
トロポロンをCu(II)と複合化させることを含むトロポロンを検出する方法
生物学的サンプル由来の微量のトロポロンを回収および分析する方法が考案される。
【発明の詳細な説明】
【発明の開示】
【0001】
本発明は、分析化学の分野および細胞培養からの組換えタンパク質の生産に関する。本発明は、動物細胞培養のための細胞培養添加物であり、かつ細胞培養上清から精製された製品タンパク質を汚染するおそれのあるトロポロンの分析量を決定するための方法である。
【0002】
トロポロン、2-ヒドロキシ-2,4,6-シクロヘプタトリエン-1-オンは、EP-610 474 Blに記載されたように、タンパク質フリーの動物細胞培養液に添加されて、細胞による鉄の取り込みを可能にする周知の鉄キレート剤である。トロポロンは、その意味では細菌で知られた天然のシデロフォアのように振舞う。トロポロンは、適切な可逆性の鉄キレート性質と非毒性の両方に優れ、適切な投与量では任意の細胞機能を妨害せず、かつ動物細胞の増殖に悪影響を与えない。
【0003】
化学的合成が行われるとき、トロポロンを添加された細胞培養物から収集された任意の生物薬学的製品タンパク質には、精製過程におけるトロポロンの混入による微量の汚染についての試験を受ける必要がある。トロポロンの十分な分離を可能にする高感度の分析方法と大雑把な分析方法が必要とされる。
【0004】
本発明の目的は、細胞培養の上清またはタンパク質の溶液(proteinaceous solution)、特に、1 mg/mL以上の濃度に富んだ製品タンパク質を含むタンパク質の溶液からトロポロンを分析するための分析学的方法を提供することである。
【0005】
この目的は、十分な量の製品タンパク質を含む動物細胞培養上清またはタンパク質の溶液からトロポロンまたはその誘導体を分析するための本発明の方法によって解決される。
【0006】
前記方法は、以下の工程を含む:
a.トロポロンまたはその誘導体をタンパク質から分離する工程、および前記工程の前または後に、
b.溶液中でトロポロンまたはその誘導体をCu(II)イオンと複合化させる工程、および
c.疎水性固定相と移動相(好ましくは水混和性移動相)(移動相はCu(II)イオンとイオン対試薬の両方を含む)を備える逆相HPLCによってトロポロンまたはその誘導体を検出する工程。
【0007】
トロポロンは、2-ヒドロキシ-2,4,6-シクロヘプタトリエニル-1-オンである。トロポロンの合成は、Doering et al. , J. Am. Chem. Soc. (1951), 73:828-838に記載されている。本発明によれば、トロポロンの適切な誘導体は、第一鉄または第二鉄をキレートすることができる誘導体であり、かつ細胞機能に悪影響を与えずに(すなわち、毒性の副作用がない)動物細胞培養における膜透過性のシデロフォア鉄キレート物質として使用することができる。本発明の範囲外である毒性のトロポロン誘導体の例には、例えば、主要な細胞機能を激しく乱す細胞に対する強い毒性のあるコルヒチンまたはコルヒセインがある(これらをシドロフォア性質について生細胞で使用しまたは試験することはできない)。好ましくは、キレート性質を与えるために、本発明による好適なトロポロン誘導体は、上述の定義を満たし、かつ以下の構造Iを有する。
【化1】

【0008】
R1, R2, R3, R4は、単独または組み合わせで、水素、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラケニル、アラルシニル、ヘテロアリール、ヘテロアラルキル、ヘテロアラルケニル、またはヘテロアラルキニル基とすることができる(但し、R1=R2=R3=R4= Hは除かれる−この場合は誘導体ではなくトロポロンそのものである)。R1〜R4基は、さらに例えば、必然的に両親媒性のシデロフォアの膜透過性質を妨害しない、ハロゲン、ケト、アミド官能基で置換されていてもよい。組み合わせとは、R1, R2, R3および/またはR4が、上述した化学種で環状の置換基を形成することを意味する。
【0009】
さらに好ましい実施形態において、本発明の方法によって分析されるのはトロポロンのみである。トロポロンの誘導体は、この方法において除かれる。トロポロンは鉄キレート性質については非常によく知られているが、Cuキレート性質についてはあまりよく知られていない。トロポロン誘導体と明示的に関係しない、本明細書中に記載されたサンプル製剤のアッセイ方法および側面の全ての好ましい実施形態は、同様にこの好ましい実施形態に適用する。
【0010】
逆相HPLCは、当業者に周知であり、例えばペプチドのHPLCが日常的に使用されている。逆相HPLCにおいて、固定相は、強い機械的強度をもつ不活性な支持材料、例えば典型的には、シリカベースの支持材料、被覆アルミナベースの支持材料、またはメタクリルポリマーの支持材料からなる。逆相のために、疎水性官能基が支持体またはマトリックス材料に共有結合している。逆相の官能基は、例えばブチル、シアノ、ジビニルフェニル、フェニル、ヘキシル、デシル、ドデカシル、オクタデシルとすることができる。また、当業者に周知である、十分な疎水性と分離性質を有するいわゆる遮蔽化または包埋化逆相カラムを使用することができる。HPLCのための固定相を提供する好適なHPLCカラムは、例えばSupelco/Fluka Chemie, Switzerland, または Waters/MA, U.S.A.から商業的に利用可能である。
【0011】
好ましくは、逆相HPLC固定相は、アルカニル、好ましくはC10〜C20アルカニル、最も好ましくはC18オクタデシルアルカニルで官能化されたマトリックスから作製される。さらに好ましくは、好ましい官能基との結合において、マトリックス材料はシリカである。本発明による好ましいHPLC固定相はアルキルシラン、より好ましくはC10〜C20アルキルシラン、最も好ましくはC18オクタデシルシランである。これらの好ましい態様は、イオン対試薬を加える本発明の方法において使用されるとき(特に、以下に記載の好ましい実施形態において使用されるとき)、トロポロンピークの分離、カラム容積、および固定相への吸着が原因の感度喪失の回避の観点において得に有利である。
【0012】
本発明によるイオン対試薬は、逆相固定相に対する一定の親和力を有する間に、Cu-トロポロン複合体と少なくとも一時的に結合することができる、少なくとも一つのイオン化可能なカルボキシル基を含む試薬であり、固定相上にCu-トロポロン複合体を保持することができる。つまり、本発明によるイオン対試薬は、カルボキシル基部分を有し、かつ20℃での0.1 % (w/v)CuSO4の存在下における10%アセトニトリル水の逆相C18 HPLCカラム上でのCu(II)-トロポロン複合体の保持時間を長くすることによって定義される。
【0013】
好ましくは、イオン対試薬は、トリフルオロ酢酸(TFA)よりも疎水性である。TFAは、ペプチドの逆相HPLCにおいて日常的に使用され、ほぼ世界的基準と考えられている。そして、ここでは逆相HPLCのためのイオン対試薬が必要である。より疎水性であるイオン対化合物は、その酸形態において、TFAと比較したその誘電体材料定数のより低い数値によって特徴づけられる。好適なイオン対試薬には、例えばテトラブチルアンモニウムフタレート、ヘプタフルオロブチル酸、メチルスルホン酸、ヘキシルスルホン酸またはこれらの塩がある。より好ましくは、イオン対試薬は、その特定の誘電率の実数値によって判定されたとき、メチルスルホン酸およびヘキシルスルホン酸の疎水性と同等かまたはこれらの酸の疎水性の範囲内にある疎水性(hydrobobicity)を有する。より好ましくは、イオン対試薬は、ブチルスルホン酸、ペンチルスルホン酸、およびヘキシルスルホン酸またはこれらの塩からなる群から選択される(アルキル置換基は、枝分れしていてもしていなくてもよいが、好ましくは枝分れしていない)。特に好ましい実施形態において、イオン対試薬は、ヘキシルスルホン酸、より好ましくはn-ヘキシル-スルホン酸、最も好ましくは移動相中において0.1〜5%(w/v)の濃度で使用される。
【0014】
本発明の目的のために、標準物質としてTFA、メチルスルホンおよびヘキシルスルホン酸を使用し、本発明の所定の逆相HPLC実施形態について分類する保持指数を与えることによって、逆相クロマトグラフィーの目的に適した疎水性性質をより正確に規定することが可能である。このような指数システムは、例えば溶媒の極性を定義するために使用される。(極性指数、 L. R. Snyder, J. Chromatogr., 92,223 (1974), J. Chromatogr. Sci., 16,223(1978)、およびイオン対試薬の極性/疎水性を規定する適切な指数が採用されてもよい)。
【0015】
好ましくは、イオン対試薬の濃度は、0.01〜10%(w/v)、より好ましくは0.1〜5%(w/v)、最も好ましくは0.11〜1%(w/v)である。特に好ましい実施形態において、ヘキシルスルホン酸が、0.1〜0.5%(w/v)の濃度で使用される。さらに特に好ましい実施形態において、このパラグラフに記載された上記濃度範囲内のヘキシルスルホン酸が、アルキルシラン固定相、好ましくはC18オクタデシル固定相との組み合わせにおいて使用される。
【0016】
HPLCの移動相は、極性の水混和性液体である。好ましくは、移動相は、1〜30%(v/v)、より好ましくは5〜20%(v/v)のアセトニトリルを含む。アセトニトリルは、少なくとも一つのさらなる極性溶媒と混合する。移動相はまた、前記極性溶媒とアセトニトリルに加えてさらなる溶媒を含んでいてもよい。
【0017】
好ましくは、さらなる極性溶媒は水であり、より好ましくは、移動相は少なくとも60%(v/v)水を含む。
【0018】
好ましくは、移動相は、Cu(II)イオンの供給源としてCuSO4を含み、Cu(II)-トロポロン複合体の形成を促進し、HPLC上での可逆的な複合体の解離を回避する。より好ましくは、CuSO4またはCu(II)イオンの濃度は、一般には移動相中において0.05%〜0.2%(w/v)、最も好ましくは0.7〜0.14 %(w/v)の範囲内にある。
【0019】
上述したように、タンパク質溶液からトロポロンまたはその誘導体を検出することが好ましい。より好ましくは、1mg/L以上の濃度に富んだ製品タンパク質を含むタンパク質溶液からトロポロンまたはその誘導体を検出する。
【0020】
さらなる好ましい実施形態において、タンパク質からの分離は、Cu(II)塩、好ましくはCuSO4を含むタンパク質とともにトロポロンまたはこれらの誘導体を沈殿させ、次に、該沈殿物を回収し、最後に、限外ろ過によって該沈殿物からタンパク質を除去することによって達成される。限外ろ過は、タンパク質または生化学者にとってよく知られた装置(すなわち、Amiconフィルター膜または該膜を使用するスピンフィルター)で行うことができる。この好ましい実施形態は、分離を含む単一工程において複合体形成を可能にし、オリジナルサンプルからのトロポロンまたはその誘導体の非常に効率的で完全な回収を可能にする。最も重要なのは、本発明の方法は、変性していないタンパク質を保管するために使用されるバッファー中に通常存在する妨害イオン化学種(リン酸塩、塩化物)(これらのイオン化学種は、トロポロンのためのHPLCアッセイを妨害する)を除去することができる。サンプルの脱塩はHPLCの操作に不要である。さらに、サンプルをHPLC上に搭載する前に脱塩する必要もない。対照的に、当該技術においてタンパク質の沈殿のために共通して使用される硫酸アンモニウム沈殿物は、トロポロンの不完全な回収をもたらす。沈殿は、0.5〜10% Cu(II)塩、より好ましくは1〜5%(w/v)Cu(II)塩で行われる。
【0021】
実験
標準的なPBSバッファー(生理学的リン酸緩衝液)で作製され、2000ng/mLのトロポロンを添加(spiked)された精製された完全なIgG抗体溶液由来のサンプル製剤について、異なる経路で試験を行った。細胞培養液は、一般に細胞培養の開始時点から約10μMのトロポロンを含む。
【0022】
異なるサンプルを処理する異なる方法を比較することによって、オリジナルサンプルから最大量のトロポロンを得る方法が決定される。最良の処理は、Cu(II)による沈殿である。この点について以下に詳述する。
【0023】
細胞培養物が30μMトロポロンの存在中で増殖するNSO細胞培養上清からの粗精製IgG抗体から誘導された処理中サンプルで十分に等しく行う方法が見出された。前記上清は、タンパク質Aセファロースクロマトグラフィーでのみ処理され、添加および未添加サンプルの両方について試験した。
【化2】
【0024】
Ultravap窒素乾燥装置中で乾燥した後、処理されたサンプルを、上述したRP HPLCによる分析のために200μL移動相中で再構成した。SPE: 500μLエタノールと500μL 5mM H2SO4で予め調整された、ポリスチレンジビニルベンゼン固相ポリマーから作製された重力操作 Strata X 3mL 固相抽出カートリッジ。
【0025】
1.CuS04サンプル沈殿
上述したように調製された標準トロポロン溶液を、2% CuSO4(w/v)の最終濃度になるまでCuSO4溶液と混合した。沈殿した塩を15000×rcfで15分間にわたる遠心分離によって取り除いた。
【0026】
Microcon(登録商標)(Millipore, USA) 10 kDA MW-カットオフ500μLフィルター(YM-10)を、400μL H2Oの添加によって平衡化し、続いて13000×rcfで40分間にわたって遠心分離した。その後、サンプル上清をMicroconフィルターに加えた。前記フィルターを13 000×rcfで40分間にわたって遠心分離した。その後、前記フィルターを100μL H2Oで洗浄し、ろ液を貯留した。貯留されたろ液を、窒素雰囲気下でガラス製バイアル中で乾燥させ、200μL移動相中で再構成させ、RP HPLCによって分析した。該方法は84%の収量を得た。一般的には、100〜2000ng/mL トロポロンを添加されたサンプルについて、これは証明された再現性のある回収を意味する。20〜40ng/mL範囲内の非常に少量のトロポロンを添加されたサンプルでは、回収率は94%程度であった。
【0027】
代わりに、この手順を、サンプルを乾燥しないで行った(乾燥は最適な手順で必要とされないことが解った)。ろ液をRP HPLCによって生で分析した。
【0028】
上記に簡単に記載した任意の処理方法は、<20%(固相抽出)および65%(Microcon限外ろ過)の平均回収率を得た。
【0029】
2.RP HPLC
定組成溶離、流速0.2 mL/分 25℃、242nmで検出および100μL注入容積の条件下において、固定相として新型クロマトグラフィー技術ACE C18 2.1mmカラムと、0.1% CuS04(w/v)/0.3%ヘキサンスルホン酸/水中10%アセトニトリル移動相とで、RP HPLCを行った。HPLCシステムは、Chemstationソフトウェア バージョンA6.04以上とともに、真空デガッサー、双対ポンプ、サーモスタット区画カラムおよび自動サンプラーを含むAgilent 1100 HPLCシステムまたはこれと同等なものとした。
【0030】
この方法で分析されたトロポロン標準体についての溶出曲線を図1aおよび1dに示す。図1bおよび1cは、異なるイオン対試薬を使用したことを除いて(代わりにTFAおよびMSA s. section 4)、同じ移動相および固定相を用いた溶出曲線を示す。
【0031】
図2a〜2bは、大規模生産から誘導された処理中の大量の完全なIgG抗体の分析を示す。前記抗体を、タンパク質Aセファロースクロマトグラフィーによってのみ部分的に精製した。更なる説明は図の表題に示す。
【0032】
ACE C-18カラムと、Hamilton(会社の所在地?)PRPポリスチレン ジビニルベンゼン4.6 mm逆相カラムとの比較を行った。ポリスチレン ジビニルベンゼン固定相の水溶液についての耐用性は、トロポロン溶出の保持時間を潜在的に増加させるより高い極性の移動相(C18カラムと比較して)の使用を可能にするので、予め評価されたPRP-1重合カラムをCuSO4移動相での使用について再調査した。トロポロンは、アセトニトリル欠損中では15分後に0.1%CuSO4移動相で溶出させることができず(データは示していない)、様々なパーセンテージのアセトニトリル(定組成5%(続いて有機カラム洗浄工程)、21%、40%および3%〜11.5%勾配)について評価を行った。使用された流速は、25℃の温度で0.8mL/分で一定とした。
【0033】
その後、移動相を様々なアセトニトリル含量で評価し、これらの条件下の分析からの結果を表3に要約した。5%アセトニトリルの濃度で、許容可能な保持(k>1.0)、許容可能なテーリング(TF 3.02)および0.1μg/mLの再現可能な検出が達成された。しかしながら、2で示された0.1μg/mLの標準化されたピーク高さは、これらの条件を使用する検出の限界に近かった(表3)。さらに、この濃度でのピークのテーリングは、2.0μg/mL(TF 1.61)についてのものよりもかなり高かった。アセトニトリルのより高い濃度(21%および40%)では、トロポロンの保持はk>0.5であり、認容できなかった。
【表3】
【0034】
3.HPLCの実行
以下の全ての研究は、実験2のRP HPLC方法と実験1のサンプル調製方法を使用して行った。
【0035】
(a)直線性
トロポロン溶液を、0, 1, 5,10, 20,50, 100,500, 2000 および 4000 ng/mLトロポロンの濃度で、上述した抗体含有バッファー中において調製した。バッファー塩の除去は、2% CuSO4での沈殿によって行った。トロポロンサンプルをRP HPLCによって分析した。ピーク領域を、トロポロン濃度に対して記録およびプロットし、直線状の回帰プロットを構築した。直線状の作用範囲は、許容可能な正確性(≦5% CV)を有する直線状の反応(r2≧0.99)を与える濃度範囲として定義された。トロポロン量についての校正曲線を、5, 10, 20, 50, 100, 500, 2000 および 4000 ng/mLトロポロンの濃度で分析されたサンプルから構築した。各トロポロン濃度の測定の精度が分析された。直線状の反応が、1 ng/mL〜4000 ng/mLで回帰分析によって視覚的に観察された。相関係数の二乗値(r2)は許容可能な直線性を示した(r2=0.9997)。
【0036】
日常的な計量プロセスのために、校正曲線を0 ng/mL〜2000 ng/mLの濃度で構築した(r2>0.99)。直線性を5つの異なる状況で調査し、回帰データを表1に要約した。
【表1】
【0037】
(b)特異性
サンプルバッファーの一連のピークからのトロポロンピークの分離(Rs)を、標準トロポロン溶液(2000ng/mL)について決定した。三重分析からのRs、sdおよび%CVの平均値を5つのアッセイについて記録した。
【0038】
予め議論したように、トロポロンから視覚的に分離されたサンプルバッファーの一連のピークは、トロポロンの直前に溶出した(セクション4.5.2; 図18 および 19)。このバッファーの一連のピークからのトロポロンの分離を、USPで定義された方法を使用する5つの別個のアッセイ中の三つのシステム適合性サンプル注入液について計算した。平均トロポロン分離因子(Rs)、sdおよび%CVを計算し、その結果を表2に要約した。
【表2】
【0039】
平均分離因子は全て≧1.5であり、前述のバッファー関連ピークからの許容可能な分離を示し、許容可能な再現性(CV<3.0%)を実証した。妨害ピークは、サンプル製剤または最終製品サンプルから観察されなかった。これは、トロポロンの検出および定量化のためのRP HPLCアッセイの特異性を確認した。
【0040】
(c)検出/定量化(LOD/LOQ)の限界
直線性について分析されたトロポロンサンプルを使用して、トロポロン分析のためにLODおよびLOQを推定した。検出可能なシグナルを与える試験対象のトロポロンの最低濃度は、LODとして割り当てられた。許容可能な再現性(≦5% CV)で測定され得るトロポロンの最低濃度は、LOQとして定義された。これは、LOD約1 ng/mL の量で見出された; LOQは約5ng/mLとして決定され、この感度が本発明の検出方法を制限する。
【0041】
4.イオン対(IP)試薬とともに銅イオン複合体を使用するACE C18 2.1mmカラム上でのHPLC条件の評価
C18 HPLCカラム上での銅イオントロポロン複合体の分析は、様々な疎水性の三つの異なる試薬を使用する逆相イオン対で試験した。これらはトリフルオロ酢酸(TFA)、最も極性のメチルスルホン酸(MSA)、中程度の極性のヘキサンスルホン酸(HAS)、最も疎水性のものであった。イオン対試薬、0.1% CuSO4およびアセトニトリルを含む移動相が調製された。使用されたHPLCパラメータは、得られた一連のピーク高さとクロマトグラム試験測定値とともに表4に要約される。
【0042】
4.1 TFAイオン対
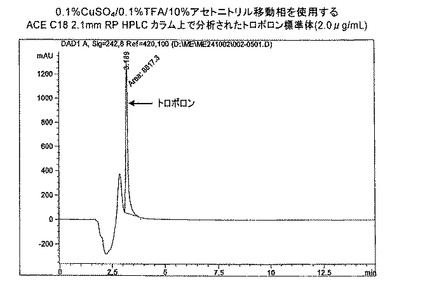
許容される移動相中の0.1% TFAを使用する標準トロポロンの保持は、十分には増加しなかった(k<1.0)(表4、図10)。これらの条件下での分析は、トロポロンのアッセイについて適切ではないと結論付けられた。
【0043】
4.2 MSAイオン対
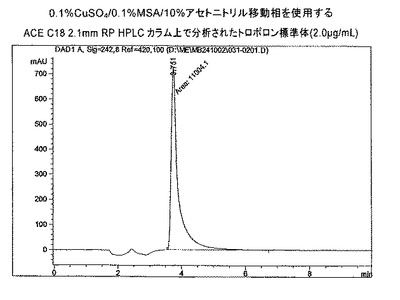
0.1% MSAを含む移動相は、5%アセトニトリルおよび0.1% MSAでの定組成溶出のもとで向上した保持時間を与えた(k≧2)。しかしながら、高いトロポロンピークのテーリングは、試験された標準の両濃度(0.1μg/mLおよび2.0μg/mL)について観察された(TF≧3.4)。0.1%MSAおよび10%アセトニトリルを含む移動相の使用(図11)は、5%アセトニトリリル/0.1%MSA(2の標準化されたピーク高さ)よりも0.1μg/mL標準トロポロンの向上した検出を実証したが(14の標準化されたピーク高さ、表4)、ピークのテーリングは広がった(TF12.05)。
【0044】
4.3 HASイオン対
トロポロン保持時間の顕著な増加は、0.1%HAS/10%アセトニトリル移動相(k≧3.4、表4)での定組成溶出を使用して達成された。さらに、0.1μg/mLの低トロポロン濃度についてのピーク高さの増加が観察され(27の標準化されたピーク高さ、表4)、試験対象の両トロポロン濃度についてピークのテーリングは許容可能かつ比較可能であった。さらに、ピーク領域の再現可能性は、2.0μg/mL(0.9% CV)および0.1 μg/mL(3.2% CV)トロポロンの濃度で許容可能であった(表4)。しかしながら、溶出したトロポロンは移動相ピークに近く、2つのピークはベースラインでの分離を実証しなかった(図12および13、Rs≦1.1)。トロポロンおよび移動相ピークは、アセトニトリルの勾配(10%〜42%)を使用して分離することはできず、分析温度を40℃まで増加(Rs≦1.0、表4)または温度を20℃、流速を0.1mL/分まで減少させて(Rs≦1.3)分離することもできなかった。
【0045】
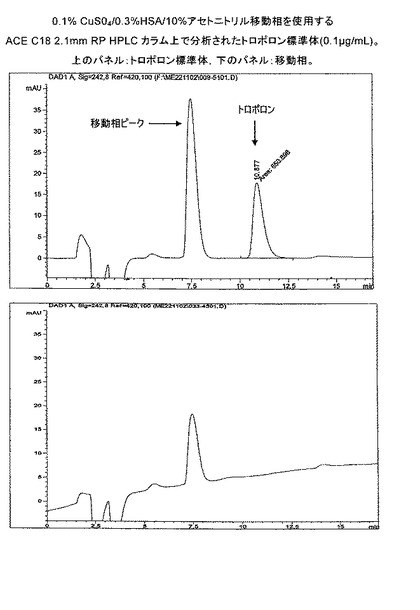
様々なイオン対(HSA)移動相の濃度について評価した。0.3% HSA/10% アセトニトリル/0.1% CuSO4移動相(流速0.2mL/分、25℃)は、両トロポロン濃度での移動相ピークからのトロポロンのベースラインでの分離を達成するために有効であった(Rs≧3.5、図14および15、表4)。流速の更なる減少(0.1mL/分)は、トロポロン分離の更なる向上を誘導しなかった(Rs≧3.8)。結論として、検出の許容可能な制限と、ピークの対称性と、標準トロポロンのアッセイについての再現性を備えたRP HPLC方法が確立された。
【表4】
【図面の簡単な説明】
【0046】
【図1a】0.1%CuSO4/0.3%HSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1b】0.1%CuSO4/0.1%TFA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1c】0.1%CuSO4/0.1%MSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1d】0.1% CuS04/0.3%HSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(0.1 μg/mL)。
【図2a】未添加IgG Lot 11859およびバッファー対照のクロマトグラムの例。
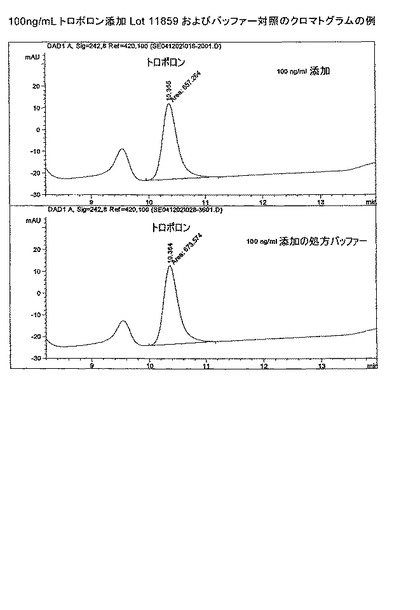
【図2b】トロポロン添加(10ng/mL)IgG Lot 11859およびバッファー対照のクロマトグラムの例。
【図2c】100ng/mLトロポロン添加IgG抗体Lot 11859およびバッファー対照のクロマトグラムの例。
【発明の開示】
【0001】
本発明は、分析化学の分野および細胞培養からの組換えタンパク質の生産に関する。本発明は、動物細胞培養のための細胞培養添加物であり、かつ細胞培養上清から精製された製品タンパク質を汚染するおそれのあるトロポロンの分析量を決定するための方法である。
【0002】
トロポロン、2-ヒドロキシ-2,4,6-シクロヘプタトリエン-1-オンは、EP-610 474 Blに記載されたように、タンパク質フリーの動物細胞培養液に添加されて、細胞による鉄の取り込みを可能にする周知の鉄キレート剤である。トロポロンは、その意味では細菌で知られた天然のシデロフォアのように振舞う。トロポロンは、適切な可逆性の鉄キレート性質と非毒性の両方に優れ、適切な投与量では任意の細胞機能を妨害せず、かつ動物細胞の増殖に悪影響を与えない。
【0003】
化学的合成が行われるとき、トロポロンを添加された細胞培養物から収集された任意の生物薬学的製品タンパク質には、精製過程におけるトロポロンの混入による微量の汚染についての試験を受ける必要がある。トロポロンの十分な分離を可能にする高感度の分析方法と大雑把な分析方法が必要とされる。
【0004】
本発明の目的は、細胞培養の上清またはタンパク質の溶液(proteinaceous solution)、特に、1 mg/mL以上の濃度に富んだ製品タンパク質を含むタンパク質の溶液からトロポロンを分析するための分析学的方法を提供することである。
【0005】
この目的は、十分な量の製品タンパク質を含む動物細胞培養上清またはタンパク質の溶液からトロポロンまたはその誘導体を分析するための本発明の方法によって解決される。
【0006】
前記方法は、以下の工程を含む:
a.トロポロンまたはその誘導体をタンパク質から分離する工程、および前記工程の前または後に、
b.溶液中でトロポロンまたはその誘導体をCu(II)イオンと複合化させる工程、および
c.疎水性固定相と移動相(好ましくは水混和性移動相)(移動相はCu(II)イオンとイオン対試薬の両方を含む)を備える逆相HPLCによってトロポロンまたはその誘導体を検出する工程。
【0007】
トロポロンは、2-ヒドロキシ-2,4,6-シクロヘプタトリエニル-1-オンである。トロポロンの合成は、Doering et al. , J. Am. Chem. Soc. (1951), 73:828-838に記載されている。本発明によれば、トロポロンの適切な誘導体は、第一鉄または第二鉄をキレートすることができる誘導体であり、かつ細胞機能に悪影響を与えずに(すなわち、毒性の副作用がない)動物細胞培養における膜透過性のシデロフォア鉄キレート物質として使用することができる。本発明の範囲外である毒性のトロポロン誘導体の例には、例えば、主要な細胞機能を激しく乱す細胞に対する強い毒性のあるコルヒチンまたはコルヒセインがある(これらをシドロフォア性質について生細胞で使用しまたは試験することはできない)。好ましくは、キレート性質を与えるために、本発明による好適なトロポロン誘導体は、上述の定義を満たし、かつ以下の構造Iを有する。
【化1】
【0008】
R1, R2, R3, R4は、単独または組み合わせで、水素、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラケニル、アラルシニル、ヘテロアリール、ヘテロアラルキル、ヘテロアラルケニル、またはヘテロアラルキニル基とすることができる(但し、R1=R2=R3=R4= Hは除かれる−この場合は誘導体ではなくトロポロンそのものである)。R1〜R4基は、さらに例えば、必然的に両親媒性のシデロフォアの膜透過性質を妨害しない、ハロゲン、ケト、アミド官能基で置換されていてもよい。組み合わせとは、R1, R2, R3および/またはR4が、上述した化学種で環状の置換基を形成することを意味する。
【0009】
さらに好ましい実施形態において、本発明の方法によって分析されるのはトロポロンのみである。トロポロンの誘導体は、この方法において除かれる。トロポロンは鉄キレート性質については非常によく知られているが、Cuキレート性質についてはあまりよく知られていない。トロポロン誘導体と明示的に関係しない、本明細書中に記載されたサンプル製剤のアッセイ方法および側面の全ての好ましい実施形態は、同様にこの好ましい実施形態に適用する。
【0010】
逆相HPLCは、当業者に周知であり、例えばペプチドのHPLCが日常的に使用されている。逆相HPLCにおいて、固定相は、強い機械的強度をもつ不活性な支持材料、例えば典型的には、シリカベースの支持材料、被覆アルミナベースの支持材料、またはメタクリルポリマーの支持材料からなる。逆相のために、疎水性官能基が支持体またはマトリックス材料に共有結合している。逆相の官能基は、例えばブチル、シアノ、ジビニルフェニル、フェニル、ヘキシル、デシル、ドデカシル、オクタデシルとすることができる。また、当業者に周知である、十分な疎水性と分離性質を有するいわゆる遮蔽化または包埋化逆相カラムを使用することができる。HPLCのための固定相を提供する好適なHPLCカラムは、例えばSupelco/Fluka Chemie, Switzerland, または Waters/MA, U.S.A.から商業的に利用可能である。
【0011】
好ましくは、逆相HPLC固定相は、アルカニル、好ましくはC10〜C20アルカニル、最も好ましくはC18オクタデシルアルカニルで官能化されたマトリックスから作製される。さらに好ましくは、好ましい官能基との結合において、マトリックス材料はシリカである。本発明による好ましいHPLC固定相はアルキルシラン、より好ましくはC10〜C20アルキルシラン、最も好ましくはC18オクタデシルシランである。これらの好ましい態様は、イオン対試薬を加える本発明の方法において使用されるとき(特に、以下に記載の好ましい実施形態において使用されるとき)、トロポロンピークの分離、カラム容積、および固定相への吸着が原因の感度喪失の回避の観点において得に有利である。
【0012】
本発明によるイオン対試薬は、逆相固定相に対する一定の親和力を有する間に、Cu-トロポロン複合体と少なくとも一時的に結合することができる、少なくとも一つのイオン化可能なカルボキシル基を含む試薬であり、固定相上にCu-トロポロン複合体を保持することができる。つまり、本発明によるイオン対試薬は、カルボキシル基部分を有し、かつ20℃での0.1 % (w/v)CuSO4の存在下における10%アセトニトリル水の逆相C18 HPLCカラム上でのCu(II)-トロポロン複合体の保持時間を長くすることによって定義される。
【0013】
好ましくは、イオン対試薬は、トリフルオロ酢酸(TFA)よりも疎水性である。TFAは、ペプチドの逆相HPLCにおいて日常的に使用され、ほぼ世界的基準と考えられている。そして、ここでは逆相HPLCのためのイオン対試薬が必要である。より疎水性であるイオン対化合物は、その酸形態において、TFAと比較したその誘電体材料定数のより低い数値によって特徴づけられる。好適なイオン対試薬には、例えばテトラブチルアンモニウムフタレート、ヘプタフルオロブチル酸、メチルスルホン酸、ヘキシルスルホン酸またはこれらの塩がある。より好ましくは、イオン対試薬は、その特定の誘電率の実数値によって判定されたとき、メチルスルホン酸およびヘキシルスルホン酸の疎水性と同等かまたはこれらの酸の疎水性の範囲内にある疎水性(hydrobobicity)を有する。より好ましくは、イオン対試薬は、ブチルスルホン酸、ペンチルスルホン酸、およびヘキシルスルホン酸またはこれらの塩からなる群から選択される(アルキル置換基は、枝分れしていてもしていなくてもよいが、好ましくは枝分れしていない)。特に好ましい実施形態において、イオン対試薬は、ヘキシルスルホン酸、より好ましくはn-ヘキシル-スルホン酸、最も好ましくは移動相中において0.1〜5%(w/v)の濃度で使用される。
【0014】
本発明の目的のために、標準物質としてTFA、メチルスルホンおよびヘキシルスルホン酸を使用し、本発明の所定の逆相HPLC実施形態について分類する保持指数を与えることによって、逆相クロマトグラフィーの目的に適した疎水性性質をより正確に規定することが可能である。このような指数システムは、例えば溶媒の極性を定義するために使用される。(極性指数、 L. R. Snyder, J. Chromatogr., 92,223 (1974), J. Chromatogr. Sci., 16,223(1978)、およびイオン対試薬の極性/疎水性を規定する適切な指数が採用されてもよい)。
【0015】
好ましくは、イオン対試薬の濃度は、0.01〜10%(w/v)、より好ましくは0.1〜5%(w/v)、最も好ましくは0.11〜1%(w/v)である。特に好ましい実施形態において、ヘキシルスルホン酸が、0.1〜0.5%(w/v)の濃度で使用される。さらに特に好ましい実施形態において、このパラグラフに記載された上記濃度範囲内のヘキシルスルホン酸が、アルキルシラン固定相、好ましくはC18オクタデシル固定相との組み合わせにおいて使用される。
【0016】
HPLCの移動相は、極性の水混和性液体である。好ましくは、移動相は、1〜30%(v/v)、より好ましくは5〜20%(v/v)のアセトニトリルを含む。アセトニトリルは、少なくとも一つのさらなる極性溶媒と混合する。移動相はまた、前記極性溶媒とアセトニトリルに加えてさらなる溶媒を含んでいてもよい。
【0017】
好ましくは、さらなる極性溶媒は水であり、より好ましくは、移動相は少なくとも60%(v/v)水を含む。
【0018】
好ましくは、移動相は、Cu(II)イオンの供給源としてCuSO4を含み、Cu(II)-トロポロン複合体の形成を促進し、HPLC上での可逆的な複合体の解離を回避する。より好ましくは、CuSO4またはCu(II)イオンの濃度は、一般には移動相中において0.05%〜0.2%(w/v)、最も好ましくは0.7〜0.14 %(w/v)の範囲内にある。
【0019】
上述したように、タンパク質溶液からトロポロンまたはその誘導体を検出することが好ましい。より好ましくは、1mg/L以上の濃度に富んだ製品タンパク質を含むタンパク質溶液からトロポロンまたはその誘導体を検出する。
【0020】
さらなる好ましい実施形態において、タンパク質からの分離は、Cu(II)塩、好ましくはCuSO4を含むタンパク質とともにトロポロンまたはこれらの誘導体を沈殿させ、次に、該沈殿物を回収し、最後に、限外ろ過によって該沈殿物からタンパク質を除去することによって達成される。限外ろ過は、タンパク質または生化学者にとってよく知られた装置(すなわち、Amiconフィルター膜または該膜を使用するスピンフィルター)で行うことができる。この好ましい実施形態は、分離を含む単一工程において複合体形成を可能にし、オリジナルサンプルからのトロポロンまたはその誘導体の非常に効率的で完全な回収を可能にする。最も重要なのは、本発明の方法は、変性していないタンパク質を保管するために使用されるバッファー中に通常存在する妨害イオン化学種(リン酸塩、塩化物)(これらのイオン化学種は、トロポロンのためのHPLCアッセイを妨害する)を除去することができる。サンプルの脱塩はHPLCの操作に不要である。さらに、サンプルをHPLC上に搭載する前に脱塩する必要もない。対照的に、当該技術においてタンパク質の沈殿のために共通して使用される硫酸アンモニウム沈殿物は、トロポロンの不完全な回収をもたらす。沈殿は、0.5〜10% Cu(II)塩、より好ましくは1〜5%(w/v)Cu(II)塩で行われる。
【0021】
実験
標準的なPBSバッファー(生理学的リン酸緩衝液)で作製され、2000ng/mLのトロポロンを添加(spiked)された精製された完全なIgG抗体溶液由来のサンプル製剤について、異なる経路で試験を行った。細胞培養液は、一般に細胞培養の開始時点から約10μMのトロポロンを含む。
【0022】
異なるサンプルを処理する異なる方法を比較することによって、オリジナルサンプルから最大量のトロポロンを得る方法が決定される。最良の処理は、Cu(II)による沈殿である。この点について以下に詳述する。
【0023】
細胞培養物が30μMトロポロンの存在中で増殖するNSO細胞培養上清からの粗精製IgG抗体から誘導された処理中サンプルで十分に等しく行う方法が見出された。前記上清は、タンパク質Aセファロースクロマトグラフィーでのみ処理され、添加および未添加サンプルの両方について試験した。
【化2】
【0024】
Ultravap窒素乾燥装置中で乾燥した後、処理されたサンプルを、上述したRP HPLCによる分析のために200μL移動相中で再構成した。SPE: 500μLエタノールと500μL 5mM H2SO4で予め調整された、ポリスチレンジビニルベンゼン固相ポリマーから作製された重力操作 Strata X 3mL 固相抽出カートリッジ。
【0025】
1.CuS04サンプル沈殿
上述したように調製された標準トロポロン溶液を、2% CuSO4(w/v)の最終濃度になるまでCuSO4溶液と混合した。沈殿した塩を15000×rcfで15分間にわたる遠心分離によって取り除いた。
【0026】
Microcon(登録商標)(Millipore, USA) 10 kDA MW-カットオフ500μLフィルター(YM-10)を、400μL H2Oの添加によって平衡化し、続いて13000×rcfで40分間にわたって遠心分離した。その後、サンプル上清をMicroconフィルターに加えた。前記フィルターを13 000×rcfで40分間にわたって遠心分離した。その後、前記フィルターを100μL H2Oで洗浄し、ろ液を貯留した。貯留されたろ液を、窒素雰囲気下でガラス製バイアル中で乾燥させ、200μL移動相中で再構成させ、RP HPLCによって分析した。該方法は84%の収量を得た。一般的には、100〜2000ng/mL トロポロンを添加されたサンプルについて、これは証明された再現性のある回収を意味する。20〜40ng/mL範囲内の非常に少量のトロポロンを添加されたサンプルでは、回収率は94%程度であった。
【0027】
代わりに、この手順を、サンプルを乾燥しないで行った(乾燥は最適な手順で必要とされないことが解った)。ろ液をRP HPLCによって生で分析した。
【0028】
上記に簡単に記載した任意の処理方法は、<20%(固相抽出)および65%(Microcon限外ろ過)の平均回収率を得た。
【0029】
2.RP HPLC
定組成溶離、流速0.2 mL/分 25℃、242nmで検出および100μL注入容積の条件下において、固定相として新型クロマトグラフィー技術ACE C18 2.1mmカラムと、0.1% CuS04(w/v)/0.3%ヘキサンスルホン酸/水中10%アセトニトリル移動相とで、RP HPLCを行った。HPLCシステムは、Chemstationソフトウェア バージョンA6.04以上とともに、真空デガッサー、双対ポンプ、サーモスタット区画カラムおよび自動サンプラーを含むAgilent 1100 HPLCシステムまたはこれと同等なものとした。
【0030】
この方法で分析されたトロポロン標準体についての溶出曲線を図1aおよび1dに示す。図1bおよび1cは、異なるイオン対試薬を使用したことを除いて(代わりにTFAおよびMSA s. section 4)、同じ移動相および固定相を用いた溶出曲線を示す。
【0031】
図2a〜2bは、大規模生産から誘導された処理中の大量の完全なIgG抗体の分析を示す。前記抗体を、タンパク質Aセファロースクロマトグラフィーによってのみ部分的に精製した。更なる説明は図の表題に示す。
【0032】
ACE C-18カラムと、Hamilton(会社の所在地?)PRPポリスチレン ジビニルベンゼン4.6 mm逆相カラムとの比較を行った。ポリスチレン ジビニルベンゼン固定相の水溶液についての耐用性は、トロポロン溶出の保持時間を潜在的に増加させるより高い極性の移動相(C18カラムと比較して)の使用を可能にするので、予め評価されたPRP-1重合カラムをCuSO4移動相での使用について再調査した。トロポロンは、アセトニトリル欠損中では15分後に0.1%CuSO4移動相で溶出させることができず(データは示していない)、様々なパーセンテージのアセトニトリル(定組成5%(続いて有機カラム洗浄工程)、21%、40%および3%〜11.5%勾配)について評価を行った。使用された流速は、25℃の温度で0.8mL/分で一定とした。
【0033】
その後、移動相を様々なアセトニトリル含量で評価し、これらの条件下の分析からの結果を表3に要約した。5%アセトニトリルの濃度で、許容可能な保持(k>1.0)、許容可能なテーリング(TF 3.02)および0.1μg/mLの再現可能な検出が達成された。しかしながら、2で示された0.1μg/mLの標準化されたピーク高さは、これらの条件を使用する検出の限界に近かった(表3)。さらに、この濃度でのピークのテーリングは、2.0μg/mL(TF 1.61)についてのものよりもかなり高かった。アセトニトリルのより高い濃度(21%および40%)では、トロポロンの保持はk>0.5であり、認容できなかった。
【表3】
【0034】
3.HPLCの実行
以下の全ての研究は、実験2のRP HPLC方法と実験1のサンプル調製方法を使用して行った。
【0035】
(a)直線性
トロポロン溶液を、0, 1, 5,10, 20,50, 100,500, 2000 および 4000 ng/mLトロポロンの濃度で、上述した抗体含有バッファー中において調製した。バッファー塩の除去は、2% CuSO4での沈殿によって行った。トロポロンサンプルをRP HPLCによって分析した。ピーク領域を、トロポロン濃度に対して記録およびプロットし、直線状の回帰プロットを構築した。直線状の作用範囲は、許容可能な正確性(≦5% CV)を有する直線状の反応(r2≧0.99)を与える濃度範囲として定義された。トロポロン量についての校正曲線を、5, 10, 20, 50, 100, 500, 2000 および 4000 ng/mLトロポロンの濃度で分析されたサンプルから構築した。各トロポロン濃度の測定の精度が分析された。直線状の反応が、1 ng/mL〜4000 ng/mLで回帰分析によって視覚的に観察された。相関係数の二乗値(r2)は許容可能な直線性を示した(r2=0.9997)。
【0036】
日常的な計量プロセスのために、校正曲線を0 ng/mL〜2000 ng/mLの濃度で構築した(r2>0.99)。直線性を5つの異なる状況で調査し、回帰データを表1に要約した。
【表1】
【0037】
(b)特異性
サンプルバッファーの一連のピークからのトロポロンピークの分離(Rs)を、標準トロポロン溶液(2000ng/mL)について決定した。三重分析からのRs、sdおよび%CVの平均値を5つのアッセイについて記録した。
【0038】
予め議論したように、トロポロンから視覚的に分離されたサンプルバッファーの一連のピークは、トロポロンの直前に溶出した(セクション4.5.2; 図18 および 19)。このバッファーの一連のピークからのトロポロンの分離を、USPで定義された方法を使用する5つの別個のアッセイ中の三つのシステム適合性サンプル注入液について計算した。平均トロポロン分離因子(Rs)、sdおよび%CVを計算し、その結果を表2に要約した。
【表2】
【0039】
平均分離因子は全て≧1.5であり、前述のバッファー関連ピークからの許容可能な分離を示し、許容可能な再現性(CV<3.0%)を実証した。妨害ピークは、サンプル製剤または最終製品サンプルから観察されなかった。これは、トロポロンの検出および定量化のためのRP HPLCアッセイの特異性を確認した。
【0040】
(c)検出/定量化(LOD/LOQ)の限界
直線性について分析されたトロポロンサンプルを使用して、トロポロン分析のためにLODおよびLOQを推定した。検出可能なシグナルを与える試験対象のトロポロンの最低濃度は、LODとして割り当てられた。許容可能な再現性(≦5% CV)で測定され得るトロポロンの最低濃度は、LOQとして定義された。これは、LOD約1 ng/mL の量で見出された; LOQは約5ng/mLとして決定され、この感度が本発明の検出方法を制限する。
【0041】
4.イオン対(IP)試薬とともに銅イオン複合体を使用するACE C18 2.1mmカラム上でのHPLC条件の評価
C18 HPLCカラム上での銅イオントロポロン複合体の分析は、様々な疎水性の三つの異なる試薬を使用する逆相イオン対で試験した。これらはトリフルオロ酢酸(TFA)、最も極性のメチルスルホン酸(MSA)、中程度の極性のヘキサンスルホン酸(HAS)、最も疎水性のものであった。イオン対試薬、0.1% CuSO4およびアセトニトリルを含む移動相が調製された。使用されたHPLCパラメータは、得られた一連のピーク高さとクロマトグラム試験測定値とともに表4に要約される。
【0042】
4.1 TFAイオン対
許容される移動相中の0.1% TFAを使用する標準トロポロンの保持は、十分には増加しなかった(k<1.0)(表4、図10)。これらの条件下での分析は、トロポロンのアッセイについて適切ではないと結論付けられた。
【0043】
4.2 MSAイオン対
0.1% MSAを含む移動相は、5%アセトニトリルおよび0.1% MSAでの定組成溶出のもとで向上した保持時間を与えた(k≧2)。しかしながら、高いトロポロンピークのテーリングは、試験された標準の両濃度(0.1μg/mLおよび2.0μg/mL)について観察された(TF≧3.4)。0.1%MSAおよび10%アセトニトリルを含む移動相の使用(図11)は、5%アセトニトリリル/0.1%MSA(2の標準化されたピーク高さ)よりも0.1μg/mL標準トロポロンの向上した検出を実証したが(14の標準化されたピーク高さ、表4)、ピークのテーリングは広がった(TF12.05)。
【0044】
4.3 HASイオン対
トロポロン保持時間の顕著な増加は、0.1%HAS/10%アセトニトリル移動相(k≧3.4、表4)での定組成溶出を使用して達成された。さらに、0.1μg/mLの低トロポロン濃度についてのピーク高さの増加が観察され(27の標準化されたピーク高さ、表4)、試験対象の両トロポロン濃度についてピークのテーリングは許容可能かつ比較可能であった。さらに、ピーク領域の再現可能性は、2.0μg/mL(0.9% CV)および0.1 μg/mL(3.2% CV)トロポロンの濃度で許容可能であった(表4)。しかしながら、溶出したトロポロンは移動相ピークに近く、2つのピークはベースラインでの分離を実証しなかった(図12および13、Rs≦1.1)。トロポロンおよび移動相ピークは、アセトニトリルの勾配(10%〜42%)を使用して分離することはできず、分析温度を40℃まで増加(Rs≦1.0、表4)または温度を20℃、流速を0.1mL/分まで減少させて(Rs≦1.3)分離することもできなかった。
【0045】
様々なイオン対(HSA)移動相の濃度について評価した。0.3% HSA/10% アセトニトリル/0.1% CuSO4移動相(流速0.2mL/分、25℃)は、両トロポロン濃度での移動相ピークからのトロポロンのベースラインでの分離を達成するために有効であった(Rs≧3.5、図14および15、表4)。流速の更なる減少(0.1mL/分)は、トロポロン分離の更なる向上を誘導しなかった(Rs≧3.8)。結論として、検出の許容可能な制限と、ピークの対称性と、標準トロポロンのアッセイについての再現性を備えたRP HPLC方法が確立された。
【表4】
【図面の簡単な説明】
【0046】
【図1a】0.1%CuSO4/0.3%HSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1b】0.1%CuSO4/0.1%TFA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1c】0.1%CuSO4/0.1%MSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(2.0 μg/mL)。
【図1d】0.1% CuS04/0.3%HSA/10%アセトニトリル移動相を使用するACE C18 2.1mm RP HPLCカラム上で分析されたトロポロン標準体(0.1 μg/mL)。
【図2a】未添加IgG Lot 11859およびバッファー対照のクロマトグラムの例。
【図2b】トロポロン添加(10ng/mL)IgG Lot 11859およびバッファー対照のクロマトグラムの例。
【図2c】100ng/mLトロポロン添加IgG抗体Lot 11859およびバッファー対照のクロマトグラムの例。
【特許請求の範囲】
【請求項1】
以下の工程を含む、豊富な製品タンパク質を含む動物細胞培養上清またはタンパク質溶液からトロポロンまたはその誘導体を検出する方法。
a.トロポロンまたはその誘導体をタンパク質から分離する工程と、前記工程の前または後に、
b.溶液中で前記トロポロンまたはその誘導体をCu(II)イオンと複合化させる工程と、
c.疎水性固定相と移動相(移動相はCu(II)イオンとイオン対試薬の両方を含む)を備える逆相HPLCによってトロポロンまたはその誘導体を検出する工程。
【請求項2】
前記疎水性固定相が、アルキルシラン固定相であることを特徴とする請求項1に記載の方法。
【請求項3】
前記アルキルシラン固定相が、枝分かれのないアルキルシラン相、好ましくはC18アルキルシランであることを特徴とする請求項2に記載の方法。
【請求項4】
前記イオン対試薬が、イオン対カルボン酸またはその塩(すなわち、トリフルオロ酢酸よりも疎水性である)であることを特徴とする請求項1に記載の方法。
【請求項5】
前記イオン対試薬が、メチルスルホン酸またはヘキシルスルホン酸の誘電率と等しい誘電率を有するか、またはメチルスルホン酸またはヘキシルスルホン酸の誘電率によって定義された範囲内の誘電率を有し、かつプロピルスルホン酸、ブチルスルホン酸、ペンチルスルホン酸、ヘキサンスルホン酸およびこれらの塩からなる群から選択されることを特徴とする請求項4に記載の方法。
【請求項6】
前記移動相が、少なくとも一つの更なる極性溶媒と混合した1〜30%のアセトニトリルを含むことを特徴とする請求項11または1に記載の方法。
【請求項7】
前記極性溶媒が、水、メタノール、エタノールまたはこれらの混合物であり、好ましくは前記移動相が、少なくとも60%の水を含むことを特徴とする請求項6に記載の方法。
【請求項8】
前記タンパク質からの分離が、第一に、トロポロンまたはその誘導体をCuSO4とともに沈殿させて該沈殿を回収し、第二に、限外ろ過によって回収された沈殿からタンパク質を取り除くことによって達成されることを特徴とする請求項1に記載の方法。
【請求項9】
前記移動相が、Cu(II)イオン源としてCuSO4を含むことを特徴とする請求項1または8に記載の方法。
【請求項10】
前記移動相中のCuSO4の濃度が、0.05%(w/v)〜0.2%(w/v)の範囲内にあることを特徴とする請求項9に記載の方法。
【請求項11】
前記移動相が、水混和性であることを特徴とする請求項1または10に記載の方法。
【請求項12】
前記タンパク質を含む動物細胞培養上清またはタンパク質溶液が、1mg/ml以上の濃度に富んでいることを特徴とする請求項1に記載の方法。
【請求項13】
前記豊富なタンパク質が、製品タンパク質であることを特徴とする請求項12に記載の方法。
【請求項1】
以下の工程を含む、豊富な製品タンパク質を含む動物細胞培養上清またはタンパク質溶液からトロポロンまたはその誘導体を検出する方法。
a.トロポロンまたはその誘導体をタンパク質から分離する工程と、前記工程の前または後に、
b.溶液中で前記トロポロンまたはその誘導体をCu(II)イオンと複合化させる工程と、
c.疎水性固定相と移動相(移動相はCu(II)イオンとイオン対試薬の両方を含む)を備える逆相HPLCによってトロポロンまたはその誘導体を検出する工程。
【請求項2】
前記疎水性固定相が、アルキルシラン固定相であることを特徴とする請求項1に記載の方法。
【請求項3】
前記アルキルシラン固定相が、枝分かれのないアルキルシラン相、好ましくはC18アルキルシランであることを特徴とする請求項2に記載の方法。
【請求項4】
前記イオン対試薬が、イオン対カルボン酸またはその塩(すなわち、トリフルオロ酢酸よりも疎水性である)であることを特徴とする請求項1に記載の方法。
【請求項5】
前記イオン対試薬が、メチルスルホン酸またはヘキシルスルホン酸の誘電率と等しい誘電率を有するか、またはメチルスルホン酸またはヘキシルスルホン酸の誘電率によって定義された範囲内の誘電率を有し、かつプロピルスルホン酸、ブチルスルホン酸、ペンチルスルホン酸、ヘキサンスルホン酸およびこれらの塩からなる群から選択されることを特徴とする請求項4に記載の方法。
【請求項6】
前記移動相が、少なくとも一つの更なる極性溶媒と混合した1〜30%のアセトニトリルを含むことを特徴とする請求項11または1に記載の方法。
【請求項7】
前記極性溶媒が、水、メタノール、エタノールまたはこれらの混合物であり、好ましくは前記移動相が、少なくとも60%の水を含むことを特徴とする請求項6に記載の方法。
【請求項8】
前記タンパク質からの分離が、第一に、トロポロンまたはその誘導体をCuSO4とともに沈殿させて該沈殿を回収し、第二に、限外ろ過によって回収された沈殿からタンパク質を取り除くことによって達成されることを特徴とする請求項1に記載の方法。
【請求項9】
前記移動相が、Cu(II)イオン源としてCuSO4を含むことを特徴とする請求項1または8に記載の方法。
【請求項10】
前記移動相中のCuSO4の濃度が、0.05%(w/v)〜0.2%(w/v)の範囲内にあることを特徴とする請求項9に記載の方法。
【請求項11】
前記移動相が、水混和性であることを特徴とする請求項1または10に記載の方法。
【請求項12】
前記タンパク質を含む動物細胞培養上清またはタンパク質溶液が、1mg/ml以上の濃度に富んでいることを特徴とする請求項1に記載の方法。
【請求項13】
前記豊富なタンパク質が、製品タンパク質であることを特徴とする請求項12に記載の方法。
【図1a】


【図1b】
【図1c】
【図1d】
【図2a】

【図2b】

【図2c】
【図1b】
【図1c】
【図1d】
【図2a】
【図2b】
【図2c】
【公表番号】特表2007−502970(P2007−502970A)
【公表日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願番号】特願2006−523575(P2006−523575)
【出願日】平成16年8月11日(2004.8.11)
【国際出願番号】PCT/EP2004/008985
【国際公開番号】WO2005/017519
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(504120486)ロンザ・バイオロジクス・ピーエルシー (12)
【Fターム(参考)】
【公表日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願日】平成16年8月11日(2004.8.11)
【国際出願番号】PCT/EP2004/008985
【国際公開番号】WO2005/017519
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(504120486)ロンザ・バイオロジクス・ピーエルシー (12)
【Fターム(参考)】
[ Back to top ]