
インスリングラルギンの長時間作用型製剤
【課題】 インスリングラルギン100Uの製剤と曝露および活性で等価な、高単位インスリングラルギンを含有する水性医薬製剤、およびそれを使用する患者のI型およびII型糖尿病を治療する方法の提供。
【解決手段】 200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含むが、ただし製剤のインスリングラルギンの濃度は684U/mLではない水性医薬製剤、およびその使用。
【解決手段】 200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含むが、ただし製剤のインスリングラルギンの濃度は684U/mLではない水性医薬製剤、およびその使用。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含む水性医薬製剤であるが、ただし該製剤の濃度が684U/mLのインスリングラルギンではない、水性医薬製剤、およびその使用に関する。
【背景技術】
【0002】
インスリングラルギンは、31B−32B−Di−Arg ヒトインスリン(ヒトインスリンのアナログ)であり、さらにA21位のアスバラギンがグリシンでさらに置換されている。
【0003】
ランタス(登録商標)は、単回投与の皮下注射後、24時間の基礎インスリン供給を提供するインスリングラルギンを含有するインスリン製品である。
【0004】
ランタス(登録商標)の糖動的効果は、皮下注射部位からのインスリングラルギンの遅延吸収および予測可能な吸収によって明らかなピークを示さずにスムーズな24時間の濃度および作用プロファイルがもたらされるという点で現在市販されている他のインスリン製品と区別される。ランタス(登録商標)は、長時間作用型インスリン製品についての医療的な必要性を満たすために開発され、24時間にわたり可能な限りスムーズな基礎インスリンプロファイルによって正常または正常に近い血中グルコースコントロールを得るために一日一回の注射として投与することができる。このような製剤は、一日中血中グルコースの良好なコントロールを提供し、その上より明らかな「ピーク」作用を持つ他のインスリン製剤で見られる低血糖症をもたらす傾向を最小にする。
【0005】
多数の患者、特に肥満によってインスリン抵抗性が増加している患者は、血中グルコースを調節するために投与量の大きいものを用いている。例えば、100単位(100U)の用量は、1mLのランタス(登録商標)U100の注射が必要であり、これはいくらかの不快感を覚える場合がある;ランタス(登録商標)U100 1mLには、100U(3.6378mg)のインスリングラルギンが含まれている。注入量を減らすために、1mLあたり300Uのインスリングラルギンを含む製剤が開発されてきた。本発明はインスリングラルギンU300の製剤に限定されないが、本明細書中に記載される臨床試験は、インスリングラルギン300U製剤を用いて行った;インスリングラルギンU300 1mLあたり300U(10.9134mg)のインスリングラルギンが含まれている。この製剤は、3分の1の注入量で同じ単位数のインスリングラルギンを患者に注入することができる。
【0006】
インスリングラルギン製剤U100とU300は両方同じインスリン曝露および同じ効果、すなわちタイムプロファイルを提供するはずであると考えられた。
【発明の概要】
【発明が解決しようとする課題】
【0007】
試験(T)の治験薬であるインスリングラルギンU300の曝露および活性を、認可されている参照(R)製品であるランタスU100の曝露および活性と等価なものについて、正常血糖クランプにて非肥満体の健常な被験者で試験した。インスリングラルギンの作用の長期的な持続時間を評価するための、皮下投与30時間後を選択した。皮下投与後のインスリングラルギン濃度のタイムプロファイルより曝露を評価し、活性はインスリン1単位当たりのグルコース利用として同時に評価した。
【0008】
反復デザイン(replicate design)により、限定数の患者で、FDAガイドライン「Guidance for Industry、Statistical Approaches to Establishing Bioequivalence」に推奨されているような生物学的同等性および変動性(variability)を評価することができた。
【0009】
それぞれの臨床試験により、曝露および活性の等価を確立することが期待された。
【0010】
0.4U/kgの用量を本試験のために選択した;この量は、患者における平均基礎インスリン用量に対応している。非肥満体の健常な被験者において、この用量は、正常血糖クランプの設定で測ることのできる血清インスリン濃度のかなりの上昇、持続的なグルコース低下効果をもたらす。
【0011】
ガイドラインで支持される反復デザインは、無作為化プランによって割り当てられる予め定義された4通りのクロスオーバーシーケンス(RTTRまたはTRRT)で、いずれかのIP(R:ランタス(登録商標)U100、T:インスリングラルギンU300)の2つの反復単回投与を必要とする。これは、4つの期間(P)1〜4において実行された。結果として、各被験者は、2つの向かい合う臍周囲領域を交互に0.4U/kgのランタス(登録商標)U100(R)およびインスリングラルギンU300(T)の2つの反復単回皮下投与を受けた。
【0012】
4〜18日間の休薬期間は各投薬日から分離される。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、4日間というのは参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間というのは、クランプ期間の間に、被験者が試験に関係のない義務を自由に果たすことを可能にする3週間の休止期間を再び与える。
【0013】
正常血糖クランプ来院の前に、SCR(スクリーニング来院)において被験者は適格性についてスクリーニングされ、EOS(試験終了時)来院において被験者は正常な健康状態を保障するための最終試験に参加する。スクリーニングおよびP1は21日間より長く離してはならないが、EOS来院は次に続く週であるP4のD1と同じ週日(すなわち、さらなる4日後)より前、かつP4のD2後の2週間よりも後(すなわち、さらなる14日後)にはしなかった。
【0014】
これは、計4回の反復投与を伴う単回投与試験である。IPの効果は約24時間続き、このことが、被験者が2日間施設に拘束される理由である。被験者は治療に4度曝露された。
【0015】
本試験の主要な目的は、正常血糖クランプ技術を用いて、バイオアベイラビリティ(曝露)および生物有効性(活性)におけるランタス(登録商標)U100(市販製剤)およびインスリングラルギン U300の平均生物学的同等性(ABE)を評価することであった。
【0016】
本試験の二次的な目的は、インスリングラルギンU300の安全性および忍容性を評価することであった。
【0017】
上述のように、U100とU300両方のインスリングラルギン製剤は、同じインスリン曝露およびインスリン効果を提供すると予測された。しかしながら驚いたことにインスリン曝露および効果は同じではないことが示された。インスリングラルギンU100およびインスリングラルギンU300はバイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。インスリングラルギンU300投与後の曝露および活性は同量(0.4U/kg)のインスリングラルギンU100の投与後の曝露および活性と比較した場合約40%低かった。
【0018】
しかし、インスリングラルギンU300は、インスリングラルギンU100よりも、基礎インスリンに望ましいよりフラットなPK(曝露)およびPD(活性)プロファイルを示した。健常な被験者に対する同じ皮下投与後の、インスリングラルギンU100製剤とインスリングラルギンU300製剤との間の曝露および活性におけるこれらの驚くべきかつ予測できない差異は、以下の図に効果的に示されている。注目すべきは、同時に血中グルコースが一定になったということである。
【0019】
インスリングラルギンの血中グルコースの低下効果は、健常な正常血糖のビーグル犬においてさらに評価された。インスリングラルギン濃度の増加に伴い、作用の平均時間はそれぞれ6.8h(U100)から7.69h(U300)まで増加した。
【0020】
イヌにおいて、100から300U/mLまでグラルギン濃度を増加することによって、血中グルコース低下の時間作用プロファイルは、よりフラットかつ延長された活性に向けて変化した。イヌにおける現在のデータはヒトにもあてはまり、インスリングラルギンのより高い薬物濃度がプロファイルおよび作用のより長い持続時間に積極的に関与することを示している。
【0021】
さらに、100U/mL、300U/mL、500U/mL、700U/mLおよび1000U/mLの濃度を有するインスリングラルギン製剤の沈殿物を顕微鏡で調べた。これらの研究により、濃度の増加に伴って明らかにより大きい粒子がもたらされるという沈殿物特性の違いが明らかとなった。
【0022】
さらに、溶解特性に関しては、より高い濃度のインスリングラルギン製剤の影響がインビトロ試験系を用いることによって調査される。それを行うために、沈殿の研究が、インビボ条件を模擬するpH7.4のリン酸緩衝液を用いて実施される。
【0023】
沈殿したインスリンの上清は、インスリングラルギン含有量を測定するためにHPLC技術を用いて試験される。
【0024】
WO2008/013938 A2は、684U/mLの濃度でインスリングラルギンを含有する水性医薬製剤を開示している。
【課題を解決するための手段】
【0025】
本発明はインスリングラルギンU300製剤に限定されず、本明細書中で詳述しているような他のより高い濃度のインスリングラルギン製剤を用いても有効であるが、本明細書中に記載される臨床試験は、インスリングラルギンU300製剤を用いて実施した。
【0026】
1mLのインスリングラルギンU300製剤は、10.913mgの21A−Gly−30Ba−L−Arg−30Bb−L−Arg ヒトインスリン[300IUのヒトインスリンと等モル]、90μgの亜鉛、2.7mgのm−クレゾール、20mgのグリセリン 85%、HClおよびNaOHを含有し(pH4.0);比重は1.006g/mLである。
【0027】
しかしながら、賦形剤の種類およびそれらの濃度に関しては変更が可能である。
【0028】
この医薬製剤は、200−1000U/mLのインスリングラルギン[200−1000IU ヒトインスリンと等モル](この中で、当該製剤の濃度は684U/mLではない)、好ましくは250−500U/mLのインスリングラルギン[250−500IU ヒトインスリンと等モル]、より好ましくは270−330U/mLのインスリングラルギン[270−330IU ヒトインスリンと等モル]、およびさらにより好ましくは300U/mLのインスリングラルギン[300IU ヒトインスリンと等モル]を含有する。
【0029】
界面活性剤、例えば、とりわけ非イオン性界面活性剤を医薬製剤に加えることができる。特に、調剤用に市販されている界面活性剤が好ましく、例えば以下のようなものである:グリセリン、ソルビトールなどのような高アルコールの部分エステルおよびエーテルならびに脂肪酸エステルおよびエーテル(Span(登録商標)、Tween(登録商標)、特にTween(登録商標)20およびTween(登録商標)80、Myrj(登録商標)、Brij(登録商標))、Cremophor(登録商標)またはポロキサマー。これらの界面活性剤は、5−200μg/ml、好ましくは5−120μg/ml、そして特に好ましくは20−75μg/mlの濃度で医薬組成物中に存在している。
【0030】
この製剤は、防腐剤(例えば、フェノール、m−クレゾール、p−クレゾール、パラベン)、等張化剤(isotonic agent)(例えば、マンニトール、ソルビトール、ラクトース、デキストロース、トレハロース、塩化ナトリウム、グリセリン)、緩衝物質、塩、酸およびアルカリをさらに含んでよく、さらなる賦形剤も含んでよい。これらの物質は各場合に個々にかまたは混合物として存在し得る。
【0031】
グリセリン、デキストロース、ラクトース、ソルビトールおよびマンニトールは、100−250mMの濃度で、NaClは150mMまでの濃度で医薬製剤中に存在し得る。緩衝物質(例えば、リン酸エステル、酢酸エステル、クエン酸エステル、アルギニン、グリシルグリシンまたはTRIS(すなわち2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオール)緩衝液および対応する塩)は、5−250mM、好ましくは10−100mMの濃度で存在し得る。さらに賦形剤はとりわけ塩類またはアルギニンであり得る。
【0032】
この製剤の亜鉛含有量は、0−1000μg/mL、好ましくは20−400μg/mLの亜鉛、最も好ましくは90μg/mLの存在量によって到達する濃度範囲である。この亜鉛は塩化亜鉛の形態で存在していてもよいが、その塩は塩化亜鉛に限定されない。
【0033】
この医薬製剤において、グリセリンおよび/もしくはマンニトールは、100−250mmol/Lの濃度で存在し得、ならびに/またはNaClは、150mmol/Lまでの濃度で存在し得る。
【0034】
この医薬製剤において、緩衝物質は、5−250mmol/Lの濃度で存在し得る。
【0035】
本発明のさらなる目的は、例えばインスリンの放出を遅らせる塩のようなさらなる添加剤を含む医薬品インスリン製剤である。このような遅延放出インスリンと上記の製剤との混合物は、本発明に含まれる。
【0036】
本発明のさらなる目的は、このような医薬製剤の製造方法である。これらの製剤を製造するために、これらの成分を水に溶解し、HClおよび/またはNaOHを用いてpHを調製する。同様に、本発明のさらなる目的は、糖尿病の治療のためのこのような製剤の使用である。
【0037】
本発明のさらなる目的は、インスリン、インスリンアナログもしくはインスリン誘導体またはこれらの製剤の製造工程の間の、安定剤としての界面活性剤の使用または添加である。
【0038】
本発明はさらに、グルカゴン様ペプチド−1(GLP1)またはそのアナログもしくは誘導体、あるいはエキセンジン−3もしくは−4またはそのアナログもしくは誘導体、好ましくはエキセンジン−4をもさらに含む、上記の製剤に関する。
【0039】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
H−desPro36−エキセンジン−4−Lys6−NH2、
H−des(Pro36,37)−エキセンジン−4−Lys4−NH2および
H−des(Pro36,37)−エキセンジン−4−Lys5−NH2、
またはこれらの薬理学的に許容される塩。
【0040】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
desPro36[Asp28]エキセンジン−4(1−39)、
desPro36[IsoAsp28]エキセンジン−4(1−39)、
desPro36[Met(O)14,Asp28]エキセンジン−4(1−39)、
desPro36[Met(O)14,IsoAsp28]エキセンジン−4(1−39)、
desPro36[Trp(O2)25,Asp28]エキセンジン−2(1−39)、
desPro36[Trp(O2)25,IsoAsp28]エキセンジン−2(1−39)、
desPro36[Met(O)14Trp(O2)25,Asp28]エキセンジン−4(1−39)および
desPro36[Met(O)14Trp(O2)25,IsoAsp28]エキセンジン−4(1−39)、
またはこれらの薬理学的に許容される塩。
【0041】
本発明はさらに、ペプチド−Lys6−NH2がエキセンジン−4のアナログのC末端に結合している、前出の段落で記載されたような製剤に関する。
【0042】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
H−(Lys)6−des Pro36[Asp28]エキセンジン−4(1−39)−Lys6−NH2
des Asp28Pro36,Pro37,Pro38 エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5 des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Trp(O2)25,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
H− des Asp28 Pro36,Pro37,Pro38[Trp(O2)25]エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Met(O)14,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
des Met(O)14 Asp28 Pro 36,Pro37,Pro38 エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro 37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
H−Asn−(Glu)5 des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
des Asp28 Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25]エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28] エキセンジン−4(1−39)−(Lys)6−NH2、
またはその薬理学的に許容される塩。
【0043】
本発明はさらに、Arg34、Lys26(Nε(γ−グルタミル(Nα−ヘキサデカノイル))) GLP−1(7−37)[リラグルチド]またはその薬理学的に許容される塩をさらに含む、上記の製剤に関する。
【0044】
一実施態様において、本発明は200−1000U/mL[200−1000IU ヒトインスリンと等モル]、好ましくは200U/mL〜650U/mL、さらに好ましくは700U/mL〜1000U/mL、より好ましくは270−330U/mLの範囲で、そして最も好ましくは300U/mLの濃度でインスリングラルギンを含む水性医薬製剤に関するが、ただし該製剤のインスリングラルギンの濃度は、684U/mLではない。
【0045】
さらに、その製剤は、エキセンジン−4のアナログ、例えば、リキシセナチド(lixisentatide)、エクセナチドおよびリラグルチドをも含み得る。これらのエキセンジン−4アナログは、インスリングラルギン1Uあたり0.1μg〜10μg、好ましくはインスリングラルギン1Uあたり0.2〜1μg、およびより好ましくはインスリングラルギン1Uあたり0.25μg〜0.7μgの範囲でその製剤中に存在する。リキシセナチドが好ましい。
【0046】
さらに、その水性医薬製剤は、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される1種またはそれ以上の賦形剤を含み得る。具体的には、その水性医薬製剤は、90μg/mLの亜鉛、2.7mg/mlのm−クレゾールおよび20mg/mlのグリセリン 85%を含み得る。場合により、その水性医薬製剤は、20μg/mLのポリソルベート20を含み得る。
【0047】
その水性医薬製剤のpHは、μμ3.4〜4.6の間、好ましくは4または4.5である。
【0048】
本発明は、I型およびII型糖尿病を治療する方法に関し、該患者に本発明の水性医薬組成物を投与することを含む。
【0049】
開示された種々の濃度範囲で好ましいのは300U/mLの濃度であり、好ましいインスリンアナログはインスリングラルギンである。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0050】
このインスリンは、1日1度投与されるのが好ましいが、必要に応じて1日に2度投与することも可能である。投薬の必要性は、正常または許容可能な血中グルコース濃度の達成によって決定される個々の患者の必要性による。
【0051】
本発明はまた、該患者に本発明の水性医薬製剤を投与することを含む、I型およびII型糖尿病患者の治療においてインスリングラルギンの曝露の持続時間を延長する方法に関する。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0052】
本発明はまた、インスリングラルギンを用いて、I型およびII型糖尿病患者の治療において低血糖の発生を低減する方法にも関し、該患者に本発明の水性医薬製剤を投与することを含む。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0053】
本発明はまた、I型およびII型糖尿病患者の治療において、インスリングラルギンによってピークのない長時間作用型基礎インスリンを提供する方法に関し、該患者に本発明の水性医薬製剤を投与することを含む。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0054】
I型糖尿病およびII型糖尿病の治療における前出の項目のいずれかに従う水性製剤の使用。
【0055】
本願を、いくつかの実施例を用いて以下に説明するが、これらの例は決して限定することを目的としていない。
【0056】
実施例1:プロトコルの記載
本試験は、6度の来院での健常な被験者における単一施設、無作為化、対照群あり(controlled)、単盲検、4期、2治療、2−シーケンスクロスオーバー試験であった:
来院1:スクリーニング(SCR)
来院2〜5、期間(Period:P)1−4:治療、正常血糖クランプ期間
来院6:試験終了時(EOS)
被験者に、0.4U/kgのインスリングラルギンU100の単回皮下用量を投与、および代替的にはインスリングラルギンU300を、2点の向かい合う臍周囲領域(左、右、左、右)の部位に異なる4日で注射した。この治験薬を治療RとTを2シーケンスで反復(RTTRまたはTRRT)して、P1〜P4期に投与した。4〜18日間の休薬期間により各投薬日を離した。
R:0.4U/kg−体重のインスリングラルギンU100(市販の製剤;基準薬)
T:0.4U/kg−体重のインスリングラルギンU300(試験薬)
P1は、SCR後多くとも3〜21日に行わねばならない。EOS来院は、P4後の4〜14日になされなければならない。
【0057】
P1〜P4の期間中、人工膵(Biostator)を被験者に連結して、血中グルコースの測定とグルコース注入率の調整を行った。血中グルコース濃度およびグルコース注入率(GIR)を、当該治験薬の皮下注射前の90分間(ベースライン期間)、および治験薬投与後の30時間にわたってモニタリングした。治験薬投与の60分、30分および5分前に測定された3つの空腹時血中グルコース値の平均値として決定された個々の空腹時血中グルコース濃度を5%下回る血中グルコース濃度を維持するために、20%のグルコース液剤の注入を開始した。GIRのプロファイルが得られた。血液サンプルを、正常血糖クランプ期間中に所定の回数採取し、血清インスリングラルギン濃度を決定した。グルコースクランプ期間中、被験者は水道水を除いて絶食状態とした。
【0058】
個体の本試験の所要期間は、SCRからEOS来院の間が最大13週間までの見込みであった。
【0059】
このプロトコルを、検討および書面による承認のために、個々の倫理委員会および/または治験審査委員会に提出した。このプロトコルは、18th World Health Congress(ヘルシンキ、1964)の勧告および全ての準拠する改正事項を満たしている。このプロトコルはまた、本試験が行なわれたドイツの法律、条例、ならびに全ての準拠するガイドラインをも満たしていた。全ての試験関連手順を行なう前に、インフームドコンセントを得た。
【0060】
実施例2:被験者の選択
20名の試験完了者を確保するために、24人の健常な被験者に対して治療するように計画した。
【0061】
以下の基準の全てを満たしている被験者が、本試験への登録を考慮された:
デモグラフィー
・18歳〜50歳の年齢のいずれかの性別である被験者:
・体重は50kg〜110kgであり、ボディマス指数(BMI)は18〜28kg/m2である;
健康状態
・以下の包括的な臨床評価(詳細な病歴および完全な身体検査)により健常であると証明されていること;
・少なくとも3ヶ月間、喫煙していないこと;
・12誘導心電図、およびバイタルサイン、但し、治験責任医師が、異常を臨床上無関係であると判断した場合を除く;
・背臥位で5分休止後の正常バイタルサイン:
95mMHg≦収縮期血圧≦140mMHg;
45mMHg≦拡張期血圧≦90mMHg;
40bpm≦心拍数≦100bpm;
・正常な12誘導ECG;120ms<PR<220ms、QRS<120ms、QTc≦430ms
(女性の場合:QTc≦450ms);
・健常な被験者の各臨床検査項目は正常範囲内にあること、但し、治験責任医師が異常を臨床的に無関係であると判断する場合を除く;
しかしながら、血清レアチニンおよび肝酵素(AST、ALT)は、臨床検査ノルムの上限を厳密に下回らなければならない;
・空腹時血清グルコース(≦100mg/dL)およびグルコシル化ヘモグロビン(HbA1c≦6.1%)として定義される正常代謝コントロールを有すること;
・被験者は、本試験に参加する前に、少なくとも4週間、日常的に用いている処方薬治療を中止せねばならない;
【0062】
女性の被験者についての義務
・妊娠する可能性があり(閉経前および外科手術による不妊処理を行っていないかまたは閉経後2年未満として定義される)、性的に活発である女性被験者は、適切な避妊をおこなわなければならない。適切な避妊とは、インプラント、経口避妊薬またはホルモン剤と組み合わせた注射剤、IUD(子宮内避妊器具)のような高度に効果的な避妊法(パールインデックス< 1%)として定義される。本臨床試験の目的のための閉経後とは2年以上の無月経または外科的に不妊であることを含む;
・女性被験者は、前試験スクリーニングの間、および最初のクランプ前に、尿β−ヒト絨毛性ゴナドトロピン(beta−HCG)妊娠テストが陰性でなければならない。
【0063】
規則(Regulations)
・本試験に関する全ての手順の前に書面によるインフォームドコンセントを行う;
・健康保険制度でカバーされており、そして/または生物医学研究に関して施行された国内法令の勧告を遵守している;
・いかなる行政または法的な監督下にはない。
【0064】
以下の項目のいずれかにあてはまる被験者は、本試験に含められていない:
病歴および臨床状態
・臨床上関連のある心血管、肺、胃腸、肝臓、腎臓、代謝、血液、神経、精神、全身性、眼または感染の疾患のいずれかの病歴または存在;いずれかの急性感染性疾患または急性疾患の兆候;
・薬物アレルギーの存在もしくは病歴、または医師によりアレルギー性疾患と診断され治療を受けた;
・キサンチン塩基を含む飲料の過剰摂取(>4カップまたはグラス/日);
・以下からの禁忌(正常範囲に従う−値が正常範囲外である場合は、治験責任医師がこの異常値が臨床的に無関係と理解した場合、その被験者は含まれ得る):
−病歴/手術歴および身体検査
−臨床検査(血液、臨床化学およびディップスティックによる検尿)
−標準的な12誘導心電図
−血圧および心拍数
・試験への参加前4週中の所定の薬物で継続中の治療または所定の薬物で定期的な治療
・本試験の目的を妨げ得る、試験前3ヶ月中の臨床的に有意な疾患の徴候、または試験前4週中のいずれかの主要な内科的疾患の兆候(治験責任医師の意見に従う)
・薬物の吸収、分布、代謝または排泄を妨げることが知られている疾患の存在もしくは続発症または他の病態
・薬物またはアルコールの乱用歴
・類似した化学構造を持つ治験薬または薬物に対する過敏症既往歴
・進行性致死的疾患
・本試験中に事前に計画された手術
・前3ヶ月中の500mLを超える献血
被験者は1度より多くの本試験への登録は許可されない。
【0065】
一般的条件
・治験責任医師の判断において、本試験中、服薬遵守しそうにないか、または言語の問題もしくは精神発達遅滞のため、もしくは被験者が本試験の性質、範囲および起こりうる結果を理解できなくする精神状態に起因して協力できない被験者
・適用し得る規則に従って、以前の研究の排除期間内にある被験者;
・被験者が本プロトコルの実施に直接的に関与する治験責任医師もしくはいずれかの治験責任分担医師、研究助手、薬剤師、治験コーディネーター、または他のそのスタッフである;
・SCR前の以前の30日以内の治験薬の受領
【0066】
生物学的状態
・以下のテストのいずれかに陽性反応である:HBs抗原、抗HCV抗体、抗HIV1抗体、抗HIV2抗体;
・SCRで尿の薬物スクリーニングでの陽性結果(アンフェタミン/メトアンフェタミン類、バルビツレート類、ベンゾジアゼピン類、カンナビノイド類、コカイン、オピエート類);
・アルコール呼気テスト陽性
【0067】
実施例3:治療
試験治療の詳細
【表1】

【0068】
ランタス(登録商標)/インスリングラルギン製剤の用量の算出
各被験者に投与するインスリングラルギンの量(0.4U/kg)を算出するために、体重(kg)は少数点以下1桁で測定し、以下の実施例に示すように算出したインスリンの量は切り上げるかまたは切り捨てして丸めている:75.3kgの体重の被験者に30Uのインスリンを投与した(75.3×0.4=30.12 切り捨てして30とする);74.4kgの体重の被験者に30Uのインスリンを投与した(74.4×0.4=29.76 切り上げして30とする)。期間1の日1に記録した体重を、期間1の体重から2kgより多く体重が変化しない限り、期間2、3および4についても治験薬用量の算出に用いた。
【0069】
ユニット中の量は、インスリングラルギンU100とインスリングラルギンU300の両方とも同じであった。この比重は、両方の薬品について同じである。しかし、インスリングラルギンU100と比較して、インスリングラルギンU300はインスリングラルギンの濃度が3倍高いことを考慮して、インスリングラルギンU300については注射すべき容量従って重量を1/3とした。個々の用量を投与する注射器を秤量して調製した。正味質量は、治験責任医師のソース文書(source-documentation)にのみ記録した。
【0070】
注入剤の用量の算出および調製
【表2】
【0071】
グルコース液剤:個々の被験者の血中グルコースを決定した目標レベルに維持するために、20% グルコース液剤を人工膵(Biostator)を用いて注入した。第2の注入ポンプ(人工膵(Biostator)の一部)は、0.9% 塩化ナトリウム溶液を送達して、ラインの開存(line patent)を維持した。必要な20% グルコース液剤の量が人工膵(Biostator)の注入容量を超える場合、第2のグルコース注入ポンプを係合した。
【0072】
ヘパリン: 100mLの0.9% 塩化ナトリウム溶液中の10000IU ヘパリンを、二腔カテーテルに約2mL/hの速度で注入し、人工膵(Biostator)による血中グルコースの測定のためのその開存を維持した。
【0073】
盲検法(blinding methods)の説明:
本試験は、単盲検(single-blinde)である。容量の異なる注射は、投薬の盲検を不可能にする。注射は、権限のある医師であるか、さもなければ本試験に関与していない医師によってなされた。治験責任医師は無作為化コードにアクセスした。
【0074】
治療群への被験者の割り当て法
手順を臨床試験プロトコルで定めた後、本治験薬を本試験に含めた被験者にのみに投与した。
【0075】
P1〜P4で注射すべき2つのランタス(登録商標)製剤の治療シーケンスのために無作為化スケジュールを、乱数表とリンクさせ、性別により階層化して作製した。
【0076】
期間1の日1の朝、被験者がプロトコルに定められている基準を満たしていることを治験責任医師が確認すると直ぐに、適格な被験者をその施設により無作為化した。その後その乱数を、P1前に被験者の適格性が確認された順に被験者番号に割り当てた。SCR後の性別階層認定のために、第一の被験者に適切な性別階層に対する第一の乱数を付与した。階層内に認定される次の被験者には、その階層内の次の乱数を付与した。
【0077】
その乱数は、被験者に治療キットを割り当てるための治療キット番号として用いた。各被験者は、割り当てられた治療キット番号を持つ治験薬を投与された。この治療キットには、IP付き一般情報、治療キット番号、期間番号、コンテナボックス上に被験者番号を書くためのフィールド、および地方条例によって要求される追加記載が含まれている。
【0078】
試験を永続的に中止した被験者は、既に与えられている場合、被験者番号および無作為化番号を保持しておいた。
【0079】
包装およびラベリング
本治験薬は、無作為化プランに従って、サノフィ−アベンティス ドイチュランド GmbH、フランクフルト アム マイン ドイツにより包装された。本治験薬を含むカートリッジおよびそれらをパックしたカートンは、試験番号、無作為化番号、バッチ番号、貯蔵条件、治験依頼者およびP番号でラベルされた。
【0080】
治験薬の供給は、一度の出荷のものを受けた。全てのコンテナは、同一形式のラベルが付されていた。さらに、注射器用のラベル1セットが供給された。治験薬と予備薬剤は、異なる冷蔵庫に保管した。
【0081】
治験薬投与の前に、薬剤師または薬剤師に指示された人が適切な治験薬を入れた注射器を調製し、その注射器に、この治験薬のコンテナに従う、被験者番号、無作為化番号および適切な期間をラベル付した。
【0082】
このラベル付の内容は、地方条例の仕様および要件に従って行った。
【0083】
貯蔵条件
本治験薬を+2℃〜+8℃の温度で光から保護して保管した。本試験薬は、凍結しないようにした。調製中は、その薬剤を光から保護する必要はなかった。
【0084】
予備サンプル(ランタス(登録商標)U100を300カートリッジ、およびインスリングラルギンU300を300カートリッジ)を、この試験施設レベルで同じ条件で保管確保した。
【0085】
実施例4: 治験製品の評価
活性または薬力学
インスリングラルギンによるインスリン受容体の刺激は、作用様式である。それに引き続く末梢グルコース取り込みおよび内因性グルコー産生の抑制は、血中グルコース濃度の減少がもたらす糖動的効果を含む。結果としてもたらされたグルコース利用は、血中グルコース濃度を一定に維持するために必要とされるグルコースのゲージ(gauge)によって最もよく特徴づけられる。
この正常血糖クランプ技術は、インスリングラルギンの注射後、ベースラインレベルより5%低く血中グルコース濃度を維持するのに必要なグルコースの量を評価するために用いられている。
【0086】
臨床評価方法
オンラインでのグルコース測定を、グルコースオキシダーゼ法を用いて人工膵(Biostator)(Life Sciences instruments、Elkhart、IN、USA)により行った。
オフサイトの血中グルコースもまた、グルコースオキシダーゼ法を用いてSuper GLグルコース分析装置により測定した。
【0087】
薬力学変数/エンドポイント
皮下注射されたインスリンの単位(用量)あたりの利用されたグルコース量が、糖動的効果の尺度(measure)である。
連続的に記録されたグルコース注入率(GIR)は、注射したインスリンの経時作用プロファイルを反映している。
【0088】
主要変数/エンドポイント
主要薬力学変数は、24時間以内のグルコース注入率時間曲線下面積[GIR−AUC0-24h(mg・kg-1)]である。
【0089】
副次変数/エンドポイント
副次薬力学変数は、50% GIR−AUC0-24hまでの時間[T50%−GIR−AUC(0-24h)(h)]である。
【0090】
薬物動態学
サンプリング時間
血清インスリングラルギンおよびC−ペプチドの濃度の評価用に血液サンプルを治験薬の皮下注射の1時間前、30分前および直前に採取し、その後は、注射の30分後、1時間後、2時間後、その後は最大で24時間までの2時間おきに、そして30時間後に採取した。
インスリングラルギンサンプルの番号を、P00、P01、P02、P03、P04などと付け、C−ペプチドサンプルの番号をC00、C01、C02、C03、C04、などと付けた(試験のフローチャートも参照のこと)。
【0091】
薬物動態学サンプリング数
最低18のサンプルをクランプ来院(P1〜P4)ごとに採取した。1被験者あたり総計72サンプルを採取した。
【0092】
PK取り扱い手順
サンプル採取の正確な時間は、CRFで必ず記録する。薬物動態学サンプル(インスリングラルギン、C−ペプチド)の保管および配送については、特別な手順を用いた。
【0093】
生物学的分析法
生物学的分析を、医薬品の安全性に関する非臨床試験の実施基準(Good Laboratory Practice(GLP))のOECD原則(1997年に改定)、ENV/MC/CHEM(98)17およびその地域に適用されるGLP規則にて認められている、このタイプの試験に適用可能なGLP条件を基に実施した。
予備サンプルは利用できないので、インスリングラルギンの測定を優先する。
【0094】
インスリングラルギン
血清インスリングラルギン濃度を、インスリングラルギン用に校正したヒトインスリン用のラジオイムノアッセイ(RIA)(インスリンRIAキット、ADALTIS、イタリア)を用いて測定した。キットREF 10624。
このアッセイでの定量下限(LLOQ)は、4.92μU/mLであった。
【0095】
C−ペプチド
血清C−ペプチド濃度をC−ペプチド用のラジオイムノアッセイ(RIA)(C−ペプチド RIAキット、ADALTIS、イタリア)を用いて測定した。キットREF C−ペプチド10282。
定量下限(LLOQ)は、0.090nmol/Lであった。
【0096】
生物学的分析法の要約
被検体 インスリン、C−ペプチド
マトリックス 血清
分析技術 RIA
定量下限 インスリン 4.92μU/mL;C−ペプチド 0.090nmol/L
アッセイ容量 インスリン 100μL;C−ペプチド 100μL
方法参照 Adaltis S.p.A.イタリア;キットREF 10624 インスリン(方法No.435VAL02)およびキットREF C−ペプチド10282(方法No.DMPK/FRA/2003−0002)
【0097】
薬物動態学変数/エンドポイント
インスリングラルギン濃度の経時曲線が、皮下注射したIPの全身インスリン曝露の尺度(measure)であった。
【0098】
主要変数/エンドポイント
主要薬物動態学変数は、血清インスリングラルギン濃度の時間曲線下面積[INS−AUC0-24h(μU・h・mL-1)]であった。
【0099】
副次変数/エンドポイント
副次薬物動態学変数は、50% INS−AUC0-24hまでの時間[T50%−INS−AUC(0-24h)(h)]であった。
【0100】
サンプリングした血液容量
【表3】
【0101】
本試験の盲検を保護するための測定
本試験は単盲検試験である。生物学的分析測定は、臨床試験が完了した後に行なった。治療コードは、予測できない全ての重篤な有害事象(SAE)を報告するために知られており、治験責任医師および/または治験依頼者のいずれかの判断に従ってIPの使用と合理的に関連付けられている。
【0102】
実施例5:試験手順
来院スケジュール
スクリーニング手順
各被験候補者の診療録を試験の開始前にチェックし、参加への適格性を決定した。被験者は、SCRでのスクリーニング試験前に10時間絶食(水を除く)した。
【0103】
以下の項目/検査を評価した:
・年齢、および人種
・身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系を含む)
・関連する病歴および手術歴(本試験に関連する所見のみを記録すべきである)
・人体計測:身長および体重、BMI[体重(kg)・(身長(m))-2]の計算
・血圧および心拍数(背臥位で5分後、および立位で3分後)
・深部体温(鼓膜)
・標準12誘導ECG
・血液学的状態、臨床化学、および検尿(ディップスティック)
・凝固状態(INR、aPPT)
・尿の薬物スクリーニング
・アルコールスクリーニング(呼気検知器)
・空腹時血中グルコース(≦100mg.dL-1)およびグリコシル化ヘモグロビン(HbA1c≦6.1%)として定義される正常な代謝コントロール
・B型/C型肝炎およびHIV試験
【0104】
被験者がスクリーニングできない場合、スクリーニング試験の臨床結果を含むSCRで得られた全てのデータをその被験者の診断録に記載した。
【0105】
来院のタイプによる記述
期間
各試験期間(P1〜P4)は日1および日2の2日間である。日1は、正常血糖クランプと治験薬の投与の開始日であった。日2は、治験薬の投与後30時間にわたる正常血糖クランプの終了の日であった。試験期間(P1〜P4)の間には、4〜18日の休薬期間を設けた。各治験薬投与前2日間は、激しい運動(例えば、マウンテンバイク、重労働の庭作業など)をさせなかった。アルコール飲料、グレープフルーツジュース、およびキサンチン誘導体を含む刺激性飲料(お茶、チョコレート、コーヒー、コカコーラ(商標)のような飲料など)およびグレープフルーツの摂取は、正常血糖クランプの24時間前から完了まで許可しなかった。被験者は各試験期間(P1〜P4)の日1の10時間前から絶食(水を除く)し、正常血糖クランプの終了まで絶食(水を除く)を続けた。被験者は、各クランプの来院につき約32時間クリニックに滞在せねばならなかった。
【0106】
期間1の日1の朝、276001001で始まる9桁の被験者番号を被験者に割り当てた。SCRに入る資格を与えられた次の被験者には、被験者番号276001002を付与した。最初の被験者には無作為化番号101を与えた。資格を与えられた次の被験者には無作為化番号102を与えた。
【0107】
被験者に、彼らの体調に臨床的に有意な変化がなく、その前の期間からのプロトコル中で定められたような一般事項および食事制限を遵守することを確実にするよう依頼した。試験規則に違反した被験者は、本試験への参加から除外した。違反の種類により、被験者はその特定の期間のみ除外されることが可能であり、試験日の計画を立て直した。どのプロトコル違反も、前もってケースバイケースに基づいて治験依頼者と議論した。
【0108】
最期の期間以来の被験者の健康状態のいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0109】
血圧、心拍数および深部体温(鼓膜)を、日1の朝、各治験薬投与(日2)30時間後のクランプ手順の前および後に、少なくとも5分間安静にしてから背臥位で測定した。体重、アルコールスクリーニングおよびRBC、Hb、HcT(P3およびP4のクランプ期間の前のみ)は、日1の朝、クランプを開始する前にのみ評価を行った。
【0110】
各期間の日1、被験者は午前6時30分にクリニックに入院した。上記の検査を通過後、被験者は3本の静脈ラインを準備された。左腕の手の甲の静脈または手首外側の静脈に逆行様式でカニューレを導入し、人工膵(Biostator)(Life Sciences instruments、Elkhart、IN、USA)に連結して血中グルコースの測定のために動脈血化した静脈血を連続的に記録した。動脈血化を達成するために、左手を約55℃の「ホットボックス(Hot−Box)」に入れた。第2の静脈ラインは、左腕の肘前静脈でとり、血清インスリングラルギンおよび参照血中グルコースの測定のためのサンプルを採取するために用いた。第3の静脈ラインは反対側の前腕にカニューレを導入し、人工膵(Biostator)を用いて20%のグルコース液剤および0.9%の生理食塩水を注入した。
【0111】
人工膵(Biostator)により血中グルコース濃度を測定し、グルコース注入率を調節して血中グルコース濃度を治験薬投与の60分前、30分前および5分前に測定した3回の空腹時血中グルコースの値の平均値として算出した個々の空腹時血中グルコースを5%下回る値に維持した。血中グルコースの測定のために追加の血液サンプル0.3mLを治験薬の投与60分前、30分前および5分前に採取し、グルコースオキシダーゼ法に基づいて、臨床基準値と照らし合わせて確認した。
【0112】
午前9時頃、インスリングラルギンU100(市販製剤)またはインスリングラルギンU300のいずれかを、臍から5cm(左、右、左、右)の臍周囲領域に標準化された皮下脂肪厚法(skin fold technique)を用いて注射した。0.30mm×8mm(30G)の針を備えた0.5mL容量のU100インスリン注射器(製造元:Beckton & Dickinson)を用いた。
【0113】
治験薬に彼らそれぞれの治療キット番号、被験者番号(無作為化後にコンテナボックスに記載したもの)、および期間番号のラベルを付した(8.5章の包装およびラベリングを参照のこと)。
【0114】
治験薬の投与後、20%グルコース液剤の注入を、一旦血中グルコース濃度を個々の空腹時濃度から5%下げ、そのレベルを維持する可変速度で開始した。このクランプ期間の所要時間は30時間であった。グルコース送達速度は、予め定めてあったアルゴリズムを用いて1分間隔で血中グルコースの変化に応じて人工膵(Biostator)によって調整した。この人工膵(Biostator)からの血中グルコース値は、クランプ全体にわたり30分間隔でグルコースオキシダーゼ法に基づいた臨床検査基準値と照らし合わせて確認した。必要に応じ、人工膵(Biostator)を臨床検査基準値法の結果に従って再校正した。被験者は、クランプ期間中背臥位のままであった。
【0115】
血清インスリングラルギンおよびC−ペプチド濃度の測定のための血液サンプルを投薬1時間前、30分前および直前、その後治験薬投与の30分後、1時間後、2時間後、その後は最大で24時間までの2時間おきに、そして30時間後に採取した。
【0116】
各試験期間(P1〜P4)の日2に、正常血糖クランプが完了した後食事を提供した。血圧、心拍数および深部体温(鼓膜)を記録し、血中グルコース測定用のサンプルを採取した。被験者は、治験責任医師によって安全が確認された後、クリニックから退院した。
【0117】
注射部位は、全クランプ期間中観察した。被験者の健康状態におけるいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0118】
血液学的な安全性(Safety Hematology)
P3におけるRBC、HbおよびHctを、P4での貧血に備えて、分析した。陽性であった場合、P3とP4の間隔を許される最大の18日まで延長し、P4の前に追加のRBC、HbおよびHct評価を行った。
【0119】
退院手順
被験者は、P4後、4〜14日間の間に、EOS来院のために再来院した。被験者は10時間絶食(水を除く)した。最終期間以降の被験者の健康状態におけるいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0120】
以下の項目/検査を評価した:
・身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系を含む)
・体重
・血圧および心拍数(背臥位で5分の後)
・深部体温(鼓膜)
・標準12誘導ECG
・血液学的状態、臨床化学、および検尿(ディップスティックによる)
・尿中のβ−HCGテスト(女性に対してのみ)
被験者は、有効な安全性データの治験責任医師によるレビューが完了した後、各期間の日2に退院した。
【0121】
生体サンプルの採取スケジュール
血液
SCR(スクリーニング):
・血液学、臨床化学、HbA1c、血清学(B/C型肝炎テスト、HIV試験):約20mLの血液を採取した。
P1〜P4(日1および日2)
・血中グルコース
人工膵(Biostator)は、治験前の期間を含む全クランプ期間にわたり1分間隔で血中グルコースを自動測定する。人工膵(Biostator)に必要とされる血液量は、2mL・h-1である。およそ252mLの血液量が、4期間に人工膵(Biostator)でのグルコース測定に必要とされる。人工膵(Biostator)から、血中グルコース値をチェックするための血液サンプル(0.3mL)を投薬の60分前、30分前、5分前および0分前、ならびに投薬後クランプの終わり(30時間)までは30分間隔で採取した。およそ41mLの血液量が4期間に採取された。
・血清インスリングラルギンおよびC−ペプチドの濃度
静脈血液サンプル(3.5mL)を、投薬1時間前、30分前、および直前と、投薬30分後、1時間後、2時間後および24時間までは2時間ごとに、そして30時間後に採取した。およそ252mLの血液量を4期間で採取した。インスリングラルギンの測定を優先的に行った。予備サンプルは、C−ペプチド濃度の測定のためにのみ用いた。
・RBC、Hb、Hct
クランプ期間3および4を開始する前に静脈血液を採取した。約4mLの血液を2期間に採取した。
【0122】
試験終了時(EOS)来院:
・血液学、臨床化学:約12mLの血液を採取した。
・尿中の尿中のβ−HCGテスト(女性に対してのみ)
【0123】
SCR−EOSの総血液量:
試験期間全体で、各被験者あたり総計約585mLの血液を採取した。
【0124】
尿
定性的な尿の薬物スクリーニングをSCRおよびEOSに実施した。尿の薬物スクリーニングは、アンフェタミン/メトアンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエートからなる。定性的な安全性検尿(safety urinalysis)をSCRおよびEOSでディップスティックを用いて行った。安全性検尿は、以下の分析項目からなる:pH、タンパク質、グルコース、血液、赤血球、白血球、ビリルビン、ウロビリノーゲン、ケトン、比重、および亜硝酸塩。
【0125】
他の試験変数の測定スケジュール
身体検査は、SCRおよびEOSにて実施した。
深部体温(鼓膜)は、SCR、クランプ期間の前および後のP1〜P4、およびEOSに記録した。
血圧および心拍数は、SCRおよびEOSで、背臥位で約5分間安静にした後、および立位で3分間安静した後にも測定した。P1〜P4においては、血圧および心拍数を日1の朝にクランプ手順を開始する前に少なくとも5分間背臥位で安静にした後と、各治験薬投与30時間後のクランプ手順が完了した後(日2)に記録した。
心電図(標準12誘導)は、SCRおよびEOSに記録した。
体重および身長は、SCRで測定した。体重は、P1〜P4の日1の朝(治験薬の投与前)およびEOSに記録した。
アルコールスクリーニング(エタノール、呼気検知器)は、SCRおよびEOS、ならびにP1〜P4の日1の朝(治験薬の投与前)に行った。
【0126】
試験における制限事項
日1の夜(P1〜P4)から、その期間(クランプ期間)を通して、被験者はアルコール、茶、コーヒー、柑橘系飲料またはコーラ飲料の摂取、喫煙を控えた。柑橘系果実の摂取も試験期間を通して禁止した。被験者は、この試験の継続期間を通して、最終の管理まで、激しい身体活動をせず安定したライフスタイルを守ることを求められた。
【0127】
原データ(source data)の定義
CRFに記録される以下に列挙した全ての評価は、以下に関する適切に署名され確認された原文書により裏付けられた:
・被験者の身元確認、病歴;
・臨床検査、バイタルサイン、体重および身長;
・臨床検査評価(laboratory assessments)、ECG;
・薬物動態の時点(Pharmacokinetic time points);
・来院および評価の日時;
・投与日時、および注射部位;
・AE;
・クランプの所要期間(開始および終了時)
・その他
このCRFは、他の項目についての原文書として考慮された。
【0128】
実施例6: 統計的考察
本実施例は、本試験のための統計学的分析プランの情報を提供する。統計学的分析プランは、被験者の参加前に立案された。
【0129】
被験者数の決定
INS−AUC(0-24h)が初期パラメータであり、それに従って被験者数の算出を実施した。
【0130】
この被験者数算出の目的のために、0.125と0.225の間で、自然対数変換したINS−AUC(0-24h)のいくつかの被験者内標準偏差(SDwithin)を考察した。平均生物学的同等性アプローチのための被験者数の算出法を、4期、2治療、2シーケンスクロスオーバーデザインのために用いた。製剤比率について、90%CIが完全に[0.80−1.25]内に含まれる場合、平均生物学的同等性がそのパラメータにより結論付けられた。
【0131】
試験HOE901/1022を、可変性における推算値のベースとした。試験HOE901/1022の統計解析に基づいて、0.175の値が、自然対数変換したスケールにおける被験者内標準偏差(SDwithin)を予測することができた。
【0132】
以下の表は、90%検出力(power)で0.85と1.15の間の真比率を推算する生物学的同等性対照間隔:[0.80−1.25]を用いて修正幾何平均値の平均生物学的同等性を表すのに必要とされる被験者の数を示している(対照製剤に対する試験)。
【0133】
【表4】
【0134】
このデザインでは、自然対数スケールでの真のSDwithinが0.175である場合、0.9の真比率を可能にする90%の能力で2つのインスリングラルギン製剤の同等性を示すためには、20名の被験体(1シーケンスあたり10名)が必要とされる。
総計24名の無作為化された被験者が、治験中止の可能性のある場合のために計上される。
【0135】
被験者の説明(Subject description)
被験者の内訳
関与した、無作為化された、曝露された(すなわち任意の量の治験薬投与を受けた)、完了した(すなわち、全ての治験期間を完了した被験者)、発生した中止の主な理由に従って中止した、被験者の数を含む、被験者の説明責任の詳細な要約が、各シーケンスおよび全ての被験者について作製される。
最終来院での被験者の内訳は、シーケンス群、本試験の終了時と治験薬の最終投与の日の内訳状態、最終来院の日付、中断の理由を含む一覧表で提示される。最初の治験薬投与の開始時または開始後に起こる本試験からの撤退は全て、治験総括報告書(CSR)の中に完全に記録される。
【0136】
プロトコルの逸脱
データベースのロックの前に、本プロトコルのコンプライアンスが、組み入れおよび除外基準、治療コンプライアンス、禁止された治療ならびに計画された評価のタイミングおよび有効性に関して調査される。プロトコルの逸脱はデータベースのロックの前に本試験チームによって確認され、欠損データおよびIPの中断を含むデータ審査報告書(Data Review Report)中に一覧にされ、大したことのない逸脱または主要な逸脱として分類される。
治験責任医師によって報告されるような組み入れおよび除外基準に対する個別の逸脱は一覧表にされている。
他の逸脱はCSRの本体中に一覧表にされ、および/または記載されている。
【0137】
解析母集団
解析されるべき母集団
任意の解析母集団から除外された被験者は、治療シーケンスおよび除外理由と共に列挙される。関連のある全ての情報がCSRに完全に記録される。
無作為化スケジュールに従って割り当てられた治療とは異なる治療を受けた被験者の事象においては、解析は無作為化治療に従うのではなく実際に受けた治療に従って行われる。
【0138】
薬物動態学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PKパラメータが有効である被験者は全て、薬物動態学母集団に含まれる。全てではなくいくつかの試験日においてPKプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0139】
薬力学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PDパラメータが有効である被験者は全て、薬物動態学母集団に含まれる、全てではなくいくつかの試験日においてGIRプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0140】
安全性の母集団
安全性の評価は、投与された治験薬の量にかかわらず、早くに中止した被験者も含めた、治験の投与を受けた被験者(曝露された母集団)をベースにした。
【0141】
人口統計学および基準値の特性
被験者の人口統計学特性、病歴および診断
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)は、体重および身長のデータより算出した:
BMI=体重[kg]・(身長[m])-2
【0142】
人口統計学および背景特徴に関する全ての変数を個別に列挙しまとめた。病歴および診断に関する組み入れ基準からの逸脱を個別に一覧にして記載した。
【0143】
ベースライン薬力学パラメータ
ベースライン血中グルコース濃度をシーケンスごとにまとめた。
【0144】
ベースライン安全性パラメータ
安全性変数について、変数として適用可能であっても、期間内または試験内の治験薬投与前に最期に組み込まれた値をベースライン値とした。そのベースラインの投与前値を投与前に再確認する場合、その再確認した値をベースラインとして認め、統計量に用いた。
【0145】
治験薬曝露の程度およびコンプライアンス
しかるべき場合には治験薬投与の詳細および補足情報を個別に列挙しまとめた。
【0146】
前/併用 治療/療法
前治療および併用療法(ある場合)を、世界保健機関−薬物文献リスト(Drug Reference List)(WHO−DRL)に従ってコード化し、個別に列挙した。
【0147】
薬力学変数の解析
薬力学変数の記載
インスリン投与に依存する、体重による被験者間の同等性を達成するために、解析用にGIRに関する全ての値を被験者の体重(kg)で割った。それにより以下のGIRは常に体重で標準化されたフルコース注入量を言及する。
【0148】
主要PD変数:
・体重で標準化されたグルコース注入率の時間曲線下面積
[GIR−AUC(0-24h)(mg・kg-1)]
副次PD変数:
・GIR−AUC(0-24h)の50%までの時間(h)[T50%−GIR−AUC(0-24h)(h)]
以下の追加PD変数を導出した:
・体重で標準化したグルコース注入率の、クランプの終了までの時間曲線下面積[GIR−AUC(0-end)(mg・kg-1)]
・体重で標準化したグルコース注入率の時間曲線下画分面積 [GIR−AUC(4-20h)、GIR−AUC(0-12h)、GIR−AUC(12-24h)(mg・kg-1)]
・体重で標準化した最大グルコース注入率 [GIRmax(mg・kg-1・min-1)]
・GIRmax までの時間 [GIR−tmax(h)]
有意義かつ信頼性のあるデータを提供するためにGIRmaxおよび対応するGIRmaxまでの時間の値は、各被験体について平滑化したGIR曲線より導出した。
【0149】
一次解析
GIR−AUC(0-24h)(mg・kg-1)についての相対的な生物有効性(活性)を推定するために、線形混合効果モデルを用いて未変換パラメータを解析した。
この混合モデルは、シーケンス、期間、製剤に関する固定項、ならびに製剤特異的(formulation specific)な被験者間および被験者内分散、および被験者付随製剤分散(subject−by−formulation variance)を伴うシーケンス内の被験者についてのランダム項を含む。その後製剤比率(T/R)についての点推定および90%信頼区間をフィエラーの定理(Fieller’s theorem)[Fieller、1954]に基づいて得た。
製剤比率についての信頼区間が[0.80−1.25]である場合、生物有効性(活性)が同等であると結論付けた。
その変数の分散についての仮定を確認した。
【0150】
二次解析/副次変数解析
個別の体重および平均体重で標準化したGIRプロファイルならびに時間に対する平均パーセンテージ累積プロファイルをプロットした。
PDパラメータは個別に列挙され、記述統計が作製される。
信頼限界を伴う製剤比率(T/R)を、1次解析について記載したような、対応する線形混合効果モデルを用いて、画分GIR−AUCs(mg・kg-1)および標準化グルコース最大注入速度[GIRmax(mg・kg-1・min-1)]について導いた。
50%−GIR−AUC(h)までの時間およびGIRmaxまでの時間[GIR−tmax(h)]は、ノンパラメトリックに解析した。
【0151】
クランプの実施
血中グルコース濃度の個別プロファイルをプロットした。
【0152】
安全性データの解析
まとめた全ての安全性データは、安全性母集団をベースにした。
安全性データの解析用の個別の治験薬投与期(on−treatment phase)は治験薬の最初の投与で始まり、EOS来院により終了した。
【0153】
有害事象(AE)
全てのAEは、MedDRA(使用時のバージョン)を用いてコード化される。
【0154】
定義
治験薬投与中のAE有害事象
全てのAEを以下のように分類した:
・試験治療下で発現した有害事象(TEAE):治療期間に初めて生じたかまたはそれ以前に発症していた場合治療中期間に悪化したAE;
・非試験治療下で発現した有害事象(NTEAE):治療期間に悪化することなく、治療期間外に生じたAE;
【0155】
製剤への割り当て
解析の目的のために、各TEAEをAEの発症および/または悪化前に与えられた最期の製剤に割り当てた。TEAEがある製剤において発生し、その後の製剤下で悪化した場合、両方の製剤に対するTEAEであると考える。
【0156】
欠測情報
情報の欠測または情報が矛盾していた場合、(例えば、部分的なデータまたは他の情報から)そのAEはTEAEではないと明らかに除外できる場合でない限り、AEはTEAEとしてカウントした。
AEの開始日が不完全または欠測していた場合、不完全な日が、AEが治験前に始まっていたと示している場合を除いて、最初の治験薬投与後に生じたと推定した。
【0157】
試験治療下で発現した有害事象
全てのAEを個別に列挙した。それらを、器官別大分類による概要を含め、製剤ごとにまとめた。
【0158】
死亡、重篤な有害事象および他の重要な有害事象
死亡、重篤なAEおよび他の重要なAEのような場合は全て個別に列挙し、総括報告書に詳細に記載した。
【0159】
治験の中止につながる有害事象
治療の中止につながるAEは、個別に列挙し、総括報告書に詳細に記載した。
【0160】
臨床検査値の評価
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。潜在的な臨床的に重要な異常(PCSA)および範囲外基準の定義は、パラメータにより報告した。
個別データを被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
正常範囲外の値の被験者およびPCSAのある被験者は、製剤ごとに、および治験評価の終了まで全期にわたり解析した。
ベースライン後PCSAのある被験者を列挙した。
【0161】
バイタルサイン
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。PCSAおよび範囲外基準の定義は、パラメータにより報告した。
PCSAのある被験者は、製剤ごとに、および治験評価の終了まで全期にわたりまとめた。ベースライン後PCSAのある被験者を列挙した。
生の値および導出されたパラメータは、製剤ごとに、および治験評価の終了まで全期にわたり解析した。個別データを、異常についてフラグを立てた上で、被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
【0162】
ECG
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。PCSAおよび範囲外基準の定義は、パラメータにより報告した。
SCRおよびEOSでのPCSAのある被験者を、全期にわたりまとめた。
個別データを、異常についてフラグを立てた上で、被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
【0163】
薬物動態学データの解析
薬物動態学パラメータ
事実上の相対時刻をPKパラメータを導出するのに用いた。
【0164】
主要変数:
・INS−AUC(0-24h)(μU・h.mL-1)
副次PK変数:
・INS−AUC(0-24h)の50%までの時間(h)[T50%−INS−AUC(0-24h)(h)]
以下の追加PK変数を導出した:
・画分INS−AUCs[INS−AUC(4-20h)、INS−AUC(0-12h)、INS−AUC(12-24h)(μU・h・mL-1)]
・クランプの終了までのINS−AUC [INS−AUC(0-end)(μU・h・mL-1)]
・最大血清インスリン濃度 [INS−Cmax(μU・mL-1)]
・INS−Cmaxまでの時間[INS−Tmax(h)]
【0165】
統計解析
記述統計
濃度データの記述統計は、プロトコル時間によって表わした。
個別のおよび平均の血清インスリン濃度プロファイルをプロットした。
血清インスリン濃度を個別に列挙し、時点あたりの記述統計を作製した。
PKパラメータの記述統計を製剤ごとに作製した。
C−ペプチドのプロファイルをプロットし、記述的に特徴づけた。
【0166】
一次解析
INS−AUC(0-24h)についての相対的なバイオアベイラビリティーを推定するために、線形混合効果モデルを用いて、対数変換したパラメータを解析した。
この混合モデルは、シーケンス、期間、製剤に関する固定項、ならびに製剤特異的(formulation specific)な被験者間および被験者内分散、および被験者付随製剤分散(subject−by−formulation variance)を伴うシーケンス内の被験者についてのランダム項を含む。
INS−AUC(0-24h)について、演算推定および製剤間の差異についての90%信頼区間より得られた、製剤比率(T/R)に対する点推定および90%信頼区間は、その混合効果モデルのフレームワーク内であることを意味し、その後その比率スケールを真数変換により変換した。
製剤比率についての信頼区間が[0.80−1.25]である場合、バイオアベイラビリティが同等であると結論付けた。
【0167】
副次PKパラメータおよび追加PKパラメータの解析
50%−INS−AUC(h)までの時間および最大濃度までの時間[INS−Tmax(h)]をノンパラメトリックに解析した。
対数変換した画分INS−AUCおよびINS−AUC(0-end)(μU・h・mL-1)ならびに最大血清インスリングラルギン濃度[INS−Cmax(μU・mL-1)]を、1次解析について記載したような対応する線形混合効果モデルを用いて解析した。点推定および信頼区間を報告した。
【0168】
C−ペプチド
入手できる場合、C−ペプチドのプロファイルをプロットし、記述的に特徴づけた。
【0169】
PK/PD解析
しかるべき場合、PK/PD解析を探索的に実行した。
【0170】
実施例6:治験結果
被験者の内訳
総数35名(女性11名、男性24名)の被験者がスクリーニングされ、そのうち24名の健康で適格な被験者が登録され、無作為化され、少なくとも1度の治験薬投与を受けた。24名の無作為化された被験者のうち1名の被験者が最初の治療期間後に自身の要求に基づいて本治験から退いた。23名の被験者はプロトコルに従って本治験を完了し、薬力学(PD)および薬物動態学(PK)解析に組み入れられた。安全性評価には24名の治験被験者全てを含めた。
主なプロトコル逸脱はなかった。
【0171】
人口統計学特性
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)を、体重および身長のデータより算出した:
BMI=体重[kg]・(身長[m])-2
【0172】
【表5】
【0173】
クランプの実施
2つの治験群、ランタスU100およびランタスU300は、個別のグルコースクランプレベルを定義するのに役立つ個別の絶食時ベースライン血中グルコース濃度に関して類似していた。投薬後のクランプの所要時間は30時間であり、これは全ての治療期間において同じであった。
【0174】
主要エンドポイント
ランタスU100とランタスU300についてのバイオアベイラビリティ(曝露)の同等性は確立されなかった。ランタスU100とランタスU300についての生物有効性(活性)の同等性は確立されなかった。
【0175】
主要変数
0〜24時間までの血清インスリングラルギン濃度の時間曲線下面積(INS−AUC(0-24h))は、ランタスU100およびランタスU300について等価でなかった。曝露はU300を用いると約40%少なかった。0〜24時間までの時間曲線に対するGIRの曲線下面積(GIR−AUC(0-24h))は、ランタスU100およびランタスU300について等価ではなかった。活性はU300を用いると約40%少なかった。
【0176】
副次変数
INS−AUC(0-24h)(h)の50%までの時間は、ランタスU100とランタスU300で類似していた。GIR−AUC(0-24h)(h)の50%までの時間は、ランタスU300が0.545(h)(0.158−1.030)長く、この値は統計学的に重要である。
【0177】
安全性
重篤な有害事象(AE)は報告されなかった。治療(治験薬および参照薬)あたり5名の被験者が計14のTEAEを報告し、それらの全てが軽度〜中程度であり、後遺症もなく解消した。最も高頻度に報告された事象は頭痛(治験あたり4名の被験者)であり、その後吐き気、嘔吐および発熱(U100において1名の被験者)、および処置上の痛み(U300において1名の被験者)であった。注目すべきは、頭痛はクランプ試験の間の一般的な意見であり、高浸透性グルコース液剤の注入に関連しているということである。しかしながら、治験薬との関連も排除できなかった。注射部位の反応は報告されなかった。
【0178】
結論
インスリングラルギンU100およびインスリングラルギンU300は、バイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。インスリングラルギンU300後の曝露および活性は同量(0.4U/kg)のインスリングラルギンU100の投与後の曝露および活性と比較した場合約40%低かった。
【0179】
しかし、インスリングラルギンU300は、インスリングラルギンU100よりも、基礎インスリンに望ましいよりフラットなPK(曝露)およびPD(活性)プロファイルを示した。健常な被験者に対する同じ皮下投与後の、インスリングラルギンU100製剤とインスリングラルギンU300製剤との間の曝露および活性におけるこれらの驚くべきかつ予測できない差異は、以下の図に効果的に示されている。注目すべきは、同時に血中グルコースが一定になったということである。
【0180】
インスリングラルギンU300の投与は、安全性および忍容性に問題はなかった。
【0181】
実施例7:インスリングラルギンU300の3つの異なる皮下用量のグルコース動的活性および曝露を比較する試験のための、理論的根拠
健常な被験者における試験からの結果(実施例1〜6を参照のこと)は、ランタス(登録商標)U100とインスリングラルギンU300との間の曝露および効果における非同等性を示した。被験者はU100およびU300について、同じ用量のインスリングラルギン(0.4U/kg)を受けたが、U300からの同じ単位量の送達は、U100からの送達よりも曝露および効果が約40%低かった。しかしながら、インスリングラルギンU300は、ランタス(登録商標)U100よりも、基礎インスリンに望ましいよりフラットな薬力学プロファイルを示した。
【0182】
従って、以下の実施例において記載される新規の試験は、I型糖尿病患者による正常血糖クランプ設定におけるコンパレータとして、ランタス(登録商標)U100の標準用量に対して、インスリングラルギンU300の3つの異なる皮下用量のグルコース動的活性および曝露を比較する。
【0183】
この試験は、等効果なU300の用量を、クランプ技術によって提供される血中グルコース処理のパラメータによって評価されるような、0.4U/kgのランタス(登録商標)U100まで近づけることを目的とする。
【0184】
インスリングラルギン曝露は、皮下投与後の濃度−時間プロファイルおよび単位インスリンあたりのグルコース利用としての活性より評価される。
【0185】
本試験は、I型糖尿病の被験者の正常血糖クランプ設定において、ランタス(登録商標)U100の標準用量と比較して、異なる用量のインスリングラルギンU300の代謝性効果および曝露を評価するためにデザインされた。この試験は、4治療(R、T1、T2およびT3)、4治療期間(TP1−4)および4シーケンスを構成する。1度のスクリーニング来院(D−28〜D−3)、4度の治療来院(TP1〜TP4においてD1〜D2)、および1度の試験終了時来院(最終投与後D5〜D14)があり、安全性パラメータの最終評価を伴う。
【0186】
被験者は、ラテン方格デザインに従い、R、T1、T2およびT3の各治療に対して一旦、クロスオーバー、二重盲検および無作為化様式で曝露された。このデザインは、薬理科学効果およびランタス(登録商標)U100と比較して異なる用量のインスリングラルギンU300の曝露を評価するのに適切であると考えられる。
【0187】
本治験用に選択されたランタス(登録商標)U100の用量 0.4U/kgは、I型糖尿病患者に正常血糖を提供することが特徴であり、I型糖尿病患者による他のクランプ試験にて容易に研究された。
【0188】
インスリングラルギンU300について、3つの異なる用量0.4、0.6および0.9U/kgを試験する。この用量範囲は、0.4U/kgのランタス(登録商標)U100に等効果なおよその用量を補完する(intrapolating)ことができる。インスリングラルギンU300の用量0.4U/kgはすでに健常なボランティアで試験しており(実施例1〜6を参照のこと)、予め設定した観察期間終了の30時間内で0.4U/kgのランタス(登録商標)U100よりも活性が低いことが明らかとなっている。総グルコースディスポジションによって測定される0.4U/kgのインスリングラルギンU300の生理活性は、参照治験薬(0.4U/kg ランタス(登録商標)U100)よりも39.4%低かった。相応して、より高い用量のインスリングラルギンU300、例えば0.6U/kgのインスリングラルギンU300は、0.4U/kgのランタス(登録商標)U100と比較してほぼ等価なグルコース動的活性をもたらすことが期待された。さらに、比例的に用量を上げて行くことで、用量に比例する曝露および効果のプロファイルを調査することができる。
【0189】
I型糖尿病の患者における試験は、内因性インスリンの混乱効果(confounding impact)を避け、曝露および作用の持続性の評価をより可能にする。さらに、インスリングラルギンに特有のアッセイがないため、全ての内因性インスリンを読み取るアッセイを用いることが強いられる。従って、外因性インスリングラルギン以外の、加えられた全てのインスリン源が過剰なインスリン濃度を不当に引き起こす。
【0190】
本試験は、クロスオーバーデザインであり;実践的および倫理的理由により、多くても3種類のU300用量をランタス(登録商標)U100と比較する。長時間作用型インスリン製剤のグルコース動的活性の評価は、延長した作用持続時間に起因して、正常血糖クランプの設定を36時間まで伸ばすことを必要とする。
【0191】
活性な医薬品成分であるインスリングラルギンは、U100とU300の両方の製剤において同じである。本試験に用いられる用量は、日常用途の範囲内である。低血糖の全体的なリスクは完全に排除できないが、正常血糖クランプ技術によって制御される。
【0192】
薬力学
インスリングラルギンの薬力学活性は、I型糖尿病患者における正常血糖クランプ技術(これは、血中グルコース処理に対する投与された外因性インスリン製剤の効果を評価するために確立された標準手順である)によって評価される。
【0193】
正常血糖クランプ設定におけるグルコースディスポジションの評価に特有のパラメータは、体重で標準化された注入量(GIR)、処理された総グルコース、GIR−AUC0-36、およびGIR−AUC0-36の所定のパーセンテージまでの時間(例えば、GIR−A
UC0-36の50%までの時間)である。
【0194】
補助的なパラメータは、平滑化体重で標準化したGIRの最大値GIRmax、および、GIRmaxまでの時間GIR−Tmaxである。
【0195】
インスリングラルギンの作用持続時間は、投薬と正常血糖(クランプ)レベルを超える予め特定された逸脱との間の時間から導出される。
【0196】
グルコースのモニタリングは、皮下投与後、インスリングラルギンの長時間持続作用に起因して36時間実施される。
【0197】
薬物動態学
インスリングラルギンの徐放性質により、濃度プロファイルにおいて目立ったピークに欠けている。従って、INS−AUCの50%までの時間(T50% INS−AUC0-36)は、インスリングラルギンの曝露プロファイルの時間位置についての測定値として算出され、INS−CmaxおよびINS−Tmaxは、追加測定値として役に立つ。
【0198】
主要試験目的
本試験の主要な目的は、0.4U/kg ランタス(登録商標)U100に対する、3つの異なるインスリングラルギンU300用量の代謝性効果比率を評価することである。
【0199】
副次試験目的
本試験の副次的な目的は、0.4U/kgのランタス(登録商標)U100に対して、3つの異なるインスリングラルギンU300用量の曝露比率を評価すること、0.4U/kgのランタス(登録商標)U100に対して用量の異なるインスリングラルギンU300の作用持続時間を比較すること、インスリングラルギンU300の用量応答および用量曝露の関係を調査すること、ならびにI型糖尿病患者におけるインスリングラルギンU300の安全性および忍容性を評価することである。
【0200】
実施例8:試験デザイン、プロトコルの記載
I型糖尿病を患う男性および女性の被験者における、第1相、単一施設、二重盲検、無作為化、クロスオーバー(4−治療、4−治療期間および4−シーケンス;ラテン方格)、アクティブコントロール、治療期間中(5〜18日、好ましくは7日)の休薬期間を伴い、以下のインスリングラルギンの単回投与を受ける
・0.4U/kg ランタス(登録商標)U100(= 対照試験 R)
・0.4U/kg インスリングラルギン U300(= 試験 T1)
・0.6U/kg インスリングラルギン U300(= 試験 T2)
・0.9U/kg インスリングラルギン U300(= 試験 T3)
この4つの治療RおよびT1-3は、被験者に無作為に割り当てられた以下の4−シーケンス(1:1:1:1の比)
・R−T1−T2−T3
・T3−R−T1−T2
・T2−T3−R−T1
・T1−T2−T3−R
での、4つの治療期間(TP1〜TP4)における所定のクロスオーバー法である。
【0201】
試験参加の所要期間
・1人の被験者に対する総試験期間:約4〜11週間(最低−最長期間、休薬期間に依存する、スクリーニングを除く)
・1人の被験者に対する試験の各期間の所要期間:
- スクリーニング: 3〜28日間(D−28〜D−3)
- 治療期間 1−4: 2日間(1晩の滞在)
- 休薬: 5〜18日間(連続した投与の間に、優先的には7日間)
- 試験終了時来院: 治験薬投与後のD5〜D14の間の1日
【0202】
実施例9: 被験者の選択
計画された被験者の数:20名の評価可能な被験者を確保するために、少なくとも24名の被験者を登録するべきである。
【0203】
組み入れ基準
患者基本情報
I01 年齢18歳〜65歳の男性または女性被験者であって、米国糖尿病学会で定義されているように(American Diabetic Association. Report of the Expert Committee on the Diagnosis and Classification of diabete. Diabetes Care 1998;21:5−19)、1年より長く糖尿病を患っている
I02 総インスリン用量< 1.2U/kg/日
I03 体重は、男性であれば50.0kg〜95.0kg、女性であれば50.0kg〜85.0kgであり、ボディマス指数が18.0〜30.0kg/m2である
健康状態
I04 空腹時陰性血清C−ペプチド(<0.3nmol/L)
I05 グリコヘモグロビン(HbA1c)≦9.0%
I06 試験前に少なくとも2ヶ月間、安定したインスリン管理ができている(被験者の安全性および試験の科学的な完全性に関する)
I07 治験責任医師が、異常が臨床的に無関係であり、本試験の実施に干渉しないと判断した場合を除いて、病歴および身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに骨格筋系)において正常な所見である(被験者の安全性および試験の科学的な完全性に関する)
I08 背臥位での10分間の安静後の正常なバイタルサイン:95mMHg < 収縮期血圧 < 140mMHg; 45mMHg < 拡張期血圧 < 90mMHg;
40bpm < 心拍数 < 100 bpm
I09 背臥位での10分間の安静後の正常な標準12誘導ECG; 120ms <PQ <220ms、QRS < 120ms、QTc≦440ms(男性の場合)、≦ 450 ms(女性の場合)
I10 治験責任医師が、異常が糖尿病患者にとって臨床的に無関係であると判断した場合を除いて、正常範囲(または治験責任医療機関向けに定義されたスクリーニング閾値)内の臨床検査項目;しかし、血清クレアチニンは、臨床検査ノルムの上限を厳密に下回らねばならない;肝臓酵素(AST、ALT)およびビリルビン(被験者がギルバート症候群であるという文書を有している場合を除く)は、1.5 ULN を上回ってはならない
【0204】
女性被験者のみ
I11 妊娠する可能性のある(閉経後2年未満であるかまたは3ヶ月以上外科的に不妊ではない)女性は、スクリーニング時に血清β−HCG妊娠テストで陰性、およびTP1〜TP4の日1の尿β−HCG 妊娠テストで陰性でなければならず、また医薬品の臨床試験のための非臨床試験の実施時期についてのガイドライン(CPMP/ICH/286/95、改定)の注記に従って、失敗率が低い(すなわち1年あたり1%未満)と定義されているバースコントロールの非常に有効な方法を用いなければならない。全試験期間中、妊娠する可能性のある女性被験者は、2つの独立した避妊法(例えばダイアフラムおよび殺精子剤をコーティングしたコンドーム)を用いなければならない。コンドームと殺精子クリームの使用では十分に信頼できない。
閉経後2年未満であり、3ヶ月以上外科的に不妊ではない閉経後の女性は、ホルモン状態を測定する(FSH >30 IU/L、エストラジオール < 20 pg/mL)
【0205】
除外基準
病歴および臨床状態
E01 臨床的に関連のある心血管、肺、胃腸、肝臓、腎臓、代謝性(I型糖尿病とは区別して)、血液、神経性、精神、全身性(全体として身体に影響を及ぼす)、眼、婦人科の(女性の場合)、または感染性疾患の病歴または存在;急性感染性疾患または急性の病気の兆候全て
E02 過去6ヶ月間に、発作、昏睡または他人の助けを必要とするような重篤な低血糖の発症が1回より多くあった場合
E03 頻繁な重篤な頭痛および/または偏頭痛、反復性の吐き気および/または嘔吐(月に2度より多く)
E04 参加前の3ヶ月間の間の血液損失(≧300mL)
E05 症候性低血圧(血圧が低下すること全て)、または背臥位から立位に変えたとき、3分以内に20mMHg以上のSBPが低下することにより定義される無症候性起立性低血圧
E06 薬物アレルギーの存在もしくは病歴または治験責任医師の判断に従う臨床的に重要なアレルギー疾患
E07 試験期間中に、本治験プロトコルで許可されない薬物による治療を必要とする可能性があること
E08 過去3ヶ月以内に任意の治験薬による試験に参加したこと
E09 本試験前3ヶ月間における臨床的に重要な病気の症状、これは治験責任医師の意見に従い、本試験の目的に影響を及ぼし得るもの
E10 薬物またはアルコールの乱用の存在(アルコール摂取量> 40グラム/日
E11 1日あたり5本または同等量よりも多く喫煙しており、本試験期間中に喫煙を中断できない
E12 キサンチンベースの飲料の過剰摂取(>4杯/日)
E13 女性である場合、妊娠(β−HCG試験陽性として定義される)、授乳中
【0206】
干渉物質
E14 組み入れ前の14日間以内またはその薬物の排出半減期もしくは薬力学半減期の5倍以内におけるあらゆる薬剤(John’s Wortを含む);試験開始前の最後の1ヶ月内におけるインスリン以外のあらゆる薬剤で最も長いものおよび定期的に使用しているもののどちらも(甲状腺ホルモン、脂質低下薬および降圧剤を除き、女性である場合はホルモン不妊法または閉経期ホルモン補充療法を除く);最後の28日間内でのあらゆるワクチン接種
【0207】
一般的条件
E15 治験責任医師の判断において、本試験期間中、非従順でありそうかまたは言語の問題もしくは乏しい精神面での発達が理由で協力ができないであろう被験者
E16 適用し得る法令に従う、以前の研究の排除期間内である被験者
E17 緊急の場合に連絡不可能な被験者
E18 被験者が本プロトコルの実施に直接的に関与する治験責任医師もしくは任意の補助治験責任医師、研究助手、薬剤師、治験コーディネーター、または他のこれらのスタッフである
【0208】
生物学的状態
E19 以下のテストのどれに対しても陽性反応である:B型肝炎表面抗原(HBs Ag)、抗B型肝炎コア抗体(抗HBc Ab)(化合物が陽性の免疫活性を有する場合)、抗C型肝炎ウイルス(抗HCV2)抗体、抗ヒト免疫不全ウイルス1および2抗体(抗HIV1および抗HIV2 Ab)
E20 尿の薬物スクリーニングで陽性結果(アンフェタミン/メタミンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエート)
E21 アルコールテスト陽性
【0209】
本試験特有事項
E22 インスリングラルギンおよび賦形剤に対する過敏性がわかっている
E23 深部脚静脈血栓の病歴もしくは存在全てまたは第1度親族(両親、兄妹または子供)に深部脚静脈血栓が頻繁に発症している
【0210】
実施例10:治療
治験薬
・インスリングラルギン
異なる2つのインスリングラルギン製剤を用いる:
- 100U/mLのインスリングラルギンを含有する注射用のランタス(登録商標)U100液剤(市販の製品)
- 300U/mLのインスリングラルギンを含有する注射用のインスリングラルギン U300液剤
・用量:
- ランタス(登録商標)U100: 0.4U/kg(= 参照試験 R)
- インスリングラルギン U300: 0.4、0.6および0.9U/kg(=試験 T1−T3)
・容器: 3mLのガラスカートリッジ
・施用経路:臍から横に5cm右と左の皮下に
・状態: 絶食時
・治療の所要期間:各期間につき1日、単回投与
・開始: 治療期間1〜4(TP1−4)の日1(D1)の09:00
・参加した被験者の100%が追加治療を受ける
【0211】
【表6】
【0212】
投与
本試験は、計4回の治験薬投与を伴う単回投与試験である。被験者は、各被験者が参照治療(R)および各試験治療(T1-3)を一度受けるように、参照治療と試験治療の異なるシーケンスに無作為化される。
【0213】
注射は臍の左または右に行い、両部位は別々の注射に用いられる。5〜18日間の休薬期間は、連続的な投薬日期間から切り離し、好ましくは7日間(連続的な投薬期間の間の7日間)である。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、5日間は参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間は、投薬期間の間に避けられない場合に被験者に試験に関係のない義務を果たすための自由を可能にする3週間の休止期間を再び与える。
【0214】
IP投与は、絶食状態で投与され;被験者は、全クランプ期間を通して絶食を続ける。
【0215】
血中グルコース濃度は、前クランプ期間の投薬前1時間の間に任意のグルコース注入なしで5.5mmol/L(100mg/dL) ±20%の範囲内である。血中グルコースが任意のグルコース注入なしで少なくとも1時間安定している場合、IPが投与される。IP投与は、治療期間1〜4の日1の9:00より早い時間および14:00より遅い時間には行わない。血中グルコースが14:00前に安定しない場合投薬は行わない。その来院は終了し、被験者は後に新しい投薬来院1〜7日間を計画される。
【0216】
被験者ごとおよび投薬ごとに新しいカートリッジが用いられる。
【0217】
IP投与は、本試験にその他の点で関与していないかまたはCROでの本試験チームの一員ではない人物によってなされる。この人物はオープンなランダムリストに従って、IP投与を準備するためのランダムコードを得、それに従って被験者に投薬する。この準備および投薬は、第2の独立した人物によってチェックされる。投薬準備および治療シーケンスのそれぞれの文書は厳密に機密とし、任意の他の人物には開示されない。
【0218】
IP(インスリングラルギン)の用量の算出
各被験者に投与されるインスリングラルギンの量を算出するために、体重(kg)を小数点1ケタまで測定し、0.6U/kgのインスリングラルギン用量について以下の例に示したように、算出したインスリンの量を整数まで切り上げまたは切り捨てた:
・体重75.3kgの被験者は、45Uのインスリンを投与される(75.3×0.6 = 45.18 、切り捨てにより45とする);
・体重74.4kg被験者は、45Uのインスリンを投与される(74.4×0.6 = 44.64、切り上げにより45とする)。
【0219】
TP1 D1に記録された体重を全ての治療期間の治験薬用量の算出に用いる。この治験薬用量は、被験者の体重がTP1からその後のTPのうちの1つまでの間に2kg以下でしか変化しなかった場合、変更されない。被験者の体重がTP1からその後のTPのうちの1つまでの間に2kgより多く変化した場合には、治験薬用量はそれぞれの治療期間のD1での体重に基づいて再計算される。
【0220】
シリンジおよび針
少量の注射液剤を正確に投与するのに適切に取り付けられた針を備えるシリンジのみが用いられる(例えば、Becton Dickinson、Ref 305502、大きさ:1mL 27G 3/8 0.40x10)。このシリンジは、治験責任医師より提供される。
【0221】
他の製品
クランプ手順期間中に用いられた他の製品を表5に記載する。
【0222】
【表7】
【0223】
グルコース液剤、塩化ナトリウム液剤、ヘパリンおよびインスリングルリジンは、治験責任医師によって提供される。
【0224】
グルコース液剤: 20% グルコース液剤は、被験者の個別の血中グルコースを設定した目的レベルに維持するために人工膵(BiostatorTM)を用いて注入される。第2の注入ポンプ(人工膵(BiostatorTM)の一部)は、ラインの開存を維持するために0.9% 塩化ナトリウム溶液を輸液する。必要な20% グルコース液剤の量が人工膵(BiostatorTM)の注入容量を超える場合、第2のグルコース注入ポンプを連動させる。
【0225】
ヘパリン:低用量ヘパリン液剤(10.000単位ヘパリン/100mL生理食塩水)を、二腔カテーテルを介して注入する。このヘパリン液剤は、カテーテルの他の管腔において、人工膵(BiostatorTM)の血中グルコース測定のために用いられる血液と共に流され、系内の血液凝固を防止する目的がある。
【0226】
インスリングルリジン: 15U アピドラ(登録商標)[100U/mL]を49mLの生理食塩水溶液に溶かし、癒着を防止するために1mLの患者自身の血液を加えて0.3U/mLの濃度で作製し、これを正常血糖を達成する個別の速度で注入する。
【0227】
盲検法の記載
被験者は、無作為化、盲検およびクロスオーバーのデザインで異なる4つの治療(R、T1、T2およびT3)を受ける。盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験またはCROでの本試験チームの一員に別な方法では関与せず、どのような情報も誰にも開示せず、本試験の盲検状態を維持することを保証する。彼/彼女は、ランダムコードを入手し、それに従って被験者に投薬する。IPの調製および投薬後、ランダムコードへのアクセス権を持つが同じように守秘義務のある第二の独立した人物によりチェックされる。
【0228】
治療群への被験者の割り付け法
IPは、文書によりインフォームドコンセントを受けた被験者に対してのみ、本治験プロトコルに従って投与される。
【0229】
全ての組み入れ/除外基準を満たす被験者は、治療期間1の日1、治験薬投与の直前に割り当てられる:
・治療期間1のD1の朝における組み入れの経時順に従う増分被験者番号。9桁の被験者番号は、3つの部分(例えば、276 001 001、276 001 002、276 001 003、など)から構成されており、その最初の3桁(276)は国番号、真ん中の3桁は治験機関番号および最後の3桁はその機関内での被験者増分番号である。この被験者番号は全試験期間中変更せずに、被験者の同一性を確認する。
・予め計画した順番での治療番号の後、次の適格な被験者についての無作為化リストは、常に無作為化リストに従う次の治療番号を受ける
IP投与は、無作為化治療シーケンスに従ってなされる。
【0230】
本研究から退いた被験者は、すでに割り当てられている場合には彼らの被験者番号および治療番号を保持しておく。代替の被験者は、異なる同定番号(すなわち500+本研究を中止した被験者の番号)を有する。各被験者は本治験を中止した被験者と同じ治療シーケンスを受ける。
【0231】
スクリーニングで不合格であった被験者は、異なる番号を割り当てられる(例えば、901、902(インフォームドコンセントの署名後のスクリーニング期間中にAEが生じた場合のみ、CRFに記録するため))
注記:被験者の無作為化は、治験責任医師が本研究に関しての被験者の適格性を確認した後に行う。ベースラインパラメータは、投薬前直前に有効なパラメータである。
【0232】
包装およびラベリング
インスリングラルギン U300液剤は、3mLカートリッジの再編成されたボックスでサノフィ−アベンティスより提供される。
【0233】
それぞれの番号のIPは、医薬品製造品質管理基準および地域の法的規制に従ってサノフィ−アベンティスの責任下で包装され、CROに提供される。
【0234】
ラベル表示の内容は地域の法的規格および規制に従ってなされる。
【0235】
ランタス(登録商標)U100は市販品であり、CROにより注文される。
【0236】
貯蔵条件
全てのIPは、治験責任医師の責任下において適切に施錠された室内で保管され、権限を与えられた人物以外が近づいてはいけない。
【0237】
このIPは+2℃〜+8℃で保管し、光から保護し、凍結してはならない。
【0238】
本試験の間の無作為化コードへのアクセス
盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験および/またはCROでの本試験チームの一員に別な方法では関与せず、いかなる情報も誰にも開示せず、本試験の盲検状態を維持することを保証する。彼/彼女は、ランダムコードを入手し、それに従って被験者に投薬する。IPの調製および投薬後、ランダムコードへのアクセス権を持つが同じように守秘義務のある第二の独立した人物によりチェックされる。
【0239】
有害事象の場合、その治験薬の知識が被験者の治療のために重要である場合の状況を除いては、そのコードは解読されない。各被験者について、治療の名前を含む解読資料(code−breaking material)は、封緘されて配布される。それは、本臨床期間を通して治験機関内の安全な場所に置いておかれる。治験依頼者は、臨床試験の完了時に全ての解読資料(開封されているかまたは封をされている)を回収する。
【0240】
盲検状態が破られた場合、治験責任医師は、源データに開封の日付とコードを解読した理由を記録する。
【0241】
治験責任医師、治験施設薬剤師、またはIPの貯蔵および投与を行ったその他の人物は、本試験に用いられたIPが治験依頼者に指定されたように、かつ適切な法的規制に従って厳重に維持されることを保証する責任がある。
【0242】
全てのIPは、治験プロトコルに従ってなされ、配布され、再び戻されたIPの正確な記録が維持されることを保証するのが治験責任医師の責任である。
【0243】
併用治療
併用薬の使用は、除外基準No. E14に明示されているように、その基準下に述べられている薬物を除いて本試験期間中許可されず、治療期間1の日1の、被験者の組み入れ前に所定のタイムフレーム内で停止される(E14を参照のこと)。
【0244】
被験者の標準インスリン治療とクランプ測定との干渉を避けるために、被験者は基礎インスリンの使用を控え、以下に切り替えねばならない:
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0245】
短時間作用型インスリンの最終皮下注射は、治験薬投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0246】
被験者の安全性を危険にさらさないような症候性の有害事象(例えば、頭痛)については、長期間持続する重篤なまたは中程度の有害事象のために併用薬を残しておく。特に、アセトアミノフェン/パラセタモールの使用は、肝毒性のリスクの知見がある場合、または肝臓酵素の異常が生じればすぐに禁止される。
【0247】
しかしながら、いかなる理由であっても特定の治療が必要とされる場合、正確な記録を適切な記録形式で取らねばならず、その記録には薬剤の名称(国際一般名)、1日の服用量およびこのような使用の継続期間を含む。治験依頼者は、頭痛の治療を除き、電子メールまたはファックスによって48時間以内に通知されねばならない。
【0248】
潜在的なアレルギー反応の治療は、他で出版されているような推奨事項に従う(Samspon HA、Munoz−Furlong A、Campbell RL et al. Second symposium on the definition and management of anaphylaxis: summary report−Second National Institute of Allergy and infective Disease/Food Allergy and
Anaphylaxis Network symposium. Journal of Allergy and Clinical Immunology 2006;117(2):391−397)。アレルギー反応の重篤度に応じて、抗ヒスタミン剤、副腎皮質ホルモンおよびエピネフリンによるによる治療が考慮される。
【0249】
治療の説明責任およびコンプライアンス
・IP コンプライアンス:
- IPは、医師の直接的な指導の下で投与され、適切な記録は、IPの分配および投与に責任のある人物または彼/彼女の代理人によって仕上げられる;治療シーケンスまたは用量における全ての情報は開示されず、文書は本試験に関与する他の人物がアクセスできないよう施錠される
- IP摂取は、測定可能な薬物アッセイ結果により確認される
・IPの説明責任:
- IPの分配および投与に責任のある人物または彼/彼女の代理人は、戻されたパック中に残っているカートリッジを数え、その後治療記録用紙(Treatment Log Form)に記入する
- 治験責任医師は、症例記録用紙(Case Report Form)(CRF)の適切なページに、投薬の日時に関する情報を記録する
- その後本試験を管理するモニターチームが、(本試験の非盲検を防ぐために)データベースをロックした後にIPと適切な説明責任様式でそれらを比較することにより、CRFデータをチェックする。
【0250】
用いられたカートリッジは、データベースがロックされた後、本試験の終わりに治験依頼者によって完全に文書化された調整項目(reconciliation)が実施されるまで治験責任医師によって保管される。
【0251】
実施例11:治験薬の評価
本試験は、0.4U/kg ランタス(登録商標)U100に対する、異なるインスリングラルギンU300用量の代謝性効果および曝露比の評価、0.4U/kg ランタス(登録商標)U100に対する、異なる3つのインスリングラルギンU300用量の作用持続時間の比較、インスリングラルギンU300の用量応答性および用量と曝露の関係の説明、ならびにI型糖尿病の被験者における正常血糖クランプ設定時のインスリングラルギンU300の安全性および忍容性の評価のためにデザインされる。
【0252】
薬力学
正常血糖クランプ
インスリングラルギンの薬力学効果、主に総グルコース処理およびインスリン作用の持続時間は、正常血糖クランプ技術によって評価される。
【0253】
正常血糖クランプの間、動脈血化した静脈の血中グルコース濃度(これは、全ての組織の総グルコース利用への供給、および被験者の血中グルコース濃度をその目的レベル(クランプレベル)に維持するのに必要とされるグルコース注入率(GIR)を反映する)は、人工膵(Biostator)装置(連続グルコースモニタリングシステム、Life Sciences Instruments、Elkhart、IN、USA)を用いて連続的に測定され、記録される。
【0254】
グルコース要求量(GIR−AUC)は、外因性インスリン過剰により媒介される組織へのグルコース取り込み(グルコース処理またはグルコース低下活性)の尺度である。人工膵(Biostator)は、1分間隔で血中グルコースレベルを測定し、予め設定したアルゴリズムを用いて血中グルコース中の変化に応じてそのグルコース注入率を調節する。
【0255】
クランプ手順
被験者の標準インスリン治療とクランプ測定との干渉を避けるために、被験者は基礎インスリンの使用を控え、以下に切り替えねばならない:
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0256】
短時間作用型インスリンの最終皮下注射は、IP投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0257】
治療期間1〜4(TP1〜TP4)の間、被験者は少なくとも10時間、一晩絶食した後、D1の朝にクリニックに入る。
【0258】
D1の朝、前クランプ手順を開始し、被験者に人工膵(Biostator)を連結する。血中グルコース濃度を4.4−6.6mmol/L(80−120mg/dL)に調節し、速効型インスリンアナログ(例えばインスリングルリジン)の静脈内ボーラス投与方法、および必要に応じて引き続くインスリンの個別注入により、これらの限界内を維持する。
【0259】
その後、治験薬投与の60分前、投薬の1時間前からいかなるグルコース注入もなしに血中グルコースを5.5mmol/L(100mg/dL) ±20%(正常血糖クランプレベル)に調節する。このインスリングルリジン注入は治験薬の投与直前には中止する。
【0260】
血中グルコースがいかなるグルコース注入もなしに5.5mmol/L(100mg/dL) ±20%の範囲内で少なくとも1時間安定であればIPを投与する(TP1〜TP4のD1のT0、9:00頃)。被験者は、参照薬または治験薬(R、T1-3、表4を参照のこと)を無作為化によって割り当てられたとおりに投与される。注射は臍の左または右になされる。
【0261】
IP投与は、治療期間1〜4の日1の9:00より早い時間および14:00より遅い時間には行わない。血中グルコースが14:00前に安定しない場合投薬は行わない。その来院は終了し、被験者は後に新しい投薬来院1〜7日間を計画される。
【0262】
IP投与は、絶食状態下で投与され;被験者は全クランプ期間を通して絶食を続ける。
【0263】
この正常血糖クランプ血中グルコースレベルはクランプの終了までグルコース液剤の静脈内注入法により連続的に維持される。
【0264】
任意の基礎インスリン補充の目的は、食間の代替内因性インスリン分泌に追加するかまたは分泌を平らにすることである。内因性インスリン分泌のない被験者において、本試験への参加を求められる場合、外因性インスリンが、肝臓グルコース生成物を処理するのに必要とされるインスリンの量をまかなうべきである。
【0265】
完璧に釣り合うと、過剰インスリンを補うための外部グルコースは不必要である。その結果グルコース注入率は約ゼロとなる。一旦インスリン作用が終わると、血中グルコース濃度は上昇する。この上昇までの時間と血中グルコース濃度が所定の閾値を越える時間を人工膵(BiostatorTM)で読み取る。
【0266】
ランタス(登録商標)U100およびインスリングラルギン U300の選択された用量は平均基礎要求(average basal need)を超え、36時間までに相当な大きさのGIRに反映されるいくらかのグルコース要求の精製に向けられる。
【0267】
クランプのパフォーマンスを示す対応パラメータ(すなわちクランプのベースラインレベルで血中グルコースを維持するための精度)は、クランプ期間にわたる血中グルコースの変動性である。血中グルコースの変動性の尺度は、個々のクランプあたりの変動係数(CV%)である。
【0268】
血中グルコース中の低い変動係数は、クランプ設定におけるインスリン効果を正確に評価するために欠くことのできないものである。
【0269】
クランプ期間は治験薬注射の36時間後である予め定めておいたクランプエンドを超えない。
【0270】
被験者は全グルコースクランプ(前クランプおよびクランプ)期間中絶食を続けるが、水は自由にとることができる。
【0271】
血中グルコースが、グルコース注入の中止後30分後のクランプの終了前に11.1mmol/L(200mg/dL)を通過し、かつ治験責任医師が上記の11.1mmol/L(200mg/dL)を超える誤った血中グルコースレベルを導くような可能性のあるエラーが全て除外されていることを確認した場合、クランプの前IP投与時間に用いられるインスリングルリジンが、36時間前にその所見を拡張するために与えられる。この場合、治験依頼者はその情報を得なければならない。
【0272】
被験者は、血中グルコースが十分等血糖(isoglycemic)範囲内である場合、クランプ設定から離れる。
【0273】
参加者は、TP1〜TP4の開放日(すなわち日2)に彼らの前治験薬を再開する。
【0274】
IPの効果は約24〜36時間続き、このことが参加者を2日間施設にとどめる理由である。
【0275】
5〜18日間の休薬期間は、連続的な投薬日期間から切り離し、好ましくは7日間(連続的な投薬期間の間の7日間)である。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、5日間は参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間は、投薬期間の間に避けられない場合に被験者に試験に関係のない義務を果たすための自由を可能にする3週間の休止期間を再び与える。
【0276】
スクリーニングおよびTP1のD1は28日を超えて切り離されないが、EOSは最終の投薬後D5より早くまたはD14より遅くには生じない。
【0277】
薬力学サンプリング時間
前クランプ(IP投与の前)およびクランプ期間(IP投与後36時間まで)の間、1分おきの動脈血中グルコース濃度の測定のために、動脈血化した静脈血を2mL/hの速度で連続的に採決する。
【0278】
人工膵(BiostatorTM)校正用の動脈血化静脈血サンプル(0.2mL)(これは技術的な要求量である)を人工膵(BiostatorTM)に連結した後、治験薬投与後36時間まで少なくとも30分間隔で採取する。
【0279】
薬力学サンプルの数
血中グルコースはクランプ手順の間連続的に測定される。さらに、被験者1人の1治療期間につき少なくとも74のサンプルをIP投与後人工膵(BiostatorTM)の校正用に採取する。計74×4×24サンプル、すなわち7104サンプルを採取する(以下の表を参照のこと)。
【0280】
【表8】
【0281】
薬力学取り扱い手順
【表9】
【0282】
薬力学パラメータ
36時間以内の体重で標準化したGIRの曲線下面積(GIR−AUC0-36)および36時間以内の総GIR−AUCの50%までの時間(T50%−GIR−AUC0-36)を算出する。
【0283】
血中グルコースコントロールの持続時間は、正常血糖において、投薬からクランプグルコースレベル(100mg/dL)を超える逸脱までの時間として採用される。予め定義された限界内での制御された血中グルコースの時間は、投薬から特定の閾値(例えば、110、130および150mg/dLの血中グルコースレベル)までが採用される。
【0284】
さらに、最大平滑化体重補正GIR(GIRmax)およびGIRmaxまでの時間GIR−Tmaxを評価する。
【0285】
さらなる補足パラメータは必要に応じて導出される。
【0286】
安全性
ベースライン人口統計学特性
このベースライン人口統計学特性は以下から構成される:
・年齢(歳)
・体重(kg)
・身長(cm)
・ボディマス指数(BMI)(kg/m2)
ベースラインおよび本試験中の安全性評価
・スクリーニング時身体検査: 心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、および筋骨格系ならびに関連する病歴および手術歴、糖尿病の病歴(糖尿病の診断、インスリン治療の開始、その後の合併症);本試験に関連する知見のみが文書に記録される
・過去および現在の喫煙状況
・投薬前および試験期間中の身体検査:心血管系、腹部および肺;本試験に関連する知見のみが文書に記録される
・体温(耳)
・バイタルサイン:背臥位で10分間の安静後測定された心拍数、呼吸数ならびに収縮期血圧および拡張期血圧、心拍数ならびに収縮期血圧および拡張期血圧は、立位で3分間の後にも測定する(人工膵(Biostator)に連結されている場合は予定外の測定は除く)
臨床試験(血液サンプルの場合は絶食状態で):
・血液学:赤血球数(RBC)、ヘマトクリット(Hct)、ヘモグロビン(Hb)、白血球百分率数(WBC)(好中球、好酸球、好塩基球、単球およびリンパ球)、血小板、INRおよびaPTT
・生化学:
−電解質: ナトリウム、カリウム、重炭酸塩、塩化物、カルシウム
−肝機能: AST、ALT、アルカリホスファターゼ、γ−グルタミルトランスフェラーゼ (γGT)、総ビリルビンおよび抱合型ビリルビン
−腎機能:クレアチニン、BUN
−代謝:グルコース、アルブミン、総タンパク質、総コレステロール、トリグリセリド、HbA1c(スクリーニング、D1 TP1およびEOS時)、LDH、アミラーゼ、リパーゼ、C−ペプチド(スクリーニングのみ)
−潜在的筋肉毒性:クレアチニンホスホキナーゼ(CPK)
−血清学:B型肝炎抗原(HBs Ag)、抗B型肝炎コア抗体(anti−HBc Ab)、抗C型肝炎抗体(anti−HCV2)、抗HIV1および抗HIV2抗体
・保管用血液サンプル: 5mLの血液サンプルを乾燥した赤い蓋のついたチューブに採取し、約1500gで10分間、4℃で遠心分離する;その後血清を3つの保管用チューブに移し、すぐに蓋をしめて−20℃で立てた状態で凍結させる。このサンプルは、予期しない安全性の問題が生じた場合、薬物投与前のベースライン値が、事前に評価されなかったパラメータ(例えば、血清学)用に確実に利用するために用いられる。このサンプルが用いられない場合は、治験依頼者の同意後に治験責任医師が破棄する
・検尿:タンパク質、グルコース、血液、ケトン体、pH
−定性検査:試験紙を用いて、定性検出のために新鮮な排尿試料に対してディップスティックを実施する;
−定量検査: 尿サンプル試験が、尿のディップスティックにより上記のパラメータのいずれかについて陽性である事象においては、グルコース、タンパク質、赤血球および白血球の数の定量測定が必要とされる(例えば、定量測定により任意の陽性ディップスティックパラメータを確認するため)。
・尿の薬物スクリーニング: アンフェタミン/メトアンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエート
・アルコール酒気検査
・妊娠/ホルモン検査(女性の場合):
−スクリーニング時、血中β−HCG
−TP1〜TP4、日1には尿β−HCG
−閉経後2年以上経過の場合は、スクリーニング時にのみFSH/エストラジオール
・有害事象:被験者による自発的な報告または治験責任医師による観察
・ECG テレメトリー(シングルリード)
・12誘導ECG(自動)
・抗インスリン抗体
【0287】
臨床試験用の血液サンプルは絶食下状態で採取される。
【0288】
ECG 方法論
ECG テレメトリー
・ECG テレメトリーは、医師により連続的にモニタリングされる。全ての不整脈イベントがプリンターによって記録され、被験者のCRFに記載される。この文書はそのイベント、発生の時間および継続時間の診断を可能にし、治験責任医師またはその代理人によりサインされる。ECGテレメトリーは、このECGテレメトリー記録は、治験薬曝露を考慮した潜在的な再分析のために保管される。
【0289】
12誘導 ECG
・12誘導ECGは、心電図装置(MAC 5500TM)を使用して、背臥位で少なくとも10分経過後に記録される。電極は本試験を通して各ECG記録について同じ位置に配置される(リードの連結場所は消えないペンで印をつける)。
【0290】
・ECGは、(たとえあるとしても)PKのサンプリング前に常に記録される。PKサンプルはECG後できる限り早く(15分以内)に描かれる。
・各ECGは、同時12誘導の、10秒間の読み取りで構成される:
−シングル12誘導ECG(25mM/s、10mm/mV)は、日時、被験者のイニシャルおよび番号、治験責任医師の署名、ならびに各誘導につき少なくとも3つの波形(complex)を含む、HR、PR、QRS、QT、QTc 自動校正評価を印刷する。治験責任医師の意見および自動値はCRFに記録される。この印刷物は、施設レベルで保管される。
−ECG中央検査部(ECG central lab)による最終的な追加リーディングを可能にするデジタル記録:各デジタルファイルは、理論上時間(日付および時間DxxTxxHxx)、実際の日付および実際の時間(記録時間)、治験依頼者研究コード、被験者番号(すなわち3桁)ならびに施設および国番号により同定される(関連する場合)。
・このデジタル記録、データ記憶および送信(要求のある場合は)は、全ての適用すべき法的規制(すなわちFDA 21 CFR、第11部)に従う。
【0291】
バイタルサイン、ECG、および血液サンプルが治験薬投与および/または食事と同時に計画される場合、それらは治験薬投与および/または食事前になされる。PK、PD、または安全性のためのバイタルサイン、ECG、および血液サンプルの測定が同時であるときは常に、以下の順番が重んじられる:ECG、バイタルサイン、PD、PK、および安全性サンプル;PKサンプルの正確なタイミング(PKサンプルに対する時間窓許容についてのフローチャートを参照する)を重んじるために、他の測定は計画された時間の前になされる。この評価スケジュールは、本試験のデザインに適応される。
【0292】
注射部位の局所忍容性
注射部位における知見(例えば紅斑、浮腫、丘疹、硬化、小疱、疱疹)は、主に全体的刺激性評点(Global Irritation Score)に従って採点される。評定尺度に従って≧3のスコアである局所注射部位反応は、有害事象としてさらに書き加えられる。
【0293】
被験者は、注射部位における感覚を報告することを求められる。
【0294】
薬物動態学
インスリングラルギンの薬物動態学評価のために、36時間までのインスリン濃度曲線下面積(INS−AUC)であるINS−AUC0-36、およびINS−AUC0-36の50%までの時間が導出される。さらに、最大インスリン濃度INS−Cmax、およびCmax(INS−Tmax)までの時間が得られる。
【0295】
サンプリング時間
治験薬の注射後0H、1H、2H、4H、6H、8H、12H、16H、20H、24H、28H、32Hおよび36Hの時点において、インスリングラルギン濃度の測定のために血液を採血する。
【0296】
薬物動態学サンプルの数
【表10】
【0297】
薬物動態学取り扱い手順
IP投与およびサンプル採取の正確な時間はCRFに記録しなければならない。
【0298】
薬物動態学パラメータ
単回投与後、以下の薬物動態学パラメータをインスリングラルギン濃度についての非分画法を用いて算出する。これらのパラメータは以下を含むがこれらに限定されない。
【0299】
【表11】
【0300】
サンプリングされた血液量
【表12】
【0301】
本治験の盲検を保護するための測定
盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験および/もしくはCROでの本試験チームの一員に別な方法では関与しないかまたは治験依頼者ではない。彼/彼女はサノフィ−アベンティスよりランダムコードを入手し、そのランダムコードまたは他の全ての情報を他の人物に開示しない。安全上の理由で、治療の無作為化コードは重篤未知疑いのある有害事象(Suspected Unexpected Adverse Drug Reaction(SUSAR))全ての保健当局(Health Authority)への報告ならびに治験責任医師および/または治験依頼者いずれかの判断に従ってIPの使用と合理的に関連付けるために盲にされない。
【0302】
被験者の安全性
治験責任医師は安全性の問題の場合において全ての臨床的に関係のある決定を採用する責任のある主要人物である。
【0303】
判断が必要な場合、専門医の意見は迅速に把握されるべきである(例えば、急性腎不全、痙攣、吹き出物、血管性浮腫、心停止、心電図の変形など)。
【0304】
実施例12:試験手順
来院スケジュール
スクリーニング手順
スクリーニング手順は、参加への被験者の適格性を決定するために組み入れの3日前までに、28日間以内で実施される。被験者は、治験責任医師より本試験の目的および手順の情報を受ける。被験者は本試験に関する実行の前にインフォームドコンセントに署名する。有害事象の記録はその後開始する。
【0305】
スクリーニングの前に、被験者は10時間(必要である場合は、低血糖の対応策として少量の炭水化物を除いて)絶食(水以外)する。
【0306】
スクリーニング来院は以下の調査を含む:
1.人口統計学(年齢、性別、人種、過去および現在の喫煙状況、身長、体重、BMI)
2.身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系)ならびに病歴および手術歴、糖尿病の病歴(糖尿病の診断、インスリン治療の開始、その後の合併症);本試験に関連する知見のみが文書に記録される
3.関連のある先行治療および全ての併用治療、試験介入の直近2カ月前における平均的なインスリン療法
4.ECG(標準12誘導)、バイタルサイン測定(脈拍、背臥位で10分間安静後および立位で3分間の後の収縮期血圧および拡張期血圧)、および深部体温(耳)
5.血液学、HbA1c、C−ペプチド、臨床化学、血清学、検尿、尿の薬物スクリーニング、アルコール酒気検査、β−HCGおよびFSH/エストラジオール血液検査(女性のみ、妥当な場合)による臨床試験
結論的な最終検査の結果に対して、1週間以内に1度の再検査が許可される。
【0307】
全ての組み入れ基準を満たし、除外基準にかからない被験者は、組み入れ来院の資格が得られる。
【0308】
スクリーニングで不合格である場合は、そのスクリーニング試験の基本結果が源文書に記録される。
【0309】
組み入れ手順(治療期間1の日1)
本試験への登録の資格を得た被験者は、TP1のD1の朝、7:00頃に絶食状態でクリニックに入る。
【0310】
この組み入れ試験は、最初の投薬日(D1、TP1)に実施され、以下の調査を含む:
最新の病歴(AE)に対する身体検査、以前の薬/併用薬および耳の体温
体重、BMI(スクリーニング時で測定した身長)
ECG(標準12誘導)、バイタルサイン測定(背臥位で10分安静した後、立位で3分経過後の心拍数、呼吸数、収縮期血圧および拡張期血圧)
血液学、臨床化学、検尿、尿の薬物スクリーニング、アルコール酒気検査、β−HCG検尿(女性のみ、妥当な場合)による臨床試験。
【0311】
各被験者は、本試験への彼/彼女の組み入れの時間順に従って増加していく同定番号を与えられる。
【0312】
無作為化は、治験責任医師による被験者の適格性が確認された後、D1/TP1で行う。1人より多くの被験者が同時に無作為化される場合、その被験者らはD1/TP1の朝の組み入れの経時順に従って連続して無作為化される(すなわち、最も低い番号の被験者が次に利用可能な無作為化番号を与えられる)。
【0313】
D1/TP1の臨床試験の結果がベースライン値であり、陰性でなければならないβ−HCG検尿(スクリーニング来院時に採取されたサンプルに基づく)を除いて、確認試験であると考えられる。
【0314】
被験者が最終的に登録される場合、血液サンプルは、記録用および抗インスリン抗体の検出用に採血される(D1/TP1のみ)。
【0315】
来院のタイプ別記載
治療期間 1−4(D1〜D2)
被験者のクランプ測定による標準的なインスリン治療の干渉を防ぐために、被験者は、基礎インスリンの使用を控え、以下に切り替える
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ、
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0316】
短時間作用型インスリンの最終皮下注射は、治験薬投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0317】
来院の際、被験者は以前の来院以降、彼らの体調に臨床的に重要な変化が起こっていないことを保証すること、プロトコルにおいて定義されている一般的および食事の制限を遵守すること、および必要な場合には彼らのインスリン治療を変更することを求められる。本試験基準に違反した場合、その被験者は本試験へのさらなる参加から排除される。違反の種類により、被験者は特定の期間のみ除外される場合があり、一度の試験または全試験の計画を立て直す。
【0318】
最終来院以降の被験者の健康状態および併用薬におけるいかなる変化も被験者の診療録(原本)およびCRFに記録される。
【0319】
治験薬投与(各TPのD1)直前の朝、体重、バイタルサイン、12誘導ECG、ECGモニタリングおよび深部体温を記録し、検尿ならびに尿薬物およびアルコールのスクリーニングを実施する。
【0320】
注射に必要とされるインスリングラルギンの量は、被験者の体重に従って算出される。
【0321】
貧血症に備えるために治療期間3のD1に血液学を分析する。陽性である場合、治療期間3と4の間の休薬間隔を可能な最大の18日間まで延長するかまたはTP4の開始を血液パラメータが正常になるまで延期する。追加の血液評価は治療期間4のD1になされる。
【0322】
被験者は、正常血糖クランプの終了まで絶食状態(水を除く)を維持する。
【0323】
その後、被験者は自動グルコース読み取り装置(人工膵(BiostatorTM))に連結される3本の静脈ラインによって前クランプ手順を介する準備をし、サンプリング期間の全継続期間中、半座位(semi−recumbent position)のままになる。
【0324】
7:30頃、左手の手背静脈または手首外側の静脈にカニューレを導入し、人工膵(BiostatorTM)に連結して血中グルコース濃度の測定のために動脈血化した静脈血を連続的に採取する。左手を約55℃の空気温度を提供する加熱されたボックス(「ホットボックス(Hot−Box)」)に入れ、動脈血化する。第2の静脈ラインは、左腕の肘前静脈でとり、インスリンおよび参照薬の血中グルコース測定のためのサンプルを採取するために用いた。第3の静脈ラインは反対側の前腕にカニューレを導入し、人工膵(BiostatorTM)のポンプで0.9%の生理食塩水および20%のグルコース液剤、または外部ポンプを用いてインスリングラルジンを注入する。
【0325】
血管カテーテルの挿入からD1の9:00頃に治験薬を投与する60分前まで、血中グルコースレベルは4.4〜6.6mmol/L(80−120mg/dL、前クランプ)内に維持される。血中グルコースレベルにより、さらなるインスリングルリジンの静脈内ボーラス注射を行い、その血中グルコースを目的範囲内に維持する。治験薬投与の1時間前には、クランプ終了まで静脈内ボーラス注射はなされない。
【0326】
血中グルコースを測定するために、さらなる血液サンプルを少なくとも30分間隔で採取し、グルコースオキシダーゼ法をベースにした臨床基準値と照らし合わせて確認する。必要な場合、人工膵(Biostator)をその臨床検査基準値法の結果に従って再校正する。
【0327】
インスリン注入量は個別に調節される。血中グルコースが目的レベルで維持されている間、インスリンとグルコース両方の注入量は、クランプの導入期中、最小にされる。インスリングルリジン液剤は高精度の注入ポンプ(Terumo Spritzenpumpe TE 311TM)により注入され、20%グルコース液剤は高精度の注入ポンプ(Terumo Infusionspumpe TE 171TM)によって適用される。
【0328】
クランプレベルは、治験薬投与の60分前に血中グルコースをクランプ期間の終わりまで約5.5mmol/L(100mg/dL)に維持するように調節される。この前クランプは延長され、IP投与は、導入期(前クランプ)中に目的のグルコースレベルを満たしていない場合には14:00まで延期される。目的のグルコースレベルが14:00までに確立されない場合には、その来院は終了し、被験者は1〜7日後に新しい投薬来院を計画される。
【0329】
インスリングルリジン注入は、治験薬投与の直前に中止される。PK用の最初のインスリンサンプルはその後すぐに採取される。9:00頃、無作為化計画に従い、参照治療(R、0.4U/kg ランタス(登録商標)U100)または試験治療(T1-3)のいずれかの治験薬が標準化皮下脂肪技術(standardized skin−fold technique)を用いて臍側部に投与される(表4)。
【0330】
クランプの間、12誘導ECGはIPの注射2時間後および12時間後、ならびにクランプ終了時に行われる。
【0331】
治験薬は、全試験期間の間、同じ人物によって投与されることが好ましい。注射の終了時を時間ゼロ(T0)と定義し、その後のクランプ期間およびPKサンプリングの開始時間を定義する。
【0332】
全てのクランプ観察期間は36時間続くため、予め定義されたクランプの終了時であるD2の21:00頃に終了する。その後、被験者は、血中グルコースが十分等血糖(isoglycemic)範囲である場合正常血糖クランプ設定から離れ、食事および日常のインスリン治療を受ける。
【0333】
血中グルコースが、グルコース注入の中止後30分間のクランプの期間に11.1mmol/L(200mg/dL)を通過し、かつ治験責任医師が上記の11.1mmol/L(200mg/dL)を超える誤った血中グルコースレベルを導くような可能性のあるエラーが全て除外されていることを確認した場合、薬物動態学の血液サンプリングのために、クランプ期間を36時間まで延長するため、クランプの前IP投与時間に用いられる速効型インスリンアナログ(例えば、インスリングルリジン)が与えられる。この場合、治験依頼者はその情報を得なければならない。その後、被験者は、血中グルコースが十分等血糖(isoglycemic)範囲内である場合、正常血糖クランプ設定から離れ、食事および彼らの通常のインスリン治療を受ける。
【0334】
注射部位の反応は治験薬の注射後15分はもちろん1時間後にも評価し、評価尺度に従って≧3のスコアが観察された場合はAEとして記録される。
【0335】
解放前に、自由な食事が提供され、通常のインスリン治療が再開される。バイタルサイン(心拍数;背臥位で10分安静にした後、および3分間立位の後に測定される収縮期血圧および拡張期血圧)が繰り返され、血中グルコースが測定される(この血中グルコースの測定値は80mg/dLを上回らねばならない)。被験者は、治験医師によってかれらの健常な状態が保障された後、TP1〜TP4のD2に解放される。
【0336】
試験終了時来院
被験者は、TP4の最終投薬後、D5とD14の間に試験終了時来院のために戻る。被験者は10時間絶食(水を除く)しておく。このEOSは以下の調査を含む:
病歴が更新されたことに伴う身体検査(体重、体温)
ECG、バイタルサイン測定
血液学、HbA1c、生化学、検尿、および女性の場合にはβ−HCG 血液検査による臨床試験
発生した全てのAEまたはTP4以降に摂取した併用薬
抗インスリン抗体測定のための血液サンプル
治験責任医師は、全ての有効な臨床試験結果に基づいて、被験者が安全に本試験から解放されることを保証する。
【0337】
試験の制限事項
被験者は、日常のインスリン治療を、用いているインスリンのタイプ(長時間作用型、NPH、中間作用型)に応じて−2〜−1日で中止する。その後血中グルコースレベルは通常の単時間作用型インスリンの複合皮下注射によって単独でコントロールされる。
【0338】
日常のインスリン治療は、TP1〜TP4のD2に解放された後、再開される。
【0339】
被験者は、本試験を通して、および試験前の2週間は、被験者の代謝性のコントロールまたはインスリン感度に関与するいかなる併用薬も摂ってはならない。
【0340】
アルコール飲料、グレープフルーツジュース、およびキサンチン誘導体を含有する刺激性飲料(茶、コーヒー、コカコーラ(登録商標)のような飲料、チョコレート)の摂取は、クランプの完了まで、各治験薬投与の24時間前から許可されない。
【0341】
オレンジジュースまたは同様の炭水化物は、クランプの間の低血糖に対する調整的手段として与えられるか、そうでなければ人工膵(BiostatorTM)に連結されている場合は静脈内グルコース注入によって適切に打ち消される。
【0342】
各治験薬投与前の2日間は、激しい運動は許されない。
【0343】
1日あたり5本以下の喫煙をする被験者は本試験に含まれ、TP1〜TP4のD1およびD2を除いて本試験期間中喫煙をしてもよい。
【0344】
スクリーニングの日、被験者は少なくとも10時間の一晩の絶食(必要な場合は低血糖に他する対策手段として少量の炭水化物を除く)後に施設を訪れる。
【0345】
TP1〜TP4のD1の朝、被験者は少なくとも10時間の一晩の絶食後にクリニックに入り、D2のクランプ期間の終了まで絶食を続ける。自由な食事は、そのクランプの終了後に提供される。
【0346】
液体の供給は、各36時間の期間につき少なくとも2500mLである。
【0347】
原データ(source data)の定義
CRFに記録される以下に列挙した全ての評価は、以下に関する適切に署名され確認された原文書により裏付けられる:
・被験者の身元確認
・病歴(アレルギー反応の場合)
・臨床検査、バイタルサイン、体重および身長、体温;
・臨床検査評価(laboratory assessments)、ECG
・薬物動態の時点(Pharmacokinetic time points)
・来院および評価の日付および時間
・有害事象
・IP 投薬
・先行薬/併用薬
・クランプ手順の開始/終了、クランプデータ
【0348】
実施例13: 統計的考察
被験者数の決定
本試験の主要な目的は、U100(R)の1用量およびU300の3つの異なる用量(T1〜T3)として与えられるインスリングラルギンについての比較代謝性効果を評価することである。
【0349】
試験PKD10086のデータに基づき、約0.375の値が、自然対数変換されたスケールでGIR−AUCend of clampのSDwithinであると予測される。
【0350】
サンプル数算出の目的のために、0.325と0.425の間の被験者内SDが用いられた。
【0351】
表11は、修正幾何平均のペアワイズ治療比率(pairwise treatment ratio)についての最大の不確実性(90%信頼区間の点より)は90%の確実性で得られ、16〜24の間の総被験者数Nに対して、log GIR−AUC0-36についての真の被験者内SD値は0.325〜0.425の間であると仮定することを示す。
【0352】
【表13】
【0353】
20人の被験者について、GIR−AUC0-36の真の被験者内SDが0.375程度である場合、治療比率は19.9%の最大不確実性(すなわち、90%CIが0.80であり1/0.80=1.25倍の観測比)、および90%の確実性を有すると推定される。
【0354】
20名の完了する被験者を確保するために24名の被験者が含まれる。
【0355】
被験者の説明
被験者の内訳(disposition)
関与した、無作為化された、曝露された(任意の量の治験薬を投与された)、完了した(すなわち全ての治験期間を完了した被験者)、中断についての主な理由に従って中断した被験者の数を含む、被験者の説明責任の詳細な要約が作製される。
【0356】
最終来院での被験者の内訳は、シーケンス群、本試験の終了時と治験薬の最終投与の日の内訳状態、最終来院の日付、中断の理由を含む一覧表で提示される。最初の治験薬投与の開始時または開始後に起こる本試験からの全ての撤退は、治験総括報告書(CSR)の中に完全に記録される。
【0357】
プロトコルの逸脱
本試験のデータをロックする前に、集団の定義および以下を含む他の試験基準について定義された基準に関して、臨床試験プロトコルの逸脱が調査される:
・組み入れおよび除外基準;
・治療のコンプライアンス;
・禁止された治療に関する、本臨床試験プロトコルのコンプライアンス;
・来院間隔および総治療所要期間に関する本臨床試験プロトコルのコンプライアンス;ならびに
・計画された活動および安全性評価が実施されたか否か、など。
【0358】
逸脱は、以下を含むがこれらに限定されない:
・無作為化後に(全ての変数の)いかなる評価も受けていない被験者;
・曝露されない被験者;
・(適切な場合)主要変数の全ての評価を受けていない被験者;
・組み入れ基準を満たしていなかったにもかかわらず本試験に介入した被験者;
・本試験の間に離脱基準が発生したが、離脱しなかった被験者;
・誤った治療または不正確な投薬を受けた被験者;
・禁止された併用薬を受けた被験者。
【0359】
主要な逸脱は列挙され、まとめられる。
【0360】
解析の母集団
任意の解析母集団(薬力学、薬物動態学および/または安全性)からの除外は全て、CSRに完全に記録される。
【0361】
任意の解析母集団から除外された被験者は、治療シーケンスおよび除外理由と共に列挙される。関連のある全ての情報がCSRに完全に記録される。解析母集団について、全体および治療ごとの被験者の頻度が作表される。
【0362】
無作為化スケジュールに従って割り当てられた治療とは異なる治療を受けた被験者の事象については、解析は無作為化治療に従うのではなく実際に受けた治療に従って行われる。
【0363】
薬力学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PDパラメータが有効である被験者は全て、薬物動態学母集団に含まれる、両方ではなく1つの期間においてPDプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0364】
IPの投与後36時間の観察期間内にインスリングルリジンを受けた(安全性の理由で)被験者については、薬力学データはインスリングルリジンの投与時までのみを考慮に入れる。
【0365】
薬力学解析からの除外
薬力学解析からの全ての除外事項は、その理由と共に列挙される。除外事項は、データベースのロックおよび盲検前にデータのレビューに基づいて決定され記録される。
【0366】
安全性の母集団
あらゆる比較治験薬に曝露された全ての被験者は、投与された治験薬の量にかかわらず、安全性母集団に含まれる。
【0367】
薬物動態学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、インスリンPKパラメータが有効である被験者は全て、薬物動態学母集団に含まれる。一度PKプロファイルが不十分であったが、それが全ての治療期間ではなかった被験者については、十分なプロファイルのパラメータがその解析に含まれる。
【0368】
インスリングラルギンの生物学的分析アッセイは、インスリングルリジンのような他のインスリン類に干渉される。従って、IP投与後36時間のクランプ観察期間内にインスリングルリジンを投与された(安全性の理由で)被験者のインスリングラルギンについての薬物動態学データは、評価から除外される。
【0369】
人口統計学およびベースラインの特性
被験者の人口統計学特性、病歴および診断
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)は、体重および身長のデータより算出する:
BMI = 体重[kg]/(身長[m])2
【0370】
人口統計学および背景特徴に関する全ての変数を、安全性母集団に関して個別に列挙しまとめる。
【0371】
病歴および診断に関する組み入れ基準からの逸脱を列挙し、個別に記載する。
【0372】
ベースライン安全性パラメータ
安全性変数について、変数として適用可能であっても、期間内または試験内の治験薬投与前に最期に組み込まれた値をベースライン値とする。そのベースラインの投与前値は、投与前に再確認する場合、その再確認した値をベースラインとして認め、統計量に用いる。
【0373】
治験曝露の程度および遵守
しかるべき場合には治験薬投与の詳細および補足情報を個別に列挙しまとめた。
【0374】
インスリングラルギンの個別総用量を治療ごとにまとめる。
【0375】
前/併用 治療/療法
前治療および併用療法(ある場合)を、世界保健機関−薬物文献リスト(Drug Reference List)(WHO−DRL、データベースをロックする時点で使用される最新バージョン)に従ってコード化し、個別に列挙した
併用インスリン薬(皮下)は、分けて列挙される。
【0376】
本クランプ手順中の任意の時間に与えられるインスリン注入またはボーラスは列挙されるかまたは、個人ベースで時間に対してプロットされる。本クランプ手順の間の投薬後に与えられるインスリン注入またはボーラスは、個人ベースで列挙される。
【0377】
薬力学変数の解析
全ての薬力学解析は、この薬力学母集団のデータを包含する。αレベルの調節は、複合解析に対してはなされない。
【0378】
インスリングラルギンの薬力学について、血中グルコース濃度およびグルコース注入率(GIR)がこのクランプ手順の間連続的に記録される。
【0379】
統計解析は、試験治療(T1〜T3)を参照治療(R)と比較する。
【0380】
薬力学変数の記載
被験者間の同等性を達成するために、体重でインスリン投与量を調節し、解析用にGIRに関する全ての値を被験者の体重(kg)で割った。それにより以下において他に言及されない限りGIRは常に体重で標準化されたフルコース注入量を言及する。
【0381】
主要PD変数
以下のPD変数が主要変数であると考えられる。
【0382】
・体重で標準化されたグルコース注入率の時間曲線下面積
[GIR−AUC0-36(mg/kg)]
GIR−AUC0-36は、分刻みの時間尺度で段階的な定数関数について、矩形法則(rectangular rule)に従って算出される。
【0383】
副次PD変数:
以下のPD変数が導出され、副次変数として考慮される:
・GIR−AUC0-36の50%までの時間(h)[T50%−GIR−AUC0-36(h)]
・平滑化した体重で標準化した最大グルコース注入率[GIRmax(mg*min/kg)]
・投薬後、GIRmaxに到達する最初の時間[GIR−Tmax(h)]
・正常血糖の所要時間(クランプレベルを超える平滑化血中グルコースプロファイルの上昇までの時間)は、投薬から、105mg/dL以下での平滑化血中グルコース濃度曲線の最終値までの時間として算出される
・予め定めておいた限度にコントロールされる血中グルコースの持続時間は、投薬から、以下の濃度以下での平滑化血中グルコース濃度の最終値までの時間として定義される
- 110mg/dL
- 130mg/dL
- 150mg/dL
【0384】
平滑化
生の体重で標準化したGIRの最大値は、GIR修正においてノイズを受けやすい。従って、GIRmaxおよびGIRmaxまでの時間の導出は、生の体重で標準化したGIRデータに対して、LOESS(locally weighted regression in smoothing scatterplots:平滑化散布点における局所重みづけ回帰)平滑化技術に基づく。ランタス(登録商標)のもとに公知であるようなGIRプロファイルの予測されるモルホロジーにより、6%の平滑化因子が用いられる(SASR、ProC LOESS、因子 0.06)。
【0385】
血中グルコースレベルは、ノイズを非常に受けやすい。従って、正常血糖の持続時間および血中グルコースコントロールの継続時間は、生の血中グルコースレベルに対して、LOESS(locally weighted regression in smoothing scatterplots:平滑化散布点における局所重みづけ回帰)平滑化技術に基づく。モルホロジーにより、6%の平滑化因子が用いられる(SASR、PC
LOESS、因子 0.06)。
【0386】
不十分な平滑化の場合には、異なる平滑化因子が追加解析のために用いられる。
【0387】
追加PD変数
さらなるパラメータを以下のとおりに導出する:
・グルコース注入の終わりまでの時間(投薬後、ゼロを超えるGIRの最後の時間として)
追加PD変数は、結果の解釈のために必要であるとみなされる場合に導出される。
【0388】
一次PD解析
以下に記載される解析の前に、GIR−AUC0-36が対数変換(自然対数)される。
【0389】
対数変換したGIR−AUC0-36は、SAS PROC MIXEDを用いて、シーケンス、期間および治療についての固定項、ならびに、シーケンスブロック内の被験者についての治療の未構築Rマトリックス(i,i)分散および共分散を用いて、線形混合効果モデルにより解析される。
log(パラメータ)= シーケンス+期間+治療+誤差
【0390】
治療幾何平均の比(T1/R、T2/R、T3/R)についての90%信頼区間(CI)は、線形混合効果モデルフレームワーク内の治療手段間の差異についての推定値および90%CIをコンピュータにより計算し、真数変換により幾何平均の比に変換することにより得られる。同等性は、その比の90%CIが完全に参照間隔と同等な0.80〜1.25の範囲内である場合に結論付けられる。
【0391】
個別の比率(参照治療に対する試験治療)の一覧表は、対応する記述統計と共に提供される。
【0392】
二次解析/副次変数の解析
GIRプロファイルについての記述的な説明
個別の体重で標準化したGIR(mg*min/kg)は、生データ、平滑化データおよび累積生データについてプロットされる。
【0393】
時間に対しての、平均および中央値の体重で標準化したGIRプロファイルならびに中央値割合累積プロファイルは治療ごとにプロットされる。
【0394】
累積プロットは投薬からクランプの終了までの時間をカバーする。
【0395】
導出されたPDパラメータについての記述的説明
PDパラメータは個別に一覧表にされ、治療ごと尾に記述統計が作製される。
【0396】
副次PDパラメータの治療比
信頼限界を伴う製剤比率(T1/R、T2/R、T3/R)は、一次解析について上に記載したような、対応する線形混合効果モデルを用いて、標準化グルコース最大注入量[GIRmax(mg*min/kg)]について導出される。治療間の診査比較は、従来の生物同等性基準(90% 信頼限界 0.80〜1.25)に基づく。
【0397】
GIR−Tmax値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、試験治療および参照治療間のGIR−Tmaxにおける差異のヒストグラムが提供される。
【0398】
副次PDパラメータについての治療差異
T50%−GIR−AUC0-36(h)は、対になった治療比較について、Hodges−
Lehmann法をベースにノンパラメトリックに解析される。中央値におけるペアワイズ治療間差異(T1−R、T2−R、T3−R)についてのCIが導出される。T50%−GIR−AUC0-36値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、治療間(T1−R、T2−R、T3−R)のT50%−GIR−AUC0-36における差異のヒストグラムが提供される。
【0399】
GIR−Tmax値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、試験治療および参照治療間のGIR−Tmaxにおける差異のヒストグラムが提供される。
【0400】
正常血糖および血中グルコースコントロールの持続時間は、ヒストグラムプロットによって表わされる。治療の比較は、ノンパラメトリックに行われる。
【0401】
クランプの実行
血中グルコース濃度の個別プロファイルをプロットした。
【0402】
クランプの所要時間は、投薬からクランプ終了までの間の時間(hr)としてクランプごとに得られる。
【0403】
クランプごとの血中グルコースの個別可変性は、クランプの個別開始時と個別終了時(またはクランプ中のインスリングルリジンの最初の投与)の間の血中グルコース値の変動係数(CV%)として導出される。クランプごとの個別平均血中グルコースレベルは、クランプの個別開始時と個別終了時(またはクランプ中のインスリングルリジンの最初の投与)の間の算術平均として導出される。
【0404】
パラメータは治療内で、個別に一覧表にされ記述的にまとめられる。
【0405】
安全性データの解析
安全性評価は個別の値(潜在的な臨床的に重要な異常)、記述統計(要約表、図)および必要な場合には統計解析(適切な推定値、信頼区間)のレビューに基づく。「潜在的な臨床的に重要な異常」(PCSA) 基準は、サノフィー・アベンティスの標準的基準に従って用いられる。基準は、本試験の統計解析プランに記録される。この安全性解析は、臨床試験からの安全性データの解析および報告に関するサノフィー・アベンティス基準に従って行われる。
【0406】
全ての安全性解析は、安全性母集団のデータを包含する。
【0407】
全ての安全性データについて、観察期間は3つの異なる分画に分けられる:
・前治療期間は、被験者がインフォームドコンセントを認めたときと、最初の比較治験薬投与との間の時間として定義される。
・治療中期間は、(最初の)治験薬投与からその後72時間までの時間として定義される。
・後治療期間は、治療中期間後から、次の期間の(最初の)治験薬投与かまたはフォローアップ期間の終わりいずれかまでの時間として定義される。
【0408】
有害事象(AE)
全ての有害事象は、MedDRA(データベースロック時の使用する際の最新バージョン)を用いてコード化される。
【0409】
以下の項目が、全ての有害事象について提供される:
・(被験者による)全ての有害事象の一覧
・有害事象に関連するコメントの一覧
【0410】
定義
安全性データに関して、観察期間は3つの異なる区分に分けられる:
・前治療期間は、被験者がインフォームドコンセントを認めたときと、最初の比較治験薬投与との間の時間として定義される。
・各期間あたりの治療中期間は、(最初の)治験薬投与からその後72時間までの時間として定義される。
・後治療期間は、治療中期間後から、次の期間の(最初の)治験薬投与かまたはフォローアップ期間の終わりいずれかまでの時間として定義される。
【0411】
試験治療下で発現した有害事象
全てのAEを以下のように分類した:
・試験治療下で発現した有害事象(TEAE)は、治療期間に生じた(悪化を含む)全てのAEである;
・非試験治療下で発現した有害事象(NTEAEs)は、TEAEとして分類されない全てのAEである:
- 前治療AE、治験薬の最初の投与前の前治療期間の間に発症した(または悪化した)AEと定義される
- 後治療AE、治療中期間には悪化せず、後治療期間の間に発症したAEと定義される。
【0412】
治療への割り当て
解析の目的のために、各TEAEをAEの発症(または悪化)に与えられた最期の治療に割り当てた。TEAEがある治療において発生し、その後の治療下で悪化した場合、両方の治療に対するTEAEであると考える。
【0413】
欠測情報
情報の欠測または情報が矛盾していた場合、(例えば、部分的なデータまたは他の情報から)そのAEはTEAEではないと明らかに除外できる場合でない限り、AEはTEAEとしてカウントされる。
【0414】
AEの開始データが不完全または欠測している場合、不完全なデータがAEが治療前に始まっていたと示している場合を除いて、最初の治験薬投与後に生じたと推定される。
【0415】
試験治療下で発現した有害事象
試験治療下で発現した有害事象は列挙され、治療ごとにまとめられる:
・TEAEの概要(少なくとも1つのTEAE、重篤なTEAE、中断につながるTEAE、死亡(ある場合)の被験者数および割合
・主要な器官別大分類および基本語(preferred term)による、全ての試験治療下で発現した有害事象の要約(少なくとも1つのTEAEを伴う被験者の数および割合)(「インテキストテーブル」)
- 事象の数を伴わない表(治験総括報告書の本体用)
- 事象の数を含めた表(治験総括報告書の付表用)
- 製剤(U100、U300)ごとの被験者の数および被験者全体の数を含めた表(治験総括報告書の付表用)
・治療、器官別大分類および基本語による、被験者が示す試験治療下で発現した有害事象の列挙
【0416】
死亡、重篤な有害事象および他の重要な有害事象
死亡、重篤なAEおよび他の重要なAEのような場合は全て個別に列挙し、総括報告書に詳細に記載される。
【0417】
治療の中止につながる有害事象
いかなる発生の場合においても、治療の中止につながる全ての有害事象について個別の被験者の一覧表が作製される。
【0418】
臨床検査値の評価
血液学および生化学データ
臨床安全性パラメータは、治療期間1のD1およびEOS時に測定される。スケジュールごとに、これらの安全性パラメータは治療中期間に評価される(TP3およびTP4での血液学を除く)。
【0419】
ベースラインとして用いられるべき値(血液学および生化学)は、最初の治療期間のD1前投薬で採取された値である。任意の計画されたベースライン試験が、任意の被験者について反復される場合、最終的に再チェックされた値がベースラインとして考慮される。ただしそれらは最初のIP投与の前になされた。
【0420】
以下の表および列挙が提供される:
・ベースラインからの生データおよび変更についての記述統計(クレアチニンについての変化割合(%)を含む)
・後ベースラインのPCSAを伴う被験者から個別データの特定の一覧が提供され、機能および測定の時間によりソートされる
・計画された血液学および生化学についての再確認された値を含む全ての個別データは、生物学的機能および測定の時間ごとに列挙される。ある場合には、計画されていなかった臨床試験からのデータがこの一覧に含まれる。これらの一覧表において、個別データは下限または上限の臨床試験限界値よりも低いかもしくは高い場合、および/または定義されている場合、PCSA基準の絶対限界に達する場合にはフラグ化される
・以下のうち少なくとも1つを経験した被験者についての肝機能データの一覧表:
- 本試験期間中にALT>3ULNを少なくとも1回発生し、かつ総ビリルビン>2ULNを少なくとも1回発生し、それらの少なくとも1つは最初の投与後である
- 治療中期間の定義にかかわらず、抱合型ビリルビン>35%総ビリルビン、かつ総ビリルビン>1.5ULNが最初の投与後に同じサンプルで提供される
・特に、薬物取り込み、病歴および手術歴、アルコール習慣、トリガーファクター、ALT値、関連する徴候および症状に伴う事象詳細な情報を含む、ALT≧2ULNの増加に関する一覧表が提供される
・範囲外の定義の一覧表が提供される。
【0421】
PCSAを伴う被験者の一覧表において、肝機能データ、CPK、および好酸球が対応するULNの多発性(multiple)として説明される。
【0422】
検尿データ
再検査値を含む全ての定性検尿結果(ディップスティック)が一覧にされる。
【0423】
バイタルサイン
血圧および心拍数
心拍数ならびに収縮期血圧および拡張期血圧(SBPおよびDBP)は、人工膵(Biostator)に連結されている場合を除き、背臥位での10分間安静後、および立位での3分間経過後に測定される。
【0424】
ベースラインとして用いられるべき値は、各治療期間のD1前投与評価値である。任意の計画されたベースライン試験が任意の被験者について繰り返される場合、最後の再検査値がベースラインとして考慮される。ただしそれらはIP投与前になされた。
【0425】
心拍数および血圧については、起立性差異が背臥位から立位への変化として計算される。
【0426】
全てのパラメータについて、全ての未計画値および再検査値を含む「治療中」解析を実施する。
【0427】
以下の表および一覧が提供される:
・ベースラインの正常な状態または異常な状態にかかわらず、PCSAを伴う被験者数の要約表がベースライン後PCSAの発生表として提供される
・心拍数および血圧(背臥位および立位)について、計画された前投与測定および定義されたベースラインを基にした測定のタイプ(位置)、各パラメータおよび時点についての生データおよびベースラインからの変化(背臥位のみ)が記述統計にまとめられる
・無計画値および再検査値を含む全ての個別データが一覧表にされる(背臥位、立位、起立性差異)。その一覧表において、定義されている場合はPCSA基準の限界に到達している場合、印をつけられる
・個別の後ベースラインPCSAのデータ一覧が提供される
・ある場合は、バイタルサイン評価に関するコメントも付表に載せる。
【0428】
体重、ボディマス指数、および体温
体重およびBMIのベースラインとして用いられるべき値は、TP1のD1に収集された値である。
【0429】
体温のベースラインとして用いられるべき値は、各TPのD1に収集された値である。
【0430】
個別データは、PCSA基準の限度に到達している場合にはその値に対する印(体重のみ)を含めて一覧表にされる。
【0431】
ECG
心拍数、PQ−、QRS−、およびQT−間隔ならびに自動読み取り機から収集されたQT(QTc)は、生パラメータ値およびベースラインからの変化として解析される。
【0432】
ベースラインとして用いられるべき値は各期間のD1投与前値である。いくつかの計画されたベースラインテストが任意の被験者について繰り返される場合、再検査値がベースラインとして考慮される。ただしそれらは期間の薬物投与前になされる。
【0433】
全てのパラメータについて、再検査値を含む、治療中期間になされた全てのベースライン後評価を用いて治療中解析が実施される。ベースライン後PCSAを伴う被験者の数は、ベースラインの正常または異常な状態にかかわらず治療群ごとに要約表で提供される。
【0434】
全てのパラメータおよびベースラインからの変化についての生データは、パラメータ、治療、および測定時間ごとに記述統計でまとめられる。
【0435】
再検査値を含む個別データは一覧表にされ、治療、被験者、来院および測定時間によりソートされる。これらの一覧表において、PCSA基準の限度に到達している値は印がつけられる。
【0436】
ベースライン後PCSAを伴う被験者からの個別データの一覧表が提供され、測定のタイプによってソートされ、被験者、期間および測定時間によってソートされる。
【0437】
さらに、延長したQTc(男性>450ms、女性>470ms)またはQTc>60msでのベースラインからの変化(男性および女性)を伴う被験者についての心臓のプロファイルの別々の一覧表、および最初の投薬後の定性評価において少なくとも1つの異常がある(すなわち異常なECG)被験者の一覧表もまた提供される。
【0438】
他の関連する安全性パラメータ
身体検査
ある場合には、身体検査に関するコメントの一覧表が提供される。
【0439】
注射部位の局所忍容性
治療による頻度分布が、注射部位の局所忍容性のレベルについて提供される。個別データが一覧表にされる。各基準および治療において、被験者は最も重篤な結果を用いてカウントされる。
【0440】
アレルギー反応
アレルギー反応の一覧表
全てのアレルギー反応症例が、詳細な補足情報を含めて有害事象として記録される。全ての症例は治験総括報告書に詳細に記載される。
個別の症例および全ての補足データが一覧表にされる。
【0441】
アレルギー病歴および家族歴
アレルギー病歴および家族歴は、潜在的なアレルギー反応の任意の発症を伴う被験者について記録される。アレルギー病歴およびアレルギー家族歴の全ての詳細が個人ベースで一覧表にされる。
【0442】
抗インスリン抗体
本試験期間中および試験後調査からの抗インスリン抗体結果について、被験者の数を伴う要約表が提供される。個別の被験者一覧表が提供される。
【0443】
薬物動態学データの解析
薬物動態学パラメータ
PKパラメータのリストは、上に示される。さらに、インスリンについてのT50%−AUC0-36が統計解析の項で導出される。
【0444】
統計解析
インスリングラルギンの薬物動態学パラメータは、各治療について、少なくとも算術平均および幾何平均、標準偏差(SD)、平均の標準誤差(SEM)、変動係数(CV%)、最小値、中央値および最大値を用いて一覧表にされまとめられる。
【0445】
全ての薬物動態学解析は、上記に定義したような対応する薬物動態学母集団のデータを包含する。αレベルの調整は、複数解析にはなされない。
【0446】
統計解析は、試験治療(T1〜T3)と参照治療(R)を比較する。
【0447】
治療比の解析
この解析は、インスリングラルギンのAUC0-36について実施される。以下に記載される全ての解析の前に、AUC0-36値が対数変換(自然対数)される。
【0448】
対数変換したパラメータは、SAS PROC MIXEDを用いて、シーケンス、期間および治療についての固定項、ならびに、シーケンスブロック内の被験者についての治療の未構築Rマトリックス(i,i)分散および共分散を用いて、線形混合効果モデルにより解析される。
log(パラメータ)= シーケンス+期間+治療+誤差。
【0449】
治療幾何平均の比(T1/R、T2/R、T3/R)についての推定値および90%信頼区間(CI)は、線形混合効果モデルフレームワーク内の治療手段間の差異についての推定値および90%CIをコンピュータにより計算し、真数変換により幾何平均の比に変換することにより得られる。生物同等性は、その比の90%CIが完全に参照間隔と同等な0.80〜1.25の範囲内である場合に結論付けられる。
【0450】
個別の治療比(T1/R、T2/R、T3/R)の一覧表は、対応する記述統計と共に提供される。
【0451】
インスリンについてのT50%−AUC0-36
インスリンについてのT50%−AUC0-36の分散は、各治療についてのヒストグラムプロットにて提示される。さらに、治療間(T1−R、T2−R、T3−R)のT50%−AUC0-36における差異のヒストグラムが提供される。
【0452】
T50%−AUC0-36(h)は、ノンパラメトリックに解析される。
【0453】
インスリングラルギンU300の用量−曝露関係
用量−曝露関係の記述的解析
インスリングラルギンU300の用量−曝露関係は、以下により図表を用いて記載される
・被験者あたりの総用量に対する、曝露の被験者あたりのプロット
・体重(kg)あたりの用量に対する、曝露の被験者あたりのプロット
・体重(kg)あたりの用量に対する、用量で正規化された曝露の被験者あたりのプロット(0.6U/kgでの用量正規化)
結果の解析が必要であるとみなされる場合、さらなる記述的解析が追加される。
【0454】
用量−曝露関係の統計解析
試験治療T1−T3について算出されたインスリングラルギンのAUCについて、用量−曝露関係は、Gough et al.(Gough K、Hutchison M、Keene O et al.Assessment of dose proportionality:report from the pharmaceutical industry.Drug Information Journal 1995;29:1039−1048)における推奨に従って、「推定値」の解釈と共に経験パワーモデル(empirical power model)(PK−パラメータ=a*用量b)を用いて評価される。
【0455】
この経験パワーモデルは、比例関係の確認と、薬物動態学および全ての離脱の臨床的重要性を評価することの両方に用いることのできる、非比例関係の程度の尺度を容易にかつ理解可能に提供する。しかしながら、用量比例関係研究の解析は、任意の非比例関係の薬物動態学および臨床的重要性が評価されることを可能にするために、有意な試験よりもむしろ推定を必要とする。
【0456】
このパワーモデルは、用量(U/kg体重)に対するランダム係数パワーモデルを用いた対数変換スケールで合致する:
log(パラメータ)=(log(α)+α[i])+(β+β[i])*log(用量)
ここで、log(α)およびβは、それぞれ母集団の切片および勾配であり、α[i]およびβ[i]は、i番目の被験者についてのαおよびβからのランダムな偏差である。
【0457】
βについての推定値および90%信頼区間は、共分散パラメータの制限付き最尤法(restricted maximum likelihood(REML))推定による、SASR/PROC MIXED手段での一般最小2乗推定(estimated generalized least squares)により得られる。βについての推定値および90%信頼区間はさらに、rを、β推定値および信頼限界の冪に累乗することによる、r倍の増加(r=1.5およびr=2.25[すなわち 高用量/低用量])に伴うPKパラメータの増加についての推定値および90%信頼区間を得るために用いられる。
【0458】
モデルの不適合(lack−of−fit)が明らかに存在する場合、(治療比の解析に用いられたような)混合効果モデルが解析に用いられる。ペアワイズ用量の増加に関連するパラメータの増加についての推定値および90%CIは、混合効果モデルのフレームワーク内の投薬間のペアワイズ差異についての仮定およびCIを最初に演算子、その後真数変換により比率に変換することにより得られる。
【0459】
PK/PD解析
適当な場合には、グラフィックディスプレイ(点図表プロット)がPK/PD関係の説明のために説明される。
【0460】
実施例14: 試験結果
被験者の内訳
I型糖尿病の被験者計24名が登録され、無作為化され、少なくとも1用量の治験薬を受けた。24名の無作為化された被験者のうち2名の被験者は自身の要求により本試験から離脱した。22名の被験者が本プロトコルに従って試験を完了し、薬力学(PD)および薬物動態学(PK)解析に含まれた。治療された24名全ての被験者が安全性評価に含まれた。
【0461】
主要なプロトコルの逸脱はなかった。
【0462】
人口統計学特性
以下のデータ(表12)を収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)を体重および身長のデータから算出した:BMI=体重[kg]・(身長[m])-2。
【0463】
【表14】
【0464】
クランプの実行
各被験者について4つの治療期間、R(LantusU100)、T1(0.4U/kg HOE901−U300)、T2(0.6U/kg HOE901−U300)およびT3(0.9U/kg HOE901−U300) において、インスリン薬物治療の前の個別のベースライン血中グルコース濃度は、100mg/dL のクランプレベルの定義と類似していた。投薬後、本クランプの観察期間の所要期間は36時間であり、全ての治療期間で同じであった。
【0465】
主要エンドポイント
RおよびTについてのバイオアベイラビリティ(曝露)および生物有効性(活性)における同等性は、立証されなかった。
【0466】
主要変数
血清インスリングラルギン濃度の0〜36時間までの時間曲線下面積(INS−AUC(0-36h))は、RおよびT1およびT2については等価ではなく、T3ではほぼ等価であった。曝露は、Rと比較して、T1では約37%未満、T2では約43%未満およびT3でも同様であると推定された。
【0467】
0〜36時間までの時間に対するGIRの曲線下面積(GIR−AUC(0-36h))は、RおよびT1およびT2については等価ではなく、T3ではほぼ等価であった。血中グルコースコントロールを保つために必要とされる外因性グルコース利用は、T1では約88%未満、T2では67%でありT3でもほぼ同じであると推定された。
【0468】
副次変数
RでのINS−AUC(0-36h)(h)の50%までの時間は約14時間であり、ゆえにT1、T2およびT3それぞれでの約16時間、16時間および19時間と比較して短かった。RでのGIR−AUC(0-36h)(h)の50%までの時間は約12時間であり、ゆえにT1、T2およびT3それぞれでの約17時間、18時間および20時間と比較して短かった。
【0469】
安全性
重篤な有害事象(AE)またはAEに起因する離脱は報告されなかった。Rにおいて2名の被験者、T1において2名、およびT3において3名が計8件のTEAEを報告し、それらの全ては軽度〜中程度であって後遺症なしに解決した。最も高頻度に報告された事象は頭痛であった。注目すべきは、頭痛はクランプ試験の間の一般的な意見であり、高浸透性グルコース液剤の注入に関連しているということである。しかしながら、治験薬との関連も排除できない。T1、T2およびT3では注射部位の反応は報告されなかったが、Rにおいて2名の被験者が注射部位にほとんど確認できないような紅斑が現れた。
【0470】
結論
同じ用量のRおよびT(U300)は、単回投与後のバイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。T1(0.4U/kg)およびT2(0.6U/kg)後の曝露および活性は、R(0.4U/kg)の投与後の曝露および活性と比較して少なかった。RおよびT3は曝露および外因性グルコース利用に関して実質的に同等であった。
【0471】
しかしながら、T1、T2およびT3は、Rに比べて平均付近でより変動の少ない、いっそうフラットなPK(曝露)およびPD(活性)プロファイル、すなわち基礎インスリン供給に望まれるようなプロファイルを示す。このことは、異なるプロファイルによって名目上等価な総曝露および総グルコース利用を提供するRとT3を比較する場合に特に明らかである。
【0472】
I型糖尿病の被験者におけるR(LantusU100)およびT(HOE901−U300)の製剤間での曝露および活性におけるこれらの驚くべきかつ予想外の差異は、以下の図に効果的に示される。
【0473】
その上、T(HOE901−U300)の投与は安全性および忍容性の問題を伴わなかった。
【0474】
実施例15: I型糖尿病の患者において、異なる2つの皮下用量の(HOE901−U300)のグルコース動的活性および曝露をランタスU100と比較するための、試験の理論的根拠
健常な被験者およびI型糖尿病の被験者における試験結果(前出の実施例を参照のこと)により、ランタス(登録商標)U100とインスリングラルギン U300とでは曝露および有効性が同等ではないことが示された。被験者は、U100およびU300について同じ用量のインスリングラルギン(0.4U/kg)を投与されたが、U300からの同じ単位量のU300送達が引き起こした血中グルコースコントロールを維持するための曝露および外因性グルコース利用は、U100からの送達よりも少なかった。ランタスU100は、平均付近での著しい変動のない曝露および薬力学プロファイルを示すが、HOE901−U300は、基礎インスリン供給にとって望ましい曝露および薬力学プロファイルにおけるさらに少ない変動、それに加えより長い作用の持続時間を示した。
【0475】
定常状態条件下での薬物動態学および薬力学のプロファイルを評価するために、以下の実施例に記載される新しい試験は、I型糖尿病の患者における最終的な正常血糖クランプ設定とのコンパレータとして、ランタス(登録商標)U100の標準用量に対して、異なる2つの皮下用量のインスリングラルギンU300を比較する。本試験は、クランプ技術によってもたらされる血中グルコースコントロールおよび血中グルコース処理のパラメータにより評価されるように、0.4U/kgのランタス(登録商標)U100と等効果なU300の用量を推定することを目的とする。インスリングラルギン曝露は定常状態での反復皮下投与後の濃度−時間プロファイル、および定常状態での単位インスリンあたりのグルコース利用としての活性より評価される。
【0476】
本試験は、2つの並列グループでの2つのクロスオーバー治療(RおよびT1、ならびにRおよびT2)、2−治療期間(TP1、TP2)および2−シーケンスをそれぞれ構成する。1度のスクリーニング来院(D21〜D3)、院内期間(クランプ評価ごとに、D1〜D4朝、およびD8朝〜D10夕方)を伴う治療来院(TP1およびTP2のD1〜D10、夕方の投薬を伴う)、および安全性パラメータの最終評価を伴う1度の試験終了時来院(最終投薬後D7〜D10の間)がある。
【0477】
本試験のために選択されたランタスU100の0.4U/kg用量は、I型糖尿病患者の正常血糖血中グルコースコントロールを提供することを特徴とし、I型糖尿病患者での他のクランプ研究において、容易に調査されている。
【0478】
インスリングラルギンU300について試験される2つの異なる用量は、0.4および0.6 U/kgである。この用量範囲は、0.4U/kgのランタス(登録商標)U100に等効果なおよその用量を補完する(intrapolating)ことができる。インスリングラルギンU300の用量0.4U/kgはすでに健常なボランティアおよびI型糖尿病患者で試験しており(前出の実施例を参照のこと)、予め設定した観察期間終了の30時間および36時間内で、0.4U/kgのランタス(登録商標)U100よりも活性が低いことが明らかとなっている。0.4U/kgのインスリングラルギン U300での血中グルコースコントロールが必要とする総グルコースディスポジションは、参照治験薬(0.4U/kg ランタス(登録商標)U100)よりも低かった。相応して、より高い用量のインスリングラルギンU300、例えば0.6U/kgのインスリングラルギンU300は、より少ない総グルコースディスポジションでよりしっかりとした血中グルコースコントロールをもたらすことが期待された。さらに、比例的に用量を上げて行くことで、用量に比例する曝露および効果のプロファイルを調査することができる。
【0479】
I型糖尿病の患者における試験は、内因性インスリンの混乱効果(confounding impact)を避け、曝露および作用の持続性の評価をより可能にする。
【0480】
本試験は、多くても2種類のHOE901−U300 用量の以前の試験の結果に基づいたクロスオーバーデザインを採用しており、ランタス(登録商標)U100と比較する。長時間作用型インスリン製品のグルコース動的活性の評価は、24時間を超える正常血糖クランプ設定を必要とし、予め定義した注射間隔は、作用の延長された持続時間によった。
【0481】
活性な医薬品成分であるインスリングラルギンは、U100とU300の両方の製剤において同じである。本試験に用いられる用量は、日常用途の範囲内である。低血糖の全体的なリスクは完全に排除できないが、正常血糖クランプ技術によって制御される。
【0482】
薬力学
インスリングラルギンの薬力学活性は、I型糖尿病患者における正常血糖クランプ技術(これは、血中グルコース処理に対する投与された外因性インスリン製剤の効果を評価するために確立された標準手順である)によって評価される。
【0483】
正常血糖クランプ設定におけるグルコースディスポジションの評価に特有のパラメータは、体重で標準化されたグルコース注入率(GIR)、24時間および36時間内に処理された総グルコース、それぞれGIR−AUC0-24 およびGIR−AUC0-36、ならびにGIR−AUC0-24 およびGIR−AUC0-36の所定のパーセンテージまでの時間(例えば、GIR−AUC0-36の50%までの時間)である。
【0484】
補助的なパラメータは、平滑化体重で標準化したGIRの最大値GIRmax、および、GIRmaxまでの時間GIR−Tmaxである。
【0485】
インスリングラルギンの作用持続時間は、投薬と正常血糖(クランプ)レベルを超える予め特定された逸脱との間の時間から導出される。
【0486】
グルコースのモニタリングは、皮下投与後、インスリングラルギンの長時間持続作用に起因して36時間実施される。
【0487】
薬物動態学
インスリングラルギンの徐放性質により、濃度プロファイルにおいて目立ったピークに欠けている。従って、INS−AUCの50%までの時間(例えば、T50% INS−AUC0-36)は、インスリングラルギンの曝露プロファイルの時間位置についての測定値として算出され、INS−CmaxおよびINS−Tmaxは、追加測定値として役に立つ。
【0488】
主要試験目的
本試験の主要な目的は、定常状態において、0.4U/kg ランタス(登録商標)U100に対する、2つの異なるインスリングラルギンU300用量の血中グルコースコントロールおよび必要とされる外因性グルコース利用を評価することである。
【0489】
副次試験目的
本試験の副次的な目的は、定常状態において、0.4U/kgのランタス(登録商標)U100に対して、2つの異なるインスリングラルギンU300用量の曝露比率を評価すること、0.4U/kgのランタス(登録商標)U100に対して2つの異なるインスリングラルギンU300用量の作用持続時間を比較すること、インスリングラルギンU300の用量応答および用量曝露の関係を調査すること、ならびにI型糖尿病患者におけるインスリングラルギンU300の安全性および忍容性を評価することである。
【0490】
実施例16: より高い濃度での長時間作用型インスリンの酸性製剤の溶解特性の変化
溶解特性に関しては、より高い濃度のインスリングラルギン製剤の影響がインビトロ試験系を用いることによって調査される。そうするために、沈殿の研究が、インビボ条件を模擬するpH7.4のリン酸緩衝液を用いて実施される。
【0491】
沈殿したインスリンの上澄みは、インスリングラルギン含有量を測定するためにHPLC技術を用いて調査される。
【0492】
本試験の詳細な記載:
沈殿緩衝溶液の調製:
19.32mgのリン酸2水素ナトリウム1水和物(M:137.98g/mol)を1mLの水に溶解した。0.1Mの水酸化ナトリウムまたは0.1Mの塩酸を用いてpHを7.4に調整した。
【0493】
沈殿研究の実施:
1000U/mL までの濃度のインスリングラルギン薬剤溶液、ならびに同量のインスリングラルギンおよび緩衝液を含む溶液をプラスチックチューブに入れ、わずかに振とうした。インスリングラルギンの沈殿後、分散物を予め定義した時間、ゆっくりとした回転速度で遠心分離した。所定量の溶解培地を取り出し、新しい緩衝培地に移した。
【0494】
インスリン含有量の測定:
上清サンプル中のインスリングラルギンの含有量を、水に溶解したリン酸2水素ナトリウム緩衝液、塩化ナトリウム(NaCl)および異なる量のアセトニトリルを含む、2つの移動相系を用いた逆相HPLCによって、それぞれのインスリン参照標準に対して定量した。
【0495】
固定相として、オクタドデシルカラムを用い、検出波長は215nmであった。
【0496】
より高い濃度の溶液(例えば、U500およびU1000)からのインスリングラルギンの放出プロファイルはランタスU100と比較してよりフラットで延長した。
【0497】
実施例17: 沈殿物の顕微鏡分析
100U/mL、300U/mL、500U/mL、700U/mLおよび1000U/mLの濃度を有するインスリングラルギン製剤の沈殿物を顕微鏡で調べた。200μLのリン酸緩衝液(pH7.4)中で沈殿した前記製剤(60Uのインスリングラルギンの量と同じ)を、100倍の大きさで透過型光学顕微鏡(Olympus Model BX61)で調べ、写真を以下に示し、また最大直径も示した。これらの研究により、濃度の増加に伴って明らかにより大きい粒子がもたらされるという沈殿物特性の違いが明らかとなった。これらの結果は図8に示した。
【0498】
実施例18: イヌにおけるインスリングラルギンの血中グルコース低下効果
インスリングラルギンの血中グルコース低下効果を健常な正常血糖のビーグル犬で評価した。このイヌは、0.3 IU/kgの単回皮下注射を受けた。静脈血中グルコースを最初の注射前とその後の24時間まで測定した。
【0499】
動物は、元々ハーラン(Harlan)より得た、30匹の健常な正常血糖のオスのビーグル犬群より取り出した。これらのイヌを標準化した条件下で、犬小屋の中で管理した。試験開始前日、それらのイヌを無作為に試験ケージに分配した。それらを、水道水へのアクセスは自由にして、実験開始前および実験中、18時間絶食させた。本試験におけるイヌの体重は13〜27kgであった。各実験後、それらのイヌを少なくとも2週間回復させた。
【0500】
これらの動物をn=6のグループに無作為化した。0時点において、これらの動物を試験化合物の単回投与で処理した。インスリングラルギンを、0.3 IU/kgの用量で単回皮下注射として投与した。
【0501】
血液のサンプリングは、薬物投与(0h)前およびその後24時間まで、前腕静脈(橈側皮静脈)の穴を介して連続的に行った。血中グルコースを酵素処理で測定した(Gluco−quant(R) グルコース/HKキット Roche/Hitachi 912)。
【0502】
インスリングラルギンの異なる濃度(100および300単位/mL)の調剤薬の皮下注射後の血中グルコースに対する影響を健常な正常血糖のビーグル犬において試験した。インスリングラルギン濃度の増加に伴い、作用の平均時間は、それぞれ6.8h(U100)〜7.69h(U300)まで増加した。
【0503】
グラルギン濃度を100から300U/mLまで増加させることによって、血中グルコース減少の時間作用プロファイルを、イヌにおいてよりフラットで延長した活性に向けて変化させた。
【0504】
イヌにおける現在のデータは、ヒトにおけるデータと一致し、インスリングラルギンのより高い薬物濃度が、作用のプロファイルおよびより長い持続時間に積極的に関連していることを示している。
【0505】
図
以下の図は、健常な被験者に対する同じ皮下投与後の、血中グルコース(PD)が一定である同時点における、ランタスU100製剤およびランタスU300製剤(インスリングラルギンU100製剤およびインスリングラルギンU300製剤)間の曝露(PK)および活性(PD)における驚くべきかつ予想外の差を効果的に示している。
【図面の簡単な説明】
【0506】
【図1】グルコース注入率(GIR) ランタスU100
【図2】グルコース注入率(GIR) ランタス U300
【図3】血清インスリン濃度; ランタスU100(左)およびU300(右)
【図4】血中グルコース(1/2)
【図5】血中グルコース(2/2)
【図6−1】正常血糖クランプ技術を用いた、I型糖尿病の患者における、0.4U/kg ランタス(登録商標)U100(インスリングラルギンU100)と比較した、0.4、0.6および0.9U/kgのHOE−901−U300(インスリングラルギン U300)の無作為化、4−シーケンス、クロスオーバー、二重盲検、用量応答試験の結果。上パネル: インスリングラルギン濃度(mU/L)、中パネル:血中グルコース(BG、mg/dL)、下パネル:グルコース注入率(GIR、mg.kg-1.min-1)。これらの曲線は、全ての被験者の全てのデータポイントのLOWESS平滑化平均を示す(母集団平均);LOWESSは、ノイズで悪影響を及ぼされている時系列から、または2つの変数間の「ノイズの入った」関係の点図表プロットから、「平滑な」選択値群(set of values)を作製するデータ解析技術である。
【図6−2】図6−1の続きである。
【図7】グルコース注入率(GIR、mg.kg-1.min-1)。これらの曲線は、全ての被験者の全てのデータポイントのLOWESS平滑化平均を示す(母集団平均);LOWESSは、ノイズで悪影響を及ぼされている時系列から、または2つの変数間の「ノイズの入った」関係の点図表プロットから、「平滑な」選択値群(set of values)を作製するデータ解析技術である。 凡例: プロファイル1〜3(上から下に) 正常血糖クランプ技術を用いた、I型糖尿病患者における0.4、0.6および1.2U/kgのランタス(登録商標)U100(インスリングラルギンU100)の、無作為化、二重盲検、並行群間用量応答試験の結果 凡例:プロファイル4〜7 (上から下に) 正常血糖クランプ技術を用いた、I型糖尿病患者における、0.4U/kg ランタス(登録商標)U100(インスリングラルギンU100)と比較した、0.4、0.6および0.9U/kg HOE−901−U300(インスリングラルギン U300の無作為化、4−シーケンス、クロスオーバー、二重盲検、用量応答試験の結果
【図8】インスリングラルギンの沈殿物の光学顕微鏡写真 濃度増加に伴う処方: A: 100U/mL、B: 300U/mL、C: 500U/mL、D: 700U/mLおよびE: 1000U/mL、 倍率100×であり、最大直径を含む。 全ての沈殿物は、インスリングラルギンの60Uを用いて実施される
【図9】正常血糖のイヌにおけるインスリングラルギンU−100対U−300の時間作用プロファイル
【0507】
略語のリスト
℃ 摂氏度
ABE 平均生物学的同等性
AE 有害事象
ALT アラニンアミノトランスフェラーゼ
aPPT 活性化部分トロンボプラスチン時間
ARF 急性腎不全
AST アスパラギン酸アミノトランスフェラーゼ
β−HCG β−ヒト絨毛性ゴナドトロピン
bpm 1分あたりの拍数
cm センチメートル
CPK クレアチニンホスホキナーゼ
CRF 症例報告書
DRF 矛盾解決フォーム(Discrepancy Resolution Form)
ECG 心電図
EOS 試験終了時(来院)
GCP 医薬品の臨床試験の実施基準
GGT γ−グルタミルトランスフェラーゼ
Hb ヘモグロビン
HbA1c グリコシル化ヘモグロビン
HBs B型肝炎表面
Hct ヘマトクリット
HCV C型肝炎ウイルス
HIV ヒト免疫不全ウイルス
HR 心拍数
INN 国際一般名称
INR 国際標準化比 (プロトロンビン時間)
IP 治験薬
IRB/I 治験審査委員会/独立倫理委員会
Kg キログラム
LOQ 定量限界
PT プロトロンビン時間
QTc ECG機械によって自動的に補正されるQT間隔
QTcB Bazettの式によって補正されるQT間隔
QTcF Fridericiaの式によって補正されるQT間隔
QtcN 母集団アプローチによって補正されるQT間隔
QtcNi 個体の母集団アプローチによって補正されるQT間隔
RBC 赤血球数
SBP 収縮期血圧
SCR スクリーニング(来院)
UDS 尿の薬物スクリーニング
ULN 正常範囲の上限
WBC 白血球数
【技術分野】
【0001】
本出願は、200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含む水性医薬製剤であるが、ただし該製剤の濃度が684U/mLのインスリングラルギンではない、水性医薬製剤、およびその使用に関する。
【背景技術】
【0002】
インスリングラルギンは、31B−32B−Di−Arg ヒトインスリン(ヒトインスリンのアナログ)であり、さらにA21位のアスバラギンがグリシンでさらに置換されている。
【0003】
ランタス(登録商標)は、単回投与の皮下注射後、24時間の基礎インスリン供給を提供するインスリングラルギンを含有するインスリン製品である。
【0004】
ランタス(登録商標)の糖動的効果は、皮下注射部位からのインスリングラルギンの遅延吸収および予測可能な吸収によって明らかなピークを示さずにスムーズな24時間の濃度および作用プロファイルがもたらされるという点で現在市販されている他のインスリン製品と区別される。ランタス(登録商標)は、長時間作用型インスリン製品についての医療的な必要性を満たすために開発され、24時間にわたり可能な限りスムーズな基礎インスリンプロファイルによって正常または正常に近い血中グルコースコントロールを得るために一日一回の注射として投与することができる。このような製剤は、一日中血中グルコースの良好なコントロールを提供し、その上より明らかな「ピーク」作用を持つ他のインスリン製剤で見られる低血糖症をもたらす傾向を最小にする。
【0005】
多数の患者、特に肥満によってインスリン抵抗性が増加している患者は、血中グルコースを調節するために投与量の大きいものを用いている。例えば、100単位(100U)の用量は、1mLのランタス(登録商標)U100の注射が必要であり、これはいくらかの不快感を覚える場合がある;ランタス(登録商標)U100 1mLには、100U(3.6378mg)のインスリングラルギンが含まれている。注入量を減らすために、1mLあたり300Uのインスリングラルギンを含む製剤が開発されてきた。本発明はインスリングラルギンU300の製剤に限定されないが、本明細書中に記載される臨床試験は、インスリングラルギン300U製剤を用いて行った;インスリングラルギンU300 1mLあたり300U(10.9134mg)のインスリングラルギンが含まれている。この製剤は、3分の1の注入量で同じ単位数のインスリングラルギンを患者に注入することができる。
【0006】
インスリングラルギン製剤U100とU300は両方同じインスリン曝露および同じ効果、すなわちタイムプロファイルを提供するはずであると考えられた。
【発明の概要】
【発明が解決しようとする課題】
【0007】
試験(T)の治験薬であるインスリングラルギンU300の曝露および活性を、認可されている参照(R)製品であるランタスU100の曝露および活性と等価なものについて、正常血糖クランプにて非肥満体の健常な被験者で試験した。インスリングラルギンの作用の長期的な持続時間を評価するための、皮下投与30時間後を選択した。皮下投与後のインスリングラルギン濃度のタイムプロファイルより曝露を評価し、活性はインスリン1単位当たりのグルコース利用として同時に評価した。
【0008】
反復デザイン(replicate design)により、限定数の患者で、FDAガイドライン「Guidance for Industry、Statistical Approaches to Establishing Bioequivalence」に推奨されているような生物学的同等性および変動性(variability)を評価することができた。
【0009】
それぞれの臨床試験により、曝露および活性の等価を確立することが期待された。
【0010】
0.4U/kgの用量を本試験のために選択した;この量は、患者における平均基礎インスリン用量に対応している。非肥満体の健常な被験者において、この用量は、正常血糖クランプの設定で測ることのできる血清インスリン濃度のかなりの上昇、持続的なグルコース低下効果をもたらす。
【0011】
ガイドラインで支持される反復デザインは、無作為化プランによって割り当てられる予め定義された4通りのクロスオーバーシーケンス(RTTRまたはTRRT)で、いずれかのIP(R:ランタス(登録商標)U100、T:インスリングラルギンU300)の2つの反復単回投与を必要とする。これは、4つの期間(P)1〜4において実行された。結果として、各被験者は、2つの向かい合う臍周囲領域を交互に0.4U/kgのランタス(登録商標)U100(R)およびインスリングラルギンU300(T)の2つの反復単回皮下投与を受けた。
【0012】
4〜18日間の休薬期間は各投薬日から分離される。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、4日間というのは参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間というのは、クランプ期間の間に、被験者が試験に関係のない義務を自由に果たすことを可能にする3週間の休止期間を再び与える。
【0013】
正常血糖クランプ来院の前に、SCR(スクリーニング来院)において被験者は適格性についてスクリーニングされ、EOS(試験終了時)来院において被験者は正常な健康状態を保障するための最終試験に参加する。スクリーニングおよびP1は21日間より長く離してはならないが、EOS来院は次に続く週であるP4のD1と同じ週日(すなわち、さらなる4日後)より前、かつP4のD2後の2週間よりも後(すなわち、さらなる14日後)にはしなかった。
【0014】
これは、計4回の反復投与を伴う単回投与試験である。IPの効果は約24時間続き、このことが、被験者が2日間施設に拘束される理由である。被験者は治療に4度曝露された。
【0015】
本試験の主要な目的は、正常血糖クランプ技術を用いて、バイオアベイラビリティ(曝露)および生物有効性(活性)におけるランタス(登録商標)U100(市販製剤)およびインスリングラルギン U300の平均生物学的同等性(ABE)を評価することであった。
【0016】
本試験の二次的な目的は、インスリングラルギンU300の安全性および忍容性を評価することであった。
【0017】
上述のように、U100とU300両方のインスリングラルギン製剤は、同じインスリン曝露およびインスリン効果を提供すると予測された。しかしながら驚いたことにインスリン曝露および効果は同じではないことが示された。インスリングラルギンU100およびインスリングラルギンU300はバイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。インスリングラルギンU300投与後の曝露および活性は同量(0.4U/kg)のインスリングラルギンU100の投与後の曝露および活性と比較した場合約40%低かった。
【0018】
しかし、インスリングラルギンU300は、インスリングラルギンU100よりも、基礎インスリンに望ましいよりフラットなPK(曝露)およびPD(活性)プロファイルを示した。健常な被験者に対する同じ皮下投与後の、インスリングラルギンU100製剤とインスリングラルギンU300製剤との間の曝露および活性におけるこれらの驚くべきかつ予測できない差異は、以下の図に効果的に示されている。注目すべきは、同時に血中グルコースが一定になったということである。
【0019】
インスリングラルギンの血中グルコースの低下効果は、健常な正常血糖のビーグル犬においてさらに評価された。インスリングラルギン濃度の増加に伴い、作用の平均時間はそれぞれ6.8h(U100)から7.69h(U300)まで増加した。
【0020】
イヌにおいて、100から300U/mLまでグラルギン濃度を増加することによって、血中グルコース低下の時間作用プロファイルは、よりフラットかつ延長された活性に向けて変化した。イヌにおける現在のデータはヒトにもあてはまり、インスリングラルギンのより高い薬物濃度がプロファイルおよび作用のより長い持続時間に積極的に関与することを示している。
【0021】
さらに、100U/mL、300U/mL、500U/mL、700U/mLおよび1000U/mLの濃度を有するインスリングラルギン製剤の沈殿物を顕微鏡で調べた。これらの研究により、濃度の増加に伴って明らかにより大きい粒子がもたらされるという沈殿物特性の違いが明らかとなった。
【0022】
さらに、溶解特性に関しては、より高い濃度のインスリングラルギン製剤の影響がインビトロ試験系を用いることによって調査される。それを行うために、沈殿の研究が、インビボ条件を模擬するpH7.4のリン酸緩衝液を用いて実施される。
【0023】
沈殿したインスリンの上清は、インスリングラルギン含有量を測定するためにHPLC技術を用いて試験される。
【0024】
WO2008/013938 A2は、684U/mLの濃度でインスリングラルギンを含有する水性医薬製剤を開示している。
【課題を解決するための手段】
【0025】
本発明はインスリングラルギンU300製剤に限定されず、本明細書中で詳述しているような他のより高い濃度のインスリングラルギン製剤を用いても有効であるが、本明細書中に記載される臨床試験は、インスリングラルギンU300製剤を用いて実施した。
【0026】
1mLのインスリングラルギンU300製剤は、10.913mgの21A−Gly−30Ba−L−Arg−30Bb−L−Arg ヒトインスリン[300IUのヒトインスリンと等モル]、90μgの亜鉛、2.7mgのm−クレゾール、20mgのグリセリン 85%、HClおよびNaOHを含有し(pH4.0);比重は1.006g/mLである。
【0027】
しかしながら、賦形剤の種類およびそれらの濃度に関しては変更が可能である。
【0028】
この医薬製剤は、200−1000U/mLのインスリングラルギン[200−1000IU ヒトインスリンと等モル](この中で、当該製剤の濃度は684U/mLではない)、好ましくは250−500U/mLのインスリングラルギン[250−500IU ヒトインスリンと等モル]、より好ましくは270−330U/mLのインスリングラルギン[270−330IU ヒトインスリンと等モル]、およびさらにより好ましくは300U/mLのインスリングラルギン[300IU ヒトインスリンと等モル]を含有する。
【0029】
界面活性剤、例えば、とりわけ非イオン性界面活性剤を医薬製剤に加えることができる。特に、調剤用に市販されている界面活性剤が好ましく、例えば以下のようなものである:グリセリン、ソルビトールなどのような高アルコールの部分エステルおよびエーテルならびに脂肪酸エステルおよびエーテル(Span(登録商標)、Tween(登録商標)、特にTween(登録商標)20およびTween(登録商標)80、Myrj(登録商標)、Brij(登録商標))、Cremophor(登録商標)またはポロキサマー。これらの界面活性剤は、5−200μg/ml、好ましくは5−120μg/ml、そして特に好ましくは20−75μg/mlの濃度で医薬組成物中に存在している。
【0030】
この製剤は、防腐剤(例えば、フェノール、m−クレゾール、p−クレゾール、パラベン)、等張化剤(isotonic agent)(例えば、マンニトール、ソルビトール、ラクトース、デキストロース、トレハロース、塩化ナトリウム、グリセリン)、緩衝物質、塩、酸およびアルカリをさらに含んでよく、さらなる賦形剤も含んでよい。これらの物質は各場合に個々にかまたは混合物として存在し得る。
【0031】
グリセリン、デキストロース、ラクトース、ソルビトールおよびマンニトールは、100−250mMの濃度で、NaClは150mMまでの濃度で医薬製剤中に存在し得る。緩衝物質(例えば、リン酸エステル、酢酸エステル、クエン酸エステル、アルギニン、グリシルグリシンまたはTRIS(すなわち2−アミノ−2−ヒドロキシメチル−1,3−プロパンジオール)緩衝液および対応する塩)は、5−250mM、好ましくは10−100mMの濃度で存在し得る。さらに賦形剤はとりわけ塩類またはアルギニンであり得る。
【0032】
この製剤の亜鉛含有量は、0−1000μg/mL、好ましくは20−400μg/mLの亜鉛、最も好ましくは90μg/mLの存在量によって到達する濃度範囲である。この亜鉛は塩化亜鉛の形態で存在していてもよいが、その塩は塩化亜鉛に限定されない。
【0033】
この医薬製剤において、グリセリンおよび/もしくはマンニトールは、100−250mmol/Lの濃度で存在し得、ならびに/またはNaClは、150mmol/Lまでの濃度で存在し得る。
【0034】
この医薬製剤において、緩衝物質は、5−250mmol/Lの濃度で存在し得る。
【0035】
本発明のさらなる目的は、例えばインスリンの放出を遅らせる塩のようなさらなる添加剤を含む医薬品インスリン製剤である。このような遅延放出インスリンと上記の製剤との混合物は、本発明に含まれる。
【0036】
本発明のさらなる目的は、このような医薬製剤の製造方法である。これらの製剤を製造するために、これらの成分を水に溶解し、HClおよび/またはNaOHを用いてpHを調製する。同様に、本発明のさらなる目的は、糖尿病の治療のためのこのような製剤の使用である。
【0037】
本発明のさらなる目的は、インスリン、インスリンアナログもしくはインスリン誘導体またはこれらの製剤の製造工程の間の、安定剤としての界面活性剤の使用または添加である。
【0038】
本発明はさらに、グルカゴン様ペプチド−1(GLP1)またはそのアナログもしくは誘導体、あるいはエキセンジン−3もしくは−4またはそのアナログもしくは誘導体、好ましくはエキセンジン−4をもさらに含む、上記の製剤に関する。
【0039】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
H−desPro36−エキセンジン−4−Lys6−NH2、
H−des(Pro36,37)−エキセンジン−4−Lys4−NH2および
H−des(Pro36,37)−エキセンジン−4−Lys5−NH2、
またはこれらの薬理学的に許容される塩。
【0040】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
desPro36[Asp28]エキセンジン−4(1−39)、
desPro36[IsoAsp28]エキセンジン−4(1−39)、
desPro36[Met(O)14,Asp28]エキセンジン−4(1−39)、
desPro36[Met(O)14,IsoAsp28]エキセンジン−4(1−39)、
desPro36[Trp(O2)25,Asp28]エキセンジン−2(1−39)、
desPro36[Trp(O2)25,IsoAsp28]エキセンジン−2(1−39)、
desPro36[Met(O)14Trp(O2)25,Asp28]エキセンジン−4(1−39)および
desPro36[Met(O)14Trp(O2)25,IsoAsp28]エキセンジン−4(1−39)、
またはこれらの薬理学的に許容される塩。
【0041】
本発明はさらに、ペプチド−Lys6−NH2がエキセンジン−4のアナログのC末端に結合している、前出の段落で記載されたような製剤に関する。
【0042】
本発明はさらに、エキセンジン−4のアナログが以下からなる群より選択される、上記の製剤に関する:
H−(Lys)6−des Pro36[Asp28]エキセンジン−4(1−39)−Lys6−NH2
des Asp28Pro36,Pro37,Pro38 エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5 des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Trp(O2)25,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
H− des Asp28 Pro36,Pro37,Pro38[Trp(O2)25]エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Met(O)14,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
des Met(O)14 Asp28 Pro 36,Pro37,Pro38 エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro 37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
H−Asn−(Glu)5 des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−Lys6−NH2、
des Asp28 Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25]エキセンジン−4(1−39)−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14,Asp28] エキセンジン−4(1−39)−NH2、
des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−(Lys)6−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28]エキセンジン−4(1−39)−(Lys)6−NH2、
H−Asn−(Glu)5−des Pro36,Pro37,Pro38[Met(O)14、Trp(O2)25,Asp28] エキセンジン−4(1−39)−(Lys)6−NH2、
またはその薬理学的に許容される塩。
【0043】
本発明はさらに、Arg34、Lys26(Nε(γ−グルタミル(Nα−ヘキサデカノイル))) GLP−1(7−37)[リラグルチド]またはその薬理学的に許容される塩をさらに含む、上記の製剤に関する。
【0044】
一実施態様において、本発明は200−1000U/mL[200−1000IU ヒトインスリンと等モル]、好ましくは200U/mL〜650U/mL、さらに好ましくは700U/mL〜1000U/mL、より好ましくは270−330U/mLの範囲で、そして最も好ましくは300U/mLの濃度でインスリングラルギンを含む水性医薬製剤に関するが、ただし該製剤のインスリングラルギンの濃度は、684U/mLではない。
【0045】
さらに、その製剤は、エキセンジン−4のアナログ、例えば、リキシセナチド(lixisentatide)、エクセナチドおよびリラグルチドをも含み得る。これらのエキセンジン−4アナログは、インスリングラルギン1Uあたり0.1μg〜10μg、好ましくはインスリングラルギン1Uあたり0.2〜1μg、およびより好ましくはインスリングラルギン1Uあたり0.25μg〜0.7μgの範囲でその製剤中に存在する。リキシセナチドが好ましい。
【0046】
さらに、その水性医薬製剤は、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される1種またはそれ以上の賦形剤を含み得る。具体的には、その水性医薬製剤は、90μg/mLの亜鉛、2.7mg/mlのm−クレゾールおよび20mg/mlのグリセリン 85%を含み得る。場合により、その水性医薬製剤は、20μg/mLのポリソルベート20を含み得る。
【0047】
その水性医薬製剤のpHは、μμ3.4〜4.6の間、好ましくは4または4.5である。
【0048】
本発明は、I型およびII型糖尿病を治療する方法に関し、該患者に本発明の水性医薬組成物を投与することを含む。
【0049】
開示された種々の濃度範囲で好ましいのは300U/mLの濃度であり、好ましいインスリンアナログはインスリングラルギンである。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0050】
このインスリンは、1日1度投与されるのが好ましいが、必要に応じて1日に2度投与することも可能である。投薬の必要性は、正常または許容可能な血中グルコース濃度の達成によって決定される個々の患者の必要性による。
【0051】
本発明はまた、該患者に本発明の水性医薬製剤を投与することを含む、I型およびII型糖尿病患者の治療においてインスリングラルギンの曝露の持続時間を延長する方法に関する。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0052】
本発明はまた、インスリングラルギンを用いて、I型およびII型糖尿病患者の治療において低血糖の発生を低減する方法にも関し、該患者に本発明の水性医薬製剤を投与することを含む。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0053】
本発明はまた、I型およびII型糖尿病患者の治療において、インスリングラルギンによってピークのない長時間作用型基礎インスリンを提供する方法に関し、該患者に本発明の水性医薬製剤を投与することを含む。開示された種々の濃度範囲の中で好ましいのは、300U/mLの濃度である。さらにその水性医薬製剤は亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムならびにそれらの混合物をも、本発明の水性医薬製剤に関して本明細書中で開示した範囲で含むことができる。好ましい実施態様において、その水性医薬製剤はインスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドも含む。
【0054】
I型糖尿病およびII型糖尿病の治療における前出の項目のいずれかに従う水性製剤の使用。
【0055】
本願を、いくつかの実施例を用いて以下に説明するが、これらの例は決して限定することを目的としていない。
【0056】
実施例1:プロトコルの記載
本試験は、6度の来院での健常な被験者における単一施設、無作為化、対照群あり(controlled)、単盲検、4期、2治療、2−シーケンスクロスオーバー試験であった:
来院1:スクリーニング(SCR)
来院2〜5、期間(Period:P)1−4:治療、正常血糖クランプ期間
来院6:試験終了時(EOS)
被験者に、0.4U/kgのインスリングラルギンU100の単回皮下用量を投与、および代替的にはインスリングラルギンU300を、2点の向かい合う臍周囲領域(左、右、左、右)の部位に異なる4日で注射した。この治験薬を治療RとTを2シーケンスで反復(RTTRまたはTRRT)して、P1〜P4期に投与した。4〜18日間の休薬期間により各投薬日を離した。
R:0.4U/kg−体重のインスリングラルギンU100(市販の製剤;基準薬)
T:0.4U/kg−体重のインスリングラルギンU300(試験薬)
P1は、SCR後多くとも3〜21日に行わねばならない。EOS来院は、P4後の4〜14日になされなければならない。
【0057】
P1〜P4の期間中、人工膵(Biostator)を被験者に連結して、血中グルコースの測定とグルコース注入率の調整を行った。血中グルコース濃度およびグルコース注入率(GIR)を、当該治験薬の皮下注射前の90分間(ベースライン期間)、および治験薬投与後の30時間にわたってモニタリングした。治験薬投与の60分、30分および5分前に測定された3つの空腹時血中グルコース値の平均値として決定された個々の空腹時血中グルコース濃度を5%下回る血中グルコース濃度を維持するために、20%のグルコース液剤の注入を開始した。GIRのプロファイルが得られた。血液サンプルを、正常血糖クランプ期間中に所定の回数採取し、血清インスリングラルギン濃度を決定した。グルコースクランプ期間中、被験者は水道水を除いて絶食状態とした。
【0058】
個体の本試験の所要期間は、SCRからEOS来院の間が最大13週間までの見込みであった。
【0059】
このプロトコルを、検討および書面による承認のために、個々の倫理委員会および/または治験審査委員会に提出した。このプロトコルは、18th World Health Congress(ヘルシンキ、1964)の勧告および全ての準拠する改正事項を満たしている。このプロトコルはまた、本試験が行なわれたドイツの法律、条例、ならびに全ての準拠するガイドラインをも満たしていた。全ての試験関連手順を行なう前に、インフームドコンセントを得た。
【0060】
実施例2:被験者の選択
20名の試験完了者を確保するために、24人の健常な被験者に対して治療するように計画した。
【0061】
以下の基準の全てを満たしている被験者が、本試験への登録を考慮された:
デモグラフィー
・18歳〜50歳の年齢のいずれかの性別である被験者:
・体重は50kg〜110kgであり、ボディマス指数(BMI)は18〜28kg/m2である;
健康状態
・以下の包括的な臨床評価(詳細な病歴および完全な身体検査)により健常であると証明されていること;
・少なくとも3ヶ月間、喫煙していないこと;
・12誘導心電図、およびバイタルサイン、但し、治験責任医師が、異常を臨床上無関係であると判断した場合を除く;
・背臥位で5分休止後の正常バイタルサイン:
95mMHg≦収縮期血圧≦140mMHg;
45mMHg≦拡張期血圧≦90mMHg;
40bpm≦心拍数≦100bpm;
・正常な12誘導ECG;120ms<PR<220ms、QRS<120ms、QTc≦430ms
(女性の場合:QTc≦450ms);
・健常な被験者の各臨床検査項目は正常範囲内にあること、但し、治験責任医師が異常を臨床的に無関係であると判断する場合を除く;
しかしながら、血清レアチニンおよび肝酵素(AST、ALT)は、臨床検査ノルムの上限を厳密に下回らなければならない;
・空腹時血清グルコース(≦100mg/dL)およびグルコシル化ヘモグロビン(HbA1c≦6.1%)として定義される正常代謝コントロールを有すること;
・被験者は、本試験に参加する前に、少なくとも4週間、日常的に用いている処方薬治療を中止せねばならない;
【0062】
女性の被験者についての義務
・妊娠する可能性があり(閉経前および外科手術による不妊処理を行っていないかまたは閉経後2年未満として定義される)、性的に活発である女性被験者は、適切な避妊をおこなわなければならない。適切な避妊とは、インプラント、経口避妊薬またはホルモン剤と組み合わせた注射剤、IUD(子宮内避妊器具)のような高度に効果的な避妊法(パールインデックス< 1%)として定義される。本臨床試験の目的のための閉経後とは2年以上の無月経または外科的に不妊であることを含む;
・女性被験者は、前試験スクリーニングの間、および最初のクランプ前に、尿β−ヒト絨毛性ゴナドトロピン(beta−HCG)妊娠テストが陰性でなければならない。
【0063】
規則(Regulations)
・本試験に関する全ての手順の前に書面によるインフォームドコンセントを行う;
・健康保険制度でカバーされており、そして/または生物医学研究に関して施行された国内法令の勧告を遵守している;
・いかなる行政または法的な監督下にはない。
【0064】
以下の項目のいずれかにあてはまる被験者は、本試験に含められていない:
病歴および臨床状態
・臨床上関連のある心血管、肺、胃腸、肝臓、腎臓、代謝、血液、神経、精神、全身性、眼または感染の疾患のいずれかの病歴または存在;いずれかの急性感染性疾患または急性疾患の兆候;
・薬物アレルギーの存在もしくは病歴、または医師によりアレルギー性疾患と診断され治療を受けた;
・キサンチン塩基を含む飲料の過剰摂取(>4カップまたはグラス/日);
・以下からの禁忌(正常範囲に従う−値が正常範囲外である場合は、治験責任医師がこの異常値が臨床的に無関係と理解した場合、その被験者は含まれ得る):
−病歴/手術歴および身体検査
−臨床検査(血液、臨床化学およびディップスティックによる検尿)
−標準的な12誘導心電図
−血圧および心拍数
・試験への参加前4週中の所定の薬物で継続中の治療または所定の薬物で定期的な治療
・本試験の目的を妨げ得る、試験前3ヶ月中の臨床的に有意な疾患の徴候、または試験前4週中のいずれかの主要な内科的疾患の兆候(治験責任医師の意見に従う)
・薬物の吸収、分布、代謝または排泄を妨げることが知られている疾患の存在もしくは続発症または他の病態
・薬物またはアルコールの乱用歴
・類似した化学構造を持つ治験薬または薬物に対する過敏症既往歴
・進行性致死的疾患
・本試験中に事前に計画された手術
・前3ヶ月中の500mLを超える献血
被験者は1度より多くの本試験への登録は許可されない。
【0065】
一般的条件
・治験責任医師の判断において、本試験中、服薬遵守しそうにないか、または言語の問題もしくは精神発達遅滞のため、もしくは被験者が本試験の性質、範囲および起こりうる結果を理解できなくする精神状態に起因して協力できない被験者
・適用し得る規則に従って、以前の研究の排除期間内にある被験者;
・被験者が本プロトコルの実施に直接的に関与する治験責任医師もしくはいずれかの治験責任分担医師、研究助手、薬剤師、治験コーディネーター、または他のそのスタッフである;
・SCR前の以前の30日以内の治験薬の受領
【0066】
生物学的状態
・以下のテストのいずれかに陽性反応である:HBs抗原、抗HCV抗体、抗HIV1抗体、抗HIV2抗体;
・SCRで尿の薬物スクリーニングでの陽性結果(アンフェタミン/メトアンフェタミン類、バルビツレート類、ベンゾジアゼピン類、カンナビノイド類、コカイン、オピエート類);
・アルコール呼気テスト陽性
【0067】
実施例3:治療
試験治療の詳細
【表1】
【0068】
ランタス(登録商標)/インスリングラルギン製剤の用量の算出
各被験者に投与するインスリングラルギンの量(0.4U/kg)を算出するために、体重(kg)は少数点以下1桁で測定し、以下の実施例に示すように算出したインスリンの量は切り上げるかまたは切り捨てして丸めている:75.3kgの体重の被験者に30Uのインスリンを投与した(75.3×0.4=30.12 切り捨てして30とする);74.4kgの体重の被験者に30Uのインスリンを投与した(74.4×0.4=29.76 切り上げして30とする)。期間1の日1に記録した体重を、期間1の体重から2kgより多く体重が変化しない限り、期間2、3および4についても治験薬用量の算出に用いた。
【0069】
ユニット中の量は、インスリングラルギンU100とインスリングラルギンU300の両方とも同じであった。この比重は、両方の薬品について同じである。しかし、インスリングラルギンU100と比較して、インスリングラルギンU300はインスリングラルギンの濃度が3倍高いことを考慮して、インスリングラルギンU300については注射すべき容量従って重量を1/3とした。個々の用量を投与する注射器を秤量して調製した。正味質量は、治験責任医師のソース文書(source-documentation)にのみ記録した。
【0070】
注入剤の用量の算出および調製
【表2】
【0071】
グルコース液剤:個々の被験者の血中グルコースを決定した目標レベルに維持するために、20% グルコース液剤を人工膵(Biostator)を用いて注入した。第2の注入ポンプ(人工膵(Biostator)の一部)は、0.9% 塩化ナトリウム溶液を送達して、ラインの開存(line patent)を維持した。必要な20% グルコース液剤の量が人工膵(Biostator)の注入容量を超える場合、第2のグルコース注入ポンプを係合した。
【0072】
ヘパリン: 100mLの0.9% 塩化ナトリウム溶液中の10000IU ヘパリンを、二腔カテーテルに約2mL/hの速度で注入し、人工膵(Biostator)による血中グルコースの測定のためのその開存を維持した。
【0073】
盲検法(blinding methods)の説明:
本試験は、単盲検(single-blinde)である。容量の異なる注射は、投薬の盲検を不可能にする。注射は、権限のある医師であるか、さもなければ本試験に関与していない医師によってなされた。治験責任医師は無作為化コードにアクセスした。
【0074】
治療群への被験者の割り当て法
手順を臨床試験プロトコルで定めた後、本治験薬を本試験に含めた被験者にのみに投与した。
【0075】
P1〜P4で注射すべき2つのランタス(登録商標)製剤の治療シーケンスのために無作為化スケジュールを、乱数表とリンクさせ、性別により階層化して作製した。
【0076】
期間1の日1の朝、被験者がプロトコルに定められている基準を満たしていることを治験責任医師が確認すると直ぐに、適格な被験者をその施設により無作為化した。その後その乱数を、P1前に被験者の適格性が確認された順に被験者番号に割り当てた。SCR後の性別階層認定のために、第一の被験者に適切な性別階層に対する第一の乱数を付与した。階層内に認定される次の被験者には、その階層内の次の乱数を付与した。
【0077】
その乱数は、被験者に治療キットを割り当てるための治療キット番号として用いた。各被験者は、割り当てられた治療キット番号を持つ治験薬を投与された。この治療キットには、IP付き一般情報、治療キット番号、期間番号、コンテナボックス上に被験者番号を書くためのフィールド、および地方条例によって要求される追加記載が含まれている。
【0078】
試験を永続的に中止した被験者は、既に与えられている場合、被験者番号および無作為化番号を保持しておいた。
【0079】
包装およびラベリング
本治験薬は、無作為化プランに従って、サノフィ−アベンティス ドイチュランド GmbH、フランクフルト アム マイン ドイツにより包装された。本治験薬を含むカートリッジおよびそれらをパックしたカートンは、試験番号、無作為化番号、バッチ番号、貯蔵条件、治験依頼者およびP番号でラベルされた。
【0080】
治験薬の供給は、一度の出荷のものを受けた。全てのコンテナは、同一形式のラベルが付されていた。さらに、注射器用のラベル1セットが供給された。治験薬と予備薬剤は、異なる冷蔵庫に保管した。
【0081】
治験薬投与の前に、薬剤師または薬剤師に指示された人が適切な治験薬を入れた注射器を調製し、その注射器に、この治験薬のコンテナに従う、被験者番号、無作為化番号および適切な期間をラベル付した。
【0082】
このラベル付の内容は、地方条例の仕様および要件に従って行った。
【0083】
貯蔵条件
本治験薬を+2℃〜+8℃の温度で光から保護して保管した。本試験薬は、凍結しないようにした。調製中は、その薬剤を光から保護する必要はなかった。
【0084】
予備サンプル(ランタス(登録商標)U100を300カートリッジ、およびインスリングラルギンU300を300カートリッジ)を、この試験施設レベルで同じ条件で保管確保した。
【0085】
実施例4: 治験製品の評価
活性または薬力学
インスリングラルギンによるインスリン受容体の刺激は、作用様式である。それに引き続く末梢グルコース取り込みおよび内因性グルコー産生の抑制は、血中グルコース濃度の減少がもたらす糖動的効果を含む。結果としてもたらされたグルコース利用は、血中グルコース濃度を一定に維持するために必要とされるグルコースのゲージ(gauge)によって最もよく特徴づけられる。
この正常血糖クランプ技術は、インスリングラルギンの注射後、ベースラインレベルより5%低く血中グルコース濃度を維持するのに必要なグルコースの量を評価するために用いられている。
【0086】
臨床評価方法
オンラインでのグルコース測定を、グルコースオキシダーゼ法を用いて人工膵(Biostator)(Life Sciences instruments、Elkhart、IN、USA)により行った。
オフサイトの血中グルコースもまた、グルコースオキシダーゼ法を用いてSuper GLグルコース分析装置により測定した。
【0087】
薬力学変数/エンドポイント
皮下注射されたインスリンの単位(用量)あたりの利用されたグルコース量が、糖動的効果の尺度(measure)である。
連続的に記録されたグルコース注入率(GIR)は、注射したインスリンの経時作用プロファイルを反映している。
【0088】
主要変数/エンドポイント
主要薬力学変数は、24時間以内のグルコース注入率時間曲線下面積[GIR−AUC0-24h(mg・kg-1)]である。
【0089】
副次変数/エンドポイント
副次薬力学変数は、50% GIR−AUC0-24hまでの時間[T50%−GIR−AUC(0-24h)(h)]である。
【0090】
薬物動態学
サンプリング時間
血清インスリングラルギンおよびC−ペプチドの濃度の評価用に血液サンプルを治験薬の皮下注射の1時間前、30分前および直前に採取し、その後は、注射の30分後、1時間後、2時間後、その後は最大で24時間までの2時間おきに、そして30時間後に採取した。
インスリングラルギンサンプルの番号を、P00、P01、P02、P03、P04などと付け、C−ペプチドサンプルの番号をC00、C01、C02、C03、C04、などと付けた(試験のフローチャートも参照のこと)。
【0091】
薬物動態学サンプリング数
最低18のサンプルをクランプ来院(P1〜P4)ごとに採取した。1被験者あたり総計72サンプルを採取した。
【0092】
PK取り扱い手順
サンプル採取の正確な時間は、CRFで必ず記録する。薬物動態学サンプル(インスリングラルギン、C−ペプチド)の保管および配送については、特別な手順を用いた。
【0093】
生物学的分析法
生物学的分析を、医薬品の安全性に関する非臨床試験の実施基準(Good Laboratory Practice(GLP))のOECD原則(1997年に改定)、ENV/MC/CHEM(98)17およびその地域に適用されるGLP規則にて認められている、このタイプの試験に適用可能なGLP条件を基に実施した。
予備サンプルは利用できないので、インスリングラルギンの測定を優先する。
【0094】
インスリングラルギン
血清インスリングラルギン濃度を、インスリングラルギン用に校正したヒトインスリン用のラジオイムノアッセイ(RIA)(インスリンRIAキット、ADALTIS、イタリア)を用いて測定した。キットREF 10624。
このアッセイでの定量下限(LLOQ)は、4.92μU/mLであった。
【0095】
C−ペプチド
血清C−ペプチド濃度をC−ペプチド用のラジオイムノアッセイ(RIA)(C−ペプチド RIAキット、ADALTIS、イタリア)を用いて測定した。キットREF C−ペプチド10282。
定量下限(LLOQ)は、0.090nmol/Lであった。
【0096】
生物学的分析法の要約
被検体 インスリン、C−ペプチド
マトリックス 血清
分析技術 RIA
定量下限 インスリン 4.92μU/mL;C−ペプチド 0.090nmol/L
アッセイ容量 インスリン 100μL;C−ペプチド 100μL
方法参照 Adaltis S.p.A.イタリア;キットREF 10624 インスリン(方法No.435VAL02)およびキットREF C−ペプチド10282(方法No.DMPK/FRA/2003−0002)
【0097】
薬物動態学変数/エンドポイント
インスリングラルギン濃度の経時曲線が、皮下注射したIPの全身インスリン曝露の尺度(measure)であった。
【0098】
主要変数/エンドポイント
主要薬物動態学変数は、血清インスリングラルギン濃度の時間曲線下面積[INS−AUC0-24h(μU・h・mL-1)]であった。
【0099】
副次変数/エンドポイント
副次薬物動態学変数は、50% INS−AUC0-24hまでの時間[T50%−INS−AUC(0-24h)(h)]であった。
【0100】
サンプリングした血液容量
【表3】
【0101】
本試験の盲検を保護するための測定
本試験は単盲検試験である。生物学的分析測定は、臨床試験が完了した後に行なった。治療コードは、予測できない全ての重篤な有害事象(SAE)を報告するために知られており、治験責任医師および/または治験依頼者のいずれかの判断に従ってIPの使用と合理的に関連付けられている。
【0102】
実施例5:試験手順
来院スケジュール
スクリーニング手順
各被験候補者の診療録を試験の開始前にチェックし、参加への適格性を決定した。被験者は、SCRでのスクリーニング試験前に10時間絶食(水を除く)した。
【0103】
以下の項目/検査を評価した:
・年齢、および人種
・身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系を含む)
・関連する病歴および手術歴(本試験に関連する所見のみを記録すべきである)
・人体計測:身長および体重、BMI[体重(kg)・(身長(m))-2]の計算
・血圧および心拍数(背臥位で5分後、および立位で3分後)
・深部体温(鼓膜)
・標準12誘導ECG
・血液学的状態、臨床化学、および検尿(ディップスティック)
・凝固状態(INR、aPPT)
・尿の薬物スクリーニング
・アルコールスクリーニング(呼気検知器)
・空腹時血中グルコース(≦100mg.dL-1)およびグリコシル化ヘモグロビン(HbA1c≦6.1%)として定義される正常な代謝コントロール
・B型/C型肝炎およびHIV試験
【0104】
被験者がスクリーニングできない場合、スクリーニング試験の臨床結果を含むSCRで得られた全てのデータをその被験者の診断録に記載した。
【0105】
来院のタイプによる記述
期間
各試験期間(P1〜P4)は日1および日2の2日間である。日1は、正常血糖クランプと治験薬の投与の開始日であった。日2は、治験薬の投与後30時間にわたる正常血糖クランプの終了の日であった。試験期間(P1〜P4)の間には、4〜18日の休薬期間を設けた。各治験薬投与前2日間は、激しい運動(例えば、マウンテンバイク、重労働の庭作業など)をさせなかった。アルコール飲料、グレープフルーツジュース、およびキサンチン誘導体を含む刺激性飲料(お茶、チョコレート、コーヒー、コカコーラ(商標)のような飲料など)およびグレープフルーツの摂取は、正常血糖クランプの24時間前から完了まで許可しなかった。被験者は各試験期間(P1〜P4)の日1の10時間前から絶食(水を除く)し、正常血糖クランプの終了まで絶食(水を除く)を続けた。被験者は、各クランプの来院につき約32時間クリニックに滞在せねばならなかった。
【0106】
期間1の日1の朝、276001001で始まる9桁の被験者番号を被験者に割り当てた。SCRに入る資格を与えられた次の被験者には、被験者番号276001002を付与した。最初の被験者には無作為化番号101を与えた。資格を与えられた次の被験者には無作為化番号102を与えた。
【0107】
被験者に、彼らの体調に臨床的に有意な変化がなく、その前の期間からのプロトコル中で定められたような一般事項および食事制限を遵守することを確実にするよう依頼した。試験規則に違反した被験者は、本試験への参加から除外した。違反の種類により、被験者はその特定の期間のみ除外されることが可能であり、試験日の計画を立て直した。どのプロトコル違反も、前もってケースバイケースに基づいて治験依頼者と議論した。
【0108】
最期の期間以来の被験者の健康状態のいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0109】
血圧、心拍数および深部体温(鼓膜)を、日1の朝、各治験薬投与(日2)30時間後のクランプ手順の前および後に、少なくとも5分間安静にしてから背臥位で測定した。体重、アルコールスクリーニングおよびRBC、Hb、HcT(P3およびP4のクランプ期間の前のみ)は、日1の朝、クランプを開始する前にのみ評価を行った。
【0110】
各期間の日1、被験者は午前6時30分にクリニックに入院した。上記の検査を通過後、被験者は3本の静脈ラインを準備された。左腕の手の甲の静脈または手首外側の静脈に逆行様式でカニューレを導入し、人工膵(Biostator)(Life Sciences instruments、Elkhart、IN、USA)に連結して血中グルコースの測定のために動脈血化した静脈血を連続的に記録した。動脈血化を達成するために、左手を約55℃の「ホットボックス(Hot−Box)」に入れた。第2の静脈ラインは、左腕の肘前静脈でとり、血清インスリングラルギンおよび参照血中グルコースの測定のためのサンプルを採取するために用いた。第3の静脈ラインは反対側の前腕にカニューレを導入し、人工膵(Biostator)を用いて20%のグルコース液剤および0.9%の生理食塩水を注入した。
【0111】
人工膵(Biostator)により血中グルコース濃度を測定し、グルコース注入率を調節して血中グルコース濃度を治験薬投与の60分前、30分前および5分前に測定した3回の空腹時血中グルコースの値の平均値として算出した個々の空腹時血中グルコースを5%下回る値に維持した。血中グルコースの測定のために追加の血液サンプル0.3mLを治験薬の投与60分前、30分前および5分前に採取し、グルコースオキシダーゼ法に基づいて、臨床基準値と照らし合わせて確認した。
【0112】
午前9時頃、インスリングラルギンU100(市販製剤)またはインスリングラルギンU300のいずれかを、臍から5cm(左、右、左、右)の臍周囲領域に標準化された皮下脂肪厚法(skin fold technique)を用いて注射した。0.30mm×8mm(30G)の針を備えた0.5mL容量のU100インスリン注射器(製造元:Beckton & Dickinson)を用いた。
【0113】
治験薬に彼らそれぞれの治療キット番号、被験者番号(無作為化後にコンテナボックスに記載したもの)、および期間番号のラベルを付した(8.5章の包装およびラベリングを参照のこと)。
【0114】
治験薬の投与後、20%グルコース液剤の注入を、一旦血中グルコース濃度を個々の空腹時濃度から5%下げ、そのレベルを維持する可変速度で開始した。このクランプ期間の所要時間は30時間であった。グルコース送達速度は、予め定めてあったアルゴリズムを用いて1分間隔で血中グルコースの変化に応じて人工膵(Biostator)によって調整した。この人工膵(Biostator)からの血中グルコース値は、クランプ全体にわたり30分間隔でグルコースオキシダーゼ法に基づいた臨床検査基準値と照らし合わせて確認した。必要に応じ、人工膵(Biostator)を臨床検査基準値法の結果に従って再校正した。被験者は、クランプ期間中背臥位のままであった。
【0115】
血清インスリングラルギンおよびC−ペプチド濃度の測定のための血液サンプルを投薬1時間前、30分前および直前、その後治験薬投与の30分後、1時間後、2時間後、その後は最大で24時間までの2時間おきに、そして30時間後に採取した。
【0116】
各試験期間(P1〜P4)の日2に、正常血糖クランプが完了した後食事を提供した。血圧、心拍数および深部体温(鼓膜)を記録し、血中グルコース測定用のサンプルを採取した。被験者は、治験責任医師によって安全が確認された後、クリニックから退院した。
【0117】
注射部位は、全クランプ期間中観察した。被験者の健康状態におけるいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0118】
血液学的な安全性(Safety Hematology)
P3におけるRBC、HbおよびHctを、P4での貧血に備えて、分析した。陽性であった場合、P3とP4の間隔を許される最大の18日まで延長し、P4の前に追加のRBC、HbおよびHct評価を行った。
【0119】
退院手順
被験者は、P4後、4〜14日間の間に、EOS来院のために再来院した。被験者は10時間絶食(水を除く)した。最終期間以降の被験者の健康状態におけるいかなる変化も被験者の診療録(原本)およびCRFに記録した。
【0120】
以下の項目/検査を評価した:
・身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系を含む)
・体重
・血圧および心拍数(背臥位で5分の後)
・深部体温(鼓膜)
・標準12誘導ECG
・血液学的状態、臨床化学、および検尿(ディップスティックによる)
・尿中のβ−HCGテスト(女性に対してのみ)
被験者は、有効な安全性データの治験責任医師によるレビューが完了した後、各期間の日2に退院した。
【0121】
生体サンプルの採取スケジュール
血液
SCR(スクリーニング):
・血液学、臨床化学、HbA1c、血清学(B/C型肝炎テスト、HIV試験):約20mLの血液を採取した。
P1〜P4(日1および日2)
・血中グルコース
人工膵(Biostator)は、治験前の期間を含む全クランプ期間にわたり1分間隔で血中グルコースを自動測定する。人工膵(Biostator)に必要とされる血液量は、2mL・h-1である。およそ252mLの血液量が、4期間に人工膵(Biostator)でのグルコース測定に必要とされる。人工膵(Biostator)から、血中グルコース値をチェックするための血液サンプル(0.3mL)を投薬の60分前、30分前、5分前および0分前、ならびに投薬後クランプの終わり(30時間)までは30分間隔で採取した。およそ41mLの血液量が4期間に採取された。
・血清インスリングラルギンおよびC−ペプチドの濃度
静脈血液サンプル(3.5mL)を、投薬1時間前、30分前、および直前と、投薬30分後、1時間後、2時間後および24時間までは2時間ごとに、そして30時間後に採取した。およそ252mLの血液量を4期間で採取した。インスリングラルギンの測定を優先的に行った。予備サンプルは、C−ペプチド濃度の測定のためにのみ用いた。
・RBC、Hb、Hct
クランプ期間3および4を開始する前に静脈血液を採取した。約4mLの血液を2期間に採取した。
【0122】
試験終了時(EOS)来院:
・血液学、臨床化学:約12mLの血液を採取した。
・尿中の尿中のβ−HCGテスト(女性に対してのみ)
【0123】
SCR−EOSの総血液量:
試験期間全体で、各被験者あたり総計約585mLの血液を採取した。
【0124】
尿
定性的な尿の薬物スクリーニングをSCRおよびEOSに実施した。尿の薬物スクリーニングは、アンフェタミン/メトアンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエートからなる。定性的な安全性検尿(safety urinalysis)をSCRおよびEOSでディップスティックを用いて行った。安全性検尿は、以下の分析項目からなる:pH、タンパク質、グルコース、血液、赤血球、白血球、ビリルビン、ウロビリノーゲン、ケトン、比重、および亜硝酸塩。
【0125】
他の試験変数の測定スケジュール
身体検査は、SCRおよびEOSにて実施した。
深部体温(鼓膜)は、SCR、クランプ期間の前および後のP1〜P4、およびEOSに記録した。
血圧および心拍数は、SCRおよびEOSで、背臥位で約5分間安静にした後、および立位で3分間安静した後にも測定した。P1〜P4においては、血圧および心拍数を日1の朝にクランプ手順を開始する前に少なくとも5分間背臥位で安静にした後と、各治験薬投与30時間後のクランプ手順が完了した後(日2)に記録した。
心電図(標準12誘導)は、SCRおよびEOSに記録した。
体重および身長は、SCRで測定した。体重は、P1〜P4の日1の朝(治験薬の投与前)およびEOSに記録した。
アルコールスクリーニング(エタノール、呼気検知器)は、SCRおよびEOS、ならびにP1〜P4の日1の朝(治験薬の投与前)に行った。
【0126】
試験における制限事項
日1の夜(P1〜P4)から、その期間(クランプ期間)を通して、被験者はアルコール、茶、コーヒー、柑橘系飲料またはコーラ飲料の摂取、喫煙を控えた。柑橘系果実の摂取も試験期間を通して禁止した。被験者は、この試験の継続期間を通して、最終の管理まで、激しい身体活動をせず安定したライフスタイルを守ることを求められた。
【0127】
原データ(source data)の定義
CRFに記録される以下に列挙した全ての評価は、以下に関する適切に署名され確認された原文書により裏付けられた:
・被験者の身元確認、病歴;
・臨床検査、バイタルサイン、体重および身長;
・臨床検査評価(laboratory assessments)、ECG;
・薬物動態の時点(Pharmacokinetic time points);
・来院および評価の日時;
・投与日時、および注射部位;
・AE;
・クランプの所要期間(開始および終了時)
・その他
このCRFは、他の項目についての原文書として考慮された。
【0128】
実施例6: 統計的考察
本実施例は、本試験のための統計学的分析プランの情報を提供する。統計学的分析プランは、被験者の参加前に立案された。
【0129】
被験者数の決定
INS−AUC(0-24h)が初期パラメータであり、それに従って被験者数の算出を実施した。
【0130】
この被験者数算出の目的のために、0.125と0.225の間で、自然対数変換したINS−AUC(0-24h)のいくつかの被験者内標準偏差(SDwithin)を考察した。平均生物学的同等性アプローチのための被験者数の算出法を、4期、2治療、2シーケンスクロスオーバーデザインのために用いた。製剤比率について、90%CIが完全に[0.80−1.25]内に含まれる場合、平均生物学的同等性がそのパラメータにより結論付けられた。
【0131】
試験HOE901/1022を、可変性における推算値のベースとした。試験HOE901/1022の統計解析に基づいて、0.175の値が、自然対数変換したスケールにおける被験者内標準偏差(SDwithin)を予測することができた。
【0132】
以下の表は、90%検出力(power)で0.85と1.15の間の真比率を推算する生物学的同等性対照間隔:[0.80−1.25]を用いて修正幾何平均値の平均生物学的同等性を表すのに必要とされる被験者の数を示している(対照製剤に対する試験)。
【0133】
【表4】
【0134】
このデザインでは、自然対数スケールでの真のSDwithinが0.175である場合、0.9の真比率を可能にする90%の能力で2つのインスリングラルギン製剤の同等性を示すためには、20名の被験体(1シーケンスあたり10名)が必要とされる。
総計24名の無作為化された被験者が、治験中止の可能性のある場合のために計上される。
【0135】
被験者の説明(Subject description)
被験者の内訳
関与した、無作為化された、曝露された(すなわち任意の量の治験薬投与を受けた)、完了した(すなわち、全ての治験期間を完了した被験者)、発生した中止の主な理由に従って中止した、被験者の数を含む、被験者の説明責任の詳細な要約が、各シーケンスおよび全ての被験者について作製される。
最終来院での被験者の内訳は、シーケンス群、本試験の終了時と治験薬の最終投与の日の内訳状態、最終来院の日付、中断の理由を含む一覧表で提示される。最初の治験薬投与の開始時または開始後に起こる本試験からの撤退は全て、治験総括報告書(CSR)の中に完全に記録される。
【0136】
プロトコルの逸脱
データベースのロックの前に、本プロトコルのコンプライアンスが、組み入れおよび除外基準、治療コンプライアンス、禁止された治療ならびに計画された評価のタイミングおよび有効性に関して調査される。プロトコルの逸脱はデータベースのロックの前に本試験チームによって確認され、欠損データおよびIPの中断を含むデータ審査報告書(Data Review Report)中に一覧にされ、大したことのない逸脱または主要な逸脱として分類される。
治験責任医師によって報告されるような組み入れおよび除外基準に対する個別の逸脱は一覧表にされている。
他の逸脱はCSRの本体中に一覧表にされ、および/または記載されている。
【0137】
解析母集団
解析されるべき母集団
任意の解析母集団から除外された被験者は、治療シーケンスおよび除外理由と共に列挙される。関連のある全ての情報がCSRに完全に記録される。
無作為化スケジュールに従って割り当てられた治療とは異なる治療を受けた被験者の事象においては、解析は無作為化治療に従うのではなく実際に受けた治療に従って行われる。
【0138】
薬物動態学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PKパラメータが有効である被験者は全て、薬物動態学母集団に含まれる。全てではなくいくつかの試験日においてPKプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0139】
薬力学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PDパラメータが有効である被験者は全て、薬物動態学母集団に含まれる、全てではなくいくつかの試験日においてGIRプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0140】
安全性の母集団
安全性の評価は、投与された治験薬の量にかかわらず、早くに中止した被験者も含めた、治験の投与を受けた被験者(曝露された母集団)をベースにした。
【0141】
人口統計学および基準値の特性
被験者の人口統計学特性、病歴および診断
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)は、体重および身長のデータより算出した:
BMI=体重[kg]・(身長[m])-2
【0142】
人口統計学および背景特徴に関する全ての変数を個別に列挙しまとめた。病歴および診断に関する組み入れ基準からの逸脱を個別に一覧にして記載した。
【0143】
ベースライン薬力学パラメータ
ベースライン血中グルコース濃度をシーケンスごとにまとめた。
【0144】
ベースライン安全性パラメータ
安全性変数について、変数として適用可能であっても、期間内または試験内の治験薬投与前に最期に組み込まれた値をベースライン値とした。そのベースラインの投与前値を投与前に再確認する場合、その再確認した値をベースラインとして認め、統計量に用いた。
【0145】
治験薬曝露の程度およびコンプライアンス
しかるべき場合には治験薬投与の詳細および補足情報を個別に列挙しまとめた。
【0146】
前/併用 治療/療法
前治療および併用療法(ある場合)を、世界保健機関−薬物文献リスト(Drug Reference List)(WHO−DRL)に従ってコード化し、個別に列挙した。
【0147】
薬力学変数の解析
薬力学変数の記載
インスリン投与に依存する、体重による被験者間の同等性を達成するために、解析用にGIRに関する全ての値を被験者の体重(kg)で割った。それにより以下のGIRは常に体重で標準化されたフルコース注入量を言及する。
【0148】
主要PD変数:
・体重で標準化されたグルコース注入率の時間曲線下面積
[GIR−AUC(0-24h)(mg・kg-1)]
副次PD変数:
・GIR−AUC(0-24h)の50%までの時間(h)[T50%−GIR−AUC(0-24h)(h)]
以下の追加PD変数を導出した:
・体重で標準化したグルコース注入率の、クランプの終了までの時間曲線下面積[GIR−AUC(0-end)(mg・kg-1)]
・体重で標準化したグルコース注入率の時間曲線下画分面積 [GIR−AUC(4-20h)、GIR−AUC(0-12h)、GIR−AUC(12-24h)(mg・kg-1)]
・体重で標準化した最大グルコース注入率 [GIRmax(mg・kg-1・min-1)]
・GIRmax までの時間 [GIR−tmax(h)]
有意義かつ信頼性のあるデータを提供するためにGIRmaxおよび対応するGIRmaxまでの時間の値は、各被験体について平滑化したGIR曲線より導出した。
【0149】
一次解析
GIR−AUC(0-24h)(mg・kg-1)についての相対的な生物有効性(活性)を推定するために、線形混合効果モデルを用いて未変換パラメータを解析した。
この混合モデルは、シーケンス、期間、製剤に関する固定項、ならびに製剤特異的(formulation specific)な被験者間および被験者内分散、および被験者付随製剤分散(subject−by−formulation variance)を伴うシーケンス内の被験者についてのランダム項を含む。その後製剤比率(T/R)についての点推定および90%信頼区間をフィエラーの定理(Fieller’s theorem)[Fieller、1954]に基づいて得た。
製剤比率についての信頼区間が[0.80−1.25]である場合、生物有効性(活性)が同等であると結論付けた。
その変数の分散についての仮定を確認した。
【0150】
二次解析/副次変数解析
個別の体重および平均体重で標準化したGIRプロファイルならびに時間に対する平均パーセンテージ累積プロファイルをプロットした。
PDパラメータは個別に列挙され、記述統計が作製される。
信頼限界を伴う製剤比率(T/R)を、1次解析について記載したような、対応する線形混合効果モデルを用いて、画分GIR−AUCs(mg・kg-1)および標準化グルコース最大注入速度[GIRmax(mg・kg-1・min-1)]について導いた。
50%−GIR−AUC(h)までの時間およびGIRmaxまでの時間[GIR−tmax(h)]は、ノンパラメトリックに解析した。
【0151】
クランプの実施
血中グルコース濃度の個別プロファイルをプロットした。
【0152】
安全性データの解析
まとめた全ての安全性データは、安全性母集団をベースにした。
安全性データの解析用の個別の治験薬投与期(on−treatment phase)は治験薬の最初の投与で始まり、EOS来院により終了した。
【0153】
有害事象(AE)
全てのAEは、MedDRA(使用時のバージョン)を用いてコード化される。
【0154】
定義
治験薬投与中のAE有害事象
全てのAEを以下のように分類した:
・試験治療下で発現した有害事象(TEAE):治療期間に初めて生じたかまたはそれ以前に発症していた場合治療中期間に悪化したAE;
・非試験治療下で発現した有害事象(NTEAE):治療期間に悪化することなく、治療期間外に生じたAE;
【0155】
製剤への割り当て
解析の目的のために、各TEAEをAEの発症および/または悪化前に与えられた最期の製剤に割り当てた。TEAEがある製剤において発生し、その後の製剤下で悪化した場合、両方の製剤に対するTEAEであると考える。
【0156】
欠測情報
情報の欠測または情報が矛盾していた場合、(例えば、部分的なデータまたは他の情報から)そのAEはTEAEではないと明らかに除外できる場合でない限り、AEはTEAEとしてカウントした。
AEの開始日が不完全または欠測していた場合、不完全な日が、AEが治験前に始まっていたと示している場合を除いて、最初の治験薬投与後に生じたと推定した。
【0157】
試験治療下で発現した有害事象
全てのAEを個別に列挙した。それらを、器官別大分類による概要を含め、製剤ごとにまとめた。
【0158】
死亡、重篤な有害事象および他の重要な有害事象
死亡、重篤なAEおよび他の重要なAEのような場合は全て個別に列挙し、総括報告書に詳細に記載した。
【0159】
治験の中止につながる有害事象
治療の中止につながるAEは、個別に列挙し、総括報告書に詳細に記載した。
【0160】
臨床検査値の評価
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。潜在的な臨床的に重要な異常(PCSA)および範囲外基準の定義は、パラメータにより報告した。
個別データを被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
正常範囲外の値の被験者およびPCSAのある被験者は、製剤ごとに、および治験評価の終了まで全期にわたり解析した。
ベースライン後PCSAのある被験者を列挙した。
【0161】
バイタルサイン
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。PCSAおよび範囲外基準の定義は、パラメータにより報告した。
PCSAのある被験者は、製剤ごとに、および治験評価の終了まで全期にわたりまとめた。ベースライン後PCSAのある被験者を列挙した。
生の値および導出されたパラメータは、製剤ごとに、および治験評価の終了まで全期にわたり解析した。個別データを、異常についてフラグを立てた上で、被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
【0162】
ECG
潜在的な臨床的に重要な異常(PCSA)および範囲外基準は、本治験の統計解析プランにおいて定義した。PCSAおよび範囲外基準の定義は、パラメータにより報告した。
SCRおよびEOSでのPCSAのある被験者を、全期にわたりまとめた。
個別データを、異常についてフラグを立てた上で、被験者ごとおよび来院ごとに、加えて補足情報を列挙した。
【0163】
薬物動態学データの解析
薬物動態学パラメータ
事実上の相対時刻をPKパラメータを導出するのに用いた。
【0164】
主要変数:
・INS−AUC(0-24h)(μU・h.mL-1)
副次PK変数:
・INS−AUC(0-24h)の50%までの時間(h)[T50%−INS−AUC(0-24h)(h)]
以下の追加PK変数を導出した:
・画分INS−AUCs[INS−AUC(4-20h)、INS−AUC(0-12h)、INS−AUC(12-24h)(μU・h・mL-1)]
・クランプの終了までのINS−AUC [INS−AUC(0-end)(μU・h・mL-1)]
・最大血清インスリン濃度 [INS−Cmax(μU・mL-1)]
・INS−Cmaxまでの時間[INS−Tmax(h)]
【0165】
統計解析
記述統計
濃度データの記述統計は、プロトコル時間によって表わした。
個別のおよび平均の血清インスリン濃度プロファイルをプロットした。
血清インスリン濃度を個別に列挙し、時点あたりの記述統計を作製した。
PKパラメータの記述統計を製剤ごとに作製した。
C−ペプチドのプロファイルをプロットし、記述的に特徴づけた。
【0166】
一次解析
INS−AUC(0-24h)についての相対的なバイオアベイラビリティーを推定するために、線形混合効果モデルを用いて、対数変換したパラメータを解析した。
この混合モデルは、シーケンス、期間、製剤に関する固定項、ならびに製剤特異的(formulation specific)な被験者間および被験者内分散、および被験者付随製剤分散(subject−by−formulation variance)を伴うシーケンス内の被験者についてのランダム項を含む。
INS−AUC(0-24h)について、演算推定および製剤間の差異についての90%信頼区間より得られた、製剤比率(T/R)に対する点推定および90%信頼区間は、その混合効果モデルのフレームワーク内であることを意味し、その後その比率スケールを真数変換により変換した。
製剤比率についての信頼区間が[0.80−1.25]である場合、バイオアベイラビリティが同等であると結論付けた。
【0167】
副次PKパラメータおよび追加PKパラメータの解析
50%−INS−AUC(h)までの時間および最大濃度までの時間[INS−Tmax(h)]をノンパラメトリックに解析した。
対数変換した画分INS−AUCおよびINS−AUC(0-end)(μU・h・mL-1)ならびに最大血清インスリングラルギン濃度[INS−Cmax(μU・mL-1)]を、1次解析について記載したような対応する線形混合効果モデルを用いて解析した。点推定および信頼区間を報告した。
【0168】
C−ペプチド
入手できる場合、C−ペプチドのプロファイルをプロットし、記述的に特徴づけた。
【0169】
PK/PD解析
しかるべき場合、PK/PD解析を探索的に実行した。
【0170】
実施例6:治験結果
被験者の内訳
総数35名(女性11名、男性24名)の被験者がスクリーニングされ、そのうち24名の健康で適格な被験者が登録され、無作為化され、少なくとも1度の治験薬投与を受けた。24名の無作為化された被験者のうち1名の被験者が最初の治療期間後に自身の要求に基づいて本治験から退いた。23名の被験者はプロトコルに従って本治験を完了し、薬力学(PD)および薬物動態学(PK)解析に組み入れられた。安全性評価には24名の治験被験者全てを含めた。
主なプロトコル逸脱はなかった。
【0171】
人口統計学特性
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)を、体重および身長のデータより算出した:
BMI=体重[kg]・(身長[m])-2
【0172】
【表5】
【0173】
クランプの実施
2つの治験群、ランタスU100およびランタスU300は、個別のグルコースクランプレベルを定義するのに役立つ個別の絶食時ベースライン血中グルコース濃度に関して類似していた。投薬後のクランプの所要時間は30時間であり、これは全ての治療期間において同じであった。
【0174】
主要エンドポイント
ランタスU100とランタスU300についてのバイオアベイラビリティ(曝露)の同等性は確立されなかった。ランタスU100とランタスU300についての生物有効性(活性)の同等性は確立されなかった。
【0175】
主要変数
0〜24時間までの血清インスリングラルギン濃度の時間曲線下面積(INS−AUC(0-24h))は、ランタスU100およびランタスU300について等価でなかった。曝露はU300を用いると約40%少なかった。0〜24時間までの時間曲線に対するGIRの曲線下面積(GIR−AUC(0-24h))は、ランタスU100およびランタスU300について等価ではなかった。活性はU300を用いると約40%少なかった。
【0176】
副次変数
INS−AUC(0-24h)(h)の50%までの時間は、ランタスU100とランタスU300で類似していた。GIR−AUC(0-24h)(h)の50%までの時間は、ランタスU300が0.545(h)(0.158−1.030)長く、この値は統計学的に重要である。
【0177】
安全性
重篤な有害事象(AE)は報告されなかった。治療(治験薬および参照薬)あたり5名の被験者が計14のTEAEを報告し、それらの全てが軽度〜中程度であり、後遺症もなく解消した。最も高頻度に報告された事象は頭痛(治験あたり4名の被験者)であり、その後吐き気、嘔吐および発熱(U100において1名の被験者)、および処置上の痛み(U300において1名の被験者)であった。注目すべきは、頭痛はクランプ試験の間の一般的な意見であり、高浸透性グルコース液剤の注入に関連しているということである。しかしながら、治験薬との関連も排除できなかった。注射部位の反応は報告されなかった。
【0178】
結論
インスリングラルギンU100およびインスリングラルギンU300は、バイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。インスリングラルギンU300後の曝露および活性は同量(0.4U/kg)のインスリングラルギンU100の投与後の曝露および活性と比較した場合約40%低かった。
【0179】
しかし、インスリングラルギンU300は、インスリングラルギンU100よりも、基礎インスリンに望ましいよりフラットなPK(曝露)およびPD(活性)プロファイルを示した。健常な被験者に対する同じ皮下投与後の、インスリングラルギンU100製剤とインスリングラルギンU300製剤との間の曝露および活性におけるこれらの驚くべきかつ予測できない差異は、以下の図に効果的に示されている。注目すべきは、同時に血中グルコースが一定になったということである。
【0180】
インスリングラルギンU300の投与は、安全性および忍容性に問題はなかった。
【0181】
実施例7:インスリングラルギンU300の3つの異なる皮下用量のグルコース動的活性および曝露を比較する試験のための、理論的根拠
健常な被験者における試験からの結果(実施例1〜6を参照のこと)は、ランタス(登録商標)U100とインスリングラルギンU300との間の曝露および効果における非同等性を示した。被験者はU100およびU300について、同じ用量のインスリングラルギン(0.4U/kg)を受けたが、U300からの同じ単位量の送達は、U100からの送達よりも曝露および効果が約40%低かった。しかしながら、インスリングラルギンU300は、ランタス(登録商標)U100よりも、基礎インスリンに望ましいよりフラットな薬力学プロファイルを示した。
【0182】
従って、以下の実施例において記載される新規の試験は、I型糖尿病患者による正常血糖クランプ設定におけるコンパレータとして、ランタス(登録商標)U100の標準用量に対して、インスリングラルギンU300の3つの異なる皮下用量のグルコース動的活性および曝露を比較する。
【0183】
この試験は、等効果なU300の用量を、クランプ技術によって提供される血中グルコース処理のパラメータによって評価されるような、0.4U/kgのランタス(登録商標)U100まで近づけることを目的とする。
【0184】
インスリングラルギン曝露は、皮下投与後の濃度−時間プロファイルおよび単位インスリンあたりのグルコース利用としての活性より評価される。
【0185】
本試験は、I型糖尿病の被験者の正常血糖クランプ設定において、ランタス(登録商標)U100の標準用量と比較して、異なる用量のインスリングラルギンU300の代謝性効果および曝露を評価するためにデザインされた。この試験は、4治療(R、T1、T2およびT3)、4治療期間(TP1−4)および4シーケンスを構成する。1度のスクリーニング来院(D−28〜D−3)、4度の治療来院(TP1〜TP4においてD1〜D2)、および1度の試験終了時来院(最終投与後D5〜D14)があり、安全性パラメータの最終評価を伴う。
【0186】
被験者は、ラテン方格デザインに従い、R、T1、T2およびT3の各治療に対して一旦、クロスオーバー、二重盲検および無作為化様式で曝露された。このデザインは、薬理科学効果およびランタス(登録商標)U100と比較して異なる用量のインスリングラルギンU300の曝露を評価するのに適切であると考えられる。
【0187】
本治験用に選択されたランタス(登録商標)U100の用量 0.4U/kgは、I型糖尿病患者に正常血糖を提供することが特徴であり、I型糖尿病患者による他のクランプ試験にて容易に研究された。
【0188】
インスリングラルギンU300について、3つの異なる用量0.4、0.6および0.9U/kgを試験する。この用量範囲は、0.4U/kgのランタス(登録商標)U100に等効果なおよその用量を補完する(intrapolating)ことができる。インスリングラルギンU300の用量0.4U/kgはすでに健常なボランティアで試験しており(実施例1〜6を参照のこと)、予め設定した観察期間終了の30時間内で0.4U/kgのランタス(登録商標)U100よりも活性が低いことが明らかとなっている。総グルコースディスポジションによって測定される0.4U/kgのインスリングラルギンU300の生理活性は、参照治験薬(0.4U/kg ランタス(登録商標)U100)よりも39.4%低かった。相応して、より高い用量のインスリングラルギンU300、例えば0.6U/kgのインスリングラルギンU300は、0.4U/kgのランタス(登録商標)U100と比較してほぼ等価なグルコース動的活性をもたらすことが期待された。さらに、比例的に用量を上げて行くことで、用量に比例する曝露および効果のプロファイルを調査することができる。
【0189】
I型糖尿病の患者における試験は、内因性インスリンの混乱効果(confounding impact)を避け、曝露および作用の持続性の評価をより可能にする。さらに、インスリングラルギンに特有のアッセイがないため、全ての内因性インスリンを読み取るアッセイを用いることが強いられる。従って、外因性インスリングラルギン以外の、加えられた全てのインスリン源が過剰なインスリン濃度を不当に引き起こす。
【0190】
本試験は、クロスオーバーデザインであり;実践的および倫理的理由により、多くても3種類のU300用量をランタス(登録商標)U100と比較する。長時間作用型インスリン製剤のグルコース動的活性の評価は、延長した作用持続時間に起因して、正常血糖クランプの設定を36時間まで伸ばすことを必要とする。
【0191】
活性な医薬品成分であるインスリングラルギンは、U100とU300の両方の製剤において同じである。本試験に用いられる用量は、日常用途の範囲内である。低血糖の全体的なリスクは完全に排除できないが、正常血糖クランプ技術によって制御される。
【0192】
薬力学
インスリングラルギンの薬力学活性は、I型糖尿病患者における正常血糖クランプ技術(これは、血中グルコース処理に対する投与された外因性インスリン製剤の効果を評価するために確立された標準手順である)によって評価される。
【0193】
正常血糖クランプ設定におけるグルコースディスポジションの評価に特有のパラメータは、体重で標準化された注入量(GIR)、処理された総グルコース、GIR−AUC0-36、およびGIR−AUC0-36の所定のパーセンテージまでの時間(例えば、GIR−A
UC0-36の50%までの時間)である。
【0194】
補助的なパラメータは、平滑化体重で標準化したGIRの最大値GIRmax、および、GIRmaxまでの時間GIR−Tmaxである。
【0195】
インスリングラルギンの作用持続時間は、投薬と正常血糖(クランプ)レベルを超える予め特定された逸脱との間の時間から導出される。
【0196】
グルコースのモニタリングは、皮下投与後、インスリングラルギンの長時間持続作用に起因して36時間実施される。
【0197】
薬物動態学
インスリングラルギンの徐放性質により、濃度プロファイルにおいて目立ったピークに欠けている。従って、INS−AUCの50%までの時間(T50% INS−AUC0-36)は、インスリングラルギンの曝露プロファイルの時間位置についての測定値として算出され、INS−CmaxおよびINS−Tmaxは、追加測定値として役に立つ。
【0198】
主要試験目的
本試験の主要な目的は、0.4U/kg ランタス(登録商標)U100に対する、3つの異なるインスリングラルギンU300用量の代謝性効果比率を評価することである。
【0199】
副次試験目的
本試験の副次的な目的は、0.4U/kgのランタス(登録商標)U100に対して、3つの異なるインスリングラルギンU300用量の曝露比率を評価すること、0.4U/kgのランタス(登録商標)U100に対して用量の異なるインスリングラルギンU300の作用持続時間を比較すること、インスリングラルギンU300の用量応答および用量曝露の関係を調査すること、ならびにI型糖尿病患者におけるインスリングラルギンU300の安全性および忍容性を評価することである。
【0200】
実施例8:試験デザイン、プロトコルの記載
I型糖尿病を患う男性および女性の被験者における、第1相、単一施設、二重盲検、無作為化、クロスオーバー(4−治療、4−治療期間および4−シーケンス;ラテン方格)、アクティブコントロール、治療期間中(5〜18日、好ましくは7日)の休薬期間を伴い、以下のインスリングラルギンの単回投与を受ける
・0.4U/kg ランタス(登録商標)U100(= 対照試験 R)
・0.4U/kg インスリングラルギン U300(= 試験 T1)
・0.6U/kg インスリングラルギン U300(= 試験 T2)
・0.9U/kg インスリングラルギン U300(= 試験 T3)
この4つの治療RおよびT1-3は、被験者に無作為に割り当てられた以下の4−シーケンス(1:1:1:1の比)
・R−T1−T2−T3
・T3−R−T1−T2
・T2−T3−R−T1
・T1−T2−T3−R
での、4つの治療期間(TP1〜TP4)における所定のクロスオーバー法である。
【0201】
試験参加の所要期間
・1人の被験者に対する総試験期間:約4〜11週間(最低−最長期間、休薬期間に依存する、スクリーニングを除く)
・1人の被験者に対する試験の各期間の所要期間:
- スクリーニング: 3〜28日間(D−28〜D−3)
- 治療期間 1−4: 2日間(1晩の滞在)
- 休薬: 5〜18日間(連続した投与の間に、優先的には7日間)
- 試験終了時来院: 治験薬投与後のD5〜D14の間の1日
【0202】
実施例9: 被験者の選択
計画された被験者の数:20名の評価可能な被験者を確保するために、少なくとも24名の被験者を登録するべきである。
【0203】
組み入れ基準
患者基本情報
I01 年齢18歳〜65歳の男性または女性被験者であって、米国糖尿病学会で定義されているように(American Diabetic Association. Report of the Expert Committee on the Diagnosis and Classification of diabete. Diabetes Care 1998;21:5−19)、1年より長く糖尿病を患っている
I02 総インスリン用量< 1.2U/kg/日
I03 体重は、男性であれば50.0kg〜95.0kg、女性であれば50.0kg〜85.0kgであり、ボディマス指数が18.0〜30.0kg/m2である
健康状態
I04 空腹時陰性血清C−ペプチド(<0.3nmol/L)
I05 グリコヘモグロビン(HbA1c)≦9.0%
I06 試験前に少なくとも2ヶ月間、安定したインスリン管理ができている(被験者の安全性および試験の科学的な完全性に関する)
I07 治験責任医師が、異常が臨床的に無関係であり、本試験の実施に干渉しないと判断した場合を除いて、病歴および身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに骨格筋系)において正常な所見である(被験者の安全性および試験の科学的な完全性に関する)
I08 背臥位での10分間の安静後の正常なバイタルサイン:95mMHg < 収縮期血圧 < 140mMHg; 45mMHg < 拡張期血圧 < 90mMHg;
40bpm < 心拍数 < 100 bpm
I09 背臥位での10分間の安静後の正常な標準12誘導ECG; 120ms <PQ <220ms、QRS < 120ms、QTc≦440ms(男性の場合)、≦ 450 ms(女性の場合)
I10 治験責任医師が、異常が糖尿病患者にとって臨床的に無関係であると判断した場合を除いて、正常範囲(または治験責任医療機関向けに定義されたスクリーニング閾値)内の臨床検査項目;しかし、血清クレアチニンは、臨床検査ノルムの上限を厳密に下回らねばならない;肝臓酵素(AST、ALT)およびビリルビン(被験者がギルバート症候群であるという文書を有している場合を除く)は、1.5 ULN を上回ってはならない
【0204】
女性被験者のみ
I11 妊娠する可能性のある(閉経後2年未満であるかまたは3ヶ月以上外科的に不妊ではない)女性は、スクリーニング時に血清β−HCG妊娠テストで陰性、およびTP1〜TP4の日1の尿β−HCG 妊娠テストで陰性でなければならず、また医薬品の臨床試験のための非臨床試験の実施時期についてのガイドライン(CPMP/ICH/286/95、改定)の注記に従って、失敗率が低い(すなわち1年あたり1%未満)と定義されているバースコントロールの非常に有効な方法を用いなければならない。全試験期間中、妊娠する可能性のある女性被験者は、2つの独立した避妊法(例えばダイアフラムおよび殺精子剤をコーティングしたコンドーム)を用いなければならない。コンドームと殺精子クリームの使用では十分に信頼できない。
閉経後2年未満であり、3ヶ月以上外科的に不妊ではない閉経後の女性は、ホルモン状態を測定する(FSH >30 IU/L、エストラジオール < 20 pg/mL)
【0205】
除外基準
病歴および臨床状態
E01 臨床的に関連のある心血管、肺、胃腸、肝臓、腎臓、代謝性(I型糖尿病とは区別して)、血液、神経性、精神、全身性(全体として身体に影響を及ぼす)、眼、婦人科の(女性の場合)、または感染性疾患の病歴または存在;急性感染性疾患または急性の病気の兆候全て
E02 過去6ヶ月間に、発作、昏睡または他人の助けを必要とするような重篤な低血糖の発症が1回より多くあった場合
E03 頻繁な重篤な頭痛および/または偏頭痛、反復性の吐き気および/または嘔吐(月に2度より多く)
E04 参加前の3ヶ月間の間の血液損失(≧300mL)
E05 症候性低血圧(血圧が低下すること全て)、または背臥位から立位に変えたとき、3分以内に20mMHg以上のSBPが低下することにより定義される無症候性起立性低血圧
E06 薬物アレルギーの存在もしくは病歴または治験責任医師の判断に従う臨床的に重要なアレルギー疾患
E07 試験期間中に、本治験プロトコルで許可されない薬物による治療を必要とする可能性があること
E08 過去3ヶ月以内に任意の治験薬による試験に参加したこと
E09 本試験前3ヶ月間における臨床的に重要な病気の症状、これは治験責任医師の意見に従い、本試験の目的に影響を及ぼし得るもの
E10 薬物またはアルコールの乱用の存在(アルコール摂取量> 40グラム/日
E11 1日あたり5本または同等量よりも多く喫煙しており、本試験期間中に喫煙を中断できない
E12 キサンチンベースの飲料の過剰摂取(>4杯/日)
E13 女性である場合、妊娠(β−HCG試験陽性として定義される)、授乳中
【0206】
干渉物質
E14 組み入れ前の14日間以内またはその薬物の排出半減期もしくは薬力学半減期の5倍以内におけるあらゆる薬剤(John’s Wortを含む);試験開始前の最後の1ヶ月内におけるインスリン以外のあらゆる薬剤で最も長いものおよび定期的に使用しているもののどちらも(甲状腺ホルモン、脂質低下薬および降圧剤を除き、女性である場合はホルモン不妊法または閉経期ホルモン補充療法を除く);最後の28日間内でのあらゆるワクチン接種
【0207】
一般的条件
E15 治験責任医師の判断において、本試験期間中、非従順でありそうかまたは言語の問題もしくは乏しい精神面での発達が理由で協力ができないであろう被験者
E16 適用し得る法令に従う、以前の研究の排除期間内である被験者
E17 緊急の場合に連絡不可能な被験者
E18 被験者が本プロトコルの実施に直接的に関与する治験責任医師もしくは任意の補助治験責任医師、研究助手、薬剤師、治験コーディネーター、または他のこれらのスタッフである
【0208】
生物学的状態
E19 以下のテストのどれに対しても陽性反応である:B型肝炎表面抗原(HBs Ag)、抗B型肝炎コア抗体(抗HBc Ab)(化合物が陽性の免疫活性を有する場合)、抗C型肝炎ウイルス(抗HCV2)抗体、抗ヒト免疫不全ウイルス1および2抗体(抗HIV1および抗HIV2 Ab)
E20 尿の薬物スクリーニングで陽性結果(アンフェタミン/メタミンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエート)
E21 アルコールテスト陽性
【0209】
本試験特有事項
E22 インスリングラルギンおよび賦形剤に対する過敏性がわかっている
E23 深部脚静脈血栓の病歴もしくは存在全てまたは第1度親族(両親、兄妹または子供)に深部脚静脈血栓が頻繁に発症している
【0210】
実施例10:治療
治験薬
・インスリングラルギン
異なる2つのインスリングラルギン製剤を用いる:
- 100U/mLのインスリングラルギンを含有する注射用のランタス(登録商標)U100液剤(市販の製品)
- 300U/mLのインスリングラルギンを含有する注射用のインスリングラルギン U300液剤
・用量:
- ランタス(登録商標)U100: 0.4U/kg(= 参照試験 R)
- インスリングラルギン U300: 0.4、0.6および0.9U/kg(=試験 T1−T3)
・容器: 3mLのガラスカートリッジ
・施用経路:臍から横に5cm右と左の皮下に
・状態: 絶食時
・治療の所要期間:各期間につき1日、単回投与
・開始: 治療期間1〜4(TP1−4)の日1(D1)の09:00
・参加した被験者の100%が追加治療を受ける
【0211】
【表6】
【0212】
投与
本試験は、計4回の治験薬投与を伴う単回投与試験である。被験者は、各被験者が参照治療(R)および各試験治療(T1-3)を一度受けるように、参照治療と試験治療の異なるシーケンスに無作為化される。
【0213】
注射は臍の左または右に行い、両部位は別々の注射に用いられる。5〜18日間の休薬期間は、連続的な投薬日期間から切り離し、好ましくは7日間(連続的な投薬期間の間の7日間)である。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、5日間は参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間は、投薬期間の間に避けられない場合に被験者に試験に関係のない義務を果たすための自由を可能にする3週間の休止期間を再び与える。
【0214】
IP投与は、絶食状態で投与され;被験者は、全クランプ期間を通して絶食を続ける。
【0215】
血中グルコース濃度は、前クランプ期間の投薬前1時間の間に任意のグルコース注入なしで5.5mmol/L(100mg/dL) ±20%の範囲内である。血中グルコースが任意のグルコース注入なしで少なくとも1時間安定している場合、IPが投与される。IP投与は、治療期間1〜4の日1の9:00より早い時間および14:00より遅い時間には行わない。血中グルコースが14:00前に安定しない場合投薬は行わない。その来院は終了し、被験者は後に新しい投薬来院1〜7日間を計画される。
【0216】
被験者ごとおよび投薬ごとに新しいカートリッジが用いられる。
【0217】
IP投与は、本試験にその他の点で関与していないかまたはCROでの本試験チームの一員ではない人物によってなされる。この人物はオープンなランダムリストに従って、IP投与を準備するためのランダムコードを得、それに従って被験者に投薬する。この準備および投薬は、第2の独立した人物によってチェックされる。投薬準備および治療シーケンスのそれぞれの文書は厳密に機密とし、任意の他の人物には開示されない。
【0218】
IP(インスリングラルギン)の用量の算出
各被験者に投与されるインスリングラルギンの量を算出するために、体重(kg)を小数点1ケタまで測定し、0.6U/kgのインスリングラルギン用量について以下の例に示したように、算出したインスリンの量を整数まで切り上げまたは切り捨てた:
・体重75.3kgの被験者は、45Uのインスリンを投与される(75.3×0.6 = 45.18 、切り捨てにより45とする);
・体重74.4kg被験者は、45Uのインスリンを投与される(74.4×0.6 = 44.64、切り上げにより45とする)。
【0219】
TP1 D1に記録された体重を全ての治療期間の治験薬用量の算出に用いる。この治験薬用量は、被験者の体重がTP1からその後のTPのうちの1つまでの間に2kg以下でしか変化しなかった場合、変更されない。被験者の体重がTP1からその後のTPのうちの1つまでの間に2kgより多く変化した場合には、治験薬用量はそれぞれの治療期間のD1での体重に基づいて再計算される。
【0220】
シリンジおよび針
少量の注射液剤を正確に投与するのに適切に取り付けられた針を備えるシリンジのみが用いられる(例えば、Becton Dickinson、Ref 305502、大きさ:1mL 27G 3/8 0.40x10)。このシリンジは、治験責任医師より提供される。
【0221】
他の製品
クランプ手順期間中に用いられた他の製品を表5に記載する。
【0222】
【表7】
【0223】
グルコース液剤、塩化ナトリウム液剤、ヘパリンおよびインスリングルリジンは、治験責任医師によって提供される。
【0224】
グルコース液剤: 20% グルコース液剤は、被験者の個別の血中グルコースを設定した目的レベルに維持するために人工膵(BiostatorTM)を用いて注入される。第2の注入ポンプ(人工膵(BiostatorTM)の一部)は、ラインの開存を維持するために0.9% 塩化ナトリウム溶液を輸液する。必要な20% グルコース液剤の量が人工膵(BiostatorTM)の注入容量を超える場合、第2のグルコース注入ポンプを連動させる。
【0225】
ヘパリン:低用量ヘパリン液剤(10.000単位ヘパリン/100mL生理食塩水)を、二腔カテーテルを介して注入する。このヘパリン液剤は、カテーテルの他の管腔において、人工膵(BiostatorTM)の血中グルコース測定のために用いられる血液と共に流され、系内の血液凝固を防止する目的がある。
【0226】
インスリングルリジン: 15U アピドラ(登録商標)[100U/mL]を49mLの生理食塩水溶液に溶かし、癒着を防止するために1mLの患者自身の血液を加えて0.3U/mLの濃度で作製し、これを正常血糖を達成する個別の速度で注入する。
【0227】
盲検法の記載
被験者は、無作為化、盲検およびクロスオーバーのデザインで異なる4つの治療(R、T1、T2およびT3)を受ける。盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験またはCROでの本試験チームの一員に別な方法では関与せず、どのような情報も誰にも開示せず、本試験の盲検状態を維持することを保証する。彼/彼女は、ランダムコードを入手し、それに従って被験者に投薬する。IPの調製および投薬後、ランダムコードへのアクセス権を持つが同じように守秘義務のある第二の独立した人物によりチェックされる。
【0228】
治療群への被験者の割り付け法
IPは、文書によりインフォームドコンセントを受けた被験者に対してのみ、本治験プロトコルに従って投与される。
【0229】
全ての組み入れ/除外基準を満たす被験者は、治療期間1の日1、治験薬投与の直前に割り当てられる:
・治療期間1のD1の朝における組み入れの経時順に従う増分被験者番号。9桁の被験者番号は、3つの部分(例えば、276 001 001、276 001 002、276 001 003、など)から構成されており、その最初の3桁(276)は国番号、真ん中の3桁は治験機関番号および最後の3桁はその機関内での被験者増分番号である。この被験者番号は全試験期間中変更せずに、被験者の同一性を確認する。
・予め計画した順番での治療番号の後、次の適格な被験者についての無作為化リストは、常に無作為化リストに従う次の治療番号を受ける
IP投与は、無作為化治療シーケンスに従ってなされる。
【0230】
本研究から退いた被験者は、すでに割り当てられている場合には彼らの被験者番号および治療番号を保持しておく。代替の被験者は、異なる同定番号(すなわち500+本研究を中止した被験者の番号)を有する。各被験者は本治験を中止した被験者と同じ治療シーケンスを受ける。
【0231】
スクリーニングで不合格であった被験者は、異なる番号を割り当てられる(例えば、901、902(インフォームドコンセントの署名後のスクリーニング期間中にAEが生じた場合のみ、CRFに記録するため))
注記:被験者の無作為化は、治験責任医師が本研究に関しての被験者の適格性を確認した後に行う。ベースラインパラメータは、投薬前直前に有効なパラメータである。
【0232】
包装およびラベリング
インスリングラルギン U300液剤は、3mLカートリッジの再編成されたボックスでサノフィ−アベンティスより提供される。
【0233】
それぞれの番号のIPは、医薬品製造品質管理基準および地域の法的規制に従ってサノフィ−アベンティスの責任下で包装され、CROに提供される。
【0234】
ラベル表示の内容は地域の法的規格および規制に従ってなされる。
【0235】
ランタス(登録商標)U100は市販品であり、CROにより注文される。
【0236】
貯蔵条件
全てのIPは、治験責任医師の責任下において適切に施錠された室内で保管され、権限を与えられた人物以外が近づいてはいけない。
【0237】
このIPは+2℃〜+8℃で保管し、光から保護し、凍結してはならない。
【0238】
本試験の間の無作為化コードへのアクセス
盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験および/またはCROでの本試験チームの一員に別な方法では関与せず、いかなる情報も誰にも開示せず、本試験の盲検状態を維持することを保証する。彼/彼女は、ランダムコードを入手し、それに従って被験者に投薬する。IPの調製および投薬後、ランダムコードへのアクセス権を持つが同じように守秘義務のある第二の独立した人物によりチェックされる。
【0239】
有害事象の場合、その治験薬の知識が被験者の治療のために重要である場合の状況を除いては、そのコードは解読されない。各被験者について、治療の名前を含む解読資料(code−breaking material)は、封緘されて配布される。それは、本臨床期間を通して治験機関内の安全な場所に置いておかれる。治験依頼者は、臨床試験の完了時に全ての解読資料(開封されているかまたは封をされている)を回収する。
【0240】
盲検状態が破られた場合、治験責任医師は、源データに開封の日付とコードを解読した理由を記録する。
【0241】
治験責任医師、治験施設薬剤師、またはIPの貯蔵および投与を行ったその他の人物は、本試験に用いられたIPが治験依頼者に指定されたように、かつ適切な法的規制に従って厳重に維持されることを保証する責任がある。
【0242】
全てのIPは、治験プロトコルに従ってなされ、配布され、再び戻されたIPの正確な記録が維持されることを保証するのが治験責任医師の責任である。
【0243】
併用治療
併用薬の使用は、除外基準No. E14に明示されているように、その基準下に述べられている薬物を除いて本試験期間中許可されず、治療期間1の日1の、被験者の組み入れ前に所定のタイムフレーム内で停止される(E14を参照のこと)。
【0244】
被験者の標準インスリン治療とクランプ測定との干渉を避けるために、被験者は基礎インスリンの使用を控え、以下に切り替えねばならない:
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0245】
短時間作用型インスリンの最終皮下注射は、治験薬投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0246】
被験者の安全性を危険にさらさないような症候性の有害事象(例えば、頭痛)については、長期間持続する重篤なまたは中程度の有害事象のために併用薬を残しておく。特に、アセトアミノフェン/パラセタモールの使用は、肝毒性のリスクの知見がある場合、または肝臓酵素の異常が生じればすぐに禁止される。
【0247】
しかしながら、いかなる理由であっても特定の治療が必要とされる場合、正確な記録を適切な記録形式で取らねばならず、その記録には薬剤の名称(国際一般名)、1日の服用量およびこのような使用の継続期間を含む。治験依頼者は、頭痛の治療を除き、電子メールまたはファックスによって48時間以内に通知されねばならない。
【0248】
潜在的なアレルギー反応の治療は、他で出版されているような推奨事項に従う(Samspon HA、Munoz−Furlong A、Campbell RL et al. Second symposium on the definition and management of anaphylaxis: summary report−Second National Institute of Allergy and infective Disease/Food Allergy and
Anaphylaxis Network symposium. Journal of Allergy and Clinical Immunology 2006;117(2):391−397)。アレルギー反応の重篤度に応じて、抗ヒスタミン剤、副腎皮質ホルモンおよびエピネフリンによるによる治療が考慮される。
【0249】
治療の説明責任およびコンプライアンス
・IP コンプライアンス:
- IPは、医師の直接的な指導の下で投与され、適切な記録は、IPの分配および投与に責任のある人物または彼/彼女の代理人によって仕上げられる;治療シーケンスまたは用量における全ての情報は開示されず、文書は本試験に関与する他の人物がアクセスできないよう施錠される
- IP摂取は、測定可能な薬物アッセイ結果により確認される
・IPの説明責任:
- IPの分配および投与に責任のある人物または彼/彼女の代理人は、戻されたパック中に残っているカートリッジを数え、その後治療記録用紙(Treatment Log Form)に記入する
- 治験責任医師は、症例記録用紙(Case Report Form)(CRF)の適切なページに、投薬の日時に関する情報を記録する
- その後本試験を管理するモニターチームが、(本試験の非盲検を防ぐために)データベースをロックした後にIPと適切な説明責任様式でそれらを比較することにより、CRFデータをチェックする。
【0250】
用いられたカートリッジは、データベースがロックされた後、本試験の終わりに治験依頼者によって完全に文書化された調整項目(reconciliation)が実施されるまで治験責任医師によって保管される。
【0251】
実施例11:治験薬の評価
本試験は、0.4U/kg ランタス(登録商標)U100に対する、異なるインスリングラルギンU300用量の代謝性効果および曝露比の評価、0.4U/kg ランタス(登録商標)U100に対する、異なる3つのインスリングラルギンU300用量の作用持続時間の比較、インスリングラルギンU300の用量応答性および用量と曝露の関係の説明、ならびにI型糖尿病の被験者における正常血糖クランプ設定時のインスリングラルギンU300の安全性および忍容性の評価のためにデザインされる。
【0252】
薬力学
正常血糖クランプ
インスリングラルギンの薬力学効果、主に総グルコース処理およびインスリン作用の持続時間は、正常血糖クランプ技術によって評価される。
【0253】
正常血糖クランプの間、動脈血化した静脈の血中グルコース濃度(これは、全ての組織の総グルコース利用への供給、および被験者の血中グルコース濃度をその目的レベル(クランプレベル)に維持するのに必要とされるグルコース注入率(GIR)を反映する)は、人工膵(Biostator)装置(連続グルコースモニタリングシステム、Life Sciences Instruments、Elkhart、IN、USA)を用いて連続的に測定され、記録される。
【0254】
グルコース要求量(GIR−AUC)は、外因性インスリン過剰により媒介される組織へのグルコース取り込み(グルコース処理またはグルコース低下活性)の尺度である。人工膵(Biostator)は、1分間隔で血中グルコースレベルを測定し、予め設定したアルゴリズムを用いて血中グルコース中の変化に応じてそのグルコース注入率を調節する。
【0255】
クランプ手順
被験者の標準インスリン治療とクランプ測定との干渉を避けるために、被験者は基礎インスリンの使用を控え、以下に切り替えねばならない:
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0256】
短時間作用型インスリンの最終皮下注射は、IP投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0257】
治療期間1〜4(TP1〜TP4)の間、被験者は少なくとも10時間、一晩絶食した後、D1の朝にクリニックに入る。
【0258】
D1の朝、前クランプ手順を開始し、被験者に人工膵(Biostator)を連結する。血中グルコース濃度を4.4−6.6mmol/L(80−120mg/dL)に調節し、速効型インスリンアナログ(例えばインスリングルリジン)の静脈内ボーラス投与方法、および必要に応じて引き続くインスリンの個別注入により、これらの限界内を維持する。
【0259】
その後、治験薬投与の60分前、投薬の1時間前からいかなるグルコース注入もなしに血中グルコースを5.5mmol/L(100mg/dL) ±20%(正常血糖クランプレベル)に調節する。このインスリングルリジン注入は治験薬の投与直前には中止する。
【0260】
血中グルコースがいかなるグルコース注入もなしに5.5mmol/L(100mg/dL) ±20%の範囲内で少なくとも1時間安定であればIPを投与する(TP1〜TP4のD1のT0、9:00頃)。被験者は、参照薬または治験薬(R、T1-3、表4を参照のこと)を無作為化によって割り当てられたとおりに投与される。注射は臍の左または右になされる。
【0261】
IP投与は、治療期間1〜4の日1の9:00より早い時間および14:00より遅い時間には行わない。血中グルコースが14:00前に安定しない場合投薬は行わない。その来院は終了し、被験者は後に新しい投薬来院1〜7日間を計画される。
【0262】
IP投与は、絶食状態下で投与され;被験者は全クランプ期間を通して絶食を続ける。
【0263】
この正常血糖クランプ血中グルコースレベルはクランプの終了までグルコース液剤の静脈内注入法により連続的に維持される。
【0264】
任意の基礎インスリン補充の目的は、食間の代替内因性インスリン分泌に追加するかまたは分泌を平らにすることである。内因性インスリン分泌のない被験者において、本試験への参加を求められる場合、外因性インスリンが、肝臓グルコース生成物を処理するのに必要とされるインスリンの量をまかなうべきである。
【0265】
完璧に釣り合うと、過剰インスリンを補うための外部グルコースは不必要である。その結果グルコース注入率は約ゼロとなる。一旦インスリン作用が終わると、血中グルコース濃度は上昇する。この上昇までの時間と血中グルコース濃度が所定の閾値を越える時間を人工膵(BiostatorTM)で読み取る。
【0266】
ランタス(登録商標)U100およびインスリングラルギン U300の選択された用量は平均基礎要求(average basal need)を超え、36時間までに相当な大きさのGIRに反映されるいくらかのグルコース要求の精製に向けられる。
【0267】
クランプのパフォーマンスを示す対応パラメータ(すなわちクランプのベースラインレベルで血中グルコースを維持するための精度)は、クランプ期間にわたる血中グルコースの変動性である。血中グルコースの変動性の尺度は、個々のクランプあたりの変動係数(CV%)である。
【0268】
血中グルコース中の低い変動係数は、クランプ設定におけるインスリン効果を正確に評価するために欠くことのできないものである。
【0269】
クランプ期間は治験薬注射の36時間後である予め定めておいたクランプエンドを超えない。
【0270】
被験者は全グルコースクランプ(前クランプおよびクランプ)期間中絶食を続けるが、水は自由にとることができる。
【0271】
血中グルコースが、グルコース注入の中止後30分後のクランプの終了前に11.1mmol/L(200mg/dL)を通過し、かつ治験責任医師が上記の11.1mmol/L(200mg/dL)を超える誤った血中グルコースレベルを導くような可能性のあるエラーが全て除外されていることを確認した場合、クランプの前IP投与時間に用いられるインスリングルリジンが、36時間前にその所見を拡張するために与えられる。この場合、治験依頼者はその情報を得なければならない。
【0272】
被験者は、血中グルコースが十分等血糖(isoglycemic)範囲内である場合、クランプ設定から離れる。
【0273】
参加者は、TP1〜TP4の開放日(すなわち日2)に彼らの前治験薬を再開する。
【0274】
IPの効果は約24〜36時間続き、このことが参加者を2日間施設にとどめる理由である。
【0275】
5〜18日間の休薬期間は、連続的な投薬日期間から切り離し、好ましくは7日間(連続的な投薬期間の間の7日間)である。休薬期間の長さは、参加者と治験責任医師の両方が彼らのニーズに合わせ個々に変える。経験により、5日間は参加者に1週間あたりのクランプを1度にできる回復のための最小期間を構成し、一方18日間は、投薬期間の間に避けられない場合に被験者に試験に関係のない義務を果たすための自由を可能にする3週間の休止期間を再び与える。
【0276】
スクリーニングおよびTP1のD1は28日を超えて切り離されないが、EOSは最終の投薬後D5より早くまたはD14より遅くには生じない。
【0277】
薬力学サンプリング時間
前クランプ(IP投与の前)およびクランプ期間(IP投与後36時間まで)の間、1分おきの動脈血中グルコース濃度の測定のために、動脈血化した静脈血を2mL/hの速度で連続的に採決する。
【0278】
人工膵(BiostatorTM)校正用の動脈血化静脈血サンプル(0.2mL)(これは技術的な要求量である)を人工膵(BiostatorTM)に連結した後、治験薬投与後36時間まで少なくとも30分間隔で採取する。
【0279】
薬力学サンプルの数
血中グルコースはクランプ手順の間連続的に測定される。さらに、被験者1人の1治療期間につき少なくとも74のサンプルをIP投与後人工膵(BiostatorTM)の校正用に採取する。計74×4×24サンプル、すなわち7104サンプルを採取する(以下の表を参照のこと)。
【0280】
【表8】
【0281】
薬力学取り扱い手順
【表9】
【0282】
薬力学パラメータ
36時間以内の体重で標準化したGIRの曲線下面積(GIR−AUC0-36)および36時間以内の総GIR−AUCの50%までの時間(T50%−GIR−AUC0-36)を算出する。
【0283】
血中グルコースコントロールの持続時間は、正常血糖において、投薬からクランプグルコースレベル(100mg/dL)を超える逸脱までの時間として採用される。予め定義された限界内での制御された血中グルコースの時間は、投薬から特定の閾値(例えば、110、130および150mg/dLの血中グルコースレベル)までが採用される。
【0284】
さらに、最大平滑化体重補正GIR(GIRmax)およびGIRmaxまでの時間GIR−Tmaxを評価する。
【0285】
さらなる補足パラメータは必要に応じて導出される。
【0286】
安全性
ベースライン人口統計学特性
このベースライン人口統計学特性は以下から構成される:
・年齢(歳)
・体重(kg)
・身長(cm)
・ボディマス指数(BMI)(kg/m2)
ベースラインおよび本試験中の安全性評価
・スクリーニング時身体検査: 心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、および筋骨格系ならびに関連する病歴および手術歴、糖尿病の病歴(糖尿病の診断、インスリン治療の開始、その後の合併症);本試験に関連する知見のみが文書に記録される
・過去および現在の喫煙状況
・投薬前および試験期間中の身体検査:心血管系、腹部および肺;本試験に関連する知見のみが文書に記録される
・体温(耳)
・バイタルサイン:背臥位で10分間の安静後測定された心拍数、呼吸数ならびに収縮期血圧および拡張期血圧、心拍数ならびに収縮期血圧および拡張期血圧は、立位で3分間の後にも測定する(人工膵(Biostator)に連結されている場合は予定外の測定は除く)
臨床試験(血液サンプルの場合は絶食状態で):
・血液学:赤血球数(RBC)、ヘマトクリット(Hct)、ヘモグロビン(Hb)、白血球百分率数(WBC)(好中球、好酸球、好塩基球、単球およびリンパ球)、血小板、INRおよびaPTT
・生化学:
−電解質: ナトリウム、カリウム、重炭酸塩、塩化物、カルシウム
−肝機能: AST、ALT、アルカリホスファターゼ、γ−グルタミルトランスフェラーゼ (γGT)、総ビリルビンおよび抱合型ビリルビン
−腎機能:クレアチニン、BUN
−代謝:グルコース、アルブミン、総タンパク質、総コレステロール、トリグリセリド、HbA1c(スクリーニング、D1 TP1およびEOS時)、LDH、アミラーゼ、リパーゼ、C−ペプチド(スクリーニングのみ)
−潜在的筋肉毒性:クレアチニンホスホキナーゼ(CPK)
−血清学:B型肝炎抗原(HBs Ag)、抗B型肝炎コア抗体(anti−HBc Ab)、抗C型肝炎抗体(anti−HCV2)、抗HIV1および抗HIV2抗体
・保管用血液サンプル: 5mLの血液サンプルを乾燥した赤い蓋のついたチューブに採取し、約1500gで10分間、4℃で遠心分離する;その後血清を3つの保管用チューブに移し、すぐに蓋をしめて−20℃で立てた状態で凍結させる。このサンプルは、予期しない安全性の問題が生じた場合、薬物投与前のベースライン値が、事前に評価されなかったパラメータ(例えば、血清学)用に確実に利用するために用いられる。このサンプルが用いられない場合は、治験依頼者の同意後に治験責任医師が破棄する
・検尿:タンパク質、グルコース、血液、ケトン体、pH
−定性検査:試験紙を用いて、定性検出のために新鮮な排尿試料に対してディップスティックを実施する;
−定量検査: 尿サンプル試験が、尿のディップスティックにより上記のパラメータのいずれかについて陽性である事象においては、グルコース、タンパク質、赤血球および白血球の数の定量測定が必要とされる(例えば、定量測定により任意の陽性ディップスティックパラメータを確認するため)。
・尿の薬物スクリーニング: アンフェタミン/メトアンフェタミン、バルビツレート、ベンゾジアゼピン、カンナビノイド、コカイン、オピエート
・アルコール酒気検査
・妊娠/ホルモン検査(女性の場合):
−スクリーニング時、血中β−HCG
−TP1〜TP4、日1には尿β−HCG
−閉経後2年以上経過の場合は、スクリーニング時にのみFSH/エストラジオール
・有害事象:被験者による自発的な報告または治験責任医師による観察
・ECG テレメトリー(シングルリード)
・12誘導ECG(自動)
・抗インスリン抗体
【0287】
臨床試験用の血液サンプルは絶食下状態で採取される。
【0288】
ECG 方法論
ECG テレメトリー
・ECG テレメトリーは、医師により連続的にモニタリングされる。全ての不整脈イベントがプリンターによって記録され、被験者のCRFに記載される。この文書はそのイベント、発生の時間および継続時間の診断を可能にし、治験責任医師またはその代理人によりサインされる。ECGテレメトリーは、このECGテレメトリー記録は、治験薬曝露を考慮した潜在的な再分析のために保管される。
【0289】
12誘導 ECG
・12誘導ECGは、心電図装置(MAC 5500TM)を使用して、背臥位で少なくとも10分経過後に記録される。電極は本試験を通して各ECG記録について同じ位置に配置される(リードの連結場所は消えないペンで印をつける)。
【0290】
・ECGは、(たとえあるとしても)PKのサンプリング前に常に記録される。PKサンプルはECG後できる限り早く(15分以内)に描かれる。
・各ECGは、同時12誘導の、10秒間の読み取りで構成される:
−シングル12誘導ECG(25mM/s、10mm/mV)は、日時、被験者のイニシャルおよび番号、治験責任医師の署名、ならびに各誘導につき少なくとも3つの波形(complex)を含む、HR、PR、QRS、QT、QTc 自動校正評価を印刷する。治験責任医師の意見および自動値はCRFに記録される。この印刷物は、施設レベルで保管される。
−ECG中央検査部(ECG central lab)による最終的な追加リーディングを可能にするデジタル記録:各デジタルファイルは、理論上時間(日付および時間DxxTxxHxx)、実際の日付および実際の時間(記録時間)、治験依頼者研究コード、被験者番号(すなわち3桁)ならびに施設および国番号により同定される(関連する場合)。
・このデジタル記録、データ記憶および送信(要求のある場合は)は、全ての適用すべき法的規制(すなわちFDA 21 CFR、第11部)に従う。
【0291】
バイタルサイン、ECG、および血液サンプルが治験薬投与および/または食事と同時に計画される場合、それらは治験薬投与および/または食事前になされる。PK、PD、または安全性のためのバイタルサイン、ECG、および血液サンプルの測定が同時であるときは常に、以下の順番が重んじられる:ECG、バイタルサイン、PD、PK、および安全性サンプル;PKサンプルの正確なタイミング(PKサンプルに対する時間窓許容についてのフローチャートを参照する)を重んじるために、他の測定は計画された時間の前になされる。この評価スケジュールは、本試験のデザインに適応される。
【0292】
注射部位の局所忍容性
注射部位における知見(例えば紅斑、浮腫、丘疹、硬化、小疱、疱疹)は、主に全体的刺激性評点(Global Irritation Score)に従って採点される。評定尺度に従って≧3のスコアである局所注射部位反応は、有害事象としてさらに書き加えられる。
【0293】
被験者は、注射部位における感覚を報告することを求められる。
【0294】
薬物動態学
インスリングラルギンの薬物動態学評価のために、36時間までのインスリン濃度曲線下面積(INS−AUC)であるINS−AUC0-36、およびINS−AUC0-36の50%までの時間が導出される。さらに、最大インスリン濃度INS−Cmax、およびCmax(INS−Tmax)までの時間が得られる。
【0295】
サンプリング時間
治験薬の注射後0H、1H、2H、4H、6H、8H、12H、16H、20H、24H、28H、32Hおよび36Hの時点において、インスリングラルギン濃度の測定のために血液を採血する。
【0296】
薬物動態学サンプルの数
【表10】
【0297】
薬物動態学取り扱い手順
IP投与およびサンプル採取の正確な時間はCRFに記録しなければならない。
【0298】
薬物動態学パラメータ
単回投与後、以下の薬物動態学パラメータをインスリングラルギン濃度についての非分画法を用いて算出する。これらのパラメータは以下を含むがこれらに限定されない。
【0299】
【表11】
【0300】
サンプリングされた血液量
【表12】
【0301】
本治験の盲検を保護するための測定
盲検を維持するために、第三者の非盲検人物をIPの調合および投与に関与させる。この人物は本試験および/もしくはCROでの本試験チームの一員に別な方法では関与しないかまたは治験依頼者ではない。彼/彼女はサノフィ−アベンティスよりランダムコードを入手し、そのランダムコードまたは他の全ての情報を他の人物に開示しない。安全上の理由で、治療の無作為化コードは重篤未知疑いのある有害事象(Suspected Unexpected Adverse Drug Reaction(SUSAR))全ての保健当局(Health Authority)への報告ならびに治験責任医師および/または治験依頼者いずれかの判断に従ってIPの使用と合理的に関連付けるために盲にされない。
【0302】
被験者の安全性
治験責任医師は安全性の問題の場合において全ての臨床的に関係のある決定を採用する責任のある主要人物である。
【0303】
判断が必要な場合、専門医の意見は迅速に把握されるべきである(例えば、急性腎不全、痙攣、吹き出物、血管性浮腫、心停止、心電図の変形など)。
【0304】
実施例12:試験手順
来院スケジュール
スクリーニング手順
スクリーニング手順は、参加への被験者の適格性を決定するために組み入れの3日前までに、28日間以内で実施される。被験者は、治験責任医師より本試験の目的および手順の情報を受ける。被験者は本試験に関する実行の前にインフォームドコンセントに署名する。有害事象の記録はその後開始する。
【0305】
スクリーニングの前に、被験者は10時間(必要である場合は、低血糖の対応策として少量の炭水化物を除いて)絶食(水以外)する。
【0306】
スクリーニング来院は以下の調査を含む:
1.人口統計学(年齢、性別、人種、過去および現在の喫煙状況、身長、体重、BMI)
2.身体検査(心血管系、胸部および肺、甲状腺、腹部、神経系、皮膚および粘膜、ならびに筋骨格系)ならびに病歴および手術歴、糖尿病の病歴(糖尿病の診断、インスリン治療の開始、その後の合併症);本試験に関連する知見のみが文書に記録される
3.関連のある先行治療および全ての併用治療、試験介入の直近2カ月前における平均的なインスリン療法
4.ECG(標準12誘導)、バイタルサイン測定(脈拍、背臥位で10分間安静後および立位で3分間の後の収縮期血圧および拡張期血圧)、および深部体温(耳)
5.血液学、HbA1c、C−ペプチド、臨床化学、血清学、検尿、尿の薬物スクリーニング、アルコール酒気検査、β−HCGおよびFSH/エストラジオール血液検査(女性のみ、妥当な場合)による臨床試験
結論的な最終検査の結果に対して、1週間以内に1度の再検査が許可される。
【0307】
全ての組み入れ基準を満たし、除外基準にかからない被験者は、組み入れ来院の資格が得られる。
【0308】
スクリーニングで不合格である場合は、そのスクリーニング試験の基本結果が源文書に記録される。
【0309】
組み入れ手順(治療期間1の日1)
本試験への登録の資格を得た被験者は、TP1のD1の朝、7:00頃に絶食状態でクリニックに入る。
【0310】
この組み入れ試験は、最初の投薬日(D1、TP1)に実施され、以下の調査を含む:
最新の病歴(AE)に対する身体検査、以前の薬/併用薬および耳の体温
体重、BMI(スクリーニング時で測定した身長)
ECG(標準12誘導)、バイタルサイン測定(背臥位で10分安静した後、立位で3分経過後の心拍数、呼吸数、収縮期血圧および拡張期血圧)
血液学、臨床化学、検尿、尿の薬物スクリーニング、アルコール酒気検査、β−HCG検尿(女性のみ、妥当な場合)による臨床試験。
【0311】
各被験者は、本試験への彼/彼女の組み入れの時間順に従って増加していく同定番号を与えられる。
【0312】
無作為化は、治験責任医師による被験者の適格性が確認された後、D1/TP1で行う。1人より多くの被験者が同時に無作為化される場合、その被験者らはD1/TP1の朝の組み入れの経時順に従って連続して無作為化される(すなわち、最も低い番号の被験者が次に利用可能な無作為化番号を与えられる)。
【0313】
D1/TP1の臨床試験の結果がベースライン値であり、陰性でなければならないβ−HCG検尿(スクリーニング来院時に採取されたサンプルに基づく)を除いて、確認試験であると考えられる。
【0314】
被験者が最終的に登録される場合、血液サンプルは、記録用および抗インスリン抗体の検出用に採血される(D1/TP1のみ)。
【0315】
来院のタイプ別記載
治療期間 1−4(D1〜D2)
被験者のクランプ測定による標準的なインスリン治療の干渉を防ぐために、被験者は、基礎インスリンの使用を控え、以下に切り替える
・長時間作用型インスリン製品(すなわち、ランタス(登録商標)(インスリングラルギン)、Levemir(登録商標)(デテミル)またはウルトラレンテインスリン)に対する場合、TP1〜TP4のD1における投薬の48時間前より中間型または短時間作用型のインスリン製品へ、
・中間型インスリン製品(すなわち、NPH−インスリン)に対する場合、TP1〜TP4のD1における投薬の24時間前より短時間作用型へ
【0316】
短時間作用型インスリンの最終皮下注射は、治験薬投与の9時間前までである。ポンプ療法の被験者は、日1の朝、各IP投与の少なくとも6時間前にインスリン注入を中断する(9:00にIP投与を開始するとすれば3:00頃に)。
【0317】
来院の際、被験者は以前の来院以降、彼らの体調に臨床的に重要な変化が起こっていないことを保証すること、プロトコルにおいて定義されている一般的および食事の制限を遵守すること、および必要な場合には彼らのインスリン治療を変更することを求められる。本試験基準に違反した場合、その被験者は本試験へのさらなる参加から排除される。違反の種類により、被験者は特定の期間のみ除外される場合があり、一度の試験または全試験の計画を立て直す。
【0318】
最終来院以降の被験者の健康状態および併用薬におけるいかなる変化も被験者の診療録(原本)およびCRFに記録される。
【0319】
治験薬投与(各TPのD1)直前の朝、体重、バイタルサイン、12誘導ECG、ECGモニタリングおよび深部体温を記録し、検尿ならびに尿薬物およびアルコールのスクリーニングを実施する。
【0320】
注射に必要とされるインスリングラルギンの量は、被験者の体重に従って算出される。
【0321】
貧血症に備えるために治療期間3のD1に血液学を分析する。陽性である場合、治療期間3と4の間の休薬間隔を可能な最大の18日間まで延長するかまたはTP4の開始を血液パラメータが正常になるまで延期する。追加の血液評価は治療期間4のD1になされる。
【0322】
被験者は、正常血糖クランプの終了まで絶食状態(水を除く)を維持する。
【0323】
その後、被験者は自動グルコース読み取り装置(人工膵(BiostatorTM))に連結される3本の静脈ラインによって前クランプ手順を介する準備をし、サンプリング期間の全継続期間中、半座位(semi−recumbent position)のままになる。
【0324】
7:30頃、左手の手背静脈または手首外側の静脈にカニューレを導入し、人工膵(BiostatorTM)に連結して血中グルコース濃度の測定のために動脈血化した静脈血を連続的に採取する。左手を約55℃の空気温度を提供する加熱されたボックス(「ホットボックス(Hot−Box)」)に入れ、動脈血化する。第2の静脈ラインは、左腕の肘前静脈でとり、インスリンおよび参照薬の血中グルコース測定のためのサンプルを採取するために用いた。第3の静脈ラインは反対側の前腕にカニューレを導入し、人工膵(BiostatorTM)のポンプで0.9%の生理食塩水および20%のグルコース液剤、または外部ポンプを用いてインスリングラルジンを注入する。
【0325】
血管カテーテルの挿入からD1の9:00頃に治験薬を投与する60分前まで、血中グルコースレベルは4.4〜6.6mmol/L(80−120mg/dL、前クランプ)内に維持される。血中グルコースレベルにより、さらなるインスリングルリジンの静脈内ボーラス注射を行い、その血中グルコースを目的範囲内に維持する。治験薬投与の1時間前には、クランプ終了まで静脈内ボーラス注射はなされない。
【0326】
血中グルコースを測定するために、さらなる血液サンプルを少なくとも30分間隔で採取し、グルコースオキシダーゼ法をベースにした臨床基準値と照らし合わせて確認する。必要な場合、人工膵(Biostator)をその臨床検査基準値法の結果に従って再校正する。
【0327】
インスリン注入量は個別に調節される。血中グルコースが目的レベルで維持されている間、インスリンとグルコース両方の注入量は、クランプの導入期中、最小にされる。インスリングルリジン液剤は高精度の注入ポンプ(Terumo Spritzenpumpe TE 311TM)により注入され、20%グルコース液剤は高精度の注入ポンプ(Terumo Infusionspumpe TE 171TM)によって適用される。
【0328】
クランプレベルは、治験薬投与の60分前に血中グルコースをクランプ期間の終わりまで約5.5mmol/L(100mg/dL)に維持するように調節される。この前クランプは延長され、IP投与は、導入期(前クランプ)中に目的のグルコースレベルを満たしていない場合には14:00まで延期される。目的のグルコースレベルが14:00までに確立されない場合には、その来院は終了し、被験者は1〜7日後に新しい投薬来院を計画される。
【0329】
インスリングルリジン注入は、治験薬投与の直前に中止される。PK用の最初のインスリンサンプルはその後すぐに採取される。9:00頃、無作為化計画に従い、参照治療(R、0.4U/kg ランタス(登録商標)U100)または試験治療(T1-3)のいずれかの治験薬が標準化皮下脂肪技術(standardized skin−fold technique)を用いて臍側部に投与される(表4)。
【0330】
クランプの間、12誘導ECGはIPの注射2時間後および12時間後、ならびにクランプ終了時に行われる。
【0331】
治験薬は、全試験期間の間、同じ人物によって投与されることが好ましい。注射の終了時を時間ゼロ(T0)と定義し、その後のクランプ期間およびPKサンプリングの開始時間を定義する。
【0332】
全てのクランプ観察期間は36時間続くため、予め定義されたクランプの終了時であるD2の21:00頃に終了する。その後、被験者は、血中グルコースが十分等血糖(isoglycemic)範囲である場合正常血糖クランプ設定から離れ、食事および日常のインスリン治療を受ける。
【0333】
血中グルコースが、グルコース注入の中止後30分間のクランプの期間に11.1mmol/L(200mg/dL)を通過し、かつ治験責任医師が上記の11.1mmol/L(200mg/dL)を超える誤った血中グルコースレベルを導くような可能性のあるエラーが全て除外されていることを確認した場合、薬物動態学の血液サンプリングのために、クランプ期間を36時間まで延長するため、クランプの前IP投与時間に用いられる速効型インスリンアナログ(例えば、インスリングルリジン)が与えられる。この場合、治験依頼者はその情報を得なければならない。その後、被験者は、血中グルコースが十分等血糖(isoglycemic)範囲内である場合、正常血糖クランプ設定から離れ、食事および彼らの通常のインスリン治療を受ける。
【0334】
注射部位の反応は治験薬の注射後15分はもちろん1時間後にも評価し、評価尺度に従って≧3のスコアが観察された場合はAEとして記録される。
【0335】
解放前に、自由な食事が提供され、通常のインスリン治療が再開される。バイタルサイン(心拍数;背臥位で10分安静にした後、および3分間立位の後に測定される収縮期血圧および拡張期血圧)が繰り返され、血中グルコースが測定される(この血中グルコースの測定値は80mg/dLを上回らねばならない)。被験者は、治験医師によってかれらの健常な状態が保障された後、TP1〜TP4のD2に解放される。
【0336】
試験終了時来院
被験者は、TP4の最終投薬後、D5とD14の間に試験終了時来院のために戻る。被験者は10時間絶食(水を除く)しておく。このEOSは以下の調査を含む:
病歴が更新されたことに伴う身体検査(体重、体温)
ECG、バイタルサイン測定
血液学、HbA1c、生化学、検尿、および女性の場合にはβ−HCG 血液検査による臨床試験
発生した全てのAEまたはTP4以降に摂取した併用薬
抗インスリン抗体測定のための血液サンプル
治験責任医師は、全ての有効な臨床試験結果に基づいて、被験者が安全に本試験から解放されることを保証する。
【0337】
試験の制限事項
被験者は、日常のインスリン治療を、用いているインスリンのタイプ(長時間作用型、NPH、中間作用型)に応じて−2〜−1日で中止する。その後血中グルコースレベルは通常の単時間作用型インスリンの複合皮下注射によって単独でコントロールされる。
【0338】
日常のインスリン治療は、TP1〜TP4のD2に解放された後、再開される。
【0339】
被験者は、本試験を通して、および試験前の2週間は、被験者の代謝性のコントロールまたはインスリン感度に関与するいかなる併用薬も摂ってはならない。
【0340】
アルコール飲料、グレープフルーツジュース、およびキサンチン誘導体を含有する刺激性飲料(茶、コーヒー、コカコーラ(登録商標)のような飲料、チョコレート)の摂取は、クランプの完了まで、各治験薬投与の24時間前から許可されない。
【0341】
オレンジジュースまたは同様の炭水化物は、クランプの間の低血糖に対する調整的手段として与えられるか、そうでなければ人工膵(BiostatorTM)に連結されている場合は静脈内グルコース注入によって適切に打ち消される。
【0342】
各治験薬投与前の2日間は、激しい運動は許されない。
【0343】
1日あたり5本以下の喫煙をする被験者は本試験に含まれ、TP1〜TP4のD1およびD2を除いて本試験期間中喫煙をしてもよい。
【0344】
スクリーニングの日、被験者は少なくとも10時間の一晩の絶食(必要な場合は低血糖に他する対策手段として少量の炭水化物を除く)後に施設を訪れる。
【0345】
TP1〜TP4のD1の朝、被験者は少なくとも10時間の一晩の絶食後にクリニックに入り、D2のクランプ期間の終了まで絶食を続ける。自由な食事は、そのクランプの終了後に提供される。
【0346】
液体の供給は、各36時間の期間につき少なくとも2500mLである。
【0347】
原データ(source data)の定義
CRFに記録される以下に列挙した全ての評価は、以下に関する適切に署名され確認された原文書により裏付けられる:
・被験者の身元確認
・病歴(アレルギー反応の場合)
・臨床検査、バイタルサイン、体重および身長、体温;
・臨床検査評価(laboratory assessments)、ECG
・薬物動態の時点(Pharmacokinetic time points)
・来院および評価の日付および時間
・有害事象
・IP 投薬
・先行薬/併用薬
・クランプ手順の開始/終了、クランプデータ
【0348】
実施例13: 統計的考察
被験者数の決定
本試験の主要な目的は、U100(R)の1用量およびU300の3つの異なる用量(T1〜T3)として与えられるインスリングラルギンについての比較代謝性効果を評価することである。
【0349】
試験PKD10086のデータに基づき、約0.375の値が、自然対数変換されたスケールでGIR−AUCend of clampのSDwithinであると予測される。
【0350】
サンプル数算出の目的のために、0.325と0.425の間の被験者内SDが用いられた。
【0351】
表11は、修正幾何平均のペアワイズ治療比率(pairwise treatment ratio)についての最大の不確実性(90%信頼区間の点より)は90%の確実性で得られ、16〜24の間の総被験者数Nに対して、log GIR−AUC0-36についての真の被験者内SD値は0.325〜0.425の間であると仮定することを示す。
【0352】
【表13】
【0353】
20人の被験者について、GIR−AUC0-36の真の被験者内SDが0.375程度である場合、治療比率は19.9%の最大不確実性(すなわち、90%CIが0.80であり1/0.80=1.25倍の観測比)、および90%の確実性を有すると推定される。
【0354】
20名の完了する被験者を確保するために24名の被験者が含まれる。
【0355】
被験者の説明
被験者の内訳(disposition)
関与した、無作為化された、曝露された(任意の量の治験薬を投与された)、完了した(すなわち全ての治験期間を完了した被験者)、中断についての主な理由に従って中断した被験者の数を含む、被験者の説明責任の詳細な要約が作製される。
【0356】
最終来院での被験者の内訳は、シーケンス群、本試験の終了時と治験薬の最終投与の日の内訳状態、最終来院の日付、中断の理由を含む一覧表で提示される。最初の治験薬投与の開始時または開始後に起こる本試験からの全ての撤退は、治験総括報告書(CSR)の中に完全に記録される。
【0357】
プロトコルの逸脱
本試験のデータをロックする前に、集団の定義および以下を含む他の試験基準について定義された基準に関して、臨床試験プロトコルの逸脱が調査される:
・組み入れおよび除外基準;
・治療のコンプライアンス;
・禁止された治療に関する、本臨床試験プロトコルのコンプライアンス;
・来院間隔および総治療所要期間に関する本臨床試験プロトコルのコンプライアンス;ならびに
・計画された活動および安全性評価が実施されたか否か、など。
【0358】
逸脱は、以下を含むがこれらに限定されない:
・無作為化後に(全ての変数の)いかなる評価も受けていない被験者;
・曝露されない被験者;
・(適切な場合)主要変数の全ての評価を受けていない被験者;
・組み入れ基準を満たしていなかったにもかかわらず本試験に介入した被験者;
・本試験の間に離脱基準が発生したが、離脱しなかった被験者;
・誤った治療または不正確な投薬を受けた被験者;
・禁止された併用薬を受けた被験者。
【0359】
主要な逸脱は列挙され、まとめられる。
【0360】
解析の母集団
任意の解析母集団(薬力学、薬物動態学および/または安全性)からの除外は全て、CSRに完全に記録される。
【0361】
任意の解析母集団から除外された被験者は、治療シーケンスおよび除外理由と共に列挙される。関連のある全ての情報がCSRに完全に記録される。解析母集団について、全体および治療ごとの被験者の頻度が作表される。
【0362】
無作為化スケジュールに従って割り当てられた治療とは異なる治療を受けた被験者の事象については、解析は無作為化治療に従うのではなく実際に受けた治療に従って行われる。
【0363】
薬力学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、PDパラメータが有効である被験者は全て、薬物動態学母集団に含まれる、両方ではなく1つの期間においてPDプロファイルが不十分な被験者については、十分なプロファイルのパラメータをその解析に含んだ。
【0364】
IPの投与後36時間の観察期間内にインスリングルリジンを受けた(安全性の理由で)被験者については、薬力学データはインスリングルリジンの投与時までのみを考慮に入れる。
【0365】
薬力学解析からの除外
薬力学解析からの全ての除外事項は、その理由と共に列挙される。除外事項は、データベースのロックおよび盲検前にデータのレビューに基づいて決定され記録される。
【0366】
安全性の母集団
あらゆる比較治験薬に曝露された全ての被験者は、投与された治験薬の量にかかわらず、安全性母集団に含まれる。
【0367】
薬物動態学の母集団
治験薬投与に関するいかなる主要な逸脱も伴わず、インスリンPKパラメータが有効である被験者は全て、薬物動態学母集団に含まれる。一度PKプロファイルが不十分であったが、それが全ての治療期間ではなかった被験者については、十分なプロファイルのパラメータがその解析に含まれる。
【0368】
インスリングラルギンの生物学的分析アッセイは、インスリングルリジンのような他のインスリン類に干渉される。従って、IP投与後36時間のクランプ観察期間内にインスリングルリジンを投与された(安全性の理由で)被験者のインスリングラルギンについての薬物動態学データは、評価から除外される。
【0369】
人口統計学およびベースラインの特性
被験者の人口統計学特性、病歴および診断
以下のデータを収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)は、体重および身長のデータより算出する:
BMI = 体重[kg]/(身長[m])2
【0370】
人口統計学および背景特徴に関する全ての変数を、安全性母集団に関して個別に列挙しまとめる。
【0371】
病歴および診断に関する組み入れ基準からの逸脱を列挙し、個別に記載する。
【0372】
ベースライン安全性パラメータ
安全性変数について、変数として適用可能であっても、期間内または試験内の治験薬投与前に最期に組み込まれた値をベースライン値とする。そのベースラインの投与前値は、投与前に再確認する場合、その再確認した値をベースラインとして認め、統計量に用いる。
【0373】
治験曝露の程度および遵守
しかるべき場合には治験薬投与の詳細および補足情報を個別に列挙しまとめた。
【0374】
インスリングラルギンの個別総用量を治療ごとにまとめる。
【0375】
前/併用 治療/療法
前治療および併用療法(ある場合)を、世界保健機関−薬物文献リスト(Drug Reference List)(WHO−DRL、データベースをロックする時点で使用される最新バージョン)に従ってコード化し、個別に列挙した
併用インスリン薬(皮下)は、分けて列挙される。
【0376】
本クランプ手順中の任意の時間に与えられるインスリン注入またはボーラスは列挙されるかまたは、個人ベースで時間に対してプロットされる。本クランプ手順の間の投薬後に与えられるインスリン注入またはボーラスは、個人ベースで列挙される。
【0377】
薬力学変数の解析
全ての薬力学解析は、この薬力学母集団のデータを包含する。αレベルの調節は、複合解析に対してはなされない。
【0378】
インスリングラルギンの薬力学について、血中グルコース濃度およびグルコース注入率(GIR)がこのクランプ手順の間連続的に記録される。
【0379】
統計解析は、試験治療(T1〜T3)を参照治療(R)と比較する。
【0380】
薬力学変数の記載
被験者間の同等性を達成するために、体重でインスリン投与量を調節し、解析用にGIRに関する全ての値を被験者の体重(kg)で割った。それにより以下において他に言及されない限りGIRは常に体重で標準化されたフルコース注入量を言及する。
【0381】
主要PD変数
以下のPD変数が主要変数であると考えられる。
【0382】
・体重で標準化されたグルコース注入率の時間曲線下面積
[GIR−AUC0-36(mg/kg)]
GIR−AUC0-36は、分刻みの時間尺度で段階的な定数関数について、矩形法則(rectangular rule)に従って算出される。
【0383】
副次PD変数:
以下のPD変数が導出され、副次変数として考慮される:
・GIR−AUC0-36の50%までの時間(h)[T50%−GIR−AUC0-36(h)]
・平滑化した体重で標準化した最大グルコース注入率[GIRmax(mg*min/kg)]
・投薬後、GIRmaxに到達する最初の時間[GIR−Tmax(h)]
・正常血糖の所要時間(クランプレベルを超える平滑化血中グルコースプロファイルの上昇までの時間)は、投薬から、105mg/dL以下での平滑化血中グルコース濃度曲線の最終値までの時間として算出される
・予め定めておいた限度にコントロールされる血中グルコースの持続時間は、投薬から、以下の濃度以下での平滑化血中グルコース濃度の最終値までの時間として定義される
- 110mg/dL
- 130mg/dL
- 150mg/dL
【0384】
平滑化
生の体重で標準化したGIRの最大値は、GIR修正においてノイズを受けやすい。従って、GIRmaxおよびGIRmaxまでの時間の導出は、生の体重で標準化したGIRデータに対して、LOESS(locally weighted regression in smoothing scatterplots:平滑化散布点における局所重みづけ回帰)平滑化技術に基づく。ランタス(登録商標)のもとに公知であるようなGIRプロファイルの予測されるモルホロジーにより、6%の平滑化因子が用いられる(SASR、ProC LOESS、因子 0.06)。
【0385】
血中グルコースレベルは、ノイズを非常に受けやすい。従って、正常血糖の持続時間および血中グルコースコントロールの継続時間は、生の血中グルコースレベルに対して、LOESS(locally weighted regression in smoothing scatterplots:平滑化散布点における局所重みづけ回帰)平滑化技術に基づく。モルホロジーにより、6%の平滑化因子が用いられる(SASR、PC
LOESS、因子 0.06)。
【0386】
不十分な平滑化の場合には、異なる平滑化因子が追加解析のために用いられる。
【0387】
追加PD変数
さらなるパラメータを以下のとおりに導出する:
・グルコース注入の終わりまでの時間(投薬後、ゼロを超えるGIRの最後の時間として)
追加PD変数は、結果の解釈のために必要であるとみなされる場合に導出される。
【0388】
一次PD解析
以下に記載される解析の前に、GIR−AUC0-36が対数変換(自然対数)される。
【0389】
対数変換したGIR−AUC0-36は、SAS PROC MIXEDを用いて、シーケンス、期間および治療についての固定項、ならびに、シーケンスブロック内の被験者についての治療の未構築Rマトリックス(i,i)分散および共分散を用いて、線形混合効果モデルにより解析される。
log(パラメータ)= シーケンス+期間+治療+誤差
【0390】
治療幾何平均の比(T1/R、T2/R、T3/R)についての90%信頼区間(CI)は、線形混合効果モデルフレームワーク内の治療手段間の差異についての推定値および90%CIをコンピュータにより計算し、真数変換により幾何平均の比に変換することにより得られる。同等性は、その比の90%CIが完全に参照間隔と同等な0.80〜1.25の範囲内である場合に結論付けられる。
【0391】
個別の比率(参照治療に対する試験治療)の一覧表は、対応する記述統計と共に提供される。
【0392】
二次解析/副次変数の解析
GIRプロファイルについての記述的な説明
個別の体重で標準化したGIR(mg*min/kg)は、生データ、平滑化データおよび累積生データについてプロットされる。
【0393】
時間に対しての、平均および中央値の体重で標準化したGIRプロファイルならびに中央値割合累積プロファイルは治療ごとにプロットされる。
【0394】
累積プロットは投薬からクランプの終了までの時間をカバーする。
【0395】
導出されたPDパラメータについての記述的説明
PDパラメータは個別に一覧表にされ、治療ごと尾に記述統計が作製される。
【0396】
副次PDパラメータの治療比
信頼限界を伴う製剤比率(T1/R、T2/R、T3/R)は、一次解析について上に記載したような、対応する線形混合効果モデルを用いて、標準化グルコース最大注入量[GIRmax(mg*min/kg)]について導出される。治療間の診査比較は、従来の生物同等性基準(90% 信頼限界 0.80〜1.25)に基づく。
【0397】
GIR−Tmax値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、試験治療および参照治療間のGIR−Tmaxにおける差異のヒストグラムが提供される。
【0398】
副次PDパラメータについての治療差異
T50%−GIR−AUC0-36(h)は、対になった治療比較について、Hodges−
Lehmann法をベースにノンパラメトリックに解析される。中央値におけるペアワイズ治療間差異(T1−R、T2−R、T3−R)についてのCIが導出される。T50%−GIR−AUC0-36値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、治療間(T1−R、T2−R、T3−R)のT50%−GIR−AUC0-36における差異のヒストグラムが提供される。
【0399】
GIR−Tmax値の分散は各治療についてヒストグラムプロットによって表わされる。さらに、試験治療および参照治療間のGIR−Tmaxにおける差異のヒストグラムが提供される。
【0400】
正常血糖および血中グルコースコントロールの持続時間は、ヒストグラムプロットによって表わされる。治療の比較は、ノンパラメトリックに行われる。
【0401】
クランプの実行
血中グルコース濃度の個別プロファイルをプロットした。
【0402】
クランプの所要時間は、投薬からクランプ終了までの間の時間(hr)としてクランプごとに得られる。
【0403】
クランプごとの血中グルコースの個別可変性は、クランプの個別開始時と個別終了時(またはクランプ中のインスリングルリジンの最初の投与)の間の血中グルコース値の変動係数(CV%)として導出される。クランプごとの個別平均血中グルコースレベルは、クランプの個別開始時と個別終了時(またはクランプ中のインスリングルリジンの最初の投与)の間の算術平均として導出される。
【0404】
パラメータは治療内で、個別に一覧表にされ記述的にまとめられる。
【0405】
安全性データの解析
安全性評価は個別の値(潜在的な臨床的に重要な異常)、記述統計(要約表、図)および必要な場合には統計解析(適切な推定値、信頼区間)のレビューに基づく。「潜在的な臨床的に重要な異常」(PCSA) 基準は、サノフィー・アベンティスの標準的基準に従って用いられる。基準は、本試験の統計解析プランに記録される。この安全性解析は、臨床試験からの安全性データの解析および報告に関するサノフィー・アベンティス基準に従って行われる。
【0406】
全ての安全性解析は、安全性母集団のデータを包含する。
【0407】
全ての安全性データについて、観察期間は3つの異なる分画に分けられる:
・前治療期間は、被験者がインフォームドコンセントを認めたときと、最初の比較治験薬投与との間の時間として定義される。
・治療中期間は、(最初の)治験薬投与からその後72時間までの時間として定義される。
・後治療期間は、治療中期間後から、次の期間の(最初の)治験薬投与かまたはフォローアップ期間の終わりいずれかまでの時間として定義される。
【0408】
有害事象(AE)
全ての有害事象は、MedDRA(データベースロック時の使用する際の最新バージョン)を用いてコード化される。
【0409】
以下の項目が、全ての有害事象について提供される:
・(被験者による)全ての有害事象の一覧
・有害事象に関連するコメントの一覧
【0410】
定義
安全性データに関して、観察期間は3つの異なる区分に分けられる:
・前治療期間は、被験者がインフォームドコンセントを認めたときと、最初の比較治験薬投与との間の時間として定義される。
・各期間あたりの治療中期間は、(最初の)治験薬投与からその後72時間までの時間として定義される。
・後治療期間は、治療中期間後から、次の期間の(最初の)治験薬投与かまたはフォローアップ期間の終わりいずれかまでの時間として定義される。
【0411】
試験治療下で発現した有害事象
全てのAEを以下のように分類した:
・試験治療下で発現した有害事象(TEAE)は、治療期間に生じた(悪化を含む)全てのAEである;
・非試験治療下で発現した有害事象(NTEAEs)は、TEAEとして分類されない全てのAEである:
- 前治療AE、治験薬の最初の投与前の前治療期間の間に発症した(または悪化した)AEと定義される
- 後治療AE、治療中期間には悪化せず、後治療期間の間に発症したAEと定義される。
【0412】
治療への割り当て
解析の目的のために、各TEAEをAEの発症(または悪化)に与えられた最期の治療に割り当てた。TEAEがある治療において発生し、その後の治療下で悪化した場合、両方の治療に対するTEAEであると考える。
【0413】
欠測情報
情報の欠測または情報が矛盾していた場合、(例えば、部分的なデータまたは他の情報から)そのAEはTEAEではないと明らかに除外できる場合でない限り、AEはTEAEとしてカウントされる。
【0414】
AEの開始データが不完全または欠測している場合、不完全なデータがAEが治療前に始まっていたと示している場合を除いて、最初の治験薬投与後に生じたと推定される。
【0415】
試験治療下で発現した有害事象
試験治療下で発現した有害事象は列挙され、治療ごとにまとめられる:
・TEAEの概要(少なくとも1つのTEAE、重篤なTEAE、中断につながるTEAE、死亡(ある場合)の被験者数および割合
・主要な器官別大分類および基本語(preferred term)による、全ての試験治療下で発現した有害事象の要約(少なくとも1つのTEAEを伴う被験者の数および割合)(「インテキストテーブル」)
- 事象の数を伴わない表(治験総括報告書の本体用)
- 事象の数を含めた表(治験総括報告書の付表用)
- 製剤(U100、U300)ごとの被験者の数および被験者全体の数を含めた表(治験総括報告書の付表用)
・治療、器官別大分類および基本語による、被験者が示す試験治療下で発現した有害事象の列挙
【0416】
死亡、重篤な有害事象および他の重要な有害事象
死亡、重篤なAEおよび他の重要なAEのような場合は全て個別に列挙し、総括報告書に詳細に記載される。
【0417】
治療の中止につながる有害事象
いかなる発生の場合においても、治療の中止につながる全ての有害事象について個別の被験者の一覧表が作製される。
【0418】
臨床検査値の評価
血液学および生化学データ
臨床安全性パラメータは、治療期間1のD1およびEOS時に測定される。スケジュールごとに、これらの安全性パラメータは治療中期間に評価される(TP3およびTP4での血液学を除く)。
【0419】
ベースラインとして用いられるべき値(血液学および生化学)は、最初の治療期間のD1前投薬で採取された値である。任意の計画されたベースライン試験が、任意の被験者について反復される場合、最終的に再チェックされた値がベースラインとして考慮される。ただしそれらは最初のIP投与の前になされた。
【0420】
以下の表および列挙が提供される:
・ベースラインからの生データおよび変更についての記述統計(クレアチニンについての変化割合(%)を含む)
・後ベースラインのPCSAを伴う被験者から個別データの特定の一覧が提供され、機能および測定の時間によりソートされる
・計画された血液学および生化学についての再確認された値を含む全ての個別データは、生物学的機能および測定の時間ごとに列挙される。ある場合には、計画されていなかった臨床試験からのデータがこの一覧に含まれる。これらの一覧表において、個別データは下限または上限の臨床試験限界値よりも低いかもしくは高い場合、および/または定義されている場合、PCSA基準の絶対限界に達する場合にはフラグ化される
・以下のうち少なくとも1つを経験した被験者についての肝機能データの一覧表:
- 本試験期間中にALT>3ULNを少なくとも1回発生し、かつ総ビリルビン>2ULNを少なくとも1回発生し、それらの少なくとも1つは最初の投与後である
- 治療中期間の定義にかかわらず、抱合型ビリルビン>35%総ビリルビン、かつ総ビリルビン>1.5ULNが最初の投与後に同じサンプルで提供される
・特に、薬物取り込み、病歴および手術歴、アルコール習慣、トリガーファクター、ALT値、関連する徴候および症状に伴う事象詳細な情報を含む、ALT≧2ULNの増加に関する一覧表が提供される
・範囲外の定義の一覧表が提供される。
【0421】
PCSAを伴う被験者の一覧表において、肝機能データ、CPK、および好酸球が対応するULNの多発性(multiple)として説明される。
【0422】
検尿データ
再検査値を含む全ての定性検尿結果(ディップスティック)が一覧にされる。
【0423】
バイタルサイン
血圧および心拍数
心拍数ならびに収縮期血圧および拡張期血圧(SBPおよびDBP)は、人工膵(Biostator)に連結されている場合を除き、背臥位での10分間安静後、および立位での3分間経過後に測定される。
【0424】
ベースラインとして用いられるべき値は、各治療期間のD1前投与評価値である。任意の計画されたベースライン試験が任意の被験者について繰り返される場合、最後の再検査値がベースラインとして考慮される。ただしそれらはIP投与前になされた。
【0425】
心拍数および血圧については、起立性差異が背臥位から立位への変化として計算される。
【0426】
全てのパラメータについて、全ての未計画値および再検査値を含む「治療中」解析を実施する。
【0427】
以下の表および一覧が提供される:
・ベースラインの正常な状態または異常な状態にかかわらず、PCSAを伴う被験者数の要約表がベースライン後PCSAの発生表として提供される
・心拍数および血圧(背臥位および立位)について、計画された前投与測定および定義されたベースラインを基にした測定のタイプ(位置)、各パラメータおよび時点についての生データおよびベースラインからの変化(背臥位のみ)が記述統計にまとめられる
・無計画値および再検査値を含む全ての個別データが一覧表にされる(背臥位、立位、起立性差異)。その一覧表において、定義されている場合はPCSA基準の限界に到達している場合、印をつけられる
・個別の後ベースラインPCSAのデータ一覧が提供される
・ある場合は、バイタルサイン評価に関するコメントも付表に載せる。
【0428】
体重、ボディマス指数、および体温
体重およびBMIのベースラインとして用いられるべき値は、TP1のD1に収集された値である。
【0429】
体温のベースラインとして用いられるべき値は、各TPのD1に収集された値である。
【0430】
個別データは、PCSA基準の限度に到達している場合にはその値に対する印(体重のみ)を含めて一覧表にされる。
【0431】
ECG
心拍数、PQ−、QRS−、およびQT−間隔ならびに自動読み取り機から収集されたQT(QTc)は、生パラメータ値およびベースラインからの変化として解析される。
【0432】
ベースラインとして用いられるべき値は各期間のD1投与前値である。いくつかの計画されたベースラインテストが任意の被験者について繰り返される場合、再検査値がベースラインとして考慮される。ただしそれらは期間の薬物投与前になされる。
【0433】
全てのパラメータについて、再検査値を含む、治療中期間になされた全てのベースライン後評価を用いて治療中解析が実施される。ベースライン後PCSAを伴う被験者の数は、ベースラインの正常または異常な状態にかかわらず治療群ごとに要約表で提供される。
【0434】
全てのパラメータおよびベースラインからの変化についての生データは、パラメータ、治療、および測定時間ごとに記述統計でまとめられる。
【0435】
再検査値を含む個別データは一覧表にされ、治療、被験者、来院および測定時間によりソートされる。これらの一覧表において、PCSA基準の限度に到達している値は印がつけられる。
【0436】
ベースライン後PCSAを伴う被験者からの個別データの一覧表が提供され、測定のタイプによってソートされ、被験者、期間および測定時間によってソートされる。
【0437】
さらに、延長したQTc(男性>450ms、女性>470ms)またはQTc>60msでのベースラインからの変化(男性および女性)を伴う被験者についての心臓のプロファイルの別々の一覧表、および最初の投薬後の定性評価において少なくとも1つの異常がある(すなわち異常なECG)被験者の一覧表もまた提供される。
【0438】
他の関連する安全性パラメータ
身体検査
ある場合には、身体検査に関するコメントの一覧表が提供される。
【0439】
注射部位の局所忍容性
治療による頻度分布が、注射部位の局所忍容性のレベルについて提供される。個別データが一覧表にされる。各基準および治療において、被験者は最も重篤な結果を用いてカウントされる。
【0440】
アレルギー反応
アレルギー反応の一覧表
全てのアレルギー反応症例が、詳細な補足情報を含めて有害事象として記録される。全ての症例は治験総括報告書に詳細に記載される。
個別の症例および全ての補足データが一覧表にされる。
【0441】
アレルギー病歴および家族歴
アレルギー病歴および家族歴は、潜在的なアレルギー反応の任意の発症を伴う被験者について記録される。アレルギー病歴およびアレルギー家族歴の全ての詳細が個人ベースで一覧表にされる。
【0442】
抗インスリン抗体
本試験期間中および試験後調査からの抗インスリン抗体結果について、被験者の数を伴う要約表が提供される。個別の被験者一覧表が提供される。
【0443】
薬物動態学データの解析
薬物動態学パラメータ
PKパラメータのリストは、上に示される。さらに、インスリンについてのT50%−AUC0-36が統計解析の項で導出される。
【0444】
統計解析
インスリングラルギンの薬物動態学パラメータは、各治療について、少なくとも算術平均および幾何平均、標準偏差(SD)、平均の標準誤差(SEM)、変動係数(CV%)、最小値、中央値および最大値を用いて一覧表にされまとめられる。
【0445】
全ての薬物動態学解析は、上記に定義したような対応する薬物動態学母集団のデータを包含する。αレベルの調整は、複数解析にはなされない。
【0446】
統計解析は、試験治療(T1〜T3)と参照治療(R)を比較する。
【0447】
治療比の解析
この解析は、インスリングラルギンのAUC0-36について実施される。以下に記載される全ての解析の前に、AUC0-36値が対数変換(自然対数)される。
【0448】
対数変換したパラメータは、SAS PROC MIXEDを用いて、シーケンス、期間および治療についての固定項、ならびに、シーケンスブロック内の被験者についての治療の未構築Rマトリックス(i,i)分散および共分散を用いて、線形混合効果モデルにより解析される。
log(パラメータ)= シーケンス+期間+治療+誤差。
【0449】
治療幾何平均の比(T1/R、T2/R、T3/R)についての推定値および90%信頼区間(CI)は、線形混合効果モデルフレームワーク内の治療手段間の差異についての推定値および90%CIをコンピュータにより計算し、真数変換により幾何平均の比に変換することにより得られる。生物同等性は、その比の90%CIが完全に参照間隔と同等な0.80〜1.25の範囲内である場合に結論付けられる。
【0450】
個別の治療比(T1/R、T2/R、T3/R)の一覧表は、対応する記述統計と共に提供される。
【0451】
インスリンについてのT50%−AUC0-36
インスリンについてのT50%−AUC0-36の分散は、各治療についてのヒストグラムプロットにて提示される。さらに、治療間(T1−R、T2−R、T3−R)のT50%−AUC0-36における差異のヒストグラムが提供される。
【0452】
T50%−AUC0-36(h)は、ノンパラメトリックに解析される。
【0453】
インスリングラルギンU300の用量−曝露関係
用量−曝露関係の記述的解析
インスリングラルギンU300の用量−曝露関係は、以下により図表を用いて記載される
・被験者あたりの総用量に対する、曝露の被験者あたりのプロット
・体重(kg)あたりの用量に対する、曝露の被験者あたりのプロット
・体重(kg)あたりの用量に対する、用量で正規化された曝露の被験者あたりのプロット(0.6U/kgでの用量正規化)
結果の解析が必要であるとみなされる場合、さらなる記述的解析が追加される。
【0454】
用量−曝露関係の統計解析
試験治療T1−T3について算出されたインスリングラルギンのAUCについて、用量−曝露関係は、Gough et al.(Gough K、Hutchison M、Keene O et al.Assessment of dose proportionality:report from the pharmaceutical industry.Drug Information Journal 1995;29:1039−1048)における推奨に従って、「推定値」の解釈と共に経験パワーモデル(empirical power model)(PK−パラメータ=a*用量b)を用いて評価される。
【0455】
この経験パワーモデルは、比例関係の確認と、薬物動態学および全ての離脱の臨床的重要性を評価することの両方に用いることのできる、非比例関係の程度の尺度を容易にかつ理解可能に提供する。しかしながら、用量比例関係研究の解析は、任意の非比例関係の薬物動態学および臨床的重要性が評価されることを可能にするために、有意な試験よりもむしろ推定を必要とする。
【0456】
このパワーモデルは、用量(U/kg体重)に対するランダム係数パワーモデルを用いた対数変換スケールで合致する:
log(パラメータ)=(log(α)+α[i])+(β+β[i])*log(用量)
ここで、log(α)およびβは、それぞれ母集団の切片および勾配であり、α[i]およびβ[i]は、i番目の被験者についてのαおよびβからのランダムな偏差である。
【0457】
βについての推定値および90%信頼区間は、共分散パラメータの制限付き最尤法(restricted maximum likelihood(REML))推定による、SASR/PROC MIXED手段での一般最小2乗推定(estimated generalized least squares)により得られる。βについての推定値および90%信頼区間はさらに、rを、β推定値および信頼限界の冪に累乗することによる、r倍の増加(r=1.5およびr=2.25[すなわち 高用量/低用量])に伴うPKパラメータの増加についての推定値および90%信頼区間を得るために用いられる。
【0458】
モデルの不適合(lack−of−fit)が明らかに存在する場合、(治療比の解析に用いられたような)混合効果モデルが解析に用いられる。ペアワイズ用量の増加に関連するパラメータの増加についての推定値および90%CIは、混合効果モデルのフレームワーク内の投薬間のペアワイズ差異についての仮定およびCIを最初に演算子、その後真数変換により比率に変換することにより得られる。
【0459】
PK/PD解析
適当な場合には、グラフィックディスプレイ(点図表プロット)がPK/PD関係の説明のために説明される。
【0460】
実施例14: 試験結果
被験者の内訳
I型糖尿病の被験者計24名が登録され、無作為化され、少なくとも1用量の治験薬を受けた。24名の無作為化された被験者のうち2名の被験者は自身の要求により本試験から離脱した。22名の被験者が本プロトコルに従って試験を完了し、薬力学(PD)および薬物動態学(PK)解析に含まれた。治療された24名全ての被験者が安全性評価に含まれた。
【0461】
主要なプロトコルの逸脱はなかった。
【0462】
人口統計学特性
以下のデータ(表12)を収集した:性別、スクリーニング時の年齢、身長、体重、および人種。被験者ごとのボディマス指数(BMI)を体重および身長のデータから算出した:BMI=体重[kg]・(身長[m])-2。
【0463】
【表14】
【0464】
クランプの実行
各被験者について4つの治療期間、R(LantusU100)、T1(0.4U/kg HOE901−U300)、T2(0.6U/kg HOE901−U300)およびT3(0.9U/kg HOE901−U300) において、インスリン薬物治療の前の個別のベースライン血中グルコース濃度は、100mg/dL のクランプレベルの定義と類似していた。投薬後、本クランプの観察期間の所要期間は36時間であり、全ての治療期間で同じであった。
【0465】
主要エンドポイント
RおよびTについてのバイオアベイラビリティ(曝露)および生物有効性(活性)における同等性は、立証されなかった。
【0466】
主要変数
血清インスリングラルギン濃度の0〜36時間までの時間曲線下面積(INS−AUC(0-36h))は、RおよびT1およびT2については等価ではなく、T3ではほぼ等価であった。曝露は、Rと比較して、T1では約37%未満、T2では約43%未満およびT3でも同様であると推定された。
【0467】
0〜36時間までの時間に対するGIRの曲線下面積(GIR−AUC(0-36h))は、RおよびT1およびT2については等価ではなく、T3ではほぼ等価であった。血中グルコースコントロールを保つために必要とされる外因性グルコース利用は、T1では約88%未満、T2では67%でありT3でもほぼ同じであると推定された。
【0468】
副次変数
RでのINS−AUC(0-36h)(h)の50%までの時間は約14時間であり、ゆえにT1、T2およびT3それぞれでの約16時間、16時間および19時間と比較して短かった。RでのGIR−AUC(0-36h)(h)の50%までの時間は約12時間であり、ゆえにT1、T2およびT3それぞれでの約17時間、18時間および20時間と比較して短かった。
【0469】
安全性
重篤な有害事象(AE)またはAEに起因する離脱は報告されなかった。Rにおいて2名の被験者、T1において2名、およびT3において3名が計8件のTEAEを報告し、それらの全ては軽度〜中程度であって後遺症なしに解決した。最も高頻度に報告された事象は頭痛であった。注目すべきは、頭痛はクランプ試験の間の一般的な意見であり、高浸透性グルコース液剤の注入に関連しているということである。しかしながら、治験薬との関連も排除できない。T1、T2およびT3では注射部位の反応は報告されなかったが、Rにおいて2名の被験者が注射部位にほとんど確認できないような紅斑が現れた。
【0470】
結論
同じ用量のRおよびT(U300)は、単回投与後のバイオアベイラビリティ(曝露)および生物有効性(活性)において等価ではない。T1(0.4U/kg)およびT2(0.6U/kg)後の曝露および活性は、R(0.4U/kg)の投与後の曝露および活性と比較して少なかった。RおよびT3は曝露および外因性グルコース利用に関して実質的に同等であった。
【0471】
しかしながら、T1、T2およびT3は、Rに比べて平均付近でより変動の少ない、いっそうフラットなPK(曝露)およびPD(活性)プロファイル、すなわち基礎インスリン供給に望まれるようなプロファイルを示す。このことは、異なるプロファイルによって名目上等価な総曝露および総グルコース利用を提供するRとT3を比較する場合に特に明らかである。
【0472】
I型糖尿病の被験者におけるR(LantusU100)およびT(HOE901−U300)の製剤間での曝露および活性におけるこれらの驚くべきかつ予想外の差異は、以下の図に効果的に示される。
【0473】
その上、T(HOE901−U300)の投与は安全性および忍容性の問題を伴わなかった。
【0474】
実施例15: I型糖尿病の患者において、異なる2つの皮下用量の(HOE901−U300)のグルコース動的活性および曝露をランタスU100と比較するための、試験の理論的根拠
健常な被験者およびI型糖尿病の被験者における試験結果(前出の実施例を参照のこと)により、ランタス(登録商標)U100とインスリングラルギン U300とでは曝露および有効性が同等ではないことが示された。被験者は、U100およびU300について同じ用量のインスリングラルギン(0.4U/kg)を投与されたが、U300からの同じ単位量のU300送達が引き起こした血中グルコースコントロールを維持するための曝露および外因性グルコース利用は、U100からの送達よりも少なかった。ランタスU100は、平均付近での著しい変動のない曝露および薬力学プロファイルを示すが、HOE901−U300は、基礎インスリン供給にとって望ましい曝露および薬力学プロファイルにおけるさらに少ない変動、それに加えより長い作用の持続時間を示した。
【0475】
定常状態条件下での薬物動態学および薬力学のプロファイルを評価するために、以下の実施例に記載される新しい試験は、I型糖尿病の患者における最終的な正常血糖クランプ設定とのコンパレータとして、ランタス(登録商標)U100の標準用量に対して、異なる2つの皮下用量のインスリングラルギンU300を比較する。本試験は、クランプ技術によってもたらされる血中グルコースコントロールおよび血中グルコース処理のパラメータにより評価されるように、0.4U/kgのランタス(登録商標)U100と等効果なU300の用量を推定することを目的とする。インスリングラルギン曝露は定常状態での反復皮下投与後の濃度−時間プロファイル、および定常状態での単位インスリンあたりのグルコース利用としての活性より評価される。
【0476】
本試験は、2つの並列グループでの2つのクロスオーバー治療(RおよびT1、ならびにRおよびT2)、2−治療期間(TP1、TP2)および2−シーケンスをそれぞれ構成する。1度のスクリーニング来院(D21〜D3)、院内期間(クランプ評価ごとに、D1〜D4朝、およびD8朝〜D10夕方)を伴う治療来院(TP1およびTP2のD1〜D10、夕方の投薬を伴う)、および安全性パラメータの最終評価を伴う1度の試験終了時来院(最終投薬後D7〜D10の間)がある。
【0477】
本試験のために選択されたランタスU100の0.4U/kg用量は、I型糖尿病患者の正常血糖血中グルコースコントロールを提供することを特徴とし、I型糖尿病患者での他のクランプ研究において、容易に調査されている。
【0478】
インスリングラルギンU300について試験される2つの異なる用量は、0.4および0.6 U/kgである。この用量範囲は、0.4U/kgのランタス(登録商標)U100に等効果なおよその用量を補完する(intrapolating)ことができる。インスリングラルギンU300の用量0.4U/kgはすでに健常なボランティアおよびI型糖尿病患者で試験しており(前出の実施例を参照のこと)、予め設定した観察期間終了の30時間および36時間内で、0.4U/kgのランタス(登録商標)U100よりも活性が低いことが明らかとなっている。0.4U/kgのインスリングラルギン U300での血中グルコースコントロールが必要とする総グルコースディスポジションは、参照治験薬(0.4U/kg ランタス(登録商標)U100)よりも低かった。相応して、より高い用量のインスリングラルギンU300、例えば0.6U/kgのインスリングラルギンU300は、より少ない総グルコースディスポジションでよりしっかりとした血中グルコースコントロールをもたらすことが期待された。さらに、比例的に用量を上げて行くことで、用量に比例する曝露および効果のプロファイルを調査することができる。
【0479】
I型糖尿病の患者における試験は、内因性インスリンの混乱効果(confounding impact)を避け、曝露および作用の持続性の評価をより可能にする。
【0480】
本試験は、多くても2種類のHOE901−U300 用量の以前の試験の結果に基づいたクロスオーバーデザインを採用しており、ランタス(登録商標)U100と比較する。長時間作用型インスリン製品のグルコース動的活性の評価は、24時間を超える正常血糖クランプ設定を必要とし、予め定義した注射間隔は、作用の延長された持続時間によった。
【0481】
活性な医薬品成分であるインスリングラルギンは、U100とU300の両方の製剤において同じである。本試験に用いられる用量は、日常用途の範囲内である。低血糖の全体的なリスクは完全に排除できないが、正常血糖クランプ技術によって制御される。
【0482】
薬力学
インスリングラルギンの薬力学活性は、I型糖尿病患者における正常血糖クランプ技術(これは、血中グルコース処理に対する投与された外因性インスリン製剤の効果を評価するために確立された標準手順である)によって評価される。
【0483】
正常血糖クランプ設定におけるグルコースディスポジションの評価に特有のパラメータは、体重で標準化されたグルコース注入率(GIR)、24時間および36時間内に処理された総グルコース、それぞれGIR−AUC0-24 およびGIR−AUC0-36、ならびにGIR−AUC0-24 およびGIR−AUC0-36の所定のパーセンテージまでの時間(例えば、GIR−AUC0-36の50%までの時間)である。
【0484】
補助的なパラメータは、平滑化体重で標準化したGIRの最大値GIRmax、および、GIRmaxまでの時間GIR−Tmaxである。
【0485】
インスリングラルギンの作用持続時間は、投薬と正常血糖(クランプ)レベルを超える予め特定された逸脱との間の時間から導出される。
【0486】
グルコースのモニタリングは、皮下投与後、インスリングラルギンの長時間持続作用に起因して36時間実施される。
【0487】
薬物動態学
インスリングラルギンの徐放性質により、濃度プロファイルにおいて目立ったピークに欠けている。従って、INS−AUCの50%までの時間(例えば、T50% INS−AUC0-36)は、インスリングラルギンの曝露プロファイルの時間位置についての測定値として算出され、INS−CmaxおよびINS−Tmaxは、追加測定値として役に立つ。
【0488】
主要試験目的
本試験の主要な目的は、定常状態において、0.4U/kg ランタス(登録商標)U100に対する、2つの異なるインスリングラルギンU300用量の血中グルコースコントロールおよび必要とされる外因性グルコース利用を評価することである。
【0489】
副次試験目的
本試験の副次的な目的は、定常状態において、0.4U/kgのランタス(登録商標)U100に対して、2つの異なるインスリングラルギンU300用量の曝露比率を評価すること、0.4U/kgのランタス(登録商標)U100に対して2つの異なるインスリングラルギンU300用量の作用持続時間を比較すること、インスリングラルギンU300の用量応答および用量曝露の関係を調査すること、ならびにI型糖尿病患者におけるインスリングラルギンU300の安全性および忍容性を評価することである。
【0490】
実施例16: より高い濃度での長時間作用型インスリンの酸性製剤の溶解特性の変化
溶解特性に関しては、より高い濃度のインスリングラルギン製剤の影響がインビトロ試験系を用いることによって調査される。そうするために、沈殿の研究が、インビボ条件を模擬するpH7.4のリン酸緩衝液を用いて実施される。
【0491】
沈殿したインスリンの上澄みは、インスリングラルギン含有量を測定するためにHPLC技術を用いて調査される。
【0492】
本試験の詳細な記載:
沈殿緩衝溶液の調製:
19.32mgのリン酸2水素ナトリウム1水和物(M:137.98g/mol)を1mLの水に溶解した。0.1Mの水酸化ナトリウムまたは0.1Mの塩酸を用いてpHを7.4に調整した。
【0493】
沈殿研究の実施:
1000U/mL までの濃度のインスリングラルギン薬剤溶液、ならびに同量のインスリングラルギンおよび緩衝液を含む溶液をプラスチックチューブに入れ、わずかに振とうした。インスリングラルギンの沈殿後、分散物を予め定義した時間、ゆっくりとした回転速度で遠心分離した。所定量の溶解培地を取り出し、新しい緩衝培地に移した。
【0494】
インスリン含有量の測定:
上清サンプル中のインスリングラルギンの含有量を、水に溶解したリン酸2水素ナトリウム緩衝液、塩化ナトリウム(NaCl)および異なる量のアセトニトリルを含む、2つの移動相系を用いた逆相HPLCによって、それぞれのインスリン参照標準に対して定量した。
【0495】
固定相として、オクタドデシルカラムを用い、検出波長は215nmであった。
【0496】
より高い濃度の溶液(例えば、U500およびU1000)からのインスリングラルギンの放出プロファイルはランタスU100と比較してよりフラットで延長した。
【0497】
実施例17: 沈殿物の顕微鏡分析
100U/mL、300U/mL、500U/mL、700U/mLおよび1000U/mLの濃度を有するインスリングラルギン製剤の沈殿物を顕微鏡で調べた。200μLのリン酸緩衝液(pH7.4)中で沈殿した前記製剤(60Uのインスリングラルギンの量と同じ)を、100倍の大きさで透過型光学顕微鏡(Olympus Model BX61)で調べ、写真を以下に示し、また最大直径も示した。これらの研究により、濃度の増加に伴って明らかにより大きい粒子がもたらされるという沈殿物特性の違いが明らかとなった。これらの結果は図8に示した。
【0498】
実施例18: イヌにおけるインスリングラルギンの血中グルコース低下効果
インスリングラルギンの血中グルコース低下効果を健常な正常血糖のビーグル犬で評価した。このイヌは、0.3 IU/kgの単回皮下注射を受けた。静脈血中グルコースを最初の注射前とその後の24時間まで測定した。
【0499】
動物は、元々ハーラン(Harlan)より得た、30匹の健常な正常血糖のオスのビーグル犬群より取り出した。これらのイヌを標準化した条件下で、犬小屋の中で管理した。試験開始前日、それらのイヌを無作為に試験ケージに分配した。それらを、水道水へのアクセスは自由にして、実験開始前および実験中、18時間絶食させた。本試験におけるイヌの体重は13〜27kgであった。各実験後、それらのイヌを少なくとも2週間回復させた。
【0500】
これらの動物をn=6のグループに無作為化した。0時点において、これらの動物を試験化合物の単回投与で処理した。インスリングラルギンを、0.3 IU/kgの用量で単回皮下注射として投与した。
【0501】
血液のサンプリングは、薬物投与(0h)前およびその後24時間まで、前腕静脈(橈側皮静脈)の穴を介して連続的に行った。血中グルコースを酵素処理で測定した(Gluco−quant(R) グルコース/HKキット Roche/Hitachi 912)。
【0502】
インスリングラルギンの異なる濃度(100および300単位/mL)の調剤薬の皮下注射後の血中グルコースに対する影響を健常な正常血糖のビーグル犬において試験した。インスリングラルギン濃度の増加に伴い、作用の平均時間は、それぞれ6.8h(U100)〜7.69h(U300)まで増加した。
【0503】
グラルギン濃度を100から300U/mLまで増加させることによって、血中グルコース減少の時間作用プロファイルを、イヌにおいてよりフラットで延長した活性に向けて変化させた。
【0504】
イヌにおける現在のデータは、ヒトにおけるデータと一致し、インスリングラルギンのより高い薬物濃度が、作用のプロファイルおよびより長い持続時間に積極的に関連していることを示している。
【0505】
図
以下の図は、健常な被験者に対する同じ皮下投与後の、血中グルコース(PD)が一定である同時点における、ランタスU100製剤およびランタスU300製剤(インスリングラルギンU100製剤およびインスリングラルギンU300製剤)間の曝露(PK)および活性(PD)における驚くべきかつ予想外の差を効果的に示している。
【図面の簡単な説明】
【0506】
【図1】グルコース注入率(GIR) ランタスU100
【図2】グルコース注入率(GIR) ランタス U300
【図3】血清インスリン濃度; ランタスU100(左)およびU300(右)
【図4】血中グルコース(1/2)
【図5】血中グルコース(2/2)
【図6−1】正常血糖クランプ技術を用いた、I型糖尿病の患者における、0.4U/kg ランタス(登録商標)U100(インスリングラルギンU100)と比較した、0.4、0.6および0.9U/kgのHOE−901−U300(インスリングラルギン U300)の無作為化、4−シーケンス、クロスオーバー、二重盲検、用量応答試験の結果。上パネル: インスリングラルギン濃度(mU/L)、中パネル:血中グルコース(BG、mg/dL)、下パネル:グルコース注入率(GIR、mg.kg-1.min-1)。これらの曲線は、全ての被験者の全てのデータポイントのLOWESS平滑化平均を示す(母集団平均);LOWESSは、ノイズで悪影響を及ぼされている時系列から、または2つの変数間の「ノイズの入った」関係の点図表プロットから、「平滑な」選択値群(set of values)を作製するデータ解析技術である。
【図6−2】図6−1の続きである。
【図7】グルコース注入率(GIR、mg.kg-1.min-1)。これらの曲線は、全ての被験者の全てのデータポイントのLOWESS平滑化平均を示す(母集団平均);LOWESSは、ノイズで悪影響を及ぼされている時系列から、または2つの変数間の「ノイズの入った」関係の点図表プロットから、「平滑な」選択値群(set of values)を作製するデータ解析技術である。 凡例: プロファイル1〜3(上から下に) 正常血糖クランプ技術を用いた、I型糖尿病患者における0.4、0.6および1.2U/kgのランタス(登録商標)U100(インスリングラルギンU100)の、無作為化、二重盲検、並行群間用量応答試験の結果 凡例:プロファイル4〜7 (上から下に) 正常血糖クランプ技術を用いた、I型糖尿病患者における、0.4U/kg ランタス(登録商標)U100(インスリングラルギンU100)と比較した、0.4、0.6および0.9U/kg HOE−901−U300(インスリングラルギン U300の無作為化、4−シーケンス、クロスオーバー、二重盲検、用量応答試験の結果
【図8】インスリングラルギンの沈殿物の光学顕微鏡写真 濃度増加に伴う処方: A: 100U/mL、B: 300U/mL、C: 500U/mL、D: 700U/mLおよびE: 1000U/mL、 倍率100×であり、最大直径を含む。 全ての沈殿物は、インスリングラルギンの60Uを用いて実施される
【図9】正常血糖のイヌにおけるインスリングラルギンU−100対U−300の時間作用プロファイル
【0507】
略語のリスト
℃ 摂氏度
ABE 平均生物学的同等性
AE 有害事象
ALT アラニンアミノトランスフェラーゼ
aPPT 活性化部分トロンボプラスチン時間
ARF 急性腎不全
AST アスパラギン酸アミノトランスフェラーゼ
β−HCG β−ヒト絨毛性ゴナドトロピン
bpm 1分あたりの拍数
cm センチメートル
CPK クレアチニンホスホキナーゼ
CRF 症例報告書
DRF 矛盾解決フォーム(Discrepancy Resolution Form)
ECG 心電図
EOS 試験終了時(来院)
GCP 医薬品の臨床試験の実施基準
GGT γ−グルタミルトランスフェラーゼ
Hb ヘモグロビン
HbA1c グリコシル化ヘモグロビン
HBs B型肝炎表面
Hct ヘマトクリット
HCV C型肝炎ウイルス
HIV ヒト免疫不全ウイルス
HR 心拍数
INN 国際一般名称
INR 国際標準化比 (プロトロンビン時間)
IP 治験薬
IRB/I 治験審査委員会/独立倫理委員会
Kg キログラム
LOQ 定量限界
PT プロトロンビン時間
QTc ECG機械によって自動的に補正されるQT間隔
QTcB Bazettの式によって補正されるQT間隔
QTcF Fridericiaの式によって補正されるQT間隔
QtcN 母集団アプローチによって補正されるQT間隔
QtcNi 個体の母集団アプローチによって補正されるQT間隔
RBC 赤血球数
SBP 収縮期血圧
SCR スクリーニング(来院)
UDS 尿の薬物スクリーニング
ULN 正常範囲の上限
WBC 白血球数
【特許請求の範囲】
【請求項1】
200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含むが、ただし製剤のインスリングラルギンの濃度は684U/mLではない水性医薬製剤。
【請求項2】
200U/mL〜650U/mLのインスリングラルギンを含む、請求項1に記載の水性製剤。
【請求項3】
700U/mL〜1000U/mLのインスリングラルギンを含む、請求項1に記載の水性製剤。
【請求項4】
270−330U/mLのインスリングラルギン[270−330IU ヒトインスリンと等モル]を含む、請求項2に記載の水性製剤。
【請求項5】
300U/mLのインスリングラルギン[300IUヒトインスリンと等モル]を含む、請求項4に記載の水性製剤。
【請求項6】
エキセンジン−4のアナログを含む、請求項1〜5のいずれか1項に記載の水性医薬製剤。
【請求項7】
エキセンジン−4のアナログが、リキシセナチド(lixisentatide)、エクセナチドおよびリラグルチドを含む群より選択される、請求項6に記載の水性製剤。
【請求項8】
インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドを含む、請求項7に記載の水性製剤。
【請求項9】
インスリングラルギン1Uあたり0.2μg〜1μgのリキシセナチドを含む、請求項8に記載の水性製剤。
【請求項10】
インスリングラルギン1Uあたり0.25μg〜0.7μgのリキシセナチドを含む、請求項9に記載の水性製剤。
【請求項11】
亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムを含む群より選択される1種またはそれ以上の賦形剤を含む請求項1〜10のいずれか1項に記載の水性製剤。
【請求項12】
90μg/mLの亜鉛、2.7mg/mlのm−クレゾールおよび20mg/mlのグリセリン85%を含む、請求項11に記載の水性製剤。
【請求項13】
90μg/mLの亜鉛、2.7mg/mlのm−クレゾール、20μg/mLのポリソルベート20および20mg/mlのグリセリン85%を含む、請求項11に記載の水性製剤。
【請求項14】
pHが3.4〜4.6である、請求項1〜13のいずれか1項に記載の水性製剤。
【請求項15】
pHが4である、請求項14に記載の水性製剤。
【請求項16】
pHが4.5である、請求項14に記載の水性製剤。
【請求項17】
患者のI型およびII型糖尿病を治療する方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項18】
請求項17に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項19】
請求項17に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項20】
患者のI型およびII型糖尿病の治療において、長時間作用型インスリン曝露の持続時間を延長する方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項21】
請求項20に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項22】
請求項20に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項23】
長時間作用型インスリンによる患者のI型およびII型糖尿病の治療において、低血糖の発生を減少させる方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、方法。
【請求項24】
請求項23に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項25】
請求項23に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項26】
患者のI型およびII型糖尿病の治療において、ピークレス長時間作用型基礎インスリンを与える方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項27】
請求項26に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項28】
請求項23に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項29】
I型糖尿病およびII型糖尿病の治療における、請求項1〜28のいずれか1項に記載の水性製剤の使用。
【請求項1】
200−1000U/mL[200−1000IU ヒトインスリンと等モル]のインスリングラルギンを含むが、ただし製剤のインスリングラルギンの濃度は684U/mLではない水性医薬製剤。
【請求項2】
200U/mL〜650U/mLのインスリングラルギンを含む、請求項1に記載の水性製剤。
【請求項3】
700U/mL〜1000U/mLのインスリングラルギンを含む、請求項1に記載の水性製剤。
【請求項4】
270−330U/mLのインスリングラルギン[270−330IU ヒトインスリンと等モル]を含む、請求項2に記載の水性製剤。
【請求項5】
300U/mLのインスリングラルギン[300IUヒトインスリンと等モル]を含む、請求項4に記載の水性製剤。
【請求項6】
エキセンジン−4のアナログを含む、請求項1〜5のいずれか1項に記載の水性医薬製剤。
【請求項7】
エキセンジン−4のアナログが、リキシセナチド(lixisentatide)、エクセナチドおよびリラグルチドを含む群より選択される、請求項6に記載の水性製剤。
【請求項8】
インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドを含む、請求項7に記載の水性製剤。
【請求項9】
インスリングラルギン1Uあたり0.2μg〜1μgのリキシセナチドを含む、請求項8に記載の水性製剤。
【請求項10】
インスリングラルギン1Uあたり0.25μg〜0.7μgのリキシセナチドを含む、請求項9に記載の水性製剤。
【請求項11】
亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムを含む群より選択される1種またはそれ以上の賦形剤を含む請求項1〜10のいずれか1項に記載の水性製剤。
【請求項12】
90μg/mLの亜鉛、2.7mg/mlのm−クレゾールおよび20mg/mlのグリセリン85%を含む、請求項11に記載の水性製剤。
【請求項13】
90μg/mLの亜鉛、2.7mg/mlのm−クレゾール、20μg/mLのポリソルベート20および20mg/mlのグリセリン85%を含む、請求項11に記載の水性製剤。
【請求項14】
pHが3.4〜4.6である、請求項1〜13のいずれか1項に記載の水性製剤。
【請求項15】
pHが4である、請求項14に記載の水性製剤。
【請求項16】
pHが4.5である、請求項14に記載の水性製剤。
【請求項17】
患者のI型およびII型糖尿病を治療する方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項18】
請求項17に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項19】
請求項17に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項20】
患者のI型およびII型糖尿病の治療において、長時間作用型インスリン曝露の持続時間を延長する方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項21】
請求項20に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項22】
請求項20に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項23】
長時間作用型インスリンによる患者のI型およびII型糖尿病の治療において、低血糖の発生を減少させる方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、方法。
【請求項24】
請求項23に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項25】
請求項23に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項26】
患者のI型およびII型糖尿病の治療において、ピークレス長時間作用型基礎インスリンを与える方法であって、300U/mLの濃度のインスリングラルギンを含む水性医薬組成物を該患者に投与することを含む、上記方法。
【請求項27】
請求項26に記載の方法であって、医薬組成物が、亜鉛、m−クレゾール、グリセリン、ポリソルベート20およびナトリウムからなる群より選択される賦形剤をさらに含む、上記方法。
【請求項28】
請求項23に記載の方法であって、医薬組成物が、インスリングラルギン1Uあたり0.1μg〜10μgのリキシセナチドをさらに含む、上記方法。
【請求項29】
I型糖尿病およびII型糖尿病の治療における、請求項1〜28のいずれか1項に記載の水性製剤の使用。
【図1】

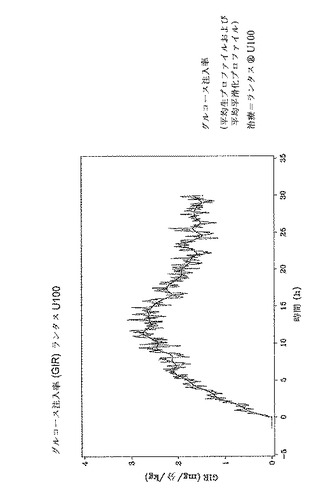
【図2】

【図3】
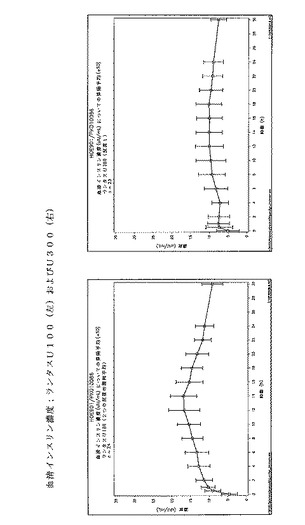
【図4】
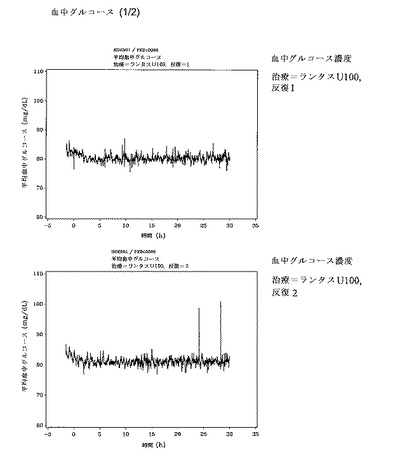
【図5】

【図6−1】
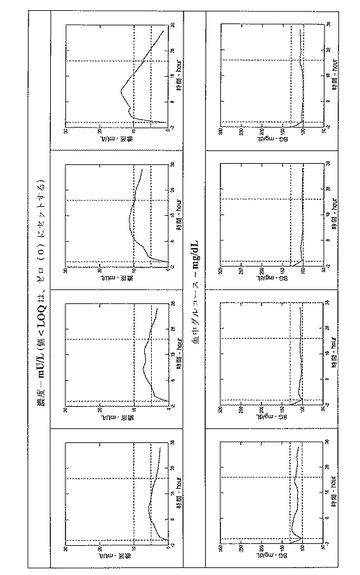
【図6−2】
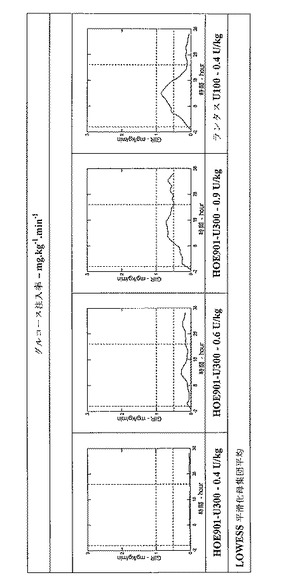
【図7】
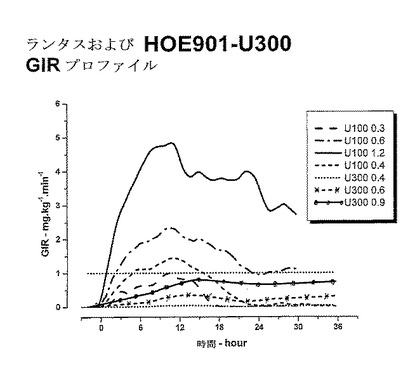
【図8】

【図9】
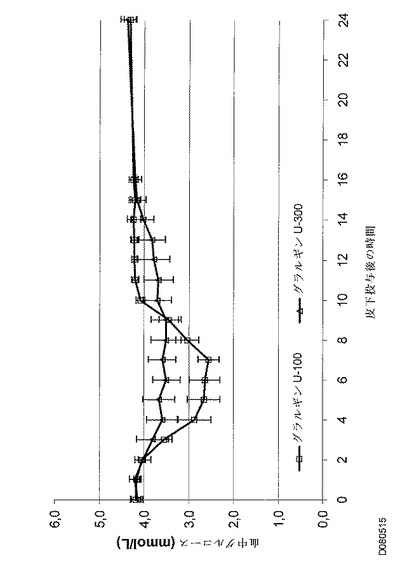
【図2】
【図3】
【図4】
【図5】
【図6−1】
【図6−2】
【図7】
【図8】
【図9】
【公開番号】特開2011−241213(P2011−241213A)
【公開日】平成23年12月1日(2011.12.1)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−110037(P2011−110037)
【出願日】平成23年5月17日(2011.5.17)
【出願人】(399050909)サノフィ (225)
【Fターム(参考)】
【公開日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願番号】特願2011−110037(P2011−110037)
【出願日】平成23年5月17日(2011.5.17)
【出願人】(399050909)サノフィ (225)
【Fターム(参考)】
[ Back to top ]