新しい種類のHDAC阻害剤の開発を示し、この阻害剤において、短鎖脂肪酸が疎水性結合を介してZn2+キレートモチーフに係留された。TSAおよびSAHAによるHDAC阻害の独特な様式により提供された作用モデルについて、これらの化合物を開発する際の本発明者らのストラテジーが立てられた(Finnin et al.,Nature 401:188−193(1999))。本発明者らの新しい係留ストラテジーからHTPB(すなわち化合物19)の発見が導かれ、これはSAHAについて報告された濃度(Richon et al.,Proc Natl Acad Sci USA 95:3003−3007(1998))に一致する準μM濃度で、HDAC阻害活性および抗増殖活性を示す。
【0114】
本発明者らは、いくつかの癌細胞株において、HTPBがHDAC活性を標的とする2つの系列の証拠を得た。具体的には、DU−145前立腺癌細胞を、0.5μMもの低濃度でHTPB処理すると、用量依存的な様式でヒストンH3並びにH4の過アセチル化が引き起こされた。同様に、HTPBに応答して、p21WAF/CIP1発現が実質的に上方制御された。対照的に、ヒストンアセチル化およびp21WAF/CIP1発現に対する同様の細胞内効果を達成するのに、親分子であるフェニルブチレートは、少なくとも2.5mMを必要とした。
【0115】
HTPBは、構造的に既存のHDAC阻害剤と区別され、その多くは、キャップ基が極性平面構造から構成されている。例えば、TSA、SAHAおよびMS−275のキャップ基は、それぞれジメチルアミノフェニル基、フェニルアミノ基およびピリジン−3−イル−メトキシカルボニル基を含有する。従って、活性部位ポケットは、キャップ基に異なる立体電子特性を与える高度な柔軟性を示すと、本発明者らはさらに結論づけた。本発明者らのデータは、結合体の活性部位ポケットへの結合促進において、フェニルブチリル基およびフェニルアセチル基は脂肪族アシル部分より効果的であったことを示している。この矛盾は、部分的に、電子密度の違いおよび/または分岐した側鎖による立体化学的障害に起因すると推測される。芳香族リンカーに関しては、4−アミノ安息香酸が、フェニルブチリル基とヒドロキサマトとを係留するのに最適であるようであり、その長さはポケットの両端に接触するのに十分なものであった。
【0116】
(第二世代HDAC阻害剤の設計および合成−化合物19の構造に基づく最適化)
化合物19(HTPB)は、構造的に既存のHDAC阻害剤とは区別され、多くは例えばTSA、ジメチルアミノフェニル;SAHA、フェニルアミノ;およびMS−27−275、ピリジン−3−イル−メトキシカルボニルのような、極性の平面構造から成るキャップ基を有する。この知見は、HDAC活性部位ポケットがキャップ基に異なる立体電子特性を与える、高度な柔軟性を示していることを示唆する。
【0117】
リガンド結合を予測するため、本発明者らは、化合物19(図9、赤字)およびTSA(図9、黄字)の、HDLP活性部位ポケットへの分子ドッキングを行い、その後個々のリガンドの認識様式を比較するためにエネルギーを最小化した。図9に示したように、双方のリガンドともポケットへの結合は類似した配置をとっている。化合物19の芳香族リンカーである4−アミノ安息香酸は、フェニルブチリル基とヒドロキサマトを係留するのに最適な長さを提供し、これによって双方の官能基はポケットの両端に接触することを可能にする。ヒドロキサム酸基[C(O)NH−OH]によって、化合物19のHDAC阻害効力はフェニルブチレートの4桁の大きさの増加に寄与するようである。
【0118】
図9のモデリングデータは、リガンド結合におけるヒドロキサマトの役割に2要素があることを示唆している。1つは、Zn2+陽イオンをキレートする。2つ目は、ヒドロキサマトは、His131−Asp173電荷リレー系の助けと共に、NH−OHからHis−131のNτへのプロトン移動を促進する(図10)。(TSAのリガンド結合研究については、Vanommeslaeghe,K.,Van Alsenoy,C.,De Proft,F.,Martins,J.C.,Tourwe,D.,and Geerlings,P.Ab initio study of the binding
of Trichostatin A(TSA) in the active site of histone deacetylase like protein(HDLP).Org Biomol Chem,1:2951−2957,2003を参照する。)
結論として、ヒドロキサマトHDAC阻害剤である、TSAあるいは化合物19のいずれかの結合によってAspからヒドロキサマトへの負電荷の転移が生じ、その結果、負荷電ヒドロキサマトとZn2+の正電荷およびイミダゾール環との間に塩架橋が形成される(図10、中央パネル)。機構的に、この塩架橋は、ヒドロキサマトベースのリガンドと活性部位ポケットとの結合に寄与する主要な力となっており、フェニルブチレートから化合物19への転換に伴う、HDAC阻害効力の104倍の増加に必要とされる微分自由エネルギー変化(ΔΔG‡=5.4kcal/mol)の根底となっている(IC50、0.4mMに対し44nM)。
【0119】
結合親和力におけるこの塩架橋の役割は、化合物19のヒドロキサマト部分をフェニレンジアミン基に置換した(化合物55)ときにHDAC阻害効果が10倍低下する(すなわち、IC50、0.044μMに対して0.4μM)ことによって、さらに強調される。ヒドロキサマトとは対照的に、フェニレンジアミン基の活性部位への結合には、His131のNτへのプロトン転移が関係しない。代わりに、His131との荷電‐荷電相互作用を形成することなく、電子に富むジアミンのみがZn2+陽イオンをキレートする。その結果、フェニレンジアミンベース(例えば、化合物55)のリガンドとの結合親和性は、対応するヒドロキサマトと比較して著しく低下する。
【0120】
この分子ドッキングはまた、化合物19の次なる改変のための有用な指針を提供した。図9に示したように、TSAおよび化合物19のキャップ基は、Tyr91、Glu92、Gly140、およびPhe141に囲まれた溝近くに位置する。本来この溝は、様々な程度のかさ高さを有するキャップ基を適合させる柔軟性を提供し、HDAC阻害効力を増大させるのに活用され得る。
【0121】
従って、本発明者らは、化合物19のフェニルブチリル部分を、様々なα分岐芳香族脂肪族アシル基に置換した。その原理は、2つであった。第1に、フェニルブチリル基と化合物19内のリンカーとの間のアミド結合が、タンパク質分解性消化に感受性であり得る。アシル基の嵩高さが増加することによって、アミド結合がより立体的に妨げられたものに変わることにより、代謝安定性が増大し得る。第2に、アシル基の大きさが増したことで、前記溝との疎水結合が増大し、これによって結合親和力が増し得る。
【0122】
このストラテジーから、化合物19(すなわち、HTPB)よりも大きな作用効力を有する、多数のHDAC阻害剤が導かれた。いくつかの代表的な誘導体の構造およびIC50を表1にまとめる。
【0123】
表1 代表的なフェニルブチレートベースのHDAC阻害剤
【0124】
【表1】
合成した80を超える誘導体の中で、化合物42がTSAに匹敵するIC50を持つ最適な薬剤である。分子モデリング解析によって、化合物42は、活性部位ポケット内側でTSAの原子配置と並立する配置をとっていることが示されている(図11を参照のこと)。さらに、イソプロピル部分が溝の内側にあり、隣接する疎水性残基と相互作用し得る。それゆえ、さらなるインビトロおよびインビボでの特徴付けのために、化合物42を選択した。
【0125】
(化合物42およびTSAは、抗増殖効果を後成的かつ細胞レベルで媒介する−新規の細胞標的の同定)
PC−3アンドロゲン非依存性前立腺癌細胞にて、p21WAF/CIP1発現およびヒストンH4過アセチル化に対する影響についての試験に、化合物42、SAHA、およびTSAを供した。図12は、これらのバイオマーカーの誘導における化合物42の効力がTSAに匹敵し、SAHAよりも約5倍高いことを示している。
【0126】
PC−3細胞(PTEN−/−)におけるAkt活性化状態に対するこれらの薬剤の影響もまた、試験した。Aktは、アンドロゲン非依存性およびこれらの細胞の化学治療耐性に寄与する機能性PTENが存在しないため、PC−3細胞にて構造的に活性化されている。PC−3細胞にて、化合物42とTSAのいずれもが、1μMもの低濃度で著しいAkt脱リン酸化を引き起こしたことに注目すべきである(図13)。対照的に、リン酸化Aktに対するSAHAの影響は認められなかった。これはSAHAと化合物42/TSAとの間には、作用様式に僅かな差異があることを示唆している。
【0127】
PC−3細胞におけるリン酸化Aktに対する化合物42の影響は、多くがPTEN変異を示すホルモン不応性前立腺癌細胞におけるこのHDAC阻害剤の臨床投与の点から注目に値する。しかし、この脱リン酸化効果はリン酸化酵素特異的なものである。試験した他の一連のシグナル伝達リン酸化酵素の中で、FAK(局所接着リン酸化酵素)およびERK類のリン酸化レベルは投与量依存的に減少したが、p38あるいはJNKのリン酸化レベルは影響を受けないままであった(図14)。
【0128】
本発明者らは、HDACアイソザイムと複合体を形成してその核への局在化を促進するプロテインホスファターゼ1(PP1)により、この脱リン酸化が仲介されると仮説を立てる。本発明者らは、PC−3細胞を化合物42あるいはTSAで処理すると核内でHDAC−PP1複合体の分解が起こり、PP1の細胞質内への再局在を誘導して標的リン酸化酵素の脱リン酸化を媒介することを提唱する。あるいは、この効果は熱ショックタンパク質(HSP)−90のアセチル化に起因し、その結果Aktへの結合性が減少してその後Aktが分解されるとも考えられる(Fuino,L.,Bali,P.,Wittmann,S.,Donapaty,S.,Guo,F.,Yamaguchi,H.,Wang,H.G.,Atadja,P.,and Bhalla,K.Histone deacetylase inhibitor LAQ824 down−regulates Her−2 and sensitizes human breast cancer cells to trastuzumab,taxotere,gemcitabine,and epothilone B.Mol Cancer Ther,2:971−984,2003)。
【0129】
(前立腺癌に対する化合物42の抗腫瘍効果のインビボでの特徴付け)
定着したPC−3異種移植片腫瘍の増殖に対する経口投与した化合物42(1日当たり50mg/kgおよび100mg/kg)の影響を、腹腔内注射したSAHA(1日当たり50g/kg)と比較して調べた。さらに本発明者らは、化合物42のHDAC阻害効力に匹敵する阻害効力を持つという点から、化合物44(1日当たり50mg/kgおよび100mg/kg)も取り入れた(IC50、32nMに対し25nM)。化合物42と44の双方とも、用量依存的に、10%FBS含有培地にてPC−3細胞の増殖をインビボで抑制するのに有効であり、IC50値はそれぞれ0.6μMと0.8μMであった(図15、72時間処理)。
【0130】
図16は、上記のようにHDAC阻害剤を処理した胸腺欠損マウスにおける皮下PC−3異種移植片腫瘍の増殖を表す(パネルA、平均腫瘍容積±SDとして表したデータ;パネルB、ビヒクル、化合物44並びに化合物42で処理したグループの代表的なマウスの写真)。示しているように、化合物42と44のいずれも、いずれかの投与量で、経口経路を介して、定着したPC−3腫瘍の増殖を抑制するのに有効であった。1日当たり50mg/kgで経口的に投与した化合物42のインビボでの効力は、同じ量を腹腔内投与したSAHAの効力に匹敵し、腫瘍の増殖を、ビヒクルで処理したコントロールグループと比較して、それぞれ70.4%並びに69.0%減少させた。1日当たり100mg/kgで、化合物42はPC−3腫瘍の増殖をほぼ完全に抑制し(91.7%の減少)、体重の継続測定並びに病理的検査によって確認したように明白な副作用もみられなかった。
【0131】
本研究の結論として、Ohio State University College
of Veterinary Medicine(オハイオ州立大獣医学部)の、専門医師会認定の獣医病理学者による、各処理群の1匹のマウスの完全病理検査を実施した。この検査は、これは臨床病理学(血液学、血清化学)ならびに完全剖検(27種類以上の組織および器官の肉眼および顕微鏡検査)を包含した。剖検で観察された肉眼的病理学的異常は、SAHA処理マウス、およびDMSO処理コントロールマウスの腹膜内の小さな接着に限られており、これは毎日腹腔内注射を行った副作用のようである。血液パラメーターは、好中球の増加を除いて正常範囲内であり、これはビヒクル処理コントロールマウス群、ならびにSAHAおよび化合物44(50mg/kg)処理群の双方のマウスのみに観察された。血清化学値は、ビヒクル処理マウスでは脱水症状に一致する値となったが、他の全てのマウスでは正常範囲内であった。
【0132】
生物学的応答とインビトロで同定された提唱される作用機構とを相関させるため、経口投与した化合物42および腹腔内投与したSAHAが、PC−3異種移植片腫瘍内のヒストンH−3のアセチル化を調節する作用を、免疫ブロットによって分析した。図17は、ビヒクル、1日当たり50もしくは100mg/kgの経口化合物42、あるいは1日当たり50mg/kgの腹腔内SAHAで28日間処理した腫瘍保有マウス由来の、2つの代表的なPC−3腫瘍のホモジネート内のヒストンH3とアセチル化H3のウエスタンブロットを、様々な容量で表す。薬剤処理グループにおいて、インビボHDAC阻害の特性である、ヒストンH3のアセチル化レベルの著しい増加が認められた。
【0133】
インビボ実験にて、等量の血清を含まない培地とマトリゲル基底膜マトリックスとに懸濁したPC−3細胞を、5〜7週齢のオスNCr胸腺欠損ヌードマウス(nu/nu)の脇腹に皮下注射した(0.5×106 細胞/0.1ml/マウス)。腫瘍容積が170〜200mm3に達したときに、強制経口投与によって化合物42もしくは44を(1日当たり50および100mg/kg)、あるいは腹腔内注射によってSAHA(1日当たり50mg/kg)を毎日処理することを開始した(各グループにつきN=6)。コントロールグループには、経口あるいは腹腔内経由でビヒクルのみ(それぞれ0.1%メチルセルロース/0.05%TWEEN80水溶液、およびDMSO)を与えた。処理は、コントロール群の平均腫瘍容積が約1,500mm3に達するまで継続した。ノギスを使用して腫瘍を毎週測定し、標準公式:(幅)2×(長さ)×0.52を用いて腫瘍の容積を算出した。体重を毎週測定し、実験期間中一定であった。
【0134】
乳癌細胞株、肺癌細胞株、および甲状腺癌細胞株における、化合物42のインビトロ抗増殖効果。NCI開発途上治療プログラム(DPT)による60細胞株スクリーニングによると、化合物42は、60細胞株全てに対し、平均GI50(細胞増殖の50%抑制)値が0.2μMという強力なインビトロ抗増殖活性を示した。さらに、本発明者らは、甲状腺癌細胞株において化合物42を試験した。代表的な乳癌細胞株、肺癌細胞株、および甲状腺癌細胞株における化合物42の用量依存的抗増殖効果を、図18に示す。
【0135】
(慢性リンパ球性白血病(CLL)における化合物42のインビトロおよびインビボ抗増殖効果)
HDAC抑制能並びに細胞毒性促進能について、化合物42を初代CLL細胞にて試験した。図19に示したように、HDACが抑制される1μM以上の濃度で、著しい細胞毒性が認められた(N=6)。さらに、TLC−1遺伝子移入マウスモデルを使用したインビボ予備試験によって、経口胃管注入によって100mg/kgの化合物42を4週間、5日投与/2日非投与し、生存率が向上する傾向と顕著な毒性がないことが示された。
【0136】
(立体選択性)
驚くべきことに、化合物42のHDAC阻害活性が立体選択的であることを発見した。S異性体は、R異性体対応物よりも強力である(IC50、15nMに対し80nM)。
【0137】
まとめると、分子モデリングとコンビナトリアル化学技術とを組み合わせることにより、短鎖脂肪酸を骨格として使用して新たな種類の強力なHDAC阻害剤を開発した。最適な薬剤である化合物42(NSC−D−731438)は、25nMのIC50でHDAC活性を阻害し、これは親化合物フェニルブチレートの10,000倍以上の増大である。化合物42は、これが臨床へ持ち込まれる有望な候補にする理由となるいくつかの独特の特性を示している。第一に、化合物42は、後生的機構および細胞性機構の両方を介して、インビトロで抗増殖効果を媒介する。この特性はTSAと同様であるが、SAHAにはないものである。第二に、これは、MS−275およびSAHAよりも優れたインビトロおよび/またはインビボでの効力を有しながら経口的に生物学的利用可能である。第三に、1日当たり50あるいは100mg/kgの投与量で28日間の過程を終えても、腫瘍保有マウスにて明らかな毒性を示さない。このことは、長期投与および他の標的治療との組み合わせのための機会を与える。最後に、化合物42は単純な構造を有し、大規模な合成に適する。
【0138】
(化合物42の生成)
本発明者らは、数グラム単位で多数回、化合物42を合成した。基本的に、4段階合成を経て調製し、全体の収量は52%であった(図20)。出発物質は全て容易に入手できるものであり、この合成は実験室の設定にて数百グラム単位のスケールアップが容易である。その手順を以下に記載する。中間生成物および最終産物は、クロマトグラフィーによる分離を用いなくとも99%以上の純度で、結晶化によって反応混合物から単離され得る。全体として、合成および精製の手順は単純明快であり、この化合物の大規模生産に関して特別な問題点は無い。
【0139】
N−(4−アセチル−フェニル−3−メチル−2−フェニル−ブチルアミド(1))。(α−イソプロピル)−フェニル酢酸(4g、22.3mmol)を、塩化チオニル(30ml)に溶解し、50℃で1時間加熱した。溶媒を除去した後、残渣をTHF(50ml)に溶解し、p−アミノ安息香酸メチルエステル(3.4g、22.5mmol)およびEt3N(3.5ml、25mmol)のTHF(50ml)溶液を、室温で撹拌しながら加えた。4時間後、真空下でTHFを除去して酢酸エチル(200ml)に溶解し、水(100ml)と食塩水(100ml)で続けて洗浄した。Na2SO4上で有機相を乾燥し、真空下で濃縮して化合物1を得た(5.6g;収量85%)。その後精製することなく、この粗生成物を次の工程に使用した。
【0140】
4−(3−メチル−2−フェニル−ブチリルアミノ)安息香酸(2)。化合物1(5.6g;20mmol)を、KOH(21g)を含有するメタノール(150ml)に溶解した。この混合物を2時間還流して0℃に冷却し、12N HCl(12N)を滴下して生成物を沈殿させた。真空下で溶媒を除去し、冷水(120ml)を加えた。ろ過によって沈殿物を回収し、水で洗浄して乾燥し、化合物2を得た(5g;収量84%)
N−ベンジルオキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド(3)。N2下で、2(5g;16.8mmol)の無水THF(120ml)溶液に、トリエチルアミン(TEA、2.5ml;16.8mmol)を加えた。この混合物を室温で10分間撹拌し、bis(2−オキソ−3−オキサゾリジニル)塩化ホスホルジアミド(BOP−Cl)(4.7g;18.7mmol)、塩酸O−ベンジルヒドロキシアミン(2.7g;17mmol)、およびTEA(7.5ml)を加えた。室温で一晩撹拌後、この溶液を真空下で濃縮し、酢酸エチル(200ml)、次いで3%NaHCO3(80ml)を加えた。有機相を分離して、それぞれ100mlの水および飽和食塩水で続けて洗浄し、Na2SO4上で乾燥して真空下で濃縮した。この残渣(6.1g、収量90%)をその後精製することなく、直接水素化分解に使用した。
【0141】
N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド(4)。化合物3(6.1g;15.2mmol)をメタノール/THFの1:1溶液(120ml)に混合し、木炭上の10%パラジウム(0.6g、10% w/w)を加えた。この混合物を大気圧下で2時間、水素で処理し、ろ過した。溶媒をエバポレートし、残渣を酢酸エチルで再結晶化した。収量は4.2gであった(収量90%)。
【0142】
本発明に従うHDAC阻害剤の代替的な合成スキームを、図21に図示する。
【0143】
本発明の他の実施形態は、この明細の考察並びにこの明細にて開示されている発明の実践から、当業者には明白であろう。この明細および実施例は、以下の請求項によって示される本発明の真の範囲および本質を伴う例示にすぎないものであるとみなされることが意図される。
【技術分野】
【0001】
(発明の説明)
本発明は、Army Grant DAMD17−02−1−0117およびNational Institute of Health Grant CA94829によって支援された。政府は、本発明において一定の権利を有する。本願は、2002年12月2日に出願された米国仮特許出願60/526,348号に対する優先権を主張する。
【0002】
(発明の分野)
本発明はヒストン脱アセチル化酵素阻害剤に関係し、特にZn2+キレートモチーフを包含するヒストン脱アセチル化酵素阻害剤に関する。より詳細には、本発明は、短鎖脂肪酸を基本とする、Zn2+キレートモチーフを包含するヒストン脱アセチル化酵素阻害剤に関する。
【背景技術】
【0003】
(発明の背景)
コアヒストンのアセチル化状態は、DNAのヌクレオソーム・パッケージングの調節を介して、遺伝子の転写調節において中枢的役割を果たしている(非特許文献1;非特許文献2;非特許文献3)。低アセチル化状態では、ヌクレオソームは緊密に凝縮されており、その結果転写因子の標的DNAへの接触が制限されることに起因して、転写が抑制される。反対に、ヒストンがアセチル化されるとヌクレオソーム構造は緩み、クロマチンは転写許容状態となる。どちらも配列特異的転写活性因子との複合体として標的遺伝子へ動員される、ヒストンアセチル基転移酵素(HAT)の活性とヒストン脱アセチル化酵素(HDAC)の活性との間の動力学的なバランスが、この翻訳後修飾のレベルを維持している。この後成的なマーキング(marking)システムの調節異常が、多くの形態の癌の主要な病因である不適切な遺伝子発現を引き起こすことが示されている(非特許文献4;非特許文献5;非特許文献6)。さらに、多くの種類の腫瘍細胞でHDACを阻害すると、少数の遺伝子の転写が再活性化されることによって、成長停止、分化および/またはアポトーシスが誘引されることを例証する多くの証拠がある(非特許文献7;非特許文献8;非特許文献9;非特許文献10;非特許文献11)。異種移植片モデルでもまた、これらインビトロでの知見が確認されており、これらのことはHDAC機能の調節が、癌の予防的および/または治療的介入のための標的となることを示唆している。
【0004】
現時点で、構造的に異なる種類のいくつかのHDAC阻害剤が報告されており(非特許文献8;非特許文献9;非特許文献10;非特許文献11)、これとしては短鎖脂肪酸(例えば、ブチレート(butyrate)、バルプロエート(valproate)、フェニルアセテート(phenylacetate)およびフェニルブチレート(phenylbutyrate))(非特許文献12;非特許文献13;非特許文献14;非特許文献15)、ベンズアミド誘導体類(例えばMS−27−275)(非特許文献16;非特許文献17)、トリコスタチンA(TSA)および類似体(非特許文献18;非特許文献19;非特許文献20)、ハイブリッド極性化合物(例えばスベロイルアニリドヒドロキサム酸(SAHA))(非特許文献21;非特許文献22)、環状テトラペプチド類(例えばアピシジン(apicidin))(非特許文献23;非特許文献24;非特許文献25;非特許文献26)、並びにデプシペプチドFR901228(非特許文献26)が挙げられる。これらの薬剤の内、短鎖脂肪酸は、他の種類のHDAC阻害剤のIC50がμMまたはnM単位ですらあることと比較して、IC50がmM単位である最も弱い阻害剤である。短鎖脂肪酸を癌治療に使用することが報告されているが、低い抗増殖活性、速い代謝、および非特異的様式での作用のため、その治療効果は限られている。
【0005】
最近、細菌性のHDAC相同体であるHDLP(ヒストン脱アセチル化酵素様タンパク質)のX線結晶分析から、タンパク質−リガンド相互作用(それによりTSAおよびSAHAが酵素阻害を媒介する)の特異な様式が示唆された(非特許文献27)。HDAC触媒ドメインは、炭素数4から6の直鎖に相当する長さにわたる、細い管状ポケット構造から成ることが明白となっている。この酵素のポケット構造の底部近くにZn2+陽イオンが位置し、これが、2つのHis−Asp電荷伝達系と協同して脱アセチル化触媒を促進していると考えられている。
【0006】
当該分野の他の研究を慎重に考察し、短鎖脂肪酸のHDAC阻害効力が弱いのは、短鎖脂肪酸が、本発明者らが脱アセチル化触媒の中心的役割を果たしていると考えている、活性部位ポケット内のZn2+陽イオンに接近できないことに一部起因すると、本発明者らは理解した。この考察およびさらなる研究を基に、本発明者らは、芳香族リンカーを介してZn2+キレートモチーフに短鎖脂肪酸を係留(tethering)することによって、短鎖脂肪酸を構造的に改変した。本発明者らの発見および研究によって、本発明者らは、新しい種類のZn2+キレートモチーフに係留された短鎖脂肪酸を発明するに至った。これらのうちのいくつかはnMの単位でHDAC活性並びに癌細胞増殖の阻害を示すものもあり、これは親化合物と比較して3桁の作用効力向上である。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Kouzarides,「Histone acetylases and deacetylases in cell proliferation.」Curr Opin Genet Dev(1999)9:40−48
【非特許文献2】Gray and Ekstrom,「The human histone deacetylase family.」Exp Cell Res(2001)262:75−83
【非特許文献3】Jenuwein and Allis,「Translating the histone code.」Science(2001)293:1074−1080
【非特許文献4】Wade,「Transcriptional control at regulatory checkpoints by histone deacetylases:molecular connections between cancer and chromatin.」Hum Mol Genet(2001)10:693−698
【非特許文献5】Cress and Seto,「Histone deacetylases,treanscriptional control,and cancer.」J Cell Physiol(2000)184:1−16
【非特許文献6】Marks et al.,「Histone deacetylases and cancer: causes and thrapies.」Natl Rev Cancer(2001)1:194−202
【非特許文献7】Jung,「Inhibitors of histone deacetylases as new anticancer agents.」Curr Med Chem(2001)8 1505−1511
【非特許文献8】Grozinger and Schreiber,「Deacetylase enzymes:biological functions and the use of small−molecule inhibitors.」Chem Biol(2002)9:3−16
【非特許文献9】Johnstone,「Histone−deacetylase inhibitors:novel drugs for the treatment of cancer.」Nat Rev Drug Discov(2002)1:287−299
【非特許文献10】Kramer et al.,「Histone deacetylase as a thrapeutic target.」Trends Endocrinol Metab(2001)12:294−300
【非特許文献11】Marks et al.,「Histone deacetylase inhibitors:inducers of differentiation or apoptosis of transformed cells」J Natl Cancer Inst(2000)92:1210−1216
【非特許文献12】Lea and Tulsyan,「Discordant effects of butylate anologues on erythroleukemia cell proliferation,differentiation and histone deacetylase.」Anticancer Res(1995)15:879:883
【非特許文献13】Kruh,「Effects of sodium butyrate,a new pharmacological agent,on cells in culture.」Mol Cell Biochem(1982)42:65−82
【非特許文献14】Newmark and Young,「Butyrate and phenylacetate as differentiating agents:practical problems and opportunities.」J Cell Biochem Suppl(1995)22:247−253
【非特許文献15】Phiel et al.,「Histone deacetylase is a direct target of valproic acid,a potent anticonvulsant,mood stabilizer,and teratogen.」J Biol Chem(2001)276:36734−36741
【非特許文献16】Suzuki et al.,「Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives.」J Med Chem(1999)42:3001−3003
【非特許文献17】Saito et al.,「A synthetic inhibitor of histone deacetylase,MS−27−275,with marked in vivo antitumor activity against human tumors.」Proc Natl Acad Sci USA(1999)96:4592−4597
【非特許文献18】Tsuji et al.,「A new antifungal antibiotic,trichostatin.」J Antibiot(Tokyo)(1976)29:1−6
【非特許文献19】Jung et al.「Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation.」J Med Chem(1999)42:4669−4679
【非特許文献20】Furumai et al.「Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin.」Proc Natl Acad Sci USA(2001)98:87−92
【非特許文献21】Richon et al.,「A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases.」Proc Natl Acad Sci USA(1998)95:3003−3007
【非特許文献22】Remiszewski et al.,「Inhibitors of human histone deacetylase:synthesis and enzyme and cellular activity of straight chain hydroxamates.」J Med Chem(2002)45:753−757
【非特許文献23】Kijima et al.,「Trapoxin,an antitumor cyclic tetrapeptide,is an inrreversible inhibitor of mammalian histone deacetylase.」J Biol Chem(1993)268:22429−22435
【非特許文献24】Shute et al.,「Analogues of the cytostatic and antimitogenic agents chlamydocin and HC−toxin:synthesis and biological activity of chloromethyl ketone and diazomethyl ketone functionalized cyclic tetrapeptides.」J Med Chem(1987)30:71−78
【非特許文献25】Han et al.,「Apicidin,a histone deacetylase inhibitor,inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin.」Cancer Res(2000)60:6068−6074
【非特許文献26】Nakajima et al.「FR901228,a potent antitumor antibiotic,is a novel histone deacetylase inhibitor.」Exp Cell Res(1998)241:126−133
【非特許文献27】Finnin et al.,「Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors」Nature(1999)401:188−193
【発明の概要】
【課題を解決するための手段】
【0008】
(本発明の要旨)
本発明者らは、短鎖脂肪酸のヒストン脱アセチル化酵素(HDAC)阻害剤としての効力が弱いのは、脱アセチル化触媒に重要と考えられるHDAC活性部位ポケット内の亜鉛陽イオンに短鎖脂肪酸が接近できないことに一部起因すると理解した。本発明は、例えばバルプロエート、ブチレート、フェニルアセテートおよびフェニルブチレートなどの短鎖脂肪酸を含む脂肪酸の構造的な改変に基づく。本発明は一般に、脂肪酸を、芳香族ωアミノ酸リンカーを介してZn2+キレートモチーフ(ヒドロキサム酸、ο‐フェニレンジアミンが挙げられるが、これらに限定されない)と結合することを含む。新しい種類のZn2+キレートモチーフ係留短鎖脂肪酸を含む本発明は、このストラテジーから導かれた。
【0009】
本発明の化合物のHDAC阻害における効力は、HDLPリガンド複合体の結晶構造から提供されるフレームワークに基づいて、強力なHDAC阻害剤が設計され得ることを示している。本発明は、短鎖脂肪酸、ωアミノ酸、ヒドロキサマト(hydroxamato)などの亜鉛キレート剤の様々な組み合わせにより、化合物の大きなライブラリを創出することを可能にする係留ストラテジーに基づいている。
【0010】
本発明は、以下の式:
【0011】
【化6】
を有するヒストン脱アセチル化酵素阻害剤を含む。ここで:
Xは、HおよびCH3から選択され;
Yは、nが0〜2の(CH2)nであり;
Zは、mが0〜3である(CH2)mおよび(CH)2から選択され;
Aは、ヒドロカルビル基であり;
Bは、ο‐アミノフェニル基またはヒドロキシ基であり;
Qは、ハロゲン、水素、またはメチル基である。
【0012】
一実施例において、Aは脂肪族を含み得、この脂肪族は分岐であり得る。Aはまた、置換されていても置換されていなくてもよい芳香族も含む。この式において、Bはο‐アミノフェニル基またはヒドロキシ基である。いくつかの実施例において、Yはnが0である(CH2)nであり得、Aは芳香族基を含み、Bはヒドロキシで、かつQは水素である。
【0013】
いくつかの実施例では、mは0かつXはHである。これらの特徴を有する化合物としては以下のものが含まれるが、これらに限定されない。
【0014】
【化7】
【0015】
【化8】
いくつかの実施例では、XはHであり、かつAは以下:
【0016】
【化9】
から選択され、ここでRは分岐または非分岐の、置換または非置換の、飽和または不飽和の、脂肪族または芳香族を含む。
【0017】
本発明は、本発明の阻害剤を含む組成物を包含し、この組成物はR立体異性体と比較してS立体異性体が富化されている。
【0018】
本発明の具体的な阻害剤としては、N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;2−プロピル−ペンタン酸−{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;2−プロピル−ペンタン酸−{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;2−プロピル−ペンタン酸{4−2−[2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;2−プロピル−ペンタン酸−[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミド;N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド;N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(3−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロシキ−ベンズアミド;4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;ピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミド;N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;4−(2,2−ジメチル−4−フェニル‐ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−メトキシ−2−フェニル−アセチルアミノ)−ベンズアミド;4−ジフェニルアセチルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;N−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミド;N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;ならびにN−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミド;N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;N−[4−(2−ヒドロキシカルバモイルーエチル)−フェニル]−2−(S)−フェニル−ブチリルアミド;N−[4−(2−ヒドロキシカルバモイルーエチル)−フェニル]−2−(R)−フェニルブチルアミド;N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミドが含まれる。
【0019】
本発明はまた、これらの阻害剤と1つ以上の薬学的に受容可能な賦形剤とを含む薬学的組成物を含む。さらになお、本発明は、動物(例えば、ヒト)内で腫瘍性細胞増殖を阻害する方法を含み、、これは治療的有効量の少なくとも1種の本発明の阻害剤を投与する工程を包含する。
【0020】
本発明のさらなる特性および利点は、以下に続く説明にてその一部が記述される。その一部はこの説明から明白となるか、または本発明の実施によって習得され得る。本発明の特性および利点は、添付の特許請求の範囲において具体的に示される要素および組み合わせによって理解され、達成されることとなる。
【0021】
前述の一般的な説明および以下の詳細な説明はいずれも、例示であって説明にすぎず、特許請求の範囲のように本発明を制限するものではないことが、理解されるべきである。この明細に添付され、この明細の一部を構成する付属の図面は、本発明の実施形態を実証し、説明と一緒になって本発明の本質を説明するのに役立つものである。
例えば、本発明は以下の項目を提供する。
(項目1)
式
【化1】
を有する、ヒストン脱アセチル化酵素阻害剤であって、ここで:
Xは、HおよびCH3から選択され;
Yは、nが0〜2である、(CH2)nから選択され;
Zは、mが0〜3である(CH2)mおよび(CH)2から選択され;
Aは、ヒドロカルビル基であり;
Bは、o−アミノフェニル基またはヒドロキシル基であり;そして
Qが、ハロゲン、水素、またはメチルである、阻害剤。
(項目2)
請求項1に記載の阻害剤であって、Aが脂肪族基を含む、阻害剤。
(項目3)
請求項2に記載の阻害剤であって、前記脂肪族基が分枝である、阻害剤。
(項目4)
請求項1に記載の阻害剤であって、Aが芳香族基を含む、阻害剤。
(項目5)
請求項4に記載の阻害剤であって、Aが、置換した芳香族基を含む、阻害剤。
(項目6)
請求項1に記載の阻害剤であって、Bがo−アミノフェニルである、阻害剤。
(項目7)
請求項1に記載の阻害剤であって、Bがヒドロキシである、阻害剤。
(項目8)
請求項1に記載の阻害剤であって、nが0であり、Aが芳香族基を含み、Bがヒドロキシであり、そしてQが水素である、阻害剤。
(項目9)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;2−プロピル−ペンタン酸{4−2−(2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;および2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミドから選択される、阻害剤。
(項目10)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;およびN−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミドから選択される、阻害剤。
(項目11)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;およびN−ヒドロキシ−4−[(3−フェニルーブチリルアミノ)−メチル]−ベンズアミドから選択される、阻害剤。
(項目12)
請求項1に記載の阻害剤であって、該阻害剤が、4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミドおよびピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミドから選択される、阻害剤。
(項目13)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;N−ヒドロキシ−4−(2−メトキシ−2−フェニルーアセチルアミノ)−ベンズアミド;4−ジフェニルアセチルアミノ−N−ヒドロキシ−ベンズアミド;N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;およびN−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミドから選択される、阻害剤。
(項目14)
請求項1に記載の阻害剤であって、該阻害剤が、N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミドから選択される、阻害剤。
(項目15)
請求項1に記載の阻害剤であって、該阻害剤が、N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(S)−フェニル−ブチルアミド;N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(R)−フェニル−ブチルアミド;N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;およびN−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミドから選択される、阻害剤。
(項目16)
請求項1に記載の阻害剤であって、該阻害剤が、エステルまたは塩である、阻害剤。
(項目17)
請求項1に記載の阻害剤と、少なくとも一つ薬学的に受容可能な賦形剤とを含む、薬学的組成物。
(項目18)
動物における腫瘍性細胞増殖を阻害する方法であって、治療上効果的な量の、請求項1に記載される少なくとも一つの阻害剤を投与する工程、を包含する、方法。
(項目19)
請求項18に記載の方法であって、前記動物がヒトである、方法。
(項目20)
請求項8に記載の阻害剤であって、m=0であり、そしてX=Hである、方法。
(項目21)
請求項20に記載の阻害剤であって、前記化合物が、
【化2】
である、阻害剤。
(項目22)
請求項20に記載の阻害剤であって、前記化合物が、
【化3】
である、阻害剤。
(項目23)
請求項20に記載の阻害剤であって、前記化合物が、
【化4】
である、阻害剤。
(項目24)
請求項21に記載の阻害剤を含む組成物であって、該組成物が、R−立体異性体と比べて、S−立体異性体が富化されている、組成物。
(項目25)
請求項1に記載の阻害剤であって、X=Hであり、そしてAが、
【化5】
から選択され、Rが、分枝または非分枝の、置換または非置換の、飽和または不飽和の、脂肪族基または芳香族基である、阻害剤。
【図面の簡単な説明】
【0022】
【図1】図1は、化合物1〜10を生成するための、バルプロ酸と5つの芳香族ωアミノ酸および2つのZn2+キレート部分との様々な結合体を示す。
【図2】図2は、化合物1〜10のHDAC阻害効力を示す。インビトロHDACアッセイは、本明細書中で記載されている市販の酵素アッセイキットを使用して行った。データは、平均値±S.D.(n=3)である。
【図3】図3は、化合物11〜22の構造およびHDAC阻害効力を示す。値は、平均値±S.D.(n=3)である。
【図4】図4は、DU−145細胞におけるヒストンアセチル化およびp21WAF/CIP1の発現に対する、HTPB、TSAおよびフェニルブチレートの影響を示す。10%FBS添加RPMI 1640培地中で、DU−145細胞を、HTPB、TSAおよびフェニルブチレートに、示された濃度で24時間曝露した。それぞれの溶解液からの等量のタンパク質を電気泳動し、それぞれの抗体を使用するウエスタンブロットによって試験した。内部参照タンパク質としてアクチンを使用した。
【図5】図5は、DU−145細胞に対するHTPBの増殖阻害効果を示す。(A)細胞生存率に対するHTPB用量依存的な影響の時間経過。DU−145細胞を、10%FBS含有RPMI 1640培地中の0〜2.5μMのHTPBで、示された時間、処理した。MTTアッセイによって生細胞を試験した。データは平均値±S.D.である(n=6)。(B)24時間曝露後のヌクレオソームDNAの形成に対する、HTPB用量依存的な影響。ヌクレオソームの形成は、各アッセイについて2×103の細胞に相当する溶解物を用いたCell Death Detection ELISAによって、定量的に測定した。データは、2回の独立した定量の平均である。
【図6】図6は、本発明の化合物を合成するためのスキームの例を示す。
【図7−1】図7(フレーム1)は、本発明に従う種々の化合物の例を表示する。
【図7−2】図7(フレーム2)は、本発明に従う種々の化合物の例を表示する。
【図7−3】図7(フレーム3)は、本発明に従う種々の化合物の例を表示する。
【図7−4】図7(フレーム4)は、本発明に従う種々の化合物の例を表示する。
【図7−5】図7(フレーム5)は、本発明に従う種々の化合物の例を表示する。
【図7−6】図7(フレーム6)は、本発明に従う種々の化合物の例を表示する。
【図7−7】図7(フレーム7)は、本発明に従う種々の化合物の例を表示する。
【図7−8】図7(フレーム8)は、本発明に従う種々の化合物の例を表示する。
【図7−9】図7(フレーム9)は、本発明に従う種々の化合物の例を表示する。
【図7−10】図7(フレーム10)は、本発明に従う種々の化合物の例を表示する。
【図7−11】図7(フレーム11)は、本発明に従う種々の化合物の例を表示する。
【図8−1】図8(フレーム1)は、本発明に従って使用され得る亜鉛キレートモチーフの例を表示する。
【図8−2】図8(フレーム2)は、本発明に従って使用され得る亜鉛キレートモチーフの例を表示する。
【図9】図9は、化合物19のリガンドドッキングの分子モデリング研究である。
【図10】図10は、化合物19とその結合部位との相互作用の過程で起こると考えられるプロトン移動を図示する。
【図11】図11は、化合物42のリガンドドッキングの分子モデリング研究である。
【図12】図12は、PC−3アンドロゲン非依存性前立腺癌細胞における、ヒストンH−4過アセチル化およびp21WAF/CIP1発現に対する、化合物42、SAHA、およびTSAの影響を示す。
【図13】図13は、PC−3細胞におけるAktの活性化状態に対する、化合物42、SAHA、およびTSAの影響を示す。
【図14】図14は、いくつかのリン酸化酵素に対する化合物42の影響を示す。
【図15】図15は、定着したPC−3異種移植片腫瘍の増殖に対する、経口投与した化合物42の影響を示す。
【図16】図16は、胸腺欠損マウスにおける皮下PC−3異種移植片腫瘍の増殖に対する、化合物42、化合物44、およびSAHAの影響を示す。
【図17】図17は、化合物42、SAHA、またはコントロールで処理した2つの代表的なPC−3腫瘍ホモジネートの、ヒストンH3並びにアセチル化H3のウエスタンブロットを示す。
【図18】図18は、代表的な乳癌細胞株、肺癌細胞株および甲状腺癌細胞株に対する、化合物42の用量依存的な抗増殖効果を示す。
【図19】図19は、初代CLL細胞に対する、化合物42の影響を示す。
【図20】図20は、化合物42の化学合成スキームを図示する。
【図21】図21は、本発明に従う化合物に関する一般的な化学合成スキームを図示する。
【発明を実施するための形態】
【0023】
(発明の実施形態の説明)
一般に、短鎖脂肪酸は、アシル鎖の構造に係わらず、mM範囲でHDAC阻害および抗増殖活性を示す(Grozinger and Schreiber,Chem Biol 9:3−16(2002);Johnstone,Nat Rev Drug Discov 1:287−299(2002);Kramer et al.,Trends Endocrinol Metab 12:294−300(2001))。本発明はとりわけ、これらの脂肪酸が、酵素ポケット入口に位置する表面残基および/またはこの管状ポケット内の疎水性部分との非特異的な疎水性相互作用を介して、HDAC阻害を発揮するという発見に基づく。本発明者らは、疎水性スペーサーを介してZn2+キレート部分を係留することによって、これら短鎖脂肪酸のHDAC阻害効力を増大させた。
【0024】
ヒストン脱アセチル化酵素の酵素活性の測定は、公知の方法論を用いて達成され得る。例えば、Yoshidaら(J.Biol.Chem.265:17174−17179(1990))は、トリコスタチンAで処理した細胞においてアセチル化ヒストンを検出することによる、ヒストン脱アセチル化酵素の酵素活性の評価を記載している。同様に、Tauntonら(Science 272:408−411(1996))は、内在性HDAC−1および組換えHDAC−1を使用してヒストン脱アセチル化酵素の酵素活性を測定する方法を記載している。Yoshidaら(J.Biol.Chem.265:17174−17179,1990)およびTauntonら(Science 272:408−411,1996)はいずれも、本明細書中に参考として援用される。
【0025】
この開示を通じて、本発明に従う化合物に対する参照がなされる。この明細書および特許請求の範囲においてそのような化合物に対する参照は、その化合物のエステルおよび塩を包含する。従って、たとえ明確に列挙されていなくても、その化合物自体への参照によって、そのようなエステルおよび塩は企図および包含される。
【0026】
本発明に従って使用され得る脂肪酸は、ヒドロカルビル部分およびカルボン酸部分を含む。本明細書中で使用される場合、用語「ヒドロカルビル」は、「脂肪族」、「環式脂肪族」、および「芳香族」が含まれると理解される。このヒドロカルビル基には、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アリール基、アラルキル基、およびアルカリル基が含まれると理解される。さらに、「ヒドロカルビル」には、非置換ヒドロカルビル基と置換ヒドロカルビル基とのいずれもが含まれると理解され、後者は炭素および水素以外の付加置換基を有する炭化水素部分を指す。さらに、「カルボン酸」はこの化合物を指すために使用されるが、そのような酸の塩(例えば、カルボン酸塩など)も明確に企図される。さらに、本明細書中においてカルボン酸とカルボン酸塩は交換可能なものとして使用され得る。
【0027】
脂肪酸には、特に、約3個〜約14個の炭素の長さの非分枝脂肪酸の長さに匹敵する長さの分子鎖を有するものが含まれるが、これらに限定されない。従って、この分子鎖は、例えば約3個、4個、5個、6個、7個、8個、9個、10個、11個、12個または13個の炭素の長さであり得る。この鎖は、最大で、例えば約14個、13個、12個、11個、9個、8個、7個、6個、5個または4個の炭素の長さであり得る。この脂肪酸は直鎖であっても分枝であってもよく、単結合、二重結合および/または三重結合を含み得る。脂肪酸の非限定的な例としては、バルプロエート、ブチレート、フェニルアセテートおよびフェニルブチレートが含まれる。
【0028】
本発明に従って企図されるZn2+キレートモチーフとしては、ヒドロキサム酸およびο−フェニレンジアミンが挙げられるが、これらに限定されない。他の例としては、トリフルオロメチルケトン、α−ケトアミド、α−ケトチアゾール、2−ケト1−メチル−1H−イミダゾール、α−ケト1H−テトラゾール、α−ケト1H−イミダゾール、5−ケト1メチル−1H−イミダゾール、α−ケトオキサゾール、α−ケト4,5−シヒドロ−オキサゾール、α−ケトベンゾオキサゾール、α−ケトオキサゾール[4,5−b]ピリジンおよびα−ケトピリジンが含まれる。これらのモチーフの構造は、図8に示される。
【0029】
スペーサーは任意のヒドロカルビルスペーサーであり得るが、好ましくは芳香族成分を含む。芳香族リンカーは、以下の利点を有していると考えられている:1)上記結合体の構造硬性を増大させる、および2)ポケットの管様疎水領域とのファンデルワールス接触を増大させて結合親和力を向上させる。リンカーの例としては、芳香族ω‐アミノ酸が挙げられるが、これに限定されない。
【0030】
このリンカーは、炭素数4から8の直鎖(例えば炭素数4、5、6、7、あるいは8の直鎖)と同じ長さを示す。従って、その長さは炭素数4〜7または4〜6の直鎖と同じであり得る。その長さは、結合ポケットの疎水領域の深さに基づき得る。本発明のリンカーの例としては、4−(アミノメチル)安息香酸、4−アミノ安息香酸、(4−アミノフェニル)酢酸、3−(4−アミノフェニル)プロピオン酸、および3−(4−アミノフェニル)−アクリル酸が挙げられるが、これらに限定されない。とりわけ、4−(アミノメチル)安息香酸は、MS−27−275(Saito et al.,Proc Natl
Acd Sci USA 96:4592−4597(1999))のリンカーとして使用されている。
【0031】
以下の化合物が具体的に企図される:
N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;
N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド;
2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド;
2−プロピル−ペンタン酸(4−ヒドロキシカルバモイル−メチル−フェニル)−アミド;
2−プロピル−ペンタン酸{4−[2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド;
2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−アミド;
2−プロピル−ペンタン酸{4−[2−(2−アミノ−フェニルカルバモイル)−ビニル]−フェニル}−アミド;
2−プロピル−ペンタン酸[4−(2−ヒドロキシカルバモイル−ビニル)−フェニル]−アミド;
N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;
N−(2−アミノ−フェニル)−4−[(4−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド;
N−ヒドロキシ−4−[(4−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド;
N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド;
N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド;
N−(2−アミノ−フェニル)−4−[(2−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
N−(2−アミノ−フェニル)−4−[(3−フェニル−ブチリルアミノ−メチル]−ベンズアミド;
N−ヒドロキシ−4−(2−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−フェニルブチリルアミノ)−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−フェニル−ブチルアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−3−フェニル−ブチルアミド;
N−ヒドロキシ−4−[(2−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
N−ヒドロキシ−4−[(3−フェニル−ブチリルアミノ)−メチル]−ベンズアミド;
4−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−メチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−クロロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−ブロモ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;]
4−(4−tert−ブチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−フェニル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−メトキシル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−トリフルオロメチル)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
4−(4−ニトロ)−ベンゾイルアミノ−N−ヒドロキシ−ベンズアミド;
ピリジン−2−カルボン酸(4−ヒドロキシカルバモイル−フェニル)−アミド;
N−ヒドロキシ−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;
4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−N−ヒドロキシ−ベンズアミド;
N−ヒドロキシ−4−[メチル−(4−フェニル−ブチリル)−アミノ]−ベンズアミド;
N−ヒドロキシ−4−(2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(2−メトキシ−2−フェニル−アセチルアミノ)−ベンズアミド;
4−ジフェニルアセチルアミノ−N−ヒドロキキシ−ベンズアミド;
N−ヒドロキシ−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;
N−(2−アミノ−フェニル)−4−フェニルアセチルアミノ−ベンズアミド;
N−(2−アミノ−フェニル)−4−(5−フェニル−ペンタノイルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2,2−ジメチル−4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−プロピオニルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(4−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−(2−メチル−2−フェニル−プロピオニルアミノ)−ベンズアミド;
N−(2−アミノ−フェニル)−4−[2−(4−イソブチル−フェニル)−プロピオニルアミノ]−ベンズアミド;
N−ヒドロキシ−4−[2−(S)−フェニルブチリルアミノ]−ベンズアミド;
N−ヒドロキシ−4−[2−(R)−フェニルブチリルアミノ]−ベンズアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(S)−フェニル−ブチルアミド;
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−2−(R)−フェニル−ブチルアミド;
N−ヒドロキシ−4−(3−(S)−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−(3−(R)−フェニルブチリルアミノ)−ベンズアミド;
N−ヒドロキシ−4−[3−(S)−フェニルブチリルアミノ]−ベンズアミド;および
N−ヒドロキシ−4−[3−(R)−フェニルブチリルアミノ]−ベンズアミド。
【0032】
本発明の化合物は、ラセミ化合物あるいはラセミ混合物である場合がある。本明細書中で使用される場合、用語「ラセミ」は、本発明の化合物の(R)鏡像異性体と(S)鏡像異性体との混合物、あるいは立体異性体混合物を意味し、ここで鏡像異性体もしくは立体異性体はいずれも、実質的に他方から精製されていない。
【0033】
本発明の(R)立体異性体または(S)立体異性体を記述するために本明細書中で使用される「富化」という語は、(S)立体異性体よりも(R)立体異性体の量が多いか、またはその逆の組成物を指す。例えば、この組成物は、化合物42の全重量に対して、50重量%、55重量%、あるいは60重量%以上の化合物42の(S)立体異性体を含有し得る。一実施例において、富化された(S)化合物42の量はより多いいものであり得、例えば、化合物42の全重量に対し、65重量%、70重量%、75重量%、80重量%、81重量%、82重量%、83重量%、84重量%、85重量%、86重量%、87重量%、88重量%、89重量%、90重量%、91重量%、92重量%、93重量%、94重量%、95重量%、96重量%、97重量%、98重量%、99重量%以上、もしくはこれらの任意の分数量(例えば90.1重量%、90.2重量%等)の(S)‐化合物42であり得る。特定の実施例において、富化された(S)化合物42の量は、化合物42の全重量に対し、99重量%、99.1重量%、99.2重量%、99.3重量%、99.4重量%、99.5重量%、99.6重量%、99.7重量%、99.8重量%、99.9重量%よりも多いものであってもよいし、100重量%であってもよい。これらの用語はまた、(S)化合物42の薬学的に受容可能な任意の塩の量を定義する。これらは非限定的な例であり、本発明の他のラセミ化合物に対しても、同じ富化が達成され得る。
【0034】
本発明の鏡像異性体富化した組成物を投与することにより、所望の治療効果が得られる。即ち、鏡像異性体富化した組成物は、低い合計濃度で治療効果をもたらし得るか、または所望されな鏡像異性体の存在に起因する副作用を軽減し得る。これらの利点は特に企図される。
【0035】
本発明の方法にて用いられる任意の本発明の化合物は、経口的に、非経口的に(IV、IM、デポーIMS、SQ、およびデポーSQ)、舌下に、経鼻的に(吸入)、髄腔内に、局所的に、あるいは直腸的に投与され得る。当業者に公知の投与形態は、本発明の方法において用いられる本発明の化合物の送達に適している。
【0036】
治療的有効量の、本発明の方法において用いられる本発明の化合物を含有する組成物が提供される。この化合物は、錠剤、カプセル、あるいはエリキシル等経口投与に適切な薬学的調製物に、あるいは非経口投与のための滅菌溶液もしくは滅菌懸濁液に処方することができる。本明細書中に記載される化合物は、当該分野で周知の技術および手順を用いて薬学的組成物に処方することができる。
【0037】
約0.1mgから1000mgの、本発明の方法において用いられる本発明の化合物あるいは発明の化合物の混合物、あるいは生理学的に受容可能な塩もしくはエステルは、認可された製薬業務に必要な単位投与量形態内で、生理学的に許容できるビヒクル、キャリア、賦形剤、結合剤、保存剤、安定剤、香味料等と混合される。これらの組成物あるいは調製物中の活性物質量は、記載した単位の適切な投与量が得られる量である。この組成物は単位投薬形態に処方され得、各投与形態物は、約1〜約500mg、または約10〜約100mgの活性成分を含有する。「単位用量形態」という語は、ヒト被験体および他の哺乳類のための単位投与物として適切な、物理的に別々の単位を指し、各単位物は、適切な賦形剤と共に、所望する治療効果が生じるよう計算された、前決定された量の活性物質を含有する。
【0038】
組成物を調製するため、本発明の方法で用いられる1つ以上の発明の化合物は、適切な薬学的に受容可能なキャリアと混合される。この化合物を混合あるいは添加すると、溶液、懸濁液、乳液などが生じ得る。薬学的に適切なキャリアとして、リポソーム懸濁液もまた、使用され得る。これらは、当業者に周知の方法に従って調製され得る。得られる混合物の形態は、多数の因子に依存し、この因子としては、意図する投与様式および選択したキャリアまたはビヒクルにおけるこの化合物の溶解性が挙げられる。有効濃度は、処置される疾患、障害または状態の1つ以上の症状が軽減または改善されるのに十分であり、経験的に決定され得る。
【0039】
本明細書中で提供される化合物の投与に適した薬学的キャリアまたは薬学的ビヒクルとしては、特定の投与様式に適する任意のキャリアが含まれる。さらに、活性物質もまた、所望の活性を損なわない他の活性物質と、または所望の活性を補う他の活性物質と、または他の活性を有する活性物質と、混合され得る。この化合物は、組成物内で唯一の薬学的活性成分として処方されてもよいし、他の活性成分と組み合わせられてもよい。
【0040】
この化合物が不十分な溶解性を示す場合、可溶化のための方法が使用され得る。そのような方法は公知であり、これとしてはジメチルスルホキシド(DNSO)のような共溶媒の使用、TWEENのような界面活性剤の使用、および炭酸水素ナトリウム水溶液への溶解が含まれるが、これらに限定されない。有効な薬学的組成物の処方において、塩またはプロドラッグのような、この化合物の誘導体もまた使用され得る。
【0041】
化合物の濃度は、化合物投与の対象である障害の少なくとも1つの症状を軽減または改善する投与量の送達に有効なものである。代表的には、この組成物は、単回用量投与のために処方される。
【0042】
本発明の方法において用いられる本発明の化合物は、徐放性処方物またはコーティングのような、体内からの急速な排出に対して化合物を保護するキャリアと一緒に、調製され得る。そのようなキャリアとしては、マイクロカプセル化された送達系などの徐放性処方物が挙げられるが、これに限定されない。この活性化合物は、治療を受ける患者に有害な副作用を与えずに、治療的に有用な効果を発揮するのに十分な量で、薬学的に受容可能なキャリア中に含有され得る。治療的に有効な濃度は、処置される疾患に対するインビトロモデル系およびインビボモデル系において、公知の化合物を試験することによって、経験的に決定され得る。
【0043】
本発明の化合物および組成物は、複数回投与容器あるいは1回投与容器内に収められ得る。この封入された化合物および組成物は、例えば、使用のために組み立てられ得る成分部分を含有するキットにおいて、提供され得る。例えば、凍結乾燥形態の発明の化合物と適切な稀釈剤とが、使用前に混合するために別々の成分部分として提供され得る。キットは、本発明の化合物と、共投与のための第二の治療剤とを包含し得る。本発明の化合物および第二の治療剤は、別々の成分部分として提供され得る。キットは複数の容器を備え得、各容器は、本発明の方法において用いられる本発明の化合物の1単位用量以上を含む。この容器は所望の投与様式に適合され得、これとしては、経口投与のための錠剤、ゲルカプセル、徐放性カプセルなど;非経口投与のためのデポー製品、充填済み注射器、アンプル、バイアルなど;局所投与のためのパッチ、医療用パッド、クリームなど、が挙げられるが、これらに限定されない。
【0044】
薬物組成物中の本発明の活性化合物の濃度は、その活性化合物の吸収性、不活性化性および排出速度、投与スケジュール、投与量、ならびに当業者に公知の他の要因に依存する。
【0045】
この活性成分は、一度に投与されてもよいし、時間間隔ごとに投与される、より小さないくつかの投与量に分割されてもよい。正確な用量および処置期間は、処置される疾患の関数であり、公知の試験プロトコルを用いて、あるいはインビボもしくはインビトロの試験データから外挿することによって、経験的に決定され得る。また、濃度および投与値は、緩和されるべき状態の重篤度によって変動し得ることに、注意するべきである。任意の特定の患者に関して、特定の投薬レジメンは、その患者の必要性および、この組成物を投与するかまたは組成物の投与を監督する者である専門家による判断に従って、時間経過と共に調整するべきであること、そして、この明細で記載される濃度の範囲は例示というだけであって、特許請求される組成物の範囲または実施の制限を意図したものでないことが、さらに理解されるべきである。
【0046】
経口投与が所望される場合、胃の酸性環境から化合物を保護する組成で、この化合物は提供され得る。例えば、この組成物は、胃の中でその保全性を維持し、そして腸内でこの活性化合物を放出する腸溶コーティング内に処方され得る。この組成物はまた、制酸薬あるいは他の同様の成分と組み合わせて処方され得る。
【0047】
経口組成物は、一般に、不活性の希釈剤あるいは食用のキャリアが含み、そして圧縮されて錠剤となるか、またはゼラチンカプセル内に封入され得る。経口治療投与の目的のために、その活性化合物を賦形剤と混合し、錠剤、カプセル、あるいはトローチの形態にて使用され得る。この組成物の一部として、薬学的に適合性の結合剤および補助物質が含有され得る。
【0048】
この錠剤、丸剤、カプセル、トローチなどは、任意の以下の成分あるいは同様の性質の化合物を含有し得る:結合剤(例えば、トラガカントゴム、アカシアゴム、トウモロコシ澱粉またはゼラチンが挙げられるが、これらに限定されない);賦形剤(例えば、ミクロクリスタリンセルロース、澱粉、あるいは乳糖);崩壊剤(例えば、アルギン酸およびトウモロコシ澱粉が挙げられるが、これらに限定されない);潤滑剤(lubricant)(例えば、ステアリン酸マグネシウムが挙げられるが、これに限定されない);潤滑剤(glidant)(例えば、二酸化ケイ素コロイドが挙げられるが、これに限定されない);甘味料(例えば、ショ糖またはサッカリン);香味料(例えば、ペパーミント、サルチル酸メチルまたはフルーツ香料)。
【0049】
投与単位形態がカプセルである場合、これは上記の種類の物質に加えて脂肪油などの液体キャリアを含有し得る。さらに、投与単位形態は、例えば糖衣あるいは他の腸溶剤のような、この投与単位形態の物理的形態を改変する様々な他の物質を含有し得る。この化合物はまた、エリキシル剤、懸濁液、シロップ、カシェ剤、チューイングガムなどの成分として投与され得る。シロップは、この活性成分に加えて、甘味料、およびある種の保存料としてショ糖、色素および着色剤、ならびに香味料を含有し得る。
【0050】
この活性物質はまた、所望の作用を損なわない他の活性物質と、あるいは所望の作用を補う物質と、混合され得る。本発明の化合物は、例えば抗がん剤、ホルモン、ステロイド、またはレチノイドと併用して使用され得る。抗がん剤は、アルキル化剤、代謝拮抗剤、ホルモン剤、抗生剤、コルヒチン、ビンカアルカロイド、L−アスパラギナーゼ、プロカルバジン、ヒドロキシ尿素、ミトタン、ニトロソウレア類、あるいはイミダゾールカルボキサミドのような多数の化学治療剤のうちの1つであり得る。適切な薬剤としては、チューブリンの脱分極を促進する薬剤が挙げられる。例としては、コルヒチンならびにビンカアルカロイド(例えば、ビンブラスチンおよびビンクリスチン)が挙げられる。
【0051】
経口、皮内、皮下、あるいは局所投与に使用する溶液または懸濁液は、以下の任意の成分を含有し得る:滅菌希釈剤(注射用水、生理食塩水、固定油、例えばゴマ油、ココナッツ油、ピーナッツ油、綿果油などのような天然植物油、あるいはオレイン酸エチルなどの合成脂質性ビヒクル、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒);抗菌剤(例えば、ベンジルアルコールおよびメチルパラベン);抗酸化剤(例えば、アスコルビン酸および重亜硫酸ナトリウム);キレート剤(例えば、エチレンジアミンテトラ酢酸(EDTA));緩衝液(例えば、酢酸、クエン酸、およびリン酸);浸透圧を調整するための薬剤(例えば、塩化ナトリウムおよびデキストロース)。非経口調製物は、アンプル、使い捨て注射器、あるいはガラス、プラスチック、もしくは他の適切な物質からできた複数回投与用バイアル内に、封入することができる。必要に応じて、緩衝液、保存剤、抗酸化剤などが組み入れられ得る。
【0052】
静脈経由で投与する場合、適切なキャリアとしては、生理食塩水、リン酸緩衝化生理食塩水(PBS)、ならびに増粘剤および溶解剤を含有する溶液(例えば、グルコース、ポリエチレングリコール、ポリプロピレングリコールおよびこれらの混合物)が挙げられるが、これらに限定されない。組織標的リポソームを含むリポソーム懸濁液もまた、薬学的に受容可能なキャリアであり得る。これらは、当該分野で公知な方法に従って調製され得る。
【0053】
本発明の化合物は、時間放出処方あるいはコーティングのような、体外への急速な排出から化合物を保護するキャリアと共に調製され得る。そのようなキャリアとしては、インプラントおよびマイクロカプセル送達系、ならびに生分解性生体適合ポリマー(例えば、コラーゲン、エチレンビニル酢酸、ポリ無水物、ポリグリコール酸、ポリオルトエステル、ポリ乳酸など)のような徐放性組成物が挙げられるが、これらに限定されない。そのような組成物を調製するための方法は、当業者に公知である。
【0054】
本発明の方法において用いられる化合物は、経腸的にあるいは非経口的に投与され得る。経口的に投与される場合、本発明の方法で用いられる化合物は、当業者に周知の、経口投与のための通常の投薬形態で投与され得る。これらの投薬形態としては、錠剤並びにカプセルの通常の固形単位用量形態、ならびに溶液、懸濁液、およびエリキシルなどの液体投与形態が挙げられる。固形投与形態を使用する場合、その形態は徐放性型であり得、その結果、本発明の方法で用いられる化合物は1日1、2回の投与のみが必要とされる。
【0055】
経口投与形態は、1日1回、2回、3回、または4回患者に投与され得る。本発明の方法で用いられる本発明の化合物は、1日3回以下、もしくは1日1回あるいは2回のみ投与され得る。つまり、本発明の方法で用いられる本発明の化合物は、経口投与形態にて投与される。どのような経口投与形態を用いる場合でも、本発明の方法で用いられる化合物を胃の酸性環境から保護するように、これらは設計され得る。腸溶性コーティング錠剤が、当業者に周知である。さらに、酸性である胃から保護するよう一つ一つがコーティングされた小球体が充填されたカプセルもまた、当業者に周知である。
【0056】
本発明の方法で用いられる本発明の化合物はまた、ナノ結晶分散処方物にて、有利に送達され得る。そのような処方物の調製は、例えば米国特許第5,145,684号に記載されており、その全内容が、参考として援用される。HIVプロテアーゼ阻害剤のナノ結晶分散物およびその使用法は、米国特許第6,045,829号に記載されており、その全内容が、参考として援用される。ナノ結晶処方物は、代表的には薬物化合物の生物学的有用性を増大させる。
【0057】
本発明の化合物および方法は、動物において腫瘍性細胞の増殖を抑制するために使用され得る。この方法は、体内に1つ以上の腫瘍性細胞を有する動物に、治療的有効量の1種以上の本発明の化合物を、上記のような組成物にて投与する工程を包含する。この動物は哺乳類(家畜を含む)であり得る。この動物はヒトであり得る。
【0058】
用語「腫瘍性細胞」は、異常な細胞増殖を示す細胞を指すために使用される。腫瘍性細胞の異常な増殖には、細胞増殖の増大が含まれる。例えば、腫瘍性細胞は、過増殖性細胞、またはインビトロにて増殖の接触抑制の欠如を示す細胞、インビボで転移することのできない良性腫瘍細胞、インビボで転移することができかつ除去の試み後に再発し得る癌細胞であり得る。用語「腫瘍形成」は、腫瘍性増殖の発症へと導く細胞増殖の誘導を示すために使用される。
【0059】
用語「治療的有効量」および「治療的有効期間」は、腫瘍性細胞増殖の軽減に有効な投与量で腫瘍性細胞増殖の軽減に有効な期間に渡る処置を示すために使用される。上記のように、そのような投与は、非経口的、経口的、舌下的、経皮的、局所的、経鼻的、あるいは直腸内的であり得る。全身投与する場合、本発明の化合物の血中濃度が約0.1μM〜約100mMを達成するのに十分な投与量で、その治療的組成物は投与され得る。局所的な投与については、これよりもはるかに少量が有効であり得、またはるかに高い濃度が許容される場合がある。当業者は、ヒストン脱アセチル化酵素阻害剤の有効濃度の低下をもたらすそのような治療効果は、本発明に従って処置される組織、器官、あるいは特定の動物もしくは患者によって著しく変動し得ることを認識する。また、患者がある投与量で開始しても、その患者の状態が変化するため、投与量は経時的に変化し得ることもまた、理解される。
【0060】
本発明は、動物の細胞増殖性疾患あるいは状態を治療するための組成物並びに方法を提供し、この方法は、そのような処置を必要とする動物に治療的有効量の、本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。記述したように、この動物は哺乳動物(家畜を含む)であり得る。この動物はヒトであり得る。
【0061】
用語「細胞増殖性疾患または細胞増殖性状態」は、異常な細胞増殖(好ましくは、異常に増大した細胞増殖)によって特徴づけられる任意の状態を指すことが意味される。そのような細胞増殖性疾患または状態の例としては、癌、再狭窄および乾癬が挙げられるが、これらに限定されない。いくつかの実施例において、本発明は、動物内の腫瘍性細胞増殖を阻害するための方法を提供し、この方法は、体内に1つ以上の腫瘍性細胞を有する動物に、治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。本発明に従って治療することのできる癌としては、前立腺癌、肺癌、急性白血病、多発性硬化症、膀胱癌腫、腎臓癌腫、乳癌腫、結腸直腸癌腫、神経芽腫、あるいは黒色腫が挙げられるが、これらに限定されない。
【0062】
本発明の化合物のいくつかは、原生動物由来のヒストン脱アセチル化酵素に対する阻害活性を有する。従って、本発明はまた、原生動物性疾患または原生動物性感染を処置もしくは予防するための方法を提供し、この方法は、そのような処置を必要とする動物に、治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。ここでも、動物は哺乳動物であり得、そしてヒトであり得る。いくつかの実施形態において、このヒストン脱アセチル化酵素阻害剤は、哺乳動物ヒストン脱アセチル化酵素、特にヒトヒストン脱アセチル化酵素阻害剤を阻害するよりも高い程度で、原生動物ヒストン脱アセチル化酵素を阻害する。
【0063】
本発明はさらに、真菌性疾患または真菌性感染を処置するための方法を提供し、この方法は、そのような処置を必要とする動物に治療的有効量の本発明のヒストン脱アセチル化酵素阻害剤を投与する工程を包含する。この動物は、ヒトを含む哺乳動物であり得る。本発明のこの実施形態に従って使用されるヒストン脱アセチル化酵素阻害剤は、哺乳類ヒストン脱アセチル化酵素、特にヒトヒストン脱アセチル化酵素阻害剤を阻害するよりも高い程度で、真菌のヒストン脱アセチル化酵素を阻害し得る。
【0064】
当業者である投与を行う医療従事者に周知なように、正確な投与量および投与の頻度が、本発明の方法で用いられる投与される特定の化合物、処置される患者の具体的な状態、処置される状態の重篤度、特定の患者の年齢、体重、全身の身体状態、およびその患者が受けている可能性のある他の医薬に依存することは、当業者には明白であるはずである。
【実施例】
【0065】
(実験)
化学試薬および有機溶媒は、他に記載のない限り、Aldrichから購入した。核磁気共鳴スペクトル(1H NMR)は、Bruker 250 MHzで測定した。化学シフト(δ)は、TMSピークに対する100万分の1(ppm)で記載する。エレクトロスプレーイオン化(ESI)質量分析は、3−Tesla Finnigan FTMS−2000 フーリエ変換質量分析器で実行した。元素分析値は、計算値の±0.4%以内であった。
【0066】
シリカゲル(230−400メッシュ)を用いて、フラッシュカラムクロマトグラフィーを行った。ωアミノ酸メチルエステルを、メタノール/TMSClを使用して市販の酸から調製し、(2−アミノ−フェニル)カルバミン酸ベンジルエステルを、標準的な手順に従ってο−フェニレンジアミンおよびギ酸ベンジルクロリドから合成した。ウサギ抗アセチルヒストンH3ポリクローナル抗体およびウサギ抗アセチルヒストンH4ポリクローナル抗体は、Upstate Biotechnology(Lake Placid,
NY)から購入し、ウサギ抗p21抗体はSanta Cruz Biotechnology(Santa Cruz,CA)から購入した。マウス抗アクチンモノクローナル抗体は、ICN Biomedicals(Irvine,CA)製であった。HRP結合対化ヤギ抗ウサギIgGおよびHRP結合体化ヤギ抗マウスIgGは、Jackson ImmunoResearch(West Grove,PA)から購入した。
【0067】
化合物1〜8および11〜22は、以下に記載する方法(図6、スキーム1A)に従って合成し、化合物9および10は、3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリル酸から、それぞれ方法fおよび方法g(図6、スキーム1B)によって調製した。これらは表題化合物の下に別々に記載している。
【0068】
方法a 塩酸[1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(EDC)カップリング]
個々の短鎖脂肪酸の無水THF(5〜10 mmol/mL)溶液に、N2下で、様々なωアミノ酸メチルエステル(1当量)を加え、続いてEDC(1.3当量)を加えた。一晩撹拌した後、減圧下でTHFを除去し、残渣を酢酸エチル(100mL)に溶解した。この混合物を、水(50mL)で2回、さらに飽和食塩水(50mL)で続けて洗浄した。有機相をNa2SO4上で乾燥し、真空下で濃縮した。生じた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0069】
方法b(エステル開裂)
方法aから得られたエステルを2M KOH/MeOH溶液に溶解した。この混合物を80℃で1時間撹拌して0℃まで冷却した。さらに2N HClでpH 3まで酸性化し、真空下で濃縮して、酢酸エチル(100mL)およびH2O(50mL)を加えた。この有機相を分離して、それぞれ50mLの水および飽和食塩水で続けて洗浄した。さらにNa2SO4上で乾燥し、真空下で濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0070】
方法c [bis(2−オキソ−3−オキザゾリジニル)ホスホルジアミド(BOP−Cl)カップリング]
方法bから得られる酸の無水THF(5〜10mmol/mL)溶液に、N2下で、トリエチルアミン(TEA、1当量)を加えた。この混合物を室温で10分間撹拌し、BOP−Cl(1.1当量)、O−ベンジルヒドロキシルアミン塩酸(1当量)、およびTEA(3当量)を加えた。室温で一晩撹拌した後、この溶液を真空下で濃縮し、酢酸エチル(100mL)を加え、次に3%NaHCO3(50mL)を加えた。この有機相を分離して、それぞれ50mLの水および飽和食塩水で続けて洗浄した。さらにNa2SO4上で乾燥し、真空下で濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0071】
方法d(EDCカップリング)
方法bから得られる個々の酸の無水THF(5〜10mmol/mL)溶液に、N2下で、(2−アミノフェニル)カルバミン酸ベンジルエステル(1当量)を加え、次にEDC(1.3当量)を加えた。一晩撹拌した後、この混合物を真空下で濃縮し、酢酸エチル(100mL)を加えた。有機相を水(50mL)で2回、次に飽和食塩水(50mL)で続けて洗浄し、さらにNa2SO4上で乾燥して濃縮した。得られた残渣をシリカゲルフラッシュクロマトグラフィーによって精製した。
【0072】
方法e(水素化分解)
方法cまたは方法dから得られたN−ベンジルオキシもしくはN−Cbz誘導体を、メタノール/THF(5〜10mmol/mL)1:1溶液に溶解し、木炭上10%パラジウム(10% w/w)を加えた。この混合物を大気圧下で2時間、水素で処理し、ろ過した。溶媒をエバポレートし、残渣を酢酸エチルで再結晶化した。
【0073】
N−(2−アミノ−フェニル)−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド(1)
【0074】
【化10−1】
【0075】
【化10−2】
N−ヒドロキシ−4−[(2−プロピル−ペンタノイルアミノ)−メチル]−ベンズアミド(2)
【0076】
【化10−3】
N−(2−アミノ−フェニル)−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド(3)
【0077】
【化10−4】
N−ヒドロキシ−4−(2−プロピル−ペンタノイルアミノ)−ベンズアミド(4)
【0078】
【化10−5】
2−プロピル−ペンタン酸{4−[(2−アミノ−フェニルカルバモイル)−メチル]−フェニル}−アミド(5)
【0079】
【化10−6】
2−プロピル−ペンタン酸(4−ヒドロキシフェニルカルバモイルメチル−フェニル)−アミド(6)
【0080】
【化10−7】
【0081】
【化10−8】
2−プロピル−ペンタン酸{4−[2−(2−アミノ−フェニルカルバモイル)−エチル]−フェニル}−アミド(7)
【0082】
【化10−9】
2−プロピル−ペンタン酸[4−(2−ヒドロキシフェニルカルバモイル)−エチル)−フェニル]−アミド(8)
【0083】
【化10−10】
3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸
化合物9および10の前駆体であるこの化合物は、2−プロピル−ペンタン酸(0.78mL、4.9mmol)および3−(4−アミノ−フェニル)−アクリル酸メチルエステル(0.86g、4.9mmol)から、前述の方法aおよび方法bに従って合成した。全収量、1.05g(2工程で70%);
【0084】
【化10−11】
N−(2−アミノ−フェニル)−3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリルアミド(9)
3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸(200mg、0.7mmol)の無水THF溶液に、N2下で、ベンゼン−1,2−ジアミン(450mg、4.2mmol)を、次にEDC(180mg、0.9mmol)を加えた。一晩撹拌した後、この混合物を真空下で濃縮し、酢酸エチル(50mL)を加え、水(30mL)で2回、そして飽和食塩水(30mL)で続けて洗浄した。有機相をNa2SO4上で乾燥し、真空下で濃縮した。この粗生成物をフラッシュクロマトグラフィー(酢酸エチル−ヘキサン、1:1)によって精製し、白色の固体として化合物9(200mg、収量76%)を得た。
【0085】
【化10−12】
N−ヒドロキシ−3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリルアミド(10)
3−[4−(2−プロピル−ペンタノイルアミノ)−フェニル]−アクリル酸(100mg、0.35mmol)の無水DMF(3mL)溶液に、窒素下で、EDC(79mg、0.53mmol)およびヒドロキシベンゾトリアゾール水和物(HOBT)(62mg、0.46mmol)を加えた。この混合物を1時間撹拌し、塩酸ヒドロキシルアミン(27.4mg、0.39mmol)およびTEA(54μL)を加え、さらに12時間撹拌し、真空下で濃縮した。さらに、酢酸エチル(40mL)および飽和NaHCO3溶液(15mL)を加えた。有機相を分離し、それぞれ20mLの水および飽和食塩水で続けて洗浄した。この有機相をNa2SO4上で乾燥し、真空下で濃縮した。この粗生成物をフラッシュクロマトグラフィー[酢酸エチル−MeOH(9:1)]によって精製し、白色固体の化合物10(45mg、収量40%)を得た。
【0086】
【化10−13】
N−(2−アミノ−フェニル)−4−(ブチリルアミノ−メチル)−ベンズアミド(11)
【0087】
【化10−14】
N−(2−アミノ−フェニル)−4−(フェニルアセチルアミノ−メチル)−ベンズアミド(12)
【0088】
【化10−15】
N−(2−アミノ−フェニル)−4−[(4−フェニルブチリルアミノ)−メチル]−ベンズアミド(13)
【0089】
【化10−16】
4−(ブチリルアミノ−メチル)−N−ヒドロキシ−ベンズアミド(14)
【0090】
【化10−17】
N−ヒドロキシ−4−(フェニルアセチルアミノ−メチル)−ベンズアミド(15)
【0091】
【化10−18】
【0092】
【化10−19】
N−ヒドロキシ−4−[(4−フェニルブチリルアミノ)−メチル]−ベンズアミド(16)
【0093】
【化10−20】
4−ブチリルアミノ−N−ヒドロキシ−ベンズアミド(17)
【0094】
【化10−21】
N−ヒドロキシ−4−フェニルアセチルアミノ−ベンズアミド(18)
【0095】
【化10−22】
N−ヒドロキシ−4−(4−フェニルブチリルアミノ)−ベンズアミド(19)
【0096】
【化10−23】
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−ブチルアミド(20)
【0097】
【化10−24】
【0098】
【化10−25】
N−ヒドロキシ−3−(4−フェニルアセチルアミノ−フェニル)−プロピオンアミド(21)
【0099】
【化10−26】
N−[4−(2−ヒドロキシカルバモイル−エチル)−フェニル]−4−フェニル−ブチルアミド(22)
【0100】
【化10−27】
さらに別の化合物23〜67を含む、全ての化合物の一覧を図6に示す。
【0101】
(インビトロHDACアッセイ)
ヒストン脱アセチル化酵素アッセイキット(Upstate Biotechnology,Lake Placid,NY)を使用して、製造者による使用説明書にわずかな改変を加えてこれに従い、HDAC活性を測定した。このアッセイは、ヒストン脱アセチル化酵素活性に富むDU−145核抽出物が、ストレプトアビジンアガロースビーズに結合させたビオチン化[3H]−アセチルヒストンH4ペプチドの脱アセチル化を媒介する能力に基づく。上清への遊離した[3H]−アセテートの放出を測定し、HDAC活性を計算した。ナトリウムブチレート(0.25〜1mM)を陽性コントロールとして使用した。
【0102】
(細胞生存率アッセイ)
各ウェル当たり4,000個のDU−145細胞を播種した96ウェルの平底プレートにて、細胞生存率に対するHTPBの影響を、MTT{[3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニル−2H−臭化テトラゾリウム]}アッセイによって分析した。細胞を、5%CO2下37℃で、記載した時間、10% FBS添加RPMI−1640培地に記載した濃度にて溶解したHTPBに曝露した。この培地を取り除いて、RPMI−1640培地に溶解した0.5mg/ml MTT 150μlに置き換え、細胞を、37℃で2時間、CO2インキュベーターにてインキュベートした。ウェルから上清を除去し、還元されたMTT色素を200μl/ウェルのDMSOで可溶化した。プレートリーダーで、570nmにて吸光度を測定した。各処理は6ウェルずつ繰り返した。
【0103】
(酵素結合免疫吸着測定法(ELISA)によるアポトーシスの検出)
Cell Death ELISA(Roche Diagnostics,Manheim,Germany)を製造者の使用説明書に従って使用して、アポトーシスの誘導を評価した。この試験は、アポトーシス性細胞死誘導後の、モノヌクレオソームおよびオリゴヌクレオソーム形態の細胞質性ヒストン結合DNAフラグメントの定量的測定に基づいている。簡潔に述べると、実験の24時間前に1×106のDU−145細胞をT−75フラスコ内で培養した。細胞を、10% FBS添加RPMI−1640培地に、記載した濃度のHTPBで処理した。浮遊細胞および吸着細胞の双方を回収し、2×103個の細胞に相当する細胞溶解物をELISAに使用した。
【0104】
(ウェエスタンブロット分析)
10% FBS添加RPMI−1640培地中で、記載した濃度のHTPBで24時間処理したDU−145細胞(1×106個)を回収し、超音波処理した。Bradfordタンパク質アッセイキット(Bio−Rad,Hercules,CA)を使用して、この溶解物のタンパク質濃度を測定した;各溶解物由来の等量のタンパク質を10% SDSポリアクリルアミドゲルに溶解し、次いで半乾式トランスファーセルにてイモビロン−ニトロセルロース膜(Millipore,Bellerica,MA)上へ移した。0.1%のTWEEN20含有トリス緩衝化生理食塩水(TBS)(TBST)で、このトランスブロットした膜を2回洗浄した。5%ノンファットミルク含有TBSTで40分間ブロッキングした後、この膜をTBST−1%ノンファットミルクに溶解した一次抗体(1:1000希釈)と共に4℃で一晩インキュベートした。一次抗体で処理した後、この膜を計15分間、TBSTで3回洗浄し、続いてヤギ抗ウサギ、もしくは抗マウスIgG西洋ワサビペルオキシダーゼ結合体(1:3000稀釈)を室温で1時間処理し、さらにTBSTで3回、計1時間洗浄した。この免疫ブロットを高感度化学発光によって可視化した。
【0105】
(結果)
第一のシリーズの化合物に関して、本発明者らは、スキーム1(A、化合物1〜8;B、化合物9および10)にて記載した手順に従って、Zn2+係留結合体(図1)を合成する先導化合物としてバルプロ酸を用いた。化合物1〜8に関して、EDC活性化を介して、バルプロ酸を4つの異なるωアミノ酸メチルエステルスペーサーと結合させた。得られたエステルを、アルカリ加水分解により酸に分解した。代表的なペプチドカップリング条件(BOPClまたはEDC)の下で、得られた酸を、Bn保護ヒドロキシアミンおよびCbz保護ο‐フェニレンジアミンで処理し、水素化分解後に、それぞれアニリドおよびヒドロキサム酸を形成した(スキーム1A)。化合物9および10は、3−[4−(2−プロピル−ペンタノイル)−フェニル]−アクリル酸を、それぞれο‐フェニレンジアミンおよびヒドロキシルアミンと、代表的なEDCカップリング条件下で直接カップリングさせることによって合成した(スキーム1B)。
【0106】
(Zn2+キレートモチーフ係留バルプロエート誘導)
バルプロ酸を5種類の芳香族リンカーと、続いて2つのZn2+結合モチーフと様々に結合体化することによって、化合物1〜10を得た。これらの係留結合体は、様々な程度のHDAC阻害効力を示し(図2)、そのIC50値は5μM(化合物8)から80μM(化合物5)の範囲にわたった。この抑効力は、親分子(IC50、0.4mM)の8倍から5倍改善された。これらの結合体のうちのいずれかからバルプロイル部分もしくはZn2+キレートモチーフを除去すると、HDAC阻害活性は完全に失われ(データは示していない)、このことはタンパク質‐リガンド相互作用において、アシル基およびZn2+キレートモチーフが重要であることを示している。
【0107】
試験した10個の係留結合体の中で、化合物1、2、4および8は最適な誘導体(IC50、5〜8μM)であり、3、9、および10がこれに次ぐ(IC50、10〜20μM)。相対的に、ヒドロキサマト(化合物2、4、6、8および10)は、一般に、対応するフェニレンジアミン(化合物1、3、5、7および9)よりも作用効力が強かった。さらに、芳香族リンカーは、HDAC阻害活性に対して微小な影響を示した。試験した5つの芳香族ωアミノ酸の中で、(4−アミノフェニル)アセテートは、HDAC阻害効力が最も弱い結合体(5および6)となったが、4−(アミノメチル)ベンゾエートは最適であるようであった。さらなる構造的改変については、化合物1、2、4および8が、全てIC50<10μMを示したので、これらを先導化合物として使用した。
【0108】
(構造的改変)
バルプロイル基を除去すると、この結合体の阻害活性が完全に失われるという発見によって、活性部位ポケットと相互作用する際のアシル部分の重要性が強調された。従って、本発明者らは、化合物1、2、4および8のバルプロイル基を、ブチリル基、フェニルアセチル基またはフェニルブチリル基に置換して、HDAC阻害に対する立体電子効果を増大させた(図3)。これら全ての誘導体は、バルプロイル対応物と比較して、向上したHDAC阻害作用効力を示した。試験した様々なアシル官能基の中で、同一のスペーサーおよびZn2+キレートモチーフと結合させた場合、相対的効力は、フェニルブチリル基>フェニルアセチル基>ブチリル基>バルプロイル基の順であった。
【0109】
これらの12個の誘導体の中で、化合物19は特に注目すべきものであった。このヒドロキサマト係留フェニルブチレート(HTPB)(すなわち、化合物19)は、44nMというIC50を示し、これはフェニルブチレートに対して強度4倍の向上である。この化合物を使用して、DU−145前立腺癌細胞におけるHDAC活性に対する影響を調べた。
【0110】
(DU−145前立腺癌細胞におけるヒストンアセチル化およびp21WAF/CIP1発現に対するHTPBの影響)
ヒストンの過アセチル化(hyperacetylation)およびサイクリン依存性リン酸化酵素阻害因子であるp21WAF/CIP1の発現増加は、細胞内HDAC阻害に関する二つの特性である(Marks et al.,Nat Rev Cancer 1:194−202(2001))。従って、本発明者らは、ヒストンアセチル化およびp21WAF/CIP1発現の状態の特性を明らかにすることによって、DU−145前立腺癌細胞におけるHDAC活性に対するHTPBの影響を、TSAおよびフェニルブチレートと比較して試験した。
【0111】
DU−145細胞を、10%FBS添加RPMI 1640培地中で24時間、0.5、1、2.5μMのHTPB、0.25および0.5μMのTSA、または1、2.5、および5mMのフェニルブチレートに曝露した。細胞溶解物のウエスタンブロット分析は、これらの薬剤の処理によってアセチル化ヒストンH3およびH4、ならびにp21WAF/CIP1のレベルが上昇したことを示している(図4)。これらのバイオマーカーに対する1μM HTPBの影響は、0.25μM TSA、もしくは2.5mMフェニルブチレートの影響に近かった。DU−145細胞は、少量だが有意な量の内在性p21WAF/CIP1を示し、そのレベルは、0.5μMという低濃度のHTPBへ曝露した後に、実質的に増加した。まとめると、これらのデータによって、HTPBはDU−145細胞にてHDAC活性を標的としていることを確認した。
【0112】
(DU−145細胞の生存率に対するHTPBの影響)
10%FBS添加RPMI 1640培地中のDU−145細胞において、癌細胞生存率に対するHTPBの影響を調べた。これらの細胞は、HTPBに対して高度の感受性を示し、増殖阻害におけるIC50は、約0.5μMと1μMの間であった(図5A)。DNAフラグメント化によって証明されたように、HTPBは用量依存的様式でDU−145細胞のアポトーシスに対する感受性を増加させた(図5B)。示したように、薬物の濃度が1μMを超えたときに、24時間にて広範囲のアポトーシスが発生し、これは少なくとも部分的には、この細胞毒性効果がHDAC阻害によってアポトーシスが誘導されたことに起因することを示している。試験した他の細胞株としては、AN3CA子宮内膜癌細胞、ならびにSW−48およびHCT−15結腸直腸癌細胞が挙げられる。これらの癌細胞もまた、同程度の効力でHTPBの細胞毒性効果に対して感受性であった(データは示さず)。
【0113】
(考察)
本明細書中で、本発明者らは、新しい種類のHDAC阻害剤の開発を示し、この阻害剤において、短鎖脂肪酸が疎水性結合を介してZn2+キレートモチーフに係留された。TSAおよびSAHAによるHDAC阻害の独特な様式により提供された作用モデルについて、これらの化合物を開発する際の本発明者らのストラテジーが立てられた(Finnin et al.,Nature 401:188−193(1999))。本発明者らの新しい係留ストラテジーからHTPB(すなわち化合物19)の発見が導かれ、これはSAHAについて報告された濃度(Richon et al.,Proc Natl Acad Sci USA 95:3003−3007(1998))に一致する準μM濃度で、HDAC阻害活性および抗増殖活性を示す。
【0114】
本発明者らは、いくつかの癌細胞株において、HTPBがHDAC活性を標的とする2つの系列の証拠を得た。具体的には、DU−145前立腺癌細胞を、0.5μMもの低濃度でHTPB処理すると、用量依存的な様式でヒストンH3並びにH4の過アセチル化が引き起こされた。同様に、HTPBに応答して、p21WAF/CIP1発現が実質的に上方制御された。対照的に、ヒストンアセチル化およびp21WAF/CIP1発現に対する同様の細胞内効果を達成するのに、親分子であるフェニルブチレートは、少なくとも2.5mMを必要とした。
【0115】
HTPBは、構造的に既存のHDAC阻害剤と区別され、その多くは、キャップ基が極性平面構造から構成されている。例えば、TSA、SAHAおよびMS−275のキャップ基は、それぞれジメチルアミノフェニル基、フェニルアミノ基およびピリジン−3−イル−メトキシカルボニル基を含有する。従って、活性部位ポケットは、キャップ基に異なる立体電子特性を与える高度な柔軟性を示すと、本発明者らはさらに結論づけた。本発明者らのデータは、結合体の活性部位ポケットへの結合促進において、フェニルブチリル基およびフェニルアセチル基は脂肪族アシル部分より効果的であったことを示している。この矛盾は、部分的に、電子密度の違いおよび/または分岐した側鎖による立体化学的障害に起因すると推測される。芳香族リンカーに関しては、4−アミノ安息香酸が、フェニルブチリル基とヒドロキサマトとを係留するのに最適であるようであり、その長さはポケットの両端に接触するのに十分なものであった。
【0116】
(第二世代HDAC阻害剤の設計および合成−化合物19の構造に基づく最適化)
化合物19(HTPB)は、構造的に既存のHDAC阻害剤とは区別され、多くは例えばTSA、ジメチルアミノフェニル;SAHA、フェニルアミノ;およびMS−27−275、ピリジン−3−イル−メトキシカルボニルのような、極性の平面構造から成るキャップ基を有する。この知見は、HDAC活性部位ポケットがキャップ基に異なる立体電子特性を与える、高度な柔軟性を示していることを示唆する。
【0117】
リガンド結合を予測するため、本発明者らは、化合物19(図9、赤字)およびTSA(図9、黄字)の、HDLP活性部位ポケットへの分子ドッキングを行い、その後個々のリガンドの認識様式を比較するためにエネルギーを最小化した。図9に示したように、双方のリガンドともポケットへの結合は類似した配置をとっている。化合物19の芳香族リンカーである4−アミノ安息香酸は、フェニルブチリル基とヒドロキサマトを係留するのに最適な長さを提供し、これによって双方の官能基はポケットの両端に接触することを可能にする。ヒドロキサム酸基[C(O)NH−OH]によって、化合物19のHDAC阻害効力はフェニルブチレートの4桁の大きさの増加に寄与するようである。
【0118】
図9のモデリングデータは、リガンド結合におけるヒドロキサマトの役割に2要素があることを示唆している。1つは、Zn2+陽イオンをキレートする。2つ目は、ヒドロキサマトは、His131−Asp173電荷リレー系の助けと共に、NH−OHからHis−131のNτへのプロトン移動を促進する(図10)。(TSAのリガンド結合研究については、Vanommeslaeghe,K.,Van Alsenoy,C.,De Proft,F.,Martins,J.C.,Tourwe,D.,and Geerlings,P.Ab initio study of the binding
of Trichostatin A(TSA) in the active site of histone deacetylase like protein(HDLP).Org Biomol Chem,1:2951−2957,2003を参照する。)
結論として、ヒドロキサマトHDAC阻害剤である、TSAあるいは化合物19のいずれかの結合によってAspからヒドロキサマトへの負電荷の転移が生じ、その結果、負荷電ヒドロキサマトとZn2+の正電荷およびイミダゾール環との間に塩架橋が形成される(図10、中央パネル)。機構的に、この塩架橋は、ヒドロキサマトベースのリガンドと活性部位ポケットとの結合に寄与する主要な力となっており、フェニルブチレートから化合物19への転換に伴う、HDAC阻害効力の104倍の増加に必要とされる微分自由エネルギー変化(ΔΔG‡=5.4kcal/mol)の根底となっている(IC50、0.4mMに対し44nM)。
【0119】
結合親和力におけるこの塩架橋の役割は、化合物19のヒドロキサマト部分をフェニレンジアミン基に置換した(化合物55)ときにHDAC阻害効果が10倍低下する(すなわち、IC50、0.044μMに対して0.4μM)ことによって、さらに強調される。ヒドロキサマトとは対照的に、フェニレンジアミン基の活性部位への結合には、His131のNτへのプロトン転移が関係しない。代わりに、His131との荷電‐荷電相互作用を形成することなく、電子に富むジアミンのみがZn2+陽イオンをキレートする。その結果、フェニレンジアミンベース(例えば、化合物55)のリガンドとの結合親和性は、対応するヒドロキサマトと比較して著しく低下する。
【0120】
この分子ドッキングはまた、化合物19の次なる改変のための有用な指針を提供した。図9に示したように、TSAおよび化合物19のキャップ基は、Tyr91、Glu92、Gly140、およびPhe141に囲まれた溝近くに位置する。本来この溝は、様々な程度のかさ高さを有するキャップ基を適合させる柔軟性を提供し、HDAC阻害効力を増大させるのに活用され得る。
【0121】
従って、本発明者らは、化合物19のフェニルブチリル部分を、様々なα分岐芳香族脂肪族アシル基に置換した。その原理は、2つであった。第1に、フェニルブチリル基と化合物19内のリンカーとの間のアミド結合が、タンパク質分解性消化に感受性であり得る。アシル基の嵩高さが増加することによって、アミド結合がより立体的に妨げられたものに変わることにより、代謝安定性が増大し得る。第2に、アシル基の大きさが増したことで、前記溝との疎水結合が増大し、これによって結合親和力が増し得る。
【0122】
このストラテジーから、化合物19(すなわち、HTPB)よりも大きな作用効力を有する、多数のHDAC阻害剤が導かれた。いくつかの代表的な誘導体の構造およびIC50を表1にまとめる。
【0123】
表1 代表的なフェニルブチレートベースのHDAC阻害剤
【0124】
【表1】
合成した80を超える誘導体の中で、化合物42がTSAに匹敵するIC50を持つ最適な薬剤である。分子モデリング解析によって、化合物42は、活性部位ポケット内側でTSAの原子配置と並立する配置をとっていることが示されている(図11を参照のこと)。さらに、イソプロピル部分が溝の内側にあり、隣接する疎水性残基と相互作用し得る。それゆえ、さらなるインビトロおよびインビボでの特徴付けのために、化合物42を選択した。
【0125】
(化合物42およびTSAは、抗増殖効果を後成的かつ細胞レベルで媒介する−新規の細胞標的の同定)
PC−3アンドロゲン非依存性前立腺癌細胞にて、p21WAF/CIP1発現およびヒストンH4過アセチル化に対する影響についての試験に、化合物42、SAHA、およびTSAを供した。図12は、これらのバイオマーカーの誘導における化合物42の効力がTSAに匹敵し、SAHAよりも約5倍高いことを示している。
【0126】
PC−3細胞(PTEN−/−)におけるAkt活性化状態に対するこれらの薬剤の影響もまた、試験した。Aktは、アンドロゲン非依存性およびこれらの細胞の化学治療耐性に寄与する機能性PTENが存在しないため、PC−3細胞にて構造的に活性化されている。PC−3細胞にて、化合物42とTSAのいずれもが、1μMもの低濃度で著しいAkt脱リン酸化を引き起こしたことに注目すべきである(図13)。対照的に、リン酸化Aktに対するSAHAの影響は認められなかった。これはSAHAと化合物42/TSAとの間には、作用様式に僅かな差異があることを示唆している。
【0127】
PC−3細胞におけるリン酸化Aktに対する化合物42の影響は、多くがPTEN変異を示すホルモン不応性前立腺癌細胞におけるこのHDAC阻害剤の臨床投与の点から注目に値する。しかし、この脱リン酸化効果はリン酸化酵素特異的なものである。試験した他の一連のシグナル伝達リン酸化酵素の中で、FAK(局所接着リン酸化酵素)およびERK類のリン酸化レベルは投与量依存的に減少したが、p38あるいはJNKのリン酸化レベルは影響を受けないままであった(図14)。
【0128】
本発明者らは、HDACアイソザイムと複合体を形成してその核への局在化を促進するプロテインホスファターゼ1(PP1)により、この脱リン酸化が仲介されると仮説を立てる。本発明者らは、PC−3細胞を化合物42あるいはTSAで処理すると核内でHDAC−PP1複合体の分解が起こり、PP1の細胞質内への再局在を誘導して標的リン酸化酵素の脱リン酸化を媒介することを提唱する。あるいは、この効果は熱ショックタンパク質(HSP)−90のアセチル化に起因し、その結果Aktへの結合性が減少してその後Aktが分解されるとも考えられる(Fuino,L.,Bali,P.,Wittmann,S.,Donapaty,S.,Guo,F.,Yamaguchi,H.,Wang,H.G.,Atadja,P.,and Bhalla,K.Histone deacetylase inhibitor LAQ824 down−regulates Her−2 and sensitizes human breast cancer cells to trastuzumab,taxotere,gemcitabine,and epothilone B.Mol Cancer Ther,2:971−984,2003)。
【0129】
(前立腺癌に対する化合物42の抗腫瘍効果のインビボでの特徴付け)
定着したPC−3異種移植片腫瘍の増殖に対する経口投与した化合物42(1日当たり50mg/kgおよび100mg/kg)の影響を、腹腔内注射したSAHA(1日当たり50g/kg)と比較して調べた。さらに本発明者らは、化合物42のHDAC阻害効力に匹敵する阻害効力を持つという点から、化合物44(1日当たり50mg/kgおよび100mg/kg)も取り入れた(IC50、32nMに対し25nM)。化合物42と44の双方とも、用量依存的に、10%FBS含有培地にてPC−3細胞の増殖をインビボで抑制するのに有効であり、IC50値はそれぞれ0.6μMと0.8μMであった(図15、72時間処理)。
【0130】
図16は、上記のようにHDAC阻害剤を処理した胸腺欠損マウスにおける皮下PC−3異種移植片腫瘍の増殖を表す(パネルA、平均腫瘍容積±SDとして表したデータ;パネルB、ビヒクル、化合物44並びに化合物42で処理したグループの代表的なマウスの写真)。示しているように、化合物42と44のいずれも、いずれかの投与量で、経口経路を介して、定着したPC−3腫瘍の増殖を抑制するのに有効であった。1日当たり50mg/kgで経口的に投与した化合物42のインビボでの効力は、同じ量を腹腔内投与したSAHAの効力に匹敵し、腫瘍の増殖を、ビヒクルで処理したコントロールグループと比較して、それぞれ70.4%並びに69.0%減少させた。1日当たり100mg/kgで、化合物42はPC−3腫瘍の増殖をほぼ完全に抑制し(91.7%の減少)、体重の継続測定並びに病理的検査によって確認したように明白な副作用もみられなかった。
【0131】
本研究の結論として、Ohio State University College
of Veterinary Medicine(オハイオ州立大獣医学部)の、専門医師会認定の獣医病理学者による、各処理群の1匹のマウスの完全病理検査を実施した。この検査は、これは臨床病理学(血液学、血清化学)ならびに完全剖検(27種類以上の組織および器官の肉眼および顕微鏡検査)を包含した。剖検で観察された肉眼的病理学的異常は、SAHA処理マウス、およびDMSO処理コントロールマウスの腹膜内の小さな接着に限られており、これは毎日腹腔内注射を行った副作用のようである。血液パラメーターは、好中球の増加を除いて正常範囲内であり、これはビヒクル処理コントロールマウス群、ならびにSAHAおよび化合物44(50mg/kg)処理群の双方のマウスのみに観察された。血清化学値は、ビヒクル処理マウスでは脱水症状に一致する値となったが、他の全てのマウスでは正常範囲内であった。
【0132】
生物学的応答とインビトロで同定された提唱される作用機構とを相関させるため、経口投与した化合物42および腹腔内投与したSAHAが、PC−3異種移植片腫瘍内のヒストンH−3のアセチル化を調節する作用を、免疫ブロットによって分析した。図17は、ビヒクル、1日当たり50もしくは100mg/kgの経口化合物42、あるいは1日当たり50mg/kgの腹腔内SAHAで28日間処理した腫瘍保有マウス由来の、2つの代表的なPC−3腫瘍のホモジネート内のヒストンH3とアセチル化H3のウエスタンブロットを、様々な容量で表す。薬剤処理グループにおいて、インビボHDAC阻害の特性である、ヒストンH3のアセチル化レベルの著しい増加が認められた。
【0133】
インビボ実験にて、等量の血清を含まない培地とマトリゲル基底膜マトリックスとに懸濁したPC−3細胞を、5〜7週齢のオスNCr胸腺欠損ヌードマウス(nu/nu)の脇腹に皮下注射した(0.5×106 細胞/0.1ml/マウス)。腫瘍容積が170〜200mm3に達したときに、強制経口投与によって化合物42もしくは44を(1日当たり50および100mg/kg)、あるいは腹腔内注射によってSAHA(1日当たり50mg/kg)を毎日処理することを開始した(各グループにつきN=6)。コントロールグループには、経口あるいは腹腔内経由でビヒクルのみ(それぞれ0.1%メチルセルロース/0.05%TWEEN80水溶液、およびDMSO)を与えた。処理は、コントロール群の平均腫瘍容積が約1,500mm3に達するまで継続した。ノギスを使用して腫瘍を毎週測定し、標準公式:(幅)2×(長さ)×0.52を用いて腫瘍の容積を算出した。体重を毎週測定し、実験期間中一定であった。
【0134】
乳癌細胞株、肺癌細胞株、および甲状腺癌細胞株における、化合物42のインビトロ抗増殖効果。NCI開発途上治療プログラム(DPT)による60細胞株スクリーニングによると、化合物42は、60細胞株全てに対し、平均GI50(細胞増殖の50%抑制)値が0.2μMという強力なインビトロ抗増殖活性を示した。さらに、本発明者らは、甲状腺癌細胞株において化合物42を試験した。代表的な乳癌細胞株、肺癌細胞株、および甲状腺癌細胞株における化合物42の用量依存的抗増殖効果を、図18に示す。
【0135】
(慢性リンパ球性白血病(CLL)における化合物42のインビトロおよびインビボ抗増殖効果)
HDAC抑制能並びに細胞毒性促進能について、化合物42を初代CLL細胞にて試験した。図19に示したように、HDACが抑制される1μM以上の濃度で、著しい細胞毒性が認められた(N=6)。さらに、TLC−1遺伝子移入マウスモデルを使用したインビボ予備試験によって、経口胃管注入によって100mg/kgの化合物42を4週間、5日投与/2日非投与し、生存率が向上する傾向と顕著な毒性がないことが示された。
【0136】
(立体選択性)
驚くべきことに、化合物42のHDAC阻害活性が立体選択的であることを発見した。S異性体は、R異性体対応物よりも強力である(IC50、15nMに対し80nM)。
【0137】
まとめると、分子モデリングとコンビナトリアル化学技術とを組み合わせることにより、短鎖脂肪酸を骨格として使用して新たな種類の強力なHDAC阻害剤を開発した。最適な薬剤である化合物42(NSC−D−731438)は、25nMのIC50でHDAC活性を阻害し、これは親化合物フェニルブチレートの10,000倍以上の増大である。化合物42は、これが臨床へ持ち込まれる有望な候補にする理由となるいくつかの独特の特性を示している。第一に、化合物42は、後生的機構および細胞性機構の両方を介して、インビトロで抗増殖効果を媒介する。この特性はTSAと同様であるが、SAHAにはないものである。第二に、これは、MS−275およびSAHAよりも優れたインビトロおよび/またはインビボでの効力を有しながら経口的に生物学的利用可能である。第三に、1日当たり50あるいは100mg/kgの投与量で28日間の過程を終えても、腫瘍保有マウスにて明らかな毒性を示さない。このことは、長期投与および他の標的治療との組み合わせのための機会を与える。最後に、化合物42は単純な構造を有し、大規模な合成に適する。
【0138】
(化合物42の生成)
本発明者らは、数グラム単位で多数回、化合物42を合成した。基本的に、4段階合成を経て調製し、全体の収量は52%であった(図20)。出発物質は全て容易に入手できるものであり、この合成は実験室の設定にて数百グラム単位のスケールアップが容易である。その手順を以下に記載する。中間生成物および最終産物は、クロマトグラフィーによる分離を用いなくとも99%以上の純度で、結晶化によって反応混合物から単離され得る。全体として、合成および精製の手順は単純明快であり、この化合物の大規模生産に関して特別な問題点は無い。
【0139】
N−(4−アセチル−フェニル−3−メチル−2−フェニル−ブチルアミド(1))。(α−イソプロピル)−フェニル酢酸(4g、22.3mmol)を、塩化チオニル(30ml)に溶解し、50℃で1時間加熱した。溶媒を除去した後、残渣をTHF(50ml)に溶解し、p−アミノ安息香酸メチルエステル(3.4g、22.5mmol)およびEt3N(3.5ml、25mmol)のTHF(50ml)溶液を、室温で撹拌しながら加えた。4時間後、真空下でTHFを除去して酢酸エチル(200ml)に溶解し、水(100ml)と食塩水(100ml)で続けて洗浄した。Na2SO4上で有機相を乾燥し、真空下で濃縮して化合物1を得た(5.6g;収量85%)。その後精製することなく、この粗生成物を次の工程に使用した。
【0140】
4−(3−メチル−2−フェニル−ブチリルアミノ)安息香酸(2)。化合物1(5.6g;20mmol)を、KOH(21g)を含有するメタノール(150ml)に溶解した。この混合物を2時間還流して0℃に冷却し、12N HCl(12N)を滴下して生成物を沈殿させた。真空下で溶媒を除去し、冷水(120ml)を加えた。ろ過によって沈殿物を回収し、水で洗浄して乾燥し、化合物2を得た(5g;収量84%)
N−ベンジルオキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド(3)。N2下で、2(5g;16.8mmol)の無水THF(120ml)溶液に、トリエチルアミン(TEA、2.5ml;16.8mmol)を加えた。この混合物を室温で10分間撹拌し、bis(2−オキソ−3−オキサゾリジニル)塩化ホスホルジアミド(BOP−Cl)(4.7g;18.7mmol)、塩酸O−ベンジルヒドロキシアミン(2.7g;17mmol)、およびTEA(7.5ml)を加えた。室温で一晩撹拌後、この溶液を真空下で濃縮し、酢酸エチル(200ml)、次いで3%NaHCO3(80ml)を加えた。有機相を分離して、それぞれ100mlの水および飽和食塩水で続けて洗浄し、Na2SO4上で乾燥して真空下で濃縮した。この残渣(6.1g、収量90%)をその後精製することなく、直接水素化分解に使用した。
【0141】
N−ヒドロキシ−4−(3−メチル−2−フェニル−ブチリルアミノ)−ベンズアミド(4)。化合物3(6.1g;15.2mmol)をメタノール/THFの1:1溶液(120ml)に混合し、木炭上の10%パラジウム(0.6g、10% w/w)を加えた。この混合物を大気圧下で2時間、水素で処理し、ろ過した。溶媒をエバポレートし、残渣を酢酸エチルで再結晶化した。収量は4.2gであった(収量90%)。
【0142】
本発明に従うHDAC阻害剤の代替的な合成スキームを、図21に図示する。
【0143】
本発明の他の実施形態は、この明細の考察並びにこの明細にて開示されている発明の実践から、当業者には明白であろう。この明細および実施例は、以下の請求項によって示される本発明の真の範囲および本質を伴う例示にすぎないものであるとみなされることが意図される。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7−1】

【図7−2】

【図7−3】

【図7−4】

【図7−5】

【図7−6】

【図7−7】

【図7−8】

【図7−9】

【図7−10】

【図7−11】

【図8−1】

【図8−2】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7−1】
【図7−2】
【図7−3】
【図7−4】
【図7−5】
【図7−6】
【図7−7】
【図7−8】
【図7−9】
【図7−10】
【図7−11】
【図8−1】
【図8−2】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2011−190283(P2011−190283A)
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−144993(P2011−144993)
【出願日】平成23年6月29日(2011.6.29)
【分割の表示】特願2006−542704(P2006−542704)の分割
【原出願日】平成16年12月1日(2004.12.1)
【出願人】(504325287)ザ オハイオ ステート ユニバーシティー リサーチ ファウンデーション (24)
【Fターム(参考)】
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【出願番号】特願2011−144993(P2011−144993)
【出願日】平成23年6月29日(2011.6.29)
【分割の表示】特願2006−542704(P2006−542704)の分割
【原出願日】平成16年12月1日(2004.12.1)
【出願人】(504325287)ザ オハイオ ステート ユニバーシティー リサーチ ファウンデーション (24)
【Fターム(参考)】
[ Back to top ]