
25個を超えるヌクレオチドを含む認識タグを単離するためのIII型制限酵素の使用
【課題】ESTデータベースが存在しないような生物についても、正確で迅速な定量的遺伝子発現解析を可能にする。
【解決手段】発現遺伝子のcDNAから、25個を超えるヌクレオチドを含み、前記発現遺伝子を同定しうるタグを単離するためのIII型制限酵素の使用であって、前記タグの3'末端が前記III型制限酵素の切断部位によって規定され、前記タグの5'末端が前記発現遺伝子のcDNAの3'末端に最も近い他の制限酵素の切断部位によって規定されることを特徴とする使用。
【解決手段】発現遺伝子のcDNAから、25個を超えるヌクレオチドを含み、前記発現遺伝子を同定しうるタグを単離するためのIII型制限酵素の使用であって、前記タグの3'末端が前記III型制限酵素の切断部位によって規定され、前記タグの5'末端が前記発現遺伝子のcDNAの3'末端に最も近い他の制限酵素の切断部位によって規定されることを特徴とする使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、分子生物学および遺伝子発現解析の分野に関する。これに関連して、本発明は、転写産物の定められた領域を単離するためのIII型制限酵素の使用に関する。
【背景技術】
【0002】
遺伝子工学による遺伝子の構造および発現の改変は、様々な医学、薬学および農学への適用に莫大な潜在的可能性をもたらす。標的遺伝子を検出し選択するために、様々な発生段階、様々な環境条件下(ストレスなど)の細胞、組織、臓器または生物におけるスクリーニングにおいて、発現プロファイリングが広範に適用されている。遺伝子発現解析で最も強力な技術の1つは、Velculescuら、Science 270:484-487、1995によって開発されたSAGE(商標)(遺伝子発現逐次解析)である。
【0003】
最初のSAGE(商標)のプロトコール(図1)では、定められた細胞、組織または臓器由来のメッセンジャーRNA(mRNA)のプールを供給源物質として使用し、それから、ビオチン化オリゴdTプライマーを用いて逆転写酵素により相補的DNA(cDNA)を逆転写する。得られた一本鎖cDNAを二本鎖DNAに転換し、配列モチーフ5'-CATG-3'を認識する制限酵素NlaIIIで消化する。ストレプトアビジン被覆マグネティックビーズを使用して、二本鎖cDNAの3'末端の断片を回収する。cDNAを2分割する。次いで、2種のリンカー(リンカー1およびリンカー2)を、分離したそれぞれのcDNAと連結する。リンカーは、配列モチーフ5'-GGGAC-3'を含む。これはII型制限酵素BsmFIの認識部位であり、この酵素は認識部位から3'方向に13bp離れた部位を切断する。したがって、リンカーの付いたcDNAをBsmFIで処理することにより、「タグ」配列と呼ばれるcDNAの13bpの断片が、リンカー断片と一緒に(「リンカータグ」断片)遊離される。次いで、分離した2つのcDNAから得られた2つのリンカータグ断片を、2つのタグの位置が隣り合ってダイタグ(ditag)を形成するような形で互いに連結し、その後リンカー配列に特異的なプライマーを用いてPCRを行う。NlaIII消化によりリンカー断片を除去した後、ダイタグをコンカテマー化し、適当なプラスミドにクローン化する。プラスミド挿入物のシーケンシングから、4bpの5'-CATG-3'配列と隣接した一連の9bpのタグが明らかとなる。13bpのタグ配列を使用すると、利用可能な発現配列タグ(EST)データベースを調べることによって、多くの場合でタグ配列が由来する遺伝子を同定することが可能である。したがって、数千個のタグの配列を決定した後、試料中の各タグの数をカウントし、さらにそれに対応する遺伝子を同定することも可能である。
【0004】
したがって、上記に記載のSAGE(商標)のプロトコールは、包括的遺伝子発現を研究するのに有効な方法である。しかし、そのタグ配列の限定されたサイズ(13bpのみ)は、そのタグが由来する遺伝子を明確に同定するには十分ではない。単一のタグ配列が複数の異なるEST配列と対応する可能性があり、さらなる解析を混乱させる可能性がある。
【0005】
その状況を改善するために、II型制限エンドヌクレアーゼMmeIを使用する、いわゆる「LongSAGE(商標)」のプロトコールが最近開発された。(最初のSAGE(商標)のプロトコールでのBsmFIの代わりに)MmeIを使用することで、19〜21bpの長さのタグを回収することが可能となる(Sahaら、Nature Biotechnology 20:508-512、2002)。
【0006】
MmeI消化により、3'突出末端(2塩基突出)が生じる。MmeI消化の後からダイタグ連結前までに平滑末端化するためには、3'突出を除去しなければならない。現在のところ、3'突出の充填に使用可能な単一の酵素は存在しない。平滑化に必要な3'突出の除去により、タグ配列の長さが17〜19bpに短縮され、タグ配列に含まれる情報が減少する。
【0007】
したがって、公表されているLongSAGE(商標)のプロトコール(Sahaら、Nature Biotechnology 20:508-512、2002)では、MmeI消化後の末端を平滑化せず、3'突出を有する断片の間で連結する。これは、リンカータグ断片がそれ自体、適合する3'末端を有する他のリンカータグ断片としか連結しないことを意味する。このため、連結後形成されるダイタグはタグの無作為な結合の結果物とはならない。この手順により、得られるダイタグ中の各タグが表すものを理論的にそれるものとなり、LongSAGE(商標)の最終結果は各遺伝子の発現量を正確に反映しない可能性がある。したがって、LongSAGE(商標)は、遺伝子発現の正確な定量分析に適用できない。このことは、LongSAGE(商標)の重大な欠点となる。この理由で、現在のLongSAGE(商標)は、従来のSAGE(商標)のプロトコールによって得られた13bpのタグ配列に注釈付けを補助するためにしか使用されない。
【0008】
ESTおよび/またはゲノムDNA配列の情報が利用可能であるとすると、LongSAGE(商標)のプロトコールが、タグに対応する遺伝子の同定が成功する可能性を高めるため、それは一歩前進である。しかし、ESTまたはゲノムDNAのデータベースが利用できない生物にSAGE(商標)のプロトコールを適用するには、タグ配列をプライマーとして使用して、PCRによってより長いcDNAを回収し、またはオリゴヌクレオチドプローブとして使用して、ハイブリダイゼーションに基づく技術によって関連するcDNAライブラリーをスクリーニングが不可避となる。これらの目的で、19〜21bpのタグの長さは依然として短すぎて、タグ配列が由来する遺伝子をはっきりと同定することができない。
【0009】
したがって、発現の正確な定量分析を行うことができる、少なくとも長さ25bpの顕著に長いタグを単離する方法が早急に必要である。しかし、そのような方法は今までに得られていない。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、DNAの定められた位置から、長さが25bpを超える、好ましくは長さ26〜50bpの、最も好ましくは長さ26〜28bpの「タグ」配列を単離し、それによって、従来のSAGE(商標)解析により対応する遺伝子を確実に同定する効率が高まる方法を提供する。さらに、この方法によって得られる遺伝子発現プロファイルは、ダイタグがタグの無作為な結合によって作製されるため、LongSAGE解析から得られるものより理論的に正確である。以下、長さが25bpを超える新しいタグ断片を用いる、この改善されたSAGE(商標)の手順を「SuperSAGE」と称する。
【課題を解決するための手段】
【0011】
本明細書に記載の方法は、cDNAの定められた位置から、長さが25bpを超えるタグ配列を単離するためのIII型制限酵素の使用に基づいている。本発明では、タグという用語は、発現遺伝子を同定しうる特定のヌクレオチド配列を指す。タグの3'末端は、III型制限酵素の切断部位によって規定され、タグの5'末端は、cDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定される。
【0012】
「制限酵素」とは、二本鎖DNAにおける4〜8ヌクレオチドの特定の配列を認識し、それを切断しうるエンドヌクレアーゼの一般的な用語である。制限酵素は現在、I型、II型、III型およびIV型と呼ばれる異なる4群に分類される(Robertsら、2003、Nucleic Acids Res.31:1805-1812)。III型制限酵素は、メチラーゼおよびエンドヌクレアーゼのサブユニットからなり、標的DNA中の非回文構造のヌクレオチド配列を認識する複合体タンパク質である。エンドヌクレアーゼ活性では、それらは、2コピーの認識配列との機能的な協調作用を必要とし、さらに、この2コピーは、DNA二本鎖内で逆向きであることが必要である(Mueckeら、2001、J.Molec.Biol.312:687-698)。
【0013】
本発明では、上記のIII型制限酵素を、長さが25bpを超えるタグ配列の単離に使用する。そのようなIII型酵素の例は、http://rebase.neb.com/cgi-bin/azlist?re3に開示されている。本発明で使用する好ましいIII型酵素には、EcoPI、EcoP15Iなどがある。
【0014】
本発明の最も好ましい実施形態では、III型制限酵素はEcoP15Iである。III型制限酵素EcoP15Iは、標的DNA分子中の2つの非メチル化逆向き5'-CAGCAG-3'部位を認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を消化する。したがって、EcoP15Iは、従来の3'充填反応を用いて容易に平滑末端化される、5'末端が突出した断片をもたらす。
【0015】
他の酵素の好ましい例は、以下のような、cDNAを切断してそれぞれ長さが平均200bp〜300bpである断片にすることができる酵素である:
【0016】
認識配列 酵素名(市販されているもののみ)
CATG^ NlaIII、Hsp92II
^CATG FatI
C^TAG BfaI、MaeI、XspI
A^CGT HpyCH4IV、MaeII,
ACGT^ Tail、TscI
AG^CT AluI
T^CGA TaqI
^GATC BfuCI、Bsp143I、BstENII、DpnII、Kzo9I、MboI、NdeII、Sau3AI
GAT^C BstKTI,
G^TAC Csp6I
【0017】
最も好ましい酵素はNlaIIIである。
本発明はまた、25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグをも提供し、そのタグの3'末端は、III型制限酵素の切断部位によって規定され、そのタグの5'末端は、発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定される。
【0018】
本発明の好ましい実施形態では、上記に記載のように、III型酵素はEcoP15Iであり、他の制限酵素は、cDNAを切断してそれぞれ長さが平均200bp〜300bpである断片にすることができる酵素である。最も好ましい酵素はNlaIIIである。
【0019】
本発明のタグの例を、下記の表1に示す。これらのタグは、従来のSAGEタグまたはLongSAGEタグより長く、対応する遺伝子のより正確な同定を可能とする。
【0020】
本発明はさらに、2個のタグを含み、そのそれぞれが異なる発現遺伝子に由来するダイタグオリゴヌクレオチドを提供する。そのダイタグオリゴヌクレオチドは、下記のステップを含む方法によって生成される:
1)オリゴdT、およびIII型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、他の制限酵素で前記cDNAプールの消化を行うステップ;
2)上記のcDNAプールからポリA配列を含む断片を精製し、その断片をIII型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップ;
3)III型制限酵素で上記の断片を消化し、3'充填反応を行った後、得られたリンカーAを含む断片とリンカーBを含む断片とを連結するステップ;および
4)上記の連結した断片をステップ1)の他の制限酵素で消化してリンカー配列を切断して、ダイタグオリゴヌクレオチドを得るステップ。
【0021】
上記のステップで示したように、III型制限酵素の第1の認識部位は、mRNAからのcDNAの逆転写に使用するRTプライマーによって標的cDNA中に組み込まれ、III型制限酵素の第2の認識部位は、リンカー配列によって標的cDNA中に組み込まれている。
【0022】
RTプライマーは、配列5'-N18〜25-CAGCAG-T15〜25-3'によって定義され、N18〜25は、任意の18〜25ヌクレオチドの配列であり、配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない。RTプライマーの5'末端は、例えばビオチンで修飾してもよい。
【0023】
本発明のIII型制限酵素は、従来の3'充填反応を用いて容易に平滑末端化できる、5'末端が突出した断片をもたらす。すなわち、上記のステップ3)では、リンカーAを含む断片、およびリンカーBを含む断片を容易に平滑末端化することができ、それによって、タグのサイズをまったく縮小することなく前記断片の無作為な結合が可能となる。
【0024】
本発明の好ましい実施形態では、III型酵素はEcoP15Iであり、他の制限酵素は、NlaIIIなど、cDNAを切断してそれぞれ長さが平均200bp〜300bpの断片にすることができる酵素である。
【0025】
本発明のダイタグオリゴヌクレオチドを生成する方法では、2種のリンカー(リンカーAおよびリンカーB)を使用する。リンカーAおよびリンカーBは、互いに異なる二本鎖DNAである。連結に関与する二本鎖リンカー断片の1つの末端は、連結する配列と隣接したIII型制限酵素の認識配列を含む。例えば、III型制限酵素がEcoP15Iであり、他の制限酵素がNlaIIIであるとき、どちらのリンカーも、その第1鎖の3'末端に5'-CAGCAGCATG-3'配列を含む。下線の配列5'-CAGCAG-3'は、EcoP15Iの認識部位の1つを構成し、5'-CATG-3'配列は、NlaIII消化で生じた断片と連結する、3'が突出した一本鎖領域を構成する。
【0026】
連結に関与しない二本鎖リンカー断片の他の末端を、FITC(フルオレセインイソチオシアネート;第1鎖の5'末端)などの標識試薬、およびアミノ部分(第2鎖の3'末端)で修飾してもよい。
【0027】
したがって、リンカーは、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製する;
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
【0028】
上記で、DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40塩基のヌクレオチド配列であり、それらは互いに相補的であり、DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよい。
【0029】
本発明はさらに、上記のダイタグオリゴヌクレオチドの連結によって得られたポリヌクレオチドを提供する。各ダイタグオリゴヌクレオチドをクローン化し、PCRによって増幅することができる。そのポリヌクレオチドは、少なくとも2個のダイタグオリゴヌクレオチドを含み、好ましくは2〜200個のダイタグオリゴヌクレオチドを含む。本発明のダイタグオリゴヌクレオチドは、2個のタグの無作為な結合によって作製され、したがってポリヌクレオチドはまた、タグの無作為なコンカテマー化によって作製される。
【0030】
本発明はさらに、ポリヌクレオチドのヌクレオチド配列の解析、およびポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づく発現遺伝子の発現レベルの定量を含む遺伝子発現解析法を提供する。
【0031】
例えば、本発明の方法は、下記のステップを含む:
1)オリゴdT、およびIII型制限酵素の認識配列を含むRTプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、その後他の制限酵素でcDNAプールの消化を行うステップ;
2)上記のcDNAプールからポリA配列を含む断片を精製し、その断片を、III型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップ;
3)III型制限酵素で上記の断片を消化し、3'充填反応を行った後、得られたリンカーAを含む断片とリンカーBを含む断片とを連結するステップ;
4)上記の連結した断片をステップ1)の他の制限酵素で消化してリンカー配列を切断して、25個を超えるヌクレオチドからなる2個のタグを含み、発現遺伝子を同定しうるダイタグオリゴヌクレオチドを得るステップ;
5)ダイタグオリゴヌクレオチドを連結して、ポリヌクレオチドを生成するステップ;および
6)上記のポリヌクレオチドのヌクレオチド配列を解析し、ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて発現遺伝子の発現レベルを定量するステップ。
【0032】
この方法の好ましい実施形態では、III型酵素はEcoP15Iであり、他の制限酵素は、NlaIIIなど、cDNAを切断してそれぞれ長さが平均200bp〜300bpの断片にすることができる酵素である。リンカーに関して、上記に記載の二本鎖DNAを好ましくは使用することができる。
【0033】
本発明はさらに、25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグを単離するためのキットを提供し、それは下記の成分を含む:
a)配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない配列5'-N18〜25によって定義され、5'末端をビオチン化してもよいRTプライマー。
b)互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製するリンカーAおよびリンカーB:
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
【0034】
上記で、(1)のN30〜40および(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよい。
c)上記のリンカーAまたはリンカーBとハイブリダイズしうるプライマー。
【0035】
そのキットはまた、EcoP15I酵素などのIII型制限酵素、および/またはNlaIIIなどの他の制限酵素をも含む。そのキットはさらに、前記の成分に加えて、本発明の遺伝子発現解析を実施するのに必要な他の成分を含んでもよい。その例としては、標識試薬、緩衝液、マグネティックビーズなどがある。
【発明を実施するための最良の形態】
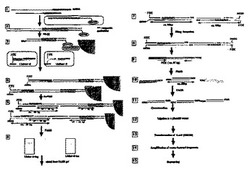
【0036】
図2を参照して、EcoP15IをIII型制限酵素として用いた本発明の好ましい実施形態を下記に説明する。
【0037】
III型制限改変酵素EcoP15Iは、標的DNA分子中の2つの非メチル化逆向き5'-CAGCAG-3'部位を認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を消化する。本発明は、cDNAの定められた位置から25〜28bpのタグ配列を単離するためのEcoP15I酵素の潜在的な適用すべてを指すものである。
【0038】
ビオチン化オリゴdTアンカープライマー(以下「RTプライマー」または逆転写プライマーと称する)を用いて、mRNAから二本鎖cDNAを合成する。このRTプライマーは、18〜25塩基の任意のヌクレオチド配列、および5'-CAGCAG-3'配列、その後に15〜25塩基のオリゴdT配列を含む。RTプライマー中に含まれる5'-CAGCAG-3'配列は、EcoP15Iの認識部位の1つを構成する(図2、ステップ1)。例えば、22ヌクレオチドの配列、および5'-CAGCAG-3'配列、その後に19個のdT配列を含むRTプライマーは、以下の通りである:
5'-CTGATCTAGAGGTACCGGATCCCAGCAGTTTTTTTTTTTTTTTTTTT-3'(配列番号1)。
【0039】
合成したcDNAを、配列モチーフ5'-CATG-3'を認識する制限エンドヌクレアーゼNlaIIIで消化する。RTプライマー配列(ビオチン標識)を含む消化断片だけを、ストレプトアビジン被覆マグネティックビーズによって捕捉する(図2、ステップ2および3)。
【0040】
二本鎖リンカー断片(46bp)を、マグネティックビーズによって捕捉した(ポリA配列を含む)cDNA断片の末端と連結する。連結に関与するこのリンカー断片の1末端は、第1鎖中の5'-CATG-3'と隣接した5'-CAGCAG-3'配列を含む(図2、ステップ4)。5'-CAGCAG-3'配列は、EcoP15Iの認識部位の1つを構成し、5'-CATG-3'配列は、NlaIII消化で生じた断片の粘着末端と連結する3'が突出した一本鎖領域を構成する。連結に関与しない二本鎖リンカー断片の他の末端を、FITC(フルオレセインイソチオシアネート;第1鎖の5'末端)、およびアミノ部分(第2鎖の3'末端)で修飾する。
【0041】
本発明では、2種のリンカーを使用する。例えば、リンカーAは、下記の2つのオリゴヌクレオチドをアニールすることによって作製される:
FITC-5'-TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATACAGCAGCATG-3'(配列番号2)、および
5'-CTGCTGTATTAAGCCTAGTTGTACTGCACCAGCAAATCCAAA-3'-NH2(配列番号3)。
【0042】
リンカーBは、下記の2つのオリゴヌクレオチドをアニールすることによって作製される:
FITC-5'-TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGCAGCAGCATG-3'(配列番号4)、および
5'-CTGCTGCGTACATCGTTAGAAGCTTGAATTCGAGCAGAAA-3'-NH2(配列番号5)。
【0043】
cDNAプールを2分割し、その一方をリンカーAと連結し、他方をリンカーBと連結することにより、「リンカーA連結cDNA」および「リンカーB連結cDNA」を得る。
【0044】
リンカーA連結cDNAでもリンカーB連結cDNAでも、ビーズと結合したDNA断片は、EcoP15Iで消化する(図2、ステップ5)。EcoP15Iは、配列5'-CAGCAG-3'の1対の逆向きモチーフを認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を切断する。消化後、2つの断片がビーズから遊離する。一方は、リンカーおよび27または28bpのタグ断片を含む断片(合計サイズ69または70bp)であり、他方は、二本鎖cDNA断片の中間に位置する様々なサイズの断片である。ポリA配列を含む断片は、マグネティックビーズに結合したままであり、以下の手順に関与しない。
【0045】
UV照射下でのFITCの蛍光によって、リンカーおよび27または28bpのタグ断片を含む69または70bpの断片を視覚化し、ポリアクリルアミドゲルから、ゲルの切り出しによってそれが容易に単離される。
【0046】
EcoP15Iは、従来の3'充填反応によって容易に平滑末端化される、5'末端が突出した断片をもたらし、それによって断片の無作為な結合が可能となる。したがって、それぞれリンカーA連結cDNAおよびリンカーB連結cDNAに由来する69または70bpの断片(リンカータグ断片)を、3'充填反応によってそれぞれ平滑末端化し、互いに連結して、2個のタグの無作為な結合によりダイタグを形成させる。末端を平滑化したcDNAタグ配列の側でのみ連結が行われるように、リンカー断片の3'末端をアミノ修飾により遮断する(図2、ステップ6、7)。
【0047】
得られたダイタグ分子をPCR増幅する(図2、ステップ9)。リンカー配列から設計したPCRプライマーの例を以下に示す:
ダイタグプライマー1E:ビオチン-5'-CAACTAGGCTTAATACAGCAGCA-3'(配列番号6)
ダイタグプライマー2E:ビオチン-5'-CTAACGATGTACGCAGCAGCA-3'(配列番号7)。
【0048】
上記のプライマーを用いたPCRによって得られるPCR産物の予想サイズは、約97bpである。
その約97bpのPCR産物をNlaIIIで消化し(図2、ステップ10)、それによって、約52bpのダイタグ断片が遊離する。これらの断片をゲルから回収し精製する。
【0049】
連結反応によってダイタグ断片をコンカテマー化する(図2、ステップ11)。アガロースゲル電気泳動によって、コンカテマーを分離する。500bpより大きい断片をゲルから溶出させ回収する。
【0050】
サイズ分離したコンカテマー断片を、SphIで予め消化しウシ小腸ホスファターゼで処理した適当なプラスミドベクターと連結し(図2、ステップ12)、そのプラスミドで大腸菌(E.coli)を形質転換する(図2、ステップ13)。
【0051】
そのプラスミドの挿入断片をPCR増幅する(図2、ステップ14)。
そのPCR産物を直接シーケンシングする(図2、ステップ15)。一連の約44bpのダイタグ配列は、NlaIII認識配列のCATGと隣接する。この約52(44+8)bpの配列情報から、各cDNAの定められた位置から単離された2つの26〜28bpのタグ配列が得られる。
【0052】
試料中の26bpのタグの数をカウントした後、最初のSAGE(商標)のプロトコールに記載のように、遺伝子発現プロファイルを得ることが可能となる。本発明のダイタグは平滑末端タグの無作為な結合によって形成されるため、その結果は、各遺伝子の発現を正確に反映する。
【0053】
26bpのタグ配列は、タグが由来する遺伝子を一意的に同定するのに十分な情報を含む。26bpのDNA配列の情報内容を用いると、対応する遺伝子のインシリコ(in silico)での同定が促進される。全Genbank配列に対して26bpのタグ配列をBLAST検索すると、タグが由来する遺伝子と正しい整合を示す(図3)。
【0054】
記載した方法をDNA配列データが利用できない生物に適用する場合、26bpのタグ配列を3'-RACE用のPCRプライマーとして直接使用して、cDNAの3'領域を回収することができる。そのようなcDNA配列をBLAST検索に使用して、その遺伝子を同定することができる(図3)。
【0055】
SuperSAGEによって明らかとなった試料間の遺伝子発現の違いを実証するために、26bpのタグ配列を用いた3'-RACEをRT-PCRとして直接行うことにより、メッセージの量を定量することができる(図3)。
【0056】
26bpのタグ配列は、「RNA干渉」(RNAi)を引き起こすのに必要なDNA配列の最小サイズ(21bp)(Elbashirら、Nature 411:494-498、2001)より長い。したがって、タグ配列を含む二本鎖RNAを、タグに対応する遺伝子をノックアウトする機能解析に直ちに使用することができる。これは、SuperSAGEによって行われる遺伝子発現解析を、26bpのタグの単離を用いた遺伝子機能解析に直接結び付けることができることを意味する。
【0057】
植物種における適用では、上記に記載の3'-RACE断片を植物ウイルスベクター中にクローン化し、「ウイルス誘導遺伝子サイレンシング(VIGS)」(Baulcombe、Curr.Opin.Plant Biol.2:109-113、1999)に使用することができ、したがって、前記SuperSAGE法は、同様に植物でも遺伝子発現解析を遺伝子機能解析と結び付ける。
[実施例]
【0058】
下記の実施例を参照して、本発明をより詳細に説明するが、本発明の範囲は、これらの実施例に限定されるものではない。
【実施例1】
【0059】
原理の証明として、26および27bpのタグ配列を、イネの擬似病斑突然変異体IB2020(Oryza sativa品種名かけはし(Kakehashi))の葉から、上記に記載の方法によって単離した。
【0060】
1)mRNAの調製
従来のRNA単離の方法によって、イネの葉身から総RNAを単離した。このRNAから、「mRNA精製キット(mRNA Purification Kit)」(Amersham Pharmacia)を用いてmRNA5μgを単離した。そのmRNAを、DEPC水29μl中に溶解させ、供給源物質として使用した。
【0061】
2)オリゴdTプライマーを用いたcDNA合成
「cDNA合成システム(cDNA Synthesis System)」(Invitrogen)を用いてこのmRNAを逆転写して、酵素EcoP15Iの認識配列である5'-CAGCAG-3'モチーフを含む下記の逆転写プライマーを用いた一本鎖cDNAを得た。
逆転写プライマー:5'-CTGATCTAGAGGTACCGGATCCCAGCAGTTTTTTTTTTTTTTTTTTT-3'(配列番号1)
同じキットを用いて、この産物を二本鎖cDNAに転換し、エタノール沈殿させ、LoTE緩衝液(3mM Tris-HCl pH7.5、0.2mM EDTA)20μl中に溶解させた。
【0062】
3)NlaIII消化
0.1mg/mlのBSAを含む1×NEB緩衝液4(New England BioLabs;NEB)中にNlaIII50単位(NEB)を含む反応緩衝液200μl中で、得られた二本鎖cDNA(20μl)を37℃で90分間消化した。消化後、TE平衡化フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)でcDNAを抽出し、エタノール沈殿させ、LoTE緩衝液20μl中に溶解させた。
【0063】
2本のエッペンドルフ(Eppendorf)チューブそれぞれに、ストレプトアビジンマグネティックビーズ懸濁液(Streptavidin Magnesphere Paramagnetic particle、Promega)1mlを移した。マグネティックビーズを、1×B&W緩衝液(5mM Tris-HCl pH7.5、0.5mM EDTA、1M NaCl)200μlで、磁性分離台(Magnesphere Technology Magnetic Separation Stand)を用いて1回洗浄した。洗浄後、1×B&W緩衝液100μl、および上記のステップから得られたcDNA10μl、および水90μlを各チューブに添加し、混合し、室温で30分間インキュベートした。マグネティックビーズと結合したcDNAを1×B&W緩衝液で3回洗浄し、LoTE緩衝液で3回洗浄した。
【0064】
4)リンカー連結
業者に委託して、以下のオリゴヌクレオチドを合成した(Qiagen)。
リンカーA1:FITC-5'-TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATACAGCAGCATG-3'(配列番号2)
リンカーA2:5'-CTGCTGTATTAAGCCTAGTTGTACTGCACCAGCAAATCCAAA-3'-NH2(配列番号3)
リンカーB1:FITC5'-TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGCAGCAGCATG-3'(配列番号4)
リンカーB2:5'-CTGCTGCGTACATCGTTAGAAGCTTGAATTCGAGCAGAAA-3'-NH2(配列番号5)
【0065】
リンカーA2およびリンカーB2の5'末端は、T4ポリヌクレオチドキナーゼ(NEB)によってリン酸化した。リンカーA1とリン酸化リンカーA2をアニールすることによってリンカーAを、リンカーB1とリン酸化リンカーB2をアニールすることによってリンカーBを調製した。リンカーAもリンカーBも、EcoP15I認識配列(5'-CAGCAG-3')を有する。
【0066】
マグネティックビーズと結合したcDNAを含む2本のチューブそれぞれに、LoTE 17μl、5×リガーゼ緩衝液(Invitrogen)3μlおよびリンカーAまたはリンカーB 1μgを添加した。混合した後、チューブを50℃で2分間インキュベートし、室温で15分間放置した。続いて、T4 DNAリガーゼ2μl(5単位/μl、Invitrogen)をチューブに添加し、16℃で90分間インキュベートした。リンカー連結の後、1×B&Wで4回、LoTE緩衝液で3回、滅菌蒸留水で1回ビーズを洗浄した。
【0067】
5)EcoP15I消化
マグネティックビーズ上のリンカー連結cDNAを、反応混合物(10mM Tris-HCl pH8.0、10mM KCl、10mM MgCl2、0.1mM EDTA、0.1mM DTT、5μg/ml BSA、2mM ATP)100μl中で、EcoP15I 10単位で消化した。チューブを37℃で90分間インキュベートした。
【0068】
6)リンカータグ断片の精製
EcoP15I消化後、チューブを磁性台上に置いた。ストレプトアビジン被覆マグネティックビーズによるビオチン化された最外部の3'末端断片の除去後、リンカータグ断片を含む非結合分画を収集し、新しいチューブに移した。フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で収集した緩衝液を抽出し、エタノール沈殿させ、LoTE緩衝液10μl中に溶解させ、8%ポリアクリルアミドゲル電気泳動によって分離した。隣接するレーン中に、SYBRグリーン(FMC)で予め染色したサイズマーカー(20bpラダー、TAKARA)を添加した。電気泳動後、ゲルをUV照明器上に置き、FITCが媒介するその蛍光によって、サイズが約69bpのリンカータグ断片を視覚化した(図4)。リンカーとリンカーの連結体(約90bp)および単一のリンカー断片(46bp)に由来するであろうさらなる2断片も視覚化した。FITCの蛍光の示すシグナルが弱すぎる場合、ゲルをSYBRグリーン(FMC)で染色して、リンカータグ断片を視覚化することができる。約69bpのリンカータグ断片をゲルから切り出し、注射針で底に針穴を開けた0.5mlチューブ中に移した。その0.5mlチューブを2mlチューブの内部に入れ、15000rpmで2分間遠心した。2mlチューブの底で収集された小さなゲル片に、LoTE緩衝液(300μl)を添加し、それを37℃で2時間インキュベートし、その後65℃で15分間インキュベートした。ゲルの懸濁液をSpinXカラム(Coaster)に移し、15000rpmで2分間遠心した。回収された溶液を、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で1回抽出し、エタノール沈殿させ、LoTE緩衝液8μl中に溶解させた。
【0069】
7)タグ断片の平滑化
リンカータグ断片の5'突出を、キット(Blunting High;TOYOBO)を用いて、DNAポリメラーゼで充填した。リンカータグ断片を含む溶液(8μl)に、反応緩衝液(KOD緩衝液;TOYOBO)1μl、およびKODポリメラーゼ(TOYOBO)1μlを添加し、それを72℃で2分間インキュベートした。
【0070】
8)ダイタグ形成のための連結
リンカーAおよびリンカーBに由来するリンカータグ断片を連結して、ダイタグ断片を形成させた。同量(2μl)の平滑末端化したリンカーAタグ溶液とリンカーBタグ溶液を混合し、LoTE緩衝液6μlおよび連結反応用混合液(Ligation High、TOYOBO)10μlを添加した。連結反応溶液を16℃で4時間〜1晩インキュベートした。
【0071】
9)ダイタグPCR
得られたダイタグ溶液を5倍および10倍に希釈し、ダイタグPCR用の鋳型として使用した。希釈したダイタグ鋳型1μl、Taqポリメラーゼ(AmpliTaq Gold;Applied Biosystems)3単位、1×AmpliTaq Gold緩衝液およびPCRプライマーを含む溶液50μlをそれぞれ含む288(=96×3)本の0.2mlチューブ中でPCR反応を行った。PCRプライマーの5'末端は、以下の通りにビオチン化した(Qiagenにより、業者を介して合成した)。
【0072】
ダイタグプライマー1E:ビオチン-5'-CAACTAGGCTTAATACAGCAGCA-3'(配列番号6)
ダイタグプライマー2E:ビオチン-5'-CTAACGATGTACGCAGCAGCA-3'(配列番号7)
PCRは、最初の95℃、12分の変性、その後の94℃、40秒および60℃、40秒の反応27〜29回からなった。増幅したダイタグ断片の予想サイズは約97bpであった(図5)。
【0073】
10)ダイタグPCR産物の精製
ダイタグPCR産物(チューブ約300本)を、10mlプラスチックチューブ中に一緒にした。フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)抽出およびエタノール沈殿後、それをLoTE緩衝液100μl中に溶解させた。このPCR産物を、1.5%の低融点アガロース(SeaPlaque、FMC)ゲル上で泳動し、約97bpの断片をゲルから切り出し、それをQiagenゲル抽出キット(Qiagen)で精製した。
【0074】
11)NlaIIIでの消化によるリンカー除去
LoTE緩衝液121μl中に溶出させた約97bpの精製断片を、0.1mg/mlのBSAを含む1×NEB緩衝液4(NEB)中で、NlaIII(NEB)120単位で消化した。ゲル電気泳動で消化されたことを確認した(サイズが52bp、22bpおよび23bpの3種の断片が視覚化された、図6)後、リンカー断片を除去するために、ストレプトアビジンマグネティックビーズで、消化液を室温で30分間処理した。チューブ中のマグネティックビーズを磁性台によって収集した後、その上清を新しいチューブに移し、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)でDNAを抽出し、エタノール沈殿させ、LoTE緩衝液10μl中に溶解させた。
【0075】
12)52bpのダイタグ断片の精製
回収したDNAを、12%ポリアクリドアミドゲルで電気泳動した。断片を視覚化するために、ゲルをSYBRグリーン(FMC)で染色した。サイズが約52bpの断片をUV照明器上でゲルから切り出した。上記に記載のように、ゲルからDNAを溶出させた。ストレプトアビジンマグネティックビーズで、ゲルから溶出させたDNAを室温で30分間処理した。上清を収集し、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で抽出し、エタノール沈殿させ、LoTE緩衝液7μl中に溶解させた。
【0076】
13)コンカテマー形成
ダイタグ断片をコンカテマー化するために、ダイタグ溶液7μl、5×リガーゼ緩衝液2μl、およびT4リガーゼ(Invitrogen)5単位を混合し、16℃で1〜4時間インキュベートした。その連結溶液を65℃で10分間インキュベートし、1%アガロースゲルに添加した(図7)。サイズが500bpを超えるDNA断片をゲルから切り出し溶出させた。精製したコンカテマーをLoTE緩衝液6μl中に溶解させた。
【0077】
14)ベクタークローン化
クローニングベクター(pGEM3Z、Promega)5μgをSphIで消化し、ウシ小腸アルカリホスファターゼ(CIAP)で処理した。コンカテマーをクローン化するために、この消化したpGEM3ZベクターとダイタグコンカテマーをT4リガーゼ(Invitrogen)を用いて連結した。連結反応を行ってから4時間後、反応溶液をフェノール/クロロホルム/イソアミルアルコール(25:24:1)で抽出し、エタノール沈殿させた。そのペレットを70%エタノールで4回洗浄して塩を完全に除去し、滅菌蒸留水10μl中に溶解させた。
【0078】
15)形質転換
エレクトロポレーション用コンピテント大腸菌細胞(DH10B、Invitrogen)を、ダイタグコンカテマーを含むプラスミドのクローニングに使用した。チューブ中に、コンピテント細胞懸濁液40μlと、精製した連結DNA1μlを氷上で混合し、エレクトロポレーション用キュベット(間隔0.1cm;BioRad)中に移した。エレクトロポレーションの条件は、コンピテント細胞の製造業者が推奨する通りであった(2.5kV、25μFD、100Ω)。エレクトロポレーション後、SOC培地(Invitrogen)1mlを添加し、その溶液を37℃で1時間インキュベートした。得られた大腸菌の懸濁液を100μg/mlのアンピシリン、20μg/mlのX-galおよび0.1mMのIPTGを含むLB培地プレート上に播いた。プレートを37℃で14時間インキュベートした。
【0079】
16)コロニーPCRおよびシーケンシング
プレート上に生じた白色コロニーを爪楊枝で拾い上げ、1×PCR緩衝液、0.4mM dNTP、0.05単位/μl Taqポリメラーゼ(TAKARA)、2pmol/μl M13-20プライマーおよび2pmol/μl M13RVプライマーを含むPCR混合液中に再懸濁させた。プラスミド中のクローン化断片をPCRで増幅し、断片のサイズをアガロースゲル電気泳動によって確認した(図8)。増幅したPCR断片を、PCR精製キット(Qiagen)を用いて精製した。
【0080】
17)コンカテマーのシーケンシング
BigDyeターミネーター(Applied Biosystems)およびM13-20プライマーを用いて、精製したPCR断片(コンカテマー)の配列を決定した。シーケンシング反応の後、「Montage SEQ96 Sequencing Reaction Cleanup Kit」(Millipore)によってDNAを精製した。RISA384キャピラリーDNAシークエンサー(Shimadzu)によってDNA配列を解析した。クローン化した断片のDNA配列の例を図9に示す。2つの26または27bpの断片を含む一連の52〜54bpのダイタグは、CATGというNlaIII部位によって境界付けられている。
【0081】
18)データ解析
ジョンスホプキンス大学(Johns Hopkins University)により供給されたSAGE(商標)2000ソフトウェアを用いて、コンカテマーのDNA配列から26bpのタグ配列を抽出し各タグの数をカウントした。いくつかのクローンについてシーケンシングした後、表1に示す遺伝子発現プロファイル作成を行った。
【0082】
【表1】

【0083】
したがって、cDNAのNlaIII部位と隣接する26または27bpの配列をイネ葉から単離することに成功した。本明細書に記載の実験手順を適用することによって、26または27bpの配列を含む認識タグをどんなcDNAからも得ることができる。26または27bpの配列を含むタグの単離に基づくこのプロトコールは、18〜21bpの配列を含むタグを使用する(「LongSAGE(商標)」として知られる)ほとんどの進歩型SAGE(商標)の手順を超えるかなりの質的な改善を表すものである。
【0084】
SuperSAGE法では、EcoP15I酵素が、タグ中に、DNAポリメラーゼによって容易に平滑末端化される5'突出末端を生じさせる。したがって、平滑末端化したタグの無作為な結合によってダイタグ形成が行われ、それによって、タグの数がタグと対応する遺伝子の発現を正確に表すことが確実となる。この特性は、LongSAGE(商標)のプロトコールにおいて、適合する2bpの3'突出末端を含む21bpのタグの間でしかダイタグが形成されないときには利用できない。Long SAGE(商標)でタグの無作為な結合を確実に行うためには、3'突出領域を除去することによって、タグを平滑末端化すべきである。この結果、タグのサイズが2bp短縮され、それによってタグの情報内容が減少する。したがって、本明細書に記載のSuperSAGE法は、タグの情報内容がより多く、遺伝子発現プロファイルをより正確に表す点から、LongSAGE(商標)法より有利である。
【実施例2】
【0085】
SuperSAGEのタグのサイズが26bpであることによって、遺伝子に対するタグの非常に信頼性の高い注釈付けが可能となることを実証するために、以下の実験を行った。
【0086】
26bpのタグ30個を表1から無作為に選択した。このDNA配列を、タグのサイズがそれぞれ20、18および15bpとなるように、3'末端から切断した。15bpのタグのサイズは、従来のSAGE(商標)で使用するものに対応する。18bpまたは20bpのタグは、LongSAGE(商標)のタグから、それぞれ末端平滑化を行ったものおよび末端平滑化を行わないものに対応する。異なるサイズのタグそれぞれを用いて、130,000を超える種由来のDNA配列を表すGenbank中に寄託されているDNA配列全体からBLAST検索を行った。所与のサイズのタグと完全な整合を示す、DNA配列を含んだ種の数をカウントし、タグ配列30個全体から種の平均数と最大数を得た。表2にその検索結果の概要を示す。
【0087】
【表2】
【0088】
いくつかのDNA配列がタグ配列と完全な整合を示し、タグのサイズが増大すると、遺伝子に対するタグの注釈付けの不明確性が低下した。
【0089】
従来のSAGE(商標)のタグ(15bp)は、平均で4を超える種の、最大で9種のDNA配列と整合した。タグ30個すべてが2つ以上の種と相関した(表2)。18bpのタグは、平均で1.8種と、最大で7種と整合した。30個のうち10個のタグが2つ以上の種と相関した。20bpのタグは、平均で1.16種と、最大で4種と整合した。30個のうち3個のタグしか2つ以上の種と相関せず、このことから最初のSAGE(商標)のタグの長さ(15bp)より大きく改善されたことが示唆される。しかし、20bpのタグは、末端を平滑化せずにリンカータグが連結するときしか抽出することができず、その結果、この方法の最終結果が必ずしも正確な遺伝子発現を表すわけではないことに留意されたい。SuperSAGE法の26bpのタグは、平均で1.06種と整合し、最大では2種としか整合しなかった。30個のうち2個のタグしか2つ以上の種と相関しなかった。これらの結果は、26bpのDNA配列中の情報内容がタグの遺伝子注釈付けの効率において大きな改善をもたらすことを明らかに示すものである。
【0090】
26bpのタグは、平均で1種のみのDNA配列と整合し、ほとんどの場合で特定の種の単一遺伝子と整合した。したがって、タグ配列の注釈付けをほとんど完全に実施することができる。EST配列データベースに対して、ならびにゲノム配列全体に対してSuperSAGEでのタグの注釈付けを行うことができる。
【0091】
SuperSAGEにおける26bpのタグの情報内容が大きいことにより、2種の生物の遺伝子発現解析を同時に行うことが可能となる。一例として、記載の方法を、真菌のイネいもち病菌(Magnaporthe grisea)によって引き起こされるいもち病に感染したイネ植物の遺伝子発現プロファイルの研究に適用した。いもち病に感染したイネ葉から合計12,119個のタグを単離した後、イネおよびイネいもち病菌(M.grisea)のゲノム配列すべてに対するBLAST検索によって各タグを注釈付けた。予想通り、大多数のタグはイネ遺伝子に注釈付けられ(表3)、その一方で74個のタグがイネの配列と整合しなかったが、いもち配列と整合した(表4)。
【0092】
【表3】
【0093】
【表4】
【0094】
SuperSAGEによって得られた結果を確認するために、以下のアンカーオリゴdTプライマーを用いて、上記と同じRNAから逆転写によりcDNAを合成した:
5'-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3'(配列番号8)。
【0095】
このcDNAを鋳型として用いて、ハイドロフォビン、ヌクレオシド二リン酸キナーゼおよび60Sリボソームタンパク質の26bpのタグ(表4)を、以下のプライマーと一緒に3'-RACEに使用した:
5'-GGCCACGCGTCGACTAGTAC-3'(配列番号9)。
【0096】
3種の遺伝子すべてについて特異的なPCR産物が得られた(図10、左)。模擬接種したいもち感受性品種(かけはし)、いもち接種した感受性品種(かけはし)、模擬接種したいもち抵抗性品種(ヒメノモチ(Himenomochi))およびいもち接種した抵抗性品種(ヒメノモチ)におけるRT-PCRに同じプライマーの組を使用した。予想通り、いもち真菌を接種した感受性品種からのみ、3種のいもち遺伝子に対応するPCR産物が増幅され(図10、右)、このことから、SuperSAGEの注釈付けが真正であることが実証された。
【0097】
本明細書で例示するように、1試料中での2つの種の遺伝子発現の定量分析は、以前の技術で行うことができなかった。遺伝子発現解析(「SuperSAGE」)に26bpのタグを使用すると、宿主と病原体など2種以上の相互作用する生物の同時遺伝子発現解析が大いに促進される。
【実施例3】
【0098】
DNA配列データを利用できない生物の遺伝子発現解析に、26bpのタグを用いるSuperSAGEを使用することができることを実証するために、以下の実験を行った。ジャガイモ疫病菌(Phytophthora infestans)由来のタンパク質エリシターINF1(Kamounら、Mol.Plant-Microbe Interact.10:13-20、1997)または水で処理した植物種ベンサミアナタバコ(Nicotiana benthamiana)の遺伝子発現解析に、SuperSAGEを適用した。エリシターまたは水(冠水)を浸透させてから1時間後にその葉を収集し、それからRNAを単離した。
【0099】
それぞれの試料から5000個を超えるタグを単離することに成功した(表5)。表5中に列挙した、最も多数認められたタグ10個を直接3'-RACEに使用し、得られたcDNAの配列を決定した。cDNAのBLAST検索から、表5に示すタグと対応する遺伝子が同定された。
【0100】
【表5】
【0101】
INF1処理試料および冠水処理試料における各タグの頻度を比較することによって、2つの試料中で示されるタグに統計上有意な差があることが確認された(表6)。これらのタグを直接使用して3'-RACEを行い、回収したcDNAを使用してBLAST検索を行った。このことにより、ほとんどのタグを注釈付けることができた。
【0102】
【表6】
【0103】
26bpのタグ配列を、アンカープライマーと一緒にRT-PCRに使用して、表6に示すSuperSAGEの結果を確認した。RT-PCRの結果(図11)は、SuperSAGEの結果がINF1処理試料と冠水処理試料との遺伝子発現の差を正確に反映することを明らかに示すものである。
【0104】
各遺伝子発現の動態研究のためのRT-PCRに、同じ26bpのタグプライマーを容易に使用することができる(図12)。試験遺伝子4種(葉緑素a/b結合タンパク質、光化学系IIタンパク質、ホスホグリセリン酸キナーゼ、およびATP合成酵素の遺伝子)の発現がINF1で処理してから15分後に遮断されたことが判明した。
【0105】
この実施例は、DNA配列データを利用できない生物における遺伝子発現解析についてのSuperSAGEの大きな潜在的可能性を実証するものである。この潜在的可能性は、タグが特異的なPCRプライマーに十分なサイズであり、SuperSAGEによって得られる遺伝子発現プロファイルが実際の遺伝子発現を正確に表すことに由来する。
【図面の簡単な説明】
【0106】
【図1】従来のSAGE(商標)のプロトコール(Velculescuら、Science 270:484-487、1995)の概略図である。
【図2A】EcoP15Iを用いて26bpのcDNAのSuperSAGEタグを単離する概略的な手順を示す図である。
【図2B】EcoP15Iを用いて26bpのcDNAのSuperSAGEタグを単離する概略的な手順を示す図である。
【図3】EcoP15Iによって得られた26bpのタグを用いたSuperSAGE解析の適用の概要を示す図である。
【図4】FITCの蛍光によって視覚化した、EcoP15I消化(ステップ6、図2)後の産物の電気泳動の例を示す図である。各断片の構造をこのパネルの右側に示す。この例では、イネ葉由来のmRNA分子を鋳型として用いてcDNAを合成した。
【図5】図2のステップ9からのPCR産物の電気泳動(PAGE)の例を示す図である。予想されるPCR産物のサイズは約97bpである。
【図6】PCR産物のNlaIII消化(ステップ10)から得られた断片の電気泳動(PAGE)の例を示す図である。ダイタグのサイズは約52bpである。
【図7】ダイタグのコンカテマー化断片(ステップ11)の電気泳動の例を示す図である。
【図8】コロニーPCR産物(ステップ14)の電気泳動の例を示す図である。
【図9】クローン化したコンカテマー中に含まれているDNA配列(ステップ15)の例を示す図である。
【図10】26bpのタグ配列をPCRプライマーとして使用する、イネいもち病菌感染イネ葉から単離したRNAを用いたRT-PCRの結果を示す図である。
【図11】ジャガイモ疫病菌由来のINF1エリシタータンパク質で、または対照である水で処理したベンサミアナタバコにおけるRT-PCRの結果を示す図である。
【図12】SuperSAGEによって同定された遺伝子4種の遺伝子発現のRT-PCR動態研究を示す図である。
【配列表フリーテキスト】
【0107】
配列番号1:RTプライマー
配列番号2:リンカーAの第1鎖
配列番号3:リンカーAの第2鎖
配列番号4:リンカーBの第1鎖
配列番号5:リンカーBの第2鎖
配列番号6:ダイタグプライマー(順向)
配列番号7:ダイタグプライマー(逆向)
配列番号8:RTプライマー
配列番号9:3'-RACE用のプライマー
配列番号10-250:合成DNA(タグ配列)
【技術分野】
【0001】
本発明は、分子生物学および遺伝子発現解析の分野に関する。これに関連して、本発明は、転写産物の定められた領域を単離するためのIII型制限酵素の使用に関する。
【背景技術】
【0002】
遺伝子工学による遺伝子の構造および発現の改変は、様々な医学、薬学および農学への適用に莫大な潜在的可能性をもたらす。標的遺伝子を検出し選択するために、様々な発生段階、様々な環境条件下(ストレスなど)の細胞、組織、臓器または生物におけるスクリーニングにおいて、発現プロファイリングが広範に適用されている。遺伝子発現解析で最も強力な技術の1つは、Velculescuら、Science 270:484-487、1995によって開発されたSAGE(商標)(遺伝子発現逐次解析)である。
【0003】
最初のSAGE(商標)のプロトコール(図1)では、定められた細胞、組織または臓器由来のメッセンジャーRNA(mRNA)のプールを供給源物質として使用し、それから、ビオチン化オリゴdTプライマーを用いて逆転写酵素により相補的DNA(cDNA)を逆転写する。得られた一本鎖cDNAを二本鎖DNAに転換し、配列モチーフ5'-CATG-3'を認識する制限酵素NlaIIIで消化する。ストレプトアビジン被覆マグネティックビーズを使用して、二本鎖cDNAの3'末端の断片を回収する。cDNAを2分割する。次いで、2種のリンカー(リンカー1およびリンカー2)を、分離したそれぞれのcDNAと連結する。リンカーは、配列モチーフ5'-GGGAC-3'を含む。これはII型制限酵素BsmFIの認識部位であり、この酵素は認識部位から3'方向に13bp離れた部位を切断する。したがって、リンカーの付いたcDNAをBsmFIで処理することにより、「タグ」配列と呼ばれるcDNAの13bpの断片が、リンカー断片と一緒に(「リンカータグ」断片)遊離される。次いで、分離した2つのcDNAから得られた2つのリンカータグ断片を、2つのタグの位置が隣り合ってダイタグ(ditag)を形成するような形で互いに連結し、その後リンカー配列に特異的なプライマーを用いてPCRを行う。NlaIII消化によりリンカー断片を除去した後、ダイタグをコンカテマー化し、適当なプラスミドにクローン化する。プラスミド挿入物のシーケンシングから、4bpの5'-CATG-3'配列と隣接した一連の9bpのタグが明らかとなる。13bpのタグ配列を使用すると、利用可能な発現配列タグ(EST)データベースを調べることによって、多くの場合でタグ配列が由来する遺伝子を同定することが可能である。したがって、数千個のタグの配列を決定した後、試料中の各タグの数をカウントし、さらにそれに対応する遺伝子を同定することも可能である。
【0004】
したがって、上記に記載のSAGE(商標)のプロトコールは、包括的遺伝子発現を研究するのに有効な方法である。しかし、そのタグ配列の限定されたサイズ(13bpのみ)は、そのタグが由来する遺伝子を明確に同定するには十分ではない。単一のタグ配列が複数の異なるEST配列と対応する可能性があり、さらなる解析を混乱させる可能性がある。
【0005】
その状況を改善するために、II型制限エンドヌクレアーゼMmeIを使用する、いわゆる「LongSAGE(商標)」のプロトコールが最近開発された。(最初のSAGE(商標)のプロトコールでのBsmFIの代わりに)MmeIを使用することで、19〜21bpの長さのタグを回収することが可能となる(Sahaら、Nature Biotechnology 20:508-512、2002)。
【0006】
MmeI消化により、3'突出末端(2塩基突出)が生じる。MmeI消化の後からダイタグ連結前までに平滑末端化するためには、3'突出を除去しなければならない。現在のところ、3'突出の充填に使用可能な単一の酵素は存在しない。平滑化に必要な3'突出の除去により、タグ配列の長さが17〜19bpに短縮され、タグ配列に含まれる情報が減少する。
【0007】
したがって、公表されているLongSAGE(商標)のプロトコール(Sahaら、Nature Biotechnology 20:508-512、2002)では、MmeI消化後の末端を平滑化せず、3'突出を有する断片の間で連結する。これは、リンカータグ断片がそれ自体、適合する3'末端を有する他のリンカータグ断片としか連結しないことを意味する。このため、連結後形成されるダイタグはタグの無作為な結合の結果物とはならない。この手順により、得られるダイタグ中の各タグが表すものを理論的にそれるものとなり、LongSAGE(商標)の最終結果は各遺伝子の発現量を正確に反映しない可能性がある。したがって、LongSAGE(商標)は、遺伝子発現の正確な定量分析に適用できない。このことは、LongSAGE(商標)の重大な欠点となる。この理由で、現在のLongSAGE(商標)は、従来のSAGE(商標)のプロトコールによって得られた13bpのタグ配列に注釈付けを補助するためにしか使用されない。
【0008】
ESTおよび/またはゲノムDNA配列の情報が利用可能であるとすると、LongSAGE(商標)のプロトコールが、タグに対応する遺伝子の同定が成功する可能性を高めるため、それは一歩前進である。しかし、ESTまたはゲノムDNAのデータベースが利用できない生物にSAGE(商標)のプロトコールを適用するには、タグ配列をプライマーとして使用して、PCRによってより長いcDNAを回収し、またはオリゴヌクレオチドプローブとして使用して、ハイブリダイゼーションに基づく技術によって関連するcDNAライブラリーをスクリーニングが不可避となる。これらの目的で、19〜21bpのタグの長さは依然として短すぎて、タグ配列が由来する遺伝子をはっきりと同定することができない。
【0009】
したがって、発現の正確な定量分析を行うことができる、少なくとも長さ25bpの顕著に長いタグを単離する方法が早急に必要である。しかし、そのような方法は今までに得られていない。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明は、DNAの定められた位置から、長さが25bpを超える、好ましくは長さ26〜50bpの、最も好ましくは長さ26〜28bpの「タグ」配列を単離し、それによって、従来のSAGE(商標)解析により対応する遺伝子を確実に同定する効率が高まる方法を提供する。さらに、この方法によって得られる遺伝子発現プロファイルは、ダイタグがタグの無作為な結合によって作製されるため、LongSAGE解析から得られるものより理論的に正確である。以下、長さが25bpを超える新しいタグ断片を用いる、この改善されたSAGE(商標)の手順を「SuperSAGE」と称する。
【課題を解決するための手段】
【0011】
本明細書に記載の方法は、cDNAの定められた位置から、長さが25bpを超えるタグ配列を単離するためのIII型制限酵素の使用に基づいている。本発明では、タグという用語は、発現遺伝子を同定しうる特定のヌクレオチド配列を指す。タグの3'末端は、III型制限酵素の切断部位によって規定され、タグの5'末端は、cDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定される。
【0012】
「制限酵素」とは、二本鎖DNAにおける4〜8ヌクレオチドの特定の配列を認識し、それを切断しうるエンドヌクレアーゼの一般的な用語である。制限酵素は現在、I型、II型、III型およびIV型と呼ばれる異なる4群に分類される(Robertsら、2003、Nucleic Acids Res.31:1805-1812)。III型制限酵素は、メチラーゼおよびエンドヌクレアーゼのサブユニットからなり、標的DNA中の非回文構造のヌクレオチド配列を認識する複合体タンパク質である。エンドヌクレアーゼ活性では、それらは、2コピーの認識配列との機能的な協調作用を必要とし、さらに、この2コピーは、DNA二本鎖内で逆向きであることが必要である(Mueckeら、2001、J.Molec.Biol.312:687-698)。
【0013】
本発明では、上記のIII型制限酵素を、長さが25bpを超えるタグ配列の単離に使用する。そのようなIII型酵素の例は、http://rebase.neb.com/cgi-bin/azlist?re3に開示されている。本発明で使用する好ましいIII型酵素には、EcoPI、EcoP15Iなどがある。
【0014】
本発明の最も好ましい実施形態では、III型制限酵素はEcoP15Iである。III型制限酵素EcoP15Iは、標的DNA分子中の2つの非メチル化逆向き5'-CAGCAG-3'部位を認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を消化する。したがって、EcoP15Iは、従来の3'充填反応を用いて容易に平滑末端化される、5'末端が突出した断片をもたらす。
【0015】
他の酵素の好ましい例は、以下のような、cDNAを切断してそれぞれ長さが平均200bp〜300bpである断片にすることができる酵素である:
【0016】
認識配列 酵素名(市販されているもののみ)
CATG^ NlaIII、Hsp92II
^CATG FatI
C^TAG BfaI、MaeI、XspI
A^CGT HpyCH4IV、MaeII,
ACGT^ Tail、TscI
AG^CT AluI
T^CGA TaqI
^GATC BfuCI、Bsp143I、BstENII、DpnII、Kzo9I、MboI、NdeII、Sau3AI
GAT^C BstKTI,
G^TAC Csp6I
【0017】
最も好ましい酵素はNlaIIIである。
本発明はまた、25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグをも提供し、そのタグの3'末端は、III型制限酵素の切断部位によって規定され、そのタグの5'末端は、発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定される。
【0018】
本発明の好ましい実施形態では、上記に記載のように、III型酵素はEcoP15Iであり、他の制限酵素は、cDNAを切断してそれぞれ長さが平均200bp〜300bpである断片にすることができる酵素である。最も好ましい酵素はNlaIIIである。
【0019】
本発明のタグの例を、下記の表1に示す。これらのタグは、従来のSAGEタグまたはLongSAGEタグより長く、対応する遺伝子のより正確な同定を可能とする。
【0020】
本発明はさらに、2個のタグを含み、そのそれぞれが異なる発現遺伝子に由来するダイタグオリゴヌクレオチドを提供する。そのダイタグオリゴヌクレオチドは、下記のステップを含む方法によって生成される:
1)オリゴdT、およびIII型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、他の制限酵素で前記cDNAプールの消化を行うステップ;
2)上記のcDNAプールからポリA配列を含む断片を精製し、その断片をIII型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップ;
3)III型制限酵素で上記の断片を消化し、3'充填反応を行った後、得られたリンカーAを含む断片とリンカーBを含む断片とを連結するステップ;および
4)上記の連結した断片をステップ1)の他の制限酵素で消化してリンカー配列を切断して、ダイタグオリゴヌクレオチドを得るステップ。
【0021】
上記のステップで示したように、III型制限酵素の第1の認識部位は、mRNAからのcDNAの逆転写に使用するRTプライマーによって標的cDNA中に組み込まれ、III型制限酵素の第2の認識部位は、リンカー配列によって標的cDNA中に組み込まれている。
【0022】
RTプライマーは、配列5'-N18〜25-CAGCAG-T15〜25-3'によって定義され、N18〜25は、任意の18〜25ヌクレオチドの配列であり、配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない。RTプライマーの5'末端は、例えばビオチンで修飾してもよい。
【0023】
本発明のIII型制限酵素は、従来の3'充填反応を用いて容易に平滑末端化できる、5'末端が突出した断片をもたらす。すなわち、上記のステップ3)では、リンカーAを含む断片、およびリンカーBを含む断片を容易に平滑末端化することができ、それによって、タグのサイズをまったく縮小することなく前記断片の無作為な結合が可能となる。
【0024】
本発明の好ましい実施形態では、III型酵素はEcoP15Iであり、他の制限酵素は、NlaIIIなど、cDNAを切断してそれぞれ長さが平均200bp〜300bpの断片にすることができる酵素である。
【0025】
本発明のダイタグオリゴヌクレオチドを生成する方法では、2種のリンカー(リンカーAおよびリンカーB)を使用する。リンカーAおよびリンカーBは、互いに異なる二本鎖DNAである。連結に関与する二本鎖リンカー断片の1つの末端は、連結する配列と隣接したIII型制限酵素の認識配列を含む。例えば、III型制限酵素がEcoP15Iであり、他の制限酵素がNlaIIIであるとき、どちらのリンカーも、その第1鎖の3'末端に5'-CAGCAGCATG-3'配列を含む。下線の配列5'-CAGCAG-3'は、EcoP15Iの認識部位の1つを構成し、5'-CATG-3'配列は、NlaIII消化で生じた断片と連結する、3'が突出した一本鎖領域を構成する。
【0026】
連結に関与しない二本鎖リンカー断片の他の末端を、FITC(フルオレセインイソチオシアネート;第1鎖の5'末端)などの標識試薬、およびアミノ部分(第2鎖の3'末端)で修飾してもよい。
【0027】
したがって、リンカーは、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製する;
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
【0028】
上記で、DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40塩基のヌクレオチド配列であり、それらは互いに相補的であり、DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよい。
【0029】
本発明はさらに、上記のダイタグオリゴヌクレオチドの連結によって得られたポリヌクレオチドを提供する。各ダイタグオリゴヌクレオチドをクローン化し、PCRによって増幅することができる。そのポリヌクレオチドは、少なくとも2個のダイタグオリゴヌクレオチドを含み、好ましくは2〜200個のダイタグオリゴヌクレオチドを含む。本発明のダイタグオリゴヌクレオチドは、2個のタグの無作為な結合によって作製され、したがってポリヌクレオチドはまた、タグの無作為なコンカテマー化によって作製される。
【0030】
本発明はさらに、ポリヌクレオチドのヌクレオチド配列の解析、およびポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づく発現遺伝子の発現レベルの定量を含む遺伝子発現解析法を提供する。
【0031】
例えば、本発明の方法は、下記のステップを含む:
1)オリゴdT、およびIII型制限酵素の認識配列を含むRTプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、その後他の制限酵素でcDNAプールの消化を行うステップ;
2)上記のcDNAプールからポリA配列を含む断片を精製し、その断片を、III型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップ;
3)III型制限酵素で上記の断片を消化し、3'充填反応を行った後、得られたリンカーAを含む断片とリンカーBを含む断片とを連結するステップ;
4)上記の連結した断片をステップ1)の他の制限酵素で消化してリンカー配列を切断して、25個を超えるヌクレオチドからなる2個のタグを含み、発現遺伝子を同定しうるダイタグオリゴヌクレオチドを得るステップ;
5)ダイタグオリゴヌクレオチドを連結して、ポリヌクレオチドを生成するステップ;および
6)上記のポリヌクレオチドのヌクレオチド配列を解析し、ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて発現遺伝子の発現レベルを定量するステップ。
【0032】
この方法の好ましい実施形態では、III型酵素はEcoP15Iであり、他の制限酵素は、NlaIIIなど、cDNAを切断してそれぞれ長さが平均200bp〜300bpの断片にすることができる酵素である。リンカーに関して、上記に記載の二本鎖DNAを好ましくは使用することができる。
【0033】
本発明はさらに、25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグを単離するためのキットを提供し、それは下記の成分を含む:
a)配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない配列5'-N18〜25によって定義され、5'末端をビオチン化してもよいRTプライマー。
b)互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製するリンカーAおよびリンカーB:
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
【0034】
上記で、(1)のN30〜40および(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよい。
c)上記のリンカーAまたはリンカーBとハイブリダイズしうるプライマー。
【0035】
そのキットはまた、EcoP15I酵素などのIII型制限酵素、および/またはNlaIIIなどの他の制限酵素をも含む。そのキットはさらに、前記の成分に加えて、本発明の遺伝子発現解析を実施するのに必要な他の成分を含んでもよい。その例としては、標識試薬、緩衝液、マグネティックビーズなどがある。
【発明を実施するための最良の形態】
【0036】
図2を参照して、EcoP15IをIII型制限酵素として用いた本発明の好ましい実施形態を下記に説明する。
【0037】
III型制限改変酵素EcoP15Iは、標的DNA分子中の2つの非メチル化逆向き5'-CAGCAG-3'部位を認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を消化する。本発明は、cDNAの定められた位置から25〜28bpのタグ配列を単離するためのEcoP15I酵素の潜在的な適用すべてを指すものである。
【0038】
ビオチン化オリゴdTアンカープライマー(以下「RTプライマー」または逆転写プライマーと称する)を用いて、mRNAから二本鎖cDNAを合成する。このRTプライマーは、18〜25塩基の任意のヌクレオチド配列、および5'-CAGCAG-3'配列、その後に15〜25塩基のオリゴdT配列を含む。RTプライマー中に含まれる5'-CAGCAG-3'配列は、EcoP15Iの認識部位の1つを構成する(図2、ステップ1)。例えば、22ヌクレオチドの配列、および5'-CAGCAG-3'配列、その後に19個のdT配列を含むRTプライマーは、以下の通りである:
5'-CTGATCTAGAGGTACCGGATCCCAGCAGTTTTTTTTTTTTTTTTTTT-3'(配列番号1)。
【0039】
合成したcDNAを、配列モチーフ5'-CATG-3'を認識する制限エンドヌクレアーゼNlaIIIで消化する。RTプライマー配列(ビオチン標識)を含む消化断片だけを、ストレプトアビジン被覆マグネティックビーズによって捕捉する(図2、ステップ2および3)。
【0040】
二本鎖リンカー断片(46bp)を、マグネティックビーズによって捕捉した(ポリA配列を含む)cDNA断片の末端と連結する。連結に関与するこのリンカー断片の1末端は、第1鎖中の5'-CATG-3'と隣接した5'-CAGCAG-3'配列を含む(図2、ステップ4)。5'-CAGCAG-3'配列は、EcoP15Iの認識部位の1つを構成し、5'-CATG-3'配列は、NlaIII消化で生じた断片の粘着末端と連結する3'が突出した一本鎖領域を構成する。連結に関与しない二本鎖リンカー断片の他の末端を、FITC(フルオレセインイソチオシアネート;第1鎖の5'末端)、およびアミノ部分(第2鎖の3'末端)で修飾する。
【0041】
本発明では、2種のリンカーを使用する。例えば、リンカーAは、下記の2つのオリゴヌクレオチドをアニールすることによって作製される:
FITC-5'-TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATACAGCAGCATG-3'(配列番号2)、および
5'-CTGCTGTATTAAGCCTAGTTGTACTGCACCAGCAAATCCAAA-3'-NH2(配列番号3)。
【0042】
リンカーBは、下記の2つのオリゴヌクレオチドをアニールすることによって作製される:
FITC-5'-TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGCAGCAGCATG-3'(配列番号4)、および
5'-CTGCTGCGTACATCGTTAGAAGCTTGAATTCGAGCAGAAA-3'-NH2(配列番号5)。
【0043】
cDNAプールを2分割し、その一方をリンカーAと連結し、他方をリンカーBと連結することにより、「リンカーA連結cDNA」および「リンカーB連結cDNA」を得る。
【0044】
リンカーA連結cDNAでもリンカーB連結cDNAでも、ビーズと結合したDNA断片は、EcoP15Iで消化する(図2、ステップ5)。EcoP15Iは、配列5'-CAGCAG-3'の1対の逆向きモチーフを認識し、その認識部位の1つの3'末端から25〜28bp離れた部位を切断する。消化後、2つの断片がビーズから遊離する。一方は、リンカーおよび27または28bpのタグ断片を含む断片(合計サイズ69または70bp)であり、他方は、二本鎖cDNA断片の中間に位置する様々なサイズの断片である。ポリA配列を含む断片は、マグネティックビーズに結合したままであり、以下の手順に関与しない。
【0045】
UV照射下でのFITCの蛍光によって、リンカーおよび27または28bpのタグ断片を含む69または70bpの断片を視覚化し、ポリアクリルアミドゲルから、ゲルの切り出しによってそれが容易に単離される。
【0046】
EcoP15Iは、従来の3'充填反応によって容易に平滑末端化される、5'末端が突出した断片をもたらし、それによって断片の無作為な結合が可能となる。したがって、それぞれリンカーA連結cDNAおよびリンカーB連結cDNAに由来する69または70bpの断片(リンカータグ断片)を、3'充填反応によってそれぞれ平滑末端化し、互いに連結して、2個のタグの無作為な結合によりダイタグを形成させる。末端を平滑化したcDNAタグ配列の側でのみ連結が行われるように、リンカー断片の3'末端をアミノ修飾により遮断する(図2、ステップ6、7)。
【0047】
得られたダイタグ分子をPCR増幅する(図2、ステップ9)。リンカー配列から設計したPCRプライマーの例を以下に示す:
ダイタグプライマー1E:ビオチン-5'-CAACTAGGCTTAATACAGCAGCA-3'(配列番号6)
ダイタグプライマー2E:ビオチン-5'-CTAACGATGTACGCAGCAGCA-3'(配列番号7)。
【0048】
上記のプライマーを用いたPCRによって得られるPCR産物の予想サイズは、約97bpである。
その約97bpのPCR産物をNlaIIIで消化し(図2、ステップ10)、それによって、約52bpのダイタグ断片が遊離する。これらの断片をゲルから回収し精製する。
【0049】
連結反応によってダイタグ断片をコンカテマー化する(図2、ステップ11)。アガロースゲル電気泳動によって、コンカテマーを分離する。500bpより大きい断片をゲルから溶出させ回収する。
【0050】
サイズ分離したコンカテマー断片を、SphIで予め消化しウシ小腸ホスファターゼで処理した適当なプラスミドベクターと連結し(図2、ステップ12)、そのプラスミドで大腸菌(E.coli)を形質転換する(図2、ステップ13)。
【0051】
そのプラスミドの挿入断片をPCR増幅する(図2、ステップ14)。
そのPCR産物を直接シーケンシングする(図2、ステップ15)。一連の約44bpのダイタグ配列は、NlaIII認識配列のCATGと隣接する。この約52(44+8)bpの配列情報から、各cDNAの定められた位置から単離された2つの26〜28bpのタグ配列が得られる。
【0052】
試料中の26bpのタグの数をカウントした後、最初のSAGE(商標)のプロトコールに記載のように、遺伝子発現プロファイルを得ることが可能となる。本発明のダイタグは平滑末端タグの無作為な結合によって形成されるため、その結果は、各遺伝子の発現を正確に反映する。
【0053】
26bpのタグ配列は、タグが由来する遺伝子を一意的に同定するのに十分な情報を含む。26bpのDNA配列の情報内容を用いると、対応する遺伝子のインシリコ(in silico)での同定が促進される。全Genbank配列に対して26bpのタグ配列をBLAST検索すると、タグが由来する遺伝子と正しい整合を示す(図3)。
【0054】
記載した方法をDNA配列データが利用できない生物に適用する場合、26bpのタグ配列を3'-RACE用のPCRプライマーとして直接使用して、cDNAの3'領域を回収することができる。そのようなcDNA配列をBLAST検索に使用して、その遺伝子を同定することができる(図3)。
【0055】
SuperSAGEによって明らかとなった試料間の遺伝子発現の違いを実証するために、26bpのタグ配列を用いた3'-RACEをRT-PCRとして直接行うことにより、メッセージの量を定量することができる(図3)。
【0056】
26bpのタグ配列は、「RNA干渉」(RNAi)を引き起こすのに必要なDNA配列の最小サイズ(21bp)(Elbashirら、Nature 411:494-498、2001)より長い。したがって、タグ配列を含む二本鎖RNAを、タグに対応する遺伝子をノックアウトする機能解析に直ちに使用することができる。これは、SuperSAGEによって行われる遺伝子発現解析を、26bpのタグの単離を用いた遺伝子機能解析に直接結び付けることができることを意味する。
【0057】
植物種における適用では、上記に記載の3'-RACE断片を植物ウイルスベクター中にクローン化し、「ウイルス誘導遺伝子サイレンシング(VIGS)」(Baulcombe、Curr.Opin.Plant Biol.2:109-113、1999)に使用することができ、したがって、前記SuperSAGE法は、同様に植物でも遺伝子発現解析を遺伝子機能解析と結び付ける。
[実施例]
【0058】
下記の実施例を参照して、本発明をより詳細に説明するが、本発明の範囲は、これらの実施例に限定されるものではない。
【実施例1】
【0059】
原理の証明として、26および27bpのタグ配列を、イネの擬似病斑突然変異体IB2020(Oryza sativa品種名かけはし(Kakehashi))の葉から、上記に記載の方法によって単離した。
【0060】
1)mRNAの調製
従来のRNA単離の方法によって、イネの葉身から総RNAを単離した。このRNAから、「mRNA精製キット(mRNA Purification Kit)」(Amersham Pharmacia)を用いてmRNA5μgを単離した。そのmRNAを、DEPC水29μl中に溶解させ、供給源物質として使用した。
【0061】
2)オリゴdTプライマーを用いたcDNA合成
「cDNA合成システム(cDNA Synthesis System)」(Invitrogen)を用いてこのmRNAを逆転写して、酵素EcoP15Iの認識配列である5'-CAGCAG-3'モチーフを含む下記の逆転写プライマーを用いた一本鎖cDNAを得た。
逆転写プライマー:5'-CTGATCTAGAGGTACCGGATCCCAGCAGTTTTTTTTTTTTTTTTTTT-3'(配列番号1)
同じキットを用いて、この産物を二本鎖cDNAに転換し、エタノール沈殿させ、LoTE緩衝液(3mM Tris-HCl pH7.5、0.2mM EDTA)20μl中に溶解させた。
【0062】
3)NlaIII消化
0.1mg/mlのBSAを含む1×NEB緩衝液4(New England BioLabs;NEB)中にNlaIII50単位(NEB)を含む反応緩衝液200μl中で、得られた二本鎖cDNA(20μl)を37℃で90分間消化した。消化後、TE平衡化フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)でcDNAを抽出し、エタノール沈殿させ、LoTE緩衝液20μl中に溶解させた。
【0063】
2本のエッペンドルフ(Eppendorf)チューブそれぞれに、ストレプトアビジンマグネティックビーズ懸濁液(Streptavidin Magnesphere Paramagnetic particle、Promega)1mlを移した。マグネティックビーズを、1×B&W緩衝液(5mM Tris-HCl pH7.5、0.5mM EDTA、1M NaCl)200μlで、磁性分離台(Magnesphere Technology Magnetic Separation Stand)を用いて1回洗浄した。洗浄後、1×B&W緩衝液100μl、および上記のステップから得られたcDNA10μl、および水90μlを各チューブに添加し、混合し、室温で30分間インキュベートした。マグネティックビーズと結合したcDNAを1×B&W緩衝液で3回洗浄し、LoTE緩衝液で3回洗浄した。
【0064】
4)リンカー連結
業者に委託して、以下のオリゴヌクレオチドを合成した(Qiagen)。
リンカーA1:FITC-5'-TTTGGATTTGCTGGTGCAGTACAACTAGGCTTAATACAGCAGCATG-3'(配列番号2)
リンカーA2:5'-CTGCTGTATTAAGCCTAGTTGTACTGCACCAGCAAATCCAAA-3'-NH2(配列番号3)
リンカーB1:FITC5'-TTTCTGCTCGAATTCAAGCTTCTAACGATGTACGCAGCAGCATG-3'(配列番号4)
リンカーB2:5'-CTGCTGCGTACATCGTTAGAAGCTTGAATTCGAGCAGAAA-3'-NH2(配列番号5)
【0065】
リンカーA2およびリンカーB2の5'末端は、T4ポリヌクレオチドキナーゼ(NEB)によってリン酸化した。リンカーA1とリン酸化リンカーA2をアニールすることによってリンカーAを、リンカーB1とリン酸化リンカーB2をアニールすることによってリンカーBを調製した。リンカーAもリンカーBも、EcoP15I認識配列(5'-CAGCAG-3')を有する。
【0066】
マグネティックビーズと結合したcDNAを含む2本のチューブそれぞれに、LoTE 17μl、5×リガーゼ緩衝液(Invitrogen)3μlおよびリンカーAまたはリンカーB 1μgを添加した。混合した後、チューブを50℃で2分間インキュベートし、室温で15分間放置した。続いて、T4 DNAリガーゼ2μl(5単位/μl、Invitrogen)をチューブに添加し、16℃で90分間インキュベートした。リンカー連結の後、1×B&Wで4回、LoTE緩衝液で3回、滅菌蒸留水で1回ビーズを洗浄した。
【0067】
5)EcoP15I消化
マグネティックビーズ上のリンカー連結cDNAを、反応混合物(10mM Tris-HCl pH8.0、10mM KCl、10mM MgCl2、0.1mM EDTA、0.1mM DTT、5μg/ml BSA、2mM ATP)100μl中で、EcoP15I 10単位で消化した。チューブを37℃で90分間インキュベートした。
【0068】
6)リンカータグ断片の精製
EcoP15I消化後、チューブを磁性台上に置いた。ストレプトアビジン被覆マグネティックビーズによるビオチン化された最外部の3'末端断片の除去後、リンカータグ断片を含む非結合分画を収集し、新しいチューブに移した。フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で収集した緩衝液を抽出し、エタノール沈殿させ、LoTE緩衝液10μl中に溶解させ、8%ポリアクリルアミドゲル電気泳動によって分離した。隣接するレーン中に、SYBRグリーン(FMC)で予め染色したサイズマーカー(20bpラダー、TAKARA)を添加した。電気泳動後、ゲルをUV照明器上に置き、FITCが媒介するその蛍光によって、サイズが約69bpのリンカータグ断片を視覚化した(図4)。リンカーとリンカーの連結体(約90bp)および単一のリンカー断片(46bp)に由来するであろうさらなる2断片も視覚化した。FITCの蛍光の示すシグナルが弱すぎる場合、ゲルをSYBRグリーン(FMC)で染色して、リンカータグ断片を視覚化することができる。約69bpのリンカータグ断片をゲルから切り出し、注射針で底に針穴を開けた0.5mlチューブ中に移した。その0.5mlチューブを2mlチューブの内部に入れ、15000rpmで2分間遠心した。2mlチューブの底で収集された小さなゲル片に、LoTE緩衝液(300μl)を添加し、それを37℃で2時間インキュベートし、その後65℃で15分間インキュベートした。ゲルの懸濁液をSpinXカラム(Coaster)に移し、15000rpmで2分間遠心した。回収された溶液を、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で1回抽出し、エタノール沈殿させ、LoTE緩衝液8μl中に溶解させた。
【0069】
7)タグ断片の平滑化
リンカータグ断片の5'突出を、キット(Blunting High;TOYOBO)を用いて、DNAポリメラーゼで充填した。リンカータグ断片を含む溶液(8μl)に、反応緩衝液(KOD緩衝液;TOYOBO)1μl、およびKODポリメラーゼ(TOYOBO)1μlを添加し、それを72℃で2分間インキュベートした。
【0070】
8)ダイタグ形成のための連結
リンカーAおよびリンカーBに由来するリンカータグ断片を連結して、ダイタグ断片を形成させた。同量(2μl)の平滑末端化したリンカーAタグ溶液とリンカーBタグ溶液を混合し、LoTE緩衝液6μlおよび連結反応用混合液(Ligation High、TOYOBO)10μlを添加した。連結反応溶液を16℃で4時間〜1晩インキュベートした。
【0071】
9)ダイタグPCR
得られたダイタグ溶液を5倍および10倍に希釈し、ダイタグPCR用の鋳型として使用した。希釈したダイタグ鋳型1μl、Taqポリメラーゼ(AmpliTaq Gold;Applied Biosystems)3単位、1×AmpliTaq Gold緩衝液およびPCRプライマーを含む溶液50μlをそれぞれ含む288(=96×3)本の0.2mlチューブ中でPCR反応を行った。PCRプライマーの5'末端は、以下の通りにビオチン化した(Qiagenにより、業者を介して合成した)。
【0072】
ダイタグプライマー1E:ビオチン-5'-CAACTAGGCTTAATACAGCAGCA-3'(配列番号6)
ダイタグプライマー2E:ビオチン-5'-CTAACGATGTACGCAGCAGCA-3'(配列番号7)
PCRは、最初の95℃、12分の変性、その後の94℃、40秒および60℃、40秒の反応27〜29回からなった。増幅したダイタグ断片の予想サイズは約97bpであった(図5)。
【0073】
10)ダイタグPCR産物の精製
ダイタグPCR産物(チューブ約300本)を、10mlプラスチックチューブ中に一緒にした。フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)抽出およびエタノール沈殿後、それをLoTE緩衝液100μl中に溶解させた。このPCR産物を、1.5%の低融点アガロース(SeaPlaque、FMC)ゲル上で泳動し、約97bpの断片をゲルから切り出し、それをQiagenゲル抽出キット(Qiagen)で精製した。
【0074】
11)NlaIIIでの消化によるリンカー除去
LoTE緩衝液121μl中に溶出させた約97bpの精製断片を、0.1mg/mlのBSAを含む1×NEB緩衝液4(NEB)中で、NlaIII(NEB)120単位で消化した。ゲル電気泳動で消化されたことを確認した(サイズが52bp、22bpおよび23bpの3種の断片が視覚化された、図6)後、リンカー断片を除去するために、ストレプトアビジンマグネティックビーズで、消化液を室温で30分間処理した。チューブ中のマグネティックビーズを磁性台によって収集した後、その上清を新しいチューブに移し、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)でDNAを抽出し、エタノール沈殿させ、LoTE緩衝液10μl中に溶解させた。
【0075】
12)52bpのダイタグ断片の精製
回収したDNAを、12%ポリアクリドアミドゲルで電気泳動した。断片を視覚化するために、ゲルをSYBRグリーン(FMC)で染色した。サイズが約52bpの断片をUV照明器上でゲルから切り出した。上記に記載のように、ゲルからDNAを溶出させた。ストレプトアビジンマグネティックビーズで、ゲルから溶出させたDNAを室温で30分間処理した。上清を収集し、フェノール/クロロホルム/イソアミルアルコール(25:24:1;pH8.0)で抽出し、エタノール沈殿させ、LoTE緩衝液7μl中に溶解させた。
【0076】
13)コンカテマー形成
ダイタグ断片をコンカテマー化するために、ダイタグ溶液7μl、5×リガーゼ緩衝液2μl、およびT4リガーゼ(Invitrogen)5単位を混合し、16℃で1〜4時間インキュベートした。その連結溶液を65℃で10分間インキュベートし、1%アガロースゲルに添加した(図7)。サイズが500bpを超えるDNA断片をゲルから切り出し溶出させた。精製したコンカテマーをLoTE緩衝液6μl中に溶解させた。
【0077】
14)ベクタークローン化
クローニングベクター(pGEM3Z、Promega)5μgをSphIで消化し、ウシ小腸アルカリホスファターゼ(CIAP)で処理した。コンカテマーをクローン化するために、この消化したpGEM3ZベクターとダイタグコンカテマーをT4リガーゼ(Invitrogen)を用いて連結した。連結反応を行ってから4時間後、反応溶液をフェノール/クロロホルム/イソアミルアルコール(25:24:1)で抽出し、エタノール沈殿させた。そのペレットを70%エタノールで4回洗浄して塩を完全に除去し、滅菌蒸留水10μl中に溶解させた。
【0078】
15)形質転換
エレクトロポレーション用コンピテント大腸菌細胞(DH10B、Invitrogen)を、ダイタグコンカテマーを含むプラスミドのクローニングに使用した。チューブ中に、コンピテント細胞懸濁液40μlと、精製した連結DNA1μlを氷上で混合し、エレクトロポレーション用キュベット(間隔0.1cm;BioRad)中に移した。エレクトロポレーションの条件は、コンピテント細胞の製造業者が推奨する通りであった(2.5kV、25μFD、100Ω)。エレクトロポレーション後、SOC培地(Invitrogen)1mlを添加し、その溶液を37℃で1時間インキュベートした。得られた大腸菌の懸濁液を100μg/mlのアンピシリン、20μg/mlのX-galおよび0.1mMのIPTGを含むLB培地プレート上に播いた。プレートを37℃で14時間インキュベートした。
【0079】
16)コロニーPCRおよびシーケンシング
プレート上に生じた白色コロニーを爪楊枝で拾い上げ、1×PCR緩衝液、0.4mM dNTP、0.05単位/μl Taqポリメラーゼ(TAKARA)、2pmol/μl M13-20プライマーおよび2pmol/μl M13RVプライマーを含むPCR混合液中に再懸濁させた。プラスミド中のクローン化断片をPCRで増幅し、断片のサイズをアガロースゲル電気泳動によって確認した(図8)。増幅したPCR断片を、PCR精製キット(Qiagen)を用いて精製した。
【0080】
17)コンカテマーのシーケンシング
BigDyeターミネーター(Applied Biosystems)およびM13-20プライマーを用いて、精製したPCR断片(コンカテマー)の配列を決定した。シーケンシング反応の後、「Montage SEQ96 Sequencing Reaction Cleanup Kit」(Millipore)によってDNAを精製した。RISA384キャピラリーDNAシークエンサー(Shimadzu)によってDNA配列を解析した。クローン化した断片のDNA配列の例を図9に示す。2つの26または27bpの断片を含む一連の52〜54bpのダイタグは、CATGというNlaIII部位によって境界付けられている。
【0081】
18)データ解析
ジョンスホプキンス大学(Johns Hopkins University)により供給されたSAGE(商標)2000ソフトウェアを用いて、コンカテマーのDNA配列から26bpのタグ配列を抽出し各タグの数をカウントした。いくつかのクローンについてシーケンシングした後、表1に示す遺伝子発現プロファイル作成を行った。
【0082】
【表1】
【0083】
したがって、cDNAのNlaIII部位と隣接する26または27bpの配列をイネ葉から単離することに成功した。本明細書に記載の実験手順を適用することによって、26または27bpの配列を含む認識タグをどんなcDNAからも得ることができる。26または27bpの配列を含むタグの単離に基づくこのプロトコールは、18〜21bpの配列を含むタグを使用する(「LongSAGE(商標)」として知られる)ほとんどの進歩型SAGE(商標)の手順を超えるかなりの質的な改善を表すものである。
【0084】
SuperSAGE法では、EcoP15I酵素が、タグ中に、DNAポリメラーゼによって容易に平滑末端化される5'突出末端を生じさせる。したがって、平滑末端化したタグの無作為な結合によってダイタグ形成が行われ、それによって、タグの数がタグと対応する遺伝子の発現を正確に表すことが確実となる。この特性は、LongSAGE(商標)のプロトコールにおいて、適合する2bpの3'突出末端を含む21bpのタグの間でしかダイタグが形成されないときには利用できない。Long SAGE(商標)でタグの無作為な結合を確実に行うためには、3'突出領域を除去することによって、タグを平滑末端化すべきである。この結果、タグのサイズが2bp短縮され、それによってタグの情報内容が減少する。したがって、本明細書に記載のSuperSAGE法は、タグの情報内容がより多く、遺伝子発現プロファイルをより正確に表す点から、LongSAGE(商標)法より有利である。
【実施例2】
【0085】
SuperSAGEのタグのサイズが26bpであることによって、遺伝子に対するタグの非常に信頼性の高い注釈付けが可能となることを実証するために、以下の実験を行った。
【0086】
26bpのタグ30個を表1から無作為に選択した。このDNA配列を、タグのサイズがそれぞれ20、18および15bpとなるように、3'末端から切断した。15bpのタグのサイズは、従来のSAGE(商標)で使用するものに対応する。18bpまたは20bpのタグは、LongSAGE(商標)のタグから、それぞれ末端平滑化を行ったものおよび末端平滑化を行わないものに対応する。異なるサイズのタグそれぞれを用いて、130,000を超える種由来のDNA配列を表すGenbank中に寄託されているDNA配列全体からBLAST検索を行った。所与のサイズのタグと完全な整合を示す、DNA配列を含んだ種の数をカウントし、タグ配列30個全体から種の平均数と最大数を得た。表2にその検索結果の概要を示す。
【0087】
【表2】
【0088】
いくつかのDNA配列がタグ配列と完全な整合を示し、タグのサイズが増大すると、遺伝子に対するタグの注釈付けの不明確性が低下した。
【0089】
従来のSAGE(商標)のタグ(15bp)は、平均で4を超える種の、最大で9種のDNA配列と整合した。タグ30個すべてが2つ以上の種と相関した(表2)。18bpのタグは、平均で1.8種と、最大で7種と整合した。30個のうち10個のタグが2つ以上の種と相関した。20bpのタグは、平均で1.16種と、最大で4種と整合した。30個のうち3個のタグしか2つ以上の種と相関せず、このことから最初のSAGE(商標)のタグの長さ(15bp)より大きく改善されたことが示唆される。しかし、20bpのタグは、末端を平滑化せずにリンカータグが連結するときしか抽出することができず、その結果、この方法の最終結果が必ずしも正確な遺伝子発現を表すわけではないことに留意されたい。SuperSAGE法の26bpのタグは、平均で1.06種と整合し、最大では2種としか整合しなかった。30個のうち2個のタグしか2つ以上の種と相関しなかった。これらの結果は、26bpのDNA配列中の情報内容がタグの遺伝子注釈付けの効率において大きな改善をもたらすことを明らかに示すものである。
【0090】
26bpのタグは、平均で1種のみのDNA配列と整合し、ほとんどの場合で特定の種の単一遺伝子と整合した。したがって、タグ配列の注釈付けをほとんど完全に実施することができる。EST配列データベースに対して、ならびにゲノム配列全体に対してSuperSAGEでのタグの注釈付けを行うことができる。
【0091】
SuperSAGEにおける26bpのタグの情報内容が大きいことにより、2種の生物の遺伝子発現解析を同時に行うことが可能となる。一例として、記載の方法を、真菌のイネいもち病菌(Magnaporthe grisea)によって引き起こされるいもち病に感染したイネ植物の遺伝子発現プロファイルの研究に適用した。いもち病に感染したイネ葉から合計12,119個のタグを単離した後、イネおよびイネいもち病菌(M.grisea)のゲノム配列すべてに対するBLAST検索によって各タグを注釈付けた。予想通り、大多数のタグはイネ遺伝子に注釈付けられ(表3)、その一方で74個のタグがイネの配列と整合しなかったが、いもち配列と整合した(表4)。
【0092】
【表3】
【0093】
【表4】
【0094】
SuperSAGEによって得られた結果を確認するために、以下のアンカーオリゴdTプライマーを用いて、上記と同じRNAから逆転写によりcDNAを合成した:
5'-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3'(配列番号8)。
【0095】
このcDNAを鋳型として用いて、ハイドロフォビン、ヌクレオシド二リン酸キナーゼおよび60Sリボソームタンパク質の26bpのタグ(表4)を、以下のプライマーと一緒に3'-RACEに使用した:
5'-GGCCACGCGTCGACTAGTAC-3'(配列番号9)。
【0096】
3種の遺伝子すべてについて特異的なPCR産物が得られた(図10、左)。模擬接種したいもち感受性品種(かけはし)、いもち接種した感受性品種(かけはし)、模擬接種したいもち抵抗性品種(ヒメノモチ(Himenomochi))およびいもち接種した抵抗性品種(ヒメノモチ)におけるRT-PCRに同じプライマーの組を使用した。予想通り、いもち真菌を接種した感受性品種からのみ、3種のいもち遺伝子に対応するPCR産物が増幅され(図10、右)、このことから、SuperSAGEの注釈付けが真正であることが実証された。
【0097】
本明細書で例示するように、1試料中での2つの種の遺伝子発現の定量分析は、以前の技術で行うことができなかった。遺伝子発現解析(「SuperSAGE」)に26bpのタグを使用すると、宿主と病原体など2種以上の相互作用する生物の同時遺伝子発現解析が大いに促進される。
【実施例3】
【0098】
DNA配列データを利用できない生物の遺伝子発現解析に、26bpのタグを用いるSuperSAGEを使用することができることを実証するために、以下の実験を行った。ジャガイモ疫病菌(Phytophthora infestans)由来のタンパク質エリシターINF1(Kamounら、Mol.Plant-Microbe Interact.10:13-20、1997)または水で処理した植物種ベンサミアナタバコ(Nicotiana benthamiana)の遺伝子発現解析に、SuperSAGEを適用した。エリシターまたは水(冠水)を浸透させてから1時間後にその葉を収集し、それからRNAを単離した。
【0099】
それぞれの試料から5000個を超えるタグを単離することに成功した(表5)。表5中に列挙した、最も多数認められたタグ10個を直接3'-RACEに使用し、得られたcDNAの配列を決定した。cDNAのBLAST検索から、表5に示すタグと対応する遺伝子が同定された。
【0100】
【表5】
【0101】
INF1処理試料および冠水処理試料における各タグの頻度を比較することによって、2つの試料中で示されるタグに統計上有意な差があることが確認された(表6)。これらのタグを直接使用して3'-RACEを行い、回収したcDNAを使用してBLAST検索を行った。このことにより、ほとんどのタグを注釈付けることができた。
【0102】
【表6】
【0103】
26bpのタグ配列を、アンカープライマーと一緒にRT-PCRに使用して、表6に示すSuperSAGEの結果を確認した。RT-PCRの結果(図11)は、SuperSAGEの結果がINF1処理試料と冠水処理試料との遺伝子発現の差を正確に反映することを明らかに示すものである。
【0104】
各遺伝子発現の動態研究のためのRT-PCRに、同じ26bpのタグプライマーを容易に使用することができる(図12)。試験遺伝子4種(葉緑素a/b結合タンパク質、光化学系IIタンパク質、ホスホグリセリン酸キナーゼ、およびATP合成酵素の遺伝子)の発現がINF1で処理してから15分後に遮断されたことが判明した。
【0105】
この実施例は、DNA配列データを利用できない生物における遺伝子発現解析についてのSuperSAGEの大きな潜在的可能性を実証するものである。この潜在的可能性は、タグが特異的なPCRプライマーに十分なサイズであり、SuperSAGEによって得られる遺伝子発現プロファイルが実際の遺伝子発現を正確に表すことに由来する。
【図面の簡単な説明】
【0106】
【図1】従来のSAGE(商標)のプロトコール(Velculescuら、Science 270:484-487、1995)の概略図である。
【図2A】EcoP15Iを用いて26bpのcDNAのSuperSAGEタグを単離する概略的な手順を示す図である。
【図2B】EcoP15Iを用いて26bpのcDNAのSuperSAGEタグを単離する概略的な手順を示す図である。
【図3】EcoP15Iによって得られた26bpのタグを用いたSuperSAGE解析の適用の概要を示す図である。
【図4】FITCの蛍光によって視覚化した、EcoP15I消化(ステップ6、図2)後の産物の電気泳動の例を示す図である。各断片の構造をこのパネルの右側に示す。この例では、イネ葉由来のmRNA分子を鋳型として用いてcDNAを合成した。
【図5】図2のステップ9からのPCR産物の電気泳動(PAGE)の例を示す図である。予想されるPCR産物のサイズは約97bpである。
【図6】PCR産物のNlaIII消化(ステップ10)から得られた断片の電気泳動(PAGE)の例を示す図である。ダイタグのサイズは約52bpである。
【図7】ダイタグのコンカテマー化断片(ステップ11)の電気泳動の例を示す図である。
【図8】コロニーPCR産物(ステップ14)の電気泳動の例を示す図である。
【図9】クローン化したコンカテマー中に含まれているDNA配列(ステップ15)の例を示す図である。
【図10】26bpのタグ配列をPCRプライマーとして使用する、イネいもち病菌感染イネ葉から単離したRNAを用いたRT-PCRの結果を示す図である。
【図11】ジャガイモ疫病菌由来のINF1エリシタータンパク質で、または対照である水で処理したベンサミアナタバコにおけるRT-PCRの結果を示す図である。
【図12】SuperSAGEによって同定された遺伝子4種の遺伝子発現のRT-PCR動態研究を示す図である。
【配列表フリーテキスト】
【0107】
配列番号1:RTプライマー
配列番号2:リンカーAの第1鎖
配列番号3:リンカーAの第2鎖
配列番号4:リンカーBの第1鎖
配列番号5:リンカーBの第2鎖
配列番号6:ダイタグプライマー(順向)
配列番号7:ダイタグプライマー(逆向)
配列番号8:RTプライマー
配列番号9:3'-RACE用のプライマー
配列番号10-250:合成DNA(タグ配列)
【特許請求の範囲】
【請求項1】
発現遺伝子のcDNAから、25個を超えるヌクレオチドを含み、前記発現遺伝子を同定しうるタグを単離するためのIII型制限酵素の使用であって、前記タグの3'末端が前記III型制限酵素の切断部位によって規定され、前記タグの5'末端が前記発現遺伝子のcDNAの3'末端に最も近い他の制限酵素の切断部位によって規定されることを特徴とする使用。
【請求項2】
前記III型制限酵素がEcoP15Iである、請求項1に記載の使用。
【請求項3】
前記他の制限酵素がNlaIIIである、請求項2に記載の使用。
【請求項4】
25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグであって、前記タグの3'末端が、III型制限酵素の切断部位によって規定され、前記タグの5'末端が、前記発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定されるタグ。
【請求項5】
前記III型制限酵素がEcoP15Iである、請求項4に記載のタグ。
【請求項6】
前記他の制限酵素がNlaIIIである、請求項5に記載のタグ。
【請求項7】
2個のタグを含み、そのそれぞれが異なる発現遺伝子に由来するダイタグオリゴヌクレオチドであって、各タグが25個を超えるヌクレオチドを含み、発現遺伝子を同定することができ、前記タグの3'末端が、III型制限酵素の切断部位によって規定され、前記タグの5'末端が、前記発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定されるダイタグオリゴヌクレオチド。
【請求項8】
以下のステップを含む方法によって生成されることを特徴とする、請求項7に記載のダイタグオリゴヌクレオチド:
1)オリゴdT、および前記III型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、他の制限酵素で前記cDNAプールの消化を行うステップと、
2)前記cDNAプールからポリA配列を含む断片を精製し、前記断片を、前記III型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップと、
3)前記III型制限酵素で前記断片を消化し、3'充填反応を行った後、得られた前記リンカーAを含む断片と前記リンカーBを含む断片とを連結するステップと、
4)前記連結した断片をステップ1)の他の制限酵素で消化して前記リンカー配列を切断して、前記ダイタグオリゴヌクレオチドを得るステップ。
【請求項9】
前記ダイタグオリゴヌクレオチドが、前記異なる発現遺伝子に由来する2個のタグの無作為な結合によって作製されるものである、請求項7または8に記載のダイタグオリゴヌクレオチド。
【請求項10】
前記III型制限酵素がEcoP15Iである、請求項7、8または9に記載のダイタグオリゴヌクレオチド。
【請求項11】
前記他の制限酵素がNlaIIIである、請求項10に記載のダイタグオリゴヌクレオチド。
【請求項12】
前記リンカーAおよびリンカーBが互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製されることを特徴とする、請求項11に記載のダイタグオリゴヌクレオチド:
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
(但し、上記DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、前記DNA(1)の5'末端は標識してもよく、前記DNA(2)の3'末端はアミノ修飾してもよい)。
【請求項13】
請求項7から12のいずれか一項に記載の少なくとも2個のダイタグオリゴヌクレオチドを含むポリヌクレオチド。
【請求項14】
前記ポリヌクレオチドが2〜200個のダイタグオリゴヌクレオチドを含む、請求項13に記載のポリヌクレオチド。
【請求項15】
請求項13または14に記載のポリヌクレオチドのヌクレオチド配列を解析するステップと、前記ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて前記発現遺伝子の発現レベルを定量するステップとを含む遺伝子発現解析法。
【請求項16】
1)オリゴdT、およびIII型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、その後他の制限酵素で前記cDNAプールの消化を行うステップと、
2)前記cDNAプールからポリA配列を含む断片を精製し、前記断片を、そのどちらも前記III型制限酵素の認識配列を含むリンカーAまたはリンカーBと連結するステップと、
3)前記III型制限酵素で前記断片を消化し、3'充填反応を行った後、前記リンカーAを含む得られた断片を、前記リンカーBを含む得られた断片と連結するステップと、
4)前記連結した断片をステップ1)の前記他の制限酵素で消化して前記リンカー配列を切断し、それによって25個を超えるヌクレオチドからなる2個のタグを含み、前記発現遺伝子を同定しうるダイタグオリゴヌクレオチドを得るステップと、
5)前記ダイタグオリゴヌクレオチドを連結して、ポリヌクレオチドを生成するステップと、
6)前記ポリヌクレオチドのヌクレオチド配列を解析し、前記ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて前記発現遺伝子の発現レベルを定量するステップとを含む、遺伝子発現解析法。
【請求項17】
前記III型制限酵素がEcoP15Iである、請求項16に記載の方法。
【請求項18】
前記他の制限酵素がNlaIIIである、請求項17に記載の方法。
【請求項19】
前記リンカーAおよびリンカーBが互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製され、
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
上記で、DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、前記DNA(1)の5'末端は標識してもよく、前記DNA(2)の3'末端はアミノ修飾してもよい、請求項18に記載の方法。
【請求項20】
25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグを単離するためのキットであって、
a)配列5'-N18〜25-CAGCAG-T15〜25-3'によって定義される逆転写プライマーであって、N18〜25は配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない任意の18〜25ヌクレオチドの配列であり、前記RTプライマーの5'末端をビオチン化してもよい逆転写プライマーの成分と、
b)互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製するリンカーAおよびリンカーBであって、
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
上記で、(1)のN30〜40および(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、
DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよいリンカーAおよびリンカーBの成分と、
c)前記リンカーAまたはリンカーBとハイブリダイズしうるプライマーの成分とを含むキット。
【請求項21】
EcoP15Iおよび/またはNlaIIIをさらに含む、請求項20に記載のキット。
【請求項1】
発現遺伝子のcDNAから、25個を超えるヌクレオチドを含み、前記発現遺伝子を同定しうるタグを単離するためのIII型制限酵素の使用であって、前記タグの3'末端が前記III型制限酵素の切断部位によって規定され、前記タグの5'末端が前記発現遺伝子のcDNAの3'末端に最も近い他の制限酵素の切断部位によって規定されることを特徴とする使用。
【請求項2】
前記III型制限酵素がEcoP15Iである、請求項1に記載の使用。
【請求項3】
前記他の制限酵素がNlaIIIである、請求項2に記載の使用。
【請求項4】
25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグであって、前記タグの3'末端が、III型制限酵素の切断部位によって規定され、前記タグの5'末端が、前記発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定されるタグ。
【請求項5】
前記III型制限酵素がEcoP15Iである、請求項4に記載のタグ。
【請求項6】
前記他の制限酵素がNlaIIIである、請求項5に記載のタグ。
【請求項7】
2個のタグを含み、そのそれぞれが異なる発現遺伝子に由来するダイタグオリゴヌクレオチドであって、各タグが25個を超えるヌクレオチドを含み、発現遺伝子を同定することができ、前記タグの3'末端が、III型制限酵素の切断部位によって規定され、前記タグの5'末端が、前記発現遺伝子のcDNAの3'末端に最も近い、他の制限酵素の切断部位によって規定されるダイタグオリゴヌクレオチド。
【請求項8】
以下のステップを含む方法によって生成されることを特徴とする、請求項7に記載のダイタグオリゴヌクレオチド:
1)オリゴdT、および前記III型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、他の制限酵素で前記cDNAプールの消化を行うステップと、
2)前記cDNAプールからポリA配列を含む断片を精製し、前記断片を、前記III型制限酵素の認識配列をともに含むリンカーAまたはリンカーBと連結するステップと、
3)前記III型制限酵素で前記断片を消化し、3'充填反応を行った後、得られた前記リンカーAを含む断片と前記リンカーBを含む断片とを連結するステップと、
4)前記連結した断片をステップ1)の他の制限酵素で消化して前記リンカー配列を切断して、前記ダイタグオリゴヌクレオチドを得るステップ。
【請求項9】
前記ダイタグオリゴヌクレオチドが、前記異なる発現遺伝子に由来する2個のタグの無作為な結合によって作製されるものである、請求項7または8に記載のダイタグオリゴヌクレオチド。
【請求項10】
前記III型制限酵素がEcoP15Iである、請求項7、8または9に記載のダイタグオリゴヌクレオチド。
【請求項11】
前記他の制限酵素がNlaIIIである、請求項10に記載のダイタグオリゴヌクレオチド。
【請求項12】
前記リンカーAおよびリンカーBが互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製されることを特徴とする、請求項11に記載のダイタグオリゴヌクレオチド:
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
(但し、上記DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、前記DNA(1)の5'末端は標識してもよく、前記DNA(2)の3'末端はアミノ修飾してもよい)。
【請求項13】
請求項7から12のいずれか一項に記載の少なくとも2個のダイタグオリゴヌクレオチドを含むポリヌクレオチド。
【請求項14】
前記ポリヌクレオチドが2〜200個のダイタグオリゴヌクレオチドを含む、請求項13に記載のポリヌクレオチド。
【請求項15】
請求項13または14に記載のポリヌクレオチドのヌクレオチド配列を解析するステップと、前記ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて前記発現遺伝子の発現レベルを定量するステップとを含む遺伝子発現解析法。
【請求項16】
1)オリゴdT、およびIII型制限酵素の認識配列を含むプライマーを用いて、発現遺伝子のmRNAからcDNAプールを合成し、その後他の制限酵素で前記cDNAプールの消化を行うステップと、
2)前記cDNAプールからポリA配列を含む断片を精製し、前記断片を、そのどちらも前記III型制限酵素の認識配列を含むリンカーAまたはリンカーBと連結するステップと、
3)前記III型制限酵素で前記断片を消化し、3'充填反応を行った後、前記リンカーAを含む得られた断片を、前記リンカーBを含む得られた断片と連結するステップと、
4)前記連結した断片をステップ1)の前記他の制限酵素で消化して前記リンカー配列を切断し、それによって25個を超えるヌクレオチドからなる2個のタグを含み、前記発現遺伝子を同定しうるダイタグオリゴヌクレオチドを得るステップと、
5)前記ダイタグオリゴヌクレオチドを連結して、ポリヌクレオチドを生成するステップと、
6)前記ポリヌクレオチドのヌクレオチド配列を解析し、前記ポリヌクレオチド中に含まれる発現遺伝子に対応するタグの数に基づいて前記発現遺伝子の発現レベルを定量するステップとを含む、遺伝子発現解析法。
【請求項17】
前記III型制限酵素がEcoP15Iである、請求項16に記載の方法。
【請求項18】
前記他の制限酵素がNlaIIIである、請求項17に記載の方法。
【請求項19】
前記リンカーAおよびリンカーBが互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製され、
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
上記で、DNA(1)のN30〜40およびDNA(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、前記DNA(1)の5'末端は標識してもよく、前記DNA(2)の3'末端はアミノ修飾してもよい、請求項18に記載の方法。
【請求項20】
25個を超えるヌクレオチドを含み、発現遺伝子を同定しうるタグを単離するためのキットであって、
a)配列5'-N18〜25-CAGCAG-T15〜25-3'によって定義される逆転写プライマーであって、N18〜25は配列5'-CAGCAG-3'および配列5'-CATG-3'を含まない任意の18〜25ヌクレオチドの配列であり、前記RTプライマーの5'末端をビオチン化してもよい逆転写プライマーの成分と、
b)互いに異なる二本鎖DNAであり、下記のDNA第1鎖(1)とDNA第2鎖(2)をアニールすることによって作製するリンカーAおよびリンカーBであって、
DNA(1):5'-N30〜40-CAGCAGCATG-3'
DNA(2):3'-N30〜40-GTCGTC-5'
上記で、(1)のN30〜40および(2)のN30〜40は、任意の30〜40ヌクレオチドの配列であり、それらは互いに相補的であり、
DNA(1)の5'末端は標識してもよく、DNA(2)の3'末端はアミノ修飾してもよいリンカーAおよびリンカーBの成分と、
c)前記リンカーAまたはリンカーBとハイブリダイズしうるプライマーの成分とを含むキット。
【請求項21】
EcoP15Iおよび/またはNlaIIIをさらに含む、請求項20に記載のキット。
【図1】


【図2】

【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図2】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2007−528193(P2007−528193A)
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願番号】特願2004−571572(P2004−571572)
【出願日】平成15年5月9日(2003.5.9)
【国際出願番号】PCT/JP2003/005840
【国際公開番号】WO2004/099445
【国際公開日】平成16年11月18日(2004.11.18)
【出願人】(390025793)岩手県 (38)
【Fターム(参考)】
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願日】平成15年5月9日(2003.5.9)
【国際出願番号】PCT/JP2003/005840
【国際公開番号】WO2004/099445
【国際公開日】平成16年11月18日(2004.11.18)
【出願人】(390025793)岩手県 (38)
【Fターム(参考)】
[ Back to top ]