
ペプチド合成のための方法および組成物
【課題】ペプチド合成のための方法および組成物の提供。
【解決手段】第一に、ペプチド、特にT-20(「DP-178」ともいう)およびT-20様ペプチドの合成方法。前記方法では、固相および液相の合成法を使用して特定のペプチド断片のグループを合成し、それらを結合させることにより目的のペプチドを生成する方法。さらに、目的のペプチド(例:T-20)を合成する際の中間体として役立つ個々のペプチド断片。また、全長T-20およびT-20様ペプチドを製造するために一緒に利用することができる、そのようなペプチド断片中間体のグループ。
【解決手段】第一に、ペプチド、特にT-20(「DP-178」ともいう)およびT-20様ペプチドの合成方法。前記方法では、固相および液相の合成法を使用して特定のペプチド断片のグループを合成し、それらを結合させることにより目的のペプチドを生成する方法。さらに、目的のペプチド(例:T-20)を合成する際の中間体として役立つ個々のペプチド断片。また、全長T-20およびT-20様ペプチドを製造するために一緒に利用することができる、そのようなペプチド断片中間体のグループ。
【発明の詳細な説明】
【技術分野】
【0001】
1.序
本発明は、第1に、ペプチド、特にT-20(「DP-178」とも呼ばれる;配列番号1)及びT-20様ペプチドの合成方法に関する。該方法では、固相及び液相合成方法を利用して、特定のペプチド断片のグループを合成し、それらを結合させて目的のペプチドを得る。本発明はさらに、目的のペプチド(例えば、T-20)の合成の中間体として有用な個々のペプチド断片に関する。本発明は、さらにまた、全長のT-20及びT-20様ペプチドを製造するために一緒に用いることができる前記ペプチド中間体断片のグループに関する。本発明は、さらにまた、ペプチド、特にT-20及びT-20様ペプチドの精製方法、並びに目的ペプチドの合成の中間体として有用な個々のペプチド断片に関する。
【背景技術】
【0002】
2.背景
近年、融合に関連した現象を抑制する能力を示し、さらに、重要なことには、強力な抗ウイルス活性をも示す多数のペプチドが同定されている。例えば、米国特許第5,464,933号;第5,656,480号及び国際公開公報第WO96/19495T-20号を参照されたい。これらのペプチドは治療薬として広く用いられているので、例えば、大規模量での合成能力に対する要請が高まっている。
【0003】
ペプチド合成のための技術(例えば、Merglerら, 1988, Tetrahedron Letters 29:4005-4008;Merglerら, 1988, Tetrahedron Letters 29:4009-4012;Kamberら(編集), 「ペプチド、化学及び生物学(Peptides, Chemistry and Biology)」, ESCOM, Leiden, 1992, 525-526;及びRinikerら, 1993, Tetrahedron Letters 49:9307-9320を参照されたい。)は存在するが、T-20及びT-20様ペプチドなどの容易に精製されたペプチドの大規模で経済的な製造に利用できる技術は現在のところ存在しない。
【発明の概要】
【0004】
3.発明の概要
本発明は、第1に、ペプチド、特にT-20(「DP-178」とも呼ばれる;配列番号1)及びT-20様ペプチドの合成方法に関する。該方法では、固相及び液相合成方法を利用して、特定のペプチド断片のグループを合成し、それらを結合させて目的のペプチドを得る。一般に、本発明の方法は、固相支持体上のT-20又はT-20様ペプチドの側鎖が保護された特定のペプチド断片中間体を合成し、溶液中で保護された断片を結合させて、保護されたT-20又はT-20様ペプチドを形成し、次いで、側鎖を脱保護して、最終のT-20又はT-20様ペプチドを得ることを含む。本発明の好ましい実施形態は、配列番号1に示されるアミノ酸配列を有するT-20ペプチドの合成に関係する。
【0005】
本発明はさらに、目的のペプチド(例えば、T-20)の合成において中間体として有用な個々のペプチド断片に関する。本発明のペプチド断片としては、限定するものではないが、下記表1に示されるアミノ酸配列を有するものが挙げられる。
【0006】
【表1】

本発明は、さらにまた、目的のペプチドの合成において中間体として有用なペプチド断片の特定のグループに関する。本発明のペプチド断片のグループとしては、下記表2に指定される、グループ1〜20が挙げられる。
【0007】
【表2】
この発明は、一部には、固相液相合成反応の特定の組合せによって初めて、高効率且つ高収量で大規模に高純度のT-20及びT-20様ペプチドの製造が可能になるという本発明者らの予期せざる発見に基づいている。特に、本発明の方法によれば、T-20及びT-20様ペプチドが、1キログラム以上の規模で合成できる。固相合成のための本発明の特定のT-20ペプチド断片を選択することによって、固相技術の高効率の結合(カップリング)が、3-、4-又は5-倍もの過剰のアミノ酸及び固相合成で通常必要とされる試薬を用いる必要なしに利用され得ることが見出された。本発明の方法では、本発明のペプチド断片の固相合成においてたった1.5倍ほどのアミノ酸しか使用しない。アミノ酸及び試薬の量におけるこのコスト低減により、本発明の方法はT-20及びT-20様ペプチドの大規模合成に適している。
【0008】
さらに、本発明者らは、驚くべきことに、特定のペプチド断片が、約0.8〜1 mmol/g(固相樹脂)のローディング量(loading)で固相合成され得ることを見出した。このローディング量は、固相ペプチド合成で一般に達成される0.25〜0.4 mmol/g(樹脂)のローディング範囲よりも顕著に高い。さらに、本発明者らは、超酸感受性樹脂を用いた固相で所定のペプチド断片を合成することが、異常に高純度のペプチド断片をもたらすことを見出した。クロマトグラフィー技術は、本発明によって製造されたペプチド断片を精製するのに必要ではなく、断片を使用の前に単に沈殿及び/又は粉砕(trituration)工程に付すか、又は樹脂から直接取得したままで使用する。超酸感受性樹脂を使用することによって、本発明の合成された保護ペプチドを、側鎖保護基の同時除去を行なうことなく樹脂から切断することが可能になる。これは、不純物を減らし、10個以上のアミノ酸を含むペプチドを、高純度で合成することを可能にする。本発明によって製造された高純度のペプチド断片を結合させることにより、本発明の方法に従って液相中で合成されるT-20及びT-20様ペプチドの不純物プロフィールは、主として、密接に関係した類似体よりもむしろ、結合しなかった断片からなる。従って、本発明によって製造されたT-20及びT-20様ペプチドは、従来技術によって製造されたものよりも精製するのがかなり容易である。下記第9及び11節に示す実施例では、T-20全長ペプチドのそのようなコンビナトリアル(組合せ)合成を実証する。第11節に示す実施例では、T-20及びT-20中間体ペプチドの大規模合成及び精製を実証する。従って、本発明の方法によれば、T-20及びT-20ペプチド中間体が、1キログラム以上の規模で製造できる。
【0009】
本発明者らはまた、予期せざることに、T-20及び他のT-20様ペプチドなどのペプチド、並びに本明細書中に記載された特定のペプチド断片が、塩基性pH範囲で使用できる高容量材料(high capacity materials)を用いて精製できることを見出した。すなわち、本発明は、さらにまた、ペプチド、特にT-20及びT-20様ペプチド、並びに目的ペプチドの合成の中間体として有用な個々のペプチド断片の精製方法に関する。
【0010】
3.1 定義
本明細書中で用いるアミノ酸表記は、慣用のものであり、以下の通りである:
一般的なアミノ酸の略号
アミノ酸 1文字表記 一般的略号
アラニン A Ala
アスパラギン N Asn
アスパラギン酸 D Asp
グルタミン Q Gln
グルタミン酸 E Glu
ヒスチジン H His
イソロイシン I Ile
ロイシン L Leu
リシン K Lys
フェニルアラニン F Phe
セリン S Ser
トレオニン T Thr
トリプトファン W Trp
チロシン Y Tyr
4.図面の簡単な説明
【図面の簡単な説明】
【0011】
【図1】図1:T-20の4断片アプローチ。この図面は、上記表2中に示される、中間体ペプチド断片グループ6から出発して全長T-20を合成するための、下記第9.1節に示される実施例に従うスキームを示す。
【図2】図2:T-20の4断片アプローチ、第2経路。この図面は、全長T-20を合成するための、上記表2に示されるペプチド中間体グループ6を結合させる追加的な4断片スキームを示す。
【図3】図3:T-20の3断片アプローチ。この図面は、全長T-20を合成するための、下記第9.1節に示される実施例に従うスキームを示す。
【図4】図4:T-20の3断片アプローチ、第2経路。この図面は、全長T-20を合成するための、下記第9.2、9.3、9.4及び9.5節に示される実施例に従うスキームを示す。
【図5】図5:T-20の2断片アプローチ。この図面は、全長T-20を合成するための、上記表2に示されるペプチド中間体グループ18を結合させるスキームを示す。
【発明を実施するための形態】
【0012】
5.発明の詳細な説明
5.1 全長ペプチド
本発明は、T-20、あるいは、DP-178として知られるペプチドを合成するために使用できる方法、ペプチド断片、ペプチド断片のグループに関する。T-20は、HIV-1LAI分離物由来の膜貫通タンパク質gp41のアミノ酸残基638-673に相当するペプチドであり、36個のアミノ酸配列(アミノNH2末端からカルボキシCOOH末端へと読む)を有する:
NH2−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−COOH
本発明の方法、断片及び断片のグループ、並びに断片及び断片のグループの選択に利用される技術は、T-20に加えてT-20様断片を合成するのにも使用できることが理解されるだろう。本明細書中で用いる語句「T-20様」とは、米国特許第5,464,933号;第5,656,480号又は国際公開公報第WO96/19495号(それぞれその全体が参照によって本明細書中に組み入れられる)に列挙されるいずれかのHIV又は非HIVペプチドを意味する。
【0013】
上記のT-20及びT-20様ペプチドに加えて、本発明の方法、断片及び断片のグループは、修飾されたアミノ及び/又はカルボキシル末端を有するペプチドを合成するのに使用できる。T-20を例にとると、該ペプチドは、
式: X−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−Z
(式中、Xは、アミノ基;カルボベンゾキシル、ダンシル、及びt−ブチルオキシカルボニルからなる群から選択される疎水性基;アセチル基;9−フルオレニル−メトキシ−カルボニル(FMOC)基;又は脂質−脂肪酸コンジュゲート、ポリエチレングリコール、及び炭水化物からなる群から選択される巨大分子キャリアー基を示し;そしてZは、カルボキシル基;アミド基;t−ブチルオキシカルボニル基;p−ニトロベンジルエステル基;又は脂質−脂肪酸コンジュゲート、ポリエチレングリコール及び炭水化物からなる群から選択される巨大分子キャリアー基を示す)で示される。前記「X」及び「Z」基の付加技術は、当業者に周知である。
【0014】
好ましい実施形態では、本発明の方法は、上記式においてXがアセチル基であり、Zがアミド基であるペプチドを合成するのに用いられる。下記第9節に示される実施例は、下記第5.2節に記載されるペプチド中間体の結合によるT-20ペプチドの成功した合成を実証する。好ましい方法では、T-20及びT-20様ペプチド並びに中間体は、限定するものではないが、高い(7より高い)pH範囲で安定なジルコニウム系充填剤、ポリスチレン、ポリアクリル又は他のポリマー系充填剤を含めて(ローディング容量を最大化するための)非シリカ系充填剤を用いて精製できる。例えば、広範なpH範囲を示す非シリカ系カラム充填剤は、Tosohaus社(Montgomeryville, PA)によって販売されているものよりも高いpH値を含む。前記材料で充填されたカラムは、低、中又は高圧クロマトグラフィーで作動する。例えば、下記第10節に示される精製法を参照されたい。
【0015】
5.2 ペプチド中間体
本発明は、限定するものではないが、上記表1に列挙された特定のアミノ酸配列を有するT-20及びT-20様ペプチドのペプチド断片中間体、及び表2に列挙されたペプチド断片中間体のグループを包含する。該ペプチド中間体、特に下記表2に列挙されるグループに含まれるものは、T-20及びT-20様ペプチドを製造するのに利用できる。
【0016】
表1又は2に列挙されたペプチド断片のアミノ酸残基の側鎖のいずれか1個以上は、t−ブチル(t-Bu)、トリチル(trt)及びt−ブチルオキシカルボニル(Boc)などの標準的な保護基によって保護できる。t-Bu基は、アミノ酸残基Tyr(Y)、Thr(T)、Ser(S)及びAsp(D)の好ましい側鎖保護基であり;trt基は、アミノ酸残基His(H)、Gln(Q)及びAsn(N)の好ましい側鎖保護基であり;Boc基は、アミノ酸残基Lys(K)及びTrp(W)の好ましい側鎖保護基である。
【0017】
表1に列挙された断片1、2、3及び4の合成の間、ヒスチジン残基の側鎖は、好ましくはトリチル(trt)保護基によって保護されていなければならない。もしそれが保護されていないと、樹脂からペプチド断片を切断するために使われる酸が、ヒスチジン残基と有害に反応し、ペプチド断片の分解を引き起こすだろう。
【0018】
本発明のペプチド断片のグルタミン残基は、トリチル(trt)基によって保護されているのが好ましい。しかしながら、断片1-16及び9-16のカルボキシル末端ではグルタミン残基を保護しないことが好ましい。1-16断片のカルボキシ末端のグルタミン残基上に保護基が存在しないと、1-16断片と17-36断片との反応が促進され、たった約2%のラセミ化しか伴わずに断片同士を結合できることがわかった。さらに、有機溶媒中での本発明のいずれかのペプチド断片のより低い溶解性が望まれる場合は、トリチル保護基を、その断片のいずれか1個以上の他のグルタミン残基から除去し得る。
【0019】
本発明の各ペプチド断片の全てのアスパラギン残基は保護されているのが好ましい。さらに、トリプトファン残基はBoc基によって保護されているのが好ましい。
【0020】
上記表1に列挙されたペプチド式1〜18に従う保護されたペプチド断片としては、限定するものではないが、下記表3に列挙される化合物が挙げられる。
【0021】
【表3】
上記表3に列挙されたペプチドのアミノ酸残基のいずれか1個以上の側鎖を、上記のように、tBu、trt及びBocなどの標準的な側鎖保護基によって保護できる。表3のペプチドの代表的な合成を下記第7及び8節に示し、そこでは下記第5.4節で述べる一般的技術を用いる。
【0022】
5.3 ペプチド合成
上述のように、本発明の個々のペプチド断片の幾つかは、固相合成法を用いて作られるのが好ましいが、本発明の他のペプチドは、固相および液相合成法を組み合わせて用いて作られるのが好ましく、前記合成は本明細書中に記載されるようにT-20またはT-20様ペプチドの製造で完了する。しかしながら、本発明のペプチド断片は、当業界で周知の技術によって合成または製造できることが理解されるであろう。例えば、参照によってその全体が本明細書中に組み入れられる、Creighton, 1983, タンパク質:構造および分子原理(Proteins: Structures and Molecular Principles), W. H. Freeman and Co., NYを参照されたい。
【0023】
あるいは、本発明のペプチドは、ペプチドのアミノ酸残基を連結する1個以上の結合が非ペプチド結合であるように合成できる。これらの他の非ペプチド結合は、当業者に周知の反応を利用することによって形成でき、以下に限定されないが、2〜3の例を挙げると、イミノ、エステル、ヒドラジド、セミカルバジド、およびアゾ結合を挙げることができる。
【0024】
本発明のまたもう1つの実施形態では、上記配列を含むT-20およびT-20様ペプチドは、例えば、ペプチドの安定性、反応性および/または溶解性が増強されるように、ペプチドのアミノおよび/またはカルボキシ末端に存在する付加的な化学基によって合成できる。例えば、カルボベンゾキシル、ダンシル、アセチルまたはt−ブチルオキシカルボニル基などの疎水性基を、ペプチドのアミノ末端に付加することができる。同様に、アセチル基または9−フルオロエニルメトキシ−カルボニル基を、ペプチドのアミノ末端に配置することができる。(上記のT-20の「X」修飾を参照されたい。)さらに、疎水性基、t−ブチルオキシカルボニル、またはアミド基を、ペプチドのカルボキシ末端に付加することができる。同様に、p−ニトロベンジルエステル基をペプチドのカルボキシ末端に配置することができる。(上記のT-20の「Z」修飾を参照されたい。)そのような修飾を導入する技術は、当業者に周知である。
【0025】
さらに、T-20およびT-20様ペプチドは、その立体配置が変わるように合成することができる。例えば、ペプチドの1個以上のアミノ酸残基の、通常のL−アイソマーよりもむしろD−アイソマーを用いることができる。
【0026】
さらにまた、本発明のペプチドの少なくとも1個のアミノ酸残基は、周知の自然に存在しないアミノ酸残基の1つによって置換できる。これらのような変更は、本発明のペプチドの安定性、反応性および/または溶解性を増加させるように働くことができる。
【0027】
T-20またはT-20様ペプチドのいずれも、それらのアミノおよび/またはカルボキシ末端に共有結合によって結合する巨大分子キャリアー基を追加的に有するように合成できる。そのような巨大分子キャリアー基としては、例えば、脂質−脂肪酸コンジュゲート、ポリエチレングリコール、炭水化物または付加的なペプチドが挙げられる。従って、上記のT-20の「X」修飾は、ペプチドのアミノ末端に共有結合によって結合する上記の巨大分子キャリアー基のいずれかを追加的に示すことができ、好ましくは付加的なペプチド基である。同様に、上記のT-20の「Z」修飾は、上記の巨大分子キャリアー基のいずれかを追加的に示すことができる。
【0028】
本発明のペプチド断片は、標準FMOCプロトコルを用いて固相ペプチド合成(SPPS)法によって合成されるのが好ましい。例えば、Carpinoら, 1970, J. Am. Chem. Soc. 92(19):5748-5749;Carpinoら, 1972, J. Org. Chem. 37(22):3404-3409を参照されたい。好ましい実施形態では、本発明のペプチド断片の固相合成は、以下に限定されないが、塩化2−クロロトリチル樹脂(例えば、Barlosら, 1989, Tetrahedron Letters 30(30):3943-3946を参照されたい。)および4−ヒドロキシメチル−3−メトキシフェノキシブチル酸樹脂(例えば、Seiber, 1987, Tetrahedron Letters 28(49):6147-6150、およびRichterら, 1994, Tetrahedron Letters 35(27):4705-4706を参照されたい。)を含む超酸感受性固相支持体上で行われる。塩化2−クロロトリチル樹脂および4−ヒドロキシメチル−3−メトキシフェノキシブチル酸樹脂の両者は、Calbiochem-Novabiochem Corp., San Diego, CAから購入できる。
【0029】
固相ペプチド合成で利用できる一般的な製造工程および樹脂の装填は、本明細書中に記載されている。樹脂の装填は、例えば、次の方法によって行うことができる:樹脂、好ましくは2−クロロトリチル樹脂などの超酸感受性樹脂を、反応チャンバーにローディングする。ジクロロメタン(DCM)などの塩素化溶媒で該樹脂を洗浄する。ベッドの液体を排出し(brained)、約8〜10倍容量(volume)のジクロロエタン(DCE)中1.5当量のアミノ酸および2.7当量のジイソプロピルエチルアミン(DIEA)からなる溶液を添加する。該アミノ酸のN末端は、好ましくはFmocによって、保護されているべきであり、該アミノ酸の側鎖は、必要または適当である場合には、保護されているべきである。この混合物を窒素通気によって2時間攪拌する。
【0030】
2−クロロトリチル樹脂を適度に膨潤させるために塩素化溶媒が必要であることに注意すべきである。文献によれば、DCEは、より高い装填効率を与えるが、DCMは、殆どまたは全く装填の低下を伴わずに置換できる。
【0031】
攪拌後、ベッドの液体を排出し、DCMで洗浄する。樹脂の活性部位を、9:1のMeOH:DIEAの溶液で、約20〜30分間末端キャッピング(endcapped)する。ベッドの液体を排出し、DCMで4回洗浄し、窒素パージによって乾燥し、装填された樹脂を得る。
【0032】
Fmocは、アミノ酸のN末端に対して好ましい保護基である。どのアミノ酸が装填されるかによって、その側鎖は、保護されていてもよいし、保護されていなくてもよい。例えば、Trpが装填されているときは、その側鎖は、Bocで保護されているべきである。同様に、Glnの側鎖は、trtで保護できる。しかしながら、1〜16ペプチド断片の合成のための調製物中にGlnが装填されているときは、その側鎖は、保護されるべきではない。Leuの側鎖は保護する必要がない。
【0033】
樹脂装填およびペプチド合成で用いられるFmoc保護アミノ酸は、必要に応じて側鎖保護基を有するまたは有しないものが、Sean社またはGenzyme社から入手できる。上記工程の代わりに、適当なアミノ酸がすでに装填されている樹脂を購入できる。
【0034】
下記第6節に示される実施例は、典型的な樹脂調製物を記載している。
【0035】
固相ペプチド合成法は、例えば、次の方法に従って行うことができる:装填された樹脂を反応チャンバーに添加し、溶媒、好ましくは塩化メチレン(DCM;好ましくは約10倍容量)で窒素攪拌下、約15分間調整して、樹脂ビーズを膨潤させる。2−クロロトリチル樹脂を適度に膨潤させるためにDCMが必要である。樹脂容量は、ビーズが膨潤し、活性部位が折りたたまれずに、反応に利用できるようになるにつれて、反応チャンバー中で2倍または3倍になるだろう。樹脂が膨潤した後、溶媒を反応チャンバーから排出する。
【0036】
末端アミンまたは樹脂からのFmoc(9−フルロエニル−メチルオキシカルボニル)保護基の除去は、その樹脂をN−メチル−2−ピロリジノン(NMP)中ピペリジンの20%溶液のアリコートで2回、それぞれ約10分間処理することによって達成される。各アリコートに必要な20%のピペリジンのNMP溶液の容量は、実施される反応規模に依存するだろう。次いで、NMP(約10倍容量)のアリコートで5〜7回樹脂を洗浄して、Fmoc副生成物(すなわち、ジベンゾフルベン(dibenzofulvene)およびそのピペリジン付加物)並びに残ったピペリジンを除去する。
【0037】
クロルアニル試験を用いて、Fmoc副生成物および残りのピペリジンの除去が完了したか否かを決定することができる。クロルアニル試験溶液を、トルエン中のクロルアニルの飽和溶液の液滴を約1mLのアセトンに添加することによって調製する。NMP洗浄は、その洗浄液の液滴をクロルアニル試験溶液に添加することによって試験できる。青色または紫色は、第2級アミンの存在について陽性であることを示し、Fmoc副生成物および/または残りのピペリジンが未だ存在することを示す。NMP洗浄を、青色または紫色がもはや観察されなくなるまで繰り返す。
【0038】
一方、樹脂に添加される、配列中の次のアミノ酸は、そのカルボキシ末端が反応のために活性化される。各アミノ酸のアミン末端は、Fmocで保護されているはずである。どのアミノ酸が添加されるかによって、その側鎖は、保護されていてもよいし、保護されていなくてもよい。好ましくは、tyr(Y)、Thr(T)、Ser(S)およびAsp(P)の側鎖はt-Buで保護され、His(H)、Gln(Q)およびAsn(N)の側鎖はtrtで保護され、そしてLys(K)およびTrp(W)の側鎖はBocで保護される。しかしながら、上記で考察したように、Hisの側鎖は保護されていなければならない。さらに、断片1〜16および9〜16のカルボキシ末端のGln残基の側鎖は保護されないことが好ましい。LeuまたはIleの側鎖は、保護する必要がない。
【0039】
アミノ酸は、次のように活性化される。Fmoc保護アミノ酸(1.5当量)、1−ヒドロキシベンゾトリアゾール水和物(HOBT)(1.5当量)、およびジイソプロピル−エチルアミン(DIEA)(1.5当量)を、室温でNMP(約7.5倍容量)に溶解する。この溶液を0〜5℃に冷却し、次いで、O−ベンゾトリアゾール−1−イル−N,N,N',N'−テトラメチルウロニウム・ヘキサフルオロホスフェート(HBTU)(1.5当量)を添加し、次いで、5〜15分間攪拌して溶解させる。活性化は、低温で行い、アミノ酸のラセミ化を最小限にすることが重要である。活性化およびラセミ化は、HBTUの非存在下では起きないので、HBTUは、冷たい溶液に添加される最後の試薬である。
【0040】
活性化されたアミノ酸の溶液を、液体を排出した樹脂にローディングし、DCM(約2.5倍容量)で洗浄する。アミノ酸の活性化は、HBTUがDCMに不溶性なので、NMP中で行うことに注意すべきである。しかしながら、樹脂ビーズの適度な膨潤を維持するために、DCMをこの時点で反応物に添加する。該反応物を窒素通気によって、約1時間攪拌する。結合の完了は、下記に記載される定性ニンヒドリン試験でモニターできる。
【0041】
定性ニンヒドリン試験を用いて反応の完了を確認するには、2〜20 mgの樹脂サンプルを回収し、メタノールできれいに洗浄する。このサンプルに、76%のフェノールのエタノール溶液を3滴、0.2 mM KCNのピリジン溶液を4または5滴、および0.28 Mニンヒドリンのエタノール溶液を3滴添加する。このサンプルをエタノールで希釈して、約0.5 mL容量にし、約75℃の加熱ブロック中に5〜10分間置く。青色または紫色は、遊離アミンの存在について陽性であることを示し、反応が未だ完結していないことを示す。濃縮サンプル中の変色程度をより容易に判断するために、このサンプルをさらに希釈して約3mL容量とする。
【0042】
1時間後にニンヒドリン試験が陽性なら、結合反応をさらに1時間続ける。2時間後にニンヒドリン試験の陽性が続くなら、樹脂の液体を排出し、約10倍容量のNMPで3回洗浄し、1当量の活性化アミノ酸を用いて結合反応を繰り返す。
【0043】
結合サイクルの間、樹脂を一晩保存すべきなら、樹脂ベッドの液体を排出し、窒素ブランケット(nitrogen blanket)下でDCMで被覆することができる。あるいは、該ベッドの液体を排出し、窒素ブランケット下で保存し、次いで、次の結合サイクルを進める前にDCM洗浄で調整する。できた断片を切断する前に一晩保存すべきなら、顕著なFmoc脱保護がNMP中で起きるので、樹脂ベッドをDCMで洗浄してNMPを除去すべきである。
【0044】
結合が完了したと判定された後、前記樹脂の液体を排出し、NMPのアリコート(約10倍容量)で3回洗浄する。ペプチド断片の次のアミノ酸の結合(mer)のために、このサイクルを繰り返す。最終の結合反応に続いて、樹脂をNMPのアリコート(約10倍容量)で4回洗浄し、次いで、DCMのアリコート(約10倍容量)で4回洗浄する。樹脂結合ペプチドは、窒素パージによって乾燥できる。
【0045】
固相合成法によって合成されたペプチドを切断し、例えば、次の方法に従って単離できる:ペプチドは、当業者に周知の方法を用いて樹脂から切断できる。例えば、トリフルオロ酢酸(TFA)の1%または2%DCM溶液あるいはTFAの1%および2%DCM溶液を組み合わせて用いてペプチドを切断できる。酢酸(HOAC)も、ペプチドの切断に使用できる。切断に必要な特異的な切断試薬、溶媒および時間は、切断される特定のペプチドに依存するだろう。切断後、切断画分を標準的な後処理工程に付し、ペプチドを単離する。一般的には、混合された切断画分を減圧下で濃縮し、次いで、エタノール、メタノールまたはヘプタンなどの溶媒で再構成する(reconstitution)。一般に、水を添加してペプチドを沈殿させ、減圧濾過によって回収する。あるいは、ペプチドの単離の前に該生成物を粉砕することができる。
【0046】
下記第7.1〜7.6節に示される実施例は、表1、2および/または3に示されるペプチド中間体の固相合成を示す。
【0047】
完全長T-20ペプチドの合成のためには、上記表1のペプチド中間体を一緒に結合させて、T-20ペプチドを得ることができる。例えば、上記表2に列挙されたペプチド中間体のグループを一緒に結合させ、T-20の完全長ペプチドを製造できる。中間体ペプチド断片からの完全長T-20のそのような合成の代表例は、下記第9節に示されており、図1〜5に概略的に記載されている。
【0048】
特定の実施形態では、T-20合成の4断片アプローチに従うことができる。例えば、「4断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合した4つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示される中間体ペプチド断片グループ5、6、8、9および12〜15は、好ましいグループを示す。図1および図2は、表2のペプチド中間体グループ6を利用し、完全長T-20を合成する、2種の4断片アプローチを示す。このグループについては、アミノ酸残基36(T-20カルボキシ1−末端アミノ酸残基)が、断片結合プロセスの間に個々に導入されることに注意する。図1に示されるT-20合成スキームの最高点(culmination)は、第9.1節で示される実施例で実証される。
【0049】
さらに、実施形態、T-20合成のための3断片アプローチに従うことができる。「3断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合した3つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示される中間体断片グループ2〜4、7、10および11は、好ましい3つの断片グループを示す。図3および図4は、表2のペプチド中間体グループ3を利用して完全長T-20を合成する、2種の3断片アプローチを示す。このグループに関しては、アミノ酸残基36(T-20カルボキシル末端アミノ酸残基)は、断片結合プロセスの間に個々に導入されることに注意する。図3に示されるT-20合成図式の最高点は、下記第9.1に示される実施例で実証される。図4に示されるT-20合成スキームの最高点は、下記第9.2〜9.5節に示される実施例で実証される。
【0050】
更なる実施形態では、T-20合成のための2断片アプローチに従うことができる。「2断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合する2つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示された中間体断片グループ1および16〜20は、好ましい断片グループを示す。図5は、表2のペプチド中間体グループ20を利用して、完全長T-20を合成する、2断片アプローチを示す。このグループについては、アミノ酸残基(T-20カルボキシル末端アミノ酸残基)は、断片結合プロセスの間に個々に導入されることに注意する。
【0051】
当業者に周知の液相ペプチド合成法は、本発明のペプチド中間体断片の合成に利用できる。第8.1〜8.11節に示される実施例は、表1、2および/または3に列挙されたペプチド中間体の典型的な液相ペプチド合成を記載している。例えば、広いpH範囲を示す非シリカ装填カラムパッキングには、Tosohaus社(Montgomeryville, PA)によって販売されるものよりも高いpH値を含むものがある。
【実施例】
【0052】
6.実施例:樹脂合成
本明細書第6.1〜6.3節に記載する実施例では、本明細書中に記載されたペプチドおよびペプチド中間体の固相合成と共に利用できるクロロトリチル樹脂を合成する。
【0053】
6.1 Fmoc−Trp(Boc)−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 25 25 −
樹脂
Fmoc−Trp(Boc)−OH − 526.60 1.5 37.5 19.7
ジイソプロピルエチル 129.25 1.7 42.5 5.5 7.4
アミン(DIEA)
ジクロロエタン(DCE) − − − − 250
ジクロロメタン(DCM) − − − − 6×250
9:1メタノール:DIEA − − − − 200
工程:
塩化2−クロロトリチル樹脂(25 g, 1当量)を、500 mLのペプチドチャンバーにローディングし、250 mLのDCMで洗浄した。ベッドの液体を排出し、10倍容量のDCE中のFmoc−Trp(Boc)−OH(1.5当量)およびDIEA(1.7当量)からなる溶液を添加した。この混合物を窒素通気によって2時間攪拌した。
【0054】
ベッドの液体を排出し、250 mLのDCMで洗浄した。樹脂の活性部位を、200 mLの9:1のMeOH:DIEA溶液で20分間末端キャッピングした。このベッドの液体を排出し、250 mLのDCMで4回洗浄し、窒素パージによって乾燥し、34.3 gの装填された樹脂を得た。
【0055】
樹脂からFmocアミノ酸を切断し、標準に対してアッセイすることによって、定量的HPLC分析を行った。材料のHPLCアッセイは、0.68 mmol/gで樹脂に装填されていることを示した。
【0056】
カラム:Phenomenox Jupiter C18;300Å;5μ
流速:1 mL/分
検出:260 nm紫外線
移動相: A:0.1%TFA水溶液
B:アセトニトリル中0.1%TFA
65% Bイソクラティック
保持時間:〜14分
6.2 Fmoc−Gln−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 25 25 −
樹脂
FmocGlnOH 368.39 1.5 37.5 13.8 −
ジイソプロピルエチル 129.25 1.7 42.5 5.5 7.4
アミン(DIEA)
ジクロロエタン(DCE) − − − − 75
N,N−ジメチルホルム − − − − 200
アミド(DMF)
ジクロロメタン(DCM) − − − − 6×250
9:1メタノール:DIEA − − − − 200
工程:
75 mLのDCEおよび200 mLのDMFからなる混合物中でFmocGlnOH(1.5当量)およびDIEA(1.7当量)からなる溶液を用いて上記第6.1節に示す実施例で用いた工程を繰り返した。DMFの添加はFmocGlnOHを安定化する。反応により33.8 gの装填された樹脂を得た。0.74 mmol/gの樹脂の理論装填量を仮定し、材料を進展させた(carried forward)。
【0057】
6.3 Fmoc−Leu−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 250 250 −
樹脂
FmocLeuOH 353.42 1.5 375 132.5 −
ジイソプロピルエチル 129.25 1.7 425 55 75
アミン(DIEA)
ジクロロエタン(DCE) − − − − 2000
ジクロロメタン(DCM) − − − − 6×1500
9:1メタノール:DIEA − − − − 1500
工程:
3Lのペプチドチャンバーに樹脂をローディングし、1.5LのDCMで洗浄した。ベッドの液体を排出し、8倍容量のDCE中のFmocLeuOH(1.5当量)およびDIEA(1.7当量)からなる溶液を添加した。この混合物を、窒素通気によって2時間攪拌した。
【0058】
ベッドの液体を排出し、1.5LのDCMで洗浄した。樹脂の活性部位を1.5Lの9:1のMeOH:DIEA溶液で30分間末端キャッピングした。このベッドの液体を排出し、1.5LのDCMで4回洗浄し、窒素パージによって乾燥して345 gの装填樹脂を得た。
【0059】
樹脂からFmocアミノ酸を切断し、標準に対してアッセイすることによって、定量的HPLC分析を行った。材料のHPLCアッセイは、0.72 mmol/gでの樹脂の装填を示した。
【0060】
カラム:Phenomenox Jupiter C18;300Å;5μ
流速:1 mL/分
検出:260 nmの紫外線
移動相: A:0.1%TFA水溶液
B:アセトニトリル中の0.1%TFA
65% Bイソクラティック
保持時間:〜8分
7.実施例:ペプチドの固相合成
下記に示す、第7.1〜7.6節は、表1、2、および/または3に列挙されたペプチド中間体の固相合成の例である。
【0061】
7.1 断片Fmoc−アミノ酸(1〜8)−OH(断片1b)構造の製造
Fmoc−Tyr(tBu)−Thr(tBu)−Ser(tBu)−Leu−Ile−His(trt)−Ser(tBu)−Leu−OH(配列番号2)
C93H121N10O15
分子量1619.06
材料: 分子量 当量 mmole グラム mL
Fmoc−Leu−2−クロロ− − 1.0 15.6 20.0 −
トリチル樹脂
Fmoc−アミノ酸* − 1.5 23.4 − −
1-ヒドロキシベンゾトリ- 153.15 1.5 23.4 3.6 −
アゾール(HOBT)水和物*
O-ベンゾトリアゾール-1- 379.25 1.5 23.4 8.9 −
イル-N,N,N',N'-テトラ
メチルウロニウム ヘキサ
フルオロホスフェート
(HBTU)*
ジイソプロピルエチル 129.25 1.5 23.4 3.0 4.1
アミン(DIEA)*
N−メチル−2−ピロリジノン − − − − 150
(NMP)*
塩化メチレン(DCM)* − − − − 50
20%ピペリジン/NMP* − − − − 2×200
すすぎ用のNMP* − − − − (洗浄毎)200
DCM中1%トリフルオロ酢酸 − − − − 300
(TFA)
0.5%TFA/DCM − − − − 200
ピリジン − − − −
エチルアルコール − − − − 110
水 − − − − 200+100
*結合サイクル当たり
理論収量:25.3 g 予想収量:80〜90%
実収量:20.0 g
工程:
1Lのペプチド反応チャンバーに、20 gのFmoc−Leu−2−クロロトリチル樹脂をローディングした。窒素攪拌下で200 mL(〜10倍容量)のDCM中で約15分間樹脂を調整し、ビーズを膨潤させ、次いで液体を排出した。
【0062】
末端アミンからのFmoc(9−フルオロエニルメチルオキシカルボニル)の除去は、NMP中ピペリジンの20%溶液200 mLで2回それぞれ10分間で達成した。次いで、樹脂を、200 mL(〜10倍容量)のNMPで5〜7回洗浄して、クロルアニル試験の陰性によって決定して、Fmoc副生成物(ジベンゾフルベンおよびそのピペリジン付加物)および残りのピペリジンを除去した。
【0063】
一方、配列中の次のアミノ酸であるFmoc−Ser(tBu)を、カルボキシル末端で反応させるために活性化した。Fmoc保護アミノ酸(1.5当量)、HOBT(1.5当量)、およびDIEA(1.5当量)を、室温で、150 mL(〜7.5倍容量)のNMPに溶解した。この溶液を0〜5℃に冷却し、次いでHBTU(1.5当量)を添加し、5〜15分攪拌して溶解させた。活性化された酸の溶液を、液体を排出した樹脂にローディングし、50 mLのDCM(〜2.5倍容量)で洗浄した。反応物を窒素通気によって1時間攪拌した。結合の完了は、定性ニンヒドリン試験でモニターした。結合反応が完結したと判定された後、樹脂の液体を排出し、200 mL(1倍容量)のNMPで3回洗浄した。
【0064】
それぞれ1.5当量のFmoc保護アミノ酸His(trt)、Ile、Leu、Ser(tBu)、Thr(tBu)およびTyr(tBu)を用いて、ペプチド断片の次の結合(mer)のためにサイクルを繰り返した。最終の結合反応に続いて、樹脂を200 mL(10倍容量)のNMPで4回、次いで、200 mL(10倍容量)のDCMで4回洗浄した。この樹脂を窒素パージによって乾燥して42 gの樹脂結合ペプチドを得た。
【0065】
300 mLのDCM中1%TFAで約2分間、次いで、200 mLのDCM中0.5%TFAを用いて、このペプチドを21 gの量の樹脂から切断した。切断画分をピリジン(TFAに対して1:1の容量比)上に回収した。切断洗浄液を合わせ、減圧下に濃縮して、容量を約50 mLとし、次いで、濃縮を続けて残りのDCMを除去し、最終容量を〜250 mLにしながら、110 mLのエタノールで再構成した。生成物を200mLの無図を添加して沈殿させた。そのスラリーを室温で30分間攪拌した。この固体を減圧濾過によって回収し、約100 mLの水で洗浄した。生成物を空気乾燥して、95%のHPLC純度を有する20.0 g(79%)のFmoc−アミノ酸(1〜8)−OHを得た。
【0066】
カラム:Phenomenox Jupiter C18
流速:1 mL/分
検出:260 nm紫外線
移動相: A:0.1%TFA水溶液
B:20分で80%B〜99%Bのアセトニトリル勾配中0.1%TFA
保持時間:約23分
7.2 断片Fmoc−アミノ酸(9〜15)−OH(断片5b)構造の製造
Fmoc−Ile−Glu(tBu)−Glu(tBu)−Ser(tBu)−Gln(trt)−Asn(trt)−G
ln(trt)−OH(配列番号6)
C117H129N10O18
分子量1963.39
材料: 分子量 当量 mmole グラム mL
Fmoc-Gln(trt)-2-クロロ- − 1.0 12.0 20.0 −
トリチル樹脂
Fmoc−アミノ酸* − 1.5 18.0 − −
1-ヒドロキシベンゾトリ- 153.15 1.5 18.0 2.8 −
アゾール(HOBT)水和物*
O-ベンゾトリアゾール-1- 379.25 1.5 18.0 6.8 −
イル-N,N,N',N'-テトラメチル-
ウロニウム ヘキサフルオロ-
ホスフェート(HBTU)*
ジイソプロピルエチルアミン 129.25 1.5 18.0 2.3 3.1
(DIEA)*
N-メチル-2-ピロリジノン − − − − 150
(NMP)*
塩化メチレン(DCM)* − − − − 50
20%ピペリジン/NMP* − − − − 2×200
すすぎ用のNMP* − − − − (洗浄毎)200
切断用DCM − − − − 160
酢酸(HOAc) − − − − 20
トリフルオロエタノール − − − − 20
ヘプタン − − − −250+250+100
メチル t-ブチルエーテル − − − − 100
イソプロパノール − − − − 60
水 − − − − 60+50
*結合サイクル当たり
理論収量:23.6 g 予想収量:89〜95%
実収量:21.1 g
工程:
20.0 gのFmoc−Gln(trt)−2−クロロトリチル樹脂、およびFmoc保護アミノ酸Asn(trt)、Gln(trt)、Ser(tBu)、Glu(tBu)、Glu(tBu)およびIleを用いて上記第7.1節に示される実施例で用いた工程を繰り返した。
【0067】
最終のカップリング反応に続き、樹脂を4 X 200mL(10容)のNMP、次いで4 X 200mL(10容)のDCMで洗浄した。
【0068】
ペプチドを200mLの8:1:1 DCM:TFE:HOAcを用いて2時間で樹脂から開裂させ、続いて2 X 100mLのDCMで洗浄した。溶出液を合わせて真空下で容量が約100mLになるまで濃縮し、次いで250mLのヘプタンで再構成する一方で、最終容量が約250mLになるまで濃縮を続けて残存しているDCMを除去した。形成した二相混合物からヘプタン層を分離した。250mLのヘプタンおよび100mLのMTBEを添加して生成物を沈殿させ、次いで室温で一晩粉砕すると所望の粘稠度の物質が得られた。固体を真空濾過によって回収して約100mLのヘプタンで洗浄した。生成物を再び後処理して残存している酢酸を除去した。濾過した固体を50℃で60mLのイソプロパノールに溶かした。溶液を氷浴中で0〜5℃に冷やし、次いで60mLの水を迅速な滴下速度で加えた。生成スラリーを氷浴中で約1時間攪拌しながら粉砕した。固体を真空濾過によって単離して約50mLの水で洗浄した。生成物を風乾すると95%HPLC純度のFmoc-AA(9-15)-OHが21.1g(90%)得られた。
【0069】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に80% B〜99% Bの勾配
保持時間:約23分
7.3 断片Fmoc-AA(1-16)-OH(断片3d)の調製
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を32.5gのFmoc-Gln-2-クロロトリチル樹脂および必要なFmoc保護アミノ酸を用いて繰り返した。上記の原料の節で示されたものとは若干異なる容量の溶媒を用いることを除けば、上記第6.1節で提示した実施例に記載された通りに反応を行った。
【0070】
最終のカップリング反応に続き、樹脂を4 X 250mL(8容)のNMP、次いで4 X 250mL(8容)のDCMで洗浄した。樹脂を窒素パージ下で乾燥させると97.4gの結合ペプチドが得られた。
【0071】
17.7gのスケールで、樹脂に結合したペプチドを2 X 190mLのDCM中の1% TFAを用いて1〜2分で樹脂から開裂させ、続いて1 X 120mLのDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)に回収した。画分および洗液を合わせて真空下で容量が約50mLになるまで濃縮し、次いで200mLのメタノールで再構成した。最終容量が約50mLになるまで濃縮を続けて残存しているDCMを除去した。250mLの水を添加して生成物を沈殿させ、室温で約30分間攪拌した。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると12.8g(84%)が得られた。生成物を再び後処理してピリジニウム塩を除去した。濾過した固体を室温で150mLのメタノールに溶かした。室温で200mLの水を添加すると生成物が沈殿した。生成物を真空濾過によって単離して約50mLの水で洗浄した。物質を風乾すると12.8g(84%)のFmoc-AA(1-16)-OHが得られた。
【0072】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に75% B〜99% Bの勾配
保持時間:約25分
7.4 断片Ac-AA(1-16)-OH(断片3c)の調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を32.5gのFmoc-Gln-2-クロロトリチル樹脂および必要なFmoc保護アミノ酸、ならびに上記の原料の節で示された溶媒容量を用いて繰り返した。
【0073】
最終のカップリング反応に続き、樹脂を4 X 250mL(8容)のNMP、次いで4 X 250mL(8容)のDCMで洗浄した。樹脂を窒素パージ下で乾燥させると97.4gの結合ペプチドが得られた。
【0074】
10gのスケールで、樹脂に結合したペプチドを100mLの3:1 NMP:DCM中で30分間無水酢酸およびピリジン(各5等量)でアセチル化し、次いで2 X 25mLのDCMで洗浄した。ペプチドを3 X 50mLのDCM中の1% TFAを用いて樹脂から開裂させ、続いて2 X 50mLのDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)中に回収した。画分および洗液を合わせて真空下で容量が約100mLになるまで濃縮し、次いで3 X 50mLのヘプタンを少量ずつ加えて再構成する一方で、最終容量が約50mLになるまで濃縮を続けて残存しているDCMを除去した。生成物は最初はややべとつく粘稠度で沈殿したが、0〜5℃の氷浴中で約30分間攪拌しながら粉砕すると濾過可能な固体となった。固体を真空濾過によって回収して約150mLのヘプタンで洗浄した。生成物を再び後処理してピリジニウム塩を除去した。濾過した固体を室温で50mLのメタノールに溶かした。溶液を氷浴中で0〜5℃に冷やし、次いで50mLの水を迅速な滴下速度で加えた。生成物は最初は粘稠な固体として沈殿し、それを氷浴中で約1時間攪拌しながら粉砕すると濾過可能な粘稠度となった。生成物を真空濾過によって単離して約25mLの水で洗浄した。生成物を風乾すると7.0g(90%)のAcAA(1-16)-OHが得られた。続いて生成物を前述のように再び後処理すると96% HPLC純度の物質が6.2g(回収率89%)得られた。
【0075】
カラム:Zorbax LP C8, 100Å, 20μ
流速:1mL/分
検出:220nmの紫外線
移動相:A: 0.1% 水性TFA
B: 1:1 ACN:IPA中の0.05%TFA
20分間に80% B〜99% Bの勾配
保持時間:約15分
7.5 断片Fmoc-AA(17-26)-OH(断片10b)の調製
構造:
Fmoc-Glu(tBu)-Lys(Boc)-Asn(trt)-Glu(tBu)-Gln(trt)-Glu(tBu)-Leu-Leu-Glu(tBu)-Leu-OH (配列番号11)
理論収量:44.4g 期待収率:90-105%
実際の収率:46.9(105%)
操作:
上記第7.1節で提示した実施例において用いられた操作を25.0gのFmoc-Leu-2-クロロトリチル樹脂、必要なFmoc保護アミノ酸、および上記の原料の節で示された溶媒容量を用いて繰り返した。
【0076】
最終のカップリング反応に続き、樹脂を4 X 250mL(10容)のNMP、次いで4 X 250mL(10容)のDCMで洗浄した。
【0077】
ペプチドを3 X 400mL(約15容)のDCM中の1% TFAを用いて樹脂から開裂させ、次いで1 X 200mL(7.5容)のDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)中に回収し、次いで画分および洗液について生成物含量を分析した。生成物を含む画分を合わせて真空下で容量が約100mLになるまで濃縮し、次いで300mLのエタノールで再構成した。最終容量が約250mLになるまで濃縮を続けて残存しているDCMを除去した。攪拌した溶液に300mLの水を加えて生成物を沈殿させた。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると97% HPLC純度のFmoc-AA(17-26)-OHが46.4g(105%)得られた。
【0078】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に75% B〜99% Bの勾配
保持時間:約25分
7.6 断片Fmoc-AA(27-35)-OH(断片16b)の調製
構造:
Fmoc-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-OH (配列番号17)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を33.0gのFmoc-Trp(Boc)-2-クロロトリチル樹脂、必要なFmoc保護アミノ酸、および上記の原料の節で示された物質を用いて繰り返した。
【0079】
最終のカップリング反応に続き、樹脂を4 X 250mL(7.5容)のNMP、次いで4 X 250mL(7.5容)のDCMで洗浄した。
【0080】
ペプチドを8:1:1 DCM:TFE:HOAcの300mL(約10容)の溶液で2時間処理することによって樹脂から開裂させた。樹脂の水分を切り、2 X 250mLのDCMで洗浄した。開裂溶液および洗液を合わせて約50mLの容量になるまで濃縮し、次いで250mLのエタノールで再構成した。溶液を氷浴中で攪拌しながら0〜5℃に冷やした。攪拌した溶液に125mLの水を加えて生成物を沈殿させた。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると95% HPLC純度のFmoc-AA(27-35)-OHが32.0g(65.4%)得られた。
【0081】
樹脂を2 X 250mLのDCM中のTFAの1%溶液で処理し、続いて100mLのDCMで洗浄した。開裂画分をTFAと1:1の容量比であるピリジン中に回収した。溶出液および洗液を合わせて容量が約50mLになるまで濃縮した。この溶液に100mLのエタノール、次いで150mLの水を加えた。生成スラリーを真空濾過すると10.7g(21.9%)(95% HPLC純度)の二次収量が得られ、合わせた収率は87.3%となった。1% TFA/DCM開裂が、より高い効率およびより低い容量であるために好ましい。
【0082】
カラム:Phenomenox Jupiter C5, 300Å, 5μ
流速:0.75mL/分
検出:260nmの紫外線
移動相:A: 0.05% 水性TFA
B: 1:1 IPA:MeOH中の0.1%TFA
10分間に70% B〜97% Bの勾配
保持時間:約25分
8. 実施例:ペプチド断片の液相合成
下記の第8.1−8.11節において提示するのは表1、2、および/または3に挙げたペプチド中間体の液相合成の例である。
【0083】
8.1 グルタミンのパラニトロベンジルエステル(OPNB)のFmoc-AA9-15OHへのカップリングによる断片Fmoc-AA9-16OPNBの調製
構造:
Fmoc-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号7)
理論収量:21.5g 期待収率:90-105%
操作:
FmocAA9-15OH(上記第7.2節で合成)、HBrGlnOPNB、HOATおよびEtPr2Nを磁気攪拌棒を備えた1Lの丸底フラスコ中で混合してNMP(200mL)を添加した。得られた溶液を窒素雰囲気下に置き、攪拌しながら0〜5℃に冷却した。冷却した溶液にHBTUを加えた。溶液を0〜5℃で15分間攪拌し、氷浴を外して攪拌を2.5時間続けた(注1)。
【0084】
反応混合物を0〜5℃に冷却し、0.5N 水性HCl(250mL)を添加して保護されたペプチドを沈殿させた。固体を真空濾過によって回収し、濾過フラスコ中で乾燥させると24gの粗FmocAA9-16OPNBが得られた。この固体を酢酸エチル(250mL)に溶かし、硫酸マグネシウム(10g)で乾燥させ、濾過して容量が100mLになるまで濃縮した。溶液を0〜5℃に冷却し、ヘキサン(250mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収して乾燥させると21.5gのFmocAA9-16OPNBが100%の収率および91-94% HPLC純度で得られた(注2)。
【0085】
注:
1. 薄層クロマトグラフィー(TLC)による工程内管理(IPC)。
90/10 クロロホルム/エタノール
紫外線、ヨウ素検出
Rt FmocAA9-15OH 0.46
Rt FmocAA9-16OPNB 0.57
2. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:13.3分
【0086】
8.2 断片HCl HAA9-16OPNBの調製
構造:
HCl H-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号7)
理論収量:19.2g 期待収率:85-105%
操作:
磁気攪拌棒を備えた1Lの丸底フラスコにFmocAA9-16OPNB(上記第8.2節で合成)および20:1 テトラヒドロフラン/ピペリジンを仕込んだ。得られた溶液を窒素雰囲気下室温で60分間攪拌した(注1)。ヘキサン(350mL)を添加してペプチドを沈殿させた。溶媒を粘性のある固体からデカントした。室温で18時間固体をMTBE(200mL)を用いて粉砕した。この固体を真空濾過によって回収して乾燥させると18.9gのHAA9-16OPNBが得られた(注2)。
【0087】
この固体をメタノール(150mL)に溶かし、攪拌しながら0〜5℃に冷却した。0.5N 水性塩酸(100mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(50mL)、次いで2-プロパノール(50mL)で洗浄して乾燥させると17.7gのHCl HAA9-16OPNBが92%の収率および92A% HPLC純度で得られた(注3)。
【0088】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:FmocAA9-16OPNB, 13.3分; HAA9-16OPNB, 10.7分
2. この時点で単離されたHAA9-16OPNBは微量のピペリジンおよびベン
ジルフルベンピペリジン付加物を含んでいる。両方をRAA1-8OHとカ
ップリングする前に除去する。
3. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B
保持時間:HAA9-16OPNB, 7.2分
TLC条件:
90/10 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rf: HAA9-16OPNB, 0.64
【0089】
8.3 断片AcAA1-8OHとHAA9-16OPMBとの液相カップリングによる断片AcAA1-16OPNBの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.38 期待収率:85-105%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにAA1-8OH(上記第7.1節で合成)、HCl HAA9-16OPNB(上記第8.2節で合成)およびHOATを仕込んだ。固体をEtPr2Nを含む9:1 DMF:DMSO(5mL)に溶かし、次いで窒素雰囲気下0〜5℃に冷却した(注1)。冷却した溶液にHBTUを加えた。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに60分間攪拌した(注2)。水(7mL)の添加によってペプチドを溶液から沈殿させた。固体を真空濾過によって回収し、水(10mL)で洗浄して乾燥させると0.36gの粗AcAA1-16OPNBが得られた。この固体を室温で1.5時間MTBE(3.5mL)を用いて粉砕し、真空濾過によって回収して乾燥させると0.335gのAcAA1-16OPNBが88%の収率および82A% HPLC純度で得られた(注3)。
【0090】
注:
1. 0〜5℃に冷却してHBTUを添加する前にすべての固体を溶液にする
ことが重要である。
2. 工程内管理, TLC, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:HAA9-16OPNB, 7.2分(Ac1-8OHは260nmに吸光度をもたな
い).
保持時間:AcAA1-16OPNB, 12.45
TLC条件:
90/10 クロロホルム/エタノール
紫外線、ヨウ素検出
Rf: HAA9-16OPNB, 0.64
Rf: Ac1-8OH, 0.35
Rf: Ac1-16OH, 0.48
3. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:Ac1-16OPNB, 16.4.
【0091】
8.4 断片FmocAA1-8OHとHCl HAA9-16OPNBとの液相カップリングによる断片FmocAA1-16OPNBの調製
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:28.3g 期待収率:85-100%
操作:
磁気攪拌棒を備えた1Lの丸底フラスコにFmocAA1-8OH(上記第7.1節で合成)、HCl HAA9-16OPNB(上記第8.2節で合成)、HOATおよびDMF(250mL)を仕込んだ。溶液にEtPr2Nを添加した。この溶液を0〜5℃に冷却し、HBTUを加えた。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに70分間攪拌した(注1)。反応混合物を0〜5℃に冷却し、10% 塩化ナトリウム/水(200mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(50mL)で洗浄して乾燥させると27gの粗FmocAA1-16OPNBが得られた。固体をメタノール(200mL)に溶かして、攪拌した塩化ナトリウムの水溶液(10% wt/vol, 300mL)に加えた。この固体を真空濾過によって回収し、水(50mL)で洗浄して乾燥させると26gのFmocAA1-16OPNBが92%の収率およびHPLCでは90A%純度で得られた(注1)。
【0092】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:HAA9-16OPNB, 7.2分, FmocAA1-8OH, 7.9分
保持時間:FmocAA1-16OPNB, 14.5分
8.5 断片H-AA1-16OPNBの調製
構造:
H-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.94g 期待収率:90-105%
操作:
磁気攪拌棒を備えた50mLの丸底フラスコにFmocAA1-16OPNB(上記第8.4節で合成)、ジクロロメタン(11.4mL)およびピペリジン(0.6mL)を仕込んだ。溶液を窒素雰囲気下、室温で90分間攪拌した(注1)。ヘキサン(45mL)を反応混合物に添加し、溶媒の容量を真空蒸留によって25mLに減らした。得られた固体を真空濾過によって回収し、乾燥させると0.96gのHAA1-16OPNBが102%の収率で得られた。この固体のHPLC分析によって72A%のHAA1-16OPNBおよび18A%のフルベンおよびピペリジン−フルベン付加物が示された。
【0093】
注:
1. Phenomenox Jupiter, C5, 5μ, 300Å
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:FmocAA1-16OPNB, 14.1分
保持時間:HAA1-16OPNB, 11.6分
保持時間:フルベンおよびピペリジン−フルベン付加物, 5.5分および4.8分。
【0094】
8.6 HAA1-16OPNBのN末端のアセチル化による断片Ac-AA1-16OPNBの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.96g 期待収率:80-100%
操作:
磁気攪拌棒を備えた50mLの丸底フラスコにHAA1-16OPNB(上記第8.5節で合成)、DMF(10mL)、無水酢酸およびピリジンを仕込んだ。反応混合物を窒素雰囲気下、室温で60分間攪拌した(注1)。水(20mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(10mL)で洗浄して乾燥させると0.87gのAcAA1-16OPNBが得られた。残存しているフルベンおよびピペリジン−フルベン付加物を除去するために、この固体を室温で4.5時間1:1 MTBE/ヘキサン(20mL)を用いて粉砕した。固体を真空濾過によって回収して乾燥させると0.82gのAcAA1-16OPNBが85%の収率およびHPLCでは90A%以上の純度で得られた(注1)。
【0095】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、15分で100%B.
保持時間:HAA1-16OPNB, 11.6分
保持時間:AcAA1-16OPNB, 12.1分
TLC, ジクロロメタン中の10%エタノール
紫外線、ヨウ素検出
Rf: AcAA1-16OPNB, 0.69
8.7 His(trt)の存在下でAcAA1-16OPNBからパラニトロベンジル保護基を選択的に除去することによる断片AcAA1-16OHの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:0.76g 期待収率:70〜85%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにAcAA1-16OPNB(上記の第8.6節で合成)およびDMF(10mL)を仕込んだ。この溶液にギ酸アンモニウム水溶液(0.5mL)、次いで湿らせたパラジウムカーボン(Degussa, 10%, 50%水) を添加した。スラリーを窒素雰囲気下、室温で120分間攪拌した(注1)。このスラリーをセライトの密な充填層で濾過し、90mLの水に取った。濾過ケーキをDMF(5mL)で洗浄した。この水性懸濁液を酢酸エチル(100mL)で洗浄した。次いで酢酸エチルを容量が20mLになるまで濃縮した(注意2)。ヘキサン(40mL)を添加して沈殿を完了させ、溶媒を固体からデカンテーションした。固体をメタノール(10mL)に溶かし、4:1の 水/塩化ナトリウム飽和水溶液(25mL)を添加してペプチドを沈殿させた。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると0.62gのAcAA1-16OHが得られた。この固体を室温で15時間50% MTBE/ヘキサン(8mL)とともに粉砕し、回収して乾燥させると0.59gのAcAA1-16OHが77%の収率およびHPLCでは90A%の純度で得られた(注3)。
【0096】
注:
1. 工程内管理, TLC
80/20 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rf: AcAA1-16OPNB, 0.90
Rf: Ac1-16OH, 69
2. AcAA1-16OHは溶媒容量を減じるにつれ沈殿し始める。
3. Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで5分間
保持時間:AcAA1-16OH, 10.73分
【0097】
8.8 FmocAA27-35OHとHPheNH2との液相カップリングによる断片FmocAA27-36NH2の調製
構造:
Fmoc-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号18)
理論収量:42.8g 期待収率:90〜105%
操作:
磁気攪拌棒を備えた2Lの丸底フラスコにFmocAA27-35OH(上記の第7.6節で合成)、HOAT、HpheNH2およびDMF(500mL)を仕込んだ。EtPr2Nを添加して溶液を0〜5℃に冷却し、次いでHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに70分間攪拌した(注1)。溶液を0〜5℃に冷却し、水(500mL)を添加してペプチドを沈殿させた。固体を減圧濾過によって回収し、水(100mL)で洗浄して乾燥させると43gのFmocAA27-36NH2が100%の収率およびHPLCでは93A%の純度で得られた(注2)。
【0098】
注:
1. 工程内管理, TLC
88/12 ジクロロメタン/メタノール
紫外線、ヨウ素検出
Rf: FmocAA27-35OH, 0.49
Rf: FmocAA27-36NH2, 0.63
2. Phenomenox Jupiter, C18, 5μ, 300オームストロング
1.0mL/分, 260nm
A H2O/0.1% TFA
B ACN
20分以上で75〜99%B、99%Bで5分間
保持時間:29.4分
8.9 断片H-AA(27-36)-NH2の調製
構造:
H-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-NH2 (配列番号17)
理論収量:19.6g 期待収率:85〜95%
操作:
磁気攪拌棒および窒素ブランケットを備えた250mLの丸底フラスコに上記の第8.8節で合成したFmoc断片、塩化メチレン(約5容)、およびピペリジンを仕込んだ。溶液が得られ、室温で1.5時間攪拌した(注1)。
【0099】
この溶液を2 X 100mLの水で洗浄した。層が分離し、有機層を減圧下で元の容量の約2分の1になるまで濃縮した。MTBEをわけて(2 X 50mL)加えつつ、濃縮を続け、重沈殿点までおよび最終ポット容量が約150mLになるまでDCMを除去した。
【0100】
生成スラリーを攪拌し、0〜5℃の氷浴中で約1時間冷却した。固体を減圧濾過によって単離し、2 X 15mLのMTBEで洗浄した。生成物を空気乾燥させると95% HPLC純度のH-AA(27-36)NH2が17.6g(89.6%)得られた(注2)。
【0101】
注:
1) 反応の完了はHPLCによってモニターした。
カラム:Phenomenox Jupiter, C18; 300オームストロング; 5μ
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% TFA水溶液
B: アセトニトリル中の0.1% TFA
20分間で75%B〜99%Bの勾配
保持時間:約18分
【0102】
2) Fmoc副生成物であるジベンゾフルベンおよびそのピペリジン付加物は双方とも後処理において効果的に除去される。しかしながら、この物質を先へ進める前に使用テストを行わねばならない。物質が使用テストを通らなければ、DCM(5容)に固体を溶かし、次いで前述の工程を続けることにより後処理操作を繰り返す。
【0103】
8.10 FmocAA17-26OHとHAA27-36NH2との液相カップリングによる断片FmocAA17-36NH2の調製
構造:
Fmoc-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号12)
理論収量:41.5g 期待収率:80〜85%
操作:
磁気攪拌棒を備えた2Lの丸底フラスコにFmocAA17-26OH(上記の第7.5節で合成)、HAA27-36NH2(上記の第8.9節で合成)、HOATおよびDMF(400mL)を仕込んだ。EtPr2Nを添加し、攪拌した溶液を窒素雰囲気下0〜5℃に冷却してHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに2.5時間攪拌した(注1)。水(500mL)を反応混合物に添加してペプチドを沈殿させた(注2)。得られたスラリーを45分間攪拌し、固体を減圧濾過によって回収し、水(100mL)で洗浄して乾燥させた。固体を磁気攪拌棒を備えた2Lの丸底フラスコに戻し、60℃の2-プロパノール(1.1L)を添加した。スラリーを室温まで冷ましながら窒素雰囲気下で攪拌した(一晩)。固体を減圧濾過によって回収して乾燥させると37.5gのFmocAA17-36NH2が90%の収率およびHPLCでは95.5A%の純度で得られた(注1)。
【0104】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで25分間
保持時間:FmocAA17-26OH, 8.2分, HAA26-36 NH2, 8.4分
保持時間:FmocAA17-36NH2, 13.3分
2. 水を添加するときに反応混合物を38℃に温めた。
【0105】
8.11 断片H-AA(17-36)-NH2の調製
構造:
H-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-NH2 (配列番号19)
理論収量:21.6g 期待収率:95〜100%
操作:
磁気攪拌棒および窒素ブランケットを備えた250mLの丸底フラスコに上記の第8.10節で合成したFmoc断片、THF(約7.5容)、および5N NaOH(約2.5容)を仕込んだ。2相溶液が得られ、室温で10〜15分間攪拌した(注1)。
【0106】
層を分離し、有機相を1N HClでpH 2〜3に調整した。次いで溶液を2 X 55mL(2.5容)の飽和食塩水で洗浄した(注2)。層を分離し、有機層を減圧下、15〜20℃で元の容量の約2分の1になるまで濃縮した。ヘプタンをわけて(3 X 25mL)加えつつ、濃縮を続け、重沈殿点までおよび最終ポット容量が約100mLになるまでTHFを除去した(注2)。
【0107】
生成スラリーを室温で約2時間攪拌した。固体を減圧濾過によって単離し、約50mLのヘプタンで洗浄した。生成物を空気乾燥させると21.0g(97.5%)H-AA(17-36)NH2が得られた。
【0108】
残存しているジベンゾフルベン副生成物を除去するために再度後処理を行ってもよい。生成物を室温で3時間200mLのヘプタン中でスラリー化した。物質を濾過し、約50mLのヘプタンで洗浄して空気乾燥させると95%以上のHPLC純度の生成物が20.8g(96.6%)得られた。
【0109】
注:
1) 反応の完了はHPLCによってモニターした。
カラム:Zorbax LP C8; 1 OOオームストロング; 2011 流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% TFA水溶液
B: 1:1 ACN:IPA中の0.05% TFA
20分間で80%B〜99%Bの勾配
保持時間:約18分
【0110】
2) 固体は最初は、ろう状ゴムとして沈殿するが、それは依然として攪拌可能であり、さらに濃縮しながら粉砕すると濾過可能な固体となる。
【0111】
9. 実施例:全長T-20ペプチドの合成
本明細書の下記の第9.1〜9.5節において提示されているのは、全長T-20ペプチドを産生するために中間体ペプチド断片を利用する実施例である。
【0112】
この節において提示された実施例は中間体ペプチド断片から全長T-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。
【0113】
9.1 AcAA1-16OHとHAA17-36NH2との液相カップリングによる断片AcAA1-36NH2の調製
ここに記載した合成経路は図1および3に図示したT-20の4つの断片からのアプローチの最終段階を表している。AcAA1-16OHが固相技術によって合成される場合には、図1のアプローチに従い、AcAA1-16OHが液相技術によって合成される場合には、図3のアプローチに従う。ここで合成されたT-20全長ペプチドはそのアミノ末端にアセチル基修飾(すなわち、「X」)およびそのカルボキシル末端にアミド基修飾(すなわち、「Z」)をもつ配列番号1のアミノ酸配列を有する。
【0114】
構造:
AC-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:1.77g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた100mLの丸底フラスコにAcAA1-16OH(固相技術により上記の第7.4節で、または液相技術により上記の第8.7節のいずれかで合成)、HOAT、DMF(20mL)を仕込み、続いてEtPr2N(0.074mL)を仕込んだ。溶液を窒素雰囲気下で0〜5℃に冷却してHBTUを添加した。溶液を0〜5℃で15分間攪拌し、HCl HAA17-36NH2(上記の第8.11節で合成)を添加し、続いてさらに0.041mLのEtPr2Nを添加した。冷却浴を外し、反応混合物を2時間攪拌した(注1)。ペプチドを沈殿させるために、水(25mL)および飽和食塩水(5mL)を添加した。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると1.74gの粗製AcAA1-36NH2が得られた(注2)。この固体を室温で2.5時間50% MTBE/ヘキサンとともに粉砕し、減圧濾過によって回収して乾燥させると96%の収率およびHPLCでは92A%の純度で1.70gが得られた。
【0115】
注:
1)工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:AcAA1-16OH, 11.8分
保持時間:HCl HAA17-36 NH2, 12.7分
保持時間:AcAA1-36NH2, 22.9分
2) 脱水によって非常に微細な沈殿が生成する。濾過を2回行うことが必要かもしれない。
【0116】
9.2 FmocAA1-16OHとHAA17-36NH2との液相カップリングによる断片FmocAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でFmocAA1-36NH2が合成されるまでを示している。
【0117】
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.91g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにFmocAA1-16OH(上記の第7.3節で合成)、HCl HAA17-36NH2(上記の第8.11節で合成)、HOAT、DMF(10mL)を仕込み、EtPr2Nを添加した。溶液を窒素雰囲気下で0〜5℃に冷却してHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めて1.5時間攪拌した(注1)。ペプチドを沈殿させるために水を添加し、固体を減圧濾過によって回収して乾燥させた。固体を室温で15時間、2プロパノール(14mL)とともに粉砕し、次いで水(3mL)を添加して溶液から所望の生成物を得た。固体を減圧濾過によって回収して乾燥させると0.80gのFmocAA1-36NH2が88%の収率および85A%のHPLC純度で得られた。
【0118】
注:
1) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:FmocAA1-16OH, 11.4分
保持時間:HCl HAA17-36 NH2, 12.分
保持時間:FmocAA1-36NH2, 20.4分
TLC, 9/1 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rft: FmocAA1-36NH2, 0.71
9.3 断片HAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生するための固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でH-AA1-36NH2が合成されるまでを示している。
【0119】
構造:
H-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.77g 期待収率:85〜95%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにFmocAA1-36NH2(上記の第9.2節で合成)、DMF(9.5mL)およびピペリジン(0.5mL)を仕込んだ。溶液を窒素雰囲気下、室温で2時間攪拌した(注1、2)。水(20mL)および塩化ナトリウム飽和水溶液(5mL)を添加して保護されたペプチドを沈殿させた。固体を減圧濾過によって回収して乾燥させるとフルベンおよびピペリジン−フルベン付加物が混在した0.77gのHAA1-36NH2が得られた。固体を室温で15時間50% MTBE/ヘキサンとともに粉砕してフルベンおよびピペリジン−フルベン付加物を除去した。固体を減圧濾過によって回収して乾燥させると0.73gのHAA1-36NH2が95%の収率およびHPLCでは90A%の純度で得られた。
【0120】
注:
1) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:FmocAA1-36OH, 20.4分
保持時間:HCl HAA1-36 NH2, 19.9分
保持時間:フルベンおよびピペリジン−フルベン付加物, 5分
TLC, 9/1 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rt: FmocAA1-36NH2, 0.71
2) 生成物および出発原料はTLCまたは逆相HPLCでは十分に分離しない。生成物の形成の後、フルベンおよびピペリジン−フルベン付加物を測定する。
【0121】
9.4 HAA1-36NH2のN末端アセチル化による断片AcAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でAc-AA1-36NH2が合成されるまでを示している。
【0122】
構造:
AC-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.71g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにHAA1-36NH2(上記の第9.3節で合成)、DMF(10mL)およびピリジン(0.046mL)および無水酢酸(0.027mL)を仕込んだ(注1)。溶液を窒素雰囲気下、室温で4時間攪拌した(注2)。水(7.5mL)および塩化ナトリウム飽和水溶液(7.5mL)を添加して保護されたペプチドを沈殿させた。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると0.65gのAcAA1-36NH2が91%の収率およびHPLCでは90A%の純度で得られた(注3)。
【0123】
注:
1) ジクロロメタンをこの反応の溶媒として使用してもよい。
2) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:HAA1-36OH, 23.3分
保持時間:AcAA1-36 NH2, 22.7分
3) 生成物および出発原料はTLCまたは逆相HPLCでは十分に分離しない。
【0124】
9.5 AcAA1-36NH2の側鎖脱保護によるT-20側の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に示したT-20の3つの断片からのアプローチを表している。
【0125】
構造:
Ac-Tyr-Thr-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lys-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Phe-NH2(配列番号1)
理論収量:157mg 期待収率:25〜50%
操作:
90:5:5(v/v/wt %)トリフルオロ酢酸/水/ジチオスレイトール溶液から窒素を脱気して0〜5℃に冷却した。冷却した溶液にAcAA1-36NH2(上記の第9.4節で合成)を添加した。固体が溶けるまで(約5分間)スラリーを0〜5℃で攪拌し、次いで室温まで温めて2.5時間攪拌した。溶液を0〜5℃のMTBE(70mL)に添加してペプチドを沈殿させた。スラリーを遠心分離機にて2200rpmで5分間回転させ、MTBEを固体からデカンテーションした。固体を再びMTBE(50mL)に懸濁させ、遠心分離機において2200rpmで5分間回転させ、MTBEをデカンテーションした。この工程をもう1度繰り返し、次いで固体を1容量%酢酸を含む1:1水/アセトニトリル(30mL)に溶かして室温で24時間保持した(注1)。溶液を凍結させ、次いで凍結乾燥機を用いて凍結乾燥させると155mgの粗製T-20が得られた。分取HPLCによる精製によって55mgの全長T-20ペプチドがHPLCでは95A%の純度で得られた(注2)。
【0126】
注:
1)Trp(Boc)のtBu側鎖を速やかに除去してTrpCOOHにする。TrpCOOHの脱カルボキシル化には、周囲温度、酢酸水溶液中において最低24時間を必要とする。
2)分取HPLC
2” YMC, 120A, 10μ, C18
220nm, 50mL/分
A H2O/0〜1% TFA
B ACN/0.1% TFA
39〜49% B/40分
【0127】
10 実施例:T-20ペプチドの精製
本明細書中に提示された実施例は、全体を通してペプチド精製を著しく増加させる条件下でT-20およびT-20様ペプチドを精製することができる方法を記載している。
【0128】
材料
使用カラム:Amberchrom CG-300S(Tosohaus; Montgomeryville, PA)の35μm粒子を充填した20 x 30cmのもの。
【0129】
バッファーの調製
バッファーA=NH4OHでpH 8.5に調整した100mM酢酸アンモニウム。
【0130】
バッファーB=アセトニトリル。
【0131】
1. 約6カラム容の20%Bを有するカラム。
【0132】
2. T-20を50〜100mLのペプチド1グラム当たり85%A/15%Bに溶かす。2M K2CO3でpHを8〜10に調整する。T-20サンプル中のアセトニトリル濃度は15〜20%を超えない。
【0133】
3. カラム圧をモニターしながら、T-20溶液を500mL/分でロードする。
【0134】
4. T-20溶液をロードした後に3カラム容(10L)の85%A/15%Bの溶液をロードしてラインを洗い落とす。
【0135】
5. カラム溶出液を303nmでモニターし、全ロード工程中の溶出液を回収する。
目盛り上にピークを維持するために実験中に波長またはアテニュエーションを調整する。吸光度は波長とセルの経路長との関数である。
【0136】
6. 全実験中に圧力をモニターしながら勾配操作を以下のように始める(B/時で1.6%の変化)。
【0137】
時間(分) %B 流速(mL/分)
0 15 330
788 36 330
7. 主要ピークが溶出し始めると、10分の画分を回収する(3.3L)。すべてのT-20が35%Bにおいて溶出するはずである。平均して35〜40の画分が回収される。プールする画分の決定がなされるまで画分を0〜5℃で貯蔵する。
【0138】
8. 検出器の吸光度が0.1AU未満になるまで、または主要ピークが溶出した後に主要変曲点が到達するまで画分を回収する。
【0139】
9. 各画分の純度は分析逆相HPLCによってモニターする。
【0140】
結果
T-20ペプチドは7より高いpH範囲で非常に可溶性が高い。ペプチドの精製に通常用いられるカラム支持体はシリカベースのものであり、シリカ支持体はより高いpH範囲で溶解する傾向にあるので、それゆえ一定のpH範囲でのみ使用することができる。
【0141】
本明細書中に記載された方法では、広いpH範囲で(pH 1〜14)安定なポリスチレンベースの樹脂支持体を利用する。この方法はカラム能力を著しく増加させ、ここでT-20の溶出量は10gから250〜450gまで増加した(直径8” x 30cmのカラムに関して)。
【0142】
11. 実施例:T-20ペプチド樹脂ローディングの大規模合成及び精製
FmocTrp(Boc)OHは、原料樹脂1グラムあたり1.2ミリモル以上のレベルで、非常に活性な2-CTC樹脂上にローディングすることができる。FmocTrp(Boc)OHの量が原料樹脂1グラムあたり1.1ミリモルを超えると(ローディングされた樹脂で1グラムあたり0.72ミリモルを超える量)、当該断片のアミノ酸のうち最後の3個について、ニンヒドリン試験が陰性となるのは難しい。同様に、FmocLeuO-樹脂を1グラムあたり0.85ミリモルより多くローディングすると、FmocAA17-26O-樹脂を作るのは困難になり、FmocGlnOH-樹脂を1グラムあたり0.75ミリモルより多くローディングすると、AcAA1-16O樹脂を作るのが難しくなる。
【0143】
ベンダーから樹脂を受領する際には、ローディングされるアミノ酸それぞれについて、2-CTC約1グラムで使用テストを行う。使用テストの目的は、ローディング中に使用されるアミノ酸量を決定して所望のローディング範囲を得ることにある。それぞれについて0.5当量過剰(報告された樹脂活性に対して)のDIEAと共に、0.8、1.0及び1.5当量のFmocGlnOH、1.0、1.2及び1.5当量のFmocTrp(Boc)OH、並びに0.8、1.0及び1.5当量のFmocLeuOHを使用する。2-CTC樹脂1キログラムに1モルのFmocLeuOHをローディングできない場合には、ベンダーから2-CTCを受け取るべきではない。以下、FmocLeuOH、FmocGlnOH及びFmocTrp(Boc)OHに対する標的樹脂について記述する。
【0144】
2-CTC樹脂には各キログラムあたり1.0〜1.1モルのFmocLeuOHをローディングすることができる。HClの損失を考慮すると、FmocLeuOHローディング後の樹脂乾燥重量は、原料樹脂重量の1.32〜1.35倍でなければならず、実測ローディングは1グラムあたり0.75〜0.81ミリモルでなければならない。
【0145】
2-CTC樹脂には各キログラムあたり0.8〜1.0モルのFmocGlnOHをローディングすることができる。HCLの損失を考慮すると、FmocGlnOHローディング後の樹脂乾燥重量は、原料樹脂重量の1.27〜1.33倍でなければならず、実測ローディングは1グラムあたり0.63〜0.75ミリモルでなければならない。
【0146】
2-CTC樹脂には各キログラムあたり0.9〜1.1モルのFmocTrp(Boc)OHをローディングすることができる。HCLの損失を考慮すると、FmocTrp(Boc)OHローディング後の樹脂乾燥重量は、原料樹脂重量の1.44〜1.54倍でなければならず、実測ローディングは1グラムあたり0.63〜0.72ミリモルでなければならない。
【0147】
重量増加を正確に解析するには、出発原料となる樹脂の乾燥損失(LOD)を測定しなければならない。LODは1%を超えてはならない。(ローディング後の)乾燥中、樹脂から完全に残留溶媒(すなわちDIEA/HCl)を除去しないと、単離される質量が多くなり、実測ローディングは低くなる。しかしながら、ローディングされる全モル数(質量×実測ローディング)は上記範囲内に収まるようにするべきである。上記アミノ酸のいずれに対してもローディング量(すなわち出発樹脂1キログラムあたりにローディングされる総モル数)の上限を10%以上超える場合には、より少ないアミノ酸を用いて使用テストをする。FmocGlnOHの場合、これは特に重要である。
【0148】
11.1 Fmoc-AA(Boc)-2-クロロトリチル樹脂の大規模製造
以下に続くセクションの目的は、固相ペプチド合成(solid phase peptide synthesis: SPPS)用のペプチド類を大量に製造するのに適した樹脂の大規模ローディングを行うことにある。以下のFmocTrp(Boc)OH、FmocGln(Boc)OH及びFmocLeu(Boc)OHそれぞれに樹脂を添加する。2-CTC樹脂約4キログラムを各出発残基に添加した。
【0149】
ローディングされた 2-CTC ローディング 置換 ローディング
AA (Kg) 後の質量(Kg) (mmol/g)1 総モル数1
FmocTrp(Boc)OH 3.7 4.493 0.56 2.52
(0.56) (2.52)
FmocLeu(Boc)OH 2.2 2.436 1.07 2.6
(0.647) (1.58)
FmocGln(Boc)OH 3.65 3.856 0.55 2.12
(0.52) (2.0)
1. 最初の数はFmoc-AA標準に対する重量ベースHPLCアッセイにより計算した。カッコ内の数値は出発物質2-CTC樹脂のLODが12%であることを考慮して、重量増加により算出した。
【0150】
出発の2-CTC樹脂はコロラド・バイオテク社(Colorado BioTech)から入手した。ローディングは1.4 meq/gと公表されている。1.5当量のFmocTrp(Boc)OH、DCM (5 vol)中1.7当量のDIEA、周囲温度で2時間という標準的なローディング手法を用い、当初の2-CTC量を3.7キログラムとした。こうしてFmocTrp(Boc)O-樹脂4.493キログラムを得たが、これは、樹脂開裂後のFmocTrp(Boc)OHの重量ベースHPLCアッセイによる0.56 mmol/gでの理論ローディングをもっていた(合計2.52モル)。製造した合計1.62モルについて、ローディングは樹脂の重量増加により1グラムあたり0.36ミリモルになると算出されている。しかしながら、合計2.52モルは樹脂4キログラムあたり4モルのFmocTrp(Boc)OHという目的のローディングからはほど遠いが、出発物質である2-CTC樹脂のLODが12%であることを考慮した場合、重量増加により算出されたローディングは0.56 mmole/gでは同じである。
【0151】
標準的なローディング法を用いた2実験で、2-CTC 3.9キログラムから合計4.59キログラムのFmocLeuO-樹脂を製造した。
【0152】
しかしながら、予想したよりも実質的に低いローディングが得られた。樹脂開裂後のFmocLeuOHを重量ベースHPLCによりアッセイしたところ、最初のバッチは1グラムあたり1.07ミリモルの理論ローディングを示した。出発物質2-CTC樹脂のLODが12%であることを考慮すると、重量増加による理論ローディングは1グラムあたり0.647ミリモル(500グラム)であるから、これは明らかに不可能である。ローディングされた合計1.577モルに対して実際のローディングは1グラムあたり0.647ミリモルに近いようである。
【0153】
2-CTC 1.7キログラムから製造した第2バッチのFmocLeuO-樹脂(2.154キログラム)は、0.68ミリモル/グラムの理論ローディングを有していたのに対し、重量増加ローディングは0.66ミリモル/グラムであった。
【0154】
0.8当量のFmocGlnOH及び7容量の5/2 DMF/DCM中1当量のDIEAを用い、3.65キログラムの2-CTCを出発物質として、シングルバッチ法によりFmocGlnO-樹脂を合計3.856キログラム作った。合計2.12モルのFmocGlnO-樹脂に対し、重量ベースHPLCアッセイによる理論置換量は1グラムあたり0.55ミリモルであった。出発2-CTC樹脂のLODが12%であることを考慮すると、合計2.0モルのFmocGlnO-樹脂に対し、重量増加による理論ローディングは、1グラムあたり0.52ミリモルである。
【0155】
一般に、重量増加によるローディングは、ローディングしたAAもしくは除去したフルベンについては、重量ベースHPLCアッセイによって算出されたローディングよりも大きいかまたは同等でなければならない。出発の2-CTCはそのLODが1%より小さくなければならず、単離されたFmocAAO樹脂は、少量のNMP、塩及び/または残留FmocAAOHを含む可能性がある。
【0156】
11.1.1 Fmoc-AA(Boc)-2-クロロトリチル樹脂の好ましい大規模製造法
FmocTrp(Boc)OH及びFmocLeuOH
空気感受性2-クロロトリチルクロリド樹脂(1当量、3.7キログラム、5.18モル)をSPPSチェンバーに入れ、DCM5容量で洗浄する。溶媒を除去し、FmocTrp(Boc)OHもしくはFmocLeu(Boc)OH(1.5当量)溶液及びDCM (5容量)中DIEA(1.7当量)を加える(注1)。このスラリーを2時間撹拌しながらかき混ぜる。溶媒を除去した後、樹脂に残留する活性部位を30分間9:1 MeOH:DIEA (5容量)でエンドキャッピングする。溶媒を除去した後、樹脂を5容量のDCMで6回洗浄する。恒量になるまで樹脂を乾燥した後、これをサンプリングし、ローディングについて分析する(注2)。同じ方法を用いてFmocLeuOHをローディングする。
【0157】
FmocGluOH
窒素雰囲気下、空気感受性2-CTC(3.65キログラム、1.4ミリモル/グラム)樹脂を40LのSPPSチェンバーに入れ、DCM(5容量)で洗浄する。20Lの反応容器にFmocGlnOH(1.506キログラム、0.8当量)、DMF (5容量)及びDIEA(1.0当量)を入れる。SPPSチェンバーをドレーンし、DMF溶液を添加する。DCM(2容量)で反応容器を洗浄し、洗液はSPPSチェンバーに加える。窒素雰囲気下2時間SPPSチェンバーで樹脂を撹拌した後、ドレーンを行う。樹脂に残留する活性部位はすべて30分間9:1 MeOH:DIEA (5容量)でキャッピングする。樹脂をドレーンした後、DCM(5容量、6回)で洗浄する。恒量になるまで樹脂を乾燥した後(3.856kg)、これをサンプリングし、ローディングレベルについて試験する(注2)。
【0158】
注:
1. 2-クロロトリチルクロリド樹脂は湿度には非常に感受性が高い。水は樹脂の表面を覆うとHClを与える。溶媒が乾燥している限り、DCEとDCMとの間にはほとんど差がない。
【0159】
2. 樹脂ローディングは樹脂からFmocアミノ酸を開裂し、定量wt/wt HPLC分析により標準に対してアッセイすることにより決定する。
【0160】
フェノメネックス・ジュピターC18、300A、Sm
1 mL/min、260 nm
A: 0.1% 水性TFA
B: アセトニトリル中0.1% TFA
65%B、アイソクラチック(isocratic)
保持時間: 約8分
樹脂乾燥前の洗浄回数が不十分で樹脂からNMPを完全に除去できない場合には、樹脂の重量増加によるローディング量の測定は不正確になることがある。計算は以下の通りである。
【0161】
(Fmoc-AA-OH樹脂質量−出発2-CTC質量)/Fmoc-AA-OH樹脂質量(ローディングされたAAの分子量−HClの分子量)
DCM中長期間(日)本発明のペプチド断片を保存すると、樹脂から実質的な開裂が起こることが観察された。例えば、FmocAA17-26OHを構築する際、部分的に構築された断片をDCM中で2度週末にかけて保存した。合成を終了して樹脂から取り出した後、8%の収率でFmocAA17-26OHが得られた。週末にかけての保存中、DCMに存在する微量のHClが、部分的に構築された断片を樹脂から徐々に開裂したのではないかと考えられる。
【0162】
FmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂のサンプルを室温で1週間DCM及びIPA中に放置した。各樹脂の上清をHPLCにより分析した。定量的な結果は得られなかったが、DCM中ではすべての断片が有意な程度まで樹脂から開裂したのに対し、IPA中では開裂は観察されなかった。部分的に構築された断片を2〜3日(週末にかけて)保存する必要がある場合には、固形物をNMPで洗浄し、ベッドをドレーンして窒素雰囲気下NMPで飽和させておいた方がよい。
【0163】
FmocLeuO-樹脂、FmocGlnO-樹脂、FmocTrp(Boc)O-樹脂、FmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂を最終洗浄するため、DCMをIPAで置換した。IPAで飽和したFmocLeuO-樹脂、FmocGlnO-樹脂及びFmocTrp(Boc)O-樹脂を40℃で乾燥した。4℃で乾燥したそれらローディング樹脂からFmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂を構築した。合成終了時に、樹脂結合断片をきれいになるまでDOMで、次にIPAで洗った。IPAで飽和した樹脂結合断片を分けて周囲温度及び40℃で乾燥した。断片を樹脂から開裂し、HPLCにより分析した。断片の純度に差は観察されなかった。IPAで飽和した樹脂結合断片を40℃で乾燥した場合、乾燥は単離された断片の性質や量には影響しない。
【0164】
NMP/DCMに対しDMF中で断片のSPPSを調べた(樹脂1グラムは両溶媒系で5 mLに膨潤する)。溶媒としてDMFを用い、カップリング試薬としてTBTU対HBTUを使用してFmocAA27-35OH断片を合成した。収率77%、純度89.5A%でFmocAA27-35OHを単離した。
【0165】
FmocAA17-36NH2断片及びAcAA1-36NH2断片の仕上げ(work up)手順によりカップリングしなかった断片を除去する。固相合成を行う間、これらの断片を欠失部位でアセチル化(エンドキャッピング)すると、欠失断片は確実に溶液相カップリングでカップリングしなくなる。FmocAA17-36NH2及びAcAA1-36NH2両断片の合成中カップリングしなかった断片はIPA及びACN仕上げの際に除去しなければならない。
【0166】
11.2 AcAA(1-16)-OH断片(3c断片)の大規模製造
SPPSによるAcAA1-16-OH断片の大規模製造を2回行って、FmocGlnOH-樹脂3.53キログラムから合計7.941キログラムの樹脂結合断片を得た。2回とも出発物質であるFmocGlnOH-樹脂のローディング値は、重量ベースHPLCアッセイにより得られた若干過大な0.55ミリモル/グラムという値であり、むしろ出発物質2-CTCの12%LODを考慮した重量増加により得られる0.52ミリモル/グラムのほうがより正確な値である。望ましい値より低い樹脂ローディングを持つほか、アミノ酸はSPPSの間それぞれ6%過剰量を使用した。
【0167】
SPPSでは樹脂に第1のFmocGln(trt)OHを2.5当量使用し、断片において次に続くアミノ酸についてはそれぞれ1.7当量を使用した(0.55ミリモル/グラムローディングに対し)。8容量の3:1 NMP:DCM中でカップリング反応を行ったところ、32カップリング反応(アセチル化を含む)の内2例を除いて残り全部の反応は2時間以内に完了した。洗浄はすべて5容量のNMPで行った。
【0168】
3.4容量(乾燥AcAA1-16O-樹脂の重量に対し)の1% TFA/DCM中0〜5℃で、1洗浄あたり5分間ずつ5、6回、樹脂結合断片を洗浄する。各洗液を1.36容量の0.33 M NaHCO3水溶液に集めてTFAを中和する。3相洗液を合わせて水(2容量)を加える。DCMを溜去すると重炭酸ナトリウム水溶液中に濾過可能な沈澱が残る。蒸留する間に多相スラリーは粘稠となってクリーム状を呈した後、DCMが最後まで除去されると濾過可能なスラリーになる。このスラリーを0〜5℃まで冷却し、pHを0.1 N HClで3.0に調整する。スラリーを0〜5℃で1〜2時間撹拌し、濾過して集め、洗浄して乾燥する。続く実験で固形物を集め、反応容器に戻し、水で1〜2時間トリチュレーションした後、集めて乾燥する。
【0169】
DCM中1% TFAを用い、4実験で樹脂からのAcAA1-16OH開裂を行った。3.53キログラムのFmocGlnO-樹脂から合計4.814キログラムのAcAA1-16OHが93〜96A%の純度で得られた。出発物質である2-CTC(理論量5.83キログラム)のLODが12%であることを考慮して重量増加により決定した0.52ミリモル/グラムのローディングにもとづく総収率は83%である。
【0170】
AcAA1-16OHのナトリウム塩はDMFにもNMPにも不溶であるので、カルボキシル末端をプロトン化するにはpH調整が必要である。水洗により生成物から確実にトリフルオロ酢酸ナトリウムを除去する。wt/wtベースでごく少量のトリフルオロ酢酸ナトリウムが存在すると、HAA17-36NH2との溶液相カップリングは妨害される。
【0171】
この断片は周囲温度で乾燥しなければならない。湿ったAcAA1-16OHを40℃で乾燥する安定性検査を行ったところ、3日間で約4%の分解を示した。興味深いことに、乾燥した固形物は80℃で24時間安定である。
【0172】
11.2.1 Ac-AA(1-16)-OH断片(3c断片)の好ましい大規模製造法
40 Lのペプチドチェンバーに樹脂結合FmocGlnOH(1.53キログラム、0.55ミリモル/グラム、0.84モル)を加える。15分間撹拌しながら、窒素下でDCM(5容量)により樹脂のコンディショニングを行った後、ドレーンする。20%ピペリジンNMP溶液(5容量)を加え、得られた懸濁液を窒素下で20分間撹拌する。溶液をドレーンし、このプロセスを繰り返す。5容量のNMPで5回樹脂を洗浄してジベンゾフルベンを除去し、クロロアニル試験により判定されるピペリジンを除去する(注1)。
【0173】
樹脂を洗浄してピペリジンを除去しながら、配列中の次のアミノ酸(1.5当量)、HOBT (1.5当量)及びDIEA(1.5当量)をNMP(6〜7容量)中で混合して0℃に冷却する。冷却した溶液にHBTU (1.5当量)を加え、溶液を10〜15分間撹拌してHBTUを溶解する。活性アミノ酸の冷却溶液を樹脂に加えた後、DCMで洗浄する(2.5容量)(注2)。窒素パージ下で2時間、懸濁液を撹拌する。その後、定性ニンヒドリン試験用に樹脂サンプルを取り出す(注3)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄する(注4)。そして配列中の次のアミノ酸についてこのサイクルを繰り返す。ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、アミノ酸1当量と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性になれば、NMP(10容量)中5当量(Seq)の無水酢酸と5当量(Seq)のピリジンで1時間エンドキャッピングを行う。
【0174】
断片合成終了後、記載したように最後のアミノ酸からFmocを除去した後、3:1 NMP:DCM(10容量)における無水酢酸とピリジン(それぞれ5当量)の溶液中で、20〜30分間、もしくは、ニンヒドリン試験が陰性になるまで、樹脂を撹拌する。樹脂をドレーンした後、5容量のNMPで2回、5容量のDCMで5回洗浄して乾燥すると、3.49キログラムのAcAA1-16O-樹脂が得られる。
【0175】
40 LのSPPSチェンバーに乾燥した樹脂結合AcAA1-16(3.49キログラム)を入れる。6 x 3.4容量の1% TFA/DCM を用いて、樹脂結合ペプチドをその樹脂から開裂する(注7)。各開裂洗液を1.36容量の0.33 M 重炭酸ナトリウム水溶液に集める。2相フラクションを合わせて2容量の水で希釈する。減圧下(15Hg、20℃)2相混合物を濃縮してDCMを除去する。撹拌を続けるのに必要であれば蒸留中さらに水(2容量)を加える(注6)。DCMを除去する場合(数時間)には、懸濁液を0℃に冷却し、1N HCl水溶液でpHを3に調整する。スラリーは0℃で1時間撹拌した後集める。なお湿っている固形物は反応容器に戻し、水(7容量)でトリチュレーションして残っているTFAとトリフルオロ酢酸ナトリウムを除去する。減圧濾過により固形物を集めて恒量になるまで乾燥する(1.12キログラム、95A%)。0.52ミリモル/グラムローディングに基づくAcAA1-16OHの収率は86%である(注7)。
【0176】
注:
1. アセトン約1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。ベッド高が高くなればなるほど、ピペリジンを洗い流すのにはさらに大量のNMPが必要になる。
【0177】
2. 樹脂の適当な膨潤を確実にするため、DCMを添加する。
【0178】
3. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出し、メタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。定量ニンヒドリン試験方法の文献: Sarin, V.K., Kent, S.B.H., Tam, J.P., & Merrifield, R.B. (1981) Analytical Biochem. iii, 147-157。
【0179】
4. 樹脂を1昼夜保存する場合には、5容量のNMPで2回洗浄した後、ドレーンして窒素下に保存のこと。DCM下に保存してはならない。この場合樹脂からペプチドが開裂して実質的な量損失を生じる。
【0180】
5. 各フラクションについては、254 nmでTLC可視化により生成物の内容を調べる。生成物の大部分は最初の5回洗浄により除去される。
【0181】
6. DCMを除去するにつれ、反応物はマシュマロクリームの固さを呈するようになる。蒸留を続けると、懸濁液は徐々に固いスラリーへと変わっていく。生成物からオイルが出るのを防ぐためDCMを徐々に除去することが重要である。
7. バイダク(Vydac) C8、5μ、300A
1 mL/min、30℃、230 nm
A 1000:1 水/TFA
B 800:200:1 IPA:ACN:TFA
60-95%B/30 min
保持時間: 13.1分
【0182】
11.3 FmocAA(17-26)-OH断片(10b断片)の大規模製造
同等サイズの2実験でFmocLeuO樹脂2.4キログラムからFmocAA17-26O-樹脂を合計5.3キログラム作った。同等サイズの4実験で、この樹脂からFmocLeuO-樹脂5.3キログラムを開裂したところ、平均して純度94.4A%、収率90%のFmocAA17-26OHが3.184キログラム得られた。
【0183】
ローディングファクターに対しアミノ酸をそれぞれ1.5当量用いて反応を行ったが、各カップリングサイクルについて65%過剰のアミノ酸を使用する結果となった。カップリング反応はすべて2時間以内に完了した(ニンヒドリン試験陰性)。
【0184】
20%ピペリジンNMP(5容量)に20分間保ってFmoc保護基を外し、これを繰り返した。5容量のNMPで5回洗ってピペリジンを樹脂から洗い流した。カップリング反応は8容量の3:1 NMP:DCM中で行った。カップリング溶液を除去し、固形物を5容量のNMPで3回洗浄した。配列の次のアミノ酸について、このサイクルを繰り返した。断片完了後、樹脂ベッドを5容量のNMPで2回、5容量のDCMで5回洗った後、恒量になるまで乾燥した。
【0185】
樹脂から断片を除去するため、DCM中1% TFAで数回洗浄する。1% TFAのDCM溶液は冷却する必要はない。この断片は1% TFA/DCM(1%/日)中ではAcAA1-16OHよりもはるかに安定だからである。生成物含有酸性DCM洗液を集め、ピリジンで中和する。合わせた洗液を蒸留してDCMを除去し、エタノールを添加して溶液を維持すると共に、蒸留する間に残留DCMを追い出す。DCMを除去した後(蒸留物が除去される温度の上昇によって判断する)、水を加えて断片を沈澱させる。減圧濾過により固形物を集める(60分以内)。なお湿っている固形物は反応容器に戻し、80/20のエタノール/水(5容量)で60分間0〜5℃でトリチュレーションする。減圧濾過により沈澱したFmocAA17-26OHを集め、最小量の80/20エタノール/水で洗浄して乾燥する(減圧、加熱なし、10日間)。
【0186】
11.3.1 FmocAA(17-26)-OH断片(10b断片)の好ましい大規模製造法
40 LのSPPSチェンバーにFmocLeuO-樹脂(1.2キログラム、0.776モル)を入れた後、DCM(5容量)で樹脂を膨潤させる。DCMは乾燥した原料樹脂を確実に完全膨潤させるのに必要である。懸濁液を20〜30分間撹拌した後、液体を除去する。20%ピペリジンNMP溶液を加え(5容量)、溶液を20分間撹拌する。溶媒を除去した後、このプロセスを繰り返す。溶媒を除去し、樹脂ベッドを5容量のNMPで5回洗浄してピペリジンを除去する(注1)。
【0187】
脱保護しながら、配列中の次のアミノ酸(1.95当量)、HOBT (1.95当量)、DIEA(1.95当量)及びNMP(6容量)を機械的撹拌機を備えた20リットルの丸底フラスコに入れる。固形物が溶解するまで溶液を撹拌し、次いで、0〜5℃に冷却して、HBTU (1.95当量)を加える。HBTUが溶解するまで溶液を撹拌するか、またはピペリジンを含まない場合には樹脂を撹拌(どちらが最初でもよい)した後、樹脂に加える。反応容器をDCM(2容量)で洗浄し、これをSPPS反応容器へ移す。注: アミノ酸と試薬の化学量論は1.5当量でなければならない。
【0188】
穏やかに撹拌しながら、樹脂をカップリング溶液に懸濁する。2時間後、樹脂試料をSPPSチェンバーから取り出して定性ニンヒドリン試験を行う(注2)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄する。そして配列中の次のアミノ酸についてこのサイクルを繰り返す(DCMによる膨潤はローディングされた最初のアミノ酸についてだけ行う)。
【0189】
ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、1当量のアミノ酸と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性の場合、NMP(10容量)中の無水酢酸5当量とピリジン5当量で1時間エンドキャッピングを行う。
【0190】
断片合成終了後、樹脂をドレーンし、5容量のNMPで2回、5容量のDCMで5回洗浄した後乾燥すると、2.67キログラムのFmocAA17-26O-樹脂が得られる。
【0191】
1.7容量のDCM中1% TFAを、1回5分間で5〜6回用いて、樹脂(1.33キログラム)からFmocAA17-26OHを開裂する。1% TFA/DCM洗液をピリジン含有フラスコに集める(洗液中のTFAについては容量比1:1)。生成物含有洗液を合わせ(約14リットル)、最小ポット容量になるまでDCMを溜去する(元の容量の約3分の1)。減圧度を調整してポット温度を15〜25℃に保つ。エタノール(6.5容量)を加え、DCMがなくなるまで蒸留を続ける(蒸留物の温度によって判断する;注3)。再び減圧度を調整してポット温度を15〜20℃に保つ。最終ポット容量は約8〜10容量でなければならない。溶液を5〜10℃に冷却し、30分間にわたって水(6.5容量)を加え、FmocAA17-26OHを沈澱させる。減圧濾過により固形物を集めて水洗する(2〜3容量)。残存するピリジン及び/又は塩を除去するため、なお湿っている固形物は反応容器に戻し、予め0℃まで冷却した80/20エタノール/水(5容量)を加える。懸濁液を60分間0℃で撹拌する。減圧濾過により固形物を集め、最小量の80/20エタノール/水で洗浄し、恒量になるまで乾燥すると、収率90.6%、純度96A%のFmocAA17-26OHが0.806キログラム得られる(注4)。
【0192】
注:
1. アセトン1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。
【0193】
2. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出しメタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。
【0194】
3. DCMの減圧蒸留中の頭部温度は10〜15℃であった。DCMを除去すると、頭部温度は3500まで上昇した。
4. バイダクC8、5μ、300A
1 mL/min、262 nm、30℃
A 水/0.1% TFA
B ACN/0.1% TFA
80-99%B/20 min
保持時間: 15.2分
【0195】
11.4 FmocAA(27-35)-OH断片(16b断片)の大規模製造
FmocTrp(Boc)O-樹脂4.45キログラムから合計4.694キログラムのFmocAA27-35-OHを合成した。固相合成は2個のバッチで行い、樹脂からの開裂は4バッチであった。1バッチから得られたFmocAA27-35-OHは他のバッチから得られた物質よりも約5%純度が低かった。これはFmocAA17-36-NH2へ処理する際に除去された、同定されていない不純物5A%によるものであった。樹脂にローディングされたFmocTrp(Boc)OHの合計量は2.5モルであった。約63%の容量でローディングされた樹脂について予想される通りに、固相合成が進行した。カップリングはすべて最初の2時間のチェックポイントで完了した。SPPS及び開裂に使用した反応及び洗浄量はFmocAA17-26OH合成に使用した量と同じであった。樹脂からの開裂は上記の通りであった。濾過は迅速であった(15分)。90/10エタノール/水トリチュレーション後の固形物乾燥には3日間かかった(減圧、加熱なし)。断片は40℃の乾燥にも安定である。
【0196】
11.4.1 Ac-AA(27-35)-OH断片(16b断片)の好ましい大規模製造法
FmocTrp(Boc)-樹脂(1当量、2.2キログラム、1.23モル)を含有するSPPSチェンバーにDCM(5容量)を加える。得られた懸濁液を15分間撹拌した後ドレーンする。20%ピペリジンのNMP溶液(5容量)を加え、得られた懸濁液を10〜15分間撹拌してFmoc保護基を外す。このプロセスを繰り返した後、クロロアニル試験が陰性を示すまで、樹脂をNMP(5容量)で5〜7回洗浄する(注1)。
【0197】
NMP(6容量)中で、次に続くアミノ酸(1.5当量)、HOBT (1.5当量)及びDIEA(1.7当量)を合わせ、0〜5℃に冷却する(注2)。HBTU を加え、溶液を10〜15分間撹拌してHBTUを溶解する。活性アミノ酸の溶液を樹脂に加える。DCMで反応容器を洗浄した後樹脂に加える(注3)。窒素雰囲気下1〜2時間この懸濁液を撹拌する。カップリングが完了したかどうかは定性ニンヒドリン試験によりモニターする(注4)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄し、配列中の次のアミノ酸についてこのサイクルを繰り返す(ローディングされた最初のアミノ酸についてのみDCMによる膨潤を行う)。
【0198】
ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、1当量のアミノ酸と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性になれば、NMP(10容量)中で1時間無水酢酸5当量とピリジン5当量のエンドキャッピングを行う。
【0199】
断片合成終了後、樹脂をドレーンし、5容量のNMPで2回、5容量のDCMで5回洗浄して乾燥すると、4.11キログラムのFmocAA27-35O-樹脂が得られる。
【0200】
樹脂から断片を開裂するには、1.7容量のDCM中1% TFAを用い、1回5分間で6回、樹脂(2.05キログラム)から樹脂を開裂する。1% TFA/DCM洗液をピリジン(洗液中のTFAに対して容量比1:1)含有フラスコに集める。生成物含有洗液を合わせ、元のポット容量の約半量になるまでDCMを溜去する(ポット温度は減圧度を調整して約15℃に保つ)。徐々にエタノール(5容量)を加え、DCMがなくなるまで(蒸留物の温度によって判断する。注5)蒸留(減圧度を調整してポット温度を約20℃に保つ)を続ける。ポット容量は約6〜7容量でなければならない。濁った溶液を10〜15℃に冷却し、素早く撹拌しながら30分間にわたって水(3.5容量)を加えると、FmocAA27-35-OHが沈澱する。減圧濾過(15分間)により固形物を集めて水(1容量)で洗浄する。残留ピリジンを除去するため、湿っている固形物を反応容器に戻し、予め0〜5℃に冷却した90/10エタノール/水(10容量)を加える。スラリーを0〜5℃で60分間撹拌する。減圧濾過により固形物を集め、90/10エタノール/水(0.5容量)で洗浄し、恒量になるまで乾燥すると、収率89%、純度89.2A%のFmocAA27-35-OHが1.19キログラム得られた(注6)。
【0201】
このプロトコールは、12時間撹拌しながら固形物を9:1エタノール/水15容量でトリチュレーションした後、集めて乾燥することにより再処理することができる。
【0202】
注:
1. アセトン1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。
【0203】
2. 溶解を補助するためHBTUを除いた試薬を室温で添加する。HBTUを冷却溶液に加えるとラセミ化が低減する。
【0204】
3. HBTUはDCMに溶けないため、活性化はNMP中で行う。DCMは反応容器を洗浄するのに使用されると同時に、適当な樹脂膨潤を維持するため樹脂に添加される。
【0205】
4. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出し、メタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。
【0206】
5. DCMを減圧蒸留する間の頭部温度は10〜15℃に保った。DCMが除去されると頭部温度は35℃まで上昇した。
6. バイダクC8、5μ、300A
1 mL/min、262 nm、30℃
A 水/0.1% TFA
B ACN/0.1% TFA
80-99%B/20 min
保持時間: 15.3分
【0207】
11.5 FmocAA(27-35)-OHとHPheNH2の固相カップリングによるFmocAA(27-36)-OH断片の大規模製造
4.676キログラムのFmocAA27-35-OHから4実験で合計5.226キログラムのFmocAA27-36NH2を作った。残留するカップリング試薬もしくは溶媒は収率100%以上になる。これらは次の段階で除去される。
【0208】
水のドロップアウト後反応容器壁に付着している固形物はこの段階で重要な問題となった。減圧濾過により粗固形物を分離するのに30分を要した。これら実験の平均乾燥時間(減圧、加熱なし)は8日間であった。固形物はオーブンへ入れる前に濾紙上で圧縮して過剰の水を除去する必要がある。この断片は40℃の乾燥に安定であり、減圧オーブンは加熱する必要がある。
【0209】
使用テストは、使用する出発物質1ロットあたりのバッチ0.5〜1グラムについて行い、性質及び同一性を確定する助けとする。薄層クロマトグラフィ(TCL)及びHPLCを用いて出発物質から生成物への変換をモニターする。FmocAA27-35OH断片において探さなければならない最初の不純物はトリフルオロ酢酸(またはその塩)である。FmocAA27-35OHに存在するトリフルオロ酢酸のフェニルアラニンアミドによる活性化及び反応は迅速なプロセスである。存在するトリフルオロ酢酸を消費するのに小過剰のフェニルアラニンアミドを使用することができる。さらに重要な性質の問題は購入したフェニルアラニンアミドについてである。ほとんどのベンダーはこれを塩酸塩として販売する。フェニルアラニンアミド塩酸塩のロットのうちいくつかでは、HPLCの際出発物質と一緒に共溶出する不純物(5〜15A%)が生成している。この不純物はTLCによって出発原料断片及び生成物から分離することができる。フェニルアラニンアミド塩酸塩に存在する塩化アンモニウムから生じるFmocAA27-35NH2は不純物である。
【0210】
11.5.1 FmocAA(27-35)-OHとHPheNH2の液相カップリングによるFmocAA(27-36)-OH断片の大規模製造
機械的攪拌機を備えた40リットルのジャケット反応容器にFmocAA27-35OH(1当量、1.185キログラム)、HOAT (1.1当量)、HCl.HPheNH2(1.15当量)及びDMF(12.5容量)を入れる。DIEA (2.1当量)を加え、溶液を0〜5℃に冷却してHBTU(1.2当量)を添加する。反応混合物を0〜5℃で15分間撹拌した後、室温まで加温し、さらに70分間撹拌する(注1、プロセスチェックのためHPLCを用いた)。HPLCにより反応が完了したと判断できた後、水(12.5容量)を15〜30分かけて加えるとペプチドが沈澱する。スラリーを周囲温度で15分間撹拌する。減圧濾過によって固形物を集め、水(3容量)で洗浄後乾燥すると、FmocAA27-36NH2が得られる(収率107%で1.357キログラム、HPLCによる純度87.1A%)(注2)。
【0211】
12時間撹拌しながら固形物を9:1アセトニトリル/水15容量でトリチュレーションした後集めて乾燥することにより、再度反応を行うこともできる。
【0212】
注:
1. プロセス制御において、TLC:
88/12ジクロロメタン/メタノール
UV、ヨード検出
Rf: FmocAA27-35OH、0.49
Rf: FmocAA27-36NH2、0.63
バイダクC8、5 m、300A
30℃、1 mL/min、262 nm
A 水/0.1% TFA
B ACN/0.15 TFA
80-99%B/20 分
保持時間: FmocAA27-35OH、15.8
保持時間: FmocAA27-36NH2、17.12
2. 一般に、単離された固形物を反応容器に戻して水でトリチュレーションをしない限り、得られる収率は理論収率より大きい。
【0213】
11.6 FmocAA(27-36)OHからのHAA(27-36)-OH断片の大規模製造
5.221キログラムのFmocAA27-36NH2から平均2段階収率86.4%で、4実験で合計3.897キログラムのHAA27-36NH2を製造した。これらの実験で観察された収率の範囲、74〜97%は1実験から次の実験への持ち込みを反映していると考えられる。
【0214】
この段階では製造も生成物単離も非常にうまくいく。正確な量のDCMがMTBEに残っており、固形物の物理的な性質が迅速な濾過・乾燥をもたらす。この生成物単離法は、以下のセクション11.6.2に記載の新段階に組み込まれている。
【0215】
DCMは、FmocAA27-36NH2の脱保護の際に使用する溶媒である。脱保護の完了時に、有機溶液を水で2回洗浄することにより、過剰ピペリジンのほとんどを除去し、次に、元の容量の約1/3に濃縮する。MTBEを添加して、フラグメントを沈殿させる。蒸留を続けることにより、残ったDCMの大部分を除去し、フラグメントの沈殿を完了する。沈殿中の質量損失を防止する上で、DCMのほとんどを除去することが重要である。また、生成物の清浄化を確実にするために、MTBE中にDCMをいくらか残すことも重要である。ジベンゾフルベン、ピペリジン/フルベン付加物および残留ピペリジンは、MTBEに可溶性である。HAA27-36NH2を回収し、乾燥させる。必要であれば、MTBEを用いた該固体の2次粉砕(12時間)により、残留ジベンゾフルベン、およびさらに重要なことにはHAA27-36NH2に存在する可能性のあるピペリジン/フルベン付加物を除去する。ヘキサンを用いた粉砕では、ジベンゾフルベンは除去されるが、ピペリジン/フルベン付加物は除去されない。MTBEから単離した固体の乾燥時間は、1日である。
【0216】
HAA27-36NH2およびDCM/MTBE中の関連不純物の高い可溶性のため、分析方法には、溶媒交換の終点を正確に決定することが求められた。MTBE中のDCMを定量するために、GC法が開発されている。蒸留は、MTBE中のDCM含有率が、6%を下回る時点で、完了する。
【0217】
11.6.1 FmocAA(27-36)-OHからのフラグメントHAA(27-36)-OHの大規模製造
撹拌機と温度計を備えた40Lのガラスジャケット付き反応器に、FmocAA27-36NH2(1当量、1.356kg)、DCM(5容)およびピペリジン(0.2容)を導入する。該溶液を周囲温度で1.5時間撹拌する(注1)。有機溶液を水(2x5容)で洗浄する。蒸留(約25mmHg、尚、固体の融解を防ぐため、ジャケット温度は45℃未満)により、上記有機層の容量を元の有機容量の約1/3に減少させる。MTBE(5容)を反応器に徐々に導入しながら、重沈殿点まで濃縮を続け、ポット容量を約5容とする。DCM含有率が6%を下回った時点で、溶媒交換を停止する。該スラリーを0〜5℃に冷却し、60分間撹拌した後、減圧濾過(急速)により回収する。固体をMTBE(2x0.5容)で洗浄し、恒量まで乾燥させて、1.022kgのHAA27-36NH2を得た。収率は89.2%(2ステップ)で、純度は、91.1A%であった(注1)。
【0218】
MTBE(12.5容)を用いた、23℃、13時間の粉砕により該反応を再実施し、減圧濾過および乾燥により、回収してもよい。
【0219】
注:
1.HPLCにより測定するIPCおよび純度:
Vydac、C8、5、300A
1mL/分、262nm、30℃
A 水/0.1%TFA
B ACN/0.1%TFA
80〜99%B/20分
保持時間:FmocAA27-36NH2、16.5, HAA27-36NH2、8.4
11.6.2 FmocAA(27-36)-OHとHPheNH2の液相カップリングによる、フラグメントHAA(27-36)-OHの改良された製造
FmocAA27-35OHから直接HAA27-36NH2を得る第11.5節から第11.6.1節を組み合わせた新しい方法が開発されている。この新しい方法は、FmocAA27-36NH2の単離に付随する問題と長い乾燥時間を解消する。以下に、詳しく説明する。この新しい方法は、合成工程から、長い乾燥時間と、一段階を排除する。
【0220】
磁気撹拌器および窒素供給口を備えた100mLの丸底フラスコに、FmocAA27-35OH(5.0g、1当量)、HOAT(0.459、1.2当量)およびPhe-NH2(0.389、1.2当量)を導入する。該フラスコに、10容のNMP(50mL)とDIEA(0.45g、1.5当量)を添加し、窒素雰囲気下で、固体が溶解するまで撹拌する。該溶液を0〜5℃に冷却した後、HBTU(1.04g、1.2当量)を添加する。窒素雰囲気下で、0〜5℃にて30分間撹拌する。反応混合物を20℃まで暖め、撹拌を継続する。HBTUを添加してから90分後に、プロセス中検査(IPC)を実施する。
【0221】
プロセス中検査(IPC)により、反応の完了が明らかにされた後、反応混合物にピペリジン(1.379、7当量)を添加し、窒素雰囲気下で1.5時間撹拌する。IPCを実施する(注1)。Fmoc除去が完全ではない場合には、さらに30分間撹拌し、IPCを実施する。
【0222】
完全であれば、この反応混合物を、温度が35℃を超えないような速度で、5%酢酸水溶液に添加する(30容)。得られたスラリーを1〜2時間撹拌し、減圧濾過により回収する(注2)。その固体を10容(50mL)の水で洗浄する。
【0223】
湿った固体を反応器に戻し、20容の水(100mL)を添加し、周囲温度で1〜2時間撹拌する。減圧濾過により固体を回収し、10容の水(50mL)で洗浄した後、乾燥する(注3)。
【0224】
乾燥した(注4)固体を、MTBE(20容)を用いて、周囲温度で2〜5時間粉砕することにより、ジベンゾフルベンとジベンゾフルベンピペリジン付加物を除去する。減圧濾過により固体を回収し、恒量まで乾燥する。その結果、4.55g(94.3%)のHAA27-36NH2が得られ、純度は85〜90A%であった(注1)。
【0225】
Fmoc副産物(ジベンゾフルベンおよびそのピペリジン付加物)は、一般に、このプロトコルで除去されるが、再処理が必要な場合には、10容のMTBE中の固体を20℃で2〜3時間撹拌し、回収および乾燥する。
【0226】
注:
1.逆相HPLCによるプロセス中検査(IPC):
カラム:Vydac C8、300A、5μ、30℃
流量:1mL/分
検出:262nmのUV
移動相:A.水/0.1%TFA
B.アセトニトリル/0.1%TFA
方法:20分間にわたり、80〜99%B
保持時間:FmocAA27-36NH2(16.5分、HAA27-36NH2(8.4分)
2.合計濾過時間は10分であった。
【0227】
3.湿ったフィルターケークは反応器に戻し、直ちに水で洗浄することにより、固体の油状化またはゴム質化を防がなければならない。
【0228】
4.MTBE粉砕によるジベンゾフルベン副産物の除去を確実にするため、粗生成物は、完全に乾燥させなければならない。
【0229】
11.7 FmocAA(17-26)-OHとHAA(27-36)-OHの液相合成融合によるフラグメントFmocAA(17-36)-OHの大規模製造
4回の実施で、2.993kgのHAA27-36NH2と3.176kgのFmocAA17-26OHから、合計5.115kgのFmocAA17-36NH2を製造した。平均収率は84.3%であった。反応混合物からの水ドロップアウト(dropout)後の粗生成物の濾過には、平均40〜50分かかった。
【0230】
カップリング反応を実施する前に、使用されるHBTUおよびフラグメントの両方を試験する使用試験を1〜2gスケールで行う。使用試験から、出発物質および試薬の計算量を決定する。使用試験では、出発物質の可溶性も記録した。濁りのある溶液は、FmocAA17-26OH中の塩の存在を示し、該フラグメントの水洗浄の根拠となるものである。ラセミ化および副産物の生成が最小限であるように出発物を完全に生成物に転化させるようにするため、HBTUの添加前に、反応混合物の全成分が、明らかに溶液中に存在していなければならない。
【0231】
水ドロップアウトから単離した粗FmocAA17-36NH2の純度を、IPAからの沈殿により大幅に増加させる。FmocAA17-36NH2は、IPAに部分的に可溶性である。予め60〜70℃に保温した約15容の95/5のIPA/水を用いて、FmocAA17-36NH2を粉砕した後、撹拌しながら、数時間にわたり室温に冷却し、フラグメントの純度を高める。出発物質、ならびに、FmocAA17-26OHのピペリジンアミドおよびHAA27-36NH2のエナミン尿素付加物は共に、95%IPAに可溶性である。室温で95%IPAを用いた粗FmocAA17-36NH2の粉砕にかける時間を延長しても、不純物の除去にはそれほど効果的ではない。60〜70℃での安定性試験を完了した。IPA溶液を52℃を上回る温度に加熱しない場合、単離した生成物の品質に及ぼす影響が最小限に抑えられるようだ。
【0232】
11.7.1 FmocAA(17-26)OHとHAA(27-36)-OHの液相合成融合によるフラグメントFmocAA(17-26)-OHの改良された大規模製造の好ましい方法
撹拌機と窒素供給口を備えた40Lのジャケット付き反応器に、FmocAA17-26OH(1当量、0.770kg)、HAA27-36NH2(1当量、0.720kg)、HOAT(1.5当量)およびDMF(FmocAA17-26OHに対して12.5容)を導入する。EtPr2N(1.5当量)を添加し、この懸濁液を、室温で、固体が溶解するまで(見た目で)撹拌する。この溶液を窒素雰囲気下で0〜5℃に冷却した後、HBTU(1〜1.05当量)を添加する。この反応混合物を、0〜5℃で、HBTUが溶解するまで(見た目で、約15分間))撹拌する。反応混合物を25℃に温め、2時間撹拌を継続する(注1)。DMF溶液を0〜5℃に冷却した後、25℃を超えないような速度で、低温のプロセス水(12.5容)を添加する。得られたスラリーを1〜24時間撹拌し、固体を減圧濾過により回収した後、水で洗浄する(3x4容)。該固体をフィルター上で16〜24時間乾燥させ、理論質量の2倍にする。40L反応器に、95/5のイソプロパノール/水(25容)を導入し、45℃に温める。急速に撹拌しながら、半乾燥状態のFmocAA17-36NH2をIPA溶液に添加する。この懸濁液を52℃に温めた後、12〜16時間撹拌しながら、室温まで冷却させる(注2)。固体を減圧濾過により回収した後、最小量のIPAで洗浄して、乾燥し、1.261kgのFmocAA17-36NH2を得た。収率は、85%、HPLCによる純度は90.SA%であった(注1)。95/5のIPA/水粉砕を繰り返すことにより、反応を再実施してもよい。
【0233】
注:
1.プロセス中検査および純度、HPLC:
Vydac C8、5μ、300A
1mL/分、262nm、30℃
A.水/0.1%TFA
B.80/20 IPA/ACN/0.1%TFA
60〜95%B/20分
保持時間:HAA27-36NH2、6.73分
FmocAA17-26OH、10.67分
FmocAA17-36NH2、20.2分、
2.FmocAA17-36NH2は、この温度では、この容量中に完全に溶解しない。固体を回収する前に、撹拌を停止し、固体を沈降させる。濾液のサンプルを採取し、HPLCにより分析する。濾液が多量のFmocAA17-36NH2を含んでいる場合には、固体を回収する前に0℃まで冷却する。
【0234】
11.8 FmocAA(17-36)-OHからのフラグメントHAA(17-36)-OHの大規模製造
4回の実施で、5.112kgのFmocAA17-36NH2から、合計4.965kgのHAA17-36NH2を製造した。単離した収率およびHPLCトレースはどちらも、ジベンゾフルベン、および恐らくは溶媒の存在を示している。存在するジベンゾフルベンは、ピペリジンではなく、炭酸カリウムで除去するため、次のカップリング反応を妨害しない。従って、ジベンゾフルベンのピペリジン付加物は、化学的には問題ではない。
【0235】
この方法には、いくつかの問題が付随する。この反応は、12容のDMF中で実施する。Fmoc基を除去する基剤は、1容の1.1M炭酸カリウムであり、これは、除去が困難な、ジベンゾフルベンとの塩基性付加物を生成することはできない。脱保護は、室温で、約90分間進行させる。
【0236】
予め0℃に冷却した1:1で飽和した塩化ナトリウム:水-水溶液を添加し、フラグメントとジベンゾフルベンを共沈殿させる。これによって、濾過するのに数(5〜8)時間を要して、微細な乳白色の固体が生成された。単離した後、湿った固体を完全に乾燥させて(1%LOD未満)、ジベンゾフルベン不純物を除去しなければならない。乾燥には10日間かける(減圧、加熱なし)。乾燥したら、固体を反応器に戻し、3:1のヘプタン:MTBE(20容)を用いて、周囲温度で18時間粉砕することにより、ジベンゾフルベンを除去する。これより粉砕時間が短いと、ジベンゾフルベンを完全に除去することはできず、また、固体に水が少しでも存在すると、除去することができない。ジベンゾフルベンが残っている場合には、3:1のヘプタン:MTBE(20容)による再処理を実施することができる。
【0237】
これらの問題を解決するために、次のような新しい方法が開発された。FmocAA17-36NH2(2.0g、1当量)およびヘプタン(8容)を、撹拌機、温度調節器および還流コンデンサーを備えた100mLの丸底フラスコに添加する。2容のMTBE(4mL)を添加し、該スラリーを45〜50℃に加熱する(注1)。ピペリジン(1 0当量)を添加する。窒素雰囲気下で、45〜50℃にて24〜36時間撹拌する(注2)。反応混合物を45〜50℃で、保温濾過し(注3)、次に、5容(10mL)の60:40のヘプタン:MTBEで、ケークを洗浄する。
【0238】
注:
1.MTBEが反応器から漏れ、そのために、ヘプタン:MTBE比が変化するようなことがある場合には、反応が遅くなる恐れがある。反応温度が50℃を超える場合には、反応固体のゴム質化が起こる可能性がある。Fmocは、5時間で大部分が除去される。
【0239】
2.反応の完了は、RP-HPLCにより監視する:
カラム: Vydac C8、300A、5、30℃
流量: 1mL/分
検出: 262nmのUV
移動相: A.水/0.1%TFA
B.80:20 IPA:CAN 0.1%TFA
方法: 30分間にわたる60〜95%B
保温濾過は、ジベンゾフルベンのピペリジン付加物の除去に役立つ。
【0240】
11.8.1 FmocAA(17-36)-OHからのフラグメントHAA(17-36)-OHの大規模製造
40Lのジャケット付き反応器に、FmocAA17-36NH2(1当量、1.26kg)およびDMF(12容)を室温で導入する。1.11M炭酸カリウム水溶液(1容、5当量)を一度に添加する(注1)。この反応混合物を、室温で、3.5時間撹拌する(注2)。反応混合物を、予め0℃に冷却した飽和塩化ナトリウム/水(10容)の1:1水溶液に徐々に添加するが、その速度は、温度10℃を超えないようなものとする(約1〜2時間)。ラバーダム(rubber dam)を用いて、固体を減圧濾過により回収し、フィルターケークから水を圧搾する(注3)。湿ったケークを反応容器に戻し、水(10容)を用いて1〜2時間粉砕(撹拌)し、残留する無機塩を除去する。固体を減圧濾過により回収し、ラバーダムで圧縮して、減圧オーブンに移し、恒量まで乾燥させる。乾燥した固体(注4)を、3:1のヘプタン:MTBE(20容)で、室温にて最低14時間粉砕(撹拌)し、ジベンゾフルベンを除去する。固体を減圧濾過により回収した後、ヘプタン(2容)で洗浄して、乾燥し、1.20kgのHAA17-36NH2を得た。収率は、99.5%、純度は75A%であった。このHAA17-36NH2は、5A%ジベンゾフルベンを含有していた。へプタン:MTBE粉砕を繰り返すことにより、反応を再実施する。
【0241】
注:
1.炭酸カリウム水溶液を添加すると、溶液は濁るが、撹拌を続けると透明になる。
【0242】
4〜5℃の発熱量を記録した。
【0243】
2.IPCおよび純度測定のために用いたHPLC:
Vydac C8、260nm
1mL/分、262nm、30℃
A.水/0.1%TFA
B.80/20 IPA/アセトニトリル/0.1%TFA
60〜95%B/30分
3.微細な粒子サイズの生成物が沈殿するため、濾過が遅くなる。
【0244】
4.ヘプタンが、ジベンゾフルベンを効果的に除去するように、この物質は無水でなければならない。
【0245】
11.9 AcAA(1-16)-OHおよびFmocAA(17-36)-OHからの液相合成によるフラグメントAcAA(1-36)-OHの大規模製造
AcAA1-16-OHを、HOATおよびDIEAと共に、DMFに溶解して0℃に冷却した後、HBTUを添加した。これを15分間、もしくはHBTUが溶解するまで撹拌し、HAA17-36NH2を添加する。HAA17-36NH2の不在におけるAcAA1-16-OHの前活性化は、予備措置であったが、後に必要がないことがわかった。さらに、活性化AcAA1-16-OHを含む0℃のDMF溶液へのHAA17-36NH2の溶解は、規模が大きくなると、遅くなることが証明された。
【0246】
4回の実施で、3.92kgのAcAA1-16-OHと、4.96kgのHAA17-36NH2から、合計6.972kgのAcAA1-36NH2を製造したが、平均収率は80.1%であった。この段階での2回の実施の平均収率は、87%であり、2回の実施の平均収率は、73%であった。
【0247】
カップリング反応を実施する前に、使用されるHBTUおよびフラグメントの両方を試験する使用試験を1〜2gスケールで行う。使用試験から、出発物質および試薬の計算量を決定する。また、出発物質の可溶性も使用試験に記録する。濁りのある溶液は、AcAA1-16-OH中の塩の存在を示し、フラグメントの水洗浄の根拠となるものである。ラセミ化および副産物の生成が最小限であるように出発物質を生成物に完全に転化させるためには、HBTUの添加前に、反応混合物の全成分が、明らかに溶液中に存在していなければならない。
【0248】
フラグメントAcAA1-16-OHおよびHAA17-36NH2、ならびに試薬HOATおよびDIEAをDMFに溶解させ、0℃に冷却し、HBTUを添加する。全実施のうち1回は、HAA17-36NH2に対しHClをピックアップするために、追加当量のDIEAを必要とした。水ドロップアウトにより、反応混合物から粗生成物を単離する(濾過時間、10分)。まだ湿っている固体を、55℃に予め保温したアセトニトリルを含む反応器に戻し、急速に撹拌しながら、3時間かけて35℃まで冷却した後、20℃で一晩放置する。このスラリーを0〜5℃に冷却し、1〜2時間さらに撹拌した後、固体を濾過により回収する。この工程の間に、AcAA1-36NH2は、55℃で溶液へとほぼ移行した。溶液が冷却するにつれ、AcAA1-36NH2は、溶液から沈殿していった(油状化した)。溶液が冷却するにつれて、油は凝固し、最終的には、鮮やかな白色の固体へと変化する。この変化の最中に、反応器の壁や撹拌棒上に多量の固体沈殿が生成する可能性がある。これらのほとんどは、スラリーを一晩20℃で撹拌するにつれて取れる。固体を回収した後、反応器に、反応器壁や撹拌棒に付着した固体全てを浸すのに十分な水を充填する。
【0249】
壁や撹拌棒から残りの固体がすべて落ちるまで(約1時間)、スラリーを撹拌した後、フィルターケークの洗浄物(wash)として用いる。アセトニトリル洗浄により、未反応の出発物質と、20アミノ酸に満たない切端フラグメントを除去する。
【0250】
11.9.1 AcAA(1-16)-OHおよびFmocAA(17-36)-OHからの液相合成によるフラグメントAcAA(1-36)-OHの好ましい大規模製造
40リットルの反応器に、AcAA1-16-OH(938kg、1当量)、HAA17-36NH2(1.19kg、1当量)、HOAT(1当量)、DMF(AcAA1-16-OHに対して19容)およびDIEA(1当量)を導入する。このスラリーを、固体が溶解するまで撹拌し、0〜5℃に冷却した後、HBTU(1.03当量)を添加する。この溶液を、0〜5℃で、15分間、もしくは固体が溶解するまで撹拌した後、20℃に保温し、2時間撹拌する。反応混合物から、IPCに付すためのサンプルを採取する(注1)。反応が完全であると判断したら、温度が35℃を超えないような速度で、水(19容)を添加する(注2)。
【0251】
得られたスラリーを1〜24時間撹拌し、固体を真空濾過(10分)により回収した後、水で洗浄する(2x3容量)。濾過ケークを圧縮し、水を出来る限り除去する。その間、反応器に、95%アセトニトリル/水(AcAA1-16OHの導入量に対して、30容量)を導入し、撹拌しながら、55℃に保温する。
【0252】
湿った固体を、凝集しないように、少しずつ反応器に添加する。スラリーを撹拌しながら、3時間かけて35℃に冷却し、その後、20℃で一晩放置する。0〜5℃に冷却し、2時間撹拌して、撹拌を停止した後、溶液をサンプリングする。
【0253】
固体を真空濾過により回収する。反応器および管路を90%アセトニトリル/水(3.6容量)で洗浄する。
【0254】
反応器に、反応器壁や撹拌棒に付着した固体を浸すのに十分な量の水を充填する(約30容量)。反応器壁や撹拌棒に付着した固体が落ちるまで、室温で、撹拌する(約1時間)。固体を残り部分と共に回収し、恒量まで乾燥させる(1.83kg、収率86%)。
【0255】
90/10のアセトニトリル/水を用いて、固体の摩砕を繰り返す。
【0256】
注:
1.IPCおよび純度、HPLC:
YMC ODS-A、150 x 4.6mm、5μ、120A
1mL/分、260nm、30℃
A.水/0.1%TFA
B.THF/0.1%TFA
60〜90%B/30分、90〜95%B/1分、95%B/5分
AcAA1-16-OH 6.37分
HAA27-36NH2 8.91分
AcAA1-36NH2 18.07分、
2.水を添加する間、温度が35℃を超えると、沈殿する固体が融解し、凝集および/または反応器壁への付着を起こす恐れがある。
【0257】
11.10 AcAA(1-16)-OHの側鎖の脱保護
この実験の目的は、沈殿物へのMTBEの添加速度を変化させて、温度を20℃以下に維持し、粗T-20固体を単離し、脱炭酸反応させる(すなわち、溶液脱炭酸反応を排除する)ことである。
【0258】
HPLCカラムに供給する前の固体として、ACN/水から、沈殿により粗T20を単離した。固体のT20含有率(%)を、重量に基づくHPLC検定により測定した。
【0259】
側鎖保護基を、90/5/5のTFA/水/ジチオトレイトール中で除去した後、溶液を0℃に冷却し、MTBE(AcAA1-36NH2導入量に対して45容量)を添加する。これは、T20の完全な沈殿を確実にするのに必要なMTBEの最小量である。混合の際に発熱があるため、温度を20℃以下に保持するように、最初の5容量はゆっくりと添加しなければならない(約60分)。さらにMTBEを添加するにつれて、添加速度を高めることができる。沈殿中、温度を20℃以下に維持することにより、固体が溶液から沈殿する際の凝集を防止する。
【0260】
上記開裂/精製プロトコルは、クロマトグラフィーによる精製に適合する規模で脱保護を実施することを含む。TFA塩として脱保護混合物から単離した粗T20を、pH5のアセトニトリル/水に溶解させ、濾過し、15時間静置することにより、トリプトファンインドールの脱炭酸反応を実施する。脱炭酸反応が完了した後、溶液を希釈し、精製装置に移して、カラムに供給した。希釈の間、溶液には曇りが生じ、この濁った溶液をポンプによりカラムに供給する。この結果、精製工程中に、固体がカラムのヘッドに付着し、徐々にカラムの性能が低下した。pH5のアセトニトリル/水中で、数時間T-20を脱炭酸反応させると、極めて不溶性の凝集体が形成される。
【0261】
溶液中の脱炭酸反応を回避するための方法が開発されている。MTBE沈殿物からTFA塩として単離した粗T-20を、室温で約5〜7日にわたり、減圧下で脱炭酸反応させる。減圧下40℃で、3日のうちに脱炭酸反応の完了が認められた。単離した固体のTFA含有率は、9%であった。材料は、0℃で、1カ月まで、1%重量/重量損失で貯蔵することができる。
【0262】
HOAcを用いたエタノール/水(1:9)における塩交換は、MTBEからのT-20-TFAの単離後に実施することができる。粗T-20の酢酸塩は、有意により安定であり、好ましいものであろう。いずれの単離方法でも、側鎖脱保護をこれまでより大きな規模で実施することが可能であり、T-20を、凝集を引き起こすことが知られている脱炭酸反応に使用されるアセトニトリル/水/HOAc条件にさらす必要はない。
【0263】
AcAA(1-36)-OHの側鎖の脱保護のための新しい方法:
90/5/5のTFA/水/ジチオトレイトール溶液(13容量)を作製し、3分間、窒素でパージする。窒素雰囲気下で、周囲温度にて、AcAA1-36NH2の一部を90/5/5のTFA/水/ジチオトレイトール溶液(13容量)に添加する。溶液状にしたら、周囲温度にて4時間撹拌し、次に0℃に冷却する。T-20を沈殿させるために、0〜5℃に冷却したMTBE(45容量)を、初めは、温度を20℃以下に維持しながら、遅い速度で滴下しながら添加する(添加時間は、約1〜2時間)。固体を減圧濾過により回収する(注1)。反応器、管路および濾過ケークを直ちに3x5容量のMTBEで洗浄する。固体を除去し、t=0のIPC(注2)用にサンプルを採取し、5日間、もしくは、HPLCが変化を示さなくなるまで、減圧下で乾燥させる。
【0264】
注:
1.脱保護溶液は吸湿性であるため、この濾過の間、固体に空気を通過させないようにする。TFA溶液を洗浄する前に固体に空気を通過させると、粘着質または油状の固体が得られる恐れがある。
【0265】
2.HPLC方法TM2-0003-01(TFA法)を使用する。酢酸アンモニウム法(TM2-0006-01)では、様々なカルボキシル化中間体を分離できない。
【0266】
AcAA(1-36)-OHの側鎖の脱保護
12回の工程で、合計7.226kgのAcAA1-36NH2を脱保護した。製造した6.972kgのAcAA1-36NH2だけが、中間体が吸湿性であることを示した。MTBEドロップアウトから単離した固体を、10容量の1:1ACN/水に溶解し、濾過した後、炭酸水素ナトリウムでpH3.5に調整した。酢酸(1.5容量%)を添加し(pH4.5〜5)、溶液を周囲温度で15時間撹拌することにより、脱炭酸反応を実施した。溶液を25容量の水で希釈し、合計40容量の85/15の水/ACN溶液を得た。この溶液は、いずれの場合も濁っており、中には、微細な固体(凝集したT-20)を含むものもあった。IPC用のサンプルを採取し、溶液を精製装置に移した。工程あたりの粗T-20の収率を、重量/重量HPLC検定を用いて計算した。
【0267】
AcAA1-36NH2の脱保護は、室温の90/5/5のTFA/水/DTTで、4〜5時間で完了する。8時間後には、顕著な分解(2%)が起こった。5℃の同じ混合物では、脱保護は8時間では完了しないが、23時間で完了する。室温で5時間後に得た粗T-20と、5℃で23時間後に得たものとの間に、純度の差はなかった。単離容量(約55容量)は、反応(脱保護)容量(13容量)よりはるかに大きいため、20Lカーボイ中のTFA溶液にAcAA1-36NH2を溶解させてから、この溶液を40L反応器に移す必要があった。
【0268】
脱保護混合物からの、粗カルボキシル化T-20(TFA塩として)の単離を、MTBEを用いた沈殿により達成する。ペプチドを沈殿させるのに必要なMTBEの量は、脱保護混合物量の3〜4倍である。MTBEを開裂混合物の容量の2倍使用することにより、ペプチドが残留する。
【0269】
開裂混合物にMTBEを添加すると、容易に濾過される固体が得られるのに対し、MTBEに開裂混合物を添加すると、濾過が難しい微細な沈殿物が生成する。TFA溶液にMTBEを添加する間、5℃から40〜45℃への温度上昇がある。温度上昇は、単離した粗T-20の純度(187/117、119)に影響を与えない。MTBE添加の間、いくらか固体の凝集が生じた。スラリーを1〜2時間撹拌して、凝集物を破壊した。MTBE添加の間、温度を20℃以下に維持することにより、より均質な固体が生成し、この固体は濾過が容易である(196/31)。MTBEの添加速度は、将来制御されるであろう。窒素雰囲気下での濾過により、粗T-20を回収した。TFAを洗浄する前の濾過中に、湿った空気にさらすと、固体は粘着質になる。TFAを洗浄した後、固体は、空気に対して安定する。
【0270】
方法工程:
90/5/5のTFA/水/ジチオトレイトール溶液(13容量)を20Lカーボイ中に製造し、窒素で3分間パージする。凝集を防ぐため、AcAA1-36NH2(650g)を少量ずつ添加する。固体が液状になったら、溶液を70L反応器に移す。カーボイと管路を最小量の90/5/5のTFA/水/ジチオトレイトールで洗浄する。窒素雰囲気下、周囲温度で溶液を4時間撹拌し、その後、0〜5℃に冷却する。MTBE(45容量)を、内部温度が30℃を超えないような速度で添加する。窒素流の下、減圧濾過により固体を回収する。2x2容量のMTBEで反応器、管路および固体を洗浄し、恒量まで乾燥させる(594g、70A%)。
【0271】
固体(594g)を50%ACN/水(AcAA1-36NH2導入量に対して8容量)に溶解させ、濾過する。50%ACN/水(2容量)で容器と管路を洗浄する。炭酸水素ナトリウムを用いて溶液のpHを3.5〜4.0に調整した後、酢酸(0.18容量)を添加して、pHを4.9にする。溶液を周囲温度で15時間撹拌し(IPC、注1)、1M炭酸カリウムでpHを9〜9.5に調整する。水(25容量)を添加することにより、溶液を85/15の水/ACNに希釈する。このようにして得られた濁った溶液から、重量/重量HPLC分析用のサンプルを採取し、精製装置に移す。
【0272】
注:
1.方法TM2-0003-01
2.方法TM2-0006-01
11.11 HAA(1-36)-OHの精製
精製、凍結乾燥法およびパッケージングを、3段階に分類する:
%wt/wt=100 − %不純物 − %酢酸塩 − %水 − %TFA。バッチ中の酢酸塩の含有率は6〜8%であった。
【0273】
合計2.08kgのT-20(正味)を6.97kgのAcAA1-36NH2から単離した。これは、AcAA1-36NH2からの収率が49.3%であることを示す。クロマトグラフィー精製における平均収率は、55%であった。個々の工程についての収率は、41〜55%までと差があった。最低収率は、カラムまたはポンプの故障により、複数箇所で通過(passes)が生じたためである。一般に、クロマトグラフィーが機能すれば、収率は、50〜55%の範囲である。単離したT-20の純度は93〜95A%であった。
【0274】
工程時間を最小限にする目的で、方法を比較するために、精製中、四つの勾配を検定した:
1.15〜22%ACN/60分、330mL/分で、26〜36%ACN/525分。
【0275】
2.16〜26%ACN/60分、330mL/分で、26〜40%のACN/525分。
【0276】
3.第2トリメリス(Trimeris)勾配。330mL/分で、15〜36%ACN1788分
4.元のトリメリス(Trimeris)勾配20〜23%ACN/1 12分、330mL/分で、23〜36%ACN/488分。
【0277】
20センチメートル(cm)のカラムをアンバークロム(Amberchrom)樹脂(台高さ35cm)で充填した(軸方向圧縮)。500〜700gのAcAA1-36NH2のバッチを脱保護し、溶液中で脱炭酸反応させた。この規模では、約400グラムのペプチド(約75A%T-20)を含有するカラム供給原材料が生成される。原液は、典型的には、曇っており、懸濁した固体を含むこともある。濁った溶液を、15%Bを用いて、500mL/分でカラムに供給する。いずれの工程のカラム供給中にも、圧力増大はなかった。流速を330mL/分に減少させ、指示された時間、特定の勾配を実施する。画分を回収し、78%以上のT-20を含有する画分をプールした(100〜110L)後、水で希釈することにより、アセトニトリル含有率を約15〜20%(約140リットル)にする。カラムを洗浄した後、15%Bで平衡させる。T-20含有溶液を、8インチカラムに、900mL/分で送り返す。%Bを50%に増加し、T-20をカラムから約25Lでフラッシュする。
【0278】
溶液を1リットルガラス鐘中で凍結し、凍結乾燥して、粉末にする。単離したT-20は、典型的には、HPLCにより、92〜94A%であり、6〜8%の酢酸塩と3〜4%の水を含有する。
【0279】
好ましい方法:
pH9.2で、85/15の水/アセトニトリル(27L)中のT-20溶液を、500mL/分で、8インチHPLCカラムにポンプ供給した。流速を30分かけて330mL/分に減少させた(5分毎に30〜40mL/分の増加量)。20%Bから36%Bまでの線状勾配を600分にわたり開始する。吸光度が0.2AUFSを下回るまで、画分を回収する。画分を、含有率について、HPLCにより分析する(注1)。カラムを80%Bで900 mL/分にて10分間洗浄し、15%Bに戻し、900mL/分で平衡化する。78A%以上のT-20を含有する画分をプールし(113L)、バッファー(28.2L)で希釈することにより、アセトニトリル濃度を15〜20%にする。溶液を900mL/分でカラムにポンプ供給する。%Bを50%に増加し、T-20を、900mL/分でカラムから約25L溶出させる。T-20の濃度は、約9〜10mg/mLである。溶液を1リットル凍結乾燥ジャーに移し、凍結させ、凍結乾燥して、収率54.9%、純度94.1A%の216.29gのT-20を得る。
【0280】
注:
1.画分は、含有率および純度について、HPLCにより分析する。
YMC ODS-A、150x4.6mm、5μm、120A
1mL/分、220nm、30℃
A 70/30、50mM NH4OAc @ pH7(NH4OHで調整する):ACN
B 5:95 50mM NH4OAc @ pH7(NH4OHで調整する):ACN
0〜15%B/50分、15〜100%B/2分、100%B/3分
T-20保持時間、29.0分
【0281】
11.12 ペプチドの熱安定性
一連の実験を実施して、中間T-20フラグメントの熱安定性を特性評価した。T-20フラグメントを水で飽和させ、濾過した後、密閉容器に導入して、高温に暴露した。いくつかの時点で、サンプルを回収し、HPLC法により分析することによって、純度変化を得た。また、乾燥したT-20フラグメントの安定性は、熱重量分析(TGA)により特性評価した。
【0282】
AcAA1-16OHを除くすべてのフラグメントを40℃で3日間乾燥させることができる。80℃で24時間後、2つのフラグメント、すなわちFmocAA27-35OH(3%)とFmocAA27-36NH2(1.4%)に、ほんのわずかな分解が認められる。フラグメントAcAA1-16OHは、初め97A%の純度であった。40℃で3日間後、93.4A%の純度および乾燥度になった。さらに80℃で24時間後にも、93.9A%を維持した。尚、湿ったAcAA1-16OHは、周囲温度で乾燥させなければならない。
【0283】
安定性試験の要点は、フラグメントを、周囲温度ではなく、40℃で乾燥することができるかどうかを決定することであった。この試験から、フラグメントが、40℃の乾燥で安定であることがわかった。これにより、乾燥時間が大幅に短縮されるであろう。
【0284】
単離および精製を模倣した様々な溶剤および温度条件の下で、フラグメントAc(1-16)-OH、Fmoc(27-35)-OH、Fmoc(17-26)-OH、Fmoc(27-36)-NH2、Fmoc(17-36)-NH2、H(17-36)-NH2、ならびにAc(1-36)-NH2の安定性を検定するために設計された試験が完了した。フラグメントはすべて、それらが単離および/または精製された溶剤系において、安定である。尚、安定性はXで表す。
【0285】
本発明は、本明細書に記載した特定の実施形態によって範囲を制限されるわけではない。これらの実施形態は、本発明の個々の態様を示すことを目的とするに過ぎず、機能的に同等の方法および要素は、本発明の範囲内に含まれるものとする。実際に、当業者には、本明細書に記載したもの以外にも、本発明の様々な変更が、これまでの説明および添付の図面から明らかとなるであろう。このような変更は、添付した請求の範囲内にあるものとする。
【技術分野】
【0001】
1.序
本発明は、第1に、ペプチド、特にT-20(「DP-178」とも呼ばれる;配列番号1)及びT-20様ペプチドの合成方法に関する。該方法では、固相及び液相合成方法を利用して、特定のペプチド断片のグループを合成し、それらを結合させて目的のペプチドを得る。本発明はさらに、目的のペプチド(例えば、T-20)の合成の中間体として有用な個々のペプチド断片に関する。本発明は、さらにまた、全長のT-20及びT-20様ペプチドを製造するために一緒に用いることができる前記ペプチド中間体断片のグループに関する。本発明は、さらにまた、ペプチド、特にT-20及びT-20様ペプチドの精製方法、並びに目的ペプチドの合成の中間体として有用な個々のペプチド断片に関する。
【背景技術】
【0002】
2.背景
近年、融合に関連した現象を抑制する能力を示し、さらに、重要なことには、強力な抗ウイルス活性をも示す多数のペプチドが同定されている。例えば、米国特許第5,464,933号;第5,656,480号及び国際公開公報第WO96/19495T-20号を参照されたい。これらのペプチドは治療薬として広く用いられているので、例えば、大規模量での合成能力に対する要請が高まっている。
【0003】
ペプチド合成のための技術(例えば、Merglerら, 1988, Tetrahedron Letters 29:4005-4008;Merglerら, 1988, Tetrahedron Letters 29:4009-4012;Kamberら(編集), 「ペプチド、化学及び生物学(Peptides, Chemistry and Biology)」, ESCOM, Leiden, 1992, 525-526;及びRinikerら, 1993, Tetrahedron Letters 49:9307-9320を参照されたい。)は存在するが、T-20及びT-20様ペプチドなどの容易に精製されたペプチドの大規模で経済的な製造に利用できる技術は現在のところ存在しない。
【発明の概要】
【0004】
3.発明の概要
本発明は、第1に、ペプチド、特にT-20(「DP-178」とも呼ばれる;配列番号1)及びT-20様ペプチドの合成方法に関する。該方法では、固相及び液相合成方法を利用して、特定のペプチド断片のグループを合成し、それらを結合させて目的のペプチドを得る。一般に、本発明の方法は、固相支持体上のT-20又はT-20様ペプチドの側鎖が保護された特定のペプチド断片中間体を合成し、溶液中で保護された断片を結合させて、保護されたT-20又はT-20様ペプチドを形成し、次いで、側鎖を脱保護して、最終のT-20又はT-20様ペプチドを得ることを含む。本発明の好ましい実施形態は、配列番号1に示されるアミノ酸配列を有するT-20ペプチドの合成に関係する。
【0005】
本発明はさらに、目的のペプチド(例えば、T-20)の合成において中間体として有用な個々のペプチド断片に関する。本発明のペプチド断片としては、限定するものではないが、下記表1に示されるアミノ酸配列を有するものが挙げられる。
【0006】
【表1】
本発明は、さらにまた、目的のペプチドの合成において中間体として有用なペプチド断片の特定のグループに関する。本発明のペプチド断片のグループとしては、下記表2に指定される、グループ1〜20が挙げられる。
【0007】
【表2】
この発明は、一部には、固相液相合成反応の特定の組合せによって初めて、高効率且つ高収量で大規模に高純度のT-20及びT-20様ペプチドの製造が可能になるという本発明者らの予期せざる発見に基づいている。特に、本発明の方法によれば、T-20及びT-20様ペプチドが、1キログラム以上の規模で合成できる。固相合成のための本発明の特定のT-20ペプチド断片を選択することによって、固相技術の高効率の結合(カップリング)が、3-、4-又は5-倍もの過剰のアミノ酸及び固相合成で通常必要とされる試薬を用いる必要なしに利用され得ることが見出された。本発明の方法では、本発明のペプチド断片の固相合成においてたった1.5倍ほどのアミノ酸しか使用しない。アミノ酸及び試薬の量におけるこのコスト低減により、本発明の方法はT-20及びT-20様ペプチドの大規模合成に適している。
【0008】
さらに、本発明者らは、驚くべきことに、特定のペプチド断片が、約0.8〜1 mmol/g(固相樹脂)のローディング量(loading)で固相合成され得ることを見出した。このローディング量は、固相ペプチド合成で一般に達成される0.25〜0.4 mmol/g(樹脂)のローディング範囲よりも顕著に高い。さらに、本発明者らは、超酸感受性樹脂を用いた固相で所定のペプチド断片を合成することが、異常に高純度のペプチド断片をもたらすことを見出した。クロマトグラフィー技術は、本発明によって製造されたペプチド断片を精製するのに必要ではなく、断片を使用の前に単に沈殿及び/又は粉砕(trituration)工程に付すか、又は樹脂から直接取得したままで使用する。超酸感受性樹脂を使用することによって、本発明の合成された保護ペプチドを、側鎖保護基の同時除去を行なうことなく樹脂から切断することが可能になる。これは、不純物を減らし、10個以上のアミノ酸を含むペプチドを、高純度で合成することを可能にする。本発明によって製造された高純度のペプチド断片を結合させることにより、本発明の方法に従って液相中で合成されるT-20及びT-20様ペプチドの不純物プロフィールは、主として、密接に関係した類似体よりもむしろ、結合しなかった断片からなる。従って、本発明によって製造されたT-20及びT-20様ペプチドは、従来技術によって製造されたものよりも精製するのがかなり容易である。下記第9及び11節に示す実施例では、T-20全長ペプチドのそのようなコンビナトリアル(組合せ)合成を実証する。第11節に示す実施例では、T-20及びT-20中間体ペプチドの大規模合成及び精製を実証する。従って、本発明の方法によれば、T-20及びT-20ペプチド中間体が、1キログラム以上の規模で製造できる。
【0009】
本発明者らはまた、予期せざることに、T-20及び他のT-20様ペプチドなどのペプチド、並びに本明細書中に記載された特定のペプチド断片が、塩基性pH範囲で使用できる高容量材料(high capacity materials)を用いて精製できることを見出した。すなわち、本発明は、さらにまた、ペプチド、特にT-20及びT-20様ペプチド、並びに目的ペプチドの合成の中間体として有用な個々のペプチド断片の精製方法に関する。
【0010】
3.1 定義
本明細書中で用いるアミノ酸表記は、慣用のものであり、以下の通りである:
一般的なアミノ酸の略号
アミノ酸 1文字表記 一般的略号
アラニン A Ala
アスパラギン N Asn
アスパラギン酸 D Asp
グルタミン Q Gln
グルタミン酸 E Glu
ヒスチジン H His
イソロイシン I Ile
ロイシン L Leu
リシン K Lys
フェニルアラニン F Phe
セリン S Ser
トレオニン T Thr
トリプトファン W Trp
チロシン Y Tyr
4.図面の簡単な説明
【図面の簡単な説明】
【0011】
【図1】図1:T-20の4断片アプローチ。この図面は、上記表2中に示される、中間体ペプチド断片グループ6から出発して全長T-20を合成するための、下記第9.1節に示される実施例に従うスキームを示す。
【図2】図2:T-20の4断片アプローチ、第2経路。この図面は、全長T-20を合成するための、上記表2に示されるペプチド中間体グループ6を結合させる追加的な4断片スキームを示す。
【図3】図3:T-20の3断片アプローチ。この図面は、全長T-20を合成するための、下記第9.1節に示される実施例に従うスキームを示す。
【図4】図4:T-20の3断片アプローチ、第2経路。この図面は、全長T-20を合成するための、下記第9.2、9.3、9.4及び9.5節に示される実施例に従うスキームを示す。
【図5】図5:T-20の2断片アプローチ。この図面は、全長T-20を合成するための、上記表2に示されるペプチド中間体グループ18を結合させるスキームを示す。
【発明を実施するための形態】
【0012】
5.発明の詳細な説明
5.1 全長ペプチド
本発明は、T-20、あるいは、DP-178として知られるペプチドを合成するために使用できる方法、ペプチド断片、ペプチド断片のグループに関する。T-20は、HIV-1LAI分離物由来の膜貫通タンパク質gp41のアミノ酸残基638-673に相当するペプチドであり、36個のアミノ酸配列(アミノNH2末端からカルボキシCOOH末端へと読む)を有する:
NH2−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−COOH
本発明の方法、断片及び断片のグループ、並びに断片及び断片のグループの選択に利用される技術は、T-20に加えてT-20様断片を合成するのにも使用できることが理解されるだろう。本明細書中で用いる語句「T-20様」とは、米国特許第5,464,933号;第5,656,480号又は国際公開公報第WO96/19495号(それぞれその全体が参照によって本明細書中に組み入れられる)に列挙されるいずれかのHIV又は非HIVペプチドを意味する。
【0013】
上記のT-20及びT-20様ペプチドに加えて、本発明の方法、断片及び断片のグループは、修飾されたアミノ及び/又はカルボキシル末端を有するペプチドを合成するのに使用できる。T-20を例にとると、該ペプチドは、
式: X−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−Z
(式中、Xは、アミノ基;カルボベンゾキシル、ダンシル、及びt−ブチルオキシカルボニルからなる群から選択される疎水性基;アセチル基;9−フルオレニル−メトキシ−カルボニル(FMOC)基;又は脂質−脂肪酸コンジュゲート、ポリエチレングリコール、及び炭水化物からなる群から選択される巨大分子キャリアー基を示し;そしてZは、カルボキシル基;アミド基;t−ブチルオキシカルボニル基;p−ニトロベンジルエステル基;又は脂質−脂肪酸コンジュゲート、ポリエチレングリコール及び炭水化物からなる群から選択される巨大分子キャリアー基を示す)で示される。前記「X」及び「Z」基の付加技術は、当業者に周知である。
【0014】
好ましい実施形態では、本発明の方法は、上記式においてXがアセチル基であり、Zがアミド基であるペプチドを合成するのに用いられる。下記第9節に示される実施例は、下記第5.2節に記載されるペプチド中間体の結合によるT-20ペプチドの成功した合成を実証する。好ましい方法では、T-20及びT-20様ペプチド並びに中間体は、限定するものではないが、高い(7より高い)pH範囲で安定なジルコニウム系充填剤、ポリスチレン、ポリアクリル又は他のポリマー系充填剤を含めて(ローディング容量を最大化するための)非シリカ系充填剤を用いて精製できる。例えば、広範なpH範囲を示す非シリカ系カラム充填剤は、Tosohaus社(Montgomeryville, PA)によって販売されているものよりも高いpH値を含む。前記材料で充填されたカラムは、低、中又は高圧クロマトグラフィーで作動する。例えば、下記第10節に示される精製法を参照されたい。
【0015】
5.2 ペプチド中間体
本発明は、限定するものではないが、上記表1に列挙された特定のアミノ酸配列を有するT-20及びT-20様ペプチドのペプチド断片中間体、及び表2に列挙されたペプチド断片中間体のグループを包含する。該ペプチド中間体、特に下記表2に列挙されるグループに含まれるものは、T-20及びT-20様ペプチドを製造するのに利用できる。
【0016】
表1又は2に列挙されたペプチド断片のアミノ酸残基の側鎖のいずれか1個以上は、t−ブチル(t-Bu)、トリチル(trt)及びt−ブチルオキシカルボニル(Boc)などの標準的な保護基によって保護できる。t-Bu基は、アミノ酸残基Tyr(Y)、Thr(T)、Ser(S)及びAsp(D)の好ましい側鎖保護基であり;trt基は、アミノ酸残基His(H)、Gln(Q)及びAsn(N)の好ましい側鎖保護基であり;Boc基は、アミノ酸残基Lys(K)及びTrp(W)の好ましい側鎖保護基である。
【0017】
表1に列挙された断片1、2、3及び4の合成の間、ヒスチジン残基の側鎖は、好ましくはトリチル(trt)保護基によって保護されていなければならない。もしそれが保護されていないと、樹脂からペプチド断片を切断するために使われる酸が、ヒスチジン残基と有害に反応し、ペプチド断片の分解を引き起こすだろう。
【0018】
本発明のペプチド断片のグルタミン残基は、トリチル(trt)基によって保護されているのが好ましい。しかしながら、断片1-16及び9-16のカルボキシル末端ではグルタミン残基を保護しないことが好ましい。1-16断片のカルボキシ末端のグルタミン残基上に保護基が存在しないと、1-16断片と17-36断片との反応が促進され、たった約2%のラセミ化しか伴わずに断片同士を結合できることがわかった。さらに、有機溶媒中での本発明のいずれかのペプチド断片のより低い溶解性が望まれる場合は、トリチル保護基を、その断片のいずれか1個以上の他のグルタミン残基から除去し得る。
【0019】
本発明の各ペプチド断片の全てのアスパラギン残基は保護されているのが好ましい。さらに、トリプトファン残基はBoc基によって保護されているのが好ましい。
【0020】
上記表1に列挙されたペプチド式1〜18に従う保護されたペプチド断片としては、限定するものではないが、下記表3に列挙される化合物が挙げられる。
【0021】
【表3】
上記表3に列挙されたペプチドのアミノ酸残基のいずれか1個以上の側鎖を、上記のように、tBu、trt及びBocなどの標準的な側鎖保護基によって保護できる。表3のペプチドの代表的な合成を下記第7及び8節に示し、そこでは下記第5.4節で述べる一般的技術を用いる。
【0022】
5.3 ペプチド合成
上述のように、本発明の個々のペプチド断片の幾つかは、固相合成法を用いて作られるのが好ましいが、本発明の他のペプチドは、固相および液相合成法を組み合わせて用いて作られるのが好ましく、前記合成は本明細書中に記載されるようにT-20またはT-20様ペプチドの製造で完了する。しかしながら、本発明のペプチド断片は、当業界で周知の技術によって合成または製造できることが理解されるであろう。例えば、参照によってその全体が本明細書中に組み入れられる、Creighton, 1983, タンパク質:構造および分子原理(Proteins: Structures and Molecular Principles), W. H. Freeman and Co., NYを参照されたい。
【0023】
あるいは、本発明のペプチドは、ペプチドのアミノ酸残基を連結する1個以上の結合が非ペプチド結合であるように合成できる。これらの他の非ペプチド結合は、当業者に周知の反応を利用することによって形成でき、以下に限定されないが、2〜3の例を挙げると、イミノ、エステル、ヒドラジド、セミカルバジド、およびアゾ結合を挙げることができる。
【0024】
本発明のまたもう1つの実施形態では、上記配列を含むT-20およびT-20様ペプチドは、例えば、ペプチドの安定性、反応性および/または溶解性が増強されるように、ペプチドのアミノおよび/またはカルボキシ末端に存在する付加的な化学基によって合成できる。例えば、カルボベンゾキシル、ダンシル、アセチルまたはt−ブチルオキシカルボニル基などの疎水性基を、ペプチドのアミノ末端に付加することができる。同様に、アセチル基または9−フルオロエニルメトキシ−カルボニル基を、ペプチドのアミノ末端に配置することができる。(上記のT-20の「X」修飾を参照されたい。)さらに、疎水性基、t−ブチルオキシカルボニル、またはアミド基を、ペプチドのカルボキシ末端に付加することができる。同様に、p−ニトロベンジルエステル基をペプチドのカルボキシ末端に配置することができる。(上記のT-20の「Z」修飾を参照されたい。)そのような修飾を導入する技術は、当業者に周知である。
【0025】
さらに、T-20およびT-20様ペプチドは、その立体配置が変わるように合成することができる。例えば、ペプチドの1個以上のアミノ酸残基の、通常のL−アイソマーよりもむしろD−アイソマーを用いることができる。
【0026】
さらにまた、本発明のペプチドの少なくとも1個のアミノ酸残基は、周知の自然に存在しないアミノ酸残基の1つによって置換できる。これらのような変更は、本発明のペプチドの安定性、反応性および/または溶解性を増加させるように働くことができる。
【0027】
T-20またはT-20様ペプチドのいずれも、それらのアミノおよび/またはカルボキシ末端に共有結合によって結合する巨大分子キャリアー基を追加的に有するように合成できる。そのような巨大分子キャリアー基としては、例えば、脂質−脂肪酸コンジュゲート、ポリエチレングリコール、炭水化物または付加的なペプチドが挙げられる。従って、上記のT-20の「X」修飾は、ペプチドのアミノ末端に共有結合によって結合する上記の巨大分子キャリアー基のいずれかを追加的に示すことができ、好ましくは付加的なペプチド基である。同様に、上記のT-20の「Z」修飾は、上記の巨大分子キャリアー基のいずれかを追加的に示すことができる。
【0028】
本発明のペプチド断片は、標準FMOCプロトコルを用いて固相ペプチド合成(SPPS)法によって合成されるのが好ましい。例えば、Carpinoら, 1970, J. Am. Chem. Soc. 92(19):5748-5749;Carpinoら, 1972, J. Org. Chem. 37(22):3404-3409を参照されたい。好ましい実施形態では、本発明のペプチド断片の固相合成は、以下に限定されないが、塩化2−クロロトリチル樹脂(例えば、Barlosら, 1989, Tetrahedron Letters 30(30):3943-3946を参照されたい。)および4−ヒドロキシメチル−3−メトキシフェノキシブチル酸樹脂(例えば、Seiber, 1987, Tetrahedron Letters 28(49):6147-6150、およびRichterら, 1994, Tetrahedron Letters 35(27):4705-4706を参照されたい。)を含む超酸感受性固相支持体上で行われる。塩化2−クロロトリチル樹脂および4−ヒドロキシメチル−3−メトキシフェノキシブチル酸樹脂の両者は、Calbiochem-Novabiochem Corp., San Diego, CAから購入できる。
【0029】
固相ペプチド合成で利用できる一般的な製造工程および樹脂の装填は、本明細書中に記載されている。樹脂の装填は、例えば、次の方法によって行うことができる:樹脂、好ましくは2−クロロトリチル樹脂などの超酸感受性樹脂を、反応チャンバーにローディングする。ジクロロメタン(DCM)などの塩素化溶媒で該樹脂を洗浄する。ベッドの液体を排出し(brained)、約8〜10倍容量(volume)のジクロロエタン(DCE)中1.5当量のアミノ酸および2.7当量のジイソプロピルエチルアミン(DIEA)からなる溶液を添加する。該アミノ酸のN末端は、好ましくはFmocによって、保護されているべきであり、該アミノ酸の側鎖は、必要または適当である場合には、保護されているべきである。この混合物を窒素通気によって2時間攪拌する。
【0030】
2−クロロトリチル樹脂を適度に膨潤させるために塩素化溶媒が必要であることに注意すべきである。文献によれば、DCEは、より高い装填効率を与えるが、DCMは、殆どまたは全く装填の低下を伴わずに置換できる。
【0031】
攪拌後、ベッドの液体を排出し、DCMで洗浄する。樹脂の活性部位を、9:1のMeOH:DIEAの溶液で、約20〜30分間末端キャッピング(endcapped)する。ベッドの液体を排出し、DCMで4回洗浄し、窒素パージによって乾燥し、装填された樹脂を得る。
【0032】
Fmocは、アミノ酸のN末端に対して好ましい保護基である。どのアミノ酸が装填されるかによって、その側鎖は、保護されていてもよいし、保護されていなくてもよい。例えば、Trpが装填されているときは、その側鎖は、Bocで保護されているべきである。同様に、Glnの側鎖は、trtで保護できる。しかしながら、1〜16ペプチド断片の合成のための調製物中にGlnが装填されているときは、その側鎖は、保護されるべきではない。Leuの側鎖は保護する必要がない。
【0033】
樹脂装填およびペプチド合成で用いられるFmoc保護アミノ酸は、必要に応じて側鎖保護基を有するまたは有しないものが、Sean社またはGenzyme社から入手できる。上記工程の代わりに、適当なアミノ酸がすでに装填されている樹脂を購入できる。
【0034】
下記第6節に示される実施例は、典型的な樹脂調製物を記載している。
【0035】
固相ペプチド合成法は、例えば、次の方法に従って行うことができる:装填された樹脂を反応チャンバーに添加し、溶媒、好ましくは塩化メチレン(DCM;好ましくは約10倍容量)で窒素攪拌下、約15分間調整して、樹脂ビーズを膨潤させる。2−クロロトリチル樹脂を適度に膨潤させるためにDCMが必要である。樹脂容量は、ビーズが膨潤し、活性部位が折りたたまれずに、反応に利用できるようになるにつれて、反応チャンバー中で2倍または3倍になるだろう。樹脂が膨潤した後、溶媒を反応チャンバーから排出する。
【0036】
末端アミンまたは樹脂からのFmoc(9−フルロエニル−メチルオキシカルボニル)保護基の除去は、その樹脂をN−メチル−2−ピロリジノン(NMP)中ピペリジンの20%溶液のアリコートで2回、それぞれ約10分間処理することによって達成される。各アリコートに必要な20%のピペリジンのNMP溶液の容量は、実施される反応規模に依存するだろう。次いで、NMP(約10倍容量)のアリコートで5〜7回樹脂を洗浄して、Fmoc副生成物(すなわち、ジベンゾフルベン(dibenzofulvene)およびそのピペリジン付加物)並びに残ったピペリジンを除去する。
【0037】
クロルアニル試験を用いて、Fmoc副生成物および残りのピペリジンの除去が完了したか否かを決定することができる。クロルアニル試験溶液を、トルエン中のクロルアニルの飽和溶液の液滴を約1mLのアセトンに添加することによって調製する。NMP洗浄は、その洗浄液の液滴をクロルアニル試験溶液に添加することによって試験できる。青色または紫色は、第2級アミンの存在について陽性であることを示し、Fmoc副生成物および/または残りのピペリジンが未だ存在することを示す。NMP洗浄を、青色または紫色がもはや観察されなくなるまで繰り返す。
【0038】
一方、樹脂に添加される、配列中の次のアミノ酸は、そのカルボキシ末端が反応のために活性化される。各アミノ酸のアミン末端は、Fmocで保護されているはずである。どのアミノ酸が添加されるかによって、その側鎖は、保護されていてもよいし、保護されていなくてもよい。好ましくは、tyr(Y)、Thr(T)、Ser(S)およびAsp(P)の側鎖はt-Buで保護され、His(H)、Gln(Q)およびAsn(N)の側鎖はtrtで保護され、そしてLys(K)およびTrp(W)の側鎖はBocで保護される。しかしながら、上記で考察したように、Hisの側鎖は保護されていなければならない。さらに、断片1〜16および9〜16のカルボキシ末端のGln残基の側鎖は保護されないことが好ましい。LeuまたはIleの側鎖は、保護する必要がない。
【0039】
アミノ酸は、次のように活性化される。Fmoc保護アミノ酸(1.5当量)、1−ヒドロキシベンゾトリアゾール水和物(HOBT)(1.5当量)、およびジイソプロピル−エチルアミン(DIEA)(1.5当量)を、室温でNMP(約7.5倍容量)に溶解する。この溶液を0〜5℃に冷却し、次いで、O−ベンゾトリアゾール−1−イル−N,N,N',N'−テトラメチルウロニウム・ヘキサフルオロホスフェート(HBTU)(1.5当量)を添加し、次いで、5〜15分間攪拌して溶解させる。活性化は、低温で行い、アミノ酸のラセミ化を最小限にすることが重要である。活性化およびラセミ化は、HBTUの非存在下では起きないので、HBTUは、冷たい溶液に添加される最後の試薬である。
【0040】
活性化されたアミノ酸の溶液を、液体を排出した樹脂にローディングし、DCM(約2.5倍容量)で洗浄する。アミノ酸の活性化は、HBTUがDCMに不溶性なので、NMP中で行うことに注意すべきである。しかしながら、樹脂ビーズの適度な膨潤を維持するために、DCMをこの時点で反応物に添加する。該反応物を窒素通気によって、約1時間攪拌する。結合の完了は、下記に記載される定性ニンヒドリン試験でモニターできる。
【0041】
定性ニンヒドリン試験を用いて反応の完了を確認するには、2〜20 mgの樹脂サンプルを回収し、メタノールできれいに洗浄する。このサンプルに、76%のフェノールのエタノール溶液を3滴、0.2 mM KCNのピリジン溶液を4または5滴、および0.28 Mニンヒドリンのエタノール溶液を3滴添加する。このサンプルをエタノールで希釈して、約0.5 mL容量にし、約75℃の加熱ブロック中に5〜10分間置く。青色または紫色は、遊離アミンの存在について陽性であることを示し、反応が未だ完結していないことを示す。濃縮サンプル中の変色程度をより容易に判断するために、このサンプルをさらに希釈して約3mL容量とする。
【0042】
1時間後にニンヒドリン試験が陽性なら、結合反応をさらに1時間続ける。2時間後にニンヒドリン試験の陽性が続くなら、樹脂の液体を排出し、約10倍容量のNMPで3回洗浄し、1当量の活性化アミノ酸を用いて結合反応を繰り返す。
【0043】
結合サイクルの間、樹脂を一晩保存すべきなら、樹脂ベッドの液体を排出し、窒素ブランケット(nitrogen blanket)下でDCMで被覆することができる。あるいは、該ベッドの液体を排出し、窒素ブランケット下で保存し、次いで、次の結合サイクルを進める前にDCM洗浄で調整する。できた断片を切断する前に一晩保存すべきなら、顕著なFmoc脱保護がNMP中で起きるので、樹脂ベッドをDCMで洗浄してNMPを除去すべきである。
【0044】
結合が完了したと判定された後、前記樹脂の液体を排出し、NMPのアリコート(約10倍容量)で3回洗浄する。ペプチド断片の次のアミノ酸の結合(mer)のために、このサイクルを繰り返す。最終の結合反応に続いて、樹脂をNMPのアリコート(約10倍容量)で4回洗浄し、次いで、DCMのアリコート(約10倍容量)で4回洗浄する。樹脂結合ペプチドは、窒素パージによって乾燥できる。
【0045】
固相合成法によって合成されたペプチドを切断し、例えば、次の方法に従って単離できる:ペプチドは、当業者に周知の方法を用いて樹脂から切断できる。例えば、トリフルオロ酢酸(TFA)の1%または2%DCM溶液あるいはTFAの1%および2%DCM溶液を組み合わせて用いてペプチドを切断できる。酢酸(HOAC)も、ペプチドの切断に使用できる。切断に必要な特異的な切断試薬、溶媒および時間は、切断される特定のペプチドに依存するだろう。切断後、切断画分を標準的な後処理工程に付し、ペプチドを単離する。一般的には、混合された切断画分を減圧下で濃縮し、次いで、エタノール、メタノールまたはヘプタンなどの溶媒で再構成する(reconstitution)。一般に、水を添加してペプチドを沈殿させ、減圧濾過によって回収する。あるいは、ペプチドの単離の前に該生成物を粉砕することができる。
【0046】
下記第7.1〜7.6節に示される実施例は、表1、2および/または3に示されるペプチド中間体の固相合成を示す。
【0047】
完全長T-20ペプチドの合成のためには、上記表1のペプチド中間体を一緒に結合させて、T-20ペプチドを得ることができる。例えば、上記表2に列挙されたペプチド中間体のグループを一緒に結合させ、T-20の完全長ペプチドを製造できる。中間体ペプチド断片からの完全長T-20のそのような合成の代表例は、下記第9節に示されており、図1〜5に概略的に記載されている。
【0048】
特定の実施形態では、T-20合成の4断片アプローチに従うことができる。例えば、「4断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合した4つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示される中間体ペプチド断片グループ5、6、8、9および12〜15は、好ましいグループを示す。図1および図2は、表2のペプチド中間体グループ6を利用し、完全長T-20を合成する、2種の4断片アプローチを示す。このグループについては、アミノ酸残基36(T-20カルボキシ1−末端アミノ酸残基)が、断片結合プロセスの間に個々に導入されることに注意する。図1に示されるT-20合成スキームの最高点(culmination)は、第9.1節で示される実施例で実証される。
【0049】
さらに、実施形態、T-20合成のための3断片アプローチに従うことができる。「3断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合した3つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示される中間体断片グループ2〜4、7、10および11は、好ましい3つの断片グループを示す。図3および図4は、表2のペプチド中間体グループ3を利用して完全長T-20を合成する、2種の3断片アプローチを示す。このグループに関しては、アミノ酸残基36(T-20カルボキシル末端アミノ酸残基)は、断片結合プロセスの間に個々に導入されることに注意する。図3に示されるT-20合成図式の最高点は、下記第9.1に示される実施例で実証される。図4に示されるT-20合成スキームの最高点は、下記第9.2〜9.5節に示される実施例で実証される。
【0050】
更なる実施形態では、T-20合成のための2断片アプローチに従うことができる。「2断片アプローチ」合成とは、固相および液相合成法を用いて合成および結合する2つのT-20中間体ペプチド断片で始まる、完全長T-20ペプチドまでのT-20合成スキームをいう。上記表2に示された中間体断片グループ1および16〜20は、好ましい断片グループを示す。図5は、表2のペプチド中間体グループ20を利用して、完全長T-20を合成する、2断片アプローチを示す。このグループについては、アミノ酸残基(T-20カルボキシル末端アミノ酸残基)は、断片結合プロセスの間に個々に導入されることに注意する。
【0051】
当業者に周知の液相ペプチド合成法は、本発明のペプチド中間体断片の合成に利用できる。第8.1〜8.11節に示される実施例は、表1、2および/または3に列挙されたペプチド中間体の典型的な液相ペプチド合成を記載している。例えば、広いpH範囲を示す非シリカ装填カラムパッキングには、Tosohaus社(Montgomeryville, PA)によって販売されるものよりも高いpH値を含むものがある。
【実施例】
【0052】
6.実施例:樹脂合成
本明細書第6.1〜6.3節に記載する実施例では、本明細書中に記載されたペプチドおよびペプチド中間体の固相合成と共に利用できるクロロトリチル樹脂を合成する。
【0053】
6.1 Fmoc−Trp(Boc)−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 25 25 −
樹脂
Fmoc−Trp(Boc)−OH − 526.60 1.5 37.5 19.7
ジイソプロピルエチル 129.25 1.7 42.5 5.5 7.4
アミン(DIEA)
ジクロロエタン(DCE) − − − − 250
ジクロロメタン(DCM) − − − − 6×250
9:1メタノール:DIEA − − − − 200
工程:
塩化2−クロロトリチル樹脂(25 g, 1当量)を、500 mLのペプチドチャンバーにローディングし、250 mLのDCMで洗浄した。ベッドの液体を排出し、10倍容量のDCE中のFmoc−Trp(Boc)−OH(1.5当量)およびDIEA(1.7当量)からなる溶液を添加した。この混合物を窒素通気によって2時間攪拌した。
【0054】
ベッドの液体を排出し、250 mLのDCMで洗浄した。樹脂の活性部位を、200 mLの9:1のMeOH:DIEA溶液で20分間末端キャッピングした。このベッドの液体を排出し、250 mLのDCMで4回洗浄し、窒素パージによって乾燥し、34.3 gの装填された樹脂を得た。
【0055】
樹脂からFmocアミノ酸を切断し、標準に対してアッセイすることによって、定量的HPLC分析を行った。材料のHPLCアッセイは、0.68 mmol/gで樹脂に装填されていることを示した。
【0056】
カラム:Phenomenox Jupiter C18;300Å;5μ
流速:1 mL/分
検出:260 nm紫外線
移動相: A:0.1%TFA水溶液
B:アセトニトリル中0.1%TFA
65% Bイソクラティック
保持時間:〜14分
6.2 Fmoc−Gln−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 25 25 −
樹脂
FmocGlnOH 368.39 1.5 37.5 13.8 −
ジイソプロピルエチル 129.25 1.7 42.5 5.5 7.4
アミン(DIEA)
ジクロロエタン(DCE) − − − − 75
N,N−ジメチルホルム − − − − 200
アミド(DMF)
ジクロロメタン(DCM) − − − − 6×250
9:1メタノール:DIEA − − − − 200
工程:
75 mLのDCEおよび200 mLのDMFからなる混合物中でFmocGlnOH(1.5当量)およびDIEA(1.7当量)からなる溶液を用いて上記第6.1節に示す実施例で用いた工程を繰り返した。DMFの添加はFmocGlnOHを安定化する。反応により33.8 gの装填された樹脂を得た。0.74 mmol/gの樹脂の理論装填量を仮定し、材料を進展させた(carried forward)。
【0057】
6.3 Fmoc−Leu−2−クロロトリチル樹脂の製造
材料: 分子量 当量 mmole グラム mL
塩化2−クロロトリチル − 1.0 250 250 −
樹脂
FmocLeuOH 353.42 1.5 375 132.5 −
ジイソプロピルエチル 129.25 1.7 425 55 75
アミン(DIEA)
ジクロロエタン(DCE) − − − − 2000
ジクロロメタン(DCM) − − − − 6×1500
9:1メタノール:DIEA − − − − 1500
工程:
3Lのペプチドチャンバーに樹脂をローディングし、1.5LのDCMで洗浄した。ベッドの液体を排出し、8倍容量のDCE中のFmocLeuOH(1.5当量)およびDIEA(1.7当量)からなる溶液を添加した。この混合物を、窒素通気によって2時間攪拌した。
【0058】
ベッドの液体を排出し、1.5LのDCMで洗浄した。樹脂の活性部位を1.5Lの9:1のMeOH:DIEA溶液で30分間末端キャッピングした。このベッドの液体を排出し、1.5LのDCMで4回洗浄し、窒素パージによって乾燥して345 gの装填樹脂を得た。
【0059】
樹脂からFmocアミノ酸を切断し、標準に対してアッセイすることによって、定量的HPLC分析を行った。材料のHPLCアッセイは、0.72 mmol/gでの樹脂の装填を示した。
【0060】
カラム:Phenomenox Jupiter C18;300Å;5μ
流速:1 mL/分
検出:260 nmの紫外線
移動相: A:0.1%TFA水溶液
B:アセトニトリル中の0.1%TFA
65% Bイソクラティック
保持時間:〜8分
7.実施例:ペプチドの固相合成
下記に示す、第7.1〜7.6節は、表1、2、および/または3に列挙されたペプチド中間体の固相合成の例である。
【0061】
7.1 断片Fmoc−アミノ酸(1〜8)−OH(断片1b)構造の製造
Fmoc−Tyr(tBu)−Thr(tBu)−Ser(tBu)−Leu−Ile−His(trt)−Ser(tBu)−Leu−OH(配列番号2)
C93H121N10O15
分子量1619.06
材料: 分子量 当量 mmole グラム mL
Fmoc−Leu−2−クロロ− − 1.0 15.6 20.0 −
トリチル樹脂
Fmoc−アミノ酸* − 1.5 23.4 − −
1-ヒドロキシベンゾトリ- 153.15 1.5 23.4 3.6 −
アゾール(HOBT)水和物*
O-ベンゾトリアゾール-1- 379.25 1.5 23.4 8.9 −
イル-N,N,N',N'-テトラ
メチルウロニウム ヘキサ
フルオロホスフェート
(HBTU)*
ジイソプロピルエチル 129.25 1.5 23.4 3.0 4.1
アミン(DIEA)*
N−メチル−2−ピロリジノン − − − − 150
(NMP)*
塩化メチレン(DCM)* − − − − 50
20%ピペリジン/NMP* − − − − 2×200
すすぎ用のNMP* − − − − (洗浄毎)200
DCM中1%トリフルオロ酢酸 − − − − 300
(TFA)
0.5%TFA/DCM − − − − 200
ピリジン − − − −
エチルアルコール − − − − 110
水 − − − − 200+100
*結合サイクル当たり
理論収量:25.3 g 予想収量:80〜90%
実収量:20.0 g
工程:
1Lのペプチド反応チャンバーに、20 gのFmoc−Leu−2−クロロトリチル樹脂をローディングした。窒素攪拌下で200 mL(〜10倍容量)のDCM中で約15分間樹脂を調整し、ビーズを膨潤させ、次いで液体を排出した。
【0062】
末端アミンからのFmoc(9−フルオロエニルメチルオキシカルボニル)の除去は、NMP中ピペリジンの20%溶液200 mLで2回それぞれ10分間で達成した。次いで、樹脂を、200 mL(〜10倍容量)のNMPで5〜7回洗浄して、クロルアニル試験の陰性によって決定して、Fmoc副生成物(ジベンゾフルベンおよびそのピペリジン付加物)および残りのピペリジンを除去した。
【0063】
一方、配列中の次のアミノ酸であるFmoc−Ser(tBu)を、カルボキシル末端で反応させるために活性化した。Fmoc保護アミノ酸(1.5当量)、HOBT(1.5当量)、およびDIEA(1.5当量)を、室温で、150 mL(〜7.5倍容量)のNMPに溶解した。この溶液を0〜5℃に冷却し、次いでHBTU(1.5当量)を添加し、5〜15分攪拌して溶解させた。活性化された酸の溶液を、液体を排出した樹脂にローディングし、50 mLのDCM(〜2.5倍容量)で洗浄した。反応物を窒素通気によって1時間攪拌した。結合の完了は、定性ニンヒドリン試験でモニターした。結合反応が完結したと判定された後、樹脂の液体を排出し、200 mL(1倍容量)のNMPで3回洗浄した。
【0064】
それぞれ1.5当量のFmoc保護アミノ酸His(trt)、Ile、Leu、Ser(tBu)、Thr(tBu)およびTyr(tBu)を用いて、ペプチド断片の次の結合(mer)のためにサイクルを繰り返した。最終の結合反応に続いて、樹脂を200 mL(10倍容量)のNMPで4回、次いで、200 mL(10倍容量)のDCMで4回洗浄した。この樹脂を窒素パージによって乾燥して42 gの樹脂結合ペプチドを得た。
【0065】
300 mLのDCM中1%TFAで約2分間、次いで、200 mLのDCM中0.5%TFAを用いて、このペプチドを21 gの量の樹脂から切断した。切断画分をピリジン(TFAに対して1:1の容量比)上に回収した。切断洗浄液を合わせ、減圧下に濃縮して、容量を約50 mLとし、次いで、濃縮を続けて残りのDCMを除去し、最終容量を〜250 mLにしながら、110 mLのエタノールで再構成した。生成物を200mLの無図を添加して沈殿させた。そのスラリーを室温で30分間攪拌した。この固体を減圧濾過によって回収し、約100 mLの水で洗浄した。生成物を空気乾燥して、95%のHPLC純度を有する20.0 g(79%)のFmoc−アミノ酸(1〜8)−OHを得た。
【0066】
カラム:Phenomenox Jupiter C18
流速:1 mL/分
検出:260 nm紫外線
移動相: A:0.1%TFA水溶液
B:20分で80%B〜99%Bのアセトニトリル勾配中0.1%TFA
保持時間:約23分
7.2 断片Fmoc−アミノ酸(9〜15)−OH(断片5b)構造の製造
Fmoc−Ile−Glu(tBu)−Glu(tBu)−Ser(tBu)−Gln(trt)−Asn(trt)−G
ln(trt)−OH(配列番号6)
C117H129N10O18
分子量1963.39
材料: 分子量 当量 mmole グラム mL
Fmoc-Gln(trt)-2-クロロ- − 1.0 12.0 20.0 −
トリチル樹脂
Fmoc−アミノ酸* − 1.5 18.0 − −
1-ヒドロキシベンゾトリ- 153.15 1.5 18.0 2.8 −
アゾール(HOBT)水和物*
O-ベンゾトリアゾール-1- 379.25 1.5 18.0 6.8 −
イル-N,N,N',N'-テトラメチル-
ウロニウム ヘキサフルオロ-
ホスフェート(HBTU)*
ジイソプロピルエチルアミン 129.25 1.5 18.0 2.3 3.1
(DIEA)*
N-メチル-2-ピロリジノン − − − − 150
(NMP)*
塩化メチレン(DCM)* − − − − 50
20%ピペリジン/NMP* − − − − 2×200
すすぎ用のNMP* − − − − (洗浄毎)200
切断用DCM − − − − 160
酢酸(HOAc) − − − − 20
トリフルオロエタノール − − − − 20
ヘプタン − − − −250+250+100
メチル t-ブチルエーテル − − − − 100
イソプロパノール − − − − 60
水 − − − − 60+50
*結合サイクル当たり
理論収量:23.6 g 予想収量:89〜95%
実収量:21.1 g
工程:
20.0 gのFmoc−Gln(trt)−2−クロロトリチル樹脂、およびFmoc保護アミノ酸Asn(trt)、Gln(trt)、Ser(tBu)、Glu(tBu)、Glu(tBu)およびIleを用いて上記第7.1節に示される実施例で用いた工程を繰り返した。
【0067】
最終のカップリング反応に続き、樹脂を4 X 200mL(10容)のNMP、次いで4 X 200mL(10容)のDCMで洗浄した。
【0068】
ペプチドを200mLの8:1:1 DCM:TFE:HOAcを用いて2時間で樹脂から開裂させ、続いて2 X 100mLのDCMで洗浄した。溶出液を合わせて真空下で容量が約100mLになるまで濃縮し、次いで250mLのヘプタンで再構成する一方で、最終容量が約250mLになるまで濃縮を続けて残存しているDCMを除去した。形成した二相混合物からヘプタン層を分離した。250mLのヘプタンおよび100mLのMTBEを添加して生成物を沈殿させ、次いで室温で一晩粉砕すると所望の粘稠度の物質が得られた。固体を真空濾過によって回収して約100mLのヘプタンで洗浄した。生成物を再び後処理して残存している酢酸を除去した。濾過した固体を50℃で60mLのイソプロパノールに溶かした。溶液を氷浴中で0〜5℃に冷やし、次いで60mLの水を迅速な滴下速度で加えた。生成スラリーを氷浴中で約1時間攪拌しながら粉砕した。固体を真空濾過によって単離して約50mLの水で洗浄した。生成物を風乾すると95%HPLC純度のFmoc-AA(9-15)-OHが21.1g(90%)得られた。
【0069】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に80% B〜99% Bの勾配
保持時間:約23分
7.3 断片Fmoc-AA(1-16)-OH(断片3d)の調製
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を32.5gのFmoc-Gln-2-クロロトリチル樹脂および必要なFmoc保護アミノ酸を用いて繰り返した。上記の原料の節で示されたものとは若干異なる容量の溶媒を用いることを除けば、上記第6.1節で提示した実施例に記載された通りに反応を行った。
【0070】
最終のカップリング反応に続き、樹脂を4 X 250mL(8容)のNMP、次いで4 X 250mL(8容)のDCMで洗浄した。樹脂を窒素パージ下で乾燥させると97.4gの結合ペプチドが得られた。
【0071】
17.7gのスケールで、樹脂に結合したペプチドを2 X 190mLのDCM中の1% TFAを用いて1〜2分で樹脂から開裂させ、続いて1 X 120mLのDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)に回収した。画分および洗液を合わせて真空下で容量が約50mLになるまで濃縮し、次いで200mLのメタノールで再構成した。最終容量が約50mLになるまで濃縮を続けて残存しているDCMを除去した。250mLの水を添加して生成物を沈殿させ、室温で約30分間攪拌した。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると12.8g(84%)が得られた。生成物を再び後処理してピリジニウム塩を除去した。濾過した固体を室温で150mLのメタノールに溶かした。室温で200mLの水を添加すると生成物が沈殿した。生成物を真空濾過によって単離して約50mLの水で洗浄した。物質を風乾すると12.8g(84%)のFmoc-AA(1-16)-OHが得られた。
【0072】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に75% B〜99% Bの勾配
保持時間:約25分
7.4 断片Ac-AA(1-16)-OH(断片3c)の調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を32.5gのFmoc-Gln-2-クロロトリチル樹脂および必要なFmoc保護アミノ酸、ならびに上記の原料の節で示された溶媒容量を用いて繰り返した。
【0073】
最終のカップリング反応に続き、樹脂を4 X 250mL(8容)のNMP、次いで4 X 250mL(8容)のDCMで洗浄した。樹脂を窒素パージ下で乾燥させると97.4gの結合ペプチドが得られた。
【0074】
10gのスケールで、樹脂に結合したペプチドを100mLの3:1 NMP:DCM中で30分間無水酢酸およびピリジン(各5等量)でアセチル化し、次いで2 X 25mLのDCMで洗浄した。ペプチドを3 X 50mLのDCM中の1% TFAを用いて樹脂から開裂させ、続いて2 X 50mLのDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)中に回収した。画分および洗液を合わせて真空下で容量が約100mLになるまで濃縮し、次いで3 X 50mLのヘプタンを少量ずつ加えて再構成する一方で、最終容量が約50mLになるまで濃縮を続けて残存しているDCMを除去した。生成物は最初はややべとつく粘稠度で沈殿したが、0〜5℃の氷浴中で約30分間攪拌しながら粉砕すると濾過可能な固体となった。固体を真空濾過によって回収して約150mLのヘプタンで洗浄した。生成物を再び後処理してピリジニウム塩を除去した。濾過した固体を室温で50mLのメタノールに溶かした。溶液を氷浴中で0〜5℃に冷やし、次いで50mLの水を迅速な滴下速度で加えた。生成物は最初は粘稠な固体として沈殿し、それを氷浴中で約1時間攪拌しながら粉砕すると濾過可能な粘稠度となった。生成物を真空濾過によって単離して約25mLの水で洗浄した。生成物を風乾すると7.0g(90%)のAcAA(1-16)-OHが得られた。続いて生成物を前述のように再び後処理すると96% HPLC純度の物質が6.2g(回収率89%)得られた。
【0075】
カラム:Zorbax LP C8, 100Å, 20μ
流速:1mL/分
検出:220nmの紫外線
移動相:A: 0.1% 水性TFA
B: 1:1 ACN:IPA中の0.05%TFA
20分間に80% B〜99% Bの勾配
保持時間:約15分
7.5 断片Fmoc-AA(17-26)-OH(断片10b)の調製
構造:
Fmoc-Glu(tBu)-Lys(Boc)-Asn(trt)-Glu(tBu)-Gln(trt)-Glu(tBu)-Leu-Leu-Glu(tBu)-Leu-OH (配列番号11)
理論収量:44.4g 期待収率:90-105%
実際の収率:46.9(105%)
操作:
上記第7.1節で提示した実施例において用いられた操作を25.0gのFmoc-Leu-2-クロロトリチル樹脂、必要なFmoc保護アミノ酸、および上記の原料の節で示された溶媒容量を用いて繰り返した。
【0076】
最終のカップリング反応に続き、樹脂を4 X 250mL(10容)のNMP、次いで4 X 250mL(10容)のDCMで洗浄した。
【0077】
ペプチドを3 X 400mL(約15容)のDCM中の1% TFAを用いて樹脂から開裂させ、次いで1 X 200mL(7.5容)のDCMで洗浄した。開裂画分をピリジン(TFAに対して1:1の容量比)中に回収し、次いで画分および洗液について生成物含量を分析した。生成物を含む画分を合わせて真空下で容量が約100mLになるまで濃縮し、次いで300mLのエタノールで再構成した。最終容量が約250mLになるまで濃縮を続けて残存しているDCMを除去した。攪拌した溶液に300mLの水を加えて生成物を沈殿させた。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると97% HPLC純度のFmoc-AA(17-26)-OHが46.4g(105%)得られた。
【0078】
カラム:Phenomenox Jupiter C18
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% 水性TFA
B:アセトニトリル中の0.1% TFA
20分間に75% B〜99% Bの勾配
保持時間:約25分
7.6 断片Fmoc-AA(27-35)-OH(断片16b)の調製
構造:
Fmoc-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-OH (配列番号17)
理論収量:48.9g 期待収率:85-90%
操作:
上記第7.1節で提示した実施例において用いられた操作を33.0gのFmoc-Trp(Boc)-2-クロロトリチル樹脂、必要なFmoc保護アミノ酸、および上記の原料の節で示された物質を用いて繰り返した。
【0079】
最終のカップリング反応に続き、樹脂を4 X 250mL(7.5容)のNMP、次いで4 X 250mL(7.5容)のDCMで洗浄した。
【0080】
ペプチドを8:1:1 DCM:TFE:HOAcの300mL(約10容)の溶液で2時間処理することによって樹脂から開裂させた。樹脂の水分を切り、2 X 250mLのDCMで洗浄した。開裂溶液および洗液を合わせて約50mLの容量になるまで濃縮し、次いで250mLのエタノールで再構成した。溶液を氷浴中で攪拌しながら0〜5℃に冷やした。攪拌した溶液に125mLの水を加えて生成物を沈殿させた。固体を真空濾過によって回収して約50mLの水で洗浄した。生成物を風乾すると95% HPLC純度のFmoc-AA(27-35)-OHが32.0g(65.4%)得られた。
【0081】
樹脂を2 X 250mLのDCM中のTFAの1%溶液で処理し、続いて100mLのDCMで洗浄した。開裂画分をTFAと1:1の容量比であるピリジン中に回収した。溶出液および洗液を合わせて容量が約50mLになるまで濃縮した。この溶液に100mLのエタノール、次いで150mLの水を加えた。生成スラリーを真空濾過すると10.7g(21.9%)(95% HPLC純度)の二次収量が得られ、合わせた収率は87.3%となった。1% TFA/DCM開裂が、より高い効率およびより低い容量であるために好ましい。
【0082】
カラム:Phenomenox Jupiter C5, 300Å, 5μ
流速:0.75mL/分
検出:260nmの紫外線
移動相:A: 0.05% 水性TFA
B: 1:1 IPA:MeOH中の0.1%TFA
10分間に70% B〜97% Bの勾配
保持時間:約25分
8. 実施例:ペプチド断片の液相合成
下記の第8.1−8.11節において提示するのは表1、2、および/または3に挙げたペプチド中間体の液相合成の例である。
【0083】
8.1 グルタミンのパラニトロベンジルエステル(OPNB)のFmoc-AA9-15OHへのカップリングによる断片Fmoc-AA9-16OPNBの調製
構造:
Fmoc-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号7)
理論収量:21.5g 期待収率:90-105%
操作:
FmocAA9-15OH(上記第7.2節で合成)、HBrGlnOPNB、HOATおよびEtPr2Nを磁気攪拌棒を備えた1Lの丸底フラスコ中で混合してNMP(200mL)を添加した。得られた溶液を窒素雰囲気下に置き、攪拌しながら0〜5℃に冷却した。冷却した溶液にHBTUを加えた。溶液を0〜5℃で15分間攪拌し、氷浴を外して攪拌を2.5時間続けた(注1)。
【0084】
反応混合物を0〜5℃に冷却し、0.5N 水性HCl(250mL)を添加して保護されたペプチドを沈殿させた。固体を真空濾過によって回収し、濾過フラスコ中で乾燥させると24gの粗FmocAA9-16OPNBが得られた。この固体を酢酸エチル(250mL)に溶かし、硫酸マグネシウム(10g)で乾燥させ、濾過して容量が100mLになるまで濃縮した。溶液を0〜5℃に冷却し、ヘキサン(250mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収して乾燥させると21.5gのFmocAA9-16OPNBが100%の収率および91-94% HPLC純度で得られた(注2)。
【0085】
注:
1. 薄層クロマトグラフィー(TLC)による工程内管理(IPC)。
90/10 クロロホルム/エタノール
紫外線、ヨウ素検出
Rt FmocAA9-15OH 0.46
Rt FmocAA9-16OPNB 0.57
2. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:13.3分
【0086】
8.2 断片HCl HAA9-16OPNBの調製
構造:
HCl H-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号7)
理論収量:19.2g 期待収率:85-105%
操作:
磁気攪拌棒を備えた1Lの丸底フラスコにFmocAA9-16OPNB(上記第8.2節で合成)および20:1 テトラヒドロフラン/ピペリジンを仕込んだ。得られた溶液を窒素雰囲気下室温で60分間攪拌した(注1)。ヘキサン(350mL)を添加してペプチドを沈殿させた。溶媒を粘性のある固体からデカントした。室温で18時間固体をMTBE(200mL)を用いて粉砕した。この固体を真空濾過によって回収して乾燥させると18.9gのHAA9-16OPNBが得られた(注2)。
【0087】
この固体をメタノール(150mL)に溶かし、攪拌しながら0〜5℃に冷却した。0.5N 水性塩酸(100mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(50mL)、次いで2-プロパノール(50mL)で洗浄して乾燥させると17.7gのHCl HAA9-16OPNBが92%の収率および92A% HPLC純度で得られた(注3)。
【0088】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:FmocAA9-16OPNB, 13.3分; HAA9-16OPNB, 10.7分
2. この時点で単離されたHAA9-16OPNBは微量のピペリジンおよびベン
ジルフルベンピペリジン付加物を含んでいる。両方をRAA1-8OHとカ
ップリングする前に除去する。
3. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B
保持時間:HAA9-16OPNB, 7.2分
TLC条件:
90/10 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rf: HAA9-16OPNB, 0.64
【0089】
8.3 断片AcAA1-8OHとHAA9-16OPMBとの液相カップリングによる断片AcAA1-16OPNBの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.38 期待収率:85-105%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにAA1-8OH(上記第7.1節で合成)、HCl HAA9-16OPNB(上記第8.2節で合成)およびHOATを仕込んだ。固体をEtPr2Nを含む9:1 DMF:DMSO(5mL)に溶かし、次いで窒素雰囲気下0〜5℃に冷却した(注1)。冷却した溶液にHBTUを加えた。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに60分間攪拌した(注2)。水(7mL)の添加によってペプチドを溶液から沈殿させた。固体を真空濾過によって回収し、水(10mL)で洗浄して乾燥させると0.36gの粗AcAA1-16OPNBが得られた。この固体を室温で1.5時間MTBE(3.5mL)を用いて粉砕し、真空濾過によって回収して乾燥させると0.335gのAcAA1-16OPNBが88%の収率および82A% HPLC純度で得られた(注3)。
【0090】
注:
1. 0〜5℃に冷却してHBTUを添加する前にすべての固体を溶液にする
ことが重要である。
2. 工程内管理, TLC, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:HAA9-16OPNB, 7.2分(Ac1-8OHは260nmに吸光度をもたな
い).
保持時間:AcAA1-16OPNB, 12.45
TLC条件:
90/10 クロロホルム/エタノール
紫外線、ヨウ素検出
Rf: HAA9-16OPNB, 0.64
Rf: Ac1-8OH, 0.35
Rf: Ac1-16OH, 0.48
3. Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で70-97%B、8分で97%B
保持時間:Ac1-16OPNB, 16.4.
【0091】
8.4 断片FmocAA1-8OHとHCl HAA9-16OPNBとの液相カップリングによる断片FmocAA1-16OPNBの調製
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:28.3g 期待収率:85-100%
操作:
磁気攪拌棒を備えた1Lの丸底フラスコにFmocAA1-8OH(上記第7.1節で合成)、HCl HAA9-16OPNB(上記第8.2節で合成)、HOATおよびDMF(250mL)を仕込んだ。溶液にEtPr2Nを添加した。この溶液を0〜5℃に冷却し、HBTUを加えた。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに70分間攪拌した(注1)。反応混合物を0〜5℃に冷却し、10% 塩化ナトリウム/水(200mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(50mL)で洗浄して乾燥させると27gの粗FmocAA1-16OPNBが得られた。固体をメタノール(200mL)に溶かして、攪拌した塩化ナトリウムの水溶液(10% wt/vol, 300mL)に加えた。この固体を真空濾過によって回収し、水(50mL)で洗浄して乾燥させると26gのFmocAA1-16OPNBが92%の収率およびHPLCでは90A%純度で得られた(注1)。
【0092】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:HAA9-16OPNB, 7.2分, FmocAA1-8OH, 7.9分
保持時間:FmocAA1-16OPNB, 14.5分
8.5 断片H-AA1-16OPNBの調製
構造:
H-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.94g 期待収率:90-105%
操作:
磁気攪拌棒を備えた50mLの丸底フラスコにFmocAA1-16OPNB(上記第8.4節で合成)、ジクロロメタン(11.4mL)およびピペリジン(0.6mL)を仕込んだ。溶液を窒素雰囲気下、室温で90分間攪拌した(注1)。ヘキサン(45mL)を反応混合物に添加し、溶媒の容量を真空蒸留によって25mLに減らした。得られた固体を真空濾過によって回収し、乾燥させると0.96gのHAA1-16OPNBが102%の収率で得られた。この固体のHPLC分析によって72A%のHAA1-16OPNBおよび18A%のフルベンおよびピペリジン−フルベン付加物が示された。
【0093】
注:
1. Phenomenox Jupiter, C5, 5μ, 300Å
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、5分で100%B.
保持時間:FmocAA1-16OPNB, 14.1分
保持時間:HAA1-16OPNB, 11.6分
保持時間:フルベンおよびピペリジン−フルベン付加物, 5.5分および4.8分。
【0094】
8.6 HAA1-16OPNBのN末端のアセチル化による断片Ac-AA1-16OPNBの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(tBu)-Glu(tBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OPNB (配列番号4)
理論収量:0.96g 期待収率:80-100%
操作:
磁気攪拌棒を備えた50mLの丸底フラスコにHAA1-16OPNB(上記第8.5節で合成)、DMF(10mL)、無水酢酸およびピリジンを仕込んだ。反応混合物を窒素雰囲気下、室温で60分間攪拌した(注1)。水(20mL)を添加してペプチドを沈殿させた。固体を真空濾過によって回収し、水(10mL)で洗浄して乾燥させると0.87gのAcAA1-16OPNBが得られた。残存しているフルベンおよびピペリジン−フルベン付加物を除去するために、この固体を室温で4.5時間1:1 MTBE/ヘキサン(20mL)を用いて粉砕した。固体を真空濾過によって回収して乾燥させると0.82gのAcAA1-16OPNBが85%の収率およびHPLCでは90A%以上の純度で得られた(注1)。
【0095】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300Å
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80-100%B、15分で100%B.
保持時間:HAA1-16OPNB, 11.6分
保持時間:AcAA1-16OPNB, 12.1分
TLC, ジクロロメタン中の10%エタノール
紫外線、ヨウ素検出
Rf: AcAA1-16OPNB, 0.69
8.7 His(trt)の存在下でAcAA1-16OPNBからパラニトロベンジル保護基を選択的に除去することによる断片AcAA1-16OHの調製
構造:
Ac-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-OH (配列番号4)
理論収量:0.76g 期待収率:70〜85%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにAcAA1-16OPNB(上記の第8.6節で合成)およびDMF(10mL)を仕込んだ。この溶液にギ酸アンモニウム水溶液(0.5mL)、次いで湿らせたパラジウムカーボン(Degussa, 10%, 50%水) を添加した。スラリーを窒素雰囲気下、室温で120分間攪拌した(注1)。このスラリーをセライトの密な充填層で濾過し、90mLの水に取った。濾過ケーキをDMF(5mL)で洗浄した。この水性懸濁液を酢酸エチル(100mL)で洗浄した。次いで酢酸エチルを容量が20mLになるまで濃縮した(注意2)。ヘキサン(40mL)を添加して沈殿を完了させ、溶媒を固体からデカンテーションした。固体をメタノール(10mL)に溶かし、4:1の 水/塩化ナトリウム飽和水溶液(25mL)を添加してペプチドを沈殿させた。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると0.62gのAcAA1-16OHが得られた。この固体を室温で15時間50% MTBE/ヘキサン(8mL)とともに粉砕し、回収して乾燥させると0.59gのAcAA1-16OHが77%の収率およびHPLCでは90A%の純度で得られた(注3)。
【0096】
注:
1. 工程内管理, TLC
80/20 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rf: AcAA1-16OPNB, 0.90
Rf: Ac1-16OH, 69
2. AcAA1-16OHは溶媒容量を減じるにつれ沈殿し始める。
3. Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで5分間
保持時間:AcAA1-16OH, 10.73分
【0097】
8.8 FmocAA27-35OHとHPheNH2との液相カップリングによる断片FmocAA27-36NH2の調製
構造:
Fmoc-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号18)
理論収量:42.8g 期待収率:90〜105%
操作:
磁気攪拌棒を備えた2Lの丸底フラスコにFmocAA27-35OH(上記の第7.6節で合成)、HOAT、HpheNH2およびDMF(500mL)を仕込んだ。EtPr2Nを添加して溶液を0〜5℃に冷却し、次いでHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに70分間攪拌した(注1)。溶液を0〜5℃に冷却し、水(500mL)を添加してペプチドを沈殿させた。固体を減圧濾過によって回収し、水(100mL)で洗浄して乾燥させると43gのFmocAA27-36NH2が100%の収率およびHPLCでは93A%の純度で得られた(注2)。
【0098】
注:
1. 工程内管理, TLC
88/12 ジクロロメタン/メタノール
紫外線、ヨウ素検出
Rf: FmocAA27-35OH, 0.49
Rf: FmocAA27-36NH2, 0.63
2. Phenomenox Jupiter, C18, 5μ, 300オームストロング
1.0mL/分, 260nm
A H2O/0.1% TFA
B ACN
20分以上で75〜99%B、99%Bで5分間
保持時間:29.4分
8.9 断片H-AA(27-36)-NH2の調製
構造:
H-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-NH2 (配列番号17)
理論収量:19.6g 期待収率:85〜95%
操作:
磁気攪拌棒および窒素ブランケットを備えた250mLの丸底フラスコに上記の第8.8節で合成したFmoc断片、塩化メチレン(約5容)、およびピペリジンを仕込んだ。溶液が得られ、室温で1.5時間攪拌した(注1)。
【0099】
この溶液を2 X 100mLの水で洗浄した。層が分離し、有機層を減圧下で元の容量の約2分の1になるまで濃縮した。MTBEをわけて(2 X 50mL)加えつつ、濃縮を続け、重沈殿点までおよび最終ポット容量が約150mLになるまでDCMを除去した。
【0100】
生成スラリーを攪拌し、0〜5℃の氷浴中で約1時間冷却した。固体を減圧濾過によって単離し、2 X 15mLのMTBEで洗浄した。生成物を空気乾燥させると95% HPLC純度のH-AA(27-36)NH2が17.6g(89.6%)得られた(注2)。
【0101】
注:
1) 反応の完了はHPLCによってモニターした。
カラム:Phenomenox Jupiter, C18; 300オームストロング; 5μ
流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% TFA水溶液
B: アセトニトリル中の0.1% TFA
20分間で75%B〜99%Bの勾配
保持時間:約18分
【0102】
2) Fmoc副生成物であるジベンゾフルベンおよびそのピペリジン付加物は双方とも後処理において効果的に除去される。しかしながら、この物質を先へ進める前に使用テストを行わねばならない。物質が使用テストを通らなければ、DCM(5容)に固体を溶かし、次いで前述の工程を続けることにより後処理操作を繰り返す。
【0103】
8.10 FmocAA17-26OHとHAA27-36NH2との液相カップリングによる断片FmocAA17-36NH2の調製
構造:
Fmoc-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号12)
理論収量:41.5g 期待収率:80〜85%
操作:
磁気攪拌棒を備えた2Lの丸底フラスコにFmocAA17-26OH(上記の第7.5節で合成)、HAA27-36NH2(上記の第8.9節で合成)、HOATおよびDMF(400mL)を仕込んだ。EtPr2Nを添加し、攪拌した溶液を窒素雰囲気下0〜5℃に冷却してHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めてさらに2.5時間攪拌した(注1)。水(500mL)を反応混合物に添加してペプチドを沈殿させた(注2)。得られたスラリーを45分間攪拌し、固体を減圧濾過によって回収し、水(100mL)で洗浄して乾燥させた。固体を磁気攪拌棒を備えた2Lの丸底フラスコに戻し、60℃の2-プロパノール(1.1L)を添加した。スラリーを室温まで冷ましながら窒素雰囲気下で攪拌した(一晩)。固体を減圧濾過によって回収して乾燥させると37.5gのFmocAA17-36NH2が90%の収率およびHPLCでは95.5A%の純度で得られた(注1)。
【0104】
注:
1. 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.75mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで25分間
保持時間:FmocAA17-26OH, 8.2分, HAA26-36 NH2, 8.4分
保持時間:FmocAA17-36NH2, 13.3分
2. 水を添加するときに反応混合物を38℃に温めた。
【0105】
8.11 断片H-AA(17-36)-NH2の調製
構造:
H-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-NH2 (配列番号19)
理論収量:21.6g 期待収率:95〜100%
操作:
磁気攪拌棒および窒素ブランケットを備えた250mLの丸底フラスコに上記の第8.10節で合成したFmoc断片、THF(約7.5容)、および5N NaOH(約2.5容)を仕込んだ。2相溶液が得られ、室温で10〜15分間攪拌した(注1)。
【0106】
層を分離し、有機相を1N HClでpH 2〜3に調整した。次いで溶液を2 X 55mL(2.5容)の飽和食塩水で洗浄した(注2)。層を分離し、有機層を減圧下、15〜20℃で元の容量の約2分の1になるまで濃縮した。ヘプタンをわけて(3 X 25mL)加えつつ、濃縮を続け、重沈殿点までおよび最終ポット容量が約100mLになるまでTHFを除去した(注2)。
【0107】
生成スラリーを室温で約2時間攪拌した。固体を減圧濾過によって単離し、約50mLのヘプタンで洗浄した。生成物を空気乾燥させると21.0g(97.5%)H-AA(17-36)NH2が得られた。
【0108】
残存しているジベンゾフルベン副生成物を除去するために再度後処理を行ってもよい。生成物を室温で3時間200mLのヘプタン中でスラリー化した。物質を濾過し、約50mLのヘプタンで洗浄して空気乾燥させると95%以上のHPLC純度の生成物が20.8g(96.6%)得られた。
【0109】
注:
1) 反応の完了はHPLCによってモニターした。
カラム:Zorbax LP C8; 1 OOオームストロング; 2011 流速:1mL/分
検出:260nmの紫外線
移動相:A: 0.1% TFA水溶液
B: 1:1 ACN:IPA中の0.05% TFA
20分間で80%B〜99%Bの勾配
保持時間:約18分
【0110】
2) 固体は最初は、ろう状ゴムとして沈殿するが、それは依然として攪拌可能であり、さらに濃縮しながら粉砕すると濾過可能な固体となる。
【0111】
9. 実施例:全長T-20ペプチドの合成
本明細書の下記の第9.1〜9.5節において提示されているのは、全長T-20ペプチドを産生するために中間体ペプチド断片を利用する実施例である。
【0112】
この節において提示された実施例は中間体ペプチド断片から全長T-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。
【0113】
9.1 AcAA1-16OHとHAA17-36NH2との液相カップリングによる断片AcAA1-36NH2の調製
ここに記載した合成経路は図1および3に図示したT-20の4つの断片からのアプローチの最終段階を表している。AcAA1-16OHが固相技術によって合成される場合には、図1のアプローチに従い、AcAA1-16OHが液相技術によって合成される場合には、図3のアプローチに従う。ここで合成されたT-20全長ペプチドはそのアミノ末端にアセチル基修飾(すなわち、「X」)およびそのカルボキシル末端にアミド基修飾(すなわち、「Z」)をもつ配列番号1のアミノ酸配列を有する。
【0114】
構造:
AC-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:1.77g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた100mLの丸底フラスコにAcAA1-16OH(固相技術により上記の第7.4節で、または液相技術により上記の第8.7節のいずれかで合成)、HOAT、DMF(20mL)を仕込み、続いてEtPr2N(0.074mL)を仕込んだ。溶液を窒素雰囲気下で0〜5℃に冷却してHBTUを添加した。溶液を0〜5℃で15分間攪拌し、HCl HAA17-36NH2(上記の第8.11節で合成)を添加し、続いてさらに0.041mLのEtPr2Nを添加した。冷却浴を外し、反応混合物を2時間攪拌した(注1)。ペプチドを沈殿させるために、水(25mL)および飽和食塩水(5mL)を添加した。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると1.74gの粗製AcAA1-36NH2が得られた(注2)。この固体を室温で2.5時間50% MTBE/ヘキサンとともに粉砕し、減圧濾過によって回収して乾燥させると96%の収率およびHPLCでは92A%の純度で1.70gが得られた。
【0115】
注:
1)工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:AcAA1-16OH, 11.8分
保持時間:HCl HAA17-36 NH2, 12.7分
保持時間:AcAA1-36NH2, 22.9分
2) 脱水によって非常に微細な沈殿が生成する。濾過を2回行うことが必要かもしれない。
【0116】
9.2 FmocAA1-16OHとHAA17-36NH2との液相カップリングによる断片FmocAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でFmocAA1-36NH2が合成されるまでを示している。
【0117】
構造:
Fmoc-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.91g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにFmocAA1-16OH(上記の第7.3節で合成)、HCl HAA17-36NH2(上記の第8.11節で合成)、HOAT、DMF(10mL)を仕込み、EtPr2Nを添加した。溶液を窒素雰囲気下で0〜5℃に冷却してHBTUを添加した。反応混合物を0〜5℃で15分間攪拌し、次いで室温まで温めて1.5時間攪拌した(注1)。ペプチドを沈殿させるために水を添加し、固体を減圧濾過によって回収して乾燥させた。固体を室温で15時間、2プロパノール(14mL)とともに粉砕し、次いで水(3mL)を添加して溶液から所望の生成物を得た。固体を減圧濾過によって回収して乾燥させると0.80gのFmocAA1-36NH2が88%の収率および85A%のHPLC純度で得られた。
【0118】
注:
1) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:FmocAA1-16OH, 11.4分
保持時間:HCl HAA17-36 NH2, 12.分
保持時間:FmocAA1-36NH2, 20.4分
TLC, 9/1 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rft: FmocAA1-36NH2, 0.71
9.3 断片HAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生するための固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でH-AA1-36NH2が合成されるまでを示している。
【0119】
構造:
H-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.77g 期待収率:85〜95%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにFmocAA1-36NH2(上記の第9.2節で合成)、DMF(9.5mL)およびピペリジン(0.5mL)を仕込んだ。溶液を窒素雰囲気下、室温で2時間攪拌した(注1、2)。水(20mL)および塩化ナトリウム飽和水溶液(5mL)を添加して保護されたペプチドを沈殿させた。固体を減圧濾過によって回収して乾燥させるとフルベンおよびピペリジン−フルベン付加物が混在した0.77gのHAA1-36NH2が得られた。固体を室温で15時間50% MTBE/ヘキサンとともに粉砕してフルベンおよびピペリジン−フルベン付加物を除去した。固体を減圧濾過によって回収して乾燥させると0.73gのHAA1-36NH2が95%の収率およびHPLCでは90A%の純度で得られた。
【0120】
注:
1) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:FmocAA1-36OH, 20.4分
保持時間:HCl HAA1-36 NH2, 19.9分
保持時間:フルベンおよびピペリジン−フルベン付加物, 5分
TLC, 9/1 ジクロロメタン/エタノール
紫外線、ヨウ素検出
Rt: FmocAA1-36NH2, 0.71
2) 生成物および出発原料はTLCまたは逆相HPLCでは十分に分離しない。生成物の形成の後、フルベンおよびピペリジン−フルベン付加物を測定する。
【0121】
9.4 HAA1-36NH2のN末端アセチル化による断片AcAA1-36NH2(T-20)の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に図示したT-20の3つの断片からのアプローチを表しており、その図の中でAc-AA1-36NH2が合成されるまでを示している。
【0122】
構造:
AC-Tyr(tBu)-Thr(tBu)-Ser(tBu)-Leu-Ile-His(trt)-Ser(tBu)-Leu-Ile-Glu(OtBu)-Glu(OtBu)-Ser(tBu)-Gln(trt)-Asn(trt)-Gln(trt)-Gln-Glu(OtBu)-Lys(Boc)-Asn(trt)-Glu(OtBu)-Gln(trt)-Glu(OtBu)-Leu-Leu-Glu(OtBu)-Leu-Asp(tBu)-Lys(Boc)-Trp(Boc)-Ala-Ser(tBu)-Leu-Trp(Boc)-Asn(trt)-Trp(Boc)-Phe-NH2 (配列番号1)
理論収量:0.71g 期待収率:85〜100%
操作:
磁気攪拌棒を備えた25mLの丸底フラスコにHAA1-36NH2(上記の第9.3節で合成)、DMF(10mL)およびピリジン(0.046mL)および無水酢酸(0.027mL)を仕込んだ(注1)。溶液を窒素雰囲気下、室温で4時間攪拌した(注2)。水(7.5mL)および塩化ナトリウム飽和水溶液(7.5mL)を添加して保護されたペプチドを沈殿させた。固体を減圧濾過によって回収し、水(10mL)で洗浄して乾燥させると0.65gのAcAA1-36NH2が91%の収率およびHPLCでは90A%の純度で得られた(注3)。
【0123】
注:
1) ジクロロメタンをこの反応の溶媒として使用してもよい。
2) 工程内管理, HPLC
Phenomenox Jupiter, C5, 5μ, 300オームストロング
0.8mL/分, 260nm
A H2O/0.05% TFA
B 50% IPA/MeOH/0.05% TFA
10分以上で80〜100%B、100%Bで15分間
保持時間:HAA1-36OH, 23.3分
保持時間:AcAA1-36 NH2, 22.7分
3) 生成物および出発原料はTLCまたは逆相HPLCでは十分に分離しない。
【0124】
9.5 AcAA1-36NH2の側鎖脱保護によるT-20側の調製
この節において提示された実施例は中間体ペプチド断片からT-20ペプチドを産生する固相および液相の首尾よくいったカップリング合成技術を実証する。特に、ここに記載した合成経路は図4に示したT-20の3つの断片からのアプローチを表している。
【0125】
構造:
Ac-Tyr-Thr-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lys-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Phe-NH2(配列番号1)
理論収量:157mg 期待収率:25〜50%
操作:
90:5:5(v/v/wt %)トリフルオロ酢酸/水/ジチオスレイトール溶液から窒素を脱気して0〜5℃に冷却した。冷却した溶液にAcAA1-36NH2(上記の第9.4節で合成)を添加した。固体が溶けるまで(約5分間)スラリーを0〜5℃で攪拌し、次いで室温まで温めて2.5時間攪拌した。溶液を0〜5℃のMTBE(70mL)に添加してペプチドを沈殿させた。スラリーを遠心分離機にて2200rpmで5分間回転させ、MTBEを固体からデカンテーションした。固体を再びMTBE(50mL)に懸濁させ、遠心分離機において2200rpmで5分間回転させ、MTBEをデカンテーションした。この工程をもう1度繰り返し、次いで固体を1容量%酢酸を含む1:1水/アセトニトリル(30mL)に溶かして室温で24時間保持した(注1)。溶液を凍結させ、次いで凍結乾燥機を用いて凍結乾燥させると155mgの粗製T-20が得られた。分取HPLCによる精製によって55mgの全長T-20ペプチドがHPLCでは95A%の純度で得られた(注2)。
【0126】
注:
1)Trp(Boc)のtBu側鎖を速やかに除去してTrpCOOHにする。TrpCOOHの脱カルボキシル化には、周囲温度、酢酸水溶液中において最低24時間を必要とする。
2)分取HPLC
2” YMC, 120A, 10μ, C18
220nm, 50mL/分
A H2O/0〜1% TFA
B ACN/0.1% TFA
39〜49% B/40分
【0127】
10 実施例:T-20ペプチドの精製
本明細書中に提示された実施例は、全体を通してペプチド精製を著しく増加させる条件下でT-20およびT-20様ペプチドを精製することができる方法を記載している。
【0128】
材料
使用カラム:Amberchrom CG-300S(Tosohaus; Montgomeryville, PA)の35μm粒子を充填した20 x 30cmのもの。
【0129】
バッファーの調製
バッファーA=NH4OHでpH 8.5に調整した100mM酢酸アンモニウム。
【0130】
バッファーB=アセトニトリル。
【0131】
1. 約6カラム容の20%Bを有するカラム。
【0132】
2. T-20を50〜100mLのペプチド1グラム当たり85%A/15%Bに溶かす。2M K2CO3でpHを8〜10に調整する。T-20サンプル中のアセトニトリル濃度は15〜20%を超えない。
【0133】
3. カラム圧をモニターしながら、T-20溶液を500mL/分でロードする。
【0134】
4. T-20溶液をロードした後に3カラム容(10L)の85%A/15%Bの溶液をロードしてラインを洗い落とす。
【0135】
5. カラム溶出液を303nmでモニターし、全ロード工程中の溶出液を回収する。
目盛り上にピークを維持するために実験中に波長またはアテニュエーションを調整する。吸光度は波長とセルの経路長との関数である。
【0136】
6. 全実験中に圧力をモニターしながら勾配操作を以下のように始める(B/時で1.6%の変化)。
【0137】
時間(分) %B 流速(mL/分)
0 15 330
788 36 330
7. 主要ピークが溶出し始めると、10分の画分を回収する(3.3L)。すべてのT-20が35%Bにおいて溶出するはずである。平均して35〜40の画分が回収される。プールする画分の決定がなされるまで画分を0〜5℃で貯蔵する。
【0138】
8. 検出器の吸光度が0.1AU未満になるまで、または主要ピークが溶出した後に主要変曲点が到達するまで画分を回収する。
【0139】
9. 各画分の純度は分析逆相HPLCによってモニターする。
【0140】
結果
T-20ペプチドは7より高いpH範囲で非常に可溶性が高い。ペプチドの精製に通常用いられるカラム支持体はシリカベースのものであり、シリカ支持体はより高いpH範囲で溶解する傾向にあるので、それゆえ一定のpH範囲でのみ使用することができる。
【0141】
本明細書中に記載された方法では、広いpH範囲で(pH 1〜14)安定なポリスチレンベースの樹脂支持体を利用する。この方法はカラム能力を著しく増加させ、ここでT-20の溶出量は10gから250〜450gまで増加した(直径8” x 30cmのカラムに関して)。
【0142】
11. 実施例:T-20ペプチド樹脂ローディングの大規模合成及び精製
FmocTrp(Boc)OHは、原料樹脂1グラムあたり1.2ミリモル以上のレベルで、非常に活性な2-CTC樹脂上にローディングすることができる。FmocTrp(Boc)OHの量が原料樹脂1グラムあたり1.1ミリモルを超えると(ローディングされた樹脂で1グラムあたり0.72ミリモルを超える量)、当該断片のアミノ酸のうち最後の3個について、ニンヒドリン試験が陰性となるのは難しい。同様に、FmocLeuO-樹脂を1グラムあたり0.85ミリモルより多くローディングすると、FmocAA17-26O-樹脂を作るのは困難になり、FmocGlnOH-樹脂を1グラムあたり0.75ミリモルより多くローディングすると、AcAA1-16O樹脂を作るのが難しくなる。
【0143】
ベンダーから樹脂を受領する際には、ローディングされるアミノ酸それぞれについて、2-CTC約1グラムで使用テストを行う。使用テストの目的は、ローディング中に使用されるアミノ酸量を決定して所望のローディング範囲を得ることにある。それぞれについて0.5当量過剰(報告された樹脂活性に対して)のDIEAと共に、0.8、1.0及び1.5当量のFmocGlnOH、1.0、1.2及び1.5当量のFmocTrp(Boc)OH、並びに0.8、1.0及び1.5当量のFmocLeuOHを使用する。2-CTC樹脂1キログラムに1モルのFmocLeuOHをローディングできない場合には、ベンダーから2-CTCを受け取るべきではない。以下、FmocLeuOH、FmocGlnOH及びFmocTrp(Boc)OHに対する標的樹脂について記述する。
【0144】
2-CTC樹脂には各キログラムあたり1.0〜1.1モルのFmocLeuOHをローディングすることができる。HClの損失を考慮すると、FmocLeuOHローディング後の樹脂乾燥重量は、原料樹脂重量の1.32〜1.35倍でなければならず、実測ローディングは1グラムあたり0.75〜0.81ミリモルでなければならない。
【0145】
2-CTC樹脂には各キログラムあたり0.8〜1.0モルのFmocGlnOHをローディングすることができる。HCLの損失を考慮すると、FmocGlnOHローディング後の樹脂乾燥重量は、原料樹脂重量の1.27〜1.33倍でなければならず、実測ローディングは1グラムあたり0.63〜0.75ミリモルでなければならない。
【0146】
2-CTC樹脂には各キログラムあたり0.9〜1.1モルのFmocTrp(Boc)OHをローディングすることができる。HCLの損失を考慮すると、FmocTrp(Boc)OHローディング後の樹脂乾燥重量は、原料樹脂重量の1.44〜1.54倍でなければならず、実測ローディングは1グラムあたり0.63〜0.72ミリモルでなければならない。
【0147】
重量増加を正確に解析するには、出発原料となる樹脂の乾燥損失(LOD)を測定しなければならない。LODは1%を超えてはならない。(ローディング後の)乾燥中、樹脂から完全に残留溶媒(すなわちDIEA/HCl)を除去しないと、単離される質量が多くなり、実測ローディングは低くなる。しかしながら、ローディングされる全モル数(質量×実測ローディング)は上記範囲内に収まるようにするべきである。上記アミノ酸のいずれに対してもローディング量(すなわち出発樹脂1キログラムあたりにローディングされる総モル数)の上限を10%以上超える場合には、より少ないアミノ酸を用いて使用テストをする。FmocGlnOHの場合、これは特に重要である。
【0148】
11.1 Fmoc-AA(Boc)-2-クロロトリチル樹脂の大規模製造
以下に続くセクションの目的は、固相ペプチド合成(solid phase peptide synthesis: SPPS)用のペプチド類を大量に製造するのに適した樹脂の大規模ローディングを行うことにある。以下のFmocTrp(Boc)OH、FmocGln(Boc)OH及びFmocLeu(Boc)OHそれぞれに樹脂を添加する。2-CTC樹脂約4キログラムを各出発残基に添加した。
【0149】
ローディングされた 2-CTC ローディング 置換 ローディング
AA (Kg) 後の質量(Kg) (mmol/g)1 総モル数1
FmocTrp(Boc)OH 3.7 4.493 0.56 2.52
(0.56) (2.52)
FmocLeu(Boc)OH 2.2 2.436 1.07 2.6
(0.647) (1.58)
FmocGln(Boc)OH 3.65 3.856 0.55 2.12
(0.52) (2.0)
1. 最初の数はFmoc-AA標準に対する重量ベースHPLCアッセイにより計算した。カッコ内の数値は出発物質2-CTC樹脂のLODが12%であることを考慮して、重量増加により算出した。
【0150】
出発の2-CTC樹脂はコロラド・バイオテク社(Colorado BioTech)から入手した。ローディングは1.4 meq/gと公表されている。1.5当量のFmocTrp(Boc)OH、DCM (5 vol)中1.7当量のDIEA、周囲温度で2時間という標準的なローディング手法を用い、当初の2-CTC量を3.7キログラムとした。こうしてFmocTrp(Boc)O-樹脂4.493キログラムを得たが、これは、樹脂開裂後のFmocTrp(Boc)OHの重量ベースHPLCアッセイによる0.56 mmol/gでの理論ローディングをもっていた(合計2.52モル)。製造した合計1.62モルについて、ローディングは樹脂の重量増加により1グラムあたり0.36ミリモルになると算出されている。しかしながら、合計2.52モルは樹脂4キログラムあたり4モルのFmocTrp(Boc)OHという目的のローディングからはほど遠いが、出発物質である2-CTC樹脂のLODが12%であることを考慮した場合、重量増加により算出されたローディングは0.56 mmole/gでは同じである。
【0151】
標準的なローディング法を用いた2実験で、2-CTC 3.9キログラムから合計4.59キログラムのFmocLeuO-樹脂を製造した。
【0152】
しかしながら、予想したよりも実質的に低いローディングが得られた。樹脂開裂後のFmocLeuOHを重量ベースHPLCによりアッセイしたところ、最初のバッチは1グラムあたり1.07ミリモルの理論ローディングを示した。出発物質2-CTC樹脂のLODが12%であることを考慮すると、重量増加による理論ローディングは1グラムあたり0.647ミリモル(500グラム)であるから、これは明らかに不可能である。ローディングされた合計1.577モルに対して実際のローディングは1グラムあたり0.647ミリモルに近いようである。
【0153】
2-CTC 1.7キログラムから製造した第2バッチのFmocLeuO-樹脂(2.154キログラム)は、0.68ミリモル/グラムの理論ローディングを有していたのに対し、重量増加ローディングは0.66ミリモル/グラムであった。
【0154】
0.8当量のFmocGlnOH及び7容量の5/2 DMF/DCM中1当量のDIEAを用い、3.65キログラムの2-CTCを出発物質として、シングルバッチ法によりFmocGlnO-樹脂を合計3.856キログラム作った。合計2.12モルのFmocGlnO-樹脂に対し、重量ベースHPLCアッセイによる理論置換量は1グラムあたり0.55ミリモルであった。出発2-CTC樹脂のLODが12%であることを考慮すると、合計2.0モルのFmocGlnO-樹脂に対し、重量増加による理論ローディングは、1グラムあたり0.52ミリモルである。
【0155】
一般に、重量増加によるローディングは、ローディングしたAAもしくは除去したフルベンについては、重量ベースHPLCアッセイによって算出されたローディングよりも大きいかまたは同等でなければならない。出発の2-CTCはそのLODが1%より小さくなければならず、単離されたFmocAAO樹脂は、少量のNMP、塩及び/または残留FmocAAOHを含む可能性がある。
【0156】
11.1.1 Fmoc-AA(Boc)-2-クロロトリチル樹脂の好ましい大規模製造法
FmocTrp(Boc)OH及びFmocLeuOH
空気感受性2-クロロトリチルクロリド樹脂(1当量、3.7キログラム、5.18モル)をSPPSチェンバーに入れ、DCM5容量で洗浄する。溶媒を除去し、FmocTrp(Boc)OHもしくはFmocLeu(Boc)OH(1.5当量)溶液及びDCM (5容量)中DIEA(1.7当量)を加える(注1)。このスラリーを2時間撹拌しながらかき混ぜる。溶媒を除去した後、樹脂に残留する活性部位を30分間9:1 MeOH:DIEA (5容量)でエンドキャッピングする。溶媒を除去した後、樹脂を5容量のDCMで6回洗浄する。恒量になるまで樹脂を乾燥した後、これをサンプリングし、ローディングについて分析する(注2)。同じ方法を用いてFmocLeuOHをローディングする。
【0157】
FmocGluOH
窒素雰囲気下、空気感受性2-CTC(3.65キログラム、1.4ミリモル/グラム)樹脂を40LのSPPSチェンバーに入れ、DCM(5容量)で洗浄する。20Lの反応容器にFmocGlnOH(1.506キログラム、0.8当量)、DMF (5容量)及びDIEA(1.0当量)を入れる。SPPSチェンバーをドレーンし、DMF溶液を添加する。DCM(2容量)で反応容器を洗浄し、洗液はSPPSチェンバーに加える。窒素雰囲気下2時間SPPSチェンバーで樹脂を撹拌した後、ドレーンを行う。樹脂に残留する活性部位はすべて30分間9:1 MeOH:DIEA (5容量)でキャッピングする。樹脂をドレーンした後、DCM(5容量、6回)で洗浄する。恒量になるまで樹脂を乾燥した後(3.856kg)、これをサンプリングし、ローディングレベルについて試験する(注2)。
【0158】
注:
1. 2-クロロトリチルクロリド樹脂は湿度には非常に感受性が高い。水は樹脂の表面を覆うとHClを与える。溶媒が乾燥している限り、DCEとDCMとの間にはほとんど差がない。
【0159】
2. 樹脂ローディングは樹脂からFmocアミノ酸を開裂し、定量wt/wt HPLC分析により標準に対してアッセイすることにより決定する。
【0160】
フェノメネックス・ジュピターC18、300A、Sm
1 mL/min、260 nm
A: 0.1% 水性TFA
B: アセトニトリル中0.1% TFA
65%B、アイソクラチック(isocratic)
保持時間: 約8分
樹脂乾燥前の洗浄回数が不十分で樹脂からNMPを完全に除去できない場合には、樹脂の重量増加によるローディング量の測定は不正確になることがある。計算は以下の通りである。
【0161】
(Fmoc-AA-OH樹脂質量−出発2-CTC質量)/Fmoc-AA-OH樹脂質量(ローディングされたAAの分子量−HClの分子量)
DCM中長期間(日)本発明のペプチド断片を保存すると、樹脂から実質的な開裂が起こることが観察された。例えば、FmocAA17-26OHを構築する際、部分的に構築された断片をDCM中で2度週末にかけて保存した。合成を終了して樹脂から取り出した後、8%の収率でFmocAA17-26OHが得られた。週末にかけての保存中、DCMに存在する微量のHClが、部分的に構築された断片を樹脂から徐々に開裂したのではないかと考えられる。
【0162】
FmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂のサンプルを室温で1週間DCM及びIPA中に放置した。各樹脂の上清をHPLCにより分析した。定量的な結果は得られなかったが、DCM中ではすべての断片が有意な程度まで樹脂から開裂したのに対し、IPA中では開裂は観察されなかった。部分的に構築された断片を2〜3日(週末にかけて)保存する必要がある場合には、固形物をNMPで洗浄し、ベッドをドレーンして窒素雰囲気下NMPで飽和させておいた方がよい。
【0163】
FmocLeuO-樹脂、FmocGlnO-樹脂、FmocTrp(Boc)O-樹脂、FmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂を最終洗浄するため、DCMをIPAで置換した。IPAで飽和したFmocLeuO-樹脂、FmocGlnO-樹脂及びFmocTrp(Boc)O-樹脂を40℃で乾燥した。4℃で乾燥したそれらローディング樹脂からFmocAA17-26O-樹脂、AcAA1-16O-樹脂及びFrmoAA27-35O-樹脂を構築した。合成終了時に、樹脂結合断片をきれいになるまでDOMで、次にIPAで洗った。IPAで飽和した樹脂結合断片を分けて周囲温度及び40℃で乾燥した。断片を樹脂から開裂し、HPLCにより分析した。断片の純度に差は観察されなかった。IPAで飽和した樹脂結合断片を40℃で乾燥した場合、乾燥は単離された断片の性質や量には影響しない。
【0164】
NMP/DCMに対しDMF中で断片のSPPSを調べた(樹脂1グラムは両溶媒系で5 mLに膨潤する)。溶媒としてDMFを用い、カップリング試薬としてTBTU対HBTUを使用してFmocAA27-35OH断片を合成した。収率77%、純度89.5A%でFmocAA27-35OHを単離した。
【0165】
FmocAA17-36NH2断片及びAcAA1-36NH2断片の仕上げ(work up)手順によりカップリングしなかった断片を除去する。固相合成を行う間、これらの断片を欠失部位でアセチル化(エンドキャッピング)すると、欠失断片は確実に溶液相カップリングでカップリングしなくなる。FmocAA17-36NH2及びAcAA1-36NH2両断片の合成中カップリングしなかった断片はIPA及びACN仕上げの際に除去しなければならない。
【0166】
11.2 AcAA(1-16)-OH断片(3c断片)の大規模製造
SPPSによるAcAA1-16-OH断片の大規模製造を2回行って、FmocGlnOH-樹脂3.53キログラムから合計7.941キログラムの樹脂結合断片を得た。2回とも出発物質であるFmocGlnOH-樹脂のローディング値は、重量ベースHPLCアッセイにより得られた若干過大な0.55ミリモル/グラムという値であり、むしろ出発物質2-CTCの12%LODを考慮した重量増加により得られる0.52ミリモル/グラムのほうがより正確な値である。望ましい値より低い樹脂ローディングを持つほか、アミノ酸はSPPSの間それぞれ6%過剰量を使用した。
【0167】
SPPSでは樹脂に第1のFmocGln(trt)OHを2.5当量使用し、断片において次に続くアミノ酸についてはそれぞれ1.7当量を使用した(0.55ミリモル/グラムローディングに対し)。8容量の3:1 NMP:DCM中でカップリング反応を行ったところ、32カップリング反応(アセチル化を含む)の内2例を除いて残り全部の反応は2時間以内に完了した。洗浄はすべて5容量のNMPで行った。
【0168】
3.4容量(乾燥AcAA1-16O-樹脂の重量に対し)の1% TFA/DCM中0〜5℃で、1洗浄あたり5分間ずつ5、6回、樹脂結合断片を洗浄する。各洗液を1.36容量の0.33 M NaHCO3水溶液に集めてTFAを中和する。3相洗液を合わせて水(2容量)を加える。DCMを溜去すると重炭酸ナトリウム水溶液中に濾過可能な沈澱が残る。蒸留する間に多相スラリーは粘稠となってクリーム状を呈した後、DCMが最後まで除去されると濾過可能なスラリーになる。このスラリーを0〜5℃まで冷却し、pHを0.1 N HClで3.0に調整する。スラリーを0〜5℃で1〜2時間撹拌し、濾過して集め、洗浄して乾燥する。続く実験で固形物を集め、反応容器に戻し、水で1〜2時間トリチュレーションした後、集めて乾燥する。
【0169】
DCM中1% TFAを用い、4実験で樹脂からのAcAA1-16OH開裂を行った。3.53キログラムのFmocGlnO-樹脂から合計4.814キログラムのAcAA1-16OHが93〜96A%の純度で得られた。出発物質である2-CTC(理論量5.83キログラム)のLODが12%であることを考慮して重量増加により決定した0.52ミリモル/グラムのローディングにもとづく総収率は83%である。
【0170】
AcAA1-16OHのナトリウム塩はDMFにもNMPにも不溶であるので、カルボキシル末端をプロトン化するにはpH調整が必要である。水洗により生成物から確実にトリフルオロ酢酸ナトリウムを除去する。wt/wtベースでごく少量のトリフルオロ酢酸ナトリウムが存在すると、HAA17-36NH2との溶液相カップリングは妨害される。
【0171】
この断片は周囲温度で乾燥しなければならない。湿ったAcAA1-16OHを40℃で乾燥する安定性検査を行ったところ、3日間で約4%の分解を示した。興味深いことに、乾燥した固形物は80℃で24時間安定である。
【0172】
11.2.1 Ac-AA(1-16)-OH断片(3c断片)の好ましい大規模製造法
40 Lのペプチドチェンバーに樹脂結合FmocGlnOH(1.53キログラム、0.55ミリモル/グラム、0.84モル)を加える。15分間撹拌しながら、窒素下でDCM(5容量)により樹脂のコンディショニングを行った後、ドレーンする。20%ピペリジンNMP溶液(5容量)を加え、得られた懸濁液を窒素下で20分間撹拌する。溶液をドレーンし、このプロセスを繰り返す。5容量のNMPで5回樹脂を洗浄してジベンゾフルベンを除去し、クロロアニル試験により判定されるピペリジンを除去する(注1)。
【0173】
樹脂を洗浄してピペリジンを除去しながら、配列中の次のアミノ酸(1.5当量)、HOBT (1.5当量)及びDIEA(1.5当量)をNMP(6〜7容量)中で混合して0℃に冷却する。冷却した溶液にHBTU (1.5当量)を加え、溶液を10〜15分間撹拌してHBTUを溶解する。活性アミノ酸の冷却溶液を樹脂に加えた後、DCMで洗浄する(2.5容量)(注2)。窒素パージ下で2時間、懸濁液を撹拌する。その後、定性ニンヒドリン試験用に樹脂サンプルを取り出す(注3)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄する(注4)。そして配列中の次のアミノ酸についてこのサイクルを繰り返す。ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、アミノ酸1当量と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性になれば、NMP(10容量)中5当量(Seq)の無水酢酸と5当量(Seq)のピリジンで1時間エンドキャッピングを行う。
【0174】
断片合成終了後、記載したように最後のアミノ酸からFmocを除去した後、3:1 NMP:DCM(10容量)における無水酢酸とピリジン(それぞれ5当量)の溶液中で、20〜30分間、もしくは、ニンヒドリン試験が陰性になるまで、樹脂を撹拌する。樹脂をドレーンした後、5容量のNMPで2回、5容量のDCMで5回洗浄して乾燥すると、3.49キログラムのAcAA1-16O-樹脂が得られる。
【0175】
40 LのSPPSチェンバーに乾燥した樹脂結合AcAA1-16(3.49キログラム)を入れる。6 x 3.4容量の1% TFA/DCM を用いて、樹脂結合ペプチドをその樹脂から開裂する(注7)。各開裂洗液を1.36容量の0.33 M 重炭酸ナトリウム水溶液に集める。2相フラクションを合わせて2容量の水で希釈する。減圧下(15Hg、20℃)2相混合物を濃縮してDCMを除去する。撹拌を続けるのに必要であれば蒸留中さらに水(2容量)を加える(注6)。DCMを除去する場合(数時間)には、懸濁液を0℃に冷却し、1N HCl水溶液でpHを3に調整する。スラリーは0℃で1時間撹拌した後集める。なお湿っている固形物は反応容器に戻し、水(7容量)でトリチュレーションして残っているTFAとトリフルオロ酢酸ナトリウムを除去する。減圧濾過により固形物を集めて恒量になるまで乾燥する(1.12キログラム、95A%)。0.52ミリモル/グラムローディングに基づくAcAA1-16OHの収率は86%である(注7)。
【0176】
注:
1. アセトン約1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。ベッド高が高くなればなるほど、ピペリジンを洗い流すのにはさらに大量のNMPが必要になる。
【0177】
2. 樹脂の適当な膨潤を確実にするため、DCMを添加する。
【0178】
3. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出し、メタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。定量ニンヒドリン試験方法の文献: Sarin, V.K., Kent, S.B.H., Tam, J.P., & Merrifield, R.B. (1981) Analytical Biochem. iii, 147-157。
【0179】
4. 樹脂を1昼夜保存する場合には、5容量のNMPで2回洗浄した後、ドレーンして窒素下に保存のこと。DCM下に保存してはならない。この場合樹脂からペプチドが開裂して実質的な量損失を生じる。
【0180】
5. 各フラクションについては、254 nmでTLC可視化により生成物の内容を調べる。生成物の大部分は最初の5回洗浄により除去される。
【0181】
6. DCMを除去するにつれ、反応物はマシュマロクリームの固さを呈するようになる。蒸留を続けると、懸濁液は徐々に固いスラリーへと変わっていく。生成物からオイルが出るのを防ぐためDCMを徐々に除去することが重要である。
7. バイダク(Vydac) C8、5μ、300A
1 mL/min、30℃、230 nm
A 1000:1 水/TFA
B 800:200:1 IPA:ACN:TFA
60-95%B/30 min
保持時間: 13.1分
【0182】
11.3 FmocAA(17-26)-OH断片(10b断片)の大規模製造
同等サイズの2実験でFmocLeuO樹脂2.4キログラムからFmocAA17-26O-樹脂を合計5.3キログラム作った。同等サイズの4実験で、この樹脂からFmocLeuO-樹脂5.3キログラムを開裂したところ、平均して純度94.4A%、収率90%のFmocAA17-26OHが3.184キログラム得られた。
【0183】
ローディングファクターに対しアミノ酸をそれぞれ1.5当量用いて反応を行ったが、各カップリングサイクルについて65%過剰のアミノ酸を使用する結果となった。カップリング反応はすべて2時間以内に完了した(ニンヒドリン試験陰性)。
【0184】
20%ピペリジンNMP(5容量)に20分間保ってFmoc保護基を外し、これを繰り返した。5容量のNMPで5回洗ってピペリジンを樹脂から洗い流した。カップリング反応は8容量の3:1 NMP:DCM中で行った。カップリング溶液を除去し、固形物を5容量のNMPで3回洗浄した。配列の次のアミノ酸について、このサイクルを繰り返した。断片完了後、樹脂ベッドを5容量のNMPで2回、5容量のDCMで5回洗った後、恒量になるまで乾燥した。
【0185】
樹脂から断片を除去するため、DCM中1% TFAで数回洗浄する。1% TFAのDCM溶液は冷却する必要はない。この断片は1% TFA/DCM(1%/日)中ではAcAA1-16OHよりもはるかに安定だからである。生成物含有酸性DCM洗液を集め、ピリジンで中和する。合わせた洗液を蒸留してDCMを除去し、エタノールを添加して溶液を維持すると共に、蒸留する間に残留DCMを追い出す。DCMを除去した後(蒸留物が除去される温度の上昇によって判断する)、水を加えて断片を沈澱させる。減圧濾過により固形物を集める(60分以内)。なお湿っている固形物は反応容器に戻し、80/20のエタノール/水(5容量)で60分間0〜5℃でトリチュレーションする。減圧濾過により沈澱したFmocAA17-26OHを集め、最小量の80/20エタノール/水で洗浄して乾燥する(減圧、加熱なし、10日間)。
【0186】
11.3.1 FmocAA(17-26)-OH断片(10b断片)の好ましい大規模製造法
40 LのSPPSチェンバーにFmocLeuO-樹脂(1.2キログラム、0.776モル)を入れた後、DCM(5容量)で樹脂を膨潤させる。DCMは乾燥した原料樹脂を確実に完全膨潤させるのに必要である。懸濁液を20〜30分間撹拌した後、液体を除去する。20%ピペリジンNMP溶液を加え(5容量)、溶液を20分間撹拌する。溶媒を除去した後、このプロセスを繰り返す。溶媒を除去し、樹脂ベッドを5容量のNMPで5回洗浄してピペリジンを除去する(注1)。
【0187】
脱保護しながら、配列中の次のアミノ酸(1.95当量)、HOBT (1.95当量)、DIEA(1.95当量)及びNMP(6容量)を機械的撹拌機を備えた20リットルの丸底フラスコに入れる。固形物が溶解するまで溶液を撹拌し、次いで、0〜5℃に冷却して、HBTU (1.95当量)を加える。HBTUが溶解するまで溶液を撹拌するか、またはピペリジンを含まない場合には樹脂を撹拌(どちらが最初でもよい)した後、樹脂に加える。反応容器をDCM(2容量)で洗浄し、これをSPPS反応容器へ移す。注: アミノ酸と試薬の化学量論は1.5当量でなければならない。
【0188】
穏やかに撹拌しながら、樹脂をカップリング溶液に懸濁する。2時間後、樹脂試料をSPPSチェンバーから取り出して定性ニンヒドリン試験を行う(注2)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄する。そして配列中の次のアミノ酸についてこのサイクルを繰り返す(DCMによる膨潤はローディングされた最初のアミノ酸についてだけ行う)。
【0189】
ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、1当量のアミノ酸と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性の場合、NMP(10容量)中の無水酢酸5当量とピリジン5当量で1時間エンドキャッピングを行う。
【0190】
断片合成終了後、樹脂をドレーンし、5容量のNMPで2回、5容量のDCMで5回洗浄した後乾燥すると、2.67キログラムのFmocAA17-26O-樹脂が得られる。
【0191】
1.7容量のDCM中1% TFAを、1回5分間で5〜6回用いて、樹脂(1.33キログラム)からFmocAA17-26OHを開裂する。1% TFA/DCM洗液をピリジン含有フラスコに集める(洗液中のTFAについては容量比1:1)。生成物含有洗液を合わせ(約14リットル)、最小ポット容量になるまでDCMを溜去する(元の容量の約3分の1)。減圧度を調整してポット温度を15〜25℃に保つ。エタノール(6.5容量)を加え、DCMがなくなるまで蒸留を続ける(蒸留物の温度によって判断する;注3)。再び減圧度を調整してポット温度を15〜20℃に保つ。最終ポット容量は約8〜10容量でなければならない。溶液を5〜10℃に冷却し、30分間にわたって水(6.5容量)を加え、FmocAA17-26OHを沈澱させる。減圧濾過により固形物を集めて水洗する(2〜3容量)。残存するピリジン及び/又は塩を除去するため、なお湿っている固形物は反応容器に戻し、予め0℃まで冷却した80/20エタノール/水(5容量)を加える。懸濁液を60分間0℃で撹拌する。減圧濾過により固形物を集め、最小量の80/20エタノール/水で洗浄し、恒量になるまで乾燥すると、収率90.6%、純度96A%のFmocAA17-26OHが0.806キログラム得られる(注4)。
【0192】
注:
1. アセトン1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。
【0193】
2. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出しメタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。
【0194】
3. DCMの減圧蒸留中の頭部温度は10〜15℃であった。DCMを除去すると、頭部温度は3500まで上昇した。
4. バイダクC8、5μ、300A
1 mL/min、262 nm、30℃
A 水/0.1% TFA
B ACN/0.1% TFA
80-99%B/20 min
保持時間: 15.2分
【0195】
11.4 FmocAA(27-35)-OH断片(16b断片)の大規模製造
FmocTrp(Boc)O-樹脂4.45キログラムから合計4.694キログラムのFmocAA27-35-OHを合成した。固相合成は2個のバッチで行い、樹脂からの開裂は4バッチであった。1バッチから得られたFmocAA27-35-OHは他のバッチから得られた物質よりも約5%純度が低かった。これはFmocAA17-36-NH2へ処理する際に除去された、同定されていない不純物5A%によるものであった。樹脂にローディングされたFmocTrp(Boc)OHの合計量は2.5モルであった。約63%の容量でローディングされた樹脂について予想される通りに、固相合成が進行した。カップリングはすべて最初の2時間のチェックポイントで完了した。SPPS及び開裂に使用した反応及び洗浄量はFmocAA17-26OH合成に使用した量と同じであった。樹脂からの開裂は上記の通りであった。濾過は迅速であった(15分)。90/10エタノール/水トリチュレーション後の固形物乾燥には3日間かかった(減圧、加熱なし)。断片は40℃の乾燥にも安定である。
【0196】
11.4.1 Ac-AA(27-35)-OH断片(16b断片)の好ましい大規模製造法
FmocTrp(Boc)-樹脂(1当量、2.2キログラム、1.23モル)を含有するSPPSチェンバーにDCM(5容量)を加える。得られた懸濁液を15分間撹拌した後ドレーンする。20%ピペリジンのNMP溶液(5容量)を加え、得られた懸濁液を10〜15分間撹拌してFmoc保護基を外す。このプロセスを繰り返した後、クロロアニル試験が陰性を示すまで、樹脂をNMP(5容量)で5〜7回洗浄する(注1)。
【0197】
NMP(6容量)中で、次に続くアミノ酸(1.5当量)、HOBT (1.5当量)及びDIEA(1.7当量)を合わせ、0〜5℃に冷却する(注2)。HBTU を加え、溶液を10〜15分間撹拌してHBTUを溶解する。活性アミノ酸の溶液を樹脂に加える。DCMで反応容器を洗浄した後樹脂に加える(注3)。窒素雰囲気下1〜2時間この懸濁液を撹拌する。カップリングが完了したかどうかは定性ニンヒドリン試験によりモニターする(注4)。ニンヒドリン試験が陰性であれば、反応容器をドレーンし、5容量のNMPで3回洗浄し、配列中の次のアミノ酸についてこのサイクルを繰り返す(ローディングされた最初のアミノ酸についてのみDCMによる膨潤を行う)。
【0198】
ニンヒドリン試験が陽性である場合には、懸濁液をさらに1時間撹拌して再試験を行う。ニンヒドリン試験が陰性であれば、次のサイクルに進む。ニンヒドリン試験がなお陽性の場合には、1当量のアミノ酸と試薬との再カップリングを行う。1時間後ニンヒドリン試験が陽性になれば、NMP(10容量)中で1時間無水酢酸5当量とピリジン5当量のエンドキャッピングを行う。
【0199】
断片合成終了後、樹脂をドレーンし、5容量のNMPで2回、5容量のDCMで5回洗浄して乾燥すると、4.11キログラムのFmocAA27-35O-樹脂が得られる。
【0200】
樹脂から断片を開裂するには、1.7容量のDCM中1% TFAを用い、1回5分間で6回、樹脂(2.05キログラム)から樹脂を開裂する。1% TFA/DCM洗液をピリジン(洗液中のTFAに対して容量比1:1)含有フラスコに集める。生成物含有洗液を合わせ、元のポット容量の約半量になるまでDCMを溜去する(ポット温度は減圧度を調整して約15℃に保つ)。徐々にエタノール(5容量)を加え、DCMがなくなるまで(蒸留物の温度によって判断する。注5)蒸留(減圧度を調整してポット温度を約20℃に保つ)を続ける。ポット容量は約6〜7容量でなければならない。濁った溶液を10〜15℃に冷却し、素早く撹拌しながら30分間にわたって水(3.5容量)を加えると、FmocAA27-35-OHが沈澱する。減圧濾過(15分間)により固形物を集めて水(1容量)で洗浄する。残留ピリジンを除去するため、湿っている固形物を反応容器に戻し、予め0〜5℃に冷却した90/10エタノール/水(10容量)を加える。スラリーを0〜5℃で60分間撹拌する。減圧濾過により固形物を集め、90/10エタノール/水(0.5容量)で洗浄し、恒量になるまで乾燥すると、収率89%、純度89.2A%のFmocAA27-35-OHが1.19キログラム得られた(注6)。
【0201】
このプロトコールは、12時間撹拌しながら固形物を9:1エタノール/水15容量でトリチュレーションした後、集めて乾燥することにより再処理することができる。
【0202】
注:
1. アセトン1ミリリットルにクロロアニルのトルエン飽和溶液1滴を加えた後、流出液を1滴加える。青もしくは紫色はピペリジンの存在について陽性であることを示す。
【0203】
2. 溶解を補助するためHBTUを除いた試薬を室温で添加する。HBTUを冷却溶液に加えるとラセミ化が低減する。
【0204】
3. HBTUはDCMに溶けないため、活性化はNMP中で行う。DCMは反応容器を洗浄するのに使用されると同時に、適当な樹脂膨潤を維持するため樹脂に添加される。
【0205】
4. カップリング効率をモニターするには定量ニンヒドリン試験が適切である。樹脂試料2〜20ミリグラムを取り出し、メタノールできれいになるまで洗浄する。試料に76%フェノールのエタノール液を3滴、0.2 mM KCNピリジン液を4滴及び0.28 Mニンヒドリンのエタノール液を3滴加える。溶液をエタノールで0.5〜1ミリリットルに希釈し、5〜10分間75℃の加熱ブロックに置く。青もしくは紫色は遊離アミンを示す(陽性)。澄んだ青色または淡青色は陰性の結果を示す。
【0206】
5. DCMを減圧蒸留する間の頭部温度は10〜15℃に保った。DCMが除去されると頭部温度は35℃まで上昇した。
6. バイダクC8、5μ、300A
1 mL/min、262 nm、30℃
A 水/0.1% TFA
B ACN/0.1% TFA
80-99%B/20 min
保持時間: 15.3分
【0207】
11.5 FmocAA(27-35)-OHとHPheNH2の固相カップリングによるFmocAA(27-36)-OH断片の大規模製造
4.676キログラムのFmocAA27-35-OHから4実験で合計5.226キログラムのFmocAA27-36NH2を作った。残留するカップリング試薬もしくは溶媒は収率100%以上になる。これらは次の段階で除去される。
【0208】
水のドロップアウト後反応容器壁に付着している固形物はこの段階で重要な問題となった。減圧濾過により粗固形物を分離するのに30分を要した。これら実験の平均乾燥時間(減圧、加熱なし)は8日間であった。固形物はオーブンへ入れる前に濾紙上で圧縮して過剰の水を除去する必要がある。この断片は40℃の乾燥に安定であり、減圧オーブンは加熱する必要がある。
【0209】
使用テストは、使用する出発物質1ロットあたりのバッチ0.5〜1グラムについて行い、性質及び同一性を確定する助けとする。薄層クロマトグラフィ(TCL)及びHPLCを用いて出発物質から生成物への変換をモニターする。FmocAA27-35OH断片において探さなければならない最初の不純物はトリフルオロ酢酸(またはその塩)である。FmocAA27-35OHに存在するトリフルオロ酢酸のフェニルアラニンアミドによる活性化及び反応は迅速なプロセスである。存在するトリフルオロ酢酸を消費するのに小過剰のフェニルアラニンアミドを使用することができる。さらに重要な性質の問題は購入したフェニルアラニンアミドについてである。ほとんどのベンダーはこれを塩酸塩として販売する。フェニルアラニンアミド塩酸塩のロットのうちいくつかでは、HPLCの際出発物質と一緒に共溶出する不純物(5〜15A%)が生成している。この不純物はTLCによって出発原料断片及び生成物から分離することができる。フェニルアラニンアミド塩酸塩に存在する塩化アンモニウムから生じるFmocAA27-35NH2は不純物である。
【0210】
11.5.1 FmocAA(27-35)-OHとHPheNH2の液相カップリングによるFmocAA(27-36)-OH断片の大規模製造
機械的攪拌機を備えた40リットルのジャケット反応容器にFmocAA27-35OH(1当量、1.185キログラム)、HOAT (1.1当量)、HCl.HPheNH2(1.15当量)及びDMF(12.5容量)を入れる。DIEA (2.1当量)を加え、溶液を0〜5℃に冷却してHBTU(1.2当量)を添加する。反応混合物を0〜5℃で15分間撹拌した後、室温まで加温し、さらに70分間撹拌する(注1、プロセスチェックのためHPLCを用いた)。HPLCにより反応が完了したと判断できた後、水(12.5容量)を15〜30分かけて加えるとペプチドが沈澱する。スラリーを周囲温度で15分間撹拌する。減圧濾過によって固形物を集め、水(3容量)で洗浄後乾燥すると、FmocAA27-36NH2が得られる(収率107%で1.357キログラム、HPLCによる純度87.1A%)(注2)。
【0211】
12時間撹拌しながら固形物を9:1アセトニトリル/水15容量でトリチュレーションした後集めて乾燥することにより、再度反応を行うこともできる。
【0212】
注:
1. プロセス制御において、TLC:
88/12ジクロロメタン/メタノール
UV、ヨード検出
Rf: FmocAA27-35OH、0.49
Rf: FmocAA27-36NH2、0.63
バイダクC8、5 m、300A
30℃、1 mL/min、262 nm
A 水/0.1% TFA
B ACN/0.15 TFA
80-99%B/20 分
保持時間: FmocAA27-35OH、15.8
保持時間: FmocAA27-36NH2、17.12
2. 一般に、単離された固形物を反応容器に戻して水でトリチュレーションをしない限り、得られる収率は理論収率より大きい。
【0213】
11.6 FmocAA(27-36)OHからのHAA(27-36)-OH断片の大規模製造
5.221キログラムのFmocAA27-36NH2から平均2段階収率86.4%で、4実験で合計3.897キログラムのHAA27-36NH2を製造した。これらの実験で観察された収率の範囲、74〜97%は1実験から次の実験への持ち込みを反映していると考えられる。
【0214】
この段階では製造も生成物単離も非常にうまくいく。正確な量のDCMがMTBEに残っており、固形物の物理的な性質が迅速な濾過・乾燥をもたらす。この生成物単離法は、以下のセクション11.6.2に記載の新段階に組み込まれている。
【0215】
DCMは、FmocAA27-36NH2の脱保護の際に使用する溶媒である。脱保護の完了時に、有機溶液を水で2回洗浄することにより、過剰ピペリジンのほとんどを除去し、次に、元の容量の約1/3に濃縮する。MTBEを添加して、フラグメントを沈殿させる。蒸留を続けることにより、残ったDCMの大部分を除去し、フラグメントの沈殿を完了する。沈殿中の質量損失を防止する上で、DCMのほとんどを除去することが重要である。また、生成物の清浄化を確実にするために、MTBE中にDCMをいくらか残すことも重要である。ジベンゾフルベン、ピペリジン/フルベン付加物および残留ピペリジンは、MTBEに可溶性である。HAA27-36NH2を回収し、乾燥させる。必要であれば、MTBEを用いた該固体の2次粉砕(12時間)により、残留ジベンゾフルベン、およびさらに重要なことにはHAA27-36NH2に存在する可能性のあるピペリジン/フルベン付加物を除去する。ヘキサンを用いた粉砕では、ジベンゾフルベンは除去されるが、ピペリジン/フルベン付加物は除去されない。MTBEから単離した固体の乾燥時間は、1日である。
【0216】
HAA27-36NH2およびDCM/MTBE中の関連不純物の高い可溶性のため、分析方法には、溶媒交換の終点を正確に決定することが求められた。MTBE中のDCMを定量するために、GC法が開発されている。蒸留は、MTBE中のDCM含有率が、6%を下回る時点で、完了する。
【0217】
11.6.1 FmocAA(27-36)-OHからのフラグメントHAA(27-36)-OHの大規模製造
撹拌機と温度計を備えた40Lのガラスジャケット付き反応器に、FmocAA27-36NH2(1当量、1.356kg)、DCM(5容)およびピペリジン(0.2容)を導入する。該溶液を周囲温度で1.5時間撹拌する(注1)。有機溶液を水(2x5容)で洗浄する。蒸留(約25mmHg、尚、固体の融解を防ぐため、ジャケット温度は45℃未満)により、上記有機層の容量を元の有機容量の約1/3に減少させる。MTBE(5容)を反応器に徐々に導入しながら、重沈殿点まで濃縮を続け、ポット容量を約5容とする。DCM含有率が6%を下回った時点で、溶媒交換を停止する。該スラリーを0〜5℃に冷却し、60分間撹拌した後、減圧濾過(急速)により回収する。固体をMTBE(2x0.5容)で洗浄し、恒量まで乾燥させて、1.022kgのHAA27-36NH2を得た。収率は89.2%(2ステップ)で、純度は、91.1A%であった(注1)。
【0218】
MTBE(12.5容)を用いた、23℃、13時間の粉砕により該反応を再実施し、減圧濾過および乾燥により、回収してもよい。
【0219】
注:
1.HPLCにより測定するIPCおよび純度:
Vydac、C8、5、300A
1mL/分、262nm、30℃
A 水/0.1%TFA
B ACN/0.1%TFA
80〜99%B/20分
保持時間:FmocAA27-36NH2、16.5, HAA27-36NH2、8.4
11.6.2 FmocAA(27-36)-OHとHPheNH2の液相カップリングによる、フラグメントHAA(27-36)-OHの改良された製造
FmocAA27-35OHから直接HAA27-36NH2を得る第11.5節から第11.6.1節を組み合わせた新しい方法が開発されている。この新しい方法は、FmocAA27-36NH2の単離に付随する問題と長い乾燥時間を解消する。以下に、詳しく説明する。この新しい方法は、合成工程から、長い乾燥時間と、一段階を排除する。
【0220】
磁気撹拌器および窒素供給口を備えた100mLの丸底フラスコに、FmocAA27-35OH(5.0g、1当量)、HOAT(0.459、1.2当量)およびPhe-NH2(0.389、1.2当量)を導入する。該フラスコに、10容のNMP(50mL)とDIEA(0.45g、1.5当量)を添加し、窒素雰囲気下で、固体が溶解するまで撹拌する。該溶液を0〜5℃に冷却した後、HBTU(1.04g、1.2当量)を添加する。窒素雰囲気下で、0〜5℃にて30分間撹拌する。反応混合物を20℃まで暖め、撹拌を継続する。HBTUを添加してから90分後に、プロセス中検査(IPC)を実施する。
【0221】
プロセス中検査(IPC)により、反応の完了が明らかにされた後、反応混合物にピペリジン(1.379、7当量)を添加し、窒素雰囲気下で1.5時間撹拌する。IPCを実施する(注1)。Fmoc除去が完全ではない場合には、さらに30分間撹拌し、IPCを実施する。
【0222】
完全であれば、この反応混合物を、温度が35℃を超えないような速度で、5%酢酸水溶液に添加する(30容)。得られたスラリーを1〜2時間撹拌し、減圧濾過により回収する(注2)。その固体を10容(50mL)の水で洗浄する。
【0223】
湿った固体を反応器に戻し、20容の水(100mL)を添加し、周囲温度で1〜2時間撹拌する。減圧濾過により固体を回収し、10容の水(50mL)で洗浄した後、乾燥する(注3)。
【0224】
乾燥した(注4)固体を、MTBE(20容)を用いて、周囲温度で2〜5時間粉砕することにより、ジベンゾフルベンとジベンゾフルベンピペリジン付加物を除去する。減圧濾過により固体を回収し、恒量まで乾燥する。その結果、4.55g(94.3%)のHAA27-36NH2が得られ、純度は85〜90A%であった(注1)。
【0225】
Fmoc副産物(ジベンゾフルベンおよびそのピペリジン付加物)は、一般に、このプロトコルで除去されるが、再処理が必要な場合には、10容のMTBE中の固体を20℃で2〜3時間撹拌し、回収および乾燥する。
【0226】
注:
1.逆相HPLCによるプロセス中検査(IPC):
カラム:Vydac C8、300A、5μ、30℃
流量:1mL/分
検出:262nmのUV
移動相:A.水/0.1%TFA
B.アセトニトリル/0.1%TFA
方法:20分間にわたり、80〜99%B
保持時間:FmocAA27-36NH2(16.5分、HAA27-36NH2(8.4分)
2.合計濾過時間は10分であった。
【0227】
3.湿ったフィルターケークは反応器に戻し、直ちに水で洗浄することにより、固体の油状化またはゴム質化を防がなければならない。
【0228】
4.MTBE粉砕によるジベンゾフルベン副産物の除去を確実にするため、粗生成物は、完全に乾燥させなければならない。
【0229】
11.7 FmocAA(17-26)-OHとHAA(27-36)-OHの液相合成融合によるフラグメントFmocAA(17-36)-OHの大規模製造
4回の実施で、2.993kgのHAA27-36NH2と3.176kgのFmocAA17-26OHから、合計5.115kgのFmocAA17-36NH2を製造した。平均収率は84.3%であった。反応混合物からの水ドロップアウト(dropout)後の粗生成物の濾過には、平均40〜50分かかった。
【0230】
カップリング反応を実施する前に、使用されるHBTUおよびフラグメントの両方を試験する使用試験を1〜2gスケールで行う。使用試験から、出発物質および試薬の計算量を決定する。使用試験では、出発物質の可溶性も記録した。濁りのある溶液は、FmocAA17-26OH中の塩の存在を示し、該フラグメントの水洗浄の根拠となるものである。ラセミ化および副産物の生成が最小限であるように出発物を完全に生成物に転化させるようにするため、HBTUの添加前に、反応混合物の全成分が、明らかに溶液中に存在していなければならない。
【0231】
水ドロップアウトから単離した粗FmocAA17-36NH2の純度を、IPAからの沈殿により大幅に増加させる。FmocAA17-36NH2は、IPAに部分的に可溶性である。予め60〜70℃に保温した約15容の95/5のIPA/水を用いて、FmocAA17-36NH2を粉砕した後、撹拌しながら、数時間にわたり室温に冷却し、フラグメントの純度を高める。出発物質、ならびに、FmocAA17-26OHのピペリジンアミドおよびHAA27-36NH2のエナミン尿素付加物は共に、95%IPAに可溶性である。室温で95%IPAを用いた粗FmocAA17-36NH2の粉砕にかける時間を延長しても、不純物の除去にはそれほど効果的ではない。60〜70℃での安定性試験を完了した。IPA溶液を52℃を上回る温度に加熱しない場合、単離した生成物の品質に及ぼす影響が最小限に抑えられるようだ。
【0232】
11.7.1 FmocAA(17-26)OHとHAA(27-36)-OHの液相合成融合によるフラグメントFmocAA(17-26)-OHの改良された大規模製造の好ましい方法
撹拌機と窒素供給口を備えた40Lのジャケット付き反応器に、FmocAA17-26OH(1当量、0.770kg)、HAA27-36NH2(1当量、0.720kg)、HOAT(1.5当量)およびDMF(FmocAA17-26OHに対して12.5容)を導入する。EtPr2N(1.5当量)を添加し、この懸濁液を、室温で、固体が溶解するまで(見た目で)撹拌する。この溶液を窒素雰囲気下で0〜5℃に冷却した後、HBTU(1〜1.05当量)を添加する。この反応混合物を、0〜5℃で、HBTUが溶解するまで(見た目で、約15分間))撹拌する。反応混合物を25℃に温め、2時間撹拌を継続する(注1)。DMF溶液を0〜5℃に冷却した後、25℃を超えないような速度で、低温のプロセス水(12.5容)を添加する。得られたスラリーを1〜24時間撹拌し、固体を減圧濾過により回収した後、水で洗浄する(3x4容)。該固体をフィルター上で16〜24時間乾燥させ、理論質量の2倍にする。40L反応器に、95/5のイソプロパノール/水(25容)を導入し、45℃に温める。急速に撹拌しながら、半乾燥状態のFmocAA17-36NH2をIPA溶液に添加する。この懸濁液を52℃に温めた後、12〜16時間撹拌しながら、室温まで冷却させる(注2)。固体を減圧濾過により回収した後、最小量のIPAで洗浄して、乾燥し、1.261kgのFmocAA17-36NH2を得た。収率は、85%、HPLCによる純度は90.SA%であった(注1)。95/5のIPA/水粉砕を繰り返すことにより、反応を再実施してもよい。
【0233】
注:
1.プロセス中検査および純度、HPLC:
Vydac C8、5μ、300A
1mL/分、262nm、30℃
A.水/0.1%TFA
B.80/20 IPA/ACN/0.1%TFA
60〜95%B/20分
保持時間:HAA27-36NH2、6.73分
FmocAA17-26OH、10.67分
FmocAA17-36NH2、20.2分、
2.FmocAA17-36NH2は、この温度では、この容量中に完全に溶解しない。固体を回収する前に、撹拌を停止し、固体を沈降させる。濾液のサンプルを採取し、HPLCにより分析する。濾液が多量のFmocAA17-36NH2を含んでいる場合には、固体を回収する前に0℃まで冷却する。
【0234】
11.8 FmocAA(17-36)-OHからのフラグメントHAA(17-36)-OHの大規模製造
4回の実施で、5.112kgのFmocAA17-36NH2から、合計4.965kgのHAA17-36NH2を製造した。単離した収率およびHPLCトレースはどちらも、ジベンゾフルベン、および恐らくは溶媒の存在を示している。存在するジベンゾフルベンは、ピペリジンではなく、炭酸カリウムで除去するため、次のカップリング反応を妨害しない。従って、ジベンゾフルベンのピペリジン付加物は、化学的には問題ではない。
【0235】
この方法には、いくつかの問題が付随する。この反応は、12容のDMF中で実施する。Fmoc基を除去する基剤は、1容の1.1M炭酸カリウムであり、これは、除去が困難な、ジベンゾフルベンとの塩基性付加物を生成することはできない。脱保護は、室温で、約90分間進行させる。
【0236】
予め0℃に冷却した1:1で飽和した塩化ナトリウム:水-水溶液を添加し、フラグメントとジベンゾフルベンを共沈殿させる。これによって、濾過するのに数(5〜8)時間を要して、微細な乳白色の固体が生成された。単離した後、湿った固体を完全に乾燥させて(1%LOD未満)、ジベンゾフルベン不純物を除去しなければならない。乾燥には10日間かける(減圧、加熱なし)。乾燥したら、固体を反応器に戻し、3:1のヘプタン:MTBE(20容)を用いて、周囲温度で18時間粉砕することにより、ジベンゾフルベンを除去する。これより粉砕時間が短いと、ジベンゾフルベンを完全に除去することはできず、また、固体に水が少しでも存在すると、除去することができない。ジベンゾフルベンが残っている場合には、3:1のヘプタン:MTBE(20容)による再処理を実施することができる。
【0237】
これらの問題を解決するために、次のような新しい方法が開発された。FmocAA17-36NH2(2.0g、1当量)およびヘプタン(8容)を、撹拌機、温度調節器および還流コンデンサーを備えた100mLの丸底フラスコに添加する。2容のMTBE(4mL)を添加し、該スラリーを45〜50℃に加熱する(注1)。ピペリジン(1 0当量)を添加する。窒素雰囲気下で、45〜50℃にて24〜36時間撹拌する(注2)。反応混合物を45〜50℃で、保温濾過し(注3)、次に、5容(10mL)の60:40のヘプタン:MTBEで、ケークを洗浄する。
【0238】
注:
1.MTBEが反応器から漏れ、そのために、ヘプタン:MTBE比が変化するようなことがある場合には、反応が遅くなる恐れがある。反応温度が50℃を超える場合には、反応固体のゴム質化が起こる可能性がある。Fmocは、5時間で大部分が除去される。
【0239】
2.反応の完了は、RP-HPLCにより監視する:
カラム: Vydac C8、300A、5、30℃
流量: 1mL/分
検出: 262nmのUV
移動相: A.水/0.1%TFA
B.80:20 IPA:CAN 0.1%TFA
方法: 30分間にわたる60〜95%B
保温濾過は、ジベンゾフルベンのピペリジン付加物の除去に役立つ。
【0240】
11.8.1 FmocAA(17-36)-OHからのフラグメントHAA(17-36)-OHの大規模製造
40Lのジャケット付き反応器に、FmocAA17-36NH2(1当量、1.26kg)およびDMF(12容)を室温で導入する。1.11M炭酸カリウム水溶液(1容、5当量)を一度に添加する(注1)。この反応混合物を、室温で、3.5時間撹拌する(注2)。反応混合物を、予め0℃に冷却した飽和塩化ナトリウム/水(10容)の1:1水溶液に徐々に添加するが、その速度は、温度10℃を超えないようなものとする(約1〜2時間)。ラバーダム(rubber dam)を用いて、固体を減圧濾過により回収し、フィルターケークから水を圧搾する(注3)。湿ったケークを反応容器に戻し、水(10容)を用いて1〜2時間粉砕(撹拌)し、残留する無機塩を除去する。固体を減圧濾過により回収し、ラバーダムで圧縮して、減圧オーブンに移し、恒量まで乾燥させる。乾燥した固体(注4)を、3:1のヘプタン:MTBE(20容)で、室温にて最低14時間粉砕(撹拌)し、ジベンゾフルベンを除去する。固体を減圧濾過により回収した後、ヘプタン(2容)で洗浄して、乾燥し、1.20kgのHAA17-36NH2を得た。収率は、99.5%、純度は75A%であった。このHAA17-36NH2は、5A%ジベンゾフルベンを含有していた。へプタン:MTBE粉砕を繰り返すことにより、反応を再実施する。
【0241】
注:
1.炭酸カリウム水溶液を添加すると、溶液は濁るが、撹拌を続けると透明になる。
【0242】
4〜5℃の発熱量を記録した。
【0243】
2.IPCおよび純度測定のために用いたHPLC:
Vydac C8、260nm
1mL/分、262nm、30℃
A.水/0.1%TFA
B.80/20 IPA/アセトニトリル/0.1%TFA
60〜95%B/30分
3.微細な粒子サイズの生成物が沈殿するため、濾過が遅くなる。
【0244】
4.ヘプタンが、ジベンゾフルベンを効果的に除去するように、この物質は無水でなければならない。
【0245】
11.9 AcAA(1-16)-OHおよびFmocAA(17-36)-OHからの液相合成によるフラグメントAcAA(1-36)-OHの大規模製造
AcAA1-16-OHを、HOATおよびDIEAと共に、DMFに溶解して0℃に冷却した後、HBTUを添加した。これを15分間、もしくはHBTUが溶解するまで撹拌し、HAA17-36NH2を添加する。HAA17-36NH2の不在におけるAcAA1-16-OHの前活性化は、予備措置であったが、後に必要がないことがわかった。さらに、活性化AcAA1-16-OHを含む0℃のDMF溶液へのHAA17-36NH2の溶解は、規模が大きくなると、遅くなることが証明された。
【0246】
4回の実施で、3.92kgのAcAA1-16-OHと、4.96kgのHAA17-36NH2から、合計6.972kgのAcAA1-36NH2を製造したが、平均収率は80.1%であった。この段階での2回の実施の平均収率は、87%であり、2回の実施の平均収率は、73%であった。
【0247】
カップリング反応を実施する前に、使用されるHBTUおよびフラグメントの両方を試験する使用試験を1〜2gスケールで行う。使用試験から、出発物質および試薬の計算量を決定する。また、出発物質の可溶性も使用試験に記録する。濁りのある溶液は、AcAA1-16-OH中の塩の存在を示し、フラグメントの水洗浄の根拠となるものである。ラセミ化および副産物の生成が最小限であるように出発物質を生成物に完全に転化させるためには、HBTUの添加前に、反応混合物の全成分が、明らかに溶液中に存在していなければならない。
【0248】
フラグメントAcAA1-16-OHおよびHAA17-36NH2、ならびに試薬HOATおよびDIEAをDMFに溶解させ、0℃に冷却し、HBTUを添加する。全実施のうち1回は、HAA17-36NH2に対しHClをピックアップするために、追加当量のDIEAを必要とした。水ドロップアウトにより、反応混合物から粗生成物を単離する(濾過時間、10分)。まだ湿っている固体を、55℃に予め保温したアセトニトリルを含む反応器に戻し、急速に撹拌しながら、3時間かけて35℃まで冷却した後、20℃で一晩放置する。このスラリーを0〜5℃に冷却し、1〜2時間さらに撹拌した後、固体を濾過により回収する。この工程の間に、AcAA1-36NH2は、55℃で溶液へとほぼ移行した。溶液が冷却するにつれ、AcAA1-36NH2は、溶液から沈殿していった(油状化した)。溶液が冷却するにつれて、油は凝固し、最終的には、鮮やかな白色の固体へと変化する。この変化の最中に、反応器の壁や撹拌棒上に多量の固体沈殿が生成する可能性がある。これらのほとんどは、スラリーを一晩20℃で撹拌するにつれて取れる。固体を回収した後、反応器に、反応器壁や撹拌棒に付着した固体全てを浸すのに十分な水を充填する。
【0249】
壁や撹拌棒から残りの固体がすべて落ちるまで(約1時間)、スラリーを撹拌した後、フィルターケークの洗浄物(wash)として用いる。アセトニトリル洗浄により、未反応の出発物質と、20アミノ酸に満たない切端フラグメントを除去する。
【0250】
11.9.1 AcAA(1-16)-OHおよびFmocAA(17-36)-OHからの液相合成によるフラグメントAcAA(1-36)-OHの好ましい大規模製造
40リットルの反応器に、AcAA1-16-OH(938kg、1当量)、HAA17-36NH2(1.19kg、1当量)、HOAT(1当量)、DMF(AcAA1-16-OHに対して19容)およびDIEA(1当量)を導入する。このスラリーを、固体が溶解するまで撹拌し、0〜5℃に冷却した後、HBTU(1.03当量)を添加する。この溶液を、0〜5℃で、15分間、もしくは固体が溶解するまで撹拌した後、20℃に保温し、2時間撹拌する。反応混合物から、IPCに付すためのサンプルを採取する(注1)。反応が完全であると判断したら、温度が35℃を超えないような速度で、水(19容)を添加する(注2)。
【0251】
得られたスラリーを1〜24時間撹拌し、固体を真空濾過(10分)により回収した後、水で洗浄する(2x3容量)。濾過ケークを圧縮し、水を出来る限り除去する。その間、反応器に、95%アセトニトリル/水(AcAA1-16OHの導入量に対して、30容量)を導入し、撹拌しながら、55℃に保温する。
【0252】
湿った固体を、凝集しないように、少しずつ反応器に添加する。スラリーを撹拌しながら、3時間かけて35℃に冷却し、その後、20℃で一晩放置する。0〜5℃に冷却し、2時間撹拌して、撹拌を停止した後、溶液をサンプリングする。
【0253】
固体を真空濾過により回収する。反応器および管路を90%アセトニトリル/水(3.6容量)で洗浄する。
【0254】
反応器に、反応器壁や撹拌棒に付着した固体を浸すのに十分な量の水を充填する(約30容量)。反応器壁や撹拌棒に付着した固体が落ちるまで、室温で、撹拌する(約1時間)。固体を残り部分と共に回収し、恒量まで乾燥させる(1.83kg、収率86%)。
【0255】
90/10のアセトニトリル/水を用いて、固体の摩砕を繰り返す。
【0256】
注:
1.IPCおよび純度、HPLC:
YMC ODS-A、150 x 4.6mm、5μ、120A
1mL/分、260nm、30℃
A.水/0.1%TFA
B.THF/0.1%TFA
60〜90%B/30分、90〜95%B/1分、95%B/5分
AcAA1-16-OH 6.37分
HAA27-36NH2 8.91分
AcAA1-36NH2 18.07分、
2.水を添加する間、温度が35℃を超えると、沈殿する固体が融解し、凝集および/または反応器壁への付着を起こす恐れがある。
【0257】
11.10 AcAA(1-16)-OHの側鎖の脱保護
この実験の目的は、沈殿物へのMTBEの添加速度を変化させて、温度を20℃以下に維持し、粗T-20固体を単離し、脱炭酸反応させる(すなわち、溶液脱炭酸反応を排除する)ことである。
【0258】
HPLCカラムに供給する前の固体として、ACN/水から、沈殿により粗T20を単離した。固体のT20含有率(%)を、重量に基づくHPLC検定により測定した。
【0259】
側鎖保護基を、90/5/5のTFA/水/ジチオトレイトール中で除去した後、溶液を0℃に冷却し、MTBE(AcAA1-36NH2導入量に対して45容量)を添加する。これは、T20の完全な沈殿を確実にするのに必要なMTBEの最小量である。混合の際に発熱があるため、温度を20℃以下に保持するように、最初の5容量はゆっくりと添加しなければならない(約60分)。さらにMTBEを添加するにつれて、添加速度を高めることができる。沈殿中、温度を20℃以下に維持することにより、固体が溶液から沈殿する際の凝集を防止する。
【0260】
上記開裂/精製プロトコルは、クロマトグラフィーによる精製に適合する規模で脱保護を実施することを含む。TFA塩として脱保護混合物から単離した粗T20を、pH5のアセトニトリル/水に溶解させ、濾過し、15時間静置することにより、トリプトファンインドールの脱炭酸反応を実施する。脱炭酸反応が完了した後、溶液を希釈し、精製装置に移して、カラムに供給した。希釈の間、溶液には曇りが生じ、この濁った溶液をポンプによりカラムに供給する。この結果、精製工程中に、固体がカラムのヘッドに付着し、徐々にカラムの性能が低下した。pH5のアセトニトリル/水中で、数時間T-20を脱炭酸反応させると、極めて不溶性の凝集体が形成される。
【0261】
溶液中の脱炭酸反応を回避するための方法が開発されている。MTBE沈殿物からTFA塩として単離した粗T-20を、室温で約5〜7日にわたり、減圧下で脱炭酸反応させる。減圧下40℃で、3日のうちに脱炭酸反応の完了が認められた。単離した固体のTFA含有率は、9%であった。材料は、0℃で、1カ月まで、1%重量/重量損失で貯蔵することができる。
【0262】
HOAcを用いたエタノール/水(1:9)における塩交換は、MTBEからのT-20-TFAの単離後に実施することができる。粗T-20の酢酸塩は、有意により安定であり、好ましいものであろう。いずれの単離方法でも、側鎖脱保護をこれまでより大きな規模で実施することが可能であり、T-20を、凝集を引き起こすことが知られている脱炭酸反応に使用されるアセトニトリル/水/HOAc条件にさらす必要はない。
【0263】
AcAA(1-36)-OHの側鎖の脱保護のための新しい方法:
90/5/5のTFA/水/ジチオトレイトール溶液(13容量)を作製し、3分間、窒素でパージする。窒素雰囲気下で、周囲温度にて、AcAA1-36NH2の一部を90/5/5のTFA/水/ジチオトレイトール溶液(13容量)に添加する。溶液状にしたら、周囲温度にて4時間撹拌し、次に0℃に冷却する。T-20を沈殿させるために、0〜5℃に冷却したMTBE(45容量)を、初めは、温度を20℃以下に維持しながら、遅い速度で滴下しながら添加する(添加時間は、約1〜2時間)。固体を減圧濾過により回収する(注1)。反応器、管路および濾過ケークを直ちに3x5容量のMTBEで洗浄する。固体を除去し、t=0のIPC(注2)用にサンプルを採取し、5日間、もしくは、HPLCが変化を示さなくなるまで、減圧下で乾燥させる。
【0264】
注:
1.脱保護溶液は吸湿性であるため、この濾過の間、固体に空気を通過させないようにする。TFA溶液を洗浄する前に固体に空気を通過させると、粘着質または油状の固体が得られる恐れがある。
【0265】
2.HPLC方法TM2-0003-01(TFA法)を使用する。酢酸アンモニウム法(TM2-0006-01)では、様々なカルボキシル化中間体を分離できない。
【0266】
AcAA(1-36)-OHの側鎖の脱保護
12回の工程で、合計7.226kgのAcAA1-36NH2を脱保護した。製造した6.972kgのAcAA1-36NH2だけが、中間体が吸湿性であることを示した。MTBEドロップアウトから単離した固体を、10容量の1:1ACN/水に溶解し、濾過した後、炭酸水素ナトリウムでpH3.5に調整した。酢酸(1.5容量%)を添加し(pH4.5〜5)、溶液を周囲温度で15時間撹拌することにより、脱炭酸反応を実施した。溶液を25容量の水で希釈し、合計40容量の85/15の水/ACN溶液を得た。この溶液は、いずれの場合も濁っており、中には、微細な固体(凝集したT-20)を含むものもあった。IPC用のサンプルを採取し、溶液を精製装置に移した。工程あたりの粗T-20の収率を、重量/重量HPLC検定を用いて計算した。
【0267】
AcAA1-36NH2の脱保護は、室温の90/5/5のTFA/水/DTTで、4〜5時間で完了する。8時間後には、顕著な分解(2%)が起こった。5℃の同じ混合物では、脱保護は8時間では完了しないが、23時間で完了する。室温で5時間後に得た粗T-20と、5℃で23時間後に得たものとの間に、純度の差はなかった。単離容量(約55容量)は、反応(脱保護)容量(13容量)よりはるかに大きいため、20Lカーボイ中のTFA溶液にAcAA1-36NH2を溶解させてから、この溶液を40L反応器に移す必要があった。
【0268】
脱保護混合物からの、粗カルボキシル化T-20(TFA塩として)の単離を、MTBEを用いた沈殿により達成する。ペプチドを沈殿させるのに必要なMTBEの量は、脱保護混合物量の3〜4倍である。MTBEを開裂混合物の容量の2倍使用することにより、ペプチドが残留する。
【0269】
開裂混合物にMTBEを添加すると、容易に濾過される固体が得られるのに対し、MTBEに開裂混合物を添加すると、濾過が難しい微細な沈殿物が生成する。TFA溶液にMTBEを添加する間、5℃から40〜45℃への温度上昇がある。温度上昇は、単離した粗T-20の純度(187/117、119)に影響を与えない。MTBE添加の間、いくらか固体の凝集が生じた。スラリーを1〜2時間撹拌して、凝集物を破壊した。MTBE添加の間、温度を20℃以下に維持することにより、より均質な固体が生成し、この固体は濾過が容易である(196/31)。MTBEの添加速度は、将来制御されるであろう。窒素雰囲気下での濾過により、粗T-20を回収した。TFAを洗浄する前の濾過中に、湿った空気にさらすと、固体は粘着質になる。TFAを洗浄した後、固体は、空気に対して安定する。
【0270】
方法工程:
90/5/5のTFA/水/ジチオトレイトール溶液(13容量)を20Lカーボイ中に製造し、窒素で3分間パージする。凝集を防ぐため、AcAA1-36NH2(650g)を少量ずつ添加する。固体が液状になったら、溶液を70L反応器に移す。カーボイと管路を最小量の90/5/5のTFA/水/ジチオトレイトールで洗浄する。窒素雰囲気下、周囲温度で溶液を4時間撹拌し、その後、0〜5℃に冷却する。MTBE(45容量)を、内部温度が30℃を超えないような速度で添加する。窒素流の下、減圧濾過により固体を回収する。2x2容量のMTBEで反応器、管路および固体を洗浄し、恒量まで乾燥させる(594g、70A%)。
【0271】
固体(594g)を50%ACN/水(AcAA1-36NH2導入量に対して8容量)に溶解させ、濾過する。50%ACN/水(2容量)で容器と管路を洗浄する。炭酸水素ナトリウムを用いて溶液のpHを3.5〜4.0に調整した後、酢酸(0.18容量)を添加して、pHを4.9にする。溶液を周囲温度で15時間撹拌し(IPC、注1)、1M炭酸カリウムでpHを9〜9.5に調整する。水(25容量)を添加することにより、溶液を85/15の水/ACNに希釈する。このようにして得られた濁った溶液から、重量/重量HPLC分析用のサンプルを採取し、精製装置に移す。
【0272】
注:
1.方法TM2-0003-01
2.方法TM2-0006-01
11.11 HAA(1-36)-OHの精製
精製、凍結乾燥法およびパッケージングを、3段階に分類する:
%wt/wt=100 − %不純物 − %酢酸塩 − %水 − %TFA。バッチ中の酢酸塩の含有率は6〜8%であった。
【0273】
合計2.08kgのT-20(正味)を6.97kgのAcAA1-36NH2から単離した。これは、AcAA1-36NH2からの収率が49.3%であることを示す。クロマトグラフィー精製における平均収率は、55%であった。個々の工程についての収率は、41〜55%までと差があった。最低収率は、カラムまたはポンプの故障により、複数箇所で通過(passes)が生じたためである。一般に、クロマトグラフィーが機能すれば、収率は、50〜55%の範囲である。単離したT-20の純度は93〜95A%であった。
【0274】
工程時間を最小限にする目的で、方法を比較するために、精製中、四つの勾配を検定した:
1.15〜22%ACN/60分、330mL/分で、26〜36%ACN/525分。
【0275】
2.16〜26%ACN/60分、330mL/分で、26〜40%のACN/525分。
【0276】
3.第2トリメリス(Trimeris)勾配。330mL/分で、15〜36%ACN1788分
4.元のトリメリス(Trimeris)勾配20〜23%ACN/1 12分、330mL/分で、23〜36%ACN/488分。
【0277】
20センチメートル(cm)のカラムをアンバークロム(Amberchrom)樹脂(台高さ35cm)で充填した(軸方向圧縮)。500〜700gのAcAA1-36NH2のバッチを脱保護し、溶液中で脱炭酸反応させた。この規模では、約400グラムのペプチド(約75A%T-20)を含有するカラム供給原材料が生成される。原液は、典型的には、曇っており、懸濁した固体を含むこともある。濁った溶液を、15%Bを用いて、500mL/分でカラムに供給する。いずれの工程のカラム供給中にも、圧力増大はなかった。流速を330mL/分に減少させ、指示された時間、特定の勾配を実施する。画分を回収し、78%以上のT-20を含有する画分をプールした(100〜110L)後、水で希釈することにより、アセトニトリル含有率を約15〜20%(約140リットル)にする。カラムを洗浄した後、15%Bで平衡させる。T-20含有溶液を、8インチカラムに、900mL/分で送り返す。%Bを50%に増加し、T-20をカラムから約25Lでフラッシュする。
【0278】
溶液を1リットルガラス鐘中で凍結し、凍結乾燥して、粉末にする。単離したT-20は、典型的には、HPLCにより、92〜94A%であり、6〜8%の酢酸塩と3〜4%の水を含有する。
【0279】
好ましい方法:
pH9.2で、85/15の水/アセトニトリル(27L)中のT-20溶液を、500mL/分で、8インチHPLCカラムにポンプ供給した。流速を30分かけて330mL/分に減少させた(5分毎に30〜40mL/分の増加量)。20%Bから36%Bまでの線状勾配を600分にわたり開始する。吸光度が0.2AUFSを下回るまで、画分を回収する。画分を、含有率について、HPLCにより分析する(注1)。カラムを80%Bで900 mL/分にて10分間洗浄し、15%Bに戻し、900mL/分で平衡化する。78A%以上のT-20を含有する画分をプールし(113L)、バッファー(28.2L)で希釈することにより、アセトニトリル濃度を15〜20%にする。溶液を900mL/分でカラムにポンプ供給する。%Bを50%に増加し、T-20を、900mL/分でカラムから約25L溶出させる。T-20の濃度は、約9〜10mg/mLである。溶液を1リットル凍結乾燥ジャーに移し、凍結させ、凍結乾燥して、収率54.9%、純度94.1A%の216.29gのT-20を得る。
【0280】
注:
1.画分は、含有率および純度について、HPLCにより分析する。
YMC ODS-A、150x4.6mm、5μm、120A
1mL/分、220nm、30℃
A 70/30、50mM NH4OAc @ pH7(NH4OHで調整する):ACN
B 5:95 50mM NH4OAc @ pH7(NH4OHで調整する):ACN
0〜15%B/50分、15〜100%B/2分、100%B/3分
T-20保持時間、29.0分
【0281】
11.12 ペプチドの熱安定性
一連の実験を実施して、中間T-20フラグメントの熱安定性を特性評価した。T-20フラグメントを水で飽和させ、濾過した後、密閉容器に導入して、高温に暴露した。いくつかの時点で、サンプルを回収し、HPLC法により分析することによって、純度変化を得た。また、乾燥したT-20フラグメントの安定性は、熱重量分析(TGA)により特性評価した。
【0282】
AcAA1-16OHを除くすべてのフラグメントを40℃で3日間乾燥させることができる。80℃で24時間後、2つのフラグメント、すなわちFmocAA27-35OH(3%)とFmocAA27-36NH2(1.4%)に、ほんのわずかな分解が認められる。フラグメントAcAA1-16OHは、初め97A%の純度であった。40℃で3日間後、93.4A%の純度および乾燥度になった。さらに80℃で24時間後にも、93.9A%を維持した。尚、湿ったAcAA1-16OHは、周囲温度で乾燥させなければならない。
【0283】
安定性試験の要点は、フラグメントを、周囲温度ではなく、40℃で乾燥することができるかどうかを決定することであった。この試験から、フラグメントが、40℃の乾燥で安定であることがわかった。これにより、乾燥時間が大幅に短縮されるであろう。
【0284】
単離および精製を模倣した様々な溶剤および温度条件の下で、フラグメントAc(1-16)-OH、Fmoc(27-35)-OH、Fmoc(17-26)-OH、Fmoc(27-36)-NH2、Fmoc(17-36)-NH2、H(17-36)-NH2、ならびにAc(1-36)-NH2の安定性を検定するために設計された試験が完了した。フラグメントはすべて、それらが単離および/または精製された溶剤系において、安定である。尚、安定性はXで表す。
【0285】
本発明は、本明細書に記載した特定の実施形態によって範囲を制限されるわけではない。これらの実施形態は、本発明の個々の態様を示すことを目的とするに過ぎず、機能的に同等の方法および要素は、本発明の範囲内に含まれるものとする。実際に、当業者には、本明細書に記載したもの以外にも、本発明の様々な変更が、これまでの説明および添付の図面から明らかとなるであろう。このような変更は、添付した請求の範囲内にあるものとする。
【特許請求の範囲】
【請求項1】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドを式:NH2-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(e) 工程(d)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項2】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドを式:NH2-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項3】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端を脱保護し、
(d) 工程(c)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(e) 工程(d)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項4】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項5】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-DKWASLWNW-COOH (配列番号17) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項6】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-LELDKWASLWNW-COOH (配列番号15) の側鎖保護ペプチドをHPheNH2 と反応させて、式:Fmoc-LELDKWASLWNWF-NH2 (配列番号16) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-EKNEQEL-COOH (配列番号10) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項7】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-DKWASLWNWF-COOH (配列番号16) のペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-EKNEQELLEL-COOH (配列番号10) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項8】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-EKNEQELLELDKWASLWNW-COOH (配列番号19) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項9】
式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドが、
(a) 式:Fmoc-IEESQNQQ-NH2 (配列番号7) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られた側鎖保護ペプチドを式:Fmoc-YTSLIHSL-COOH (配列番号2) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項1または3記載の方法。
【請求項10】
式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドが、
(a) 式:Fmoc-IEESQNQ-NH2 (配列番号6) の側鎖保護ペプチドをHGlnOPNBと反応させて、式:Fmoc-IEESQNQQ-OPNB (配列番号7) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られた側鎖保護ペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られた側鎖保護ペプチドを式:Ac-YTSLIHSL-COOH (配列番号2) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQ-OPNB (配列番号4) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られた側鎖保護ペプチドのカルボキシル末端を脱保護して、式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項2または4記載の方法。
【請求項11】
式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保
護ペプチドが、
(a) 式:Fmoc-YTSLIHSLIEESQNQ-COOH (配列番号3) の側鎖保護ペプチドをHGlnOPNBと反応させて、式:Fmoc-YTSLIHSLIEESQNQQ-OPNB (配列番号4) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのカルボキシル末端を脱保護して、式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項1または3記載の方法。
【請求項12】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQ-COOH (配列番号3) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項13】
式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドが、
(a) 式:Fmoc-QEKNEQELLELDKWASLWNW-NH2 (配列番号8) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項12記載の方法。
【請求項14】
式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドの合成方法であって、
(a) 式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQEK-COOH (配列番号5) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項15】
式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドが、
(a) 式:Fmoc-NEQELLELDKWASLWNW-NH2 (配列番号13) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項14記載の方法。
【請求項16】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項1、2、3、4、12または14記載の方法。
【請求項17】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項1、2、3または4記載の方法。
【請求項18】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項5記載の方法。
【請求項19】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項6記載の方法。
【請求項20】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項7記載の方法。
【請求項21】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項8記載の方法。
【請求項22】
式:Fmoc-YTSLIHSLIEESQNQQ-NH2 (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項9記載の方法。
【請求項23】
式:Ac-YTSLIHSLIEESQNQQ (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項10記載の方法。
【請求項24】
式:Ac-YTSLIHSLIEESQNQQ-NH2 (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項11記載の方法。
【請求項25】
式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項13記載の方法。
【請求項26】
式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項15記載の方法。
【請求項27】
次のセット:
(a) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLELDKWASLWNWF (配列番号12);
(b) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(c) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(d) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLELDKWASLWNWF (配列番号12);
(e) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(f) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(g) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLELDKWASLWNWF (配列番号12);
(h) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(i) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(j) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(k) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(l) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(m) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(n) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(o) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(p) YTSLIHSLIEESQNQ (配列番号3),
QEKNEQELLELDKWASLWNW (配列番号8);
(q) YTSLIHSLIEESQNQ (配列番号3),
QEKNEQELLELDKWASLWNWF (配列番号9);
(r) YTSLIHSLIEESQNQQEK (配列番号5),
NEQELLELDKWASLWNWF (配列番号14);
(s) YTSLIHSLIEESQNQQEK (配列番号5),
NEQELLELDKWASLWNW (配列番号13);および
(t) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLELDKWASLWNW (配列番号19)
からなる群より選択されるペプチド断片のセット。
【請求項28】
次のペプチド:
YTSLIHL (配列番号2);
IEESQNQ (配列番号6);
IEESQNQQ (配列番号7);
QEKNEQELLELDKWASLWNW (配列番号8);
EKNEQEL (配列番号10);
EKNEQELLEL (配列番号11);
NEQELLELDKWASLWNW (配列番号13);
LELDKWASLWNW (配列番号15);
LELDKWASLWNWF (配列番号16);
DKWASLWNW (配列番号17);
DKWASLWNWF (配列番号18);および
EKNEQELLELDKWASLWNW (配列番号19)
からなる群より選択されるペプチド。
【請求項1】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドを式:NH2-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(e) 工程(d)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項2】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドを式:NH2-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項3】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端を脱保護し、
(d) 工程(c)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(e) 工程(d)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項4】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項5】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-DKWASLWNW-COOH (配列番号17) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-DKWASLWNWF-NH2 (配列番号18) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-EKNEQELLEL-COOH (配列番号11) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項6】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-LELDKWASLWNW-COOH (配列番号15) の側鎖保護ペプチドをHPheNH2 と反応させて、式:Fmoc-LELDKWASLWNWF-NH2 (配列番号16) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られたペプチドを式:Fmoc-EKNEQEL-COOH (配列番号10) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項7】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-DKWASLWNWF-COOH (配列番号16) のペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-EKNEQELLEL-COOH (配列番号10) の側鎖保護ペプチドと反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項8】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが、
(a) 式:Fmoc-EKNEQELLELDKWASLWNW-COOH (配列番号19) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項3または4記載の方法。
【請求項9】
式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドが、
(a) 式:Fmoc-IEESQNQQ-NH2 (配列番号7) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られた側鎖保護ペプチドを式:Fmoc-YTSLIHSL-COOH (配列番号2) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項1または3記載の方法。
【請求項10】
式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドが、
(a) 式:Fmoc-IEESQNQ-NH2 (配列番号6) の側鎖保護ペプチドをHGlnOPNBと反応させて、式:Fmoc-IEESQNQQ-OPNB (配列番号7) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られた側鎖保護ペプチドのアミノ末端を脱保護し、
(c) 工程(b)で得られた側鎖保護ペプチドを式:Ac-YTSLIHSL-COOH (配列番号2) の側鎖保護ペプチドと反応させて、式:Ac-YTSLIHSLIEESQNQQ-OPNB (配列番号4) の側鎖保護ペプチドを生成し、
(d) 工程(c)で得られた側鎖保護ペプチドのカルボキシル末端を脱保護して、式:Ac-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項2または4記載の方法。
【請求項11】
式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保
護ペプチドが、
(a) 式:Fmoc-YTSLIHSLIEESQNQ-COOH (配列番号3) の側鎖保護ペプチドをHGlnOPNBと反応させて、式:Fmoc-YTSLIHSLIEESQNQQ-OPNB (配列番号4) の側鎖保護ペプチドを生成し、
(b) 工程(a)で得られたペプチドのカルボキシル末端を脱保護して、式:Fmoc-YTSLIHSLIEESQNQQ-COOH (配列番号4) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項1または3記載の方法。
【請求項12】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) を有するペプチドの合成方法であって、
(a) 式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQ-COOH (配列番号3) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項13】
式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドが、
(a) 式:Fmoc-QEKNEQELLELDKWASLWNW-NH2 (配列番号8) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項12記載の方法。
【請求項14】
式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドの合成方法であって、
(a) 式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドのアミノ末端を脱保護し、
(b) 工程(a)で得られたペプチドを式:Fmoc-YTSLIHSLIEESQNQQEK-COOH (配列番号5) の側鎖保護ペプチドと反応させて、式:Fmoc-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドを生成し、
(c) 工程(b)で得られたペプチドのアミノ末端をアセチル基修飾へと修飾し、
(d) 工程(c)で得られた側鎖保護ペプチドの側鎖を脱保護して、式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) のペプチドを得る、
ことを含んでなる方法。
【請求項15】
式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドが、
(a) 式:Fmoc-NEQELLELDKWASLWNW-NH2 (配列番号13) の側鎖保護ペプチドをHPheNH2と反応させて、式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドを生成する、
ことからなる方法により合成される、請求項14記載の方法。
【請求項16】
式:Ac-YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF-NH2 (配列番号1) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項1、2、3、4、12または14記載の方法。
【請求項17】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項1、2、3または4記載の方法。
【請求項18】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項5記載の方法。
【請求項19】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項6記載の方法。
【請求項20】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項7記載の方法。
【請求項21】
式:Fmoc-EKNEQELLELDKWASLWNWF-NH2 (配列番号12) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項8記載の方法。
【請求項22】
式:Fmoc-YTSLIHSLIEESQNQQ-NH2 (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項9記載の方法。
【請求項23】
式:Ac-YTSLIHSLIEESQNQQ (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項10記載の方法。
【請求項24】
式:Ac-YTSLIHSLIEESQNQQ-NH2 (配列番号4) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項11記載の方法。
【請求項25】
式:Fmoc-QEKNEQELLELDKWASLWNWF-NH2 (配列番号9) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項13記載の方法。
【請求項26】
式:Fmoc-NEQELLELDKWASLWNWF-NH2 (配列番号14) の側鎖保護ペプチドが約1kg以上の量で合成される、請求項15記載の方法。
【請求項27】
次のセット:
(a) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLELDKWASLWNWF (配列番号12);
(b) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(c) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(d) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLELDKWASLWNWF (配列番号12);
(e) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(f) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(g) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLELDKWASLWNWF (配列番号12);
(h) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLEL (配列番号11),
DKWASLWNWF (配列番号18);
(i) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQELLEL (配列番号11),
DKWASLWNW (配列番号17);
(j) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(k) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(l) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(m) YTSLIHSL (配列番号2),
IEESQNQ (配列番号6),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(n) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQEL (配列番号10),
LELDKWASLWNWF (配列番号16);
(o) YTSLIHSL (配列番号2),
IEESQNQQ (配列番号7),
EKNEQEL (配列番号10),
LELDKWASLWNW (配列番号15);
(p) YTSLIHSLIEESQNQ (配列番号3),
QEKNEQELLELDKWASLWNW (配列番号8);
(q) YTSLIHSLIEESQNQ (配列番号3),
QEKNEQELLELDKWASLWNWF (配列番号9);
(r) YTSLIHSLIEESQNQQEK (配列番号5),
NEQELLELDKWASLWNWF (配列番号14);
(s) YTSLIHSLIEESQNQQEK (配列番号5),
NEQELLELDKWASLWNW (配列番号13);および
(t) YTSLIHSLIEESQNQQ (配列番号4),
EKNEQELLELDKWASLWNW (配列番号19)
からなる群より選択されるペプチド断片のセット。
【請求項28】
次のペプチド:
YTSLIHL (配列番号2);
IEESQNQ (配列番号6);
IEESQNQQ (配列番号7);
QEKNEQELLELDKWASLWNW (配列番号8);
EKNEQEL (配列番号10);
EKNEQELLEL (配列番号11);
NEQELLELDKWASLWNW (配列番号13);
LELDKWASLWNW (配列番号15);
LELDKWASLWNWF (配列番号16);
DKWASLWNW (配列番号17);
DKWASLWNWF (配列番号18);および
EKNEQELLELDKWASLWNW (配列番号19)
からなる群より選択されるペプチド。
【図1】


【図2】
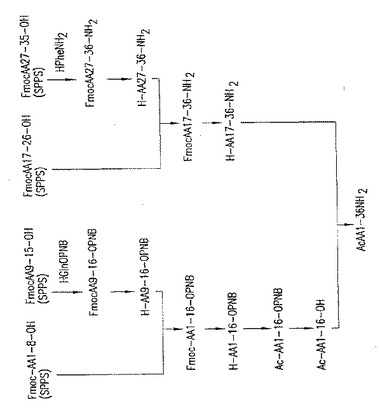
【図3】
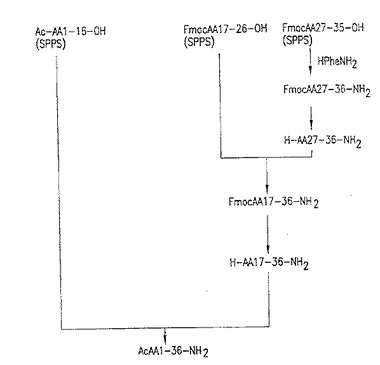
【図4】

【図5】
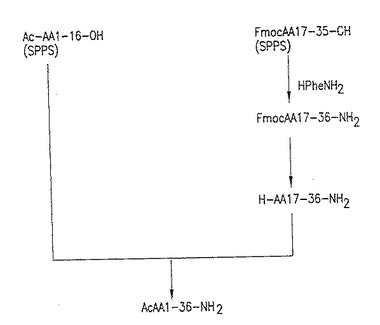
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2010−65044(P2010−65044A)
【公開日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願番号】特願2009−247579(P2009−247579)
【出願日】平成21年10月28日(2009.10.28)
【分割の表示】特願2000−537561(P2000−537561)の分割
【原出願日】平成11年3月22日(1999.3.22)
【出願人】(500447071)トリメリス,インコーポレーテッド (3)
【Fターム(参考)】
【公開日】平成22年3月25日(2010.3.25)
【国際特許分類】
【出願日】平成21年10月28日(2009.10.28)
【分割の表示】特願2000−537561(P2000−537561)の分割
【原出願日】平成11年3月22日(1999.3.22)
【出願人】(500447071)トリメリス,インコーポレーテッド (3)
【Fターム(参考)】
[ Back to top ]