
核酸
本発明は,非特異的プロモーターと,少なくとも1つの異種T細胞エピトープを有するが制御性T細胞エピトープを有しないポリペプチドをコードする少なくとも1つの配列と,を含む核酸を提供する。ポリペプチドは,ヘテロ二量体の一方の鎖であり,異種T細胞エピトープは,ヘテロ二量体の他方の鎖と結合することができないようなヘテロ二量体の破断を引き起こす。核酸は,前記少なくとも1つの異種T細胞に対するT細胞反応を引き起こすのに利用しうる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は,核酸,及び核酸のワクチンとしての利用に関し,該核酸は,免疫反応が引き起こされうるT細胞エピトープをコードする。該ワクチンは,腫瘍の治療に利用することができる。
【背景技術】
【0002】
癌ワクチン及び慢性ウイルス感染の分野において,頻度以外の要因,例えば,腫瘍特異的T細胞の機能的活性やプライミングの経路,がワクチンの有効性を最大化する重要な決定要因であることが近年明らかになりつつある。高活性のCD8+ T細胞が優れた抗腫瘍活性を示すことを多くのグループが示している(Alexander−Miller, Immunologic Research, 2005;31: 13−24, Hodge et al, J Immunol 2005;174: 5994−6004, Valmori et al, J Immunol 2002;168: 4231−40, Zeh et al, J Immunol 1999;162: 989−94)。高活性のT細胞が患者の腫瘍退縮において重要な役割を果たすことが示唆されている。これは,高活性の抗原特異的腫瘍浸潤リンパ球(TIL)が腫瘍が劇的に退縮した患者において検出された一研究に例示されている(Khong & Rosenberg, J Immunol 2002;168: 951−6)。インビトロ刺激による自己腫瘍特異的T細胞の養子免疫伝達が成功するのは,おそらくインビトロ刺激が高活性のT細胞の選択を可能にするためであるということを示す証拠も出てきている(Vignard et al., J Immunol 2005 ;175: 4797−805, Dudley et al., J lmmunother 2001 ;24: 363−73, Morgan et al, J Immunol 2003;171: 3287−95, Rosenberg & Dudley, Proceedings of the National Academy of Sciences of the United States of America 2004;101 Suppl 2: 14639−45)。
【発明の概要】
【発明が解決しようとする課題】
【0003】
これまでに,多くのグループが,抗体をエピトープのキャリアとして用いて,所定のエピトープに対する細胞性免疫反応を引き起こそうと試みてきた。例えば,国際公開第96/19584号パンフレット(Bona et al.)は,T細胞エピトープが抗体の相補性決定領域(CDR)に挿入されるキメラ抗体を開示し,該キメラ抗体が細胞傷害性T細胞(CTL)反応を引き起こすのに適している旨主張している。しかしながら,該文献は,DNAが機能タンパク質をコードしなければならないことを説いている。よって,要約書には,「エピトープ及び親免疫グロブリンの機能性が保持される」旨記載されている。同様に,21ページ目には,所望のエピトープの挿入は,親免疫グロブリン分子の発現や機能にとって不可欠ではない親免疫グロブリン分子をコードする核酸の領域でなされるべきである旨記載されている。さらに,国際公開第96/19584号パンフレットに記載のすべての実施例は,無傷免疫グロブリンがT細胞エピトープの挿入後に生成されることを示している。
【0004】
米国特許第7,067,110号明細書は,ポリペプチド結合により抗原と融合する免疫グロブリン可変領域ドメインを欠く抗体の融合タンパク質を用いて抗原に対する免疫反応を引き起こす方法を開示している。該融合タンパク質は,Fcに結合する能力を保持する。
【0005】
欧州特許第0759944号明細書は,無傷免疫グロブリンタンパク質として分泌され,腫瘍に対するCTLエピトープを標的とすることでCTLの良好な標的とすることができるT細胞エピトープを抗体分子内に取り込む方法を開示している。
【0006】
国際公開第00/64488号パンフレットは,核酸がB細胞における発現を指向していれば,CDR(但し,その可変領域を除く。)に挿入される異種T細胞エピトープを有するキメラ抗体をコードする核酸によってCTL反応を引き起こすことができる旨開示している。B細胞はナイーブT細胞反応を刺激することができないので,国際公開第00/64488号パンフレットに記載のワクチンは,既存のT細胞反応を促進するのに有用であるだけであろう。
【0007】
国際公開第02/092126号パンフレットは,異種T細胞と高アフィニティCD64レセプターに結合するヒトFcの一部とを含むポリペプチドによってCTL反応を引き起こすことができる旨開示している。しかしながら,本願発明者(ら)は,T細胞エピトープを,例えば,不適切なCDRや抗体の可変領域に挿入することによる抗体配列の破断は,重鎖及び軽鎖の結合を防ぎ,機能性抗体が分泌されないことを示している。これらのミスフォールドした抗体をコードするDNAは,突然強力なT細胞反応を引き起こす。さらに,これは,CD64により媒介されない。なぜなら,ヒトIgG2は,マウス又はヒトCD64に結合せず,ヒトIgG1同様効率的に働くためである。
【課題を解決するための手段】
【0008】
本発明の一側面において,非特異的プロモーターと,少なくとも1つの異種T細胞エピトープを有するが制御性T細胞エピトープを有しないポリペプチドをコードする少なくとも1つの配列と,を含む核酸が提供される。
【0009】
該ポリペプチドは,好ましくは,同種キャリアである。例えば,ヒトにT細胞反応を起こすのに用いられる場合,ヒトタンパク質,又は異種タンパク質,又はすべてのT制御性エピトープを同定及び除去したヒト/異種キメラタンパク質でありうる。
【0010】
該ポリペプチドは,好ましくは,ヘテロ二量体の一方の鎖であり,異種T細胞エピトープは,ヘテロ二量体の他方の鎖と結合できないようなヘテロ二量体の鎖の破断を引き起こす。多くの分子は,一方の鎖がフォールディング及び分泌を他方の鎖に依存したヘテロ二量体である。異種T細胞エピトープの挿入により二次構造が破断されると,フォールディング及び分泌が抑制される。ある実施形態において,一方の鎖は,分泌され異種CTLエピトープを含み,他方の鎖は,異種ヘルパーエピトープを含むが,二次フォールディングの破断により,分泌されない。このため,核酸は,ヘテロ二量体の両方の鎖をコードしうる。この場合,一方の鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,他方の鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない。あるいは,各々のヘテロ二量体の鎖は,別の核酸分子上にコードされうる。
【0011】
ヘテロ二量体は,免疫グロブリン分子でありうる。免疫グロブリン分子の重鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,免疫グロブリン分子の軽鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない。
【0012】
本発明の第1の側面の核酸は,制御性T細胞(Treg)エピトープを含まないポリペプチドをコードする。該ポリペプチドは,T細胞エピトープの不活性キャリアとして作用し,免疫システムにより免疫反応を刺激するのに用いられうる分子又は分子の一部でありうる。なぜなら,これらの分子は,その名の通り,競合Tregエピトープを発現しないためである。適切な分子には,HLA分子,T細胞レセプター,TOLレセプター,TOLリガンド,サイトカインレセプター,ケモカイン,ケモカインレセプターが含まれる。分子は,抗体又はその一部であることが好ましい。
【0013】
理論に束縛されるものではないが,本発明は,少なくとも一部,T細胞エピトープを含むが制御性T細胞エピトープを含まないポリペプチドをコードする核酸の投与によって,特異的T細胞エピトープ(CTLエピトープなど)に対するT細胞反応を生成することができる,という概念に基づく。核酸は,抗原提示細胞(APC)に取り込まれ,リンパ節に移動し,直接提示されるか,又は,発現されて,分泌されその後他のAPCに取り込まれるポリペプチドを生成する,と考えられている。前者の核酸は,ヘルパーT細胞エピトープを刺激するのに適しており,後者は,CTL反応を刺激するのに適している。核酸によってコードされるポリペプチドは,理想的にはナチュラルT細胞エピトープを有しない。この点において適切なポリペプチドは,抗体などの免疫分子である。軽鎖がAPCにとどまり,重鎖が分泌されるように結合することができない抗体の重鎖と軽鎖が,本発明の実施に適しているが,本発明は,T細胞エピトープのキャリアとしての抗体の利用に限定されるものではない。
【0014】
サプレッサーT細胞集団がおよそ40年前に同定されたが,細胞を同定する特別な技術の欠如により,また,抑制の存在に関する科学的懐疑主義のために進展が阻まれた。しかしながら,Sakaguchi et alは,CD4+CD25+T細胞を枯渇したリンパ球の無胸腺マウスへの移入が,受容マウスにさまざまな自己免疫疾患の発症を引き起こすこと,また,CD4+CD25+T細胞による再構成は,該マウスの自己免疫反応を阻止することを証明することによって1995年にサプレッサー細胞への関心を復活させた(Sakaguchi et al J. Immunol 1995;155:1151−1164)。その後,制御性活性を有するさまざまなT細胞集団が,自己抗原(自己寛容制御)(Sakaguchi et al J Immunol 1995;155:1151−1164)及び異種抗原(Shevach, Immunity 2006;25: 195−201, Coleman et al, J. Cell Mol. Med. 2007;11: 1291−1325)に対する免疫反応(先天性免疫反応及び適応的免疫反応の両方)の抑制において重要な役割を果たすことが多数のマウス及びヒトにおける研究により示されてきている。癌のマウスモデルにおけるTreg細胞の枯渇は,内因性免疫介在性腫瘍拒絶(Shimizu, et al, J. Immunol. 1999; 163: 5211 −5218, Onizuka et al, Cancer Research 1999; 59: 3128−3133)及び抗原特異的抗腫瘍免疫(Tanaka, et al, J. Immunother. 2002;25:207−217)を改善することが証明されている。また,Treg細胞の枯渇は,ワクチン接種を含む腫瘍免疫療法(Tanaka, et al, J. Immunother. 2002;25:207−217, Dannull et al, J. Clin. Invest. 2005;115:3623−3633)及びCTLA−4遮断(Sutmuller et al, J. Exp. Med. 2001 ;194:823−832)を増強させる。さらに,多数のTreg細胞は,末梢血において増加し,さまざまな癌患者の腫瘍微小環境及び流入領域リンパ節を占めさせる(populate)。胃癌(Sasada et al, Cancer 2003;98:1089−1099, Ichihara et al, Clinical Cancer Research 2003;9:4404−4408)及び卵巣癌(Curiel et al, Nature Medicine 2004;10:942−949)の患者において,予後不良及び生存率の低下は,Treg細胞の高頻度と関連していた。Treg細胞はまた,腫瘍特異的CD8+(Liyanage et al, J. Immunology 2002;169:2756−2761, Piccirillo et al, J. Immunology 2001;167:1137−1140, Mempel et al, Immunity 206;25:129−141, Annacker et al, J. Immunology 2001;166:3008−3018, Woo et al, J. Immunology 2002;168:4272−4276)及びCD4+(Liyanage et al, J. Immunology 2002;169:2756−2761, Ichihara et al, Clinical Cancer Research 2003;9:4404− 4408, Nishikawa et al, Blood 2005;106:1008−1011)T細胞の増殖,サイトカイン生成(IFNγ, IL−2),及び細胞溶解活性を抑制することが証明されている。また,Treg細胞は,樹枝状細胞(Romagnani et al, Eur. J. Immunol. 2005;35:2452−2458),NK細胞(Ralainirina et al, J. Leukoc. Biol. 2007;81:144−153)及びB細胞(Lim et al, J. Immunology 2005;175:4180−4183)の機能を抑制しうる。まとめると,これらの研究は,腫瘍免疫病理学におけるTreg細胞の重要な役割を示唆し,Treg細胞の頻度と腫瘍の成長との間の密接な相関関係を示している。
【0015】
Treg細胞は,ナチュラルCD4+CD25+ T細胞及び誘発/適応Treg細胞のさまざまな集団に分けられる(Shevach, Immunity 2006;25: 195−201, Bluestone et al, Nat. Immunol. 2005;6:345−352)(表1)。マウス及びヒトのCD4+ T細胞の約5%〜10%は,ナチュラルTreg細胞である(Sakaguchi et al, Nat. Immunology 2005;6:345−352)。ナチュラルTreg細胞は,自己ペプチドとの強力なTCR相互作用によって胸腺に発生し(Picca et al, Current Opinion in Immunology 2005;17:131−136, Jordan et al, Nature Immunology 2001;2(4):301−306, Picca et al, Immunological Reviews 2006;212:74−85),一方,誘発Treg細胞は,非制御性T細胞から末梢に発生する。該胸腺外変換は,低用量抗原への連続暴露,全身性末梢抗原への暴露,TGFβへの暴露といった特殊な免疫学的条件を必要とする(Shevach, Immunity 2006;25: 195−201, Akbar et al, Nat. Rev. Immunol. 2007;7:231−237)。Treg細胞は,以下のメカニズム:i)細胞間接触依存性メカニズム,ii)IL−10やTGFβなどの免疫抑制サイトカインの分泌によって,iii)標的細胞パーフォリン・グランザイム経路(perforin−granzyme pathway)の直接殺傷,の1つ又は組み合わせによって抑制を媒介しうる(von Boehmer, Nature Immunology 2005;6(4):338−344)。
【0016】
これまで,ヒトTreg細胞の抗原特異性についてはほとんど知られていない。Wang et alは,癌患者におけるLAGE−1特異的CD4+CD25+GITR+機能性Treg細胞クローンの同定を報告した(Wang et al, Immunity 2004;20:107−118)。Vence et alは,転移性メラノーマ患者の末梢血における腫瘍抗原特異的CD4+ Treg細胞の存在を証明した。該Treg細胞は,TRP1,NY−ESO−1,gp100,及びスルビビンを含むさまざまな腫瘍抗原を識別した。また,Vence et alは,NY−ESO−1分子内におけるNY−ESO−1特異的Treg細胞エピトープの存在を最初に証明した。さらに,合成ペプチド又は腫瘍溶解物を含む樹枝状細胞を有するメラノーマ患者のワクチンは,Treg細胞の頻度増加を誘発し,腫瘍特異的CD8+ T細胞の増殖を伴うことが分かった。これは,該ワクチンが,未同定Treg細胞エピトープ及びCD8+ T細胞エピトープを含み,Treg細胞T細胞レセプター(TCR)を通じたリガンド特異的活性化によるTreg細胞のインビボでの増殖につながる可能性を示唆している。Treg細胞は,TCR識別/関与を通じた抗原特異的活性化を必要とするが,抗原非特異的バイスタンダー抑制を媒介することが広く認められている(Thorton & Shevach, J. Immunology 2000;164: 183190)。さらに,Li et alは,感染患者のHCV特異的Treg細胞を刺激するC型肝炎ウイルスコアタンパク質内の優性Tregエピトープの存在を示唆した(Li et al, Immunol. Cell Biol. 2007;85(3):197−204)。まとめると,これらの研究,及びHHDトランスジェニックマウスの抗内皮DNAコンストラクトC200Fcによる免疫は,有意なTie−21−196特異的抗腫瘍免疫反応を刺激しないとの最近の発見,並びに(400μgのPC61モノクローナル抗体の投与による)CD4+CD25+ Treg細胞の枯渇後,C200Fc DNA免疫前に(Middleton, PhD Thesis. University of Nottingham, November 2007)HHDマウスの脾細胞から細胞を分泌するTie−21−196特異的IFNγの頻度の増加は,DNAワクチン内のTie−21−196が未同定のTreg細胞エピトープ及びCD8+エピトープを含んでいることを示す。これは,ワクチンが自己抗原Tie−2に対するトレランスを破壊しないこと,及び細胞媒介性抗腫瘍免疫反応を抑制する豊富な抗原特異的増殖Treg細胞によってHHDマウスに抗腫瘍免疫を引き起こさないことを説明するものである。よって,エフェクターエピトープに対する免疫反応を管理して優勢Treg反応の刺激を阻止するTregエピトープを発現しない不活性免疫キャリアでTエフェクターエピトープを発現することには利点がある。
【0017】
有利には,本発明の核酸は,発現ポリヌクレオチドを分泌可能にする,リーダー配列などの配列をコードする配列を含む。これは,ポリヌクレオチドの抗原提示細胞(APC)への移入を可能にする。配列は,ポリヌクレオチドで自然に発現されるリーダー配列や,免疫グロブリンリーダー配列などの添加される異種リーダー配列でありうる。後者は,ポリヌクレオチドが膜結合分子をコードする場合に特に適している。
【0018】
本発明の別の側面によれば,非特異的プロモーターと,免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列を含む核酸が提供され,ここで,重鎖は,核酸が発現されると重鎖が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを含む。
【0019】
本発明の該側面の核酸は,免疫グロブリン分子の組み換え重鎖をコードする。該重鎖の構造は,当業者に知られており,通常,可変領域と定常領域とを含む。重鎖は,抗体由来のものでありうる。抗体は,モノクローナル又はポリクローナルであり,IgA,IgD,IgE,IgG,又はIgMでありうるが,IgGが好ましい。IgG抗体は,任意のIgGサブクラス,例えば,ヒトIgG1,IgG2,IgG3,IgG4,又は,マウスIgG1,IgG2a,IgG2b,IgG3,でありうる。IgG抗体は,ヒトIgG2 Fc結合ドメインを有するヒトIgG1抗体,又はIgG1 Fc結合ドメインを有するヒトIgG2抗体でありうる。重鎖は,ヒト抗体の定常領域,及び異種T細胞エピトープが挿入されたマウスモノクローナル抗体の可変又は超可変(CDR)領域を有しうる。超可変領域を除く可変領域もまた,ヒト抗体の可変領域に由来しうる。抗体に適用されると(すなわち,重鎖と軽鎖とを含む),該抗体は,ヒト化と呼ばれる。ヒト化抗体の作製方法は,当業者に周知である。該方法は,例えば,Winterによる米国特許第5,225,539号明細書に記載されている。マウス超可変領域を除く重鎖の可変領域もまた,マウスモノクローナル抗体に由来しうる。この場合,全可変領域は,マウスモノクローナル抗体に由来し,抗体に適用されると,該抗体は,キメラ化と呼ばれる。キメラ化抗体の作製方法は,当業者に周知である。該方法には,例えば,Boss(Celltech)及びCabilly(Genentech)による米国特許明細書に記載のものが含まれる。それぞれ,米国特許第4,816,397号明細書及び第4,816,567号明細書を参照のこと。
【0020】
ある実施形態において,本発明の核酸は,免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列をさらに含む。あるいは,免疫グロブリン分子の軽鎖をコードする別の核酸が提供されうる。軽鎖は,少なくとも1つの異種T細胞エピトープを有しうる。T細胞エピトープは,核酸が発現されるても軽鎖が未変性コンフォメーションをとることができないようなものでありうる。軽鎖は,軽鎖に関して本明細書に記載した特徴のいずれかを有しうる。したがって,本発明はまた,免疫グロブリン分子の組み換え軽鎖をコードする核酸を提供し,ここで,軽鎖は,核酸が発現されるても軽鎖が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを有する。核酸は,非特異的プロモーターを含みうる。該核酸は,抗体などの免疫グロブリン分子をコードする。
【0021】
このように,本発明の別の側面によれば,非特異的プロモーターと,組み換え免疫グロブリン分子をコードする少なくとも1つの配列とを含む核酸が提供され,ここで,免疫グロブリン分子は,核酸が発現されても免疫グロブリン分子が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを有する。組み換え免疫グロブリン分子並びに上記の重鎖及び軽鎖は,制御性T細胞エピトープを有しないことが好ましい。
【0022】
本発明はまた,以下を提供する:
・本発明の核酸とアジュバントとを含むワクチン;
・本発明の核酸と,薬学的に許容されるキャリア,賦形剤,又は希釈剤とを含む医薬組成物;
・医薬用の本発明の核酸;
・少なくとも1つのT細胞エピトープに対する免疫反応を刺激するための薬剤の製造における本発明の前記核酸の利用;
・少なくとも1つのT細胞エピトープに対する免疫反応を刺激するための本発明の核酸;及び
・T細胞エピトープに対する免疫反応を刺激するための方法であって,該免疫反応を必要とする被験者に本発明の核酸の治療効果のある量を投与するステップを含む方法
【0023】
驚くべきことに,本願発明者(ら)は,ヒトか非ヒトかを問わず,一次抗体構造を破断し,フォールディングを抑制し,及び/又は,重鎖又はごく少量の無傷抗体への分泌を制限するように可変領域でクローニングされた所定のT細胞エピトープを有する抗体が,強力なヘルパー及び抗原特異的T細胞反応を刺激することを発見した。本願発明者(ら)はまた,該効果が該抗体の重鎖をコードする核酸を用いて得られることを発見した。T細胞エピトープは,免疫プロテアソームによって処理されるが破壊はされないと考えられている。ある実施形態において,本発明は,T細胞反応の頻度及び活性を強化する変性免疫グロブリン内の所定のT細胞エピトープを提示するDNAワクチンを提供する。本発明の核酸によってコードされるポリペプチドを,本明細書において「ImmunoBody」と呼ぶことにする。
【0024】
T細胞エピトープに対する免疫反応が,免疫グロブリンが未変性コンフォメーションをとることができないようにT細胞エピトープが挿入される免疫グロブリンの重鎖を少なくともコードする核酸によって刺激されうるとの発見は,抗体は機能的な形で発現されなければならないと説く当該技術分野の予想に反するものである。例えば,上述のように,国際公開第96/19584号パンフレットは,核酸がT細胞エピトープが抗体のCDRに挿入される抗体をコードする場合,該核酸は,機能的抗体をコードしなければならない旨説いている。同様に,欧州特許第0759944号明細書は,無傷免疫グロブリンタンパク質として分泌されるT細胞エピトープを抗体分子へ取り込む方法を記載している。米国特許第7,067,110号明細書は,抗体の融合タンパク質によって抗原に対する免疫反応を引き起こすことができる旨開示しているが,該抗体は,免疫グロブリン可変領域を欠くものとして開示されている。また,該融合タンパク質は,抗原中に制御性T細胞エピトープを有する。よって,該タンパク質は,抗体反応を刺激しうるが,抗原中の制御性T細胞エピトープにより高活性T細胞反応を刺激しないであろう。
【0025】
上述したように,国際公開第00/64488号パンフレットは,CDR(その可変領域を除く)に挿入される異種T細胞エピトープを有するキメラ抗体をコードする核酸を開示しており,該核酸は,B細胞における発現を指向している。本発明の核酸は,B細胞における発現を指向しておらず,したがって,インビトロでもインビボでも特にB細胞を標的としない。本発明の核酸は,樹枝状核酸を含む任意の抗原提示細胞に取り込まれうり,したがって,ナイーブCTL及びヘルパーT細胞反応をプライミングすることができるのに対し,国際公開第00/64488号パンフレットに記載のワクチンは,既存のT細胞反応を促進する上でしか有用でないであろう。
【0026】
本発明による核酸によって誘発される反応の機能的活性の分析は,合成ペプチドによる免疫と比べて高活性反応を生成しうることを示した。これはまた,インビトロ及びインビボで腫瘍細胞を識別し殺傷する能力の強化と相関関係があった。該観察は,良好な抗腫瘍活性が高活性TRP2特異的CTLにより示される他の研究における報告に匹敵するものである(Zeh et al, J Immunol 1999;162: 989−94, Harada et al, Immunology 2001;104: 67−74)。
【0027】
本発明の核酸は,非特異的プロモーター,すなわち,核酸の発現を促進するが発現が促進される細胞に対する特異性は有さないプロモーター,を有する。該プロモーターは,好ましくは,樹枝状細胞及び/又はケラチン生成細胞における核酸の発現を引き起こす。適切なプロモーターの例には,CMVプロモーター,SV40プロモーター,及び当業者に周知の他の非特異的プロモーターが含まれる。あるいは,本発明の核酸は,樹枝状細胞(例えば,Cd11bプロモーター)及びケラチン生成細胞(例えば,MHCIIプロモーター, Chin et al., 2001 J. Immunol. 167, 5549−5557)において特異的発現を引き起こす1つ以上のプロモーターを有しうる。
【0028】
本発明のある側面の核酸は,免疫グロブリン分子,好ましくは,抗体の主要な特徴のすべて,すなわち,可変領域及び定常領域を含む重鎖及び軽鎖,を含む抗体をコードする。該抗体は,モノクローナル又はポリクローナルであり,IgA,IgD,IgE,IgG,又はIgMでありうるが,IgGが好ましい。IgG抗体は,任意のIgGサブクラス,例えば,ヒトIgG1,IgG2,IgG3,IgG4,又は,マウスIgG1,IgG2a,IgG2b,IgG3,でありうる。IgG抗体は,ヒトIgG2 Fc結合ドメインを有するヒトIgG1抗体でありうる。該抗体は,ヒト抗体の定常領域,及び異種T細胞エピトープが挿入されたマウスモノクローナル抗体の可変又は超可変(CDR)領域を有しうる。超可変領域を除く可変領域もまた,ヒト抗体の可変領域に由来しうる。該抗体は,ヒト化と呼ばれる。ヒト化抗体の作製方法は,当業者に周知である。該方法は,例えば,Winterによる米国特許第5,225,539号明細書に記載されている。マウス超可変領域を除く抗体の可変領域もまた,マウスモノクローナル抗体に由来しうる。この場合,全可変領域は,マウスモノクローナル抗体に由来し,該抗体は,キメラ化と呼ばれる。キメラ化抗体の作製方法は,当業者に周知である。該方法には,例えば,Boss(Celltech)及びCabilly(Genentech)による米国特許明細書に記載のものが含まれる。それぞれ,米国特許第4,816,397号明細書及び第4,816,567号明細書を参照のこと。
【0029】
本発明のある側面の核酸は,重鎖,軽鎖,又は発現される免疫グロブリン分子が,重鎖,軽鎖,又は免疫グロブリン分子が未変性コンフォメーションをとることができないように,少なくとも1つの異種T細胞エピトープを有する。該T細胞エピトープは,例えば,免疫グロブリンの重鎖が抗原に結合することができないように,又は,重鎖及び軽鎖(存在する場合)が結合することができないように,又は,重鎖又は免疫グロブリン分子が適切に分泌されえないように,発現タンパク質を破断しうる。該破断は,免疫グロブリン分子の三次構造で起こりうり,ジスルフィド結合(disulphide bond)の形成を阻止しうる。
【0030】
以下でより詳細に述べるように,免疫グロブリン分子が抗体である場合,T細胞エピトープは,抗体のCDR1領域及びCDR2領域に挿入されるか,置換されうる。CDR1及びCDR2は,抗体のβシート構造の一部を形成し,フォールドされた分子内に一部が沈殿している。長さ,アミノ酸組成や荷電のいかなる変化も,この構造を破断し,重鎖及び軽鎖のフォールディング及び結合を阻止する。CDRH3は,免疫グロブリン分子の表面に暴露され,したがって,変化に対して許容状態である。本発明において,CDR1及び/又はCDR2がT細胞エピトープで置換されるのが好ましい。事実,ある実施形態において,CDRHの接点におけるフレームワーク領域の欠損は,抗体のフォールディングを完全に破断するが,これらの領域におけるエピトープの挿入は,良好なT細胞反応を与える。CDRH1(5アミノ酸の長さ)又はCDRH2(17アミノ酸の長さ)へのエピトープの取り込みは,軽鎖が天然配列を有していても,ごく少量の無傷抗体を除く重鎖の分泌を可能にするほどの破断を引き起こす。これは,二次構造が重鎖と軽鎖の組み合わせにとって重要であることを示している。軽鎖のCDRL1内へのエピトープの取り込みは,重鎖のCDRH3へ取り込まれる唯一のエピトープが存在するとしても,軽鎖の分泌は低レベルとなる。
【0031】
「異種T細胞エピトープ」とは,抗体にとって異種であるT細胞エピトープを意味している。例えば,異種T細胞エピトープは,抗体中にこれまでに存在していなかったものでありうる。異種T細胞エピトープは,挿入されるアミノ酸配列から構成されうるが,第2の部分の隣接アミノ酸とともに,全体として挿入されうる。これは,挿入されるエピトープが,異種核酸に元の抗原由来のものと同様の処理プロファイルを有することを確実にするためである。一つ以上のCTL/ヘルパーエピトープを同じ可変領域に挿入しうる。
【0032】
T細胞エピトープは,重鎖又は軽鎖のどこにでも挿入しうる。該又は各エピトープは,重鎖及び/又は軽鎖の可変領域に挿入されるのが好ましいが,重鎖及び/又は軽鎖の定常領域にのみ,又は定常領域と可変領域に挿入されるT細胞エピトープを有する抗体や重鎖をコードする核酸が本発明に含まれる。本発明の核酸において,T細胞エピトープをコードする配列は,重鎖及び/又は軽鎖をコードする配列に挿入される(すなわち,追加される),又は重鎖及び/又は軽鎖をコードする配列に置換されうる。
【0033】
可変領域において,T細胞エピトープは,重鎖及び/又は軽鎖のいずれか1つ以上のCDR,すなわち,L1,L2,L3,H1,H2,又はH3,に挿入されるか,又は置換されうる。当然ながら,L1,H1及びH2が目下好ましい。ある実施形態において,T細胞エピトープは,CDRL1及び/又はH1及び/又はH2に挿入されるか,又は置換される。好ましくは,取り込まれるT細胞エピトープは,抗体が未変性コンフォメーションをとらないように,すなわち,フォールドせず,また,正しく分泌されないように,抗体の元のCDRのアミノ酸と同じ大きさ及び荷電ではない。あるいは又は加えて,それらは,CDRを取り囲むフレームワーク領域に挿入されるか,置換されうる。
【0034】
挿入されるT細胞エピトープは,好ましくは,細胞傷害性T細胞(CTL又はCD8)エピトープである。あるいは又は加えて,ヘルパーT細胞(CD4)エピトープが挿入されうる。T細胞エピトープは,周知のT細胞アルゴリズムを用いて予測されうる,又は,ペプチドとして合成て標準的なT細胞アッセイを用いてスクリーニングされうる。T細胞エピトープは,5〜50,7〜40,8〜30,又は9〜20アミノ酸,例えば,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,又は30の範囲のアミノ酸長を有しうる。エピトープは,抗原エピトープをコードする相補オリゴヌクレオチドを用いて挿入されうり,それらはアニールされ,CDR(又は他の領域)が特異的制限酵素部位に置換された抗体フレームワークの特異的部位にクローニングされる。組み換え抗体がヘルパー及び細胞傷害性T細胞反応を刺激する能力は,本明細書に例示されているようなかたちでスクリーニングすることができる。
【0035】
本発明の範囲内においてさまざまな組み合わせが可能である。ある実施形態において,1つ以上のCD8エピトープが,重鎖又は抗体のCDR H1及び/又はH2又は非CDRの可変領域に挿入されうる及び/又は置換されうる。あるいは又は加えて,1つ又は複数のCD4エピトープが,軽鎖又は抗体のCDR L1又は非CDR可変領域に挿入されうる及び/又は置換されうる。複数のT細胞エピトープが存在する場合,T細胞エピトープは,同じである又は異なりうる。当業者であれば,以下を含め,多数の組み合わせが可能であることを理解するであろう。
・CDR H1のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H2のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H1及びCDR H2のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H1の2つのCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H2の2つのCD8エピトープ,及びCDR L1のCD4エピトープ;など
【0036】
本発明の核酸は,MHCクラスI分子及びMHCクラスII分子両方の大部分と結合しうる単一の標的抗原からの複数のT細胞エピトープを取り込みうる。これにより,広範囲の集団ワクチン接種において利用可能なワクチンを製造しうる。あるいは,本発明に有用な核酸は,最も一般的なクラスI及びクラスII表現型に結合可能な複数の標的抗原からの複数のT細胞エピトープを取り込みうる。これにより,抗原が欠損した変異体の選択を阻止しうるワクチンを製造しうる。標的抗原は,単一の病原体又は腫瘍型由来であるか,又は様々な病原体又は癌に対して免疫反応を示すように選択されうる。特異的共通HLA表現型を標的とする本発明に有用な核酸は,さまざまな癌及び/又は病原体からの多数のT細胞エピトープを取り込み,病気を予防する単一のワクチンを提供しうる。
【0037】
ヘルパー及び/又は細胞傷害性T細胞反応を刺激するものであれば,任意のT細胞エピトープを挿入しうる。HIV,C型肝炎,及びその他の感染症などの潜伏感染を除去するのにCTLを必要とする病原体由来のT細胞エピトープを用いうるが,該エピトープは,「自己ペプチド」であること,すなわち,癌などの細胞増殖に関連する状態/疾患に関連する場合が好ましい。好ましくは,T細胞エピトープは,重鎖又は抗体が正しくフォールドされえず,分泌されえない。したがって,挿入されるエピトープは,元の可変領域と同様の大きさ及びアミノ酸組成ではないことが好ましい。核酸は,さまざまなT細胞反応を引き起こすように複数の異なるT細胞エピトープを有しうる。核酸は,単一の抗原由来の複数のエピトープを取り込みうり,それにより,異なるHLA型を有する個人の大多数が単一のワクチンに反応することを確実にしうる。あるいは,制限された範囲のHLA型を標的とする複数の抗原由来の複数のT細胞エピトープを用いうる。本発明の核酸分子は,単一の病原体又は癌型由来のさまざまな抗原を含みうる,又は,それらは,さまざまな固形腫瘍又は病原体を標的とする異種抗原を含みうる。本発明の核酸分子は,腫瘍内の異なる細胞集団,例えば,上皮腫瘍及び内皮抗原,を標的とするように設計されうる。
【0038】
驚くべきことに,本願発明者(ら)は,T細胞エピトープが重鎖の構造的に制限されたCDR又は非CDR領域に挿入されると,それらは,優れたCTR反応を示すことを発見した。これは,巨大なエピトープの可変領域への挿入により軽鎖と弱くしか結合することができない大量の重鎖の分泌によるものと思われる。これは,「抗原提示細胞により内因性に合成されたタンパク質のみがMHCクラスI分子上に提示されCTLにより識別される」(国際公開第96/19584号パンフレット)旨記述する教義に反している。外因性抗原の取り込み及びMHCクラスI上の提示は,交差提示として知られるプロセスであり,通常,特異的レセプターによる取り込みを必要とする。これは,ヒトFcγ1抗体のCD64レセプターでありうる。しかしながら,大量の無傷抗体又は抗原抗体複合体がこのレセプターをうまく標的とするであろうことは予測されるであろう。一方,本明細書に提示された結果は,CD64に結合せずにすぐれたCTL反応を示す非常に低レベルの無傷抗体及び大量の自由重鎖を明確に示している。実際,CTLエピトープが,CD64に結合することができない抗体,例えば,CD64結合領域がIgG2由来の非CD64結合領域で置換されたIgG2抗体やIgG1分子,に挿入されると,CTL反応を刺激しうることが本明細書において示されている。
【0039】
重鎖をコードする核酸は,好ましくは,分泌を可能にするリーダー配列を含む。本願発明者(ら)は,重鎖のリーダー配列が,分泌を阻止し,内因性タンパク質の生成を可能にするために除去されると,これによりCTL反応が低減することを発見した。これは,完全に予想に反するのものである。発明者(ら)は,理論に束縛されることを望んではいないが,これは,核酸が,高レベルの重鎖及び後に抗原提示細胞に取り込まれうる微量の天然タンパク質を分泌する非抗原提示細胞において発現されることを意味するものと考えている。あるいは,核酸は,排出リンパ節へ移動する抗原提示細胞を直接トランスフェクトしうる。該排出リンパ節において,抗原提示細胞は,微量の天然タンパク質,及び同じ又は隣接する抗原提示細胞に取り込まれ,ナイーブCTLに対するMHCクラスI上に提示される大量の重鎖を分泌する。したがって,核酸ワクチンが,効果的なCTL反応を刺激するために,核酸は,好ましくは,非常に低レベルで分泌される及び/又は同時に大量の変性タンパク質を分泌するタンパク質内のCTLエピトープをコードする。しかしながら,CTL反応は,ヘルパー反応非存在下では,高アフィニティ記憶反応に発達しえない。したがって,Tヘルパーエピトープが重鎖又は免疫グロブリン分子,好ましくは,抗体の軽鎖の可変領域,に挿入されるのが好ましい。さらに,驚くべきことに,また,「標的細胞に外因性に取り込まれるタンパク質のみがMHCクラスII分子によって提示され,ヘルパーT細胞によって識別される」旨述べる教義に反して,軽鎖は,ごく少量しか分泌されなかった。軽鎖の分泌を阻止するリーダー配列の除去は,ヘルパー反応に何ら影響を及ぼさなかった。したがって,本発明の核酸は,抗体の軽鎖のリーダー配列を有する場合もあれば有しない場合もある。これらの結果は,核酸は,MHCクラスIIとの関連で,内因性に合成されたタンパク質由来のTヘルパーエピトープを提示する抗原提示細胞,場合によってはオートファジー,に取り込まれることを暗示している。ヘルパーT細胞がCTL反応を助けるために,ヘルパーT細胞が識別する両T細胞エピトープは,結合T細胞ヘルプとして知られるプロセスにおいて同じ抗原提示細胞上に発現される必要がある。これは,核酸によってコードされる,軽鎖を合成する抗原提示細胞は,CTLエピトープ自体をも合成,分泌及び交差提示するか,又は隣接APCから重鎖を取り込む必要があることを暗示している。
【0040】
本発明はまた,内因性に生成された軽鎖由来の異種ヘルパーT細胞エピトープ,及び交差提示された重鎖由来の異種CTLエピトープをMHCクラスII上に提示する単離樹枝状細胞を提供する。該樹枝状細胞は,本明細書に記載の治療に利用しうる。
【0041】
本発明の核酸は,T細胞反応の頻度と活性の両方を増大させる変性抗体をコードすることにより,既存のT細胞エピトープをより免疫原性のあるものにしうる。
【0042】
本発明の核酸は,クローニングにより得られる,又は,化学合成によって全体的又は部分的に生成される,DNA,cDNA,又はmRNAなどのRNAでありうる。治療用として,核酸は,治療を受ける被験者において発現されうるかたちであることが好ましい。
【0043】
本発明の核酸は,組み換え型であるか,又は,分離された及び/又は精製された形で提供されうる。それは,1つ以上の発現用調節配列を除き,ヒトゲノム中に核酸隣接遺伝子を含まないか,又は実質上含まないことがある。本発明による核酸がRNAを含む場合,本明細書に示される配列への言及は,UをTと置き換えたRNA等価物への言及と解釈されるべきである。
【0044】
本発明の核酸は,核酸配列及びクローンが入手可能であることを所与として,例えば,本明細書に記載の情報及び参考文献や当技術分野で周知の技術(例えば,Sambrook, Fritsch and Maniatis, “Molecular Cloning”, A Laboratory Manual, Cold Spring Harbor Laboratory Press, 1989, 及び Ausubel et al, Short Protocols in Molecular Biology, John Wiley and Sons, 1992を参照のこと)を用いて,当業者が容易に作製することができる。これらの技術は,(i)例えば,ゲノムソースからの該核酸のサンプルを増幅するポリメラーゼ連鎖反応(PCR)の利用,(ii)化学合成,又は(iii)cDNA配列の作製,を含む。ポリペプチドをコードするDNAは,当業者に周知の任意の適切な方法を用いて生成し使用することができ,例えば,コードするDNAの取り込み,発現される部位の両側における適切な制限酵素認識部位の同定,及びDNAからの該部位の除去,がある。該部位は,後に,標準的な市販の発現系における適切なプロモーターと動作可能に結合しうる。他の組み換えアプローチは,DNAの関連部位を適切なPCRプライマーで増幅することである。例えば,部位特異的突然変異誘発法を用いて,修飾ペプチドの発現につながるように,又は,核酸の発現に使用される宿主細胞のコドン選択を考慮するように,配列の修飾を行うことができる。
【0045】
核酸配列の発現を得るために,該配列を,動作可能に核酸に結合してその発現を制御する1つ以上の制御配列を有するベクターに取り込むことができる。ベクターは,例えば,挿入される核酸の発現を促進するプロモーターやエンハンサーなどの他の配列,ポリペプチドを溶解として生成するような核酸配列,及び/又は宿主細胞で生成されるポリペプチドが細胞から分泌されるような分泌信号をコードする核酸を含みうる。必要に応じ,ベクターをベクターが機能する宿主細胞に変え,ポリペプチドが生成されるように宿主細胞を培養し,宿主細胞又は周囲媒質からポリペプチドを回収することによってポリペプチドを生成しうる。原核細胞及び真核細胞が当技術分野において該目的のために用いられる。その中には,大腸菌株,酵母菌株,及び,昆虫細胞や動物細胞などの真核細胞,例えば,COS細胞,CHO細胞,ボウズメラノーマ細胞,及びその他の適切なヒト細胞,が含まれる。本発明が抗体の重鎖及び軽鎖をコードする核酸に関連する場合,個々の核酸は,同じ又は異なるプロモーターに刺激されて,同じ発現ベクター又は別の発現ベクターに存在しうる。
【0046】
本発明の核酸は,ヒトを含む哺乳動物などの患者の少なくとも1つのT細胞エピトープに対する免疫反応を刺激するために利用されうる。ヘルパー及び/又は細胞傷害性T細胞反応を刺激しうる。本発明により得られる特定のエピトープに対するT細胞反応は,単純なペプチドと同じエピトープによる免疫によって,又は,ペプチド又は核酸と同じ抗体内でコードされるエピトープによる免疫によって得られるものよりも高い結合活性を有しうる。本発明の核酸は,併用療法,すなわち,軽鎖をコードする核酸及び重鎖をコードする核酸,として投与されうる。核酸は,静脈内投与,経皮投与,筋肉内投与,経口投与,又は他の投与法で投与されうる。経皮投与又は筋肉内投与が好ましい。というのも,これらの組織は,樹枝状細胞を含むからである。
【0047】
本明細書において,「治療」の語は,ヒト又は非ヒト動物のためになりうるいかなる養生法を含む。治療は,遺伝性疾患又は後天性疾患のためのものでありうる。好ましくは,治療は,癌や感染症といった細胞増殖に関連する状態/疾患のためのものである。核酸で治療可能な癌の種類の例には,固形腫瘍,結腸直腸癌,肺腫瘍,乳房腫瘍,胃腫瘍,卵巣腫瘍,子宮腫瘍,肝腫瘍,腎腫瘍,膵臓腫瘍,メラノーマ腫瘍,膀胱腫瘍,頭頸部腫瘍,脳腫瘍,食道腫瘍,膵臓腫瘍,骨腫瘍,並びに軟部組織癌及び白血病,が含まれる。核酸で治療可能な感染症の例には,HIV感染症,C型肝炎感染症や,クリアランス用T細胞免疫を必要とする慢性感染症,が含まれる。
【0048】
核酸は,薬学的に許容されるキャリアと併用しうる。該キャリアには,これらに限定されるものではないが,生理食塩水,緩衝生理食塩水,デキストロース,リポソーム,水,グリセロール,エタノール,及びこれらの組み合わせ,が含まれる。
【0049】
宿主の免疫反応の刺激を促進するためにアジュバントを用いうる。アジュバントには,水酸化アルミニウム,リゾレシチン,プルロニック,ポリオール,ポリアニオン,ペプチド,タンパク質,及び油乳剤,が含まれる。
【0050】
本発明に有用な核酸は,医薬組成物で処方しうる。これらの組成は,上記物質の一つに加え,薬学的に許容される賦形剤,キャリア,緩衝剤,安定剤,又は当業者に周知の他の物質を含みうる。キャリアや他の物質の正確な特質は,投与経路,例えば,経皮経路,経口経路,静脈内経路,皮膚経路又は皮下経路,経鼻経路,筋肉内経路,腹腔内経路,に依存しうる。処方は,好ましくは,微小な金粒子の表面上に沈殿し,遺伝子銃による注入に適した安定乾燥粉末としての核酸である。処方は,エレクトロポレーションを用いた経皮又は筋肉内投与に適しうる。
【0051】
核酸を含む,又は核酸の送達のための組成物は,好ましくは,「治療効果のある量」個人に投与され,これは,個人にとっての利益を示すのに十分である。投与される実際の量,及び投与の割合及び経時変化は,治療対象の特質及び重症度に依存する。治療の処方,例えば,投与量に関する決定など,は,一般開業医その他の医師の責任内にあり,通常,治療対象の疾患,個々の患者の状態,送達部位,投与方法,及び開業医に周知の他の要因を考慮に入れる。本発明の核酸は,特に,既存の癌の治療,及び最初の治療又は手術後の癌の再発予防に関連している。上述の技術及び手順の例は,Remington’s Pharmaceutical Sciences, 16th edition, Oslo, A.(ed), 1980に見出すことができる。
【0052】
好ましくは,本発明の核酸は,効果のある量ヒトに投与すると腫瘍細胞の成長を有意に抑制しうるヘルパー及び/又は細胞傷害性T細胞を刺激する。適量は,多数のパラメーター,例えば,年齢,性別,体重,治療対象の状態の重症度,投与される活性成分,及び投与経路,に基づいて医師により決定されうる。例えば,1〜1000μgのDNAの投与量は,ヘルパー及び細胞傷害性T細胞反応の両方を刺激するのに十分である。
【0053】
本発明の核酸は,追加の薬学的に許容される成分とともに投与されうる。該成分には,例えば,免疫系刺激剤が含まれる。
【0054】
組成物は,治療対象の状態に応じて,単独で,又は他の治療と組み合わせて,同時に又は連続して投与されうる。他の癌治療には,当技術分野において周知の,他のモノクローナル抗体,他の化学療法剤,他の放射線治療技術や免疫療法が含まれる。本発明の組成物の一つの特定の用途は,手術の補助,すなわち,腫瘍除去後の癌再発のリスクを低下させる手助けをすることである。
【0055】
注射(id)は,本発明の核酸の治療的投与のための第一の経路でありうる。
【0056】
核酸は,局所的に腫瘍部位や他の所望の部位に投与され,又は腫瘍や他の細胞を標的とするような方法で送達されうる。
【0057】
核酸の投与量は,利用される作用物質の特性,例えば,その結合活性及びインビボ血中濃度半減期,製剤中のポリペプチド濃度,投与経路,投与部位及び割合,関係患者の臨床的耐性,患者を苦しめている病的状態,など,十分に医師の技能の範囲内にあるもの,に依存する。例えば,一患者一回につき100μgの核酸の投与量が好ましいが,投与量は,一回あたり約10μgから1mgの幅がありうる。一連の連続的接種中に異なる摂取量が用いられる。開業医は,最初の接種を行い,次に,比較的少量の核酸で促進しうる。
【0058】
他の実施形態において,本発明は,標的抗原からのT細胞エピトープを可変領域の抗体へ改変する方法,及び,ヘルパー及び細胞傷害性T細胞反応の両方を刺激するワクチンとしての該改変抗体の利用に関する。
【0059】
本発明のさらなる側面は,本明細書に開示される核酸を含む宿主細胞を提供する。本発明の核酸は,宿主細胞のゲノム(例えば,染色体)に組み込まれうる。組み込みは,標準的な技術に従ってゲノムとの組み換えを促進する配列を含むことにより促進されうる。核酸は,細胞内の染色体外ベクター上にありうる,あるいは,細胞に対し識別可能に異種又は異質でありうる。
【0060】
さらなる側面は,本発明の核酸を宿主細胞に導入するステップを含む方法を提供する。該導入は,(特にインビトロ導入の場合は)一般的に制限なく「形質転換」と呼ぶことができるが,任意の利用可能な技術を利用しうる。真核細胞の場合,適切な技術には,リン酸カルシウムトランスフェクション,DEAEデキストラン,エレクトロポレーション,リポソーム媒介トランスフェクション,及びレトロウイルス又は他のウイルス,例えば,ワクシニア,昆虫細胞の場合はバキュロウイルス,を用いた形質導入が含まれうる。
【0061】
細菌性細胞の場合,適切な技術には,塩化カルシウム形質転換,エレクトロポレーション,及びバクテリオファージを用いたトランスフェクションが含まれる。他の方法として,核酸の直接注入を用いることができる。
【0062】
抗生物質抵抗性遺伝子や感受性遺伝子といった標識遺伝子を,当技術分野において周知のように,関心のある核酸を含むクローンを同定するのに利用しうる。
【0063】
導入に続いて,核酸からの発現が引き起こされるか可能となりうる。例えば,コードされたポリペプチド(又はペプチド)が生成されるように,遺伝子発現の条件下で宿主細胞(実際に形質転換された細胞を含みうる。おそらく,該細胞は,形質転換された細胞の子孫であるだろうが)を培養することによって。ポリペプチドが適切なシグナルリーダーペプチドを結合して発現される場合,細胞から培養培地へ分泌されうる。発現による生成の後に,ポリペプチド又はペプチドは,宿主細胞,及び/又は場合によっては培養培地から単離及び/又は精製されうり,その後,所望するように,例えば,1種類以上の追加の成分を含みうる組成,例えば,1種類以上の薬学的に許容される賦形剤,媒体,又はキャリア(例えば,以下を参照のこと)を含む医薬組成物など,の製剤において利用されうる。
【0064】
本発明はまた,候補抗原のT細胞エピトープを同定する方法を提供する。該方法は,非ヒト動物のT制御性細胞を枯渇させるステップと,非ヒト動物を候補抗原で免疫するステップと,T細胞反応が,候補抗原の予測エピトープに対するペプチド又は候補抗原内のすべての可能性のある重複ペプチドに対して引き起こされたかどうかを確かめるためにスクリーニングするステップと,を含む。
【0065】
該方法は,マウスやラットなどの非ヒト動物において実施されうる。T制御性細胞は,Ontakなどの毒素と随意に接合しうる抗CD25抗体を用いて,又は,T制御性細胞を選択的に殺すシクロフォスファミドなどの化学療法によって非ヒト動物において枯渇されうる。T制御性細胞が枯渇すると,非ヒト動物は,候補抗原をコードするDNAによって,又は候補抗原自体によって免疫されうる。候補抗原は,抗原Fc融合タンパク質として提供されるのが好ましい。スクリーニングステップにおいて,非ヒト動物においてT細胞反応が刺激されるペプチドが同定される。これは,ELISPOTなどの技術を用いてインビトロで行うことができる。T細胞反応が候補エピトープに対して引き起こされる場合,このエピトープを非ヒト動物に免疫するのに用いることができる。このペプチドがT細胞反応を引き起こす場合,結合活性及び頻度は,本発明による核酸内のエピトープをコードすることにより強化することができる。この方法は,免疫プロテアソームによって処理されるT細胞エピトープの同定を可能にする。
【0066】
本発明の各側面の好ましい特徴は,各々の他の側面に変更すべきところは変更して関係する。本明細書に記載の先行技術文献は,法律が許す最大限の範囲で組み込まれる。
【図面の簡単な説明】
【0067】
以下,本発明を以下の実施例(これらに限定されない)においてさらに述べる。以下の図面を参照する。
【0068】
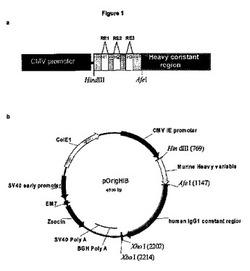
【図1】重鎖ベクターpOrigHIBの特徴を示す図 抗体SC100の野生型脱免疫重可変領域を,ヒトIgG1Fc定常領域のフレームのHindIII/AfeIを用いてクローニングした。Fc領域は,CH1領域,CH2領域,CH3領域及びヒンジ領域を含む。哺乳類細胞における高レベル発現は,ヒトサイトメガロウィルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,Orig HIBヒトIgG1鎖の下流に信号を送る。EM7は,大腸菌における抗生物質選択を可能にするzeocin耐性遺伝子の発現を制御する細菌プロモーターであり,一方,該耐性遺伝子のSV40早期プロモーター上流は,哺乳類細胞における選択を可能にする。SV40ポリアデニル化は,zeoΓmRNAの3’末端の適切な処理を管理するために該耐性遺伝子の下流に信号を送る。ベクターはまた,その内部に,細菌における増殖のためのColE1複製起点を含む。相補性決定DNA配列は,効果的に除去され,制限部位RE1,RE2,RE3(それぞれ,FspI,MscI,SrfI)と単独で及び組み合わさって交換される。
【0069】
【図2】重鎖ベクターpOrigLIBの特徴を示す図 抗体SC100の野生型脱免疫された軽可変領域を,ヒトカッパ定常領域のフレームのBamHI/BsiWIを用いてクローニングした。哺乳類細胞における高レベル発現は,ヒトサイトメガロウィルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,Orig LIB鎖の下流に信号を送る。ベクターはまた,細菌における増殖及び選択を可能にするアンピシリン用抗体耐性遺伝子及びColE1複製起点を含む。相補性決定領域は,効果的に除去され,制限部位RE4,RE5,RE6(それぞれ,EcoRV,SspI,HpaI)と単独で及び組み合わさって交換される。
【0070】
【図3】野生型ImmunoBodyキメラ重鎖の配列 ヌクレオチド及び翻訳中のアミノ酸配列が完全長キメラIgG1重鎖に図示されている。CDRの位置は,カバット番号付与体系によって定義されるボックス内にある。ストップコンドンは赤のアスタリスクで描かれている。重可変領域の転移に利用されるHindlll/Afel制限部位が強調されている。
【0071】
【図4】野生型ImmunoBodyキメラカッパ鎖の配列 ヌクレオチド及び翻訳中のアミノ酸配列が完全長キメラカッパ鎖に図示されている。CDRの位置は,カバット番号付与体系によって定義されるボックス内にある。ストップコンドンはアスタリスクで描かれている。軽可変領域の転移に利用されるBamHI/BsiWI制限部位が強調されている。
【0072】
【図5】重複伸長PCR CDRを取り除き,重複PCRによって独特の制限部位に置換した。フォワードプライマーH1,H2,H3,L1,L2,及びL3(表2)を,重鎖及び軽鎖の各々の可変領域内のCDR1,2,3を置換するように設計した。中央に配置された各プライマーは,取り除かれ(緑色部分)10〜20bpの野生型配列の横に配置するようにCDR配列を欠く選択された独特の酵素認識配列を含んでいた。フォワードプライマーを,野生型コンストラクトpOrigHIB及びpOrigLIBの各々の内部のヒト重・軽定常領域にアニールする,一般的なリバースプライマーhuHeClonR又はhuLiClonR(表2)とともにPCRの第一段階において使用した。生成されたフラグメントは,野生型CDR配列(赤色部分)を含んでいないが,制限部位と効果的に交換される。可変重・軽領域全体を増幅するために,PCRの第二段階は,単一のプラスミド内のCMVプロモーターにアニールする一般的なCMVフォワードプライマーとともにリバースプライマーとして第一段階から生成されるPCR生成物を用いることが要求される。第二段階のPCR生成物を,pcr2.1(Invitrogen)にサブクローニングし,配列確認後,H1,H2,H3,L1,L2,L3バージョンを単独,組み合わせて及び一緒に含む重/軽(VH及びVL)可変領域を,単一のコンストラクトpOrigHIB及びpOrigLIBに挿入し,野生型領域をHindIII/Afel及びBamHI/BsiWIそれぞれを用いて交換した。
【0073】
【図6A】ImmunoBody重鎖可変領域の配列 CDRが対応する酵素部位H1,H2,H3(単独,組み合わせて及び一緒に)で置換された重可変領域のヌクレオチド及びアミノ酸配列。独特の制限酵素部位が強調されている。CDR1,2,3を,それぞれ,FspI,MscI,SrfIと置換した。
【図6B】続き
【図6C】続き
【0074】
【図7A】ImmunoBodyカッパ鎖可変領域の配列 CDRが対応する酵素部位L1,L2,L3(単独,組み合わせて及び一緒に)で置換された重可変領域のヌクレオチド及びアミノ酸配列。独特の制限酵素部位が強調されている。CDR1,2,3を,それぞれ,EcoRV,SspI,HpaIと置換した。
【図7B】続き
【図7C】続き
【0075】
【図8】二重発現ベクターpDCOrigの特徴を示す図 すべてのエピトープが単一ベクター内の可変重部位及び可変軽部位に組み込まれると,それらは,各々ヒト定常領域のフレームの強調されたHindIII/AfeI及びBamHI/BsiWIを用いて二重発現に導入される。重鎖のFc領域は,CH1,CH2,CH3領域及びヒンジ領域からなる。哺乳類細胞における重鎖及び軽鎖の両方の高レベルの発現は,ヒトサイトメガロウイルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,両鎖の下流に信号を送る。EM7は,大腸菌における抗生物質選択を可能にするzeocin耐性遺伝子の発現を制御する細菌プロモーターであり,一方,該耐性遺伝子のSV40早期プロモーター上流は,哺乳類細胞における選択を可能にする。SV40ポリアデニル化は,zeoΓmRNAの3’末端の適切な処理を管理するために該耐性遺伝子の下流に信号を送る。ベクターはまた,そのバックボーンの内部に,細菌における増殖のためのColE1複製起点を含む。
【0076】
【図9】FC領域の合成を防止するストップコンドンを含むImmunoBodyIB15重鎖の配列 キメラ重鎖pDCOrig IB15 CH1ストップのヌクレオチド及びアミノ酸配列。ストップコンドンは,アスタリスクで描かれるヒトIgG1 Fc定常領域のCH1の後に部位特異的な突然変異生成により挿入される。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。単一のコンストラクトからの可変重領域の移転に利用されるHindIII/AfeI制限部位が強調されている。
【0077】
【図10】リーダーを有さないDCIB15重可変領域のヌクレオチド及びアミノ酸配列 リーダーは,重可変(VH)領域を効果的に再増幅するヒトIgG1 CH1領域に結合するリバースプライマーhuHeClonR(表2)とともにフォワードプライマーpOrig重非リーダーを用いてPCRにより取り除かれた。配列確認後,リーダーを除くVH領域を,ヒトIgG1定常領域のフレームのHindIII/AfeIを用いて二重発現コンストラクトDCIB15にクローニングした。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。可変重領域の移転に利用されるHindIII/AfeI制限部位が強調されている。
【0078】
【図11】リーダーを有さないDCIB15カッパ可変領域のヌクレオチド及びアミノ酸配列 リーダーを,軽可変(VL)領域を効果的に再増幅するリバースプライマーhuLiClonR(表2)とともにフォワードプライマーpOrig軽非リーダーを用いてPCRにより取り除いた。配列確認後,リーダーを除くVL領域を,ヒトカッパ定常領域のフレームのBamHI/BsiWIを用いて二重発現コンストラクトDCIB15にクローニングした。ボックス内のアミノ酸は,H1のHepB CD4エピトープ(TPPAYRPPNAPIL)を表す。可変軽領域の移転に利用されるBamHI/BsiWI制限部位が強調されている。
【0079】
【図12】ヒトIgG2定常領域の配列 増幅された重ヒトIgG2定常領域のヌクレオチド及びアミノ酸配列。二重発現ベクターDCIB15のhuigG1定常領域の移転及び置換に利用されるAfeI及びSapI制限部位が強調されている。
【0080】
【図13】ヒトigG3定常領域の配列 増幅される重ヒトigG2定常領域のヌクレオチド及びアミノ酸配列。二重発現ベクターDCIB15のhuigG1定常領域の移転及び置換に利用されるAfeI及びSapI制限部位が強調されている。
【0081】
【図14】ImmunoBody二重発現ベクターのヒトアイソタイプ 図Aは,二重発現ベクターpDCOrigIB15 huigG2を示す。図Bは,二重発現ベクターpDCOrigIB15 huigG3を示す。可変重領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0082】
【図15】G2モチーフを含むDCIB66重鎖の配列 キメラ重鎖のヌクレオチド及びアミノ酸配列。高アフィニティFcγR1(CD64)との相互作用のための臨界結合モチーフ内のアミノ酸E233 L234 L235は,ヒトIgG2からのP233 V234 A235(ボックス内にボールドで強調)で置換されている。ボックス内の他のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。pDCOrigIB15 huigG1への置換を含む部分の移転に利用されるAgeI/AhdI部位が強調されている。可変重領域の移転に利用されるHindIII/AfeI制限部位はボールドで示されている。
【0083】
【図16】G1結合モチーフを含むDCIB67重鎖の配列 キメラ重鎖のヌクレオチド及びアミノ酸配列。ヒトIgG2定常領域内のアミノ酸P233 V234 A235は,ヒトigG1からの高アフィニティFcγR1(CD64)E233 L234 L235 G236(ボックス内にボールドで強調)との相互作用のための臨界結合モチーフで置換されている。ボックス内の他のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。pDCOrigIB15 huigG2への置換を含む部分の移転に利用されるAgeI/AhdI部位が強調されている。可変重領域の移転に利用されるHindIII/AfeI制限部位はボールドで示されている。
【0084】
【図17】マウスIgG2aImmunoBody発現ベクター 図Aは,一本鎖pMoOrigHIBベクターを示す。図Bは,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を含む二重発現ベクターDCIB53を示す。図Cは,H1のHLA−DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を含む二重発現ベクターDCIB63を示す。利用される制限部位が示されている。
【0085】
【図18】規制準拠プラスミドpVAXDCIB54の構造を示す概略図 重一本鎖ベクターpVaxIB54 HIB(図A)は,Nrulを用いて線形化された。pOrigLIBからの軽鎖発現カセット(図B)は,Nrul及びHpalを用いて摘出され,線形化されたプラスミドにクローン化され,二重発現ベクターpVaxDCIB54(図C)を生成した。可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0086】
【図19】DCIB15の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0087】
【図20A】低レベルの無傷タンパク質を生成するImmunoBodyコンストラクト A:gp100/H1,TRP2/H2,及びHepB CD4/L1(DCIB15)を含むImmunoBodyを導入されたCHO−S細胞の上清からのサンドウィッチElisaによるImmunoBody重鎖のレベルの定量化。上清は,そのまま使用し,培地で1/3,1/10,及び1/30に希釈し,ヒトIgG陽性対照と比較した。 B:陽性対照と比較したサンドウィッチElisaによるgp100/H1,TRP2/H2,及びHepB help/L1(DCIB15)を含む精製ImmunoBodyの分析 C及びD:サンドウィッチElisaによるCHO−Sトランスフェクションの上清からの重鎖及び無傷ImmunoBodyの発現の測定。プレートを抗ヒトFc特異抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を使用し,無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を使用した。 E:サンドウィッチElisaによるCHO−Sトランスフェクション(DCIB15,DCIB31,DCIB32,DCIB36,DCIB48,DCIB49,DCIB52,DCIB54)の上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。
【図20B】続き
【0088】
【図21】DCIB24の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のオボアルブミンエピトープ(SIINFEKL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0089】
【図22】DCIB25の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0090】
【図23】DCIB31の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のTRP2エピトープ(SVYDFFVWL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0091】
【図24】DCIB32の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のTRP2エピトープ(SVYDFFVWL)及びL3のHepB CD4エピトープを示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0092】
【図25】DCIB36の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,L3のTRP2エピトープ(SVYDFFVWL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0093】
【図26】DCIB349の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びH3のHLA−DR4制限gp100 CD4エピトープ(WBRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0094】
【図27】DCIB52の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0095】
【図28】DCIB54の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びH3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0096】
【図29】DCIB36の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のHLA―DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0097】
【図30】DCIB18の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0098】
【図31A】ImmunoBodyフレームワークに取り込まれるCTLエピトープは,処理され,発現され,インビボで免疫反応を引き起こす。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H2のTRP2エピトープ及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB18)で免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:免疫マウスからの脾細胞からCD8 T細胞を枯渇させ,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対してIFNγ elispotアッセイにおけるエピトープ特異反応の有無を分析した。反応は,spots/million脾細胞として測定した。 D:インビトロTRP2ペプチド刺激の6日後のB16F10,B16F10 IFNα,及びB16F10 siKbメラノーマ細胞(株)に対する4時間におよぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 E:C57BI/6又はHLA−DR4トランスジェニックマウスに0日目,7日目,14日目にImmunoBody DNA(DCIB15,DCIB31,DCIB32,DCIB36,DCIB48,DCIB52,及びDCIB54)で免疫した。19日目に,IFNγelispotアッセイにより,脾細胞を,TRP2ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 F:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 G:C57BI/6又はHLA−DR4マウスに0日目,7日目,14日目にImmunoBody DNA(DCIB15,DCIB48,DCIB49,DCIB52,及びDCIB54)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepBヘルパーペプチド(DCIB15,DCIB49,及びDCIB52)又はgp100 DR4ヘルパーペプチド(DCIB48及びDCIB54),及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【図31B】続き
【0099】
【図32A】ImmunoBody DNA免疫は,ペプチド免疫又は全抗原による免疫よりもよい。 A:ImmunoBodyDNA免疫(DCIB18)を,フロイント不完全アジュバントのペプチドエピトープによるSC免疫又はTRP2抗原を発現するDNAによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド(■),HepBヘルパーペプチド(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,ImmunoBody DNA(◇)及びペプチド(◆)免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:インビトロTRP2ペプチド刺激の6日後のB16F10(■),B16F10 IFNα(■(薄灰色)),及びB16F10 siKb(□)メラノーマ細胞株に対する4時間におよぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 D:ImmunoBody DNA免疫(DCIB18)を,TRP2ペプチドパルスDCによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のTRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:ImmunoBody DNA免疫(DCIB18)を,TRP2ペプチドパルスDCによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,脾細胞を,TRP2ペプチドパルスLPS Bblastによりインビトロで刺激した。刺激の6日後,クロム遊離アッセイにより,CTL株のB16F10又はB16F10 siKbメラノーマ株を溶解させる能力を評価した。反応は,%細胞毒性として測定した。 F:ImmunoBody DNA免疫(DCIB24)を,SIIFEKLペプチドによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,SIINFEKLペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 G:ImmunoBody DNA免疫(DCIB15)を,gp100 210Mペプチドによる免疫と比較した。HHDIIマウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のgp100 210Mペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 H:ImmunoBody DNA免疫(DCIB24)を,SIIFEKLペプチドによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のSIINFEKLペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 I:ImmunoBody DNA免疫(DCIB15)を,gp100 210Mペプチドによる免疫と比較した。HHDIIマウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のgp100 210Mペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【図32B】続き
【図32C】続き
【0100】
【図33】DCIB21の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のHepB S Agエピトープ(IPQSLDSWWTSL)及びL1のI−Ad制限Flu HA CD4エピトープ(FERFEIFPKE)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0101】
【図34】複数のエピトープは,CDR H2部位から処理されうる。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H2のSIINFEKLエピトープ及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB24)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,SIINFEKLペプチド,無関係ペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:Balb/cマウスに0日目,7日目,14日目にCDR H2のHepB CD8エピトープ及びCDR L1のFlu HA CD4エピトープを含むImmunoBodyコンストラクト(DCIB21)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepB CD8ペプチド,無関係ペプチド,Flu HA CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0102】
【図35】DCIB17の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0103】
【図36】DCIB26の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のTie−2 Z84エピトープ(FLPATLTMV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0104】
【図37】複数のCTLエピトープは,可変領域から処理されうる。 A:HHDIIマウスに0日目,7日目,14日目にCDR H1のgp100 IMDQVPFSVエピトープ(フレームワーク部分を除く。)及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB17)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100 IMDQVPFSVペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:HHDIIマウスに0日目,7日目,14日目にCDR H1のTie2エピトープ(フレームワーク部分を除く。)及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB26)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,Tie2ペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0105】
【図38】複数のCTL反応は,同じImmunoBodyコンストラクト内の異なるエピトープから生成されうる。 A:HHDIIマウスに0日目,7日目,14日目にImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100ペプチド,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のgp100修飾IMDQVPFSV(◆)エピトープ,gp100 wt ITDQVPFSVエピトープ(▲),及びTRP2ペプチド(■)TRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:gp100 IMDQVPFSVペプチドでパルスされたT2細胞,TRP2ペプチド又は対照,並びに,B16F10及びB16F10 HHDメラノーマ細胞株に対する4時間に及ぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 D:HHDIIマウスに0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),又は,ii)CDR H2のTRP2エピトープ及びHepB CD4エピトープ(DCIB18),を含むImmunoBody DNAで免疫した。19日目に,IFNγelispotアッセイにより,脾細胞を,gp100ペプチド(■),TRP2ペプチド(■(濃灰色)),HepBヘルパーペプチド(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 E:C57BI/6マウスに,10μgのDNA溶液をエレクトロポレーションを併用して筋肉内免疫した。免疫は,頸骨筋に週間隔で3回行った。DCIB24又はDCIB18単体で,両者を混合して同じ部位に,又は両者を同時ではあるが別々の部位に,マウスに免疫した。19日目に,TRP2,SIINFEKLペプチド特異的免疫反応の有無に関し脾細胞を分析した。反応は,spots/million脾細胞として測定した。
【0106】
【図39】DCIB37の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Lエピトープ(TITDQVPLSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0107】
【図40】DCIB40の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Iエピトープ(TITDQVPISV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0108】
【図41】DCIB41の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100野生型エピトープ(TITDQVPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0109】
【図42】DCIB42の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Yエピトープ(TITDQVPYSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0110】
【図43】DCIB43の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 V5Lエピトープ(TITDQLPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0111】
【図44】非係留性残基における改変はエピトープ免疫原性を強化しうる。 HHDIIマウスに0日目,7日目,14日目にCDR H1領域の改変gp100エピトープImmunoBodyコンストラクト(DCIB37,DCIB40,DCIB41,DCIB42,及びDCIB43)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100野生型ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0112】
【図45】DCIB35の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0113】
【図46】複数のCD4ヘルパー反応は,インビボで免疫反応を引き起こすように処理及び提供されうる。 A:HHDII又はC57BI/6マウスに0日目,7日目,14日目にCDR L1領域のI−Ab制限HepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB15)で免疫した。 B:Balb/cマウスに0日目,7日目,14日目にCDR L1領域のI−Ab制限Flu HA CD4エピトープを含むImmunoBodyコンストラクト(DCIB21)で免疫した。 C:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目にCDR L1のHLA−DR4制限gp100 CD4エピトープを含むImmunoBodyコンストラクト(DCIB35)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,対応ペプチド,無関係ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 D:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目にCDR L1,CDR H3(DCIB54),及びCDR L3(DCIB50)のHLA−DR4制限gp100 CD4エピトープを含むImmunoBodyコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,対応ペプチド,無関係ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0114】
【図47】DCIB50の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0115】
【図48】CD8 T細胞反応は,分泌重鎖に部分的に依存するが,ヘルパー反応は,分泌軽鎖を必要としない。 A:HHDIIマウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし),iii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(軽鎖上にリーダー配列なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100(■)及びHepB CD4(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 B:サンドウィッチElisaによるCHO−Sトランスフェクションの上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。 C:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし,を含むImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定した。 D:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし,を含むImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepBヘルパーペプチドに対して分析した。反応は,spots/million脾細胞として測定した。
【0116】
【図49A】ImmunoBody Fc領域は,効果的な免疫反応を確立するのに有用である。 A:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(Fc領域なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2(■(濃灰色)),培地制御(□),B16F10メラノーマ株(■),及びB16F10siKb陰性対照細胞株(■(薄灰色))に対して分析した。反応は,spots/million脾細胞として測定した。 B:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(Fc領域なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定した。 C:同じマウスを,HepBヘルパーペプチドに特異的な反応に対して分析した。反応は,spots/million脾細胞として測定した。 D:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB15又はFc領域を有さないDCIB15(DCIB15 FcStop)での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)ヒトIgG2定常領域を有する同じコンストラクト(DCIB33),iii)ヒトIgG3定常領域を有する同じコンストラクト(DCIB65),iv)ヒトIgG2からの結合モチーフと置換されたヒトIgG1結合モチーフを有する同じコンストラクト(DCIB66),及びv)ヒトIgG1(DCIB67)からのモチーフに置換された結合モチーフを有するDCIB33,を含むImmunoBody DNAコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド(■),培地制御(□),及びHepBヘルパーペプチド(■(薄灰色))に対して分析した。反応は,spots/million脾細胞として測定した。 F:サンドウィッチElisaによるCHO−Sトランスフェクション(DCIB15,DCIB33,DCIB65,DCIB66,及びDCIB67)の上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。 G:サンドウィッチElisaによるDCIB53を導入されたCHO−Sトランスフェクションの上清からの重鎖ImmunoBodyの測定。プレートを抗マウスFc特異抗体でコーティングした。重鎖を検出するために,抗マウスIgG2a特異HRP抗体を使用した。
【図49B】続き
【0117】
【図50A】ImmunoBody免疫は,免疫反応を強化し,全抗原から観察される制御を克服する。 A:HLA−A2トランスジェニックマウス(HHDII)に,0日目,7日目,14日目に,ImmunoBody DNAコンストラクトDCIB15又はpcDNA3ベクターの全gp100抗原で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100ペプチド又は対照に対して分析した。反応は,spots/million脾細胞として測定した。 B:抗CD25抗体(PC61)400μgを腹腔内に注入することにより,C57BI/6マウスからCD25陽性細胞を枯渇させた。続いて,CD25枯渇マウス及び非枯渇動物の両方に,4日目,11日目,18日目に,ImmunoBody DNAコンストラクトDCIB15又はpOrigベクターの全TRP2抗原で免疫した。23日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド又は対照に対して分析した。反応は,spots/million脾細胞として測定した。 C及びD:HHDIIマウスを,未処理(c),又は,400μgのPC61mAbを腹腔内投与にて処理した(d)。4日後,全てのマウスにTie2 C200HFc DNAコンストラクトで免疫した。DNA免疫を7日間隔で計3回繰り返した。最後の免疫の6日後,脾細胞を採取し,インビトロIFNγ ELISPOTアッセイにより,1μg/mlのTie−2からの各予測CTLエピトープで再び刺激した。バーは,背景制御に正規化された個々のマウスの三重値の平均を示し,エラーバーは,平均からの標準偏差を示す。 E及びF:HHDIIマウスを,未処理(e)(n=3),又は,400μgのPC61抗体を腹腔内投与にて処理した(f)(n=2)。4日後,全てのマウスに100μgのZ12ペプチド及び100μgのZ48ペプチドをIFAで1:1に混合して(皮下)免疫した。最初のペプチド免疫の7日後,反復ペプチド免疫を施した。最後の免疫の14日後,脾細胞を採取し,インビトロIFNγ ELISPOTアッセイにより,1μg/mlのZ12ペプチド(ブラックバー)又は培地単体(オープンバー)で再び刺激した。バーは,三重値の平均を示し,エラーバーは,平均からの標準偏差を示す。 G:HHDIIマウスに,IFA(皮下)で1:1に混合した100μgのZ12ペプチドで免疫した。最初のペプチド免疫の7日後及び14日後に,反復ペプチド免疫を施した。最後の免疫の7日後,脾細胞を採取し,インビトロIFNγ elispotアッセイにより,ペプチド濃度の増加に対するエピトープ特異的反応の有無を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 H:HHDIIマウスに,0日目,7日目,14日目に,遺伝子銃により,ImmunoBody DNAコンストラクトDCIB71で免疫した。最後の免疫の7日後,脾細胞を採取し,インビトロIFNγ elispotアッセイにより,ペプチド濃度の増加に対するエピトープ特異的反応の有無を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【図50B】続き
【0118】
【図51】DCIB71の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のTie−2 Z12エピトープ(ILINSLPLV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0119】
【図52】DCIB72の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTie−2 Z12エピトープ(ILINSLPLV及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0120】
【図53】ImmunoBodyフレームワークに取り込まれるCTLエピトープは,処理され,発現され,インビボで免疫反応を引き起こす。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H1のgp100エピトープ又はCDR H2のTRP2エピトープ(それぞれ,IB17及びIB18)を含むImmunoBodyDNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,gp100ペプチド又はTRP2ペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,CDR H2のTRP2エピトープを含むImmunoBody重鎖での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:C57BI/6マウスに0日目,7日目,14日目に,CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1ヒトIgG1のHepB CD4(DCIB15),又は,CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びマウスIgG2a定常領域を有するCDR L1のHepB CD4エピトープ(DCIB53),を含むImmunoBodyDNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,HebBヘルパーペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 D:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB15又はDCIB53での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目に,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びCDR H3ヒトIgG1のgp100DR7(DCIB54),又は,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びマウスIgG2a定常領域を有するCDR H3のgp100DR7エピトープ(DCIB64),を含むImmunoBody DNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,gp100DR4ヘルパーペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 F:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB54又はDCIB64での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【0121】
【図54A】DCIB53の配列 発現ベクターpDCOrig moigG2a内のマウス重・軽完全長鎖のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【図54B】続き
【0122】
【図55A】DCIB64の配列 発現ベクターpDCOrig moigG2a内のマウス重・軽完全長鎖のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のgp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【図55B】続き
【0123】
【図56】免疫プロテアソーム処理は,ImmunoBodyコンストラクト内のエピトープからの反応の生成において重要である。 HHDIIマウスに0日目,7日目,14日目に,CDR H1のgp100209−217エピトープ(DCIB41),又は修飾型CDR H1のgp100210Mエピトープ(DCIB15),を含むImmunoBodyコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100209−217ペプチド又はgp100210Mペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。
【0124】
【図57】異なる免疫方法は,ImmunoBodyワクチンからの免疫反応を引き起こすのに効果的である。 A:C57BI/6マウスに0日目,7日目,14日目に,遺伝子銃により,エレクトロポレーションとともに又は伴わずに筋肉内で,又はエレクトロポレーションとともに又は伴わずに皮肉内で,ImmunoBody DNA(DCIB15)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,異なるルートによる免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【0125】
【図58】ImmunoBody免疫は,白斑様色素脱失を引き起こし,腫瘍攻撃を阻止する A:CDR H2のTRP2エピトープ及びCDR L1のHepB CD4エピトープ(DCIB18)を含むImmunoBody DNAによる免疫C57BI/6マウスは,免疫部位における発毛において色素脱失を示す。 B:免疫C57BI/6マウスを,2x104のB16F10 INFα細胞による3回目と4回目の静脈内免疫の間に攻撃した。肺の全身腫瘍組織量を腫瘍攻撃後49日目に評価した。全身腫瘍組織量は,全肺面積に対する平均腫瘍面積で表される。免疫マウスを,2x104のB16F10 INFα細胞による最後の皮下免疫後7日目に攻撃した。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが限度を超えたらマウスを安楽死させた。 C:腫瘍注入後46日目に評価した腫瘍の大きさ D:生存
【0126】
【図59】ImmunoBody免疫は,腫瘍の成長を著しく遅らせる。 A:C57B16マウスに,2x104のB16F10細胞を皮下注入した。腫瘍注入後4日目に,マウスにDCIB52 ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後11日目及び18日目に施した。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。経時的な腫瘍の体積をプロットした。 B:C57B16マウスに,2x104のB16F10 IFNα細胞を皮下注入した。腫瘍注入後14日目に,マウスにDCIB52 ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後21日目及び28日目に施した。11日目の免疫は,抗CTLA−4抗体の腹腔内注入と適宜組み合わせて行った。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。腫瘍移植後47日目における腫瘍の体積が示されている。 C:C57B16マウスに,2x104のB16F10細胞を皮下にて,及び抗CD25抗体を腹腔内にて,適宜注入した。腫瘍注入後4日目に,マウスにDCIB52 ImmunoBody DNA又は対照ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後11日目及び18日目に施した。11日目の免疫は,抗CTLA−4抗体の腹腔内注入と適宜組み合わせて行った。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。経時的な腫瘍の体積をプロットした。
【0127】
【図60】DCIB68の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1及びL3のHLA−DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3及びL1のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0128】
【図61】免疫反応は,異なるベクターバックボーンから発現されたImmunoBodyコンストラクトから生成されうる。 C57BI/6マウスに,0日目,7日目,14日目に,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びpVaxベクター(VaxDCIB54,C1−3)の等価コンストラクトであるCDR H3ヒトIgG1(DCIB54,B1−3)のgp100DR7エピトープ,を含むImmunoBody DNAコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【発明を実施するための形態】
【0129】
[実施例]
[方法]
DNAベクターの生成
ベクターpSVgptHuigG1及びpSVhygHuCk(Biovation Ltd)内のSC100クローンVHd VKb(WO01/88138)の脱免疫されたマウスの重・軽可変領域をPCRによって増幅した。VH及びVL領域PCR生成物を,HindIII/AfeI及びBamHI/BsiWI部位を用いてヒトIgG1及びカッパ定常領域のフレームにクローニングし,単一鎖コンストラクトpOrigHIB及びpOrigLIB(図1及び図2を参照)を生成した。完全長キメラ重鎖及びカッパ鎖の配列をジデオキシ鎖終結法によって確認した(Sanger et al, Proceedings of the National Academy of Sciences of the United States of America 1977;74: 5463−7)。キメラ重・鎖鎖のDNA及び翻訳されたタンパク質配列を,それぞれ,図3及び図4に示す。相補性決定領域(CDR)の位置が示されている。
【0130】
6つのアミノ酸を保持する重CDR2領域を除いて,重・軽鎖のCDRを完全に取り除き,独特な制限酵素部位と交換した。これは,制限酵素部位の生成を可能にする除去用配列の片側の領域を入念に検討することによって達成した。これらの独特な制限部位を使用してDNAを開き,抗原エピトープをコードするオリゴヌクレオチドを挿入することができる。制限部位を生成し損なったほとんどのフレームワーク配列は,エピトープにプライマーを含むことで置換され,翻訳中にアミノ酸が保持されること,及び配列がフレーム中にとどまることを確実にする。表1は,すべてのCDRの選択された酵素部位及びエピトープオリゴヌクレオチド配列を記載している。
【0131】
CDR領域を取り除き,図5に示すように重複伸長PCRによって独特な制限部位を置換した。重可変領域の場合,オリゴヌクレオチドH1,H2,及びH3(表2を参照)を3つのCDRの各々を置換するように設計した。各特異的プライマーは,酵素部位の両側が取り込まれる10〜20bpの配列を含む。ヒトIgG1定常領域に結合する一般的なリバースプライマーhuHeClonR(表2を参照)と併用して,一回目のPCRは,1μlの鋳型プラスミドpOrigHIB,2μlのdNTPS(2.5mM),5μlの10 x taqポリメラーゼ緩衝剤,1μlのフォワード・リバースプライマー(25pmols),滅菌蒸留水で最終体積が50μlとなるように作り上げた5ユニットのtaqポリメラーゼ(New England Biolabs)から構成した。反応は,最初の変性を95℃で5分受け,その後,95℃で30秒が35サイクル,55℃で1分(アニーリング),72℃で1分(伸長)と続いた。最後のサイクルは,Techne PHC−1プログラム可能循環反応器を用いた10分の伸長を含んだ。同様に,軽可変領域の場合,オリゴヌクレオチドL1,L2,及びL3を3つのCDRの各々を置換するように設計した(表2を参照)。一回目のPCRは,ヒトカッパ鎖及び鋳型pOrigLIBの定常領域に結合するリバースプライマーhuLiClonR(表2を参照)を用いた以外は,上述のとおり構成した。
【0132】
(表1)CDR置換酵素及びエピトープオリゴヌクレオチド配列のリスト

【0133】
次に,1μlの結果として生じるPCR生成物を,上記のように構成したCMVフォワードプライマーとともに,リバースプライマーとして後続のPCRで使用した。450bpの増幅されたDNA断片を,TA TOPOベクターpCR2.1(Invitrogen)並びにCDRを欠くVH及びVL領域の増幅及び制限部位の置換を確認するよう配列したクローンへ直接クローニングした。
【0134】
可変重・軽内のCDRを対応酵素H1,H2,H3,L1,L2,及びL3単体,組み合わせ,及び全体と置換した(図6及び図7)。次に,HindIII/AfeI及びBamHI/BsiWIを用いて,親の野生型脱免疫SC100 VH及びVL領域の直接置換により,異なるバージョンをpOrigHIB及びpOrigLIBに挿入した。これにより,単一又は複数のエピトープ(同じ又は異なる抗原からのもの)を含む分子の生成が可能になった。
【0135】
(表2)プライマー
【0136】
抗原エピトープの一本鎖ベクターのCDR部位への挿入
多数のCD8 CTL及びCD4ヘルパーエピトープが表3にリストアップされている。もっとも,任意のエピトープを一本鎖ベクター内の任意の部位に容易に挿入することができるが。例えば,TRP2エピトープのpOrigHIBのH2部位への挿入を以下のように行った。
【0137】
相補オリゴヌクレオチドを,翻訳中エピトープを発現するヌクレオチド配列をコードするように設計した。エピトープをコードするDNA配列の横には,対応CDRヌクレオチドが配置され,翻訳中アミノ酸が保持されること,及び配列がフレームにとどまることを確実にする(表1を参照)プライマーは,合成(MWG)及び5’末端リン酸化のために送られる。
【0138】
【0139】
相補オリゴヌクレオチドを滅菌再蒸留水で最終濃度1mg/mlまで再懸濁し,10μlの各プライマーとの反応を起こすことにより,TE緩衝剤とともに最終体積が50μlになるまでアニールした。反応は,95℃で5分(0.1℃/秒),72℃で20分(0.1℃/秒),55℃で20分,と繰り返し,その後4℃で保持した。
【0140】
H2部位への挿入のため,ベクターpOrigHIB H2及び/又はpOrigHIB H1H2をMscl制限消化を引き起こすことにより線状化し,37℃で一晩培養した。該消化を1.5%のアガロースゲルで電気泳動し,切断ベクターをゲル抽出により精製した。線状化ベクターのセルフライゲーション(self ligation)を防止するために,仔牛小腸由来アルカリホスファターゼ(CIAP)5ユニット及び滅菌蒸留水で最終体積100μlに作製した10 x NEB Buffer 3により処理及び37℃で一晩培養することでベクターの5’末端のリン酸基を除去した。脱リン酸化ベクターを精製し,。ユニバーサルプライマーCMVフォワードを使用して単一ベクター内に配列することにより,エピトープの挿入を確認した。
【0141】
(表3)CTL及びヘルパーエピトープ
【0142】
二重発現ベクターpDCOrigへの移転
すべてのエピトープが単一ベクター内のVH及びVL部位に取り込まれると,各々のヒト定常領域のフレームのHindIII/AfeIを用いて二重発現ベクターpDCOrigに移転される。ImmunoBody二重発現ベクターpDCOrigを生成するために,CMVプロモーターに隣接位置にある平滑末端制限エンドヌクレアーゼNrulを用いて線状化した。pOrigLIBを平滑末端Nrul及びHpalエンドヌクレアーゼにより消化して,CMVプロモーター,脱免疫ヒトカッパ鎖及びBGH polyAシグナルからなる全軽鎖発現カセットを摘出した。ゲル電気泳動,並びに線状化ベクターpOrigHIB及び軽鎖発現カセットの単離及びゲル抽出の後に,ベクターを脱リン酸化し,軽鎖発現カセットを結合してコンストラクトpDCOrigを形成した(図8)。pDCOrig内の軽鎖カセットの配向を制限分析により確認した。
【0143】
pDCOrigは,同じコンストラクト内に結合した重・軽鎖両方の遺伝子コード配列を含み,イントロン配列及び2つのベクター系を除去する。発現は,高レベルCMV前初期プロモーターその他のDNA制御要素(ウシ成長ホルモン)により促進される。選択マーカーzeocinもまた,発現及び生成効率を最大化するために含まれている。該ベクターの慎重な設計は,可変領域及び定常領域の接点における独特の制限酵素を保持し,可変領域の異なる組み合わせを作る手早く簡単な方法を提供する(エピトープ挿入。図8を参照のこと)。
【0144】
(表4)pDCOrigコンストラクト
【0145】
pDCOrig IB15 CH1ストップの生成
Quik change site directed mutagenesis kit (Stratagene)並びに相補オリゴヌクレオチドorigstophuHeCH1 Forward及びOrigstophuHeCh1リバースプライマー(表2を参照のこと)を製造者の指示する通りに用いて,ストップコンドンを,コンストラクトpDCOrig IB15内のヒトIgG1定常領域のCH1ドメインの後に取り込んだ。ストップコンドンの取り込みは,DNAシーケンシングにより確認した(図9)。
【0146】
pDCOrig IB15からのリーダー配列の除去
ベクターpDCOrig IB15の重・軽鎖のリーダー配列を除去するために,鋳型pDCOrig IB15を,リバースプライマーhuHeClonR及びhiLiClonRそれぞれと結合したフォワードプライマーpOrig軽ノーリーダー及びpOrig重ノーリーダーとともに使用してPCRを引き起こした。増幅断片は,ベクターpCR2.1(Invitrogen)に結合したTA TOPO及びシーケンシングにより確認したクローンであった。リーダーを欠くIB15 VH及びVL領域の両方を,HindIII/Afel及びBamHI/BsiWI部位を用いてpDCOrig IB15にクローニングした。VH及びVL領域のDNA配列及び翻訳を図10及び図11それぞれに示す。
【0147】
ImmunoBody二重発現ベクターpDCOrigのヒトIgG2及びIgG3アイソタイプの設計
AfeI及びEcoRVそれぞれを鋳型pOTBhuigG3(イメージクローン4566267MGC45809)とともに取り込むhuigg3フォワード及びリバースプライマー(表2)を用いてPCRにより,ヒトIgG3定常領域を増幅した。同様に,鋳型pTOB7 huiG2(イメージクローン6281452MGC71314)とともにigG2フォワード及びリバースプライマー(表2)を用いて,ヒトIgG2定常領域を増幅した。
【0148】
両断片は,pCR2.1に結合したTOPO及び確認した配列であった(表12及び表13)。コンストラクトpDCOrigIB15内のhuigG1定常領域を,AfeI及びSapI部位を用いて重可変のフレームにクローニングされたhuigG2及びhuigG3の両方と効果的に置換し,pDCOrigIb15 huigG2及びpDCOrigIB15 huigG3(図14)を生成した。両ベクターは,可変・定常領域の接点に同じ独特の制限部位を保持する。これにより,すべてのヒトアイソトープ単一及び二重鎖ImmunoBodyベクター間の可変領域の容易な交換が可能となる。
【0149】
ヒトIgG1 Fcγ及びヒトIgG2レセプター結合ドメインの突然変異
CH2ドメイン内のhuigG1結合モチーフのアミノ酸E233 L234 L235をhuig2のP233 V234 A235と置換するために,突然変異を組み込んだPCRによって短い部分を再増幅した。置換及び構成的制限部位AhdIを含むリバースプライマーhuigG1 PVA RevをフォーワードプライマーHIBF(表2)及び鋳型pDCOrig IB15とともに利用した。結果として生じる断片はベクターpCR2.1(Invitrogen)に結合した。配列確認後,プラスミドpDCOrig IB15 huigG1(図15)の単一カッターAgeI/AhdI部位に挿入することにより,野生型配列を突然変異を含む部分と効果的に置換した。
【0150】
コンストラクトpDCOrig IB15 huigG2のhuigG2定常領域内のアミノ酸P233 V234 A235もまた,huigG1結合モチーフELLGと置換した。前述同様に,置換及び構成的制限部位AhdIを含むリバースプライマーhuigG2 ELLG Rev(表2)をフォーワードプライマーHIBF(表2)及び鋳型pDCOrig IB15ヒトIgG2とともに利用した。断片は,ベクターpCR2.1に結合したTA TOPOであった。配列確認後,プラスミドpDCOrig IB15 huigG2(図16)のAgeI/AhdI部位を用いて,野生型配列をhuigG1結合モチーフを含む部分と置換した。
【0151】
pDCOrigマウスIgG2aプラスミドDCIB53及びDCIB63の生成
二重発現ベクターpDCOrigのマウスIgG2a型を生成するために,ハイブリドーマ細胞株337から単離したtotal RNAからcDNAを合成した。マウスIgG2a定常領域の増幅のために,制限部位AfeIを含むフォワードプライマーmigG2aC1AfeF2を,ストップコンドンの後のXbal部位を含むリバースプライマーmigG2aXbaRAとともに用いた。PCR断片は,ベクターpCR2.1に結合したTOPOであった。配列確認後,マウスIgG2a定常領域を摘出し,マウス重可変領域のフレームでヒトIgG1を効果的に置換するベクターpOrigHIBのAfeI/XbaI部位へクローニングした。BamHI及びXhoI部位を,翻訳中,マウスIgG2a定常領域のアミノ酸配列を変更することなく,連続してQuik chage site directed mutagenesis kit (Incitrogen)及び相補プライマーMoigG2BamHIFOR及びREV,MoigG2XholFOR及びREVそれぞれを用いて部位特異的突然変異誘発法により除去した。これは,ファンいつ鎖ImmunoBodyベクターpMoOrigHIB(図17a)を生成した。MoigG2a定常領域を含むpMogOrigHIBの部分は,AfeI及びSV40プロモーターに位置する単一カッターAvrIIを用いて,単一コンストラクトからマウス重可変領域のフレームの二重発現ベクターpDCOrig IB15へ移動し,ヒトカッパ領域をなお含む中間ベクターpDCOrigIB15MoigG2a hukappaを生成した。
【0152】
マウスカッパ領域の増幅のために,ストップコンドンの後のXhoI部位を取り込むBsiWI部位及びMoLCXhoIを含むプライマーMoLC1BsiF1とともに,cDNAを鋳型として用いた。増幅断片は,前述同様に,ベクターpCR2.1にクローニングしたTOPOであった。マウスカッパ領域を摘出し,BsiWI/XhoIを用いてヒトカッパ領域を置換するImmunoBodyベクターpDCOrigLIB L1及びpDCOrigLIB hepB help/L1へ結合し,中間ベクターpMoLIBL1Bsi及びpMoLIB HepB help/L1Bsiを生成した。Immnobodyシステムは,可変領域及び定常領域の接点における独特の制限部位を用いた可変領域の移動を伴う。一方,マウス重可変及びmoigG2a定常間の接点は,AfeI部位(すべてのヒトImunoBodyベクターに存在)に適応することができ,翻訳中アミノ酸配列を変更しない。マウス可変及びカッパ間の領域は疑わしい。この接点における配列を分析すると,アミノ酸配列を変更しない独特の制限部位を全く取り込むことができない。この接点のBsiWI部位を除去して野生型配列に戻した。これは,全マウス完全長鎖を重複PCRによって増幅することにより実現した。最初のPCRは,接点及び隣接領域に野生型配列を含むフォワードプライマーMoKappaSDMforを用いて構成し,BsiWI,BGHリバースプライマー,及び鋳型としての中間軽鎖ベクターpMOLIB1Bsi及びpMoLIB hepB help/L1 Bsi,のそれぞれを除去した。一回目のPCRのおよそ430bpの増幅断片をBamHI部位を含むフォワードプライマーImmunoLikozForとともにリバースプライマーとして用いた。増幅した完全長マウスカッパ鎖は,pCR2.1に結合したTOPO及び確認した配列であった。pCR2.1のL1部位にhepB helpを含む完全長マウスカッパ鎖を摘出して,ヒトカッパ鎖を置換する中間二重発現ベクターpDCOrigIB15MoigG2aH hukappaのBamHI/XhoI部位にクローニングし,マウス二重発現ベクターpDCOrigIB GP200210m/H1 TRP2/H2 HepB help/L1 moigG2a(DCIB53,図17B及び図54)を生成した。
【0153】
同様に,L1部位を含む完全長マウスカッパ鎖を摘出して,ヒトカッパ鎖を置換する中間二重発現ベクターpDCOrigIB15MoigG2aH hukappaのBamHI/XhoI部位にクローニングし,空のL1部位を有するマウス二重発現ベクターpDCOrigIB15moIgG2aを生成した。野生型軽可変領域を有するコンストラクトを生成するために,相補5’リン酸化プライマーwtkappavarL1for及びrev(表2)をアニールして,上述したEcoRVによる線状化後にL1部位に挿入した。最後に,GP100DR7/H1 TRP2/H2及びGP100DR4/H3を含むDCIB54の重可変領域を,HindIII/AfeIを用いて移動し,pDCOrig GP100DR7/H1 TRP2/H2 GP100DR4/H3 moigG2a野生型カッパ(DCIB68,図17c及び図60)を生成した。
【0154】
調節DNAワクチン要件のためのImmunobody二重発現ベクターpDCOrigからの真核SV40プロモーターの除去
Nhe1部位を取り込むフォワードプライマーSV40PremFOR及び鋳型pOrigHIBを有するSV40remREVリバースプライマー(表2)を用いて,EM7細菌プロモーター及びzeocin遺伝子を増幅した。結果として生じる511bpPCR断片は,結合しシーケンシングにより確認したpCR2.1 TOPOであった。EM7プロモーター及びzeocin遺伝子の一部をpCR2.1のNheI及びFseIを用いて摘出し,pOrigHIB H1に直接クローニングし,効果的にSV40プロモーターを除去した。NheI部位は,SV40プロモーターの前に存在し,一方,FseI認識配列は,ベクターのzeocin遺伝子内の単一カッターである。配列確認後,より大きな部分が単一ベクターからhuigG1,BGH polyA,EM7の末端をコードするpDCOrig IB68ベクター並びにSapI及びFseIで消化するzeocin遺伝子の一部に移動し,二重発現ベクターからSV40プロモーターを効果的に除去する。
【0155】
FDA規制準拠pVax1(Invitrogen)用pDCOrig骨格の変更
Immunobody完全長ヒトIgG1重鎖をHindIII及びXbalを用いてコンストラクトDCIB54から摘出し,ベクターpVax1のMCS内のこれらの部位(図18A)に挿入した。二重鎖発現ベクターのpVax型を生成するために,CMVプロモーターの隣接位置にある平滑末端制限エンドヌクレアーゼNrulを用いてpVaxIB54HIBを線状化した。平滑末端Nrul及びHpalエンドヌクレアーゼによりpOrigLIB(図18B)を消化し,CMVプロモーター,Immunobodyヒトカッパ鎖,及びBGH polyAシグナルからなる全軽鎖発現カセットを摘出した。ゲル電気泳動,並びに線状化ベクターpVaxIB54HIB及び軽鎖発現カセットの単離及びゲル抽出の後に,ベクターはリン酸化し,軽鎖発現カセットは結合してコンストラクトpVaxDCIB54(図18C)を形成した。pVaxDCIB54内の軽鎖カセットの配向は,制限分析により確認した。pVaxDCIB54は可変/定常領域の接点に同じ独特の制限部位を保持し,全てのヒトアイソタイプ単一及び二重鎖Immunobodyベクター間の可変領域の容易な交換を可能にする。例えば,pVaxDCIB68(図60)を生成するために,Gp100DR4/L1及びGp100DR7/L3を含むマウス軽可変領域をBamHI/BsiWIを用いてDCIB68から摘出し,pVaxDCIB54にクローニングし,軽野生型可変領域を効果的に置換した。
【0156】
pOrigマウスTRP2及びpCDNA3 Gp100の生成
pOrigマウスTRP2を生成するために,細胞株B16F10から単離したtotal RNA5μgから合成したcDNAを,プライマーマウスTRP2フォワード及びリバース(表2)をHindIII又はEcoRV部位それぞれとともに用いて,完全長マウスチロシナーゼ関連タンパク質2の増幅用鋳型として用いた。完全長TRP2は,ベクターpOrigHIBのHindIII/EcoRV複数クローニング部位に結合した。完全長マウスGP100もまた,EcoRV及びXhoI部位をそれぞれ含む設計されたマウスGP100フォワード及びリバースプライマーを用いてcDNAから増幅した。PCR生成物は,哺乳類発現ベクターpCDNA3(Invitrogen)のEcoRV/XhoI部位にクローニングした。両プラスミドは,制限分析により同定し,DNAシーケンシングにより確認した。
【0157】
サンドウィッチElisa
Falcon社製の96−ウェルフレキシブルプレートを50μlの抗ヒトIgG,Fc特異抗体(Sigma I2136)又はPBSで10μg/mlの抗ヒトカッパ軽鎖抗体(Dako A0191)で一晩4℃でコーティングした。プレートは,Skan Washer 400(Molecular Devices)を用いて200μl/well PBS−Tween 20(0.05%)で3回洗浄し,ウェルは,PBS(1% FSG/PBS)の1% Ffish skin gelatin(Sigma)で塞いだ。発現ImmnoBody又は精製ImmunoBodyタンパク質(50μl)を含む組織培養上清を三重にウェルに添加し,プレートを室温で1時間培養した。プレートを1%FSG/PBSで洗浄し,結合したImmunoBodyを50μl/wellのペルオキシダーゼ抱合抗ヒトIgG,Fc特異抗体(Sigma A0170)又は抗ヒトカッパ軽鎖抗体(Sigma A7164)を添加することにより検出し,1%FSG/PBSで1/2000に希釈し,室温で1時間培養した。プレートを1%FSG/PBSで洗浄し,TMB基質(R&D Systems)を50μl/wellで添加することにより育成した。吸光度をVERSAマックスマイクロプレートリーダー(Molecular Devices)により650nmで測定した。
【0158】
マウス及び免疫
英国内務省認定のプロジェクトライセンスの下,動物実験を行った。6週齢〜12週齢の雌雄のC57BI/6(Harlan)又はHLA−A2トランスジェニック(HHDII)(Pasteur Institute,Paris)を用いた。合成ペプチド(manufactured by John Keyte, Department of Biomedical Sciences, Nottingham University, UK)をフロイント不完全アジュバントにより乳状にし,皮下投与により注入した。製造者の使用説明書を用いて1.0μmの金粒子上にDNAをコーティングし,Helios Gene Gun(BioRad)により皮内投与した。各マウスは,1μgのDNA/免疫を受けた。極短電気パルスにより直後注射とともに裸のDNA溶液をも皮内又は筋肉内(10μg/免疫)投与した。マウスに0,1,2週間隔で免疫し,3週目に脾臓を除去した。免疫の4日前における400μgの抗CD25抗体(PC61)の腹腔内注入により,又は,2回目の免疫と同時の200μgの抗CTLA−4抗体の腹腔内注入により,インビボでの細胞サブセットの枯渇を行った。
【0159】
インビトロ再刺激
最後の免疫の5日後,10%FBS,2mMのグルタミン,20mMのHEPESバッファ,100ユニット/mlのペニシリン,100μG/ml-1のストレプトマイシン,及び10-5Mの2−メルカプトエタノールを有する2mlのRPMI−1644のペプチドパルスリポ多糖類(LPS)ブラストとともに24ウェルプレートにて37℃で共培養した。37℃で3日間,25μg/mlのLPS(Sigma)及び7μg/mlの硫酸デキストラン(Pharmacia,Milton Keynes,UK)により脾細胞(1.5x106細胞/ml)を活性化させることにより,LPSブラストを得た。使用前に,2x107のLPSブラストを100μg/mlの合成ペプチドで1時間培養した。6日目に51Cr放出アッセイにより培養を分析した。
【0160】
51Cr放出アッセイ
標的細胞を,100μg/mlのペプチドとともに又は含まずに,1.85Mbqのクロム酸(51Cr)ナトリウム(Amersham,Essex,UK)で1時間標識化した。培養後,RPMIで3回洗浄し,100μg/mlのペプチドとともにさらに1時間培養した。5x103target/wellの96ウェルV底プレートをセットし,最終容積200μlの異なる密度のエフェクター細胞で共培養した。37℃で4時間後,50μlの上清を各ウェルから除去し,Lumapplate(Packard,Rigaweg,the Netherlands)に移動させた。Topcount Microplate Scintillation Counter (Packard)でプレートを読み取った。特異的溶解度を以下の式を用いて計算した。
特異的溶解=100×[(実験的放出−自然放出)/(最大放出−自然放出)]
【0161】
インビトロElispotアッセイ
製造者の使用説明書(Mabtech,Sweden)に従って,マウスIFNγ捕捉・検出試薬を用いてElispotアッセイを行った。手短に説明すると,抗IFNγ抗体を96ウェルImmunobilin−Pプレートのウェル上にコーティングし,複製ウェルに5x105の脾細胞を播種した。(さまざまな濃度の)合成ペプチド又は5x104の標的メラノーマ細胞をこれらのウェルに添加し,37℃で40時間培養した。培養後,ビオチン化抗IFNγ抗体,並びにストレプトアビジンアルカリホスファターゼ(strepatavidin alkaline phosphatase)及び発色基質の成長により捕捉IFNγを検出した。自動プレートリーダー(CTL)を用いてスポットを分析し数えた。エフェクター機能対ペプチド濃度のグラフを用いて,機能活性を50%最大エフェクター機能を媒介する濃度として計算した。製造者の使用説明書に従って,CD8 Dynabeads(Dynal)を用いてCD8 T細胞の脾細胞集団からの枯渇を行い,次に,インビトロelispotアッセイに追加した。
【0162】
腫瘍の研究
C57BI/6マウスを治療群に無作為に抽出し,週間隔で5週間免疫した。3回目と4回目の免疫の間に,尾静脈に1x104のB16F10 IFNαメラノーマ細胞で静脈注入することによりマウスを攻撃した。静脈注入すると,B16F10細胞は,肺へ移動し,転移を形成した。腫瘍の増殖及び苦痛に関しマウスを観察した。腫瘍攻撃後49日目に,マウスを安楽死させ,転移の有無について肺を分析した。インビトロelispotアッセイによりエピトープ及び腫瘍特異的免疫反応の有無について脾臓を分析した。
【0163】
HHDIIマウスを週間隔で3週間免疫し,最後の免疫後7日間,2x104のB16F10 HHDメラノーマ細胞で右側腹部への皮下注射により攻撃した。腫瘍の増殖を3〜4日間隔で観察し,腫瘍の大きさをキャリパーを用いて測定した。
【0164】
実施例1 ImmunoBodyコンストラクトは低レベルの無傷抗体を生成する。
CDR H1及びCDR H2それぞれのgp100 エピトープIMDQVPFSV及びTRP2エピトープSVYDFFVWL及びCDR L1のHepB CD4エピトープTPPAYRPPNAPIL(DCIB15,図19)により,安定CHO−S細胞トランスフェクタントを生成した。
【0165】
これらのトランスフェクタントの上清を,サンドウィッチelisaにより,ImmunoBodyタンパク質の発現について分析した。プレートを,抗ヒトIgG Fc特異抗体及び添加上清でコーティングした。結合ImmunoBodyを重鎖を検出する抗ヒトFc特異HRP抗体を用いて検出した。対照と比較して,およそ1μg/mlの濃度の上清に重鎖を検出した(図20a)。タンパク質Aアフィニティカラムを用いて上清からImmunoBodyを精製し,ImmunoBodyno有無を分析した。ImmunoBodyの精製は,対照と比較して,予め予想したよりはるかに低い量のタンパク質を生じた(図20b)。このように低い量の無傷タンパク質が精製されうる可能性のため,ImmunoBodyコンストラクトを,サンドウィッチElisaにより,トランスフェクト細胞の上清における重鎖及び無傷抗体の両方の発現について分析した。CDR L1のHepB CD4エピトープ及びCDR H2のSIINKEFLエピトープ(DCIB24,図21)又はCDR H1及びCDR H2それぞれにgp100エピトープIMDQVPFSV及びTRP2エピトープSVYDFFVELをCDR L3のHepB CD4エピトープTPPAYRPPNAPIL(DCIB25,図22)とともに有するコンストラクトもまた検査した。プレートを抗ヒトIgG Fc特異抗体及び添加上清でコーティングした。結合ImmunoBodyを,重鎖を検出する抗ヒトFc特異HRP抗体,又は無傷ImmunoBodyを検出する抗ヒトカッパ鎖特異HRP抗体を用いて検出した。ImmunoBodyトランスフェクタントは,高レベルの重鎖分泌を示しているが,非常に低レベルの無傷ImmunoBodyを示している(図20c及び図20d)。
【0166】
このデータは,CD8及びCD4 T細胞エピトープの重・軽鎖可変領域への取り込みが,無傷抗体の形成を防止するImmunoBodyの全体構造を崩壊させることを示している。
【0167】
トランスフェ区とCHO−S細胞の上清の分析に関する追加データは,CDRH3又はCDRL3に取り込まれたCTLエピトープを有するコンストラクトのみが無傷抗体として分泌されるということを示している(図20e)。対照的に,CDRH1又はCDRH2内のエピトープの取り込みは重の分泌を可能にはするが,軽鎖に取り込まれるものはなく分泌されても無傷抗体の量は少ない。CDRL1内のエピトープの取り込みは,重鎖のCDRH3に取り込まれるエピトープしか存在しない場合,軽鎖の低レベルの分泌をもたらす。
【0168】
実施例2 ImmunoBodyフレームワークに取り込まれたCTLエピトープはインビボで免疫反応を引き起こすように処理され発現される
既刊のTRP2のCTLエピトープaa280−288(Boom et al, The Journal of Experimental Medicine 1997;185:453−9)をCDR L1のB型肝炎ユニバーサルCD4エピトープと一緒にImmunoBodyコンストラクトのCDR H2領域に改変した。C57BI/6マウスに遺伝子銃によりImmunoBody DNAを週間隔で三度経皮で免疫した。その後,脾細胞を,IFNγ elispotにより,TRP2特異的反応について分析した。ImmunoBody DNAを免疫したマウスは,対照と比べてかなりのTRP2ペプチド特異的反応を示す一方,HepB CD4ペプチドに特異的な反応は低レベルであった(図31a)。TRP2特異的反応の活性もまた,IFNγ elispotでペプチド滴定により研究した。5つの異なる実験で試験した15匹のマウスにおいて,反応の活性は,10−9〜10−11Mペプチドに及んでいる。代表例を図31bに示す。
【0169】
TRP2特異的反応がCD8 T細胞により媒介されることを確認するために,C57BI/6マウスに週間隔で3回ImmunoBody DNAを免疫した。最後の免疫の6日後,脾細胞を単離し,IFNγ elispotにより特異的反応についてインビトロで分析した。TRP2特異的反応がCD8 T細胞により媒介されるかどうかを判定するために,CD8 T細胞をelispotアッセイによる分析前に枯渇させた。CD8 T細胞の枯渇は,TRP2特異的反応の廃止につながった。しかしながら,CD8の枯渇は,HepB CD4ペプチド反応に影響を与えなかった。これは,CD4 T細胞により媒介される可能性が高いことを示している(図31c)。
【0170】
ImmunoBody DNA免疫により起こされる反応がインビトロで標的細胞を殺傷することができるかどうかを判定するために,TRP2ペプチドパルスLPSブラストによりインビトロで刺激を与え,B16F10メラノーマ細胞に対するクロム遊離アッセイにより分析した。ImmunoBody DNA免疫マウスの脾細胞は,H−2Kb分子(B16F10 siKb)を発現しないB16F10に比べ,低レベルの表面MHCクラスI発現を有するB16F10細胞,及び高い表面MHCクラスI発現を有するB16F10 IFNα細胞の両方のすぐれた溶解を示した。B16F10 siKb細胞株に対する殺傷の廃止は,殺傷がCD8に依存し,H−2Kbにより制限されることを示す(図31d)。
【0171】
これらの結果は,ImmunoBodyフレームワークのCDR H2領域に組み込まれるTRP2(SVYDFFVWL)CD8エピトープは,MHCクラスIにより媒介される高頻度の反応を引き起こすよう処理され発現されることを示す。HepB CD4エピトープもまた,MHCクラスIIとの関係で,DNA免疫の良好なCD4媒介反応を引き起こすように処理され発現される。
【0172】
TRP2エピトープ特異的反応もまた,同じ方法を用いてコンストラクトを含む他のTRO2エピトープから分析した。TRP2エピトープの重鎖内のCDRへの取り込みは,高頻度のペプチド特異的反応につながった(図31e)。対照的に,CTLエピトープの軽鎖への取り込みは,CTL頻度の有意な減少につながった(DCIB36)。TRP2エピトープ特異的反応の活性の分析は,該反応が重鎖内のエピトープから引き起こされた場合には高い活性を示すが,軽鎖からのエピトープの発現の場合にはこれが著しく低いことを明らかにしている(図31f)。すべてのコンストラクトについて観察された高頻度・高活性ヘルパー反応(図31g)は,重鎖の分泌は,CTL反応を刺激するには好都合であるが,ヘルパー反応には好都合でないことを示している。
【0173】
実施例3 ImmunoBody DNA免疫はペプチド免疫又は全抗原免疫よりも好ましい
ImmunoBody DNA免疫の効率を分析するために,これを,フロイント不完全アジュバントにおけるペプチドエピトープによる皮下免疫,又はTRP2抗原を発現するDNAによる免疫と比較した。
【0174】
C57BI/6マウスは,IFAのユニバーサルヘルパーエピトープに結合したTRP2エピトープからなるペプチド又はDNAによる免疫を週間隔で3回受けた。ImmunoBody免疫マウスに引き起こされたTRP2及びヘルパーペプチド特異的反応は,ペプチド免疫又は全TRP2抗原による免疫により起こされる反応よりも規模の点ではるかに上回っていた。これらのペプチド特異的反応の活性のさらなる分析は,ImmunoBody DNAによる免疫マウスにより引き起こされる反応は,ペプチド免疫個体の反応よりもログ活性が高いことを明らかにした(図32b)。その後,C57BI/6マウスに引き起こされる反応を,B16F10細胞株,及び陰性対照としてB16F10 siKb細胞株に対するインビトロでの細胞傷害能力について分析した。図32cは,ImmunoBody DNA免疫マウスは,H−2Kb拘束性のインビトロでの抗腫瘍活性を示し,ペプチド免疫マウス及び全抗原免疫マウスは,同じメラノーマ細胞株の殺傷に失敗することを示している。
【0175】
ImmunoBody免疫をDC+ペプチドによる免疫とも比較した。C57BI/6マウスは,DNA又はDC+ペプチドによる免疫を週間隔で3回受けた。TRP2ペプチド特異的反応は,同等の頻度であったが,ImmunoBody免疫マウスは,DC+ペプチド免疫マウスに比べ,高い活性反応を引き起こした(図32d)。これらの反応をインビトロでB16F10メラノーマ細胞を殺傷する能力において分析した場合もこのことが証明された。ImmunoBody免疫による引き起こされる反応は,DC+ペプチド免疫マウスの反応よりも目標比率に対して低いエフェクターでの高いB16F10メラノーマの殺傷を示した。これらはまた、H−2Kbのレベルを下げたB16F20 siKbメラノーマ株の高い特異的溶解を示した。
【0176】
H−2Kb拘束性オボアルブミンエピトープ,SIINFEKL,及びアンカー修飾HLA−A2拘束性gp100エピトープ,IMDQVPFSV(210M)を含むImmunoBodyコンストラクトを,C57BI/6又はHHDIIマウスそれぞれにおける対応するエピトープペプチド免疫と比較した。マウスは,IFAのDNA又はペプチドによる免疫を週間隔で3回受けた。最後の免疫後の反応の分析は,ImmunoBody DNA免疫マウスは,ペプチド免疫マウスに比べ,高頻度のペプチド特異的反応を引き起こすことを明らかにしている(図32f及び図32g)。これらの反応を,ペプチド滴定による活性についても分析した。ImmunoBody免疫は,ペプチド免疫より著しく高い活性反応を引き起こす(図32h及び図32i)。
【0177】
ImmunoBody DNAワクチンにより引き起こされるTRP2特異的反応の大きさは,合成ペプチド又は全TRP2抗原により引き起こされる反応をはるかに上回る。しかしながら,臨床試験による証拠は,高頻度の腫瘍特異的CD8 T細胞の存在が必ずしも腫瘍退縮にはつながらず,一般的にワクチンの試用において客観的な臨床的反応率は非常に低いことを示している(Rosenberg et al, J Immunol 2005;175:6169−76; Rosenberg et al, Nature Medicine 2004;10:909−15)。腫瘍特異的T細胞の機能活性やプライミング経路などの頻度以外の要因は、ワクチンの有効性を最大化する上で主要な決定要因であることが現在明らかになりつつある。多数のグループが、高活性CD8 T細胞が抗腫瘍活性を示すことを示している(Alexander− Miller, Immunologic research, 2005;31 : 13−24; Hodge et al, J Immunol 2005;174: 5994−6004; Valmori et al, J Immunol 2002;168: 4231 −40; Zeh et al, J Immunol 1999;162: 989−94; Alexander−Miller et al, Proceedings of the National Academy of Sciences of the United States of America 1996; 93: 4102−7)。我々の研究では,ImmunoBody誘発TRP2特異的反応の機能活性の分析は,合成ペプチドによる免疫に比べ,高活性反応を生成することができることを実証した。この高活性反応はまた,インビトロで腫瘍細胞を識別し殺傷する能力の増大と相互に関連があった。APCからの信号又は反応のプライミングの経路もまた,高活性免疫反応の誘発にとって極めて重要である(Oh et al, J Immunol 2003;170: 2523−30)。
【0178】
実施例4 複数のエピトープはCDR H2部位から処理されうる
複数のエピトープを免疫反応を引き起こすようにCDR H2から処理され発現されうることを実証するために,オボアルブミンのH−2Kb拘束性エピトープSIINFEKL(DCIB24:図21)及びH−2Kd拘束性B型肝炎エピトープIPQSLDSWWTSL(DCIB21,図33)を重可変領域のH2部位に改変した。これらのImmunoBodyコンストラクトはまた,軽可変領域にI−Ab拘束性(TPPAYRPPNAPIL)エピトープB型肝炎CD4エピトープ又はI−Ad拘束性インフルエンザ赤血球凝集素(FERFEIFPKE)エピトープを含んでいた。
【0179】
C57BI/6又はBalb/cマウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0180】
C57BI/6マウスは,高頻度SIINFEKL特異的反応を示す一方,低いヘルパーエピトープ特異的反応を示した(図34a)。Balb/cマウスもまた,ヘルパーエピトープと同程度の反応を有する高頻度B型肝炎エピトープ特異的CD8反応を引き起こした。
【0181】
このデータは,CDR H2からのCD8エピトープの処理及び発現は,特異的エピトープの配列又は長さに制限されないことを示している。
【0182】
実施例5 複数のCTLエピトープは可変領域から処理されうる
エピトープがCDR領域だけでなく可変領域から処理及び発現されうることを実証すべく,フレームワーク領域の一部の除去とともにエピトープをCDR H1部位に組み込んだ。
【0183】
エピトープの例は,改変されたHLA−A2拘束性のエピトープであるgp100のIMDQVPFSV(DCIB17,図35)及びTie−2のFLPATLTMV(DCIB26,図36)である。ImmunoBodyコンストラクトはまた,CDR L1部位にB型肝炎CD4エピトープを含んでいた。
【0184】
HLA−A2トランスジェニックマウス(HHDII)に,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0185】
HHDIIマウスは,HepB CD4エピトープに対する相当の反応とともに高頻度gp100 210M特異的反応を引き起こした(図37a)。コンストラクトを含むTie2エピトープで免疫したHHDIIマウスにおける反応は,高頻度のものではなかったが,Tie2エピトープ及びHepB CD4エピトープの両方に特異的なかなりの反応を引き起こした(図37b)。
【0186】
この例のデータは,可変領域に挿入されたエピトープは,インビボで免疫反応を起こすよう処理及び発現されうることを示している。これは1つのエピトープ配列に制限されないことも明らかである。
【0187】
実施例6 複数のCTL反応は同じImmunoBodyコンストラクト内の異なるエピトープから生成されうる
前述のHLA−A2拘束性gp100エピトープIMDQVPFSVを,同じコンストラクトのCDR H2部位のHLA−A2により同様に拘束されているTRP2エピトープSVYDFFVWLとともにCDR H1に改変した。HepB CD4エピトープはCDR L1部位に存在した(DCIB15,図19)。
【0188】
HHDIIマウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0189】
図38aは,gp100及びTRP2エピトープの両方に特異的な反応が生成される様子を示すが,TRP2特異的反応の頻度は少ない。HepB CD4ペプチドに対する反応もまた生成される。TRP2特異的反応の活性もまた,IFNγ elispotでペプチド摘定により研究した。反応の活性は,gp100エピトープの場合,10−10M〜10−11Mペプチドの範囲であり,TRP2エピトープの場合,10−9M〜10−10Mペプチドの範囲である。代表例を図38bに示す。反応がインビトロで標的細胞を殺傷することができるかどうかを判定するために,脾細胞をTRP2及びgp100ペプチドパルスLPSブラスにより6日間インビトロで刺激し,ペプチド標識T2細胞及びB16F10 HHDメラノーマ細胞に対してクロム遊離アッセイで分析した。対照B16F10メラノーマ株に比べ,B16F10 HHDメラノーマ株の特異的殺傷。反応はまた,対照と比較したペプチド標識T細胞の特異的溶解を実証した(図38c)。
【0190】
単一のImmunoBodyコンストラクトの2つのCD8エピトープを結合させることは,エピトープ間のある程度の免疫優勢をもたらすように思われる。免疫優勢エピトープは,MHCクラスIに対する最高のアフィニティを有するエピトープである。gp100及びTRP2 CD8エピトープの両方を含むコンストラクトにより免疫されたマウスとTRP2 CD8エピトープのみを含むコンストラクトで免疫されたマウスとを比較すると,TRP2反応の頻度は減少する(図38d)。
【0191】
このデータは,エピトープ特異的免疫反応が2つの異なるCD8エピトープに特異的な同じDNAコンストラクトから生成されうることを実証している。これらはまた,インビトロでの抗腫瘍活性を示す。しかしながら,サブドミナントエピトープに対する反応の頻度を抑制するある程度の免疫優勢が存在する。
【0192】
CDRH2(DCIB18)のTRP2エピトープ又はCDRH2(DCIB24)のSIINFEKLを含む個別のImmunoBodyコンストラクトを用いて同様の研究を行った。マウスに,DCIB18又はDCIB24を単体で,DCIB及びDCIB24を組み合わせて同じ部位に,又はDCIB18とDCIB24を同時にしかし別の部位にて,免疫した。免疫は週間隔で3回行い,DNAをエレクトロポレーションと併用して頚骨筋に筋肉内注入した。生成された免疫反応の分析は,マウスをDCIB18又はDCIB単体で免疫した場合に高頻度ペプチド特異的反応を引き起こすことができることを示している(図38e)。同じ部位においてこれらのコンストラクトによりマウスに免疫することは,TRP2ペプチド特異的反応の顕著な減少につながる。これは,SIINFEKLエピトープがTRP2エピトープに対して優勢であることを示す。TRP2特異的反応は,マウスに別々の部位(p=0.0026)のコンストラクトにより免疫すると回復することができる。このデータは,免疫優勢は,IB免疫により生成される免疫反応に実際影響を与えるが,これは空間的に離れた部位における免疫により解決されうることを示す。
【0193】
実施例7 無アンカー残留物改変はT細胞識別を強化しうる
前述の例は,改変gp100エピトープIMDQVPFSVが免疫優勢であり,HLA−A2に対する高いアフィニティを有することを示している(SYFPEITHIアルゴリズムを用いて予測したように。また,T2安定化アッセイで実証したように。表5)。野生型gp100エピトープITDQVPFSVは,免疫原性を持たないため,野生型エピトープと同様のHLA−A2結合活性を有しながらなおかつ免疫原性を高める無アンカー残留物で改変を行った。これらの改変エピトープをImmunoBodyコンストラクトのCDR H1部位に改変し,野生型エピトープとともに試験した(DCIB37,DCIB40,DCIB41,DCIB42,DCIB43,図39〜図43)。
【0194】
HHDIIマウスに,遺伝子銃により,ImmunoBody重鎖DNA単体で週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8反応の有無について分析した。HLA−A2(表5)に対するアフィニティを保持する野生型gp100エピトープへの2つの改変(F7L及びF7I,DCIB37,図39,DCIB40,図40)は,野生型エピトープに比べ,エピトープ特異的免疫反応を誘発する優れた能力を実証した(図44a)。
【0195】
(表5)
【0196】
実施例8 複数のCD4ヘルパー反応はインビボで免疫反応を起こすように処理及び発現されうる
CD4ヘルパーエピトープをインビボで免疫反応を起こすように処理及び発現されうるかどうかを分析するために,異なるエピトープをImmunoBodyコンストラクトのCDR L1部位に独立に改変した。これらは,インフルエンザ赤血球凝集素のI−Ad拘束性エピトープFERFEIFPKE(DCIB21,図33),HBcAgのI−Ab拘束性エピトープTPPAYRPPNAPIL(DCIB15,図19),及びgp100のHLA−DR4拘束性エピトープWNRQLYPEWTEAQRLD(DCIB35,図45)を含んでいた。
【0197】
Balb/c,C57BI/6又はHHDII及びDR4トランスジェニックマウスに,遺伝子銃により,ImmunoBody DNAで週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ eslispotによりエピトープ特異的CD4反応の有無について分析した。図46a,b,cは,3つのCD4ヘルパーエピトープのすべてがエピトープ特異的免疫反応をインビボで起こすようにCDR L1部位から処理及び発現されうることを示している。
【0198】
gp100HLA−DR4拘束性エピトープを異なるCDRからの処理及び発現についても試験した。エピトープをCDRL1(DCIB35,図45),DCRH3(DCIB54,図29),又はCDRL3(DCIB50,図47)に組み込むコンストラクトを使用して,HLA−DR4トランスジェニックマウスに週間隔で3回免疫した。図46dは,ヘルパーエピトープは高頻度ヘルパー反応を起こすように異なるCDRから効率よく処理されうることを示している。
【0199】
実施例9 CTL反応は分泌重鎖に部分的に依存するが,ヘルパー反応は分泌軽鎖を必要としない
古典的に,CD4 T細胞エピトープは,体外から獲得したタンパク質から処理され,CD8 T細胞エピトープは,体外で生成されたタンパク質から処理される。体外から獲得した抗原からのエピトープの交差提示がCD8 T細胞媒介反応を起こす証拠が現在存在する。このプライミング経路はまた,CD8 T細胞媒免疫反応の開発の点でより効率的なものであることが提案されている。最近,CD4媒介反応についても同様の発見があった。山のような証拠は,細胞内タンパク質由来のCD4 T細胞エピトープはMHCクラスIIとの関連で処理及び発現されうることを示している。
【0200】
分泌ImmunoBodyがCD8及びCD4 T細胞反応に必要であるかどうかを判定するために,CDR H1部位のHLA−A2拘束性gp100エピトープIMDQVPFSV及びCDR L1部位のI−Ab拘束性HepBヘルパーエピトープTOOAYRPPNAPILを含むImmunoBodyコンストラクトを重鎖又は軽鎖上にリーダー配列なしで作った(図10及び図11)。
【0201】
HHDIIマウスに,遺伝子銃により,ImmunoBody DNAにより週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4 T細胞反応の有無について分析した。反応をgp100特異的CD8反応について分析すると,ImmunoBodyコンストラクトの重鎖からのリーダー配列の除去は,エピトープ特異的反応の減少につながるが,CD44反応には影響を及ぼさないことが観察された(図48a)。重鎖からのリーダー配列の除去は,トランスフェクトCHO−S細胞による重鎖の分泌に影響を及ぼした(図48b)。軽鎖からのリーダー配列の除去,これによる軽鎖の分泌防止,は,エピトープ特異的CD8又はCD4反応に影響を与えないように思われる(図48c)。CD8反応は,重鎖上にリーダー配列が存在しない場合顕著に減少したが,CD4反応は,影響を受けなかった(図48c及び図48d)。
【0202】
このデータは,重鎖の分泌がCD8 T細胞反応の効率的誘発にとって重要であることを意味し,CD8エピトープは交差提示を受けていることを示している。第二に,それは,CD4エピトープは免疫反応を起こす細胞内ImmunoBodyに由来することを意味している。
【0203】
実施例10 タンパク質分泌の欠乏によるFcのない低下したCTL反応
この実験は,Fc領域の存在が効率的な免疫反応を達成する上で有用であるかどうかを観察するものである。転写及び翻訳を防止するストップコンドンをFcの前に組み込むことにより,CDR H2のH−2Kb拘束性TRP2エピトープSVYDFFVWL及びI−Ab拘束性HepB CD4エピトープTPPAYRPPNAPIL(DCIB15)を含むImmunoBodyコンストラクトからFc領域を除去した(図9)。
【0204】
C57BI/6マウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回経皮で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4 T細胞反応の有無について分析した。
【0205】
Fc領域を有さないImmunoBodyコンストラクトにより免疫したマウスは,Fc領域を有するコンストラクトに比べ,非常に低レベルの腫瘍細胞株B16F10の識別が可能な低レベルのTRP2ペプチド特異的反応を生成した(図49a)。多数の実験からのTRP2及びHepBヘルパーペプチド特異的反応の両方の分析は,Fc領域を有さないコンストラクトは著しく低いTRP2ペプチド特異的反応を生成することを示している(図49b)。しかしながら,HepBヘルパー反応は,Fc領域の除去により影響を受けない(図49c)。これは,ヘルパーが分泌されず,よって直接提示により機能している軽鎖においてヘルパーが最もよく機能することを示す我々の前述の結果と一致するものである。対照的に,CTL反応は,直接提示及び間接提示の両方により刺激され,後者はFc標的の恩恵を受けうる。あるいは,Fcストップコンストラクトは,低下した反応を説明しうる欠失重鎖の低い分泌につながる。したがって,TRP−2をコードするImmunoBodyは,IgG2(DCIB33)及びIgG3定常領域(DCIB65)により改変される。前者は,CD64に結合しないはずだが,CD32に結合しうる。また,マウスのFcレセプターIVにも結合しうる。ヒトIgG3は,CD32及びCD64の両方に結合しうる。両ImmunoBodyは,強力なCTL反応を刺激した(図49e)。これは,Fc標的が間接提示の強力な要因ではないことを示している。この問題をさらに検証するために,IgG1のFc標的ドメインを同等のIgG2ドメインと置換し,又はその逆を行った(DCIB66,67,図15及び図16)。両コンストラクトは,強力なCTL反応を刺激した(図49e)。これは,Fc結合に関連せずに可能にする重鎖のみを分泌するImmunoBodyワクチンのためでありうる。
【0206】
実施例11 ImmunoBody免疫は,免疫反応を強化し全抗原で観察された規制を克服する。それはまた,新しいT細胞エピトープの同定を可能にする
これは,ほとんどの自己抗原と対照的に,制御性エピトープを発現しない不活性担体であるT細胞エピトープをコードするヒト抗体により免疫することの第二の利点につながる。gp100エピトープ又はTRP−2エピトープを発現するImmunoBodyは,高頻度・高活性のT細胞反応(頻度1/103,活性10−10M)を刺激する一方,TRP−2のgp100全抗原は,低頻度かつ低活性でT細胞を刺激した(頻度1/105,活性10−7M)。CD25の枯渇は,抗原に対する反応を部分的に回復させたが,ImmunoBodyはなお100倍優れていた(図50a及び図50b)。
【0207】
同様に,Fcに結合するTie−2の最初の200アミノ酸をコードするDNAによる免疫は,上位10の予測エピトープに対する免疫反応を刺激しなかった。Tie−2の最初の196アミノ酸の配列をEpijen及びNetCTLオンライン予測アルゴリズムに入れた。これらの方法は両方ともHLA−A*0201結合アフィニティに加え,プロテアソーム切断及びTAP輸送を考慮に入れている。MHCpred及びSyfpeithiアルゴリズムをも予測MHC結合アフィニティのみを考慮に入れる古い予測アルゴリズムの例として用いた。全Tie−2分子は,最初の196アミノ酸に存在するエピトープに対して免疫優勢の影響を及ぼしうる追加のCTLエピトープを含みえた。したがって,全分子からの各予測エピトープのランクを得るために,Tie−2の全配列も同じアルゴリズムに入れた。ヒトとマウスで相同でないペプチドは考慮に入れなかった。複数の異なる予測アルゴリズムにより良好なCTLエピトープを発現すると確実に予測された残存ペプチドのうちの6つを選択した。これらのペプチドの各々に対して異なるアルゴリズムにより得られた相対スコアを,Z83(これまでに同定されたエピトープ)の結果とともに,表6にまとめている。
【0208】
トランスフェクトCHO−S細胞からの上清の分析に関する追加データは,CDRH3又はCDRL3に組み込まれるCTLエピトープを有するコンストラクトのみが無傷抗体として分泌されることを示している(図20e)。対照的に,CDRH1又はCDRH2内の任意のエピトープの組み込みは,軽鎖内に組み込まれるものがなく分泌されても重だが少量の無傷抗体の分泌を可能にした。任意の軽鎖のCDRL1内の任意のエピトープの組み込みは,重鎖のCDRH3に組み込まれるエピトープしか存在しない場合でも,軽鎖の低レベルの分泌につながった。
【0209】
T細胞レパートリーが任意のTie−2からの予測CTLエピトープを識別するHLA−A*0201トランスジェニックマウスに存在するかどうかを判定するために,動物にnative Tie2 C200hFc DNAコンストラクトで免疫し(Ramage et al, Int. J. Cancer 2004:110:245−250),脾細胞をELISPOTアッセイでペプチド特異的IFNγ反応についてスクリーニングした。別のグループのマウスには,DNA免疫の4日前に,前述同様に,PC61 mAbによる処理を行った後に,C200hFcにより免疫した。
【0210】
native C200hFc DNAにより免疫したマウスは,該動物が免疫の前にCD25+制御性T細胞を枯渇していたか否かにかかわらず,Z83を識別するIFNγ反応を開始しなかった。免疫前に制御性T細胞を枯渇していなかった動物から試験した任意の新しいペプチドに対する著しいIFNγ反応は見られなかったが,例外として,Z284は,平均69SFC/million脾細胞で1つの動物(M3)において反応を刺激しているように見えた(図50c及び図50d)。DNA免疫前に制御性T細胞を枯渇した動物からは,2/3の動物(M1及びM3)がZ282ペプチドによる再刺激に対するIFNγ反応を示し,平均値は,それぞれ,320及び94SFC/million脾細胞であった。M1はまた,Z285による再刺激に対する部分的反応を示し,平均が85SFC/million脾細胞であった。
【0211】
Tie−2からの予測CD8+エピトープのインビボスクリーニングの明らかに矛盾する結果は,免疫優勢の結果であろう。なぜなら,CD25+細胞がない場合native C200HFcにより免疫したマウスからのIFNγ反応は,1つの優勢ペプチドに偏るように思われるためである。他の潜在CD8+エピトープからの競争がない場合,Z282エピトープに反応することのできるT細胞レパートリーをさらに調査するために,HHDマウスのグループに,CD25+制御性T細胞の存在下又は非存在下で,IFAのZ282ペプチドにより免疫した。
【0212】
Z282を免疫したマウスはすべて,CD25+制御性T細胞の存在下で免疫された場合でも,ペプチド特異的IFNγ反応を開始した。非枯渇動物のマウス3は,最高の反応を開始し,平均値が215SFC/million脾細胞であった。枯渇動物からの最高の反応はマウス2から観察され,平均値が137SFC/million脾細胞であった(図50e及び図50f)。
【0213】
ペプチド免疫により誘発された反応は低頻度のままである。エピトープを全抗原により生成される制御作用から除去した場合に高頻度反応が生成されるかどうかを観察するために,z282(z12としても知られる)エピトープをL1のHepB CD4とともにImmunoBodyコンストラクトのH1部位に改変した(DCIB71,図51)。その後,HLA−A2トランスジェニックマウスに(遺伝子銃により)z12ペプチド又はImmunoBody DNAで週間隔で3回免疫し,その後,エピトープ特異的免疫反応について分析した。z12ペプチドにより免疫したすべてのマウスは,低頻度及び低活性エピトープ特異的反応を示す(図50g)。しかしながら,z12エピトープをImmunoBodyコンストラクトに改変すると,高頻度及び高活性反応がすべてのマウスに誘発される(図50h)。
【0214】
要約すると,免疫前にCD25細胞を枯渇させると,免疫反応がTie2エピトープの3/10にまで刺激された。同様に,これらのエピトープの1つがペプチドとして発現されると,弱い免疫反応が生成されえた。しかしながら,このエピトープがImmunoBodyコンストラクト内で発現されると,高頻度及び高活性のT細胞反応が生成された。これらの結果は,Tie−2の最初の200アミノ酸内にはCTL反応を抑制するTregエピトープが存在することを示している。これらのTreg又はそのエピトープを除去すると,ImmunoBody内での発現によりさらに強化されうる自己抗原に対する反応を発見することが可能である。
【0215】
(表6)予測されたTie−2からのHLA−A*0201拘束性CTLエピトープ
1:ペプチド名。
2:Tie−2分子内のアミノ酸残基開始位置。
3:ペプチド配列。
4:Epijenウェブサーバを用いた予測。スコアは,IC50nMの単位で与えられ,スコアが低いほど高アフィニティのペプチドであることを表す。
5:NetCTL 1.2ウェブサーバを用いた予測。スコアは,3つの個々の予測方法の加重和を表し,1に対するMHC結合の相対的重み付けを伴う。*は,周知のエピトープに対して得られるデータセットからのCTLエピトープのカットオフ値として認定される閾値0.75以上のスコアを示す。
6:SYFPEITHIプログラムを用いた予測。HLA−A*0201結合ペプチドの最大スコアは36である。
7:MHC及びTAPに対するペプチドアフィニティを予測するMHCPred相加法を用いた予測。スコアは再びIC50nMの単位で与えられ,スコアが低いほど高いアフィニティのペプチドであることを表す。推奨されるIC50値は,0.01〜5000nMである。
すべての予測方法において,ランク値は,196アミノ酸断片からエピトープが予測される順序を示し,括弧内の値は,全Tie−2分子からのランク予測を表す。インフルエンザA型ウイルスのマトリクスタンパク質由来の周知のZ18 CTLエピトープに対して得られる値が比較のために含まれている。
【0216】
実施例12 T細胞ヘルプの提供における異種Fcの役割及び抗原特異的T細胞ヘルプの要件
高活性T細胞反応の刺激は,通常,プライミング中T細胞ヘルプを必要とする。これは,軽鎖内にコードされるHep B外来ヘルパーエピトープによって提供されるであろうと従来考えられていた。実際,強力なヘルパー反応は,このエピトープに対して生成された。しかしながら,重鎖と軽鎖の両方の鎖が同じAPCにより生成される場合,Hep Bエピトープは直接提示のためのヘルプを提供することができたが,重鎖は分泌され,軽鎖は分泌されなかったため,間接提示重鎖のためのヘルプを提供しえたとは考えにくい。なぜなら,これは,同じ抗原提示細胞によって取り込まれるとは考えにくいからである。したがって,マウスに,重鎖のみをコードするDNAベクターにより免疫した。高頻度・高活性のCTL反応がなお生成された(図53a及び図53b)。これは,ヘルプを必要としないか,あるいは,マウスにおいて移出であるヒトFcが結合外来ヘルプを提供していることを意味する。したがって,マウスIgG2aコンストラクトを重鎖及び軽鎖について分析し(図49g),免疫反応の生成についてスクリーニングした(DCIB53,図54)。なお高頻度・高活性のT細胞反応を生成したが,ヒトコンストラクトと同程度には強力でなく,異種Fcは結合ヘルプを提供していた(図53c及び図53d)。その後,CTL生成のための結合ヘルプ,及び腫瘍環境内の炎症を刺激する抗原特異的T細胞ヘルプの両方を提供するようにHLA−DR4 gp100エピトープをマウスIgG2aコンストラクト(DCIB64,図55)に組み込んだ。これらのコンストラクトは,高頻度・高活性のCTL及びヘルパー反応を刺激する(図53e及び図53f)。同じエピトープを発現するgIgG1コンストラクトをヒト患者に用いることができる。
【0217】
実施例13 免疫プロテアソーム処理はImmunoBodyコンストラクト内のエピトープからの反応の生成において重要である
免疫プロテアソームは,異なるパターンの分裂を有するため,自己抗原から生成されるエピトープの配列を変更する能力を有することが示唆されている。ある場合には,新しいエピトープは,免疫プロテアソームの上方調節により生成され,またある場合には,エピトープは破壊される。免疫プロテアソームは,メラノーマ抗原,すなわち,MelanA/MART−1,gp100209−217,及びTyrosinase369−377に由来する複数のエピトープを生成することができないという証拠がある(Chapiro et al 2006. J Immunol; 176: 1053−61)。Chapiro及び同僚は,gp100エピトープを処理及び発現する能力は,免疫プロテアソームの上方調節に関連があることを示唆した。成熟DCは,免疫反応のプライミングに関与すると考えられており,また,免疫プロテアソームを構成的に発現することが知られている(Macagno et al, 2001. Eur J Immunol; 31: 3271−80)。そこで,gp100209−217エピトープをImmunoBodyコンストラクトのCDRH1部位に改変し,HLA−A2トランスジェニックマウスにおけるペプチド特異的免疫反応を誘発する能力を試験した。このコンストラクトからはペプチド特異的反応は観察されなかった(図56)。しかしながら,該エピトープを210(210M)の位置においてトレオニンの代わりにメチオニンを有するように改変すると,これは,免疫プロテアソームによる分裂を防止し,エピトープ特異的反応が観察された(図56)。
【0218】
VEGFR2(aa773−781 VIAMFFWLL)由来のHLA−A2拘束性ペプチド及び2つの改変hTERTペプチド(aa572−580 YLFFYRKSV及びaa988−997 YLQVNSLQTV)もまたImmunoBodyコンストラクトからの反応の生成について試験した。これらのエピトープは最初コンピュータによるエピトープ予測及びペプチド免疫により発見されたため,プロテアソーム処理の要件を否定する。しかしながら,これらは,宿主内皮/腫瘍細胞の表面にて発現され,このことは,これらが構成的プロテアソームにより全抗原から処理されることを示唆している。これらのエピトープのいずれもが,ImmunoBodyコンストラクトに改変された場合に反応を生成しなかったが,このことは,プロテアソームによる処理が免疫反応の効率的な生成に必要とされうることを示唆している。
【0219】
実施例14 異なる免疫方法は,ImmunoBodyワクチンからの免疫反応を引き起こすのに効率的である
ImmunoBodyワクチンは,遺伝子銃により投与した場合に,高頻度・高活性のCD8及びCD4反応を引き起こすのに効率的であることが示されている。その後,ImmunoBodyワクチンを他の免疫方法を用いてT細胞反応の生成について試験した。
【0220】
C57BI/6マウスに,CDRH2のTRP2エピトープを含むImmunoBody DNAにより皮内又は筋肉内経路で免疫した。免疫は,エレクトロポレーションと組み合わせて又は組み合わせずに,週間隔で3回行った。
【0221】
遺伝子銃で免疫したマウスは,高頻度TRP2ペプチド特異的反応を示す。これらは,エレクトロポレーションと組み合わせて皮内又は筋肉内経路で免疫したマウスと同程度である。エレクトロポレーションを伴わずに皮内又は筋肉内経路で免疫した場合は,低頻度のTRP2ペプチド特異的反応を生成した(図57a)。すべてのTRP2ペプチド特異的反応は,ペプチド摘定により測定した(図57b)。
【0222】
実施例15 ImmunoBody免疫は白斑様色素脱失を誘発し腫瘍攻撃から保護する
ImmunoBody DNAにより免疫したマウスは,高転移性かつ低免疫原性の腫瘍細胞株B16F10に対する細胞毒性能力のある免疫反応を生成するため,ワクチンをインビボでの予防効果について試験した。
【0223】
マウスに遺伝子銃で週間隔で5回腹部の毛を剃った皮膚にIB DNA(DCIB18,図30)により免疫した。免疫スケジュールの途中,マウスに,肺に転移性腫瘍を形成するIFNαを発現する1x104のB16F10細胞により静脈内注入した。最後の免疫後,毛を元の状態に生えさせると,ImmunoBody DNAにより免疫したマウスの免疫に部位に白い毛が生えてくるのが観察された(図58a)。腫瘍細胞注入の7週間後,マウスを犠牲にし,内部及び外部の肺転移数を分析した。ImmunoBody DNAを免疫したマウスは,非処理の対照マウスに比べ,肺転移の数に著しい減少が見られた。
【0224】
また,マウスに遺伝子銃で週間隔で3回IB DNA(DCIB18)により免疫した。最後の免疫の7日後,マウスを皮下でIFNαを発現する2x104のB16F10細胞で攻撃した。マウスを腫瘍の成長及び生存について監視した。腫瘍が内務省の規制による最大限度に達するとマウスを安楽死させた。ImmunoBody DNA免疫マウスは,著しく低い腫瘍の成長及び長期生存を示した。
【0225】
TRP2特異的反応は,CD8+細胞が反応を無効にするので,CD8に媒介される。CD8 T細胞は,抗腫瘍免疫において主役であると見なされており,我々の結果は,ImmunoBody DNA免疫がマウスモデルにおいてインビボ抗腫瘍免疫を引き起こすことを示している。病気の兆候がないすべての免疫マウスは,免疫の部位において毛皮の白斑様色素脱失を示した。これまで白斑はしばしばマウスの腫瘍保護と関連があり,転移性メラノーマの患者における良好なIL−2免疫療法と大いに相関性がある(Overwijk et al, Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 2982−7; Lane et al, Cancer Research 2004;64: 1509−14; Steitz et al, Cancer Immunol lmmunother 2006; 55: 246−53; Rosenberg & White, J lmmunother Emphasis Tumor Immunol 1996;19: 81 −4)。
【0226】
実施例16 ImmunoBody免疫は腫瘍の成長を著しく遅らせる
ImmunoBody免疫は,腫瘍攻撃を有意に阻止することがこれまで示されている。その後,ワクチンを治療環境における有効性について試験した。
【0227】
C57BI/6マウスに2x104のB16F10腫瘍細胞で皮下注入した。注入の4日後,マウスに,CDRH2のTRP2エピトープを含むImmunoBody DNA又は対照ImmunoBody DNAで免疫した。腫瘍注入後11日目及び18日目に反復免疫を行った。腫瘍の成長を3〜4日間隔で監視した。ImmunoBody免疫マウスは,対照免疫マウスに比べ,攻撃的B16F10メラノーマの成長に有意な遅延を示した。
【0228】
また,攻撃性の弱いB16F10 IFNα腫瘍株を用いて同様の研究を行った。C57BI/6マウスに,2x104の腫瘍細胞を皮下で注入し,14日目にImuunoBody DNAまたは対照DNAで免疫した。腫瘍注入後21日目及び28日目に反復免疫を行った。ImmunoBody免疫マウスは,腫瘍注入後47日目に対照免疫マウスに比べ優位に低い腫瘍の成長を示した。以前のデータは,T制御性細胞の枯渇が免疫反応の生成を強化し,よって,抗腫瘍の研究が行われることを示唆していた。この研究において,マウスに2x104のB16F10腫瘍細胞を皮下で注入し,4日目,11日目,18日目にImmunoBody DNA又は対照DNAで免疫した。0日目に抗CD25抗体(PC61)の注入によりマウスからT制御性細胞を枯渇させた。2回目の免疫と同時に,マウスには抗CTLA−4抗体も注入した。なぜなら,CTLA−4の遮断もまた制御性T細胞の抑制に有用であることが分かったためである。腫瘍の成長を監視した。ImmunoBody免疫は,腫瘍の成長(p=0.0188)を有意に遅らせたが、これは、抗CD25及び抗CTLA−4抗体(p=0.001)による処理によりさらに強化された(図59c)。抗CD25抗体による処理は、ImmunoBody免疫マウスで観察された腫瘍の成長を有意に遅らせないようであった。
【0229】
実施例17 免疫反応は異なるベクターバックボーンから発現されるImmunoBodyコンストラクトから生成されうる
ImmunoBodyコンストラクトが異なるベクターバックボーンから発現される場合の反応を分析した。CDRH1のgp100DR7エピトープ,CDRH2のTRP2エピトープ,及びCDRH3のgp100DR4を野生型軽鎖とともに含むImmunoBodyコンストラクトを二重発現ベクターDCOrig及びDCVaxに改変した(DCIB54,図18及び図29)。HLA−DR4トランスジェニックマウスに遺伝子銃により週間隔で3回免疫し,反応をIFNγ elispotアッセイによりインビトロで分析した。
【0230】
CDRH1及びCDRL3のgp100DR7,CDRH2のTRP22エピトープ,CDRH3及びCDRL1のgp100DR4エピトープを含むImmunoBodyコンストラクトを用いて同様の実験を行った(DCIB68,図60)。このコンストラクトを,DCOrig,及びSV40プロモーター及びDCVaxベクターバックボーンを欠くDCOrigの両方に改変した。
【0231】
Origベクター(B1−3)のImmunoBodyコンストラクトで免疫したマウスは,pVaxベクター(C1−3)のImmunoBodyコンストラクトに比べ,同様の頻度のエピトープ反応を示した(図61)。
【0232】
要約すると,ImmunoBody技術は,非免疫原性の抗体フレームワークから高頻度・高活性のCD8・CD4免疫反応を引き起こす優れた能力を有し,インビボでの腫瘍の成長を効果的に防ぐことができる。それは,6つの異なる抗原を同時に標的にする能力を有し,また,全抗原免疫原を使用する場合にしばしば発生する制御性T細胞の問題を回避する能力を有する。この技術は,ワクチン接種への新しいアプローチを提供し,他の多くの癌型及び微生物関連疾患用の多価ワクチンとして用いられるImmunoBodyシステムの可能性を示す。
【技術分野】
【0001】
本発明は,核酸,及び核酸のワクチンとしての利用に関し,該核酸は,免疫反応が引き起こされうるT細胞エピトープをコードする。該ワクチンは,腫瘍の治療に利用することができる。
【背景技術】
【0002】
癌ワクチン及び慢性ウイルス感染の分野において,頻度以外の要因,例えば,腫瘍特異的T細胞の機能的活性やプライミングの経路,がワクチンの有効性を最大化する重要な決定要因であることが近年明らかになりつつある。高活性のCD8+ T細胞が優れた抗腫瘍活性を示すことを多くのグループが示している(Alexander−Miller, Immunologic Research, 2005;31: 13−24, Hodge et al, J Immunol 2005;174: 5994−6004, Valmori et al, J Immunol 2002;168: 4231−40, Zeh et al, J Immunol 1999;162: 989−94)。高活性のT細胞が患者の腫瘍退縮において重要な役割を果たすことが示唆されている。これは,高活性の抗原特異的腫瘍浸潤リンパ球(TIL)が腫瘍が劇的に退縮した患者において検出された一研究に例示されている(Khong & Rosenberg, J Immunol 2002;168: 951−6)。インビトロ刺激による自己腫瘍特異的T細胞の養子免疫伝達が成功するのは,おそらくインビトロ刺激が高活性のT細胞の選択を可能にするためであるということを示す証拠も出てきている(Vignard et al., J Immunol 2005 ;175: 4797−805, Dudley et al., J lmmunother 2001 ;24: 363−73, Morgan et al, J Immunol 2003;171: 3287−95, Rosenberg & Dudley, Proceedings of the National Academy of Sciences of the United States of America 2004;101 Suppl 2: 14639−45)。
【発明の概要】
【発明が解決しようとする課題】
【0003】
これまでに,多くのグループが,抗体をエピトープのキャリアとして用いて,所定のエピトープに対する細胞性免疫反応を引き起こそうと試みてきた。例えば,国際公開第96/19584号パンフレット(Bona et al.)は,T細胞エピトープが抗体の相補性決定領域(CDR)に挿入されるキメラ抗体を開示し,該キメラ抗体が細胞傷害性T細胞(CTL)反応を引き起こすのに適している旨主張している。しかしながら,該文献は,DNAが機能タンパク質をコードしなければならないことを説いている。よって,要約書には,「エピトープ及び親免疫グロブリンの機能性が保持される」旨記載されている。同様に,21ページ目には,所望のエピトープの挿入は,親免疫グロブリン分子の発現や機能にとって不可欠ではない親免疫グロブリン分子をコードする核酸の領域でなされるべきである旨記載されている。さらに,国際公開第96/19584号パンフレットに記載のすべての実施例は,無傷免疫グロブリンがT細胞エピトープの挿入後に生成されることを示している。
【0004】
米国特許第7,067,110号明細書は,ポリペプチド結合により抗原と融合する免疫グロブリン可変領域ドメインを欠く抗体の融合タンパク質を用いて抗原に対する免疫反応を引き起こす方法を開示している。該融合タンパク質は,Fcに結合する能力を保持する。
【0005】
欧州特許第0759944号明細書は,無傷免疫グロブリンタンパク質として分泌され,腫瘍に対するCTLエピトープを標的とすることでCTLの良好な標的とすることができるT細胞エピトープを抗体分子内に取り込む方法を開示している。
【0006】
国際公開第00/64488号パンフレットは,核酸がB細胞における発現を指向していれば,CDR(但し,その可変領域を除く。)に挿入される異種T細胞エピトープを有するキメラ抗体をコードする核酸によってCTL反応を引き起こすことができる旨開示している。B細胞はナイーブT細胞反応を刺激することができないので,国際公開第00/64488号パンフレットに記載のワクチンは,既存のT細胞反応を促進するのに有用であるだけであろう。
【0007】
国際公開第02/092126号パンフレットは,異種T細胞と高アフィニティCD64レセプターに結合するヒトFcの一部とを含むポリペプチドによってCTL反応を引き起こすことができる旨開示している。しかしながら,本願発明者(ら)は,T細胞エピトープを,例えば,不適切なCDRや抗体の可変領域に挿入することによる抗体配列の破断は,重鎖及び軽鎖の結合を防ぎ,機能性抗体が分泌されないことを示している。これらのミスフォールドした抗体をコードするDNAは,突然強力なT細胞反応を引き起こす。さらに,これは,CD64により媒介されない。なぜなら,ヒトIgG2は,マウス又はヒトCD64に結合せず,ヒトIgG1同様効率的に働くためである。
【課題を解決するための手段】
【0008】
本発明の一側面において,非特異的プロモーターと,少なくとも1つの異種T細胞エピトープを有するが制御性T細胞エピトープを有しないポリペプチドをコードする少なくとも1つの配列と,を含む核酸が提供される。
【0009】
該ポリペプチドは,好ましくは,同種キャリアである。例えば,ヒトにT細胞反応を起こすのに用いられる場合,ヒトタンパク質,又は異種タンパク質,又はすべてのT制御性エピトープを同定及び除去したヒト/異種キメラタンパク質でありうる。
【0010】
該ポリペプチドは,好ましくは,ヘテロ二量体の一方の鎖であり,異種T細胞エピトープは,ヘテロ二量体の他方の鎖と結合できないようなヘテロ二量体の鎖の破断を引き起こす。多くの分子は,一方の鎖がフォールディング及び分泌を他方の鎖に依存したヘテロ二量体である。異種T細胞エピトープの挿入により二次構造が破断されると,フォールディング及び分泌が抑制される。ある実施形態において,一方の鎖は,分泌され異種CTLエピトープを含み,他方の鎖は,異種ヘルパーエピトープを含むが,二次フォールディングの破断により,分泌されない。このため,核酸は,ヘテロ二量体の両方の鎖をコードしうる。この場合,一方の鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,他方の鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない。あるいは,各々のヘテロ二量体の鎖は,別の核酸分子上にコードされうる。
【0011】
ヘテロ二量体は,免疫グロブリン分子でありうる。免疫グロブリン分子の重鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,免疫グロブリン分子の軽鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない。
【0012】
本発明の第1の側面の核酸は,制御性T細胞(Treg)エピトープを含まないポリペプチドをコードする。該ポリペプチドは,T細胞エピトープの不活性キャリアとして作用し,免疫システムにより免疫反応を刺激するのに用いられうる分子又は分子の一部でありうる。なぜなら,これらの分子は,その名の通り,競合Tregエピトープを発現しないためである。適切な分子には,HLA分子,T細胞レセプター,TOLレセプター,TOLリガンド,サイトカインレセプター,ケモカイン,ケモカインレセプターが含まれる。分子は,抗体又はその一部であることが好ましい。
【0013】
理論に束縛されるものではないが,本発明は,少なくとも一部,T細胞エピトープを含むが制御性T細胞エピトープを含まないポリペプチドをコードする核酸の投与によって,特異的T細胞エピトープ(CTLエピトープなど)に対するT細胞反応を生成することができる,という概念に基づく。核酸は,抗原提示細胞(APC)に取り込まれ,リンパ節に移動し,直接提示されるか,又は,発現されて,分泌されその後他のAPCに取り込まれるポリペプチドを生成する,と考えられている。前者の核酸は,ヘルパーT細胞エピトープを刺激するのに適しており,後者は,CTL反応を刺激するのに適している。核酸によってコードされるポリペプチドは,理想的にはナチュラルT細胞エピトープを有しない。この点において適切なポリペプチドは,抗体などの免疫分子である。軽鎖がAPCにとどまり,重鎖が分泌されるように結合することができない抗体の重鎖と軽鎖が,本発明の実施に適しているが,本発明は,T細胞エピトープのキャリアとしての抗体の利用に限定されるものではない。
【0014】
サプレッサーT細胞集団がおよそ40年前に同定されたが,細胞を同定する特別な技術の欠如により,また,抑制の存在に関する科学的懐疑主義のために進展が阻まれた。しかしながら,Sakaguchi et alは,CD4+CD25+T細胞を枯渇したリンパ球の無胸腺マウスへの移入が,受容マウスにさまざまな自己免疫疾患の発症を引き起こすこと,また,CD4+CD25+T細胞による再構成は,該マウスの自己免疫反応を阻止することを証明することによって1995年にサプレッサー細胞への関心を復活させた(Sakaguchi et al J. Immunol 1995;155:1151−1164)。その後,制御性活性を有するさまざまなT細胞集団が,自己抗原(自己寛容制御)(Sakaguchi et al J Immunol 1995;155:1151−1164)及び異種抗原(Shevach, Immunity 2006;25: 195−201, Coleman et al, J. Cell Mol. Med. 2007;11: 1291−1325)に対する免疫反応(先天性免疫反応及び適応的免疫反応の両方)の抑制において重要な役割を果たすことが多数のマウス及びヒトにおける研究により示されてきている。癌のマウスモデルにおけるTreg細胞の枯渇は,内因性免疫介在性腫瘍拒絶(Shimizu, et al, J. Immunol. 1999; 163: 5211 −5218, Onizuka et al, Cancer Research 1999; 59: 3128−3133)及び抗原特異的抗腫瘍免疫(Tanaka, et al, J. Immunother. 2002;25:207−217)を改善することが証明されている。また,Treg細胞の枯渇は,ワクチン接種を含む腫瘍免疫療法(Tanaka, et al, J. Immunother. 2002;25:207−217, Dannull et al, J. Clin. Invest. 2005;115:3623−3633)及びCTLA−4遮断(Sutmuller et al, J. Exp. Med. 2001 ;194:823−832)を増強させる。さらに,多数のTreg細胞は,末梢血において増加し,さまざまな癌患者の腫瘍微小環境及び流入領域リンパ節を占めさせる(populate)。胃癌(Sasada et al, Cancer 2003;98:1089−1099, Ichihara et al, Clinical Cancer Research 2003;9:4404−4408)及び卵巣癌(Curiel et al, Nature Medicine 2004;10:942−949)の患者において,予後不良及び生存率の低下は,Treg細胞の高頻度と関連していた。Treg細胞はまた,腫瘍特異的CD8+(Liyanage et al, J. Immunology 2002;169:2756−2761, Piccirillo et al, J. Immunology 2001;167:1137−1140, Mempel et al, Immunity 206;25:129−141, Annacker et al, J. Immunology 2001;166:3008−3018, Woo et al, J. Immunology 2002;168:4272−4276)及びCD4+(Liyanage et al, J. Immunology 2002;169:2756−2761, Ichihara et al, Clinical Cancer Research 2003;9:4404− 4408, Nishikawa et al, Blood 2005;106:1008−1011)T細胞の増殖,サイトカイン生成(IFNγ, IL−2),及び細胞溶解活性を抑制することが証明されている。また,Treg細胞は,樹枝状細胞(Romagnani et al, Eur. J. Immunol. 2005;35:2452−2458),NK細胞(Ralainirina et al, J. Leukoc. Biol. 2007;81:144−153)及びB細胞(Lim et al, J. Immunology 2005;175:4180−4183)の機能を抑制しうる。まとめると,これらの研究は,腫瘍免疫病理学におけるTreg細胞の重要な役割を示唆し,Treg細胞の頻度と腫瘍の成長との間の密接な相関関係を示している。
【0015】
Treg細胞は,ナチュラルCD4+CD25+ T細胞及び誘発/適応Treg細胞のさまざまな集団に分けられる(Shevach, Immunity 2006;25: 195−201, Bluestone et al, Nat. Immunol. 2005;6:345−352)(表1)。マウス及びヒトのCD4+ T細胞の約5%〜10%は,ナチュラルTreg細胞である(Sakaguchi et al, Nat. Immunology 2005;6:345−352)。ナチュラルTreg細胞は,自己ペプチドとの強力なTCR相互作用によって胸腺に発生し(Picca et al, Current Opinion in Immunology 2005;17:131−136, Jordan et al, Nature Immunology 2001;2(4):301−306, Picca et al, Immunological Reviews 2006;212:74−85),一方,誘発Treg細胞は,非制御性T細胞から末梢に発生する。該胸腺外変換は,低用量抗原への連続暴露,全身性末梢抗原への暴露,TGFβへの暴露といった特殊な免疫学的条件を必要とする(Shevach, Immunity 2006;25: 195−201, Akbar et al, Nat. Rev. Immunol. 2007;7:231−237)。Treg細胞は,以下のメカニズム:i)細胞間接触依存性メカニズム,ii)IL−10やTGFβなどの免疫抑制サイトカインの分泌によって,iii)標的細胞パーフォリン・グランザイム経路(perforin−granzyme pathway)の直接殺傷,の1つ又は組み合わせによって抑制を媒介しうる(von Boehmer, Nature Immunology 2005;6(4):338−344)。
【0016】
これまで,ヒトTreg細胞の抗原特異性についてはほとんど知られていない。Wang et alは,癌患者におけるLAGE−1特異的CD4+CD25+GITR+機能性Treg細胞クローンの同定を報告した(Wang et al, Immunity 2004;20:107−118)。Vence et alは,転移性メラノーマ患者の末梢血における腫瘍抗原特異的CD4+ Treg細胞の存在を証明した。該Treg細胞は,TRP1,NY−ESO−1,gp100,及びスルビビンを含むさまざまな腫瘍抗原を識別した。また,Vence et alは,NY−ESO−1分子内におけるNY−ESO−1特異的Treg細胞エピトープの存在を最初に証明した。さらに,合成ペプチド又は腫瘍溶解物を含む樹枝状細胞を有するメラノーマ患者のワクチンは,Treg細胞の頻度増加を誘発し,腫瘍特異的CD8+ T細胞の増殖を伴うことが分かった。これは,該ワクチンが,未同定Treg細胞エピトープ及びCD8+ T細胞エピトープを含み,Treg細胞T細胞レセプター(TCR)を通じたリガンド特異的活性化によるTreg細胞のインビボでの増殖につながる可能性を示唆している。Treg細胞は,TCR識別/関与を通じた抗原特異的活性化を必要とするが,抗原非特異的バイスタンダー抑制を媒介することが広く認められている(Thorton & Shevach, J. Immunology 2000;164: 183190)。さらに,Li et alは,感染患者のHCV特異的Treg細胞を刺激するC型肝炎ウイルスコアタンパク質内の優性Tregエピトープの存在を示唆した(Li et al, Immunol. Cell Biol. 2007;85(3):197−204)。まとめると,これらの研究,及びHHDトランスジェニックマウスの抗内皮DNAコンストラクトC200Fcによる免疫は,有意なTie−21−196特異的抗腫瘍免疫反応を刺激しないとの最近の発見,並びに(400μgのPC61モノクローナル抗体の投与による)CD4+CD25+ Treg細胞の枯渇後,C200Fc DNA免疫前に(Middleton, PhD Thesis. University of Nottingham, November 2007)HHDマウスの脾細胞から細胞を分泌するTie−21−196特異的IFNγの頻度の増加は,DNAワクチン内のTie−21−196が未同定のTreg細胞エピトープ及びCD8+エピトープを含んでいることを示す。これは,ワクチンが自己抗原Tie−2に対するトレランスを破壊しないこと,及び細胞媒介性抗腫瘍免疫反応を抑制する豊富な抗原特異的増殖Treg細胞によってHHDマウスに抗腫瘍免疫を引き起こさないことを説明するものである。よって,エフェクターエピトープに対する免疫反応を管理して優勢Treg反応の刺激を阻止するTregエピトープを発現しない不活性免疫キャリアでTエフェクターエピトープを発現することには利点がある。
【0017】
有利には,本発明の核酸は,発現ポリヌクレオチドを分泌可能にする,リーダー配列などの配列をコードする配列を含む。これは,ポリヌクレオチドの抗原提示細胞(APC)への移入を可能にする。配列は,ポリヌクレオチドで自然に発現されるリーダー配列や,免疫グロブリンリーダー配列などの添加される異種リーダー配列でありうる。後者は,ポリヌクレオチドが膜結合分子をコードする場合に特に適している。
【0018】
本発明の別の側面によれば,非特異的プロモーターと,免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列を含む核酸が提供され,ここで,重鎖は,核酸が発現されると重鎖が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを含む。
【0019】
本発明の該側面の核酸は,免疫グロブリン分子の組み換え重鎖をコードする。該重鎖の構造は,当業者に知られており,通常,可変領域と定常領域とを含む。重鎖は,抗体由来のものでありうる。抗体は,モノクローナル又はポリクローナルであり,IgA,IgD,IgE,IgG,又はIgMでありうるが,IgGが好ましい。IgG抗体は,任意のIgGサブクラス,例えば,ヒトIgG1,IgG2,IgG3,IgG4,又は,マウスIgG1,IgG2a,IgG2b,IgG3,でありうる。IgG抗体は,ヒトIgG2 Fc結合ドメインを有するヒトIgG1抗体,又はIgG1 Fc結合ドメインを有するヒトIgG2抗体でありうる。重鎖は,ヒト抗体の定常領域,及び異種T細胞エピトープが挿入されたマウスモノクローナル抗体の可変又は超可変(CDR)領域を有しうる。超可変領域を除く可変領域もまた,ヒト抗体の可変領域に由来しうる。抗体に適用されると(すなわち,重鎖と軽鎖とを含む),該抗体は,ヒト化と呼ばれる。ヒト化抗体の作製方法は,当業者に周知である。該方法は,例えば,Winterによる米国特許第5,225,539号明細書に記載されている。マウス超可変領域を除く重鎖の可変領域もまた,マウスモノクローナル抗体に由来しうる。この場合,全可変領域は,マウスモノクローナル抗体に由来し,抗体に適用されると,該抗体は,キメラ化と呼ばれる。キメラ化抗体の作製方法は,当業者に周知である。該方法には,例えば,Boss(Celltech)及びCabilly(Genentech)による米国特許明細書に記載のものが含まれる。それぞれ,米国特許第4,816,397号明細書及び第4,816,567号明細書を参照のこと。
【0020】
ある実施形態において,本発明の核酸は,免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列をさらに含む。あるいは,免疫グロブリン分子の軽鎖をコードする別の核酸が提供されうる。軽鎖は,少なくとも1つの異種T細胞エピトープを有しうる。T細胞エピトープは,核酸が発現されるても軽鎖が未変性コンフォメーションをとることができないようなものでありうる。軽鎖は,軽鎖に関して本明細書に記載した特徴のいずれかを有しうる。したがって,本発明はまた,免疫グロブリン分子の組み換え軽鎖をコードする核酸を提供し,ここで,軽鎖は,核酸が発現されるても軽鎖が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを有する。核酸は,非特異的プロモーターを含みうる。該核酸は,抗体などの免疫グロブリン分子をコードする。
【0021】
このように,本発明の別の側面によれば,非特異的プロモーターと,組み換え免疫グロブリン分子をコードする少なくとも1つの配列とを含む核酸が提供され,ここで,免疫グロブリン分子は,核酸が発現されても免疫グロブリン分子が未変性コンフォメーションをとることができないように少なくとも1つの異種T細胞エピトープを有する。組み換え免疫グロブリン分子並びに上記の重鎖及び軽鎖は,制御性T細胞エピトープを有しないことが好ましい。
【0022】
本発明はまた,以下を提供する:
・本発明の核酸とアジュバントとを含むワクチン;
・本発明の核酸と,薬学的に許容されるキャリア,賦形剤,又は希釈剤とを含む医薬組成物;
・医薬用の本発明の核酸;
・少なくとも1つのT細胞エピトープに対する免疫反応を刺激するための薬剤の製造における本発明の前記核酸の利用;
・少なくとも1つのT細胞エピトープに対する免疫反応を刺激するための本発明の核酸;及び
・T細胞エピトープに対する免疫反応を刺激するための方法であって,該免疫反応を必要とする被験者に本発明の核酸の治療効果のある量を投与するステップを含む方法
【0023】
驚くべきことに,本願発明者(ら)は,ヒトか非ヒトかを問わず,一次抗体構造を破断し,フォールディングを抑制し,及び/又は,重鎖又はごく少量の無傷抗体への分泌を制限するように可変領域でクローニングされた所定のT細胞エピトープを有する抗体が,強力なヘルパー及び抗原特異的T細胞反応を刺激することを発見した。本願発明者(ら)はまた,該効果が該抗体の重鎖をコードする核酸を用いて得られることを発見した。T細胞エピトープは,免疫プロテアソームによって処理されるが破壊はされないと考えられている。ある実施形態において,本発明は,T細胞反応の頻度及び活性を強化する変性免疫グロブリン内の所定のT細胞エピトープを提示するDNAワクチンを提供する。本発明の核酸によってコードされるポリペプチドを,本明細書において「ImmunoBody」と呼ぶことにする。
【0024】
T細胞エピトープに対する免疫反応が,免疫グロブリンが未変性コンフォメーションをとることができないようにT細胞エピトープが挿入される免疫グロブリンの重鎖を少なくともコードする核酸によって刺激されうるとの発見は,抗体は機能的な形で発現されなければならないと説く当該技術分野の予想に反するものである。例えば,上述のように,国際公開第96/19584号パンフレットは,核酸がT細胞エピトープが抗体のCDRに挿入される抗体をコードする場合,該核酸は,機能的抗体をコードしなければならない旨説いている。同様に,欧州特許第0759944号明細書は,無傷免疫グロブリンタンパク質として分泌されるT細胞エピトープを抗体分子へ取り込む方法を記載している。米国特許第7,067,110号明細書は,抗体の融合タンパク質によって抗原に対する免疫反応を引き起こすことができる旨開示しているが,該抗体は,免疫グロブリン可変領域を欠くものとして開示されている。また,該融合タンパク質は,抗原中に制御性T細胞エピトープを有する。よって,該タンパク質は,抗体反応を刺激しうるが,抗原中の制御性T細胞エピトープにより高活性T細胞反応を刺激しないであろう。
【0025】
上述したように,国際公開第00/64488号パンフレットは,CDR(その可変領域を除く)に挿入される異種T細胞エピトープを有するキメラ抗体をコードする核酸を開示しており,該核酸は,B細胞における発現を指向している。本発明の核酸は,B細胞における発現を指向しておらず,したがって,インビトロでもインビボでも特にB細胞を標的としない。本発明の核酸は,樹枝状核酸を含む任意の抗原提示細胞に取り込まれうり,したがって,ナイーブCTL及びヘルパーT細胞反応をプライミングすることができるのに対し,国際公開第00/64488号パンフレットに記載のワクチンは,既存のT細胞反応を促進する上でしか有用でないであろう。
【0026】
本発明による核酸によって誘発される反応の機能的活性の分析は,合成ペプチドによる免疫と比べて高活性反応を生成しうることを示した。これはまた,インビトロ及びインビボで腫瘍細胞を識別し殺傷する能力の強化と相関関係があった。該観察は,良好な抗腫瘍活性が高活性TRP2特異的CTLにより示される他の研究における報告に匹敵するものである(Zeh et al, J Immunol 1999;162: 989−94, Harada et al, Immunology 2001;104: 67−74)。
【0027】
本発明の核酸は,非特異的プロモーター,すなわち,核酸の発現を促進するが発現が促進される細胞に対する特異性は有さないプロモーター,を有する。該プロモーターは,好ましくは,樹枝状細胞及び/又はケラチン生成細胞における核酸の発現を引き起こす。適切なプロモーターの例には,CMVプロモーター,SV40プロモーター,及び当業者に周知の他の非特異的プロモーターが含まれる。あるいは,本発明の核酸は,樹枝状細胞(例えば,Cd11bプロモーター)及びケラチン生成細胞(例えば,MHCIIプロモーター, Chin et al., 2001 J. Immunol. 167, 5549−5557)において特異的発現を引き起こす1つ以上のプロモーターを有しうる。
【0028】
本発明のある側面の核酸は,免疫グロブリン分子,好ましくは,抗体の主要な特徴のすべて,すなわち,可変領域及び定常領域を含む重鎖及び軽鎖,を含む抗体をコードする。該抗体は,モノクローナル又はポリクローナルであり,IgA,IgD,IgE,IgG,又はIgMでありうるが,IgGが好ましい。IgG抗体は,任意のIgGサブクラス,例えば,ヒトIgG1,IgG2,IgG3,IgG4,又は,マウスIgG1,IgG2a,IgG2b,IgG3,でありうる。IgG抗体は,ヒトIgG2 Fc結合ドメインを有するヒトIgG1抗体でありうる。該抗体は,ヒト抗体の定常領域,及び異種T細胞エピトープが挿入されたマウスモノクローナル抗体の可変又は超可変(CDR)領域を有しうる。超可変領域を除く可変領域もまた,ヒト抗体の可変領域に由来しうる。該抗体は,ヒト化と呼ばれる。ヒト化抗体の作製方法は,当業者に周知である。該方法は,例えば,Winterによる米国特許第5,225,539号明細書に記載されている。マウス超可変領域を除く抗体の可変領域もまた,マウスモノクローナル抗体に由来しうる。この場合,全可変領域は,マウスモノクローナル抗体に由来し,該抗体は,キメラ化と呼ばれる。キメラ化抗体の作製方法は,当業者に周知である。該方法には,例えば,Boss(Celltech)及びCabilly(Genentech)による米国特許明細書に記載のものが含まれる。それぞれ,米国特許第4,816,397号明細書及び第4,816,567号明細書を参照のこと。
【0029】
本発明のある側面の核酸は,重鎖,軽鎖,又は発現される免疫グロブリン分子が,重鎖,軽鎖,又は免疫グロブリン分子が未変性コンフォメーションをとることができないように,少なくとも1つの異種T細胞エピトープを有する。該T細胞エピトープは,例えば,免疫グロブリンの重鎖が抗原に結合することができないように,又は,重鎖及び軽鎖(存在する場合)が結合することができないように,又は,重鎖又は免疫グロブリン分子が適切に分泌されえないように,発現タンパク質を破断しうる。該破断は,免疫グロブリン分子の三次構造で起こりうり,ジスルフィド結合(disulphide bond)の形成を阻止しうる。
【0030】
以下でより詳細に述べるように,免疫グロブリン分子が抗体である場合,T細胞エピトープは,抗体のCDR1領域及びCDR2領域に挿入されるか,置換されうる。CDR1及びCDR2は,抗体のβシート構造の一部を形成し,フォールドされた分子内に一部が沈殿している。長さ,アミノ酸組成や荷電のいかなる変化も,この構造を破断し,重鎖及び軽鎖のフォールディング及び結合を阻止する。CDRH3は,免疫グロブリン分子の表面に暴露され,したがって,変化に対して許容状態である。本発明において,CDR1及び/又はCDR2がT細胞エピトープで置換されるのが好ましい。事実,ある実施形態において,CDRHの接点におけるフレームワーク領域の欠損は,抗体のフォールディングを完全に破断するが,これらの領域におけるエピトープの挿入は,良好なT細胞反応を与える。CDRH1(5アミノ酸の長さ)又はCDRH2(17アミノ酸の長さ)へのエピトープの取り込みは,軽鎖が天然配列を有していても,ごく少量の無傷抗体を除く重鎖の分泌を可能にするほどの破断を引き起こす。これは,二次構造が重鎖と軽鎖の組み合わせにとって重要であることを示している。軽鎖のCDRL1内へのエピトープの取り込みは,重鎖のCDRH3へ取り込まれる唯一のエピトープが存在するとしても,軽鎖の分泌は低レベルとなる。
【0031】
「異種T細胞エピトープ」とは,抗体にとって異種であるT細胞エピトープを意味している。例えば,異種T細胞エピトープは,抗体中にこれまでに存在していなかったものでありうる。異種T細胞エピトープは,挿入されるアミノ酸配列から構成されうるが,第2の部分の隣接アミノ酸とともに,全体として挿入されうる。これは,挿入されるエピトープが,異種核酸に元の抗原由来のものと同様の処理プロファイルを有することを確実にするためである。一つ以上のCTL/ヘルパーエピトープを同じ可変領域に挿入しうる。
【0032】
T細胞エピトープは,重鎖又は軽鎖のどこにでも挿入しうる。該又は各エピトープは,重鎖及び/又は軽鎖の可変領域に挿入されるのが好ましいが,重鎖及び/又は軽鎖の定常領域にのみ,又は定常領域と可変領域に挿入されるT細胞エピトープを有する抗体や重鎖をコードする核酸が本発明に含まれる。本発明の核酸において,T細胞エピトープをコードする配列は,重鎖及び/又は軽鎖をコードする配列に挿入される(すなわち,追加される),又は重鎖及び/又は軽鎖をコードする配列に置換されうる。
【0033】
可変領域において,T細胞エピトープは,重鎖及び/又は軽鎖のいずれか1つ以上のCDR,すなわち,L1,L2,L3,H1,H2,又はH3,に挿入されるか,又は置換されうる。当然ながら,L1,H1及びH2が目下好ましい。ある実施形態において,T細胞エピトープは,CDRL1及び/又はH1及び/又はH2に挿入されるか,又は置換される。好ましくは,取り込まれるT細胞エピトープは,抗体が未変性コンフォメーションをとらないように,すなわち,フォールドせず,また,正しく分泌されないように,抗体の元のCDRのアミノ酸と同じ大きさ及び荷電ではない。あるいは又は加えて,それらは,CDRを取り囲むフレームワーク領域に挿入されるか,置換されうる。
【0034】
挿入されるT細胞エピトープは,好ましくは,細胞傷害性T細胞(CTL又はCD8)エピトープである。あるいは又は加えて,ヘルパーT細胞(CD4)エピトープが挿入されうる。T細胞エピトープは,周知のT細胞アルゴリズムを用いて予測されうる,又は,ペプチドとして合成て標準的なT細胞アッセイを用いてスクリーニングされうる。T細胞エピトープは,5〜50,7〜40,8〜30,又は9〜20アミノ酸,例えば,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,又は30の範囲のアミノ酸長を有しうる。エピトープは,抗原エピトープをコードする相補オリゴヌクレオチドを用いて挿入されうり,それらはアニールされ,CDR(又は他の領域)が特異的制限酵素部位に置換された抗体フレームワークの特異的部位にクローニングされる。組み換え抗体がヘルパー及び細胞傷害性T細胞反応を刺激する能力は,本明細書に例示されているようなかたちでスクリーニングすることができる。
【0035】
本発明の範囲内においてさまざまな組み合わせが可能である。ある実施形態において,1つ以上のCD8エピトープが,重鎖又は抗体のCDR H1及び/又はH2又は非CDRの可変領域に挿入されうる及び/又は置換されうる。あるいは又は加えて,1つ又は複数のCD4エピトープが,軽鎖又は抗体のCDR L1又は非CDR可変領域に挿入されうる及び/又は置換されうる。複数のT細胞エピトープが存在する場合,T細胞エピトープは,同じである又は異なりうる。当業者であれば,以下を含め,多数の組み合わせが可能であることを理解するであろう。
・CDR H1のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H2のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H1及びCDR H2のCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H1の2つのCD8エピトープ,及びCDR L1のCD4エピトープ;
・CDR H2の2つのCD8エピトープ,及びCDR L1のCD4エピトープ;など
【0036】
本発明の核酸は,MHCクラスI分子及びMHCクラスII分子両方の大部分と結合しうる単一の標的抗原からの複数のT細胞エピトープを取り込みうる。これにより,広範囲の集団ワクチン接種において利用可能なワクチンを製造しうる。あるいは,本発明に有用な核酸は,最も一般的なクラスI及びクラスII表現型に結合可能な複数の標的抗原からの複数のT細胞エピトープを取り込みうる。これにより,抗原が欠損した変異体の選択を阻止しうるワクチンを製造しうる。標的抗原は,単一の病原体又は腫瘍型由来であるか,又は様々な病原体又は癌に対して免疫反応を示すように選択されうる。特異的共通HLA表現型を標的とする本発明に有用な核酸は,さまざまな癌及び/又は病原体からの多数のT細胞エピトープを取り込み,病気を予防する単一のワクチンを提供しうる。
【0037】
ヘルパー及び/又は細胞傷害性T細胞反応を刺激するものであれば,任意のT細胞エピトープを挿入しうる。HIV,C型肝炎,及びその他の感染症などの潜伏感染を除去するのにCTLを必要とする病原体由来のT細胞エピトープを用いうるが,該エピトープは,「自己ペプチド」であること,すなわち,癌などの細胞増殖に関連する状態/疾患に関連する場合が好ましい。好ましくは,T細胞エピトープは,重鎖又は抗体が正しくフォールドされえず,分泌されえない。したがって,挿入されるエピトープは,元の可変領域と同様の大きさ及びアミノ酸組成ではないことが好ましい。核酸は,さまざまなT細胞反応を引き起こすように複数の異なるT細胞エピトープを有しうる。核酸は,単一の抗原由来の複数のエピトープを取り込みうり,それにより,異なるHLA型を有する個人の大多数が単一のワクチンに反応することを確実にしうる。あるいは,制限された範囲のHLA型を標的とする複数の抗原由来の複数のT細胞エピトープを用いうる。本発明の核酸分子は,単一の病原体又は癌型由来のさまざまな抗原を含みうる,又は,それらは,さまざまな固形腫瘍又は病原体を標的とする異種抗原を含みうる。本発明の核酸分子は,腫瘍内の異なる細胞集団,例えば,上皮腫瘍及び内皮抗原,を標的とするように設計されうる。
【0038】
驚くべきことに,本願発明者(ら)は,T細胞エピトープが重鎖の構造的に制限されたCDR又は非CDR領域に挿入されると,それらは,優れたCTR反応を示すことを発見した。これは,巨大なエピトープの可変領域への挿入により軽鎖と弱くしか結合することができない大量の重鎖の分泌によるものと思われる。これは,「抗原提示細胞により内因性に合成されたタンパク質のみがMHCクラスI分子上に提示されCTLにより識別される」(国際公開第96/19584号パンフレット)旨記述する教義に反している。外因性抗原の取り込み及びMHCクラスI上の提示は,交差提示として知られるプロセスであり,通常,特異的レセプターによる取り込みを必要とする。これは,ヒトFcγ1抗体のCD64レセプターでありうる。しかしながら,大量の無傷抗体又は抗原抗体複合体がこのレセプターをうまく標的とするであろうことは予測されるであろう。一方,本明細書に提示された結果は,CD64に結合せずにすぐれたCTL反応を示す非常に低レベルの無傷抗体及び大量の自由重鎖を明確に示している。実際,CTLエピトープが,CD64に結合することができない抗体,例えば,CD64結合領域がIgG2由来の非CD64結合領域で置換されたIgG2抗体やIgG1分子,に挿入されると,CTL反応を刺激しうることが本明細書において示されている。
【0039】
重鎖をコードする核酸は,好ましくは,分泌を可能にするリーダー配列を含む。本願発明者(ら)は,重鎖のリーダー配列が,分泌を阻止し,内因性タンパク質の生成を可能にするために除去されると,これによりCTL反応が低減することを発見した。これは,完全に予想に反するのものである。発明者(ら)は,理論に束縛されることを望んではいないが,これは,核酸が,高レベルの重鎖及び後に抗原提示細胞に取り込まれうる微量の天然タンパク質を分泌する非抗原提示細胞において発現されることを意味するものと考えている。あるいは,核酸は,排出リンパ節へ移動する抗原提示細胞を直接トランスフェクトしうる。該排出リンパ節において,抗原提示細胞は,微量の天然タンパク質,及び同じ又は隣接する抗原提示細胞に取り込まれ,ナイーブCTLに対するMHCクラスI上に提示される大量の重鎖を分泌する。したがって,核酸ワクチンが,効果的なCTL反応を刺激するために,核酸は,好ましくは,非常に低レベルで分泌される及び/又は同時に大量の変性タンパク質を分泌するタンパク質内のCTLエピトープをコードする。しかしながら,CTL反応は,ヘルパー反応非存在下では,高アフィニティ記憶反応に発達しえない。したがって,Tヘルパーエピトープが重鎖又は免疫グロブリン分子,好ましくは,抗体の軽鎖の可変領域,に挿入されるのが好ましい。さらに,驚くべきことに,また,「標的細胞に外因性に取り込まれるタンパク質のみがMHCクラスII分子によって提示され,ヘルパーT細胞によって識別される」旨述べる教義に反して,軽鎖は,ごく少量しか分泌されなかった。軽鎖の分泌を阻止するリーダー配列の除去は,ヘルパー反応に何ら影響を及ぼさなかった。したがって,本発明の核酸は,抗体の軽鎖のリーダー配列を有する場合もあれば有しない場合もある。これらの結果は,核酸は,MHCクラスIIとの関連で,内因性に合成されたタンパク質由来のTヘルパーエピトープを提示する抗原提示細胞,場合によってはオートファジー,に取り込まれることを暗示している。ヘルパーT細胞がCTL反応を助けるために,ヘルパーT細胞が識別する両T細胞エピトープは,結合T細胞ヘルプとして知られるプロセスにおいて同じ抗原提示細胞上に発現される必要がある。これは,核酸によってコードされる,軽鎖を合成する抗原提示細胞は,CTLエピトープ自体をも合成,分泌及び交差提示するか,又は隣接APCから重鎖を取り込む必要があることを暗示している。
【0040】
本発明はまた,内因性に生成された軽鎖由来の異種ヘルパーT細胞エピトープ,及び交差提示された重鎖由来の異種CTLエピトープをMHCクラスII上に提示する単離樹枝状細胞を提供する。該樹枝状細胞は,本明細書に記載の治療に利用しうる。
【0041】
本発明の核酸は,T細胞反応の頻度と活性の両方を増大させる変性抗体をコードすることにより,既存のT細胞エピトープをより免疫原性のあるものにしうる。
【0042】
本発明の核酸は,クローニングにより得られる,又は,化学合成によって全体的又は部分的に生成される,DNA,cDNA,又はmRNAなどのRNAでありうる。治療用として,核酸は,治療を受ける被験者において発現されうるかたちであることが好ましい。
【0043】
本発明の核酸は,組み換え型であるか,又は,分離された及び/又は精製された形で提供されうる。それは,1つ以上の発現用調節配列を除き,ヒトゲノム中に核酸隣接遺伝子を含まないか,又は実質上含まないことがある。本発明による核酸がRNAを含む場合,本明細書に示される配列への言及は,UをTと置き換えたRNA等価物への言及と解釈されるべきである。
【0044】
本発明の核酸は,核酸配列及びクローンが入手可能であることを所与として,例えば,本明細書に記載の情報及び参考文献や当技術分野で周知の技術(例えば,Sambrook, Fritsch and Maniatis, “Molecular Cloning”, A Laboratory Manual, Cold Spring Harbor Laboratory Press, 1989, 及び Ausubel et al, Short Protocols in Molecular Biology, John Wiley and Sons, 1992を参照のこと)を用いて,当業者が容易に作製することができる。これらの技術は,(i)例えば,ゲノムソースからの該核酸のサンプルを増幅するポリメラーゼ連鎖反応(PCR)の利用,(ii)化学合成,又は(iii)cDNA配列の作製,を含む。ポリペプチドをコードするDNAは,当業者に周知の任意の適切な方法を用いて生成し使用することができ,例えば,コードするDNAの取り込み,発現される部位の両側における適切な制限酵素認識部位の同定,及びDNAからの該部位の除去,がある。該部位は,後に,標準的な市販の発現系における適切なプロモーターと動作可能に結合しうる。他の組み換えアプローチは,DNAの関連部位を適切なPCRプライマーで増幅することである。例えば,部位特異的突然変異誘発法を用いて,修飾ペプチドの発現につながるように,又は,核酸の発現に使用される宿主細胞のコドン選択を考慮するように,配列の修飾を行うことができる。
【0045】
核酸配列の発現を得るために,該配列を,動作可能に核酸に結合してその発現を制御する1つ以上の制御配列を有するベクターに取り込むことができる。ベクターは,例えば,挿入される核酸の発現を促進するプロモーターやエンハンサーなどの他の配列,ポリペプチドを溶解として生成するような核酸配列,及び/又は宿主細胞で生成されるポリペプチドが細胞から分泌されるような分泌信号をコードする核酸を含みうる。必要に応じ,ベクターをベクターが機能する宿主細胞に変え,ポリペプチドが生成されるように宿主細胞を培養し,宿主細胞又は周囲媒質からポリペプチドを回収することによってポリペプチドを生成しうる。原核細胞及び真核細胞が当技術分野において該目的のために用いられる。その中には,大腸菌株,酵母菌株,及び,昆虫細胞や動物細胞などの真核細胞,例えば,COS細胞,CHO細胞,ボウズメラノーマ細胞,及びその他の適切なヒト細胞,が含まれる。本発明が抗体の重鎖及び軽鎖をコードする核酸に関連する場合,個々の核酸は,同じ又は異なるプロモーターに刺激されて,同じ発現ベクター又は別の発現ベクターに存在しうる。
【0046】
本発明の核酸は,ヒトを含む哺乳動物などの患者の少なくとも1つのT細胞エピトープに対する免疫反応を刺激するために利用されうる。ヘルパー及び/又は細胞傷害性T細胞反応を刺激しうる。本発明により得られる特定のエピトープに対するT細胞反応は,単純なペプチドと同じエピトープによる免疫によって,又は,ペプチド又は核酸と同じ抗体内でコードされるエピトープによる免疫によって得られるものよりも高い結合活性を有しうる。本発明の核酸は,併用療法,すなわち,軽鎖をコードする核酸及び重鎖をコードする核酸,として投与されうる。核酸は,静脈内投与,経皮投与,筋肉内投与,経口投与,又は他の投与法で投与されうる。経皮投与又は筋肉内投与が好ましい。というのも,これらの組織は,樹枝状細胞を含むからである。
【0047】
本明細書において,「治療」の語は,ヒト又は非ヒト動物のためになりうるいかなる養生法を含む。治療は,遺伝性疾患又は後天性疾患のためのものでありうる。好ましくは,治療は,癌や感染症といった細胞増殖に関連する状態/疾患のためのものである。核酸で治療可能な癌の種類の例には,固形腫瘍,結腸直腸癌,肺腫瘍,乳房腫瘍,胃腫瘍,卵巣腫瘍,子宮腫瘍,肝腫瘍,腎腫瘍,膵臓腫瘍,メラノーマ腫瘍,膀胱腫瘍,頭頸部腫瘍,脳腫瘍,食道腫瘍,膵臓腫瘍,骨腫瘍,並びに軟部組織癌及び白血病,が含まれる。核酸で治療可能な感染症の例には,HIV感染症,C型肝炎感染症や,クリアランス用T細胞免疫を必要とする慢性感染症,が含まれる。
【0048】
核酸は,薬学的に許容されるキャリアと併用しうる。該キャリアには,これらに限定されるものではないが,生理食塩水,緩衝生理食塩水,デキストロース,リポソーム,水,グリセロール,エタノール,及びこれらの組み合わせ,が含まれる。
【0049】
宿主の免疫反応の刺激を促進するためにアジュバントを用いうる。アジュバントには,水酸化アルミニウム,リゾレシチン,プルロニック,ポリオール,ポリアニオン,ペプチド,タンパク質,及び油乳剤,が含まれる。
【0050】
本発明に有用な核酸は,医薬組成物で処方しうる。これらの組成は,上記物質の一つに加え,薬学的に許容される賦形剤,キャリア,緩衝剤,安定剤,又は当業者に周知の他の物質を含みうる。キャリアや他の物質の正確な特質は,投与経路,例えば,経皮経路,経口経路,静脈内経路,皮膚経路又は皮下経路,経鼻経路,筋肉内経路,腹腔内経路,に依存しうる。処方は,好ましくは,微小な金粒子の表面上に沈殿し,遺伝子銃による注入に適した安定乾燥粉末としての核酸である。処方は,エレクトロポレーションを用いた経皮又は筋肉内投与に適しうる。
【0051】
核酸を含む,又は核酸の送達のための組成物は,好ましくは,「治療効果のある量」個人に投与され,これは,個人にとっての利益を示すのに十分である。投与される実際の量,及び投与の割合及び経時変化は,治療対象の特質及び重症度に依存する。治療の処方,例えば,投与量に関する決定など,は,一般開業医その他の医師の責任内にあり,通常,治療対象の疾患,個々の患者の状態,送達部位,投与方法,及び開業医に周知の他の要因を考慮に入れる。本発明の核酸は,特に,既存の癌の治療,及び最初の治療又は手術後の癌の再発予防に関連している。上述の技術及び手順の例は,Remington’s Pharmaceutical Sciences, 16th edition, Oslo, A.(ed), 1980に見出すことができる。
【0052】
好ましくは,本発明の核酸は,効果のある量ヒトに投与すると腫瘍細胞の成長を有意に抑制しうるヘルパー及び/又は細胞傷害性T細胞を刺激する。適量は,多数のパラメーター,例えば,年齢,性別,体重,治療対象の状態の重症度,投与される活性成分,及び投与経路,に基づいて医師により決定されうる。例えば,1〜1000μgのDNAの投与量は,ヘルパー及び細胞傷害性T細胞反応の両方を刺激するのに十分である。
【0053】
本発明の核酸は,追加の薬学的に許容される成分とともに投与されうる。該成分には,例えば,免疫系刺激剤が含まれる。
【0054】
組成物は,治療対象の状態に応じて,単独で,又は他の治療と組み合わせて,同時に又は連続して投与されうる。他の癌治療には,当技術分野において周知の,他のモノクローナル抗体,他の化学療法剤,他の放射線治療技術や免疫療法が含まれる。本発明の組成物の一つの特定の用途は,手術の補助,すなわち,腫瘍除去後の癌再発のリスクを低下させる手助けをすることである。
【0055】
注射(id)は,本発明の核酸の治療的投与のための第一の経路でありうる。
【0056】
核酸は,局所的に腫瘍部位や他の所望の部位に投与され,又は腫瘍や他の細胞を標的とするような方法で送達されうる。
【0057】
核酸の投与量は,利用される作用物質の特性,例えば,その結合活性及びインビボ血中濃度半減期,製剤中のポリペプチド濃度,投与経路,投与部位及び割合,関係患者の臨床的耐性,患者を苦しめている病的状態,など,十分に医師の技能の範囲内にあるもの,に依存する。例えば,一患者一回につき100μgの核酸の投与量が好ましいが,投与量は,一回あたり約10μgから1mgの幅がありうる。一連の連続的接種中に異なる摂取量が用いられる。開業医は,最初の接種を行い,次に,比較的少量の核酸で促進しうる。
【0058】
他の実施形態において,本発明は,標的抗原からのT細胞エピトープを可変領域の抗体へ改変する方法,及び,ヘルパー及び細胞傷害性T細胞反応の両方を刺激するワクチンとしての該改変抗体の利用に関する。
【0059】
本発明のさらなる側面は,本明細書に開示される核酸を含む宿主細胞を提供する。本発明の核酸は,宿主細胞のゲノム(例えば,染色体)に組み込まれうる。組み込みは,標準的な技術に従ってゲノムとの組み換えを促進する配列を含むことにより促進されうる。核酸は,細胞内の染色体外ベクター上にありうる,あるいは,細胞に対し識別可能に異種又は異質でありうる。
【0060】
さらなる側面は,本発明の核酸を宿主細胞に導入するステップを含む方法を提供する。該導入は,(特にインビトロ導入の場合は)一般的に制限なく「形質転換」と呼ぶことができるが,任意の利用可能な技術を利用しうる。真核細胞の場合,適切な技術には,リン酸カルシウムトランスフェクション,DEAEデキストラン,エレクトロポレーション,リポソーム媒介トランスフェクション,及びレトロウイルス又は他のウイルス,例えば,ワクシニア,昆虫細胞の場合はバキュロウイルス,を用いた形質導入が含まれうる。
【0061】
細菌性細胞の場合,適切な技術には,塩化カルシウム形質転換,エレクトロポレーション,及びバクテリオファージを用いたトランスフェクションが含まれる。他の方法として,核酸の直接注入を用いることができる。
【0062】
抗生物質抵抗性遺伝子や感受性遺伝子といった標識遺伝子を,当技術分野において周知のように,関心のある核酸を含むクローンを同定するのに利用しうる。
【0063】
導入に続いて,核酸からの発現が引き起こされるか可能となりうる。例えば,コードされたポリペプチド(又はペプチド)が生成されるように,遺伝子発現の条件下で宿主細胞(実際に形質転換された細胞を含みうる。おそらく,該細胞は,形質転換された細胞の子孫であるだろうが)を培養することによって。ポリペプチドが適切なシグナルリーダーペプチドを結合して発現される場合,細胞から培養培地へ分泌されうる。発現による生成の後に,ポリペプチド又はペプチドは,宿主細胞,及び/又は場合によっては培養培地から単離及び/又は精製されうり,その後,所望するように,例えば,1種類以上の追加の成分を含みうる組成,例えば,1種類以上の薬学的に許容される賦形剤,媒体,又はキャリア(例えば,以下を参照のこと)を含む医薬組成物など,の製剤において利用されうる。
【0064】
本発明はまた,候補抗原のT細胞エピトープを同定する方法を提供する。該方法は,非ヒト動物のT制御性細胞を枯渇させるステップと,非ヒト動物を候補抗原で免疫するステップと,T細胞反応が,候補抗原の予測エピトープに対するペプチド又は候補抗原内のすべての可能性のある重複ペプチドに対して引き起こされたかどうかを確かめるためにスクリーニングするステップと,を含む。
【0065】
該方法は,マウスやラットなどの非ヒト動物において実施されうる。T制御性細胞は,Ontakなどの毒素と随意に接合しうる抗CD25抗体を用いて,又は,T制御性細胞を選択的に殺すシクロフォスファミドなどの化学療法によって非ヒト動物において枯渇されうる。T制御性細胞が枯渇すると,非ヒト動物は,候補抗原をコードするDNAによって,又は候補抗原自体によって免疫されうる。候補抗原は,抗原Fc融合タンパク質として提供されるのが好ましい。スクリーニングステップにおいて,非ヒト動物においてT細胞反応が刺激されるペプチドが同定される。これは,ELISPOTなどの技術を用いてインビトロで行うことができる。T細胞反応が候補エピトープに対して引き起こされる場合,このエピトープを非ヒト動物に免疫するのに用いることができる。このペプチドがT細胞反応を引き起こす場合,結合活性及び頻度は,本発明による核酸内のエピトープをコードすることにより強化することができる。この方法は,免疫プロテアソームによって処理されるT細胞エピトープの同定を可能にする。
【0066】
本発明の各側面の好ましい特徴は,各々の他の側面に変更すべきところは変更して関係する。本明細書に記載の先行技術文献は,法律が許す最大限の範囲で組み込まれる。
【図面の簡単な説明】
【0067】
以下,本発明を以下の実施例(これらに限定されない)においてさらに述べる。以下の図面を参照する。
【0068】
【図1】重鎖ベクターpOrigHIBの特徴を示す図 抗体SC100の野生型脱免疫重可変領域を,ヒトIgG1Fc定常領域のフレームのHindIII/AfeIを用いてクローニングした。Fc領域は,CH1領域,CH2領域,CH3領域及びヒンジ領域を含む。哺乳類細胞における高レベル発現は,ヒトサイトメガロウィルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,Orig HIBヒトIgG1鎖の下流に信号を送る。EM7は,大腸菌における抗生物質選択を可能にするzeocin耐性遺伝子の発現を制御する細菌プロモーターであり,一方,該耐性遺伝子のSV40早期プロモーター上流は,哺乳類細胞における選択を可能にする。SV40ポリアデニル化は,zeoΓmRNAの3’末端の適切な処理を管理するために該耐性遺伝子の下流に信号を送る。ベクターはまた,その内部に,細菌における増殖のためのColE1複製起点を含む。相補性決定DNA配列は,効果的に除去され,制限部位RE1,RE2,RE3(それぞれ,FspI,MscI,SrfI)と単独で及び組み合わさって交換される。
【0069】
【図2】重鎖ベクターpOrigLIBの特徴を示す図 抗体SC100の野生型脱免疫された軽可変領域を,ヒトカッパ定常領域のフレームのBamHI/BsiWIを用いてクローニングした。哺乳類細胞における高レベル発現は,ヒトサイトメガロウィルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,Orig LIB鎖の下流に信号を送る。ベクターはまた,細菌における増殖及び選択を可能にするアンピシリン用抗体耐性遺伝子及びColE1複製起点を含む。相補性決定領域は,効果的に除去され,制限部位RE4,RE5,RE6(それぞれ,EcoRV,SspI,HpaI)と単独で及び組み合わさって交換される。
【0070】
【図3】野生型ImmunoBodyキメラ重鎖の配列 ヌクレオチド及び翻訳中のアミノ酸配列が完全長キメラIgG1重鎖に図示されている。CDRの位置は,カバット番号付与体系によって定義されるボックス内にある。ストップコンドンは赤のアスタリスクで描かれている。重可変領域の転移に利用されるHindlll/Afel制限部位が強調されている。
【0071】
【図4】野生型ImmunoBodyキメラカッパ鎖の配列 ヌクレオチド及び翻訳中のアミノ酸配列が完全長キメラカッパ鎖に図示されている。CDRの位置は,カバット番号付与体系によって定義されるボックス内にある。ストップコンドンはアスタリスクで描かれている。軽可変領域の転移に利用されるBamHI/BsiWI制限部位が強調されている。
【0072】
【図5】重複伸長PCR CDRを取り除き,重複PCRによって独特の制限部位に置換した。フォワードプライマーH1,H2,H3,L1,L2,及びL3(表2)を,重鎖及び軽鎖の各々の可変領域内のCDR1,2,3を置換するように設計した。中央に配置された各プライマーは,取り除かれ(緑色部分)10〜20bpの野生型配列の横に配置するようにCDR配列を欠く選択された独特の酵素認識配列を含んでいた。フォワードプライマーを,野生型コンストラクトpOrigHIB及びpOrigLIBの各々の内部のヒト重・軽定常領域にアニールする,一般的なリバースプライマーhuHeClonR又はhuLiClonR(表2)とともにPCRの第一段階において使用した。生成されたフラグメントは,野生型CDR配列(赤色部分)を含んでいないが,制限部位と効果的に交換される。可変重・軽領域全体を増幅するために,PCRの第二段階は,単一のプラスミド内のCMVプロモーターにアニールする一般的なCMVフォワードプライマーとともにリバースプライマーとして第一段階から生成されるPCR生成物を用いることが要求される。第二段階のPCR生成物を,pcr2.1(Invitrogen)にサブクローニングし,配列確認後,H1,H2,H3,L1,L2,L3バージョンを単独,組み合わせて及び一緒に含む重/軽(VH及びVL)可変領域を,単一のコンストラクトpOrigHIB及びpOrigLIBに挿入し,野生型領域をHindIII/Afel及びBamHI/BsiWIそれぞれを用いて交換した。
【0073】
【図6A】ImmunoBody重鎖可変領域の配列 CDRが対応する酵素部位H1,H2,H3(単独,組み合わせて及び一緒に)で置換された重可変領域のヌクレオチド及びアミノ酸配列。独特の制限酵素部位が強調されている。CDR1,2,3を,それぞれ,FspI,MscI,SrfIと置換した。
【図6B】続き
【図6C】続き
【0074】
【図7A】ImmunoBodyカッパ鎖可変領域の配列 CDRが対応する酵素部位L1,L2,L3(単独,組み合わせて及び一緒に)で置換された重可変領域のヌクレオチド及びアミノ酸配列。独特の制限酵素部位が強調されている。CDR1,2,3を,それぞれ,EcoRV,SspI,HpaIと置換した。
【図7B】続き
【図7C】続き
【0075】
【図8】二重発現ベクターpDCOrigの特徴を示す図 すべてのエピトープが単一ベクター内の可変重部位及び可変軽部位に組み込まれると,それらは,各々ヒト定常領域のフレームの強調されたHindIII/AfeI及びBamHI/BsiWIを用いて二重発現に導入される。重鎖のFc領域は,CH1,CH2,CH3領域及びヒンジ領域からなる。哺乳類細胞における重鎖及び軽鎖の両方の高レベルの発現は,ヒトサイトメガロウイルス前初期プロモーターから排除される。BGHポリアデニル化は,mRNA安定性と有効な終止を確保するために,両鎖の下流に信号を送る。EM7は,大腸菌における抗生物質選択を可能にするzeocin耐性遺伝子の発現を制御する細菌プロモーターであり,一方,該耐性遺伝子のSV40早期プロモーター上流は,哺乳類細胞における選択を可能にする。SV40ポリアデニル化は,zeoΓmRNAの3’末端の適切な処理を管理するために該耐性遺伝子の下流に信号を送る。ベクターはまた,そのバックボーンの内部に,細菌における増殖のためのColE1複製起点を含む。
【0076】
【図9】FC領域の合成を防止するストップコンドンを含むImmunoBodyIB15重鎖の配列 キメラ重鎖pDCOrig IB15 CH1ストップのヌクレオチド及びアミノ酸配列。ストップコンドンは,アスタリスクで描かれるヒトIgG1 Fc定常領域のCH1の後に部位特異的な突然変異生成により挿入される。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。単一のコンストラクトからの可変重領域の移転に利用されるHindIII/AfeI制限部位が強調されている。
【0077】
【図10】リーダーを有さないDCIB15重可変領域のヌクレオチド及びアミノ酸配列 リーダーは,重可変(VH)領域を効果的に再増幅するヒトIgG1 CH1領域に結合するリバースプライマーhuHeClonR(表2)とともにフォワードプライマーpOrig重非リーダーを用いてPCRにより取り除かれた。配列確認後,リーダーを除くVH領域を,ヒトIgG1定常領域のフレームのHindIII/AfeIを用いて二重発現コンストラクトDCIB15にクローニングした。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。可変重領域の移転に利用されるHindIII/AfeI制限部位が強調されている。
【0078】
【図11】リーダーを有さないDCIB15カッパ可変領域のヌクレオチド及びアミノ酸配列 リーダーを,軽可変(VL)領域を効果的に再増幅するリバースプライマーhuLiClonR(表2)とともにフォワードプライマーpOrig軽非リーダーを用いてPCRにより取り除いた。配列確認後,リーダーを除くVL領域を,ヒトカッパ定常領域のフレームのBamHI/BsiWIを用いて二重発現コンストラクトDCIB15にクローニングした。ボックス内のアミノ酸は,H1のHepB CD4エピトープ(TPPAYRPPNAPIL)を表す。可変軽領域の移転に利用されるBamHI/BsiWI制限部位が強調されている。
【0079】
【図12】ヒトIgG2定常領域の配列 増幅された重ヒトIgG2定常領域のヌクレオチド及びアミノ酸配列。二重発現ベクターDCIB15のhuigG1定常領域の移転及び置換に利用されるAfeI及びSapI制限部位が強調されている。
【0080】
【図13】ヒトigG3定常領域の配列 増幅される重ヒトigG2定常領域のヌクレオチド及びアミノ酸配列。二重発現ベクターDCIB15のhuigG1定常領域の移転及び置換に利用されるAfeI及びSapI制限部位が強調されている。
【0081】
【図14】ImmunoBody二重発現ベクターのヒトアイソタイプ 図Aは,二重発現ベクターpDCOrigIB15 huigG2を示す。図Bは,二重発現ベクターpDCOrigIB15 huigG3を示す。可変重領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0082】
【図15】G2モチーフを含むDCIB66重鎖の配列 キメラ重鎖のヌクレオチド及びアミノ酸配列。高アフィニティFcγR1(CD64)との相互作用のための臨界結合モチーフ内のアミノ酸E233 L234 L235は,ヒトIgG2からのP233 V234 A235(ボックス内にボールドで強調)で置換されている。ボックス内の他のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。pDCOrigIB15 huigG1への置換を含む部分の移転に利用されるAgeI/AhdI部位が強調されている。可変重領域の移転に利用されるHindIII/AfeI制限部位はボールドで示されている。
【0083】
【図16】G1結合モチーフを含むDCIB67重鎖の配列 キメラ重鎖のヌクレオチド及びアミノ酸配列。ヒトIgG2定常領域内のアミノ酸P233 V234 A235は,ヒトigG1からの高アフィニティFcγR1(CD64)E233 L234 L235 G236(ボックス内にボールドで強調)との相互作用のための臨界結合モチーフで置換されている。ボックス内の他のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びH2のTRP2エピトープ(SVYDFFVWL)を表す。pDCOrigIB15 huigG2への置換を含む部分の移転に利用されるAgeI/AhdI部位が強調されている。可変重領域の移転に利用されるHindIII/AfeI制限部位はボールドで示されている。
【0084】
【図17】マウスIgG2aImmunoBody発現ベクター 図Aは,一本鎖pMoOrigHIBベクターを示す。図Bは,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を含む二重発現ベクターDCIB53を示す。図Cは,H1のHLA−DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を含む二重発現ベクターDCIB63を示す。利用される制限部位が示されている。
【0085】
【図18】規制準拠プラスミドpVAXDCIB54の構造を示す概略図 重一本鎖ベクターpVaxIB54 HIB(図A)は,Nrulを用いて線形化された。pOrigLIBからの軽鎖発現カセット(図B)は,Nrul及びHpalを用いて摘出され,線形化されたプラスミドにクローン化され,二重発現ベクターpVaxDCIB54(図C)を生成した。可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0086】
【図19】DCIB15の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0087】
【図20A】低レベルの無傷タンパク質を生成するImmunoBodyコンストラクト A:gp100/H1,TRP2/H2,及びHepB CD4/L1(DCIB15)を含むImmunoBodyを導入されたCHO−S細胞の上清からのサンドウィッチElisaによるImmunoBody重鎖のレベルの定量化。上清は,そのまま使用し,培地で1/3,1/10,及び1/30に希釈し,ヒトIgG陽性対照と比較した。 B:陽性対照と比較したサンドウィッチElisaによるgp100/H1,TRP2/H2,及びHepB help/L1(DCIB15)を含む精製ImmunoBodyの分析 C及びD:サンドウィッチElisaによるCHO−Sトランスフェクションの上清からの重鎖及び無傷ImmunoBodyの発現の測定。プレートを抗ヒトFc特異抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を使用し,無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を使用した。 E:サンドウィッチElisaによるCHO−Sトランスフェクション(DCIB15,DCIB31,DCIB32,DCIB36,DCIB48,DCIB49,DCIB52,DCIB54)の上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。
【図20B】続き
【0088】
【図21】DCIB24の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のオボアルブミンエピトープ(SIINFEKL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0089】
【図22】DCIB25の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0090】
【図23】DCIB31の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のTRP2エピトープ(SVYDFFVWL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0091】
【図24】DCIB32の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のTRP2エピトープ(SVYDFFVWL)及びL3のHepB CD4エピトープを示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0092】
【図25】DCIB36の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,L3のTRP2エピトープ(SVYDFFVWL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0093】
【図26】DCIB349の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びH3のHLA−DR4制限gp100 CD4エピトープ(WBRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0094】
【図27】DCIB52の配列 ヒトIgG1 Fcのフレームにクローン化された重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0095】
【図28】DCIB54の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びH3のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0096】
【図29】DCIB36の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のHLA―DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0097】
【図30】DCIB18の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTRP2エピトープ(SVYDFFVWL)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0098】
【図31A】ImmunoBodyフレームワークに取り込まれるCTLエピトープは,処理され,発現され,インビボで免疫反応を引き起こす。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H2のTRP2エピトープ及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB18)で免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:免疫マウスからの脾細胞からCD8 T細胞を枯渇させ,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対してIFNγ elispotアッセイにおけるエピトープ特異反応の有無を分析した。反応は,spots/million脾細胞として測定した。 D:インビトロTRP2ペプチド刺激の6日後のB16F10,B16F10 IFNα,及びB16F10 siKbメラノーマ細胞(株)に対する4時間におよぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 E:C57BI/6又はHLA−DR4トランスジェニックマウスに0日目,7日目,14日目にImmunoBody DNA(DCIB15,DCIB31,DCIB32,DCIB36,DCIB48,DCIB52,及びDCIB54)で免疫した。19日目に,IFNγelispotアッセイにより,脾細胞を,TRP2ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 F:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 G:C57BI/6又はHLA−DR4マウスに0日目,7日目,14日目にImmunoBody DNA(DCIB15,DCIB48,DCIB49,DCIB52,及びDCIB54)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepBヘルパーペプチド(DCIB15,DCIB49,及びDCIB52)又はgp100 DR4ヘルパーペプチド(DCIB48及びDCIB54),及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【図31B】続き
【0099】
【図32A】ImmunoBody DNA免疫は,ペプチド免疫又は全抗原による免疫よりもよい。 A:ImmunoBodyDNA免疫(DCIB18)を,フロイント不完全アジュバントのペプチドエピトープによるSC免疫又はTRP2抗原を発現するDNAによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド(■),HepBヘルパーペプチド(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,ImmunoBody DNA(◇)及びペプチド(◆)免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:インビトロTRP2ペプチド刺激の6日後のB16F10(■),B16F10 IFNα(■(薄灰色)),及びB16F10 siKb(□)メラノーマ細胞株に対する4時間におよぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 D:ImmunoBody DNA免疫(DCIB18)を,TRP2ペプチドパルスDCによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のTRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:ImmunoBody DNA免疫(DCIB18)を,TRP2ペプチドパルスDCによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,脾細胞を,TRP2ペプチドパルスLPS Bblastによりインビトロで刺激した。刺激の6日後,クロム遊離アッセイにより,CTL株のB16F10又はB16F10 siKbメラノーマ株を溶解させる能力を評価した。反応は,%細胞毒性として測定した。 F:ImmunoBody DNA免疫(DCIB24)を,SIIFEKLペプチドによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,SIINFEKLペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 G:ImmunoBody DNA免疫(DCIB15)を,gp100 210Mペプチドによる免疫と比較した。HHDIIマウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のgp100 210Mペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 H:ImmunoBody DNA免疫(DCIB24)を,SIIFEKLペプチドによる免疫と比較した。C57BI/6マウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のSIINFEKLペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 I:ImmunoBody DNA免疫(DCIB15)を,gp100 210Mペプチドによる免疫と比較した。HHDIIマウスに0日目,7日目,14日目に免疫し,19日目に,IFNγ elispotアッセイにより,脾細胞を,滴定量のgp100 210Mペプチドに対して分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【図32B】続き
【図32C】続き
【0100】
【図33】DCIB21の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のHepB S Agエピトープ(IPQSLDSWWTSL)及びL1のI−Ad制限Flu HA CD4エピトープ(FERFEIFPKE)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0101】
【図34】複数のエピトープは,CDR H2部位から処理されうる。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H2のSIINFEKLエピトープ及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB24)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,SIINFEKLペプチド,無関係ペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:Balb/cマウスに0日目,7日目,14日目にCDR H2のHepB CD8エピトープ及びCDR L1のFlu HA CD4エピトープを含むImmunoBodyコンストラクト(DCIB21)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepB CD8ペプチド,無関係ペプチド,Flu HA CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0102】
【図35】DCIB17の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0103】
【図36】DCIB26の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のTie−2 Z84エピトープ(FLPATLTMV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0104】
【図37】複数のCTLエピトープは,可変領域から処理されうる。 A:HHDIIマウスに0日目,7日目,14日目にCDR H1のgp100 IMDQVPFSVエピトープ(フレームワーク部分を除く。)及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB17)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100 IMDQVPFSVペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:HHDIIマウスに0日目,7日目,14日目にCDR H1のTie2エピトープ(フレームワーク部分を除く。)及びCDR L1のHepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB26)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,Tie2ペプチド,HepB CD4ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0105】
【図38】複数のCTL反応は,同じImmunoBodyコンストラクト内の異なるエピトープから生成されうる。 A:HHDIIマウスに0日目,7日目,14日目にImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100ペプチド,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,免疫マウスからの脾細胞のgp100修飾IMDQVPFSV(◆)エピトープ,gp100 wt ITDQVPFSVエピトープ(▲),及びTRP2ペプチド(■)TRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:gp100 IMDQVPFSVペプチドでパルスされたT2細胞,TRP2ペプチド又は対照,並びに,B16F10及びB16F10 HHDメラノーマ細胞株に対する4時間に及ぶ51Cr放出アッセイにおける免疫マウスからの脾細胞の細胞毒性 D:HHDIIマウスに0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),又は,ii)CDR H2のTRP2エピトープ及びHepB CD4エピトープ(DCIB18),を含むImmunoBody DNAで免疫した。19日目に,IFNγelispotアッセイにより,脾細胞を,gp100ペプチド(■),TRP2ペプチド(■(濃灰色)),HepBヘルパーペプチド(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 E:C57BI/6マウスに,10μgのDNA溶液をエレクトロポレーションを併用して筋肉内免疫した。免疫は,頸骨筋に週間隔で3回行った。DCIB24又はDCIB18単体で,両者を混合して同じ部位に,又は両者を同時ではあるが別々の部位に,マウスに免疫した。19日目に,TRP2,SIINFEKLペプチド特異的免疫反応の有無に関し脾細胞を分析した。反応は,spots/million脾細胞として測定した。
【0106】
【図39】DCIB37の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Lエピトープ(TITDQVPLSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0107】
【図40】DCIB40の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Iエピトープ(TITDQVPISV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0108】
【図41】DCIB41の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100野生型エピトープ(TITDQVPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0109】
【図42】DCIB42の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 F7Yエピトープ(TITDQVPYSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0110】
【図43】DCIB43の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100 V5Lエピトープ(TITDQLPFSV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0111】
【図44】非係留性残基における改変はエピトープ免疫原性を強化しうる。 HHDIIマウスに0日目,7日目,14日目にCDR H1領域の改変gp100エピトープImmunoBodyコンストラクト(DCIB37,DCIB40,DCIB41,DCIB42,及びDCIB43)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100野生型ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0112】
【図45】DCIB35の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0113】
【図46】複数のCD4ヘルパー反応は,インビボで免疫反応を引き起こすように処理及び提供されうる。 A:HHDII又はC57BI/6マウスに0日目,7日目,14日目にCDR L1領域のI−Ab制限HepB CD4エピトープを含むImmunoBodyコンストラクト(DCIB15)で免疫した。 B:Balb/cマウスに0日目,7日目,14日目にCDR L1領域のI−Ab制限Flu HA CD4エピトープを含むImmunoBodyコンストラクト(DCIB21)で免疫した。 C:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目にCDR L1のHLA−DR4制限gp100 CD4エピトープを含むImmunoBodyコンストラクト(DCIB35)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,対応ペプチド,無関係ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 D:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目にCDR L1,CDR H3(DCIB54),及びCDR L3(DCIB50)のHLA−DR4制限gp100 CD4エピトープを含むImmunoBodyコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,対応ペプチド,無関係ペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【0114】
【図47】DCIB50の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0115】
【図48】CD8 T細胞反応は,分泌重鎖に部分的に依存するが,ヘルパー反応は,分泌軽鎖を必要としない。 A:HHDIIマウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし),iii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(軽鎖上にリーダー配列なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100(■)及びHepB CD4(■(薄灰色)),及び培地制御(□)に対して分析した。反応は,spots/million脾細胞として測定した。 B:サンドウィッチElisaによるCHO−Sトランスフェクションの上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。 C:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし,を含むImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定した。 D:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(重鎖上にリーダー配列なし,を含むImmunoBodyDNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,HepBヘルパーペプチドに対して分析した。反応は,spots/million脾細胞として測定した。
【0116】
【図49A】ImmunoBody Fc領域は,効果的な免疫反応を確立するのに有用である。 A:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(Fc領域なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2(■(濃灰色)),培地制御(□),B16F10メラノーマ株(■),及びB16F10siKb陰性対照細胞株(■(薄灰色))に対して分析した。反応は,spots/million脾細胞として測定した。 B:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(Fc領域なし),を含むImmunoBody DNAで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチドに対して分析した。反応は,spots/million脾細胞として測定した。 C:同じマウスを,HepBヘルパーペプチドに特異的な反応に対して分析した。反応は,spots/million脾細胞として測定した。 D:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB15又はFc領域を有さないDCIB15(DCIB15 FcStop)での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:C57BI/6マウスに,0日目,7日目,14日目に,i)CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1のHepB CD4エピトープ(DCIB15),ii)ヒトIgG2定常領域を有する同じコンストラクト(DCIB33),iii)ヒトIgG3定常領域を有する同じコンストラクト(DCIB65),iv)ヒトIgG2からの結合モチーフと置換されたヒトIgG1結合モチーフを有する同じコンストラクト(DCIB66),及びv)ヒトIgG1(DCIB67)からのモチーフに置換された結合モチーフを有するDCIB33,を含むImmunoBody DNAコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド(■),培地制御(□),及びHepBヘルパーペプチド(■(薄灰色))に対して分析した。反応は,spots/million脾細胞として測定した。 F:サンドウィッチElisaによるCHO−Sトランスフェクション(DCIB15,DCIB33,DCIB65,DCIB66,及びDCIB67)の上清からの重鎖,軽鎖,及び無傷ImmunoBodyの測定。プレートを抗ヒトFc特異抗体又は抗ヒトカッパ鎖抗体でコーティングした。重鎖を検出するために,抗ヒトIgG Fc特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。無傷ImmunoBodyを検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトFc特異コーティング抗体と組み合わせて使用した。軽鎖を検出するために,抗ヒトカッパ鎖特異HRP抗体を抗ヒトカッパ鎖特異抗体を組み合わせて使用した。 G:サンドウィッチElisaによるDCIB53を導入されたCHO−Sトランスフェクションの上清からの重鎖ImmunoBodyの測定。プレートを抗マウスFc特異抗体でコーティングした。重鎖を検出するために,抗マウスIgG2a特異HRP抗体を使用した。
【図49B】続き
【0117】
【図50A】ImmunoBody免疫は,免疫反応を強化し,全抗原から観察される制御を克服する。 A:HLA−A2トランスジェニックマウス(HHDII)に,0日目,7日目,14日目に,ImmunoBody DNAコンストラクトDCIB15又はpcDNA3ベクターの全gp100抗原で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100ペプチド又は対照に対して分析した。反応は,spots/million脾細胞として測定した。 B:抗CD25抗体(PC61)400μgを腹腔内に注入することにより,C57BI/6マウスからCD25陽性細胞を枯渇させた。続いて,CD25枯渇マウス及び非枯渇動物の両方に,4日目,11日目,18日目に,ImmunoBody DNAコンストラクトDCIB15又はpOrigベクターの全TRP2抗原で免疫した。23日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド又は対照に対して分析した。反応は,spots/million脾細胞として測定した。 C及びD:HHDIIマウスを,未処理(c),又は,400μgのPC61mAbを腹腔内投与にて処理した(d)。4日後,全てのマウスにTie2 C200HFc DNAコンストラクトで免疫した。DNA免疫を7日間隔で計3回繰り返した。最後の免疫の6日後,脾細胞を採取し,インビトロIFNγ ELISPOTアッセイにより,1μg/mlのTie−2からの各予測CTLエピトープで再び刺激した。バーは,背景制御に正規化された個々のマウスの三重値の平均を示し,エラーバーは,平均からの標準偏差を示す。 E及びF:HHDIIマウスを,未処理(e)(n=3),又は,400μgのPC61抗体を腹腔内投与にて処理した(f)(n=2)。4日後,全てのマウスに100μgのZ12ペプチド及び100μgのZ48ペプチドをIFAで1:1に混合して(皮下)免疫した。最初のペプチド免疫の7日後,反復ペプチド免疫を施した。最後の免疫の14日後,脾細胞を採取し,インビトロIFNγ ELISPOTアッセイにより,1μg/mlのZ12ペプチド(ブラックバー)又は培地単体(オープンバー)で再び刺激した。バーは,三重値の平均を示し,エラーバーは,平均からの標準偏差を示す。 G:HHDIIマウスに,IFA(皮下)で1:1に混合した100μgのZ12ペプチドで免疫した。最初のペプチド免疫の7日後及び14日後に,反復ペプチド免疫を施した。最後の免疫の7日後,脾細胞を採取し,インビトロIFNγ elispotアッセイにより,ペプチド濃度の増加に対するエピトープ特異的反応の有無を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 H:HHDIIマウスに,0日目,7日目,14日目に,遺伝子銃により,ImmunoBody DNAコンストラクトDCIB71で免疫した。最後の免疫の7日後,脾細胞を採取し,インビトロIFNγ elispotアッセイにより,ペプチド濃度の増加に対するエピトープ特異的反応の有無を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【図50B】続き
【0118】
【図51】DCIB71の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のTie−2 Z12エピトープ(ILINSLPLV)及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0119】
【図52】DCIB72の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H2のTie−2 Z12エピトープ(ILINSLPLV及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0120】
【図53】ImmunoBodyフレームワークに取り込まれるCTLエピトープは,処理され,発現され,インビボで免疫反応を引き起こす。 A:C57BI/6マウスに0日目,7日目,14日目にCDR H1のgp100エピトープ又はCDR H2のTRP2エピトープ(それぞれ,IB17及びIB18)を含むImmunoBodyDNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,gp100ペプチド又はTRP2ペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,CDR H2のTRP2エピトープを含むImmunoBody重鎖での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 C:C57BI/6マウスに0日目,7日目,14日目に,CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びCDR L1ヒトIgG1のHepB CD4(DCIB15),又は,CDR H1のgp100エピトープ,CDR H2のTRP2エピトープ,及びマウスIgG2a定常領域を有するCDR L1のHepB CD4エピトープ(DCIB53),を含むImmunoBodyDNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,HebBヘルパーペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 D:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB15又はDCIB53での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。 E:HLA−DR4トランスジェニックマウスに0日目,7日目,14日目に,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びCDR H3ヒトIgG1のgp100DR7(DCIB54),又は,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びマウスIgG2a定常領域を有するCDR H3のgp100DR7エピトープ(DCIB64),を含むImmunoBody DNAコンストラクトで免疫した。19日目に,elispotアッセイにより,IFNγ脾細胞を,TRP2ペプチド,gp100DR4ヘルパーペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。 F:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,DCIB54又はDCIB64での免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【0121】
【図54A】DCIB53の配列 発現ベクターpDCOrig moigG2a内のマウス重・軽完全長鎖のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のGP100210Mエピトープ(TIMDQVPFSV),H2のTRP2エピトープ(SVYDFFVWL),及びL1のHepB CD4エピトープ(TPPAYRPPNAPIL)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【図54B】続き
【0122】
【図55A】DCIB64の配列 発現ベクターpDCOrig moigG2a内のマウス重・軽完全長鎖のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1のgp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【図55B】続き
【0123】
【図56】免疫プロテアソーム処理は,ImmunoBodyコンストラクト内のエピトープからの反応の生成において重要である。 HHDIIマウスに0日目,7日目,14日目に,CDR H1のgp100209−217エピトープ(DCIB41),又は修飾型CDR H1のgp100210Mエピトープ(DCIB15),を含むImmunoBodyコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,gp100209−217ペプチド又はgp100210Mペプチド,及び対照に対して分析した。反応は,spots/million脾細胞として測定した。
【0124】
【図57】異なる免疫方法は,ImmunoBodyワクチンからの免疫反応を引き起こすのに効果的である。 A:C57BI/6マウスに0日目,7日目,14日目に,遺伝子銃により,エレクトロポレーションとともに又は伴わずに筋肉内で,又はエレクトロポレーションとともに又は伴わずに皮肉内で,ImmunoBody DNA(DCIB15)で免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド,HepBヘルパーペプチド,及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。 B:IFNγ elispotアッセイにおけるペプチド濃度の増加に対する反応を測定することによって,異なるルートによる免疫マウスからの脾細胞のTRP2エピトープに対する活性を分析した。反応は,spots/million脾細胞として測定し,50%の最大エフェクター機能を与える濃度として活性を与えた。
【0125】
【図58】ImmunoBody免疫は,白斑様色素脱失を引き起こし,腫瘍攻撃を阻止する A:CDR H2のTRP2エピトープ及びCDR L1のHepB CD4エピトープ(DCIB18)を含むImmunoBody DNAによる免疫C57BI/6マウスは,免疫部位における発毛において色素脱失を示す。 B:免疫C57BI/6マウスを,2x104のB16F10 INFα細胞による3回目と4回目の静脈内免疫の間に攻撃した。肺の全身腫瘍組織量を腫瘍攻撃後49日目に評価した。全身腫瘍組織量は,全肺面積に対する平均腫瘍面積で表される。免疫マウスを,2x104のB16F10 INFα細胞による最後の皮下免疫後7日目に攻撃した。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが限度を超えたらマウスを安楽死させた。 C:腫瘍注入後46日目に評価した腫瘍の大きさ D:生存
【0126】
【図59】ImmunoBody免疫は,腫瘍の成長を著しく遅らせる。 A:C57B16マウスに,2x104のB16F10細胞を皮下注入した。腫瘍注入後4日目に,マウスにDCIB52 ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後11日目及び18日目に施した。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。経時的な腫瘍の体積をプロットした。 B:C57B16マウスに,2x104のB16F10 IFNα細胞を皮下注入した。腫瘍注入後14日目に,マウスにDCIB52 ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後21日目及び28日目に施した。11日目の免疫は,抗CTLA−4抗体の腹腔内注入と適宜組み合わせて行った。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。腫瘍移植後47日目における腫瘍の体積が示されている。 C:C57B16マウスに,2x104のB16F10細胞を皮下にて,及び抗CD25抗体を腹腔内にて,適宜注入した。腫瘍注入後4日目に,マウスにDCIB52 ImmunoBody DNA又は対照ImmunoBody DNAで免疫した。反復免疫を腫瘍注入後11日目及び18日目に施した。11日目の免疫は,抗CTLA−4抗体の腹腔内注入と適宜組み合わせて行った。腫瘍の大きさを3〜4日間隔で測定し,腫瘍の大きさが最大許容限度を超えたらマウスを安楽死させた。経時的な腫瘍の体積をプロットした。
【0127】
【図60】DCIB68の配列 ヒトIgG1 Fcのフレームにクローニングした重・軽可変領域及び発現ベクターpDCOrig内のカッパ定常領域のヌクレオチド及びアミノ酸配列。ボックス内のアミノ酸は,H1及びL3のHLA−DR7制限gp100 CD4エピトープ(GTGRAMLGTHTMEVTVYH),H2のTRP2エピトープ(SVYDFFVWL),及びH3及びL1のHLA−DR4制限gp100 CD4エピトープ(WNRQLYPEWTEAQRLD)を示す。単一のコンストラクトからの可変重・軽領域の移転に利用されるHindIII/AfeI制限部位及びBamHI/BsiWI制限部位が強調されている。
【0128】
【図61】免疫反応は,異なるベクターバックボーンから発現されたImmunoBodyコンストラクトから生成されうる。 C57BI/6マウスに,0日目,7日目,14日目に,CDR H1のgp100DR4エピトープ,CDR H2のTRP2エピトープ,及びpVaxベクター(VaxDCIB54,C1−3)の等価コンストラクトであるCDR H3ヒトIgG1(DCIB54,B1−3)のgp100DR7エピトープ,を含むImmunoBody DNAコンストラクトで免疫した。19日目に,IFNγ elispotアッセイにより,脾細胞を,TRP2ペプチド及び培地制御に対して分析した。反応は,spots/million脾細胞として測定した。
【発明を実施するための形態】
【0129】
[実施例]
[方法]
DNAベクターの生成
ベクターpSVgptHuigG1及びpSVhygHuCk(Biovation Ltd)内のSC100クローンVHd VKb(WO01/88138)の脱免疫されたマウスの重・軽可変領域をPCRによって増幅した。VH及びVL領域PCR生成物を,HindIII/AfeI及びBamHI/BsiWI部位を用いてヒトIgG1及びカッパ定常領域のフレームにクローニングし,単一鎖コンストラクトpOrigHIB及びpOrigLIB(図1及び図2を参照)を生成した。完全長キメラ重鎖及びカッパ鎖の配列をジデオキシ鎖終結法によって確認した(Sanger et al, Proceedings of the National Academy of Sciences of the United States of America 1977;74: 5463−7)。キメラ重・鎖鎖のDNA及び翻訳されたタンパク質配列を,それぞれ,図3及び図4に示す。相補性決定領域(CDR)の位置が示されている。
【0130】
6つのアミノ酸を保持する重CDR2領域を除いて,重・軽鎖のCDRを完全に取り除き,独特な制限酵素部位と交換した。これは,制限酵素部位の生成を可能にする除去用配列の片側の領域を入念に検討することによって達成した。これらの独特な制限部位を使用してDNAを開き,抗原エピトープをコードするオリゴヌクレオチドを挿入することができる。制限部位を生成し損なったほとんどのフレームワーク配列は,エピトープにプライマーを含むことで置換され,翻訳中にアミノ酸が保持されること,及び配列がフレーム中にとどまることを確実にする。表1は,すべてのCDRの選択された酵素部位及びエピトープオリゴヌクレオチド配列を記載している。
【0131】
CDR領域を取り除き,図5に示すように重複伸長PCRによって独特な制限部位を置換した。重可変領域の場合,オリゴヌクレオチドH1,H2,及びH3(表2を参照)を3つのCDRの各々を置換するように設計した。各特異的プライマーは,酵素部位の両側が取り込まれる10〜20bpの配列を含む。ヒトIgG1定常領域に結合する一般的なリバースプライマーhuHeClonR(表2を参照)と併用して,一回目のPCRは,1μlの鋳型プラスミドpOrigHIB,2μlのdNTPS(2.5mM),5μlの10 x taqポリメラーゼ緩衝剤,1μlのフォワード・リバースプライマー(25pmols),滅菌蒸留水で最終体積が50μlとなるように作り上げた5ユニットのtaqポリメラーゼ(New England Biolabs)から構成した。反応は,最初の変性を95℃で5分受け,その後,95℃で30秒が35サイクル,55℃で1分(アニーリング),72℃で1分(伸長)と続いた。最後のサイクルは,Techne PHC−1プログラム可能循環反応器を用いた10分の伸長を含んだ。同様に,軽可変領域の場合,オリゴヌクレオチドL1,L2,及びL3を3つのCDRの各々を置換するように設計した(表2を参照)。一回目のPCRは,ヒトカッパ鎖及び鋳型pOrigLIBの定常領域に結合するリバースプライマーhuLiClonR(表2を参照)を用いた以外は,上述のとおり構成した。
【0132】
(表1)CDR置換酵素及びエピトープオリゴヌクレオチド配列のリスト
【0133】
次に,1μlの結果として生じるPCR生成物を,上記のように構成したCMVフォワードプライマーとともに,リバースプライマーとして後続のPCRで使用した。450bpの増幅されたDNA断片を,TA TOPOベクターpCR2.1(Invitrogen)並びにCDRを欠くVH及びVL領域の増幅及び制限部位の置換を確認するよう配列したクローンへ直接クローニングした。
【0134】
可変重・軽内のCDRを対応酵素H1,H2,H3,L1,L2,及びL3単体,組み合わせ,及び全体と置換した(図6及び図7)。次に,HindIII/AfeI及びBamHI/BsiWIを用いて,親の野生型脱免疫SC100 VH及びVL領域の直接置換により,異なるバージョンをpOrigHIB及びpOrigLIBに挿入した。これにより,単一又は複数のエピトープ(同じ又は異なる抗原からのもの)を含む分子の生成が可能になった。
【0135】
(表2)プライマー
【0136】
抗原エピトープの一本鎖ベクターのCDR部位への挿入
多数のCD8 CTL及びCD4ヘルパーエピトープが表3にリストアップされている。もっとも,任意のエピトープを一本鎖ベクター内の任意の部位に容易に挿入することができるが。例えば,TRP2エピトープのpOrigHIBのH2部位への挿入を以下のように行った。
【0137】
相補オリゴヌクレオチドを,翻訳中エピトープを発現するヌクレオチド配列をコードするように設計した。エピトープをコードするDNA配列の横には,対応CDRヌクレオチドが配置され,翻訳中アミノ酸が保持されること,及び配列がフレームにとどまることを確実にする(表1を参照)プライマーは,合成(MWG)及び5’末端リン酸化のために送られる。
【0138】
【0139】
相補オリゴヌクレオチドを滅菌再蒸留水で最終濃度1mg/mlまで再懸濁し,10μlの各プライマーとの反応を起こすことにより,TE緩衝剤とともに最終体積が50μlになるまでアニールした。反応は,95℃で5分(0.1℃/秒),72℃で20分(0.1℃/秒),55℃で20分,と繰り返し,その後4℃で保持した。
【0140】
H2部位への挿入のため,ベクターpOrigHIB H2及び/又はpOrigHIB H1H2をMscl制限消化を引き起こすことにより線状化し,37℃で一晩培養した。該消化を1.5%のアガロースゲルで電気泳動し,切断ベクターをゲル抽出により精製した。線状化ベクターのセルフライゲーション(self ligation)を防止するために,仔牛小腸由来アルカリホスファターゼ(CIAP)5ユニット及び滅菌蒸留水で最終体積100μlに作製した10 x NEB Buffer 3により処理及び37℃で一晩培養することでベクターの5’末端のリン酸基を除去した。脱リン酸化ベクターを精製し,。ユニバーサルプライマーCMVフォワードを使用して単一ベクター内に配列することにより,エピトープの挿入を確認した。
【0141】
(表3)CTL及びヘルパーエピトープ
【0142】
二重発現ベクターpDCOrigへの移転
すべてのエピトープが単一ベクター内のVH及びVL部位に取り込まれると,各々のヒト定常領域のフレームのHindIII/AfeIを用いて二重発現ベクターpDCOrigに移転される。ImmunoBody二重発現ベクターpDCOrigを生成するために,CMVプロモーターに隣接位置にある平滑末端制限エンドヌクレアーゼNrulを用いて線状化した。pOrigLIBを平滑末端Nrul及びHpalエンドヌクレアーゼにより消化して,CMVプロモーター,脱免疫ヒトカッパ鎖及びBGH polyAシグナルからなる全軽鎖発現カセットを摘出した。ゲル電気泳動,並びに線状化ベクターpOrigHIB及び軽鎖発現カセットの単離及びゲル抽出の後に,ベクターを脱リン酸化し,軽鎖発現カセットを結合してコンストラクトpDCOrigを形成した(図8)。pDCOrig内の軽鎖カセットの配向を制限分析により確認した。
【0143】
pDCOrigは,同じコンストラクト内に結合した重・軽鎖両方の遺伝子コード配列を含み,イントロン配列及び2つのベクター系を除去する。発現は,高レベルCMV前初期プロモーターその他のDNA制御要素(ウシ成長ホルモン)により促進される。選択マーカーzeocinもまた,発現及び生成効率を最大化するために含まれている。該ベクターの慎重な設計は,可変領域及び定常領域の接点における独特の制限酵素を保持し,可変領域の異なる組み合わせを作る手早く簡単な方法を提供する(エピトープ挿入。図8を参照のこと)。
【0144】
(表4)pDCOrigコンストラクト
【0145】
pDCOrig IB15 CH1ストップの生成
Quik change site directed mutagenesis kit (Stratagene)並びに相補オリゴヌクレオチドorigstophuHeCH1 Forward及びOrigstophuHeCh1リバースプライマー(表2を参照のこと)を製造者の指示する通りに用いて,ストップコンドンを,コンストラクトpDCOrig IB15内のヒトIgG1定常領域のCH1ドメインの後に取り込んだ。ストップコンドンの取り込みは,DNAシーケンシングにより確認した(図9)。
【0146】
pDCOrig IB15からのリーダー配列の除去
ベクターpDCOrig IB15の重・軽鎖のリーダー配列を除去するために,鋳型pDCOrig IB15を,リバースプライマーhuHeClonR及びhiLiClonRそれぞれと結合したフォワードプライマーpOrig軽ノーリーダー及びpOrig重ノーリーダーとともに使用してPCRを引き起こした。増幅断片は,ベクターpCR2.1(Invitrogen)に結合したTA TOPO及びシーケンシングにより確認したクローンであった。リーダーを欠くIB15 VH及びVL領域の両方を,HindIII/Afel及びBamHI/BsiWI部位を用いてpDCOrig IB15にクローニングした。VH及びVL領域のDNA配列及び翻訳を図10及び図11それぞれに示す。
【0147】
ImmunoBody二重発現ベクターpDCOrigのヒトIgG2及びIgG3アイソタイプの設計
AfeI及びEcoRVそれぞれを鋳型pOTBhuigG3(イメージクローン4566267MGC45809)とともに取り込むhuigg3フォワード及びリバースプライマー(表2)を用いてPCRにより,ヒトIgG3定常領域を増幅した。同様に,鋳型pTOB7 huiG2(イメージクローン6281452MGC71314)とともにigG2フォワード及びリバースプライマー(表2)を用いて,ヒトIgG2定常領域を増幅した。
【0148】
両断片は,pCR2.1に結合したTOPO及び確認した配列であった(表12及び表13)。コンストラクトpDCOrigIB15内のhuigG1定常領域を,AfeI及びSapI部位を用いて重可変のフレームにクローニングされたhuigG2及びhuigG3の両方と効果的に置換し,pDCOrigIb15 huigG2及びpDCOrigIB15 huigG3(図14)を生成した。両ベクターは,可変・定常領域の接点に同じ独特の制限部位を保持する。これにより,すべてのヒトアイソトープ単一及び二重鎖ImmunoBodyベクター間の可変領域の容易な交換が可能となる。
【0149】
ヒトIgG1 Fcγ及びヒトIgG2レセプター結合ドメインの突然変異
CH2ドメイン内のhuigG1結合モチーフのアミノ酸E233 L234 L235をhuig2のP233 V234 A235と置換するために,突然変異を組み込んだPCRによって短い部分を再増幅した。置換及び構成的制限部位AhdIを含むリバースプライマーhuigG1 PVA RevをフォーワードプライマーHIBF(表2)及び鋳型pDCOrig IB15とともに利用した。結果として生じる断片はベクターpCR2.1(Invitrogen)に結合した。配列確認後,プラスミドpDCOrig IB15 huigG1(図15)の単一カッターAgeI/AhdI部位に挿入することにより,野生型配列を突然変異を含む部分と効果的に置換した。
【0150】
コンストラクトpDCOrig IB15 huigG2のhuigG2定常領域内のアミノ酸P233 V234 A235もまた,huigG1結合モチーフELLGと置換した。前述同様に,置換及び構成的制限部位AhdIを含むリバースプライマーhuigG2 ELLG Rev(表2)をフォーワードプライマーHIBF(表2)及び鋳型pDCOrig IB15ヒトIgG2とともに利用した。断片は,ベクターpCR2.1に結合したTA TOPOであった。配列確認後,プラスミドpDCOrig IB15 huigG2(図16)のAgeI/AhdI部位を用いて,野生型配列をhuigG1結合モチーフを含む部分と置換した。
【0151】
pDCOrigマウスIgG2aプラスミドDCIB53及びDCIB63の生成
二重発現ベクターpDCOrigのマウスIgG2a型を生成するために,ハイブリドーマ細胞株337から単離したtotal RNAからcDNAを合成した。マウスIgG2a定常領域の増幅のために,制限部位AfeIを含むフォワードプライマーmigG2aC1AfeF2を,ストップコンドンの後のXbal部位を含むリバースプライマーmigG2aXbaRAとともに用いた。PCR断片は,ベクターpCR2.1に結合したTOPOであった。配列確認後,マウスIgG2a定常領域を摘出し,マウス重可変領域のフレームでヒトIgG1を効果的に置換するベクターpOrigHIBのAfeI/XbaI部位へクローニングした。BamHI及びXhoI部位を,翻訳中,マウスIgG2a定常領域のアミノ酸配列を変更することなく,連続してQuik chage site directed mutagenesis kit (Incitrogen)及び相補プライマーMoigG2BamHIFOR及びREV,MoigG2XholFOR及びREVそれぞれを用いて部位特異的突然変異誘発法により除去した。これは,ファンいつ鎖ImmunoBodyベクターpMoOrigHIB(図17a)を生成した。MoigG2a定常領域を含むpMogOrigHIBの部分は,AfeI及びSV40プロモーターに位置する単一カッターAvrIIを用いて,単一コンストラクトからマウス重可変領域のフレームの二重発現ベクターpDCOrig IB15へ移動し,ヒトカッパ領域をなお含む中間ベクターpDCOrigIB15MoigG2a hukappaを生成した。
【0152】
マウスカッパ領域の増幅のために,ストップコンドンの後のXhoI部位を取り込むBsiWI部位及びMoLCXhoIを含むプライマーMoLC1BsiF1とともに,cDNAを鋳型として用いた。増幅断片は,前述同様に,ベクターpCR2.1にクローニングしたTOPOであった。マウスカッパ領域を摘出し,BsiWI/XhoIを用いてヒトカッパ領域を置換するImmunoBodyベクターpDCOrigLIB L1及びpDCOrigLIB hepB help/L1へ結合し,中間ベクターpMoLIBL1Bsi及びpMoLIB HepB help/L1Bsiを生成した。Immnobodyシステムは,可変領域及び定常領域の接点における独特の制限部位を用いた可変領域の移動を伴う。一方,マウス重可変及びmoigG2a定常間の接点は,AfeI部位(すべてのヒトImunoBodyベクターに存在)に適応することができ,翻訳中アミノ酸配列を変更しない。マウス可変及びカッパ間の領域は疑わしい。この接点における配列を分析すると,アミノ酸配列を変更しない独特の制限部位を全く取り込むことができない。この接点のBsiWI部位を除去して野生型配列に戻した。これは,全マウス完全長鎖を重複PCRによって増幅することにより実現した。最初のPCRは,接点及び隣接領域に野生型配列を含むフォワードプライマーMoKappaSDMforを用いて構成し,BsiWI,BGHリバースプライマー,及び鋳型としての中間軽鎖ベクターpMOLIB1Bsi及びpMoLIB hepB help/L1 Bsi,のそれぞれを除去した。一回目のPCRのおよそ430bpの増幅断片をBamHI部位を含むフォワードプライマーImmunoLikozForとともにリバースプライマーとして用いた。増幅した完全長マウスカッパ鎖は,pCR2.1に結合したTOPO及び確認した配列であった。pCR2.1のL1部位にhepB helpを含む完全長マウスカッパ鎖を摘出して,ヒトカッパ鎖を置換する中間二重発現ベクターpDCOrigIB15MoigG2aH hukappaのBamHI/XhoI部位にクローニングし,マウス二重発現ベクターpDCOrigIB GP200210m/H1 TRP2/H2 HepB help/L1 moigG2a(DCIB53,図17B及び図54)を生成した。
【0153】
同様に,L1部位を含む完全長マウスカッパ鎖を摘出して,ヒトカッパ鎖を置換する中間二重発現ベクターpDCOrigIB15MoigG2aH hukappaのBamHI/XhoI部位にクローニングし,空のL1部位を有するマウス二重発現ベクターpDCOrigIB15moIgG2aを生成した。野生型軽可変領域を有するコンストラクトを生成するために,相補5’リン酸化プライマーwtkappavarL1for及びrev(表2)をアニールして,上述したEcoRVによる線状化後にL1部位に挿入した。最後に,GP100DR7/H1 TRP2/H2及びGP100DR4/H3を含むDCIB54の重可変領域を,HindIII/AfeIを用いて移動し,pDCOrig GP100DR7/H1 TRP2/H2 GP100DR4/H3 moigG2a野生型カッパ(DCIB68,図17c及び図60)を生成した。
【0154】
調節DNAワクチン要件のためのImmunobody二重発現ベクターpDCOrigからの真核SV40プロモーターの除去
Nhe1部位を取り込むフォワードプライマーSV40PremFOR及び鋳型pOrigHIBを有するSV40remREVリバースプライマー(表2)を用いて,EM7細菌プロモーター及びzeocin遺伝子を増幅した。結果として生じる511bpPCR断片は,結合しシーケンシングにより確認したpCR2.1 TOPOであった。EM7プロモーター及びzeocin遺伝子の一部をpCR2.1のNheI及びFseIを用いて摘出し,pOrigHIB H1に直接クローニングし,効果的にSV40プロモーターを除去した。NheI部位は,SV40プロモーターの前に存在し,一方,FseI認識配列は,ベクターのzeocin遺伝子内の単一カッターである。配列確認後,より大きな部分が単一ベクターからhuigG1,BGH polyA,EM7の末端をコードするpDCOrig IB68ベクター並びにSapI及びFseIで消化するzeocin遺伝子の一部に移動し,二重発現ベクターからSV40プロモーターを効果的に除去する。
【0155】
FDA規制準拠pVax1(Invitrogen)用pDCOrig骨格の変更
Immunobody完全長ヒトIgG1重鎖をHindIII及びXbalを用いてコンストラクトDCIB54から摘出し,ベクターpVax1のMCS内のこれらの部位(図18A)に挿入した。二重鎖発現ベクターのpVax型を生成するために,CMVプロモーターの隣接位置にある平滑末端制限エンドヌクレアーゼNrulを用いてpVaxIB54HIBを線状化した。平滑末端Nrul及びHpalエンドヌクレアーゼによりpOrigLIB(図18B)を消化し,CMVプロモーター,Immunobodyヒトカッパ鎖,及びBGH polyAシグナルからなる全軽鎖発現カセットを摘出した。ゲル電気泳動,並びに線状化ベクターpVaxIB54HIB及び軽鎖発現カセットの単離及びゲル抽出の後に,ベクターはリン酸化し,軽鎖発現カセットは結合してコンストラクトpVaxDCIB54(図18C)を形成した。pVaxDCIB54内の軽鎖カセットの配向は,制限分析により確認した。pVaxDCIB54は可変/定常領域の接点に同じ独特の制限部位を保持し,全てのヒトアイソタイプ単一及び二重鎖Immunobodyベクター間の可変領域の容易な交換を可能にする。例えば,pVaxDCIB68(図60)を生成するために,Gp100DR4/L1及びGp100DR7/L3を含むマウス軽可変領域をBamHI/BsiWIを用いてDCIB68から摘出し,pVaxDCIB54にクローニングし,軽野生型可変領域を効果的に置換した。
【0156】
pOrigマウスTRP2及びpCDNA3 Gp100の生成
pOrigマウスTRP2を生成するために,細胞株B16F10から単離したtotal RNA5μgから合成したcDNAを,プライマーマウスTRP2フォワード及びリバース(表2)をHindIII又はEcoRV部位それぞれとともに用いて,完全長マウスチロシナーゼ関連タンパク質2の増幅用鋳型として用いた。完全長TRP2は,ベクターpOrigHIBのHindIII/EcoRV複数クローニング部位に結合した。完全長マウスGP100もまた,EcoRV及びXhoI部位をそれぞれ含む設計されたマウスGP100フォワード及びリバースプライマーを用いてcDNAから増幅した。PCR生成物は,哺乳類発現ベクターpCDNA3(Invitrogen)のEcoRV/XhoI部位にクローニングした。両プラスミドは,制限分析により同定し,DNAシーケンシングにより確認した。
【0157】
サンドウィッチElisa
Falcon社製の96−ウェルフレキシブルプレートを50μlの抗ヒトIgG,Fc特異抗体(Sigma I2136)又はPBSで10μg/mlの抗ヒトカッパ軽鎖抗体(Dako A0191)で一晩4℃でコーティングした。プレートは,Skan Washer 400(Molecular Devices)を用いて200μl/well PBS−Tween 20(0.05%)で3回洗浄し,ウェルは,PBS(1% FSG/PBS)の1% Ffish skin gelatin(Sigma)で塞いだ。発現ImmnoBody又は精製ImmunoBodyタンパク質(50μl)を含む組織培養上清を三重にウェルに添加し,プレートを室温で1時間培養した。プレートを1%FSG/PBSで洗浄し,結合したImmunoBodyを50μl/wellのペルオキシダーゼ抱合抗ヒトIgG,Fc特異抗体(Sigma A0170)又は抗ヒトカッパ軽鎖抗体(Sigma A7164)を添加することにより検出し,1%FSG/PBSで1/2000に希釈し,室温で1時間培養した。プレートを1%FSG/PBSで洗浄し,TMB基質(R&D Systems)を50μl/wellで添加することにより育成した。吸光度をVERSAマックスマイクロプレートリーダー(Molecular Devices)により650nmで測定した。
【0158】
マウス及び免疫
英国内務省認定のプロジェクトライセンスの下,動物実験を行った。6週齢〜12週齢の雌雄のC57BI/6(Harlan)又はHLA−A2トランスジェニック(HHDII)(Pasteur Institute,Paris)を用いた。合成ペプチド(manufactured by John Keyte, Department of Biomedical Sciences, Nottingham University, UK)をフロイント不完全アジュバントにより乳状にし,皮下投与により注入した。製造者の使用説明書を用いて1.0μmの金粒子上にDNAをコーティングし,Helios Gene Gun(BioRad)により皮内投与した。各マウスは,1μgのDNA/免疫を受けた。極短電気パルスにより直後注射とともに裸のDNA溶液をも皮内又は筋肉内(10μg/免疫)投与した。マウスに0,1,2週間隔で免疫し,3週目に脾臓を除去した。免疫の4日前における400μgの抗CD25抗体(PC61)の腹腔内注入により,又は,2回目の免疫と同時の200μgの抗CTLA−4抗体の腹腔内注入により,インビボでの細胞サブセットの枯渇を行った。
【0159】
インビトロ再刺激
最後の免疫の5日後,10%FBS,2mMのグルタミン,20mMのHEPESバッファ,100ユニット/mlのペニシリン,100μG/ml-1のストレプトマイシン,及び10-5Mの2−メルカプトエタノールを有する2mlのRPMI−1644のペプチドパルスリポ多糖類(LPS)ブラストとともに24ウェルプレートにて37℃で共培養した。37℃で3日間,25μg/mlのLPS(Sigma)及び7μg/mlの硫酸デキストラン(Pharmacia,Milton Keynes,UK)により脾細胞(1.5x106細胞/ml)を活性化させることにより,LPSブラストを得た。使用前に,2x107のLPSブラストを100μg/mlの合成ペプチドで1時間培養した。6日目に51Cr放出アッセイにより培養を分析した。
【0160】
51Cr放出アッセイ
標的細胞を,100μg/mlのペプチドとともに又は含まずに,1.85Mbqのクロム酸(51Cr)ナトリウム(Amersham,Essex,UK)で1時間標識化した。培養後,RPMIで3回洗浄し,100μg/mlのペプチドとともにさらに1時間培養した。5x103target/wellの96ウェルV底プレートをセットし,最終容積200μlの異なる密度のエフェクター細胞で共培養した。37℃で4時間後,50μlの上清を各ウェルから除去し,Lumapplate(Packard,Rigaweg,the Netherlands)に移動させた。Topcount Microplate Scintillation Counter (Packard)でプレートを読み取った。特異的溶解度を以下の式を用いて計算した。
特異的溶解=100×[(実験的放出−自然放出)/(最大放出−自然放出)]
【0161】
インビトロElispotアッセイ
製造者の使用説明書(Mabtech,Sweden)に従って,マウスIFNγ捕捉・検出試薬を用いてElispotアッセイを行った。手短に説明すると,抗IFNγ抗体を96ウェルImmunobilin−Pプレートのウェル上にコーティングし,複製ウェルに5x105の脾細胞を播種した。(さまざまな濃度の)合成ペプチド又は5x104の標的メラノーマ細胞をこれらのウェルに添加し,37℃で40時間培養した。培養後,ビオチン化抗IFNγ抗体,並びにストレプトアビジンアルカリホスファターゼ(strepatavidin alkaline phosphatase)及び発色基質の成長により捕捉IFNγを検出した。自動プレートリーダー(CTL)を用いてスポットを分析し数えた。エフェクター機能対ペプチド濃度のグラフを用いて,機能活性を50%最大エフェクター機能を媒介する濃度として計算した。製造者の使用説明書に従って,CD8 Dynabeads(Dynal)を用いてCD8 T細胞の脾細胞集団からの枯渇を行い,次に,インビトロelispotアッセイに追加した。
【0162】
腫瘍の研究
C57BI/6マウスを治療群に無作為に抽出し,週間隔で5週間免疫した。3回目と4回目の免疫の間に,尾静脈に1x104のB16F10 IFNαメラノーマ細胞で静脈注入することによりマウスを攻撃した。静脈注入すると,B16F10細胞は,肺へ移動し,転移を形成した。腫瘍の増殖及び苦痛に関しマウスを観察した。腫瘍攻撃後49日目に,マウスを安楽死させ,転移の有無について肺を分析した。インビトロelispotアッセイによりエピトープ及び腫瘍特異的免疫反応の有無について脾臓を分析した。
【0163】
HHDIIマウスを週間隔で3週間免疫し,最後の免疫後7日間,2x104のB16F10 HHDメラノーマ細胞で右側腹部への皮下注射により攻撃した。腫瘍の増殖を3〜4日間隔で観察し,腫瘍の大きさをキャリパーを用いて測定した。
【0164】
実施例1 ImmunoBodyコンストラクトは低レベルの無傷抗体を生成する。
CDR H1及びCDR H2それぞれのgp100 エピトープIMDQVPFSV及びTRP2エピトープSVYDFFVWL及びCDR L1のHepB CD4エピトープTPPAYRPPNAPIL(DCIB15,図19)により,安定CHO−S細胞トランスフェクタントを生成した。
【0165】
これらのトランスフェクタントの上清を,サンドウィッチelisaにより,ImmunoBodyタンパク質の発現について分析した。プレートを,抗ヒトIgG Fc特異抗体及び添加上清でコーティングした。結合ImmunoBodyを重鎖を検出する抗ヒトFc特異HRP抗体を用いて検出した。対照と比較して,およそ1μg/mlの濃度の上清に重鎖を検出した(図20a)。タンパク質Aアフィニティカラムを用いて上清からImmunoBodyを精製し,ImmunoBodyno有無を分析した。ImmunoBodyの精製は,対照と比較して,予め予想したよりはるかに低い量のタンパク質を生じた(図20b)。このように低い量の無傷タンパク質が精製されうる可能性のため,ImmunoBodyコンストラクトを,サンドウィッチElisaにより,トランスフェクト細胞の上清における重鎖及び無傷抗体の両方の発現について分析した。CDR L1のHepB CD4エピトープ及びCDR H2のSIINKEFLエピトープ(DCIB24,図21)又はCDR H1及びCDR H2それぞれにgp100エピトープIMDQVPFSV及びTRP2エピトープSVYDFFVELをCDR L3のHepB CD4エピトープTPPAYRPPNAPIL(DCIB25,図22)とともに有するコンストラクトもまた検査した。プレートを抗ヒトIgG Fc特異抗体及び添加上清でコーティングした。結合ImmunoBodyを,重鎖を検出する抗ヒトFc特異HRP抗体,又は無傷ImmunoBodyを検出する抗ヒトカッパ鎖特異HRP抗体を用いて検出した。ImmunoBodyトランスフェクタントは,高レベルの重鎖分泌を示しているが,非常に低レベルの無傷ImmunoBodyを示している(図20c及び図20d)。
【0166】
このデータは,CD8及びCD4 T細胞エピトープの重・軽鎖可変領域への取り込みが,無傷抗体の形成を防止するImmunoBodyの全体構造を崩壊させることを示している。
【0167】
トランスフェ区とCHO−S細胞の上清の分析に関する追加データは,CDRH3又はCDRL3に取り込まれたCTLエピトープを有するコンストラクトのみが無傷抗体として分泌されるということを示している(図20e)。対照的に,CDRH1又はCDRH2内のエピトープの取り込みは重の分泌を可能にはするが,軽鎖に取り込まれるものはなく分泌されても無傷抗体の量は少ない。CDRL1内のエピトープの取り込みは,重鎖のCDRH3に取り込まれるエピトープしか存在しない場合,軽鎖の低レベルの分泌をもたらす。
【0168】
実施例2 ImmunoBodyフレームワークに取り込まれたCTLエピトープはインビボで免疫反応を引き起こすように処理され発現される
既刊のTRP2のCTLエピトープaa280−288(Boom et al, The Journal of Experimental Medicine 1997;185:453−9)をCDR L1のB型肝炎ユニバーサルCD4エピトープと一緒にImmunoBodyコンストラクトのCDR H2領域に改変した。C57BI/6マウスに遺伝子銃によりImmunoBody DNAを週間隔で三度経皮で免疫した。その後,脾細胞を,IFNγ elispotにより,TRP2特異的反応について分析した。ImmunoBody DNAを免疫したマウスは,対照と比べてかなりのTRP2ペプチド特異的反応を示す一方,HepB CD4ペプチドに特異的な反応は低レベルであった(図31a)。TRP2特異的反応の活性もまた,IFNγ elispotでペプチド滴定により研究した。5つの異なる実験で試験した15匹のマウスにおいて,反応の活性は,10−9〜10−11Mペプチドに及んでいる。代表例を図31bに示す。
【0169】
TRP2特異的反応がCD8 T細胞により媒介されることを確認するために,C57BI/6マウスに週間隔で3回ImmunoBody DNAを免疫した。最後の免疫の6日後,脾細胞を単離し,IFNγ elispotにより特異的反応についてインビトロで分析した。TRP2特異的反応がCD8 T細胞により媒介されるかどうかを判定するために,CD8 T細胞をelispotアッセイによる分析前に枯渇させた。CD8 T細胞の枯渇は,TRP2特異的反応の廃止につながった。しかしながら,CD8の枯渇は,HepB CD4ペプチド反応に影響を与えなかった。これは,CD4 T細胞により媒介される可能性が高いことを示している(図31c)。
【0170】
ImmunoBody DNA免疫により起こされる反応がインビトロで標的細胞を殺傷することができるかどうかを判定するために,TRP2ペプチドパルスLPSブラストによりインビトロで刺激を与え,B16F10メラノーマ細胞に対するクロム遊離アッセイにより分析した。ImmunoBody DNA免疫マウスの脾細胞は,H−2Kb分子(B16F10 siKb)を発現しないB16F10に比べ,低レベルの表面MHCクラスI発現を有するB16F10細胞,及び高い表面MHCクラスI発現を有するB16F10 IFNα細胞の両方のすぐれた溶解を示した。B16F10 siKb細胞株に対する殺傷の廃止は,殺傷がCD8に依存し,H−2Kbにより制限されることを示す(図31d)。
【0171】
これらの結果は,ImmunoBodyフレームワークのCDR H2領域に組み込まれるTRP2(SVYDFFVWL)CD8エピトープは,MHCクラスIにより媒介される高頻度の反応を引き起こすよう処理され発現されることを示す。HepB CD4エピトープもまた,MHCクラスIIとの関係で,DNA免疫の良好なCD4媒介反応を引き起こすように処理され発現される。
【0172】
TRP2エピトープ特異的反応もまた,同じ方法を用いてコンストラクトを含む他のTRO2エピトープから分析した。TRP2エピトープの重鎖内のCDRへの取り込みは,高頻度のペプチド特異的反応につながった(図31e)。対照的に,CTLエピトープの軽鎖への取り込みは,CTL頻度の有意な減少につながった(DCIB36)。TRP2エピトープ特異的反応の活性の分析は,該反応が重鎖内のエピトープから引き起こされた場合には高い活性を示すが,軽鎖からのエピトープの発現の場合にはこれが著しく低いことを明らかにしている(図31f)。すべてのコンストラクトについて観察された高頻度・高活性ヘルパー反応(図31g)は,重鎖の分泌は,CTL反応を刺激するには好都合であるが,ヘルパー反応には好都合でないことを示している。
【0173】
実施例3 ImmunoBody DNA免疫はペプチド免疫又は全抗原免疫よりも好ましい
ImmunoBody DNA免疫の効率を分析するために,これを,フロイント不完全アジュバントにおけるペプチドエピトープによる皮下免疫,又はTRP2抗原を発現するDNAによる免疫と比較した。
【0174】
C57BI/6マウスは,IFAのユニバーサルヘルパーエピトープに結合したTRP2エピトープからなるペプチド又はDNAによる免疫を週間隔で3回受けた。ImmunoBody免疫マウスに引き起こされたTRP2及びヘルパーペプチド特異的反応は,ペプチド免疫又は全TRP2抗原による免疫により起こされる反応よりも規模の点ではるかに上回っていた。これらのペプチド特異的反応の活性のさらなる分析は,ImmunoBody DNAによる免疫マウスにより引き起こされる反応は,ペプチド免疫個体の反応よりもログ活性が高いことを明らかにした(図32b)。その後,C57BI/6マウスに引き起こされる反応を,B16F10細胞株,及び陰性対照としてB16F10 siKb細胞株に対するインビトロでの細胞傷害能力について分析した。図32cは,ImmunoBody DNA免疫マウスは,H−2Kb拘束性のインビトロでの抗腫瘍活性を示し,ペプチド免疫マウス及び全抗原免疫マウスは,同じメラノーマ細胞株の殺傷に失敗することを示している。
【0175】
ImmunoBody免疫をDC+ペプチドによる免疫とも比較した。C57BI/6マウスは,DNA又はDC+ペプチドによる免疫を週間隔で3回受けた。TRP2ペプチド特異的反応は,同等の頻度であったが,ImmunoBody免疫マウスは,DC+ペプチド免疫マウスに比べ,高い活性反応を引き起こした(図32d)。これらの反応をインビトロでB16F10メラノーマ細胞を殺傷する能力において分析した場合もこのことが証明された。ImmunoBody免疫による引き起こされる反応は,DC+ペプチド免疫マウスの反応よりも目標比率に対して低いエフェクターでの高いB16F10メラノーマの殺傷を示した。これらはまた、H−2Kbのレベルを下げたB16F20 siKbメラノーマ株の高い特異的溶解を示した。
【0176】
H−2Kb拘束性オボアルブミンエピトープ,SIINFEKL,及びアンカー修飾HLA−A2拘束性gp100エピトープ,IMDQVPFSV(210M)を含むImmunoBodyコンストラクトを,C57BI/6又はHHDIIマウスそれぞれにおける対応するエピトープペプチド免疫と比較した。マウスは,IFAのDNA又はペプチドによる免疫を週間隔で3回受けた。最後の免疫後の反応の分析は,ImmunoBody DNA免疫マウスは,ペプチド免疫マウスに比べ,高頻度のペプチド特異的反応を引き起こすことを明らかにしている(図32f及び図32g)。これらの反応を,ペプチド滴定による活性についても分析した。ImmunoBody免疫は,ペプチド免疫より著しく高い活性反応を引き起こす(図32h及び図32i)。
【0177】
ImmunoBody DNAワクチンにより引き起こされるTRP2特異的反応の大きさは,合成ペプチド又は全TRP2抗原により引き起こされる反応をはるかに上回る。しかしながら,臨床試験による証拠は,高頻度の腫瘍特異的CD8 T細胞の存在が必ずしも腫瘍退縮にはつながらず,一般的にワクチンの試用において客観的な臨床的反応率は非常に低いことを示している(Rosenberg et al, J Immunol 2005;175:6169−76; Rosenberg et al, Nature Medicine 2004;10:909−15)。腫瘍特異的T細胞の機能活性やプライミング経路などの頻度以外の要因は、ワクチンの有効性を最大化する上で主要な決定要因であることが現在明らかになりつつある。多数のグループが、高活性CD8 T細胞が抗腫瘍活性を示すことを示している(Alexander− Miller, Immunologic research, 2005;31 : 13−24; Hodge et al, J Immunol 2005;174: 5994−6004; Valmori et al, J Immunol 2002;168: 4231 −40; Zeh et al, J Immunol 1999;162: 989−94; Alexander−Miller et al, Proceedings of the National Academy of Sciences of the United States of America 1996; 93: 4102−7)。我々の研究では,ImmunoBody誘発TRP2特異的反応の機能活性の分析は,合成ペプチドによる免疫に比べ,高活性反応を生成することができることを実証した。この高活性反応はまた,インビトロで腫瘍細胞を識別し殺傷する能力の増大と相互に関連があった。APCからの信号又は反応のプライミングの経路もまた,高活性免疫反応の誘発にとって極めて重要である(Oh et al, J Immunol 2003;170: 2523−30)。
【0178】
実施例4 複数のエピトープはCDR H2部位から処理されうる
複数のエピトープを免疫反応を引き起こすようにCDR H2から処理され発現されうることを実証するために,オボアルブミンのH−2Kb拘束性エピトープSIINFEKL(DCIB24:図21)及びH−2Kd拘束性B型肝炎エピトープIPQSLDSWWTSL(DCIB21,図33)を重可変領域のH2部位に改変した。これらのImmunoBodyコンストラクトはまた,軽可変領域にI−Ab拘束性(TPPAYRPPNAPIL)エピトープB型肝炎CD4エピトープ又はI−Ad拘束性インフルエンザ赤血球凝集素(FERFEIFPKE)エピトープを含んでいた。
【0179】
C57BI/6又はBalb/cマウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0180】
C57BI/6マウスは,高頻度SIINFEKL特異的反応を示す一方,低いヘルパーエピトープ特異的反応を示した(図34a)。Balb/cマウスもまた,ヘルパーエピトープと同程度の反応を有する高頻度B型肝炎エピトープ特異的CD8反応を引き起こした。
【0181】
このデータは,CDR H2からのCD8エピトープの処理及び発現は,特異的エピトープの配列又は長さに制限されないことを示している。
【0182】
実施例5 複数のCTLエピトープは可変領域から処理されうる
エピトープがCDR領域だけでなく可変領域から処理及び発現されうることを実証すべく,フレームワーク領域の一部の除去とともにエピトープをCDR H1部位に組み込んだ。
【0183】
エピトープの例は,改変されたHLA−A2拘束性のエピトープであるgp100のIMDQVPFSV(DCIB17,図35)及びTie−2のFLPATLTMV(DCIB26,図36)である。ImmunoBodyコンストラクトはまた,CDR L1部位にB型肝炎CD4エピトープを含んでいた。
【0184】
HLA−A2トランスジェニックマウス(HHDII)に,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0185】
HHDIIマウスは,HepB CD4エピトープに対する相当の反応とともに高頻度gp100 210M特異的反応を引き起こした(図37a)。コンストラクトを含むTie2エピトープで免疫したHHDIIマウスにおける反応は,高頻度のものではなかったが,Tie2エピトープ及びHepB CD4エピトープの両方に特異的なかなりの反応を引き起こした(図37b)。
【0186】
この例のデータは,可変領域に挿入されたエピトープは,インビボで免疫反応を起こすよう処理及び発現されうることを示している。これは1つのエピトープ配列に制限されないことも明らかである。
【0187】
実施例6 複数のCTL反応は同じImmunoBodyコンストラクト内の異なるエピトープから生成されうる
前述のHLA−A2拘束性gp100エピトープIMDQVPFSVを,同じコンストラクトのCDR H2部位のHLA−A2により同様に拘束されているTRP2エピトープSVYDFFVWLとともにCDR H1に改変した。HepB CD4エピトープはCDR L1部位に存在した(DCIB15,図19)。
【0188】
HHDIIマウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4反応の有無について分析した。
【0189】
図38aは,gp100及びTRP2エピトープの両方に特異的な反応が生成される様子を示すが,TRP2特異的反応の頻度は少ない。HepB CD4ペプチドに対する反応もまた生成される。TRP2特異的反応の活性もまた,IFNγ elispotでペプチド摘定により研究した。反応の活性は,gp100エピトープの場合,10−10M〜10−11Mペプチドの範囲であり,TRP2エピトープの場合,10−9M〜10−10Mペプチドの範囲である。代表例を図38bに示す。反応がインビトロで標的細胞を殺傷することができるかどうかを判定するために,脾細胞をTRP2及びgp100ペプチドパルスLPSブラスにより6日間インビトロで刺激し,ペプチド標識T2細胞及びB16F10 HHDメラノーマ細胞に対してクロム遊離アッセイで分析した。対照B16F10メラノーマ株に比べ,B16F10 HHDメラノーマ株の特異的殺傷。反応はまた,対照と比較したペプチド標識T細胞の特異的溶解を実証した(図38c)。
【0190】
単一のImmunoBodyコンストラクトの2つのCD8エピトープを結合させることは,エピトープ間のある程度の免疫優勢をもたらすように思われる。免疫優勢エピトープは,MHCクラスIに対する最高のアフィニティを有するエピトープである。gp100及びTRP2 CD8エピトープの両方を含むコンストラクトにより免疫されたマウスとTRP2 CD8エピトープのみを含むコンストラクトで免疫されたマウスとを比較すると,TRP2反応の頻度は減少する(図38d)。
【0191】
このデータは,エピトープ特異的免疫反応が2つの異なるCD8エピトープに特異的な同じDNAコンストラクトから生成されうることを実証している。これらはまた,インビトロでの抗腫瘍活性を示す。しかしながら,サブドミナントエピトープに対する反応の頻度を抑制するある程度の免疫優勢が存在する。
【0192】
CDRH2(DCIB18)のTRP2エピトープ又はCDRH2(DCIB24)のSIINFEKLを含む個別のImmunoBodyコンストラクトを用いて同様の研究を行った。マウスに,DCIB18又はDCIB24を単体で,DCIB及びDCIB24を組み合わせて同じ部位に,又はDCIB18とDCIB24を同時にしかし別の部位にて,免疫した。免疫は週間隔で3回行い,DNAをエレクトロポレーションと併用して頚骨筋に筋肉内注入した。生成された免疫反応の分析は,マウスをDCIB18又はDCIB単体で免疫した場合に高頻度ペプチド特異的反応を引き起こすことができることを示している(図38e)。同じ部位においてこれらのコンストラクトによりマウスに免疫することは,TRP2ペプチド特異的反応の顕著な減少につながる。これは,SIINFEKLエピトープがTRP2エピトープに対して優勢であることを示す。TRP2特異的反応は,マウスに別々の部位(p=0.0026)のコンストラクトにより免疫すると回復することができる。このデータは,免疫優勢は,IB免疫により生成される免疫反応に実際影響を与えるが,これは空間的に離れた部位における免疫により解決されうることを示す。
【0193】
実施例7 無アンカー残留物改変はT細胞識別を強化しうる
前述の例は,改変gp100エピトープIMDQVPFSVが免疫優勢であり,HLA−A2に対する高いアフィニティを有することを示している(SYFPEITHIアルゴリズムを用いて予測したように。また,T2安定化アッセイで実証したように。表5)。野生型gp100エピトープITDQVPFSVは,免疫原性を持たないため,野生型エピトープと同様のHLA−A2結合活性を有しながらなおかつ免疫原性を高める無アンカー残留物で改変を行った。これらの改変エピトープをImmunoBodyコンストラクトのCDR H1部位に改変し,野生型エピトープとともに試験した(DCIB37,DCIB40,DCIB41,DCIB42,DCIB43,図39〜図43)。
【0194】
HHDIIマウスに,遺伝子銃により,ImmunoBody重鎖DNA単体で週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8反応の有無について分析した。HLA−A2(表5)に対するアフィニティを保持する野生型gp100エピトープへの2つの改変(F7L及びF7I,DCIB37,図39,DCIB40,図40)は,野生型エピトープに比べ,エピトープ特異的免疫反応を誘発する優れた能力を実証した(図44a)。
【0195】
(表5)
【0196】
実施例8 複数のCD4ヘルパー反応はインビボで免疫反応を起こすように処理及び発現されうる
CD4ヘルパーエピトープをインビボで免疫反応を起こすように処理及び発現されうるかどうかを分析するために,異なるエピトープをImmunoBodyコンストラクトのCDR L1部位に独立に改変した。これらは,インフルエンザ赤血球凝集素のI−Ad拘束性エピトープFERFEIFPKE(DCIB21,図33),HBcAgのI−Ab拘束性エピトープTPPAYRPPNAPIL(DCIB15,図19),及びgp100のHLA−DR4拘束性エピトープWNRQLYPEWTEAQRLD(DCIB35,図45)を含んでいた。
【0197】
Balb/c,C57BI/6又はHHDII及びDR4トランスジェニックマウスに,遺伝子銃により,ImmunoBody DNAで週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ eslispotによりエピトープ特異的CD4反応の有無について分析した。図46a,b,cは,3つのCD4ヘルパーエピトープのすべてがエピトープ特異的免疫反応をインビボで起こすようにCDR L1部位から処理及び発現されうることを示している。
【0198】
gp100HLA−DR4拘束性エピトープを異なるCDRからの処理及び発現についても試験した。エピトープをCDRL1(DCIB35,図45),DCRH3(DCIB54,図29),又はCDRL3(DCIB50,図47)に組み込むコンストラクトを使用して,HLA−DR4トランスジェニックマウスに週間隔で3回免疫した。図46dは,ヘルパーエピトープは高頻度ヘルパー反応を起こすように異なるCDRから効率よく処理されうることを示している。
【0199】
実施例9 CTL反応は分泌重鎖に部分的に依存するが,ヘルパー反応は分泌軽鎖を必要としない
古典的に,CD4 T細胞エピトープは,体外から獲得したタンパク質から処理され,CD8 T細胞エピトープは,体外で生成されたタンパク質から処理される。体外から獲得した抗原からのエピトープの交差提示がCD8 T細胞媒介反応を起こす証拠が現在存在する。このプライミング経路はまた,CD8 T細胞媒免疫反応の開発の点でより効率的なものであることが提案されている。最近,CD4媒介反応についても同様の発見があった。山のような証拠は,細胞内タンパク質由来のCD4 T細胞エピトープはMHCクラスIIとの関連で処理及び発現されうることを示している。
【0200】
分泌ImmunoBodyがCD8及びCD4 T細胞反応に必要であるかどうかを判定するために,CDR H1部位のHLA−A2拘束性gp100エピトープIMDQVPFSV及びCDR L1部位のI−Ab拘束性HepBヘルパーエピトープTOOAYRPPNAPILを含むImmunoBodyコンストラクトを重鎖又は軽鎖上にリーダー配列なしで作った(図10及び図11)。
【0201】
HHDIIマウスに,遺伝子銃により,ImmunoBody DNAにより週間隔で3回皮内で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4 T細胞反応の有無について分析した。反応をgp100特異的CD8反応について分析すると,ImmunoBodyコンストラクトの重鎖からのリーダー配列の除去は,エピトープ特異的反応の減少につながるが,CD44反応には影響を及ぼさないことが観察された(図48a)。重鎖からのリーダー配列の除去は,トランスフェクトCHO−S細胞による重鎖の分泌に影響を及ぼした(図48b)。軽鎖からのリーダー配列の除去,これによる軽鎖の分泌防止,は,エピトープ特異的CD8又はCD4反応に影響を与えないように思われる(図48c)。CD8反応は,重鎖上にリーダー配列が存在しない場合顕著に減少したが,CD4反応は,影響を受けなかった(図48c及び図48d)。
【0202】
このデータは,重鎖の分泌がCD8 T細胞反応の効率的誘発にとって重要であることを意味し,CD8エピトープは交差提示を受けていることを示している。第二に,それは,CD4エピトープは免疫反応を起こす細胞内ImmunoBodyに由来することを意味している。
【0203】
実施例10 タンパク質分泌の欠乏によるFcのない低下したCTL反応
この実験は,Fc領域の存在が効率的な免疫反応を達成する上で有用であるかどうかを観察するものである。転写及び翻訳を防止するストップコンドンをFcの前に組み込むことにより,CDR H2のH−2Kb拘束性TRP2エピトープSVYDFFVWL及びI−Ab拘束性HepB CD4エピトープTPPAYRPPNAPIL(DCIB15)を含むImmunoBodyコンストラクトからFc領域を除去した(図9)。
【0204】
C57BI/6マウスに,遺伝子銃により,ImmunoBody DNAを週間隔で3回経皮で免疫した。その後,脾細胞をIFNγ elispotによりエピトープ特異的CD8及びCD4 T細胞反応の有無について分析した。
【0205】
Fc領域を有さないImmunoBodyコンストラクトにより免疫したマウスは,Fc領域を有するコンストラクトに比べ,非常に低レベルの腫瘍細胞株B16F10の識別が可能な低レベルのTRP2ペプチド特異的反応を生成した(図49a)。多数の実験からのTRP2及びHepBヘルパーペプチド特異的反応の両方の分析は,Fc領域を有さないコンストラクトは著しく低いTRP2ペプチド特異的反応を生成することを示している(図49b)。しかしながら,HepBヘルパー反応は,Fc領域の除去により影響を受けない(図49c)。これは,ヘルパーが分泌されず,よって直接提示により機能している軽鎖においてヘルパーが最もよく機能することを示す我々の前述の結果と一致するものである。対照的に,CTL反応は,直接提示及び間接提示の両方により刺激され,後者はFc標的の恩恵を受けうる。あるいは,Fcストップコンストラクトは,低下した反応を説明しうる欠失重鎖の低い分泌につながる。したがって,TRP−2をコードするImmunoBodyは,IgG2(DCIB33)及びIgG3定常領域(DCIB65)により改変される。前者は,CD64に結合しないはずだが,CD32に結合しうる。また,マウスのFcレセプターIVにも結合しうる。ヒトIgG3は,CD32及びCD64の両方に結合しうる。両ImmunoBodyは,強力なCTL反応を刺激した(図49e)。これは,Fc標的が間接提示の強力な要因ではないことを示している。この問題をさらに検証するために,IgG1のFc標的ドメインを同等のIgG2ドメインと置換し,又はその逆を行った(DCIB66,67,図15及び図16)。両コンストラクトは,強力なCTL反応を刺激した(図49e)。これは,Fc結合に関連せずに可能にする重鎖のみを分泌するImmunoBodyワクチンのためでありうる。
【0206】
実施例11 ImmunoBody免疫は,免疫反応を強化し全抗原で観察された規制を克服する。それはまた,新しいT細胞エピトープの同定を可能にする
これは,ほとんどの自己抗原と対照的に,制御性エピトープを発現しない不活性担体であるT細胞エピトープをコードするヒト抗体により免疫することの第二の利点につながる。gp100エピトープ又はTRP−2エピトープを発現するImmunoBodyは,高頻度・高活性のT細胞反応(頻度1/103,活性10−10M)を刺激する一方,TRP−2のgp100全抗原は,低頻度かつ低活性でT細胞を刺激した(頻度1/105,活性10−7M)。CD25の枯渇は,抗原に対する反応を部分的に回復させたが,ImmunoBodyはなお100倍優れていた(図50a及び図50b)。
【0207】
同様に,Fcに結合するTie−2の最初の200アミノ酸をコードするDNAによる免疫は,上位10の予測エピトープに対する免疫反応を刺激しなかった。Tie−2の最初の196アミノ酸の配列をEpijen及びNetCTLオンライン予測アルゴリズムに入れた。これらの方法は両方ともHLA−A*0201結合アフィニティに加え,プロテアソーム切断及びTAP輸送を考慮に入れている。MHCpred及びSyfpeithiアルゴリズムをも予測MHC結合アフィニティのみを考慮に入れる古い予測アルゴリズムの例として用いた。全Tie−2分子は,最初の196アミノ酸に存在するエピトープに対して免疫優勢の影響を及ぼしうる追加のCTLエピトープを含みえた。したがって,全分子からの各予測エピトープのランクを得るために,Tie−2の全配列も同じアルゴリズムに入れた。ヒトとマウスで相同でないペプチドは考慮に入れなかった。複数の異なる予測アルゴリズムにより良好なCTLエピトープを発現すると確実に予測された残存ペプチドのうちの6つを選択した。これらのペプチドの各々に対して異なるアルゴリズムにより得られた相対スコアを,Z83(これまでに同定されたエピトープ)の結果とともに,表6にまとめている。
【0208】
トランスフェクトCHO−S細胞からの上清の分析に関する追加データは,CDRH3又はCDRL3に組み込まれるCTLエピトープを有するコンストラクトのみが無傷抗体として分泌されることを示している(図20e)。対照的に,CDRH1又はCDRH2内の任意のエピトープの組み込みは,軽鎖内に組み込まれるものがなく分泌されても重だが少量の無傷抗体の分泌を可能にした。任意の軽鎖のCDRL1内の任意のエピトープの組み込みは,重鎖のCDRH3に組み込まれるエピトープしか存在しない場合でも,軽鎖の低レベルの分泌につながった。
【0209】
T細胞レパートリーが任意のTie−2からの予測CTLエピトープを識別するHLA−A*0201トランスジェニックマウスに存在するかどうかを判定するために,動物にnative Tie2 C200hFc DNAコンストラクトで免疫し(Ramage et al, Int. J. Cancer 2004:110:245−250),脾細胞をELISPOTアッセイでペプチド特異的IFNγ反応についてスクリーニングした。別のグループのマウスには,DNA免疫の4日前に,前述同様に,PC61 mAbによる処理を行った後に,C200hFcにより免疫した。
【0210】
native C200hFc DNAにより免疫したマウスは,該動物が免疫の前にCD25+制御性T細胞を枯渇していたか否かにかかわらず,Z83を識別するIFNγ反応を開始しなかった。免疫前に制御性T細胞を枯渇していなかった動物から試験した任意の新しいペプチドに対する著しいIFNγ反応は見られなかったが,例外として,Z284は,平均69SFC/million脾細胞で1つの動物(M3)において反応を刺激しているように見えた(図50c及び図50d)。DNA免疫前に制御性T細胞を枯渇した動物からは,2/3の動物(M1及びM3)がZ282ペプチドによる再刺激に対するIFNγ反応を示し,平均値は,それぞれ,320及び94SFC/million脾細胞であった。M1はまた,Z285による再刺激に対する部分的反応を示し,平均が85SFC/million脾細胞であった。
【0211】
Tie−2からの予測CD8+エピトープのインビボスクリーニングの明らかに矛盾する結果は,免疫優勢の結果であろう。なぜなら,CD25+細胞がない場合native C200HFcにより免疫したマウスからのIFNγ反応は,1つの優勢ペプチドに偏るように思われるためである。他の潜在CD8+エピトープからの競争がない場合,Z282エピトープに反応することのできるT細胞レパートリーをさらに調査するために,HHDマウスのグループに,CD25+制御性T細胞の存在下又は非存在下で,IFAのZ282ペプチドにより免疫した。
【0212】
Z282を免疫したマウスはすべて,CD25+制御性T細胞の存在下で免疫された場合でも,ペプチド特異的IFNγ反応を開始した。非枯渇動物のマウス3は,最高の反応を開始し,平均値が215SFC/million脾細胞であった。枯渇動物からの最高の反応はマウス2から観察され,平均値が137SFC/million脾細胞であった(図50e及び図50f)。
【0213】
ペプチド免疫により誘発された反応は低頻度のままである。エピトープを全抗原により生成される制御作用から除去した場合に高頻度反応が生成されるかどうかを観察するために,z282(z12としても知られる)エピトープをL1のHepB CD4とともにImmunoBodyコンストラクトのH1部位に改変した(DCIB71,図51)。その後,HLA−A2トランスジェニックマウスに(遺伝子銃により)z12ペプチド又はImmunoBody DNAで週間隔で3回免疫し,その後,エピトープ特異的免疫反応について分析した。z12ペプチドにより免疫したすべてのマウスは,低頻度及び低活性エピトープ特異的反応を示す(図50g)。しかしながら,z12エピトープをImmunoBodyコンストラクトに改変すると,高頻度及び高活性反応がすべてのマウスに誘発される(図50h)。
【0214】
要約すると,免疫前にCD25細胞を枯渇させると,免疫反応がTie2エピトープの3/10にまで刺激された。同様に,これらのエピトープの1つがペプチドとして発現されると,弱い免疫反応が生成されえた。しかしながら,このエピトープがImmunoBodyコンストラクト内で発現されると,高頻度及び高活性のT細胞反応が生成された。これらの結果は,Tie−2の最初の200アミノ酸内にはCTL反応を抑制するTregエピトープが存在することを示している。これらのTreg又はそのエピトープを除去すると,ImmunoBody内での発現によりさらに強化されうる自己抗原に対する反応を発見することが可能である。
【0215】
(表6)予測されたTie−2からのHLA−A*0201拘束性CTLエピトープ
1:ペプチド名。
2:Tie−2分子内のアミノ酸残基開始位置。
3:ペプチド配列。
4:Epijenウェブサーバを用いた予測。スコアは,IC50nMの単位で与えられ,スコアが低いほど高アフィニティのペプチドであることを表す。
5:NetCTL 1.2ウェブサーバを用いた予測。スコアは,3つの個々の予測方法の加重和を表し,1に対するMHC結合の相対的重み付けを伴う。*は,周知のエピトープに対して得られるデータセットからのCTLエピトープのカットオフ値として認定される閾値0.75以上のスコアを示す。
6:SYFPEITHIプログラムを用いた予測。HLA−A*0201結合ペプチドの最大スコアは36である。
7:MHC及びTAPに対するペプチドアフィニティを予測するMHCPred相加法を用いた予測。スコアは再びIC50nMの単位で与えられ,スコアが低いほど高いアフィニティのペプチドであることを表す。推奨されるIC50値は,0.01〜5000nMである。
すべての予測方法において,ランク値は,196アミノ酸断片からエピトープが予測される順序を示し,括弧内の値は,全Tie−2分子からのランク予測を表す。インフルエンザA型ウイルスのマトリクスタンパク質由来の周知のZ18 CTLエピトープに対して得られる値が比較のために含まれている。
【0216】
実施例12 T細胞ヘルプの提供における異種Fcの役割及び抗原特異的T細胞ヘルプの要件
高活性T細胞反応の刺激は,通常,プライミング中T細胞ヘルプを必要とする。これは,軽鎖内にコードされるHep B外来ヘルパーエピトープによって提供されるであろうと従来考えられていた。実際,強力なヘルパー反応は,このエピトープに対して生成された。しかしながら,重鎖と軽鎖の両方の鎖が同じAPCにより生成される場合,Hep Bエピトープは直接提示のためのヘルプを提供することができたが,重鎖は分泌され,軽鎖は分泌されなかったため,間接提示重鎖のためのヘルプを提供しえたとは考えにくい。なぜなら,これは,同じ抗原提示細胞によって取り込まれるとは考えにくいからである。したがって,マウスに,重鎖のみをコードするDNAベクターにより免疫した。高頻度・高活性のCTL反応がなお生成された(図53a及び図53b)。これは,ヘルプを必要としないか,あるいは,マウスにおいて移出であるヒトFcが結合外来ヘルプを提供していることを意味する。したがって,マウスIgG2aコンストラクトを重鎖及び軽鎖について分析し(図49g),免疫反応の生成についてスクリーニングした(DCIB53,図54)。なお高頻度・高活性のT細胞反応を生成したが,ヒトコンストラクトと同程度には強力でなく,異種Fcは結合ヘルプを提供していた(図53c及び図53d)。その後,CTL生成のための結合ヘルプ,及び腫瘍環境内の炎症を刺激する抗原特異的T細胞ヘルプの両方を提供するようにHLA−DR4 gp100エピトープをマウスIgG2aコンストラクト(DCIB64,図55)に組み込んだ。これらのコンストラクトは,高頻度・高活性のCTL及びヘルパー反応を刺激する(図53e及び図53f)。同じエピトープを発現するgIgG1コンストラクトをヒト患者に用いることができる。
【0217】
実施例13 免疫プロテアソーム処理はImmunoBodyコンストラクト内のエピトープからの反応の生成において重要である
免疫プロテアソームは,異なるパターンの分裂を有するため,自己抗原から生成されるエピトープの配列を変更する能力を有することが示唆されている。ある場合には,新しいエピトープは,免疫プロテアソームの上方調節により生成され,またある場合には,エピトープは破壊される。免疫プロテアソームは,メラノーマ抗原,すなわち,MelanA/MART−1,gp100209−217,及びTyrosinase369−377に由来する複数のエピトープを生成することができないという証拠がある(Chapiro et al 2006. J Immunol; 176: 1053−61)。Chapiro及び同僚は,gp100エピトープを処理及び発現する能力は,免疫プロテアソームの上方調節に関連があることを示唆した。成熟DCは,免疫反応のプライミングに関与すると考えられており,また,免疫プロテアソームを構成的に発現することが知られている(Macagno et al, 2001. Eur J Immunol; 31: 3271−80)。そこで,gp100209−217エピトープをImmunoBodyコンストラクトのCDRH1部位に改変し,HLA−A2トランスジェニックマウスにおけるペプチド特異的免疫反応を誘発する能力を試験した。このコンストラクトからはペプチド特異的反応は観察されなかった(図56)。しかしながら,該エピトープを210(210M)の位置においてトレオニンの代わりにメチオニンを有するように改変すると,これは,免疫プロテアソームによる分裂を防止し,エピトープ特異的反応が観察された(図56)。
【0218】
VEGFR2(aa773−781 VIAMFFWLL)由来のHLA−A2拘束性ペプチド及び2つの改変hTERTペプチド(aa572−580 YLFFYRKSV及びaa988−997 YLQVNSLQTV)もまたImmunoBodyコンストラクトからの反応の生成について試験した。これらのエピトープは最初コンピュータによるエピトープ予測及びペプチド免疫により発見されたため,プロテアソーム処理の要件を否定する。しかしながら,これらは,宿主内皮/腫瘍細胞の表面にて発現され,このことは,これらが構成的プロテアソームにより全抗原から処理されることを示唆している。これらのエピトープのいずれもが,ImmunoBodyコンストラクトに改変された場合に反応を生成しなかったが,このことは,プロテアソームによる処理が免疫反応の効率的な生成に必要とされうることを示唆している。
【0219】
実施例14 異なる免疫方法は,ImmunoBodyワクチンからの免疫反応を引き起こすのに効率的である
ImmunoBodyワクチンは,遺伝子銃により投与した場合に,高頻度・高活性のCD8及びCD4反応を引き起こすのに効率的であることが示されている。その後,ImmunoBodyワクチンを他の免疫方法を用いてT細胞反応の生成について試験した。
【0220】
C57BI/6マウスに,CDRH2のTRP2エピトープを含むImmunoBody DNAにより皮内又は筋肉内経路で免疫した。免疫は,エレクトロポレーションと組み合わせて又は組み合わせずに,週間隔で3回行った。
【0221】
遺伝子銃で免疫したマウスは,高頻度TRP2ペプチド特異的反応を示す。これらは,エレクトロポレーションと組み合わせて皮内又は筋肉内経路で免疫したマウスと同程度である。エレクトロポレーションを伴わずに皮内又は筋肉内経路で免疫した場合は,低頻度のTRP2ペプチド特異的反応を生成した(図57a)。すべてのTRP2ペプチド特異的反応は,ペプチド摘定により測定した(図57b)。
【0222】
実施例15 ImmunoBody免疫は白斑様色素脱失を誘発し腫瘍攻撃から保護する
ImmunoBody DNAにより免疫したマウスは,高転移性かつ低免疫原性の腫瘍細胞株B16F10に対する細胞毒性能力のある免疫反応を生成するため,ワクチンをインビボでの予防効果について試験した。
【0223】
マウスに遺伝子銃で週間隔で5回腹部の毛を剃った皮膚にIB DNA(DCIB18,図30)により免疫した。免疫スケジュールの途中,マウスに,肺に転移性腫瘍を形成するIFNαを発現する1x104のB16F10細胞により静脈内注入した。最後の免疫後,毛を元の状態に生えさせると,ImmunoBody DNAにより免疫したマウスの免疫に部位に白い毛が生えてくるのが観察された(図58a)。腫瘍細胞注入の7週間後,マウスを犠牲にし,内部及び外部の肺転移数を分析した。ImmunoBody DNAを免疫したマウスは,非処理の対照マウスに比べ,肺転移の数に著しい減少が見られた。
【0224】
また,マウスに遺伝子銃で週間隔で3回IB DNA(DCIB18)により免疫した。最後の免疫の7日後,マウスを皮下でIFNαを発現する2x104のB16F10細胞で攻撃した。マウスを腫瘍の成長及び生存について監視した。腫瘍が内務省の規制による最大限度に達するとマウスを安楽死させた。ImmunoBody DNA免疫マウスは,著しく低い腫瘍の成長及び長期生存を示した。
【0225】
TRP2特異的反応は,CD8+細胞が反応を無効にするので,CD8に媒介される。CD8 T細胞は,抗腫瘍免疫において主役であると見なされており,我々の結果は,ImmunoBody DNA免疫がマウスモデルにおいてインビボ抗腫瘍免疫を引き起こすことを示している。病気の兆候がないすべての免疫マウスは,免疫の部位において毛皮の白斑様色素脱失を示した。これまで白斑はしばしばマウスの腫瘍保護と関連があり,転移性メラノーマの患者における良好なIL−2免疫療法と大いに相関性がある(Overwijk et al, Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 2982−7; Lane et al, Cancer Research 2004;64: 1509−14; Steitz et al, Cancer Immunol lmmunother 2006; 55: 246−53; Rosenberg & White, J lmmunother Emphasis Tumor Immunol 1996;19: 81 −4)。
【0226】
実施例16 ImmunoBody免疫は腫瘍の成長を著しく遅らせる
ImmunoBody免疫は,腫瘍攻撃を有意に阻止することがこれまで示されている。その後,ワクチンを治療環境における有効性について試験した。
【0227】
C57BI/6マウスに2x104のB16F10腫瘍細胞で皮下注入した。注入の4日後,マウスに,CDRH2のTRP2エピトープを含むImmunoBody DNA又は対照ImmunoBody DNAで免疫した。腫瘍注入後11日目及び18日目に反復免疫を行った。腫瘍の成長を3〜4日間隔で監視した。ImmunoBody免疫マウスは,対照免疫マウスに比べ,攻撃的B16F10メラノーマの成長に有意な遅延を示した。
【0228】
また,攻撃性の弱いB16F10 IFNα腫瘍株を用いて同様の研究を行った。C57BI/6マウスに,2x104の腫瘍細胞を皮下で注入し,14日目にImuunoBody DNAまたは対照DNAで免疫した。腫瘍注入後21日目及び28日目に反復免疫を行った。ImmunoBody免疫マウスは,腫瘍注入後47日目に対照免疫マウスに比べ優位に低い腫瘍の成長を示した。以前のデータは,T制御性細胞の枯渇が免疫反応の生成を強化し,よって,抗腫瘍の研究が行われることを示唆していた。この研究において,マウスに2x104のB16F10腫瘍細胞を皮下で注入し,4日目,11日目,18日目にImmunoBody DNA又は対照DNAで免疫した。0日目に抗CD25抗体(PC61)の注入によりマウスからT制御性細胞を枯渇させた。2回目の免疫と同時に,マウスには抗CTLA−4抗体も注入した。なぜなら,CTLA−4の遮断もまた制御性T細胞の抑制に有用であることが分かったためである。腫瘍の成長を監視した。ImmunoBody免疫は,腫瘍の成長(p=0.0188)を有意に遅らせたが、これは、抗CD25及び抗CTLA−4抗体(p=0.001)による処理によりさらに強化された(図59c)。抗CD25抗体による処理は、ImmunoBody免疫マウスで観察された腫瘍の成長を有意に遅らせないようであった。
【0229】
実施例17 免疫反応は異なるベクターバックボーンから発現されるImmunoBodyコンストラクトから生成されうる
ImmunoBodyコンストラクトが異なるベクターバックボーンから発現される場合の反応を分析した。CDRH1のgp100DR7エピトープ,CDRH2のTRP2エピトープ,及びCDRH3のgp100DR4を野生型軽鎖とともに含むImmunoBodyコンストラクトを二重発現ベクターDCOrig及びDCVaxに改変した(DCIB54,図18及び図29)。HLA−DR4トランスジェニックマウスに遺伝子銃により週間隔で3回免疫し,反応をIFNγ elispotアッセイによりインビトロで分析した。
【0230】
CDRH1及びCDRL3のgp100DR7,CDRH2のTRP22エピトープ,CDRH3及びCDRL1のgp100DR4エピトープを含むImmunoBodyコンストラクトを用いて同様の実験を行った(DCIB68,図60)。このコンストラクトを,DCOrig,及びSV40プロモーター及びDCVaxベクターバックボーンを欠くDCOrigの両方に改変した。
【0231】
Origベクター(B1−3)のImmunoBodyコンストラクトで免疫したマウスは,pVaxベクター(C1−3)のImmunoBodyコンストラクトに比べ,同様の頻度のエピトープ反応を示した(図61)。
【0232】
要約すると,ImmunoBody技術は,非免疫原性の抗体フレームワークから高頻度・高活性のCD8・CD4免疫反応を引き起こす優れた能力を有し,インビボでの腫瘍の成長を効果的に防ぐことができる。それは,6つの異なる抗原を同時に標的にする能力を有し,また,全抗原免疫原を使用する場合にしばしば発生する制御性T細胞の問題を回避する能力を有する。この技術は,ワクチン接種への新しいアプローチを提供し,他の多くの癌型及び微生物関連疾患用の多価ワクチンとして用いられるImmunoBodyシステムの可能性を示す。
【特許請求の範囲】
【請求項1】
非特異的プロモーターと,
少なくとも1つの異種T細胞エピトープを有するが,制御性T細胞(regulatory T cell)エピトープを有しないポリペプチドをコードする少なくとも1つの配列と,
を含む核酸。
【請求項2】
前記ポリペプチドは,ヘテロ二量体の一方の鎖であり,
前記異種T細胞エピトープは,該ヘテロ二量体の鎖が該ヘテロ二量体の他方の鎖と結合することができないような該ヘテロ二量体の鎖の破断を引き起こす,
請求項1に記載の核酸。
【請求項3】
前記ヘテロ二量体の両方の鎖をコードし,
一方の鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,
他方の鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない,
請求項2に記載の核酸。
【請求項4】
前記ヘテロ二量体は,免疫グロブリン分子である,
請求項1又は2に記載の核酸。
【請求項5】
前記免疫グロブリン分子の重鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,
前記免疫グロブリン分子の軽鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない,
請求項4に記載の核酸。
【請求項6】
非特異的プロモーターと,
免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列と,
を含む核酸であって,
該重鎖は,核酸が発現されても該重鎖が未変性コンフォメーションをとることができないような少なくとも1つの異種T細胞エピトープを有する,
核酸。
【請求項7】
免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列をさらに含む,
請求項6に記載の核酸。
【請求項8】
免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列を含む核酸と結合した,
請求項6に記載の核酸。
【請求項9】
前記軽鎖は,少なくとも1つの異種T細胞エピトープを有する,
請求項7又は8に記載の核酸。
【請求項10】
前記T細胞エピトープは,前記核酸が発現されても前記軽鎖が未変性コンフォメーションをとることができない,
請求項9に記載の核酸。
【請求項11】
前記少なくとも1つのT細胞エピトープは,前記重鎖及び/又は前記軽鎖の可変領域に存在する,
請求項4〜10のいずれか1項に記載の核酸。
【請求項12】
前記少なくとも1つのT細胞エピトープは,前記抗体の前記重鎖及び/又は前記軽鎖の前記少なくとも1つの相補性決定領域(CDR)に存在する,
請求項11に記載の核酸。
【請求項13】
前記相補性決定領域は,CDRL1,CDRH1,及び/又はCDRH2である,
請求項12に記載の核酸。
【請求項14】
前記少なくとも1つのT細胞エピトープをコードする前記配列は,前記重鎖又は前記軽鎖をコードする前記配列に挿入される,
請求項4〜13のいずれか1項に記載の核酸。
【請求項15】
前記少なくとも1つのT細胞エピトープをコードする前記配列は,前記重鎖又は前記軽鎖をコードする前記配列に置換される,
請求項4〜13のいずれか1項に記載の核酸。
【請求項16】
抗体をコードする,
請求項1〜15のいずれか1項に記載の核酸。
【請求項17】
前記抗体は,モノクローナル抗体である,
請求項16に記載の核酸。
【請求項18】
前記少なくとも1つのT細胞エピトープは,細胞傷害性T細胞エピトープである,
請求項1〜17のいずれか1項に記載の核酸。
【請求項19】
前記少なくとも1つのT細胞エピトープは,ヘルパーT細胞エピトープである,
請求項1〜18のいずれか1項に記載の核酸
【請求項20】
前記重鎖及び/又は前記軽鎖は,
少なくとも1つの細胞傷害性T細胞エピトープと,
少なくとも1つのヘルパーT細胞エピトープと,
を有する,
請求項4〜19のいずれか1項に記載の核酸
【請求項21】
非特異的プロモーターと,
免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列と,
を含む核酸であって,
該重鎖は,核酸が発現されても該重鎖が未変性コンフォメーションをとることができないような少なくとも1つの異種T細胞エピトープを有する,
核酸。
【請求項22】
請求項6〜20のいずれか1項の特徴により改変される,
請求項21に記載の核酸。
【請求項23】
請求項1〜22のいずれか1項に記載の核酸と,
アジュバントと,
を含むワクチン。
【請求項24】
請求項1〜22のいずれか1項に記載の核酸と,
薬学的に許容されるキャリア,賦形剤,又は希釈剤と,
を含む医薬組成物。
【請求項25】
医薬用の請求項1〜22のいずれか1項に記載の核酸。
【請求項26】
少なくくとも1つの前記T細胞エピトープに対する免疫反応を刺激するのに用いるための請求項25に記載の核酸。
【請求項27】
少なくとも1つの前記T細胞エピトープに対する免疫反応を刺激するための薬剤の製造における請求項1〜22のいずれか1項に記載の核酸の利用。
【請求項28】
T細胞エピトープに対する免疫反応を刺激するための方法であって,
該方法は,該免疫反応を必要とする被験者に請求項1〜22のいずれか1項に記載の核酸の治療効果のある量を投与するステップを含む,
方法。
【請求項1】
非特異的プロモーターと,
少なくとも1つの異種T細胞エピトープを有するが,制御性T細胞(regulatory T cell)エピトープを有しないポリペプチドをコードする少なくとも1つの配列と,
を含む核酸。
【請求項2】
前記ポリペプチドは,ヘテロ二量体の一方の鎖であり,
前記異種T細胞エピトープは,該ヘテロ二量体の鎖が該ヘテロ二量体の他方の鎖と結合することができないような該ヘテロ二量体の鎖の破断を引き起こす,
請求項1に記載の核酸。
【請求項3】
前記ヘテロ二量体の両方の鎖をコードし,
一方の鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,
他方の鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない,
請求項2に記載の核酸。
【請求項4】
前記ヘテロ二量体は,免疫グロブリン分子である,
請求項1又は2に記載の核酸。
【請求項5】
前記免疫グロブリン分子の重鎖は,異種細胞傷害性T細胞(CTL)エピトープを含み,発現されると分泌され,
前記免疫グロブリン分子の軽鎖は,異種ヘルパーエピトープを含み,発現されても分泌されない,
請求項4に記載の核酸。
【請求項6】
非特異的プロモーターと,
免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列と,
を含む核酸であって,
該重鎖は,核酸が発現されても該重鎖が未変性コンフォメーションをとることができないような少なくとも1つの異種T細胞エピトープを有する,
核酸。
【請求項7】
免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列をさらに含む,
請求項6に記載の核酸。
【請求項8】
免疫グロブリン分子の軽鎖をコードする少なくとも1つの配列を含む核酸と結合した,
請求項6に記載の核酸。
【請求項9】
前記軽鎖は,少なくとも1つの異種T細胞エピトープを有する,
請求項7又は8に記載の核酸。
【請求項10】
前記T細胞エピトープは,前記核酸が発現されても前記軽鎖が未変性コンフォメーションをとることができない,
請求項9に記載の核酸。
【請求項11】
前記少なくとも1つのT細胞エピトープは,前記重鎖及び/又は前記軽鎖の可変領域に存在する,
請求項4〜10のいずれか1項に記載の核酸。
【請求項12】
前記少なくとも1つのT細胞エピトープは,前記抗体の前記重鎖及び/又は前記軽鎖の前記少なくとも1つの相補性決定領域(CDR)に存在する,
請求項11に記載の核酸。
【請求項13】
前記相補性決定領域は,CDRL1,CDRH1,及び/又はCDRH2である,
請求項12に記載の核酸。
【請求項14】
前記少なくとも1つのT細胞エピトープをコードする前記配列は,前記重鎖又は前記軽鎖をコードする前記配列に挿入される,
請求項4〜13のいずれか1項に記載の核酸。
【請求項15】
前記少なくとも1つのT細胞エピトープをコードする前記配列は,前記重鎖又は前記軽鎖をコードする前記配列に置換される,
請求項4〜13のいずれか1項に記載の核酸。
【請求項16】
抗体をコードする,
請求項1〜15のいずれか1項に記載の核酸。
【請求項17】
前記抗体は,モノクローナル抗体である,
請求項16に記載の核酸。
【請求項18】
前記少なくとも1つのT細胞エピトープは,細胞傷害性T細胞エピトープである,
請求項1〜17のいずれか1項に記載の核酸。
【請求項19】
前記少なくとも1つのT細胞エピトープは,ヘルパーT細胞エピトープである,
請求項1〜18のいずれか1項に記載の核酸
【請求項20】
前記重鎖及び/又は前記軽鎖は,
少なくとも1つの細胞傷害性T細胞エピトープと,
少なくとも1つのヘルパーT細胞エピトープと,
を有する,
請求項4〜19のいずれか1項に記載の核酸
【請求項21】
非特異的プロモーターと,
免疫グロブリン分子の組み換え重鎖をコードする少なくとも1つの配列と,
を含む核酸であって,
該重鎖は,核酸が発現されても該重鎖が未変性コンフォメーションをとることができないような少なくとも1つの異種T細胞エピトープを有する,
核酸。
【請求項22】
請求項6〜20のいずれか1項の特徴により改変される,
請求項21に記載の核酸。
【請求項23】
請求項1〜22のいずれか1項に記載の核酸と,
アジュバントと,
を含むワクチン。
【請求項24】
請求項1〜22のいずれか1項に記載の核酸と,
薬学的に許容されるキャリア,賦形剤,又は希釈剤と,
を含む医薬組成物。
【請求項25】
医薬用の請求項1〜22のいずれか1項に記載の核酸。
【請求項26】
少なくくとも1つの前記T細胞エピトープに対する免疫反応を刺激するのに用いるための請求項25に記載の核酸。
【請求項27】
少なくとも1つの前記T細胞エピトープに対する免疫反応を刺激するための薬剤の製造における請求項1〜22のいずれか1項に記載の核酸の利用。
【請求項28】
T細胞エピトープに対する免疫反応を刺激するための方法であって,
該方法は,該免疫反応を必要とする被験者に請求項1〜22のいずれか1項に記載の核酸の治療効果のある量を投与するステップを含む,
方法。
【図1】

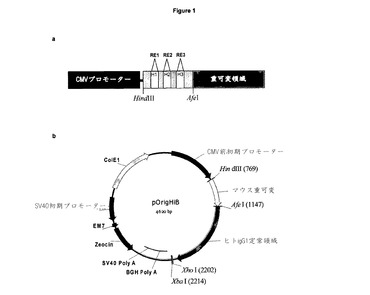
【図2】
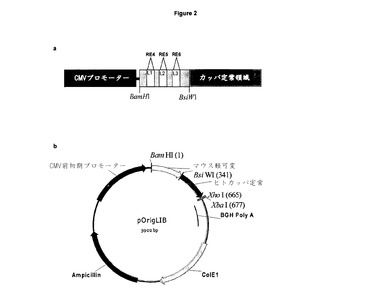
【図3】

【図4】

【図5】
【図6A】

【図6B】

【図6C】

【図7A】

【図7B】

【図7C】

【図8】

【図9】
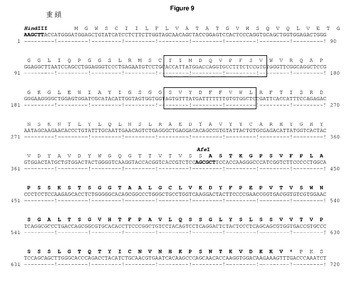
【図10】

【図11】
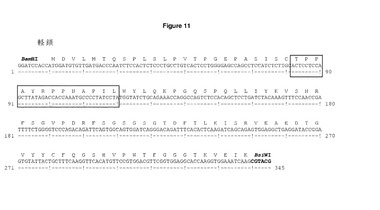
【図12】

【図13】

【図14】


【図15】

【図16】

【図17】

【図18】
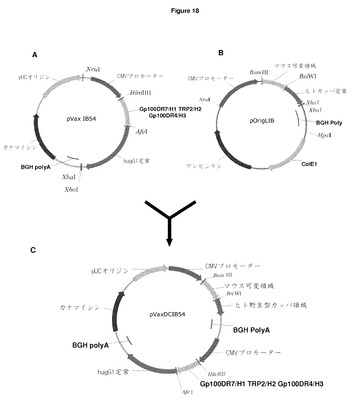
【図19】
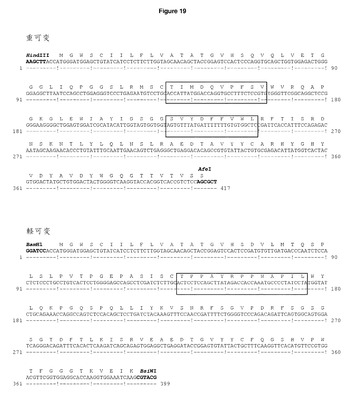
【図20A】
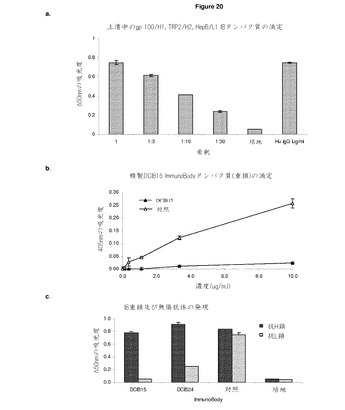
【図20B】
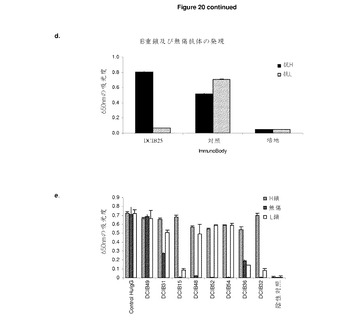
【図21】

【図22】

【図23】

【図24】

【図25】

【図26】

【図27】

【図28】
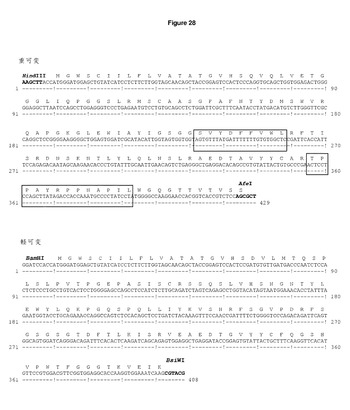
【図29】

【図30】

【図32B】

【図32C】

【図33】

【図34】

【図35】

【図36】

【図37】

【図38】
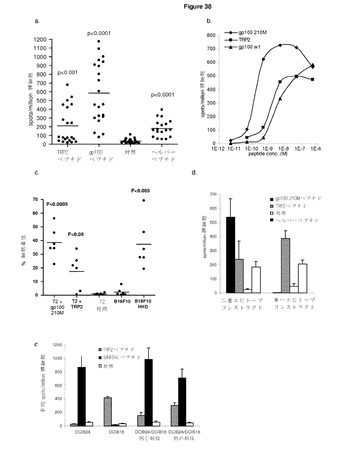
【図39】

【図40】

【図41】

【図42】

【図43】

【図44】
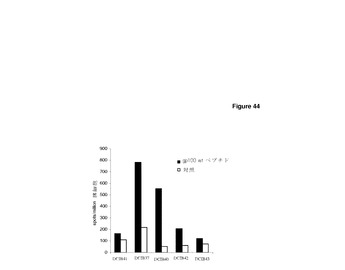
【図45】
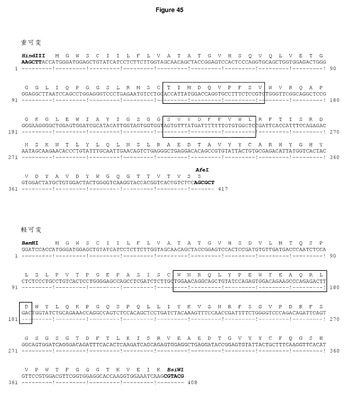
【図46】
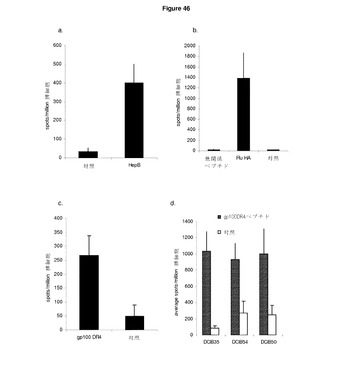
【図47】

【図48】
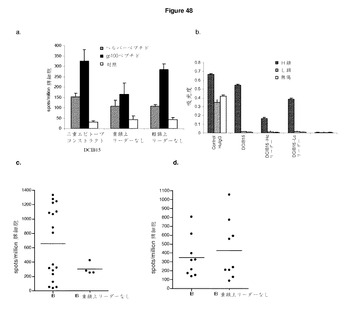
【図49A】
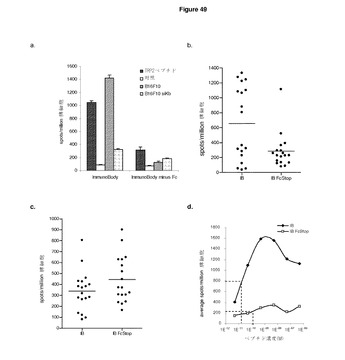
【図49B】

【図50A】
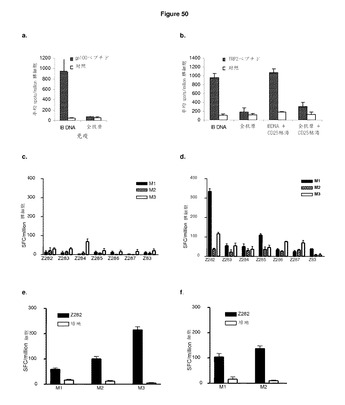
【図50B】

【図51】
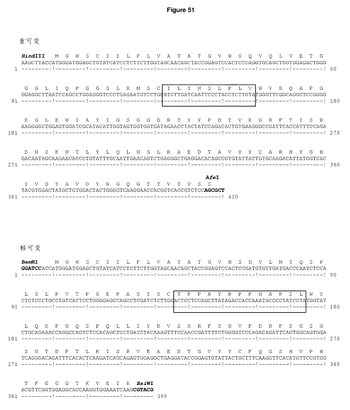
【図52】

【図53】
【図54A】


【図54B】
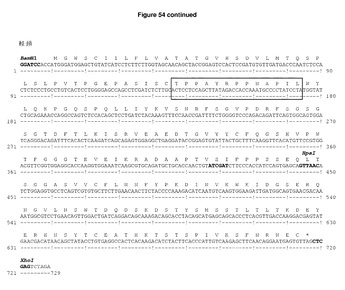
【図55A】

【図55B】

【図56】
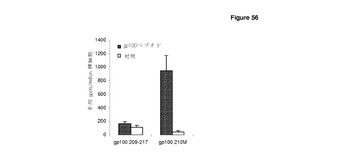
【図57】
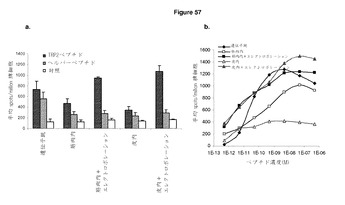
【図58】
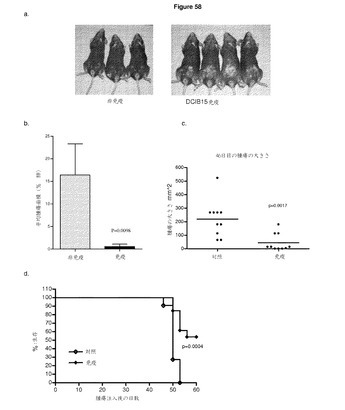
【図59】
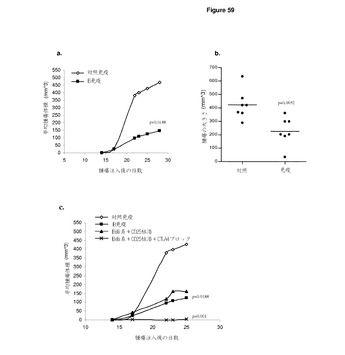
【図60】
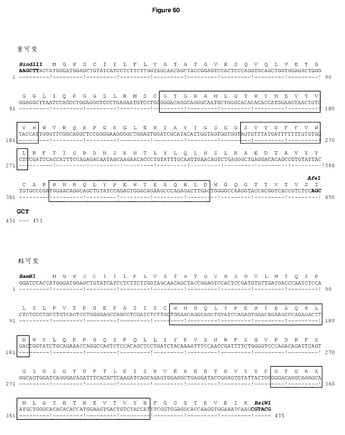
【図61】
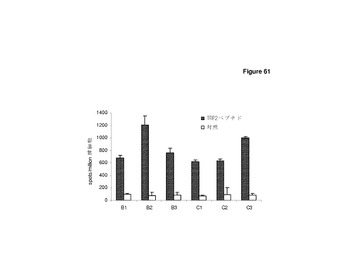
【図31A】
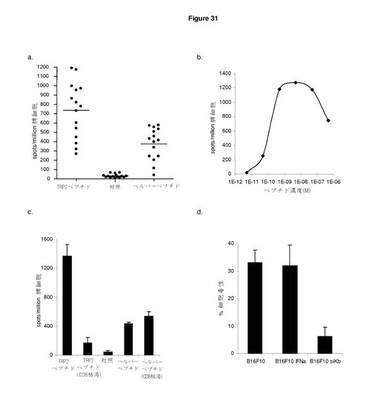
【図31B】
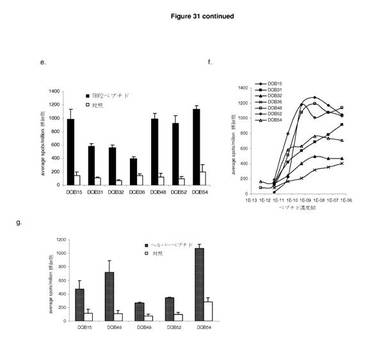
【図32A】
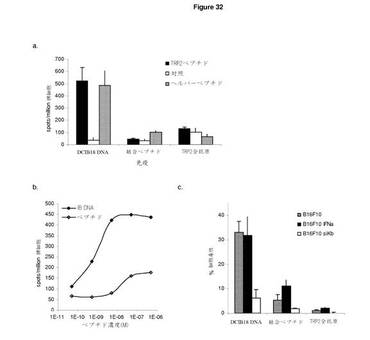
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7A】
【図7B】
【図7C】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20A】
【図20B】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図32B】
【図32C】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49A】
【図49B】
【図50A】
【図50B】
【図51】
【図52】
【図53】
【図54A】
【図54B】
【図55A】
【図55B】
【図56】
【図57】
【図58】
【図59】
【図60】
【図61】
【図31A】
【図31B】
【図32A】
【公表番号】特表2010−522550(P2010−522550A)
【公表日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願番号】特願2010−500296(P2010−500296)
【出願日】平成20年3月28日(2008.3.28)
【国際出願番号】PCT/EP2008/053761
【国際公開番号】WO2008/116937
【国際公開日】平成20年10月2日(2008.10.2)
【出願人】(509258599)スキャンセル リミテッド (1)
【氏名又は名称原語表記】SCANCELL LIMITED
【Fターム(参考)】
【公表日】平成22年7月8日(2010.7.8)
【国際特許分類】
【出願日】平成20年3月28日(2008.3.28)
【国際出願番号】PCT/EP2008/053761
【国際公開番号】WO2008/116937
【国際公開日】平成20年10月2日(2008.10.2)
【出願人】(509258599)スキャンセル リミテッド (1)
【氏名又は名称原語表記】SCANCELL LIMITED
【Fターム(参考)】
[ Back to top ]