
サイクリックGMP結合性、サイクリックGMP特異的ホスホジエステラーゼの材料および方法
【課題】 ヒトに由来する新規のサイクリックGMP結合性、サイクリックGMP特異的ホスホジエステラーゼ(cGB-PDE)ポリペプチドおよびその酵素活性を調節する化合物を同定するための方法を提供する。
【解決手段】 被験候補化合物の存在下または不在下で、cGB-PDEポリペプチドのホスホジエステラーゼ活性を分析し、および被験候補化合物の不在下でのホスホジエステラーゼ活性と比較して、その存在下でのホスホジエステラーゼ活性に変化を及ぼす化合物を調節化合物として同定する。
【解決手段】 被験候補化合物の存在下または不在下で、cGB-PDEポリペプチドのホスホジエステラーゼ活性を分析し、および被験候補化合物の不在下でのホスホジエステラーゼ活性と比較して、その存在下でのホスホジエステラーゼ活性に変化を及ぼす化合物を調節化合物として同定する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は一般に、cGB-PDEと名付けたサイクリックグアノシンモノホスフェート結合性、サイクリックグアノシンモノホスフェート特異的ホスホジエステラーゼに関し、さらに詳細にはcGB-PDEポリペプチドをコードする新規な精製および単離されたポリヌクレオチド、cGB-PDEポリヌクレオチドの組換え製造のための方法および材料、ならびにcGB-PDE活性のモジュレーターを同定するための方法に関する。
【背景技術】
【0002】
サイクリックグアノシンモノホスフェート(cGMP)およびサイクリックアデノシンモノホスフェート(cAMP)などの3'5'サイクリックヌクレオチドの対応するヌクレオシド5'モノホスフェートへの加水分解を触媒するサイクリックヌクレオチドホスホジエステラーゼ(PDE)は、酵素の複合ファミリーを構成している。 サイクリックヌクレオチドの細胞内濃度を媒介することによって、サイクリックヌクレオチドセカンドメッセンジャーを含むシグナルトランスダクションにおいてPDEアイソザイムは機能する。
【0003】
異なる組織供給源から種々のPDEが単離され、今日までに特徴付けされたPDEの多くは、物理化学的性質、基質特異性、阻害剤に対する感度、免疫学的反応性および調節の態様を含む生物学的性質において差異を呈する。 (ビーボ(Beavo)ら、サイクリック・ヌクレオチド・ホスホジエステラーゼズ:ストラクチャー、レギュレーション・アンド・ドラッグ・アクション(Cyclic Nucleotide Phosphodiesterases: Structure, Regulation and Drug Action)、ジョーン・ウィレイ・アンド・サンズ(John Wiley & Sons)、チチェスター(Chichester)、英国)を参照されたい)種々のPDEの既知のアミノ酸配列の比較により、ほとんどのPDEが別の触媒および調節ドメインを有するキメラ性マルチドメインタンパク質であることが示される。 (シャーボンニュー(Charbonneau)、ビーボら、前出、267〜296頁を参照されたい)今日までに特徴付けがなされたすべての哺乳動物のPDEは、触媒部位を含んでなると考えられ、酵素のカルボキシル末端領域に位置する長さおよそ250アミノ酸残基の配列を共有している。 アロステリック分子または調節分子と相互作用するPDEドメインは、アイソザイムのアミノ末端領域に位置すると思われる。 それらの生物学的性質に基づき、PDEは以下の6つの概括的なファミリーに分類され得る:Ca2+/カルモジュリンで刺激されるPDE(I型)、cGMPで刺激されるPDE(II型)、cGMPで阻害されるPDE(III型)、cAMP特異的PDE(IV型)、本発明の主題であるcGMP特異的ホスホジエステラーゼcGB-PDE(V型)およびcGMP特異的フォトレセプターPDE(VI型)。 cGMP結合性PDE(II型、V型およびVI型PDE)は、それらのカルボキシル末端近傍に相同な触媒ドメインを有するに加えて、それらのアミノ末端により近く位置し、アロステリックなcGMP結合ドメインを含み得る第2の保存された配列を有する。 シャーボンニューら、プロシーディング・オブ・ナショナル・アカデミー・オブ・サイエンス・ユー・エス・エー(Proc. Natl. Acad. Sci. USA)、87巻、288〜292頁(1990年)を参照されたい。
【0004】
II型のcGMPで刺激されるPDE(cGs-PDE)は、異なる組織のタイプに広く分布し、100〜105kDaのサブユニットのホモダイマーとして存在すると考えられる。 cGs-PDEは生理的条件下においてcAMP加水分解の速度を高めることによって上昇したcGMP濃度に応答する。 ウシ心臓cGs-PDEのアミノ酸配列およびウシ副腎cGs-PDEの部分cDNA配列はレトロング(LeTrong)ら、Biochemistry、29巻、10280〜10288頁(1990)に報告され、さらにウシ副腎およびヒト胎児脳cGB-PDEの全長のcDNA配列が1992年10月29日公開のPCT国際公開第 WO92/18541号に記載されている。 ウシ副腎の全長のcDNA配列はソネンバーグ(Sonnenburg)ら、J. Biol. Chem.、266巻、17655〜17661頁(1991)にも記載されている。
【0005】
フォトレセプターPDEおよびcGB-PDEはcAMPよりもcGMPに対して、加水分解について50倍またはそれより多くの選択性を呈するので、cGMP特異的なPDEとして記載されている。
【0006】
フォトレセプターPDEは、桿状体外部セグメントPDE(ROS-PDE)および円錐体(cone)PDEである。 ROS-PDEのホロ酵素構造は、いずれも触媒的に活性を有する2つの大きなサブユニットα(88kDa)およびβ(84kDa)ならびに2つのもっと小さなγ調節サブユニット(いずれも11kDa)からなる。 α、β、およびγサブユニットならびにCOS-PDEの15kDaサブユニットと同じであるらしいδサブユニット(15kDa)を含むROS-PDEの可溶性型もまた同定されている。 ウシの膜会合性ROS-PDE α−サブユニットに対応する全長のcDNAは、オブチニコフ(Ovchinnikov)ら、FEBS Lett.、223巻169〜173頁(1987)に記載されており、ウシ桿状体外部セグメントPDE β−サブユニットに対応する全長のcDNAは、リプキン(Lipkin)ら、J. Biol. Chem.、265巻、12955〜12959頁(1990)に記載されている。 オブチニコフら、FEBS Latt.、204巻、169〜173頁(1986)は、ウシROS-PDE γ−サブユニットに対応する全長のcDNAおよびそのδサブユニットのアミノ酸配列を示している。 ROS-PDEの発現が、脳においてもコリンズ(Collins)ら、ゲノミックス(Genomics)、13巻、698〜704頁(1992)にて報告されている。 COS-PDEは、2つの同一のα'(94kDA)サブユニットならびに11kDa、13kDaおよび15kDaの3つのもっと小さなサブユニットで構成される。 ウシCOS-PDE α'-サブユニットに対応する全長のcDNAは、リー(Li)ら、Proc. Natl. Acad. Sci. USA、87巻、293〜297頁(1990)に報告されている。
【0007】
cGB-PDEは、ラット(フランシス(Francis)ら、メソッズ・イン・エンザイモロジー(Methods Enzymol.)、159巻、722〜729頁(1988))およびウシ肺組織(トーマス(Thomas)ら、J. Biol. Chem.、265巻14964〜14970頁(1990)、以下「トーマスI」と記載)から均質に精製されている。 この酵素または類似の酵素の存在が、ラットならびにヒトの血小板(ハメット(Hamet)ら、Adv. Cyclic Nucleotide Protein Phosphorylation Res.、16巻、119〜136頁(1984))、ラット脾臓(コンクイル(Conquil)ら、Biochem. Biophys.Res.Commun.、127巻、226〜231頁(1985))、モルモット肺(デイビス(Davis)ら、J. Biol. Chem.、252巻4078〜4084頁(1977))、管平滑筋(コンクイルら、Biocehm.Biophys.Acta、61巻、148〜165頁(1980))、およびウニ精子(フランシスら、J.Biol.Chem.、255巻、620〜626頁(1979))を含めた種々の組織および種において報告されている。 cGB-PDEは2つの93kDaサブユニットからなるホモダイマーかもしれない。 (トーマスI、前出、参照)cGB-PDEは、cGMP依存性プロテインキナーゼ(cGK)によりリン酸化され、より低い親和性でcAMP依存性プロテインキナーゼによりリン酸化される、他の既知cGMP結合性PDEにおいては見出されない単一の部位を含むことが示されている。 (トーマスら、J. Biol. Chem.、265巻、14971〜14978頁(1990)、以下「トーマスII」と記載、参照)当該リン酸化部位およびcGB-PDEのキモトリプシン消化により作製された断片のアミノ末端部の一次アミノ酸配列は、トーマスII、前出およびトーマスI、前出にそれぞれ記載されている。 しかしながら、cGB-PDEのアミノ酸配列の大半はこれまでに報告されていない。
【0008】
異なる型のPDEの阻害剤が、文献に記載されている。 V型PDEにいくらか特異性を呈する2つの阻害剤は、ザプリナスト(zaprinast)およびジピリダモル(dipyridamole)である。 ビーボら、前出中、117〜140頁、フランシスら、を参照のこと。
【0009】
cGB-PDEをコードするDNAおよびアミノ酸配列の解明ならびに組換え法によるcGB-PDEポリペプチドの製造により、cGB-PDEの活性を選択的に修飾する新規物質の同定を可能ならしめる情報および材料が提供されるであろう。 PDEアイソザイムの別々の型またはファミリーがあること、および異なる組織が異なるPDEの異なる補体を発現することが認められることが、セカンドメッセンジャーとしてサイクリックヌクレオチドを用いるシグナルトランスダクション経路に関連する病状に対する治療上の示唆を有し得るPDEモジュレーターの開発における興味をもたらしている。 PDE活性の種々の選択的および非選択的阻害剤が、ムレイ(Murray)ら、Biochem. Soc. Trans.、202(2)巻、460〜464頁(1992)において議論されている。 組換えDNA技術による、特異的なPDE産生能をもたないPDEモジュレーターの開発は、あらゆるPDEが同じ基本的な反応を触媒し、重複する基質特異性を有し、且つ微量のみしか生じないために困難である。 結果的に、多くのPDEを均質にまで精製するのは冗長で困難な過程である。
【発明の開示】
【発明が解決しようとする課題】
【0010】
かくしてcGB-PDEに対するDNAおよびアミノ酸配列の情報、cGB-PDEポリペプチドの組換え製造のための方法および材料ならびにcGB-PDE活性の特異的なモジュレーターを同定するための方法の必要性が、当該技術において現存し続けている。
【課題を解決するための手段および発明の効果】
【0011】
本発明は、cGB-PDEと称されるcGMP結合性、cGMP特異的PDEをコードする新規な精製され単離されたポリヌクレオチド(例えば、センスおよびアンチセンスストランドのいずれものDNA配列およびRNA転写産物、それらのスプライス変異体も含めて)を提供する。 本発明の好ましいDNA配列は、ゲノムおよびcDNA配列のみならず全体的または部分的に化学的に合成されたDNA配列を含む。 配列番号:9または20で示されるcGB-PDEをコードするDNA配列およびストリンジェント条件下で当該配列とハイブリダイズするDNA配列または遺伝コードの縮重なしに当該配列とハイブリダイズするであろうDNA配列が、本発明により企図される。 本発明により、本発明のDNA配列の生物学的複製物(例えば、生体内または生体外で作製された、単離されたDNA配列)もまた企図される。 cGB-PDE配列を組込んだプラスミドおよびウイルスDNAベクター、ならびに、特にcGB-PDEをコードするDNAが内在または外来の発現制御DNA配列および転写ターミネーターなどに作動可能に連結されたベクターなどの、自律的に複製する組換え構築体もまた提供される。 特に例証となる本発明の発現プラスミドは、アメリカン・タイプ・カルチャー・コレクション(ATCC)、12301, Parklawn Drive Rockville, Maryland 20852に、1993年5月4日に受託番号69296として寄託された大腸菌株JM109中のプラスミドhcgbmet156-2 6nである。
【0012】
本発明の他の特徴において、原核細胞および真核細胞を含む宿主細胞が、その細胞内で所望のペプチドが発現されることを許容する方法で本発明のDNA配列を用いて形質転換される。 cGB-PDE産物を発現している宿主細胞は、種々の有用な目的で役立つことができる。 このような細胞は、cGB-PDEと特異的に免疫反応性を有する抗体物質を作製するための免疫源用の貴重な供給源を構成する。 本発明の宿主細胞は、cGB-PDEポリペプチドの大量産生のための方法において顕著に有用である。 この方法において、細胞が好適な培養培地中で生育され、当該細胞からまたは当該細胞が生育された培地から、たとえばイムノアフィニティー精製により所望のポリペプチド産物が単離される。
【0013】
cGB-PDE産物は、天然細胞供給源から単離物として得られてもよく、または化学的に合成されてもよいが、好ましくは本発明の宿主細胞を包含する組換え手法により製造される。
【0014】
哺乳動物宿主細胞を用いることで、本発明の組換え発現産物に至適な生物学的活性を付与するために必要とされ得るような転写後の修飾(例えば、グリコシレーション、トランケーション、リピデーションおよびチロシン、セリンまたはスレオニンのリン酸化)を提供することが期待される。 本発明のcGB-PDE産物は、全長のポリペプチド、断片または変異体であってよい。 変異体は、(1)cGB-PDEに特異的な生物学的活性または免疫学的性質の1以上を喪失することなく;または(2)cGB-PDEの特定の生物学的活性を特に損なわせた:1以上の特定の(すなわち、天然にコードされた)アミノ酸が欠失されたもしくは置換された、または1以上の特定されないアミノ酸が付加された、cGB-PDEポリペプチド類似体からなり得る。
【0015】
本発明にさらに包含されるのは、抗体物質(例えば、モノクローナルおよびポリクローナル抗体、単鎖抗体、キメラ抗体、CDR-移植(grafted)抗体など)ならびにcGB-PDEに特異的な他の結合タンパク質である。 特異的な結合タンパク質は、単離されたもしくは組換えcGB-PDEまたはcGB-PDE変異体もしくはこのような産物を発現している細胞を用いて作製することができる。 結合タンパク質はさらに、免疫のためのみならず、cGB-PDEポリペプチドの精製ならびに既知の免疫学的手法による体液および組織試料におけるcGB-PDEポリペプチドの検出または定量のための組成物においても有用である。 それらはまた、cGB-PDEの生化学的活性、特にシグナルトランスダクションに関連するそれらの活性の修飾(すなわち、ブロッキング、阻害または促進)において極めて有用である。 抗cGB-PDE抗体物質に対して特異的な抗イデオタイプ抗体も企図される。
【0016】
本発明のDNAおよびアミノ酸配列の開示を通して与えられる情報の科学的な価値が明示される。 一連の例として、cGB-PDEに対するcDNAの配列を知ることで、DNA/DNAハイブリダイゼーションによって、cGB-PDEをコードするゲノムDNA配列を単離すること、ならびにプロモーター、オペレーターなどのcGB-PDE発現制御調節配列を特定することが可能となる。 ストリンジェント条件下で本発明のDNA配列を用いて行われるDNA/DNAハイブリダイゼーションにより、cGB-PDEの対立変異体、cGB-PDEに特異的な生化学的および/または免疫学的特性を共有する、他の構造的に関連するタンパク質、ならびにcGB-PDEと相同な非ヒト種のタンパク質をコードするDNAの単離を可能とすることが期待される。 本発明のポリヌクレオチドは、好適に標識された場合、cGB-PDEを合成する細胞の能力を検出するためのハイブリダイゼーションアッセイにおいて有用である。 本発明のポリヌクレオチドはまた、1または複数の疾病状態の根底にあるcGB-PDE遺伝子座における遺伝的変化を同定するために有用な診断法の基礎となるかもしれない。 本発明により利用可能となるのはまた、通常はcGB-PDEを発現している細胞による、cGB-PDEの発現調節に関連のあるアンチセンスポリヌクレオチドである。
【0017】
本発明によって提供されるDNAおよびアミノ酸配列情報はまた、cGB-PDEの構造および機能のシステム分析ならびにcGB-PDEが相互作用するであろう分子の定義付けをも可能とする。 cGB-PDE活性を修飾する薬剤は、推定モジュレーターを組換えcGB-PDEを発現する真核細胞からの溶解物とインキュベートし、次いでcGB-PDEホスホジエステラーゼ活性に対する推定モジュレーターの効果を決定することによって同定され得る。 好ましい実施態様において、真核細胞は内在性のサイクリックヌクレオチドホスホジエステラーゼ活性を欠くものである。 特に例証となるこのような真核細胞は、受託番号74225として1993年5月19日にATCCに寄託された酵母株YKS45である。 cGB-PDEの活性を修飾する化合物の選択性は、cGB-PDEに対する当該化合物の活性を他のPDEアイソザイムに対する活性と比較することにより評価することができる。 一連の独立した分析における、本発明の組換えcGB-PDE産物と他の組換えPDE産物との組合せによって、cGB-PDEの選択的なモジュレーターを開発するためのシステムが提供される。
【0018】
選択的モジュレーターには、たとえば、cGB-PDEまたはcGB-PDE核酸に選択的に結合する抗体および他のタンパク質またはペプチド、cGB-PDEまたはcGB-PDE核酸に選択的に結合するオリゴヌクレオチドならびにcGB-PDEまたはcGB-PDE核酸に選択的に結合する他の非ペプチド化合物(たとえば、単離された、または合成の有機分子)が含まれ得る。 野生型cGB-PDEの酵素活性または細胞局在に影響を及ぼすcGB-PDEの変異型もまた、本発明により企図される。 選択的なモジュレーターの開発のための好ましい標的には、たとえば、(1)他のタンパク質と接触し、および/または細胞内にてcGB-PDEが局在するcGB-PDEの領域、(2)基質が結合するcGB-PDEの領域、(3)cGB-PDEの1または複数のアロステリックなcGMP結合部位、(4)cGB-PDEの1または複数のリン酸化部位並びに(5)cGB-PDEサブユニットのダイマー形成に関与するcGB-PDEの領域が含まれる。 cGB-PDE活性のモジュレーターは、広範囲の疾病および生理学的条件の処置において治療上有用であるかもしれない。
【発明を実施するための最良の形態】
【0019】
本発明を、以下の実施例に従って詳細に説明する。
【0020】
実施例1では、PCRによるウシcGB-PDE cDNA断片の単離および引続いてそのPCR断片をプローブとして用いた全長のcGB-PDEcDNAの単離を記載する。 実施例2は、種々の他のPDEに対して報告された配列と、ウシcGB-PDEアミノ酸配列の関係の分析を示す。 種々のウシ組織中のcGB-PDE mRNAのノザンブロット分析を、実施例3に示す。 COS細胞におけるウシcGB-PDE cDNAの発現を、実施例4に記載する。 実施例5には、cGB-PDE COS細胞発現産物のホスホジエステラーゼ活性、cGMP結合活性およびZn2+加水分解酵素についての分析の結果を示す。 実施例6は、ウシcGB-PDE cDNAと相同性を有するヒトcDNAの単離を記載する。 酵母細胞内でのヒトcGB-PDE cDNAの発現を、実施例7に示す。 ヒト組織におけるcGB-PDEを検出するためのRNaseプロテクションアッセイを、実施例8に記載する。 実施例9には、ヒトcGB-PDE cDNAの細菌における発現および当該細菌のcGB-PDE発現産物と反応性を有する抗体の作製を記載する。 実施例10には、cGB-PDE類似体と断片を記載する。 cGB-PDEを認識するモノクローナル抗体の作製を、実施例11に記載する。 実施例12は、cGB-PDEの生物学的活性を選択的に修飾する薬剤を開発するための本発明の組換えcGB-PDE産物の利用に関する。
【実施例】
【0021】
実施例1
ポリメラーゼ連鎖反応(PCR)を、ウシ肺第1ストランドから、cGB-PDEの一部をコードするcDNA断片を単離するために利用した。 トーマスI、前出に記載の部分的なcGB-PDEアミノ酸配列および新規な部分アミノ酸配列情報に基づいて、充分に縮重したセンスおよびアンチセンスPCRプライマーを設計した。
【0022】
A.cGB-PDEタンパク質の精製
cGB-PDEは、トーマスI、前出に記載のように、またはその方法を下記のように変更することによって精製した。
【0023】
新鮮なウシ肺(5〜10kg)を屠殺場から得、直ちに氷上に置いた。 組織を細切し、冷PEM緩衝液(2mM EDTAおよび25mM β−メルカプトエタノールを含む20mMリン酸ナトリウム、pH6.8)と合わせた。 ホモジナイズして遠心した後、得られた上清を4〜7リットルのDEAE-セルロース(ワットマン(Whatman)、英国)と3〜4時間インキュベートした。次いでそのDEAEスラリーを真空下で濾過し、冷PEMを大量に用いて濯いだ。 その樹脂をガラスカラムに注ぎ、3〜4倍容量のPEMを用いて洗浄した。タンパク質は、100mM NaClを含むPEMを用いて溶出し、12の1リットル画分を集めた。 画分を、トーマスら、前出に記載された標準法によってIBMX-刺激cGMP結合およびcGMPホスホジエステラーゼ活性について分析した。 適切な画分をプールし、冷脱イオン水を用いて2倍に希釈し、次いでブルーセファロース(登録商標)CL-6B(ファルマシア・エル・ケー・ビー・バイオテクノロジー(Phermacia LKB Biotechnology)社、ピスカタウェイ(Piscataway)、NJ)クロマトグラフィーに付した。 次にアガロースまたはセファロースベースのゲルマトリックスを用いて、亜鉛キレートアフィニティー吸収クロマトグラフィーを行った。 亜鉛キレート工程から得られたタンパク質のプールを、トーマスI、前出に記載のように処理するか、または変更を施した精製法に付した。
【0024】
トーマスI、前出に記載のようにタンパク質のプールは、4℃でPEM中に平衡化したHPLC Bio-Sil TSK-545 DEAEカラム(150×21.5mm)(バイオラッド・ラボラトリーズ(BioRad Laboratories)、ハーカルズ(Hercules)、CA)を複数回付した。 平衡化の後、50mM NaCLを含むPEM 120-mlで洗浄し、続いて2ml/分の流速にて120-mlの直線濃度勾配(50〜200mM NaClを含むPEM)での溶出を行った。 適切な画分をプールし、透析チューブ中でセファデックスG-200(ベーリンガー・マンハイム・バイオケミカルズ(Boehringer Mannheim Biochemicals)、英国)に対して、1.5mlの最終容量までに濃縮した。 濃縮されたcGB-PDEプールは、100mMリン酸ナトリウム、pH 6.8、2mM EDTA、25mM β−メルカプトエタノール中に平衡化したHPLCゲル濾過カラム(Bio-Sil TSK-250、500×21.5mm)に供し、4℃で2ml/分の流速にて溶出した。
【0025】
より煩わしくない変法を行う場合は、タンパク質プールをPEMに対して2時間透析し、PEM緩衝液中に平衡化した10mlの調製用DEAEセファセルカラム(ファルマシア)にかけた。タンパク質を0.5M NaClを含むPEMを用いてバッチ法にて溶出し、およそ10〜15倍濃度のタンパク質を得た。 濃縮されたタンパク質試料を、0.1M NaClを含むPEM中に平衡化した800ml(2.5cm×154cm)のセファクリルS400ゲル濾過カラム(ベーリンガー)にかけ、1.7ml/分の流速で溶出した。
【0026】
タンパク質の純度は、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)の後、クマシー染色により評価した。 ウシ肺10kg当たりおよそ0.5〜3.0mgの純粋なcGB-PDEが得られた。
【0027】
精製されたウシcGB-PDEに特異的なウサギポリクローナル抗体を、標準法によって作製した。
【0028】
B.cGB-PDEのアミノ酸配列決定
cGB-PDEは、[32P]ATPを用いてリン酸化し、次いで32Pで標識されたホスホペプチドを得るため、プロテアーゼで消化した。 およそ100μgの精製されたcGB-PDEを、9mM MgCl2、9μM[32P]ATP、10μM cGMP、および4.2μgのcAMP依存性プロテインキナーゼ(cAK)の、精製されたウシの触媒能を有するサブユニットを含む反応混液(900μlの最終容量)中でリン酸化した。 cAKの触媒能を有するサブユニットは、マランゴス(Marangos)ら、Brain Receptor Methodologies, Part A、Academic Press、Orlando、フロリダ(1984)、209〜215頁、フロックハート(Flockhart)らの方法に従って調製した。 反応は30℃にて30分インキュベートを行い、60μlの200mM EDTAを添加することによって停止した。
【0029】
cGB-PDEから第1のペプチド配列を得るために、KPE緩衝液中のα−キモトリプシン1mg/ml溶液(2mM EDTAを含む10mMリン酸ナトリウム、pH 6.8)3.7μlを、100μgの精製されリン酸化されたcGB-PDEに添加し、混液を30℃にて30分インキュベートした。 タンパク質分解は、50μlの10% SDSおよび25μlのβ−メルカプトエタノールを添加することで停止した。 容量が400μlより少なく減じられるまで試料を沸騰させ、8%の調製用SDS-ポリアクリルアミドゲルにかけ、50mAmpにて電気泳動に付した。 分離された消化産物は、マツダイラ(Matsudaira)、J. Biol. Chem.、262巻、10035〜10038頁(1987)の方法に従ってイモビロン・ポリビニリデン・ジフルオリド(ミリポア、ベドフォード、MA)上に電気的にブロットした。 転写したタンパク質は、クマシーブルー染色によって同定し、自動ガス相アミノ酸配列決定用に、50kDaのバンドを膜から切り出した。 α−キモトリプシン消化の手順により得られたペプチドの配列は、以下の配列番号:1に示す。
【0030】
配列番号:1 REXDANRINYMYAQYVKNTM
第2の配列は、V8タンパク分解により作製されたcGB-PDEペプチド断片から得られた。
【0031】
およそ200μgの精製されたペプチド断片を、10mM MgCl2、10μM[32P]ATP、100μM cGMP、および1μg/mlの精製されたcAKの、最終容量1.4ml中に添加した。 反応は、30℃にて30分インキュベートを行い、160μlの0.2M EDTAを添加することによって停止した。次に、KPE中に希釈した9μlの1mg/mlのStaphylococcal aureus V8プロテアーゼ(インターナショナル・ケミカル・ヌクレアー・バイオメディカルズ(International Chemical Nuclear Biomedicals)、コスタ・メサ(Costa Mesa)、CA)を添加し、続いて30℃にて15分インキュベートした。 タンパク質分解は、88μlの10%SDSおよび45μlのβ−メルカプトエタノールを添加することにより停止した。 消化産物は、25mAmpで4.5時間行った調製用10% SDSポリアクリルアミドゲルの電気泳動により分離した。 タンパク質は電気的にブロットし、前記したように染色した。 28kDaのタンパク質のバンドを膜から切り出し、自動ガス相アミノ酸配列決定に付した。 得られた配列を、以下の配列番号:2に示す。
【0032】
配列番号:2 QSLAAAVVP
C.ウシcDNAのPCR増幅
プライマーを設計するために用いられる部分アミノ酸配列(配列番号:3、下記、配列番号:1のアミノ酸9〜20)ならびに対応するPCRプライマー(IUPAC命名法で)に対応する配列を以下に示す。 ここで配列番号:3は、トーマスI、前出において報告された配列である。
【0033】
配列番号:3 F D N D E G E Q
5’TTY GAY AAY GAY GAR GGN GAR CA 3’(配列番号:4)
3’AAR CTR TTR CTR CTY CCN CTY GT 5’(配列番号:5)
配列番号:1、アミノ酸9〜20
N Y M Y A Q Y V K N T M
5’AAY TAY ATG TAY GCN CAR TAY GT3’(配列番号:6)
3’TTR ATR TAC ATR CGN GTY ATR CA5’(配列番号:7)
3’TTR ATR TAC ATR CGN GTY ATR CAN TTY TTR TGN TAC5’(配列番号:8)
アプライド・バイオシステムズ(Applied Biosystems)モデル380Aシンセサイザー(フォスター・シティー(Foster City)、CA)を用いて合成されたセンスおよびアンチセンスプライマーを、下記のようにウシ肺第1ストランドcDNAからcGB-PDE特異的な配列を増幅するために可能なあらゆる組合せで用いた。
【0034】
エタノール沈殿の後、オリゴヌクレオチドの対を1つのPCR反応においてそれぞれ400nMで組合わせた(配列番号:4または5を配列番号:6、7または8と組合わせた)。 50ngのウシ肺第1ストランドのcDNA(オリゴdTで選択したウシ肺mRNAについてAMV逆転写酵素およびランダムプライマーを用いて作製した)、200μM dNTP、および2単位のTaqポリメラーゼを用いて反応を行った。 初めの変性工程は、94℃にて5分間行い、引き続き94℃における1分の変性工程、50℃における2分のアニーリング工程、72℃における2分の伸長(extension)工程を30サイクル行った。 PCRは、Hybaid Thermal Reactor (ENK Scientific Products, Saratoga, CA)を用いて実施し、産物を40mMトリス−酢酸、2mM EDTAで流した1%低融点アガロースゲルでのゲル電気泳動により分離した。 約800〜840bpの弱いバンドが、配列番号:4および7で示されるプライマーを用いた場合および配列番号:4および8で示されるプライマーを用いた場合に認められた。 配列番号:4および7で示されるプライマーを用いた増幅により作製されるPCR産物を、ジーン・クリーン(Gene Clean(登録商標))(Bio101、ラ・ヨラ(La jolla)、CA)DNA精製キットを用い、製造業者のプロトコルに従って単離した。 PCR産物(20ng)は、200ngの直鎖状としたpBluescript KS(+)(ストラタジーン、ラ・ヨラ、CA)へと連結し、そして得られたプラスミド構築体を、大腸菌XL1 Blue Cell(ストラタジーン・クローニング・システムズ、ラ・ヨラ、CA)を形質転換するために用いた。 形質転換陽性と推定されるものを、配列決定によりスクリーニングした。 得られた配列は、既知のいかなる配列ともまたは既知のcGB-PDE部分配列とも相同でなかった。
【0035】
配列番号:4および7で示されるプライマーを用いて、ウシ肺の第1ストランドcDNAについて再びPCRを行った。 単一の大きなオープンリーディングフレームを有する0.8Kbのインサートを含むクローンが同定された。 オープンリーディングフレームは、アミノ酸KNTM(配列番号:7で示されるプライマー配列を設計するのには用いられなかった配列番号:1のアミノ酸17〜200)を含み、かつcGs-、ROS-およびCOS-PDEの演繹されたアミノ酸配列と高い程度の相同性をもつポリペプチドをコードしていた。 同定されたクローンは、配列番号:9のヌクレオチド489〜1312に対応する。
【0036】
D.ウシcDNAライブラリーの構築およびハイブリダイゼーションスクリーニング
全長のcGB-PDEをコードするcDNAを得るために、ウシ肺cDNAライブラリーを、プローブとして32Pで標識されたPCRで作製されたcDNAインサートを用いてスクリーニングした。
【0037】
ポリアデニル化RNAは、ソネンバーグ(Sonnenburg)ら、J. Biol. Chem.、266巻、17655〜17661頁(1991)に記載のとおりにウシ肺から調製した。 第1ストランドcDNAは、アウスベル(Ausubel)ら、Current Protocols in Molecular Biology, John Wiley & Sons、ニューヨーク(1987)に記載のとおりにランダムヘキサヌクレオチドプライマーとAMV逆転写酵素(ライフ・サイエンシズ(Life Sciences)、St. Petersberg、FL)を用いて合成した。 第2ストランドcDNAは、大腸菌DNAリガーゼおよび大腸菌RNAse Hの存在下で大腸菌DNAポリメラーゼIを用いて合成した。 500bpより大きいcDNAの選択は、セファロース(登録商標)CL-4B(ミリポア)クロマトグラフィーによって行った。 EcoRIアダプター(プロメガ(Promega)、マディソン(Madison)、WI)を、T4 DNAリガーゼを用いて当該cDNAに連結した。 熱によるリガーゼの不活性化に続いて、T4ポリヌクレオチドキナーゼを用いてcDNAをリン酸化した。 連結されなかったアダプターは、セファロース(登録商標)CL-4Bクロマトグラフィー(ファルマシア(Pharmacia)、ピスカタウェイ(Piscataway)、NJ)によって除去した。 cDNAはEcoRIで消化し、脱リン酸化したラムダZap(登録商標)IIアーム(ストラタジーン)へと連結し、製造業者のプロトコルに従ってGigapack(登録商標)Gold(ストラタジーン)を用いてパッケージングした。 未増幅のライブラリーのタイターは、18%の非組換え体で、9.9×105であった。 ライブラリーは、20の150mmプレートに対して50,000プラーク形成単位(pfu)を播くことにより増幅し、最終的なタイターとして21%の非組換え体で5.95×106pfu/mlが得られた。
【0038】
ライブラリーを50,000 pfu/mlで24枚の150mmプレートに播き、32Pで標識したcDNAクローンを用いてスクリーニングした。 プローブはファインバーグ(Feinberg)ら、Anal.Biochem.、137巻、266〜267頁(1984)の方法を用いて調製し、32Pで標識されたDNAはElutip-D(登録商標)カラム(シュライヒャー・アンド・シュエル(Schleicher and Schuell)社、キーン(Keene)、NH)を用い、製造業者のプロトコルを用いて精製した。 プラーク−リフトは、15cmニトロセルロースフィルターを用いて行った。 変性および中和に続いて、80℃にて2時間ベーキングすることにより、DNAをフィルター上に固定した。 ハイブリダイゼーションは、50%ホルムアミド、5×SSC(0.75M NaCl、0.75M クエン酸ナトリウム、pH7)、25mM リン酸ナトリウム(pH7.0)、2×デンハーツ溶液、10%デキストラン硫酸、90μg/ml酵母tRNA、およびおよそ106cpm/mlの32Pで標識されたプローブ(5×108cpm/μg)を含む溶液中で、42℃にて一晩行った。 フィルターは、1回の洗浄について15分間、室温にて0.1×SSC、1% SDS中で2回洗浄し、続いて45℃にて0.1×SSC、1%SDS中で20分間1度、洗浄した。 次にフィルターを−70℃で、数日間X線フィルムに露光した。
【0039】
標識したプローブとハイブリダイズしたプラークを再度播種し、再度のスクリーニングを数回行うことによって精製した。 cDNAインサートは、製造業者のプロトコルにより記載されたインビボの切出し方法によってpBluescript SK(-)ベクター(ストラタジーン)の中にサブクローン化した。 救出されたcDNAがPCRプローブとハイブリダイズすることを明らかにするため、サザンブロットを行った。 推定されるcGB-PDE cDNAを、Sequanse(登録商標)バージョン2.0(ユナイテッド・ステイツ・バイオケミカル・コーポレーション(United States Biochemical Corporation)、クリーブランド、オハイオ)またはTaqTrack(登録商標)キット(プロメガ)を用いて配列決定した。
【0040】
cGB-2、cGB-8およびcGB-10と名付けた3つの異なるcDNAクローンを単離した。 クローンcGB-8のDNAおよび演繹されたアミノ酸配列を配列番号:9および10に示す。 ヌクレオチド2686の下流のDNA配列は、クローニングのアーティファクトを表すのかもしれない。 cGB-10のDNA配列は、1つのヌクレオチドを例外としてcGB-8の配列と同一である。クローンcGB-2のDNA配列は、クローンcGB-8の5'からクローンcgb-8(配列番号:9参照)のヌクレオチド219までの配列より分岐しており、異なるアミノ末端を有するタンパク質をコードし得た。
【0041】
cGB-8 cDNAクローンは長さ4474bpであり、2625bpの大きなオープンリーディングフレームを含む。 ヌクレオチド配列中、第99〜101位のトリプレットATGは、枠内終止コドンの前にあり、且つ周囲の塩基が真核細胞のmRNAに対するコザックのコンセンサス開始部位に適合するので、cGB-PDE遺伝子の翻訳開始部位であることが予測される。 終止コドンTAGは2724〜2726位に位置し、3'非翻訳配列の1748bpが続く。 cGB-8の配列は、転写終了コンセンサス配列を含まず、従ってクローンは、対応するmRNAの全体の3'非翻訳領域は示さないのかもしれない。
【0042】
cGB-8 cDNAのオープンリーディングフレームは、875アミノ酸のポリヌクレオチドをコードし、計算された分子量は99.5kDである。 この計算された分子量は、SDS-PAGE分析によると約93kDaであると見積もられた、精製されたcGB-PDEの分子量の報告値よりわずかに少しだけ大きい。 cGB-8の演繹されたアミノ酸配列は、精製されたウシ肺cGB-PDEから得られたすべてのペプチド配列に正確に対応しており、cGB-8がcGB-PDEをコードする強い証拠を提供するものである。
【0043】
実施例2
Genetics Computer Group(GCG)Software Package (Madison, Wisconsin)が提供しているFASTAプログラムを用いて行ったSWISS-PROTおよびGEnEmb1データバンク(1992年2月リリース)の検索により、他のPDEに対して報告されたDNAおよびアミノ酸配列のみがクローンcGB-8のDNAおよび演繹されたアミノ酸と有意な類似性を呈することが明らかになった。
【0044】
cGB-PDEの演繹されたアミノ酸配列を8つの他のPDEの配列とペアで比較することが、ALIGN(デイホフ(Dayhoff)ら、Methods Enzymol. 、92巻、524〜545頁(1983))およびBESTFIT(ウィルバー(Wilbur)ら、Proc.Natl.Acad.Sci.USA、80巻、726〜730頁(1983))プログラムを用いて行われた。 今日までに配列決定がなされたすべての哺乳動物ホスホジエステラーゼのように、cGB-PDEは、触媒活性に必須であると思われる、タンパク質のカルボキシル末端の半分のおよそ250アミノ酸の保存された触媒ドメイン配列を含んでいる。 このセグメントは、配列番号:9のアミノ酸578〜812を含んでなり、他のPDEの対応する領域と配列の保存性を呈する。 以下の表1に、他のPDEのcGB-PDEのアミノ酸578〜812とのペアでの比較において得られる特定の同一性値を示す。 ここで「ラットダンス(dunce)」はラットcAMP特異的PDEであり、「61kCaM」はウシ61kDaカルシウム/カルモジュリン依存性PDEであり、「63kCaM」はウシ63kDaカルシウム/カルモジュリン依存性PDEであり、「ドロスダンス」はドロソフィアcAMP特異的ダンスPDEであり、「ROS-α」はウシROS-PDE α−サブユニットであり、「ROS-β」はウシROS-PDE β−サブユニットであり、「COS-α」はウシCOS-PDE α’サブユニットであって、さらに「cGs」はウシcGs-PDEである(612〜844)。
【0045】
【表1】

【0046】
PILEUPプログラム(GCGソフトウェア)で実行される、Progressive Alignment Algorithm(フェン(Feng)ら、Methods Enzymol.、183巻、375〜387頁(1990))を用いて、複数の配列のアラインメントを行った。 図1A〜1Cに、cGB-PDEの提唱される触媒ドメインの、表1のPDEのすべての対応する領域との複数の配列アラインメントを示す。 28残基(図1Aから1Cの「保存された」線での1文字のアミノ酸略号により示される残基を参照されたい)が、触媒において機能的な役割を果たすと予測されるいくつかの保存されたヒスチジン残基を含めて、アイソザイム間において不変である。 シャーボンニューら、Proc. Natl. Acad USA、前出を参照されたい。 cGB-PDEの触媒ドメインは、他のPDEアイソザイムでの対応する領域よりROS-PDEおよびCOS-PDEの触媒ドメインに、より類似している。 フォトレセプターPDEとcGB-PDEとの間に、他のPDEが共有しないいくつかの保存領域がある。 フォトレセプターPDEとcGB-PDE配列において不変なこれらの領域におけるアミノ酸の位置は、図1A〜1Cの「保存された」線の星印により示される。 cGB-PDEならびにROS-およびCOS-PDEの間の相同性領域は、cAMP加水分解に比してcGMP加水分解に対する特異性を賦与すること、または特定の生理学的薬剤への感受性を賦与することにおいて重要な役割を果たすかもしれない。
【0047】
cGB-PDE、cGs-PDEおよびフォトレセプターPDE間の配列の類似性は、保存された触媒ドメインに限定されず、タンパク質のアミノ末端半分における非触媒cGMP結合ドメインにも含まれる。 cGB-PDE、cGs-PDEおよびフォトレセプターPDE間のアラインメントの至適化により、アミノ末端保存セグメントが配列番号:9のアミノ酸142〜526を含めて存在するかもしれないことが示される。 cGB-PDEの提唱されるcGMP結合ドメインと、フォトレセプターPDEおよびcGs-PDEの対応する領域との配列のペアでの分析によって、26〜28%の配列の同一性が明らかとなった。 当該提唱されるcGMP結合ドメインと、cGMP結合性PDEとの複数の配列とのアラインメントを図2A〜2Cに示し、ここで略号は表1に示したと同様である。 この非触媒ドメインにおける38の位置が、すべてのcGMP結合性PDEの間で不変であることがわかる(図2A〜2Cの「保存された」線での1文字のアミノ酸の略号により示される位置を参照されたい)。
【0048】
cGMP結合性PDEのcGMP結合ドメインは、2つの類似するが別異のサブユニット間またはサブユニット内のcGMP結合部位を形成するかもしれない、内的に相同性のあるリピートを含む。 図3に、cGMP結合PDEのリピートa(cGB-PDEのアミノ酸228〜311に対応する)およびb(cGB-PDEのアミノ酸410〜500に対応する)の複数の配列アラインメントを示す。7つの残基が、各AおよびB領域において不変である(図3の「保存された」線で1文字のアミノ酸の略号により示される残基を参照されたい)。 AおよびB領域において化学的に保存されている残基を、図3の「保存された」線で星印により示す。 cGB-PDEのcGMP類似体研究により、cGB-PDE上のサイクリックヌクレオチド結合部位とcGMPの2’OHとの間に水素結合が存在することが裏付けられる。
【0049】
cGB-PDEの3つの領域は、他のPDEアイソザイムと有意な配列の類似性を有していない。これらの領域には、触媒ドメインのカルボキシル末端のフランキング配列(アミノ酸812〜875)、cGMP結合ドメインと触媒ドメインを分かつ配列(アミノ酸527〜577)およびアミノ酸1〜141にわたるアミノ末端配列が含まれる。 cGKによるcGB-PDEのリン酸化の部位(配列番号:10の92位のセリン)は、この配列のアミノ末端領域に位置する。cGB-PDE上のアロステリック部位へのcGMPの結合が、そのリン酸化に必要とされる。
【0050】
前記した他のPDEアイソザイムとの比較に基づき提唱されるcGB-PDEのドメイン構造を図4に示す。 このドメイン構造は、ウシ肺から精製されたcGB-PDEの生化学的研究により裏付けられる。
【0051】
実施例3
種々のウシ組織におけるcGB-PDE mRNAの存在を、ノザンブロットハイブリダイゼーションにより実験した。
【0052】
ポリアデニル化RNAは、製造業者のプロトコルに従ってPoly(A)Quick(登録商標)mRNA精製キット(スタラタジーン(Stratagene)を用いて総RNA調製物から精製した。 RNA試料(5μg)を1.2%アガロース、6.7%ホルムアルデヒドゲル上にのせた。 電気泳動およびRNA転写は、ソネンバーグら、前出に以前報告されたとおりに実施した。 RNAブロットのプレハイブリダイゼーションは、50%ホルムアミド、5×SSC、25mMリン酸ナトリウム、pH7、2×デンハーツ溶液、10%デキストラン硫酸、および0.1mg/ml酵母tRNAを含む溶液中で45℃にて4時間行った。 ランダムヘキサヌクレオチド−プライマー−標識プローブ(5×108cpm/μg)は、AccIおよびSacIIを用いた消化により切出した実施例2の4.7kbのcGB-8 cDNAクローンを用いて、フェインバーグら、前出に記載のとおりに調製した。プローブを加熱変性し、プレハイブリダイゼーションに続いてブロッティングバッグの中に注入した(6×105cpm/ml)。 ノザンブロットは、45℃にて一晩ハイブリダイズし、続いて室温で2×SSC、0.1%SDSを用いて、15分間1回洗浄し、次いで45℃にて0.1×SSC、0.1%SDSを用いて20分間、3回洗浄した。 ブロットは−70℃で24時間、X線フィルムに露光した。 cGB-PDEプローブとハイブリダイズするRNAのサイズは、臭化エチジウムで染色し、UV光で目視化した、0.24〜9.5kbのRNAラダーを用いて評価した。
【0053】
32Pで標識されたcGB-PDE cDNAは、単一の6.8kbウシ肺RNA種とハイブリダイズした。 同一のサイズのmRNAが、ウシの気管、大動脈、腎臓および脾臓から単離されたポリアデニル化RNAにおいても検出された。
【0054】
実施例4
実施例2のクローンcGB-8におけるcGB-PDE cDNAをCOS-7細胞(ATCC CRL1651)内で発現させた。
【0055】
制限酵素XbaIを用いた消化に続いて、cGB-PDEの一部を単離した。 XbaIは、cGB-8インサートの5'末端の30bp上流に位置するpBluescriptポリリンカー配列における位置で、および当該cGB-8インサートの中の3359位で切断する。 この結果得られた3389bp断片(cGB-8の全体のコード領域を含む)を、次いで発現ベクターpCDM8(インビトロゲン(Invitrogen)、サンジエゴ、CA)の独特なXba8Iクローニング部位に連結した。 pCDM8プラスミドはサイトメガロウイルスプロモーターおよびエンハンサー、SV-40由来の複製の起点、ポリアデニル化シグナル、複製の原核性起点(pBR322由来)および原核性遺伝マーカー(supF)を含む4.5kbの真核性発現ベクターである。 大腸菌MC1061/C3細胞(インビトロゲン)を、得られた連結産物を用いて形質転換し、形質転換陽性のコロニーをPCRおよび制限酵素分析を用いて、cGB-8が正しい方向となっているかについてスクリーニングした。 正しい方向でcGB-8インサートを含む、得られた発現構築体をpCDM8-cGB-PDEと言及する。
【0056】
pCDM8-cGB-PDEのDNAはQiagen pack-500カラム(チャツウォース(Chatsworth)、CA)を用い、製造業者のプロトコルに従って大量のプラスミド調製物から精製した。 COS-7細胞は、10%ウシ胎児血清、50μg/mlペニシリンおよび50μg/mlストレプトマイシンを含むダルベッコ変法イーグル培地(Dulbecco's modified Eagle's medium)(DMEM)中で、37℃にて、給湿した5%CO2大気中で培養した。 トランスフェクションのおよそ24時間前に、集密な細胞の100mmディッシュを、もとの密度の4分の1または5分の1で再度播いた。 典型的なトランスフェクション実験において、細胞は137mM NaCl、2.7mM KCl、1.1mM リン酸カリウム、および8.1mMリン酸ナトリウム、pH7.2(PBS)を含む緩衝液で洗浄した。 次いで10% NuSerum(コラボレーティブ・バイオメディカル・プロダクツ(Collaborative Biomedical Products)、 ベドフォード、MA)を含む4〜5mlのDMEMを各プレートに添加した。 60μl TBS(トリス緩衝生理食塩水:25mM Tris-HCl(pH7.4)、137mM NaCl、5mM KCl、0.6mM Na2HPO4、0.7mM CaCl2、および0.5mM MgCl2)中、400μgのDEAE-デキストラン(ファルマシア)と混合した10μgのpCDM8-cGB-PDEのDNAまたはpCDM8ベクターDNAでのトランスフェクションを、各プレートに混合物を滴下により添加して行った。 細胞を37℃にて5%CO2で4時間インキュベートし、次いで10%ジメチルスルホキシド含有PBSで1分間処理した。 2分後、ジメチルスルホキシドを除去し、細胞をPBSで洗浄して完全培地中でインキュベートした。 48時間後、細胞のプレートあたり0.5〜1mlの冷ホモジナイズ緩衝液(40mM トリス−塩酸(pH7.5)、15mM ベンズアミジン、15mM β−メルカプトエタノール、0.7μg/ml ペプスタチンA、0.5μg/ml ロイペプチン、および5μM EDTA)に細胞を懸濁し、ダウンス(Dounce)ホモジナイザーを用いて破砕した。 その結果得られた全細胞抽出物を、ホスホジエステラーゼ活性、cGMP結合活性および総タンパク質濃度について以下の実施例5に記載するようにアッセイした。
【0057】
実施例5
実施例4のトランスフェクトされたCOS細胞の抽出物又はモックトランスフェクトされたCOS細胞の抽出物におけるホスホジエステラーゼ活性を、Martins et al., J. Biol. Chem.,257:1973-1979(1982)のcGs-PDEについて記載されているアッセイ手法を改良したものを用いて測定した。 細胞を採取し、トランスフェクション後48時間で抽出物を調製した。インキュベーション混合物は、全容量250μl中に、40mMのMOPS緩衝液(pH7)、0.8mMのEDTA、15mMの酢酸マグネシウム、2mg/mlのウシ血清アルブミン、20μM、[3H]cGMP又は[3H]cAMP(100,000-200,000cpm/アッセイ)及びCOS-7細胞抽出物を含んでいる。 この反応混合物を30℃にて10分間インキュベートした。 次に、10mg/mlのCrotalus atrox venom (Sigma)を10μl加え、さらに30℃で10分間インキュベートした。 ヌクレオシド産物は、Martins et al.、前出に記載されているとおりに未反応のヌクレオチドから分離した。 全ての実験において、全[3H]環状ヌクレオチドの15%以下が、反応中に加水分解されていた。
【0058】
アッセイの結果を第5図に示すが、ここでは3つの別々のトランスフェクションの平均が示されている。 COS-7細胞のpCDM8-cGB-PDE DNAによるトランスフェクションは、モックトランスフェクト細胞又はpCDM8ベクター単独でトランスフェクトされた細胞より、cGMPホスホジエステラーゼ活性が15倍高いレベルとなっている。 モック又はベクター単独トランスフェクト細胞におけるcAMPホスホジエステラーゼ活性の増大は、pCDM8-cGB-PDE DNAでトランスフェクトした細胞からの抽出物中では検出されなかった。 これらの結果により、cGB-PDEウシcDNAはcGMP特異的ホスホジエステラーゼをコードしていることが確認された。
【0059】
また、実施例4のトランスフェクトされたCOS細胞からの抽出物を、PDE阻害剤、ザプリナスト、ジピリダモル (Sigma), イソブチル-1-メチル-8-メトキシメチルキサンタン(MeOxMeMIX)及びロリプラム(Rolipram)の一連の濃度の存在下におけるcGMP PDE活性のアッセイに供した。 アッセイの結果は第6図に示されており、阻害剤のない場合の活性を100%とし、各データポイントは2つの別々の測定の平均を示している。 発現されたcGB-BPDE cDNAタンパク産物によるcGMP加水分解の阻害に対するPDE阻害剤の相対的効能は、ウシ肺(トーマスI、前出)から精製したcGB-PDE本来のものについて報告されているこれらの相対的効能と一致している。 第6図の曲線から計算したIC50値は以下のとおりである。 ザプリナスト(黒塗り円),2μM;ジピリダモル(黒塗り四角),3.5μM;MeOxMeMIX(黒塗り三角)、30μM;及びロリプラム(白抜き円)、>300μM。 cGMP特異的ホスホジエステラーゼの比較的特異的な阻害剤であるザプリナストのIC50値は、cGs-PDE又はcGMP阻害ホスホジエステラーゼ(cGi-PDE)(Reeves et al.,pp.300-316 Beavo et al.,前出)のホスホジエステラーゼ活性の阻害について報告されているものより、少なくとも2オーダー低かった。 選択されたcAMP及びcGMP特異的ホスホジエステラーゼの効果的な阻害剤であるジピリダモルは、発現されたcGB-PDEの強力な阻害剤でもある。 相対的に選択的な、カルシウム/カルモジュリンにより刺激されるホスホジエステラーゼ(CaM-PDE),MeOxMeMIXの選択的阻害剤は、ザプリナスト及びジピリダモルより10倍低い効能であり、これはウシ肺から精製されたcGB-PDE活性体を使用した結果と一致している。
【0060】
低いKmのcAMPホスホジエステラーゼであるロリプラムは、発現されたcGB-BPDE cDNAタンパク産物に対しては低い阻害剤である。 これらの結果は、cGB-PDE cDNAが、ウシ組織から単離されたcGB-PDEの触媒活性特性を有するホスホジエステラーゼをコードしていることを示しており、これにより、cGB-8 cDNAクローンがcGB-PDEと同一であることが確認された。
【0061】
cGMP加水分解の阻害に対するPDE阻害剤の相対的な効能は、組換え遺伝子及びウシ単離cGB-PDEに対する場合と同様であるものの、全ての阻害剤についての絶対値IC50値は、組換えcGB-PDEより2〜7倍高いことは興味深い。 この違いは、COS-7細胞抽出物に存在する因子のcGMP加水分解活性に対する影響に起因すると考えることはできない。 なぜなら、ウシ組織から単離されたcGB-PDEは、純粋な酵素と同様か、又はモックトランスフェクトされたCOS-7細胞の抽出物に戻した場合と同様の阻害速度を示すからである。 この薬理学的感度の明らかな違いは、触媒部位及びその近傍における翻訳後修飾などの組換えcGB-PDE cDNAタンパク産物及びウシ肺cGB-PDEの構造におけるわずかな違いによっている。
【0062】
この違いはまた、いくつかの精製工程におけるウシ肺cGB-PDEの触媒活性の変化によっている。
【0063】
細胞抽出物を0.2mMの3−イソブチル-1-メチルキサンチン(IBMX)(Sigma)の存在下及び非存在下における[3H]cGMP結合活性のアッセイに供した。 トーマスI、前出に記載されているアッセイを改変したcGMP結合活性のアッセイは、80μlの総容量で行った。 その混合物の成分の最終濃度が、1μM[3H]cGMP,5μM cAMP、及び10μM 8−ブロモ-cBMPとなるように、60μlの細胞抽出物を20μlの結合カクテルに加えた。 cAMP及び8-ブロモ-cBMPは、それぞれ[3H]cGMPがcAK及びcGKに結合するのを阻止するために加えた。 アッセイは、0.2mMのIBMXの存在下及び非存在下で行った。 反応は細胞抽出物の添加により開始し、0℃で60分間インキュベートした。 トーマスI、前出に記載されているとおりに反応混合物を濾過した。 細胞抽出物に代えて均質化緩衝液、又は非標識cGMPの100倍量を用いて並行してインキュベーションを行い、ブランクを決定した。 同様の結果が、両方の方法で得られた。 細胞抽出物の総タンパク質濃度は、Bradford, Anal. Biochem., 72:248-254(1976)により、ウシ血清アルブミンを標準として定量した。
【0064】
アッセイの結果を、第7図に示した。 0.2mMのIBMXの存在下、1μMの[3H]cGMPで測定した場合、pCDM8-cGB-PDEでトランスフェクトされたCOS-7細胞からの抽出物は、モックトランスフェクトされた細胞からの抽出物より8倍高いcGMP結合活性を示した。 バックグラウンドcGMP結合のIBMXの促進は観察されず、内在性のcGB-PDEは、COS-7細胞抽出物には、ほんのわずか又は全く存在していないことを示している。 pCDM8-cGB-PDEトランスフェクトされた細胞の抽出物中では、cGMP特異活性は0.2mMのIBMXの添加により、約1.8倍に促進されている。 IBMXのcGMP結合を2〜5倍に促進する能力は、cGMP結合ホスホジエステラーゼ特有の性質である。
【0065】
上述したように、細胞抽出物を、過剰の非標識cAMP又はcGMPの存在下で、[3H]cGMP結合活性のアッセイ(ここでの[3H]cGMPの濃度は2.5μMである)に供した。 結果を第8図に示し、ここでは非標識の競合剤の非存在下におけるcGMP結合を100%とし、各データポイントは3つの別々の測定の平均を表している。 cGB-PDEによってコードされたタンパク産物の結合活性は、cGMPがcAMPに比較して特異的である。 10倍未満の高濃度の非標識cGMPが、[3H]cGMP結合活性を50%まで阻害するのに必要であったが、約100倍の濃度のcAMPがこれと同程度の阻害に必要であった。
【0066】
本実施例におけるこの結果は、cGB-PDE cDNAが、ネイティブなcGMB-PDEの生化学的活性特性を有するホスホジエステラーゼをコードすることを示している。
【0067】
哺乳類PDE及びドロソフィアPDEの触媒ドメインは、サーモリシン(thermolysin、Vallee and Auld, Biochem., 29:5647-5659(1990))などの、Zn2+加水分解酵素における典型的なZn2+結合モチーフである2つの直列保存配列(HX3HX24-25E)を含んでいる。 cGB-PDEは過剰のMg2+、Mn2+、Fe2+、Fe3+、Ca2+又はCd2+の存在下でZn2+と結合する。 添加金属の非存在下では、cGB-PDEは40mMのMg2+の存在下で見られる最大の活性の約20%のPDE活性を有し、この基礎活性は1、10-フェナントロリン又はEDTAにより阻害される。 このことは、金属を除去する完全な処理にも関わらず、痕跡量の金属がPDE活性に影響していることを示唆している。 PDE活性は、Zn2+(0.02−1μM)又はCo2+(1−20μM)の添加により促進されるが、Fe2+、Fe3+、Ca2+、Cd2+又はCu2+によっては阻害されない。 Zn2+は、40mMのMg2+によってもたらされる最大の促進の約70%まで基礎PDE活性を増大させる。 これらのアッセイにおけるZn2+の促進効果は、1>1μMのZn2+濃度による阻害効果の妥協によるものである。 Zn2+によって支持されたPDE活性及びcGB-PDEによるZn2+結合は、Zn2+の同様の濃度においても現れる。 このように、cGB-PDEはZn2+加水分解酵素であると考えられ、Zn2+は、この酵素の活性における重要な役割を演じていると考えられる。 Colbran et al., The FASEB J., 8: Abstruct 2148(March 15, 1994)を参照されたい。
【0068】
実施例6
cGB-PDEをコードするウシcDNAクロ−ンと相同のいくつかのヒトcDNAクローンが、ウシcGB-8クローン(配列番号:9のヌクレオチド489-1312)の部分に相当する核酸プローブを使用するストリンジェント条件下でのハイブリダイゼーションによって単離された。
【0069】
ヒトcGB-PDEをコードするcDNA断片の単離
ベクターラムダZapの、3つのヒトcDNAライブラリー(2つのグリア芽細胞腫及び1つの肺)を、ウシcGB-PDE配列によりプローブ探針した。 実施例1に記載した配列番号:9のヌクレオチド484〜1312に相当する、PCRで作製したクローンを、EcoRI及びSalIで消化し、得られた0.8kbのcDNAインサートを単離して、アガロース電気泳動により精製した。この断片を、ランダムプライムドDNAラベリングキット(Boehringer)を用いて放射性ヌクレオチドで標識した。
【0070】
cDNAライブラリーは、プレート当たり約50,000プラークの密度となるように150mmのペトリ皿に播種した。 二重のニトロセルロースフィルターレプリカを調製した。 プレハイブリダイゼーション緩衝液は、3×SSC、0.1%サルコシル、10×デンハーツ、20mMリン酸ナトリウム(pH 6.8)および50μg/mlのサケ精巣DNAである。 プレハイブリダイゼーションは、1〜5×105cpm/mlのプローブを加えた同様の組成の緩衝液中で、65℃で一夜行った。 フィルターを65℃で2×SSC、0.1%SDSで洗浄した。 ハイブリダイズしたプラークを、オートラジオグラフィで検出した。 ウシプローブにハイブリダイズしたcDNAの数、およびスクリーニングされたcDNAの数を表2に示す。
【0071】
【表2】
【0072】
cgbS2.1、cgbS3.1、cgbL23.1、cgbL27.1及びcgbS27.1と命名されたプラスミドを、ラムダZapクローン及び配列から、in vivoで試験した。
【0073】
クローンcgbS3.1は、推定イントロンに続くPDEオープンリーディングフレームの2060bpを含んでいる。 cgbS2.1の分析は、これがcgbS3.1の位置664から2060に相当し、読み取り前の付加的な585bpのPDEオープンリーディングフレームを推定イントロンまで伸ばすものである。 推定5'非翻訳領域の配列およびcgbS2.1、cgbS3.1クローンの部分をコードするタンパク質は、それぞれ配列番号:11及び12で示される。 2つのcDNAを結合することにより、PDEのオープンリーディングをコードする約2.7kbを含む配列が得られる。 他の3つのsDNAは、cDNA cgbS3.1又はcgbS2.1を5'又は3'よりさらに伸長しているものではなかった。
【0074】
さらなるcDNAを単離するために、クローンcgbS3.1の5'末端およびクローンcgbS2.1の3'末端を調製し、SW1088グリア芽細胞腫cDNAライブラリーおよびヒト大動脈cDNAライブラリーをスクリーニングするために使用した。 5'プローブは、プライマーgbS3.1S311及びcgbL23.1A1286を用いたPCRにより、クローンcgbS3.1に由来し、上記プライマーgbS3.1S311及び上記cgbL23.1A1286はそれぞれ、配列番号:8及び9、並びに下記によって示されている。
【0075】
プライマーcgbS3.1S311(配列番号:13)
5’GCCACCAGAGAAATGGTC3’
プライマーcgbL23.1A1286(配列番号:14)
5’ACAATGGGTCTAAGAGGC3’
PCR反応は、cgbS3.1 cDNAを50pg、dNTPを0.2mM、各プライマーを10μg/ml、KClを50mM、トリス塩酸pH8.2を10mM、MgCl2を1.5mMおよびTaqポリメラーゼを含む50μlの反応容量中で行った。 最初の94℃での4分間の変性後、94℃で1分、50℃で2分及び72℃で4分の、サイクルを30回行った。 PCR反応によって約0.2kbの断片が生成し、これはクローンcgbS3.1のヌクレオチド300〜496に相当する。
【0076】
3'プローブは、下記の配列を有するオリゴcgbL23.1S1190およびcgbS2.1A231を用いたPCRにより、cDNAcgbS2.1に由来するものであった。
【0077】
プライマーcgbL23.1S1190(配列番号:15)
5’TCAGTGCATGTTTGCTGC3’
プライマーcgbS2.1A231(配列番号:16)
5’ACAATGGGTCTAAGAGGC3’
PCR反応は、5'プローブの生成で上述したと同様に行い、cDNA cgbS2.1のヌクレオチド1358〜2139に相当する約0.8kbの断片を得た。 PCR断片(配列番号:12には示していない)の3' 157ヌクレオチドは、推定イントロン内にある。
【0078】
2つのPCR断片を、アガロースゲル電気泳動により精製および単離し、ランダムプライムにより放射性ヌクレオチドで標識した。 ランダムプライムSW1088cDNAライブラリー(1.5×106プラーク)を、上記標識された断片でスクリーニングし、19のハイブリダイジングプラークを単離した。 さらに50のハイブリダイズするグプラークが、ヒト大動脈cDNAライブラリー(dT及びランダムプライム、Clontech, Palo Alto, CA)から単離された。
【0079】
プラスミドを、陽性のラムダZapクローン及び配列のいくつかから、in vivo で切り出した。 cgbS53.2と命名された一つのクローン(その配列は、配列番号:17で示される)は、その配列がcgbS3.1の最後の61bpに重複する約1.1kbのインサートを含んでおり、さらなる135bpのPDEオープンリーディングフレームを、cgbS2.1に見出されるものを越えて伸長するものである。 このクローンは、終止コドンと、推定3'非翻訳配列の約0.3kbを含んでいる。
【0080】
ヒトcGB-PDEをコードする複合cDNAの生成
クローンcgbS3.1、cgbS2.1およびcgbS53.2を、以下のパラグラフに記載されているように、完全なヒトcGB-PDEのオープンリーディングフレームを含む複合cDNAを構築するために使用した。 複合cDNAをcgbmet156-2と命名し、酵母ADH1発現ベクターpBNY6Nに挿入した。
【0081】
先ず、cGB-PDEオープンリーディングフレームの3'末端を含むcgbstop-2と命名されたプラスミドを生成した。 プラスミドのインサートの一部を、鋳型としてクローンcgbS53.2を使用して、PCRによって作製した。 使用したプライマーはcgbS2.1S1700及びcgbstop-2であった。
【0082】
プライマーcgbS2.1S1700(配列番号:18)
5’TTTGGAAGATCCTCATCA3’
プライマーcgbstop-2(配列番号:19)
5’ATGTCTCGAGTCAGTTCCGCTTGGCCTG3’
PCR反応は、鋳型DNAを50pg、dNTPを0.2mM、トリス塩酸pH8.2を20mM、KClを10mM、(NH4)2SO4を6mM、MgCl2を1.5mM、0.1%Triton-X-100、各プライマー500ngおよび Pfuポリメラーゼ(Stratagene)の0.5単位を含む50μl中で行った。 反応液は94℃で4分間加熱し、94℃で1分、50℃で2分及び72℃で4分の30回のサイクルを行った。 ポリメラーゼは最初のサイクルで50℃の間に添加した。 得られたPCR産物は、フェノール/クロロホルム抽出し、クロロホルム抽出し、エタノール沈殿を行い、そして制限酵素BclI及びXhoIで切断した。 制限断片は、アガロースゲルで精製して溶出させた。
【0083】
この断片を、dam E. coli内で成長させたcDNA cgbS2.1に結合させ、制限酵素Bcl1及びXhoIで切断し、及びPromega magic PCR Kit を用いてゲル精製を行った。 結果として得られるプラスミドは、cgbstop-2がcGB-PDEオープンリーディングフレームの3'部分を含んでいることを確認するために配列決定された。
【0084】
二番目に、ヒトcGB-PDEオープンリーディングフレームの5'末端を担持するプラスミドを生成させた。 そのインサートは、鋳型としてクローンcgbS3.1を用いてPCRにより生成させた。 PCRは、プライマーcgbmet156及びcgbS2.1A2150を用い、上述と同様にして行った。
【0085】
プライマーcgbmet156(配列番号:20)
5’TACAGAATTCTGACCATGGAGCGGGCCGGC3’
プライマーcgbS2.1A2150(配列番号:21)
5’CATTCTAAGCGGATACAG3’
結果として得られるPCR断片は、フェノール/クロロホルム抽出し、クロロホルム抽出し、エタノール沈殿を行い、Sepharose CL-6Bカラムで精製した。 この断片を、制限酵素EcoRV及びEcoRIで切断してアガロースゲルを通し、そしてガラスウールを通すスピニングにより精製した。 フェノール/クロロホルム抽出、クロロホルム抽出、及びエタノール沈殿の後に、この断片はEcoRI/EcoRVで消化されたBluescriptII SK(+)に連結し、プラスミドcgbmet156を作製した。 インサートと接合部のDNA配列を決定した。 インサートは新たなEcoRI部位を含み、開始コドンの元の155ヌクレオチド5'を互いに置換した付加的な5ヌクレオチドを含んでいる。 インサートは、開始コドンより531ヌクレオチドから始まるEcoRV部位まで伸びている。
【0086】
cGB-PDEオープンリーディングフレームの5'および3'の部分を、次いてベクターpBNY6aに集めた。 ベクターpBNY6aを、EcoRVおよびXhoIで切断し、ゲルにより単離し、アガロースゲルで精製したcgbmet156からのEcoRI/EcoRV断片と、アガロースゲルで精製したcgbstop-2からのEcoRV/XhoIとに結合させた。 インサートの接合部を配列決定し、そして、構築体を、hcbgmet156-26aと命名した。
【0087】
次に、hcbgmet156-26aからのcGB-PDEインサートを、発現ベクターpBNY6nに移動させた。このベクターに挿入されたDNAの発現は、酵母ADH1プロモータおよびターミネーターより導かれる。 このベクターは、酵母の2ミクロンの複製起点、複製のpUC19起点、およびアンピシリン耐性遺伝子を含んでいる。 ベクターpBNY6nをEcoRIおよびXhoIで切断し、ゲル精製を行った。 hcgbmet156-26aからのEcoRI/XhoIインサートをPromega Magic PCR カラムを用いてゲル精製し、pBNY6nに結合させた。 結果として得られる構築体hcgbmet156-26nにおける全ての新しい接合部を配列決定した。 複合ヒトcGB-PDEをコードするhcgbmet156-26nのインサートのDNAと推定アミノ酸配列を、配列番号:22及び23に示す。
【0088】
インサートは、クローンcgbS3.1(ヌクレオチド156)の最初のメチオニンから複合cDNAの終止コドン(ヌクレオチド2781)まで伸びている。 そのメチオニンは、クローンcgbS3.1にて最も5'側のメチオニンであるので、また、メチオニンを有するフレームには、終止コドンが存在しないので、pBNY6nにおけるインサートは、オープンリーディングフレームの端を切欠した形状を現しているのかもしれない。
【0089】
変異cDNA
hcgbmet156-26n複合cDNAとは異なる4つのヒトcGB-PDE cDNAが単離されている。 一つのcDNA、cgbL23.1は、hcgbmet156-26nの内部領域(ヌクレオチド997〜1000から1444〜1447)が欠損しているものである。 欠損部分の正確な終端は、それらの位置におけるcDNA配列から決定することはできない。 4つの変異cDNAのうちの3つは、hcgbmet156-26n配列のヌクレオチド151(cDNA cgbA7f、cgbA5C、cgbI2)の上流から分岐する5'末端配列を有している。 これらのcDNAは、選択的スプライシングを受けた又はスプライシングを受けていないmRNAを表しているのかもしれない。
【0090】
実施例7
複合ヒトcGB-PDE cDNA構築体、hcgbmet156-26nを、酵母菌株YKS45(ATCC74225)(MATαhis3 trp1 ura3 leu3 pde1::HIS3 pde2::TRP1)へ形質転換したが、これは2つの内在性PDE遺伝子が欠損してたものである。 YKS45菌株のleu-欠損を補足する形質転換体を選択し、cGB-PDE活性についてアッセイを行ったプラスミドhcgbmet156-26nを産出する細胞からの抽出物は、以下に示すアッセイにより、サイクリックGMP特異的ホスホジエステラーゼ活性を呈することを調べた。
【0091】
プラスミドcgbmet156-26nで形質転換されSC-leu培地で1〜2×107細胞/mlの密度まで成長したYKS45細胞の1リットルを遠心分離により採集し、脱イオン水で一度洗浄し、ドライアイス/エタノールで凍結させて−70℃で保存した。 細胞ペレット(1〜1.5ml)を、等容量の25mMトリス-Cl(pH 8.0)/5mM EDTA/5mM EGTA/1mM オルトフェナントロリン/0.5mM AEBSF(Calbiochem)/0.1% β−メルカプトエタノールおよび10μg/mlのアプロチニン、ロイペプチン及びペプスタチンAのそれぞれの存在下に、氷上で解凍した。解凍した細胞は、15mlのCorexチューブ内の酸洗浄したガラスビーズ(425〜600μM、Sigma)2mlに加えた。 1にセットした30秒間のボルテックスと、次に60秒間の氷上でのインキュベーションとからなるサイクルを4回行うことにより、細胞を破壊した。 細胞溶解液を12,000×gで10分間遠心分離し、上清を0.8μのフィルターに通した。 上清を以下に示すようにcGMP PDE活性のアッセイに供した。 45mMトリス-Cl(pH 8.0)、2mM EGTA、1mM EDTA、0.2mg/ml BSA、5mM MgCl2、0.2mM オルトフェナントロリン、2μg/mlのペプスタチンA、ロイペプチン及びアプロチニンのそれぞれ、0.1mM AEBSF、0.02% β−メルカプトエタノールおよび基質として0.1mMの[3H]cGMPの存在下に、試料を30℃で20分間インキュベートした。 [14C]-AMP(0.5Ci/アッセイ)を回収標準物として加えた。 反応は停止緩衝液(0.1MエタノールアミンpH 9.0、0.5M硫酸アンモニウム、10mM EDTA、最終濃度0.05%のSDS)で停止した。 生成物は、BioRad Affi-Gel601上のクロマトグラフィーにより、サイクリックヌクレオチド基質から分離した。 カラム緩衝液(0.5M硫酸アンモニウムを含む0.1MエタノールアミンpH 9.0)で平衡化したAffi-Gel601の約0.25mlを含むカラムを適用した。 カラムはカラム緩衝液の0.5mlで5回洗浄した。 生成物は、0.25の酢酸の0.5mlで4回溶出させ、5mlのEcolume(ICN Biochemicals)と混合した。 放射性生成物をシンチレーション計数により測定した。
【0092】
実施例8
ヒト組織におけるcGB-PDE mRNAの発現の分析を、RNaseプロテクションアッセイにより行った。
【0093】
cGB-PDE(配列番号:13のヌクレオチド1450〜1851に相当する402bp)の推定cGMP結合ドメインの部分に相当するプローブを、PCRにより生成させた。 このPCR断片をプラスミドpBSII SK(-)のEcoRI部位に挿入してプラスミドRP3を作製した。 RP3プラスミドDNAをXbalIで直鎖状化し、アンチセンスプローブを Stratagene T7 RNA ポリメラーゼキットの改変によって作製した。 25ngの直鎖状化されたプラスミドを、20μキュリーのアルファ32PrUTP(800Ci/mmol、10mCi/ml)、1×転写緩衝液(40mMトリスCl、pH8、8mM MgCl2、2mM スペリミジン、50mM NaCl)、0.25mMの各rATP、rGTPおよびrCTP、0.1単位のRNase BlockII、5mM DTT、8μM rUTPおよび全容量5μlの5単位のT7RNAポリメラーゼと合わせた。 室温で1時間反応を行い、RNase不含のDNaseでDNA鋳型を消化させた。
【0094】
反応液を100μlの40mM トリスCl、pH8、6mMのMgCl2、及び10mMのNaClで希釈した。
【0095】
RNase不含の5単位のDNaseを加え、更に15分間、37℃で反応を継続した。 フェノール抽出によって反応を停止させ、次にフェノールクロロホルム抽出を行った。 7.5MのNH4OAcの半分の容量を加え、プローブをエタノール沈殿させた。
【0096】
RNaseプロテクションアッセイを、Ambion RNase Protection kit(Austin,TX)と、酸グアニジニン抽出法によるヒト組織から単離した10μgのRNAとを用いて行った。 cGB-PDE mRNAの発現は、骨格筋、子宮、気管支、皮膚、右の伏在静脈、大動脈およびSW1088グリア芽細胞腫細胞から抽出したRNAで容易に検出された。 右心房、右心室、腎臓皮質および腎臓髄質ではわずかに検出し得る発現が見られた。 PR3プローブの完全な保護のみが見られた。 部分的保護がないことは、少なくともこれらのRNA試料で主要な転写を表すcDNA cgbL23.1(実施例7に記載した変異cDNA)と反対の結論である。
【0097】
実施例9
ポリクロナール抗血清を、ヒトcGB-PDEの大腸菌生成断片に起用した。
【0098】
ヒトcGB-PDE cDNA(配列番号:22のヌクレオチド1668〜2612、配列番号:23のアミノ酸515〜819)の部分をPCRにより増幅し、BamHI/EcoRIとしてE. coli発現ベクターpGEX2T(Pharmacia)に挿入した。 pGEX2Tプラスミドは、アンピシリンR因子(耐性遺伝子)、大腸菌laq Iq因子、及び日本住血吸虫(Schistosoma japonicum) グルタチオン-S-転写(GST)因子を運ぶ。 プラスミドに挿入されたDNAは、GSTとの融合タンパク質として発現され、トロンビンによりこのタンパクのGST部分から分割されることができる。 この結果得られるcgbPE3と表されるプラスミドを、大腸菌株LE392(Stratagene)に形質転換した。 形質転換された細胞は、37℃で、0.6のOD600までに成長させた。 IPTG(イソプロピルチオアラクトピラノシド)を0.1mMまで加え、細胞を37℃で更に2時間成長させた。細胞を遠心分離により集め、超音波で溶解させた。 細胞の残滓を遠心分離により除去し、SDS-PAGEにより分画した。 ゲルを冷0.4M KClで着色し、GTS-cgb融合タンパクのバンドを切り出し、電気溶出させた。 タンパク質のPDE部分は、スロンビンの消化によりGTS部分から分離された。 消化物をSDS-PAGEにより分画し、PDEタンパク質を電気溶出して、ウサギに皮下注射した。 結果として得られる抗血清は、抗原として使用したウシcGB-PDE断片と、酵母に発現された(実施例8を参照されたい)ヒトcGB-PDEタンパクの全長との両方を認識する。
【0099】
実施例10
種々のcGB-PDE類似体をコードするポリヌクレオチドおよびcGB-PDE断片を標準の方法により生成させた。
【0100】
A.cGB-PDE類似体
公知のcGMP結合PDEの全ては、それらの推定cGMP結合ドメインに2つの内的に一致する直列のリピートを含んでいる。 本発明のウシcGB-PDEでは、リピートは少なくとも配列番号:10の残基228〜311(リピートA)及び410〜500(リピートB)にわたる。 公知のcGMP結合PDEの全てのリピートA及びBに保持されているアスパラギン酸の部位特異的突然変異誘発を、Ala(D289A)で置換したAsp-289か、又はAla(D478A)で置換したAsp-478を有する類似のcGB-PDEを作製するのに使用した。 組換え体の野生型(WT)のウシ及び突然変異ウシcGB-PDEを、COS-7細胞にて発現させた。 ウシ肺(自然のcGB-PDE)から精製されたcGB-PDEと、WT cGB-PDEとは同じcGMP結合動力学を示して、Kdは約2μMであり、曲直線(curvlinear)解離プロファイル(4℃でt1/2=1.3時間)を示した。 この曲直線性は、cGMP結合の明らかに高い親和性と低い親和性の部位の存在によるのかもしれない。 D289Aの突然変異体は、cGMPに対して明らかに減少した親和性を有し(Kd>20μM)、cGMP群のの単一の速度(t1/2=0.5時間)を有しており、これは、WTおよび自然のcGB-PDEの速い部位について計算したものと同じである。 このことは、突然変異体のリピートAにおける遅いcGMP結合部位の損失を示唆している。 逆に、D478A突然変異体はcGMPに対して高い親和性と(約0.5μMのKd)、単一のcGMP解離速度(t1/2=2.8時間)を示し、これは、WTおよび自然のcGB-PDEの遅い部位について計算したものと同様である。このことは、リピートBの改変に際して、速い部位が損失されることを示唆している。 これらの結果は、2量体cGMPは2つの相同であるが動力学的に区別されるcGMP結合部位を有しており、この部位は各部位でcGMPと相互作用するのに重要なアスパラギン酸を保持している。 Colbran et al., FASEB J., 8:Abstract 2149(May 15, 1994)を参照されたい。
【0101】
B.アミノ末端を切欠したcGB-PDEポリペプチド
配列番号:23のアミノ酸516〜875を含む末端を切欠したヒトcGB-PDEポリペプチドを、酵母において発現させた。 配列番号:22のヌクレオチド1555のNcoIから、配列番号:22の3’末端のXhoI部位まで伸びるcDNAインサートを、NcoI及びSalIで消化しておいたADH2酵母発現ベクターYEpC-PADH2d(Price et al., Meth. Enzymol., 18:308-318(1990))に挿入して、プラスミドYEpC-PADH2d HcGBを作製した。 このプラスミドは、酵母菌株yBJ2-54(prcl-407 prbl-1122 pep4-3 leu2 trpl ura3-52 Δpdel::URA3,HIS3 Δpde2::TRP1 cir)のスフェロプラストへと形質転換した。 内在性のPDE因子は、この菌株では欠落している。 細胞を、2%のグルコースを含むSC-leu培地で107個まで生育し、濾過によって集め、3%グリセロールを含むYEP培地で24時間生育した。 細胞は遠心分離によりペレット化し、凍結保存した。 細胞をガラスビーズで粉砕し、細胞懸濁液を、Prpic et al., Anal.Biochem., 208:155-160(1993)に本質的に記載されているホスホジエステラーゼ活性のためのアッセイに供した。 末端を切欠したヒトcGB-PDEポリペプチドは、ホスホジエステラーゼ活性を示した。
【0102】
C.カルボキシ末端を切欠したcGB-PDEポリペプチド
カルボキシ末端を切欠したcGB-PDEポリペプチドをコードする2つの異なるプラスミドを構築した。
【0103】
配列番号:23のアミノ酸1〜494をコードするcDNAを含むプラスミドpBJ6-84Hinを、ベクターYEpC-PADH2dのNcoI及びSalI部位に挿入した。 cDNAのインサートは、配列番号:22のヌクレオチドの第10位のNcoI部位から、リンカーに続く配列番号:22のヌクレオチドの第1494位のHindIII部位とYEpC-PADH2dのSalI部位まで伸びている。
【0104】
プラスミドpBJ6-84Banは、ベクターYEpC-PADH2dのNcoI及びSalI部位に挿入された、配列番号:23のアミノ酸1〜549をコードするcDNAを含んでいる。 このcDNAインサートは、配列番号:22のヌクレオチドの位置10のNcoI部位から、リンカーに続く配列番号:22のヌクレオチドの位置1654のBanI部位とYEpC-PADH2dのSalI部位まで伸びている。
【0105】
末端を切欠したcGB-PDEポリペプチドは、cGB-PDE活性のモジュレーターをスクリーニングするのに有用である。
【0106】
実施例11
ヒトcGB-PDEと反応するモノクロナール抗体を生成した。
【0107】
プラスミドYEpADH2 HcGBを含む酵母yBJ2-54(実施例10B)を、New Brunswick Scientificの10リットルのMicrofarm内で発酵させた。 プラスミドYEpADH2 HcGB内のcGB-PDE cDNAインサートは、配列番号:22のヌクレオチドの第12位のNcoI部位から、配列番号:22の3'末端のXhoI部位まで伸びている。 4×109個の細胞の接種物を、SC-leu、5%グルコース、微量金属、及び微量ビタミンを含む8リットルの培地に加えた。 発酵は26℃を保ち、標準の微生物用の撹拌翼を用いて600rpmで撹拌し、圧搾空気で1分間当たり10倍容積のエアレーションを行った。 接種後24時間でグルコースが0.3%に減少したとき、培養液に15%のグリセロールを含む2リットルの5×YEP培地を注入した。 接種後66時間で、4℃、4,000×gで30分間遠心分離することにより、発酵液から酵母を収穫した。 この発酵におけるバイオマスの収量は、湿重量で350gに達した。
【0108】
ヒトcGB-PDE酵素を、酵母細胞ペレットから精製した。 基質として1mMのcGMPを使用するPDE活性のためのアッセイを、この酵素のクロマトグラフィーに続いて採用した。 全てのクロマトグラフィーの操作は、4℃で行った。
【0109】
酵母(湿重量で29g)を、再び70mlの緩衝液A(25mMのトリス pH8.0、0.25mMのDTT、5mMのMgCl2、10μMのZnSO4、1mMのベンズアミジン)に分散させ、22−24,000psiでマイクロフリュイダイザーを通して溶解させた。 溶解液は、10,000×gで30分間遠心分離し、20mMのビストリス−プロパンpH6.8、0.25mMのDTT、1mMのMgCl2、及び10μMのZnSO4を含む緩衝液Bで平衡化したPharmacia Fast Flow Q noアニオン交換樹脂を充填した2.6×28cmのカラムに上清を通した。 このカラムをカラム容量の5倍の、0.125MのNaClを含む緩衝液Bで洗浄し、次に0.125M〜1.0MまでのNaClの直線勾配で展開した。 酵素を含む画分をプールし、20mMのビストリス−プロパンpH 6.8、0.25mMのDTT、0.25MのKCl、1mMのMgCl2、及び10μMのZnSO4を含む緩衝液Cで平衡化したセラミックヒドロキシアパタイト(BioRad)の5×20cmのカラムに直接付した。 このカラムをカラム容量の5倍の緩衝液Cで洗浄し、緩衝液C中で0〜250mMまでのリン酸カリウムの直線勾配で溶出させた。 プールした酵素を限外濾過(YM30メンブレン、Amicon)で8倍まで濃縮した。 濃縮した酵素を、25mMのビストリス−プロパン pH6.8、0.25mMのDTT、0.25MのNaCl、1mMのMgCl2、及び20μMのZnSO4で平衡化した Pharmacia Sephacryl S300(Piscataway, NJ)の2.6×90cmのカラムでのクラマトグラフィーにかけた。 約4mgのタンパクが得られた。 組換えヒトcGB-PDE酵素は、SDSポリアクリルアミドゲルの電気泳動と、次のクマシーブルー染色とにより、得られたタンパクの約90%であると計算された。
【0110】
精製したタンパク質を、モノクロナール抗体を惹起するための抗原として使用した。 19週齢のBalb/cマウス(Charles River Biotechnical Service, Inc., Wilmington, Mass)の皮下に、50%のフロイント完全アジュバント(Sigma Chemical Co.)からなる200μlのエマルジョン中の50μgの精製ヒトcGB-PDE酵素を免疫投与した。 続く追加免疫を、20日と43日に不完全フロイントアジュバントで投与した。 PBS中の50μgの酵素を用いて、前融合の後追加免疫を86日に行った。 融合は90日に行った。 マウス#1817からの脾臓を無菌的に取り出し、10mlの無血清RPMI1640中に入れた。 単細胞懸濁液を形成し、無菌の70メッシュの Nitex 細胞ストレーナ(Becton Dickinson, Parsippany, New Jersey)に通して、200gで5分間遠心分離により2回洗浄し、20mlの無血清RPMI中にペレットを再び分散させた。 3匹の無処置Balb/cマウスから取り出した胸腺細胞を同様の方法で調製した。
【0111】
11%の胎児クローン(Fetalclone)(FBS)(Hyclone Laboratories, Inc., Logan, Utah)を含むRPMI中に、融合に先立って3日間、対数増殖期で保存しておいたNS-1骨髄腫細胞を、200gで5分間遠心分離し、ペレットを前述のパラグラフに記載したようにして2回洗浄した。 洗浄後、各細胞懸濁液を最終的に10mlの無血清RPMI容量となるように加え、20μlを1mlの無血清RPMIに1:50で希釈した。 各希釈液の20μlを取り出し、0.85%シラン(Gibco)中の0.4%トリパンブルー染色液の20μlと混合し、血球計算器(Baxter Healthcare Corp., Deerfield, Illinois)上に付して計数を行った。
【0112】
2×108個の脾臓細胞を4.0×107個のNS-1細胞と合わせ、遠心分離して上清をアスピレータで吸引した。 細胞ペレットは、チューブをタッピングすることにより移動させ、37℃のPEG1500(75mMのHepe pH8.0中50%)2mlを撹拌しながら1分間にわたって加え、次に14mlの無血清RPMIを7分間にわたって加えた。 更に16mlのRPMIを加え、細胞を200gで10分間遠心分離した。 上清を捨てた後、15%のFBS、100μMのヒポキサンチンナトリウム、0.4μMのアミノプテリン、16μMのチミジン(HAT)(Gibco)、25単位/mlのIL-6(Boehringer Mannheim)及び1.5×106の 胸腺細胞/mlを含む200mlのRPMIにペレットを再分散させた。 先ず、懸濁液を96のウェルの平底組織培養プレート10枚に、1ウェル当たり200μlで分配する前の2時間、T225フラスコ(Corning, United Kingdom)に37℃で2時間入れておいた。 プレート中の細胞は、融合後3、4、5日で、20Gの針を用いて各ウェルから約100μlを吸引することにより、そして、10単位/mlのIL-6を含み胸腺細胞を含まないことを除いて上述と同様の1ウェル当たり100μlの播種培地を加えることにより、栄養供給した。
【0113】
融合体を、先ずELISAによりスクリーニングした。 Immunone4プレート(Dynatech)を、組換え精製ヒトcGB-PDE酵素(50mMのカーボネート緩衝液pH9.6中、100ng/ウェル)で一晩、4℃にて被覆した。 このプレートを、0.05%のTween20(PBST)を含むPBSで3回洗浄した。 個々のハイブリドーマウェルからの上清を、酵素で被覆したウェルに加えた(50μl/ウェル)。 37℃で30分間インキュベート後、上述と同様に洗浄し、セイヨウワサビペルオキシダーゼ接合ヤギ抗-マウスIgG(fc)(Jackson ImmunoReserch, West Grove, Pennsylvania)をPBSTで1:3500に希釈したものを50μl加えた。 プレートを上述と同様にインキュベートし、PBSTで4回洗浄し、1mg/mlのオルトフェニレンジアミン(Sigma)、及び100mMのクエン酸塩溶液中の30%H2O2の0.1μ/mlからなる100μlの基質を加えた。 着色反応を5分で15%H2SO4のμlを加えて停止した。 プレートリーダ(Dynatech)にて、A490を読み取った。
【0114】
ウェルC5G、E4D、F1G、F9H、F11G、J4A、及びJ5Dを拾い、それぞれ102A、102B、102C、102D、102E、102F、及び102Gと名付け、RPMI、15%のFBS、100μMのヒポキサンチンナトリウム、16μMのチミジン(HAT)(Gibco)、および10単位/mlのIL-6に2回希釈することにより、2又は3回連続してクローニングを行った。 クローンプレートのウェルを、4日後には目視に評価し、最も密度の低いウェルのコロニーの数を記録した。 各クローニングの選択されたウェルを、ELISAによってテストした。
【0115】
上述のハイブリドーマによって生成したモノクロナール抗体を、ELISAアッセイによってアイソタイプ判定した。 その結果、モノクロナール抗体102Aから102EはIgG1、102FはIgG2b、102GはIgG2aであることが示された。
【0116】
ウェスタン分析によって判定したところ、7つの全てのモノクロナール抗体がヒトcGS-PDEと反応した。
【0117】
実施例12
特異的PDEの生物学的活性のモジュレーターを開発するためには、特定のアッセイの調製物に存在するPDEアイソザイムを識別することが必要である。 天然の組織源からPDEを単離し、新しいアイソザイムのそれぞれを研究する伝統的な酵素学的アプローチは、精製技術の限界と、アイソザイムの完全な分離が為されるかどうかを明確に評価するのが不可能であることによって阻まれている。 一つのアイソザイムの寄与に助力し、調製における他の寄与を最小限にするアッセイ条件を同定する他のアプローチもある。 更にまた、PDEの分離を免疫学的手段によって分離する他のアプローチもある。 先に述べたPDEアイソザイムを識別するアプローチのそれぞれは、時間を要し、技術的に困難である。 結果的に、選択的PDEモジュレーターを開発する多くの試みが、1以上のアイソザイム調製物を用いて為されてきた。 更に、天然組織源からのPDEの調製物は、限定されたタンパク分解に感受性が高く、そして全長のPDEとは異なる動力学、異なる調節、及び異なる物理学的性質を有する活性なタンパク分解産物の混合物を含んでいるかもしれない。
【0118】
本発明の組換えcGB-PDEポリペプチド産物は、新たなそして特異的cGB-PDEモジュレーターの開発を大いに促進する。 モジュレーターのスクリーニングのためのヒト組換え酵素の使用は、多くの固有の利点を有している。 内在性のホスホジエステラーゼ活性を欠いている宿主細胞(例えば、ATCC 74225として寄託した酵母菌株YKS45)において組換え法により発現させることによって、アイソザイムの精製の必要性を回避することができる。 ヒトタンパク質に対する化合物をスクリーニングすることで、非ヒトタンパクについて至適化された化合物がヒトタンパク質に対して特異的ではないか又はヒトタンパク質と反応しない場合に、非ヒトタンパク質に対するスクリーニングから起こることの多い複雑さを回避することができる。 例えば、ヒトと齧歯類5HT1Bセロトニンレセプタとの間の単一のアミノ酸の違いが、受容体に対する化合物の結合の差の原因となっている。(Oskenberg et al., Nature, 360:161-163(1992)を参照されたい)。 ひとたびcGB-PDEの活性をモジュレートする化合物が発見されると、その選択性は、cGB-PDEにおけるその活性を他のPDEアイソザイムに対するその活性と比較することにより評価され得る。 このように、一連の独立したアッセイでの、本発明の組換えcGB-PDE産物と他の組換えPDEとの組み合わせは、cGB-PDEの選択的モジュレーターを開発するシステムを提供する。 選択的モジュレーターには、例えば、抗体及び他のタンパク、又はcGB-PDE若しくはcGB-PDE核酸、又はオリゴヌクレオチドに特異的に結合するペプチドが含まれ得、上記オリゴヌクレオチドは、cGB-PDE(特許協力条約国際公開第WO 93/05182、1993年3月18日、これには、目的生物分子に選択的に結合するオリゴヌクレオチドを選択する方法が記載されている)に、又はcGB-PDE核酸(即ち、アンチセンスオリゴヌクレオチド)に、及びcGB-PDE若しくはcGB-PDE核酸に特異的に結合する他の非ペプチド天然若しくは合成化合物に、特異的に結合する。 cGB-PDEの酵素活性又は局在性を変化させるcGB-PDEの突然変異体も企図される。組換えcGB-PDE単独、およびモジュレーターとの組み合わせの結晶化、X線結晶学による原子構造の分析、並びにこれらのコンピュータモデリングは、非ペプチド選択的モジュレーターの設計と最適化に有用な方法である。 例えば、構造に基づくドラッグデザインの一般的総説である、Erickson et al., Ann. Rep. Med. Chem., 27:271-289(1992)を参照されたい。
【0119】
選択的モジュレーターの開発の目的は、例えば、以下のものを含んでいる:(1)他のタンパクと接触する、及び/又は細胞内でのcGB-PDEを局在化させるcGB-PDEの領域、(2)基質を結合するcGB-PDEの領域、(3)cGB-PDEのアロステリックなcGMP結合部位、(4)cGB-PDEの金属結合領域、(5)cGB-PDEのリン酸化部位(1以上)、ならびに(6)cGB-PDEサブユニットの2量化に関係するcGB-PDEの領域。
【産業上の利用可能性】
【0120】
このように、本発明によって明らかにされたDNAおよびアミノ酸配列は、cGB-PDEに対するcDNAの配列のみならず、cGB-PDEをコードするゲノムDNA配列、ならびにプロモーター、オペレーターなどのcGB-PDE発現制御調節配列を特定する。 また、ストリンジェント条件下で本発明のDNA配列を用いて行われるDNA/DNAハイブリダイゼーションを利用することで、cGB-PDEの対立変異体、cGB-PDEに特異的な生化学的および/または免疫学的特性を共有する、他の構造的に関連するタンパク質、ならびにcGB-PDEと相同な非ヒト種のタンパク質をコードするDNAの単離も可能となる。 本発明のポリヌクレオチドは、好適に標識された場合、cGB-PDEを合成する細胞の能力を検出するためのハイブリダイゼーションアッセイにおいて有用である。 本発明のポリヌクレオチドはまた、1または複数の疾病状態の根底にあるcGB-PDE遺伝子座における遺伝的変化を同定するために有用な診断法を提供する。 通常は、cGB-PDEを発現している細胞において、cGB-PDEの発現調節に関連のあるアンチセンスポリヌクレオチドが利用可能である。 また、本発明によって明らかにされたDNAおよびアミノ酸配列は、cGB-PDEの構造および機能のシステム分析ならびにcGB-PDEが相互作用するであろう分子の特定も可能とする。
【0121】
さらに、cGB-PDE活性を修飾する薬剤は、推定モジュレーターを組換えcGB-PDEを発現する真核細胞、好ましくは、内在性のサイクリックヌクレオチドホスホジエステラーゼ活性を欠いた真核細胞からの溶解物とインキュベートし、次いでcGB-PDEホスホジエステラーゼ活性に対する推定モジュレーターの効果を決定することによって同定できる。 cGB-PDEの活性を修飾する化合物の選択性は、cGB-PDEに対する当該化合物の活性を他のPDEアイソザイムに対する活性と比較することにより評価することができる。 cGB-PDE活性のモジュレーターは、広範囲の疾病および生理学的条件の処置において治療上有用である。
【図面の簡単な説明】
【0122】
【図1A】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第578位〜第674位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図1B】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第675位〜第764位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図1C】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第765位〜第812位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図2A】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第139位〜第287位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図2B】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第288位〜第411位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図2C】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第412位〜第526位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図3】種々のPDEアイソザイムにおいて認められる内的に相同なリピートのアラインメントを示す図である。
【図4】cGB-PDEのドメインの構成を示す模式図である。
【図5】ウシcGB-PDE配列でトランスフェクトされたCOS細胞から得た抽出物が保有するホスホジエステラーゼ活性に関する分析結果を示す棒グラフである。
【図6】ウシcGB-PDE配列でトランスフェクトされた細胞から得た抽出物について認められた、ホスホジエステラーゼ阻害剤の存在下でのcGMPホスホジエステラーゼ活性に関する分析結果を示すグラフである。
【図7】ウシcGB-PDE配列でトランスフェクトされたCOS細胞から得た抽出物について認められた、0.2mMのIBMX存在下でのcGMP結合活性に関する分析結果を示す棒グラフである。
【図8】ウシcGB-PDE配列でトランスフェクトされた細胞から得た抽出物について認められた、過剰の非標識cAMPまたはcGMPの存在下での[3H]cGMP結合活性に関する分析結果を示す棒グラフである。
【技術分野】
【0001】
本発明は一般に、cGB-PDEと名付けたサイクリックグアノシンモノホスフェート結合性、サイクリックグアノシンモノホスフェート特異的ホスホジエステラーゼに関し、さらに詳細にはcGB-PDEポリペプチドをコードする新規な精製および単離されたポリヌクレオチド、cGB-PDEポリヌクレオチドの組換え製造のための方法および材料、ならびにcGB-PDE活性のモジュレーターを同定するための方法に関する。
【背景技術】
【0002】
サイクリックグアノシンモノホスフェート(cGMP)およびサイクリックアデノシンモノホスフェート(cAMP)などの3'5'サイクリックヌクレオチドの対応するヌクレオシド5'モノホスフェートへの加水分解を触媒するサイクリックヌクレオチドホスホジエステラーゼ(PDE)は、酵素の複合ファミリーを構成している。 サイクリックヌクレオチドの細胞内濃度を媒介することによって、サイクリックヌクレオチドセカンドメッセンジャーを含むシグナルトランスダクションにおいてPDEアイソザイムは機能する。
【0003】
異なる組織供給源から種々のPDEが単離され、今日までに特徴付けされたPDEの多くは、物理化学的性質、基質特異性、阻害剤に対する感度、免疫学的反応性および調節の態様を含む生物学的性質において差異を呈する。 (ビーボ(Beavo)ら、サイクリック・ヌクレオチド・ホスホジエステラーゼズ:ストラクチャー、レギュレーション・アンド・ドラッグ・アクション(Cyclic Nucleotide Phosphodiesterases: Structure, Regulation and Drug Action)、ジョーン・ウィレイ・アンド・サンズ(John Wiley & Sons)、チチェスター(Chichester)、英国)を参照されたい)種々のPDEの既知のアミノ酸配列の比較により、ほとんどのPDEが別の触媒および調節ドメインを有するキメラ性マルチドメインタンパク質であることが示される。 (シャーボンニュー(Charbonneau)、ビーボら、前出、267〜296頁を参照されたい)今日までに特徴付けがなされたすべての哺乳動物のPDEは、触媒部位を含んでなると考えられ、酵素のカルボキシル末端領域に位置する長さおよそ250アミノ酸残基の配列を共有している。 アロステリック分子または調節分子と相互作用するPDEドメインは、アイソザイムのアミノ末端領域に位置すると思われる。 それらの生物学的性質に基づき、PDEは以下の6つの概括的なファミリーに分類され得る:Ca2+/カルモジュリンで刺激されるPDE(I型)、cGMPで刺激されるPDE(II型)、cGMPで阻害されるPDE(III型)、cAMP特異的PDE(IV型)、本発明の主題であるcGMP特異的ホスホジエステラーゼcGB-PDE(V型)およびcGMP特異的フォトレセプターPDE(VI型)。 cGMP結合性PDE(II型、V型およびVI型PDE)は、それらのカルボキシル末端近傍に相同な触媒ドメインを有するに加えて、それらのアミノ末端により近く位置し、アロステリックなcGMP結合ドメインを含み得る第2の保存された配列を有する。 シャーボンニューら、プロシーディング・オブ・ナショナル・アカデミー・オブ・サイエンス・ユー・エス・エー(Proc. Natl. Acad. Sci. USA)、87巻、288〜292頁(1990年)を参照されたい。
【0004】
II型のcGMPで刺激されるPDE(cGs-PDE)は、異なる組織のタイプに広く分布し、100〜105kDaのサブユニットのホモダイマーとして存在すると考えられる。 cGs-PDEは生理的条件下においてcAMP加水分解の速度を高めることによって上昇したcGMP濃度に応答する。 ウシ心臓cGs-PDEのアミノ酸配列およびウシ副腎cGs-PDEの部分cDNA配列はレトロング(LeTrong)ら、Biochemistry、29巻、10280〜10288頁(1990)に報告され、さらにウシ副腎およびヒト胎児脳cGB-PDEの全長のcDNA配列が1992年10月29日公開のPCT国際公開第 WO92/18541号に記載されている。 ウシ副腎の全長のcDNA配列はソネンバーグ(Sonnenburg)ら、J. Biol. Chem.、266巻、17655〜17661頁(1991)にも記載されている。
【0005】
フォトレセプターPDEおよびcGB-PDEはcAMPよりもcGMPに対して、加水分解について50倍またはそれより多くの選択性を呈するので、cGMP特異的なPDEとして記載されている。
【0006】
フォトレセプターPDEは、桿状体外部セグメントPDE(ROS-PDE)および円錐体(cone)PDEである。 ROS-PDEのホロ酵素構造は、いずれも触媒的に活性を有する2つの大きなサブユニットα(88kDa)およびβ(84kDa)ならびに2つのもっと小さなγ調節サブユニット(いずれも11kDa)からなる。 α、β、およびγサブユニットならびにCOS-PDEの15kDaサブユニットと同じであるらしいδサブユニット(15kDa)を含むROS-PDEの可溶性型もまた同定されている。 ウシの膜会合性ROS-PDE α−サブユニットに対応する全長のcDNAは、オブチニコフ(Ovchinnikov)ら、FEBS Lett.、223巻169〜173頁(1987)に記載されており、ウシ桿状体外部セグメントPDE β−サブユニットに対応する全長のcDNAは、リプキン(Lipkin)ら、J. Biol. Chem.、265巻、12955〜12959頁(1990)に記載されている。 オブチニコフら、FEBS Latt.、204巻、169〜173頁(1986)は、ウシROS-PDE γ−サブユニットに対応する全長のcDNAおよびそのδサブユニットのアミノ酸配列を示している。 ROS-PDEの発現が、脳においてもコリンズ(Collins)ら、ゲノミックス(Genomics)、13巻、698〜704頁(1992)にて報告されている。 COS-PDEは、2つの同一のα'(94kDA)サブユニットならびに11kDa、13kDaおよび15kDaの3つのもっと小さなサブユニットで構成される。 ウシCOS-PDE α'-サブユニットに対応する全長のcDNAは、リー(Li)ら、Proc. Natl. Acad. Sci. USA、87巻、293〜297頁(1990)に報告されている。
【0007】
cGB-PDEは、ラット(フランシス(Francis)ら、メソッズ・イン・エンザイモロジー(Methods Enzymol.)、159巻、722〜729頁(1988))およびウシ肺組織(トーマス(Thomas)ら、J. Biol. Chem.、265巻14964〜14970頁(1990)、以下「トーマスI」と記載)から均質に精製されている。 この酵素または類似の酵素の存在が、ラットならびにヒトの血小板(ハメット(Hamet)ら、Adv. Cyclic Nucleotide Protein Phosphorylation Res.、16巻、119〜136頁(1984))、ラット脾臓(コンクイル(Conquil)ら、Biochem. Biophys.Res.Commun.、127巻、226〜231頁(1985))、モルモット肺(デイビス(Davis)ら、J. Biol. Chem.、252巻4078〜4084頁(1977))、管平滑筋(コンクイルら、Biocehm.Biophys.Acta、61巻、148〜165頁(1980))、およびウニ精子(フランシスら、J.Biol.Chem.、255巻、620〜626頁(1979))を含めた種々の組織および種において報告されている。 cGB-PDEは2つの93kDaサブユニットからなるホモダイマーかもしれない。 (トーマスI、前出、参照)cGB-PDEは、cGMP依存性プロテインキナーゼ(cGK)によりリン酸化され、より低い親和性でcAMP依存性プロテインキナーゼによりリン酸化される、他の既知cGMP結合性PDEにおいては見出されない単一の部位を含むことが示されている。 (トーマスら、J. Biol. Chem.、265巻、14971〜14978頁(1990)、以下「トーマスII」と記載、参照)当該リン酸化部位およびcGB-PDEのキモトリプシン消化により作製された断片のアミノ末端部の一次アミノ酸配列は、トーマスII、前出およびトーマスI、前出にそれぞれ記載されている。 しかしながら、cGB-PDEのアミノ酸配列の大半はこれまでに報告されていない。
【0008】
異なる型のPDEの阻害剤が、文献に記載されている。 V型PDEにいくらか特異性を呈する2つの阻害剤は、ザプリナスト(zaprinast)およびジピリダモル(dipyridamole)である。 ビーボら、前出中、117〜140頁、フランシスら、を参照のこと。
【0009】
cGB-PDEをコードするDNAおよびアミノ酸配列の解明ならびに組換え法によるcGB-PDEポリペプチドの製造により、cGB-PDEの活性を選択的に修飾する新規物質の同定を可能ならしめる情報および材料が提供されるであろう。 PDEアイソザイムの別々の型またはファミリーがあること、および異なる組織が異なるPDEの異なる補体を発現することが認められることが、セカンドメッセンジャーとしてサイクリックヌクレオチドを用いるシグナルトランスダクション経路に関連する病状に対する治療上の示唆を有し得るPDEモジュレーターの開発における興味をもたらしている。 PDE活性の種々の選択的および非選択的阻害剤が、ムレイ(Murray)ら、Biochem. Soc. Trans.、202(2)巻、460〜464頁(1992)において議論されている。 組換えDNA技術による、特異的なPDE産生能をもたないPDEモジュレーターの開発は、あらゆるPDEが同じ基本的な反応を触媒し、重複する基質特異性を有し、且つ微量のみしか生じないために困難である。 結果的に、多くのPDEを均質にまで精製するのは冗長で困難な過程である。
【発明の開示】
【発明が解決しようとする課題】
【0010】
かくしてcGB-PDEに対するDNAおよびアミノ酸配列の情報、cGB-PDEポリペプチドの組換え製造のための方法および材料ならびにcGB-PDE活性の特異的なモジュレーターを同定するための方法の必要性が、当該技術において現存し続けている。
【課題を解決するための手段および発明の効果】
【0011】
本発明は、cGB-PDEと称されるcGMP結合性、cGMP特異的PDEをコードする新規な精製され単離されたポリヌクレオチド(例えば、センスおよびアンチセンスストランドのいずれものDNA配列およびRNA転写産物、それらのスプライス変異体も含めて)を提供する。 本発明の好ましいDNA配列は、ゲノムおよびcDNA配列のみならず全体的または部分的に化学的に合成されたDNA配列を含む。 配列番号:9または20で示されるcGB-PDEをコードするDNA配列およびストリンジェント条件下で当該配列とハイブリダイズするDNA配列または遺伝コードの縮重なしに当該配列とハイブリダイズするであろうDNA配列が、本発明により企図される。 本発明により、本発明のDNA配列の生物学的複製物(例えば、生体内または生体外で作製された、単離されたDNA配列)もまた企図される。 cGB-PDE配列を組込んだプラスミドおよびウイルスDNAベクター、ならびに、特にcGB-PDEをコードするDNAが内在または外来の発現制御DNA配列および転写ターミネーターなどに作動可能に連結されたベクターなどの、自律的に複製する組換え構築体もまた提供される。 特に例証となる本発明の発現プラスミドは、アメリカン・タイプ・カルチャー・コレクション(ATCC)、12301, Parklawn Drive Rockville, Maryland 20852に、1993年5月4日に受託番号69296として寄託された大腸菌株JM109中のプラスミドhcgbmet156-2 6nである。
【0012】
本発明の他の特徴において、原核細胞および真核細胞を含む宿主細胞が、その細胞内で所望のペプチドが発現されることを許容する方法で本発明のDNA配列を用いて形質転換される。 cGB-PDE産物を発現している宿主細胞は、種々の有用な目的で役立つことができる。 このような細胞は、cGB-PDEと特異的に免疫反応性を有する抗体物質を作製するための免疫源用の貴重な供給源を構成する。 本発明の宿主細胞は、cGB-PDEポリペプチドの大量産生のための方法において顕著に有用である。 この方法において、細胞が好適な培養培地中で生育され、当該細胞からまたは当該細胞が生育された培地から、たとえばイムノアフィニティー精製により所望のポリペプチド産物が単離される。
【0013】
cGB-PDE産物は、天然細胞供給源から単離物として得られてもよく、または化学的に合成されてもよいが、好ましくは本発明の宿主細胞を包含する組換え手法により製造される。
【0014】
哺乳動物宿主細胞を用いることで、本発明の組換え発現産物に至適な生物学的活性を付与するために必要とされ得るような転写後の修飾(例えば、グリコシレーション、トランケーション、リピデーションおよびチロシン、セリンまたはスレオニンのリン酸化)を提供することが期待される。 本発明のcGB-PDE産物は、全長のポリペプチド、断片または変異体であってよい。 変異体は、(1)cGB-PDEに特異的な生物学的活性または免疫学的性質の1以上を喪失することなく;または(2)cGB-PDEの特定の生物学的活性を特に損なわせた:1以上の特定の(すなわち、天然にコードされた)アミノ酸が欠失されたもしくは置換された、または1以上の特定されないアミノ酸が付加された、cGB-PDEポリペプチド類似体からなり得る。
【0015】
本発明にさらに包含されるのは、抗体物質(例えば、モノクローナルおよびポリクローナル抗体、単鎖抗体、キメラ抗体、CDR-移植(grafted)抗体など)ならびにcGB-PDEに特異的な他の結合タンパク質である。 特異的な結合タンパク質は、単離されたもしくは組換えcGB-PDEまたはcGB-PDE変異体もしくはこのような産物を発現している細胞を用いて作製することができる。 結合タンパク質はさらに、免疫のためのみならず、cGB-PDEポリペプチドの精製ならびに既知の免疫学的手法による体液および組織試料におけるcGB-PDEポリペプチドの検出または定量のための組成物においても有用である。 それらはまた、cGB-PDEの生化学的活性、特にシグナルトランスダクションに関連するそれらの活性の修飾(すなわち、ブロッキング、阻害または促進)において極めて有用である。 抗cGB-PDE抗体物質に対して特異的な抗イデオタイプ抗体も企図される。
【0016】
本発明のDNAおよびアミノ酸配列の開示を通して与えられる情報の科学的な価値が明示される。 一連の例として、cGB-PDEに対するcDNAの配列を知ることで、DNA/DNAハイブリダイゼーションによって、cGB-PDEをコードするゲノムDNA配列を単離すること、ならびにプロモーター、オペレーターなどのcGB-PDE発現制御調節配列を特定することが可能となる。 ストリンジェント条件下で本発明のDNA配列を用いて行われるDNA/DNAハイブリダイゼーションにより、cGB-PDEの対立変異体、cGB-PDEに特異的な生化学的および/または免疫学的特性を共有する、他の構造的に関連するタンパク質、ならびにcGB-PDEと相同な非ヒト種のタンパク質をコードするDNAの単離を可能とすることが期待される。 本発明のポリヌクレオチドは、好適に標識された場合、cGB-PDEを合成する細胞の能力を検出するためのハイブリダイゼーションアッセイにおいて有用である。 本発明のポリヌクレオチドはまた、1または複数の疾病状態の根底にあるcGB-PDE遺伝子座における遺伝的変化を同定するために有用な診断法の基礎となるかもしれない。 本発明により利用可能となるのはまた、通常はcGB-PDEを発現している細胞による、cGB-PDEの発現調節に関連のあるアンチセンスポリヌクレオチドである。
【0017】
本発明によって提供されるDNAおよびアミノ酸配列情報はまた、cGB-PDEの構造および機能のシステム分析ならびにcGB-PDEが相互作用するであろう分子の定義付けをも可能とする。 cGB-PDE活性を修飾する薬剤は、推定モジュレーターを組換えcGB-PDEを発現する真核細胞からの溶解物とインキュベートし、次いでcGB-PDEホスホジエステラーゼ活性に対する推定モジュレーターの効果を決定することによって同定され得る。 好ましい実施態様において、真核細胞は内在性のサイクリックヌクレオチドホスホジエステラーゼ活性を欠くものである。 特に例証となるこのような真核細胞は、受託番号74225として1993年5月19日にATCCに寄託された酵母株YKS45である。 cGB-PDEの活性を修飾する化合物の選択性は、cGB-PDEに対する当該化合物の活性を他のPDEアイソザイムに対する活性と比較することにより評価することができる。 一連の独立した分析における、本発明の組換えcGB-PDE産物と他の組換えPDE産物との組合せによって、cGB-PDEの選択的なモジュレーターを開発するためのシステムが提供される。
【0018】
選択的モジュレーターには、たとえば、cGB-PDEまたはcGB-PDE核酸に選択的に結合する抗体および他のタンパク質またはペプチド、cGB-PDEまたはcGB-PDE核酸に選択的に結合するオリゴヌクレオチドならびにcGB-PDEまたはcGB-PDE核酸に選択的に結合する他の非ペプチド化合物(たとえば、単離された、または合成の有機分子)が含まれ得る。 野生型cGB-PDEの酵素活性または細胞局在に影響を及ぼすcGB-PDEの変異型もまた、本発明により企図される。 選択的なモジュレーターの開発のための好ましい標的には、たとえば、(1)他のタンパク質と接触し、および/または細胞内にてcGB-PDEが局在するcGB-PDEの領域、(2)基質が結合するcGB-PDEの領域、(3)cGB-PDEの1または複数のアロステリックなcGMP結合部位、(4)cGB-PDEの1または複数のリン酸化部位並びに(5)cGB-PDEサブユニットのダイマー形成に関与するcGB-PDEの領域が含まれる。 cGB-PDE活性のモジュレーターは、広範囲の疾病および生理学的条件の処置において治療上有用であるかもしれない。
【発明を実施するための最良の形態】
【0019】
本発明を、以下の実施例に従って詳細に説明する。
【0020】
実施例1では、PCRによるウシcGB-PDE cDNA断片の単離および引続いてそのPCR断片をプローブとして用いた全長のcGB-PDEcDNAの単離を記載する。 実施例2は、種々の他のPDEに対して報告された配列と、ウシcGB-PDEアミノ酸配列の関係の分析を示す。 種々のウシ組織中のcGB-PDE mRNAのノザンブロット分析を、実施例3に示す。 COS細胞におけるウシcGB-PDE cDNAの発現を、実施例4に記載する。 実施例5には、cGB-PDE COS細胞発現産物のホスホジエステラーゼ活性、cGMP結合活性およびZn2+加水分解酵素についての分析の結果を示す。 実施例6は、ウシcGB-PDE cDNAと相同性を有するヒトcDNAの単離を記載する。 酵母細胞内でのヒトcGB-PDE cDNAの発現を、実施例7に示す。 ヒト組織におけるcGB-PDEを検出するためのRNaseプロテクションアッセイを、実施例8に記載する。 実施例9には、ヒトcGB-PDE cDNAの細菌における発現および当該細菌のcGB-PDE発現産物と反応性を有する抗体の作製を記載する。 実施例10には、cGB-PDE類似体と断片を記載する。 cGB-PDEを認識するモノクローナル抗体の作製を、実施例11に記載する。 実施例12は、cGB-PDEの生物学的活性を選択的に修飾する薬剤を開発するための本発明の組換えcGB-PDE産物の利用に関する。
【実施例】
【0021】
実施例1
ポリメラーゼ連鎖反応(PCR)を、ウシ肺第1ストランドから、cGB-PDEの一部をコードするcDNA断片を単離するために利用した。 トーマスI、前出に記載の部分的なcGB-PDEアミノ酸配列および新規な部分アミノ酸配列情報に基づいて、充分に縮重したセンスおよびアンチセンスPCRプライマーを設計した。
【0022】
A.cGB-PDEタンパク質の精製
cGB-PDEは、トーマスI、前出に記載のように、またはその方法を下記のように変更することによって精製した。
【0023】
新鮮なウシ肺(5〜10kg)を屠殺場から得、直ちに氷上に置いた。 組織を細切し、冷PEM緩衝液(2mM EDTAおよび25mM β−メルカプトエタノールを含む20mMリン酸ナトリウム、pH6.8)と合わせた。 ホモジナイズして遠心した後、得られた上清を4〜7リットルのDEAE-セルロース(ワットマン(Whatman)、英国)と3〜4時間インキュベートした。次いでそのDEAEスラリーを真空下で濾過し、冷PEMを大量に用いて濯いだ。 その樹脂をガラスカラムに注ぎ、3〜4倍容量のPEMを用いて洗浄した。タンパク質は、100mM NaClを含むPEMを用いて溶出し、12の1リットル画分を集めた。 画分を、トーマスら、前出に記載された標準法によってIBMX-刺激cGMP結合およびcGMPホスホジエステラーゼ活性について分析した。 適切な画分をプールし、冷脱イオン水を用いて2倍に希釈し、次いでブルーセファロース(登録商標)CL-6B(ファルマシア・エル・ケー・ビー・バイオテクノロジー(Phermacia LKB Biotechnology)社、ピスカタウェイ(Piscataway)、NJ)クロマトグラフィーに付した。 次にアガロースまたはセファロースベースのゲルマトリックスを用いて、亜鉛キレートアフィニティー吸収クロマトグラフィーを行った。 亜鉛キレート工程から得られたタンパク質のプールを、トーマスI、前出に記載のように処理するか、または変更を施した精製法に付した。
【0024】
トーマスI、前出に記載のようにタンパク質のプールは、4℃でPEM中に平衡化したHPLC Bio-Sil TSK-545 DEAEカラム(150×21.5mm)(バイオラッド・ラボラトリーズ(BioRad Laboratories)、ハーカルズ(Hercules)、CA)を複数回付した。 平衡化の後、50mM NaCLを含むPEM 120-mlで洗浄し、続いて2ml/分の流速にて120-mlの直線濃度勾配(50〜200mM NaClを含むPEM)での溶出を行った。 適切な画分をプールし、透析チューブ中でセファデックスG-200(ベーリンガー・マンハイム・バイオケミカルズ(Boehringer Mannheim Biochemicals)、英国)に対して、1.5mlの最終容量までに濃縮した。 濃縮されたcGB-PDEプールは、100mMリン酸ナトリウム、pH 6.8、2mM EDTA、25mM β−メルカプトエタノール中に平衡化したHPLCゲル濾過カラム(Bio-Sil TSK-250、500×21.5mm)に供し、4℃で2ml/分の流速にて溶出した。
【0025】
より煩わしくない変法を行う場合は、タンパク質プールをPEMに対して2時間透析し、PEM緩衝液中に平衡化した10mlの調製用DEAEセファセルカラム(ファルマシア)にかけた。タンパク質を0.5M NaClを含むPEMを用いてバッチ法にて溶出し、およそ10〜15倍濃度のタンパク質を得た。 濃縮されたタンパク質試料を、0.1M NaClを含むPEM中に平衡化した800ml(2.5cm×154cm)のセファクリルS400ゲル濾過カラム(ベーリンガー)にかけ、1.7ml/分の流速で溶出した。
【0026】
タンパク質の純度は、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)の後、クマシー染色により評価した。 ウシ肺10kg当たりおよそ0.5〜3.0mgの純粋なcGB-PDEが得られた。
【0027】
精製されたウシcGB-PDEに特異的なウサギポリクローナル抗体を、標準法によって作製した。
【0028】
B.cGB-PDEのアミノ酸配列決定
cGB-PDEは、[32P]ATPを用いてリン酸化し、次いで32Pで標識されたホスホペプチドを得るため、プロテアーゼで消化した。 およそ100μgの精製されたcGB-PDEを、9mM MgCl2、9μM[32P]ATP、10μM cGMP、および4.2μgのcAMP依存性プロテインキナーゼ(cAK)の、精製されたウシの触媒能を有するサブユニットを含む反応混液(900μlの最終容量)中でリン酸化した。 cAKの触媒能を有するサブユニットは、マランゴス(Marangos)ら、Brain Receptor Methodologies, Part A、Academic Press、Orlando、フロリダ(1984)、209〜215頁、フロックハート(Flockhart)らの方法に従って調製した。 反応は30℃にて30分インキュベートを行い、60μlの200mM EDTAを添加することによって停止した。
【0029】
cGB-PDEから第1のペプチド配列を得るために、KPE緩衝液中のα−キモトリプシン1mg/ml溶液(2mM EDTAを含む10mMリン酸ナトリウム、pH 6.8)3.7μlを、100μgの精製されリン酸化されたcGB-PDEに添加し、混液を30℃にて30分インキュベートした。 タンパク質分解は、50μlの10% SDSおよび25μlのβ−メルカプトエタノールを添加することで停止した。 容量が400μlより少なく減じられるまで試料を沸騰させ、8%の調製用SDS-ポリアクリルアミドゲルにかけ、50mAmpにて電気泳動に付した。 分離された消化産物は、マツダイラ(Matsudaira)、J. Biol. Chem.、262巻、10035〜10038頁(1987)の方法に従ってイモビロン・ポリビニリデン・ジフルオリド(ミリポア、ベドフォード、MA)上に電気的にブロットした。 転写したタンパク質は、クマシーブルー染色によって同定し、自動ガス相アミノ酸配列決定用に、50kDaのバンドを膜から切り出した。 α−キモトリプシン消化の手順により得られたペプチドの配列は、以下の配列番号:1に示す。
【0030】
配列番号:1 REXDANRINYMYAQYVKNTM
第2の配列は、V8タンパク分解により作製されたcGB-PDEペプチド断片から得られた。
【0031】
およそ200μgの精製されたペプチド断片を、10mM MgCl2、10μM[32P]ATP、100μM cGMP、および1μg/mlの精製されたcAKの、最終容量1.4ml中に添加した。 反応は、30℃にて30分インキュベートを行い、160μlの0.2M EDTAを添加することによって停止した。次に、KPE中に希釈した9μlの1mg/mlのStaphylococcal aureus V8プロテアーゼ(インターナショナル・ケミカル・ヌクレアー・バイオメディカルズ(International Chemical Nuclear Biomedicals)、コスタ・メサ(Costa Mesa)、CA)を添加し、続いて30℃にて15分インキュベートした。 タンパク質分解は、88μlの10%SDSおよび45μlのβ−メルカプトエタノールを添加することにより停止した。 消化産物は、25mAmpで4.5時間行った調製用10% SDSポリアクリルアミドゲルの電気泳動により分離した。 タンパク質は電気的にブロットし、前記したように染色した。 28kDaのタンパク質のバンドを膜から切り出し、自動ガス相アミノ酸配列決定に付した。 得られた配列を、以下の配列番号:2に示す。
【0032】
配列番号:2 QSLAAAVVP
C.ウシcDNAのPCR増幅
プライマーを設計するために用いられる部分アミノ酸配列(配列番号:3、下記、配列番号:1のアミノ酸9〜20)ならびに対応するPCRプライマー(IUPAC命名法で)に対応する配列を以下に示す。 ここで配列番号:3は、トーマスI、前出において報告された配列である。
【0033】
配列番号:3 F D N D E G E Q
5’TTY GAY AAY GAY GAR GGN GAR CA 3’(配列番号:4)
3’AAR CTR TTR CTR CTY CCN CTY GT 5’(配列番号:5)
配列番号:1、アミノ酸9〜20
N Y M Y A Q Y V K N T M
5’AAY TAY ATG TAY GCN CAR TAY GT3’(配列番号:6)
3’TTR ATR TAC ATR CGN GTY ATR CA5’(配列番号:7)
3’TTR ATR TAC ATR CGN GTY ATR CAN TTY TTR TGN TAC5’(配列番号:8)
アプライド・バイオシステムズ(Applied Biosystems)モデル380Aシンセサイザー(フォスター・シティー(Foster City)、CA)を用いて合成されたセンスおよびアンチセンスプライマーを、下記のようにウシ肺第1ストランドcDNAからcGB-PDE特異的な配列を増幅するために可能なあらゆる組合せで用いた。
【0034】
エタノール沈殿の後、オリゴヌクレオチドの対を1つのPCR反応においてそれぞれ400nMで組合わせた(配列番号:4または5を配列番号:6、7または8と組合わせた)。 50ngのウシ肺第1ストランドのcDNA(オリゴdTで選択したウシ肺mRNAについてAMV逆転写酵素およびランダムプライマーを用いて作製した)、200μM dNTP、および2単位のTaqポリメラーゼを用いて反応を行った。 初めの変性工程は、94℃にて5分間行い、引き続き94℃における1分の変性工程、50℃における2分のアニーリング工程、72℃における2分の伸長(extension)工程を30サイクル行った。 PCRは、Hybaid Thermal Reactor (ENK Scientific Products, Saratoga, CA)を用いて実施し、産物を40mMトリス−酢酸、2mM EDTAで流した1%低融点アガロースゲルでのゲル電気泳動により分離した。 約800〜840bpの弱いバンドが、配列番号:4および7で示されるプライマーを用いた場合および配列番号:4および8で示されるプライマーを用いた場合に認められた。 配列番号:4および7で示されるプライマーを用いた増幅により作製されるPCR産物を、ジーン・クリーン(Gene Clean(登録商標))(Bio101、ラ・ヨラ(La jolla)、CA)DNA精製キットを用い、製造業者のプロトコルに従って単離した。 PCR産物(20ng)は、200ngの直鎖状としたpBluescript KS(+)(ストラタジーン、ラ・ヨラ、CA)へと連結し、そして得られたプラスミド構築体を、大腸菌XL1 Blue Cell(ストラタジーン・クローニング・システムズ、ラ・ヨラ、CA)を形質転換するために用いた。 形質転換陽性と推定されるものを、配列決定によりスクリーニングした。 得られた配列は、既知のいかなる配列ともまたは既知のcGB-PDE部分配列とも相同でなかった。
【0035】
配列番号:4および7で示されるプライマーを用いて、ウシ肺の第1ストランドcDNAについて再びPCRを行った。 単一の大きなオープンリーディングフレームを有する0.8Kbのインサートを含むクローンが同定された。 オープンリーディングフレームは、アミノ酸KNTM(配列番号:7で示されるプライマー配列を設計するのには用いられなかった配列番号:1のアミノ酸17〜200)を含み、かつcGs-、ROS-およびCOS-PDEの演繹されたアミノ酸配列と高い程度の相同性をもつポリペプチドをコードしていた。 同定されたクローンは、配列番号:9のヌクレオチド489〜1312に対応する。
【0036】
D.ウシcDNAライブラリーの構築およびハイブリダイゼーションスクリーニング
全長のcGB-PDEをコードするcDNAを得るために、ウシ肺cDNAライブラリーを、プローブとして32Pで標識されたPCRで作製されたcDNAインサートを用いてスクリーニングした。
【0037】
ポリアデニル化RNAは、ソネンバーグ(Sonnenburg)ら、J. Biol. Chem.、266巻、17655〜17661頁(1991)に記載のとおりにウシ肺から調製した。 第1ストランドcDNAは、アウスベル(Ausubel)ら、Current Protocols in Molecular Biology, John Wiley & Sons、ニューヨーク(1987)に記載のとおりにランダムヘキサヌクレオチドプライマーとAMV逆転写酵素(ライフ・サイエンシズ(Life Sciences)、St. Petersberg、FL)を用いて合成した。 第2ストランドcDNAは、大腸菌DNAリガーゼおよび大腸菌RNAse Hの存在下で大腸菌DNAポリメラーゼIを用いて合成した。 500bpより大きいcDNAの選択は、セファロース(登録商標)CL-4B(ミリポア)クロマトグラフィーによって行った。 EcoRIアダプター(プロメガ(Promega)、マディソン(Madison)、WI)を、T4 DNAリガーゼを用いて当該cDNAに連結した。 熱によるリガーゼの不活性化に続いて、T4ポリヌクレオチドキナーゼを用いてcDNAをリン酸化した。 連結されなかったアダプターは、セファロース(登録商標)CL-4Bクロマトグラフィー(ファルマシア(Pharmacia)、ピスカタウェイ(Piscataway)、NJ)によって除去した。 cDNAはEcoRIで消化し、脱リン酸化したラムダZap(登録商標)IIアーム(ストラタジーン)へと連結し、製造業者のプロトコルに従ってGigapack(登録商標)Gold(ストラタジーン)を用いてパッケージングした。 未増幅のライブラリーのタイターは、18%の非組換え体で、9.9×105であった。 ライブラリーは、20の150mmプレートに対して50,000プラーク形成単位(pfu)を播くことにより増幅し、最終的なタイターとして21%の非組換え体で5.95×106pfu/mlが得られた。
【0038】
ライブラリーを50,000 pfu/mlで24枚の150mmプレートに播き、32Pで標識したcDNAクローンを用いてスクリーニングした。 プローブはファインバーグ(Feinberg)ら、Anal.Biochem.、137巻、266〜267頁(1984)の方法を用いて調製し、32Pで標識されたDNAはElutip-D(登録商標)カラム(シュライヒャー・アンド・シュエル(Schleicher and Schuell)社、キーン(Keene)、NH)を用い、製造業者のプロトコルを用いて精製した。 プラーク−リフトは、15cmニトロセルロースフィルターを用いて行った。 変性および中和に続いて、80℃にて2時間ベーキングすることにより、DNAをフィルター上に固定した。 ハイブリダイゼーションは、50%ホルムアミド、5×SSC(0.75M NaCl、0.75M クエン酸ナトリウム、pH7)、25mM リン酸ナトリウム(pH7.0)、2×デンハーツ溶液、10%デキストラン硫酸、90μg/ml酵母tRNA、およびおよそ106cpm/mlの32Pで標識されたプローブ(5×108cpm/μg)を含む溶液中で、42℃にて一晩行った。 フィルターは、1回の洗浄について15分間、室温にて0.1×SSC、1% SDS中で2回洗浄し、続いて45℃にて0.1×SSC、1%SDS中で20分間1度、洗浄した。 次にフィルターを−70℃で、数日間X線フィルムに露光した。
【0039】
標識したプローブとハイブリダイズしたプラークを再度播種し、再度のスクリーニングを数回行うことによって精製した。 cDNAインサートは、製造業者のプロトコルにより記載されたインビボの切出し方法によってpBluescript SK(-)ベクター(ストラタジーン)の中にサブクローン化した。 救出されたcDNAがPCRプローブとハイブリダイズすることを明らかにするため、サザンブロットを行った。 推定されるcGB-PDE cDNAを、Sequanse(登録商標)バージョン2.0(ユナイテッド・ステイツ・バイオケミカル・コーポレーション(United States Biochemical Corporation)、クリーブランド、オハイオ)またはTaqTrack(登録商標)キット(プロメガ)を用いて配列決定した。
【0040】
cGB-2、cGB-8およびcGB-10と名付けた3つの異なるcDNAクローンを単離した。 クローンcGB-8のDNAおよび演繹されたアミノ酸配列を配列番号:9および10に示す。 ヌクレオチド2686の下流のDNA配列は、クローニングのアーティファクトを表すのかもしれない。 cGB-10のDNA配列は、1つのヌクレオチドを例外としてcGB-8の配列と同一である。クローンcGB-2のDNA配列は、クローンcGB-8の5'からクローンcgb-8(配列番号:9参照)のヌクレオチド219までの配列より分岐しており、異なるアミノ末端を有するタンパク質をコードし得た。
【0041】
cGB-8 cDNAクローンは長さ4474bpであり、2625bpの大きなオープンリーディングフレームを含む。 ヌクレオチド配列中、第99〜101位のトリプレットATGは、枠内終止コドンの前にあり、且つ周囲の塩基が真核細胞のmRNAに対するコザックのコンセンサス開始部位に適合するので、cGB-PDE遺伝子の翻訳開始部位であることが予測される。 終止コドンTAGは2724〜2726位に位置し、3'非翻訳配列の1748bpが続く。 cGB-8の配列は、転写終了コンセンサス配列を含まず、従ってクローンは、対応するmRNAの全体の3'非翻訳領域は示さないのかもしれない。
【0042】
cGB-8 cDNAのオープンリーディングフレームは、875アミノ酸のポリヌクレオチドをコードし、計算された分子量は99.5kDである。 この計算された分子量は、SDS-PAGE分析によると約93kDaであると見積もられた、精製されたcGB-PDEの分子量の報告値よりわずかに少しだけ大きい。 cGB-8の演繹されたアミノ酸配列は、精製されたウシ肺cGB-PDEから得られたすべてのペプチド配列に正確に対応しており、cGB-8がcGB-PDEをコードする強い証拠を提供するものである。
【0043】
実施例2
Genetics Computer Group(GCG)Software Package (Madison, Wisconsin)が提供しているFASTAプログラムを用いて行ったSWISS-PROTおよびGEnEmb1データバンク(1992年2月リリース)の検索により、他のPDEに対して報告されたDNAおよびアミノ酸配列のみがクローンcGB-8のDNAおよび演繹されたアミノ酸と有意な類似性を呈することが明らかになった。
【0044】
cGB-PDEの演繹されたアミノ酸配列を8つの他のPDEの配列とペアで比較することが、ALIGN(デイホフ(Dayhoff)ら、Methods Enzymol. 、92巻、524〜545頁(1983))およびBESTFIT(ウィルバー(Wilbur)ら、Proc.Natl.Acad.Sci.USA、80巻、726〜730頁(1983))プログラムを用いて行われた。 今日までに配列決定がなされたすべての哺乳動物ホスホジエステラーゼのように、cGB-PDEは、触媒活性に必須であると思われる、タンパク質のカルボキシル末端の半分のおよそ250アミノ酸の保存された触媒ドメイン配列を含んでいる。 このセグメントは、配列番号:9のアミノ酸578〜812を含んでなり、他のPDEの対応する領域と配列の保存性を呈する。 以下の表1に、他のPDEのcGB-PDEのアミノ酸578〜812とのペアでの比較において得られる特定の同一性値を示す。 ここで「ラットダンス(dunce)」はラットcAMP特異的PDEであり、「61kCaM」はウシ61kDaカルシウム/カルモジュリン依存性PDEであり、「63kCaM」はウシ63kDaカルシウム/カルモジュリン依存性PDEであり、「ドロスダンス」はドロソフィアcAMP特異的ダンスPDEであり、「ROS-α」はウシROS-PDE α−サブユニットであり、「ROS-β」はウシROS-PDE β−サブユニットであり、「COS-α」はウシCOS-PDE α’サブユニットであって、さらに「cGs」はウシcGs-PDEである(612〜844)。
【0045】
【表1】
【0046】
PILEUPプログラム(GCGソフトウェア)で実行される、Progressive Alignment Algorithm(フェン(Feng)ら、Methods Enzymol.、183巻、375〜387頁(1990))を用いて、複数の配列のアラインメントを行った。 図1A〜1Cに、cGB-PDEの提唱される触媒ドメインの、表1のPDEのすべての対応する領域との複数の配列アラインメントを示す。 28残基(図1Aから1Cの「保存された」線での1文字のアミノ酸略号により示される残基を参照されたい)が、触媒において機能的な役割を果たすと予測されるいくつかの保存されたヒスチジン残基を含めて、アイソザイム間において不変である。 シャーボンニューら、Proc. Natl. Acad USA、前出を参照されたい。 cGB-PDEの触媒ドメインは、他のPDEアイソザイムでの対応する領域よりROS-PDEおよびCOS-PDEの触媒ドメインに、より類似している。 フォトレセプターPDEとcGB-PDEとの間に、他のPDEが共有しないいくつかの保存領域がある。 フォトレセプターPDEとcGB-PDE配列において不変なこれらの領域におけるアミノ酸の位置は、図1A〜1Cの「保存された」線の星印により示される。 cGB-PDEならびにROS-およびCOS-PDEの間の相同性領域は、cAMP加水分解に比してcGMP加水分解に対する特異性を賦与すること、または特定の生理学的薬剤への感受性を賦与することにおいて重要な役割を果たすかもしれない。
【0047】
cGB-PDE、cGs-PDEおよびフォトレセプターPDE間の配列の類似性は、保存された触媒ドメインに限定されず、タンパク質のアミノ末端半分における非触媒cGMP結合ドメインにも含まれる。 cGB-PDE、cGs-PDEおよびフォトレセプターPDE間のアラインメントの至適化により、アミノ末端保存セグメントが配列番号:9のアミノ酸142〜526を含めて存在するかもしれないことが示される。 cGB-PDEの提唱されるcGMP結合ドメインと、フォトレセプターPDEおよびcGs-PDEの対応する領域との配列のペアでの分析によって、26〜28%の配列の同一性が明らかとなった。 当該提唱されるcGMP結合ドメインと、cGMP結合性PDEとの複数の配列とのアラインメントを図2A〜2Cに示し、ここで略号は表1に示したと同様である。 この非触媒ドメインにおける38の位置が、すべてのcGMP結合性PDEの間で不変であることがわかる(図2A〜2Cの「保存された」線での1文字のアミノ酸の略号により示される位置を参照されたい)。
【0048】
cGMP結合性PDEのcGMP結合ドメインは、2つの類似するが別異のサブユニット間またはサブユニット内のcGMP結合部位を形成するかもしれない、内的に相同性のあるリピートを含む。 図3に、cGMP結合PDEのリピートa(cGB-PDEのアミノ酸228〜311に対応する)およびb(cGB-PDEのアミノ酸410〜500に対応する)の複数の配列アラインメントを示す。7つの残基が、各AおよびB領域において不変である(図3の「保存された」線で1文字のアミノ酸の略号により示される残基を参照されたい)。 AおよびB領域において化学的に保存されている残基を、図3の「保存された」線で星印により示す。 cGB-PDEのcGMP類似体研究により、cGB-PDE上のサイクリックヌクレオチド結合部位とcGMPの2’OHとの間に水素結合が存在することが裏付けられる。
【0049】
cGB-PDEの3つの領域は、他のPDEアイソザイムと有意な配列の類似性を有していない。これらの領域には、触媒ドメインのカルボキシル末端のフランキング配列(アミノ酸812〜875)、cGMP結合ドメインと触媒ドメインを分かつ配列(アミノ酸527〜577)およびアミノ酸1〜141にわたるアミノ末端配列が含まれる。 cGKによるcGB-PDEのリン酸化の部位(配列番号:10の92位のセリン)は、この配列のアミノ末端領域に位置する。cGB-PDE上のアロステリック部位へのcGMPの結合が、そのリン酸化に必要とされる。
【0050】
前記した他のPDEアイソザイムとの比較に基づき提唱されるcGB-PDEのドメイン構造を図4に示す。 このドメイン構造は、ウシ肺から精製されたcGB-PDEの生化学的研究により裏付けられる。
【0051】
実施例3
種々のウシ組織におけるcGB-PDE mRNAの存在を、ノザンブロットハイブリダイゼーションにより実験した。
【0052】
ポリアデニル化RNAは、製造業者のプロトコルに従ってPoly(A)Quick(登録商標)mRNA精製キット(スタラタジーン(Stratagene)を用いて総RNA調製物から精製した。 RNA試料(5μg)を1.2%アガロース、6.7%ホルムアルデヒドゲル上にのせた。 電気泳動およびRNA転写は、ソネンバーグら、前出に以前報告されたとおりに実施した。 RNAブロットのプレハイブリダイゼーションは、50%ホルムアミド、5×SSC、25mMリン酸ナトリウム、pH7、2×デンハーツ溶液、10%デキストラン硫酸、および0.1mg/ml酵母tRNAを含む溶液中で45℃にて4時間行った。 ランダムヘキサヌクレオチド−プライマー−標識プローブ(5×108cpm/μg)は、AccIおよびSacIIを用いた消化により切出した実施例2の4.7kbのcGB-8 cDNAクローンを用いて、フェインバーグら、前出に記載のとおりに調製した。プローブを加熱変性し、プレハイブリダイゼーションに続いてブロッティングバッグの中に注入した(6×105cpm/ml)。 ノザンブロットは、45℃にて一晩ハイブリダイズし、続いて室温で2×SSC、0.1%SDSを用いて、15分間1回洗浄し、次いで45℃にて0.1×SSC、0.1%SDSを用いて20分間、3回洗浄した。 ブロットは−70℃で24時間、X線フィルムに露光した。 cGB-PDEプローブとハイブリダイズするRNAのサイズは、臭化エチジウムで染色し、UV光で目視化した、0.24〜9.5kbのRNAラダーを用いて評価した。
【0053】
32Pで標識されたcGB-PDE cDNAは、単一の6.8kbウシ肺RNA種とハイブリダイズした。 同一のサイズのmRNAが、ウシの気管、大動脈、腎臓および脾臓から単離されたポリアデニル化RNAにおいても検出された。
【0054】
実施例4
実施例2のクローンcGB-8におけるcGB-PDE cDNAをCOS-7細胞(ATCC CRL1651)内で発現させた。
【0055】
制限酵素XbaIを用いた消化に続いて、cGB-PDEの一部を単離した。 XbaIは、cGB-8インサートの5'末端の30bp上流に位置するpBluescriptポリリンカー配列における位置で、および当該cGB-8インサートの中の3359位で切断する。 この結果得られた3389bp断片(cGB-8の全体のコード領域を含む)を、次いで発現ベクターpCDM8(インビトロゲン(Invitrogen)、サンジエゴ、CA)の独特なXba8Iクローニング部位に連結した。 pCDM8プラスミドはサイトメガロウイルスプロモーターおよびエンハンサー、SV-40由来の複製の起点、ポリアデニル化シグナル、複製の原核性起点(pBR322由来)および原核性遺伝マーカー(supF)を含む4.5kbの真核性発現ベクターである。 大腸菌MC1061/C3細胞(インビトロゲン)を、得られた連結産物を用いて形質転換し、形質転換陽性のコロニーをPCRおよび制限酵素分析を用いて、cGB-8が正しい方向となっているかについてスクリーニングした。 正しい方向でcGB-8インサートを含む、得られた発現構築体をpCDM8-cGB-PDEと言及する。
【0056】
pCDM8-cGB-PDEのDNAはQiagen pack-500カラム(チャツウォース(Chatsworth)、CA)を用い、製造業者のプロトコルに従って大量のプラスミド調製物から精製した。 COS-7細胞は、10%ウシ胎児血清、50μg/mlペニシリンおよび50μg/mlストレプトマイシンを含むダルベッコ変法イーグル培地(Dulbecco's modified Eagle's medium)(DMEM)中で、37℃にて、給湿した5%CO2大気中で培養した。 トランスフェクションのおよそ24時間前に、集密な細胞の100mmディッシュを、もとの密度の4分の1または5分の1で再度播いた。 典型的なトランスフェクション実験において、細胞は137mM NaCl、2.7mM KCl、1.1mM リン酸カリウム、および8.1mMリン酸ナトリウム、pH7.2(PBS)を含む緩衝液で洗浄した。 次いで10% NuSerum(コラボレーティブ・バイオメディカル・プロダクツ(Collaborative Biomedical Products)、 ベドフォード、MA)を含む4〜5mlのDMEMを各プレートに添加した。 60μl TBS(トリス緩衝生理食塩水:25mM Tris-HCl(pH7.4)、137mM NaCl、5mM KCl、0.6mM Na2HPO4、0.7mM CaCl2、および0.5mM MgCl2)中、400μgのDEAE-デキストラン(ファルマシア)と混合した10μgのpCDM8-cGB-PDEのDNAまたはpCDM8ベクターDNAでのトランスフェクションを、各プレートに混合物を滴下により添加して行った。 細胞を37℃にて5%CO2で4時間インキュベートし、次いで10%ジメチルスルホキシド含有PBSで1分間処理した。 2分後、ジメチルスルホキシドを除去し、細胞をPBSで洗浄して完全培地中でインキュベートした。 48時間後、細胞のプレートあたり0.5〜1mlの冷ホモジナイズ緩衝液(40mM トリス−塩酸(pH7.5)、15mM ベンズアミジン、15mM β−メルカプトエタノール、0.7μg/ml ペプスタチンA、0.5μg/ml ロイペプチン、および5μM EDTA)に細胞を懸濁し、ダウンス(Dounce)ホモジナイザーを用いて破砕した。 その結果得られた全細胞抽出物を、ホスホジエステラーゼ活性、cGMP結合活性および総タンパク質濃度について以下の実施例5に記載するようにアッセイした。
【0057】
実施例5
実施例4のトランスフェクトされたCOS細胞の抽出物又はモックトランスフェクトされたCOS細胞の抽出物におけるホスホジエステラーゼ活性を、Martins et al., J. Biol. Chem.,257:1973-1979(1982)のcGs-PDEについて記載されているアッセイ手法を改良したものを用いて測定した。 細胞を採取し、トランスフェクション後48時間で抽出物を調製した。インキュベーション混合物は、全容量250μl中に、40mMのMOPS緩衝液(pH7)、0.8mMのEDTA、15mMの酢酸マグネシウム、2mg/mlのウシ血清アルブミン、20μM、[3H]cGMP又は[3H]cAMP(100,000-200,000cpm/アッセイ)及びCOS-7細胞抽出物を含んでいる。 この反応混合物を30℃にて10分間インキュベートした。 次に、10mg/mlのCrotalus atrox venom (Sigma)を10μl加え、さらに30℃で10分間インキュベートした。 ヌクレオシド産物は、Martins et al.、前出に記載されているとおりに未反応のヌクレオチドから分離した。 全ての実験において、全[3H]環状ヌクレオチドの15%以下が、反応中に加水分解されていた。
【0058】
アッセイの結果を第5図に示すが、ここでは3つの別々のトランスフェクションの平均が示されている。 COS-7細胞のpCDM8-cGB-PDE DNAによるトランスフェクションは、モックトランスフェクト細胞又はpCDM8ベクター単独でトランスフェクトされた細胞より、cGMPホスホジエステラーゼ活性が15倍高いレベルとなっている。 モック又はベクター単独トランスフェクト細胞におけるcAMPホスホジエステラーゼ活性の増大は、pCDM8-cGB-PDE DNAでトランスフェクトした細胞からの抽出物中では検出されなかった。 これらの結果により、cGB-PDEウシcDNAはcGMP特異的ホスホジエステラーゼをコードしていることが確認された。
【0059】
また、実施例4のトランスフェクトされたCOS細胞からの抽出物を、PDE阻害剤、ザプリナスト、ジピリダモル (Sigma), イソブチル-1-メチル-8-メトキシメチルキサンタン(MeOxMeMIX)及びロリプラム(Rolipram)の一連の濃度の存在下におけるcGMP PDE活性のアッセイに供した。 アッセイの結果は第6図に示されており、阻害剤のない場合の活性を100%とし、各データポイントは2つの別々の測定の平均を示している。 発現されたcGB-BPDE cDNAタンパク産物によるcGMP加水分解の阻害に対するPDE阻害剤の相対的効能は、ウシ肺(トーマスI、前出)から精製したcGB-PDE本来のものについて報告されているこれらの相対的効能と一致している。 第6図の曲線から計算したIC50値は以下のとおりである。 ザプリナスト(黒塗り円),2μM;ジピリダモル(黒塗り四角),3.5μM;MeOxMeMIX(黒塗り三角)、30μM;及びロリプラム(白抜き円)、>300μM。 cGMP特異的ホスホジエステラーゼの比較的特異的な阻害剤であるザプリナストのIC50値は、cGs-PDE又はcGMP阻害ホスホジエステラーゼ(cGi-PDE)(Reeves et al.,pp.300-316 Beavo et al.,前出)のホスホジエステラーゼ活性の阻害について報告されているものより、少なくとも2オーダー低かった。 選択されたcAMP及びcGMP特異的ホスホジエステラーゼの効果的な阻害剤であるジピリダモルは、発現されたcGB-PDEの強力な阻害剤でもある。 相対的に選択的な、カルシウム/カルモジュリンにより刺激されるホスホジエステラーゼ(CaM-PDE),MeOxMeMIXの選択的阻害剤は、ザプリナスト及びジピリダモルより10倍低い効能であり、これはウシ肺から精製されたcGB-PDE活性体を使用した結果と一致している。
【0060】
低いKmのcAMPホスホジエステラーゼであるロリプラムは、発現されたcGB-BPDE cDNAタンパク産物に対しては低い阻害剤である。 これらの結果は、cGB-PDE cDNAが、ウシ組織から単離されたcGB-PDEの触媒活性特性を有するホスホジエステラーゼをコードしていることを示しており、これにより、cGB-8 cDNAクローンがcGB-PDEと同一であることが確認された。
【0061】
cGMP加水分解の阻害に対するPDE阻害剤の相対的な効能は、組換え遺伝子及びウシ単離cGB-PDEに対する場合と同様であるものの、全ての阻害剤についての絶対値IC50値は、組換えcGB-PDEより2〜7倍高いことは興味深い。 この違いは、COS-7細胞抽出物に存在する因子のcGMP加水分解活性に対する影響に起因すると考えることはできない。 なぜなら、ウシ組織から単離されたcGB-PDEは、純粋な酵素と同様か、又はモックトランスフェクトされたCOS-7細胞の抽出物に戻した場合と同様の阻害速度を示すからである。 この薬理学的感度の明らかな違いは、触媒部位及びその近傍における翻訳後修飾などの組換えcGB-PDE cDNAタンパク産物及びウシ肺cGB-PDEの構造におけるわずかな違いによっている。
【0062】
この違いはまた、いくつかの精製工程におけるウシ肺cGB-PDEの触媒活性の変化によっている。
【0063】
細胞抽出物を0.2mMの3−イソブチル-1-メチルキサンチン(IBMX)(Sigma)の存在下及び非存在下における[3H]cGMP結合活性のアッセイに供した。 トーマスI、前出に記載されているアッセイを改変したcGMP結合活性のアッセイは、80μlの総容量で行った。 その混合物の成分の最終濃度が、1μM[3H]cGMP,5μM cAMP、及び10μM 8−ブロモ-cBMPとなるように、60μlの細胞抽出物を20μlの結合カクテルに加えた。 cAMP及び8-ブロモ-cBMPは、それぞれ[3H]cGMPがcAK及びcGKに結合するのを阻止するために加えた。 アッセイは、0.2mMのIBMXの存在下及び非存在下で行った。 反応は細胞抽出物の添加により開始し、0℃で60分間インキュベートした。 トーマスI、前出に記載されているとおりに反応混合物を濾過した。 細胞抽出物に代えて均質化緩衝液、又は非標識cGMPの100倍量を用いて並行してインキュベーションを行い、ブランクを決定した。 同様の結果が、両方の方法で得られた。 細胞抽出物の総タンパク質濃度は、Bradford, Anal. Biochem., 72:248-254(1976)により、ウシ血清アルブミンを標準として定量した。
【0064】
アッセイの結果を、第7図に示した。 0.2mMのIBMXの存在下、1μMの[3H]cGMPで測定した場合、pCDM8-cGB-PDEでトランスフェクトされたCOS-7細胞からの抽出物は、モックトランスフェクトされた細胞からの抽出物より8倍高いcGMP結合活性を示した。 バックグラウンドcGMP結合のIBMXの促進は観察されず、内在性のcGB-PDEは、COS-7細胞抽出物には、ほんのわずか又は全く存在していないことを示している。 pCDM8-cGB-PDEトランスフェクトされた細胞の抽出物中では、cGMP特異活性は0.2mMのIBMXの添加により、約1.8倍に促進されている。 IBMXのcGMP結合を2〜5倍に促進する能力は、cGMP結合ホスホジエステラーゼ特有の性質である。
【0065】
上述したように、細胞抽出物を、過剰の非標識cAMP又はcGMPの存在下で、[3H]cGMP結合活性のアッセイ(ここでの[3H]cGMPの濃度は2.5μMである)に供した。 結果を第8図に示し、ここでは非標識の競合剤の非存在下におけるcGMP結合を100%とし、各データポイントは3つの別々の測定の平均を表している。 cGB-PDEによってコードされたタンパク産物の結合活性は、cGMPがcAMPに比較して特異的である。 10倍未満の高濃度の非標識cGMPが、[3H]cGMP結合活性を50%まで阻害するのに必要であったが、約100倍の濃度のcAMPがこれと同程度の阻害に必要であった。
【0066】
本実施例におけるこの結果は、cGB-PDE cDNAが、ネイティブなcGMB-PDEの生化学的活性特性を有するホスホジエステラーゼをコードすることを示している。
【0067】
哺乳類PDE及びドロソフィアPDEの触媒ドメインは、サーモリシン(thermolysin、Vallee and Auld, Biochem., 29:5647-5659(1990))などの、Zn2+加水分解酵素における典型的なZn2+結合モチーフである2つの直列保存配列(HX3HX24-25E)を含んでいる。 cGB-PDEは過剰のMg2+、Mn2+、Fe2+、Fe3+、Ca2+又はCd2+の存在下でZn2+と結合する。 添加金属の非存在下では、cGB-PDEは40mMのMg2+の存在下で見られる最大の活性の約20%のPDE活性を有し、この基礎活性は1、10-フェナントロリン又はEDTAにより阻害される。 このことは、金属を除去する完全な処理にも関わらず、痕跡量の金属がPDE活性に影響していることを示唆している。 PDE活性は、Zn2+(0.02−1μM)又はCo2+(1−20μM)の添加により促進されるが、Fe2+、Fe3+、Ca2+、Cd2+又はCu2+によっては阻害されない。 Zn2+は、40mMのMg2+によってもたらされる最大の促進の約70%まで基礎PDE活性を増大させる。 これらのアッセイにおけるZn2+の促進効果は、1>1μMのZn2+濃度による阻害効果の妥協によるものである。 Zn2+によって支持されたPDE活性及びcGB-PDEによるZn2+結合は、Zn2+の同様の濃度においても現れる。 このように、cGB-PDEはZn2+加水分解酵素であると考えられ、Zn2+は、この酵素の活性における重要な役割を演じていると考えられる。 Colbran et al., The FASEB J., 8: Abstruct 2148(March 15, 1994)を参照されたい。
【0068】
実施例6
cGB-PDEをコードするウシcDNAクロ−ンと相同のいくつかのヒトcDNAクローンが、ウシcGB-8クローン(配列番号:9のヌクレオチド489-1312)の部分に相当する核酸プローブを使用するストリンジェント条件下でのハイブリダイゼーションによって単離された。
【0069】
ヒトcGB-PDEをコードするcDNA断片の単離
ベクターラムダZapの、3つのヒトcDNAライブラリー(2つのグリア芽細胞腫及び1つの肺)を、ウシcGB-PDE配列によりプローブ探針した。 実施例1に記載した配列番号:9のヌクレオチド484〜1312に相当する、PCRで作製したクローンを、EcoRI及びSalIで消化し、得られた0.8kbのcDNAインサートを単離して、アガロース電気泳動により精製した。この断片を、ランダムプライムドDNAラベリングキット(Boehringer)を用いて放射性ヌクレオチドで標識した。
【0070】
cDNAライブラリーは、プレート当たり約50,000プラークの密度となるように150mmのペトリ皿に播種した。 二重のニトロセルロースフィルターレプリカを調製した。 プレハイブリダイゼーション緩衝液は、3×SSC、0.1%サルコシル、10×デンハーツ、20mMリン酸ナトリウム(pH 6.8)および50μg/mlのサケ精巣DNAである。 プレハイブリダイゼーションは、1〜5×105cpm/mlのプローブを加えた同様の組成の緩衝液中で、65℃で一夜行った。 フィルターを65℃で2×SSC、0.1%SDSで洗浄した。 ハイブリダイズしたプラークを、オートラジオグラフィで検出した。 ウシプローブにハイブリダイズしたcDNAの数、およびスクリーニングされたcDNAの数を表2に示す。
【0071】
【表2】
【0072】
cgbS2.1、cgbS3.1、cgbL23.1、cgbL27.1及びcgbS27.1と命名されたプラスミドを、ラムダZapクローン及び配列から、in vivoで試験した。
【0073】
クローンcgbS3.1は、推定イントロンに続くPDEオープンリーディングフレームの2060bpを含んでいる。 cgbS2.1の分析は、これがcgbS3.1の位置664から2060に相当し、読み取り前の付加的な585bpのPDEオープンリーディングフレームを推定イントロンまで伸ばすものである。 推定5'非翻訳領域の配列およびcgbS2.1、cgbS3.1クローンの部分をコードするタンパク質は、それぞれ配列番号:11及び12で示される。 2つのcDNAを結合することにより、PDEのオープンリーディングをコードする約2.7kbを含む配列が得られる。 他の3つのsDNAは、cDNA cgbS3.1又はcgbS2.1を5'又は3'よりさらに伸長しているものではなかった。
【0074】
さらなるcDNAを単離するために、クローンcgbS3.1の5'末端およびクローンcgbS2.1の3'末端を調製し、SW1088グリア芽細胞腫cDNAライブラリーおよびヒト大動脈cDNAライブラリーをスクリーニングするために使用した。 5'プローブは、プライマーgbS3.1S311及びcgbL23.1A1286を用いたPCRにより、クローンcgbS3.1に由来し、上記プライマーgbS3.1S311及び上記cgbL23.1A1286はそれぞれ、配列番号:8及び9、並びに下記によって示されている。
【0075】
プライマーcgbS3.1S311(配列番号:13)
5’GCCACCAGAGAAATGGTC3’
プライマーcgbL23.1A1286(配列番号:14)
5’ACAATGGGTCTAAGAGGC3’
PCR反応は、cgbS3.1 cDNAを50pg、dNTPを0.2mM、各プライマーを10μg/ml、KClを50mM、トリス塩酸pH8.2を10mM、MgCl2を1.5mMおよびTaqポリメラーゼを含む50μlの反応容量中で行った。 最初の94℃での4分間の変性後、94℃で1分、50℃で2分及び72℃で4分の、サイクルを30回行った。 PCR反応によって約0.2kbの断片が生成し、これはクローンcgbS3.1のヌクレオチド300〜496に相当する。
【0076】
3'プローブは、下記の配列を有するオリゴcgbL23.1S1190およびcgbS2.1A231を用いたPCRにより、cDNAcgbS2.1に由来するものであった。
【0077】
プライマーcgbL23.1S1190(配列番号:15)
5’TCAGTGCATGTTTGCTGC3’
プライマーcgbS2.1A231(配列番号:16)
5’ACAATGGGTCTAAGAGGC3’
PCR反応は、5'プローブの生成で上述したと同様に行い、cDNA cgbS2.1のヌクレオチド1358〜2139に相当する約0.8kbの断片を得た。 PCR断片(配列番号:12には示していない)の3' 157ヌクレオチドは、推定イントロン内にある。
【0078】
2つのPCR断片を、アガロースゲル電気泳動により精製および単離し、ランダムプライムにより放射性ヌクレオチドで標識した。 ランダムプライムSW1088cDNAライブラリー(1.5×106プラーク)を、上記標識された断片でスクリーニングし、19のハイブリダイジングプラークを単離した。 さらに50のハイブリダイズするグプラークが、ヒト大動脈cDNAライブラリー(dT及びランダムプライム、Clontech, Palo Alto, CA)から単離された。
【0079】
プラスミドを、陽性のラムダZapクローン及び配列のいくつかから、in vivo で切り出した。 cgbS53.2と命名された一つのクローン(その配列は、配列番号:17で示される)は、その配列がcgbS3.1の最後の61bpに重複する約1.1kbのインサートを含んでおり、さらなる135bpのPDEオープンリーディングフレームを、cgbS2.1に見出されるものを越えて伸長するものである。 このクローンは、終止コドンと、推定3'非翻訳配列の約0.3kbを含んでいる。
【0080】
ヒトcGB-PDEをコードする複合cDNAの生成
クローンcgbS3.1、cgbS2.1およびcgbS53.2を、以下のパラグラフに記載されているように、完全なヒトcGB-PDEのオープンリーディングフレームを含む複合cDNAを構築するために使用した。 複合cDNAをcgbmet156-2と命名し、酵母ADH1発現ベクターpBNY6Nに挿入した。
【0081】
先ず、cGB-PDEオープンリーディングフレームの3'末端を含むcgbstop-2と命名されたプラスミドを生成した。 プラスミドのインサートの一部を、鋳型としてクローンcgbS53.2を使用して、PCRによって作製した。 使用したプライマーはcgbS2.1S1700及びcgbstop-2であった。
【0082】
プライマーcgbS2.1S1700(配列番号:18)
5’TTTGGAAGATCCTCATCA3’
プライマーcgbstop-2(配列番号:19)
5’ATGTCTCGAGTCAGTTCCGCTTGGCCTG3’
PCR反応は、鋳型DNAを50pg、dNTPを0.2mM、トリス塩酸pH8.2を20mM、KClを10mM、(NH4)2SO4を6mM、MgCl2を1.5mM、0.1%Triton-X-100、各プライマー500ngおよび Pfuポリメラーゼ(Stratagene)の0.5単位を含む50μl中で行った。 反応液は94℃で4分間加熱し、94℃で1分、50℃で2分及び72℃で4分の30回のサイクルを行った。 ポリメラーゼは最初のサイクルで50℃の間に添加した。 得られたPCR産物は、フェノール/クロロホルム抽出し、クロロホルム抽出し、エタノール沈殿を行い、そして制限酵素BclI及びXhoIで切断した。 制限断片は、アガロースゲルで精製して溶出させた。
【0083】
この断片を、dam E. coli内で成長させたcDNA cgbS2.1に結合させ、制限酵素Bcl1及びXhoIで切断し、及びPromega magic PCR Kit を用いてゲル精製を行った。 結果として得られるプラスミドは、cgbstop-2がcGB-PDEオープンリーディングフレームの3'部分を含んでいることを確認するために配列決定された。
【0084】
二番目に、ヒトcGB-PDEオープンリーディングフレームの5'末端を担持するプラスミドを生成させた。 そのインサートは、鋳型としてクローンcgbS3.1を用いてPCRにより生成させた。 PCRは、プライマーcgbmet156及びcgbS2.1A2150を用い、上述と同様にして行った。
【0085】
プライマーcgbmet156(配列番号:20)
5’TACAGAATTCTGACCATGGAGCGGGCCGGC3’
プライマーcgbS2.1A2150(配列番号:21)
5’CATTCTAAGCGGATACAG3’
結果として得られるPCR断片は、フェノール/クロロホルム抽出し、クロロホルム抽出し、エタノール沈殿を行い、Sepharose CL-6Bカラムで精製した。 この断片を、制限酵素EcoRV及びEcoRIで切断してアガロースゲルを通し、そしてガラスウールを通すスピニングにより精製した。 フェノール/クロロホルム抽出、クロロホルム抽出、及びエタノール沈殿の後に、この断片はEcoRI/EcoRVで消化されたBluescriptII SK(+)に連結し、プラスミドcgbmet156を作製した。 インサートと接合部のDNA配列を決定した。 インサートは新たなEcoRI部位を含み、開始コドンの元の155ヌクレオチド5'を互いに置換した付加的な5ヌクレオチドを含んでいる。 インサートは、開始コドンより531ヌクレオチドから始まるEcoRV部位まで伸びている。
【0086】
cGB-PDEオープンリーディングフレームの5'および3'の部分を、次いてベクターpBNY6aに集めた。 ベクターpBNY6aを、EcoRVおよびXhoIで切断し、ゲルにより単離し、アガロースゲルで精製したcgbmet156からのEcoRI/EcoRV断片と、アガロースゲルで精製したcgbstop-2からのEcoRV/XhoIとに結合させた。 インサートの接合部を配列決定し、そして、構築体を、hcbgmet156-26aと命名した。
【0087】
次に、hcbgmet156-26aからのcGB-PDEインサートを、発現ベクターpBNY6nに移動させた。このベクターに挿入されたDNAの発現は、酵母ADH1プロモータおよびターミネーターより導かれる。 このベクターは、酵母の2ミクロンの複製起点、複製のpUC19起点、およびアンピシリン耐性遺伝子を含んでいる。 ベクターpBNY6nをEcoRIおよびXhoIで切断し、ゲル精製を行った。 hcgbmet156-26aからのEcoRI/XhoIインサートをPromega Magic PCR カラムを用いてゲル精製し、pBNY6nに結合させた。 結果として得られる構築体hcgbmet156-26nにおける全ての新しい接合部を配列決定した。 複合ヒトcGB-PDEをコードするhcgbmet156-26nのインサートのDNAと推定アミノ酸配列を、配列番号:22及び23に示す。
【0088】
インサートは、クローンcgbS3.1(ヌクレオチド156)の最初のメチオニンから複合cDNAの終止コドン(ヌクレオチド2781)まで伸びている。 そのメチオニンは、クローンcgbS3.1にて最も5'側のメチオニンであるので、また、メチオニンを有するフレームには、終止コドンが存在しないので、pBNY6nにおけるインサートは、オープンリーディングフレームの端を切欠した形状を現しているのかもしれない。
【0089】
変異cDNA
hcgbmet156-26n複合cDNAとは異なる4つのヒトcGB-PDE cDNAが単離されている。 一つのcDNA、cgbL23.1は、hcgbmet156-26nの内部領域(ヌクレオチド997〜1000から1444〜1447)が欠損しているものである。 欠損部分の正確な終端は、それらの位置におけるcDNA配列から決定することはできない。 4つの変異cDNAのうちの3つは、hcgbmet156-26n配列のヌクレオチド151(cDNA cgbA7f、cgbA5C、cgbI2)の上流から分岐する5'末端配列を有している。 これらのcDNAは、選択的スプライシングを受けた又はスプライシングを受けていないmRNAを表しているのかもしれない。
【0090】
実施例7
複合ヒトcGB-PDE cDNA構築体、hcgbmet156-26nを、酵母菌株YKS45(ATCC74225)(MATαhis3 trp1 ura3 leu3 pde1::HIS3 pde2::TRP1)へ形質転換したが、これは2つの内在性PDE遺伝子が欠損してたものである。 YKS45菌株のleu-欠損を補足する形質転換体を選択し、cGB-PDE活性についてアッセイを行ったプラスミドhcgbmet156-26nを産出する細胞からの抽出物は、以下に示すアッセイにより、サイクリックGMP特異的ホスホジエステラーゼ活性を呈することを調べた。
【0091】
プラスミドcgbmet156-26nで形質転換されSC-leu培地で1〜2×107細胞/mlの密度まで成長したYKS45細胞の1リットルを遠心分離により採集し、脱イオン水で一度洗浄し、ドライアイス/エタノールで凍結させて−70℃で保存した。 細胞ペレット(1〜1.5ml)を、等容量の25mMトリス-Cl(pH 8.0)/5mM EDTA/5mM EGTA/1mM オルトフェナントロリン/0.5mM AEBSF(Calbiochem)/0.1% β−メルカプトエタノールおよび10μg/mlのアプロチニン、ロイペプチン及びペプスタチンAのそれぞれの存在下に、氷上で解凍した。解凍した細胞は、15mlのCorexチューブ内の酸洗浄したガラスビーズ(425〜600μM、Sigma)2mlに加えた。 1にセットした30秒間のボルテックスと、次に60秒間の氷上でのインキュベーションとからなるサイクルを4回行うことにより、細胞を破壊した。 細胞溶解液を12,000×gで10分間遠心分離し、上清を0.8μのフィルターに通した。 上清を以下に示すようにcGMP PDE活性のアッセイに供した。 45mMトリス-Cl(pH 8.0)、2mM EGTA、1mM EDTA、0.2mg/ml BSA、5mM MgCl2、0.2mM オルトフェナントロリン、2μg/mlのペプスタチンA、ロイペプチン及びアプロチニンのそれぞれ、0.1mM AEBSF、0.02% β−メルカプトエタノールおよび基質として0.1mMの[3H]cGMPの存在下に、試料を30℃で20分間インキュベートした。 [14C]-AMP(0.5Ci/アッセイ)を回収標準物として加えた。 反応は停止緩衝液(0.1MエタノールアミンpH 9.0、0.5M硫酸アンモニウム、10mM EDTA、最終濃度0.05%のSDS)で停止した。 生成物は、BioRad Affi-Gel601上のクロマトグラフィーにより、サイクリックヌクレオチド基質から分離した。 カラム緩衝液(0.5M硫酸アンモニウムを含む0.1MエタノールアミンpH 9.0)で平衡化したAffi-Gel601の約0.25mlを含むカラムを適用した。 カラムはカラム緩衝液の0.5mlで5回洗浄した。 生成物は、0.25の酢酸の0.5mlで4回溶出させ、5mlのEcolume(ICN Biochemicals)と混合した。 放射性生成物をシンチレーション計数により測定した。
【0092】
実施例8
ヒト組織におけるcGB-PDE mRNAの発現の分析を、RNaseプロテクションアッセイにより行った。
【0093】
cGB-PDE(配列番号:13のヌクレオチド1450〜1851に相当する402bp)の推定cGMP結合ドメインの部分に相当するプローブを、PCRにより生成させた。 このPCR断片をプラスミドpBSII SK(-)のEcoRI部位に挿入してプラスミドRP3を作製した。 RP3プラスミドDNAをXbalIで直鎖状化し、アンチセンスプローブを Stratagene T7 RNA ポリメラーゼキットの改変によって作製した。 25ngの直鎖状化されたプラスミドを、20μキュリーのアルファ32PrUTP(800Ci/mmol、10mCi/ml)、1×転写緩衝液(40mMトリスCl、pH8、8mM MgCl2、2mM スペリミジン、50mM NaCl)、0.25mMの各rATP、rGTPおよびrCTP、0.1単位のRNase BlockII、5mM DTT、8μM rUTPおよび全容量5μlの5単位のT7RNAポリメラーゼと合わせた。 室温で1時間反応を行い、RNase不含のDNaseでDNA鋳型を消化させた。
【0094】
反応液を100μlの40mM トリスCl、pH8、6mMのMgCl2、及び10mMのNaClで希釈した。
【0095】
RNase不含の5単位のDNaseを加え、更に15分間、37℃で反応を継続した。 フェノール抽出によって反応を停止させ、次にフェノールクロロホルム抽出を行った。 7.5MのNH4OAcの半分の容量を加え、プローブをエタノール沈殿させた。
【0096】
RNaseプロテクションアッセイを、Ambion RNase Protection kit(Austin,TX)と、酸グアニジニン抽出法によるヒト組織から単離した10μgのRNAとを用いて行った。 cGB-PDE mRNAの発現は、骨格筋、子宮、気管支、皮膚、右の伏在静脈、大動脈およびSW1088グリア芽細胞腫細胞から抽出したRNAで容易に検出された。 右心房、右心室、腎臓皮質および腎臓髄質ではわずかに検出し得る発現が見られた。 PR3プローブの完全な保護のみが見られた。 部分的保護がないことは、少なくともこれらのRNA試料で主要な転写を表すcDNA cgbL23.1(実施例7に記載した変異cDNA)と反対の結論である。
【0097】
実施例9
ポリクロナール抗血清を、ヒトcGB-PDEの大腸菌生成断片に起用した。
【0098】
ヒトcGB-PDE cDNA(配列番号:22のヌクレオチド1668〜2612、配列番号:23のアミノ酸515〜819)の部分をPCRにより増幅し、BamHI/EcoRIとしてE. coli発現ベクターpGEX2T(Pharmacia)に挿入した。 pGEX2Tプラスミドは、アンピシリンR因子(耐性遺伝子)、大腸菌laq Iq因子、及び日本住血吸虫(Schistosoma japonicum) グルタチオン-S-転写(GST)因子を運ぶ。 プラスミドに挿入されたDNAは、GSTとの融合タンパク質として発現され、トロンビンによりこのタンパクのGST部分から分割されることができる。 この結果得られるcgbPE3と表されるプラスミドを、大腸菌株LE392(Stratagene)に形質転換した。 形質転換された細胞は、37℃で、0.6のOD600までに成長させた。 IPTG(イソプロピルチオアラクトピラノシド)を0.1mMまで加え、細胞を37℃で更に2時間成長させた。細胞を遠心分離により集め、超音波で溶解させた。 細胞の残滓を遠心分離により除去し、SDS-PAGEにより分画した。 ゲルを冷0.4M KClで着色し、GTS-cgb融合タンパクのバンドを切り出し、電気溶出させた。 タンパク質のPDE部分は、スロンビンの消化によりGTS部分から分離された。 消化物をSDS-PAGEにより分画し、PDEタンパク質を電気溶出して、ウサギに皮下注射した。 結果として得られる抗血清は、抗原として使用したウシcGB-PDE断片と、酵母に発現された(実施例8を参照されたい)ヒトcGB-PDEタンパクの全長との両方を認識する。
【0099】
実施例10
種々のcGB-PDE類似体をコードするポリヌクレオチドおよびcGB-PDE断片を標準の方法により生成させた。
【0100】
A.cGB-PDE類似体
公知のcGMP結合PDEの全ては、それらの推定cGMP結合ドメインに2つの内的に一致する直列のリピートを含んでいる。 本発明のウシcGB-PDEでは、リピートは少なくとも配列番号:10の残基228〜311(リピートA)及び410〜500(リピートB)にわたる。 公知のcGMP結合PDEの全てのリピートA及びBに保持されているアスパラギン酸の部位特異的突然変異誘発を、Ala(D289A)で置換したAsp-289か、又はAla(D478A)で置換したAsp-478を有する類似のcGB-PDEを作製するのに使用した。 組換え体の野生型(WT)のウシ及び突然変異ウシcGB-PDEを、COS-7細胞にて発現させた。 ウシ肺(自然のcGB-PDE)から精製されたcGB-PDEと、WT cGB-PDEとは同じcGMP結合動力学を示して、Kdは約2μMであり、曲直線(curvlinear)解離プロファイル(4℃でt1/2=1.3時間)を示した。 この曲直線性は、cGMP結合の明らかに高い親和性と低い親和性の部位の存在によるのかもしれない。 D289Aの突然変異体は、cGMPに対して明らかに減少した親和性を有し(Kd>20μM)、cGMP群のの単一の速度(t1/2=0.5時間)を有しており、これは、WTおよび自然のcGB-PDEの速い部位について計算したものと同じである。 このことは、突然変異体のリピートAにおける遅いcGMP結合部位の損失を示唆している。 逆に、D478A突然変異体はcGMPに対して高い親和性と(約0.5μMのKd)、単一のcGMP解離速度(t1/2=2.8時間)を示し、これは、WTおよび自然のcGB-PDEの遅い部位について計算したものと同様である。このことは、リピートBの改変に際して、速い部位が損失されることを示唆している。 これらの結果は、2量体cGMPは2つの相同であるが動力学的に区別されるcGMP結合部位を有しており、この部位は各部位でcGMPと相互作用するのに重要なアスパラギン酸を保持している。 Colbran et al., FASEB J., 8:Abstract 2149(May 15, 1994)を参照されたい。
【0101】
B.アミノ末端を切欠したcGB-PDEポリペプチド
配列番号:23のアミノ酸516〜875を含む末端を切欠したヒトcGB-PDEポリペプチドを、酵母において発現させた。 配列番号:22のヌクレオチド1555のNcoIから、配列番号:22の3’末端のXhoI部位まで伸びるcDNAインサートを、NcoI及びSalIで消化しておいたADH2酵母発現ベクターYEpC-PADH2d(Price et al., Meth. Enzymol., 18:308-318(1990))に挿入して、プラスミドYEpC-PADH2d HcGBを作製した。 このプラスミドは、酵母菌株yBJ2-54(prcl-407 prbl-1122 pep4-3 leu2 trpl ura3-52 Δpdel::URA3,HIS3 Δpde2::TRP1 cir)のスフェロプラストへと形質転換した。 内在性のPDE因子は、この菌株では欠落している。 細胞を、2%のグルコースを含むSC-leu培地で107個まで生育し、濾過によって集め、3%グリセロールを含むYEP培地で24時間生育した。 細胞は遠心分離によりペレット化し、凍結保存した。 細胞をガラスビーズで粉砕し、細胞懸濁液を、Prpic et al., Anal.Biochem., 208:155-160(1993)に本質的に記載されているホスホジエステラーゼ活性のためのアッセイに供した。 末端を切欠したヒトcGB-PDEポリペプチドは、ホスホジエステラーゼ活性を示した。
【0102】
C.カルボキシ末端を切欠したcGB-PDEポリペプチド
カルボキシ末端を切欠したcGB-PDEポリペプチドをコードする2つの異なるプラスミドを構築した。
【0103】
配列番号:23のアミノ酸1〜494をコードするcDNAを含むプラスミドpBJ6-84Hinを、ベクターYEpC-PADH2dのNcoI及びSalI部位に挿入した。 cDNAのインサートは、配列番号:22のヌクレオチドの第10位のNcoI部位から、リンカーに続く配列番号:22のヌクレオチドの第1494位のHindIII部位とYEpC-PADH2dのSalI部位まで伸びている。
【0104】
プラスミドpBJ6-84Banは、ベクターYEpC-PADH2dのNcoI及びSalI部位に挿入された、配列番号:23のアミノ酸1〜549をコードするcDNAを含んでいる。 このcDNAインサートは、配列番号:22のヌクレオチドの位置10のNcoI部位から、リンカーに続く配列番号:22のヌクレオチドの位置1654のBanI部位とYEpC-PADH2dのSalI部位まで伸びている。
【0105】
末端を切欠したcGB-PDEポリペプチドは、cGB-PDE活性のモジュレーターをスクリーニングするのに有用である。
【0106】
実施例11
ヒトcGB-PDEと反応するモノクロナール抗体を生成した。
【0107】
プラスミドYEpADH2 HcGBを含む酵母yBJ2-54(実施例10B)を、New Brunswick Scientificの10リットルのMicrofarm内で発酵させた。 プラスミドYEpADH2 HcGB内のcGB-PDE cDNAインサートは、配列番号:22のヌクレオチドの第12位のNcoI部位から、配列番号:22の3'末端のXhoI部位まで伸びている。 4×109個の細胞の接種物を、SC-leu、5%グルコース、微量金属、及び微量ビタミンを含む8リットルの培地に加えた。 発酵は26℃を保ち、標準の微生物用の撹拌翼を用いて600rpmで撹拌し、圧搾空気で1分間当たり10倍容積のエアレーションを行った。 接種後24時間でグルコースが0.3%に減少したとき、培養液に15%のグリセロールを含む2リットルの5×YEP培地を注入した。 接種後66時間で、4℃、4,000×gで30分間遠心分離することにより、発酵液から酵母を収穫した。 この発酵におけるバイオマスの収量は、湿重量で350gに達した。
【0108】
ヒトcGB-PDE酵素を、酵母細胞ペレットから精製した。 基質として1mMのcGMPを使用するPDE活性のためのアッセイを、この酵素のクロマトグラフィーに続いて採用した。 全てのクロマトグラフィーの操作は、4℃で行った。
【0109】
酵母(湿重量で29g)を、再び70mlの緩衝液A(25mMのトリス pH8.0、0.25mMのDTT、5mMのMgCl2、10μMのZnSO4、1mMのベンズアミジン)に分散させ、22−24,000psiでマイクロフリュイダイザーを通して溶解させた。 溶解液は、10,000×gで30分間遠心分離し、20mMのビストリス−プロパンpH6.8、0.25mMのDTT、1mMのMgCl2、及び10μMのZnSO4を含む緩衝液Bで平衡化したPharmacia Fast Flow Q noアニオン交換樹脂を充填した2.6×28cmのカラムに上清を通した。 このカラムをカラム容量の5倍の、0.125MのNaClを含む緩衝液Bで洗浄し、次に0.125M〜1.0MまでのNaClの直線勾配で展開した。 酵素を含む画分をプールし、20mMのビストリス−プロパンpH 6.8、0.25mMのDTT、0.25MのKCl、1mMのMgCl2、及び10μMのZnSO4を含む緩衝液Cで平衡化したセラミックヒドロキシアパタイト(BioRad)の5×20cmのカラムに直接付した。 このカラムをカラム容量の5倍の緩衝液Cで洗浄し、緩衝液C中で0〜250mMまでのリン酸カリウムの直線勾配で溶出させた。 プールした酵素を限外濾過(YM30メンブレン、Amicon)で8倍まで濃縮した。 濃縮した酵素を、25mMのビストリス−プロパン pH6.8、0.25mMのDTT、0.25MのNaCl、1mMのMgCl2、及び20μMのZnSO4で平衡化した Pharmacia Sephacryl S300(Piscataway, NJ)の2.6×90cmのカラムでのクラマトグラフィーにかけた。 約4mgのタンパクが得られた。 組換えヒトcGB-PDE酵素は、SDSポリアクリルアミドゲルの電気泳動と、次のクマシーブルー染色とにより、得られたタンパクの約90%であると計算された。
【0110】
精製したタンパク質を、モノクロナール抗体を惹起するための抗原として使用した。 19週齢のBalb/cマウス(Charles River Biotechnical Service, Inc., Wilmington, Mass)の皮下に、50%のフロイント完全アジュバント(Sigma Chemical Co.)からなる200μlのエマルジョン中の50μgの精製ヒトcGB-PDE酵素を免疫投与した。 続く追加免疫を、20日と43日に不完全フロイントアジュバントで投与した。 PBS中の50μgの酵素を用いて、前融合の後追加免疫を86日に行った。 融合は90日に行った。 マウス#1817からの脾臓を無菌的に取り出し、10mlの無血清RPMI1640中に入れた。 単細胞懸濁液を形成し、無菌の70メッシュの Nitex 細胞ストレーナ(Becton Dickinson, Parsippany, New Jersey)に通して、200gで5分間遠心分離により2回洗浄し、20mlの無血清RPMI中にペレットを再び分散させた。 3匹の無処置Balb/cマウスから取り出した胸腺細胞を同様の方法で調製した。
【0111】
11%の胎児クローン(Fetalclone)(FBS)(Hyclone Laboratories, Inc., Logan, Utah)を含むRPMI中に、融合に先立って3日間、対数増殖期で保存しておいたNS-1骨髄腫細胞を、200gで5分間遠心分離し、ペレットを前述のパラグラフに記載したようにして2回洗浄した。 洗浄後、各細胞懸濁液を最終的に10mlの無血清RPMI容量となるように加え、20μlを1mlの無血清RPMIに1:50で希釈した。 各希釈液の20μlを取り出し、0.85%シラン(Gibco)中の0.4%トリパンブルー染色液の20μlと混合し、血球計算器(Baxter Healthcare Corp., Deerfield, Illinois)上に付して計数を行った。
【0112】
2×108個の脾臓細胞を4.0×107個のNS-1細胞と合わせ、遠心分離して上清をアスピレータで吸引した。 細胞ペレットは、チューブをタッピングすることにより移動させ、37℃のPEG1500(75mMのHepe pH8.0中50%)2mlを撹拌しながら1分間にわたって加え、次に14mlの無血清RPMIを7分間にわたって加えた。 更に16mlのRPMIを加え、細胞を200gで10分間遠心分離した。 上清を捨てた後、15%のFBS、100μMのヒポキサンチンナトリウム、0.4μMのアミノプテリン、16μMのチミジン(HAT)(Gibco)、25単位/mlのIL-6(Boehringer Mannheim)及び1.5×106の 胸腺細胞/mlを含む200mlのRPMIにペレットを再分散させた。 先ず、懸濁液を96のウェルの平底組織培養プレート10枚に、1ウェル当たり200μlで分配する前の2時間、T225フラスコ(Corning, United Kingdom)に37℃で2時間入れておいた。 プレート中の細胞は、融合後3、4、5日で、20Gの針を用いて各ウェルから約100μlを吸引することにより、そして、10単位/mlのIL-6を含み胸腺細胞を含まないことを除いて上述と同様の1ウェル当たり100μlの播種培地を加えることにより、栄養供給した。
【0113】
融合体を、先ずELISAによりスクリーニングした。 Immunone4プレート(Dynatech)を、組換え精製ヒトcGB-PDE酵素(50mMのカーボネート緩衝液pH9.6中、100ng/ウェル)で一晩、4℃にて被覆した。 このプレートを、0.05%のTween20(PBST)を含むPBSで3回洗浄した。 個々のハイブリドーマウェルからの上清を、酵素で被覆したウェルに加えた(50μl/ウェル)。 37℃で30分間インキュベート後、上述と同様に洗浄し、セイヨウワサビペルオキシダーゼ接合ヤギ抗-マウスIgG(fc)(Jackson ImmunoReserch, West Grove, Pennsylvania)をPBSTで1:3500に希釈したものを50μl加えた。 プレートを上述と同様にインキュベートし、PBSTで4回洗浄し、1mg/mlのオルトフェニレンジアミン(Sigma)、及び100mMのクエン酸塩溶液中の30%H2O2の0.1μ/mlからなる100μlの基質を加えた。 着色反応を5分で15%H2SO4のμlを加えて停止した。 プレートリーダ(Dynatech)にて、A490を読み取った。
【0114】
ウェルC5G、E4D、F1G、F9H、F11G、J4A、及びJ5Dを拾い、それぞれ102A、102B、102C、102D、102E、102F、及び102Gと名付け、RPMI、15%のFBS、100μMのヒポキサンチンナトリウム、16μMのチミジン(HAT)(Gibco)、および10単位/mlのIL-6に2回希釈することにより、2又は3回連続してクローニングを行った。 クローンプレートのウェルを、4日後には目視に評価し、最も密度の低いウェルのコロニーの数を記録した。 各クローニングの選択されたウェルを、ELISAによってテストした。
【0115】
上述のハイブリドーマによって生成したモノクロナール抗体を、ELISAアッセイによってアイソタイプ判定した。 その結果、モノクロナール抗体102Aから102EはIgG1、102FはIgG2b、102GはIgG2aであることが示された。
【0116】
ウェスタン分析によって判定したところ、7つの全てのモノクロナール抗体がヒトcGS-PDEと反応した。
【0117】
実施例12
特異的PDEの生物学的活性のモジュレーターを開発するためには、特定のアッセイの調製物に存在するPDEアイソザイムを識別することが必要である。 天然の組織源からPDEを単離し、新しいアイソザイムのそれぞれを研究する伝統的な酵素学的アプローチは、精製技術の限界と、アイソザイムの完全な分離が為されるかどうかを明確に評価するのが不可能であることによって阻まれている。 一つのアイソザイムの寄与に助力し、調製における他の寄与を最小限にするアッセイ条件を同定する他のアプローチもある。 更にまた、PDEの分離を免疫学的手段によって分離する他のアプローチもある。 先に述べたPDEアイソザイムを識別するアプローチのそれぞれは、時間を要し、技術的に困難である。 結果的に、選択的PDEモジュレーターを開発する多くの試みが、1以上のアイソザイム調製物を用いて為されてきた。 更に、天然組織源からのPDEの調製物は、限定されたタンパク分解に感受性が高く、そして全長のPDEとは異なる動力学、異なる調節、及び異なる物理学的性質を有する活性なタンパク分解産物の混合物を含んでいるかもしれない。
【0118】
本発明の組換えcGB-PDEポリペプチド産物は、新たなそして特異的cGB-PDEモジュレーターの開発を大いに促進する。 モジュレーターのスクリーニングのためのヒト組換え酵素の使用は、多くの固有の利点を有している。 内在性のホスホジエステラーゼ活性を欠いている宿主細胞(例えば、ATCC 74225として寄託した酵母菌株YKS45)において組換え法により発現させることによって、アイソザイムの精製の必要性を回避することができる。 ヒトタンパク質に対する化合物をスクリーニングすることで、非ヒトタンパクについて至適化された化合物がヒトタンパク質に対して特異的ではないか又はヒトタンパク質と反応しない場合に、非ヒトタンパク質に対するスクリーニングから起こることの多い複雑さを回避することができる。 例えば、ヒトと齧歯類5HT1Bセロトニンレセプタとの間の単一のアミノ酸の違いが、受容体に対する化合物の結合の差の原因となっている。(Oskenberg et al., Nature, 360:161-163(1992)を参照されたい)。 ひとたびcGB-PDEの活性をモジュレートする化合物が発見されると、その選択性は、cGB-PDEにおけるその活性を他のPDEアイソザイムに対するその活性と比較することにより評価され得る。 このように、一連の独立したアッセイでの、本発明の組換えcGB-PDE産物と他の組換えPDEとの組み合わせは、cGB-PDEの選択的モジュレーターを開発するシステムを提供する。 選択的モジュレーターには、例えば、抗体及び他のタンパク、又はcGB-PDE若しくはcGB-PDE核酸、又はオリゴヌクレオチドに特異的に結合するペプチドが含まれ得、上記オリゴヌクレオチドは、cGB-PDE(特許協力条約国際公開第WO 93/05182、1993年3月18日、これには、目的生物分子に選択的に結合するオリゴヌクレオチドを選択する方法が記載されている)に、又はcGB-PDE核酸(即ち、アンチセンスオリゴヌクレオチド)に、及びcGB-PDE若しくはcGB-PDE核酸に特異的に結合する他の非ペプチド天然若しくは合成化合物に、特異的に結合する。 cGB-PDEの酵素活性又は局在性を変化させるcGB-PDEの突然変異体も企図される。組換えcGB-PDE単独、およびモジュレーターとの組み合わせの結晶化、X線結晶学による原子構造の分析、並びにこれらのコンピュータモデリングは、非ペプチド選択的モジュレーターの設計と最適化に有用な方法である。 例えば、構造に基づくドラッグデザインの一般的総説である、Erickson et al., Ann. Rep. Med. Chem., 27:271-289(1992)を参照されたい。
【0119】
選択的モジュレーターの開発の目的は、例えば、以下のものを含んでいる:(1)他のタンパクと接触する、及び/又は細胞内でのcGB-PDEを局在化させるcGB-PDEの領域、(2)基質を結合するcGB-PDEの領域、(3)cGB-PDEのアロステリックなcGMP結合部位、(4)cGB-PDEの金属結合領域、(5)cGB-PDEのリン酸化部位(1以上)、ならびに(6)cGB-PDEサブユニットの2量化に関係するcGB-PDEの領域。
【産業上の利用可能性】
【0120】
このように、本発明によって明らかにされたDNAおよびアミノ酸配列は、cGB-PDEに対するcDNAの配列のみならず、cGB-PDEをコードするゲノムDNA配列、ならびにプロモーター、オペレーターなどのcGB-PDE発現制御調節配列を特定する。 また、ストリンジェント条件下で本発明のDNA配列を用いて行われるDNA/DNAハイブリダイゼーションを利用することで、cGB-PDEの対立変異体、cGB-PDEに特異的な生化学的および/または免疫学的特性を共有する、他の構造的に関連するタンパク質、ならびにcGB-PDEと相同な非ヒト種のタンパク質をコードするDNAの単離も可能となる。 本発明のポリヌクレオチドは、好適に標識された場合、cGB-PDEを合成する細胞の能力を検出するためのハイブリダイゼーションアッセイにおいて有用である。 本発明のポリヌクレオチドはまた、1または複数の疾病状態の根底にあるcGB-PDE遺伝子座における遺伝的変化を同定するために有用な診断法を提供する。 通常は、cGB-PDEを発現している細胞において、cGB-PDEの発現調節に関連のあるアンチセンスポリヌクレオチドが利用可能である。 また、本発明によって明らかにされたDNAおよびアミノ酸配列は、cGB-PDEの構造および機能のシステム分析ならびにcGB-PDEが相互作用するであろう分子の特定も可能とする。
【0121】
さらに、cGB-PDE活性を修飾する薬剤は、推定モジュレーターを組換えcGB-PDEを発現する真核細胞、好ましくは、内在性のサイクリックヌクレオチドホスホジエステラーゼ活性を欠いた真核細胞からの溶解物とインキュベートし、次いでcGB-PDEホスホジエステラーゼ活性に対する推定モジュレーターの効果を決定することによって同定できる。 cGB-PDEの活性を修飾する化合物の選択性は、cGB-PDEに対する当該化合物の活性を他のPDEアイソザイムに対する活性と比較することにより評価することができる。 cGB-PDE活性のモジュレーターは、広範囲の疾病および生理学的条件の処置において治療上有用である。
【図面の簡単な説明】
【0122】
【図1A】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第578位〜第674位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図1B】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第675位〜第764位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図1C】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第765位〜第812位のアミノ酸に対応する)触媒ドメインのアラインメントを示す図である。
【図2A】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第139位〜第287位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図2B】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第288位〜第411位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図2C】種々のPDEアイソザイムにおいて保存されている(cGB-PDEでの第412位〜第526位のアミノ酸に対応する)cGMP結合ドメインのアラインメントを示す図である。
【図3】種々のPDEアイソザイムにおいて認められる内的に相同なリピートのアラインメントを示す図である。
【図4】cGB-PDEのドメインの構成を示す模式図である。
【図5】ウシcGB-PDE配列でトランスフェクトされたCOS細胞から得た抽出物が保有するホスホジエステラーゼ活性に関する分析結果を示す棒グラフである。
【図6】ウシcGB-PDE配列でトランスフェクトされた細胞から得た抽出物について認められた、ホスホジエステラーゼ阻害剤の存在下でのcGMPホスホジエステラーゼ活性に関する分析結果を示すグラフである。
【図7】ウシcGB-PDE配列でトランスフェクトされたCOS細胞から得た抽出物について認められた、0.2mMのIBMX存在下でのcGMP結合活性に関する分析結果を示す棒グラフである。
【図8】ウシcGB-PDE配列でトランスフェクトされた細胞から得た抽出物について認められた、過剰の非標識cAMPまたはcGMPの存在下での[3H]cGMP結合活性に関する分析結果を示す棒グラフである。
【特許請求の範囲】
【請求項1】
基質が結合する配列番号:23に記載のアミノ酸配列の領域を含み、かつホスホジエステラーゼ活性を奏することを特徴とするヒトのcGB-PDEポリペプチド断片。
【請求項2】
基質が結合する配列番号:23に記載のヒトのcGB-PDEポリペプチドの領域を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項3】
配列番号:23に記載のアミノ酸配列の第1位〜第494位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項4】
配列番号:23に記載のアミノ酸配列の第1位〜第549位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項5】
配列番号:23に記載のアミノ酸配列の第515位〜第819位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項6】
65℃で、3×SSCを含む溶液にてハイブリダイゼーションし、かつ、65℃で、2×SSCを含む溶液で洗浄する条件下で、請求項2乃至5のいずれかに記載のポリヌクレオチドの非コード鎖とハイブリダイズする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項7】
配列番号:23に記載のアミノ酸配列を含むサイクリックGMP結合性、サイクリックGMP特異的ホスホジエステラーゼ(cGB-PDE)ポリペプチドのホスホジエステラーゼ活性を調節する化合物を同定する方法であって、以下の工程、すなわち;
(a) 配列番号:23に記載のアミノ酸配列で表されるcGB-PDEポリペプチドの断片を含むポリペプチドを、被験候補化合物と接触せしめて、当該断片に結合する化合物を同定し、
(b) 工程(a)で同定された結合化合物の存在下または不在下で、配列番号:23に記載のアミノ酸配列で表されるcGB-PDEポリペプチドまたは配列番号:23に記載のアミノ酸配列の第516位〜第875位の連続するアミノ酸を含むポリペプチドのcGMPホスホジエステラーゼ活性を分析し、および
(c) 工程(b)で分析された化合物の内、その不在下でのホスホジエステラーゼ活性と比較して、その存在下でのホスホジエステラーゼ活性に変化を及ぼす化合物をcGB-PDEの調節化合物として同定する、
工程を含む、ことを特徴とするヒトのcGB-PDEポリペプチドのホスホジエステラーゼ活性を調節する化合物の同定方法。
【請求項8】
前記調節化合物が、cGB-PDE活性を阻害する請求項7に記載の方法。
【請求項9】
前記調節化合物が、cGB-PDE活性を強化する請求項7に記載の方法。
【請求項10】
前記工程(a)のポリペプチドが、基質が結合する配列番号:23に記載のアミノ酸配列の領域を含む請求項7に記載の方法。
【請求項11】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第516位〜第875位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項12】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第1位〜第494位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項13】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第1位〜第549位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項14】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第515位〜第819位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項1】
基質が結合する配列番号:23に記載のアミノ酸配列の領域を含み、かつホスホジエステラーゼ活性を奏することを特徴とするヒトのcGB-PDEポリペプチド断片。
【請求項2】
基質が結合する配列番号:23に記載のヒトのcGB-PDEポリペプチドの領域を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項3】
配列番号:23に記載のアミノ酸配列の第1位〜第494位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項4】
配列番号:23に記載のアミノ酸配列の第1位〜第549位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項5】
配列番号:23に記載のアミノ酸配列の第515位〜第819位の連続するアミノ酸を含むヒトのcGB-PDEポリペプチド断片を含むポリペプチドをコードする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項6】
65℃で、3×SSCを含む溶液にてハイブリダイゼーションし、かつ、65℃で、2×SSCを含む溶液で洗浄する条件下で、請求項2乃至5のいずれかに記載のポリヌクレオチドの非コード鎖とハイブリダイズする、ことを特徴とする精製および単離されたポリヌクレオチド。
【請求項7】
配列番号:23に記載のアミノ酸配列を含むサイクリックGMP結合性、サイクリックGMP特異的ホスホジエステラーゼ(cGB-PDE)ポリペプチドのホスホジエステラーゼ活性を調節する化合物を同定する方法であって、以下の工程、すなわち;
(a) 配列番号:23に記載のアミノ酸配列で表されるcGB-PDEポリペプチドの断片を含むポリペプチドを、被験候補化合物と接触せしめて、当該断片に結合する化合物を同定し、
(b) 工程(a)で同定された結合化合物の存在下または不在下で、配列番号:23に記載のアミノ酸配列で表されるcGB-PDEポリペプチドまたは配列番号:23に記載のアミノ酸配列の第516位〜第875位の連続するアミノ酸を含むポリペプチドのcGMPホスホジエステラーゼ活性を分析し、および
(c) 工程(b)で分析された化合物の内、その不在下でのホスホジエステラーゼ活性と比較して、その存在下でのホスホジエステラーゼ活性に変化を及ぼす化合物をcGB-PDEの調節化合物として同定する、
工程を含む、ことを特徴とするヒトのcGB-PDEポリペプチドのホスホジエステラーゼ活性を調節する化合物の同定方法。
【請求項8】
前記調節化合物が、cGB-PDE活性を阻害する請求項7に記載の方法。
【請求項9】
前記調節化合物が、cGB-PDE活性を強化する請求項7に記載の方法。
【請求項10】
前記工程(a)のポリペプチドが、基質が結合する配列番号:23に記載のアミノ酸配列の領域を含む請求項7に記載の方法。
【請求項11】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第516位〜第875位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項12】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第1位〜第494位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項13】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第1位〜第549位の連続するアミノ酸を含む請求項7に記載の方法。
【請求項14】
前記工程(a)のポリペプチドが、配列番号:23に記載のアミノ酸配列の第515位〜第819位の連続するアミノ酸を含む請求項7に記載の方法。
【図1A】


【図1B】

【図1C】

【図2A】

【図2B】

【図2C】

【図3】

【図4】

【図5】

【図6】

【図7】
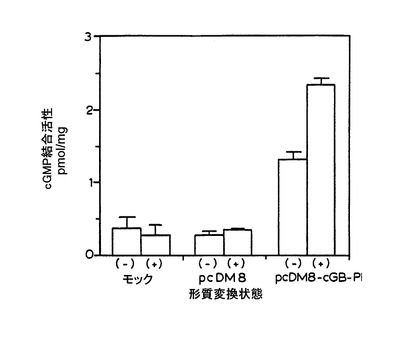
【図8】

【図1B】
【図1C】
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2010−42035(P2010−42035A)
【公開日】平成22年2月25日(2010.2.25)
【国際特許分類】
【出願番号】特願2009−267135(P2009−267135)
【出願日】平成21年11月25日(2009.11.25)
【分割の表示】特願2005−150760(P2005−150760)の分割
【原出願日】平成6年5月27日(1994.5.27)
【出願人】(505154510)ボード オブ リージェンツ オブ ザ ユニヴァーシティ オブ ワシントン (1)
【出願人】(500246762)イコス コーポレイション (3)
【Fターム(参考)】
【公開日】平成22年2月25日(2010.2.25)
【国際特許分類】
【出願日】平成21年11月25日(2009.11.25)
【分割の表示】特願2005−150760(P2005−150760)の分割
【原出願日】平成6年5月27日(1994.5.27)
【出願人】(505154510)ボード オブ リージェンツ オブ ザ ユニヴァーシティ オブ ワシントン (1)
【出願人】(500246762)イコス コーポレイション (3)
【Fターム(参考)】
[ Back to top ]