
アミロイドーシスを処置するための製剤および方法
【課題】アミロイドーシスを処置するための方法、製剤、および組成物の提供。
【解決手段】腎機能の安定化もしくは改善、または腎疾患の進行を遅らせるために、式(I)を有する治療的有効量の化合物が適用される:

式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3、または4であり;およびmは1または2でありうる。但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【解決手段】腎機能の安定化もしくは改善、または腎疾患の進行を遅らせるために、式(I)を有する治療的有効量の化合物が適用される:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3、または4であり;およびmは1または2でありうる。但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、その全内容物が参照により本明細書に組み入れられる、2005年4月15日に提出された米国特許仮出願第60/671,866号に対する優先権を主張する。
【背景技術】
【0002】
発明の背景
アミロイドーシスは、特異的臓器における不溶性の原線維タンパク質(アミロイド)の細胞外沈着に関連し、最終的に罹患臓器の不全に至る多数の疾患に関する一般用語である。R.H. Falk et al., The Systemic Amyloidosis, 337 N ENGL J MED 898-909 (1997)(非特許文献1), P.N. Hawkins, Amyloidosis, 9 BLOOD REV 135-42 (1995)(非特許文献2), J.D. Sipe, Amyloidosis, 31 CR REV CLIN LAB SCI 325-54 (1994)(非特許文献3); A.S. Cohen, Amyloidosis, 40(2) BULL RHEUM DISEASES 1-12 (1991)(非特許文献4)。アミロイド沈着は、一つの臓器に限定されたままとなりうる(限局性アミロイドーシス)、またはより広く分布する可能性がある(全身性アミロイドーシス)。全身性アミロイドーシスは一般的に、原線維沈着の性質に基づいて四つのタイプに分類される:(i)特発性または原発性アミロイドーシス(ALアミロイドーシス);(ii)反応性、二次性、またはアミロイドA(AA)アミロイドーシス;(iii)家族性アミロイド性多発神経障害;および(iv)透析関連アミロイドーシス。その発生は多様であるが、全てのアミロイド沈着は共通の形態学的特性を有し、特異的色素(例えば、コンゴーレッド)によって染色され、染色後に偏光において特徴的な複屈折外観を有する。それらはまた、共通の超音波特色を有し、共通のX-線回折および赤外線スペクトルを有する。
【0003】
AAアミロイドーシスは、炎症に反応して肝細胞から産生および分泌される急性期タンパク質である、前駆体の血清アミロイドA(SAA)から形成されたアミロイドA(AA)タンパク質に関連すると考えられている。AAアミロイドーシスは、慢性炎症状態(例えば、リウマチ性関節炎、強直性脊椎炎、炎症性腸疾患等)、慢性感染症(例えば、結核、骨髄炎等)、および遺伝性の発熱、例えば家族性地中海熱に関連する(R.H. Falk et al., 337 N ENGL J MED 898-909 (1997)(非特許文献5), A.S. Cohen, 40(2) BULL RHEUM DISEASES 1-12 (1991)(非特許文献6), G. Grateau, 12 CURRENT OPINION IN RHEUMATOL 61-64 (2000)(非特許文献7))。リウマチ性関節炎は、西欧および北米におけるAAアミロイドーシスの主な原因である(M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)(非特許文献8), M.A. Gertz, Secondary amyloidosis, 232 J INT MED 517-18 (1992)(非特許文献9))。
【0004】
AAアミロイドーシスは、腎臓、脾臓、肝臓、および副腎のような実質様臓器に主に罹患する。AAアミロイドーシスの最も一般的な臨床特色は、ネフローゼ範囲のタンパク尿症として発現する腎機能障害、または診断時の腎機能不全である。末期腎不全は、症例の40〜60%における死因である(M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)(非特許文献8), M.A. Gertz, 232 J INT MED 517-18 (1992)(非特許文献10), M.A. Gertz and R.A. Kyle, 70 MEDICINE 246-256 (1991)(非特許文献11))。胃腸管の罹患もまた頻繁であり、通常慢性の下痢、体重減少、および吸収不良として現れる。肝臓および脾臓の肥大も同様に何人かの被験体において起こる可能性がある。心臓の罹患はまれであり、疾患の後期に起こる。診断からの生存期間の中央値は、診断時の疾患の進行期に応じて2〜8年である(M.A. Gertz and R. A. Kyle, 70 MEDICINE 246-256 (1991)(非特許文献11))。
【0005】
AAアミロイドーシスは通常、慢性感染症(結核のような)、または慢性炎症(リウマチ性関節炎または遺伝性発熱)に関連して認められる。家族性型のAAアミロイドーシスは、家族性地中海熱(FMF)において認められる。この家族型アミロイドーシスは遺伝され、特異的集団群において認められる。ALおよびAAアミロイドーシスの双方において、沈着はいくつかの臓器において見いだされ、このように全身性アミロイド疾患であると考えられる。
【0006】
「局所アミロイドーシス」は、一つの臓器系に罹患する傾向があるアミロイドーシスである。異なるアミロイドーシスはまた、沈着物に存在するタンパク質のタイプを特徴とする。例えば、スクレイピー、ウシ海綿状脳症、クロイツフェルト-ヤコブ病等のような神経変性疾患は、中枢神経系におけるプロテアーゼ抵抗性型のプリオンタンパク質(AScrまたはPrP-27と呼ばれる)の出現および蓄積を特徴とする。同様に、もう一つの神経変性障害であるアルツハイマー病は、神経炎プラークおよび神経原線維のもつれを特徴とする。この場合、実質および血管において認められるアミロイド斑は、原線維Aβアミロイドタンパク質の沈着によって形成される。成人発症型糖尿病(II型糖尿病)のような他の疾患は、膵臓におけるアミロイド原線維の局所蓄積を特徴とする。
【0007】
これらのアミロイドがひとたび形成されると、その場でアミロイド沈着を有意に溶解する、さらなるアミロイドの沈着を予防する、またはアミロイド沈着の開始を予防する公知の広く容認された治療または処置はない。
【0008】
それぞれのアミロイド原性タンパク質は、コンフォメーションの変化を受ける能力、β-シートを構築する能力、および細胞内または細胞外で沈着される可能性がある不溶性の原線維を形成する能力を有する。それぞれのアミロイド原性タンパク質は、アミノ酸配列が異なるが、原線維を形成して、プロテオグリカン、アミロイドP、および補体成分のような他の要素に結合するという同じ特性を有する。その上、それぞれのアミロイド原性タンパク質は、配列は異なるが、β-シート形成を促進する他の領域と共に、プロテオグリカンのグリコサミノグリカン(GAG)部分に対する結合能を有する領域(GAG結合部位と呼ばれる)のような類似性を示すアミノ酸配列を有する。プロテオグリカンは、体内のほぼ至る所に分布する様々な大きさおよび構造の高分子である。それらは、細胞内区画において、細胞表面上で、および細胞外マトリクスの一部として見いだされうる。全てのプロテオグリカンの基本構造は、コアタンパク質と、コアタンパク質に付着する少なくとも一つ、しかし頻繁に複数の多糖類鎖(GAGs)を含む。コンドロイチン硫酸、デルマタン硫酸、ケラタン硫酸、ヘパリン、およびヒアルロナンを含む異なる多くのGAGsが発見されている。
【0009】
いくつかのGAG模倣体は、アミロイド沈着を阻害するために、および/またはいくつかの型のアミロイドーシスを処置するために有用であることが知られている。WO 94/22437(特許文献1)、WO 96/28187(特許文献2)、およびWO 00/64420(特許文献3)を参照されたい。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO 94/22437
【特許文献2】WO 96/28187
【特許文献3】WO 00/64420
【非特許文献】
【0011】
【非特許文献1】R.H. Falk et al., The Systemic Amyloidosis, 337 N ENGL J MED 898-909 (1997)
【非特許文献2】P.N. Hawkins, Amyloidosis, 9 BLOOD REV 135-42 (1995)
【非特許文献3】J.D. Sipe, Amyloidosis, 31 CR REV CLIN LAB SCI 325-54 (1994)
【非特許文献4】A.S. Cohen, Amyloidosis, 40(2) BULL RHEUM DISEASES 1-12 (1991)
【非特許文献5】R.H. Falk et al., 337 N ENGL J MED 898-909 (1997)
【非特許文献6】A.S. Cohen, 40(2) BULL RHEUM DISEASES 1-12 (1991)
【非特許文献7】G. Grateau, 12 CURRENT OPINION IN RHEUMATOL 61-64 (2000)
【非特許文献8】M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)
【非特許文献9】M.A. Gertz, Secondary amyloidosis, 232 J INT MED 517-18 (1992)
【非特許文献10】M.A. Gertz, 232 J INT MED 517-18 (1992)
【非特許文献11】M.A. Gertz and R.A. Kyle, 70 MEDICINE 246-256 (1991)
【発明の概要】
【0012】
発明の概要
一つの態様において、本発明は、腎障害(PRI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、以下の式の化合物の治療的有効量を標的被験体に投与することによって、標的被験体におけるAAアミロイドーシスを処置するかまたは予防する方法に関する:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2である。さらに、この態様において、標的被験体はAAアミロイドーシスに関して処置され、腎障害に関連したパラメータを有する、またはそれに対して感受性がある。さらなる態様において、式(I)の化合物は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩である。
【0013】
もう一つの態様において、本発明には、胃腸管障害(PGI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を標的被験体に投与することによって、標的被験体におけるAAアミロイドーシスを処置または予防する方法が含まれる。さらに、この態様において、標的被験体はAAアミロイドーシスに関して処置され、胃腸管障害に関連するパラメータを有する、またはそれに対して感受性がある。
【0014】
もう一つのさらなる態様において、本発明には、アミロイド関連疾患が処置または予防されるように、クレアチニンクリアランス速度に基づいて選択された用量の式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与することによって、被験体におけるアミロイド関連疾患を処置または予防する方法に関する。
【0015】
本発明はまた、少なくとも部分的に、例えばAUC、Cmax、AUCss、Css、Tmax等によって測定した場合に、被験体において有効な曝露が提供されるような用量で投与される、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体におけるAAアミロイドーシスを処置または予防するための方法に関する。
【0016】
さらに、本発明はまた、被験体における腎および/または胃腸管機能を安定化するかまたは改善する方法にも関する。方法には、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0017】
もう一つの態様において、本発明は、被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、AAアミロイドーシスが処置または予防されるように、第二の物質と併用して、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与する段階が含まれる。
【0018】
なおもう一つの態様において、本発明は、少なくとも部分的に、被験体における化合物の経口生物学的利用率が増加するように、食物なしで薬学的組成物において式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与することによって、被験体における化合物の経口生物学的利用率を増加させる方法に関する。
【0019】
本発明はまた、少なくとも部分的に、炎症疾患が被験体において処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体における炎症疾患を処置する方法にも関する。
【0020】
本発明はまた、少なくとも部分的に、被験体における遺伝性の発熱が処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体における遺伝性発熱を処置する方法にも関する。
【0021】
本発明はまた、少なくとも部分的に、被験体におけるリウマチ性関節炎を処置するための方法にも関する。方法には、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0022】
さらに、本発明にはまた、被験体における悪性新生物を処置する方法が含まれる。方法には、悪性新生物が被験体において処置されるように、第二の物質と併用して、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与する段階が含まれる。
【0023】
さらなる態様において、本発明は、少なくとも部分的に、被験体における慢性感染症、例えば微生物またはウイルス感染症を処置する方法に関する。方法には、慢性感染症が被験体において処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0024】
もう一つのさらなる態様において、本発明は、少なくとも部分的に、炎症性疾患、悪性新生物、慢性感染症、または遺伝性の発熱を有する被験体における腎機能を安定化するかもしくは改善させる、または腎疾患の進行を遅らせる方法に関する。方法には、腎機能を安定化するかもしくは改善するように、または腎疾患の進行が遅れるように、1,3-プロパンジスルホン酸またはその薬学的に許容される塩の治療的有効量を被験体に投与する段階が含まれる。
【0025】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるESRD/透析への進行を予防するかまたは遅らせるための方法に関する。方法には、ESRD/透析への進行が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、被験体、例えばAAアミロイドーシスを有する被験体に投与する段階が含まれる。
【0026】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における血清クレアチニン倍加までの時間を予防するかまたは遅らせるための方法に関する。方法には、血清クレアチニン倍加までの時間が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0027】
なおもう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%減少までの時間を予防するかまたは遅らせるための方法に関する。方法には、クレアチニンクリアランスの少なくとも50%減少までの時間が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0028】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%増加までの時間を減少させるための方法に関する。方法には、クレアチニンクリアランスの少なくとも50%増加までの時間が減少するように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0029】
さらにもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの勾配によって測定した腎疾患の進行速度を低減させるための方法が含まれる。方法には、腎疾患の進行速度が低減されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0030】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるタンパク尿症を安定化するかまたは低減させるための方法に関する。方法には、被験体におけるタンパク尿症が安定化されるかまたは低減されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0031】
なおもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体における腎機能を安定化するか、または腎疾患の進行を遅らせる方法が含まれる。方法には、腎機能を安定化するか、または腎疾患の進行が遅れるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。一つの局面において、腎疾患の進行は、クレアチニンクリアランス(CrCl)の50%減少、血清クレアチニンの倍加(SCr)、および/またはESRDへの進行によって測定してもよい。
【0032】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における腎機能障害を処置するための方法に関する。方法には、腎機能障害が処置されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0033】
本発明はまた、少なくとも部分的に、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量と、第二の物質とを含む薬学的組成物にも関する。
【0034】
さらなる態様において、本発明は、包装された薬学的組成物に関する。包装された薬学的組成物には、組成物を第二の物質と併用して投与することを勧める表示または添付文書と共に包装された、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量が含まれる。
【0035】
なおもう一つのさらなる態様において、本発明は、組成物を式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩と併用して投与するように勧める表示または添付文書と共に包装された第二の物質の治療的有効量を含む、包装された薬学的組成物に関する。
【0036】
なおもう一つの態様において、本発明は、組成物を食事なしで投与することを勧める表示または添付文書と共に、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を含む薬学的組成物を保持する容器を含む、包装された薬学的組成物に関する。
【0037】
なおもう一つの態様において、本発明はAAアミロイドーシスを処置するための薬学的製剤に関する。製剤は、被験体に投与した場合に少なくとも一つの都合のよい生物学的特性(FBP)を有する、製剤において式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を含む。
【0038】
本発明はまた、少なくとも部分的に、抗アミロイド原性物質含有製剤が、それが生物学的に都合のよい製剤となるように、被験体に投与した場合に、少なくとも一つの都合のよい生物学的特性を有するように予め決定された標準的な製剤と同等である、製剤における抗アミロイド原性物質に関する。
【0039】
もう一つの態様において、本発明にはまた、式(I)の化合物と一つまたは複数の薬学的に許容される担体とを含む、薬学的製剤が含まれる。この態様において、薬学的組成物は、それを必要とする被験体に投与した場合に、Cmax約200〜約2000 ng/mlを提供する。
【0040】
なおもう一つの態様において、本発明はまた、式(I)の化合物と、一つまたは複数の薬学的に許容される担体とを含む、薬学的製剤にも関する。この態様において、薬学的製剤は、それを必要とする被験体に投与した場合に、AUC∞約2,000〜約44,000 ng/mlを提供する。
【0041】
本発明はまた、少なくとも部分的に、それを必要とする被験体に化合物を投与する方法にも関する。方法には、Cmax約200〜約3,400 ng/mlを達成するために十分な量で被験体に式(I)の化合物を投与する段階が含まれる。Cmaxは投与後約0.25〜約9.00時間で起こる可能性がある。
【0042】
もう一つの態様において、本発明はまた、少なくとも部分的に、それを必要とする被験体に式(I)の化合物を投与する方法にも関する。方法には、AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の式(I)の化合物を被験体に投与する段階が含まれる。
【0043】
なおもう一つの態様において、本発明は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む薬学的製剤に関する。薬学的製剤は、それを必要とする被験体に1回投与した場合に、Cmax約200〜約2000 ng/mlを提供する。
【0044】
なおもう一つの態様において、本発明にはまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む薬学的製剤に関する。薬学的製剤は、それを必要とする被験体に投与した場合に、AUC∞約2,000〜約44,000 ng/mlを提供する。
【0045】
なおもう一つの態様において、本発明はまた、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法に関する。方法には、投与後約0.25〜約9.00時間でCmax約200〜約3,400 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階が含まれる。
【0046】
もう一つの態様において、本発明はまた、AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与することによって、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法にも関する。
【0047】
なおもう一つの態様において、本発明は、少なくとも部分的に、薬学的製剤に関する。薬学的製剤は、AAアミロイドーシスを処置するかまたは予防するために有効な量の活性物質(例えば、1,3-プロパンジスルホン酸、二ナトリウム塩(PDSとも呼ばれる))と、薬学的に許容される担体とを含み、製剤を健康な被験体に経口投与した場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0048】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを処置するかまたは予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、健康被験体に経口投与した場合、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、薬学的製剤にも関する。
【0049】
なおもう一つのさらなる態様において、本発明はまた、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、健康被験体に経口投与した場合、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、薬学的製剤に関する。
【0050】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体には活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0051】
なおもう一つのさらなる態様において、本発明は、活性物質(例えば、PDS)800 mgと薬学的に許容される担体とを含む薬学的製剤であって、被験体に経口投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0052】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に24ヶ月間経口投与した場合:活性物質の用量が400 mgの場合、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0053】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回経口投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回経口投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回経口投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0054】
なおもう一つの態様において、本発明はまた、AAアミロイドーシスを有する被験体における腎機能を安定化するかもしくは改善させる、または腎疾患の進行を遅らせる方法に関する。方法には、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体を含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階が含まれる。例えば、製剤を用量400 mgで投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を800 mgの用量で投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を1200 mgの用量で投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0055】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体である活性化物質とを含む薬学的製剤に関する。さらに、この製剤を、AAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体に活性物質の用量400 mgを経口投与すると、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを経口投与すると、平均AUC∞約3,000〜20,000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを経口投与すると、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0056】
なおもう一つの態様において、本発明は、少なくとも部分的に、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階を含む、AAアミロイドーシスを有する被験体における腎機能を安定化するかもしくは改善する、または腎疾患の進行を遅らせる方法に関する。さらに、製剤を用量400 mgで投与した場合、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を用量800 mgで投与した場合、平均AUC∞約3,000〜20,000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を用量1200 mgで投与した場合、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【図面の簡単な説明】
【0057】
【図1】PDS対プラセボを投与した被験体に関する「悪化」事象に関するカプラン-マイヤー曲線を描写するグラフである。
【図2】PDS対プラセボを投与した被験体に関するクレアチニンクリアランスの勾配を示す折れ線グラフである。
【図3】PDS対プラセボを投与した被験体に関するクレアチニンクリアランスの50%減少までの時間に関するカプラン-マイヤー曲線を描写するグラフである。
【図4】PDS対プラセボを投与した被験体に関するESRD/透析までの時間に関するカプラン-マイヤー曲線を描写するグラフである。
【発明を実施するための形態】
【0058】
発明の詳細な説明
A.本発明の化合物を用いて標的被験体を処置する方法
一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスのために処置され、および腎障害に関連したパラメータを有する、またはそれに対して感受性がある標的被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、腎障害(PRI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、以下の式の化合物の治療的有効量を標的被験体に投与する段階が含まれる:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2である。
【0059】
もう一つの態様において、本発明には、AAアミロイドーシスのために処置され、および胃腸管障害に関連した二次的障害または病態を有する、またはそれに対して感受性を有する標的被験体におけるAAアミロイドーシスを処置または予防する方法が含まれる。方法には、胃腸管障害(PGI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、式(I)の化合物の治療的有効量を標的被験体に投与する段階が含まれる。
【0060】
一般的に、AAアミロイドーシスは、持続的な急性期反応を誘発する多数の疾患の徴候である。そのような疾患には、慢性炎症性疾患、慢性局所または全身性微生物感染症、および悪性新生物が含まれる。反応性または二次性(AA)アミロイドーシスの最も一般的な型は、長期間の炎症病態の結果として認められる。例えば、リウマチ性関節炎または家族性地中海熱(遺伝病である)を有する被験体はAAアミロイドーシスを発症しうる。「AAアミロイドーシス」、「二次性アミロイドーシス」、および「二次性(AA)アミロイドーシス」という用語は互換的に用いられる。
【0061】
AA原線維は一般的に、IL-1、IL-6、およびTNFのようなサイトカインに反応して肝細胞において主に合成される循環中のアポリポタンパク質である、血清アミロイドAタンパク質(ApoSAA)のタンパク質分解切断によって形成された8,000ダルトンの断片(AAペプチドまたは断片)で構成される。一度分泌されると、ApoSAAは、HDLと複合体を形成する。AA原線維の沈着は、実質臓器に選択的に、体内に広がりうる。腎臓は通常、沈着部位であり、肝臓および脾臓も罹患する可能性がある。沈着はまた、心臓、胃腸管、および皮膚においても認められる。
【0062】
AAアミロイドーシスの発症に至りうる基礎疾患には、慢性炎症疾患、リウマチ性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、若年性慢性関節炎、若年性リウマチ性関節炎、潰瘍性大腸炎、慢性発熱、気管支狭窄、マラリア、血管炎、静脈内薬物乱用、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、およびクローン病のような炎症疾患が含まれるがこれらに限定されるわけではない。AA沈着はまた、慢性感染症、例えばAIDS、HIV、B型肝炎、C型肝炎、慢性微生物感染症、例えば、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、肺結核、肺感染症、再発性膿瘍、ベーチェット病、およびホイップル病の結果として産生される。特定の悪性新生物によってもまた、AA原線維アミロイド沈着が起こりうる。これらには、ホジキンリンパ腫、腎癌、腸管、肺、および尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、およびヘアリーセル白血病のような病態が含まれる。
【0063】
「被験体」という用語には、AAアミロイドーシスもしくはアミロイド関連疾患が起こりうるか、またはAAアミロイドーシスもしくはアミロイド関連疾患に対して感受性がある生きている生物が含まれる。「被験体」という用語には、動物(例えば、哺乳動物、例えばネコ、イヌ、ウマ、ブタ、ウシ、ヤギ、ヒツジ、齧歯類、例えばマウスまたはラット、ウサギ、リス、クマ、霊長類(例えば、チンパンジー、サル、ゴリラ、およびヒト))と共にニワトリ、アヒル、北京ダック、ガチョウ、およびそのトランスジェニック種が含まれる。
【0064】
「標的被験体」という用語は、式(I)の組成物または化合物を投与されるように特に選択された被験体、例えばヒトを指す。したがって、いくつかの態様において、標的被験体には、AAアミロイド関連疾患、例えばAAアミロイドーシスのリスクを有する、またはそれらであると診断された被験体が含まれる。AAアミロイドーシスを発症するリスクを有する被験体には、炎症疾患、感染症、遺伝性発熱、または新生物のような基礎疾患を有する被験体が含まれる。他の態様において、標的被験体には、腎障害および/または胃腸管障害に関連するパラメータを有するか、またはパラメータに対して感受性を有する被験体が含まれる。標的被験体にはまた、AAアミロイド関連疾患であると診断されると共に、腎障害および/または胃腸管障害に関連するパラメータを有することが公知である被験体が含まれてもよい。好ましい標的被験体はヒトである。
【0065】
「許容認容性指数」および「ATI」という用語は、被験体を苦しめる疾患または障害における所定の時点で十分だと見なされる、被験体における病気のレベルを指すために互換的に用いられる。いくつかの態様において、ATIは本明細書において記述されるように被験体における病気の改善または安定化である。他の態様において、ATIは先の時点と比較して被験体における病気の悪化がより低いことであり、例えば被験体が血清クレアチニンレベルの急激な増加を経験している場合、ATIは血清クレアチニンレベルのより遅い増加であるかもしれない。したがって、一つの態様において、ATIは、被験体における腎障害または胃腸管障害に関連する少なくとも一つのパラメータの悪化がより低いことである。もう一つの態様において、ATIは、被験体における腎障害および/または胃腸管障害に関連した少なくとも二つのパラメータの悪化がより低いことである。なおもう一つの態様において、ATIは、被験体における腎障害および/または胃腸管障害に関連した少なくとも三つ、四つ、または五つのパラメータの悪化がより低いことである。
【0066】
「腎障害に関連するパラメータ」および「PRI」という用語は、クレアチニンクリアランスの減少、血清クレアチニンレベルの増加、タンパク尿症、透析/末期腎疾患(ESRD)への進行、低アルブミン血症、および/または浮腫のような、しかしこれらに限定されない異常な腎機能に一般的に関連するパラメータを含めるために互換的に用いられる。いくつかの態様において、腎障害に関連するパラメータは少なくとも部分的に、AAアミロイドーシスまたは体内のアミロイドAタンパク質の存在によって引き起こされる。
【0067】
「胃腸管障害に関連するパラメータ」および「PGI」という用語には、慢性的な下痢および/または体重減少のような、しかしこれらに限定されない異常な胃腸管機能に一般的に関連するパラメータが含まれる。いくつかの態様において、胃腸管障害に関連するパラメータは、少なくとも部分的に、AAアミロイドーシスまたは体内のアミロイドAタンパク質の存在によって引き起こされる。
【0068】
被験体の「処置」または「処置する」という用語には、疾患もしくは病態、疾患もしくは病態の症状、または疾患もしくは病態のリスク(または感受性)を安定化、治癒(curing)、治癒(healing)、緩和、軽減、変化、救済、より低い悪化、回復、改善、または影響を及ぼすことを目的として、被験体に本発明の化合物を適用または投与すること(または被験体からの細胞または組織に本発明の化合物を適用または投与すること)が含まれる。「処置する」という用語は、軽減;寛解;悪化速度の減弱;安定化、症状または損傷発生の減少、被験体により認容可能な病理または病態、変性速度の遅延もしくは低下;最終的な変性点をより消耗性でなくなるようにする;または被験体の身体的もしくは精神的安寧を改善することのような、任意の客観的または主観的パラメータを含む、損傷、病理、または病態の処置または回復における任意の成功の指標を指す。一つの態様において、「処置する」という用語には、被験体の寿命を増加させる段階が含まれうる。
【0069】
「慢性的な下痢の寛解」という用語は、慢性的な下痢の事例がないこと、および少なくとも連続して4ヶ月間下痢止め薬を慢性的に用いていないことを指す。
【0070】
一つの態様において、透析への進行は、被験体、例えばAAアミロイドーシスを有する被験体において遅れるかまたは予防される。例えば、被験体の透析への進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、11ヶ月もしくはそれより長く、1年もしくはそれより長く、1.5年もしくはそれより長く、2年もしくはそれより長く、3年もしくはそれより長く、4年もしくはそれより長く、5年もしくはそれより長く、7.5年もしくはそれより長く、10年もしくはそれより長く、15年もしくはそれより長く、または20年もしくはそれより長く遅れる可能性がある。特定の態様において、進行は約6ヶ月遅れる。
【0071】
もう一つの態様において、「処置する」という用語には、腎臓の低下の任意の「悪化」事象(実施例3を参照されたい)または全ての原因の死亡率のリスクを少なくとも5%もしくはそれより大きく、少なくとも10%もしくはそれより大きく、少なくとも15%もしくはそれより大きく、少なくとも20%もしくはそれより大きく、少なくとも30%もしくはそれより大きく、少なくとも40%もしくはそれより大きく、少なくとも50%もしくはそれより大きく、少なくとも60%もしくはそれより大きく、または少なくとも63%もしくはそれより大きく減少させる段階が含まれる。もう一つの態様において、腎臓の低下または全ての原因の死亡率に関する任意の「悪化」事象のリスクは7%〜63%減少する。
【0072】
もう一つの態様において、「処置する」という用語にはまた、第一の「悪化」事象までの平均時間を増加させる段階が含まれる。増加は、約0.5ヶ月もしくはそれより長く、約1ヶ月もしくはそれより長く、約2ヶ月もしくはそれより長く、約3ヶ月もしくはそれより長く、約4ヶ月もしくはそれより長く、約5ヶ月もしくはそれより長く、約6ヶ月もしくはそれより長く、約7ヶ月もしくはそれより長く、約8ヶ月もしくはそれより長く、約9ヶ月もしくはそれより長く、約10ヶ月もしくはそれより長く、または約11ヶ月もしくはそれより長くてもよい。もう一つの態様において、期間は、PDS処置被験体において約2.8ヶ月±7.5ヶ月またはそれより大きく増加する。
【0073】
もう一つの態様において、被験体のクレアチニンクリアランス速度が安定化するかまたは改善される。例えば、被験体のクレアチニンクリアランス速度は、本発明の化合物を処置する前の被験体のレベルと比較して、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく増加する可能性がある。
【0074】
もう一つの態様において、クレアチニンクリアランスが50%またはそれより大きく減少するリスクは、少なくとも約5%もしくはそれより多く、少なくとも約10%もしくはそれより多く、少なくとも約15%もしくはそれより多く、または少なくとも約18%もしくはそれより多く低減される。さらなる態様において、クレアチニンクリアランスが50%またはそれより大きく減少するリスクは、約18%〜72%低減される。
【0075】
もう一つの態様において、被験体の血清クレアチニン、血清アルブミンレベル、および/または血清アルカリホスファターゼレベルは安定化されるかまたは改善される。例えば、被験体の血清クレアチニン、血清アルブミンレベル、および/または血清アルカリホスファターゼレベルは、本発明の化合物による処置の前の被験体のレベルと比較して約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく増加する可能性がある。
【0076】
さらなる態様において、血清クレアチニンが倍加するリスクは少なくとも5%もしくはそれより大きく、少なくとも10%もしくはそれより大きく、少なくとも11%もしくはそれより大きく、少なくとも12%もしくはそれより大きく、少なくとも13%もしくはそれより大きく、または少なくとも14%もしくはそれより大きく低減される。さらなる態様において、被験体の血清クレアチニンが倍加するリスクは約14%〜約81%低減される。
【0077】
もう一つの態様において、「処置する」という用語にはまた、血清クレアチニンの倍加までの平均時間を増加させる段階が含まれる。増加は約0.5ヶ月もしくはそれより長く、約1ヶ月もしくはそれより長く、約2ヶ月もしくはそれより長く、約3ヶ月もしくはそれより長く、約4ヶ月もしくはそれより長く、約5ヶ月もしくはそれより長く、約6ヶ月もしくはそれより長く、約7ヶ月もしくはそれより長く、約8ヶ月もしくはそれより長く、約9ヶ月もしくはそれより長く、約10ヶ月もしくはそれより長く、約11ヶ月もしくはそれより長く、または1年もしくはそれより長くてもよい。
【0078】
もう一つの態様において、被験体のタンパク尿レベル、内臓アミロイド負荷、および/または吸引脂肪組織におけるアミロイド含有量は、安定化されるかまたは改善される。例えば、被験体のタンパク尿レベル、内臓アミロイド負荷、および/または吸引脂肪組織におけるアミロイド含有量は、本発明の化合物による処置前の被験体のレベルと比較して、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく低減される可能性がある。
【0079】
もう一つの態様において、被験体の内臓アミロイド負荷を低減するかまたは安定化する。被験体の内臓アミロイド負荷は、例えば、123I-放射標識血清アミロイドP成分(SAP)のシンチグラフィーを用いて査定することができる。SAPは、アミロイド原線維に対して特異的に結合して、組織アミロイド沈着物において長期間にわたって保持され、それが循環中に受ける正常で急速な異化から明らかに保護される。放射標識SAPのシンチグラフィーによる撮像法は、内臓アミロイド負荷を査定するための特異的非侵襲的方法として開発されている(Hawkins PN et al. N Engl J Med, 1990; 323:508-13)。内臓のアミロイド負荷は、例えば、放射性核種の注射後24時間で得られた全身シンチグラフの肉眼的査定によって定量することができる。
【0080】
もう一つの態様において、吸引脂肪組織における被験体のアミロイド含有量は、低減されるかまたは安定化される。「吸引脂肪組織におけるアミロイド含有量」という用語は、吸引された脂肪組織におけるアミロイドAの含有量を指す。吸引脂肪組織におけるアミロイドA含有量の変化は、コンゴーレッド染色によって半定量的に測定することができる。脂肪組織におけるアミロイドA含有量は、例えば被験体から採取した脂肪組織を用いるモノクローナル抗体に基づくサンドイッチELISAを用いることによって、定量的に測定することができる(Hazenberg B et al. Ann Rheum Dis, 1999; 58: 96-102)。
【0081】
「起立性低血圧」という用語は、人が立つ姿勢をとる場合に起こる血圧の突然の下降を指す。突然立ち上がった後に一般的に起こる症状には、めまい、立ちくらみ、目のかすみ、および失神(意識の一時的な喪失)が含まれる。自律神経系(ANS)は時に、AAアミロイドーシスにおいて影響を受ける。姿勢による血圧の減少(例えば、仰臥位から起立姿勢の際に収縮期圧の>20 mmHgの下降、または拡張期圧の10 mmHgの下降が少なくとも3分間持続する)は、ANS機能障害の兆候である。
【0082】
もう一つの態様において、被験体の体重減少は改善もしくは安定化され、または被験体の体重は増加する。例えば、被験体は本発明の化合物による処置の前の体重の5%もしくはそれより多く、約10%もしくはそれより多く、約20%もしくはそれより多く、約30%もしくはそれより多く、約40%もしくはそれより多く、約50%もしくはそれより多く、または約60%もしくはそれより多く増加する可能性がある。
【0083】
もう一つの態様において、被験体のネフローゼ症候群は、安定化されるかまたは寛解される可能性がある。もう一つの態様において、被験体の浮腫は消散または緩和される可能性がある。もう一つの態様において、被験体における下痢の安定化、改善、治癒、または寛解が起こる可能性がある。なおもう一つの態様において、被験体における起立性低血圧、脾腫、および/または肝腫の安定化または低減が起こる可能性がある。
【0084】
一つの態様において、ネフローゼ症候群の寛解には、タンパク尿の<1 g/24時間への減少、および血清アルブミンの3.4 g/dLより大きい増加または浮腫の緩解および/または浮腫の改善に反応した利尿薬の中止のいずれかが含まれる。
【0085】
「治療的有効量」という用語は、被験体を処置するために、例えばAAアミロイドーシスもしくはアミロイド関連疾患に関して被験体を処置するために、または炎症性疾患、悪性新生物、もしくは慢性微生物感染症のような、しかしこれらに限定されない基礎疾患を有する被験体を処置するために有効である化合物の量を指す。治療的有効量は、被験体が苦しんでいる特定の障害、特定の被験体の年齢、体重、および生活様式に基づいて変化してもよい。さらに、治療的有効量は、病態の重症度、臓器機能、腎機能、または基礎疾患(例えば、被験体が炎症疾患、悪性新生物、慢性感染症に苦しんでいてもよい)に依存してもよい。一つの態様において、被験体はネフローゼである。もう一つの態様において、被験体は非ネフローゼである。
【0086】
「ネフローゼ」という用語は、ネフローゼ症候群に苦しむ被験体を指す。ネフローゼ症候群は、一般的に重度のタンパク尿症(例えば、尿中タンパク質>3 g/24時間)と共に以下の二つの腎臓外特色であると定義される:1)低アルブミン血症(例えば、血清アルブミン<3.4 g/dL);および2)健康診査による末梢の浮腫および/または浮腫を処置するための利尿剤の使用。
【0087】
「非ネフローゼ」という用語は、ネフローゼ症候群にまだ進行していない、またはネフローゼ症候群が寛解した被験体を指す。ネフローゼ症候群の寛解は、タンパク尿症の<1 g/24時間への減少および以下の二つの腎臓外特色の一つの改善である:1)血清アルブミンの>3.4 g/dLへの増加、または2)浮腫の消散および/または浮腫の改善に反応した利尿剤の中止。ネフローゼ症候群への進行は、タンパク尿症の>3 g/24時間への増加、および以下の二つの腎臓外特色の発生である:1)低アルブミン血症(例えば、血清アルブミン<3.4 g/dL);および2)浮腫および/または浮腫を処置するための利尿剤の使用。
【0088】
もう一つのさらなる態様において、本発明はまた、被験体のクレアチニンクリアランス速度、タンパク尿レベル、および/または血清アルブミンレベルに基づいて選択された用量の式(I)の化合物の治療的有効量を被験体に投与することによって、被験体におけるアミロイド関連疾患を処置または予防する方法に関する。
【0089】
「クレアチニンクリアランス」という用語は、当技術分野で認識され、腎臓が血液中からクレアチニンを消失させる速度を指す。クレアチニンは、健康な被験体において腎臓によって容易に排泄される物質である。クレアチニンクリアランスは一般的に、尿中のクレアチニンレベルを血液中のクレアチニンレベルと比較する。クリアランスはしばしば、ミリリットル/分(ml/分)として測定される。
【0090】
本発明の方法において投与される用量は、クレアチニンクリアランス速度に基づいて選択してもよい。例えば、式(I)の化合物の用量は、クレアチニンクリアランス速度が>80 ml/分の場合、約1200 mgを1日2回であるように選択されてもよい。クレアチニンクリアランス速度が約30〜80 ml/分の場合、式(I)の化合物の用量は、約800 mgを1日2回であるように選択されてもよい。クレアチニンクリアランス速度が約20〜30 ml/分の場合、式(I)の化合物の用量は、約400 mgを1日2回であるように選択されてもよい。さらに、用量は、被験体におけるクレアチニンクリアランス速度の変化に基づいて調節してもよい。
【0091】
一つの態様において、本発明の化合物を被験体に投与した後に、所望の薬物動態パラメータおよび/または生物学的に都合のよいパラメータが得られるように、用量を選択してもよい。一つの態様において、用量は、被験体に1回投与後、被験体における平均AUCssが約7,000〜約26,000 ng.h/mlとなるように、および平均定常状態濃度が約500〜約1200 ng/mlとなるように選択される。もう一つの態様において、用量は被験体に1回投与した後、被験体におけるCmaxが約1,200〜約3,100 ng/mlとなるように、およびAUC∞が約5,000〜約43,000 ng.h/mlとなるように選択される。腎機能障害を有する被験体では、特定のAUCss、AUC∞、Cmaxおよび定常状態平均濃度を達成するために必要な用量を調節する必要がある可能性がある。
【0092】
さらなる態様において、Cmax、AUC0-tlast、および/またはAUC∞は、表1において示される値と比較して、特定の被験体に関して約±10%、約±20%、約±30%、または約±40%変化してもよい。
【0093】
「アミロイド関連疾患」という言語は、アミロイド線維の存在を特徴とする病態を指す。「アミロイド」は、多くの異なる疾患において認められる、多様であるが特異的なタンパク質沈着(細胞内または細胞外)の群を指す一般用語である。その発生は多様であるが、全てのアミロイド沈着物は、共通の形態学的特性を有し、特異的色素(例えば、コンゴーレッド)で染色され、染色後に偏光において特徴的な赤-緑複屈折外観を有する。それらはまた、共通の超微細構造、共通のX-線回折、および赤外線スペクトルを共有する。
【0094】
本発明はまた、少なくとも部分的に、被験体におけるAAアミロイドーシスを処置または予防するためのもう一つの方法に関する。本方法には、例えばAUC、Cmax、AUCss、Css、Tmax等によって測定した場合に、被験体において有効な全身曝露が提供されるような用量で投与される式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0095】
「標的血漿濃度」という用語は、それによってAAアミロイドーシスに関する被験体の処置が起こる、被験体における本発明の化合物の濃度範囲を指す。一つの態様において、被験体は、約500〜約1200 ng/mlの定常状態濃度(Css)を維持する。もう一つの態様において、被験体は、約7000〜約26,000 ng.h/mlのAUCssを維持する。例えば、被験体は、定常状態濃度約600〜約700 ng/mlまたは約900〜約1100 ng/ml、および/またはAUCss約8000〜約9000 ng.h/ml、約11,000〜約13,000 ng.h/ml,、約23,000〜約26,000 ng.h/ml、または約15,500〜約16,500 ng.h/mlを維持してもよい。さらなる態様において、AUCssまたは定常状態濃度はこれらの値の±20%以内である。
【0096】
さらに、本発明は、少なくとも部分的に、被験体における腎および/または胃腸管機能を安定化するかまたは改善する方法に関する。方法には、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0097】
さらなる態様において、本発明はまた、少なくとも部分的に薬学的製剤にも関する。製剤は、AAアミロイドーシスを処置または予防するために有効な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と、薬学的に許容される担体とを含む。さらに、製剤を健康被験体に経口投与すると、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。代わりの態様において、製剤を健康被験体に経口投与すると、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。もう一つの代わりの態様において、製剤を健康被験体に経口投与すると、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0098】
もう一つの態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と、薬学的に許容される担体とを含む薬学的製剤に関する。この態様において、製剤をAAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体に活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0099】
もう一つの態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質800 mgと薬学的に許容される担体とを含む薬学的製剤に関する。さらに、本製剤を被験体に経口投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0100】
もう一つのさらなる態様において、本発明は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と薬学的に許容される担体とを含む薬学的製剤に関する。さらに本製剤を、AAアミロイドーシスを有する被験体に24ヶ月間経口投与する場合、活性物質の用量400 mgでは、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0101】
もう一つのさらなる態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と薬学的に許容される担体とを含む薬学的製剤に関する。さらに本製剤を健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0102】
もう一つのさらなる態様において、本発明はまた、腎機能を安定化するかもしくは改善させる、またはAAアミロイドーシスを有する被験体における腎疾患の進行を遅らせる方法にも関する。方法には、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階が含まれる。さらに、製剤を用量400 mgで投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を800 mgの用量で投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を1200 mgの用量で投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0103】
さらなる態様において、被験体のクレアチニンクリアランス速度が約30 ml/分未満である場合、用量は400 mgであり、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合、用量は800 mgであり、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合、用量は1200 mgである。もう一つのさらなる態様において、被験体のクレアチニンクリアランス速度は、約60〜約90 ml/分であり、用量1200 mgが投与される。
【0104】
もう一つの態様において、本発明は、被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、AAアミロイドーシスが処置または予防されるように、第二の物質と併用して、式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0105】
「併用する」という用語は、式(I)の化合物と第二の物質との同時投与;第二の物質の投与前の式(I)の化合物の投与;または式(I)の化合物の投与前の第二の物質の投与を指す。
【0106】
「第二の物質」という用語には、基礎疾患、例えば炎症疾患(例えば、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫症、マックル-ウェルズ症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、およびクローン病等)、慢性感染症に関連した疾患(例えば、AIDS、HIV、B型肝炎、C型肝炎等)、慢性微生物感染症に関連した疾患(例えば、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、ホイップル病、集族性ざ瘡、分類不能型免疫不全、肺結核、肺感染症、再発性膿瘍、ベーチェット病、低γグロブリン血症、嚢胞性線維症)、または特定の悪性新生物(例えば、ホジキンリンパ腫、腎癌、腸管、肺、および尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、およびヘアリーセル白血病)を処置することが知られている薬物が含まれる。「第二の物質」という用語にはまた、救済薬、化学療法剤、抗炎症剤、例えば非ステロイド性抗炎症剤等、および抗生物質が含まれる。第二の物質の例には、メソトレキセート、コルヒチン、抗TNF抗体、および抗インターロイキン1または6抗体が含まれる。
【0107】
非ステロイド性抗炎症剤(「NSAID」)の例には、イブプロフェン、ナプロキセン、サリンダック、およびインドメタシンが含まれる。他の抗炎症剤には、COX-2阻害剤(Vioxx(商標)およびCelebrex(商標)のような)、サイトカイン阻害剤(WO 95/04533において開示されるサリドマイドおよびデキサナビノールのような)、補体阻害剤、ロイコトリエン受容体拮抗剤およびその組み合わせが含まれる。例には、酢酸誘導体サリンダック(Clinoril(商標), Merck & Co., Inc., Rahway, New Jersey)、インドメタシン(Indocin(商標), Merck & Co., Inc., Rahway, New Jersey);エトドラック(Lodine(商標), Wyeth, Madison, New Jersey)、ナブメトン(Relafen(商標), GlaxoSmithKline, Middlesex, England)、トルメチンナトリウム(Tolectin(商標), McNeil Pharmaceuticals, Spring House, Pennsylvania);アントラニル酸誘導体:メクロフェナム酸ナトリウム(Meclomen(商標), Pfizer, New York, New York)、メフェナム酸(Ponstel(商標), Pfizer, New York, New York);エノール酸誘導体:ピロキシカム(Feldene(商標), Pfizer, New York, New York)、Mobic(商標)(メロキシカム);フェニル酢酸誘導体:arthrotec(ジクロフェナク/ミソプロストール)、Voltaren(商標)(ジクロフェナク);プロピオン酸誘導体:ナプロキセンナトリウム(Anaprox(商標), Naprosyn(商標), Hoffmann-La Roche Inc. (Roche), Nutley, N.J.)、フルビプロフェン(Ansaid(商標), Upjohn、現在はPfizer, New York, New York)、オキサプロジン(Daypro(商標), G.D Searle, 現在はPfizer, New York, New York);イブプロフェン(Motrin(商標), Upjohn、現在はPfizer, New York, New York)、フェノプロフェンカルシウム(Nalfon(商標), Dista, Ranbaxy, Princeton, NJ)、ケトプロフェン(Oruvail(商標)またはOrudis(商標), Wyeth, Madison, New Jersey)、ケトロラックトロメタミン(Toradol(商標), Syntex Laboratories, Hoffmann-La Roche Inc. (Roche), Nutley, N.J.);サリチル酸誘導体:ジフルニサル(Dolobid(商標), Merck & Co., Inc., Rahway, New Jersey);およびCOX-2選択的阻害剤:Bextra(商標)(バルデコキシブ)、Celebrex(商標)(セレコキシブ、Pfizer, New York, New York)、およびVioxx(商標)(ロフェコキシブ、Merck & Co., Inc., Rahway, New Jersey)、およびシクロスポリン(Maas BiolAB, Albuquerque, New Mexico)が含まれる。
【0108】
「化学療法剤」という言語には、細胞または組織の成長が望ましくない、増殖しつつある細胞または組織の成長を阻害する物質、またはそのような成長によって起こる少なくとも一つの症状を処置する物質が含まれる。化学療法剤の例には:ブレオマイシン、ドセタキセル(Taxotere)、ドキソルビシン、エダトレキセート、エトポシド、フィナステリド(Proscar)、フルタミド(Eulexin)、ゲンシタビン(Gemzar)、酢酸ゴセレリン(Zoladex)、グラニセトロン(Kytril)、イリノテカン(Campto/Camptosar)、オンダンセトロン(Zofran)、パクリタキセル(Taxol)、ペガスパルガーゼ(Oncaspar)、塩酸ピロカルピン(Salagen)、ポルフィマーナトリウム(Photofrin)、インターロイキン-2(Proleukin)、リツキシマブ(Rituxan)、トポテカン(Hycamtin)、トラスツズマブ(Herceptin)、トレチノイン(Retin-A)、トリアピン、ビンクリスチン、および酒石酸ビノレルビン(Navelbine)が含まれる。
【0109】
化学療法剤の他の例には、ナイトロジェンマスタード(例えば、メクロレタミン(HN2))、シクロホスファミド、イフォスファミド、メルファラン(L-サルコリシン)、クロラムブシル等のようなアルキル化剤;エチレンイミン、メチルメラミン(例えば、ヘキサメチルメラミン、チオテパ等)、アルキルスルホネート(例えば、ブスルファン等)、ニトロソウレア(例えば、カルムスチン(BCNU)、ロムスチン(CCNU)、セムスチン(メチル-CCNU)、ストレプトゾシン(ストレプトゾトシン)等)、トリアゼン(例えば、デカルバジン(DTIC;ジメチルトリアゼノイミダゾールカルボキサミド)、アルキル化剤(例えば、シス-ジアミンジクロロプラチナII(CDDP))が含まれる。
【0110】
化学療法剤の他の例には、葉酸類似体(例えば、メソトレキセート(アメトプテリン));ピリミジン類似体(例えば、フルオロウラシル('5-フルオロウラシル;5-FU);フロクスリジン(フルオロデオキシウリジン);FUdr;シタラビン(シトシンアラビノシド)等);プリン類似体(例えば、メルカプトプリン(6-メルカプトプリン;6-MP);チオグアニン(6-チオグアニン;TG);およびペントスタチン(2'-デオキシコフォマイシン)等)のような抗代謝剤が含まれる。
【0111】
化学療法剤の他の例にはまた、ビンカアルカロイド(例えば、ビンブラスチン(VLB)およびビンクリスチン);トポイソメラーゼ阻害剤(例えば、エトポシド、テニポシド、カンプトテシン、トポテカン、9-アミノ-カンプトテシンCPT-11等);抗生物質(例えば、ダクチノマイシン(アクチノマイシンD)、アドリアマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、プリカマイシン(ミスラマイシン)、マイトマイシン(マイトマイシンC)、タキソール、タキソテール等);酵素(例えば、L-アスパラギナーゼ);および生物反応改変剤(例えば、インターフェロン-α;インターロイキン2等)が含まれる。他の化学療法剤には、シス-ジアミンジクロロプラチナII(CDDP);カルボプラチン;アントラセンジオン(例えば、ミトキサントロン);ヒドロキシウレア;プロカルバジン(N-メチルヒドラジン);および副腎皮質抑制剤(例えば、ミトーテン、アミノグルテチミド等)が含まれる。
【0112】
他の化学療法剤には、副腎皮質ステロイド(例えば、プレドニゾン);プロゲスチン(例えば、カプロン酸ヒドロキシプロゲステロン;酢酸メドロキシプロゲステロン、酢酸メゲステロール等);エストロゲン(例えば、ジエチルスチルベストロール;エテニルエストラジオール等);抗エストロゲン(例えば、タモキシフェン等);アンドロゲン(例えば、プロピオン酸テストステロン、フルオキシメステロン等)、抗アンドロゲン(例えば、フルタミド);および性腺刺激ホルモン放出ホルモン類似体(例えば、リュープロリド)が含まれる。
【0113】
「抗生物質」という用語には、微生物感染症を処置するために当技術分野において公知の抗生物質が含まれる。抗生物質の例には、アモキシシリン、アミノグリコシド、アミノグリコシド類似体、β-ラクタム、β-ラクタマーゼ、β-ラクタマーゼ類似体、クリンダマイシン、クロラムフェニコール、セファロスポリン、セファロスポリン類似体、シプロフロキサシン、シプロフロキサシン類似体、エリスロマイシン、フルオロキノロン、フルオロキノロン類似体、マクロライド、マクロライド類似体、メトロニダゾール、ペニシリン、ペニシリン類似体、キノロン、キノロン類似体、リファンピン、ストレプトマイシン、スルホンアミド、テトラサイクリン、テトラサイクリン類似体、トリメトプリム、トリメトプリム-スルファメトキサゾール、およびバンコマイシンが含まれるがこれらに限定されるわけではない。
【0114】
「救済薬」という用語には、基礎疾患を抑制する能力を有するが、進行性AAアミロイドーシスの特色を回復する主な指標と共に導入される、処置のあいだに行われる任意の投薬を指す。そのような投薬には、コルヒチン、細胞障害剤、および抗TNF-剤が含まれるがこれらに限定されるわけではない。
【0115】
抗TNF剤の例には、TNFを阻害する物質、例えば抗TNFα抗体が含まれる。抗TNF物質の例には、エタネルセプト(Enbrel(商標), Amgen)、インフリキシマブ(Remicade(商標), Johnson and Johnson、例えば米国特許第6,790,444号を参照されたい)、ヒト抗-TNFモノクローナル抗体(D2E7/HUMIRA(商標), Abbott Laboratories)、CDP 571 (Celltech)、およびCDP 870 (Celltech)が含まれる。
【0116】
他の第二の物質の例には、免疫抑制剤、コルチコステロイド(全身投与されるコルチコステロイドを含む)、スルファサラジン、レニン-アンジオテンシン系遮断剤または拮抗剤、利尿剤(例えば、フロセミド)、カルシウムチャンネル遮断剤、β遮断剤、抗リウマチ薬、アンジオテンシン変換酵素阻害剤(ACEi)、アンジオテンシンII受容体遮断剤(ARBs)、アセチルサリチル酸、アモキシシリン、カルシウム、炭酸カルシウム、クロラムブシル、コルヒチン、シクロホスファミド、ジクロフェナク、エナラプリル、葉酸、メソトレキセート、メチルプレドニゾロン、オメプラゾール、パラセタモール、プレドニゾロン、およびプレドニゾンが含まれる。
【0117】
もう一つの態様において、本発明は、少なくとも部分的に、被験体における炎症疾患を処置する方法に関する。方法には、被験体における炎症疾患が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。さらなる態様において、第二の物質は抗炎症剤である。
【0118】
「炎症疾患」という用語には、炎症に関連し、本発明の化合物を用いて処置されうる疾患または障害が含まれる。炎症疾患には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それを引き起こす、それによって引き起こされる、それが原因で起こる、またはそうでなければアミロイドーシスに関連する疾患が含まれてもよい。そのような炎症疾患の例には、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、およびクローン病が含まれるがこれらに限定されるわけではない。
【0119】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置する、予防する、またはその発症を遅らせるために有効であり、第二の物質は、基礎となる障害、例えば基礎となる炎症性疾患を処置するために有効な量で投与される。
【0120】
なおもう一つの態様において、本発明には、被験体におけるリウマチ性関節炎を処置する方法が含まれる。方法には、被験体におけるリウマチ性関節炎が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。さらなる態様において、第二の物質は抗炎症剤である。さらなる態様において、第二の物質はインフリキシマブであり、これは例えば参照により本明細書に組み入れられる米国特許第6,790,444号において記述される技法によって投与されてもよい。
【0121】
さらなる態様において、第二の物質は、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、家族性地中海熱、成人スティル病、ベーチェット症候群、炎症性腸疾患、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、遺伝性周期性発熱、またはクローン病のような炎症疾患を処置することが知られている物質である。被験体に投与されてもよい物質の例には、例えば、抗TNF剤、メソトレキセート、抗炎症剤、およびその組み合わせが含まれる。
【0122】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅らせるために有効であり、第二の物質はリウマチ性関節炎を処置するために有効量で投与される。
【0123】
さらなる態様において、本発明は、少なくとも部分的に、被験体における悪性新生物を処置する方法に関する。方法には、被験体における悪性新生物が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0124】
「悪性新生物」という用語には、本発明の化合物を用いて処置されうる新生物が含まれる。悪性新生物には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それによって引き起こされる、それを引き起こす、それが原因で起こる、またはそうれなければアミロイドーシスに関連する新生物が含まれてもよい。そのような悪性新生物の例には、ホジキンリンパ腫、腎癌、腸管癌、肺癌、尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、またはヘアリーセル白血病が含まれるがこれらに限定されるわけではない。
【0125】
悪性新生物の処置にとって有用となる可能性がある第二の物質の例には、ホジキンリンパ腫、腎癌、腸管癌、肺癌、尿性器管の癌、基底細胞癌、またはヘアリーセル白血病を処置することが知られている物質が含まれる。用いてもよい第二の物質のさらなる例には、化学療法剤または細胞障害剤が含まれる。
【0126】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅延させるために有効であり、第二の物質は悪性新生物を処置するために有効量で投与される。
【0127】
なおもう一つの態様において、本発明は、慢性感染症を処置する方法に関する。方法には、慢性感染症が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0128】
「慢性感染症」という用語には、本発明の化合物を用いて処置されうる慢性的なウイルス、細菌、真菌、および微生物感染症が含まれうる。感染症には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それを引き起こす、それによって引き起こされる、それが原因で起こる、またはそうれなければアミロイドーシスに関連する感染症が含まれてもよい。微生物感染症は局所または全身性であってもよい。そのような微生物感染症の例には、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、肺結核、肺感染症、再発性膿瘍、ベーチェット病、およびホイップル病が含まれるがこれらに限定されるわけではない。他の慢性感染症には、AIDS、HIV、B型肝炎およびC型肝炎が含まれる。
【0129】
感染症を処置するために有用となる可能性がある第二の物質の例には、AIDS、HIV、B型肝炎、C型肝炎、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、肺結核、肺感染症、再発性膿瘍、ベーチェット病、またはホイップル病を処置することが知られている物質が含まれる。被験体に投与してもよい物質の例には、例えば、抗炎症剤および抗生物質が含まれる。
【0130】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅らせるために有効であり、第二の物質は慢性感染症を処置するために有効量で投与される。
【0131】
なおもう一つの態様において、本発明は、少なくとも部分的に、被験体における化合物の経口生物学的利用率が増加するように、食物なしで薬学的組成物における式(I)の化合物の治療的有効量を被験体に投与することによって、被験体における化合物の経口生物学的利用率を増加させる方法に関する。
【0132】
「経口生物学的利用率」は、経口投与後に血流に達する薬物の量を指す。「経口生物学的利用率の増加」という用語は、生物学的利用率の約5%もしくはそれより大きい、約10%もしくはそれより大きい、約15%もしくはそれより大きい、約20%もしくはそれより大きい、約25%もしくはそれより大きい、約30%もしくはそれより大きい、約35%もしくはそれより大きい、約40%もしくはそれより大きい、約50%もしくはそれより大きい、約55%もしくはそれより大きい、約60%もしくはそれより大きい、約65%もしくはそれより大きい、約70%もしくはそれより大きい、約75%もしくはそれより大きい、約80%もしくはそれより大きい、約85%もしくはそれより大きい、約90%もしくはそれより大きい、約95%もしくはそれより大きい、または約100%もしくはそれより大きい増加を指す。
【0133】
さらなる態様において、食物なしでの本発明の化合物の投与によって、食物と共に投与した場合と比較して、最高血漿濃度(Cmax)の増加および化合物の吸収の程度(AUC)の増加が起こる。Cmaxおよび/またはAUCの増加は、食物と共に化合物を投与した場合と比較して、約5%もしくはそれより大きい、約10%もしくはそれより大きい、約15%もしくはそれより大きい、約20%もしくはそれより大きい、約25%もしくはそれより大きい、約30%もしくはそれより大きい、約35%もしくはそれより大きい、約40%もしくはそれより大きい、約50%もしくはそれより大きい、約55%もしくはそれより大きい、約60%もしくはそれより大きい、約65%もしくはそれより大きい、約70%もしくはそれより大きい、約75%もしくはそれより大きい、約80%もしくはそれより大きい、約85%もしくはそれより大きい、約90%もしくはそれより大きい、約95%もしくはそれより大きい、または約100%もしくはそれより大きい可能性がある。さらなる態様において、食物と共に投与した場合と比較して、投与によって化合物の最高血漿濃度(Cmax)および吸収の程度(AUC)の増加が起こることが、被験体に通知される(医師もしくは薬剤師による説明によって、または本発明の化合物に添付される表示もしくは添付文書によって)。
【0134】
「食物なし」という用語は、薬剤または本発明の組成物を実質的に空の胃に投与することを指す。したがって、いくつかの態様において、食物なしの投与には、最も近い食物摂取より15分、30分、60分、90分、120分、150分、3時間、4時間、5時間、6時間、7時間、または8時間より後での投与が含まれる。他の態様において、食物なしの投与には、次の食物摂取の少なくとも15分、30分、60分、90分、120分、150分、3時間、4時間、5時間、6時間、7時間、または8時間前での投与が含まれる。一つの態様において、「食物なし」という用語は、食事前少なくとも1時間、または食後少なくとも2時間に本発明の化合物を投与することである。この態様において、「約」という用語には、表記の期間の±10〜20%の値が含まれる。
【0135】
もう一つの態様において、本発明は、例えば、血清クレアチニンの倍加の発生、クレアチニンクリアランスの50%減少より大きいまたはそれに等しい、透析/末期腎疾患、および/または全ての原因の死亡率によって測定した場合に、それを必要とする被験体における腎症の進行速度を低減させる方法に関する。例えば、AAアミロイドーシス、リウマチ性関節炎、慢性炎症、慢性感染症、遺伝性発熱等を有してもよい。
【0136】
もう一つの態様において、本発明は、AAアミロイドーシスを有する被験体における末期腎疾患(ESRD)および/または透析への進行を予防するかまたは遅らせるための方法に関する。方法には、ESRDおよび/または透析への進行が遅れるまたは予防されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0137】
さらなる態様において、ESRDおよび/または透析への進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。なおさらなる態様において、透析および/またはESRDは、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して6ヶ月間遅れる。
【0138】
もう一つの態様において、ESRDへの進行のリスクは約0〜78%低減される。もう一つの態様において、リスクは約5%、約10%、約15%、約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、約60%、約65%、約70%、約75%、または約78%低減される。この態様において、「約」という用語には±5%の値が含まれる。
【0139】
さらなる態様において、透析までの期間の中央値は、少なくとも約1ヶ月、約2ヶ月、約3ヶ月、約4ヶ月、約5ヶ月、約6ヶ月、約7ヶ月、約8ヶ月、または約9ヶ月遅れる。この態様において、「約」という用語には、表記の期間の±0.5ヶ月の範囲が含まれる。さらなる態様において、透析までの期間の中央値は、式(I)の化合物、例えばPDSによって被験体において3.5ヶ月±5.5、または9ヶ月まで長い。
【0140】
さらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における血清クレアチニンの倍加までの時間を予防するかまたは遅らせるための方法に関する。方法には、血清クレアチニンの倍加までの時間が遅れるまたは予防されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0141】
さらなる態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%減少までの時間を予防するかまたは遅らせるための方法が含まれる。方法には、クレアチニンクリアランスの少なくとも50%減少までの時間が遅れるまたは予防されるように、化合物の治療的有効量を被験体に投与する段階が含まれる。さらなる態様において、クレアチニンクリアランスの少なくとも50%減少までの時間の中央値は、PDSのような式(I)の化合物によって処置した被験体において約1ヶ月、約2ヶ月、約3ヶ月、約4ヶ月、約5ヶ月、約6ヶ月、約7ヶ月、約8ヶ月、約9ヶ月、約10ヶ月、約11ヶ月、または約12ヶ月である。この態様において、「約」という用語には、表記の期間の±0.5ヶ月の範囲が含まれる。さらなる態様において、クレアチニンクリアランスの少なくとも50%減少までの時間の中央値は、PDS処置患者において12ヶ月まで長い。
【0142】
さらなる態様において、血清クレアチニンの倍加までの時間および/またはクレアチニンクリアランスの少なくとも50%減少は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。もう一つの態様において、クレアチニンクリアランスの50%減少は、約3〜約5ヶ月、または約4ヶ月遅れる。なおさらなる態様において、血清クレアチニンの倍加および/またはクレアチニンクリアランスの少なくとも50%減少は、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して少なくとも約6ヶ月遅れる。さらなる態様において、血清クレアチニンの倍加までの期間は、約3ヶ月〜約5ヶ月、または約4ヶ月遅れる。
【0143】
なおもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%増加までの時間を減少させる方法に関する。方法には、クレアチニンクリアランスの少なくとも50%の増加までの時間が減少するように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0144】
さらなる態様において、クレアチニンクリアランスの少なくとも50%の増加までの時間は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く減少される。なおさらなる態様において、クレアチニンクリアランスの少なくとも50%増加は、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して6ヶ月間減少する。
【0145】
もう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの勾配によって測定した場合に、腎疾患の進行速度を低減させる方法が含まれる。方法には、腎疾患の進行速度が、例えばクレアチニンクリアランスの減少速度の低下によって測定した場合に低減されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0146】
さらなる態様において、クレアチニンクリアランスの勾配は、約0〜10 ml/分/1.73m2/年低減される。もう一つのさらなる態様において、クレアチニンクリアランスの勾配は、約1 ml/分/1.73m2/年、約2 ml/分/1.73m2/年、約3 ml/分/1.73m2/年、約4 ml/分/1.73m2/年、約5 ml/分/1.73m2/年、約6 ml/分/1.73m2/年、約7 ml/分/1.73m2/年、約8 ml/分/1.73m2/年、約9 ml/分/1.73m2/年、または約10 ml/分/1.73m2/年低減される。さらなる態様において、クレアチニンクリアランスの勾配は、約4.7±5 ml/分/1.73m2/年低減される。この態様において、「約」という用語には、値±0.5 ml/分/1.73m2/年が含まれる。
【0147】
さらなる態様において、腎疾患の進行速度は、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、または約60%もしくはそれより大きく低減される。特定の態様において、腎疾患の進行速度は、約30%〜約40%低減される。
【0148】
「クレアチニンクリアランスの変化率」という用語は、経時的な被験体の体表面積に関して標準化されるクレアチニンクリアランスの変化率を指す。例えば、被験体のクレアチニンクリアランスは、例えば指定の時点での24時間尿採取によって測定されうる。このクレアチニンクリアランスは、体表面積に関して標準化され、その被験体に関して利用可能なクレアチニンクリアランス測定を用いて、被験体内の勾配の最小二乗法推定値を計算してもよい。一般的に、クレアチニンクリアランスの勾配は、年間変化率として表記される。必要であれば、勾配の計算の前に、適した変換(すなわち、対数変換)を適用してもよい。
【0149】
さらなる態様において、被験体のクレアチニンクリアランスの変化率は、約1 ml/分/年もしくはそれより多く、約2 ml/分/年もしくはそれより多く、約3 ml/分/年もしくはそれより多く、約4 ml/分/年もしくはそれより多く、約5 ml/分/年もしくはそれより多く、約6 ml/分/年もしくはそれより多く、約7 ml/分/年もしくはそれより多く、約8 ml/分/年もしくはそれより多く、約9 ml/分/年もしくはそれより多く、または約10 ml/分/年もしくはそれより多く改善される。さらなる態様において、クレアチニンクリアランス速度の減少は約2〜約5 ml/分/年減弱される。
【0150】
さらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるタンパク尿症を安定化するかまたは低減させるための方法に関する。方法には、被験体におけるタンパク尿症が安定化されるかまたは低減されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。一つの態様において、タンパク尿症は、約0.5 g/24時間もしくはそれより多く、約1 g/24時間もしくはそれより多く、約1.5 g/24時間もしくはそれより多く、または約2 g/24時間もしくはそれより多く低減される。一つの態様において、タンパク尿症は1 g/24時間未満、またはそれに等しい値で安定化される。
【0151】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における腎機能を安定化するため、または腎疾患の進行を遅らせるための方法に関する。方法には、被験体の腎機能が安定化される、または腎疾患の進行が遅れるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0152】
さらなる態様において、腎疾患の進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。
【0153】
なおもう一つの態様において、本発明は、AAアミロイドーシスを有する被験体における腎障害を処置するための方法に関する。方法には、腎障害が処置されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0154】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群への進行を予防するかまたは遅らせるための方法に関する。方法には、ネフローゼ症候群への進行が予防するかまたは遅れるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0155】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群を処置するための方法に関する。方法には、ネフローゼ症候群に関連するパラメータが改善される、またはネフローゼ症候群が寛解されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0156】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群の寛解を維持するための方法に関する。方法には、ネフローゼ症候群の寛解が例えば、約4、6、8、10、または12ヶ月間持続するように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。特定の態様において、ネフローゼ症候群の寛解は、約6〜約8ヶ月間患者において持続される。
【0157】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるGFRを安定化するかまたは増加させるための方法に関する。方法には、GFRを安定化するかまたは増加させるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0158】
「糸球体濾過速度」および「GFR」という用語は、本明細書において互換的に用いられ、腎機能の指標である。例えば、被験体のGFRの一つの手段は、クレアチニンクリアランス速度である。腎機能および/またはGFRは、例えば血清クレアチニンレベル、尿中クレアチニンレベル、尿中アルブミンレベル、尿中微小タンパク質レベル(例えば、レチノール結合タンパク質、N-アセチル-β-D-グルコサミニダーゼ、ミクロアルブミン等)、血漿インスリンクリアランス、クレアチニンクリアランス、タンパク尿症等のような、多数の基準を用いて評価することができる。
【0159】
さらに、被験体は軽度、中等度、または重度腎障害を有してもよい。例えば、健康被験体は典型的に、約100 ml/分より大きいGFRを有する。「軽度の」腎障害を有する被験体は、例えばGFR約50〜約80 ml/分、またはGFR 100未満、またはクレアチニンクリアランス速度約60〜約90 ml/分を有してもよい。「中等度の」腎障害を有する被験体は、例えば、GFR約30〜約50 ml/分、またはクレアチニンクリアランス速度約30〜約60 ml/分を有してもよい。「重度の」腎障害を有する被験体は、例えばGFR約30 ml/分未満、またはクレアチニンクリアランス速度約15〜約30 ml/分を有してもよい。被験体はまた、本明細書に記述の実施例において記述されるように、または当技術分野で公知の基準を用いて、軽度、中等度、または重度であると分類されてもよい(例えば、McCullough, P. A., Rev. Cardiovasc. Med. 2003;4(suppl. 1): S2-S6;K/DOQI guidelines at www.kidney.org/ professionalsを参照されたい)。
【0160】
他の態様において、被験体は、処置前(例えば、ベースライン時)、約50〜約120 ml/分、約60〜約100 ml/分、約70〜約110 ml/分、または約70〜約100 ml/分のクレアチニンクリアランス速度を有してもよい。
【0161】
さらなる態様において、被験体はネフローゼである。もう一つのさらなる態様において、被験体は非ネフローゼである。被験体は、例えば炎症性疾患、悪性新生物、または慢性感染症のような障害に苦しんでもよい。
【0162】
B.本発明の化合物
一つの態様において、本発明は式(I)の化合物に関する:
式中、Yは各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3、または4であり;mは1または2であり、ただし、mが2の場合、-(CH2)n-基の一つの水素は存在しない。
【0163】
「陽イオン基」という用語には、陽電荷と水素原子とを有する基が含まれる。陽イオンの例には、SO3-またはOSO3-の薬学的に許容される塩が含まれる。陽イオン基の例には、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム、およびアルミニウム等のようなアルカリまたはアルカリ土類金属イオンが含まれる。さらなる態様において、陽イオン基はH+またはNa+である。
【0164】
本発明の化合物の例には、以下の化合物およびその薬学的に許容される塩が含まれる。
1,2-エタンジスルホン酸 HO3SCH2CH2SO3H
1,2-エタンジスルホン酸ナトリウム NaO3SCH2CH2SO3NA
1,3-プロパンジスルホン酸 HO3SCH2CH2CH2SO3H
1,3-プロパンジスルホン酸ナトリウム(1,3-プロパンジスルホン酸、二ナトリウム塩) NaO3SCH2CH2CH2SO3Na
1,2-エタンジオールビス(硫酸水素塩) HO3SOCH2CH2OSO3H
1,2-エタンジオールジスルファート、二ナトリウム塩 NaO3SOCH2CH2OSO3Na
1,3-プロパンジオールビス(硫酸水素塩) HO3SOCH2CH2CH2OSO3H
1,3-プロパンジオールジスルファート、二ナトリウム塩NaO3SOCH2CH2CH2OSO3Na
2-スルホメチル-1,4-ブタンジスルホン酸 HO3SCH2CH2CH(CH2SO3H)2
2-スルホメチルブタン-1,4-ジスルホン酸、三ナトリウム塩 NaO3SCH2CH2CH(CH2SO3Na)2
【0165】
一つの態様において、化合物または抗アミロイド原性物質は、1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸ではない。もう一つの態様において、化合物または抗アミロイド原性物質は、1,3-プロパンジスルホン酸二ナトリウム塩ではない。
【0166】
「化合物」という用語には、化学実体が含まれる。化合物は、固体、液体、または気体様の相に存在してもよい。化合物という用語には、式(I)の化合物およびその薬学的に許容される塩が含まれる。本発明の化合物は、本明細書においてその化学構造および/または化学名によって同定される。化合物が化学構造および化学名の双方によって参照され、その化学構造と化学名が一致しない場合、化学構造が化合物の同一性を決定する。本発明の化合物は、キラル中心を含んでもよく、したがって、立体異性体として存在してもよい。本明細書において定義されるように、化合物は天然起源から精製してもよく、販売元から購入してもよく、または当技術分野において認識された技術を用いて化学合成してもよい。
【0167】
さらに、本発明の化合物はまた、水和型および無水型として存在してもよい。式(I)の化合物の水和型は、式(I)の化合物として含まれる。さらなる態様において、式(I)の化合物は一水和物である。一つの態様において、式(I)の化合物は、水を重量で約10%もしくはそれ未満、約9%もしくはそれ未満、約8%もしくはそれ未満、約7%もしくはそれ未満、約6%もしくはそれ未満、約5%もしくはそれ未満、約4%もしくはそれ未満、約3%もしくはそれ未満、約2%もしくはそれ未満、約1%もしくはそれ未満、約0.5%もしくはそれ未満、約0.1%もしくはそれ未満含む。もう一つの態様において、本発明の化合物は、水を重量で、約0.1%もしくはそれより多く、約0.5%もしくはそれより多く、約1%もしくはそれより多く、約2%もしくはそれより多く、約3%もしくはそれより多く、約4%もしくはそれより多く、約5%もしくはそれより多く、または約6%もしくはそれより多く含む。
【0168】
さらに、本発明の化合物はまた、一つより多い多形型、水和型等を含んでもよい。例えば、一つの型、型Iは本発明の化合物、例えば1,3-プロパンジスルホン酸、二ナトリウム塩の直接再結晶によって調製されうる。化合物は、16:1エタノール:水(v/v)の溶液によって沈殿する。再結晶産物は微細白色粉末として回収され、次にこれを65℃、4 mmHgで16時間乾燥させる。得られた非水和型は、水分含有量0.2%および見かけの密度0.64 g/mlを有する。さらなる態様において、式(I)の化合物は約0.2%の水分含有量を有する。
【0169】
さらに、もう一つの型IIは、型Iと同様の方法で市販の1,3-プロパンジスルホン酸、二ナトリウム塩の直接再結晶により調製されうる。化合物は、8:1エタノール:水(v/v)の溶液から沈殿する。再結晶産物は白色固体として回収され、次にこれを20〜25℃、4 mmHgで16時間乾燥させる。得られた非水和型は、水分含有量約7% w/wおよび見かけの密度0.46 g/mlを有する。さらなる態様において、式(I)の化合物は水分含有量約7%を有する。
【0170】
型Iはまた、減圧下で持続的に加熱することによって、型IIの多形型からも調製されうる。第一に、型IIの多形型(水分含有量6.8%)を4 mm Hgの真空において65℃で16時間乾燥させる。この初回乾燥によって、先の水和多形型の水分含有量は2.3%に低減される。65℃でさらに24時間後、先の一水和多形型の水分含有量は1%に低減される。化合物は77℃でさらに48時間乾燥した後に型I多形型のみに完全に変換される。
【0171】
本発明の化合物は、一つまたは複数の酸性官能基を含み、このように、薬学的に許容される塩基と薬学的に許容される塩を形成することができる。これらの事例における「薬学的に許容される塩」という用語は、本発明の化合物の比較的非毒性の無機および有機塩基付加塩を指す。
【0172】
これらの塩も同様に、物質の最終単離および精製の際にインサイチューで、またはその遊離の酸型の精製物質を、薬学的に許容される金属陽イオンの水酸化物、炭酸塩、もしくは重炭酸塩のような適した塩基、アンモニア、または薬学的に許容される有機1級、2級、もしくは3級アミンと個別に反応させることによって調製されうる。代表的なアルカリまたはアルカリ土類塩には、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム、およびアルミニウム塩等が含まれる。塩基付加塩の形成にとって有用な代表的な有機アミンには、エチルアミン、ジエチルアミン、エチレンジアミン、エタノールアミン、ジエタノールアミン、ピペラジン等が含まれる。
【0173】
「薬学的に許容される塩」にはまた、例えば以下で詳しく記述されるようにおよび本出願において他所で記述されるように、その塩基性塩を作製することによって改変された物質の誘導体が含まれる。薬学的に許容される塩の例には、スルホネートのような酸性残基のアルカリまたは有機塩が含まれる。薬学的に許容される塩には、例えば非毒性の無機または有機酸から形成された親物質の通常の非毒性の塩または4級アンモニウム塩が含まれる。そのような通常の非毒性の塩には、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸、および硝酸のような無機酸に由来する塩;酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パルモ酸、マレイン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、メシル酸、安息香酸、サリチル酸、スルファニル酸、2-アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、およびイセチオン酸のような有機酸から調製された塩が含まれる。薬学的に許容される塩は、通常の化学法によって塩基性または酸性部分を含む親物質から合成してもよい。一般的に、そのような塩は、これらの物質の遊離の酸または塩基型を、水もしくは有機溶媒、または両者の混合物において、適当な塩基または酸の化学量論的量と反応させることによって調製してもよい。
【0174】
記述の化合物の全ての酸、塩、塩基、ならびに他のイオンおよび非イオン型が本発明の化合物として含まれる。例えば、化合物が本明細書において酸として示される場合、化合物の塩型も同様に含まれる。同様に、化合物が塩として示される場合、酸および/または塩基型も同様に含まれる。
【0175】
さらなる態様において、式(I)の化合物は1,3-プロパンジスルホン酸二ナトリウム塩または1,3-プロパンジスルホン酸ではない。
【0176】
C.本発明の製剤
もう一つの態様において、本発明は、製剤が被験体への投与時に少なくとも一つの都合のよい生物学的特性(FBP)を有するように、製剤において式(I)の化合物の治療的有効量を含む、AAアミロイドーシスを処置するための薬学的製剤に関する。
【0177】
「薬学的製剤」という用語には、以下に記述される薬学的組成物が含まれる。さらなる態様において、薬学的製剤は、本発明の化合物のAAアミロイドーシスおよび/またはアミロイド関連疾患の処置能を増強する、都合のよい生物学的特性を有するように設計される。製剤の都合のよい生物学的特性は、臨床試験の際に被験体に本発明の化合物を投与することによって発見された。
【0178】
「都合のよい生物学的特性」という用語には、本発明の化合物がその意図する機能を行う能力、例えばAAアミロイドーシスおよび/またはアミロイド関連疾患を処置する能力を増強する、本発明の化合物がAAアミロイドーシスを阻害するおよび/またはアミロイド関連疾患を処置する能力以外の生物学的特性が含まれる。一つの態様において、都合のよい生物学的特性は、薬物動態プロフィールとなりうる。用いてもよいそのようなパラメータの例には、Cmax、Css、Tmax、AUC0-t、AUC∞、およびT1/2が含まれるがこれらに限定されるわけではない。これらのパラメータ(Cmax、Tmax、AUC0-t、AUC∞、およびT1/2)は、例えばWinNonlin(登録商標)(Pharsight Corporation, Mountain View, CA)またはWindows(登録商標)用SAS(登録商標)(SAS Institute Inc., Cary, NC)を用いる非コンパートメント分析によって誘導してもよい。さらなる態様において、都合のよい生物学的特性は、標的血漿濃度または標的全身曝露である。
【0179】
「Cmax」という用語は、特定の被験体における本発明の化合物の観察された最高血漿濃度を指す。
【0180】
「Css」という用語は、特定の被験体における本発明の化合物の定常状態血漿濃度を指す。
【0181】
「Tmax」という用語は、Cmaxに達するまでの時間を指す。
【0182】
「AUC0-t」という用語は、線形台形則によって計算した、ゼロ時間から、濃度が定量限界であるまたはそれより上である最後の試料採取時間までの血漿濃度対時間曲線下面積を指す。
【0183】
「AUC∞」という用語は、AUC0-t+(Clast/λz)から計算した、ゼロ時間から無限大までの血漿濃度対時間曲線下面積を指し、Clastは最後に観察された定量可能な濃度であり、λzは見かけの終末速度定数である。
【0184】
「T1/2」という用語は、ln 2λzから計算された見かけの終末半減期である。
【0185】
本発明にはまた、薬物動態パラメータまたはその組み合わせのような、二つまたはそれより多い都合のよい生物学的特性を合わせる製剤および組成物が含まれる。これらの薬物動態パラメータの例には、AUC0-t、AUC∞、Cmax、およびTmaxが含まれる。例えば、本発明の製剤は、健康な被験体(または腎障害を有しない被験体)に投与した場合に、選択された製剤が被験体に一つまたは複数の所望の薬物動態パラメータを提供するように選択されてもよい。または、本発明の製剤は、腎機能障害を有する被験体および/またはAAアミロイドーシスを有する、もしくはAAアミロイドーシスのリスクを有する被験体に投与した場合に、選択された製剤が所望の薬物動態パラメータの一つまたは複数を被験体に提供するように選択されてもよい。
【0186】
さらなる態様において、製剤は実施例7に記述されたとおりではない。もう一つのさらなる態様において、少なくとも一つの成分は、実施例7において記述された成分ではない。
【0187】
もう一つのさらなる態様において、製剤は乳糖一水和物ではない希釈剤を有する。乳糖一水和物ではない希釈剤の例には、例えば糖(例えば、グルコース、蔗糖、果糖等)、デンプン(例えば、コーンスターチ、ジャガイモデンプン等)、セルロース、セルロース誘導体(例えば、カルボキシメチルセルロースナトリウム、エチルセルロース、酢酸セルロース等)、粉末トラガカント、麦芽、ゼラチン、タルク、およびその混合物が含まれる。もう一つのさらなる態様において、希釈剤および/または潤滑剤の吸湿性は、得られたカプセルがFDA規則に許容されるように選択される。
【0188】
もう一つのさらなる態様において、製剤は、ステアリン酸マグネシウムではない潤滑剤を有する。ステアリン酸マグネシウムではない潤滑剤の例には、例えばステアリン酸粉末、タルク、ステアリン酸カルシウム、ポリエチレングリコール、ラウリル硫酸ナトリウム、およびその混合物が含まれる。
【0189】
もう一つの態様において、製剤は、約0.5%(w/w)未満の任意の一つの公知の不純物、約1.5%(w/w)未満の総硫酸塩、約0.1%(w/w)未満の未知の総不純物、および約5.0%(w/w)未満の全不純物を含む。
【0190】
もう一つの態様において、式(I)の化合物は、1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸の参照標準IR(赤外線)スペクトルおよび/またはIC(イオンクロマトグラフィー)に適合する。もう一つの態様において、式(I)の化合物は、約1.0%w/w未満の水を含む。なおもう一つの態様において、式(I)の化合物は、約20 ppm未満の重金属、および/または約0.5%w/w未満の残留溶媒を有する。
【0191】
もう一つの態様において、本発明は、抗アミロイド原性物質含有製剤が、それが生物学的に都合のよい製剤であるように、被験体に投与した場合に少なくとも一つの都合のよい生物学的特性を有することが予め決定された標準的な製剤と同等であるように、製剤中に抗アミロイド原性物質を含む、AAアミロイドーシスを処置するための生物学的に都合のよい製剤に関する。
【0192】
「生物学的に都合のよい製剤」という用語は、少なくとも一つの都合のよい生物学的特性を有する薬学的製剤を指す。さらなる態様において、製剤は、二つもしくはそれより多い、三つもしくはそれより多い、または四つもしくはそれより多い生物学的に都合のよい特性を有する。一つの態様において、生物学的に都合のよい製剤は、本発明の化合物の標的血漿濃度が、化合物の被験体への投与後30分もしくはそれ未満、1時間もしくはそれ未満、2時間もしくはそれ未満、または5時間もしくはそれ未満で被験体において達成されるように製剤される。
【0193】
一つの態様において、生物学的に都合のよい製剤は、特定のAUCssおよび/またはCssを達成するために、被験体のクレアチニンクリアランス速度に基づいて選択される。
【0194】
例えば、都合のよい生物学的特性は、約5000 ng.h/mLもしくはそれより上;約6000 ng.h/mLもしくはそれより上;約6500 ng.h/mLもしくはそれより上;約7000 ng.h/mLもしくはそれより上;約8000 ng.h/mLもしくはそれより上;約9000 ng.h/mLもしくはそれより上;約10,000 ng.h/mLもしくはそれより上;約11,000 ng.h/mLもしくはそれより上;約12,000 ng.h/mLもしくはそれより上;約13,000 ng.h/mLもしくはそれより上;約14,000 ng.h/mLもしくはそれより上;約15,000 ng.h/mLもしくはそれより上;約16,000 ng.h/mLもしくはそれより上;約17,000 ng.h/mLもしくはそれより上;約18,000 ng.h/mLもしくはそれより上;約19,000 ng.h/mLもしくはそれより上;約20,000 ng.h/mLもしくはそれより上;約21,000 ng.h/mLもしくはそれより上;約22,000 ng.h/mLもしくはそれより上;約23,000 ng.h/mLもしくはそれより上;約24,000 ng.h/mLもしくはそれより上;または約25,000 ng.h/mLもしくはそれより上のAUCssであってもよい。もう一つの態様において、都合のよい生物学的特性は、約7000 ng.h/mL; 約8000 ng.h/mLもしくはそれ未満;約9000 ng.h/mLもしくはそれ未満;約10,000 ng.h/mLもしくはそれ未満;約11,000 ng.h/mLもしくはそれ未満;約12,000 ng.h/mLもしくはそれ未満;約13,000 ng.h/mLもしくはそれ未満;約14,000 ng.h/mLもしくはそれ未満;約15,000 ng.h/mLもしくはそれ未満;約16,000 ng.h/mLもしくはそれ未満;約17,000 ng.h/mLもしくはそれ未満;約18,000 ng.h/mLもしくはそれ未満;約19,000 ng.h/mLもしくはそれ未満;約20,000 ng.h/mLもしくはそれ未満;約21,000 ng.h/mLもしくはそれ未満;約22,000 ng.h/mLもしくはそれ未満;約23,000 ng.h/mLもしくはそれ未満;約24,000 ng.h/mLもしくはそれ未満;約25,000 ng.h/mLもしくはそれ未満;または約26,000 ng.h/mLもしくはそれ未満のAUCssである。
【0195】
生物学的に都合のよい特性は、望ましい定常状態濃度(Css)であってもよい。例えば、都合のよい生物学的特性は、約500 ng/mLもしくはそれより上;約600 ng/mLもしくはそれより上;約700 ng/mLもしくはそれより上;約800 ng/mLもしくはそれより上;約900 ng/mLもしくはそれより上;約950もしくはそれより上ng/mL; 約1000 ng/mLもしくはそれより上;約1100 ng/mLもしくはそれより上;または約1200 ng/mLのCssであってもよい。さらに、生物学的に都合のよい特性は、約1200 ng/mLもしくはそれ未満、約1100 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約800 ng/mLもしくはそれ未満;約700 ng/mLもしくはそれ未満;約600 ng/mLもしくはそれ未満;または約500 ng/mL未満のCssであってもよい。
【0196】
もう一つの態様において、都合のよい生物学的特性は、約250〜約2000 ng/mlのCmax(化合物の1回経口投与後の)であってもよい。さらなる態様において、Cmaxは、約250 ng/mlもしくはそれより大きい;約300 ng/mLもしくはそれより大きい;約350 ng/mLもしくはそれより大きい;約400 ng/mLもしくはそれより大きい;約500 ng/mLもしくはそれより大きい;約600 ng/mLもしくはそれより大きい;約700 ng/mLもしくはそれより大きい;約800 ng/mLもしくはそれより大きい;約900 ng/mLもしくはそれより大きい;約1000 ng/mLもしくはそれより大きい;約1100 ng/mLもしくはそれより大きい;約1200 ng/mLもしくはそれより大きい;約1300 ng/mLもしくはそれより大きい;約1400 ng/mLもしくはそれより大きい;約1500 ng/mLもしくはそれより大きい;約1600 ng/mLもしくはそれより大きい;約1700 ng/mLもしくはそれより大きい;約1800 ng/mLもしくはそれより大きい;約1900 ng/mLもしくはそれより大きい;または約2000 ng/mLもしくはそれより大きい。この態様において「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0197】
さらなる態様において、1回経口投与後のCmaxは、約850 ng/mL; 約1700 ng/mLもしくはそれ未満;約1600 ng/mLもしくはそれ未満;約1500 ng/mLもしくはそれ未満;約1400 ng/mLもしくはそれ未満;約1300 ng/mLもしくはそれ未満;約1200 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約800 ng/mLもしくはそれ未満;約700もしくはそれ未満;約700 ng/mLもしくはそれ未満;約500 ng/mLもしくはそれ未満;約400 ng/mLもしくはそれ未満;または約300 ng/mLもしくはそれ未満である。もう一つの態様において、化合物の1回経口投与後のCmaxは、少なくとも200 ng/mLである。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0198】
もう一つの態様において、都合のよい生物学的特性は、約400〜約3800 ng/mlのCmax(化合物の複数回経口投与後の)であってもよい。さらなる態様において、Cmaxは、約400 ng/mLもしくはそれより大きい;約500 ng/mLもしくはそれより大きい;約600 ng/mLもしくはそれより大きい;約700 ng/mLもしくはそれより大きい;約800 ng/mLもしくはそれより大きい;約900 ng/mLもしくはそれより大きい;約1000 ng/mLもしくはそれより大きい;約1100 ng/mLもしくはそれより大きい;約1200 ng/mLもしくはそれより大きい;約1300 ng/mLもしくはそれより大きい;約1400 ng/mLもしくはそれより大きい;約1500 ng/mLもしくはそれより大きい;約1600 ng/mLもしくはそれより大きい;約1700 ng/mLもしくはそれより大きい;約1800 ng/mLもしくはそれより大きい;約1900 ng/mLもしくはそれより大きい;約2000 ng/mLもしくはそれより大きい;約2100 ng/mLもしくはそれより大きい;約2200もしくはそれより大きい;約2300 ng/mLもしくはそれより大きい;約2400 ng/mLもしくはそれより大きい;約2500 ng/mLもしくはそれより大きい;約2600 ng/mLもしくはそれより大きい;約2700 ng/mLもしくはそれより大きい;約2800 ng/mLもしくはそれより大きい;約2900 ng/mLもしくはそれより大きい;約3000 ng/mLもしくはそれより大きい;約3100 ng/mLもしくはそれより大きい;約3200もしくはそれより大きい;約3300 ng/mLもしくはそれより大きい;約3400 ng/mLもしくはそれより大きい;約3500 ng/mLもしくはそれより大きい;約3600 ng/mLもしくはそれより大きい;約3700 ng/mLもしくはそれより大きい;約3800 ng/mLもしくはそれより大きい;または約3900 ng/mLもしくはそれより大きい。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0199】
さらなる態様において、Cmax(化合物の複数回経口投与後の)は、約500〜約3900 ng/mlである。さらなる態様において、Cmaxは、約500 ng/mLもしくはそれ未満約600 ng/mLもしくはそれ未満;約700 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約1100 ng/mLもしくはそれ未満;約1200 ng/mL; 約1300 ng/mLもしくはそれ未満;約1400 ng/mLもしくはそれ未満;約1500 ng/mLもしくはそれ未満;約1600 ng/mLもしくはそれ未満;約1700 ng/mLもしくはそれ未満;約1800 ng/mLもしくはそれ未満;約1900 ng/mLもしくはそれ未満;約2000 ng/mLもしくはそれ未満;約2100 ng/mLもしくはそれ未満;約2200もしくはそれ未満;約2300 ng/mLもしくはそれ未満;約2400 ng/mLもしくはそれ未満;約2500 ng/mLもしくはそれ未満;約2600 ng/mLもしくはそれ未満;約2700 ng/mLもしくはそれ未満;約2800 ng/mLもしくはそれ未満;約2900 ng/mLもしくはそれ未満;約3000 ng/mLもしくはそれ未満;約3100 ng/mLもしくはそれ未満;約3200もしくはそれ未満;約3300 ng/mLもしくはそれ未満;約3400 ng/mLもしくはそれ未満;約3500 ng/mLもしくはそれ未満;約3600 ng/mLもしくはそれ未満;約3700 ng/mLもしくはそれ未満;約3800 ng/mLもしくはそれ未満;または約3900 ng/mLもしくはそれ未満である。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0200】
例えば、都合のよい生物学的特性は、約2000 ng.h/mLもしくはそれより上;約3000 ng.h/mLもしくはそれより上;約4000 ng.h/mLもしくはそれより上;約5000 ng.h/mLもしくはそれより上;約6000 ng.h/mLもしくはそれより上;約6500 ng.h/mLもしくはそれより上;約7000 ng.h/mLもしくはそれより上;約8000 ng.h/mLもしくはそれより上;約9000 ng.h/mLもしくはそれより上;約10,000 ng.h/mLもしくはそれより上;約11,000 ng.h/mLもしくはそれより上;約12,000 ng.h/mLもしくはそれより上;約13,000 ng.h/mLもしくはそれより上;約14,000 ng.h/mLもしくはそれより上;約15,000 ng.h/mLもしくはそれより上;約16,000 ng.h/mLもしくはそれより上;約17,000 ng.h/mLもしくはそれより上;約18,000 ng.h/mLもしくはそれより上;約19,000 ng.h/mLもしくはそれより上;約20,000 ng.h/mLもしくはそれより上;約21,000 ng.h/mLもしくはそれより上;約22,000 ng.h/mLもしくはそれより上;約23,000 ng.h/mLもしくはそれより上;約24,000 ng.h/mLもしくはそれより上;約25,000 ng.h/mLもしくはそれより上;約26,000 ng.h/mLもしくはそれより上;約27,000 ng.h/mLもしくはそれより上;約28,000 ng.h/mLもしくはそれより上;約29,000 ng.h/mLもしくはそれより上;約30,000 ng.h/mLもしくはそれより上;約31,000 ng.h/mLもしくはそれより上;約32,000 ng.h/mLもしくはそれより上;約33,000 ng.h/mLもしくはそれより上;約34,000 ng.h/mLもしくはそれより上;約35,000 ng.h/mLもしくはそれより上;約36,000 ng.h/mLもしくはそれより上;約37,000 ng.h/mLもしくはそれより上;約38,000 ng.h/mLもしくはそれより上;約39,000 ng.h/mLもしくはそれより上;約40,000 ng.h/mLもしくはそれより上;約41,000 ng.h/mLもしくはそれより上;約42,000 ng.h/mLもしくはそれより上;約43,000 ng.h/mL; 約44,000 ng.h/mLもしくはそれより上;約45,000 ng.h/mLもしくはそれより上;または約46,000 ng.h/mLのAUC∞であってもよい。この態様において、「約」という用語には、表記の範囲の±750 ng/mlの値が含まれる。
【0201】
もう一つの態様において、都合のよい生物学的特性は、約2000 ng.h/ml;約3000 ng.h/mLもしくはそれ未満;約4000 ng.h/mLもしくはそれ未満;約5000 ng.h/mLもしくはそれ未満;約5000 ng.h/mLもしくはそれ未満;約6000 ng.h/mLもしくはそれ未満;約6500 ng.h/mLもしくはそれ未満;約7000 ng.h/mLもしくはそれ未満;約8000 ng.h/mLもしくはそれ未満;約9000 ng.h/mLもしくはそれ未満;約10,000 ng.h/mLもしくはそれ未満;約11,000 ng.h/mLもしくはそれ未満;約12,000 ng.h/mLもしくはそれ未満;約13,000 ng.h/mLもしくはそれ未満;約14,000 ng.h/mLもしくはそれ未満;約15,000 ng.h/mLもしくはそれ未満;約16,000 ng.h/mLもしくはそれ未満;約17,000 ng.h/mLもしくはそれ未満;約18,000 ng.h/mLもしくはそれ未満;約19,000 ng.h/mLもしくはそれ未満;約20,000 ng.h/mLもしくはそれ未満;約21,000 ng.h/mLもしくはそれ未満;約22,000 ng.h/mLもしくはそれ未満;約23,000 ng.h/mLもしくはそれ未満;約24,000 ng.h/mLもしくはそれ未満;約25,000 ng.h/mLもしくはそれ未満;約26,000 ng.h/mLもしくはそれ未満;約27,000 ng.h/mLもしくはそれ未満;約28,000 ng.h/mLもしくはそれ未満;約29,000 ng.h/mLもしくはそれ未満;約30,000 ng.h/mLもしくはそれ未満;約31,000 ng.h/mLもしくはそれ未満;約32,000 ng.h/mLもしくはそれ未満;約33,000 ng.h/mLもしくはそれ未満;約34,000 ng.h/mLもしくはそれ未満;約35,000 ng.h/mLもしくはそれ未満;約36,000 ng.h/mLもしくはそれ未満;約37,000 ng.h/mLもしくはそれ未満;約38,000 ng.h/mLもしくはそれ未満;約39,000 ng.h/mLもしくはそれ未満;約40,000 ng.h/mLもしくはそれ未満;約41,000 ng.h/mLもしくはそれ未満;約42,000 ng.h/mLもしくはそれ未満;約43,000 ng.h/mLもしくはそれ未満;約44,000 ng.h/mLもしくはそれ未満;約45,000 ng.h/mLもしくはそれ未満;または約46,000 ng.h/mLもしくはそれ未満のAUC∞である。この態様において、「約」という用語には、表記の範囲の±750 ng.h/mlの値が含まれる。
【0202】
なおもう一つの態様において、本発明は、少なくとも部分的に、薬学的製剤に関する。薬学的製剤は、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、製剤を健康な被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0203】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、健康被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される薬学的製剤にも関する。
【0204】
なおもう一つのさらなる態様において、本発明はまた、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、健康被験体に経口投与する場合、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される薬学的製剤に関する。
【0205】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に経口投与する場合、活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0206】
なおもう一つのさらなる態様において、本発明は、活性物質(例えば、PDS)800 mgと薬学的に許容される担体とを含む薬学的製剤であって、製剤を被験体に投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0207】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、製剤をAAアミロイドーシスを有する被験体に24ヶ月間経口投与する場合、活性物質の用量が400 mgの場合、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0208】
もう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、製剤を健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0209】
「同等」という用語は、本発明の製剤と生物学的に同等または機能的に同等であると見なされる製剤を指す。さらなる態様において、製剤は、1,3-プロパンジスルホン酸二ナトリウム塩または1,3-プロパンジスルホン酸を含む製剤と同等である。さらなる態様において、用語には、21 U.S.C.§255および/または21 CFR§320の下で生物学的同等性に関する定義を満たす製剤が含まれる。もう一つの態様において、「同等」は、被験体に投与した場合に、例えば本明細書において開示した値の±10%、±20%、±30%、または±40%以内である薬物動態パラメータ、例えばCmax、AUC∞が得られる製剤を指す。
【0210】
さらなる態様において、「同等」という用語には、類似の実験条件で投与した場合に、同等である可能性がある製剤の吸収速度および程度が、標準的な製剤の吸収速度および程度と有意差を示さない、例えば±10%、±20%、±30%、または±40%以内である、同等である可能性がある製剤が含まれる。もう一つの態様において、おそらく同等である製剤の吸収の程度は、類似の実験条件で投与した場合に、同等である可能性がある製剤の吸収速度および程度が、標準的な製剤の吸収速度および程度と有意差を示さない、例えば±10%、±20%、±30%、または±40%以内であり、標準製剤からの差は、慢性的な使用の際に有効な体薬物濃度を達成するために本質的ではなく、および/または薬物にとって医学的に重要ではないと見なされる。一つの局面において、同じモル用量を投与してもよい。
【0211】
「標準製剤」という用語は、式(I)の化合物、例えば少なくとも一つの都合のよい生物学的特性を有する1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸の製剤を指す。他の態様において、標準製剤は、薬物製剤の仕様書の必要条件を満たす、またはそれを超える。さらなる態様において、標準製剤は実施例7に記載される製剤である。
【0212】
「薬物製剤の仕様書」という用語は、米国FDAの薬物製剤仕様書を満たす製剤を指す。さらなる態様において、製剤は、多くて約0.5%w/wの任意の一つの公知の不純物、多くて約1.5%w/wの総硫酸塩、多くて0.1%w/wの任意の一つの未知不純物、および多くて約5.0%w/wの総不純物を含む。
【0213】
「抗アミロイド原性物質」は、AAアミロイド結合アッセイ(AAABA)において試験陽性である化合物を指す。さらなる態様において、抗アミロイド原性物質は、AAABAにおいて測定した場合AAアミロイド濃度400μMで30%もしくはそれより大きい、45%もしくはそれより大きい、60%もしくはそれより大きい、70%もしくはそれより大きい、80%もしくはそれより大きい、または90%もしくはそれより大きい割合でAAに結合する。もう一つの態様において、抗アミロイド原性化合物は、AAABAにおいて測定した場合AAアミロイド濃度200μMで30%もしくはそれより大きい、45%もしくはそれより大きい、60%もしくはそれより大きい、70%もしくはそれより大きい、80%もしくはそれより大きい、または90%もしくはそれより大きい割合でAAに結合する。
【0214】
「抗アミロイド原性物質」という用語にはまた、ジスルホン化化合物およびスルホン化アルキル化合物が含まれる。さらなる態様において、この用語には、1,3-プロパンジスルホン酸の類似体またはその薬学的に許容される塩である化合物が含まれる。
【0215】
さらなる態様において、抗アミロイド原性物質は、先に記述したように式(I)の化合物である。抗アミロイド原性物質の例には、1,2-エタンジスルホン酸、1,2-エタンジスルホン酸ナトリウム、1,2-エタンジオールビス(硫酸水素塩)、1,2-エタンジオールジスルファート二ナトリウム塩、1,3-プロパンジオールビス(硫酸水素塩)、1,3-プロパンジオールジスルファート二ナトリウム塩、2-スルホメチル-1,4-ブタンジスルホン酸、または2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩のような化合物が含まれる。さらなる態様において、抗アミロイド原性物質は、1,3-プロパンジスルホン酸または1,3-プロパンジスルホン酸二ナトリウム塩である。
【0216】
AAアミロイド結合アッセイ(AAABA)
AAに関するMSアッセイにおいて、試料を水溶液(必要であれば水に溶解するために20%エタノールを加える)、200μM試験化合物および20μM可溶化AA、または400μM試験化合物および40μM可溶化AAとして調製する。各試料のpH値を、0.1%水酸化ナトリウム水溶液を加えることによって7.4(±0.2)に調節する。次に、溶液を、Waters ZQ 4000質量分析計を用いて、エレクトロスプレーイオン化質量分析計によって分析する。試料を、試料調製後2時間以内に流速25μL/分で直接注入によって導入する。全ての分析に関して、ソース温度を70℃に維持して、コーン電圧は20 Vである。データはMasslynx 3.5ソフトウェアを用いて処理する。MSアッセイは、可溶性AAに対する化合物の結合能に基づくデータを生じる。
【0217】
D.本発明の化合物を含む薬学的組成物
本発明はまた、少なくとも部分的に、式(I)の化合物の治療的有効量と第二の物質とを含む薬学的組成物に関する。さらなる態様において、治療的有効量はAAアミロイドーシスを処置するために有効である。
【0218】
さらなる態様において、本発明は、包装された薬学的組成物に関する。包装された薬学的組成物には、組成物を第二の物質と併用して投与することを勧める表示または添付文書と共に包装された式(I)の化合物の治療的有効量が含まれる。さらなる態様において、治療的有効量はAAアミロイドーシスを処置するために有効である。
【0219】
なおもう一つのさらなる態様において、本発明は、組成物を式(I)の化合物と併用して投与することを勧める表示または添付文書と共に包装された第二の物質の治療的有効量を含む、包装された薬学的組成物に関する。
【0220】
「表示または添付文書」という用語には、被験体、または本発明の組成物の投与に関して被験体の医療に実質的に責任を有する任意の人との全ての書面、電子的、または会話による伝達が含まれるがこれらに限定されるわけではない。添付文書にはさらに、本発明の組成物を他の化合物または組成物、例えば第二の物質と同時投与することに関する情報が含まれる。さらに、添付文書には、食物なしで本発明の組成物の投与に関する説明書が含まれてもよい。
【0221】
なおもう一つの態様において、本発明は、組成物を食物なしで投与することを勧める表示または添付文書と共に、式(I)の化合物の治療的有効量を含む薬学的組成物を保持する容器が含まれる、包装された薬学的組成物に関する。
【0222】
式(I)の化合物は、適当な溶媒による溶液で、または溶媒を含まない型(例えば、凍結乾燥型)で供給されてもよい。本発明のもう一つの局面において、物質および本発明の方法を行うために必要な物質および緩衝液はキットとして包装されてもよい。キットは、本明細書に記述の方法に従って商業的に用いられてもよく、本発明の方法において用いるための説明書が含まれてもよい。さらなるキットの成分には、酸、塩基、緩衝剤、無機塩、溶媒、抗酸化剤、保存剤、または金属キレート剤が含まれてもよい。さらなるキットの成分は純粋な組成物として、または一つもしくは複数のさらなるキット成分を組み入れる水溶液もしくは有機溶液として存在する。任意のまたは全てのキット成分は任意でさらに緩衝液を含む。
【0223】
式(I)の化合物はまた、非経口、腹腔内、脊柱内、または脳内に投与してもよい。グリセロール、液体ポリエチレングリコール、およびその混合物において、ならびに油において分散剤を調製することができる。通常の保存および使用条件において、これらの調製物は、微生物の成長を予防するために保存剤を含んでもよい。
【0224】
本発明の化合物を非経口投与以外で投与するために、その不活化を予防するための材料によって物質をコーティングする、またはその材料を物質と同時投与する必要がある可能性がある。例えば、本発明の化合物は、適当な担体、例えばリポソーム、または希釈剤において被験体に投与してもよい。薬学的に許容される希釈剤には、生理食塩液および水性緩衝液が含まれる。リポソームには、水中油中水型CGF乳剤と共に通常のリポソームが含まれる(Strejan et al., J. Neuroimmunol. 7, 27 (1984))。「薬学的組成物」という用語には、先に記述した「薬学的製剤」が含まれることに注意すべきである。
【0225】
注射での使用に適した薬学的組成物には、滅菌水溶液(水溶性の場合)または分散液、および滅菌注射用溶液または分散液を即時調製するための滅菌粉末が含まれる。全ての場合において、組成物は無菌的でなければならず、容易なシリンジ作動性が存在する程度に流動性でなければならない。これは製造および保存条件で安定でなければならず、細菌および真菌のような微生物の混入作用に対して保護されなければならない。
【0226】
適した薬学的に許容される溶媒には、リン酸緩衝生理食塩液(PBS)のような、経口、非経口、鼻腔内、粘膜、経皮、血管内(IV)、動脈内(IA)、筋肉内(IM)、および皮下(SC)投与経路にとって適した任意の非免疫原性の薬学的補助剤が含まれるがこれらに限定されるわけではない。
【0227】
媒体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコール等)、その適した混合物、および植物油を含む溶媒または分散培地となりうる。例えばレシチンのようなコーティングを用いることによって、分散液の場合には必要な粒子径を維持することによって、および界面活性剤を用いることによって、適切な流動性を維持することができる。微生物の作用の予防は、様々な抗菌および抗真菌剤、例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサル等によって得ることができる。多くの場合において、組成物には、等張剤、例えば糖、塩化ナトリウム、またはマンニトールおよびソルビトールのような多価アルコールが含まれる。注射可能な組成物の持続的な吸収は、吸収を遅らせる物質、例えばモノステアリン酸アルミニウムまたはゼラチンを組成物に含めることによってもたらされうる。
【0228】
滅菌注射液は、先に列挙した成分の一つまたは組み合わせと共に適当な溶媒において必要量の治療物質を組み入れて、必要に応じてその後濾過滅菌することによって調製されうる。一般的に、分散液は、基本分散培地と、先に列挙した中からの必要な他の成分とを含む滅菌媒体に治療物質を組み入れることによって調製される。滅菌注射液を調製するための滅菌粉末の場合、調製法は、活性成分(すなわち本発明の化合物)プラス予め濾過滅菌したその溶液からの任意のさらなる所望の成分の粉末を生じる真空乾燥および凍結乾燥である。
【0229】
本発明の化合物は、例えば不活性希釈剤または同化可能な食用担体と共に経口投与されうる。本発明の化合物および他の成分はまた、硬または軟シェルゼラチンカプセルに封入してもよく、錠剤に圧縮してもよく、または被験体の食事に直接組み入れてもよい。経口治療的投与の場合、本発明の化合物は、賦形剤と共に組み入れて、摂取可能な錠剤、口腔内錠剤、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、ウェーハ等の剤形で用いてもよい。組成物および調製物における本発明の化合物の百分率は、当然変化してもよい。そのような治療的に有用な組成物における本発明の化合物の量は、適した用量が得られるような量である。
【0230】
したがって、本発明には、エアロゾル、経口および非経口投与のための薬学的に許容される媒体において、その薬学的に許容される塩を含む、本発明の化合物を含む薬学的製剤が含まれる。同様に、本発明には、静脈内、筋肉内、または皮下注射によるような、凍結乾燥されて、投与のための薬学的に許容される製剤を形成するように再構成されてもよい、化合物またはその塩が含まれる。投与はまた、皮内または経皮であってもよい。
【0231】
本発明に従って、本明細書において記述される式(I)の物質、およびその薬学的に許容される塩は、固体として経口もしくは吸入によって投与されてもよく、または溶液、懸濁液、もしくは乳液として筋肉内もしくは静脈内に投与されてもよい。または、物質または塩はまた、リポソーム懸濁液として吸入、静脈内、または筋肉内に投与されてもよい。
【0232】
吸入によって、エアロゾルとしての投与に適した薬学的組成物または製剤も同様に提供される。これらの製剤は、本発明の所望の化合物もしくはその塩、または物質もしくは塩の複数の固体粒子の溶液もしくは懸濁液を含む。所望の製剤を、小さいチャンバーに入れて噴霧してもよい。噴霧は、物質または塩を含む複数の液体小滴または固体粒子を形成するために圧縮空気または超音波エネルギーによって行ってもよい。液体小滴または固体粒子は、約0.5〜約5ミクロンの粒子径を有しなければならない。固体粒子は、本発明の任意の化合物またはその塩の固体物質を、微粉化のような当技術分野で公知の適当な方法で処理することによって得ることができる。固体粒子または小滴の大きさは、例えば約1〜約2ミクロンであろう。この局面において、この目的を達成するために市販のネブライザーを利用することができる。
【0233】
エアロゾルとしての投与に適した薬学的製剤は、液体剤形であってもよく、製剤は、水を含む担体において、本発明の化合物またはその塩の水溶性物質を含むであろう。噴霧に供した場合に所望の大きさの範囲内の小滴が形成されるために十分に製剤の表面張力を低下させる界面活性剤が存在してもよい。
【0234】
経口組成物にはまた、液体溶液、乳剤、懸濁液等が含まれる。そのような組成物の調製にとって適した薬学的に許容される媒体は、当技術分野で周知である。シロップ、エリキシル剤、乳剤および懸濁剤にとって典型的な担体成分には、エタノール、グリセロール、プロピレングリコール、ポリエチレングリコール、液体蔗糖、ソルビトール、および水が含まれる。懸濁液の場合、典型的な懸濁剤には、メチルセルロース、カルボキシメチルセルロースナトリウム、トラガカント、およびアルギン酸ナトリウムが含まれ;典型的な湿潤剤には、レシチンおよびポリソルベート80が含まれ;ならびに典型的な保存剤には、メチルパラベンおよび安息香酸ナトリウムが含まれる。経口液体組成物はまた、先に開示した甘味料、着香料、および着色剤のような一つまたは複数の成分を含んでもよい。
【0235】
薬学的組成物はまた、被験物質が所望の局所適用の近傍で、または望ましい作用を持続させるために様々な時間、胃腸管において放出されるように、通常の方法によって、典型的にpHまたは時間依存的コーティングによってコーティングしてもよい。そのような投与剤形には典型的に、酢酸フタル酸セルロース、ポリビニル酢酸フタレート、ヒドロキシプロピルメチルセルロースフタレート、エチルセルロース、ロウおよびシェラックの一つまたは複数が含まれるがこれらに限定されるわけではない。
【0236】
被験物質の全身送達を達成するために有用な他の組成物には、舌下、口腔内、および鼻腔内剤形が含まれる。そのような組成物は典型的に、蔗糖、ソルビトール、およびマンニトールのような可溶性増量物質;ならびにアカシア、微結晶セルロース、カルボキシメチルセルロース、およびヒドロキシプロピルメチルセルロースのような結合材の一つまたは複数を含む。先に開示したグライダント、潤滑剤、甘味料、着色料、抗酸化剤、および着香料もまた含まれてもよい。
【0237】
本発明の組成物はまた、被験体の表皮もしくは上皮組織上に組成物を直接載せるもしくは広げることによって、または「パッチ」によって経皮的に被験体に局所投与されうる。そのような組成物には、例えば、ローション、クリーム、溶液、ゲル、および固体が含まれる。これらの局所組成物は本発明の化合物の有効量、通常少なくとも約0.1%、または約1%〜約5%さえ含んでもよい。局所投与のための適した担体は、典型的に、持続的な被膜として皮膚に留まっていて、発汗または水にぬれることによっても除去されない。一般的に、担体は本質的に有機性であり、その中に治療物質を分散または溶解することができる。担体には、薬学的に許容される皮膚軟化剤、乳化剤、濃化剤、溶媒等が含まれてもよい。
【0238】
そのような物質の毒性および治療効率は、例えばLD50(集団の50%に対して致死的な用量)およびED50(集団の50%において治療的に有効な用量)を決定するための細胞培養または実験動物における標準的な薬学的技法によって決定されうる。毒性効果と治療効果の用量比が治療指数であり、比LD50/ED50として表記することができ、通常、より大きい治療指数がより有効である。毒性の副作用を示す化合物を用いてもよいが、非罹患細胞に対する起こりうる障害を最小限にして、それによって副作用を低減するために、罹患組織部位にそのような物質を標的化する送達系を設計するように注意しなければならい。
【0239】
適当な用量は、当業者の医師、獣医師、または研究者に理解できる多数の要因に依存すると理解される。低分子の用量は、例えば被験体または処置される試料の同一性、大きさ、および状態に応じて、さらに適用可能であれば組成物が投与される経路、および被験体に及ぼしてほしいと医師が願う低分子の効果に応じて、変化するであろう。例としての用量には、被験体または試料の重量1 kgあたり低分子のミリグラムまたはマイクログラム量が含まれる(例えば、約1μg/kg〜約500 mg/kg、約100μg/kg〜約5 mg/kg、または約1μg/kg〜約50μg/kg)。適当な用量は効力に依存するとさらに理解される。そのような適当な用量は、当技術分野で公知のアッセイを用いて決定してもよい。これらの化合物の一つまたは複数を動物(例えば、ヒト)に投与する場合、医師、獣医師、または研究者は、例えば比較的低用量を最初に製剤した後、適当な反応が得られるまで用量を増加させてもよい。さらに、任意の特定の動物被験体に関する特異的用量レベルは、用いる特異的化合物の活性、被験体の年齢、体重、全身健康、性別、および食事、投与期間、投与経路、排泄速度、および任意の薬物併用を含む多数の要因に依存するであろうと理解される。
【0240】
AAアミロイドーシスまたは腎障害を有する被験体に関して、用量は、例えば被験体からの化合物のクリアランス速度に影響を及ぼす可能性があるクレアチニンクリアランス速度によって測定した被験体における腎機能の状態に依存してもよい。この場合、より低いクレアチニンクリアランス速度を有する被験体は、より高いクレアチニンクリアランス速度を有する被験体より、低い用量で特定の血漿濃度に達すると予想されるであろう。
【0241】
非経口組成物は、投与の容易さおよび用量の均一性のために単位用量で製剤されてもよい。本明細書において用いられるように、単位投与剤形は、処置される被験体に関する単位用量として適した物理的に個別の単位を指す;各単位は必要な薬学的媒体に関連して所望の治療効果を生じるように計算された本発明の化合物の既定量を含む。本発明の単位投与剤形の明細は、(a)治療物質の独自の特徴および達成される特定の治療効果、および(b)AAアミロイドーシスまたはアミロイド関連疾患の処置のために本発明のそのような化合物を合成する技術分野における固有の制限、によって指図され、直接依存する。
【0242】
例えば、式(I)の化合物の治療的有効量は約100〜2500 mg毎日であってもよい。本発明の化合物は、本発明の化合物の用量200 mg、400 mg、または800 mgを有するカプセルにおいて製造されてもよい。または、本発明の化合物は、用量400 mgで1日2回、800 mgで1日2回、または1200 mgで1日2回投与されてもよい。
【0243】
当業者は、本明細書に記述の特異的技法、態様、特許請求の範囲、および実施例に対する多数の同等物を単なる日常的な実験を用いて認識し、または確認することができるであろう。そのような同等物は本発明の範囲内であると見なされ、本明細書に添付される特許請求の範囲に含まれる。本出願を通して引用した全ての参考文献、交付された特許、および公開された特許出願は、参照により本明細書に組み入れられる。本発明はさらに、さらに制限的であると解釈されてはならない以下の実施例によって説明される。
【実施例】
【0244】
本発明の具体例
実施例1
様々な程度の腎障害を有する被験体における1,3-プロパンジスルホン酸二ナトリウム塩の1回経口用量800 mgの薬物動態プロフィールを、健康ボランティアと比較して査定するために、オープンラベルの非無作為平行群試験を実施した。血液および尿試料を投与後24時間採取した。1,3-プロパンジスルホン酸の血漿および尿中濃度を、確認されたHPLC法を用いて決定する。全体として、腎障害は、健康被験体と比較して、より低い腎クリアランスおよびPDSに対するより大きい全身曝露(AUCおよびCmaxによって特徴付けられる)に関連する。その結果、用量の減少は、腎機能障害を有する患者において許容される全身曝露を維持するために必要であるように思われる。結果を表1に示す。
【0245】
(表1)
値は平均値であり、括弧内に範囲を示す(個々の患者の最小値および最大値)、ただしTmaxでは中央値を表し、括弧内に範囲を表す。
1 インスリンクリアランスによって決定。
2 消失速度定数を推定することができる被験体に限って決定
【0246】
実施例2
二次性(AA)アミロイドーシスを有する被験体における1,3-プロパンジスルホン酸二ナトリウム塩(PDS)の有効性および安全性を査定するための多施設多国間無作為二重盲験プラセボ対照平行デザイン試験を実施する。全体で被験体183人を無作為化して、PDSまたはプラセボを1日2回24ヶ月間投与する。投与は被験体における腎障害の重症度に依存する:クレアチニンクリアランス(ClCr)>80 ml/分を有する被験体には1200 mgを1日2回;ClCrが30〜80 ml/分の場合、被験体には800 mgを1日2回投与し;およびClCrが20〜30 ml/分であって、おそらく透析を開始する前の場合、被験体には400 mgを1日2回投与する。ClCrが次のより低い、またはより高い範囲のレベルまで減少または増加する場合、投与レジメをそれに従って調節する。被験体の投薬は、経口投与である(カプセル剤)。各患者を0、1、4、8、12、16、20、および24ヶ月目での現場での診察を含め、16回評価する。
【0247】
血清クレアチニンおよび体表面積に関して標準化したクレアチニンクリアランスのベースラインからの変化を、試験を通して査定する(スクリーニング、ベースライン、4、8、12、16、20および24ヶ月での診察)。被験体に、予定診察の前日に指示した通り24時間尿を採取するように要請する。尿の容積を、試験施設での症例報告書に記録して、尿の少量を尿中クレアチニンの決定のために中心となる研究所に送る。血液試料を採取して血清クレアチニンを測定する。以下の式を用いてクレアチニンクリアランス(ClCr)を計算して症例報告書に記録する。
【0248】
体表面積に関して標準化したクレアチニンクリアランスは、クレアチニンクリアランスを体表面積によって除することによって計算される。体表面積は以下の等式によって計算される:Wt0.425×Ht0.725×0.007184(DuBois D, Clinical Calorimetry, Arch Intern Med 1916; 17:87)。
【0249】
24ヶ月間の処置期間終了時に、ベースラインと比較した腎機能の複合査定に基づいて被験体を3つの範疇に分類する。被験体は、以下の悪化の臨床指標の少なくとも一つを満たす場合「悪化」であると分類される:クレアチニンクリアランスの50%低減、血清クレアチニンレベルの倍加、ESRD/透析への進行、または死亡。被験体は、以下の改善の臨床指標を満たす場合「改善」であると分類される:ClCrの>50%増加、および悪化に関する臨床指標を認めない。被験体は悪化または改善に関する臨床指標のいずれも満たさない場合「安定」であると分類される。この基準を用いて、PDSを投与された13.4%より少ない被験体がプラセボを投与された被験体と比較して悪化して、PDSを投与された13.4%より多い被験体が、プラセボと比較して安定または改善である(p値=0.06)。
【0250】
実施例3
有効性の分析
全ての統計分析は記述の被験体集団に基づくであろう。「安全な集団」は、試験薬(PDS)の少なくとも1回用量を服用した処置群に無作為化された全ての被験体の組であろう。スクリーニング期間の際に無作為化されたが、ベースラインで参加基準の一つを満たさず、したがってPDSを服用しなかった被験体は安全集団には含まれないであろう。「Intent-to-Treat(ITT)集団」は、安全集団、すなわち、無作為化されてPDSの任意の量を服用した全ての被験体からなるであろう。「有効性評価可能な(プロトコールあたり;PP)集団」は、ITT被験体のサブセットであろう。これは処置期間を終了し、一次有効性エンドポイントに関して査定されうる全ての被験体からなるであろう。以下の状況下では被験体は除外されるであろう:試験中のアンジオテンシン変換酵素(ACE)阻害剤またはアンジオテンシンII受容体拮抗剤治療の開始;救済薬の使用;ESRD/透析への進行または死亡以外の理由による中止;主なプロトコール違反;および低いコンプライアンス率。
【0251】
一次有効性エンドポイント
被験体は全て、その疾患の進行状況に従って分類される(「悪化」、「安定」、または「改善」)。これは試験の一次有効性エンドポイントを構成し、二つの予め明記された統計学的方法論に従ってIntent-to-Treat(ITT)集団において分析する:1)ベースラインの状態と比較した、試験終了時で各範疇に達する被験体の割合を比較するためのコクラン-マンテル-ヘンツェル行平均スコア(row mean score)検定(CMH検定)、および2)第一の「悪化」事象の数と共にそれらが起こった時期の双方を処置群のあいだで比較して、このように利用可能な全てのデータをより利用するための比例ハザード回帰モデル(Cox分析)。いずれの方法論も予め明記されたベースラインでのネフローゼの状態(ネフローゼ対非ネフローゼ)の階層化に関して調節される。
【0252】
CMH行平均スコア試験を用いる一次エンドポイントの場合、試験終了時にこの複合査定の各範疇に達する患者の数および百分率のような、要約統計値を処置群毎に表す。反応レベルは、必ずしも等しい間隔で観察される必要はないが、明確な順序を有し、改変riditスコア選択肢を用いる。この採点法は、その相対的順序によって暗示される以外の反応レベルのスケーリングを必要としない。CMH検定の統計値は、母集団の大きさと比較した試験施設の数が大きい(27)ために施設毎に階層化されない。したがって、施設の効果は、記述的統計法のみを用いて調べられる。国毎の効果も同様に記述的に調べられる。
【0253】
特定の細胞における母集団の大きさが任意の統計検定の必要条件にとって小さすぎる場合、複合変数の「改善」および「安定」範疇をなくして、「悪化」対「安定または改善」転帰を作製することが予め明記される。これはサブグループ分析または他の診査的分析にも当てはまる。
【0254】
Cox比例有害回帰モデルを用いる一次エンドポイントの場合、処置と複合エンドポイントとの関連を、いくつかのベースライン変数に関して調節することができるが、CMH検定方法論との比較を可能にするために、ベースラインでのネフローゼ状態(ネフローゼ対非ネフローゼ)のみを、調節のために最初に用いる。Coxモデルを用いる他の分析に含まれうるであろう他の変数は:処置群、年齢、ベースラインCrCl、ベースラインタンパク尿、ベースラインSCr、および基礎疾患(家族性地中海熱、リウマチ性炎症疾患、およびその他)である。
【0255】
一次有効性エンドポイントは、腎機能の臨床的改善/悪化の複合査定である。試験終了時、被験体を三つの範疇、すなわち「悪化」および死亡(全ての原因の)、「安定」、または「改善」に分類する。悪化に関する以下の臨床指標の少なくとも一つを満たす場合、被験体は「悪化」であると分類される:ベースラインから24ヶ月までの血清クレアチニンの倍加;ベースラインから試験終了時までの体表面積に関して標準化したクレアチニンクリアランスの50%もしくはそれより大きい減少;透析/ESRDへの進行;または死亡。悪化または改善に関する臨床指標のいずれも満たさない場合、被験体は「安定」であると分類される。改善に関する以下の臨床指標を満たす場合、被験体は「改善」であると分類される:ベースラインから試験終了時までの体表面積に関して標準化したクレアチニンクリアランスの50%もしくはそれより大きい増加;および悪化に関する臨床指標なし。「試験終了時」という用語は、試験を終了した被験体の場合には24ヶ月目、および早期に試験を中止した被験体の場合には最後に利用可能な測定として定義される。
【0256】
試験終了時、腎機能低下または全ての原因の死亡率に関する任意の「悪化」事象のリスクがプラセボのリスクの42%に低減されることが見いだされる。これは、処置群における「悪化」患者の数がより少ない結果と一致する。さらに、第一の「悪化」事象までの期間の中央値は、PDSによって処置した被験体では6.4ヶ月長い(処置群に関して14.5ヶ月対プラセボ群に関して8.1ヶ月)。第一の「悪化」事象までの時間に関するカプラン-マイヤー曲線を描写するグラフを図1に示す。
【0257】
二次有効性エンドポイント
二次有効性エンドポイントは、腎機能と胃腸管機能の双方の臨床的改善/悪化の査定である。試験終了時、被験体はその病態に基づいて三つの範疇に分類される:悪化、安定、または改善。二次有効性エンドポイントの例には、1)体表面積に関して標準化したクレアチニンクリアランスの経時的勾配;2)血清クレアチニンの逆数(1/血清クレアチニン)の経時的勾配;3)腎事象までの時間(例えば、血清クレアチニンの倍加までの時間;体表面積に対して標準化したクレアチニンクリアランスの>50%増加までの時間;体表面積に対して標準化したクレアチニンクリアランスの>50%減少までの時間;透析/ESRDまでの時間;および/または死亡までの時間);および4)ベースラインから試験終了時までのタンパク尿の変化および体表面積に関して標準化したクレアチニンクリアランスの変化、が含まれる。
【0258】
定量的二次有効性パラメータに関して、観察数(n)、平均値、SD、中央値、最小値、および最大値のような要約統計値を、各評価診察時に処置群毎に示す。実際の値およびベースラインからの百分率変化も同様に、各評価診察時に処置群毎に示す。その上、処置群を二元共分散分析(ANCOVA)を用いて比較して、ベースラインおよびベースラインでの腎状態に関して調節する。
【0259】
試験される最初の完全なモデルは以下の通りであろう:
ベースラインからの変化=処置+ベースラインの値+ベースラインでの腎状態+(ベースラインでの腎状態*処置)+(ベースラインの値*処置)
式中、
処置=PDS対プラセボ
ベースライン値=試験パラメータに関する被験体のベースライン値
ベースラインでの腎状態=ネフローゼ対非ネフローゼ
ベースラインでの腎状態*処置=処置によるベースラインでの腎状態に関する交互作用の項
ベースライン値*処置=処置毎のベースライン値に関する交互作用の項
【0260】
二つの交互作用の項は、そのそれぞれの有意性に関する試験の結果、10%より大きいp値が得られれば、一次モデルから脱落するであろう。次に、得られたモデルを用いて処置の差に関する一次試験が行われるであろう。ANCOVAモデルの基礎となる仮定を満足しない場合、階数変換アプローチ(Inman and Conover)が用いられるであろう。Iman and Conover技法の後、完全な組の観察を最小値から最大値まで階数にして、最小の観察値には階数1、第二の最小値には階数2等を与える。同値の場合、平均階数が用いられるであろう。適当な変換の後、ベースラインが共変しない技法ANCOVAモデルを、先に記述の方法と類似の方法で変換データに適用するであろう。
【0261】
定量的二次有効性パラメータに関して、分析したパラメータの各範疇における被験体の数および百分は、各評価診察時に処置群毎に示されるであろう。その上、処置群は、ベースラインでの腎状態(ネフローゼ対非ネフローゼ)に関して調節したコクラン-マンテル-ヘンツェル(CMH)行平均スコア試験を用いて一次有効性エンドポイントと同様に比較されるであろう。
【0262】
試験終了時、血清クレアチニン倍加のリスクはプラセボを処置した被験体と比較して59%低減されることが見いだされる。同様に、クレアチニンクリアランスの>50%減少のリスクは52%低減され、透析/ESRDのリスクはプラセボを処置した被験体と比較して46%低減されることが見いだされる。
【0263】
体表面積に関して標準化したクレアチニンクリアランスの経時的勾配
経時的なクレアチニンクリアランスの勾配は、長期間の腎転帰およびESRDまでの時間を予測するために、腎臓学者によって一般的に臨床的に用いられている。負の勾配は、腎機能の喪失を示している。勾配がより負であれば、腎機能の喪失はより速い。
【0264】
クレアチニンクリアランスを、各時点での24時間尿の採取を通して、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。このパラメータを体表面積に関して標準化して、被験体内の勾配の最小二乗法による推定値が、利用可能な全てのクレアチニンクリアランス測定値から計算されるであろう。試験は2年であるが、勾配は年間変化率で表記する。データが経時的に非直線的変化を示す場合、勾配の計算前に適した変換(すなわち、対数変換)が適用されるであろう。
【0265】
被験体内勾配を査定するために、各被験体に関して回帰を行い、このように各被験体に関する勾配を提供する。その後、被験体内勾配値の要約統計値が、ITTおよび有効性評価可能な集団の双方に関して各診察時に処置群毎に示されるであろう。
【0266】
試験終了時、経時的なクレアチニンクリアランスの勾配によって測定すると、PDSによる処置によって、AAアミロイドーシス被験体における腎機能の喪失速度が低減されることが見いだされる。表2および図2において示されるように、PDS処置被験体(処置群対プラセボに関して平均値の差は4.7 ml/分/1.73 m2/年)において腎機能の喪失速度は30.1%低減される。
【0267】
(表2)
【0268】
血清クレアチニンの逆数の経時的勾配
血清クレアチニンは、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。利用可能な全ての血清クレアチニン測定を用いて被験体内勾配の最小二乗法による推定値を計算する。試験は2年であるが、勾配は年間変化率で表記する。データが経時的に非直線的変化を示す場合、適した変換(すなわち、対数変換)が勾配の計算に適用されるであろう。これらの査定は、ITTおよび有効性評価可能な集団の双方に関して行われるであろう。
【0269】
被験体内勾配を査定するために、各被験体に関して回帰を行い、このように各被験体に関する勾配を提供する。その後、被験体内勾配値の要約統計値は、ITTおよび有効性評価可能な集団の双方に関して各診察時に処置群毎に示されるであろう。
【0270】
時間(月)の計算
以下の計算に関して、99%CIを有する生存カプラン-マイヤー推定値、四分位数、および生存期間の中央値のような要約統計値は、ITT集団に関して処置群毎に示される。99%CIはグリーンウッド分散公式に基づく。KM推定値は以下の時間間隔で表される:0〜112日、113〜224日、225〜336日、337〜448日、449〜560日、および560日より長い。
【0271】
二つの処置群を階層化ログランク検定を用いて比較して、階層化因子は、ベースラインでの腎状態である(ネフローゼ対非ネフローゼ)。各処置群における事象までの時間を描写するために、四つの群をカプラン-マイヤープロットにおいて比較する:ベースラインネフローゼ(処置対無処置)およびベースライン非ネフローゼ(処置対無処置)。
* 体表面積に関して標準化
【0272】
試験終了時、SCrの倍加を有する患者において、血清クレアチニンの倍加までの時間の中央値は、プラセボ処置被験体と比較して、PDS処置被験体において3.6ヶ月長いことが見いだされる。さらに、CrClの>50%減少を有する被験体では、クレアチニンクリアランスの少なくとも50%減少までの期間の中央値は、プラセボ処置被験体と比較してPDS処置被験体において4.4ヶ月長い(カプラン-マイヤー曲線に関しては図3を参照されたい)。さらに、透析まで進行した患者において、同様に透析までの期間の中央値は、プラセボ処置被験体と比較してPDS処置被験体では5.3ヶ月長いことが見いだされる(カプラン-マイヤー曲線に関しては図4を参照されたい)。
【0273】
タンパク尿およびクレアチニンクリアランスのベースラインからの変化
タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。ベースラインから試験終了時までのタンパク尿およびクレアチニンクリアランスの変化は、二次有効性エンドポイントとして分析されるであろう。試験の終了は、試験を完了した被験体に関しては24ヶ月目、早期に中止した被験体に関しては最後に利用可能な診察として定義される。スクリーニング時、タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、試験登録(ベースライン診察)前3ヶ月以内に少なくとも1週間離して2回の異なる査定から採取する。クレアチニンクリアランスパラメータに関して、標準化された値が用いられるであろう。
【0274】
タンパク尿/クレアチニンクリアランス/血清クレアチニン/血清アルブミン/血清アルカリホスファターゼ
タンパク尿、クレアチニンクリアランス、血清クレアチニン、血清アルブミン、および血清アルカリホスファターゼを、スクリーニング、ベースライン、4、8、12、16、20(血清アルブミンおよび血清アルカリホスファターゼを除く)、24ヶ月目での診察時に査定する。スクリーニング時、タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、試験登録前3ヶ月以内に少なくとも1週間あけなければならない2回の異なる査定から採取する。
【0275】
血清クレアチニン(SCr)パラメータに関して、値とその逆数、すなわち1/SCrの双方を分析する。クレアチニンクリアランスパラメータに関して、標準化された値が用いられるであろう。これらのパラメータのそれぞれの実際の値の要約統計値をITT集団に関して各診察時に処置群毎に表す。要約統計値はまた、これらのパラメータのそれぞれに関する各ベースライン後査定時のベースラインからの変化およびベースラインからの変化百分率に関して提供されるであろう。
【0276】
タンパク尿および体表面積に関して標準化したクレアチニンクリアランスに関して、二次有効性定量的パラメータに関して記述したように、ベースラインから4、8、12、16、および20ヶ月までの変化に関して処置群の比較を行う。類似のモデルは、ベースラインから4、8、12、16、20、および24ヶ月までの血清クレアチニン、血清アルブミン、および血清アルカリホスファターゼの変化に関して表されるであろう。
【0277】
試験終了時、PDS処置被験体は、プラセボ処置患者に対して-14.7±4.2 ml/分/1.73 m2対-22.3±4.1 ml/分/1.73 m2のCrClの平均値の差を示した(推定の差は7.7 ml/分/1.73 m2)。表3は、ベースラインから試験終了までの変化を示す、この試験からのデータを描写する。
【0278】
(表3)
【0279】
ネフローゼ症候群の状態:進行と寛解
ネフローゼ症候群は、本実施例において、以下の二つの腎臓外特色に関連した重度のタンパク尿症(尿中タンパク質>3 g/24時間)として定義される:1)低アルブミン血症(血清アルブミン<3.4 g/dL)および2)健康診査による末梢の浮腫および/または浮腫を処置するための利尿薬の使用。
【0280】
ベースラインでネフローゼ症候群を有しない被験体におけるネフローゼ症候群への進行は、以下のように定義される:タンパク尿症の>3 g/24時間への増加(ベースラインで<3 g/24時間である必要がある)および以下の二つの腎臓外特色の発生:1)低アルブミン血症(ベースラインでは存在しない)および2)浮腫および/または浮腫を処置するための利尿薬の使用(ベースラインでは浮腫を認めず利尿薬の使用を認めない)。被験体が試験終了時に三つ全ての基準を満たしていない場合、彼らは試験分析の目的に関してネフローゼであると見なされないであろう。被験体がESRD/透析に進行して、血清アルブミンまたは浮腫のいずれかに関する情報を紛失している場合、彼れはネフローゼ症候群に進行しているとは見なされないが、それにもかかわらずその悪化に関する情報は一次エンドポイント分析において獲得されるであろう。
【0281】
ベースラインでネフローゼ症候群を有する被験体におけるネフローゼ症候群の寛解は、以下のように定義される:タンパク尿症の<1 g/24時間(ベースラインで>3 g/24時間である必要がある)への減少および以下の二つの腎臓外特色の一つの改善:1)ベースラインまたは試験中の他の任意の時点で血清アルブミン>3.4 g/dL(ベースラインまたは試験中の他の任意の時点で血清アルブミン<3.4 g/dL)、または2)浮腫の消散および/または浮腫の改善に反応した利尿薬の中止(浮腫が主な症状である場合、または患者がベースラインもしくは試験中の他の任意の時点で利尿薬を用いている場合)。
【0282】
起立性低血圧症
自律神経系(ANS)は、AAアミロイドーシスにおいて影響を受ける可能性がある。血圧の姿勢による減少は、仰臥位から起立位への収縮期圧(SBP)の>20 mmHgの下降、または拡張期圧(DBP)での10 mmHgの低下として定義される。収縮期または拡張期圧の低下が少なくとも3分間持続する場合、これはANS機能障害の兆候である。血圧は、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月での診察時に仰臥位、起立位、および起立して3分後の状態で測定する。
【0283】
各範疇における被験体の数および百分率のような記述統計値(はい対いいえ)は、ITT集団において処置群毎に毎回の試験診察時に示される。処置群は、ベースラインでの腎状態に関して調節したCMH一般関連試験を用いて比較される。
【0284】
脾腫/肝腫
脾腫は、脾臓の肥大として定義される。脾臓の大きさは、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月目での診察時に測定されるであろう。これは定規を用いて前腋窩腺で左肋骨端からセンチメートルで測定される。測定は、被験体が横になり、吸気終了時に行う。
【0285】
肝腫は、肝臓の肥大として定義される。肝臓の大きさは、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月目での健康診査時に測定されるであろう。これは定規を用いて鎖骨中線で右肋骨端からセンチメートルで測定する。測定は、被験体が横になり、吸気終了時に行う。
【0286】
薬物動態分析
以下の実施例において、用語を以下のように定義する。薬物動態パラメータは、WinNonlin(登録商標)(Pharsight Corp., Mountain View, CA, USA)を用いて非コンパートメント分析によって誘導した。
Cmax− 観察された最高血漿濃度
Tmax− Cmaxが起こった時間。
AUC0-t− 線形台形則によって計算した、ゼロ時間から、濃度が定量限界またはそれを超えている最後の試料採取時間までの血漿濃度対時間曲線下面積。
AUC∞− AUC0-t+(Clast/λz)から計算した、ゼロ時間から無限大までの血漿濃度対時間曲線下面積を指し、Clastは最後に観察された定量可能な濃度であり、λzは見かけの終末速度定数である。
t1/2− ln 2/λzから計算された見かけの終末半減期である。
【0287】
実施例4
健康な男性成人ボランティアにおける1回経口投与後のPDSの安全性、認容性、および薬物動態を決定する。PDSの薬物動態に及ぼす食物の効果も同様に決定する。試験の第一の部分は、健康な男性被験体におけるPDSの6つの増加経口用量の薬物動態プロフィールを査定するための無作為二重盲験プラセボ対照試験である。この試験の第二の部分は、絶食時および食事条件でのPDSの1回経口投与の薬物動態プロフィールに及ぼす食物の効果を調べるためのオープンラベルの二元交叉試験である。血液試料を投与後36時間の間に採取する。PDSの血漿濃度を確認されたHPLC法を用いて決定する。PDSの1回経口投与後、最高血漿濃度は、一般的に投与後0.5〜1時間以内に達し、平均半減期(T1/2)は2〜26時間の範囲である。PDSに対する全身曝露(AUCおよびCmax)の程度は、用量が増加すれば増加する。この増加は、投与された用量と妥当に比例する。その上PDSを食事条件で投与した場合の全身利用率の速度および程度の双方に減少が認められる。したがって、PDSは、高脂肪食と同時に与えてはならない。結果を表4に示す。
【0288】
(表4)健康な男性被験体への100、200、400、800、1600、および2400 mgの1回経口投与および健康な男性被験体(絶食および食後状態)への1600 mgの1回経口投与を示すPDSの薬物動態パラメータ
値は、中央値であるTmaxを除き、平均値であり、括弧内に範囲(すなわち、個体に関する最小値と最大値)示す。
NC:計算していない。
【0289】
実施例5
健康な男性成人ボランティアに複数の増加経口用量を投与後PDSの安全性、認容性、および薬物動態を決定する。これは、健康な男性被験体において400、800、1600 mgのPDSを1回および反復経口投与した場合の薬物動態プロフィールを査定するための無作為二重盲験プラセボ対照試験である。PDSの用量400、800、および1600 mgを1および7日目に健康男性被験体に1回経口投与する。1日目に1回経口投与後、PDSを2、3、4、5および6日目に400、800、および1600 mgを1日4回投与する、または1600 mgを1日3回投与する。血液試料を1日目の投与後24時間を通して採取し、2日目から6日目では最初と最後の投与前、および7日目の最後の投与の48時間後に採取した。PDSの血漿濃度は、確認されたHPLC法を用いて決定する。PDSの1回および繰り返し経口投与後、一般的に投与後0.25〜1時間に、最高血漿濃度に達する。1回または複数回投与後の平均半減期(T1/2)は5〜20時間の範囲である。複数回投与後の蓄積は、T1/2および投与回数と一貫する。PDSに対する全身曝露の程度(AUC0-tおよびCmax)は、用量が増加すると増加して、全身曝露のこの増加は、投与された用量とほぼ比例する。結果を表5に示す。
【0290】
(表5)健康男性被験体に400、800、および1600 mgを複数回経口投与後のPDSの薬物動態パラメータ
値は、中央値であるTmaxを除き、平均値であり、括弧内に範囲(すなわち、個体に関する最小値と最大値)示す。
NC:計算していない。
1日目は1回経口投与後のパラメータに対応する。
7日目は反復経口投与後のパラメータに対応する。
【0291】
実施例6
AAアミロイドーシスを有する被験体における1回および複数回経口投与後のPDSの安全性、有効性、および薬物動態を決定する。これは、PDSの1回および反復投与の薬物動態プロフィールを査定するための、多施設多国間無作為二重盲験プラセボ対照平行デザイン試験である。投与は、被験体における腎障害の重症度に依存する:クレアチニンクリアランス(ClCr)>80 ml/分を有する被験体には、1200 mgを1日2回;ClCrが30〜80 ml/分の被験体には、800 mgを1日2回;およびClCrが20〜30 ml/分の被験体には400 mgを1日2回投与する。ClCrが次のより低い範囲レベルまで減少する場合、投与レジメをそれに従って調節する。PDSは、0ヶ月目および24回の診察時にAAアミロイドーシス患者のサブセットに関して1回経口用量として投与される。0ヶ月目での1回投与後、PDSを1日2回、24ヶ月間投与する。血液試料を0ヶ月および24ヶ月目の投与後24時間のあいだに採取して、4、8、12ヶ月での診察時では投与後8時間のあいだに採取する。PDSの血漿濃度は確認されたHPLC法を用いて決定する。PDSの複数回経口投与後、被験体における蓄積の範囲は1.75〜3.44である。蓄積因子に基づいて、計算された平均T1/2は15時間であり、範囲は10〜24時間である。結果を表6に示す。
【0292】
(表6)AAアミロイドーシス患者に1回(0ヶ月)および複数回(24ヶ月)経口投与後のPDSの薬物動態パラメータ
値は平均値であり、範囲(すなわち個体に関する最小値および最大値)を括弧内に示す。
0ヶ月目は、1回経口投与後のパラメータに対応する。
24ヶ月目は、反復経口投与後のパラメータに対応する。
n=各投与群の患者数
NA=適用可能でない
【0293】
実施例7
1,3プロパンジスルホン酸二ナトリウム塩の400 mgカプセルの製剤の例を以下に記述する。
【0294】
1,3-プロパンジスルホン酸二ナトリウム塩の400 mgカプセルは、0番の白色の不透明な硬ゼラチンカプセルに1,3-プロパンジスルホン酸二ナトリウム塩400 mgおよび賦形剤40 mgを含む白色粉末を充填することによって製造される。
* MHS−製造元の社内標準物質
【0295】
実施例8:
1,2-エタンジスルホン酸を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0296】
実施例9:
1,2-エタンジスルホン酸ナトリウムを活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0297】
実施例10:
1,2-エタンジオールビス(硫酸水素塩)を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0298】
実施例11:
1,2-エタンジオールジスルファート二ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0299】
実施例12:
1,3-プロパンジオールビス(硫酸水素塩)を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0300】
実施例13:
1,3-プロパンジオールジスルファート二ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0301】
実施例14:
2-スルホメチル-1,4-ブタンジスルホン酸を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0302】
実施例15:
2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0303】
同等物
当業者は、本明細書に記述の特異的技法に対する多数の同等物を単なる日常的な実験を用いて認識し、または確認することができるであろう。そのような同等物は、本発明の範囲内であると見なされ、以下の特許請求の範囲に含まれる。本出願を通して引用した全ての参考文献、特許、および特許出願の内容物は、参照により本明細書に組み入れられる。それらの特許、出願、および他の文書の適当な成分、プロセス、および方法は、本発明およびその態様のために選択されてもよい。
【技術分野】
【0001】
関連出願
本出願は、その全内容物が参照により本明細書に組み入れられる、2005年4月15日に提出された米国特許仮出願第60/671,866号に対する優先権を主張する。
【背景技術】
【0002】
発明の背景
アミロイドーシスは、特異的臓器における不溶性の原線維タンパク質(アミロイド)の細胞外沈着に関連し、最終的に罹患臓器の不全に至る多数の疾患に関する一般用語である。R.H. Falk et al., The Systemic Amyloidosis, 337 N ENGL J MED 898-909 (1997)(非特許文献1), P.N. Hawkins, Amyloidosis, 9 BLOOD REV 135-42 (1995)(非特許文献2), J.D. Sipe, Amyloidosis, 31 CR REV CLIN LAB SCI 325-54 (1994)(非特許文献3); A.S. Cohen, Amyloidosis, 40(2) BULL RHEUM DISEASES 1-12 (1991)(非特許文献4)。アミロイド沈着は、一つの臓器に限定されたままとなりうる(限局性アミロイドーシス)、またはより広く分布する可能性がある(全身性アミロイドーシス)。全身性アミロイドーシスは一般的に、原線維沈着の性質に基づいて四つのタイプに分類される:(i)特発性または原発性アミロイドーシス(ALアミロイドーシス);(ii)反応性、二次性、またはアミロイドA(AA)アミロイドーシス;(iii)家族性アミロイド性多発神経障害;および(iv)透析関連アミロイドーシス。その発生は多様であるが、全てのアミロイド沈着は共通の形態学的特性を有し、特異的色素(例えば、コンゴーレッド)によって染色され、染色後に偏光において特徴的な複屈折外観を有する。それらはまた、共通の超音波特色を有し、共通のX-線回折および赤外線スペクトルを有する。
【0003】
AAアミロイドーシスは、炎症に反応して肝細胞から産生および分泌される急性期タンパク質である、前駆体の血清アミロイドA(SAA)から形成されたアミロイドA(AA)タンパク質に関連すると考えられている。AAアミロイドーシスは、慢性炎症状態(例えば、リウマチ性関節炎、強直性脊椎炎、炎症性腸疾患等)、慢性感染症(例えば、結核、骨髄炎等)、および遺伝性の発熱、例えば家族性地中海熱に関連する(R.H. Falk et al., 337 N ENGL J MED 898-909 (1997)(非特許文献5), A.S. Cohen, 40(2) BULL RHEUM DISEASES 1-12 (1991)(非特許文献6), G. Grateau, 12 CURRENT OPINION IN RHEUMATOL 61-64 (2000)(非特許文献7))。リウマチ性関節炎は、西欧および北米におけるAAアミロイドーシスの主な原因である(M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)(非特許文献8), M.A. Gertz, Secondary amyloidosis, 232 J INT MED 517-18 (1992)(非特許文献9))。
【0004】
AAアミロイドーシスは、腎臓、脾臓、肝臓、および副腎のような実質様臓器に主に罹患する。AAアミロイドーシスの最も一般的な臨床特色は、ネフローゼ範囲のタンパク尿症として発現する腎機能障害、または診断時の腎機能不全である。末期腎不全は、症例の40〜60%における死因である(M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)(非特許文献8), M.A. Gertz, 232 J INT MED 517-18 (1992)(非特許文献10), M.A. Gertz and R.A. Kyle, 70 MEDICINE 246-256 (1991)(非特許文献11))。胃腸管の罹患もまた頻繁であり、通常慢性の下痢、体重減少、および吸収不良として現れる。肝臓および脾臓の肥大も同様に何人かの被験体において起こる可能性がある。心臓の罹患はまれであり、疾患の後期に起こる。診断からの生存期間の中央値は、診断時の疾患の進行期に応じて2〜8年である(M.A. Gertz and R. A. Kyle, 70 MEDICINE 246-256 (1991)(非特許文献11))。
【0005】
AAアミロイドーシスは通常、慢性感染症(結核のような)、または慢性炎症(リウマチ性関節炎または遺伝性発熱)に関連して認められる。家族性型のAAアミロイドーシスは、家族性地中海熱(FMF)において認められる。この家族型アミロイドーシスは遺伝され、特異的集団群において認められる。ALおよびAAアミロイドーシスの双方において、沈着はいくつかの臓器において見いだされ、このように全身性アミロイド疾患であると考えられる。
【0006】
「局所アミロイドーシス」は、一つの臓器系に罹患する傾向があるアミロイドーシスである。異なるアミロイドーシスはまた、沈着物に存在するタンパク質のタイプを特徴とする。例えば、スクレイピー、ウシ海綿状脳症、クロイツフェルト-ヤコブ病等のような神経変性疾患は、中枢神経系におけるプロテアーゼ抵抗性型のプリオンタンパク質(AScrまたはPrP-27と呼ばれる)の出現および蓄積を特徴とする。同様に、もう一つの神経変性障害であるアルツハイマー病は、神経炎プラークおよび神経原線維のもつれを特徴とする。この場合、実質および血管において認められるアミロイド斑は、原線維Aβアミロイドタンパク質の沈着によって形成される。成人発症型糖尿病(II型糖尿病)のような他の疾患は、膵臓におけるアミロイド原線維の局所蓄積を特徴とする。
【0007】
これらのアミロイドがひとたび形成されると、その場でアミロイド沈着を有意に溶解する、さらなるアミロイドの沈着を予防する、またはアミロイド沈着の開始を予防する公知の広く容認された治療または処置はない。
【0008】
それぞれのアミロイド原性タンパク質は、コンフォメーションの変化を受ける能力、β-シートを構築する能力、および細胞内または細胞外で沈着される可能性がある不溶性の原線維を形成する能力を有する。それぞれのアミロイド原性タンパク質は、アミノ酸配列が異なるが、原線維を形成して、プロテオグリカン、アミロイドP、および補体成分のような他の要素に結合するという同じ特性を有する。その上、それぞれのアミロイド原性タンパク質は、配列は異なるが、β-シート形成を促進する他の領域と共に、プロテオグリカンのグリコサミノグリカン(GAG)部分に対する結合能を有する領域(GAG結合部位と呼ばれる)のような類似性を示すアミノ酸配列を有する。プロテオグリカンは、体内のほぼ至る所に分布する様々な大きさおよび構造の高分子である。それらは、細胞内区画において、細胞表面上で、および細胞外マトリクスの一部として見いだされうる。全てのプロテオグリカンの基本構造は、コアタンパク質と、コアタンパク質に付着する少なくとも一つ、しかし頻繁に複数の多糖類鎖(GAGs)を含む。コンドロイチン硫酸、デルマタン硫酸、ケラタン硫酸、ヘパリン、およびヒアルロナンを含む異なる多くのGAGsが発見されている。
【0009】
いくつかのGAG模倣体は、アミロイド沈着を阻害するために、および/またはいくつかの型のアミロイドーシスを処置するために有用であることが知られている。WO 94/22437(特許文献1)、WO 96/28187(特許文献2)、およびWO 00/64420(特許文献3)を参照されたい。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO 94/22437
【特許文献2】WO 96/28187
【特許文献3】WO 00/64420
【非特許文献】
【0011】
【非特許文献1】R.H. Falk et al., The Systemic Amyloidosis, 337 N ENGL J MED 898-909 (1997)
【非特許文献2】P.N. Hawkins, Amyloidosis, 9 BLOOD REV 135-42 (1995)
【非特許文献3】J.D. Sipe, Amyloidosis, 31 CR REV CLIN LAB SCI 325-54 (1994)
【非特許文献4】A.S. Cohen, Amyloidosis, 40(2) BULL RHEUM DISEASES 1-12 (1991)
【非特許文献5】R.H. Falk et al., 337 N ENGL J MED 898-909 (1997)
【非特許文献6】A.S. Cohen, 40(2) BULL RHEUM DISEASES 1-12 (1991)
【非特許文献7】G. Grateau, 12 CURRENT OPINION IN RHEUMATOL 61-64 (2000)
【非特許文献8】M. Skinner Amyloidosis, CURRENT THERAPY IN ALLERGY, IMMUNOLOGY, AND RHEUMATOLOGY 235-40 (Mosby-Year Book Inc., 1996)
【非特許文献9】M.A. Gertz, Secondary amyloidosis, 232 J INT MED 517-18 (1992)
【非特許文献10】M.A. Gertz, 232 J INT MED 517-18 (1992)
【非特許文献11】M.A. Gertz and R.A. Kyle, 70 MEDICINE 246-256 (1991)
【発明の概要】
【0012】
発明の概要
一つの態様において、本発明は、腎障害(PRI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、以下の式の化合物の治療的有効量を標的被験体に投与することによって、標的被験体におけるAAアミロイドーシスを処置するかまたは予防する方法に関する:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2である。さらに、この態様において、標的被験体はAAアミロイドーシスに関して処置され、腎障害に関連したパラメータを有する、またはそれに対して感受性がある。さらなる態様において、式(I)の化合物は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩である。
【0013】
もう一つの態様において、本発明には、胃腸管障害(PGI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を標的被験体に投与することによって、標的被験体におけるAAアミロイドーシスを処置または予防する方法が含まれる。さらに、この態様において、標的被験体はAAアミロイドーシスに関して処置され、胃腸管障害に関連するパラメータを有する、またはそれに対して感受性がある。
【0014】
もう一つのさらなる態様において、本発明には、アミロイド関連疾患が処置または予防されるように、クレアチニンクリアランス速度に基づいて選択された用量の式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与することによって、被験体におけるアミロイド関連疾患を処置または予防する方法に関する。
【0015】
本発明はまた、少なくとも部分的に、例えばAUC、Cmax、AUCss、Css、Tmax等によって測定した場合に、被験体において有効な曝露が提供されるような用量で投与される、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体におけるAAアミロイドーシスを処置または予防するための方法に関する。
【0016】
さらに、本発明はまた、被験体における腎および/または胃腸管機能を安定化するかまたは改善する方法にも関する。方法には、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0017】
もう一つの態様において、本発明は、被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、AAアミロイドーシスが処置または予防されるように、第二の物質と併用して、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与する段階が含まれる。
【0018】
なおもう一つの態様において、本発明は、少なくとも部分的に、被験体における化合物の経口生物学的利用率が増加するように、食物なしで薬学的組成物において式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与することによって、被験体における化合物の経口生物学的利用率を増加させる方法に関する。
【0019】
本発明はまた、少なくとも部分的に、炎症疾患が被験体において処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体における炎症疾患を処置する方法にも関する。
【0020】
本発明はまた、少なくとも部分的に、被験体における遺伝性の発熱が処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与することによって、被験体における遺伝性発熱を処置する方法にも関する。
【0021】
本発明はまた、少なくとも部分的に、被験体におけるリウマチ性関節炎を処置するための方法にも関する。方法には、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0022】
さらに、本発明にはまた、被験体における悪性新生物を処置する方法が含まれる。方法には、悪性新生物が被験体において処置されるように、第二の物質と併用して、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量をそれを必要とする被験体に投与する段階が含まれる。
【0023】
さらなる態様において、本発明は、少なくとも部分的に、被験体における慢性感染症、例えば微生物またはウイルス感染症を処置する方法に関する。方法には、慢性感染症が被験体において処置されるように、第二の物質と併用して式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0024】
もう一つのさらなる態様において、本発明は、少なくとも部分的に、炎症性疾患、悪性新生物、慢性感染症、または遺伝性の発熱を有する被験体における腎機能を安定化するかもしくは改善させる、または腎疾患の進行を遅らせる方法に関する。方法には、腎機能を安定化するかもしくは改善するように、または腎疾患の進行が遅れるように、1,3-プロパンジスルホン酸またはその薬学的に許容される塩の治療的有効量を被験体に投与する段階が含まれる。
【0025】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるESRD/透析への進行を予防するかまたは遅らせるための方法に関する。方法には、ESRD/透析への進行が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を、被験体、例えばAAアミロイドーシスを有する被験体に投与する段階が含まれる。
【0026】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における血清クレアチニン倍加までの時間を予防するかまたは遅らせるための方法に関する。方法には、血清クレアチニン倍加までの時間が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0027】
なおもう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%減少までの時間を予防するかまたは遅らせるための方法に関する。方法には、クレアチニンクリアランスの少なくとも50%減少までの時間が遅れるまたは予防されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0028】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%増加までの時間を減少させるための方法に関する。方法には、クレアチニンクリアランスの少なくとも50%増加までの時間が減少するように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0029】
さらにもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの勾配によって測定した腎疾患の進行速度を低減させるための方法が含まれる。方法には、腎疾患の進行速度が低減されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0030】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるタンパク尿症を安定化するかまたは低減させるための方法に関する。方法には、被験体におけるタンパク尿症が安定化されるかまたは低減されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0031】
なおもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体における腎機能を安定化するか、または腎疾患の進行を遅らせる方法が含まれる。方法には、腎機能を安定化するか、または腎疾患の進行が遅れるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。一つの局面において、腎疾患の進行は、クレアチニンクリアランス(CrCl)の50%減少、血清クレアチニンの倍加(SCr)、および/またはESRDへの進行によって測定してもよい。
【0032】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における腎機能障害を処置するための方法に関する。方法には、腎機能障害が処置されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0033】
本発明はまた、少なくとも部分的に、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量と、第二の物質とを含む薬学的組成物にも関する。
【0034】
さらなる態様において、本発明は、包装された薬学的組成物に関する。包装された薬学的組成物には、組成物を第二の物質と併用して投与することを勧める表示または添付文書と共に包装された、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量が含まれる。
【0035】
なおもう一つのさらなる態様において、本発明は、組成物を式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩と併用して投与するように勧める表示または添付文書と共に包装された第二の物質の治療的有効量を含む、包装された薬学的組成物に関する。
【0036】
なおもう一つの態様において、本発明は、組成物を食事なしで投与することを勧める表示または添付文書と共に、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を含む薬学的組成物を保持する容器を含む、包装された薬学的組成物に関する。
【0037】
なおもう一つの態様において、本発明はAAアミロイドーシスを処置するための薬学的製剤に関する。製剤は、被験体に投与した場合に少なくとも一つの都合のよい生物学的特性(FBP)を有する、製剤において式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を含む。
【0038】
本発明はまた、少なくとも部分的に、抗アミロイド原性物質含有製剤が、それが生物学的に都合のよい製剤となるように、被験体に投与した場合に、少なくとも一つの都合のよい生物学的特性を有するように予め決定された標準的な製剤と同等である、製剤における抗アミロイド原性物質に関する。
【0039】
もう一つの態様において、本発明にはまた、式(I)の化合物と一つまたは複数の薬学的に許容される担体とを含む、薬学的製剤が含まれる。この態様において、薬学的組成物は、それを必要とする被験体に投与した場合に、Cmax約200〜約2000 ng/mlを提供する。
【0040】
なおもう一つの態様において、本発明はまた、式(I)の化合物と、一つまたは複数の薬学的に許容される担体とを含む、薬学的製剤にも関する。この態様において、薬学的製剤は、それを必要とする被験体に投与した場合に、AUC∞約2,000〜約44,000 ng/mlを提供する。
【0041】
本発明はまた、少なくとも部分的に、それを必要とする被験体に化合物を投与する方法にも関する。方法には、Cmax約200〜約3,400 ng/mlを達成するために十分な量で被験体に式(I)の化合物を投与する段階が含まれる。Cmaxは投与後約0.25〜約9.00時間で起こる可能性がある。
【0042】
もう一つの態様において、本発明はまた、少なくとも部分的に、それを必要とする被験体に式(I)の化合物を投与する方法にも関する。方法には、AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の式(I)の化合物を被験体に投与する段階が含まれる。
【0043】
なおもう一つの態様において、本発明は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む薬学的製剤に関する。薬学的製剤は、それを必要とする被験体に1回投与した場合に、Cmax約200〜約2000 ng/mlを提供する。
【0044】
なおもう一つの態様において、本発明にはまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む薬学的製剤に関する。薬学的製剤は、それを必要とする被験体に投与した場合に、AUC∞約2,000〜約44,000 ng/mlを提供する。
【0045】
なおもう一つの態様において、本発明はまた、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法に関する。方法には、投与後約0.25〜約9.00時間でCmax約200〜約3,400 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階が含まれる。
【0046】
もう一つの態様において、本発明はまた、AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与することによって、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法にも関する。
【0047】
なおもう一つの態様において、本発明は、少なくとも部分的に、薬学的製剤に関する。薬学的製剤は、AAアミロイドーシスを処置するかまたは予防するために有効な量の活性物質(例えば、1,3-プロパンジスルホン酸、二ナトリウム塩(PDSとも呼ばれる))と、薬学的に許容される担体とを含み、製剤を健康な被験体に経口投与した場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0048】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを処置するかまたは予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、健康被験体に経口投与した場合、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、薬学的製剤にも関する。
【0049】
なおもう一つのさらなる態様において、本発明はまた、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、健康被験体に経口投与した場合、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、薬学的製剤に関する。
【0050】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体には活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0051】
なおもう一つのさらなる態様において、本発明は、活性物質(例えば、PDS)800 mgと薬学的に許容される担体とを含む薬学的製剤であって、被験体に経口投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0052】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に24ヶ月間経口投与した場合:活性物質の用量が400 mgの場合、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0053】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回経口投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回経口投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回経口投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0054】
なおもう一つの態様において、本発明はまた、AAアミロイドーシスを有する被験体における腎機能を安定化するかもしくは改善させる、または腎疾患の進行を遅らせる方法に関する。方法には、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体を含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階が含まれる。例えば、製剤を用量400 mgで投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を800 mgの用量で投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を1200 mgの用量で投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0055】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体である活性化物質とを含む薬学的製剤に関する。さらに、この製剤を、AAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体に活性物質の用量400 mgを経口投与すると、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを経口投与すると、平均AUC∞約3,000〜20,000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを経口投与すると、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0056】
なおもう一つの態様において、本発明は、少なくとも部分的に、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階を含む、AAアミロイドーシスを有する被験体における腎機能を安定化するかもしくは改善する、または腎疾患の進行を遅らせる方法に関する。さらに、製剤を用量400 mgで投与した場合、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を用量800 mgで投与した場合、平均AUC∞約3,000〜20,000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を用量1200 mgで投与した場合、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【図面の簡単な説明】
【0057】
【図1】PDS対プラセボを投与した被験体に関する「悪化」事象に関するカプラン-マイヤー曲線を描写するグラフである。
【図2】PDS対プラセボを投与した被験体に関するクレアチニンクリアランスの勾配を示す折れ線グラフである。
【図3】PDS対プラセボを投与した被験体に関するクレアチニンクリアランスの50%減少までの時間に関するカプラン-マイヤー曲線を描写するグラフである。
【図4】PDS対プラセボを投与した被験体に関するESRD/透析までの時間に関するカプラン-マイヤー曲線を描写するグラフである。
【発明を実施するための形態】
【0058】
発明の詳細な説明
A.本発明の化合物を用いて標的被験体を処置する方法
一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスのために処置され、および腎障害に関連したパラメータを有する、またはそれに対して感受性がある標的被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、腎障害(PRI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、AAアミロイドーシスが処置または予防されるように、以下の式の化合物の治療的有効量を標的被験体に投与する段階が含まれる:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2である。
【0059】
もう一つの態様において、本発明には、AAアミロイドーシスのために処置され、および胃腸管障害に関連した二次的障害または病態を有する、またはそれに対して感受性を有する標的被験体におけるAAアミロイドーシスを処置または予防する方法が含まれる。方法には、胃腸管障害(PGI)に関連するパラメータに関する許容認容性指数(ATI)を維持しながら、式(I)の化合物の治療的有効量を標的被験体に投与する段階が含まれる。
【0060】
一般的に、AAアミロイドーシスは、持続的な急性期反応を誘発する多数の疾患の徴候である。そのような疾患には、慢性炎症性疾患、慢性局所または全身性微生物感染症、および悪性新生物が含まれる。反応性または二次性(AA)アミロイドーシスの最も一般的な型は、長期間の炎症病態の結果として認められる。例えば、リウマチ性関節炎または家族性地中海熱(遺伝病である)を有する被験体はAAアミロイドーシスを発症しうる。「AAアミロイドーシス」、「二次性アミロイドーシス」、および「二次性(AA)アミロイドーシス」という用語は互換的に用いられる。
【0061】
AA原線維は一般的に、IL-1、IL-6、およびTNFのようなサイトカインに反応して肝細胞において主に合成される循環中のアポリポタンパク質である、血清アミロイドAタンパク質(ApoSAA)のタンパク質分解切断によって形成された8,000ダルトンの断片(AAペプチドまたは断片)で構成される。一度分泌されると、ApoSAAは、HDLと複合体を形成する。AA原線維の沈着は、実質臓器に選択的に、体内に広がりうる。腎臓は通常、沈着部位であり、肝臓および脾臓も罹患する可能性がある。沈着はまた、心臓、胃腸管、および皮膚においても認められる。
【0062】
AAアミロイドーシスの発症に至りうる基礎疾患には、慢性炎症疾患、リウマチ性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、若年性慢性関節炎、若年性リウマチ性関節炎、潰瘍性大腸炎、慢性発熱、気管支狭窄、マラリア、血管炎、静脈内薬物乱用、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、およびクローン病のような炎症疾患が含まれるがこれらに限定されるわけではない。AA沈着はまた、慢性感染症、例えばAIDS、HIV、B型肝炎、C型肝炎、慢性微生物感染症、例えば、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、肺結核、肺感染症、再発性膿瘍、ベーチェット病、およびホイップル病の結果として産生される。特定の悪性新生物によってもまた、AA原線維アミロイド沈着が起こりうる。これらには、ホジキンリンパ腫、腎癌、腸管、肺、および尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、およびヘアリーセル白血病のような病態が含まれる。
【0063】
「被験体」という用語には、AAアミロイドーシスもしくはアミロイド関連疾患が起こりうるか、またはAAアミロイドーシスもしくはアミロイド関連疾患に対して感受性がある生きている生物が含まれる。「被験体」という用語には、動物(例えば、哺乳動物、例えばネコ、イヌ、ウマ、ブタ、ウシ、ヤギ、ヒツジ、齧歯類、例えばマウスまたはラット、ウサギ、リス、クマ、霊長類(例えば、チンパンジー、サル、ゴリラ、およびヒト))と共にニワトリ、アヒル、北京ダック、ガチョウ、およびそのトランスジェニック種が含まれる。
【0064】
「標的被験体」という用語は、式(I)の組成物または化合物を投与されるように特に選択された被験体、例えばヒトを指す。したがって、いくつかの態様において、標的被験体には、AAアミロイド関連疾患、例えばAAアミロイドーシスのリスクを有する、またはそれらであると診断された被験体が含まれる。AAアミロイドーシスを発症するリスクを有する被験体には、炎症疾患、感染症、遺伝性発熱、または新生物のような基礎疾患を有する被験体が含まれる。他の態様において、標的被験体には、腎障害および/または胃腸管障害に関連するパラメータを有するか、またはパラメータに対して感受性を有する被験体が含まれる。標的被験体にはまた、AAアミロイド関連疾患であると診断されると共に、腎障害および/または胃腸管障害に関連するパラメータを有することが公知である被験体が含まれてもよい。好ましい標的被験体はヒトである。
【0065】
「許容認容性指数」および「ATI」という用語は、被験体を苦しめる疾患または障害における所定の時点で十分だと見なされる、被験体における病気のレベルを指すために互換的に用いられる。いくつかの態様において、ATIは本明細書において記述されるように被験体における病気の改善または安定化である。他の態様において、ATIは先の時点と比較して被験体における病気の悪化がより低いことであり、例えば被験体が血清クレアチニンレベルの急激な増加を経験している場合、ATIは血清クレアチニンレベルのより遅い増加であるかもしれない。したがって、一つの態様において、ATIは、被験体における腎障害または胃腸管障害に関連する少なくとも一つのパラメータの悪化がより低いことである。もう一つの態様において、ATIは、被験体における腎障害および/または胃腸管障害に関連した少なくとも二つのパラメータの悪化がより低いことである。なおもう一つの態様において、ATIは、被験体における腎障害および/または胃腸管障害に関連した少なくとも三つ、四つ、または五つのパラメータの悪化がより低いことである。
【0066】
「腎障害に関連するパラメータ」および「PRI」という用語は、クレアチニンクリアランスの減少、血清クレアチニンレベルの増加、タンパク尿症、透析/末期腎疾患(ESRD)への進行、低アルブミン血症、および/または浮腫のような、しかしこれらに限定されない異常な腎機能に一般的に関連するパラメータを含めるために互換的に用いられる。いくつかの態様において、腎障害に関連するパラメータは少なくとも部分的に、AAアミロイドーシスまたは体内のアミロイドAタンパク質の存在によって引き起こされる。
【0067】
「胃腸管障害に関連するパラメータ」および「PGI」という用語には、慢性的な下痢および/または体重減少のような、しかしこれらに限定されない異常な胃腸管機能に一般的に関連するパラメータが含まれる。いくつかの態様において、胃腸管障害に関連するパラメータは、少なくとも部分的に、AAアミロイドーシスまたは体内のアミロイドAタンパク質の存在によって引き起こされる。
【0068】
被験体の「処置」または「処置する」という用語には、疾患もしくは病態、疾患もしくは病態の症状、または疾患もしくは病態のリスク(または感受性)を安定化、治癒(curing)、治癒(healing)、緩和、軽減、変化、救済、より低い悪化、回復、改善、または影響を及ぼすことを目的として、被験体に本発明の化合物を適用または投与すること(または被験体からの細胞または組織に本発明の化合物を適用または投与すること)が含まれる。「処置する」という用語は、軽減;寛解;悪化速度の減弱;安定化、症状または損傷発生の減少、被験体により認容可能な病理または病態、変性速度の遅延もしくは低下;最終的な変性点をより消耗性でなくなるようにする;または被験体の身体的もしくは精神的安寧を改善することのような、任意の客観的または主観的パラメータを含む、損傷、病理、または病態の処置または回復における任意の成功の指標を指す。一つの態様において、「処置する」という用語には、被験体の寿命を増加させる段階が含まれうる。
【0069】
「慢性的な下痢の寛解」という用語は、慢性的な下痢の事例がないこと、および少なくとも連続して4ヶ月間下痢止め薬を慢性的に用いていないことを指す。
【0070】
一つの態様において、透析への進行は、被験体、例えばAAアミロイドーシスを有する被験体において遅れるかまたは予防される。例えば、被験体の透析への進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、11ヶ月もしくはそれより長く、1年もしくはそれより長く、1.5年もしくはそれより長く、2年もしくはそれより長く、3年もしくはそれより長く、4年もしくはそれより長く、5年もしくはそれより長く、7.5年もしくはそれより長く、10年もしくはそれより長く、15年もしくはそれより長く、または20年もしくはそれより長く遅れる可能性がある。特定の態様において、進行は約6ヶ月遅れる。
【0071】
もう一つの態様において、「処置する」という用語には、腎臓の低下の任意の「悪化」事象(実施例3を参照されたい)または全ての原因の死亡率のリスクを少なくとも5%もしくはそれより大きく、少なくとも10%もしくはそれより大きく、少なくとも15%もしくはそれより大きく、少なくとも20%もしくはそれより大きく、少なくとも30%もしくはそれより大きく、少なくとも40%もしくはそれより大きく、少なくとも50%もしくはそれより大きく、少なくとも60%もしくはそれより大きく、または少なくとも63%もしくはそれより大きく減少させる段階が含まれる。もう一つの態様において、腎臓の低下または全ての原因の死亡率に関する任意の「悪化」事象のリスクは7%〜63%減少する。
【0072】
もう一つの態様において、「処置する」という用語にはまた、第一の「悪化」事象までの平均時間を増加させる段階が含まれる。増加は、約0.5ヶ月もしくはそれより長く、約1ヶ月もしくはそれより長く、約2ヶ月もしくはそれより長く、約3ヶ月もしくはそれより長く、約4ヶ月もしくはそれより長く、約5ヶ月もしくはそれより長く、約6ヶ月もしくはそれより長く、約7ヶ月もしくはそれより長く、約8ヶ月もしくはそれより長く、約9ヶ月もしくはそれより長く、約10ヶ月もしくはそれより長く、または約11ヶ月もしくはそれより長くてもよい。もう一つの態様において、期間は、PDS処置被験体において約2.8ヶ月±7.5ヶ月またはそれより大きく増加する。
【0073】
もう一つの態様において、被験体のクレアチニンクリアランス速度が安定化するかまたは改善される。例えば、被験体のクレアチニンクリアランス速度は、本発明の化合物を処置する前の被験体のレベルと比較して、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく増加する可能性がある。
【0074】
もう一つの態様において、クレアチニンクリアランスが50%またはそれより大きく減少するリスクは、少なくとも約5%もしくはそれより多く、少なくとも約10%もしくはそれより多く、少なくとも約15%もしくはそれより多く、または少なくとも約18%もしくはそれより多く低減される。さらなる態様において、クレアチニンクリアランスが50%またはそれより大きく減少するリスクは、約18%〜72%低減される。
【0075】
もう一つの態様において、被験体の血清クレアチニン、血清アルブミンレベル、および/または血清アルカリホスファターゼレベルは安定化されるかまたは改善される。例えば、被験体の血清クレアチニン、血清アルブミンレベル、および/または血清アルカリホスファターゼレベルは、本発明の化合物による処置の前の被験体のレベルと比較して約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく増加する可能性がある。
【0076】
さらなる態様において、血清クレアチニンが倍加するリスクは少なくとも5%もしくはそれより大きく、少なくとも10%もしくはそれより大きく、少なくとも11%もしくはそれより大きく、少なくとも12%もしくはそれより大きく、少なくとも13%もしくはそれより大きく、または少なくとも14%もしくはそれより大きく低減される。さらなる態様において、被験体の血清クレアチニンが倍加するリスクは約14%〜約81%低減される。
【0077】
もう一つの態様において、「処置する」という用語にはまた、血清クレアチニンの倍加までの平均時間を増加させる段階が含まれる。増加は約0.5ヶ月もしくはそれより長く、約1ヶ月もしくはそれより長く、約2ヶ月もしくはそれより長く、約3ヶ月もしくはそれより長く、約4ヶ月もしくはそれより長く、約5ヶ月もしくはそれより長く、約6ヶ月もしくはそれより長く、約7ヶ月もしくはそれより長く、約8ヶ月もしくはそれより長く、約9ヶ月もしくはそれより長く、約10ヶ月もしくはそれより長く、約11ヶ月もしくはそれより長く、または1年もしくはそれより長くてもよい。
【0078】
もう一つの態様において、被験体のタンパク尿レベル、内臓アミロイド負荷、および/または吸引脂肪組織におけるアミロイド含有量は、安定化されるかまたは改善される。例えば、被験体のタンパク尿レベル、内臓アミロイド負荷、および/または吸引脂肪組織におけるアミロイド含有量は、本発明の化合物による処置前の被験体のレベルと比較して、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、約60%もしくはそれより大きく、約70%もしくはそれより大きく、約80%もしくはそれより大きく、約90%もしくはそれより大きく、または約100%もしくはそれより大きく低減される可能性がある。
【0079】
もう一つの態様において、被験体の内臓アミロイド負荷を低減するかまたは安定化する。被験体の内臓アミロイド負荷は、例えば、123I-放射標識血清アミロイドP成分(SAP)のシンチグラフィーを用いて査定することができる。SAPは、アミロイド原線維に対して特異的に結合して、組織アミロイド沈着物において長期間にわたって保持され、それが循環中に受ける正常で急速な異化から明らかに保護される。放射標識SAPのシンチグラフィーによる撮像法は、内臓アミロイド負荷を査定するための特異的非侵襲的方法として開発されている(Hawkins PN et al. N Engl J Med, 1990; 323:508-13)。内臓のアミロイド負荷は、例えば、放射性核種の注射後24時間で得られた全身シンチグラフの肉眼的査定によって定量することができる。
【0080】
もう一つの態様において、吸引脂肪組織における被験体のアミロイド含有量は、低減されるかまたは安定化される。「吸引脂肪組織におけるアミロイド含有量」という用語は、吸引された脂肪組織におけるアミロイドAの含有量を指す。吸引脂肪組織におけるアミロイドA含有量の変化は、コンゴーレッド染色によって半定量的に測定することができる。脂肪組織におけるアミロイドA含有量は、例えば被験体から採取した脂肪組織を用いるモノクローナル抗体に基づくサンドイッチELISAを用いることによって、定量的に測定することができる(Hazenberg B et al. Ann Rheum Dis, 1999; 58: 96-102)。
【0081】
「起立性低血圧」という用語は、人が立つ姿勢をとる場合に起こる血圧の突然の下降を指す。突然立ち上がった後に一般的に起こる症状には、めまい、立ちくらみ、目のかすみ、および失神(意識の一時的な喪失)が含まれる。自律神経系(ANS)は時に、AAアミロイドーシスにおいて影響を受ける。姿勢による血圧の減少(例えば、仰臥位から起立姿勢の際に収縮期圧の>20 mmHgの下降、または拡張期圧の10 mmHgの下降が少なくとも3分間持続する)は、ANS機能障害の兆候である。
【0082】
もう一つの態様において、被験体の体重減少は改善もしくは安定化され、または被験体の体重は増加する。例えば、被験体は本発明の化合物による処置の前の体重の5%もしくはそれより多く、約10%もしくはそれより多く、約20%もしくはそれより多く、約30%もしくはそれより多く、約40%もしくはそれより多く、約50%もしくはそれより多く、または約60%もしくはそれより多く増加する可能性がある。
【0083】
もう一つの態様において、被験体のネフローゼ症候群は、安定化されるかまたは寛解される可能性がある。もう一つの態様において、被験体の浮腫は消散または緩和される可能性がある。もう一つの態様において、被験体における下痢の安定化、改善、治癒、または寛解が起こる可能性がある。なおもう一つの態様において、被験体における起立性低血圧、脾腫、および/または肝腫の安定化または低減が起こる可能性がある。
【0084】
一つの態様において、ネフローゼ症候群の寛解には、タンパク尿の<1 g/24時間への減少、および血清アルブミンの3.4 g/dLより大きい増加または浮腫の緩解および/または浮腫の改善に反応した利尿薬の中止のいずれかが含まれる。
【0085】
「治療的有効量」という用語は、被験体を処置するために、例えばAAアミロイドーシスもしくはアミロイド関連疾患に関して被験体を処置するために、または炎症性疾患、悪性新生物、もしくは慢性微生物感染症のような、しかしこれらに限定されない基礎疾患を有する被験体を処置するために有効である化合物の量を指す。治療的有効量は、被験体が苦しんでいる特定の障害、特定の被験体の年齢、体重、および生活様式に基づいて変化してもよい。さらに、治療的有効量は、病態の重症度、臓器機能、腎機能、または基礎疾患(例えば、被験体が炎症疾患、悪性新生物、慢性感染症に苦しんでいてもよい)に依存してもよい。一つの態様において、被験体はネフローゼである。もう一つの態様において、被験体は非ネフローゼである。
【0086】
「ネフローゼ」という用語は、ネフローゼ症候群に苦しむ被験体を指す。ネフローゼ症候群は、一般的に重度のタンパク尿症(例えば、尿中タンパク質>3 g/24時間)と共に以下の二つの腎臓外特色であると定義される:1)低アルブミン血症(例えば、血清アルブミン<3.4 g/dL);および2)健康診査による末梢の浮腫および/または浮腫を処置するための利尿剤の使用。
【0087】
「非ネフローゼ」という用語は、ネフローゼ症候群にまだ進行していない、またはネフローゼ症候群が寛解した被験体を指す。ネフローゼ症候群の寛解は、タンパク尿症の<1 g/24時間への減少および以下の二つの腎臓外特色の一つの改善である:1)血清アルブミンの>3.4 g/dLへの増加、または2)浮腫の消散および/または浮腫の改善に反応した利尿剤の中止。ネフローゼ症候群への進行は、タンパク尿症の>3 g/24時間への増加、および以下の二つの腎臓外特色の発生である:1)低アルブミン血症(例えば、血清アルブミン<3.4 g/dL);および2)浮腫および/または浮腫を処置するための利尿剤の使用。
【0088】
もう一つのさらなる態様において、本発明はまた、被験体のクレアチニンクリアランス速度、タンパク尿レベル、および/または血清アルブミンレベルに基づいて選択された用量の式(I)の化合物の治療的有効量を被験体に投与することによって、被験体におけるアミロイド関連疾患を処置または予防する方法に関する。
【0089】
「クレアチニンクリアランス」という用語は、当技術分野で認識され、腎臓が血液中からクレアチニンを消失させる速度を指す。クレアチニンは、健康な被験体において腎臓によって容易に排泄される物質である。クレアチニンクリアランスは一般的に、尿中のクレアチニンレベルを血液中のクレアチニンレベルと比較する。クリアランスはしばしば、ミリリットル/分(ml/分)として測定される。
【0090】
本発明の方法において投与される用量は、クレアチニンクリアランス速度に基づいて選択してもよい。例えば、式(I)の化合物の用量は、クレアチニンクリアランス速度が>80 ml/分の場合、約1200 mgを1日2回であるように選択されてもよい。クレアチニンクリアランス速度が約30〜80 ml/分の場合、式(I)の化合物の用量は、約800 mgを1日2回であるように選択されてもよい。クレアチニンクリアランス速度が約20〜30 ml/分の場合、式(I)の化合物の用量は、約400 mgを1日2回であるように選択されてもよい。さらに、用量は、被験体におけるクレアチニンクリアランス速度の変化に基づいて調節してもよい。
【0091】
一つの態様において、本発明の化合物を被験体に投与した後に、所望の薬物動態パラメータおよび/または生物学的に都合のよいパラメータが得られるように、用量を選択してもよい。一つの態様において、用量は、被験体に1回投与後、被験体における平均AUCssが約7,000〜約26,000 ng.h/mlとなるように、および平均定常状態濃度が約500〜約1200 ng/mlとなるように選択される。もう一つの態様において、用量は被験体に1回投与した後、被験体におけるCmaxが約1,200〜約3,100 ng/mlとなるように、およびAUC∞が約5,000〜約43,000 ng.h/mlとなるように選択される。腎機能障害を有する被験体では、特定のAUCss、AUC∞、Cmaxおよび定常状態平均濃度を達成するために必要な用量を調節する必要がある可能性がある。
【0092】
さらなる態様において、Cmax、AUC0-tlast、および/またはAUC∞は、表1において示される値と比較して、特定の被験体に関して約±10%、約±20%、約±30%、または約±40%変化してもよい。
【0093】
「アミロイド関連疾患」という言語は、アミロイド線維の存在を特徴とする病態を指す。「アミロイド」は、多くの異なる疾患において認められる、多様であるが特異的なタンパク質沈着(細胞内または細胞外)の群を指す一般用語である。その発生は多様であるが、全てのアミロイド沈着物は、共通の形態学的特性を有し、特異的色素(例えば、コンゴーレッド)で染色され、染色後に偏光において特徴的な赤-緑複屈折外観を有する。それらはまた、共通の超微細構造、共通のX-線回折、および赤外線スペクトルを共有する。
【0094】
本発明はまた、少なくとも部分的に、被験体におけるAAアミロイドーシスを処置または予防するためのもう一つの方法に関する。本方法には、例えばAUC、Cmax、AUCss、Css、Tmax等によって測定した場合に、被験体において有効な全身曝露が提供されるような用量で投与される式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0095】
「標的血漿濃度」という用語は、それによってAAアミロイドーシスに関する被験体の処置が起こる、被験体における本発明の化合物の濃度範囲を指す。一つの態様において、被験体は、約500〜約1200 ng/mlの定常状態濃度(Css)を維持する。もう一つの態様において、被験体は、約7000〜約26,000 ng.h/mlのAUCssを維持する。例えば、被験体は、定常状態濃度約600〜約700 ng/mlまたは約900〜約1100 ng/ml、および/またはAUCss約8000〜約9000 ng.h/ml、約11,000〜約13,000 ng.h/ml,、約23,000〜約26,000 ng.h/ml、または約15,500〜約16,500 ng.h/mlを維持してもよい。さらなる態様において、AUCssまたは定常状態濃度はこれらの値の±20%以内である。
【0096】
さらに、本発明は、少なくとも部分的に、被験体における腎および/または胃腸管機能を安定化するかまたは改善する方法に関する。方法には、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0097】
さらなる態様において、本発明はまた、少なくとも部分的に薬学的製剤にも関する。製剤は、AAアミロイドーシスを処置または予防するために有効な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と、薬学的に許容される担体とを含む。さらに、製剤を健康被験体に経口投与すると、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。代わりの態様において、製剤を健康被験体に経口投与すると、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。もう一つの代わりの態様において、製剤を健康被験体に経口投与すると、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0098】
もう一つの態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と、薬学的に許容される担体とを含む薬学的製剤に関する。この態様において、製剤をAAアミロイドーシスを有する被験体に経口投与する場合、クレアチニンクリアランス速度約30 ml/分未満を有する被験体に活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0099】
もう一つの態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質800 mgと薬学的に許容される担体とを含む薬学的製剤に関する。さらに、本製剤を被験体に経口投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0100】
もう一つのさらなる態様において、本発明は、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と薬学的に許容される担体とを含む薬学的製剤に関する。さらに本製剤を、AAアミロイドーシスを有する被験体に24ヶ月間経口投与する場合、活性物質の用量400 mgでは、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0101】
もう一つのさらなる態様において、本発明はまた、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である活性物質と薬学的に許容される担体とを含む薬学的製剤に関する。さらに本製剤を健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0102】
もう一つのさらなる態様において、本発明はまた、腎機能を安定化するかもしくは改善させる、またはAAアミロイドーシスを有する被験体における腎疾患の進行を遅らせる方法にも関する。方法には、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、一つまたは複数の薬学的に許容される担体とを含む製剤を、被験体のクレアチニンクリアランス速度に従って決定された量で経口投与する段階が含まれる。さらに、製剤を用量400 mgで投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが得られ、製剤を800 mgの用量で投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または製剤を1200 mgの用量で投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる。
【0103】
さらなる態様において、被験体のクレアチニンクリアランス速度が約30 ml/分未満である場合、用量は400 mgであり、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合、用量は800 mgであり、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合、用量は1200 mgである。もう一つのさらなる態様において、被験体のクレアチニンクリアランス速度は、約60〜約90 ml/分であり、用量1200 mgが投与される。
【0104】
もう一つの態様において、本発明は、被験体におけるAAアミロイドーシスを処置または予防する方法に関する。方法には、AAアミロイドーシスが処置または予防されるように、第二の物質と併用して、式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0105】
「併用する」という用語は、式(I)の化合物と第二の物質との同時投与;第二の物質の投与前の式(I)の化合物の投与;または式(I)の化合物の投与前の第二の物質の投与を指す。
【0106】
「第二の物質」という用語には、基礎疾患、例えば炎症疾患(例えば、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫症、マックル-ウェルズ症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、およびクローン病等)、慢性感染症に関連した疾患(例えば、AIDS、HIV、B型肝炎、C型肝炎等)、慢性微生物感染症に関連した疾患(例えば、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、ホイップル病、集族性ざ瘡、分類不能型免疫不全、肺結核、肺感染症、再発性膿瘍、ベーチェット病、低γグロブリン血症、嚢胞性線維症)、または特定の悪性新生物(例えば、ホジキンリンパ腫、腎癌、腸管、肺、および尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、およびヘアリーセル白血病)を処置することが知られている薬物が含まれる。「第二の物質」という用語にはまた、救済薬、化学療法剤、抗炎症剤、例えば非ステロイド性抗炎症剤等、および抗生物質が含まれる。第二の物質の例には、メソトレキセート、コルヒチン、抗TNF抗体、および抗インターロイキン1または6抗体が含まれる。
【0107】
非ステロイド性抗炎症剤(「NSAID」)の例には、イブプロフェン、ナプロキセン、サリンダック、およびインドメタシンが含まれる。他の抗炎症剤には、COX-2阻害剤(Vioxx(商標)およびCelebrex(商標)のような)、サイトカイン阻害剤(WO 95/04533において開示されるサリドマイドおよびデキサナビノールのような)、補体阻害剤、ロイコトリエン受容体拮抗剤およびその組み合わせが含まれる。例には、酢酸誘導体サリンダック(Clinoril(商標), Merck & Co., Inc., Rahway, New Jersey)、インドメタシン(Indocin(商標), Merck & Co., Inc., Rahway, New Jersey);エトドラック(Lodine(商標), Wyeth, Madison, New Jersey)、ナブメトン(Relafen(商標), GlaxoSmithKline, Middlesex, England)、トルメチンナトリウム(Tolectin(商標), McNeil Pharmaceuticals, Spring House, Pennsylvania);アントラニル酸誘導体:メクロフェナム酸ナトリウム(Meclomen(商標), Pfizer, New York, New York)、メフェナム酸(Ponstel(商標), Pfizer, New York, New York);エノール酸誘導体:ピロキシカム(Feldene(商標), Pfizer, New York, New York)、Mobic(商標)(メロキシカム);フェニル酢酸誘導体:arthrotec(ジクロフェナク/ミソプロストール)、Voltaren(商標)(ジクロフェナク);プロピオン酸誘導体:ナプロキセンナトリウム(Anaprox(商標), Naprosyn(商標), Hoffmann-La Roche Inc. (Roche), Nutley, N.J.)、フルビプロフェン(Ansaid(商標), Upjohn、現在はPfizer, New York, New York)、オキサプロジン(Daypro(商標), G.D Searle, 現在はPfizer, New York, New York);イブプロフェン(Motrin(商標), Upjohn、現在はPfizer, New York, New York)、フェノプロフェンカルシウム(Nalfon(商標), Dista, Ranbaxy, Princeton, NJ)、ケトプロフェン(Oruvail(商標)またはOrudis(商標), Wyeth, Madison, New Jersey)、ケトロラックトロメタミン(Toradol(商標), Syntex Laboratories, Hoffmann-La Roche Inc. (Roche), Nutley, N.J.);サリチル酸誘導体:ジフルニサル(Dolobid(商標), Merck & Co., Inc., Rahway, New Jersey);およびCOX-2選択的阻害剤:Bextra(商標)(バルデコキシブ)、Celebrex(商標)(セレコキシブ、Pfizer, New York, New York)、およびVioxx(商標)(ロフェコキシブ、Merck & Co., Inc., Rahway, New Jersey)、およびシクロスポリン(Maas BiolAB, Albuquerque, New Mexico)が含まれる。
【0108】
「化学療法剤」という言語には、細胞または組織の成長が望ましくない、増殖しつつある細胞または組織の成長を阻害する物質、またはそのような成長によって起こる少なくとも一つの症状を処置する物質が含まれる。化学療法剤の例には:ブレオマイシン、ドセタキセル(Taxotere)、ドキソルビシン、エダトレキセート、エトポシド、フィナステリド(Proscar)、フルタミド(Eulexin)、ゲンシタビン(Gemzar)、酢酸ゴセレリン(Zoladex)、グラニセトロン(Kytril)、イリノテカン(Campto/Camptosar)、オンダンセトロン(Zofran)、パクリタキセル(Taxol)、ペガスパルガーゼ(Oncaspar)、塩酸ピロカルピン(Salagen)、ポルフィマーナトリウム(Photofrin)、インターロイキン-2(Proleukin)、リツキシマブ(Rituxan)、トポテカン(Hycamtin)、トラスツズマブ(Herceptin)、トレチノイン(Retin-A)、トリアピン、ビンクリスチン、および酒石酸ビノレルビン(Navelbine)が含まれる。
【0109】
化学療法剤の他の例には、ナイトロジェンマスタード(例えば、メクロレタミン(HN2))、シクロホスファミド、イフォスファミド、メルファラン(L-サルコリシン)、クロラムブシル等のようなアルキル化剤;エチレンイミン、メチルメラミン(例えば、ヘキサメチルメラミン、チオテパ等)、アルキルスルホネート(例えば、ブスルファン等)、ニトロソウレア(例えば、カルムスチン(BCNU)、ロムスチン(CCNU)、セムスチン(メチル-CCNU)、ストレプトゾシン(ストレプトゾトシン)等)、トリアゼン(例えば、デカルバジン(DTIC;ジメチルトリアゼノイミダゾールカルボキサミド)、アルキル化剤(例えば、シス-ジアミンジクロロプラチナII(CDDP))が含まれる。
【0110】
化学療法剤の他の例には、葉酸類似体(例えば、メソトレキセート(アメトプテリン));ピリミジン類似体(例えば、フルオロウラシル('5-フルオロウラシル;5-FU);フロクスリジン(フルオロデオキシウリジン);FUdr;シタラビン(シトシンアラビノシド)等);プリン類似体(例えば、メルカプトプリン(6-メルカプトプリン;6-MP);チオグアニン(6-チオグアニン;TG);およびペントスタチン(2'-デオキシコフォマイシン)等)のような抗代謝剤が含まれる。
【0111】
化学療法剤の他の例にはまた、ビンカアルカロイド(例えば、ビンブラスチン(VLB)およびビンクリスチン);トポイソメラーゼ阻害剤(例えば、エトポシド、テニポシド、カンプトテシン、トポテカン、9-アミノ-カンプトテシンCPT-11等);抗生物質(例えば、ダクチノマイシン(アクチノマイシンD)、アドリアマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、プリカマイシン(ミスラマイシン)、マイトマイシン(マイトマイシンC)、タキソール、タキソテール等);酵素(例えば、L-アスパラギナーゼ);および生物反応改変剤(例えば、インターフェロン-α;インターロイキン2等)が含まれる。他の化学療法剤には、シス-ジアミンジクロロプラチナII(CDDP);カルボプラチン;アントラセンジオン(例えば、ミトキサントロン);ヒドロキシウレア;プロカルバジン(N-メチルヒドラジン);および副腎皮質抑制剤(例えば、ミトーテン、アミノグルテチミド等)が含まれる。
【0112】
他の化学療法剤には、副腎皮質ステロイド(例えば、プレドニゾン);プロゲスチン(例えば、カプロン酸ヒドロキシプロゲステロン;酢酸メドロキシプロゲステロン、酢酸メゲステロール等);エストロゲン(例えば、ジエチルスチルベストロール;エテニルエストラジオール等);抗エストロゲン(例えば、タモキシフェン等);アンドロゲン(例えば、プロピオン酸テストステロン、フルオキシメステロン等)、抗アンドロゲン(例えば、フルタミド);および性腺刺激ホルモン放出ホルモン類似体(例えば、リュープロリド)が含まれる。
【0113】
「抗生物質」という用語には、微生物感染症を処置するために当技術分野において公知の抗生物質が含まれる。抗生物質の例には、アモキシシリン、アミノグリコシド、アミノグリコシド類似体、β-ラクタム、β-ラクタマーゼ、β-ラクタマーゼ類似体、クリンダマイシン、クロラムフェニコール、セファロスポリン、セファロスポリン類似体、シプロフロキサシン、シプロフロキサシン類似体、エリスロマイシン、フルオロキノロン、フルオロキノロン類似体、マクロライド、マクロライド類似体、メトロニダゾール、ペニシリン、ペニシリン類似体、キノロン、キノロン類似体、リファンピン、ストレプトマイシン、スルホンアミド、テトラサイクリン、テトラサイクリン類似体、トリメトプリム、トリメトプリム-スルファメトキサゾール、およびバンコマイシンが含まれるがこれらに限定されるわけではない。
【0114】
「救済薬」という用語には、基礎疾患を抑制する能力を有するが、進行性AAアミロイドーシスの特色を回復する主な指標と共に導入される、処置のあいだに行われる任意の投薬を指す。そのような投薬には、コルヒチン、細胞障害剤、および抗TNF-剤が含まれるがこれらに限定されるわけではない。
【0115】
抗TNF剤の例には、TNFを阻害する物質、例えば抗TNFα抗体が含まれる。抗TNF物質の例には、エタネルセプト(Enbrel(商標), Amgen)、インフリキシマブ(Remicade(商標), Johnson and Johnson、例えば米国特許第6,790,444号を参照されたい)、ヒト抗-TNFモノクローナル抗体(D2E7/HUMIRA(商標), Abbott Laboratories)、CDP 571 (Celltech)、およびCDP 870 (Celltech)が含まれる。
【0116】
他の第二の物質の例には、免疫抑制剤、コルチコステロイド(全身投与されるコルチコステロイドを含む)、スルファサラジン、レニン-アンジオテンシン系遮断剤または拮抗剤、利尿剤(例えば、フロセミド)、カルシウムチャンネル遮断剤、β遮断剤、抗リウマチ薬、アンジオテンシン変換酵素阻害剤(ACEi)、アンジオテンシンII受容体遮断剤(ARBs)、アセチルサリチル酸、アモキシシリン、カルシウム、炭酸カルシウム、クロラムブシル、コルヒチン、シクロホスファミド、ジクロフェナク、エナラプリル、葉酸、メソトレキセート、メチルプレドニゾロン、オメプラゾール、パラセタモール、プレドニゾロン、およびプレドニゾンが含まれる。
【0117】
もう一つの態様において、本発明は、少なくとも部分的に、被験体における炎症疾患を処置する方法に関する。方法には、被験体における炎症疾患が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。さらなる態様において、第二の物質は抗炎症剤である。
【0118】
「炎症疾患」という用語には、炎症に関連し、本発明の化合物を用いて処置されうる疾患または障害が含まれる。炎症疾患には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それを引き起こす、それによって引き起こされる、それが原因で起こる、またはそうでなければアミロイドーシスに関連する疾患が含まれてもよい。そのような炎症疾患の例には、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、成人スティル病、ベーチェット症候群、家族性地中海熱、炎症性腸疾患、遺伝性周期性発熱、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、およびクローン病が含まれるがこれらに限定されるわけではない。
【0119】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置する、予防する、またはその発症を遅らせるために有効であり、第二の物質は、基礎となる障害、例えば基礎となる炎症性疾患を処置するために有効な量で投与される。
【0120】
なおもう一つの態様において、本発明には、被験体におけるリウマチ性関節炎を処置する方法が含まれる。方法には、被験体におけるリウマチ性関節炎が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。さらなる態様において、第二の物質は抗炎症剤である。さらなる態様において、第二の物質はインフリキシマブであり、これは例えば参照により本明細書に組み入れられる米国特許第6,790,444号において記述される技法によって投与されてもよい。
【0121】
さらなる態様において、第二の物質は、慢性炎症疾患、リウマチ性関節炎、若年性慢性関節炎、強直性脊椎炎、乾癬、乾癬性関節症、ライター症候群、家族性地中海熱、成人スティル病、ベーチェット症候群、炎症性腸疾患、乾癬性関節炎、紅斑性狼瘡関節炎、結節性関節周囲炎、ヴェグナー肉芽腫、マックル-ウェルズ症候群、遺伝性周期性発熱、またはクローン病のような炎症疾患を処置することが知られている物質である。被験体に投与されてもよい物質の例には、例えば、抗TNF剤、メソトレキセート、抗炎症剤、およびその組み合わせが含まれる。
【0122】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅らせるために有効であり、第二の物質はリウマチ性関節炎を処置するために有効量で投与される。
【0123】
さらなる態様において、本発明は、少なくとも部分的に、被験体における悪性新生物を処置する方法に関する。方法には、被験体における悪性新生物が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0124】
「悪性新生物」という用語には、本発明の化合物を用いて処置されうる新生物が含まれる。悪性新生物には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それによって引き起こされる、それを引き起こす、それが原因で起こる、またはそうれなければアミロイドーシスに関連する新生物が含まれてもよい。そのような悪性新生物の例には、ホジキンリンパ腫、腎癌、腸管癌、肺癌、尿性器管の癌、基底細胞癌、肝癌、キャッスルマン病、シュニッツラー症候群、ヴァルデンストレーム病、またはヘアリーセル白血病が含まれるがこれらに限定されるわけではない。
【0125】
悪性新生物の処置にとって有用となる可能性がある第二の物質の例には、ホジキンリンパ腫、腎癌、腸管癌、肺癌、尿性器管の癌、基底細胞癌、またはヘアリーセル白血病を処置することが知られている物質が含まれる。用いてもよい第二の物質のさらなる例には、化学療法剤または細胞障害剤が含まれる。
【0126】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅延させるために有効であり、第二の物質は悪性新生物を処置するために有効量で投与される。
【0127】
なおもう一つの態様において、本発明は、慢性感染症を処置する方法に関する。方法には、慢性感染症が処置されるように、第二の物質と併用して式(I)の化合物の治療的有効量を、それを必要とする被験体に投与する段階が含まれる。
【0128】
「慢性感染症」という用語には、本発明の化合物を用いて処置されうる慢性的なウイルス、細菌、真菌、および微生物感染症が含まれうる。感染症には、アミロイドーシス、例えばAAアミロイドーシスに関連する、それを引き起こす、それによって引き起こされる、それが原因で起こる、またはそうれなければアミロイドーシスに関連する感染症が含まれてもよい。微生物感染症は局所または全身性であってもよい。そのような微生物感染症の例には、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、肺結核、肺感染症、再発性膿瘍、ベーチェット病、およびホイップル病が含まれるがこれらに限定されるわけではない。他の慢性感染症には、AIDS、HIV、B型肝炎およびC型肝炎が含まれる。
【0129】
感染症を処置するために有用となる可能性がある第二の物質の例には、AIDS、HIV、B型肝炎、C型肝炎、らい、結核、気管支拡張、褥瘡性潰瘍、腎盂腎炎、骨髄炎、集族性ざ瘡、分類不能型免疫不全、低γグロブリン血症、嚢胞性線維症、肺結核、肺感染症、再発性膿瘍、ベーチェット病、またはホイップル病を処置することが知られている物質が含まれる。被験体に投与してもよい物質の例には、例えば、抗炎症剤および抗生物質が含まれる。
【0130】
さらなる態様において、本発明の化合物の治療的有効量は、AAアミロイドーシスを処置するか、予防するか、またはその発症を遅らせるために有効であり、第二の物質は慢性感染症を処置するために有効量で投与される。
【0131】
なおもう一つの態様において、本発明は、少なくとも部分的に、被験体における化合物の経口生物学的利用率が増加するように、食物なしで薬学的組成物における式(I)の化合物の治療的有効量を被験体に投与することによって、被験体における化合物の経口生物学的利用率を増加させる方法に関する。
【0132】
「経口生物学的利用率」は、経口投与後に血流に達する薬物の量を指す。「経口生物学的利用率の増加」という用語は、生物学的利用率の約5%もしくはそれより大きい、約10%もしくはそれより大きい、約15%もしくはそれより大きい、約20%もしくはそれより大きい、約25%もしくはそれより大きい、約30%もしくはそれより大きい、約35%もしくはそれより大きい、約40%もしくはそれより大きい、約50%もしくはそれより大きい、約55%もしくはそれより大きい、約60%もしくはそれより大きい、約65%もしくはそれより大きい、約70%もしくはそれより大きい、約75%もしくはそれより大きい、約80%もしくはそれより大きい、約85%もしくはそれより大きい、約90%もしくはそれより大きい、約95%もしくはそれより大きい、または約100%もしくはそれより大きい増加を指す。
【0133】
さらなる態様において、食物なしでの本発明の化合物の投与によって、食物と共に投与した場合と比較して、最高血漿濃度(Cmax)の増加および化合物の吸収の程度(AUC)の増加が起こる。Cmaxおよび/またはAUCの増加は、食物と共に化合物を投与した場合と比較して、約5%もしくはそれより大きい、約10%もしくはそれより大きい、約15%もしくはそれより大きい、約20%もしくはそれより大きい、約25%もしくはそれより大きい、約30%もしくはそれより大きい、約35%もしくはそれより大きい、約40%もしくはそれより大きい、約50%もしくはそれより大きい、約55%もしくはそれより大きい、約60%もしくはそれより大きい、約65%もしくはそれより大きい、約70%もしくはそれより大きい、約75%もしくはそれより大きい、約80%もしくはそれより大きい、約85%もしくはそれより大きい、約90%もしくはそれより大きい、約95%もしくはそれより大きい、または約100%もしくはそれより大きい可能性がある。さらなる態様において、食物と共に投与した場合と比較して、投与によって化合物の最高血漿濃度(Cmax)および吸収の程度(AUC)の増加が起こることが、被験体に通知される(医師もしくは薬剤師による説明によって、または本発明の化合物に添付される表示もしくは添付文書によって)。
【0134】
「食物なし」という用語は、薬剤または本発明の組成物を実質的に空の胃に投与することを指す。したがって、いくつかの態様において、食物なしの投与には、最も近い食物摂取より15分、30分、60分、90分、120分、150分、3時間、4時間、5時間、6時間、7時間、または8時間より後での投与が含まれる。他の態様において、食物なしの投与には、次の食物摂取の少なくとも15分、30分、60分、90分、120分、150分、3時間、4時間、5時間、6時間、7時間、または8時間前での投与が含まれる。一つの態様において、「食物なし」という用語は、食事前少なくとも1時間、または食後少なくとも2時間に本発明の化合物を投与することである。この態様において、「約」という用語には、表記の期間の±10〜20%の値が含まれる。
【0135】
もう一つの態様において、本発明は、例えば、血清クレアチニンの倍加の発生、クレアチニンクリアランスの50%減少より大きいまたはそれに等しい、透析/末期腎疾患、および/または全ての原因の死亡率によって測定した場合に、それを必要とする被験体における腎症の進行速度を低減させる方法に関する。例えば、AAアミロイドーシス、リウマチ性関節炎、慢性炎症、慢性感染症、遺伝性発熱等を有してもよい。
【0136】
もう一つの態様において、本発明は、AAアミロイドーシスを有する被験体における末期腎疾患(ESRD)および/または透析への進行を予防するかまたは遅らせるための方法に関する。方法には、ESRDおよび/または透析への進行が遅れるまたは予防されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0137】
さらなる態様において、ESRDおよび/または透析への進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。なおさらなる態様において、透析および/またはESRDは、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して6ヶ月間遅れる。
【0138】
もう一つの態様において、ESRDへの進行のリスクは約0〜78%低減される。もう一つの態様において、リスクは約5%、約10%、約15%、約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、約60%、約65%、約70%、約75%、または約78%低減される。この態様において、「約」という用語には±5%の値が含まれる。
【0139】
さらなる態様において、透析までの期間の中央値は、少なくとも約1ヶ月、約2ヶ月、約3ヶ月、約4ヶ月、約5ヶ月、約6ヶ月、約7ヶ月、約8ヶ月、または約9ヶ月遅れる。この態様において、「約」という用語には、表記の期間の±0.5ヶ月の範囲が含まれる。さらなる態様において、透析までの期間の中央値は、式(I)の化合物、例えばPDSによって被験体において3.5ヶ月±5.5、または9ヶ月まで長い。
【0140】
さらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における血清クレアチニンの倍加までの時間を予防するかまたは遅らせるための方法に関する。方法には、血清クレアチニンの倍加までの時間が遅れるまたは予防されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0141】
さらなる態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%減少までの時間を予防するかまたは遅らせるための方法が含まれる。方法には、クレアチニンクリアランスの少なくとも50%減少までの時間が遅れるまたは予防されるように、化合物の治療的有効量を被験体に投与する段階が含まれる。さらなる態様において、クレアチニンクリアランスの少なくとも50%減少までの時間の中央値は、PDSのような式(I)の化合物によって処置した被験体において約1ヶ月、約2ヶ月、約3ヶ月、約4ヶ月、約5ヶ月、約6ヶ月、約7ヶ月、約8ヶ月、約9ヶ月、約10ヶ月、約11ヶ月、または約12ヶ月である。この態様において、「約」という用語には、表記の期間の±0.5ヶ月の範囲が含まれる。さらなる態様において、クレアチニンクリアランスの少なくとも50%減少までの時間の中央値は、PDS処置患者において12ヶ月まで長い。
【0142】
さらなる態様において、血清クレアチニンの倍加までの時間および/またはクレアチニンクリアランスの少なくとも50%減少は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。もう一つの態様において、クレアチニンクリアランスの50%減少は、約3〜約5ヶ月、または約4ヶ月遅れる。なおさらなる態様において、血清クレアチニンの倍加および/またはクレアチニンクリアランスの少なくとも50%減少は、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して少なくとも約6ヶ月遅れる。さらなる態様において、血清クレアチニンの倍加までの期間は、約3ヶ月〜約5ヶ月、または約4ヶ月遅れる。
【0143】
なおもう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの少なくとも50%増加までの時間を減少させる方法に関する。方法には、クレアチニンクリアランスの少なくとも50%の増加までの時間が減少するように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0144】
さらなる態様において、クレアチニンクリアランスの少なくとも50%の増加までの時間は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く減少される。なおさらなる態様において、クレアチニンクリアランスの少なくとも50%増加は、本発明の化合物によって処置していない類似の障害を有する標準的な被験体と比較して6ヶ月間減少する。
【0145】
もう一つの態様において、本発明には、AAアミロイドーシスを有する被験体におけるクレアチニンクリアランスの勾配によって測定した場合に、腎疾患の進行速度を低減させる方法が含まれる。方法には、腎疾患の進行速度が、例えばクレアチニンクリアランスの減少速度の低下によって測定した場合に低減されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0146】
さらなる態様において、クレアチニンクリアランスの勾配は、約0〜10 ml/分/1.73m2/年低減される。もう一つのさらなる態様において、クレアチニンクリアランスの勾配は、約1 ml/分/1.73m2/年、約2 ml/分/1.73m2/年、約3 ml/分/1.73m2/年、約4 ml/分/1.73m2/年、約5 ml/分/1.73m2/年、約6 ml/分/1.73m2/年、約7 ml/分/1.73m2/年、約8 ml/分/1.73m2/年、約9 ml/分/1.73m2/年、または約10 ml/分/1.73m2/年低減される。さらなる態様において、クレアチニンクリアランスの勾配は、約4.7±5 ml/分/1.73m2/年低減される。この態様において、「約」という用語には、値±0.5 ml/分/1.73m2/年が含まれる。
【0147】
さらなる態様において、腎疾患の進行速度は、約10%もしくはそれより大きく、約20%もしくはそれより大きく、約30%もしくはそれより大きく、約40%もしくはそれより大きく、約50%もしくはそれより大きく、または約60%もしくはそれより大きく低減される。特定の態様において、腎疾患の進行速度は、約30%〜約40%低減される。
【0148】
「クレアチニンクリアランスの変化率」という用語は、経時的な被験体の体表面積に関して標準化されるクレアチニンクリアランスの変化率を指す。例えば、被験体のクレアチニンクリアランスは、例えば指定の時点での24時間尿採取によって測定されうる。このクレアチニンクリアランスは、体表面積に関して標準化され、その被験体に関して利用可能なクレアチニンクリアランス測定を用いて、被験体内の勾配の最小二乗法推定値を計算してもよい。一般的に、クレアチニンクリアランスの勾配は、年間変化率として表記される。必要であれば、勾配の計算の前に、適した変換(すなわち、対数変換)を適用してもよい。
【0149】
さらなる態様において、被験体のクレアチニンクリアランスの変化率は、約1 ml/分/年もしくはそれより多く、約2 ml/分/年もしくはそれより多く、約3 ml/分/年もしくはそれより多く、約4 ml/分/年もしくはそれより多く、約5 ml/分/年もしくはそれより多く、約6 ml/分/年もしくはそれより多く、約7 ml/分/年もしくはそれより多く、約8 ml/分/年もしくはそれより多く、約9 ml/分/年もしくはそれより多く、または約10 ml/分/年もしくはそれより多く改善される。さらなる態様において、クレアチニンクリアランス速度の減少は約2〜約5 ml/分/年減弱される。
【0150】
さらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるタンパク尿症を安定化するかまたは低減させるための方法に関する。方法には、被験体におけるタンパク尿症が安定化されるかまたは低減されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。一つの態様において、タンパク尿症は、約0.5 g/24時間もしくはそれより多く、約1 g/24時間もしくはそれより多く、約1.5 g/24時間もしくはそれより多く、または約2 g/24時間もしくはそれより多く低減される。一つの態様において、タンパク尿症は1 g/24時間未満、またはそれに等しい値で安定化される。
【0151】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体における腎機能を安定化するため、または腎疾患の進行を遅らせるための方法に関する。方法には、被験体の腎機能が安定化される、または腎疾患の進行が遅れるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0152】
さらなる態様において、腎疾患の進行は、1ヶ月もしくはそれより長く、2ヶ月もしくはそれより長く、3ヶ月もしくはそれより長く、4ヶ月もしくはそれより長く、5ヶ月もしくはそれより長く、6ヶ月もしくはそれより長く、7ヶ月もしくはそれより長く、8ヶ月もしくはそれより長く、9ヶ月もしくはそれより長く、10ヶ月もしくはそれより長く、または12ヶ月もしくはそれより長く遅れる。
【0153】
なおもう一つの態様において、本発明は、AAアミロイドーシスを有する被験体における腎障害を処置するための方法に関する。方法には、腎障害が処置されるように、式(I)の化合物の治療的有効量を被験体に投与する段階が含まれる。
【0154】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群への進行を予防するかまたは遅らせるための方法に関する。方法には、ネフローゼ症候群への進行が予防するかまたは遅れるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0155】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群を処置するための方法に関する。方法には、ネフローゼ症候群に関連するパラメータが改善される、またはネフローゼ症候群が寛解されるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0156】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるネフローゼ症候群の寛解を維持するための方法に関する。方法には、ネフローゼ症候群の寛解が例えば、約4、6、8、10、または12ヶ月間持続するように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。特定の態様において、ネフローゼ症候群の寛解は、約6〜約8ヶ月間患者において持続される。
【0157】
もう一つの態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを有する被験体におけるGFRを安定化するかまたは増加させるための方法に関する。方法には、GFRを安定化するかまたは増加させるように、式(I)の化合物、例えば1,3-プロパンジスルホン酸またはその薬学的に許容される塩、例えば二ナトリウム塩の治療的有効量を被験体に投与する段階が含まれる。
【0158】
「糸球体濾過速度」および「GFR」という用語は、本明細書において互換的に用いられ、腎機能の指標である。例えば、被験体のGFRの一つの手段は、クレアチニンクリアランス速度である。腎機能および/またはGFRは、例えば血清クレアチニンレベル、尿中クレアチニンレベル、尿中アルブミンレベル、尿中微小タンパク質レベル(例えば、レチノール結合タンパク質、N-アセチル-β-D-グルコサミニダーゼ、ミクロアルブミン等)、血漿インスリンクリアランス、クレアチニンクリアランス、タンパク尿症等のような、多数の基準を用いて評価することができる。
【0159】
さらに、被験体は軽度、中等度、または重度腎障害を有してもよい。例えば、健康被験体は典型的に、約100 ml/分より大きいGFRを有する。「軽度の」腎障害を有する被験体は、例えばGFR約50〜約80 ml/分、またはGFR 100未満、またはクレアチニンクリアランス速度約60〜約90 ml/分を有してもよい。「中等度の」腎障害を有する被験体は、例えば、GFR約30〜約50 ml/分、またはクレアチニンクリアランス速度約30〜約60 ml/分を有してもよい。「重度の」腎障害を有する被験体は、例えばGFR約30 ml/分未満、またはクレアチニンクリアランス速度約15〜約30 ml/分を有してもよい。被験体はまた、本明細書に記述の実施例において記述されるように、または当技術分野で公知の基準を用いて、軽度、中等度、または重度であると分類されてもよい(例えば、McCullough, P. A., Rev. Cardiovasc. Med. 2003;4(suppl. 1): S2-S6;K/DOQI guidelines at www.kidney.org/ professionalsを参照されたい)。
【0160】
他の態様において、被験体は、処置前(例えば、ベースライン時)、約50〜約120 ml/分、約60〜約100 ml/分、約70〜約110 ml/分、または約70〜約100 ml/分のクレアチニンクリアランス速度を有してもよい。
【0161】
さらなる態様において、被験体はネフローゼである。もう一つのさらなる態様において、被験体は非ネフローゼである。被験体は、例えば炎症性疾患、悪性新生物、または慢性感染症のような障害に苦しんでもよい。
【0162】
B.本発明の化合物
一つの態様において、本発明は式(I)の化合物に関する:
式中、Yは各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3、または4であり;mは1または2であり、ただし、mが2の場合、-(CH2)n-基の一つの水素は存在しない。
【0163】
「陽イオン基」という用語には、陽電荷と水素原子とを有する基が含まれる。陽イオンの例には、SO3-またはOSO3-の薬学的に許容される塩が含まれる。陽イオン基の例には、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム、およびアルミニウム等のようなアルカリまたはアルカリ土類金属イオンが含まれる。さらなる態様において、陽イオン基はH+またはNa+である。
【0164】
本発明の化合物の例には、以下の化合物およびその薬学的に許容される塩が含まれる。
1,2-エタンジスルホン酸 HO3SCH2CH2SO3H
1,2-エタンジスルホン酸ナトリウム NaO3SCH2CH2SO3NA
1,3-プロパンジスルホン酸 HO3SCH2CH2CH2SO3H
1,3-プロパンジスルホン酸ナトリウム(1,3-プロパンジスルホン酸、二ナトリウム塩) NaO3SCH2CH2CH2SO3Na
1,2-エタンジオールビス(硫酸水素塩) HO3SOCH2CH2OSO3H
1,2-エタンジオールジスルファート、二ナトリウム塩 NaO3SOCH2CH2OSO3Na
1,3-プロパンジオールビス(硫酸水素塩) HO3SOCH2CH2CH2OSO3H
1,3-プロパンジオールジスルファート、二ナトリウム塩NaO3SOCH2CH2CH2OSO3Na
2-スルホメチル-1,4-ブタンジスルホン酸 HO3SCH2CH2CH(CH2SO3H)2
2-スルホメチルブタン-1,4-ジスルホン酸、三ナトリウム塩 NaO3SCH2CH2CH(CH2SO3Na)2
【0165】
一つの態様において、化合物または抗アミロイド原性物質は、1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸ではない。もう一つの態様において、化合物または抗アミロイド原性物質は、1,3-プロパンジスルホン酸二ナトリウム塩ではない。
【0166】
「化合物」という用語には、化学実体が含まれる。化合物は、固体、液体、または気体様の相に存在してもよい。化合物という用語には、式(I)の化合物およびその薬学的に許容される塩が含まれる。本発明の化合物は、本明細書においてその化学構造および/または化学名によって同定される。化合物が化学構造および化学名の双方によって参照され、その化学構造と化学名が一致しない場合、化学構造が化合物の同一性を決定する。本発明の化合物は、キラル中心を含んでもよく、したがって、立体異性体として存在してもよい。本明細書において定義されるように、化合物は天然起源から精製してもよく、販売元から購入してもよく、または当技術分野において認識された技術を用いて化学合成してもよい。
【0167】
さらに、本発明の化合物はまた、水和型および無水型として存在してもよい。式(I)の化合物の水和型は、式(I)の化合物として含まれる。さらなる態様において、式(I)の化合物は一水和物である。一つの態様において、式(I)の化合物は、水を重量で約10%もしくはそれ未満、約9%もしくはそれ未満、約8%もしくはそれ未満、約7%もしくはそれ未満、約6%もしくはそれ未満、約5%もしくはそれ未満、約4%もしくはそれ未満、約3%もしくはそれ未満、約2%もしくはそれ未満、約1%もしくはそれ未満、約0.5%もしくはそれ未満、約0.1%もしくはそれ未満含む。もう一つの態様において、本発明の化合物は、水を重量で、約0.1%もしくはそれより多く、約0.5%もしくはそれより多く、約1%もしくはそれより多く、約2%もしくはそれより多く、約3%もしくはそれより多く、約4%もしくはそれより多く、約5%もしくはそれより多く、または約6%もしくはそれより多く含む。
【0168】
さらに、本発明の化合物はまた、一つより多い多形型、水和型等を含んでもよい。例えば、一つの型、型Iは本発明の化合物、例えば1,3-プロパンジスルホン酸、二ナトリウム塩の直接再結晶によって調製されうる。化合物は、16:1エタノール:水(v/v)の溶液によって沈殿する。再結晶産物は微細白色粉末として回収され、次にこれを65℃、4 mmHgで16時間乾燥させる。得られた非水和型は、水分含有量0.2%および見かけの密度0.64 g/mlを有する。さらなる態様において、式(I)の化合物は約0.2%の水分含有量を有する。
【0169】
さらに、もう一つの型IIは、型Iと同様の方法で市販の1,3-プロパンジスルホン酸、二ナトリウム塩の直接再結晶により調製されうる。化合物は、8:1エタノール:水(v/v)の溶液から沈殿する。再結晶産物は白色固体として回収され、次にこれを20〜25℃、4 mmHgで16時間乾燥させる。得られた非水和型は、水分含有量約7% w/wおよび見かけの密度0.46 g/mlを有する。さらなる態様において、式(I)の化合物は水分含有量約7%を有する。
【0170】
型Iはまた、減圧下で持続的に加熱することによって、型IIの多形型からも調製されうる。第一に、型IIの多形型(水分含有量6.8%)を4 mm Hgの真空において65℃で16時間乾燥させる。この初回乾燥によって、先の水和多形型の水分含有量は2.3%に低減される。65℃でさらに24時間後、先の一水和多形型の水分含有量は1%に低減される。化合物は77℃でさらに48時間乾燥した後に型I多形型のみに完全に変換される。
【0171】
本発明の化合物は、一つまたは複数の酸性官能基を含み、このように、薬学的に許容される塩基と薬学的に許容される塩を形成することができる。これらの事例における「薬学的に許容される塩」という用語は、本発明の化合物の比較的非毒性の無機および有機塩基付加塩を指す。
【0172】
これらの塩も同様に、物質の最終単離および精製の際にインサイチューで、またはその遊離の酸型の精製物質を、薬学的に許容される金属陽イオンの水酸化物、炭酸塩、もしくは重炭酸塩のような適した塩基、アンモニア、または薬学的に許容される有機1級、2級、もしくは3級アミンと個別に反応させることによって調製されうる。代表的なアルカリまたはアルカリ土類塩には、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム、およびアルミニウム塩等が含まれる。塩基付加塩の形成にとって有用な代表的な有機アミンには、エチルアミン、ジエチルアミン、エチレンジアミン、エタノールアミン、ジエタノールアミン、ピペラジン等が含まれる。
【0173】
「薬学的に許容される塩」にはまた、例えば以下で詳しく記述されるようにおよび本出願において他所で記述されるように、その塩基性塩を作製することによって改変された物質の誘導体が含まれる。薬学的に許容される塩の例には、スルホネートのような酸性残基のアルカリまたは有機塩が含まれる。薬学的に許容される塩には、例えば非毒性の無機または有機酸から形成された親物質の通常の非毒性の塩または4級アンモニウム塩が含まれる。そのような通常の非毒性の塩には、塩酸、臭化水素酸、硫酸、スルファミン酸、リン酸、および硝酸のような無機酸に由来する塩;酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パルモ酸、マレイン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、メシル酸、安息香酸、サリチル酸、スルファニル酸、2-アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、およびイセチオン酸のような有機酸から調製された塩が含まれる。薬学的に許容される塩は、通常の化学法によって塩基性または酸性部分を含む親物質から合成してもよい。一般的に、そのような塩は、これらの物質の遊離の酸または塩基型を、水もしくは有機溶媒、または両者の混合物において、適当な塩基または酸の化学量論的量と反応させることによって調製してもよい。
【0174】
記述の化合物の全ての酸、塩、塩基、ならびに他のイオンおよび非イオン型が本発明の化合物として含まれる。例えば、化合物が本明細書において酸として示される場合、化合物の塩型も同様に含まれる。同様に、化合物が塩として示される場合、酸および/または塩基型も同様に含まれる。
【0175】
さらなる態様において、式(I)の化合物は1,3-プロパンジスルホン酸二ナトリウム塩または1,3-プロパンジスルホン酸ではない。
【0176】
C.本発明の製剤
もう一つの態様において、本発明は、製剤が被験体への投与時に少なくとも一つの都合のよい生物学的特性(FBP)を有するように、製剤において式(I)の化合物の治療的有効量を含む、AAアミロイドーシスを処置するための薬学的製剤に関する。
【0177】
「薬学的製剤」という用語には、以下に記述される薬学的組成物が含まれる。さらなる態様において、薬学的製剤は、本発明の化合物のAAアミロイドーシスおよび/またはアミロイド関連疾患の処置能を増強する、都合のよい生物学的特性を有するように設計される。製剤の都合のよい生物学的特性は、臨床試験の際に被験体に本発明の化合物を投与することによって発見された。
【0178】
「都合のよい生物学的特性」という用語には、本発明の化合物がその意図する機能を行う能力、例えばAAアミロイドーシスおよび/またはアミロイド関連疾患を処置する能力を増強する、本発明の化合物がAAアミロイドーシスを阻害するおよび/またはアミロイド関連疾患を処置する能力以外の生物学的特性が含まれる。一つの態様において、都合のよい生物学的特性は、薬物動態プロフィールとなりうる。用いてもよいそのようなパラメータの例には、Cmax、Css、Tmax、AUC0-t、AUC∞、およびT1/2が含まれるがこれらに限定されるわけではない。これらのパラメータ(Cmax、Tmax、AUC0-t、AUC∞、およびT1/2)は、例えばWinNonlin(登録商標)(Pharsight Corporation, Mountain View, CA)またはWindows(登録商標)用SAS(登録商標)(SAS Institute Inc., Cary, NC)を用いる非コンパートメント分析によって誘導してもよい。さらなる態様において、都合のよい生物学的特性は、標的血漿濃度または標的全身曝露である。
【0179】
「Cmax」という用語は、特定の被験体における本発明の化合物の観察された最高血漿濃度を指す。
【0180】
「Css」という用語は、特定の被験体における本発明の化合物の定常状態血漿濃度を指す。
【0181】
「Tmax」という用語は、Cmaxに達するまでの時間を指す。
【0182】
「AUC0-t」という用語は、線形台形則によって計算した、ゼロ時間から、濃度が定量限界であるまたはそれより上である最後の試料採取時間までの血漿濃度対時間曲線下面積を指す。
【0183】
「AUC∞」という用語は、AUC0-t+(Clast/λz)から計算した、ゼロ時間から無限大までの血漿濃度対時間曲線下面積を指し、Clastは最後に観察された定量可能な濃度であり、λzは見かけの終末速度定数である。
【0184】
「T1/2」という用語は、ln 2λzから計算された見かけの終末半減期である。
【0185】
本発明にはまた、薬物動態パラメータまたはその組み合わせのような、二つまたはそれより多い都合のよい生物学的特性を合わせる製剤および組成物が含まれる。これらの薬物動態パラメータの例には、AUC0-t、AUC∞、Cmax、およびTmaxが含まれる。例えば、本発明の製剤は、健康な被験体(または腎障害を有しない被験体)に投与した場合に、選択された製剤が被験体に一つまたは複数の所望の薬物動態パラメータを提供するように選択されてもよい。または、本発明の製剤は、腎機能障害を有する被験体および/またはAAアミロイドーシスを有する、もしくはAAアミロイドーシスのリスクを有する被験体に投与した場合に、選択された製剤が所望の薬物動態パラメータの一つまたは複数を被験体に提供するように選択されてもよい。
【0186】
さらなる態様において、製剤は実施例7に記述されたとおりではない。もう一つのさらなる態様において、少なくとも一つの成分は、実施例7において記述された成分ではない。
【0187】
もう一つのさらなる態様において、製剤は乳糖一水和物ではない希釈剤を有する。乳糖一水和物ではない希釈剤の例には、例えば糖(例えば、グルコース、蔗糖、果糖等)、デンプン(例えば、コーンスターチ、ジャガイモデンプン等)、セルロース、セルロース誘導体(例えば、カルボキシメチルセルロースナトリウム、エチルセルロース、酢酸セルロース等)、粉末トラガカント、麦芽、ゼラチン、タルク、およびその混合物が含まれる。もう一つのさらなる態様において、希釈剤および/または潤滑剤の吸湿性は、得られたカプセルがFDA規則に許容されるように選択される。
【0188】
もう一つのさらなる態様において、製剤は、ステアリン酸マグネシウムではない潤滑剤を有する。ステアリン酸マグネシウムではない潤滑剤の例には、例えばステアリン酸粉末、タルク、ステアリン酸カルシウム、ポリエチレングリコール、ラウリル硫酸ナトリウム、およびその混合物が含まれる。
【0189】
もう一つの態様において、製剤は、約0.5%(w/w)未満の任意の一つの公知の不純物、約1.5%(w/w)未満の総硫酸塩、約0.1%(w/w)未満の未知の総不純物、および約5.0%(w/w)未満の全不純物を含む。
【0190】
もう一つの態様において、式(I)の化合物は、1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸の参照標準IR(赤外線)スペクトルおよび/またはIC(イオンクロマトグラフィー)に適合する。もう一つの態様において、式(I)の化合物は、約1.0%w/w未満の水を含む。なおもう一つの態様において、式(I)の化合物は、約20 ppm未満の重金属、および/または約0.5%w/w未満の残留溶媒を有する。
【0191】
もう一つの態様において、本発明は、抗アミロイド原性物質含有製剤が、それが生物学的に都合のよい製剤であるように、被験体に投与した場合に少なくとも一つの都合のよい生物学的特性を有することが予め決定された標準的な製剤と同等であるように、製剤中に抗アミロイド原性物質を含む、AAアミロイドーシスを処置するための生物学的に都合のよい製剤に関する。
【0192】
「生物学的に都合のよい製剤」という用語は、少なくとも一つの都合のよい生物学的特性を有する薬学的製剤を指す。さらなる態様において、製剤は、二つもしくはそれより多い、三つもしくはそれより多い、または四つもしくはそれより多い生物学的に都合のよい特性を有する。一つの態様において、生物学的に都合のよい製剤は、本発明の化合物の標的血漿濃度が、化合物の被験体への投与後30分もしくはそれ未満、1時間もしくはそれ未満、2時間もしくはそれ未満、または5時間もしくはそれ未満で被験体において達成されるように製剤される。
【0193】
一つの態様において、生物学的に都合のよい製剤は、特定のAUCssおよび/またはCssを達成するために、被験体のクレアチニンクリアランス速度に基づいて選択される。
【0194】
例えば、都合のよい生物学的特性は、約5000 ng.h/mLもしくはそれより上;約6000 ng.h/mLもしくはそれより上;約6500 ng.h/mLもしくはそれより上;約7000 ng.h/mLもしくはそれより上;約8000 ng.h/mLもしくはそれより上;約9000 ng.h/mLもしくはそれより上;約10,000 ng.h/mLもしくはそれより上;約11,000 ng.h/mLもしくはそれより上;約12,000 ng.h/mLもしくはそれより上;約13,000 ng.h/mLもしくはそれより上;約14,000 ng.h/mLもしくはそれより上;約15,000 ng.h/mLもしくはそれより上;約16,000 ng.h/mLもしくはそれより上;約17,000 ng.h/mLもしくはそれより上;約18,000 ng.h/mLもしくはそれより上;約19,000 ng.h/mLもしくはそれより上;約20,000 ng.h/mLもしくはそれより上;約21,000 ng.h/mLもしくはそれより上;約22,000 ng.h/mLもしくはそれより上;約23,000 ng.h/mLもしくはそれより上;約24,000 ng.h/mLもしくはそれより上;または約25,000 ng.h/mLもしくはそれより上のAUCssであってもよい。もう一つの態様において、都合のよい生物学的特性は、約7000 ng.h/mL; 約8000 ng.h/mLもしくはそれ未満;約9000 ng.h/mLもしくはそれ未満;約10,000 ng.h/mLもしくはそれ未満;約11,000 ng.h/mLもしくはそれ未満;約12,000 ng.h/mLもしくはそれ未満;約13,000 ng.h/mLもしくはそれ未満;約14,000 ng.h/mLもしくはそれ未満;約15,000 ng.h/mLもしくはそれ未満;約16,000 ng.h/mLもしくはそれ未満;約17,000 ng.h/mLもしくはそれ未満;約18,000 ng.h/mLもしくはそれ未満;約19,000 ng.h/mLもしくはそれ未満;約20,000 ng.h/mLもしくはそれ未満;約21,000 ng.h/mLもしくはそれ未満;約22,000 ng.h/mLもしくはそれ未満;約23,000 ng.h/mLもしくはそれ未満;約24,000 ng.h/mLもしくはそれ未満;約25,000 ng.h/mLもしくはそれ未満;または約26,000 ng.h/mLもしくはそれ未満のAUCssである。
【0195】
生物学的に都合のよい特性は、望ましい定常状態濃度(Css)であってもよい。例えば、都合のよい生物学的特性は、約500 ng/mLもしくはそれより上;約600 ng/mLもしくはそれより上;約700 ng/mLもしくはそれより上;約800 ng/mLもしくはそれより上;約900 ng/mLもしくはそれより上;約950もしくはそれより上ng/mL; 約1000 ng/mLもしくはそれより上;約1100 ng/mLもしくはそれより上;または約1200 ng/mLのCssであってもよい。さらに、生物学的に都合のよい特性は、約1200 ng/mLもしくはそれ未満、約1100 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約800 ng/mLもしくはそれ未満;約700 ng/mLもしくはそれ未満;約600 ng/mLもしくはそれ未満;または約500 ng/mL未満のCssであってもよい。
【0196】
もう一つの態様において、都合のよい生物学的特性は、約250〜約2000 ng/mlのCmax(化合物の1回経口投与後の)であってもよい。さらなる態様において、Cmaxは、約250 ng/mlもしくはそれより大きい;約300 ng/mLもしくはそれより大きい;約350 ng/mLもしくはそれより大きい;約400 ng/mLもしくはそれより大きい;約500 ng/mLもしくはそれより大きい;約600 ng/mLもしくはそれより大きい;約700 ng/mLもしくはそれより大きい;約800 ng/mLもしくはそれより大きい;約900 ng/mLもしくはそれより大きい;約1000 ng/mLもしくはそれより大きい;約1100 ng/mLもしくはそれより大きい;約1200 ng/mLもしくはそれより大きい;約1300 ng/mLもしくはそれより大きい;約1400 ng/mLもしくはそれより大きい;約1500 ng/mLもしくはそれより大きい;約1600 ng/mLもしくはそれより大きい;約1700 ng/mLもしくはそれより大きい;約1800 ng/mLもしくはそれより大きい;約1900 ng/mLもしくはそれより大きい;または約2000 ng/mLもしくはそれより大きい。この態様において「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0197】
さらなる態様において、1回経口投与後のCmaxは、約850 ng/mL; 約1700 ng/mLもしくはそれ未満;約1600 ng/mLもしくはそれ未満;約1500 ng/mLもしくはそれ未満;約1400 ng/mLもしくはそれ未満;約1300 ng/mLもしくはそれ未満;約1200 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約800 ng/mLもしくはそれ未満;約700もしくはそれ未満;約700 ng/mLもしくはそれ未満;約500 ng/mLもしくはそれ未満;約400 ng/mLもしくはそれ未満;または約300 ng/mLもしくはそれ未満である。もう一つの態様において、化合物の1回経口投与後のCmaxは、少なくとも200 ng/mLである。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0198】
もう一つの態様において、都合のよい生物学的特性は、約400〜約3800 ng/mlのCmax(化合物の複数回経口投与後の)であってもよい。さらなる態様において、Cmaxは、約400 ng/mLもしくはそれより大きい;約500 ng/mLもしくはそれより大きい;約600 ng/mLもしくはそれより大きい;約700 ng/mLもしくはそれより大きい;約800 ng/mLもしくはそれより大きい;約900 ng/mLもしくはそれより大きい;約1000 ng/mLもしくはそれより大きい;約1100 ng/mLもしくはそれより大きい;約1200 ng/mLもしくはそれより大きい;約1300 ng/mLもしくはそれより大きい;約1400 ng/mLもしくはそれより大きい;約1500 ng/mLもしくはそれより大きい;約1600 ng/mLもしくはそれより大きい;約1700 ng/mLもしくはそれより大きい;約1800 ng/mLもしくはそれより大きい;約1900 ng/mLもしくはそれより大きい;約2000 ng/mLもしくはそれより大きい;約2100 ng/mLもしくはそれより大きい;約2200もしくはそれより大きい;約2300 ng/mLもしくはそれより大きい;約2400 ng/mLもしくはそれより大きい;約2500 ng/mLもしくはそれより大きい;約2600 ng/mLもしくはそれより大きい;約2700 ng/mLもしくはそれより大きい;約2800 ng/mLもしくはそれより大きい;約2900 ng/mLもしくはそれより大きい;約3000 ng/mLもしくはそれより大きい;約3100 ng/mLもしくはそれより大きい;約3200もしくはそれより大きい;約3300 ng/mLもしくはそれより大きい;約3400 ng/mLもしくはそれより大きい;約3500 ng/mLもしくはそれより大きい;約3600 ng/mLもしくはそれより大きい;約3700 ng/mLもしくはそれより大きい;約3800 ng/mLもしくはそれより大きい;または約3900 ng/mLもしくはそれより大きい。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0199】
さらなる態様において、Cmax(化合物の複数回経口投与後の)は、約500〜約3900 ng/mlである。さらなる態様において、Cmaxは、約500 ng/mLもしくはそれ未満約600 ng/mLもしくはそれ未満;約700 ng/mLもしくはそれ未満;約900 ng/mLもしくはそれ未満;約1000 ng/mLもしくはそれ未満;約1100 ng/mLもしくはそれ未満;約1200 ng/mL; 約1300 ng/mLもしくはそれ未満;約1400 ng/mLもしくはそれ未満;約1500 ng/mLもしくはそれ未満;約1600 ng/mLもしくはそれ未満;約1700 ng/mLもしくはそれ未満;約1800 ng/mLもしくはそれ未満;約1900 ng/mLもしくはそれ未満;約2000 ng/mLもしくはそれ未満;約2100 ng/mLもしくはそれ未満;約2200もしくはそれ未満;約2300 ng/mLもしくはそれ未満;約2400 ng/mLもしくはそれ未満;約2500 ng/mLもしくはそれ未満;約2600 ng/mLもしくはそれ未満;約2700 ng/mLもしくはそれ未満;約2800 ng/mLもしくはそれ未満;約2900 ng/mLもしくはそれ未満;約3000 ng/mLもしくはそれ未満;約3100 ng/mLもしくはそれ未満;約3200もしくはそれ未満;約3300 ng/mLもしくはそれ未満;約3400 ng/mLもしくはそれ未満;約3500 ng/mLもしくはそれ未満;約3600 ng/mLもしくはそれ未満;約3700 ng/mLもしくはそれ未満;約3800 ng/mLもしくはそれ未満;または約3900 ng/mLもしくはそれ未満である。この態様において、「約」という用語には、表記の範囲の±50 ng/mlの値が含まれる。
【0200】
例えば、都合のよい生物学的特性は、約2000 ng.h/mLもしくはそれより上;約3000 ng.h/mLもしくはそれより上;約4000 ng.h/mLもしくはそれより上;約5000 ng.h/mLもしくはそれより上;約6000 ng.h/mLもしくはそれより上;約6500 ng.h/mLもしくはそれより上;約7000 ng.h/mLもしくはそれより上;約8000 ng.h/mLもしくはそれより上;約9000 ng.h/mLもしくはそれより上;約10,000 ng.h/mLもしくはそれより上;約11,000 ng.h/mLもしくはそれより上;約12,000 ng.h/mLもしくはそれより上;約13,000 ng.h/mLもしくはそれより上;約14,000 ng.h/mLもしくはそれより上;約15,000 ng.h/mLもしくはそれより上;約16,000 ng.h/mLもしくはそれより上;約17,000 ng.h/mLもしくはそれより上;約18,000 ng.h/mLもしくはそれより上;約19,000 ng.h/mLもしくはそれより上;約20,000 ng.h/mLもしくはそれより上;約21,000 ng.h/mLもしくはそれより上;約22,000 ng.h/mLもしくはそれより上;約23,000 ng.h/mLもしくはそれより上;約24,000 ng.h/mLもしくはそれより上;約25,000 ng.h/mLもしくはそれより上;約26,000 ng.h/mLもしくはそれより上;約27,000 ng.h/mLもしくはそれより上;約28,000 ng.h/mLもしくはそれより上;約29,000 ng.h/mLもしくはそれより上;約30,000 ng.h/mLもしくはそれより上;約31,000 ng.h/mLもしくはそれより上;約32,000 ng.h/mLもしくはそれより上;約33,000 ng.h/mLもしくはそれより上;約34,000 ng.h/mLもしくはそれより上;約35,000 ng.h/mLもしくはそれより上;約36,000 ng.h/mLもしくはそれより上;約37,000 ng.h/mLもしくはそれより上;約38,000 ng.h/mLもしくはそれより上;約39,000 ng.h/mLもしくはそれより上;約40,000 ng.h/mLもしくはそれより上;約41,000 ng.h/mLもしくはそれより上;約42,000 ng.h/mLもしくはそれより上;約43,000 ng.h/mL; 約44,000 ng.h/mLもしくはそれより上;約45,000 ng.h/mLもしくはそれより上;または約46,000 ng.h/mLのAUC∞であってもよい。この態様において、「約」という用語には、表記の範囲の±750 ng/mlの値が含まれる。
【0201】
もう一つの態様において、都合のよい生物学的特性は、約2000 ng.h/ml;約3000 ng.h/mLもしくはそれ未満;約4000 ng.h/mLもしくはそれ未満;約5000 ng.h/mLもしくはそれ未満;約5000 ng.h/mLもしくはそれ未満;約6000 ng.h/mLもしくはそれ未満;約6500 ng.h/mLもしくはそれ未満;約7000 ng.h/mLもしくはそれ未満;約8000 ng.h/mLもしくはそれ未満;約9000 ng.h/mLもしくはそれ未満;約10,000 ng.h/mLもしくはそれ未満;約11,000 ng.h/mLもしくはそれ未満;約12,000 ng.h/mLもしくはそれ未満;約13,000 ng.h/mLもしくはそれ未満;約14,000 ng.h/mLもしくはそれ未満;約15,000 ng.h/mLもしくはそれ未満;約16,000 ng.h/mLもしくはそれ未満;約17,000 ng.h/mLもしくはそれ未満;約18,000 ng.h/mLもしくはそれ未満;約19,000 ng.h/mLもしくはそれ未満;約20,000 ng.h/mLもしくはそれ未満;約21,000 ng.h/mLもしくはそれ未満;約22,000 ng.h/mLもしくはそれ未満;約23,000 ng.h/mLもしくはそれ未満;約24,000 ng.h/mLもしくはそれ未満;約25,000 ng.h/mLもしくはそれ未満;約26,000 ng.h/mLもしくはそれ未満;約27,000 ng.h/mLもしくはそれ未満;約28,000 ng.h/mLもしくはそれ未満;約29,000 ng.h/mLもしくはそれ未満;約30,000 ng.h/mLもしくはそれ未満;約31,000 ng.h/mLもしくはそれ未満;約32,000 ng.h/mLもしくはそれ未満;約33,000 ng.h/mLもしくはそれ未満;約34,000 ng.h/mLもしくはそれ未満;約35,000 ng.h/mLもしくはそれ未満;約36,000 ng.h/mLもしくはそれ未満;約37,000 ng.h/mLもしくはそれ未満;約38,000 ng.h/mLもしくはそれ未満;約39,000 ng.h/mLもしくはそれ未満;約40,000 ng.h/mLもしくはそれ未満;約41,000 ng.h/mLもしくはそれ未満;約42,000 ng.h/mLもしくはそれ未満;約43,000 ng.h/mLもしくはそれ未満;約44,000 ng.h/mLもしくはそれ未満;約45,000 ng.h/mLもしくはそれ未満;または約46,000 ng.h/mLもしくはそれ未満のAUC∞である。この態様において、「約」という用語には、表記の範囲の±750 ng.h/mlの値が含まれる。
【0202】
なおもう一つの態様において、本発明は、少なくとも部分的に、薬学的製剤に関する。薬学的製剤は、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、製剤を健康な被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される。
【0203】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、健康被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される薬学的製剤にも関する。
【0204】
なおもう一つのさらなる態様において、本発明はまた、AAアミロイドーシスを処置または予防するために有効な量の活性物質(例えば、PDS)と、薬学的に許容される担体とを含み、健康被験体に経口投与する場合、平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される薬学的製剤に関する。
【0205】
なおもう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と、薬学的に許容される担体とを含む薬学的製剤であって、AAアミロイドーシスを有する被験体に経口投与する場合、活性物質の用量400 mgを投与すると、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量800 mgを投与すると、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量1200 mgを投与すると、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0206】
なおもう一つのさらなる態様において、本発明は、活性物質(例えば、PDS)800 mgと薬学的に許容される担体とを含む薬学的製剤であって、製剤を被験体に投与する場合、被験体が健康である場合、平均AUC∞約4,000〜6,000 ng.h/ml±20%、および平均Cmax約1,200〜1,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が軽度の腎障害を有する場合、平均AUC∞約12,000〜14,000 ng.h/ml±20%、および平均Cmax約2,500〜3,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、被験体が中等度の腎障害を有する場合、平均AUC∞約9,000〜11,000 ng.h/ml±20%、および平均Cmax約2,000〜2,200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または被験体が重度の腎障害を有する場合、平均AUC∞約40,000〜46,000 ng.h/ml±20%、および平均Cmax約2,100〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0207】
なおもう一つのさらなる態様において、本発明はまた、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、製剤をAAアミロイドーシスを有する被験体に24ヶ月間経口投与する場合、活性物質の用量が400 mgの場合、平均AUC∞約25,000〜26,000 ng.h/ml±20%、および平均Cmax約2,000〜2,300 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;活性物質の用量が800 mgの場合、平均AUC∞約20,000〜22,000 ng.h/ml±20%、および平均Cmax約1,600〜2,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ;または活性物質の用量が1200 mgの場合、平均AUC∞約8,000〜10,000 ng.h/ml±20%、および平均Cmax約800〜1,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0208】
もう一つのさらなる態様において、本発明は、少なくとも部分的に、活性物質(例えば、PDS)と薬学的に許容される担体とを含む薬学的製剤であって、製剤を健康な男性被験体に7日間経口投与する場合、活性物質400 mgを1日4回投与すると、平均AUC∞約10,000〜11,500 ng.h/ml±20%、および平均Cmax約900〜1100 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質800 mgを1日4回投与すると、平均AUC∞約19,000〜21,000 ng.h/ml±20%、および平均Cmax約1,600〜1,800 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、活性物質1600 mgを1日3回投与すると、平均AUC∞約25,000〜27,000 ng.h/ml±20%、および平均Cmax約4,000〜6,000 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られ、または活性物質1600 mgを1日4回投与すると、平均AUC∞約23,000〜25,500 ng.h/ml±20%、および平均Cmax約4,500〜6,500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが得られる、薬学的製剤に関する。
【0209】
「同等」という用語は、本発明の製剤と生物学的に同等または機能的に同等であると見なされる製剤を指す。さらなる態様において、製剤は、1,3-プロパンジスルホン酸二ナトリウム塩または1,3-プロパンジスルホン酸を含む製剤と同等である。さらなる態様において、用語には、21 U.S.C.§255および/または21 CFR§320の下で生物学的同等性に関する定義を満たす製剤が含まれる。もう一つの態様において、「同等」は、被験体に投与した場合に、例えば本明細書において開示した値の±10%、±20%、±30%、または±40%以内である薬物動態パラメータ、例えばCmax、AUC∞が得られる製剤を指す。
【0210】
さらなる態様において、「同等」という用語には、類似の実験条件で投与した場合に、同等である可能性がある製剤の吸収速度および程度が、標準的な製剤の吸収速度および程度と有意差を示さない、例えば±10%、±20%、±30%、または±40%以内である、同等である可能性がある製剤が含まれる。もう一つの態様において、おそらく同等である製剤の吸収の程度は、類似の実験条件で投与した場合に、同等である可能性がある製剤の吸収速度および程度が、標準的な製剤の吸収速度および程度と有意差を示さない、例えば±10%、±20%、±30%、または±40%以内であり、標準製剤からの差は、慢性的な使用の際に有効な体薬物濃度を達成するために本質的ではなく、および/または薬物にとって医学的に重要ではないと見なされる。一つの局面において、同じモル用量を投与してもよい。
【0211】
「標準製剤」という用語は、式(I)の化合物、例えば少なくとも一つの都合のよい生物学的特性を有する1,3-プロパンジスルホン酸二ナトリウム塩、または1,3-プロパンジスルホン酸の製剤を指す。他の態様において、標準製剤は、薬物製剤の仕様書の必要条件を満たす、またはそれを超える。さらなる態様において、標準製剤は実施例7に記載される製剤である。
【0212】
「薬物製剤の仕様書」という用語は、米国FDAの薬物製剤仕様書を満たす製剤を指す。さらなる態様において、製剤は、多くて約0.5%w/wの任意の一つの公知の不純物、多くて約1.5%w/wの総硫酸塩、多くて0.1%w/wの任意の一つの未知不純物、および多くて約5.0%w/wの総不純物を含む。
【0213】
「抗アミロイド原性物質」は、AAアミロイド結合アッセイ(AAABA)において試験陽性である化合物を指す。さらなる態様において、抗アミロイド原性物質は、AAABAにおいて測定した場合AAアミロイド濃度400μMで30%もしくはそれより大きい、45%もしくはそれより大きい、60%もしくはそれより大きい、70%もしくはそれより大きい、80%もしくはそれより大きい、または90%もしくはそれより大きい割合でAAに結合する。もう一つの態様において、抗アミロイド原性化合物は、AAABAにおいて測定した場合AAアミロイド濃度200μMで30%もしくはそれより大きい、45%もしくはそれより大きい、60%もしくはそれより大きい、70%もしくはそれより大きい、80%もしくはそれより大きい、または90%もしくはそれより大きい割合でAAに結合する。
【0214】
「抗アミロイド原性物質」という用語にはまた、ジスルホン化化合物およびスルホン化アルキル化合物が含まれる。さらなる態様において、この用語には、1,3-プロパンジスルホン酸の類似体またはその薬学的に許容される塩である化合物が含まれる。
【0215】
さらなる態様において、抗アミロイド原性物質は、先に記述したように式(I)の化合物である。抗アミロイド原性物質の例には、1,2-エタンジスルホン酸、1,2-エタンジスルホン酸ナトリウム、1,2-エタンジオールビス(硫酸水素塩)、1,2-エタンジオールジスルファート二ナトリウム塩、1,3-プロパンジオールビス(硫酸水素塩)、1,3-プロパンジオールジスルファート二ナトリウム塩、2-スルホメチル-1,4-ブタンジスルホン酸、または2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩のような化合物が含まれる。さらなる態様において、抗アミロイド原性物質は、1,3-プロパンジスルホン酸または1,3-プロパンジスルホン酸二ナトリウム塩である。
【0216】
AAアミロイド結合アッセイ(AAABA)
AAに関するMSアッセイにおいて、試料を水溶液(必要であれば水に溶解するために20%エタノールを加える)、200μM試験化合物および20μM可溶化AA、または400μM試験化合物および40μM可溶化AAとして調製する。各試料のpH値を、0.1%水酸化ナトリウム水溶液を加えることによって7.4(±0.2)に調節する。次に、溶液を、Waters ZQ 4000質量分析計を用いて、エレクトロスプレーイオン化質量分析計によって分析する。試料を、試料調製後2時間以内に流速25μL/分で直接注入によって導入する。全ての分析に関して、ソース温度を70℃に維持して、コーン電圧は20 Vである。データはMasslynx 3.5ソフトウェアを用いて処理する。MSアッセイは、可溶性AAに対する化合物の結合能に基づくデータを生じる。
【0217】
D.本発明の化合物を含む薬学的組成物
本発明はまた、少なくとも部分的に、式(I)の化合物の治療的有効量と第二の物質とを含む薬学的組成物に関する。さらなる態様において、治療的有効量はAAアミロイドーシスを処置するために有効である。
【0218】
さらなる態様において、本発明は、包装された薬学的組成物に関する。包装された薬学的組成物には、組成物を第二の物質と併用して投与することを勧める表示または添付文書と共に包装された式(I)の化合物の治療的有効量が含まれる。さらなる態様において、治療的有効量はAAアミロイドーシスを処置するために有効である。
【0219】
なおもう一つのさらなる態様において、本発明は、組成物を式(I)の化合物と併用して投与することを勧める表示または添付文書と共に包装された第二の物質の治療的有効量を含む、包装された薬学的組成物に関する。
【0220】
「表示または添付文書」という用語には、被験体、または本発明の組成物の投与に関して被験体の医療に実質的に責任を有する任意の人との全ての書面、電子的、または会話による伝達が含まれるがこれらに限定されるわけではない。添付文書にはさらに、本発明の組成物を他の化合物または組成物、例えば第二の物質と同時投与することに関する情報が含まれる。さらに、添付文書には、食物なしで本発明の組成物の投与に関する説明書が含まれてもよい。
【0221】
なおもう一つの態様において、本発明は、組成物を食物なしで投与することを勧める表示または添付文書と共に、式(I)の化合物の治療的有効量を含む薬学的組成物を保持する容器が含まれる、包装された薬学的組成物に関する。
【0222】
式(I)の化合物は、適当な溶媒による溶液で、または溶媒を含まない型(例えば、凍結乾燥型)で供給されてもよい。本発明のもう一つの局面において、物質および本発明の方法を行うために必要な物質および緩衝液はキットとして包装されてもよい。キットは、本明細書に記述の方法に従って商業的に用いられてもよく、本発明の方法において用いるための説明書が含まれてもよい。さらなるキットの成分には、酸、塩基、緩衝剤、無機塩、溶媒、抗酸化剤、保存剤、または金属キレート剤が含まれてもよい。さらなるキットの成分は純粋な組成物として、または一つもしくは複数のさらなるキット成分を組み入れる水溶液もしくは有機溶液として存在する。任意のまたは全てのキット成分は任意でさらに緩衝液を含む。
【0223】
式(I)の化合物はまた、非経口、腹腔内、脊柱内、または脳内に投与してもよい。グリセロール、液体ポリエチレングリコール、およびその混合物において、ならびに油において分散剤を調製することができる。通常の保存および使用条件において、これらの調製物は、微生物の成長を予防するために保存剤を含んでもよい。
【0224】
本発明の化合物を非経口投与以外で投与するために、その不活化を予防するための材料によって物質をコーティングする、またはその材料を物質と同時投与する必要がある可能性がある。例えば、本発明の化合物は、適当な担体、例えばリポソーム、または希釈剤において被験体に投与してもよい。薬学的に許容される希釈剤には、生理食塩液および水性緩衝液が含まれる。リポソームには、水中油中水型CGF乳剤と共に通常のリポソームが含まれる(Strejan et al., J. Neuroimmunol. 7, 27 (1984))。「薬学的組成物」という用語には、先に記述した「薬学的製剤」が含まれることに注意すべきである。
【0225】
注射での使用に適した薬学的組成物には、滅菌水溶液(水溶性の場合)または分散液、および滅菌注射用溶液または分散液を即時調製するための滅菌粉末が含まれる。全ての場合において、組成物は無菌的でなければならず、容易なシリンジ作動性が存在する程度に流動性でなければならない。これは製造および保存条件で安定でなければならず、細菌および真菌のような微生物の混入作用に対して保護されなければならない。
【0226】
適した薬学的に許容される溶媒には、リン酸緩衝生理食塩液(PBS)のような、経口、非経口、鼻腔内、粘膜、経皮、血管内(IV)、動脈内(IA)、筋肉内(IM)、および皮下(SC)投与経路にとって適した任意の非免疫原性の薬学的補助剤が含まれるがこれらに限定されるわけではない。
【0227】
媒体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコール等)、その適した混合物、および植物油を含む溶媒または分散培地となりうる。例えばレシチンのようなコーティングを用いることによって、分散液の場合には必要な粒子径を維持することによって、および界面活性剤を用いることによって、適切な流動性を維持することができる。微生物の作用の予防は、様々な抗菌および抗真菌剤、例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサル等によって得ることができる。多くの場合において、組成物には、等張剤、例えば糖、塩化ナトリウム、またはマンニトールおよびソルビトールのような多価アルコールが含まれる。注射可能な組成物の持続的な吸収は、吸収を遅らせる物質、例えばモノステアリン酸アルミニウムまたはゼラチンを組成物に含めることによってもたらされうる。
【0228】
滅菌注射液は、先に列挙した成分の一つまたは組み合わせと共に適当な溶媒において必要量の治療物質を組み入れて、必要に応じてその後濾過滅菌することによって調製されうる。一般的に、分散液は、基本分散培地と、先に列挙した中からの必要な他の成分とを含む滅菌媒体に治療物質を組み入れることによって調製される。滅菌注射液を調製するための滅菌粉末の場合、調製法は、活性成分(すなわち本発明の化合物)プラス予め濾過滅菌したその溶液からの任意のさらなる所望の成分の粉末を生じる真空乾燥および凍結乾燥である。
【0229】
本発明の化合物は、例えば不活性希釈剤または同化可能な食用担体と共に経口投与されうる。本発明の化合物および他の成分はまた、硬または軟シェルゼラチンカプセルに封入してもよく、錠剤に圧縮してもよく、または被験体の食事に直接組み入れてもよい。経口治療的投与の場合、本発明の化合物は、賦形剤と共に組み入れて、摂取可能な錠剤、口腔内錠剤、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、ウェーハ等の剤形で用いてもよい。組成物および調製物における本発明の化合物の百分率は、当然変化してもよい。そのような治療的に有用な組成物における本発明の化合物の量は、適した用量が得られるような量である。
【0230】
したがって、本発明には、エアロゾル、経口および非経口投与のための薬学的に許容される媒体において、その薬学的に許容される塩を含む、本発明の化合物を含む薬学的製剤が含まれる。同様に、本発明には、静脈内、筋肉内、または皮下注射によるような、凍結乾燥されて、投与のための薬学的に許容される製剤を形成するように再構成されてもよい、化合物またはその塩が含まれる。投与はまた、皮内または経皮であってもよい。
【0231】
本発明に従って、本明細書において記述される式(I)の物質、およびその薬学的に許容される塩は、固体として経口もしくは吸入によって投与されてもよく、または溶液、懸濁液、もしくは乳液として筋肉内もしくは静脈内に投与されてもよい。または、物質または塩はまた、リポソーム懸濁液として吸入、静脈内、または筋肉内に投与されてもよい。
【0232】
吸入によって、エアロゾルとしての投与に適した薬学的組成物または製剤も同様に提供される。これらの製剤は、本発明の所望の化合物もしくはその塩、または物質もしくは塩の複数の固体粒子の溶液もしくは懸濁液を含む。所望の製剤を、小さいチャンバーに入れて噴霧してもよい。噴霧は、物質または塩を含む複数の液体小滴または固体粒子を形成するために圧縮空気または超音波エネルギーによって行ってもよい。液体小滴または固体粒子は、約0.5〜約5ミクロンの粒子径を有しなければならない。固体粒子は、本発明の任意の化合物またはその塩の固体物質を、微粉化のような当技術分野で公知の適当な方法で処理することによって得ることができる。固体粒子または小滴の大きさは、例えば約1〜約2ミクロンであろう。この局面において、この目的を達成するために市販のネブライザーを利用することができる。
【0233】
エアロゾルとしての投与に適した薬学的製剤は、液体剤形であってもよく、製剤は、水を含む担体において、本発明の化合物またはその塩の水溶性物質を含むであろう。噴霧に供した場合に所望の大きさの範囲内の小滴が形成されるために十分に製剤の表面張力を低下させる界面活性剤が存在してもよい。
【0234】
経口組成物にはまた、液体溶液、乳剤、懸濁液等が含まれる。そのような組成物の調製にとって適した薬学的に許容される媒体は、当技術分野で周知である。シロップ、エリキシル剤、乳剤および懸濁剤にとって典型的な担体成分には、エタノール、グリセロール、プロピレングリコール、ポリエチレングリコール、液体蔗糖、ソルビトール、および水が含まれる。懸濁液の場合、典型的な懸濁剤には、メチルセルロース、カルボキシメチルセルロースナトリウム、トラガカント、およびアルギン酸ナトリウムが含まれ;典型的な湿潤剤には、レシチンおよびポリソルベート80が含まれ;ならびに典型的な保存剤には、メチルパラベンおよび安息香酸ナトリウムが含まれる。経口液体組成物はまた、先に開示した甘味料、着香料、および着色剤のような一つまたは複数の成分を含んでもよい。
【0235】
薬学的組成物はまた、被験物質が所望の局所適用の近傍で、または望ましい作用を持続させるために様々な時間、胃腸管において放出されるように、通常の方法によって、典型的にpHまたは時間依存的コーティングによってコーティングしてもよい。そのような投与剤形には典型的に、酢酸フタル酸セルロース、ポリビニル酢酸フタレート、ヒドロキシプロピルメチルセルロースフタレート、エチルセルロース、ロウおよびシェラックの一つまたは複数が含まれるがこれらに限定されるわけではない。
【0236】
被験物質の全身送達を達成するために有用な他の組成物には、舌下、口腔内、および鼻腔内剤形が含まれる。そのような組成物は典型的に、蔗糖、ソルビトール、およびマンニトールのような可溶性増量物質;ならびにアカシア、微結晶セルロース、カルボキシメチルセルロース、およびヒドロキシプロピルメチルセルロースのような結合材の一つまたは複数を含む。先に開示したグライダント、潤滑剤、甘味料、着色料、抗酸化剤、および着香料もまた含まれてもよい。
【0237】
本発明の組成物はまた、被験体の表皮もしくは上皮組織上に組成物を直接載せるもしくは広げることによって、または「パッチ」によって経皮的に被験体に局所投与されうる。そのような組成物には、例えば、ローション、クリーム、溶液、ゲル、および固体が含まれる。これらの局所組成物は本発明の化合物の有効量、通常少なくとも約0.1%、または約1%〜約5%さえ含んでもよい。局所投与のための適した担体は、典型的に、持続的な被膜として皮膚に留まっていて、発汗または水にぬれることによっても除去されない。一般的に、担体は本質的に有機性であり、その中に治療物質を分散または溶解することができる。担体には、薬学的に許容される皮膚軟化剤、乳化剤、濃化剤、溶媒等が含まれてもよい。
【0238】
そのような物質の毒性および治療効率は、例えばLD50(集団の50%に対して致死的な用量)およびED50(集団の50%において治療的に有効な用量)を決定するための細胞培養または実験動物における標準的な薬学的技法によって決定されうる。毒性効果と治療効果の用量比が治療指数であり、比LD50/ED50として表記することができ、通常、より大きい治療指数がより有効である。毒性の副作用を示す化合物を用いてもよいが、非罹患細胞に対する起こりうる障害を最小限にして、それによって副作用を低減するために、罹患組織部位にそのような物質を標的化する送達系を設計するように注意しなければならい。
【0239】
適当な用量は、当業者の医師、獣医師、または研究者に理解できる多数の要因に依存すると理解される。低分子の用量は、例えば被験体または処置される試料の同一性、大きさ、および状態に応じて、さらに適用可能であれば組成物が投与される経路、および被験体に及ぼしてほしいと医師が願う低分子の効果に応じて、変化するであろう。例としての用量には、被験体または試料の重量1 kgあたり低分子のミリグラムまたはマイクログラム量が含まれる(例えば、約1μg/kg〜約500 mg/kg、約100μg/kg〜約5 mg/kg、または約1μg/kg〜約50μg/kg)。適当な用量は効力に依存するとさらに理解される。そのような適当な用量は、当技術分野で公知のアッセイを用いて決定してもよい。これらの化合物の一つまたは複数を動物(例えば、ヒト)に投与する場合、医師、獣医師、または研究者は、例えば比較的低用量を最初に製剤した後、適当な反応が得られるまで用量を増加させてもよい。さらに、任意の特定の動物被験体に関する特異的用量レベルは、用いる特異的化合物の活性、被験体の年齢、体重、全身健康、性別、および食事、投与期間、投与経路、排泄速度、および任意の薬物併用を含む多数の要因に依存するであろうと理解される。
【0240】
AAアミロイドーシスまたは腎障害を有する被験体に関して、用量は、例えば被験体からの化合物のクリアランス速度に影響を及ぼす可能性があるクレアチニンクリアランス速度によって測定した被験体における腎機能の状態に依存してもよい。この場合、より低いクレアチニンクリアランス速度を有する被験体は、より高いクレアチニンクリアランス速度を有する被験体より、低い用量で特定の血漿濃度に達すると予想されるであろう。
【0241】
非経口組成物は、投与の容易さおよび用量の均一性のために単位用量で製剤されてもよい。本明細書において用いられるように、単位投与剤形は、処置される被験体に関する単位用量として適した物理的に個別の単位を指す;各単位は必要な薬学的媒体に関連して所望の治療効果を生じるように計算された本発明の化合物の既定量を含む。本発明の単位投与剤形の明細は、(a)治療物質の独自の特徴および達成される特定の治療効果、および(b)AAアミロイドーシスまたはアミロイド関連疾患の処置のために本発明のそのような化合物を合成する技術分野における固有の制限、によって指図され、直接依存する。
【0242】
例えば、式(I)の化合物の治療的有効量は約100〜2500 mg毎日であってもよい。本発明の化合物は、本発明の化合物の用量200 mg、400 mg、または800 mgを有するカプセルにおいて製造されてもよい。または、本発明の化合物は、用量400 mgで1日2回、800 mgで1日2回、または1200 mgで1日2回投与されてもよい。
【0243】
当業者は、本明細書に記述の特異的技法、態様、特許請求の範囲、および実施例に対する多数の同等物を単なる日常的な実験を用いて認識し、または確認することができるであろう。そのような同等物は本発明の範囲内であると見なされ、本明細書に添付される特許請求の範囲に含まれる。本出願を通して引用した全ての参考文献、交付された特許、および公開された特許出願は、参照により本明細書に組み入れられる。本発明はさらに、さらに制限的であると解釈されてはならない以下の実施例によって説明される。
【実施例】
【0244】
本発明の具体例
実施例1
様々な程度の腎障害を有する被験体における1,3-プロパンジスルホン酸二ナトリウム塩の1回経口用量800 mgの薬物動態プロフィールを、健康ボランティアと比較して査定するために、オープンラベルの非無作為平行群試験を実施した。血液および尿試料を投与後24時間採取した。1,3-プロパンジスルホン酸の血漿および尿中濃度を、確認されたHPLC法を用いて決定する。全体として、腎障害は、健康被験体と比較して、より低い腎クリアランスおよびPDSに対するより大きい全身曝露(AUCおよびCmaxによって特徴付けられる)に関連する。その結果、用量の減少は、腎機能障害を有する患者において許容される全身曝露を維持するために必要であるように思われる。結果を表1に示す。
【0245】
(表1)
値は平均値であり、括弧内に範囲を示す(個々の患者の最小値および最大値)、ただしTmaxでは中央値を表し、括弧内に範囲を表す。
1 インスリンクリアランスによって決定。
2 消失速度定数を推定することができる被験体に限って決定
【0246】
実施例2
二次性(AA)アミロイドーシスを有する被験体における1,3-プロパンジスルホン酸二ナトリウム塩(PDS)の有効性および安全性を査定するための多施設多国間無作為二重盲験プラセボ対照平行デザイン試験を実施する。全体で被験体183人を無作為化して、PDSまたはプラセボを1日2回24ヶ月間投与する。投与は被験体における腎障害の重症度に依存する:クレアチニンクリアランス(ClCr)>80 ml/分を有する被験体には1200 mgを1日2回;ClCrが30〜80 ml/分の場合、被験体には800 mgを1日2回投与し;およびClCrが20〜30 ml/分であって、おそらく透析を開始する前の場合、被験体には400 mgを1日2回投与する。ClCrが次のより低い、またはより高い範囲のレベルまで減少または増加する場合、投与レジメをそれに従って調節する。被験体の投薬は、経口投与である(カプセル剤)。各患者を0、1、4、8、12、16、20、および24ヶ月目での現場での診察を含め、16回評価する。
【0247】
血清クレアチニンおよび体表面積に関して標準化したクレアチニンクリアランスのベースラインからの変化を、試験を通して査定する(スクリーニング、ベースライン、4、8、12、16、20および24ヶ月での診察)。被験体に、予定診察の前日に指示した通り24時間尿を採取するように要請する。尿の容積を、試験施設での症例報告書に記録して、尿の少量を尿中クレアチニンの決定のために中心となる研究所に送る。血液試料を採取して血清クレアチニンを測定する。以下の式を用いてクレアチニンクリアランス(ClCr)を計算して症例報告書に記録する。
【0248】
体表面積に関して標準化したクレアチニンクリアランスは、クレアチニンクリアランスを体表面積によって除することによって計算される。体表面積は以下の等式によって計算される:Wt0.425×Ht0.725×0.007184(DuBois D, Clinical Calorimetry, Arch Intern Med 1916; 17:87)。
【0249】
24ヶ月間の処置期間終了時に、ベースラインと比較した腎機能の複合査定に基づいて被験体を3つの範疇に分類する。被験体は、以下の悪化の臨床指標の少なくとも一つを満たす場合「悪化」であると分類される:クレアチニンクリアランスの50%低減、血清クレアチニンレベルの倍加、ESRD/透析への進行、または死亡。被験体は、以下の改善の臨床指標を満たす場合「改善」であると分類される:ClCrの>50%増加、および悪化に関する臨床指標を認めない。被験体は悪化または改善に関する臨床指標のいずれも満たさない場合「安定」であると分類される。この基準を用いて、PDSを投与された13.4%より少ない被験体がプラセボを投与された被験体と比較して悪化して、PDSを投与された13.4%より多い被験体が、プラセボと比較して安定または改善である(p値=0.06)。
【0250】
実施例3
有効性の分析
全ての統計分析は記述の被験体集団に基づくであろう。「安全な集団」は、試験薬(PDS)の少なくとも1回用量を服用した処置群に無作為化された全ての被験体の組であろう。スクリーニング期間の際に無作為化されたが、ベースラインで参加基準の一つを満たさず、したがってPDSを服用しなかった被験体は安全集団には含まれないであろう。「Intent-to-Treat(ITT)集団」は、安全集団、すなわち、無作為化されてPDSの任意の量を服用した全ての被験体からなるであろう。「有効性評価可能な(プロトコールあたり;PP)集団」は、ITT被験体のサブセットであろう。これは処置期間を終了し、一次有効性エンドポイントに関して査定されうる全ての被験体からなるであろう。以下の状況下では被験体は除外されるであろう:試験中のアンジオテンシン変換酵素(ACE)阻害剤またはアンジオテンシンII受容体拮抗剤治療の開始;救済薬の使用;ESRD/透析への進行または死亡以外の理由による中止;主なプロトコール違反;および低いコンプライアンス率。
【0251】
一次有効性エンドポイント
被験体は全て、その疾患の進行状況に従って分類される(「悪化」、「安定」、または「改善」)。これは試験の一次有効性エンドポイントを構成し、二つの予め明記された統計学的方法論に従ってIntent-to-Treat(ITT)集団において分析する:1)ベースラインの状態と比較した、試験終了時で各範疇に達する被験体の割合を比較するためのコクラン-マンテル-ヘンツェル行平均スコア(row mean score)検定(CMH検定)、および2)第一の「悪化」事象の数と共にそれらが起こった時期の双方を処置群のあいだで比較して、このように利用可能な全てのデータをより利用するための比例ハザード回帰モデル(Cox分析)。いずれの方法論も予め明記されたベースラインでのネフローゼの状態(ネフローゼ対非ネフローゼ)の階層化に関して調節される。
【0252】
CMH行平均スコア試験を用いる一次エンドポイントの場合、試験終了時にこの複合査定の各範疇に達する患者の数および百分率のような、要約統計値を処置群毎に表す。反応レベルは、必ずしも等しい間隔で観察される必要はないが、明確な順序を有し、改変riditスコア選択肢を用いる。この採点法は、その相対的順序によって暗示される以外の反応レベルのスケーリングを必要としない。CMH検定の統計値は、母集団の大きさと比較した試験施設の数が大きい(27)ために施設毎に階層化されない。したがって、施設の効果は、記述的統計法のみを用いて調べられる。国毎の効果も同様に記述的に調べられる。
【0253】
特定の細胞における母集団の大きさが任意の統計検定の必要条件にとって小さすぎる場合、複合変数の「改善」および「安定」範疇をなくして、「悪化」対「安定または改善」転帰を作製することが予め明記される。これはサブグループ分析または他の診査的分析にも当てはまる。
【0254】
Cox比例有害回帰モデルを用いる一次エンドポイントの場合、処置と複合エンドポイントとの関連を、いくつかのベースライン変数に関して調節することができるが、CMH検定方法論との比較を可能にするために、ベースラインでのネフローゼ状態(ネフローゼ対非ネフローゼ)のみを、調節のために最初に用いる。Coxモデルを用いる他の分析に含まれうるであろう他の変数は:処置群、年齢、ベースラインCrCl、ベースラインタンパク尿、ベースラインSCr、および基礎疾患(家族性地中海熱、リウマチ性炎症疾患、およびその他)である。
【0255】
一次有効性エンドポイントは、腎機能の臨床的改善/悪化の複合査定である。試験終了時、被験体を三つの範疇、すなわち「悪化」および死亡(全ての原因の)、「安定」、または「改善」に分類する。悪化に関する以下の臨床指標の少なくとも一つを満たす場合、被験体は「悪化」であると分類される:ベースラインから24ヶ月までの血清クレアチニンの倍加;ベースラインから試験終了時までの体表面積に関して標準化したクレアチニンクリアランスの50%もしくはそれより大きい減少;透析/ESRDへの進行;または死亡。悪化または改善に関する臨床指標のいずれも満たさない場合、被験体は「安定」であると分類される。改善に関する以下の臨床指標を満たす場合、被験体は「改善」であると分類される:ベースラインから試験終了時までの体表面積に関して標準化したクレアチニンクリアランスの50%もしくはそれより大きい増加;および悪化に関する臨床指標なし。「試験終了時」という用語は、試験を終了した被験体の場合には24ヶ月目、および早期に試験を中止した被験体の場合には最後に利用可能な測定として定義される。
【0256】
試験終了時、腎機能低下または全ての原因の死亡率に関する任意の「悪化」事象のリスクがプラセボのリスクの42%に低減されることが見いだされる。これは、処置群における「悪化」患者の数がより少ない結果と一致する。さらに、第一の「悪化」事象までの期間の中央値は、PDSによって処置した被験体では6.4ヶ月長い(処置群に関して14.5ヶ月対プラセボ群に関して8.1ヶ月)。第一の「悪化」事象までの時間に関するカプラン-マイヤー曲線を描写するグラフを図1に示す。
【0257】
二次有効性エンドポイント
二次有効性エンドポイントは、腎機能と胃腸管機能の双方の臨床的改善/悪化の査定である。試験終了時、被験体はその病態に基づいて三つの範疇に分類される:悪化、安定、または改善。二次有効性エンドポイントの例には、1)体表面積に関して標準化したクレアチニンクリアランスの経時的勾配;2)血清クレアチニンの逆数(1/血清クレアチニン)の経時的勾配;3)腎事象までの時間(例えば、血清クレアチニンの倍加までの時間;体表面積に対して標準化したクレアチニンクリアランスの>50%増加までの時間;体表面積に対して標準化したクレアチニンクリアランスの>50%減少までの時間;透析/ESRDまでの時間;および/または死亡までの時間);および4)ベースラインから試験終了時までのタンパク尿の変化および体表面積に関して標準化したクレアチニンクリアランスの変化、が含まれる。
【0258】
定量的二次有効性パラメータに関して、観察数(n)、平均値、SD、中央値、最小値、および最大値のような要約統計値を、各評価診察時に処置群毎に示す。実際の値およびベースラインからの百分率変化も同様に、各評価診察時に処置群毎に示す。その上、処置群を二元共分散分析(ANCOVA)を用いて比較して、ベースラインおよびベースラインでの腎状態に関して調節する。
【0259】
試験される最初の完全なモデルは以下の通りであろう:
ベースラインからの変化=処置+ベースラインの値+ベースラインでの腎状態+(ベースラインでの腎状態*処置)+(ベースラインの値*処置)
式中、
処置=PDS対プラセボ
ベースライン値=試験パラメータに関する被験体のベースライン値
ベースラインでの腎状態=ネフローゼ対非ネフローゼ
ベースラインでの腎状態*処置=処置によるベースラインでの腎状態に関する交互作用の項
ベースライン値*処置=処置毎のベースライン値に関する交互作用の項
【0260】
二つの交互作用の項は、そのそれぞれの有意性に関する試験の結果、10%より大きいp値が得られれば、一次モデルから脱落するであろう。次に、得られたモデルを用いて処置の差に関する一次試験が行われるであろう。ANCOVAモデルの基礎となる仮定を満足しない場合、階数変換アプローチ(Inman and Conover)が用いられるであろう。Iman and Conover技法の後、完全な組の観察を最小値から最大値まで階数にして、最小の観察値には階数1、第二の最小値には階数2等を与える。同値の場合、平均階数が用いられるであろう。適当な変換の後、ベースラインが共変しない技法ANCOVAモデルを、先に記述の方法と類似の方法で変換データに適用するであろう。
【0261】
定量的二次有効性パラメータに関して、分析したパラメータの各範疇における被験体の数および百分は、各評価診察時に処置群毎に示されるであろう。その上、処置群は、ベースラインでの腎状態(ネフローゼ対非ネフローゼ)に関して調節したコクラン-マンテル-ヘンツェル(CMH)行平均スコア試験を用いて一次有効性エンドポイントと同様に比較されるであろう。
【0262】
試験終了時、血清クレアチニン倍加のリスクはプラセボを処置した被験体と比較して59%低減されることが見いだされる。同様に、クレアチニンクリアランスの>50%減少のリスクは52%低減され、透析/ESRDのリスクはプラセボを処置した被験体と比較して46%低減されることが見いだされる。
【0263】
体表面積に関して標準化したクレアチニンクリアランスの経時的勾配
経時的なクレアチニンクリアランスの勾配は、長期間の腎転帰およびESRDまでの時間を予測するために、腎臓学者によって一般的に臨床的に用いられている。負の勾配は、腎機能の喪失を示している。勾配がより負であれば、腎機能の喪失はより速い。
【0264】
クレアチニンクリアランスを、各時点での24時間尿の採取を通して、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。このパラメータを体表面積に関して標準化して、被験体内の勾配の最小二乗法による推定値が、利用可能な全てのクレアチニンクリアランス測定値から計算されるであろう。試験は2年であるが、勾配は年間変化率で表記する。データが経時的に非直線的変化を示す場合、勾配の計算前に適した変換(すなわち、対数変換)が適用されるであろう。
【0265】
被験体内勾配を査定するために、各被験体に関して回帰を行い、このように各被験体に関する勾配を提供する。その後、被験体内勾配値の要約統計値が、ITTおよび有効性評価可能な集団の双方に関して各診察時に処置群毎に示されるであろう。
【0266】
試験終了時、経時的なクレアチニンクリアランスの勾配によって測定すると、PDSによる処置によって、AAアミロイドーシス被験体における腎機能の喪失速度が低減されることが見いだされる。表2および図2において示されるように、PDS処置被験体(処置群対プラセボに関して平均値の差は4.7 ml/分/1.73 m2/年)において腎機能の喪失速度は30.1%低減される。
【0267】
(表2)
【0268】
血清クレアチニンの逆数の経時的勾配
血清クレアチニンは、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。利用可能な全ての血清クレアチニン測定を用いて被験体内勾配の最小二乗法による推定値を計算する。試験は2年であるが、勾配は年間変化率で表記する。データが経時的に非直線的変化を示す場合、適した変換(すなわち、対数変換)が勾配の計算に適用されるであろう。これらの査定は、ITTおよび有効性評価可能な集団の双方に関して行われるであろう。
【0269】
被験体内勾配を査定するために、各被験体に関して回帰を行い、このように各被験体に関する勾配を提供する。その後、被験体内勾配値の要約統計値は、ITTおよび有効性評価可能な集団の双方に関して各診察時に処置群毎に示されるであろう。
【0270】
時間(月)の計算
以下の計算に関して、99%CIを有する生存カプラン-マイヤー推定値、四分位数、および生存期間の中央値のような要約統計値は、ITT集団に関して処置群毎に示される。99%CIはグリーンウッド分散公式に基づく。KM推定値は以下の時間間隔で表される:0〜112日、113〜224日、225〜336日、337〜448日、449〜560日、および560日より長い。
【0271】
二つの処置群を階層化ログランク検定を用いて比較して、階層化因子は、ベースラインでの腎状態である(ネフローゼ対非ネフローゼ)。各処置群における事象までの時間を描写するために、四つの群をカプラン-マイヤープロットにおいて比較する:ベースラインネフローゼ(処置対無処置)およびベースライン非ネフローゼ(処置対無処置)。
* 体表面積に関して標準化
【0272】
試験終了時、SCrの倍加を有する患者において、血清クレアチニンの倍加までの時間の中央値は、プラセボ処置被験体と比較して、PDS処置被験体において3.6ヶ月長いことが見いだされる。さらに、CrClの>50%減少を有する被験体では、クレアチニンクリアランスの少なくとも50%減少までの期間の中央値は、プラセボ処置被験体と比較してPDS処置被験体において4.4ヶ月長い(カプラン-マイヤー曲線に関しては図3を参照されたい)。さらに、透析まで進行した患者において、同様に透析までの期間の中央値は、プラセボ処置被験体と比較してPDS処置被験体では5.3ヶ月長いことが見いだされる(カプラン-マイヤー曲線に関しては図4を参照されたい)。
【0273】
タンパク尿およびクレアチニンクリアランスのベースラインからの変化
タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、スクリーニング、ベースライン、4、8、12、16、20、24ヶ月目での診察時に査定する。ベースラインから試験終了時までのタンパク尿およびクレアチニンクリアランスの変化は、二次有効性エンドポイントとして分析されるであろう。試験の終了は、試験を完了した被験体に関しては24ヶ月目、早期に中止した被験体に関しては最後に利用可能な診察として定義される。スクリーニング時、タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、試験登録(ベースライン診察)前3ヶ月以内に少なくとも1週間離して2回の異なる査定から採取する。クレアチニンクリアランスパラメータに関して、標準化された値が用いられるであろう。
【0274】
タンパク尿/クレアチニンクリアランス/血清クレアチニン/血清アルブミン/血清アルカリホスファターゼ
タンパク尿、クレアチニンクリアランス、血清クレアチニン、血清アルブミン、および血清アルカリホスファターゼを、スクリーニング、ベースライン、4、8、12、16、20(血清アルブミンおよび血清アルカリホスファターゼを除く)、24ヶ月目での診察時に査定する。スクリーニング時、タンパク尿、クレアチニンクリアランス、および血清クレアチニンを、試験登録前3ヶ月以内に少なくとも1週間あけなければならない2回の異なる査定から採取する。
【0275】
血清クレアチニン(SCr)パラメータに関して、値とその逆数、すなわち1/SCrの双方を分析する。クレアチニンクリアランスパラメータに関して、標準化された値が用いられるであろう。これらのパラメータのそれぞれの実際の値の要約統計値をITT集団に関して各診察時に処置群毎に表す。要約統計値はまた、これらのパラメータのそれぞれに関する各ベースライン後査定時のベースラインからの変化およびベースラインからの変化百分率に関して提供されるであろう。
【0276】
タンパク尿および体表面積に関して標準化したクレアチニンクリアランスに関して、二次有効性定量的パラメータに関して記述したように、ベースラインから4、8、12、16、および20ヶ月までの変化に関して処置群の比較を行う。類似のモデルは、ベースラインから4、8、12、16、20、および24ヶ月までの血清クレアチニン、血清アルブミン、および血清アルカリホスファターゼの変化に関して表されるであろう。
【0277】
試験終了時、PDS処置被験体は、プラセボ処置患者に対して-14.7±4.2 ml/分/1.73 m2対-22.3±4.1 ml/分/1.73 m2のCrClの平均値の差を示した(推定の差は7.7 ml/分/1.73 m2)。表3は、ベースラインから試験終了までの変化を示す、この試験からのデータを描写する。
【0278】
(表3)
【0279】
ネフローゼ症候群の状態:進行と寛解
ネフローゼ症候群は、本実施例において、以下の二つの腎臓外特色に関連した重度のタンパク尿症(尿中タンパク質>3 g/24時間)として定義される:1)低アルブミン血症(血清アルブミン<3.4 g/dL)および2)健康診査による末梢の浮腫および/または浮腫を処置するための利尿薬の使用。
【0280】
ベースラインでネフローゼ症候群を有しない被験体におけるネフローゼ症候群への進行は、以下のように定義される:タンパク尿症の>3 g/24時間への増加(ベースラインで<3 g/24時間である必要がある)および以下の二つの腎臓外特色の発生:1)低アルブミン血症(ベースラインでは存在しない)および2)浮腫および/または浮腫を処置するための利尿薬の使用(ベースラインでは浮腫を認めず利尿薬の使用を認めない)。被験体が試験終了時に三つ全ての基準を満たしていない場合、彼らは試験分析の目的に関してネフローゼであると見なされないであろう。被験体がESRD/透析に進行して、血清アルブミンまたは浮腫のいずれかに関する情報を紛失している場合、彼れはネフローゼ症候群に進行しているとは見なされないが、それにもかかわらずその悪化に関する情報は一次エンドポイント分析において獲得されるであろう。
【0281】
ベースラインでネフローゼ症候群を有する被験体におけるネフローゼ症候群の寛解は、以下のように定義される:タンパク尿症の<1 g/24時間(ベースラインで>3 g/24時間である必要がある)への減少および以下の二つの腎臓外特色の一つの改善:1)ベースラインまたは試験中の他の任意の時点で血清アルブミン>3.4 g/dL(ベースラインまたは試験中の他の任意の時点で血清アルブミン<3.4 g/dL)、または2)浮腫の消散および/または浮腫の改善に反応した利尿薬の中止(浮腫が主な症状である場合、または患者がベースラインもしくは試験中の他の任意の時点で利尿薬を用いている場合)。
【0282】
起立性低血圧症
自律神経系(ANS)は、AAアミロイドーシスにおいて影響を受ける可能性がある。血圧の姿勢による減少は、仰臥位から起立位への収縮期圧(SBP)の>20 mmHgの下降、または拡張期圧(DBP)での10 mmHgの低下として定義される。収縮期または拡張期圧の低下が少なくとも3分間持続する場合、これはANS機能障害の兆候である。血圧は、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月での診察時に仰臥位、起立位、および起立して3分後の状態で測定する。
【0283】
各範疇における被験体の数および百分率のような記述統計値(はい対いいえ)は、ITT集団において処置群毎に毎回の試験診察時に示される。処置群は、ベースラインでの腎状態に関して調節したCMH一般関連試験を用いて比較される。
【0284】
脾腫/肝腫
脾腫は、脾臓の肥大として定義される。脾臓の大きさは、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月目での診察時に測定されるであろう。これは定規を用いて前腋窩腺で左肋骨端からセンチメートルで測定される。測定は、被験体が横になり、吸気終了時に行う。
【0285】
肝腫は、肝臓の肥大として定義される。肝臓の大きさは、スクリーニング、ベースライン、4、8、12、16、20、および24ヶ月目での健康診査時に測定されるであろう。これは定規を用いて鎖骨中線で右肋骨端からセンチメートルで測定する。測定は、被験体が横になり、吸気終了時に行う。
【0286】
薬物動態分析
以下の実施例において、用語を以下のように定義する。薬物動態パラメータは、WinNonlin(登録商標)(Pharsight Corp., Mountain View, CA, USA)を用いて非コンパートメント分析によって誘導した。
Cmax− 観察された最高血漿濃度
Tmax− Cmaxが起こった時間。
AUC0-t− 線形台形則によって計算した、ゼロ時間から、濃度が定量限界またはそれを超えている最後の試料採取時間までの血漿濃度対時間曲線下面積。
AUC∞− AUC0-t+(Clast/λz)から計算した、ゼロ時間から無限大までの血漿濃度対時間曲線下面積を指し、Clastは最後に観察された定量可能な濃度であり、λzは見かけの終末速度定数である。
t1/2− ln 2/λzから計算された見かけの終末半減期である。
【0287】
実施例4
健康な男性成人ボランティアにおける1回経口投与後のPDSの安全性、認容性、および薬物動態を決定する。PDSの薬物動態に及ぼす食物の効果も同様に決定する。試験の第一の部分は、健康な男性被験体におけるPDSの6つの増加経口用量の薬物動態プロフィールを査定するための無作為二重盲験プラセボ対照試験である。この試験の第二の部分は、絶食時および食事条件でのPDSの1回経口投与の薬物動態プロフィールに及ぼす食物の効果を調べるためのオープンラベルの二元交叉試験である。血液試料を投与後36時間の間に採取する。PDSの血漿濃度を確認されたHPLC法を用いて決定する。PDSの1回経口投与後、最高血漿濃度は、一般的に投与後0.5〜1時間以内に達し、平均半減期(T1/2)は2〜26時間の範囲である。PDSに対する全身曝露(AUCおよびCmax)の程度は、用量が増加すれば増加する。この増加は、投与された用量と妥当に比例する。その上PDSを食事条件で投与した場合の全身利用率の速度および程度の双方に減少が認められる。したがって、PDSは、高脂肪食と同時に与えてはならない。結果を表4に示す。
【0288】
(表4)健康な男性被験体への100、200、400、800、1600、および2400 mgの1回経口投与および健康な男性被験体(絶食および食後状態)への1600 mgの1回経口投与を示すPDSの薬物動態パラメータ
値は、中央値であるTmaxを除き、平均値であり、括弧内に範囲(すなわち、個体に関する最小値と最大値)示す。
NC:計算していない。
【0289】
実施例5
健康な男性成人ボランティアに複数の増加経口用量を投与後PDSの安全性、認容性、および薬物動態を決定する。これは、健康な男性被験体において400、800、1600 mgのPDSを1回および反復経口投与した場合の薬物動態プロフィールを査定するための無作為二重盲験プラセボ対照試験である。PDSの用量400、800、および1600 mgを1および7日目に健康男性被験体に1回経口投与する。1日目に1回経口投与後、PDSを2、3、4、5および6日目に400、800、および1600 mgを1日4回投与する、または1600 mgを1日3回投与する。血液試料を1日目の投与後24時間を通して採取し、2日目から6日目では最初と最後の投与前、および7日目の最後の投与の48時間後に採取した。PDSの血漿濃度は、確認されたHPLC法を用いて決定する。PDSの1回および繰り返し経口投与後、一般的に投与後0.25〜1時間に、最高血漿濃度に達する。1回または複数回投与後の平均半減期(T1/2)は5〜20時間の範囲である。複数回投与後の蓄積は、T1/2および投与回数と一貫する。PDSに対する全身曝露の程度(AUC0-tおよびCmax)は、用量が増加すると増加して、全身曝露のこの増加は、投与された用量とほぼ比例する。結果を表5に示す。
【0290】
(表5)健康男性被験体に400、800、および1600 mgを複数回経口投与後のPDSの薬物動態パラメータ
値は、中央値であるTmaxを除き、平均値であり、括弧内に範囲(すなわち、個体に関する最小値と最大値)示す。
NC:計算していない。
1日目は1回経口投与後のパラメータに対応する。
7日目は反復経口投与後のパラメータに対応する。
【0291】
実施例6
AAアミロイドーシスを有する被験体における1回および複数回経口投与後のPDSの安全性、有効性、および薬物動態を決定する。これは、PDSの1回および反復投与の薬物動態プロフィールを査定するための、多施設多国間無作為二重盲験プラセボ対照平行デザイン試験である。投与は、被験体における腎障害の重症度に依存する:クレアチニンクリアランス(ClCr)>80 ml/分を有する被験体には、1200 mgを1日2回;ClCrが30〜80 ml/分の被験体には、800 mgを1日2回;およびClCrが20〜30 ml/分の被験体には400 mgを1日2回投与する。ClCrが次のより低い範囲レベルまで減少する場合、投与レジメをそれに従って調節する。PDSは、0ヶ月目および24回の診察時にAAアミロイドーシス患者のサブセットに関して1回経口用量として投与される。0ヶ月目での1回投与後、PDSを1日2回、24ヶ月間投与する。血液試料を0ヶ月および24ヶ月目の投与後24時間のあいだに採取して、4、8、12ヶ月での診察時では投与後8時間のあいだに採取する。PDSの血漿濃度は確認されたHPLC法を用いて決定する。PDSの複数回経口投与後、被験体における蓄積の範囲は1.75〜3.44である。蓄積因子に基づいて、計算された平均T1/2は15時間であり、範囲は10〜24時間である。結果を表6に示す。
【0292】
(表6)AAアミロイドーシス患者に1回(0ヶ月)および複数回(24ヶ月)経口投与後のPDSの薬物動態パラメータ
値は平均値であり、範囲(すなわち個体に関する最小値および最大値)を括弧内に示す。
0ヶ月目は、1回経口投与後のパラメータに対応する。
24ヶ月目は、反復経口投与後のパラメータに対応する。
n=各投与群の患者数
NA=適用可能でない
【0293】
実施例7
1,3プロパンジスルホン酸二ナトリウム塩の400 mgカプセルの製剤の例を以下に記述する。
【0294】
1,3-プロパンジスルホン酸二ナトリウム塩の400 mgカプセルは、0番の白色の不透明な硬ゼラチンカプセルに1,3-プロパンジスルホン酸二ナトリウム塩400 mgおよび賦形剤40 mgを含む白色粉末を充填することによって製造される。
* MHS−製造元の社内標準物質
【0295】
実施例8:
1,2-エタンジスルホン酸を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0296】
実施例9:
1,2-エタンジスルホン酸ナトリウムを活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0297】
実施例10:
1,2-エタンジオールビス(硫酸水素塩)を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0298】
実施例11:
1,2-エタンジオールジスルファート二ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0299】
実施例12:
1,3-プロパンジオールビス(硫酸水素塩)を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0300】
実施例13:
1,3-プロパンジオールジスルファート二ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0301】
実施例14:
2-スルホメチル-1,4-ブタンジスルホン酸を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0302】
実施例15:
2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩を活性物質として、実施例7において記述されたように薬学的組成物を製剤する。
【0303】
同等物
当業者は、本明細書に記述の特異的技法に対する多数の同等物を単なる日常的な実験を用いて認識し、または確認することができるであろう。そのような同等物は、本発明の範囲内であると見なされ、以下の特許請求の範囲に含まれる。本出願を通して引用した全ての参考文献、特許、および特許出願の内容物は、参照により本明細書に組み入れられる。それらの特許、出願、および他の文書の適当な成分、プロセス、および方法は、本発明およびその態様のために選択されてもよい。
【特許請求の範囲】
【請求項1】
被験体における腎機能の安定化もしくは改善、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項2】
式(I)の化合物の治療的有効量が毎日約100〜2500 mgの間である、請求項1記載の使用。
【請求項3】
医薬が、200 mg、400 mg、または800 mgカプセルを含む、請求項1記載の使用。
【請求項4】
医薬が、クレアチニンクリアランス速度>80 ml/分の場合約1200 mgを1日2回、クレアチニンクリアランスが約30〜80 ml/分の場合約800 mgを1日2回、またはクレアチニンクリアランス速度が約20〜30 ml/分の場合約400 mgを1日2回である用量を有する、請求項1記載の使用。
【請求項5】
医薬が、被験体において透析またはESRDへの進行を遅らすか、または予防する、請求項1〜4のいずれか一項記載の使用。
【請求項6】
医薬が、被験体において血清クレアチンの倍加への進行を遅らすか、または予防する、請求項1〜5のいずれか一項記載の使用。
【請求項7】
医薬が、被験体におけるクレアチニンクリアランス速度を安定化するか、または改善する、請求項1〜6のいずれか一項記載の使用。
【請求項8】
医薬が、被験体における内臓のアミロイド負荷を安定化するか、または低減する、請求項1〜7のいずれか一項記載の使用。
【請求項9】
医薬が食物なしの投与のためである、請求項1〜8のいずれか一項記載の使用。
【請求項10】
医薬が、第二の物質との併用投与のためである、請求項1〜8のいずれか一項記載の使用。
【請求項11】
第二の物質が細胞障害剤、化学療法剤、抗炎症剤、または抗生物質である、請求項10記載の使用。
【請求項12】
式(I)の化合物が、1,2-エタンジスルホン酸、1,2-エタンジスルホン酸ナトリウム、1,3-プロパンジスルホン酸、1,3-プロパンジスルホン酸二ナトリウム塩、1,2-エタンジオールビス(硫酸水素塩)、1,2-エタンジオールジスルファート二ナトリウム塩、1,3-プロパンジオールビス(硫酸水素塩)、1,3-プロパンジオールジスルファート二ナトリウム塩、2-スルホメチル-1,4-ブタンジスルホン酸、または2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩である、請求項1〜11のいずれか一項記載の使用。
【請求項13】
式(I)の化合物が、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である、請求項1〜12のいずれか一項記載の使用。
【請求項14】
式(I)の化合物が、1,3-プロパンジスルホン酸二ナトリウム塩である、請求項1〜12のいずれか一項記載の使用。
【請求項15】
医薬が、被験体において約7000〜約26,000 ng.h/mlのAUCssをもたらすのに十分な量の式(I)の化合物の投与のためである、請求項1〜14のいずれか一項記載の使用。
【請求項16】
医薬が、被験体において約500〜約1200 ng/mlのCmaxをもたらすのに十分な量の式(I)の化合物の投与のためである、請求項1〜15のいずれか一項記載の使用。
【請求項17】
炎症性疾患、悪性新生物、慢性感染症、または遺伝性発熱を有する被験体における腎機能の安定化もしくは改善、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩の使用。
【請求項18】
薬学的に許容される塩が、1,3-プロパンジスルホン酸二ナトリウム塩である、請求項17記載の使用。
【請求項19】
AAアミロイドーシスを有する被験体におけるESRD/透析への進行を予防するか、または遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項20】
AAアミロイドーシスを有する被験体における腎機能を安定化する、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項21】
医薬が、約30〜約50 ml/分、約50〜約80 ml/分、約30 ml/分未満、または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体における使用のためである、請求項17〜20のいずれか一項記載の使用。
【請求項22】
医薬が、約60〜約90 ml/分のクレアチニンクリアランス速度を有する被験体における使用のためである、請求項17〜20のいずれか一項記載の使用。
【請求項23】
投与後約0.25〜約9.00時間でCmax約200〜約3,400 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階を含む、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法。
【請求項24】
AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階を含む、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法。
【請求項25】
薬学的に許容される塩が二ナトリウム塩である、請求項23または24記載の方法。
【請求項26】
健康な被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、AAアミロイドーシスを処置または予防するために有効な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む、薬学的製剤。
【請求項27】
AAアミロイドーシスを有する被験体に経口投与する場合、
クレアチニンクリアランス速度約30 ml/分未満を有する被験体に対して活性物質の用量400 mgでは、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;
クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に対して活性物質の用量800 mgでは、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に対して活性物質の用量1200 mgでは、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む薬学的製剤。
【請求項28】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量400 mgで投与する場合、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが達成され;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量800 mgで投与する場合、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量1200 mgで投与する場合、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
被験体のクレアチニンクリアランス速度に従って決定された量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を、AAアミロイドーシスを有する被験体に投与する際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、1つまたは複数の薬学的に許容される担体の使用。
【請求項29】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、被験体のクレアチニンクリアランス速度が約30 ml/分未満である場合400 mgの用量で、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合800 mgの用量で、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合1200 mgの用量で投与される、請求項28記載の使用。
【請求項30】
被験体のクレアチニンクリアランス速度が約60〜約90 ml/分である場合、1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、1200 mgの用量で投与される、請求項28記載の使用。
【請求項31】
医薬が、被験体における血清クレアチニンの倍加を遅らせるか、または予防する、請求項28〜30のいずれか一項記載の使用。
【請求項32】
医薬が、被験体におけるクレアチニンクリアランス速度の50%減少を遅らせるか、または予防する、請求項28〜30のいずれか一項記載の使用。
【請求項33】
医薬が、被験体における透析またはESRDへの進行を遅らせるか、または予防する、請求項28〜32のいずれか一項記載の使用。
【請求項34】
AAアミロイドーシスを有する被験体に経口投与する場合、
クレアチニンクリアランス速度約30 ml/分未満を有する被験体に対して活性物質の用量400 mgでは、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;
クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に対して活性物質の用量800 mgでは、平均AUC∞約3000〜20000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に対して活性物質の用量1200 mgでは、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む、薬学的製剤。
【請求項35】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量400 mgで投与する場合、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが達成され;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量800 mgで投与する場合、平均AUC∞約3000〜20000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量1200 mgで投与する場合、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を、それを必要とする被験体に投与する際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、1つまたは複数の薬学的に許容される担体の使用。
【請求項36】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、被験体のクレアチニンクリアランス速度が約30 ml/分より大きい場合400 mの用量で、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合800 mgの用量で、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合1200 mgの用量で投与される、請求項35記載の使用。
【請求項37】
被験体のクレアチニンクリアランス速度が約60〜約90 ml/分である場合、1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、1200 mgの用量で投与される、請求項35記載の使用。
【請求項38】
医薬が、被験体における血清クレアチニンの倍加を遅らせるか、または予防する、請求項35〜37のいずれか一項記載の使用。
【請求項39】
医薬が、被験体におけるクレアチニンクリアランス速度の50%減少を遅らせるか、または予防する、請求項35〜38のいずれか一項記載の使用。
【請求項40】
医薬が、被験体における透析またはESRDへの進行を遅らせるか、または予防する、請求項35〜39のいずれか一項記載の使用。
【請求項1】
被験体における腎機能の安定化もしくは改善、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項2】
式(I)の化合物の治療的有効量が毎日約100〜2500 mgの間である、請求項1記載の使用。
【請求項3】
医薬が、200 mg、400 mg、または800 mgカプセルを含む、請求項1記載の使用。
【請求項4】
医薬が、クレアチニンクリアランス速度>80 ml/分の場合約1200 mgを1日2回、クレアチニンクリアランスが約30〜80 ml/分の場合約800 mgを1日2回、またはクレアチニンクリアランス速度が約20〜30 ml/分の場合約400 mgを1日2回である用量を有する、請求項1記載の使用。
【請求項5】
医薬が、被験体において透析またはESRDへの進行を遅らすか、または予防する、請求項1〜4のいずれか一項記載の使用。
【請求項6】
医薬が、被験体において血清クレアチンの倍加への進行を遅らすか、または予防する、請求項1〜5のいずれか一項記載の使用。
【請求項7】
医薬が、被験体におけるクレアチニンクリアランス速度を安定化するか、または改善する、請求項1〜6のいずれか一項記載の使用。
【請求項8】
医薬が、被験体における内臓のアミロイド負荷を安定化するか、または低減する、請求項1〜7のいずれか一項記載の使用。
【請求項9】
医薬が食物なしの投与のためである、請求項1〜8のいずれか一項記載の使用。
【請求項10】
医薬が、第二の物質との併用投与のためである、請求項1〜8のいずれか一項記載の使用。
【請求項11】
第二の物質が細胞障害剤、化学療法剤、抗炎症剤、または抗生物質である、請求項10記載の使用。
【請求項12】
式(I)の化合物が、1,2-エタンジスルホン酸、1,2-エタンジスルホン酸ナトリウム、1,3-プロパンジスルホン酸、1,3-プロパンジスルホン酸二ナトリウム塩、1,2-エタンジオールビス(硫酸水素塩)、1,2-エタンジオールジスルファート二ナトリウム塩、1,3-プロパンジオールビス(硫酸水素塩)、1,3-プロパンジオールジスルファート二ナトリウム塩、2-スルホメチル-1,4-ブタンジスルホン酸、または2-スルホメチルブタン-1,4-ジスルホン酸三ナトリウム塩である、請求項1〜11のいずれか一項記載の使用。
【請求項13】
式(I)の化合物が、1,3-プロパンジスルホン酸またはその薬学的に許容される塩である、請求項1〜12のいずれか一項記載の使用。
【請求項14】
式(I)の化合物が、1,3-プロパンジスルホン酸二ナトリウム塩である、請求項1〜12のいずれか一項記載の使用。
【請求項15】
医薬が、被験体において約7000〜約26,000 ng.h/mlのAUCssをもたらすのに十分な量の式(I)の化合物の投与のためである、請求項1〜14のいずれか一項記載の使用。
【請求項16】
医薬が、被験体において約500〜約1200 ng/mlのCmaxをもたらすのに十分な量の式(I)の化合物の投与のためである、請求項1〜15のいずれか一項記載の使用。
【請求項17】
炎症性疾患、悪性新生物、慢性感染症、または遺伝性発熱を有する被験体における腎機能の安定化もしくは改善、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩の使用。
【請求項18】
薬学的に許容される塩が、1,3-プロパンジスルホン酸二ナトリウム塩である、請求項17記載の使用。
【請求項19】
AAアミロイドーシスを有する被験体におけるESRD/透析への進行を予防するか、または遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項20】
AAアミロイドーシスを有する被験体における腎機能を安定化する、または腎疾患の進行を遅らせる際の使用のための医薬の製造のための、下記式を有する化合物の使用:
式中、Yは、各発生に対して独立して選択されるSO3XまたはOSO3Xであり;Xは各発生に対して独立して選択される陽イオン基であり;nは1、2、3または4であり;およびmは1または2であり、但しmが2である場合、-(CH2)n-基の一つの水素を欠く。
【請求項21】
医薬が、約30〜約50 ml/分、約50〜約80 ml/分、約30 ml/分未満、または約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体における使用のためである、請求項17〜20のいずれか一項記載の使用。
【請求項22】
医薬が、約60〜約90 ml/分のクレアチニンクリアランス速度を有する被験体における使用のためである、請求項17〜20のいずれか一項記載の使用。
【請求項23】
投与後約0.25〜約9.00時間でCmax約200〜約3,400 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階を含む、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法。
【請求項24】
AUC∞約2,000〜約44,000 ng/mlを達成するために十分な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を被験体に投与する段階を含む、それを必要とする被験体に1,3-プロパンジスルホン酸またはその薬学的に許容される塩を投与する方法。
【請求項25】
薬学的に許容される塩が二ナトリウム塩である、請求項23または24記載の方法。
【請求項26】
健康な被験体に経口投与する場合、平均AUC∞約2900〜約9000 ng.h/ml±20%および平均Cmax約450〜約2150 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される、AAアミロイドーシスを処置または予防するために有効な量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む、薬学的製剤。
【請求項27】
AAアミロイドーシスを有する被験体に経口投与する場合、
クレアチニンクリアランス速度約30 ml/分未満を有する被験体に対して活性物質の用量400 mgでは、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;
クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に対して活性物質の用量800 mgでは、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に対して活性物質の用量1200 mgでは、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む薬学的製剤。
【請求項28】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量400 mgで投与する場合、平均AUC∞約10,000〜12,000 ng.h/ml±20%、および平均Cmax約800〜900 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが達成され;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量800 mgで投与する場合、平均AUC∞約9,000〜10,500 ng.h/ml±20%、および平均Cmax約750〜875 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量1200 mgで投与する場合、平均AUC∞約5,000〜6,000 ng.h/ml±20%、および平均Cmax約800〜925 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
被験体のクレアチニンクリアランス速度に従って決定された量の1,3-プロパンジスルホン酸またはその薬学的に許容される塩を、AAアミロイドーシスを有する被験体に投与する際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、1つまたは複数の薬学的に許容される担体の使用。
【請求項29】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、被験体のクレアチニンクリアランス速度が約30 ml/分未満である場合400 mgの用量で、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合800 mgの用量で、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合1200 mgの用量で投与される、請求項28記載の使用。
【請求項30】
被験体のクレアチニンクリアランス速度が約60〜約90 ml/分である場合、1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、1200 mgの用量で投与される、請求項28記載の使用。
【請求項31】
医薬が、被験体における血清クレアチニンの倍加を遅らせるか、または予防する、請求項28〜30のいずれか一項記載の使用。
【請求項32】
医薬が、被験体におけるクレアチニンクリアランス速度の50%減少を遅らせるか、または予防する、請求項28〜30のいずれか一項記載の使用。
【請求項33】
医薬が、被験体における透析またはESRDへの進行を遅らせるか、または予防する、請求項28〜32のいずれか一項記載の使用。
【請求項34】
AAアミロイドーシスを有する被験体に経口投与する場合、
クレアチニンクリアランス速度約30 ml/分未満を有する被験体に対して活性物質の用量400 mgでは、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;
クレアチニンクリアランス速度約30〜約80 ml/分を有する被験体に対して活性物質の用量800 mgでは、平均AUC∞約3000〜20000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
約80 ml/分より大きいクレアチニンクリアランス速度を有する被験体に対して活性物質の用量1200 mgでは、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、薬学的に許容される担体とを含む、薬学的製剤。
【請求項35】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量400 mgで投与する場合、平均AUC∞約6,000〜17,000 ng.h/ml±20%、および平均Cmax約500〜1200 ng/ml±20%を有する1,3-プロパンジスルホン酸の平均血漿濃度プロフィールが達成され;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量800 mgで投与する場合、平均AUC∞約3000〜20000 ng.h/ml±20%、および平均Cmax約300〜1200 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成され;または
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を用量1200 mgで投与する場合、平均AUC∞約2,000〜11,000 ng.h/ml±20%、および平均Cmax約400〜1500 ng/ml±20%を有する活性物質の平均血漿濃度プロフィールが達成される;
1,3-プロパンジスルホン酸またはその薬学的に許容される塩を、それを必要とする被験体に投与する際の使用のための医薬の製造のための、1,3-プロパンジスルホン酸またはその薬学的に許容される塩と、1つまたは複数の薬学的に許容される担体の使用。
【請求項36】
1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、被験体のクレアチニンクリアランス速度が約30 ml/分より大きい場合400 mの用量で、被験体のクレアチニンクリアランス速度が約30〜約80 ml/分である場合800 mgの用量で、および被験体のクレアチニンクリアランス速度が約80 ml/分より大きい場合1200 mgの用量で投与される、請求項35記載の使用。
【請求項37】
被験体のクレアチニンクリアランス速度が約60〜約90 ml/分である場合、1,3-プロパンジスルホン酸またはその薬学的に許容される塩が、1200 mgの用量で投与される、請求項35記載の使用。
【請求項38】
医薬が、被験体における血清クレアチニンの倍加を遅らせるか、または予防する、請求項35〜37のいずれか一項記載の使用。
【請求項39】
医薬が、被験体におけるクレアチニンクリアランス速度の50%減少を遅らせるか、または予防する、請求項35〜38のいずれか一項記載の使用。
【請求項40】
医薬が、被験体における透析またはESRDへの進行を遅らせるか、または予防する、請求項35〜39のいずれか一項記載の使用。
【図1】


【図2】
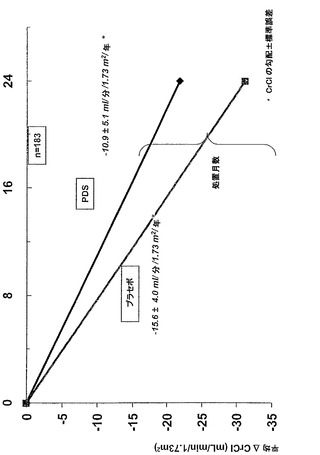
【図3】
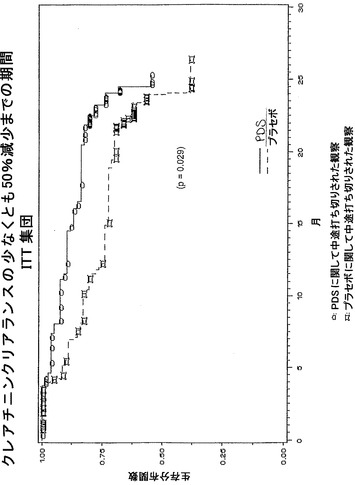
【図4】
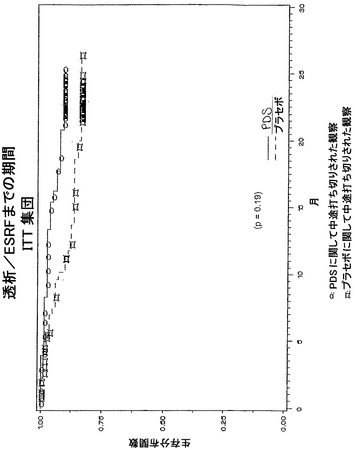
【図2】
【図3】
【図4】
【公開番号】特開2013−79260(P2013−79260A)
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願番号】特願2012−271057(P2012−271057)
【出願日】平成24年12月12日(2012.12.12)
【分割の表示】特願2008−505996(P2008−505996)の分割
【原出願日】平成18年4月17日(2006.4.17)
【出願人】(506365038)ベルス ヘルス (インターナショナル) リミティッド (4)
【Fターム(参考)】
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願日】平成24年12月12日(2012.12.12)
【分割の表示】特願2008−505996(P2008−505996)の分割
【原出願日】平成18年4月17日(2006.4.17)
【出願人】(506365038)ベルス ヘルス (インターナショナル) リミティッド (4)
【Fターム(参考)】
[ Back to top ]