
ヒト癌原遺伝子及びこれによってコードされるタンパク質
本発明は、既存の癌遺伝子とは相同性を示さない新規の癌原遺伝子HPP1(Human proto−oncogene1)及びこれによってコードされるタンパク質に関するものである。この新規癌原遺伝子は、多様な癌の診断、形質転換動物の製造及びアンチセンス(anti‐sense)遺伝子治療などに有効に利用され得る。

【発明の詳細な説明】
【発明の詳細な説明】
【0001】
〔技術分野〕
本発明は、新規癌原遺伝子及びこれによってコードされるタンパク質に関するものであり、より詳しくは、多様な癌の診断に用いられるヒト癌原遺伝子(以下、HPPI癌原遺伝子と称する)及びこれによってコードされるタンパク質に関する。
【0002】
〔背景技術〕
ヒトを含む高等動物は、約100、000個の遺伝子を有しているが、これらのうち約15%のみが各々の個体で発現される。従って、いかなる遺伝子が選択されて発現されるかによって生命のすべての現象、即ち、発生、分化、恒常性(homeostasis)、刺激に対する反応、細胞分裂周期の調節、老化及びアポプトシス(apoptosis; programmed cell death)などが決められる(Liang,P.and A.B.Pardee,Science 257: 967‐971,1992)。
【0003】
腫瘍発生のような病理学的現象は、遺伝子変異過程によって誘発され結局遺伝子発現の変化をもたらす。従って、相異なる細胞の間で現れる遺伝子発現同士の比較は多様な生物学的現象を理解する上で基本的で効果的な接近方法であるといえる。例えば、リアンとパーディー(Liang,P.and A.B.Pardee,Science 257: 967‐971,1992)によって提案されたmRNA鑑別展開(differential display)方法は、現在腫瘍抑制遺伝子や細胞分裂周期(cell cycle)に係る遺伝子及びアポプトシスに係る転写調節遺伝子(transcriptional regulatory gene)などを探索する上で有効に用いられており、また一つの細胞で起こる多様な遺伝子の相互関連性の究明にも多様に活用されている。
【0004】
腫瘍発生に対する多様な研究結果をまとめると、特定染色体部位の喪失(loss of chromosomal heterozygosity)、癌原遺伝子の活性化及びp53遺伝子を含む他の腫瘍抑制遺伝子の不活性化などのような種々の遺伝的変化が腫瘍組織に蓄積されてヒト腫瘍を起こすと報告された(Bishop,J.M.,Cell 64: 235‐248,1991; Hunter,T.,Cell 64: 249‐270,1991)。さらに、癌の10ないし30%は癌原遺伝子が増幅されることによって活性化され起こると報告されており、癌原遺伝子の活性化は多くの癌の病因学研究に重要な役割を果たし、これを明らかにすることが要求されている。
【0005】
そこで、本発明者らは、子宮頸部癌の発生メカニズムを癌原遺伝子の水準で接近した結果、ヒト癌原遺伝子タンパク質1(human protooncogene protein 1;HPP1)と名付けられた癌原遺伝子が、癌細胞にのみ特異に発現が増加されるということがわかった。上記癌原遺伝子は、白血病、リンパ腫、大腸癌、肺癌、皮膚癌、 腎臓癌、乳房癌、大腸癌、卵巣癌、胃癌、肝癌、及び子宮頸部癌などのような種々の癌の診断、予防及び治療に効果的に用いられ得る。
【0006】
〔発明の概要〕
本発明の主たる目的は、新規な癌原遺伝子を提供することである。
【0007】
本発明の他の目的は、上記癌原遺伝子を含む組換えベクター及びそれによって形質転換された微生物を提供することである。
【0008】
本発明のさらに他の目的は、上記癌原遺伝子によってコードされるタンパク質を提供することである。
【0009】
本発明のさらに他の目的は、上記癌原遺伝子を含む癌診断用キットを提供することである。
【0010】
本発明のさらに他の目的は、上記タンパク質を含む癌診断用キットを提供することである。
【0011】
本発明のさらに他の目的は、上記癌原遺伝子から誘導されるものに相補的な塩基配列を有するアンチセンス遺伝子を提供することである。
【0012】
本発明のさらに他の目的は、上記アンチセンス遺伝子を用いて癌及びその転移を予防するか治療する方法を提供することである。
【0013】
上記目的によって、本発明では配列番号のヌクレオチド配列を有する新規癌原遺伝子を提供する。
【0014】
上記の他の目的に従って、本発明では上記癌原遺伝子を含む組換えベクター及びそれによって形質転換された微生物を提供する。
【0015】
上記のさらに他の目的に従って、上記癌原遺伝子由来の配列番号2のアミノ酸配列を有するタンパク質を提供する。
【0016】
〔発明の詳細な説明〕
本発明の新規癌原遺伝子(oncogene)であるヒト癌原遺伝子タンパク質1 (Human protooncogene protein 1,HPP1)は、配列番号:1で記載される1、792bp長さのDNA配列を有する。
【0017】
配列番号:1の塩基配列において、ヌクレオチド番号16ないし1620部位(1618‐1620:終了コドン)に該当する開放型読み枠(open reading frame)は、全体タンパク質コーディング領域であって、それから誘導される予測アミノ酸配列は配列番号:2に示されており、534個のアミノ酸からなっている(以下、「HPP1タンパク質」と称する)。
【0018】
コドンの縮退性(degeneracy)によってまたは本発明の上記癌原遺伝子を発現させようとする特定生物で好まれるコドンを考慮して、配列番号:1のDNA配列はコーディング領域から発現される発癌タンパク質のアミノ酸配列を変化させない範囲内でコーディング領域に多様な変形が行われることができ、コーディング領域を除いた部分でも遺伝子の発現に影響を及ぼさない範囲内で多様な変形または修正が行われることができて、そのような変形遺伝子も本発明の範囲に含まれる。従って、本発明は上記配列番号:1の癌原遺伝子と実質的に同一の塩基配列を有するポリヌクレオチド及び上記遺伝子の断片をも含む。ここで、「実質的に同一のポリヌクレオチド」とは、本発明の癌原遺伝子と80%以上、望ましくは90%以上、最も望ましくは95%以上の配列相同性を有することを意味する。
本発明の癌原遺伝子から発現されるタンパク質は534個のアミノ酸からなっており配列番号:2で記載されたアミノ酸配列を有して大きさは約60kDaである。しかし、上記タンパク質のアミノ酸配列でも同様にタンパク質の機能に影響を及ぼさない範囲内でアミノ酸の置換、付加または欠失が行われることができ、目的によってタンパク質の一部のみが使用されることもある。そのような変形されたアミノ酸配列及びそれらの断片も本発明の範囲に含まれる。従って、本発明は上記発癌タンパク質と実質的に同一のアミノ酸配列を有するポリペプチド及びその断片をも含む。ここで、「実質的に同一のポリペプチド」とは、配列番号:2のアミノ酸配列と80%以上、望ましくは90%以上、最も望ましくは95%以上の配列相同性を有することを意味する。
【0019】
本発明の癌原遺伝子HPP1及びタンパク質はヒトの癌組織から分離したり、公知のDNAまたはペプチド合成方法によって合成することもできる。また、このように製造された遺伝子を当分野に公知された微生物発現用ベクターに挿入して発現ベクターを製造した後これを適切な宿主細胞、例えば、大腸菌または酵母細胞などに導入し得る。
【0020】
例えば、癌原遺伝子HPP1で大腸菌DH5αを形質感染(transfection)させ、これから得られたDH5α/HCC‐11/pCEV‐LACを、2002年12月5日付で、特許手続のための微生物寄託の国際的承認に関するブダペスト条約に従い、韓国生命工学研究所遺伝子銀行(Korea Collection for Type Cultures; KCTC、住所:Korean Research Institute of Bioscience and Biotechnology(KRIBB),#52 Oun−dong,Yusong−ku,Taejon 305−333,Republic of Korea)に寄託番号第KCTC 10397BP号として寄託した。
【0021】
ベクター制作の際には、用いられた宿主細胞の種類によってプロモーター、ターミネータなどのような発現調節配列、自家複製配列及び分泌シグナルなどを適宜選択して組み合わせることができる。
【0022】
本発明の遺伝子は、ノーザンブロット(northern blot)などの分析方法で正常子宮頸部組織では発現がほとんど確認されなかった反面、子宮頸部癌組織と子宮癌細胞株ではその過発現が確認されるので、子宮癌を誘発する強力な発癌遺伝子として判断される。また、癌転移リンパ節組織で発現が増加されることから癌転移を誘発する癌転移関連遺伝子とも判断される。子宮頸部癌のような上皮性組織の他にも、本発明の癌原遺伝子は、白血病、リンパ腫、大腸癌、皮膚癌、肺癌、腎臓癌、乳房癌、卵巣癌、胃癌、肝癌及び子宮頸部癌などのような多くの他の癌腫瘍で高く発現される。従って、本発明の癌原遺伝子は多様な癌の発生に共通した発癌遺伝子であると判断され、多様な癌の診断、形質転換された動物の製造及びアンチセンス(anti‐sense)遺伝子治療などに有効に利用され得る。
【0023】
上記癌原遺伝子を用いた癌の診断方法は、例えば、上記癌原遺伝子の全部または一部をプローブを用いて対象者の体液から分離した核酸とハイブリッド化した後、当分野で公知の多様な方法でこれを検出することによって、対象者が本発明の癌原遺伝子を有しているかどうかを判断する過程を含む。上記プローブを放射線同位元素または酵素などで標識することによって容易に癌原遺伝子HPP1の存在を確認することができる。従って、本発明では上記癌原遺伝子の全部または一部を含む癌診断用キットをも提供する。
【0024】
形質転換動物は本発明の癌原遺伝子を哺乳動物、例えば、鼠などの哺乳類動物に導入することによって製造でき、この遺伝子を少なくとも8細胞期以前の受精卵段階で導入することが望ましい。このように製造された形質転換動物は、発癌性物質または化学療法の薬物のような抗癌性物質の探索などに有用に用いられ得る。
【0025】
本発明は遺伝子治療にも有効であり、従って本発明は上記新規癌原遺伝子HPP1により誘導されたmRNA相補的塩基配列を含むアンチセンス遺伝子をも提供する。
【0026】
本発明は、また、遺伝子治療に効果的なアンチセンス遺伝子を提供する。本願において、「アンチセンス遺伝子(anti‐sense gene)」とは、配列番号:1の癌原遺伝子またはその一部の塩基配列を有する癌原遺伝子HPP1から転写されるmRNAの一部または全部と相補的な配列を有するポリヌクレオチであって、上記mRNAのタンパク質結合部位に結合して癌原遺伝子の開放型読み枠(open reading frame;ORF)の発現を抑制できる配列を有するポリヌクレオチドを意味する。
【0027】
本発明はまた、その範囲内に本発明のアンチセンス遺伝子の治療効果量を患者に投与することによって癌を治療または予防する方法を含む。
【0028】
本発明のアンチセンス遺伝子治療において、本発明のアンチセンス遺伝子は通常の方法で患者に投与されて癌原遺伝子の発現を予防する。例えば、文献(J.S.Kim et al.,J.Controlled Release 53:175‐182,1998)の方法に従ってアンチセンスオリゴデオキシヌクレオチド(ODN)をポリ‐L‐リシン誘導体と静電気的引力によって混合し、この混合体を患者に静脈投与する。
【0029】
本発明の範囲にはまた、薬学的に許容可能な担体、賦形剤または必要に応じて他の添加剤とともに本発明のアンチセンス遺伝子を活性成分として含む抗癌組成物が含まれる。本発明の薬剤学的組成物を注射剤として製剤化することが望ましい。
【0030】
実際に投与されるHPP1アンチセンス遺伝子の量は、治療される症状、投与経路、患者の年齢及び体重、症状の重症度のような種々の関連因子を考慮して決める。
【0031】
本発明の癌原遺伝子HPP1から発現されるタンパク質は、診断道具として有用な抗体を生産するのに有用に有用できる。本発明の抗体は、本発明の癌原遺伝子から発現された配列番号:2のアミノ酸配列を有するタンパク質またはその断片を用いて当分野で公知の方法にとってモノクロナルの抗体またはポリクロナルの抗体として製造でき、このような抗体を用いて当分野で公知の酵素免疫測定法(enzyme linked immunosorbentassay; ELISA)、放射線免疫測定法(radioimmunoassay; RIA)、サンドイッチ測定法(sandwich assay)、免疫組織化学染色(Immunohistochemical staining)、ポリアクリルアミドゲル上のウエスタンブロットまたは免疫ブロットなどの方法によって対象者の体液試料の中に上記タンパク質が発現されたかを確認することによって癌を診断することができる。従って、配列番号:2のアミノ酸配列を有するタンパク質またはその断片を含む癌診断キットも本発明の範囲に含まれる。
【0032】
また、本発明の癌原遺伝子を用いて持続的に増殖し得る癌細胞株を確立することができ、このような細胞株は、例えば、上記癌原遺伝子が形質導入された繊維母細胞を用いてヌードマウスなどに形成させた腫瘍組織から製造できる。このような癌細胞株は抗癌剤などの探索に有用に利用することができる。
【0033】
下記実施例は本発明をより詳しく説明するものであって、本発明の内容が下記実施例に限定されるのではない。
【0034】
〔実施例1〕:腫瘍細胞の培養及び総RNAの分離
(段階1)腫瘍細胞培養
子宮摘出術(hysterectomy)を受けた子宮筋腫患者から正常の子宮頸部(exocervical)組織、そして以前に抗癌療法と放射線治療を受けなかった子宮癌患者から手術の際に原発性子宮頸部腫瘍組織及び転移リンパ節腫瘍組織を取った。ヒト子宮頸部癌細胞株としてCUMC‐6(Kim,J.W.et al.,Gynecol.Oncol.62: 230‐240,1996)を用いた。
【0035】
上記組織及びCUMC‐6細胞株から得られた細胞を2mM グルタミン、100IU/ml ペニシリン、100μg/ml ストレプトマイシン及び10% ウシ胎児血清(Gibco,USA)が含有されたウエイマウス(Waymouth’s) MB 752/1培養液(Gibco)で増殖させた。実験に用いた培養細胞は指数的増殖過程の細胞であってトリパンブルー(trypan blue)染色によって95%以上の生存度を示す細胞を用いた(Freshney,「Culture of Animal Cells: A Manual of Basic Technique」 2nd Ed.,A.R.Liss,New York,1987)。
(段階2)RNAの分離及びmRNAの鑑別展開法
商業的に販売されるシステムであるRNeasy総RNAキット(Qiagen Inc.,ドイツ)を用いて、段階1で得られた組織標本及び細胞から総RNAを抽出した。メッセージクリーンキット(Message clean kit,GenHunter Corp.,Brookline,MA)を用いてRNAからDNA汚染源を除去した。
【0036】
〔実施例2〕:鑑別展開逆転写重合酵素連鎖反応(differential display reverse transcription‐PCR)
鑑別展開逆転写は、上記リアンとパーディーによって記述された逆転写‐重合酵素連鎖反応(RT‐PCR)を若干変形して行った。
【0037】
まず、実施例1の段階1で得られた総RNA 0.2μgずつに固定されたオリゴ‐dTプライマーとして配列番号:3で記載される塩基配列を有するH−T11A固定プライマー(RNAimage kit,Genhunter,Cor.,MA,USA)を用いて逆転写を行った。
【0038】
次いで、0.5mM[α‐35S] dATP(1200 Ci/mmole)の存在の下で、同一の固定プライマー及び無作為5’11プライマー(RNAimage primer sets 1‐5)H‐AP1ないし40の中、配列番号:4で記載される塩基配列を有するH‐AP33プライマーを用いてPCRを行った。PCR反応の条件は、95℃で40秒間反応後40℃で2分、72℃で40秒を40回反応させて最終延長(final extension)のために72℃で5分間さらに反応させた。得られたPCR産物を6% ポリアクリルアミドシーケンシングゲルに溶解した後、放射能写真を用いて相違な発現(differentially expressed)程度を示すバンドの位置を確認した。
【0039】
乾燥したゲルから181塩基対(bp)大きさのバンド、CA336 cDNA(配列番号:1の1569‐1749 bp)を切取った。15分間加熱してCA336 cDNAを溶出させた後、[α‐35S]標識されたdATP(1200 Ci/mmole)及び20μM dNTPを用いなかったことを除いては上記と同一のプライマー及び同一の条件でPCRを行なって再増幅させた。
【0040】
〔実施例3〕:クローニング
上記で得られた再増幅されたCA336 PCR産物をTAクローニングシステム(Promega、USA)を用いて製造社の方法に従ってpGEM‐T EASYベクターに挿入した。
(段階1)連結反応
実施例2で得られた再増幅されたCA336 PCR産物2μL、pGEM‐T EASYベクター(50ng)1μL、T4 DNA リガーゼ 10X 緩衝液 1μL及びT4 リガーゼー(3 weiss units/μL; T4 DNA ligase,Promega)1μLを0.5ml試験管に入れた後、蒸留水を加えて最終体積が10μLになるようにした。連結反応混合物は14℃で一晩中培養した。
(段階2)TAクローニング形質転換
TAクローニング形質転換は次のように行われた。
【0041】
大腸菌JM109(Promega,WI,USA)を10mlのLBブロス(バクト‐トリプトン 10g、バクト‐酵母抽出物 5g、NaCl 5g)中で600nmでの光学密度が約0.3ないし0.6になるまで培養した。培養混合物を氷に約10分間放置した後、4℃で10分間4000rpmで遠心分離して、上澄液を取除き、細胞を収集した。収集した細胞ペレットを10mlの氷冷0.1M CaCl2に30分ないし1時間露出させてコンペテント(competent)細胞を製造した。結果物を4℃で4000rpmで10分間さらに遠心分離して上澄液を取除き、細胞を収集して2mlの氷冷0.1M CaCl2に懸濁した。
【0042】
コンペテント細胞懸濁液200μLを新しいマイクロフュズ(microfuge)チューブに移し、これに段階1で製造した連結反応産物2μLを加えた。混合物を42℃の 水浴槽(water bath)で90秒間培養した後、0℃で急冷した。これに、SOC培地(バクト‐トリプトン2.0g,バクト‐酵母抽出物 0.5g,1M NaCl 1ml,1M KCl 0.25ml,TDW 97ml,2M Mg2+ 1ml,2M グルコース 1ml)800μLを加え、混合物を37℃で220rpmの回転振盪培養器で45分間培養した。
【0043】
37℃培養器に予め入れておいた、エンピシリンを含むLBプレートにX‐gal (40mg/ml ジメチルホルムアミドに貯蔵)25μLをガラス棒で拡散させた後、これに25μLの形質転換された細胞を入れてさらにガラス棒で拡散させ、37℃で一晩中培養した。培養後形成された白色コロニーを3〜4個選択してエンピシリンを含むLBプレートに各々の選択されたクローンを植えた。プラスミドを製造するためにこれらのうち挿入が確認されたコロニー、即ち、形質転換された大腸菌 JM109/CA336を選択して10mlのテリフィックブロス(terrific broth; TDW 900ml、バクト‐トリプトン 12g、バクト‐酵母抽出物 24g、グリセロール 4ml、0.17M KH2PO4、0.72N K2HPO4 100ml)で培養した。
【0044】
〔実施例4〕:組換えプラスミドDNAの分離
ウイザードフラスミニプレップスDNA精製キット(WizardTMPlus Minipreps DNA Purification Kit,Promega,USA)を用いて製造社の指針書に従って形質転換された大腸菌からCA336プラスミドDNAを分離した。
【0045】
分離されたDNAの一部をECoRI酵素で処理した後、2% ゲルで電気泳動を行ってCA336部分配列がプラスミドに挿入されたことを確認した。
【0046】
〔実施例5〕:DNA塩基配列分析
実施例2で得られたCA336 PCR産物を通常の方法に従って増幅させた後、クローニングして再増幅させたCA336 PCR断片をシケナーゼバージョン2.0DNA配列分析キット(Sequenase version 2.0 DNA Sequencing kit; United States Biochemical,Cleveland,OH,USA)を用いてジデオキシ鎖終結法(dideoxy chain termination)に従って配列分析を行った。
【0047】
上記DNAの塩基配列は配列番号: 1のヌクレオチド番号 1569 ないし1749に該当し、本願に「CA336」と命名した。
【0048】
5’無作為プライマーH‐AP33及び3’H‐T11A固定プライマーを用いて上記で得られた181 bp cDNA 断片、即ち、CA336を鑑別展開逆転写重合酵素連鎖反応(DDRT‐PCR)を行い、電気泳動で確認した。
【0049】
図1に示されたように、cDNA断片(CA336)は、転移リンパ節組織及びCUMC‐6細胞では発現されたが、正常組織では発現されなかった。
【0050】
〔実施例6〕:HPP1 癌原遺伝子の全体cDNA配列分析
32P‐標識されたCA336 cDNAをプローブを用いたプラークハイブリッド化(plaque hybridization)でバクテリオファージλgt11 ヒト肺胎児繊維母細胞cDNAライブラリー(Miki,T.et al.,Gene 83: 137‐146,1989)をスクリーニングした。上記ヒト肺胎児繊維母細胞 cDNAライブラリーからpCEV‐LAC ベクター内の全体HPP1 cDNA クローンを得た。このベクターは2001年5月5日付で登録番号第AY197612号としてアメリカ遺伝子銀行(GeneBank)に登録された(公開日:2004年5月1日)。
HPP1は配列番号:1の1792bp配列を有し、配列番号:1の塩基配列において、HPP1癌原遺伝子の全体開放型読み枠は配列番号:2のタンパク質をコードするヌクレオチド番号16から1620に対応すると仮定した。
【0051】
ファージからλpCEV ベクターに挿入されたHPP1 クローンをNot I切断によってエンピシリン耐性pCEV‐LACファージミド(phagemid)ベクターの形態に分離した(Miki,T.et al.,Gene 83: 137‐146,1989)。
【0052】
上記HPP1遺伝子を含むpCEV‐LACベクターをT4 DNAリガーゼーで連結してHPP1プラスミドDNAを製造し、連結されたクローンで大腸菌DH5αを形質転換させた。
【0053】
上記から得られた大腸菌形質転換体DH5α/HCC‐11/pCEV‐LACを特許手続のための微生物寄託の国際的承認に関するブダペスト条約の規定により、2002年12月5日付で韓国生命工学研究所遺伝子銀行(Korea Collection for Type Cultures,KCTC、住所:Korean Research Institute of Bioscience and Biotechnology(KRIBB),#52 Oun−dong,Yusong−ku,Taejon 305−333,Republic of Korea )に寄託番号第KCTC 10397BP号として寄託した。
【0054】
〔実施例7〕:ノーザンブロット分析
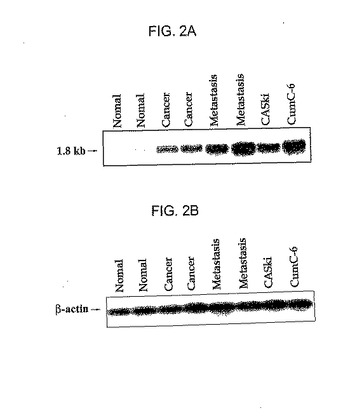
実施例1と同様に、正常子宮頸部組織、子宮頸部癌組織、子宮癌転移リンパ節組織及びヒト子宮頸部癌細胞株CaSki(ATCC CRL 1550)及びCUMC‐6から総RNAを抽出した。各々の組織及び細胞株から抽出した変性された総RNA20μgずつを1%ホルムアルデヒドアガロースゲルで電気泳動した後、ナイロン膜(Boehringer‐Mannheim、ドイツ)に移した。レジプライムII(Rediprime II)無作為プライム標識システム(Amersham、英国)を用いて製造した32P‐標識された無作為プライムされたHPP1全体cDNAプローブでブロットをハイブリッド化した。ノーザンブロット分析を2回繰り返して行い、その結果は濃度計(densitometry)を用いて定量化し、β‐アクチン(actin)で標準化した。
【0055】
図2aは、正常子宮頸部組織、子宮頸部癌組織、子宮頸部癌転移リンパ節組織及び子宮頸部癌細胞株(CaSki及びCUMC‐6)におけるHPP1 cDNAプローブを用いてノーザンブロット分析結果を示したものであり、図2bは同一の試料をβ‐アクチンプローブを用いてハイブリッド化したものである。図2aからわかるように、HPP1癌原遺伝子は子宮頸部癌組織、及び子宮頸部癌細胞株においては顕著に遺伝子発現が増加したが、正常子宮頸部組織においては子宮頸部発現程度が非常に低かった。
【0056】
図3aは、正常ヒトの脳、心臓、横紋筋、大腸、胸腺、脾臓、腎臓、肝、小腸、胎盤、肺及び抹消血液白血球(Clontech)におけるHPP1 cDNAプローブを用いたノーザンブロット分析結果を示したものである。図3bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。図3aからわかるように、HPP1 mRNA (約1.8kb)は正常組織においては殆ど発現されなかったり弱く発現されている。
【0057】
図4aは、ヒト癌細胞株、HL‐60、HeLa、K‐562、MOLT‐4、Raji、SW480、A549及びG361(Clontech)からHPP1癌原遺伝子の発現の可否を確認したノーザンブロット分析結果を示したものである。図4bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。図4aからわかるように、HPP1は大腸癌細胞SW480、肺癌細胞株A549及び皮膚癌細胞株G361で過発現され、特に前骨髄細胞白血病細胞株HL‐60、HeLa子宮癌細胞株、慢性骨髄白血病細胞株K‐562、リンパ球白血病細胞株MOLT‐4、バーキット(Burkitt)リンプ種細胞株Raji において高い水準で転写された。
【0058】
図5aは、ヒトの腎腸、乳房、大腸、卵巣、胃、肝癌組織及びこれらの各々の正常組織において本発明のHPP1癌原遺伝子の発現の可否を確認したノーザンブロット分析の結果を示したものである。 図5bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。HPP1はヒト癌組織において高い水準で発現される反面、正常組織においては非常に弱く発現されることが観察された。
【0059】
〔実施例8〕:HPP1癌原遺伝子を大腸菌に形質感染させた後発現されるタンパク質の大きさの決定
配列番号:1のHPP1癌原遺伝子の全体をpET‐32b(+)ベクター(Novagen,Germany)の多重‐クローニング部位内BamHI/SalI制限酵素 部位に挿入した後、生成されたpET‐32b(+)/HPP1ベクターを大腸菌BL21(ATCC 47092)に形質感染させた。pET‐32b(+)ベクターの多重‐クローニング部位の前部位には発現タンパク質であるTrx・Tag、His・Tag及びS・Tagが挿入されている。形質感染された大腸菌をLBブロスで振盪培養した後、これを1/100に希釈し、さらに3時間培養した。これに1mMのイソプロピルβ‐D‐チオガラクトピラノシド(Isopropyl beta‐D‐thiogalacto‐pyranoside; IPTG,Sigma)を添加してタンパク質生産を誘導した。
【0060】
IPTG誘導前後の培養液中の大腸菌細胞を超音波粉砕した後、12%ドデシル硫酸ナトリウム‐ポリアクリルアミドゲルで電気泳動した(SDS‐PAGE)。図6は、pET‐32b(+)/HPP1ベクターで形質感染された大腸菌BL21菌株のタンパク質発現様子をSDS‐PAGEで確認した結果であって、IPTG誘導後約80kDaの融合タンパク質バンドが明確に観察された。この80kDa融合タンパク質はpET‐32b(+)/HPP1 ベクターに挿入されている約21kDa大きさのTrx・Tag、His・Tag及びS・Tagタンパク質と約60kDaのHPP1タンパク質を含有している。
以上のように、本発明の実施の形態が説明されたが、本発明の属する分野の通常の知識を持つ者により多様な変形または修正が行われ得ることは明らかであり、これらは添付の請求の範囲により定められる本発明の範囲に含まれることと解釈すべきである。
【図面の簡単な説明】
【0061】
【図1】図1は、正常の子宮頸部組織、子宮頸部腫瘍組織、転移リンパ節腫瘍組織及びCUMC‐6癌細胞におけるCA336 DNA断片の発現の可否を確認した鑑別展開の逆転写重合酵素連鎖反応(DDRT‐PCR)の結果である。
【図2a】図2aは、子宮頸部癌組織における本発明のHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図2b】図2bは、図2aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図3a】正常のヒト12‐列多重組織におけるHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図3b】図3bは、図3aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図4a】図4aは、ヒト癌細胞株におけるHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図4b】図4bは、図4aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図5a】図5aは、ヒト腎臓、乳房、大腸、卵巣、胃、肝及び子宮頸部の腫瘍組織及び各々の正常組織における発現されたHPP1癌原遺伝子に対するノーザンブロットの分析結果である。
【図5b】図5bは、図5aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図6】図6は、HPP1癌原遺伝子を大腸菌に形質導入した後、IPTG誘導以前及び以後に発現されるタンパク質の大きさをドデシル硫酸ナトリウム‐ポリアクリルアミドゲル(SDS‐PAGE)電気泳動結果である。
【発明の詳細な説明】
【0001】
〔技術分野〕
本発明は、新規癌原遺伝子及びこれによってコードされるタンパク質に関するものであり、より詳しくは、多様な癌の診断に用いられるヒト癌原遺伝子(以下、HPPI癌原遺伝子と称する)及びこれによってコードされるタンパク質に関する。
【0002】
〔背景技術〕
ヒトを含む高等動物は、約100、000個の遺伝子を有しているが、これらのうち約15%のみが各々の個体で発現される。従って、いかなる遺伝子が選択されて発現されるかによって生命のすべての現象、即ち、発生、分化、恒常性(homeostasis)、刺激に対する反応、細胞分裂周期の調節、老化及びアポプトシス(apoptosis; programmed cell death)などが決められる(Liang,P.and A.B.Pardee,Science 257: 967‐971,1992)。
【0003】
腫瘍発生のような病理学的現象は、遺伝子変異過程によって誘発され結局遺伝子発現の変化をもたらす。従って、相異なる細胞の間で現れる遺伝子発現同士の比較は多様な生物学的現象を理解する上で基本的で効果的な接近方法であるといえる。例えば、リアンとパーディー(Liang,P.and A.B.Pardee,Science 257: 967‐971,1992)によって提案されたmRNA鑑別展開(differential display)方法は、現在腫瘍抑制遺伝子や細胞分裂周期(cell cycle)に係る遺伝子及びアポプトシスに係る転写調節遺伝子(transcriptional regulatory gene)などを探索する上で有効に用いられており、また一つの細胞で起こる多様な遺伝子の相互関連性の究明にも多様に活用されている。
【0004】
腫瘍発生に対する多様な研究結果をまとめると、特定染色体部位の喪失(loss of chromosomal heterozygosity)、癌原遺伝子の活性化及びp53遺伝子を含む他の腫瘍抑制遺伝子の不活性化などのような種々の遺伝的変化が腫瘍組織に蓄積されてヒト腫瘍を起こすと報告された(Bishop,J.M.,Cell 64: 235‐248,1991; Hunter,T.,Cell 64: 249‐270,1991)。さらに、癌の10ないし30%は癌原遺伝子が増幅されることによって活性化され起こると報告されており、癌原遺伝子の活性化は多くの癌の病因学研究に重要な役割を果たし、これを明らかにすることが要求されている。
【0005】
そこで、本発明者らは、子宮頸部癌の発生メカニズムを癌原遺伝子の水準で接近した結果、ヒト癌原遺伝子タンパク質1(human protooncogene protein 1;HPP1)と名付けられた癌原遺伝子が、癌細胞にのみ特異に発現が増加されるということがわかった。上記癌原遺伝子は、白血病、リンパ腫、大腸癌、肺癌、皮膚癌、 腎臓癌、乳房癌、大腸癌、卵巣癌、胃癌、肝癌、及び子宮頸部癌などのような種々の癌の診断、予防及び治療に効果的に用いられ得る。
【0006】
〔発明の概要〕
本発明の主たる目的は、新規な癌原遺伝子を提供することである。
【0007】
本発明の他の目的は、上記癌原遺伝子を含む組換えベクター及びそれによって形質転換された微生物を提供することである。
【0008】
本発明のさらに他の目的は、上記癌原遺伝子によってコードされるタンパク質を提供することである。
【0009】
本発明のさらに他の目的は、上記癌原遺伝子を含む癌診断用キットを提供することである。
【0010】
本発明のさらに他の目的は、上記タンパク質を含む癌診断用キットを提供することである。
【0011】
本発明のさらに他の目的は、上記癌原遺伝子から誘導されるものに相補的な塩基配列を有するアンチセンス遺伝子を提供することである。
【0012】
本発明のさらに他の目的は、上記アンチセンス遺伝子を用いて癌及びその転移を予防するか治療する方法を提供することである。
【0013】
上記目的によって、本発明では配列番号のヌクレオチド配列を有する新規癌原遺伝子を提供する。
【0014】
上記の他の目的に従って、本発明では上記癌原遺伝子を含む組換えベクター及びそれによって形質転換された微生物を提供する。
【0015】
上記のさらに他の目的に従って、上記癌原遺伝子由来の配列番号2のアミノ酸配列を有するタンパク質を提供する。
【0016】
〔発明の詳細な説明〕
本発明の新規癌原遺伝子(oncogene)であるヒト癌原遺伝子タンパク質1 (Human protooncogene protein 1,HPP1)は、配列番号:1で記載される1、792bp長さのDNA配列を有する。
【0017】
配列番号:1の塩基配列において、ヌクレオチド番号16ないし1620部位(1618‐1620:終了コドン)に該当する開放型読み枠(open reading frame)は、全体タンパク質コーディング領域であって、それから誘導される予測アミノ酸配列は配列番号:2に示されており、534個のアミノ酸からなっている(以下、「HPP1タンパク質」と称する)。
【0018】
コドンの縮退性(degeneracy)によってまたは本発明の上記癌原遺伝子を発現させようとする特定生物で好まれるコドンを考慮して、配列番号:1のDNA配列はコーディング領域から発現される発癌タンパク質のアミノ酸配列を変化させない範囲内でコーディング領域に多様な変形が行われることができ、コーディング領域を除いた部分でも遺伝子の発現に影響を及ぼさない範囲内で多様な変形または修正が行われることができて、そのような変形遺伝子も本発明の範囲に含まれる。従って、本発明は上記配列番号:1の癌原遺伝子と実質的に同一の塩基配列を有するポリヌクレオチド及び上記遺伝子の断片をも含む。ここで、「実質的に同一のポリヌクレオチド」とは、本発明の癌原遺伝子と80%以上、望ましくは90%以上、最も望ましくは95%以上の配列相同性を有することを意味する。
本発明の癌原遺伝子から発現されるタンパク質は534個のアミノ酸からなっており配列番号:2で記載されたアミノ酸配列を有して大きさは約60kDaである。しかし、上記タンパク質のアミノ酸配列でも同様にタンパク質の機能に影響を及ぼさない範囲内でアミノ酸の置換、付加または欠失が行われることができ、目的によってタンパク質の一部のみが使用されることもある。そのような変形されたアミノ酸配列及びそれらの断片も本発明の範囲に含まれる。従って、本発明は上記発癌タンパク質と実質的に同一のアミノ酸配列を有するポリペプチド及びその断片をも含む。ここで、「実質的に同一のポリペプチド」とは、配列番号:2のアミノ酸配列と80%以上、望ましくは90%以上、最も望ましくは95%以上の配列相同性を有することを意味する。
【0019】
本発明の癌原遺伝子HPP1及びタンパク質はヒトの癌組織から分離したり、公知のDNAまたはペプチド合成方法によって合成することもできる。また、このように製造された遺伝子を当分野に公知された微生物発現用ベクターに挿入して発現ベクターを製造した後これを適切な宿主細胞、例えば、大腸菌または酵母細胞などに導入し得る。
【0020】
例えば、癌原遺伝子HPP1で大腸菌DH5αを形質感染(transfection)させ、これから得られたDH5α/HCC‐11/pCEV‐LACを、2002年12月5日付で、特許手続のための微生物寄託の国際的承認に関するブダペスト条約に従い、韓国生命工学研究所遺伝子銀行(Korea Collection for Type Cultures; KCTC、住所:Korean Research Institute of Bioscience and Biotechnology(KRIBB),#52 Oun−dong,Yusong−ku,Taejon 305−333,Republic of Korea)に寄託番号第KCTC 10397BP号として寄託した。
【0021】
ベクター制作の際には、用いられた宿主細胞の種類によってプロモーター、ターミネータなどのような発現調節配列、自家複製配列及び分泌シグナルなどを適宜選択して組み合わせることができる。
【0022】
本発明の遺伝子は、ノーザンブロット(northern blot)などの分析方法で正常子宮頸部組織では発現がほとんど確認されなかった反面、子宮頸部癌組織と子宮癌細胞株ではその過発現が確認されるので、子宮癌を誘発する強力な発癌遺伝子として判断される。また、癌転移リンパ節組織で発現が増加されることから癌転移を誘発する癌転移関連遺伝子とも判断される。子宮頸部癌のような上皮性組織の他にも、本発明の癌原遺伝子は、白血病、リンパ腫、大腸癌、皮膚癌、肺癌、腎臓癌、乳房癌、卵巣癌、胃癌、肝癌及び子宮頸部癌などのような多くの他の癌腫瘍で高く発現される。従って、本発明の癌原遺伝子は多様な癌の発生に共通した発癌遺伝子であると判断され、多様な癌の診断、形質転換された動物の製造及びアンチセンス(anti‐sense)遺伝子治療などに有効に利用され得る。
【0023】
上記癌原遺伝子を用いた癌の診断方法は、例えば、上記癌原遺伝子の全部または一部をプローブを用いて対象者の体液から分離した核酸とハイブリッド化した後、当分野で公知の多様な方法でこれを検出することによって、対象者が本発明の癌原遺伝子を有しているかどうかを判断する過程を含む。上記プローブを放射線同位元素または酵素などで標識することによって容易に癌原遺伝子HPP1の存在を確認することができる。従って、本発明では上記癌原遺伝子の全部または一部を含む癌診断用キットをも提供する。
【0024】
形質転換動物は本発明の癌原遺伝子を哺乳動物、例えば、鼠などの哺乳類動物に導入することによって製造でき、この遺伝子を少なくとも8細胞期以前の受精卵段階で導入することが望ましい。このように製造された形質転換動物は、発癌性物質または化学療法の薬物のような抗癌性物質の探索などに有用に用いられ得る。
【0025】
本発明は遺伝子治療にも有効であり、従って本発明は上記新規癌原遺伝子HPP1により誘導されたmRNA相補的塩基配列を含むアンチセンス遺伝子をも提供する。
【0026】
本発明は、また、遺伝子治療に効果的なアンチセンス遺伝子を提供する。本願において、「アンチセンス遺伝子(anti‐sense gene)」とは、配列番号:1の癌原遺伝子またはその一部の塩基配列を有する癌原遺伝子HPP1から転写されるmRNAの一部または全部と相補的な配列を有するポリヌクレオチであって、上記mRNAのタンパク質結合部位に結合して癌原遺伝子の開放型読み枠(open reading frame;ORF)の発現を抑制できる配列を有するポリヌクレオチドを意味する。
【0027】
本発明はまた、その範囲内に本発明のアンチセンス遺伝子の治療効果量を患者に投与することによって癌を治療または予防する方法を含む。
【0028】
本発明のアンチセンス遺伝子治療において、本発明のアンチセンス遺伝子は通常の方法で患者に投与されて癌原遺伝子の発現を予防する。例えば、文献(J.S.Kim et al.,J.Controlled Release 53:175‐182,1998)の方法に従ってアンチセンスオリゴデオキシヌクレオチド(ODN)をポリ‐L‐リシン誘導体と静電気的引力によって混合し、この混合体を患者に静脈投与する。
【0029】
本発明の範囲にはまた、薬学的に許容可能な担体、賦形剤または必要に応じて他の添加剤とともに本発明のアンチセンス遺伝子を活性成分として含む抗癌組成物が含まれる。本発明の薬剤学的組成物を注射剤として製剤化することが望ましい。
【0030】
実際に投与されるHPP1アンチセンス遺伝子の量は、治療される症状、投与経路、患者の年齢及び体重、症状の重症度のような種々の関連因子を考慮して決める。
【0031】
本発明の癌原遺伝子HPP1から発現されるタンパク質は、診断道具として有用な抗体を生産するのに有用に有用できる。本発明の抗体は、本発明の癌原遺伝子から発現された配列番号:2のアミノ酸配列を有するタンパク質またはその断片を用いて当分野で公知の方法にとってモノクロナルの抗体またはポリクロナルの抗体として製造でき、このような抗体を用いて当分野で公知の酵素免疫測定法(enzyme linked immunosorbentassay; ELISA)、放射線免疫測定法(radioimmunoassay; RIA)、サンドイッチ測定法(sandwich assay)、免疫組織化学染色(Immunohistochemical staining)、ポリアクリルアミドゲル上のウエスタンブロットまたは免疫ブロットなどの方法によって対象者の体液試料の中に上記タンパク質が発現されたかを確認することによって癌を診断することができる。従って、配列番号:2のアミノ酸配列を有するタンパク質またはその断片を含む癌診断キットも本発明の範囲に含まれる。
【0032】
また、本発明の癌原遺伝子を用いて持続的に増殖し得る癌細胞株を確立することができ、このような細胞株は、例えば、上記癌原遺伝子が形質導入された繊維母細胞を用いてヌードマウスなどに形成させた腫瘍組織から製造できる。このような癌細胞株は抗癌剤などの探索に有用に利用することができる。
【0033】
下記実施例は本発明をより詳しく説明するものであって、本発明の内容が下記実施例に限定されるのではない。
【0034】
〔実施例1〕:腫瘍細胞の培養及び総RNAの分離
(段階1)腫瘍細胞培養
子宮摘出術(hysterectomy)を受けた子宮筋腫患者から正常の子宮頸部(exocervical)組織、そして以前に抗癌療法と放射線治療を受けなかった子宮癌患者から手術の際に原発性子宮頸部腫瘍組織及び転移リンパ節腫瘍組織を取った。ヒト子宮頸部癌細胞株としてCUMC‐6(Kim,J.W.et al.,Gynecol.Oncol.62: 230‐240,1996)を用いた。
【0035】
上記組織及びCUMC‐6細胞株から得られた細胞を2mM グルタミン、100IU/ml ペニシリン、100μg/ml ストレプトマイシン及び10% ウシ胎児血清(Gibco,USA)が含有されたウエイマウス(Waymouth’s) MB 752/1培養液(Gibco)で増殖させた。実験に用いた培養細胞は指数的増殖過程の細胞であってトリパンブルー(trypan blue)染色によって95%以上の生存度を示す細胞を用いた(Freshney,「Culture of Animal Cells: A Manual of Basic Technique」 2nd Ed.,A.R.Liss,New York,1987)。
(段階2)RNAの分離及びmRNAの鑑別展開法
商業的に販売されるシステムであるRNeasy総RNAキット(Qiagen Inc.,ドイツ)を用いて、段階1で得られた組織標本及び細胞から総RNAを抽出した。メッセージクリーンキット(Message clean kit,GenHunter Corp.,Brookline,MA)を用いてRNAからDNA汚染源を除去した。
【0036】
〔実施例2〕:鑑別展開逆転写重合酵素連鎖反応(differential display reverse transcription‐PCR)
鑑別展開逆転写は、上記リアンとパーディーによって記述された逆転写‐重合酵素連鎖反応(RT‐PCR)を若干変形して行った。
【0037】
まず、実施例1の段階1で得られた総RNA 0.2μgずつに固定されたオリゴ‐dTプライマーとして配列番号:3で記載される塩基配列を有するH−T11A固定プライマー(RNAimage kit,Genhunter,Cor.,MA,USA)を用いて逆転写を行った。
【0038】
次いで、0.5mM[α‐35S] dATP(1200 Ci/mmole)の存在の下で、同一の固定プライマー及び無作為5’11プライマー(RNAimage primer sets 1‐5)H‐AP1ないし40の中、配列番号:4で記載される塩基配列を有するH‐AP33プライマーを用いてPCRを行った。PCR反応の条件は、95℃で40秒間反応後40℃で2分、72℃で40秒を40回反応させて最終延長(final extension)のために72℃で5分間さらに反応させた。得られたPCR産物を6% ポリアクリルアミドシーケンシングゲルに溶解した後、放射能写真を用いて相違な発現(differentially expressed)程度を示すバンドの位置を確認した。
【0039】
乾燥したゲルから181塩基対(bp)大きさのバンド、CA336 cDNA(配列番号:1の1569‐1749 bp)を切取った。15分間加熱してCA336 cDNAを溶出させた後、[α‐35S]標識されたdATP(1200 Ci/mmole)及び20μM dNTPを用いなかったことを除いては上記と同一のプライマー及び同一の条件でPCRを行なって再増幅させた。
【0040】
〔実施例3〕:クローニング
上記で得られた再増幅されたCA336 PCR産物をTAクローニングシステム(Promega、USA)を用いて製造社の方法に従ってpGEM‐T EASYベクターに挿入した。
(段階1)連結反応
実施例2で得られた再増幅されたCA336 PCR産物2μL、pGEM‐T EASYベクター(50ng)1μL、T4 DNA リガーゼ 10X 緩衝液 1μL及びT4 リガーゼー(3 weiss units/μL; T4 DNA ligase,Promega)1μLを0.5ml試験管に入れた後、蒸留水を加えて最終体積が10μLになるようにした。連結反応混合物は14℃で一晩中培養した。
(段階2)TAクローニング形質転換
TAクローニング形質転換は次のように行われた。
【0041】
大腸菌JM109(Promega,WI,USA)を10mlのLBブロス(バクト‐トリプトン 10g、バクト‐酵母抽出物 5g、NaCl 5g)中で600nmでの光学密度が約0.3ないし0.6になるまで培養した。培養混合物を氷に約10分間放置した後、4℃で10分間4000rpmで遠心分離して、上澄液を取除き、細胞を収集した。収集した細胞ペレットを10mlの氷冷0.1M CaCl2に30分ないし1時間露出させてコンペテント(competent)細胞を製造した。結果物を4℃で4000rpmで10分間さらに遠心分離して上澄液を取除き、細胞を収集して2mlの氷冷0.1M CaCl2に懸濁した。
【0042】
コンペテント細胞懸濁液200μLを新しいマイクロフュズ(microfuge)チューブに移し、これに段階1で製造した連結反応産物2μLを加えた。混合物を42℃の 水浴槽(water bath)で90秒間培養した後、0℃で急冷した。これに、SOC培地(バクト‐トリプトン2.0g,バクト‐酵母抽出物 0.5g,1M NaCl 1ml,1M KCl 0.25ml,TDW 97ml,2M Mg2+ 1ml,2M グルコース 1ml)800μLを加え、混合物を37℃で220rpmの回転振盪培養器で45分間培養した。
【0043】
37℃培養器に予め入れておいた、エンピシリンを含むLBプレートにX‐gal (40mg/ml ジメチルホルムアミドに貯蔵)25μLをガラス棒で拡散させた後、これに25μLの形質転換された細胞を入れてさらにガラス棒で拡散させ、37℃で一晩中培養した。培養後形成された白色コロニーを3〜4個選択してエンピシリンを含むLBプレートに各々の選択されたクローンを植えた。プラスミドを製造するためにこれらのうち挿入が確認されたコロニー、即ち、形質転換された大腸菌 JM109/CA336を選択して10mlのテリフィックブロス(terrific broth; TDW 900ml、バクト‐トリプトン 12g、バクト‐酵母抽出物 24g、グリセロール 4ml、0.17M KH2PO4、0.72N K2HPO4 100ml)で培養した。
【0044】
〔実施例4〕:組換えプラスミドDNAの分離
ウイザードフラスミニプレップスDNA精製キット(WizardTMPlus Minipreps DNA Purification Kit,Promega,USA)を用いて製造社の指針書に従って形質転換された大腸菌からCA336プラスミドDNAを分離した。
【0045】
分離されたDNAの一部をECoRI酵素で処理した後、2% ゲルで電気泳動を行ってCA336部分配列がプラスミドに挿入されたことを確認した。
【0046】
〔実施例5〕:DNA塩基配列分析
実施例2で得られたCA336 PCR産物を通常の方法に従って増幅させた後、クローニングして再増幅させたCA336 PCR断片をシケナーゼバージョン2.0DNA配列分析キット(Sequenase version 2.0 DNA Sequencing kit; United States Biochemical,Cleveland,OH,USA)を用いてジデオキシ鎖終結法(dideoxy chain termination)に従って配列分析を行った。
【0047】
上記DNAの塩基配列は配列番号: 1のヌクレオチド番号 1569 ないし1749に該当し、本願に「CA336」と命名した。
【0048】
5’無作為プライマーH‐AP33及び3’H‐T11A固定プライマーを用いて上記で得られた181 bp cDNA 断片、即ち、CA336を鑑別展開逆転写重合酵素連鎖反応(DDRT‐PCR)を行い、電気泳動で確認した。
【0049】
図1に示されたように、cDNA断片(CA336)は、転移リンパ節組織及びCUMC‐6細胞では発現されたが、正常組織では発現されなかった。
【0050】
〔実施例6〕:HPP1 癌原遺伝子の全体cDNA配列分析
32P‐標識されたCA336 cDNAをプローブを用いたプラークハイブリッド化(plaque hybridization)でバクテリオファージλgt11 ヒト肺胎児繊維母細胞cDNAライブラリー(Miki,T.et al.,Gene 83: 137‐146,1989)をスクリーニングした。上記ヒト肺胎児繊維母細胞 cDNAライブラリーからpCEV‐LAC ベクター内の全体HPP1 cDNA クローンを得た。このベクターは2001年5月5日付で登録番号第AY197612号としてアメリカ遺伝子銀行(GeneBank)に登録された(公開日:2004年5月1日)。
HPP1は配列番号:1の1792bp配列を有し、配列番号:1の塩基配列において、HPP1癌原遺伝子の全体開放型読み枠は配列番号:2のタンパク質をコードするヌクレオチド番号16から1620に対応すると仮定した。
【0051】
ファージからλpCEV ベクターに挿入されたHPP1 クローンをNot I切断によってエンピシリン耐性pCEV‐LACファージミド(phagemid)ベクターの形態に分離した(Miki,T.et al.,Gene 83: 137‐146,1989)。
【0052】
上記HPP1遺伝子を含むpCEV‐LACベクターをT4 DNAリガーゼーで連結してHPP1プラスミドDNAを製造し、連結されたクローンで大腸菌DH5αを形質転換させた。
【0053】
上記から得られた大腸菌形質転換体DH5α/HCC‐11/pCEV‐LACを特許手続のための微生物寄託の国際的承認に関するブダペスト条約の規定により、2002年12月5日付で韓国生命工学研究所遺伝子銀行(Korea Collection for Type Cultures,KCTC、住所:Korean Research Institute of Bioscience and Biotechnology(KRIBB),#52 Oun−dong,Yusong−ku,Taejon 305−333,Republic of Korea )に寄託番号第KCTC 10397BP号として寄託した。
【0054】
〔実施例7〕:ノーザンブロット分析
実施例1と同様に、正常子宮頸部組織、子宮頸部癌組織、子宮癌転移リンパ節組織及びヒト子宮頸部癌細胞株CaSki(ATCC CRL 1550)及びCUMC‐6から総RNAを抽出した。各々の組織及び細胞株から抽出した変性された総RNA20μgずつを1%ホルムアルデヒドアガロースゲルで電気泳動した後、ナイロン膜(Boehringer‐Mannheim、ドイツ)に移した。レジプライムII(Rediprime II)無作為プライム標識システム(Amersham、英国)を用いて製造した32P‐標識された無作為プライムされたHPP1全体cDNAプローブでブロットをハイブリッド化した。ノーザンブロット分析を2回繰り返して行い、その結果は濃度計(densitometry)を用いて定量化し、β‐アクチン(actin)で標準化した。
【0055】
図2aは、正常子宮頸部組織、子宮頸部癌組織、子宮頸部癌転移リンパ節組織及び子宮頸部癌細胞株(CaSki及びCUMC‐6)におけるHPP1 cDNAプローブを用いてノーザンブロット分析結果を示したものであり、図2bは同一の試料をβ‐アクチンプローブを用いてハイブリッド化したものである。図2aからわかるように、HPP1癌原遺伝子は子宮頸部癌組織、及び子宮頸部癌細胞株においては顕著に遺伝子発現が増加したが、正常子宮頸部組織においては子宮頸部発現程度が非常に低かった。
【0056】
図3aは、正常ヒトの脳、心臓、横紋筋、大腸、胸腺、脾臓、腎臓、肝、小腸、胎盤、肺及び抹消血液白血球(Clontech)におけるHPP1 cDNAプローブを用いたノーザンブロット分析結果を示したものである。図3bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。図3aからわかるように、HPP1 mRNA (約1.8kb)は正常組織においては殆ど発現されなかったり弱く発現されている。
【0057】
図4aは、ヒト癌細胞株、HL‐60、HeLa、K‐562、MOLT‐4、Raji、SW480、A549及びG361(Clontech)からHPP1癌原遺伝子の発現の可否を確認したノーザンブロット分析結果を示したものである。図4bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。図4aからわかるように、HPP1は大腸癌細胞SW480、肺癌細胞株A549及び皮膚癌細胞株G361で過発現され、特に前骨髄細胞白血病細胞株HL‐60、HeLa子宮癌細胞株、慢性骨髄白血病細胞株K‐562、リンパ球白血病細胞株MOLT‐4、バーキット(Burkitt)リンプ種細胞株Raji において高い水準で転写された。
【0058】
図5aは、ヒトの腎腸、乳房、大腸、卵巣、胃、肝癌組織及びこれらの各々の正常組織において本発明のHPP1癌原遺伝子の発現の可否を確認したノーザンブロット分析の結果を示したものである。 図5bは、同一の試料をβ‐アクチンプローブでハイブリッド化してmRNAの存在を確認したものである。HPP1はヒト癌組織において高い水準で発現される反面、正常組織においては非常に弱く発現されることが観察された。
【0059】
〔実施例8〕:HPP1癌原遺伝子を大腸菌に形質感染させた後発現されるタンパク質の大きさの決定
配列番号:1のHPP1癌原遺伝子の全体をpET‐32b(+)ベクター(Novagen,Germany)の多重‐クローニング部位内BamHI/SalI制限酵素 部位に挿入した後、生成されたpET‐32b(+)/HPP1ベクターを大腸菌BL21(ATCC 47092)に形質感染させた。pET‐32b(+)ベクターの多重‐クローニング部位の前部位には発現タンパク質であるTrx・Tag、His・Tag及びS・Tagが挿入されている。形質感染された大腸菌をLBブロスで振盪培養した後、これを1/100に希釈し、さらに3時間培養した。これに1mMのイソプロピルβ‐D‐チオガラクトピラノシド(Isopropyl beta‐D‐thiogalacto‐pyranoside; IPTG,Sigma)を添加してタンパク質生産を誘導した。
【0060】
IPTG誘導前後の培養液中の大腸菌細胞を超音波粉砕した後、12%ドデシル硫酸ナトリウム‐ポリアクリルアミドゲルで電気泳動した(SDS‐PAGE)。図6は、pET‐32b(+)/HPP1ベクターで形質感染された大腸菌BL21菌株のタンパク質発現様子をSDS‐PAGEで確認した結果であって、IPTG誘導後約80kDaの融合タンパク質バンドが明確に観察された。この80kDa融合タンパク質はpET‐32b(+)/HPP1 ベクターに挿入されている約21kDa大きさのTrx・Tag、His・Tag及びS・Tagタンパク質と約60kDaのHPP1タンパク質を含有している。
以上のように、本発明の実施の形態が説明されたが、本発明の属する分野の通常の知識を持つ者により多様な変形または修正が行われ得ることは明らかであり、これらは添付の請求の範囲により定められる本発明の範囲に含まれることと解釈すべきである。
【図面の簡単な説明】
【0061】
【図1】図1は、正常の子宮頸部組織、子宮頸部腫瘍組織、転移リンパ節腫瘍組織及びCUMC‐6癌細胞におけるCA336 DNA断片の発現の可否を確認した鑑別展開の逆転写重合酵素連鎖反応(DDRT‐PCR)の結果である。
【図2a】図2aは、子宮頸部癌組織における本発明のHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図2b】図2bは、図2aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図3a】正常のヒト12‐列多重組織におけるHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図3b】図3bは、図3aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図4a】図4aは、ヒト癌細胞株におけるHPP1癌原遺伝子の発現の可否を確認したノーザンブロットの分析結果である。
【図4b】図4bは、図4aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図5a】図5aは、ヒト腎臓、乳房、大腸、卵巣、胃、肝及び子宮頸部の腫瘍組織及び各々の正常組織における発現されたHPP1癌原遺伝子に対するノーザンブロットの分析結果である。
【図5b】図5bは、図5aと同様なサンプルをβ‐アクチンでハイブリッド化して得られた結果である。
【図6】図6は、HPP1癌原遺伝子を大腸菌に形質導入した後、IPTG誘導以前及び以後に発現されるタンパク質の大きさをドデシル硫酸ナトリウム‐ポリアクリルアミドゲル(SDS‐PAGE)電気泳動結果である。
【特許請求の範囲】
【請求項1】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質を含む癌診断用組成物。
【請求項2】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質をコーディングするヒト癌原遺伝子を含む癌診断用組成物。
【請求項3】
上記ヒト癌原遺伝子は配列番号:1の塩基番号16ないし1620に該当する塩基配列を含むことを特徴とする請求項2に記載の癌診断用組成物。
【請求項4】
上記ヒト癌原遺伝子は配列番号:1の塩基配列であることを特徴とする請求項2に記載の癌診断用組成物。
【請求項5】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質を含む癌診断用キット。
【請求項6】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質をコーディングする癌原遺伝子を含む癌診断用キット。
【請求項7】
上記癌原遺伝子は配列番号:1の塩基番号16ないし1620に該当する塩基配列及び配列番号:1の塩基配列からなる群より選択された何れか一つであることを特徴とする請求項6に記載の癌診断用キット。
【請求項8】
配列番号:1の塩基番号16ないし1620に該当する塩基配列及び配列番号:1の塩基配列からなる群より選択された癌原遺伝子から転写されたmRNAの配列に相補的な塩基配列を有し、上記癌原遺伝子の発現を抑制するためにmRNAに結合する癌治療または予防用アンチセンス遺伝子。
【請求項9】
請求項8のアンチセンス遺伝子の治療学的有効量及び薬剤学的に許容可能な担体を含有する癌及び転移の予防又は治療用組成物。
【請求項1】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質を含む癌診断用組成物。
【請求項2】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質をコーディングするヒト癌原遺伝子を含む癌診断用組成物。
【請求項3】
上記ヒト癌原遺伝子は配列番号:1の塩基番号16ないし1620に該当する塩基配列を含むことを特徴とする請求項2に記載の癌診断用組成物。
【請求項4】
上記ヒト癌原遺伝子は配列番号:1の塩基配列であることを特徴とする請求項2に記載の癌診断用組成物。
【請求項5】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質を含む癌診断用キット。
【請求項6】
癌診断のためのマーカーとして配列番号:2のアミノ酸配列を有するヒト癌原タンパク質をコーディングする癌原遺伝子を含む癌診断用キット。
【請求項7】
上記癌原遺伝子は配列番号:1の塩基番号16ないし1620に該当する塩基配列及び配列番号:1の塩基配列からなる群より選択された何れか一つであることを特徴とする請求項6に記載の癌診断用キット。
【請求項8】
配列番号:1の塩基番号16ないし1620に該当する塩基配列及び配列番号:1の塩基配列からなる群より選択された癌原遺伝子から転写されたmRNAの配列に相補的な塩基配列を有し、上記癌原遺伝子の発現を抑制するためにmRNAに結合する癌治療または予防用アンチセンス遺伝子。
【請求項9】
請求項8のアンチセンス遺伝子の治療学的有効量及び薬剤学的に許容可能な担体を含有する癌及び転移の予防又は治療用組成物。
【図1】





【図6】

【図6】
【公表番号】特表2007−528483(P2007−528483A)
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願番号】特願2006−502690(P2006−502690)
【出願日】平成16年1月27日(2004.1.27)
【国際出願番号】PCT/KR2004/000137
【国際公開番号】WO2004/067562
【国際公開日】平成16年8月12日(2004.8.12)
【出願人】(501241818)
【Fターム(参考)】
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願日】平成16年1月27日(2004.1.27)
【国際出願番号】PCT/KR2004/000137
【国際公開番号】WO2004/067562
【国際公開日】平成16年8月12日(2004.8.12)
【出願人】(501241818)
【Fターム(参考)】
[ Back to top ]