
組換え哺乳動物細胞、組換え哺乳動物細胞の製造方法、および目的タンパク質の生産方法
【課題】組換え哺乳類動物細胞により目的タンパク質を高レベルで、かつ選択薬剤の非存在下においても安定に長期間発現させる方法を提供する。
【解決手段】ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座に外来性の目的タンパク質遺伝子発現ユニットの複数コピーが組込まれてなる組換え哺乳類動物細胞を用意し、該細胞を培養して前記目的タンパク質を産生させる。目的タンパク質遺伝子のコピー数が2以上である。目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれている。目的タンパク質遺伝子または発現ユニットが、タンデムに並んで繰り返し組込まれている。
【解決手段】ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座に外来性の目的タンパク質遺伝子発現ユニットの複数コピーが組込まれてなる組換え哺乳類動物細胞を用意し、該細胞を培養して前記目的タンパク質を産生させる。目的タンパク質遺伝子のコピー数が2以上である。目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれている。目的タンパク質遺伝子または発現ユニットが、タンデムに並んで繰り返し組込まれている。
【発明の詳細な説明】
【関連出願】
【0001】
本特許出願は、2007年8月10日に出願された日本国特許出願2007−210122号、および2008年7月24日に出願された日本国特許出願2008−191278号に基づく優先権の主張を伴うものであり、かかる先の特許出願における全開示内容は、引用することにより本明細書の一部とされる。
【発明の背景】
【0002】
発明の分野
本発明は、組換え哺乳類動物培養細胞、および該細胞を利用したタンパク質の生産方法に関する。
【0003】
背景技術
原核生物や真核生物を宿主とした、種々の組換えタンパク質生産システムが知られている。哺乳類動物細胞を宿主とした組換えタンパク質生産システムによれば、ヒトを始めとする高等動物由来のタンパク質に対し、糖鎖付加、フォールディング、リン酸化などの翻訳後修飾を、より生体で作られるものと同じように施すことが可能である。
【0004】
この翻訳後修飾は、タンパク質の本来有する生理活性を組換えタンパク質で再現するために必要なものである。従って、そのような生理活性が特に必要とされる医薬品などに用いられる組換えタンパク質の生産系には、哺乳類動物細胞を宿主としたタンパク質生産システムが好ましく用いられている。
【0005】
医薬タンパク質の工業的生産においては、高いタンパク質発現レベルでかつ発現レベルが安定に維持されることが重要である。特に発現レベルが安定に維持されることに関しては、コスト面のみならず、医薬タンパク質としての同一性および安全性を証明するためにも重要である。組換えタンパク質生産細胞を工業的スケールでの生産に用いるためには生産細胞クローンの培養スケールの拡大を図る必要がある。これには通常少なくとも樹立直後のクローンから約60回の細胞分裂を経なければならないと見積られており(Brown, M. E. et al. (1992) Cytotechnology, 9, 231-236.)、この間、発現レベルは一定に保たれていなければならない。
【0006】
また、培養スケールの拡大の期間中または期間後、組換え細胞クローンの比生産性が減少することにより、生産細胞としての使用が難しくなることがあり、この場合、数ヶ月にも及ぶ開発期間が無駄となる(Barnes, L. M. et al. (2003) Biotechnol. Bioeng. 81, 631-639.)。
【0007】
また、スケールアップ期間及び実生産期間中に、選択薬剤を使用することは培養コストを上昇させることのみならず、医薬品への毒物混入リスク回避のために行われる精製プロセスのコストも上昇させる。このため、選択薬剤は添加せずに行われるのが一般的である。
【0008】
このような背景から、目的タンパク質を生産する組換え哺乳類動物細胞クローンには、高い比生産性レベルを有し、選択薬剤を添加することなく比生産性レベルを安定に維持できる特性を有することが求められる。
【0009】
高い比生産性レベルの達成に関しては、目的タンパク質をコードする外来性遺伝子のコピー数を遺伝子増幅技術により増加させる方法が一般的であり、CHO-DHFRシステムやGS-NS0システムなどが実用技術として確立されている(特公平7-40933、Werner R. G. et al. (1998) Arzneim.-Forsch./Drug Res 48, 870-880.など)。
【0010】
しかしながら、比生産性レベルが増大した細胞クローンの選択後、選択薬剤を含まない培地で選択細胞クローンの培養を継続する場合、ほとんどのクローンにおいて比生産性レベルの減少または消失が確認されている。さらに、組込み遺伝子のコピー数の増加に必ずしも比例して目的タンパク質の発現レベルが増大しないことも報告されている(特表2002-541854公報、Kim, N. S. et al. (1998) Biotechnol. Bioeng., 60, 679-688など)。
【0011】
このような比生産性レベルの減少または目的タンパク質生産の消失は、遺伝子コピー数の減少が主な原因であることが報告されている(Kim, N. S. et al. (1998) Biotechnol. Bioeng., 60, 679-688、Kim, S. J. (1998) Biotechnol. Bioeng. 58, 73-84.、Yoshikawa, T. et al. (2000) Biotechnol. Progr. 16, 710-715.など)。
【0012】
また、他の原因として、複数の同一配列を有する遺伝子コピーがタンデムに繰り返して組込まれた場合に生じる、Repeat-Induced Gene Silencing(RIGS)が報告されている(Henikoff, S. (1998) Bioessays 20, 532-535.、Garrick, D. et al. (1998) Nat. Genet. 18, 56-59.)。RIGSは遺伝子コピー数が2〜3コピー程度の少ない場合においても生じることが示されている(McBurney, M. W. (2002) Exp. Cell Res. 274, 1-8)。
【0013】
そして、これまでのところ、目的タンパク質の比生産性レベルの安定性に関して十分な解決策が提示されているとはいえない。クローン選択は長期培養時における成長速度並びに生産性に関して蓄積されたデータを元に、経験的に行なわれているのが現状である。そして、この経験的な方法では、目的タンパク質の比生産性レベルの安定な細胞クローンを取得できることは希であり、偶然によるところが大きい(Barnes, L. M. et al. (2003) Biotechnol. Bioeng. 81, 631-639.)。
【0014】
本発明者らは、先に、ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座にシングルコピーのGreen fluorescent protein(GFP)の発現ユニットを相同組換えにより組込み、組換え細胞を取得する手法を報告している(Biotechnol Bioeng 95(6): 1052-1060. 2006.。hprt遺伝子座への位置特異的な組込みにより作製された組換え細胞クローンは、選択薬剤非添加条件下において、長期間の培養期間中も比生産性レベルを安定に保持する。
【0015】
通常のランダム組換え細胞クローンの取得においては、複数コピーの外来性遺伝子が同時に同一染色体位置に組み込まれる場合が多い(Martin, D. I. K. and Whitelaw, E. (1996) Bioessays 18, 919-923.)。これはRIGSの標的になりうる。これに対し、相同組換えなどにより、1コピーを組込む方法ではRIGSの回避が可能である(Whitelaw, E. et al. (2001) Methods in Mol. Biol. 158, 351-368.)。
【0016】
一方で、1コピーの組込みにおいても、組込まれる染色体位置により比生産性レベルの安定性は大きく異なることが知られている(Walters, M. C. et al. (2007) Genes Dev. 10, 185-195.)。
【0017】
このような事実から、次のようなことが分かる。すなわち、hprt遺伝子位置が、外来性遺伝子を1コピー組込んだ際に、その比生産性レベルの安定性の保持において優れたものである。しかしながら、一方で、hprt遺伝子位置に複数の外来性遺伝子コピーを組込んだ際の比生産性レベルの安定性については、RIGSを回避可能か否かについても含め、予測はできない。
【0018】
また、特表平7-500969公報には、ヒト線維肉腫由来のHT1080細胞のhprt遺伝子座にエリスロポエチン遺伝子発現ユニットを相同組換えにより組込んだとの開示がある。しかし、エリスロポエチン遺伝子の発現は確認されておらず、さらにその複数コピーを組込んだとの記載もない。
【発明の概要】
【0019】
本発明者らは、今般、hprt遺伝子座に目的タンパク質遺伝子を複数コピー組込むことにより得られた哺乳動物細胞は、長期間に渡り目的タンパク質の比生産性レベルを安定に維持させることができ、さらにその比生産性レベルが組込まれた目的タンパク質遺伝子のコピー数に比例するとの極めて意外な知見を得た。本発明は、かかる知見に基づくものである。
【0020】
したがって、本発明は、目的タンパク質を高レベルで安定に製造できる組換え細胞およびその製造方法ならびにその組換え細胞を用いた目的タンパク質の製造方法の提供をその目的としている。
【0021】
そして、本発明による哺乳動物細胞は、hprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーが組込まれてなるものである。
また、本発明による上記哺乳動物細胞の製造方法は、宿主細胞のhprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーを組込む工程を含んでなるものである。
また、本発明による目的タンパク質の製造方法は、上記哺乳動物細胞を培養して前記目的タンパク質を産生することを含んでなるものである。
【図面の簡単な説明】
【0022】
【図1】シングルコピーのエリスロポエチン遺伝子を含有するphprt-GT-EPO geneプラスミドベクターの模式図である。
【図2】組換えクローン(#14-5E, #19-3B)におけるサザンハイブリダイゼーションの結果を示す写真である。
【図3】(A)は、hprt遺伝子における相同組換えターゲット部位を示す。(B)は、1コピーのphprt-GT-EPO gene導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。また、(C)は、2コピーのphprt-GT-EPO gene導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。
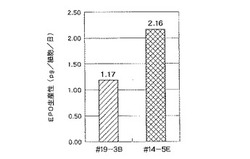
【図4】樹立直後にG418および6TG不含培地で培養した組換えクローン(#14-5E, #19-3B)のEPO生産性を示すグラフである。
【図5】G418および6TG不含培地で96日間培養を継続した場合の、組換えクローン(#14-5E)のEPO生産性の推移を示すグラフである。
【図6】G418および6TG不含培地で96日間培養を継続した前後における、組換えクローン(#14-5E)のサザンハイブリダイゼーションの結果を示す写真である。
【図7】1コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-1 プラスミドベクターの模式図である。
【図8】2コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-2 プラスミドベクターの模式図である。
【図9】3コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-3 プラスミドベクターの模式図である。
【図10】(A)は、ゲノムPCRによる相同組み換え反応の解析の詳細を説明する模式図である。 (B)は、導入ベクターのホモロジーアーム領域由来のDNA(3209bp)を指標とした、ゲノムPCR産物の電気泳動の結果を示す写真である。(C)は、導入ベクターのホモロジーアーム領域由来のDNA(1443bp)を指標とした、ゲノムPCR産物の電気泳動の結果を示す写真である。
【図11】エリスロポエチン遺伝子およびgs遺伝子増幅マーカーを含有するpSV-GS-neo-GT-EPO #22プラスミドベクターの模式図である。
【図12】(A)は、hprt遺伝子座における相同組換えターゲット部位を示す。(B)は、1コピーのpSV-GS-neo-GT-EPO #22導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。
【図13】組換えクローン(118S-1、118S-2、118S-3、118S-5)におけるサザンハイブリダイゼーションの結果を示す写真である。
【図14】Real-time定量PCR法によりゲノム中のgs遺伝子コピー数を解析した結果である。2-1F8および1-2E6はMSX 5 μMに対して耐性を示した細胞、S-5(Glu-)およびS-5(Glu+)はそれぞれグルタミン非添加培地またはグルタミン添加培地中で維持していた118S-5細胞を示す。結果は、S-5(Glu+)の値に対する相対値で示してある。
【発明の具体的説明】
【0023】
hprt遺伝子座/目的タンパク質遺伝子の複数コピーの導入
本発明による目的タンパク質の製造方法にあっては、hprt遺伝子座を複数コピーの目的タンパク質遺伝子組換えのターゲット領域とする。このhprt遺伝子は、ヒト等のX染色体長腕に存在するハウスキーピング遺伝子の一つとして知られている。このhprt遺伝子座に複数コピーの外来性の目的タンパク質を組込んで発現させる場合、目的タンパク質の比生産性レベルがそのコピー数にほぼ比例して増加する。しかも、このタンパク質発現量を長期間に渡り安定に維持することが可能となる。本発明の好ましい態様によれば、この安定性は、選択薬剤の非存在下にあっても長期にわたり維持させることができる。
【0024】
ここで、外来性遺伝子の複数コピーとは、2以上の実質的に同一の遺伝子または遺伝子発現ユニットが、hprt遺伝子座上に存在することを意味する。ここで、複数の発現ユニットの転写方向は必ずしも同一方向でなくともよく、またすべて同一方向にタンデムに繰り返して存在することもできる。繰り返し存在している遺伝子または発現ユニット間には、他のDNA配列が含まれていてもよい。また、比例するとは、比生産性レベルが遺伝子コピー数と正の比例関係にあることを意味し、好ましくは、遺伝子コピー数の増加に応じて比生産性レベルが加算的に上昇することを意味する。
【0025】
従来、目的タンパク質遺伝子の複数コピーを、その発現量の増加を期待して組込むことは行われていたが、多くの場合、組込まれたコピー数に比例するとまでは言い難い。よって、本発明における、組込まれたコピー数に比例して発現量が増加するとの効果は、目的タンパク質の生産に極めて有利であり、かつ意外なものである。
【0026】
本発明による製造方法にあっては、hprt遺伝子座に組込まれる外来性の目的タンパク質遺伝子のコピー数は2以上であり、好ましい上限は500程度であり、より好ましい上限は100であり、さらに好ましい上限は10であり、さらに好ましい上限は3である。また、目的タンパク質遺伝子の複数コピーは、後述するベクター等によりhprt遺伝子座に組込まれうる。
【0027】
目的タンパク質遺伝子
上記目的タンパク質遺伝子は、cDNAに由来する配列であっても、ゲノムDNAに由来する天然イントロンを含む構造遺伝子であっても、好適に利用可能である。また、目的タンパク質遺伝子は、医薬として有用なタンパク質をコードしていることが好ましい。該タンパク質は細胞内に蓄積されるものであっても、細胞外に分泌されるものであっても本発明において利用することが可能である。また、かかる目的タンパク質としては、酵素、サイトカイン、ホルモン、抗体、凝固因子、調節タンパク質、レセプター等が挙げられるが、より具体的な例としては、エリスロポエチン、モノクローナル抗体、組織特異的プラスミノーゲン活性化因子、顆粒球コロニー活性化因子等が挙げられる。
【0028】
発現ユニット
上記目的タンパク質遺伝子は、hprt遺伝子座において、プロモーター配列や転写終結シグナル配列等の発現に必要な要素を含む発現ユニットとして組込まれることが好ましい。したがって、本発明の一つの態様によれば、目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる。
【0029】
また、上記発現ユニットにおけるプロモーターや転写終結シグナル等の発現に必要な要素は、目的タンパク質遺伝子の種類、性質等に応じて適宜決定してよく、プロモーター配列の好適な例としては、CMVプロモーター、SV40 プロモーター等が挙げられる。また、転写終結シグナル配列の好適な例としては、BGHポリAシグナル配列、SV40ポリAシグナル配列等が挙げられる。
【0030】
また、上記発現ユニットにおいて、プロモーター配列、転写終結シグナル配列の他の発現に必要な要素としては、例えば、目的遺伝子を効率的に発現させるための調節エレメント(例えば、エンハンサー、IRES(internal ribosome entry site)配列)等が挙げられる。調節エレメントはその性質に応じて、発現ユニットにおける適切な位置に配置することが可能である。これら発現に必要な要素は、宿主との組み合わせおよび目的タンパク質の生産性を勘案して選択することが好ましい。
【0031】
また、上記発現に必要な要素は、翻訳反応の増強を勘案して、目的タンパク質遺伝子以外のイントロン配列を含んでもよい。イントロン配列は発現ユニット中の転写開始部位から転写終結部位の間に配置されていればよく、好ましくは、ウイルス由来のもの、哺乳類動物ゲノム由来のもの等が挙げられるが、宿主内でスプライシング効率の高いイントロン配列であることが好ましい。
【0032】
複数コピーの発現ユニットのhprt遺伝子座への組換え
本発明の一つの態様によれば、複数コピーの発現ユニットをhprt遺伝子座へ組換える場合、複数コピーの発現ユニットを含むベクターを用いることが好適である。また、シングルコピーの発現ユニットを含むベクターを哺乳動物細胞に導入し、複数コピーの発現ユニットが組換えられた細胞を選択・取得してもよく、本発明にはかかる態様も包含される。また、本発明の別の態様によれば、遺伝子増幅マーカー遺伝子と目的タンパク質遺伝子の発現ユニットとを含むベクターを用いてhprt遺伝子座へ組換えた後、適切な遺伝子増幅プロセスにより、複数コピーの発現ユニットが組込まれた細胞を選択することもまた好適である。
【0033】
本発明の好ましい態様によれば、目的タンパク質遺伝子の発現ユニットは、hprt遺伝子座に相同組換えにより組換えられたものである。かかる相同組換えの利用は、組換え細胞を迅速に構築し、さらには、目的タンパク質遺伝子を高レベルで安定に発現させる上で有利である。
【0034】
また、相同組換えの標的となるhprt遺伝子座中の位置は、目的タンパク質遺伝子の効率的な発現を妨げない限り適宜設定してよく、例えば、本発明の一つの態様によれば、hprt遺伝子のエクソン配列(例えば、エクソン3等)上に設定する。エクソン配列上にターゲット領域を設定することは、hprt遺伝子自体の発現を阻害するため、6−チオグアニン(6-TG)や8−アザグアニン(8-AG)による薬剤選択によって、効率的に組換え細胞を取得する上で有利である。
【0035】
ベクター
本発明に用いられるベクターとしては、哺乳動物細胞染色体へ目的遺伝子を組換えうるものであれば特に限定されず、例えば、プラスミドベクター、コスミドベクター、ファージベクター、人工染色体ベクター等が挙げられるが、好ましくは、プラスミドベクターである。また、上記ベクターは、直鎖状または環状のいずれで構成してもよい。
【0036】
上記ベクターは、当技術分野で周知となっている標準的方法を利用して構築することができ、例えば、Sambrook, J.らの「Molecular Cloning: a laboratory manual」、Cold Spring Harbor Laboratory Press、New York(1989)に記載の方法に従って構築することができる。
【0037】
相同組換えにより発現ユニットをhprt遺伝子座に組換える場合、該発現ユニットを組換えるためのベクターには、hprt遺伝子座の一部と相同組換え可能な相同性を有する相同DNA配列を配置する。ベクターに含まれる相同DNA配列は、目的タンパク質遺伝子の効率的な組換えおよび発現を妨げない限り、単数であっても複数であってもよいが、好ましくは2つである。さらに、かかる2つの相同DNA配列は、ベクターの5’末端および3’末端に配置されていることが好ましい。したがって、本発明の一つの態様によれば、ベクターは、5’末端に配置された相同DNA配列と、目的タンパク質遺伝子の発現ユニットと、3’末端に配置された相同DNA配列とを少なくとも含んでなる。
【0038】
また、相同DNA配列は、hprt遺伝子座と相同組換え可能な相同性および長さを有する。相同組換え反応の生じやすさまたはその確実性を考慮すれば、相同DNA配列とhprt遺伝子座との相同性は十分に高い方が好ましく、好ましくは99%以上、より好ましくは99.9%以上が相同であり、さらに好ましく両者は同一配列である。また、二つの相同DNA断片の長さは各々が、好ましくは数百塩基対(bp)以上であり、より好ましくは500bp以上であり、さらに好ましくは1000bp以上である。
【0039】
複数コピーの発現ユニットを含むベクターを構築する場合、かかるベクターは、制限酵素切断反応およびライゲーション反応を組み合わせて好適に構築される。例えば、発現ユニットの両末端にII型制限酵素の認識配列を含有させ、該認識配列用の制限酵素で切断反応を行い、不要なDNA配列(大腸菌内での操作用配列等)をゲル切り出し等の処理によって除去し、得られた発現ユニットのライゲーション反応を行うことにより、複数の発現ユニットがタンデムに並んだベクターDNAを構築しうる。
【0040】
上記制限酵素のすべての認識配列を同一のものにする場合には、複数の目的タンパク質遺伝子の転写方向がランダムであるベクターDNAを構築しうる。また、制限酵素認識配列として異なるものを選択する場合、DNA配列中の目的タンパク質遺伝子の向きを制御することが可能であり、例えば、すべての目的タンパク質遺伝子が同方向に配置されたベクターDNAや、目的タンパク質遺伝子の向きが交互に異なるベクターDNA等を構築しうる。
【0041】
使用可能な上記制限酵素は平滑末端を生じるものであっても付着末端を生じるものであってもよいが、ライゲーション効率を勘案すれば、付着末端を生じる制限酵素が好ましい。また、目的タンパク質遺伝子の向きの制御を勘案すれば、種々の認識配列を有する制限酵素を利用することが好ましく、例えば、Biotech. Appl. Biochem. 20: 157-171. 1994.や特表2003-530886等に記載されるIIS型の制限酵素、例えばSfi I制限酵素等が挙げられる。Sfi I制限酵素は、1つの制限酵素の利用により64通りの異なる付着末端を生じさせることから、多数の目的タンパク質遺伝子の向きを制御する上で好ましい。制限酵素に関する上述の文献の内容は、引用することにより本明細書の一部とされる。
【0042】
ライゲーション反応により構築した複数コピーの発現ユニットを含むベクターDNAは、フェノール・クロロホルム抽出等により精製し、ベクターの種類等を勘案して選択した大腸菌、酵母等の宿主細胞において増殖させることができる。また、適当な宿主細胞が利用できない場合、複数コピーの目的タンパク質遺伝子を含むベクターDNAは、フェノール・クロロホルム抽出処理後、直接哺乳類動物細胞へと導入することもできる。
【0043】
また、遺伝子増幅を行うためのベクターにおいては、目的タンパク質遺伝子の発現ユニットに加え、遺伝子増幅マーカー遺伝子が配置される。遺伝子増幅マーカー遺伝子は適切に発現するよう、目的タンパク質遺伝子の発現ユニットの近傍に配置されていればよい。また、目的タンパク質遺伝子の発現ユニットはシングルコピーで含有されていても複数コピーで含有されていてもよい。
【0044】
また、上述したベクターいずれにおいても、上記の他、組換え細胞の選択を勘案し、薬剤耐性による優性選択が可能なマーカー遺伝子(ネオマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、ゼオシン耐性遺伝子、ピューロマイシン耐性遺伝子)等を適宜組み込むことが可能である。
【0045】
ベクターの細胞への導入方法
上記ベクターの哺乳動物細胞への導入は、当該技術分野において公知の方法を用いることが可能であり、例えば、エレクトロポレーション法、マイクロインジェクション法、リン酸カルシウム法、リポフェクション法などが挙げられる。かかる導入方法は、哺乳動物細胞の種類、ベクターのサイズ、導入効率等を勘案して当業者により適宜選択される。ここで、ベクターが環状である場合、公知の方法により線状化して細胞に導入されてもよい。
【0046】
組換え細胞の選択
組換え哺乳動物細胞は、薬剤選択等の当該分野で周知となっている方法で選択し、取得することができる。例えば、目的タンパク質の発現ユニットを含むプラスミドベクターが優性選択が可能な薬剤耐性遺伝子等の選択マーカーを有する場合、選択薬剤を添加した培地で細胞の培養を行うことにより、組換え細胞を選択し、取得することが可能である。また、hprt遺伝子の発現をブロックする位置において目的タンパク質遺伝子が組込まれた場合、6-TGまたは8-AGを培地に添加することにより、組換え哺乳動物細胞を効率的に選択しうる。
【0047】
さらに、組換え哺乳動物細胞の選択においては、PCR反応等による組換え細胞のゲノムDNA解析や、サザンブロッティング法等を用いることにより、複数コピーの目的タンパク質遺伝子の発現ユニットを含む組換え細胞を正確に選択することが好ましい。また、バックグラウンドの低減を勘案すれば、ベクターの導入前に、哺乳動物細胞をHAT(ヒポキサンチン、アミノプテリン、チミジン)含有培地で前培養することが好ましい。
【0048】
遺伝子増幅による複数コピーの組込み
適切な遺伝子増幅マーカー遺伝子と外来性遺伝子(好ましくはそれを発現ユニットとして)とを含むベクターをhprt遺伝子座に組換えた細胞を用意し、適切な遺伝子増幅プロセスを行うことにより、hprt遺伝子座に外来性遺伝子発現ユニットが複数コピー組込まれた組換え細胞を取得することができる。
【0049】
遺伝子増幅技術では、遺伝子増幅マーカー近傍(〜10kbp程度)のDNA配列も同時にコピー数が増幅されることが知られている。この増幅は同一染色体位置で生じるため、本技術における遺伝子増幅の利用により、hprt遺伝子座内で目的タンパク質遺伝子のコピー数を増加させることができる。また、遺伝子増幅を利用した場合、増加したコピーの配向は制御できず、必ずしも発現ユニットが同一転写方向に揃ったタンデムな配置とはならない。
【0050】
本発明にあっては、公知となっている遺伝子増幅マーカー遺伝子であればいずれも好適に利用可能であるが、好ましくは、ジヒドロ葉酸還元酵素遺伝子(DHFR)またはグルタミン合成酵素遺伝子(GS)であり、より好ましくはGS遺伝子である。また、遺伝子増幅プロセスとしては、DHFR阻害剤であるメトトレキセート(MTX)を添加した培地での耐性細胞のスクリーニングや、GS阻害剤であるメチオニンスルフォキシミン(MSX)を添加した培地でのスクリーニング等の周知の方法が好適に利用可能である。また、MTXやMSXの添加濃度を段階的に引き上げるという周知の方法がより好適に利用可能である。
【0051】
組換え哺乳動物細胞
また、本発明による哺乳動物細胞は、上述の手法により製造されるものであり、hprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーが組込まれてなることを特徴とする。かかる組換え哺乳動物細胞にあっては、目的タンパク質遺伝子の比生産性レベルはその遺伝子コピー数に比例して増加し、この比生産性レベルは長期間に渡り安定に維持しうる。
さらに、この複数コピーの目的タンパク質遺伝子の長期的な発現安定性は、選択薬剤の非存在下であっても維持され、培養コストや異物混入リスク回避のために行われる精製プロセスのコストを低減する上で有利である。
【0052】
そして、上記組換え哺乳動物細胞において、目的タンパク質遺伝子のコピー数は2以上である。また、本発明のより好ましい態様によれば、上記組換え哺乳動物細胞における目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる。また、上記組換え哺乳動物細胞における目的タンパク質遺伝子または前記発現ユニットは、タンデムに並んで繰り返し組込まれてもよい。また、一つの態様によれば、上記組換え哺乳動物細胞における目的タンパク質遺伝子は、相同組換えにより組込まれたものである。
【0053】
また、本発明による組換え哺乳動物細胞にあっては、上記目的タンパク質の比生産性レベルはhprt遺伝子座に組込まれた目的タンパク質遺伝子のコピー数等に応じて増加するものであり、例えば、目的タンパク質遺伝子のコピー数が2つである場合、1細胞あたり約0.4〜20pg/日とすることができる。
【0054】
また、本発明による組換え哺乳動物細胞は、上記目的タンパク質の比生産レベルを、少なくとも97日間維持することができる。
上記目的タンパク質遺伝子の長期的な発現安定性は、選択薬剤の非存在下であっても維持されるものであり、かかる選択薬剤としては、細胞選択において用いられる公知薬剤が挙げられるが、例えば、ネオマイシン、6TG、MTX、MSX等である。
【0055】
宿主細胞
また、本発明において、宿主細胞として用いられる哺乳動物細胞は、好ましくはヒト由来のものであり、かかる哺乳動物細胞の具体的な例としては、例えば、ヒト線維肉腫由来のHT1080細胞株等が挙げられるが、hprt遺伝子の哺乳動物における普遍性を鑑みれば、これらに限定されるものではない。
また、相同組換えによりhprt遺伝子座へ目的タンパク質遺伝子を組込む場合、6TGや8AGなどの選択薬剤による相同組換え細胞の選択を勘案すれば、宿主細胞としてはhprt遺伝子座を全ゲノム中に1ヶ所のみ有する細胞が好ましい。そのような細胞の例としては、上述したHT1080株や、男性由来のX染色体を1本しか持たない細胞株などが挙げられる。
【0056】
培養/目的タンパク質の単離
また、本発明による目的タンパク質の製造方法にあっては、hprt遺伝子座に複数コピーの目的タンパク質遺伝子が組込まれた組換え哺乳動物細胞を培地中で培養し、目的タンパク質を産生させることができる。上記哺乳動物細胞の培養条件の詳細は、細胞の性質および状態等に応じて当業者によって適宜決定されるが、培養コストを勘案すれば、培地は、好ましくは無血清培地であり、より好ましくはChemically defined(CD)培地である。
【0057】
また、本発明の一つの態様によれば、目的タンパク質は、組換え哺乳動物細胞の培養物から単離することが好ましい。単離手法については、目的タンパク質の性質等に応じて、遠心分離、ゲルろ過、フィルターろ過などの公知の手法を用いることができる。
【実施例】
【0058】
以下、本発明を実施例によって具体的に説明するが、本発明はこれら実施例に限定されるものではない。
なお、以下の実験において、制限酵素による反応、PCR反応、ライゲーション反応等の各反応条件は、メーカーの推奨する反応条件、あるいはMolecular Cloning 2nd edition Sambrook et.al. Cold Spring Harbor Laboratory Pressに記載の方法に従って設定した。また、得られた種々のプラスミドベクターDNA等については、自動DNAシークエンサー(310 Genetic Analyzer、Applied Bio Systems, Inc.)を用いてDNA配列を決定した。
【0059】
例1:hprt遺伝子座に複数のエリスロポエチン遺伝子発現ユニットが組込まれた組換え細胞の製造1
1−1:プラスミドベクターの構築
以下、複数コピーのエリスロポエチン(EPO)遺伝子をhprt遺伝子座に対して組込むため、シングルコピーのEPO遺伝子を含む、図1で表されるベクター(phprt-GT-EPO gene)を構築した。
【0060】
相同DNA配列の取得
JCRBセルバンクから入手したヒト由来細胞株HT1080細胞株(カタログ番号:IF050354)をGFX(商品名) Genomic Blood DNA Purification Kit (Amersham Biosciences)により処理し、ゲノムDNAを得た。次に、このゲノムDNAを鋳型とし、PCR反応(KOD-Plus-、TOYOBO)によって、ターゲットとなるhprt遺伝子の相同DNA配列(HA1およびHA2)をクローニングした。HA1およびHA2は、後述する図3でも示される通り、hprt遺伝子のエクソン3を含む領域と相同な配列に設定した。PCR反応に用いたプライマー配列は以下に示す通りである。
【0061】
HA1 sense プライマー:
5’-CCTGCAGGTCGCGATTGGTACTTGTTCAGCTTTATTCAAG-3’(配列番号1)
HA1 antisense プライマー:
5’-GTCGACAAGGACGCGTTCTGATAAAATCTACAGTCATAGGA-3’(配列番号2)
HA2 sense プライマー:
5’GTCGACCTCTAGCTAGAGCTTGGCGTAATCATGGTCTCCGGAGACTGAAGAGCTATTGTGTGAGTAT-3’(配列番号3)
HA2 antisense プライマー:
5’-ACATGTTCTCTTAAGTCGCGAAGTAGTGTTATGATGTATGGGCATA-3’(配列番号4)
【0062】
上記PCR反応においては、HA1センスプライマーの5’末端には、制限酵素Sse 8387IおよびNru Iの認識サイトを付加した。同様に、HA1アンチセンスプライマーの5’末端にはSal IおよびMlu Iの認識サイト、HA2センスプライマーの5’末端にはSal IおよびAcc IIIの認識サイト、HA2アンチセンスプライマーの5’末端にはPci IおよびNru Iの認識サイトをそれぞれ付加した。
【0063】
pQBI25プラスミドベクター(和光純薬株式会社)のDNAから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマーの5’末端および3’末端に、制限酵素Pci IおよびSse 8387Iの認識サイトを付加した。
【0064】
Ecoli sense プライマー:
5’-ACATGTGAGCAAAAGGCCAGCAAAAGGCCAGGAAC-3’(配列番号5)
Ecoli antisense プライマー:
5’-CCTGCAGGGACGTCAGGTGGCACTTTTCGGGGAAATGTGC-3’(配列番号6)
【0065】
HA1、HA2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Pci I、Sse 8387IおよびSal Iにより切断し、ライゲーション反応を行うことによりpHA12プラスミドベクターを得た。次に、pHA12プラスミドベクターを制限酵素Mlu IおよびSal Iにより切断し、5’末端から順に、HA2と、ori配列およびアンピシリン耐性遺伝子を含むDNA配列と、HA1とが連結したDNA配列1を得た。
【0066】
また、pQBI25プラスミドベクターを、制限酵素Not IおよびSal Iにより切断し、BGH ポリAシグナル配列(BGH pA)およびネオマイシン耐性遺伝子を含むDNA配列2(SV40, neoR, SV40pAを含む発現カセット)を取得した。
【0067】
また、pcDNA3.1プラスミドベクター(インビトロジェン)を、制限酵素Mlu IおよびNot Iにより切断し、CMVプロモーター・エンハンサー配列(CMV)およびマルチクローニングサイトを含むDNA配列3を取得した。
次に、DNA配列1〜3をライゲートしてphprt-GT-MCSプラスミドベクターを得た。
【0068】
次に、HT1080細胞株から抽出したゲノムDNAを鋳型とし、PCR反応によってエリスロポエチン遺伝子を増幅した。PCR反応に用いたセンスプライマーの5’末端には、制限酵素Nhe Iの認識配列を付加し、およびアンチセンスプライマーの5’末端には制限酵素Eco RIの認識配列を付加した。プライマーの塩基配列は以下の通りである。
【0069】
EPO gene ss プライマー:
5'- CCTTGCTAGCATGGGGGTGCACGGTGAGTA -3'(配列番号7)
EPO gene as プライマー:
5'- CCTTGAATTCTCATCTGTCCCCTGTCCTGC -3'(配列番号8)
【0070】
上記PCR反応により増幅されたEPO 遺伝子と、制限酵素Nhe IおよびEco RI により処理されたphprt-GT-MCSプラスミドとのライゲーションにより、EPO 遺伝子をphprt-GT-MCSプラスミドのマルチクローニングサイトのNhe I−Eco RIサイト間に挿入させ、図1に示されるphprt-GT-EPO gene(9116bp)を得た。得られたphprt-GT-EPO geneは大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0071】
1−2:細胞へのベクターの導入
プラスミド線状化
プラスミドベクターphprt-GT-EPO geneを、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0072】
トランスフェクション
ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)を1×107細胞/mLに調製し、線状化したプラスミドベクターphprt-GT-EPO gene、2μgと混合した。次に、得られた混合液を用いて、エレクトロポレーション法によりphprt-GT-EPO geneを細胞株HT-1080へトランスフェクトした。エレクトロポレーションは、Gene Pulser (BioRad社)を用いて、950μFの条件で行った。この実験のさらなる詳細は、Biotech. Bioeng. 2006. 95: 1052-1060.に記載の条件に従った。トランスフェクトした細胞は、500 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養し(培地: Advanced MEM(GIBCO社)に5% FBS、1x Glutamax(GIBCO社)を添加したもの)、トランスフェクションから24時間後にG418(インビトロジェン社)を加えた(最終濃度; 500 μg/ mL)。
【0073】
スクリーニング
培養8-12日後、上記プレートにG418耐性コロニーが現れたことを確認した。この段階で、6TG(最終濃度; 50 μM)(和光純薬工業)が添加された新しい培地を加え、さらに8日間培養した。培養後、全ウェルを確認し、6TG耐性コロニーを単離した。
【0074】
1−3:サザンハイブリダイゼーションによる解析/目的細胞の取得
以下、サザンハイブリダイゼーションを用い、hprt遺伝子座に複数コピーのEPO遺伝子が組込まれた組換え細胞を取得するため、6TG耐性コロニーのスクリーニングを行った。
プローブの調製
phprt-GT-EPO gene中のネオマイシン耐性遺伝子に相補的な配列を有するNRプローブを、以下の手順で合成した。まず、phprt-GT-EPO gene中のネオマイシン耐性遺伝子コード配列の全長をPCRにより増幅し、pGEM T プラスミドベクター(Promega社)にTAクローニングした。次に、PCR DIGプローブ合成キット(ロシュ社、プライマー:M13 Forward/Reverseプライマー)を用いて、DIG(Digoxigein)標識されたプローブを作製した。
【0075】
メンブレンの調製
6TG耐性コロニーから各細胞クローンのゲノムDNAをGFX Genomic Blood DNA purification kit(Amersham Biosciences社)を用いて抽出し、Bgl II制限酵素により切断した。切断したゲノムDNA10μgを、0.6%アガロースゲルを用いて電気泳動し、ナイロンメンブレン(Hybond N+ membrane、AmaershamBiosciences社)へ転写した。得られたメンブレンは80℃で2時間インキュベートし、DNAをメンブレン上に固定した。
【0076】
ハイブリダイゼーション
上記メンブレンに対し、上記NRプローブをハイブリダイズさせた。この際、プレハイブリダイゼーション、ハイブリダイゼーションおよびプローブの検出は、DIGアプリケーション マニュアル(ロシュ社)に従って行った。また、プローブのストリッピイングは、ストリッピングバッファー(0.2M NaOH, 0.1% SDS)を用い、37℃で15分の条件で2回行った。
【0077】
図2に示される通り、組換えクローン#14-5Eにおいて、NRプローブにより15822 bpのDNA断片が検出された。この組換えクローンは、約30クローンのスクリーニングの結果、1クローンのみ取得された。一方、参照として、組換えクローン#19-3Bを選択したところ、8546 bpのDNA断片が検出された。
【0078】
以下、図3(A)〜(C)に示される模式図および図2の結果に基づき、例1の結
果を説明する。
図3(A)で示される通り、hprt遺伝子(1)の相同組換えのターゲット部位(2)は、エクソン3(3)を含む領域に設定され、ホモロジーアーム1(HA1)(4)およびホモロジーアーム2(HA2)(5)は、ターゲット部位における二つの隣接した領域に相同であるように設定されている。そして、Bgl II制限酵素サイトは、ターゲット部位(2)を挟むように位置し、その内側には存在しない。一方、図3(B)および(C)に示されるphprt-GT-EPO geneDNA配列(6)にはBgl II制限酵素サイトは存在しない。そして、phprt-GT-EPO geneDNA配列(6)には、NRプローブ(7)がハイブリダイズしうるように設計されている。
【0079】
図3(C)に示される通り、Bgl II制限酵素サイトの位置、およびDNA配列の鎖長に基づき、15822 bpのDNA断片が検出された組換えクローン#14-5Eは、2コピーのphprt-GT-EPO geneDNA配列(6)がタンデムに繰り返し組込まれた目的細胞であることが確認された。
一方、図3(B)に示される通り、参照細胞であって8546 bpのDNA断片が検出された組換えクローン#19-3Bは、1コピーのphprt-GT-EPO geneDNA配列(6)が組込まれたものであることが確認された。
【0080】
例2:EPO比生産性の検討
2−1:組換えクローンの培養
組換えクローン#14-5Eおよび#19-3Bを樹立直後に、選択薬剤G418および6TG不存在下で培養を行った。
培養は、5 % FBS(Japan Bioserum社製)含有Advanced MEM(GIBCO社製)中、5 % CO2存在下37 ℃で行われた。トリプシン処理による継代は、約4〜5日の間隔で、細胞数および細胞密度が70-80 %に達した時点で行った。定期的に対数増殖期において、細胞数のカウントと培地のサンプリングを行い、以下のEPOの生産性解析に用いた。
【0081】
2−2:組換えクローンおよび培地のサンプリング
上記方法により培養した組換えクローン#14-5E および#19-3Bについて、以下の手順に従い、サンプル細胞を取得した。
まず、細胞サンプルを1シャーレあたり5 x 104 細胞播種し、FBS(Japan Bio Serum社製)を5 %含んだAdvanced MEM(GIBCO社製)中、5 % CO2存在下37 ℃で培養した。培養を開始してから3日目および4日目(対数増殖期)に、培地を回収し、さらに、培地回収後、細胞をトリプシンで処理し、細胞を回収した。回収した培地は、以下のELISA解析に用い、回収した組換えクローンの細胞数は血球計算板を用いてカウントした。なお、上記トリプシン処理は、0.25% Trypsin-EDTA溶液(GIBCO社製)を加え、室温で3〜5分間行った。トリプシン処理後、細胞を回収前には、血清添加培地を加えて反応を停止させた。
【0082】
2−3:ELISA
回収した培地中のEPO蓄積量は、固定化抗体としてMonoclonal anti-human EPO(R&D Systems)、1次抗体としてPolyclonal anti-human EPO(R&D Systems)、2次抗体としてAnti-mouse Ig, horseradish peroxidase linked whole antibody(from donkey)(Amersham Biosciences社製)を用い、TMB No Hydrogen Peroxide 1 Component HRP Microwell substrate(BioFX社製)により、450nmの吸光度を測定することにより解析した。
【0083】
2−4:組換えクローン#14-5Eと#19-3BとのEPO比生産性の比較
樹立直後の組換えクローン #14-5Eおよび#19-3Bに関し、細胞あたりの1日あたりのEPO比生産性を、回収した培地中のEPO蓄積量、細胞数、培養時間に基づき、次式にしたがって算出した。
(EPO比生産性)=(EPO蓄積量4日目-EPO蓄積量3日目)/((細胞数3日目+細胞数4日目)/2)/((サンプリング間の培養時間)/24)
【0084】
結果は、図4に示される通りであった。
hprt遺伝子座に2コピーのEPO遺伝子を含有する#14-5Eでは比生産性が2.16 pg/細胞/日であった。一方、1コピーのEPO遺伝子を含有する#19-3Bでは1.17 pg/細胞/日であった。#14-5EクローンのEPO生産性は#19-3Bクローンの約2倍であり、組込まれたコピー数に対して加算的に、EPO比生産性レベルが増加していた。
【0085】
例3:選択薬剤非添加培地における長期培養の影響評価
3−1:EPO生産性の測定
組換えクローン#14-5Eを、例2と同様の手法により、G418および6TG不含培地中で97日間継続培養を行った。継続培養中、定期的にEPO生産性の測定を行った。
【0086】
結果は、図5に示される通りであった。組換えクローン#14-5Eでは、EPO比生産性レベルは少なくとも97日間、一定に保たれていた。
【0087】
3−2:サザンハイブリダイゼーション
また、hprt遺伝子座に並列して組込まれた2コピーのEPO遺伝子が長期培養後も安定に保持されているか否かを例1と同様の手法により、サザンハイブリダイゼーションによって確認した。
【0088】
図6に示される通り、培養開始日(0日)および97日後において、15822 bpのDNA断片が検出された。少なくとも97日間、hprt遺伝子座にタンデムに繰り返し組込まれた2コピーのEPO遺伝子配列が安定に保持されていることが確認された。
【0089】
以上の結果に示される通り、hprt遺伝子座に2コピーのEPO遺伝子がタンデムに繰り返し組込まれた細胞株において、EPO遺伝子コピー数の増加に伴いEPO比生産性レベルが加算的に増加し、両者はほぼ正比例していることが確認された。さらに、選択圧が存在しない条件での培養を継続しても、EPO遺伝子のコピー数の減少やジーンサイレンシング、比生産性レベルの減少は認められなかった。
【0090】
例4:hprt遺伝子に複数のエリスロポエチン遺伝子発現ユニットが組込まれた組換え細胞の製造2
以下の手順に従い、効率的に複数のエリスロポエチン(EPO)遺伝子をhprt遺伝子に対して組換えるため、複数コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3)を構築した。
【0091】
4−1.プラスミドベクターの構築
1コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-1)の構築
複数コピーのEPO遺伝子を含むベクターの構築用材料として、まず、図7で示される1コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-1)を以下の手順により構築した。
phprt-GT-MCSプラスミドをEco RVおよびApa I制限酵素で切断し、Klenow酵素を作用させた後、セルフライゲーション反応を行い、MCS中の不要な制限酵素認識配列を除去したphprt-GT-MCSプラスミドを得た。
また、pIRESプラスミド(BDバイオサイエンス)を鋳型とし、PCR反応によりCMVプロモーター・エンハンサー配列、イントロン配列を含むDNA断片をクローニングし、PCR増幅産物を得た。PCR反応に用いたプライマー配列は以下の通りである。
【0092】
CMV−IVS−sense プライマー(配列番号9):
5’-CCTTACGCGTTCAATATTGGCCATTAGCCA-3’
CMV−IVS−antisense プライマー(配列番号10):
5’-CCTTGCTAGCCTATAGTGAGTCGTATTAAG-3’
【0093】
センスプライマーおよびアンチセンスプライマーの5’末端にはそれぞれMlu IおよびNhe I制限酵素認識配列を付加した。phprt-GT-MCSプラスミドを、Nhe IおよびMlu I制限酵素切断し、上記PCR増幅産物とライゲートしてphprt-IVS-GT-MCSプラスミドを得た。
次に、相同組換えHT1080細胞クローン#19-3Bより抽出した全RNAを鋳型とし、RT-PCR反応により、EPO cDNA配列をクローニングした。RT-PCR反応に用いたプライマー配列は以下の通りであった。
【0094】
EPO sense プライマー(配列番号11):
5’-CCTTGCTAGCATGGGGGTGCACGAATGTCC-3’
EPO gene as プライマー(配列番号8):
5'- CCTTGAATTCTCATCTGTCCCCTGTCCTGC -3'
【0095】
次に、EPO cDNAをNhe IおよびEco RI制限酵素サイトにおいて、phprt-IVS-GT-MCSプラスミドのMCSに組み込み、phprt-IVS-GT-EPOプラスミドを構築した。
phprt-IVS-GT-EPOプラスミド中、ネオマイシン耐性遺伝子ユニットとHA2との間に存在するユニークな制限酵素サイト(Acc IIIおよびSal I)の間に、5’末端側から順に制限酵素サイトBgl II、Not I、Xba I、およびApa Iを含むDNA断片をライゲートし、図7に示されるphprt-IVS-GT-EPO-1プラスミドを得た。このphprt-IVS-GT-EPO-1プラスミドは、以下の複数コピーのEPO遺伝子を含むベクターの構築において用いた。
【0096】
2コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-2)の構築
次に、図8に示される2コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2)を以下の手順により構築した。
まず、phprt-IVS-GT-EPOプラスミドをSal IおよびMlu I制限酵素で切断し、得られたプラスミドからHA1、HA2、および大腸菌内での操作配列を除去したEPO遺伝子発現ユニットを含む配列を得た。次に、pQBI25プラスミドベクター(和光純薬工業)を鋳型とし、Ecoli-BセンスプライマーとEcoli-Nアンチセンスプライマーを用いたPCR反応、およびEcoli-XセンスプライマーとEcoli-Aアンチセンスプライマーを用いたPCR反応により、大腸菌内における操作配列2種を得た。この操作配列2種をそれぞれ、上記EPO遺伝子発現ユニットを含む配列にライゲートし、両端にBgl IIおよびNot I制限酵素認識配列を含むEPO-BNプラスミド、および、両端にXba IおよびApa I制限酵素認識配列を含むEPO-XAプラスミドを得た。
【0097】
Ecoli-B sense プライマー(Bgl IIサイト用:配列番号12):
5’- CTACGCGTAGATCTGACGTCAGGTGGCACT -3’
Ecoli-N antisense プライマー(Not Iサイト用:配列番号13):
5’- CTGTCGACGCGGCCGCACATGTGAGCAAAA -3'
Ecoli-X sense プライマー(Xba Iサイト用:配列番号14):
5’- CTACGCGTTCTAGAGACGTCAGGTGGCACT -3'
Ecoli-A antisense プライマー(Apa Iサイト用:配列番号15):
5’- CTGTCGACGGGCCCACATGTGAGCAAAAGG -3'
【0098】
次に、phprt-IVS-GT-EPO-1プラスミド、およびEPO-BNプラスミドをそれぞれBgl IIおよびNot I制限酵素で切断し、ライゲートし、EPO遺伝子発現ユニットが2コピー同方向に並んで繰り返し含有するphprt-IVS-GT-EPO-2プラスミドを得た(図8)。
【0099】
3コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-3)の構築
次に、phprt-IVS-GT-EPO-2プラスミド、およびEPO-XAプラスミドをそれぞれXba IおよびApa I制限酵素で切断し、ライゲートし、EPO遺伝子発現ユニットが3コピー同方向に並んで繰り返し含有するphprt-IVS-GT-EPO-3プラスミドを得た(図9)。
なお、得られたphprt-IVS-GT-EPO-1、phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3は、それぞれ大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0100】
4−2:細胞へのベクターの導入
プラスミド線状化
複数のEPO遺伝子を含む上記プラスミドベクターphprt-IVS-GT-EPO-2、phprt-IVS-GT-EPO-3は、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0101】
トランスフェクションおよびスクリーニング
線状化したプラスミドベクターphprt-IVS-GT-EPO-2、および、hprt-IVS-GT-EPO-3を、ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)に導入し、相同組換え細胞株の取得を行った。トランスフェクションおよびスクリーニングの条件は、例1に記載した方法と同様にして行った。
【0102】
phprt-IVS-GT-EPO-2のトランスフェクションによりG418および6TGに耐性の3個のコロニーを得た。一方、phprt-IVS-GT-EPO-3のトランスフェクションにおいては、同条件でエレクトロポレーションを2回行うことにより、1個のG418および6TG耐性コロニーを得た。
【0103】
4−3:ゲノムPCRによる解析
GFX Genomic Blood DNA purification kit(Amersham Biosciences社)を用い、phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3のトランスフェクションにより得たG418および6TGに耐性のコロニーからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。なお、参考として、シングルコピーのEPO遺伝子を含むphprt-IVS-GT-EPO-1についても、線上化、トランスフェクションおよびスクリーニングをphprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3と同様に行い、ゲノムPCRによる解析を行った。
【0104】
図10(A)は、ゲノムPCRによる相同組み換え反応の解析の詳細を説明する模式図である。hprt遺伝子(1)の相同組換えのターゲット部位(2)は、エクソン3(3)を含んで設定されており、この領域にベクターDNA(8)中のHA1(4)、HA2(5)およびこれに挟まれたEPO発現ユニットを含む繰返し配列(9)は相同組み換えにより組込まれる。ここで、nはEPO発現ユニットを含む繰返し配列の数を意味し、例4では1〜3である。
HA1(4)またはHA2(5)とEPO発現ユニットを含む繰返し配列(9)の一部とを含むDNA配列(3209 bp 、1443 bpのDNA配列)は相同組み換えによってのみゲノムから取得することができ、相同組み換えの指標となる。
よって、HA1とターゲット部位との相同組換えの指標として、図10(A)に示す3209 bpのDNA配列を設定し、このDNA配列は、以下に示すプライマーHPRTs2/Bgh-scによるPCR反応により検出した。一方、HA2と標的hprt遺伝子座との相同組換えの指標として、図10(A)に示す1443 bpのDNA配列を設定し、このDNA配列は、以下に示すプライマーHPRTas1/Neo-seqSCによるPCR反応により検出した。
【0105】
HPRTs2プライマー(配列番号16):
5’- AAAGTTCTCTCCTTTCAGCCTTCTGTACAC -3’
Bgh-scプライマー(配列番号17):
5’- GCACCTTCCAGGGTCAAGGA -3'
HPRTas1プライマー(配列番号18):
5’- ACAAGTTAAAAGGAGCTTATGGGTAGGAAG -3'
Neo-seqSCプライマー(配列番号19):
5’- CCTTCTATCGCCTTCTTGAC -3’
【0106】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図10(B)および(C)に示される通りであった。
図10(B)および(C)において、2-1、2-2、2-3はphprt-IVS-GT-EPO-2プラスミドベクターにより得られたクローンを示し、3-1はphprt-IVS-GT-EPO-3プラスミドベクターにより得られたクローンを示し、1-1は、phprt-IVS-GT-EPO-1プラスミドベクターにより得られたクローンを示す。取得したクローンすべてにおいて、3209 bpおよび1443 bpのPCR増幅産物が確認され、相同組換え反応が確認された。
【0107】
以上の通り、複数コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3)を用いた場合、2コピー組込まれた組換え細胞クローンの出現頻度は、1回のエレクトロポレーションに対して3クローンであり、3コピー組込まれたクローンについては、2回のエレクトロポレーション操作に対して1クローンであった。
【0108】
例5:hprt遺伝子座にepo遺伝子発現ユニットおよびgs遺伝子増幅マーカーが1コピー組込まれた組換え細胞の製造
5−1:プラスミドベクターの構築
以下、エリスロポエチン(EPO)遺伝子およびグルタミン合成酵素(GS)遺伝子増幅マーカーをhprt遺伝子座に組込むため、図11で表されるベクター(pSV-GS-neo-GT-EPO #22)を構築した。
【0109】
ベクター基本骨格の構築
市販されているpIRESベクター(BD Biosciences)を鋳型とし、NEO-ssプライマーおよびNEO-asプライマーを用いたPCR反応により、5’-端にSma I制限酵素サイト、3’-端にNot I制限酵素サイトを含むネオマイシン耐性遺伝子コード配列のPCR増幅産物1を得た。このPCR増幅産物1およびpIRESベクターをSma IおよびNot I制限酵素で切断し、ライゲート反応することでプラスミド1を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0110】
NEO-ssプライマー:
5’-CCTTCCCGGGATGATTGAACAAGATGGAT-3’(配列番号20)
NEO-asプライマー:
5’-CCTTGCGGCCGCTCAGAAGAACTCGTCA-3’(配列番号21)
【0111】
次にpQBI25ベクター(和光純薬工業)を鋳型とし、BGH-ssプライマーおよびBGH-asプライマーを用いたPCR反応により、5’-端にSal I制限酵素サイト、3’-端にBam HI制限酵素サイトを含むPCR増幅産物2を得た。同様に、pIRESベクターを鋳型とし、MCS-ssプライマーおよびMCS-asプライマーを用いたPCR反応により、5’-端にStu I制限酵素サイト、3’-端にSal I制限酵素サイトを含むPCR増幅産物3を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0112】
BGH-ssプライマー:
5’-GTCGACGATATCTCTAGATGTGCCTTCTAG-3’(配列番号22)
BGH-asプライマー:
5’-CCTTGGATCCTCCGGAAGCCATAGAGCCCA-3’(配列番号23)
MCS-ssプライマー:
5’-CCTTAGGCCTAGGCTTTTGCAAAAAGCTTTATTGCGGTAGT-3’(配列番号24)
MCS-asプライマー:
5’-TCTAGAGATATCGTCGACCTATAGTGAGTC-3’(配列番号25)
【0113】
前記プラスミド1をStu IおよびBam HI制限酵素反応により切断し、991 bpと5876 bpの切断断片を得た。このうち、5876 bpの切断断片をアガロースゲル電気泳動およびゲル切り出し操作によって精製し、DNA断片1を得た。また、前記PCR増幅産物2をSal IおよびBam HI制限酵素反応により切断し、DNA断片2を得た。同様に、PCR増幅産物3をStu IおよびSal I制限酵素反応により切断し、DNA断片3を得た。このDNA断片1〜3をライゲート反応して、プラスミド2を得た。
【0114】
JCRBセルバンクから入手したヒト由来細胞株HT1080細胞株(カタログ番号:IFO50354)から、ISOGEN(ニッポンジーン)を用いて抽出した全RNAを鋳型とし、GS-ssプライマーおよびGS-asプライマーを用いたRT-PCR反応により、gs コード配列の5’-端にSal I制限酵素サイト、3’-端にXba I制限酵素サイトを含むPCR増幅産物4を得た。RT-PCR反応にはOne Step RT-PCR Kit(QIAGEN)を用いた。また、ヒトHT1080細胞株からGFXTM Genomic Blood DNA Purification Kit(Amersham Biosciences)を用いて抽出したゲノムDNAを鋳型とし、EPO-ssプライマーおよびEPO-asプライマーを用いたPCR反応により、epo遺伝子の5’-端にNhe I制限酵素サイト、3’-端にEco RI制限酵素サイトを含むPCR増幅産物5を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0115】
GS-ssプライマー:
5’-CCTTGTCGACCACCATGACCACCTCAGCAA-3’(配列番号26)
GS-asプライマー:
5’-CCTTTCTAGATTAATTTTTGTACTGGAAGG-3’(配列番号27)
EPO-ssプライマー:
5’-CCTTGCTAGCATGGGGGTGCACGGTGAGTA-3’(配列番号28)
EPO-asプライマー:
5’-CCTTGAATTCTCATCTGTCCCCTGTCCTGC-3’(配列番号29)
【0116】
プラスミド2およびPCR増幅産物4をSal IおよびXba I制限酵素反応により切断し、ライゲートすることでプラスミド3を得た。次にプラスミド3およびPCR増幅産物5をNhe IおよびEco RI制限酵素反応により切断し、ライゲートすることで、EPO発現ユニットおよびgs増幅マーカーを含むプラスミド4を得た。
【0117】
相同DNA配列の取得
HT1080株のゲノムDNAを鋳型とし、PCR反応(KOD-Plus-、TOYOBO)によって、ターゲットとなるhprt遺伝子の相同DNA配列(HA1およびHA2)をクローニングした。HA1およびHA2は、図12に示される通り、hprt遺伝子のエクソン3を含む領域と相同な配列に設定した。PCR反応に用いたプライマー配列は以下に示す通りである。
【0118】
HA1 sense プライマー:
5’-CCTGCAGGTCGCGATTGGTACTTGTTCAGCTTTATTCAAG-3’(配列番号1)
HA1-SB antisense プライマー:
5’-GTCGACAAGGAGATCTACGCGTTCTGATAAAATCTACAGTCATAGGA-3’(配列番号30)
HA2 sense プライマー:
5’-GTCGACCTCTAGCTAGAGCTTGGCGTAATCATGGTCTCCGGAGACTGAAGAGCTATTGTGTGAGTAT-3’(配列番号3)
HA2 antisense プライマー:
5’-ACATGTTCTCTTAAGTCGCGAAGTAGTGTTATGATGTATGGGCATA-3’(配列番号4)
【0119】
上記PCR反応においては、HA1センスプライマーの5’末端には、制限酵素Sse 8387IおよびNru Iの認識サイトを付加した。同様に、HA1アンチセンスプライマーの5’末端にはSal IおよびBgl IIの認識サイト、HA2センスプライマーの5’末端にはSal IおよびAcc IIIの認識サイト、HA2アンチセンスプライマーの5’末端にはPci IおよびNru Iの認識サイトをそれぞれ付加した。
【0120】
pQBI25プラスミドベクターから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Pci I、およびアンチセンスプライマーの5’末端に制限酵素Sse 8387Iの認識サイトを付加した。
【0121】
Ecoli sense プライマー:
5’-ACATGTGAGCAAAAGGCCAGCAAAAGGCCAGGAAC-3’(配列番号5)
Ecoli antisense プライマー:
5’-CCTGCAGGGACGTCAGGTGGCACTTTTCGGGGAAATGTGC-3’(配列番号6)
【0122】
HA1、HA2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Pci I、Sse 8387IおよびSal Iにより切断し、ライゲーション反応を行うことによりpHA12プラスミドベクターを得た。次に、pHA12プラスミドベクターを制限酵素Bgl IIおよびAcc IIIにより切断し、5’末端から順に、HA2と、ori配列およびアンピシリン耐性遺伝子を含むDNA配列と、HA1とが連結したDNA配列1を得た。
【0123】
EPO発現ユニットおよびgs増幅マーカーを含むプラスミド4を同様に制限酵素Bgl IIおよびAcc IIIにより切断し、約2 kbpと約6.2 kbpの切断断片とした。アガロースゲル電気泳動およびゲル切り出し操作により精製し、約6.2 kbpの断片を得た。これを前記DNA配列1とライゲートすることにより、図11で表されるベクターpSV-GS-neo-GT-EPO #22を得た。得られたpSV-GS-neo-GT-EPO #22は大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0124】
5−2:細胞へのベクターの導入
プラスミド線状化
プラスミドベクターpSV-GS-neo-GT-EPO #22を、Endofree Plasmid Maxi kit(QIAGEN)を用いて精製し、Nru Iにて切断した。フェノール・クロロホルム抽出により精製した後、2 g/ Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0125】
トランスフェクション
ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)を1×107細胞/mLに調製し、線状化したプラスミドベクターpSV-GS-neo-GT-EPO #22、2μgと混合した。次に、得られた混合液を用いて、エレクトロポレーション法によりpSV-GS-neo-GT-EPO #22を細胞株HT-1080へトランスフェクトした。エレクトロポレーションは、GenePulser(BioRad)を用いて、950 μFの条件で行った。この実験のさらなる詳細は、Biotech. Bioeng. 2006. 95: 1052-1060.に記載の条件に従った。トランスフェクトした細胞は、500 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養し(培地: Advanced MEM(GIBCO)に5% FBS、1x Glutamax(GIBCO)を添加したもの)、トランスフェクションから48時間後にG418(インビトロジェン)を加えた(最終濃度; 500 μg/ mL)。
【0126】
スクリーニング
培養8-12日後、上記プレートにG418耐性コロニーが現れたことを確認した。この段階で、6TG(最終濃度; 50 μM)(和光純薬工業)が添加された新しい培地を加え、さらに8日間培養した。培養後、全ウェルを確認し、6TG耐性コロニーを単離した。
【0127】
5−3:サザンハイブリダイゼーションによる解析
以下、サザンハイブリダイゼーションにより、6TG耐性コロニーのスクリーニングを行った。
プローブの調製
pSV-GS-neo-GT-EPO #22中のネオマイシン耐性遺伝子に相補的な配列を有するNRプローブを、以下の手順で合成した。まず、pSV-GS-neo-GT-EPO #22中のネオマイシン耐性遺伝子コード配列の全長をPCRにより増幅し、pGEM T プラスミドベクター(Promega)にTAクローニングした。次に、PCR DIGプローブ合成キット(ロシュ、プライマー:M13 Forward/Reverseプライマー)を用いて、DIG(Digoxigein)標識されたプローブを作製した。
【0128】
メンブレンの調製
6TG耐性コロニーから各細胞クローンのゲノムDNAをgenomic prep cells and tissue DNA isolation kit(GE healthcare)を用いて抽出し、Eco RI制限酵素により切断した。切断したゲノムDNA 10μgを、0.6%アガロースゲルを用いて電気泳動し、ナイロンメンブレン(Hybond N+ membrane、AmaershamBiosciences)へ転写した。得られたメンブレンは80℃で2時間インキュベートし、DNAをメンブレン上に固定した。
【0129】
ハイブリダイゼーション
上記メンブレンに対し、上記NRプローブをハイブリダイズさせた。この際、プレハイブリダイゼーション、ハイブリダイゼーションおよびプローブの検出は、DIGアプリケーション マニュアル(ロシュ)に従って行った。
【0130】
図13に示される通り、組換えクローン118S-1、118S-2、118S-3、118S-5において、NRプローブにより約8 kbpのDNA断片が検出された。制限酵素Eco RIサイトはpSV-GS-neo-GT-EPO #22中および標的hprt遺伝子座中のいずれにも存在しており、相同組換え反応が首尾よく生じていればNRプローブによって7781 bpのEco RI断片が検出される。この結果は図13に示した4クローンがいずれも目的とするhprt遺伝子座にepo遺伝子発現ユニットおよびgs遺伝子増幅マーカーが1コピー組込まれた組換え細胞であることを示している。
【0131】
例6:遺伝子増幅によってhprt遺伝子座に複数のepo遺伝子発現ユニットを組込んだ組換え細胞の製造
6−1:グルタミンフリー培地での増殖能検討
培地へのMSX添加による遺伝子増幅細胞のスクリーニングに先立って、グルタミン非添加培地での増殖能を評価した。グルタミン添加培地(培地組成: Advanced MEMに5% FBS、1x Glutamaxを添加したもの)で維持していた前記細胞クローンを、70〜80%コンフルエントになった段階でトリプシン処理し、細胞を回収した。回収した細胞懸濁液の細胞密度は、トリパンブルー染色法によって求めた。培養ディッシュ(直径10 cm)1枚あたり1.7X105 cellsの細胞を播種し、10 mlのグルタミン非添加培地(培地組成: Advanced MEMに2% FBS、1x GS supplement(ニチレイ)を添加したもの)を加え、CO2インキュベーター内で培養した。6日間培養後、同じ手順で細胞を回収し、回収した細胞懸濁液の液量および細胞密度から総細胞数を算出し、増殖能を評価した。同様の手順で1.7X105 cellsの細胞を新しい培養ディッシュに移し、10 mlのグルタミン非添加培地を加えてCO2インキュベーター内で培養した。この操作を2回繰り返し、グルタミンフリー培地での増殖を検証した。
【0132】
馴化クローンの選択
118S-2および118S-5クローンが3回めの培養期間中に、それぞれ2倍および4倍に細胞数が増加していた。非組換えHT1080細胞では、3回めの培養期間中での増殖がみられなかったことから、これら2クローンは外来性gs遺伝子の発現によりグルタミン非添加培地での増殖が支持されているものと考えられた。以降のMSXスクリーニングには、最も増殖のよかった118S-5クローンを用いた。
【0133】
6−2:MSXスクリーニング
グルタミン非添加培地で維持していた118S-5クローンを、70〜80%コンフルエントになった段階でトリプシン処理により回収した。MSX濃度を 5または10 μMに調整した選択培地(培地組成: Advanced MEMに2% FBS、1x GS supplement、5または10 μM MSXを含有するもの)で、1×104 cells/ mlと2×104 cells/ mlの細胞懸濁液を調整した。この細胞懸濁液を96ウェルマイクロテストプレート(BD Falcon)に、1ウェルあたり100 μLづつ播種し、37℃、5% CO2条件下で培養を行った。培養開始から1週間後に培地をフレッシュなものに交換した。さらに1週間の培養後、増殖のみられるウェルを顕微鏡観察により判別した。MSX濃度を10 μMとした選択培地では耐性を示した細胞は存在せず、細胞増殖がみられるウェルは存在していなかった。一方、MSX濃度を5 μMとした選択培地では、播種したウェルのおおよそ30%のウェルに耐性細胞が存在していた。次にこれらのウェル中のEPO蓄積量をELISAによって測定した。
【0134】
6−3:EPO生産量による遺伝子増幅細胞の選択
ELISA
固定化抗体としてMonoclonal anti-human EPO(R&D Systems)、1次抗体としてPolyclonal anti-human EPO(R&D Systems)、2次抗体としてAnti-mouse Ig, horseradish peroxidase linked whole antibody(from donkey)(Amersham Biosciences)を用い、TMB No Hydrogen Peroxide 1 Component HRP Microwell substrate(BioFX)により、450nmの吸光度を測定することにより解析した。
【0135】
EPO蓄積量の解析
ELISAによるEPO蓄積量測定の2日前に、耐性細胞が存在する96ウェルプレートの選択培地をフレッシュなものに完全交換した。各ウェルあたり50 μLの培地を回収し、ELISAにより測定を行った結果、2日間の培養で0.3〜37 ng/ mlのEPOが蓄積していた。次に、EPO蓄積量が最大値37.3 ng/ mlを示した2-1F8クローン、中程度の値17.7 ng/mlを示した1-2E6クローンに対してgs遺伝子コピー数の解析を行った。
【0136】
6−4:gs遺伝子コピー数の解析
ゲノムサンプルの調整
細胞からのゲノム抽出は、DNA Isolation kit for cells and tissuesキット(Roche)を用いて行った。抽出したゲノムを制限酵素Hind III(TaKaRa)で切断した後、エタノール沈殿でゲノムを回収し、滅菌水に溶解させた。吸光度測定によりDNA濃度を測定した。
【0137】
Real-time定量PCR解析
調整したゲノムDNAを用いて増幅マーカーであるgs遺伝子のコピー数の測定をReal-time定量PCRにより行った。Real-time定量PCR はABI PRISM 7300(ABI)を用い、Taqman Universal PCR Master Mix(ABI)により反応を行った。PCR反応に用いたプライマーおよびプローブの配列を以下に示す。
【0138】
hGS Forプライマー:
5’-ACCCCTTTTCGGTGACAGAA-3’(配列番号31)
hGS Revプライマー:
5’-TCGCCGGTTTCATTGAGAAG-3’(配列番号32)
hGS cDNA-Taqmanプローブ:
5’-CCCTCATCCGCACGTG-3’(配列番号33)
【0139】
コピー数解析
定量PCR結果の解析は装置付属のソフト7300 system softwareで行った。コピー数の算出は以下の手順に従い行った。各ゲノムサンプルに対し、hGSプライマーおよびプローブで測定したCt値と、濃度既知のベクター(pSV-GS-neo-GT-EPO#22)による検量線からコピー数を算出した。サンプル中のゲノム量を補正するため、ゲノム中の他部位としてベータアクチンのコピー数を各サンプルについてさらに測定し、この値が一定となるよう補正を行った。
【0140】
結果を図14に示す。2-1F8細胞と1-2E6細胞は、118S-5クローンからMSX 5 μMへ耐性を示す細胞として取得したものである。S-5(Glu-)とした細胞は、グルタミン非添加培地で維持していた118S-5クローンである。S-5(Glu+)はグルタミン添加培地で維持していた118S-5クローンである。このS-5(Glu+)を対照として、それぞれの細胞におけるRealtime PCR法によるコピー数解析結果をS-5(Glu+)に対する相対値で算出し、コピー数比としてグラフに示した。2-1F8細胞は2.67、1-2E6細胞は1.43という値を示した。これらの結果から、MSX耐性細胞株である2-1F8細胞では遺伝子増幅によりコピー数が増加していると判断できる。遺伝子増幅技術の原理を鑑みれば、組込み位置近傍のhprt遺伝子座内でコピー数が増加した細胞株であると判断できる。また、ELISA解析によるEPO蓄積量は、2-1F8細胞で37.3 ng/ ml、1-2E6細胞で17.7 ng/mlであり、コピー数比の値と相関していた。
【関連出願】
【0001】
本特許出願は、2007年8月10日に出願された日本国特許出願2007−210122号、および2008年7月24日に出願された日本国特許出願2008−191278号に基づく優先権の主張を伴うものであり、かかる先の特許出願における全開示内容は、引用することにより本明細書の一部とされる。
【発明の背景】
【0002】
発明の分野
本発明は、組換え哺乳類動物培養細胞、および該細胞を利用したタンパク質の生産方法に関する。
【0003】
背景技術
原核生物や真核生物を宿主とした、種々の組換えタンパク質生産システムが知られている。哺乳類動物細胞を宿主とした組換えタンパク質生産システムによれば、ヒトを始めとする高等動物由来のタンパク質に対し、糖鎖付加、フォールディング、リン酸化などの翻訳後修飾を、より生体で作られるものと同じように施すことが可能である。
【0004】
この翻訳後修飾は、タンパク質の本来有する生理活性を組換えタンパク質で再現するために必要なものである。従って、そのような生理活性が特に必要とされる医薬品などに用いられる組換えタンパク質の生産系には、哺乳類動物細胞を宿主としたタンパク質生産システムが好ましく用いられている。
【0005】
医薬タンパク質の工業的生産においては、高いタンパク質発現レベルでかつ発現レベルが安定に維持されることが重要である。特に発現レベルが安定に維持されることに関しては、コスト面のみならず、医薬タンパク質としての同一性および安全性を証明するためにも重要である。組換えタンパク質生産細胞を工業的スケールでの生産に用いるためには生産細胞クローンの培養スケールの拡大を図る必要がある。これには通常少なくとも樹立直後のクローンから約60回の細胞分裂を経なければならないと見積られており(Brown, M. E. et al. (1992) Cytotechnology, 9, 231-236.)、この間、発現レベルは一定に保たれていなければならない。
【0006】
また、培養スケールの拡大の期間中または期間後、組換え細胞クローンの比生産性が減少することにより、生産細胞としての使用が難しくなることがあり、この場合、数ヶ月にも及ぶ開発期間が無駄となる(Barnes, L. M. et al. (2003) Biotechnol. Bioeng. 81, 631-639.)。
【0007】
また、スケールアップ期間及び実生産期間中に、選択薬剤を使用することは培養コストを上昇させることのみならず、医薬品への毒物混入リスク回避のために行われる精製プロセスのコストも上昇させる。このため、選択薬剤は添加せずに行われるのが一般的である。
【0008】
このような背景から、目的タンパク質を生産する組換え哺乳類動物細胞クローンには、高い比生産性レベルを有し、選択薬剤を添加することなく比生産性レベルを安定に維持できる特性を有することが求められる。
【0009】
高い比生産性レベルの達成に関しては、目的タンパク質をコードする外来性遺伝子のコピー数を遺伝子増幅技術により増加させる方法が一般的であり、CHO-DHFRシステムやGS-NS0システムなどが実用技術として確立されている(特公平7-40933、Werner R. G. et al. (1998) Arzneim.-Forsch./Drug Res 48, 870-880.など)。
【0010】
しかしながら、比生産性レベルが増大した細胞クローンの選択後、選択薬剤を含まない培地で選択細胞クローンの培養を継続する場合、ほとんどのクローンにおいて比生産性レベルの減少または消失が確認されている。さらに、組込み遺伝子のコピー数の増加に必ずしも比例して目的タンパク質の発現レベルが増大しないことも報告されている(特表2002-541854公報、Kim, N. S. et al. (1998) Biotechnol. Bioeng., 60, 679-688など)。
【0011】
このような比生産性レベルの減少または目的タンパク質生産の消失は、遺伝子コピー数の減少が主な原因であることが報告されている(Kim, N. S. et al. (1998) Biotechnol. Bioeng., 60, 679-688、Kim, S. J. (1998) Biotechnol. Bioeng. 58, 73-84.、Yoshikawa, T. et al. (2000) Biotechnol. Progr. 16, 710-715.など)。
【0012】
また、他の原因として、複数の同一配列を有する遺伝子コピーがタンデムに繰り返して組込まれた場合に生じる、Repeat-Induced Gene Silencing(RIGS)が報告されている(Henikoff, S. (1998) Bioessays 20, 532-535.、Garrick, D. et al. (1998) Nat. Genet. 18, 56-59.)。RIGSは遺伝子コピー数が2〜3コピー程度の少ない場合においても生じることが示されている(McBurney, M. W. (2002) Exp. Cell Res. 274, 1-8)。
【0013】
そして、これまでのところ、目的タンパク質の比生産性レベルの安定性に関して十分な解決策が提示されているとはいえない。クローン選択は長期培養時における成長速度並びに生産性に関して蓄積されたデータを元に、経験的に行なわれているのが現状である。そして、この経験的な方法では、目的タンパク質の比生産性レベルの安定な細胞クローンを取得できることは希であり、偶然によるところが大きい(Barnes, L. M. et al. (2003) Biotechnol. Bioeng. 81, 631-639.)。
【0014】
本発明者らは、先に、ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座にシングルコピーのGreen fluorescent protein(GFP)の発現ユニットを相同組換えにより組込み、組換え細胞を取得する手法を報告している(Biotechnol Bioeng 95(6): 1052-1060. 2006.。hprt遺伝子座への位置特異的な組込みにより作製された組換え細胞クローンは、選択薬剤非添加条件下において、長期間の培養期間中も比生産性レベルを安定に保持する。
【0015】
通常のランダム組換え細胞クローンの取得においては、複数コピーの外来性遺伝子が同時に同一染色体位置に組み込まれる場合が多い(Martin, D. I. K. and Whitelaw, E. (1996) Bioessays 18, 919-923.)。これはRIGSの標的になりうる。これに対し、相同組換えなどにより、1コピーを組込む方法ではRIGSの回避が可能である(Whitelaw, E. et al. (2001) Methods in Mol. Biol. 158, 351-368.)。
【0016】
一方で、1コピーの組込みにおいても、組込まれる染色体位置により比生産性レベルの安定性は大きく異なることが知られている(Walters, M. C. et al. (2007) Genes Dev. 10, 185-195.)。
【0017】
このような事実から、次のようなことが分かる。すなわち、hprt遺伝子位置が、外来性遺伝子を1コピー組込んだ際に、その比生産性レベルの安定性の保持において優れたものである。しかしながら、一方で、hprt遺伝子位置に複数の外来性遺伝子コピーを組込んだ際の比生産性レベルの安定性については、RIGSを回避可能か否かについても含め、予測はできない。
【0018】
また、特表平7-500969公報には、ヒト線維肉腫由来のHT1080細胞のhprt遺伝子座にエリスロポエチン遺伝子発現ユニットを相同組換えにより組込んだとの開示がある。しかし、エリスロポエチン遺伝子の発現は確認されておらず、さらにその複数コピーを組込んだとの記載もない。
【発明の概要】
【0019】
本発明者らは、今般、hprt遺伝子座に目的タンパク質遺伝子を複数コピー組込むことにより得られた哺乳動物細胞は、長期間に渡り目的タンパク質の比生産性レベルを安定に維持させることができ、さらにその比生産性レベルが組込まれた目的タンパク質遺伝子のコピー数に比例するとの極めて意外な知見を得た。本発明は、かかる知見に基づくものである。
【0020】
したがって、本発明は、目的タンパク質を高レベルで安定に製造できる組換え細胞およびその製造方法ならびにその組換え細胞を用いた目的タンパク質の製造方法の提供をその目的としている。
【0021】
そして、本発明による哺乳動物細胞は、hprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーが組込まれてなるものである。
また、本発明による上記哺乳動物細胞の製造方法は、宿主細胞のhprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーを組込む工程を含んでなるものである。
また、本発明による目的タンパク質の製造方法は、上記哺乳動物細胞を培養して前記目的タンパク質を産生することを含んでなるものである。
【図面の簡単な説明】
【0022】
【図1】シングルコピーのエリスロポエチン遺伝子を含有するphprt-GT-EPO geneプラスミドベクターの模式図である。
【図2】組換えクローン(#14-5E, #19-3B)におけるサザンハイブリダイゼーションの結果を示す写真である。
【図3】(A)は、hprt遺伝子における相同組換えターゲット部位を示す。(B)は、1コピーのphprt-GT-EPO gene導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。また、(C)は、2コピーのphprt-GT-EPO gene導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。
【図4】樹立直後にG418および6TG不含培地で培養した組換えクローン(#14-5E, #19-3B)のEPO生産性を示すグラフである。
【図5】G418および6TG不含培地で96日間培養を継続した場合の、組換えクローン(#14-5E)のEPO生産性の推移を示すグラフである。
【図6】G418および6TG不含培地で96日間培養を継続した前後における、組換えクローン(#14-5E)のサザンハイブリダイゼーションの結果を示す写真である。
【図7】1コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-1 プラスミドベクターの模式図である。
【図8】2コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-2 プラスミドベクターの模式図である。
【図9】3コピーのエリスロポエチン遺伝子を含有するphprt-IVS-GT-EPO-3 プラスミドベクターの模式図である。
【図10】(A)は、ゲノムPCRによる相同組み換え反応の解析の詳細を説明する模式図である。 (B)は、導入ベクターのホモロジーアーム領域由来のDNA(3209bp)を指標とした、ゲノムPCR産物の電気泳動の結果を示す写真である。(C)は、導入ベクターのホモロジーアーム領域由来のDNA(1443bp)を指標とした、ゲノムPCR産物の電気泳動の結果を示す写真である。
【図11】エリスロポエチン遺伝子およびgs遺伝子増幅マーカーを含有するpSV-GS-neo-GT-EPO #22プラスミドベクターの模式図である。
【図12】(A)は、hprt遺伝子座における相同組換えターゲット部位を示す。(B)は、1コピーのpSV-GS-neo-GT-EPO #22導入DNA配列が、相同組換えにより細胞クローンのゲノムDNAに組込まれた場合の模式図である。
【図13】組換えクローン(118S-1、118S-2、118S-3、118S-5)におけるサザンハイブリダイゼーションの結果を示す写真である。
【図14】Real-time定量PCR法によりゲノム中のgs遺伝子コピー数を解析した結果である。2-1F8および1-2E6はMSX 5 μMに対して耐性を示した細胞、S-5(Glu-)およびS-5(Glu+)はそれぞれグルタミン非添加培地またはグルタミン添加培地中で維持していた118S-5細胞を示す。結果は、S-5(Glu+)の値に対する相対値で示してある。
【発明の具体的説明】
【0023】
hprt遺伝子座/目的タンパク質遺伝子の複数コピーの導入
本発明による目的タンパク質の製造方法にあっては、hprt遺伝子座を複数コピーの目的タンパク質遺伝子組換えのターゲット領域とする。このhprt遺伝子は、ヒト等のX染色体長腕に存在するハウスキーピング遺伝子の一つとして知られている。このhprt遺伝子座に複数コピーの外来性の目的タンパク質を組込んで発現させる場合、目的タンパク質の比生産性レベルがそのコピー数にほぼ比例して増加する。しかも、このタンパク質発現量を長期間に渡り安定に維持することが可能となる。本発明の好ましい態様によれば、この安定性は、選択薬剤の非存在下にあっても長期にわたり維持させることができる。
【0024】
ここで、外来性遺伝子の複数コピーとは、2以上の実質的に同一の遺伝子または遺伝子発現ユニットが、hprt遺伝子座上に存在することを意味する。ここで、複数の発現ユニットの転写方向は必ずしも同一方向でなくともよく、またすべて同一方向にタンデムに繰り返して存在することもできる。繰り返し存在している遺伝子または発現ユニット間には、他のDNA配列が含まれていてもよい。また、比例するとは、比生産性レベルが遺伝子コピー数と正の比例関係にあることを意味し、好ましくは、遺伝子コピー数の増加に応じて比生産性レベルが加算的に上昇することを意味する。
【0025】
従来、目的タンパク質遺伝子の複数コピーを、その発現量の増加を期待して組込むことは行われていたが、多くの場合、組込まれたコピー数に比例するとまでは言い難い。よって、本発明における、組込まれたコピー数に比例して発現量が増加するとの効果は、目的タンパク質の生産に極めて有利であり、かつ意外なものである。
【0026】
本発明による製造方法にあっては、hprt遺伝子座に組込まれる外来性の目的タンパク質遺伝子のコピー数は2以上であり、好ましい上限は500程度であり、より好ましい上限は100であり、さらに好ましい上限は10であり、さらに好ましい上限は3である。また、目的タンパク質遺伝子の複数コピーは、後述するベクター等によりhprt遺伝子座に組込まれうる。
【0027】
目的タンパク質遺伝子
上記目的タンパク質遺伝子は、cDNAに由来する配列であっても、ゲノムDNAに由来する天然イントロンを含む構造遺伝子であっても、好適に利用可能である。また、目的タンパク質遺伝子は、医薬として有用なタンパク質をコードしていることが好ましい。該タンパク質は細胞内に蓄積されるものであっても、細胞外に分泌されるものであっても本発明において利用することが可能である。また、かかる目的タンパク質としては、酵素、サイトカイン、ホルモン、抗体、凝固因子、調節タンパク質、レセプター等が挙げられるが、より具体的な例としては、エリスロポエチン、モノクローナル抗体、組織特異的プラスミノーゲン活性化因子、顆粒球コロニー活性化因子等が挙げられる。
【0028】
発現ユニット
上記目的タンパク質遺伝子は、hprt遺伝子座において、プロモーター配列や転写終結シグナル配列等の発現に必要な要素を含む発現ユニットとして組込まれることが好ましい。したがって、本発明の一つの態様によれば、目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる。
【0029】
また、上記発現ユニットにおけるプロモーターや転写終結シグナル等の発現に必要な要素は、目的タンパク質遺伝子の種類、性質等に応じて適宜決定してよく、プロモーター配列の好適な例としては、CMVプロモーター、SV40 プロモーター等が挙げられる。また、転写終結シグナル配列の好適な例としては、BGHポリAシグナル配列、SV40ポリAシグナル配列等が挙げられる。
【0030】
また、上記発現ユニットにおいて、プロモーター配列、転写終結シグナル配列の他の発現に必要な要素としては、例えば、目的遺伝子を効率的に発現させるための調節エレメント(例えば、エンハンサー、IRES(internal ribosome entry site)配列)等が挙げられる。調節エレメントはその性質に応じて、発現ユニットにおける適切な位置に配置することが可能である。これら発現に必要な要素は、宿主との組み合わせおよび目的タンパク質の生産性を勘案して選択することが好ましい。
【0031】
また、上記発現に必要な要素は、翻訳反応の増強を勘案して、目的タンパク質遺伝子以外のイントロン配列を含んでもよい。イントロン配列は発現ユニット中の転写開始部位から転写終結部位の間に配置されていればよく、好ましくは、ウイルス由来のもの、哺乳類動物ゲノム由来のもの等が挙げられるが、宿主内でスプライシング効率の高いイントロン配列であることが好ましい。
【0032】
複数コピーの発現ユニットのhprt遺伝子座への組換え
本発明の一つの態様によれば、複数コピーの発現ユニットをhprt遺伝子座へ組換える場合、複数コピーの発現ユニットを含むベクターを用いることが好適である。また、シングルコピーの発現ユニットを含むベクターを哺乳動物細胞に導入し、複数コピーの発現ユニットが組換えられた細胞を選択・取得してもよく、本発明にはかかる態様も包含される。また、本発明の別の態様によれば、遺伝子増幅マーカー遺伝子と目的タンパク質遺伝子の発現ユニットとを含むベクターを用いてhprt遺伝子座へ組換えた後、適切な遺伝子増幅プロセスにより、複数コピーの発現ユニットが組込まれた細胞を選択することもまた好適である。
【0033】
本発明の好ましい態様によれば、目的タンパク質遺伝子の発現ユニットは、hprt遺伝子座に相同組換えにより組換えられたものである。かかる相同組換えの利用は、組換え細胞を迅速に構築し、さらには、目的タンパク質遺伝子を高レベルで安定に発現させる上で有利である。
【0034】
また、相同組換えの標的となるhprt遺伝子座中の位置は、目的タンパク質遺伝子の効率的な発現を妨げない限り適宜設定してよく、例えば、本発明の一つの態様によれば、hprt遺伝子のエクソン配列(例えば、エクソン3等)上に設定する。エクソン配列上にターゲット領域を設定することは、hprt遺伝子自体の発現を阻害するため、6−チオグアニン(6-TG)や8−アザグアニン(8-AG)による薬剤選択によって、効率的に組換え細胞を取得する上で有利である。
【0035】
ベクター
本発明に用いられるベクターとしては、哺乳動物細胞染色体へ目的遺伝子を組換えうるものであれば特に限定されず、例えば、プラスミドベクター、コスミドベクター、ファージベクター、人工染色体ベクター等が挙げられるが、好ましくは、プラスミドベクターである。また、上記ベクターは、直鎖状または環状のいずれで構成してもよい。
【0036】
上記ベクターは、当技術分野で周知となっている標準的方法を利用して構築することができ、例えば、Sambrook, J.らの「Molecular Cloning: a laboratory manual」、Cold Spring Harbor Laboratory Press、New York(1989)に記載の方法に従って構築することができる。
【0037】
相同組換えにより発現ユニットをhprt遺伝子座に組換える場合、該発現ユニットを組換えるためのベクターには、hprt遺伝子座の一部と相同組換え可能な相同性を有する相同DNA配列を配置する。ベクターに含まれる相同DNA配列は、目的タンパク質遺伝子の効率的な組換えおよび発現を妨げない限り、単数であっても複数であってもよいが、好ましくは2つである。さらに、かかる2つの相同DNA配列は、ベクターの5’末端および3’末端に配置されていることが好ましい。したがって、本発明の一つの態様によれば、ベクターは、5’末端に配置された相同DNA配列と、目的タンパク質遺伝子の発現ユニットと、3’末端に配置された相同DNA配列とを少なくとも含んでなる。
【0038】
また、相同DNA配列は、hprt遺伝子座と相同組換え可能な相同性および長さを有する。相同組換え反応の生じやすさまたはその確実性を考慮すれば、相同DNA配列とhprt遺伝子座との相同性は十分に高い方が好ましく、好ましくは99%以上、より好ましくは99.9%以上が相同であり、さらに好ましく両者は同一配列である。また、二つの相同DNA断片の長さは各々が、好ましくは数百塩基対(bp)以上であり、より好ましくは500bp以上であり、さらに好ましくは1000bp以上である。
【0039】
複数コピーの発現ユニットを含むベクターを構築する場合、かかるベクターは、制限酵素切断反応およびライゲーション反応を組み合わせて好適に構築される。例えば、発現ユニットの両末端にII型制限酵素の認識配列を含有させ、該認識配列用の制限酵素で切断反応を行い、不要なDNA配列(大腸菌内での操作用配列等)をゲル切り出し等の処理によって除去し、得られた発現ユニットのライゲーション反応を行うことにより、複数の発現ユニットがタンデムに並んだベクターDNAを構築しうる。
【0040】
上記制限酵素のすべての認識配列を同一のものにする場合には、複数の目的タンパク質遺伝子の転写方向がランダムであるベクターDNAを構築しうる。また、制限酵素認識配列として異なるものを選択する場合、DNA配列中の目的タンパク質遺伝子の向きを制御することが可能であり、例えば、すべての目的タンパク質遺伝子が同方向に配置されたベクターDNAや、目的タンパク質遺伝子の向きが交互に異なるベクターDNA等を構築しうる。
【0041】
使用可能な上記制限酵素は平滑末端を生じるものであっても付着末端を生じるものであってもよいが、ライゲーション効率を勘案すれば、付着末端を生じる制限酵素が好ましい。また、目的タンパク質遺伝子の向きの制御を勘案すれば、種々の認識配列を有する制限酵素を利用することが好ましく、例えば、Biotech. Appl. Biochem. 20: 157-171. 1994.や特表2003-530886等に記載されるIIS型の制限酵素、例えばSfi I制限酵素等が挙げられる。Sfi I制限酵素は、1つの制限酵素の利用により64通りの異なる付着末端を生じさせることから、多数の目的タンパク質遺伝子の向きを制御する上で好ましい。制限酵素に関する上述の文献の内容は、引用することにより本明細書の一部とされる。
【0042】
ライゲーション反応により構築した複数コピーの発現ユニットを含むベクターDNAは、フェノール・クロロホルム抽出等により精製し、ベクターの種類等を勘案して選択した大腸菌、酵母等の宿主細胞において増殖させることができる。また、適当な宿主細胞が利用できない場合、複数コピーの目的タンパク質遺伝子を含むベクターDNAは、フェノール・クロロホルム抽出処理後、直接哺乳類動物細胞へと導入することもできる。
【0043】
また、遺伝子増幅を行うためのベクターにおいては、目的タンパク質遺伝子の発現ユニットに加え、遺伝子増幅マーカー遺伝子が配置される。遺伝子増幅マーカー遺伝子は適切に発現するよう、目的タンパク質遺伝子の発現ユニットの近傍に配置されていればよい。また、目的タンパク質遺伝子の発現ユニットはシングルコピーで含有されていても複数コピーで含有されていてもよい。
【0044】
また、上述したベクターいずれにおいても、上記の他、組換え細胞の選択を勘案し、薬剤耐性による優性選択が可能なマーカー遺伝子(ネオマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、ゼオシン耐性遺伝子、ピューロマイシン耐性遺伝子)等を適宜組み込むことが可能である。
【0045】
ベクターの細胞への導入方法
上記ベクターの哺乳動物細胞への導入は、当該技術分野において公知の方法を用いることが可能であり、例えば、エレクトロポレーション法、マイクロインジェクション法、リン酸カルシウム法、リポフェクション法などが挙げられる。かかる導入方法は、哺乳動物細胞の種類、ベクターのサイズ、導入効率等を勘案して当業者により適宜選択される。ここで、ベクターが環状である場合、公知の方法により線状化して細胞に導入されてもよい。
【0046】
組換え細胞の選択
組換え哺乳動物細胞は、薬剤選択等の当該分野で周知となっている方法で選択し、取得することができる。例えば、目的タンパク質の発現ユニットを含むプラスミドベクターが優性選択が可能な薬剤耐性遺伝子等の選択マーカーを有する場合、選択薬剤を添加した培地で細胞の培養を行うことにより、組換え細胞を選択し、取得することが可能である。また、hprt遺伝子の発現をブロックする位置において目的タンパク質遺伝子が組込まれた場合、6-TGまたは8-AGを培地に添加することにより、組換え哺乳動物細胞を効率的に選択しうる。
【0047】
さらに、組換え哺乳動物細胞の選択においては、PCR反応等による組換え細胞のゲノムDNA解析や、サザンブロッティング法等を用いることにより、複数コピーの目的タンパク質遺伝子の発現ユニットを含む組換え細胞を正確に選択することが好ましい。また、バックグラウンドの低減を勘案すれば、ベクターの導入前に、哺乳動物細胞をHAT(ヒポキサンチン、アミノプテリン、チミジン)含有培地で前培養することが好ましい。
【0048】
遺伝子増幅による複数コピーの組込み
適切な遺伝子増幅マーカー遺伝子と外来性遺伝子(好ましくはそれを発現ユニットとして)とを含むベクターをhprt遺伝子座に組換えた細胞を用意し、適切な遺伝子増幅プロセスを行うことにより、hprt遺伝子座に外来性遺伝子発現ユニットが複数コピー組込まれた組換え細胞を取得することができる。
【0049】
遺伝子増幅技術では、遺伝子増幅マーカー近傍(〜10kbp程度)のDNA配列も同時にコピー数が増幅されることが知られている。この増幅は同一染色体位置で生じるため、本技術における遺伝子増幅の利用により、hprt遺伝子座内で目的タンパク質遺伝子のコピー数を増加させることができる。また、遺伝子増幅を利用した場合、増加したコピーの配向は制御できず、必ずしも発現ユニットが同一転写方向に揃ったタンデムな配置とはならない。
【0050】
本発明にあっては、公知となっている遺伝子増幅マーカー遺伝子であればいずれも好適に利用可能であるが、好ましくは、ジヒドロ葉酸還元酵素遺伝子(DHFR)またはグルタミン合成酵素遺伝子(GS)であり、より好ましくはGS遺伝子である。また、遺伝子増幅プロセスとしては、DHFR阻害剤であるメトトレキセート(MTX)を添加した培地での耐性細胞のスクリーニングや、GS阻害剤であるメチオニンスルフォキシミン(MSX)を添加した培地でのスクリーニング等の周知の方法が好適に利用可能である。また、MTXやMSXの添加濃度を段階的に引き上げるという周知の方法がより好適に利用可能である。
【0051】
組換え哺乳動物細胞
また、本発明による哺乳動物細胞は、上述の手法により製造されるものであり、hprt遺伝子座に、外来性の目的タンパク質遺伝子の複数コピーが組込まれてなることを特徴とする。かかる組換え哺乳動物細胞にあっては、目的タンパク質遺伝子の比生産性レベルはその遺伝子コピー数に比例して増加し、この比生産性レベルは長期間に渡り安定に維持しうる。
さらに、この複数コピーの目的タンパク質遺伝子の長期的な発現安定性は、選択薬剤の非存在下であっても維持され、培養コストや異物混入リスク回避のために行われる精製プロセスのコストを低減する上で有利である。
【0052】
そして、上記組換え哺乳動物細胞において、目的タンパク質遺伝子のコピー数は2以上である。また、本発明のより好ましい態様によれば、上記組換え哺乳動物細胞における目的タンパク質遺伝子は、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる。また、上記組換え哺乳動物細胞における目的タンパク質遺伝子または前記発現ユニットは、タンデムに並んで繰り返し組込まれてもよい。また、一つの態様によれば、上記組換え哺乳動物細胞における目的タンパク質遺伝子は、相同組換えにより組込まれたものである。
【0053】
また、本発明による組換え哺乳動物細胞にあっては、上記目的タンパク質の比生産性レベルはhprt遺伝子座に組込まれた目的タンパク質遺伝子のコピー数等に応じて増加するものであり、例えば、目的タンパク質遺伝子のコピー数が2つである場合、1細胞あたり約0.4〜20pg/日とすることができる。
【0054】
また、本発明による組換え哺乳動物細胞は、上記目的タンパク質の比生産レベルを、少なくとも97日間維持することができる。
上記目的タンパク質遺伝子の長期的な発現安定性は、選択薬剤の非存在下であっても維持されるものであり、かかる選択薬剤としては、細胞選択において用いられる公知薬剤が挙げられるが、例えば、ネオマイシン、6TG、MTX、MSX等である。
【0055】
宿主細胞
また、本発明において、宿主細胞として用いられる哺乳動物細胞は、好ましくはヒト由来のものであり、かかる哺乳動物細胞の具体的な例としては、例えば、ヒト線維肉腫由来のHT1080細胞株等が挙げられるが、hprt遺伝子の哺乳動物における普遍性を鑑みれば、これらに限定されるものではない。
また、相同組換えによりhprt遺伝子座へ目的タンパク質遺伝子を組込む場合、6TGや8AGなどの選択薬剤による相同組換え細胞の選択を勘案すれば、宿主細胞としてはhprt遺伝子座を全ゲノム中に1ヶ所のみ有する細胞が好ましい。そのような細胞の例としては、上述したHT1080株や、男性由来のX染色体を1本しか持たない細胞株などが挙げられる。
【0056】
培養/目的タンパク質の単離
また、本発明による目的タンパク質の製造方法にあっては、hprt遺伝子座に複数コピーの目的タンパク質遺伝子が組込まれた組換え哺乳動物細胞を培地中で培養し、目的タンパク質を産生させることができる。上記哺乳動物細胞の培養条件の詳細は、細胞の性質および状態等に応じて当業者によって適宜決定されるが、培養コストを勘案すれば、培地は、好ましくは無血清培地であり、より好ましくはChemically defined(CD)培地である。
【0057】
また、本発明の一つの態様によれば、目的タンパク質は、組換え哺乳動物細胞の培養物から単離することが好ましい。単離手法については、目的タンパク質の性質等に応じて、遠心分離、ゲルろ過、フィルターろ過などの公知の手法を用いることができる。
【実施例】
【0058】
以下、本発明を実施例によって具体的に説明するが、本発明はこれら実施例に限定されるものではない。
なお、以下の実験において、制限酵素による反応、PCR反応、ライゲーション反応等の各反応条件は、メーカーの推奨する反応条件、あるいはMolecular Cloning 2nd edition Sambrook et.al. Cold Spring Harbor Laboratory Pressに記載の方法に従って設定した。また、得られた種々のプラスミドベクターDNA等については、自動DNAシークエンサー(310 Genetic Analyzer、Applied Bio Systems, Inc.)を用いてDNA配列を決定した。
【0059】
例1:hprt遺伝子座に複数のエリスロポエチン遺伝子発現ユニットが組込まれた組換え細胞の製造1
1−1:プラスミドベクターの構築
以下、複数コピーのエリスロポエチン(EPO)遺伝子をhprt遺伝子座に対して組込むため、シングルコピーのEPO遺伝子を含む、図1で表されるベクター(phprt-GT-EPO gene)を構築した。
【0060】
相同DNA配列の取得
JCRBセルバンクから入手したヒト由来細胞株HT1080細胞株(カタログ番号:IF050354)をGFX(商品名) Genomic Blood DNA Purification Kit (Amersham Biosciences)により処理し、ゲノムDNAを得た。次に、このゲノムDNAを鋳型とし、PCR反応(KOD-Plus-、TOYOBO)によって、ターゲットとなるhprt遺伝子の相同DNA配列(HA1およびHA2)をクローニングした。HA1およびHA2は、後述する図3でも示される通り、hprt遺伝子のエクソン3を含む領域と相同な配列に設定した。PCR反応に用いたプライマー配列は以下に示す通りである。
【0061】
HA1 sense プライマー:
5’-CCTGCAGGTCGCGATTGGTACTTGTTCAGCTTTATTCAAG-3’(配列番号1)
HA1 antisense プライマー:
5’-GTCGACAAGGACGCGTTCTGATAAAATCTACAGTCATAGGA-3’(配列番号2)
HA2 sense プライマー:
5’GTCGACCTCTAGCTAGAGCTTGGCGTAATCATGGTCTCCGGAGACTGAAGAGCTATTGTGTGAGTAT-3’(配列番号3)
HA2 antisense プライマー:
5’-ACATGTTCTCTTAAGTCGCGAAGTAGTGTTATGATGTATGGGCATA-3’(配列番号4)
【0062】
上記PCR反応においては、HA1センスプライマーの5’末端には、制限酵素Sse 8387IおよびNru Iの認識サイトを付加した。同様に、HA1アンチセンスプライマーの5’末端にはSal IおよびMlu Iの認識サイト、HA2センスプライマーの5’末端にはSal IおよびAcc IIIの認識サイト、HA2アンチセンスプライマーの5’末端にはPci IおよびNru Iの認識サイトをそれぞれ付加した。
【0063】
pQBI25プラスミドベクター(和光純薬株式会社)のDNAから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマーの5’末端および3’末端に、制限酵素Pci IおよびSse 8387Iの認識サイトを付加した。
【0064】
Ecoli sense プライマー:
5’-ACATGTGAGCAAAAGGCCAGCAAAAGGCCAGGAAC-3’(配列番号5)
Ecoli antisense プライマー:
5’-CCTGCAGGGACGTCAGGTGGCACTTTTCGGGGAAATGTGC-3’(配列番号6)
【0065】
HA1、HA2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Pci I、Sse 8387IおよびSal Iにより切断し、ライゲーション反応を行うことによりpHA12プラスミドベクターを得た。次に、pHA12プラスミドベクターを制限酵素Mlu IおよびSal Iにより切断し、5’末端から順に、HA2と、ori配列およびアンピシリン耐性遺伝子を含むDNA配列と、HA1とが連結したDNA配列1を得た。
【0066】
また、pQBI25プラスミドベクターを、制限酵素Not IおよびSal Iにより切断し、BGH ポリAシグナル配列(BGH pA)およびネオマイシン耐性遺伝子を含むDNA配列2(SV40, neoR, SV40pAを含む発現カセット)を取得した。
【0067】
また、pcDNA3.1プラスミドベクター(インビトロジェン)を、制限酵素Mlu IおよびNot Iにより切断し、CMVプロモーター・エンハンサー配列(CMV)およびマルチクローニングサイトを含むDNA配列3を取得した。
次に、DNA配列1〜3をライゲートしてphprt-GT-MCSプラスミドベクターを得た。
【0068】
次に、HT1080細胞株から抽出したゲノムDNAを鋳型とし、PCR反応によってエリスロポエチン遺伝子を増幅した。PCR反応に用いたセンスプライマーの5’末端には、制限酵素Nhe Iの認識配列を付加し、およびアンチセンスプライマーの5’末端には制限酵素Eco RIの認識配列を付加した。プライマーの塩基配列は以下の通りである。
【0069】
EPO gene ss プライマー:
5'- CCTTGCTAGCATGGGGGTGCACGGTGAGTA -3'(配列番号7)
EPO gene as プライマー:
5'- CCTTGAATTCTCATCTGTCCCCTGTCCTGC -3'(配列番号8)
【0070】
上記PCR反応により増幅されたEPO 遺伝子と、制限酵素Nhe IおよびEco RI により処理されたphprt-GT-MCSプラスミドとのライゲーションにより、EPO 遺伝子をphprt-GT-MCSプラスミドのマルチクローニングサイトのNhe I−Eco RIサイト間に挿入させ、図1に示されるphprt-GT-EPO gene(9116bp)を得た。得られたphprt-GT-EPO geneは大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0071】
1−2:細胞へのベクターの導入
プラスミド線状化
プラスミドベクターphprt-GT-EPO geneを、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0072】
トランスフェクション
ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)を1×107細胞/mLに調製し、線状化したプラスミドベクターphprt-GT-EPO gene、2μgと混合した。次に、得られた混合液を用いて、エレクトロポレーション法によりphprt-GT-EPO geneを細胞株HT-1080へトランスフェクトした。エレクトロポレーションは、Gene Pulser (BioRad社)を用いて、950μFの条件で行った。この実験のさらなる詳細は、Biotech. Bioeng. 2006. 95: 1052-1060.に記載の条件に従った。トランスフェクトした細胞は、500 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養し(培地: Advanced MEM(GIBCO社)に5% FBS、1x Glutamax(GIBCO社)を添加したもの)、トランスフェクションから24時間後にG418(インビトロジェン社)を加えた(最終濃度; 500 μg/ mL)。
【0073】
スクリーニング
培養8-12日後、上記プレートにG418耐性コロニーが現れたことを確認した。この段階で、6TG(最終濃度; 50 μM)(和光純薬工業)が添加された新しい培地を加え、さらに8日間培養した。培養後、全ウェルを確認し、6TG耐性コロニーを単離した。
【0074】
1−3:サザンハイブリダイゼーションによる解析/目的細胞の取得
以下、サザンハイブリダイゼーションを用い、hprt遺伝子座に複数コピーのEPO遺伝子が組込まれた組換え細胞を取得するため、6TG耐性コロニーのスクリーニングを行った。
プローブの調製
phprt-GT-EPO gene中のネオマイシン耐性遺伝子に相補的な配列を有するNRプローブを、以下の手順で合成した。まず、phprt-GT-EPO gene中のネオマイシン耐性遺伝子コード配列の全長をPCRにより増幅し、pGEM T プラスミドベクター(Promega社)にTAクローニングした。次に、PCR DIGプローブ合成キット(ロシュ社、プライマー:M13 Forward/Reverseプライマー)を用いて、DIG(Digoxigein)標識されたプローブを作製した。
【0075】
メンブレンの調製
6TG耐性コロニーから各細胞クローンのゲノムDNAをGFX Genomic Blood DNA purification kit(Amersham Biosciences社)を用いて抽出し、Bgl II制限酵素により切断した。切断したゲノムDNA10μgを、0.6%アガロースゲルを用いて電気泳動し、ナイロンメンブレン(Hybond N+ membrane、AmaershamBiosciences社)へ転写した。得られたメンブレンは80℃で2時間インキュベートし、DNAをメンブレン上に固定した。
【0076】
ハイブリダイゼーション
上記メンブレンに対し、上記NRプローブをハイブリダイズさせた。この際、プレハイブリダイゼーション、ハイブリダイゼーションおよびプローブの検出は、DIGアプリケーション マニュアル(ロシュ社)に従って行った。また、プローブのストリッピイングは、ストリッピングバッファー(0.2M NaOH, 0.1% SDS)を用い、37℃で15分の条件で2回行った。
【0077】
図2に示される通り、組換えクローン#14-5Eにおいて、NRプローブにより15822 bpのDNA断片が検出された。この組換えクローンは、約30クローンのスクリーニングの結果、1クローンのみ取得された。一方、参照として、組換えクローン#19-3Bを選択したところ、8546 bpのDNA断片が検出された。
【0078】
以下、図3(A)〜(C)に示される模式図および図2の結果に基づき、例1の結
果を説明する。
図3(A)で示される通り、hprt遺伝子(1)の相同組換えのターゲット部位(2)は、エクソン3(3)を含む領域に設定され、ホモロジーアーム1(HA1)(4)およびホモロジーアーム2(HA2)(5)は、ターゲット部位における二つの隣接した領域に相同であるように設定されている。そして、Bgl II制限酵素サイトは、ターゲット部位(2)を挟むように位置し、その内側には存在しない。一方、図3(B)および(C)に示されるphprt-GT-EPO geneDNA配列(6)にはBgl II制限酵素サイトは存在しない。そして、phprt-GT-EPO geneDNA配列(6)には、NRプローブ(7)がハイブリダイズしうるように設計されている。
【0079】
図3(C)に示される通り、Bgl II制限酵素サイトの位置、およびDNA配列の鎖長に基づき、15822 bpのDNA断片が検出された組換えクローン#14-5Eは、2コピーのphprt-GT-EPO geneDNA配列(6)がタンデムに繰り返し組込まれた目的細胞であることが確認された。
一方、図3(B)に示される通り、参照細胞であって8546 bpのDNA断片が検出された組換えクローン#19-3Bは、1コピーのphprt-GT-EPO geneDNA配列(6)が組込まれたものであることが確認された。
【0080】
例2:EPO比生産性の検討
2−1:組換えクローンの培養
組換えクローン#14-5Eおよび#19-3Bを樹立直後に、選択薬剤G418および6TG不存在下で培養を行った。
培養は、5 % FBS(Japan Bioserum社製)含有Advanced MEM(GIBCO社製)中、5 % CO2存在下37 ℃で行われた。トリプシン処理による継代は、約4〜5日の間隔で、細胞数および細胞密度が70-80 %に達した時点で行った。定期的に対数増殖期において、細胞数のカウントと培地のサンプリングを行い、以下のEPOの生産性解析に用いた。
【0081】
2−2:組換えクローンおよび培地のサンプリング
上記方法により培養した組換えクローン#14-5E および#19-3Bについて、以下の手順に従い、サンプル細胞を取得した。
まず、細胞サンプルを1シャーレあたり5 x 104 細胞播種し、FBS(Japan Bio Serum社製)を5 %含んだAdvanced MEM(GIBCO社製)中、5 % CO2存在下37 ℃で培養した。培養を開始してから3日目および4日目(対数増殖期)に、培地を回収し、さらに、培地回収後、細胞をトリプシンで処理し、細胞を回収した。回収した培地は、以下のELISA解析に用い、回収した組換えクローンの細胞数は血球計算板を用いてカウントした。なお、上記トリプシン処理は、0.25% Trypsin-EDTA溶液(GIBCO社製)を加え、室温で3〜5分間行った。トリプシン処理後、細胞を回収前には、血清添加培地を加えて反応を停止させた。
【0082】
2−3:ELISA
回収した培地中のEPO蓄積量は、固定化抗体としてMonoclonal anti-human EPO(R&D Systems)、1次抗体としてPolyclonal anti-human EPO(R&D Systems)、2次抗体としてAnti-mouse Ig, horseradish peroxidase linked whole antibody(from donkey)(Amersham Biosciences社製)を用い、TMB No Hydrogen Peroxide 1 Component HRP Microwell substrate(BioFX社製)により、450nmの吸光度を測定することにより解析した。
【0083】
2−4:組換えクローン#14-5Eと#19-3BとのEPO比生産性の比較
樹立直後の組換えクローン #14-5Eおよび#19-3Bに関し、細胞あたりの1日あたりのEPO比生産性を、回収した培地中のEPO蓄積量、細胞数、培養時間に基づき、次式にしたがって算出した。
(EPO比生産性)=(EPO蓄積量4日目-EPO蓄積量3日目)/((細胞数3日目+細胞数4日目)/2)/((サンプリング間の培養時間)/24)
【0084】
結果は、図4に示される通りであった。
hprt遺伝子座に2コピーのEPO遺伝子を含有する#14-5Eでは比生産性が2.16 pg/細胞/日であった。一方、1コピーのEPO遺伝子を含有する#19-3Bでは1.17 pg/細胞/日であった。#14-5EクローンのEPO生産性は#19-3Bクローンの約2倍であり、組込まれたコピー数に対して加算的に、EPO比生産性レベルが増加していた。
【0085】
例3:選択薬剤非添加培地における長期培養の影響評価
3−1:EPO生産性の測定
組換えクローン#14-5Eを、例2と同様の手法により、G418および6TG不含培地中で97日間継続培養を行った。継続培養中、定期的にEPO生産性の測定を行った。
【0086】
結果は、図5に示される通りであった。組換えクローン#14-5Eでは、EPO比生産性レベルは少なくとも97日間、一定に保たれていた。
【0087】
3−2:サザンハイブリダイゼーション
また、hprt遺伝子座に並列して組込まれた2コピーのEPO遺伝子が長期培養後も安定に保持されているか否かを例1と同様の手法により、サザンハイブリダイゼーションによって確認した。
【0088】
図6に示される通り、培養開始日(0日)および97日後において、15822 bpのDNA断片が検出された。少なくとも97日間、hprt遺伝子座にタンデムに繰り返し組込まれた2コピーのEPO遺伝子配列が安定に保持されていることが確認された。
【0089】
以上の結果に示される通り、hprt遺伝子座に2コピーのEPO遺伝子がタンデムに繰り返し組込まれた細胞株において、EPO遺伝子コピー数の増加に伴いEPO比生産性レベルが加算的に増加し、両者はほぼ正比例していることが確認された。さらに、選択圧が存在しない条件での培養を継続しても、EPO遺伝子のコピー数の減少やジーンサイレンシング、比生産性レベルの減少は認められなかった。
【0090】
例4:hprt遺伝子に複数のエリスロポエチン遺伝子発現ユニットが組込まれた組換え細胞の製造2
以下の手順に従い、効率的に複数のエリスロポエチン(EPO)遺伝子をhprt遺伝子に対して組換えるため、複数コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3)を構築した。
【0091】
4−1.プラスミドベクターの構築
1コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-1)の構築
複数コピーのEPO遺伝子を含むベクターの構築用材料として、まず、図7で示される1コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-1)を以下の手順により構築した。
phprt-GT-MCSプラスミドをEco RVおよびApa I制限酵素で切断し、Klenow酵素を作用させた後、セルフライゲーション反応を行い、MCS中の不要な制限酵素認識配列を除去したphprt-GT-MCSプラスミドを得た。
また、pIRESプラスミド(BDバイオサイエンス)を鋳型とし、PCR反応によりCMVプロモーター・エンハンサー配列、イントロン配列を含むDNA断片をクローニングし、PCR増幅産物を得た。PCR反応に用いたプライマー配列は以下の通りである。
【0092】
CMV−IVS−sense プライマー(配列番号9):
5’-CCTTACGCGTTCAATATTGGCCATTAGCCA-3’
CMV−IVS−antisense プライマー(配列番号10):
5’-CCTTGCTAGCCTATAGTGAGTCGTATTAAG-3’
【0093】
センスプライマーおよびアンチセンスプライマーの5’末端にはそれぞれMlu IおよびNhe I制限酵素認識配列を付加した。phprt-GT-MCSプラスミドを、Nhe IおよびMlu I制限酵素切断し、上記PCR増幅産物とライゲートしてphprt-IVS-GT-MCSプラスミドを得た。
次に、相同組換えHT1080細胞クローン#19-3Bより抽出した全RNAを鋳型とし、RT-PCR反応により、EPO cDNA配列をクローニングした。RT-PCR反応に用いたプライマー配列は以下の通りであった。
【0094】
EPO sense プライマー(配列番号11):
5’-CCTTGCTAGCATGGGGGTGCACGAATGTCC-3’
EPO gene as プライマー(配列番号8):
5'- CCTTGAATTCTCATCTGTCCCCTGTCCTGC -3'
【0095】
次に、EPO cDNAをNhe IおよびEco RI制限酵素サイトにおいて、phprt-IVS-GT-MCSプラスミドのMCSに組み込み、phprt-IVS-GT-EPOプラスミドを構築した。
phprt-IVS-GT-EPOプラスミド中、ネオマイシン耐性遺伝子ユニットとHA2との間に存在するユニークな制限酵素サイト(Acc IIIおよびSal I)の間に、5’末端側から順に制限酵素サイトBgl II、Not I、Xba I、およびApa Iを含むDNA断片をライゲートし、図7に示されるphprt-IVS-GT-EPO-1プラスミドを得た。このphprt-IVS-GT-EPO-1プラスミドは、以下の複数コピーのEPO遺伝子を含むベクターの構築において用いた。
【0096】
2コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-2)の構築
次に、図8に示される2コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2)を以下の手順により構築した。
まず、phprt-IVS-GT-EPOプラスミドをSal IおよびMlu I制限酵素で切断し、得られたプラスミドからHA1、HA2、および大腸菌内での操作配列を除去したEPO遺伝子発現ユニットを含む配列を得た。次に、pQBI25プラスミドベクター(和光純薬工業)を鋳型とし、Ecoli-BセンスプライマーとEcoli-Nアンチセンスプライマーを用いたPCR反応、およびEcoli-XセンスプライマーとEcoli-Aアンチセンスプライマーを用いたPCR反応により、大腸菌内における操作配列2種を得た。この操作配列2種をそれぞれ、上記EPO遺伝子発現ユニットを含む配列にライゲートし、両端にBgl IIおよびNot I制限酵素認識配列を含むEPO-BNプラスミド、および、両端にXba IおよびApa I制限酵素認識配列を含むEPO-XAプラスミドを得た。
【0097】
Ecoli-B sense プライマー(Bgl IIサイト用:配列番号12):
5’- CTACGCGTAGATCTGACGTCAGGTGGCACT -3’
Ecoli-N antisense プライマー(Not Iサイト用:配列番号13):
5’- CTGTCGACGCGGCCGCACATGTGAGCAAAA -3'
Ecoli-X sense プライマー(Xba Iサイト用:配列番号14):
5’- CTACGCGTTCTAGAGACGTCAGGTGGCACT -3'
Ecoli-A antisense プライマー(Apa Iサイト用:配列番号15):
5’- CTGTCGACGGGCCCACATGTGAGCAAAAGG -3'
【0098】
次に、phprt-IVS-GT-EPO-1プラスミド、およびEPO-BNプラスミドをそれぞれBgl IIおよびNot I制限酵素で切断し、ライゲートし、EPO遺伝子発現ユニットが2コピー同方向に並んで繰り返し含有するphprt-IVS-GT-EPO-2プラスミドを得た(図8)。
【0099】
3コピーのエリスロポエチン遺伝子を含むベクター(phprt-IVS-GT-EPO-3)の構築
次に、phprt-IVS-GT-EPO-2プラスミド、およびEPO-XAプラスミドをそれぞれXba IおよびApa I制限酵素で切断し、ライゲートし、EPO遺伝子発現ユニットが3コピー同方向に並んで繰り返し含有するphprt-IVS-GT-EPO-3プラスミドを得た(図9)。
なお、得られたphprt-IVS-GT-EPO-1、phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3は、それぞれ大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0100】
4−2:細胞へのベクターの導入
プラスミド線状化
複数のEPO遺伝子を含む上記プラスミドベクターphprt-IVS-GT-EPO-2、phprt-IVS-GT-EPO-3は、Endofree Plasmid Maxi kit(QIAGEN社製)を用いて精製し、Nru Iにて切断した。2g/Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0101】
トランスフェクションおよびスクリーニング
線状化したプラスミドベクターphprt-IVS-GT-EPO-2、および、hprt-IVS-GT-EPO-3を、ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)に導入し、相同組換え細胞株の取得を行った。トランスフェクションおよびスクリーニングの条件は、例1に記載した方法と同様にして行った。
【0102】
phprt-IVS-GT-EPO-2のトランスフェクションによりG418および6TGに耐性の3個のコロニーを得た。一方、phprt-IVS-GT-EPO-3のトランスフェクションにおいては、同条件でエレクトロポレーションを2回行うことにより、1個のG418および6TG耐性コロニーを得た。
【0103】
4−3:ゲノムPCRによる解析
GFX Genomic Blood DNA purification kit(Amersham Biosciences社)を用い、phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3のトランスフェクションにより得たG418および6TGに耐性のコロニーからゲノムDNAを抽出し、このゲノムDNAを鋳型とした以下に示すPCR反応により、標的hprt遺伝子座への位置特異的な組換えの確認を行った。なお、参考として、シングルコピーのEPO遺伝子を含むphprt-IVS-GT-EPO-1についても、線上化、トランスフェクションおよびスクリーニングをphprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3と同様に行い、ゲノムPCRによる解析を行った。
【0104】
図10(A)は、ゲノムPCRによる相同組み換え反応の解析の詳細を説明する模式図である。hprt遺伝子(1)の相同組換えのターゲット部位(2)は、エクソン3(3)を含んで設定されており、この領域にベクターDNA(8)中のHA1(4)、HA2(5)およびこれに挟まれたEPO発現ユニットを含む繰返し配列(9)は相同組み換えにより組込まれる。ここで、nはEPO発現ユニットを含む繰返し配列の数を意味し、例4では1〜3である。
HA1(4)またはHA2(5)とEPO発現ユニットを含む繰返し配列(9)の一部とを含むDNA配列(3209 bp 、1443 bpのDNA配列)は相同組み換えによってのみゲノムから取得することができ、相同組み換えの指標となる。
よって、HA1とターゲット部位との相同組換えの指標として、図10(A)に示す3209 bpのDNA配列を設定し、このDNA配列は、以下に示すプライマーHPRTs2/Bgh-scによるPCR反応により検出した。一方、HA2と標的hprt遺伝子座との相同組換えの指標として、図10(A)に示す1443 bpのDNA配列を設定し、このDNA配列は、以下に示すプライマーHPRTas1/Neo-seqSCによるPCR反応により検出した。
【0105】
HPRTs2プライマー(配列番号16):
5’- AAAGTTCTCTCCTTTCAGCCTTCTGTACAC -3’
Bgh-scプライマー(配列番号17):
5’- GCACCTTCCAGGGTCAAGGA -3'
HPRTas1プライマー(配列番号18):
5’- ACAAGTTAAAAGGAGCTTATGGGTAGGAAG -3'
Neo-seqSCプライマー(配列番号19):
5’- CCTTCTATCGCCTTCTTGAC -3’
【0106】
次に、得られたPCR増幅産物を1.0%アガロースゲル電気泳動により解析した。結果は、図10(B)および(C)に示される通りであった。
図10(B)および(C)において、2-1、2-2、2-3はphprt-IVS-GT-EPO-2プラスミドベクターにより得られたクローンを示し、3-1はphprt-IVS-GT-EPO-3プラスミドベクターにより得られたクローンを示し、1-1は、phprt-IVS-GT-EPO-1プラスミドベクターにより得られたクローンを示す。取得したクローンすべてにおいて、3209 bpおよび1443 bpのPCR増幅産物が確認され、相同組換え反応が確認された。
【0107】
以上の通り、複数コピーのEPO遺伝子を含むベクター(phprt-IVS-GT-EPO-2およびphprt-IVS-GT-EPO-3)を用いた場合、2コピー組込まれた組換え細胞クローンの出現頻度は、1回のエレクトロポレーションに対して3クローンであり、3コピー組込まれたクローンについては、2回のエレクトロポレーション操作に対して1クローンであった。
【0108】
例5:hprt遺伝子座にepo遺伝子発現ユニットおよびgs遺伝子増幅マーカーが1コピー組込まれた組換え細胞の製造
5−1:プラスミドベクターの構築
以下、エリスロポエチン(EPO)遺伝子およびグルタミン合成酵素(GS)遺伝子増幅マーカーをhprt遺伝子座に組込むため、図11で表されるベクター(pSV-GS-neo-GT-EPO #22)を構築した。
【0109】
ベクター基本骨格の構築
市販されているpIRESベクター(BD Biosciences)を鋳型とし、NEO-ssプライマーおよびNEO-asプライマーを用いたPCR反応により、5’-端にSma I制限酵素サイト、3’-端にNot I制限酵素サイトを含むネオマイシン耐性遺伝子コード配列のPCR増幅産物1を得た。このPCR増幅産物1およびpIRESベクターをSma IおよびNot I制限酵素で切断し、ライゲート反応することでプラスミド1を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0110】
NEO-ssプライマー:
5’-CCTTCCCGGGATGATTGAACAAGATGGAT-3’(配列番号20)
NEO-asプライマー:
5’-CCTTGCGGCCGCTCAGAAGAACTCGTCA-3’(配列番号21)
【0111】
次にpQBI25ベクター(和光純薬工業)を鋳型とし、BGH-ssプライマーおよびBGH-asプライマーを用いたPCR反応により、5’-端にSal I制限酵素サイト、3’-端にBam HI制限酵素サイトを含むPCR増幅産物2を得た。同様に、pIRESベクターを鋳型とし、MCS-ssプライマーおよびMCS-asプライマーを用いたPCR反応により、5’-端にStu I制限酵素サイト、3’-端にSal I制限酵素サイトを含むPCR増幅産物3を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0112】
BGH-ssプライマー:
5’-GTCGACGATATCTCTAGATGTGCCTTCTAG-3’(配列番号22)
BGH-asプライマー:
5’-CCTTGGATCCTCCGGAAGCCATAGAGCCCA-3’(配列番号23)
MCS-ssプライマー:
5’-CCTTAGGCCTAGGCTTTTGCAAAAAGCTTTATTGCGGTAGT-3’(配列番号24)
MCS-asプライマー:
5’-TCTAGAGATATCGTCGACCTATAGTGAGTC-3’(配列番号25)
【0113】
前記プラスミド1をStu IおよびBam HI制限酵素反応により切断し、991 bpと5876 bpの切断断片を得た。このうち、5876 bpの切断断片をアガロースゲル電気泳動およびゲル切り出し操作によって精製し、DNA断片1を得た。また、前記PCR増幅産物2をSal IおよびBam HI制限酵素反応により切断し、DNA断片2を得た。同様に、PCR増幅産物3をStu IおよびSal I制限酵素反応により切断し、DNA断片3を得た。このDNA断片1〜3をライゲート反応して、プラスミド2を得た。
【0114】
JCRBセルバンクから入手したヒト由来細胞株HT1080細胞株(カタログ番号:IFO50354)から、ISOGEN(ニッポンジーン)を用いて抽出した全RNAを鋳型とし、GS-ssプライマーおよびGS-asプライマーを用いたRT-PCR反応により、gs コード配列の5’-端にSal I制限酵素サイト、3’-端にXba I制限酵素サイトを含むPCR増幅産物4を得た。RT-PCR反応にはOne Step RT-PCR Kit(QIAGEN)を用いた。また、ヒトHT1080細胞株からGFXTM Genomic Blood DNA Purification Kit(Amersham Biosciences)を用いて抽出したゲノムDNAを鋳型とし、EPO-ssプライマーおよびEPO-asプライマーを用いたPCR反応により、epo遺伝子の5’-端にNhe I制限酵素サイト、3’-端にEco RI制限酵素サイトを含むPCR増幅産物5を得た。PCR反応に用いたプライマー配列は以下に示す通りである。
【0115】
GS-ssプライマー:
5’-CCTTGTCGACCACCATGACCACCTCAGCAA-3’(配列番号26)
GS-asプライマー:
5’-CCTTTCTAGATTAATTTTTGTACTGGAAGG-3’(配列番号27)
EPO-ssプライマー:
5’-CCTTGCTAGCATGGGGGTGCACGGTGAGTA-3’(配列番号28)
EPO-asプライマー:
5’-CCTTGAATTCTCATCTGTCCCCTGTCCTGC-3’(配列番号29)
【0116】
プラスミド2およびPCR増幅産物4をSal IおよびXba I制限酵素反応により切断し、ライゲートすることでプラスミド3を得た。次にプラスミド3およびPCR増幅産物5をNhe IおよびEco RI制限酵素反応により切断し、ライゲートすることで、EPO発現ユニットおよびgs増幅マーカーを含むプラスミド4を得た。
【0117】
相同DNA配列の取得
HT1080株のゲノムDNAを鋳型とし、PCR反応(KOD-Plus-、TOYOBO)によって、ターゲットとなるhprt遺伝子の相同DNA配列(HA1およびHA2)をクローニングした。HA1およびHA2は、図12に示される通り、hprt遺伝子のエクソン3を含む領域と相同な配列に設定した。PCR反応に用いたプライマー配列は以下に示す通りである。
【0118】
HA1 sense プライマー:
5’-CCTGCAGGTCGCGATTGGTACTTGTTCAGCTTTATTCAAG-3’(配列番号1)
HA1-SB antisense プライマー:
5’-GTCGACAAGGAGATCTACGCGTTCTGATAAAATCTACAGTCATAGGA-3’(配列番号30)
HA2 sense プライマー:
5’-GTCGACCTCTAGCTAGAGCTTGGCGTAATCATGGTCTCCGGAGACTGAAGAGCTATTGTGTGAGTAT-3’(配列番号3)
HA2 antisense プライマー:
5’-ACATGTTCTCTTAAGTCGCGAAGTAGTGTTATGATGTATGGGCATA-3’(配列番号4)
【0119】
上記PCR反応においては、HA1センスプライマーの5’末端には、制限酵素Sse 8387IおよびNru Iの認識サイトを付加した。同様に、HA1アンチセンスプライマーの5’末端にはSal IおよびBgl IIの認識サイト、HA2センスプライマーの5’末端にはSal IおよびAcc IIIの認識サイト、HA2アンチセンスプライマーの5’末端にはPci IおよびNru Iの認識サイトをそれぞれ付加した。
【0120】
pQBI25プラスミドベクターから、大腸菌内での複製起点であるori配列およびアンピシリン耐性遺伝子を含むDNA配列を、PCR反応によりクローニングした。PCR反応に用いたプライマー配列は以下に示す通りである。このPCR反応において、センスプライマーの5’末端に制限酵素Pci I、およびアンチセンスプライマーの5’末端に制限酵素Sse 8387Iの認識サイトを付加した。
【0121】
Ecoli sense プライマー:
5’-ACATGTGAGCAAAAGGCCAGCAAAAGGCCAGGAAC-3’(配列番号5)
Ecoli antisense プライマー:
5’-CCTGCAGGGACGTCAGGTGGCACTTTTCGGGGAAATGTGC-3’(配列番号6)
【0122】
HA1、HA2、およびori配列およびアンピシリン耐性遺伝子を含むDNA配列をそれぞれ、PCR反応によりクローニングした。これら3つのDNA配列を制限酵素Pci I、Sse 8387IおよびSal Iにより切断し、ライゲーション反応を行うことによりpHA12プラスミドベクターを得た。次に、pHA12プラスミドベクターを制限酵素Bgl IIおよびAcc IIIにより切断し、5’末端から順に、HA2と、ori配列およびアンピシリン耐性遺伝子を含むDNA配列と、HA1とが連結したDNA配列1を得た。
【0123】
EPO発現ユニットおよびgs増幅マーカーを含むプラスミド4を同様に制限酵素Bgl IIおよびAcc IIIにより切断し、約2 kbpと約6.2 kbpの切断断片とした。アガロースゲル電気泳動およびゲル切り出し操作により精製し、約6.2 kbpの断片を得た。これを前記DNA配列1とライゲートすることにより、図11で表されるベクターpSV-GS-neo-GT-EPO #22を得た。得られたpSV-GS-neo-GT-EPO #22は大腸菌DH5α(New England Biolabs社製)中で増殖させた。
【0124】
5−2:細胞へのベクターの導入
プラスミド線状化
プラスミドベクターpSV-GS-neo-GT-EPO #22を、Endofree Plasmid Maxi kit(QIAGEN)を用いて精製し、Nru Iにて切断した。フェノール・クロロホルム抽出により精製した後、2 g/ Lの濃度になるように滅菌水に溶解し、以下のトランスフェクション実験に用いた。
【0125】
トランスフェクション
ヒト線維肉腫細胞株HT-1080(JCRBセルバンクID:IFO50354)を1×107細胞/mLに調製し、線状化したプラスミドベクターpSV-GS-neo-GT-EPO #22、2μgと混合した。次に、得られた混合液を用いて、エレクトロポレーション法によりpSV-GS-neo-GT-EPO #22を細胞株HT-1080へトランスフェクトした。エレクトロポレーションは、GenePulser(BioRad)を用いて、950 μFの条件で行った。この実験のさらなる詳細は、Biotech. Bioeng. 2006. 95: 1052-1060.に記載の条件に従った。トランスフェクトした細胞は、500 細胞/ウェルにて96ウェルプレートに播種し、インキュベーター中37 ℃、5 % CO2にて培養し(培地: Advanced MEM(GIBCO)に5% FBS、1x Glutamax(GIBCO)を添加したもの)、トランスフェクションから48時間後にG418(インビトロジェン)を加えた(最終濃度; 500 μg/ mL)。
【0126】
スクリーニング
培養8-12日後、上記プレートにG418耐性コロニーが現れたことを確認した。この段階で、6TG(最終濃度; 50 μM)(和光純薬工業)が添加された新しい培地を加え、さらに8日間培養した。培養後、全ウェルを確認し、6TG耐性コロニーを単離した。
【0127】
5−3:サザンハイブリダイゼーションによる解析
以下、サザンハイブリダイゼーションにより、6TG耐性コロニーのスクリーニングを行った。
プローブの調製
pSV-GS-neo-GT-EPO #22中のネオマイシン耐性遺伝子に相補的な配列を有するNRプローブを、以下の手順で合成した。まず、pSV-GS-neo-GT-EPO #22中のネオマイシン耐性遺伝子コード配列の全長をPCRにより増幅し、pGEM T プラスミドベクター(Promega)にTAクローニングした。次に、PCR DIGプローブ合成キット(ロシュ、プライマー:M13 Forward/Reverseプライマー)を用いて、DIG(Digoxigein)標識されたプローブを作製した。
【0128】
メンブレンの調製
6TG耐性コロニーから各細胞クローンのゲノムDNAをgenomic prep cells and tissue DNA isolation kit(GE healthcare)を用いて抽出し、Eco RI制限酵素により切断した。切断したゲノムDNA 10μgを、0.6%アガロースゲルを用いて電気泳動し、ナイロンメンブレン(Hybond N+ membrane、AmaershamBiosciences)へ転写した。得られたメンブレンは80℃で2時間インキュベートし、DNAをメンブレン上に固定した。
【0129】
ハイブリダイゼーション
上記メンブレンに対し、上記NRプローブをハイブリダイズさせた。この際、プレハイブリダイゼーション、ハイブリダイゼーションおよびプローブの検出は、DIGアプリケーション マニュアル(ロシュ)に従って行った。
【0130】
図13に示される通り、組換えクローン118S-1、118S-2、118S-3、118S-5において、NRプローブにより約8 kbpのDNA断片が検出された。制限酵素Eco RIサイトはpSV-GS-neo-GT-EPO #22中および標的hprt遺伝子座中のいずれにも存在しており、相同組換え反応が首尾よく生じていればNRプローブによって7781 bpのEco RI断片が検出される。この結果は図13に示した4クローンがいずれも目的とするhprt遺伝子座にepo遺伝子発現ユニットおよびgs遺伝子増幅マーカーが1コピー組込まれた組換え細胞であることを示している。
【0131】
例6:遺伝子増幅によってhprt遺伝子座に複数のepo遺伝子発現ユニットを組込んだ組換え細胞の製造
6−1:グルタミンフリー培地での増殖能検討
培地へのMSX添加による遺伝子増幅細胞のスクリーニングに先立って、グルタミン非添加培地での増殖能を評価した。グルタミン添加培地(培地組成: Advanced MEMに5% FBS、1x Glutamaxを添加したもの)で維持していた前記細胞クローンを、70〜80%コンフルエントになった段階でトリプシン処理し、細胞を回収した。回収した細胞懸濁液の細胞密度は、トリパンブルー染色法によって求めた。培養ディッシュ(直径10 cm)1枚あたり1.7X105 cellsの細胞を播種し、10 mlのグルタミン非添加培地(培地組成: Advanced MEMに2% FBS、1x GS supplement(ニチレイ)を添加したもの)を加え、CO2インキュベーター内で培養した。6日間培養後、同じ手順で細胞を回収し、回収した細胞懸濁液の液量および細胞密度から総細胞数を算出し、増殖能を評価した。同様の手順で1.7X105 cellsの細胞を新しい培養ディッシュに移し、10 mlのグルタミン非添加培地を加えてCO2インキュベーター内で培養した。この操作を2回繰り返し、グルタミンフリー培地での増殖を検証した。
【0132】
馴化クローンの選択
118S-2および118S-5クローンが3回めの培養期間中に、それぞれ2倍および4倍に細胞数が増加していた。非組換えHT1080細胞では、3回めの培養期間中での増殖がみられなかったことから、これら2クローンは外来性gs遺伝子の発現によりグルタミン非添加培地での増殖が支持されているものと考えられた。以降のMSXスクリーニングには、最も増殖のよかった118S-5クローンを用いた。
【0133】
6−2:MSXスクリーニング
グルタミン非添加培地で維持していた118S-5クローンを、70〜80%コンフルエントになった段階でトリプシン処理により回収した。MSX濃度を 5または10 μMに調整した選択培地(培地組成: Advanced MEMに2% FBS、1x GS supplement、5または10 μM MSXを含有するもの)で、1×104 cells/ mlと2×104 cells/ mlの細胞懸濁液を調整した。この細胞懸濁液を96ウェルマイクロテストプレート(BD Falcon)に、1ウェルあたり100 μLづつ播種し、37℃、5% CO2条件下で培養を行った。培養開始から1週間後に培地をフレッシュなものに交換した。さらに1週間の培養後、増殖のみられるウェルを顕微鏡観察により判別した。MSX濃度を10 μMとした選択培地では耐性を示した細胞は存在せず、細胞増殖がみられるウェルは存在していなかった。一方、MSX濃度を5 μMとした選択培地では、播種したウェルのおおよそ30%のウェルに耐性細胞が存在していた。次にこれらのウェル中のEPO蓄積量をELISAによって測定した。
【0134】
6−3:EPO生産量による遺伝子増幅細胞の選択
ELISA
固定化抗体としてMonoclonal anti-human EPO(R&D Systems)、1次抗体としてPolyclonal anti-human EPO(R&D Systems)、2次抗体としてAnti-mouse Ig, horseradish peroxidase linked whole antibody(from donkey)(Amersham Biosciences)を用い、TMB No Hydrogen Peroxide 1 Component HRP Microwell substrate(BioFX)により、450nmの吸光度を測定することにより解析した。
【0135】
EPO蓄積量の解析
ELISAによるEPO蓄積量測定の2日前に、耐性細胞が存在する96ウェルプレートの選択培地をフレッシュなものに完全交換した。各ウェルあたり50 μLの培地を回収し、ELISAにより測定を行った結果、2日間の培養で0.3〜37 ng/ mlのEPOが蓄積していた。次に、EPO蓄積量が最大値37.3 ng/ mlを示した2-1F8クローン、中程度の値17.7 ng/mlを示した1-2E6クローンに対してgs遺伝子コピー数の解析を行った。
【0136】
6−4:gs遺伝子コピー数の解析
ゲノムサンプルの調整
細胞からのゲノム抽出は、DNA Isolation kit for cells and tissuesキット(Roche)を用いて行った。抽出したゲノムを制限酵素Hind III(TaKaRa)で切断した後、エタノール沈殿でゲノムを回収し、滅菌水に溶解させた。吸光度測定によりDNA濃度を測定した。
【0137】
Real-time定量PCR解析
調整したゲノムDNAを用いて増幅マーカーであるgs遺伝子のコピー数の測定をReal-time定量PCRにより行った。Real-time定量PCR はABI PRISM 7300(ABI)を用い、Taqman Universal PCR Master Mix(ABI)により反応を行った。PCR反応に用いたプライマーおよびプローブの配列を以下に示す。
【0138】
hGS Forプライマー:
5’-ACCCCTTTTCGGTGACAGAA-3’(配列番号31)
hGS Revプライマー:
5’-TCGCCGGTTTCATTGAGAAG-3’(配列番号32)
hGS cDNA-Taqmanプローブ:
5’-CCCTCATCCGCACGTG-3’(配列番号33)
【0139】
コピー数解析
定量PCR結果の解析は装置付属のソフト7300 system softwareで行った。コピー数の算出は以下の手順に従い行った。各ゲノムサンプルに対し、hGSプライマーおよびプローブで測定したCt値と、濃度既知のベクター(pSV-GS-neo-GT-EPO#22)による検量線からコピー数を算出した。サンプル中のゲノム量を補正するため、ゲノム中の他部位としてベータアクチンのコピー数を各サンプルについてさらに測定し、この値が一定となるよう補正を行った。
【0140】
結果を図14に示す。2-1F8細胞と1-2E6細胞は、118S-5クローンからMSX 5 μMへ耐性を示す細胞として取得したものである。S-5(Glu-)とした細胞は、グルタミン非添加培地で維持していた118S-5クローンである。S-5(Glu+)はグルタミン添加培地で維持していた118S-5クローンである。このS-5(Glu+)を対照として、それぞれの細胞におけるRealtime PCR法によるコピー数解析結果をS-5(Glu+)に対する相対値で算出し、コピー数比としてグラフに示した。2-1F8細胞は2.67、1-2E6細胞は1.43という値を示した。これらの結果から、MSX耐性細胞株である2-1F8細胞では遺伝子増幅によりコピー数が増加していると判断できる。遺伝子増幅技術の原理を鑑みれば、組込み位置近傍のhprt遺伝子座内でコピー数が増加した細胞株であると判断できる。また、ELISA解析によるEPO蓄積量は、2-1F8細胞で37.3 ng/ ml、1-2E6細胞で17.7 ng/mlであり、コピー数比の値と相関していた。
【特許請求の範囲】
【請求項1】
ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座に外来性の目的タンパク質遺伝子の複数コピーが導入されてなる哺乳動物細胞を用意し、該細胞を培養して前記目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法。
【請求項2】
前記目的タンパク質遺伝子のコピー数が2以上である、請求項1に記載の方法。
【請求項3】
前記目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる、請求項1または2に記載の方法。
【請求項4】
前記目的タンパク質遺伝子または前記発現ユニットが、タンデムに並んで繰り返し組込まれてなる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
宿主細胞がヒト由来の細胞である、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記宿主細胞がヒト線維肉腫由来のHT1080細胞株である、請求項5に記載の方法。
【請求項7】
hprt遺伝子座に、外来性のタンパク質遺伝子の複数コピーが組込まれてなる、哺乳動物細胞。
【請求項8】
前記目的タンパク質遺伝子のコピー数が2以上である、請求項7に記載の細胞。
【請求項9】
前記目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に導入されてなる、請求項7または8に記載の細胞。
【請求項10】
前記目的タンパク質遺伝子または前記発現ユニットが、タンデムに並んで繰り返し導入されてなる、請求項7〜9のいずれか一項に記載の細胞。
【請求項11】
宿主細胞がヒト由来の細胞である、請求項7〜10のいずれか一項に記載の細胞。
【請求項12】
前記宿主細胞がヒト線維肉腫由来のHT1080細胞株である、請求項11に記載の細胞。
【請求項13】
宿主細胞のhprt遺伝子座に外来性の目的タンパク質遺伝子の複数コピーを組込む工程を含んでなる、請求項7〜12のいずれか一項に記載の細胞の製造方法。
【請求項14】
前記目的タンパク質遺伝子が、相同組換えにより導入される、請求項13に記載の方法。
【請求項15】
宿主細胞のhprt遺伝子座に目的タンパク質遺伝子が組込まれ、かつ遺伝子増幅マーカー遺伝子が組込まれた細胞を用意し、
該細胞を、遺伝子増幅を生じさせる条件に付し、hprt遺伝子座に組込まれた外来性の目的タンパク質遺伝子のコピー数を増加させること
を含んでなる、請求項7〜12のいずれか一項に記載の細胞の製造方法。
【請求項1】
ヒポキサンチン−ホスホリボシルトランスフェラーゼ酵素(hprt)遺伝子座に外来性の目的タンパク質遺伝子の複数コピーが導入されてなる哺乳動物細胞を用意し、該細胞を培養して前記目的タンパク質を産生することを含んでなる、目的タンパク質の製造方法。
【請求項2】
前記目的タンパク質遺伝子のコピー数が2以上である、請求項1に記載の方法。
【請求項3】
前記目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に組込まれてなる、請求項1または2に記載の方法。
【請求項4】
前記目的タンパク質遺伝子または前記発現ユニットが、タンデムに並んで繰り返し組込まれてなる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
宿主細胞がヒト由来の細胞である、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記宿主細胞がヒト線維肉腫由来のHT1080細胞株である、請求項5に記載の方法。
【請求項7】
hprt遺伝子座に、外来性のタンパク質遺伝子の複数コピーが組込まれてなる、哺乳動物細胞。
【請求項8】
前記目的タンパク質遺伝子のコピー数が2以上である、請求項7に記載の細胞。
【請求項9】
前記目的タンパク質遺伝子が、少なくともプロモーター配列および転写終結シグナル配列を含んだ発現ユニットとしてhprt遺伝子座に導入されてなる、請求項7または8に記載の細胞。
【請求項10】
前記目的タンパク質遺伝子または前記発現ユニットが、タンデムに並んで繰り返し導入されてなる、請求項7〜9のいずれか一項に記載の細胞。
【請求項11】
宿主細胞がヒト由来の細胞である、請求項7〜10のいずれか一項に記載の細胞。
【請求項12】
前記宿主細胞がヒト線維肉腫由来のHT1080細胞株である、請求項11に記載の細胞。
【請求項13】
宿主細胞のhprt遺伝子座に外来性の目的タンパク質遺伝子の複数コピーを組込む工程を含んでなる、請求項7〜12のいずれか一項に記載の細胞の製造方法。
【請求項14】
前記目的タンパク質遺伝子が、相同組換えにより導入される、請求項13に記載の方法。
【請求項15】
宿主細胞のhprt遺伝子座に目的タンパク質遺伝子が組込まれ、かつ遺伝子増幅マーカー遺伝子が組込まれた細胞を用意し、
該細胞を、遺伝子増幅を生じさせる条件に付し、hprt遺伝子座に組込まれた外来性の目的タンパク質遺伝子のコピー数を増加させること
を含んでなる、請求項7〜12のいずれか一項に記載の細胞の製造方法。
【図1】



【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2010−46066(P2010−46066A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−214833(P2009−214833)
【出願日】平成21年9月16日(2009.9.16)
【分割の表示】特願2008−555549(P2008−555549)の分割
【原出願日】平成20年8月8日(2008.8.8)
【出願人】(000010087)TOTO株式会社 (3,889)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成21年9月16日(2009.9.16)
【分割の表示】特願2008−555549(P2008−555549)の分割
【原出願日】平成20年8月8日(2008.8.8)
【出願人】(000010087)TOTO株式会社 (3,889)
【Fターム(参考)】
[ Back to top ]