
ビタミン受容体結合性薬剤送達結合体
【課題】ビタミン受容体結合性薬剤送達結合体、およびその調製法の提供。
【解決手段】ビタミン受容体結合成分、二価のリンカー(L)、および薬剤から成る。ビタミン受容体結合成分は、ビタミン、または、そのビタミン受容体結合性類縁体または誘導体であり、薬剤は、その類縁体または誘導体を含む。ビタミン受容体結合成分は、二価リンカーに共有結合され、薬剤またはその類縁体または誘導体は、二価リンカーに共有結合され、二価リンカー(L)は、一つ以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーを含む。この薬剤送達結合体を用いて病原細胞集団を除去ことができる。
【解決手段】ビタミン受容体結合成分、二価のリンカー(L)、および薬剤から成る。ビタミン受容体結合成分は、ビタミン、または、そのビタミン受容体結合性類縁体または誘導体であり、薬剤は、その類縁体または誘導体を含む。ビタミン受容体結合成分は、二価リンカーに共有結合され、薬剤またはその類縁体または誘導体は、二価リンカーに共有結合され、二価リンカー(L)は、一つ以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーを含む。この薬剤送達結合体を用いて病原細胞集団を除去ことができる。
【発明の詳細な説明】
【技術分野】
【0001】
===関連出願の相互参照===
本出願は、米国特許法119条(e)の下に、2003年1月27日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/442,845号、2003年8月1日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/492,119号、および、2003年10月31日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/516,188号に対する優先権を主張する。上記各出願の開示の全体を参照することにより本出願に含める。
【0002】
本発明は、標的に向けた薬剤送達に使用される組成物および方法に関する。さらに具体的には、本発明は、病原性細胞集団によって引き起こされる病的状態の治療に用いられるビタミン受容体結合性薬剤送達結合体、および、そのための方法および製薬組成物に向けられる。
【背景技術】
【0003】
哺乳類免疫系は、腫瘍細胞、その他の病原性細胞、および、侵入する外来病原体を認識し、除去するための手段を与える。正常時、免疫系は強力な防衛線を供給するが、ガン細胞、その他の病原細胞、および、感染源が、宿主の免疫反応を回避して、増殖または残存して、同時に宿主の病原となる場合が数多くある。例えば、増殖する新生物を除去するために、化学療法剤および放射線療法が開発されている。しかしながら、現在利用可能な化学療法剤および放射線治療処方の多くは副作用を持つ。なぜなら、これらの薬剤および療法は病原細胞を破壊するだけでなく、造血系の細胞のような正常な宿主細胞をも侵すからである。これらの抗ガン剤の副作用は、病原性細胞集団に対して選択的で、宿主に対する毒性の低い新規療法の開発の必要を強調する。
【0004】
研究者達は、病原性細胞に対して細胞傷害性化合物を標的化することによってその病原性細胞を破壊するための治療プロトコールを開発している。これらのプロトコールの多くは、毒素を利用するのであるが、その毒素の正常細胞に対する送達を極小にするために、病原細胞に対して固有の、または、病原細胞によって過剰発現される抗原に結合する抗体に結合させた毒素を利用する。この方法を用いて、いくつかの免疫毒素が開発された。これらのものは、病原細胞上の特異的抗原に向けられた抗体から成り、抗体は、リシン、シュードモナス体外毒素、および腫瘍壊死因子のような毒素に連結される。これらの免疫毒素は、例えば腫瘍細胞のような、その抗体によって認識される特異的抗原を帯びる病原細胞を標的とする(非特許文献1、2および特許文献1)。
【0005】
宿主における病原細胞集団、例えば、ガン細胞または外来病原体から成る集団を標的とするもう一つの方法は、それ自体も独立した宿主毒性を示しかねない化合物投与の必要を回避するために、病原細胞に対する宿主の免疫反応を強化することである。免疫療法のための一つの報告されている戦略は、抗体を、例えば、遺伝子工学的に加工された多量体抗体を、腫瘍細胞の表面に結合させ、それらの抗体の定常域を細胞表面において提示させ、各種免疫系仲介過程を通じて腫瘍細胞殺戮を誘発することである(非特許文献3、および特許文献2)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】PCT公報WO91/07418、1991年5月30日発行
【特許文献2】米国特許第5,672,486号、Soulillou, J.P.に付与
【非特許文献】
【0007】
【非特許文献1】Olsnes, S., Immunol. Today, 10, pp.291-295, 1989
【非特許文献2】Melby, E.L., Cancer Res., 53(8), pp.1755-1760, 1993
【非特許文献3】De Vita, V.T., 「ガンの生物学的治療法(”Biologic Therapy of Cancer”)」、第2版、フィラデルフィア、Lippincott、1995
【発明の概要】
【0008】
病原細胞に対して特異的であり、正常細胞に対しては毒性を極小とした効果的な療法を開発するために、ビタミン受容体結合薬剤送達結合体が開発されてきた。本発明は、宿主動物において様々な病理現象を引き起こす病原細胞の集団に適用が可能である。本発明の薬剤送達結合体によって治療が可能な病原細胞としては、ガン細胞、細菌およびウィルスのような感染源、細菌またはウィルス感染細胞、およびその他の、ビタミン受容体、または、ビタミン類縁体または誘導体に結合する受容体を特異的に発現するか、優先的に発現するか、または、過剰に発現する、任意の病原細胞が挙げられる。
【0009】
一つの実施態様では、ビタミン受容体結合性薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価のリンカー、および薬剤を含む。本明細書で用いる”V”は、ビタミン受容体結合成分を指し、ビタミン、および、ビタミン受容体結合性類縁体または誘導体を含み、「ビタミン、またはその類縁体または誘導体」という用語は、ビタミン受容体に結合することが可能なビタミン、および、その類縁体および誘導体を指す。本明細書で用いる”D”とは薬剤を指し、その類縁体または誘導体を含む。ビタミン、またはその類縁体または誘導体は、二価のリンカー(L)に共有結合され、薬剤、またはその類縁体または誘導体も二価リンカー(L)に共有結合される。二価リンカー(L)は、複数のリンカーを含むことが可能である。例えば、二価リンカー(L)は、スペーサーリンカー(ls)、放出リンカー(lr)、およびヘテロ原子リンカー(lH)のうちの1つ以上、および、それらの、順番は任意とする組み合わせを含む。
【0010】
本実施態様を例示する薬剤送達結合体としては、
V-L-D
V-(lr)c-D
V-(ls)a-D
V-(ls)a-(lr)c-D
V-(lr)c-(ls)a-D
V-(lH)b-(lr)c-D
V-(lr)c-(lH)b-D
V-(lH)d-(lr)c-(lH)e-D
V-(ls)a-(lH)b-(lr)c-D
V-(lr)c-(lH)b-(ls)a-D
V-(lH)d-(ls)a-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(ls)a-(lH)e-D
V-(lH)d-(ls)a-(lH)b-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(lH)b-(ls)a-(lH)e-D
V-(ls)a-(lr)c-(lH)b-D
V-[(ls)a-(lH)b]d-(lr)c-(lH)e-D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)は本明細書に定義した通りであり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体であり、式中、二価のLは、1個の、または、各種の(ls)、(lH)、または(lr)を任意の順序で任意の組み合わせで含む。二価リンカーLの上記例は、二価リンカーの含む、(lH)、(ls)、(lr)の広範な集合を例示することを意図したものであって、限定することを意図したものではないことが理解される。
【0011】
スペーサー、ヘテロ原子、および放出リンカーはそれぞれ二価であることが理解される。さらに、本明細書で定義した、各種スペーサー、ヘテロ原子、および放出リンカーの各要素間の結合性、および、各種スペーサー、ヘテロ原子、および放出リンカーと、Dおよび/またはVとの間の結合性は、それら各種のスペーサー、ヘテロ原子および放出リンカー中に見られる任意の原子で起こってもよく、各種スペーサー、ヘテロ原子および放出リンカーの内の任意のものの任意の見かけ上の末端に必ずしも起こらなくてもよいことを理解しなければならない。例えば、例示の実施態様において、二価のリンカーは、

すなわち、式中、二価のリンカーLは、-(lH)-(ls)5-(lr-lH)2-Dであって、それぞれ、(lH)は窒素であり、(ls)5は、 Ala-Glu-Lys-Asp-Asp であり、(lr-lH)2は、 −(CH2)2-S-S-(CH2)2-O-C(O)-O- である。この実施態様において、(lr-lH)2リンカーは、(ls)5リンカーの中間部分に接続する。
【0012】
別の実施態様では、ビタミン受容体結合薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、および薬剤を含み、二価リンカー(L)は1種以上のヘテロ原子リンカー(lH)を含む。ビタミン受容体結合成分は、第1ヘテロ原子リンカー(lH)dを通じて、二価のリンカー(L)に共有結合し、薬剤は、第2ヘテロ原子リンカー(lH)eを通じて、二価のリンカー(L)に共有結合する。二価のリンカー(L)は1種以上のスペーサーリンカーおよび放出リンカーも含み、このスペーサーリンカーと放出リンカーは、第3のヘテロ原子リンカー(lH)bを通じて互いに共有結合されてもよい。この実施態様を例示する薬剤送達結合体は下記の通りである、すなわち、
V−(lH)d−(ls)a−(lH)b−(lr)c−(lH)e−D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)、VおよびDは本明細書に定義した通りであり、式中、二価のLは、図示のように(ls)、(lH)、および(lr)を含む。
【0013】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、および薬剤を含み、二価リンカー(L)はヘテロ原子リンカー(lH)を含む。ビタミン受容体結合成分は、ビタミン、またはその類縁体または誘導体であり、薬剤は、その類縁体または誘導体も含む。ビタミン、またはその類縁体または誘導体は、二価リンカー(L)に共有結合し、薬剤、またはその類縁体または誘導体は二価リンカー(L)に共有結合する。二価リンカー(L)は、スペーサーリンカーおよび放出リンカーも含み、このスペーサーリンカーと放出リンカーは、ヘテロ原子リンカーを通じて互いに共有結合されてもよい。この実施態様を図示する薬剤送達結合体は下記の通りである、すなわち、
V−(ls)a−(lH)b−(lr)c−D
式中、a、b、およびcは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)、VおよびDは本明細書に定義した通りであり、式中、二価のLは、図示のように(ls)、(lH)、および(lr)を含む。
【0014】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が提供される。この実施態様では、Lは1種以上のリンカー(lr)c、(ls)a、と(lH)b、およびそれらの任意の順序の組み合わせから構築される。ここに、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、bおよびcはそれぞれ独立に0、1、2、3、または4であり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体である。本明細書に記載される薬剤送達結合体は、1個を越えるスペーサーリンカー、放出リンカー、またはヘテロ原子リンカーを持つ二価リンカーを含んでもよいことが理解される。例えば、2個以上の放出リンカー(lr)を含む二価リンカーが考慮される。さらに、この放出リンカーの構成は、二価リンカーであって、複数の放出リンカーが共有結合され、この複数の放出リンカーが、1個以上のヘテロ原子リンカーおよび/またはスペーサーリンカーによって相互に隔てられている二価リンカーを含む。
【0015】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が記載される。式中、Lは、(ls)aと(lH)b、およびそれらの任意の順序の組み合わせを含む二価のリンカーであり、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りである。この実施態様では、薬剤送達結合体の薬剤は、ハプテン、例えば、と言ってそれらに限定されないが、フルオレセイン、ジニトロフェニル等であることが可能である。
【0016】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が記載される。式中、Lは、(ls)a、(lH)bと(lr)c、およびそれらの任意の順序の組み合わせを含む二価のリンカーであり、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りであり、(lr)の少なくとも一つはジスルフィドではない。この実施態様の二価リンカーは、1個を越える(lr)を持つ、すなわち、cは1よりも大であるが、もう一つの、または、別の放出リンカーに加えて、ジスルフィド放出リンカーを含んでもよいことが理解される。
【0017】
本明細書に記載される各種ビタミン受容体結合性薬剤送達結合体の一つの局面では、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルオキシメチルオキシを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、メチルは、要すれば随意に、アルキル、または、置換アリールによって置換される。
【0018】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルカルボニルを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とアシルアジリジンを形成する。
【0019】
別の局面で、二価リンカーは、まとまって1-アルコキシシクロアルキレンオキシを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。
【0020】
別の局面で、二価リンカーは、まとまってアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシレートを形成するスペーサーリンカー、ヘテロ原子リンカー、および放出リンカーを含む。
【0021】
別の局面で、二価リンカーは、まとまってジチオアルキルカルボニルヒドラジドを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成する。
【0022】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルカルボニルヒドラジドを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成する。
【0023】
別の局面で、二価リンカーは、まとまって3-チオアルキルスルフォニルアルキル(二置換シリル)オキシを形成するヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、二置換シリルは、アルキル、または、要すれば随意に置換されるアリールによって置換される。
【0024】
別の局面で、二価リンカーは、天然アミノ酸およびその立体異性体から成るグループから選ばれる複数のスペーサーリンカーを含む。
【0025】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成する。
【0026】
別の局面で、二価リンカーは、まとまって3-ジチオアリールアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成し、アリールは要すれば随意に置換される。
【0027】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルオキシアルキルオキシアルキリデンを形成するヘテロ原子リンカー、スペーサーリンカー、放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アルキリデンは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成し、それぞれのアルキルは独立に選択され、オキシアルキルオキシは要すれば随意に、アルキル、または要すれば随意に置換されたアリールに置換される。
【0028】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルオキシカルボニルヒドラジドを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。
【0029】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アミノは、薬剤、またはその類縁体または誘導体とビニール性アミドを形成する。
【0030】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アミノは、薬剤、またはその類縁体または誘導体とビニール性アミドを形成し、アルキルはエチルである。
【0031】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成する。
【0032】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成し、アルキルはエチルである。
【0033】
別の局面で、二価リンカーは、まとまって3-ジチオアリールアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメート、またはカルバモイルアジリジンを形成する。
【0034】
一つの局面で、放出、スペーサー、およびヘテロ原子リンカーは、二価リンカーの結合分断に次いで、放出された官能基が、隣接基支援分断または破壊とも呼ばれる、さらに別の結合の破壊または分断を化学的に支援するように配置されていてもよい。このような二価リンカーまたはその部分の例示の実施態様は、下記の式を持つ化合物を含む、すなわち、
式中、Xは、窒素、酸素、または硫黄のようなヘテロ原子、nは、0、1、2および3から選ばれる整数、Rは水素、または、アリール環の陽電荷を誘導的にまたは共鳴によって安定化することが可能な置換基を含む置換基、例えば、アルコキシ等であり、記号(*)は、二価リンカーを形成する、さらに別のスペーサー、ヘテロ原子、または放出リンカーの付着部位、あるいは、薬剤、またはその類縁体または誘導体、または、ビタミン、またはその類縁体または誘導体の付着部位を示す。その他の置換基も、アリール環、ベンジル炭素、アルカン酸、または、メチレン架橋に存在することが可能であり、そのような置換基は、例えば、と言ってそれらに限定されないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロ等を含むことが理解される。支援分断としては、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ除去等を含む機構を含む。さらに、放出リンカーの分断に続く断片化の他に、放出リンカーの初期分断を、隣接基支援機構によって促進するようにしてもよいことが理解される。
【0035】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体中間体が提供される。この中間体は、ビタミン受容体結合性成分、第1末端と第2末端を有する二価リンカー、および結合基を含む。ビタミン受容体結合性成分は、ビタミン、またはその類縁体または誘導体であり、結合基は、求核要素、求電子要素、またはその前駆物質である。ビタミン受容体結合成分は、二価リンカーの第1末端において二価リンカーに共有結合し、結合基は、二価リンカーの第2末端において二価リンカーに共有結合し、二価リンカーは、1個以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、および、順序は任意のそれらの組み合わせを含む。
【0036】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体中間体が記載される。この中間体は、第1末端と第2末端を有する二価リンカー、薬剤、またはその類縁体または誘導体、および結合基を含む。二価リンカーは、本明細書で記載するスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーから選ばれる1種以上の成分を含む。結合基は、二価リンカーの第1末端において二価リンカーに共有結合し、薬剤およびその類縁体または誘導体は、二価リンカーの第2末端において二価リンカーに共有結合する。さらに、結合基は、ビタミン受容体結合成分と共有結合を形成することが可能な求核要素、求電子要素、またはその前駆物質であり、ビタミン受容体結合性成分は、ビタミン、またはその類縁体または誘導体である。
【0037】
本明細書で記載される、ビタミン受容体結合性薬剤送達結合体中間体の別の例示の実施態様では、結合基はマイケルアクセプターであり、二価リンカーは、式-C(O)NHN=、-NHC(O)NHN=、または-CH2C(O)NHN=を有する放出リンカーを含む。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の一つの例示の局面では、結合基と二価リンカーは共同して、下記の式を持つ化合物、またはその保護された誘導体を形成する、すなわち、
式中、Dは、本明細書で示すようにヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、nは、1、2、3、または4のような整数である。本明細書で記載される、ビタミン受容体結合性薬剤送達結合体中間体の別の例示の局面では、ビタミン、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0038】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の別の例示の実施態様では、結合基は、酸素、窒素、または硫黄のようなヘテロ原子であり、二価リンカーは、ビタミン、またはその類縁体または誘導体を、結合基に共有結合によって接続する、1個以上のヘテロ原子リンカー、および1個以上のスペーサーリンカーを含む。一つの例示の局面では、本明細書に記載されるビタミン受容体結合性送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Xは、酸素、窒素、または硫黄であり、mは、1、2、または3のような整数であり、V、ls、およびlHは上に定義した通りである。
【0039】
別の局面では、本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Xは、窒素または硫黄であり、Vおよびlsは、上に定義した通りである。
【0040】
別の局面では、本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Yは、水素または置換基であり、例示としては、電子吸引置換基で、例えば、と言ってそれらに限定されないが、ニトロ、シアノ、ハロ、アルキルスルフォニル、カルボン酸誘導体等を含む置換基であり、Vおよびlsは、上に定義した通りである。
【0041】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の、別の例示の実施態様では、結合基はマイケルアクセプターであり、二価リンカーは、ビタミン、またはその類縁体または誘導体を、結合基に共有結合によって接続する、1個以上のヘテロ原子リンカー、および1個以上のスペーサーリンカーを含む。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の一つの例示の局面では、結合基と二価リンカーは共同して、下記の式を持つ化合物、またはその保護された誘導体を形成する、すなわち、
式中、Xは、酸素、窒素、または硫黄であり、mおよびnは、1、2、または3のような独立に選択された整数であり、V、ls、およびlHは上に定義した通りである。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の別の例示の局面では、薬剤、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0042】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の、別の例示の局面では、中間体は、下記の式を持つ化合物、またはその保護された誘導体を含む、すなわち、
式中、Vはビタミン、またはその類縁体または誘導体であり、AAは、例示として天然アミノ酸およびその立体異性体から成るグループから選ばれるアミノ酸であり、Xは、窒素、酸素、または硫黄であり、Yは、水素または置換基であり、例示としては、電子吸引置換基で、例えば、と言ってそれらに限定されないが、ニトロ、シアノ、ハロ、アルキルスルフォニル、カルボン酸誘導体等を含む置換基であり、nおよびmは、1、2、または3のような独立に選択された整数であり、および、pは、1、2、3、4、または、5のような整数である。AAは、他の任意のアミノ酸、例えば、下記の一般式を有する任意のアミノ酸であってもよい、すなわち、
−N(R)−(CR’R’’)q−C(O)−
式中、Rは、水素、アルキル、アシル、または、適当な窒素保護基であり、R’およびR’’は、それぞれの出現において互いに独立に選択される水素または置換基であり、および、qは、1、2、3、4、または、5のような整数である。例示的には、R’および/またはR’’はそれぞれ独立して、水素、または、天然のアミノ酸に存在する側鎖、例えば、メチル、ベンジル、ヒドロキシメチル、チオメチル、カルボキシル、カルボキシルメチル、グアニジノプロピル等、および、それらの誘導体および保護誘導体に一致するが、ただしそれらに限定されない。前述の式は、立体異性変種を全て含む。例えば、アミノ酸は、アスパラギン、アスパラギン酸、システイン、グルタミン酸、リシン、グルタミン、アルギニン、セリン、オルニチン、トレオニン等から選ばれてもよい。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の別の局面では、薬剤、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0043】
別の実施態様では、下式を持つ化合物、またはその保護された誘導体を調製する方法が記載される:
式中、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせを含むリンカーであり、Dは、ヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、および、(lr)c、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りである。当該方法は、
(a)下式:
を持つ化合物またはその保護された誘導体を、下式:
を持つ化合物またはその保護された誘導体と反応させて、チオスクシニミド誘導体を形成させる工程、および、
(b)チオスクシニミド誘導体によって、薬剤またはその類縁体または誘導体のヒドラゾン誘導体を形成させる工程、
を含む。
【0044】
別の実施態様では、下式を持つ化合物、またはその保護された誘導体を調製する方法が記載される:
式中、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせを含むリンカーであり、Dは、ヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、および、(lr)c、(ls)aと(lH)b、およびVとは本明細書に定義する通りである。当該方法は、
下式:
を持つ化合物またはその保護された誘導体を、下式:
を持つ化合物またはその保護された誘導体と反応させる工程、
を含む。
【0045】
別の実施態様では、製薬組成物が記載される。製薬組成物は、本発明による薬剤送達結合体、および、製薬学的に受容可能な担体を含む。
【0046】
別の実施態様では、病原細胞の集団を抱える宿主動物において病原細胞集団を除去する方法であって、病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体のの接近可能な結合部位を有しており、結合部位は、病原細胞によって特異的に発現されるか、過剰に発現されるか、または優先的に発現されることを特徴とする方法が記載される。この方法は、本発明による薬剤送達結合体、またはその製薬組成物を宿主に投与する工程を含む。
【図面の簡単な説明】
【0047】
【図1】図1は、EC112(実施例9c)によるM109腫瘍成長の抑制を示す。
【図2】図2は、EC112(実施例9c)の動物体重に対する作用を示す。
【図3】図3は、EC105(実施例10a)によるM109腫瘍成長の抑制を示す。
【図4】図4は、EC105(実施例10a)の動物体重に対する作用を示す。
【図5】図5は、EC105(実施例10a)による4T1腫瘍成長抑制の欠如を示す。
【図6】図6は、EC145(実施例16b)によるM109腫瘍成長の抑制を示す。
【図7】図7は、EC140(実施例17a)によるM109腫瘍成長の抑制を示す。
【図8】図8は、EC136(実施例10b)によるL1210腫瘍成長の抑制を示す。
【図9】図9は、EC135(実施例17b)による細胞DNA合成の抑制を示す。
【図10】図10は、EC136(実施例10b)による細胞DNA合成の抑制を示す。
【図11】図11は、EC137(実施例16a)による細胞DNA合成の抑制を示す。
【図12】図12は、EC138(実施例10c)による細胞DNA合成の抑制を示す。
【図13】図13は、EC140(実施例17a)による細胞DNA合成の抑制を示す。
【図14】図14は、EC145(実施例16b)による細胞DNA合成の抑制を示す。
【図15】図15は、EC158(実施例14e)による細胞DNA合成の抑制を示す。
【図16】図16は、EC159(実施例15)による細胞DNA合成の抑制を示す。
【発明を実施するための形態】
【0048】
本発明は、ビタミン受容体結合性成分、二価リンカー(L)、および、薬剤を含むビタミン受容体結合性薬剤送達結合体であって、ビタミン受容体結合性成分と薬剤とが、それぞれ、要すれば随意にヘテロ原子リンカーを介して、二価リンカー(L)に結合するビタミン受容体結合性薬剤送達結合体に関する。二価リンカー(L)は、1個以上のスペーサーリンカー、ヘテロ原子リンカー、および、放出(すなわち分断可能)リンカー、および、順序は任意のそれらの組み合わせを含む。
【0049】
本明細書で用いる「放出リンカー」という用語は、生理的条件下で破壊することが可能な少なくとも一つの結合(例えば、pH易変性、酸易変性、酸化易変性、または酵素易変性の結合)を含むリンカーを指す。結合破壊をもたらすこのような生理的条件は、例えば、生理的pH下で起こる、または、細胞原形質pHよりも低いpHを持つエンドソームのような細胞内小器官への封入の結果として起こる、標準的化学的加水分解反応が挙げられる。
【0050】
分断可能な結合は、放出リンカー内の二つの隣接原子を接続することが可能であり、および/または、放出リンカーの一方の端に、または、その両端に、他のリンカー、または本明細書に記述するVおよび/またはDを接続することが可能である。分断可能な結合が、放出リンカー内の二つの原子を接続している場合、結合の破壊の次に、放出リンカーは、2個以上の断片に分解される。あるいは、分断可能な結合が、放出リンカーと別の成分、例えば、ヘテロ原子リンカー、スペーサーリンカー、別の放出リンカー、薬剤またはその類縁体または誘導体、または、ビタミンまたはその類縁体または誘導体の間にある場合、結合の破壊の次に、放出リンカーは、他の成分とは分離される。
【0051】
分断可能結合の易変性は、例えば、分断可能結合の、または、その近傍の置換的な改変、例えば、分断性ジスルフィド結合近傍のα分枝や、加水分解可能なシリコン-酸素結合を有する成分のシリコンの置換基の疎水性の増大、加水分解可能なケタールまたはアセタールの一部を形成するアルコキシ基を均一化すること等を含む改変によって調節することが可能である。
【0052】
本発明によれば、ビタミン受容体結合性薬剤送達結合体を用いて、宿主における病原細胞集団の存在によって特徴付けられる病状であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体の接近可能な結合部位を有しており、前記結合部位は、病原細胞によって特異的に発現される、過剰に発現される、または優先的に発現されることを特徴とする、病状を治療することが可能である。病原細胞の選択的除去は、ビタミン受容体結合性薬剤送達結合体のビタミン成分を、ビタミンまたはその類縁体または誘導体に特異的に結合し、かつ、病原細胞によって特異的に、過剰に、または優先的に発現されるビタミン受容体、トランスポーター、またはその他の表面発現性蛋白質に結合させることによって仲介される。病原細胞によって、特異的に、過剰に、または優先的に発現される表面発現性蛋白質は、非病原性細胞には存在しないか、または低濃度でしか存在しない受容体であり、これが、病原細胞の選択的除去手段を提供する。
【0053】
例えば、高親和度葉酸受容体のような表面発現性ビタミン受容体は、ガン細胞において過剰に発現される。卵巣、乳腺、結腸、肺、鼻、喉、および脳の上皮ガンは全て、高レベルの葉酸受容体を発現することが報告されている。事実、全てのヒトの卵巣腫瘍の90%を越えるものが、大量のこの受容体を発現することが知られている。従って、本発明の薬剤送達結合体は、各種の腫瘍細胞タイプを始め、優先的にビタミン受容体を発現し、従って、ビタミンまたはその類縁体または誘導体に対して表面的結合部位を有する、他のタイプの病原細胞、例えば、感染媒介物をも治療するために使用が可能である。
【0054】
本明細書に記述されるビタミンの他に、他のリガンドも、本明細書に記載・考察される薬剤およびリンカーに結合して、薬剤の、所望の標的に対する送達を促進可能とするリガンド-リンカー-薬剤結合体を形成することが可能であることが理解される。上記他のリンガンドも、記載のビタミンおよびその類縁体および誘導体に加えて、標的細胞に結合が可能な薬剤送達結合体を形成するのに使用することが可能である。一般に、細胞表面受容体のリガンドであればいずれのものでも、それに対してリンカー-薬剤結合体を調製可能な標的指向性リガンドとして好適に利用することが可能である。本明細書で考察される例示の、その他のリガンドとしては、ライブラリースクリーニングによって特定されるペプチドリガンド、腫瘍細胞特異的ペプチド、腫瘍細胞特異的アプタマー、腫瘍細胞特異的炭水化物、腫瘍細胞特異的モノクロナール抗体またはポリクロナール抗体、抗体のFabまたはscFv(すなわち、1本鎖可変域)断片、例えば、EphA2、または、転移ガン細胞において特異的に発現される、または、特異的に接近可能なその他の蛋白質に向けられた抗体のFab断片、組み合わせライブラリーから得られた小型有機分子、成長因子、例えば、EGF、FGF、インスリン、およびインスリン様成長因子、および相同ポリペプチド、ソマトスタチンとその類縁体、トランスフェリン、リポ蛋白質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種ガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に対して特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β-ラクタム抗生物質、例えばペニシリン、抗菌剤を含む小型有機分子、および、腫瘍細胞または感染性生物の表面に優先的に発現される受容体に特異的に結合するその他の分子、受容体または他の細胞表面蛋白質の結晶構造に基づいてある特定の受容体の結合ポケットにフィットするように設計された抗菌剤およびその他の薬剤、腫瘍細胞上に優先的に発現される腫瘍抗原またはその他の分子のリガンド、または、上記分子の内の任意のものの断片が挙げられる。リガンド-免疫原複合体のための結合部位として機能する可能性のある腫瘍特異的抗原の例としては、Ephrin族蛋白質のメンバー、例えば、EphA2、の細胞外エピトープが挙げられる。EphA2発現は、正常細胞では細胞・細胞接合部に限定されるが、EphA2は、転移腫瘍細胞では全細胞表面に広がる。従って、転移細胞上のEphA2は、例えば、免疫原と複合体化した抗体のFab断片の結合にとって接触可能となるが、一方、この蛋白質は、正常細胞上ではFab断片に対しては接触不能であるから、結果として、リガンド免疫原複合体を転移ガン細胞特異的なものとする。
【0055】
本発明はさらに、除去される病原細胞に対する標的化効果を最大にするための、リガンド-リンカー-薬剤結合体の組み合わせの使用法を考察する。
【0056】
例示の薬剤送達結合体は下記の通りである、すなわち、
V-L-D
V-(lr)c-D
V-(ls)a-D
V-(ls)a-(lr)c-D
V-(lr)c-(ls)a-D
V-(lH)b-(lr)c-D
V-(lr)c-(lH)b-D
V-(lH)d-(lr)c-(lH)e-D
V-(ls)a-(lH)b-(lr)c-D
V-(lr)c-(lH)b-(ls)a-D
V-(lH)d-(ls)a-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(ls)a-(lH)e-D
V-(lH)d-(ls)a-(lH)b-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(lH)b-(ls)a-(lH)e-D
V-(ls)a-(lr)c-(lH)b-D
V-[(ls)a-(lH)b]d-(lr)c-(lH)e-D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカー、および(lr)は放出リンカーであり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体であり、式中、二価のLは、1個の、または、各種の(ls)、(lH)、または(lr)を任意の順序で任意の組み合わせで含む。二価リンカーLの上記例は、二価リンカーの含む、(lH)、(ls)、(lr)の広範な集合を例示することを意図したものであって、限定することを意図したものではないことが理解される。
【0057】
薬剤送達結合体V-L-Dの一つの実施態様では、下記の式に従って、Vはビタミン葉酸であり、Lはエチレンジアミンではない、すなわち、
薬剤送達結合体V-L-Dの別の実施態様では、下記の式に従って、Vはビタミン葉酸であり、Dは薬剤マイトマイシンCであり、LはL-Cys-(S-チオエチル)ではない、すなわち、
Lは、下記の式に従って、L-Asp-L-Arg-L-Asp-L-Cys-(S-チオエチル)ではない、すなわち、
また、Lは、下式に従って、L-Arg-L-Cys-(S-チオエチル)-L-Ala-L-Gly-OHではない、すなわち、
【0058】
さらに本発明に従って考察されるものは、ビタミン、またはその類縁体または誘導体は放出リンカーに付着され、放出リンカーは、スペーサーリンカーを介して薬剤に付着される、薬剤送達結合体である。さらに、薬剤、および、ビタミンまたはその類縁体または誘導体の両方がそれぞれスペーサーリンカーに付着が可能であり、スペーサーリンカー同士は、相互に、放出リンカーを通じて付着される。加えて、薬剤、および、ビタミンまたはその類縁体または誘導体の両方がそれぞれ放出リンカーに付着が可能であり、放出リンカー同士は、相互に、スペーサーリンカーを通じて付着される。ヘテロ原子リンカーは、任意の二つのリンカーの間に、または、任意のリンカーと、ビタミンまたはその類縁体または誘導体の間に、または、任意のリンカーと、薬剤またはその類縁体または誘導体の間に設置してもよい。その他可能な全ての順列と組み合わせが考察される。
【0059】
一つの実施態様では、本発明は、ビタミン受容体結合性薬剤送達結合体を提供する。薬剤送達結合体は、ビタミン受容体結合成分、二価のリンカー(L)、および薬剤から成る。ビタミン受容体結合成分は、ビタミン受容体に結合することが可能なビタミン、またはその類縁体または誘導体であり、薬剤は、薬剤活性を示す類縁体または誘導体を含む。ビタミン、またはその類縁体または誘導体は、二価リンカー(L)に共有結合され、薬剤、またはその類縁体または誘導体も、二価リンカー(L)に共有結合される。二価リンカー(L)は、1個以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、および、順序は任意のそれらの組み合わせを含む。例えば、ヘテロ原子リンカーは窒素であってもよく、放出リンカーとヘテロ原子リンカーはまとめられて二価の基、例えば、アルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルフォキシルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、スルフォニルアルキルアジリジン-1-イル、またはアルキレンスルフォニルアジリジン-1-イルを含む二価の基を形成することが可能であり、各放出リンカーは、要すれば随意に、下記に定義する置換基X2によって置換される。あるいは別に、ヘテロ原子リンカーは、窒素、酸素、硫黄、および、式-(NHR1NHR2)-、-SO-、-(SO2)-、-N(R3)O-であることが可能である。式中、R1、R2、およびR3は、それぞれ独立に、水素、アルキル、アリール、アリールアルキル、アリール置換体、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、および、アルコキシアルキルから選ばれる。別の実施態様では、ヘテロ原子リンカーは酸素であってもよく、スペーサーリンカーは、要すれば随意に下に定義される置換基X1によって置換されてもよい1-アルキレンスクシニミド-3-イルであってもよく、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、スペーサーリンカーと放出リンカーとは、それぞれ、ヘテロ原子リンカーに結合されて、スクシニミド-1-イルアルキルアセタルまたはケタルを形成する。
【0060】
スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イル、アルキレンスルフォキシル、スルフォニルアルキル、アルキレンスルフォキシルアルキル、アルキレンスルフォニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、下に定義される置換基X1によって、要すれば随意に置換されてもよい。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、下に定義される置換基X1によって、要すれば随意に置換されてもよく、スペーサーリンカーは前記窒素に結合してアミドを形成する。あるいは別に、ヘテロ原子リンカーは硫黄であってもよく、スペーサーリンカーはアルキレンおよびシクロアルキレンであってもよく、各スペーサーリンカーは、カルボキシル基によって要すれば随意に置換されてもよく、スペーサーリンカーは前記硫黄に結合してチオールを形成する。別の実施態様では、ヘテロ原子リンカーは硫黄であってもよく、スペーサーリンカーは、1-アルキレンスクシニミド-3-イルおよび1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、スペーサーリンカーは、前記硫黄に結合してスクシニミド-3-イルチオールを形成する。
【0061】
前述の実施態様に対する別態様では、ヘテロ原子リンカーは窒素であってもよく、放出リンカーとヘテロ原子リンカーはまとめられて二価の基、例えば、アルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、またはスルフォニルアルキルアジリジン-1-イルを含む二価の基を形成することが可能であり、各放出リンカーは、要すれば随意に、下記に定義する置換基X2によって置換される。この別態様では、スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、要すれば随意に、下記に定義する置換基X1によって置換され、スペーサーリンカーは放出リンカーに結合してアジリジンアミドを形成する。
【0062】
置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、置換基X1とヘテロ原子リンカーは結合するスペーサーリンカーとまとめられて複素環を形成することも可能である。
【0063】
放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルフォニルオキシ、オキシスルフォニルアルキル、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデニミニル、アルキレンチオ、アルキレンアリールチオ、および、カルボニルアルキルチオであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換される。
【0064】
前述の実施態様では、ヘテロ原子リンカーは酸素であってもよく、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記酸素に結合されてアセタルまたはケタルを形成する。あるいは別に、ヘテロ原子リンカーは酸素であってもよく、放出リンカーはメチレンであってもよく、メチレンは、随意に置換されてもよいアリールによって置換され、放出リンカーは酸素に結合してアセタールまたはケタールを形成する。さらに、ヘテロ原子リンカーは酸素であってもよく、放出リンカーはスルフォニルアルキルであってもよく、放出リンカーは酸素に結合してアルキルスルフォネートを形成する。
【0065】
前述の放出リンカー態様の別態様として、ヘテロ原子リンカーは窒素であってもよく、放出リンカーは、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、および、カルボニルシクロアルキリデニミニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記窒素に結合されてヒドラゾンを形成する。別形態として、ヒドラゾンは、カルボン酸誘導体、オルトギ酸塩誘導体、またはカルバモイル誘導体によってアシル化されて、各種アシルヒドラゾン放出リンカーを形成してもよい。
【0066】
別態様として、ヘテロ原子リンカーは酸素であってもよく、放出リンカーは、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、および(ジアリールシリル)アリールであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記窒素に結合されてシラノールを形成する。
【0067】
前述の放出リンカー実施態様では、薬剤は窒素原子を含んでもよく、ヘテロ原子リンカーは窒素であってもよく、放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルであってもよく、放出リンカーはヘテロ原子窒素に結合してアミドを形成してもよく、薬剤酸素に結合してアミドを形成してもよい。
【0068】
前述の放出リンカー実施態様では、薬剤は酸素原子を含んでもよく、ヘテロ原子リンカーは窒素でもよく、放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルであってもよく、放出リンカーは、ヘテロ原子リンカー窒素に結合してアミドを形成してもよく、薬剤窒素に結合してエステルを形成してもよい。
【0069】
置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってもよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、置換基X2とヘテロ原子リンカーは結合する放出リンカーとまとめられて複素環を形成することも可能である。
【0070】
複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、および、スクシニミドであってもよい。
【0071】
薬剤は、マイトマイシン、マイトマイシン誘導体、またはマイトマイシン類縁体であってもよく、本実施態様では、放出リンカーは、カルボニルアルキルチオ、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシミニド-3-イルであってもよく、各放出リンカーは、置換基X2によって、要すれば随意に置換されてもよく、マイトマイシンのアジリジンは、放出リンカーに結合されてアシルアジリジンを形成する。
【0072】
薬剤は窒素原子を含んでもよく、放出リンカーは、要すれば随意に置換基X2によって置換されるハロアルキレンカルボニルであってもよく、放出リンカーは薬剤窒素に結合してアミドを形成する。
【0073】
薬剤は酸素原子を含んでもよく、放出リンカーは、要すれば随意に置換基X2によって置換されるハロアルキレンカルボニルであってもよく、放出リンカーは薬剤酸素に結合してエステルを形成する。
【0074】
薬剤は、二重結合窒素原子を含んでもよく、本実施態様では、放出リンカーはアルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシニミド-3-イルであってもよく、放出リンカーは薬剤窒素と結合してヒドラゾンを形成することが可能である。
【0075】
薬剤は硫黄原子を含んでもよく、本実施態様では、放出リンカーは、アルキレンチオ、およびカルボニルアルキルチオであってもよく、放出リンカーは薬剤硫黄に結合してジスルフィドを形成することが可能である。
【0076】
ビタミンは、窒素を含む葉酸塩であってもよく、本実施態様では、スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、置換基X1によって要すれば随意に置換され、スペーサーリンカーは、葉酸窒素に結合してイミドまたはアルキルアミドを形成する。本実施態様では、置換基X1は、アルキル、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、カルボキシ、カルボキシアルキル、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってもよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。
【0077】
本明細書で用いる「アルキル」という用語は、要すれば随意に分枝されてもよい、一価の炭素原子直鎖、例えば、メチル、エチル、プロピル、3-メチルペンチル等を指す。
【0078】
本明細書で用いる「シクロアルキル」という用語は、その一部が環を形成する、一価の炭素原子鎖、例えば、シクロプロピル、シクロヘキシル、3-エチルシクロペンチル等を指す。
【0079】
本明細書で用いる「アルキレン」という用語は、要すれば随意に分枝されてもよい、二価の炭素原子直鎖、例えば、メチレン、エチレン、プロピレン、3-メチルペンチレン等を指す。
【0080】
本明細書で用いる「シクロアルキレン」という用語は、その一部が環を形成する、二価の炭素原子鎖、例えば、シクロプロプ-1,1-ジイル、シクロプロプ-1,2-ジイル、シクロヘクス-1,4-ジイル、3-エチルシクロペント-1,2-ジイル、1-メチレンシクロヘクス-4-イル等を指す。
【0081】
本明細書で用いる「複素環」という用語は、炭素と、窒素、酸素および硫黄から選ばれるヘテロ原子とから成る一価の鎖であって、少なくとも1個のヘテロ原子を含むその一部が環を形成する鎖を指し、例えば、アジリジン、ピロリジン、オキサゾリジン、3-メトキシピロリジン、3-メチルピペラジン等を指す。
【0082】
本明細書で用いる「アルコキシ」という用語は、末端酸素と結合した、本明細書で定義するアルキル、例えば、メトキシ、エトキシ、プロポキシ、3-メチルペントキシ等を指す。
【0083】
「ハロ」または「ハロゲン」という用語は、フルオロ、クロロ、ブロモ、およびイオドを指す。
【0084】
本明細書で用いる「アリール」という用語は、炭素原子から成る、単環または多環芳香族環、例えば、フェニル、ナフチル等を指す。
【0085】
本明細書で用いる「ヘテロアリール」という用語は、炭素原子および、窒素、酸素、および硫黄から選ばれる少なくとも1個のヘテロ原子から成る、単環または多環芳香族環、を指し、例えば、ピリジニル、ピリミジニル、インドリル、ベンゾキサゾリル等を指す。
【0086】
本明細書で用いる「アリール置換体」または「ヘテロアリール置換体」という用語は、ハロ、ヒドロキシ、アミノ、アルキルまたはジアルキルアミノ、アルコキシ、アルキルスルフォニル、シアノ、ニトロ等から選ばれる1種以上の置換基によって置換されるアリールまたはヘテロアリールを指す。
【0087】
本明細書で用いる「イミノアルキリデニル」という用語は、本明細書で定義するアルキレン、および窒素原子を含む二価の基であって、アルキレンの末端炭素が、例えば、式-(CH)=N-、-(CH2)2(CH)=N-、-CH2C(Me)=N-等のように窒素原子と二重結合するものを指す。
【0088】
本明細書で用いる「アミノ酸」という用語は、一般に、アミノアルキルカルボキシレートであって、アルキル基は、要すれば随意にアルキル、ヒドロキシアルキル、スルフヒドリルアルキル、アミノアルキル、カルボキシアルキル等によって置換されてもよい、アミノアルキルカルボキシレートを指し、天然アミノ酸、例えば、セリン、システイン、メチオニン、アスパラギン酸、グルタミン酸等と一致するグループを含む。
【0089】
「アリールアルキル」という用語は、本明細書で定義されるアルキレン基によって置換された、本明細書で定義されるアリールを指し、例えば、ベンジル、フェネチル、α-メチルベンジル等を指す。
【0090】
前述の用語は組み合わせて、化学的関連グループ、例えば、メチルオキシメチル、エチルオキシエチル等を指す「アルコキシアルキル」、および、トリフルオロメチルオキシエチル、1,2-ジフルオロ-2-クロロエト-1-イルオキシプロピル等を指す「ハロアルコキシアルキル」、等を生成することが可能であることを理解しなければならない。
【0091】
本明細書で用いる「アミノ酸誘導体」という用語は、一般に、アミノアルキルカルボキシレートであって、アミノ基またはカルボキシレート基は、それぞれ独立に、アルキル、カルボキシアルキル、アルキルアミノ等によって要すれば随意に置換され、あるいは要すれば随意に保護され、介在する二価のアルキル断片は、アルキル、ヒドロキシアルキル、スルフヒドリルアルキル、アミノアルキル、カルボキシアルキル等によって要すれば随意に置換される、アミノアルキルカルボキシレートを指し、天然アミノ酸に見られる側鎖、例えば、セリン、システイン、メチオニン、アスパラギン酸、グルタミン酸等に見られるものと一致する基を含む。
【0092】
本明細書で用いる「ペプチド」という用語は、一般に、相互にアミド結合によって共有結合された一連のアミノ酸、およびその類縁体および誘導体を指す。
【0093】
放出リンカーは、生理的条件下で破壊または分断が可能な、少なくとも1個の結合(例えば、pH易変性、酸易変性、酸化易変性、または酵素易変性結合)を含む。この分断可能な結合(単数または複数)は、分断リンカーの内部にあってもよいし、および/または、分断リンカーの一端または両端にあってもよい。分断可能結合の易変性は、隣接基支援とも呼ばれる、結合破壊を支援、または促進することが可能な官能基または断片を、二価リンカーLの内部に含ませることによって調節が可能であることが理解される。さらに、放出リンカーの結合破壊の後で、ビタミン受容体結合性薬剤結合体のさらにそれ以上の断片化を支援または促進することが可能な、別の官能基または断片を二価リンカーLの内部に含めてもよいことが理解される。
【0094】
放出リンカーの結合分断の例示の機構として、下記の、オキソニウム支援分断が挙げられる、すなわち、
式中、Zはビタミン、またはその類縁体または誘導体、あるいは、薬剤、またはその類縁体または誘導体、または、それぞれ、二価リンカーの他の部分と結合するビタミンまたは薬剤成分であり、例えば、1種以上のスペーサーリンカー、ヘテロ原子リンカー、および/または、他の放出リンカーを含む、薬剤またはビタミン成分である。本実施態様では、酸触媒によるカルバメート除去によって、CO2とZに付着する窒素含有基の放出、およびベンジル陽イオンの形成がもたらされ、後者は、水、または他の任意のルイス塩基によって捕捉される。
【0095】
放出リンカーに接続され、または、放出リンカーの中に含まれる結合であって、二価リンカーLの一部を形成してもよい結合の、別の分断の例示の機構として、下記の、ベータ除去、およびビニール的ベータ除去機構が挙げられる、すなわち、
式中、Xは求核要素、GSH、グルタチオン、または生体還元因子等であり、ZまたはZ’のどちらかは、ビタミンまたはその類縁体または誘導体、あるいは、薬剤またはその類縁体または誘導体、あるいは、二価リンカーの他の部分と結合するビタミンまたは薬剤成分である。結合分断は、酸触媒によるカルバメート基の除去によっても起こってもよく、カルバメート基の除去は、上述の例で示されたベータ硫黄またはジスルフィドのアリール基によって実現される安定化によって隣接基支援を受けてもよいことが理解される。。本実施態様のこれらの変種では、放出リンカーはカルバメート基である。
【0096】
別の例示の機構は、放出リンカー、スペーサー、および、ヘテロ原子リンカーを適当なやり方で配置することを含む。その配置は、二価リンカーの一つの結合の分断に次いで、放出された官能基がさらに別の結合の破壊または分断を化学的に支援するように行う。後者は、隣接基支援分断または破壊とも呼ばれる。このような二価リンカーまたは部分の例示の実施態様は、下式の化合物を含む、すなわち、
式中、Xはヘテロ原子、例えば、窒素、酸素、または硫黄であり、nは0、1、2および3から選ばれる整数であり、Rは水素、または置換基、例えば、誘電的に、または共鳴によって、アリール環上の陽性電荷を安定化することのできる置換基を含む置換基、例えば、アルコキシ等であり、ZまたはZ’のどちらかは、ビタミンまたはその類縁体または誘導体、あるいは、薬剤またはその類縁体または誘導体、あるいは、二価リンカーの他の部分と結合するビタミンまたは薬剤成分である。アリール環、ベンジル炭素、カルバメート窒素、アルカン酸、またはメチレン架橋には、他の置換基も、例えば、と言ってそれらに限定されるものではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロ等を含む、他の置換基も存在してよいことが理解される。支援による分断としては、ベンジリウム中間体、ベンジン中間体、ラクトン環状化、オキソニウム中間体、ベータ除去等を含む機構が挙げられる。放出リンカーの分断に次ぐ断片化に加えてさらに、放出リンカーの最初の分断が、隣接基支援機構によって促進されてもよいことが理解される。
【0097】
本実施態様では、環状化してもよいヒドロキシアルカン酸は、例えば、オキソニウムイオンによってメチレン架橋の分断を促進するし、また、放出リンカーの結合分断、または、結合分断後の断片化を促進する。あるいは別に、酸触媒オキソニウムイオン支援メチレン架橋分断が、この例示の二価リンカー、またはその断片の、カスケード的に継続する断片化を起動してもよい。あるいは別に、酸触媒による、カルバメートの加水分解が、環状化してもよいヒドロキシアルカン酸のベータ除去を促進してもよいし、また、例えば、オキソニウムイオンによるメチレン架橋の分断を促進してもよい。結合破壊または分断の、その他の化学的機構も、本明細書に記述される代謝的、生理的、または細胞条件下で、このようなカスケード的に継続する断片化を起動してもよいことが理解される。
【0098】
本明細書に記載される薬剤送達結合体は、従来技術で認知済みの合成法によって調製が可能である。合成法は、ヘテロ原子リンカーの選択、および、スペーサーリンカーおよび放出リンカーに存在する官能基に応じて選ばれる。一般に、関連する結合形成反応は、Richard C. Larock、「有機変換総論、官能基調製入門(”Comprehensive Organic Transformations, a guide to functional group preparations”)」、VCH Publishers, Inc.ニューヨーク(1989)、および、Theodora E. Greene & Peter G.M. Wuts, 「有機合成における保護基(”Protective Groups in Organic Synthesis”)」、第2版、John Wiley & Sons, Inc.ニューヨーク(1991)に記載されている。この開示を、引用することにより本明細書に含める。
【0099】
===アミドおよびエステルの一般的形成===
例えば、ヘテロ原子リンカーが窒素原子で、スペーサーリンカーまたは放出リンカーに存在する末端官能基がカルボニル基である場合、必要なアミド基は、対応するカルボン酸またはその誘導体と、アミンとの結合反応またはアシル化反応によって、例えば、スキーム1に図示するようにして実現が可能である。式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフレート、ペンタフルオロフェノキシ、トリメチルシリルオキシ、スクシニミド-N-オキシ等である。
スキーム1
【0100】
結合試薬としては、DCC、EDC、RRDQ、CGI、HBTU、TBTU、HOBT/DCC、HOBT/EDC、BOP-C1、PyBOP、PyBroP等が挙げられる。あるいは別に、親酸は、活性化カルボニル誘導体、例えば、酸塩化物、N-ヒドロキシスクシニミジルエステル、ペンタフルオロフェニルエステル等に変換されてもよい。このアミド形成反応は、塩基、例えば、トリエチルアミン、ジイソプロピルエチルアミン、N,N-ジメチル-4-アミノピリジン等の存在下に実行することも可能である。本明細書に記載されるアミドを形成するのに好適な溶媒としては、CH2Cl2、CHCl3、THF、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。例示として、アミドは、約-15℃から約80℃、または約0℃から約45℃の範囲の温度で調製することが可能である。アミドは、例えば、窒素含有アジリジン環、炭水化物、およびα-ハロゲン化カルボン酸から形成することが可能である。アミドを形成するのに好適な例示のカルボン酸として下記の式を持つ化合物等が挙げられる、すなわち、
等で、式中、nは、1、2、3または4のような整数である。
【0101】
同様に、ヘテロ原子リンカーは酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する末端官能基はカルボニル基である場合には、必要なエステル基は、対応するカルボン酸または誘導体と、アルコールとの結合反応によって得ることが可能である。
【0102】
結合試薬としては、DCC、EDC、CDI、BOP、PyBOP、イソプロペニルクロロギ酸、EEDQ、DEAD、PPh3等が挙げられる。溶媒としては、CH2Cl2、CHCl3、THF、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。塩基としては、トリエチルアミン、ジイソプロピル-エチルアミン、および、N,N-ジメチル-4-アミノピリジンが挙げられる。あるいは別に、親酸は、活性化カルボニル誘導体、例えば、酸塩化物、N-ヒドロキシスクシニミジルエステル、ペンタフルオロフェニルエステル等に変換されてもよい。
【0103】
===ケタールおよびアセタールの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子で、スペーサーリンカーまたは放出リンカーに存在する官能基が1-アルコキシアルキルである場合、必要なアセタールまたはケタール基は、スキーム2に示すように、対応するアルコールとエノールエーテルとのケタールおよびアセタール形成反応によって形成することが可能である。
スキーム2
【0104】
溶媒としては、アルコール、CH2Cl2、CHCl3、THF、ジエチルエーテル、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。このようなアセタールおよびケタールの形成は、酸触媒によって実現することが可能である。ヘテロ原子リンカーが二つの酸素原子を含み、放出リンカーが、要すれば随意に本明細書で記載するX2基で置換されてよいメチレンである場合、必要な対称型アセタールまたはケタール基は、例示として、スキーム3で示すように、対応するアルコールとアルデヒドまたはケトンによるアセタールおよびケタール形成反応によって形成することが可能である。
スキーム3
【0105】
あるいは別に、メチレンが、随意に置換されてよいアリール基で置換されている場合、必要なアセタールまたはケタールは、スキーム4に図示するように段階的に調製されてもよく、式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフルオロアセトキシ、トリフレート等である。スキーム4に示される過程は、従来型の調製法であり、一般的にR.R. Schmidt et al., Chem. Rev., 2000, 100, 4423-42によって総説される過程に従う。なお、この開示を引用することにより本明細書に含める。
スキーム4
【0106】
得られたアリールアルキルエーテルは、酸化剤、例えば、DDQ等によって処理され、中間体オキソニウムイオンを形成し、このイオンは次に別のアルコールによって処理されアセタールまたはケタールを生成する。
【0107】
===スクシニミドの一般的形成===
さらに、ヘテロ原子リンカーが、例えば、窒素、酸素、または硫黄原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がスクシニミド誘導体である場合、得られる炭素ヘテロ原子結合は、スキーム5に示すように、対応するアミン、アルコール、またはチオールと、マレイミド誘導体とのマイケル付加によって形成が可能であり、式中、Xは、ヘテロ原子リンカーである。
スキーム5
【0108】
マイケル添加を実行するための溶媒としては、THF、EtOAc、CH2Cl2、DMF、DMSO、H2O等が挙げられる。このようなマイケル付加物の形成は、等モル量の塩基、例えば、トリエチルアミン、ヒューニッヒ塩基(Hunig’s base)を添加することによって、または、水溶液のpHを6.0-7.4に調節することによって実現することが可能である。ヘテロ原子リンカーが酸素または窒素原子である場合、マイケル添加を促進するように、例えば、より高い反応温度を用いる、触媒を添加する、より極性の高い溶媒、例えば、DMF、DMSO等を用いる、マレイミドをシリル化試薬で活性化すること等によって、反応条件を調整してもよいことが理解される。
【0109】
===シリルオキシの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がシリル誘導体である場合、必要なシリルオキシ基は、対応するシリル誘導体とアルコールとを反応させることによって、例えば、スキーム6に図示するようにして生成してもよい。式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフルオロアセトキシ、トリフレート等である。
スキーム6
【0110】
シリル誘導体は、好適に官能化されたシリル誘導体、例えば、ビニールスルフォノアルキルジアリール、またはジアリール、またはアルキルアリールシリルクロリドを含む。ビニールスルフォノアルキル基の代わりに、β-クロロエチルスルフォノアルキル前駆物質を用いてもよい。非プロトン性の無水溶媒と窒素含有塩基であれば、いずれのものでも反応溶媒として使用が可能である。この変換に採用が可能な温度範囲は、-78℃と80℃の間を変動してもよい。
【0111】
===ヒドラゾンの一般的形成===
さらに、ヘテロ原子リンカーが窒素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がイミニル誘導体である場合、必要なヒドラゾン基は、スキーム7のそれぞれ式(1)と(2)に示されるように、対応するアルデヒドまたはケトンと、ヒドラジンまたはアシルヒドラジン誘導体とを反応させることによって形成することが可能である。
スキーム7
【0112】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO、MeOH等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。任意の酸性触媒、例えば、鉱酸、H3CCOOH、F3CCOOH、p-TsOH・H2O、ピリジニウムp-トルエンスルフォネート等を使用することが可能である。式(2)のアシルヒドラゾンの場合、アシルヒドラゾンは、スキーム1で一般的に上述したように、最初ヒドラジンを、適当なカルボン酸または誘導体でアシル化し、その後アシルヒドラジドを、対応するアルデヒドまたはケトンと反応させて、アシルヒドラゾンを形成してもよい。あるいは別に、ヒドラゾン官能基を、最初に、ヒドラジンを対応するアルデヒドまたはケトンと反応させることによって形成してもよい。得られたヒドラゾンは、次に、スキーム1で一般的に上述したように、適当なカルボン酸または誘導体によってアシル化してもよい。
【0113】
===ジスルフィドの一般的形成===
さらに、ヘテロ原子リンカーが硫黄原子であり、放出リンカーに存在する官能基がアルキレンチオール誘導体である場合、必要なジスルフィド基は、スキーム8に示されるように、対応するアルキルまたはアリールスルフォニルチオアルキル誘導体、あるいは、対応するヘテロアリールジチオアルキル誘導体、例えば、ピリジン-2-イルジチオアルキル誘導体等と、アルキレンチオール誘導体とを反応させることによって形成することが可能である。
スキーム8
【0114】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。必要なアルキルまたはアリールスルフォニルチオアルキル誘導体は、従来技術で認知済みのプロトコールを用いて、また、Ranasinghe & Fuchs, Synth. Commun. 18(3), 227-32 (1988)の方法に従って調製してもよい。なお、上記開示を、引用することにより本明細書に含める。非対称のジアルキルジスルフィドのその他の調製法は、国際特許出願WO88/01622、欧州特許出願0116208A1、および米国特許第4,691,024号に記載されているように、非対称型のヘテロアリール-アルキルジスルフィド、例えば、2-チオピリジニル、3-ニトロ-2-チオピリジニル等のジスルフィドに対する、アルキルチオールによるチオール変換にもとづく。なお、上記開示を、引用することにより本明細書に含める。
【0115】
===炭酸塩の一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がアルコキシカルボニル誘導体である場合、必要な炭酸基は、スキーム9に示されるように、対応するヒドロキシ置換化合物を、活性化アルコキシカルボニル誘導体と反応させることによって形成することが可能である。式中、Lは適当な脱離基である。
スキーム9
【0116】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。反応を促進するために、任意の塩基性触媒、例えば、無機塩基、アミン塩基、ポリマー結合塩基等を使用することが可能である。
【0117】
===セミカルバゾンの一般的形成===
さらに、ヘテロ原子リンカーが窒素原子であり、一方のスペーサーリンカーまたは放出リンカーに存在する官能基がイミニル誘導体であり、他方のスペーサーリンカーまたは他方の放出リンカーに存在する官能基がアルキルアミノまたはアリールアミノカルボニル誘導体である場合、必要なセミカルバゾン基は、スキーム10に示されるように、対応するアルデヒドまたはケトンと、セミカルバジドとを反応させることによって形成することが可能である。
スキーム10
【0118】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO、MeOH等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。任意の酸性触媒、例えば、鉱酸、H3CCOOH、F3CCOOH、p-TsOH・H2O、ピリジニウムp-トルエンスルフォネート等を使用することが可能である。さらに、セミカルバゾンを形成するに際して、ヒドラゾン官能基を、最初に、ヒドラジンを対応するアルデヒドまたはケトンと反応させることによって形成してもよい。得られたヒドラゾンは、次に、イソシアネート、またはカルバモイル誘導体、例えば、カルバモイルハロゲン化物でアシル化することによってセミカルバゾンを形成してもよい。あるいは別に、対応するセミカルバジドを、ヒドラジンを、イソシアネート、またはカルバモイル誘導体、例えば、カルバモイルハロゲン化物と反応させることによって形成してもよい。次に、このセミカルバジドを、対応するアルデヒドまたはケトンと反応させてセミカルバゾンを形成してもよい。
【0119】
===スルフォネートの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がスルフォニル誘導体である場合、必要なスルフォネート基は、スキーム11に示されるように、対応するヒドロキシ置換化合物と、活性化スルフォニル誘導体とを反応させることによって形成することが可能である。式中、Lは適当な脱離基、例えば、ハロ等である。
スキーム11
【0120】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。反応を促進するために、任意の塩基性触媒、例えば、無機塩基、アミン塩基、ポリマー結合塩基等を使用することが可能である。
【0121】
===葉酸ペプチドの一般的形成===
葉酸含有ペプチジル断片Pte-Glu-(AA)n-NH(CHR2)CO2H(3)は、スキーム12に示すように、標準法によるポリマー支持連続法、例えば、酸感受性Fmoc-AA-Wang樹脂(1)上でFmoc策を実施することによって調製される。
スキーム12
(a)20%ピペリジン/DMF;(b)Fmoc-AA-OH、PyBop、DIPEA、DMF;(c)Fmoc-Glu(O-t-Bu)-OH、PyBop、DIPEA、DMF;(d)1.N10(TFA)-Pte-OH;PyBop、DIPEA、DMSO;(e)TFAA、(CH2SH)2、i-Pr3SiH;(f)NH4OH、pH10.3
【0122】
この例示の過程に関する実施態様では、R1はFmocであり、R2は、適当に保護された所望のアミノ酸側鎖であり、DIPEAはジイソプロピルエチルアミンである。効率的な結合を確保するために、結合剤、例えば、PyBOP、および、本明細書で記載される、または従来技術で既知の、その他の結合剤が活性化試薬として与えられる場合、標準的な結合工程が用いられる。Fmoc保護基は、各結合工程後、標準条件下で、例えば、ピペリジン、テトラブチルアンモニウムフロリド(TBAF)等による処理によって除去される。適当に保護されたアミノ酸建設ブロック、例えば、Fmoc-Glu-OtBu、N10-TFA-Pte-OH等は、スキーム12で記載し、工程(b)でFmoc-AA-OHで示されるように用いられる。従って、AAは、適当に保護された、任意のアミノ酸の開始材料を指す。本明細書で用いられるアミノ酸という用語は、アミンとカルボン酸官能基が、1個以上の炭素で隔てられた任意の試薬を指し、天然のアルファーおよびベータアミノ酸を始め、それらアミノ酸のアミノ酸誘導体および類縁体を含むことを理解しなければならない。特に、保護された側鎖を有するアミノ酸、例えば、保護されたセリン、トレオニン、システイン、アスパラギン酸等も、本明細書に記載する葉酸-ペプチド合成に使用してよい。さらに、ガンマ、デルタ、またはさらに長い相同のアミノ酸もまた本明細書に記載する葉酸‐ペプチド合成の開始材料に含めてもよい。さらに、相同側鎖を有する、または、交互分枝構造を有するアミノ酸類縁体、例えば、ノルロイシン、イソバリン、β-メチルスレオニン、β-メチルシステイン、β、β-ジメチルシステイン等を、本明細書に記載する葉酸-ペプチド合成において開始材料に含めてもよい。
【0123】
Fmoc-AA-OHを含む結合順序(工程(a)と(b))は、”n”回実行されて、固相支持体ペプチド2を調製する。nは整数であり、0から約100に等しくともよい。最後の結合工程後、残余のFmoc基は除去され(工程(a))、このペプチドは、順次、グルタミン酸誘導体に結合され(工程(c))、脱保護され、TFA保護プテロイン酸に結合される(工程(d))。次に、このペプチドは、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランによる処理によってポリマー支持体から分離される(工程(e))。これらの反応条件によって、適当に保護されるアミノ酸側鎖の一部を形成してもよいt-Bu、t-Boc、およびTrt保護基の同時除去が実現される。TFA保護基が、塩基による処理によって除去されて(工程(f))、葉酸含有ペプチジル断片3を与える。
【0124】
スペーサーおよび放出リンカー、およびヘテロ原子リンカーは様々なやり方で組み合わせることが可能である。例示として、リンカー同士は、互いに、ヘテロ原子リンカーを介して付着される、例えば、下式で示されるように、アルキレン-アミノ-アルキレンカルボニル、アルキレン-チオ-カルボニルアルキルスクシニミド-3-イル等のようである。式中、整数xとyは1、2、3、4、または5である。すなわち、
【0125】
本明細書に記載されるリンカーの別の例示の実施態様は、本明細書に記載される条件下で、ベータ除去の関与する化学機構によって分断される放出リンカーを含む。一つの局面で、このような放出リンカーは、ベータ-チオ、ベータ-ヒドロキシ、および、ベータ-アミノ置換カルボン酸、およびその誘導体、例えば、エステル、アミド、炭酸塩、カルバメート、および尿素を含む。別の局面では、このような放出リンカーは、2-および4-チオアリールエステル、カルバメート、および炭酸塩を含む。
【0126】
さらに、ビタミンまたは薬剤の、ヘテロ原子リンカーに対する付着は、下式に示されるように、ヘテロ原子リンカーにあらかじめ変換された、例えば、アクラマイシンケトンの対応するヒドラゾンへの変換、葉酸の対応するアミドへの変換等によって生じた、薬剤またはビタミン上の反応性官能基を介して実現することが可能である。すなわち、
【0127】
二価リンカー(L)は、スペーサーリンカー、放出リンカー、ヘテロ原子リンカー、および、順序は任意のそれらの組み合わせから選ばれる、1種以上の成分を含む。例えば、表1および2に示す、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせが考えられる。これらのリンカーリストは包括的なものではなく、単に例示的なものであり、本明細書に記載される発明を限定するものと解釈してはならない。前述の構造に存在する星印は、表1および2のものも含めて、追加スペーサー、放出またはヘテロ原子リンカーの、または、ビタミン受容体結合性薬剤送達結合体の薬剤またはビタミン成分の、例示の付着点を特定する。二価リンカーLは、表1および2に示したものを含めた1種以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーを含むこと、および、このスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーは任意の順序で組み合わせて二価リンカーLを形成することが可能であることが理解される。
【0128】
<表1>
リンカー、および、いくつかのスペーサーおよびヘテロ原子リンカーの組み合わせの考察例
【0129】
<表2>
リンカー、および、いくつかの放出およびヘテロ原子リンカーの組み合わせの考察例
【0130】
本発明の薬剤送達結合体は、中間体から製造することも可能である。一つの実施態様では、下式:
V−L−Z1
の化合物の製造が可能である。式中、Z1は、薬剤、またはその類縁体または誘導体の付着を促進するのに好適な、求電子要素、求核要素、または前駆物質である。
【0131】
一つの局面では、Z1は、薬剤またはその類縁体または誘導体に存在する求核残基を介して薬剤の付着を可能とする脱離基、例えば、窒素等のヘテロ原子であってもよい。
【0132】
別の局面では、Z1は、薬剤またはその類縁体または誘導体に存在する脱離基、例えば、酸塩化物等のカルボン酸誘導体等、を移動させることが可能な求核要素、例えば、窒素等のヘテロ原子であってもよい。
【0133】
別の局面では、Z1は、前駆物質、例えば、還元反応を通じて求核性窒素に加工することが可能なニトロ基や、加水分解と塩素化の順次反応によって求電子性酸塩化物に加工することが可能なエステル等、であってもよい。Z1は、ヘテロ原子リンカーであってもよいことを理解しなければならない。
【0134】
別の実施態様では、薬剤送達結合体は、下記のような中間体から製造することが可能である。すなわち、
Z2−L−D
式中、Z2は、ビタミン、またはその類縁体または誘導体の付着を促進するのに好適な、求電子要素、求核要素、または前駆物質である。
【0135】
一つの局面では、Z1は、ビタミンまたはその類縁体または誘導体に存在する求核残基を介してビタミンの付着を可能とする脱離基、例えば、窒素等のヘテロ原子であってもよい。
【0136】
別の局面では、Z1は、ビタミンまたはその類縁体または誘導体に存在する脱離基、例えば、酸塩化物等のカルボン酸誘導体、を移動させることが可能な求核要素、例えば、窒素等のヘテロ原子であってもよい。
【0137】
別の局面では、Z1は、前駆物質、例えば、還元反応を通じて求核性窒素に加工することが可能なニトロ基や、加水分解と塩素化の順次反応によって求電子性酸塩化物に加工することが可能なエステル等、であってもよい。Z2は、ヘテロ原子リンカーであってもよいことを理解しなければならない。
【0138】
別の実施態様では、二価リンカー(L)は、別に合成され、次に、例えば、下式のような化合物を中間体として調製することによって、後続工程においてビタミンと薬剤に付着されてもよい。すなわち、
Z1−L−Z2
式中、Z1とZ2はそれぞれ独立に選択され、上に定義した通りである。
【0139】
本発明の薬剤送達結合体は、ヒトの臨床医学と獣医学応用の両方のために使用することが可能である。従って、病原細胞集団を抱え、本ビタミン受容体結合性薬剤送達結合体によって治療される宿主動物は、ヒトであってもよいし、獣医学応用の場合は、実験動物、農業動物、家畜動物、または野生動物であってもよい。本発明は、宿主動物、例えば、と言ってそれらに限定されるわけではないが、ヒト、実験動物、例えば、げっ歯類(例えば、マウス、ラット、ハムスター等)、ウサギ、サル、チンパンジー等、家畜動物、例えば、イヌ、ネコ、およびウサギ等、農業動物、例えば、ウシ、ウマ、ブタ、ヒツジ、ヤギ等、および、拘束された野生動物、例えば、クマ、パンダ、ライオン、トラ、ヒョウ、ゾウ、シマウマ、キリン、ゴリラ、イルカ、およびクジラ等、を含む宿主動物、に施すことが可能である。
【0140】
本発明は、上記宿主動物において各種の病態を招く病原細胞集団に対して適用することが可能である。本発明によれば、「病原細胞」とは、ガン細胞、感染病媒介因子、例えば、細菌およびウィルス、細菌またはウィルス感染細胞、病的状態をもたらすことが可能な活性化マクロファージ、およびその他の、ビタミン受容体、または、ビタミンの類縁体または誘導体に結合する受容体を、特異的に発現する、優先的に発現する、または過剰発現する、任意のタイプの病原細胞を意味する。病原細胞はまた、本発明のビタミン受容体結合性薬剤送達結合体による治療が、その細胞の引き起こす病気の症状の寛解をもたらす、そのような病気の原因細胞全てを含むことが可能である。例えば、病原細胞は、ある状況下では病原性となる宿主細胞、例えば、移植片対宿主疾患の原因とはなるが、別の状況下では病原性ではない免疫系細胞、であることも可能である。
【0141】
従って、病原細胞集団は、腫瘍形成性、例えば、良性腫瘍および悪性腫瘍を含む腫瘍形成性のガン細胞集団であってもよいし、または、非腫瘍形成性であってもよい。ガン細胞集団は自発性に生じてもよいし、あるいは、宿主動物の生殖細胞系に存在する突然変異または体細胞突然変異のような過程によって生じてもよいし、あるいは、化学的、ウィルス的、または放射線誘発的に生じてもよい。本発明は、ガン、例えば、上皮ガン、肉腫、リンパ腫、ホジキン病、メラノーマ、中皮腫、バーキットリンパ腫、鼻咽頭ガン、白血病、および骨髄腫、を治療するのに利用が可能である。ガン細胞集団としては、口腔、甲状腺、内分泌腺、皮膚、胃、食道、喉頭、膵臓、結腸、膀胱、骨、卵巣、子宮頸部、子宮、乳房、睾丸、前立腺、直腸、腎臓、肝臓、および、肺の各ガンが挙げられるが、ただしそれらに限定されない。
【0142】
病原細胞集団がガン細胞集団である場合、結合体投与の効果は、腫瘍塊の減少または除去、あるいは、腫瘍細胞増殖の抑制によって測定される治療反応である。腫瘍の場合、除去とは、原発腫瘍細胞の除去、あるいは、転移した、または、原発腫瘍から解離する過程にある細胞の除去であることが可能である。何かの治療法、例えば、腫瘍の外科切除、放射線治療、化学療法、または生物学療法等、によって腫瘍を除去した後に腫瘍の復帰を防止するための、ビタミン受容体結合性薬剤送達結合体による予防療法も、本発明において考慮されている。予防療法は、薬剤送達結合体による初回治療、例えば、1日当たり複数回投与スケジュールの治療であってもよいし、および/または、初回の治療(単数または複数)後、数日または数ヶ月の間隔を置いて為される1回の追加治療または一連の治療であってもよい。従って、本発明に従って治療される病原細胞集団の除去は、病原細胞数の減少、病原細胞増殖の抑制、病原細胞復帰を防止する予防療法、病気の症状の低下をもたらす病原細胞の処置を含む。
【0143】
ガン細胞が除去されつつある場合、本発明の方法は、腫瘍の外科的切除、放射線療法、化学療法、または生物学的療法、例えば、他の免疫療法、例えば、と言ってそれらに限定されないが、モノクロナール抗体療法、免疫修飾剤による治療、免疫エフェクター細胞の養子移入、造血成長因子、サイトカインによる治療、および、ワクチン療法等を含む免疫療法等、と組み合わせて使用することが可能である。
【0144】
本発明はまた、各種感染症を引き起こす病原細胞集団に適用することも可能である。例えば、本発明は、病原細胞集団、例えば、細菌、酵母を含む真菌、ウィルス、ウィルス感染細胞、マイコプラズマ、および寄生生物に対して適用が可能である。本発明の薬剤送達結合体によって治療が可能な感染性生物は、動物に病原性をもたらすものであれば、従来技術で認知済みの任意の感染性生物、例えば、グラム陰性またはグラム陽性の球菌または桿菌である細菌である。例えば、プロテウス種、クレブシエラ種、プロビデンシア種、イェルシニア種、エルウィニア種、エンテロバクター種、サルモネラ種、セラチア種、アエロバクター種、エシェリキア種、シュードモナス種、シゲラ種、ビブリオ種、アエロモナス種、カンピロバクター種、ストレプトコッカス種、スタフィロコッカス種、ラクトバチルス種、ミクロコッカス種、モラキセラ種、バチルス種、クロストリジウム種、コリネバクテリウム種、エベルセラ種、ミクロコッカス種、マイコバクテリウム種、ナイセリア種、ヘモフィルス種、バクテロイデス種、リステリア種、エリシペロトリックス種、アシネトバクター種、ブルセラ種、パスツレラ種、ビブリオ種、フラボバクテリウム種、フソバクテリウム種、ストレプトバチルス種、カリマトバクテリウム種、レジオネラ種、トレポネーマ種、ボレリア種、レプトスピラ種、アクチノミセス種、ノカルジア種、リケッチア種、および、宿主に病気を起こす、その他の任意の細菌種は、本発明の薬剤送達結合体によって治療することが可能である。
【0145】
特に関心があるのは、抗生物質に対して耐性を持つ細菌、例えば、抗生物質耐性ストレプトコッカス種およびスタフィロコッカス種、あるいは、抗生物質に対して感受性を持つが、抗生物質で治療されると繰り返し感染を招き、最終的には耐性微生物を生ずるに至る細菌である。抗生物質に対して感受性を持つが、抗生物質で治療されると繰り返し感染を招き、最終的には耐性微生物を生ずるに至る細菌は、抗生物質の不在下に、または、抗生物質耐性細菌株の発生を回避するために、患者に通常投与されるものよりも低い用量の抗生物質と組み合わせて、本発明の薬剤送達結合体によって治療することが可能である。
【0146】
ウィルス、例えば、DNAおよびRNAウィルスも、本発明に従って処置することが可能である。そのようなウィルスとしては、例えば、と言ってそれらに限定されないが、DNAウィルス、例えば、パピローマウィルス、パルボウィルス、アデノウィルス、ヘルペスウィルス、および、ワクシニアウィルス等、および、RNAウィルス、例えば、アレナウィルス、コロナウィルス、ライノウィルス、呼吸器系合胞体ウィルス、インフルエンザウィルス、ピコルナウィルス、パラミクソウィルス、レオウィルス、レトロウィルス、レンチウィルス、およびラブドウィルス等、が挙げられる。
【0147】
本発明はまた、酵母を含む任意の真菌、マイコプラズマ種、寄生生物、またはその他の、動物に病気を引き起こす感染性微生物に対して適用が可能である。本発明の方法および組成物によって治療することが可能な真菌の例としては、例えば、カビのように成長する真菌、または酵母様真菌であって、例えば、病気、例えば、白癬、ヒストプラスマ症、ブラストミセス症、アスペルギルス症、クリプトコックス症、スポロトリクス症、コクシジオイディス真菌症、パラコクシジオイディス真菌症、ムコール菌症、クロモブラストミコーシス、皮膚糸状菌症、プロトテコーシス、フザリオーシス、ひこう疹、菌腫、パラコクシジオイディス真菌症、フェオフィホ真菌症、シュードアレシェリア症、スポロトリクス症、砂毛症、ニューモシスティス感染、および、カンジダ症を引き起こす真菌である。
【0148】
本発明はまた、寄生生物感染、例えば、と言ってそれらに限定されないが、条虫類、例えば、サナダムシ、膜様条虫、裂頭条虫、およびエキノコッカス種、吸虫類、例えば、肥大吸虫、異形吸虫、メタゴニムス、肝吸虫、ファスキオラ、肺吸虫、および住血吸虫種、回虫類、例えば、蟯虫、鞭虫、回虫、鉤虫、アメリカ鉤虫、円虫、旋毛虫、糸状虫、ブルギア、ロアオンコセルカ、およびドラクンクルス種、アメーバ類、例えば、ネグレリア、およびアカントアメーバ種、および、原虫類、例えば、プラスモディウム、トリパノソーマ、リーシュマニア、トキソプラスマ、エントアメーバ、ジアルジア、イソスポラ、クリプトスポリジウム、およびエンテルシトゾーン種、によって引き起こされる感染を治療するのに利用することが可能である。
【0149】
本発明の薬剤送達結合体が向けられる病原細胞は、それらの細胞が優先的にビタミン受容体を発現するのであれば、内在性病原体を抱える細胞、例えば、ウィルス、マイコプラズマ、寄生生物、または細菌によって感染された細胞であってもよい。
【0150】
一つの実施態様では、ビタミン受容体結合性薬剤送達結合体は、そのビタミン成分が、ビタミンに対して特異的に結合し、病原細胞において優先的に発現されるビタミン受容体、トランスポーター、またはその他の表面発現蛋白質に結合して、標的病原細胞の内部に取り込まれることがあり得る。このような内部への取り込みは、例えば、受容体介在性エンドサイトーシスによって起こることが可能である。薬剤送達結合体が放出リンカーを含む場合、ビタミン成分と薬剤が細胞内で解離し、薬剤が細胞内標的に作用することが可能となる。
【0151】
別の実施態様では、薬剤送達結合体のビタミン成分は、病原細胞に結合し、薬剤を、その病原細胞表面に近接させることが可能である。次に、薬剤は、放出リンカーの分断によって放出させることが可能である。例えば、放出リンカーがジスルフィド基である場合、薬剤は、ジスルフィドイソメラーゼ蛋白質によって放出することが可能である。次に、この薬剤は、ビタミン受容体結合性薬剤送達結合体が結合した病原細胞によって摂取されるか、あるいは、薬剤は、それに近接した別の細胞によって摂取されることが可能である。あるいは別に、放出リンカーがジスルフィド基である場合、薬剤は、細胞内部でジスルフィドイソメラーゼ蛋白質によって放出されることが可能である。薬剤はまた、あるベータ除去機構に関連して上述したように、加水分解機構、例えば、酸触媒加水分解によって放出してもよいし、あるいは、オキソニウムイオンまたはラクトニウムイオン産生機構を介する隣接基支援分断によって放出されてもよい。放出リンカー(単数または複数)の選択に応じて、薬剤が結合体から解離される機構が決められる。このような選択は、薬剤結合体の使用が予想される条件によってあらかじめ定義することが可能であることが理解される。
【0152】
別の実施態様で、リンカーが放出リンカーを含まない場合、薬剤送達結合体のビタミン成分は、病原細胞に結合し、薬剤を病原細胞の表面に設置し、薬剤に結合が可能な他の分子による攻撃に備えて病原細胞を標的化することが可能である。あるいは別に、この実施態様では薬剤送達結合体は、結合後に標的細胞に取り込まれ、ビタミン成分と薬剤は細胞内で会合を持続し、薬剤は、ビタミン成分から解離すること無しにその効果を発揮することも可能である。
【0153】
さらに別の実施態様で、または、上述の実施態様と組み合わせて、ビタミン受容体結合性薬剤送達結合体は、細胞のビタミン受容体とは独立した機構を通じて作用することが可能である。例えば、薬剤送達結合体は、血清中に存在する可溶性ビタミン受容体、または、血清タンパク、例えば、アルブミンに結合し、結果として、未結合の薬剤よりも長期の結合体の循環を実現し、病原細胞集団に対して未結合の薬剤よりも高い結合体の活性をもたらすことができる。
【0154】
本発明の別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が提供される。Lは、(ls)aおよび(lH)b、およびそれらの組み合わせから選ばれ、(ls)a、(lH)bおよびVは本明細書に定義する通りであり、Dは、免疫原物質のような薬剤である。免疫原物質は、ハプテン、例えば、フルオレセイン、ジニトロフェニル等であってもよい。本実施態様では、ビタミン受容体結合性薬剤送達結合体は、病原細胞の表面に結合し、その細胞を免疫原物質によって「標識し」、その標識された病原細胞集団に向けられた免疫反応を起動する。受動免疫化において宿主に投与された抗体、または、あらかじめ存在する先天的または後天的免疫によって宿主の中に存在する抗体が、免疫原物質に結合して、内在的免疫反応を起動する。細胞に結合したビタミン−免疫原物質複合体への抗体の結合は、結果として、補体介在性細胞傷害、抗体依存性細胞介在性細胞傷害、抗体オプソニン化と食作用、細胞死または不活性化信号を伝える抗体誘発性受容体凝集、または、細胞に結合したリガンド免疫原複合体への抗体結合によって刺激される、その他の任意の体液性または細胞性免疫反応をもたらす。免疫原物質が、事前の抗体オプソニン化無しに、免疫細胞によって直接認識されることが可能な場合、病原細胞の直接殺戮が起こる可能性がある。この実施態様は、米国特許出願第09/822,379号にさらに精しく記載されており、この出願を引用することにより本明細書に含める。薬剤が免疫原物質である本実施態様のある変種では、二価リンカーは、上述のように放出リンカー、例えば、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体を含んでもよいことが理解される。式中、Lは、(ls)a、(lH)b、(lr)c、およびそれらの組み合わせから選ばれ、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカー、(lr)は放出リンカーであり、Vは、ビタミン、またはその類縁体または誘導体であり、a、bおよびcは整数である。
【0155】
本明細書に記載されるビタミン受容体結合性薬剤送達結合体は、ビタミン受容体結合性成分、二価リンカー(L)、薬剤、および、要すれば随意に、ビタミン受容体結合性成分と薬剤とを二価リンカー(L)に連結するヘテロ原子リンカーを含む。二価リンカー(L)は、スペーサーリンカー、放出(すなわち分断可能)リンカー、およびヘテロ原子リンカー、またはそれらの組み合わせを含むことが可能である。
【0156】
本明細書に記載されるビタミン受容体結合性薬剤送達結合体は、多種多様なビタミン、または受容体結合性ビタミン類縁体/誘導体、リンカー、および薬剤から形成することが可能である。本発明の薬剤送達結合体は、病原細胞では、ビタミン結合が可能な、ビタミン受容体が優先的に発現されるために、宿主動物における病原細胞集団を選択的に標的化することが可能である。例示のビタミン成分としては、カルニチン、イノシトール、リポ酸、ピリドキサル、アスコルビン酸、ナイアシン、パントテン酸、葉酸、リボフラビン、チアミン、ビオチン、ビタミンB12、および、脂溶性ビタミンA、D、EおよびKが挙げられる。これらのビタミン、および受容体結合性類縁体および誘導体は、二価リンカー(L)によって薬剤に結合される標的体を構成し、本明細書に記載されるビタミン受容体結合性薬剤送達結合体を形成する。従って、「ビタミン」という用語は、ビタミン類縁体および/または誘導体(例えば、葉酸の誘導体であるプテロイン酸、ビオチン類縁体、例えば、ビオシチン、ビオチンスルフォキシド、オキシビオチン、およびその他のビオチン受容体結合性化合物等)を含む。本発明によれば、ビタミン類縁体または誘導体とは、ビタミン類縁体または誘導体が、それを介して二価リンカー(L)に対して共有結合されるヘテロ原子を組み込んだビタミンを意味することが可能であることが理解される。
【0157】
例示のビタミン成分としては、葉酸、ビオチン、リボフラビン、チアミン、ビタミンB12、および、これらビタミン分子の受容体結合性類縁体および誘導体、および、その他の関連ビタミン受容体結合性分子が挙げられる。ビタミン類縁体および/または誘導体の具体例としては、葉酸塩の類縁体および誘導体、例えば、フォリン酸、プテロポリグルタミン酸、および、葉酸受容体結合性プテリジン、例えば、テトラヒドロプテリン、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、および、それらのデアザおよびジデアザ類縁体が挙げられる。「デアザ」および「ジデアザ」類縁体という用語は、天然の葉酸構造、またはその類縁体または誘導体において、1個または2個の窒素原子を置換する炭素原子を有する、従来技術で認知済みの類縁体を指す。例えば、デアザ類縁体としては、葉酸の、1-デアザ、3-デアザ、5-デアザ、8-デアザ、および、10-デアザ類縁体が挙げられる。ジデアザ類縁体としては、例えば、葉酸の、1,5-ジデアザ、5,10-ジデアザ、8,10-ジデアザ、および、5,8-ジデアザ類縁体が挙げられる。本発明において複合体形成リガンドとして有用なその他の葉酸塩は、葉酸受容体結合性類縁体である、アミノプテリン、アメトプテリン(メトトレキセート)、N10-メチル葉酸塩、2-デアミノヒドロキシ葉酸塩、デアザ類縁体、例えば、1-デアザメトプテリンまたは3-デアザメトプテリン、および、3’,5’-ジクロロ-4-アミノ-4-デオキシ-N10-メチルプテロイルグルタミン酸(ジクロロメトトレキセート)である。前述の葉酸類縁体および/または誘導体は、それらが葉酸受容体に結合する能力を有することを反映して、通常「葉酸塩」と呼ばれるが、このようなリガンドは、外来分子に結合された場合、膜貫通輸送、例えば、本明細書に記載される葉酸仲介性エンドサイトーシス経由輸送を強化するのに効果的である。葉酸受容体に結合して、複合体の受容体介在エンドサイトーシス輸送を起動することが可能な、その他の好適なリガンドとしては、葉酸受容体に対する抗イディオタイプ抗体が挙げられる。葉酸受容体に対する抗イディオタイプ抗体を有する複合体における外来分子は、本発明に従って、その複合体の膜貫通輸送を起動するために使用される。
【0158】
ビタミン類縁体および/または誘導体の具体例としてはさらに、ビオチンの類縁体および誘導体、例えば、ビオシチン、ビオチンスルフォキシド、オキシビオチン、および、その他のビオチン受容体結合性化合物等が挙げられる。本明細書に記載される他のビタミンの類縁体および誘導体も、本明細書では考慮されることが理解される。一つの実施態様では、本明細書で記載される薬剤送達結合体で使用されるビタミンは、活性化マクロファージにおいて特異的に発現されるビタミン受容体、例えば、本明細書に記載されるような葉酸またはその類縁体または誘導体に結合する葉酸受容体、に結合するものが挙げられる。
【0159】
ビタミンに対する結合部位は、受容体であって、病原細胞集団によって特異的に発現される、過剰に発現される、または優先的に発現される受容体に対して特異的に結合することが可能な、任意のビタミン分子、またはその誘導体または類縁体に対する、そのような受容体が挙げられる。病原細胞によって特異的に発現される、過剰に発現される、または優先的に発現される表面発現性蛋白質は、通常、非病原細胞では存在しないか、低濃度で存在する受容体であって、これが、病原細胞の選択除去手段を提供する。ビタミン受容体結合性薬剤送達結合体は、ガン細胞または、その他のタイプの病原細胞上の受容体に対して高い親和度をもって結合することが可能であってよい。高親和度結合は、ビタミン成分には元々備わっているものであることもできるし、あるいは、結合親和度を、化学的に修飾されたビタミン(すなわち、類縁体または誘導体)を用いることによって強化することも可能である。
【0160】
薬剤は、製薬学的活性化合物を含め、細胞機能を変調させる、またはその他のやり方で修飾することが可能ならば、いずれの分子であってもよい。好適な分子としては、例えば、と言ってそれらに限定されるわけではないが、ペプチド、オリゴペプチド、レトロ逆転(retro-inverso)オリゴペプチド、蛋白質、蛋白質類縁体であって、少なくとも一つの非ペプチド結合がペプチド結合を置換する類縁体、アポ蛋白質、糖蛋白質、酵素、補酵素、酵素阻害剤、アミノ酸とその誘導体、受容体、およびその他の膜蛋白質;抗原、およびそれに対する抗体;ハプテン、およびそれに対する抗体;ホルモン、脂質、リン脂質、リポソーム;毒素;抗生物質;鎮痛剤;気管支拡張剤;ベータブロッカー;抗菌剤;抗高血圧剤;循環系薬剤、例えば、抗不整脈剤、グリコシド心臓薬、抗狭心症剤、および、血管拡張剤等;中枢神経系薬剤、例えば、刺激剤、向精神薬、抗躁剤、および、抑制剤等;抗ウィルス剤;抗ヒスタミン剤;ガン治療薬、例えば、化学療法剤等;精神安定剤;抗うつ剤;H-2拮抗剤;抗痙攣剤;抗嘔吐薬;プロスタグランジンおよびプロスタグランジン類縁体;筋弛緩剤;抗炎症物質;刺激剤;うっ血除去剤;鎮吐剤;利尿剤;鎮痙剤;抗喘息薬;抗パーキンソン薬;去痰剤;鎮咳剤;粘液溶解剤;および、ミネラルおよび栄養添加物が挙げられる。
【0161】
さらに、薬剤は、細胞傷害性を持つ、腫瘍透過性を強化する、腫瘍細胞増殖を抑制する、アポトーシスを増進する、標的細胞の抗アポトーシス活性を下げる、感染媒介物による病気を治療するのに使用される、病原細胞に向けた内在的免疫反応を強化する、または、任意のタイプの病原細胞による病的状態の治療に使用される、従来技術で既知の、任意の薬剤であることが可能である。本発明に従って使用するのに好適な薬剤としては、アドレノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝剤、例えば、シトシンアラビノシド、プリン類縁体、ピリミジン類縁体、および、メトトレキセート、ブスルファン、カルボプラチン、クロラムブシル、シスプラチンおよび他の白金化合物、タモキシフェン、タキソール、パクリタクセル、パクリタクセル誘導体、タキソテール(登録商標)、シクロフォスファミド、ダウノマイシン、リゾキシン、T2トキシン、植物アルカロイド、プレドニソン、ヒドロキシ尿素、テニポシド、マイトマイシン、ディスコデルモリド、微小管阻害剤、エポチロン、チューブリシン、シクロプロピルベンゾ[e]インドロン、seco-シクロプロピルベンゾ[e]インドロン、O-Ac-seco-シクロプロピルベンゾ[e]インドロン、ブレオマイシンおよび他の任意の抗生物質、窒素マスタード、ニトロスウレア、ビンクリスチン、ビンブラスチン、およびその類縁体と誘導体、例えば、デアセチルビンブラスチンモノヒドラジド等、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン類、例えば、ドラスタチン10、アマニチン類、例えば、α-アマニチン等、カンプトテシン、イリノテカン、および他のカンプトテシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エストラムスチン、ノコダゾール、MAP4、コルセミド、炎症および炎症誘発剤、ペプチドおよびペプチド様シグナル伝達阻害剤、および、その他、従来技術で認知済みの任意の薬剤または毒素が挙げられる。本発明に従って使用が可能なその他の薬剤としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンフォテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、および、その他、従来技術で認知済みの任意の抗菌化合物が挙げられる。
【0162】
一つの実施態様では、本発明に従って用いられる薬剤は、血清中で少なくとも4時間安定であり続ける。別の実施態様では、薬剤は、ナノモル範囲のIC50を持ち、また別の実施態様では、薬剤は水溶性である。薬剤が水溶性でない場合、二価リンカー(L)を、水溶性を強化するように誘導体形成することも可能である。「薬剤」という用語はまた、上述の薬剤類縁体または誘導体、例えば、と言ってそれらに限定されないが、ドラスタチン類例えばドラスタチン10、アマニチン類例えばα-アマニチン、カムプトテシンおよびイリノテカン、および、他のカムプトテシンおよびイリノテカン誘導体を含む類縁体または誘導体等、を意味する。本発明によれば、薬剤類縁体または誘導体とは、それを介して薬剤類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだ薬剤を意味することも可能であることを理解しなければならない。
【0163】
本発明のビタミン受容体結合性薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、薬剤、および、要すれば随意に、ビタミン受容体結合成分と薬剤とを二価リンカー(L)に連結するヘテロ原子リンカーを含むことができる。本発明によれば、ビタミン類縁体または誘導体とは、それを介してビタミン類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだビタミンを意味することも可能であることを理解しなければならない。従って、ビタミンは、ヘテロ原子リンカーを介して二価リンカー(L)に共有結合することも可能であるし、あるいは、ビタミン類縁体または誘導体(すなわち、ヘテロ原子を組み込んだもの)が直接二価リンカー(L)に結合することも可能である。同様に、薬剤類縁体または誘導体は、本発明による薬剤であり、薬剤類縁体または誘導体とは、それを介して薬剤類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだ薬剤を意味してもよい。従って、薬剤は、ヘテロ原子リンカーを介して二価リンカー(L)に共有結合することが可能であるし、あるいは、薬剤類縁体または誘導体(すなわち、ヘテロ原子を組み込んだもの)は、二価リンカー(L)に直接結合することも可能である。二価リンカー(L)は、スペーサーリンカー、放出(すなわち、分断可能)リンカー、および、上記二つのタイプのリンカーを含む結合体においてスペーサーリンカーを放出リンカーに連結するヘテロ原子リンカーを含むことが可能である。
【0164】
従って、本発明によれば、二価リンカー(L)は、ビタミンを薬剤に会合する手段、例えば、ヘテロ原子リンカー(すなわち、スペーサーアームまたは架橋分子)による接続、または、二価リンカー(L)の、ビタミンまたは薬剤類縁体または誘導体に対する直接共有結合による接続、を含むことが可能である。いずれの会合手段も、本発明の方法の動作のためには、ビタミン、またはビタミン受容体結合性誘導体または類縁体の、細胞膜上のビタミン受容体に対する結合を阻止するものであってはならない。
【0165】
一般に、二価リンカー(L)と、ビタミンまたはその類縁体または誘導体の間の複合体や、二価リンカー(L)と、薬剤またはその類縁体または誘導体の間の複合体を形成するためには、たとえば、介在ヘテロ原子リンカーなど、任意の手段が本発明に従って利用が可能である。さらに、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカーの間に複合体を形成し、二価リンカー(L)を形成するために、従来技術で認知済みの任意の手段を用いることが可能である。複合体は、上記の分子の中から任意に選ばれたものを、例えば、水素、イオン、または、共有結合等の結合を介して直接結合することによって形成することが可能である。共有結合は、例えば、酸、アルデヒド、ヒドロキシ、アミノ、スルフヒドリル、またはヒドラゾ基の間に、アミド、エステル、ジスルフィド、またはイミノ結合を形成することによって実現することが可能である。
【0166】
スペーサーおよび/または放出リンカー(すなわち分断可能リンカー)は、生物適合性であるいずれのリンカーであってもよい。分断可能リンカーは、例えば、細胞内の、または、細胞表面の還元または酸化条件下における分断に対して感受性を持つリンカー、酸易変性または塩基易変性を持つリンカーであるpH感受性リンカー、または、生化学的または代謝的過程によって分断されるリンカー、例えば、酵素易変性リンカーであってもよい。典型的には、スペーサーおよび/または放出リンカーは、約1から約30個の炭素原子を含むが、より典型的には、約2から約20個の炭素原子を含む。典型的には、より低分子量のリンカー(すなわち、約30から約300の分子量を持つもの)が用いられる。このようなリンカーの前駆物質は、通常は、求核性、または求電子性官能基のいずれかまたはその両方を持つように選ばれ、要すれば随意に、中間体合成における利便性を増すために、簡単に分断可能な保護基を持つ保護形として選ばれる。
【0167】
本発明はまた、1回以上の用量で投与された場合、宿主動物の病原細胞集団を除去するのに十分に有効な量のビタミン受容体結合性薬剤送達結合体を含む、製薬組成物に向けられる。この薬剤送達結合体は、宿主動物に対して、好ましくは非経口的に、例えば、皮内、皮下、筋肉内、腹腔内、静脈内、または、硬膜下腔内に、投与される。あるいは別に、この薬剤送達結合体は、他の医学的に有用な過程を通じて、例えば、経口的に投与することが可能であり、任意の効果的な用量、および、持続放出剤形を含めた、任意の好適な治療剤形の使用が可能である。
【0168】
非経口剤形の例としては、活性剤を、等張生食液、5%ブドウ糖、または、その他の既知の製薬学的に受容可能な液性担体、例えば、液性アルコール、グリコール、エステル、および、アミドに溶解させた溶液が挙げられる。本発明による非経口剤形は、薬剤送達結合体用量を含む、再構成可能な凍結乾燥体の形を取ることも可能である。本実施態様の一つの局面では、従来技術で既知のいくつかの持続放出剤形の内から任意に選ばれるもの、例えば、米国特許第4,713,249、5,266,333および5,417,982号に記載される、生体分解性炭水化物基質、を投与することが可能である。なお、この特許の開示を引用することにより本明細書に含める。あるいは別に、徐放ポンプ(例えば、浸透圧ポンプ)を使用することも可能である。
【0169】
治療因子を含む少なくとも一つの添加組成物を、、薬剤送達結合体仲介による病原細胞集団の除去を強化するために、前述の方法と組み合わせて、または、補助剤として投与することが可能であるし、あるいは、1種を越える追加治療因子を投与することが可能である。この治療因子は、内在的免疫反応を刺激することが可能な化合物、化学療法剤、または投与薬剤送達結合体の効力を補佐することが可能な別の治療因子から選んでもよい。本発明の方法は、前述の結合体の他に、内在的免疫反応を刺激することが可能な化合物または組成物(例えば、サイトカイン)、例えば、と言ってそれらに限定されないが、サイトカインまたは免疫細胞成長因子、例えば、インターロイキン1-18、幹細胞因子、塩基性FGF、EGF、G-CSF、GM-CSF、FLK-2リガンド、HILDA、MIP-1α、TGF-α、TGF-β、M-CSF、IFN-α、IFN-β、IFN-γ、可溶性CD23、LIF、および、それらの組み合わせを含む化合物または組成物、を宿主に対し投与することによって実行が可能である。
【0170】
これらの因子の治療的に有効な組み合わせも使用が可能である。一つの実施態様では、例えば、IL-2の治療的有効量、例えば、約0.1 MIU/m2/用量/日から約15 MIU/m2/用量/日の範囲の量を、1日複数回投与のスケジュールで、かつ、IFN-αを、例えば、0.1 MIU/m2/用量/日から約7.5 MIU/m2/用量/日の範囲の量を、1日複数回投与のスケジュールで、病原細胞を抱える宿主動物の病原細胞を除去する、低減させる、または中和させるために、薬剤送達結合体と同時に使用することが可能である(MIU=百万国際単位、m2=平均人のおよその体表面積)。別の実施態様では、IL-12およびIFN-αが、インターロイキンおよびインターフェロンの前述の治療有効量として使用される。さらに別の実施態様では、IL-15およびIFN-αが、インターロイキンおよびインターフェロンの前述の治療有効量として使用される。別の実施態様では、IL-2およびIFN-αまたはIFN-γ、およびGM-CSFが、前述の治療有効量において併用される。本発明はまた、サイトカイン類の、他の任意の有効な組み合わせ、例えば、他のインターロイキンおよびインターフェロン、およびコロニー刺激因子の組み合わせを含めた併用を考慮する。
【0171】
例えば、それ自体が細胞傷害性である、または、腫瘍透過性を強化するように作用することが可能な化学療法剤も、薬剤送達結合体と組み合わせて本発明の方法に用いるのに好適である。このような化学療法剤としては、アドレノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝剤類、例えば、シトシンアラビノシド、プリン類縁体、ピリミジン類縁体、および、メトトレキセート等、ブスルファン、カルボプラチン、クロラムブシル、シスプラチンおよび他の白金化合物、タモキシフェン、タキソール、パクリタクセル、パクリタクセル誘導体、タキソテール(登録商標)、シクロフォスファミド、ダウノマイシン、リゾキシン、T2トキシン、植物アルカロイド、プレドニソン、ヒドロキシウレア、テニポシド、マイトマイシン、ディスコデルモリド、微小管阻害剤、エポチロン、チューブリシン、シクロプロピルべンジンドロン、seco-シクロプロピルベンジンドロン、O-Ac-seco-シクロプロピルベンジンドロン、ブレオマイシンおよび他の任意の抗生物質、ナイトロジェンマスタード、ニトロス尿素、ビンクリスチン、ビンブラスチン、およびその類縁体と誘導体、例えば、デアセチルビンブラスチンモノヒドラジド等、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン類、例えば、ドラスタチン10等、アマニチン類、例えば、α-アマニチン等、カンプトテシン、イリノテカン、および他のカンプトテシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エストラムスチン、ノコダゾール、MAP4、コルセミド、炎症および炎症誘発剤、ペプチドおよびペプチド様シグナル伝達阻害剤、および、その他、従来技術で認知済みの任意の薬剤または毒素が挙げられる。本発明に従って使用が可能なその他の薬剤としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンフォテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、マイタンシン類およびその類縁体および誘導体、ゲムシタビン、および、その他、従来技術で認知済みの任意の抗菌化合物が挙げられる。
【0172】
治療因子は、ビタミン受容体結合性薬剤送達結合体の前にか、後にか、または同時に宿主動物に投与することが可能であり、また、治療因子は、薬剤送達結合体を含む同じ組成物の一部として、または、薬剤送達結合体とは別の組成物の一部として投与することが可能である。治療因子を治療有効量として含む治療組成物は、いずれのものでも本発明において使用が可能である。
【0173】
さらに、1種を越える薬剤送達結合体を使用することが可能である。例えば、宿主動物を、ビタミンでは異なり、薬剤は同じ複数の結合体(例えば、葉酸-マイトマイシン結合体とビタミンB12-マイトマイシン結合体)の同時投与プロトコールで治療することも可能である。別の実施態様では、宿主動物を、異なる薬剤に連結した同じビタミンを含む、複数の結合体、または、各種薬剤に連結した各種ビタミンを含む、複数の結合体で治療することも可能である。例えば、宿主動物は、葉酸-マイトマイシン結合体と葉酸-シスプラチン結合体によって治療することも可能であるし、あるいは、葉酸-マイトマイシン結合体とビタミンB12-シスプラチン結合体によって治療することも可能である。さらに、同じ薬剤送達結合体の一部として、同じか、異なるビタミンおよび、同じか、異なる薬剤で、複数のビタミンと複数の薬剤を含む薬剤送達結合体も使用が可能である。
【0174】
薬剤送達結合体の、1日当たりの単位用量は、宿主の状態、治療される病状、結合体の分子量、投与ルートと組織分布、および、他の治療処置、例えば、放射線治療の併用の可能性に応じて相当に変動することがあり得る。患者に投与される有効量は、体表面積、患者の体重、患者の状態に関する医師の評価に基づいて決められる。有効用量は、例えば、約1 ng/kgから1mg/kg、約1 μg/kgから約500 μg/kg、約1 μg/kgから100 μg/kgの範囲で変動することが可能である。
【0175】
薬剤送達結合体を投与するために効果的なスケジュールであればどのようなスケジュールを用いてもよい。例えば、薬剤送達結合体は、1日当たり、1回用量として投与してもよく、分割して、複数回用量として投与してもよい。さらに、交互スケジュール、例えば、連日治療に代わるものとして、週当たり1から3日を用いてもよく、本発明を定義するために、このような断続的または交互的連日スケジュールは、毎日治療と等価と考えられ、本発明の範囲内とされる。本発明の一つの実施態様では、宿主は、病原細胞集団を除去するために、薬剤送達結合体の複数注入によって治療される。一つの実施態様では、宿主は、薬剤送達結合体を複数回(好ましくは、約2回から約50回まで)、例えば、12-72時間間隔で、または、48-72時間間隔で注入される。患者に対し、薬剤送達結合体の追加注入を、最初の注入(単複)後、数日または数ヶ月の間隔を置いて投与することが可能である。追加注入は、病原細胞によって引き起こされる病状の復帰を防止する。
【0176】
一つの実施態様では、本発明の薬剤送達結合体で用いることのできるビタミン、またはその類縁体または誘導体は、活性化マクロファージ上に特異的に発現される受容体、例えば、葉酸、またはその類縁体または誘導体に結合する葉酸受容体、に結合するものが挙げられる。例えば、葉酸連結結合体は、宿主に病態を引き起こす活性化マクロファージを殺す、または、その活性を抑えるのに使用される。このようなマクロファージ標的化結合体は、活性化マクロファージ仲介性病態に苦しむ患者に投与されると、結合体化薬剤を、活性化マクロファージ集団に集中させ、会合させるように働き、活性化マクロファージを殺すか、マクロファージ機能を抑圧する。活性化マクロファージ集団の除去、低減、または不活性化は、治療される病状に特徴的な活性化マクロファージ仲介性の病原性を停止または低減させるように働く。活性化マクロファージによって仲介されることが知られる病気の例としては、関節リューマチ、潰瘍性大腸炎、クローン病、乾癬、骨髄炎、多発性硬化症、アテローム硬化症、肺線維症、サルコイドーシス、全身硬化症、臓器移植拒絶反応(GVHD)、および慢性炎症が挙げられる。薬剤送達結合体の投与は、典型的には、病態の症状が低減または除去されるまで続けられる。
【0177】
活性化マクロファージを殺すために、または、活性化マクロファージの機能を抑えるために投与される薬剤送達結合体は、その病態に苦しむ動物または患者に対して非経口的に、例えば、皮内、皮下、筋肉内、腹腔内、または、静脈内に、製薬学的に受容可能な担体と組み合わせて投与することが可能である。あるいは別に、薬剤送達結合体は、他の、医学的に有用な過程を通じて動物または患者に投与することが可能であり、有効用量を、標準的または持続放出剤形として投与することが可能である。この治療法は、単独で用いてもよいし、活性化マクロファージによって仲介される病態治療用として認められている他の治療法と組み合わせて用いてもよい。
【0178】
下記の薬剤送達結合体は、本明細書に記載される本発明の範囲内に収まると考えられる薬剤送達結合体の例である。これらの薬剤送達結合体は、従来技術で認知済みのプロトコールに加えて本明細書に記載される過程を用いることによって本発明に従って調製することが可能である。
【0179】
さらに、下記の薬剤送達結合体も、本明細書に記載される本発明の範囲内に収まると考えられる薬剤送達結合体の例である。付属の合成手順は、本明細書に記載される薬剤送達結合体を調製するのに使用が可能なものを例示する。
【0180】
ジアセトキシシルペノール(DAS)のアセトニトリル溶液に、1.0 eg.の1,2,4,5-ベンゼンテトラカルボン酸二無水物を加え、その後、1 eg.のヒューニッヒ塩基を加える。この反応混合物を、アルゴンの下で室温で1.5時間攪拌する。いくらかのDASが未反応のまま残っている場合は、さらに0.2 eg.の二無水物を加え、攪拌を1時間続ける。1.2 eg.のプテロイルヒドラジドの無水DMSO溶液(J. Am. Chem. Soc., 1997, 119, 10004に従って調製、なおこの開示を引用することによって本明細書に含める)を加え、その後、1.0 eg.のヒューニッヒ塩基を加える。反応混合物を1時間攪拌し、ジエチルエーテルで沈殿させる。得られた沈殿を分取HPLCによってさらに精製する。
【0181】
実施例10aに一般的に記述されているように、Boc-ヒドラジドは、コハク酸無水物と反応させられ、得られた産物は、縮合剤としてのEDCの存在下に5’’-アミノ-ビス-インドリル-seco-CBIと反応させられる。Boc除去および、遊離レブリン酸とのアシルヒドラゾン形成により、NHS-エステル活性化後、ペプチド断片Pte-γ-Glu-Asp-Arg-Asp-Dap-OHに対する反応相手が得られる。このペプチド断片は、スキーム12に一般的に記述されるように、Fmoc-Dap(Boc)-Wang樹脂でスタートするFmoc策を用いるポリマー支持継続法によって調製される。
【0182】
Fmoc-ヒドラジドは、3-(2-ピリジルジチオ)プロピオン酸と反応させられて、Fmoc-ヒドラジド-[3-(2-ピリジルジチオ)プロピオネート]を生ずる。マイトマイシンC誘導体(マイトマイシンC、N-(CH2)2SH)との反応により、マイトマイシンCのジスルフィド含有誘導体が得られる。標準プロトコールによるFmoc-除去、および、レブリン酸によるアシルヒドラゾン形成により、連続的NHS-エステル活性化後、ペプチド断片Pte-γ-Glu-Dap-OHに対する反応相手が得られる。このペプチド断片は、一般にスキーム12に記載する通りに調製される。
【実施例】
【0183】
下記の例示の実施例は、制限的であることを意図するものではなく、また制限的なものと考えてはならない。例えば、本明細書に示される各化合物において、リンカーを形成する際に使用されるアミノ酸の立体化学は、随意に、天然のL型から、または、非天然のD型から選ばれてよい。各実施例は、表示されるようにNMR、MS、および/またはUV分光光度計測、および/またはHPLCによってその特性が解明され、選択された特性信号は適宜注記される。
【0184】
[実施例1]
ジエチレントリアミン葉酸、γ-アミド(DETA-葉酸塩)は、P. Fuchs et al., J. Am. Chem. Soc., 1997, 119, 10004によって記載される工程に従って合成された。なお、この開示を引用することにより本明細書に含める。この化合物(100 mg)を、2 mLの0.1N HClに溶解した。得られた溶液を、1 mLの0.1N HClに溶解したK2PtCl4(158 mg)溶液に攪拌しながら加えた。3 mLのDMSOを加え、攪拌を3日間続け、溶液をろ過し、ろ液をアセトニトリルで沈殿させたところ、170 mgの黄色粉末が得られた。MS(MALDI) 1249.92, 1286.27; 1H NMR(D2O)δ1.05(t, 1H), 2.3(t, 2H), 3.1(t, 2H), 4.45(m, 1H), 6.65(d, 2H), 7.5(d, 2H), 8.65(s, 1H)。
【0185】
[実施例2a]
N10-トリフルオロアセチル保護葉酸含有ペプチジル断片N10-TFA-Pte-Glu-Glu-Lys-OHを、スキーム12に一般的に示すように、Fmoc-策を用いたポリマー支持継続法によって調製した。これは、酸感受性のFmoc-Lys(Boc)-Wang樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。保護されたアミノ酸建設ブロックとしてFmoc-Glu-OtBuおよびN10-TFA-Pte-OHを用いた。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Buとt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCによって精製したところ、N10-TFA-Pte-γGlu-γGlu-Lys-OHがTFA塩として得られた。このペプチド81 mg(0.1 mmol)を2 mL DMSOに溶解した溶液を、15 μL(0.11 mmol)のEt3Nおよび35 mg(0.1 mmol)のマイトマイシンAで処理した。マイトマイシンAは、M. Matsui, Y. Yamada, K. Uzu, and T. Hirata, J. Antibiot. 21, 189-198(1968)およびD. Vias, D. Benigni, R. Partyka, and T. Doyle, J. Org. Chem. 51, 4307-4309(1986)の処理工程に従ってマイトマイシンCから調製してもよい。なお、これらの開示を引用することによって本明細書に含める。反応混合物は、室温で48時間攪拌し、溶媒を凍結乾燥によって除去した。別様に注記しない限り、溶媒の蒸発は全て減圧下で行った。最後に、トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)で分離し、産物をアセトニトリル中で沈殿させたところ102 mgの結合体が黄色固体として得られた。1H NMR(D2O)δ2.45(q, 1H), 2.95(m, 2H), 3.35(dd, 1H), 3.5(d, 1H), 6.5(d, 2H), 7.55(d, 2H), 8.55(s, 1H)。
【0186】
[実施例2b]
N10-トリフルオロアセチル保護葉酸含有ペプチジル断片N10-TFA-Pte-Glu-Glu-Cys-OHを、スキーム12に一般的に示すように、また、実施例2aに記載されるようにFmoc-策を用いたポリマー支持継続法によって調製した。シスタミンをマイトマイシンAと反応させたところ(Matsui, et al., J. Antibiot. 21, 189-198(1968); Vias, et al., J. Org. Chem. 51, 4307-4309(1986)参照)、末端に遊離アミノ酸を有するジスルフィド含有マイトマイシンC誘導体が得られた。これをレブリン酸と結合させ、その後、カルボニル基をマレイニド誘導アシルヒドラジドと反応させた。得られたマイケルアクセプターとN10-TFA-Pte-Glu-Cys-OHを反応させ、水酸化アンモニウム水溶液(pH=10.0)でトリフルオロアセチル保護基を除去し、アセトニトロル中で沈殿させたところ、最終的な結合体が得られた。MS(MALDI) 1059.04, 1148.44, 1225.32, 1300.8; 1H NMR(D2O)δ1.8(d, 2H), 1.9(s, 1H), 2.3(q, 1H), 2.45(q, 1H), 2.9(t, 1H), 3.35(dd, 1H), 4.45(s, 1H), 4.5(dd, 1H), 6.65(d, 2H), 7.55(d, 2H), 8.6(s, 1H)。
【0187】
[実施例3]
T-2トキシンの中間体マレイミドp-メトキシベンジリデンアセタールを、市販のN-(2-ヒドロキシエチル)マレイミドから出発して合成した。そのヒドロキシル基を、穏やかな塩基として塩化メチレン中で酸化銀(I)の存在下にp-メトキシベンジルクロリド(1.2 eg.)と反応させた。未精製産物をシリカカラムで精製した。得られたp-メトキシベンジルエーテルを、OH含有T-2トキシン(1 eg.)の存在下に1.5 eg.の2,3-ジクロロ-5,6-ジシアノ-ベンゾキノン(DDQ)によって酸化処理したところ、所望のp-メトキシベンジリデンアセタールが、安定化p-メトキシベンジルカルベニウム中間体を介して得られた。
【0188】
もう一方の反応相手、Pte-γ-Glu-Arg-Asp-Cys-OHを、Fmoc策を用いたポリマー支持継続法によって調製した。これを、酸感受性H-Cys(4-メトキシトリチル)-2-クロロトリチル樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Fmoc-Arg(Pbf)-OH、Fmoc-Glu-OtBu)およびN10-TFA-Pte-OHを用いた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Buとt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCによって精製したところ、N10-TFA-Pte-γ-Glu-Arg-Asp-Cys-OHが得られた。トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)中で分離した。
【0189】
最後に、標的とするp-メトキシベンジリデンアセタール連結葉酸-薬剤結合体を、アルゴン下に、ペプチドのバッファー水溶液(pH=7.0)を、T2トキシンのマレイミド含有アセタールの等モルアセトニトリル溶液と混合することによって調製した。室温で1時間攪拌後、最終結合体を分取HPLCで精製したところ、収集分画の凍結乾燥後に黄色粉末が得られた。MS(m+H)+ 1541.3; 1H NMR(DMSO-d6)δ0.1(s, 1H), 0.55(d, 2H), 0.9(dd, 3H), 1.65(s, 1H), 2.0(d, 1H), 3.75(d, 2H), 5.25(d, 1H), 6.65(d, 2H), 6.9(d, 2H), 7.3(t, 2H), 7.65(d, 2H), 8.65(s, 1H)。
【0190】
[実施例4a]
[実施例4b]
[実施例4c]
実施例4a、4bおよび4cの化合物は、アシルアジリジンは、市販の適当なN-(アルカン酸)マレイミドによってマイトマイシンAをアシル化すること(スキーム1参照)によって調製されたことを除いては、一般に実施例3で記載した工程に従って調製した。
【0191】
[実施例5]
塩基として2.2 eg. NaHCO3、および、溶媒としてアセトニトリル/水(l/l)の存在下に、trans-4-アミノシクロヘキサノール塩酸を、等モル量のFmoc-OSuと反応させると、N-Fmoc保護アミノアルコールが得られた。これをSwernの条件(Synthesis, 1981, 165)を用いて酸化して対応するN-Fmoc-保護アミノケトンを得た。4 eq.のメチルオルトギ酸および触媒量のトリフルオロ酢酸によってケタール化したところ、N-Fmoc-保護アミノケタールが定量的収率で得られた。このケタールを、等モル量のトリメチルシリルトリフルオロメタンスルフォネートおよび2,4,6-トリ-t-ブチル-ピリジンで処理したところ、産物として、4-Fmoc-アミノシクロヘキシルエノールエーテルが得られた。次の工程で、薬剤T-2トキシンを、分子ふるい(3オングストローム)および触媒量のトリフルオロ酢酸の存在下に、4倍過剰量のエノールエーテルで処理した。得られた非対称混合ケタールをシリカで精製した。Fmoc保護基は、DMF中で樹脂結合ピペリジンによる処理で除去した。解放されたアミノ酸は、1.1 eg.のヒューニッヒ塩基の存在下に1.1 eg.のマレイミド酢酸-NHS-エステルと反応させた。T-2トキシンのマレイミド含有ケタールをシリカの上で精製した。
【0192】
葉酸含有ペプチド断片、Pte-γ-Glu-β-Dap-Asp-Cys-OHは、スキーム12に一般的に記載されるように、Fmoc策を用いたポリマー支持継続法によって調製した。Fmoc-L-Cys(Trt)-OHを負荷した、酸感受性Wang樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Boc-Dap(Fmoc)-OH、Fmoc-Glu-OtBu、およびN10-TFA-Pte-OHを用いた。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Bu、t-Boc、およびトリチル保護基の同時除去も実現された。最後に、トリフルオロアセチル基を水酸化アンモニウム水溶液中で分離したところ、所望のチオール含有ペプチドが得られた。この未精製ペプチドを、分取HPLCにて精製した。
【0193】
最後に、標的とするケタール連結葉酸-薬剤結合体を、アルゴン下に、ペプチドのバッファー水溶液(pH=7.0)と、T-2トキシンのマレイミド含有ケタールの等モル量アセトニトリル溶液とを混合することによって調製した。室温で1時間攪拌した後、最終結合体を分取HPLCで精製したところ、収集画分の凍結乾燥後に黄色粉末が得られた。ES MS(m-H)- 1474.5, (m+H)+ 1476.2, (m+Na)+ 1498.3。
【0194】
[実施例6]
エチレンジアミン葉酸、γ-アミド(EDA-葉酸塩)は、P. Fuchs et al.(J. Am. Chem. Soc., 1997, 119, 10004、例えば、本論文に記載される化合物52の合成を参照)によって記載される工程に従って合成された。なお、この開示を引用することにより本明細書に含める。EDA葉酸塩(600 mg)を、5 mLの無水DMSOに懸濁させた。60℃で4時間攪拌後、得られた溶液を20℃に冷却し、4 eg.の1,2,4,5-ベンゼンテトラカルボン酸無水物(無水BTCA)を加えた。5分後、反応混合液を、よく攪拌した無水アセトニトリルに注入した。得られた沈殿を遠心によって単離したところ、657mgのBTCA(一無水物)-EDA-葉酸塩が得られた。
【0195】
乾燥DMSOに溶解したダウノマイシンのよく攪拌された溶液に、固体のBTCA(一無水物)-EDA-葉酸塩(1.5 eg.)を添加した。さらに14時間攪拌後、50%のダウノマイシンが未反応のまま残り(HPLC)、1.5 eg. BTCA(一無水物)-EDA-葉酸を加えた。さらに4時間攪拌した後、ダウノマイシンは全て消費された。HPLCプロフィールにおいて、二つの新しいピークが観察された。これらは、保持時間が近接しており、最終結合体の二つの位置異性体を表していた。未精製産物をアセトニトリル中の沈殿後に単離し、逆相HPLCによってさらに精製した。産物の構造は、ES MS(m-H)- 1227.1に一致した。
【0196】
[実施例7]
アルゴン下0℃にて、4 mLの無水ジクロロメタンに溶解した250 mg(0.25 mmol)のパクリタクセルおよび130 μL(0.73 mmol)のヒューニッヒ塩基のよく攪拌された溶液に、85 μL(0.8 mmol)のAlloc-Clをゆっくりと加えた。さらに12時間攪拌を続け、産物を、標準的抽出法によって単離した。この白色粉末、2’-alloc-パクリタクセルを、これ以上精製することなく次の工程に用いた。
【0197】
この工程では、108 mg(0.117 mmol)の2’-alloc-パクリタクセルを、1.0 mLの無水アセトニトリルに溶解した。アルゴン下攪拌しながら、25 mg(0.117 mmol)の1,2,4,5-ベンゼンテトラカルボン酸無水物(無水BTCA)および21 μL(0.120 mmol)のヒューニッヒ塩基をこの溶液に加えた。攪拌をさらに2.5時間続けた。別の反応フラスコで、52 mgのEDA-葉酸を60℃で、全物質が溶解するまで攪拌した(約60分)。室温に冷却後、前の反応混合液をこの溶液に添加し、攪拌をさらに2時間続けた。反応混合液を、アセトニトリル/ジエチルエーテル(20:80)のよく攪拌された混合液に滴下により加えた。黄色沈殿を遠心によって分離し、さらに分取HPLCによって精製した。この産物の構造は、1Dおよび2D(COSY)1H-NMRスペクトル;ES MS(m+H)+ 1555.5と一致した。
【0198】
[実施例8]
1.0 eg.のアクラマイシン、2.0 eg.のヒドラジド-[3-(2-ピリジルジチオ)プロピオネート](SPDP-ヒドラゾン)、および、数結晶のピリジニウムp-トルエンスルフォネートから成る混合物を、アルゴン下、無水メタノールに攪拌しながら溶解した。この反応混合液を室温で8時間攪拌した。溶媒を蒸留乾固した。残渣を、あらかじめクロロフォルム/メタノール(90:10)に溶解させた1.5%トリエチルアミンで前処理したシリカカラムにて精製した。得られたアクラマイシンアシルヒドラゾンを、最少量のアセトニトリルに溶解した。得られた溶液に、等モル量のPte-γ-Glu-Cys-OH(水に溶解させ、pH=6.8に調整)をアルゴン下ゆっくりと加えた。このPte-γ-Glu-Cys-OHの調製は、実施例2aに記載する工程と似ており、一般にスキーム12に記載される。ジスルフィド交換反応は10分を要した。この反応溶液を、過剰量のアセトニトリルにゆっくりと加え、得られた沈殿を遠心にて単離した。沈殿を、もう一度アセトニトリルに再懸濁し、15分攪拌後遠心にて分離した。高真空にて一晩乾燥した後では、最終結合体は十分に純粋であった(HPLC); ES MS(m+H)+ 1474.1。
【0199】
[実施例9a]
1.0 eg.のアクラマイシンと1.2 eg.のβ-マレイミドプロピオン酸・TFAの混合物を、アルゴン下、無水メタノールに攪拌しながら溶解した。この反応混合液を室温で1時間攪拌した。溶媒を蒸留乾固した。残渣を、あらかじめクロロフォルム/メタノール(90:10)に溶解させた1.5%トリエチルアミンで前処理した短いシリカカラムにて精製した。別のフラスコで、ペプチド断片Pte-γ-Glu-γ-Glu-Cys-OHを、pHを6.8に調節しながら、アルゴン下に水に溶解した。このPte-γ-Glu-γ-Glu-Cys-OHの調製は、実施例2aに記載する工程と似ており、一般にスキーム12に記載される。得られた黄色味を帯びた溶液に、最少量のメタノールに溶解させたアクラマイシンのマレイミドヒドラゾンをゆっくりと加えた。この反応混合液をアルゴン下1時間攪拌した。メタノールを除去し、残渣を、HPLCの分取カラムにて精製し、その後凍結乾燥した; ES MS(m+H)+ 1722.3。
【0200】
[実施例9b]
[実施例9c]
実施例9bと9cの化合物は、実施例9aに一般的に記載される工程に従って、ドキソルビシン(14-ヒドロキシダウノマイシン)誘導体から調製された。
【0201】
[実施例10a]
極めて強力な細胞傷害性薬剤ビス-インドリル-seco-1,2,9,9a-テトラヒドロシクロプロパ[c]ベンズ[e]インドール-4-オン(ビス-インドイル-seco-CBI)の5’’-(N-Boc)アミノ類縁体を、最初に、D. Boger et al., J. Org. Chem., 1992, 57, 2873によって記載されたもののやや修飾された工程に従って調製した。なお、この開示を引用することにより本明細書に含める。
【0202】
ペプチド断片、Pte-γ-Glu-Asp-Arg-Asp-Cys-OHは、スキーム12に一般的に記載されるように、Fmoc策を用いたポリマー支持継続法によって酸感受性H-Cys(4-メトキシトリチル)-2-クロロトリチル樹脂上で調製した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Fmoc-Arg(Pbf)-OH、Fmoc-Glu-OtBu、およびN10-TFA-Pte-OHを用いた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Bu、およびt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCにて精製したところ、N10-Pte-γ-Glu-Asp-Arg-Asp-Cys-OHが得られた。トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)中で分離した。
【0203】
Lev-Val-OHは、バリンメチルエステル塩酸を、EDCとヒューニッヒ塩基の存在下に縮合し、その後メチルエステルを、水酸化リチウムと水で加水分解することを含む、標準的プロトコールを用いて合成した。
【0204】
複合した結合体の最終集合は、5’’-(N-Boc)アミノ-ビス-インドリル-seco-CBIのN-Boc基の除去と、EDCの存在下に、解放されたアミノ基とLev-Val-OHのカルボキシ官能基との結合で始まった。アシルヒドラゾン形成は、テトラヒドロフランにおけるレブリン酸成分のケトン官能基と1.2 eg.のβ-マレイミドプロピオン酸・TFAとのテトラヒドロフラン中での反応によって実現された。クロマトグラフィーによる精製後(シリカ、THF/ヘキサン=1/1)、前述の反応の産物をDMSOに溶解した。この溶液に、アルゴン下、0.9 eg.のPte-γ-Glu-Asp-Arg-Asp-Cys-OHを加え、反応混合液を18時間攪拌した。溶媒を凍結乾燥にて除去し、残渣をHPLCにて精製した。
【0205】
[実施例10b]
[実施例10c]
実施例10cおよび10cの化合物は、実施例10aに一般的に記載した工程に従って、5’’-(N-Boc)アミノ-ビス-インドリル-seco-CBIの誘導体から調製した。
【0206】
[実施例11]
炭酸カリウムの存在下に、2-メルカプトエタノールのS-アルキル化を臭化アリルによって実現した。得られたアリルβ-ヒドロキシエチルスルフィドのヒドロキシル基を、塩化チオニルによって塩素と交換した。この産物を、酢酸と無水酢酸の存在下に過酸化水素で酸化すると(J. Am. Chem. Soc., 1950, 72, 59、なお、この開示を引用することにより本明細書に含める)、アリルβ-クロロエチルスルフォンが得られた。この産物を、触媒量の白金6塩化水素酸(IV)の存在下に高温でクロロジメチルシランと反応させると、分留後、3-(β-クロロエチルスルフォニル)プロピルジメチルシリルクロリドが得られた。このクロロシランは、1 eg.のピリジンを塩基として用いると、高い細胞傷害性を持つ化合物リゾキシンのヒドロキシル基をシリル化した。この分子のβ-クロロエチルスルフォン基を、過剰なトリエチルアミンによって処理すると、塩化水素の滑らかなβ-除去が達成され、それぞれのビニールスルフォンが形成された。
【0207】
もう一方の反応相手、ペプチド断片Pte-γ-Glu-Arg-Asp-Cys-OHは、実施例2aとスキーム12に一般的に記載されるように、Fmocプロトコールを用いたポリマー支持継続法によって調製された。
【0208】
複合した結合体の最終集合は、リゾキシンに付着したシリコンリンカーのビニールスルフォン基に対して、ペプチド断片のチオール基をマイケル付加することによって実現された。この変換の反応溶媒は、50:50アセトニトリル/水(pH=7.2)であった。室温で24時間攪拌後、最終結合体を、HPLCの分取カラムにて単離した。ES MS(m+H)+ 1631.6; (m-H)- 1629.6。
【0209】
[実施例12]
このリゾキシンのシリコン連結結合体は、クロロジメチルシランの代わりに市販のクロロメチルフェニルシランを用いたことを除いては、実施例11に記載するプロトコールを用いて合成した。
【0210】
[実施例13]
===システインジスルフィド結合を含む化合物の一般的調製===
Ranasinghe and Fuchs, Synth. Commun. 18(3), 227-32(1988)の方法(この開示を引用することにより本明細書に含める)に従って調製されたチオスルフォネート4(1 eq.)を、薬剤、薬剤類縁体、または、薬剤誘導体5(1 eq.)と反応させて、スキーム13に示すように、メタノール溶液として薬剤チオスルフォネート6を調製する。式中、Rはアルキルまたはアリールであり、Lは適当な脱離基、例えば、ハロゲン、ペンタフルオロフェニル等であり、nは1から4までの整数であり、Xは-O-、-NH-、-C(O)O-、または-C(O)NH-である。変換は、各開始材料の消失をTLC(シリカゲル;CHCl3/MeOH=9/1)で観察することによって監視される。
スキーム13
【0211】
葉酸含有ペプチジル断片Pte-Glu-(AA)n-Cys-OH(9)は、スキーム14に示すように、酸感受性Fmoc-Cys(Trt)-Wang樹脂(7)上でFmoc策を用いるポリマー支持継続法によって調製した。R1はFmocであり、R2はトリチルであり、DIPEAはジイソプロピルエチルアミンである。PyBopは、効率的結合を確保するための活性化試薬として与えられる。各結合工程の後、標準条件下でFmoc保護基を除去する。適当に保護されたアミノ酸建設ブロック、例えば、Fmoc-Glu-OtBu、N10-TFA-Pte-OH等が、スキーム14に記載するように、また、工程(b)においてFmoc-AA-OHによって表されるように用いられる。従って、AAは、適当に保護されたアミノ酸開始材料を指す。Fmoc-AA-OHを含む結合手順(工程(a)および(b))は、”n”回繰り返されて、固相支持ペプチド8を調製する。式中nは整数であり、0から約100に等しい。最後の結合工程後、残余のFmoc基を除去し、順次ペプチドを、グルタミン酸誘導体に結合させ(工程(c))、脱保護し、TFA保護プテロイン酸に結合させる(工程(d))。次に、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分離する(工程(e))。この反応条件により、t-Bu、t-Boc、およびTrt保護基の同時除去が実現される。TFA保護基は、塩基処理によって除去され(工程(f))、葉酸含有Cys含有ペプチジル断片9を与える。
スキーム14
(a)20%ピペリジン/DMF; Fmoc-AA-OH, PyBop DIPEA, DMF;(c)Fmoc-Glu(O-t-Bu)-OH, PyBop,DIPEA, DMF; (d)1. N10(TFA)-Pte-OH; PyBop,DIPEA, DNSO; (e) TFAA, (CH2SH2)2, i-Pr3SiH; (f) NH4OH, pH10.3。
【0212】
薬剤結合体は、葉酸誘導体9(0.9-0.95 eq.)を、脱イオン水に溶解させた薬剤チオスルフォネート6(0.04M、pHを0.1 N NaHCO3で7に調節)と、アルゴン下で約30分反応させ、ジスルフィド結合を形成させて調製する。メタノールを減圧蒸留後、結合体を、分取HPLC(Prep Novapak HR C18 19 X 300 mMカラム;展開相(A)−1.0mMリン酸バッファー、pH=6;有機相(B)−アセトニトリル;条件−勾配、99%Aと1%Bから50%Aと50%Bまで30分;流速=15 mL/分)にて精製してもよい。
【0213】
[実施例14a]
1H NMR(DMSO-d6)δ4.7(d, 1H), 4.95(d, 1H), 6.7(d, 4H), 6.9(t, 1H), 7.95(d, 2H), 8.1(d, 2H), 8.2(m, 1H), 8.3(s, 1H), 8.4(s, 1H), 8.7(s, 1H), 10.2(s, 1H), 11.8(d, 2H)。
【0214】
[実施例14b]
ES MS(m-H)- 1436.4, (m+H)+ 1438.3。
【0215】
[実施例14c]
1H NMR(DMSO-d6/D2O)δ1.0(s, 1H), 1.1(s, 1H), 1.6(s, 1H), 1.8(s, 1H), 2.1(s, 1H), 2.25(s, 3H), 2.65(dd, 2H), 3.7(d, 1H), 4.4(t, 1H), 4.55(q, 2H), 4.6(d, 2H), 4.95(d, 1H), 5.9(t, 1H), 6.15(s, 1H), 6.6(d, 2H), 7.85(d, 2H), 7.95(d, 2H), 8.6(s, 1H), 8.95(d, 1H)。
【0216】
[実施例14d]
1H NMR(DMSO-d6/D2O)δ1.0(s, 1H), 1.1(s, 1H), 1.65(s, 1H), 2.1(s, 1H), 2.25(s, 3H), 2.6(dd, 2H), 3.25(dd, 1H), 3.6(t, 2H), 3.7(d, 1H), 4.4(t, 1H), 4.6(d, 1H), 4.95(d, 1H), 5.9(t, 1H), 6.2(s, 1H), 6.6(d, 2H), 7.7(t, 1H), 7.9(d, 2H), 7.95(d, 2H), 8.6(s, 1H), 9.1(d, 2H)。
【0217】
[実施例14e]
1H NMR(DMSO-d6/D2O)δ10.85(d, 2H), 1.05(d, 2H), 1.2(d, 2H), 1.7(d, 2H), 3.95(d, 1H), 4.05(dd, 1H), 5.4(dd, 1H), 5.7(dd, 1H), 6.65(d, 2H), 7.6(d, 2H), 7.95(s, 1H), 8.65(s, 1H)。
【0218】
[実施例14f]
ES MS (m+H)+ 1487.23; 1H NMR(DMSO-d6/D2O)δ0.9(t, 2H), 1.3(t, 2H), 2.15(t, 2H), 3.2(dd, 1H), 4.0(t, 1H), 4.15(q, 1H), 5.3(s, 2H), 5.5(s, 2H), 6.6(d, 2H), 7.0(s, 1H), 7.4(m, 2H), 7.55(d, 1H), 8.0(d, 2H), 8.6(s, 1H)。
【0219】
実施例14a、14b、14c、14d、14e、および14fは、下記の一般的手順によって調製した。-OH基を有する対応薬剤のよく攪拌された溶液(乾燥CH2Cl2または乾燥THFに溶解した1 eq.)に、6-(トリフルオロメチル)ベンゾトリアゾリル2-(2’-ピリジルジチオエチルカルボネート(1.3 eq.)およびNN-ジメチルアミノピリジン(1.5 eq.)をアルゴン下に加えた。この反応混合液を3時間攪拌し、ピリジルジチオ-誘導体薬剤をシリカクロマトグラフィーにて単離した(各サンプルについて>65%)。スキーム12に概略した一般的方法によって調製した対応ペプチジル断片(0.5 eq.)をDMSOに溶解した。得られた黄色透明液に、ピリジル-ジチオ誘導体薬剤を加えた。30分後、反応は完了し、結合体はHPLCにて精製した。実施例14eの場合、先ず、ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OHを水に溶解し、溶液のpHを、0.1N HClによって2.5に調節した。これによって、ペプチジル断片は沈殿した。このペプチジル断片を遠心によって収集し、乾燥し、DMSOに溶解して、つぎの、ピリジルジチオ-誘導体薬剤との反応に備えた。
【0220】
[実施例15]
SN38(10-ヒドロキシ-7-エチルカンプトテシン)の、中間体4-(2-ピリジニルジチオ)ベンジルカルボネートを、P. Senter et al., J. Org. Chem. 1990, 55, 2875によって記載される手順に従って調製した。なお、この開示を引用することにより本明細書に含める。ペプチジル断片Pte-Glu-Asp-Arg-Asp-Cys-OHをDMSOに溶解し、得られた黄色透明溶液に、ピリジルジチオ誘導体薬剤を加えた。30分後反応は完了し、結合体はHPLCにて精製した。ES MS (m+H)+ 1425.38; 1H NMR(DMSO-d6/D2O)δ0.9(t), 1.15(t), 3.9(t), 4.0(t), 4.25(t), 5.1(m), 5.2(s), 5.4(s), 6.55(d), 7.25(d), 7.35(d), 7.5(d), 7.9(d), 8.55(s)。
【0221】
[実施例16a]
[実施例16b]
実施例16aと16bの化合物は、スキーム12に記載される一般的手順に従って、ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OHから調製した。このペプチジル断片を、seco-CBI-ビス-インドールのマレイミド誘導体にマイケル添加したところ、葉酸結合体実施例16aが得られた。このペプチジル断片はさらに、チオスルフォネート、またはピリジルジチオ活性化ビンブラスチンと反応して実施例16bを形成した。seco-CBI-ビス-インドールのマレイミド誘導体、および、チオスルフォネートおよびピリジルジチオ活性化ビンブラスチン中間体は、他の実施例について本明細書で記載する方法を用いて調製した。
【0222】
[実施例17a]
デアセチルビンブラスチンモノヒドラジド(1 eq.)(Barnett et al., J. Med. Chem. 1978, 21, 88を参照、なお、この開示を引用することにより本明細書に含める)を、新鮮蒸留THF中で、1 eq.のトリフルオロ酢酸で処理した。10分間攪拌後、溶液を1.05 eq.のN-(4-アセチルフェニル)マレイミドで処理した。アシルヒドラゾン形成は45分で完了し、溶媒は留去した。スキーム12に概説した一般的方法に従って調製されたペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(0.85 eq.)を水に溶解し、溶液のpHを、0.1N HClによって2.5に調節した。これによって、ペプチドは沈殿した。このペプチジル断片を遠心によって収集し、乾燥し、DMSOに溶解した。得られた黄色透明液に、ヒューニッヒ塩基(15 eq.)およびアシルヒドラゾンマイケル添加物を加えた。1時間後、最終的結合体をHPLCにて精製した。
【0223】
[実施例17b]
[実施例17c]
実施例17bと17cは、実施例17aに記載される手順に従って、対応するペプチジル断片とCBIのモノヒドラジド誘導体から調製した。
【0224】
実施例18-41の化合物は、実施例13に一般的に記載される手順に従って調製された。実施例18-41は、エレクトロスプレー質量分析(ES MS)、および、1Dおよび2D NMRおよびUVを含めた、その他の分光光度法によってその特性を明らかにした。
【0225】
[実施例18]
ES MS (m+H)+ 1071.9, (m+Na)+ 1093.9; 1H NMR(D2O)δ2.6(t, 4H), 2.7(t, 4H), 4.15(s, 2H), 5.45(s, 2H), 7.75(d, 2H), 8.15(d, 2H), 8.9(s, 1H)。
【0226】
[実施例19]
UV(nm)233(max), 255, 280; 1H NMR(D2O, NaOD, CD3CN)δ1.15(d, 3H), 2.3(s, 3H), 3.6(s, 1H), 3.85(s, 3H), 4.9(s, 1H), 5.3(s, 1H), 6.5(d, 2H), 7.3(m, 1H), 7.5(d, 2H), 7.65(d, 2H), 8.4(s, 1H)。
【0227】
[実施例20]
ES MS (m-H)- 935.6, (m+H)+ 937.4, (m+Na)+ 959.5。
【0228】
[実施例21]
1H NMR(D2O, NaOD, CD3CN)δ0.1(s, 1H), 1.1(s, 3H), 1.2(s, 3H), 1.75(s, 3H), 1.9(s, 3H), 2.05(s, 3H), 2.35(s, 3H), 3.3(dd, 2H), 3.8(d, 1H), 4.3(q, 2H), 4.9(d, 1H), 5.1(d, 1H), 5.4(q, 1H), 5.55(d, 1H), 5.65(d, 1H), 6.1(t, 1H), 6.35(s, 1H), 6.9(d, 2H), 7.9(d, 2H), 8.15(d, 2H), 8.7(s, 1H)。
【0229】
[実施例22]
【0230】
[実施例23]
ES MS (m-H)- 1136.5。
【0231】
[実施例24]
ES MS (m-H)- 1136.3, (m+H)+ 1138.0。
【0232】
[実施例25]
ES MS (m+H)+ 1382.3, (m+Na)+ 1405.4。
【0233】
[実施例26]
ES MS (m-H)- 1379.2, (m+H)+ 1381.2。
【0234】
[実施例27]
ES MS (mH)- 949.2; 1H NMR(D2O)δ1.55(s, 3H), 1.95(m, 2H), 2.05(s, 3H), 2.45(s, 3H), 2.75(dd, 2H), 2.95(dd, 2H), 3.05(s, 3H), 3.3(dd, 2H), 3.35(d, 2H), 3.45(t, 2H), 4.85(q, 2H), 6.5(d, 2H), 7.45(d, 2H), 8.5(s, 1H)。
【0235】
[実施例28]
1H NMR(DMSO-d6)δ1.5(s), 2.25(t), 2.75(m), 3.9(q), 4.6(d), 4.85(t), 6.6(d), 7.6(d), 7.9(d), 8.15(d), 8.25(t), 8.65(s), 8.7(m), 9.3(m), 10.2(t)。
【0236】
[実施例29]
ES MS (m-H)- 1413.5, (m+H)+ 1415.3。
【0237】
[実施例30]
ES MS (m+H)+ 1530.2; 1H NMR(DMSO-d6/D2O)δ1.2(s, 1H), 2.9(t, 1H), 3.65(t, 1H), 4.15(t, 1H), 4.25(t, 1H), 4.35(t, 1H), 6.7(d, 2H), 7.0(s, 1H), 8.1(d, 2H), 8.25(s, 1H), 8.7(s, 1H)。
【0238】
[実施例31]
1H NMR(DMSO-d6)δ1.75(s, 1H), 1.85(s, 1H), 2.1(t, 2H), 4.3(t, 1H), 4.6(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 8.15(s, 2H), 8.6(s, 1H)。
【0239】
[実施例32]
ES MS (m+H)+ 1408.4。
【0240】
[実施例33]
ES MS (m-H)- 1491.1, (m+H)+ 1493.1; 1H NMR(DMSO-d6/D2O)δ4.15(q, 1H), 4.6(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 7.25(s, 1H), 7.4(d, 1H), 7.9(d, 1H), 7.95(d, 2H), 8.15(d, 2H), 8.6(s, 1H)。
【0241】
[実施例34]
1H NMR(DMSO-d6/D2O)δ2.1(t, 2H), 2.75(q, 2H), 4.3(t, 1H), 4.65(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 7.9(d, 1H), 8.0(d, 2H), 8.2(t, 2H), 8.6(s, 1H)。
【0242】
[実施例35]
【0243】
[実施例36]
ES MS (m+H)+ 1680.4; 1H NMR(DMSO-d6/D2O)δ0.3(s, 3H), 0.35(s, 3H), 1.05(s, 9H), 2.15(t, 2H), 4.15(t, 1H), 4.85(t, 1H), 6.6(d, 2H), 7.55(t, 4H), 7.9(d, 1H), 8.0(s, 1H), 8.15(s, 1H), 8.6(s, 1H)。
【0244】
[実施例37]
1H NMR(DMSO-d6/D2O)δ1.1(s, 3H), 1.8(s, 1H), 4.55(d, 1H), 4.8(t, 1H), 6.6(d, 2H), 7.8(d, 1H), 8.1(d, 1H), 8.15(s, 1H), 8.6(s, 1H)。
【0245】
[実施例38]
【0246】
[実施例39]
【0247】
[実施例40]
【0248】
[実施例41]
【0249】
[実施例42]
===EC112治療マウスにおける腫瘍成長の抑制===
薬剤がダウノルビシンである、実施例9b(EC111)および9c(EC112)の化合物の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の4日後、マウス(グループ当たり5匹)に、実施例9bまたは実施例9cの化合物2-10 μmol/kg、あるいは、結合されていないダウノルビシンまたはPBSを、週に2度4週間静注した。腫瘍成長は、各治療グループにおいて、3日または4日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。動物の体重も、3日または4日間隔で測定した。
図1および2に示すように、実施例9cの化合物による治療は、外見上毒性もなく(動物の体重に基づく)、M109腫瘍成長を効果的に遅らせた。未結合のドキソルビシンも抗腫瘍反応を与えたが、同時に体重に基づく毒性作用を示した。
【0250】
[実施例43]
===EC105治療マウスにおける腫瘍成長の抑制===
実施例10aの化合物(EC105)を使用したことを除いては、プロトコールは実施例42に記載した通りであった。この化合物では、薬剤は、ビス-インドリル-seco-CBIであった。実施例10aの化合物を0.3 μmol/kgの用量で注入した。さらに、M109モデル(葉酸受容体陽性)および4T1モデル(葉酸受容体陰性)を含む二つの皮下腫瘍モデルを試験した。数匹の動物では、67倍過剰の遊離葉酸(20 μmol/kg, FA)を、結合体(すなわち、実施例10aの化合物)と同時投与した。
実施例10aの化合物において、動物体重に基づく毒性を外見上示すことなく、著明な抗腫瘍反応が観察された(図3および4参照)。実施例10aの化合物による抗腫瘍反応は、過剰な遊離葉酸によって阻止され、反応の特異性を示した(図3参照)。図5に示すように、4T1モデル(葉酸受容体陰性)では抗腫瘍作用が観察されず、これも反応の特異性を示した。
【0251】
[実施例44]
===EC145治療マウスにおける腫瘍成長の抑制===
薬剤がデアセチルビンブラスチンモノヒドラジドである、実施例16bの化合物(EC145)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の約11日後(t0における平均腫瘍容量=60 mm3)、マウス(グループ当たり5匹)に、EC145の1500 nmol/kg、あるいは、等投与容量のPBS(コントロール)を、週に2度(BIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図6に示すように、EC145による治療は、生食液治療動物におけるM109腫瘍の成長と比べると、M109腫瘍成長を効果的に遅らせた。
【0252】
[実施例45]
===EC140治療マウスにおける腫瘍成長の抑制===
薬剤がデアセチルビンブラスチンモノヒドラジドである、実施例17aの化合物(EC140)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の約11日後(t0における平均腫瘍容量=60 mm3)、マウス(グループ当たり5匹)に、EC140の1500 nmol/kg、あるいは、等投与容量のPBS(コントロール)を、週に3度(TIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図7に示すように、EC140による治療は、生食液治療動物におけるM109腫瘍の成長と比べると、M109腫瘍成長を効果的に遅らせた。
【0253】
[実施例46]
===EC136治療マウスにおける腫瘍成長の抑制===
薬剤がCBIである、実施例10bの化合物(EC136)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にL1210A腫瘍を負荷させたDBAマウスで評価した。右腋窩皮下に0.25x105 L1210A細胞注入による腫瘍接種の約5日後(t0における平均腫瘍容量は~50 mm3、グループ当たり5匹)に、EC136の400 nmol/kg、あるいは、等投与容量のPBS単独(コントロール)を、週に3度(TIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図8に示すように、EC136による治療は、生食液治療動物におけるL1210A腫瘍の成長と比べると、L1210A腫瘍成長を効果的に遅らせた。
【0254】
[実施例47]
===各種葉酸-薬剤結合体による細胞DNA合成の抑制===
実施例17b、10b、16a、10c、17a、16b、14e、および15の化合物(それぞれ、EC135、EC136、EC137、EC138、EC140、EC145、EC158、およびEC159)を、薬剤の、葉酸受容体陽性KB細胞の成長抑制能力を予測する、インビトロ細胞傷害性アッセイを用いて評価した。化合物は、本明細書に記載するプロトコールに従って調製された、それぞれの化学療法剤に連結された葉酸塩から成っていた。KB細胞は、表示の濃度の葉酸-薬剤結合体(図9-16のx軸参照)に対して、少なくとも100倍過剰量の葉酸の不在下に、または存在下に37℃で最大7時間暴露した。次に、細胞を、新鮮な培養液で濯ぎ、新鮮な培養液で37℃で72時間インキュベートした。細胞生存率を、3H-チミジン取り込みアッセイによって評価した。
【0255】
図9-16に示すように、用量依存性細胞傷害性が測定され、ほとんどの場合、IC50値(新たに合成されるDNAへの3H-チミジン取り込みを50%低下させるのに必要な、薬剤結合体の濃度)は、低ナノモル範囲であった。さらに、これら結合体の細胞傷害性は、過剰な遊離葉酸の存在下では低下した。このことは、観察された細胞殺戮は、葉酸受容体結合によって仲介されたことを示す。
【0256】
EC158、および、細胞系統、例えば、IGROV(既知の細胞系統)、A549-クローン-4(ヒト葉酸受容体cDNAをトランスフェクトされたA549細胞)、新規系統-01(インビボにおいて葉酸受容体発現について選択された系統-01突然変異株)、M109、4T1クローン-2(げっ歯類葉酸受容体cDNAをトランスフェクトされた4T1細胞)、および、HeLa細胞を含む細胞系統等、を用いた、このタイプのアッセイにおいても同様の結果が得られた。
【技術分野】
【0001】
===関連出願の相互参照===
本出願は、米国特許法119条(e)の下に、2003年1月27日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/442,845号、2003年8月1日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/492,119号、および、2003年10月31日出願、名称「ビタミン受容体結合性薬剤送達結合体(Vitamin-Receptor Binding Drug Delivery Conjugates)」の米国出願第60/516,188号に対する優先権を主張する。上記各出願の開示の全体を参照することにより本出願に含める。
【0002】
本発明は、標的に向けた薬剤送達に使用される組成物および方法に関する。さらに具体的には、本発明は、病原性細胞集団によって引き起こされる病的状態の治療に用いられるビタミン受容体結合性薬剤送達結合体、および、そのための方法および製薬組成物に向けられる。
【背景技術】
【0003】
哺乳類免疫系は、腫瘍細胞、その他の病原性細胞、および、侵入する外来病原体を認識し、除去するための手段を与える。正常時、免疫系は強力な防衛線を供給するが、ガン細胞、その他の病原細胞、および、感染源が、宿主の免疫反応を回避して、増殖または残存して、同時に宿主の病原となる場合が数多くある。例えば、増殖する新生物を除去するために、化学療法剤および放射線療法が開発されている。しかしながら、現在利用可能な化学療法剤および放射線治療処方の多くは副作用を持つ。なぜなら、これらの薬剤および療法は病原細胞を破壊するだけでなく、造血系の細胞のような正常な宿主細胞をも侵すからである。これらの抗ガン剤の副作用は、病原性細胞集団に対して選択的で、宿主に対する毒性の低い新規療法の開発の必要を強調する。
【0004】
研究者達は、病原性細胞に対して細胞傷害性化合物を標的化することによってその病原性細胞を破壊するための治療プロトコールを開発している。これらのプロトコールの多くは、毒素を利用するのであるが、その毒素の正常細胞に対する送達を極小にするために、病原細胞に対して固有の、または、病原細胞によって過剰発現される抗原に結合する抗体に結合させた毒素を利用する。この方法を用いて、いくつかの免疫毒素が開発された。これらのものは、病原細胞上の特異的抗原に向けられた抗体から成り、抗体は、リシン、シュードモナス体外毒素、および腫瘍壊死因子のような毒素に連結される。これらの免疫毒素は、例えば腫瘍細胞のような、その抗体によって認識される特異的抗原を帯びる病原細胞を標的とする(非特許文献1、2および特許文献1)。
【0005】
宿主における病原細胞集団、例えば、ガン細胞または外来病原体から成る集団を標的とするもう一つの方法は、それ自体も独立した宿主毒性を示しかねない化合物投与の必要を回避するために、病原細胞に対する宿主の免疫反応を強化することである。免疫療法のための一つの報告されている戦略は、抗体を、例えば、遺伝子工学的に加工された多量体抗体を、腫瘍細胞の表面に結合させ、それらの抗体の定常域を細胞表面において提示させ、各種免疫系仲介過程を通じて腫瘍細胞殺戮を誘発することである(非特許文献3、および特許文献2)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】PCT公報WO91/07418、1991年5月30日発行
【特許文献2】米国特許第5,672,486号、Soulillou, J.P.に付与
【非特許文献】
【0007】
【非特許文献1】Olsnes, S., Immunol. Today, 10, pp.291-295, 1989
【非特許文献2】Melby, E.L., Cancer Res., 53(8), pp.1755-1760, 1993
【非特許文献3】De Vita, V.T., 「ガンの生物学的治療法(”Biologic Therapy of Cancer”)」、第2版、フィラデルフィア、Lippincott、1995
【発明の概要】
【0008】
病原細胞に対して特異的であり、正常細胞に対しては毒性を極小とした効果的な療法を開発するために、ビタミン受容体結合薬剤送達結合体が開発されてきた。本発明は、宿主動物において様々な病理現象を引き起こす病原細胞の集団に適用が可能である。本発明の薬剤送達結合体によって治療が可能な病原細胞としては、ガン細胞、細菌およびウィルスのような感染源、細菌またはウィルス感染細胞、およびその他の、ビタミン受容体、または、ビタミン類縁体または誘導体に結合する受容体を特異的に発現するか、優先的に発現するか、または、過剰に発現する、任意の病原細胞が挙げられる。
【0009】
一つの実施態様では、ビタミン受容体結合性薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価のリンカー、および薬剤を含む。本明細書で用いる”V”は、ビタミン受容体結合成分を指し、ビタミン、および、ビタミン受容体結合性類縁体または誘導体を含み、「ビタミン、またはその類縁体または誘導体」という用語は、ビタミン受容体に結合することが可能なビタミン、および、その類縁体および誘導体を指す。本明細書で用いる”D”とは薬剤を指し、その類縁体または誘導体を含む。ビタミン、またはその類縁体または誘導体は、二価のリンカー(L)に共有結合され、薬剤、またはその類縁体または誘導体も二価リンカー(L)に共有結合される。二価リンカー(L)は、複数のリンカーを含むことが可能である。例えば、二価リンカー(L)は、スペーサーリンカー(ls)、放出リンカー(lr)、およびヘテロ原子リンカー(lH)のうちの1つ以上、および、それらの、順番は任意とする組み合わせを含む。
【0010】
本実施態様を例示する薬剤送達結合体としては、
V-L-D
V-(lr)c-D
V-(ls)a-D
V-(ls)a-(lr)c-D
V-(lr)c-(ls)a-D
V-(lH)b-(lr)c-D
V-(lr)c-(lH)b-D
V-(lH)d-(lr)c-(lH)e-D
V-(ls)a-(lH)b-(lr)c-D
V-(lr)c-(lH)b-(ls)a-D
V-(lH)d-(ls)a-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(ls)a-(lH)e-D
V-(lH)d-(ls)a-(lH)b-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(lH)b-(ls)a-(lH)e-D
V-(ls)a-(lr)c-(lH)b-D
V-[(ls)a-(lH)b]d-(lr)c-(lH)e-D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)は本明細書に定義した通りであり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体であり、式中、二価のLは、1個の、または、各種の(ls)、(lH)、または(lr)を任意の順序で任意の組み合わせで含む。二価リンカーLの上記例は、二価リンカーの含む、(lH)、(ls)、(lr)の広範な集合を例示することを意図したものであって、限定することを意図したものではないことが理解される。
【0011】
スペーサー、ヘテロ原子、および放出リンカーはそれぞれ二価であることが理解される。さらに、本明細書で定義した、各種スペーサー、ヘテロ原子、および放出リンカーの各要素間の結合性、および、各種スペーサー、ヘテロ原子、および放出リンカーと、Dおよび/またはVとの間の結合性は、それら各種のスペーサー、ヘテロ原子および放出リンカー中に見られる任意の原子で起こってもよく、各種スペーサー、ヘテロ原子および放出リンカーの内の任意のものの任意の見かけ上の末端に必ずしも起こらなくてもよいことを理解しなければならない。例えば、例示の実施態様において、二価のリンカーは、
すなわち、式中、二価のリンカーLは、-(lH)-(ls)5-(lr-lH)2-Dであって、それぞれ、(lH)は窒素であり、(ls)5は、 Ala-Glu-Lys-Asp-Asp であり、(lr-lH)2は、 −(CH2)2-S-S-(CH2)2-O-C(O)-O- である。この実施態様において、(lr-lH)2リンカーは、(ls)5リンカーの中間部分に接続する。
【0012】
別の実施態様では、ビタミン受容体結合薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、および薬剤を含み、二価リンカー(L)は1種以上のヘテロ原子リンカー(lH)を含む。ビタミン受容体結合成分は、第1ヘテロ原子リンカー(lH)dを通じて、二価のリンカー(L)に共有結合し、薬剤は、第2ヘテロ原子リンカー(lH)eを通じて、二価のリンカー(L)に共有結合する。二価のリンカー(L)は1種以上のスペーサーリンカーおよび放出リンカーも含み、このスペーサーリンカーと放出リンカーは、第3のヘテロ原子リンカー(lH)bを通じて互いに共有結合されてもよい。この実施態様を例示する薬剤送達結合体は下記の通りである、すなわち、
V−(lH)d−(ls)a−(lH)b−(lr)c−(lH)e−D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)、VおよびDは本明細書に定義した通りであり、式中、二価のLは、図示のように(ls)、(lH)、および(lr)を含む。
【0013】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体が提供される。この薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、および薬剤を含み、二価リンカー(L)はヘテロ原子リンカー(lH)を含む。ビタミン受容体結合成分は、ビタミン、またはその類縁体または誘導体であり、薬剤は、その類縁体または誘導体も含む。ビタミン、またはその類縁体または誘導体は、二価リンカー(L)に共有結合し、薬剤、またはその類縁体または誘導体は二価リンカー(L)に共有結合する。二価リンカー(L)は、スペーサーリンカーおよび放出リンカーも含み、このスペーサーリンカーと放出リンカーは、ヘテロ原子リンカーを通じて互いに共有結合されてもよい。この実施態様を図示する薬剤送達結合体は下記の通りである、すなわち、
V−(ls)a−(lH)b−(lr)c−D
式中、a、b、およびcは、それぞれ独立に、0、1、2、3、または4であり、(ls)、(lH)、および(lr)、VおよびDは本明細書に定義した通りであり、式中、二価のLは、図示のように(ls)、(lH)、および(lr)を含む。
【0014】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が提供される。この実施態様では、Lは1種以上のリンカー(lr)c、(ls)a、と(lH)b、およびそれらの任意の順序の組み合わせから構築される。ここに、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、bおよびcはそれぞれ独立に0、1、2、3、または4であり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体である。本明細書に記載される薬剤送達結合体は、1個を越えるスペーサーリンカー、放出リンカー、またはヘテロ原子リンカーを持つ二価リンカーを含んでもよいことが理解される。例えば、2個以上の放出リンカー(lr)を含む二価リンカーが考慮される。さらに、この放出リンカーの構成は、二価リンカーであって、複数の放出リンカーが共有結合され、この複数の放出リンカーが、1個以上のヘテロ原子リンカーおよび/またはスペーサーリンカーによって相互に隔てられている二価リンカーを含む。
【0015】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が記載される。式中、Lは、(ls)aと(lH)b、およびそれらの任意の順序の組み合わせを含む二価のリンカーであり、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りである。この実施態様では、薬剤送達結合体の薬剤は、ハプテン、例えば、と言ってそれらに限定されないが、フルオレセイン、ジニトロフェニル等であることが可能である。
【0016】
別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が記載される。式中、Lは、(ls)a、(lH)bと(lr)c、およびそれらの任意の順序の組み合わせを含む二価のリンカーであり、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りであり、(lr)の少なくとも一つはジスルフィドではない。この実施態様の二価リンカーは、1個を越える(lr)を持つ、すなわち、cは1よりも大であるが、もう一つの、または、別の放出リンカーに加えて、ジスルフィド放出リンカーを含んでもよいことが理解される。
【0017】
本明細書に記載される各種ビタミン受容体結合性薬剤送達結合体の一つの局面では、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルオキシメチルオキシを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、メチルは、要すれば随意に、アルキル、または、置換アリールによって置換される。
【0018】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルカルボニルを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とアシルアジリジンを形成する。
【0019】
別の局面で、二価リンカーは、まとまって1-アルコキシシクロアルキレンオキシを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。
【0020】
別の局面で、二価リンカーは、まとまってアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシレートを形成するスペーサーリンカー、ヘテロ原子リンカー、および放出リンカーを含む。
【0021】
別の局面で、二価リンカーは、まとまってジチオアルキルカルボニルヒドラジドを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成する。
【0022】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルカルボニルヒドラジドを形成するヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成する。
【0023】
別の局面で、二価リンカーは、まとまって3-チオアルキルスルフォニルアルキル(二置換シリル)オキシを形成するヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含む。式中、二置換シリルは、アルキル、または、要すれば随意に置換されるアリールによって置換される。
【0024】
別の局面で、二価リンカーは、天然アミノ酸およびその立体異性体から成るグループから選ばれる複数のスペーサーリンカーを含む。
【0025】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成する。
【0026】
別の局面で、二価リンカーは、まとまって3-ジチオアリールアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成し、アリールは要すれば随意に置換される。
【0027】
別の局面で、二価リンカーは、まとまって3-チオスクシニミド-1-イルアルキルオキシアルキルオキシアルキリデンを形成するヘテロ原子リンカー、スペーサーリンカー、放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アルキリデンは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成し、それぞれのアルキルは独立に選択され、オキシアルキルオキシは要すれば随意に、アルキル、または要すれば随意に置換されたアリールに置換される。
【0028】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルオキシカルボニルヒドラジドを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。
【0029】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アミノは、薬剤、またはその類縁体または誘導体とビニール性アミドを形成する。
【0030】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、アミノは、薬剤、またはその類縁体または誘導体とビニール性アミドを形成し、アルキルはエチルである。
【0031】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成する。
【0032】
別の局面で、二価リンカーは、まとまって3-ジチオアルキルアミノカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成し、アルキルはエチルである。
【0033】
別の局面で、二価リンカーは、まとまって3-ジチオアリールアルキルオキシカルボニルを形成する放出リンカー、スペーサーリンカー、および放出リンカーを含む。式中、カルボニルは、薬剤、またはその類縁体または誘導体とカルバメート、またはカルバモイルアジリジンを形成する。
【0034】
一つの局面で、放出、スペーサー、およびヘテロ原子リンカーは、二価リンカーの結合分断に次いで、放出された官能基が、隣接基支援分断または破壊とも呼ばれる、さらに別の結合の破壊または分断を化学的に支援するように配置されていてもよい。このような二価リンカーまたはその部分の例示の実施態様は、下記の式を持つ化合物を含む、すなわち、
式中、Xは、窒素、酸素、または硫黄のようなヘテロ原子、nは、0、1、2および3から選ばれる整数、Rは水素、または、アリール環の陽電荷を誘導的にまたは共鳴によって安定化することが可能な置換基を含む置換基、例えば、アルコキシ等であり、記号(*)は、二価リンカーを形成する、さらに別のスペーサー、ヘテロ原子、または放出リンカーの付着部位、あるいは、薬剤、またはその類縁体または誘導体、または、ビタミン、またはその類縁体または誘導体の付着部位を示す。その他の置換基も、アリール環、ベンジル炭素、アルカン酸、または、メチレン架橋に存在することが可能であり、そのような置換基は、例えば、と言ってそれらに限定されないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロ等を含むことが理解される。支援分断としては、ベンジリウム中間体、ベンジン中間体、ラクトン環化、オキソニウム中間体、ベータ除去等を含む機構を含む。さらに、放出リンカーの分断に続く断片化の他に、放出リンカーの初期分断を、隣接基支援機構によって促進するようにしてもよいことが理解される。
【0035】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体中間体が提供される。この中間体は、ビタミン受容体結合性成分、第1末端と第2末端を有する二価リンカー、および結合基を含む。ビタミン受容体結合性成分は、ビタミン、またはその類縁体または誘導体であり、結合基は、求核要素、求電子要素、またはその前駆物質である。ビタミン受容体結合成分は、二価リンカーの第1末端において二価リンカーに共有結合し、結合基は、二価リンカーの第2末端において二価リンカーに共有結合し、二価リンカーは、1個以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、および、順序は任意のそれらの組み合わせを含む。
【0036】
別の実施態様では、ビタミン受容体結合性薬剤送達結合体中間体が記載される。この中間体は、第1末端と第2末端を有する二価リンカー、薬剤、またはその類縁体または誘導体、および結合基を含む。二価リンカーは、本明細書で記載するスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーから選ばれる1種以上の成分を含む。結合基は、二価リンカーの第1末端において二価リンカーに共有結合し、薬剤およびその類縁体または誘導体は、二価リンカーの第2末端において二価リンカーに共有結合する。さらに、結合基は、ビタミン受容体結合成分と共有結合を形成することが可能な求核要素、求電子要素、またはその前駆物質であり、ビタミン受容体結合性成分は、ビタミン、またはその類縁体または誘導体である。
【0037】
本明細書で記載される、ビタミン受容体結合性薬剤送達結合体中間体の別の例示の実施態様では、結合基はマイケルアクセプターであり、二価リンカーは、式-C(O)NHN=、-NHC(O)NHN=、または-CH2C(O)NHN=を有する放出リンカーを含む。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の一つの例示の局面では、結合基と二価リンカーは共同して、下記の式を持つ化合物、またはその保護された誘導体を形成する、すなわち、
式中、Dは、本明細書で示すようにヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、nは、1、2、3、または4のような整数である。本明細書で記載される、ビタミン受容体結合性薬剤送達結合体中間体の別の例示の局面では、ビタミン、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0038】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の別の例示の実施態様では、結合基は、酸素、窒素、または硫黄のようなヘテロ原子であり、二価リンカーは、ビタミン、またはその類縁体または誘導体を、結合基に共有結合によって接続する、1個以上のヘテロ原子リンカー、および1個以上のスペーサーリンカーを含む。一つの例示の局面では、本明細書に記載されるビタミン受容体結合性送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Xは、酸素、窒素、または硫黄であり、mは、1、2、または3のような整数であり、V、ls、およびlHは上に定義した通りである。
【0039】
別の局面では、本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Xは、窒素または硫黄であり、Vおよびlsは、上に定義した通りである。
【0040】
別の局面では、本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体は、下式を有する化合物、または、その保護された誘導体を含む、すなわち、
式中、Yは、水素または置換基であり、例示としては、電子吸引置換基で、例えば、と言ってそれらに限定されないが、ニトロ、シアノ、ハロ、アルキルスルフォニル、カルボン酸誘導体等を含む置換基であり、Vおよびlsは、上に定義した通りである。
【0041】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の、別の例示の実施態様では、結合基はマイケルアクセプターであり、二価リンカーは、ビタミン、またはその類縁体または誘導体を、結合基に共有結合によって接続する、1個以上のヘテロ原子リンカー、および1個以上のスペーサーリンカーを含む。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の一つの例示の局面では、結合基と二価リンカーは共同して、下記の式を持つ化合物、またはその保護された誘導体を形成する、すなわち、
式中、Xは、酸素、窒素、または硫黄であり、mおよびnは、1、2、または3のような独立に選択された整数であり、V、ls、およびlHは上に定義した通りである。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の別の例示の局面では、薬剤、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0042】
本明細書で記載されるビタミン受容体結合性薬剤送達結合体中間体の、別の例示の局面では、中間体は、下記の式を持つ化合物、またはその保護された誘導体を含む、すなわち、
式中、Vはビタミン、またはその類縁体または誘導体であり、AAは、例示として天然アミノ酸およびその立体異性体から成るグループから選ばれるアミノ酸であり、Xは、窒素、酸素、または硫黄であり、Yは、水素または置換基であり、例示としては、電子吸引置換基で、例えば、と言ってそれらに限定されないが、ニトロ、シアノ、ハロ、アルキルスルフォニル、カルボン酸誘導体等を含む置換基であり、nおよびmは、1、2、または3のような独立に選択された整数であり、および、pは、1、2、3、4、または、5のような整数である。AAは、他の任意のアミノ酸、例えば、下記の一般式を有する任意のアミノ酸であってもよい、すなわち、
−N(R)−(CR’R’’)q−C(O)−
式中、Rは、水素、アルキル、アシル、または、適当な窒素保護基であり、R’およびR’’は、それぞれの出現において互いに独立に選択される水素または置換基であり、および、qは、1、2、3、4、または、5のような整数である。例示的には、R’および/またはR’’はそれぞれ独立して、水素、または、天然のアミノ酸に存在する側鎖、例えば、メチル、ベンジル、ヒドロキシメチル、チオメチル、カルボキシル、カルボキシルメチル、グアニジノプロピル等、および、それらの誘導体および保護誘導体に一致するが、ただしそれらに限定されない。前述の式は、立体異性変種を全て含む。例えば、アミノ酸は、アスパラギン、アスパラギン酸、システイン、グルタミン酸、リシン、グルタミン、アルギニン、セリン、オルニチン、トレオニン等から選ばれてもよい。本明細書に記載されるビタミン受容体結合性薬剤送達結合体中間体の別の局面では、薬剤、またはその類縁体または誘導体は、アルキルチオール求核要素を含む。
【0043】
別の実施態様では、下式を持つ化合物、またはその保護された誘導体を調製する方法が記載される:
式中、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせを含むリンカーであり、Dは、ヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、および、(lr)c、(ls)aと(lH)b、およびVとDとは本明細書に定義する通りである。当該方法は、
(a)下式:
を持つ化合物またはその保護された誘導体を、下式:
を持つ化合物またはその保護された誘導体と反応させて、チオスクシニミド誘導体を形成させる工程、および、
(b)チオスクシニミド誘導体によって、薬剤またはその類縁体または誘導体のヒドラゾン誘導体を形成させる工程、
を含む。
【0044】
別の実施態様では、下式を持つ化合物、またはその保護された誘導体を調製する方法が記載される:
式中、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせを含むリンカーであり、Dは、ヒドラゾンを形成することが可能な薬剤、またはその類縁体または誘導体であり、および、(lr)c、(ls)aと(lH)b、およびVとは本明細書に定義する通りである。当該方法は、
下式:
を持つ化合物またはその保護された誘導体を、下式:
を持つ化合物またはその保護された誘導体と反応させる工程、
を含む。
【0045】
別の実施態様では、製薬組成物が記載される。製薬組成物は、本発明による薬剤送達結合体、および、製薬学的に受容可能な担体を含む。
【0046】
別の実施態様では、病原細胞の集団を抱える宿主動物において病原細胞集団を除去する方法であって、病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体のの接近可能な結合部位を有しており、結合部位は、病原細胞によって特異的に発現されるか、過剰に発現されるか、または優先的に発現されることを特徴とする方法が記載される。この方法は、本発明による薬剤送達結合体、またはその製薬組成物を宿主に投与する工程を含む。
【図面の簡単な説明】
【0047】
【図1】図1は、EC112(実施例9c)によるM109腫瘍成長の抑制を示す。
【図2】図2は、EC112(実施例9c)の動物体重に対する作用を示す。
【図3】図3は、EC105(実施例10a)によるM109腫瘍成長の抑制を示す。
【図4】図4は、EC105(実施例10a)の動物体重に対する作用を示す。
【図5】図5は、EC105(実施例10a)による4T1腫瘍成長抑制の欠如を示す。
【図6】図6は、EC145(実施例16b)によるM109腫瘍成長の抑制を示す。
【図7】図7は、EC140(実施例17a)によるM109腫瘍成長の抑制を示す。
【図8】図8は、EC136(実施例10b)によるL1210腫瘍成長の抑制を示す。
【図9】図9は、EC135(実施例17b)による細胞DNA合成の抑制を示す。
【図10】図10は、EC136(実施例10b)による細胞DNA合成の抑制を示す。
【図11】図11は、EC137(実施例16a)による細胞DNA合成の抑制を示す。
【図12】図12は、EC138(実施例10c)による細胞DNA合成の抑制を示す。
【図13】図13は、EC140(実施例17a)による細胞DNA合成の抑制を示す。
【図14】図14は、EC145(実施例16b)による細胞DNA合成の抑制を示す。
【図15】図15は、EC158(実施例14e)による細胞DNA合成の抑制を示す。
【図16】図16は、EC159(実施例15)による細胞DNA合成の抑制を示す。
【発明を実施するための形態】
【0048】
本発明は、ビタミン受容体結合性成分、二価リンカー(L)、および、薬剤を含むビタミン受容体結合性薬剤送達結合体であって、ビタミン受容体結合性成分と薬剤とが、それぞれ、要すれば随意にヘテロ原子リンカーを介して、二価リンカー(L)に結合するビタミン受容体結合性薬剤送達結合体に関する。二価リンカー(L)は、1個以上のスペーサーリンカー、ヘテロ原子リンカー、および、放出(すなわち分断可能)リンカー、および、順序は任意のそれらの組み合わせを含む。
【0049】
本明細書で用いる「放出リンカー」という用語は、生理的条件下で破壊することが可能な少なくとも一つの結合(例えば、pH易変性、酸易変性、酸化易変性、または酵素易変性の結合)を含むリンカーを指す。結合破壊をもたらすこのような生理的条件は、例えば、生理的pH下で起こる、または、細胞原形質pHよりも低いpHを持つエンドソームのような細胞内小器官への封入の結果として起こる、標準的化学的加水分解反応が挙げられる。
【0050】
分断可能な結合は、放出リンカー内の二つの隣接原子を接続することが可能であり、および/または、放出リンカーの一方の端に、または、その両端に、他のリンカー、または本明細書に記述するVおよび/またはDを接続することが可能である。分断可能な結合が、放出リンカー内の二つの原子を接続している場合、結合の破壊の次に、放出リンカーは、2個以上の断片に分解される。あるいは、分断可能な結合が、放出リンカーと別の成分、例えば、ヘテロ原子リンカー、スペーサーリンカー、別の放出リンカー、薬剤またはその類縁体または誘導体、または、ビタミンまたはその類縁体または誘導体の間にある場合、結合の破壊の次に、放出リンカーは、他の成分とは分離される。
【0051】
分断可能結合の易変性は、例えば、分断可能結合の、または、その近傍の置換的な改変、例えば、分断性ジスルフィド結合近傍のα分枝や、加水分解可能なシリコン-酸素結合を有する成分のシリコンの置換基の疎水性の増大、加水分解可能なケタールまたはアセタールの一部を形成するアルコキシ基を均一化すること等を含む改変によって調節することが可能である。
【0052】
本発明によれば、ビタミン受容体結合性薬剤送達結合体を用いて、宿主における病原細胞集団の存在によって特徴付けられる病状であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体の接近可能な結合部位を有しており、前記結合部位は、病原細胞によって特異的に発現される、過剰に発現される、または優先的に発現されることを特徴とする、病状を治療することが可能である。病原細胞の選択的除去は、ビタミン受容体結合性薬剤送達結合体のビタミン成分を、ビタミンまたはその類縁体または誘導体に特異的に結合し、かつ、病原細胞によって特異的に、過剰に、または優先的に発現されるビタミン受容体、トランスポーター、またはその他の表面発現性蛋白質に結合させることによって仲介される。病原細胞によって、特異的に、過剰に、または優先的に発現される表面発現性蛋白質は、非病原性細胞には存在しないか、または低濃度でしか存在しない受容体であり、これが、病原細胞の選択的除去手段を提供する。
【0053】
例えば、高親和度葉酸受容体のような表面発現性ビタミン受容体は、ガン細胞において過剰に発現される。卵巣、乳腺、結腸、肺、鼻、喉、および脳の上皮ガンは全て、高レベルの葉酸受容体を発現することが報告されている。事実、全てのヒトの卵巣腫瘍の90%を越えるものが、大量のこの受容体を発現することが知られている。従って、本発明の薬剤送達結合体は、各種の腫瘍細胞タイプを始め、優先的にビタミン受容体を発現し、従って、ビタミンまたはその類縁体または誘導体に対して表面的結合部位を有する、他のタイプの病原細胞、例えば、感染媒介物をも治療するために使用が可能である。
【0054】
本明細書に記述されるビタミンの他に、他のリガンドも、本明細書に記載・考察される薬剤およびリンカーに結合して、薬剤の、所望の標的に対する送達を促進可能とするリガンド-リンカー-薬剤結合体を形成することが可能であることが理解される。上記他のリンガンドも、記載のビタミンおよびその類縁体および誘導体に加えて、標的細胞に結合が可能な薬剤送達結合体を形成するのに使用することが可能である。一般に、細胞表面受容体のリガンドであればいずれのものでも、それに対してリンカー-薬剤結合体を調製可能な標的指向性リガンドとして好適に利用することが可能である。本明細書で考察される例示の、その他のリガンドとしては、ライブラリースクリーニングによって特定されるペプチドリガンド、腫瘍細胞特異的ペプチド、腫瘍細胞特異的アプタマー、腫瘍細胞特異的炭水化物、腫瘍細胞特異的モノクロナール抗体またはポリクロナール抗体、抗体のFabまたはscFv(すなわち、1本鎖可変域)断片、例えば、EphA2、または、転移ガン細胞において特異的に発現される、または、特異的に接近可能なその他の蛋白質に向けられた抗体のFab断片、組み合わせライブラリーから得られた小型有機分子、成長因子、例えば、EGF、FGF、インスリン、およびインスリン様成長因子、および相同ポリペプチド、ソマトスタチンとその類縁体、トランスフェリン、リポ蛋白質複合体、胆汁塩、セレクチン、ステロイドホルモン、Arg-Gly-Asp含有ペプチド、レチノイド、各種ガレクチン、δ-オピオイド受容体リガンド、コレシストキニンA受容体リガンド、アンギオテンシンAT1またはAT2受容体に対して特異的なリガンド、ペルオキシソーム増殖因子活性化受容体λリガンド、β-ラクタム抗生物質、例えばペニシリン、抗菌剤を含む小型有機分子、および、腫瘍細胞または感染性生物の表面に優先的に発現される受容体に特異的に結合するその他の分子、受容体または他の細胞表面蛋白質の結晶構造に基づいてある特定の受容体の結合ポケットにフィットするように設計された抗菌剤およびその他の薬剤、腫瘍細胞上に優先的に発現される腫瘍抗原またはその他の分子のリガンド、または、上記分子の内の任意のものの断片が挙げられる。リガンド-免疫原複合体のための結合部位として機能する可能性のある腫瘍特異的抗原の例としては、Ephrin族蛋白質のメンバー、例えば、EphA2、の細胞外エピトープが挙げられる。EphA2発現は、正常細胞では細胞・細胞接合部に限定されるが、EphA2は、転移腫瘍細胞では全細胞表面に広がる。従って、転移細胞上のEphA2は、例えば、免疫原と複合体化した抗体のFab断片の結合にとって接触可能となるが、一方、この蛋白質は、正常細胞上ではFab断片に対しては接触不能であるから、結果として、リガンド免疫原複合体を転移ガン細胞特異的なものとする。
【0055】
本発明はさらに、除去される病原細胞に対する標的化効果を最大にするための、リガンド-リンカー-薬剤結合体の組み合わせの使用法を考察する。
【0056】
例示の薬剤送達結合体は下記の通りである、すなわち、
V-L-D
V-(lr)c-D
V-(ls)a-D
V-(ls)a-(lr)c-D
V-(lr)c-(ls)a-D
V-(lH)b-(lr)c-D
V-(lr)c-(lH)b-D
V-(lH)d-(lr)c-(lH)e-D
V-(ls)a-(lH)b-(lr)c-D
V-(lr)c-(lH)b-(ls)a-D
V-(lH)d-(ls)a-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(ls)a-(lH)e-D
V-(lH)d-(ls)a-(lH)b-(lr)c-(lH)e-D
V-(lH)d-(lr)c-(lH)b-(ls)a-(lH)e-D
V-(ls)a-(lr)c-(lH)b-D
V-[(ls)a-(lH)b]d-(lr)c-(lH)e-D
式中、a、b、c、d、およびeは、それぞれ独立に、0、1、2、3、または4であり、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカー、および(lr)は放出リンカーであり、Vはビタミン、またはその類縁体または誘導体であり、Dは薬剤、またはその類縁体または誘導体であり、式中、二価のLは、1個の、または、各種の(ls)、(lH)、または(lr)を任意の順序で任意の組み合わせで含む。二価リンカーLの上記例は、二価リンカーの含む、(lH)、(ls)、(lr)の広範な集合を例示することを意図したものであって、限定することを意図したものではないことが理解される。
【0057】
薬剤送達結合体V-L-Dの一つの実施態様では、下記の式に従って、Vはビタミン葉酸であり、Lはエチレンジアミンではない、すなわち、
薬剤送達結合体V-L-Dの別の実施態様では、下記の式に従って、Vはビタミン葉酸であり、Dは薬剤マイトマイシンCであり、LはL-Cys-(S-チオエチル)ではない、すなわち、
Lは、下記の式に従って、L-Asp-L-Arg-L-Asp-L-Cys-(S-チオエチル)ではない、すなわち、
また、Lは、下式に従って、L-Arg-L-Cys-(S-チオエチル)-L-Ala-L-Gly-OHではない、すなわち、
【0058】
さらに本発明に従って考察されるものは、ビタミン、またはその類縁体または誘導体は放出リンカーに付着され、放出リンカーは、スペーサーリンカーを介して薬剤に付着される、薬剤送達結合体である。さらに、薬剤、および、ビタミンまたはその類縁体または誘導体の両方がそれぞれスペーサーリンカーに付着が可能であり、スペーサーリンカー同士は、相互に、放出リンカーを通じて付着される。加えて、薬剤、および、ビタミンまたはその類縁体または誘導体の両方がそれぞれ放出リンカーに付着が可能であり、放出リンカー同士は、相互に、スペーサーリンカーを通じて付着される。ヘテロ原子リンカーは、任意の二つのリンカーの間に、または、任意のリンカーと、ビタミンまたはその類縁体または誘導体の間に、または、任意のリンカーと、薬剤またはその類縁体または誘導体の間に設置してもよい。その他可能な全ての順列と組み合わせが考察される。
【0059】
一つの実施態様では、本発明は、ビタミン受容体結合性薬剤送達結合体を提供する。薬剤送達結合体は、ビタミン受容体結合成分、二価のリンカー(L)、および薬剤から成る。ビタミン受容体結合成分は、ビタミン受容体に結合することが可能なビタミン、またはその類縁体または誘導体であり、薬剤は、薬剤活性を示す類縁体または誘導体を含む。ビタミン、またはその類縁体または誘導体は、二価リンカー(L)に共有結合され、薬剤、またはその類縁体または誘導体も、二価リンカー(L)に共有結合される。二価リンカー(L)は、1個以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、および、順序は任意のそれらの組み合わせを含む。例えば、ヘテロ原子リンカーは窒素であってもよく、放出リンカーとヘテロ原子リンカーはまとめられて二価の基、例えば、アルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルフォキシルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、スルフォニルアルキルアジリジン-1-イル、またはアルキレンスルフォニルアジリジン-1-イルを含む二価の基を形成することが可能であり、各放出リンカーは、要すれば随意に、下記に定義する置換基X2によって置換される。あるいは別に、ヘテロ原子リンカーは、窒素、酸素、硫黄、および、式-(NHR1NHR2)-、-SO-、-(SO2)-、-N(R3)O-であることが可能である。式中、R1、R2、およびR3は、それぞれ独立に、水素、アルキル、アリール、アリールアルキル、アリール置換体、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、および、アルコキシアルキルから選ばれる。別の実施態様では、ヘテロ原子リンカーは酸素であってもよく、スペーサーリンカーは、要すれば随意に下に定義される置換基X1によって置換されてもよい1-アルキレンスクシニミド-3-イルであってもよく、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、スペーサーリンカーと放出リンカーとは、それぞれ、ヘテロ原子リンカーに結合されて、スクシニミド-1-イルアルキルアセタルまたはケタルを形成する。
【0060】
スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イル、アルキレンスルフォキシル、スルフォニルアルキル、アルキレンスルフォキシルアルキル、アルキレンスルフォニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、下に定義される置換基X1によって、要すれば随意に置換されてもよい。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、下に定義される置換基X1によって、要すれば随意に置換されてもよく、スペーサーリンカーは前記窒素に結合してアミドを形成する。あるいは別に、ヘテロ原子リンカーは硫黄であってもよく、スペーサーリンカーはアルキレンおよびシクロアルキレンであってもよく、各スペーサーリンカーは、カルボキシル基によって要すれば随意に置換されてもよく、スペーサーリンカーは前記硫黄に結合してチオールを形成する。別の実施態様では、ヘテロ原子リンカーは硫黄であってもよく、スペーサーリンカーは、1-アルキレンスクシニミド-3-イルおよび1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、スペーサーリンカーは、前記硫黄に結合してスクシニミド-3-イルチオールを形成する。
【0061】
前述の実施態様に対する別態様では、ヘテロ原子リンカーは窒素であってもよく、放出リンカーとヘテロ原子リンカーはまとめられて二価の基、例えば、アルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、またはスルフォニルアルキルアジリジン-1-イルを含む二価の基を形成することが可能であり、各放出リンカーは、要すれば随意に、下記に定義する置換基X2によって置換される。この別態様では、スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、要すれば随意に、下記に定義する置換基X1によって置換され、スペーサーリンカーは放出リンカーに結合してアジリジンアミドを形成する。
【0062】
置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、置換基X1とヘテロ原子リンカーは結合するスペーサーリンカーとまとめられて複素環を形成することも可能である。
【0063】
放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルフォニルオキシ、オキシスルフォニルアルキル、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデニミニル、アルキレンチオ、アルキレンアリールチオ、および、カルボニルアルキルチオであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換される。
【0064】
前述の実施態様では、ヘテロ原子リンカーは酸素であってもよく、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記酸素に結合されてアセタルまたはケタルを形成する。あるいは別に、ヘテロ原子リンカーは酸素であってもよく、放出リンカーはメチレンであってもよく、メチレンは、随意に置換されてもよいアリールによって置換され、放出リンカーは酸素に結合してアセタールまたはケタールを形成する。さらに、ヘテロ原子リンカーは酸素であってもよく、放出リンカーはスルフォニルアルキルであってもよく、放出リンカーは酸素に結合してアルキルスルフォネートを形成する。
【0065】
前述の放出リンカー態様の別態様として、ヘテロ原子リンカーは窒素であってもよく、放出リンカーは、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、および、カルボニルシクロアルキリデニミニルであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記窒素に結合されてヒドラゾンを形成する。別形態として、ヒドラゾンは、カルボン酸誘導体、オルトギ酸塩誘導体、またはカルバモイル誘導体によってアシル化されて、各種アシルヒドラゾン放出リンカーを形成してもよい。
【0066】
別態様として、ヘテロ原子リンカーは酸素であってもよく、放出リンカーは、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、および(ジアリールシリル)アリールであってもよく、各放出リンカーは、下に定義される置換基X2によって、要すれば随意に置換されてもよく、放出リンカーは、前記窒素に結合されてシラノールを形成する。
【0067】
前述の放出リンカー実施態様では、薬剤は窒素原子を含んでもよく、ヘテロ原子リンカーは窒素であってもよく、放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルであってもよく、放出リンカーはヘテロ原子窒素に結合してアミドを形成してもよく、薬剤酸素に結合してアミドを形成してもよい。
【0068】
前述の放出リンカー実施態様では、薬剤は酸素原子を含んでもよく、ヘテロ原子リンカーは窒素でもよく、放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルであってもよく、放出リンカーは、ヘテロ原子リンカー窒素に結合してアミドを形成してもよく、薬剤窒素に結合してエステルを形成してもよい。
【0069】
置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってもよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。この実施態様では、ヘテロ原子リンカーは窒素であってもよく、置換基X2とヘテロ原子リンカーは結合する放出リンカーとまとめられて複素環を形成することも可能である。
【0070】
複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、および、スクシニミドであってもよい。
【0071】
薬剤は、マイトマイシン、マイトマイシン誘導体、またはマイトマイシン類縁体であってもよく、本実施態様では、放出リンカーは、カルボニルアルキルチオ、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシミニド-3-イルであってもよく、各放出リンカーは、置換基X2によって、要すれば随意に置換されてもよく、マイトマイシンのアジリジンは、放出リンカーに結合されてアシルアジリジンを形成する。
【0072】
薬剤は窒素原子を含んでもよく、放出リンカーは、要すれば随意に置換基X2によって置換されるハロアルキレンカルボニルであってもよく、放出リンカーは薬剤窒素に結合してアミドを形成する。
【0073】
薬剤は酸素原子を含んでもよく、放出リンカーは、要すれば随意に置換基X2によって置換されるハロアルキレンカルボニルであってもよく、放出リンカーは薬剤酸素に結合してエステルを形成する。
【0074】
薬剤は、二重結合窒素原子を含んでもよく、本実施態様では、放出リンカーはアルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシニミド-3-イルであってもよく、放出リンカーは薬剤窒素と結合してヒドラゾンを形成することが可能である。
【0075】
薬剤は硫黄原子を含んでもよく、本実施態様では、放出リンカーは、アルキレンチオ、およびカルボニルアルキルチオであってもよく、放出リンカーは薬剤硫黄に結合してジスルフィドを形成することが可能である。
【0076】
ビタミンは、窒素を含む葉酸塩であってもよく、本実施態様では、スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イルであってもよく、各スペーサーリンカーは、置換基X1によって要すれば随意に置換され、スペーサーリンカーは、葉酸窒素に結合してイミドまたはアルキルアミドを形成する。本実施態様では、置換基X1は、アルキル、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、カルボキシ、カルボキシアルキル、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルであってもよく、R4とR5は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立にアミノ酸、アミノ酸誘導体、およびペプチドから選ばれる。
【0077】
本明細書で用いる「アルキル」という用語は、要すれば随意に分枝されてもよい、一価の炭素原子直鎖、例えば、メチル、エチル、プロピル、3-メチルペンチル等を指す。
【0078】
本明細書で用いる「シクロアルキル」という用語は、その一部が環を形成する、一価の炭素原子鎖、例えば、シクロプロピル、シクロヘキシル、3-エチルシクロペンチル等を指す。
【0079】
本明細書で用いる「アルキレン」という用語は、要すれば随意に分枝されてもよい、二価の炭素原子直鎖、例えば、メチレン、エチレン、プロピレン、3-メチルペンチレン等を指す。
【0080】
本明細書で用いる「シクロアルキレン」という用語は、その一部が環を形成する、二価の炭素原子鎖、例えば、シクロプロプ-1,1-ジイル、シクロプロプ-1,2-ジイル、シクロヘクス-1,4-ジイル、3-エチルシクロペント-1,2-ジイル、1-メチレンシクロヘクス-4-イル等を指す。
【0081】
本明細書で用いる「複素環」という用語は、炭素と、窒素、酸素および硫黄から選ばれるヘテロ原子とから成る一価の鎖であって、少なくとも1個のヘテロ原子を含むその一部が環を形成する鎖を指し、例えば、アジリジン、ピロリジン、オキサゾリジン、3-メトキシピロリジン、3-メチルピペラジン等を指す。
【0082】
本明細書で用いる「アルコキシ」という用語は、末端酸素と結合した、本明細書で定義するアルキル、例えば、メトキシ、エトキシ、プロポキシ、3-メチルペントキシ等を指す。
【0083】
「ハロ」または「ハロゲン」という用語は、フルオロ、クロロ、ブロモ、およびイオドを指す。
【0084】
本明細書で用いる「アリール」という用語は、炭素原子から成る、単環または多環芳香族環、例えば、フェニル、ナフチル等を指す。
【0085】
本明細書で用いる「ヘテロアリール」という用語は、炭素原子および、窒素、酸素、および硫黄から選ばれる少なくとも1個のヘテロ原子から成る、単環または多環芳香族環、を指し、例えば、ピリジニル、ピリミジニル、インドリル、ベンゾキサゾリル等を指す。
【0086】
本明細書で用いる「アリール置換体」または「ヘテロアリール置換体」という用語は、ハロ、ヒドロキシ、アミノ、アルキルまたはジアルキルアミノ、アルコキシ、アルキルスルフォニル、シアノ、ニトロ等から選ばれる1種以上の置換基によって置換されるアリールまたはヘテロアリールを指す。
【0087】
本明細書で用いる「イミノアルキリデニル」という用語は、本明細書で定義するアルキレン、および窒素原子を含む二価の基であって、アルキレンの末端炭素が、例えば、式-(CH)=N-、-(CH2)2(CH)=N-、-CH2C(Me)=N-等のように窒素原子と二重結合するものを指す。
【0088】
本明細書で用いる「アミノ酸」という用語は、一般に、アミノアルキルカルボキシレートであって、アルキル基は、要すれば随意にアルキル、ヒドロキシアルキル、スルフヒドリルアルキル、アミノアルキル、カルボキシアルキル等によって置換されてもよい、アミノアルキルカルボキシレートを指し、天然アミノ酸、例えば、セリン、システイン、メチオニン、アスパラギン酸、グルタミン酸等と一致するグループを含む。
【0089】
「アリールアルキル」という用語は、本明細書で定義されるアルキレン基によって置換された、本明細書で定義されるアリールを指し、例えば、ベンジル、フェネチル、α-メチルベンジル等を指す。
【0090】
前述の用語は組み合わせて、化学的関連グループ、例えば、メチルオキシメチル、エチルオキシエチル等を指す「アルコキシアルキル」、および、トリフルオロメチルオキシエチル、1,2-ジフルオロ-2-クロロエト-1-イルオキシプロピル等を指す「ハロアルコキシアルキル」、等を生成することが可能であることを理解しなければならない。
【0091】
本明細書で用いる「アミノ酸誘導体」という用語は、一般に、アミノアルキルカルボキシレートであって、アミノ基またはカルボキシレート基は、それぞれ独立に、アルキル、カルボキシアルキル、アルキルアミノ等によって要すれば随意に置換され、あるいは要すれば随意に保護され、介在する二価のアルキル断片は、アルキル、ヒドロキシアルキル、スルフヒドリルアルキル、アミノアルキル、カルボキシアルキル等によって要すれば随意に置換される、アミノアルキルカルボキシレートを指し、天然アミノ酸に見られる側鎖、例えば、セリン、システイン、メチオニン、アスパラギン酸、グルタミン酸等に見られるものと一致する基を含む。
【0092】
本明細書で用いる「ペプチド」という用語は、一般に、相互にアミド結合によって共有結合された一連のアミノ酸、およびその類縁体および誘導体を指す。
【0093】
放出リンカーは、生理的条件下で破壊または分断が可能な、少なくとも1個の結合(例えば、pH易変性、酸易変性、酸化易変性、または酵素易変性結合)を含む。この分断可能な結合(単数または複数)は、分断リンカーの内部にあってもよいし、および/または、分断リンカーの一端または両端にあってもよい。分断可能結合の易変性は、隣接基支援とも呼ばれる、結合破壊を支援、または促進することが可能な官能基または断片を、二価リンカーLの内部に含ませることによって調節が可能であることが理解される。さらに、放出リンカーの結合破壊の後で、ビタミン受容体結合性薬剤結合体のさらにそれ以上の断片化を支援または促進することが可能な、別の官能基または断片を二価リンカーLの内部に含めてもよいことが理解される。
【0094】
放出リンカーの結合分断の例示の機構として、下記の、オキソニウム支援分断が挙げられる、すなわち、
式中、Zはビタミン、またはその類縁体または誘導体、あるいは、薬剤、またはその類縁体または誘導体、または、それぞれ、二価リンカーの他の部分と結合するビタミンまたは薬剤成分であり、例えば、1種以上のスペーサーリンカー、ヘテロ原子リンカー、および/または、他の放出リンカーを含む、薬剤またはビタミン成分である。本実施態様では、酸触媒によるカルバメート除去によって、CO2とZに付着する窒素含有基の放出、およびベンジル陽イオンの形成がもたらされ、後者は、水、または他の任意のルイス塩基によって捕捉される。
【0095】
放出リンカーに接続され、または、放出リンカーの中に含まれる結合であって、二価リンカーLの一部を形成してもよい結合の、別の分断の例示の機構として、下記の、ベータ除去、およびビニール的ベータ除去機構が挙げられる、すなわち、
式中、Xは求核要素、GSH、グルタチオン、または生体還元因子等であり、ZまたはZ’のどちらかは、ビタミンまたはその類縁体または誘導体、あるいは、薬剤またはその類縁体または誘導体、あるいは、二価リンカーの他の部分と結合するビタミンまたは薬剤成分である。結合分断は、酸触媒によるカルバメート基の除去によっても起こってもよく、カルバメート基の除去は、上述の例で示されたベータ硫黄またはジスルフィドのアリール基によって実現される安定化によって隣接基支援を受けてもよいことが理解される。。本実施態様のこれらの変種では、放出リンカーはカルバメート基である。
【0096】
別の例示の機構は、放出リンカー、スペーサー、および、ヘテロ原子リンカーを適当なやり方で配置することを含む。その配置は、二価リンカーの一つの結合の分断に次いで、放出された官能基がさらに別の結合の破壊または分断を化学的に支援するように行う。後者は、隣接基支援分断または破壊とも呼ばれる。このような二価リンカーまたは部分の例示の実施態様は、下式の化合物を含む、すなわち、
式中、Xはヘテロ原子、例えば、窒素、酸素、または硫黄であり、nは0、1、2および3から選ばれる整数であり、Rは水素、または置換基、例えば、誘電的に、または共鳴によって、アリール環上の陽性電荷を安定化することのできる置換基を含む置換基、例えば、アルコキシ等であり、ZまたはZ’のどちらかは、ビタミンまたはその類縁体または誘導体、あるいは、薬剤またはその類縁体または誘導体、あるいは、二価リンカーの他の部分と結合するビタミンまたは薬剤成分である。アリール環、ベンジル炭素、カルバメート窒素、アルカン酸、またはメチレン架橋には、他の置換基も、例えば、と言ってそれらに限定されるものではないが、ヒドロキシ、アルキル、アルコキシ、アルキルチオ、ハロ等を含む、他の置換基も存在してよいことが理解される。支援による分断としては、ベンジリウム中間体、ベンジン中間体、ラクトン環状化、オキソニウム中間体、ベータ除去等を含む機構が挙げられる。放出リンカーの分断に次ぐ断片化に加えてさらに、放出リンカーの最初の分断が、隣接基支援機構によって促進されてもよいことが理解される。
【0097】
本実施態様では、環状化してもよいヒドロキシアルカン酸は、例えば、オキソニウムイオンによってメチレン架橋の分断を促進するし、また、放出リンカーの結合分断、または、結合分断後の断片化を促進する。あるいは別に、酸触媒オキソニウムイオン支援メチレン架橋分断が、この例示の二価リンカー、またはその断片の、カスケード的に継続する断片化を起動してもよい。あるいは別に、酸触媒による、カルバメートの加水分解が、環状化してもよいヒドロキシアルカン酸のベータ除去を促進してもよいし、また、例えば、オキソニウムイオンによるメチレン架橋の分断を促進してもよい。結合破壊または分断の、その他の化学的機構も、本明細書に記述される代謝的、生理的、または細胞条件下で、このようなカスケード的に継続する断片化を起動してもよいことが理解される。
【0098】
本明細書に記載される薬剤送達結合体は、従来技術で認知済みの合成法によって調製が可能である。合成法は、ヘテロ原子リンカーの選択、および、スペーサーリンカーおよび放出リンカーに存在する官能基に応じて選ばれる。一般に、関連する結合形成反応は、Richard C. Larock、「有機変換総論、官能基調製入門(”Comprehensive Organic Transformations, a guide to functional group preparations”)」、VCH Publishers, Inc.ニューヨーク(1989)、および、Theodora E. Greene & Peter G.M. Wuts, 「有機合成における保護基(”Protective Groups in Organic Synthesis”)」、第2版、John Wiley & Sons, Inc.ニューヨーク(1991)に記載されている。この開示を、引用することにより本明細書に含める。
【0099】
===アミドおよびエステルの一般的形成===
例えば、ヘテロ原子リンカーが窒素原子で、スペーサーリンカーまたは放出リンカーに存在する末端官能基がカルボニル基である場合、必要なアミド基は、対応するカルボン酸またはその誘導体と、アミンとの結合反応またはアシル化反応によって、例えば、スキーム1に図示するようにして実現が可能である。式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフレート、ペンタフルオロフェノキシ、トリメチルシリルオキシ、スクシニミド-N-オキシ等である。
スキーム1
【0100】
結合試薬としては、DCC、EDC、RRDQ、CGI、HBTU、TBTU、HOBT/DCC、HOBT/EDC、BOP-C1、PyBOP、PyBroP等が挙げられる。あるいは別に、親酸は、活性化カルボニル誘導体、例えば、酸塩化物、N-ヒドロキシスクシニミジルエステル、ペンタフルオロフェニルエステル等に変換されてもよい。このアミド形成反応は、塩基、例えば、トリエチルアミン、ジイソプロピルエチルアミン、N,N-ジメチル-4-アミノピリジン等の存在下に実行することも可能である。本明細書に記載されるアミドを形成するのに好適な溶媒としては、CH2Cl2、CHCl3、THF、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。例示として、アミドは、約-15℃から約80℃、または約0℃から約45℃の範囲の温度で調製することが可能である。アミドは、例えば、窒素含有アジリジン環、炭水化物、およびα-ハロゲン化カルボン酸から形成することが可能である。アミドを形成するのに好適な例示のカルボン酸として下記の式を持つ化合物等が挙げられる、すなわち、
等で、式中、nは、1、2、3または4のような整数である。
【0101】
同様に、ヘテロ原子リンカーは酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する末端官能基はカルボニル基である場合には、必要なエステル基は、対応するカルボン酸または誘導体と、アルコールとの結合反応によって得ることが可能である。
【0102】
結合試薬としては、DCC、EDC、CDI、BOP、PyBOP、イソプロペニルクロロギ酸、EEDQ、DEAD、PPh3等が挙げられる。溶媒としては、CH2Cl2、CHCl3、THF、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。塩基としては、トリエチルアミン、ジイソプロピル-エチルアミン、および、N,N-ジメチル-4-アミノピリジンが挙げられる。あるいは別に、親酸は、活性化カルボニル誘導体、例えば、酸塩化物、N-ヒドロキシスクシニミジルエステル、ペンタフルオロフェニルエステル等に変換されてもよい。
【0103】
===ケタールおよびアセタールの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子で、スペーサーリンカーまたは放出リンカーに存在する官能基が1-アルコキシアルキルである場合、必要なアセタールまたはケタール基は、スキーム2に示すように、対応するアルコールとエノールエーテルとのケタールおよびアセタール形成反応によって形成することが可能である。
スキーム2
【0104】
溶媒としては、アルコール、CH2Cl2、CHCl3、THF、ジエチルエーテル、DMF、DMSO、アセトニトリル、EtOAc等が挙げられる。このようなアセタールおよびケタールの形成は、酸触媒によって実現することが可能である。ヘテロ原子リンカーが二つの酸素原子を含み、放出リンカーが、要すれば随意に本明細書で記載するX2基で置換されてよいメチレンである場合、必要な対称型アセタールまたはケタール基は、例示として、スキーム3で示すように、対応するアルコールとアルデヒドまたはケトンによるアセタールおよびケタール形成反応によって形成することが可能である。
スキーム3
【0105】
あるいは別に、メチレンが、随意に置換されてよいアリール基で置換されている場合、必要なアセタールまたはケタールは、スキーム4に図示するように段階的に調製されてもよく、式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフルオロアセトキシ、トリフレート等である。スキーム4に示される過程は、従来型の調製法であり、一般的にR.R. Schmidt et al., Chem. Rev., 2000, 100, 4423-42によって総説される過程に従う。なお、この開示を引用することにより本明細書に含める。
スキーム4
【0106】
得られたアリールアルキルエーテルは、酸化剤、例えば、DDQ等によって処理され、中間体オキソニウムイオンを形成し、このイオンは次に別のアルコールによって処理されアセタールまたはケタールを生成する。
【0107】
===スクシニミドの一般的形成===
さらに、ヘテロ原子リンカーが、例えば、窒素、酸素、または硫黄原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がスクシニミド誘導体である場合、得られる炭素ヘテロ原子結合は、スキーム5に示すように、対応するアミン、アルコール、またはチオールと、マレイミド誘導体とのマイケル付加によって形成が可能であり、式中、Xは、ヘテロ原子リンカーである。
スキーム5
【0108】
マイケル添加を実行するための溶媒としては、THF、EtOAc、CH2Cl2、DMF、DMSO、H2O等が挙げられる。このようなマイケル付加物の形成は、等モル量の塩基、例えば、トリエチルアミン、ヒューニッヒ塩基(Hunig’s base)を添加することによって、または、水溶液のpHを6.0-7.4に調節することによって実現することが可能である。ヘテロ原子リンカーが酸素または窒素原子である場合、マイケル添加を促進するように、例えば、より高い反応温度を用いる、触媒を添加する、より極性の高い溶媒、例えば、DMF、DMSO等を用いる、マレイミドをシリル化試薬で活性化すること等によって、反応条件を調整してもよいことが理解される。
【0109】
===シリルオキシの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がシリル誘導体である場合、必要なシリルオキシ基は、対応するシリル誘導体とアルコールとを反応させることによって、例えば、スキーム6に図示するようにして生成してもよい。式中、Lは、好適に選ばれた脱離基、例えば、ハロ、トリフルオロアセトキシ、トリフレート等である。
スキーム6
【0110】
シリル誘導体は、好適に官能化されたシリル誘導体、例えば、ビニールスルフォノアルキルジアリール、またはジアリール、またはアルキルアリールシリルクロリドを含む。ビニールスルフォノアルキル基の代わりに、β-クロロエチルスルフォノアルキル前駆物質を用いてもよい。非プロトン性の無水溶媒と窒素含有塩基であれば、いずれのものでも反応溶媒として使用が可能である。この変換に採用が可能な温度範囲は、-78℃と80℃の間を変動してもよい。
【0111】
===ヒドラゾンの一般的形成===
さらに、ヘテロ原子リンカーが窒素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がイミニル誘導体である場合、必要なヒドラゾン基は、スキーム7のそれぞれ式(1)と(2)に示されるように、対応するアルデヒドまたはケトンと、ヒドラジンまたはアシルヒドラジン誘導体とを反応させることによって形成することが可能である。
スキーム7
【0112】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO、MeOH等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。任意の酸性触媒、例えば、鉱酸、H3CCOOH、F3CCOOH、p-TsOH・H2O、ピリジニウムp-トルエンスルフォネート等を使用することが可能である。式(2)のアシルヒドラゾンの場合、アシルヒドラゾンは、スキーム1で一般的に上述したように、最初ヒドラジンを、適当なカルボン酸または誘導体でアシル化し、その後アシルヒドラジドを、対応するアルデヒドまたはケトンと反応させて、アシルヒドラゾンを形成してもよい。あるいは別に、ヒドラゾン官能基を、最初に、ヒドラジンを対応するアルデヒドまたはケトンと反応させることによって形成してもよい。得られたヒドラゾンは、次に、スキーム1で一般的に上述したように、適当なカルボン酸または誘導体によってアシル化してもよい。
【0113】
===ジスルフィドの一般的形成===
さらに、ヘテロ原子リンカーが硫黄原子であり、放出リンカーに存在する官能基がアルキレンチオール誘導体である場合、必要なジスルフィド基は、スキーム8に示されるように、対応するアルキルまたはアリールスルフォニルチオアルキル誘導体、あるいは、対応するヘテロアリールジチオアルキル誘導体、例えば、ピリジン-2-イルジチオアルキル誘導体等と、アルキレンチオール誘導体とを反応させることによって形成することが可能である。
スキーム8
【0114】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。必要なアルキルまたはアリールスルフォニルチオアルキル誘導体は、従来技術で認知済みのプロトコールを用いて、また、Ranasinghe & Fuchs, Synth. Commun. 18(3), 227-32 (1988)の方法に従って調製してもよい。なお、上記開示を、引用することにより本明細書に含める。非対称のジアルキルジスルフィドのその他の調製法は、国際特許出願WO88/01622、欧州特許出願0116208A1、および米国特許第4,691,024号に記載されているように、非対称型のヘテロアリール-アルキルジスルフィド、例えば、2-チオピリジニル、3-ニトロ-2-チオピリジニル等のジスルフィドに対する、アルキルチオールによるチオール変換にもとづく。なお、上記開示を、引用することにより本明細書に含める。
【0115】
===炭酸塩の一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がアルコキシカルボニル誘導体である場合、必要な炭酸基は、スキーム9に示されるように、対応するヒドロキシ置換化合物を、活性化アルコキシカルボニル誘導体と反応させることによって形成することが可能である。式中、Lは適当な脱離基である。
スキーム9
【0116】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。反応を促進するために、任意の塩基性触媒、例えば、無機塩基、アミン塩基、ポリマー結合塩基等を使用することが可能である。
【0117】
===セミカルバゾンの一般的形成===
さらに、ヘテロ原子リンカーが窒素原子であり、一方のスペーサーリンカーまたは放出リンカーに存在する官能基がイミニル誘導体であり、他方のスペーサーリンカーまたは他方の放出リンカーに存在する官能基がアルキルアミノまたはアリールアミノカルボニル誘導体である場合、必要なセミカルバゾン基は、スキーム10に示されるように、対応するアルデヒドまたはケトンと、セミカルバジドとを反応させることによって形成することが可能である。
スキーム10
【0118】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4、DMF、DMSO、MeOH等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。任意の酸性触媒、例えば、鉱酸、H3CCOOH、F3CCOOH、p-TsOH・H2O、ピリジニウムp-トルエンスルフォネート等を使用することが可能である。さらに、セミカルバゾンを形成するに際して、ヒドラゾン官能基を、最初に、ヒドラジンを対応するアルデヒドまたはケトンと反応させることによって形成してもよい。得られたヒドラゾンは、次に、イソシアネート、またはカルバモイル誘導体、例えば、カルバモイルハロゲン化物でアシル化することによってセミカルバゾンを形成してもよい。あるいは別に、対応するセミカルバジドを、ヒドラジンを、イソシアネート、またはカルバモイル誘導体、例えば、カルバモイルハロゲン化物と反応させることによって形成してもよい。次に、このセミカルバジドを、対応するアルデヒドまたはケトンと反応させてセミカルバゾンを形成してもよい。
【0119】
===スルフォネートの一般的形成===
さらに、ヘテロ原子リンカーが酸素原子であり、スペーサーリンカーまたは放出リンカーに存在する官能基がスルフォニル誘導体である場合、必要なスルフォネート基は、スキーム11に示されるように、対応するヒドロキシ置換化合物と、活性化スルフォニル誘導体とを反応させることによって形成することが可能である。式中、Lは適当な脱離基、例えば、ハロ等である。
スキーム11
【0120】
使用が可能な溶媒としては、THF、EtOAc、CH2Cl2、CHCl3、CCl4等が挙げられる。この変換に採用される温度範囲は、0℃と80℃の間を変動してもよい。反応を促進するために、任意の塩基性触媒、例えば、無機塩基、アミン塩基、ポリマー結合塩基等を使用することが可能である。
【0121】
===葉酸ペプチドの一般的形成===
葉酸含有ペプチジル断片Pte-Glu-(AA)n-NH(CHR2)CO2H(3)は、スキーム12に示すように、標準法によるポリマー支持連続法、例えば、酸感受性Fmoc-AA-Wang樹脂(1)上でFmoc策を実施することによって調製される。
スキーム12
(a)20%ピペリジン/DMF;(b)Fmoc-AA-OH、PyBop、DIPEA、DMF;(c)Fmoc-Glu(O-t-Bu)-OH、PyBop、DIPEA、DMF;(d)1.N10(TFA)-Pte-OH;PyBop、DIPEA、DMSO;(e)TFAA、(CH2SH)2、i-Pr3SiH;(f)NH4OH、pH10.3
【0122】
この例示の過程に関する実施態様では、R1はFmocであり、R2は、適当に保護された所望のアミノ酸側鎖であり、DIPEAはジイソプロピルエチルアミンである。効率的な結合を確保するために、結合剤、例えば、PyBOP、および、本明細書で記載される、または従来技術で既知の、その他の結合剤が活性化試薬として与えられる場合、標準的な結合工程が用いられる。Fmoc保護基は、各結合工程後、標準条件下で、例えば、ピペリジン、テトラブチルアンモニウムフロリド(TBAF)等による処理によって除去される。適当に保護されたアミノ酸建設ブロック、例えば、Fmoc-Glu-OtBu、N10-TFA-Pte-OH等は、スキーム12で記載し、工程(b)でFmoc-AA-OHで示されるように用いられる。従って、AAは、適当に保護された、任意のアミノ酸の開始材料を指す。本明細書で用いられるアミノ酸という用語は、アミンとカルボン酸官能基が、1個以上の炭素で隔てられた任意の試薬を指し、天然のアルファーおよびベータアミノ酸を始め、それらアミノ酸のアミノ酸誘導体および類縁体を含むことを理解しなければならない。特に、保護された側鎖を有するアミノ酸、例えば、保護されたセリン、トレオニン、システイン、アスパラギン酸等も、本明細書に記載する葉酸-ペプチド合成に使用してよい。さらに、ガンマ、デルタ、またはさらに長い相同のアミノ酸もまた本明細書に記載する葉酸‐ペプチド合成の開始材料に含めてもよい。さらに、相同側鎖を有する、または、交互分枝構造を有するアミノ酸類縁体、例えば、ノルロイシン、イソバリン、β-メチルスレオニン、β-メチルシステイン、β、β-ジメチルシステイン等を、本明細書に記載する葉酸-ペプチド合成において開始材料に含めてもよい。
【0123】
Fmoc-AA-OHを含む結合順序(工程(a)と(b))は、”n”回実行されて、固相支持体ペプチド2を調製する。nは整数であり、0から約100に等しくともよい。最後の結合工程後、残余のFmoc基は除去され(工程(a))、このペプチドは、順次、グルタミン酸誘導体に結合され(工程(c))、脱保護され、TFA保護プテロイン酸に結合される(工程(d))。次に、このペプチドは、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランによる処理によってポリマー支持体から分離される(工程(e))。これらの反応条件によって、適当に保護されるアミノ酸側鎖の一部を形成してもよいt-Bu、t-Boc、およびTrt保護基の同時除去が実現される。TFA保護基が、塩基による処理によって除去されて(工程(f))、葉酸含有ペプチジル断片3を与える。
【0124】
スペーサーおよび放出リンカー、およびヘテロ原子リンカーは様々なやり方で組み合わせることが可能である。例示として、リンカー同士は、互いに、ヘテロ原子リンカーを介して付着される、例えば、下式で示されるように、アルキレン-アミノ-アルキレンカルボニル、アルキレン-チオ-カルボニルアルキルスクシニミド-3-イル等のようである。式中、整数xとyは1、2、3、4、または5である。すなわち、
【0125】
本明細書に記載されるリンカーの別の例示の実施態様は、本明細書に記載される条件下で、ベータ除去の関与する化学機構によって分断される放出リンカーを含む。一つの局面で、このような放出リンカーは、ベータ-チオ、ベータ-ヒドロキシ、および、ベータ-アミノ置換カルボン酸、およびその誘導体、例えば、エステル、アミド、炭酸塩、カルバメート、および尿素を含む。別の局面では、このような放出リンカーは、2-および4-チオアリールエステル、カルバメート、および炭酸塩を含む。
【0126】
さらに、ビタミンまたは薬剤の、ヘテロ原子リンカーに対する付着は、下式に示されるように、ヘテロ原子リンカーにあらかじめ変換された、例えば、アクラマイシンケトンの対応するヒドラゾンへの変換、葉酸の対応するアミドへの変換等によって生じた、薬剤またはビタミン上の反応性官能基を介して実現することが可能である。すなわち、
【0127】
二価リンカー(L)は、スペーサーリンカー、放出リンカー、ヘテロ原子リンカー、および、順序は任意のそれらの組み合わせから選ばれる、1種以上の成分を含む。例えば、表1および2に示す、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせが考えられる。これらのリンカーリストは包括的なものではなく、単に例示的なものであり、本明細書に記載される発明を限定するものと解釈してはならない。前述の構造に存在する星印は、表1および2のものも含めて、追加スペーサー、放出またはヘテロ原子リンカーの、または、ビタミン受容体結合性薬剤送達結合体の薬剤またはビタミン成分の、例示の付着点を特定する。二価リンカーLは、表1および2に示したものを含めた1種以上のスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーを含むこと、および、このスペーサーリンカー、放出リンカー、およびヘテロ原子リンカーは任意の順序で組み合わせて二価リンカーLを形成することが可能であることが理解される。
【0128】
<表1>
リンカー、および、いくつかのスペーサーおよびヘテロ原子リンカーの組み合わせの考察例
【0129】
<表2>
リンカー、および、いくつかの放出およびヘテロ原子リンカーの組み合わせの考察例
【0130】
本発明の薬剤送達結合体は、中間体から製造することも可能である。一つの実施態様では、下式:
V−L−Z1
の化合物の製造が可能である。式中、Z1は、薬剤、またはその類縁体または誘導体の付着を促進するのに好適な、求電子要素、求核要素、または前駆物質である。
【0131】
一つの局面では、Z1は、薬剤またはその類縁体または誘導体に存在する求核残基を介して薬剤の付着を可能とする脱離基、例えば、窒素等のヘテロ原子であってもよい。
【0132】
別の局面では、Z1は、薬剤またはその類縁体または誘導体に存在する脱離基、例えば、酸塩化物等のカルボン酸誘導体等、を移動させることが可能な求核要素、例えば、窒素等のヘテロ原子であってもよい。
【0133】
別の局面では、Z1は、前駆物質、例えば、還元反応を通じて求核性窒素に加工することが可能なニトロ基や、加水分解と塩素化の順次反応によって求電子性酸塩化物に加工することが可能なエステル等、であってもよい。Z1は、ヘテロ原子リンカーであってもよいことを理解しなければならない。
【0134】
別の実施態様では、薬剤送達結合体は、下記のような中間体から製造することが可能である。すなわち、
Z2−L−D
式中、Z2は、ビタミン、またはその類縁体または誘導体の付着を促進するのに好適な、求電子要素、求核要素、または前駆物質である。
【0135】
一つの局面では、Z1は、ビタミンまたはその類縁体または誘導体に存在する求核残基を介してビタミンの付着を可能とする脱離基、例えば、窒素等のヘテロ原子であってもよい。
【0136】
別の局面では、Z1は、ビタミンまたはその類縁体または誘導体に存在する脱離基、例えば、酸塩化物等のカルボン酸誘導体、を移動させることが可能な求核要素、例えば、窒素等のヘテロ原子であってもよい。
【0137】
別の局面では、Z1は、前駆物質、例えば、還元反応を通じて求核性窒素に加工することが可能なニトロ基や、加水分解と塩素化の順次反応によって求電子性酸塩化物に加工することが可能なエステル等、であってもよい。Z2は、ヘテロ原子リンカーであってもよいことを理解しなければならない。
【0138】
別の実施態様では、二価リンカー(L)は、別に合成され、次に、例えば、下式のような化合物を中間体として調製することによって、後続工程においてビタミンと薬剤に付着されてもよい。すなわち、
Z1−L−Z2
式中、Z1とZ2はそれぞれ独立に選択され、上に定義した通りである。
【0139】
本発明の薬剤送達結合体は、ヒトの臨床医学と獣医学応用の両方のために使用することが可能である。従って、病原細胞集団を抱え、本ビタミン受容体結合性薬剤送達結合体によって治療される宿主動物は、ヒトであってもよいし、獣医学応用の場合は、実験動物、農業動物、家畜動物、または野生動物であってもよい。本発明は、宿主動物、例えば、と言ってそれらに限定されるわけではないが、ヒト、実験動物、例えば、げっ歯類(例えば、マウス、ラット、ハムスター等)、ウサギ、サル、チンパンジー等、家畜動物、例えば、イヌ、ネコ、およびウサギ等、農業動物、例えば、ウシ、ウマ、ブタ、ヒツジ、ヤギ等、および、拘束された野生動物、例えば、クマ、パンダ、ライオン、トラ、ヒョウ、ゾウ、シマウマ、キリン、ゴリラ、イルカ、およびクジラ等、を含む宿主動物、に施すことが可能である。
【0140】
本発明は、上記宿主動物において各種の病態を招く病原細胞集団に対して適用することが可能である。本発明によれば、「病原細胞」とは、ガン細胞、感染病媒介因子、例えば、細菌およびウィルス、細菌またはウィルス感染細胞、病的状態をもたらすことが可能な活性化マクロファージ、およびその他の、ビタミン受容体、または、ビタミンの類縁体または誘導体に結合する受容体を、特異的に発現する、優先的に発現する、または過剰発現する、任意のタイプの病原細胞を意味する。病原細胞はまた、本発明のビタミン受容体結合性薬剤送達結合体による治療が、その細胞の引き起こす病気の症状の寛解をもたらす、そのような病気の原因細胞全てを含むことが可能である。例えば、病原細胞は、ある状況下では病原性となる宿主細胞、例えば、移植片対宿主疾患の原因とはなるが、別の状況下では病原性ではない免疫系細胞、であることも可能である。
【0141】
従って、病原細胞集団は、腫瘍形成性、例えば、良性腫瘍および悪性腫瘍を含む腫瘍形成性のガン細胞集団であってもよいし、または、非腫瘍形成性であってもよい。ガン細胞集団は自発性に生じてもよいし、あるいは、宿主動物の生殖細胞系に存在する突然変異または体細胞突然変異のような過程によって生じてもよいし、あるいは、化学的、ウィルス的、または放射線誘発的に生じてもよい。本発明は、ガン、例えば、上皮ガン、肉腫、リンパ腫、ホジキン病、メラノーマ、中皮腫、バーキットリンパ腫、鼻咽頭ガン、白血病、および骨髄腫、を治療するのに利用が可能である。ガン細胞集団としては、口腔、甲状腺、内分泌腺、皮膚、胃、食道、喉頭、膵臓、結腸、膀胱、骨、卵巣、子宮頸部、子宮、乳房、睾丸、前立腺、直腸、腎臓、肝臓、および、肺の各ガンが挙げられるが、ただしそれらに限定されない。
【0142】
病原細胞集団がガン細胞集団である場合、結合体投与の効果は、腫瘍塊の減少または除去、あるいは、腫瘍細胞増殖の抑制によって測定される治療反応である。腫瘍の場合、除去とは、原発腫瘍細胞の除去、あるいは、転移した、または、原発腫瘍から解離する過程にある細胞の除去であることが可能である。何かの治療法、例えば、腫瘍の外科切除、放射線治療、化学療法、または生物学療法等、によって腫瘍を除去した後に腫瘍の復帰を防止するための、ビタミン受容体結合性薬剤送達結合体による予防療法も、本発明において考慮されている。予防療法は、薬剤送達結合体による初回治療、例えば、1日当たり複数回投与スケジュールの治療であってもよいし、および/または、初回の治療(単数または複数)後、数日または数ヶ月の間隔を置いて為される1回の追加治療または一連の治療であってもよい。従って、本発明に従って治療される病原細胞集団の除去は、病原細胞数の減少、病原細胞増殖の抑制、病原細胞復帰を防止する予防療法、病気の症状の低下をもたらす病原細胞の処置を含む。
【0143】
ガン細胞が除去されつつある場合、本発明の方法は、腫瘍の外科的切除、放射線療法、化学療法、または生物学的療法、例えば、他の免疫療法、例えば、と言ってそれらに限定されないが、モノクロナール抗体療法、免疫修飾剤による治療、免疫エフェクター細胞の養子移入、造血成長因子、サイトカインによる治療、および、ワクチン療法等を含む免疫療法等、と組み合わせて使用することが可能である。
【0144】
本発明はまた、各種感染症を引き起こす病原細胞集団に適用することも可能である。例えば、本発明は、病原細胞集団、例えば、細菌、酵母を含む真菌、ウィルス、ウィルス感染細胞、マイコプラズマ、および寄生生物に対して適用が可能である。本発明の薬剤送達結合体によって治療が可能な感染性生物は、動物に病原性をもたらすものであれば、従来技術で認知済みの任意の感染性生物、例えば、グラム陰性またはグラム陽性の球菌または桿菌である細菌である。例えば、プロテウス種、クレブシエラ種、プロビデンシア種、イェルシニア種、エルウィニア種、エンテロバクター種、サルモネラ種、セラチア種、アエロバクター種、エシェリキア種、シュードモナス種、シゲラ種、ビブリオ種、アエロモナス種、カンピロバクター種、ストレプトコッカス種、スタフィロコッカス種、ラクトバチルス種、ミクロコッカス種、モラキセラ種、バチルス種、クロストリジウム種、コリネバクテリウム種、エベルセラ種、ミクロコッカス種、マイコバクテリウム種、ナイセリア種、ヘモフィルス種、バクテロイデス種、リステリア種、エリシペロトリックス種、アシネトバクター種、ブルセラ種、パスツレラ種、ビブリオ種、フラボバクテリウム種、フソバクテリウム種、ストレプトバチルス種、カリマトバクテリウム種、レジオネラ種、トレポネーマ種、ボレリア種、レプトスピラ種、アクチノミセス種、ノカルジア種、リケッチア種、および、宿主に病気を起こす、その他の任意の細菌種は、本発明の薬剤送達結合体によって治療することが可能である。
【0145】
特に関心があるのは、抗生物質に対して耐性を持つ細菌、例えば、抗生物質耐性ストレプトコッカス種およびスタフィロコッカス種、あるいは、抗生物質に対して感受性を持つが、抗生物質で治療されると繰り返し感染を招き、最終的には耐性微生物を生ずるに至る細菌である。抗生物質に対して感受性を持つが、抗生物質で治療されると繰り返し感染を招き、最終的には耐性微生物を生ずるに至る細菌は、抗生物質の不在下に、または、抗生物質耐性細菌株の発生を回避するために、患者に通常投与されるものよりも低い用量の抗生物質と組み合わせて、本発明の薬剤送達結合体によって治療することが可能である。
【0146】
ウィルス、例えば、DNAおよびRNAウィルスも、本発明に従って処置することが可能である。そのようなウィルスとしては、例えば、と言ってそれらに限定されないが、DNAウィルス、例えば、パピローマウィルス、パルボウィルス、アデノウィルス、ヘルペスウィルス、および、ワクシニアウィルス等、および、RNAウィルス、例えば、アレナウィルス、コロナウィルス、ライノウィルス、呼吸器系合胞体ウィルス、インフルエンザウィルス、ピコルナウィルス、パラミクソウィルス、レオウィルス、レトロウィルス、レンチウィルス、およびラブドウィルス等、が挙げられる。
【0147】
本発明はまた、酵母を含む任意の真菌、マイコプラズマ種、寄生生物、またはその他の、動物に病気を引き起こす感染性微生物に対して適用が可能である。本発明の方法および組成物によって治療することが可能な真菌の例としては、例えば、カビのように成長する真菌、または酵母様真菌であって、例えば、病気、例えば、白癬、ヒストプラスマ症、ブラストミセス症、アスペルギルス症、クリプトコックス症、スポロトリクス症、コクシジオイディス真菌症、パラコクシジオイディス真菌症、ムコール菌症、クロモブラストミコーシス、皮膚糸状菌症、プロトテコーシス、フザリオーシス、ひこう疹、菌腫、パラコクシジオイディス真菌症、フェオフィホ真菌症、シュードアレシェリア症、スポロトリクス症、砂毛症、ニューモシスティス感染、および、カンジダ症を引き起こす真菌である。
【0148】
本発明はまた、寄生生物感染、例えば、と言ってそれらに限定されないが、条虫類、例えば、サナダムシ、膜様条虫、裂頭条虫、およびエキノコッカス種、吸虫類、例えば、肥大吸虫、異形吸虫、メタゴニムス、肝吸虫、ファスキオラ、肺吸虫、および住血吸虫種、回虫類、例えば、蟯虫、鞭虫、回虫、鉤虫、アメリカ鉤虫、円虫、旋毛虫、糸状虫、ブルギア、ロアオンコセルカ、およびドラクンクルス種、アメーバ類、例えば、ネグレリア、およびアカントアメーバ種、および、原虫類、例えば、プラスモディウム、トリパノソーマ、リーシュマニア、トキソプラスマ、エントアメーバ、ジアルジア、イソスポラ、クリプトスポリジウム、およびエンテルシトゾーン種、によって引き起こされる感染を治療するのに利用することが可能である。
【0149】
本発明の薬剤送達結合体が向けられる病原細胞は、それらの細胞が優先的にビタミン受容体を発現するのであれば、内在性病原体を抱える細胞、例えば、ウィルス、マイコプラズマ、寄生生物、または細菌によって感染された細胞であってもよい。
【0150】
一つの実施態様では、ビタミン受容体結合性薬剤送達結合体は、そのビタミン成分が、ビタミンに対して特異的に結合し、病原細胞において優先的に発現されるビタミン受容体、トランスポーター、またはその他の表面発現蛋白質に結合して、標的病原細胞の内部に取り込まれることがあり得る。このような内部への取り込みは、例えば、受容体介在性エンドサイトーシスによって起こることが可能である。薬剤送達結合体が放出リンカーを含む場合、ビタミン成分と薬剤が細胞内で解離し、薬剤が細胞内標的に作用することが可能となる。
【0151】
別の実施態様では、薬剤送達結合体のビタミン成分は、病原細胞に結合し、薬剤を、その病原細胞表面に近接させることが可能である。次に、薬剤は、放出リンカーの分断によって放出させることが可能である。例えば、放出リンカーがジスルフィド基である場合、薬剤は、ジスルフィドイソメラーゼ蛋白質によって放出することが可能である。次に、この薬剤は、ビタミン受容体結合性薬剤送達結合体が結合した病原細胞によって摂取されるか、あるいは、薬剤は、それに近接した別の細胞によって摂取されることが可能である。あるいは別に、放出リンカーがジスルフィド基である場合、薬剤は、細胞内部でジスルフィドイソメラーゼ蛋白質によって放出されることが可能である。薬剤はまた、あるベータ除去機構に関連して上述したように、加水分解機構、例えば、酸触媒加水分解によって放出してもよいし、あるいは、オキソニウムイオンまたはラクトニウムイオン産生機構を介する隣接基支援分断によって放出されてもよい。放出リンカー(単数または複数)の選択に応じて、薬剤が結合体から解離される機構が決められる。このような選択は、薬剤結合体の使用が予想される条件によってあらかじめ定義することが可能であることが理解される。
【0152】
別の実施態様で、リンカーが放出リンカーを含まない場合、薬剤送達結合体のビタミン成分は、病原細胞に結合し、薬剤を病原細胞の表面に設置し、薬剤に結合が可能な他の分子による攻撃に備えて病原細胞を標的化することが可能である。あるいは別に、この実施態様では薬剤送達結合体は、結合後に標的細胞に取り込まれ、ビタミン成分と薬剤は細胞内で会合を持続し、薬剤は、ビタミン成分から解離すること無しにその効果を発揮することも可能である。
【0153】
さらに別の実施態様で、または、上述の実施態様と組み合わせて、ビタミン受容体結合性薬剤送達結合体は、細胞のビタミン受容体とは独立した機構を通じて作用することが可能である。例えば、薬剤送達結合体は、血清中に存在する可溶性ビタミン受容体、または、血清タンパク、例えば、アルブミンに結合し、結果として、未結合の薬剤よりも長期の結合体の循環を実現し、病原細胞集団に対して未結合の薬剤よりも高い結合体の活性をもたらすことができる。
【0154】
本発明の別の実施態様では、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体が提供される。Lは、(ls)aおよび(lH)b、およびそれらの組み合わせから選ばれ、(ls)a、(lH)bおよびVは本明細書に定義する通りであり、Dは、免疫原物質のような薬剤である。免疫原物質は、ハプテン、例えば、フルオレセイン、ジニトロフェニル等であってもよい。本実施態様では、ビタミン受容体結合性薬剤送達結合体は、病原細胞の表面に結合し、その細胞を免疫原物質によって「標識し」、その標識された病原細胞集団に向けられた免疫反応を起動する。受動免疫化において宿主に投与された抗体、または、あらかじめ存在する先天的または後天的免疫によって宿主の中に存在する抗体が、免疫原物質に結合して、内在的免疫反応を起動する。細胞に結合したビタミン−免疫原物質複合体への抗体の結合は、結果として、補体介在性細胞傷害、抗体依存性細胞介在性細胞傷害、抗体オプソニン化と食作用、細胞死または不活性化信号を伝える抗体誘発性受容体凝集、または、細胞に結合したリガンド免疫原複合体への抗体結合によって刺激される、その他の任意の体液性または細胞性免疫反応をもたらす。免疫原物質が、事前の抗体オプソニン化無しに、免疫細胞によって直接認識されることが可能な場合、病原細胞の直接殺戮が起こる可能性がある。この実施態様は、米国特許出願第09/822,379号にさらに精しく記載されており、この出願を引用することにより本明細書に含める。薬剤が免疫原物質である本実施態様のある変種では、二価リンカーは、上述のように放出リンカー、例えば、一般式V-L-Dのビタミン受容体結合性薬剤送達結合体を含んでもよいことが理解される。式中、Lは、(ls)a、(lH)b、(lr)c、およびそれらの組み合わせから選ばれ、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカー、(lr)は放出リンカーであり、Vは、ビタミン、またはその類縁体または誘導体であり、a、bおよびcは整数である。
【0155】
本明細書に記載されるビタミン受容体結合性薬剤送達結合体は、ビタミン受容体結合性成分、二価リンカー(L)、薬剤、および、要すれば随意に、ビタミン受容体結合性成分と薬剤とを二価リンカー(L)に連結するヘテロ原子リンカーを含む。二価リンカー(L)は、スペーサーリンカー、放出(すなわち分断可能)リンカー、およびヘテロ原子リンカー、またはそれらの組み合わせを含むことが可能である。
【0156】
本明細書に記載されるビタミン受容体結合性薬剤送達結合体は、多種多様なビタミン、または受容体結合性ビタミン類縁体/誘導体、リンカー、および薬剤から形成することが可能である。本発明の薬剤送達結合体は、病原細胞では、ビタミン結合が可能な、ビタミン受容体が優先的に発現されるために、宿主動物における病原細胞集団を選択的に標的化することが可能である。例示のビタミン成分としては、カルニチン、イノシトール、リポ酸、ピリドキサル、アスコルビン酸、ナイアシン、パントテン酸、葉酸、リボフラビン、チアミン、ビオチン、ビタミンB12、および、脂溶性ビタミンA、D、EおよびKが挙げられる。これらのビタミン、および受容体結合性類縁体および誘導体は、二価リンカー(L)によって薬剤に結合される標的体を構成し、本明細書に記載されるビタミン受容体結合性薬剤送達結合体を形成する。従って、「ビタミン」という用語は、ビタミン類縁体および/または誘導体(例えば、葉酸の誘導体であるプテロイン酸、ビオチン類縁体、例えば、ビオシチン、ビオチンスルフォキシド、オキシビオチン、およびその他のビオチン受容体結合性化合物等)を含む。本発明によれば、ビタミン類縁体または誘導体とは、ビタミン類縁体または誘導体が、それを介して二価リンカー(L)に対して共有結合されるヘテロ原子を組み込んだビタミンを意味することが可能であることが理解される。
【0157】
例示のビタミン成分としては、葉酸、ビオチン、リボフラビン、チアミン、ビタミンB12、および、これらビタミン分子の受容体結合性類縁体および誘導体、および、その他の関連ビタミン受容体結合性分子が挙げられる。ビタミン類縁体および/または誘導体の具体例としては、葉酸塩の類縁体および誘導体、例えば、フォリン酸、プテロポリグルタミン酸、および、葉酸受容体結合性プテリジン、例えば、テトラヒドロプテリン、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、および、それらのデアザおよびジデアザ類縁体が挙げられる。「デアザ」および「ジデアザ」類縁体という用語は、天然の葉酸構造、またはその類縁体または誘導体において、1個または2個の窒素原子を置換する炭素原子を有する、従来技術で認知済みの類縁体を指す。例えば、デアザ類縁体としては、葉酸の、1-デアザ、3-デアザ、5-デアザ、8-デアザ、および、10-デアザ類縁体が挙げられる。ジデアザ類縁体としては、例えば、葉酸の、1,5-ジデアザ、5,10-ジデアザ、8,10-ジデアザ、および、5,8-ジデアザ類縁体が挙げられる。本発明において複合体形成リガンドとして有用なその他の葉酸塩は、葉酸受容体結合性類縁体である、アミノプテリン、アメトプテリン(メトトレキセート)、N10-メチル葉酸塩、2-デアミノヒドロキシ葉酸塩、デアザ類縁体、例えば、1-デアザメトプテリンまたは3-デアザメトプテリン、および、3’,5’-ジクロロ-4-アミノ-4-デオキシ-N10-メチルプテロイルグルタミン酸(ジクロロメトトレキセート)である。前述の葉酸類縁体および/または誘導体は、それらが葉酸受容体に結合する能力を有することを反映して、通常「葉酸塩」と呼ばれるが、このようなリガンドは、外来分子に結合された場合、膜貫通輸送、例えば、本明細書に記載される葉酸仲介性エンドサイトーシス経由輸送を強化するのに効果的である。葉酸受容体に結合して、複合体の受容体介在エンドサイトーシス輸送を起動することが可能な、その他の好適なリガンドとしては、葉酸受容体に対する抗イディオタイプ抗体が挙げられる。葉酸受容体に対する抗イディオタイプ抗体を有する複合体における外来分子は、本発明に従って、その複合体の膜貫通輸送を起動するために使用される。
【0158】
ビタミン類縁体および/または誘導体の具体例としてはさらに、ビオチンの類縁体および誘導体、例えば、ビオシチン、ビオチンスルフォキシド、オキシビオチン、および、その他のビオチン受容体結合性化合物等が挙げられる。本明細書に記載される他のビタミンの類縁体および誘導体も、本明細書では考慮されることが理解される。一つの実施態様では、本明細書で記載される薬剤送達結合体で使用されるビタミンは、活性化マクロファージにおいて特異的に発現されるビタミン受容体、例えば、本明細書に記載されるような葉酸またはその類縁体または誘導体に結合する葉酸受容体、に結合するものが挙げられる。
【0159】
ビタミンに対する結合部位は、受容体であって、病原細胞集団によって特異的に発現される、過剰に発現される、または優先的に発現される受容体に対して特異的に結合することが可能な、任意のビタミン分子、またはその誘導体または類縁体に対する、そのような受容体が挙げられる。病原細胞によって特異的に発現される、過剰に発現される、または優先的に発現される表面発現性蛋白質は、通常、非病原細胞では存在しないか、低濃度で存在する受容体であって、これが、病原細胞の選択除去手段を提供する。ビタミン受容体結合性薬剤送達結合体は、ガン細胞または、その他のタイプの病原細胞上の受容体に対して高い親和度をもって結合することが可能であってよい。高親和度結合は、ビタミン成分には元々備わっているものであることもできるし、あるいは、結合親和度を、化学的に修飾されたビタミン(すなわち、類縁体または誘導体)を用いることによって強化することも可能である。
【0160】
薬剤は、製薬学的活性化合物を含め、細胞機能を変調させる、またはその他のやり方で修飾することが可能ならば、いずれの分子であってもよい。好適な分子としては、例えば、と言ってそれらに限定されるわけではないが、ペプチド、オリゴペプチド、レトロ逆転(retro-inverso)オリゴペプチド、蛋白質、蛋白質類縁体であって、少なくとも一つの非ペプチド結合がペプチド結合を置換する類縁体、アポ蛋白質、糖蛋白質、酵素、補酵素、酵素阻害剤、アミノ酸とその誘導体、受容体、およびその他の膜蛋白質;抗原、およびそれに対する抗体;ハプテン、およびそれに対する抗体;ホルモン、脂質、リン脂質、リポソーム;毒素;抗生物質;鎮痛剤;気管支拡張剤;ベータブロッカー;抗菌剤;抗高血圧剤;循環系薬剤、例えば、抗不整脈剤、グリコシド心臓薬、抗狭心症剤、および、血管拡張剤等;中枢神経系薬剤、例えば、刺激剤、向精神薬、抗躁剤、および、抑制剤等;抗ウィルス剤;抗ヒスタミン剤;ガン治療薬、例えば、化学療法剤等;精神安定剤;抗うつ剤;H-2拮抗剤;抗痙攣剤;抗嘔吐薬;プロスタグランジンおよびプロスタグランジン類縁体;筋弛緩剤;抗炎症物質;刺激剤;うっ血除去剤;鎮吐剤;利尿剤;鎮痙剤;抗喘息薬;抗パーキンソン薬;去痰剤;鎮咳剤;粘液溶解剤;および、ミネラルおよび栄養添加物が挙げられる。
【0161】
さらに、薬剤は、細胞傷害性を持つ、腫瘍透過性を強化する、腫瘍細胞増殖を抑制する、アポトーシスを増進する、標的細胞の抗アポトーシス活性を下げる、感染媒介物による病気を治療するのに使用される、病原細胞に向けた内在的免疫反応を強化する、または、任意のタイプの病原細胞による病的状態の治療に使用される、従来技術で既知の、任意の薬剤であることが可能である。本発明に従って使用するのに好適な薬剤としては、アドレノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝剤、例えば、シトシンアラビノシド、プリン類縁体、ピリミジン類縁体、および、メトトレキセート、ブスルファン、カルボプラチン、クロラムブシル、シスプラチンおよび他の白金化合物、タモキシフェン、タキソール、パクリタクセル、パクリタクセル誘導体、タキソテール(登録商標)、シクロフォスファミド、ダウノマイシン、リゾキシン、T2トキシン、植物アルカロイド、プレドニソン、ヒドロキシ尿素、テニポシド、マイトマイシン、ディスコデルモリド、微小管阻害剤、エポチロン、チューブリシン、シクロプロピルベンゾ[e]インドロン、seco-シクロプロピルベンゾ[e]インドロン、O-Ac-seco-シクロプロピルベンゾ[e]インドロン、ブレオマイシンおよび他の任意の抗生物質、窒素マスタード、ニトロスウレア、ビンクリスチン、ビンブラスチン、およびその類縁体と誘導体、例えば、デアセチルビンブラスチンモノヒドラジド等、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン類、例えば、ドラスタチン10、アマニチン類、例えば、α-アマニチン等、カンプトテシン、イリノテカン、および他のカンプトテシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エストラムスチン、ノコダゾール、MAP4、コルセミド、炎症および炎症誘発剤、ペプチドおよびペプチド様シグナル伝達阻害剤、および、その他、従来技術で認知済みの任意の薬剤または毒素が挙げられる。本発明に従って使用が可能なその他の薬剤としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンフォテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、および、その他、従来技術で認知済みの任意の抗菌化合物が挙げられる。
【0162】
一つの実施態様では、本発明に従って用いられる薬剤は、血清中で少なくとも4時間安定であり続ける。別の実施態様では、薬剤は、ナノモル範囲のIC50を持ち、また別の実施態様では、薬剤は水溶性である。薬剤が水溶性でない場合、二価リンカー(L)を、水溶性を強化するように誘導体形成することも可能である。「薬剤」という用語はまた、上述の薬剤類縁体または誘導体、例えば、と言ってそれらに限定されないが、ドラスタチン類例えばドラスタチン10、アマニチン類例えばα-アマニチン、カムプトテシンおよびイリノテカン、および、他のカムプトテシンおよびイリノテカン誘導体を含む類縁体または誘導体等、を意味する。本発明によれば、薬剤類縁体または誘導体とは、それを介して薬剤類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだ薬剤を意味することも可能であることを理解しなければならない。
【0163】
本発明のビタミン受容体結合性薬剤送達結合体は、ビタミン受容体結合成分、二価リンカー(L)、薬剤、および、要すれば随意に、ビタミン受容体結合成分と薬剤とを二価リンカー(L)に連結するヘテロ原子リンカーを含むことができる。本発明によれば、ビタミン類縁体または誘導体とは、それを介してビタミン類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだビタミンを意味することも可能であることを理解しなければならない。従って、ビタミンは、ヘテロ原子リンカーを介して二価リンカー(L)に共有結合することも可能であるし、あるいは、ビタミン類縁体または誘導体(すなわち、ヘテロ原子を組み込んだもの)が直接二価リンカー(L)に結合することも可能である。同様に、薬剤類縁体または誘導体は、本発明による薬剤であり、薬剤類縁体または誘導体とは、それを介して薬剤類縁体または誘導体が二価リンカー(L)に共有結合されるヘテロ原子を組み込んだ薬剤を意味してもよい。従って、薬剤は、ヘテロ原子リンカーを介して二価リンカー(L)に共有結合することが可能であるし、あるいは、薬剤類縁体または誘導体(すなわち、ヘテロ原子を組み込んだもの)は、二価リンカー(L)に直接結合することも可能である。二価リンカー(L)は、スペーサーリンカー、放出(すなわち、分断可能)リンカー、および、上記二つのタイプのリンカーを含む結合体においてスペーサーリンカーを放出リンカーに連結するヘテロ原子リンカーを含むことが可能である。
【0164】
従って、本発明によれば、二価リンカー(L)は、ビタミンを薬剤に会合する手段、例えば、ヘテロ原子リンカー(すなわち、スペーサーアームまたは架橋分子)による接続、または、二価リンカー(L)の、ビタミンまたは薬剤類縁体または誘導体に対する直接共有結合による接続、を含むことが可能である。いずれの会合手段も、本発明の方法の動作のためには、ビタミン、またはビタミン受容体結合性誘導体または類縁体の、細胞膜上のビタミン受容体に対する結合を阻止するものであってはならない。
【0165】
一般に、二価リンカー(L)と、ビタミンまたはその類縁体または誘導体の間の複合体や、二価リンカー(L)と、薬剤またはその類縁体または誘導体の間の複合体を形成するためには、たとえば、介在ヘテロ原子リンカーなど、任意の手段が本発明に従って利用が可能である。さらに、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカーの間に複合体を形成し、二価リンカー(L)を形成するために、従来技術で認知済みの任意の手段を用いることが可能である。複合体は、上記の分子の中から任意に選ばれたものを、例えば、水素、イオン、または、共有結合等の結合を介して直接結合することによって形成することが可能である。共有結合は、例えば、酸、アルデヒド、ヒドロキシ、アミノ、スルフヒドリル、またはヒドラゾ基の間に、アミド、エステル、ジスルフィド、またはイミノ結合を形成することによって実現することが可能である。
【0166】
スペーサーおよび/または放出リンカー(すなわち分断可能リンカー)は、生物適合性であるいずれのリンカーであってもよい。分断可能リンカーは、例えば、細胞内の、または、細胞表面の還元または酸化条件下における分断に対して感受性を持つリンカー、酸易変性または塩基易変性を持つリンカーであるpH感受性リンカー、または、生化学的または代謝的過程によって分断されるリンカー、例えば、酵素易変性リンカーであってもよい。典型的には、スペーサーおよび/または放出リンカーは、約1から約30個の炭素原子を含むが、より典型的には、約2から約20個の炭素原子を含む。典型的には、より低分子量のリンカー(すなわち、約30から約300の分子量を持つもの)が用いられる。このようなリンカーの前駆物質は、通常は、求核性、または求電子性官能基のいずれかまたはその両方を持つように選ばれ、要すれば随意に、中間体合成における利便性を増すために、簡単に分断可能な保護基を持つ保護形として選ばれる。
【0167】
本発明はまた、1回以上の用量で投与された場合、宿主動物の病原細胞集団を除去するのに十分に有効な量のビタミン受容体結合性薬剤送達結合体を含む、製薬組成物に向けられる。この薬剤送達結合体は、宿主動物に対して、好ましくは非経口的に、例えば、皮内、皮下、筋肉内、腹腔内、静脈内、または、硬膜下腔内に、投与される。あるいは別に、この薬剤送達結合体は、他の医学的に有用な過程を通じて、例えば、経口的に投与することが可能であり、任意の効果的な用量、および、持続放出剤形を含めた、任意の好適な治療剤形の使用が可能である。
【0168】
非経口剤形の例としては、活性剤を、等張生食液、5%ブドウ糖、または、その他の既知の製薬学的に受容可能な液性担体、例えば、液性アルコール、グリコール、エステル、および、アミドに溶解させた溶液が挙げられる。本発明による非経口剤形は、薬剤送達結合体用量を含む、再構成可能な凍結乾燥体の形を取ることも可能である。本実施態様の一つの局面では、従来技術で既知のいくつかの持続放出剤形の内から任意に選ばれるもの、例えば、米国特許第4,713,249、5,266,333および5,417,982号に記載される、生体分解性炭水化物基質、を投与することが可能である。なお、この特許の開示を引用することにより本明細書に含める。あるいは別に、徐放ポンプ(例えば、浸透圧ポンプ)を使用することも可能である。
【0169】
治療因子を含む少なくとも一つの添加組成物を、、薬剤送達結合体仲介による病原細胞集団の除去を強化するために、前述の方法と組み合わせて、または、補助剤として投与することが可能であるし、あるいは、1種を越える追加治療因子を投与することが可能である。この治療因子は、内在的免疫反応を刺激することが可能な化合物、化学療法剤、または投与薬剤送達結合体の効力を補佐することが可能な別の治療因子から選んでもよい。本発明の方法は、前述の結合体の他に、内在的免疫反応を刺激することが可能な化合物または組成物(例えば、サイトカイン)、例えば、と言ってそれらに限定されないが、サイトカインまたは免疫細胞成長因子、例えば、インターロイキン1-18、幹細胞因子、塩基性FGF、EGF、G-CSF、GM-CSF、FLK-2リガンド、HILDA、MIP-1α、TGF-α、TGF-β、M-CSF、IFN-α、IFN-β、IFN-γ、可溶性CD23、LIF、および、それらの組み合わせを含む化合物または組成物、を宿主に対し投与することによって実行が可能である。
【0170】
これらの因子の治療的に有効な組み合わせも使用が可能である。一つの実施態様では、例えば、IL-2の治療的有効量、例えば、約0.1 MIU/m2/用量/日から約15 MIU/m2/用量/日の範囲の量を、1日複数回投与のスケジュールで、かつ、IFN-αを、例えば、0.1 MIU/m2/用量/日から約7.5 MIU/m2/用量/日の範囲の量を、1日複数回投与のスケジュールで、病原細胞を抱える宿主動物の病原細胞を除去する、低減させる、または中和させるために、薬剤送達結合体と同時に使用することが可能である(MIU=百万国際単位、m2=平均人のおよその体表面積)。別の実施態様では、IL-12およびIFN-αが、インターロイキンおよびインターフェロンの前述の治療有効量として使用される。さらに別の実施態様では、IL-15およびIFN-αが、インターロイキンおよびインターフェロンの前述の治療有効量として使用される。別の実施態様では、IL-2およびIFN-αまたはIFN-γ、およびGM-CSFが、前述の治療有効量において併用される。本発明はまた、サイトカイン類の、他の任意の有効な組み合わせ、例えば、他のインターロイキンおよびインターフェロン、およびコロニー刺激因子の組み合わせを含めた併用を考慮する。
【0171】
例えば、それ自体が細胞傷害性である、または、腫瘍透過性を強化するように作用することが可能な化学療法剤も、薬剤送達結合体と組み合わせて本発明の方法に用いるのに好適である。このような化学療法剤としては、アドレノコルチコイドおよびコルチコステロイド、アルキル化剤、抗アンドロゲン、抗エストロゲン、アンドロゲン、アクラマイシンおよびアクラマイシン誘導体、エストロゲン、抗代謝剤類、例えば、シトシンアラビノシド、プリン類縁体、ピリミジン類縁体、および、メトトレキセート等、ブスルファン、カルボプラチン、クロラムブシル、シスプラチンおよび他の白金化合物、タモキシフェン、タキソール、パクリタクセル、パクリタクセル誘導体、タキソテール(登録商標)、シクロフォスファミド、ダウノマイシン、リゾキシン、T2トキシン、植物アルカロイド、プレドニソン、ヒドロキシウレア、テニポシド、マイトマイシン、ディスコデルモリド、微小管阻害剤、エポチロン、チューブリシン、シクロプロピルべンジンドロン、seco-シクロプロピルベンジンドロン、O-Ac-seco-シクロプロピルベンジンドロン、ブレオマイシンおよび他の任意の抗生物質、ナイトロジェンマスタード、ニトロス尿素、ビンクリスチン、ビンブラスチン、およびその類縁体と誘導体、例えば、デアセチルビンブラスチンモノヒドラジド等、コルヒチン、コルヒチン誘導体、アロコルヒチン、チオコルヒチン、トリチルシステイン、ハリコンドリンB、ドラスタチン類、例えば、ドラスタチン10等、アマニチン類、例えば、α-アマニチン等、カンプトテシン、イリノテカン、および他のカンプトテシン誘導体、ゲルダナマイシンおよびゲルダナマイシン誘導体、エストラムスチン、ノコダゾール、MAP4、コルセミド、炎症および炎症誘発剤、ペプチドおよびペプチド様シグナル伝達阻害剤、および、その他、従来技術で認知済みの任意の薬剤または毒素が挙げられる。本発明に従って使用が可能なその他の薬剤としては、ペニシリン、セファロスポリン、バンコマイシン、エリスロマイシン、クリンダマイシン、リファンピン、クロラムフェニコール、アミノグリコシド抗生物質、ゲンタマイシン、アンフォテリシンB、アシクロビル、トリフルリジン、ガンシクロビル、ジドブジン、アマンタジン、リバビリン、マイタンシン類およびその類縁体および誘導体、ゲムシタビン、および、その他、従来技術で認知済みの任意の抗菌化合物が挙げられる。
【0172】
治療因子は、ビタミン受容体結合性薬剤送達結合体の前にか、後にか、または同時に宿主動物に投与することが可能であり、また、治療因子は、薬剤送達結合体を含む同じ組成物の一部として、または、薬剤送達結合体とは別の組成物の一部として投与することが可能である。治療因子を治療有効量として含む治療組成物は、いずれのものでも本発明において使用が可能である。
【0173】
さらに、1種を越える薬剤送達結合体を使用することが可能である。例えば、宿主動物を、ビタミンでは異なり、薬剤は同じ複数の結合体(例えば、葉酸-マイトマイシン結合体とビタミンB12-マイトマイシン結合体)の同時投与プロトコールで治療することも可能である。別の実施態様では、宿主動物を、異なる薬剤に連結した同じビタミンを含む、複数の結合体、または、各種薬剤に連結した各種ビタミンを含む、複数の結合体で治療することも可能である。例えば、宿主動物は、葉酸-マイトマイシン結合体と葉酸-シスプラチン結合体によって治療することも可能であるし、あるいは、葉酸-マイトマイシン結合体とビタミンB12-シスプラチン結合体によって治療することも可能である。さらに、同じ薬剤送達結合体の一部として、同じか、異なるビタミンおよび、同じか、異なる薬剤で、複数のビタミンと複数の薬剤を含む薬剤送達結合体も使用が可能である。
【0174】
薬剤送達結合体の、1日当たりの単位用量は、宿主の状態、治療される病状、結合体の分子量、投与ルートと組織分布、および、他の治療処置、例えば、放射線治療の併用の可能性に応じて相当に変動することがあり得る。患者に投与される有効量は、体表面積、患者の体重、患者の状態に関する医師の評価に基づいて決められる。有効用量は、例えば、約1 ng/kgから1mg/kg、約1 μg/kgから約500 μg/kg、約1 μg/kgから100 μg/kgの範囲で変動することが可能である。
【0175】
薬剤送達結合体を投与するために効果的なスケジュールであればどのようなスケジュールを用いてもよい。例えば、薬剤送達結合体は、1日当たり、1回用量として投与してもよく、分割して、複数回用量として投与してもよい。さらに、交互スケジュール、例えば、連日治療に代わるものとして、週当たり1から3日を用いてもよく、本発明を定義するために、このような断続的または交互的連日スケジュールは、毎日治療と等価と考えられ、本発明の範囲内とされる。本発明の一つの実施態様では、宿主は、病原細胞集団を除去するために、薬剤送達結合体の複数注入によって治療される。一つの実施態様では、宿主は、薬剤送達結合体を複数回(好ましくは、約2回から約50回まで)、例えば、12-72時間間隔で、または、48-72時間間隔で注入される。患者に対し、薬剤送達結合体の追加注入を、最初の注入(単複)後、数日または数ヶ月の間隔を置いて投与することが可能である。追加注入は、病原細胞によって引き起こされる病状の復帰を防止する。
【0176】
一つの実施態様では、本発明の薬剤送達結合体で用いることのできるビタミン、またはその類縁体または誘導体は、活性化マクロファージ上に特異的に発現される受容体、例えば、葉酸、またはその類縁体または誘導体に結合する葉酸受容体、に結合するものが挙げられる。例えば、葉酸連結結合体は、宿主に病態を引き起こす活性化マクロファージを殺す、または、その活性を抑えるのに使用される。このようなマクロファージ標的化結合体は、活性化マクロファージ仲介性病態に苦しむ患者に投与されると、結合体化薬剤を、活性化マクロファージ集団に集中させ、会合させるように働き、活性化マクロファージを殺すか、マクロファージ機能を抑圧する。活性化マクロファージ集団の除去、低減、または不活性化は、治療される病状に特徴的な活性化マクロファージ仲介性の病原性を停止または低減させるように働く。活性化マクロファージによって仲介されることが知られる病気の例としては、関節リューマチ、潰瘍性大腸炎、クローン病、乾癬、骨髄炎、多発性硬化症、アテローム硬化症、肺線維症、サルコイドーシス、全身硬化症、臓器移植拒絶反応(GVHD)、および慢性炎症が挙げられる。薬剤送達結合体の投与は、典型的には、病態の症状が低減または除去されるまで続けられる。
【0177】
活性化マクロファージを殺すために、または、活性化マクロファージの機能を抑えるために投与される薬剤送達結合体は、その病態に苦しむ動物または患者に対して非経口的に、例えば、皮内、皮下、筋肉内、腹腔内、または、静脈内に、製薬学的に受容可能な担体と組み合わせて投与することが可能である。あるいは別に、薬剤送達結合体は、他の、医学的に有用な過程を通じて動物または患者に投与することが可能であり、有効用量を、標準的または持続放出剤形として投与することが可能である。この治療法は、単独で用いてもよいし、活性化マクロファージによって仲介される病態治療用として認められている他の治療法と組み合わせて用いてもよい。
【0178】
下記の薬剤送達結合体は、本明細書に記載される本発明の範囲内に収まると考えられる薬剤送達結合体の例である。これらの薬剤送達結合体は、従来技術で認知済みのプロトコールに加えて本明細書に記載される過程を用いることによって本発明に従って調製することが可能である。
【0179】
さらに、下記の薬剤送達結合体も、本明細書に記載される本発明の範囲内に収まると考えられる薬剤送達結合体の例である。付属の合成手順は、本明細書に記載される薬剤送達結合体を調製するのに使用が可能なものを例示する。
【0180】
ジアセトキシシルペノール(DAS)のアセトニトリル溶液に、1.0 eg.の1,2,4,5-ベンゼンテトラカルボン酸二無水物を加え、その後、1 eg.のヒューニッヒ塩基を加える。この反応混合物を、アルゴンの下で室温で1.5時間攪拌する。いくらかのDASが未反応のまま残っている場合は、さらに0.2 eg.の二無水物を加え、攪拌を1時間続ける。1.2 eg.のプテロイルヒドラジドの無水DMSO溶液(J. Am. Chem. Soc., 1997, 119, 10004に従って調製、なおこの開示を引用することによって本明細書に含める)を加え、その後、1.0 eg.のヒューニッヒ塩基を加える。反応混合物を1時間攪拌し、ジエチルエーテルで沈殿させる。得られた沈殿を分取HPLCによってさらに精製する。
【0181】
実施例10aに一般的に記述されているように、Boc-ヒドラジドは、コハク酸無水物と反応させられ、得られた産物は、縮合剤としてのEDCの存在下に5’’-アミノ-ビス-インドリル-seco-CBIと反応させられる。Boc除去および、遊離レブリン酸とのアシルヒドラゾン形成により、NHS-エステル活性化後、ペプチド断片Pte-γ-Glu-Asp-Arg-Asp-Dap-OHに対する反応相手が得られる。このペプチド断片は、スキーム12に一般的に記述されるように、Fmoc-Dap(Boc)-Wang樹脂でスタートするFmoc策を用いるポリマー支持継続法によって調製される。
【0182】
Fmoc-ヒドラジドは、3-(2-ピリジルジチオ)プロピオン酸と反応させられて、Fmoc-ヒドラジド-[3-(2-ピリジルジチオ)プロピオネート]を生ずる。マイトマイシンC誘導体(マイトマイシンC、N-(CH2)2SH)との反応により、マイトマイシンCのジスルフィド含有誘導体が得られる。標準プロトコールによるFmoc-除去、および、レブリン酸によるアシルヒドラゾン形成により、連続的NHS-エステル活性化後、ペプチド断片Pte-γ-Glu-Dap-OHに対する反応相手が得られる。このペプチド断片は、一般にスキーム12に記載する通りに調製される。
【実施例】
【0183】
下記の例示の実施例は、制限的であることを意図するものではなく、また制限的なものと考えてはならない。例えば、本明細書に示される各化合物において、リンカーを形成する際に使用されるアミノ酸の立体化学は、随意に、天然のL型から、または、非天然のD型から選ばれてよい。各実施例は、表示されるようにNMR、MS、および/またはUV分光光度計測、および/またはHPLCによってその特性が解明され、選択された特性信号は適宜注記される。
【0184】
[実施例1]
ジエチレントリアミン葉酸、γ-アミド(DETA-葉酸塩)は、P. Fuchs et al., J. Am. Chem. Soc., 1997, 119, 10004によって記載される工程に従って合成された。なお、この開示を引用することにより本明細書に含める。この化合物(100 mg)を、2 mLの0.1N HClに溶解した。得られた溶液を、1 mLの0.1N HClに溶解したK2PtCl4(158 mg)溶液に攪拌しながら加えた。3 mLのDMSOを加え、攪拌を3日間続け、溶液をろ過し、ろ液をアセトニトリルで沈殿させたところ、170 mgの黄色粉末が得られた。MS(MALDI) 1249.92, 1286.27; 1H NMR(D2O)δ1.05(t, 1H), 2.3(t, 2H), 3.1(t, 2H), 4.45(m, 1H), 6.65(d, 2H), 7.5(d, 2H), 8.65(s, 1H)。
【0185】
[実施例2a]
N10-トリフルオロアセチル保護葉酸含有ペプチジル断片N10-TFA-Pte-Glu-Glu-Lys-OHを、スキーム12に一般的に示すように、Fmoc-策を用いたポリマー支持継続法によって調製した。これは、酸感受性のFmoc-Lys(Boc)-Wang樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。保護されたアミノ酸建設ブロックとしてFmoc-Glu-OtBuおよびN10-TFA-Pte-OHを用いた。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Buとt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCによって精製したところ、N10-TFA-Pte-γGlu-γGlu-Lys-OHがTFA塩として得られた。このペプチド81 mg(0.1 mmol)を2 mL DMSOに溶解した溶液を、15 μL(0.11 mmol)のEt3Nおよび35 mg(0.1 mmol)のマイトマイシンAで処理した。マイトマイシンAは、M. Matsui, Y. Yamada, K. Uzu, and T. Hirata, J. Antibiot. 21, 189-198(1968)およびD. Vias, D. Benigni, R. Partyka, and T. Doyle, J. Org. Chem. 51, 4307-4309(1986)の処理工程に従ってマイトマイシンCから調製してもよい。なお、これらの開示を引用することによって本明細書に含める。反応混合物は、室温で48時間攪拌し、溶媒を凍結乾燥によって除去した。別様に注記しない限り、溶媒の蒸発は全て減圧下で行った。最後に、トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)で分離し、産物をアセトニトリル中で沈殿させたところ102 mgの結合体が黄色固体として得られた。1H NMR(D2O)δ2.45(q, 1H), 2.95(m, 2H), 3.35(dd, 1H), 3.5(d, 1H), 6.5(d, 2H), 7.55(d, 2H), 8.55(s, 1H)。
【0186】
[実施例2b]
N10-トリフルオロアセチル保護葉酸含有ペプチジル断片N10-TFA-Pte-Glu-Glu-Cys-OHを、スキーム12に一般的に示すように、また、実施例2aに記載されるようにFmoc-策を用いたポリマー支持継続法によって調製した。シスタミンをマイトマイシンAと反応させたところ(Matsui, et al., J. Antibiot. 21, 189-198(1968); Vias, et al., J. Org. Chem. 51, 4307-4309(1986)参照)、末端に遊離アミノ酸を有するジスルフィド含有マイトマイシンC誘導体が得られた。これをレブリン酸と結合させ、その後、カルボニル基をマレイニド誘導アシルヒドラジドと反応させた。得られたマイケルアクセプターとN10-TFA-Pte-Glu-Cys-OHを反応させ、水酸化アンモニウム水溶液(pH=10.0)でトリフルオロアセチル保護基を除去し、アセトニトロル中で沈殿させたところ、最終的な結合体が得られた。MS(MALDI) 1059.04, 1148.44, 1225.32, 1300.8; 1H NMR(D2O)δ1.8(d, 2H), 1.9(s, 1H), 2.3(q, 1H), 2.45(q, 1H), 2.9(t, 1H), 3.35(dd, 1H), 4.45(s, 1H), 4.5(dd, 1H), 6.65(d, 2H), 7.55(d, 2H), 8.6(s, 1H)。
【0187】
[実施例3]
T-2トキシンの中間体マレイミドp-メトキシベンジリデンアセタールを、市販のN-(2-ヒドロキシエチル)マレイミドから出発して合成した。そのヒドロキシル基を、穏やかな塩基として塩化メチレン中で酸化銀(I)の存在下にp-メトキシベンジルクロリド(1.2 eg.)と反応させた。未精製産物をシリカカラムで精製した。得られたp-メトキシベンジルエーテルを、OH含有T-2トキシン(1 eg.)の存在下に1.5 eg.の2,3-ジクロロ-5,6-ジシアノ-ベンゾキノン(DDQ)によって酸化処理したところ、所望のp-メトキシベンジリデンアセタールが、安定化p-メトキシベンジルカルベニウム中間体を介して得られた。
【0188】
もう一方の反応相手、Pte-γ-Glu-Arg-Asp-Cys-OHを、Fmoc策を用いたポリマー支持継続法によって調製した。これを、酸感受性H-Cys(4-メトキシトリチル)-2-クロロトリチル樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Fmoc-Arg(Pbf)-OH、Fmoc-Glu-OtBu)およびN10-TFA-Pte-OHを用いた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Buとt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCによって精製したところ、N10-TFA-Pte-γ-Glu-Arg-Asp-Cys-OHが得られた。トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)中で分離した。
【0189】
最後に、標的とするp-メトキシベンジリデンアセタール連結葉酸-薬剤結合体を、アルゴン下に、ペプチドのバッファー水溶液(pH=7.0)を、T2トキシンのマレイミド含有アセタールの等モルアセトニトリル溶液と混合することによって調製した。室温で1時間攪拌後、最終結合体を分取HPLCで精製したところ、収集分画の凍結乾燥後に黄色粉末が得られた。MS(m+H)+ 1541.3; 1H NMR(DMSO-d6)δ0.1(s, 1H), 0.55(d, 2H), 0.9(dd, 3H), 1.65(s, 1H), 2.0(d, 1H), 3.75(d, 2H), 5.25(d, 1H), 6.65(d, 2H), 6.9(d, 2H), 7.3(t, 2H), 7.65(d, 2H), 8.65(s, 1H)。
【0190】
[実施例4a]
[実施例4b]
[実施例4c]
実施例4a、4bおよび4cの化合物は、アシルアジリジンは、市販の適当なN-(アルカン酸)マレイミドによってマイトマイシンAをアシル化すること(スキーム1参照)によって調製されたことを除いては、一般に実施例3で記載した工程に従って調製した。
【0191】
[実施例5]
塩基として2.2 eg. NaHCO3、および、溶媒としてアセトニトリル/水(l/l)の存在下に、trans-4-アミノシクロヘキサノール塩酸を、等モル量のFmoc-OSuと反応させると、N-Fmoc保護アミノアルコールが得られた。これをSwernの条件(Synthesis, 1981, 165)を用いて酸化して対応するN-Fmoc-保護アミノケトンを得た。4 eq.のメチルオルトギ酸および触媒量のトリフルオロ酢酸によってケタール化したところ、N-Fmoc-保護アミノケタールが定量的収率で得られた。このケタールを、等モル量のトリメチルシリルトリフルオロメタンスルフォネートおよび2,4,6-トリ-t-ブチル-ピリジンで処理したところ、産物として、4-Fmoc-アミノシクロヘキシルエノールエーテルが得られた。次の工程で、薬剤T-2トキシンを、分子ふるい(3オングストローム)および触媒量のトリフルオロ酢酸の存在下に、4倍過剰量のエノールエーテルで処理した。得られた非対称混合ケタールをシリカで精製した。Fmoc保護基は、DMF中で樹脂結合ピペリジンによる処理で除去した。解放されたアミノ酸は、1.1 eg.のヒューニッヒ塩基の存在下に1.1 eg.のマレイミド酢酸-NHS-エステルと反応させた。T-2トキシンのマレイミド含有ケタールをシリカの上で精製した。
【0192】
葉酸含有ペプチド断片、Pte-γ-Glu-β-Dap-Asp-Cys-OHは、スキーム12に一般的に記載されるように、Fmoc策を用いたポリマー支持継続法によって調製した。Fmoc-L-Cys(Trt)-OHを負荷した、酸感受性Wang樹脂の上で合成した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Boc-Dap(Fmoc)-OH、Fmoc-Glu-OtBu、およびN10-TFA-Pte-OHを用いた。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Bu、t-Boc、およびトリチル保護基の同時除去も実現された。最後に、トリフルオロアセチル基を水酸化アンモニウム水溶液中で分離したところ、所望のチオール含有ペプチドが得られた。この未精製ペプチドを、分取HPLCにて精製した。
【0193】
最後に、標的とするケタール連結葉酸-薬剤結合体を、アルゴン下に、ペプチドのバッファー水溶液(pH=7.0)と、T-2トキシンのマレイミド含有ケタールの等モル量アセトニトリル溶液とを混合することによって調製した。室温で1時間攪拌した後、最終結合体を分取HPLCで精製したところ、収集画分の凍結乾燥後に黄色粉末が得られた。ES MS(m-H)- 1474.5, (m+H)+ 1476.2, (m+Na)+ 1498.3。
【0194】
[実施例6]
エチレンジアミン葉酸、γ-アミド(EDA-葉酸塩)は、P. Fuchs et al.(J. Am. Chem. Soc., 1997, 119, 10004、例えば、本論文に記載される化合物52の合成を参照)によって記載される工程に従って合成された。なお、この開示を引用することにより本明細書に含める。EDA葉酸塩(600 mg)を、5 mLの無水DMSOに懸濁させた。60℃で4時間攪拌後、得られた溶液を20℃に冷却し、4 eg.の1,2,4,5-ベンゼンテトラカルボン酸無水物(無水BTCA)を加えた。5分後、反応混合液を、よく攪拌した無水アセトニトリルに注入した。得られた沈殿を遠心によって単離したところ、657mgのBTCA(一無水物)-EDA-葉酸塩が得られた。
【0195】
乾燥DMSOに溶解したダウノマイシンのよく攪拌された溶液に、固体のBTCA(一無水物)-EDA-葉酸塩(1.5 eg.)を添加した。さらに14時間攪拌後、50%のダウノマイシンが未反応のまま残り(HPLC)、1.5 eg. BTCA(一無水物)-EDA-葉酸を加えた。さらに4時間攪拌した後、ダウノマイシンは全て消費された。HPLCプロフィールにおいて、二つの新しいピークが観察された。これらは、保持時間が近接しており、最終結合体の二つの位置異性体を表していた。未精製産物をアセトニトリル中の沈殿後に単離し、逆相HPLCによってさらに精製した。産物の構造は、ES MS(m-H)- 1227.1に一致した。
【0196】
[実施例7]
アルゴン下0℃にて、4 mLの無水ジクロロメタンに溶解した250 mg(0.25 mmol)のパクリタクセルおよび130 μL(0.73 mmol)のヒューニッヒ塩基のよく攪拌された溶液に、85 μL(0.8 mmol)のAlloc-Clをゆっくりと加えた。さらに12時間攪拌を続け、産物を、標準的抽出法によって単離した。この白色粉末、2’-alloc-パクリタクセルを、これ以上精製することなく次の工程に用いた。
【0197】
この工程では、108 mg(0.117 mmol)の2’-alloc-パクリタクセルを、1.0 mLの無水アセトニトリルに溶解した。アルゴン下攪拌しながら、25 mg(0.117 mmol)の1,2,4,5-ベンゼンテトラカルボン酸無水物(無水BTCA)および21 μL(0.120 mmol)のヒューニッヒ塩基をこの溶液に加えた。攪拌をさらに2.5時間続けた。別の反応フラスコで、52 mgのEDA-葉酸を60℃で、全物質が溶解するまで攪拌した(約60分)。室温に冷却後、前の反応混合液をこの溶液に添加し、攪拌をさらに2時間続けた。反応混合液を、アセトニトリル/ジエチルエーテル(20:80)のよく攪拌された混合液に滴下により加えた。黄色沈殿を遠心によって分離し、さらに分取HPLCによって精製した。この産物の構造は、1Dおよび2D(COSY)1H-NMRスペクトル;ES MS(m+H)+ 1555.5と一致した。
【0198】
[実施例8]
1.0 eg.のアクラマイシン、2.0 eg.のヒドラジド-[3-(2-ピリジルジチオ)プロピオネート](SPDP-ヒドラゾン)、および、数結晶のピリジニウムp-トルエンスルフォネートから成る混合物を、アルゴン下、無水メタノールに攪拌しながら溶解した。この反応混合液を室温で8時間攪拌した。溶媒を蒸留乾固した。残渣を、あらかじめクロロフォルム/メタノール(90:10)に溶解させた1.5%トリエチルアミンで前処理したシリカカラムにて精製した。得られたアクラマイシンアシルヒドラゾンを、最少量のアセトニトリルに溶解した。得られた溶液に、等モル量のPte-γ-Glu-Cys-OH(水に溶解させ、pH=6.8に調整)をアルゴン下ゆっくりと加えた。このPte-γ-Glu-Cys-OHの調製は、実施例2aに記載する工程と似ており、一般にスキーム12に記載される。ジスルフィド交換反応は10分を要した。この反応溶液を、過剰量のアセトニトリルにゆっくりと加え、得られた沈殿を遠心にて単離した。沈殿を、もう一度アセトニトリルに再懸濁し、15分攪拌後遠心にて分離した。高真空にて一晩乾燥した後では、最終結合体は十分に純粋であった(HPLC); ES MS(m+H)+ 1474.1。
【0199】
[実施例9a]
1.0 eg.のアクラマイシンと1.2 eg.のβ-マレイミドプロピオン酸・TFAの混合物を、アルゴン下、無水メタノールに攪拌しながら溶解した。この反応混合液を室温で1時間攪拌した。溶媒を蒸留乾固した。残渣を、あらかじめクロロフォルム/メタノール(90:10)に溶解させた1.5%トリエチルアミンで前処理した短いシリカカラムにて精製した。別のフラスコで、ペプチド断片Pte-γ-Glu-γ-Glu-Cys-OHを、pHを6.8に調節しながら、アルゴン下に水に溶解した。このPte-γ-Glu-γ-Glu-Cys-OHの調製は、実施例2aに記載する工程と似ており、一般にスキーム12に記載される。得られた黄色味を帯びた溶液に、最少量のメタノールに溶解させたアクラマイシンのマレイミドヒドラゾンをゆっくりと加えた。この反応混合液をアルゴン下1時間攪拌した。メタノールを除去し、残渣を、HPLCの分取カラムにて精製し、その後凍結乾燥した; ES MS(m+H)+ 1722.3。
【0200】
[実施例9b]
[実施例9c]
実施例9bと9cの化合物は、実施例9aに一般的に記載される工程に従って、ドキソルビシン(14-ヒドロキシダウノマイシン)誘導体から調製された。
【0201】
[実施例10a]
極めて強力な細胞傷害性薬剤ビス-インドリル-seco-1,2,9,9a-テトラヒドロシクロプロパ[c]ベンズ[e]インドール-4-オン(ビス-インドイル-seco-CBI)の5’’-(N-Boc)アミノ類縁体を、最初に、D. Boger et al., J. Org. Chem., 1992, 57, 2873によって記載されたもののやや修飾された工程に従って調製した。なお、この開示を引用することにより本明細書に含める。
【0202】
ペプチド断片、Pte-γ-Glu-Asp-Arg-Asp-Cys-OHは、スキーム12に一般的に記載されるように、Fmoc策を用いたポリマー支持継続法によって酸感受性H-Cys(4-メトキシトリチル)-2-クロロトリチル樹脂上で調製した。低当量のアミノ酸による効率的結合を確保するためにPyBopを活性化試薬として与えた。保護されたアミノ酸建設ブロックとしてFmoc-Asp(OtBu)-OH、Fmoc-Arg(Pbf)-OH、Fmoc-Glu-OtBu、およびN10-TFA-Pte-OHを用いた。各結合工程の後で、標準条件下(DMFに溶解した20%ピペリジン液)にFmoc保護基を除去した。最後の集合工程の後で、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分断した。この反応では、t-Bu、およびt-Boc保護基の同時除去も実現された。この未精製ペプチドを、分取HPLCにて精製したところ、N10-Pte-γ-Glu-Asp-Arg-Asp-Cys-OHが得られた。トリフルオロアセチル保護基は、水酸化アンモニウム水溶液(pH=10.0)中で分離した。
【0203】
Lev-Val-OHは、バリンメチルエステル塩酸を、EDCとヒューニッヒ塩基の存在下に縮合し、その後メチルエステルを、水酸化リチウムと水で加水分解することを含む、標準的プロトコールを用いて合成した。
【0204】
複合した結合体の最終集合は、5’’-(N-Boc)アミノ-ビス-インドリル-seco-CBIのN-Boc基の除去と、EDCの存在下に、解放されたアミノ基とLev-Val-OHのカルボキシ官能基との結合で始まった。アシルヒドラゾン形成は、テトラヒドロフランにおけるレブリン酸成分のケトン官能基と1.2 eg.のβ-マレイミドプロピオン酸・TFAとのテトラヒドロフラン中での反応によって実現された。クロマトグラフィーによる精製後(シリカ、THF/ヘキサン=1/1)、前述の反応の産物をDMSOに溶解した。この溶液に、アルゴン下、0.9 eg.のPte-γ-Glu-Asp-Arg-Asp-Cys-OHを加え、反応混合液を18時間攪拌した。溶媒を凍結乾燥にて除去し、残渣をHPLCにて精製した。
【0205】
[実施例10b]
[実施例10c]
実施例10cおよび10cの化合物は、実施例10aに一般的に記載した工程に従って、5’’-(N-Boc)アミノ-ビス-インドリル-seco-CBIの誘導体から調製した。
【0206】
[実施例11]
炭酸カリウムの存在下に、2-メルカプトエタノールのS-アルキル化を臭化アリルによって実現した。得られたアリルβ-ヒドロキシエチルスルフィドのヒドロキシル基を、塩化チオニルによって塩素と交換した。この産物を、酢酸と無水酢酸の存在下に過酸化水素で酸化すると(J. Am. Chem. Soc., 1950, 72, 59、なお、この開示を引用することにより本明細書に含める)、アリルβ-クロロエチルスルフォンが得られた。この産物を、触媒量の白金6塩化水素酸(IV)の存在下に高温でクロロジメチルシランと反応させると、分留後、3-(β-クロロエチルスルフォニル)プロピルジメチルシリルクロリドが得られた。このクロロシランは、1 eg.のピリジンを塩基として用いると、高い細胞傷害性を持つ化合物リゾキシンのヒドロキシル基をシリル化した。この分子のβ-クロロエチルスルフォン基を、過剰なトリエチルアミンによって処理すると、塩化水素の滑らかなβ-除去が達成され、それぞれのビニールスルフォンが形成された。
【0207】
もう一方の反応相手、ペプチド断片Pte-γ-Glu-Arg-Asp-Cys-OHは、実施例2aとスキーム12に一般的に記載されるように、Fmocプロトコールを用いたポリマー支持継続法によって調製された。
【0208】
複合した結合体の最終集合は、リゾキシンに付着したシリコンリンカーのビニールスルフォン基に対して、ペプチド断片のチオール基をマイケル付加することによって実現された。この変換の反応溶媒は、50:50アセトニトリル/水(pH=7.2)であった。室温で24時間攪拌後、最終結合体を、HPLCの分取カラムにて単離した。ES MS(m+H)+ 1631.6; (m-H)- 1629.6。
【0209】
[実施例12]
このリゾキシンのシリコン連結結合体は、クロロジメチルシランの代わりに市販のクロロメチルフェニルシランを用いたことを除いては、実施例11に記載するプロトコールを用いて合成した。
【0210】
[実施例13]
===システインジスルフィド結合を含む化合物の一般的調製===
Ranasinghe and Fuchs, Synth. Commun. 18(3), 227-32(1988)の方法(この開示を引用することにより本明細書に含める)に従って調製されたチオスルフォネート4(1 eq.)を、薬剤、薬剤類縁体、または、薬剤誘導体5(1 eq.)と反応させて、スキーム13に示すように、メタノール溶液として薬剤チオスルフォネート6を調製する。式中、Rはアルキルまたはアリールであり、Lは適当な脱離基、例えば、ハロゲン、ペンタフルオロフェニル等であり、nは1から4までの整数であり、Xは-O-、-NH-、-C(O)O-、または-C(O)NH-である。変換は、各開始材料の消失をTLC(シリカゲル;CHCl3/MeOH=9/1)で観察することによって監視される。
スキーム13
【0211】
葉酸含有ペプチジル断片Pte-Glu-(AA)n-Cys-OH(9)は、スキーム14に示すように、酸感受性Fmoc-Cys(Trt)-Wang樹脂(7)上でFmoc策を用いるポリマー支持継続法によって調製した。R1はFmocであり、R2はトリチルであり、DIPEAはジイソプロピルエチルアミンである。PyBopは、効率的結合を確保するための活性化試薬として与えられる。各結合工程の後、標準条件下でFmoc保護基を除去する。適当に保護されたアミノ酸建設ブロック、例えば、Fmoc-Glu-OtBu、N10-TFA-Pte-OH等が、スキーム14に記載するように、また、工程(b)においてFmoc-AA-OHによって表されるように用いられる。従って、AAは、適当に保護されたアミノ酸開始材料を指す。Fmoc-AA-OHを含む結合手順(工程(a)および(b))は、”n”回繰り返されて、固相支持ペプチド8を調製する。式中nは整数であり、0から約100に等しい。最後の結合工程後、残余のFmoc基を除去し、順次ペプチドを、グルタミン酸誘導体に結合させ(工程(c))、脱保護し、TFA保護プテロイン酸に結合させる(工程(d))。次に、ペプチドを、トリフルオロ酢酸、エタンジチオール、およびトリイソプロピルシランで処理することによってポリマー支持体から分離する(工程(e))。この反応条件により、t-Bu、t-Boc、およびTrt保護基の同時除去が実現される。TFA保護基は、塩基処理によって除去され(工程(f))、葉酸含有Cys含有ペプチジル断片9を与える。
スキーム14
(a)20%ピペリジン/DMF; Fmoc-AA-OH, PyBop DIPEA, DMF;(c)Fmoc-Glu(O-t-Bu)-OH, PyBop,DIPEA, DMF; (d)1. N10(TFA)-Pte-OH; PyBop,DIPEA, DNSO; (e) TFAA, (CH2SH2)2, i-Pr3SiH; (f) NH4OH, pH10.3。
【0212】
薬剤結合体は、葉酸誘導体9(0.9-0.95 eq.)を、脱イオン水に溶解させた薬剤チオスルフォネート6(0.04M、pHを0.1 N NaHCO3で7に調節)と、アルゴン下で約30分反応させ、ジスルフィド結合を形成させて調製する。メタノールを減圧蒸留後、結合体を、分取HPLC(Prep Novapak HR C18 19 X 300 mMカラム;展開相(A)−1.0mMリン酸バッファー、pH=6;有機相(B)−アセトニトリル;条件−勾配、99%Aと1%Bから50%Aと50%Bまで30分;流速=15 mL/分)にて精製してもよい。
【0213】
[実施例14a]
1H NMR(DMSO-d6)δ4.7(d, 1H), 4.95(d, 1H), 6.7(d, 4H), 6.9(t, 1H), 7.95(d, 2H), 8.1(d, 2H), 8.2(m, 1H), 8.3(s, 1H), 8.4(s, 1H), 8.7(s, 1H), 10.2(s, 1H), 11.8(d, 2H)。
【0214】
[実施例14b]
ES MS(m-H)- 1436.4, (m+H)+ 1438.3。
【0215】
[実施例14c]
1H NMR(DMSO-d6/D2O)δ1.0(s, 1H), 1.1(s, 1H), 1.6(s, 1H), 1.8(s, 1H), 2.1(s, 1H), 2.25(s, 3H), 2.65(dd, 2H), 3.7(d, 1H), 4.4(t, 1H), 4.55(q, 2H), 4.6(d, 2H), 4.95(d, 1H), 5.9(t, 1H), 6.15(s, 1H), 6.6(d, 2H), 7.85(d, 2H), 7.95(d, 2H), 8.6(s, 1H), 8.95(d, 1H)。
【0216】
[実施例14d]
1H NMR(DMSO-d6/D2O)δ1.0(s, 1H), 1.1(s, 1H), 1.65(s, 1H), 2.1(s, 1H), 2.25(s, 3H), 2.6(dd, 2H), 3.25(dd, 1H), 3.6(t, 2H), 3.7(d, 1H), 4.4(t, 1H), 4.6(d, 1H), 4.95(d, 1H), 5.9(t, 1H), 6.2(s, 1H), 6.6(d, 2H), 7.7(t, 1H), 7.9(d, 2H), 7.95(d, 2H), 8.6(s, 1H), 9.1(d, 2H)。
【0217】
[実施例14e]
1H NMR(DMSO-d6/D2O)δ10.85(d, 2H), 1.05(d, 2H), 1.2(d, 2H), 1.7(d, 2H), 3.95(d, 1H), 4.05(dd, 1H), 5.4(dd, 1H), 5.7(dd, 1H), 6.65(d, 2H), 7.6(d, 2H), 7.95(s, 1H), 8.65(s, 1H)。
【0218】
[実施例14f]
ES MS (m+H)+ 1487.23; 1H NMR(DMSO-d6/D2O)δ0.9(t, 2H), 1.3(t, 2H), 2.15(t, 2H), 3.2(dd, 1H), 4.0(t, 1H), 4.15(q, 1H), 5.3(s, 2H), 5.5(s, 2H), 6.6(d, 2H), 7.0(s, 1H), 7.4(m, 2H), 7.55(d, 1H), 8.0(d, 2H), 8.6(s, 1H)。
【0219】
実施例14a、14b、14c、14d、14e、および14fは、下記の一般的手順によって調製した。-OH基を有する対応薬剤のよく攪拌された溶液(乾燥CH2Cl2または乾燥THFに溶解した1 eq.)に、6-(トリフルオロメチル)ベンゾトリアゾリル2-(2’-ピリジルジチオエチルカルボネート(1.3 eq.)およびNN-ジメチルアミノピリジン(1.5 eq.)をアルゴン下に加えた。この反応混合液を3時間攪拌し、ピリジルジチオ-誘導体薬剤をシリカクロマトグラフィーにて単離した(各サンプルについて>65%)。スキーム12に概略した一般的方法によって調製した対応ペプチジル断片(0.5 eq.)をDMSOに溶解した。得られた黄色透明液に、ピリジル-ジチオ誘導体薬剤を加えた。30分後、反応は完了し、結合体はHPLCにて精製した。実施例14eの場合、先ず、ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OHを水に溶解し、溶液のpHを、0.1N HClによって2.5に調節した。これによって、ペプチジル断片は沈殿した。このペプチジル断片を遠心によって収集し、乾燥し、DMSOに溶解して、つぎの、ピリジルジチオ-誘導体薬剤との反応に備えた。
【0220】
[実施例15]
SN38(10-ヒドロキシ-7-エチルカンプトテシン)の、中間体4-(2-ピリジニルジチオ)ベンジルカルボネートを、P. Senter et al., J. Org. Chem. 1990, 55, 2875によって記載される手順に従って調製した。なお、この開示を引用することにより本明細書に含める。ペプチジル断片Pte-Glu-Asp-Arg-Asp-Cys-OHをDMSOに溶解し、得られた黄色透明溶液に、ピリジルジチオ誘導体薬剤を加えた。30分後反応は完了し、結合体はHPLCにて精製した。ES MS (m+H)+ 1425.38; 1H NMR(DMSO-d6/D2O)δ0.9(t), 1.15(t), 3.9(t), 4.0(t), 4.25(t), 5.1(m), 5.2(s), 5.4(s), 6.55(d), 7.25(d), 7.35(d), 7.5(d), 7.9(d), 8.55(s)。
【0221】
[実施例16a]
[実施例16b]
実施例16aと16bの化合物は、スキーム12に記載される一般的手順に従って、ペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OHから調製した。このペプチジル断片を、seco-CBI-ビス-インドールのマレイミド誘導体にマイケル添加したところ、葉酸結合体実施例16aが得られた。このペプチジル断片はさらに、チオスルフォネート、またはピリジルジチオ活性化ビンブラスチンと反応して実施例16bを形成した。seco-CBI-ビス-インドールのマレイミド誘導体、および、チオスルフォネートおよびピリジルジチオ活性化ビンブラスチン中間体は、他の実施例について本明細書で記載する方法を用いて調製した。
【0222】
[実施例17a]
デアセチルビンブラスチンモノヒドラジド(1 eq.)(Barnett et al., J. Med. Chem. 1978, 21, 88を参照、なお、この開示を引用することにより本明細書に含める)を、新鮮蒸留THF中で、1 eq.のトリフルオロ酢酸で処理した。10分間攪拌後、溶液を1.05 eq.のN-(4-アセチルフェニル)マレイミドで処理した。アシルヒドラゾン形成は45分で完了し、溶媒は留去した。スキーム12に概説した一般的方法に従って調製されたペプチジル断片Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH(0.85 eq.)を水に溶解し、溶液のpHを、0.1N HClによって2.5に調節した。これによって、ペプチドは沈殿した。このペプチジル断片を遠心によって収集し、乾燥し、DMSOに溶解した。得られた黄色透明液に、ヒューニッヒ塩基(15 eq.)およびアシルヒドラゾンマイケル添加物を加えた。1時間後、最終的結合体をHPLCにて精製した。
【0223】
[実施例17b]
[実施例17c]
実施例17bと17cは、実施例17aに記載される手順に従って、対応するペプチジル断片とCBIのモノヒドラジド誘導体から調製した。
【0224】
実施例18-41の化合物は、実施例13に一般的に記載される手順に従って調製された。実施例18-41は、エレクトロスプレー質量分析(ES MS)、および、1Dおよび2D NMRおよびUVを含めた、その他の分光光度法によってその特性を明らかにした。
【0225】
[実施例18]
ES MS (m+H)+ 1071.9, (m+Na)+ 1093.9; 1H NMR(D2O)δ2.6(t, 4H), 2.7(t, 4H), 4.15(s, 2H), 5.45(s, 2H), 7.75(d, 2H), 8.15(d, 2H), 8.9(s, 1H)。
【0226】
[実施例19]
UV(nm)233(max), 255, 280; 1H NMR(D2O, NaOD, CD3CN)δ1.15(d, 3H), 2.3(s, 3H), 3.6(s, 1H), 3.85(s, 3H), 4.9(s, 1H), 5.3(s, 1H), 6.5(d, 2H), 7.3(m, 1H), 7.5(d, 2H), 7.65(d, 2H), 8.4(s, 1H)。
【0227】
[実施例20]
ES MS (m-H)- 935.6, (m+H)+ 937.4, (m+Na)+ 959.5。
【0228】
[実施例21]
1H NMR(D2O, NaOD, CD3CN)δ0.1(s, 1H), 1.1(s, 3H), 1.2(s, 3H), 1.75(s, 3H), 1.9(s, 3H), 2.05(s, 3H), 2.35(s, 3H), 3.3(dd, 2H), 3.8(d, 1H), 4.3(q, 2H), 4.9(d, 1H), 5.1(d, 1H), 5.4(q, 1H), 5.55(d, 1H), 5.65(d, 1H), 6.1(t, 1H), 6.35(s, 1H), 6.9(d, 2H), 7.9(d, 2H), 8.15(d, 2H), 8.7(s, 1H)。
【0229】
[実施例22]
【0230】
[実施例23]
ES MS (m-H)- 1136.5。
【0231】
[実施例24]
ES MS (m-H)- 1136.3, (m+H)+ 1138.0。
【0232】
[実施例25]
ES MS (m+H)+ 1382.3, (m+Na)+ 1405.4。
【0233】
[実施例26]
ES MS (m-H)- 1379.2, (m+H)+ 1381.2。
【0234】
[実施例27]
ES MS (mH)- 949.2; 1H NMR(D2O)δ1.55(s, 3H), 1.95(m, 2H), 2.05(s, 3H), 2.45(s, 3H), 2.75(dd, 2H), 2.95(dd, 2H), 3.05(s, 3H), 3.3(dd, 2H), 3.35(d, 2H), 3.45(t, 2H), 4.85(q, 2H), 6.5(d, 2H), 7.45(d, 2H), 8.5(s, 1H)。
【0235】
[実施例28]
1H NMR(DMSO-d6)δ1.5(s), 2.25(t), 2.75(m), 3.9(q), 4.6(d), 4.85(t), 6.6(d), 7.6(d), 7.9(d), 8.15(d), 8.25(t), 8.65(s), 8.7(m), 9.3(m), 10.2(t)。
【0236】
[実施例29]
ES MS (m-H)- 1413.5, (m+H)+ 1415.3。
【0237】
[実施例30]
ES MS (m+H)+ 1530.2; 1H NMR(DMSO-d6/D2O)δ1.2(s, 1H), 2.9(t, 1H), 3.65(t, 1H), 4.15(t, 1H), 4.25(t, 1H), 4.35(t, 1H), 6.7(d, 2H), 7.0(s, 1H), 8.1(d, 2H), 8.25(s, 1H), 8.7(s, 1H)。
【0238】
[実施例31]
1H NMR(DMSO-d6)δ1.75(s, 1H), 1.85(s, 1H), 2.1(t, 2H), 4.3(t, 1H), 4.6(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 8.15(s, 2H), 8.6(s, 1H)。
【0239】
[実施例32]
ES MS (m+H)+ 1408.4。
【0240】
[実施例33]
ES MS (m-H)- 1491.1, (m+H)+ 1493.1; 1H NMR(DMSO-d6/D2O)δ4.15(q, 1H), 4.6(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 7.25(s, 1H), 7.4(d, 1H), 7.9(d, 1H), 7.95(d, 2H), 8.15(d, 2H), 8.6(s, 1H)。
【0241】
[実施例34]
1H NMR(DMSO-d6/D2O)δ2.1(t, 2H), 2.75(q, 2H), 4.3(t, 1H), 4.65(d, 1H), 4.9(t, 1H), 6.6(d, 2H), 7.9(d, 1H), 8.0(d, 2H), 8.2(t, 2H), 8.6(s, 1H)。
【0242】
[実施例35]
【0243】
[実施例36]
ES MS (m+H)+ 1680.4; 1H NMR(DMSO-d6/D2O)δ0.3(s, 3H), 0.35(s, 3H), 1.05(s, 9H), 2.15(t, 2H), 4.15(t, 1H), 4.85(t, 1H), 6.6(d, 2H), 7.55(t, 4H), 7.9(d, 1H), 8.0(s, 1H), 8.15(s, 1H), 8.6(s, 1H)。
【0244】
[実施例37]
1H NMR(DMSO-d6/D2O)δ1.1(s, 3H), 1.8(s, 1H), 4.55(d, 1H), 4.8(t, 1H), 6.6(d, 2H), 7.8(d, 1H), 8.1(d, 1H), 8.15(s, 1H), 8.6(s, 1H)。
【0245】
[実施例38]
【0246】
[実施例39]
【0247】
[実施例40]
【0248】
[実施例41]
【0249】
[実施例42]
===EC112治療マウスにおける腫瘍成長の抑制===
薬剤がダウノルビシンである、実施例9b(EC111)および9c(EC112)の化合物の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の4日後、マウス(グループ当たり5匹)に、実施例9bまたは実施例9cの化合物2-10 μmol/kg、あるいは、結合されていないダウノルビシンまたはPBSを、週に2度4週間静注した。腫瘍成長は、各治療グループにおいて、3日または4日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。動物の体重も、3日または4日間隔で測定した。
図1および2に示すように、実施例9cの化合物による治療は、外見上毒性もなく(動物の体重に基づく)、M109腫瘍成長を効果的に遅らせた。未結合のドキソルビシンも抗腫瘍反応を与えたが、同時に体重に基づく毒性作用を示した。
【0250】
[実施例43]
===EC105治療マウスにおける腫瘍成長の抑制===
実施例10aの化合物(EC105)を使用したことを除いては、プロトコールは実施例42に記載した通りであった。この化合物では、薬剤は、ビス-インドリル-seco-CBIであった。実施例10aの化合物を0.3 μmol/kgの用量で注入した。さらに、M109モデル(葉酸受容体陽性)および4T1モデル(葉酸受容体陰性)を含む二つの皮下腫瘍モデルを試験した。数匹の動物では、67倍過剰の遊離葉酸(20 μmol/kg, FA)を、結合体(すなわち、実施例10aの化合物)と同時投与した。
実施例10aの化合物において、動物体重に基づく毒性を外見上示すことなく、著明な抗腫瘍反応が観察された(図3および4参照)。実施例10aの化合物による抗腫瘍反応は、過剰な遊離葉酸によって阻止され、反応の特異性を示した(図3参照)。図5に示すように、4T1モデル(葉酸受容体陰性)では抗腫瘍作用が観察されず、これも反応の特異性を示した。
【0251】
[実施例44]
===EC145治療マウスにおける腫瘍成長の抑制===
薬剤がデアセチルビンブラスチンモノヒドラジドである、実施例16bの化合物(EC145)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の約11日後(t0における平均腫瘍容量=60 mm3)、マウス(グループ当たり5匹)に、EC145の1500 nmol/kg、あるいは、等投与容量のPBS(コントロール)を、週に2度(BIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図6に示すように、EC145による治療は、生食液治療動物におけるM109腫瘍の成長と比べると、M109腫瘍成長を効果的に遅らせた。
【0252】
[実施例45]
===EC140治療マウスにおける腫瘍成長の抑制===
薬剤がデアセチルビンブラスチンモノヒドラジドである、実施例17aの化合物(EC140)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にM109腫瘍を負荷させたBalb/cマウスで評価した。右腋窩皮下に1x106 M109細胞注入による腫瘍接種の約11日後(t0における平均腫瘍容量=60 mm3)、マウス(グループ当たり5匹)に、EC140の1500 nmol/kg、あるいは、等投与容量のPBS(コントロール)を、週に3度(TIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図7に示すように、EC140による治療は、生食液治療動物におけるM109腫瘍の成長と比べると、M109腫瘍成長を効果的に遅らせた。
【0253】
[実施例46]
===EC136治療マウスにおける腫瘍成長の抑制===
薬剤がCBIである、実施例10bの化合物(EC136)の、腫瘍負荷動物に静注投与した場合の抗腫瘍活性を、皮下にL1210A腫瘍を負荷させたDBAマウスで評価した。右腋窩皮下に0.25x105 L1210A細胞注入による腫瘍接種の約5日後(t0における平均腫瘍容量は~50 mm3、グループ当たり5匹)に、EC136の400 nmol/kg、あるいは、等投与容量のPBS単独(コントロール)を、週に3度(TIW)、3週間静注した。腫瘍成長は、各治療グループにおいて、2日または3日間隔でキャリパーにて測定した。腫瘍容量は、方程式V=axb2/2を用いて計算した。式中、”a”は腫瘍の、ミリメートルで表した長さであり、”b”は幅である。
図8に示すように、EC136による治療は、生食液治療動物におけるL1210A腫瘍の成長と比べると、L1210A腫瘍成長を効果的に遅らせた。
【0254】
[実施例47]
===各種葉酸-薬剤結合体による細胞DNA合成の抑制===
実施例17b、10b、16a、10c、17a、16b、14e、および15の化合物(それぞれ、EC135、EC136、EC137、EC138、EC140、EC145、EC158、およびEC159)を、薬剤の、葉酸受容体陽性KB細胞の成長抑制能力を予測する、インビトロ細胞傷害性アッセイを用いて評価した。化合物は、本明細書に記載するプロトコールに従って調製された、それぞれの化学療法剤に連結された葉酸塩から成っていた。KB細胞は、表示の濃度の葉酸-薬剤結合体(図9-16のx軸参照)に対して、少なくとも100倍過剰量の葉酸の不在下に、または存在下に37℃で最大7時間暴露した。次に、細胞を、新鮮な培養液で濯ぎ、新鮮な培養液で37℃で72時間インキュベートした。細胞生存率を、3H-チミジン取り込みアッセイによって評価した。
【0255】
図9-16に示すように、用量依存性細胞傷害性が測定され、ほとんどの場合、IC50値(新たに合成されるDNAへの3H-チミジン取り込みを50%低下させるのに必要な、薬剤結合体の濃度)は、低ナノモル範囲であった。さらに、これら結合体の細胞傷害性は、過剰な遊離葉酸の存在下では低下した。このことは、観察された細胞殺戮は、葉酸受容体結合によって仲介されたことを示す。
【0256】
EC158、および、細胞系統、例えば、IGROV(既知の細胞系統)、A549-クローン-4(ヒト葉酸受容体cDNAをトランスフェクトされたA549細胞)、新規系統-01(インビボにおいて葉酸受容体発現について選択された系統-01突然変異株)、M109、4T1クローン-2(げっ歯類葉酸受容体cDNAをトランスフェクトされた4T1細胞)、および、HeLa細胞を含む細胞系統等、を用いた、このタイプのアッセイにおいても同様の結果が得られた。
【特許請求の範囲】
【請求項1】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン受容体結合性成分、
(b)二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに共有結合し、かつ、
二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、
二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとする、
ことを特徴とする薬剤送達結合体。
【請求項2】
前記ビタミン受容体結合性成分は、ビタミン、および、その、ビタミン結合性類縁体および誘導体から成るグループから選ばれることを特徴とする、請求項1の薬剤送達結合体。
【請求項3】
前記ヘテロ原子リンカーは、窒素、酸素、または硫黄原子であるか、あるいは、式-(NHR1NHR2)-、-SO-、-S(O)2-、および-NR3O-の式であって、式中、R1、R2、およびR3は、それぞれ独立に、水素、アルキル、アリール、アリールアルキル、アリール置換体、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、および、アルコキシアルキルから選ばれる、複数の式から成るグループから選ばれることを特徴とする、請求項1の薬剤送達結合体。
【請求項4】
前記スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イル、アルキレンスルフォキシル、スルフォニルアルキル、アルキレンスルフォキシルアルキル、アルキレンスルフォニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシニミド-3-イルから成るグループから選ばれ、
前記各スペーサーリンカーは、一つ以上の置換基X1によって、要すれば随意に置換され、
各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項5】
前記ヘテロ原子リンカーは窒素であり、前記置換基X1とヘテロ原子リンカーは、結合するスペーサーリンカーと共同して複素環を形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項6】
前記複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシニミドから成るグループから選ばれることを特徴とする、請求項5の薬剤送達結合体。
【請求項7】
前記放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルフォニルアルキル、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデニミニル、アルキレンスルフォニル、アルキレンチオ、アルキレンアリールチオ、および、カルボニルアルキルチオから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって、要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項8】
前記ヘテロ原子リンカーは窒素であり、前記置換基X2とヘテロ原子リンカーは、結合する放出リンカーと共同して複素環を形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項9】
前記複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシニミドから成るグループから選ばれることを特徴とする、請求項8の薬剤送達結合体。
【請求項10】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーとヘテロ原子リンカーは共同して二価の基、例えば、アルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルフォキシルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、スルフォニルアルキルアジリジン-1-イル、またはアルキレンスルフォニルアジリジン-1-イルを含む二価の基を形成し、前記各放出リンカーは、要すれば随意に、一つ以上の置換基X2によって置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項11】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーとヘテロ原子リンカーは共同して二価の基、例えば、アルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、またはスルフォニルアルキルアジリジン-1-イルを含む二価の基を形成することを特徴とする、請求項10の薬剤送達結合体。
【請求項12】
前記スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記各スペーサーリンカーは、一つ以上の置換基X1によって、要すれば随意に置換され、
各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
スペーサーリンカーは放出リンカーと結合してアジリジンアミドを形成する、
ことを特徴とする、請求項11の薬剤送達結合体。
【請求項13】
前記薬剤は、マイトマイシン、マイトマイシン誘導体、またはマイトマイシン類縁体であり、前記放出リンカーは、カルボニルアルキルチオ、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシミニド-3-イルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって、要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
マイトマイシンのアジリジンは、放出リンカーに結合されてアシルアジリジンを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項14】
前記薬剤は、二重結合窒素原子を含み、前記放出リンカーは、アルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシニミド-3-イルから成るグループから選ばれ、前記放出リンカーは薬剤窒素と結合してヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項15】
前記薬剤は硫黄原子を含み、前記放出リンカーは、アルキレンチオ、およびカルボニルアルキルチオから成るグループから選ばれ、前記放出リンカーは薬剤硫黄に結合してジスルフィドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項16】
前記ビタミン受容体結合成分は、窒素を含む葉酸塩であり、前記スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記スペーサーリンカーは、葉酸窒素に結合してイミドまたはアルキルアミドを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項17】
各置換基X1は、アルキル、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、カルボキシ、カルボキシアルキル、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから選ばれる、
ことを特徴とする、請求項16の薬剤送達結合体。
【請求項18】
前記ヘテロ原子リンカーは窒素であり、前記スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記各スペーサーリンカーは、一つ以上の置換基X1によって要すれば随意に置換され、スペーサーリンカーは前記窒素に結合してアミドを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項19】
前記ヘテロ原子リンカーは硫黄であり、前記スペーサーリンカーは、アルキレンおよびシクロアルキレンから成るグループから選ばれ、前記各スペーサーリンカーは要すれば随意にカルボキシによって置換され、スペーサーリンカーは前記硫黄に結合してチオールを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項20】
前記ヘテロ原子リンカーは硫黄であり、前記スペーサーリンカーは、1-アルキレンスクシニミド-3-イルおよび1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、スペーサーリンカーは前記硫黄に結合してスクシニミド-3-イルチオールを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項21】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、および1-アルコキシシクロアルキレンカルボニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記酸素に結合してアセタールまたはケタールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項22】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーはメチレンであり、前記メチレンは、要すれば随意に置換されるアリールによって置換され、放出リンカーは前記酸素に結合してアセタールまたはケタールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項23】
前記薬剤は窒素原子を含み、前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルから成るグループから選ばれ、放出リンカーは、前記ヘテロ原子リンカー窒素に結合してアミドを形成し、さらに前記薬剤窒素にも結合してアミドを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項24】
前記薬剤は酸素原子を含み、前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルから成るグループから選ばれ、放出リンカーは、前記ヘテロ原子リンカー窒素に結合してアミドを形成し、さらに前記薬剤酸素にも結合してエステルを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項25】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、およびカルボニルシクロアルキリデニミニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記窒素に結合してヒドラゾンを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項26】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーは、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、および(ジアリールシリル)アリールから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記酸素に結合してシラノールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項27】
前記薬剤は窒素原子を含み、前記放出リンカーは、要すれば随意に一つ以上の置換基X2によって置換されるハロアルキレンカルボニルであり、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記放出リンカーは前記薬剤窒素に結合してアミドを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項28】
前記薬剤は酸素原子を含み、前記放出リンカーは、要すれば随意に一つ以上の置換基X2によって置換されるアルキレンオキシカルボニルまたはハロアルキレンカルボニルであり、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記放出リンカーは前記薬剤酸素に結合して炭酸塩またはエステルを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項29】
前記ヘテロ原子リンカーは酸素であり、前記スペーサーリンカーは、要すれば随意に一つ以上の置換基X1によって置換される1-アルキレンスクシニミド-3-イルであり、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記スペーサーリンカーと放出リンカーとはそれぞれ、前記ヘテロ原子リンカーに結合してスクシニミド-1-イルアルキルアセタールまたはケタールを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項30】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルオキシメチルオキシを形成し、前記メチルは、要すれば随意にアルキルまたはアリール置換体によって置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項31】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とアシルアジリジンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項32】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して1-アルコキシシクロアルキレンオキシを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項33】
前記二価リンカーは、スペーサーリンカー、ヘテロ原子リンカー、および放出リンカーを含み、これらは共同してアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシレートを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項34】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同してジチオアルキルカルボニルヒドラジドを形成し、前記ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項35】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルカルボニルヒドラジドを形成し、前記ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項36】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオアルキルスルフォニルアルキル(二置換シリル)オキシを形成し、前記二置換シリルは、アルキル、または要すれば随意に置換されるアリールによって置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項37】
前記二価リンカーは、天然アミノ酸、およびその立体異性体から成るグループから選ばれる複数のスペーサーリンカーを含むことを特徴とする、請求項1の薬剤送達結合体。
【請求項38】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項39】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアリールアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成し、前記アリールは要すれば随意に置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項40】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルオキシアルキルオキシアルキリデンを形成し、前記アルキリデンは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成し、各アルキルは独立に選択され、前記オキシアルキルオキシは、アルキル、または要すれば随意に置換されるアリールによって要すれば随意に置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項41】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルオキシカルボニルヒドラジドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項42】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルアミノを形成し、前記アミノは、薬剤、またはその類縁体または誘導体とビニール様アミドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項43】
前記アルキルはエチルであることを特徴とする、請求項42の薬剤送達結合体。
【請求項44】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルアミノカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項45】
前記アルキルはエチルであることを特徴とする、請求項44の薬剤送達結合体。
【請求項46】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアリールアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とカルバメート、またはカルバモイルアジリジンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項47】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン、および、その、ビタミン受容体結合性類縁体および誘導体から成るグループから選ばれるビタミン受容体結合性成分、
(b)第1、第2、および第3ヘテロ原子リンカー、スペーサーリンカー、および、少なくとも一つの、ジスルフィドではない放出リンカーを含む二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに、第1ヘテロ原子リンカーを介して共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに、第2ヘテロ原子リンカーを介して共有結合し、かつ、
スペーサーリンカーと放出リンカーとは、互いに、第3ヘテロ原子リンカーを介して共有結合する
ことを特徴とする薬剤送達結合体。
【請求項48】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン、および、その、ビタミン受容体結合性類縁体および誘導体から成るグループから選ばれるビタミン受容体結合性成分、
(b)ヘテロ原子リンカー、スペーサーリンカー、および、少なくとも一つの、ジスルフィドではない放出リンカーを含む二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに共有結合し、かつ、
スペーサーリンカーと放出リンカーとは、互いに、ヘテロ原子リンカーを介して共有結合する
ことを特徴とする薬剤送達結合体。
【請求項49】
請求項1から48のいずれか1項による薬剤送達結合体、および、その製薬学的に受容可能な担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項50】
病原細胞集団を抱える宿主動物において病原細胞集団を除去する方法であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体に対して接触可能な結合部位を持ち、結合部位は、病原細胞によって特異的に発現されるか、過剰発現されるか、または優先的に発現され、前記方法は、請求項1から48のいずれか1項による薬剤送達結合体、またはその製薬組成物を、前記宿主に投与する工程を含む方法。
【請求項51】
ビタミン受容体結合性薬剤送達結合体中間体であって、
(a)ビタミン受容体結合性成分、
(b)第1末端と第2末端とを有する二価のリンカー、および、
(c)結合基を含み、
結合基は、薬剤、またはその類縁体または誘導体と共有結合を形成することが可能な求核基、求電子基、またはその前駆物質であり、
ビタミン受容体結合性成分は、二価リンカーの第1末端において二価リンカーに共有結合し、結合基は、二価リンカーの第2末端において二価リンカーに共有結合し、
かつ、二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとする、
ことを特徴とする薬剤送達結合体中間体。
【請求項52】
下式:
V−L−D
を持つビタミン受容体結合性薬剤送達結合体であって、式中、
Lは、(ls)a、(lH)b、および(lr)c、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、
Vはビタミン受容体結合性成分であり、Dは薬剤、またはその類縁体または誘導体であり、かつ、
a、b、およびcは、それぞれ独立に、0、1、2、3、または4であり、
Lは、少なくとも一つの、ジスルフィドではない(lr)を含むものとする、
ことを特徴とする薬剤送達結合体。
【請求項53】
下式:
V−L−D
を持つビタミン受容体結合性薬剤送達結合体であって、式中、
Lは、(ls)a、および(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、aおよびbは、それぞれ独立に、0、1、2、3、または4であり、かつ、
Vはビタミン受容体結合性成分であり、Dは薬剤、またはその類縁体または誘導体である、
ことを特徴とする薬剤送達結合体。
【請求項54】
前記薬剤はハプテンであることを特徴とする、請求項53の薬剤送達結合体。
【請求項55】
前記ハプテンは、フルオレセインおよびジニトロフェニルから成るグループから選ばれることを特徴とする、請求項54の薬剤送達結合体。
【請求項56】
請求項53から55のいずれか1項による薬剤送達結合体、および、その製薬学的に受容可能な担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項57】
病原細胞集団を抱える宿主動物において病原細胞集団を除去する方法であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体に対して接触可能な結合部位を持ち、結合部位は、病原細胞によって特異的に発現されるか、過剰発現されるか、または優先的に発現され、前記方法は、請求項53から55のいずれか1項による薬剤送達結合体、またはその製薬組成物を、前記宿主に投与する工程を含む方法。
【請求項58】
ビタミン受容体結合性薬剤送達結合体中間体であって、
(a)第1末端と第2末端とを有する二価のリンカー、
(b)薬剤、またはその類縁体または誘導体、および、
(c)結合基を含み、
二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとし、
結合基は、二価リンカーの第1末端において二価リンカーに共有結合し、薬剤、またはその類縁体または誘導体は、二価リンカーの第2末端において二価リンカーに共有結合し、かつ、
結合基は、ビタミン受容体結合性成分と共有結合を形成することが可能な求核基、求電子基、またはその前駆物質であるものとする、
ことを特徴とする中間体。
【請求項59】
前記ビタミン受容体結合性成分は、ビタミン、または、その、ビタミン受容体結合性類縁体または誘導体であり、前記結合基はマイケルアクセプターであり、前記二価リンカーは、式-C(O)NHN=, -NHC(O)NHN=、および-CH2C(O)NHN=から成るグループから選ばれる式を有する放出リンカーを含むことを特徴とする、請求項58の中間体。
【請求項60】
前記二価リンカーは、第1放出リンカーおよび第1スペーサーリンカーを含み、前記第1放出リンカー、第1スペーサーリンカーおよび結合基とは共同して下記の式:
(式中、Dは、薬剤、またはその類縁体または誘導体であり、nは、1、2、3、および4から成るグループから選ばれる整数である)を持つ化合物を形成することを特徴とする、請求項58の中間体。
【請求項61】
前記ビタミン受容体結合成分は、アルキルチオール求核基を含むことを特徴とする、請求項60の中間体。
【請求項62】
下記の式:
(式中、Vはビタミン受容体結合成分であり、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、b、およびcは、0、1、2、3、および4から成るグループから選ばれる整数であり、Dは、薬剤、またはその類縁体または誘導体である)を持つ化合物の調製法であって、
(a)下記の式:
を持つ化合物を、下記の式:
を持つ化合物と反応させること、および、
(b)薬剤、またはその類縁体または誘導体のヒドラゾン誘導体を形成させること、
を含む調製法。
【請求項63】
下記の式:
(式中、Vはビタミン受容体結合成分であり、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、b、およびcは、0、1、2、3、および4から成るグループから選ばれる整数であり、Dは、薬剤、またはその類縁体または誘導体である)を持つ化合物の調製法であって、
下記の式:
を持つ化合物を、下記の式:
を持つ化合物と反応させること、
を含む調製法。
【請求項64】
前記薬剤、またはその類縁体または誘導体は、ダウノマイシン類、ダウノマイシノン類、ダウノルビシン類、アクラマイシン類、タキソール類、クロモマイシン類、およびアウリル酸類(aurilic acids)から成るグループから選ばれることを特徴とする、請求項63の調製法。
【請求項1】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン受容体結合性成分、
(b)二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに共有結合し、かつ、
二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、
二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとする、
ことを特徴とする薬剤送達結合体。
【請求項2】
前記ビタミン受容体結合性成分は、ビタミン、および、その、ビタミン結合性類縁体および誘導体から成るグループから選ばれることを特徴とする、請求項1の薬剤送達結合体。
【請求項3】
前記ヘテロ原子リンカーは、窒素、酸素、または硫黄原子であるか、あるいは、式-(NHR1NHR2)-、-SO-、-S(O)2-、および-NR3O-の式であって、式中、R1、R2、およびR3は、それぞれ独立に、水素、アルキル、アリール、アリールアルキル、アリール置換体、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、および、アルコキシアルキルから選ばれる、複数の式から成るグループから選ばれることを特徴とする、請求項1の薬剤送達結合体。
【請求項4】
前記スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレン、シクロアルキレン、アルキレンシクロアルキル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-アルキレンスクシニミド-3-イル、1-(カルボニルアルキル)スクシニミド-3-イル、アルキレンスルフォキシル、スルフォニルアルキル、アルキレンスルフォキシルアルキル、アルキレンスルフォニルアルキル、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシニミド-3-イルから成るグループから選ばれ、
前記各スペーサーリンカーは、一つ以上の置換基X1によって、要すれば随意に置換され、
各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項5】
前記ヘテロ原子リンカーは窒素であり、前記置換基X1とヘテロ原子リンカーは、結合するスペーサーリンカーと共同して複素環を形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項6】
前記複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシニミドから成るグループから選ばれることを特徴とする、請求項5の薬剤送達結合体。
【請求項7】
前記放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニル、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニル、ハロアルキレンカルボニル、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、(ジアリールシリル)アリール、オキシカルボニルオキシ、オキシカルボニルオキシアルキル、スルフォニルアルキル、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、カルボニルシクロアルキリデニミニル、アルキレンスルフォニル、アルキレンチオ、アルキレンアリールチオ、および、カルボニルアルキルチオから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって、要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項8】
前記ヘテロ原子リンカーは窒素であり、前記置換基X2とヘテロ原子リンカーは、結合する放出リンカーと共同して複素環を形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項9】
前記複素環は、ピロリジン、ピペリジン、オキサゾリジン、イソキサゾリジン、チアゾリジン、イソチアゾリジン、ピロリジノン、ピペリジノン、オキサゾリジノン、イソキサゾリジノン、チアゾリジノン、イソチアゾリジノン、およびスクシニミドから成るグループから選ばれることを特徴とする、請求項8の薬剤送達結合体。
【請求項10】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーとヘテロ原子リンカーは共同して二価の基、例えば、アルキレンアジリジン-1-イル、アルキレンカルボニルアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、アルキレンスルフォキシルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、スルフォニルアルキルアジリジン-1-イル、またはアルキレンスルフォニルアジリジン-1-イルを含む二価の基を形成し、前記各放出リンカーは、要すれば随意に、一つ以上の置換基X2によって置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれる、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項11】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーとヘテロ原子リンカーは共同して二価の基、例えば、アルキレンアジリジン-1-イル、カルボニルアルキルアジリジン-1-イル、スルフォキシルアルキルアジリジン-1-イル、またはスルフォニルアルキルアジリジン-1-イルを含む二価の基を形成することを特徴とする、請求項10の薬剤送達結合体。
【請求項12】
前記スペーサーリンカーは、カルボニル、チオノカルボニル、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記各スペーサーリンカーは、一つ以上の置換基X1によって、要すれば随意に置換され、
各置換基X1は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
スペーサーリンカーは放出リンカーと結合してアジリジンアミドを形成する、
ことを特徴とする、請求項11の薬剤送達結合体。
【請求項13】
前記薬剤は、マイトマイシン、マイトマイシン誘導体、またはマイトマイシン類縁体であり、前記放出リンカーは、カルボニルアルキルチオ、カルボニルテトラヒドロ-2H-ピラニル、カルボニルテトラヒドロフラニル、1-(カルボニルテトラヒドロ-2H-ピラニル)スクシニミド-3-イル、および、1-(カルボニルテトラヒドロフラニル)スクシミニド-3-イルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって、要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
マイトマイシンのアジリジンは、放出リンカーに結合されてアシルアジリジンを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項14】
前記薬剤は、二重結合窒素原子を含み、前記放出リンカーは、アルキレンカルボニルアミノおよび1-(アルキレンカルボニルアミノ)スクシニミド-3-イルから成るグループから選ばれ、前記放出リンカーは薬剤窒素と結合してヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項15】
前記薬剤は硫黄原子を含み、前記放出リンカーは、アルキレンチオ、およびカルボニルアルキルチオから成るグループから選ばれ、前記放出リンカーは薬剤硫黄に結合してジスルフィドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項16】
前記ビタミン受容体結合成分は、窒素を含む葉酸塩であり、前記スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記スペーサーリンカーは、葉酸窒素に結合してイミドまたはアルキルアミドを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項17】
各置換基X1は、アルキル、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、カルボキシ、カルボキシアルキル、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから選ばれる、
ことを特徴とする、請求項16の薬剤送達結合体。
【請求項18】
前記ヘテロ原子リンカーは窒素であり、前記スペーサーリンカーは、アルキレンカルボニル、シクロアルキレンカルボニル、カルボニルアルキルカルボニル、および1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、前記各スペーサーリンカーは、一つ以上の置換基X1によって要すれば随意に置換され、スペーサーリンカーは前記窒素に結合してアミドを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項19】
前記ヘテロ原子リンカーは硫黄であり、前記スペーサーリンカーは、アルキレンおよびシクロアルキレンから成るグループから選ばれ、前記各スペーサーリンカーは要すれば随意にカルボキシによって置換され、スペーサーリンカーは前記硫黄に結合してチオールを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項20】
前記ヘテロ原子リンカーは硫黄であり、前記スペーサーリンカーは、1-アルキレンスクシニミド-3-イルおよび1-(カルボニルアルキル)スクシニミド-3-イルから成るグループから選ばれ、スペーサーリンカーは前記硫黄に結合してスクシニミド-3-イルチオールを形成することを特徴とする、請求項4の薬剤送達結合体。
【請求項21】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、および1-アルコキシシクロアルキレンカルボニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記酸素に結合してアセタールまたはケタールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項22】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーはメチレンであり、前記メチレンは、要すれば随意に置換されるアリールによって置換され、放出リンカーは前記酸素に結合してアセタールまたはケタールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項23】
前記薬剤は窒素原子を含み、前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルから成るグループから選ばれ、放出リンカーは、前記ヘテロ原子リンカー窒素に結合してアミドを形成し、さらに前記薬剤窒素にも結合してアミドを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項24】
前記薬剤は酸素原子を含み、前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、カルボニルアリールカルボニル、カルボニル(カルボキシアリール)カルボニル、カルボニル(ビスカルボキシアリール)カルボニルから成るグループから選ばれ、放出リンカーは、前記ヘテロ原子リンカー窒素に結合してアミドを形成し、さらに前記薬剤酸素にも結合してエステルを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項25】
前記ヘテロ原子リンカーは窒素であり、前記放出リンカーは、イミノアルキリデニル、カルボニルアルキリデニミニル、イミノシクロアルキリデニル、およびカルボニルシクロアルキリデニミニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記窒素に結合してヒドラゾンを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項26】
前記ヘテロ原子リンカーは酸素であり、前記放出リンカーは、アルキレン(ジアルキルシリル)、アルキレン(アルキルアリールシリル)、アルキレン(ジアリールシリル)、(ジアルキルシリル)アリール、(アルキルアリールシリル)アリール、および(ジアリールシリル)アリールから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、放出リンカーは前記酸素に結合してシラノールを形成することを特徴とする、請求項7の薬剤送達結合体。
【請求項27】
前記薬剤は窒素原子を含み、前記放出リンカーは、要すれば随意に一つ以上の置換基X2によって置換されるハロアルキレンカルボニルであり、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記放出リンカーは前記薬剤窒素に結合してアミドを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項28】
前記薬剤は酸素原子を含み、前記放出リンカーは、要すれば随意に一つ以上の置換基X2によって置換されるアルキレンオキシカルボニルまたはハロアルキレンカルボニルであり、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記放出リンカーは前記薬剤酸素に結合して炭酸塩またはエステルを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項29】
前記ヘテロ原子リンカーは酸素であり、前記スペーサーリンカーは、要すれば随意に一つ以上の置換基X1によって置換される1-アルキレンスクシニミド-3-イルであり、放出リンカーは、メチレン、1-アルコキシアルキレン、1-アルコキシシクロアルキレン、1-アルコキシアルキレンカルボニル、1-アルコキシシクロアルキレンカルボニルから成るグループから選ばれ、前記各放出リンカーは、一つ以上の置換基X2によって要すれば随意に置換され、
各置換基X2は、アルキル、アルコキシ、アルコキシアルキル、ヒドロキシ、ヒドロキシアルキル、アミノ、アミノアルキル、アルキルアミノアルキル、ジアルキルアミノアルキル、ハロ、ハロアルキル、スルフヒドリルアルキル、アルキルチオアルキル、アリール、アリール置換体、アリールアルキル、アリールアルキル置換体、ヘテロアリール、ヘテロアリール置換体、カルボキシ、カルボキシアルキル、アルキルカルボキシレート、アルキルアルカノエート、グアニジノアルキル、R4-カルボニル、R5-カルボニルアルキル、R6-アシルアミノ、および、R7-アシルアミノアルキルから成るグループから独立に選ばれ、R4とR5は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、R6とR7は、それぞれ独立に、アミノ酸、アミノ酸誘導体、およびペプチドから成るグループから選ばれ、
前記スペーサーリンカーと放出リンカーとはそれぞれ、前記ヘテロ原子リンカーに結合してスクシニミド-1-イルアルキルアセタールまたはケタールを形成する、
ことを特徴とする、請求項1の薬剤送達結合体。
【請求項30】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルオキシメチルオキシを形成し、前記メチルは、要すれば随意にアルキルまたはアリール置換体によって置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項31】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とアシルアジリジンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項32】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して1-アルコキシシクロアルキレンオキシを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項33】
前記二価リンカーは、スペーサーリンカー、ヘテロ原子リンカー、および放出リンカーを含み、これらは共同してアルキレンアミノカルボニル(ジカルボキシルアリーレン)カルボキシレートを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項34】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同してジチオアルキルカルボニルヒドラジドを形成し、前記ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項35】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルカルボニルヒドラジドを形成し、前記ヒドラジドは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項36】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、ヘテロ原子リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオアルキルスルフォニルアルキル(二置換シリル)オキシを形成し、前記二置換シリルは、アルキル、または要すれば随意に置換されるアリールによって置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項37】
前記二価リンカーは、天然アミノ酸、およびその立体異性体から成るグループから選ばれる複数のスペーサーリンカーを含むことを特徴とする、請求項1の薬剤送達結合体。
【請求項38】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項39】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアリールアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体と炭酸塩を形成し、前記アリールは要すれば随意に置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項40】
前記二価リンカーは、ヘテロ原子リンカー、スペーサーリンカー、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-チオスクシニミド-1-イルアルキルオキシアルキルオキシアルキリデンを形成し、前記アルキリデンは、薬剤、またはその類縁体または誘導体とヒドラゾンを形成し、各アルキルは独立に選択され、前記オキシアルキルオキシは、アルキル、または要すれば随意に置換されるアリールによって要すれば随意に置換されることを特徴とする、請求項1の薬剤送達結合体。
【請求項41】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルオキシカルボニルヒドラジドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項42】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルアミノを形成し、前記アミノは、薬剤、またはその類縁体または誘導体とビニール様アミドを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項43】
前記アルキルはエチルであることを特徴とする、請求項42の薬剤送達結合体。
【請求項44】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアルキルアミノカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とカルバメートを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項45】
前記アルキルはエチルであることを特徴とする、請求項44の薬剤送達結合体。
【請求項46】
前記二価リンカーは、放出リンカー、スペーサーリンカー、および放出リンカーを含み、これらは共同して3-ジチオアリールアルキルオキシカルボニルを形成し、前記カルボニルは、薬剤、またはその類縁体または誘導体とカルバメート、またはカルバモイルアジリジンを形成することを特徴とする、請求項1の薬剤送達結合体。
【請求項47】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン、および、その、ビタミン受容体結合性類縁体および誘導体から成るグループから選ばれるビタミン受容体結合性成分、
(b)第1、第2、および第3ヘテロ原子リンカー、スペーサーリンカー、および、少なくとも一つの、ジスルフィドではない放出リンカーを含む二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに、第1ヘテロ原子リンカーを介して共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに、第2ヘテロ原子リンカーを介して共有結合し、かつ、
スペーサーリンカーと放出リンカーとは、互いに、第3ヘテロ原子リンカーを介して共有結合する
ことを特徴とする薬剤送達結合体。
【請求項48】
ビタミン受容体結合性薬剤送達結合体であって、
(a)ビタミン、および、その、ビタミン受容体結合性類縁体および誘導体から成るグループから選ばれるビタミン受容体結合性成分、
(b)ヘテロ原子リンカー、スペーサーリンカー、および、少なくとも一つの、ジスルフィドではない放出リンカーを含む二価のリンカー、および、
(c)薬剤、またはその類縁体または誘導体を含み、
ビタミン受容体結合性成分は、二価リンカーに共有結合し、
薬剤、または、その類縁体または誘導体は、二価リンカーに共有結合し、かつ、
スペーサーリンカーと放出リンカーとは、互いに、ヘテロ原子リンカーを介して共有結合する
ことを特徴とする薬剤送達結合体。
【請求項49】
請求項1から48のいずれか1項による薬剤送達結合体、および、その製薬学的に受容可能な担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項50】
病原細胞集団を抱える宿主動物において病原細胞集団を除去する方法であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体に対して接触可能な結合部位を持ち、結合部位は、病原細胞によって特異的に発現されるか、過剰発現されるか、または優先的に発現され、前記方法は、請求項1から48のいずれか1項による薬剤送達結合体、またはその製薬組成物を、前記宿主に投与する工程を含む方法。
【請求項51】
ビタミン受容体結合性薬剤送達結合体中間体であって、
(a)ビタミン受容体結合性成分、
(b)第1末端と第2末端とを有する二価のリンカー、および、
(c)結合基を含み、
結合基は、薬剤、またはその類縁体または誘導体と共有結合を形成することが可能な求核基、求電子基、またはその前駆物質であり、
ビタミン受容体結合性成分は、二価リンカーの第1末端において二価リンカーに共有結合し、結合基は、二価リンカーの第2末端において二価リンカーに共有結合し、
かつ、二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとする、
ことを特徴とする薬剤送達結合体中間体。
【請求項52】
下式:
V−L−D
を持つビタミン受容体結合性薬剤送達結合体であって、式中、
Lは、(ls)a、(lH)b、および(lr)c、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、
Vはビタミン受容体結合性成分であり、Dは薬剤、またはその類縁体または誘導体であり、かつ、
a、b、およびcは、それぞれ独立に、0、1、2、3、または4であり、
Lは、少なくとも一つの、ジスルフィドではない(lr)を含むものとする、
ことを特徴とする薬剤送達結合体。
【請求項53】
下式:
V−L−D
を持つビタミン受容体結合性薬剤送達結合体であって、式中、
Lは、(ls)a、および(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、aおよびbは、それぞれ独立に、0、1、2、3、または4であり、かつ、
Vはビタミン受容体結合性成分であり、Dは薬剤、またはその類縁体または誘導体である、
ことを特徴とする薬剤送達結合体。
【請求項54】
前記薬剤はハプテンであることを特徴とする、請求項53の薬剤送達結合体。
【請求項55】
前記ハプテンは、フルオレセインおよびジニトロフェニルから成るグループから選ばれることを特徴とする、請求項54の薬剤送達結合体。
【請求項56】
請求項53から55のいずれか1項による薬剤送達結合体、および、その製薬学的に受容可能な担体、希釈剤、または賦形剤を含む製薬組成物。
【請求項57】
病原細胞集団を抱える宿主動物において病原細胞集団を除去する方法であって、前記病原細胞集団のメンバーは、ビタミン、またはその類縁体または誘導体に対して接触可能な結合部位を持ち、結合部位は、病原細胞によって特異的に発現されるか、過剰発現されるか、または優先的に発現され、前記方法は、請求項53から55のいずれか1項による薬剤送達結合体、またはその製薬組成物を、前記宿主に投与する工程を含む方法。
【請求項58】
ビタミン受容体結合性薬剤送達結合体中間体であって、
(a)第1末端と第2末端とを有する二価のリンカー、
(b)薬剤、またはその類縁体または誘導体、および、
(c)結合基を含み、
二価リンカーは、スペーサーリンカー、放出リンカー、およびヘテロ原子リンカー、およびそれらの組み合わせから成るグループから選ばれる一つ以上の成分を含み、二価リンカーは、ジスルフィドではない少なくとも一つの放出リンカーを含むものとし、
結合基は、二価リンカーの第1末端において二価リンカーに共有結合し、薬剤、またはその類縁体または誘導体は、二価リンカーの第2末端において二価リンカーに共有結合し、かつ、
結合基は、ビタミン受容体結合性成分と共有結合を形成することが可能な求核基、求電子基、またはその前駆物質であるものとする、
ことを特徴とする中間体。
【請求項59】
前記ビタミン受容体結合性成分は、ビタミン、または、その、ビタミン受容体結合性類縁体または誘導体であり、前記結合基はマイケルアクセプターであり、前記二価リンカーは、式-C(O)NHN=, -NHC(O)NHN=、および-CH2C(O)NHN=から成るグループから選ばれる式を有する放出リンカーを含むことを特徴とする、請求項58の中間体。
【請求項60】
前記二価リンカーは、第1放出リンカーおよび第1スペーサーリンカーを含み、前記第1放出リンカー、第1スペーサーリンカーおよび結合基とは共同して下記の式:
(式中、Dは、薬剤、またはその類縁体または誘導体であり、nは、1、2、3、および4から成るグループから選ばれる整数である)を持つ化合物を形成することを特徴とする、請求項58の中間体。
【請求項61】
前記ビタミン受容体結合成分は、アルキルチオール求核基を含むことを特徴とする、請求項60の中間体。
【請求項62】
下記の式:
(式中、Vはビタミン受容体結合成分であり、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、b、およびcは、0、1、2、3、および4から成るグループから選ばれる整数であり、Dは、薬剤、またはその類縁体または誘導体である)を持つ化合物の調製法であって、
(a)下記の式:
を持つ化合物を、下記の式:
を持つ化合物と反応させること、および、
(b)薬剤、またはその類縁体または誘導体のヒドラゾン誘導体を形成させること、
を含む調製法。
【請求項63】
下記の式:
(式中、Vはビタミン受容体結合成分であり、Lは、(lr)c、(ls)aと(lH)b、およびそれらの組み合わせから成るグループから選ばれ、(lr)は放出リンカー、(ls)はスペーサーリンカー、(lH)はヘテロ原子リンカーであり、a、b、およびcは、0、1、2、3、および4から成るグループから選ばれる整数であり、Dは、薬剤、またはその類縁体または誘導体である)を持つ化合物の調製法であって、
下記の式:
を持つ化合物を、下記の式:
を持つ化合物と反応させること、
を含む調製法。
【請求項64】
前記薬剤、またはその類縁体または誘導体は、ダウノマイシン類、ダウノマイシノン類、ダウノルビシン類、アクラマイシン類、タキソール類、クロモマイシン類、およびアウリル酸類(aurilic acids)から成るグループから選ばれることを特徴とする、請求項63の調製法。
【図1】


【図2】
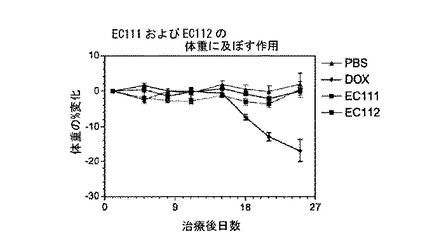
【図3】
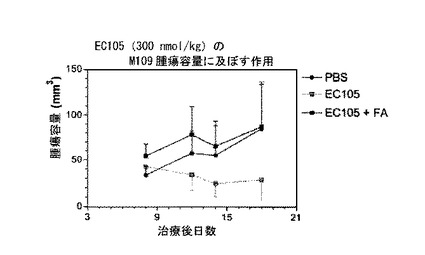
【図4】

【図5】
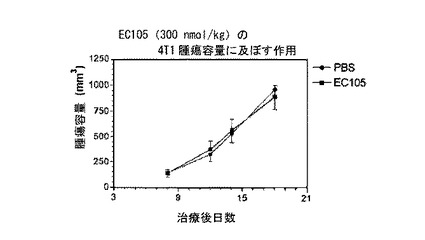
【図6】
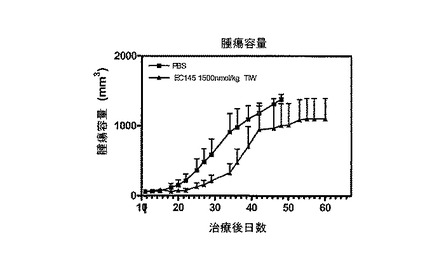
【図7】

【図8】

【図9】

【図10】

【図11】
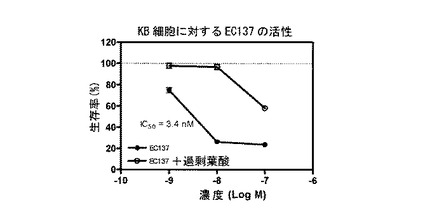
【図12】

【図13】
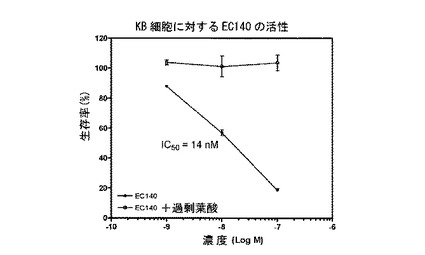
【図14】
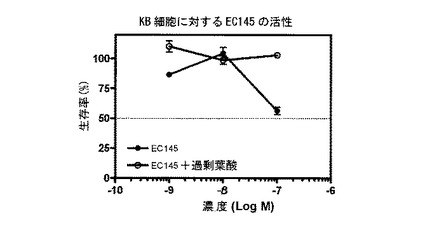
【図15】
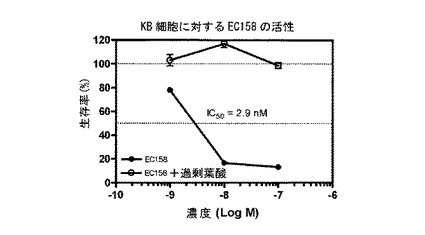
【図16】
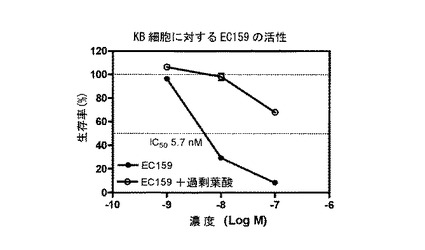
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2011−256184(P2011−256184A)
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−165553(P2011−165553)
【出願日】平成23年7月28日(2011.7.28)
【分割の表示】特願2005−518938(P2005−518938)の分割
【原出願日】平成16年1月27日(2004.1.27)
【出願人】(504389588)エンドサイト,インコーポレイテッド (16)
【Fターム(参考)】
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願番号】特願2011−165553(P2011−165553)
【出願日】平成23年7月28日(2011.7.28)
【分割の表示】特願2005−518938(P2005−518938)の分割
【原出願日】平成16年1月27日(2004.1.27)
【出願人】(504389588)エンドサイト,インコーポレイテッド (16)
【Fターム(参考)】
[ Back to top ]