
Clostridiumjosui由来のCel48Aセルラーゼ、ならびにその生産のための宿主及び方法
【課題】Clostridium josui由来Cel48Aセルラーゼ及びそれを生産するための宿主および方法の提供。
【解決手段】Clostridium josui由来Cel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。当該組換えバチルス属細菌を用いることを特徴とするClostridium josui由来Cel48Aセルラーゼの生産方法。当該生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
【解決手段】Clostridium josui由来Cel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。当該組換えバチルス属細菌を用いることを特徴とするClostridium josui由来Cel48Aセルラーゼの生産方法。当該生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、Clostridium josui由来のCel48Aセルラーゼ、ならびにそれを生産するための新規な微生物宿主及び生産方法に関する。
【背景技術】
【0002】
セルラーゼは、セルロースのβ−1,4グルコシド結合を加水分解する酵素の総称であり、アミラーゼ、プロテアーゼ、リパーゼと共に加水分解酵素の代表的存在である。これまでに、真菌類や嫌気性細菌由来の中酸性に最適反応pHを有するセルラーゼが主に見出されているが、一方、好アルカリ性バチルス属細菌由来のアルカリ性に最適反応pHを有するアルカリセルラーゼもまた見出されている。
【0003】
セルラーゼの基質となるセルロースは、植物細胞壁の主成分として年間1000億トン以上も生産されるバイオマスであり、衣料、紙、建築材料等に有効利用されている。またセルロースは、グルコースが直鎖状に結合した巨大分子であるため、分解によって燃料物質やより高付加価値の代謝物質に変換が可能である。そのため、セルロースの効率的分解や、その分解産物の有効利用に関する研究が広く行われている。セルロースを高付加価値物質に変換する場合、通常、セルロース原料を前処理によって高反応性セルロースに変換した後、グルコースなどの低分子糖に分解する。得られた低分子糖は微生物による発酵等の化学反応を経て、高付加価値物質へと変換される。近年、この一連のセルロース分解工程においては、より経済的な方法として、セルラーゼよる酵素分解が一般に利用されている。
【0004】
しかし現状では、セルラーゼによるセルロース分解の速度は充分ではなく、必要な分解速度を達成するためには多量の酵素を使用するか、あるいは多種類の酵素を混合して使用しなければならない。またこのことは必然的に、酵素に多額のコストがかかる結果となる。より効率的且つ経済的にセルロース分解産物を生産するために、セルラーゼライブラリの拡充及びより高活性のセルラーゼの開発が求められている。
【0005】
Clostridium属細菌は、偏性嫌気性のグラム陽性細菌であり、セルロソームという高分子酵素複合体を作り、セルロースを効率良く分解することが知られている。その中のセルラーゼの主たるコンポーネントとして、糖加水分解酵素(glycoside hydrolase; GH)ファミリー48に属するCel48が知られている。Clostridium属のCel48については、C. thermocellumのCelS(Cel48A)に関して多くの報告があり、遺伝子取得(非特許文献1)、大腸菌による発現(非特許文献2)、及び蛋白結晶構造解析(非特許文献3)がなされている。しかしながら、C. thermocellum以外のClostridium属のCel48Aについての報告は少ない。例えば、Clostridium josui由来のCelD(Cel48A)に関しては、遺伝子取得はなされているものの(非特許文献4)、実際に蛋白を単離・同定するには至っていない。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】J Bacteriol, 175(5), 1293-1302, 1993
【非特許文献2】J Bacteriol, 177(6), 1641-1644, 1995
【非特許文献3】J. Mol. Biol., 320, 587-596
【非特許文献4】J Bacteriol, 180(16), 4303-4308, 1998
【発明の概要】
【発明が解決しようとする課題】
【0007】
Clostridium属由来セルラーゼを製造する場合、外来タンパク質を発現させるための方法として従来一般的に行われてきた大腸菌を用いて組換え蛋白質を発現させる方法では、Clostridium josui由来Cel48Aにおいては組換え蛋白質の発現が見られていない。本発明は、Clostridium josui由来のCel48Aセルラーゼ、ならびにそのより効率的な生産を可能にする宿主微生物及び生産方法を提供する。
【課題を解決するための手段】
【0008】
本発明者らは、Clostridium josui由来のCel48Aセルラーゼをより効率的に発現させる方法について検討を行ったところ、バチルス属細菌を宿主に用いることにより、大腸菌を用いた従来法では発現させることができなかった菌株由来のセルラーゼを得ることが可能となった。
【0009】
すなわち本発明は、以下を提供するものである。
(1)Clostridium josui由来のCel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。
(2)前記Cel48Aセルラーゼ遺伝子を含むベクターを含む(1)記載の組換えバチルス属細菌。
(3)前記Cel48Aセルラーゼ遺伝子が配列番号1で示される塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、(1)又は(2)に記載の組換えバチルス属細菌。
(4)前記Cel48Aセルラーゼ遺伝子の上流に転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域から選ばれる1以上の領域が作動可能に結合されている、(1)〜(3)のいずれか1に記載の組換えバチルス属細菌。
(5)転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されている、(4)記載の組換えバチルス属細菌。
(6)分泌シグナル領域がバチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域由来のものである、(5)記載の組換えバチルス属細菌。
(7)転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、上記いずれかの塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片である、(5)記載の組換えバチルス属細菌。
(8)前記Cel48Aセルラーゼ遺伝子が配列番号1の塩基番号94〜2160の塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、(1)〜(7)のいずれか1に記載の組換えバチルス属細菌。
(9)前記バチルス属細菌が枯草菌又はその変異株である、(1)〜(8)のいずれか1に記載の組換えバチルス属細菌。
(10)(1)〜(9)のいずれか1に記載の組換えバチルス属細菌を用いることを特徴とする、Clostridium josui由来Cel48Aセルラーゼの生産方法。
(11)(10)記載の生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
(12)以下の性質を有する(11)記載のセルラーゼ:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、及びCellulose powderを分解し、還元糖を生成する;
最適反応pH:約7;
最適反応温度:約60℃(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である;
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、65℃まで安定である。
分子量:SDS−PAGEで、77〜79kDaである。
【発明の効果】
【0010】
本発明の組換えバチルス属細菌は、Clostridium josui由来Cel48Aセルラーゼを高効率で産生するための宿主微生物として有用である。当該組換えバチルス属細菌を用いる本発明のClostridium josui由来Cel48Aセルラーゼの生産方法によれば、Clostridium josui由来Cel48Aセルラーゼを効率よく生産することができる。また、本発明により提供されるClostridium josui由来Cel48Aセルラーゼは、高いセルロース分解活性を有する有用な酵素である。したがって、本発明によれば、より効率的且つ経済的なセルロース分解産物の生産を可能にするセルラーゼを効率よく安価に提供することが可能となり、ひいてはセルロースを原料とする燃料物質や高付加価値物質をより効率的且つ安価に提供することが可能となる。
【図面の簡単な説明】
【0011】
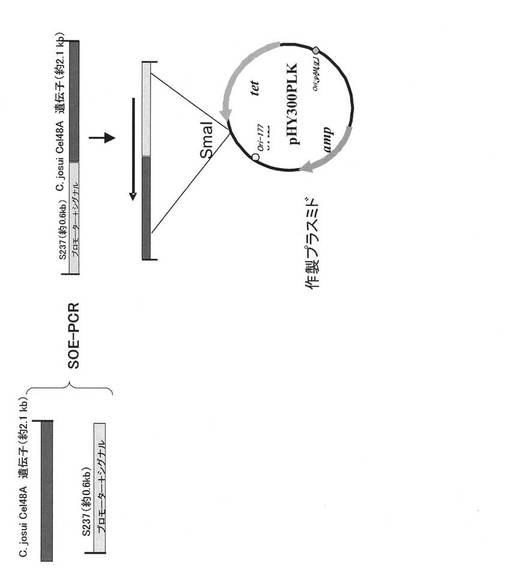
【図1】Clostridium josuiセルラーゼをコードするベクターの作製手順。
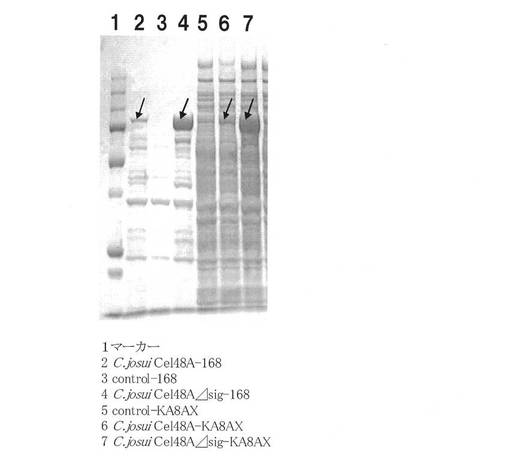
【図2】形質転換体培養液のSDS-PAGE電気泳動像。
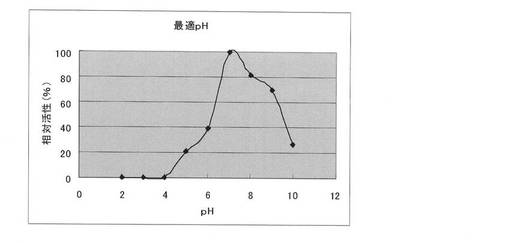
【図3】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの最適pH。
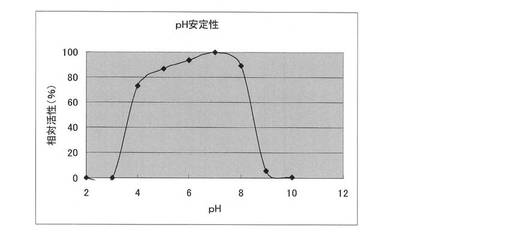
【図4】本発明の方法により取得したClostridium josui Cel48AセルラーゼのpH安定性。
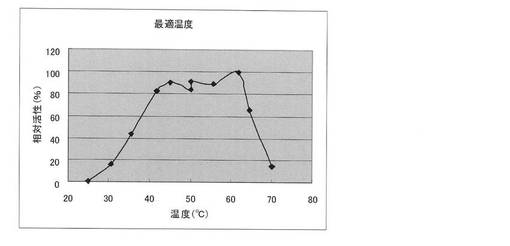
【図5】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの最適温度。
【図6】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの温度安定性。
【発明を実施するための形態】
【0012】
本発明の組換えバチルス属細菌は、バチルス属細菌にClostridium josui由来のCel48Aセルラーゼをコードする遺伝子を導入することによって得ることができる。本発明の組換えバチルス属細菌の親菌株としては、バチルス属細菌であれば特に限定されないが、好ましくは枯草菌及びその変異株が挙げられる。枯草菌及びその変異株の例としては、枯草菌168株、及び蛋白質産生のための宿主として公知の各種プロテアーゼ欠損枯草菌変異株が挙げられる。プロテアーゼ欠損枯草菌変異株の例としては、特開2006-174707に記載のプロテアーゼ9重欠損株(KA8AX)等が挙げられる。
【0013】
本発明の組換えバチルス属細菌にはClostridium josui由来のCel48Aセルラーゼをコードする遺伝子を導入する。あるいは、セルラーゼ遺伝子は、配列番号1で示される塩基配列からなるセルラーゼ遺伝子、又は当該塩基配列と70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の配列同一性を有し、且つセルラーゼ活性を有する蛋白質をコードする遺伝子である。またあるいは、セルラーゼ遺伝子は、配列番号1の塩基番号1〜2160もしくは塩基番号94〜2160で示される塩基配列からなるセルラーゼ遺伝子、又は当該塩基配列と70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の配列同一性を有し、且つセルラーゼ活性を有する蛋白質をコードする遺伝子である。
【0014】
本発明において、塩基配列及びアミノ酸配列の同一性はLipman-Pearson法 (Science,227,1435,1985)によって計算される。具体的には、遺伝情報処理ソフトウェアGenetyx-Win(ソフトウェア開発)のホモロジー解析(Search homology)プログラムを用いて、Unit size to compare(ktup)を2として解析を行うことにより算出される。
【0015】
上記セルラーゼ遺伝子は、例えばClostridium josui(FERMP-9684等)から、当該分野で用いられる任意の方法を用いて単離することができる。例えば、当該遺伝子は、Clostridium josuiから全ゲノムDNAを抽出した後、目的遺伝子の塩基配列(例えば、配列番号1の塩基配列)を元に設計したプライマーを用いたPCRにより当該遺伝子を選択的に増幅し、増幅した遺伝子を精製することで得ることができる。あるいは、当該セルラーゼ遺伝子は、公開されているその塩基配列又はそれがコードする蛋白質のアミノ酸配列に基づいて、遺伝子工学的又は化学的に合成することができる。
【0016】
次いで、上記で得られたセルラーゼ遺伝子を適切なベクターに導入する。本発明に使用されるベクターの種類としては、特に限定されず、タンパク質産生に通常用いられるベクター、例えばプラスミド、コスミド、ファージ、ウイルス、YAC、BAC等が挙げられる。プラスミドベクターが好ましく、例えば、市販のタンパク質発現用プラスミドベクター、例えばシャトルベクターpHY300PLK(TaKaRa製)、pUC19(TaKaRa製)、pUC119(TaKaRa製)、pBR322(TaKaRa製)等を好適に用いることができる。上記ベクターは、DNAの複製開始領域を含むDNA断片、複製起点を含むDNA領域を含む。また上記ベクターには、当該ベクターが適切に導入された微生物を選択するための薬剤耐性遺伝子(アンピシリン、ネオマイシン、カナマイシン、クロラムフェニコール等)がさらに組み込まれていてもよい。
【0017】
あるいは、上記ベクターにおいては、上記セルラーゼ遺伝子の上流に、当該遺伝子の転写、翻訳、分泌に関わる制御領域、即ち、プロモーター及び転写開始点を含む転写開始制御領域、リボソーム結合部位及び開始コドンを含む翻訳開始領域並びに分泌シグナルペプチド領域から選ばれる1以上の領域が作動可能に連結されていてもよい。好ましくは、転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が作動可能に連結されている。より好ましくは、分泌シグナルペプチド領域は、バチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域は、当該バチルス属細菌セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域である。
【0018】
例えば、上記Clostridium josui由来Cel48Aセルラーゼ遺伝子の上流には、特開2000−210081号公報や特開平4−190793号公報等に記載されているバチルス(Bacillus)属細菌、すなわちKSM-S237株(FERM BP-7875)、又はKSM-64株(FERM BP-2886)由来のセルラーゼ遺伝子の転写開始制御領域、翻訳開始領域及び分泌シグナルペプチド領域が作動可能に連結されていることが望ましい。より具体的には、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、配列番号4で示されるS237eglプロモーター及びシグナル配列(Biosci. Biotechnol. Biochem., 64(11):2281-9, 2000)、又は当該塩基配列に対して70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の同一性を有する塩基配列からなり、且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片、あるいは上記いずれかの塩基配列からなるDNA断片とストリンジェントな条件でハイブリダイズし、且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片が、作動可能に連結されていることが望ましい。
【0019】
本明細書において、転写開始制御領域、翻訳開始領域、又は分泌シグナルと遺伝子との「作動可能な連結」とは、上記制御領域が遺伝子の転写又は翻訳を誘導し得るように、又は遺伝子にコードされたタンパク質又はペプチドが分泌シグナルの働きにより分泌されるように、連結されていることをいう。上記制御領域又は分泌シグナルと遺伝子との「作動可能な連結」の手順は、当業者に周知である。また本明細書において、「塩基配列の一部が欠失した塩基配列からなるDNA断片」とは、上記塩基配列の一部、好ましくは数個(例えば、1〜50個、好ましくは1〜30個、より好ましくは1〜20個、さらに好ましくは1〜10個)の塩基を欠失しているが、遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片を意味する。また本明細書において、ハイブリダイゼーションにおける「ストリンジェントな条件」とは、例えば[Molecular cloning-a Laboratory manual 2nd edition(Sambrookら、1989)]に記載の条件等が挙げられる。例えば、6×SSC(1×SSCの組成:0.15M塩化ナトリウム、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mLニシン精子DNAを含む溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0020】
セルラーゼ遺伝子と上記制御配列、薬剤耐性遺伝子との連結は、SOE(splicing by overlap extension)−PCR法(Gene,77,61,1989)等の方法によって行うことができる。プラスミドベクターへの遺伝子の導入手順は当該分野で周知である。
【0021】
次いで、構築された上記ベクターをバチルス属細菌に導入すれば、本発明の組換えバチルス属細菌を得ることができる。好ましいバチルス属細菌は、枯草菌及びその変異体である。変異体としてはプロテアーゼ9重欠損株(KA8AX)等が挙げられる。ベクター導入の方法としては、プロトプラスト法、エレクトロポレーション等の当該分野で通常使用される方法を用いることができる。導入が適切に行われた株を薬剤耐性等を指標に選択することで、目的の形質転換体を得ることができる。
【0022】
あるいは、本発明の組換えバチルス属細菌は、バチルス属細菌のゲノムに上記セルラーゼ遺伝子を直接組み込むことによって得ることができる。たとえば、バチルス属細菌のゲノム上の遺伝子の転写、翻訳、分泌に関わる制御領域に相同なDNA断片と上記セルラーゼ遺伝子を含むDNA断片とを結合させたDNA断片を構築し、これを当該細菌に導入して細菌のゲノムと相同組換えすることによって、当該細菌のゲノム上の制御領域の下流に上記セルラーゼ遺伝子が組み込まれた組換えバチルス属細菌を得ることができる。上記のような相同組換えの手法は当業者に周知である。
【0023】
斯くして得られた組換えバチルス属細菌を適切な培地で培養すれば、当該細菌中でClostridium josui由来Cel48Aセルラーゼをコードする遺伝子が発現して、Clostridium josui由来Cel48Aセルラーゼ蛋白質が合成される。合成されたセルラーゼ蛋白質を該培養物から通常の方法により単離又は精製することにより、Clostridium josui由来Cel48Aセルラーゼ蛋白質を取得することができる。したがって、本発明はまた、本発明の組換えバチルス属細菌を用いることを特徴とするClostridium josui由来Cel48Aセルラーゼの生産方法を提供する。培養に使用する培地は、バチルス属細菌の培養に通常使用されるものであればよく、当業者が適宜選択することができる。このとき、組換えバチルス属細菌中で目的のセルラーゼ遺伝子と分泌シグナル配列が作動可能に連結されている場合、生成されたセルラーゼは菌体外に分泌されるため回収がより容易になる。
【0024】
本発明はまた、上記の本発明によるClostridium josui由来Cel48Aセルラーゼの生産方法によって生産されるClostridium jyosui由来Cel48Aセルラーゼを提供する。当該Clostridium josui由来Cel48Aセルラーゼは、以下の性質を有する:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、及びCellulose powderを分解し、還元糖を生成する;
最適反応pH:少なくともpH4〜10で活性を有し、pH7〜8で相対活性60%以上を有し、最適pHは約7である;
最適反応温度:少なくとも45〜65℃で相対活性60%以上を有し、最適温度は約60℃である(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である(相対活性60%以上、好ましくは70%以上を有する);
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、約65℃まで安定(相対活性40%以上)である。
分子量:SDS−PAGEで、77〜79kDaである(セルラーゼサンプル5μlにLaemmli sample buffer(Bio-Rad社)25μlを加え、100℃、5分間熱処理をした後、10%T SDS−PAGEゲルに供し、25mM Tris、192mM Glycine、及び0.1%SDS(pH8.3)を含む緩衝液中にて25分間泳動)。
【実施例】
【0025】
以下、実施例に基づき本発明をさらに詳細に説明する。
【0026】
実施例1 C. josui セルラーゼ遺伝子のクローニング
(1−1 ゲノムDNAの抽出)
Clostridium josui FERM P-9684株を、500mlのG−S培地[セロビオース 1%(w/v)、リン酸1カリウム 0.15%(w/v)、リン酸2カリウム 0.29%(w/v)、硫酸アンモニウム 0.13%(w/v)、酵母エキス 0.45%(w/v)、システイン塩酸塩 0.1%(w/v)、MOPS 2.1%(w/v)、塩化マグネシウム6水塩 0.1%(w/v)、塩化カルシウム2水塩 0.015%(w/v)、硫酸第一鉄7水塩 極微量、pH7.4]へ5.0ml前培養液を接種した。定常期まで(7日間)培養した培養液を4000×Gで10分間遠心した後、上清を除去して菌体を得た。これを9mlの50mM Tris−HCl(pH8.0)1mM EDTA溶液に懸濁した後、10mg/mlのリゾチーム溶液を1.0ml添加し軽く撹拌して氷中に10分間放置した。次に10%SDS(Sodiumlaurylsulfate)溶液を1.0ml、0.5M EDTA溶液を1.0ml加えて45℃で溶菌するまでゆっくり撹拌した。その溶菌液に等容のフェノールを加え、穏やかに撹拌後15000×G、10分間の遠心を行い、水層を抽出した。このフェノール抽出の操作を2回繰り返した。回収した水層に1/10容の3M酢酸ナトリウム溶液と2容の99.9%エタノールを加え、ゆっくり撹拌して−20℃、20分間放置した。沈殿してきた染色体DNAをガラス棒で巻取り、冷70%エタノールで洗った後に10mlのTE(10mM Tris−1mM EDTA)溶液に溶解した。これに10mg/mlのRNase溶液を1μl加え、4℃で一晩反応させた。更に溶液にプロテナーゼK(10mg/mlの水溶液)を300μl加え、37℃、2時間反応させた。反応後、等容のフェノールを加え穏やかに撹拌し、15000×G、10分間の遠心を行い、水層を回収した。続いてクロロホルムを加え穏やかに撹拌し、15000×G、10分間の遠心を行い水層を回収した。回収した水層に1/10容の3M−酢酸ナトリウム溶液と2容の99.9%エタノールを加え、ゆっくり撹拌して−20℃、20分間放置した。沈殿してきた染色体DNAをガラス棒で巻取り、冷70%エタノールで洗った後に1mlのTE溶液に溶解した。
【0027】
(1−2 遺伝子断片の取得)
1−1で得られたゲノムDNAを鋳型として、セルラーゼ遺伝子全長塩基配列(配列番号1)を元に設計した各プライマーを用いてPCRを行った。N−末端アミノ酸配列をコードする遺伝子から設計されたフォワードプライマー1(配列番号5)及び推定されたシグナル配列を除いた成熟酵素より設計したフォワードプライマー2(配列番号6)にはセンス鎖の5’末端側にS237eglシグナル配列を付加した。鋳型DNA 0.5μL、50μMフォワードプライマー1(配列番号5)若しくはフォワードプライマー2(配列番号6)0.5μL、50μMリバースプライマー1(配列番号7)0.5μL、Pyrobest DNA Polymerase(TaKaRa製)0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200(MJ Japan製)でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、58℃で1分間、72℃で3分間を1サイクルとして25サイクル反応させた。増幅した遺伝子断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した。フォワードプライマー1とリバースプライマー1から増幅されたシグナルを含む断片(Cj48-1)とフォワードプライマー2とリバースプライマー1から増幅された全長より推定シグナル配列を除いた断片(Cj48ΔSig-1)を得た。
【0028】
(1−3 S237eglプロモーター及びシグナル配列とのSOE−PCR)
pHY-S237egl(Biosci. Biotechnol. Biochem., 64(11):2281-9, 2000)(配列番号4)を鋳型として、S237eglプロモーター及びシグナル部分の塩基配列を元に設計したフォワードプライマー3(配列番号8)及びリバースプライマー2(配列番号9)を用いてPCRを行った。鋳型DNA 0.5μL、50μMフォワードプライマー3(配列番号8)0.5μL、50μMリバースプライマー2(配列番号9)0.5μL、Pyrobest DNA Polymerase 0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、55℃で1分間、72℃で3分間を1サイクルとして25サイクル反応させた。増幅したプロモーター及びシグナル配列断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した。精製したプロモーター及びシグナル配列の断片と1−2で得られた各遺伝子断片とを鋳型として、フォワードプライマー3(配列番号8)及びリバースプライマー1(配列番号7)を用いてSOE−PCR(Gene,77,61, 1989)を行った。プロモーター及びシグナル配列の断片0.5μL、各遺伝子断片0.5μL、50μMフォワードプライマー3(配列番号8)0.5μL、50μMリバースプライマー1(配列番号7)0.5μL、Pyrobest DNA Polymerase 0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、60℃で1分間、72℃で4分間を1サイクルとして25サイクル反応させた。増幅した遺伝子断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した(図1)。
【0029】
(1−4 ベクターへの連結)
1−3で得られた精製PCR産物を、BKL kit(TaKaRa製)にて平末端処理した。予めSmaIで処理したシャトルベクターpHY300PLK(TaKaRa製)にLigation High(TOYOBO製)を用いて連結した(16℃、24時間)(図1)。
【0030】
(1−5 E. coliによるプラスミド調製)
1−4で得られたライゲーション混液2μLを用いて、コンピテントセルE.coli HB101(TaKaRa製)20μLを形質転換した。形質転換体の再生培地には、アンピシリン含有寒天培地[カルボキシメチルセルロース(CMC;関東化学株式会社製)1.0%(w/v)、塩化ナトリウム(和光純薬工業株式会社製)1.0%(w/v)、バクトトリプトン(Difco製)1.0%(w/v)、酵母エキス(Difco製)0.5%(w/v)、トリパンブルー(ACROS ORGANICS製)0.005%(w/v)、アンピシリンナトリウム(和光純薬工業株式会社製)0.02%(w/v)、寒天(和光純薬工業株式会社製)1.5%(w/v)]を用いた。アンピシリン耐性株として得られた形質転換体の中から、コロニーPCRにより目的の遺伝子が挿入されたプラスミドを保持する菌株を選別した。選別した形質転換体をアンピシリン含有LB培地0.4mL[バクトトリプトン1.0%(w/v)、酵母エキス0.5%(w/v)、塩化ナトリウム1.0%(w/v)、アンピシリンナトリウム0.005%(w/v)]を用いて培養後(37℃、1,000rpm、4時間)、得られた菌体からプラスミドをHigh Pure Plasmid Isolation kit(Roche製)を用いて回収、精製した。
【0031】
(1−6 Bacillus subtilisの形質転換)
精製したプラスミドを用いて、プロトプラスト法(Mol. Gen. Genet. 168, 111-115 ,1979)により宿主菌Bacillus subtilis 168株及びプロテアーゼ9重欠損株(KA8AX)(特開2006-174707)の形質転換を行った。形質転換体の再生培地には、テトラサイクリン含有DM3再生寒天培地[CMC 1.0%(w/v)、バクトカザミノ酸(Difco製)0.5%(w/v)、酵母エキス0.5%(w/v)、DL−メチオニン(和光純薬工業株式会社製)0.01%(w/v)、L−リシン一塩酸塩(和光純薬工業株式会社製)0.01%(w/v)、L−トリプトファン(和光純薬工業株式会社製)0.01%(w/v)、コハク酸二ナトリウム六水和物(和光純薬工業株式会社製)8.1%(w/v)、リン酸一水素二カリウム(和光純薬工業株式会社製)0.35%(w/v)、リン酸二水素一カリウム(和光純薬工業株式会社製)0.15%(w/v)、グルコース(和光純薬工業株式会社製)0.5%(w/v)、塩化マグネシウム(和光純薬工業株式会社製)20mM、牛血清アルブミン(Sigma製)0.01%(w/v)、トリパンブルー0.01%(w/v)、テトラサイクリン塩酸塩(和光純薬工業株式会社製)0.005%(w/v)、寒天1.0%(w/v)]を用いた。テトラサイクリン耐性株として得られた形質転換体の中から、コロニーPCRにより目的の遺伝子が挿入されたプラスミド(プラスミドCj48-1、及びプラスミドCj48ΔSig-1)を保持する菌株を選別した。なお、遺伝子が挿入されていない空のベクターを保持する菌株をベクターコントロールとした。
【0032】
実施例2 セルラーゼの生産
DM3再生寒天培地上に生育した形質転換体又はコントロール菌株をテトラサイクリン含有LB培地5mL[バクトトリプトン1.0%(w/v)、酵母エキス0.5%(w/v)、塩化ナトリウム1.0%(w/v)、テトラサイクリン塩酸塩0.0015%(w/v)]を用いてシード培養後(30℃、250rpm、24時間)、シード培養液0.4mLをテトラサイクリン含有2×L培地20mL[バクトトリプトン2%(w/v)、酵母エキス1.0%(w/v)、塩化ナトリウム1.0%(w/v)、硫酸マンガン五水和物(和光純薬工業株式会社製)0.00075%(w/v)、マルトース一水和物(和光純薬工業株式会社製)7.5%(w/v)、テトラサイクリン塩酸塩0.0015%(w/v)]に添加し、メイン培養を行った(30℃、230rpm、3日間)。培養後、遠心分離(11,000rpm、15分間、4℃)により培養上清(セルラーゼ粗酵素溶液)を得た。
【0033】
実施例3 セルラーゼの活性発現
終濃度1.0%(w/v)のCMC(日本製紙株式会社製、製品名A01MC)及び50mMリン酸緩衝液(pH7)を含む基質溶液0.9mLに適当な濃度に希釈した粗酵素溶液0.1mLを添加し、50℃で20分間反応した。DNS(0.5% 3,5−ジニトロサリチル酸、30%酒石酸ナトリウムカリウム四水和物、1.6%水酸化ナトリウム)溶液1.0mLを添加し反応を停止させた後、沸騰浴中で5分間煮沸した。氷水中で冷却後、イオン交換水4.0mLを加えてよく攪拌し、535nmの吸光度をスペクトロフォトメーター(U-2800A;日立製作所株式会社製)で測定した。尚、基質溶液0.9mLにDNS溶液を1.0mL添加後、酵素溶液0.1mLを加え、同様の操作を行ったものをブランクとした。上記条件下で、1unit(U)は1分間に1μmolのD−グルコース相当の還元糖を遊離する酵素量とした。結果を表1に示す。
【0034】
【表1】

【0035】
実施例4 セルラーゼのSDS−PAGE
発現したセルラーゼサンプル5μlにLaemmli sample buffer(Bio-Rad社)25μlを加え、100℃、5分間熱処理をした。処理したサンプルを10%T SDS−PAGEゲルに供し、25mM Tris、192mM Glycine、及び0.1%SDS(pH8.3)を含む緩衝液中にて25分間泳動した。分子量マーカーを基準に分子量を測定したところ、Cj48-1から得られたセルラーゼは79±2kDaであり、Cj48ΔSig-1から得られたセルラーゼは77±2kDaであった(図2、矢印)。
【0036】
実施例5 セルラーゼの基質特異性
各種基質に対する活性を以下の要領で測定し、結果を表2に示した。CMC分解活性を100としたときの相対活性(%)で表記した。
【0037】
【表2】
【0038】
(4−1 CMC分解活性)
実施例3と同様の方法で行った。得られた活性を100%として、他の基質の活性の基準値とした。
【0039】
(4−2 Cellulose Powder分解活性)
終濃度0.25%(w/v)のCellulose Powder(Fluka製、製品番号22183)及び50mMリン酸緩衝液(pH7)を含む基質溶液0.9mLに適当な濃度に希釈した粗酵素溶液0.1mLを添加し、50℃で20分間反応した。ジニトロサリチル酸(DNS)溶液1.0mLを添加し反応を停止させた後、遠心分離(3,000rpm、5分間、20℃)により得られた上清1mLを沸騰浴中で5分間煮沸した。氷水中で冷却後、イオン交換水4.0mLを加えてよく攪拌し、535nmの吸光度をスペクトロフォトメーター(U-2800A;日立製作所株式会社製)で測定した。尚、基質溶液0.9mLにDNS溶液を1.0mL添加後、酵素溶液0.1mLを加え、同様の操作を行ったものをブランクとした。上記条件下で、1unit(U)は1分間に1μmolのD−グルコース相当の還元糖を遊離する酵素量とした。結果を表2に示す。
【0040】
(4−3 Avicel分解活性)
基質に終濃度0.25%(w/v)のAvicel(Fluka製、製品番号11365)を使用する以外は、実施例4−2と同様の方法で行った。結果を表2に示す。
【0041】
(4−4 Xylan分解活性)
基質に終濃度0.25%(w/v)のXylan(Fluka製、製品番号95590)を使用する以外は、実施例4−2と同様の方法で行った。また、1unit(U)は1分間に1μmolのD−キシロース相当の還元糖を遊離する酵素量とした。結果を表2に示す。
【0042】
(4−5 Xyloglucan分解活性)
基質に終濃度0.25%(w/v)のXyloglucan(Megazyme製、製品名P-XYGLN)を使用する以外は、実施例3と同様の方法で行った。結果を表2に示す。
【0043】
実施例5 セルラーゼの最適反応pH
50mMの各緩衝液[グリシン−塩酸緩衝液(pH2.0及びpH3.0)、酢酸−酢酸ナトリウム緩衝液(pH4.0及びpH5.0)、リン酸緩衝液(pH6.0、pH7.0及びpH8.0)、ならびにグリシン−水酸化ナトリウム緩衝液(pH9.0及びpH10.0)]を使用する以外は実施例3と同様の方法で行った。最大活性を示したpHでの活性値を100としたときの相対活性(%)で示した。結果を図3に示す。
【0044】
実施例6 セルラーゼのpH安定性
50mMの各緩衝液[グリシン−塩酸緩衝液(pH2.0及びpH3.0)、酢酸−酢酸ナトリウム緩衝液(pH4.0及びpH5.0)、リン酸緩衝液(pH6.0、pH7.0及びpH8.0)、ならびにグリシン−水酸化ナトリウム緩衝液(pH9.0及びpH10.0)]中、4℃で24時間処理した後は、実施例3と同様の方法で行った。最大活性を示したpHでの活性値を100としたときの相対活性(%)で示した。結果を図4に示す。
【0045】
実施例7 セルラーゼの最適反応温度
反応温度を20℃〜70℃で反応させる以外は実施例3と同様の方法で行った。最高活性を示した温度での活性値を100とした相対活性(%)で示した。結果を図5に示す。
【0046】
実施例8 セルラーゼの温度安定性
50mMのリン酸緩衝液(pH7)中、20℃〜70℃20分間処理した後は、実施例3と同様の方法で行った。熱に対して未処理の活性値を100としたときの残存活性(%)で示した。結果を図6に示す。
【技術分野】
【0001】
本発明は、Clostridium josui由来のCel48Aセルラーゼ、ならびにそれを生産するための新規な微生物宿主及び生産方法に関する。
【背景技術】
【0002】
セルラーゼは、セルロースのβ−1,4グルコシド結合を加水分解する酵素の総称であり、アミラーゼ、プロテアーゼ、リパーゼと共に加水分解酵素の代表的存在である。これまでに、真菌類や嫌気性細菌由来の中酸性に最適反応pHを有するセルラーゼが主に見出されているが、一方、好アルカリ性バチルス属細菌由来のアルカリ性に最適反応pHを有するアルカリセルラーゼもまた見出されている。
【0003】
セルラーゼの基質となるセルロースは、植物細胞壁の主成分として年間1000億トン以上も生産されるバイオマスであり、衣料、紙、建築材料等に有効利用されている。またセルロースは、グルコースが直鎖状に結合した巨大分子であるため、分解によって燃料物質やより高付加価値の代謝物質に変換が可能である。そのため、セルロースの効率的分解や、その分解産物の有効利用に関する研究が広く行われている。セルロースを高付加価値物質に変換する場合、通常、セルロース原料を前処理によって高反応性セルロースに変換した後、グルコースなどの低分子糖に分解する。得られた低分子糖は微生物による発酵等の化学反応を経て、高付加価値物質へと変換される。近年、この一連のセルロース分解工程においては、より経済的な方法として、セルラーゼよる酵素分解が一般に利用されている。
【0004】
しかし現状では、セルラーゼによるセルロース分解の速度は充分ではなく、必要な分解速度を達成するためには多量の酵素を使用するか、あるいは多種類の酵素を混合して使用しなければならない。またこのことは必然的に、酵素に多額のコストがかかる結果となる。より効率的且つ経済的にセルロース分解産物を生産するために、セルラーゼライブラリの拡充及びより高活性のセルラーゼの開発が求められている。
【0005】
Clostridium属細菌は、偏性嫌気性のグラム陽性細菌であり、セルロソームという高分子酵素複合体を作り、セルロースを効率良く分解することが知られている。その中のセルラーゼの主たるコンポーネントとして、糖加水分解酵素(glycoside hydrolase; GH)ファミリー48に属するCel48が知られている。Clostridium属のCel48については、C. thermocellumのCelS(Cel48A)に関して多くの報告があり、遺伝子取得(非特許文献1)、大腸菌による発現(非特許文献2)、及び蛋白結晶構造解析(非特許文献3)がなされている。しかしながら、C. thermocellum以外のClostridium属のCel48Aについての報告は少ない。例えば、Clostridium josui由来のCelD(Cel48A)に関しては、遺伝子取得はなされているものの(非特許文献4)、実際に蛋白を単離・同定するには至っていない。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】J Bacteriol, 175(5), 1293-1302, 1993
【非特許文献2】J Bacteriol, 177(6), 1641-1644, 1995
【非特許文献3】J. Mol. Biol., 320, 587-596
【非特許文献4】J Bacteriol, 180(16), 4303-4308, 1998
【発明の概要】
【発明が解決しようとする課題】
【0007】
Clostridium属由来セルラーゼを製造する場合、外来タンパク質を発現させるための方法として従来一般的に行われてきた大腸菌を用いて組換え蛋白質を発現させる方法では、Clostridium josui由来Cel48Aにおいては組換え蛋白質の発現が見られていない。本発明は、Clostridium josui由来のCel48Aセルラーゼ、ならびにそのより効率的な生産を可能にする宿主微生物及び生産方法を提供する。
【課題を解決するための手段】
【0008】
本発明者らは、Clostridium josui由来のCel48Aセルラーゼをより効率的に発現させる方法について検討を行ったところ、バチルス属細菌を宿主に用いることにより、大腸菌を用いた従来法では発現させることができなかった菌株由来のセルラーゼを得ることが可能となった。
【0009】
すなわち本発明は、以下を提供するものである。
(1)Clostridium josui由来のCel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。
(2)前記Cel48Aセルラーゼ遺伝子を含むベクターを含む(1)記載の組換えバチルス属細菌。
(3)前記Cel48Aセルラーゼ遺伝子が配列番号1で示される塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、(1)又は(2)に記載の組換えバチルス属細菌。
(4)前記Cel48Aセルラーゼ遺伝子の上流に転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域から選ばれる1以上の領域が作動可能に結合されている、(1)〜(3)のいずれか1に記載の組換えバチルス属細菌。
(5)転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されている、(4)記載の組換えバチルス属細菌。
(6)分泌シグナル領域がバチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域由来のものである、(5)記載の組換えバチルス属細菌。
(7)転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、上記いずれかの塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片である、(5)記載の組換えバチルス属細菌。
(8)前記Cel48Aセルラーゼ遺伝子が配列番号1の塩基番号94〜2160の塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、(1)〜(7)のいずれか1に記載の組換えバチルス属細菌。
(9)前記バチルス属細菌が枯草菌又はその変異株である、(1)〜(8)のいずれか1に記載の組換えバチルス属細菌。
(10)(1)〜(9)のいずれか1に記載の組換えバチルス属細菌を用いることを特徴とする、Clostridium josui由来Cel48Aセルラーゼの生産方法。
(11)(10)記載の生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
(12)以下の性質を有する(11)記載のセルラーゼ:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、及びCellulose powderを分解し、還元糖を生成する;
最適反応pH:約7;
最適反応温度:約60℃(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である;
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、65℃まで安定である。
分子量:SDS−PAGEで、77〜79kDaである。
【発明の効果】
【0010】
本発明の組換えバチルス属細菌は、Clostridium josui由来Cel48Aセルラーゼを高効率で産生するための宿主微生物として有用である。当該組換えバチルス属細菌を用いる本発明のClostridium josui由来Cel48Aセルラーゼの生産方法によれば、Clostridium josui由来Cel48Aセルラーゼを効率よく生産することができる。また、本発明により提供されるClostridium josui由来Cel48Aセルラーゼは、高いセルロース分解活性を有する有用な酵素である。したがって、本発明によれば、より効率的且つ経済的なセルロース分解産物の生産を可能にするセルラーゼを効率よく安価に提供することが可能となり、ひいてはセルロースを原料とする燃料物質や高付加価値物質をより効率的且つ安価に提供することが可能となる。
【図面の簡単な説明】
【0011】
【図1】Clostridium josuiセルラーゼをコードするベクターの作製手順。
【図2】形質転換体培養液のSDS-PAGE電気泳動像。
【図3】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの最適pH。
【図4】本発明の方法により取得したClostridium josui Cel48AセルラーゼのpH安定性。
【図5】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの最適温度。
【図6】本発明の方法により取得したClostridium josui Cel48Aセルラーゼの温度安定性。
【発明を実施するための形態】
【0012】
本発明の組換えバチルス属細菌は、バチルス属細菌にClostridium josui由来のCel48Aセルラーゼをコードする遺伝子を導入することによって得ることができる。本発明の組換えバチルス属細菌の親菌株としては、バチルス属細菌であれば特に限定されないが、好ましくは枯草菌及びその変異株が挙げられる。枯草菌及びその変異株の例としては、枯草菌168株、及び蛋白質産生のための宿主として公知の各種プロテアーゼ欠損枯草菌変異株が挙げられる。プロテアーゼ欠損枯草菌変異株の例としては、特開2006-174707に記載のプロテアーゼ9重欠損株(KA8AX)等が挙げられる。
【0013】
本発明の組換えバチルス属細菌にはClostridium josui由来のCel48Aセルラーゼをコードする遺伝子を導入する。あるいは、セルラーゼ遺伝子は、配列番号1で示される塩基配列からなるセルラーゼ遺伝子、又は当該塩基配列と70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の配列同一性を有し、且つセルラーゼ活性を有する蛋白質をコードする遺伝子である。またあるいは、セルラーゼ遺伝子は、配列番号1の塩基番号1〜2160もしくは塩基番号94〜2160で示される塩基配列からなるセルラーゼ遺伝子、又は当該塩基配列と70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の配列同一性を有し、且つセルラーゼ活性を有する蛋白質をコードする遺伝子である。
【0014】
本発明において、塩基配列及びアミノ酸配列の同一性はLipman-Pearson法 (Science,227,1435,1985)によって計算される。具体的には、遺伝情報処理ソフトウェアGenetyx-Win(ソフトウェア開発)のホモロジー解析(Search homology)プログラムを用いて、Unit size to compare(ktup)を2として解析を行うことにより算出される。
【0015】
上記セルラーゼ遺伝子は、例えばClostridium josui(FERMP-9684等)から、当該分野で用いられる任意の方法を用いて単離することができる。例えば、当該遺伝子は、Clostridium josuiから全ゲノムDNAを抽出した後、目的遺伝子の塩基配列(例えば、配列番号1の塩基配列)を元に設計したプライマーを用いたPCRにより当該遺伝子を選択的に増幅し、増幅した遺伝子を精製することで得ることができる。あるいは、当該セルラーゼ遺伝子は、公開されているその塩基配列又はそれがコードする蛋白質のアミノ酸配列に基づいて、遺伝子工学的又は化学的に合成することができる。
【0016】
次いで、上記で得られたセルラーゼ遺伝子を適切なベクターに導入する。本発明に使用されるベクターの種類としては、特に限定されず、タンパク質産生に通常用いられるベクター、例えばプラスミド、コスミド、ファージ、ウイルス、YAC、BAC等が挙げられる。プラスミドベクターが好ましく、例えば、市販のタンパク質発現用プラスミドベクター、例えばシャトルベクターpHY300PLK(TaKaRa製)、pUC19(TaKaRa製)、pUC119(TaKaRa製)、pBR322(TaKaRa製)等を好適に用いることができる。上記ベクターは、DNAの複製開始領域を含むDNA断片、複製起点を含むDNA領域を含む。また上記ベクターには、当該ベクターが適切に導入された微生物を選択するための薬剤耐性遺伝子(アンピシリン、ネオマイシン、カナマイシン、クロラムフェニコール等)がさらに組み込まれていてもよい。
【0017】
あるいは、上記ベクターにおいては、上記セルラーゼ遺伝子の上流に、当該遺伝子の転写、翻訳、分泌に関わる制御領域、即ち、プロモーター及び転写開始点を含む転写開始制御領域、リボソーム結合部位及び開始コドンを含む翻訳開始領域並びに分泌シグナルペプチド領域から選ばれる1以上の領域が作動可能に連結されていてもよい。好ましくは、転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が作動可能に連結されている。より好ましくは、分泌シグナルペプチド領域は、バチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域は、当該バチルス属細菌セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域である。
【0018】
例えば、上記Clostridium josui由来Cel48Aセルラーゼ遺伝子の上流には、特開2000−210081号公報や特開平4−190793号公報等に記載されているバチルス(Bacillus)属細菌、すなわちKSM-S237株(FERM BP-7875)、又はKSM-64株(FERM BP-2886)由来のセルラーゼ遺伝子の転写開始制御領域、翻訳開始領域及び分泌シグナルペプチド領域が作動可能に連結されていることが望ましい。より具体的には、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、配列番号4で示されるS237eglプロモーター及びシグナル配列(Biosci. Biotechnol. Biochem., 64(11):2281-9, 2000)、又は当該塩基配列に対して70%以上、好ましくは80%以上、より好ましくは90%以上、さらに好ましくは95%以上、さらにより好ましくは98%以上の同一性を有する塩基配列からなり、且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片、あるいは上記いずれかの塩基配列からなるDNA断片とストリンジェントな条件でハイブリダイズし、且つ遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片が、作動可能に連結されていることが望ましい。
【0019】
本明細書において、転写開始制御領域、翻訳開始領域、又は分泌シグナルと遺伝子との「作動可能な連結」とは、上記制御領域が遺伝子の転写又は翻訳を誘導し得るように、又は遺伝子にコードされたタンパク質又はペプチドが分泌シグナルの働きにより分泌されるように、連結されていることをいう。上記制御領域又は分泌シグナルと遺伝子との「作動可能な連結」の手順は、当業者に周知である。また本明細書において、「塩基配列の一部が欠失した塩基配列からなるDNA断片」とは、上記塩基配列の一部、好ましくは数個(例えば、1〜50個、好ましくは1〜30個、より好ましくは1〜20個、さらに好ましくは1〜10個)の塩基を欠失しているが、遺伝子の転写、翻訳、分泌に関わる機能を有するDNA断片を意味する。また本明細書において、ハイブリダイゼーションにおける「ストリンジェントな条件」とは、例えば[Molecular cloning-a Laboratory manual 2nd edition(Sambrookら、1989)]に記載の条件等が挙げられる。例えば、6×SSC(1×SSCの組成:0.15M塩化ナトリウム、0.015Mクエン酸ナトリウム、pH7.0)、0.5%SDS、5×デンハート及び100mg/mLニシン精子DNAを含む溶液にプローブとともに65℃で8〜16時間恒温し、ハイブリダイズさせる条件が挙げられる。
【0020】
セルラーゼ遺伝子と上記制御配列、薬剤耐性遺伝子との連結は、SOE(splicing by overlap extension)−PCR法(Gene,77,61,1989)等の方法によって行うことができる。プラスミドベクターへの遺伝子の導入手順は当該分野で周知である。
【0021】
次いで、構築された上記ベクターをバチルス属細菌に導入すれば、本発明の組換えバチルス属細菌を得ることができる。好ましいバチルス属細菌は、枯草菌及びその変異体である。変異体としてはプロテアーゼ9重欠損株(KA8AX)等が挙げられる。ベクター導入の方法としては、プロトプラスト法、エレクトロポレーション等の当該分野で通常使用される方法を用いることができる。導入が適切に行われた株を薬剤耐性等を指標に選択することで、目的の形質転換体を得ることができる。
【0022】
あるいは、本発明の組換えバチルス属細菌は、バチルス属細菌のゲノムに上記セルラーゼ遺伝子を直接組み込むことによって得ることができる。たとえば、バチルス属細菌のゲノム上の遺伝子の転写、翻訳、分泌に関わる制御領域に相同なDNA断片と上記セルラーゼ遺伝子を含むDNA断片とを結合させたDNA断片を構築し、これを当該細菌に導入して細菌のゲノムと相同組換えすることによって、当該細菌のゲノム上の制御領域の下流に上記セルラーゼ遺伝子が組み込まれた組換えバチルス属細菌を得ることができる。上記のような相同組換えの手法は当業者に周知である。
【0023】
斯くして得られた組換えバチルス属細菌を適切な培地で培養すれば、当該細菌中でClostridium josui由来Cel48Aセルラーゼをコードする遺伝子が発現して、Clostridium josui由来Cel48Aセルラーゼ蛋白質が合成される。合成されたセルラーゼ蛋白質を該培養物から通常の方法により単離又は精製することにより、Clostridium josui由来Cel48Aセルラーゼ蛋白質を取得することができる。したがって、本発明はまた、本発明の組換えバチルス属細菌を用いることを特徴とするClostridium josui由来Cel48Aセルラーゼの生産方法を提供する。培養に使用する培地は、バチルス属細菌の培養に通常使用されるものであればよく、当業者が適宜選択することができる。このとき、組換えバチルス属細菌中で目的のセルラーゼ遺伝子と分泌シグナル配列が作動可能に連結されている場合、生成されたセルラーゼは菌体外に分泌されるため回収がより容易になる。
【0024】
本発明はまた、上記の本発明によるClostridium josui由来Cel48Aセルラーゼの生産方法によって生産されるClostridium jyosui由来Cel48Aセルラーゼを提供する。当該Clostridium josui由来Cel48Aセルラーゼは、以下の性質を有する:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、及びCellulose powderを分解し、還元糖を生成する;
最適反応pH:少なくともpH4〜10で活性を有し、pH7〜8で相対活性60%以上を有し、最適pHは約7である;
最適反応温度:少なくとも45〜65℃で相対活性60%以上を有し、最適温度は約60℃である(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である(相対活性60%以上、好ましくは70%以上を有する);
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、約65℃まで安定(相対活性40%以上)である。
分子量:SDS−PAGEで、77〜79kDaである(セルラーゼサンプル5μlにLaemmli sample buffer(Bio-Rad社)25μlを加え、100℃、5分間熱処理をした後、10%T SDS−PAGEゲルに供し、25mM Tris、192mM Glycine、及び0.1%SDS(pH8.3)を含む緩衝液中にて25分間泳動)。
【実施例】
【0025】
以下、実施例に基づき本発明をさらに詳細に説明する。
【0026】
実施例1 C. josui セルラーゼ遺伝子のクローニング
(1−1 ゲノムDNAの抽出)
Clostridium josui FERM P-9684株を、500mlのG−S培地[セロビオース 1%(w/v)、リン酸1カリウム 0.15%(w/v)、リン酸2カリウム 0.29%(w/v)、硫酸アンモニウム 0.13%(w/v)、酵母エキス 0.45%(w/v)、システイン塩酸塩 0.1%(w/v)、MOPS 2.1%(w/v)、塩化マグネシウム6水塩 0.1%(w/v)、塩化カルシウム2水塩 0.015%(w/v)、硫酸第一鉄7水塩 極微量、pH7.4]へ5.0ml前培養液を接種した。定常期まで(7日間)培養した培養液を4000×Gで10分間遠心した後、上清を除去して菌体を得た。これを9mlの50mM Tris−HCl(pH8.0)1mM EDTA溶液に懸濁した後、10mg/mlのリゾチーム溶液を1.0ml添加し軽く撹拌して氷中に10分間放置した。次に10%SDS(Sodiumlaurylsulfate)溶液を1.0ml、0.5M EDTA溶液を1.0ml加えて45℃で溶菌するまでゆっくり撹拌した。その溶菌液に等容のフェノールを加え、穏やかに撹拌後15000×G、10分間の遠心を行い、水層を抽出した。このフェノール抽出の操作を2回繰り返した。回収した水層に1/10容の3M酢酸ナトリウム溶液と2容の99.9%エタノールを加え、ゆっくり撹拌して−20℃、20分間放置した。沈殿してきた染色体DNAをガラス棒で巻取り、冷70%エタノールで洗った後に10mlのTE(10mM Tris−1mM EDTA)溶液に溶解した。これに10mg/mlのRNase溶液を1μl加え、4℃で一晩反応させた。更に溶液にプロテナーゼK(10mg/mlの水溶液)を300μl加え、37℃、2時間反応させた。反応後、等容のフェノールを加え穏やかに撹拌し、15000×G、10分間の遠心を行い、水層を回収した。続いてクロロホルムを加え穏やかに撹拌し、15000×G、10分間の遠心を行い水層を回収した。回収した水層に1/10容の3M−酢酸ナトリウム溶液と2容の99.9%エタノールを加え、ゆっくり撹拌して−20℃、20分間放置した。沈殿してきた染色体DNAをガラス棒で巻取り、冷70%エタノールで洗った後に1mlのTE溶液に溶解した。
【0027】
(1−2 遺伝子断片の取得)
1−1で得られたゲノムDNAを鋳型として、セルラーゼ遺伝子全長塩基配列(配列番号1)を元に設計した各プライマーを用いてPCRを行った。N−末端アミノ酸配列をコードする遺伝子から設計されたフォワードプライマー1(配列番号5)及び推定されたシグナル配列を除いた成熟酵素より設計したフォワードプライマー2(配列番号6)にはセンス鎖の5’末端側にS237eglシグナル配列を付加した。鋳型DNA 0.5μL、50μMフォワードプライマー1(配列番号5)若しくはフォワードプライマー2(配列番号6)0.5μL、50μMリバースプライマー1(配列番号7)0.5μL、Pyrobest DNA Polymerase(TaKaRa製)0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200(MJ Japan製)でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、58℃で1分間、72℃で3分間を1サイクルとして25サイクル反応させた。増幅した遺伝子断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した。フォワードプライマー1とリバースプライマー1から増幅されたシグナルを含む断片(Cj48-1)とフォワードプライマー2とリバースプライマー1から増幅された全長より推定シグナル配列を除いた断片(Cj48ΔSig-1)を得た。
【0028】
(1−3 S237eglプロモーター及びシグナル配列とのSOE−PCR)
pHY-S237egl(Biosci. Biotechnol. Biochem., 64(11):2281-9, 2000)(配列番号4)を鋳型として、S237eglプロモーター及びシグナル部分の塩基配列を元に設計したフォワードプライマー3(配列番号8)及びリバースプライマー2(配列番号9)を用いてPCRを行った。鋳型DNA 0.5μL、50μMフォワードプライマー3(配列番号8)0.5μL、50μMリバースプライマー2(配列番号9)0.5μL、Pyrobest DNA Polymerase 0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、55℃で1分間、72℃で3分間を1サイクルとして25サイクル反応させた。増幅したプロモーター及びシグナル配列断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した。精製したプロモーター及びシグナル配列の断片と1−2で得られた各遺伝子断片とを鋳型として、フォワードプライマー3(配列番号8)及びリバースプライマー1(配列番号7)を用いてSOE−PCR(Gene,77,61, 1989)を行った。プロモーター及びシグナル配列の断片0.5μL、各遺伝子断片0.5μL、50μMフォワードプライマー3(配列番号8)0.5μL、50μMリバースプライマー1(配列番号7)0.5μL、Pyrobest DNA Polymerase 0.125μL、10×Pyrobest Buffer II 2.5μL、dNTP Mixture 2μL、滅菌脱イオン水18.875μLを混合した後、DNA Engine(商標)PTC-200でPCRを行った。PCRの条件は、95℃で3分間鋳型DNAを変性させた後、95℃で30秒間、60℃で1分間、72℃で4分間を1サイクルとして25サイクル反応させた。増幅した遺伝子断片をHigh Pure PCR Product Purification kit(Roche製)にて精製した(図1)。
【0029】
(1−4 ベクターへの連結)
1−3で得られた精製PCR産物を、BKL kit(TaKaRa製)にて平末端処理した。予めSmaIで処理したシャトルベクターpHY300PLK(TaKaRa製)にLigation High(TOYOBO製)を用いて連結した(16℃、24時間)(図1)。
【0030】
(1−5 E. coliによるプラスミド調製)
1−4で得られたライゲーション混液2μLを用いて、コンピテントセルE.coli HB101(TaKaRa製)20μLを形質転換した。形質転換体の再生培地には、アンピシリン含有寒天培地[カルボキシメチルセルロース(CMC;関東化学株式会社製)1.0%(w/v)、塩化ナトリウム(和光純薬工業株式会社製)1.0%(w/v)、バクトトリプトン(Difco製)1.0%(w/v)、酵母エキス(Difco製)0.5%(w/v)、トリパンブルー(ACROS ORGANICS製)0.005%(w/v)、アンピシリンナトリウム(和光純薬工業株式会社製)0.02%(w/v)、寒天(和光純薬工業株式会社製)1.5%(w/v)]を用いた。アンピシリン耐性株として得られた形質転換体の中から、コロニーPCRにより目的の遺伝子が挿入されたプラスミドを保持する菌株を選別した。選別した形質転換体をアンピシリン含有LB培地0.4mL[バクトトリプトン1.0%(w/v)、酵母エキス0.5%(w/v)、塩化ナトリウム1.0%(w/v)、アンピシリンナトリウム0.005%(w/v)]を用いて培養後(37℃、1,000rpm、4時間)、得られた菌体からプラスミドをHigh Pure Plasmid Isolation kit(Roche製)を用いて回収、精製した。
【0031】
(1−6 Bacillus subtilisの形質転換)
精製したプラスミドを用いて、プロトプラスト法(Mol. Gen. Genet. 168, 111-115 ,1979)により宿主菌Bacillus subtilis 168株及びプロテアーゼ9重欠損株(KA8AX)(特開2006-174707)の形質転換を行った。形質転換体の再生培地には、テトラサイクリン含有DM3再生寒天培地[CMC 1.0%(w/v)、バクトカザミノ酸(Difco製)0.5%(w/v)、酵母エキス0.5%(w/v)、DL−メチオニン(和光純薬工業株式会社製)0.01%(w/v)、L−リシン一塩酸塩(和光純薬工業株式会社製)0.01%(w/v)、L−トリプトファン(和光純薬工業株式会社製)0.01%(w/v)、コハク酸二ナトリウム六水和物(和光純薬工業株式会社製)8.1%(w/v)、リン酸一水素二カリウム(和光純薬工業株式会社製)0.35%(w/v)、リン酸二水素一カリウム(和光純薬工業株式会社製)0.15%(w/v)、グルコース(和光純薬工業株式会社製)0.5%(w/v)、塩化マグネシウム(和光純薬工業株式会社製)20mM、牛血清アルブミン(Sigma製)0.01%(w/v)、トリパンブルー0.01%(w/v)、テトラサイクリン塩酸塩(和光純薬工業株式会社製)0.005%(w/v)、寒天1.0%(w/v)]を用いた。テトラサイクリン耐性株として得られた形質転換体の中から、コロニーPCRにより目的の遺伝子が挿入されたプラスミド(プラスミドCj48-1、及びプラスミドCj48ΔSig-1)を保持する菌株を選別した。なお、遺伝子が挿入されていない空のベクターを保持する菌株をベクターコントロールとした。
【0032】
実施例2 セルラーゼの生産
DM3再生寒天培地上に生育した形質転換体又はコントロール菌株をテトラサイクリン含有LB培地5mL[バクトトリプトン1.0%(w/v)、酵母エキス0.5%(w/v)、塩化ナトリウム1.0%(w/v)、テトラサイクリン塩酸塩0.0015%(w/v)]を用いてシード培養後(30℃、250rpm、24時間)、シード培養液0.4mLをテトラサイクリン含有2×L培地20mL[バクトトリプトン2%(w/v)、酵母エキス1.0%(w/v)、塩化ナトリウム1.0%(w/v)、硫酸マンガン五水和物(和光純薬工業株式会社製)0.00075%(w/v)、マルトース一水和物(和光純薬工業株式会社製)7.5%(w/v)、テトラサイクリン塩酸塩0.0015%(w/v)]に添加し、メイン培養を行った(30℃、230rpm、3日間)。培養後、遠心分離(11,000rpm、15分間、4℃)により培養上清(セルラーゼ粗酵素溶液)を得た。
【0033】
実施例3 セルラーゼの活性発現
終濃度1.0%(w/v)のCMC(日本製紙株式会社製、製品名A01MC)及び50mMリン酸緩衝液(pH7)を含む基質溶液0.9mLに適当な濃度に希釈した粗酵素溶液0.1mLを添加し、50℃で20分間反応した。DNS(0.5% 3,5−ジニトロサリチル酸、30%酒石酸ナトリウムカリウム四水和物、1.6%水酸化ナトリウム)溶液1.0mLを添加し反応を停止させた後、沸騰浴中で5分間煮沸した。氷水中で冷却後、イオン交換水4.0mLを加えてよく攪拌し、535nmの吸光度をスペクトロフォトメーター(U-2800A;日立製作所株式会社製)で測定した。尚、基質溶液0.9mLにDNS溶液を1.0mL添加後、酵素溶液0.1mLを加え、同様の操作を行ったものをブランクとした。上記条件下で、1unit(U)は1分間に1μmolのD−グルコース相当の還元糖を遊離する酵素量とした。結果を表1に示す。
【0034】
【表1】
【0035】
実施例4 セルラーゼのSDS−PAGE
発現したセルラーゼサンプル5μlにLaemmli sample buffer(Bio-Rad社)25μlを加え、100℃、5分間熱処理をした。処理したサンプルを10%T SDS−PAGEゲルに供し、25mM Tris、192mM Glycine、及び0.1%SDS(pH8.3)を含む緩衝液中にて25分間泳動した。分子量マーカーを基準に分子量を測定したところ、Cj48-1から得られたセルラーゼは79±2kDaであり、Cj48ΔSig-1から得られたセルラーゼは77±2kDaであった(図2、矢印)。
【0036】
実施例5 セルラーゼの基質特異性
各種基質に対する活性を以下の要領で測定し、結果を表2に示した。CMC分解活性を100としたときの相対活性(%)で表記した。
【0037】
【表2】
【0038】
(4−1 CMC分解活性)
実施例3と同様の方法で行った。得られた活性を100%として、他の基質の活性の基準値とした。
【0039】
(4−2 Cellulose Powder分解活性)
終濃度0.25%(w/v)のCellulose Powder(Fluka製、製品番号22183)及び50mMリン酸緩衝液(pH7)を含む基質溶液0.9mLに適当な濃度に希釈した粗酵素溶液0.1mLを添加し、50℃で20分間反応した。ジニトロサリチル酸(DNS)溶液1.0mLを添加し反応を停止させた後、遠心分離(3,000rpm、5分間、20℃)により得られた上清1mLを沸騰浴中で5分間煮沸した。氷水中で冷却後、イオン交換水4.0mLを加えてよく攪拌し、535nmの吸光度をスペクトロフォトメーター(U-2800A;日立製作所株式会社製)で測定した。尚、基質溶液0.9mLにDNS溶液を1.0mL添加後、酵素溶液0.1mLを加え、同様の操作を行ったものをブランクとした。上記条件下で、1unit(U)は1分間に1μmolのD−グルコース相当の還元糖を遊離する酵素量とした。結果を表2に示す。
【0040】
(4−3 Avicel分解活性)
基質に終濃度0.25%(w/v)のAvicel(Fluka製、製品番号11365)を使用する以外は、実施例4−2と同様の方法で行った。結果を表2に示す。
【0041】
(4−4 Xylan分解活性)
基質に終濃度0.25%(w/v)のXylan(Fluka製、製品番号95590)を使用する以外は、実施例4−2と同様の方法で行った。また、1unit(U)は1分間に1μmolのD−キシロース相当の還元糖を遊離する酵素量とした。結果を表2に示す。
【0042】
(4−5 Xyloglucan分解活性)
基質に終濃度0.25%(w/v)のXyloglucan(Megazyme製、製品名P-XYGLN)を使用する以外は、実施例3と同様の方法で行った。結果を表2に示す。
【0043】
実施例5 セルラーゼの最適反応pH
50mMの各緩衝液[グリシン−塩酸緩衝液(pH2.0及びpH3.0)、酢酸−酢酸ナトリウム緩衝液(pH4.0及びpH5.0)、リン酸緩衝液(pH6.0、pH7.0及びpH8.0)、ならびにグリシン−水酸化ナトリウム緩衝液(pH9.0及びpH10.0)]を使用する以外は実施例3と同様の方法で行った。最大活性を示したpHでの活性値を100としたときの相対活性(%)で示した。結果を図3に示す。
【0044】
実施例6 セルラーゼのpH安定性
50mMの各緩衝液[グリシン−塩酸緩衝液(pH2.0及びpH3.0)、酢酸−酢酸ナトリウム緩衝液(pH4.0及びpH5.0)、リン酸緩衝液(pH6.0、pH7.0及びpH8.0)、ならびにグリシン−水酸化ナトリウム緩衝液(pH9.0及びpH10.0)]中、4℃で24時間処理した後は、実施例3と同様の方法で行った。最大活性を示したpHでの活性値を100としたときの相対活性(%)で示した。結果を図4に示す。
【0045】
実施例7 セルラーゼの最適反応温度
反応温度を20℃〜70℃で反応させる以外は実施例3と同様の方法で行った。最高活性を示した温度での活性値を100とした相対活性(%)で示した。結果を図5に示す。
【0046】
実施例8 セルラーゼの温度安定性
50mMのリン酸緩衝液(pH7)中、20℃〜70℃20分間処理した後は、実施例3と同様の方法で行った。熱に対して未処理の活性値を100としたときの残存活性(%)で示した。結果を図6に示す。
【特許請求の範囲】
【請求項1】
Clostridium josui由来Cel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。
【請求項2】
前記Cel48Aセルラーゼ遺伝子を含むベクターを含む請求項1記載の組換えバチルス属細菌。
【請求項3】
前記Cel48Aセルラーゼ遺伝子が配列番号1で示される塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、請求項1又は2に記載の組換えバチルス属細菌。
【請求項4】
前記Cel48Aセルラーゼ遺伝子の上流に転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域から選ばれる1以上の領域が作動可能に結合されている、請求項1〜3のいずれか1項に記載の組換えバチルス属細菌。
【請求項5】
転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されている、請求項4記載の組換えバチルス属細菌。
【請求項6】
分泌シグナル領域がバチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域由来のものである、請求項5記載の組換えバチルス属細菌。
【請求項7】
転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、上記いずれかの塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片である、請求項5記載の組換えバチルス属細菌。
【請求項8】
前記Cel48Aセルラーゼ遺伝子が配列番号1の塩基番号94〜2160の塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、請求項1〜7のいずれか1項に記載の組換えバチルス属細菌。
【請求項9】
前記バチルス属細菌が枯草菌又はその変異株である、請求項1〜8のいずれか1項に記載の組換えバチルス属細菌。
【請求項10】
請求項1〜9のいずれか1項に記載の組換えバチルス属細菌を用いることを特徴とする、Clostridium josui由来Cel48Aセルラーゼの生産方法。
【請求項11】
請求項10記載の生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
【請求項12】
以下の性質を有する請求項11記載のClostridium josui由来Cel48Aセルラーゼ:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、Cellulose powderを分解し、還元糖を生成する;
最適反応pH:約7;
最適反応温度:約60℃(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である;
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、65℃まで安定である。
分子量:SDS−PAGEで、77〜79kDaである。
【請求項1】
Clostridium josui由来Cel48Aセルラーゼ遺伝子を含む組換えバチルス属細菌。
【請求項2】
前記Cel48Aセルラーゼ遺伝子を含むベクターを含む請求項1記載の組換えバチルス属細菌。
【請求項3】
前記Cel48Aセルラーゼ遺伝子が配列番号1で示される塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、請求項1又は2に記載の組換えバチルス属細菌。
【請求項4】
前記Cel48Aセルラーゼ遺伝子の上流に転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域から選ばれる1以上の領域が作動可能に結合されている、請求項1〜3のいずれか1項に記載の組換えバチルス属細菌。
【請求項5】
転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が結合されている、請求項4記載の組換えバチルス属細菌。
【請求項6】
分泌シグナル領域がバチルス属細菌のセルラーゼ遺伝子由来のものであり、転写開始制御領域及び翻訳開始制御領域が当該セルラーゼ遺伝子の開始コドンから始まる長さ0.6〜1kbの上流領域由来のものである、請求項5記載の組換えバチルス属細菌。
【請求項7】
転写開始制御領域、翻訳開始制御領域及び分泌シグナル領域からなる3領域が、配列番号2で示される塩基配列の塩基番号1〜659の塩基配列からなるDNA断片、配列番号3で示される塩基配列からなるセルラーゼ遺伝子の塩基番号1〜696の塩基配列からなるDNA断片、当該塩基配列のいずれかと70%以上の同一性を有する塩基配列からなるDNA断片、上記いずれかの塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNA断片、又は上記いずれかの塩基配列の一部が欠失した塩基配列からなるDNA断片である、請求項5記載の組換えバチルス属細菌。
【請求項8】
前記Cel48Aセルラーゼ遺伝子が配列番号1の塩基番号94〜2160の塩基配列又は当該塩基配列と70%以上の同一性を有する塩基配列からなる遺伝子である、請求項1〜7のいずれか1項に記載の組換えバチルス属細菌。
【請求項9】
前記バチルス属細菌が枯草菌又はその変異株である、請求項1〜8のいずれか1項に記載の組換えバチルス属細菌。
【請求項10】
請求項1〜9のいずれか1項に記載の組換えバチルス属細菌を用いることを特徴とする、Clostridium josui由来Cel48Aセルラーゼの生産方法。
【請求項11】
請求項10記載の生産方法によって生産されるClostridium josui由来Cel48Aセルラーゼ。
【請求項12】
以下の性質を有する請求項11記載のClostridium josui由来Cel48Aセルラーゼ:
作用:カルボキシメチルセルロースを液化型で良好に分解する;
基質特異性:カルボキシメチルセルース、Avicel、Cellulose powderを分解し、還元糖を生成する;
最適反応pH:約7;
最適反応温度:約60℃(50mMリン酸緩衝液(pH7)で反応を行った場合);
安定pH範囲:4℃で24時間処理した場合、pH4〜8の範囲で安定である;
耐熱性:50mMリン酸緩衝液(pH7)中、20分間の処理において、65℃まで安定である。
分子量:SDS−PAGEで、77〜79kDaである。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】

【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−5404(P2012−5404A)
【公開日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願番号】特願2010−143432(P2010−143432)
【出願日】平成22年6月24日(2010.6.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構、「バイオマスエネルギー先導技術開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000000918)花王株式会社 (8,290)
【出願人】(304026696)国立大学法人三重大学 (270)
【Fターム(参考)】
【公開日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願日】平成22年6月24日(2010.6.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構、「バイオマスエネルギー先導技術開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000000918)花王株式会社 (8,290)
【出願人】(304026696)国立大学法人三重大学 (270)
【Fターム(参考)】
[ Back to top ]