
熱不安定性ホスファターゼの過剰発現、精製及び性状分析
【課題】アルカリホスファターゼのための組成並びに熱不安定性のアンタークティックホスファターゼ(TAP)の過剰発現及び精製の方法を提供すること。
【解決手段】アルカリホスファターゼのための組成並びに熱不安定性のアンタークティックホスファターゼ(TAP)の過剰発現及び精製の方法が提供される。TAPの用途には、核酸、糖、ペプチド及びタンパク質の脱リン酸化が挙げられる。本明細書に記載されたTAPは、65℃における熱不安定性及びおよそ中性のpHでの脱リン酸化活性の効率性に関してほかの供給源からのホスファターゼより多くの利点を有する。
【解決手段】アルカリホスファターゼのための組成並びに熱不安定性のアンタークティックホスファターゼ(TAP)の過剰発現及び精製の方法が提供される。TAPの用途には、核酸、糖、ペプチド及びタンパク質の脱リン酸化が挙げられる。本明細書に記載されたTAPは、65℃における熱不安定性及びおよそ中性のpHでの脱リン酸化活性の効率性に関してほかの供給源からのホスファターゼより多くの利点を有する。
【発明の詳細な説明】
【背景技術】
【0001】
精製ホスファターゼは、試験管内で線状化したDNA、RNA、ヌクレオチド及びタンパク質からリン酸基を外すのに分子生物学者らによって使用される。DNAリガーゼは、一方のヌクレオチドが5’−ホスフェートを含有し、他方が3’水酸基を含有する場合のみ隣接するヌクレオチド間のホスホジエステル結合の形成を触媒するので、線状化したDNAベクターから双方の5’−末端リン酸残基を外すとライゲーション反応におけるベクターDNAの再環化が妨げられる。しかしながら、5’末端のホスフェートを持つ外来のDNA断片を脱リン酸化したベクターに効率よく連結し、コンピテント細胞を容易に形質転換することができる2つのニックを含有する開環分子を生じることができる。
【0002】
生体分子からリン酸基を外すために幾つかの異なったホスファターゼが開発されており、それぞれ、それ自体の長所及び短所を有する。この目的で使用されるべき最初のホスファターゼは、大腸菌(E.coli)から精製された細菌性アルカリホスファターゼ(BAP)であった。BAPは、あらゆる種類のDNA末端に対して良好な活性を有するという長所を有する。しかしながら、それは熱及び界面活性剤への耐性がきわめて強いので反応物から除くのが困難である(非特許文献1)。伝えられるところによれば、熱安定性を高めた変異体BAPが調製されている(特許文献1、特許文献2)。仔ウシ腸管アルカリホスファターゼ(CIP又はCIAP)は別のホスファターゼであり、分子生物学の技術で広範に使用されている(特許文献3及び特許文献4)。CIAPは、BAPほどDNAに対して活性は高くないが、プロテイナーゼK処理その後のフェノール:クロロホルム抽出又はEDTAの存在下での加熱工程その後のフェノール:クロロホルム抽出のいずれかの使用を必要とする反応物から除くのがやや容易である(非特許文献1)。さらに最近、北極のエビ、Pandalus borealis (SAP)から単離されたホスファターゼ(特許文献5及び特許文献6)は、使用するのがさらに容易であることが判っている。それは、BAPのようにあらゆる種類のDNA末端に良好な活性を有するが、65℃にて15分間熱不活性化することによって容易に反応物から除かれるという長所を有することが報告されている(Amersham Bioscience,Piscataway,NJ)。
【0003】
文献には、そのほかの熱不安定性ホスファターゼに関するそのほかの報告もあり、熱不安定性のアンタークティックホスファターゼ(TAP)と呼ばれる南極大陸で単離された好冷性の株TAB5から精製されたものが含まれる(非特許文献2)。他のホスファターゼを超えるTAPの長所には、高温不安定性及び高い比活性が挙げられる。Rinaらは、このホスファターゼがp−ニトロフェニルリン酸(pNPP)基質に対しタンパク質のmg当たり1650単位の比活性を有し、それは、そのほかのいかなる既知のホスファターゼの活性よりも有意に高いことを報告した。しかしながら、Rinaらが報告したクローン(非特許文献2)により産生されるタンパク質は、過剰発現及び精製に関連する多数の問題があった。たとえば、Rinaらにより記載された精製プロトコール(非特許文献2)は、TAPが明らかに会合する細胞膜からTAPを抽出するのに複数の超遠心工程を必要とする。このプロトコールは、大規模な製造(たとえば、300g以上の細胞ベーストの生産に関与する製造プロトコール)には向いていない。さらに、Rinaらにより記載されたプロトコールを用いたTAP遺伝子の過剰発現は、結果として、きわめて低い且つそのために大規模製造では費用有効性がない酵素の収量を生じた。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5,891,699号
【特許文献2】欧州特許第0441252号
【特許文献3】米国特許第5,773,226号
【特許文献4】米国特許第5,707,853号
【特許文献5】米国特許第6,387,634号
【特許文献6】米国特許第6,379,940号
【非特許文献】
【0005】
【非特許文献1】Sambrook,et al.,Molecular Cloning,A Laboratory Manual Sections 1.53−1.72(1989)
【非特許文献2】Rina,et al.,Eur. J. Biochem.267:1230−1238(2000)
【図面の簡単な説明】
【0006】
【図1】pN1を含有する大腸菌株からのTAP精製の結果を示す図である。レーン1〜6は、精製における連続工程である。レーン1は、誘導した後の全粗抽出物である。レーン2は、可溶化した膜分画である。レーン3は、DEAEカラムからの通過物である。レーン4は、Qセファロースカラムからのプールした分画である。レーン5は、ハイトラップSカラムからの通過物である。レーン6は、ヒドロキシアパタイトカラムからの通過物である。レーン7は、pNIクローンから得た精製したTAPである。分子量標準(S)は、kDで示す。レーン7の47.5kDに相当するバンドが実質的に精製されたTAPを表す。
【図2】TAP遺伝子を過剰発現させるベクターを構築するのに使用した工程を示す説明図である。図の上側の細長い四角は、遺伝子のそれに相当する発現生成物のN末端部分及びC末端部分を含むTAP遺伝子の模式図である。N末端では、メチオニン開始コドン及び仮定されたシグナル配列をコードするプロセッシング部位周辺の領域の位置を示す。C末端では、星印で示した停止コドンと共に、遺伝子産物のアミノ酸を示す。TAP遺伝子のPCR増幅に使用したプライマーを、各プライマーがTAP遺伝子にハイブリッド形成する部位で、細長い四角の上(実際のサイズではない)(正方向プライマーA、B及びCについて)及び下(逆方向プライマーD及びE)に示す。プライマー上のN及びXは、制限エンドヌクレアーゼNdeI又はXhoIについて操作した切断部位を示す。プライマーCは、NdeIに続く6個のHを示す。これらは、プライマーのメチオニン開始コドンの下流に挿入され、それに隣接するヒスチジンをコードする6個のコドンに相当し、TAPと融合したN末端Hisタグを創る。「PCR」に続く次の一連の線は、指示したプライマー対を用いることによって生じる結果の産物を示す。上記のように、星印は停止コドンを示し、6個のHは、ヒスチジンタグをいう。PCR産物をpET21aに挿入することによって創った5種の異なるプラスミドを示す。pEGTAP1.1.1はHisタグのない無傷のTAPをコードする。pEGTAP1.2.1は、Hisタグのない切り詰め型TAPをコードする。pEGTAP1.3.1はC末端Hisタグを持つ無傷のTAPをコードする。pEGTAP1.4.1はC末端Hisタグを持つ切り詰め型TAPをコードする。後の実験では、N末端Hisタグを持つ切り詰め型TAPをコードするようにpEGTAP7.4.1を構築した。
【図3】Hisタグの存在下又は非存在下、TAP遺伝子が切り詰められたか又は切り詰められていない様々なクローンから生じるTAPのSDS−PAGE解析を示す図である。「試験的」精製は、1mL未満の試料サイズを言う。レーン1は、Hisタグなしの無傷のTAP遺伝子を持つクローンpEGTAP1.1.1からの可溶性粗抽出物である。レーン2は、Hisタグなしの切り詰め型TAP遺伝子を持つクローンpEGTAP1.2.1からの可溶性粗抽出物である。レーン3及びレーン4は、Hisタグに融合した無傷のTAP遺伝子を持つクローンpEGTAP1.3.1からの2種の異なった単離物から得た可溶性粗抽出物である。レーン5及びレーン6は、Hisタグに融合した切り詰め型TAP遺伝子を持つクローンpEGTAP1.4.1からの2種の異なった単離物から得た可溶性粗抽出物である。分子量標準(S)は、kDで示す。切り詰め型TAP遺伝子を有するそれらのクローン(レーン2、5及び6)はTAPに相当する最強のバンドを提供した。
【図4】C末端のHisタグと共に切り詰め型の遺伝子を含有する(pEGTAP1.4.1)大腸菌(ER2566)クローンから非変性条件下で精製した可溶性TAPのSDS−PAGE解析を示す図である。全粗抽出物(T)、清澄化した粗抽出物(装填)(L)、Ni−NTAスピンカラム(カリフォルニア州、Studio City、キアゲン)からの通過物(FT)、Ni−NTAスピンカラムの洗浄物(W)並びに溶出緩衝液でNi−NTAスピンカラムから溶出された第1の試料及び第2の試料(E1及びE2)の試料10μLを、10〜20%のトリストリシンPAG(カリフォルニア州、Carlsbad、Invitrogen Corp.)で泳動し、クマシーブリリアントブルーで染色した。分子量標準(S)は、kDで示す。
【図5】解析したクローンがER2566(pEGTAP7.4.1)であったことを除いて図4で使用したのと同様の精製プロトコールの結果を示す図である。また、細胞抽出物全体をゲル上で泳動するのに代えて、粗抽出物を遠心して得られたペレットを再懸濁させて、不溶性ペレット(P)として泳動した。ペレット、可溶性装填試料(L)、Ni−NTAスピンカラムの通過物(FT)、Ni−NTAスピンカラムの洗浄物(W)、イミダゾールによるNi−NTAスピンカラムからの溶出物(E)及びkDで示す分子量標準(S)各10μLを、10〜20%のトリストリシンPAGで泳動し、クマシーブリリアントブルーで染色した。
【図6】様々なpH及び様々な緩衝液におけるTAP活性の比較を示す図である。ホスフェート放出アッセイを用いてpHの最適性を決定した。32P標識した二本鎖DNA、1mMのMgCl2及び0.1mMのZnCl2を含有する反応混合物中で50mMの各緩衝液をインキュベートした。反応物を37℃にて5分間インキュベートし、トリクロロ酢酸(TCA)可溶性のカウントの量を測定した。
【図7】ホスフェート放出アッセイを用いた、TAP活性に対する様々な塩の影響を示す図である。100mMの各塩、KCl、NaCl、酢酸カリウム又は酢酸ナトリウムを調べた。
【図8】CIAP、SAP及びTAPの温度不安定性の比較を示す図である。DNAの存在下、65℃にて各酵素を30分までインキュベートした。インキュベート中、様々な時点で試料を取り出し、pNPPで活性を測定した。
【図9】TAPによるpNPPからの経時的なホスフェートの放出を測定することにより、37℃及び25℃における安定性を示す図である。1単位のTAP及び未反応のpNPPを含有する試験管を25℃又は35℃のいずれかでインキュベートした。最終濃度5NとなるようにNaOHを添加することにより0〜30分について5分間隔で反応を止めた。ホスフェートの放出(pNPの蓄積)を405nm(黄色)にて分光光度計で測定し、反応時間に対してグラフを作った。
【図10】様々な種類のDNA末端からのリン酸基の取り外しにおけるTAPの効率のSAPに対する比較を示す図である:5’末端突出、平滑末端及び3’末端突出を調べた。これらの末端は、ベクターをそれぞれHindIII、EcoRV及びPstIで消化することにより作製した。
【図11】TAP及びSAPについてデオキシヌクレオチダーゼ活性の反応時間経過を示す図である。dATPアーゼ反応混合物の組成は、5分間隔でCap−HPLCにより測定した。結果は、存在するヌクレオチド全体の比率として各成分を表す。
【図12】TAPによるピロリン酸からの無機ホスフェートの放出を示す図である。0.32mMのピロリン酸ナトリウムを含有する各反応混合物に量を増やしながらTAPを加えた。無機ホスフェートは、Heinonen及びLahtiの方法(Heinonen J.K.& Lahti R.J.,「無機オルトホスフェートの新しい且つ好都合な比色分析測定及び無機ピロホスファターゼの測定へのその応用」Anal.Biochem.,113(2):313−7,1981)により既知のホスフェート標準と比較することによって本質的に測定した。並行する反応で熱安定性の無機ピロホスフェート(TIPP)を用いて、同等性を実証した。TAPについては37℃で、TIPPについては75℃で10分間のアッセイについて結果を示す。
【図13】リン酸化されたミエリン塩基性タンパク質におけるCIAPと比べたTAPの比活性を示す表である。
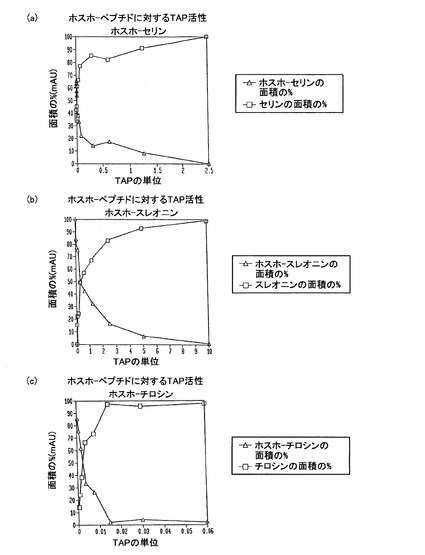
【図14】Cap−HPLCで測定した場合の、3種のホスホペプチド:ホスホセリン(グラフA)、ホスホスレオニン(グラフB)及びホスホチロシン(グラフC)に対するTAPの活性を示す図である。ホスホペプチド及びペプチドについての吸収ピークの面積が測定され、合計面積の比率としてTAPの単位数に対してグラフ上にプロットされた。
【図15】3種の異なった糖−リン酸、グルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6リン酸に対する1力価のTAPのTLC解析を示す図である。陽性対照として、3μgのグルコース(G)及びマンノース(M)のスポットを置いた。陰性対照(−)は、酵素なしでインキュベートした緩衝液と糖リン酸基質を含有する反応混合物だけを含有する。1〜6と付けたレーンは、12.5単位のTAPを含有する反応1で開始し、0.4単位のTAPを含有する反応6まで落とす、反応混合物における酵素の段階希釈(1:1)である。矢印は、糖及び糖−リン酸がTLC上を移動する距離を示す。
【図16】pN1プラスミドからの1.1kbの断片をPCRするのに使用したプライマーの配列(配列番号6及び7)である。クローニングしたPCR断片からの発現の結果、予想されるタンパク質のN末端配列(配列番号8)を正方向プライマーの配列の下に示す。プライマー内に含有されるNdeI及びXhoIのクローニング部位も示す。
【図17】無傷のTAP遺伝子のDNA配列(配列番号9)及びアミノ酸配列(配列番号10)を示す図である。四角で示すのは、N末端配列解析(Rina et al.,上記)により決定されるときのタンパク質の開始点である。矢印は、切り詰め型遺伝子をクローニングするのに使用したPCRプライマーがアニーリングする位置を示す。
【発明を実施するための形態】
【0007】
本発明の本実施態様は、中性pHで高められた高温不安定性を有し、高い活性があり、タンパク質のN末端に親水性リーダー配列を持つアルカリホスファターゼを提供する。親水性のリーダー配列は、オリゴペプチド、たとえば、Hisタグのような正味の正の電荷を持つオリゴペプチドであってもよい。このホスファターゼの長所には、ほかの反応物や生成物が影響を受けずにいられるレベルに温度を上げることによって活性を消去する能力が挙げられる。これらの特性を有するホスファターゼの例が、実施例で詳細に記載されるTAPである。本明細書で記載する方法は、タンパク質のN末端に融合したHisタグのような親水性リーダー配列の存在に頼るが、ニッケルカラムの緩衝液は、活性に悪影響を与えるので、ニッケルカラムを用いたタンパク質の精製は行わないことが好ましい。それにもかかわらず、驚くべきことに、親水性リーダー配列の存在は、他のカラム組成を用いたカラム精製で収量を高めた。
【0008】
本発明の実施態様では、切り詰め部がシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端がたとえば、Hisタグのような正に荷電するオリゴペプチドのような親水性のリーダー配列に共有結合する、切り詰められた、酵素活性のあるTAPが提供される。切り詰められたTAPは、約65℃にて15分未満で実質的に不活化することができる。特に、切り詰められたTAPの活性は、pH6より高い、さらに特にpH約7.4でのトリス塩酸中にて実質的に安定である。
【0009】
さらに、TAP(ジェンバンク(GenBank)Y18016にも記載されている)をコードするが、天然に存在するTAP遺伝子に関連するシグナル配列の一部又は全部を欠くDNAを記載する。該DNAは、好ましくは、発現されるTAP遺伝子のN末端で複数のHisアミノ酸をコードする配列をさらに含んでもよい。このDNAは、強力なプロモータ、たとえば、T7プロモータの制御下でベクターにおいて発現させてもよい。
【0010】
好ましい実施態様では、上述のDNAを含有するベクターが提供される。これらベクターの一例は、pEGTAP7.4.1.である。本発明の追加の実施態様では、少なくとも1種のベクターで形質転換された宿主細胞が提供される。他の種類の宿主細胞の使用を除外しないが、実施例では、宿主細胞は大腸菌である。
【0011】
本発明の追加の実施態様では、トリス塩酸、MgCl2、DTT及びグリセロールを含み、さらに特には、pHが約7.4である緩衝液中で37℃にて実質的に安定であるTAPの組成物が提供される。
【0012】
本発明の実施態様では、(a)たとえば、Hisタグのような親水性オリゴペプチドを発現するための配列に融合した切り詰め型TAP遺伝子をT7プロモータに操作可能に連結すること、(b)宿主細胞を形質転換すること、及び(c)TAPを過剰発現することを含むTAPを過剰発現するための方法が提供される。さらなる実施態様では、TAP遺伝子は、N末端及びC末端先端を有し、N末端先端で切り詰められ、切り詰め部がシグナル配列に相当し、該N末端先端がそこに結合された親水性オリゴペプチドを有するTAPタンパク質をコードする。
【0013】
本発明の追加の実施態様では、(a)上述のような形質転換された宿主細胞を得ること;(b)該宿主細胞を破砕して可溶性分画を得ること;及び(c)該可溶性分画からTAPを精製することを含む、精製したTAPを得る方法が提供される。さらに、該方法は、TAPがカラムから溶出されるカラム分離を追加的に含んでもよい。このような状況で、精製は、破砕した細胞混合物における全タンパク質に対するTAPタンパク質の比率の増加を言う。たとえば、TAPタンパク質の比率が混合物における全タンパク質の5%を超えた場合、精製が達成することができる。好ましくは、TAPタンパク質の比率は、全タンパク質の50%、80%又は90%を超えてもよい。特定の実施態様では、TAPタンパク質の比率は、全タンパク質の約95%まで精製される。
【0014】
本発明の追加の実施態様では、(a)約pH5.5〜7.0の緩衝液中の基質にTAPを加えること;並びに(b)規定された時間及び規定された温度で基質から外れるホスフェートの量を測定してTAPの活性を決定することを含む、TAPの活性をアッセイする方法が提供される。好ましい実施態様では、緩衝液はZnCl2塩及びMgCl2塩を含む。さらに好ましくは、緩衝液は、ビス−トリスプロパンである。
【0015】
追加の実施態様では、pH5.5〜7.0の緩衝液中の基質にTAPを加えること;並びに規定された時間及び規定された温度で基質から外されるべきホスフェートの量をもたらすことを含むリン酸化された基質を脱リン酸化する方法が提供される。
【0016】
基質は、核酸又は核酸の末端ヌクレオチド、糖リン酸、或いはリン酸化されたペプチド又はタンパク質であってもよい。
【発明を実施するための最良の形態】
【0017】
TAP遺伝子は、Rinaらにより単離され、クローニングされ(pN1)、DNA配列は公表されている。しかしながら、公表されたプロトコールを用いてこのクローンからTAPを過剰発現させ、精製することを試みた場合、試薬目的には不十分な量の精製酵素しか得られないこと、及び酵素が会合する膜断片から酵素を分離するのに複数の超遠心工程を必要とすることを含む多数の課題に遭遇した。本明細書では、TAP遺伝子を確実に過剰発現させて、大規模製造で費用有効性の高い酵素の収量を提供するプロトコールを提供する。さらに、複数の超遠心工程の必要性を回避し、大規模製造に適した精製プロトコールを開発した。核酸、ペプチド及び糖からホスフェートを外すTAPの用途が確立している。TAPの基質として作用してもよいそのほかの基質は、「The Enzymes(酵素)」,Vol.IV(1971)ch17,p373でReid & Wilsonにより記載されたものである。
【0018】
好ましい実施態様では、本明細書では「糖類」は、単糖類、オリゴ糖類又は多糖類を言う。糖類は、任意のペントース、ヘキソース又はヘプツロースの化合物を含む。ホスフェートがα異性体であろうとβ異性体であろうとそれには関係なく、ホスフェート(単数)又はホスフェート(複数)がどこに位置しようと、TAPを用いて、糖における任意の炭素からホスフェートを切断してもよい。一部の糖リン酸の例は、ウエブサイト、http://www.arabidopsis.org.で提供されている。糖リン酸の例には、マンノース、グルコース、フコース、ガラクトース、N−アセチルガラクトサミン、N−アセチルグルコサミン、キシロース及びラムノースのホスフェートが挙げられる。
【0019】
精製の収量及び容易さを改善するように公表されたプロトコールを改変して用い、pN1プラスミドを含有するER2575(ER2566pLysS)からのTAPの精製を試みた。形質転換した大腸菌株を増殖させ、誘導し、以下の例外と共に、Rinaら(上記)に記載されるようにホスファターゼを精製した。超遠心により可溶性膜分画を単離した後、タンパク質を緩衝液A(20mMのトリス、pH7.6、10mMのMgCl2、50mMのNaCl、0.2%のトリトンX−100)で透析し、次いで、緩衝液Aで平衡化したDEAEカラムに装填した。pNPPアッセイで測定したときホスファターゼ活性を有する通過物をRinaらが記載したようなQ−セファロースカラムに装填した。Q−セファロースカラムからの分画をプールして緩衝液B(リン酸カリウム緩衝液、pH6.6、25mMのNaCl、10mMのMgCl2)で透析し、緩衝液Bで平衡化したハイトラップSPカラムに装填した。ホスファターゼ活性を有する通過物を緩衝液C(リン酸カリウム緩衝液、pH7.2、50mMのNaCl、10mMのMgCl2)で透析し、緩衝液Cで平衡化したハイドロキシアパタイトカラムに装填した。ホスファターゼ活性は通過した。各工程からの試料を10〜20%のトリストリシンPAG(カリフォルニア州、Carlsbad、Invitrogen Corp.)で泳動し、クマシーブリリアントブルーで染色した。図1に見ることができるように、レーン1〜6におけるホスファターゼの収量は、期待はずれだった。
【0020】
上記プロトコールを用いて得られた期待はずれの結果のために、図2に記載したようなTAP遺伝子のクローニングを含む別のアプローチを検討した。
【0021】
具体的には、強力なプロモータ(T7プロモータ)の直後にTAP遺伝子を直接クローニングし、精製を円滑にするためにHisタグ(親和性タグ)を取り付けた。図13及び14に示すHisタグの配列は、タンパク質のN末端先端(遺伝子の5’末端)に融合した6個のヒスチジンを有する。しかしながら、ヒスチジンのこの数は重要ではない。本明細書で記載された目的を達成するには、タグにおけるヒスチジンの数は3個ほどで少なくても、6個を超えて所望のとおり多くてもよい。幾つかの異なった構築物を作成した。2種の構築物では、メチオニン開始コドンに隣接する、タンパク質のN末端の22アミノ酸から成る推定上のシグナル配列を取り除くことによってTAP遺伝子を切り詰めた。一方のクローンでは、C末端のHisタグを加えて切り詰め型ホスファターゼとの融合タンパク質を形成したが、他方ではそうはしなかった。たとえば、pEGTAP1.4.1は、Hisタグを有したが、pEGTAP1.2.1は有さなかった。2種のほかの構築物では、遺伝子は推定上のシグナル配列を保持し、いずれかがさらにC末端Hisタグを含有し(pEGTAP1.3.1)、又は含有しなかった(pEGTAP1.1.1)(図2)。
【0022】
pET21aにPCR産物を挿入することによって(実施例I、図2、11及び12)創った5種の異なるプラスミドを図2に示す。pEGTAP1.1.1はHisタグのない無傷のTAPをコードする。pEGTAP1.2.1は、Hisタグのない、切り詰め型TAPをコードする。pEGTAP1.3.1はC末端Hisタグを持つ、無傷のTAPをコードする。pEGTAP1.4.1はC末端Hisタグを持つ、切り詰め型TAPをコードする。pEGTAP7.4.1は、N末端Hisタグを持つ、切り詰め型TAPをコードする。
【0023】
C末端でHisタグを持つ又は持たない、推定上のシグナル配列を持つ無傷のTAP遺伝子を含有するプラスミドは、誘導後、SDS−PAGEにおいて、いかなる明瞭な可溶性ホスファターゼも産生することができなかった(図3、レーン1、3及び4)。しかしながら、Hisタグを持つ又は持たない、推定上のシグナル配列なしで構築されたプラスミドはSDS−PAGEにおいて正しいサイズのきわめて強いバンドを生じた(図3、レーン2、5及び6)。しかしながら、タンパク質の精製中、問題が生じた。pEGTAP1.4.1(切り詰め型TAP及びC末端His)を含有する、形質転換した宿主細胞ER2566は、非変性条件で主に不溶性TAPを産生した(図4)。変性条件下で精製を試み、その後、変性させたホスファターゼを折り畳んだ状態に戻した。しかしながら、このアプローチでは、活性のある酵素は、相対的に少ない量しか得られなかった。pEGTAP1.2.1(Hisタグを持たない切り詰め型TAP)を含有する大腸菌ER2566は可溶性のTAPを産生したが、タンパク質はほとんどのカラムに結合できなかった。
【0024】
上記の問題を克服するために、Hisタグをコードする配列をTAP遺伝子のC末端に代えてN末端に配置した第5のクローン(pEGTAP7.4.1)を構築した(図2)。この構築物は、可溶性分画で良好な収量のTAPを産生し(図5)、複数の超遠心工程なしで容易にそれを精製することができた(実施例II)。具体的には、ホスファターゼのN末端におけるHisタグの存在によって、ニッケルカラムを含まないたった2又は3回のカラムでほぼ均質までの精製が可能になった。理論によって限定されることを望まないで、Hisタグの最初の効用は、親和性タグとしてではなく、可溶性ホスファターゼの産生を促進することだったと思われる。
【0025】
DNA及びタンパク質基質を用いたアッセイ(実施例VII)によって上記方法(実施例I及びII)により調製したTAPホスファターゼの活性を、SAP及びCIAPのホスファターゼ活性と比較し、TAPが向上した活性を有することが判明した。たとえば、実施例VIIに記載したホスフェート放出アッセイを用いて、DNAの5’末端突出、3’末端突出及び平滑末端について、TAP及びSAPの線状DNAの脱リン酸化活性を測定した。これらの異なった構造の脱リン酸化を、同一のpNPP単位量を用いて比較すると、TAPは一貫してSAPよりも効率が良いことが分かった(図10)。
【0026】
TAPはまた、CIAPと同じくらい効率的に、リン酸化されたタンパク質におけるセリン/スレオニン残基及びチロシン残基からリン酸基を取り外すことができる(実施例VII、図13)。
【0027】
他のホスファターゼと比べたTAPの高い活性に加えて、TAPは、高い熱不安定性を有することが判明した(実施例IV、図8)。CIAP、SAP及びTAPの高温不安定性を比較すると、TAPが65℃で5分後に98%の活性を失ったのに比べて、SAPはこの温度で70%の活性を失い、CIAPはたった10%の活性しか失わなかったことが判明した。
【0028】
本発明のほかの実施態様では、ZnCl2及びMgCl2が、ホスファターゼの活性を上げることが判明した(実施例III)。TAPの活性は、たとえば、ビス−トリスプロパン緩衝液を用いてpH約5.5〜7.0で最適化された(実施例III)。
【0029】
以下の実施例によって本発明をさらに説明する。これら実施例は本発明の理解を助けるために提供されるのであって、その限定として解釈されるものではない。
【0030】
上記及び以下に引用した参考文献は、参考により本明細書に組み入れられる。
【実施例】
【0031】
実施例I:TAP−Hisタグ融合遺伝子の構築及び発現
分類されていない南極の株TAB5に由来する切り詰め型ホスファターゼ遺伝子をN末端の6ヒスチジン残基のタグと共にクローニングした。簡単に言えば、PCRにより、組換えプラスミドpN1(Rina,et al.,Eur.J.Biochem.,267:1230−1238,2000)から1.1kbの断片を増幅した。正方向のプライマー(図2、プライマーC)は、推定上のシグナル配列(63塩基)(図16及び17)を持たない遺伝子の5’末端に相同の配列を含有することに加えて、ATG開始コドンの一部としてのNdeI制限部位及びATG開始コドンと切り詰め型ホスファターゼ遺伝子の最初のコドンとの間に位置する6個のヒスチジン残基をコードする6個のコドンを含有した(図16及び17)。逆方向のプライマー(図2、プライマーD)は、遺伝子の3’末端に逆相補的な配列を含有することに加えて、TAA停止コドンのすぐ下流にXhoI制限部位を含有した(図16及び17)。いったん増幅し、1.1kbの断片をNdeI及びXhoIで消化して、同様にNdeI及びXhoIで消化したT7発現ベクターpET21a(ウィスコンシン州、マジソン、Novagen)に連結した(Sambrook,et al.,Molecular Cloning,A Laboratory Manual,Sections 1.53−1.72(1989))。得られたライゲーションで大腸菌株ER2688(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ)を形質転換した。形質転換のプラスミドを単離し、制限マッピング及びDNAの配列決定により特性分析し、特性分析した際、正しいサイズ及び配列の挿入物を含有することが判った。T7プロモータでホスファターゼ遺伝子を発現させるために、プラスミド構築物で大腸菌株ER2566(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ)を形質転換した。pEGTAP7.4.1が、最良の収量及び精製特性を提供することが判明した(図3〜5)。
【0032】
実施例II:TAP−Hisタグ融合タンパク質の製造及び精製
(a)TAPの小規模の精製
100mg/mLのカルベニシリンを含む5mLのリッチブロス(1リットル当たり10gのトリプトン(ミシガン州、リボニア、ディフコラボラトリーズ)、5gの酵母抽出物(ミシガン州、リボニア、ディフコラボラトリーズ)、5gのNaCl、NaOHでpH7.2)中にて単離したコロニーからER2566(pEGTAP7.4.1)を30℃で一晩増殖させた。3mLの一晩培養物を用いて、100mg/mLのカルベニシリンを含む300mLのリッチブロスに植菌した。30℃にて対数増殖期の中間(クレット70)まで培養物を増殖させ、氷上で冷却し、0.4mMのイソプロピル−β−チオガラクトシド(IPTG)で誘導し、15℃にて20時間増殖させた。4℃にて8000rpmで10分間遠心分離することにより35mLの細胞を回収した。上清を除き、細胞ペレットを秤量し、−20℃に2時間置いた。ペレットを氷上で融解し、1mLの細胞溶解緩衝液(50mMのNaH2PO4、300mMのNaCl、10mMのイミダゾール、pH8.0)に細胞を再懸濁した。リゾチームを1mL/mLまで加え、試料を氷上で30分間インキュベートした。インキュベートに続いて、細胞を、氷上で6回それぞれ10秒間、途中で1分間停止しての超音波処理により破壊した。4℃にて14,000rpmで20分間、粗抽出物を遠心し、未破砕の細胞及びそのほかの不溶性物質を除いて清澄な粗抽出物を生成した。0.6mLの細胞溶解緩衝液で平衡化したNi−NTAスピンカラムに0.6mLの清澄な粗抽出物を装填した。700xgにて2分間、試料を遠心した。通過物を除き、0.6mLの洗浄緩衝液(50mMのNaH2PO4、300mMのNaCl、20mMのイミダゾール、pH8.0)で2回カラムを洗浄した。0.2mLの溶出緩衝液(50mMのNaH2PO4、300mMのNaCl、250mMのイミダゾール、pH8.0)でタンパク質を2回溶出した。各工程における10μLをSDS−PAGEで解析した。pEGTAP7.4.1の結果を図5に示す。
【0033】
(b)ER2566におけるpEGTAP7.4.1からのTAPの大規模な精製
25℃にて発酵用のリッチブロス(100リットル当たり、500gのAmberexの酵母抽出物(ウィスコンシン州、ミルウォーキーのSensient Technologies)、1kgのCE90MSトリプトン(ニュージャージー州、Carlstadt、Marcor Development Corp.)、27gのNaOH,10gのアンピシリン、5.5gのポリプロピレングリコール消泡剤(ウエストバージニア州、サウスチャールストン、Arco Chemical Co.))2000mL中で25℃にてpEGTAP7.4.1プラスミドを収容する大腸菌株ER2566を増殖させた。この培養物を用いて100リットルの発酵用リッチブロスに植菌した。70のクレット(Klett)に達するまで25℃にて5時間、細胞を好気的に増殖させた。次いで発酵槽を15℃に冷却し、クレットが90に達したとき、最終濃度0.3mMとなるようにIPTGを加えた。次いで15℃にて細胞を好気的にさらに17時間増殖させ、細胞は最終クレット305を持つ定常期に達した。321グラム湿重量の細胞ペレットを回収するのに100リットルの発酵を要した。321グラムの細胞ペレットを963mLの緩衝液A(20mMリン酸カリウム緩衝液、pH7.4、50mMのNaCl、5%のグリセロール)に懸濁し、約12,000psigでゴーリンのホモゲナイザーを通過させた。細胞溶解物を約13,000Gにて40分間遠心し、上清を回収した(1150mL)。
【0034】
緩衝液Aで平衡化した500mLのヘパリンハイパー−Dカラム(カリフォルニア州、フレモントのCiphergen Biosystems,Inc.)に上清溶液をかけた。1Lの緩衝液Aで洗浄し、次いで、緩衝液A中0.05M〜1MのNaClの勾配2Lを流し、50mLの分画を回収した。10mMのMgCl2を含有する1Mのジエタノールアミン/HCl緩衝液(pH8.5)中の0.1MのpNPPと共に試料をインキュベートすることにより、ホスファターゼ活性について各分画を測定した。37℃にて1〜5分間反応を行い、活性は、黄色の生成として、405nmにて分光光度計で測定した。0.05〜0.35MのNaClでホスファターゼ活性は溶出された。
【0035】
ヘパリンハイパー−Dカラムのホスファターゼ活性を含有する分画をプールし、次いで、緩衝液Aに対して一晩透析した。この800mLのプールの100mLを105mLのソースQカラム(ニューヨーク州、ニューヨークのファイザー社)にかけた。210mLの緩衝液Aで洗浄した後、緩衝液A中0.05M〜1MのNaClの勾配1Lを流し、15mLの分画を回収した。上で記載したpNPPアッセイを用いて分画を測定し、ホスファターゼ活性は、0.15〜0.18MのNaClで溶出された。ヘパリンハイパー−Dでプールした残りの700mLも同様にカラムにかけ、溶出し;次いでホスファターゼ活性を含有する勾配分画をプールした。
【0036】
集め合わせたソースQのプールを緩衝液Aに対して透析し、50%グリセロールを補った。この120mLのプールの40mLを500mLの緩衝液Aで希釈し、次いで、緩衝液Aで平衡化した400mLのPEIカラム(英国、ケントのワットマン)にかけた。400mLの緩衝液Aで洗浄した後、緩衝液A中0.05M〜1MのNaClの線形勾配を流し、15mLの分画を回収した。上で記載したpNPPアッセイを用いて分画を測定した。ホスファターゼ活性は0.14〜0.2MのNaClの間で溶出された。ソースQでプールした残りの80mLも同様にカラムにかけ、溶出し;次いでホスファターゼ活性を含有する勾配分画をプールした。10mMのトリス(pH7.4)、1mMのMgCl2,1mMのDTT、50%のグリセロールを含有する保存緩衝液に対してこのプールを透析した。
【0037】
実施例III:TAPによるリン酸基取り外しのための至適pH及び塩条件の決定
実施例IIで記載されたように精製したTAPを用いて、DNAからリン酸基を外すための最適条件を決定した。以下のようなホスフェート放出アッセイによって脱リン酸化を測定した。50μLの反応物中で、2種の相補的な40量体のオリゴヌクレオチド
5’−ACGTATGTTAGGTTAGGTTAGGTTAGGTTAGGTTAGGCTC−3’(配列番号1)
3’−TGCATACAATCCAATCCAATCCAATCCAATCCAATCCGAG−5’(配列番号2)
をアニーリングし、T4ポリヌクレオチドキナーゼ(マサチューセッツ州、バーバリ、ニューイングランドバイオラボ社)及びγ32P−ATPを用いて製造元により推奨されるように末端標識した。この放射活性のある二量体を用いて、ホスファターゼ基質(最終20μg/mL)として作用するラムダHindIII断片の混合物1mgを「スパイク」した。37℃にて5分間の、基質混合物(1mMのMgCl2及び0.1mMのZnCl2を含有する緩衝液中で反応物当たり0.01単位のpNPP)とホスファターゼとのインキュベートに続く放射活性の放出によってTAP活性を測定した。TCAによるDNA基質の沈殿及び沈殿しなかった放射活性のシンチレーションカウントにより放出を測定した。TCA沈殿は、100μLのニシン精子DNA(2mg/mL)をキャリアとして各反応物に加えることから成った。次いで、150μLの20%冷却TCAを加えた。試料をボルテックスし、5分間氷上で冷却した。微量遠心機にて14,000xgで5分間、各反応物を遠心した。各上清(50%)150μLを2mLのシンチラントに加え、0.5分間カウントした。pNPP比色アッセイにより測定された0.001〜0.01単位の範囲にわたってTAP活性は線形であることが判明した。
【0038】
3種の異なった緩衝液:pH範囲が6〜9.5のトリス−塩酸、pH範囲が6〜7のビストリスプロパン緩衝液及びpH範囲が5.5〜7のビス−トリスプロパン緩衝液を用いてpH範囲が5.5〜9.5の反応緩衝液を調べた。ビス−トリスプロパン緩衝液にてDNA基質に対して最適の活性を与えたpH範囲は、5.5〜6.0の間であることが判った(図6)。
【0039】
カチオンの必要性を調べた。pNPPでのアッセイにおいてZn2+イオンが阻害活性を示すと報告したRinaら(上記、2000年)とは対照的に、上述のアッセイは、ZnCl2の存在が10倍多いホスフェートの放出を与えることを示していることを確定した。その後、TAP活性のアッセイに1mMのMgCl2、0.1mMのZnCl2を含めた。100mMの濃度でのKCl、NaCl、酢酸カリウム及び酢酸ナトリウムを含むそのほかの塩の添加は、TAP活性に影響を有さないことが認められた(図7)。
【0040】
実施例IV:TAPの熱不安定性の確定
pNPP活性の1単位は、室温にて1分間に1mLの反応容積において1μモルのpNPPをp−ニトロフェノールに加水分解するのに必要とされる酵素の量に相当する。各酵素について推奨された反応緩衝液にて、TAP、CIAP及びSAPそれぞれの10pNPP単位を1μgのラムダHindIII断片と混合した。混合物を37℃にて10分間インキュベートし、次いで氷上に置いた。次いで反応物を65℃の温水浴に入れ、5分、10分、20分及び30分後に試料を取り出し、氷上に置いた。熱処理に続いて、pNPP活性について試料を測定した。0時点の比率として残っている活性を計算した。5分後、TAPは98%を超えるその活性を喪失した。同時間では、SAPは依然として30%の活性が残っており、CIAPは、ほぼ90%の活性を残していた。30分後、SAPが依然として20%の活性を残し、CIPが40%の活性を残していたということは、TAPは、SAP又はCIAPのいずれかよりはるかに高温不安定性であることを示している(図8)。
【0041】
実施例V:37℃及び25℃におけるTAPの安定性
比色アッセイにおけるpNPPからの遊離のホスフェートの放出によって37℃及び25℃でのTAP活性の安定性を測定した。このアッセイでの活性は、pNPPがp−ニトロフェノール(pNP)に変換される際に生じる色の生成によって決定された。1Mのジエタノールアミン(pH8.5)、10mMのMgCl2、10mMのpNPP及び1mL当たり10単位のTAPを含有するホスフェート反応混合物を調製した。この混合物を3mLの試験管に、試験管当たり0.1mLずつ分注し、25℃又は37℃に試験管を置くことにより反応を開始させた。0〜30分の間5分間隔で10NのNaOHを0.1mL添加することにより反応を終了させた。各反応物を1mLの反応混合物(pNPP又はTAPなしの)で希釈して405nmの分光光度測定に好適な容積にした。蓄積させたpNPを測定し、既知のpNP標準と比較することにより放出されたホスフェートを決定した。結果は、30分の時間経過にわたる25℃又は37℃で放出されたホスフェートのナノモルとして表した(図9)。これらの結果は、TAP活性は、25℃よりも37℃の方が約2倍高く、いずれの温度でもTAP活性は少なくとも30分間安定であることを実証した。
【0042】
実施例VI:TAPの保存のための最適な条件
保存のための最適な条件を決定するためにTAPを75℃にて5分間インキュベートした。3種の異なった緩衝液、ビス−トリスプロパン緩衝液(pH6.0)、リン酸緩衝液(pH7.0)及びトリス塩酸緩衝液(pH7.4)を調べ、後者がTAPを安定化するのに最良であることが判った。様々な濃度のNaClを調べ、酵素の活性を維持するには、50mM,100mM又は200mMに比べて、NaClを含まないほうが良いことが判った。EDTAがTAPの酵素活性に阻害活性を示す場合、1mMのDTT及び200μg/mLのBSAの双方がTAP活性を安定化した。トリス塩酸(pH7.4)、1mMのMgCl2、1mMのDTT、200μg/mLのBSA及び50%グリセロールを含有する緩衝液において、TAPは活性を低下させることなく、12ヵ月にわたって安定であることが判明した。
【0043】
実施例VII:ホスファターゼ活性
(a)pNPP活性について標準化したDNAライゲーションアッセイにおけるSAPとTAPの酵素活性の比較:5’末端、3’末端及び平滑末端における脱リン酸化
【0044】
効率は、1μgのDNAを脱リン酸化できる希釈に基づいた1pNPP単位の酵素により脱リン酸化することができるDNAの量として定義される。
【0045】
pNPPアッセイを用いてμL当たり1単位にSAP及びTAPの双方を調整した。リトマス28DNA(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ社)をHindIII(5’末端突出)、EcoRV(平滑末端)又はPstI(3’末端突出)のいずれかで消化した。各切断ベクターDNAの部分試料(1mg/50μL)を、推奨される緩衝液中で37℃にて30分間、数種に希釈した各ホスファターゼで処理した。65℃にて5分間で、TAPを熱で失活させた。65℃にて15分間で、SAPを熱で失活させた。クイックリガーゼキット(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ社)を用い、指示書に従って、切断し、脱リン酸化したDNAを再環化した。次いで、連結したベクターで大腸菌を形質転換し、アンピシリン入りプレートに一晩置いた。コロニーの数が対照(ベクターを切断したが、脱リン酸化しなかった)の5%未満であれば、ホスファターゼ活性は完全であるとみなした。1pNPP単位のSAPが、5μgの5’末端突出DNAを脱リン酸化することができたのに対して、同一単位のTAPは、同一DNA50μgを脱リン酸化することができた(図10)。従って、TAPは、DNAにおける5’末端突出からリン酸基を外すとき、10倍多く効率的であることが示された。平滑末端では、TAPは、SAPよりも50倍高く効率的であり、3’末端突出では、TAPは8倍高く効率的だった。
【0046】
(b)TAPはデオキシヌクレオチドからリン酸基を外すことができる
Heinonen及びLahti(上記)により記載されたように本質的な比色アッセイにおける遊離のホスフェートの放出により、デオキシヌクレオチダーゼとしてのTAPの活性を測定した。このアッセイの活性は、既知のホスフェート標準に比べて決定された。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び1mMのdATP、dCTP、dGTP又はTTPのナトリウム塩を含有する4種の異なったデオキシヌクレオチド反応混合物を4本の別個の1.5mLの試験管に入れた。各試験管に2.5単位のTAPを加えた。試験管を37℃に置くことにより反応を開始させ、15分後、試験管を65℃に5分間置くことにより反応を停止させた。各反応混合物の試験管の内容物すべて(0.1mL)を、1.25NのH2SO4、4mMのモリブデン酸アンモニウム及び50%のアセトンを含有する測定溶液2.4mLを含有するガラス管に加えた。ガラス管を手短にボルテックスし、室温にて15分間インキュベートした。最終濃度0.33Mとなるようにクエン酸を加えることにより測定反応を停止させ、各管のOD390を記録した。0〜30mMのホスフェート標準それぞれ10μLを、処理し、TAP反応の試験管と同様に測定される模擬反応試験管に加えることにより標準曲線を生成した。dATP、dCTP,dGTP及びdTTPはTAPに対するほぼ同等の基質であり、これらの条件下で単位当たり1分当たり1〜2ナノモルのホスフェートを生成することを割り出した。
【0047】
SAP及びTAPのデオキシヌクレオチダーゼ活性を比較した。dATPを試験基質として用いたが、dCTP、dGTP又はdTTPを用いて同様の結果が得られると結論付けられた。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び0.8mMのdATPを含有するデオキシヌクレオチダーゼ反応混合物を調製した。この混合物0.1mLを1.5mLの反応管7本に分注し、5単位のTAP又はSAPを各管に加えた。試験管を37℃に置くことにより反応を開始させた。試験開始から0〜30分間後、5分間隔で各試験管を65℃に5分間置くことにより反応を停止させた。dATP、dADP、dAMP及びデオキシアデノシンについて標準を含有するプロファイルと比較するために、反応生成物をキャピラリ高速液体クロマトグラフィ(Cap−HPLC)の対象とした。自動試料注入器、カラムヒーター及びダイオードアレイ検出器を備えたアジレント(Agilent) 1100シリーズCap−HPLCを用いて、すべての解析を行った。標準のdATP、dADP、dAMP及びdAは、シグマケミカルから購入した。3μmの150x1mmのC18逆相Cap−HPLCデベロシル(Develosil)カラム(日本、愛知、野村化学株式会社)を用いて4つの種を分離した。100μLのフローセンサーを用いた、30℃、流速20μL/分での95%の0.1MのK2HPO4(KOHでpH6.0に調整)及び5%のアセトニトリルを用いたイソクラティック分離は、4種すべての良好な分離を生じることが判明した。dATP、dADP、dAMP及びdAの典型的な保持時間はそれぞれ、4.7分、5.2分、7.0分及び21.6分であった。TAP及びSAPで処理した試料を上記緩衝液で0.2mLに希釈し、1回当たり4μLを注入した。データは、254nm及び280nmでの吸収について、アジレント(Agilent)ケムステーション(ChemStation)ソフトウエア(カリフォルニア州、パロアルト、アジレントテクノロジーズ)を用いて回収し、ソフトウエアによってピークサイズを定量した。結果は、各反応成分について、存在する全ヌクレオチドの比率として表した(図11)。これらの結果は、TAPがデオキシヌクレオチダーゼとして機能することができ、dNTP、dNDP又はdNMP(Nは、アデノシン、シトシン、グアノシン又はチミジンを表す)からリン酸基を外すことができ、且つ、TAPは、デオキシヌクレオチダーゼとしてSAP(Heinonen及びLahti、上記)よりも高い活性を有することを実証した。
【0048】
(c)TAPはピロホスフェートから無機ホスフェートを放出することができる
Heinonen及びLahti(上記)により記載されたように本質的な比色アッセイにおける遊離のホスフェートの放出により、ピロホスファターゼとしてのTAPの活性を測定した。このアッセイにおける活性は、既知のピロホスファターゼを対照として用い、既知のホスフェート標準に比べて決定された。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び0.32mMのピロリン酸ナトリウムを含有するピロホスファターゼ反応混合物を調製した。この混合物を1.5mLの反応試験管に分注し、試験管当たり0.5mLで、最初の試験管が10単位で始まるTAPの2倍段階希釈を行った。試験管を37℃に置くことにより反応を開始させ、10分後、試験管を65℃に5分間置くことにより反応を停止させた。1.25NのH2SO4、4mMのモリブデン酸アンモニウム及び50%のアセトンを含有するホスフェート測定溶液を調製した。各反応混合物の試験管の内容物すべて(0.5mL)を、測定溶液2mLを含有するガラス管に加え、手短にボルテックスし、室温にて15分間インキュベートした。最終濃度0.33Mとなるようにクエン酸を加えることにより測定反応を停止させ、各試験管のOD390を記録した。0〜30mMのホスフェート標準それぞれ10μLを、TAP反応の試験管と同様に処理し測定される模擬反応試験管に加えることにより標準曲線を生成した。TIPPを含有する反応を並行して行った。TIPP反応混合物は、2単位のTIPP、50mMのトリシン(pH8.0)、1mMのMgCl2及び0.32mMのピロリン酸ナトリウムを含有し、75℃でそれをインキュベートした。10分後、TIPPが不活性である室温に試験管を置くことにより反応を停止させた。TAPと同様に放出されたホスフェートの測定を行った。この実験は、10単位のTAPが2単位のTIPPとほぼ同じ量の無機ホスフェートを放出することを示した(図12)。これらの結果は、TAPがピロホスファターゼとして機能することができ、ピロホスフェート(PPi)を無機ホスフェート(Pi)に切断することができることを実証した。
【0049】
(d)TAPは、リン酸化されたペプチドからリン酸基を外すことができる
33P標識したミエリン塩基性タンパク質基質(MyBP)の調製
【0050】
マサチューセッツ州、バーバリのニューイングランドバイオラブズのタンパク質セリン/スレオニンホスファターゼ(PSP)アッセイシステムキット(カタログ番号#P0780S)を用いて、セリン/スレオニン(Ser/Thr)がリン酸化されたMyBP基質を調製した。取り込まれた33Pを考慮して50μM(5x濃縮された)の濃度にSer/Thr標識したMyBP基質を希釈した。
【0051】
マサチューセッツ州、バーバリのニューイングランドバイオラブズのタンパク質チロシンホスファターゼ(PTP)アッセイシステムキット(カタログ番号#P0785S)を用いて、チロシン(Tyr)がリン酸化されたMyBP基質を調製した。取り込まれた33Pを考慮して25μM(5x濃縮された)の濃度にTyr標識したMyBP基質を希釈した。
【0052】
推奨された緩衝液中において、段階希釈したTAPホスファターゼ、10pNPP単位のCIAPホスファターゼと共に、又は酵素なしで37℃にて1時間、Ser/Thr(50μM)又はTyr(25μM)標識したMyBPタンパク質基質10μLをインキュベートした。200μLの冷却TCAを加え、氷上で5〜10分間インキュベートすることにより反応を停止させた。次いで、試料を12,000xgにて5分間遠心した。上清150μLを慎重に取り出し、2mLのシンチレーション液を加え、シンチレーションカウンタで計数した。結果を図13に示す。
【0053】
リン酸化されたSer/Thr残基に対するTAPの比活性は、1910ナノモル/分/mgであり、同一基質に対するCIAPの比活性よりもやや高い。リン酸化されたTyr残基に対するTAPの比活性もCIAPの比活性より高い(図13)。
【0054】
TAPの活性に関する上記実験の結果は、それぞれ単一のホスホ−アミノ酸残基を含有する3種のペプチド基質を用いて確認されたものである。標準のFMOC化学反応を用いてこれらのペプチドを調製した。ペプチド1はホスホセリン残基を含有し、ペプチド2はホスホスレオニン残基を含有し、ペプチド3はホスホチロシンを含有し、それぞれ、pSer、pThr及びpTyrで示した。以下に列記するのは各ペプチドの配列である。
【0055】
1.H−Val−Pro−Ile−Pro−Gly−Arg−Phe−Asp−Arg−Arg−Val−pSer−Val−Ala−Ala−Glu−NH2 (配列番号3)
【0056】
2.H−Thr−Ala−Asp−Ser−Gln−His−Ser−pThr−Pro−Pro−Lys−Lys−Lys−Arg−Lys−Val−Glu−OH(配列番号4)
【0057】
3.H−Glu−Trp−Met−Arg−Glu−Asn−Ala−Glu−pTyr−Leu−Arg−Val−Ala−OH(配列番号5)
【0058】
各ペプチドについて、同一配列を有するがホスフェートを欠いた対照ペプチドも合成した。
【0059】
1xTAP緩衝液(実施例VIIIを参照のこと)中で各ホスホ−ペプチド5μgを含有する反応物50μLを37℃にて10分間、段階希釈したTAPと共にインキュベートした。インキュベートの後、Cap−HPLCを用いて試料を解析した。バイダック(Vydac)5μm、C18の1x250mmカラムを流速25μL/分で用いて、3種のペプチドの脱リン酸化反応すべてを分析した。しかしながら、各ペプチドセットについて、リン酸化されたペプチドと脱リン酸化されたペプチドを分離するには最適化された勾配を必要とした。0.1%TFA水溶液(v/v)に対してアセトニトリルの比率(%B)を高めることによるCap−HPLCの勾配をプログラムした時間割は以下のとおりである。
【0060】
【表1】

【0061】
ペプチド1、ペプチド2及びペプチド3のカラムを平衡化するのにそれぞれ、80%のAと20%のB、91.5%のAと8.5%のB,及び95%のAと5%のBの比率を用いた。214nm、254nm及び280nmおける吸収についてアジレント(Agilent)ケムステーション(ChemStation)ソフトウエア(カリフォルニア州、パロアルト、アジレントテクノロジーズ)を用いてデータを回収し、ソフトウエアによってピークサイズを計算した。反応に存在する種の同一性を検証するために、マトリクス支援レーザー脱離/イオン化飛行時間型質量分析計(MALDI−TOF−MS)においても反応を調べた。α−シアノ−4−ヒドロキシ桂皮酸をマトリクスとして、加速電圧20kV及び陽イオンモードにて約150η秒の遅延時間で、ABIボイジャー(Voyager)DE質量分析計を用いた。図14は、TAPの量が増えるにつれて、量が減るホスホペプチド及び量が増える脱リン酸化されたペプチドを示すデータをまとめた3つのグラフから成る。このデータは、TAPがホスホ−チロシンペプチドに対して最も活性が高く、10分間で5μgのペプチドからホスフェートをすべて外すのに必要とした酵素はたった0.02単位であることを示している。ホスホ−セリン及びホスホ−スレオニンペプチドからホスフェートを完全に外すにはそれぞれ、2.5単位及び10単位のTAPを必要とした。このデータは、ペプチドからリン酸基を外すのにTAPを使用できることを示している。
【0062】
実施例VIII:TAPは、糖におけるホスフェートの位置にかかわりなく糖リン酸を脱リン酸化することができる
TAPが位置にかかわりなく、糖リン酸からリン酸残基を切断できるかどうかを調べるために、3種の糖リン酸、グルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6−リン酸に対するその活性を測定した。各糖リン酸について反応混合物の段階希釈物を調製したが、各反応混合物は、5mg/mLの1種の糖リン酸の溶液100μL、10xTAP緩衝液(500mMのビストリス−プロパン、10mMのMgCl2、1mMのZnCl2、pH6.0)を30μL及びH2Oを210μL含有した。50μLの反応混合物を最初の試験管に入れ、それに続く5本の試験管に25μL入れた。3種の調べるべき糖リン酸それぞれについてこれを繰り返した。これらの試験管を氷上に置いて冷却した。50μLの反応混合物を含有する最初の試験管に5μLの冷却TAP(5U/μL)をいったん加えた。混合した後、この試験管から25μLを取り出し、25μLの反応混合物を含有する次の試験管に入れた。混合した後、この第2の試験管から25μLを取り出し、次の試験管に入れた。この1:1の段階希釈を6本の試験管すべてについて行った。各糖リン酸についてTAPの段階希釈を行った後、試験管を氷から取り出し、37℃に15分間置いた。インキュベートした後、シリカゲルガラス上の薄層クロマトグラフィ(TLC)プレート上の密着したバンドに各試料3μLでスポットを作った。酵素なしでインキュベートした混合物のみを含有する陰性対照及び1mg/mLのグルコース又はマンノースを含有する陽性対照でもTLC上にスポットを作った。ホットエアガン(70℃を超えない温度)でスポットを完全に乾燥した。イソプロパノール、エタノール及び水(2.5:1:0.5、v:v:v)の中で溶媒の先頭が9cm移動するまでプレートを展開した。クロマトグラフィに続いて、プレートを乾燥させ、10%の過塩素酸を噴霧した。ヒートガンでプレートを焦がすことにより糖を視覚化した。366nmでのUVのもとで生成物を視覚化することにより感度を高めた。42μgのグルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6−リン酸の完全な消化は、6UのTAPを用いて37℃にて15分で達成された(図15)。
【0063】
糖におけるホスフェートの連結(すなわち、1又は6)は、酵素の活性に明瞭な影響を有さなかった。さらに、マンノース糖リン酸におけるTAPの活性は、グルコース糖リン酸におけるものと同様だった。
【背景技術】
【0001】
精製ホスファターゼは、試験管内で線状化したDNA、RNA、ヌクレオチド及びタンパク質からリン酸基を外すのに分子生物学者らによって使用される。DNAリガーゼは、一方のヌクレオチドが5’−ホスフェートを含有し、他方が3’水酸基を含有する場合のみ隣接するヌクレオチド間のホスホジエステル結合の形成を触媒するので、線状化したDNAベクターから双方の5’−末端リン酸残基を外すとライゲーション反応におけるベクターDNAの再環化が妨げられる。しかしながら、5’末端のホスフェートを持つ外来のDNA断片を脱リン酸化したベクターに効率よく連結し、コンピテント細胞を容易に形質転換することができる2つのニックを含有する開環分子を生じることができる。
【0002】
生体分子からリン酸基を外すために幾つかの異なったホスファターゼが開発されており、それぞれ、それ自体の長所及び短所を有する。この目的で使用されるべき最初のホスファターゼは、大腸菌(E.coli)から精製された細菌性アルカリホスファターゼ(BAP)であった。BAPは、あらゆる種類のDNA末端に対して良好な活性を有するという長所を有する。しかしながら、それは熱及び界面活性剤への耐性がきわめて強いので反応物から除くのが困難である(非特許文献1)。伝えられるところによれば、熱安定性を高めた変異体BAPが調製されている(特許文献1、特許文献2)。仔ウシ腸管アルカリホスファターゼ(CIP又はCIAP)は別のホスファターゼであり、分子生物学の技術で広範に使用されている(特許文献3及び特許文献4)。CIAPは、BAPほどDNAに対して活性は高くないが、プロテイナーゼK処理その後のフェノール:クロロホルム抽出又はEDTAの存在下での加熱工程その後のフェノール:クロロホルム抽出のいずれかの使用を必要とする反応物から除くのがやや容易である(非特許文献1)。さらに最近、北極のエビ、Pandalus borealis (SAP)から単離されたホスファターゼ(特許文献5及び特許文献6)は、使用するのがさらに容易であることが判っている。それは、BAPのようにあらゆる種類のDNA末端に良好な活性を有するが、65℃にて15分間熱不活性化することによって容易に反応物から除かれるという長所を有することが報告されている(Amersham Bioscience,Piscataway,NJ)。
【0003】
文献には、そのほかの熱不安定性ホスファターゼに関するそのほかの報告もあり、熱不安定性のアンタークティックホスファターゼ(TAP)と呼ばれる南極大陸で単離された好冷性の株TAB5から精製されたものが含まれる(非特許文献2)。他のホスファターゼを超えるTAPの長所には、高温不安定性及び高い比活性が挙げられる。Rinaらは、このホスファターゼがp−ニトロフェニルリン酸(pNPP)基質に対しタンパク質のmg当たり1650単位の比活性を有し、それは、そのほかのいかなる既知のホスファターゼの活性よりも有意に高いことを報告した。しかしながら、Rinaらが報告したクローン(非特許文献2)により産生されるタンパク質は、過剰発現及び精製に関連する多数の問題があった。たとえば、Rinaらにより記載された精製プロトコール(非特許文献2)は、TAPが明らかに会合する細胞膜からTAPを抽出するのに複数の超遠心工程を必要とする。このプロトコールは、大規模な製造(たとえば、300g以上の細胞ベーストの生産に関与する製造プロトコール)には向いていない。さらに、Rinaらにより記載されたプロトコールを用いたTAP遺伝子の過剰発現は、結果として、きわめて低い且つそのために大規模製造では費用有効性がない酵素の収量を生じた。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5,891,699号
【特許文献2】欧州特許第0441252号
【特許文献3】米国特許第5,773,226号
【特許文献4】米国特許第5,707,853号
【特許文献5】米国特許第6,387,634号
【特許文献6】米国特許第6,379,940号
【非特許文献】
【0005】
【非特許文献1】Sambrook,et al.,Molecular Cloning,A Laboratory Manual Sections 1.53−1.72(1989)
【非特許文献2】Rina,et al.,Eur. J. Biochem.267:1230−1238(2000)
【図面の簡単な説明】
【0006】
【図1】pN1を含有する大腸菌株からのTAP精製の結果を示す図である。レーン1〜6は、精製における連続工程である。レーン1は、誘導した後の全粗抽出物である。レーン2は、可溶化した膜分画である。レーン3は、DEAEカラムからの通過物である。レーン4は、Qセファロースカラムからのプールした分画である。レーン5は、ハイトラップSカラムからの通過物である。レーン6は、ヒドロキシアパタイトカラムからの通過物である。レーン7は、pNIクローンから得た精製したTAPである。分子量標準(S)は、kDで示す。レーン7の47.5kDに相当するバンドが実質的に精製されたTAPを表す。
【図2】TAP遺伝子を過剰発現させるベクターを構築するのに使用した工程を示す説明図である。図の上側の細長い四角は、遺伝子のそれに相当する発現生成物のN末端部分及びC末端部分を含むTAP遺伝子の模式図である。N末端では、メチオニン開始コドン及び仮定されたシグナル配列をコードするプロセッシング部位周辺の領域の位置を示す。C末端では、星印で示した停止コドンと共に、遺伝子産物のアミノ酸を示す。TAP遺伝子のPCR増幅に使用したプライマーを、各プライマーがTAP遺伝子にハイブリッド形成する部位で、細長い四角の上(実際のサイズではない)(正方向プライマーA、B及びCについて)及び下(逆方向プライマーD及びE)に示す。プライマー上のN及びXは、制限エンドヌクレアーゼNdeI又はXhoIについて操作した切断部位を示す。プライマーCは、NdeIに続く6個のHを示す。これらは、プライマーのメチオニン開始コドンの下流に挿入され、それに隣接するヒスチジンをコードする6個のコドンに相当し、TAPと融合したN末端Hisタグを創る。「PCR」に続く次の一連の線は、指示したプライマー対を用いることによって生じる結果の産物を示す。上記のように、星印は停止コドンを示し、6個のHは、ヒスチジンタグをいう。PCR産物をpET21aに挿入することによって創った5種の異なるプラスミドを示す。pEGTAP1.1.1はHisタグのない無傷のTAPをコードする。pEGTAP1.2.1は、Hisタグのない切り詰め型TAPをコードする。pEGTAP1.3.1はC末端Hisタグを持つ無傷のTAPをコードする。pEGTAP1.4.1はC末端Hisタグを持つ切り詰め型TAPをコードする。後の実験では、N末端Hisタグを持つ切り詰め型TAPをコードするようにpEGTAP7.4.1を構築した。
【図3】Hisタグの存在下又は非存在下、TAP遺伝子が切り詰められたか又は切り詰められていない様々なクローンから生じるTAPのSDS−PAGE解析を示す図である。「試験的」精製は、1mL未満の試料サイズを言う。レーン1は、Hisタグなしの無傷のTAP遺伝子を持つクローンpEGTAP1.1.1からの可溶性粗抽出物である。レーン2は、Hisタグなしの切り詰め型TAP遺伝子を持つクローンpEGTAP1.2.1からの可溶性粗抽出物である。レーン3及びレーン4は、Hisタグに融合した無傷のTAP遺伝子を持つクローンpEGTAP1.3.1からの2種の異なった単離物から得た可溶性粗抽出物である。レーン5及びレーン6は、Hisタグに融合した切り詰め型TAP遺伝子を持つクローンpEGTAP1.4.1からの2種の異なった単離物から得た可溶性粗抽出物である。分子量標準(S)は、kDで示す。切り詰め型TAP遺伝子を有するそれらのクローン(レーン2、5及び6)はTAPに相当する最強のバンドを提供した。
【図4】C末端のHisタグと共に切り詰め型の遺伝子を含有する(pEGTAP1.4.1)大腸菌(ER2566)クローンから非変性条件下で精製した可溶性TAPのSDS−PAGE解析を示す図である。全粗抽出物(T)、清澄化した粗抽出物(装填)(L)、Ni−NTAスピンカラム(カリフォルニア州、Studio City、キアゲン)からの通過物(FT)、Ni−NTAスピンカラムの洗浄物(W)並びに溶出緩衝液でNi−NTAスピンカラムから溶出された第1の試料及び第2の試料(E1及びE2)の試料10μLを、10〜20%のトリストリシンPAG(カリフォルニア州、Carlsbad、Invitrogen Corp.)で泳動し、クマシーブリリアントブルーで染色した。分子量標準(S)は、kDで示す。
【図5】解析したクローンがER2566(pEGTAP7.4.1)であったことを除いて図4で使用したのと同様の精製プロトコールの結果を示す図である。また、細胞抽出物全体をゲル上で泳動するのに代えて、粗抽出物を遠心して得られたペレットを再懸濁させて、不溶性ペレット(P)として泳動した。ペレット、可溶性装填試料(L)、Ni−NTAスピンカラムの通過物(FT)、Ni−NTAスピンカラムの洗浄物(W)、イミダゾールによるNi−NTAスピンカラムからの溶出物(E)及びkDで示す分子量標準(S)各10μLを、10〜20%のトリストリシンPAGで泳動し、クマシーブリリアントブルーで染色した。
【図6】様々なpH及び様々な緩衝液におけるTAP活性の比較を示す図である。ホスフェート放出アッセイを用いてpHの最適性を決定した。32P標識した二本鎖DNA、1mMのMgCl2及び0.1mMのZnCl2を含有する反応混合物中で50mMの各緩衝液をインキュベートした。反応物を37℃にて5分間インキュベートし、トリクロロ酢酸(TCA)可溶性のカウントの量を測定した。
【図7】ホスフェート放出アッセイを用いた、TAP活性に対する様々な塩の影響を示す図である。100mMの各塩、KCl、NaCl、酢酸カリウム又は酢酸ナトリウムを調べた。
【図8】CIAP、SAP及びTAPの温度不安定性の比較を示す図である。DNAの存在下、65℃にて各酵素を30分までインキュベートした。インキュベート中、様々な時点で試料を取り出し、pNPPで活性を測定した。
【図9】TAPによるpNPPからの経時的なホスフェートの放出を測定することにより、37℃及び25℃における安定性を示す図である。1単位のTAP及び未反応のpNPPを含有する試験管を25℃又は35℃のいずれかでインキュベートした。最終濃度5NとなるようにNaOHを添加することにより0〜30分について5分間隔で反応を止めた。ホスフェートの放出(pNPの蓄積)を405nm(黄色)にて分光光度計で測定し、反応時間に対してグラフを作った。
【図10】様々な種類のDNA末端からのリン酸基の取り外しにおけるTAPの効率のSAPに対する比較を示す図である:5’末端突出、平滑末端及び3’末端突出を調べた。これらの末端は、ベクターをそれぞれHindIII、EcoRV及びPstIで消化することにより作製した。
【図11】TAP及びSAPについてデオキシヌクレオチダーゼ活性の反応時間経過を示す図である。dATPアーゼ反応混合物の組成は、5分間隔でCap−HPLCにより測定した。結果は、存在するヌクレオチド全体の比率として各成分を表す。
【図12】TAPによるピロリン酸からの無機ホスフェートの放出を示す図である。0.32mMのピロリン酸ナトリウムを含有する各反応混合物に量を増やしながらTAPを加えた。無機ホスフェートは、Heinonen及びLahtiの方法(Heinonen J.K.& Lahti R.J.,「無機オルトホスフェートの新しい且つ好都合な比色分析測定及び無機ピロホスファターゼの測定へのその応用」Anal.Biochem.,113(2):313−7,1981)により既知のホスフェート標準と比較することによって本質的に測定した。並行する反応で熱安定性の無機ピロホスフェート(TIPP)を用いて、同等性を実証した。TAPについては37℃で、TIPPについては75℃で10分間のアッセイについて結果を示す。
【図13】リン酸化されたミエリン塩基性タンパク質におけるCIAPと比べたTAPの比活性を示す表である。
【図14】Cap−HPLCで測定した場合の、3種のホスホペプチド:ホスホセリン(グラフA)、ホスホスレオニン(グラフB)及びホスホチロシン(グラフC)に対するTAPの活性を示す図である。ホスホペプチド及びペプチドについての吸収ピークの面積が測定され、合計面積の比率としてTAPの単位数に対してグラフ上にプロットされた。
【図15】3種の異なった糖−リン酸、グルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6リン酸に対する1力価のTAPのTLC解析を示す図である。陽性対照として、3μgのグルコース(G)及びマンノース(M)のスポットを置いた。陰性対照(−)は、酵素なしでインキュベートした緩衝液と糖リン酸基質を含有する反応混合物だけを含有する。1〜6と付けたレーンは、12.5単位のTAPを含有する反応1で開始し、0.4単位のTAPを含有する反応6まで落とす、反応混合物における酵素の段階希釈(1:1)である。矢印は、糖及び糖−リン酸がTLC上を移動する距離を示す。
【図16】pN1プラスミドからの1.1kbの断片をPCRするのに使用したプライマーの配列(配列番号6及び7)である。クローニングしたPCR断片からの発現の結果、予想されるタンパク質のN末端配列(配列番号8)を正方向プライマーの配列の下に示す。プライマー内に含有されるNdeI及びXhoIのクローニング部位も示す。
【図17】無傷のTAP遺伝子のDNA配列(配列番号9)及びアミノ酸配列(配列番号10)を示す図である。四角で示すのは、N末端配列解析(Rina et al.,上記)により決定されるときのタンパク質の開始点である。矢印は、切り詰め型遺伝子をクローニングするのに使用したPCRプライマーがアニーリングする位置を示す。
【発明を実施するための形態】
【0007】
本発明の本実施態様は、中性pHで高められた高温不安定性を有し、高い活性があり、タンパク質のN末端に親水性リーダー配列を持つアルカリホスファターゼを提供する。親水性のリーダー配列は、オリゴペプチド、たとえば、Hisタグのような正味の正の電荷を持つオリゴペプチドであってもよい。このホスファターゼの長所には、ほかの反応物や生成物が影響を受けずにいられるレベルに温度を上げることによって活性を消去する能力が挙げられる。これらの特性を有するホスファターゼの例が、実施例で詳細に記載されるTAPである。本明細書で記載する方法は、タンパク質のN末端に融合したHisタグのような親水性リーダー配列の存在に頼るが、ニッケルカラムの緩衝液は、活性に悪影響を与えるので、ニッケルカラムを用いたタンパク質の精製は行わないことが好ましい。それにもかかわらず、驚くべきことに、親水性リーダー配列の存在は、他のカラム組成を用いたカラム精製で収量を高めた。
【0008】
本発明の実施態様では、切り詰め部がシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端がたとえば、Hisタグのような正に荷電するオリゴペプチドのような親水性のリーダー配列に共有結合する、切り詰められた、酵素活性のあるTAPが提供される。切り詰められたTAPは、約65℃にて15分未満で実質的に不活化することができる。特に、切り詰められたTAPの活性は、pH6より高い、さらに特にpH約7.4でのトリス塩酸中にて実質的に安定である。
【0009】
さらに、TAP(ジェンバンク(GenBank)Y18016にも記載されている)をコードするが、天然に存在するTAP遺伝子に関連するシグナル配列の一部又は全部を欠くDNAを記載する。該DNAは、好ましくは、発現されるTAP遺伝子のN末端で複数のHisアミノ酸をコードする配列をさらに含んでもよい。このDNAは、強力なプロモータ、たとえば、T7プロモータの制御下でベクターにおいて発現させてもよい。
【0010】
好ましい実施態様では、上述のDNAを含有するベクターが提供される。これらベクターの一例は、pEGTAP7.4.1.である。本発明の追加の実施態様では、少なくとも1種のベクターで形質転換された宿主細胞が提供される。他の種類の宿主細胞の使用を除外しないが、実施例では、宿主細胞は大腸菌である。
【0011】
本発明の追加の実施態様では、トリス塩酸、MgCl2、DTT及びグリセロールを含み、さらに特には、pHが約7.4である緩衝液中で37℃にて実質的に安定であるTAPの組成物が提供される。
【0012】
本発明の実施態様では、(a)たとえば、Hisタグのような親水性オリゴペプチドを発現するための配列に融合した切り詰め型TAP遺伝子をT7プロモータに操作可能に連結すること、(b)宿主細胞を形質転換すること、及び(c)TAPを過剰発現することを含むTAPを過剰発現するための方法が提供される。さらなる実施態様では、TAP遺伝子は、N末端及びC末端先端を有し、N末端先端で切り詰められ、切り詰め部がシグナル配列に相当し、該N末端先端がそこに結合された親水性オリゴペプチドを有するTAPタンパク質をコードする。
【0013】
本発明の追加の実施態様では、(a)上述のような形質転換された宿主細胞を得ること;(b)該宿主細胞を破砕して可溶性分画を得ること;及び(c)該可溶性分画からTAPを精製することを含む、精製したTAPを得る方法が提供される。さらに、該方法は、TAPがカラムから溶出されるカラム分離を追加的に含んでもよい。このような状況で、精製は、破砕した細胞混合物における全タンパク質に対するTAPタンパク質の比率の増加を言う。たとえば、TAPタンパク質の比率が混合物における全タンパク質の5%を超えた場合、精製が達成することができる。好ましくは、TAPタンパク質の比率は、全タンパク質の50%、80%又は90%を超えてもよい。特定の実施態様では、TAPタンパク質の比率は、全タンパク質の約95%まで精製される。
【0014】
本発明の追加の実施態様では、(a)約pH5.5〜7.0の緩衝液中の基質にTAPを加えること;並びに(b)規定された時間及び規定された温度で基質から外れるホスフェートの量を測定してTAPの活性を決定することを含む、TAPの活性をアッセイする方法が提供される。好ましい実施態様では、緩衝液はZnCl2塩及びMgCl2塩を含む。さらに好ましくは、緩衝液は、ビス−トリスプロパンである。
【0015】
追加の実施態様では、pH5.5〜7.0の緩衝液中の基質にTAPを加えること;並びに規定された時間及び規定された温度で基質から外されるべきホスフェートの量をもたらすことを含むリン酸化された基質を脱リン酸化する方法が提供される。
【0016】
基質は、核酸又は核酸の末端ヌクレオチド、糖リン酸、或いはリン酸化されたペプチド又はタンパク質であってもよい。
【発明を実施するための最良の形態】
【0017】
TAP遺伝子は、Rinaらにより単離され、クローニングされ(pN1)、DNA配列は公表されている。しかしながら、公表されたプロトコールを用いてこのクローンからTAPを過剰発現させ、精製することを試みた場合、試薬目的には不十分な量の精製酵素しか得られないこと、及び酵素が会合する膜断片から酵素を分離するのに複数の超遠心工程を必要とすることを含む多数の課題に遭遇した。本明細書では、TAP遺伝子を確実に過剰発現させて、大規模製造で費用有効性の高い酵素の収量を提供するプロトコールを提供する。さらに、複数の超遠心工程の必要性を回避し、大規模製造に適した精製プロトコールを開発した。核酸、ペプチド及び糖からホスフェートを外すTAPの用途が確立している。TAPの基質として作用してもよいそのほかの基質は、「The Enzymes(酵素)」,Vol.IV(1971)ch17,p373でReid & Wilsonにより記載されたものである。
【0018】
好ましい実施態様では、本明細書では「糖類」は、単糖類、オリゴ糖類又は多糖類を言う。糖類は、任意のペントース、ヘキソース又はヘプツロースの化合物を含む。ホスフェートがα異性体であろうとβ異性体であろうとそれには関係なく、ホスフェート(単数)又はホスフェート(複数)がどこに位置しようと、TAPを用いて、糖における任意の炭素からホスフェートを切断してもよい。一部の糖リン酸の例は、ウエブサイト、http://www.arabidopsis.org.で提供されている。糖リン酸の例には、マンノース、グルコース、フコース、ガラクトース、N−アセチルガラクトサミン、N−アセチルグルコサミン、キシロース及びラムノースのホスフェートが挙げられる。
【0019】
精製の収量及び容易さを改善するように公表されたプロトコールを改変して用い、pN1プラスミドを含有するER2575(ER2566pLysS)からのTAPの精製を試みた。形質転換した大腸菌株を増殖させ、誘導し、以下の例外と共に、Rinaら(上記)に記載されるようにホスファターゼを精製した。超遠心により可溶性膜分画を単離した後、タンパク質を緩衝液A(20mMのトリス、pH7.6、10mMのMgCl2、50mMのNaCl、0.2%のトリトンX−100)で透析し、次いで、緩衝液Aで平衡化したDEAEカラムに装填した。pNPPアッセイで測定したときホスファターゼ活性を有する通過物をRinaらが記載したようなQ−セファロースカラムに装填した。Q−セファロースカラムからの分画をプールして緩衝液B(リン酸カリウム緩衝液、pH6.6、25mMのNaCl、10mMのMgCl2)で透析し、緩衝液Bで平衡化したハイトラップSPカラムに装填した。ホスファターゼ活性を有する通過物を緩衝液C(リン酸カリウム緩衝液、pH7.2、50mMのNaCl、10mMのMgCl2)で透析し、緩衝液Cで平衡化したハイドロキシアパタイトカラムに装填した。ホスファターゼ活性は通過した。各工程からの試料を10〜20%のトリストリシンPAG(カリフォルニア州、Carlsbad、Invitrogen Corp.)で泳動し、クマシーブリリアントブルーで染色した。図1に見ることができるように、レーン1〜6におけるホスファターゼの収量は、期待はずれだった。
【0020】
上記プロトコールを用いて得られた期待はずれの結果のために、図2に記載したようなTAP遺伝子のクローニングを含む別のアプローチを検討した。
【0021】
具体的には、強力なプロモータ(T7プロモータ)の直後にTAP遺伝子を直接クローニングし、精製を円滑にするためにHisタグ(親和性タグ)を取り付けた。図13及び14に示すHisタグの配列は、タンパク質のN末端先端(遺伝子の5’末端)に融合した6個のヒスチジンを有する。しかしながら、ヒスチジンのこの数は重要ではない。本明細書で記載された目的を達成するには、タグにおけるヒスチジンの数は3個ほどで少なくても、6個を超えて所望のとおり多くてもよい。幾つかの異なった構築物を作成した。2種の構築物では、メチオニン開始コドンに隣接する、タンパク質のN末端の22アミノ酸から成る推定上のシグナル配列を取り除くことによってTAP遺伝子を切り詰めた。一方のクローンでは、C末端のHisタグを加えて切り詰め型ホスファターゼとの融合タンパク質を形成したが、他方ではそうはしなかった。たとえば、pEGTAP1.4.1は、Hisタグを有したが、pEGTAP1.2.1は有さなかった。2種のほかの構築物では、遺伝子は推定上のシグナル配列を保持し、いずれかがさらにC末端Hisタグを含有し(pEGTAP1.3.1)、又は含有しなかった(pEGTAP1.1.1)(図2)。
【0022】
pET21aにPCR産物を挿入することによって(実施例I、図2、11及び12)創った5種の異なるプラスミドを図2に示す。pEGTAP1.1.1はHisタグのない無傷のTAPをコードする。pEGTAP1.2.1は、Hisタグのない、切り詰め型TAPをコードする。pEGTAP1.3.1はC末端Hisタグを持つ、無傷のTAPをコードする。pEGTAP1.4.1はC末端Hisタグを持つ、切り詰め型TAPをコードする。pEGTAP7.4.1は、N末端Hisタグを持つ、切り詰め型TAPをコードする。
【0023】
C末端でHisタグを持つ又は持たない、推定上のシグナル配列を持つ無傷のTAP遺伝子を含有するプラスミドは、誘導後、SDS−PAGEにおいて、いかなる明瞭な可溶性ホスファターゼも産生することができなかった(図3、レーン1、3及び4)。しかしながら、Hisタグを持つ又は持たない、推定上のシグナル配列なしで構築されたプラスミドはSDS−PAGEにおいて正しいサイズのきわめて強いバンドを生じた(図3、レーン2、5及び6)。しかしながら、タンパク質の精製中、問題が生じた。pEGTAP1.4.1(切り詰め型TAP及びC末端His)を含有する、形質転換した宿主細胞ER2566は、非変性条件で主に不溶性TAPを産生した(図4)。変性条件下で精製を試み、その後、変性させたホスファターゼを折り畳んだ状態に戻した。しかしながら、このアプローチでは、活性のある酵素は、相対的に少ない量しか得られなかった。pEGTAP1.2.1(Hisタグを持たない切り詰め型TAP)を含有する大腸菌ER2566は可溶性のTAPを産生したが、タンパク質はほとんどのカラムに結合できなかった。
【0024】
上記の問題を克服するために、Hisタグをコードする配列をTAP遺伝子のC末端に代えてN末端に配置した第5のクローン(pEGTAP7.4.1)を構築した(図2)。この構築物は、可溶性分画で良好な収量のTAPを産生し(図5)、複数の超遠心工程なしで容易にそれを精製することができた(実施例II)。具体的には、ホスファターゼのN末端におけるHisタグの存在によって、ニッケルカラムを含まないたった2又は3回のカラムでほぼ均質までの精製が可能になった。理論によって限定されることを望まないで、Hisタグの最初の効用は、親和性タグとしてではなく、可溶性ホスファターゼの産生を促進することだったと思われる。
【0025】
DNA及びタンパク質基質を用いたアッセイ(実施例VII)によって上記方法(実施例I及びII)により調製したTAPホスファターゼの活性を、SAP及びCIAPのホスファターゼ活性と比較し、TAPが向上した活性を有することが判明した。たとえば、実施例VIIに記載したホスフェート放出アッセイを用いて、DNAの5’末端突出、3’末端突出及び平滑末端について、TAP及びSAPの線状DNAの脱リン酸化活性を測定した。これらの異なった構造の脱リン酸化を、同一のpNPP単位量を用いて比較すると、TAPは一貫してSAPよりも効率が良いことが分かった(図10)。
【0026】
TAPはまた、CIAPと同じくらい効率的に、リン酸化されたタンパク質におけるセリン/スレオニン残基及びチロシン残基からリン酸基を取り外すことができる(実施例VII、図13)。
【0027】
他のホスファターゼと比べたTAPの高い活性に加えて、TAPは、高い熱不安定性を有することが判明した(実施例IV、図8)。CIAP、SAP及びTAPの高温不安定性を比較すると、TAPが65℃で5分後に98%の活性を失ったのに比べて、SAPはこの温度で70%の活性を失い、CIAPはたった10%の活性しか失わなかったことが判明した。
【0028】
本発明のほかの実施態様では、ZnCl2及びMgCl2が、ホスファターゼの活性を上げることが判明した(実施例III)。TAPの活性は、たとえば、ビス−トリスプロパン緩衝液を用いてpH約5.5〜7.0で最適化された(実施例III)。
【0029】
以下の実施例によって本発明をさらに説明する。これら実施例は本発明の理解を助けるために提供されるのであって、その限定として解釈されるものではない。
【0030】
上記及び以下に引用した参考文献は、参考により本明細書に組み入れられる。
【実施例】
【0031】
実施例I:TAP−Hisタグ融合遺伝子の構築及び発現
分類されていない南極の株TAB5に由来する切り詰め型ホスファターゼ遺伝子をN末端の6ヒスチジン残基のタグと共にクローニングした。簡単に言えば、PCRにより、組換えプラスミドpN1(Rina,et al.,Eur.J.Biochem.,267:1230−1238,2000)から1.1kbの断片を増幅した。正方向のプライマー(図2、プライマーC)は、推定上のシグナル配列(63塩基)(図16及び17)を持たない遺伝子の5’末端に相同の配列を含有することに加えて、ATG開始コドンの一部としてのNdeI制限部位及びATG開始コドンと切り詰め型ホスファターゼ遺伝子の最初のコドンとの間に位置する6個のヒスチジン残基をコードする6個のコドンを含有した(図16及び17)。逆方向のプライマー(図2、プライマーD)は、遺伝子の3’末端に逆相補的な配列を含有することに加えて、TAA停止コドンのすぐ下流にXhoI制限部位を含有した(図16及び17)。いったん増幅し、1.1kbの断片をNdeI及びXhoIで消化して、同様にNdeI及びXhoIで消化したT7発現ベクターpET21a(ウィスコンシン州、マジソン、Novagen)に連結した(Sambrook,et al.,Molecular Cloning,A Laboratory Manual,Sections 1.53−1.72(1989))。得られたライゲーションで大腸菌株ER2688(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ)を形質転換した。形質転換のプラスミドを単離し、制限マッピング及びDNAの配列決定により特性分析し、特性分析した際、正しいサイズ及び配列の挿入物を含有することが判った。T7プロモータでホスファターゼ遺伝子を発現させるために、プラスミド構築物で大腸菌株ER2566(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ)を形質転換した。pEGTAP7.4.1が、最良の収量及び精製特性を提供することが判明した(図3〜5)。
【0032】
実施例II:TAP−Hisタグ融合タンパク質の製造及び精製
(a)TAPの小規模の精製
100mg/mLのカルベニシリンを含む5mLのリッチブロス(1リットル当たり10gのトリプトン(ミシガン州、リボニア、ディフコラボラトリーズ)、5gの酵母抽出物(ミシガン州、リボニア、ディフコラボラトリーズ)、5gのNaCl、NaOHでpH7.2)中にて単離したコロニーからER2566(pEGTAP7.4.1)を30℃で一晩増殖させた。3mLの一晩培養物を用いて、100mg/mLのカルベニシリンを含む300mLのリッチブロスに植菌した。30℃にて対数増殖期の中間(クレット70)まで培養物を増殖させ、氷上で冷却し、0.4mMのイソプロピル−β−チオガラクトシド(IPTG)で誘導し、15℃にて20時間増殖させた。4℃にて8000rpmで10分間遠心分離することにより35mLの細胞を回収した。上清を除き、細胞ペレットを秤量し、−20℃に2時間置いた。ペレットを氷上で融解し、1mLの細胞溶解緩衝液(50mMのNaH2PO4、300mMのNaCl、10mMのイミダゾール、pH8.0)に細胞を再懸濁した。リゾチームを1mL/mLまで加え、試料を氷上で30分間インキュベートした。インキュベートに続いて、細胞を、氷上で6回それぞれ10秒間、途中で1分間停止しての超音波処理により破壊した。4℃にて14,000rpmで20分間、粗抽出物を遠心し、未破砕の細胞及びそのほかの不溶性物質を除いて清澄な粗抽出物を生成した。0.6mLの細胞溶解緩衝液で平衡化したNi−NTAスピンカラムに0.6mLの清澄な粗抽出物を装填した。700xgにて2分間、試料を遠心した。通過物を除き、0.6mLの洗浄緩衝液(50mMのNaH2PO4、300mMのNaCl、20mMのイミダゾール、pH8.0)で2回カラムを洗浄した。0.2mLの溶出緩衝液(50mMのNaH2PO4、300mMのNaCl、250mMのイミダゾール、pH8.0)でタンパク質を2回溶出した。各工程における10μLをSDS−PAGEで解析した。pEGTAP7.4.1の結果を図5に示す。
【0033】
(b)ER2566におけるpEGTAP7.4.1からのTAPの大規模な精製
25℃にて発酵用のリッチブロス(100リットル当たり、500gのAmberexの酵母抽出物(ウィスコンシン州、ミルウォーキーのSensient Technologies)、1kgのCE90MSトリプトン(ニュージャージー州、Carlstadt、Marcor Development Corp.)、27gのNaOH,10gのアンピシリン、5.5gのポリプロピレングリコール消泡剤(ウエストバージニア州、サウスチャールストン、Arco Chemical Co.))2000mL中で25℃にてpEGTAP7.4.1プラスミドを収容する大腸菌株ER2566を増殖させた。この培養物を用いて100リットルの発酵用リッチブロスに植菌した。70のクレット(Klett)に達するまで25℃にて5時間、細胞を好気的に増殖させた。次いで発酵槽を15℃に冷却し、クレットが90に達したとき、最終濃度0.3mMとなるようにIPTGを加えた。次いで15℃にて細胞を好気的にさらに17時間増殖させ、細胞は最終クレット305を持つ定常期に達した。321グラム湿重量の細胞ペレットを回収するのに100リットルの発酵を要した。321グラムの細胞ペレットを963mLの緩衝液A(20mMリン酸カリウム緩衝液、pH7.4、50mMのNaCl、5%のグリセロール)に懸濁し、約12,000psigでゴーリンのホモゲナイザーを通過させた。細胞溶解物を約13,000Gにて40分間遠心し、上清を回収した(1150mL)。
【0034】
緩衝液Aで平衡化した500mLのヘパリンハイパー−Dカラム(カリフォルニア州、フレモントのCiphergen Biosystems,Inc.)に上清溶液をかけた。1Lの緩衝液Aで洗浄し、次いで、緩衝液A中0.05M〜1MのNaClの勾配2Lを流し、50mLの分画を回収した。10mMのMgCl2を含有する1Mのジエタノールアミン/HCl緩衝液(pH8.5)中の0.1MのpNPPと共に試料をインキュベートすることにより、ホスファターゼ活性について各分画を測定した。37℃にて1〜5分間反応を行い、活性は、黄色の生成として、405nmにて分光光度計で測定した。0.05〜0.35MのNaClでホスファターゼ活性は溶出された。
【0035】
ヘパリンハイパー−Dカラムのホスファターゼ活性を含有する分画をプールし、次いで、緩衝液Aに対して一晩透析した。この800mLのプールの100mLを105mLのソースQカラム(ニューヨーク州、ニューヨークのファイザー社)にかけた。210mLの緩衝液Aで洗浄した後、緩衝液A中0.05M〜1MのNaClの勾配1Lを流し、15mLの分画を回収した。上で記載したpNPPアッセイを用いて分画を測定し、ホスファターゼ活性は、0.15〜0.18MのNaClで溶出された。ヘパリンハイパー−Dでプールした残りの700mLも同様にカラムにかけ、溶出し;次いでホスファターゼ活性を含有する勾配分画をプールした。
【0036】
集め合わせたソースQのプールを緩衝液Aに対して透析し、50%グリセロールを補った。この120mLのプールの40mLを500mLの緩衝液Aで希釈し、次いで、緩衝液Aで平衡化した400mLのPEIカラム(英国、ケントのワットマン)にかけた。400mLの緩衝液Aで洗浄した後、緩衝液A中0.05M〜1MのNaClの線形勾配を流し、15mLの分画を回収した。上で記載したpNPPアッセイを用いて分画を測定した。ホスファターゼ活性は0.14〜0.2MのNaClの間で溶出された。ソースQでプールした残りの80mLも同様にカラムにかけ、溶出し;次いでホスファターゼ活性を含有する勾配分画をプールした。10mMのトリス(pH7.4)、1mMのMgCl2,1mMのDTT、50%のグリセロールを含有する保存緩衝液に対してこのプールを透析した。
【0037】
実施例III:TAPによるリン酸基取り外しのための至適pH及び塩条件の決定
実施例IIで記載されたように精製したTAPを用いて、DNAからリン酸基を外すための最適条件を決定した。以下のようなホスフェート放出アッセイによって脱リン酸化を測定した。50μLの反応物中で、2種の相補的な40量体のオリゴヌクレオチド
5’−ACGTATGTTAGGTTAGGTTAGGTTAGGTTAGGTTAGGCTC−3’(配列番号1)
3’−TGCATACAATCCAATCCAATCCAATCCAATCCAATCCGAG−5’(配列番号2)
をアニーリングし、T4ポリヌクレオチドキナーゼ(マサチューセッツ州、バーバリ、ニューイングランドバイオラボ社)及びγ32P−ATPを用いて製造元により推奨されるように末端標識した。この放射活性のある二量体を用いて、ホスファターゼ基質(最終20μg/mL)として作用するラムダHindIII断片の混合物1mgを「スパイク」した。37℃にて5分間の、基質混合物(1mMのMgCl2及び0.1mMのZnCl2を含有する緩衝液中で反応物当たり0.01単位のpNPP)とホスファターゼとのインキュベートに続く放射活性の放出によってTAP活性を測定した。TCAによるDNA基質の沈殿及び沈殿しなかった放射活性のシンチレーションカウントにより放出を測定した。TCA沈殿は、100μLのニシン精子DNA(2mg/mL)をキャリアとして各反応物に加えることから成った。次いで、150μLの20%冷却TCAを加えた。試料をボルテックスし、5分間氷上で冷却した。微量遠心機にて14,000xgで5分間、各反応物を遠心した。各上清(50%)150μLを2mLのシンチラントに加え、0.5分間カウントした。pNPP比色アッセイにより測定された0.001〜0.01単位の範囲にわたってTAP活性は線形であることが判明した。
【0038】
3種の異なった緩衝液:pH範囲が6〜9.5のトリス−塩酸、pH範囲が6〜7のビストリスプロパン緩衝液及びpH範囲が5.5〜7のビス−トリスプロパン緩衝液を用いてpH範囲が5.5〜9.5の反応緩衝液を調べた。ビス−トリスプロパン緩衝液にてDNA基質に対して最適の活性を与えたpH範囲は、5.5〜6.0の間であることが判った(図6)。
【0039】
カチオンの必要性を調べた。pNPPでのアッセイにおいてZn2+イオンが阻害活性を示すと報告したRinaら(上記、2000年)とは対照的に、上述のアッセイは、ZnCl2の存在が10倍多いホスフェートの放出を与えることを示していることを確定した。その後、TAP活性のアッセイに1mMのMgCl2、0.1mMのZnCl2を含めた。100mMの濃度でのKCl、NaCl、酢酸カリウム及び酢酸ナトリウムを含むそのほかの塩の添加は、TAP活性に影響を有さないことが認められた(図7)。
【0040】
実施例IV:TAPの熱不安定性の確定
pNPP活性の1単位は、室温にて1分間に1mLの反応容積において1μモルのpNPPをp−ニトロフェノールに加水分解するのに必要とされる酵素の量に相当する。各酵素について推奨された反応緩衝液にて、TAP、CIAP及びSAPそれぞれの10pNPP単位を1μgのラムダHindIII断片と混合した。混合物を37℃にて10分間インキュベートし、次いで氷上に置いた。次いで反応物を65℃の温水浴に入れ、5分、10分、20分及び30分後に試料を取り出し、氷上に置いた。熱処理に続いて、pNPP活性について試料を測定した。0時点の比率として残っている活性を計算した。5分後、TAPは98%を超えるその活性を喪失した。同時間では、SAPは依然として30%の活性が残っており、CIAPは、ほぼ90%の活性を残していた。30分後、SAPが依然として20%の活性を残し、CIPが40%の活性を残していたということは、TAPは、SAP又はCIAPのいずれかよりはるかに高温不安定性であることを示している(図8)。
【0041】
実施例V:37℃及び25℃におけるTAPの安定性
比色アッセイにおけるpNPPからの遊離のホスフェートの放出によって37℃及び25℃でのTAP活性の安定性を測定した。このアッセイでの活性は、pNPPがp−ニトロフェノール(pNP)に変換される際に生じる色の生成によって決定された。1Mのジエタノールアミン(pH8.5)、10mMのMgCl2、10mMのpNPP及び1mL当たり10単位のTAPを含有するホスフェート反応混合物を調製した。この混合物を3mLの試験管に、試験管当たり0.1mLずつ分注し、25℃又は37℃に試験管を置くことにより反応を開始させた。0〜30分の間5分間隔で10NのNaOHを0.1mL添加することにより反応を終了させた。各反応物を1mLの反応混合物(pNPP又はTAPなしの)で希釈して405nmの分光光度測定に好適な容積にした。蓄積させたpNPを測定し、既知のpNP標準と比較することにより放出されたホスフェートを決定した。結果は、30分の時間経過にわたる25℃又は37℃で放出されたホスフェートのナノモルとして表した(図9)。これらの結果は、TAP活性は、25℃よりも37℃の方が約2倍高く、いずれの温度でもTAP活性は少なくとも30分間安定であることを実証した。
【0042】
実施例VI:TAPの保存のための最適な条件
保存のための最適な条件を決定するためにTAPを75℃にて5分間インキュベートした。3種の異なった緩衝液、ビス−トリスプロパン緩衝液(pH6.0)、リン酸緩衝液(pH7.0)及びトリス塩酸緩衝液(pH7.4)を調べ、後者がTAPを安定化するのに最良であることが判った。様々な濃度のNaClを調べ、酵素の活性を維持するには、50mM,100mM又は200mMに比べて、NaClを含まないほうが良いことが判った。EDTAがTAPの酵素活性に阻害活性を示す場合、1mMのDTT及び200μg/mLのBSAの双方がTAP活性を安定化した。トリス塩酸(pH7.4)、1mMのMgCl2、1mMのDTT、200μg/mLのBSA及び50%グリセロールを含有する緩衝液において、TAPは活性を低下させることなく、12ヵ月にわたって安定であることが判明した。
【0043】
実施例VII:ホスファターゼ活性
(a)pNPP活性について標準化したDNAライゲーションアッセイにおけるSAPとTAPの酵素活性の比較:5’末端、3’末端及び平滑末端における脱リン酸化
【0044】
効率は、1μgのDNAを脱リン酸化できる希釈に基づいた1pNPP単位の酵素により脱リン酸化することができるDNAの量として定義される。
【0045】
pNPPアッセイを用いてμL当たり1単位にSAP及びTAPの双方を調整した。リトマス28DNA(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ社)をHindIII(5’末端突出)、EcoRV(平滑末端)又はPstI(3’末端突出)のいずれかで消化した。各切断ベクターDNAの部分試料(1mg/50μL)を、推奨される緩衝液中で37℃にて30分間、数種に希釈した各ホスファターゼで処理した。65℃にて5分間で、TAPを熱で失活させた。65℃にて15分間で、SAPを熱で失活させた。クイックリガーゼキット(マサチューセッツ州、バーバリ、ニューイングランドバイオラブズ社)を用い、指示書に従って、切断し、脱リン酸化したDNAを再環化した。次いで、連結したベクターで大腸菌を形質転換し、アンピシリン入りプレートに一晩置いた。コロニーの数が対照(ベクターを切断したが、脱リン酸化しなかった)の5%未満であれば、ホスファターゼ活性は完全であるとみなした。1pNPP単位のSAPが、5μgの5’末端突出DNAを脱リン酸化することができたのに対して、同一単位のTAPは、同一DNA50μgを脱リン酸化することができた(図10)。従って、TAPは、DNAにおける5’末端突出からリン酸基を外すとき、10倍多く効率的であることが示された。平滑末端では、TAPは、SAPよりも50倍高く効率的であり、3’末端突出では、TAPは8倍高く効率的だった。
【0046】
(b)TAPはデオキシヌクレオチドからリン酸基を外すことができる
Heinonen及びLahti(上記)により記載されたように本質的な比色アッセイにおける遊離のホスフェートの放出により、デオキシヌクレオチダーゼとしてのTAPの活性を測定した。このアッセイの活性は、既知のホスフェート標準に比べて決定された。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び1mMのdATP、dCTP、dGTP又はTTPのナトリウム塩を含有する4種の異なったデオキシヌクレオチド反応混合物を4本の別個の1.5mLの試験管に入れた。各試験管に2.5単位のTAPを加えた。試験管を37℃に置くことにより反応を開始させ、15分後、試験管を65℃に5分間置くことにより反応を停止させた。各反応混合物の試験管の内容物すべて(0.1mL)を、1.25NのH2SO4、4mMのモリブデン酸アンモニウム及び50%のアセトンを含有する測定溶液2.4mLを含有するガラス管に加えた。ガラス管を手短にボルテックスし、室温にて15分間インキュベートした。最終濃度0.33Mとなるようにクエン酸を加えることにより測定反応を停止させ、各管のOD390を記録した。0〜30mMのホスフェート標準それぞれ10μLを、処理し、TAP反応の試験管と同様に測定される模擬反応試験管に加えることにより標準曲線を生成した。dATP、dCTP,dGTP及びdTTPはTAPに対するほぼ同等の基質であり、これらの条件下で単位当たり1分当たり1〜2ナノモルのホスフェートを生成することを割り出した。
【0047】
SAP及びTAPのデオキシヌクレオチダーゼ活性を比較した。dATPを試験基質として用いたが、dCTP、dGTP又はdTTPを用いて同様の結果が得られると結論付けられた。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び0.8mMのdATPを含有するデオキシヌクレオチダーゼ反応混合物を調製した。この混合物0.1mLを1.5mLの反応管7本に分注し、5単位のTAP又はSAPを各管に加えた。試験管を37℃に置くことにより反応を開始させた。試験開始から0〜30分間後、5分間隔で各試験管を65℃に5分間置くことにより反応を停止させた。dATP、dADP、dAMP及びデオキシアデノシンについて標準を含有するプロファイルと比較するために、反応生成物をキャピラリ高速液体クロマトグラフィ(Cap−HPLC)の対象とした。自動試料注入器、カラムヒーター及びダイオードアレイ検出器を備えたアジレント(Agilent) 1100シリーズCap−HPLCを用いて、すべての解析を行った。標準のdATP、dADP、dAMP及びdAは、シグマケミカルから購入した。3μmの150x1mmのC18逆相Cap−HPLCデベロシル(Develosil)カラム(日本、愛知、野村化学株式会社)を用いて4つの種を分離した。100μLのフローセンサーを用いた、30℃、流速20μL/分での95%の0.1MのK2HPO4(KOHでpH6.0に調整)及び5%のアセトニトリルを用いたイソクラティック分離は、4種すべての良好な分離を生じることが判明した。dATP、dADP、dAMP及びdAの典型的な保持時間はそれぞれ、4.7分、5.2分、7.0分及び21.6分であった。TAP及びSAPで処理した試料を上記緩衝液で0.2mLに希釈し、1回当たり4μLを注入した。データは、254nm及び280nmでの吸収について、アジレント(Agilent)ケムステーション(ChemStation)ソフトウエア(カリフォルニア州、パロアルト、アジレントテクノロジーズ)を用いて回収し、ソフトウエアによってピークサイズを定量した。結果は、各反応成分について、存在する全ヌクレオチドの比率として表した(図11)。これらの結果は、TAPがデオキシヌクレオチダーゼとして機能することができ、dNTP、dNDP又はdNMP(Nは、アデノシン、シトシン、グアノシン又はチミジンを表す)からリン酸基を外すことができ、且つ、TAPは、デオキシヌクレオチダーゼとしてSAP(Heinonen及びLahti、上記)よりも高い活性を有することを実証した。
【0048】
(c)TAPはピロホスフェートから無機ホスフェートを放出することができる
Heinonen及びLahti(上記)により記載されたように本質的な比色アッセイにおける遊離のホスフェートの放出により、ピロホスファターゼとしてのTAPの活性を測定した。このアッセイにおける活性は、既知のピロホスファターゼを対照として用い、既知のホスフェート標準に比べて決定された。50mMのビストリス−プロパン(pH6.0)、1mMのMgCl2、0.1mMのZnCl2及び0.32mMのピロリン酸ナトリウムを含有するピロホスファターゼ反応混合物を調製した。この混合物を1.5mLの反応試験管に分注し、試験管当たり0.5mLで、最初の試験管が10単位で始まるTAPの2倍段階希釈を行った。試験管を37℃に置くことにより反応を開始させ、10分後、試験管を65℃に5分間置くことにより反応を停止させた。1.25NのH2SO4、4mMのモリブデン酸アンモニウム及び50%のアセトンを含有するホスフェート測定溶液を調製した。各反応混合物の試験管の内容物すべて(0.5mL)を、測定溶液2mLを含有するガラス管に加え、手短にボルテックスし、室温にて15分間インキュベートした。最終濃度0.33Mとなるようにクエン酸を加えることにより測定反応を停止させ、各試験管のOD390を記録した。0〜30mMのホスフェート標準それぞれ10μLを、TAP反応の試験管と同様に処理し測定される模擬反応試験管に加えることにより標準曲線を生成した。TIPPを含有する反応を並行して行った。TIPP反応混合物は、2単位のTIPP、50mMのトリシン(pH8.0)、1mMのMgCl2及び0.32mMのピロリン酸ナトリウムを含有し、75℃でそれをインキュベートした。10分後、TIPPが不活性である室温に試験管を置くことにより反応を停止させた。TAPと同様に放出されたホスフェートの測定を行った。この実験は、10単位のTAPが2単位のTIPPとほぼ同じ量の無機ホスフェートを放出することを示した(図12)。これらの結果は、TAPがピロホスファターゼとして機能することができ、ピロホスフェート(PPi)を無機ホスフェート(Pi)に切断することができることを実証した。
【0049】
(d)TAPは、リン酸化されたペプチドからリン酸基を外すことができる
33P標識したミエリン塩基性タンパク質基質(MyBP)の調製
【0050】
マサチューセッツ州、バーバリのニューイングランドバイオラブズのタンパク質セリン/スレオニンホスファターゼ(PSP)アッセイシステムキット(カタログ番号#P0780S)を用いて、セリン/スレオニン(Ser/Thr)がリン酸化されたMyBP基質を調製した。取り込まれた33Pを考慮して50μM(5x濃縮された)の濃度にSer/Thr標識したMyBP基質を希釈した。
【0051】
マサチューセッツ州、バーバリのニューイングランドバイオラブズのタンパク質チロシンホスファターゼ(PTP)アッセイシステムキット(カタログ番号#P0785S)を用いて、チロシン(Tyr)がリン酸化されたMyBP基質を調製した。取り込まれた33Pを考慮して25μM(5x濃縮された)の濃度にTyr標識したMyBP基質を希釈した。
【0052】
推奨された緩衝液中において、段階希釈したTAPホスファターゼ、10pNPP単位のCIAPホスファターゼと共に、又は酵素なしで37℃にて1時間、Ser/Thr(50μM)又はTyr(25μM)標識したMyBPタンパク質基質10μLをインキュベートした。200μLの冷却TCAを加え、氷上で5〜10分間インキュベートすることにより反応を停止させた。次いで、試料を12,000xgにて5分間遠心した。上清150μLを慎重に取り出し、2mLのシンチレーション液を加え、シンチレーションカウンタで計数した。結果を図13に示す。
【0053】
リン酸化されたSer/Thr残基に対するTAPの比活性は、1910ナノモル/分/mgであり、同一基質に対するCIAPの比活性よりもやや高い。リン酸化されたTyr残基に対するTAPの比活性もCIAPの比活性より高い(図13)。
【0054】
TAPの活性に関する上記実験の結果は、それぞれ単一のホスホ−アミノ酸残基を含有する3種のペプチド基質を用いて確認されたものである。標準のFMOC化学反応を用いてこれらのペプチドを調製した。ペプチド1はホスホセリン残基を含有し、ペプチド2はホスホスレオニン残基を含有し、ペプチド3はホスホチロシンを含有し、それぞれ、pSer、pThr及びpTyrで示した。以下に列記するのは各ペプチドの配列である。
【0055】
1.H−Val−Pro−Ile−Pro−Gly−Arg−Phe−Asp−Arg−Arg−Val−pSer−Val−Ala−Ala−Glu−NH2 (配列番号3)
【0056】
2.H−Thr−Ala−Asp−Ser−Gln−His−Ser−pThr−Pro−Pro−Lys−Lys−Lys−Arg−Lys−Val−Glu−OH(配列番号4)
【0057】
3.H−Glu−Trp−Met−Arg−Glu−Asn−Ala−Glu−pTyr−Leu−Arg−Val−Ala−OH(配列番号5)
【0058】
各ペプチドについて、同一配列を有するがホスフェートを欠いた対照ペプチドも合成した。
【0059】
1xTAP緩衝液(実施例VIIIを参照のこと)中で各ホスホ−ペプチド5μgを含有する反応物50μLを37℃にて10分間、段階希釈したTAPと共にインキュベートした。インキュベートの後、Cap−HPLCを用いて試料を解析した。バイダック(Vydac)5μm、C18の1x250mmカラムを流速25μL/分で用いて、3種のペプチドの脱リン酸化反応すべてを分析した。しかしながら、各ペプチドセットについて、リン酸化されたペプチドと脱リン酸化されたペプチドを分離するには最適化された勾配を必要とした。0.1%TFA水溶液(v/v)に対してアセトニトリルの比率(%B)を高めることによるCap−HPLCの勾配をプログラムした時間割は以下のとおりである。
【0060】
【表1】
【0061】
ペプチド1、ペプチド2及びペプチド3のカラムを平衡化するのにそれぞれ、80%のAと20%のB、91.5%のAと8.5%のB,及び95%のAと5%のBの比率を用いた。214nm、254nm及び280nmおける吸収についてアジレント(Agilent)ケムステーション(ChemStation)ソフトウエア(カリフォルニア州、パロアルト、アジレントテクノロジーズ)を用いてデータを回収し、ソフトウエアによってピークサイズを計算した。反応に存在する種の同一性を検証するために、マトリクス支援レーザー脱離/イオン化飛行時間型質量分析計(MALDI−TOF−MS)においても反応を調べた。α−シアノ−4−ヒドロキシ桂皮酸をマトリクスとして、加速電圧20kV及び陽イオンモードにて約150η秒の遅延時間で、ABIボイジャー(Voyager)DE質量分析計を用いた。図14は、TAPの量が増えるにつれて、量が減るホスホペプチド及び量が増える脱リン酸化されたペプチドを示すデータをまとめた3つのグラフから成る。このデータは、TAPがホスホ−チロシンペプチドに対して最も活性が高く、10分間で5μgのペプチドからホスフェートをすべて外すのに必要とした酵素はたった0.02単位であることを示している。ホスホ−セリン及びホスホ−スレオニンペプチドからホスフェートを完全に外すにはそれぞれ、2.5単位及び10単位のTAPを必要とした。このデータは、ペプチドからリン酸基を外すのにTAPを使用できることを示している。
【0062】
実施例VIII:TAPは、糖におけるホスフェートの位置にかかわりなく糖リン酸を脱リン酸化することができる
TAPが位置にかかわりなく、糖リン酸からリン酸残基を切断できるかどうかを調べるために、3種の糖リン酸、グルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6−リン酸に対するその活性を測定した。各糖リン酸について反応混合物の段階希釈物を調製したが、各反応混合物は、5mg/mLの1種の糖リン酸の溶液100μL、10xTAP緩衝液(500mMのビストリス−プロパン、10mMのMgCl2、1mMのZnCl2、pH6.0)を30μL及びH2Oを210μL含有した。50μLの反応混合物を最初の試験管に入れ、それに続く5本の試験管に25μL入れた。3種の調べるべき糖リン酸それぞれについてこれを繰り返した。これらの試験管を氷上に置いて冷却した。50μLの反応混合物を含有する最初の試験管に5μLの冷却TAP(5U/μL)をいったん加えた。混合した後、この試験管から25μLを取り出し、25μLの反応混合物を含有する次の試験管に入れた。混合した後、この第2の試験管から25μLを取り出し、次の試験管に入れた。この1:1の段階希釈を6本の試験管すべてについて行った。各糖リン酸についてTAPの段階希釈を行った後、試験管を氷から取り出し、37℃に15分間置いた。インキュベートした後、シリカゲルガラス上の薄層クロマトグラフィ(TLC)プレート上の密着したバンドに各試料3μLでスポットを作った。酵素なしでインキュベートした混合物のみを含有する陰性対照及び1mg/mLのグルコース又はマンノースを含有する陽性対照でもTLC上にスポットを作った。ホットエアガン(70℃を超えない温度)でスポットを完全に乾燥した。イソプロパノール、エタノール及び水(2.5:1:0.5、v:v:v)の中で溶媒の先頭が9cm移動するまでプレートを展開した。クロマトグラフィに続いて、プレートを乾燥させ、10%の過塩素酸を噴霧した。ヒートガンでプレートを焦がすことにより糖を視覚化した。366nmでのUVのもとで生成物を視覚化することにより感度を高めた。42μgのグルコース−1−リン酸、グルコース−6−リン酸及びマンノース−6−リン酸の完全な消化は、6UのTAPを用いて37℃にて15分で達成された(図15)。
【0063】
糖におけるホスフェートの連結(すなわち、1又は6)は、酵素の活性に明瞭な影響を有さなかった。さらに、マンノース糖リン酸におけるTAPの活性は、グルコース糖リン酸におけるものと同様だった。
【特許請求の範囲】
【請求項1】
切り詰め部が配列番号10により表されるアミノ酸配列からなるタンパク質のシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端がヒスチジンタグと共有結合している、配列番号10により表されるアミノ酸配列を含むタンパク質から誘導された、切り詰め型の、酵素活性のある熱不安定性のアンタークティックホスファターゼ(TAP)。
【請求項2】
65℃にて30分未満でTAPが不活化される請求項1に記載のホスファターゼ。
【請求項3】
pH6より高いpHのトリス塩酸でTAP活性が安定である請求項1に記載のホスファターゼ。
【請求項4】
pH7.4のトリス塩酸緩衝液でTAP活性が安定である請求項1に記載のホスファターゼ。
【請求項5】
ヒスチジンタグをコードする配列がTAPのDNA配列の5’末端に融合される請求項1に記載のホスファターゼをコードするDNA。
【請求項6】
請求項5に記載のDNAを含むベクター。
【請求項7】
請求項6に記載のベクターで形質転換される宿主細胞。
【請求項8】
切り詰め部が配列番号10により表されるアミノ酸配列からなるタンパク質のシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端が親水性のリーダー配列と共有結合している、配列番号10により表されるアミノ酸配列を含むタンパク質から誘導された、切り詰め型の、酵素活性のある熱不安定性のアンタークティックホスファターゼ(TAP)を含み、
亜鉛イオンをさらに含む、調製物。
【請求項9】
親水性のリーダー配列がオリゴペプチドである請求項8に記載の調製物。
【請求項10】
オリゴペプチドが正に荷電したオリゴペプチドである請求項9に記載の調製物。
【請求項11】
正に荷電したオリゴペプチドがヒスチジンタグである請求項10に記載の調製物。
【請求項12】
65℃にて30分未満でTAPが不活化される請求項8に記載の調製物。
【請求項13】
pH6より高いpHのトリス塩酸でTAP活性が安定である請求項8に記載の調製物。
【請求項14】
pH7.4のトリス塩酸緩衝液でTAP活性が安定である請求項8に記載の調製物。
【請求項1】
切り詰め部が配列番号10により表されるアミノ酸配列からなるタンパク質のシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端がヒスチジンタグと共有結合している、配列番号10により表されるアミノ酸配列を含むタンパク質から誘導された、切り詰め型の、酵素活性のある熱不安定性のアンタークティックホスファターゼ(TAP)。
【請求項2】
65℃にて30分未満でTAPが不活化される請求項1に記載のホスファターゼ。
【請求項3】
pH6より高いpHのトリス塩酸でTAP活性が安定である請求項1に記載のホスファターゼ。
【請求項4】
pH7.4のトリス塩酸緩衝液でTAP活性が安定である請求項1に記載のホスファターゼ。
【請求項5】
ヒスチジンタグをコードする配列がTAPのDNA配列の5’末端に融合される請求項1に記載のホスファターゼをコードするDNA。
【請求項6】
請求項5に記載のDNAを含むベクター。
【請求項7】
請求項6に記載のベクターで形質転換される宿主細胞。
【請求項8】
切り詰め部が配列番号10により表されるアミノ酸配列からなるタンパク質のシグナル配列の欠失に相当し、ホスファターゼがC末端及びN末端先端を有し、該N末端先端が親水性のリーダー配列と共有結合している、配列番号10により表されるアミノ酸配列を含むタンパク質から誘導された、切り詰め型の、酵素活性のある熱不安定性のアンタークティックホスファターゼ(TAP)を含み、
亜鉛イオンをさらに含む、調製物。
【請求項9】
親水性のリーダー配列がオリゴペプチドである請求項8に記載の調製物。
【請求項10】
オリゴペプチドが正に荷電したオリゴペプチドである請求項9に記載の調製物。
【請求項11】
正に荷電したオリゴペプチドがヒスチジンタグである請求項10に記載の調製物。
【請求項12】
65℃にて30分未満でTAPが不活化される請求項8に記載の調製物。
【請求項13】
pH6より高いpHのトリス塩酸でTAP活性が安定である請求項8に記載の調製物。
【請求項14】
pH7.4のトリス塩酸緩衝液でTAP活性が安定である請求項8に記載の調製物。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】
【図15】

【図16】

【図17】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2011−167210(P2011−167210A)
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2011−127750(P2011−127750)
【出願日】平成23年6月7日(2011.6.7)
【分割の表示】特願2006−503728(P2006−503728)の分割
【原出願日】平成16年2月20日(2004.2.20)
【出願人】(501425360)ニュー・イングランド・バイオラブズ・インコーポレイティッド (6)
【氏名又は名称原語表記】New England Biolabs, Inc.
【Fターム(参考)】
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成23年6月7日(2011.6.7)
【分割の表示】特願2006−503728(P2006−503728)の分割
【原出願日】平成16年2月20日(2004.2.20)
【出願人】(501425360)ニュー・イングランド・バイオラブズ・インコーポレイティッド (6)
【氏名又は名称原語表記】New England Biolabs, Inc.
【Fターム(参考)】
[ Back to top ]