
静脈内投与による有効成分制御放出のための生体適合性を有するアルギン酸微粒子組成物
【課題】血中有効成分の半減期又は残存率を高めるのに十分なサイズを有するとともに、静脈内投与時に低い肝臓取込み及び迅速な細胞クリアランスを示す微粒子を提供する。
【解決手段】アルギン酸又はその塩の微粒子と有効成分とを含んでなる生体適合性組成物。より詳細には、必要な患者に静脈内投与すべく有効成分を被包化するための微粒子。
【解決手段】アルギン酸又はその塩の微粒子と有効成分とを含んでなる生体適合性組成物。より詳細には、必要な患者に静脈内投与すべく有効成分を被包化するための微粒子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はアルギン酸又はその塩の微粒子(micoparticles)と有効成分を含んでなる生体適合性組成物に関し、より詳細には、必要とする患者に静脈内投与するべき有効成分を被包化するための微粒子に関する。かかる微粒子は、血中有効成分の半減期又は残存率を高めるのに十分なサイズと、静脈内投与時の低い肝臓取込み及び迅速な細胞クリアランス(cell clearance)とを併せ持つ。本発明の組成物中の有効成分としては、天然、組換、又はトランスジェニックの、ヒト又は動物由来のペプチド、蛋白質又はホルモンが挙げられる。本発明の組成物の有効成分の例としては、第VIII因子、第IX因子、又は第VIIa因子などの血液凝固因子が挙げられる。
【背景技術】
【0002】
治療有効成分の血中半減期の延長には、所望の治療効果を得るために必要な投与回数の低減等の利点がある。かかる投与回数の低減は、特に非経口投与用の薬剤にとって、中でも静脈注射用の薬剤にとって重要であり、また、慢性疾患治療等のための長期投薬に特に深い関連を有している。
【0003】
可能なかぎり、静脈内アクセスを必要としない手段で有効成分を投与するのが、最近の傾向である。かかる方法の使用は、患者にとって煩雑且つ不便だからである。しかし、現在でも静脈投与に代わる手段がない一連の有効成分が存在する。かかる成分としては、サイズが大きく複雑な有効成分、例えば蛋白質やホルモン等の生物産物又は生物工学産物等が挙げられる。
【0004】
複雑な有効成分の静脈内反復投与が必要な慢性的治療疾患の一例に血友病がある。血友病は、血液の凝固に関連する蛋白質の全欠損又は部分欠損による内外出血の出現を特徴とする遺伝病である。血友病Aの特徴は第VIII凝固因子の欠損であり、これがトロンビン(thrombin)の正常な生成を妨げる結果、血液の正常な凝固が困難となる。血友病Bでは第IX因子の欠損が同様の病態をもたらす。
【0005】
血友病の治療における第一の選択肢となるのは、前記因子(FVIII又はFIX)を含有する治療用濃縮物を投与して欠損蛋白質を置換する方法である。血友病において的確な止血を得る別の治療上の選択肢はFVIIaの投与であり、これはFVIII又はFIXがなくてもトロンビンを生成させることができる。しかし、このタイプの治療は通常、FVIII又はFIXでの治療に問題があったり、かかる治療が有効でないと判明した場合、例えばこれらの有効成分に対して抑制的な免疫応答が働いた患者等の場合に限られる。現在まで、これらの産物は何れも、その構造上の複雑さ及び上皮透過性の低さゆえに、静脈内投与以外の方法による投与は成功していない。
【0006】
従って、血友病の患者には、血漿中半減期に応じて定まる頻度で繰り返し静脈内投与を行う必要がある。FVIIIの場合、半減期は約12時間である。これは、世界血友病連盟(the World Federation of Haemophilia)のモノグラフ(非特許文献1:Casper, CK, Hereditary Plasma Clotting Factor Disorders and Their Management 5th ed. WFH, Sam Schulman Ed., 2008)によれば、一次予防法の場合、すなわち関節損傷のない小児の出血の予防の場合、48時間毎に約20U/kgの投与量の使用を意味する。これは正常値の1%を超える血漿FVIIIレベルを維持するのに十分な量である。原則として、この治療によって、重度の血友病が、軽度又は中度の血友病に変わる。FIXの場合、半減期は約26時間であり、一次予防としては、最低1%のレベルを維持するために、約40U/kgの量を週2回投与すればよい。
【0007】
考慮しなければならないのは、若年齢(約1歳又は匍匐開始期)からの予防が、重度血友病の場合の関節損傷を避けるのに必要な標準的治療であるという点である。
【0008】
従って、血友病は、有効成分の半減期の延長が患者の生活の質(quality of life)に実質的な改善をもたらす明らかな例である。幼い子供にはことに困難な静脈内投与の回数を低減し得るからである。
【0009】
静脈内投与製品による長期治療の他の例としては、原発性免疫不全における免疫グロブリン(IgG)の使用、先天性欠損におけるアンチトロンビンIII(AT)及びアルファ−1−アンチトリプシン(AAT)の使用等が挙げられる。
【0010】
これらのタイプの有効成分の血漿中半減期の延長を目的とする技術的アプローチは多数存在する。最も研究されてきたものの一つは、ポリエチレングリコール(PEG)のように、適合性を有するポリマーで蛋白質を誘導体化することであった。この技術は、蛋白質のアミノ酸にPEG鎖を共有結合させる化学反応操作からなる。この技術は、インターフェロン等のサイズが小さいホルモンやペプチド鎖に有用であることが分かっている。このタイプの化合物の場合、主たる排除機構は腎クリアランスであり、これは単にサイズを大きくすることで容易に制御できるからである(非特許文献2:Bailon Pascal et al, Bioconjugate Chem. 2001, 12, 195-202)。しかし、これは処理対象の蛋白質構造の外的修飾に基づく手法であるため、より複雑な有効成分にも使用できるか否かはまだ分からない。また、このタイプの蛋白質との共有結合によれば、処理後のホルモン又は蛋白質の生物活性は大幅に低下する。
【0011】
半減期を改善するもう一つの代案は、蛋白質やホルモンが有する天然の糖残基の付加又は修飾であった(非特許文献3:Perlman Signe et al, The Journal of Clinical Endocrinology & Metabolism 88 (7): 3227-3235, 2003)。この手法は、その分解に関与する受容体による認識を変更することにより、蛋白質を改変するというものである。しかし、この改変には固有のリスクが存在することは、前記蛋白質に存在する糖鎖の免疫原性を考慮すれば明らかである。
【0012】
三番目の方針は、アルブミンや免疫グロブリン断片の場合のように、相当な半減期を有する血漿蛋白質の配列と結合させた状態で、所期の蛋白質の活性を有する配列を発現したキメラ蛋白質を得ることであった(非特許文献4:Dennis, Marks S. et al, The Journal of Biological Chemistry vol. 277, No. 38, Issue of September 20, pp. 35035-35043, 2002)。しかし、この技術は、天然に存在しない蛋白質に患者を暴露することによる免疫原性が予想されることに加え、構造の改変により蛋白質の効力が極めて劇的に喪失するという大きな不利益を有する。
【0013】
複雑な有効成分の半減期を延長するべく研究されたもう一つの可能性は、前記製品とPEGで安定化したリポソームとの同時投与であった。この技術は、前記有効成分のPEGへの親和性に基づいており、前記蛋白質とリポソームとの可逆的会合を可能にするものである。この一時的会合は前記有効蛋白成分の半減期の増加をもたらすはずである。PEGで安定化したリポソームは長期間循環系に滞留するからである。しかし、実際にはこの仮説を裏付けることはできなかった。このシステムでは、血友病患者でFVIIIの半減期を延長する効果がないと判明したからである(非特許文献5:Powell J.S et al, Journal of Thrombosis and Haemostasis, 6: 277-283,2007)。
【0014】
以上説明した系の何れも、現在までのところ、半減期を大幅に改善するには至っていない。例外として上述した構造上の修飾や改変の導入も、ヒト病理の治療への応用は不可能であるか、複雑さゆえに極めて困難である。
【0015】
生分解性ポリマー微小球(micropsheres)により被包化した治療剤の制御放出が、大々的に研究されている。有効成分を生分解性ポリマーでマイクロ被包化することにより、薬剤の制御放出が可能となる。このアプローチは近年、乳酸及びグリコール酸の誘導体を主とする皮下用途の制御放出処方に応用されている。かかる処方はとりわけ、細胞増殖抑制剤、抗炎症剤、ペプチド、及びホルモンを含む広範囲の有効成分の被包化に首尾よく使用されてきた(非特許文献6:Tamilvanan S. et al, PDA Journal of Pharmaceutical Science and Technology, vol. 62, No. 2, March-April 2008 pp. 125-154)。
【0016】
Pankaj(特許文献1:米国特許5,417,982)は、経口投与によるホルモン制御放出用の乳酸及びグリコール酸微小球の使用について記載する。Pankajは注射用製品の取得可能性についても言及しているが、かかる投与方法の必要条件を考慮すると、この発明が静脈投与できる可能性はきわめて低い。何れにせよ、この発明ではかかる目的でのアルギン酸塩(alginate)の使用を想定していない。
【0017】
Sivadas (非特許文献7:Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167)は、吸入投与用の蛋白質被包化のための賦形剤として、ヒドロキシプロピルセルロース、キトサン、ヒアルロン酸、ゼラチン、オボアルブミン及びグリコールポリ乳酸等の種々のポリマーの使用について記載する。
【0018】
乳酸及びグリコール酸誘導体の使用に伴う不利益の一つは、有機溶媒の存在下での生成を要することである。中には、例えばポリビニルアルコールのように毒性を有し、蛋白質やホルモン等の複雑な有効成分の生物活性の保持とは相容れない性質を示すものもある。
【0019】
また、これらのポリマーを使用すると、得られる粒子は疎水性が極めて高いものとなり、以下に述べるように、細胞の取込み機構により急速に循環系から排出される。更なる不利点として、粒子の溶解時、ひいては有効成分の放出時に、粒子の周囲には局所的に強酸性環境が形成される。これは粒子の溶解時にポリマーが乳酸及びグリコール酸に分解し、粒子の周囲に強酸性環境を形成することによるものである。この酸性環境こそが、脆弱な有効成分、特に不安定な生物活性と複雑なアミノ酸構造を有する成分に損傷を与えるのである。
【0020】
アルギン酸塩は概して、食品・医薬業界及び化学工業界において多くの用途を有する。かかる多種多様の用途を規定するのが、その親水コロイドの性質、すなわち、水和によって粘性溶液、分散液又はゲルを形成し得る能力である。かかる特徴ゆえに、アルギン酸塩は、増粘剤、安定剤、ゲル化剤及び膜形成剤として独自の性質を示すのである。
【0021】
かかるアルギン酸塩の性質を広く利用してきた領域の一つが有効成分の被包化であり、特にその溶解性の改善や、種々の投与法における薬剤投与の補助を目的としたものである(非特許文献8:Tonnesen, Hanne Hjorth et al, Drug Development and Industrial Pharmacy, 28(6), 621-630 (2002))。特に、アルギン酸塩の粘膜付着性を利用した経口投与における使用が挙げられる。皮下法も試験されてきた。しかし、静脈内投与の必要条件は厳しいため、かかる用途の先例はない。例えば、Benchabane (非特許文献9:Benchabane, Samir et al, Journal of Microencapsulation, September 2007; 24(6): pp. 565-576)は、経口投与のための「噴霧乾燥」によるアルブミンマイクロカプセルの製造におけるアルギン酸塩の使用について記載する。同様の先例においてCoppi (非特許文献10:Coppi, Gilberto et al, 2001, Drug Development and Industrial Pharmacy, 27(5), pp. 393-400)は、カルシウム及びキトサンと共重合した蛋白質経口投与用の微小球の形成を実証している。何れの場合もアルギン酸塩は、胃内消化時に生じる天然の蛋白質分解に対する蛋白質保護剤としての役割をもつ。
【0022】
さらに、Mladenovska (非特許文献11:Mladenovska, K., International Journal of Pharmaceutics 342 (2007) pp. 124-136)は、結腸投与用のアルギン酸塩/キトサン微粒子の取得について記載する。
【0023】
Sivadas (非特許文献12:Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167)も、アルギン酸塩を吸入投与用の蛋白質被包化のための賦形剤として使用することに言及している。
【0024】
有効成分の直接投与とは別に、アルギン酸塩は複雑な治療形態の投与のための賦形剤としても検討されてきた。例えば、国際公開第2006/028996号(特許文献2)には、バクテリア毒素解毒剤輸送のためのアルギン酸塩及びエマルサンの使用が開示されている。
【0025】
他の例としては、多重小胞リポソームの被包化(非特許文献13:Dai, Chuanyun, et al, Colloids and Surfaces B: Biointerfaces 47 (2006) pp. 205-210)又は生細胞の被包化(特許文献3:欧州特許公開第2159523号)におけるアルギン酸塩の使用があげられる。この場合、生細胞投与の目的は、再生医療又は遺伝子治療用途にある(特許文献4:国際公開第2007/046719号;非特許文献14:Peirone, Michael et al, J. Biomed. Mater. Res. 42, pp.587-596, 1998;非特許文献15:Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223)。興味深いことに、Garcia-Martin (非特許文献15:Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223)は、血友病A治療用に遺伝子操作した生細胞投与の適用可能性について記載しつつ、この問題と医薬との関係について触れている。この場合、生細胞を含むアルギン酸塩マイクロカプセルは、カテーテルの導入により腹腔内に埋め込まれる。この場合、治療目的も投与経路(非静脈)も、本発明からはほど遠い。
【0026】
蛋白質等の複雑な有効成分を被包化するためのポリマーの使用に関し、このように広範な知見があるにもかかわらず、これらの製品の静脈内投与に伴う問題を解決し得る参考文献はない。Wongら(非特許文献16:Wong, Joseph et al, Advanced Drug Delivery Reviews 60 (2008) pp. 939-954)が述べているように、静脈内投与に粒子懸濁液を使っている認可製品は3つしかなく、その何れもが組成中にアルギン酸塩を使用していない。何れのケースも、半減期の延長ではなく、製品の溶解性改善を目的としたものである。
【0027】
静脈内に微粒子を効果的に投与することの困難さは、a)製品設計という基礎的な側面、例えば粒子サイズと分布、有機溶媒の不在、及び、懸濁液の均一性、粘性及び「注射針通過性」(「注射針通過性」とは製品の吸引及び注入の容易性の意である);b)工業的スケールでの生産及び調製の技術的側面、例えば一回投与量(dose)の均一性、溶媒沈殿で得られる製品の場合、塩の望ましくない結晶化、製品の無菌度(sterility)と無発熱原性(apyrogenicity);並びに、c)生物学的側面、例えば薬物動態及び薬力学プロフィールの無計画な改変、生体分布の改変、ポリマーの生物蓄積、食作用の活性化、塞栓の毒性と作用、あるいは捕体の活性化、として表現される。
【0028】
これに関連して、これら製品を開発するにあたり最も大きな問題の一つは、以前は細網内皮系(RES)と呼ばれていた単核食細胞系(MPS)により迅速に除去されることであり、この系には骨髄の単球性前駆細胞由来の全ての細胞、末梢血の単球、及びさまざまな器官や組織のマクロファージ又は組織球が含まれる。後者の中では、血漿中の微粒子のクリアランスにおける重要性の点で、肝臓のクッパー細胞と、脾臓及び骨髄に分布するマクロファージに言及せねばならない(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)。
【0029】
ナノ粒子又はマイクロ粒子を静脈内投与した場合、血漿中の蛋白質によって迅速にオプソニン化されることが、広く報告されてきた。これらの表面に吸収される蛋白質は、MPS細胞による認識及び取込みを誘導する(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)。
【0030】
同様の作用はリポソームでも観察されており(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165)、同文献には加速化血中クリアランス(Accelerated Blood Clearance)(ABC)として知られる現象が記載されている。ポリマー微粒子及びリポソームの何れの場合も、オプソニン化の現象は補体系活性化に直接関連している(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165;非特許文献19:Koide, Hiroyuki et al, International Journal of Pharmaceutics 362 (2008) pp. 197-200)。
【0031】
実用に際して、この食作用現象は、静脈内投与される微粒子を用いて半減期を延長しようとする薬剤の開発を阻害する。被包化にともなうサイズの増大は、単なる増大ではなく、時として分解を加速する場合もあるからである。明らかに、この現象は動物の薬物動態研究等のインビボ実験によってしか観察できない。
【0032】
この食作用によるクリアランスと粒子サイズの関係は広く記録されてきた。Champion (非特許文献20:Champion, JA, Pharm Res. 2008 Aug; 25(8): 1815-21. Epub 2008 Mar 29)は、ポリマー微粒子が遭遇した食作用と微粒子のサイズとの関係について具体的に記載するものであるが、2〜3μmで最大の影響が観察された。インビボでのMPSによる微粒子の取込みを規定する他の特徴は、粒子の疎水性とそのゼータ電位(Z Potential)である(非特許文献21:Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0033】
Z電位は粒子の特性の一つである。具体的には、分散粒子は外相からのイオンの吸着や、それ自体の表面官能基のイオン化により帯電する傾向がある。これによる帰結の一つは、スターン層(Stern layer)と呼ばれる対イオンの層が、負に帯電した分散粒子の環境で粒子と背中合わせに出現することである。前記スターン層上に(両電荷の)可動電荷の存在を特徴とする拡散層が出現し、粒子までの距離の関数として、粒子表面の荷電を中和する。Z電位とは、対イオン層と動電学的に中性な点とのいわゆる電位差のことである。
【0034】
Z電位値は、分散系の大部分の安定化にとって極めて重要である。後者が、同じ荷電を有する分散粒子間の反発度を抑制しつつ、粒子相互間の過剰な接近を防止するとともに、癒合現象に起因する粒子間引力が支配的になるのを防止するからである。Z電位に関しては、0付近の、部分的に負のZ電位が食作用を低減するということが開示されている(非特許文献21:Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0035】
更に、疎水的性質も粒子のオプソニン化及び取込みを促進する。この事実は特に興味深い。ポリ乳酸やグリコール酸由来の粒子は、例えば、極めて疎水性が高いからである。
【0036】
微粒子及びリポソームの血漿中での半減期を延長するためのアプローチの一つは、荷電を改善して親水性表面層を形成し得る荷電ポリマーをその表面に導入し、オプソニン化や食作用を防止することであった。例としては、ポリエチレングリコール(PEG)(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165;非特許文献22:Owens III, Donald E et al, International Journal of Pharmaceutics, volume 307, issue 1, 3 January 2006, Pages 93-102)あるいはヘパリン(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)の使用が挙げられる。
【0037】
このアプローチは、系の複雑性を増加させるため、医薬製品開発を複雑、困難なものにする。また、上で論じたように、PEG−リポソームの使用は、FVIII等の複雑な蛋白質の半減期の延長には効果がないことが分かっている(非特許文献5:Powell J.S et al 2007, Journal of Thrombosis and Haemostasis, 6: pp. 277-283)。
【0038】
微粒子の場合、実現可能な静脈内投与用製品を得るためには、適切なサイズとZ電位を親水性の粒子に持たせることが必要となるだろう。
【先行技術文献】
【特許文献】
【0039】
【特許文献1】米国特許5,417,982
【特許文献2】国際公開第2006/028996号
【特許文献3】欧州特許公開第2159523号
【特許文献4】国際公開第2007/046719号
【非特許文献】
【0040】
【非特許文献1】Casper, CK, Hereditary Plasma Clotting Factor Disorders and Their Management 5th ed. WFH, Sam Schulman Ed., 2008
【非特許文献2】Bailon Pascal et al, Bioconjugate Chem. 2001, 12, 195-202
【非特許文献3】Perlman Signe et al, The Journal of Clinical Endocrinology & Metabolism 88 (7): 3227-3235, 2003
【非特許文献4】Dennis, Marks S. et al, The Journal of Biological Chemistry vol. 277, No. 38, Issue of September 20, pp. 35035-35043, 2002
【非特許文献5】Powell J.S et al, Journal of Thrombosis and Haemostasis, 6: 277-283, 2007
【非特許文献6】Tamilvanan S. et al, PDA Journal of Pharmaceutical Science and Technology, vol. 62, No. 2, March-April 2008 pp. 125-154
【非特許文献7】Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167
【非特許文献8】Tonnesen, Hanne Hjorth et al, Drug Development and Industrial Pharmacy, 28(6), 621-630 (2002)
【非特許文献9】Benchabane, Samir et al, Journal of Microencapsulation, September 2007; 24(6): pp. 565-576
【非特許文献10】Coppi, Gilberto et al, 2001, Drug Development and Industrial Pharmacy, 27(5), pp. 393-400
【非特許文献11】Mladenovska, K., International Journal of Pharmaceutics 342 (2007) pp. 124-136
【非特許文献12】Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167
【非特許文献13】Dai, Chuanyun, et al, Colloids and Surfaces B: Biointerfaces 47 (2006) pp. 205-210
【非特許文献14】Peirone, Michael et al, J. Biomed. Mater. Res. 42, pp.587-596, 1998
【非特許文献15】Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223
【非特許文献16】Wong, Joseph et al, Advanced Drug Delivery Reviews 60 (2008) pp. 939-954
【非特許文献17】Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050
【非特許文献18】Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165
【非特許文献19】Koide, Hiroyuki et al, International Journal of Pharmaceutics 362 (2008) pp. 197-200
【非特許文献20】Champion, JA, Pharm Res. 2008 Aug; 25(8): 1815-21. Epub 2008 Mar 29
【非特許文献21】Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages
【非特許文献22】Owens III, Donald E et al, International Journal of Pharmaceutics, volume 307, issue 1, 3 January 2006, Pages 93-102
【発明の概要】
【発明が解決しようとする課題】
【0041】
Terrence (欧州特許公開第2 286 040号, 欧州特許出願第00973477.3号)は、活性を有する被包化成分の半減期を増加させ得る投与の系として、ポリマーの使用について記載する。この発明ではそのために、(1)第一の水溶性ポリマー、(2)第一の錯化剤として少なくとも一つの陰イオン性多糖類、及び(3)第二の錯化剤として2価の陽イオンを使用することを要する。上で述べたように、この発明は技術的に複雑で、実際の使用が困難なものである。対照的に、本発明によれば、有効成分の制御放出をはるかに簡素な微粒子で達成することができる。かかる微粒子が要するのは、その用途に必要な全ての特性を有する一種類のポリマーの使用のみである。さらにTerrenceの発明は、静脈内用調製物のサイズによる適合性を実証しておらず、細胞の食作用を避ける方法を説明してもいない。
【0042】
アルギン酸塩は、PLAやPLGA等の他のポリマーとは異なり、親水性である。本発明において生成される粒子は、粒子の凝集を十分に防止し得る部分的に負のZ電位を有しつつ、低オプソニン化プロファイルを示し得る程度に中性であることが示されている。
【0043】
静脈内投与に許容できる最大粒子サイズはおよそ5μmである。このことは、超音波診断用に標識された平均サイズ3.0〜4.5μmのアルブミンを使用した登録薬剤(Optison, data sheet 28)の存在により実証されている。
【0044】
アルギン酸塩は生体適合性を有し、その食品業界での広範な使用に基づいて、ヒトへの経口使用に大々的に使用されてきた。非粒子型ポリマーとして静脈注射された場合、半減期4時間及び22時間という二相様式で排出され(Hagan, A et al, European journal of Pharmaceutical Sciences, Volume 4, Supplement 1, September 1996, pp. 100-100 (1))、副作用も見受けられない。アルギン酸塩は尿中に排出される。
【0045】
加えて、水溶性ポリマーであるという事実は、複雑な蛋白質との適合性に寄与する。後者が天然の溶媒となるからである。
【課題を解決するための手段】
【0046】
本発明は、アルギン酸又はその医薬的に許容し得る塩の微粒子を含んでなる組成物であって、制御放出を達成するとともに、静脈内投与された有効成分の半減期を延長することによって投与頻度の低減を達成し、並びに血中有効成分レベルの更なる安定化を達成し、ひいては、有効成分を定期的に注入した場合に典型的に生じる有効成分濃度のピーク及びトラフを低減することが可能となる。
【0047】
本発明は、静脈点滴に適したサイズと迅速な食作用を阻止するのに適した生理化学的特徴を併せ持ち、複雑な有効成分の制御放出を可能にする、アルギン酸塩の親水性微粒子について記載する。
【図面の簡単な説明】
【0048】
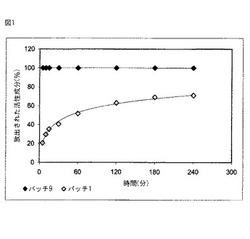
【図1】図1は、バッチ9及びバッチ1のインビトロ放出テスト結果の比較グラフである。
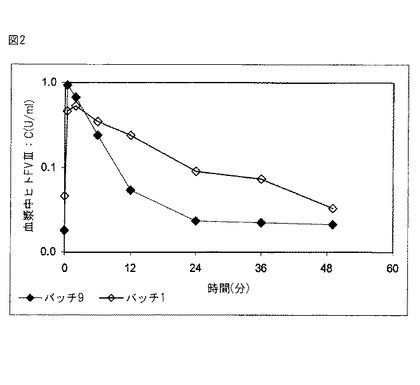
【図2】図2は、非被包化FVIIIの投与後及び本発明の組成物の適用後におけるウサギ血漿中ヒトFVIII:Cの薬物動態を示す。
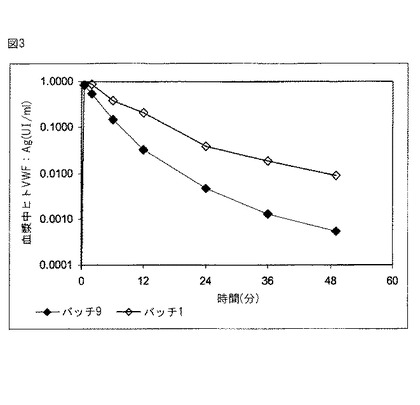
【図3】図3は、非被包化FVIIIの投与後及び本発明の組成物の適用後におけるウサギ血漿中ヒトVWF:Agの薬物動態を示す。
【発明を実施するための形態】
【0049】
アルギン酸及びその塩(アルギン酸アンモニウム、アルギン酸カルシウム、アルギン酸カリウム、アルギン酸ナトリウム及びアルギン酸プロピレングリコール)は、その物理化学的及び生化学的特性ゆえに、有効成分の被包化において最も使用及び研究されているポリマーの1つである。これらは天然由来の多糖であり、商業的には藻類や細菌から製造される。
【0050】
アルギン酸塩(alginates)はアルギン酸(alginic acid)の塩であり、β−(1−4)−D−マンヌロン酸(M)及びα−(1−4)−L−グルロン酸(G)の2つのモノマー単位からなる直鎖多糖である。種々の配列を有するブロックに分類されるが、最も一般的なものはG、M及びMGである。
【0051】
カルシウム(Ca++)等の多価カチオンの存在下では、隣接するGブロック間で強力な結合が形成され、拡大したアルギン酸塩のネットワークが形成される。カルシウムイオンはグルロン酸の負電荷を有する基の間を連結するように位置する。
【0052】
処方によっては、キトサン等の他の多糖と組み合わされる場合が多い。キトサンは、ランダムに分布したβ−(1−4)−D−グルコサミン(脱アセチル化単位)とN−アセチル−D−グルコサミン(アセチル化単位)の鎖からなる直鎖の多糖である。
【0053】
アルギン酸塩の処方によっては、アルブミン、好ましくは無菌且つ発熱物質不含有(pyrogen-free)ヒトアルブミンを、荷電物質として使用してもよい。これは製造工程時には有効成分の保護剤としても機能し、また、製品の長期保存時には安定剤としても機能する。
【0054】
血漿中の放出改善の対象となる有効成分は、複雑で不安定な有効成分であってもよい。より具体的には、有効成分は生物活性を示すことを特徴とする。この生物活性は、例えば酵素活性、輸送、分子間相互作用、又はリガンドとの結合によってもたらされる。何れの場合も、高エネルギーの製造条件、特に温度、圧力及び/又は無極性環境等に対して不安定又は感受性である有効成分が問題となるだろう。構造上の小さな変化が生物活性の不可逆な損失につながり得るからである。
【0055】
生物活性を有する有効成分の例として、ヒトペプチドホルモン、例えばメラトニン、セロトニン、チロキシン、エピネフリン、ノルエピネフリン、ドーパミン、副腎皮質刺激ホルモン、アンギオテンシノーゲン及びアンギオテンシン、バソプレッシン、心房性ナトリウム利尿ペプチド、カルシトニン、エリスロポエチン、卵胞刺激ホルモン、グルカゴン、ヒト絨毛性ゴナドトロピン、ヒト胎盤性ラクトゲン、成長ホルモン、インヒビン、インスリン、インスリン型成長因子(又はソマトメジン)、黄体形成ホルモン、メラニン細胞刺激ホルモン、オキシトシン、プロラクチン、トロンボポエチン、神経ペプチドY、ヒスタミン及びそれらの誘導体が挙げられる。
【0056】
他の例としては、生物活性蛋白質、例えば、アルブミン、アルファ1−アンチトリプシン、アルファ酸グリコプロテイン(alpha-acid glycoprotein)、アルファ−2−マクログロブリン、アンチトロンビン、ハプトグロビン、セルロプラズミン、リポ蛋白質、トランスフェリン、プラスミノーゲン、フィブリノーゲン、補体蛋白質、凝固因子、及び免疫グロブリン等も挙げられる。
【0057】
これらの有効成分は、生物活性を有するがゆえに、小さな構造上の損傷の結果として生じ得る機能喪失を被り易い。かかる構造上の損傷は、例えば温度、圧力、溶媒の極性、浸透圧、酸素の存在、攪拌等と関連し得る。
【0058】
これに関して、これらの有効成分の中でも凝固第VIII因子は、極度の不安定さゆえに際立っている。FVIIIは複雑な構造を有するため、その生物活性を十分に安定化させるのは、特に精製状態では非常に困難である。例えばParti R ら (Haemophilia 2000; 6: 513-522)は、凍結乾燥状態でさえも、40℃超の温度ではFVIIIの生物活性が減弱し始めると説明している。かかる不安定さが最も顕となるのは、FVIIIが溶液状態の場合であり、25℃でさえ不安定の兆候が見られる。第IX因子及び第VIIa因子についても、温度等の外的因子に対する感受性が知られている。
【0059】
これに関して留意すべきは、本製造方法を採用すると、FVIIIの生物活性を保持する医薬製剤が得られるという点である。これは、本方法が生物活性の安定化が困難な有効成分に対しても適用できること、従って、本発明はFVIIIのように不安定な成分に適用できることを意味する。更に言えば、本発明はFVIIIより安定な成分にも適用できる。結果として、凝固因子類は、本発明に記載した処方の適用により利益を享受し得る有効成分の明白な例となる。
【0060】
すなわち、本発明では、ポリマー微小球に含まれる有効成分は、生物活性を示すペプチド、蛋白質、又はホルモンである。好ましくは、前記活性成分は凝固因子であり、より好ましくは、前記活性成分は第VIII因子、フォンヴィレブランド(von Willebrand)因子、第VIII因子とフォンヴィレブランド因子の複合体、第IX因子又は第VIIa因子である。
【0061】
これらの成分は、ヒト、動物、組換、トランスジェニックの何れに由来するものであってよい。後者の場合には、合成分子は天然分子を再現したものでも、意図的な改変を加えたものでもよい。
【0062】
組成物の取得
マイクロ被包化(microencapsulation)は、分子、固体粒子又は液体小球を、別の性質を有する原料で被覆し、マイクロメートルサイズの粒子を作製する手法である。この加工法により生じる生成物は、微粒子(microparticles)、マイクロカプセル(microcapsules)、又は微小球(microspheres)と呼ばれる。
【0063】
幾つかのマイクロ被包化技術が存在する。
−化学的手法によるマイクロ被包化
・ 界面重合
【0064】
−物理化学的手法によるマイクロ被包化
・ 溶剤蒸散
・ 液滴形成
・ ゲル化
・ キレート化
・ 小胞形成
【0065】
−機械的手法によるマイクロ被包化
・ 押出成形
・ 共押出法
・ 噴霧乾燥
・ 噴霧冷却
【0066】
本発明記載の微粒子の製造に選択した技法は、Erdinc B.I. [Erdinc B.I. (2007) Micro/nanoencapsulation of proteins within alginate/chitosan matrix by spray drying, Degree Thesis, Queen’s University, Kingston, Canada]に記載の噴霧乾燥である。この製造法は単一工程であることを特徴とし、最終産物として微粒子が得られる。
【0067】
本発明の、有効成分を制御放出するための、アルギン酸又はその塩の微粒子を含む静脈内投与用の生体適合性組成物の製造方法は:
−有効成分及びポリマーを含む溶液/懸濁液/乳化液を、ノズルを通して送り出し、液滴形状で分散させる噴霧工程、
−熱風が前記液滴からの溶媒の蒸散を助ける、乾燥室内での乾燥工程、及び
−被包化された生成物の収集工程、
の各段階を特徴とし、かかる手順は140〜180℃の温度、35〜40m3/hの供給流速、3.5〜5ml/minの注入流速、及び4〜6psiの圧力で実施される。
【0068】
かかる条件下であれば、5μm以下、好ましくは1〜4.5μmのサイズの粒子を取得し、且つ、有効成分の活性を維持することが可能となる。更に、噴霧段階の前段に、乳化液をホモジナイズするという工程を任意で追加することにより、粒子の平均サイズを改善することができる。この追加のホモジナイズ工程は、例えば1500〜2000psiの圧力により行なわれる。
【0069】
噴霧乾燥による有効成分の被包化は、溶液又は乳化液を脱水する連続プロセスであり、微粒子によって形成される固形物をプロセスの最後に回収する。
【0070】
この目的のために、有効成分を含む流体は、所定の注入流速でノズル又は回転ディスクに向けて機械的に運ばれ、そこで無数の微小の液滴状に噴霧される。液滴のサイズは主に流体を噴霧するガス圧で決まる。このプロセスは、制御したガス流(通常は空気)が所定の吸気速度及び制御温度下で連続的に循環する密室で行なう。
【0071】
噴霧の結果、流体の空気との接触表面積は大幅に増大するので、乾燥空気流に接触した流体の溶媒(通常は水)の急速な蒸散が生じる。この急速な蒸散に伴い、状態変化に必要な熱が奪われて、各小液滴の内部が冷却される。このように、有効成分への熱ショックを最小化しつつ、迅速な乾燥を行なうことができる。このプロセスの完了後、生成物は固形として収集される。
【0072】
組成物の説明
得られた微粒子は、平均粒子サイズ、Z電位、及び生物活性の測定により識別される。粒子サイズは、Beckman Coulter LS13320装置で回折レーザーにより決定される。
【0073】
問題となる静脈内投与の場合、粒子サイズを5μm以下、好ましくは1〜4.5μmとする必要がある。粒子サイズがこれより大きいと、血栓の形成を引き起こす可能性があるからである。
【0074】
Malvern Zetasizerの装置で決定されるZ電位は、懸濁液中の粒子間相互作用を制御する基本パラメータの一つである。これは粒子表面の性質と分散溶媒によって決定される。本発明では、粒子間斥力を確保し凝集を阻止する観点から、最適値は−30mVを超えた値である。Z電位が0近く、好ましくは−30〜0mVの微粒子は、低い肝臓取込みと細胞クリアランスのレベルを有することが示されている(Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0075】
組成物の用途
調節放出又は制御放出される医薬形態とは、同様に投与した従来放出法の医薬形態に対して、有効成分放出の速度及び/又は場所を変更するべく設計されたものである。
【0076】
本発明では、蛋白質等の生物活性を示す有効成分、より具体的には凝固因子の被包化が、インビトロの放出モデルでいかに制御放出を可能にするかを観察した。第VIII因子はその構造上の複雑さゆえに、外的因子に対して高い感受性を有する点で注目に値する。実際、FVIIIをその天然基質であるヒトの血漿自体の状態で凍結しても、生物活性の部分的喪失が生じる(Bravo, M.I. et al, Pharmeuropa Scientific Notes, 2006-1 pp. 1-5)。
【0077】
そこで、本発明に規定のヒトFVIII含有微粒子を、ヒト血漿の環境と同様の連続フローセル中に入れると、非被包化製品に比して、媒質中へのFVIII放出が遅くなることが観察された。
【0078】
同様に、本発明のFVIII含有微粒子をウサギに静脈投与すると、従来品に比して、血漿中のFVIIIの半減期が大幅に、且つ安定して延長されるという結果が得られた。さらに、上記処方に伴い動物が中毒作用を示すことはなく、副作用は見られなかった。
【0079】
このインビボの薬物動態データは極めて重要である。本発明の処方ではオプソニン化及びMPSへの取込み促進の作用が適切に処理されていることを、本データは紛れもなく示しているからである。
【0080】
本発明は複雑な成分の静脈投与を必要とする多様な病状の治療に使用できる。例としては出血性疾患及び凝固障害、ホルモン障害党が挙げられる。これらのケースでは、半減期の大幅な延長が達成されるであろう。例えばFVIIIの場合、一次予防法を維持するための投与回数を、例えば週1回の投与に低減することもできるだろう。
【0081】
親水性ポリマーの使用に伴い生じ得る欠点は、無発熱原性且つ無菌注射投与用の水性媒体(例えば水)中に前記製品を懸濁してから静脈点滴までの間に、微粒子の部分的な溶解が生じ得る点である。かかる不利益は、例えば、部分的に無極性の生体適合性溶液、例としてはエタノール、プロピレングリコール、ポリエチレングリコール、ジメチルスルフォキシド、N−メチルー2−ピロリドン、グリコフロール、イソプロピリデン−グリセロール、グリセロールホルマール又はアセトン等(Mottu F et al. Journal of Pharmaceutical Science & Technology 2000 Vol. 54, No. 6, 456-469)を、本発明に記載の微粒子の再懸濁及び投与用媒体として使用することにより解消し得る。
【0082】
本発明は、例えば、真空又は不活性雰囲気中でパックした脱水又は凍結乾燥生成物の状態として製造することもできる。これにより種々の温度条件下、例えば2℃〜40℃での長期安定化が可能となる。このようにして保存した製品は、溶媒で再構成後、静脈投与することができる。かかる溶媒としては、注射用蒸留水、食塩水、又は混合物が挙げられる。或いは、エタノール、プロピレングリコール、ポリエチレングリコール、ジメチルスルフォキシド、N−メチル−2−ピロリドン、グリコフロール、イソプロピリデン−グリセロール、グリセロールホルマール又はアセトン等の生体適合性溶媒を、例えば0.5%〜50%等の様々な含量で有する水性食塩水であってもよい。
【0083】
従来技術に優る利点
本発明は、静脈点滴に適したサイズと、迅速な食作用を阻止するのに適した物理化学的特徴とを併せ持ち、複雑な有効成分の半減期を延長し得る、アルギン酸塩の親水性微粒子の製造について記載する。
【0084】
アルギン酸塩は生体適合性を有し、尿により排泄され、いかなる公知の中毒作用をも伴わない。かかる特徴ゆえ、本発明は蛋白質及び複雑な有効成分の投与に適合している。
【0085】
本発明によれば、制御投与静脈系の実用化を阻んできたあらゆる不利益を克服することができ、その結果、静脈用途の有効成分を変更することなく、治療に必要な投与回数を低減することができる。この点において、本発明は、アミノ酸配列、グリコシル化又は合成誘導体の導入など、有効成分の改変を必要としない点に留意すべきである。
【実施例1】
【0086】
微粒子の調製
Erdinc B.I. [Erdinc B.I. (2007) Micro/nanoencapsulation of proteins within alginate/chitosan matrix by spray drying, Degree Thesis, Queen’s University, Kingston, Canada]に記載のように、アルギン酸塩微粒子の生成には噴霧乾燥プロセスが使われている。基本的に、微粒子は前記ポリマーと、選択した有効成分との乳化液を生成することにより調製した。
【0087】
試料の噴霧は、Buchi Mini Spray Dryer B-290装置を用い、以下の条件で行った。噴霧温度:140℃〜180℃、吸入速度:35〜40m3/h、注入流速:3.5〜5ml/min、圧力4〜6psi。
【実施例2】
【0088】
微粒子の説明
表1、2及び3に、微粒子の製造に使用した原料と、そのサイズ、Z電位及び収量等の特性を示す。使用した製造方法及び条件は実施例1に記載の通りである。
【0089】
【表1】

【0090】
FVIII活性/FVIII抗原量比は、ある試料中の活性蛋白質の割合に想到する。このように、当初試料中の活性/抗原量比を、被包化試料について得られる値と比較すると、マイクロ被包化後も機能を保持する有効成分の割合を算出できる。実施例において、被包化プロセス中の活性収率を当初活性収率との比で表わすと、バッチ1、2、及び3はそれぞれ57.6%、33.9%及び35.7%であった。
【0091】
【表2】
【0092】
この場合、バッチ4、5及び6の被包化プロセス中の活性収率は、これらのバッチ全てで100%であった。
【0093】
【表3】
【0094】
組換由来蛋白質の場合、被包化プロセスで測定した活性収率は、バッチ7及び8でそれぞれ25%及び71%であった。
【0095】
全てのバッチで、粒子のサイズは回折レーザーによりBeckman Coulter LS13320装置で測定し、Z電位はMalvern Zetasizer装置で測定した。
【0096】
FVIIIの生物活性は、欠乏血漿凝固アッセイ又は発色によるFXa生成の評価により測定した。FVIIa及びFIXの場合、生物活性はそれぞれFVII及びFIXを含まない血漿の凝固時間(部分活性化トロンボプラスチン時間)を評価することで測定した。蛋白質濃度は、FVIII:Ag、FIX:Ag、又はFVII:Agにそれぞれ特異的な抗体を用い、酵素免疫測定法(ELISA)による免疫学的検出法で測定した。
【0097】
ある試料中の活性蛋白質の割合の指標となる活性/抗原量比は、試料中の特異有効成分の活性と抗原単位量との商により算出した。活性/抗原量・収率の算出は、出発試料の活性/抗原量比と最終被包化製品の活性/抗原量比との変動率パーセンテージを評価することで行われる。
【0098】
いずれのケースでもみられるように、平均粒子サイズは5μm以下、Z電位は負の値である。また、活性/Ag・収率の結果は、プロセス中の生物活性が保持されていることを示している。
【0099】
これら種々の表は、前記制御放出系が様々な有効成分に適していることを示している。
【実施例3】
【0100】
インビトロ放出テスト
有効成分放出の評価には、連続フローセルを用いたインビトロ放出テストを、Sotax CE1装置にて閉回路でおこなう。
【0101】
前記テストは、溶解剤として1%のヒトアルブミンを含むpH7.3のイミダゾールバッファーを用い、37℃の温度、7〜25ml/minの流速で行なった。分析用の代表試料を異なる時点(5分、10分、15分、30分、60分、120分、180分及び240分)で抽出した。抽出試料を同量の新たな媒質と置換し、量の損失を補填した。
【0102】
FVIIIの生物活性は、欠乏血漿凝固アッセイ又は発色によるFXa生成の評価により測定した。FVIIa及びFIXの場合、生物活性はそれぞれFVII及びFIXを含まない血漿の凝固時間(部分活性化トロンボプラスチン時間)を評価することで測定した。蛋白質濃度は、FVIII:Ag、FIX:Ag、又はFVII:Agにそれぞれ特異的な抗体を用い、酵素免疫測定法(ELISA)による免疫学的検出法で測定した。
【0103】
テスト終了後、以下の結果が得られた:
【0104】
【表4】
【0105】
【表5】
【0106】
有効成分に適用した微粒子の組成が、非被包化製品に比して、前記製品放出の動態を改変することがわかる。
【実施例4】
【0107】
動物での第VIII因子の薬物動態
インビボ有効成分放出への前記組成物の作用を評価するため、薬物動態テストをウサギで行なった。このために、バッチ9(非被包化)由来のヒトFVIII 50IU/kg量を、3匹の雌ニュージーランド白ウサギ(New Zealand White rabbits)に静脈内投与した。同様に、実施例1記載の方法で作製し、実施例2で説明したバッチ1由来の被包化FVIII 50IU/kg量を、別の3匹の雌ニュージーランド白ウサギに静脈投与した。表6に記載のように、各時点で血漿サンプルを得、分析によりヒトFVIII:Cの存在を検出した。ヒトFVIIIの検出は、ヒトFVIII分子の選択的免疫キャプチャー(selective immunological capture)後の発色で行なった。これにより、注入したヒトFVIII活性を、ウサギFVIII活性と区別することが可能となる。
【0108】
【表6】
【0109】
これらの結果から、本発明の組成物は血漿中有効成分の放出を遅延させることがわかる。また、これらの結果は、前記微粒子のサイズにもかかわらず、かかる微粒子を循環系から迅速に除く細胞メカニズム(肝臓、脾臓又はマクロファージ)が存在しないことを示している。
【0110】
本目的に適したソフトウェア(WinNonlin 5.2)を用いてこれらのデータを解析することにより、表7に詳述する薬物動態定数が算出された。
【0111】
【表7】
【実施例5】
【0112】
動物でのフォンヴィルブランド因子の薬物動態
バッチ9の調製物(非被包化FVIII)及びバッチ1の調製物(被包化FVIII)のいずれの場合も、FVIIIは血漿由来であり、相当量のフォンヴィルブランド因子(VWF)を含んでいた。これは、VWFの被包化がFVIIIの被包化と同時に起こることを意味する。それゆえ、これらの挙動を独立して分析することが可能となる。このため、ウサギ血漿中ヒトVWF抗原(VWF:Ag)の存在を評価して、VWF自体の薬物動態分析を進めた。結果を表8に示す。
【0113】
【表8】
【0114】
これらの結果から、本発明の組成物は血漿中有効成分の放出を遅延させることがわかる。また、これらの結果は、前記微粒子のサイズにもかかわらず、かかる微粒子を循環系から迅速に除く細胞メカニズム(肝臓、脾臓又はマクロファージ)が存在しないことを示している。
【0115】
本目的に適したソフトウェア(WinNonlin 5.2)を用いてこれらのデータを解析することにより、表9に詳述する薬物動態定数が算出された。
【0116】
【表9】
【0117】
有効成分(この場合はVWF)の被包化により、半減期が有意に延長されることがわかる。
【0118】
以上、好ましい態様の例を挙げて本発明を説明してきたが、本発明は添付の特許請求の範囲をより広く解釈して定義されるものであり、上述の例によって限定されると解すべきではない。
【技術分野】
【0001】
本発明はアルギン酸又はその塩の微粒子(micoparticles)と有効成分を含んでなる生体適合性組成物に関し、より詳細には、必要とする患者に静脈内投与するべき有効成分を被包化するための微粒子に関する。かかる微粒子は、血中有効成分の半減期又は残存率を高めるのに十分なサイズと、静脈内投与時の低い肝臓取込み及び迅速な細胞クリアランス(cell clearance)とを併せ持つ。本発明の組成物中の有効成分としては、天然、組換、又はトランスジェニックの、ヒト又は動物由来のペプチド、蛋白質又はホルモンが挙げられる。本発明の組成物の有効成分の例としては、第VIII因子、第IX因子、又は第VIIa因子などの血液凝固因子が挙げられる。
【背景技術】
【0002】
治療有効成分の血中半減期の延長には、所望の治療効果を得るために必要な投与回数の低減等の利点がある。かかる投与回数の低減は、特に非経口投与用の薬剤にとって、中でも静脈注射用の薬剤にとって重要であり、また、慢性疾患治療等のための長期投薬に特に深い関連を有している。
【0003】
可能なかぎり、静脈内アクセスを必要としない手段で有効成分を投与するのが、最近の傾向である。かかる方法の使用は、患者にとって煩雑且つ不便だからである。しかし、現在でも静脈投与に代わる手段がない一連の有効成分が存在する。かかる成分としては、サイズが大きく複雑な有効成分、例えば蛋白質やホルモン等の生物産物又は生物工学産物等が挙げられる。
【0004】
複雑な有効成分の静脈内反復投与が必要な慢性的治療疾患の一例に血友病がある。血友病は、血液の凝固に関連する蛋白質の全欠損又は部分欠損による内外出血の出現を特徴とする遺伝病である。血友病Aの特徴は第VIII凝固因子の欠損であり、これがトロンビン(thrombin)の正常な生成を妨げる結果、血液の正常な凝固が困難となる。血友病Bでは第IX因子の欠損が同様の病態をもたらす。
【0005】
血友病の治療における第一の選択肢となるのは、前記因子(FVIII又はFIX)を含有する治療用濃縮物を投与して欠損蛋白質を置換する方法である。血友病において的確な止血を得る別の治療上の選択肢はFVIIaの投与であり、これはFVIII又はFIXがなくてもトロンビンを生成させることができる。しかし、このタイプの治療は通常、FVIII又はFIXでの治療に問題があったり、かかる治療が有効でないと判明した場合、例えばこれらの有効成分に対して抑制的な免疫応答が働いた患者等の場合に限られる。現在まで、これらの産物は何れも、その構造上の複雑さ及び上皮透過性の低さゆえに、静脈内投与以外の方法による投与は成功していない。
【0006】
従って、血友病の患者には、血漿中半減期に応じて定まる頻度で繰り返し静脈内投与を行う必要がある。FVIIIの場合、半減期は約12時間である。これは、世界血友病連盟(the World Federation of Haemophilia)のモノグラフ(非特許文献1:Casper, CK, Hereditary Plasma Clotting Factor Disorders and Their Management 5th ed. WFH, Sam Schulman Ed., 2008)によれば、一次予防法の場合、すなわち関節損傷のない小児の出血の予防の場合、48時間毎に約20U/kgの投与量の使用を意味する。これは正常値の1%を超える血漿FVIIIレベルを維持するのに十分な量である。原則として、この治療によって、重度の血友病が、軽度又は中度の血友病に変わる。FIXの場合、半減期は約26時間であり、一次予防としては、最低1%のレベルを維持するために、約40U/kgの量を週2回投与すればよい。
【0007】
考慮しなければならないのは、若年齢(約1歳又は匍匐開始期)からの予防が、重度血友病の場合の関節損傷を避けるのに必要な標準的治療であるという点である。
【0008】
従って、血友病は、有効成分の半減期の延長が患者の生活の質(quality of life)に実質的な改善をもたらす明らかな例である。幼い子供にはことに困難な静脈内投与の回数を低減し得るからである。
【0009】
静脈内投与製品による長期治療の他の例としては、原発性免疫不全における免疫グロブリン(IgG)の使用、先天性欠損におけるアンチトロンビンIII(AT)及びアルファ−1−アンチトリプシン(AAT)の使用等が挙げられる。
【0010】
これらのタイプの有効成分の血漿中半減期の延長を目的とする技術的アプローチは多数存在する。最も研究されてきたものの一つは、ポリエチレングリコール(PEG)のように、適合性を有するポリマーで蛋白質を誘導体化することであった。この技術は、蛋白質のアミノ酸にPEG鎖を共有結合させる化学反応操作からなる。この技術は、インターフェロン等のサイズが小さいホルモンやペプチド鎖に有用であることが分かっている。このタイプの化合物の場合、主たる排除機構は腎クリアランスであり、これは単にサイズを大きくすることで容易に制御できるからである(非特許文献2:Bailon Pascal et al, Bioconjugate Chem. 2001, 12, 195-202)。しかし、これは処理対象の蛋白質構造の外的修飾に基づく手法であるため、より複雑な有効成分にも使用できるか否かはまだ分からない。また、このタイプの蛋白質との共有結合によれば、処理後のホルモン又は蛋白質の生物活性は大幅に低下する。
【0011】
半減期を改善するもう一つの代案は、蛋白質やホルモンが有する天然の糖残基の付加又は修飾であった(非特許文献3:Perlman Signe et al, The Journal of Clinical Endocrinology & Metabolism 88 (7): 3227-3235, 2003)。この手法は、その分解に関与する受容体による認識を変更することにより、蛋白質を改変するというものである。しかし、この改変には固有のリスクが存在することは、前記蛋白質に存在する糖鎖の免疫原性を考慮すれば明らかである。
【0012】
三番目の方針は、アルブミンや免疫グロブリン断片の場合のように、相当な半減期を有する血漿蛋白質の配列と結合させた状態で、所期の蛋白質の活性を有する配列を発現したキメラ蛋白質を得ることであった(非特許文献4:Dennis, Marks S. et al, The Journal of Biological Chemistry vol. 277, No. 38, Issue of September 20, pp. 35035-35043, 2002)。しかし、この技術は、天然に存在しない蛋白質に患者を暴露することによる免疫原性が予想されることに加え、構造の改変により蛋白質の効力が極めて劇的に喪失するという大きな不利益を有する。
【0013】
複雑な有効成分の半減期を延長するべく研究されたもう一つの可能性は、前記製品とPEGで安定化したリポソームとの同時投与であった。この技術は、前記有効成分のPEGへの親和性に基づいており、前記蛋白質とリポソームとの可逆的会合を可能にするものである。この一時的会合は前記有効蛋白成分の半減期の増加をもたらすはずである。PEGで安定化したリポソームは長期間循環系に滞留するからである。しかし、実際にはこの仮説を裏付けることはできなかった。このシステムでは、血友病患者でFVIIIの半減期を延長する効果がないと判明したからである(非特許文献5:Powell J.S et al, Journal of Thrombosis and Haemostasis, 6: 277-283,2007)。
【0014】
以上説明した系の何れも、現在までのところ、半減期を大幅に改善するには至っていない。例外として上述した構造上の修飾や改変の導入も、ヒト病理の治療への応用は不可能であるか、複雑さゆえに極めて困難である。
【0015】
生分解性ポリマー微小球(micropsheres)により被包化した治療剤の制御放出が、大々的に研究されている。有効成分を生分解性ポリマーでマイクロ被包化することにより、薬剤の制御放出が可能となる。このアプローチは近年、乳酸及びグリコール酸の誘導体を主とする皮下用途の制御放出処方に応用されている。かかる処方はとりわけ、細胞増殖抑制剤、抗炎症剤、ペプチド、及びホルモンを含む広範囲の有効成分の被包化に首尾よく使用されてきた(非特許文献6:Tamilvanan S. et al, PDA Journal of Pharmaceutical Science and Technology, vol. 62, No. 2, March-April 2008 pp. 125-154)。
【0016】
Pankaj(特許文献1:米国特許5,417,982)は、経口投与によるホルモン制御放出用の乳酸及びグリコール酸微小球の使用について記載する。Pankajは注射用製品の取得可能性についても言及しているが、かかる投与方法の必要条件を考慮すると、この発明が静脈投与できる可能性はきわめて低い。何れにせよ、この発明ではかかる目的でのアルギン酸塩(alginate)の使用を想定していない。
【0017】
Sivadas (非特許文献7:Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167)は、吸入投与用の蛋白質被包化のための賦形剤として、ヒドロキシプロピルセルロース、キトサン、ヒアルロン酸、ゼラチン、オボアルブミン及びグリコールポリ乳酸等の種々のポリマーの使用について記載する。
【0018】
乳酸及びグリコール酸誘導体の使用に伴う不利益の一つは、有機溶媒の存在下での生成を要することである。中には、例えばポリビニルアルコールのように毒性を有し、蛋白質やホルモン等の複雑な有効成分の生物活性の保持とは相容れない性質を示すものもある。
【0019】
また、これらのポリマーを使用すると、得られる粒子は疎水性が極めて高いものとなり、以下に述べるように、細胞の取込み機構により急速に循環系から排出される。更なる不利点として、粒子の溶解時、ひいては有効成分の放出時に、粒子の周囲には局所的に強酸性環境が形成される。これは粒子の溶解時にポリマーが乳酸及びグリコール酸に分解し、粒子の周囲に強酸性環境を形成することによるものである。この酸性環境こそが、脆弱な有効成分、特に不安定な生物活性と複雑なアミノ酸構造を有する成分に損傷を与えるのである。
【0020】
アルギン酸塩は概して、食品・医薬業界及び化学工業界において多くの用途を有する。かかる多種多様の用途を規定するのが、その親水コロイドの性質、すなわち、水和によって粘性溶液、分散液又はゲルを形成し得る能力である。かかる特徴ゆえに、アルギン酸塩は、増粘剤、安定剤、ゲル化剤及び膜形成剤として独自の性質を示すのである。
【0021】
かかるアルギン酸塩の性質を広く利用してきた領域の一つが有効成分の被包化であり、特にその溶解性の改善や、種々の投与法における薬剤投与の補助を目的としたものである(非特許文献8:Tonnesen, Hanne Hjorth et al, Drug Development and Industrial Pharmacy, 28(6), 621-630 (2002))。特に、アルギン酸塩の粘膜付着性を利用した経口投与における使用が挙げられる。皮下法も試験されてきた。しかし、静脈内投与の必要条件は厳しいため、かかる用途の先例はない。例えば、Benchabane (非特許文献9:Benchabane, Samir et al, Journal of Microencapsulation, September 2007; 24(6): pp. 565-576)は、経口投与のための「噴霧乾燥」によるアルブミンマイクロカプセルの製造におけるアルギン酸塩の使用について記載する。同様の先例においてCoppi (非特許文献10:Coppi, Gilberto et al, 2001, Drug Development and Industrial Pharmacy, 27(5), pp. 393-400)は、カルシウム及びキトサンと共重合した蛋白質経口投与用の微小球の形成を実証している。何れの場合もアルギン酸塩は、胃内消化時に生じる天然の蛋白質分解に対する蛋白質保護剤としての役割をもつ。
【0022】
さらに、Mladenovska (非特許文献11:Mladenovska, K., International Journal of Pharmaceutics 342 (2007) pp. 124-136)は、結腸投与用のアルギン酸塩/キトサン微粒子の取得について記載する。
【0023】
Sivadas (非特許文献12:Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167)も、アルギン酸塩を吸入投与用の蛋白質被包化のための賦形剤として使用することに言及している。
【0024】
有効成分の直接投与とは別に、アルギン酸塩は複雑な治療形態の投与のための賦形剤としても検討されてきた。例えば、国際公開第2006/028996号(特許文献2)には、バクテリア毒素解毒剤輸送のためのアルギン酸塩及びエマルサンの使用が開示されている。
【0025】
他の例としては、多重小胞リポソームの被包化(非特許文献13:Dai, Chuanyun, et al, Colloids and Surfaces B: Biointerfaces 47 (2006) pp. 205-210)又は生細胞の被包化(特許文献3:欧州特許公開第2159523号)におけるアルギン酸塩の使用があげられる。この場合、生細胞投与の目的は、再生医療又は遺伝子治療用途にある(特許文献4:国際公開第2007/046719号;非特許文献14:Peirone, Michael et al, J. Biomed. Mater. Res. 42, pp.587-596, 1998;非特許文献15:Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223)。興味深いことに、Garcia-Martin (非特許文献15:Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223)は、血友病A治療用に遺伝子操作した生細胞投与の適用可能性について記載しつつ、この問題と医薬との関係について触れている。この場合、生細胞を含むアルギン酸塩マイクロカプセルは、カテーテルの導入により腹腔内に埋め込まれる。この場合、治療目的も投与経路(非静脈)も、本発明からはほど遠い。
【0026】
蛋白質等の複雑な有効成分を被包化するためのポリマーの使用に関し、このように広範な知見があるにもかかわらず、これらの製品の静脈内投与に伴う問題を解決し得る参考文献はない。Wongら(非特許文献16:Wong, Joseph et al, Advanced Drug Delivery Reviews 60 (2008) pp. 939-954)が述べているように、静脈内投与に粒子懸濁液を使っている認可製品は3つしかなく、その何れもが組成中にアルギン酸塩を使用していない。何れのケースも、半減期の延長ではなく、製品の溶解性改善を目的としたものである。
【0027】
静脈内に微粒子を効果的に投与することの困難さは、a)製品設計という基礎的な側面、例えば粒子サイズと分布、有機溶媒の不在、及び、懸濁液の均一性、粘性及び「注射針通過性」(「注射針通過性」とは製品の吸引及び注入の容易性の意である);b)工業的スケールでの生産及び調製の技術的側面、例えば一回投与量(dose)の均一性、溶媒沈殿で得られる製品の場合、塩の望ましくない結晶化、製品の無菌度(sterility)と無発熱原性(apyrogenicity);並びに、c)生物学的側面、例えば薬物動態及び薬力学プロフィールの無計画な改変、生体分布の改変、ポリマーの生物蓄積、食作用の活性化、塞栓の毒性と作用、あるいは捕体の活性化、として表現される。
【0028】
これに関連して、これら製品を開発するにあたり最も大きな問題の一つは、以前は細網内皮系(RES)と呼ばれていた単核食細胞系(MPS)により迅速に除去されることであり、この系には骨髄の単球性前駆細胞由来の全ての細胞、末梢血の単球、及びさまざまな器官や組織のマクロファージ又は組織球が含まれる。後者の中では、血漿中の微粒子のクリアランスにおける重要性の点で、肝臓のクッパー細胞と、脾臓及び骨髄に分布するマクロファージに言及せねばならない(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)。
【0029】
ナノ粒子又はマイクロ粒子を静脈内投与した場合、血漿中の蛋白質によって迅速にオプソニン化されることが、広く報告されてきた。これらの表面に吸収される蛋白質は、MPS細胞による認識及び取込みを誘導する(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)。
【0030】
同様の作用はリポソームでも観察されており(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165)、同文献には加速化血中クリアランス(Accelerated Blood Clearance)(ABC)として知られる現象が記載されている。ポリマー微粒子及びリポソームの何れの場合も、オプソニン化の現象は補体系活性化に直接関連している(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165;非特許文献19:Koide, Hiroyuki et al, International Journal of Pharmaceutics 362 (2008) pp. 197-200)。
【0031】
実用に際して、この食作用現象は、静脈内投与される微粒子を用いて半減期を延長しようとする薬剤の開発を阻害する。被包化にともなうサイズの増大は、単なる増大ではなく、時として分解を加速する場合もあるからである。明らかに、この現象は動物の薬物動態研究等のインビボ実験によってしか観察できない。
【0032】
この食作用によるクリアランスと粒子サイズの関係は広く記録されてきた。Champion (非特許文献20:Champion, JA, Pharm Res. 2008 Aug; 25(8): 1815-21. Epub 2008 Mar 29)は、ポリマー微粒子が遭遇した食作用と微粒子のサイズとの関係について具体的に記載するものであるが、2〜3μmで最大の影響が観察された。インビボでのMPSによる微粒子の取込みを規定する他の特徴は、粒子の疎水性とそのゼータ電位(Z Potential)である(非特許文献21:Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0033】
Z電位は粒子の特性の一つである。具体的には、分散粒子は外相からのイオンの吸着や、それ自体の表面官能基のイオン化により帯電する傾向がある。これによる帰結の一つは、スターン層(Stern layer)と呼ばれる対イオンの層が、負に帯電した分散粒子の環境で粒子と背中合わせに出現することである。前記スターン層上に(両電荷の)可動電荷の存在を特徴とする拡散層が出現し、粒子までの距離の関数として、粒子表面の荷電を中和する。Z電位とは、対イオン層と動電学的に中性な点とのいわゆる電位差のことである。
【0034】
Z電位値は、分散系の大部分の安定化にとって極めて重要である。後者が、同じ荷電を有する分散粒子間の反発度を抑制しつつ、粒子相互間の過剰な接近を防止するとともに、癒合現象に起因する粒子間引力が支配的になるのを防止するからである。Z電位に関しては、0付近の、部分的に負のZ電位が食作用を低減するということが開示されている(非特許文献21:Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0035】
更に、疎水的性質も粒子のオプソニン化及び取込みを促進する。この事実は特に興味深い。ポリ乳酸やグリコール酸由来の粒子は、例えば、極めて疎水性が高いからである。
【0036】
微粒子及びリポソームの血漿中での半減期を延長するためのアプローチの一つは、荷電を改善して親水性表面層を形成し得る荷電ポリマーをその表面に導入し、オプソニン化や食作用を防止することであった。例としては、ポリエチレングリコール(PEG)(非特許文献18:Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165;非特許文献22:Owens III, Donald E et al, International Journal of Pharmaceutics, volume 307, issue 1, 3 January 2006, Pages 93-102)あるいはヘパリン(非特許文献17:Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050)の使用が挙げられる。
【0037】
このアプローチは、系の複雑性を増加させるため、医薬製品開発を複雑、困難なものにする。また、上で論じたように、PEG−リポソームの使用は、FVIII等の複雑な蛋白質の半減期の延長には効果がないことが分かっている(非特許文献5:Powell J.S et al 2007, Journal of Thrombosis and Haemostasis, 6: pp. 277-283)。
【0038】
微粒子の場合、実現可能な静脈内投与用製品を得るためには、適切なサイズとZ電位を親水性の粒子に持たせることが必要となるだろう。
【先行技術文献】
【特許文献】
【0039】
【特許文献1】米国特許5,417,982
【特許文献2】国際公開第2006/028996号
【特許文献3】欧州特許公開第2159523号
【特許文献4】国際公開第2007/046719号
【非特許文献】
【0040】
【非特許文献1】Casper, CK, Hereditary Plasma Clotting Factor Disorders and Their Management 5th ed. WFH, Sam Schulman Ed., 2008
【非特許文献2】Bailon Pascal et al, Bioconjugate Chem. 2001, 12, 195-202
【非特許文献3】Perlman Signe et al, The Journal of Clinical Endocrinology & Metabolism 88 (7): 3227-3235, 2003
【非特許文献4】Dennis, Marks S. et al, The Journal of Biological Chemistry vol. 277, No. 38, Issue of September 20, pp. 35035-35043, 2002
【非特許文献5】Powell J.S et al, Journal of Thrombosis and Haemostasis, 6: 277-283, 2007
【非特許文献6】Tamilvanan S. et al, PDA Journal of Pharmaceutical Science and Technology, vol. 62, No. 2, March-April 2008 pp. 125-154
【非特許文献7】Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167
【非特許文献8】Tonnesen, Hanne Hjorth et al, Drug Development and Industrial Pharmacy, 28(6), 621-630 (2002)
【非特許文献9】Benchabane, Samir et al, Journal of Microencapsulation, September 2007; 24(6): pp. 565-576
【非特許文献10】Coppi, Gilberto et al, 2001, Drug Development and Industrial Pharmacy, 27(5), pp. 393-400
【非特許文献11】Mladenovska, K., International Journal of Pharmaceutics 342 (2007) pp. 124-136
【非特許文献12】Sivadas Neeraj et al, International Journal of Pharmaceutics 358 (2008) pp. 159-167
【非特許文献13】Dai, Chuanyun, et al, Colloids and Surfaces B: Biointerfaces 47 (2006) pp. 205-210
【非特許文献14】Peirone, Michael et al, J. Biomed. Mater. Res. 42, pp.587-596, 1998
【非特許文献15】Garcia-Martin Carmen et al, The Journal of Gene Medicine, J Gene Med 2002; 4: pp. 215-223
【非特許文献16】Wong, Joseph et al, Advanced Drug Delivery Reviews 60 (2008) pp. 939-954
【非特許文献17】Passirane, Catherine et al, Pharmaceutical Research, Vol. 15, No. 7 1998 pp. 1046-1050
【非特許文献18】Ishida, Tatsuhiro et al, Journal of Controlled Release 126 (2008) pp. 162-165
【非特許文献19】Koide, Hiroyuki et al, International Journal of Pharmaceutics 362 (2008) pp. 197-200
【非特許文献20】Champion, JA, Pharm Res. 2008 Aug; 25(8): 1815-21. Epub 2008 Mar 29
【非特許文献21】Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages
【非特許文献22】Owens III, Donald E et al, International Journal of Pharmaceutics, volume 307, issue 1, 3 January 2006, Pages 93-102
【発明の概要】
【発明が解決しようとする課題】
【0041】
Terrence (欧州特許公開第2 286 040号, 欧州特許出願第00973477.3号)は、活性を有する被包化成分の半減期を増加させ得る投与の系として、ポリマーの使用について記載する。この発明ではそのために、(1)第一の水溶性ポリマー、(2)第一の錯化剤として少なくとも一つの陰イオン性多糖類、及び(3)第二の錯化剤として2価の陽イオンを使用することを要する。上で述べたように、この発明は技術的に複雑で、実際の使用が困難なものである。対照的に、本発明によれば、有効成分の制御放出をはるかに簡素な微粒子で達成することができる。かかる微粒子が要するのは、その用途に必要な全ての特性を有する一種類のポリマーの使用のみである。さらにTerrenceの発明は、静脈内用調製物のサイズによる適合性を実証しておらず、細胞の食作用を避ける方法を説明してもいない。
【0042】
アルギン酸塩は、PLAやPLGA等の他のポリマーとは異なり、親水性である。本発明において生成される粒子は、粒子の凝集を十分に防止し得る部分的に負のZ電位を有しつつ、低オプソニン化プロファイルを示し得る程度に中性であることが示されている。
【0043】
静脈内投与に許容できる最大粒子サイズはおよそ5μmである。このことは、超音波診断用に標識された平均サイズ3.0〜4.5μmのアルブミンを使用した登録薬剤(Optison, data sheet 28)の存在により実証されている。
【0044】
アルギン酸塩は生体適合性を有し、その食品業界での広範な使用に基づいて、ヒトへの経口使用に大々的に使用されてきた。非粒子型ポリマーとして静脈注射された場合、半減期4時間及び22時間という二相様式で排出され(Hagan, A et al, European journal of Pharmaceutical Sciences, Volume 4, Supplement 1, September 1996, pp. 100-100 (1))、副作用も見受けられない。アルギン酸塩は尿中に排出される。
【0045】
加えて、水溶性ポリマーであるという事実は、複雑な蛋白質との適合性に寄与する。後者が天然の溶媒となるからである。
【課題を解決するための手段】
【0046】
本発明は、アルギン酸又はその医薬的に許容し得る塩の微粒子を含んでなる組成物であって、制御放出を達成するとともに、静脈内投与された有効成分の半減期を延長することによって投与頻度の低減を達成し、並びに血中有効成分レベルの更なる安定化を達成し、ひいては、有効成分を定期的に注入した場合に典型的に生じる有効成分濃度のピーク及びトラフを低減することが可能となる。
【0047】
本発明は、静脈点滴に適したサイズと迅速な食作用を阻止するのに適した生理化学的特徴を併せ持ち、複雑な有効成分の制御放出を可能にする、アルギン酸塩の親水性微粒子について記載する。
【図面の簡単な説明】
【0048】
【図1】図1は、バッチ9及びバッチ1のインビトロ放出テスト結果の比較グラフである。
【図2】図2は、非被包化FVIIIの投与後及び本発明の組成物の適用後におけるウサギ血漿中ヒトFVIII:Cの薬物動態を示す。
【図3】図3は、非被包化FVIIIの投与後及び本発明の組成物の適用後におけるウサギ血漿中ヒトVWF:Agの薬物動態を示す。
【発明を実施するための形態】
【0049】
アルギン酸及びその塩(アルギン酸アンモニウム、アルギン酸カルシウム、アルギン酸カリウム、アルギン酸ナトリウム及びアルギン酸プロピレングリコール)は、その物理化学的及び生化学的特性ゆえに、有効成分の被包化において最も使用及び研究されているポリマーの1つである。これらは天然由来の多糖であり、商業的には藻類や細菌から製造される。
【0050】
アルギン酸塩(alginates)はアルギン酸(alginic acid)の塩であり、β−(1−4)−D−マンヌロン酸(M)及びα−(1−4)−L−グルロン酸(G)の2つのモノマー単位からなる直鎖多糖である。種々の配列を有するブロックに分類されるが、最も一般的なものはG、M及びMGである。
【0051】
カルシウム(Ca++)等の多価カチオンの存在下では、隣接するGブロック間で強力な結合が形成され、拡大したアルギン酸塩のネットワークが形成される。カルシウムイオンはグルロン酸の負電荷を有する基の間を連結するように位置する。
【0052】
処方によっては、キトサン等の他の多糖と組み合わされる場合が多い。キトサンは、ランダムに分布したβ−(1−4)−D−グルコサミン(脱アセチル化単位)とN−アセチル−D−グルコサミン(アセチル化単位)の鎖からなる直鎖の多糖である。
【0053】
アルギン酸塩の処方によっては、アルブミン、好ましくは無菌且つ発熱物質不含有(pyrogen-free)ヒトアルブミンを、荷電物質として使用してもよい。これは製造工程時には有効成分の保護剤としても機能し、また、製品の長期保存時には安定剤としても機能する。
【0054】
血漿中の放出改善の対象となる有効成分は、複雑で不安定な有効成分であってもよい。より具体的には、有効成分は生物活性を示すことを特徴とする。この生物活性は、例えば酵素活性、輸送、分子間相互作用、又はリガンドとの結合によってもたらされる。何れの場合も、高エネルギーの製造条件、特に温度、圧力及び/又は無極性環境等に対して不安定又は感受性である有効成分が問題となるだろう。構造上の小さな変化が生物活性の不可逆な損失につながり得るからである。
【0055】
生物活性を有する有効成分の例として、ヒトペプチドホルモン、例えばメラトニン、セロトニン、チロキシン、エピネフリン、ノルエピネフリン、ドーパミン、副腎皮質刺激ホルモン、アンギオテンシノーゲン及びアンギオテンシン、バソプレッシン、心房性ナトリウム利尿ペプチド、カルシトニン、エリスロポエチン、卵胞刺激ホルモン、グルカゴン、ヒト絨毛性ゴナドトロピン、ヒト胎盤性ラクトゲン、成長ホルモン、インヒビン、インスリン、インスリン型成長因子(又はソマトメジン)、黄体形成ホルモン、メラニン細胞刺激ホルモン、オキシトシン、プロラクチン、トロンボポエチン、神経ペプチドY、ヒスタミン及びそれらの誘導体が挙げられる。
【0056】
他の例としては、生物活性蛋白質、例えば、アルブミン、アルファ1−アンチトリプシン、アルファ酸グリコプロテイン(alpha-acid glycoprotein)、アルファ−2−マクログロブリン、アンチトロンビン、ハプトグロビン、セルロプラズミン、リポ蛋白質、トランスフェリン、プラスミノーゲン、フィブリノーゲン、補体蛋白質、凝固因子、及び免疫グロブリン等も挙げられる。
【0057】
これらの有効成分は、生物活性を有するがゆえに、小さな構造上の損傷の結果として生じ得る機能喪失を被り易い。かかる構造上の損傷は、例えば温度、圧力、溶媒の極性、浸透圧、酸素の存在、攪拌等と関連し得る。
【0058】
これに関して、これらの有効成分の中でも凝固第VIII因子は、極度の不安定さゆえに際立っている。FVIIIは複雑な構造を有するため、その生物活性を十分に安定化させるのは、特に精製状態では非常に困難である。例えばParti R ら (Haemophilia 2000; 6: 513-522)は、凍結乾燥状態でさえも、40℃超の温度ではFVIIIの生物活性が減弱し始めると説明している。かかる不安定さが最も顕となるのは、FVIIIが溶液状態の場合であり、25℃でさえ不安定の兆候が見られる。第IX因子及び第VIIa因子についても、温度等の外的因子に対する感受性が知られている。
【0059】
これに関して留意すべきは、本製造方法を採用すると、FVIIIの生物活性を保持する医薬製剤が得られるという点である。これは、本方法が生物活性の安定化が困難な有効成分に対しても適用できること、従って、本発明はFVIIIのように不安定な成分に適用できることを意味する。更に言えば、本発明はFVIIIより安定な成分にも適用できる。結果として、凝固因子類は、本発明に記載した処方の適用により利益を享受し得る有効成分の明白な例となる。
【0060】
すなわち、本発明では、ポリマー微小球に含まれる有効成分は、生物活性を示すペプチド、蛋白質、又はホルモンである。好ましくは、前記活性成分は凝固因子であり、より好ましくは、前記活性成分は第VIII因子、フォンヴィレブランド(von Willebrand)因子、第VIII因子とフォンヴィレブランド因子の複合体、第IX因子又は第VIIa因子である。
【0061】
これらの成分は、ヒト、動物、組換、トランスジェニックの何れに由来するものであってよい。後者の場合には、合成分子は天然分子を再現したものでも、意図的な改変を加えたものでもよい。
【0062】
組成物の取得
マイクロ被包化(microencapsulation)は、分子、固体粒子又は液体小球を、別の性質を有する原料で被覆し、マイクロメートルサイズの粒子を作製する手法である。この加工法により生じる生成物は、微粒子(microparticles)、マイクロカプセル(microcapsules)、又は微小球(microspheres)と呼ばれる。
【0063】
幾つかのマイクロ被包化技術が存在する。
−化学的手法によるマイクロ被包化
・ 界面重合
【0064】
−物理化学的手法によるマイクロ被包化
・ 溶剤蒸散
・ 液滴形成
・ ゲル化
・ キレート化
・ 小胞形成
【0065】
−機械的手法によるマイクロ被包化
・ 押出成形
・ 共押出法
・ 噴霧乾燥
・ 噴霧冷却
【0066】
本発明記載の微粒子の製造に選択した技法は、Erdinc B.I. [Erdinc B.I. (2007) Micro/nanoencapsulation of proteins within alginate/chitosan matrix by spray drying, Degree Thesis, Queen’s University, Kingston, Canada]に記載の噴霧乾燥である。この製造法は単一工程であることを特徴とし、最終産物として微粒子が得られる。
【0067】
本発明の、有効成分を制御放出するための、アルギン酸又はその塩の微粒子を含む静脈内投与用の生体適合性組成物の製造方法は:
−有効成分及びポリマーを含む溶液/懸濁液/乳化液を、ノズルを通して送り出し、液滴形状で分散させる噴霧工程、
−熱風が前記液滴からの溶媒の蒸散を助ける、乾燥室内での乾燥工程、及び
−被包化された生成物の収集工程、
の各段階を特徴とし、かかる手順は140〜180℃の温度、35〜40m3/hの供給流速、3.5〜5ml/minの注入流速、及び4〜6psiの圧力で実施される。
【0068】
かかる条件下であれば、5μm以下、好ましくは1〜4.5μmのサイズの粒子を取得し、且つ、有効成分の活性を維持することが可能となる。更に、噴霧段階の前段に、乳化液をホモジナイズするという工程を任意で追加することにより、粒子の平均サイズを改善することができる。この追加のホモジナイズ工程は、例えば1500〜2000psiの圧力により行なわれる。
【0069】
噴霧乾燥による有効成分の被包化は、溶液又は乳化液を脱水する連続プロセスであり、微粒子によって形成される固形物をプロセスの最後に回収する。
【0070】
この目的のために、有効成分を含む流体は、所定の注入流速でノズル又は回転ディスクに向けて機械的に運ばれ、そこで無数の微小の液滴状に噴霧される。液滴のサイズは主に流体を噴霧するガス圧で決まる。このプロセスは、制御したガス流(通常は空気)が所定の吸気速度及び制御温度下で連続的に循環する密室で行なう。
【0071】
噴霧の結果、流体の空気との接触表面積は大幅に増大するので、乾燥空気流に接触した流体の溶媒(通常は水)の急速な蒸散が生じる。この急速な蒸散に伴い、状態変化に必要な熱が奪われて、各小液滴の内部が冷却される。このように、有効成分への熱ショックを最小化しつつ、迅速な乾燥を行なうことができる。このプロセスの完了後、生成物は固形として収集される。
【0072】
組成物の説明
得られた微粒子は、平均粒子サイズ、Z電位、及び生物活性の測定により識別される。粒子サイズは、Beckman Coulter LS13320装置で回折レーザーにより決定される。
【0073】
問題となる静脈内投与の場合、粒子サイズを5μm以下、好ましくは1〜4.5μmとする必要がある。粒子サイズがこれより大きいと、血栓の形成を引き起こす可能性があるからである。
【0074】
Malvern Zetasizerの装置で決定されるZ電位は、懸濁液中の粒子間相互作用を制御する基本パラメータの一つである。これは粒子表面の性質と分散溶媒によって決定される。本発明では、粒子間斥力を確保し凝集を阻止する観点から、最適値は−30mVを超えた値である。Z電位が0近く、好ましくは−30〜0mVの微粒子は、低い肝臓取込みと細胞クリアランスのレベルを有することが示されている(Szycher, Michael, High Performance Biomaterials: A Comprehensive Guide to Medical and Pharmaceutical Applications, published by CRC Press, 1991 ISB 0877627754, 9780877627753, 812 pages)。
【0075】
組成物の用途
調節放出又は制御放出される医薬形態とは、同様に投与した従来放出法の医薬形態に対して、有効成分放出の速度及び/又は場所を変更するべく設計されたものである。
【0076】
本発明では、蛋白質等の生物活性を示す有効成分、より具体的には凝固因子の被包化が、インビトロの放出モデルでいかに制御放出を可能にするかを観察した。第VIII因子はその構造上の複雑さゆえに、外的因子に対して高い感受性を有する点で注目に値する。実際、FVIIIをその天然基質であるヒトの血漿自体の状態で凍結しても、生物活性の部分的喪失が生じる(Bravo, M.I. et al, Pharmeuropa Scientific Notes, 2006-1 pp. 1-5)。
【0077】
そこで、本発明に規定のヒトFVIII含有微粒子を、ヒト血漿の環境と同様の連続フローセル中に入れると、非被包化製品に比して、媒質中へのFVIII放出が遅くなることが観察された。
【0078】
同様に、本発明のFVIII含有微粒子をウサギに静脈投与すると、従来品に比して、血漿中のFVIIIの半減期が大幅に、且つ安定して延長されるという結果が得られた。さらに、上記処方に伴い動物が中毒作用を示すことはなく、副作用は見られなかった。
【0079】
このインビボの薬物動態データは極めて重要である。本発明の処方ではオプソニン化及びMPSへの取込み促進の作用が適切に処理されていることを、本データは紛れもなく示しているからである。
【0080】
本発明は複雑な成分の静脈投与を必要とする多様な病状の治療に使用できる。例としては出血性疾患及び凝固障害、ホルモン障害党が挙げられる。これらのケースでは、半減期の大幅な延長が達成されるであろう。例えばFVIIIの場合、一次予防法を維持するための投与回数を、例えば週1回の投与に低減することもできるだろう。
【0081】
親水性ポリマーの使用に伴い生じ得る欠点は、無発熱原性且つ無菌注射投与用の水性媒体(例えば水)中に前記製品を懸濁してから静脈点滴までの間に、微粒子の部分的な溶解が生じ得る点である。かかる不利益は、例えば、部分的に無極性の生体適合性溶液、例としてはエタノール、プロピレングリコール、ポリエチレングリコール、ジメチルスルフォキシド、N−メチルー2−ピロリドン、グリコフロール、イソプロピリデン−グリセロール、グリセロールホルマール又はアセトン等(Mottu F et al. Journal of Pharmaceutical Science & Technology 2000 Vol. 54, No. 6, 456-469)を、本発明に記載の微粒子の再懸濁及び投与用媒体として使用することにより解消し得る。
【0082】
本発明は、例えば、真空又は不活性雰囲気中でパックした脱水又は凍結乾燥生成物の状態として製造することもできる。これにより種々の温度条件下、例えば2℃〜40℃での長期安定化が可能となる。このようにして保存した製品は、溶媒で再構成後、静脈投与することができる。かかる溶媒としては、注射用蒸留水、食塩水、又は混合物が挙げられる。或いは、エタノール、プロピレングリコール、ポリエチレングリコール、ジメチルスルフォキシド、N−メチル−2−ピロリドン、グリコフロール、イソプロピリデン−グリセロール、グリセロールホルマール又はアセトン等の生体適合性溶媒を、例えば0.5%〜50%等の様々な含量で有する水性食塩水であってもよい。
【0083】
従来技術に優る利点
本発明は、静脈点滴に適したサイズと、迅速な食作用を阻止するのに適した物理化学的特徴とを併せ持ち、複雑な有効成分の半減期を延長し得る、アルギン酸塩の親水性微粒子の製造について記載する。
【0084】
アルギン酸塩は生体適合性を有し、尿により排泄され、いかなる公知の中毒作用をも伴わない。かかる特徴ゆえ、本発明は蛋白質及び複雑な有効成分の投与に適合している。
【0085】
本発明によれば、制御投与静脈系の実用化を阻んできたあらゆる不利益を克服することができ、その結果、静脈用途の有効成分を変更することなく、治療に必要な投与回数を低減することができる。この点において、本発明は、アミノ酸配列、グリコシル化又は合成誘導体の導入など、有効成分の改変を必要としない点に留意すべきである。
【実施例1】
【0086】
微粒子の調製
Erdinc B.I. [Erdinc B.I. (2007) Micro/nanoencapsulation of proteins within alginate/chitosan matrix by spray drying, Degree Thesis, Queen’s University, Kingston, Canada]に記載のように、アルギン酸塩微粒子の生成には噴霧乾燥プロセスが使われている。基本的に、微粒子は前記ポリマーと、選択した有効成分との乳化液を生成することにより調製した。
【0087】
試料の噴霧は、Buchi Mini Spray Dryer B-290装置を用い、以下の条件で行った。噴霧温度:140℃〜180℃、吸入速度:35〜40m3/h、注入流速:3.5〜5ml/min、圧力4〜6psi。
【実施例2】
【0088】
微粒子の説明
表1、2及び3に、微粒子の製造に使用した原料と、そのサイズ、Z電位及び収量等の特性を示す。使用した製造方法及び条件は実施例1に記載の通りである。
【0089】
【表1】
【0090】
FVIII活性/FVIII抗原量比は、ある試料中の活性蛋白質の割合に想到する。このように、当初試料中の活性/抗原量比を、被包化試料について得られる値と比較すると、マイクロ被包化後も機能を保持する有効成分の割合を算出できる。実施例において、被包化プロセス中の活性収率を当初活性収率との比で表わすと、バッチ1、2、及び3はそれぞれ57.6%、33.9%及び35.7%であった。
【0091】
【表2】
【0092】
この場合、バッチ4、5及び6の被包化プロセス中の活性収率は、これらのバッチ全てで100%であった。
【0093】
【表3】
【0094】
組換由来蛋白質の場合、被包化プロセスで測定した活性収率は、バッチ7及び8でそれぞれ25%及び71%であった。
【0095】
全てのバッチで、粒子のサイズは回折レーザーによりBeckman Coulter LS13320装置で測定し、Z電位はMalvern Zetasizer装置で測定した。
【0096】
FVIIIの生物活性は、欠乏血漿凝固アッセイ又は発色によるFXa生成の評価により測定した。FVIIa及びFIXの場合、生物活性はそれぞれFVII及びFIXを含まない血漿の凝固時間(部分活性化トロンボプラスチン時間)を評価することで測定した。蛋白質濃度は、FVIII:Ag、FIX:Ag、又はFVII:Agにそれぞれ特異的な抗体を用い、酵素免疫測定法(ELISA)による免疫学的検出法で測定した。
【0097】
ある試料中の活性蛋白質の割合の指標となる活性/抗原量比は、試料中の特異有効成分の活性と抗原単位量との商により算出した。活性/抗原量・収率の算出は、出発試料の活性/抗原量比と最終被包化製品の活性/抗原量比との変動率パーセンテージを評価することで行われる。
【0098】
いずれのケースでもみられるように、平均粒子サイズは5μm以下、Z電位は負の値である。また、活性/Ag・収率の結果は、プロセス中の生物活性が保持されていることを示している。
【0099】
これら種々の表は、前記制御放出系が様々な有効成分に適していることを示している。
【実施例3】
【0100】
インビトロ放出テスト
有効成分放出の評価には、連続フローセルを用いたインビトロ放出テストを、Sotax CE1装置にて閉回路でおこなう。
【0101】
前記テストは、溶解剤として1%のヒトアルブミンを含むpH7.3のイミダゾールバッファーを用い、37℃の温度、7〜25ml/minの流速で行なった。分析用の代表試料を異なる時点(5分、10分、15分、30分、60分、120分、180分及び240分)で抽出した。抽出試料を同量の新たな媒質と置換し、量の損失を補填した。
【0102】
FVIIIの生物活性は、欠乏血漿凝固アッセイ又は発色によるFXa生成の評価により測定した。FVIIa及びFIXの場合、生物活性はそれぞれFVII及びFIXを含まない血漿の凝固時間(部分活性化トロンボプラスチン時間)を評価することで測定した。蛋白質濃度は、FVIII:Ag、FIX:Ag、又はFVII:Agにそれぞれ特異的な抗体を用い、酵素免疫測定法(ELISA)による免疫学的検出法で測定した。
【0103】
テスト終了後、以下の結果が得られた:
【0104】
【表4】
【0105】
【表5】
【0106】
有効成分に適用した微粒子の組成が、非被包化製品に比して、前記製品放出の動態を改変することがわかる。
【実施例4】
【0107】
動物での第VIII因子の薬物動態
インビボ有効成分放出への前記組成物の作用を評価するため、薬物動態テストをウサギで行なった。このために、バッチ9(非被包化)由来のヒトFVIII 50IU/kg量を、3匹の雌ニュージーランド白ウサギ(New Zealand White rabbits)に静脈内投与した。同様に、実施例1記載の方法で作製し、実施例2で説明したバッチ1由来の被包化FVIII 50IU/kg量を、別の3匹の雌ニュージーランド白ウサギに静脈投与した。表6に記載のように、各時点で血漿サンプルを得、分析によりヒトFVIII:Cの存在を検出した。ヒトFVIIIの検出は、ヒトFVIII分子の選択的免疫キャプチャー(selective immunological capture)後の発色で行なった。これにより、注入したヒトFVIII活性を、ウサギFVIII活性と区別することが可能となる。
【0108】
【表6】
【0109】
これらの結果から、本発明の組成物は血漿中有効成分の放出を遅延させることがわかる。また、これらの結果は、前記微粒子のサイズにもかかわらず、かかる微粒子を循環系から迅速に除く細胞メカニズム(肝臓、脾臓又はマクロファージ)が存在しないことを示している。
【0110】
本目的に適したソフトウェア(WinNonlin 5.2)を用いてこれらのデータを解析することにより、表7に詳述する薬物動態定数が算出された。
【0111】
【表7】
【実施例5】
【0112】
動物でのフォンヴィルブランド因子の薬物動態
バッチ9の調製物(非被包化FVIII)及びバッチ1の調製物(被包化FVIII)のいずれの場合も、FVIIIは血漿由来であり、相当量のフォンヴィルブランド因子(VWF)を含んでいた。これは、VWFの被包化がFVIIIの被包化と同時に起こることを意味する。それゆえ、これらの挙動を独立して分析することが可能となる。このため、ウサギ血漿中ヒトVWF抗原(VWF:Ag)の存在を評価して、VWF自体の薬物動態分析を進めた。結果を表8に示す。
【0113】
【表8】
【0114】
これらの結果から、本発明の組成物は血漿中有効成分の放出を遅延させることがわかる。また、これらの結果は、前記微粒子のサイズにもかかわらず、かかる微粒子を循環系から迅速に除く細胞メカニズム(肝臓、脾臓又はマクロファージ)が存在しないことを示している。
【0115】
本目的に適したソフトウェア(WinNonlin 5.2)を用いてこれらのデータを解析することにより、表9に詳述する薬物動態定数が算出された。
【0116】
【表9】
【0117】
有効成分(この場合はVWF)の被包化により、半減期が有意に延長されることがわかる。
【0118】
以上、好ましい態様の例を挙げて本発明を説明してきたが、本発明は添付の特許請求の範囲をより広く解釈して定義されるものであり、上述の例によって限定されると解すべきではない。
【特許請求の範囲】
【請求項1】
静脈内投与用の生体適合性組成物であって、有効成分の制御放出用のアルギン酸又はその塩の微粒子を含んでなり、前記微粒子が5μm以下のサイズと負のZポテンシャルとを有する、生体適合性組成物。
【請求項2】
前記微粒子のサイズが1〜4.5μmである、請求項1記載の組成物。
【請求項3】
前記Zポテンシャルが−70〜0であり、0を含まない、請求項1記載の組成物。
【請求項4】
前記有効成分がペプチド、蛋白質又はホルモンである、請求項1記載の組成物。
【請求項5】
前記有効成分がヒト、動物、組換又はトランスジェニック由来である、請求項4記載の組成物。
【請求項6】
前記有効成分が不安定な生物活性を示す、請求項4記載の組成物。
【請求項7】
前記有効成分が血液凝固因子である、請求項4記載の組成物。
【請求項8】
前記有効成分が第VIII因子である、請求項4記載の組成物。
【請求項9】
前記有効成分がVWFである、請求項4記載の組成物。
【請求項10】
前記有効成分がFVIIIとVWFとで形成される複合体である、請求項4記載の組成物。
【請求項11】
前記有効成分が第IX因子である、請求項4記載の組成物。
【請求項12】
前記有効成分が第VIIa因子である、請求項4記載の組成物。
【請求項13】
請求項1〜12の何れか一項に記載の、有効成分の制御放出用のアルギン酸又はその塩の微粒子を含んでなる静脈内投与用の生体適合性組成物の製造方法であって、
−前記有効成分及びポリマーを含む溶液/懸濁液/乳化液を、ノズルを通して送り出し、液滴状に分散させる噴霧工程、
−熱風により前記液滴から溶媒を蒸散させる、乾燥室内での乾燥工程、及び
−被包化された生成物の収集工程
を含んでなるとともに、
140〜180℃の温度、35〜40m3/hの供給流速、3.5〜5ml/minの注入流速、及び4〜6psiの圧力で実施される方法。
【請求項14】
任意の工程として、前記乳化液のホモジナイズ処理が、前記噴霧工程の前段において付加的に実施される、請求項13に記載の方法。
【請求項15】
前記付加的なホモジナイズ処理工程が、1500〜2000psiの圧力下で実施される、請求項13に記載の方法。
【請求項16】
得られる前記微粒子のサイズが1〜4.5μmである、請求項13に記載の方法。
【請求項17】
得られる前記微粒子のZポテンシャルが−70〜0であり、0を含まない、請求項13に記載の方法。
【請求項18】
前記有効成分がペプチド、蛋白質又はホルモンである、請求項13に記載の方法。
【請求項19】
前記有効成分がヒト、動物、組換又はトランスジェニック由来である、請求項18に記載の方法。
【請求項20】
前記有効成分が不安定な生物活性を示す、請求項18に記載の方法。
【請求項21】
前記有効成分が血液凝固因子である、請求項18に記載の方法。
【請求項22】
前記有効成分が第VIII因子である、請求項18に記載の方法。
【請求項23】
前記有効成分がVWFである、請求項18に記載の方法。
【請求項24】
前記有効成分がFVIIIとVWFとで形成される複合体である、請求項18に記載の方法。
【請求項25】
前記有効成分が第IX因子である、請求項18に記載の方法。
【請求項26】
前記有効成分が第VIIa因子である、請求項18に記載の方法。
【請求項27】
前記出血性疾患、凝固変化、及びホルモン病の治療における、請求項1〜12の何れか一項に記載の生体適合性組成物の使用。
【請求項1】
静脈内投与用の生体適合性組成物であって、有効成分の制御放出用のアルギン酸又はその塩の微粒子を含んでなり、前記微粒子が5μm以下のサイズと負のZポテンシャルとを有する、生体適合性組成物。
【請求項2】
前記微粒子のサイズが1〜4.5μmである、請求項1記載の組成物。
【請求項3】
前記Zポテンシャルが−70〜0であり、0を含まない、請求項1記載の組成物。
【請求項4】
前記有効成分がペプチド、蛋白質又はホルモンである、請求項1記載の組成物。
【請求項5】
前記有効成分がヒト、動物、組換又はトランスジェニック由来である、請求項4記載の組成物。
【請求項6】
前記有効成分が不安定な生物活性を示す、請求項4記載の組成物。
【請求項7】
前記有効成分が血液凝固因子である、請求項4記載の組成物。
【請求項8】
前記有効成分が第VIII因子である、請求項4記載の組成物。
【請求項9】
前記有効成分がVWFである、請求項4記載の組成物。
【請求項10】
前記有効成分がFVIIIとVWFとで形成される複合体である、請求項4記載の組成物。
【請求項11】
前記有効成分が第IX因子である、請求項4記載の組成物。
【請求項12】
前記有効成分が第VIIa因子である、請求項4記載の組成物。
【請求項13】
請求項1〜12の何れか一項に記載の、有効成分の制御放出用のアルギン酸又はその塩の微粒子を含んでなる静脈内投与用の生体適合性組成物の製造方法であって、
−前記有効成分及びポリマーを含む溶液/懸濁液/乳化液を、ノズルを通して送り出し、液滴状に分散させる噴霧工程、
−熱風により前記液滴から溶媒を蒸散させる、乾燥室内での乾燥工程、及び
−被包化された生成物の収集工程
を含んでなるとともに、
140〜180℃の温度、35〜40m3/hの供給流速、3.5〜5ml/minの注入流速、及び4〜6psiの圧力で実施される方法。
【請求項14】
任意の工程として、前記乳化液のホモジナイズ処理が、前記噴霧工程の前段において付加的に実施される、請求項13に記載の方法。
【請求項15】
前記付加的なホモジナイズ処理工程が、1500〜2000psiの圧力下で実施される、請求項13に記載の方法。
【請求項16】
得られる前記微粒子のサイズが1〜4.5μmである、請求項13に記載の方法。
【請求項17】
得られる前記微粒子のZポテンシャルが−70〜0であり、0を含まない、請求項13に記載の方法。
【請求項18】
前記有効成分がペプチド、蛋白質又はホルモンである、請求項13に記載の方法。
【請求項19】
前記有効成分がヒト、動物、組換又はトランスジェニック由来である、請求項18に記載の方法。
【請求項20】
前記有効成分が不安定な生物活性を示す、請求項18に記載の方法。
【請求項21】
前記有効成分が血液凝固因子である、請求項18に記載の方法。
【請求項22】
前記有効成分が第VIII因子である、請求項18に記載の方法。
【請求項23】
前記有効成分がVWFである、請求項18に記載の方法。
【請求項24】
前記有効成分がFVIIIとVWFとで形成される複合体である、請求項18に記載の方法。
【請求項25】
前記有効成分が第IX因子である、請求項18に記載の方法。
【請求項26】
前記有効成分が第VIIa因子である、請求項18に記載の方法。
【請求項27】
前記出血性疾患、凝固変化、及びホルモン病の治療における、請求項1〜12の何れか一項に記載の生体適合性組成物の使用。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2010−150257(P2010−150257A)
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【外国語出願】
【出願番号】特願2009−286418(P2009−286418)
【出願日】平成21年12月17日(2009.12.17)
【出願人】(509348487)グリフォルス,ソシエダッド アノニマ (4)
【Fターム(参考)】
【公開日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2009−286418(P2009−286418)
【出願日】平成21年12月17日(2009.12.17)
【出願人】(509348487)グリフォルス,ソシエダッド アノニマ (4)
【Fターム(参考)】
[ Back to top ]