
ニトリルオキシド化合物、変性高分子材料並びにその製造方法及び成形体
【課題】安定性が高いニトリルオキシド化合物と、このニトリルオキシド化合物で高分子材料を変性した変性高分子材料を提供する。
【解決手段】アクリル酸エステル、メタクリル酸エステル又はアクリル酸アミドの極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端に、一般式[1]で表される末端構造が結合しているニトリルオキシド化合物をNR、EPDM、NBR又はPANの高分子材料に反応させる。

【解決手段】アクリル酸エステル、メタクリル酸エステル又はアクリル酸アミドの極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端に、一般式[1]で表される末端構造が結合しているニトリルオキシド化合物をNR、EPDM、NBR又はPANの高分子材料に反応させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ニトリルオキシド化合物、ニトリルオキシド化合物で高分子材料を変性した変性高分子材料、この変性高分子材料の製造方法及びこの変性高分子材料の成形体に関するものである。
【背景技術】
【0002】
EPDM、NR、NBR等のように分子内に炭素−炭素二重結合を有する高分子材料は、太陽光(特に紫外線)やオゾンにより劣化し易いため、用途によっては耐候性に問題が生じることがあった。また、これらの高分子材料は、特定の有機溶媒等に対しては溶解することがあり、そのような有機溶媒等と接触するおそれがある部位にも用いることができなかった。このため、これらの高分子材料の用途を広げる一策として、特許文献1、2に記載のように、ベンゼン等の芳香環にニトリルオキシド基が結合したニトリルオキシド化合物での変性(化学修飾)が考えられている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2011−52072号公報
【特許文献2】特開平11−180943号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、特許文献1、2に記載のニトリルオキシド化合物のように、芳香環にニトリルオキシド基が結合した芳香族ニトリルオキシド化合物は、安定性が低いものとなっていた(本明細書の段落0066〜段落0073参照)。このため、分解し易くなっており、高分子材料と反応する前に分解してしまうものが多い場合には、高分子材料を十分に変性できないおそれがあった。
【0005】
また、特許文献1、2に記載のニトリルオキシド化合物は、分子内に含まれる極性基が少ないことから、このニトリルオキシド化合物で変性した変性高分子材料は、求められる耐油性が高い場合には、その求めに十分な耐油性を得ることはできなかった。
【0006】
そこで、本発明は、安定性が高いニトリルオキシド化合物と、このニトリルオキシド化合物で高分子材料を変性した変性高分子材料と、この変性高分子材料の製造方法と、この変性高分子材料からなる成形体とを提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明の発明者らは、芳香族ニトリルオキシド化合物の安定性が低い理由は、芳香環の高い転位能に起因して、分子内転位反応が起こり、イソシアナートへと異性化し易いためであると考えた。
【0008】
そこで、炭素鎖の末端にニトリルオキシド基が結合した脂肪族ニトリルオキシド化合物にすることで分子内転位反応を起こり難くし、且つ、そのα位の炭素にフェニル基が二つ結合することでニトリルオキシ基を反応し難くしている脂肪族ニトリルオキシド誘導体を合成したところ、安定性が高いことを見出した(本明細書の段落0066〜段落0073参照)。
【0009】
上記課題を解決する本発明のニトリルオキシド化合物は、極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端、スチレン系単量体を重合してなる炭素鎖の片方の末端又はアルキル基に、下記の一般式[1]で表される末端構造が結合しているニトリルオキシド化合物である。
【化2】
(式中、R1、R2は、同一又は異なって、アルキル基又はアリール基を示す。)
【0010】
本発明の変性高分子材料の製造方法は、分子内にニトリルオキシドと反応する多重結合を有する高分子材料を変性した変性高分子材料の製造方法であって、上記のニトリルオキシド化合物を前記高分子材料に反応させる反応過程を備えることを特徴とする。
【0011】
本発明の変性高分子材料は、分子内にニトリルオキシドと反応する多重結合を有する高分子材料が、上記のニトリルオキシド化合物で変性されてなる。
【0012】
本発明の成形体は、上記の変性高分子材料を成形してなる。
【0013】
本発明のニトリルオキシド化合物、変性高分子材料、変性高分子材料の製造方法及び成形体における各要素の態様を以下に例示する。
【0014】
1.極性のアニオン重合性単量体
極性のアニオン重合性単量体としては、特に限定されないが、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド、メタクリル酸アミド等が例示できる。
【0015】
アクリル酸エステルとしては、特に限定されないが、アクリル酸メチル、アクリル酸エチル、アクリル酸n−プロピル、アクリル酸n−ブチル、アクリル酸n−ペンチル、アクリル酸n−ヘキシル、アクリル酸n−ヘプチル、アクリル酸n−オクチル、アクリル酸n−デシル、アクリル酸n−ドデシル、アクリル酸n−ラウリル、アクリル酸n−テトラデシル、アクリル酸n−ヘキサデシル、アクリル酸n−オクタデシル、アクリル酸イソプロピル、アクリル酸イソブチル、アクリル酸t−ブチル、アクリル酸イソペンチル、アクリル酸ネオペンチル、アクリル酸イソヘキシル、アクリル酸イソヘプチル、アクリル酸イソオクチル、アクリル酸2−エチルヘキシル、アクリル酸フェニル、アクリル酸ビフェニル、アクリル酸ジフェニルエチル、アクリル酸t−ブチルフェニル、アクリル酸シクロヘキシル、アクリル酸t−ブチルシクロヘキシル等が例示できる。
【0016】
メタクリル酸エステルとしては、特に限定されないが、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸n−プロピル、メタクリル酸n−ブチル、メタクリル酸n−ペンチル、メタクリル酸n−ヘキシル、メタクリル酸n−ヘプチル、メタクリル酸n−オクチル、メタクリル酸n−デシル、メタクリル酸n−ドデシル、メタクリル酸n−ラウリル、メタクリル酸n−テトラデシル、メタクリル酸n−ヘキサデシル、メタクリル酸n−オクタデシル、メタクリル酸イソプロピル、メタクリル酸イソブチル、メタクリル酸t−ブチル、メタクリル酸イソペンチル、メタクリル酸ネオペンチル、メタクリル酸イソヘキシル、メタクリル酸イソヘプチル、メタクリル酸イソオクチル、メタクリル酸2−エチルヘキシル、メタクリル酸フェニル、メタクリル酸ビフェニル、メタクリル酸ジフェニルエチル、メタクリル酸t−ブチルフェニル、メタクリル酸シクロヘキシル、メタクリル酸t−ブチルシクロヘキシル等が例示できる。
【0017】
アクリル酸アミドとしては、特に限定されないが、アクリルアミド、N−メチルアクリルアミド、N,N−ジメチルアクリルアミド、N−エチルアクリルアミド、N,N−ジエチルアクリルアミド、N−プロピルアクリルアミド、N,N−ジプロピルアクリルアミド、N−ブチルアクリルアミド、N,N−ジブチルアクリルアミド等が例示できる。
【0018】
メタクリル酸アミドとしては、特に限定されないが、メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−エチルメタクリルアミド、N,N−ジエチルメタクリルアミド、N−プロピルメタクリルアミド、N,N−ジプロピルメタクリルアミド、N−ブチルメタクリルアミド、N,N−ジブチルメタクリルアミド等が例示できる。
【0019】
2.スチレン系単量体
スチレン系単量体としては、特に限定されないが、スチレン、ビニルナフタレン、2−メチルスチレン、3−メチルスチレン、4−メチルスチレン、2−エチルスチレン、3−エチルスチレン、4−エチルスチレン、α−メチルスチレン等が例示できる。
【0020】
3.重合
極性のアニオン重合性単量体又はスチレン系単量体を重合させる重合方法は、特に限定されないが、これらの単量体を直鎖状に重合することができ、且つ、1−ニトロ−2,2ジフェニルエチレン、1−ニトロ−2−メチル−2−フェニルエチレン等のニトリルエチレン誘導体を用いてニトリルオキシドを合成できることから、アニオン重合であることが好ましい。
【0021】
アニオン重合の場合に用いる重合開始剤としては、特に限定されないが、メチルリチウム、n−ブチルリチウム、sec−ブチルリチウム、t−ブチルリチウム等のアルカリ金属アルキルや、ナトリウムメチラート、ナトリウムエチラート、カリウムメチラート、リチウムメチラート等のアルカリ金属アルコキシドや、グリニャール試薬等が例示でき、反応性が高いことから、アルカリ金属アルキルが好ましい。また、重合副反応を抑制できることから、ジフェニルエチレンを併用することが好ましい。
【0022】
4.アルキル基
アルキル基としては、特に限定されないが、直鎖状でもよいし、分岐状でもよいし、環状でもよい。上記の一般式[1]のR1、R2のアルキル基の場合には、ニトリルオキシド化合物が合成し易いことから、炭素数が1〜20の直鎖状又は分岐状のものが好ましく、より好ましくは、炭素数が1〜4の直鎖状又は分岐状のものである、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、sec−ブチル基、イソブチル基、t−ブチル基である。
【0023】
5.アリール基
アリール基としては、特に限定されないが、フェニル基、ナフチル基等が例示できる。
【0024】
6.ニトリルオキシド化合物
ニトリルオキシド化合物の分子量は、特に限定されないが、極性のアニオン重合性単量体又はスチレン系単量体を重合してなる炭素鎖を有するニトリルオキシド化合物の場合には、数平均分子量(Mn)が1000〜20000が例示できる。
【0025】
7.高分子材料
高分子材料の多重結合としては、特に限定されないが、C=S、N=N、P(V)=C、C=P(III)、C=As、C=C、C=N、C=Se、B=N、C≡P、C≡C、P(V)=N、C≡N、C=O等が例示できる。
【0026】
高分子材料としては、特に限定されないが、分子内にニトリル基(C≡N)を有するPAN(ポリアクリロニトリル)等の高分子や、分子内に炭素−炭素二重結合(C=C)を有するNR(天然ゴム)、EPDM(エチレン−プロピレン−ジエン共重合ゴム)、分子内にニトリル基及び炭素−炭素二重結合を有するNBR(ニトリルゴム)等の分子内に炭素−炭素二重結合を有するゴム等が例示できる。
【0027】
8.反応過程
反応過程は、特に限定されないが、有機溶媒中で行ってもよいし、無溶媒で行ってもよい。
【0028】
8−1.有機溶媒中での反応過程
有機溶媒としては、特に限定されないが、高分子材料及びニトリルオキシド化合物が共に溶解し易いものであることが好ましい。具体的には、トルエン、メシチレン、クロロホルム、DMF(N,N−ジメチルホルムアミド)等が例示できる。
【0029】
8−2.無溶媒での反応過程
無溶媒で行う場合には、空気下で行ってもよいし、不活性ガスが充填された雰囲気下で行ってもよい。
不活性ガスとしては、特に限定されないが、アルゴン、窒素等が例示できる。
反応過程が無溶媒で行われる場合には、効率よく製造できることから、反応過程を、混練装置で行う、即ち、混練装置を用いて混練装置内で反応過程を行うことが好ましい。
混練装置としては、特に限定されないが、二軸混練機、密閉式混練機、バンバリーミキサー、インターミックス等の混練機や二軸押出機、単軸押出機、多軸押出機等の押出機等が例示できる。
【0030】
8−3.反応過程の温度
反応過程の温度は、ニトリルオキシド化合物が高分子材料と反応する温度であれば、特に限定されない。敢えていうならば、化学反応であることから温度が高ければ反応が促進され、また加熱等の温度調節を行わなければ製造工程の管理が容易になることから、0〜150℃であることが好ましい。
【0031】
9.成形体
成形体の成形方法は、特に限定されないが、圧縮成形、射出成形、トランスファー成形等が例示できる。
成形体は、特に限定されないが、耐油性が高い場合には、オイルホース、ガスケット等が例示できる。
【発明の効果】
【0032】
本発明によれば、安定性が高いニトリルオキシド化合物と、このニトリルオキシド化合物で高分子材料を変性した変性高分子材料と、この変性高分子材料の製造方法と、この変性高分子材料からなる成形体を提供することができる。
【図面の簡単な説明】
【0033】
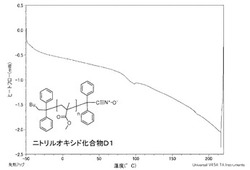
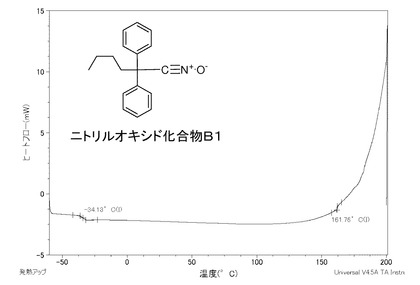
【図1】本発明の実施例B1であるニトリルオキシド化合物B1のDSC曲線のグラフである。
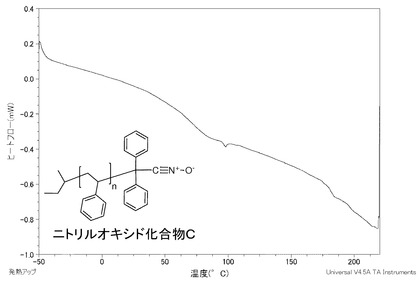
【図2】同じく実施例Cであるニトリルオキシド化合物CのDSC曲線のグラフである。
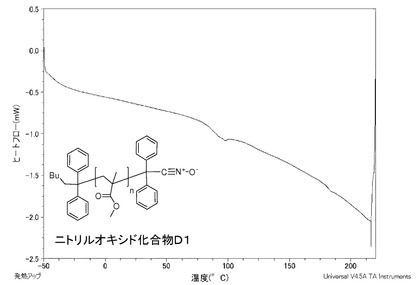
【図3】同じく実施例Dであるニトリルオキシド化合物D1のDSC曲線のグラフである。
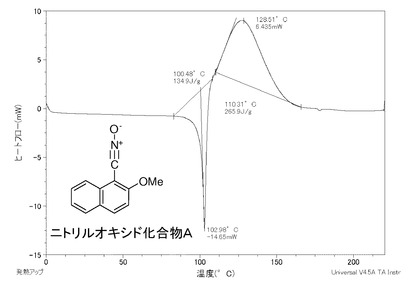
【図4】比較例Aであるニトリルオキシド化合物AのDSC曲線のグラフである。
【発明を実施するための形態】
【0034】
本発明のニトリルオキシド化合物、変性高分子材料及び変性高分子材料の製造方法の好適な実施形態について以下に説明する。
【0035】
本発明の好適な実施形態のニトリルオキシド化合物は、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、下記の構造式[3a]又は[3b]で表される末端構造が結合している化合物、即ち、ニトリルオキシド基のα位の炭素に二つのフェニル基又はメチル基とフェニル基とが結合し、且つ、このα位の炭素が、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に結合している化合物である。
【化3】
【化4】
【0036】
本発明の好適な実施形態の変性高分子材料は、分子内に炭素−炭素二重結合を有するゴムが、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、上記の構造式[3a]又は[3b]で表される末端構造が結合しているニトリルオキシド化合物で変性されてなる変性ゴムである。
【0037】
本発明の好適な実施形態の変性高分子材料の製造方法は、分子内に炭素−炭素二重結合を有するゴムを変性した変性ゴムの製造方法であって、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、上記の構造式[3a]又は[3b]で表される末端構造が結合しているニトリルオキシド化合物を、分子内に炭素−炭素二重結合を有するゴムに反応させる反応過程を備えることを特徴とする。
【実施例】
【0038】
<ニトリルオキシド化合物>
本発明の実施例の8種類のニトリルオキシド化合物について説明する。
【0039】
各ニトリルオキシド化合物の説明の前に、これらのニトリルオキシド化合物の合成において、ニトリルオキシド基を導入するために用いた、1−ニトロ−2,2−ジフェニルエチレン及び1−ニトロ−2−メチル−2−フェニルエチレンについて説明する。
【0040】
《1》1−ニトロ−2,2−ジフェニルエチレン
1−ニトロ−2,2−ジフェニルエチレンの構造式[4]を次に示す。
【化5】
【0041】
1−ニトロ−2,2−ジフェニルエチレンの合成は、次のようにして行った。
【化6】
【0042】
32.3g(200mmol)のHMDS(ヘキサメチルジシラザン)を溶かした100mLのTHF(テトラヒドロフラン)溶液を、0℃で攪拌しているところへ、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液76.9mL(n−BuLi量:200mmol)を加えて、0℃で30分間攪拌した。なお、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。
その後、18.2g(100mmol)のベンゾフェノンを加えて、室温で1日間攪拌した。
その後、この反応溶液を減圧濃縮した後に、40mLのニトロメタンを加えて、10分間超音波を照射した後に、濾過して、濾液を3日間還流した。
その後、室温に放冷した後に、減圧濃縮して粗生成物を得た。
そして、この粗生成物をヘキサン/酢酸エチルで再結晶し、さらにトルエンで再結晶して、黄色板状結晶の1−ニトロ−2,2−ジフェニルエチレンを16.3g(収率:73%)得た。
【0043】
《2》1−ニトロ−2−メチル−2−フェニルエチレン
1−ニトロ−2−メチル−2−フェニルエチレンの構造式[5]を次に示す。
【化7】
【0044】
1−ニトロ−2−メチル−2−フェニルエチレンの合成は、次のようにして行った。
【化8】
【0045】
12.0g(0.10mol)のアセトフェノンを溶かした64mLのトルエン溶液に、21.6mL(0.40mol)のニトロメタンと、3.9mL(0.04mol)のノルマルブチルアミンとを加え、ディーン・スターク(Dean Stark)トラップを用いて水を留去しながら、5日間還流した。
その後、室温まで冷却した後に、60mLの酢酸エチルを加えて希釈した。
その後、30mLの飽和塩化アンモニウム水溶液を加えて分液し、有機相を硫酸マグネシウムで乾燥した。
その後、濾過を行い、濾液を減圧濃縮した後に、シリカゲルカラムクロマトグラフィー(溶離液は、ヘキサン+1%ジエチルエーテル)で精製して、赤色オイル状の1−ニトロ−2−メチル−2−フェニルエチレンを1.4g(収率:11%)得た。
【0046】
上記のようにして合成された1−ニトロ−2,2−ジフェニルエチレン又は1−ニトロ−2−メチル−2−フェニルエチレンを用いて合成された8種類のニトリルオキシド化合物について説明する。
【0047】
〈実施例B1〉2,2−ジフェニルヘキサンニトリル−N−オキシド
2,2−ジフェニルヘキサンニトリル−N−オキシド(以下、ニトリルオキシド化合物B1と言う場合がある)の構造式[B1]を次に示す。
【化9】
【0048】
ニトリルオキシド化合物B1の合成は、次のようにして行った。
【化10】
【0049】
アルゴン雰囲気下において、−78℃の80mLのTHFに、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液3.08mL(n−BuLi量:8.00mmol)を滴下し、5分間攪拌した後に、901mg(4.00mmol)の1−ニトロ−2,2−ジフェニルエチレンを加えて、30分間攪拌した。なお、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。また、以下に説明する他のニトリルオキシド化合物の合成においても、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。
その後、この反応溶液を0℃の95%濃硫酸中に滴下し、30分間攪拌した。
その後、ジクロロメタンで抽出し、純水で洗浄した後に、MgSO4(硫酸マグネシウム)で乾燥して粗生成物を得た。
その後、この粗生成物をシリカゲルカラムクロマトグラフィー(溶離液は、ヘキサン:酢酸エチル=5:1(体積比)を使用)によって、橙色オイル状のニトリルオキシド化合物B1を1.01g(収率:95%)得た。
【0050】
〈実施例B2〉2−メチル−2−フェニルヘキサンニトリル−N−オキシド
2−メチル−2−フェニルヘキサンニトリル−N−オキシド(以下、ニトリルオキシド化合物B2と言う場合がある)の構造式[B2]を次に示す。
【化11】
【0051】
ニトリルオキシド化合物B2の合成は、1−ニトロ−2,2−ジフェニルエチレンの替わりに、652mg(4.00mmol)の1−ニトロ−2−メチル−2−フェニルエチレンを加えた点のみが、ニトリルオキシド化合物B1の合成と異なり、その他の点はニトリルオキシド化合物B1の合成と同じである。そして、赤色オイル状のニトリルオキシド化合物B2(収率:99%)が得られた。
【0052】
〈実施例C〉スチレンが重合した炭素鎖を有するニトリルオキシド化合物
スチレンが重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、sec−ブチル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Cと言う場合がある)の構造式[C]を次に示す。式中nは自然数である。
【化12】
【0053】
ニトリルオキシド化合物Cの合成は、次のようにして行った。
【化13】
【0054】
アルゴン雰囲気下において、1.02Mのsec−ブチルリチウム(sec−BuLi)のシクロヘキサン溶液3.92mL(sec−BuLi量:4.00mmol)を加えた40mLのTHFを、−78℃で攪拌しているところへ、4.58mL(40.0mmol)のスチレンを溶かした35mLのTHF溶液を加え、−78℃で10分間攪拌した。
その後、901mg(4.00mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした5.0mLのTHF溶液を加え、−78℃で30分間攪拌した。
その後、この反応液を、0℃の2.0g(20.0mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応溶液を、CH2Cl2/H2O(ジクロロメタンと水)で抽出し、有機相をMgSO4(硫酸マグネシウム)で乾燥した。
その後、溶媒を減圧濃縮して、ニトリルオキシド化合物C(数平均分子量Mn:1500(Mw/Mn:1.85))の粗生成物を得た。なお、後述する、高分子材料の変性には、この粗生成物をそのまま使用した。
【0055】
〈実施例D〉メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物
メチルメタクリレート(メタクリル酸メチル)が重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニルヘキシル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Dと言う場合がある)の構造式[D]を次に示す。式中nは自然数である。
【化14】
【0056】
ニトリルオキシド化合物Dの合成は、次のようにして行った。なお、ニトリルオキシド化合物Dは、メチルメタクリレートの重合度を変えた、ニトリルオキシド化合物D1、ニトリルオキシド化合物D2及びニトリルオキシド化合物D3の3種類を合成した。
【化15】
【0057】
アルゴン雰囲気下において、1.80g(10.0mmol)の1,1−ジフェニルエチレンを溶かした150mLのTHF溶液を、−78℃で攪拌しているところへ、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液3.84mL(n−BuLi量:10.0mmol)を加え、−78℃で20分間攪拌した。
その後、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加え、−78℃で1時間攪拌した。
その後、2.25g(10.0mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした12.5mLのTHF溶液を加え、−78℃で30分間攪拌した。
その後、この反応液を、0℃の5.42mL(100mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応溶液を、CH2Cl2/H2O(ジクロロメタンと水)で抽出し、有機相をMgSO4(硫酸マグネシウム)で乾燥した。
その後、溶媒を減圧濃縮して、ニトリルオキシド化合物D1の粗生成物を得た。
その後、この粗生成物をクロロホルムに溶解し、ヘキサンに再沈殿することによって精製を行い、白色粉末のニトリルオキシド化合物D1(数平均分子量Mn:2300(Mw/Mn:1.26))を8.5g得た。
【0058】
また、加えるメチルメタクリレートの量を変えることでメチルメタクリレートの重合度を変えたニトリルオキシド化合物D2とニトリルオキシド化合物D3とを合成した。
【0059】
ニトリルオキシド化合物D2の合成は、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液の替わりに、21.30mL(200mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加えた点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。そして、ニトリルオキシド化合物D2(数平均分子量Mn:3062(Mw/Mn:1.35))を得た。
【0060】
ニトリルオキシド化合物D3の合成は、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液の替わりに、42.60mL(400mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加えた点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。そして、ニトリルオキシド化合物D3(数平均分子量Mn:8070(Mw/Mn:1.34))を得た。
【0061】
〈実施例E〉t−ブチルアクリレートが重合した炭素鎖を有するニトリルオキシド化合物
t−ブチルアクリレート(アクリル酸t−ブチル)が重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニルヘキシル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Eと言う場合がある)の構造式[E]を次に示す。式中nは自然数である。
【化16】
【0062】
ニトリルオキシド化合物Eの合成は、メチルメタクリレートのTHF溶液の替わりに、14.5mL(100mmol)のt−ブチルアクリレートを溶かしたTHF溶液を加え、0℃に昇温して、0℃で1時間攪拌した点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。なお、0℃で1時間攪拌した後、−78℃に冷却してから、1−ニトロ−2,2−ジフェニルエチレンのTHF溶液を加え、−78℃で30分間攪拌した。そして、褐色粉末のニトリルオキシド化合物E(数平均分子量Mn:18400(Mw/Mn:1.87))を得た。
【0063】
〈実施例F〉N,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物
N,N−ジメチルアクリルアミドが重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニル−3−メチルペンチル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Fと言う場合がある)の構造式[F]を次に示す。式中nは自然数である。
【化17】
【0064】
ニトリルオキシド化合物Fの合成は、次のようにして行った。
【化18】
【0065】
アルゴン雰囲気下において、180mg(1.0mmol)の1,1−ジフェニルエチレンを溶かした80mLのTHF溶液を、−78℃で攪拌しているところへ、1.02Mのsec−ブチルリチウム(sec−BuLi)のシクロヘキサン溶液1.96mL(sec−BuLi量:2.0mmol)を加え、−78℃で20分間攪拌した。
その後、1.0MのZnEt2(ジエチル亜鉛)のヘキサン溶液24mL(ZnEt2量:24mmol)を加え、−78℃で10分間攪拌した。
その後、1.98g(20mmol)のN,N−ジメチルアクリルアミドを溶かした10mLのTHF溶液を加え、−78℃で1時間攪拌した。
その後、450mg(2.0mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした5mLのTHF溶液を加え、−20℃に昇温して、−20℃で1時間攪拌した。
その後、この反応液を、0℃の2.35mL(44mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応液を、ヘキサンに再沈殿し、沈殿物を回収した。そして、この沈殿物をメタノール/THF(1/5:体積比)溶液で洗浄してZnEt2を除去し、ニトリルオキシド化合物F(数平均分子量Mn:3320(Mw/Mn:1.21))を得た。
【0066】
このようにして合成したニトリルオキシド化合物のうちで、ニトリルオキシド化合物B1、同C及び同D1について、安定性を調べるため、DSC(示差走査熱量)測定を行った。また、比較例として、2−メトキシ−1−ナフトニトリルオキシド(以下、ニトリルオキシド化合物Aと言う場合がある)のDSC測定を行った。さらに、ニトリルオキシド化合物B1及び同Aの半減期を測定した。
【0067】
〈比較例A〉2−メトキシ−1−ナフトニトリルオキシド
2−メトキシ−1−ナフトニトリルオキシドの構造式[A]を次に示す。この2−メトキシ−1−ナフトニトリルオキシドの合成は、特許文献1の段落0020に記載の方法で行った。
【化19】
【0068】
(1)DSC測定
測定条件は、窒素雰囲気下で、−60℃から200℃まで、毎分10℃の昇温スピードで行った。
ニトリルオキシド化合物B1、同C、同D1及び同Aを測定したDSC曲線のグラフを図1〜4に示す。
【0069】
ニトリルオキシド化合物B1は、図1に示すように、約160℃を超えたところに熱分解由来の発熱のピークがあることから、このピーク温度のところで分解が起きており、約160℃までは安定している。
ニトリルオキシド化合物Cは、図2に示すように、−50〜200℃の温度範囲に発熱のピークがないことから、この温度範囲で分解が起きず、安定している。
ニトリルオキシド化合物D1は、図3に示すように、−50〜200℃の温度範囲に発熱のピークがないことから、この温度範囲で分解が起きず、安定している。
ニトリルオキシド化合物Aは、図4に示すように、約110℃を超えたところに熱分解由来の発熱のピークがあることから、このピーク温度のところで分解が起きており、約110℃までしか安定していない。
【0070】
(2)半減期の測定
60℃又は100℃の温度における半減期を測定した。
具体的には、重溶媒(60℃での測定には重クロロホルムを使用、100℃での測定には重DMSOを使用)に試料を溶解し、それを60℃又は100℃に加熱し、その状態で試料の量が半減するまでの時間(半減期)をNMR測定装置で測定した。
【0071】
ニトリルオキシド化合物B1及び同Aの測定結果を表1に示す。
【表1】
【0072】
表1に示すように、ニトリルオキシド化合物B1は、60℃及び100℃において、48時間経っても半減しなかったことから、半減期が48時間以上であった。
ニトリルオキシド化合物Aは、60℃において、半減期が2時間であった。
【0073】
以上より、本発明の実施例であるニトリルオキシド化合物は、2−メトキシ−1−ナフトニトリルオキシドより、安定性が向上している。
なお、ニトリルオキシド化合物B2、ニトリルオキシド化合物D2、ニトリルオキシド化合物D3、ニトリルオキシド化合物E及びニトリルオキシド化合物Fについては、DSC測定及び半減期の測定を行っていないが、ニトリルオキシド化合物B2はニトリルオキシド化合物B1の測定結果より、ニトリルオキシド化合物D2及びニトリルオキシド化合物D3はニトリルオキシド化合物D1の測定結果より、ニトリルオキシド化合物E及びニトリルオキシド化合物Fはニトリルオキシド化合物C及びニトリルオキシド化合物D1の測定結果より、2−メトキシ−1−ナフトニトリルオキシドより安定性が向上していると推測できる。
【0074】
<変性高分子材料>
本発明の実施例として、NR(天然ゴム)、EPDM(エチレン−プロピレン−ジエン共重合ゴム)、NBR(ニトリルゴム)及びPAN(ポリアクリロニトリル)の4種類の高分子材料を、本発明の実施例の5種類のニトリルオキシド化合物(B1、C、D1、E、F)を変性剤として用いて変性した変性高分子材料を製造した。また、比較例として、EPDMを、ニトリルオキシド化合物Aで変性した変性高分子材料を製造した。
【0075】
ニトリルオキシド化合物B1と、NBRとの反応を次に示す。
【化20】
【0076】
それぞれの実施例及び比較例の製造(反応)条件を次の表2、3に示す。また、ニトリルオキシド化合物に、ニトリルオキシド化合物A又はニトリルオキシド化合物B1を用いたもは収率と修飾率とを表2に示す。ニトリルオキシド化合物に、ニトリルオキシド化合物C、ニトリルオキシド化合物D1、ニトリルオキシド化合物E又はニトリルオキシド化合物Fを用いたもは変性率を表3に示す。なお、表2、3のニトリルオキシド化合物の欄の括弧内の数字は、それぞれのニトリルオキシド化合物の添加量(高分子材料に対する当量)である。また、NBRの修飾率は、炭素−炭素二重結合(diene)とニトリル基(CN)とのそれぞれについて示す。
【0077】
【表2】
【0078】
【表3】
【0079】
本実施例及び比較例に用いた高分子材料のうち、EPDMはジエンの質量比が10%のものを用い、NBRはアクリロニトリルの質量比が33%のものを用いた。
【0080】
次に、各実施例及び比較例について説明する。
実施例1は、トルエン(toluene)の溶媒中にNRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、70℃の温度で2時間攪拌して反応を行った。
実施例2は、攪拌又は混合して反応を行う時間(以下、反応時間と言う場合がある)を24時間に変更した以外は実施例1と同じ条件で反応を行った。
実施例3は、反応時間を48時間に変更した以外は実施例1と同じ条件で反応を行った。
【0081】
実施例4は、反応が行われる温度(以下、反応温度と言う場合がある)を100℃に変更した以外は実施例1と同じ条件で反応を行った。
実施例5は、反応時間を24時間に変更した以外は実施例4と同じ条件で反応を行った。
実施例6は、反応時間を48時間に変更した以外は実施例4と同じ条件で反応を行った。
【0082】
実施例7は、メシチレン(mesitylene)の溶媒中にNRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、120℃の温度で48時間攪拌して反応を行った。
【0083】
実施例8は、溶媒を用いず空気下において、粉砕したNRと1.0当量のニトリルオキシド化合物B1とを室温(RT)の乳鉢中で2時間加圧混合して反応を行った。
実施例9は、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例8と同じ条件で反応を行った。
【0084】
実施例10は、トルエンの溶媒中にEPDMを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、100℃の温度で48時間攪拌して反応を行った。
【0085】
実施例11は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物B1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例12は、ニトリルオキシド化合物B1の量を1.0当量に変更し、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例11と同じ条件で反応を行った。
【0086】
実施例13は、CHCl3(クロロホルム)の溶媒中にNBRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、室温で48時間攪拌して反応を行った。
実施例14は、反応温度を70℃に変更し、反応時間を24時間に変更した以外は実施例13と同じ条件で反応を行った。
実施例15は、反応温度を70℃に変更した以外は実施例13と同じ条件で反応を行った。具体的には、2.0mLのCHCl3の溶媒中に、50mg(0.93mmol)のNBRを溶解させ、そこに260mg(0.93mmol)のニトリルオキシド化合物B1を添加し、70℃で48時間攪拌した。その後、この反応溶液をメタノールに再沈殿させ、不溶部を真空乾燥して、ニトリルオキシド化合物B1によって変性された変性NBRを137mg(収率:97%)得た。
【0087】
実施例16は、溶媒を用いず空気下において、粉砕したNBRと1.0当量のニトリルオキシド化合物B1とを室温の乳鉢中で2時間加圧混合して反応を行った。
実施例17は、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例16と同じ条件で反応を行った。具体的には、凍結粉砕した50mg(0.93mmol)のNBRと、260mg(0.93mmol)のニトリルオキシド化合物B1とを、ホットプレート上で100℃に温度調整された乳鉢中で2時間加圧混合した。その後、得られた生成体をメタノールで洗浄し、不溶部を真空乾燥して、ニトリルオキシド化合物B1によって変性された変性NBRを157mg(収率:61%)得た。
【0088】
実施例18は、DMF(N,N−ジメチルホルムアミド)の溶媒中にPANを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、100℃で1時間攪拌して反応を行った。
実施例19は、反応時間を24時間に変更した以外は実施例18と同じ条件で反応を行った。
実施例20は、反応温度を70℃に変更し、反応時間を48時間に変更した以外は実施例18と同じ条件で反応を行った。
【0089】
実施例21は、溶媒を用いず空気下において、粉砕したNRと0.04当量のニトリルオキシド化合物D1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0090】
実施例22は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Cとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0091】
実施例23は、溶媒を用いず空気下において、粉砕したEPDMと0.05当量のニトリルオキシド化合物D1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例24は、ニトリルオキシド化合物D1の量を0.2当量に変更した以外は実施例23と同じ条件で反応を行った。
【0092】
実施例25は、溶媒を用いず空気下において、粉砕したEPDMと0.04当量のニトリルオキシド化合物Eとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0093】
実施例26は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Fとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0094】
実施例27は、トルエンの溶媒中にNBRを溶解させ、そこに0.1当量のニトリルオキシド化合物D1を添加し、100℃で48時間攪拌して反応を行った。
【0095】
実施例28は、溶媒を用いず空気下において、粉砕したNBRと0.1当量のニトリルオキシド化合物D1とを100℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例29は、ニトリルオキシド化合物D1の量を0.04当量に変更し、130℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例28と同じ条件で反応を行った。
【0096】
比較例1は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Aとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0097】
(3)修飾率
修飾率、即ち、高分子材料中の炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加した割合を、IR測定及び1HNMR測定により算出した。従って、修飾率が100%であると高分子材料中の全ての炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加したことになり、50%であると高分子材料中の半数の炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加したことになる。
【0098】
(4)収率
上記のようにして求められた修飾率による理論収量を求め、その理論収量に対する実際の収量の割合を算出し、その値を収率として求めた。
具体的には、実際に得られた変性高分子材料の収量(質量)Wpを、その変性高分子材料の修飾率より算出された理論収量(質量)Wtで割った百分率である。
収率[%]=Wp÷Wt×100
【0099】
(5)変性率
変性率は、次のようにして求めた。
各実施例の変性高分子材料に、パーオキサイド系加硫剤(2,5−ジメチル−2,5−ビス(t−ブチルペルオキシ)ヘキサン)を2質量%添加した後、180℃、20分間、10MPaの条件で処理することによって架橋を行い精製前架橋サンプルを作成した。その後、この精製前架橋サンプルをメタノールで洗浄して、未反応のニトリルオキシド化合物を除去した精製後架橋サンプルを作成した。
そして、精製後架橋サンプルの質量Waを、精製前架橋サンプルの質量Wbで割った値に、ニトリルオキシド化合物の添加当量Eqを掛けた百分率として算出した。
変性率[%]=Wa÷Wb×Eq×100
【0100】
実施例11と比較例1との対比より、ニトリルオキシド化合物B1、即ち、2,2−ジフェニルヘキサンニトリル−N−オキシド(実施例11)は、同じ反応条件において、ニトリルオキシド化合物A、即ち、2−メトキシ−1−ナフトニトリルオキシドを用いた場合(比較例1)より、修飾率が高いことから、高分子材料との反応の前に分解するものが少なくなった。
【0101】
(6)耐油性
ニトリルオキシド化合物でゴムを変性した本発明の実施例の変性高分子材料(8種類)、比較例1の変性高分子材料及び未変性のゴム(EPDM、NR、NBR)について、耐油性として膨潤度を測定し、その結果を次の表4に示す。
【0102】
【表4】
【0103】
試験方法は、各試料にパーオキサイド系加硫剤(2,5−ジメチル−2,5−ビス(t−ブチルペルオキシ)ヘキサン)を2質量%添加した後、180℃、20分間、10MPaの条件で処理することによって架橋を行い試験片を作成した。
そして、この試験片をNo.3油(IRM903)に100℃、72時間の条件で浸漬して試験を行った。
そして、試験前後(浸漬前後)の試験片の質量変化の量より、膨潤度を算出した。具体的には、試験(浸漬)直後の試験片の質量Taから試験(浸漬)前の試験片の質量Tbを引いた値を、試験(浸漬)前の試験片の質量Tbで割った百分率である。
膨潤度[%]=(Ta−Tb)÷Tb×100
【0104】
EPDM、NR又はNBRに、本発明の実施例のニトリルオキシド化合物を反応させたもの(実施例11、21〜26、29)は、未変性の(ニトリルオキシド化合物を反応させていない)もの(比較例2〜4)に対し、膨潤度が変化したことから、本発明の実施例のニトリルオキシド化合物によってEPDM、NR及びNBRの油に対する性質(耐油性)を変えることができた。
ニトリルオキシド化合物B1でEPDMを変性したもの(実施例11)は、ニトリルオキシド化合物AでEPDMを変性したもの(比較例1)及び変性されていないEPDM(比較例2)より膨潤度が小さくなり、耐油性が向上した。
メチルメタクリレート、t−ブチルアクリレート又はN,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物D1、同E、同FでEPDMを変性したもの(実施例23〜26)は、ニトリルオキシド化合物AでEPDMを変性したもの(比較例1)及び変性されていないEPDM(比較例2)より膨潤度が小さくなり、耐油性が向上した。
メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物D1でNRを変性したもの(実施例21)は、変性されていないNR(比較例3)より膨潤度が小さくなり、耐油性が向上した。
スチレンが重合した炭素鎖を有するニトリルオキシド化合物CでEPDMを変性したもの(実施例22)は、変性されていないEPDM(比較例2)より膨潤度がかなり大きくなり、親油性が大きく向上した。
メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物D1でNBRを変性したもの(実施例29)は、変性されていないNBR(比較例4)より膨潤度が少し大きくなり、親油性が少し向上した。
【0105】
なお、本発明は前記実施例に限定されるものではなく、発明の趣旨から逸脱しない範囲で適宜変更して具体化することもできる。
例えば、スチレン、メチルメタクリレート、t−ブチルアクリレート又はN,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物において、その合成で、1−ニトロ−2,2−ジフェニルエチレンの替わりに1−ニトロ−2−メチル−2−フェニルエチレンを用いることで、炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)にメチル基とフェニル基とが結合した末端構造が結合したニトリルオキシド化合物にする。
ニトリルオキシド化合物をNR、EPDM、NBR又はPANに反応させる反応過程を二軸押出機等の混練装置で行う。
【技術分野】
【0001】
本発明は、ニトリルオキシド化合物、ニトリルオキシド化合物で高分子材料を変性した変性高分子材料、この変性高分子材料の製造方法及びこの変性高分子材料の成形体に関するものである。
【背景技術】
【0002】
EPDM、NR、NBR等のように分子内に炭素−炭素二重結合を有する高分子材料は、太陽光(特に紫外線)やオゾンにより劣化し易いため、用途によっては耐候性に問題が生じることがあった。また、これらの高分子材料は、特定の有機溶媒等に対しては溶解することがあり、そのような有機溶媒等と接触するおそれがある部位にも用いることができなかった。このため、これらの高分子材料の用途を広げる一策として、特許文献1、2に記載のように、ベンゼン等の芳香環にニトリルオキシド基が結合したニトリルオキシド化合物での変性(化学修飾)が考えられている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2011−52072号公報
【特許文献2】特開平11−180943号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、特許文献1、2に記載のニトリルオキシド化合物のように、芳香環にニトリルオキシド基が結合した芳香族ニトリルオキシド化合物は、安定性が低いものとなっていた(本明細書の段落0066〜段落0073参照)。このため、分解し易くなっており、高分子材料と反応する前に分解してしまうものが多い場合には、高分子材料を十分に変性できないおそれがあった。
【0005】
また、特許文献1、2に記載のニトリルオキシド化合物は、分子内に含まれる極性基が少ないことから、このニトリルオキシド化合物で変性した変性高分子材料は、求められる耐油性が高い場合には、その求めに十分な耐油性を得ることはできなかった。
【0006】
そこで、本発明は、安定性が高いニトリルオキシド化合物と、このニトリルオキシド化合物で高分子材料を変性した変性高分子材料と、この変性高分子材料の製造方法と、この変性高分子材料からなる成形体とを提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明の発明者らは、芳香族ニトリルオキシド化合物の安定性が低い理由は、芳香環の高い転位能に起因して、分子内転位反応が起こり、イソシアナートへと異性化し易いためであると考えた。
【0008】
そこで、炭素鎖の末端にニトリルオキシド基が結合した脂肪族ニトリルオキシド化合物にすることで分子内転位反応を起こり難くし、且つ、そのα位の炭素にフェニル基が二つ結合することでニトリルオキシ基を反応し難くしている脂肪族ニトリルオキシド誘導体を合成したところ、安定性が高いことを見出した(本明細書の段落0066〜段落0073参照)。
【0009】
上記課題を解決する本発明のニトリルオキシド化合物は、極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端、スチレン系単量体を重合してなる炭素鎖の片方の末端又はアルキル基に、下記の一般式[1]で表される末端構造が結合しているニトリルオキシド化合物である。
【化2】
(式中、R1、R2は、同一又は異なって、アルキル基又はアリール基を示す。)
【0010】
本発明の変性高分子材料の製造方法は、分子内にニトリルオキシドと反応する多重結合を有する高分子材料を変性した変性高分子材料の製造方法であって、上記のニトリルオキシド化合物を前記高分子材料に反応させる反応過程を備えることを特徴とする。
【0011】
本発明の変性高分子材料は、分子内にニトリルオキシドと反応する多重結合を有する高分子材料が、上記のニトリルオキシド化合物で変性されてなる。
【0012】
本発明の成形体は、上記の変性高分子材料を成形してなる。
【0013】
本発明のニトリルオキシド化合物、変性高分子材料、変性高分子材料の製造方法及び成形体における各要素の態様を以下に例示する。
【0014】
1.極性のアニオン重合性単量体
極性のアニオン重合性単量体としては、特に限定されないが、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド、メタクリル酸アミド等が例示できる。
【0015】
アクリル酸エステルとしては、特に限定されないが、アクリル酸メチル、アクリル酸エチル、アクリル酸n−プロピル、アクリル酸n−ブチル、アクリル酸n−ペンチル、アクリル酸n−ヘキシル、アクリル酸n−ヘプチル、アクリル酸n−オクチル、アクリル酸n−デシル、アクリル酸n−ドデシル、アクリル酸n−ラウリル、アクリル酸n−テトラデシル、アクリル酸n−ヘキサデシル、アクリル酸n−オクタデシル、アクリル酸イソプロピル、アクリル酸イソブチル、アクリル酸t−ブチル、アクリル酸イソペンチル、アクリル酸ネオペンチル、アクリル酸イソヘキシル、アクリル酸イソヘプチル、アクリル酸イソオクチル、アクリル酸2−エチルヘキシル、アクリル酸フェニル、アクリル酸ビフェニル、アクリル酸ジフェニルエチル、アクリル酸t−ブチルフェニル、アクリル酸シクロヘキシル、アクリル酸t−ブチルシクロヘキシル等が例示できる。
【0016】
メタクリル酸エステルとしては、特に限定されないが、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸n−プロピル、メタクリル酸n−ブチル、メタクリル酸n−ペンチル、メタクリル酸n−ヘキシル、メタクリル酸n−ヘプチル、メタクリル酸n−オクチル、メタクリル酸n−デシル、メタクリル酸n−ドデシル、メタクリル酸n−ラウリル、メタクリル酸n−テトラデシル、メタクリル酸n−ヘキサデシル、メタクリル酸n−オクタデシル、メタクリル酸イソプロピル、メタクリル酸イソブチル、メタクリル酸t−ブチル、メタクリル酸イソペンチル、メタクリル酸ネオペンチル、メタクリル酸イソヘキシル、メタクリル酸イソヘプチル、メタクリル酸イソオクチル、メタクリル酸2−エチルヘキシル、メタクリル酸フェニル、メタクリル酸ビフェニル、メタクリル酸ジフェニルエチル、メタクリル酸t−ブチルフェニル、メタクリル酸シクロヘキシル、メタクリル酸t−ブチルシクロヘキシル等が例示できる。
【0017】
アクリル酸アミドとしては、特に限定されないが、アクリルアミド、N−メチルアクリルアミド、N,N−ジメチルアクリルアミド、N−エチルアクリルアミド、N,N−ジエチルアクリルアミド、N−プロピルアクリルアミド、N,N−ジプロピルアクリルアミド、N−ブチルアクリルアミド、N,N−ジブチルアクリルアミド等が例示できる。
【0018】
メタクリル酸アミドとしては、特に限定されないが、メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−エチルメタクリルアミド、N,N−ジエチルメタクリルアミド、N−プロピルメタクリルアミド、N,N−ジプロピルメタクリルアミド、N−ブチルメタクリルアミド、N,N−ジブチルメタクリルアミド等が例示できる。
【0019】
2.スチレン系単量体
スチレン系単量体としては、特に限定されないが、スチレン、ビニルナフタレン、2−メチルスチレン、3−メチルスチレン、4−メチルスチレン、2−エチルスチレン、3−エチルスチレン、4−エチルスチレン、α−メチルスチレン等が例示できる。
【0020】
3.重合
極性のアニオン重合性単量体又はスチレン系単量体を重合させる重合方法は、特に限定されないが、これらの単量体を直鎖状に重合することができ、且つ、1−ニトロ−2,2ジフェニルエチレン、1−ニトロ−2−メチル−2−フェニルエチレン等のニトリルエチレン誘導体を用いてニトリルオキシドを合成できることから、アニオン重合であることが好ましい。
【0021】
アニオン重合の場合に用いる重合開始剤としては、特に限定されないが、メチルリチウム、n−ブチルリチウム、sec−ブチルリチウム、t−ブチルリチウム等のアルカリ金属アルキルや、ナトリウムメチラート、ナトリウムエチラート、カリウムメチラート、リチウムメチラート等のアルカリ金属アルコキシドや、グリニャール試薬等が例示でき、反応性が高いことから、アルカリ金属アルキルが好ましい。また、重合副反応を抑制できることから、ジフェニルエチレンを併用することが好ましい。
【0022】
4.アルキル基
アルキル基としては、特に限定されないが、直鎖状でもよいし、分岐状でもよいし、環状でもよい。上記の一般式[1]のR1、R2のアルキル基の場合には、ニトリルオキシド化合物が合成し易いことから、炭素数が1〜20の直鎖状又は分岐状のものが好ましく、より好ましくは、炭素数が1〜4の直鎖状又は分岐状のものである、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、sec−ブチル基、イソブチル基、t−ブチル基である。
【0023】
5.アリール基
アリール基としては、特に限定されないが、フェニル基、ナフチル基等が例示できる。
【0024】
6.ニトリルオキシド化合物
ニトリルオキシド化合物の分子量は、特に限定されないが、極性のアニオン重合性単量体又はスチレン系単量体を重合してなる炭素鎖を有するニトリルオキシド化合物の場合には、数平均分子量(Mn)が1000〜20000が例示できる。
【0025】
7.高分子材料
高分子材料の多重結合としては、特に限定されないが、C=S、N=N、P(V)=C、C=P(III)、C=As、C=C、C=N、C=Se、B=N、C≡P、C≡C、P(V)=N、C≡N、C=O等が例示できる。
【0026】
高分子材料としては、特に限定されないが、分子内にニトリル基(C≡N)を有するPAN(ポリアクリロニトリル)等の高分子や、分子内に炭素−炭素二重結合(C=C)を有するNR(天然ゴム)、EPDM(エチレン−プロピレン−ジエン共重合ゴム)、分子内にニトリル基及び炭素−炭素二重結合を有するNBR(ニトリルゴム)等の分子内に炭素−炭素二重結合を有するゴム等が例示できる。
【0027】
8.反応過程
反応過程は、特に限定されないが、有機溶媒中で行ってもよいし、無溶媒で行ってもよい。
【0028】
8−1.有機溶媒中での反応過程
有機溶媒としては、特に限定されないが、高分子材料及びニトリルオキシド化合物が共に溶解し易いものであることが好ましい。具体的には、トルエン、メシチレン、クロロホルム、DMF(N,N−ジメチルホルムアミド)等が例示できる。
【0029】
8−2.無溶媒での反応過程
無溶媒で行う場合には、空気下で行ってもよいし、不活性ガスが充填された雰囲気下で行ってもよい。
不活性ガスとしては、特に限定されないが、アルゴン、窒素等が例示できる。
反応過程が無溶媒で行われる場合には、効率よく製造できることから、反応過程を、混練装置で行う、即ち、混練装置を用いて混練装置内で反応過程を行うことが好ましい。
混練装置としては、特に限定されないが、二軸混練機、密閉式混練機、バンバリーミキサー、インターミックス等の混練機や二軸押出機、単軸押出機、多軸押出機等の押出機等が例示できる。
【0030】
8−3.反応過程の温度
反応過程の温度は、ニトリルオキシド化合物が高分子材料と反応する温度であれば、特に限定されない。敢えていうならば、化学反応であることから温度が高ければ反応が促進され、また加熱等の温度調節を行わなければ製造工程の管理が容易になることから、0〜150℃であることが好ましい。
【0031】
9.成形体
成形体の成形方法は、特に限定されないが、圧縮成形、射出成形、トランスファー成形等が例示できる。
成形体は、特に限定されないが、耐油性が高い場合には、オイルホース、ガスケット等が例示できる。
【発明の効果】
【0032】
本発明によれば、安定性が高いニトリルオキシド化合物と、このニトリルオキシド化合物で高分子材料を変性した変性高分子材料と、この変性高分子材料の製造方法と、この変性高分子材料からなる成形体を提供することができる。
【図面の簡単な説明】
【0033】
【図1】本発明の実施例B1であるニトリルオキシド化合物B1のDSC曲線のグラフである。
【図2】同じく実施例Cであるニトリルオキシド化合物CのDSC曲線のグラフである。
【図3】同じく実施例Dであるニトリルオキシド化合物D1のDSC曲線のグラフである。
【図4】比較例Aであるニトリルオキシド化合物AのDSC曲線のグラフである。
【発明を実施するための形態】
【0034】
本発明のニトリルオキシド化合物、変性高分子材料及び変性高分子材料の製造方法の好適な実施形態について以下に説明する。
【0035】
本発明の好適な実施形態のニトリルオキシド化合物は、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、下記の構造式[3a]又は[3b]で表される末端構造が結合している化合物、即ち、ニトリルオキシド基のα位の炭素に二つのフェニル基又はメチル基とフェニル基とが結合し、且つ、このα位の炭素が、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に結合している化合物である。
【化3】
【化4】
【0036】
本発明の好適な実施形態の変性高分子材料は、分子内に炭素−炭素二重結合を有するゴムが、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、上記の構造式[3a]又は[3b]で表される末端構造が結合しているニトリルオキシド化合物で変性されてなる変性ゴムである。
【0037】
本発明の好適な実施形態の変性高分子材料の製造方法は、分子内に炭素−炭素二重結合を有するゴムを変性した変性ゴムの製造方法であって、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドを重合してなる炭素鎖の片方の末端に、上記の構造式[3a]又は[3b]で表される末端構造が結合しているニトリルオキシド化合物を、分子内に炭素−炭素二重結合を有するゴムに反応させる反応過程を備えることを特徴とする。
【実施例】
【0038】
<ニトリルオキシド化合物>
本発明の実施例の8種類のニトリルオキシド化合物について説明する。
【0039】
各ニトリルオキシド化合物の説明の前に、これらのニトリルオキシド化合物の合成において、ニトリルオキシド基を導入するために用いた、1−ニトロ−2,2−ジフェニルエチレン及び1−ニトロ−2−メチル−2−フェニルエチレンについて説明する。
【0040】
《1》1−ニトロ−2,2−ジフェニルエチレン
1−ニトロ−2,2−ジフェニルエチレンの構造式[4]を次に示す。
【化5】
【0041】
1−ニトロ−2,2−ジフェニルエチレンの合成は、次のようにして行った。
【化6】
【0042】
32.3g(200mmol)のHMDS(ヘキサメチルジシラザン)を溶かした100mLのTHF(テトラヒドロフラン)溶液を、0℃で攪拌しているところへ、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液76.9mL(n−BuLi量:200mmol)を加えて、0℃で30分間攪拌した。なお、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。
その後、18.2g(100mmol)のベンゾフェノンを加えて、室温で1日間攪拌した。
その後、この反応溶液を減圧濃縮した後に、40mLのニトロメタンを加えて、10分間超音波を照射した後に、濾過して、濾液を3日間還流した。
その後、室温に放冷した後に、減圧濃縮して粗生成物を得た。
そして、この粗生成物をヘキサン/酢酸エチルで再結晶し、さらにトルエンで再結晶して、黄色板状結晶の1−ニトロ−2,2−ジフェニルエチレンを16.3g(収率:73%)得た。
【0043】
《2》1−ニトロ−2−メチル−2−フェニルエチレン
1−ニトロ−2−メチル−2−フェニルエチレンの構造式[5]を次に示す。
【化7】
【0044】
1−ニトロ−2−メチル−2−フェニルエチレンの合成は、次のようにして行った。
【化8】
【0045】
12.0g(0.10mol)のアセトフェノンを溶かした64mLのトルエン溶液に、21.6mL(0.40mol)のニトロメタンと、3.9mL(0.04mol)のノルマルブチルアミンとを加え、ディーン・スターク(Dean Stark)トラップを用いて水を留去しながら、5日間還流した。
その後、室温まで冷却した後に、60mLの酢酸エチルを加えて希釈した。
その後、30mLの飽和塩化アンモニウム水溶液を加えて分液し、有機相を硫酸マグネシウムで乾燥した。
その後、濾過を行い、濾液を減圧濃縮した後に、シリカゲルカラムクロマトグラフィー(溶離液は、ヘキサン+1%ジエチルエーテル)で精製して、赤色オイル状の1−ニトロ−2−メチル−2−フェニルエチレンを1.4g(収率:11%)得た。
【0046】
上記のようにして合成された1−ニトロ−2,2−ジフェニルエチレン又は1−ニトロ−2−メチル−2−フェニルエチレンを用いて合成された8種類のニトリルオキシド化合物について説明する。
【0047】
〈実施例B1〉2,2−ジフェニルヘキサンニトリル−N−オキシド
2,2−ジフェニルヘキサンニトリル−N−オキシド(以下、ニトリルオキシド化合物B1と言う場合がある)の構造式[B1]を次に示す。
【化9】
【0048】
ニトリルオキシド化合物B1の合成は、次のようにして行った。
【化10】
【0049】
アルゴン雰囲気下において、−78℃の80mLのTHFに、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液3.08mL(n−BuLi量:8.00mmol)を滴下し、5分間攪拌した後に、901mg(4.00mmol)の1−ニトロ−2,2−ジフェニルエチレンを加えて、30分間攪拌した。なお、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。また、以下に説明する他のニトリルオキシド化合物の合成においても、THFは、ナトリウム−ベンゾフェノンで蒸留したものを使用した。
その後、この反応溶液を0℃の95%濃硫酸中に滴下し、30分間攪拌した。
その後、ジクロロメタンで抽出し、純水で洗浄した後に、MgSO4(硫酸マグネシウム)で乾燥して粗生成物を得た。
その後、この粗生成物をシリカゲルカラムクロマトグラフィー(溶離液は、ヘキサン:酢酸エチル=5:1(体積比)を使用)によって、橙色オイル状のニトリルオキシド化合物B1を1.01g(収率:95%)得た。
【0050】
〈実施例B2〉2−メチル−2−フェニルヘキサンニトリル−N−オキシド
2−メチル−2−フェニルヘキサンニトリル−N−オキシド(以下、ニトリルオキシド化合物B2と言う場合がある)の構造式[B2]を次に示す。
【化11】
【0051】
ニトリルオキシド化合物B2の合成は、1−ニトロ−2,2−ジフェニルエチレンの替わりに、652mg(4.00mmol)の1−ニトロ−2−メチル−2−フェニルエチレンを加えた点のみが、ニトリルオキシド化合物B1の合成と異なり、その他の点はニトリルオキシド化合物B1の合成と同じである。そして、赤色オイル状のニトリルオキシド化合物B2(収率:99%)が得られた。
【0052】
〈実施例C〉スチレンが重合した炭素鎖を有するニトリルオキシド化合物
スチレンが重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、sec−ブチル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Cと言う場合がある)の構造式[C]を次に示す。式中nは自然数である。
【化12】
【0053】
ニトリルオキシド化合物Cの合成は、次のようにして行った。
【化13】
【0054】
アルゴン雰囲気下において、1.02Mのsec−ブチルリチウム(sec−BuLi)のシクロヘキサン溶液3.92mL(sec−BuLi量:4.00mmol)を加えた40mLのTHFを、−78℃で攪拌しているところへ、4.58mL(40.0mmol)のスチレンを溶かした35mLのTHF溶液を加え、−78℃で10分間攪拌した。
その後、901mg(4.00mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした5.0mLのTHF溶液を加え、−78℃で30分間攪拌した。
その後、この反応液を、0℃の2.0g(20.0mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応溶液を、CH2Cl2/H2O(ジクロロメタンと水)で抽出し、有機相をMgSO4(硫酸マグネシウム)で乾燥した。
その後、溶媒を減圧濃縮して、ニトリルオキシド化合物C(数平均分子量Mn:1500(Mw/Mn:1.85))の粗生成物を得た。なお、後述する、高分子材料の変性には、この粗生成物をそのまま使用した。
【0055】
〈実施例D〉メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物
メチルメタクリレート(メタクリル酸メチル)が重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニルヘキシル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Dと言う場合がある)の構造式[D]を次に示す。式中nは自然数である。
【化14】
【0056】
ニトリルオキシド化合物Dの合成は、次のようにして行った。なお、ニトリルオキシド化合物Dは、メチルメタクリレートの重合度を変えた、ニトリルオキシド化合物D1、ニトリルオキシド化合物D2及びニトリルオキシド化合物D3の3種類を合成した。
【化15】
【0057】
アルゴン雰囲気下において、1.80g(10.0mmol)の1,1−ジフェニルエチレンを溶かした150mLのTHF溶液を、−78℃で攪拌しているところへ、2.6Mのn−ブチルリチウム(n−BuLi)のヘキサン溶液3.84mL(n−BuLi量:10.0mmol)を加え、−78℃で20分間攪拌した。
その後、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加え、−78℃で1時間攪拌した。
その後、2.25g(10.0mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした12.5mLのTHF溶液を加え、−78℃で30分間攪拌した。
その後、この反応液を、0℃の5.42mL(100mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応溶液を、CH2Cl2/H2O(ジクロロメタンと水)で抽出し、有機相をMgSO4(硫酸マグネシウム)で乾燥した。
その後、溶媒を減圧濃縮して、ニトリルオキシド化合物D1の粗生成物を得た。
その後、この粗生成物をクロロホルムに溶解し、ヘキサンに再沈殿することによって精製を行い、白色粉末のニトリルオキシド化合物D1(数平均分子量Mn:2300(Mw/Mn:1.26))を8.5g得た。
【0058】
また、加えるメチルメタクリレートの量を変えることでメチルメタクリレートの重合度を変えたニトリルオキシド化合物D2とニトリルオキシド化合物D3とを合成した。
【0059】
ニトリルオキシド化合物D2の合成は、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液の替わりに、21.30mL(200mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加えた点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。そして、ニトリルオキシド化合物D2(数平均分子量Mn:3062(Mw/Mn:1.35))を得た。
【0060】
ニトリルオキシド化合物D3の合成は、10.65mL(100mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液の替わりに、42.60mL(400mmol)のメチルメタクリレートを溶かした37.5mLのTHF溶液を加えた点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。そして、ニトリルオキシド化合物D3(数平均分子量Mn:8070(Mw/Mn:1.34))を得た。
【0061】
〈実施例E〉t−ブチルアクリレートが重合した炭素鎖を有するニトリルオキシド化合物
t−ブチルアクリレート(アクリル酸t−ブチル)が重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニルヘキシル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Eと言う場合がある)の構造式[E]を次に示す。式中nは自然数である。
【化16】
【0062】
ニトリルオキシド化合物Eの合成は、メチルメタクリレートのTHF溶液の替わりに、14.5mL(100mmol)のt−ブチルアクリレートを溶かしたTHF溶液を加え、0℃に昇温して、0℃で1時間攪拌した点のみが、ニトリルオキシド化合物D1の合成と異なり、その他の点はニトリルオキシド化合物D1の合成と同じである。なお、0℃で1時間攪拌した後、−78℃に冷却してから、1−ニトロ−2,2−ジフェニルエチレンのTHF溶液を加え、−78℃で30分間攪拌した。そして、褐色粉末のニトリルオキシド化合物E(数平均分子量Mn:18400(Mw/Mn:1.87))を得た。
【0063】
〈実施例F〉N,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物
N,N−ジメチルアクリルアミドが重合した炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)に二つのフェニル基が結合した末端構造が結合し、他方の末端に、1,1−ジフェニル−3−メチルペンチル基が結合したニトリルオキシド化合物(以下、ニトリルオキシド化合物Fと言う場合がある)の構造式[F]を次に示す。式中nは自然数である。
【化17】
【0064】
ニトリルオキシド化合物Fの合成は、次のようにして行った。
【化18】
【0065】
アルゴン雰囲気下において、180mg(1.0mmol)の1,1−ジフェニルエチレンを溶かした80mLのTHF溶液を、−78℃で攪拌しているところへ、1.02Mのsec−ブチルリチウム(sec−BuLi)のシクロヘキサン溶液1.96mL(sec−BuLi量:2.0mmol)を加え、−78℃で20分間攪拌した。
その後、1.0MのZnEt2(ジエチル亜鉛)のヘキサン溶液24mL(ZnEt2量:24mmol)を加え、−78℃で10分間攪拌した。
その後、1.98g(20mmol)のN,N−ジメチルアクリルアミドを溶かした10mLのTHF溶液を加え、−78℃で1時間攪拌した。
その後、450mg(2.0mmol)の1−ニトロ−2,2−ジフェニルエチレン(末端停止剤)を溶かした5mLのTHF溶液を加え、−20℃に昇温して、−20℃で1時間攪拌した。
その後、この反応液を、0℃の2.35mL(44mmol)の濃硫酸(>95%)中に注ぎ、0℃で30分間攪拌した。
その後、この反応液を、ヘキサンに再沈殿し、沈殿物を回収した。そして、この沈殿物をメタノール/THF(1/5:体積比)溶液で洗浄してZnEt2を除去し、ニトリルオキシド化合物F(数平均分子量Mn:3320(Mw/Mn:1.21))を得た。
【0066】
このようにして合成したニトリルオキシド化合物のうちで、ニトリルオキシド化合物B1、同C及び同D1について、安定性を調べるため、DSC(示差走査熱量)測定を行った。また、比較例として、2−メトキシ−1−ナフトニトリルオキシド(以下、ニトリルオキシド化合物Aと言う場合がある)のDSC測定を行った。さらに、ニトリルオキシド化合物B1及び同Aの半減期を測定した。
【0067】
〈比較例A〉2−メトキシ−1−ナフトニトリルオキシド
2−メトキシ−1−ナフトニトリルオキシドの構造式[A]を次に示す。この2−メトキシ−1−ナフトニトリルオキシドの合成は、特許文献1の段落0020に記載の方法で行った。
【化19】
【0068】
(1)DSC測定
測定条件は、窒素雰囲気下で、−60℃から200℃まで、毎分10℃の昇温スピードで行った。
ニトリルオキシド化合物B1、同C、同D1及び同Aを測定したDSC曲線のグラフを図1〜4に示す。
【0069】
ニトリルオキシド化合物B1は、図1に示すように、約160℃を超えたところに熱分解由来の発熱のピークがあることから、このピーク温度のところで分解が起きており、約160℃までは安定している。
ニトリルオキシド化合物Cは、図2に示すように、−50〜200℃の温度範囲に発熱のピークがないことから、この温度範囲で分解が起きず、安定している。
ニトリルオキシド化合物D1は、図3に示すように、−50〜200℃の温度範囲に発熱のピークがないことから、この温度範囲で分解が起きず、安定している。
ニトリルオキシド化合物Aは、図4に示すように、約110℃を超えたところに熱分解由来の発熱のピークがあることから、このピーク温度のところで分解が起きており、約110℃までしか安定していない。
【0070】
(2)半減期の測定
60℃又は100℃の温度における半減期を測定した。
具体的には、重溶媒(60℃での測定には重クロロホルムを使用、100℃での測定には重DMSOを使用)に試料を溶解し、それを60℃又は100℃に加熱し、その状態で試料の量が半減するまでの時間(半減期)をNMR測定装置で測定した。
【0071】
ニトリルオキシド化合物B1及び同Aの測定結果を表1に示す。
【表1】
【0072】
表1に示すように、ニトリルオキシド化合物B1は、60℃及び100℃において、48時間経っても半減しなかったことから、半減期が48時間以上であった。
ニトリルオキシド化合物Aは、60℃において、半減期が2時間であった。
【0073】
以上より、本発明の実施例であるニトリルオキシド化合物は、2−メトキシ−1−ナフトニトリルオキシドより、安定性が向上している。
なお、ニトリルオキシド化合物B2、ニトリルオキシド化合物D2、ニトリルオキシド化合物D3、ニトリルオキシド化合物E及びニトリルオキシド化合物Fについては、DSC測定及び半減期の測定を行っていないが、ニトリルオキシド化合物B2はニトリルオキシド化合物B1の測定結果より、ニトリルオキシド化合物D2及びニトリルオキシド化合物D3はニトリルオキシド化合物D1の測定結果より、ニトリルオキシド化合物E及びニトリルオキシド化合物Fはニトリルオキシド化合物C及びニトリルオキシド化合物D1の測定結果より、2−メトキシ−1−ナフトニトリルオキシドより安定性が向上していると推測できる。
【0074】
<変性高分子材料>
本発明の実施例として、NR(天然ゴム)、EPDM(エチレン−プロピレン−ジエン共重合ゴム)、NBR(ニトリルゴム)及びPAN(ポリアクリロニトリル)の4種類の高分子材料を、本発明の実施例の5種類のニトリルオキシド化合物(B1、C、D1、E、F)を変性剤として用いて変性した変性高分子材料を製造した。また、比較例として、EPDMを、ニトリルオキシド化合物Aで変性した変性高分子材料を製造した。
【0075】
ニトリルオキシド化合物B1と、NBRとの反応を次に示す。
【化20】
【0076】
それぞれの実施例及び比較例の製造(反応)条件を次の表2、3に示す。また、ニトリルオキシド化合物に、ニトリルオキシド化合物A又はニトリルオキシド化合物B1を用いたもは収率と修飾率とを表2に示す。ニトリルオキシド化合物に、ニトリルオキシド化合物C、ニトリルオキシド化合物D1、ニトリルオキシド化合物E又はニトリルオキシド化合物Fを用いたもは変性率を表3に示す。なお、表2、3のニトリルオキシド化合物の欄の括弧内の数字は、それぞれのニトリルオキシド化合物の添加量(高分子材料に対する当量)である。また、NBRの修飾率は、炭素−炭素二重結合(diene)とニトリル基(CN)とのそれぞれについて示す。
【0077】
【表2】
【0078】
【表3】
【0079】
本実施例及び比較例に用いた高分子材料のうち、EPDMはジエンの質量比が10%のものを用い、NBRはアクリロニトリルの質量比が33%のものを用いた。
【0080】
次に、各実施例及び比較例について説明する。
実施例1は、トルエン(toluene)の溶媒中にNRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、70℃の温度で2時間攪拌して反応を行った。
実施例2は、攪拌又は混合して反応を行う時間(以下、反応時間と言う場合がある)を24時間に変更した以外は実施例1と同じ条件で反応を行った。
実施例3は、反応時間を48時間に変更した以外は実施例1と同じ条件で反応を行った。
【0081】
実施例4は、反応が行われる温度(以下、反応温度と言う場合がある)を100℃に変更した以外は実施例1と同じ条件で反応を行った。
実施例5は、反応時間を24時間に変更した以外は実施例4と同じ条件で反応を行った。
実施例6は、反応時間を48時間に変更した以外は実施例4と同じ条件で反応を行った。
【0082】
実施例7は、メシチレン(mesitylene)の溶媒中にNRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、120℃の温度で48時間攪拌して反応を行った。
【0083】
実施例8は、溶媒を用いず空気下において、粉砕したNRと1.0当量のニトリルオキシド化合物B1とを室温(RT)の乳鉢中で2時間加圧混合して反応を行った。
実施例9は、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例8と同じ条件で反応を行った。
【0084】
実施例10は、トルエンの溶媒中にEPDMを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、100℃の温度で48時間攪拌して反応を行った。
【0085】
実施例11は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物B1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例12は、ニトリルオキシド化合物B1の量を1.0当量に変更し、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例11と同じ条件で反応を行った。
【0086】
実施例13は、CHCl3(クロロホルム)の溶媒中にNBRを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、室温で48時間攪拌して反応を行った。
実施例14は、反応温度を70℃に変更し、反応時間を24時間に変更した以外は実施例13と同じ条件で反応を行った。
実施例15は、反応温度を70℃に変更した以外は実施例13と同じ条件で反応を行った。具体的には、2.0mLのCHCl3の溶媒中に、50mg(0.93mmol)のNBRを溶解させ、そこに260mg(0.93mmol)のニトリルオキシド化合物B1を添加し、70℃で48時間攪拌した。その後、この反応溶液をメタノールに再沈殿させ、不溶部を真空乾燥して、ニトリルオキシド化合物B1によって変性された変性NBRを137mg(収率:97%)得た。
【0087】
実施例16は、溶媒を用いず空気下において、粉砕したNBRと1.0当量のニトリルオキシド化合物B1とを室温の乳鉢中で2時間加圧混合して反応を行った。
実施例17は、100℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例16と同じ条件で反応を行った。具体的には、凍結粉砕した50mg(0.93mmol)のNBRと、260mg(0.93mmol)のニトリルオキシド化合物B1とを、ホットプレート上で100℃に温度調整された乳鉢中で2時間加圧混合した。その後、得られた生成体をメタノールで洗浄し、不溶部を真空乾燥して、ニトリルオキシド化合物B1によって変性された変性NBRを157mg(収率:61%)得た。
【0088】
実施例18は、DMF(N,N−ジメチルホルムアミド)の溶媒中にPANを溶解させ、そこに1.0当量のニトリルオキシド化合物B1を添加し、100℃で1時間攪拌して反応を行った。
実施例19は、反応時間を24時間に変更した以外は実施例18と同じ条件で反応を行った。
実施例20は、反応温度を70℃に変更し、反応時間を48時間に変更した以外は実施例18と同じ条件で反応を行った。
【0089】
実施例21は、溶媒を用いず空気下において、粉砕したNRと0.04当量のニトリルオキシド化合物D1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0090】
実施例22は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Cとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0091】
実施例23は、溶媒を用いず空気下において、粉砕したEPDMと0.05当量のニトリルオキシド化合物D1とを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例24は、ニトリルオキシド化合物D1の量を0.2当量に変更した以外は実施例23と同じ条件で反応を行った。
【0092】
実施例25は、溶媒を用いず空気下において、粉砕したEPDMと0.04当量のニトリルオキシド化合物Eとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0093】
実施例26は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Fとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0094】
実施例27は、トルエンの溶媒中にNBRを溶解させ、そこに0.1当量のニトリルオキシド化合物D1を添加し、100℃で48時間攪拌して反応を行った。
【0095】
実施例28は、溶媒を用いず空気下において、粉砕したNBRと0.1当量のニトリルオキシド化合物D1とを100℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
実施例29は、ニトリルオキシド化合物D1の量を0.04当量に変更し、130℃に温度調整された乳鉢中での加圧混合に変更した以外は実施例28と同じ条件で反応を行った。
【0096】
比較例1は、溶媒を用いず空気下において、粉砕したEPDMと0.2当量のニトリルオキシド化合物Aとを130℃に温度調整された乳鉢中で2時間加圧混合して反応を行った。
【0097】
(3)修飾率
修飾率、即ち、高分子材料中の炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加した割合を、IR測定及び1HNMR測定により算出した。従って、修飾率が100%であると高分子材料中の全ての炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加したことになり、50%であると高分子材料中の半数の炭素−炭素二重結合及びニトリル基にニトリルオキシド化合物が付加したことになる。
【0098】
(4)収率
上記のようにして求められた修飾率による理論収量を求め、その理論収量に対する実際の収量の割合を算出し、その値を収率として求めた。
具体的には、実際に得られた変性高分子材料の収量(質量)Wpを、その変性高分子材料の修飾率より算出された理論収量(質量)Wtで割った百分率である。
収率[%]=Wp÷Wt×100
【0099】
(5)変性率
変性率は、次のようにして求めた。
各実施例の変性高分子材料に、パーオキサイド系加硫剤(2,5−ジメチル−2,5−ビス(t−ブチルペルオキシ)ヘキサン)を2質量%添加した後、180℃、20分間、10MPaの条件で処理することによって架橋を行い精製前架橋サンプルを作成した。その後、この精製前架橋サンプルをメタノールで洗浄して、未反応のニトリルオキシド化合物を除去した精製後架橋サンプルを作成した。
そして、精製後架橋サンプルの質量Waを、精製前架橋サンプルの質量Wbで割った値に、ニトリルオキシド化合物の添加当量Eqを掛けた百分率として算出した。
変性率[%]=Wa÷Wb×Eq×100
【0100】
実施例11と比較例1との対比より、ニトリルオキシド化合物B1、即ち、2,2−ジフェニルヘキサンニトリル−N−オキシド(実施例11)は、同じ反応条件において、ニトリルオキシド化合物A、即ち、2−メトキシ−1−ナフトニトリルオキシドを用いた場合(比較例1)より、修飾率が高いことから、高分子材料との反応の前に分解するものが少なくなった。
【0101】
(6)耐油性
ニトリルオキシド化合物でゴムを変性した本発明の実施例の変性高分子材料(8種類)、比較例1の変性高分子材料及び未変性のゴム(EPDM、NR、NBR)について、耐油性として膨潤度を測定し、その結果を次の表4に示す。
【0102】
【表4】
【0103】
試験方法は、各試料にパーオキサイド系加硫剤(2,5−ジメチル−2,5−ビス(t−ブチルペルオキシ)ヘキサン)を2質量%添加した後、180℃、20分間、10MPaの条件で処理することによって架橋を行い試験片を作成した。
そして、この試験片をNo.3油(IRM903)に100℃、72時間の条件で浸漬して試験を行った。
そして、試験前後(浸漬前後)の試験片の質量変化の量より、膨潤度を算出した。具体的には、試験(浸漬)直後の試験片の質量Taから試験(浸漬)前の試験片の質量Tbを引いた値を、試験(浸漬)前の試験片の質量Tbで割った百分率である。
膨潤度[%]=(Ta−Tb)÷Tb×100
【0104】
EPDM、NR又はNBRに、本発明の実施例のニトリルオキシド化合物を反応させたもの(実施例11、21〜26、29)は、未変性の(ニトリルオキシド化合物を反応させていない)もの(比較例2〜4)に対し、膨潤度が変化したことから、本発明の実施例のニトリルオキシド化合物によってEPDM、NR及びNBRの油に対する性質(耐油性)を変えることができた。
ニトリルオキシド化合物B1でEPDMを変性したもの(実施例11)は、ニトリルオキシド化合物AでEPDMを変性したもの(比較例1)及び変性されていないEPDM(比較例2)より膨潤度が小さくなり、耐油性が向上した。
メチルメタクリレート、t−ブチルアクリレート又はN,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物D1、同E、同FでEPDMを変性したもの(実施例23〜26)は、ニトリルオキシド化合物AでEPDMを変性したもの(比較例1)及び変性されていないEPDM(比較例2)より膨潤度が小さくなり、耐油性が向上した。
メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物D1でNRを変性したもの(実施例21)は、変性されていないNR(比較例3)より膨潤度が小さくなり、耐油性が向上した。
スチレンが重合した炭素鎖を有するニトリルオキシド化合物CでEPDMを変性したもの(実施例22)は、変性されていないEPDM(比較例2)より膨潤度がかなり大きくなり、親油性が大きく向上した。
メチルメタクリレートが重合した炭素鎖を有するニトリルオキシド化合物D1でNBRを変性したもの(実施例29)は、変性されていないNBR(比較例4)より膨潤度が少し大きくなり、親油性が少し向上した。
【0105】
なお、本発明は前記実施例に限定されるものではなく、発明の趣旨から逸脱しない範囲で適宜変更して具体化することもできる。
例えば、スチレン、メチルメタクリレート、t−ブチルアクリレート又はN,N−ジメチルアクリルアミドが重合した炭素鎖を有するニトリルオキシド化合物において、その合成で、1−ニトロ−2,2−ジフェニルエチレンの替わりに1−ニトロ−2−メチル−2−フェニルエチレンを用いることで、炭素鎖の一方の末端に、ニトリルオキシド基に結合した炭素(α位炭素)にメチル基とフェニル基とが結合した末端構造が結合したニトリルオキシド化合物にする。
ニトリルオキシド化合物をNR、EPDM、NBR又はPANに反応させる反応過程を二軸押出機等の混練装置で行う。
【特許請求の範囲】
【請求項1】
極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端、スチレン系単量体を重合してなる炭素鎖の片方の末端又はアルキル基に、下記の一般式[1]で表される末端構造が結合しているニトリルオキシド化合物。
【化1】
(式中、R1、R2は、同一又は異なって、アルキル基又はアリール基を示す。)
【請求項2】
前記極性のアニオン重合性単量体は、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドである請求項1記載のニトリルオキシド化合物。
【請求項3】
分子内にニトリルオキシドと反応する多重結合を有する高分子材料を変性した変性高分子材料の製造方法であって、
請求項1又は2記載のニトリルオキシド化合物を前記高分子材料に反応させる反応過程を備えることを特徴とする変性高分子材料の製造方法。
【請求項4】
前記高分子材料は、NR、EPDM、NBR又はPANである請求項3記載の変性高分子材料の製造方法。
【請求項5】
前記反応過程は、有機溶媒中又は無溶媒で行う請求項3又は4記載の変性高分子材料の製造方法。
【請求項6】
前記反応過程を無溶媒で行う場合に、前記反応過程を混練装置で行う請求項5記載の変性高分子材料の製造方法。
【請求項7】
分子内にニトリルオキシドと反応する多重結合を有する高分子材料が、請求項1又は2記載のニトリルオキシド化合物で変性されてなる変性高分子材料。
【請求項8】
前記高分子材料は、NR、EPDM、NBR又はPANである請求項7記載の変性高分子材料。
【請求項9】
請求項7又は8記載の変性高分子材料を成形してなる成形体。
【請求項1】
極性のアニオン重合性単量体を重合してなる炭素鎖の片方の末端、スチレン系単量体を重合してなる炭素鎖の片方の末端又はアルキル基に、下記の一般式[1]で表される末端構造が結合しているニトリルオキシド化合物。
【化1】
(式中、R1、R2は、同一又は異なって、アルキル基又はアリール基を示す。)
【請求項2】
前記極性のアニオン重合性単量体は、アクリル酸エステル、メタクリル酸エステル、アクリル酸アミド又はメタクリル酸アミドである請求項1記載のニトリルオキシド化合物。
【請求項3】
分子内にニトリルオキシドと反応する多重結合を有する高分子材料を変性した変性高分子材料の製造方法であって、
請求項1又は2記載のニトリルオキシド化合物を前記高分子材料に反応させる反応過程を備えることを特徴とする変性高分子材料の製造方法。
【請求項4】
前記高分子材料は、NR、EPDM、NBR又はPANである請求項3記載の変性高分子材料の製造方法。
【請求項5】
前記反応過程は、有機溶媒中又は無溶媒で行う請求項3又は4記載の変性高分子材料の製造方法。
【請求項6】
前記反応過程を無溶媒で行う場合に、前記反応過程を混練装置で行う請求項5記載の変性高分子材料の製造方法。
【請求項7】
分子内にニトリルオキシドと反応する多重結合を有する高分子材料が、請求項1又は2記載のニトリルオキシド化合物で変性されてなる変性高分子材料。
【請求項8】
前記高分子材料は、NR、EPDM、NBR又はPANである請求項7記載の変性高分子材料。
【請求項9】
請求項7又は8記載の変性高分子材料を成形してなる成形体。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2013−112741(P2013−112741A)
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願番号】特願2011−260135(P2011−260135)
【出願日】平成23年11月29日(2011.11.29)
【出願人】(000241463)豊田合成株式会社 (3,467)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願日】平成23年11月29日(2011.11.29)
【出願人】(000241463)豊田合成株式会社 (3,467)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
[ Back to top ]