
Apo−2リガンド
【課題】Fas/Apo-1、TNF-R1、又はTNF-R2レセプター等によって感知できるほどに阻害されない新規なサイトカインを提供する。
【解決手段】哺乳動物癌細胞のアポトーシスを誘発するApo-2リガンドと命名された新規なサイトカイン。Apo-2リガンドは、TNFサイトカインファミリーのメンバーに属すると考えられる。Apo-2リガンドキメラを含有する組成物、Apo-2リガンドをコードする核酸、及びApo-2リガンドに対する抗体、さらに、アポトーシスを誘発し癌などの病理学的状態を治療するためのApo-2リガンドの使用方法。
【解決手段】哺乳動物癌細胞のアポトーシスを誘発するApo-2リガンドと命名された新規なサイトカイン。Apo-2リガンドは、TNFサイトカインファミリーのメンバーに属すると考えられる。Apo-2リガンドキメラを含有する組成物、Apo-2リガンドをコードする核酸、及びApo-2リガンドに対する抗体、さらに、アポトーシスを誘発し癌などの病理学的状態を治療するためのApo-2リガンドの使用方法。
【発明の詳細な説明】
【発明の開示】
【0001】
(発明の分野)
本発明は、一般に、ここに「Apo-2リガンド」と命名する新規サイトカインの同定、単離、及び組換え生産、及びそのような組成物の使用方法に関する。
【0002】
(発明の背景)
哺乳動物における細胞数のコントロールは、一部には細胞増殖と細胞死のバランスにより決定されると考えられている。しばしば壊死性細胞死と称される細胞死の一形態は、典型的には、ある種の外傷又は細胞傷害の結果生じる細胞死の病理的形態として特徴付けられる。これに対して、通常は規則的又はコントロールされた状態で進行する細胞死の他の「生理的」形態がある。細胞死のこの規則的又はコントロールされた形態は、しばしば「アポトーシス」と称される[例えば、Barr等, Bio/Technology, 12:487-493(1994)を参照]。アポトーシス性細胞死は、免疫系におけるクローン選択と胚の発達を含む多くの生理的プロセスにおいて自然に生じる[Itohら, Cell, 66:233-243(1991)]。アポトーシス性細胞死のレベルの減少は、癌、狼瘡、ヘルペスウイスル感染を含む種々の病理的条件に関連している[Thompson, Science, 267:1456-1462(1995)]。
【0003】
アポトーシス性細胞死には、典型的には、細胞内における一又は複数の特徴的な形態学的及び生化学的変化、例えば細胞質の凝結、原形質膜の微絨毛の喪失、核の分節化、染色体DNAの分解又はミトコンドリア機能の喪失が伴う。様々な外因的及び内因的シグナルが、このような形態学的及び生化学的な細胞変化を惹起又は誘発すると考えられている[Raff, Nature, 356:397-400(1992);Steller, Science, 267: 1445-1449 (1995); Sachs等, Blood, 82:15(1993)]。例えば、ホルモンの刺激、例えば未成熟胸腺細胞に対する糖質コルチコイドホルモン、並びにある種の成長因子の退薬により惹起され得る[Watanabe-Fukunagaら, Nature, 356:314-317(1992)]。また、幾つかの同定された発癌遺伝子、例えばmyc、rel、及びE1A、及び腫瘍サプレッサーは、p53と同様に、アポトーシスの誘発においてある役割を有していることも報告されている。ある種の化学療法薬及びある種の放射線も同様にアポトーシス誘発活性を有していることも見出されている[Thompson, 上掲]。
【0004】
様々な分子、例えば腫瘍壊死因子-α(「TNF-α」)、腫瘍壊死因子-β(「TNF-β」又は「リンホトキシン」)、CD30リガンド、CD27リガンド、CD40リガンド、OX-40リガンド、4-1BBリガンド、及びApo-1リガンド(Fasリガンド又はCD95リガンドとも称される)が、サイトカインの腫瘍壊死因子(「TNF」)ファミリーのメンバーとして同定された[例えば、Gruss及びDower, Blood, 85:3378-3404(1995)]。これらの分子のなかでも、TNF-α、TNF-β、CD30リガンド、4-1BBリガンド、Apo-1リガンド、及びApo-2リガンド(TRAIL)は、アポトーシス性細胞死に関与していることが報告されている。TNF-αとTNF-βの両方とも、感受性腫瘍細胞におけるアポトーシス性死を誘発することが報告されている[Schmidら, Proc. Natl. Acad. Sci., 83:1881(1986);Dealtryら, Eur. J. Immunol., 17:689(1987)]。ゼングらは、TNF-αがCD8ポジティブT細胞のポスト刺激性アポトーシスに関与していることを報告している[Zheng等, Nature, 377:348-351(1995)]。他の研究者らは、CD30リガンドが胸腺における自己反応性T細胞の欠失に関与していることを報告している[Amakawa等, プログラム細胞死に関するコールドスプリングハーバー研究所のシンポジウム、要約集、第10巻、(1995)]。
【0005】
マウスのFas/Apo-1レセプター又はリガンド遺伝子(それぞれlpr及びgldと呼称される)における変異が幾つかの自己免疫疾患に関連しており、Apo-1リガンドが末梢の自己反応性リンパ球のクローン除去の調節においてある役割を担っていることを示している[Krammer等, Curr. Op. Immunol., 6:279-289(1994); Nagata等, Science, 267:1449-1456(1995)]。また、Apo-1リガンドは、CD4-ポジティブTリンパ球及びBリンパ球においてポスト刺激性アポトーシスを誘発することが報告されており、それらの機能がもはや必要でなくなった際の活性化リンパ球の除去に関与している[Krammer等, 上掲; Nagata等, 上掲]。Apo-1レセプターと特異的に結合するアゴニストのマウスモノクローナル抗体は、TNF-αに匹敵するか類似する細胞死滅活性を示すことが報告されている[Yonehara等, J. Exp. Med., 169:1747-1756(1989)]。
【0006】
このようなTNFファミリーのサイトカインが介在する種々の細胞反応の誘発は、それらが特異的な細胞レセプターに結合することにより開始されると考えられている。約55-kDa(TNF-R1)と75-kDa(TNF-R2)の2つの異なるTNFレセプターが同定されており[Hohman等, J. Biol. Chem., 264:14927-14934(1989);Brockhaus等, Proc. Natl. Acad. Sci., 87:3127-3131(1990);1991年3月20日に公開されたEP417,563]、双方のレセプター型に対応するヒト及びマウスcDNAが単離され、特徴付けされている[Loetscher等, Cell, 61:351(1990);Schall等, Cell, 61:361(1990);Smith等, Science, 248:1019-1023(1990);Lewis等, Proc. Natl. Acad. Sci., 88:2830-2834(1991);Goodwin等, Mol. Cell. Biol., 11:3020-3026(1991)]。
【0007】
イトー等は、Apo-1レセプターが55-kDaのTNF-R1によりシグナル化されるものと同様のアポトーシス性細胞死をシグナル化し得ることを開示している[Itoh等, 上掲]。また、Apo-1抗原の発現は、細胞をTNF-α又は抗Apo-1のマウスモノクローナル抗体で処理した場合に、TNF-R1のものと共にダウンレギュレーションされることが報告されている[Krammer等, 上掲;Nagata等, 上掲]。従って、Apo-1及びTNF-R1レセプターの双方を同時発現する株化細胞が、共通のシグナル伝達経路を通して細胞死を媒介しているとの仮説を唱える研究者もいた[同]。
【0008】
今日までに同定されているTNFファミリーのリガンドは、リンホトキシン-αを除き、II型の膜貫通タンパク質であり、そのC末端は細胞外にある。これに対して、今日までに同定されているTNFレセプター(TNFR)ファミリーのレセプター類はI型の膜貫通タンパク質である。しかしながら、TNFリガンド及びレセプターファミリーの双方において、ファミリーメンバー間で同定された相同性は、主として細胞外ドメイン(「ECD」)において見出されている。TNF-α、Apo-1リガンド及びCD40リガンドを含むTNFファミリーサイトカインのいくつかは、細胞表面においてタンパク分解的に切断され;各場合に得られたタンパク質は、典型的には、可溶性サイトカインとして機能するホモ三量体分子を形成する。また、TNFレセプターファミリーのタンパク質は、通常、タンパク分解的に切断され、同族のサイトカインの阻害剤として機能し得る可溶性リガンドのECDを放出する。サイトカインのTNFファミリー及びそれらのレセプター類の概説については、上掲のGruss及びDowerを参照されたい。
【0009】
(発明の概要)
本出願人は、「Apo-2リガンド」と命名される新規なサイトカインをコードするcDNAクローンを同定した。現在では、Apo-2リガンドはTNFサイトカインファミリーのメンバーであると信じられており;Apo-2リガンドは、Apo-1リガンドを含む幾つかの知られたTNF関連タンパク質とアミノ酸配列において関連している。しかしながら、本出願人は、Apo-2リガンドが知られた可溶性Apo-1又はTNFレセプター類、例えばFas/Apo-1、TNF-R1、又はTNF-R2レセプター等によって感知できるほどに阻害されないことを見出した。
【0010】
一実施態様において、本発明は、単離されたApo-2リガンドを提供する。特に、本発明は、図1Aの残基114〜281を含んでなるアミノ酸配列を含む単離されたApo-2リガンドを提供する。他の実施態様においては、Apo-2リガンドは、図1Aの残基92〜281を含んでなるアミノ酸配列を含む。さらなる実施態様では、Apo-2リガンドは、図1Aの残基91〜281を含んでなるアミノ酸配列を含む。さらに他の実施態様では、Apo-2リガンドは、図1Aの残基41〜281又は15〜281を含んでなるアミノ酸配列を含む。さらなる実施態様では、図1A(配列番号:1)の残基1〜281として示したアミノ酸配列を含む。
【0011】
また、本発明の単離されたApo-2リガンドは、上記の参照配列の置換変異体を含む。特に、一実施態様では、図1Aのアミノ酸91〜281を含むApo-2リガンドにおいて、位置203、218又は269の少なくとも1つがアラニンに置換された置換変異体が提供される。これらの置換変異体は特に、「D203A」;「D218A」及び「D269A」と定義される。この命名法は、例えば図1Aのアミノ酸91〜281を含むApo-2リガンドポリペプチドであって、(図1Aに示した番号を用いた)位置203、218及び/又は269のアスパラギン酸残基がアラニン残基で置換されたものを同定するのに用いられる。場合によっては、置換変異体は一又は複数のそのような置換を含んでいてもよい。
他の実施態様では、本発明は、他の異種ポリペプチドに融合したApo-2リガンドを含むキメラ分子を提供する。そのようなキメラ分子の例は、タグポリペプチド配列に融合したApo-2リガンドを含む。
【0012】
他の実施態様では、本発明は、Apo-2リガンドをコードする単離された核酸分子を提供する。一態様では、核酸分子はRNA又はDNAであり、Apo-2リガンドをコードするか、又はそのようなApo-2リガンドをコードする核酸配列に相補的であり、少なくとも中程度の厳密性条件下で、それを安定に結合させたままである。一実施態様では、核酸配列は:
(a)残基1から残基281までの全長タンパク質をコードする図1Aの核酸配列のコード領域(即ち、ヌクレオチド91〜933)、あるいは図1A(配列番号:2)に示した核酸配列の又は残基41〜281の細胞外タンパク質をコードするヌクレオチド211〜933、又は残基92〜281の細胞外タンパク質をコードするヌクレオチド364〜933、又は残基91〜281の細胞外タンパク質をコードするヌクレオチド361〜933、又は残基114〜281の細胞外タンパク質をコードするヌクレオチド430〜933;あるいは、
(b)遺伝子コードの縮退の範囲内で(a)の配列に相当する配列から選択される。
【0013】
さらなる実施態様において、本発明は、当該ベクターで形質転換された宿主細胞によって認識されるコントロール配列に作用可能に結合したApo-2リガンドをコードする核酸分子を含む複製可能なベクターを提供する。このベクター又は核酸分子を含む宿主細胞も提供される。さらに、この核酸分子を含む宿主細胞を培養し、当該宿主細胞培地からタンパク質を回収することを含んでなるApo-2リガンドの作成方法が提供される。
他の実施態様では、本発明はApo-2リガンドに結合する抗体を提供する。一態様において、この抗体はApo-2リガンドに対して抗原特異性を有するモノクローナル抗体である。
他の実施態様では、本発明はApo-2リガンド及び担体を含んでなる組成物を提供する。この組成物は、アポトーシスを誘発又は刺激するのに有用な製薬組成物であってよい。
他の実施態様では、本発明は、アポトーシスの誘発に有効な量のApo-2リガンドに、インビボ又はエキソビボで、哺乳動物細胞を暴露することを含んでなる、哺乳動物細胞においてアポトーシスを誘発する方法を提供する。
【0014】
他の実施態様では、本発明は癌を持つ哺乳動物の治療方法を提供する。この方法においては、癌を持つと診断された哺乳動物に、有効量のApo-2リガンドが投与される。Apo-2リガンドは、一又は複数の他の治療、例えば化学治療、放射線治療、又は抗腫瘍活性を示す他の薬剤とともに哺乳動物に投与してもよい。
さらなる実施態様では、本発明はApo-2リガンド又はApo-2リガンド抗体を含む製造品及びキットを提供する。この製造品及びキットは、容器、容器上のラベル、及び容器内に収容された組成物を具備する。容器上のラベルは、組成物が或る種の治療又は非治療用途に使用できることを表示する。この組成物は活性薬を含有し、この活性薬はApo-2リガンド又はApo-2リガンド抗体を含む。
【0015】
(好適な実施態様の詳細な説明)
I.定義
ここで使用される際の「Apo-2リガンド」及び「Apo-2L」という用語には、図1Aのに示したアミノ酸配列のアミノ酸残基114〜281を含む、又は残基92〜281を含む、又は残基91〜281を含む、又は残基41〜281を含む、又は残基15〜281を含む、又は残基1〜281を含むポリペプチド配列、並びに、前記配列の欠失、挿入、又は置換変異体を意味する。一実施態様では、このポリペプチド配列は、少なくとも図1Aの残基114〜281を有する。場合によっては、ポリペプチド配列は、少なくとも図1Aの残基92〜281又は残基91〜281を有する。他の好ましい実施態様では、変異体は生物学的に活性であり、上記の配列の1つと少なくとも約80%のアミノ酸配列同一性、より好ましくは少なくとも約90%の配列同一性、さらに好ましくは少なくとも約95%の配列同一性を有する。この定義は、図1Aのアミノ酸91〜281を複myApo-2リガンドの置換変異体であって、位置203、218又は269の少なくとも1つのアミノ酸がアラニンに置換されたものを含む。また、この定義はApo-2リガンド供給源から、例えばここに述べるヒト組織型(実施例8参照)から、又は他の供給源から単離された、又は組換え又は合成方法によって調製された天然配列Apo-2リガンドを含む。Apo-2リガンドのこの定義は、周知のEST配列、例えばGenBank HHEA47M、T90422、R31020、H43566、H44565、H44567、H54628、H44772、H54629、T82085、及びT10524は除外する。
【0016】
「エピトープタグ」なる用語は、ここで用いられるときは、「タグポリペプチド」に融合したApo-2リガンド、又はそれらの一部を含んでなるキメラポリペプチドを指す。タグポリペプチドは、その抗体が産生され得るエピトープを提供するに十分な数の残基を有しているが、その長さはApo-2リガンドの活性を阻害しないよう充分に短い。また、タグポリペプチドは、好ましくは、抗体が他のエピトープと実質的に交差反応をしないようにかなり独特である。適切なタグポリペプチドは、一般に、少なくとも6のアミノ酸残基、通常は約8〜約50のアミノ酸残基(好ましくは約10〜約20の残基)を有する。
【0017】
「単離された」とは、ここで開示された種々のポリペプチドを記述するために使用するときは、その自然環境の成分から同定され分離され及び/又は回収されたポリペプチドを意味する。その自然環境の汚染成分とは、ポリペプチドの診断又は治療への使用を典型的には妨害する物質であり、酵素、ホルモン、及び他のタンパク質様又は非タンパク質様溶質が含まれる。好ましい実施態様において、タンパク質は、(1)スピニングカップシークエネーターを使用することにより、少なくとも15残基のN末端あるいは内部アミノ酸配列を得るのに充分なほど、あるいは、(2)クーマシーブルーあるいは好ましくは銀染色を用いた非還元あるいは還元条件下でのSDS-PAGEによる均一性が得られるように充分なほど精製される。Apo-2リガンドの自然環境の少なくとも1つの成分が存在しないため、単離されたタンパク質には、組換え細胞内のインサイツのタンパク質が含まれる。しかしながら、通常は、単離されたタンパク質は少なくとも1つの精製工程により調製される。
【0018】
「単離された」Apo-2リガンド核酸分子は、同定され、Apo-2リガンド核酸の天然源に通常付随している少なくとも1つの汚染核酸分子から分離された核酸分子である。単離されたApo-2リガンド核酸分子は、天然に見出される形態あるいは設定以外のものである。ゆえに、単離されたApo-2リガンド核酸分子は、天然の細胞中に存在するApo-2リガンド核酸分子とは区別される。しかし、単離されたApo-2リガンド核酸分子は、例えば、核酸分子が天然の細胞のものとは異なった染色体位置にあるApo-2リガンドを通常発現する細胞に含まれるApo-2リガンド核酸分子を含む。
【0019】
ここで同定されている配列に対する「パーセント(%)アミノ酸配列同一性」は、配列を整列させ、最大のパーセント配列同一性を得るために必要ならば間隙を導入し、如何なる保存的置換も配列同一性の一部と考えないとした、Apo-2リガンド配列のアミノ酸残基と同一である候補配列中のアミノ酸残基のパーセントとして定義される。%同一性の値は、Altschul等, Methods in Enzymology, 266: 460-480 (1996);http://blast.wustl/edu/blast/README.htmlから得られるWU-BLAST-2によって算出することができる。WU-BLAST-2は幾つかの検索パラメータを使用し、その殆どが初期値に設定される。調節可能なパラメータは次の値:オーバーラップ・スパン=1、オーバーラップ・フラクション=0.125、ワードしきい値(T)=11に設定する。同様にして、Apo-2リガンドポリペプチドの核酸配列に対する「パーセント(%)核酸配列同一性」は、Apo-2リガンドコード配列のヌクレオチドと同一の候補配列中のヌクレオチドのパーセントとして定義される。同一性の値は、オーバーラップ・スパン及びオーバーラップ・フラクションが各々1及び0.125に設定された初期設定のWU-BLAST-2のBLASTNモジュールにより算出される。
【0020】
「コントロール配列」という用語は、特定の宿主生物において作用可能に結合されたコード配列を発現するために必要なDNA配列を指す。例えば原核生物に好適なコントロール配列は、プロモーター、場合によってはオペレータ配列、及びリボソーム結合部位を含む。真核生物の細胞は、プロモーター、ポリアデニル化シグナル及びエンハンサーを利用することが知られている。
核酸は、他の核酸配列と機能的な関係にあるときに「作用可能に結合され」ている。例えば、プレ配列あるいは分泌リーダーのDNAは、ポリペプチドの分泌に寄与するプレタンパク質として発現されているならそのポリペプチドのDNAに作用可能に結合されている;プロモーター又はエンハンサーは、配列の転写に影響を及ぼすならばコード配列に作用可能に結合されている;又はリボソーム結合部位は、もしそれが翻訳を容易にするような位置にあるならコード配列と作用可能に結合されている。一般的に、「作用可能に結合される」とは、結合されたDNA配列が近接しており、分泌リーダーの場合には近接していて読みフェーズにある。しかし、エンハンサーは必ずしも近接しているわけではない。結合は簡便な制限部位でのライゲーションにより達成される。そのような部位が存在しない場合は、通常の手法にしたがって、合成されたオリゴヌクレオチドアダプターあるいはリンカーが使用される。
【0021】
「抗体」という用語は最も広い意味において使用され、特に単一の抗Apo-2リガンドモノクローナル抗体(アゴニスト及びアンタゴニスト抗体を含む)、及び多エピトープ特異性を持つ抗Apo-2リガンド抗体組成物を包含している。
ここで使用される「モノクローナル抗体」という用語は、実質的に均一な抗体の集団から得られる抗体を称する、すなわち、集団を構成する個々の抗体が、少量存在しうる自然に生じる可能な突然変異を除いて同一である。モノクローナル抗体は高度に特異的であり、一つの抗原部位に対応する。さらに、異なる決定基(エピトープ)に対応する異なる抗体を典型的に含む従来の(ポリクローナル)抗体調製物とは異なり、各モノクローナル抗体は抗原の単一の決定基に対応する。
【0022】
ここで、モノクローナル抗体は、起源の種又は免疫グロブリンクラス又はサブクラスの命名に拘わらず、定常ドメインを有する抗Apo-2リガンド抗体の可変(高頻度可変を含む)ドメイン、又は重鎖を有する軽鎖、又は他の種由来の鎖を有する或る種由来の鎖、あるいは異種タンパク質との融合体をスプライシングすることによって得られるハイブリッド及び組換え抗体(例えば「ヒト化」抗体)、並びにそれが所望の生物的活性を有する限り抗体断片(例えばFab、F(ab')2及びFv)を特に含む。例えば、米国特許第4,816,567号、及びMage等, Monoclonal Antibody Productuon Techniques and Applications, pp.79-97(Marcel Dekker, Inc.:ニューヨーク, 1987)を参照。
従って、「モノクローナル」との形容は、実質的に均一な抗体集団から得られたという抗体の性質を示し、抗体を何か特定の方法で生産しなければならないことを意味するものではない。例えば、本発明に従って使用されるモノクローナル抗体は、最初にKohler及びMilsteinによって、Nature, 256:495 (1975)に記載されたハイブリドーマ法によって作ることができ、あるいは例えば米国特許第4,816,567号に記載された組換えDNA法によって作ることができる。「モノクローナル抗体」は、例えば、McCafferty等, Nature, 348:552-554(1990)に記載された技術を用いてファージライブラリから作成することができる。
【0023】
非ヒト(例えばマウス)抗体の「ヒト化」形とは、キメラ免疫グロブリン、免疫グロブリン鎖、あるいはそれらの断片(例えばFv、Fab、Fab'、F(ab')2あるいは抗体の他の抗原結合サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。大部分においてヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)である。ある場合には、ヒト免疫グロブリンのFvフレームワーク領域(FR)残基は、対応する非ヒト残基によって置換されている。更に、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでもよい。これらの修飾は抗体の特性を更に洗練し、最適化するために行われる。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリン共通配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域又はドメイン(Fc)、典型的にはヒトの免疫グロブリンの定常領域又はドメインの少なくとも一部を含んでなる。
【0024】
ここで意図している「生物学的に活性な」とは、(a)インビボ又はエキソビボで少なくとも一種類の哺乳動物細胞においてアポトーシスを誘発する能力を有しているか;(b)抗体を生成できる、即ち、免疫原性であるか;又は(c)天然又は天然発生Apo-2L活性を保持していることを意味する。
「アポトーシス」及び「アポトーシス活性」という用語は広義に使用され、典型的には、細胞質の凝縮、原形質膜の微絨毛の喪失、核の断片化、染色体DNAの分解又はミトコンドリア機能の喪失を含む一又は複数の特徴的な細胞変化を伴う、哺乳動物における細胞死の規則的又はコントロールされた形態を指す。この活性は、例えば細胞生死判別アッセイ、FACS分析又はDNA電気泳動法等、全て従来から知られている方法により決定し測定することができる。
【0025】
「癌」及び「癌性」という用語は、典型的には調節されない細胞成長を特徴とする、哺乳動物における生理学的状態を指すか記述する。癌の例には、これらに限定されるものではないが、癌腫、リンパ腫、芽細胞腫、肉腫、及び白血病が含まれる。このような癌のより特定の例には、扁平上皮細胞癌、小細胞肺癌、非小細胞肺癌、芽細胞腫、胃腸癌、腎臓癌、膵臓癌、神経膠芽細胞腫、神経芽腫、子宮頸管癌、卵巣癌、肝臓癌、胃癌、膀胱癌、肝細胞腫(hepatoma)、乳癌、大腸癌、結腸直腸癌、子宮体癌、唾液腺癌、腎臓癌、肝臓癌、前立腺癌、産卵口癌、甲状腺癌、肝癌(hepatic carcinoma)及び様々な種類の頭部及び頸部の癌が含まれる。一実施態様では、癌は濾胞性リンパ腫、p53変異を持つ癌、又はホルモン依存性癌、例えば乳癌、前立腺癌、又は卵巣癌を含む。
ここで使用される「治療する」、「治療」及び「治療法」とは、治癒的療法、予防的療法及び防護的療法を称する。
ここで使用される「哺乳動物」という用語は、ヒト、ウシ、ウマ、イヌ及びネコを含む哺乳動物として分類されるあらゆる動物を指す。本発明の好ましい実施態様においては、哺乳動物はヒトである。
【0026】
II. 本発明の組成物と方法
本発明は、TNFリガンドファミリーに関連するサイトカインであり、ここで「Apo-2リガンド」と定義されるサイトカインを提供する。ヒトApo-2リガンドの推定成熟アミノ酸配列は281アミノ酸を含み、約32.5kDaの計算された分子量及び約7.63の等電点を有する。N末端には明確なシグナル配列は無いが、ヒドロパシー分析は残基15と40との間に疎水性領域の存在を示している。シグナル配列が無いこと及び内部疎水性領域が存在することは、Apo-2リガンドがII型膜貫通タンパク質であることを示唆している。潜在的なN結合グリコシル化部位は、推定細胞外領域の残基109に位置している。図1Aに示したように、推定細胞質領域はアミノ酸残基1−14を含み、膜貫通領域はアミノ酸残基15−40を含み、細胞外領域はアミノ酸残基41−281を含む。可溶性細胞外ドメインApo-2リガンドポリペプチドは本発明の範囲内に含まれ、限定されるわけではないが、図1Aに示した細胞外領域のアミノ酸残基114−281、92−281、又は91−281を含むApo-2リガンドポリペプチドを包含する。
【0027】
また本発明はApo-2L置換変異体も提供する。ここに記載するように、幾つかの生物学的活性を有する置換変異体を同定するためにアラニンスキャンニング技術が利用される。Apo-2リガンドの特別な置換変異体は、図1Aのアミノ酸残基91−281において、位置203、218又は269の少なくとも1つのアミノ酸がアラニンに置換されたものを含む。これらの置換変異体は、「D203A」;「D218A」及び「D269A」と定義される。この命名法は、例えば図1Aのアミノ酸91〜281を含むApo-2リガンドポリペプチドであって、(図1Aに示した番号を用いた)位置203、218及び/又は269のアスパラギン酸残基がアラニン残基で置換されたものを特定するのに用いられる。場合によっては、置換変異体は一又は複数のそのような置換を含んでいてもよい。
【0028】
A.Apo-2リガンドの調製
以下の説明は、主として、Apo-2リガンド核酸を含むベクターで形質転換又は形質移入された細胞を培養し、細胞培地からポリペプチドを回収することによりApo-2リガンドを生産する方法に関する。もちろん、当該分野においてよく知られている他の方法を用いてApo-2リガンドを調製することはできると考えられる。
1. Apo-2リガンドをコードするDNAの単離
Apo-2リガンドをコードするDNAは、Apo-2リガンドmRNAを保有していてそれを検出可能なレベルで発現すると考えられる組織から調製された任意のcDNAライブラリから得ることができる。従って、ヒトApo-2リガンドDNAは、実施例1に記載されたヒト胎盤cDNAのバクテリオファージライブラリのように、ヒトの組織から調製されたcDNAライブラリから簡便に得ることができる。またApo-2リガンドコード化遺伝子は、ゲノムライブラリから又はオリゴヌクレオチド合成により得ることもできる。
【0029】
ライブラリは、対象となる遺伝子あるいはその遺伝子によりコードされるタンパク質を同定するために設計された(Apo-2リガンドに対する抗体又は少なくとも約20−80塩基のオリゴヌクレオチド等の)プローブによってスクリーニングできる。オリゴヌクレオチドプローブの例は実施例1に提供する。選択されたプローブによるcDNA又はゲノムライブラリのスクリーニングは、例えばSambrook等, Molecular Cloning: A Laboratory Manual(New York: Cold Spring Harbor Laboratory Press, 1989)に記載されている標準的な手順を使用して実施することができる。Apo-2リガンドをコードする遺伝子を単離する他の方法はPCR法を使用するものである[Sambrook等,上掲;Dieffenbach等, PCR Primer:A Laboratory Manual(Cold Spring Harbor Laboratory Press, 1995)]。
スクリーニングの一つの好ましい方法は、種々のヒト組織からcDNAライブラリをスクリーニングするために選別したオリゴヌクレオチド配列を用いることである。以下の実施例1には、2つの異なるオリゴヌクオチドプローブでのcDNAライブラリのスクリーニング技術を記載している。プローブとして選択されたオリゴヌクレオチド配列は、充分な長さで、疑陽性が最小化されるよう充分に明瞭でなければならない。オリゴヌクレオチドは、スクリーニングされるライブラリ内のDNAとのハイブリッド化時に検出可能であるように標識されていることが好ましい。標識化の方法は、当該分野において良く知られており、32P標識されたATPのような放射標識、ビオチン化あるいは酵素標識の使用が含まれる。
【0030】
全てのタンパク質コード化配列を有する核酸は、ここで開示された推定アミノ酸配列を使用し、また必要ならば、cDNAに逆転写されなかったmRNAの生成中間体及び先駆物質を検出する上掲のSambrookらにより記述されている従来のプライマー伸展法を使用し、選択されたcDNA又はゲノムライブラリをスクリーニングすることにより得られる。
Apo-2リガンドのアミノ酸配列変異体は、Apo-2リガンドDNAに適切なヌクレオチド変化を導入するか、所望のApo-2リガンドポリペプチドを合成することにより調製することができる。このような変異体は全長Apo-2リガンドについて図1Aに示したアミノ酸配列の細胞内領域、膜貫通領域、又は細胞外領域の内部あるいは一方又は両方の末端に残基の挿入、置換及び/又は欠失を示す。挿入、置換及び/又は欠失の任意の組み合わせで最終構造物に到達することができるが、この最終構造物は、例えばここに定義する所望のアポトーシス活性を有する。好ましい実施態様では、変異体は、Apo-2リガンドの細胞内、膜貫通、又は細胞外領域についてここで同定した配列、あるいはApo-2リガンドの全長配列に対して、少なくとも約80%のアミノ酸配列同一性、より好ましくは少なくとも約90%の配列同一性、さらに好ましくは少なくとも約95%の配列同一性を有する。また、アミノ酸の変化により、グリコシル化部位の数と位置の変化、膜係留特性の変更のように、Apo-2リガンドの翻訳後過程を改変し得る。
【0031】
ここで記載されているApo-2リガンド配列における変異は、米国特許第5,364,934号に記載された保存的あるいは非保存的突然変異に関する技術とガイドラインの任意のものを使用して起こすことができる。これらは、オリゴヌクレオチド媒介(部位特異的)突然変異誘発、アラニンスキャニング、及びPCR突然変異誘発などを含む。
近接配列に沿った一又は複数のアミノ酸を同定するために、スキャニングアミノ酸分析法を使用することができる。好ましいスキャニングアミノ酸は、比較的小さい中性アミノ酸である。このようなアミノ酸には、アラニン、グリシン、セリン及びシステインが含まれる。変異体の主鎖構造をあまり改変することなく、ベータ炭素を越えた側鎖が除去されるため、アラニンがこの群のなかで好ましいスキャニングアミノ酸である[Cunningham等, Science, 244: 1081 (1989)]。またアラニンは最も一般的なアミノ酸であることによっても好ましい。さらに、アラニンは埋設及び露出位置の双方に頻繁に見出される[Creighton, The Proteins, (W.H. Freeman & Co., N.Y.);Chothia, J. Mol. Biol., 150:1(1976)]。
また、本発明の範囲内に含まれるApo-2リガンド配列の変異体は、アミノ末端誘導体又は修飾形にも関連する。このようなApo-2リガンド配列は、ポリペプチド配列のN末端にメチオニン又は修飾メチオニン(例えばホルミルメチオニル又は他のブロックしたメチオニル種など)を有するここに記載した任意のApo-2リガンドポリペプチドを含む。
【0032】
2. 複製可能なベクターへの核酸の挿入
天然又は変異体Apo-2リガンドをコードする核酸(例えば、cDNA又はゲノムDNA)は、さらなるクローニング(DNAの増幅)又は発現のために、複製可能なベクター内に挿入される。様々なベクターが公的に入手可能である。ベクター成分としては、一般に、これらに制限されるものではないが、次のものの一又は複数が含まれる:シグナル配列、複製開始点、一又は複数のマーカー遺伝子、エンハンサー成分、プロモーター、及び転写終結配列であり、それぞれを以下に説明する。
(i) シグナル配列成分
Apo-2リガンドは直接的に組換え手法によって生産されるだけではなく、シグナル配列あるいは成熟タンパク質あるいはポリペプチドのN末端に特異的切断部位を有する他のポリペプチドである異種性ポリペプチドとの融合ペプチドとしても生産される。一般に、シグナル配列はベクターの成分であるか、ベクターに挿入されるApo-2リガンドDNAの一部である。好ましく選択された異種シグナル配列は宿主細胞によって認識され加工される(すなわち、シグナルペプチダーゼによって切断される)ものである。シグナル配列は、例えばアルカリホスファターゼ、ペニシリナーゼ、lppあるいは熱安定なエンテロトキシンIIリーダーの群から選択される原核生物シグナル配列であってよい。酵母の分泌に関しては、シグナル配列は、酵母インベルターゼリーダー、アルファ因子リーダー(酵母菌属(Saccharomyces)及びクルイベロマイシス(Kluyveromyces)α因子リーダーを含み、後者は米国特許第5,010,182号に記載されている)、又は酸ホスフォターゼリーダー、白体(C.albicans)グルコアミラーゼリーダー(1990年4月4日発行のEP362179)、又は1990年11月15日に公開された国際特許出願第WO 90/13646号に記載されているシグナルであり得る。哺乳動物細胞の発現においては、同一あるいは関連ある種の分泌ポリペプチド由来のシグナル配列、単純ヘルペスグリコタンパク質Dシグナルのようなウイルス分泌リーダーのような他の哺乳動物のシグナル配列をタンパク質の直接分泌に使用してもよいが、インビボにおけるヒト細胞の細胞膜へのApo-2リガンドの挿入を通常指示する天然Apo-2リガンドプレ配列が十分である。
このような前駆体領域のDNAは、好ましくは、Apo-2リガンドをコードするDNAにリーディングフレームが結合される。
【0033】
(ii)複製開始点成分
発現及びクローニングベクターは共に一又は複数の選択された宿主細胞においてベクターの複製を可能にする核酸配列を含む。一般に、この配列はクローニングベクターにおいて、宿主染色体DNAとは独立にベクターが複製することを可能にするものであり、複製開始点又は自律的複製配列を含む。そのような配列は多くの細菌、酵母及びウイルスに対してよく知られている。プラスミドpBR322に由来する複製開始点は大部分のグラム陰性細菌に好適であり、2μプラスミド開始点は酵母に適しており、様々なウイルス開始点(SV40、ポリオーマ、アデノウイルス、VSV又はBPV)は哺乳動物細胞におけるクローニングベクターに有用である。一般には、哺乳動物の発現ベクターには複製開始点成分は不要である(SV40開始点が典型的には初期プロモーターを有しているため用いられる)。
殆どの発現ベクターは「シャトル」ベクターである、すなわち、それらは少なくとも一つのクラスの生物において複製可能であるが、発現のために他の生物に形質移入され得る。例えば、大腸菌においてベクターがクローン化され、そのベクターが酵母あるいは哺乳動物細胞に形質移入され、宿主細胞染色体と独立して複製することはできないとしても、発現する。
DNAは宿主ゲノムに挿入することによって増幅され得る。これは、例えばベクターにバシラス(Bacillus)ゲノムDNAに見られる配列と相補的なDNA配列を含めることにより、宿主としてバシラス種を用いて容易に達成される。このベクターを用いたバシラスの形質移入は、ゲノムとの相同的組換え及びApo-2リガンドDNAの挿入をもたらす。しかし、Apo-2リガンドをコードするゲノムDNAの回収は、Apo-2リガンドDNAを切断するのに制限酵素による消化を必要とするために、外来的に複製したベクターの場合よりも複雑である。
【0034】
(iii)選択遺伝子成分
発現及びクローニングベクターは、典型的には、選べるマーカーとも称される選択遺伝子を含む。この遺伝子は、選択的培地で成長させた形質転換宿主細胞の生存又は成長に必要なタンパク質をコードする。選択遺伝子を含むベクターと共に形質転換されていない宿主細胞は培地で生存しない。典型的な選択遺伝子は、(a)アンピシリン、ネオマイシン、メトトレキセートあるいはテトラサイクリンのような抗生物質あるいは他の毒素に耐性を与え、(b)栄養要求性欠陥を補い、又は(c)例えばバシリに対する遺伝子コードD-アラニンラセマーゼのような、複合培地から得られない重要な栄養素を供給する、タンパク質をコードする。
選択技術の一例においては、宿主細胞の成長を抑止する薬物が用いられる。異種性遺伝子で首尾よく形質転換した細胞は、抗薬物性を付与し、選択工程を生存するタンパク質を生産する。このような優性選択の例としては、薬物ネオマイシン[Southern等, J. Molec. Appl. Genet., 1:327(1982)]、ミコフェノール酸[Mulligan等, Science, 209:1422(1980)]又はハイグロマイシン[Sugden等, Mol. Cell. Biol., 5:410-413(1985)]が使用される。上述した3つの例は、真核生物のコントロール下で細菌遺伝子を用いて、適切な薬物G418又はネオマイシン(ジェネテシン)、xgpt(ミコフェノール酸)、又はハイグロマイシンにそれぞれ耐性を付与する。
【0035】
哺乳動物細胞に適切な選べるマーカーの他の例は、DHFRあるいはチミジンキナーゼのように、Apo-2リガンド核酸を捕捉することのできる細胞成分を同定することのできるものである。哺乳動物細胞の形質転換細胞は、マーカーを捕捉することによって当該形質転換細胞のみが生存できるように独特に適応化された淘汰圧下に置かれる。淘汰圧は、培地中の選択剤の濃度が次第に変化する条件下で形質転換細胞を培養することにより課し、選択遺伝子とApo-2をコードするDNAの双方を増幅させる。増幅は、成長に重要なタンパク質の生産に対する要求度が高い遺伝子が、組換え細胞の後の世代の染色体内に直列に反復されるプロセスである。増幅されたDNAから増大したApo-2リガンド量が合成される。増幅可能な遺伝子のほかの例には、メタロチオネインI及びII、アデノシンデアミナーゼ、及びオルニチンデカルボキシラーゼが含まれる。
DHFR選択遺伝子によって形質転換された細胞は、先ず、DHFRの競合的アンタゴニストであるメトトリキセート(Mtx)を含む培地において形質転換物の全てを培養することで同定される。野生型DHFRを用いた場合の好適な宿主細胞は、Urlaub 等により, Proc. Natl. Acad. Sci. USA, 77:7216 (1980)に記載されているようにして調製され増殖されたDHFR活性に欠陥のあるチャイニーズハムスター卵巣(CHO)株化細胞である。形質転換した細胞は次に濃度の高いメトトレキセートに接触させる。これによりDHFR遺伝子の複数コピーが合成され、同時に、Apo-2リガンドをコードするDNAのような発現ベクターを含む他のDNAの複数コピーが作られる。この増幅方法は、もしMtxに高度に耐性である変異体DHFR遺伝子が使用されたような場合には内在性DHFRの存在にかかわらず、任意の他の適切な宿主、例えば、ATCC番号CCL61 CHO-K1を使用することができる(EP117,060)。
【0036】
あるいは、Apo-2リガンドをコードするDNA配列、野生型DHFRタンパク質、及びアミノグリコシド3'-ホスホトランスフェラーゼ(APH)のような他の選べるマーカーで形質転換あるいは同時形質転換した宿主細胞(特に、内在性DHFRを含む野生型宿主)は、カナマイシン、ネオマイシンあるいはG418のようなアミノグリコシド抗生物質のような選択可能マーカーの選択剤を含む培地における細胞増殖により選択することができる。米国特許第4,965,199号を参照。
酵母中での使用に好適な選択遺伝子は酵母プラスミドYRp7に存在するtrp1遺伝子である[Stinchcomb等, Nature, 282:39(1979);Kingman等, Gene, 7:141(1979);Tschemper等, Gene, 10:157(1980)]。trp1遺伝子は、例えば、ATCC番号44076あるいはPEP4-1のようなトリプトファン内で成長する能力に欠ける酵母の突然変異株に対する選択マーカーを提供する[Jones, Genetics, 85:12 (1977)]。酵母宿主細胞ゲノムにtrp1破壊が存在することは、トリプトファンの不存在下における増殖による形質転換を検出するための有効な環境を提供する。同様に、Leu2欠陥酵母株(ATCC 20,622あるいは 38,626)は、Leu2遺伝子を有する既知のプラスミドによって補完される。
さらに、1.6μmの円形プラスミドpKD1由来のベクターは、クルイヴェロマイシス(Kluyveromyces)酵母の形質転換に用いることができる[Bianchi 等., Curr. Genet., 12:185(1987)]。より最近では、組換え子ウシのキモシンの大量生産のための発現系がK.ラクティス(lactis)に対して報告されている[Van den Berg, Bio/Technology, 8:135 (1990)]。クルイヴェロマイシスの工業的な菌株からの、組換えによる成熟したヒト血清アルブミンを分泌する安定した複数コピー発現ベクターも開示されている[Fleer 等, Bio/Technology,9:968-975 (1991)]。
【0037】
(iv)プロモーター成分
発現及びクローニングベクターは、通常、宿主生物によって認識され、Apo-2リガンド核酸配列に作用可能に結合したプロモーターを含む。プロモーターは、作用可能に結合したApo-2リガンド核酸配列のような特定の核酸配列の転写及び翻訳を制御する構造的な遺伝子(一般的に約100ないし1000塩基対)の開始コドンの上流側(5')に位置する未翻訳配列である。このようなプロモーターは典型的には、誘発的なクラス及び構成的なクラスの2つのクラスに属する。誘発的なプロモーターは、養分の存在あるいは不存在、温度変化等の培養条件のある変化に対応してその制御の下でDNAからの転写レベルを上昇させるプロモーターである。現時点において多種の可能な宿主細胞により認識される非常に多くのプロモーターがよく知られている。これらのプロモーターは、制限酵素の消化によって供給源DNAからプロモーターを除去し、ベクターに単離したプロモーター配列を挿入することで、Apo-2リガンドをコードするDNAに作用的に結合している。天然のApo-2リガンドプロモーター配列及び多くの異種性プロモーターはいずれもApo-2リガンドDNAの直接増幅及び/又は発現に用いることができる。
【0038】
原核生物宿主での使用に好適なプロモーターはβラクタマーゼ及びラクトースプロモーター系[Cahng 等, Nature, 275:615 (1978), Goeddel 等, Nature, 281:544 (1979)]、アルカリホスファターゼ、トリプトファン(trp)プロモーター系[Goeddel, Nucleic Acids Res., 8:4057 (1980); EP 36,776]、及びハイブリッドプロモーター、例えばtacプロモーター[deBoer 等, Proc. Natl. Acad. Sci. USA, 80:21-25 (1983)]を含む。しかし、他の既知の細菌プロモーターも好適である。これらのヌクレオチド配列は公表されており、よって当業者は、任意の所望の制限部位を提供するためにリンカーあるいはアダプターを使用することでApo-2をコードするDNA[Siebenlist 等, Cell, 20:269 (1980)]にそれらを作用可能に結合させることが可能である。細菌系で使用するプロモータもまたApo-2リガンドをコードするDNAと作用可能に結合したシャイン・ダルガーノ(S.D.)配列を有する。
真核生物に対してもプロモーター配列が知られている。実質的に全ての真核生物の遺伝子は、転写開始部位からおよそ25ないし30塩基上流に見出されるATリッチ領域を有する。多数の遺伝子の転写開始位置から70ないし80塩基上流に見出される他の配列は、Xが任意のヌクレオチドであるCXCAAT領域である。大部分の真核生物遺伝子の3'末端には、コード配列の3'末端へのポリA尾部の付加に対するシグナルであるAATAAA配列がある。これらの配列は全て真核生物の発現ベクターに適切に挿入される。
【0039】
酵母宿主と共に用いて好適なプロモーター配列の例としては、3-ホスホグリセラートキナーゼ[Hitzeman 等, J. Biol. Chem., 255:2073 (1980)]又は他の糖分解酵素[Hess 等, J. Adv. Enzyme Reg., 7:149 (1968);Holland, Biochemistry, 17:4900(1987)]、例えばエノラーゼ、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ、デヒドロゲナーゼ、ヘキソキナーゼ、ピルビン酸デカルボキシラーゼ、ホスホフルクトキナーゼ、グルコース-6-リン酸イソメラーゼ、3-ホスホグリセレートムターゼ、ピルビン酸キナーゼ、トリオセリン酸イソメラーゼ、ホスホグルコースイソメラーゼ、及びグルコキナーゼが含まれる。
他の酵母プロモーターとしては、成長条件によって転写が制御される付加的効果を有する誘発的プロモーターであり、アルコールデヒドロゲナーゼ2、イソチトクロムC、酸ホスファターゼ、窒素代謝と関連する分解性酵素、メタロチオネイン、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ、及びマルトース及びガラクトースの利用を支配する酵素のプロモーター領域がある。酵母の発現に好適に用いられるベクターとプロモータはEP73,657に更に記載されている。また酵母エンハンサーも酵母プロモーターと共に好適に用いられる。
【0040】
哺乳動物の宿主細胞におけるベクターからのApo-2リガンド転写は、例えば、ポリオーマウィルス、伝染性上皮腫ウィルス(1989年7月5日公開のUK 2,211,504)、アデノウィルス(例えばアデノウィルス2)、ウシ乳頭腫ウィルス、トリ肉腫ウィルス、サイトメガロウィルス、レトロウィルス、B型肝炎ウィルス及び最も好ましくはサルウィルス40(SV40)のようなウィルスのゲノムから得られるプロモーター、異種性哺乳動物プロモーター、例えばアクチンプロモーター又は免疫グロブリンプロモーター、熱衝撃プロモーター、そしてApo-2配列に通常付随するプロモーターによって、このようなプロモーターが宿主細胞系に適合し得る限り、調節される。
SV40ウィルスの初期及び後期プロモーターは、SV40ウイルスの複製起点をさらに含むSV40制限断片として簡便に得られる[Fiers等, Nature, 273:113 (1978);Mulligan及びBerg, Science, 209:1422-1427 (1980); Pavlakis等, Proc. Natl. Acad. Sci. USA, 78: 7398-7402 (1981)]。ヒトサイトメガロウィルスの最初期プロモーターは、HindIIIE制限断片として簡便に得られる[Greenaway等, Gene, 18:355-360 (1982)]。ベクターとしてウシ乳頭腫ウィルスを用いて哺乳動物宿主でDNAを発現する系が、米国特許第4,419,446号に開示されている。この系の修飾は米国特許第4,601,978号に開示されている[また、サル細胞での免疫インターフェロンをコードしているcDNAの発現について、Gray等, Nature, 295:503-508(1982)を;単純ヘルペスウイルス由来のチミジンキナーゼプロモーターの調節下でのマウス細胞におけるヒトβインターフェロンcDNAの発現について、Reyes等, Nature, 297:598-601(1982)を;培養されたマウス及びウサギの細胞におけるヒトインターフェロンβ1遺伝子の発現について、Canaani及びBerg, Proc. Natl. Acad. Sci. USA, 79:5166-5170(1982)を;プロモーターとしてラウス肉腫ウィルスの長い末端反復を用いたCV-1サル腎臓細胞、ニワトリ胚線維芽細胞、チャイニーズハムスター卵巣細胞、HeLa細胞、及びマウスNIH-3T3細胞における細菌CAT配列の発現について、Gorma等, Proc. Natl. Acas. Sci. USA, 79:6777-6781 (1982)を参照のこと]。
【0041】
(v)エンハンサーエレメント成分
より高等の真核生物による本発明のApo-2リガンドをコードしているDNAの転写は、ベクター中にエンハンサー配列を挿入することによって増強され得る。エンハンサーは、通常は約10から300塩基対で、プロモーターに作用してその転写を増強するDNAのシス作動要素である。エンハンサーは、相対的に配向及び位置が独立しており、転写ユニットの5'[Laimins等, Proc. Natl. Acad. Sci. USA, 78:993 (1981)]及び 3'[Lusky等, Mol. Cell Bio., 3:1108 (1983)]、イントロン内部[Banerji等, Cell, 33:729 (1983)]並びにコード配列自身の内部[Osborne等, Mol. Cell Bio., 4:1293 (1984)]に見出されている。哺乳動物の遺伝子由来の多くのエンハンサー配列が現在知られている(グロビン、エラスターゼ、アルブミン、α-フェトプロテイン及びインスリン)。しかしながら、典型的には、真核細胞ウィルス由来のエンハンサーが用いられるであろう。例としては、複製起点の後期側のSV40エンハンサー(100-270塩基対)、サイトメガロウィルス初期プロモーターエンハンサー、複製起点の後期側のポリオーマエンハンサー及びアデノウィルスエンハンサーが含まれる。真核生物のプロモーターの活性化のための増強要素については、Yaniv, Nature, 297:17-18 (1982)もまた参照のこと。エンハンサーは、Apo-2リガンドコード配列の5'又は3'位でベクター中にスプライシングされ得るが、好ましくはプロモーターから5'位に位置している。
【0042】
(vi)転写終結成分
真核生物宿主細胞(酵母、真菌、昆虫、植物、動物、ヒト、又は他の多細胞生物由来の有核細胞)に用いられる発現ベクターは、また転写の終結及びmRNAの安定化に必要な配列を含む。このような配列は、真核生物又はウィルスのDNA又はcDNAの5'、時には3'の非翻訳領域から一般に取得できる。これらの領域は、Apo-2リガンドをコードしているmRNAの非翻訳部分にポリアデニル化断片として転写されるヌクレオチドセグメントを含む。
【0043】
(vii)ベクターの作成と分析
一又は複数の上に列挙した成分を含む適切なベクターの作成には標準的なライゲーション技術を用いる。分離されたプラスミド又はDNA断片を開裂させ、整え、そして必要とされるプラスミドの生成のために望ましい型に再ライゲーションする。
作成されたプラスミドが正しい配列であることを確認する分析のために、ライゲーション混合物を用いて、大腸菌K12菌株294(ATCC 31446)を形質転換し、適切な場合にはアンピシリン又はテトラサイクリン耐性によって、形質転換細胞を好適に選択する。形質転換細胞からプラスミドを調製し、制限エンドヌクレアーゼ消化により分析し、及び/又はMessing等, Nucleic Acids Res., 9:309 (1981)の方法又はMaxam等, Methods in Enzymology, 65:499 (1980)の方法によって配列決定する。
【0044】
(viii)一過性発現ベクター
Apo-2リガンドをコードしているDNAの哺乳動物細胞における一過性発現をもたらす発現ベクターを使用することができる。一般に、一過性発現は、宿主細胞が発現ベクターの多くのコピーを蓄積し、次にその発現ベクターによってコードされている所望のポリペプチドを高レベルで合成するように、宿主細胞中で効果的に複製できる発現ベクターを使用することを含む[Sambrook等, 上掲]。一過性発現系は、適切な発現ベクターと宿主細胞を含むが、クローニングされたDNAによりコードされているポリペプチドの簡便で確実な同定並びに所望の生物学的又は生理学的性質についてのポリペプチドの迅速なスクリーニングを可能にする。したがって、一過性発現系は、本発明において、生物学的に活性なApo-2リガンドであるApo-2リガンドの類似物及び変異体を同定する目的のために特に有用である。
【0045】
(ix)適切な例示的脊椎動物細胞ベクター
組換え脊椎動物細胞培養でのApo-2リガンドの合成に適応化するのに適切な他の方法、ベクター及び宿主細胞は、Gething等, Nature, 293:620-625 (1981); Mantei等, Nature, 281:40-46 (1979); EP117,060; 及びEP117,058に記載されている。Apo-2リガンドの哺乳動物細胞培地での発現に特に適したプラスミドは、pRK5[EP307,247;実施例1にも記載]又はpSVI6B[1991年6月13日に発行のWO 91/08291]である。
【0046】
3. 宿主細胞の選択及び形質転換
ここに記載のベクターにDNAをクローニングあるいは発現するために適切な宿主細胞は、上述の原核生物、酵母、又は高等真核生物細胞である。この目的にとって適切な原核生物は、限定するものではないが、真正細菌、例えばグラム陰性又はグラム陽性生物体、例えばエシェリチアのような腸内菌科、例えば大腸菌、エンテロバクター、エルウィニア(Erwinia)、クレブシエラ、プロテウス、サルモネラ、例えばネズミチフス菌、セラチア属、例えばセラチア・マルセスキャンス及び赤痢菌属、並びに桿菌、例えば枯草菌及びバシリ・リチェフォルミス(licheniformis)(例えば、1989年4月12日に公開されたDD266,710に開示されたバシリ・リチェニフォルミス41P)、シュードモナス属、例えば緑膿菌及びストレプトマイセス属を含む。好ましくは、宿主細胞は最小量のタンパク質分解酵素を分泌すべきである。
原核生物に加えて、糸状菌又は酵母菌のような真核微生物は、Apo-2リガンドをコードするベクターのための適切なクローニング又は発現宿主である。サッカロミセス・セレヴィシア、又は一般的なパン酵母は下等真核生物宿主微生物のなかで最も一般的に用いられる。しかしながら、多数の他の属、種及び菌株も、一般的に入手可能で有用である。
【0047】
グリコシル化Apo-2リガンドの発現に適切な宿主細胞は、多細胞生物から誘導される。このような宿主細胞は、複雑なプロセシング及びグリコシル化活動が可能である。原則的には、脊椎動物であろうと無脊椎動物培養であろうと、任意のより高等の真核生物細胞培養が使用できる。無脊椎動物細胞の例としては植物及び昆虫細胞が含まれる。多数のバキュロウィルス株及び変異体及び対応する許容可能な昆虫宿主細胞、例えばスポドプテラ・フルギペルダ(毛虫)、アエデス・アエジプティ(蚊)、アエデス・アルボピクトゥス(蚊)、ドゥロソフィラ・メラノガスター(ショウジョウバエ)、及びボンビクス・モリが同定されている[例えば、Luckow等, Bio/Technology, 6:47-55 (1988); Miller等, Genetic Engineering, Setlow等, eds., Vol. 8 (Plenum Publishing, 1986), pp.277-279; 及びMaeda等, Nature, 315:592-594 (1985)を参照のこと]。トランスフェクションのための種々のウィルス株、例えば、オートグラファ・カリフォルニカNPVのL-1変異体とボンビクス・モリ NPVのBm-5株が公に利用でき、それらのウイルスは、特に実施例2に記載するSpodopterafugiperda(「Sf9」)の形質移入のために、本発明に従うウイルスとして使用できる。
【0048】
綿花、コーン、ジャガイモ、大豆、ペチュニア、トマト、及びタバコのような植物細胞培養を宿主として利用することができる。典型的には、植物細胞は、細菌アグロバクテリウム・トゥメファシエンスの、既にApo-2リガンドコード化DNAを含むように操作された或る菌株と共にインキュベートすることによってトランスフェクトされる。A. トゥメファシエンスと共に植物細胞培養をインキュベートする間に、Apo-2リガンドをコードするDNAが、植物細胞宿主にそれが形質移入されるよう移され、そして適切な条件下でApo-2リガンドをコードするDNAが発現される。加えて、例えば、ノパリンシンターゼプロモーター及びポリアデニル化シグナル配列のような、植物細胞と適合しうる調節及びシグナル配列が利用できる[Depicker等, J. Mol. Appl. Gen., 1:561 (1982)]。また、T-DNA780遺伝子の上流領域から分離されるDNAセグメントは、組換えDNAを含む植物組織中の植物発現遺伝子の転写レベルを活性化又は増強しうる[1989年6月21日公開のEP 321,196]。
【0049】
培地(組織培地)中での脊椎動物細胞の増殖は、当該分野おいてよく知られている[例えば、Tissue Culture,Academic Press,編者Kruse及びPatterson(1973)を参照のこと]。有用な哺乳動物宿主株化細胞の例は、SV40によって形質転換されたサル腎臓CV1株 (COS-7, ATCC CRL 1651);ヒト胚腎臓株[293又は懸濁培養での増殖のためにサブクローン化された293細胞、Graham等, J. Gen Virol., 36:59 (1977)];ハムスター乳児腎細胞(BHK, ATCC CCL 10);チャイニーズハムスター卵巣細胞/-DHFR(CHO, Urlaub及びChasin, Proc. Natl. Acad. Sci. USA, 77:4216 (1980));マウスのセルトリ細胞[TM4, Mather, Biol. Reprod., 23:243-251 (1980)];サルの腎細胞 (CVI ATCC CCL 70); アフリカミドリザルの腎細胞(VERO-76, ATCC CRL-1587); ヒト子宮頸癌細胞 (HELA, ATCC CCL 2); イヌ腎細胞 (MDCK, ATCC CCL 34); バッファローラット肝細胞 (BRL 3A, ATCC CRL 1442); ヒト肺細胞 (W138, ATCC CCL 75); ヒト肝細胞 (Hep G2, HB 8065); マウス乳房腫瘍細胞 (MMT 060562, ATTC CCL51);TRI細胞[Mother等, Annals N.Y. Acad. Sci., 383:44-68 (1982)];MRC5細胞;及びFS4細胞である。
【0050】
宿主細胞をトランスフェクトし、好ましくは上述のApo-2リガンド産生のための発現又はクローニングベクターで形質転換し、プロモーターを誘導し、形質転換体を選択し、又は所望の配列をコードしている遺伝子を増幅するために適切に修飾された標準栄養培地で培養する。
形質移入は、如何なるコード配列が実際に発現されるか否かにかかわらず、宿主細胞による発現ベクターの取り込みを意味する。多数の形質移入の方法が当業者に知られている。例えば、CaPO4及びエレクトロポレーションである。このベクターの操作のあらゆる徴候が宿主細胞内で生じたときに成功した形質移入が一般に認められる。
形質転換は、染色体外の成分としてであろうと染色体成分によってであろうと、DNAが複製可能であるように生物体中にDNAを導入することを意味する。用いられる宿主細胞に応じて、そのような細胞に対して適した標準的な方法を用いて形質転換はなされる。前掲のSambrook等により記載された塩化カルシウムを用いるカルシウム処理又はエレクトロポレーションが、原核生物又は実質的な細胞壁障壁を含む他の細胞に対して用いられる。アグロバクテリウム・トゥメファシエンスによる感染が、Shaw等, Gene, 23:315 (1983)及び1989年6月29日公開の国際特許出願第WO 89/05859に記載されたように、ある種の植物細胞の形質転換に用いられる。加えて、1991年1月10日に公開された国際特許出願第WO 91/00358に記載されているように、超音波処理を用いて植物を形質移入することもできる。
【0051】
このような細胞壁のない哺乳動物細胞に対しては、Graham及びvan der Eb, Virology, 52:456-457 (1978)のリン酸カルシウム沈殿法が好ましい。哺乳動物細胞の宿主系形質転換の一般的な側面は米国特許第4,399,216号に記載されている。酵母中の形質転換は、典型的には、Van solingen等, J. Bact., 130:946 (1977)及びHsiao等, Proc. Natl. Acad. Sci. USA, 76:3829 (1979)の方法によって実施する。しかしながら、DNAを細胞中に導入する他の方法、例えば、核マイクロインジェクション、エレクトロポレーション、無傷の細胞、又はポリカチオン、例えばポリブレン、ポリオルニチン等を用いる細菌プロトプラスト融合もまた用いることもできる。哺乳動物細胞を形質転換するための種々の技術については、Keown等, Methods in Enzymology, 185:527-537 (1990)及び Mansour等, Nature, 336:348-352 (1988)を参照のこと。
【0052】
4.宿主細胞の培養
本発明のApo-2リガンドポリペプチドを生成るために用いられる原核細胞は、前掲のSambrook等により記載されているような適切な培地で培養される。
Apo-2リガンドの産生に用いられる哺乳動物の宿主細胞は種々の培地において培養することができる。市販培地の例としては、ハム(Ham)のF10(シグマ)、最小必須培地(「MEM」,シグマ)、RPMI-1640(シグマ)及びダルベッコの改良イーグル培地(「DMEM」,シグマ)が含まれる。これらの培地はいずれも、ホルモン及び/又は他の成長因子(例えばインスリン、トランスフェリン、又は表皮成長因子)、塩類(例えば、塩化ナトリウム、カルシウム、マグネシウム及びリン酸塩)、バッファー(例えばHEPES)、ヌクレオシド(例えばアデノシン及びチミジン)、抗生物質(例えば、ゲンタマイシンTM薬)、微量元素(最終濃度がマイクロモル範囲で通常存在する無機化合物として定義される)及びグルコース又は同等のエネルギー源を必要に応じて補充することができる。任意の他の必要な補充物質もまた当業者に知られている適当な濃度で含むことができる。培養条件、例えば温度、pH等々は、発現のために選ばれた宿主細胞について以前から用いられているものであり、当業者には明らかであろう。
一般に、哺乳動物の細胞培養の生産性を最大にするための原理、プロトコール、及び実用技術は、Mammalian Cell Biotechnology: a Practical Approach, M.Butler編 (IRL Press, 1991)に見出すことができる。
この明細書において言及される宿主細胞は培養中の細胞並びに宿主動物内にある細胞を包含する。
【0053】
5.遺伝子増幅/発現の検出
遺伝子の増幅及び/又は発現は、ここで提供された配列に基づき、適切に標識されたプローブを用い、例えば、従来よりのサザンブロット法、mRNAの転写を定量化するノーザンブロット法[Thomas,Proc. Natl. Acad. Sci. USA,77:5201-5205 (1980)]、ドットブロット法(DNA分析)、又はインサイツハイブリッド形成法によって、直接的に試料中で測定することができる。種々の標識を用いることができ、最も一般的なものは放射性同位元素、特に32Pである。しかしながら、他の方法、例えばポリヌクレオチド中への導入のためのビオチン修飾されたヌクレオチドもまた使用することができる。ついで、このビオチンは、例えば放射性ヌクレオチド、蛍光剤又は酵素のような広範囲の標識で標識することができるアビジン又は抗体への結合部位として作用する。また、DNA二本鎖、RNA二本鎖及びDNA−RNAハイブリッド二本鎖又はDNA-タンパク質二本鎖を含む、特異的二本鎖を認識することができる抗体を用いることもできる。ついで、抗体を標識し、アッセイを実施することができ、ここで二本鎖は表面に結合しており、その結果二本鎖の表面での形成の時点でその二本鎖に結合した抗体の存在を検出することができる。
あるいは、遺伝子の発現は、遺伝子産物の発現を直接的に定量する免疫学的な方法、例えば細胞又は組織切片の免疫組織化学的染色及び細胞培養又は体液のアッセイによって、測定することもできる。免疫組織化学的染色技術では、細胞試料を、典型的には脱水と固定によって調製し、結合した遺伝子産物に対し特異的な標識化抗体と反応させるが、この標識は通常は視覚的に検出可能であり、例えば酵素的標識、蛍光標識、又はルミネサンス標識である。
試料液の免疫組織化学的染色及び/又はアッセイに有用な抗体は、モノクローナルでもポリクローナルでもよく、任意の哺乳動物で調製することができる。簡便には、抗体は、天然配列Apo-2リガンドポリペプチドに対して、又はここで提供されるDNA配列をベースとした合成ペプチドに対して、又はApo-2リガンドDNAに融合し特異的抗体エピトープをコードする外因性配列に対して調製され得る。
【0054】
6.Apo-2リガンドポリペプチドの精製
Apo-2リガンドは、培養培地から分泌されたポリペプチドとして回収されるが、分泌シグナル無しで直接産生される場合は宿主細胞溶解液から回収してもよい。Apo-2リガンドが膜結合性であるならば、適切な洗浄液(例えばトリトン-X100)を用いて膜から引き離すか、又は酵素的切断によりその細胞外ドメインを放出させる。
Apo-2リガンドがヒト起源のもの以外の組換え細胞でつくられるときは、Apo-2リガンドはヒト起源のタンパク質又はポリペプチドを含んでいない。しかしながら、Apo-2リガンドに関して実質的に相同である調製物を得るには、組換え細胞タンパク又はポリペプチドからApo-2リガンドを精製することが望ましい。第一段階として、培地又は溶菌液を遠心分離して粒状の細胞屑を除去することができる。ついで、Apo-2リガンドを、汚染した可溶性タンパク質及びポリペプチドから、適切な精製手順の例である次の手順により精製される:すなわち、イオン交換カラムでの分画;エタノール沈殿;逆相HPLC;シリカ又はカチオン交換樹脂、例えばDEAEによるクロマトグラフィー;クロマトフォーカシング;SDS-PAGE;硫酸アンモニウム沈殿;例えばセファデックスG-75を用いるゲル濾過;及びIgGのような汚染物を除くプロテインAセファロースカラムである。
好ましい実施態様では、Apo-2リガンドは実施例3に記載するようなアフィニティクロマトグラフィによって精製される。
【0055】
残基が欠失され、挿入され、又は置換されたApo-2リガンド変異体は、その変異によってしばしば惹起される実質的な特性変化を考慮に入れて、天然Apo-2リガンドと同じようにして回収することができる。例えば、他のタンパク質又はポリペプチド、例えば細菌性もしくはウイルス性抗原とApo-2リガンドとの融合体の調製は精製を容易にする;配列に対する抗体を含む免疫アフィニティーカラムを、融合ポリペプチドを吸着するために使用することができる。好ましい実施態様では、Apo-2リガンドの細胞外配列をHis10ペプチドに融合させ、Ni2+キレートアフィニティクロマトグラフィにより精製する。
例えばフェニルメチルスルホニルフロリド(PMSF)のようなプロテアーゼインヒビターもまた精製の間のタンパク分解を阻害するのに有用であり、偶発的な汚染物質の成長を防止するために抗生物質を含めることができる。天然Apo-2リガンドに適切な精製方法は、組換え細胞培養の発現の際におけるApo-2リガンド又はその変異体の特性の変化の起因となる改変が必要となることは、当業者であれば理解するであろう。
【0056】
7.Apo-2リガンドポリペプチドの共有結合的修飾
Apo-2リガンドの共有結合的修飾は本発明の範囲内に含まれる。天然Apo-2リガンド及びApo-2リガンドのアミノ酸配列変異体の両方が共有結合的に修飾されうる。Apo-2リガンドの共有結合的修飾の一つの型は、Apo-2リガンドの標的化アミノ酸残基を、Apo-2リガンドのN末端又はC末端残基、又は選択された側鎖と反応できる有機誘導体形成剤と反応させることによって分子内に導入することができる。
二官能性試薬による誘導体形成は、抗Apo-2リガンド抗体を精製する方法に使用する水不溶性支持体マトリックス又は表面へのApo-2リガンドの架橋に有用であり、またその逆も同様である。通常使用される架橋剤には、例えば、1,1-ビス(ジアゾアセチル)-2-フェニルエタン、グルタルアルデヒド、N-ヒドロキシスクシンイミドエステル、例えば、4-アジドサリチル酸とのエステル、3,3'-ジチオビス(スクシンイミジルプロピオナート)のようなジスクシンイミジルエステルを包含するホモ二官能性イミドエステル、及びビス-N-マレイミド-1,8-オクタンのような二官能性マレイミドが含まれる。メチル-3-[(p-アジドフェニル)ジチオ]プロピオイミダートのような誘導体形成剤は、光の存在下で架橋を形成することができる光活性化中間体を生じる。また、臭化シアン活性化炭水化物のような反応性の水不溶性マトリックス及び米国特許第3,969,287号;3,691,016号;4,195,128号;4,247,642号;4,229,537号及び4,330,440号に記載されている反応物質がタンパク固定化に用いられる。
【0057】
その他の修飾は、それぞれ対応するグルタミル及びアスパルチル残基へのグルタミニル及びアスパラギニル残基の脱アミド化、プロリンとリジンのヒドロキシル化、セリル又はスレオニル残基のヒドロキシル基のリン酸化、リジン、アルギニン、及びヒスチジン側鎖のα-アミノ基のメチル化[T.E. Creighton, Proteins: Structure and Molecular Properties, W.H. Freeman & Co., San Francisco, PP.79-86 (1983)]、N末端アミンのアセチル化、及び任意のC末端カルボキシル基のアミド化を含む。残基の修飾型も本発明の範囲に入る。
本発明の範囲内に含まれるApo-2リガンドポリペプチドの共有結合的修飾の他の型は、ポリペプチドの天然グリコシル化パターンを変更することを含む。「天然グリコシル化パターンの変更」とは、天然Apo-2リガンドに見出される一又は複数の炭水化物部分の欠失、及び/又は天然Apo-2リガンドに存在しない一又は複数のグリコシル化部位を付加することを意味することをここでは意図している。
【0058】
ポリペプチドのグリコシル化は、典型的には、N結合又はO結合の何れかである。N結合とは、アスパラギン残基の側鎖への炭水化物部分の結合を指す。アスパラギン-X-セリン及びアスパラギン-X-スレオニン(ここでXはプロリンを除く任意のアミノ酸)というトリペプチド配列は、アスパラギン側鎖への炭水化物部分の酵素的結合のための認識配列である。したがって、ポリペプチド中にこれらのトリペプチド配列の何れかが存在すると、可能性のあるグリコシル化部位が作り出される。O結合グリコシル化は、ヒドロキシルアミノ酸、最も一般的にはセリン又はスレオニン(5-ヒドロキシプロリン又は5-ヒドロキシリジンもまた用いられるが)に、糖類N-アセチルガラクトサミン、ガラクトース、又はキシロースの一つが結合することを意味する。
Apo-2リガンドポリペプチドへのグリコシル化部位の付加は、アミノ酸配列を、それが一又は複数の上述したトリペプチド配列(N結合グリコシル化部位のもの)を含むように変化させることによって達成される。この変化は、天然Apo-2リガンドへの一又は複数のセリン又はスレオニン残基の付加、又はこれによる置換によってもなされる(O結合グリコシル化部位の場合)。場合によっては、Apo-2リガンドアミノ酸配列はDNAレベルでの変化によって、特に、所望のアミノ酸に翻訳するコドンが産生されるように予め選んだ塩基でApo-2リガンドポリペプチドをコードしているDNAを突然変異させることによって変更される。このDNA突然変異は、上記に記載され前掲の米国特許第5,364,934号に記載された方法を用いてなされる。
【0059】
Apo-2リガンドポリペプチド上の炭水化物部分の数を増加させる他の手段は、該ポリペプチドへのグリコシドの化学的又は酵素的結合による。用いられる結合形態に応じて、糖(類)は、(a)アルギニンとヒスチジンに、(b)遊離のカルボキシル基に、(c)遊離のスルフヒドリル基、例えばシステインのものに、(d)セリン、スレオニン又はヒドロキシプロリンのもののような遊離のヒドロキシル基に、(e)フェニルアラニン、チロシン又はトリプトファンのような芳香族残基、又は(f)グルタミンのアミノ基に結合される。これらの方法は1987年9月11日公開の国際特許出願第WO 87/05330号及びAplin及びWriston, CRC Crit. Rev. Biochem., pp259-306 (1981)に記載されている。
Apo-2リガンドポリペプチド上に存在する炭水化物部分の除去は、化学的又は酵素的になされる。例えば、化学的脱グリコシル化は、化合物トリフルオロメタンスルホン酸、又は等価な化合物へ該ポリペプチドを曝露し、該ポリペプチドを無傷のまま残しながら、結合糖(N-アセチルグルコサミン又はN-アセチルガラクトサミン)を除く殆ど又は全ての糖を開裂させる。化学的脱グリコシル化は、Hakimuddin等, Arch. Biochem Biophys., 259:52 (1987)及びEdge等, Anal. Biochem., 118:131 (1981)により記載されている。ポリペプチド上の炭水化物部分の酵素的開裂は、Thotakura等, Meth. Enzymol., 138:350 (1987)に記載されているように、種々のエンド及びエキソグリコシダーゼを使用して達成することができる。
【0060】
潜在的なグリコシル化部位でのグリコシル化は、Duskin等, J. Biol. Chem., 257:3105 (1982)に記載されているように、化合物ツニカマイシンを使用して防止することができる。ツニカマイシンはタンパク質-N-グルコシド結合の形成を阻害する。
Apo-2リガンドの共有結合的修飾の他の型は、Apo-2リガンドポリペプチドの種々の非タンパク質性ポリマー、例えばポリエチレングリコール、ポリプロピレングリコール、又はポリオキシアルキレンの1つに結合させることを含み、これは例えば米国特許第4,640,835号;第4,496,689号;第4,301,144号;第4,670,417号;第4,791,192号又は第4,179,337号に記載されているように行われる。
【0061】
8.エピトープタグApo-2リガンド
また本発明は、他の異種性ポリペプチドと融合したApo-2リガンドを含むキメラポリペプチドを提供する。一実施態様では、キメラポリペプチドは、抗タグ抗体が選択的に結合するエピトープを提供するタグポリペプチドとのApo-2リガンドの融合体を含む。エピトープタグは一般にApo-2リガンドのアミノ又はカルボキシル末端に位置する。Apo-2リガンドのこのようなエピトープタグが付けられた形の存在は、タグポリペプチドに対する抗体を用いて検出することができる。また、エピトープタグを供給すると、Apo-2リガンドを抗タグ抗体又はエピトープタグに結合する他の種類のアフィニティーマトリックスを用いたアフィニティー精製によって容易に精製することが可能になる。
【0062】
様々なタグポリペプチドとその各々の抗体はこの分野で良く知られている。例としては、fluHAタグポリペプチドとその抗体12CA5[Field等, Mol. Cell. Biol., 8:2159-2165 (1988)];c-mycタグとそれに対する8F9、3C7、6E10、G4、B7及び9E10抗体[Evan等, Molecular and Cellular Biology, 5:3610-3616 (1985)];及び単純ヘルペスウィルス糖タンパクD(gD)タグとその抗体[Paborsky等, Protein Engineering, 3(6):547-553 (1990)]が含まれる。他のタグポリペプチドには、フラッグ-ペプチド(Flag-peptide)[Hopp等, BioTechnology, 6:1204-1210 (1988)];KT3エピトープペプチド[Martin等, Science, 255:192-194 (1992)];α-チューブリンエピトープペプチド[Skinner等, J. Biol. Chem., 266:15163-15166 (1991)];及びT7遺伝子10タンパクペプチドタグ[Lutz-Freyermuth等, Proc. Natl. Acad. Sci. USA, 87:6393-6397 (1990)]が含まれる。ひとたびタグポリペプチドが選択されれば、それに対する抗体を、ここに開示した方法を用いて産生することができる。
【0063】
一般的に、エピトープタグApo-2リガンドは、天然又は変異体Apo-2リガンドに対して上述した方法に従い作成され生産される。Apo-2リガンド-タグポリペプチド融合体は、Apo-2リガンド部分をインフレームでコードしているcDNA配列をタグポリペプチドDNA配列に融合させ、得られたDNA融合作成物を適当な宿主細胞に発現させることによって好ましく作成される。通常は、本発明のApo-2リガンド-タグポリペプチドキメラを調製するときは、Apo-2リガンドをコードしている核酸を、タグポリペプチドのN末端をコードしている核酸にその3'末端で融合させるが、5'融合もまた可能である。エピトープタグApo-2リガンドの例は、下記の実施例2にさらに詳細に記載する。
エピトープタグApo-2リガンドは、抗タグ抗体を用いてアフィニティークロマトグラフィーによって精製することができる。アフィニティー抗体が固着されるマトリクスには、例えばアガロース、調整穴明きガラス又はポリ(スチレンジビニル)ベンゼンが含まれる。ついで、エピトープタグApo-2リガンドは、当該分野において知られた技術を使用して、アフィニティーカラムから溶出することができる。
【0064】
B.Apo-2の治療的用途
本明細書に開示されているように、Apo-2リガンドは治療的に用いることができる。例えばApo-2リガンド哺乳動物癌細胞にアポトーシスを誘発するために用いることができる。一般的に、哺乳動物癌細胞においてアポトーシスを誘発する方法は、当該細胞を有効量のApo-2リガンドに暴露することを含む。これは、例えば下記の実施例に記載する方法に従って、インビボ又はエキソビボで行うことができる。アポトーシスを誘発する方法は、アポトーシスレベルの低下を特徴とする特定の病理学的状態の治療に用いることができる。このような病理学的状態の例は、自己免疫疾患、例えば狼瘡及び免疫媒介糸球体腎炎、及び癌を含む。Apo-2リガンドの癌の治療のための治療的用途を以下に詳細に説明する。
癌の治療方法において、Apo-2リガンドは癌を持つと診断された哺乳動物に投与される。言うまでもなく、Apo-2リガンドは、他のアポトーシス誘発剤、化学治療、放射線治療、及び手術を含むさらに他の治療的組成物及び技術と組み合わせて用いることもできる。
【0065】
Apo-2リガンドは、好ましくは担体中で哺乳動物に投与される。好適な担体とそれらの製剤は、Oslo等により編集されたRemington's Pharmaceutical Sciences, 16th ed., 1980, Mack Publishing Co.,に記載されている。典型的には、適量の製薬的に許容可能な塩が、製剤を等浸透圧にするために製剤において使用される。製薬的に許容可能な担体の例には、生理食塩水、リンガー液及びデキストロース液が含まれる。溶液のpHは、好ましくは約5〜約8、さらに好ましくは約7.4〜約7.8である。例えば投与経路及び投与されるApo-2リガンドの濃度よっては、或る種の担体がより好ましくなることは、当業者には明らかである。
Apo-2リガンドは、注射(例えば、静脈内、腹腔内、皮下、筋肉内)、又は点滴のように、有効な形態で血流への送達を確実にする他の方法により哺乳動物に投与することができる(実施例15参照)。また、Apo-2リガンドはインビボ又はエキソビボ遺伝子治療により投与できるとも考えられる。
【0066】
Apo-2リガンド投与の有効な用量とスケジュールは経験的に決定することができ、このような決定は当業者の技量の範囲に含まれる。今日では、単独で用いられるApo-2リガンドの有効用量又は量は、1日に体重1kg当たり約1μg〜約100mg又はそれ以上の範囲であると考えられている。用量の種内スケーリングは、この分野で周知の方法、例えばMordenti等, Pharmaceut. Res., 8 1351 (1991)に記載されているように実施することができる。当業者はApo-2リガンドの投与しなければならない用量が、例えばApo-2リガンドを受容する哺乳動物、投与経路、及び哺乳動物に施される他の医薬又は治療に応じて変化することを理解するであろう。
哺乳動物に施される一又は複数の他の治療には、限定されるものではないが、化学治療及び/又は放射線治療、イムノアジュバント、サイトカイン、及び抗体ベース療法が含まれる。例としては、インターロイキン(例えば、IL-1、IL-2、IL-3、IL-6)、白血病抑制因子、インターフェロン、TGF-β、エリスロポイエチン、トロンボポイエチン、抗-VEGF抗体及びHER-2抗体が含まれる。哺乳動物細胞でアポトーシスを誘発することが知られた他の薬剤を用いてもよく、そのような薬剤にはTNF-α、TNF-β(リンホトキシン-α)、CD30リガンド、4-1BBリガンド、及びApo-1リガンドが含まれる。
【0067】
本発明において考慮される化学治療薬には、当該分野において既知であって市販されている化学物質又は薬物、例えばドキソルビシン、5-フルオロウラシル(「5-FU」)、エトポシド、カンプトテシン、ロイコボリン、シトシンアラビノシド(「Ara-C」)、シクロホスファミド、チオテパ、ブスルファン、サイトキシン、タキソール、メトトレキセート、シスプラチン、メルファラン、ビンブラスチン及びカルボプラチンが含まれる。このような化学治療薬に対する調製法及び用量スケジュールは、製造者の指示に従って使用されるか、熟練した実務家により経験的に決定される。そのような化学治療薬に対する調製法及び用量スケジュールはまた化学療法サービス(Chemotherapy Cervice), M.C.Perry編, Williams & Wilkins, Baltimore, MD (1992)にも記載されている。
化学治療薬は、Apo-2リガンドについて上述したもののような、製薬的に許容可能な担体で好ましく投与される。化学治療薬の投与方法はApo-2リガンドについて用いられるのと同様としてもよく、あるいは異なる方法を介して投与してもよい。例えば、Apo-2リガンドを注射する一方、化学治療薬を経口で哺乳動物に投与してもよい。Apo-2リガンドと組み合わせた化学治療薬の投与方法は、下記の実施例9−12に更に詳細に記載する。
【0068】
放射線治療は、この分野で通常用いられ当業者に知られたプロトコールに従って哺乳動物に施すことができる。このような治療は、セシウム、イリジウム、ヨウ素、又はコバルト照射を含む。放射線治療は、全身照射でもよく、又は身体の特定部位又は組織に局所的に向けられてもよい。典型的には、放射線治療は約1〜約2週間の期間に渡ってパルス的に施される。しかしながら、放射線治療は、より長期間にわたって施してもよい。場合によっては、放射線治療は1回照射又は多数、連続照射として施してもよい。
Apo-2リガンド及び一又は複数の他の治療は、哺乳動物に同時に又は連続的に施すことができる。Apo-2リガンド及び一又は複数の他の治療の施与に続いて、哺乳動物の癌及び生理学的状態を、熟練した実務者に知られた種々の方法で監視することができる。例えば、腫瘍の量は、生検により、又は標準的なX線画像技術により物理的に観察できる。
【0069】
Apo-2リガンドがエキソビボでの癌細胞の治療に用いられると考えられる。このような秋曽比簿治療は、骨髄移植、及び一部の自己骨髄移植において有用である。例えば、癌細胞を持つ細胞又は組織の、場合によっては上記したような一又は複数の他の治療と共に行うApo-2リガンドでの治療は、アポトーシスを誘発し、受容者哺乳動物に移植する前に実質的に癌細胞を除去するために用いることができる。
癌細胞を含む細胞又は組織は、供与者哺乳動物において最初に観察される。細胞又は組織は手術により、好ましくは無菌で得られる。移植用骨髄の治療方法では、骨髄は哺乳動物から針吸引により得る。次いで、癌細胞を含む細胞又は組織をApo-2リガンドで、場合によっては上記のような一又は複数の他の治療と共に治療する。骨髄は、好ましくは、Apo-2リガンドでの処理に先立って、単細胞分画を得るために(フィコール‐ハイパーク勾配の遠心分離などで)分画する。
処理した細胞又は組織は、次いで、受容者哺乳動物に注入又は移植する。受容者哺乳動物は、供与者哺乳動物と同じ個体でもよく、他の異種哺乳動物でもよい。自己骨髄移植については、哺乳動物は移植の前に有効用量の当該分野で知られ、例えば、Autologous Bone Marrow Transplantation: Proceedings of the Third International Symposium, Dicke等, 編, University of Texas M.D. Anderson Hospital and Tumor Institute (1987)に記載された放射線又は化学治療で処理される。
【0070】
C.Apo-2リガンドの非治療的用途
本発明のApo-2リガンドは非治療的用途においても有用性を有している。RApo-2リガンドをコードする核酸配列を、組織特異性の型分類のための診断に使用することもできる。例えば、インサイツハイブリッド形成、ノーザン及びサザンブロット法、及びPCR分析のような手順を、Apo-2リガンドをコードするDNA及び/又はRNAが評価されている細胞型に存在しているか否かを決定するために使用することができる。Apo-2リガンド核酸は、ここに記載されている組換え技術によるApo-2リガンドポリペプチドの調製にもまた有用である。
単離されたApo-2リガンドは、定量的診断アッセイで対照として用いられ、それに対して未知量のApo-2リガンドを含有するサンプルが調製される。またApo-2リガンド調製物は、抗体を産生する際、(例えば、ラジオイムノアッセイ、ラジオレセプターアッセイ又は酵素結合免疫測定法における標準としての使用のためにApo-2リガンドを標識することにより)Apo-2リガンドに対するアッセイにおける標準として、アフィニティー精製技術において、及び例えば放射性ヨウ素、酵素又はフルオロフォアで標識された場合の競合型レセプター結合アッセイにおいて有用である。
【0071】
また、Apo-2リガンドをコードする核酸は、トランスジェニック動物又は「ノックアウト」動物を産生するのに使用でき、これらは治療的に有用な試薬の開発やスクリーニングに有用である。トランスジェニック動物(例えばマウス又はラット)とは、出生前、例えば胚段階で、その動物又はその動物の祖先に導入された導入遺伝子を含む細胞を有する動物である。導入遺伝子とは、トランスジェニック動物が発生する細胞のゲノムに組み込まれたDNAである。一実施形態では、Apo-2リガンドをコードするcDNA又はその適当な配列を、確立された技術に従ってApo-2リガンドをコードするゲノムDNAをクローン化するために使用することができ、ゲノム配列は、Apo-2リガンドをコードするDNAを発現する細胞を有するトランスジェニック動物を産生するために使用することができる。トランスジェニック動物、特にマウス又はラット等の動物を産生する方法は当該分野において常套的になっており、例えば米国特許第4,736,866号や第4,870,009号に記述されている。典型的には、特定の細胞を組織特異的エンハンサーでのApo-2リガンド導入遺伝子の導入の標的にする。胚段階で動物の生殖系列に導入されたApo-2リガンドをコードする導入遺伝子のコピーを含むトランスジェニック動物はApo-2リガンドをコードするDNAの増大した発現の影響を調べるために使用できる。
【0072】
あるいは、Apo-2リガンドの非ヒト相同体は、動物の胚性細胞に導入されたApo-2リガンドをコードする変更ゲノムDNAと、Apo-2リガンドをコードする内在性遺伝子との間の相同的組換えによって、Apo-2リガンドをコードする欠陥又は変更遺伝子を有するApo-2リガンド「ノックアウト」動物を作成するために使用できる。例えば、Apo-2リガンドをコードするcDNAは、確立された技術に従い、Apo-2リガンドをコードするゲノムDNAのクローニングに使用できる。Apo-2リガンドをコードするゲノムDNAの一部を欠失したり、組み込みを監視するために使用する選択可能なマーカーをコードする遺伝子等の他の遺伝子で置換することができる。典型的には、ベクターは無変化のフランキングDNA(5'と3'末端の両方)を数キロベース含む[例えば、相同的組換えベクターについてはThomas及びCapecchi, Cell, 51:503(1987)を参照のこと]。ベクターは胚性幹細胞に(例えばエレクトロポレーション等によって)導入し、導入されたDNAが内在性DNAと相同的に組換えられた細胞を選択する[例えば、Li等, Cell, 69:915(1992)参照]。選択された細胞は次に動物(例えばマウス又はラット)の胚盤胞内に注入され、集合キメラを形成する[例えば、Bradley, Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, E. J. Robertson, ed. (IRL, Oxford, 1987), pp. 113-152参照]。その後、キメラ性胚を適切な偽妊娠の雌性乳母に移植し、期間をおいて「ノックアウト」動物をつくり出す。胚細胞に相同的に組換えられたDNAを有する子孫は標準的な技術により同定され、それらを利用して動物の全細胞が相同的に組換えられたDNAを含む動物を繁殖させることができる。ノックアウト動物は、例えば腫瘍の発達を含む、Apo-2リガンドポリペプチドが不在であることによるある種の病理的状態及びその病理的状態の発達に対する防御能力によって特徴付けられる。
【0073】
D.抗Apo-2リガンド抗体の調製
本発明は、さらに抗Apo-2リガンド抗体を提供するものである。Apo-2リガンドに対する抗体は以下のようにして調製することができる。抗体の例としては、ポリクローナル、モノクローナル、ヒト化、二重特異性及びヘテロ抱合体抗体が含まれる。
1.ポリクローナル抗体
Apo-2リガンド抗体はポリクローナル抗体を含んでよい。ポリクローナル抗体の調製方法は当業者に知られている。哺乳動物においてポリクローナル抗体は、例えば免疫化剤、及び所望するのであればアジュバントを、一又は複数回注射することで発生させることができる。典型的には、免疫化剤及び/又はアジュバントを複数回皮下又は腹腔内注射により、哺乳動物に注射する。免疫化剤は、Apo-2リガンドポリペプチド又はその融合タンパク質を含みうる。免疫化剤を、免疫化される哺乳動物で免疫原性が知られたタンパク質に抱合させるのが有用である。使用され得るそのような免疫原性タンパク質の例は、限定するものではないが、キーホール・リンペット(keyhole limpet))ヘモシアニン、血清アルブミン、ウシサイログロブリン及び大豆トリプシンインヒビターが含まれる。また、哺乳動物の免疫反応を増強するために、ミョウバンのような凝集剤を使用してもよい。使用され得るアジュバントの例には、フロイント完全アジュバント及びMPL-TDMアジュバント(モノホスホリル脂質A、合成トレハロースジコリノミコラート)が含まれる。免疫化プロトコールは、過度の実験なく当業者により選択されるであろう。哺乳動物から採血し、血清を検定して抗体価を求める。望まれるならば、抗体価が増加又は平坦化するまで哺乳動物に追加免疫を施す。
【0074】
2.モノクローナル抗体
あるいは、Apo-2リガンド抗体はモノクローナル抗体であってもよい。モノクローナル抗体は、Kohler及びMilstein, Nature, 256: 495 (1975)に記載されているようなハイブリドーマ法を使用することで調製することができる。ハイブリドーマ法では、マウス、ハムスター又は他の適切な宿主動物を典型的には免疫化剤により免疫化する(上述したようにして)ことで、免疫化剤に特異的に結合する抗体を生成するかあるいは生成可能なリンパ球を誘発する。また、リンパ球をインビトロで免疫化することもできる。
免疫化剤は、典型的にはApo-2リガンドポリペプチド又はその融合タンパク質を含む。表面にApo-2リガンドを発現する細胞もまた使用することができる。一般にヒト由来の細胞が望まれる場合には末梢血リンパ球(「PBL」)が使用され、あるいは非ヒト哺乳動物源が望まれている場合は、脾臓細胞又はリンパ節細胞が使用される。次いで、ポリエチレングリコール等の適当な融合剤を用いてリンパ球を不死化株化細胞と融合させ、ハイブリドーマ細胞を形成する[Goding, Monoclonal Antibodies: Principles and Practice, Academic Press, (1986) pp. 59-103]。不死化株化細胞は、通常は、形質転換した哺乳動物細胞、特に齧歯動物、ウシ、及びヒト由来の骨髄腫細胞である。通常、ラット及びマウスの骨髄腫細胞が使用される。ハイブリドーマ細胞は、好ましくは、未融合の不死化細胞の生存又は成長を阻害する一又は複数の物質を含有する適切な培地で培養される。例えば、親の細胞が、酵素のヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRT又はHPRT)を欠いていると、ハイブリドーマの培地は、典型的には、ヒポキサチン、アミノプチリン及びチミジンを含み(「HAT培地」)、この物質がHGPRT欠乏性細胞の増殖を阻止する。
【0075】
好ましい不死化株化細胞は、効率的に融合し、選択された抗体生成細胞による安定した高レベルの抗体発現を支援し、HAT培地のような培地に対して感受性であるものが望ましい。より好ましい不死化株化細胞はマウス骨髄腫株であり、これはカリフォルニア州サンディエゴのSalk Institute Cell Distribution Centerやバージニア州マナッサスのアメリカン・タイプ・カルチャー・コレクションより入手可能である。ヒトモノクローナル抗体を生成するためのヒト骨髄腫及びマウス-ヒト異種骨髄腫株化細胞も開示されている[Kozbor, J. Immunol., 133:3001 (1984)、Brodeur等, Monoclonal Antibody Production Techniques and Applications, Marcel Dekker, Inc., New York, (1987) pp. 51-63]。
ハイブリドーマ細胞が培養される培地を、Apo-2リガンドに対するモノクローナル抗体の存在について検定する。好ましくは、ハイブリドーマ細胞によって生成されたモノクローナル抗体の結合特異性は免疫沈降又はラジオイムノアッセイ(RIA)、蛍光活性化セルソーター(FACS)又は酵素結合免疫測定法(ELISA)等のインビトロ結合検定法によって測定する。このような技術及びアッセイは、当該分野において公知であり、下記の実施例でさらに記載される。モノクローナル抗体の結合親和性は、例えばMunson及びPollard, Anal. Biochem., 107:220 (1980)によるスキャッチャード分析法によって測定することができる。
【0076】
所望のハイブリドーマ細胞が同定された後、クローンを制限希釈工程を経てサブクローニングし、標準的な方法で増殖させることができる[Goding, 上掲]。この目的のための適当な培地には、例えば、ダルベッコの改変イーグル培地及びRPMI-1640倍地が含まれる。あるいは、ハイブリドーマ細胞は哺乳動物においてインビボで腹水として成長させることもできる。
サブクローンによって分泌されるモノクローナル抗体は、例えばプロテインAセファロース法、ヒドロキシルアパタイトクロマトグラフィー法、ゲル電気泳動法、透析法又はアフィニティークロマトグラフィー等の従来の免疫グロブリン精製方法によって培地又は腹水液から分離又は精製される。
本発明の一実施態様では、モノクローナル抗体は、ここ及び下記の実施例に記載する1D1、2G6、2E11、又は5C2抗体を含んでよい。また、モノクローナル抗体は、各々アメリカン・タイプ・カルチャー・コレクションの登録番号ATCC HB-12256, HB-12257, HB-12258, 又はHB-12259で寄託されたハイブリドーマ細胞系によって分泌された1D1、2G6、2E11、又は5C2モノクローナル抗体と同じ生物学的特性を有する抗体も含む。「生物学的活性」という用語は、モノクローナル抗体のインビトロ及び/又はインビボ活性、例えば、Apo-2リガンド誘発アポトーシスを実質的に低減又は阻害する、又はApo-2リガンドのそのレセプターへの結合を低減又は阻止する能力を意味するのに用いられる。抗体は、ここに記載した1D1、2G6、2E11、又は5C2抗体と同じエピトープ、又は実質的に同じエピトープに結合するのが好ましい。
【0077】
また、モノクローナル抗体は、組換えDNA法、例えば米国特許第4,816,567号に記載された方法により作成することができる。本発明のモノクローナル抗体をコードするDNAは、常套的な方法によって(例えば、マウス抗体の重鎖及び軽鎖をコードする遺伝子に特異的に結合可能なオリゴヌクレオチドプローブを使用して)、容易に単離し配列決定することができる。本発明のハイブリドーマ細胞はそのようなDNAの好ましい供給源となる。ひとたび単離されたら、DNAは発現ベクター内に配することができ、これが宿主細胞、例えばサルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、あるいは免疫グロブリンタンパク質を生成しない骨髄腫細胞内に形質移入され、組換え宿主細胞内でモノクローナル抗体の合成をすることができる。また、DNAは、例えば相同マウス配列に換えてヒト重鎖及び軽鎖定常ドメインのコード配列を置換することにより[米国特許第4,816,567号;Morrison等, 上掲]、又は免疫グロブリンコード配列に非免疫グロブリンポリペプチドのコード配列の一部又は全部を共有結合することにより修飾することができる。このような非免疫グロブリンポリペプチドは、本発明の抗体の定常ドメインの代わりに置換するか、本発明の抗体の一つの抗原結合部位の可変ドメインの代わりに置換し、キメラ性二価抗体を産生することができる。
【0078】
本発明の抗体は一価抗体であってもよい。一価抗体の調製方法は当該分野においてよく知られてる。例えば、一つの方法は免疫グロブリン軽鎖と修飾重鎖の組換え発現を含む。重鎖は一般的に、重鎖の架橋を防止するようにFc領域の任意のポイントで切断される。あるいは、関連したシステイン残基を他のアミノ酸残基で置換するか欠失させて架橋を防止する。
一価抗体の調製にはインビトロ法がまた適している。抗体の消化による、その断片、特にFab断片の調製は、当該分野において知られている慣用的技術を使用して達成できる。例えば、消化はパパインの使用により行うことができる。パパイン消化の例は、12/22/94に公開された国際公開第WO 94/29348号、及び米国特許第4,342,566号に記載されている。抗体のパパイン消化は、典型的には、Fab断片と呼ばれ各々が単一の抗原結合部位を有する2つの同一の抗原結合断片と、残りのFc断片を生成する。ペプシン処理により、2つの抗原結合部位を有し抗原の架橋が尚も可能なF(ab')2断片が得られる。
また、抗体の消化により生産されたFab断片は、軽鎖の定常ドメインと重鎖の第1定常ドメイン(CH1)を含む。Fab’断片は、抗体のヒンジ領域から一又は複数のシステインを含む重鎖CH1ドメインのカルボキシ末端に幾つかの残基が付加されているということで、Fab断片とは異なっている。Fab’-SHとは、定常ドメインのシステイン残基が遊離のチオール基を担持しているFab’に対するここでの命名である。F(ab’)2抗体断片は、本来は、それらの間にヒンジシステインを有するFab’断片の対として生産された。抗体断片の他の化学的結合もまた知られている。
【0079】
3.ヒト化抗体
本発明のApo-2リガンド抗体は、さらにヒト化抗体又はヒト抗体を含む。非ヒト(例えばマウス)抗体のヒト化形とは、キメラ免疫グロブリン、免疫グロブリン鎖あるいはその断片(例えばFv、Fab、Fab'、F(ab')2あるいは抗体の他の抗原結合サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。ヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト種(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)を含む。ある場合には、ヒト免疫グロブリンのFvフレームワーク残基は、対応する非ヒト残基によって置換されている。更に、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでいてもよい。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリンコンセンサス配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域(Fc)、典型的にはヒトの免疫グロブリンの定常領域の少なくとも一部を含んでなる[Jones等, Nature, 321:522-525 (1986); Riechmann 等, Nature, 332:323-329 (1988); 及びPresta, Curr. Op Struct. Biol., 2:593-596 (1992)]。
【0080】
非ヒト抗体をヒト化する方法はこの分野で良く知られている。一般的に、ヒト化抗体には非ヒト由来の一又は複数のアミノ酸残基が導入されている。これら非ヒトアミノ酸残基は、しばしば、典型的には「移入」可変ドメインから得られる「移入」残基と称される。ヒト化は基本的にウィンター及び共同研究者[Jones等, Nature, 321:522-525 (1986)、Riechmann等, Nature, 332:323-327 (1988)、Verhoeyen等, Science, 239:1534-1536 (1988)]の方法、及びコンピュータモデルを用いたQueen等, Proc Natl. Acad. Sci., 86: 10029-10033 (1989)の方法に従って、齧歯動物のCDR又はCDR配列でヒト抗体の対応する配列を置換することにより実施される。よって、このような「ヒト化」抗体は、無傷のヒト可変ドメインより実質的に少ない分が非ヒト種由来の対応する配列で置換されたキメラ抗体である(米国特許第4,816,567号)。実際には、ヒト化抗体は典型的には幾つかのCDR残基及び場合によっては幾つかのFR残基が齧歯類抗体の類似部位からの残基によって置換されたヒト抗体である。
抗原性を軽減するには、ヒト化抗体を作成するために使用するヒトの軽鎖及び重鎖可変ドメインの両方の選択が非常に重要である。「ベストフィット法」に従うと、齧歯動物抗体の可変ドメインの配列を既知のヒト可変ドメイン配列ライブラリ全体に対してスクリーニングする。齧歯動物のものと最も近いヒトの配列を次にヒト化抗体のヒトフレームワーク(FR)として受け入れる[Sims等, J. Immunol., 151:2296 (1993);Chothia及びLesk, J. Mol. Biol., 196:901 (1987)]。他の方法では、軽鎖又は重鎖の特定のサブグループのヒト抗体全てのコンセンサス配列から誘導される特定のフレームワークを使用する。同じフレームワークを幾つかの異なるヒト化抗体に使用できる[Carter等, Proc. Natl. Acad. Sci. USA, 89:4285 (1992);Presta等, J. Immunol., 151:2623 (1993)]。
【0081】
さらに、抗体は、抗原に対する高親和性や他の好ましい生物学的性質を保持してヒト化することが重要である。この目標を達成するべく、好ましい方法では、親及びヒト化配列の三次元モデルを使用して、親配列及び様々な概念的ヒト化産物の分析工程を経てヒト化抗体を調製する。三次元免疫グロブリンモデルは一般的に入手可能であり、当業者にはよく知られている。選択された候補免疫グロブリン配列の推測三次元立体配座構造を図解し、表示するコンピュータプログラムは入手可能である。これら表示を見ることで、候補免疫グロブリン配列の機能における残基の役割の分析、すなわち候補免疫グログリンの抗原と結合する能力に影響を及ぼす残基の分析が可能となる。このようにして、例えば標的抗原に対する親和性を高めるといった、望ましい抗体特徴が得られるように、FR残基をコンセンサス及び移入配列から選択し、組み合わせることができる。一般的に、CDR残基は、直接かつ最も実質的に抗原結合性に影響を及ぼしている[1994年、3月3日に公開されたWO 94/04679を参照]。
免疫化することで、内在性免疫グロブリンが生成されない状態でもヒト抗体の完全リパートリを生成することができるトランスジェニック動物(例えばマウス)を使用することが可能である。例えば、キメラ及び生殖系列変異マウスにおいて抗体重鎖結合領域(JH)遺伝子をホモ接合的に欠失させると内在性抗体生産の完全な阻害が生じることが記述されている。このような生殖系列変異マウスにヒト生殖系列免疫グロブリン遺伝子配列を移すと、抗原投与時にヒト抗体の生成が生じる[例えば、Jakobovits等, Proc. Natl. Acad. Sci. USA, 90:2551-255 (1993);Jakobovits等, Nature, 362:255-258 (1993);Bruggerman等, Year in Immuno., 7:33 (1993)を参照]。また、ヒト抗体はファージ表示ライブラリ[Hoogenboom及びWinter, J. Mol. Biol., 227:381 (1992);Marks等, J. Mol. Biol., 222:581 (1991)]において作成することもできる。また、Cole等及びBoerner等の方法も、ヒトモノクローナル抗体の調製に利用することができる[Cole等, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss. p.77(1985)及びBoerner等, J. Immunol., 147(1):86-95(1991) ]。
【0082】
4.二重特異性抗体
二重特異性抗体は、少なくとも2つの異なる抗原に対して結合特異性を有するモノクローナル抗体、好ましくはヒトもしくはヒト化抗体である。本発明の場合において、結合特異性の一方はApo-2リガンドに対してであり、他方は任意の他の抗原、好ましくは細胞表面タンパク質又はレセプター又はレセプターサブユニットに対してである。
二重特異性抗体を生成する方法は当該技術分野において周知である。伝統的には、二重特異性抗体の組換え生成方法は、二つの重鎖が異なる特異性を持つ二つの免疫グロブリン重鎖/軽鎖対の同時発現に基づく[Milstein及びCuello, Nature, 305:537-539 (1983)]。免疫グロブリンの重鎖と軽鎖を無作為に取り揃えたため、これらハイブリドーマ(クアドローマ)は10種の異なる抗体分子の潜在的混合物を作成でき、その内一種のみが正しい二重特異性構造を有する。正しい分子の精製は、アフィニティークロマトグラフィー工程によって通常達成される。同様の手順が1993年5月13日公開の国際特許出願第 WO 93/08829号、及びTraunecke等, EMBO J.,10:3655-3656 (1991)に開示されている。
【0083】
別のより好ましいアプローチによれば、所望の結合特異性(抗体-抗原結合部位)を有する抗体可変ドメインを免疫グロブリン定常ドメイン配列に融合する。融合は、好ましくは少なくともヒンジ部、CH2及びCH3領域の一部を含む免疫グロブリン重鎖定常ドメインとのものである。少なくとも一つの融合には軽鎖結合に必要な部位を含む第一の重鎖定常領域(CH1)が存在することが望ましい。免疫グロブリン重鎖融合をコードするDNA、及び望むのであれば免疫グロブリン軽鎖を、別々の発現ベクターに挿入し、適当な宿主生物に同時形質移入する。これは、作成に使用する三つのポリペプチド鎖が不等の比であるときに最高の収率が得られる実施態様において、三つのポリペプチド断片の相互比率の調節に大なるフレキシビリティをもたらす。しかし、少なくとも二つのポリペプチド鎖が同比率で発現すると高収率が得られる場合や比率が特に重要ではない場合には、一つの発現ベクターに二つ又は三つ全てのポリペプチド鎖のコード配列を挿入することができる。このアプローチの好適な形態では、二重特異性抗体は、一方のアームの第一結合特異性を有するハイブリッド免疫グロブリン重鎖と、他方のアームのハイブリッド免疫グロブリン重鎖/軽鎖対(第二の結合特異性をもたらす)からなる。このような非対称的構造は、二重特異性分子の半分にのみ免疫グロブリン軽鎖が存在すると容易な方法で分解できるので、所望の二重特異性化合物を不要な免疫グロブリン鎖の組合わせから分解し易くすることが見出された。このアプローチは1994 3月3日公開の国際公開WO 94/04690号によって開示されている。二重特異性抗体を生成するための更なる詳細については、例えばSuresh等, Methods in Enzymology, 121:210(1986)を参照されたい。
【0084】
5.ヘテロ抱合体抗体
ヘテロ抱合抗体もまた本発明の範囲に入る。ヘテロ抱合抗体は、2つの共有的に結合した抗体からなる。このような抗体は、例えば、免疫系細胞を不要な細胞に対して標的化させるため[米国特許第4,676,980号]及びHIV感染の治療のために[WO 91/00360; WO 92/200373; EP 03089]提案された。本抗体は、架橋剤に関連したものを含む合成タンパク質化学における既知の方法を使用してインビトロで調製することができると考えられる。例えば、ジスルフィド交換反応を使用するか又はチオエーテル結合を形成することにより免疫毒素を作成することができる。この目的に対して好適な試薬の例には、イミノチオレート及びメチル-4-メルカプトブチリミデート、及び例えば米国特許第4,676,980号に開示されているものが含まれる。
【0085】
E.Apo-2リガンド抗体の用途
さらに、Apo-2リガンド抗体は、Apo-2リガンドの診断アッセイ法、例えば特異的細胞、組織又は血清におけるその発現の検出に使用することができる。当該分野において知られている様々な診断アッセイ技術、例えば、競合的結合アッセイ、直接的又は間接的サンドイッチアッセイ及び不均一又は均一相の何れにおいても実施される免疫沈降検定を使用することができる[Zola, Monoclonal Antibodies: A Manual of Techniques,CRC Press,Inc.,(1987) pp.147-158]。診断アッセイ法に使用される抗体は、検出可能部分で標識することができる。検出可能成分は、直接的に又は間接的に検出可能なシグナルをつくりだすことができなければならない。例えば検出可能部分は、3H、14C、32P、35S又は125I等の放射性同位体、蛍光イソチオシアネート、ローダミン又はルシフェリン等の蛍光又は化学発光化合物、もしくはアルカリホスファターゼ、βガラクトシダーゼ又は西洋わさびペルオキシダーゼ等の酵素であってもよい。Hunter等, Nature, 144: 945 (1962)、David等, Biochemistry,13:1014(1974)、Pain等, J. Immunol. Meth.,40:219 (1981)及びNygren等, J. Histochem. and Cytochem.,30:407 (1982)などに記載されている方法を含み、検出可能部分に抗体を抱合させるための当該分野において知られている任意の方法を使用することができる。
【0086】
また、Apo-2リガンド抗体は天然供給源又は組換え細胞培養からのApo-2リガンドのアフィニティー精製にも有用である。この方法においては、Apo-2リガンドに対する抗体を、当該分野でよく知られている方法を使用して、セファデックス樹脂や濾紙のような適当な支持体に固定する。次に、固定された抗体を、精製するApo-2リガンドを含有する試料と接触させ、次いで、固定された抗体に結合したApo-2リガンド以外の試料中の物質を実質的に全て除去する適当な溶媒で支持体を洗浄する。最後に、Apo-2リガンドを抗体から離脱させる他の適当な溶媒で支持体を洗浄する。またApo-2リガンド抗体は、可溶化Apo-2レセプターのアフィニティ精製又はApo-2レセプターの発現クローニングにも有用である。
ここに記載した抗体は、治療的にも使用される。例えば、Apo-2リガンド活性(Apo-2リガンド誘発アポトーシス等)を阻止する抗-Apo-2リガンド抗体は、増大したアポトーシスに伴う病理学的状態又は疾患の治療に用いられる[Thompson, 上掲参照]。
【0087】
F.Apo-2リガンド又はApo-2リガンド抗体を含むキット
本発明のさらなる実施態様においては、例えば上述の治療的又は非治療的用途に使用可能なApo-2リガンド又はApo-2リガンド抗体を含むキット及び製造品が提供される。製造品にはラベルが付された容器が含まれる。適切な容器には、例えばボトル、バイアル、及び試験管が含まれる。容器はガラス又はプラスチックのような種々の物質から形成できる。容器は、上述のような治療的又は非治療的用途に有効な活性剤を含む組成物を収容する。組成物中の活性剤は、Apo-2リガンド又はApo-2リガンド抗体である。容器のラベルには、組成物が特定の治療的又は非治療的用途に使用されることが示され、また上述のもののような、インビボ又はインビトロのいずれかの使用の指示が示されている。
【0088】
本発明のキットは、典型的には、上述の容器と、商業上及び使用者の観点から望まれる、バッファー、希釈液、フィルター、針、注入器及び使用説明が記されたパッケージ挿入物を含む、材料を含む一又は複数の他の容器を含んでいる。
以下の実施例は例示するためにのみ提供されるものであって、本発明の範囲を決して限定することを意図するものではない。
本明細書で引用した全ての特許及び参考文献の全体を、出典明示によりここに取り込む。
【0089】
(実施例)
実施例において言及されている全ての制限酵素は、ニューイングランドバイオラボ社(New England Biolabs)から購入し、製造者の使用説明に従い使用した。実施例で言及されている全ての他の市販試薬は、特に示していない限りは、製造者の使用説明に従い使用した。ATCC登録番号により次の実施例及び明細書全体を通して特定している細胞の供給源はアメリカン・タイプ・カルチャー・コレクション(Mannasas,Virginia)である。
【0090】
(実施例1)
ヒトApo-2リガンドをコードするcDNAクローンの単離
Apo-2リガンドの全長cDNAを単離するために、ヒト胎盤cDNAのラムダgt11バクテリオファージライブラリ(約1x106クローン)(HL10756、Clontechから市販)を、ヒトFas/Apo-1リガンドに幾分の相同性を示すEST配列(GenBank locus HHEA47M)に基づく合成オリゴヌクレオチドプローブでハイブリッド形成することによりスクリーニングした。HHEA47MのEST配列は390塩基対で、その3+フレームで翻訳した場合、ヒトApo-1リガンドの34アミノ酸領域に16の同一性を示す。HHEA47Mの配列は次の通りである:
GGGACCCCAATGACGAAGAGAGTATGAACAGCCCCTGCTGGCAAGTCAAGTGGCAACTCCGTCAGCTCGTTAGAAAGATGATTTTAGATTCCTCTGAGGAAACCATTTCTACAGTTCAAGAAAAGCAACAAAATATTTCTCCCCTAGTGAGAGAAAGAGGTCCTCAGAGAGTAGCAGCTCACATAACTGGGACCAGAGGAAGAAGCAACACATTGTCTTCTCCAAACTCCAAGAATGAAAAGGCTCTGGGCCGCAAAATAAACTCCTGGGAATCATCAAGGAGTGGGCATTCATTCCTGAGCAACTTGCACTTGAGGAATGGTGAACTGGTCATCCATGAAAAAGGGTTTTACTACATCTATTCCCAAACATACTTTCGATTTCAGGAGG 配列番号:3。
以下の配列を持つ60塩基対のオリゴヌクレオチドをスクリーニングに用いた:
TGACGAAGAGAGTATGAACAGCCCCTGCTGGCAAGTCAAGTGGCAACTCCGTCAGCTCGT 配列番号:4。
ハイブリッド形成は、20%のホルムアルデヒド、5X SSC、10%のデキストラン硫酸、0.1%のNaPiPO4、0.05MのNaPO4、0.05mgのサケ精子DNA、及び0.1%のドデシル硫酸ナトリウムを含むバッファー中、室温で終夜行い、次いで42℃において5X SSC、続いて2X SSC中で数回洗浄した。cDNAライブラリ中で12の陽性クローンを同定し、陽性クローンを以下の配列を有する60塩基対の第2のオリゴヌクレオチドプローブ(第1のプローブとの重複は無い)にハイブリッド形成することによって再スクリーニングした。
GGTGAACTGGTCATCCATGAAAAAGGGTTTTACTACATCTATTCCCAAACATACTTTCGA 配列番号:5。
ハイブリッド形成は、上記のように行った。
【0091】
得られた4つの陽性クローンを、フランキング5’ベクター配列に基づき外部ClaI制限部位を付加したプライマー及び3’フランキングベクター配列に基づき外部HindIII制限部位を付加したプライマーを用いたポリメラーゼ連鎖反応(PCR)により同定した。PCR産物はゲル精製し、T-AライゲーションによりpGEM-T(Promegaから市販)にサブクローニングした。異なるPCRからの3つの独立したクローンに、次いでジデオキシDNA配列を施した。これらのクローンのDNA配列分析は、それらが5’領域に幾分の長さ変化を持つが実質的に同一であることを示した。
Apo-2リガンドのコード化領域の核酸配列を図1Aに示した。クローンの1つの下流側3’末端領域の配列分析により、特徴的なポリアデニル化部位が明らかにされた(データは示さず)。cDNAは1つのオープンリーディングフレームを含み、それはヌクレオチド位置91−93のATGコドンに割り当てられた開始部位を持つ。この部位を取り囲む配列は、開始部位について提案されたコンセンサス配列[Kozak, J. Cell Biol., 115: 887-903 (1991)]と合理的に一致した。オープンリーディングフレームは、ヌクレオチド位置934−936の停止コドンTAAで終端する。
【0092】
ヒトApo-2リガンドの推定成熟アミノ酸配列は、281アミノ酸を含み、約32.5kDaの計算された分子量及び約7.63の等電点を有する。N末端には明らかなシグナル配列は存在しないが、ヒドロパシー分析(データは示さず)は残基15と40との間に疎水性領域の存在を示している。シグナル配列が無いこと及び内部疎水性領域が存在することは、Apo-2リガンドがII型膜貫通タンパク質であることを示唆している。推定細胞内、膜貫通及び細胞外領域は、各々14、26及び241アミノ酸長である。推定膜貫通領域は、図1Aで下線を付した。潜在的なN結合グリコシル化部位は、推定細胞外領域の残基109に位置している。
Apo-2リガンドのC末端領域と、TNFサイトカインファミリーの知られたメンバーとのアミノ酸配列のアラインメントは、C末端領域内で、Apo-2リガンドがApo-1リガンドと23.2%の同一性を有することを示した(図1B)。アラインメント分析は、他のTNFファミリーメンバーとのより低い程度の同一性を示した:CD40L(20.8%)、LT-α1(20.2%)LT-β(19.6%)、TNF-α(19.0%)、CD30L及びCD27L(15.5%)、OX-40L(14.3%)、及び4-1BBL(13.7%)。TNFサイトカインファミリーの中で、TNF-α及びLT-αの構造[Eck等, J. Bio. Chem., 264: 17595-17605 (1989); Eck等, J. Bio. Chem., 267: 2119-2122 (1992)]に基づいてβ鎖を形成すると予想される領域内の残基は、予想される連結ループ内の残基よりも高度に他のTNFファミリーメンバーに保持される傾向がある。Apo-2リガンドが、予想される連結ループ内の相同性に比較して、その推定β鎖領域において他のTNFファミリーメンバーと大きな相同性を示すことが見いだされた。また、ループ連結推定β鎖、B及びB’は、Apo-2リガンドにおいて顕著に長い。
【0093】
(実施例2)
ヒトApo-2リガンドの発現 A.全長cDNA融合作成物の293細胞における発現
mycエピトープタグに融合した全長Apo-2リガンドcDNAを次のように作成した。Apo-2リガンドcDNA挿入物を親pGEM-TApo-2リガンドプラスミド(実施例1に記載)からClaI及びHindIIIによる消化により除去し、同じ制限酵素で消化されたpRK5哺乳動物発現プラスミド[Schall等, Cell, 61:361-370 (1990);Suva等, Science, 237:893-896 (1987)]に挿入した。13アミノ酸のmycエピトープタグSer Met Glu Gln Lys Leu Ile Ser Glu Glu Asp Leu Asn 配列番号:6[Evan等, Mol. Cell. Biol., 5: 3610-3616 (1985)]をコードする配列を、Apo-2リガンドコード化配列の3’末端のコドン281と停止コドン(コドン282)との間にオリゴヌクレオチド指向性突然変異誘発[Zoller等, Nucleic Acids Res., 10: 6487-6469 (1982)]により挿入し、プラスミドpRK5Apo-2リガンド-mycを与えた。
pRK5Apo-2リガンド-mycプラスミドを、リン酸カルシウム沈殿法によりヒト293細胞(ATCC CRL 1573)にネオマイシン耐性遺伝子を持つpRK5プラスミドと同時形質移入した。抗生物質G418(0.5mg/mL)(GIBCO)の存在下での50%HAM’SF12/50%DMEM(GIBCO)中での成長能力によりApo-2リガンド-mycを発現する安定なクローンを選択した。
Apo-2リガンドのトポロジーを試験するために、G418耐性クローンを、抗mycモノクローナル抗体(mAb)クローン9E10[Evans, 上掲:Oncogene Scienceから市販]、次いでフィコエリスリン(PE)抱合ヤギ抗マウス抗体(Jackson ImmunoReserchから市販)で染色した後にFACS分析した。FACS分析により、mock形質移入細胞に比較してApo-2リガンド-myc形質移入細胞では特定の正の染色シフトが明らかとなり、Apo-2リガンドが細胞表面で発現され(図1C)、そのカルボキシル末端が露出されることが示された。従って、Apo-2リガンドはII型膜貫通タンパク質であると考えられる。
【0094】
B.ECD融合作成物の293細胞及びバキュロウイルスでの発現
Apo-2リガンドのC末端領域の上流側に他の配列を融合させた2つの可溶性Apo-2リガンド細胞外ドメイン(「ECD」)融合作成物を調製した。
一方の作成物では、ヘルペスウイルス糖タンパク質D(「gD」)シグナルペプチド[Lasky等, DNA, 3: 23-29 (1984); Pennica等, Proc. Natl. Acad. Sci., 92: 1142-1146 (1995); Paborsky等, Protein Engineering, 3: 547-553 (1990)に記載]の27アミノ酸及びエピトープタグ配列
Lys Tyr Ala Leu Ala Asp Ala Ser Leu Lys Met Ala Asp Pro Asn Arg Phe Arg Gly Lys Asp Leu Pro Val Leu Asp Gln 配列番号:7をpRK5哺乳動物発現プラスミド内でApo-2リガンドのコドン114−281の上流側に融合させた。簡単に言えば、gD配列は親プラスミドpCHAD(Genentech, Lasty等, Science, 233: 209-212 (1986)に記載されたように調製)から、3’プライマーがgDの3’領域並びにApo-2リガンドのコドン114−121に相補的なPCRで増幅した。生成物は、pRK5Apo-2リガンドプラスミドがテンプレートとして用いられる引き続くPCRにおいて、Apo-2リガンドコード化領域の3’末端に相補的な3’プライマーとともに5’プライマーとして使用した。生成物は、gD-Apo-2リガンドECD融合物をコードするが、次いでpRK5プラスミドにサブクローニングしてプラスミドpRK5gD-Apo-2リガンドECDを与えた。
【0095】
ヒト胚腎臓293細胞(ATCC CRL 1573)をリン酸カルシウム沈殿法により、pRK5gD-Apo-2リガンドECDプラスミド又はpRK5で一過性形質移入した。可溶性gD-Apo-2リガンドタンパク質の発現を、形質移入細胞の35S-Cys及び35S-Metで代謝的に標識することにより試験した。細胞上清を24時間後に回収し、遠心分離で透明化した。免疫沈降法のために、5mlの上清を5B6抗-gDモノクローナル抗体(Genentech)とともに1μg/mlで終夜4℃でインキュベートした。次いで、25μlのパンソルビン(Sigma)を添加して更に4℃で1時間おいた。管を回旋させ、ペレットをPBS中で洗浄し、SDS試料バッファー中で5分間煮沸した。煮沸試料を再度回旋させ、上清にSDS-PAGE及びオートラジオグラフィを施した。
抗gD抗体での免疫沈降は、gD-Apo-2リガンドプラスミドで形質移入した細胞の上清中に3つの優勢なタンパク質バンドを明示した(図1E)。これらのバンドは、23,48及び74kDaの相対分子量(Mr)で泳動した。成熟gD-Apo-2ポリペプチドの計算分子量は約22.5kDaである;よって、観察されたバンドは融合タンパク質の単量体(23kDa)、二量体(48kDa)及び三量体(74kDa)形であり、Apo-2リガンドが哺乳動物細胞において分泌された可溶性gD融合タンパク質として発現されうることを示した。
【0096】
第2の作成物では、MetGlyHis10配列(プラスミドpET19Bから誘導、Novagen)、次いで12アミノ酸の塩テロキナーゼ切断部位
Met Gly His His His His His His His His His His Ser Ser Gly His Ile Asp Asp Asp Asp Lys His Met 配列番号:8をバキュロウイルス発現プラスミド(pVL1392、Pharmingen)内のApo-2リガンドのコドン114−281の上流側に融合させた。簡単に言えば、Apo-2リガンドコドン114−281領域を、pRK5Apo-2リガンドプラスミド(実施例1に記載)から、各々フランキングNdeI及びBamHI制限部位を導入した5’及び3’領域に相補的なプライマーでのPCRにより増幅した。生成物は、T−AライゲーションによりpGEM-T(Promega)にサブクローニングし、DNA配列を確認した。次いで、挿入物をNdeI及びBamHIでの消化により除去し、アミノ末端MetGlyHis10タグ及びエンテロキナーゼ切断部位を含む修飾バキュロウイルス発現ベクターpVL1392(Pharmingenから市販)にサブクローニングした。
組換えバキュロウイルスは、His10-Apo-2ECDプラスミド及びBaculoGold(商品名)ウイルスDNA(Pharmingen)を、Spodopterafrugiperda(「Sf9」)細胞(ATCC SRL 1711)にリポフェクチン(GIBCO-BRLから市販)を用いて同時形質移入することにより生成した。28℃で4−5日のインキュベーションの後、放出されたウイルスを回収し、さらなる増幅に用いた。ウイルス感染及びタンパク質発現は、O'Relley等, Baculovirus expression vectors: A laboratory Manual, Oxford: Oxford University Press (1994)に記載されているように実施した。タンパク質は、下記実施例3に記載するようにNi2+キレートアフィニティクロマトグラフィで精製した。
【0097】
(実施例3)
組換えヒトApo-2リガンドの精製
抽出物を、組換えウイルス感染及びmock感染Sf9細胞(実施例2、項目B参照)から、Rupert等, Nature, 362: 175-179 (1993)に記載されているように調製した。簡単に言えば、Sf9細胞を洗浄し、超音波処理バッファー(25mLのHepes、pH7.9;12.5mMのMgCl2;0.1mMのEDTA;10%のグリセロール;0.1%のNP-40;0.4MのKCl)中に再懸濁し、氷上で2回20秒間超音波処理した。超音波処理物は、遠心分離で透明化し、上清を負荷バッファー(50mMリン酸塩、300mMのNaCl、10%のグリセロール、pH7.8)中に50倍に希釈し、0.45μmフィルターを通して濾過した。Ni2+-NTAアガロースカラム(Qiagenから市販)を5mLのベッド容量で調製し、25mLの水で洗浄して25mLの負荷バッファーで平衡させた。濾過した細胞抽出物を1分当たり0.5mLでカラムに負荷した。分画回収を開始する点であるベースラインA280までカラムを負荷バッファーで洗浄した。次に、カラムを、非特異的結合したタンパク質を溶離する二次洗浄バッファー(50mMリン酸塩、300mMのNaCl、10%のグリセロール、pH6.0)で洗浄した。再度ベースラインA280に達した後、カラムを二次洗浄バッファー中の0から500mMイミダゾール勾配で展開した。1mL分画を回収し、SDS−PAGE及び銀染色又はアルカリホスファターゼ(Qiagen)に抱合したNi2+−NTAでのウェスタンブロットにより分析した。溶離したHis10−Apo-2タンパク質を含有する分画をプールし、負荷バッファーに対して透析した。
出発材料としてのmock感染Sf9細胞に同じ手法を繰り返し、同様の分画をプールし、透析し、精製したヒトApo-2の対照として使用した。
精製タンパク質のSDS−PAGE分析は、His10-Apo-2リガンド単量体について計算された分子量22.4kDaに相当するMr24kDaの優勢なバンドを明示し(図1D、レーン3);タンパク質配列マイクロ分析(データは示さず)は24kDaのバンドがHis10-Apo-2リガンドポリペプチドに相当することを確認した。少量の48kDa及び66kDaバンドも観察され、可溶性Apo-2リガンドの同種二量体及び同種三量体の存在を示した。スルホ-NHS(5mM)(Pierce Chemical)及びECD(Pierce Chemical)25mM及び50mMとのインキュベーションによる精製His10-Apo-2リガンドの化学的架橋(各々、図1D、レーン1及び2)は、タンパク質を主に66kDaのバンドにシフトさせた。これらの結果は、溶液内でのApo-2の優勢な形態は同種三量体であり、これらの三量隊がSDSの存在下で二量体又は単量体に分解することを示している。
【0098】
(実施例4)
Apo-2リガンドのヒトリンパ細胞系に対するアポトーシス活性
精製した可溶性Apo-2リガンド(実施例3に記載)のアポトーシス活性を、幾つかのヒトリンパ細胞系を用いて試験した。第1の実験では、Apo-2リガンドの、エプスタインバールウイルス(EBV)形質転換ヒト末梢血B細胞から誘導した9D細胞(Genentech, Inc.)への影響を試験した。9D細胞(5x104細胞/ウェル、PRMI1640培地プラス10%ウシ胎児血清)を、対照培地、Apo-2(3μg/ml、上記実施例3のように調製した)、又は抗Apo-1モノクローナル抗体、CH11(1μg/ml)[Yonehara等, J. Exp. Med., 169: 1747-1756 (1989)に記載;Medical and Biological Laboratories Co.から市販]のいずれかとともに24時間インキュベートした。CH11抗Apo-1抗体はFas/Apo-1リガンド活性の類似したアゴニスト抗体である。
インキュベーションの後、細胞をサイトスピンスライドガラス上に回収し、逆光顕微鏡下で写真撮影した。Apo-2リガンド及び抗Apo-1モノクローナル抗体の両方が類似のアポトーシス効果を誘発し、細胞質凝集及び細胞数の減少を特徴としていた(図2A参照)。
Apo-2リガンドの、9D細胞、並びにRaji細胞(ヒトバーキットリンパB細胞系、ATCC CCL 86)及びジャーカット細胞(ヒト急性T細胞白血病細胞系、ATCC TIB 152)に対する効果をFACSにより更に分析した。FACS分析は、アポトーシス性細胞死について確立された基準を用いて、即ち2つのマーカー(a)アポトーシス細胞は染色するが生存細胞はしないヨウ化プロピジウム([PI」)染料及び(b)アポトーシス細胞には見られるが生存細胞には見られない露出されたホスファチジルセリンに結合するタンパク質アネキシンVの蛍光誘導体での細胞の蛍光染色関係を用いて実施した[Darzykiewicz等, Methods in Cell Biol., 41: 15-38 (!994); Fadok等, J. Immunol., 14: 2207-2214 (1992); Koopman等, Blood, 84: 1415-1420 (1994)]。
9D細胞(図2B)、Raji細胞(図2C)及びジャーカット細胞(図2D)を、対照培地(左側パネル)、Apo-2リガンド(3μg/ml、実施例3に記載したように調製)(真中パネル)、又は抗Apo-1抗体(1μg/ml)(右側パネル)とともに24時間インキュベート(1x106細胞/ウェル)した。次いで細胞を洗浄し、PI及びフルオレセインチオシアネート(FITC)-抱合アネキシンV(Brand Applicationsから購入)で染色し、フローサイトメトリーで分析した。PI及びアネキシンV染色の両方に陰性の細胞(第3象限)が生存細胞を表し;PI陰性、アネキシンV陽性染色細胞(第4象限)は初期アポトーシス細胞を表し;PI陽性、アネキシンV陽性染色細胞(第2象限)は主にアポトーシスの後期段階の細胞を表す。
【0099】
Apo-2リガンド処理9D細胞は、向上した細胞外アネキシンV結合性、並びに顕著に増加したPIの取り込みを示し(図2B)、Apo-2リガンドが細胞でアポトーシスを誘発したことを示している。抗Apo-1抗体、CH11では匹敵する結果がえられた(図2B)。Apo-2リガンドは、Raji及びジャーカット細胞で抗Apo-1抗体と類似の応答を誘発した(図2C及び2D参照)。対照及び抗Apo-1抗体と比較したときのこれらの細胞系でのApo-2リガンドによるアポトーシスの誘発(アポトーシス細胞の%で測定した場合)を下記の表1に示す。
Apo-2リガンドによるヌクレオソーム間DNA断片化の活性化も分析した。ジャーカット細胞(左側レーン)及び9D細胞(右側レーン)を対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに6時間インキュベート(2x106細胞/ウェル)し、次いでDNAを細胞から抽出し、ターミナルトランスフェラーゼを用いて32P-ddATPで標識した。標識したDNA試料に電気泳動2%アガロースゲルを施し、後にオートラジオグラフィで分析した[Moore等, Cytotechnology, 17: 1-11 (1995)]。Apo-2リガンドは、ジャーカット細胞及び9D細胞の両方でヌクレオソーム間DNA断片化を誘発した(図2E)。このような断片化はアポトーシスに特徴的である[Cohen, Advances Immunol., 50: 55-85 (1991)]。
Apo-2リガンドのアポトーシス活性の時間変化を試験するために、9D細胞をミクロタイターディッシュ(5x104細胞/ウェル)で、対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに0〜50時間の範囲の期間インキュベートした。インキュベーションに続いて、死亡及び生存細胞の数を、ヘモサイトメータを用いて顕微鏡検査で測定した。
図3Aに示すように、細胞死の最大レベルは9D細胞において24時間以内に起こる。
Apo-2リガンド誘発細胞死の用量依存性を試験するために、9D細胞(5x104細胞/ウェル)を、連続的に希釈した対照培地又はApo-2(実施例3に記載したように調製)とともに24時間インキュベートした。インキュベーション後の死亡及び生存細胞の数を上記のように測定した。結果を図3Bに示した。特異的アポトーシスは、Apo-2リガンド処理細胞でのアポトーシス%を対照中でのアポトーシス%で除することにより決定した。最大値の半分のアポトーシス活性化は約0.1μg/ml(約1nM)で起こり、最大誘発は約1〜3μg/ml(約10〜30nM)で起こった。
【0100】
(実施例5)
Apo-2リガンドのヒト非リンパ腫瘍細胞系に対するアポトーシス活性
Apo-2リガンドのヒト非リンパ腫瘍細胞系に対する影響を、以下の細胞系を用いて試験した:HeLa(ヒト頸部癌から誘導、ATCC CCL 22);Me-180(ヒト頸部癌から誘導、ATCC HTB 33);MCF7(ヒト乳癌から誘導、ATCC HTB 22);U-937(ヒト組織リンパ腫から誘導、ATCC CRL 1593 ;A549(ヒト肺癌から誘導、ATCC CCL 185);及び293(アデノウイルス形質移入ヒト胚腎臓細胞から誘導、ATCC CCL 1573)。
アッセイにおいて、各細胞系の1x106細胞を、対照培地、Apo-2リガンド(3μg/ml、実施例3に記載したように調製)、又は抗Apo-1抗体CH11(1μg/ml)とともに24時間インキュベートした。インキュベーションの後、アポトーシスを実施例4に記載したようにFACS分析で測定した。結果を下記の表1に示す。
【0101】
(表1)

【0102】
HeLa細胞及びMCF7細胞は、CH11抗Apo-1抗体に比較してApo-2リガンドによるアポトーシス誘発に対して等しい感受性であった。これに対して、U-937細胞及びA549細胞は、Apo-2リガンドによるアポトーシス誘発に対して極めて大きな感受性を有していた。ME-180細胞はApo-2リガンドに対しては全く感受性であったが、抗Apo-1抗体には比較的耐性があった。293細胞はApo-2リガンドに耐性であり、抗Apo-1抗体には若干応答した。
よって、Apo-2リガンドは非リンパ由来の細胞、並びにリンパ由来の細胞(実施例4参照)でアポトーシスを誘発できる。また、十分に理解されておらず、いかなる特定の理論との結びつけるつもりはないが、本出願人は、Apo-2リガンドがApo-1とは区別されるレセプターを介して作用すると考えている。この考えは、上記の細胞系がApo-2リガンド及び抗Apo-1抗体に対して異なる感受性パターンを示すここに記載したデータによって支持されている。
【0103】
(実施例6)
Apo-2リガンドのヒト末梢血液単球に対する影響
末梢血液単核細胞(「PBMC」)は、ヒト供与者の血液から、リンパ球分離媒体(LSM(登録商標)、Organon Teknika)を用いたフィコール密度勾配遠心分離により単離した。単離下T細胞集団は、抗Igカラムへの表面Ig結合によりB細胞を除去し、IgカラムへのFcレセプターの結合を通して単球を除去することによりPBMCから調製した(R & D Systems)。単離したB細胞集団はOKT3骨髄細胞(ATCC, CRL 8001)により生成された抗CD3抗体と反応したT細胞及び4F2C13ハイブリドーマ(ATCC, HB 22)により生成された単球特異的抗体と反応した単球の補体媒介除去によりPBMCから調製した。さらなる単球除去はプラスチックへの接着によって達成した。
【0104】
新たに単離した末梢血液B又はT細胞(1x106細胞/ウェル)を、対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)の存在下で3日間培養した。活性化のために、B細胞は同時にリポ多糖(「LPS」、1μg/ml)で処理し、T細胞はホルボールミリステートアセテート(「PMA」、10ng/ml)プラスイノマイシン(1μg/ml)(Sigma)で処理した。インターロイキン-2(「IL-2」)前処理のために、T細胞は、Apo-2リガンドに暴露する前に、IL-2(50U/ml)(Genzyme)の存在下で3−5日間培養した。アポトーシスは、本質的に実施例4に記載したようにFACS分析で決定した。しかし、B細胞は、抗CD19/CD20抗体(Jackson Immunoreserch)でゲートし、T細胞は抗CD4/CD8抗体(Jackson Immunoreserch)でゲートした。結果を下記の表2に示すが、独立した実験[Bリンパ球−9実験;Tリンパ球−8実験;Tリンパ球プラスIL-2−5実験]の平均±SEで表し、各データ点毎に500,000細胞が分析された。統計的分析をスチューデントt試験を用いて実施した、表2において、個々の対照に対してa=p<0.05及びb=p<0.02である。
【0105】
(表2)
Apo-2リガンドは、非刺激B細胞、LPSで活性化したB細胞、及びPMA及びイノマイシンで活性化したT細胞においてアポトーシスを誘発した。IL-2の存在下で細胞を培養することにより末梢T細胞がアポトーシスを起こしやすくなることは既に報告されている[Lenardo等, Nature, 353: 858-861 (1991)]。本研究は、IL-2での前処理は、末梢T細胞をApo-2リガンド誘発死に対して敏感化させないことを示した。
【0106】
(実施例7)
Fas/Apo-1及びTNFレセプターを用いた阻害アッセイ
Fas/Apo-1レセプター、並びにI型及びII型TNFレセプター(TNF-R1及びTNF-R2)がApo-2リガンドのアポトーシス活性の媒介に含まれているか否かを決定するために、これらのレセプターの可溶化形態が精製した可溶性Apo-2リガンド(実施例3に記載)のアポトーシス活性を阻害するか否かを試験することにより検定した。
9D細胞(5x104細胞/ウェル)を対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに、対照バッファー、CD4-IgG対照(25μg/ml)、可溶性Apo-1-IgG(25μg/ml)、可溶性TNFR1-IgG(25μg/ml)又は可溶性TNFR2-IgG融合タンパク質(25μg/ml)の存在下で24時間インキュベートした。
Fas/Apo-1、TNF-R1及びTNF-R2レセプターの可溶性誘導体はAshkenazi等, Methods, 8: 104-115 (1995)に記載したようにIgG融合タンパク質として作成した。CD4-IgGは、Byrn等, Nature, 344: 667-670 (1990)に記載されたようにIgG融合タンパク質として作成し、対照として用いた。
図3Cに示したように、レセプター融合分子はApo-2リガンドの9D細胞に対するアポトーシス活性を阻害しなかった。これらの結果は、Apo-2リガンドのアポトーシス活性がFas/Apo-1及びTNF-R1及びTNF-R2とは独立であることを示している。
【0107】
(実施例8)
哺乳動物組織におけるApo-2リガンドmRNAの発現
ヒト組織におけるApo-2リガンドmRNAの発現を、ノーザンブロット分析によって検査した(図4)。ヒトRNAブロットを、全長Apo-2リガンドcDNAをベースにした32P標識DNAプローブ、又はGenBank EST配列、HHEA47M(実施例1参照)をベースにした32P標識RNAプローブにハイブリッド形成させた。ヒト胎児RNAブロットMTN(Clontech)及びヒト成人RNAブロットMTN-II(Clonetech)をDNAプローブと共にインキュベートし、ヒト成人RNAブロットMTN-I(Clonetech)をRNAプローブと共にインキュベートした。ブロットをハイブリッド形成用バッファー[5X SSPE;2X デンハード溶液;100mg/mLの変性剪断されたサケ精子DNA;50%のホルムアミド;2%のSDS]に入ったプローブと共に、42℃で16時間インキュベートした。ブロットを1X SSPE;2%のSDSを用い、65℃で1時間数回洗浄し、ついで50%の脱イオンホルムアミド;1X SSPE;0.2%のSDSを用い、65℃で30分間洗浄した。ブロットを一晩さらした後、リン光画像機(Fuji)を用いた展開させた。
結果を図4に示す。ヒト胎児組織では、Apo-2リガンドmRNA発現は、肺、肝臓及び腎臓で検出されたが脳組織ではされなかった。ヒト成人組織では、Apo-2リガンドmRNA発現は、脾臓、胸腺、前立腺、卵巣、小腸、末梢血液リンパ球、心臓、胎盤、肺、及び腎臓で検出された。精巣、脳、骨格筋、及び膵臓では殆ど又は全く発現は検出されなかった。上記のようなApo-2リガンドの発現パターンは、主にT細胞及び精巣で発現されるApo-1リガンドと同じではなかった[Nagata等, 上掲]。
【0108】
(実施例9)
Apo-2リガンドのヒト腫瘍細胞系に対するアポトーシス活性
Apo-2リガンド(実施例3に記載)のヒト腫瘍細胞系に対するアポトーシス活性を、幾つかの化学治療薬の存在又は不存在下でさらに試験した。
以下のヒト腫瘍細胞系をアッセイした:A549(肺癌、ATCC CCL 185);HTC116(結腸癌、ATCC CCL 247);SW480(結腸腺癌、ATCC CCL 228);MDA231(乳癌、ATCC HTB 26);HeLa(頸部癌、ATCC CCL 22);ME-180(頸部癌、ATCC HTB 33);T24(膀胱癌、ATCC HTB 4);SK-N-AS(神経芽腫、White等, Proc. Natl. Acad. Sci., 92: 5520-5524 (1995))。下記表3に示すように、これらの細胞系の幾つかは野生型p53を発現するが(WT)、他は突然変異により発現しない(mut)。細胞を2.5x105細胞/ウェルで96ウェルプレートに蒔き、終夜インキュベートした。細胞を2倍希釈のApo-2リガンド(100ng/mlから0.01nm/ml)の存在下で培養した。幾つかの培地には化学治療薬−シクロヘキサミド(「CHX」)(50μg/ml;Sigma Chemicals)、ドキソルビシン(10−100μg/ml;Pharmacia)、又は5-FU(6mg/ml;Roche)も24時間添加した。
24時間のインキュベーション後、細胞を0.5%クリスタル-ビオレチン20%メタノールで染色した。0.1Mクエン酸ナトリウム(50%メタノール中0.1Mクエン酸)で染色細胞から染料を溶離し、540nmでの吸収を測定して細胞生存を測定した。
結果を下記の表3に示す。
【0109】
(表3)
+: 100nm/mlApo-2Lで24時間後>35%死亡。
++: 100nm/mlApo-2Lで24時間後>70%死亡。
+++: 10nm/mlApo-2Lで24時間後>70%死亡。
これらの結果は、Apo-2リガンドが種々の腫瘍型から誘導された腫瘍細胞系において細胞死を誘発したことを示し、Apo-2Lが腫瘍細胞のp53状態とは無関係に細胞死を誘発したことを示している。これらの結果はまた、Apo-2リガンド誘発細胞死が幾つかの異なる化学治療薬によって増強されることも示している。
【0110】
(実施例10)
インビボでのApo-2リガンドのアポトーシス活性
腫瘍を持つヌードマウスにおけるApo-2リガンドの影響を試験した。ヌードマウス(各群当たり5−10匹のマウス)(Harlan Sprague Dawleyから購入)に、MDA231ヒト乳ガン細胞(ATCC HTB 26)(2x106細胞/マウス)を皮下注射した(第0日)。腫瘍を14日間成長させた。14及び15日目に、2μg/0.05ml/マウスのApo-2リガンド(実施例3)及び/又は10μg/0.05ml/マウスのドキソルビシン(Pharmacia)を腫瘍部位に注射した。対照動物は同様に0.05mlのPBSを注射した。21日目に動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図5に示す。データは、Apo-2リガンド処理が腫瘍成長そのものを阻害し、Apo-2リガンドが腫瘍成長に対するドキソルビシンの効果を向上させたことを示している。
【0111】
(実施例11)
インビボでのApo-2リガンドのアポトーシス活性
実施例10に記載したように、腫瘍を持つヌードマウスにおけるApo-2リガンドの抗腫瘍効果を試験した。ただし、第0日において、マウスにHCT116ヒト結腸癌細胞(ATCC CCL 247)(2x106細胞/マウス)皮下注射した。腫瘍を14日間成長させた。14及び15日目に、2μg/0.05ml/マウスのApo-2リガンド及び/又は10μg/0.05ml/マウスの5-FU(Roche)を腫瘍部位に注射した。対照動物は同様に0.05mlのPBSを注射した。21日目に動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図6に示す。これらの結果は、Apo-2リガンド処理が腫瘍成長そのものを阻害し、Apo-2リガンドが腫瘍成長に対する5-FUの阻害効果を向上させたことを示している。
【0112】
(実施例12)
インビボでのApo-2リガンドのアポトーシス活性
実施例11に記載したように、腫瘍を持つヌードマウスにおけるApo-2リガンドの抗腫瘍効果を試験した。ただし、第1及び2日目に、10μg/0.05ml/マウスのApo-2リガンド及び/又は100μg/0.05ml/マウスの5-FU(Roche)を腹膜内注射した。対照動物は同様にPBSを注射した。腫瘍サイズ(mm2)を5、9及び15日目に測定し、動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図7及び8に示す。これらの結果は、Apo-2リガンドが腹膜内注射したときでも皮下の腫瘍部位に到達でき抗腫瘍活性を発揮できることを示している。また、これらの結果は、腫瘍成長自体を阻害し、腫瘍成長に対する5-FUの阻害効果を向上させるApo-2リガンドの能力を確認した。
【0113】
(実施例13)
CrmAを用いた阻害アッセイ
ICE及びCPP32/Yama等のプロテアーゼがApo-2リガンドによるアポトーシス誘発において役割を果たしているか否かを実験するために、CrmAがApo-2リガンド誘発アポトーシスを阻止するか否化を決定するアッセイを行った[Marsters等, Current Biology, 6: 750-752 (1996)]。CrmAは、死亡プロテアーゼICE及びCPP/Yamaのポックスウイルス誘導阻害剤であり、TNFR1及びFasApo-1による死亡シグナル伝達を阻止する。さらに、Apo-1リガンド及びTNFによるアポトーシスを媒介するアダプタータンパク質FADDを含む「死亡ドメイン」[Chinnaiyan等, Cell, 81: 505-512 (1995); Hsu等, Cell, 84: 299-308 (1996)]が、Apo-2リガンド誘発アポトーシスに含まれるか否かを実験するために、FADDのドミナントネガティブ突然変異(FADD-DN)[Hsu, 上掲]がApo-2リガンド機能を阻害するか否かを決定するアッセイを行った[Marsters等, Current Biology, 6: 750-752 (1996)]。
HeLA-S3(ATCC CCL 22)細胞をpRK5-CrmA発現プラスミド(Ray等, 上掲で報告されたCrmA配列)又はpRK5-FADD-DN発現プラスミド(Hsu等, Cell, 84: 299-308 (1996))で形質移入した。pRK5は対照として使用した。細胞を、プラスミドDNA取り込みのマーカーとしてのpRK5-CD4(Smith等, Science, 238: 1704-1707 (1988))と同時形質移入した。形質移入細胞をフィコエリスリン抱合抗CD4抗体(Jackson Immunoreserch)で染色して同定し、アポトーシスは本質的に上記実施例4に記載したようにFACSで分析した。
結果を図9に示す。CrmAは、Apo-2リガンド誘発アポトーシス、並びに抗Apo-1抗体に誘発されたアポトーシスを阻止した。これに対して、FADD-NDはApo-2誘発アポトーシスには殆ど影響しなかったが、抗Apo-1抗体に誘発されたアポトーシスは実質的に阻止した。従って、このアッセイの結果は、Apo-2リガンド、TNFR1及びFas/Apo-1はアポトーシス性細胞死を活性化するのに共通の遠位シグナル伝達経路を用いることを示唆している。特に、結果は、ICE及びCPP/Yama等のプロテアーゼがApo-2リガンド誘発アポトーシスには必要であることを示している。これに対して、FADDはTNFR1及びFas/Apo-1に誘発される細胞死には必要であるがApo-2リガンドでは必要ない。
【0114】
Apo-2リガンド抗体の調製
1μgのApo-2リガンド(実施例3に記載したように調製し、Ribi Immunochemical Research Inc., Hamilton, MTから購入したMPL-TDMアジュバントで希釈)を、各々の後足の裏の柔らかい部分に、1週間の間隔で10回注射することで、Balb/cマウス(チャールズリヴァー研究所から得たもの)を免疫化した。最終の追加免疫から3日後に、膝窩リンパ節をマウスから取り除き、単細胞懸濁液を、1%のペニシリン-ストレプトマイシンが補われたDMEM培地(Biowhitakker Corp.から得たもの)中で調製した。ついで、リンパ節細胞を35%のポリエチレングリコールを使用し、マウス骨髄腫細胞P3X63AgU.1(ATCC CRL 1597)と融合させ[Laskov等, Cell. Immunol., 55:251(1980)]、96-ウェル培養プレートで培養した。融合の結果得られたハイブリドーマをHAT培地において選択した。融合から10日後に、ハイブリドーマ培養の上清をELISAでスクリーニングし[Kim等, J. Immunol. Meth., 156: 9-17 (1992)]、Apo-2リガンドタンパク質に結合するモノクローナル抗体の有無を試験した。
ELISAにおいて、各々のウェルに、PBSに0.5μg/mlのApo-2リガンド(実施例3参照)が入ったものを50μl添加して、96-ウェルマイクロタイタープレート(Nunc)をコートし、4℃で一晩インキュベートした。ついでプレートを洗浄用バッファー(0.05%のトゥイーン20を含有するPBS)で3回洗浄した。ついで、マイクロタイタープレートのウェルを、2.0%のウシ血清アルブミン(BSA)200μlでブロックし、室温で1時間インキュベートした。ついでプレートを、洗浄用バッファーで、再度3回洗浄した。
【0115】
洗浄工程後、2μg/mlのApo-2リガンド抗体50μl又は100μlのハイブリドーマ上清を指定ウェルに添加した。100μlのP3X63AgU.1骨髄腫細胞の条件培地を、対照として他の指定ウェルに添加した。プレートを振盪装置上で、室温で1時間インキュベートし、続いて洗浄用バッファーで3回洗浄した。
次に、アッセイ用バッファー(0.5%のウシ血清アルブミン、0.05%のトゥイーン20、0.01%のチメルソール(Thimersol)がPBSに入ったもの)で、1:1000に希釈されたHRP-抱合ヤギ抗マウスIgG Fc(Cappel Laboratories社から購入)を50μl、各々のウェルに添加し、プレートを振盪装置上で、室温で1時間インキュベートした。プレートを洗浄用バッファーで3回洗浄し、ついで各々のウェルに50μlの基質(TMB、3,3',5,5'-テトラメチルベンジジン;Kirkegaard & Perry, Gaithersburg, MDから入手)を添加し、室温で10分間インキュベートした。50μlの終止溶液(Kirkegaard & Perry)を各々のウェルに添加して反応を終了させ、450nmでの吸光度を自動マイクロタイタープレート読取装置で読み取った。
【0116】
ハイブリドーマ上清(選択した99)を、Apo-22リガンド誘発9D細胞死を阻止する活性について試験した。活性は、トリパンブルー染料排除を用いて処理した9D細胞の生存%を試験することにより最初に決定した。
阻止活性はFACS分析でも確認した。9D細胞(5x105細胞/0.5ml)を完全RPMI培地(RPMIプラス10%のFCS、グルタミン、非必須アミノ酸、ペニシリン、ストレプトマイシン、ピルビン酸ナトリウム)に懸濁し、24ウェルのミクロタイタープレートに配した。0.5mlのApo-2リガンド(1μg/ml)(実施例3で調製)を完全RPMI培地に懸濁し、10μgの精製モノクローナル抗体又は100μlの培地上清とともにプレインキュベートし、次いで9D細胞を含む24マクロタイターウェルに添加した。マクロタイタープレートは7%CO2の存在下で終夜37℃でインキュベートした。インキュベートした細胞は、次いで回収し、PBSで1回洗浄した。細胞の生存を製造者(Clontech)の推奨に従ってホスファチジルセリンに結合したFITCアネキシンVの染色により決定した。細胞をPBSで洗浄し、200μl結合バッファー中に再懸濁した。10μlのアネキシンV-FITC(1μg/ml)及び10μlのヨウ化プロオピジウムを細胞に添加した。暗中15分のインキュベーションの後、9D細胞をFACSで分析した。
8つの潜在的阻止、4つの非阻止抗体分泌ハイブリドーマが同定され、更に制限希釈技術によりクローニング(2回)した。
【0117】
モノクローナル抗体1D1、2G6、2E11、及び5C2と命名した4つの抗体のFACS分析を図10に例示した(後述するように、1D1、2G6、2E11、及び5C2抗体は、各々ハイブリドーマ1D1.12.4、2G6.3.4、2E11.5.5、及び5C2.4.9から生成され、それらは全てATCCに寄託されている)。Apo-2リガンドで処理した9D細胞(左上図)は、未処理の対照細胞(右上図)より50%高いのアポトーシス細胞を示した。Apo-2リガンドに、2E115C2、2G6、又は1D1抗体を加えて処理した9D細胞は、未処理の対照細胞より各々0%、6%、26%、及び48%高いアポトーシス細胞を示した。これらの結果は、5C2、2E11及び2G6抗体は阻止抗体であるが1D1抗体は非阻止抗体であることを示す。最も高い阻止活性は5C2抗体で得られた。
4つの抗体の抗原特異性もELISAで試験した。ミクロタイターウェルを2μg/mlのリンホトキシン(Genentech, Inc., EP 164,965, Gray等, Nature, 312: 721-724 (1984)も参照)、TNF-α(Genentech, Inc., Pennica等, Nature, 312: 724-729 (1984); Aggarwal等, J. BIol. Chem., 260: 2345-2354 (1985)も参照)、又はApo-2リガンド(実施例3参照)でコートした。モノクローナル抗体1D1、2G6、2E11、及び5C2は10μg/mlの濃度で試験した。
アッセイの結果は図11に示した。図11の結果は、モノクローナル抗体2G6、2E11、及び5C2はApo-2リガンドに特異的だが、モノクローナル抗体1D1はリンホトキシン及びTNF-αと弱い交差反応結合を有することを示した。
【0118】
B.アイソタイピング
1D1、2G6、2E11、及び5C2抗体(上記項目Aに記載)のアイソタイプを、アイソタイプ特異的ヤギ抗マウスIg(Fisher Biotech, Pitsburgh, PA)でミクロタイタープレートを4℃で終夜コートすることにより決定した。プレートは次いで上記の洗浄バッファーで洗浄した。ミクロタイタープレートのウェルを200μlの2%ウシ血清アルブミンでブロックし、室温で1時間インキュベートした。プレートを再度洗浄バッファーで3回洗浄した。
次に、100μlの5μg/mlの精製Apo-2リガンド抗体又は100μlのハイブリドーマ培地上清を指定ウェルに添加した。プレートを室温で30分間インキュベートし、次いで50μlのHRP-抱合ヤギ抗マウスIgG(上記)を各ウェルに添加した。プレートを室温で30分間インキュベートした。HRPのレベルを上記のTRP基質を用いて検出した。
アイソタイピング分析は、1D1及び2G6抗体はIgG2b抗体であり、2E11及び5C2抗体はIgG2a抗体であることを示した。
【0119】
C.エピトープマッピング
エピトープマッピングは、上掲のKim等に記載されているように、ビオチニル化モノクローナル抗体を用いて競合的結合ELISAを使用して実施した。選択されたモノクローナル抗体は、Antibodies, a laboratory Manual, Eds. E. Harlow及びD. Lane, p.342に記載されているようにN-ヒドロキシスクシンイミドを用いてビオチニル化した。ミクロタイタープレートウェルを、50μlのApo-2リガンド(実施例3参照、0.1μg/ml)でコートし、4℃で終夜維持し、次いで2%BSAで室温において1時間ブロックした。ミクロタイターウェルを洗浄した後、あらかじめ決定した最適濃度のビオチニル化抗体及び1000倍過剰の非標識抗体を各ウェルに添加した。室温で1時間インキュベートした後、プレートを洗浄し、ビオチニル化抗体の量をHRP-ストレプトアビジンの添加で検出した。ミクロタイターウェルを洗浄した後、結合した酵素を基質(TMB)の添加で検出し、プレートをELISAプレート読み取り機で490nmで読み取った。
結果を図12に示す。結果は、HRP-抱合抗体の結合は、それ自身の抗体の過剰量で有効に阻害されたが、アッセイした他の抗体ではされないことを示した。
モノクローナル抗体で認識されるApo-2リガンドの領域は、合成ペプチド[図1Aに示したようにApo-2リガンド配列のaa 128-143(ペプチド「APO14」);aa 144-159(ペプチド「APO15」);aa 192-204(ペプチド「APO17」);aa 230-238(ペプチド「APO18」);aa 261-272(ペプチド「APO19」)]を用いてChuntharapai等, J. Immunol., 152: 1783-1789 (1994)に記載されているようにELISAで決定した。結果を図13に示した。1D1抗体は、Apo-2リガンドのアミノ酸残基192−204を含むAPO17ペプチドに結合性を示した。
【0120】
(実施例15)
CHO細胞におけるApo-2リガンドの発現
pGEM-TApo-2リガンドプラスミド(上記実施例1及び2に記載)からの全長Apo-2リガンドcDNA挿入物をpRK5プラスミドに挿入した。このプラスミドを、次いで、リポフェクタミンプラス(Gibco/BRL)法を用いて、DHFR選択遺伝子を持つpFD11プラスミド(SV40初期プロモーター)とDP-12CHO細胞に同時形質移入した。Apo-2リガンドを発現する安定なクローンを、2.5%透析FBSを含むPS21(G1074)/280GHT-枯渇選択成長培地中での成長能力で選択した。
選択したApo-2リガンドを発現するCHO細胞を37℃で2−4日インキュベートした。次いで細胞培地を回収して濾過した。細胞培地上清中のApo-2リガンドの存在は、ウェスタンブロット分析により試験した。ウェスタン分析用のタンパク質を調製するために、培地上清を、制御された孔のガラスビーズに結合させたここに記載の5C2抗Apo-2抗体とともにインキュベートした。インキュベーションに続いて、ビーズを洗浄し、結合タンパク質を25mMのDTTを含有するSDS−PAGE試料バッファーで溶離した。次いで培地を、4−20%ポリアクリルアミド勾配SDSゲルを走らせ、ニトロセルロースフィルター上にエレクトロブロットした。フィルターをポリクローナルウサギ抗ヒトApo-2リガンドポリクローナル抗体、続いてセイヨウワサビペルオキシダーゼに抱合させた抗ウサギIgGとともにインキュベートし、製造者(NEB)の指示に従って化学発光によって展開した。Apo-2リガンドを発現するCHO細胞からの試料は、ゲル上に22,000ダルトンの単一のバンドを生じた(データは示さず)。
【0121】
形質移入したCHO細胞によって発現したApo-2リガンドの配列を決定するために、25mlの細胞培養培地上清を抗Apo-2L抗体(ここに記載した5C2抗体)を用いて精製した。精製カラムを調製するために、2.0mgの5C2抗体を0.5mlの制御孔ガラスビーズ(CPG, Inc., Fairfield, NJ)に製造者の指示に従って結合させた。結合の後、樹脂を15mlの50mMトリスpH7.5、次いで15mlの0.1M酢酸/0.15Mクエン酸ナトリウム/0.05MのNaClで洗浄した。
負荷に先立って、0.5mlカラムを15mlの50mMトリスpH7.5で平衡させた。約25mlの回収したCHO細胞培養培地、pH7.2、導電性10.2mmhoを、抗体カラムに勾配を与える負荷モードで負荷した。負荷流出物をカラムにリサイクルして全負荷時間を1時間とした。負荷に続いて、非特異的タンパク質を2mlの0.5MのTMAC/0.25MのNaClで洗い落とした。カラムを1.5mlの0.1M酢酸/0.15MNaCl、pH3.0で溶離した。1.5mlの溶離物を管に回収し、即座に50μlの3MトリスpH9.0でpH7.0に中和した。
【0122】
溶離物質のアリコートをSDSゲル上で分析し、平行して、既に大腸菌で発現された図1Aのアミノ酸残基96−281をからなる精製対照Apo-2リガンドポリペプチドを精製した。ゲルは、CHO細胞発現可溶性Apo-2リガンドポリペプチドが5C2抗体アフィニティカラム上で精製されたことを確認した。溶離物質の他のアリコートを濃縮し、SDS−PAGE上を走らせ、PVDF膜にエレクトロブロットし、次いでEdman分解で配列決定した。タンパク質配列分析により、CHO細胞が図1Aの配列の位置92におけるN末端アミノ酸を有するApo-2リガンドの可溶化形態を発現することが明らかとなった。よって、可溶化Apo-2リガンドポリペプチドは図1Aのアミノ酸92−281を含む。現在では、Apo-2リガンドのこの可溶化92−281アミノ酸形態は、Apo-2リガンドの天然に切断された形態を含むと考えられている。
CHO細胞の発現されたApo-2リガンドのアポトーシス活性を、培養したHeLa細胞(ATCC CCL 22)で試験した。HeLa細胞を10%のFBSを含むHamのF12培地中で6ウェルの培養プレートに蒔き、37℃で終夜インキュベートした。培地を吸引し、発現されたApo-2リガンド(1:10、1;20、1:40希釈)を含む2mlのCHO細胞培地上清を各資料ウェルに添加した。プレートを37℃で終夜インキュベートした。2mlの一条件培地(非形質移入CHO細胞培地上清)を含む対照ウェルを同じ条件下で平行に走らせた。
次いで、処理したHeLa細胞を、光顕微鏡下でアポトーシス形態について分析した(図14A−14D参照)。各視野でのアポトーシス細胞の数を図14Eに示す。発現されたApo-2リガンドを含有するCHO細胞培地上清で処理したHeLa細胞は、非条件培地で処理した細胞よりアポトーシス形態において2倍又はそれ以上の増加を示した。
【0123】
(実施例16)
大腸菌発現Apo-2リガンドのアポトーシス活性
Apo-2リガンドの抗腫瘍効果を腫瘍を持つヌードマウスで試験した。上記の実施例とは対照的に、可溶性Apo-2リガンドポリペプチドは大腸菌で発現され、次いで埋め込みミニポンプ装置を通して動物中に注入した。
Apo-2リガンドは、アミノ酸91−281(図1A参照)をコードするcDNAをpS1346プラスミドに挿入することにより調製した[pS1346は挿入された転写停止剤を持つhgh207-1プラスミドを含む;DeBoer等, Proc. Natl. Acad. Sci., 80: 21-25 (1983); Scholtissek等, Nucl. Acids Reserch, 15: 3185 (1987)参照]。このプラスミドは、次いで大腸菌株52A7に形質移入した。株52A7は次の遺伝子型を持つ大腸菌K12W3110株である:fhuA(tonA)lon galE rpoHts(htpRts)clpP lacIq。大腸菌の25ml培地をLB培地で光学密度約1.0OD550まで成長させ、遠心分離(5000rpmで15分間)で回収した。細胞ペレットを0.15MのNaClで1回洗浄した後2.5mlのバッファー(LBブロス、HClでpH6.1、100g/LのPEG8000、10mMのMgSO4、10mMのMgCll2及び5%DMSOを含有)中で再懸濁した。細胞懸濁物を氷上に30〜45分間放置し、アリコートを−80℃で保存した。この細胞懸濁物を形質転換プロセスの競合細胞として提供した。競合細胞のプラスミド調製物でも形質転換、形質転換物の選択及び単離は、Maniatis等, Molecular Cloning: A Laboratory manual, second Edition, vol.1: 1.74-1.84に記載された標準プロトコールで実施した。
【0124】
1mlの形質転換体を5μg/mlテトラサイクリンを含有する500mlのLB培地に播種して発酵種菌を調製した。この培地を振盪する2Lのバッフルフラスコで30又は37℃において10時間インキュベートした。次いで、得られた培地を、25g/LのNZアミンAS、6.25g/Lの酵母菌抽出物、0.125g/Lのトリプトファン、6.25g/Lのテトラサイクリン、0.94g/Lのグルコース、0.625g/LのL-イソロイシン、及び94mg/LのL-61抗発泡剤(antifoam)を含む8リットルの培地(6.25g/Lの硫酸アンモニウム、7.5g/Lの二塩基リン酸カリウム、3.75g/Lの一塩基リン酸ナトリウム二水和物、及び1.25g/Lのクエン酸ナトリウム)を含む10リットル発酵器の播種に使用した。
発酵は30℃において、強く撹拌しながら通気し、NH4OH添加でpHを7.0に調節して行った。最初のグルコースが消費された後、無菌の50%グルコース溶液を与えて培地を維持した。約30ODにおいて、温度を25℃にシフトさせて発泡を最小にした。培地のODが約50(A550)に達したとき、25mg/mlのIAA(3-ベータ-インドールアクリル酸)25mlを添加してtrpプロモータに調節されたApo-2リガンド発現を誘発した。IAA添加の6〜10時間後に遠心分離により細胞を回収した。
【0125】
発現されたポリペプチドを以下のように精製した。大腸菌から発現されたApo-2リガンドを含む細胞ペーストを、0.1Mのトリス、0.2MのNaCl、50mMのEDTA、pH8バッファーで抽出した。次いで抽出物を40%の硫酸アンモニウムを用いて沈殿させた。全てのクロマトグラフィ工程は、特に示さない場合は室温で実施した。硫酸アンモニウム沈殿物を50mMのHEPES、0.05%のTriton x-100、pH8バッファー中に溶解させ、次いで50mMのHEPES、0.05%のTriton-x100、pH8バッファーで平衡させたマクロ-prepヒドロキシアパタイトのカラムに80cm/時間の流速で適用した。カラムを平衡バッファーでA280での吸収がベースライン近くに戻るまで洗浄した。Apo-2リガンドを0から0.2Mリン酸ナトリウム平衡バッファーの線形勾配で8カラム容量によってカラムから溶離した。Apo-2リガンドを含む分画をプールし、pHを6.5に調節した。pH調節したプールを、0.35MのNaCl/PBSバッファーで平衡させたNi-NTAスーパーフローのカラムに80cm/時間の流速で負荷した。カラムを平衡バッファーで洗浄して吸収をベースライン近くにした。Apo-2リガンドを0から50mMイミダゾール/平衡バッファーの線形勾配で8カラム容量によってカラムから溶離した。Apo-2リガンドを含む分画をプールし、Millipore Lab TFF system, Biomax 8 膜を用いて濃縮した。濃縮したプールを、20mMのトリス、8%のトレハロース、0.01%トゥイーン20バッファー中のG-25カラムで調製した。調製したApo-2リガンドプールを0.22ミクロンフィルターを通して濾過した。
精製した物質の分析により、それが(約50:50の比率で)アミノ酸91−281(図1A参照))を有するApo-2リガンドポリペプチド、及びアミノ酸92−281(図1A参照を有するApo-2リガンドポリペプチドを含むことが公にされた(この比率は現在のところ潜在的N末端加工によると考えられており;図1Aに示したアミノ酸残基91はメチオニン残基である)。
【0126】
実験において、ヌードマウスにHCT116ヒト結腸癌細胞(ATCC CCL 247)(1x106細胞)を動物の各側に皮下注射した。次いで腫瘍を直径約1cmとなるまで(およそ10日間)成長させた。第0日に、腫瘍容積をノギスで測定した。容積は、a=長さ及びb=幅としたときのπ/6xab2として計算した。次いで、浸透ミニポンプ(Alza Corp.から購入、Model 1003)を(1)対照媒体(20mMのトリスpH7.5、8%のトレハロース、0.01%のトゥイーン-20)又は(2)Apo-2リガンドのいずれかで負荷した。各群の5匹の動物は対照媒体又はApo-2リガンドのいずれかを受容する。ミニポンプを較正して毎日10mg/kgのApo-2リガンド(10mg/mlx2μl/時間)又は2μl/時間の対照媒体を輸送した。
第3日目に、腫瘍容積を再度測定し、動物を犠牲にした。動物の試験は毒性の如何なる証拠も示さなかった。図15において、結果は腫瘍容積のパーセント変化を示す。媒体群には腫瘍容積の変化は無かったが、Apo-2Lを注入したマウスでは腫瘍サイズの顕著な縮小が起こった。Apo-2リガンドでの処理は、3日に渡って腫瘍を約50%に収縮させた。
【0127】
(実施例17)
Apo-2リガンド置換変異体の調製
レセプター結合及びアポトーシス誘発活性にとって重要なエピトープを発見し記述するためにアラニンスキャニング突然変異誘発を用いた。アラニンスキャニング突然変異誘発は、Kelley等, Biochemistry, 34: 10383-10392 (1995)及びCunningham及びWells, Sience, 244: 1081-1085 (1989)に記載されているように、結合性エピトープを同定するのに用いられる。一般的にアミノ酸アラニンをコードする特全変異が他の残基に代えてここに記載された方法によって導入される。アラニンは、先端を切ったような側鎖(メチル基のみ)を有するので選択される置換基である。さらに、アラニンは表面及びタンパク質内部に埋まった状態の両方で見出されるので、発現されたタンパク質の構造一体性を乱す可能性が最も低いと思われる。
アラニン置換タンパク質をコードするプラスミドをオリゴヌクレオチド指向性突然変異誘発(Kunkel, T.A.等, Methods in Enzymology, 154: 367-382 (1987))を用いてプラスミドpAPOK5の一本鎖形態上で作成した(図16参照)。トリプトファン(trp)プロモーターによるApo-2Lの91−281形態の細胞内大腸菌発現のために、このプラスミドを設計した。pAPOK5はApo-2LcDNA(残基91−281をコードする)をtrpプロモーターを持つプラスミドpS1162にクローニングするPCRを用いて作成した。全てのアラニン置換基はジデオキシヌクレオチド配列により確認した(Sanger, F.等, Proc. Natl. Acad.. Sci., 74: 5463-5467 (1977))。
アラニン置換されたApo-2Lタンパク質をコードするプラスミドは、次いで発現のために大腸菌株294に形質転換した。培地はルリアブロスに50μg/mlのカルベニシリンを追加したものの中で37℃において終夜成長させて飽和させた。続いて細胞を、Na2HPO4(6g/L)、KH2PO4(3g/L)、NaCl(0.5g/L)、NH4Cl(1g/L)、グルコース(4.9g/L)、カサミノ酸(casamino acids)(4.9g/L)、27mMのMgSO4、0.003%のチアミンHCl、及び50μg/mLのカルベニシリンの20倍希釈物を追加した無菌フィルター培地に蒔き、37℃で1時間成長させた。
【0128】
次いで、25μg/mLでの3-β-インドールアクリル酸(IAA)(Sigma, St.Louis, MO.)で発現を誘発し、細胞を30℃で振盪させながら終夜成長させた。細胞を遠心分離で回収し、以下に述べるようなApo-2Lの引き続く回収のために−20℃で凍結貯蔵した。
Apo-2Lタンパク質は、10容量(重量/容量)の100mMトリス、pH8.0/200mMのNaCl/5mlのEDTA/1mMのDTT中で、モデルM110-Fマイクロフリューダイザ(Microfluidics Corporation, Newton, MA.)を用いた均質化により凍結大腸菌細胞ペレットから抽出した。ポリエチレンイミン(PEI)を最終濃度0.5%(容量/容量)でホモジェネートに添加し、それを次いで遠心分離して細胞断片を除去した。個体硫酸アンモニウムを抽出上清に室温で撹拌しながら飽和の45%という最終濃度で添加し、ペレットを遠心分離で回収した。硫酸アンモニウムペレットを50%硫酸アンモニウム溶液で洗浄して残ったEDTAを除去し、次いで50容量(重量/容量)の50mMのEPPS、pH7.5/0.1%トリトンX-100中に再懸濁した。得られた溶液を遠心分離で透明化し、5mLのHiTrapキレート化セファロースTMがカラム(Pharmacia, Piscataway, NJ.)を用いた固定化金属アフィニティクロマトグラフィ(IMAC)により精製した。100mMのNiSO4/300mMのトリス、pH7.5中のニッケルでカラムを負荷し、リン酸緩衝塩水(PBS)中の350mMのNaClで平衡化した。負荷後、カラムをPBS中350mMのNaClで洗浄し、50mMのイミダゾール/350mMのNaCl(PBS中)で溶離した。IMAC溶離物を20mMのトリス、pH7.5に対して透析し、遠心分離で透明化し、5mLのTiTrapSPセファロースTMがカラム(Pharmacia)を用いたカチオン交換クロマトグラフィーで更に精製し、それを20mMトリス、pH7.5で平衡化して洗浄した。HiTrapSPカラムを20mMのトリス、pH7.5/0.5MのNaClで溶離した。Spカラム溶離物は2mMのDTTで還元し、続いて室温で撹拌しながら飽和の45%という最終濃度まで固体硫酸アンモニウムを添加することにより沈殿させた。硫酸アンモニウムペレットを、3.5mLの20っMトリス、pH7.5/100mMのNaClに再懸濁し、PD10カラム(Pharmacia)を用いたゲル濾過クロマトグラフィーにより20mMトリス、pH7.5/100mMのNaClという最終バッファーに交換した。精製したApo-2Lアラニン置換タンパク質は、クマシーブルー染色SDS−PAGE及びN末端配列決定によって特徴付けし、−20℃で凍結保存した。
【0129】
(実施例18)
Apo-2L置換変異体を用いたアポトーシス活性
上記実施例17に記載したように調製した幾つかのApo-2リガンド置換変異体のアポトーシス活性を決定するためにインビトロアッセイを行った。
SK-MES-1ヒト肺癌細胞(ATCC HTB 58)を、10%FBS、2mMのL-グルタミン、100U/mlのペニシリン、及び100マイクログラムのストレプトマイシンを添加したDMEM:HamのF-12(50:50)培地で培養した。SK-MES-1細胞を96ウェルのファルコン組織培地で処理したミクロプレートに、4.0x104細胞/ウェルの密度で0.1mlの容量で蒔き、37℃で終夜接着させた。種々のApo-2L置換変異体又はApo-2L(図1Aのアミノ酸残基91−281からなる;Apo-2L.2と呼ぶ)を24時間添加してアポトーシスを誘発した。試験タンパク質での処理に続いて、培地を除去し、ウェルをメタノール中のクリスタルバイオレットの0.5%溶液で15分間染色した。SLT 340 ATCプレートリーダー(Salzburg, Austria)を用いて540nmで乾燥プレートの光学密度を測定することにより細胞数を決定した。
結果を図17及び18に示した。図17は変異体D218A、D269A、及びV207AのバイオアッセイデータをApo-2L.2分子と比較して示す(V207Aは図1のアミノ酸91−281を含むが図1Aの配列の位置207におけるバリンがアラニンに置換されたと定義される置換変異体である)。変異体D218A及びD269Aは、Apo-2L.2に比較した場合、各々約3倍及び7倍増加したアポトーシス活性を示した。V207AはバイオアッセイにおいてApo-2L.2より約9倍小さな活性であった。
図18は、Apo-2L.2と比較したD203Aのバイオアッセイデータを示す。変異体D203Aは、Apo-2L.2に比較した場合、約2倍の増大したアポトーシス誘発活性を示した。
【0130】
(実施例19)
Apo-2リガンド置換変異体の結合分析
Apo-2Lアラニン-置換タンパク質(上記実施例18で参照したD203A、D218A及びD269A)を、文献に記載された種々のApo-2Lレセプター、DR4[Pan等, Science, 276: 111-113 (1997)];DR5[Sheridan等, Science, 277: 818-821 (1997)];デコイレセプターDcR2[Marsters等, Current Biology, 7: 1003-1006 (1997)];及びOPG[simonet等, Cell, 89: 309-319 (1997)]に対する結合性をBIAcore(Pharmacia)を用いて分析した。比較のため、アミノ酸91−281からなるApo-2LもBIAcoreにおいてIgG-レセプター融合分子への結合性を試験した。Apo-2Lレセプタータンパク質の各々を、Ashkenazi等, Proc. Natl. acad. Sci., 88: 10535-10539 (1991)に実質的に記載されているようにイムノアドヘシンとして調製した。IgG-レセプター融合分子は、CM5 BIAcoreチップ、研究等級(Pharmacia)にNHS-EDCアミンカップリング化学を用いて固定化した。Apo-2Lアラニン置換タンパク質をBIAcoreランニングバッファー、PBS/0.05%トゥイーン20で500nMから15.625nMまで連続的に2倍希釈して、固定化レセプターを有するセンサーチップ表面を横切って通過させた。KD値を決定するためにBIAcore評価ソフトウェアを(製造者の指示に従って)使用して特異的動的結合パラメータを用いた。
結果を図19に示す。この表は、BIAcore(Pharmacia)を用いた動的結合分析によって決定した場合のApo-2L.2と比較した変異体D203A、D218A及びD269Aの解離定数(KD)を示す。データは、Apo-2L.2と比較した場合、アラニン置換変異体類のIgG-レセプター融合タンパク質DR4、DR5、及びDcR2に対する結合性に有意な変化(>2倍)は無かった。OPGR-IgGへの結合の場合、変異体D203A、D218A、及びD269Aは、Apo-2L.2に比較してより緊密な結合性を示した(2<KDwt<KDmut<4)。
【0131】
材料の寄託
次の細胞系をアメリカン・タイプ・カルチャー・コレクション,10801 University Boulevard., Manassas, VA, USA(ATCC)に寄託した:
細胞系 ATCC寄託番号 寄託日
2935-pRK5-hApo-2L-myc clone2.1 CRL-12014 1996年1月3日
1D1.12.4 HB-12257 1997年1 月8日
2G6.3.4 HB-12259 1997年1 月8日
2E11.5.5 HB-12256 1997年1 月8日
5C2.4.9 HB-12258 1997年1 月8日
【0132】
この寄託は、特許手続き上の微生物の寄託の国際的承認に関するブダペスト条約及びその規則(ブダペスト条約)の規定に従って行われた。これは、寄託の日付から30年間、寄託の生存可能な培養が維持されることを保証するものである。寄託物はブダペスト条約の条項に従い、またジェネンテク社とATCCとの間の合意に従い、ATCCから入手することができ、これは、どれが最初であろうとも、関連した米国特許の発行時又は任意の米国又は外国特許出願の公開時に、寄託培養物の後代を永久かつ非制限的に入手可能とすることを保証し、米国特許法第122条及びそれに従う特許庁長官規則(特に参照番号886OG638の37CFR第1.14条を含む)に従って権利を有すると米国特許庁長官が決定した者に子孫を入手可能とすることを保証するものである。
本出願の譲受人は、寄託した培養物が、適切な条件下で培養されていた場合に死亡もしくは損失又は破壊されたならば、材料は通知時に同一の他のものと即座に取り替えることに同意する。寄託物質の入手可能性は、特許法に従いあらゆる政府の権限下で認められた権利に違反して、本発明を実施するライセンスであるとみなされるものではない。
上記の文書による明細書は、当業者に本発明を実施できるようにするために十分であると考えられる。寄託した態様は、本発明のある側面の一つの説明として意図されており、機能的に等価なあらゆる作成物がこの発明の範囲内にあるため、寄託された作成物により、本発明の範囲が限定されるものではない。ここでの物質の寄託は、ここに含まれる文書による説明が、そのベストモードを含む、本発明の任意の側面の実施を可能にするために不十分であることを認めるものではないし、それが表す特定の例証に対して請求の範囲を制限するものと解釈されるものでもない。実際、ここに示し記載したものに加えて、本発明を様々に改変することは、前記の記載から当業者にとっては明らかなものであり、添付の請求の範囲内に入るものである。
【図面の簡単な説明】
【0133】
【図1A】ヒトApo-2リガンドcDNAの核酸配列及びその誘導アミノ酸配列を示す図である。
【図1B】ヒトApo-2リガンドのC末端領域の、ヒトTNFサイトカインファミリーの知られたメンバー、4-1BBL、OX40L、CD27L、CD30L、TNF-α、LT-β、LT-α、CD40L、及びApo-1Lの対応する領域とのアラインメントを示す図である。
【図1C−1E】(C)抗-mycエピトープ抗体を用いたFACS分析で測定した場合の、ヒト293細胞で発現された組換え、全長、C末端mycエピトープタグApo-2リガンドの細胞トポロジーを示す図である。(D)化学的架橋有り(レーン2、3)又は無し(レーン1)に次いでSDS-PAGE及び銀染色で測定した場合の、組換えバキュロウイルス感染昆虫細胞で発現されNi2+-キレートアフィニティクロマトグラフィで精製した組換えHis10エピトープタグ可溶性Apo-2のサイズ及びサブユニット構造を示す図である。(E)抗-gDエピトープ抗体での免疫沈降に次いでSDS-PAGE及びオートラジオグラフィで測定した場合の、代謝性標識されたヒト293細胞で発現された組換えgDエピトープタグ可溶性Apo-2リガンドのサイズ及びサブユニット構造を示す図である。
【図2A】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞は特徴的な形態変化により同定した。
【図2B】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2C】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2D】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2E】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はヌクレオソーム内DNA断片化の分析(E)により同定した。
【図3A】Apo-2リガンド誘発アポトーシスの時間変化を示す図である。
【図3B】Apo-2リガンド誘発アポトーシスの用量依存性を示す図である。
【図3C】Fas/Apo-1レセプター、TNF-R1レセプター、又はTNF-R2レセプターに基づく可溶性レセプター−IgG融合タンパク質によるApo-2リガンド誘発アポトーシスの阻害欠如を示す図である。
【図4】ノーザンブロット分析で測定した場合の、ヒト胎児及びヒト成人組織におけるApo-2リガンドmRNAの発現を示す図である。
【図5】ヌードマウスにおいて成長させたヒトMDA231乳癌細胞に基づく腫瘍の重量に対する、単独又はドキソルビシンと混合して腫瘍内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図6】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍の重量に対する、単独又は5-FUと混合して腫瘍内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図7】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍のサイズに対する、単独又は5-FUと混合して腹膜内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図8】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍の重量に対する、単独又は5-FUと混合して腹膜内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図9】ドミナントネガティブFADDではなくCrmAがHeLa-S3細胞におけるApo-2リガンド誘発アポトーシスを阻止することを示す棒グラフである。
【図10】Apo-2リガンドに誘発されたアポトーシス及び4つの抗-Apo-2リガンド抗体:1D1、2G6、2E11及び5C2による影響のFACS分析を示す図である(FITC複合アネキシンVを用いて検出したアポトーシス9D細胞−太線;生存、非染色細胞−細線)。
【図11】モノクローナル抗体1D1、2G6、2E11、及び5C2の抗原特異性を示す棒グラフである。
【図12】モノクローナル抗体1D1、2G6、2E11、及び5C2のエピトープマッピングアッセイの結果を示す棒グラフである。
【図13】Apo-2リガンドの特異的アミノ酸領域からなる数種の合成ペプチドに対するモノクローナル抗体1D1が結合する能力を試験するアッセイの結果を示す棒グラフである。
【図14A−D】発現されたApo-2リガンドを含有するCHO細胞培地上清(1:10、1:20、1:40希釈)又は非条件培地で処理した培養HeLa細胞を示す図である。
【図14E】上記各視野におけるアポトーシス細胞の数を示す棒グラフである。
【図15】(実施例16に記載したように)大腸菌で発現されたApo-2リガンドポリペプチドを浸透ミニポンプを介して投与したヌードマウスにおけるHCT116結腸癌腫瘍容積におけるパーセント(%)変化を示す図である。
【図16】pAPOK5プラスミドの制限マップを示す図である。
【図17】バイオアッセイにおいて、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D218A、D269A及びD207Aの、アポトーシス細胞死を誘発する能力を示す図である。
【図18】バイオアッセイにおいて、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D203Aのアポトーシス細胞死を誘発する能力を示す図である。
【図19】BIAcore(Pharmacia)を用いた動力学的結合アッセイで測定した場合の、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D203A、D218A、及びD269Aの解離定数(KD)を示す表である。
【発明の開示】
【0001】
(発明の分野)
本発明は、一般に、ここに「Apo-2リガンド」と命名する新規サイトカインの同定、単離、及び組換え生産、及びそのような組成物の使用方法に関する。
【0002】
(発明の背景)
哺乳動物における細胞数のコントロールは、一部には細胞増殖と細胞死のバランスにより決定されると考えられている。しばしば壊死性細胞死と称される細胞死の一形態は、典型的には、ある種の外傷又は細胞傷害の結果生じる細胞死の病理的形態として特徴付けられる。これに対して、通常は規則的又はコントロールされた状態で進行する細胞死の他の「生理的」形態がある。細胞死のこの規則的又はコントロールされた形態は、しばしば「アポトーシス」と称される[例えば、Barr等, Bio/Technology, 12:487-493(1994)を参照]。アポトーシス性細胞死は、免疫系におけるクローン選択と胚の発達を含む多くの生理的プロセスにおいて自然に生じる[Itohら, Cell, 66:233-243(1991)]。アポトーシス性細胞死のレベルの減少は、癌、狼瘡、ヘルペスウイスル感染を含む種々の病理的条件に関連している[Thompson, Science, 267:1456-1462(1995)]。
【0003】
アポトーシス性細胞死には、典型的には、細胞内における一又は複数の特徴的な形態学的及び生化学的変化、例えば細胞質の凝結、原形質膜の微絨毛の喪失、核の分節化、染色体DNAの分解又はミトコンドリア機能の喪失が伴う。様々な外因的及び内因的シグナルが、このような形態学的及び生化学的な細胞変化を惹起又は誘発すると考えられている[Raff, Nature, 356:397-400(1992);Steller, Science, 267: 1445-1449 (1995); Sachs等, Blood, 82:15(1993)]。例えば、ホルモンの刺激、例えば未成熟胸腺細胞に対する糖質コルチコイドホルモン、並びにある種の成長因子の退薬により惹起され得る[Watanabe-Fukunagaら, Nature, 356:314-317(1992)]。また、幾つかの同定された発癌遺伝子、例えばmyc、rel、及びE1A、及び腫瘍サプレッサーは、p53と同様に、アポトーシスの誘発においてある役割を有していることも報告されている。ある種の化学療法薬及びある種の放射線も同様にアポトーシス誘発活性を有していることも見出されている[Thompson, 上掲]。
【0004】
様々な分子、例えば腫瘍壊死因子-α(「TNF-α」)、腫瘍壊死因子-β(「TNF-β」又は「リンホトキシン」)、CD30リガンド、CD27リガンド、CD40リガンド、OX-40リガンド、4-1BBリガンド、及びApo-1リガンド(Fasリガンド又はCD95リガンドとも称される)が、サイトカインの腫瘍壊死因子(「TNF」)ファミリーのメンバーとして同定された[例えば、Gruss及びDower, Blood, 85:3378-3404(1995)]。これらの分子のなかでも、TNF-α、TNF-β、CD30リガンド、4-1BBリガンド、Apo-1リガンド、及びApo-2リガンド(TRAIL)は、アポトーシス性細胞死に関与していることが報告されている。TNF-αとTNF-βの両方とも、感受性腫瘍細胞におけるアポトーシス性死を誘発することが報告されている[Schmidら, Proc. Natl. Acad. Sci., 83:1881(1986);Dealtryら, Eur. J. Immunol., 17:689(1987)]。ゼングらは、TNF-αがCD8ポジティブT細胞のポスト刺激性アポトーシスに関与していることを報告している[Zheng等, Nature, 377:348-351(1995)]。他の研究者らは、CD30リガンドが胸腺における自己反応性T細胞の欠失に関与していることを報告している[Amakawa等, プログラム細胞死に関するコールドスプリングハーバー研究所のシンポジウム、要約集、第10巻、(1995)]。
【0005】
マウスのFas/Apo-1レセプター又はリガンド遺伝子(それぞれlpr及びgldと呼称される)における変異が幾つかの自己免疫疾患に関連しており、Apo-1リガンドが末梢の自己反応性リンパ球のクローン除去の調節においてある役割を担っていることを示している[Krammer等, Curr. Op. Immunol., 6:279-289(1994); Nagata等, Science, 267:1449-1456(1995)]。また、Apo-1リガンドは、CD4-ポジティブTリンパ球及びBリンパ球においてポスト刺激性アポトーシスを誘発することが報告されており、それらの機能がもはや必要でなくなった際の活性化リンパ球の除去に関与している[Krammer等, 上掲; Nagata等, 上掲]。Apo-1レセプターと特異的に結合するアゴニストのマウスモノクローナル抗体は、TNF-αに匹敵するか類似する細胞死滅活性を示すことが報告されている[Yonehara等, J. Exp. Med., 169:1747-1756(1989)]。
【0006】
このようなTNFファミリーのサイトカインが介在する種々の細胞反応の誘発は、それらが特異的な細胞レセプターに結合することにより開始されると考えられている。約55-kDa(TNF-R1)と75-kDa(TNF-R2)の2つの異なるTNFレセプターが同定されており[Hohman等, J. Biol. Chem., 264:14927-14934(1989);Brockhaus等, Proc. Natl. Acad. Sci., 87:3127-3131(1990);1991年3月20日に公開されたEP417,563]、双方のレセプター型に対応するヒト及びマウスcDNAが単離され、特徴付けされている[Loetscher等, Cell, 61:351(1990);Schall等, Cell, 61:361(1990);Smith等, Science, 248:1019-1023(1990);Lewis等, Proc. Natl. Acad. Sci., 88:2830-2834(1991);Goodwin等, Mol. Cell. Biol., 11:3020-3026(1991)]。
【0007】
イトー等は、Apo-1レセプターが55-kDaのTNF-R1によりシグナル化されるものと同様のアポトーシス性細胞死をシグナル化し得ることを開示している[Itoh等, 上掲]。また、Apo-1抗原の発現は、細胞をTNF-α又は抗Apo-1のマウスモノクローナル抗体で処理した場合に、TNF-R1のものと共にダウンレギュレーションされることが報告されている[Krammer等, 上掲;Nagata等, 上掲]。従って、Apo-1及びTNF-R1レセプターの双方を同時発現する株化細胞が、共通のシグナル伝達経路を通して細胞死を媒介しているとの仮説を唱える研究者もいた[同]。
【0008】
今日までに同定されているTNFファミリーのリガンドは、リンホトキシン-αを除き、II型の膜貫通タンパク質であり、そのC末端は細胞外にある。これに対して、今日までに同定されているTNFレセプター(TNFR)ファミリーのレセプター類はI型の膜貫通タンパク質である。しかしながら、TNFリガンド及びレセプターファミリーの双方において、ファミリーメンバー間で同定された相同性は、主として細胞外ドメイン(「ECD」)において見出されている。TNF-α、Apo-1リガンド及びCD40リガンドを含むTNFファミリーサイトカインのいくつかは、細胞表面においてタンパク分解的に切断され;各場合に得られたタンパク質は、典型的には、可溶性サイトカインとして機能するホモ三量体分子を形成する。また、TNFレセプターファミリーのタンパク質は、通常、タンパク分解的に切断され、同族のサイトカインの阻害剤として機能し得る可溶性リガンドのECDを放出する。サイトカインのTNFファミリー及びそれらのレセプター類の概説については、上掲のGruss及びDowerを参照されたい。
【0009】
(発明の概要)
本出願人は、「Apo-2リガンド」と命名される新規なサイトカインをコードするcDNAクローンを同定した。現在では、Apo-2リガンドはTNFサイトカインファミリーのメンバーであると信じられており;Apo-2リガンドは、Apo-1リガンドを含む幾つかの知られたTNF関連タンパク質とアミノ酸配列において関連している。しかしながら、本出願人は、Apo-2リガンドが知られた可溶性Apo-1又はTNFレセプター類、例えばFas/Apo-1、TNF-R1、又はTNF-R2レセプター等によって感知できるほどに阻害されないことを見出した。
【0010】
一実施態様において、本発明は、単離されたApo-2リガンドを提供する。特に、本発明は、図1Aの残基114〜281を含んでなるアミノ酸配列を含む単離されたApo-2リガンドを提供する。他の実施態様においては、Apo-2リガンドは、図1Aの残基92〜281を含んでなるアミノ酸配列を含む。さらなる実施態様では、Apo-2リガンドは、図1Aの残基91〜281を含んでなるアミノ酸配列を含む。さらに他の実施態様では、Apo-2リガンドは、図1Aの残基41〜281又は15〜281を含んでなるアミノ酸配列を含む。さらなる実施態様では、図1A(配列番号:1)の残基1〜281として示したアミノ酸配列を含む。
【0011】
また、本発明の単離されたApo-2リガンドは、上記の参照配列の置換変異体を含む。特に、一実施態様では、図1Aのアミノ酸91〜281を含むApo-2リガンドにおいて、位置203、218又は269の少なくとも1つがアラニンに置換された置換変異体が提供される。これらの置換変異体は特に、「D203A」;「D218A」及び「D269A」と定義される。この命名法は、例えば図1Aのアミノ酸91〜281を含むApo-2リガンドポリペプチドであって、(図1Aに示した番号を用いた)位置203、218及び/又は269のアスパラギン酸残基がアラニン残基で置換されたものを同定するのに用いられる。場合によっては、置換変異体は一又は複数のそのような置換を含んでいてもよい。
他の実施態様では、本発明は、他の異種ポリペプチドに融合したApo-2リガンドを含むキメラ分子を提供する。そのようなキメラ分子の例は、タグポリペプチド配列に融合したApo-2リガンドを含む。
【0012】
他の実施態様では、本発明は、Apo-2リガンドをコードする単離された核酸分子を提供する。一態様では、核酸分子はRNA又はDNAであり、Apo-2リガンドをコードするか、又はそのようなApo-2リガンドをコードする核酸配列に相補的であり、少なくとも中程度の厳密性条件下で、それを安定に結合させたままである。一実施態様では、核酸配列は:
(a)残基1から残基281までの全長タンパク質をコードする図1Aの核酸配列のコード領域(即ち、ヌクレオチド91〜933)、あるいは図1A(配列番号:2)に示した核酸配列の又は残基41〜281の細胞外タンパク質をコードするヌクレオチド211〜933、又は残基92〜281の細胞外タンパク質をコードするヌクレオチド364〜933、又は残基91〜281の細胞外タンパク質をコードするヌクレオチド361〜933、又は残基114〜281の細胞外タンパク質をコードするヌクレオチド430〜933;あるいは、
(b)遺伝子コードの縮退の範囲内で(a)の配列に相当する配列から選択される。
【0013】
さらなる実施態様において、本発明は、当該ベクターで形質転換された宿主細胞によって認識されるコントロール配列に作用可能に結合したApo-2リガンドをコードする核酸分子を含む複製可能なベクターを提供する。このベクター又は核酸分子を含む宿主細胞も提供される。さらに、この核酸分子を含む宿主細胞を培養し、当該宿主細胞培地からタンパク質を回収することを含んでなるApo-2リガンドの作成方法が提供される。
他の実施態様では、本発明はApo-2リガンドに結合する抗体を提供する。一態様において、この抗体はApo-2リガンドに対して抗原特異性を有するモノクローナル抗体である。
他の実施態様では、本発明はApo-2リガンド及び担体を含んでなる組成物を提供する。この組成物は、アポトーシスを誘発又は刺激するのに有用な製薬組成物であってよい。
他の実施態様では、本発明は、アポトーシスの誘発に有効な量のApo-2リガンドに、インビボ又はエキソビボで、哺乳動物細胞を暴露することを含んでなる、哺乳動物細胞においてアポトーシスを誘発する方法を提供する。
【0014】
他の実施態様では、本発明は癌を持つ哺乳動物の治療方法を提供する。この方法においては、癌を持つと診断された哺乳動物に、有効量のApo-2リガンドが投与される。Apo-2リガンドは、一又は複数の他の治療、例えば化学治療、放射線治療、又は抗腫瘍活性を示す他の薬剤とともに哺乳動物に投与してもよい。
さらなる実施態様では、本発明はApo-2リガンド又はApo-2リガンド抗体を含む製造品及びキットを提供する。この製造品及びキットは、容器、容器上のラベル、及び容器内に収容された組成物を具備する。容器上のラベルは、組成物が或る種の治療又は非治療用途に使用できることを表示する。この組成物は活性薬を含有し、この活性薬はApo-2リガンド又はApo-2リガンド抗体を含む。
【0015】
(好適な実施態様の詳細な説明)
I.定義
ここで使用される際の「Apo-2リガンド」及び「Apo-2L」という用語には、図1Aのに示したアミノ酸配列のアミノ酸残基114〜281を含む、又は残基92〜281を含む、又は残基91〜281を含む、又は残基41〜281を含む、又は残基15〜281を含む、又は残基1〜281を含むポリペプチド配列、並びに、前記配列の欠失、挿入、又は置換変異体を意味する。一実施態様では、このポリペプチド配列は、少なくとも図1Aの残基114〜281を有する。場合によっては、ポリペプチド配列は、少なくとも図1Aの残基92〜281又は残基91〜281を有する。他の好ましい実施態様では、変異体は生物学的に活性であり、上記の配列の1つと少なくとも約80%のアミノ酸配列同一性、より好ましくは少なくとも約90%の配列同一性、さらに好ましくは少なくとも約95%の配列同一性を有する。この定義は、図1Aのアミノ酸91〜281を複myApo-2リガンドの置換変異体であって、位置203、218又は269の少なくとも1つのアミノ酸がアラニンに置換されたものを含む。また、この定義はApo-2リガンド供給源から、例えばここに述べるヒト組織型(実施例8参照)から、又は他の供給源から単離された、又は組換え又は合成方法によって調製された天然配列Apo-2リガンドを含む。Apo-2リガンドのこの定義は、周知のEST配列、例えばGenBank HHEA47M、T90422、R31020、H43566、H44565、H44567、H54628、H44772、H54629、T82085、及びT10524は除外する。
【0016】
「エピトープタグ」なる用語は、ここで用いられるときは、「タグポリペプチド」に融合したApo-2リガンド、又はそれらの一部を含んでなるキメラポリペプチドを指す。タグポリペプチドは、その抗体が産生され得るエピトープを提供するに十分な数の残基を有しているが、その長さはApo-2リガンドの活性を阻害しないよう充分に短い。また、タグポリペプチドは、好ましくは、抗体が他のエピトープと実質的に交差反応をしないようにかなり独特である。適切なタグポリペプチドは、一般に、少なくとも6のアミノ酸残基、通常は約8〜約50のアミノ酸残基(好ましくは約10〜約20の残基)を有する。
【0017】
「単離された」とは、ここで開示された種々のポリペプチドを記述するために使用するときは、その自然環境の成分から同定され分離され及び/又は回収されたポリペプチドを意味する。その自然環境の汚染成分とは、ポリペプチドの診断又は治療への使用を典型的には妨害する物質であり、酵素、ホルモン、及び他のタンパク質様又は非タンパク質様溶質が含まれる。好ましい実施態様において、タンパク質は、(1)スピニングカップシークエネーターを使用することにより、少なくとも15残基のN末端あるいは内部アミノ酸配列を得るのに充分なほど、あるいは、(2)クーマシーブルーあるいは好ましくは銀染色を用いた非還元あるいは還元条件下でのSDS-PAGEによる均一性が得られるように充分なほど精製される。Apo-2リガンドの自然環境の少なくとも1つの成分が存在しないため、単離されたタンパク質には、組換え細胞内のインサイツのタンパク質が含まれる。しかしながら、通常は、単離されたタンパク質は少なくとも1つの精製工程により調製される。
【0018】
「単離された」Apo-2リガンド核酸分子は、同定され、Apo-2リガンド核酸の天然源に通常付随している少なくとも1つの汚染核酸分子から分離された核酸分子である。単離されたApo-2リガンド核酸分子は、天然に見出される形態あるいは設定以外のものである。ゆえに、単離されたApo-2リガンド核酸分子は、天然の細胞中に存在するApo-2リガンド核酸分子とは区別される。しかし、単離されたApo-2リガンド核酸分子は、例えば、核酸分子が天然の細胞のものとは異なった染色体位置にあるApo-2リガンドを通常発現する細胞に含まれるApo-2リガンド核酸分子を含む。
【0019】
ここで同定されている配列に対する「パーセント(%)アミノ酸配列同一性」は、配列を整列させ、最大のパーセント配列同一性を得るために必要ならば間隙を導入し、如何なる保存的置換も配列同一性の一部と考えないとした、Apo-2リガンド配列のアミノ酸残基と同一である候補配列中のアミノ酸残基のパーセントとして定義される。%同一性の値は、Altschul等, Methods in Enzymology, 266: 460-480 (1996);http://blast.wustl/edu/blast/README.htmlから得られるWU-BLAST-2によって算出することができる。WU-BLAST-2は幾つかの検索パラメータを使用し、その殆どが初期値に設定される。調節可能なパラメータは次の値:オーバーラップ・スパン=1、オーバーラップ・フラクション=0.125、ワードしきい値(T)=11に設定する。同様にして、Apo-2リガンドポリペプチドの核酸配列に対する「パーセント(%)核酸配列同一性」は、Apo-2リガンドコード配列のヌクレオチドと同一の候補配列中のヌクレオチドのパーセントとして定義される。同一性の値は、オーバーラップ・スパン及びオーバーラップ・フラクションが各々1及び0.125に設定された初期設定のWU-BLAST-2のBLASTNモジュールにより算出される。
【0020】
「コントロール配列」という用語は、特定の宿主生物において作用可能に結合されたコード配列を発現するために必要なDNA配列を指す。例えば原核生物に好適なコントロール配列は、プロモーター、場合によってはオペレータ配列、及びリボソーム結合部位を含む。真核生物の細胞は、プロモーター、ポリアデニル化シグナル及びエンハンサーを利用することが知られている。
核酸は、他の核酸配列と機能的な関係にあるときに「作用可能に結合され」ている。例えば、プレ配列あるいは分泌リーダーのDNAは、ポリペプチドの分泌に寄与するプレタンパク質として発現されているならそのポリペプチドのDNAに作用可能に結合されている;プロモーター又はエンハンサーは、配列の転写に影響を及ぼすならばコード配列に作用可能に結合されている;又はリボソーム結合部位は、もしそれが翻訳を容易にするような位置にあるならコード配列と作用可能に結合されている。一般的に、「作用可能に結合される」とは、結合されたDNA配列が近接しており、分泌リーダーの場合には近接していて読みフェーズにある。しかし、エンハンサーは必ずしも近接しているわけではない。結合は簡便な制限部位でのライゲーションにより達成される。そのような部位が存在しない場合は、通常の手法にしたがって、合成されたオリゴヌクレオチドアダプターあるいはリンカーが使用される。
【0021】
「抗体」という用語は最も広い意味において使用され、特に単一の抗Apo-2リガンドモノクローナル抗体(アゴニスト及びアンタゴニスト抗体を含む)、及び多エピトープ特異性を持つ抗Apo-2リガンド抗体組成物を包含している。
ここで使用される「モノクローナル抗体」という用語は、実質的に均一な抗体の集団から得られる抗体を称する、すなわち、集団を構成する個々の抗体が、少量存在しうる自然に生じる可能な突然変異を除いて同一である。モノクローナル抗体は高度に特異的であり、一つの抗原部位に対応する。さらに、異なる決定基(エピトープ)に対応する異なる抗体を典型的に含む従来の(ポリクローナル)抗体調製物とは異なり、各モノクローナル抗体は抗原の単一の決定基に対応する。
【0022】
ここで、モノクローナル抗体は、起源の種又は免疫グロブリンクラス又はサブクラスの命名に拘わらず、定常ドメインを有する抗Apo-2リガンド抗体の可変(高頻度可変を含む)ドメイン、又は重鎖を有する軽鎖、又は他の種由来の鎖を有する或る種由来の鎖、あるいは異種タンパク質との融合体をスプライシングすることによって得られるハイブリッド及び組換え抗体(例えば「ヒト化」抗体)、並びにそれが所望の生物的活性を有する限り抗体断片(例えばFab、F(ab')2及びFv)を特に含む。例えば、米国特許第4,816,567号、及びMage等, Monoclonal Antibody Productuon Techniques and Applications, pp.79-97(Marcel Dekker, Inc.:ニューヨーク, 1987)を参照。
従って、「モノクローナル」との形容は、実質的に均一な抗体集団から得られたという抗体の性質を示し、抗体を何か特定の方法で生産しなければならないことを意味するものではない。例えば、本発明に従って使用されるモノクローナル抗体は、最初にKohler及びMilsteinによって、Nature, 256:495 (1975)に記載されたハイブリドーマ法によって作ることができ、あるいは例えば米国特許第4,816,567号に記載された組換えDNA法によって作ることができる。「モノクローナル抗体」は、例えば、McCafferty等, Nature, 348:552-554(1990)に記載された技術を用いてファージライブラリから作成することができる。
【0023】
非ヒト(例えばマウス)抗体の「ヒト化」形とは、キメラ免疫グロブリン、免疫グロブリン鎖、あるいはそれらの断片(例えばFv、Fab、Fab'、F(ab')2あるいは抗体の他の抗原結合サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。大部分においてヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)である。ある場合には、ヒト免疫グロブリンのFvフレームワーク領域(FR)残基は、対応する非ヒト残基によって置換されている。更に、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでもよい。これらの修飾は抗体の特性を更に洗練し、最適化するために行われる。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリン共通配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域又はドメイン(Fc)、典型的にはヒトの免疫グロブリンの定常領域又はドメインの少なくとも一部を含んでなる。
【0024】
ここで意図している「生物学的に活性な」とは、(a)インビボ又はエキソビボで少なくとも一種類の哺乳動物細胞においてアポトーシスを誘発する能力を有しているか;(b)抗体を生成できる、即ち、免疫原性であるか;又は(c)天然又は天然発生Apo-2L活性を保持していることを意味する。
「アポトーシス」及び「アポトーシス活性」という用語は広義に使用され、典型的には、細胞質の凝縮、原形質膜の微絨毛の喪失、核の断片化、染色体DNAの分解又はミトコンドリア機能の喪失を含む一又は複数の特徴的な細胞変化を伴う、哺乳動物における細胞死の規則的又はコントロールされた形態を指す。この活性は、例えば細胞生死判別アッセイ、FACS分析又はDNA電気泳動法等、全て従来から知られている方法により決定し測定することができる。
【0025】
「癌」及び「癌性」という用語は、典型的には調節されない細胞成長を特徴とする、哺乳動物における生理学的状態を指すか記述する。癌の例には、これらに限定されるものではないが、癌腫、リンパ腫、芽細胞腫、肉腫、及び白血病が含まれる。このような癌のより特定の例には、扁平上皮細胞癌、小細胞肺癌、非小細胞肺癌、芽細胞腫、胃腸癌、腎臓癌、膵臓癌、神経膠芽細胞腫、神経芽腫、子宮頸管癌、卵巣癌、肝臓癌、胃癌、膀胱癌、肝細胞腫(hepatoma)、乳癌、大腸癌、結腸直腸癌、子宮体癌、唾液腺癌、腎臓癌、肝臓癌、前立腺癌、産卵口癌、甲状腺癌、肝癌(hepatic carcinoma)及び様々な種類の頭部及び頸部の癌が含まれる。一実施態様では、癌は濾胞性リンパ腫、p53変異を持つ癌、又はホルモン依存性癌、例えば乳癌、前立腺癌、又は卵巣癌を含む。
ここで使用される「治療する」、「治療」及び「治療法」とは、治癒的療法、予防的療法及び防護的療法を称する。
ここで使用される「哺乳動物」という用語は、ヒト、ウシ、ウマ、イヌ及びネコを含む哺乳動物として分類されるあらゆる動物を指す。本発明の好ましい実施態様においては、哺乳動物はヒトである。
【0026】
II. 本発明の組成物と方法
本発明は、TNFリガンドファミリーに関連するサイトカインであり、ここで「Apo-2リガンド」と定義されるサイトカインを提供する。ヒトApo-2リガンドの推定成熟アミノ酸配列は281アミノ酸を含み、約32.5kDaの計算された分子量及び約7.63の等電点を有する。N末端には明確なシグナル配列は無いが、ヒドロパシー分析は残基15と40との間に疎水性領域の存在を示している。シグナル配列が無いこと及び内部疎水性領域が存在することは、Apo-2リガンドがII型膜貫通タンパク質であることを示唆している。潜在的なN結合グリコシル化部位は、推定細胞外領域の残基109に位置している。図1Aに示したように、推定細胞質領域はアミノ酸残基1−14を含み、膜貫通領域はアミノ酸残基15−40を含み、細胞外領域はアミノ酸残基41−281を含む。可溶性細胞外ドメインApo-2リガンドポリペプチドは本発明の範囲内に含まれ、限定されるわけではないが、図1Aに示した細胞外領域のアミノ酸残基114−281、92−281、又は91−281を含むApo-2リガンドポリペプチドを包含する。
【0027】
また本発明はApo-2L置換変異体も提供する。ここに記載するように、幾つかの生物学的活性を有する置換変異体を同定するためにアラニンスキャンニング技術が利用される。Apo-2リガンドの特別な置換変異体は、図1Aのアミノ酸残基91−281において、位置203、218又は269の少なくとも1つのアミノ酸がアラニンに置換されたものを含む。これらの置換変異体は、「D203A」;「D218A」及び「D269A」と定義される。この命名法は、例えば図1Aのアミノ酸91〜281を含むApo-2リガンドポリペプチドであって、(図1Aに示した番号を用いた)位置203、218及び/又は269のアスパラギン酸残基がアラニン残基で置換されたものを特定するのに用いられる。場合によっては、置換変異体は一又は複数のそのような置換を含んでいてもよい。
【0028】
A.Apo-2リガンドの調製
以下の説明は、主として、Apo-2リガンド核酸を含むベクターで形質転換又は形質移入された細胞を培養し、細胞培地からポリペプチドを回収することによりApo-2リガンドを生産する方法に関する。もちろん、当該分野においてよく知られている他の方法を用いてApo-2リガンドを調製することはできると考えられる。
1. Apo-2リガンドをコードするDNAの単離
Apo-2リガンドをコードするDNAは、Apo-2リガンドmRNAを保有していてそれを検出可能なレベルで発現すると考えられる組織から調製された任意のcDNAライブラリから得ることができる。従って、ヒトApo-2リガンドDNAは、実施例1に記載されたヒト胎盤cDNAのバクテリオファージライブラリのように、ヒトの組織から調製されたcDNAライブラリから簡便に得ることができる。またApo-2リガンドコード化遺伝子は、ゲノムライブラリから又はオリゴヌクレオチド合成により得ることもできる。
【0029】
ライブラリは、対象となる遺伝子あるいはその遺伝子によりコードされるタンパク質を同定するために設計された(Apo-2リガンドに対する抗体又は少なくとも約20−80塩基のオリゴヌクレオチド等の)プローブによってスクリーニングできる。オリゴヌクレオチドプローブの例は実施例1に提供する。選択されたプローブによるcDNA又はゲノムライブラリのスクリーニングは、例えばSambrook等, Molecular Cloning: A Laboratory Manual(New York: Cold Spring Harbor Laboratory Press, 1989)に記載されている標準的な手順を使用して実施することができる。Apo-2リガンドをコードする遺伝子を単離する他の方法はPCR法を使用するものである[Sambrook等,上掲;Dieffenbach等, PCR Primer:A Laboratory Manual(Cold Spring Harbor Laboratory Press, 1995)]。
スクリーニングの一つの好ましい方法は、種々のヒト組織からcDNAライブラリをスクリーニングするために選別したオリゴヌクレオチド配列を用いることである。以下の実施例1には、2つの異なるオリゴヌクオチドプローブでのcDNAライブラリのスクリーニング技術を記載している。プローブとして選択されたオリゴヌクレオチド配列は、充分な長さで、疑陽性が最小化されるよう充分に明瞭でなければならない。オリゴヌクレオチドは、スクリーニングされるライブラリ内のDNAとのハイブリッド化時に検出可能であるように標識されていることが好ましい。標識化の方法は、当該分野において良く知られており、32P標識されたATPのような放射標識、ビオチン化あるいは酵素標識の使用が含まれる。
【0030】
全てのタンパク質コード化配列を有する核酸は、ここで開示された推定アミノ酸配列を使用し、また必要ならば、cDNAに逆転写されなかったmRNAの生成中間体及び先駆物質を検出する上掲のSambrookらにより記述されている従来のプライマー伸展法を使用し、選択されたcDNA又はゲノムライブラリをスクリーニングすることにより得られる。
Apo-2リガンドのアミノ酸配列変異体は、Apo-2リガンドDNAに適切なヌクレオチド変化を導入するか、所望のApo-2リガンドポリペプチドを合成することにより調製することができる。このような変異体は全長Apo-2リガンドについて図1Aに示したアミノ酸配列の細胞内領域、膜貫通領域、又は細胞外領域の内部あるいは一方又は両方の末端に残基の挿入、置換及び/又は欠失を示す。挿入、置換及び/又は欠失の任意の組み合わせで最終構造物に到達することができるが、この最終構造物は、例えばここに定義する所望のアポトーシス活性を有する。好ましい実施態様では、変異体は、Apo-2リガンドの細胞内、膜貫通、又は細胞外領域についてここで同定した配列、あるいはApo-2リガンドの全長配列に対して、少なくとも約80%のアミノ酸配列同一性、より好ましくは少なくとも約90%の配列同一性、さらに好ましくは少なくとも約95%の配列同一性を有する。また、アミノ酸の変化により、グリコシル化部位の数と位置の変化、膜係留特性の変更のように、Apo-2リガンドの翻訳後過程を改変し得る。
【0031】
ここで記載されているApo-2リガンド配列における変異は、米国特許第5,364,934号に記載された保存的あるいは非保存的突然変異に関する技術とガイドラインの任意のものを使用して起こすことができる。これらは、オリゴヌクレオチド媒介(部位特異的)突然変異誘発、アラニンスキャニング、及びPCR突然変異誘発などを含む。
近接配列に沿った一又は複数のアミノ酸を同定するために、スキャニングアミノ酸分析法を使用することができる。好ましいスキャニングアミノ酸は、比較的小さい中性アミノ酸である。このようなアミノ酸には、アラニン、グリシン、セリン及びシステインが含まれる。変異体の主鎖構造をあまり改変することなく、ベータ炭素を越えた側鎖が除去されるため、アラニンがこの群のなかで好ましいスキャニングアミノ酸である[Cunningham等, Science, 244: 1081 (1989)]。またアラニンは最も一般的なアミノ酸であることによっても好ましい。さらに、アラニンは埋設及び露出位置の双方に頻繁に見出される[Creighton, The Proteins, (W.H. Freeman & Co., N.Y.);Chothia, J. Mol. Biol., 150:1(1976)]。
また、本発明の範囲内に含まれるApo-2リガンド配列の変異体は、アミノ末端誘導体又は修飾形にも関連する。このようなApo-2リガンド配列は、ポリペプチド配列のN末端にメチオニン又は修飾メチオニン(例えばホルミルメチオニル又は他のブロックしたメチオニル種など)を有するここに記載した任意のApo-2リガンドポリペプチドを含む。
【0032】
2. 複製可能なベクターへの核酸の挿入
天然又は変異体Apo-2リガンドをコードする核酸(例えば、cDNA又はゲノムDNA)は、さらなるクローニング(DNAの増幅)又は発現のために、複製可能なベクター内に挿入される。様々なベクターが公的に入手可能である。ベクター成分としては、一般に、これらに制限されるものではないが、次のものの一又は複数が含まれる:シグナル配列、複製開始点、一又は複数のマーカー遺伝子、エンハンサー成分、プロモーター、及び転写終結配列であり、それぞれを以下に説明する。
(i) シグナル配列成分
Apo-2リガンドは直接的に組換え手法によって生産されるだけではなく、シグナル配列あるいは成熟タンパク質あるいはポリペプチドのN末端に特異的切断部位を有する他のポリペプチドである異種性ポリペプチドとの融合ペプチドとしても生産される。一般に、シグナル配列はベクターの成分であるか、ベクターに挿入されるApo-2リガンドDNAの一部である。好ましく選択された異種シグナル配列は宿主細胞によって認識され加工される(すなわち、シグナルペプチダーゼによって切断される)ものである。シグナル配列は、例えばアルカリホスファターゼ、ペニシリナーゼ、lppあるいは熱安定なエンテロトキシンIIリーダーの群から選択される原核生物シグナル配列であってよい。酵母の分泌に関しては、シグナル配列は、酵母インベルターゼリーダー、アルファ因子リーダー(酵母菌属(Saccharomyces)及びクルイベロマイシス(Kluyveromyces)α因子リーダーを含み、後者は米国特許第5,010,182号に記載されている)、又は酸ホスフォターゼリーダー、白体(C.albicans)グルコアミラーゼリーダー(1990年4月4日発行のEP362179)、又は1990年11月15日に公開された国際特許出願第WO 90/13646号に記載されているシグナルであり得る。哺乳動物細胞の発現においては、同一あるいは関連ある種の分泌ポリペプチド由来のシグナル配列、単純ヘルペスグリコタンパク質Dシグナルのようなウイルス分泌リーダーのような他の哺乳動物のシグナル配列をタンパク質の直接分泌に使用してもよいが、インビボにおけるヒト細胞の細胞膜へのApo-2リガンドの挿入を通常指示する天然Apo-2リガンドプレ配列が十分である。
このような前駆体領域のDNAは、好ましくは、Apo-2リガンドをコードするDNAにリーディングフレームが結合される。
【0033】
(ii)複製開始点成分
発現及びクローニングベクターは共に一又は複数の選択された宿主細胞においてベクターの複製を可能にする核酸配列を含む。一般に、この配列はクローニングベクターにおいて、宿主染色体DNAとは独立にベクターが複製することを可能にするものであり、複製開始点又は自律的複製配列を含む。そのような配列は多くの細菌、酵母及びウイルスに対してよく知られている。プラスミドpBR322に由来する複製開始点は大部分のグラム陰性細菌に好適であり、2μプラスミド開始点は酵母に適しており、様々なウイルス開始点(SV40、ポリオーマ、アデノウイルス、VSV又はBPV)は哺乳動物細胞におけるクローニングベクターに有用である。一般には、哺乳動物の発現ベクターには複製開始点成分は不要である(SV40開始点が典型的には初期プロモーターを有しているため用いられる)。
殆どの発現ベクターは「シャトル」ベクターである、すなわち、それらは少なくとも一つのクラスの生物において複製可能であるが、発現のために他の生物に形質移入され得る。例えば、大腸菌においてベクターがクローン化され、そのベクターが酵母あるいは哺乳動物細胞に形質移入され、宿主細胞染色体と独立して複製することはできないとしても、発現する。
DNAは宿主ゲノムに挿入することによって増幅され得る。これは、例えばベクターにバシラス(Bacillus)ゲノムDNAに見られる配列と相補的なDNA配列を含めることにより、宿主としてバシラス種を用いて容易に達成される。このベクターを用いたバシラスの形質移入は、ゲノムとの相同的組換え及びApo-2リガンドDNAの挿入をもたらす。しかし、Apo-2リガンドをコードするゲノムDNAの回収は、Apo-2リガンドDNAを切断するのに制限酵素による消化を必要とするために、外来的に複製したベクターの場合よりも複雑である。
【0034】
(iii)選択遺伝子成分
発現及びクローニングベクターは、典型的には、選べるマーカーとも称される選択遺伝子を含む。この遺伝子は、選択的培地で成長させた形質転換宿主細胞の生存又は成長に必要なタンパク質をコードする。選択遺伝子を含むベクターと共に形質転換されていない宿主細胞は培地で生存しない。典型的な選択遺伝子は、(a)アンピシリン、ネオマイシン、メトトレキセートあるいはテトラサイクリンのような抗生物質あるいは他の毒素に耐性を与え、(b)栄養要求性欠陥を補い、又は(c)例えばバシリに対する遺伝子コードD-アラニンラセマーゼのような、複合培地から得られない重要な栄養素を供給する、タンパク質をコードする。
選択技術の一例においては、宿主細胞の成長を抑止する薬物が用いられる。異種性遺伝子で首尾よく形質転換した細胞は、抗薬物性を付与し、選択工程を生存するタンパク質を生産する。このような優性選択の例としては、薬物ネオマイシン[Southern等, J. Molec. Appl. Genet., 1:327(1982)]、ミコフェノール酸[Mulligan等, Science, 209:1422(1980)]又はハイグロマイシン[Sugden等, Mol. Cell. Biol., 5:410-413(1985)]が使用される。上述した3つの例は、真核生物のコントロール下で細菌遺伝子を用いて、適切な薬物G418又はネオマイシン(ジェネテシン)、xgpt(ミコフェノール酸)、又はハイグロマイシンにそれぞれ耐性を付与する。
【0035】
哺乳動物細胞に適切な選べるマーカーの他の例は、DHFRあるいはチミジンキナーゼのように、Apo-2リガンド核酸を捕捉することのできる細胞成分を同定することのできるものである。哺乳動物細胞の形質転換細胞は、マーカーを捕捉することによって当該形質転換細胞のみが生存できるように独特に適応化された淘汰圧下に置かれる。淘汰圧は、培地中の選択剤の濃度が次第に変化する条件下で形質転換細胞を培養することにより課し、選択遺伝子とApo-2をコードするDNAの双方を増幅させる。増幅は、成長に重要なタンパク質の生産に対する要求度が高い遺伝子が、組換え細胞の後の世代の染色体内に直列に反復されるプロセスである。増幅されたDNAから増大したApo-2リガンド量が合成される。増幅可能な遺伝子のほかの例には、メタロチオネインI及びII、アデノシンデアミナーゼ、及びオルニチンデカルボキシラーゼが含まれる。
DHFR選択遺伝子によって形質転換された細胞は、先ず、DHFRの競合的アンタゴニストであるメトトリキセート(Mtx)を含む培地において形質転換物の全てを培養することで同定される。野生型DHFRを用いた場合の好適な宿主細胞は、Urlaub 等により, Proc. Natl. Acad. Sci. USA, 77:7216 (1980)に記載されているようにして調製され増殖されたDHFR活性に欠陥のあるチャイニーズハムスター卵巣(CHO)株化細胞である。形質転換した細胞は次に濃度の高いメトトレキセートに接触させる。これによりDHFR遺伝子の複数コピーが合成され、同時に、Apo-2リガンドをコードするDNAのような発現ベクターを含む他のDNAの複数コピーが作られる。この増幅方法は、もしMtxに高度に耐性である変異体DHFR遺伝子が使用されたような場合には内在性DHFRの存在にかかわらず、任意の他の適切な宿主、例えば、ATCC番号CCL61 CHO-K1を使用することができる(EP117,060)。
【0036】
あるいは、Apo-2リガンドをコードするDNA配列、野生型DHFRタンパク質、及びアミノグリコシド3'-ホスホトランスフェラーゼ(APH)のような他の選べるマーカーで形質転換あるいは同時形質転換した宿主細胞(特に、内在性DHFRを含む野生型宿主)は、カナマイシン、ネオマイシンあるいはG418のようなアミノグリコシド抗生物質のような選択可能マーカーの選択剤を含む培地における細胞増殖により選択することができる。米国特許第4,965,199号を参照。
酵母中での使用に好適な選択遺伝子は酵母プラスミドYRp7に存在するtrp1遺伝子である[Stinchcomb等, Nature, 282:39(1979);Kingman等, Gene, 7:141(1979);Tschemper等, Gene, 10:157(1980)]。trp1遺伝子は、例えば、ATCC番号44076あるいはPEP4-1のようなトリプトファン内で成長する能力に欠ける酵母の突然変異株に対する選択マーカーを提供する[Jones, Genetics, 85:12 (1977)]。酵母宿主細胞ゲノムにtrp1破壊が存在することは、トリプトファンの不存在下における増殖による形質転換を検出するための有効な環境を提供する。同様に、Leu2欠陥酵母株(ATCC 20,622あるいは 38,626)は、Leu2遺伝子を有する既知のプラスミドによって補完される。
さらに、1.6μmの円形プラスミドpKD1由来のベクターは、クルイヴェロマイシス(Kluyveromyces)酵母の形質転換に用いることができる[Bianchi 等., Curr. Genet., 12:185(1987)]。より最近では、組換え子ウシのキモシンの大量生産のための発現系がK.ラクティス(lactis)に対して報告されている[Van den Berg, Bio/Technology, 8:135 (1990)]。クルイヴェロマイシスの工業的な菌株からの、組換えによる成熟したヒト血清アルブミンを分泌する安定した複数コピー発現ベクターも開示されている[Fleer 等, Bio/Technology,9:968-975 (1991)]。
【0037】
(iv)プロモーター成分
発現及びクローニングベクターは、通常、宿主生物によって認識され、Apo-2リガンド核酸配列に作用可能に結合したプロモーターを含む。プロモーターは、作用可能に結合したApo-2リガンド核酸配列のような特定の核酸配列の転写及び翻訳を制御する構造的な遺伝子(一般的に約100ないし1000塩基対)の開始コドンの上流側(5')に位置する未翻訳配列である。このようなプロモーターは典型的には、誘発的なクラス及び構成的なクラスの2つのクラスに属する。誘発的なプロモーターは、養分の存在あるいは不存在、温度変化等の培養条件のある変化に対応してその制御の下でDNAからの転写レベルを上昇させるプロモーターである。現時点において多種の可能な宿主細胞により認識される非常に多くのプロモーターがよく知られている。これらのプロモーターは、制限酵素の消化によって供給源DNAからプロモーターを除去し、ベクターに単離したプロモーター配列を挿入することで、Apo-2リガンドをコードするDNAに作用的に結合している。天然のApo-2リガンドプロモーター配列及び多くの異種性プロモーターはいずれもApo-2リガンドDNAの直接増幅及び/又は発現に用いることができる。
【0038】
原核生物宿主での使用に好適なプロモーターはβラクタマーゼ及びラクトースプロモーター系[Cahng 等, Nature, 275:615 (1978), Goeddel 等, Nature, 281:544 (1979)]、アルカリホスファターゼ、トリプトファン(trp)プロモーター系[Goeddel, Nucleic Acids Res., 8:4057 (1980); EP 36,776]、及びハイブリッドプロモーター、例えばtacプロモーター[deBoer 等, Proc. Natl. Acad. Sci. USA, 80:21-25 (1983)]を含む。しかし、他の既知の細菌プロモーターも好適である。これらのヌクレオチド配列は公表されており、よって当業者は、任意の所望の制限部位を提供するためにリンカーあるいはアダプターを使用することでApo-2をコードするDNA[Siebenlist 等, Cell, 20:269 (1980)]にそれらを作用可能に結合させることが可能である。細菌系で使用するプロモータもまたApo-2リガンドをコードするDNAと作用可能に結合したシャイン・ダルガーノ(S.D.)配列を有する。
真核生物に対してもプロモーター配列が知られている。実質的に全ての真核生物の遺伝子は、転写開始部位からおよそ25ないし30塩基上流に見出されるATリッチ領域を有する。多数の遺伝子の転写開始位置から70ないし80塩基上流に見出される他の配列は、Xが任意のヌクレオチドであるCXCAAT領域である。大部分の真核生物遺伝子の3'末端には、コード配列の3'末端へのポリA尾部の付加に対するシグナルであるAATAAA配列がある。これらの配列は全て真核生物の発現ベクターに適切に挿入される。
【0039】
酵母宿主と共に用いて好適なプロモーター配列の例としては、3-ホスホグリセラートキナーゼ[Hitzeman 等, J. Biol. Chem., 255:2073 (1980)]又は他の糖分解酵素[Hess 等, J. Adv. Enzyme Reg., 7:149 (1968);Holland, Biochemistry, 17:4900(1987)]、例えばエノラーゼ、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ、デヒドロゲナーゼ、ヘキソキナーゼ、ピルビン酸デカルボキシラーゼ、ホスホフルクトキナーゼ、グルコース-6-リン酸イソメラーゼ、3-ホスホグリセレートムターゼ、ピルビン酸キナーゼ、トリオセリン酸イソメラーゼ、ホスホグルコースイソメラーゼ、及びグルコキナーゼが含まれる。
他の酵母プロモーターとしては、成長条件によって転写が制御される付加的効果を有する誘発的プロモーターであり、アルコールデヒドロゲナーゼ2、イソチトクロムC、酸ホスファターゼ、窒素代謝と関連する分解性酵素、メタロチオネイン、グリセルアルデヒド-3-リン酸デヒドロゲナーゼ、及びマルトース及びガラクトースの利用を支配する酵素のプロモーター領域がある。酵母の発現に好適に用いられるベクターとプロモータはEP73,657に更に記載されている。また酵母エンハンサーも酵母プロモーターと共に好適に用いられる。
【0040】
哺乳動物の宿主細胞におけるベクターからのApo-2リガンド転写は、例えば、ポリオーマウィルス、伝染性上皮腫ウィルス(1989年7月5日公開のUK 2,211,504)、アデノウィルス(例えばアデノウィルス2)、ウシ乳頭腫ウィルス、トリ肉腫ウィルス、サイトメガロウィルス、レトロウィルス、B型肝炎ウィルス及び最も好ましくはサルウィルス40(SV40)のようなウィルスのゲノムから得られるプロモーター、異種性哺乳動物プロモーター、例えばアクチンプロモーター又は免疫グロブリンプロモーター、熱衝撃プロモーター、そしてApo-2配列に通常付随するプロモーターによって、このようなプロモーターが宿主細胞系に適合し得る限り、調節される。
SV40ウィルスの初期及び後期プロモーターは、SV40ウイルスの複製起点をさらに含むSV40制限断片として簡便に得られる[Fiers等, Nature, 273:113 (1978);Mulligan及びBerg, Science, 209:1422-1427 (1980); Pavlakis等, Proc. Natl. Acad. Sci. USA, 78: 7398-7402 (1981)]。ヒトサイトメガロウィルスの最初期プロモーターは、HindIIIE制限断片として簡便に得られる[Greenaway等, Gene, 18:355-360 (1982)]。ベクターとしてウシ乳頭腫ウィルスを用いて哺乳動物宿主でDNAを発現する系が、米国特許第4,419,446号に開示されている。この系の修飾は米国特許第4,601,978号に開示されている[また、サル細胞での免疫インターフェロンをコードしているcDNAの発現について、Gray等, Nature, 295:503-508(1982)を;単純ヘルペスウイルス由来のチミジンキナーゼプロモーターの調節下でのマウス細胞におけるヒトβインターフェロンcDNAの発現について、Reyes等, Nature, 297:598-601(1982)を;培養されたマウス及びウサギの細胞におけるヒトインターフェロンβ1遺伝子の発現について、Canaani及びBerg, Proc. Natl. Acad. Sci. USA, 79:5166-5170(1982)を;プロモーターとしてラウス肉腫ウィルスの長い末端反復を用いたCV-1サル腎臓細胞、ニワトリ胚線維芽細胞、チャイニーズハムスター卵巣細胞、HeLa細胞、及びマウスNIH-3T3細胞における細菌CAT配列の発現について、Gorma等, Proc. Natl. Acas. Sci. USA, 79:6777-6781 (1982)を参照のこと]。
【0041】
(v)エンハンサーエレメント成分
より高等の真核生物による本発明のApo-2リガンドをコードしているDNAの転写は、ベクター中にエンハンサー配列を挿入することによって増強され得る。エンハンサーは、通常は約10から300塩基対で、プロモーターに作用してその転写を増強するDNAのシス作動要素である。エンハンサーは、相対的に配向及び位置が独立しており、転写ユニットの5'[Laimins等, Proc. Natl. Acad. Sci. USA, 78:993 (1981)]及び 3'[Lusky等, Mol. Cell Bio., 3:1108 (1983)]、イントロン内部[Banerji等, Cell, 33:729 (1983)]並びにコード配列自身の内部[Osborne等, Mol. Cell Bio., 4:1293 (1984)]に見出されている。哺乳動物の遺伝子由来の多くのエンハンサー配列が現在知られている(グロビン、エラスターゼ、アルブミン、α-フェトプロテイン及びインスリン)。しかしながら、典型的には、真核細胞ウィルス由来のエンハンサーが用いられるであろう。例としては、複製起点の後期側のSV40エンハンサー(100-270塩基対)、サイトメガロウィルス初期プロモーターエンハンサー、複製起点の後期側のポリオーマエンハンサー及びアデノウィルスエンハンサーが含まれる。真核生物のプロモーターの活性化のための増強要素については、Yaniv, Nature, 297:17-18 (1982)もまた参照のこと。エンハンサーは、Apo-2リガンドコード配列の5'又は3'位でベクター中にスプライシングされ得るが、好ましくはプロモーターから5'位に位置している。
【0042】
(vi)転写終結成分
真核生物宿主細胞(酵母、真菌、昆虫、植物、動物、ヒト、又は他の多細胞生物由来の有核細胞)に用いられる発現ベクターは、また転写の終結及びmRNAの安定化に必要な配列を含む。このような配列は、真核生物又はウィルスのDNA又はcDNAの5'、時には3'の非翻訳領域から一般に取得できる。これらの領域は、Apo-2リガンドをコードしているmRNAの非翻訳部分にポリアデニル化断片として転写されるヌクレオチドセグメントを含む。
【0043】
(vii)ベクターの作成と分析
一又は複数の上に列挙した成分を含む適切なベクターの作成には標準的なライゲーション技術を用いる。分離されたプラスミド又はDNA断片を開裂させ、整え、そして必要とされるプラスミドの生成のために望ましい型に再ライゲーションする。
作成されたプラスミドが正しい配列であることを確認する分析のために、ライゲーション混合物を用いて、大腸菌K12菌株294(ATCC 31446)を形質転換し、適切な場合にはアンピシリン又はテトラサイクリン耐性によって、形質転換細胞を好適に選択する。形質転換細胞からプラスミドを調製し、制限エンドヌクレアーゼ消化により分析し、及び/又はMessing等, Nucleic Acids Res., 9:309 (1981)の方法又はMaxam等, Methods in Enzymology, 65:499 (1980)の方法によって配列決定する。
【0044】
(viii)一過性発現ベクター
Apo-2リガンドをコードしているDNAの哺乳動物細胞における一過性発現をもたらす発現ベクターを使用することができる。一般に、一過性発現は、宿主細胞が発現ベクターの多くのコピーを蓄積し、次にその発現ベクターによってコードされている所望のポリペプチドを高レベルで合成するように、宿主細胞中で効果的に複製できる発現ベクターを使用することを含む[Sambrook等, 上掲]。一過性発現系は、適切な発現ベクターと宿主細胞を含むが、クローニングされたDNAによりコードされているポリペプチドの簡便で確実な同定並びに所望の生物学的又は生理学的性質についてのポリペプチドの迅速なスクリーニングを可能にする。したがって、一過性発現系は、本発明において、生物学的に活性なApo-2リガンドであるApo-2リガンドの類似物及び変異体を同定する目的のために特に有用である。
【0045】
(ix)適切な例示的脊椎動物細胞ベクター
組換え脊椎動物細胞培養でのApo-2リガンドの合成に適応化するのに適切な他の方法、ベクター及び宿主細胞は、Gething等, Nature, 293:620-625 (1981); Mantei等, Nature, 281:40-46 (1979); EP117,060; 及びEP117,058に記載されている。Apo-2リガンドの哺乳動物細胞培地での発現に特に適したプラスミドは、pRK5[EP307,247;実施例1にも記載]又はpSVI6B[1991年6月13日に発行のWO 91/08291]である。
【0046】
3. 宿主細胞の選択及び形質転換
ここに記載のベクターにDNAをクローニングあるいは発現するために適切な宿主細胞は、上述の原核生物、酵母、又は高等真核生物細胞である。この目的にとって適切な原核生物は、限定するものではないが、真正細菌、例えばグラム陰性又はグラム陽性生物体、例えばエシェリチアのような腸内菌科、例えば大腸菌、エンテロバクター、エルウィニア(Erwinia)、クレブシエラ、プロテウス、サルモネラ、例えばネズミチフス菌、セラチア属、例えばセラチア・マルセスキャンス及び赤痢菌属、並びに桿菌、例えば枯草菌及びバシリ・リチェフォルミス(licheniformis)(例えば、1989年4月12日に公開されたDD266,710に開示されたバシリ・リチェニフォルミス41P)、シュードモナス属、例えば緑膿菌及びストレプトマイセス属を含む。好ましくは、宿主細胞は最小量のタンパク質分解酵素を分泌すべきである。
原核生物に加えて、糸状菌又は酵母菌のような真核微生物は、Apo-2リガンドをコードするベクターのための適切なクローニング又は発現宿主である。サッカロミセス・セレヴィシア、又は一般的なパン酵母は下等真核生物宿主微生物のなかで最も一般的に用いられる。しかしながら、多数の他の属、種及び菌株も、一般的に入手可能で有用である。
【0047】
グリコシル化Apo-2リガンドの発現に適切な宿主細胞は、多細胞生物から誘導される。このような宿主細胞は、複雑なプロセシング及びグリコシル化活動が可能である。原則的には、脊椎動物であろうと無脊椎動物培養であろうと、任意のより高等の真核生物細胞培養が使用できる。無脊椎動物細胞の例としては植物及び昆虫細胞が含まれる。多数のバキュロウィルス株及び変異体及び対応する許容可能な昆虫宿主細胞、例えばスポドプテラ・フルギペルダ(毛虫)、アエデス・アエジプティ(蚊)、アエデス・アルボピクトゥス(蚊)、ドゥロソフィラ・メラノガスター(ショウジョウバエ)、及びボンビクス・モリが同定されている[例えば、Luckow等, Bio/Technology, 6:47-55 (1988); Miller等, Genetic Engineering, Setlow等, eds., Vol. 8 (Plenum Publishing, 1986), pp.277-279; 及びMaeda等, Nature, 315:592-594 (1985)を参照のこと]。トランスフェクションのための種々のウィルス株、例えば、オートグラファ・カリフォルニカNPVのL-1変異体とボンビクス・モリ NPVのBm-5株が公に利用でき、それらのウイルスは、特に実施例2に記載するSpodopterafugiperda(「Sf9」)の形質移入のために、本発明に従うウイルスとして使用できる。
【0048】
綿花、コーン、ジャガイモ、大豆、ペチュニア、トマト、及びタバコのような植物細胞培養を宿主として利用することができる。典型的には、植物細胞は、細菌アグロバクテリウム・トゥメファシエンスの、既にApo-2リガンドコード化DNAを含むように操作された或る菌株と共にインキュベートすることによってトランスフェクトされる。A. トゥメファシエンスと共に植物細胞培養をインキュベートする間に、Apo-2リガンドをコードするDNAが、植物細胞宿主にそれが形質移入されるよう移され、そして適切な条件下でApo-2リガンドをコードするDNAが発現される。加えて、例えば、ノパリンシンターゼプロモーター及びポリアデニル化シグナル配列のような、植物細胞と適合しうる調節及びシグナル配列が利用できる[Depicker等, J. Mol. Appl. Gen., 1:561 (1982)]。また、T-DNA780遺伝子の上流領域から分離されるDNAセグメントは、組換えDNAを含む植物組織中の植物発現遺伝子の転写レベルを活性化又は増強しうる[1989年6月21日公開のEP 321,196]。
【0049】
培地(組織培地)中での脊椎動物細胞の増殖は、当該分野おいてよく知られている[例えば、Tissue Culture,Academic Press,編者Kruse及びPatterson(1973)を参照のこと]。有用な哺乳動物宿主株化細胞の例は、SV40によって形質転換されたサル腎臓CV1株 (COS-7, ATCC CRL 1651);ヒト胚腎臓株[293又は懸濁培養での増殖のためにサブクローン化された293細胞、Graham等, J. Gen Virol., 36:59 (1977)];ハムスター乳児腎細胞(BHK, ATCC CCL 10);チャイニーズハムスター卵巣細胞/-DHFR(CHO, Urlaub及びChasin, Proc. Natl. Acad. Sci. USA, 77:4216 (1980));マウスのセルトリ細胞[TM4, Mather, Biol. Reprod., 23:243-251 (1980)];サルの腎細胞 (CVI ATCC CCL 70); アフリカミドリザルの腎細胞(VERO-76, ATCC CRL-1587); ヒト子宮頸癌細胞 (HELA, ATCC CCL 2); イヌ腎細胞 (MDCK, ATCC CCL 34); バッファローラット肝細胞 (BRL 3A, ATCC CRL 1442); ヒト肺細胞 (W138, ATCC CCL 75); ヒト肝細胞 (Hep G2, HB 8065); マウス乳房腫瘍細胞 (MMT 060562, ATTC CCL51);TRI細胞[Mother等, Annals N.Y. Acad. Sci., 383:44-68 (1982)];MRC5細胞;及びFS4細胞である。
【0050】
宿主細胞をトランスフェクトし、好ましくは上述のApo-2リガンド産生のための発現又はクローニングベクターで形質転換し、プロモーターを誘導し、形質転換体を選択し、又は所望の配列をコードしている遺伝子を増幅するために適切に修飾された標準栄養培地で培養する。
形質移入は、如何なるコード配列が実際に発現されるか否かにかかわらず、宿主細胞による発現ベクターの取り込みを意味する。多数の形質移入の方法が当業者に知られている。例えば、CaPO4及びエレクトロポレーションである。このベクターの操作のあらゆる徴候が宿主細胞内で生じたときに成功した形質移入が一般に認められる。
形質転換は、染色体外の成分としてであろうと染色体成分によってであろうと、DNAが複製可能であるように生物体中にDNAを導入することを意味する。用いられる宿主細胞に応じて、そのような細胞に対して適した標準的な方法を用いて形質転換はなされる。前掲のSambrook等により記載された塩化カルシウムを用いるカルシウム処理又はエレクトロポレーションが、原核生物又は実質的な細胞壁障壁を含む他の細胞に対して用いられる。アグロバクテリウム・トゥメファシエンスによる感染が、Shaw等, Gene, 23:315 (1983)及び1989年6月29日公開の国際特許出願第WO 89/05859に記載されたように、ある種の植物細胞の形質転換に用いられる。加えて、1991年1月10日に公開された国際特許出願第WO 91/00358に記載されているように、超音波処理を用いて植物を形質移入することもできる。
【0051】
このような細胞壁のない哺乳動物細胞に対しては、Graham及びvan der Eb, Virology, 52:456-457 (1978)のリン酸カルシウム沈殿法が好ましい。哺乳動物細胞の宿主系形質転換の一般的な側面は米国特許第4,399,216号に記載されている。酵母中の形質転換は、典型的には、Van solingen等, J. Bact., 130:946 (1977)及びHsiao等, Proc. Natl. Acad. Sci. USA, 76:3829 (1979)の方法によって実施する。しかしながら、DNAを細胞中に導入する他の方法、例えば、核マイクロインジェクション、エレクトロポレーション、無傷の細胞、又はポリカチオン、例えばポリブレン、ポリオルニチン等を用いる細菌プロトプラスト融合もまた用いることもできる。哺乳動物細胞を形質転換するための種々の技術については、Keown等, Methods in Enzymology, 185:527-537 (1990)及び Mansour等, Nature, 336:348-352 (1988)を参照のこと。
【0052】
4.宿主細胞の培養
本発明のApo-2リガンドポリペプチドを生成るために用いられる原核細胞は、前掲のSambrook等により記載されているような適切な培地で培養される。
Apo-2リガンドの産生に用いられる哺乳動物の宿主細胞は種々の培地において培養することができる。市販培地の例としては、ハム(Ham)のF10(シグマ)、最小必須培地(「MEM」,シグマ)、RPMI-1640(シグマ)及びダルベッコの改良イーグル培地(「DMEM」,シグマ)が含まれる。これらの培地はいずれも、ホルモン及び/又は他の成長因子(例えばインスリン、トランスフェリン、又は表皮成長因子)、塩類(例えば、塩化ナトリウム、カルシウム、マグネシウム及びリン酸塩)、バッファー(例えばHEPES)、ヌクレオシド(例えばアデノシン及びチミジン)、抗生物質(例えば、ゲンタマイシンTM薬)、微量元素(最終濃度がマイクロモル範囲で通常存在する無機化合物として定義される)及びグルコース又は同等のエネルギー源を必要に応じて補充することができる。任意の他の必要な補充物質もまた当業者に知られている適当な濃度で含むことができる。培養条件、例えば温度、pH等々は、発現のために選ばれた宿主細胞について以前から用いられているものであり、当業者には明らかであろう。
一般に、哺乳動物の細胞培養の生産性を最大にするための原理、プロトコール、及び実用技術は、Mammalian Cell Biotechnology: a Practical Approach, M.Butler編 (IRL Press, 1991)に見出すことができる。
この明細書において言及される宿主細胞は培養中の細胞並びに宿主動物内にある細胞を包含する。
【0053】
5.遺伝子増幅/発現の検出
遺伝子の増幅及び/又は発現は、ここで提供された配列に基づき、適切に標識されたプローブを用い、例えば、従来よりのサザンブロット法、mRNAの転写を定量化するノーザンブロット法[Thomas,Proc. Natl. Acad. Sci. USA,77:5201-5205 (1980)]、ドットブロット法(DNA分析)、又はインサイツハイブリッド形成法によって、直接的に試料中で測定することができる。種々の標識を用いることができ、最も一般的なものは放射性同位元素、特に32Pである。しかしながら、他の方法、例えばポリヌクレオチド中への導入のためのビオチン修飾されたヌクレオチドもまた使用することができる。ついで、このビオチンは、例えば放射性ヌクレオチド、蛍光剤又は酵素のような広範囲の標識で標識することができるアビジン又は抗体への結合部位として作用する。また、DNA二本鎖、RNA二本鎖及びDNA−RNAハイブリッド二本鎖又はDNA-タンパク質二本鎖を含む、特異的二本鎖を認識することができる抗体を用いることもできる。ついで、抗体を標識し、アッセイを実施することができ、ここで二本鎖は表面に結合しており、その結果二本鎖の表面での形成の時点でその二本鎖に結合した抗体の存在を検出することができる。
あるいは、遺伝子の発現は、遺伝子産物の発現を直接的に定量する免疫学的な方法、例えば細胞又は組織切片の免疫組織化学的染色及び細胞培養又は体液のアッセイによって、測定することもできる。免疫組織化学的染色技術では、細胞試料を、典型的には脱水と固定によって調製し、結合した遺伝子産物に対し特異的な標識化抗体と反応させるが、この標識は通常は視覚的に検出可能であり、例えば酵素的標識、蛍光標識、又はルミネサンス標識である。
試料液の免疫組織化学的染色及び/又はアッセイに有用な抗体は、モノクローナルでもポリクローナルでもよく、任意の哺乳動物で調製することができる。簡便には、抗体は、天然配列Apo-2リガンドポリペプチドに対して、又はここで提供されるDNA配列をベースとした合成ペプチドに対して、又はApo-2リガンドDNAに融合し特異的抗体エピトープをコードする外因性配列に対して調製され得る。
【0054】
6.Apo-2リガンドポリペプチドの精製
Apo-2リガンドは、培養培地から分泌されたポリペプチドとして回収されるが、分泌シグナル無しで直接産生される場合は宿主細胞溶解液から回収してもよい。Apo-2リガンドが膜結合性であるならば、適切な洗浄液(例えばトリトン-X100)を用いて膜から引き離すか、又は酵素的切断によりその細胞外ドメインを放出させる。
Apo-2リガンドがヒト起源のもの以外の組換え細胞でつくられるときは、Apo-2リガンドはヒト起源のタンパク質又はポリペプチドを含んでいない。しかしながら、Apo-2リガンドに関して実質的に相同である調製物を得るには、組換え細胞タンパク又はポリペプチドからApo-2リガンドを精製することが望ましい。第一段階として、培地又は溶菌液を遠心分離して粒状の細胞屑を除去することができる。ついで、Apo-2リガンドを、汚染した可溶性タンパク質及びポリペプチドから、適切な精製手順の例である次の手順により精製される:すなわち、イオン交換カラムでの分画;エタノール沈殿;逆相HPLC;シリカ又はカチオン交換樹脂、例えばDEAEによるクロマトグラフィー;クロマトフォーカシング;SDS-PAGE;硫酸アンモニウム沈殿;例えばセファデックスG-75を用いるゲル濾過;及びIgGのような汚染物を除くプロテインAセファロースカラムである。
好ましい実施態様では、Apo-2リガンドは実施例3に記載するようなアフィニティクロマトグラフィによって精製される。
【0055】
残基が欠失され、挿入され、又は置換されたApo-2リガンド変異体は、その変異によってしばしば惹起される実質的な特性変化を考慮に入れて、天然Apo-2リガンドと同じようにして回収することができる。例えば、他のタンパク質又はポリペプチド、例えば細菌性もしくはウイルス性抗原とApo-2リガンドとの融合体の調製は精製を容易にする;配列に対する抗体を含む免疫アフィニティーカラムを、融合ポリペプチドを吸着するために使用することができる。好ましい実施態様では、Apo-2リガンドの細胞外配列をHis10ペプチドに融合させ、Ni2+キレートアフィニティクロマトグラフィにより精製する。
例えばフェニルメチルスルホニルフロリド(PMSF)のようなプロテアーゼインヒビターもまた精製の間のタンパク分解を阻害するのに有用であり、偶発的な汚染物質の成長を防止するために抗生物質を含めることができる。天然Apo-2リガンドに適切な精製方法は、組換え細胞培養の発現の際におけるApo-2リガンド又はその変異体の特性の変化の起因となる改変が必要となることは、当業者であれば理解するであろう。
【0056】
7.Apo-2リガンドポリペプチドの共有結合的修飾
Apo-2リガンドの共有結合的修飾は本発明の範囲内に含まれる。天然Apo-2リガンド及びApo-2リガンドのアミノ酸配列変異体の両方が共有結合的に修飾されうる。Apo-2リガンドの共有結合的修飾の一つの型は、Apo-2リガンドの標的化アミノ酸残基を、Apo-2リガンドのN末端又はC末端残基、又は選択された側鎖と反応できる有機誘導体形成剤と反応させることによって分子内に導入することができる。
二官能性試薬による誘導体形成は、抗Apo-2リガンド抗体を精製する方法に使用する水不溶性支持体マトリックス又は表面へのApo-2リガンドの架橋に有用であり、またその逆も同様である。通常使用される架橋剤には、例えば、1,1-ビス(ジアゾアセチル)-2-フェニルエタン、グルタルアルデヒド、N-ヒドロキシスクシンイミドエステル、例えば、4-アジドサリチル酸とのエステル、3,3'-ジチオビス(スクシンイミジルプロピオナート)のようなジスクシンイミジルエステルを包含するホモ二官能性イミドエステル、及びビス-N-マレイミド-1,8-オクタンのような二官能性マレイミドが含まれる。メチル-3-[(p-アジドフェニル)ジチオ]プロピオイミダートのような誘導体形成剤は、光の存在下で架橋を形成することができる光活性化中間体を生じる。また、臭化シアン活性化炭水化物のような反応性の水不溶性マトリックス及び米国特許第3,969,287号;3,691,016号;4,195,128号;4,247,642号;4,229,537号及び4,330,440号に記載されている反応物質がタンパク固定化に用いられる。
【0057】
その他の修飾は、それぞれ対応するグルタミル及びアスパルチル残基へのグルタミニル及びアスパラギニル残基の脱アミド化、プロリンとリジンのヒドロキシル化、セリル又はスレオニル残基のヒドロキシル基のリン酸化、リジン、アルギニン、及びヒスチジン側鎖のα-アミノ基のメチル化[T.E. Creighton, Proteins: Structure and Molecular Properties, W.H. Freeman & Co., San Francisco, PP.79-86 (1983)]、N末端アミンのアセチル化、及び任意のC末端カルボキシル基のアミド化を含む。残基の修飾型も本発明の範囲に入る。
本発明の範囲内に含まれるApo-2リガンドポリペプチドの共有結合的修飾の他の型は、ポリペプチドの天然グリコシル化パターンを変更することを含む。「天然グリコシル化パターンの変更」とは、天然Apo-2リガンドに見出される一又は複数の炭水化物部分の欠失、及び/又は天然Apo-2リガンドに存在しない一又は複数のグリコシル化部位を付加することを意味することをここでは意図している。
【0058】
ポリペプチドのグリコシル化は、典型的には、N結合又はO結合の何れかである。N結合とは、アスパラギン残基の側鎖への炭水化物部分の結合を指す。アスパラギン-X-セリン及びアスパラギン-X-スレオニン(ここでXはプロリンを除く任意のアミノ酸)というトリペプチド配列は、アスパラギン側鎖への炭水化物部分の酵素的結合のための認識配列である。したがって、ポリペプチド中にこれらのトリペプチド配列の何れかが存在すると、可能性のあるグリコシル化部位が作り出される。O結合グリコシル化は、ヒドロキシルアミノ酸、最も一般的にはセリン又はスレオニン(5-ヒドロキシプロリン又は5-ヒドロキシリジンもまた用いられるが)に、糖類N-アセチルガラクトサミン、ガラクトース、又はキシロースの一つが結合することを意味する。
Apo-2リガンドポリペプチドへのグリコシル化部位の付加は、アミノ酸配列を、それが一又は複数の上述したトリペプチド配列(N結合グリコシル化部位のもの)を含むように変化させることによって達成される。この変化は、天然Apo-2リガンドへの一又は複数のセリン又はスレオニン残基の付加、又はこれによる置換によってもなされる(O結合グリコシル化部位の場合)。場合によっては、Apo-2リガンドアミノ酸配列はDNAレベルでの変化によって、特に、所望のアミノ酸に翻訳するコドンが産生されるように予め選んだ塩基でApo-2リガンドポリペプチドをコードしているDNAを突然変異させることによって変更される。このDNA突然変異は、上記に記載され前掲の米国特許第5,364,934号に記載された方法を用いてなされる。
【0059】
Apo-2リガンドポリペプチド上の炭水化物部分の数を増加させる他の手段は、該ポリペプチドへのグリコシドの化学的又は酵素的結合による。用いられる結合形態に応じて、糖(類)は、(a)アルギニンとヒスチジンに、(b)遊離のカルボキシル基に、(c)遊離のスルフヒドリル基、例えばシステインのものに、(d)セリン、スレオニン又はヒドロキシプロリンのもののような遊離のヒドロキシル基に、(e)フェニルアラニン、チロシン又はトリプトファンのような芳香族残基、又は(f)グルタミンのアミノ基に結合される。これらの方法は1987年9月11日公開の国際特許出願第WO 87/05330号及びAplin及びWriston, CRC Crit. Rev. Biochem., pp259-306 (1981)に記載されている。
Apo-2リガンドポリペプチド上に存在する炭水化物部分の除去は、化学的又は酵素的になされる。例えば、化学的脱グリコシル化は、化合物トリフルオロメタンスルホン酸、又は等価な化合物へ該ポリペプチドを曝露し、該ポリペプチドを無傷のまま残しながら、結合糖(N-アセチルグルコサミン又はN-アセチルガラクトサミン)を除く殆ど又は全ての糖を開裂させる。化学的脱グリコシル化は、Hakimuddin等, Arch. Biochem Biophys., 259:52 (1987)及びEdge等, Anal. Biochem., 118:131 (1981)により記載されている。ポリペプチド上の炭水化物部分の酵素的開裂は、Thotakura等, Meth. Enzymol., 138:350 (1987)に記載されているように、種々のエンド及びエキソグリコシダーゼを使用して達成することができる。
【0060】
潜在的なグリコシル化部位でのグリコシル化は、Duskin等, J. Biol. Chem., 257:3105 (1982)に記載されているように、化合物ツニカマイシンを使用して防止することができる。ツニカマイシンはタンパク質-N-グルコシド結合の形成を阻害する。
Apo-2リガンドの共有結合的修飾の他の型は、Apo-2リガンドポリペプチドの種々の非タンパク質性ポリマー、例えばポリエチレングリコール、ポリプロピレングリコール、又はポリオキシアルキレンの1つに結合させることを含み、これは例えば米国特許第4,640,835号;第4,496,689号;第4,301,144号;第4,670,417号;第4,791,192号又は第4,179,337号に記載されているように行われる。
【0061】
8.エピトープタグApo-2リガンド
また本発明は、他の異種性ポリペプチドと融合したApo-2リガンドを含むキメラポリペプチドを提供する。一実施態様では、キメラポリペプチドは、抗タグ抗体が選択的に結合するエピトープを提供するタグポリペプチドとのApo-2リガンドの融合体を含む。エピトープタグは一般にApo-2リガンドのアミノ又はカルボキシル末端に位置する。Apo-2リガンドのこのようなエピトープタグが付けられた形の存在は、タグポリペプチドに対する抗体を用いて検出することができる。また、エピトープタグを供給すると、Apo-2リガンドを抗タグ抗体又はエピトープタグに結合する他の種類のアフィニティーマトリックスを用いたアフィニティー精製によって容易に精製することが可能になる。
【0062】
様々なタグポリペプチドとその各々の抗体はこの分野で良く知られている。例としては、fluHAタグポリペプチドとその抗体12CA5[Field等, Mol. Cell. Biol., 8:2159-2165 (1988)];c-mycタグとそれに対する8F9、3C7、6E10、G4、B7及び9E10抗体[Evan等, Molecular and Cellular Biology, 5:3610-3616 (1985)];及び単純ヘルペスウィルス糖タンパクD(gD)タグとその抗体[Paborsky等, Protein Engineering, 3(6):547-553 (1990)]が含まれる。他のタグポリペプチドには、フラッグ-ペプチド(Flag-peptide)[Hopp等, BioTechnology, 6:1204-1210 (1988)];KT3エピトープペプチド[Martin等, Science, 255:192-194 (1992)];α-チューブリンエピトープペプチド[Skinner等, J. Biol. Chem., 266:15163-15166 (1991)];及びT7遺伝子10タンパクペプチドタグ[Lutz-Freyermuth等, Proc. Natl. Acad. Sci. USA, 87:6393-6397 (1990)]が含まれる。ひとたびタグポリペプチドが選択されれば、それに対する抗体を、ここに開示した方法を用いて産生することができる。
【0063】
一般的に、エピトープタグApo-2リガンドは、天然又は変異体Apo-2リガンドに対して上述した方法に従い作成され生産される。Apo-2リガンド-タグポリペプチド融合体は、Apo-2リガンド部分をインフレームでコードしているcDNA配列をタグポリペプチドDNA配列に融合させ、得られたDNA融合作成物を適当な宿主細胞に発現させることによって好ましく作成される。通常は、本発明のApo-2リガンド-タグポリペプチドキメラを調製するときは、Apo-2リガンドをコードしている核酸を、タグポリペプチドのN末端をコードしている核酸にその3'末端で融合させるが、5'融合もまた可能である。エピトープタグApo-2リガンドの例は、下記の実施例2にさらに詳細に記載する。
エピトープタグApo-2リガンドは、抗タグ抗体を用いてアフィニティークロマトグラフィーによって精製することができる。アフィニティー抗体が固着されるマトリクスには、例えばアガロース、調整穴明きガラス又はポリ(スチレンジビニル)ベンゼンが含まれる。ついで、エピトープタグApo-2リガンドは、当該分野において知られた技術を使用して、アフィニティーカラムから溶出することができる。
【0064】
B.Apo-2の治療的用途
本明細書に開示されているように、Apo-2リガンドは治療的に用いることができる。例えばApo-2リガンド哺乳動物癌細胞にアポトーシスを誘発するために用いることができる。一般的に、哺乳動物癌細胞においてアポトーシスを誘発する方法は、当該細胞を有効量のApo-2リガンドに暴露することを含む。これは、例えば下記の実施例に記載する方法に従って、インビボ又はエキソビボで行うことができる。アポトーシスを誘発する方法は、アポトーシスレベルの低下を特徴とする特定の病理学的状態の治療に用いることができる。このような病理学的状態の例は、自己免疫疾患、例えば狼瘡及び免疫媒介糸球体腎炎、及び癌を含む。Apo-2リガンドの癌の治療のための治療的用途を以下に詳細に説明する。
癌の治療方法において、Apo-2リガンドは癌を持つと診断された哺乳動物に投与される。言うまでもなく、Apo-2リガンドは、他のアポトーシス誘発剤、化学治療、放射線治療、及び手術を含むさらに他の治療的組成物及び技術と組み合わせて用いることもできる。
【0065】
Apo-2リガンドは、好ましくは担体中で哺乳動物に投与される。好適な担体とそれらの製剤は、Oslo等により編集されたRemington's Pharmaceutical Sciences, 16th ed., 1980, Mack Publishing Co.,に記載されている。典型的には、適量の製薬的に許容可能な塩が、製剤を等浸透圧にするために製剤において使用される。製薬的に許容可能な担体の例には、生理食塩水、リンガー液及びデキストロース液が含まれる。溶液のpHは、好ましくは約5〜約8、さらに好ましくは約7.4〜約7.8である。例えば投与経路及び投与されるApo-2リガンドの濃度よっては、或る種の担体がより好ましくなることは、当業者には明らかである。
Apo-2リガンドは、注射(例えば、静脈内、腹腔内、皮下、筋肉内)、又は点滴のように、有効な形態で血流への送達を確実にする他の方法により哺乳動物に投与することができる(実施例15参照)。また、Apo-2リガンドはインビボ又はエキソビボ遺伝子治療により投与できるとも考えられる。
【0066】
Apo-2リガンド投与の有効な用量とスケジュールは経験的に決定することができ、このような決定は当業者の技量の範囲に含まれる。今日では、単独で用いられるApo-2リガンドの有効用量又は量は、1日に体重1kg当たり約1μg〜約100mg又はそれ以上の範囲であると考えられている。用量の種内スケーリングは、この分野で周知の方法、例えばMordenti等, Pharmaceut. Res., 8 1351 (1991)に記載されているように実施することができる。当業者はApo-2リガンドの投与しなければならない用量が、例えばApo-2リガンドを受容する哺乳動物、投与経路、及び哺乳動物に施される他の医薬又は治療に応じて変化することを理解するであろう。
哺乳動物に施される一又は複数の他の治療には、限定されるものではないが、化学治療及び/又は放射線治療、イムノアジュバント、サイトカイン、及び抗体ベース療法が含まれる。例としては、インターロイキン(例えば、IL-1、IL-2、IL-3、IL-6)、白血病抑制因子、インターフェロン、TGF-β、エリスロポイエチン、トロンボポイエチン、抗-VEGF抗体及びHER-2抗体が含まれる。哺乳動物細胞でアポトーシスを誘発することが知られた他の薬剤を用いてもよく、そのような薬剤にはTNF-α、TNF-β(リンホトキシン-α)、CD30リガンド、4-1BBリガンド、及びApo-1リガンドが含まれる。
【0067】
本発明において考慮される化学治療薬には、当該分野において既知であって市販されている化学物質又は薬物、例えばドキソルビシン、5-フルオロウラシル(「5-FU」)、エトポシド、カンプトテシン、ロイコボリン、シトシンアラビノシド(「Ara-C」)、シクロホスファミド、チオテパ、ブスルファン、サイトキシン、タキソール、メトトレキセート、シスプラチン、メルファラン、ビンブラスチン及びカルボプラチンが含まれる。このような化学治療薬に対する調製法及び用量スケジュールは、製造者の指示に従って使用されるか、熟練した実務家により経験的に決定される。そのような化学治療薬に対する調製法及び用量スケジュールはまた化学療法サービス(Chemotherapy Cervice), M.C.Perry編, Williams & Wilkins, Baltimore, MD (1992)にも記載されている。
化学治療薬は、Apo-2リガンドについて上述したもののような、製薬的に許容可能な担体で好ましく投与される。化学治療薬の投与方法はApo-2リガンドについて用いられるのと同様としてもよく、あるいは異なる方法を介して投与してもよい。例えば、Apo-2リガンドを注射する一方、化学治療薬を経口で哺乳動物に投与してもよい。Apo-2リガンドと組み合わせた化学治療薬の投与方法は、下記の実施例9−12に更に詳細に記載する。
【0068】
放射線治療は、この分野で通常用いられ当業者に知られたプロトコールに従って哺乳動物に施すことができる。このような治療は、セシウム、イリジウム、ヨウ素、又はコバルト照射を含む。放射線治療は、全身照射でもよく、又は身体の特定部位又は組織に局所的に向けられてもよい。典型的には、放射線治療は約1〜約2週間の期間に渡ってパルス的に施される。しかしながら、放射線治療は、より長期間にわたって施してもよい。場合によっては、放射線治療は1回照射又は多数、連続照射として施してもよい。
Apo-2リガンド及び一又は複数の他の治療は、哺乳動物に同時に又は連続的に施すことができる。Apo-2リガンド及び一又は複数の他の治療の施与に続いて、哺乳動物の癌及び生理学的状態を、熟練した実務者に知られた種々の方法で監視することができる。例えば、腫瘍の量は、生検により、又は標準的なX線画像技術により物理的に観察できる。
【0069】
Apo-2リガンドがエキソビボでの癌細胞の治療に用いられると考えられる。このような秋曽比簿治療は、骨髄移植、及び一部の自己骨髄移植において有用である。例えば、癌細胞を持つ細胞又は組織の、場合によっては上記したような一又は複数の他の治療と共に行うApo-2リガンドでの治療は、アポトーシスを誘発し、受容者哺乳動物に移植する前に実質的に癌細胞を除去するために用いることができる。
癌細胞を含む細胞又は組織は、供与者哺乳動物において最初に観察される。細胞又は組織は手術により、好ましくは無菌で得られる。移植用骨髄の治療方法では、骨髄は哺乳動物から針吸引により得る。次いで、癌細胞を含む細胞又は組織をApo-2リガンドで、場合によっては上記のような一又は複数の他の治療と共に治療する。骨髄は、好ましくは、Apo-2リガンドでの処理に先立って、単細胞分画を得るために(フィコール‐ハイパーク勾配の遠心分離などで)分画する。
処理した細胞又は組織は、次いで、受容者哺乳動物に注入又は移植する。受容者哺乳動物は、供与者哺乳動物と同じ個体でもよく、他の異種哺乳動物でもよい。自己骨髄移植については、哺乳動物は移植の前に有効用量の当該分野で知られ、例えば、Autologous Bone Marrow Transplantation: Proceedings of the Third International Symposium, Dicke等, 編, University of Texas M.D. Anderson Hospital and Tumor Institute (1987)に記載された放射線又は化学治療で処理される。
【0070】
C.Apo-2リガンドの非治療的用途
本発明のApo-2リガンドは非治療的用途においても有用性を有している。RApo-2リガンドをコードする核酸配列を、組織特異性の型分類のための診断に使用することもできる。例えば、インサイツハイブリッド形成、ノーザン及びサザンブロット法、及びPCR分析のような手順を、Apo-2リガンドをコードするDNA及び/又はRNAが評価されている細胞型に存在しているか否かを決定するために使用することができる。Apo-2リガンド核酸は、ここに記載されている組換え技術によるApo-2リガンドポリペプチドの調製にもまた有用である。
単離されたApo-2リガンドは、定量的診断アッセイで対照として用いられ、それに対して未知量のApo-2リガンドを含有するサンプルが調製される。またApo-2リガンド調製物は、抗体を産生する際、(例えば、ラジオイムノアッセイ、ラジオレセプターアッセイ又は酵素結合免疫測定法における標準としての使用のためにApo-2リガンドを標識することにより)Apo-2リガンドに対するアッセイにおける標準として、アフィニティー精製技術において、及び例えば放射性ヨウ素、酵素又はフルオロフォアで標識された場合の競合型レセプター結合アッセイにおいて有用である。
【0071】
また、Apo-2リガンドをコードする核酸は、トランスジェニック動物又は「ノックアウト」動物を産生するのに使用でき、これらは治療的に有用な試薬の開発やスクリーニングに有用である。トランスジェニック動物(例えばマウス又はラット)とは、出生前、例えば胚段階で、その動物又はその動物の祖先に導入された導入遺伝子を含む細胞を有する動物である。導入遺伝子とは、トランスジェニック動物が発生する細胞のゲノムに組み込まれたDNAである。一実施形態では、Apo-2リガンドをコードするcDNA又はその適当な配列を、確立された技術に従ってApo-2リガンドをコードするゲノムDNAをクローン化するために使用することができ、ゲノム配列は、Apo-2リガンドをコードするDNAを発現する細胞を有するトランスジェニック動物を産生するために使用することができる。トランスジェニック動物、特にマウス又はラット等の動物を産生する方法は当該分野において常套的になっており、例えば米国特許第4,736,866号や第4,870,009号に記述されている。典型的には、特定の細胞を組織特異的エンハンサーでのApo-2リガンド導入遺伝子の導入の標的にする。胚段階で動物の生殖系列に導入されたApo-2リガンドをコードする導入遺伝子のコピーを含むトランスジェニック動物はApo-2リガンドをコードするDNAの増大した発現の影響を調べるために使用できる。
【0072】
あるいは、Apo-2リガンドの非ヒト相同体は、動物の胚性細胞に導入されたApo-2リガンドをコードする変更ゲノムDNAと、Apo-2リガンドをコードする内在性遺伝子との間の相同的組換えによって、Apo-2リガンドをコードする欠陥又は変更遺伝子を有するApo-2リガンド「ノックアウト」動物を作成するために使用できる。例えば、Apo-2リガンドをコードするcDNAは、確立された技術に従い、Apo-2リガンドをコードするゲノムDNAのクローニングに使用できる。Apo-2リガンドをコードするゲノムDNAの一部を欠失したり、組み込みを監視するために使用する選択可能なマーカーをコードする遺伝子等の他の遺伝子で置換することができる。典型的には、ベクターは無変化のフランキングDNA(5'と3'末端の両方)を数キロベース含む[例えば、相同的組換えベクターについてはThomas及びCapecchi, Cell, 51:503(1987)を参照のこと]。ベクターは胚性幹細胞に(例えばエレクトロポレーション等によって)導入し、導入されたDNAが内在性DNAと相同的に組換えられた細胞を選択する[例えば、Li等, Cell, 69:915(1992)参照]。選択された細胞は次に動物(例えばマウス又はラット)の胚盤胞内に注入され、集合キメラを形成する[例えば、Bradley, Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, E. J. Robertson, ed. (IRL, Oxford, 1987), pp. 113-152参照]。その後、キメラ性胚を適切な偽妊娠の雌性乳母に移植し、期間をおいて「ノックアウト」動物をつくり出す。胚細胞に相同的に組換えられたDNAを有する子孫は標準的な技術により同定され、それらを利用して動物の全細胞が相同的に組換えられたDNAを含む動物を繁殖させることができる。ノックアウト動物は、例えば腫瘍の発達を含む、Apo-2リガンドポリペプチドが不在であることによるある種の病理的状態及びその病理的状態の発達に対する防御能力によって特徴付けられる。
【0073】
D.抗Apo-2リガンド抗体の調製
本発明は、さらに抗Apo-2リガンド抗体を提供するものである。Apo-2リガンドに対する抗体は以下のようにして調製することができる。抗体の例としては、ポリクローナル、モノクローナル、ヒト化、二重特異性及びヘテロ抱合体抗体が含まれる。
1.ポリクローナル抗体
Apo-2リガンド抗体はポリクローナル抗体を含んでよい。ポリクローナル抗体の調製方法は当業者に知られている。哺乳動物においてポリクローナル抗体は、例えば免疫化剤、及び所望するのであればアジュバントを、一又は複数回注射することで発生させることができる。典型的には、免疫化剤及び/又はアジュバントを複数回皮下又は腹腔内注射により、哺乳動物に注射する。免疫化剤は、Apo-2リガンドポリペプチド又はその融合タンパク質を含みうる。免疫化剤を、免疫化される哺乳動物で免疫原性が知られたタンパク質に抱合させるのが有用である。使用され得るそのような免疫原性タンパク質の例は、限定するものではないが、キーホール・リンペット(keyhole limpet))ヘモシアニン、血清アルブミン、ウシサイログロブリン及び大豆トリプシンインヒビターが含まれる。また、哺乳動物の免疫反応を増強するために、ミョウバンのような凝集剤を使用してもよい。使用され得るアジュバントの例には、フロイント完全アジュバント及びMPL-TDMアジュバント(モノホスホリル脂質A、合成トレハロースジコリノミコラート)が含まれる。免疫化プロトコールは、過度の実験なく当業者により選択されるであろう。哺乳動物から採血し、血清を検定して抗体価を求める。望まれるならば、抗体価が増加又は平坦化するまで哺乳動物に追加免疫を施す。
【0074】
2.モノクローナル抗体
あるいは、Apo-2リガンド抗体はモノクローナル抗体であってもよい。モノクローナル抗体は、Kohler及びMilstein, Nature, 256: 495 (1975)に記載されているようなハイブリドーマ法を使用することで調製することができる。ハイブリドーマ法では、マウス、ハムスター又は他の適切な宿主動物を典型的には免疫化剤により免疫化する(上述したようにして)ことで、免疫化剤に特異的に結合する抗体を生成するかあるいは生成可能なリンパ球を誘発する。また、リンパ球をインビトロで免疫化することもできる。
免疫化剤は、典型的にはApo-2リガンドポリペプチド又はその融合タンパク質を含む。表面にApo-2リガンドを発現する細胞もまた使用することができる。一般にヒト由来の細胞が望まれる場合には末梢血リンパ球(「PBL」)が使用され、あるいは非ヒト哺乳動物源が望まれている場合は、脾臓細胞又はリンパ節細胞が使用される。次いで、ポリエチレングリコール等の適当な融合剤を用いてリンパ球を不死化株化細胞と融合させ、ハイブリドーマ細胞を形成する[Goding, Monoclonal Antibodies: Principles and Practice, Academic Press, (1986) pp. 59-103]。不死化株化細胞は、通常は、形質転換した哺乳動物細胞、特に齧歯動物、ウシ、及びヒト由来の骨髄腫細胞である。通常、ラット及びマウスの骨髄腫細胞が使用される。ハイブリドーマ細胞は、好ましくは、未融合の不死化細胞の生存又は成長を阻害する一又は複数の物質を含有する適切な培地で培養される。例えば、親の細胞が、酵素のヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRT又はHPRT)を欠いていると、ハイブリドーマの培地は、典型的には、ヒポキサチン、アミノプチリン及びチミジンを含み(「HAT培地」)、この物質がHGPRT欠乏性細胞の増殖を阻止する。
【0075】
好ましい不死化株化細胞は、効率的に融合し、選択された抗体生成細胞による安定した高レベルの抗体発現を支援し、HAT培地のような培地に対して感受性であるものが望ましい。より好ましい不死化株化細胞はマウス骨髄腫株であり、これはカリフォルニア州サンディエゴのSalk Institute Cell Distribution Centerやバージニア州マナッサスのアメリカン・タイプ・カルチャー・コレクションより入手可能である。ヒトモノクローナル抗体を生成するためのヒト骨髄腫及びマウス-ヒト異種骨髄腫株化細胞も開示されている[Kozbor, J. Immunol., 133:3001 (1984)、Brodeur等, Monoclonal Antibody Production Techniques and Applications, Marcel Dekker, Inc., New York, (1987) pp. 51-63]。
ハイブリドーマ細胞が培養される培地を、Apo-2リガンドに対するモノクローナル抗体の存在について検定する。好ましくは、ハイブリドーマ細胞によって生成されたモノクローナル抗体の結合特異性は免疫沈降又はラジオイムノアッセイ(RIA)、蛍光活性化セルソーター(FACS)又は酵素結合免疫測定法(ELISA)等のインビトロ結合検定法によって測定する。このような技術及びアッセイは、当該分野において公知であり、下記の実施例でさらに記載される。モノクローナル抗体の結合親和性は、例えばMunson及びPollard, Anal. Biochem., 107:220 (1980)によるスキャッチャード分析法によって測定することができる。
【0076】
所望のハイブリドーマ細胞が同定された後、クローンを制限希釈工程を経てサブクローニングし、標準的な方法で増殖させることができる[Goding, 上掲]。この目的のための適当な培地には、例えば、ダルベッコの改変イーグル培地及びRPMI-1640倍地が含まれる。あるいは、ハイブリドーマ細胞は哺乳動物においてインビボで腹水として成長させることもできる。
サブクローンによって分泌されるモノクローナル抗体は、例えばプロテインAセファロース法、ヒドロキシルアパタイトクロマトグラフィー法、ゲル電気泳動法、透析法又はアフィニティークロマトグラフィー等の従来の免疫グロブリン精製方法によって培地又は腹水液から分離又は精製される。
本発明の一実施態様では、モノクローナル抗体は、ここ及び下記の実施例に記載する1D1、2G6、2E11、又は5C2抗体を含んでよい。また、モノクローナル抗体は、各々アメリカン・タイプ・カルチャー・コレクションの登録番号ATCC HB-12256, HB-12257, HB-12258, 又はHB-12259で寄託されたハイブリドーマ細胞系によって分泌された1D1、2G6、2E11、又は5C2モノクローナル抗体と同じ生物学的特性を有する抗体も含む。「生物学的活性」という用語は、モノクローナル抗体のインビトロ及び/又はインビボ活性、例えば、Apo-2リガンド誘発アポトーシスを実質的に低減又は阻害する、又はApo-2リガンドのそのレセプターへの結合を低減又は阻止する能力を意味するのに用いられる。抗体は、ここに記載した1D1、2G6、2E11、又は5C2抗体と同じエピトープ、又は実質的に同じエピトープに結合するのが好ましい。
【0077】
また、モノクローナル抗体は、組換えDNA法、例えば米国特許第4,816,567号に記載された方法により作成することができる。本発明のモノクローナル抗体をコードするDNAは、常套的な方法によって(例えば、マウス抗体の重鎖及び軽鎖をコードする遺伝子に特異的に結合可能なオリゴヌクレオチドプローブを使用して)、容易に単離し配列決定することができる。本発明のハイブリドーマ細胞はそのようなDNAの好ましい供給源となる。ひとたび単離されたら、DNAは発現ベクター内に配することができ、これが宿主細胞、例えばサルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、あるいは免疫グロブリンタンパク質を生成しない骨髄腫細胞内に形質移入され、組換え宿主細胞内でモノクローナル抗体の合成をすることができる。また、DNAは、例えば相同マウス配列に換えてヒト重鎖及び軽鎖定常ドメインのコード配列を置換することにより[米国特許第4,816,567号;Morrison等, 上掲]、又は免疫グロブリンコード配列に非免疫グロブリンポリペプチドのコード配列の一部又は全部を共有結合することにより修飾することができる。このような非免疫グロブリンポリペプチドは、本発明の抗体の定常ドメインの代わりに置換するか、本発明の抗体の一つの抗原結合部位の可変ドメインの代わりに置換し、キメラ性二価抗体を産生することができる。
【0078】
本発明の抗体は一価抗体であってもよい。一価抗体の調製方法は当該分野においてよく知られてる。例えば、一つの方法は免疫グロブリン軽鎖と修飾重鎖の組換え発現を含む。重鎖は一般的に、重鎖の架橋を防止するようにFc領域の任意のポイントで切断される。あるいは、関連したシステイン残基を他のアミノ酸残基で置換するか欠失させて架橋を防止する。
一価抗体の調製にはインビトロ法がまた適している。抗体の消化による、その断片、特にFab断片の調製は、当該分野において知られている慣用的技術を使用して達成できる。例えば、消化はパパインの使用により行うことができる。パパイン消化の例は、12/22/94に公開された国際公開第WO 94/29348号、及び米国特許第4,342,566号に記載されている。抗体のパパイン消化は、典型的には、Fab断片と呼ばれ各々が単一の抗原結合部位を有する2つの同一の抗原結合断片と、残りのFc断片を生成する。ペプシン処理により、2つの抗原結合部位を有し抗原の架橋が尚も可能なF(ab')2断片が得られる。
また、抗体の消化により生産されたFab断片は、軽鎖の定常ドメインと重鎖の第1定常ドメイン(CH1)を含む。Fab’断片は、抗体のヒンジ領域から一又は複数のシステインを含む重鎖CH1ドメインのカルボキシ末端に幾つかの残基が付加されているということで、Fab断片とは異なっている。Fab’-SHとは、定常ドメインのシステイン残基が遊離のチオール基を担持しているFab’に対するここでの命名である。F(ab’)2抗体断片は、本来は、それらの間にヒンジシステインを有するFab’断片の対として生産された。抗体断片の他の化学的結合もまた知られている。
【0079】
3.ヒト化抗体
本発明のApo-2リガンド抗体は、さらにヒト化抗体又はヒト抗体を含む。非ヒト(例えばマウス)抗体のヒト化形とは、キメラ免疫グロブリン、免疫グロブリン鎖あるいはその断片(例えばFv、Fab、Fab'、F(ab')2あるいは抗体の他の抗原結合サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。ヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト種(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)を含む。ある場合には、ヒト免疫グロブリンのFvフレームワーク残基は、対応する非ヒト残基によって置換されている。更に、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでいてもよい。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリンコンセンサス配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域(Fc)、典型的にはヒトの免疫グロブリンの定常領域の少なくとも一部を含んでなる[Jones等, Nature, 321:522-525 (1986); Riechmann 等, Nature, 332:323-329 (1988); 及びPresta, Curr. Op Struct. Biol., 2:593-596 (1992)]。
【0080】
非ヒト抗体をヒト化する方法はこの分野で良く知られている。一般的に、ヒト化抗体には非ヒト由来の一又は複数のアミノ酸残基が導入されている。これら非ヒトアミノ酸残基は、しばしば、典型的には「移入」可変ドメインから得られる「移入」残基と称される。ヒト化は基本的にウィンター及び共同研究者[Jones等, Nature, 321:522-525 (1986)、Riechmann等, Nature, 332:323-327 (1988)、Verhoeyen等, Science, 239:1534-1536 (1988)]の方法、及びコンピュータモデルを用いたQueen等, Proc Natl. Acad. Sci., 86: 10029-10033 (1989)の方法に従って、齧歯動物のCDR又はCDR配列でヒト抗体の対応する配列を置換することにより実施される。よって、このような「ヒト化」抗体は、無傷のヒト可変ドメインより実質的に少ない分が非ヒト種由来の対応する配列で置換されたキメラ抗体である(米国特許第4,816,567号)。実際には、ヒト化抗体は典型的には幾つかのCDR残基及び場合によっては幾つかのFR残基が齧歯類抗体の類似部位からの残基によって置換されたヒト抗体である。
抗原性を軽減するには、ヒト化抗体を作成するために使用するヒトの軽鎖及び重鎖可変ドメインの両方の選択が非常に重要である。「ベストフィット法」に従うと、齧歯動物抗体の可変ドメインの配列を既知のヒト可変ドメイン配列ライブラリ全体に対してスクリーニングする。齧歯動物のものと最も近いヒトの配列を次にヒト化抗体のヒトフレームワーク(FR)として受け入れる[Sims等, J. Immunol., 151:2296 (1993);Chothia及びLesk, J. Mol. Biol., 196:901 (1987)]。他の方法では、軽鎖又は重鎖の特定のサブグループのヒト抗体全てのコンセンサス配列から誘導される特定のフレームワークを使用する。同じフレームワークを幾つかの異なるヒト化抗体に使用できる[Carter等, Proc. Natl. Acad. Sci. USA, 89:4285 (1992);Presta等, J. Immunol., 151:2623 (1993)]。
【0081】
さらに、抗体は、抗原に対する高親和性や他の好ましい生物学的性質を保持してヒト化することが重要である。この目標を達成するべく、好ましい方法では、親及びヒト化配列の三次元モデルを使用して、親配列及び様々な概念的ヒト化産物の分析工程を経てヒト化抗体を調製する。三次元免疫グロブリンモデルは一般的に入手可能であり、当業者にはよく知られている。選択された候補免疫グロブリン配列の推測三次元立体配座構造を図解し、表示するコンピュータプログラムは入手可能である。これら表示を見ることで、候補免疫グロブリン配列の機能における残基の役割の分析、すなわち候補免疫グログリンの抗原と結合する能力に影響を及ぼす残基の分析が可能となる。このようにして、例えば標的抗原に対する親和性を高めるといった、望ましい抗体特徴が得られるように、FR残基をコンセンサス及び移入配列から選択し、組み合わせることができる。一般的に、CDR残基は、直接かつ最も実質的に抗原結合性に影響を及ぼしている[1994年、3月3日に公開されたWO 94/04679を参照]。
免疫化することで、内在性免疫グロブリンが生成されない状態でもヒト抗体の完全リパートリを生成することができるトランスジェニック動物(例えばマウス)を使用することが可能である。例えば、キメラ及び生殖系列変異マウスにおいて抗体重鎖結合領域(JH)遺伝子をホモ接合的に欠失させると内在性抗体生産の完全な阻害が生じることが記述されている。このような生殖系列変異マウスにヒト生殖系列免疫グロブリン遺伝子配列を移すと、抗原投与時にヒト抗体の生成が生じる[例えば、Jakobovits等, Proc. Natl. Acad. Sci. USA, 90:2551-255 (1993);Jakobovits等, Nature, 362:255-258 (1993);Bruggerman等, Year in Immuno., 7:33 (1993)を参照]。また、ヒト抗体はファージ表示ライブラリ[Hoogenboom及びWinter, J. Mol. Biol., 227:381 (1992);Marks等, J. Mol. Biol., 222:581 (1991)]において作成することもできる。また、Cole等及びBoerner等の方法も、ヒトモノクローナル抗体の調製に利用することができる[Cole等, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss. p.77(1985)及びBoerner等, J. Immunol., 147(1):86-95(1991) ]。
【0082】
4.二重特異性抗体
二重特異性抗体は、少なくとも2つの異なる抗原に対して結合特異性を有するモノクローナル抗体、好ましくはヒトもしくはヒト化抗体である。本発明の場合において、結合特異性の一方はApo-2リガンドに対してであり、他方は任意の他の抗原、好ましくは細胞表面タンパク質又はレセプター又はレセプターサブユニットに対してである。
二重特異性抗体を生成する方法は当該技術分野において周知である。伝統的には、二重特異性抗体の組換え生成方法は、二つの重鎖が異なる特異性を持つ二つの免疫グロブリン重鎖/軽鎖対の同時発現に基づく[Milstein及びCuello, Nature, 305:537-539 (1983)]。免疫グロブリンの重鎖と軽鎖を無作為に取り揃えたため、これらハイブリドーマ(クアドローマ)は10種の異なる抗体分子の潜在的混合物を作成でき、その内一種のみが正しい二重特異性構造を有する。正しい分子の精製は、アフィニティークロマトグラフィー工程によって通常達成される。同様の手順が1993年5月13日公開の国際特許出願第 WO 93/08829号、及びTraunecke等, EMBO J.,10:3655-3656 (1991)に開示されている。
【0083】
別のより好ましいアプローチによれば、所望の結合特異性(抗体-抗原結合部位)を有する抗体可変ドメインを免疫グロブリン定常ドメイン配列に融合する。融合は、好ましくは少なくともヒンジ部、CH2及びCH3領域の一部を含む免疫グロブリン重鎖定常ドメインとのものである。少なくとも一つの融合には軽鎖結合に必要な部位を含む第一の重鎖定常領域(CH1)が存在することが望ましい。免疫グロブリン重鎖融合をコードするDNA、及び望むのであれば免疫グロブリン軽鎖を、別々の発現ベクターに挿入し、適当な宿主生物に同時形質移入する。これは、作成に使用する三つのポリペプチド鎖が不等の比であるときに最高の収率が得られる実施態様において、三つのポリペプチド断片の相互比率の調節に大なるフレキシビリティをもたらす。しかし、少なくとも二つのポリペプチド鎖が同比率で発現すると高収率が得られる場合や比率が特に重要ではない場合には、一つの発現ベクターに二つ又は三つ全てのポリペプチド鎖のコード配列を挿入することができる。このアプローチの好適な形態では、二重特異性抗体は、一方のアームの第一結合特異性を有するハイブリッド免疫グロブリン重鎖と、他方のアームのハイブリッド免疫グロブリン重鎖/軽鎖対(第二の結合特異性をもたらす)からなる。このような非対称的構造は、二重特異性分子の半分にのみ免疫グロブリン軽鎖が存在すると容易な方法で分解できるので、所望の二重特異性化合物を不要な免疫グロブリン鎖の組合わせから分解し易くすることが見出された。このアプローチは1994 3月3日公開の国際公開WO 94/04690号によって開示されている。二重特異性抗体を生成するための更なる詳細については、例えばSuresh等, Methods in Enzymology, 121:210(1986)を参照されたい。
【0084】
5.ヘテロ抱合体抗体
ヘテロ抱合抗体もまた本発明の範囲に入る。ヘテロ抱合抗体は、2つの共有的に結合した抗体からなる。このような抗体は、例えば、免疫系細胞を不要な細胞に対して標的化させるため[米国特許第4,676,980号]及びHIV感染の治療のために[WO 91/00360; WO 92/200373; EP 03089]提案された。本抗体は、架橋剤に関連したものを含む合成タンパク質化学における既知の方法を使用してインビトロで調製することができると考えられる。例えば、ジスルフィド交換反応を使用するか又はチオエーテル結合を形成することにより免疫毒素を作成することができる。この目的に対して好適な試薬の例には、イミノチオレート及びメチル-4-メルカプトブチリミデート、及び例えば米国特許第4,676,980号に開示されているものが含まれる。
【0085】
E.Apo-2リガンド抗体の用途
さらに、Apo-2リガンド抗体は、Apo-2リガンドの診断アッセイ法、例えば特異的細胞、組織又は血清におけるその発現の検出に使用することができる。当該分野において知られている様々な診断アッセイ技術、例えば、競合的結合アッセイ、直接的又は間接的サンドイッチアッセイ及び不均一又は均一相の何れにおいても実施される免疫沈降検定を使用することができる[Zola, Monoclonal Antibodies: A Manual of Techniques,CRC Press,Inc.,(1987) pp.147-158]。診断アッセイ法に使用される抗体は、検出可能部分で標識することができる。検出可能成分は、直接的に又は間接的に検出可能なシグナルをつくりだすことができなければならない。例えば検出可能部分は、3H、14C、32P、35S又は125I等の放射性同位体、蛍光イソチオシアネート、ローダミン又はルシフェリン等の蛍光又は化学発光化合物、もしくはアルカリホスファターゼ、βガラクトシダーゼ又は西洋わさびペルオキシダーゼ等の酵素であってもよい。Hunter等, Nature, 144: 945 (1962)、David等, Biochemistry,13:1014(1974)、Pain等, J. Immunol. Meth.,40:219 (1981)及びNygren等, J. Histochem. and Cytochem.,30:407 (1982)などに記載されている方法を含み、検出可能部分に抗体を抱合させるための当該分野において知られている任意の方法を使用することができる。
【0086】
また、Apo-2リガンド抗体は天然供給源又は組換え細胞培養からのApo-2リガンドのアフィニティー精製にも有用である。この方法においては、Apo-2リガンドに対する抗体を、当該分野でよく知られている方法を使用して、セファデックス樹脂や濾紙のような適当な支持体に固定する。次に、固定された抗体を、精製するApo-2リガンドを含有する試料と接触させ、次いで、固定された抗体に結合したApo-2リガンド以外の試料中の物質を実質的に全て除去する適当な溶媒で支持体を洗浄する。最後に、Apo-2リガンドを抗体から離脱させる他の適当な溶媒で支持体を洗浄する。またApo-2リガンド抗体は、可溶化Apo-2レセプターのアフィニティ精製又はApo-2レセプターの発現クローニングにも有用である。
ここに記載した抗体は、治療的にも使用される。例えば、Apo-2リガンド活性(Apo-2リガンド誘発アポトーシス等)を阻止する抗-Apo-2リガンド抗体は、増大したアポトーシスに伴う病理学的状態又は疾患の治療に用いられる[Thompson, 上掲参照]。
【0087】
F.Apo-2リガンド又はApo-2リガンド抗体を含むキット
本発明のさらなる実施態様においては、例えば上述の治療的又は非治療的用途に使用可能なApo-2リガンド又はApo-2リガンド抗体を含むキット及び製造品が提供される。製造品にはラベルが付された容器が含まれる。適切な容器には、例えばボトル、バイアル、及び試験管が含まれる。容器はガラス又はプラスチックのような種々の物質から形成できる。容器は、上述のような治療的又は非治療的用途に有効な活性剤を含む組成物を収容する。組成物中の活性剤は、Apo-2リガンド又はApo-2リガンド抗体である。容器のラベルには、組成物が特定の治療的又は非治療的用途に使用されることが示され、また上述のもののような、インビボ又はインビトロのいずれかの使用の指示が示されている。
【0088】
本発明のキットは、典型的には、上述の容器と、商業上及び使用者の観点から望まれる、バッファー、希釈液、フィルター、針、注入器及び使用説明が記されたパッケージ挿入物を含む、材料を含む一又は複数の他の容器を含んでいる。
以下の実施例は例示するためにのみ提供されるものであって、本発明の範囲を決して限定することを意図するものではない。
本明細書で引用した全ての特許及び参考文献の全体を、出典明示によりここに取り込む。
【0089】
(実施例)
実施例において言及されている全ての制限酵素は、ニューイングランドバイオラボ社(New England Biolabs)から購入し、製造者の使用説明に従い使用した。実施例で言及されている全ての他の市販試薬は、特に示していない限りは、製造者の使用説明に従い使用した。ATCC登録番号により次の実施例及び明細書全体を通して特定している細胞の供給源はアメリカン・タイプ・カルチャー・コレクション(Mannasas,Virginia)である。
【0090】
(実施例1)
ヒトApo-2リガンドをコードするcDNAクローンの単離
Apo-2リガンドの全長cDNAを単離するために、ヒト胎盤cDNAのラムダgt11バクテリオファージライブラリ(約1x106クローン)(HL10756、Clontechから市販)を、ヒトFas/Apo-1リガンドに幾分の相同性を示すEST配列(GenBank locus HHEA47M)に基づく合成オリゴヌクレオチドプローブでハイブリッド形成することによりスクリーニングした。HHEA47MのEST配列は390塩基対で、その3+フレームで翻訳した場合、ヒトApo-1リガンドの34アミノ酸領域に16の同一性を示す。HHEA47Mの配列は次の通りである:
GGGACCCCAATGACGAAGAGAGTATGAACAGCCCCTGCTGGCAAGTCAAGTGGCAACTCCGTCAGCTCGTTAGAAAGATGATTTTAGATTCCTCTGAGGAAACCATTTCTACAGTTCAAGAAAAGCAACAAAATATTTCTCCCCTAGTGAGAGAAAGAGGTCCTCAGAGAGTAGCAGCTCACATAACTGGGACCAGAGGAAGAAGCAACACATTGTCTTCTCCAAACTCCAAGAATGAAAAGGCTCTGGGCCGCAAAATAAACTCCTGGGAATCATCAAGGAGTGGGCATTCATTCCTGAGCAACTTGCACTTGAGGAATGGTGAACTGGTCATCCATGAAAAAGGGTTTTACTACATCTATTCCCAAACATACTTTCGATTTCAGGAGG 配列番号:3。
以下の配列を持つ60塩基対のオリゴヌクレオチドをスクリーニングに用いた:
TGACGAAGAGAGTATGAACAGCCCCTGCTGGCAAGTCAAGTGGCAACTCCGTCAGCTCGT 配列番号:4。
ハイブリッド形成は、20%のホルムアルデヒド、5X SSC、10%のデキストラン硫酸、0.1%のNaPiPO4、0.05MのNaPO4、0.05mgのサケ精子DNA、及び0.1%のドデシル硫酸ナトリウムを含むバッファー中、室温で終夜行い、次いで42℃において5X SSC、続いて2X SSC中で数回洗浄した。cDNAライブラリ中で12の陽性クローンを同定し、陽性クローンを以下の配列を有する60塩基対の第2のオリゴヌクレオチドプローブ(第1のプローブとの重複は無い)にハイブリッド形成することによって再スクリーニングした。
GGTGAACTGGTCATCCATGAAAAAGGGTTTTACTACATCTATTCCCAAACATACTTTCGA 配列番号:5。
ハイブリッド形成は、上記のように行った。
【0091】
得られた4つの陽性クローンを、フランキング5’ベクター配列に基づき外部ClaI制限部位を付加したプライマー及び3’フランキングベクター配列に基づき外部HindIII制限部位を付加したプライマーを用いたポリメラーゼ連鎖反応(PCR)により同定した。PCR産物はゲル精製し、T-AライゲーションによりpGEM-T(Promegaから市販)にサブクローニングした。異なるPCRからの3つの独立したクローンに、次いでジデオキシDNA配列を施した。これらのクローンのDNA配列分析は、それらが5’領域に幾分の長さ変化を持つが実質的に同一であることを示した。
Apo-2リガンドのコード化領域の核酸配列を図1Aに示した。クローンの1つの下流側3’末端領域の配列分析により、特徴的なポリアデニル化部位が明らかにされた(データは示さず)。cDNAは1つのオープンリーディングフレームを含み、それはヌクレオチド位置91−93のATGコドンに割り当てられた開始部位を持つ。この部位を取り囲む配列は、開始部位について提案されたコンセンサス配列[Kozak, J. Cell Biol., 115: 887-903 (1991)]と合理的に一致した。オープンリーディングフレームは、ヌクレオチド位置934−936の停止コドンTAAで終端する。
【0092】
ヒトApo-2リガンドの推定成熟アミノ酸配列は、281アミノ酸を含み、約32.5kDaの計算された分子量及び約7.63の等電点を有する。N末端には明らかなシグナル配列は存在しないが、ヒドロパシー分析(データは示さず)は残基15と40との間に疎水性領域の存在を示している。シグナル配列が無いこと及び内部疎水性領域が存在することは、Apo-2リガンドがII型膜貫通タンパク質であることを示唆している。推定細胞内、膜貫通及び細胞外領域は、各々14、26及び241アミノ酸長である。推定膜貫通領域は、図1Aで下線を付した。潜在的なN結合グリコシル化部位は、推定細胞外領域の残基109に位置している。
Apo-2リガンドのC末端領域と、TNFサイトカインファミリーの知られたメンバーとのアミノ酸配列のアラインメントは、C末端領域内で、Apo-2リガンドがApo-1リガンドと23.2%の同一性を有することを示した(図1B)。アラインメント分析は、他のTNFファミリーメンバーとのより低い程度の同一性を示した:CD40L(20.8%)、LT-α1(20.2%)LT-β(19.6%)、TNF-α(19.0%)、CD30L及びCD27L(15.5%)、OX-40L(14.3%)、及び4-1BBL(13.7%)。TNFサイトカインファミリーの中で、TNF-α及びLT-αの構造[Eck等, J. Bio. Chem., 264: 17595-17605 (1989); Eck等, J. Bio. Chem., 267: 2119-2122 (1992)]に基づいてβ鎖を形成すると予想される領域内の残基は、予想される連結ループ内の残基よりも高度に他のTNFファミリーメンバーに保持される傾向がある。Apo-2リガンドが、予想される連結ループ内の相同性に比較して、その推定β鎖領域において他のTNFファミリーメンバーと大きな相同性を示すことが見いだされた。また、ループ連結推定β鎖、B及びB’は、Apo-2リガンドにおいて顕著に長い。
【0093】
(実施例2)
ヒトApo-2リガンドの発現 A.全長cDNA融合作成物の293細胞における発現
mycエピトープタグに融合した全長Apo-2リガンドcDNAを次のように作成した。Apo-2リガンドcDNA挿入物を親pGEM-TApo-2リガンドプラスミド(実施例1に記載)からClaI及びHindIIIによる消化により除去し、同じ制限酵素で消化されたpRK5哺乳動物発現プラスミド[Schall等, Cell, 61:361-370 (1990);Suva等, Science, 237:893-896 (1987)]に挿入した。13アミノ酸のmycエピトープタグSer Met Glu Gln Lys Leu Ile Ser Glu Glu Asp Leu Asn 配列番号:6[Evan等, Mol. Cell. Biol., 5: 3610-3616 (1985)]をコードする配列を、Apo-2リガンドコード化配列の3’末端のコドン281と停止コドン(コドン282)との間にオリゴヌクレオチド指向性突然変異誘発[Zoller等, Nucleic Acids Res., 10: 6487-6469 (1982)]により挿入し、プラスミドpRK5Apo-2リガンド-mycを与えた。
pRK5Apo-2リガンド-mycプラスミドを、リン酸カルシウム沈殿法によりヒト293細胞(ATCC CRL 1573)にネオマイシン耐性遺伝子を持つpRK5プラスミドと同時形質移入した。抗生物質G418(0.5mg/mL)(GIBCO)の存在下での50%HAM’SF12/50%DMEM(GIBCO)中での成長能力によりApo-2リガンド-mycを発現する安定なクローンを選択した。
Apo-2リガンドのトポロジーを試験するために、G418耐性クローンを、抗mycモノクローナル抗体(mAb)クローン9E10[Evans, 上掲:Oncogene Scienceから市販]、次いでフィコエリスリン(PE)抱合ヤギ抗マウス抗体(Jackson ImmunoReserchから市販)で染色した後にFACS分析した。FACS分析により、mock形質移入細胞に比較してApo-2リガンド-myc形質移入細胞では特定の正の染色シフトが明らかとなり、Apo-2リガンドが細胞表面で発現され(図1C)、そのカルボキシル末端が露出されることが示された。従って、Apo-2リガンドはII型膜貫通タンパク質であると考えられる。
【0094】
B.ECD融合作成物の293細胞及びバキュロウイルスでの発現
Apo-2リガンドのC末端領域の上流側に他の配列を融合させた2つの可溶性Apo-2リガンド細胞外ドメイン(「ECD」)融合作成物を調製した。
一方の作成物では、ヘルペスウイルス糖タンパク質D(「gD」)シグナルペプチド[Lasky等, DNA, 3: 23-29 (1984); Pennica等, Proc. Natl. Acad. Sci., 92: 1142-1146 (1995); Paborsky等, Protein Engineering, 3: 547-553 (1990)に記載]の27アミノ酸及びエピトープタグ配列
Lys Tyr Ala Leu Ala Asp Ala Ser Leu Lys Met Ala Asp Pro Asn Arg Phe Arg Gly Lys Asp Leu Pro Val Leu Asp Gln 配列番号:7をpRK5哺乳動物発現プラスミド内でApo-2リガンドのコドン114−281の上流側に融合させた。簡単に言えば、gD配列は親プラスミドpCHAD(Genentech, Lasty等, Science, 233: 209-212 (1986)に記載されたように調製)から、3’プライマーがgDの3’領域並びにApo-2リガンドのコドン114−121に相補的なPCRで増幅した。生成物は、pRK5Apo-2リガンドプラスミドがテンプレートとして用いられる引き続くPCRにおいて、Apo-2リガンドコード化領域の3’末端に相補的な3’プライマーとともに5’プライマーとして使用した。生成物は、gD-Apo-2リガンドECD融合物をコードするが、次いでpRK5プラスミドにサブクローニングしてプラスミドpRK5gD-Apo-2リガンドECDを与えた。
【0095】
ヒト胚腎臓293細胞(ATCC CRL 1573)をリン酸カルシウム沈殿法により、pRK5gD-Apo-2リガンドECDプラスミド又はpRK5で一過性形質移入した。可溶性gD-Apo-2リガンドタンパク質の発現を、形質移入細胞の35S-Cys及び35S-Metで代謝的に標識することにより試験した。細胞上清を24時間後に回収し、遠心分離で透明化した。免疫沈降法のために、5mlの上清を5B6抗-gDモノクローナル抗体(Genentech)とともに1μg/mlで終夜4℃でインキュベートした。次いで、25μlのパンソルビン(Sigma)を添加して更に4℃で1時間おいた。管を回旋させ、ペレットをPBS中で洗浄し、SDS試料バッファー中で5分間煮沸した。煮沸試料を再度回旋させ、上清にSDS-PAGE及びオートラジオグラフィを施した。
抗gD抗体での免疫沈降は、gD-Apo-2リガンドプラスミドで形質移入した細胞の上清中に3つの優勢なタンパク質バンドを明示した(図1E)。これらのバンドは、23,48及び74kDaの相対分子量(Mr)で泳動した。成熟gD-Apo-2ポリペプチドの計算分子量は約22.5kDaである;よって、観察されたバンドは融合タンパク質の単量体(23kDa)、二量体(48kDa)及び三量体(74kDa)形であり、Apo-2リガンドが哺乳動物細胞において分泌された可溶性gD融合タンパク質として発現されうることを示した。
【0096】
第2の作成物では、MetGlyHis10配列(プラスミドpET19Bから誘導、Novagen)、次いで12アミノ酸の塩テロキナーゼ切断部位
Met Gly His His His His His His His His His His Ser Ser Gly His Ile Asp Asp Asp Asp Lys His Met 配列番号:8をバキュロウイルス発現プラスミド(pVL1392、Pharmingen)内のApo-2リガンドのコドン114−281の上流側に融合させた。簡単に言えば、Apo-2リガンドコドン114−281領域を、pRK5Apo-2リガンドプラスミド(実施例1に記載)から、各々フランキングNdeI及びBamHI制限部位を導入した5’及び3’領域に相補的なプライマーでのPCRにより増幅した。生成物は、T−AライゲーションによりpGEM-T(Promega)にサブクローニングし、DNA配列を確認した。次いで、挿入物をNdeI及びBamHIでの消化により除去し、アミノ末端MetGlyHis10タグ及びエンテロキナーゼ切断部位を含む修飾バキュロウイルス発現ベクターpVL1392(Pharmingenから市販)にサブクローニングした。
組換えバキュロウイルスは、His10-Apo-2ECDプラスミド及びBaculoGold(商品名)ウイルスDNA(Pharmingen)を、Spodopterafrugiperda(「Sf9」)細胞(ATCC SRL 1711)にリポフェクチン(GIBCO-BRLから市販)を用いて同時形質移入することにより生成した。28℃で4−5日のインキュベーションの後、放出されたウイルスを回収し、さらなる増幅に用いた。ウイルス感染及びタンパク質発現は、O'Relley等, Baculovirus expression vectors: A laboratory Manual, Oxford: Oxford University Press (1994)に記載されているように実施した。タンパク質は、下記実施例3に記載するようにNi2+キレートアフィニティクロマトグラフィで精製した。
【0097】
(実施例3)
組換えヒトApo-2リガンドの精製
抽出物を、組換えウイルス感染及びmock感染Sf9細胞(実施例2、項目B参照)から、Rupert等, Nature, 362: 175-179 (1993)に記載されているように調製した。簡単に言えば、Sf9細胞を洗浄し、超音波処理バッファー(25mLのHepes、pH7.9;12.5mMのMgCl2;0.1mMのEDTA;10%のグリセロール;0.1%のNP-40;0.4MのKCl)中に再懸濁し、氷上で2回20秒間超音波処理した。超音波処理物は、遠心分離で透明化し、上清を負荷バッファー(50mMリン酸塩、300mMのNaCl、10%のグリセロール、pH7.8)中に50倍に希釈し、0.45μmフィルターを通して濾過した。Ni2+-NTAアガロースカラム(Qiagenから市販)を5mLのベッド容量で調製し、25mLの水で洗浄して25mLの負荷バッファーで平衡させた。濾過した細胞抽出物を1分当たり0.5mLでカラムに負荷した。分画回収を開始する点であるベースラインA280までカラムを負荷バッファーで洗浄した。次に、カラムを、非特異的結合したタンパク質を溶離する二次洗浄バッファー(50mMリン酸塩、300mMのNaCl、10%のグリセロール、pH6.0)で洗浄した。再度ベースラインA280に達した後、カラムを二次洗浄バッファー中の0から500mMイミダゾール勾配で展開した。1mL分画を回収し、SDS−PAGE及び銀染色又はアルカリホスファターゼ(Qiagen)に抱合したNi2+−NTAでのウェスタンブロットにより分析した。溶離したHis10−Apo-2タンパク質を含有する分画をプールし、負荷バッファーに対して透析した。
出発材料としてのmock感染Sf9細胞に同じ手法を繰り返し、同様の分画をプールし、透析し、精製したヒトApo-2の対照として使用した。
精製タンパク質のSDS−PAGE分析は、His10-Apo-2リガンド単量体について計算された分子量22.4kDaに相当するMr24kDaの優勢なバンドを明示し(図1D、レーン3);タンパク質配列マイクロ分析(データは示さず)は24kDaのバンドがHis10-Apo-2リガンドポリペプチドに相当することを確認した。少量の48kDa及び66kDaバンドも観察され、可溶性Apo-2リガンドの同種二量体及び同種三量体の存在を示した。スルホ-NHS(5mM)(Pierce Chemical)及びECD(Pierce Chemical)25mM及び50mMとのインキュベーションによる精製His10-Apo-2リガンドの化学的架橋(各々、図1D、レーン1及び2)は、タンパク質を主に66kDaのバンドにシフトさせた。これらの結果は、溶液内でのApo-2の優勢な形態は同種三量体であり、これらの三量隊がSDSの存在下で二量体又は単量体に分解することを示している。
【0098】
(実施例4)
Apo-2リガンドのヒトリンパ細胞系に対するアポトーシス活性
精製した可溶性Apo-2リガンド(実施例3に記載)のアポトーシス活性を、幾つかのヒトリンパ細胞系を用いて試験した。第1の実験では、Apo-2リガンドの、エプスタインバールウイルス(EBV)形質転換ヒト末梢血B細胞から誘導した9D細胞(Genentech, Inc.)への影響を試験した。9D細胞(5x104細胞/ウェル、PRMI1640培地プラス10%ウシ胎児血清)を、対照培地、Apo-2(3μg/ml、上記実施例3のように調製した)、又は抗Apo-1モノクローナル抗体、CH11(1μg/ml)[Yonehara等, J. Exp. Med., 169: 1747-1756 (1989)に記載;Medical and Biological Laboratories Co.から市販]のいずれかとともに24時間インキュベートした。CH11抗Apo-1抗体はFas/Apo-1リガンド活性の類似したアゴニスト抗体である。
インキュベーションの後、細胞をサイトスピンスライドガラス上に回収し、逆光顕微鏡下で写真撮影した。Apo-2リガンド及び抗Apo-1モノクローナル抗体の両方が類似のアポトーシス効果を誘発し、細胞質凝集及び細胞数の減少を特徴としていた(図2A参照)。
Apo-2リガンドの、9D細胞、並びにRaji細胞(ヒトバーキットリンパB細胞系、ATCC CCL 86)及びジャーカット細胞(ヒト急性T細胞白血病細胞系、ATCC TIB 152)に対する効果をFACSにより更に分析した。FACS分析は、アポトーシス性細胞死について確立された基準を用いて、即ち2つのマーカー(a)アポトーシス細胞は染色するが生存細胞はしないヨウ化プロピジウム([PI」)染料及び(b)アポトーシス細胞には見られるが生存細胞には見られない露出されたホスファチジルセリンに結合するタンパク質アネキシンVの蛍光誘導体での細胞の蛍光染色関係を用いて実施した[Darzykiewicz等, Methods in Cell Biol., 41: 15-38 (!994); Fadok等, J. Immunol., 14: 2207-2214 (1992); Koopman等, Blood, 84: 1415-1420 (1994)]。
9D細胞(図2B)、Raji細胞(図2C)及びジャーカット細胞(図2D)を、対照培地(左側パネル)、Apo-2リガンド(3μg/ml、実施例3に記載したように調製)(真中パネル)、又は抗Apo-1抗体(1μg/ml)(右側パネル)とともに24時間インキュベート(1x106細胞/ウェル)した。次いで細胞を洗浄し、PI及びフルオレセインチオシアネート(FITC)-抱合アネキシンV(Brand Applicationsから購入)で染色し、フローサイトメトリーで分析した。PI及びアネキシンV染色の両方に陰性の細胞(第3象限)が生存細胞を表し;PI陰性、アネキシンV陽性染色細胞(第4象限)は初期アポトーシス細胞を表し;PI陽性、アネキシンV陽性染色細胞(第2象限)は主にアポトーシスの後期段階の細胞を表す。
【0099】
Apo-2リガンド処理9D細胞は、向上した細胞外アネキシンV結合性、並びに顕著に増加したPIの取り込みを示し(図2B)、Apo-2リガンドが細胞でアポトーシスを誘発したことを示している。抗Apo-1抗体、CH11では匹敵する結果がえられた(図2B)。Apo-2リガンドは、Raji及びジャーカット細胞で抗Apo-1抗体と類似の応答を誘発した(図2C及び2D参照)。対照及び抗Apo-1抗体と比較したときのこれらの細胞系でのApo-2リガンドによるアポトーシスの誘発(アポトーシス細胞の%で測定した場合)を下記の表1に示す。
Apo-2リガンドによるヌクレオソーム間DNA断片化の活性化も分析した。ジャーカット細胞(左側レーン)及び9D細胞(右側レーン)を対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに6時間インキュベート(2x106細胞/ウェル)し、次いでDNAを細胞から抽出し、ターミナルトランスフェラーゼを用いて32P-ddATPで標識した。標識したDNA試料に電気泳動2%アガロースゲルを施し、後にオートラジオグラフィで分析した[Moore等, Cytotechnology, 17: 1-11 (1995)]。Apo-2リガンドは、ジャーカット細胞及び9D細胞の両方でヌクレオソーム間DNA断片化を誘発した(図2E)。このような断片化はアポトーシスに特徴的である[Cohen, Advances Immunol., 50: 55-85 (1991)]。
Apo-2リガンドのアポトーシス活性の時間変化を試験するために、9D細胞をミクロタイターディッシュ(5x104細胞/ウェル)で、対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに0〜50時間の範囲の期間インキュベートした。インキュベーションに続いて、死亡及び生存細胞の数を、ヘモサイトメータを用いて顕微鏡検査で測定した。
図3Aに示すように、細胞死の最大レベルは9D細胞において24時間以内に起こる。
Apo-2リガンド誘発細胞死の用量依存性を試験するために、9D細胞(5x104細胞/ウェル)を、連続的に希釈した対照培地又はApo-2(実施例3に記載したように調製)とともに24時間インキュベートした。インキュベーション後の死亡及び生存細胞の数を上記のように測定した。結果を図3Bに示した。特異的アポトーシスは、Apo-2リガンド処理細胞でのアポトーシス%を対照中でのアポトーシス%で除することにより決定した。最大値の半分のアポトーシス活性化は約0.1μg/ml(約1nM)で起こり、最大誘発は約1〜3μg/ml(約10〜30nM)で起こった。
【0100】
(実施例5)
Apo-2リガンドのヒト非リンパ腫瘍細胞系に対するアポトーシス活性
Apo-2リガンドのヒト非リンパ腫瘍細胞系に対する影響を、以下の細胞系を用いて試験した:HeLa(ヒト頸部癌から誘導、ATCC CCL 22);Me-180(ヒト頸部癌から誘導、ATCC HTB 33);MCF7(ヒト乳癌から誘導、ATCC HTB 22);U-937(ヒト組織リンパ腫から誘導、ATCC CRL 1593 ;A549(ヒト肺癌から誘導、ATCC CCL 185);及び293(アデノウイルス形質移入ヒト胚腎臓細胞から誘導、ATCC CCL 1573)。
アッセイにおいて、各細胞系の1x106細胞を、対照培地、Apo-2リガンド(3μg/ml、実施例3に記載したように調製)、又は抗Apo-1抗体CH11(1μg/ml)とともに24時間インキュベートした。インキュベーションの後、アポトーシスを実施例4に記載したようにFACS分析で測定した。結果を下記の表1に示す。
【0101】
(表1)
【0102】
HeLa細胞及びMCF7細胞は、CH11抗Apo-1抗体に比較してApo-2リガンドによるアポトーシス誘発に対して等しい感受性であった。これに対して、U-937細胞及びA549細胞は、Apo-2リガンドによるアポトーシス誘発に対して極めて大きな感受性を有していた。ME-180細胞はApo-2リガンドに対しては全く感受性であったが、抗Apo-1抗体には比較的耐性があった。293細胞はApo-2リガンドに耐性であり、抗Apo-1抗体には若干応答した。
よって、Apo-2リガンドは非リンパ由来の細胞、並びにリンパ由来の細胞(実施例4参照)でアポトーシスを誘発できる。また、十分に理解されておらず、いかなる特定の理論との結びつけるつもりはないが、本出願人は、Apo-2リガンドがApo-1とは区別されるレセプターを介して作用すると考えている。この考えは、上記の細胞系がApo-2リガンド及び抗Apo-1抗体に対して異なる感受性パターンを示すここに記載したデータによって支持されている。
【0103】
(実施例6)
Apo-2リガンドのヒト末梢血液単球に対する影響
末梢血液単核細胞(「PBMC」)は、ヒト供与者の血液から、リンパ球分離媒体(LSM(登録商標)、Organon Teknika)を用いたフィコール密度勾配遠心分離により単離した。単離下T細胞集団は、抗Igカラムへの表面Ig結合によりB細胞を除去し、IgカラムへのFcレセプターの結合を通して単球を除去することによりPBMCから調製した(R & D Systems)。単離したB細胞集団はOKT3骨髄細胞(ATCC, CRL 8001)により生成された抗CD3抗体と反応したT細胞及び4F2C13ハイブリドーマ(ATCC, HB 22)により生成された単球特異的抗体と反応した単球の補体媒介除去によりPBMCから調製した。さらなる単球除去はプラスチックへの接着によって達成した。
【0104】
新たに単離した末梢血液B又はT細胞(1x106細胞/ウェル)を、対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)の存在下で3日間培養した。活性化のために、B細胞は同時にリポ多糖(「LPS」、1μg/ml)で処理し、T細胞はホルボールミリステートアセテート(「PMA」、10ng/ml)プラスイノマイシン(1μg/ml)(Sigma)で処理した。インターロイキン-2(「IL-2」)前処理のために、T細胞は、Apo-2リガンドに暴露する前に、IL-2(50U/ml)(Genzyme)の存在下で3−5日間培養した。アポトーシスは、本質的に実施例4に記載したようにFACS分析で決定した。しかし、B細胞は、抗CD19/CD20抗体(Jackson Immunoreserch)でゲートし、T細胞は抗CD4/CD8抗体(Jackson Immunoreserch)でゲートした。結果を下記の表2に示すが、独立した実験[Bリンパ球−9実験;Tリンパ球−8実験;Tリンパ球プラスIL-2−5実験]の平均±SEで表し、各データ点毎に500,000細胞が分析された。統計的分析をスチューデントt試験を用いて実施した、表2において、個々の対照に対してa=p<0.05及びb=p<0.02である。
【0105】
(表2)
Apo-2リガンドは、非刺激B細胞、LPSで活性化したB細胞、及びPMA及びイノマイシンで活性化したT細胞においてアポトーシスを誘発した。IL-2の存在下で細胞を培養することにより末梢T細胞がアポトーシスを起こしやすくなることは既に報告されている[Lenardo等, Nature, 353: 858-861 (1991)]。本研究は、IL-2での前処理は、末梢T細胞をApo-2リガンド誘発死に対して敏感化させないことを示した。
【0106】
(実施例7)
Fas/Apo-1及びTNFレセプターを用いた阻害アッセイ
Fas/Apo-1レセプター、並びにI型及びII型TNFレセプター(TNF-R1及びTNF-R2)がApo-2リガンドのアポトーシス活性の媒介に含まれているか否かを決定するために、これらのレセプターの可溶化形態が精製した可溶性Apo-2リガンド(実施例3に記載)のアポトーシス活性を阻害するか否かを試験することにより検定した。
9D細胞(5x104細胞/ウェル)を対照培地又はApo-2リガンド(3μg/ml、実施例3に記載したように調製)とともに、対照バッファー、CD4-IgG対照(25μg/ml)、可溶性Apo-1-IgG(25μg/ml)、可溶性TNFR1-IgG(25μg/ml)又は可溶性TNFR2-IgG融合タンパク質(25μg/ml)の存在下で24時間インキュベートした。
Fas/Apo-1、TNF-R1及びTNF-R2レセプターの可溶性誘導体はAshkenazi等, Methods, 8: 104-115 (1995)に記載したようにIgG融合タンパク質として作成した。CD4-IgGは、Byrn等, Nature, 344: 667-670 (1990)に記載されたようにIgG融合タンパク質として作成し、対照として用いた。
図3Cに示したように、レセプター融合分子はApo-2リガンドの9D細胞に対するアポトーシス活性を阻害しなかった。これらの結果は、Apo-2リガンドのアポトーシス活性がFas/Apo-1及びTNF-R1及びTNF-R2とは独立であることを示している。
【0107】
(実施例8)
哺乳動物組織におけるApo-2リガンドmRNAの発現
ヒト組織におけるApo-2リガンドmRNAの発現を、ノーザンブロット分析によって検査した(図4)。ヒトRNAブロットを、全長Apo-2リガンドcDNAをベースにした32P標識DNAプローブ、又はGenBank EST配列、HHEA47M(実施例1参照)をベースにした32P標識RNAプローブにハイブリッド形成させた。ヒト胎児RNAブロットMTN(Clontech)及びヒト成人RNAブロットMTN-II(Clonetech)をDNAプローブと共にインキュベートし、ヒト成人RNAブロットMTN-I(Clonetech)をRNAプローブと共にインキュベートした。ブロットをハイブリッド形成用バッファー[5X SSPE;2X デンハード溶液;100mg/mLの変性剪断されたサケ精子DNA;50%のホルムアミド;2%のSDS]に入ったプローブと共に、42℃で16時間インキュベートした。ブロットを1X SSPE;2%のSDSを用い、65℃で1時間数回洗浄し、ついで50%の脱イオンホルムアミド;1X SSPE;0.2%のSDSを用い、65℃で30分間洗浄した。ブロットを一晩さらした後、リン光画像機(Fuji)を用いた展開させた。
結果を図4に示す。ヒト胎児組織では、Apo-2リガンドmRNA発現は、肺、肝臓及び腎臓で検出されたが脳組織ではされなかった。ヒト成人組織では、Apo-2リガンドmRNA発現は、脾臓、胸腺、前立腺、卵巣、小腸、末梢血液リンパ球、心臓、胎盤、肺、及び腎臓で検出された。精巣、脳、骨格筋、及び膵臓では殆ど又は全く発現は検出されなかった。上記のようなApo-2リガンドの発現パターンは、主にT細胞及び精巣で発現されるApo-1リガンドと同じではなかった[Nagata等, 上掲]。
【0108】
(実施例9)
Apo-2リガンドのヒト腫瘍細胞系に対するアポトーシス活性
Apo-2リガンド(実施例3に記載)のヒト腫瘍細胞系に対するアポトーシス活性を、幾つかの化学治療薬の存在又は不存在下でさらに試験した。
以下のヒト腫瘍細胞系をアッセイした:A549(肺癌、ATCC CCL 185);HTC116(結腸癌、ATCC CCL 247);SW480(結腸腺癌、ATCC CCL 228);MDA231(乳癌、ATCC HTB 26);HeLa(頸部癌、ATCC CCL 22);ME-180(頸部癌、ATCC HTB 33);T24(膀胱癌、ATCC HTB 4);SK-N-AS(神経芽腫、White等, Proc. Natl. Acad. Sci., 92: 5520-5524 (1995))。下記表3に示すように、これらの細胞系の幾つかは野生型p53を発現するが(WT)、他は突然変異により発現しない(mut)。細胞を2.5x105細胞/ウェルで96ウェルプレートに蒔き、終夜インキュベートした。細胞を2倍希釈のApo-2リガンド(100ng/mlから0.01nm/ml)の存在下で培養した。幾つかの培地には化学治療薬−シクロヘキサミド(「CHX」)(50μg/ml;Sigma Chemicals)、ドキソルビシン(10−100μg/ml;Pharmacia)、又は5-FU(6mg/ml;Roche)も24時間添加した。
24時間のインキュベーション後、細胞を0.5%クリスタル-ビオレチン20%メタノールで染色した。0.1Mクエン酸ナトリウム(50%メタノール中0.1Mクエン酸)で染色細胞から染料を溶離し、540nmでの吸収を測定して細胞生存を測定した。
結果を下記の表3に示す。
【0109】
(表3)
+: 100nm/mlApo-2Lで24時間後>35%死亡。
++: 100nm/mlApo-2Lで24時間後>70%死亡。
+++: 10nm/mlApo-2Lで24時間後>70%死亡。
これらの結果は、Apo-2リガンドが種々の腫瘍型から誘導された腫瘍細胞系において細胞死を誘発したことを示し、Apo-2Lが腫瘍細胞のp53状態とは無関係に細胞死を誘発したことを示している。これらの結果はまた、Apo-2リガンド誘発細胞死が幾つかの異なる化学治療薬によって増強されることも示している。
【0110】
(実施例10)
インビボでのApo-2リガンドのアポトーシス活性
腫瘍を持つヌードマウスにおけるApo-2リガンドの影響を試験した。ヌードマウス(各群当たり5−10匹のマウス)(Harlan Sprague Dawleyから購入)に、MDA231ヒト乳ガン細胞(ATCC HTB 26)(2x106細胞/マウス)を皮下注射した(第0日)。腫瘍を14日間成長させた。14及び15日目に、2μg/0.05ml/マウスのApo-2リガンド(実施例3)及び/又は10μg/0.05ml/マウスのドキソルビシン(Pharmacia)を腫瘍部位に注射した。対照動物は同様に0.05mlのPBSを注射した。21日目に動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図5に示す。データは、Apo-2リガンド処理が腫瘍成長そのものを阻害し、Apo-2リガンドが腫瘍成長に対するドキソルビシンの効果を向上させたことを示している。
【0111】
(実施例11)
インビボでのApo-2リガンドのアポトーシス活性
実施例10に記載したように、腫瘍を持つヌードマウスにおけるApo-2リガンドの抗腫瘍効果を試験した。ただし、第0日において、マウスにHCT116ヒト結腸癌細胞(ATCC CCL 247)(2x106細胞/マウス)皮下注射した。腫瘍を14日間成長させた。14及び15日目に、2μg/0.05ml/マウスのApo-2リガンド及び/又は10μg/0.05ml/マウスの5-FU(Roche)を腫瘍部位に注射した。対照動物は同様に0.05mlのPBSを注射した。21日目に動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図6に示す。これらの結果は、Apo-2リガンド処理が腫瘍成長そのものを阻害し、Apo-2リガンドが腫瘍成長に対する5-FUの阻害効果を向上させたことを示している。
【0112】
(実施例12)
インビボでのApo-2リガンドのアポトーシス活性
実施例11に記載したように、腫瘍を持つヌードマウスにおけるApo-2リガンドの抗腫瘍効果を試験した。ただし、第1及び2日目に、10μg/0.05ml/マウスのApo-2リガンド及び/又は100μg/0.05ml/マウスの5-FU(Roche)を腹膜内注射した。対照動物は同様にPBSを注射した。腫瘍サイズ(mm2)を5、9及び15日目に測定し、動物を犠牲にし、腫瘍を切除して秤量(グラム)した。
結果を図7及び8に示す。これらの結果は、Apo-2リガンドが腹膜内注射したときでも皮下の腫瘍部位に到達でき抗腫瘍活性を発揮できることを示している。また、これらの結果は、腫瘍成長自体を阻害し、腫瘍成長に対する5-FUの阻害効果を向上させるApo-2リガンドの能力を確認した。
【0113】
(実施例13)
CrmAを用いた阻害アッセイ
ICE及びCPP32/Yama等のプロテアーゼがApo-2リガンドによるアポトーシス誘発において役割を果たしているか否かを実験するために、CrmAがApo-2リガンド誘発アポトーシスを阻止するか否化を決定するアッセイを行った[Marsters等, Current Biology, 6: 750-752 (1996)]。CrmAは、死亡プロテアーゼICE及びCPP/Yamaのポックスウイルス誘導阻害剤であり、TNFR1及びFasApo-1による死亡シグナル伝達を阻止する。さらに、Apo-1リガンド及びTNFによるアポトーシスを媒介するアダプタータンパク質FADDを含む「死亡ドメイン」[Chinnaiyan等, Cell, 81: 505-512 (1995); Hsu等, Cell, 84: 299-308 (1996)]が、Apo-2リガンド誘発アポトーシスに含まれるか否かを実験するために、FADDのドミナントネガティブ突然変異(FADD-DN)[Hsu, 上掲]がApo-2リガンド機能を阻害するか否かを決定するアッセイを行った[Marsters等, Current Biology, 6: 750-752 (1996)]。
HeLA-S3(ATCC CCL 22)細胞をpRK5-CrmA発現プラスミド(Ray等, 上掲で報告されたCrmA配列)又はpRK5-FADD-DN発現プラスミド(Hsu等, Cell, 84: 299-308 (1996))で形質移入した。pRK5は対照として使用した。細胞を、プラスミドDNA取り込みのマーカーとしてのpRK5-CD4(Smith等, Science, 238: 1704-1707 (1988))と同時形質移入した。形質移入細胞をフィコエリスリン抱合抗CD4抗体(Jackson Immunoreserch)で染色して同定し、アポトーシスは本質的に上記実施例4に記載したようにFACSで分析した。
結果を図9に示す。CrmAは、Apo-2リガンド誘発アポトーシス、並びに抗Apo-1抗体に誘発されたアポトーシスを阻止した。これに対して、FADD-NDはApo-2誘発アポトーシスには殆ど影響しなかったが、抗Apo-1抗体に誘発されたアポトーシスは実質的に阻止した。従って、このアッセイの結果は、Apo-2リガンド、TNFR1及びFas/Apo-1はアポトーシス性細胞死を活性化するのに共通の遠位シグナル伝達経路を用いることを示唆している。特に、結果は、ICE及びCPP/Yama等のプロテアーゼがApo-2リガンド誘発アポトーシスには必要であることを示している。これに対して、FADDはTNFR1及びFas/Apo-1に誘発される細胞死には必要であるがApo-2リガンドでは必要ない。
【0114】
Apo-2リガンド抗体の調製
1μgのApo-2リガンド(実施例3に記載したように調製し、Ribi Immunochemical Research Inc., Hamilton, MTから購入したMPL-TDMアジュバントで希釈)を、各々の後足の裏の柔らかい部分に、1週間の間隔で10回注射することで、Balb/cマウス(チャールズリヴァー研究所から得たもの)を免疫化した。最終の追加免疫から3日後に、膝窩リンパ節をマウスから取り除き、単細胞懸濁液を、1%のペニシリン-ストレプトマイシンが補われたDMEM培地(Biowhitakker Corp.から得たもの)中で調製した。ついで、リンパ節細胞を35%のポリエチレングリコールを使用し、マウス骨髄腫細胞P3X63AgU.1(ATCC CRL 1597)と融合させ[Laskov等, Cell. Immunol., 55:251(1980)]、96-ウェル培養プレートで培養した。融合の結果得られたハイブリドーマをHAT培地において選択した。融合から10日後に、ハイブリドーマ培養の上清をELISAでスクリーニングし[Kim等, J. Immunol. Meth., 156: 9-17 (1992)]、Apo-2リガンドタンパク質に結合するモノクローナル抗体の有無を試験した。
ELISAにおいて、各々のウェルに、PBSに0.5μg/mlのApo-2リガンド(実施例3参照)が入ったものを50μl添加して、96-ウェルマイクロタイタープレート(Nunc)をコートし、4℃で一晩インキュベートした。ついでプレートを洗浄用バッファー(0.05%のトゥイーン20を含有するPBS)で3回洗浄した。ついで、マイクロタイタープレートのウェルを、2.0%のウシ血清アルブミン(BSA)200μlでブロックし、室温で1時間インキュベートした。ついでプレートを、洗浄用バッファーで、再度3回洗浄した。
【0115】
洗浄工程後、2μg/mlのApo-2リガンド抗体50μl又は100μlのハイブリドーマ上清を指定ウェルに添加した。100μlのP3X63AgU.1骨髄腫細胞の条件培地を、対照として他の指定ウェルに添加した。プレートを振盪装置上で、室温で1時間インキュベートし、続いて洗浄用バッファーで3回洗浄した。
次に、アッセイ用バッファー(0.5%のウシ血清アルブミン、0.05%のトゥイーン20、0.01%のチメルソール(Thimersol)がPBSに入ったもの)で、1:1000に希釈されたHRP-抱合ヤギ抗マウスIgG Fc(Cappel Laboratories社から購入)を50μl、各々のウェルに添加し、プレートを振盪装置上で、室温で1時間インキュベートした。プレートを洗浄用バッファーで3回洗浄し、ついで各々のウェルに50μlの基質(TMB、3,3',5,5'-テトラメチルベンジジン;Kirkegaard & Perry, Gaithersburg, MDから入手)を添加し、室温で10分間インキュベートした。50μlの終止溶液(Kirkegaard & Perry)を各々のウェルに添加して反応を終了させ、450nmでの吸光度を自動マイクロタイタープレート読取装置で読み取った。
【0116】
ハイブリドーマ上清(選択した99)を、Apo-22リガンド誘発9D細胞死を阻止する活性について試験した。活性は、トリパンブルー染料排除を用いて処理した9D細胞の生存%を試験することにより最初に決定した。
阻止活性はFACS分析でも確認した。9D細胞(5x105細胞/0.5ml)を完全RPMI培地(RPMIプラス10%のFCS、グルタミン、非必須アミノ酸、ペニシリン、ストレプトマイシン、ピルビン酸ナトリウム)に懸濁し、24ウェルのミクロタイタープレートに配した。0.5mlのApo-2リガンド(1μg/ml)(実施例3で調製)を完全RPMI培地に懸濁し、10μgの精製モノクローナル抗体又は100μlの培地上清とともにプレインキュベートし、次いで9D細胞を含む24マクロタイターウェルに添加した。マクロタイタープレートは7%CO2の存在下で終夜37℃でインキュベートした。インキュベートした細胞は、次いで回収し、PBSで1回洗浄した。細胞の生存を製造者(Clontech)の推奨に従ってホスファチジルセリンに結合したFITCアネキシンVの染色により決定した。細胞をPBSで洗浄し、200μl結合バッファー中に再懸濁した。10μlのアネキシンV-FITC(1μg/ml)及び10μlのヨウ化プロオピジウムを細胞に添加した。暗中15分のインキュベーションの後、9D細胞をFACSで分析した。
8つの潜在的阻止、4つの非阻止抗体分泌ハイブリドーマが同定され、更に制限希釈技術によりクローニング(2回)した。
【0117】
モノクローナル抗体1D1、2G6、2E11、及び5C2と命名した4つの抗体のFACS分析を図10に例示した(後述するように、1D1、2G6、2E11、及び5C2抗体は、各々ハイブリドーマ1D1.12.4、2G6.3.4、2E11.5.5、及び5C2.4.9から生成され、それらは全てATCCに寄託されている)。Apo-2リガンドで処理した9D細胞(左上図)は、未処理の対照細胞(右上図)より50%高いのアポトーシス細胞を示した。Apo-2リガンドに、2E115C2、2G6、又は1D1抗体を加えて処理した9D細胞は、未処理の対照細胞より各々0%、6%、26%、及び48%高いアポトーシス細胞を示した。これらの結果は、5C2、2E11及び2G6抗体は阻止抗体であるが1D1抗体は非阻止抗体であることを示す。最も高い阻止活性は5C2抗体で得られた。
4つの抗体の抗原特異性もELISAで試験した。ミクロタイターウェルを2μg/mlのリンホトキシン(Genentech, Inc., EP 164,965, Gray等, Nature, 312: 721-724 (1984)も参照)、TNF-α(Genentech, Inc., Pennica等, Nature, 312: 724-729 (1984); Aggarwal等, J. BIol. Chem., 260: 2345-2354 (1985)も参照)、又はApo-2リガンド(実施例3参照)でコートした。モノクローナル抗体1D1、2G6、2E11、及び5C2は10μg/mlの濃度で試験した。
アッセイの結果は図11に示した。図11の結果は、モノクローナル抗体2G6、2E11、及び5C2はApo-2リガンドに特異的だが、モノクローナル抗体1D1はリンホトキシン及びTNF-αと弱い交差反応結合を有することを示した。
【0118】
B.アイソタイピング
1D1、2G6、2E11、及び5C2抗体(上記項目Aに記載)のアイソタイプを、アイソタイプ特異的ヤギ抗マウスIg(Fisher Biotech, Pitsburgh, PA)でミクロタイタープレートを4℃で終夜コートすることにより決定した。プレートは次いで上記の洗浄バッファーで洗浄した。ミクロタイタープレートのウェルを200μlの2%ウシ血清アルブミンでブロックし、室温で1時間インキュベートした。プレートを再度洗浄バッファーで3回洗浄した。
次に、100μlの5μg/mlの精製Apo-2リガンド抗体又は100μlのハイブリドーマ培地上清を指定ウェルに添加した。プレートを室温で30分間インキュベートし、次いで50μlのHRP-抱合ヤギ抗マウスIgG(上記)を各ウェルに添加した。プレートを室温で30分間インキュベートした。HRPのレベルを上記のTRP基質を用いて検出した。
アイソタイピング分析は、1D1及び2G6抗体はIgG2b抗体であり、2E11及び5C2抗体はIgG2a抗体であることを示した。
【0119】
C.エピトープマッピング
エピトープマッピングは、上掲のKim等に記載されているように、ビオチニル化モノクローナル抗体を用いて競合的結合ELISAを使用して実施した。選択されたモノクローナル抗体は、Antibodies, a laboratory Manual, Eds. E. Harlow及びD. Lane, p.342に記載されているようにN-ヒドロキシスクシンイミドを用いてビオチニル化した。ミクロタイタープレートウェルを、50μlのApo-2リガンド(実施例3参照、0.1μg/ml)でコートし、4℃で終夜維持し、次いで2%BSAで室温において1時間ブロックした。ミクロタイターウェルを洗浄した後、あらかじめ決定した最適濃度のビオチニル化抗体及び1000倍過剰の非標識抗体を各ウェルに添加した。室温で1時間インキュベートした後、プレートを洗浄し、ビオチニル化抗体の量をHRP-ストレプトアビジンの添加で検出した。ミクロタイターウェルを洗浄した後、結合した酵素を基質(TMB)の添加で検出し、プレートをELISAプレート読み取り機で490nmで読み取った。
結果を図12に示す。結果は、HRP-抱合抗体の結合は、それ自身の抗体の過剰量で有効に阻害されたが、アッセイした他の抗体ではされないことを示した。
モノクローナル抗体で認識されるApo-2リガンドの領域は、合成ペプチド[図1Aに示したようにApo-2リガンド配列のaa 128-143(ペプチド「APO14」);aa 144-159(ペプチド「APO15」);aa 192-204(ペプチド「APO17」);aa 230-238(ペプチド「APO18」);aa 261-272(ペプチド「APO19」)]を用いてChuntharapai等, J. Immunol., 152: 1783-1789 (1994)に記載されているようにELISAで決定した。結果を図13に示した。1D1抗体は、Apo-2リガンドのアミノ酸残基192−204を含むAPO17ペプチドに結合性を示した。
【0120】
(実施例15)
CHO細胞におけるApo-2リガンドの発現
pGEM-TApo-2リガンドプラスミド(上記実施例1及び2に記載)からの全長Apo-2リガンドcDNA挿入物をpRK5プラスミドに挿入した。このプラスミドを、次いで、リポフェクタミンプラス(Gibco/BRL)法を用いて、DHFR選択遺伝子を持つpFD11プラスミド(SV40初期プロモーター)とDP-12CHO細胞に同時形質移入した。Apo-2リガンドを発現する安定なクローンを、2.5%透析FBSを含むPS21(G1074)/280GHT-枯渇選択成長培地中での成長能力で選択した。
選択したApo-2リガンドを発現するCHO細胞を37℃で2−4日インキュベートした。次いで細胞培地を回収して濾過した。細胞培地上清中のApo-2リガンドの存在は、ウェスタンブロット分析により試験した。ウェスタン分析用のタンパク質を調製するために、培地上清を、制御された孔のガラスビーズに結合させたここに記載の5C2抗Apo-2抗体とともにインキュベートした。インキュベーションに続いて、ビーズを洗浄し、結合タンパク質を25mMのDTTを含有するSDS−PAGE試料バッファーで溶離した。次いで培地を、4−20%ポリアクリルアミド勾配SDSゲルを走らせ、ニトロセルロースフィルター上にエレクトロブロットした。フィルターをポリクローナルウサギ抗ヒトApo-2リガンドポリクローナル抗体、続いてセイヨウワサビペルオキシダーゼに抱合させた抗ウサギIgGとともにインキュベートし、製造者(NEB)の指示に従って化学発光によって展開した。Apo-2リガンドを発現するCHO細胞からの試料は、ゲル上に22,000ダルトンの単一のバンドを生じた(データは示さず)。
【0121】
形質移入したCHO細胞によって発現したApo-2リガンドの配列を決定するために、25mlの細胞培養培地上清を抗Apo-2L抗体(ここに記載した5C2抗体)を用いて精製した。精製カラムを調製するために、2.0mgの5C2抗体を0.5mlの制御孔ガラスビーズ(CPG, Inc., Fairfield, NJ)に製造者の指示に従って結合させた。結合の後、樹脂を15mlの50mMトリスpH7.5、次いで15mlの0.1M酢酸/0.15Mクエン酸ナトリウム/0.05MのNaClで洗浄した。
負荷に先立って、0.5mlカラムを15mlの50mMトリスpH7.5で平衡させた。約25mlの回収したCHO細胞培養培地、pH7.2、導電性10.2mmhoを、抗体カラムに勾配を与える負荷モードで負荷した。負荷流出物をカラムにリサイクルして全負荷時間を1時間とした。負荷に続いて、非特異的タンパク質を2mlの0.5MのTMAC/0.25MのNaClで洗い落とした。カラムを1.5mlの0.1M酢酸/0.15MNaCl、pH3.0で溶離した。1.5mlの溶離物を管に回収し、即座に50μlの3MトリスpH9.0でpH7.0に中和した。
【0122】
溶離物質のアリコートをSDSゲル上で分析し、平行して、既に大腸菌で発現された図1Aのアミノ酸残基96−281をからなる精製対照Apo-2リガンドポリペプチドを精製した。ゲルは、CHO細胞発現可溶性Apo-2リガンドポリペプチドが5C2抗体アフィニティカラム上で精製されたことを確認した。溶離物質の他のアリコートを濃縮し、SDS−PAGE上を走らせ、PVDF膜にエレクトロブロットし、次いでEdman分解で配列決定した。タンパク質配列分析により、CHO細胞が図1Aの配列の位置92におけるN末端アミノ酸を有するApo-2リガンドの可溶化形態を発現することが明らかとなった。よって、可溶化Apo-2リガンドポリペプチドは図1Aのアミノ酸92−281を含む。現在では、Apo-2リガンドのこの可溶化92−281アミノ酸形態は、Apo-2リガンドの天然に切断された形態を含むと考えられている。
CHO細胞の発現されたApo-2リガンドのアポトーシス活性を、培養したHeLa細胞(ATCC CCL 22)で試験した。HeLa細胞を10%のFBSを含むHamのF12培地中で6ウェルの培養プレートに蒔き、37℃で終夜インキュベートした。培地を吸引し、発現されたApo-2リガンド(1:10、1;20、1:40希釈)を含む2mlのCHO細胞培地上清を各資料ウェルに添加した。プレートを37℃で終夜インキュベートした。2mlの一条件培地(非形質移入CHO細胞培地上清)を含む対照ウェルを同じ条件下で平行に走らせた。
次いで、処理したHeLa細胞を、光顕微鏡下でアポトーシス形態について分析した(図14A−14D参照)。各視野でのアポトーシス細胞の数を図14Eに示す。発現されたApo-2リガンドを含有するCHO細胞培地上清で処理したHeLa細胞は、非条件培地で処理した細胞よりアポトーシス形態において2倍又はそれ以上の増加を示した。
【0123】
(実施例16)
大腸菌発現Apo-2リガンドのアポトーシス活性
Apo-2リガンドの抗腫瘍効果を腫瘍を持つヌードマウスで試験した。上記の実施例とは対照的に、可溶性Apo-2リガンドポリペプチドは大腸菌で発現され、次いで埋め込みミニポンプ装置を通して動物中に注入した。
Apo-2リガンドは、アミノ酸91−281(図1A参照)をコードするcDNAをpS1346プラスミドに挿入することにより調製した[pS1346は挿入された転写停止剤を持つhgh207-1プラスミドを含む;DeBoer等, Proc. Natl. Acad. Sci., 80: 21-25 (1983); Scholtissek等, Nucl. Acids Reserch, 15: 3185 (1987)参照]。このプラスミドは、次いで大腸菌株52A7に形質移入した。株52A7は次の遺伝子型を持つ大腸菌K12W3110株である:fhuA(tonA)lon galE rpoHts(htpRts)clpP lacIq。大腸菌の25ml培地をLB培地で光学密度約1.0OD550まで成長させ、遠心分離(5000rpmで15分間)で回収した。細胞ペレットを0.15MのNaClで1回洗浄した後2.5mlのバッファー(LBブロス、HClでpH6.1、100g/LのPEG8000、10mMのMgSO4、10mMのMgCll2及び5%DMSOを含有)中で再懸濁した。細胞懸濁物を氷上に30〜45分間放置し、アリコートを−80℃で保存した。この細胞懸濁物を形質転換プロセスの競合細胞として提供した。競合細胞のプラスミド調製物でも形質転換、形質転換物の選択及び単離は、Maniatis等, Molecular Cloning: A Laboratory manual, second Edition, vol.1: 1.74-1.84に記載された標準プロトコールで実施した。
【0124】
1mlの形質転換体を5μg/mlテトラサイクリンを含有する500mlのLB培地に播種して発酵種菌を調製した。この培地を振盪する2Lのバッフルフラスコで30又は37℃において10時間インキュベートした。次いで、得られた培地を、25g/LのNZアミンAS、6.25g/Lの酵母菌抽出物、0.125g/Lのトリプトファン、6.25g/Lのテトラサイクリン、0.94g/Lのグルコース、0.625g/LのL-イソロイシン、及び94mg/LのL-61抗発泡剤(antifoam)を含む8リットルの培地(6.25g/Lの硫酸アンモニウム、7.5g/Lの二塩基リン酸カリウム、3.75g/Lの一塩基リン酸ナトリウム二水和物、及び1.25g/Lのクエン酸ナトリウム)を含む10リットル発酵器の播種に使用した。
発酵は30℃において、強く撹拌しながら通気し、NH4OH添加でpHを7.0に調節して行った。最初のグルコースが消費された後、無菌の50%グルコース溶液を与えて培地を維持した。約30ODにおいて、温度を25℃にシフトさせて発泡を最小にした。培地のODが約50(A550)に達したとき、25mg/mlのIAA(3-ベータ-インドールアクリル酸)25mlを添加してtrpプロモータに調節されたApo-2リガンド発現を誘発した。IAA添加の6〜10時間後に遠心分離により細胞を回収した。
【0125】
発現されたポリペプチドを以下のように精製した。大腸菌から発現されたApo-2リガンドを含む細胞ペーストを、0.1Mのトリス、0.2MのNaCl、50mMのEDTA、pH8バッファーで抽出した。次いで抽出物を40%の硫酸アンモニウムを用いて沈殿させた。全てのクロマトグラフィ工程は、特に示さない場合は室温で実施した。硫酸アンモニウム沈殿物を50mMのHEPES、0.05%のTriton x-100、pH8バッファー中に溶解させ、次いで50mMのHEPES、0.05%のTriton-x100、pH8バッファーで平衡させたマクロ-prepヒドロキシアパタイトのカラムに80cm/時間の流速で適用した。カラムを平衡バッファーでA280での吸収がベースライン近くに戻るまで洗浄した。Apo-2リガンドを0から0.2Mリン酸ナトリウム平衡バッファーの線形勾配で8カラム容量によってカラムから溶離した。Apo-2リガンドを含む分画をプールし、pHを6.5に調節した。pH調節したプールを、0.35MのNaCl/PBSバッファーで平衡させたNi-NTAスーパーフローのカラムに80cm/時間の流速で負荷した。カラムを平衡バッファーで洗浄して吸収をベースライン近くにした。Apo-2リガンドを0から50mMイミダゾール/平衡バッファーの線形勾配で8カラム容量によってカラムから溶離した。Apo-2リガンドを含む分画をプールし、Millipore Lab TFF system, Biomax 8 膜を用いて濃縮した。濃縮したプールを、20mMのトリス、8%のトレハロース、0.01%トゥイーン20バッファー中のG-25カラムで調製した。調製したApo-2リガンドプールを0.22ミクロンフィルターを通して濾過した。
精製した物質の分析により、それが(約50:50の比率で)アミノ酸91−281(図1A参照))を有するApo-2リガンドポリペプチド、及びアミノ酸92−281(図1A参照を有するApo-2リガンドポリペプチドを含むことが公にされた(この比率は現在のところ潜在的N末端加工によると考えられており;図1Aに示したアミノ酸残基91はメチオニン残基である)。
【0126】
実験において、ヌードマウスにHCT116ヒト結腸癌細胞(ATCC CCL 247)(1x106細胞)を動物の各側に皮下注射した。次いで腫瘍を直径約1cmとなるまで(およそ10日間)成長させた。第0日に、腫瘍容積をノギスで測定した。容積は、a=長さ及びb=幅としたときのπ/6xab2として計算した。次いで、浸透ミニポンプ(Alza Corp.から購入、Model 1003)を(1)対照媒体(20mMのトリスpH7.5、8%のトレハロース、0.01%のトゥイーン-20)又は(2)Apo-2リガンドのいずれかで負荷した。各群の5匹の動物は対照媒体又はApo-2リガンドのいずれかを受容する。ミニポンプを較正して毎日10mg/kgのApo-2リガンド(10mg/mlx2μl/時間)又は2μl/時間の対照媒体を輸送した。
第3日目に、腫瘍容積を再度測定し、動物を犠牲にした。動物の試験は毒性の如何なる証拠も示さなかった。図15において、結果は腫瘍容積のパーセント変化を示す。媒体群には腫瘍容積の変化は無かったが、Apo-2Lを注入したマウスでは腫瘍サイズの顕著な縮小が起こった。Apo-2リガンドでの処理は、3日に渡って腫瘍を約50%に収縮させた。
【0127】
(実施例17)
Apo-2リガンド置換変異体の調製
レセプター結合及びアポトーシス誘発活性にとって重要なエピトープを発見し記述するためにアラニンスキャニング突然変異誘発を用いた。アラニンスキャニング突然変異誘発は、Kelley等, Biochemistry, 34: 10383-10392 (1995)及びCunningham及びWells, Sience, 244: 1081-1085 (1989)に記載されているように、結合性エピトープを同定するのに用いられる。一般的にアミノ酸アラニンをコードする特全変異が他の残基に代えてここに記載された方法によって導入される。アラニンは、先端を切ったような側鎖(メチル基のみ)を有するので選択される置換基である。さらに、アラニンは表面及びタンパク質内部に埋まった状態の両方で見出されるので、発現されたタンパク質の構造一体性を乱す可能性が最も低いと思われる。
アラニン置換タンパク質をコードするプラスミドをオリゴヌクレオチド指向性突然変異誘発(Kunkel, T.A.等, Methods in Enzymology, 154: 367-382 (1987))を用いてプラスミドpAPOK5の一本鎖形態上で作成した(図16参照)。トリプトファン(trp)プロモーターによるApo-2Lの91−281形態の細胞内大腸菌発現のために、このプラスミドを設計した。pAPOK5はApo-2LcDNA(残基91−281をコードする)をtrpプロモーターを持つプラスミドpS1162にクローニングするPCRを用いて作成した。全てのアラニン置換基はジデオキシヌクレオチド配列により確認した(Sanger, F.等, Proc. Natl. Acad.. Sci., 74: 5463-5467 (1977))。
アラニン置換されたApo-2Lタンパク質をコードするプラスミドは、次いで発現のために大腸菌株294に形質転換した。培地はルリアブロスに50μg/mlのカルベニシリンを追加したものの中で37℃において終夜成長させて飽和させた。続いて細胞を、Na2HPO4(6g/L)、KH2PO4(3g/L)、NaCl(0.5g/L)、NH4Cl(1g/L)、グルコース(4.9g/L)、カサミノ酸(casamino acids)(4.9g/L)、27mMのMgSO4、0.003%のチアミンHCl、及び50μg/mLのカルベニシリンの20倍希釈物を追加した無菌フィルター培地に蒔き、37℃で1時間成長させた。
【0128】
次いで、25μg/mLでの3-β-インドールアクリル酸(IAA)(Sigma, St.Louis, MO.)で発現を誘発し、細胞を30℃で振盪させながら終夜成長させた。細胞を遠心分離で回収し、以下に述べるようなApo-2Lの引き続く回収のために−20℃で凍結貯蔵した。
Apo-2Lタンパク質は、10容量(重量/容量)の100mMトリス、pH8.0/200mMのNaCl/5mlのEDTA/1mMのDTT中で、モデルM110-Fマイクロフリューダイザ(Microfluidics Corporation, Newton, MA.)を用いた均質化により凍結大腸菌細胞ペレットから抽出した。ポリエチレンイミン(PEI)を最終濃度0.5%(容量/容量)でホモジェネートに添加し、それを次いで遠心分離して細胞断片を除去した。個体硫酸アンモニウムを抽出上清に室温で撹拌しながら飽和の45%という最終濃度で添加し、ペレットを遠心分離で回収した。硫酸アンモニウムペレットを50%硫酸アンモニウム溶液で洗浄して残ったEDTAを除去し、次いで50容量(重量/容量)の50mMのEPPS、pH7.5/0.1%トリトンX-100中に再懸濁した。得られた溶液を遠心分離で透明化し、5mLのHiTrapキレート化セファロースTMがカラム(Pharmacia, Piscataway, NJ.)を用いた固定化金属アフィニティクロマトグラフィ(IMAC)により精製した。100mMのNiSO4/300mMのトリス、pH7.5中のニッケルでカラムを負荷し、リン酸緩衝塩水(PBS)中の350mMのNaClで平衡化した。負荷後、カラムをPBS中350mMのNaClで洗浄し、50mMのイミダゾール/350mMのNaCl(PBS中)で溶離した。IMAC溶離物を20mMのトリス、pH7.5に対して透析し、遠心分離で透明化し、5mLのTiTrapSPセファロースTMがカラム(Pharmacia)を用いたカチオン交換クロマトグラフィーで更に精製し、それを20mMトリス、pH7.5で平衡化して洗浄した。HiTrapSPカラムを20mMのトリス、pH7.5/0.5MのNaClで溶離した。Spカラム溶離物は2mMのDTTで還元し、続いて室温で撹拌しながら飽和の45%という最終濃度まで固体硫酸アンモニウムを添加することにより沈殿させた。硫酸アンモニウムペレットを、3.5mLの20っMトリス、pH7.5/100mMのNaClに再懸濁し、PD10カラム(Pharmacia)を用いたゲル濾過クロマトグラフィーにより20mMトリス、pH7.5/100mMのNaClという最終バッファーに交換した。精製したApo-2Lアラニン置換タンパク質は、クマシーブルー染色SDS−PAGE及びN末端配列決定によって特徴付けし、−20℃で凍結保存した。
【0129】
(実施例18)
Apo-2L置換変異体を用いたアポトーシス活性
上記実施例17に記載したように調製した幾つかのApo-2リガンド置換変異体のアポトーシス活性を決定するためにインビトロアッセイを行った。
SK-MES-1ヒト肺癌細胞(ATCC HTB 58)を、10%FBS、2mMのL-グルタミン、100U/mlのペニシリン、及び100マイクログラムのストレプトマイシンを添加したDMEM:HamのF-12(50:50)培地で培養した。SK-MES-1細胞を96ウェルのファルコン組織培地で処理したミクロプレートに、4.0x104細胞/ウェルの密度で0.1mlの容量で蒔き、37℃で終夜接着させた。種々のApo-2L置換変異体又はApo-2L(図1Aのアミノ酸残基91−281からなる;Apo-2L.2と呼ぶ)を24時間添加してアポトーシスを誘発した。試験タンパク質での処理に続いて、培地を除去し、ウェルをメタノール中のクリスタルバイオレットの0.5%溶液で15分間染色した。SLT 340 ATCプレートリーダー(Salzburg, Austria)を用いて540nmで乾燥プレートの光学密度を測定することにより細胞数を決定した。
結果を図17及び18に示した。図17は変異体D218A、D269A、及びV207AのバイオアッセイデータをApo-2L.2分子と比較して示す(V207Aは図1のアミノ酸91−281を含むが図1Aの配列の位置207におけるバリンがアラニンに置換されたと定義される置換変異体である)。変異体D218A及びD269Aは、Apo-2L.2に比較した場合、各々約3倍及び7倍増加したアポトーシス活性を示した。V207AはバイオアッセイにおいてApo-2L.2より約9倍小さな活性であった。
図18は、Apo-2L.2と比較したD203Aのバイオアッセイデータを示す。変異体D203Aは、Apo-2L.2に比較した場合、約2倍の増大したアポトーシス誘発活性を示した。
【0130】
(実施例19)
Apo-2リガンド置換変異体の結合分析
Apo-2Lアラニン-置換タンパク質(上記実施例18で参照したD203A、D218A及びD269A)を、文献に記載された種々のApo-2Lレセプター、DR4[Pan等, Science, 276: 111-113 (1997)];DR5[Sheridan等, Science, 277: 818-821 (1997)];デコイレセプターDcR2[Marsters等, Current Biology, 7: 1003-1006 (1997)];及びOPG[simonet等, Cell, 89: 309-319 (1997)]に対する結合性をBIAcore(Pharmacia)を用いて分析した。比較のため、アミノ酸91−281からなるApo-2LもBIAcoreにおいてIgG-レセプター融合分子への結合性を試験した。Apo-2Lレセプタータンパク質の各々を、Ashkenazi等, Proc. Natl. acad. Sci., 88: 10535-10539 (1991)に実質的に記載されているようにイムノアドヘシンとして調製した。IgG-レセプター融合分子は、CM5 BIAcoreチップ、研究等級(Pharmacia)にNHS-EDCアミンカップリング化学を用いて固定化した。Apo-2Lアラニン置換タンパク質をBIAcoreランニングバッファー、PBS/0.05%トゥイーン20で500nMから15.625nMまで連続的に2倍希釈して、固定化レセプターを有するセンサーチップ表面を横切って通過させた。KD値を決定するためにBIAcore評価ソフトウェアを(製造者の指示に従って)使用して特異的動的結合パラメータを用いた。
結果を図19に示す。この表は、BIAcore(Pharmacia)を用いた動的結合分析によって決定した場合のApo-2L.2と比較した変異体D203A、D218A及びD269Aの解離定数(KD)を示す。データは、Apo-2L.2と比較した場合、アラニン置換変異体類のIgG-レセプター融合タンパク質DR4、DR5、及びDcR2に対する結合性に有意な変化(>2倍)は無かった。OPGR-IgGへの結合の場合、変異体D203A、D218A、及びD269Aは、Apo-2L.2に比較してより緊密な結合性を示した(2<KDwt<KDmut<4)。
【0131】
材料の寄託
次の細胞系をアメリカン・タイプ・カルチャー・コレクション,10801 University Boulevard., Manassas, VA, USA(ATCC)に寄託した:
細胞系 ATCC寄託番号 寄託日
2935-pRK5-hApo-2L-myc clone2.1 CRL-12014 1996年1月3日
1D1.12.4 HB-12257 1997年1 月8日
2G6.3.4 HB-12259 1997年1 月8日
2E11.5.5 HB-12256 1997年1 月8日
5C2.4.9 HB-12258 1997年1 月8日
【0132】
この寄託は、特許手続き上の微生物の寄託の国際的承認に関するブダペスト条約及びその規則(ブダペスト条約)の規定に従って行われた。これは、寄託の日付から30年間、寄託の生存可能な培養が維持されることを保証するものである。寄託物はブダペスト条約の条項に従い、またジェネンテク社とATCCとの間の合意に従い、ATCCから入手することができ、これは、どれが最初であろうとも、関連した米国特許の発行時又は任意の米国又は外国特許出願の公開時に、寄託培養物の後代を永久かつ非制限的に入手可能とすることを保証し、米国特許法第122条及びそれに従う特許庁長官規則(特に参照番号886OG638の37CFR第1.14条を含む)に従って権利を有すると米国特許庁長官が決定した者に子孫を入手可能とすることを保証するものである。
本出願の譲受人は、寄託した培養物が、適切な条件下で培養されていた場合に死亡もしくは損失又は破壊されたならば、材料は通知時に同一の他のものと即座に取り替えることに同意する。寄託物質の入手可能性は、特許法に従いあらゆる政府の権限下で認められた権利に違反して、本発明を実施するライセンスであるとみなされるものではない。
上記の文書による明細書は、当業者に本発明を実施できるようにするために十分であると考えられる。寄託した態様は、本発明のある側面の一つの説明として意図されており、機能的に等価なあらゆる作成物がこの発明の範囲内にあるため、寄託された作成物により、本発明の範囲が限定されるものではない。ここでの物質の寄託は、ここに含まれる文書による説明が、そのベストモードを含む、本発明の任意の側面の実施を可能にするために不十分であることを認めるものではないし、それが表す特定の例証に対して請求の範囲を制限するものと解釈されるものでもない。実際、ここに示し記載したものに加えて、本発明を様々に改変することは、前記の記載から当業者にとっては明らかなものであり、添付の請求の範囲内に入るものである。
【図面の簡単な説明】
【0133】
【図1A】ヒトApo-2リガンドcDNAの核酸配列及びその誘導アミノ酸配列を示す図である。
【図1B】ヒトApo-2リガンドのC末端領域の、ヒトTNFサイトカインファミリーの知られたメンバー、4-1BBL、OX40L、CD27L、CD30L、TNF-α、LT-β、LT-α、CD40L、及びApo-1Lの対応する領域とのアラインメントを示す図である。
【図1C−1E】(C)抗-mycエピトープ抗体を用いたFACS分析で測定した場合の、ヒト293細胞で発現された組換え、全長、C末端mycエピトープタグApo-2リガンドの細胞トポロジーを示す図である。(D)化学的架橋有り(レーン2、3)又は無し(レーン1)に次いでSDS-PAGE及び銀染色で測定した場合の、組換えバキュロウイルス感染昆虫細胞で発現されNi2+-キレートアフィニティクロマトグラフィで精製した組換えHis10エピトープタグ可溶性Apo-2のサイズ及びサブユニット構造を示す図である。(E)抗-gDエピトープ抗体での免疫沈降に次いでSDS-PAGE及びオートラジオグラフィで測定した場合の、代謝性標識されたヒト293細胞で発現された組換えgDエピトープタグ可溶性Apo-2リガンドのサイズ及びサブユニット構造を示す図である。
【図2A】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞は特徴的な形態変化により同定した。
【図2B】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2C】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2D】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はフローサイトメトリーで測定したヨウ化プロピジウム(PI)及びFITC複合アネキシンVでの正の蛍光染色で同定した。
【図2E】Apo-2リガンドによるB及びTリンパ球細胞系におけるアポトーシスの誘発を示す図である。アポトーシス細胞はヌクレオソーム内DNA断片化の分析(E)により同定した。
【図3A】Apo-2リガンド誘発アポトーシスの時間変化を示す図である。
【図3B】Apo-2リガンド誘発アポトーシスの用量依存性を示す図である。
【図3C】Fas/Apo-1レセプター、TNF-R1レセプター、又はTNF-R2レセプターに基づく可溶性レセプター−IgG融合タンパク質によるApo-2リガンド誘発アポトーシスの阻害欠如を示す図である。
【図4】ノーザンブロット分析で測定した場合の、ヒト胎児及びヒト成人組織におけるApo-2リガンドmRNAの発現を示す図である。
【図5】ヌードマウスにおいて成長させたヒトMDA231乳癌細胞に基づく腫瘍の重量に対する、単独又はドキソルビシンと混合して腫瘍内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図6】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍の重量に対する、単独又は5-FUと混合して腫瘍内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図7】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍のサイズに対する、単独又は5-FUと混合して腹膜内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図8】ヌードマウスにおいて成長させたヒトHCT116結腸癌細胞に基づく腫瘍の重量に対する、単独又は5-FUと混合して腹膜内注射で投与したApo-2リガンドのインビボ効果を示す図である。
【図9】ドミナントネガティブFADDではなくCrmAがHeLa-S3細胞におけるApo-2リガンド誘発アポトーシスを阻止することを示す棒グラフである。
【図10】Apo-2リガンドに誘発されたアポトーシス及び4つの抗-Apo-2リガンド抗体:1D1、2G6、2E11及び5C2による影響のFACS分析を示す図である(FITC複合アネキシンVを用いて検出したアポトーシス9D細胞−太線;生存、非染色細胞−細線)。
【図11】モノクローナル抗体1D1、2G6、2E11、及び5C2の抗原特異性を示す棒グラフである。
【図12】モノクローナル抗体1D1、2G6、2E11、及び5C2のエピトープマッピングアッセイの結果を示す棒グラフである。
【図13】Apo-2リガンドの特異的アミノ酸領域からなる数種の合成ペプチドに対するモノクローナル抗体1D1が結合する能力を試験するアッセイの結果を示す棒グラフである。
【図14A−D】発現されたApo-2リガンドを含有するCHO細胞培地上清(1:10、1:20、1:40希釈)又は非条件培地で処理した培養HeLa細胞を示す図である。
【図14E】上記各視野におけるアポトーシス細胞の数を示す棒グラフである。
【図15】(実施例16に記載したように)大腸菌で発現されたApo-2リガンドポリペプチドを浸透ミニポンプを介して投与したヌードマウスにおけるHCT116結腸癌腫瘍容積におけるパーセント(%)変化を示す図である。
【図16】pAPOK5プラスミドの制限マップを示す図である。
【図17】バイオアッセイにおいて、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D218A、D269A及びD207Aの、アポトーシス細胞死を誘発する能力を示す図である。
【図18】バイオアッセイにおいて、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D203Aのアポトーシス細胞死を誘発する能力を示す図である。
【図19】BIAcore(Pharmacia)を用いた動力学的結合アッセイで測定した場合の、図1Aのアミノ酸91−281からなるApo-2リガンド(「Apo2L.2」)と比較した場合の、Apo-2リガンド置換変異体D203A、D218A、及びD269Aの解離定数(KD)を示す表である。
【特許請求の範囲】
【請求項1】
図1A(配列番号:1)のアミノ酸残基91〜281を含んでなる単離された可溶性Apo-2リガンドポリペプチド。
【請求項2】
図1A(配列番号:1)のアミノ酸残基92〜281を含んでなる請求項1に記載のApo-2リガンドポリペプチド。
【請求項3】
図1A(配列番号:1)のアミノ酸残基91〜281からなる単離された可溶性Apo-2リガンドポリペプチド。
【請求項4】
図1A(配列番号:1)のアミノ酸残基91〜281から実質的になる単離された可溶性Apo-2リガンドポリペプチド。
【請求項5】
図1A(配列番号:1)のアミノ酸残基92〜281から実質的になる単離された可溶性Apo-2リガンドポリペプチド。
【請求項6】
請求項1に記載のApo-2リガンドポリペプチドと少なくとも約80%のアミノ酸配列同一性を有する単離されたApo-2リガンドポリペプチド。
【請求項7】
前記ポリペプチドが少なくとも約90%のアミノ酸配列同一性を有する請求項6に記載のApo-2リガンドポリペプチド。
【請求項8】
前記ポリペプチドが少なくとも約95%のアミノ酸配列同一性を有する請求項7に記載のApo-2リガンドポリペプチド。
【請求項9】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置203のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項10】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置218のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項11】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置269のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項12】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置203、218及び269のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項13】
前記ポリペプチドが非タンパク質性ポリマーに結合した請求項1に記載のApo-2リガンドポリペプチド。
【請求項14】
前記非タンパク質性ポリマーがポリエチレングリコールである請求項13に記載のApo-2リガンドポリペプチド。
【請求項15】
異種ポリペプチド配列に融合した請求項1に記載のApo-2リガンドポリペプチドを含んでなるキメラポリペプチド。
【請求項16】
異種ポリペプチド配列がタグポリペプチド配列である請求項15に記載のキメラポリペプチド。
【請求項17】
請求項1に記載のApo-2リガンドポリペプチドをコードするDNAを含んでなる単離された核酸。
【請求項18】
請求項17に記載の核酸を含んでなるベクター。
【請求項19】
請求項18に記載のベクターを含んでなる宿主細胞。
【請求項20】
前記宿主細胞がCHO細胞を含んでなる請求項19に記載の宿主細胞。
【請求項21】
前記宿主細胞が大腸菌を含んでなる請求項19に記載の宿主細胞。
【請求項22】
前記宿主細胞が酵母細胞を含んでなる請求項19に記載の宿主細胞。
【請求項23】
請求項19に記載の宿主細胞を培養し、宿主細胞培地からApo-2リガンドポリペプチド回収することを含んでなるApo-2リガンドポリペプチドの作成方法。
【請求項24】
全長ヒトApo-2リガンドをコードする遺伝子をCHO細胞で発現させることにより作成された可溶性Apo-2リガンドポリペプチド。
【請求項25】
前記ポリペプチドが天然配列Apo-2リガンドの細胞外ドメインからなる請求項24に記載のApo-2リガンドポリペプチド。
【請求項26】
前記ポリペプチドが図1Aのアミノ酸残基92〜281を含む請求項24に記載のApo-2リガンドポリペプチド。
【請求項27】
請求項1に記載のApo-2リガンドポリペプチド及び担体を含んでなる組成物。
【請求項28】
担体中に請求項1に記載のApo-2リガンドポリペプチドの有効量を含有してなる哺乳動物細胞アポトーシスの刺激に有用な組成物。
【請求項29】
請求項1に記載のApo-2リガンドポリペプチドの有効量に哺乳動物癌細胞を暴露することを含んでなる哺乳動物癌細胞におけるアポトーシスを誘発する方法。
【請求項30】
前記Apo-2リガンドポリペプチドが癌を持つと診断された哺乳動物への注入により投与される請求項29に記載の方法。
【請求項1】
図1A(配列番号:1)のアミノ酸残基91〜281を含んでなる単離された可溶性Apo-2リガンドポリペプチド。
【請求項2】
図1A(配列番号:1)のアミノ酸残基92〜281を含んでなる請求項1に記載のApo-2リガンドポリペプチド。
【請求項3】
図1A(配列番号:1)のアミノ酸残基91〜281からなる単離された可溶性Apo-2リガンドポリペプチド。
【請求項4】
図1A(配列番号:1)のアミノ酸残基91〜281から実質的になる単離された可溶性Apo-2リガンドポリペプチド。
【請求項5】
図1A(配列番号:1)のアミノ酸残基92〜281から実質的になる単離された可溶性Apo-2リガンドポリペプチド。
【請求項6】
請求項1に記載のApo-2リガンドポリペプチドと少なくとも約80%のアミノ酸配列同一性を有する単離されたApo-2リガンドポリペプチド。
【請求項7】
前記ポリペプチドが少なくとも約90%のアミノ酸配列同一性を有する請求項6に記載のApo-2リガンドポリペプチド。
【請求項8】
前記ポリペプチドが少なくとも約95%のアミノ酸配列同一性を有する請求項7に記載のApo-2リガンドポリペプチド。
【請求項9】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置203のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項10】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置218のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項11】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置269のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項12】
前記ポリぺプチドが図1A(配列番号:1)の配列における位置203、218及び269のアスパラギン酸残基に置換したアラニン残基を有する置換変異体である請求項6に記載のApo-2リガンドポリペプチド。
【請求項13】
前記ポリペプチドが非タンパク質性ポリマーに結合した請求項1に記載のApo-2リガンドポリペプチド。
【請求項14】
前記非タンパク質性ポリマーがポリエチレングリコールである請求項13に記載のApo-2リガンドポリペプチド。
【請求項15】
異種ポリペプチド配列に融合した請求項1に記載のApo-2リガンドポリペプチドを含んでなるキメラポリペプチド。
【請求項16】
異種ポリペプチド配列がタグポリペプチド配列である請求項15に記載のキメラポリペプチド。
【請求項17】
請求項1に記載のApo-2リガンドポリペプチドをコードするDNAを含んでなる単離された核酸。
【請求項18】
請求項17に記載の核酸を含んでなるベクター。
【請求項19】
請求項18に記載のベクターを含んでなる宿主細胞。
【請求項20】
前記宿主細胞がCHO細胞を含んでなる請求項19に記載の宿主細胞。
【請求項21】
前記宿主細胞が大腸菌を含んでなる請求項19に記載の宿主細胞。
【請求項22】
前記宿主細胞が酵母細胞を含んでなる請求項19に記載の宿主細胞。
【請求項23】
請求項19に記載の宿主細胞を培養し、宿主細胞培地からApo-2リガンドポリペプチド回収することを含んでなるApo-2リガンドポリペプチドの作成方法。
【請求項24】
全長ヒトApo-2リガンドをコードする遺伝子をCHO細胞で発現させることにより作成された可溶性Apo-2リガンドポリペプチド。
【請求項25】
前記ポリペプチドが天然配列Apo-2リガンドの細胞外ドメインからなる請求項24に記載のApo-2リガンドポリペプチド。
【請求項26】
前記ポリペプチドが図1Aのアミノ酸残基92〜281を含む請求項24に記載のApo-2リガンドポリペプチド。
【請求項27】
請求項1に記載のApo-2リガンドポリペプチド及び担体を含んでなる組成物。
【請求項28】
担体中に請求項1に記載のApo-2リガンドポリペプチドの有効量を含有してなる哺乳動物細胞アポトーシスの刺激に有用な組成物。
【請求項29】
請求項1に記載のApo-2リガンドポリペプチドの有効量に哺乳動物癌細胞を暴露することを含んでなる哺乳動物癌細胞におけるアポトーシスを誘発する方法。
【請求項30】
前記Apo-2リガンドポリペプチドが癌を持つと診断された哺乳動物への注入により投与される請求項29に記載の方法。
【図1A】


【図1B】

【図1C−1E】
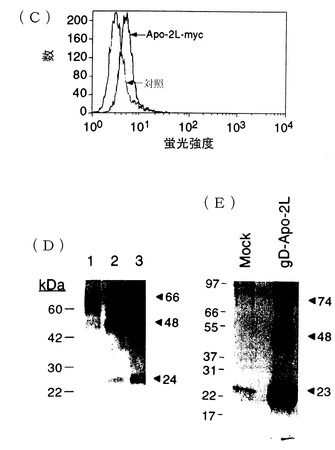
【図2A】

【図2B】
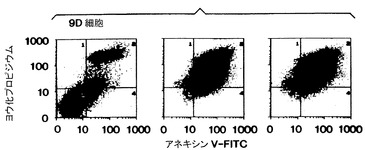
【図2C】

【図2D】

【図2E】
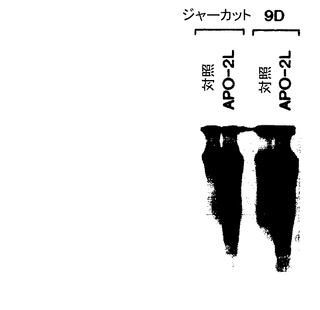
【図3A】

【図3B】
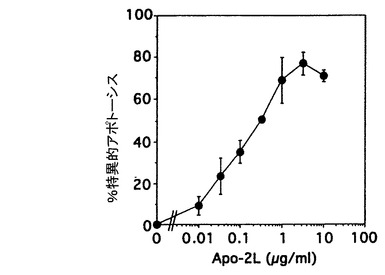
【図3C】
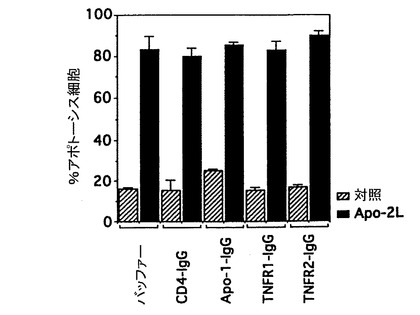
【図4】
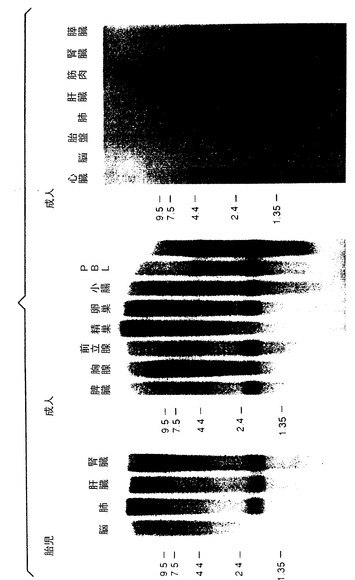
【図5】
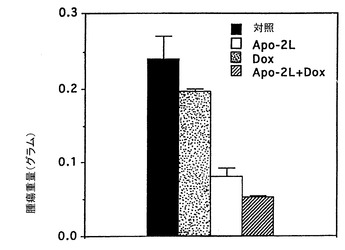
【図6】
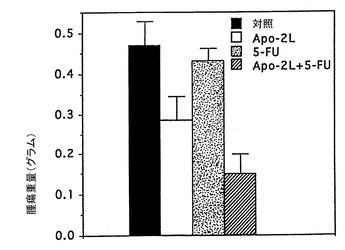
【図7】
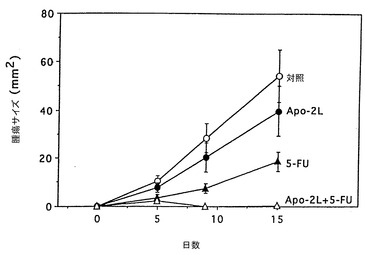
【図8】
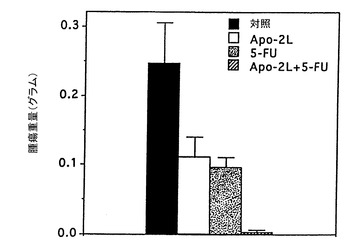
【図9】

【図10】
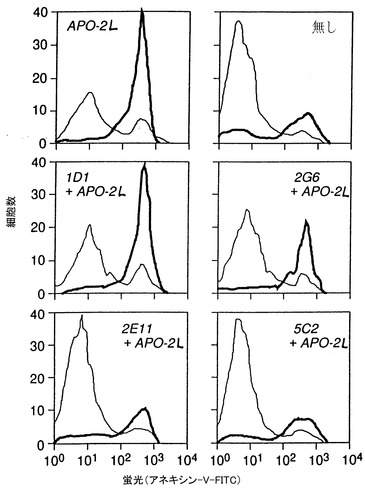
【図11】
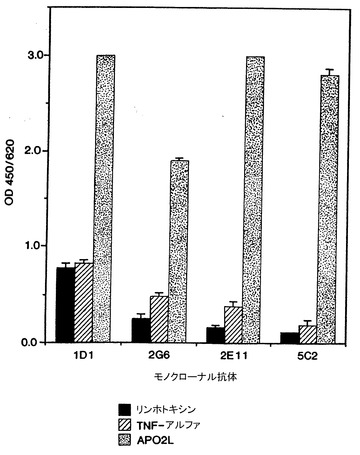
【図12】
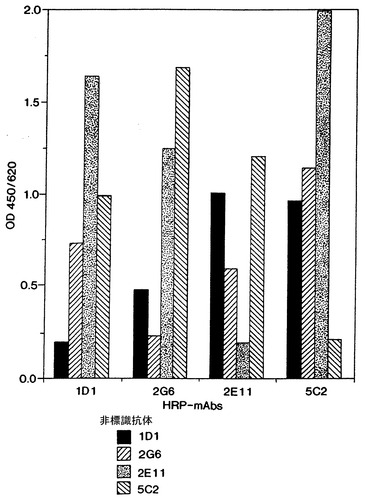
【図13】
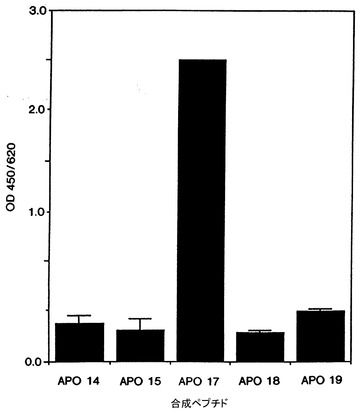
【図14A−B】

【図14C−D】
【図14E】
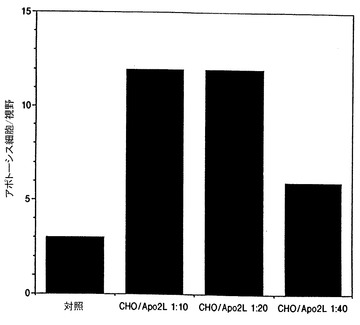
【図15】

【図16】

【図17】
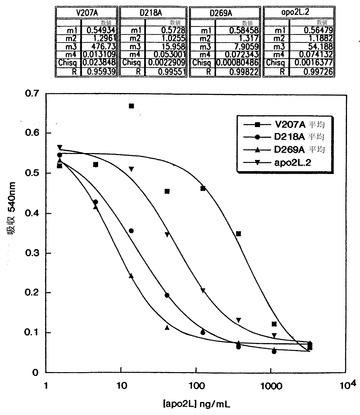
【図18】

【図19】
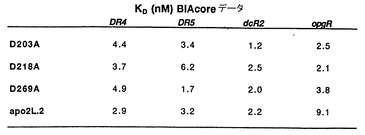
【図1B】
【図1C−1E】
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3A】
【図3B】
【図3C】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A−B】
【図14C−D】
【図14E】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2010−246560(P2010−246560A)
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−151519(P2010−151519)
【出願日】平成22年7月2日(2010.7.2)
【分割の表示】特願2000−540237(P2000−540237)の分割
【原出願日】平成11年1月15日(1999.1.15)
【出願人】(509012625)ジェネンテック, インコーポレイテッド (357)
【Fターム(参考)】
【公開日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願番号】特願2010−151519(P2010−151519)
【出願日】平成22年7月2日(2010.7.2)
【分割の表示】特願2000−540237(P2000−540237)の分割
【原出願日】平成11年1月15日(1999.1.15)
【出願人】(509012625)ジェネンテック, インコーポレイテッド (357)
【Fターム(参考)】
[ Back to top ]