
新規なシャイン・ダルガノ配列、及びそれを用いたタンパク質の製造方法

【課題】 遺伝子工学的手法を用いた高等生物由来のタンパク質の製造において、活性型タンパク質を大量に生産するための制御因子、及び前記因子を用いた製造方法を提供すること。
【解決の手段】 バチルス属細菌由来中性プロテアーゼ遺伝子の一部を含むシャイン・ダルガノ(SD)配列、及び前記配列を含む発現プラスミドを宿主に形質転換して得られる組み換え微生物を用いてタンパク質製造を行なうことで前記課題を解決した。
【解決の手段】 バチルス属細菌由来中性プロテアーゼ遺伝子の一部を含むシャイン・ダルガノ(SD)配列、及び前記配列を含む発現プラスミドを宿主に形質転換して得られる組み換え微生物を用いてタンパク質製造を行なうことで前記課題を解決した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はタンパク質を大量に発現させるのに有用なシャイン・ダルガノ配列、及びそれを用いた遺伝子工学的にタンパク質を製造する方法に関する。
【背景技術】
【0002】
今日までに産業上有用な物質を効率よく多量に生産するために多様な微生物を利用する技術が開発されている。特に大腸菌を用いた系は強力なプロモーター系、正確な制御系、多様な宿主系が開発されており、微生物由来の遺伝子以外にも植物、動物といった高等生物の遺伝子を発現制御することも可能となっている。しかしながら大腸菌は細胞壁の外側に外膜を有しているため、一般的に分泌されるタンパク質は内膜との間に存在するペリプラズム層に蓄積し、培養液中に放出されない。そのため、物理的又は化学的に菌体を破砕して、目的のタンパク質を抽出する必要があるが、前記抽出操作の際、目的のタンパク質が機械的または酵素的な破壊を受けることで活性を失う問題があった。
【0003】
近年、ペリプラズム層を介して可溶性で活性型のタンパク質を大腸菌の菌体外へ分泌生産する技術が開発された(特許文献1)。しかしながら、生産するタンパク質の量が少なく工業生産に適用するには経済性に問題があった。
【0004】
また、大腸菌では高等生物由来のタンパク質の発現の際、多くの場合活性を持たない不溶物として得られる問題があり、さらにリフォーディングとよばれる操作で活性を持つ構造のタンパク質に再構築する必要があった。そのため、工業生産への適用はさらに困難であった。
【0005】
近年、他の微生物内での分泌発現によって活性型で異種タンパク質を得る技術が開発されている。このような発現系の一つとして、タンパク質分泌能が高いバチルス属細菌を宿主として用いる方法が報告されている(非特許文献1)。バチルス属細菌の細胞内でのタンパク質発現量を調節する重要な制御因子としてプロモーター、シャイン・ダルガノ(SD)配列、シグナルペプチド、及びターミネーターと呼ばれる塩基配列が知られている。
【0006】
プロモーターはmRNA合成の開始に関与する特定の短い塩基配列のことであり、細菌の場合は転写開始点から約10塩基上流(−10領域)と約35塩基上流(−35領域)に見られる二つの短い配列から構成されている。
【0007】
SD配列は原核生物のmRNAにおいて開始コドンの上流に見られる配列で、リボソームの結合部位となる部分であり、リボソームにおけるタンパク質の合成開始に関与するといわれている。
【0008】
シグナルペプチドは細胞質内でタンパク質の局在化や膜輸送を指示するペプチドであり、N末端付近の正電荷を持つアミノ酸が局在した領域、及び疎水性のコア領域及びシグナルペプチダーゼに認識切断される領域からなる、20から50アミノ酸残基のペプチドである。当該ペプチドは、バチルス属細菌がタンパク質を細胞外分泌生産する際に中心的な役割を果たすものと考えられている。
【0009】
ターミネーターは構造遺伝子の最後に接続した塩基配列であり、mRNA合成の終了に関与する配列と考えられている。
【0010】
前記制御因子の一例として、バチルス属菌中性プロテアーゼ由来のプロモーター、SD配列、シグナルペプチド及びターミネーター配列が非特許文献2、及び特許文献2から5に開示されており、バチルス属細菌で抗体を発現させうるプロモーター及び分泌シグナル配列が特許文献6に開示されている。
【0011】
前記文献で開示の制御因子は、いずれも強力な発現因子として報告されているが、目的とするタンパク質によっては必ずしも良好に作用せず、特に高等生物由来のタンパク質について活性型が分泌されにくいという問題があった。例えばプロテアーゼのようなバチルス属細菌で一般的に高発現するタンパク質のSD配列やシグナルペプチドが必ずしもすべてのタンパク質に強力には働かず、宿主の種類および発現しようとするタンパク質ごとにプロモーター、SD配列、シグナルペプチド及びターミネーターとの間に相性があり、最適なものを選定することが必要である。近年、様々な高等生物由来の活性型タンパク質の工業生産に対する要求が高まり、これに適する新規な制御因子の提供が求められていた。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】特開平8−322586号公報
【特許文献2】特開平2−265491号公報
【特許文献3】特開平2−265492号公報
【特許文献4】特開平2−268687号公報
【特許文献5】特開平2−268688号公報
【特許文献6】特開平7−075581号公報
【特許文献7】特開平5−184363号公報
【特許文献8】特開平10−309198号公報
【非特許文献】
【0013】
【非特許文献1】Wolfgang Schuman、Adv.Appl.Microbiol.、62、137(2007)
【非特許文献2】Kubo and Imanaka、J.Gen.Microbiol.、134、1883(1988)
【非特許文献3】Hanahan、J.Mol.Biol.、16、557(1983)
【非特許文献4】Debnau and Avidoff−Abelson、J.Mol.Biol、56,209(1971)
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、遺伝子工学的手法を用いた高等生物由来のタンパク質の製造において、活性型タンパク質を大量に生産するための制御因子、及び前記因子を用いたタンパク質の製造方法を提供することである。
【課題を解決するための手段】
【0015】
発明者らは、前記課題に対し、バチルス属細菌由来中性プロテアーゼの遺伝子を詳細に検討した結果、先行文献中には特別な機能を持たない配列として公開された配列の一部がシャイン・ダルガノ(SD)配列として強力な機能を有していることを見出し、本発明を完成した。
【0016】
すなわち、本発明は、以下の発明を包含する:
第一の発明は、少なくともAAAGGAGGAからなる配列を含む、シャイン・ダルガノ(SD)配列である。
【0017】
第二の発明は、第一の発明に記載のシャイン・ダルガノ配列を含む発現プラスミドを宿主に形質転換することで得られた、遺伝子組み換え体である。
【0018】
第三の発明は、第二の発明に記載の遺伝子組み換え体を用いた、タンパク質の製造方法である。
【0019】
第四の発明は、タンパク質が抗体または抗体の断片である、第三の発明に記載のタンパク質の製造方法である。
【0020】
以下、本発明を詳細に説明する。
【0021】
本発明のシャイン・ダルガノ(SD)配列は少なくともAAAGGAGGAを含んでいればよく、一例として配列番号29で示す配列をあげることができる。なお、本発明のSD配列をコードするDNAを得る方法としては、既存のプラスミドであるpUBTZ2(バチルス・ステアロサーモフィルス由来の中性プロテアーゼ遺伝子をクローニングした大腸菌及び枯草菌間のシャトルベクター、特許文献7)よりPCR反応で当該DNA断片を増幅して得る方法、及び化学合成によって直接前記DNAを得る方法が例示できる。
【0022】
本発明の宿主はバチルス属細菌であれば特に限定がないが、この中でも特にタンパク質分泌能が高く、かつ病原性がない枯草菌(バチルス・ズブチリス、Bacillus subtilis)が好ましい。
【0023】
本発明で用いる発現プラスミドにおいて、プロモーターはバチルス属細菌内で機能するものであればよく、一例として、特許文献4で開示の、−35領域がTTTTCC、−10領域がTATTGTからそれぞれなるプロモーターがあげられる。また、プロモーター、及びその間に介在するDNAを得るには、公知の化学合成によって直接前記DNAを得る方法、天然由来のDNAから制限酵素で切り出して得る方法、既存のDNAからPCR反応により核酸増幅して得る方法等により得られる。
【0024】
本発明で用いる発現プラスミドにおいて、分泌発現に用いるシグナルペプチドはバチルス属細菌内で機能するものであればよく、一例として、特許文献3の請求項4で開示の、MKRKMKLRSFGVAAGLA(配列番号30)で示される耐熱性中性プロテアーゼのシグナルペプチド、及び非特許文献2で開示のMKRKMKMKLASFGLAAGLAAQVFLPYNALA(配列番号31)からなるペプチドがあげられる。前記シグナルペプチドをコードするDNAを得る方法としては、既存の遺伝子からPCR反応により前記DNAを核酸増幅して得る方法、また化学合成法によって直接前記DNAを得る方法を例示できる。
【0025】
本発明において分泌発現を目的とするタンパク質に特に限定はないが、アミラーゼ、プロテアーゼ、抗体といった天然においても細胞外に分泌生産されるタンパク質が好ましい。また前記タンパク質をコードする遺伝子を得る方法にも特に限定はなく、抗体を目的タンパク質とした場合の一例として、特許文献8に開示の抗エストラジオール抗体遺伝子取得法があげられる。また、特許文献8に開示の、クローニング済みの遺伝子をプラスミドから切り出す方法や、前記遺伝子断片をPCR反応等の方法により増幅させる方法も用いることもできる。
【0026】
本発明で用いる発現プラスミドにおける、もう一つの構成要素であるタンパク質発現の構造遺伝子群の終点に位置するターミネーターもバチルス属細菌内で機能するものであればよく、一例として、特許文献5の請求項3で開示の、CATCAGTGGGGGATTTTTTCCTCCACTGATG(配列番号32)を含む配列があげられる。
【0027】
本発明のタンパク質製造は、任意のプロモーターの下流に本発明のSD配列を配置し、その下流に任意のシグナルペプチドをコードするDNA配列を介して、所望のタンパク質をコードする遺伝子を持つDNA配列を配置し、さらにその下流に任意のターミネーターを配置した発現プラスミドを作成し、前記プラスミドをバチルス属細菌に導入することでタンパク質製造を行なう。即ち、一般的には、5’末端側から、プロモーター、SD配列、シグナルペプチド、目的タンパク質遺伝子、ターミネーターの順に配置し、発現プラスミドを作成する。
【0028】
発現培養に用いる条件は、バチルス属細菌の培養で通常設定する条件の中から適宜選択すればよい。培養温度は20から40℃が好ましく、特に25から30℃が好ましい。培養時のpHは6から8が好ましい。培養に用いる培地としてはバチルス属細菌が増殖し、かつ目的のタンパク質が分泌発現可能な培地を適宜選択すればよい。培養時間は、目的のタンパク質が分泌発現するのに適する時間であれば任意に設定できるが、通常は数時間から100時間の間に設定される。
【0029】
発現したタンパク質を定量する方法としては一般的なSDS−PAGEでタンパク質を分離した後に色素や免疫学的方法で染色して比色定量する方法、及びELISA法を例示でき、また酵素等を発現する場合には前記酵素の活性を測定する方法なども用いることができる。特に、抗体やレセプターなどを製造する場合はELISA法による活性定量法が簡便であるため好ましい。
【0030】
ELISA法におけるタンパク質の組み合わせは目的のタンパク質が定量できる方法であれば特に限定されないが、一例として、目的抗体に対する抗原、目的抗体、酵素標識抗目的抗体、の順に重ねたサンドイッチ法をあげることができる。ここで酵素標識抗目的抗体の一例として、アルカリフォスファターゼや西洋ワサビペルオキシダーゼ等の酵素で標識された抗目的抗体をあげることができる。
【0031】
また、ELISAの検出法についても特に限定されないが、標識に用いた酵素の特異的発色試薬、蛍光試薬又は化学発光試薬が市販されており、それらを標識に用いた酵素に応じて適宜選択し使用することができる。例えば西洋ワサビペルオキシダーゼを用いた場合は発色基質をペルオキシダーゼと過酸化水素で酸化反応させて比色定量する方法があり、例えばTMB 2−Component Microwell Peroxidase Substrate Kit(商品名、フナコシ社製)といった市販の試薬で発色させた後、マイクロタイタープレートリーダMPR4i(商品名、東ソー社製)といった市販の測定装置で比色定量することができる。
【発明の効果】
【0032】
本発明は、バチルス属細菌由来中性プロテアーゼ遺伝子中、先行文献中には特別な機能を持たない配列として公開された配列の一部をシャイン・ダルガノ(SD)配列として使用することで、バチルス属細菌に所望の高等生物由来タンパク質を分泌生産することが可能なタンパク質の製造方法を提供している。本発明により得られた、タンパク質は、菌体外に活性型の状態で分泌される。これにより、タンパク質抽出のための菌体破砕工程や、変性剤によるタンパク質の活性型への再構築が不要となり、効率よく所望のタンパク質を得ることができる。また、非特許文献2で開示のシャイン・ダルガノ(SD)配列と比較し、プロモーター部とシグナル配列部が短いDNA配列であっても分泌生産が可能となったことから、ベクターの安定化と高発現性が期待される。
【図面の簡単な説明】
【0033】
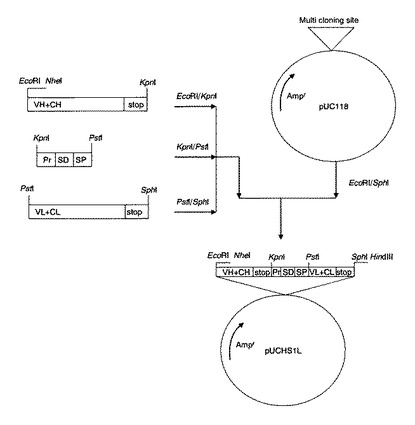
【図1】プラスミドpUCHS1L作成の工程図。図中、VH+CHは抗体のH鎖のVH領域及びCH1領域の遺伝子断片、stopはストップコドン、VL+CLは抗体L鎖のVL領域及びCL領域の遺伝子断片、Prは耐熱性中性プロテアーゼ由来のプロモーター領域、SDは本発明のシャイン・ダルガノ(SD)配列(配列番号29)、SPは耐熱性中性プロテアーゼ由来のシグナルペプチド領域、Amprはアンピシリン耐性遺伝子領域であることを示し、EcoRI、NheI、KpnI、PstI、SphIは対応する制限酵素の切断サイトを示す。
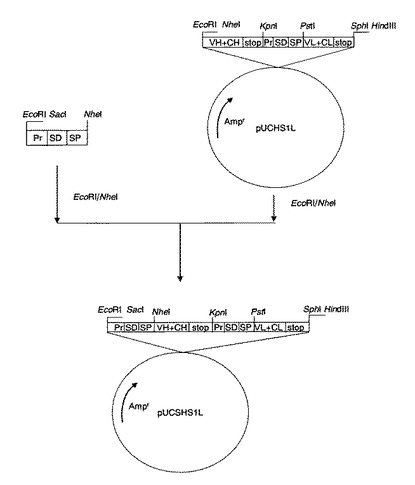
【図2】プラスミドpUCSHS1L作成の工程図。図中の略号は図1に準ずる。
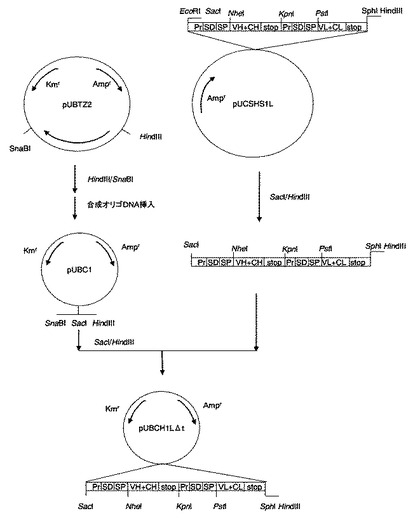
【図3】プラスミドpUBCH1LΔt作成の工程図。図中の略号のうち、Kmrはカナマイシン耐性遺伝子領域であることを示し、その他は図1に準ずる。
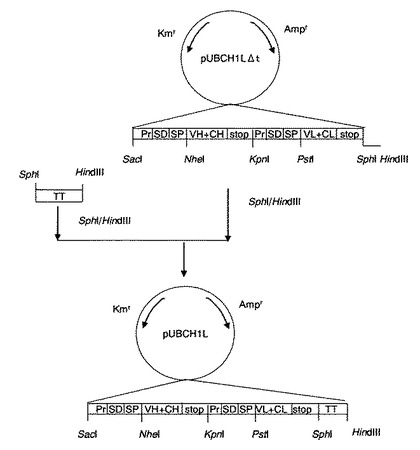
【図4】プラスミドpUBCH1L作成の工程図。図中の略号のうち、Kmrはカナマイシン耐性遺伝子領域、TTは耐熱性中性プロテアーゼ由来のターミネーター配列であることをそれぞれ示し、その他は図1に準ずる。
【図5】プラスミドpUBCH2Lの概略図。図中の略号は図4に準ずる。
【図6】プラスミドpUBCH3Lの概略図。図中の略号のうち、Supは13塩基の付加配列(配列番号33)であることを示す。その他は図4に準ずる。
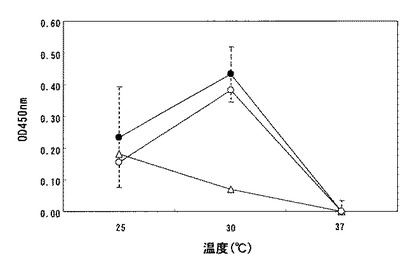
【図7】プラスミドpUBCH2Lで形質転換した枯草菌DB117株(黒丸)、DB104株(白丸)、及びMT2株(白三角)を25℃、30℃、又は37℃で24時間培養したのちの、培養上清中の抗体力価を測定した結果を示す。図中の各シンボルは各条件で24点の同一の実験を行なった際の平均値を示す。また、破線は標準偏差を示す。
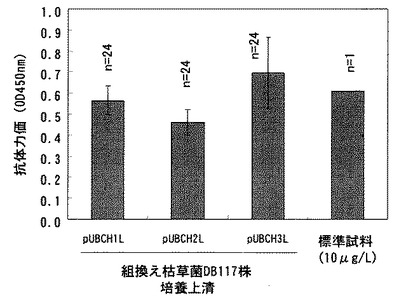
【図8】プラスミドpUBCH1L、pUBCH2L、又はpUBCH3Lで形質転換した枯草菌DB117株を30℃で24時間培養後の、培養上清中の抗体力価を測定した結果を示す。図中の棒は各形質転換体を24回培養した結果の平均値を示す。また、棒に示した縦線は標準偏差を示す。
【実施例】
【0034】
以下に本発明の実施例を示すが、本発明はこれらに限定されるものではない。
【0035】
実施例1 発現ベクターpUBCH1Lの作成
(1)インサート配列(プロモーターからシグナル配列を含む塩基配列)の合成
(1−1)配列番号1から12(配列番号1は配列番号13の1から40番目の塩基に、配列番号2は配列番号13の35から79番目の塩基の相補鎖に、配列番号3は配列番号13の73から115番目の塩基に、配列番号4は配列番号13の108から152番目の塩基の相補鎖に、配列番号5は配列番号13の144から188番目の塩基に、配列番号6は配列番号13の179から223番目の塩基の相補鎖に、配列番号7は配列番号13の217から261番目の塩基に、配列番号8は配列番号13の254から298番目の塩基の相補鎖に、配列番号9は配列番号13の291から335番目の塩基に、配列番号10は配列番号13の327から371番目の塩基の相補鎖に、配列番号11は配列番号13の366から410番目の塩基に、配列番号12は配列番号13の400から435番目の塩基の相補鎖に、それぞれ相当)に示すオリゴヌクレオチドを混合し、タカラバイオ社製DNAポリメラーゼ(商品名、PrimeSTAR HS DNA Polymerase)を用いて1回目のPCRを行ないDNAを増幅した。PCR反応は94℃で5分加熱した後、94℃での加熱(30秒)、57℃でのアニーリング(30秒)及び72度での伸張反応(45秒)のサイクルを25回繰り返した後、72℃で7分加温することにより行なった。
(1−2)1回目のPCR反応後の反応液をキアゲン社製PCR精製キットで精製して50μLのDNA溶液を得た。
(1−3)(1−2)で得られたDNA溶液のうちの1μLを鋳型とし、配列番号1及び12のオリゴヌクレオチドをプライマーとして2回目のPCR反応を、1回目と同じ条件で行なった。
(1−4)2回目のPCR反応後の反応液を、アガロースゲル電気泳動より約400bpのDNA断片を切出し、キアゲン社製ゲル抽出キットにより精製して配列番号13に示すインサート配列(5’末端付近にEcoRIサイトとSacIサイトを含み、中央部に中性プロテアーゼプロモーター配列、本発明のシャイン・ダルガノ(SD)配列(配列番号29、325から339番目の塩基)、および中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、340から429番目の塩基)を含み、3’末端付近にNheIサイトを含む)を得た。
(1−5)2回目のPCR反応用オリゴヌクレオチドとして、配列番号14及び15(配列番号14は配列番号16の1から34番目の塩基に、配列番号15は配列番号16の396から433番目の塩基の相補鎖に、それぞれ相当)を用いた他は(1−3)から(1−4)と同様な操作を行ない、配列番号16に示すインサート配列(5’末端付近にKpnIサイトを含み、中央部に中性プロテアーゼプロモーター配列、本発明のSD配列(配列番号29、319から333番目の塩基)、及び中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、334から423番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
【0036】
(2)インサート配列(SD配列からシグナル配列を含む塩基配列)の合成
(2−1)1回目のPCR反応用オリゴヌクレオチドとして、配列番号10、11、15、17(配列番号17は配列番号18の1から32番目の塩基に相当)、2回目のPCR反応用オリゴヌクレオチドとして、配列番号15及び17を用いた他は、(1−1)から(1−4)と同様な操作を行ない、配列番号18に示すインサート配列(5’末端付近にKpnIサイトを含み、中央部に本発明のSD配列(配列番号29、13から27番目の塩基)と中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、28から117番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
(2−2)2回目のPCR反応用オリゴヌクレオチドとして、配列番号15及び19(配列番号19は配列番号20の1から36番目の塩基に相当)を用いた他は、(2−1)と同様の操作を行ない、配列番号20に示すインサート配列(5’末端付近にKpnIサイトと13塩基の付加配列(配列番号33、13から25番目の塩基)を含み、中央部に本発明のSD配列(配列番号29、26から40番目の塩基)と中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、41から130番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
【0037】
(3)抗体H鎖遺伝子断片(VHからCH1領域)の増幅
特許文献8で開示の方法で調製された抗エストラジオール抗体発現プラスミドを鋳型とし、配列番号21及び22に示されるオリゴヌクレオチドをプライマーとしたPCR反応により抗体H鎖遺伝子断片(VHからCH1領域)を増幅した。PCR反応は94℃で5分加熱した後、94℃での加熱(30秒)、57℃でのアニーリング(30秒)及び72度での伸張反応(45秒)のサイクルを25回繰り返した後、72℃で7分加温することにより行なった。増幅した遺伝子はキアゲン社製PCR精製キットで精製した。
【0038】
(4)抗体L鎖遺伝子断片(VLからCL領域)の増幅
プライマーとして配列番号23及び24に示されるオリゴヌクレオチドを用いた他は、(3)と同様にPCR反応を行ない、抗体L鎖遺伝子(VLからCL領域)の増幅断片を得た。
【0039】
(5)ターミネーターの増幅
鋳型として特許文献7で開示のプラスミドpUBTZ2を、プライマーとして配列番号25及び26に示されるオリゴヌクレオチドを用いた他は、(3)と同様にPCR反応を行ない、中性プロテアーゼターミネーターの増幅断片(配列番号34、23から53番目の塩基は配列番号32と同じ)を得た。
【0040】
(6)発現ベクターの構築
(6−1)(3)で調製した抗体H鎖遺伝子断片(VHからCH1領域)を制限酵素EcoRI及びKpnIで、(4)で調製したL鎖抗体遺伝子断片(VLからCL領域)を制限酵素PstI及びSphIで、(1−5)で調製した配列番号16に示したインサート配列をKpnI及びPstIで、それぞれ切断した後、それぞれキアゲン社PCR精製キットで精製した。
(6−2)プラスミドpUC118(タカラバイオ製)をEcoRIとSphIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約3kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−3)(6−2)の断片に、(6−1)で調製した制限酵素処理済の抗体H鎖遺伝子断片、配列番号16に示すインサート配列及び抗体L鎖遺伝子断片を、T4DNAリガーゼを用いて結合し、プラスミドpUCHS1L(図1)を合成した。
(6−4)プラスミドpUCHS1L(図1)をEcoRIとNheIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約4.8kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−5)(6−4)で得られた断片に、予めEcoRIとNheIで切断してキアゲン社PCR精製キットで精製した配列番号13に示すインサート配列をT4DNAリガーゼを用いて結合し、プラスミドpUCSHS1L(図2)を合成した。
(6−6)プラスミドpUCSHS1L(図2)を制限酵素SacIとHindIIIで切断し、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約2kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製し、5’末端側から、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、抗体H鎖遺伝子断片(VHからCH1領域)、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、L鎖抗体遺伝子断片(VLからCL領域)より構成されるDNA断片を得た。
(6−7)特許文献7で開示のpUBTZ2を、SnaBI及びHindIIIで消化後、アルカリフォスファターゼによる脱リン酸化処理後、1%アガロースゲル電気泳動からの切出しにより約5kbpの断片を調製した。
(6−8)(6−7)で得られた断片に、予めT4DNAキナーゼ処理によりリン酸化した2つのオリゴDNA(配列番号27及び28)をT4DNAリガーゼにより結合させプラスミドpUBC1(図3)を合成した。
(6−9)プラスミドpUBC1(図3)を制限酵素SacI及びHindIIIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約5kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−10)(6−9)で得られた断片と、(6−6)で調製した、5’末端側から、プロモーター中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、抗体H鎖遺伝子断片(VHからCH1領域)、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、L鎖抗体遺伝子断片(VLからCL領域)より構成されるDNA断片を、T4DNAリガーゼを用いて結合し、プラスミドpUBH1LΔt(図3)を合成した。
(6−11)プラスミドpUBH1LΔt(図3)を、制限酵素SphI及びHindIIIで切断してアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約7kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。この断片に(5)で合成したターミネーター断片をT4DNAリガーゼを用いて結合し、プラスミドpUBCH1L(図4)を合成した。
【0041】
実施例2 発現ベクターpUBCH2Lの作成
配列番号16のインサート配列の代わりに(2−1)で調製した配列番号18を用いた他は、実施例1の(6)と同様の操作を行ない、プラスミドpUBCH2L(図5)を合成した。当該プラスミドと実施例1で調製したプラスミドpUBCH1Lの相違は、前者の抗体H鎖遺伝子とL鎖遺伝子をつなぐ断片が、終止コドン、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成されるのに対し、後者では終止コドン、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成される点が異なる。
【0042】
実施例3 発現ベクターpUBCH3Lの作成
配列番号16のインサート配列の代わりに(2−2)で調製した配列番号20を用いた他は、実施例1の(6)と同様の操作を行ない、プラスミドpUBCH3L(図6)を合成した。当該プラスミドと実施例1で調製したプラスミドpUBCH1Lの相違は、前者の抗体H鎖遺伝子とL鎖遺伝子をつなぐ断片が、終止コドン、13塩基の付加配列(AAATACAAAGAAT、配列番号33)、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成されるのに対し、後者では終止コドン、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成される点が異なる(図6)。
【0043】
実施例4 枯草菌への形質転換
(1)大腸菌JM103株のコンピテントセルをHanahanの方法(非特許文献3)に従って作成し、発現ベクターpUBCH1L、pUBCH2L又はpUBCH3Lで形質転換した。これらの形質転換大腸菌を100μg/mLのアンピシリンを含むLB培地で一晩培養した。この培養液からキアゲン社プラスミド抽出キットを用いて各プラスミドの水溶液30μLを調製した。
(2)枯草菌MT2株、DB104株又はDB117株のコンピテントセルを公知の方法(非特許文献4)に従って調製した。前記細胞懸濁液100μLに、(1)で調製した各プラスミド水溶液5μLを加えて、37℃で1時間、穏やかに振とうした後、LB培地900μLを加えて37℃で1.5時間、激しく振とうして培養した。(3)(2)の培養液より遠心分離で菌体を回収し、100μLの生理食塩水に再懸濁し、50μg/mLのカナマイシンを含むLB平板培地に10μLまたは90μLを塗布して37℃で一晩静置培養した。生育したカナマイシン耐性のコロニーより目的のプラスミドを持つものを選別し、目的の形質転換体枯草菌菌株を調製した。
【0044】
実施例5 抗体遺伝子の発現に対する菌株と温度の影響
(1)実施例4で調製した形質転換体のうち、プラスミドpUBCH2Lで形質転換した枯草菌MT2、DB104及びDB117株を2PY培地(ポリペプトン 40g/L、バクト酵母エキス 5g/L、グルコース 20g/L、MgSO4・7H2O 0.1g/L、FeSO4・7H2O 0.01g/L、MnSO4・4H2O 0.01g/L、ZnSO4・7H2O 0.001g/L)0.75mLを入れた96穴深穴プレート(BM6030S深穴プレート角型V底、ビーエム機器社製)に植菌し、温度25℃、30℃又は37℃にて、バイオシェーカーMBR−022UP(商品名、タイテック社製)を用いて1200rpmの振とう速度で24時間培養した。
【0045】
(2)(1)の培養液を遠心分離して菌体を除去し、上清液中の抗体力価を以下の方法によりELISA法で測定した。
(2−1)予めエストラジオール標識牛血清アルブミンを結合させた後にパーフェクトブロック(商品名、ピアス社製)でブロッキングしておいた96穴プレートに、培養上清100μLを入れて37℃で1時間抗原抗体反応を行なった後、溶液を除去した。
(2−2)西洋ワサビペルオキシダーゼ修飾抗マウス抗体抗体溶液を添加して37℃で1時間抗原抗体反応を行なった後、再度溶液を除去した。
(2−3)西洋ワサビペルオキシダーゼの発色試薬(TMB 2−Component Microwell Peroxidase Substrate Kit(商品名)、フナコシ社製)50μLを添加して適当な色調になるまで発色を行ない、50μLの10%リン酸水溶液で反応を停止し、450nmの吸光度を測定した。ブランクとして培養上清の代わりに2PY培地を用いて同様の反応を行なって450nmの吸光度を測定し、検体の450nmの吸光度からブランクの吸光度を引くことにより抗体力価とした。
【0046】
ELISAの結果を図7に示す。いずれの菌株も37℃で培養した場合には培養上清中に抗体力価は確認できなかったものの、25℃又は30℃で培養した場合には明らかな抗体力価が確認できており、本発明のSD配列を含んだ発現プラスミドを枯草菌に形質転換することにより、抗原への結合活性を保持した抗体が得られることが示された。なお、DB104株及びDB117株では30℃での抗体生産が最も多かったが、MT2株における30℃での生産性は他の菌株より低かった。
【0047】
実施例6 プラスミド構成の違いにおける抗体遺伝子の発現の影響
(1)発現プラスミドpUBCH1L、pUBCH2L又はpUBCH3Lで形質転換した枯草菌DB117株を、培養温度が30℃で実施例5と同様に培養し、遠心分離して培養上清を得た。
【0048】
(2)培養上清中の抗体力価を実施例5と同様に測定した。
【0049】
結果を図8に示す。使用した発現プラスミドは抗体H鎖遺伝子とL鎖遺伝子を連結した配列において、pUBCH1Lでは中性プロテアーゼ由来プロモーター、本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列を含み、pUBCH2Lでは本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列のみを含み、またpUBCH3Lでは13塩基の付加配列(配列番号33)と本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列を含む点で異なるが、これらの相違による抗体生産性への影響は認められなかった。図8には抗エストラジオール抗体産生ハイブリドーマ(特許文献8)の培養上清より調製した標準抗体(約10μg/mL)の測定結果も示した。発色量の比較から前記3つの組換え枯草菌による抗体生産量は約10μg/mLと推定された。
【技術分野】
【0001】
本発明はタンパク質を大量に発現させるのに有用なシャイン・ダルガノ配列、及びそれを用いた遺伝子工学的にタンパク質を製造する方法に関する。
【背景技術】
【0002】
今日までに産業上有用な物質を効率よく多量に生産するために多様な微生物を利用する技術が開発されている。特に大腸菌を用いた系は強力なプロモーター系、正確な制御系、多様な宿主系が開発されており、微生物由来の遺伝子以外にも植物、動物といった高等生物の遺伝子を発現制御することも可能となっている。しかしながら大腸菌は細胞壁の外側に外膜を有しているため、一般的に分泌されるタンパク質は内膜との間に存在するペリプラズム層に蓄積し、培養液中に放出されない。そのため、物理的又は化学的に菌体を破砕して、目的のタンパク質を抽出する必要があるが、前記抽出操作の際、目的のタンパク質が機械的または酵素的な破壊を受けることで活性を失う問題があった。
【0003】
近年、ペリプラズム層を介して可溶性で活性型のタンパク質を大腸菌の菌体外へ分泌生産する技術が開発された(特許文献1)。しかしながら、生産するタンパク質の量が少なく工業生産に適用するには経済性に問題があった。
【0004】
また、大腸菌では高等生物由来のタンパク質の発現の際、多くの場合活性を持たない不溶物として得られる問題があり、さらにリフォーディングとよばれる操作で活性を持つ構造のタンパク質に再構築する必要があった。そのため、工業生産への適用はさらに困難であった。
【0005】
近年、他の微生物内での分泌発現によって活性型で異種タンパク質を得る技術が開発されている。このような発現系の一つとして、タンパク質分泌能が高いバチルス属細菌を宿主として用いる方法が報告されている(非特許文献1)。バチルス属細菌の細胞内でのタンパク質発現量を調節する重要な制御因子としてプロモーター、シャイン・ダルガノ(SD)配列、シグナルペプチド、及びターミネーターと呼ばれる塩基配列が知られている。
【0006】
プロモーターはmRNA合成の開始に関与する特定の短い塩基配列のことであり、細菌の場合は転写開始点から約10塩基上流(−10領域)と約35塩基上流(−35領域)に見られる二つの短い配列から構成されている。
【0007】
SD配列は原核生物のmRNAにおいて開始コドンの上流に見られる配列で、リボソームの結合部位となる部分であり、リボソームにおけるタンパク質の合成開始に関与するといわれている。
【0008】
シグナルペプチドは細胞質内でタンパク質の局在化や膜輸送を指示するペプチドであり、N末端付近の正電荷を持つアミノ酸が局在した領域、及び疎水性のコア領域及びシグナルペプチダーゼに認識切断される領域からなる、20から50アミノ酸残基のペプチドである。当該ペプチドは、バチルス属細菌がタンパク質を細胞外分泌生産する際に中心的な役割を果たすものと考えられている。
【0009】
ターミネーターは構造遺伝子の最後に接続した塩基配列であり、mRNA合成の終了に関与する配列と考えられている。
【0010】
前記制御因子の一例として、バチルス属菌中性プロテアーゼ由来のプロモーター、SD配列、シグナルペプチド及びターミネーター配列が非特許文献2、及び特許文献2から5に開示されており、バチルス属細菌で抗体を発現させうるプロモーター及び分泌シグナル配列が特許文献6に開示されている。
【0011】
前記文献で開示の制御因子は、いずれも強力な発現因子として報告されているが、目的とするタンパク質によっては必ずしも良好に作用せず、特に高等生物由来のタンパク質について活性型が分泌されにくいという問題があった。例えばプロテアーゼのようなバチルス属細菌で一般的に高発現するタンパク質のSD配列やシグナルペプチドが必ずしもすべてのタンパク質に強力には働かず、宿主の種類および発現しようとするタンパク質ごとにプロモーター、SD配列、シグナルペプチド及びターミネーターとの間に相性があり、最適なものを選定することが必要である。近年、様々な高等生物由来の活性型タンパク質の工業生産に対する要求が高まり、これに適する新規な制御因子の提供が求められていた。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】特開平8−322586号公報
【特許文献2】特開平2−265491号公報
【特許文献3】特開平2−265492号公報
【特許文献4】特開平2−268687号公報
【特許文献5】特開平2−268688号公報
【特許文献6】特開平7−075581号公報
【特許文献7】特開平5−184363号公報
【特許文献8】特開平10−309198号公報
【非特許文献】
【0013】
【非特許文献1】Wolfgang Schuman、Adv.Appl.Microbiol.、62、137(2007)
【非特許文献2】Kubo and Imanaka、J.Gen.Microbiol.、134、1883(1988)
【非特許文献3】Hanahan、J.Mol.Biol.、16、557(1983)
【非特許文献4】Debnau and Avidoff−Abelson、J.Mol.Biol、56,209(1971)
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、遺伝子工学的手法を用いた高等生物由来のタンパク質の製造において、活性型タンパク質を大量に生産するための制御因子、及び前記因子を用いたタンパク質の製造方法を提供することである。
【課題を解決するための手段】
【0015】
発明者らは、前記課題に対し、バチルス属細菌由来中性プロテアーゼの遺伝子を詳細に検討した結果、先行文献中には特別な機能を持たない配列として公開された配列の一部がシャイン・ダルガノ(SD)配列として強力な機能を有していることを見出し、本発明を完成した。
【0016】
すなわち、本発明は、以下の発明を包含する:
第一の発明は、少なくともAAAGGAGGAからなる配列を含む、シャイン・ダルガノ(SD)配列である。
【0017】
第二の発明は、第一の発明に記載のシャイン・ダルガノ配列を含む発現プラスミドを宿主に形質転換することで得られた、遺伝子組み換え体である。
【0018】
第三の発明は、第二の発明に記載の遺伝子組み換え体を用いた、タンパク質の製造方法である。
【0019】
第四の発明は、タンパク質が抗体または抗体の断片である、第三の発明に記載のタンパク質の製造方法である。
【0020】
以下、本発明を詳細に説明する。
【0021】
本発明のシャイン・ダルガノ(SD)配列は少なくともAAAGGAGGAを含んでいればよく、一例として配列番号29で示す配列をあげることができる。なお、本発明のSD配列をコードするDNAを得る方法としては、既存のプラスミドであるpUBTZ2(バチルス・ステアロサーモフィルス由来の中性プロテアーゼ遺伝子をクローニングした大腸菌及び枯草菌間のシャトルベクター、特許文献7)よりPCR反応で当該DNA断片を増幅して得る方法、及び化学合成によって直接前記DNAを得る方法が例示できる。
【0022】
本発明の宿主はバチルス属細菌であれば特に限定がないが、この中でも特にタンパク質分泌能が高く、かつ病原性がない枯草菌(バチルス・ズブチリス、Bacillus subtilis)が好ましい。
【0023】
本発明で用いる発現プラスミドにおいて、プロモーターはバチルス属細菌内で機能するものであればよく、一例として、特許文献4で開示の、−35領域がTTTTCC、−10領域がTATTGTからそれぞれなるプロモーターがあげられる。また、プロモーター、及びその間に介在するDNAを得るには、公知の化学合成によって直接前記DNAを得る方法、天然由来のDNAから制限酵素で切り出して得る方法、既存のDNAからPCR反応により核酸増幅して得る方法等により得られる。
【0024】
本発明で用いる発現プラスミドにおいて、分泌発現に用いるシグナルペプチドはバチルス属細菌内で機能するものであればよく、一例として、特許文献3の請求項4で開示の、MKRKMKLRSFGVAAGLA(配列番号30)で示される耐熱性中性プロテアーゼのシグナルペプチド、及び非特許文献2で開示のMKRKMKMKLASFGLAAGLAAQVFLPYNALA(配列番号31)からなるペプチドがあげられる。前記シグナルペプチドをコードするDNAを得る方法としては、既存の遺伝子からPCR反応により前記DNAを核酸増幅して得る方法、また化学合成法によって直接前記DNAを得る方法を例示できる。
【0025】
本発明において分泌発現を目的とするタンパク質に特に限定はないが、アミラーゼ、プロテアーゼ、抗体といった天然においても細胞外に分泌生産されるタンパク質が好ましい。また前記タンパク質をコードする遺伝子を得る方法にも特に限定はなく、抗体を目的タンパク質とした場合の一例として、特許文献8に開示の抗エストラジオール抗体遺伝子取得法があげられる。また、特許文献8に開示の、クローニング済みの遺伝子をプラスミドから切り出す方法や、前記遺伝子断片をPCR反応等の方法により増幅させる方法も用いることもできる。
【0026】
本発明で用いる発現プラスミドにおける、もう一つの構成要素であるタンパク質発現の構造遺伝子群の終点に位置するターミネーターもバチルス属細菌内で機能するものであればよく、一例として、特許文献5の請求項3で開示の、CATCAGTGGGGGATTTTTTCCTCCACTGATG(配列番号32)を含む配列があげられる。
【0027】
本発明のタンパク質製造は、任意のプロモーターの下流に本発明のSD配列を配置し、その下流に任意のシグナルペプチドをコードするDNA配列を介して、所望のタンパク質をコードする遺伝子を持つDNA配列を配置し、さらにその下流に任意のターミネーターを配置した発現プラスミドを作成し、前記プラスミドをバチルス属細菌に導入することでタンパク質製造を行なう。即ち、一般的には、5’末端側から、プロモーター、SD配列、シグナルペプチド、目的タンパク質遺伝子、ターミネーターの順に配置し、発現プラスミドを作成する。
【0028】
発現培養に用いる条件は、バチルス属細菌の培養で通常設定する条件の中から適宜選択すればよい。培養温度は20から40℃が好ましく、特に25から30℃が好ましい。培養時のpHは6から8が好ましい。培養に用いる培地としてはバチルス属細菌が増殖し、かつ目的のタンパク質が分泌発現可能な培地を適宜選択すればよい。培養時間は、目的のタンパク質が分泌発現するのに適する時間であれば任意に設定できるが、通常は数時間から100時間の間に設定される。
【0029】
発現したタンパク質を定量する方法としては一般的なSDS−PAGEでタンパク質を分離した後に色素や免疫学的方法で染色して比色定量する方法、及びELISA法を例示でき、また酵素等を発現する場合には前記酵素の活性を測定する方法なども用いることができる。特に、抗体やレセプターなどを製造する場合はELISA法による活性定量法が簡便であるため好ましい。
【0030】
ELISA法におけるタンパク質の組み合わせは目的のタンパク質が定量できる方法であれば特に限定されないが、一例として、目的抗体に対する抗原、目的抗体、酵素標識抗目的抗体、の順に重ねたサンドイッチ法をあげることができる。ここで酵素標識抗目的抗体の一例として、アルカリフォスファターゼや西洋ワサビペルオキシダーゼ等の酵素で標識された抗目的抗体をあげることができる。
【0031】
また、ELISAの検出法についても特に限定されないが、標識に用いた酵素の特異的発色試薬、蛍光試薬又は化学発光試薬が市販されており、それらを標識に用いた酵素に応じて適宜選択し使用することができる。例えば西洋ワサビペルオキシダーゼを用いた場合は発色基質をペルオキシダーゼと過酸化水素で酸化反応させて比色定量する方法があり、例えばTMB 2−Component Microwell Peroxidase Substrate Kit(商品名、フナコシ社製)といった市販の試薬で発色させた後、マイクロタイタープレートリーダMPR4i(商品名、東ソー社製)といった市販の測定装置で比色定量することができる。
【発明の効果】
【0032】
本発明は、バチルス属細菌由来中性プロテアーゼ遺伝子中、先行文献中には特別な機能を持たない配列として公開された配列の一部をシャイン・ダルガノ(SD)配列として使用することで、バチルス属細菌に所望の高等生物由来タンパク質を分泌生産することが可能なタンパク質の製造方法を提供している。本発明により得られた、タンパク質は、菌体外に活性型の状態で分泌される。これにより、タンパク質抽出のための菌体破砕工程や、変性剤によるタンパク質の活性型への再構築が不要となり、効率よく所望のタンパク質を得ることができる。また、非特許文献2で開示のシャイン・ダルガノ(SD)配列と比較し、プロモーター部とシグナル配列部が短いDNA配列であっても分泌生産が可能となったことから、ベクターの安定化と高発現性が期待される。
【図面の簡単な説明】
【0033】
【図1】プラスミドpUCHS1L作成の工程図。図中、VH+CHは抗体のH鎖のVH領域及びCH1領域の遺伝子断片、stopはストップコドン、VL+CLは抗体L鎖のVL領域及びCL領域の遺伝子断片、Prは耐熱性中性プロテアーゼ由来のプロモーター領域、SDは本発明のシャイン・ダルガノ(SD)配列(配列番号29)、SPは耐熱性中性プロテアーゼ由来のシグナルペプチド領域、Amprはアンピシリン耐性遺伝子領域であることを示し、EcoRI、NheI、KpnI、PstI、SphIは対応する制限酵素の切断サイトを示す。
【図2】プラスミドpUCSHS1L作成の工程図。図中の略号は図1に準ずる。
【図3】プラスミドpUBCH1LΔt作成の工程図。図中の略号のうち、Kmrはカナマイシン耐性遺伝子領域であることを示し、その他は図1に準ずる。
【図4】プラスミドpUBCH1L作成の工程図。図中の略号のうち、Kmrはカナマイシン耐性遺伝子領域、TTは耐熱性中性プロテアーゼ由来のターミネーター配列であることをそれぞれ示し、その他は図1に準ずる。
【図5】プラスミドpUBCH2Lの概略図。図中の略号は図4に準ずる。
【図6】プラスミドpUBCH3Lの概略図。図中の略号のうち、Supは13塩基の付加配列(配列番号33)であることを示す。その他は図4に準ずる。
【図7】プラスミドpUBCH2Lで形質転換した枯草菌DB117株(黒丸)、DB104株(白丸)、及びMT2株(白三角)を25℃、30℃、又は37℃で24時間培養したのちの、培養上清中の抗体力価を測定した結果を示す。図中の各シンボルは各条件で24点の同一の実験を行なった際の平均値を示す。また、破線は標準偏差を示す。
【図8】プラスミドpUBCH1L、pUBCH2L、又はpUBCH3Lで形質転換した枯草菌DB117株を30℃で24時間培養後の、培養上清中の抗体力価を測定した結果を示す。図中の棒は各形質転換体を24回培養した結果の平均値を示す。また、棒に示した縦線は標準偏差を示す。
【実施例】
【0034】
以下に本発明の実施例を示すが、本発明はこれらに限定されるものではない。
【0035】
実施例1 発現ベクターpUBCH1Lの作成
(1)インサート配列(プロモーターからシグナル配列を含む塩基配列)の合成
(1−1)配列番号1から12(配列番号1は配列番号13の1から40番目の塩基に、配列番号2は配列番号13の35から79番目の塩基の相補鎖に、配列番号3は配列番号13の73から115番目の塩基に、配列番号4は配列番号13の108から152番目の塩基の相補鎖に、配列番号5は配列番号13の144から188番目の塩基に、配列番号6は配列番号13の179から223番目の塩基の相補鎖に、配列番号7は配列番号13の217から261番目の塩基に、配列番号8は配列番号13の254から298番目の塩基の相補鎖に、配列番号9は配列番号13の291から335番目の塩基に、配列番号10は配列番号13の327から371番目の塩基の相補鎖に、配列番号11は配列番号13の366から410番目の塩基に、配列番号12は配列番号13の400から435番目の塩基の相補鎖に、それぞれ相当)に示すオリゴヌクレオチドを混合し、タカラバイオ社製DNAポリメラーゼ(商品名、PrimeSTAR HS DNA Polymerase)を用いて1回目のPCRを行ないDNAを増幅した。PCR反応は94℃で5分加熱した後、94℃での加熱(30秒)、57℃でのアニーリング(30秒)及び72度での伸張反応(45秒)のサイクルを25回繰り返した後、72℃で7分加温することにより行なった。
(1−2)1回目のPCR反応後の反応液をキアゲン社製PCR精製キットで精製して50μLのDNA溶液を得た。
(1−3)(1−2)で得られたDNA溶液のうちの1μLを鋳型とし、配列番号1及び12のオリゴヌクレオチドをプライマーとして2回目のPCR反応を、1回目と同じ条件で行なった。
(1−4)2回目のPCR反応後の反応液を、アガロースゲル電気泳動より約400bpのDNA断片を切出し、キアゲン社製ゲル抽出キットにより精製して配列番号13に示すインサート配列(5’末端付近にEcoRIサイトとSacIサイトを含み、中央部に中性プロテアーゼプロモーター配列、本発明のシャイン・ダルガノ(SD)配列(配列番号29、325から339番目の塩基)、および中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、340から429番目の塩基)を含み、3’末端付近にNheIサイトを含む)を得た。
(1−5)2回目のPCR反応用オリゴヌクレオチドとして、配列番号14及び15(配列番号14は配列番号16の1から34番目の塩基に、配列番号15は配列番号16の396から433番目の塩基の相補鎖に、それぞれ相当)を用いた他は(1−3)から(1−4)と同様な操作を行ない、配列番号16に示すインサート配列(5’末端付近にKpnIサイトを含み、中央部に中性プロテアーゼプロモーター配列、本発明のSD配列(配列番号29、319から333番目の塩基)、及び中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、334から423番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
【0036】
(2)インサート配列(SD配列からシグナル配列を含む塩基配列)の合成
(2−1)1回目のPCR反応用オリゴヌクレオチドとして、配列番号10、11、15、17(配列番号17は配列番号18の1から32番目の塩基に相当)、2回目のPCR反応用オリゴヌクレオチドとして、配列番号15及び17を用いた他は、(1−1)から(1−4)と同様な操作を行ない、配列番号18に示すインサート配列(5’末端付近にKpnIサイトを含み、中央部に本発明のSD配列(配列番号29、13から27番目の塩基)と中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、28から117番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
(2−2)2回目のPCR反応用オリゴヌクレオチドとして、配列番号15及び19(配列番号19は配列番号20の1から36番目の塩基に相当)を用いた他は、(2−1)と同様の操作を行ない、配列番号20に示すインサート配列(5’末端付近にKpnIサイトと13塩基の付加配列(配列番号33、13から25番目の塩基)を含み、中央部に本発明のSD配列(配列番号29、26から40番目の塩基)と中性プロテアーゼシグナル配列(配列番号31のペプチドをコードする塩基配列、41から130番目の塩基)を含み、3’末端付近にPstIサイトを含む)を得た。
【0037】
(3)抗体H鎖遺伝子断片(VHからCH1領域)の増幅
特許文献8で開示の方法で調製された抗エストラジオール抗体発現プラスミドを鋳型とし、配列番号21及び22に示されるオリゴヌクレオチドをプライマーとしたPCR反応により抗体H鎖遺伝子断片(VHからCH1領域)を増幅した。PCR反応は94℃で5分加熱した後、94℃での加熱(30秒)、57℃でのアニーリング(30秒)及び72度での伸張反応(45秒)のサイクルを25回繰り返した後、72℃で7分加温することにより行なった。増幅した遺伝子はキアゲン社製PCR精製キットで精製した。
【0038】
(4)抗体L鎖遺伝子断片(VLからCL領域)の増幅
プライマーとして配列番号23及び24に示されるオリゴヌクレオチドを用いた他は、(3)と同様にPCR反応を行ない、抗体L鎖遺伝子(VLからCL領域)の増幅断片を得た。
【0039】
(5)ターミネーターの増幅
鋳型として特許文献7で開示のプラスミドpUBTZ2を、プライマーとして配列番号25及び26に示されるオリゴヌクレオチドを用いた他は、(3)と同様にPCR反応を行ない、中性プロテアーゼターミネーターの増幅断片(配列番号34、23から53番目の塩基は配列番号32と同じ)を得た。
【0040】
(6)発現ベクターの構築
(6−1)(3)で調製した抗体H鎖遺伝子断片(VHからCH1領域)を制限酵素EcoRI及びKpnIで、(4)で調製したL鎖抗体遺伝子断片(VLからCL領域)を制限酵素PstI及びSphIで、(1−5)で調製した配列番号16に示したインサート配列をKpnI及びPstIで、それぞれ切断した後、それぞれキアゲン社PCR精製キットで精製した。
(6−2)プラスミドpUC118(タカラバイオ製)をEcoRIとSphIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約3kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−3)(6−2)の断片に、(6−1)で調製した制限酵素処理済の抗体H鎖遺伝子断片、配列番号16に示すインサート配列及び抗体L鎖遺伝子断片を、T4DNAリガーゼを用いて結合し、プラスミドpUCHS1L(図1)を合成した。
(6−4)プラスミドpUCHS1L(図1)をEcoRIとNheIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約4.8kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−5)(6−4)で得られた断片に、予めEcoRIとNheIで切断してキアゲン社PCR精製キットで精製した配列番号13に示すインサート配列をT4DNAリガーゼを用いて結合し、プラスミドpUCSHS1L(図2)を合成した。
(6−6)プラスミドpUCSHS1L(図2)を制限酵素SacIとHindIIIで切断し、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約2kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製し、5’末端側から、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、抗体H鎖遺伝子断片(VHからCH1領域)、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、L鎖抗体遺伝子断片(VLからCL領域)より構成されるDNA断片を得た。
(6−7)特許文献7で開示のpUBTZ2を、SnaBI及びHindIIIで消化後、アルカリフォスファターゼによる脱リン酸化処理後、1%アガロースゲル電気泳動からの切出しにより約5kbpの断片を調製した。
(6−8)(6−7)で得られた断片に、予めT4DNAキナーゼ処理によりリン酸化した2つのオリゴDNA(配列番号27及び28)をT4DNAリガーゼにより結合させプラスミドpUBC1(図3)を合成した。
(6−9)プラスミドpUBC1(図3)を制限酵素SacI及びHindIIIで切断し、さらにアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約5kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。
(6−10)(6−9)で得られた断片と、(6−6)で調製した、5’末端側から、プロモーター中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、抗体H鎖遺伝子断片(VHからCH1領域)、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、中性プロテアーゼシグナル配列、L鎖抗体遺伝子断片(VLからCL領域)より構成されるDNA断片を、T4DNAリガーゼを用いて結合し、プラスミドpUBH1LΔt(図3)を合成した。
(6−11)プラスミドpUBH1LΔt(図3)を、制限酵素SphI及びHindIIIで切断してアルカリフォスファターゼ処理により脱リン酸化した後、アガロースゲル(ゲル濃度1%)電気泳動を行ない、約7kbの断片をキアゲン社製ゲル抽出キットを用いて切出し精製した。この断片に(5)で合成したターミネーター断片をT4DNAリガーゼを用いて結合し、プラスミドpUBCH1L(図4)を合成した。
【0041】
実施例2 発現ベクターpUBCH2Lの作成
配列番号16のインサート配列の代わりに(2−1)で調製した配列番号18を用いた他は、実施例1の(6)と同様の操作を行ない、プラスミドpUBCH2L(図5)を合成した。当該プラスミドと実施例1で調製したプラスミドpUBCH1Lの相違は、前者の抗体H鎖遺伝子とL鎖遺伝子をつなぐ断片が、終止コドン、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成されるのに対し、後者では終止コドン、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成される点が異なる。
【0042】
実施例3 発現ベクターpUBCH3Lの作成
配列番号16のインサート配列の代わりに(2−2)で調製した配列番号20を用いた他は、実施例1の(6)と同様の操作を行ない、プラスミドpUBCH3L(図6)を合成した。当該プラスミドと実施例1で調製したプラスミドpUBCH1Lの相違は、前者の抗体H鎖遺伝子とL鎖遺伝子をつなぐ断片が、終止コドン、13塩基の付加配列(AAATACAAAGAAT、配列番号33)、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成されるのに対し、後者では終止コドン、中性プロテアーゼプロモーター、本発明のSD配列(配列番号29)、及び中性プロテアーゼシグナル配列により構成される点が異なる(図6)。
【0043】
実施例4 枯草菌への形質転換
(1)大腸菌JM103株のコンピテントセルをHanahanの方法(非特許文献3)に従って作成し、発現ベクターpUBCH1L、pUBCH2L又はpUBCH3Lで形質転換した。これらの形質転換大腸菌を100μg/mLのアンピシリンを含むLB培地で一晩培養した。この培養液からキアゲン社プラスミド抽出キットを用いて各プラスミドの水溶液30μLを調製した。
(2)枯草菌MT2株、DB104株又はDB117株のコンピテントセルを公知の方法(非特許文献4)に従って調製した。前記細胞懸濁液100μLに、(1)で調製した各プラスミド水溶液5μLを加えて、37℃で1時間、穏やかに振とうした後、LB培地900μLを加えて37℃で1.5時間、激しく振とうして培養した。(3)(2)の培養液より遠心分離で菌体を回収し、100μLの生理食塩水に再懸濁し、50μg/mLのカナマイシンを含むLB平板培地に10μLまたは90μLを塗布して37℃で一晩静置培養した。生育したカナマイシン耐性のコロニーより目的のプラスミドを持つものを選別し、目的の形質転換体枯草菌菌株を調製した。
【0044】
実施例5 抗体遺伝子の発現に対する菌株と温度の影響
(1)実施例4で調製した形質転換体のうち、プラスミドpUBCH2Lで形質転換した枯草菌MT2、DB104及びDB117株を2PY培地(ポリペプトン 40g/L、バクト酵母エキス 5g/L、グルコース 20g/L、MgSO4・7H2O 0.1g/L、FeSO4・7H2O 0.01g/L、MnSO4・4H2O 0.01g/L、ZnSO4・7H2O 0.001g/L)0.75mLを入れた96穴深穴プレート(BM6030S深穴プレート角型V底、ビーエム機器社製)に植菌し、温度25℃、30℃又は37℃にて、バイオシェーカーMBR−022UP(商品名、タイテック社製)を用いて1200rpmの振とう速度で24時間培養した。
【0045】
(2)(1)の培養液を遠心分離して菌体を除去し、上清液中の抗体力価を以下の方法によりELISA法で測定した。
(2−1)予めエストラジオール標識牛血清アルブミンを結合させた後にパーフェクトブロック(商品名、ピアス社製)でブロッキングしておいた96穴プレートに、培養上清100μLを入れて37℃で1時間抗原抗体反応を行なった後、溶液を除去した。
(2−2)西洋ワサビペルオキシダーゼ修飾抗マウス抗体抗体溶液を添加して37℃で1時間抗原抗体反応を行なった後、再度溶液を除去した。
(2−3)西洋ワサビペルオキシダーゼの発色試薬(TMB 2−Component Microwell Peroxidase Substrate Kit(商品名)、フナコシ社製)50μLを添加して適当な色調になるまで発色を行ない、50μLの10%リン酸水溶液で反応を停止し、450nmの吸光度を測定した。ブランクとして培養上清の代わりに2PY培地を用いて同様の反応を行なって450nmの吸光度を測定し、検体の450nmの吸光度からブランクの吸光度を引くことにより抗体力価とした。
【0046】
ELISAの結果を図7に示す。いずれの菌株も37℃で培養した場合には培養上清中に抗体力価は確認できなかったものの、25℃又は30℃で培養した場合には明らかな抗体力価が確認できており、本発明のSD配列を含んだ発現プラスミドを枯草菌に形質転換することにより、抗原への結合活性を保持した抗体が得られることが示された。なお、DB104株及びDB117株では30℃での抗体生産が最も多かったが、MT2株における30℃での生産性は他の菌株より低かった。
【0047】
実施例6 プラスミド構成の違いにおける抗体遺伝子の発現の影響
(1)発現プラスミドpUBCH1L、pUBCH2L又はpUBCH3Lで形質転換した枯草菌DB117株を、培養温度が30℃で実施例5と同様に培養し、遠心分離して培養上清を得た。
【0048】
(2)培養上清中の抗体力価を実施例5と同様に測定した。
【0049】
結果を図8に示す。使用した発現プラスミドは抗体H鎖遺伝子とL鎖遺伝子を連結した配列において、pUBCH1Lでは中性プロテアーゼ由来プロモーター、本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列を含み、pUBCH2Lでは本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列のみを含み、またpUBCH3Lでは13塩基の付加配列(配列番号33)と本発明のSD配列(配列番号29)および中性プロテアーゼ由来シグナル配列を含む点で異なるが、これらの相違による抗体生産性への影響は認められなかった。図8には抗エストラジオール抗体産生ハイブリドーマ(特許文献8)の培養上清より調製した標準抗体(約10μg/mL)の測定結果も示した。発色量の比較から前記3つの組換え枯草菌による抗体生産量は約10μg/mLと推定された。
【特許請求の範囲】
【請求項1】
少なくともAAAGGAGGAからなる配列を含む、シャイン・ダルガノ(SD)配列。
【請求項2】
請求項1に記載のシャイン・ダルガノ配列を含む発現プラスミドを宿主に形質転換することで得られた、遺伝子組み換え体。
【請求項3】
請求項2に記載の遺伝子組み換え体を用いた、タンパク質の製造方法。
【請求項4】
タンパク質が抗体または抗体の断片である、請求項3に記載のタンパク質の製造方法。
【請求項1】
少なくともAAAGGAGGAからなる配列を含む、シャイン・ダルガノ(SD)配列。
【請求項2】
請求項1に記載のシャイン・ダルガノ配列を含む発現プラスミドを宿主に形質転換することで得られた、遺伝子組み換え体。
【請求項3】
請求項2に記載の遺伝子組み換え体を用いた、タンパク質の製造方法。
【請求項4】
タンパク質が抗体または抗体の断片である、請求項3に記載のタンパク質の製造方法。
【図1】


【図2】
【図3】
【図4】
【図5】

【図6】

【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2010−166905(P2010−166905A)
【公開日】平成22年8月5日(2010.8.5)
【国際特許分類】
【出願番号】特願2009−244954(P2009−244954)
【出願日】平成21年10月23日(2009.10.23)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成22年8月5日(2010.8.5)
【国際特許分類】
【出願日】平成21年10月23日(2009.10.23)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]