
リンホトキシン−β、リンホトキシン−β複合体、それらの薬学的な調製物および治療への使用
【課題】リンホトキシン−β、リンパ球膜タイプタンパク質を提供すること。また、リンホトキシン−βとリンホトキシン−αのような他のペプチドとの間で形成される複合体およびリンホトキシン−βの多数のサブユニットを包含する複合体を提供すること。これらのタンパク質および複合体を使用して、細胞内で形成されるLT−αを細胞表面に保持し、LT−α/LT−β複合体が炎症調節剤、腫瘍増殖阻害剤、T細胞阻害剤、T細胞活性化剤、自己免疫疾患調節剤、またはHIV阻害剤として作用させ得る。さらに、LT−α/LT−β複合体の抗腫瘍活性を、LT−βの遺伝子でトランスフェクトされた腫瘍湿潤リンパ球(TIL)によって腫瘍細胞に送達し得る。
【解決手段】配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−β。
【解決手段】配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−β。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の技術分野)
本発明は、リンパ球膜タイプポリペプチドであるリンホトキシン−βに関する。リンホトキシン−β(p33と呼ばれる)は、Tリンパ球、T細胞株、B細胞株およびリンホカイン活性化キラー細胞の表面上で同定されている。
【0002】
本発明はまた、リンホトキシン−β(LT−β)およびリンホトキシン(ここでは「リンホトキシン−α(LT−α)」と称して、LT−βと区別している)のような他のリンホトキシンタイプペプチドとの間で形成される複合体ならびにLT−βの多数のサブユニットを含有する複合体に関する。本発明のLT−βポリペプチドは、細胞中で形成されたLT−αを細胞表面上に保持する点で有益であると考えられ、該表面で、LT−βまたはLT−α/LT−β複合体のいずれかが浸潤調節剤、腫瘍成長阻害剤、T細胞抑制剤、T細胞活性化剤、免疫モジュレ一ター剤、自己免疫疾患調節剤またはHIV調節剤として作用し得る。さらに、LT−α/LT−β複合体の抗腫瘍活性は、LT−βの遺伝子でトランスフェクトされた腫瘍浸潤リンパ球(TIL)によって腫瘍細胞へ送達され得る。
【0003】
ここに記述した発明は、国立衛生研究所(the National Institutes of Health)許可番号CA35638−07−10の下での一連の研究の中の一部でなされた。米国政府はこの発明に一定の権利を有している。
【背景技術】
【0004】
(発明の背景)
免疫応答の開始は、細胞間シグナルの複雑な配列を含む。これらのシグナルは、代表的に多数の細胞−細胞接触依存シグナルでカップルした可溶性サイトカインを含有する。接触依存事象(T細胞レセプターの最も有名な活性化)は、応答に特異性を与えるが、他方可溶性メディエーターは、細胞分化および細胞増殖の維持に対して一般的に応答可能である。腫瘍壊死因子(TNF)およびLT−αは、一般的に免疫応答の開始に関与すると認識されている2つのポリペプチドである。
【0005】
TNFおよびLT−αは、元来腫瘍の成長を阻害する能力が知られている可溶性タンパク質である。[L.Old、「腫瘍壊死因子」、Science、230、630(1985)]。さらなる研究により、両方のタンパク質は広範囲な活性を有することが示された。TNFは、造血細胞および非造血細胞の両方を含有する種々の細胞タイプによって、種々の炎症性の傷害に応答して合成されるが、対照的にLT−αはリンパ球によって特異的に作られる。2つの公知のTNFレセプターは、LT−αとTNFとをはっきりと区別することができない。[T.Schallら、「ヒト腫瘍壊死因子のレセプターの分子クローニングおよび発現」、Cell、61、361−370(1990);C.Smithら、「腫瘍壊死因子のレセプターが、細胞のおよびウイルス性タンパク質の異常なファミリーを定義する」Science、248、1019(1990)]。一般に、LT−αはしばしばほとんど強力ではないが、LT−αおよびTNFは、インビトロ系で類似の活性スペクトルを示す。[J.Browningら、「いくつかのヒト腫瘍系統の成長に対する腫瘍壊死因子およびリンホトキシンの異なる影響の研究」、J.Immunol.、143、1859(1989)]。
【0006】
TNFは、内毒素ショックヘの応答、および造血細胞発達の調節において、代謝制御の特別な局面で主要な役割をつとめるようである。[B.Beutlerら、「カケクチン(cachectin)の由来、性質、および生物学的効果」、Biochemistry、27、(1988);M.Akashiら、「リンホトキシン:線維芽細胞中のコロニー促進因子の促進および調節」、Blood、74、2383(1989);G.Roodmanら、「腫瘍壊死因子−αおよび造血前駆体:赤血球前駆体CFU−EおよびBFU−Eならびに造血幹系統k562、HL60、およびHEL細胞の成長に対する腫瘍壊死因子の効果」、Exp.Hematol.、15、928(1987)]。
【0007】
IL−1およびIL−6とともに、TNFもまた、炎症性応答の主要なメディエ一ターである。[D.Cavenderら、「腫瘍壊死因子およびリンホトキシンによって誘発される内皮細胞の活性化」、Amer.Jour.Path.、134、551(1989);R.Cotranら、「炎症性および免疫反応における内皮細胞活性化の役割」、Endothelial Cell Biology、(Plenum Press、SimonescuおよびSimonescu編、1988)335]。TNFはまた、ある条件下でT細胞活性化に関与しているようである。[M.Shalabyら、「混合リンパ球反応中のヒト腫瘍壊死因子−αおよび−βの関与」、J.Immunol.、141、499(1988);N.Damleら、「ヒトT細胞活性化のCD3依存およびCD3非依存開始の間のIL−4およびTNF−αの異なる調節効果」、Lymph.Res.、8、85(1989);G.Rangesら、「IL−2依存性T細胞株の増殖シグナルとしての腫瘍壊死因子−α:活性の絶対種特異性」、Amer.Assoc.Immunol、142、1203(1989);G.Rangesら、「腫瘍壊死因子α/カケクチンは胸腺細胞の成長因子である」、J.Exp.Med.、167、1472(1988);P.Scheurichら、「組換えヒト腫瘍壊死因子(TNF)−αの免疫調節活性:ヒトT細胞のTNFレセプターの誘発およびT細胞応答のTNF−α−仲介促進」、J.Immunol.、138、1786(1987)]。
【0008】
TNFは、単球、線維芽細胞、T細胞およびナチュラルキラー(NK)細胞を含む数種のタイプの細胞によって生産される。[D.Goeddelら、「腫瘍壊死因子:遺伝子構造および生物学的活性」、Cold Spring Harbor Symposium Quant.Biol.、51、597(1986);D.Spriggsら、「ヒト上皮腫瘍細胞株での腫瘍壊死因子の発現」、J.Clin.Invest.、81、455(1988);M.Turnerら、「自己免疫および正常個体からのヒトT細胞が腫瘍壊死因子を生産し得る」、Eur.J.Immunol.、17、1807(1987)]。研究者はまた、ネズミまたはヒト形態のTNFを検出し、それらがトランスメンブランタンパク質またはレセプター結合分子のいずれかとして種々の細胞の表面に結合していることを示した。[B.Luettigら、「膜腫瘍壊死因子の2形態の存在についての証拠:内在性タンパク質およびそのレセプターに付着した分子」、J.Immunol.、143、4034(1989);M.Krieglerら、「新規形態のTNF/カケクチンは細胞表面細胞傷害性トランスメンブランタンパク質である:TNFの複合生理学についての分枝」、Cell、53、45−53頁(1988)];およびM.Kinkhabwalaら、「T細胞レパートリーヘの新規追加」、J.Exp.Med.、171、941−946頁(1990)]。
【0009】
LT−αは、TNFの活性とは一般的に類似しているものの同一ではない多くの活性、例えば、腫瘍壊死、抗ウイルス状態の誘発、多形核白血球の活性化、内皮細胞上のクラスI主要組織適合性複合抗原の誘発、内皮細胞の接着分子の誘発および成長ホルモン刺激を含む。[N.RuddleおよびR.Homer、「炎症におけるリンホトキシンの役割」、Prog.Allergy、40、162−182頁(1988)]。LT−αおよびTNFの両方とも、神経成長因子(NGF)レセプターファミリーのメンバーのリガンドである。[S−MallettおよびA.N.Barclay、「神経成長因子レセプターに関連する細胞表面タンパク質の新しいスーパーファミリー」、Immunology Today、12、7、220−223(1991)]。
【0010】
TNFとは対照的に、LT−αの分泌は、活性化されたT細胞およびある種のB−リンパ芽球腫瘍のみの特異的な性質のようである。[N.Paulら、「リンホトキシン」、Ann.Rev.Immunol.、6、407(1988)]。ある研究者たちはまた、LT−αの膜結合形態は、ある環境下でリンパ球の表面上で発現され得ることを示した。[J.Hiserodtら、「インビトロでの異種の抗LT抗血清を使用するマイトジェン活性化ヒトリンパ球上の膜結合リンホトキシン(LT)の同定」、Cell.Immunol.、34、326−339頁(1977);C.wareら、「リンパ球仲介細胞障害性の機構」、J、Immunol.、126、1927−1933頁(1981);U.Andersonら、J.Immunol.Methods.、123、233(1989);Y.Abeら、Jpn.J.Canc.Res.、82、23(1991);Y.Abeら、「インビトロでのヒトリンホカイン活性化Tキラー細胞における膜結合および可溶性(分泌)リンホトキシンの研究」、Lymphokine and Cytokine Research、11、2、115−121(1992)]。
【0011】
近年、TNFおよびLT−αの両方の遺伝子が単離され、そしてクローン化されて、完全な特徴づけがなされ両方の組換え形態のタンパク質が入手可能となった。[P.Grayら、「ヒトリンホトキシンのcDNAのクローニングおよび発現、腫瘍壊死活性を有するリンホカイン」、Nature、312、121−124頁(1984);D.Pennicaら、「ヒト腫瘍壊死因子:前駆体の構造、発現およびリンホトキシンとのホモロジー」、Nature、312、724(1984)]。
【0012】
CD40タンパク質を含有する他の「サイトカイン様」細胞表面タンパク質は、TNFおよびLT−αとある種の類似性を共有していることが近年示された。TNFおよびLT−αと同様に、CD40タンパク質は、TNF/神経成長因子(NGF)レセプターファミリーのメンバーに対するリガンドである。[S.MallettおよびN.Barclay、Immunology Today、12、220−223頁(1991)]。CD40タンパク質は、B−リンパ球、上皮細胞、およびある種の癌腫細胞様の表面に発現する277アミノ酸のタンパク質である。[R.Armitageら、Nature、357、80−82頁(1992);T.FarrahおよびC.Smith、「サイトカインファミリーの発生」、Nature、358、p.26(1992)]。
【0013】
本発明者らは、新規な表面タンパク質、リンホトキシン−β(LT−β)すなわちp33を同定した。LT−βは、種々のタイプのリンパ球の表面で同定されたが、これらのリンパ球は、OKT3刺激一次T細胞、抗原特異的IL−2依存CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマII−23.D7を含む。LT−βは、細胞の中で生産され細胞膜に向かうLT−αを標的としており、細胞膜では、LT−βおよびLT−αは複合体(本開示を通して「LT−α/LT−β」と表示する)のようにみえる。LT−α/LT−βは、活性化T細胞によるLT−αの膜発現に対する新規機構であると考えられている。[Androlewiczら、「リンホトキシンは、活性化ヒトT細胞ハイブリドーマの表面上で別の33kDa糖タンパク質とヘテロマーな複合体として発現される」、Journal of Biological Chemistry、267、2542−2547頁(1992)]。LT−α/LT−β複合体は、可溶性LT−α、TNFおよびCD40タンパク質と類似の細胞溶解および細胞調節活性を示し得る。LT−αと複合した膜結合LT−βは、複合体としてT細胞が他の細胞と相互作用をする新規リガンドを表し得、そしてまた標的細胞の溶解に有用であり得る。
【発明の開示】
【課題を解決するための手段】
【0014】
(発明の要旨)
本発明の新規タンパク質はリンホトキシン−β(LT−β)と命名された。このタンパク質は、種々のタイプのリンパ球の表面で見い出されるが、これらのリンパ球はOKT3刺激一次T細胞、抗原特異的IL−2依存CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマII−23.D7を含む。それはLT−αとの新規複合体を形成し、そして他のLT−βサブユニットとの複合体を形成する(例えば、(LT−β)2LT−α複合体)。
【0015】
LT−βは、31−35kDの分子量を有するが、これは免疫沈降およびSDS−PAGEによって測定された。LT−βは、N−結合グルコシル化を有する。リンホトキシン−βのアミノ酸配列を配列番号2で示し、そしていくつかの可溶性リンホトキシン−βペプチドのアミノ酸配列を配列番号4、配列番号6および配列番号8で示す。リンホトキシン−βをコードするDNA配列を配列番号1で示し、数種の可溶性リンホトキシン−βペプチドをコードするDNA配列を、配列番号3、配列番号5および配列番号7で示す。
【0016】
細胞膜タンパク質としてのLT−βは、合成中のLT−αと結合する。従って、LT−αを細胞膜に「標的化」する。LT−βの非存在下で、LT−αは細胞外培地に分泌される。LT−α/LT−β複合体は、CHO細胞中で発現された組換えリンホトキシン−α(rLT−α)に対して生じたポリクローナル抗血清により、または天然LT−αに対して生じたモノクローナル抗体(mAbs)により認識される。さらに、LT−α/LT−β複合体および(LT−α)3を認識する抗血清は、混合リンパ球反応(MLR)をブロックする。MLRは同種間刺激に対するTリンパ球の予想される増殖応答であり、すなわち、他の個体からのTリンパ球を導入した際に「レスポンダー」リンパ球により異物(非自己)として認識される標準免疫学的アッセイである。[例えばM.Shalabyら、J.Immunl.、141、499(1988)を参照のこと]。
【0017】
LT−βタンパク質をアフィニティクロマトグラフィにより精製し、部分配列を決定し、そして特異的オリゴヌクレオチドプローブを設計した。LT−αをコードするcDNAを、活性化II−23.D7細胞(ホルボールエステル活性化により大量の表面リンホトキシンを表すヒトT細胞ハイブリドーマ)からのcDNAライブラリーをプローブすることによって単離した。同定したアミノ酸配列は、240〜244アミノ酸配列(約25130〜25390kDaの非修飾タンパク質の分子量)をコードする。配列番号2を参照のこと。アミノ酸配列およびトランスメンブラン領域位置はタイプII膜タンパク質の典型である。
【0018】
この配列は、短い14〜18アミノ酸N−末端「細胞質」ドメインを含有する。この細胞質ドメインの後ろに、おそらく膜固定ドメインとして働く30の疎水性アミノ酸の長い広がりがある。同一の配列は利用可能なデータベースには見られなかった。細胞外ドメインには1つのシステイン残基およびC−末端の最後の17アミノ酸に2つのメチオニン残基がある。これは、このタンパク質が示す非常に制限された臭化シアン切断パターンと一致する。
【0019】
LT−β配列とTNF/NGFレセプターファミリーのメンバーに結合することが知られている公知の他のタンパク質を比較すると、かなり構造の類似性がある。TNF/NGFレセプターファミリーのメンバーに対する4つのリガンド(TNF、LT−α、LT−βおよびCD40リガンド)は、タイプII膜タンパク質に類似し、そして図14に示されるような細胞外ドメイン中の保存配列の少なくとも4つの領域を共有している。LT−βと共有する、保存されたTNFおよびLT−αドメインは、サブユニット間相互作用およびβシート構造に関連しているようである。これらの保存領域は、LT−αおよびLT−βの間の結合を説明し得る。これらホモロジー領域の存在は、ポリペプチドを設計して、例えば、TNFまたはCD40リガンドと複合体を容易に形成させ得る。このような分子は、機能を混在し得、そして注文のデザインされた薬として使用され得る。[J.Fuhら、「ヒト成長ホルモンレセプターと拮抗するアンタゴニストの合理的なデザイン」、Science、256、1677(1992)参照のこと]。
【0020】
本発明のペプチド複合体はT細胞活性化の事象に重要であり、T細胞活性化あるいはT細胞抑制のための組成物および方法、ならびに炎症の治療剤として、あるいは腫瘍細胞または新生物の増殖阻害のような細胞障害活性を必要とする適用するための治療剤として有用であると考えられる。また、ポリペプチド複合体は細胞免疫療法に有用であり、腫瘍浸潤リンパ球(「TIL」)治療における腫瘍浸潤リンパ球の殺腫瘍能力を高めることが重要であると考えられる。TIL免疫療法は、遺伝子組換え技術により改善され得る。例えば、遺伝子を、体の免疫系を誘導する目的で腫瘍細胞に加えて、効果的な腫瘍特異的免疫応答を仲介し得る。[例えば、W.F.Anderson、「ヒト遺伝子療法」、Science、256、808−813(1992)参照のこと]。
【0021】
また、「Fas」として同定される分子とTNF/NGFレセプターファミリーのメンバーとの間の類似性に基づいて、本発明のポリペプチド複合体は、プログラムされた細胞死またはアポトーシスとして知られている細胞の内部プロセスに関与し得、それゆえ、自己免疫病の仲介に関与し得ると考えられる。[例えば、N.Itohら、「ヒト細胞表面抗体FasのcDNAによってコードされるポリペプチドはアポトーシスを仲介し得る」、Cell、66、233−243(1991);R.Watanabe−Fukunagaら、「アポトーシスを仲介するFas抗原の欠陥によって説明されるマウスにおけるリンパ増殖疾患」、Nature、356、341(1992)参照のこと]。
【0022】
LT−β、その関連ポリペプチド、LT−α/LT−β複合体または本発明の他のポリペプチド複合体に対する抗体はまた、特定のレセプターとの重要なLT相互作用を破壊する。すなわち、既知のTNFレセプター形態を介して仲介される事象以外の特異的に影響するLT仲介事象を形成し得る。同様に、TNF、LT−αまたはLT−α/LT−β、またはこれらの誘導体(例えば、可溶性レセプターおよびIgG/レセプター融合タンパク質)に対するレセプターも、本発明のポリペプチドおよび複合体の阻害に使用し得る。
【0023】
本発明は、以下を提供する:
(項目1) 配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−β。
【0024】
(項目2) 上記ポリペプチドが細胞表面に結合しているポリペプチドである、項目1に記載のポリペプチド。
【0025】
(項目3) 上記ポリペプチドが、OKT3刺激一次T細胞、抗原特異的IL−2依存性CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマであるII−23.D7に結合している、項目2に記載のポリペプチド。
【0026】
(項目4) 可溶性リンホトキシン−βペプチドであって、以下からなる群から選択されるアミノ酸配列:
(a)配列番号4;
(b)配列番号6;および
(c)以下の式で示されるアミノ酸配列:
X−配列番号6、
ここで、Xは配列番号8の3’末端から始まる1つまたはそれ以上のアミノ酸残基を含有する;
を包含するペプチド。
【0027】
(項目5) 前記ペプチドが、さらに5’末端でのリーダー配列を含有する、項目4に記載のペプチド。
【0028】
(項目6) ポリペプチドであって、以下からなる群からのDNA:
(a)配列番号1を含有するDNA配列;
(b)DNA配列であって、配列番号1によって定義されるDNAにハイブリダイズし、かつリンホトキシン−βと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(c)配列番号1によって定義されるDNA配列によってコードされるポリペプチドをコードする縮重ヌクレオチド配列を含有する、DNA;
によってコードされるアミノ酸配列を含有する、ポリペプチド。
【0029】
(項目7) ポリペプチドであって、以下からなる群からのDNAによってコードされるアミノ酸配列:
(a)配列番号3を含有するDNA配列;
(b)配列番号5を含有するDNA配列;
(c)以下の式で示されるDNA配列:
X−配列番号5、
ここで、Xは、配列番号7の3’末端から始まる1つまたはそれ以上のヌクレオシドトリプレットを含有する;
(d)DNA配列であって、配列番号3、配列番号5および上記(c)部のいずれか1つとハイブリダイズし、可溶性リンホトキシン−βペプチドと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(e)DNA配列であって、配列番号3、配列番号5および上記(c)部の配列のいずれか1つによってコードされるポリペプチドをコードしている、縮重ヌクレオチド配列を含有する、DNA配列;
を含有するポリペプチド。
【0030】
(項目8) 設計されたポリペプチドであって、配列番号2で定義されるアミノ酸配列を包含し、ここで配列Leu Gly Leuが上記配列の5’末端から切断され、かつ1つのMetまたは1っのLeu残基によって置換される、ポリペプチド。
【0031】
(項目9) 単離されたDNA配列であって、以下からなる群:
(a)配列番号1で定義されるヌクレオチド配列を含有するDNA配列;
(b)DNA配列であって、配列番号1によって定義されるDNA配列とハイブリダイズし、かつリンホトキシン−βと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;
(c)リンホトキシン−βをコードする縮重ヌクレオチド配列を含有するDNA配列;から選択される、DNA配列。
【0032】
(項目10) 単離されたDNA配列であって、以下からなる群:
(a)配列番号3で定義されるヌクレオチド配列を含有するDNA配列;
(b)配列番号5で定義されるヌクレオチド配列を含有するDNA配列;
(c)項目7(c)のヌクレオチド配列を含有するDNA配列;
(d)DNA配列であって、配列番号3、配列番号5または項目7(c)の配列のいずれか1つによって定義されるDNA配列にハイブリダイズし、かつ可溶性リンホトキシン−βペプチドと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(e)可溶性リンホトキシン−βペプチドをコードする縮重ヌクレオチド配列を含有するDNA配列;
から選択される、DNA配列。
【0033】
(項目11) 設計されたDNA配列であって、配列番号1で定義されるヌクレオチド配列を含有し、ここでヌクレオチドCTGGGGCTGが上記配列の5’末端から切断され、かつ1つの開始コドンによって置換される、DNA配列。
【0034】
(項目12) 組換えDNA分子であって、以下からなる群:
(a)配列番号1で定義されるDNA配列;
(b)配列番号3で定義されるDNA配列;
(c)配列番号5で定義されるDNA配列;
(d)項目7(c)に記載のDNA配列;
(e)項目11に記載のDNA配列;
(f)DNA配列であって、配列番号1、配列番号3、配列番号5および項目7(c)に記載の配列のいずれか1つによって定義されるDNA配列とハイブリダイズし、かつリンホトキシン−βまたは可溶性リンホトキシン−βペプチドを発現するようにコードする、DNA配列;
(g)縮重ヌクレオチド配列を含有し、リンホトキシン−βに対してコードするDNA配列;および
(h)縮重ヌクレオチド配列を含有し、可溶性リンホトキシン−βペプチドに対してコードするDNA配列;
から選択されるDNA配列を含有する、組換えDNA分子。
【0035】
(項目13) 単細胞宿主、培養中の動物細胞および培養中のヒト細胞からなる群から選択される宿主であって、項目12に記載の組換えDNA分子でトランスフェクトされた、宿主。
【0036】
(項目14) 腫瘍浸潤リンパ球、リンホカイン活性化キラー細胞、キラー細胞および患者から取り出され、遺伝的に設計された腫瘍細胞からなる群から選択される項目13に記載の宿主。
【0037】
(項目15) 項目1〜8のいずれかに記載のポリペプチドを生産する方法であって、上記方法は項目13に記載の形質転換宿主を培養する工程、および該ポリペプチドを回収する工程を包含する、方法。
【0038】
(項目16) ポリペプチド複合体であって、配列番号2、配列番号4、配列番号6、項目8のポリペプチド、および項目4(c)の可溶性リンホトキシン−βペプチドからなる群から選択される第1のポリペプチド、および、リンホトキシン−α、天然ヒトまたは動物リンホトキシン、組換えリンホトキシン、可溶性リンホトキシン、分泌リンホトキシン、あるいはリンホトキシンまたは上記の任意のリンホトキシン活性フラグメントからなる群から選択される第2のポリペプチドを含有する、ポリペプチド複合体。
【0039】
(項目17) 複数のリンホトキシン−βポリペプチドユニットを含有するポリペプチド複合体。
【0040】
(項目18) 項目16に記載のポリペプチド複合体であって、上記複合体が細胞表面と結合している、ポリペプチド複合体。
【0041】
(項目19) 項目18に記載のポリペプチド複合体であって、上記第1のポリペプチドがOKT3刺激一次T細胞、抗原特異的IL−2依存性CTLクローン、およびPMA刺激非リンホトキシンヒトT細胞ハイブリドーマであるII−23.D7の表面と結合している、ポリペプチド複合体。
【0042】
(項目20) 細胞の表面上でリンホトキシンエピトープを生産する方法であって、項目12に記載の組換えDNA分子で該細胞をトランスフェクトする工程、および該細胞中でDNAを発現させる工程を包含する、方法。
【0043】
(項目21) 腫瘍浸潤リンパ球の標的殺腫瘍性活性を高める方法であって、項目12に記載の組換えDNA分子で上記リンパ球をトランスフェクトする工程、および上記形質転換されたリンパ球を患者に導入する工程を包含する、方法。
【0044】
(項目22) 項目21に記載の方法であって、上記形質転換されたリンパ球を項目12に記載の組換えDNAでトランスフェクトする前またはトランスフェクトした後に、リンホカインとともにインキュベートする、方法。
【0045】
(項目23) 項目22に記載の方法であって、前記リンホカインがIL−2である、方法。
【0046】
(項目24) HIV感染、新生物形成、炎症または炎症性の病気、ならびに自己免疫疾患の進行、重篤、または影響を、抑制、処理または軽減するための組成物であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを含有する、組成物。
【0047】
(項目25) HIV感染、新生物形成、炎症または炎症性の病気、ならびに自己免疫疾患の進行、重篤、または影響を、抑制、処理または軽減するための方法であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを投与することを含有する、方法。
【0048】
(項目26) 免疫系を抑制する組成物であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを含有する、組成物。
【0049】
(項目27) 免疫系を抑制する方法であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを投与することを包含する、方法。
【0050】
(項目28) リンホトキシン−βをコードするヌクレオチド配列であって、配列番号1により表される上記ヌクレオチド配列を含有し、さらに5’末端で設計されたヌクレオチド配列を含有し、上記設計されたヌクレオチド配列がATGまたはCTGである機能的な開始コドンを含有し、上記設計されたヌクレオチド配列中で、ロイシンをコードする他のコドンがCTGでない、ヌクレオチド配列。
【0051】
(発明の詳細な説明)
ここに記載された本発明を完全に理解するために、以下に詳細に説明する。
【0052】
本発明は、リンパ球膜タイプポリペプチドであるリンホトキシン−βに関する。リンホトキシン−βのアミノ酸配列を配列番号2に示す。
【0053】
ポリペプチド(またp33という)は、31〜35kDの分子量を有する。本発明のポリペプチドは、細胞表面に結合するか、またはそのような表面に結合しない。
【0054】
本発明はまた、リンホトキシン−βの可溶形態に関する。可溶性リンホトキシン−βペプチドは、リンホトキシン−βのアミノ酸配列で定義され、ここで、配列は、トランスメンブラン領域の端(すなわち、アミノ酸番号44付近)とアミノ酸番号95付近の最初のホモロジー領域との間の任意の位置で切断される。2つの可溶性リンホトキシン−βペプチドのアミノ酸配列は、配列番号4および6で定義されている。いくつかの付加的な可溶性リンホトキシン−βポリペプチドは、配列番号6によって定義されるアミノ酸配列の5’末端に付加的アミノ酸残基を有する配列を包含する。付加的な残基は、配列番号8によって定義される52アミノ酸残基を包含し得る。可溶性リンホトキシンはまた、配列番号6で定義される配列に3’末端から始まる1〜51のアミノ酸残基を含有する配列番号8の一部を加えたアミノ酸配列を包含する。このような可溶性ペプチドは、5’末端に周知のリーダー配列を任意の数包含し得る。このような配列により、真核系でペプチドが発現される。[例えば、Ernstら、米国特許番号第5,082,783(1992)]。
【0055】
本発明のポリペプチド複合体は、配列番号2、配列番号4、配列番号6、および配列番号6プラス上述したような配列番号8の全部または一部分によって定義される配列からなる群から選択されるアミノ酸配列を含有する第1のポリペプチドと、同一群から選択される第2のポリペプチド、および/またはリンホトキシン−α、天然ヒトまたは動物リンホトキシン−α、組換えリンホトキシン−α、可溶性リンホトキシン−α、分泌リンホトキシン−α、または上記の任意のリンホトキシン−α活性フラグメント等との第2のポリペプチドと複合した複合体に関する。
【0056】
新規LT−βペプチドはLT−αと複合体を形成し、そして他のLT−βサブユニットと複合体を形成する(例えば、(LT−β)2LT−α複合体)。これらの複合体は、細胞と結合し得、または結合し得ず、そして上述のように共通ホモロジー領域を共有する他のタイプII膜タンパク質との複合体であり得る。
【0057】
ポリペプチド複合体は、組換えLT−αを認識する多特異性抗血清によって認識され、この複合体はLT−αエピトープを示すことを示唆する。これらの抗血清は、市販の抗LT−αモノクローナル抗体(Boehring Mannheim)、およびトランスフェクトされたチャイニーズハムスター卵巣中(CHO)細胞で発現する組換えリンホトキシン−α(rLT−α)に対して生じる多特異性抗血清を包含する。これらの複合体を認識するポリクローナル抗血清はまた、混合リンパ球反応(MLR)をブロックするが、可溶性LT−αを認識するモノクローナル抗rLT−α抗体はMLRをブロックしない。従って、本発明の複合体は、T細胞活性化における重要な役割を行うようである。また、これらの複合体が可溶性LT−αまたはTNFの活性と同様にT細胞制御活性および細胞障害性を有することが予想される。
【0058】
本発明はまた、上記のアミノ酸配列を有するポリペプチドをコードするDNA配列を本質的に含有するDNA、このDNAによって特徴付けられる組換えDNA分子、本DNAでトランスフェクトされた培養物中の単細胞宿主または動物およびヒト細胞の群から選択される宿主、ならびにこのDNAおよびこれらの組換えDNA分子および宿主を使用しこれらによりコードされるポリペプチドを生産する組換え方法に関する。
【0059】
さらに詳しくは、本発明は、配列番号1によって定義されるヌクレオチド配列を含有する単離されたDNA配列に関する。
【0060】
本発明はさらに、上記配列、リンホトキシン−βと実質的に相同なポリペプチドをコードするDNAとハイブリダイズするDNA配列、およびリンホトキシン−βをコードするヌクレオチド配列を有する縮重DNA配列に関する。
【0061】
本発明はまた、可溶性リンホトキシン−βペプチドをコードするDNA配列に関する。これらのDNA配列は、配列番号3および配列番号5で定義される。本発明はまた、配列番号5のDNAの5’末端にいくつかの付加的なヌクレオチドトリプレットを有するDNAでコードされる数種の付加的な可溶性リンホトキシン−βペプチドに関する。付加的なヌクレオチドトリプレットは、配列番号7によって定義される52トリプレットを包含する。可溶性リンホトキシンペプチドはまた、配列番号5に3’末端から始まる1〜51ヌクレオチドトリプレットを含有する配列番号7の一部分が付加された配列にコードされ得る。
【0062】
本発明はまた、リンホトキシン−βまたは可溶性リンホトキシン−βペプチドを発現するようにコードする上記同定された任意の配列とハイブリダイズするDNA配列に関する。本発明はまた、リンホトキシン−βまたは可溶性リンホトキシン−βペプチドをコードする縮重ヌクレオチド配列、および可溶性リンホトキシン−βペプチドと実質的に相同なペプチドをコードするDNA配列に関する。
【0063】
リンホトキシン−βを同定し、単離しそして以下に記述する技術を使用して特徴付けた。
【0064】
(フローサイトフルオロメトリー分析)
まず、フローサイトフルオロメトリー分析を使用してT細胞の表面上のLT−αエピトープの発現を説明する。OKT3モノクローナル抗体で活性化されたヒト末梢血単核球細胞が、抗rLT−α抗血清と反応することによりLT−αエピトープの発現を示すことが判明した。抗rTNF抗血清ではなく、抗rLT−α抗血清のみが、OKT3刺激一次T細胞に結合した。
【0065】
また、ヒトT細胞ハイブリドーマ、II−23.D7[C.Wareら、「細胞障害性リンホトキシンを生産するヒトT細胞ハイブリドーマ:リンホトキシン放出の誘導および抗CD3モノクローナル抗体またはレクチンおよびホルボールエステルによるキラー細胞活性」、Lymph.Res.、5、313(1986)]が、PMA刺激によりLT−αを分泌すること、さらにまたPMA刺激により表面LT−α関連エピトープを発現することが観察された。また、PMA活性化II−23.D7細胞がウサギ抗rLT−α抗血清からLT中和抗体を除去できたが、一方コントロールU937細胞(表面LT−α形態全てが欠如している)は除去できなかったことが示された。さらに、ウサギ抗rLT−α抗血清がウサギLT−αと結合(複合体化)していた(生じた複合体が次にII−23.D7細胞上の細胞性LT−α/TNFレセプターと結合する)という可能性を排除したが、それは、細胞性レセプターを過剰の可溶性TNFまたはLT−αで飽和し、そしてこれが染色に関して影響を持たなかったことを観察したことによる。これらのアッセイの結果は、ハイブリドーマ上のLT−α関連エピトープが本当にLT−αと関係していることを示す。
【0066】
また、過剰のrLT−αによる抗血清の前処理は、抗血清がII−23.D7細胞を染色する能力を防ぐが、一方rTNFでの前処理は効果を持たなかったということが観察された。このアッセイは、LT−α関連エピトープに対する抗血清の特異性を示す。
【0067】
染色する前に刺激したII−23.D7細胞をトリプシン処理することにより、シグナルを失わせるが、これは、抗血清によって認識されるエピトープはタンパク質であったことを示した。
【0068】
また、CHO由来の混入物が活性化II−23.D7細胞の表面上の誘導タンパク質の抗血清による認識に寄与しなかったことが示されたが、それは可溶性LT−αのみを生産するLT−α遺伝子で安定にトランスフェクトされたCHO細胞が抗LT−α抗血清によって染色されなかったことによる。
【0069】
(免疫沈降)
さらに、これら表面LT−α関連タンパク質を、PMA活性化II−23.D7細胞の表面のヨウ素化(125I−標識化)または代謝標識化(35S−Metまたは35S−Cys)のいずれかにより標識し、続いてデタージェントで原形質膜を可溶化し、標識LT−α関連タンパク質の免疫沈降により特徴付けた。
【0070】
免疫沈降で結合した表面ヨウ素化によって、抗LT−α抗血清によって認識される2つのタンパク質:以降LT−αという25〜26kD形態、および以後、LT−βまたはp33という31〜35kD形態が明らかとなった。同じウサギからの免疫前抗血清も抗rTNF−ウサギ血清のいずれもヨウ素化されたPMA活性化II−23.D7細胞からのバンドを免疫沈降しなかったことを観察した。ヨウ素化し免疫沈降したバンドの一次元部分的CNBr処理ペプチドマップは、25〜26kD形態(LT−α)の切断によりヨウ素化した組換えLT−αのものと同一のパターンを示した。このことは、表面LT−αと可溶性LT−αとの相互関係を補強する。ヨウ素化された31〜35kD形態(LT−β、すなわちp33)はCNBrによって切断されず、公知のLT−α遺伝子生成物とは別であることが示された。
【0071】
さらに、PMA活性化II−23.D7細胞を35S−メチオニンまたは35S−システインで代謝標識し、LT−αおよびLT−βタンパク質を特徴付けた。システインおよびメチオニンの分布は、TNFとLT−αとの間、ならびに、シグナル配列を有する形態と有しない形態との間を区別するための方法が提供される。[M.Krieglerら、Cell、53、45(1988)]。分泌されたTNFはシステインを含有するが、メチオニンはなく、一方、分泌されたLT−αはメチオニンのみを含有し、システイン残基はない。しかし、LT−αはシグナル配列中に1つのシステイン残基を有するが、一方、TNFはシグナル配列中に2つのメチオニン残基を含有する。
【0072】
35S−メチオニンまたは35S−システインのいずれかでPMA処理II−23.D7細胞をそれぞれ培養して標識し、そして培地および細胞からの免疫反応性タンパク質を沈降した。次に、35S−メチオニンで標識した細胞の培地からの免疫沈降物をSDS−PAGE分析すると、LT−αの25kD形態が現れたが、一方35S−システインで標識した細胞の培地からの免疫沈降物では現れず、分泌LT−αに対して予測されるパターンではなかった。洗浄した細胞の分析では、25〜26kD LT−α形態および33kD LT−β形態の両方を示した。これらの結果は、表面ヨウ素化を用いて観察された膜結合形態と同じである。
【0073】
25〜26kD LT−αはシステインを欠如しており、これはリーディング配列のプロセッシングを示している。また、33kD LT−βは35S−メチオニンと35S−システインとの両方を取り込み、それ自身が25kD LT−α形態とは異なる形態であることが観察された。典型的に、そのレセプターと結合したLT−αは、BOSCOES(すなわち、ビス[2−[スクシンイミドオキシ−カルボニルオキシ]エチル]スルホン;Pierce、Rockford、IL)のような化学的架橋剤を用いてレセプターと架橋し得る。[J.S.Andrewsら、「ヒトTリンパ球上の腫瘍壊死因子(TNF)およびリンホトキシン(LT)に対するレセプターの特徴付け」、J.Immunol.、144、2582(1990)]。表面ヨウ素化II−23.D7細胞を架橋剤で処理した時、25〜26kD LT−αまたは33kD LT−β関連形態のいずれかが、他の付加的膜タンパク質と結合しなかったことが観察された。このアッセイは、レセプター結合はLT−αおよびLT−βが細胞膜と結合したままのメカニズムではないことを示した。LT−βの配列分析は、2つのTNFレセプター形態のいずれとも関係ないことを示した。[C.Smithら、Science、248、1019、(1990);T.J.Schallら、Cell、61、361(1990)]。
【0074】
(アフィニティークロマトグラフィー)
II−23.D7細胞の表面上のLT−βおよびLT−αタンパク質のさらなる特徴付けを、アフィニティークロマトグラフィーを用いて行った。PMA処理II−23.D7細胞の表面上のLT−α/LT−β複合体がレンチルレクチンと結合することが観察されたが、これは、それぞれの形態が糖タンパク質構造であることを示す。従って、精製工程としてレンチルレクチンクロマトグラフィー工程を、抗血清アフィニティークロマトグラフィーの前に使用した。デタージェントで可溶化したPMA処理II−23.D7タンパク質をレンチルレクチンセファロースに結合し、そしてα−メチルマンノシドで溶出した。コントロールIgGおよび抗LT−α−IgGアフィニティーカラムの両方を調製し、これらのタンパク質が抗LT−α抗血清によって特異的に認識されることを正確に評価した。次に、レンチルレクチンに結合したタンパク質をこれらのカラムに適用した。カラムを低pHで溶出したときに抗LT−αアフィニティーカラムからLT−βおよびLT−αタンパク質が放出されることを観察した。溶出液のSDS−PAGE分析結果は、表面ヨウ素化PMA処理II−23.D7細胞からの免疫沈降タンパク質のSDS−PAGE分析結果とかなり類似していた。この比較は、類似のタンパク質が2つの方法によって精製されたことを示した。
【0075】
アフィニティー精製の間、約25kD LT−α形態が切断されて19〜20kD形態、すなわち「デス(des)20」形態にとなるようであることを観察した。RPMI 1788腫瘍細胞株からの天然LT−αの最初の単離[B.Aggarwalら、「1788腫瘍細胞株由来のヒトリンホトキシンの一次構造」、J.Bio.Chem.、260、2334−2344頁(1985)]もまた、N−末端的に切断された20kD LT−α形態を与えていた。この「デス20」天然型LT−α中の1つのメチオニンが失われており、無傷の分子と比べると異なるCNBr切断パターンとなる。アフィニティー精製LT−αタンパク質の一次元CNBr切断は、短縮型の天然LT−α形態と一致する切断パターンを示し、そして、アフィニティー精製「デス20」LT−α形態はおそらく天然LT−α「デス20」で観測されたような類似の切断から生じたと結論した。
【0076】
さらに、LT−βが部分的CNBr切断でダブレットを生じることを観察した。LT−βタンパク質による切断パターンは、メチオニン残基が存在すること、および少なくとも1つのメチオニンがC−またはN−末端いずれかから5〜20残基の範囲内にある(このマッピング技術を使用して切断した時、いずれかの末端の1〜5残基の範囲内でメチオニン残基は検出されなかった)。このパターンは、LT−βは公知のLT−α配列の全部を含有していないことを示唆した。
【0077】
LT−βタンパク質はまた、抗原活性化一次細胞障害性Tリンパ球クローンによって発現されることが観察された。これらの細胞の代謝標識後の抗rLT−αを用いる免疫沈降の結果は、少量のLT−αと共にLT−βが現れた。これらの結果は、LT−βは一次T細胞およびII−23.D7ハイブリドーマによって作成され得ることを示した。
【0078】
(LT−βおよびLT−αタンパク質の初期精製)
以下の一般的な工程を使用してLT−αおよびLT−βタンパク質を精製した。まずホルボールミリスチンアセテート(PMA)をII−23.D7細胞に添加した。24時間後に、細胞を収集し、それらを無血清冷RPMI培地で洗浄した。冷たい細胞ペレットに、氷冷溶解緩衝液(HEPES、NP−40、EDTA、NaCl、およびアジ化ナトリウム)を加え、それにベンズアミジン、フェニルメチルスルホニルクロライド(PMSF)、ならびにN−エチルマレイミド(NEM)、ダイズトリプシンインヒビター、ペプスタチンおよびアプロチニンを新たに添加した。Dounceホモジナイザーで細胞を穏やかに均一化し、そして溶解液を遠心分離した。遠心分離し、そして上清を採取した。CaCl2およびMnCl2を添加した溶解緩衝液で予め平衡化したレンチルレクチンセファロースカラムに、上清をロードした。CaCl2およびMnCl2を有する溶解緩衝液でカラムを洗浄し、次にα−メチルマンノシドを含有する溶解緩衝液で溶出した。溶出画分をプールし、そしてウサギ非特異的IgGセファロースアフィニティーカラムの上にロードしたが、このカラムはウサギ抗rLT−αセファロースアフィニティーカラムに直接連結していた。EDTAを含有する同じ溶解緩衝液で、次にNP−40をMEGA−8(オクタノイル−N−メチルグルカミド、Boehringer−Mannheim)で置換した溶解緩衝液で両方のカラムを洗浄した。洗浄したカラムをそれぞれ別個に5mM MEGA−8、50mM グリシン、NaCl、ベンズアミジン、およびEDTA溶液で溶出したが、その方法は、Browningらの「リンホトキシンおよび関連33−kD糖タンパクは、活性化ヒトT細胞ハイブリドーマの表面上で発現される」、J.Immunol.147、1230−1237頁(1991)に記載された方法に従った。pHシフト後の第1回分をプールし、凍結乾燥し、SDSで水に再懸濁し、そしてHEPESおよびSDSの溶液に対して透析した。speed−vacで透析画分を乾燥し、そして水に再懸濁した。アリコートをLaemmliローディング緩衝液と混合し、そしてSDS−PAGE上で電気泳動した。銀染色でタンパク質を視覚化した。
【0079】
1つまたは複数のLT−αエピトープがPMA処理で生じるような細胞活性化によってのみ、II−23.D7T細胞ハイブリドーマの表面に存在することが観察された。反対に、一次T細胞上で存在する時、PMA処理により表面抗原が失われる。さらに、組換えLT−α(CHO細胞で生産される)あるいは天然LT−α(例えば、Genzyme、Boston、Mass.)のいずれかに対するウサギポリクローナル抗血清が1つまたは複数のLT−αエピトープを認識したことを見出した。
【0080】
また、LT−α/LT−β複合体を認識するこの抗血清はMLRをブロックするが、可溶性LT−αを認識する特定のモノクローナル抗体はブロックしないことを観察した[M.Shalabyら、J.Immunol.、141、499(1988)]。それゆえ、本発明のLT−α/LT−β複合体は、T細胞の活性化のメディエーターであり得る。
【0081】
細胞溶解物からの免疫沈降物中のLT−βがLT−αと共に存在することは、LT−βが免疫原性的にLT−αと関連するかあるいはLT−βがLT−αと結合されるか、または両方を示唆している。この問題を解決するために、35S−メチオニン標識細胞からの25kDおよび33kDバンドを、ウサギポリクローナル抗rLT−α血清で免疫沈降し、切り出したゲルスライスから溶出し、そして抗rLT−αポリクローナル抗血清または抗rLT−α mAbのいずれかで再免疫沈降した。LT−α(LT−βではなく)は、抗rLT−α抗体のいずれかで免疫沈降し、LT−βが免疫原性的にLT−αと関連しないことを示唆した。これらの観察は、LT−βが物理的にLT−αと結合していることを示した。
【0082】
表面LT−αおよびLT−βが複合体を形成するという仮説をさらに検証するために、変性および未変性条件下で等電点フォーカシング法(IEF)実験を行った。LT−αおよびLT−βが物理的に結合されれば、これらは未変性条件下では複合体としてフォーカスするはずであるが、変性条件下では分離した形でフォーカスするという理論的根拠に基づく。LT−αおよびLT−βに対する個々の等電点(pI)を、二次元ゲル分析(変性条件)によって決定した(図9A)。LT−αは、6.5〜7.3のpIの範囲で5つの荷電化したアイソマーを有するが、LT−βは5.5〜6.0のpIの範囲で4つの荷電化したアイソマーを有する。しかし、フォーカシング法を未変性条件下で行った時、LT−αおよびLT−βは共に6.3〜7.2のpIの範囲で広いバンド幅で集中した。それゆえ、LT−βの泳動は、未変性条件下で顕著に遅れた。
【0083】
(LT−αおよびLT−βのさらなる精製および同定)
LT−αおよびLT−βタンパク質を以下の一般的な工程を使用して精製した。ウシ胎児血清を含むRPMI培地でII−23.D7細胞を培養し、そして細胞を50lのRPMIから集め、そして培地に再懸濁し、そしてホルボールミリスチンアセテート(PMA)を添加した。6時間の活性化の後、遠心分離によって細胞を収集し、Dulbeccoのリン酸緩衝液生理食塩水で洗浄した。冷溶解緩衝液に最終細胞ペレットを懸濁し、そしてペレットをいったん窒素キャビテーターの中を通過させた。溶解した細胞を遠心分離し、そして上清を捨てた。一晩デタージェントと共に溶解緩衝液中でペレットを抽出し、次に再び遠心分離した。
【0084】
デタージェントで可溶化した膜を含有する上清を、Affi−gel(10)と結合したモノクローナル抗LT−αを含むアフィニティー樹脂に添加し、懸濁液を一晩振動した。樹脂を小さいカラム中に回収し、nonidet P40を含むHEPESで、次に1% w/v MEGA−8を含む同じ緩衝液でそれを洗浄し、MEGA−8を含むグリシン緩衝液で結合したタンパク質を溶出し、そしてその画分をトリス塩基で直ちに中和した。画分中にLT−βおよびLT−αの存在することをSDS−PAGE分析および銀染色法によって確認した。これらタンパク質を含有する画分をプールし、そしてSDSを添加し、そしてこのプールを0.l×laemmliサンプル緩衝液に対して透析した(MEGA−8デタージェントを除去するため何度も取り替えた)。透析した溶液を乾燥状態まで、凍結乾燥して、元の1/10量の水に再懸濁した。
【0085】
SDS−PAGEゲル上にサンプルを流し、ProBlotメンプラン上にプロットし、そしてクーマシーブルー染料で染色した。LT−βおよびLT−αバンドを切り出し、そしてタンパク質シーケンサーにロードした。エドマン分解によってN−末端配列を得た。膜に結合したLT−αバンドの配列は、分泌性のLT−αについて記載された配列と正確に一致することがわかった。すなわちLeu Pro Gly Val Gly Leu Thr Pro Ser(アミノ酸番号1〜9)[P.Grayら、Nature、312、121−124頁(1984)]と一致する。エドマン分解分析によって、結合LT−βタンパク質のN−末端部分のアミノ酸配列が2つのアミノ酸配列:Gly Leu Glu Gly Arg Gly Gln Arg Leu GlnまたはGly Leu Glu Gly Arg Leu Gln Arg Leu Glnを含む可能性があることを示した。引き続きより正確なcDNA技術を使用するDNA分析で、正しい配列はGly Leu Glu Gly Arg Gly Gly Arg Leu Glnであったことを確認した。
【0086】
LT−α形態が検出されたそれぞれの場合で、また、LT−βを検出し得た(すなわち、PMA活性化II−23.D7細胞、活性化CTLクローン、およびHut−78細胞の表面LT−α形態を構成的に発現する)。LT−αは表面LT−αの非存在下でトランスフェクトされたCHO細胞から分泌され、そしてLT−βの存在が表面結合LT−αと関連するので、LT−βはLT−αと複合体を形成し細胞表面でそれを標的化すると結論付けた。生化学的には、LT−βおよびLT−αは非変性等電点フォーカシングゲル上で共に泳動するが、複合体が尿素で解離すると、2つのタンパク質が分離して流れる。[図9A、10A参照のこと]。これらの観察は、LT−αおよびLT−βは細胞表面上で複合体として存在するという結論に導いた。
【0087】
(リンホトキシン−βおよび可溶性リンホトシン−βペプチドに対してコードするDNA配列の同定)
リンホトキシン−βを、上述したように免疫アフィニティークロマトグラフィーにより精製した。直接N−末端配列決定およびインサイチュトリプシン分解に続いて、分解ペプチドの逆相HPLC分離を行った。[Abersoldら、「ニトロセルロース上のインサイチュプロテアーゼ分解後の一次元または二次元ゲル電気泳動法によって分離されるタンパク質の内部アミノ酸配列分析」、PNAS、84、6970−6974(1987)参照のこと]。次に得られたN−末端および内部トリプシンフラグメントペプチドの配列を通常の方法を使用して決定した。N−末端およびT105、T87/88、T110およびT67と命名した内部ペプチドの配列決定を図13に示す。
【0088】
2つのアンチセンス17−merオリゴヌクレオチドプローブGTYTCNGGCTCYTCYTC[配列番号9]およびGTYTCNGGTTCYTCYTC[配列番号10](それぞれ1368および1369と命名した)を、ペプチドT−87/T−88の配列の一部分と一致するように合成し、そして32Pで放射能標識した。ノーザン分析の結果、1368と命名されたプローブが、0.9〜1.1kb mRNAバンドに強くハイブリダイズしたことを示した。このmRNAは、前述したようにホルボールエステルで前処理したII−23.D7細胞中で強く誘導された。
【0089】
ベクターpCDM8中のcDNAライブラリーを、PMAで6時間誘導したII−23.D7細胞から単離されたポリA+mRNAから構築した。ライブラリーを1368と命名された標識オリゴマーでスクリーニングし、そしてポジティブクローンを単離し、次に50℃で3M テトラメチルアンモニウムクロライドで洗浄した。0.8〜0.9kD挿入物を有するいくつかの(>16)クローンを、DNA配列分析にかけた。
【0090】
クローンpCDM8/LT−β−12は、配列番号1に示されるリンホトキシン−βをコードする配列を含んでいた。他のクローンは、5’末端で種々の短縮形であることを除いて、同一であった。クローン12 cDNAは、機能的なリンホトキシン−βをコードする。標準プライマー伸長法を用いて、アミノ酸残基−−MET GLY ALA−−をコードする付加的なコドンを同定した。862〜867位の終止コドン AATAAAは、3’ポリA領域のちょうど前に、あり、これは全3’末端領域が同定されたことを示す。タンパク質コード配列は、240アミノ酸をコードし非修飾分子量で約25,390kDaと計算される。
【0091】
クローン12 cDNAおよびプライマー伸長から同定されたLT−β DNA配列の5’末端、ATGGGGGCACTGGGGCTG[配列番号11]から、3つの開始コドン(下線)の可能性があることが明らかとなった。[例えば、M.Kozak、「脊椎動物mRNA配列の分析:翻訳調節の暗示」、J.Cell.Biol.、115、4、887−903(1991)]。設計されたLT−βポリペプチドおよびDNA配列は、この5’配列の全てまたは一部分を切断することならびに1つの開始コドンを置換することで誘導し得る。
【0092】
このアミノ酸配列プロフィルは、タイプII膜タンパク質に典型的である。短い(最大17)アミノ酸配列N−末端「細胞質」ドメインに続いて、膜固定ドメインとしておそらく働く30の疎水性アミノ酸が拡がる範囲がある。入手可能なデータベースでは同一の配列が発見されなかった。細胞外ドメイン中に1つのシステイン残基があり、そして最後のC−末端17アミノ酸中に2つのメチオニンがある。これは、このタンパク質で示される非常に制限された臭化シアン切断パターンと一致する。
【0093】
LT−βの配列を決定した後の比較では、LT−βは、タイプII膜タンパク質であり、TNF、LT−αおよびCD40タンパク質に対して有意に相同性を有することが明らかになった。これらのポリペプチドは、細胞外ドメイン中に4つの配列保存領域を共有している。図14参照のこと。このような保存領域は、これらのポリペプチドがお互いに複合体を形成するのを可能にするようである。[以下を参照のこと;例えば、M.EckおよびS.Sprang、「2.6Å分解能での腫瘍壊死因子−αの構造、レセプター結合との関係」、J.Bioloical Chemistry、264、29、17595−17605頁(1989);E.Jonesら、「腫瘍壊死因子の構造」、Nature、338、225−228頁(1989);M.Eckら、「1.9Å分解能でのヒトリンホトキシン(腫瘍壊死因子−β)の構造」、J.Bioloical Chemistry、267、4、2119−2112頁(1992);J.Tavernierら、「腫瘍壊死因子およびリンホトキシンの保存残基は、トリマー構造の骨組みを構成する」、Fed.Eur.Biochem.Soc.Lett.、257、2、(1989)]。
【0094】
(クローン化LT−βの発現)
pCDM8/LT−βクローン12*(*プラスミドpCDM/LT−βクローン12を有するE.coli K12は、BN1289(MC1061/P3/P33−クローン−12)と命名され、1992年11月13日にATCCに寄託された。)またはコントロールプラスミド、クローン4(非機能性LT−β cDNA挿入断片を有するpCDM8)を、エレクトロポレーションによって、安定にヒトLT−αでトランスフェクトしたCHO dhfr−細胞およびCHO細胞中に導入した。3日後、細胞を5mM EDTAを含むCa/Mgを含まないHank溶液に取り出し、そして上述したような10μg/mlコントロールIgG1または抗LT−αモノクローナル抗体(Boehringer−Mannheim)のいずれかを使用したFACS分析のために染色し、その後、FITCまたはフィコエリトリン標識ヤギ抗マウス調製物のいずれかで、結合免疫グロブリンを標識した。
【0095】
他の実験において、COS細胞を、pCDM8ベクター中でもまた等量のヒトLT−αcDNAの存在または非存在下で、pCDM8中のクローン4またはクローン12のLT−βcDNAのいずれかでエレクトポレートし、そして上記のように3日後FACS分析のために染色した。LT−αを発現するCHO細胞のみが、機能性LT−β DNA(すなわち、クローン12)でのトランスフェクションにおいて表面リンホトキシンを示した。
【0096】
クローン12は開始ATGコドンを欠損しているが、いくつかのCTG開始コドンを有しており、それゆえこの発現実験は1つまたはいくつかの5’CTGコドンが翻訳を開始しなければならないことを示す。CTGコドンは、いくつかの真核タンパク質の翻訳に対する開始部位となることは公知である。[M.Kozak、J.Cell.Biol、115、4、887−903(1991)]。LT−αおよびLT−β DNAの両方を受け入れるCOS細胞のみが、FACS分析中の実質的な表面LT−αを示すような同様の結果を、COS細胞中の2つのトランスフェクション系を使用して観測した。
【0097】
(LT−βおよびLT−αならびにLT−α/LT−βのポテンシャル使用)
上記のように、LT−β、LT−α、TNFおよびCD40リガンドの間にかなりの構造的類似性がある。LT−β、LT−α、TNFおよびCD40リガンドはタイプII膜タンパク質であり、そして細胞外ドメイン中に少なくとも4つの配列保存領域を共有している。
【0098】
構造的類似性の観点において、LT−αが、分泌される分子と同一の形態で活性化リンパ球の表面で見られるが、LT−αを表面におそらく固定する他の不可欠な膜タンパク質と複合されるという問題がある。発明者らは、この独特の複合体(すでにLT−α/LT−βであると決定された)が、LT−αのさらに関連している形態を示し、TNFと関連する特異性を与えると考えている。
【0099】
リンホカインのヘテロメトリックの複合体の存在は、免疫系に独特であるが、他の領域中のシグナル分子を暗示する(例えば、PDGFおよびインヒビン/アクチビンヘテロメトリック複合体)。LT−α/LT−β複合体の記述は、LT−αホモトリマーによって模倣できない複合体独特の免疫調節活性の可能性を提供する。複合体は、高い親和性相互作用および生化学的に関連したシグナルを導く独特のレセプターまたはレセプター鎖組合せに結合し得る。独特のレセプター複合体とのLTα−/LT−β相互作用の仮説は、多くの系でのTNF(2つの公知TNFレセプターの研究によって説明できない観測)と比較してLT−αホモトリマーの相対的に弱い活性を説明し得た。[T.Schallら、Cell、61、361−370(1990);C.Smithら、Science、248、1019(1990)]。
【0100】
LT−βとの複合体化を経て可溶性LT−αを細胞表面につなぐことは、LT−α/LT−βを介する細胞−細胞接触特異的シグナルが、免疫調節の重要な面であり得ることを示す。TNFおよび関連したLT−α形態は分泌されるので、LT−βもまた分泌され得ると考えられる。これは、抗LT−βモノクローナル抗体を使用する研究で実証し得る。このような抗体はまた、LT−βホモ−オリゴマーが自然に生じるかどうか決定するために使用され得る。
【0101】
一般的に、LT一およびTNFは、定性的に同じ活性スペクトルを示し、そしてLT−αおよびTNFは同じセットのレセプター(55および80kD TNFレセプターと呼ぶ)と相互作用すると考えられる。[C.Smithら、Science、248、1019(1990);T.Schallら、Cell、61、361(1990)]。それでもなお、LT−αおよびTNFによって示される生物学的能力の定量的パターンは劇的に異なり、LT−αはしばしばTNFよりさらに能力が少ない[例えば、Browningら、J.Immunol.、143、1859(1989)を参照のこと]。これらの観測は、存在するレセプター結合データと一致させることが難しい。LT−α/LT−β複合体は、他の現在まで不確定のレセプターと相互作用するような、独特の特性をLT−αに与える可能性がある。この場合、LT−α/LT−β複合体および本発明の他の複合体は、LT−αまたはTNFのいずれかと区別する独特の生物学的特性を有する。LT−α/LT−β複合体は、例えばLT−α/LT−βまたはLT−β特異的レセプターを同定し、そしてクローニングするために使用され得る。さらに、さらなる複合体の使用は、新規な生物学的活性を明らかにし得る。
【0102】
また、多数のT細胞およびマクロファージ細胞株がHIVウイルスによって感染可能であることは公知であるが、実際には、少数の細胞様が組織培養中のウイルスを増殖するのに有用である。例えば、H9株(Galloらによって最初に開発されたHut−78の誘導体[M.Popovicら、Science、224、497−500(1984)])、および他のヒトリンパ球株(C8166)は、HIV増殖に対して有用であった。[M.SomasundaranおよびH.Robinson、Science、242、1554−1557(1988)]。表面LT−α発現または表面LT−αの発現の能力は、所定の細胞をHIV増殖に対して良い標識にし得る。
【0103】
HIV増殖を高めることにおけるTNFの役割が提案された[L.Osbornら、Proc、Natl.Acad.Sci.USA、86、2336(1989);Z.RosenbergおよびA.Fauci、Immunol.Today、11、176(1990);C.Locardiら、J.Virology、64、5874(1990);G.Oliら、Proc.Natl.Acad、Sci.USA、87、782(1990)]。発明者らは、II−23.D7株がHIV株IIIBで感染可能であるが、PMA処理でこのウイルスによる感染が劇的に増加することを発見した。Hut−78細胞株が、表面LT形態を構成的に発現することが見出され、そしてC8166株は、その後のPMA処理で明らかとなる表面LT中でII−23.D7と似ている。[Wareら、J.Immunol.、印刷中(1992)]。
【0104】
これらのHIVによるII−23.D7の感染可能性の結果、および感染可能な細胞株と表面LT発現との関係を考慮すると、これらの株がHIV感染および複製に対し良い宿主であり得ることを提案する。なぜなら、LT−α/LT−β複合体、および本発明の他のポリペプチドおよび複合体が調節の役割を果たすからである。LT−α遺伝子がHIV転写活性化因子TATの発現によって誘導されることが証明され[K.Sastryら、J.Biol.Chem.、265、20091(1990)]、そして、さらに、HTLV−1感染もまたLT−α発現を誘導することが示された[N.Paulら、J.Virol.、64、5412(1990);E.Tschachlerら、Human Retrovirology(Raven Press 1990)、W.Blattner編、105頁]。従って、HIV感染および続いて起こるLT−α/LT−β複合体または他の複合体の発現によるLT−αの誘導、またはこれらのタンパク質を作る能力がある細胞株でのPMA処理によるLT−α/LT−βまたは他の複合体の発現の誘導は、ウイルス複製を高めるために用い得る。この理由のために、LT−α/LT−β複合体もしくは本発明の他のポリペプチド複合体、またはこれらの複合体の可溶性形態、またはLT−βおよび本発明の他のポリペプチドに対する抗体または特異的結合タンパク質(例えば、可溶性レセプター)は、HIV増殖を阻害し得、またはHIV誘導T細胞死をブロックし得る。
【0105】
LT−α/LT−βとCD40レセプターリガンドペアとの間の類似点が示され得、ここでT細胞表面CD40リガンドからのシグナルは、CD40レセプターを介してB細胞へ「ヘルプ」を与える。従って、表面LT−α/LT−βが、T細胞または他の造血系(LAKまたはNK細胞およびB−細胞など)のT細胞調節の成分であり得ると仮定し得る。さらに、この相互作用は、いくつかの自己免疫疾患中の機能不全であり得る。[例えば、R.Watanabe−Fukunaga、Nature、356、314−317頁(1992)を参照のこと]。
【0106】
さらに、Fas抗原と呼ばれる細胞表面タンパク質は、TNF、NGF、およびCD40タンパク質を含む多数の細胞表面レセプターとかなりの構造的相同性を有することを示した。Fas抗原は、アポトーシスを仲介することに関係しており、プロセスもまたプログラム細胞死として関連している[R.Watanabe−Fukunaga、Nature、356、314−317頁(1992);N.Itohら、Cell、66、233−243頁(1991)]。Fas抗原の欠損を説明するマウスの株は、全身性エリテマトーデス様自己免疫疾患を発生する。これは、構造的に類似しているLT−βまたはLT−α/LT−β複合体もまた、全身性エリテマトーデスを仲介する役割を行い得、それゆえ、この経路中の介入は、種々の自己免疫疾患を治療することに臨床的に有用であり得る。あるいは、LT−βまたはLT−α/LT−β複合体は、細胞−細胞接触依存機構を介するプログラム細胞死を誘導することに関連し得る。すでに大規模なTNF/NGFタイプレセプターのファミリーを補充するためのTNF関連リガンドのこのファミリーの出現は、免疫系中での重要な調節因子の追加配列の存在を示唆する。
【0107】
LT−βまたはLT−β/LT−α複合体は、免疫系を抑制する役割を同様に行い得、そしてアレルギーを治療し、耐性を誘発することに潜在的に有用であり得る。
【0108】
ゲノムのMHC領域のTNF/LT遺伝子座の位置は、研究者らに種々の自己免疫疾患、特にインスリン依存性真性糖尿病に対する関連を調査させた。[例えば、F.Pociotら、「モノカイン分泌とインスリン依存性真性糖尿病との関係における腫瘍壊死因子β遺伝子多形性」、Scand.J.Immunol.、33、37−49(1991);K.Badenhoopら、「(インスリン依存性)真性糖尿病タイプ1におけるTNF−α遺伝子多形性」、Diabetologia、32、445−448(1989)を参照のこと]。発明者らは、LT−β遺伝子がTNF/LT遺伝子座の次に位置することを発見したので、LT−β遺伝子またはそのレセプターがこの自己免疫条件に関連する可能性がある。従って、LT−βまたはそのレセプターまたはLT−βに対する抗体は、糖尿病のこの形態における置換療法を包含する。
【0109】
上述したように、LT−βポリペプチド、および本発明のポリペプチド複合体は、抗腫瘍、T細胞活性化、またはT細胞抑制適用を含む多数の可能性のある使用、全身性エリテマトーデスの治療を含む適用、および抗炎症性の組成物での使用および方法を有することが期待される。次に、LT−βポリペプチドをコードするDNA配列、同様なDNA配列を含む組換えDNA分子、ならびに同様の組換えDNA分子でトランスフェクトした培養物中の単細胞宿主および動物またはヒト細胞を用いて、組成物および上記の療法で使用するために、実質的に他のヒトタンパク質を含まない大量の本発明のポリペプチドを生成し得る。
【0110】
本発明のポリペプチド複合体の表面上で発現するリンパ球、および好ましくはLT−α/LT−β複合体は、腫瘍細胞を殺す能力を高め得たリンパ球のサブセットを示す。同様に、このサブセットは、LAK(リンホトキシン活性化キラー)細胞またはTIL(腫瘍浸潤リンパ球)細胞療法に有用であり得る。[H.Thomas、K.Sikora、「癌療法への生物学的アプローチ」、Jour.Int.Med.Res.、17、191(1989)]。TIL免疫療法は、遺伝子転移技法によって改善され得る。LT−βについての組換え遺伝子およびそれに基づく関連のポリペプチドは、例えばTIL療法で、治療上有用であり、ここでLT−β遺伝子は(LT−α遺伝子と共にまたはLT−α遺伝子なしのいずれかで)、腫瘍から単離したT細胞に導入し、そして患者に導入され得る。さらに好ましくは、細胞を患者から取り出し、本発明のポリペプチドを発現するようにコードするDNA配列でトランスフェクトし、トランスフェクションの前または後でリンホカイン、好ましくはIL−2でインキュベートし、そして患者に戻す。トランスフェクトしたT細胞(すでにLT−βを発現し、そしてまたその後LT−αと複合化している)は、取り出された腫瘍に戻り、そこでLT−αの殺腫瘍性作用を直接腫瘍に伝える。同様に、LAK細胞中に導入されるLT−β遺伝子が細胞上の表面複合体の数を増加させ、そしてそれらの活性を高めることが考えられる。あるいは、患者腫瘍細胞中へのLT−β遺伝子の導入は、LT−β改変腫瘍が、腫瘍自体に対する高められた免疫応答を誘発する腫瘍ワクチンを造り出すことに、有用であり得る。[例えば、W.F.Anderson、Science、256、808−813(1992)を参照のこと]。
【0111】
本発明のポリペプチドおよびポリペプチド複合体に対する抗体または抗体誘導体もまた、この細胞集団を富化するための通常の免疫学的方法(例えば、パンニングまたはフローサイトフルオロメトリー選別)に有用である。[L.J.WysockiおよびV.L.Sato、「リンパ球についてのパンニング:細胞選別の方法」、PNAS、75、2844(1978)]。
【0112】
本発明のポリペプチドおよびポリペプチド複合体、またはフラグメントまたはその誘導体は、リンホトキシン−αおよび腫瘍壊死因子が使用されるのと同様に、細胞調節または治療の適用に有用である。
【0113】
本発明の組成物は、特定の臨床状態に対して治療するために、有効な用量で投与され得る。所定の適用についての特定の用量の決定は、例えば、患者の状態および体重、所望の治療の程度および治療に対する患者の耐性を、当該技術分野では十分考慮に入れる。本発明の複合体およびポリペプチド、またはおそらくそれに由来するペプチド、またはそれからもしくはそれらのアミノ酸配列を使用して合成されるペプチド(ポリペプチドの単離および精製形態を包含する)、またはそれらの塩または薬学的に受容可能な誘導体の投与は、抗腫瘍、T細胞活性化、T細胞抑制または抗炎症活性を示す薬剤の任意の従来受容された態様を介するものであり得る。
【0114】
これらの療法で使用される組成物もまた、種々の形態であり得る。これらは、例えば、錠剤、丸剤、散剤、液体溶液または懸濁液、注入可能または浸出可能な溶液のような、固体、半固体、および液体の投与形態、および遺伝子療法を包含し得る。好ましい形態は、投与の意図した形式および療法の適用に依存する。投与の態様は、経口、非経口、皮下、静脈内、病変内、または局所投与を包含し得る。組成物はまた、好ましくは、通常の薬学的に受容可能なキャリアを包含し、そして他の医薬剤、キャリア、遺伝子キャリア、アジュバント、賦形剤など、例えば、ヒト血清アルブミンまたは血漿調製物を包含し得る。好ましくは、組成物は1単位用量の形態であり、そして通常1日1回またはそれ以上を投与される。
【0115】
以下は、本発明のLT−βおよびLT−α/LT−β複合体、およびこれらを特徴付けるために使用される方法を説明する実施例である。これらの実施例は限定して解釈されるべきものではなく:実施例は例証の目的のために含まれ、そして本発明は請求の範囲によってのみ限定される。
【実施例】
【0116】
実施例において以下の実験手順を使用した:
(抗血清)
組換えヒトLT−α(rLT−α)を、無血清条件培地中で安定にトランスフェクトしたチャイニーズハムスター卵巣(CHO)細胞株によって、発現しそして分泌した。無血清条件培地から、一連のセファロースS、レンズマメレクチンセファロースおよびFPLC MonoQカラムクロマトグラフィー工程によって、分泌rLT−αを精製した。CHO細胞由来のrLT−α調製物の特性は記載されている。[J.Browningら、J.Immunol.、143、1859(1989)]。リンパ腺手順[M.Sigelら、「リンパ腺中への接種による抗体の産生」、Met.Enz.、93、3(1983)]によって、完全フロイントアジュバンド中25μgの天然rLT−αで2匹のウサギ(4および5)を免疫した。3番目のウサギ(6)を、完全フロイントァジュバンド中25μgの変性rLT−αで同じ経路により免疫した。変性rLT−αをSDS−PAGEに続いて0.1%SDS−カーボネート緩衝液中で電気溶出によって調製した。
【0117】
上記の方法を使用して、3つの抗rLT−α抗血清を、2つを天然rLT−αに対して、3番目をSDS変性rLT−αに対して生成した。天然タンパク質(ウサギ4および5)での免疫によって生じた抗血清は、1:2000〜5000の希釈で50ユニット/ml溶液を中和し得た。変性rLT−α(ウサギ6)に対して生じた血清は、中和力価がないが、ウェスタンブロットではrLT−αと弱く反応した。抗血清は、r−ヒトTNFを中和し得ず、またウサギ6由来の抗血清中で非常に弱い力価であることを除いて、ELISAプレートに結合したr−ヒトTNFを認識し得なかった。ウサギ6由来の抗血清のみがウエスタン分析でrLTを認識可能であった。
【0118】
組換えヒトTNFで4番目のウサギを免疫した。完全フロイントアジュバント中で、続いて不完全フロイントアジュバント中のブーストにより、組換えヒトTNF(E.coli由来[D.Weirら、Handbook Of Experimental Immunology In Four Volumes、8章「実験動物の免疫」])を使用する古典的免疫スキームを経て、ポリクローナル抗rTNFウサギ血清を調製した。免疫によりrTNFに対して生じた血清は、良好な中和力価を有した。TNFに対する中和モノクローナル抗体が記述されている[Liangら、「組換えヒト腫瘍壊死因子/カケクチンに対するモノクローナル抗体の産生および特徴付け」、Biochem.Biophys.Res.Comm.、137、847(1986)]。予め免疫した血清を、コントロールとしての使用のために全ての動物から収集した。
【0119】
(細胞増殖およびT細胞活性化)
全ての細胞を、上述したLT−αトランスフェクトしたチャイニーズハムスター卵巣(CHO)細胞株を除いて、アメリカンタイプカルチャーコレクション(ATCC)から入手した[Browning、J.Immunol.、143、1859−1867頁(1989)]。
【0120】
細胞を、1%グルタミン、10mM HEPES緩衝液、pH7.5、ペニシリン/ストレプトマイシン、および10%ウシ胎児血清(Hyclone−defined)を補足したRPMI 1640(「完全RPMI」と呼ぶ)中で増殖した。ただし、上記のように補足したDulbecco改変Eagle培地中で増殖したトランスフェクトされたCHO細胞は除く。ヒトT細胞ハイブリドーマ、II−23は、活性化末梢リンパ球とヒトCEM腫瘍株との融合の結果であり、そしてさらにサブクローニングした(II−23.D7)[C.Wareら、「細胞障害性リンホカインを生産するヒトT細胞ハイブリドーマ:抗CD3モノクローナル抗体またはレクチンおよびホルボールエステルによるリンホトキシン放出およびキラー細胞活性の誘導」、Lymph.Res.、5、313(1986)]。ヒト末梢血リンパ球(PBL)をヘパリン添加ガラスチューブ中に入れ、Ficoll−Hypaque遠心分離によって単離し、洗浄し、そして完全RPMI培地で再懸濁した。1μg/ml インドメタシン存在下および、いくつかの実施例では10ng/ml rIL−2(Biogen,Inc.、Cambridge、MA)を用いて、OKT3条件培地の1:500希釈(最終的に約2ng/ml)を用いて、2×106細胞/mlで、PBLを処理した。ヒトCTL−クローンを、記述したように生成し[L.Greenら、「クローン化ヒト細胞障害性Tリンパ球によって生産される細胞障害性リンホカイン」、J、Immunol、135、4034(1985)]、そして照射刺激細胞(抗原)またはE.Reinherzによって提供された抗CD2モノクローナル抗体(Tl12+Tl13)の組合せのいずれかで活性化した。
【0121】
(フローサイトメトリー)
10%ウシ胎児血清(FBS)、0.1% アジ化ナトリウム、および0.1mg/ml ヒトIgGを含むRPMI 1640培地中で、0℃で、細胞を再懸濁した。ヒトIgGでのプレインキュベーションに続いて、所望の抗血清を含有する追加培地を添加した。代表的には、細胞を1:200の抗rLT−αおよび抗rTNF血清の最終希釈で60〜90分間インキュベートした。Dulbeccoのリン酸緩衝液生理食塩水(PBS)で細胞を2回洗浄し、次に、上記の培地中でフルオレセイン標識ヤギ抗ウサギIgG(Cappel Durham、N.C.)の1:500希釈で最低60分間これらをインキュベートした。次に細胞を1回洗浄し、そして直接に分析するか、またはいくつかの場合では0.5%パラホルムアルデヒドを用いて0℃にて10分間の固定後に分析するかのいずれかを行った。2番目の抗体段階でのフィコエリスリン標識leu−4、leu−2、leu−M3、またはleu−16もしくはleu−19(Becton−Dickinson、Mountain View、CA)を添加することを除いて、上記のように2つの色分析を行った。表面結合LT−αとIL−2レセプターレベルとの比較を、フルオレセイン標識抗IL−2レセプター(CD25)抗体(Becton−Dickinson、Mountain View、Ca.)を用いる分離単色分析で行った。分析を、FACStar機器(Becton−Dickinson)で行った。
【0122】
(活性化II−23.D7細胞による中和抗rLT−α抗体の吸着)
完全RPMI培地中で、1×106細胞/mlで8時間、10ng/mlのPMAと共に、II−23.D7およびU93前単球細胞を刺激した。3回培地で細胞(1×108個)を洗浄し、そして上清を吸引して、乾燥ペレットを得た。次に、細胞を、抗rLT−α血清(ウサギ4由来)の1:1000希釈を含有する1mlの培地で再懸濁し、そして混合しながら1.5時間氷上でインキュベートした。細胞を遠心分離によって抗血清から取り除いた。吸着した抗血清(免疫前および免疫後の両方)を、15U/mlのrLT−αを含有する等量(50μl)の培地と混合し、そして20分間室温でインキュベートした。この混合物を培地中に連続的に希釈し、そしてL929細胞(0.1ml中)に添加し、そしてさらに24時間インキュベートした。記載されたようなMTTアッセイ[L.Greenら、「細胞生存度に対する迅速な比色アッセイ:細胞障害性および増殖阻害リンホカインの定量への適用」、Jour.Immunol.Meth.、70、257−268(1984)]によって、細胞生存度を評価した。
【0123】
(T細胞の35S−メチオニンまたは35S−システインの代謝標識)
ペニシリン/ストレプトマイシン、グルタミン、10mM HEPES pH7.5、10% v/v 透析FBS、および2% v/v 通常のRPMI(冷キャリア添加)を補足したシステインを含まないまたはメチオニンを含まないRPMI 1640中に、細胞を移した。細胞濃度を2〜3×105細胞/mlに調節し、そして35S−メチオニンまたは35S−システインを100〜200μCi/mlのレベルにした適切な培地に添加した。新たに活性化したPBLの場合は、上清を穏やかに除去し、そして細胞を遠心分離し、標識培地中で再懸濁し、そして元の付着集団に戻して添加した。12〜18時間の標識期間の後、以下に記述するように細胞を洗浄し、そして溶解した。PBLを用いて、細胞をピペットにより除去し、そして付着集団をPBS中で5mMEDTAで処理することで部分的に除去した。
【0124】
(免疫沈降)
標識した細胞溶解物の0.2〜0.5mlに2〜4μlのウサギ血清を添加した。サンプルを1〜2時間、4℃で放置した。次に、洗浄したプロテインAセファロース(Pharmacia,Piscataway,N.J.)の60〜75%懸濁液の60μlアリコートを添加し、そして6〜18時間4℃でサンプルを振とうした。プロテインAセファロースペレットをカルシウム/マグネシウムを含まないPBS中 1%NP−40で3回洗浄し、それらを50μlのLaemmli SDSローディング緩衝液に再懸濁した。代表的には、1つの溶解物のサンプルを、連続した免疫沈降(免疫前抗rLT−α血清、抗rTNF抗血清、および最後に免疫後抗rLT−α抗血清)を介して循環した。1つのセットの実験で、5mM CaCl2およびMnCl2を溶解物に添加し、そして洗浄したレンズマメレクチン−セファロースの75%懸濁液の75μlと共に溶解物を一晩振とうした。このセファロースをNP−40/PBSで2回洗浄し、次に0.25M α−メチルマンノシドを含む75μlの1% NP−40/PBSの3回の連続的な添加により溶出した。プールした洗浄液を、免疫沈降プロトコールに用いた。
【0125】
(ウサギ抗rLTアフィニティーカラム)
プロテインAセファロースを用いて酸性pH溶出で抗rLT血清(ウサギ4由来)から免疫グロブリン画分を精製した。溶出したIgG含有面分をPBSで透析し、そしてアミコン濾過により濃縮した。抗rLT−α−IgG溶液(6mg/mlを15ml)を、使用説明書に従い8mlのAffi−gel 10樹脂(Biorad,Richmond,Ca.)に結合した。非特異的ウサギIgG(Cappel,Durham,N.C.)を用いて同一のアフィニティーカラムを調製した。両方のカラムを、PBS、1% NP40を含む1M 酢酸 pH3.0、そして最後にプロテアーゼインヒビターを含まない溶解緩衝液で洗浄した。
【0126】
(LT−βおよびLT−αの最初の精製)
II−2S.D7細胞(15L)を5×105細胞/mlの濃度になるまで増殖し、ホルボールミリスチンアセテート(PMA)を添加し、最終濃度25ng/mlにした。24時間後、細胞を採取し、そしてそれを冷無血清RPMI培地中で洗浄した。7×109細胞を含む凍結した細胞ペレットに、100mlの氷冷溶解緩衝液(50mM HEPES pH7.5、1% v/v NP−40、2mM EDTA、0.15M NaCl、および0.1% アジ化ナトリウム)を添加した。その溶解緩衝液には、5mM ベンズアミジン、1mM フェニルメチルスルホニルクロライド(PMSF)、および0.25mM N−エチルマレイミド(NEM)、10μg/ml大豆トリプシンインヒビター、0.7μg/ml ペプスタチンおよび10μg/ml アプロチニンが新たに添加されている。dounceホモジナイザーで細胞を穏やかにホモジナイズし、そして細胞溶解物を10,000×gで10分間遠心分離した。上清を60,000×gで90分間遠心分離し、そして上清を収集した。
【0127】
高速遠心分離からの上清に、5mM CaCl2および5mM MnCl2を添加した。次に、この上清を、CaCl2およびMnCl2を加えた溶解緩衝液で平衡化した20mlのレンズマメレクチンセファロースカラム(Pharmacia,Piscataway,N.J.)にロードした。溶解緩衝液(CaCl2およびMnCl2を含む)でカラムを洗浄し、そして次に0.25M α−メチルマンノシドを含有する溶解緩衝液でカラムを溶出した。
【0128】
レンズマメレクチン溶出画分を50mlの体積までプールし、そしてそれを2mlのウサギ非特異的IgGセファロースアフィニティーカラムに直接ロードした。このカラムを2mlのウサギ抗rLT−αセファロースアフィニティーカラムに直接連結した。両方のカラムをEDTAを含む同じ溶解緩衝液で洗浄し、次に1% NP−40が1% W/V
MEGA−8(オクタノイル−N−メチルグルカミド,Boehringer一Mannheim,Indianapolis,IN.)に置換されている溶解緩衝液で洗浄した。
【0129】
洗浄したカラムを、各々、1% MEGA−8、50mM グリシンpH2.5、0.05M NaCl、5mMベンズアミジン、および2mM EDTAで溶出した。pHが変わった後の最初の20mlをプールし、このプールを凍結乾燥し、0.05% SDSを含む水1mlに再懸濁し、そして10mM HEPES pH7.5、0.05% SDS、および0.1% MEGA−8で透析した。透析した画分をスピード真空(speed−vac)で乾燥し、そして0.15mlの水に再懸濁した。アリコートとLaemmliローディング緩衝液とを混合し、SDS−PAGEで電気泳動した。LT−βおよびLT−αタンパク質を銀染色により視覚化した。
【0130】
(II−23.D7細胞表面のヨウ素化)
コントロールまたはPMA誘導II−23.D7細胞のいずれかを、カルシウム/マグネシウムを含まないPBSで大量に洗浄し、1mM PMSFおよび0.25mM NEMで処理し、そして次に2回洗浄した。50μgのヨードジェン(iodogen)(Pierce)でコートした12×75mmガラスチューブに、0.3mlの細胞(1×107全量)および1〜2mCiの125ヨウ化ナトリウムを添加した。細胞を25分間、室温で定期的に撹拝しながら放置し、10%のFBSを含むPBSで3回洗浄し、そして上記の溶解緩衝液に再懸濁した。Eppendorf遠沈管での2分間の遠心分離により核を除去した。次に、この上清をさらに15分間遠心分離した。透明にした上清を免疫沈降のプロトコールに用いた。
【0131】
(一次元CNBrペプチドマッピング)
サンプルを、12%アクリルアミドSDS−PAGE Laemmliシステムゲル上にて短距離で電気泳動し、適切なゲル断片を切り出した。ゲルの薄片を、90%ギ酸中に新鮮な700mg/mlのCNBrを15μl含む1.0mlの0.1N HCl、0.2% 2−メルカプトエタノールに1時間浸漬した。次にこの薄片を取り出し、そして0.1M Tris−Cl pH8.0で5分間、25mM Tris−Cl pH8.0で5分間、そして最後に1×Laemmli SDS−PAGEローディング緩衝液で10分間洗浄した。12% アクリルアミドスタッキングゲルを有する15% SDS−PAGE Laemmliゲル上で薄片をロードした。ペプチドのバンドを銀染色または乾燥したゲルのオートラジオグラフィーにより視覚化した。
【0132】
(再免疫沈降)
SDS−PAGEで分離した抗原の再免疫沈降を、ゲルから標識したバンドを切り出し、TBS、0.2% SDSで10分間再水和化し、そして次にゲルの薄片を小さな切片に刻むことにより行った。タンパク質を、1ml TBS、0.2% SDS中で8時間、室温で回転しながらインキュベートすることにより溶出した。溶出後、ゲルの切片を遠心分離で取り除き、そしてNP−40を最終濃度が2%になるように上清に添加した。次に溶出されたタンパク質を上記の様に免疫沈降し、そしてSDS−PAGEで再分析した。
【0133】
(等電点電気泳動(IEF))
二次元IEFを、本質的にP.H.O’Farrell[J.Biol.Chem.,250,4007−4021(1975)]に記載されたように行った。125Iで標識した抗原を、II−23.D7細胞抽出物から免疫沈降し、そして免疫沈降したタンパク質を、9.5M尿素を含む100μlのO’Farrellサンプル緩衝液中で100℃、5分間の加熱により溶出した。次に、溶出したタンパク質を、最終濃度2%アンホリン(ampholines)(pH3〜10領域、Sigma)を有する14cm×3mmのチューブゲル上で室温で16時間定電圧(400V)で泳動した(第一次元)。第二次元は12%SDS−PAGEであった。
【0134】
天然(未変性)条件下でのIEFについて、125Iで標識した細胞抽出物を、尿素が存在することを除いて上記と同様にチューブゲルに直接電気泳動した。標識した抽出物(容積200μl)をチューブゲルでロードする前に、100,000×g(30psi、airfuge)で10分間遠心分離した。電気泳動を上記と同じ条件下で4℃にて行った。次にチューブゲルを取り出し、そして1cmの断片にスライスし、タンパク質を、1mlのTBS、2% NP−40、2mM PMSF中で、8時間室温でそれぞれの薄片を回転させながらインキュベートすることにより溶出した。次に溶出したタンパク質を含む上清を免疫沈降し、そしてSDS−PAGEにより分析した。変性および未変性のチューブゲルの両方についてのpH勾配を、並行で行ったゲルからの個々の薄片のpHの測定により決定した。
【0135】
(T細胞増殖アッセイ)
PBLを単離し、そして10%ヒト自己血清、1μg/ml インドメタシン、および50U/mlポリミキシンBを含むウシ胎児血清を除いた上記の完全RPMIに再懸濁した。MLR実験で、自己血清は応答動物の血清であった。異なるドナーからの刺激細胞を3000ラドで照射した。ウサギ血清を56℃で1時間前加熱し、そして増殖アッセイに用いる前に血清を希釈し、濾過滅菌した。丸底96ウェルプレートの0.2mlの細胞(全量1×105)を5μg/mlフィトヘマグルチニン、1〜2ng/ml OKT3または1.5〜2×105の照射した刺激細胞のどちらかで、種々の抗血清またはサイトカインの存在または非存在下で処理した。3日後(PHAまたはOKT3活性化)または5日後(MLR)、細胞を3H−チミジンでパルスし、採取しそして計数した。
【0136】
(LT−αおよびLT−βのさらなる精製)
II−23.D7細胞を10%ウシ胎児血清を含むRPMI培地で培養し、50LのRPMI培地から細胞を採取し、4×106細胞/mlの濃度になるよう培地に再懸濁し、そして50ng/ml ホルボールミリストイルアセテート(PMA)を添加した。6時間の活性化の後、遠心分離により細胞を採取し、そしてDulbeccoリン酸緩衝生理食塩水で洗浄した。200mlの冷溶解緩衝液(50mM HEPES緩衝液、pH7.0;0.1M NaCl、10mM EDTA、5mM ベンズアミジン、各10μg/mlの大豆トリプシンインヒビター(アプロチニン、キモスタチン、ロイペプチン、アンチパイン)、1μg/mlペプスタチン、および1mM フェニルメチルスルホニルフルオリド)中に最終的に4×1010細胞の細胞ペレットを懸濁し、このペレットを一回窒素キャビテ一ターに通した。溶解した細胞を、50.2 Tiローターで40,000rpm、60分間遠心分離し、上清を捨てた。ペレットを、1% w/v Nonidet
P40デタージェントを含む120mlの溶解緩衝液中で一晩抽出し、そして次に上記の様に遠心分離した。
【0137】
Affi−gel 10(BioRad)に結合しているモノクローナル抗リンホトキシン(Boehringer Mannheimから入手した抗腫瘍壊死因子−β)からなる2mlのアフィニティー樹脂に、デタージェントで可溶化した膜タンパク質を含む上清を添加し、そしてこの懸濁液を一晩振とうした。樹脂を小さなカラムに集め、1% Nonidet P40を含む50mM HEPES、pH7.0、そして次に1% w/v MEGA−8(Boehringer Mannheim)を含む同じ緩衝液で洗浄した。50mMグリシン緩衝液pH2.5中の1%MEGA−8で、結合したタンパク質を溶出し、そして画分を直ぐにTris塩基で中和した。SDS−PAGE分析および銀染色により、画分中のp33およびLTの存在を決定した。これらのタンパク質を含む画分をプールし、そしてSDSを最終濃度0.1% w/vになるまで添加し、そして0.1×Laemmliサンプル緩衝液(MEGA−8デタージェントを除去するために様々に変化している)でこのプールを透析した。透析した溶液を凍結乾燥し、そして元の水の1/10量に再懸濁した。SDS−PAGEゲル上にサンプルをロードし、ProBlotメンブラン(Applied Biosystems)にブロットし、そしてクマシーブルー染料で染色した。
【0138】
このスキームは、 ブロット上のバンドでLT−βを精製可能にする。当業者がイオン交換クロマトグラフィーによりアフィニティー樹脂から溶出したタンパク質を分離することは可能であるべきである。例えば、複合体を尿素で解離し得、そしてLT−αおよびLT−βタンパク質を、例えば1%非イオン性デタージェント(例えば、MEGA−8、Boehringer−Mannheim)および尿素を含むTris−Cl緩衝液pH8.0中で、MONO Q FPLC(Pharmacia)陰イオン交換クロマトグラフィーにより、塩勾配溶出を用いて分離し得る。このクロマトグラフィーの技術は、タンパク質の異なった電荷に基づいて分離する。2種類のタンパク質は、等電点電気泳動実験(上記参照)で電荷の差異に基づいて分離され得、このとき尿素を使用し、LT−α/LT−β複合体を解離する。このようなアフィニティークロマトグラフィー、尿素/非イオン性デタージェントでの解離、およびイオン交換クロマトグラフィーの組合せは、可溶性LT−βまたはLT−α/LT−β複合体の精製を可能にする。
【0139】
(ペプチド配列決定アッセイ)
ProblotからLT−βおよびLT−αバンドを切り出し、タンパク質シークエンサーにロードした。120A PTHアミノ酸分析機と連結した、Applied Biosystemsシークエンサーモデル470Aを用いて、Edman分解によりN末端の配列の情報を得た。LT−βを上記の免疫アフィニティークロマトグラフィーにより精製し、そしてトリプシン断片の配列を得た。[Abersoldら、「インサイチュ後の一次元または二次元ゲル電気泳動により分離したタンパク質の内部アミノ酸配列分析、ニトロセルロース上でのプロテアーゼ切断」PNAS,84,6970−6974(1987)を参照のこと]。すなわち、インサイチュでブロット上のタンパク質をトリプシンで切断し、切断したペプチドの逆相HPLC解析を行った。得られたN末端および内部のトリプシン断片ペプチドを、次にEdman分解により配列決定した。T105、T87/88、T100、およびT67と呼ばれるN末端および内部のペプチドの配列決定を図13に示す。
【0140】
(オリゴヌレオチドプローブの構築)
T87/88の配列から、以下のアンチセンスプローブを設計した:
1368 GTYTCNGGCTCYTCYTC[配列番号9]
1369 GTYTCNGGTTCYTCYTC[配列番号10]
そして標準方法により合成した。[例えば、J.Sambrookら、Molecular Cloning,A Laboratory Manual,第2版.(1989)を参照のこと]。
【0141】
(誘導されたII−23 cDNAライブラリーの調製)
cDNAサブライブラリーを以下のように調製した:
II−23.D7細胞を6時間、50ng/ml PMAで刺激し、LT−β mRNAの存在を確認した。これらの細胞からmRNAを単離し、そして当該技術分野で周知の技術を用いてそれを逆転写しcDNAにした。[B.SeedおよびA.Aruffo、「迅速免疫選択方法による、CD2抗原、T細胞赤血球レセプターの分子クローニング」PNAS、84,3365−3369(1987)]。標準手法を用いて、2本鎖cDNAを以下の配列を有するNotI−BstXIリンカー/アダプターに連結した:
【化1】

【0142】
次に4.2mlの5〜20%酢酸カリウム勾配、2mM EDTA、1μg/mlエチジウムブロマイド、Beckman(登録商標)SWローターで、50,000rpm、22℃にて3時間標準方法によりcDNAをサイズ選択した。500塩基対より大きなcDNA断片をプールした。次いで、ベクター、pCDM8(Brian Seed(Massachusetts General Hospital)から供与された)を調製した。このプラスミドをBstXIで切断した。400塩基対のスタッファー(stuffer)フラグメントを除去するために、この混合物を上記の様に酢酸カリウム勾配で遠心分離し、そしてより大きなフラグメントを単離した。このフラグメントをアガロースゲル電気泳動によりさらに精製し、そして次いでcDNAをベクターに連結した。この方法で、誘導したII−23.D7細胞で発現したmRNAに対するDNA配列を含む組換えDNA分子を作製した。これらのプラスミドを用いて、E.coli MC1061 P3を形質転換した。PMA誘導したII−23.D7 mRNAのcDNAライブラリーを有する、1×106以上の組換えクローンのコレクションを得た。
【0143】
(スリーニングおよびクローンのDNA配列決定)
pCDM8 II−23.D7ライブラリーを32P標識したオリゴマー1368でスクリーニングし、ポジティブなクローンを単離し、3M テトラメチルアンモニウムクロライドで50℃にて洗浄した。[J.Sambrookら、Molecular Cloning,A Laboratory Manual,(1989);Jacobsら、「テトラアルキルアンモニウム塩溶液中の配列非依存の二本鎖オリゴヌクレオチドの熱安定性:組換えDNAクローンの同定への適用」,Nucleic Acids Research,16,10,4637−4649(1988)]。0.9kbの挿入物を含むいくつかのクローンをジデオキシヌクレオチドDNA配列分析に用いた[同上]。
【0144】
(LT−β cDNAの発現)
pCDM8/LT−βクローン12またはコントロールプラスミドのクローン4(無関係なcDNA挿入物を含むpCDM8)を、エレクトロポレーションによりCHO dhfr−に導入し、CHO細胞をヒトLT−αで安定にトランスフェクトした。3日後、細胞を5mM EDTAを含みCa/Mgを含まないHank溶液に取り出し、10μg/ml コントロールIgG1または抗LTモノクローナル抗体(Boehringer−Mannheim)のどちらかを用いて上記の様にFACS分析のために染色し、次にFITCまたはフィコエリスリンのどちらかで標識したヤギ抗マウス調製物と結合した免疫グロブリンを標識した。その他の実験では、これもまたpCDM8ベクター中にある等量のヒトLT−α cDNAの存在または非存在下で、COS細胞を、pCDM8中のクローン4またはクローン12のいずれかのLT−β cDNAでエレクトロポレーションし、上記のように3日後FACS分析のために染色した。
【0145】
(LT−βの発現のノーザン分析)
ポリA+ RNAをII−23.D7細胞または末梢血単核細胞(PMBC)のどちらかでInvitrogenにより提供されたFast−TrackTMシステムを用いて単離した。J.Sambrookら、Molecular Cloning,A Laboratory Manual,(1989)に本質的には記載されているように、2μg/レーンのRNAおよびホルムアミドゲルでの電気泳動を用いてノーザンブロットを調製し、次にGene Screenナイロンメンブラン上へ転写し、そしてUV架橋した。ブロットを、ゲル精製したLT−β cDNAのランダムプライムBstEII/Xmn−Iフラグメント、あるいはヒトLT−αまたはアクチンのフラグメントでプローブした。II−23.D7細胞を、50ng/ml PMAで時間を変えて誘導し、そしてLT−αおよびLT−βの両方の発現が誘導されたことを見出した。PBMCを、RPMI培地のみまたは1000ユニット/mlのIL−2の存在下、あるいはT細胞を活性化するOKT3とともに培養した。
【0146】
(LT−βの5’末端の決定)
mRNAの5’の配列をプライマー伸長分析により決定した。[B.Wallnerら、Nature 320,77−81(1986).]オリゴヌクレオチドプライマー(プローブ360−121 5’GACAGTGATAGGCACCGCCAGCAACAA−3’)[配列番号13]を用いたプライマー伸長により、約128〜130bpの産物を得た。これは、MaxamおよびGilbert方法論[A.MaxamおよびW.Gilbert,「塩基特異的化学開裂を用いる末端標識DNAの配列決定」 Methods In Enzmology,65,499(1988)]を用いた配列決定により、メチオニンATGの7〜9bp上流にある転写開始部位を示した。一時的な実験でクローン12により示された発現は、1つまたは両方のLeu−4またはLeu−6の開始部位は機能的であることを示した。mRNAの5’配列を証明するために、コスミドクローン、031A[Spiesら、Science 243,214(1989)]をいくつかの制限酵素で切断し、電気泳動し、ブロッティングし、そしてLT−β cDNAのBST E2/Xmn−1フラグメントでプローブした。コスミドは、6kbのEcoRIフラグメント内にLT−β遺伝子を含有した。EcoRIフラグメントを、カナマイシン耐性遺伝子を有するpNN109と呼ぶpUC誘導体にサブクローニングした。ジデオキシ核酸配列決定により完全なゲノム配列を得た。
【0147】
(実施例1:T細胞がその表面にLT−関連エピトープを発現する)
上記の条件下で、ヒト末梢単核細胞(PMN)を、OKT3モノクローナル抗体で活性化し、培養の2日後に、フローサイトフルオロメトリー分析によって、LT−α/LT−β複合関連形態の発現について分析した。1つの実験(結果は図1に示す)で、新鮮なPBLをOKT3およびIL2とともに3日間培養し、それらを、未変性rLT−α(それぞれ、ウサギ4および5由来である、図1の「LT−4」および「LT−5」パネル)、変性rLT−α(ウサギ6由来である、図1の「LT−6」パネル)、および未変性rTNF(ウサギ7由来である、図1の「TNF」パネル)に対して1:200希釈の抗血清で染色した。細胞を、各動物からの免疫後の血清(図1のパネルの実線)あるいは免疫前の血清(図1のパネルの破線)で染色した。図1は、ウサギ4および5由来の抗rLT−α血清のみが、活性化末梢T細胞上のエピトープを認識したことを示す。
【0148】
図2に示す実験では、II−23.D7細胞を15時間、10ng/ml PMAで処理するかあるいは処理せずに、それらを、図1に記載の実験のように、ウサギ4抗rLT−α免疫後の血清(図2のパネルの実線)、あるいはウサギ4免疫前の血清(図2のパネルの破線)で染色した。図2に示すように、ホルボールエステル(PMA)刺激でLT−αを合成するT細胞ハイブリドーマII−23.D7が、PMA活性化で表面LT−関連エピトープを発現することを認めた。
【0149】
T細胞上のLT−α関連エピトープは、LTに関連し、組み換えLT−α調製物由来のCHO細胞中のなんらかの夾雑物に関連しないことを確認するために、PMA活性化し洗浄したII−23.D7あるいはU937細胞で、ウサギ4由来の抗血清の1:1000希釈物を処理した。図3に示す実験では、抗rLT−α血清(1:1000抗LT−4)の試料1mlを、細胞を含まない(−−)、1×108 U937細胞(−〇−)、1×108(−−)PMA活性化II−23.D7細胞、あるいは1×107(−−)PMA活性化II−23.D7細胞のいずれかで処理した。吸収抗血清の希釈物を、L929細胞障害性アッセイで限定量のrLT−αに加えて、最初のウエルに1:4000の最終希釈物が存在するようにした。このアッセイは、24時間以内に、マウス繊維芽細胞株L929を殺すLT−αの能力を測定する[L.Green,J.L.Reade,C.F.Ware,「細胞生存性についての迅速な比色アッセイ:細胞障害性および増殖阻害性リンホカインの定量への適用」J.Immunol.Methods,70,257(1984)]。24時間後に、細胞生存率をMTT読み取りによって評価した。図3のプロットは、吸収抗血清の希釈に対する吸光度(細胞生存率に比例する)である。データは、2つのウエルの平均であり、2つは概して、符号で示されている範囲内に存在する。図3に示されているように、標準L929細胞障害性アッセイでの吸収抗血清中和力価の分析は、U937細胞は効果がないが、活性化II−23.D7細胞が、LT−α中和抗体を除去することを実証した。これらのデータは、膜表面の抗原構造が実質的にLT−αに関連することを示す。
【0150】
ハイブリドーマII−23.D7を、T細胞表面のLT−α関連エピトープの明確な存在についての多くの平凡な説明を確かめるために、さらなる多くの処置を行った。まず最初に、抗血清中のLT−α:抗体複合体が、ハイブリドーマ上のTNF/LT−αレセプターに結合し得る可能性を除外した。TNFおよびLT−αの両方は、複合体中の抗体結合エピトープの存在を考慮し得る三量体構造を有する。しかし、細胞TNFレセプターの可溶性TNFあるいはLT−αでの前飽和は、表面染色には効果はなかった。このような飽和は、このような免疫複合体のこのようなレセプターヘの結合を妨害した。
【0151】
結合されたTNFをそのレセプターから解離し得る、pH3の乳酸処理は、シグナルには影響しなかった。このことは、LT−αがレセプター結合されないことを示唆する。しかし、II−23.D7細胞へ結合する125I−LT−αを使用する実験は、レセプター結合LT−αが、TNFよりも酸性pHではそのレセプターから解離させるのはより困難であることを示した。
【0152】
染色前の細胞のおだやかなトリプシン処理は、シグナルの消失を導き、このことは、エピトープがタンパク質であることを示す。表面結合LT−αがフォスファチジルイノシトール結合されたかどうかを決定するために、細胞をフォスファチジルイノシトール特異性フォスホリパーゼCで処理した。PI−結合抗原LFA−3が解離され得る条件下[A.Petersonら、T細胞赤血球レセプター(CD2)のモノクローナル抗体およびリガンド結合部位」Nature,329,842(1987)]で、LT−αエピトープには効果が認められなかった。
【0153】
LT−α遺伝子で安定にトランスフェクトされたCHO細胞を、(前PMA活性を伴うか、あるいは伴わない)染色し得なかった。このことは、ウサギを免疫するのに使用された、もともとのrLT−α中のCHO由来の夾雑物に対する抗体が、II−23.D7細胞の染色に寄与するのに十分な量が存在しなかったことを示す。同様に、任意のウシ胎児血清タンパク質を夾雑するLT−α調製物に対して誘起された抗体は、染色が10%ウシ胎児血清中で実施されたので、T細胞染色には影響しなかった。
【0154】
抗rLT−α血清のrLT−αでの前処理は、rTNFでの前処理では阻止しなかったが、II−23.D7細胞上のLT−α形態の染色を阻止した。
【0155】
(実施例2:T細胞ハイブリドーマII−23.D7上のLT−α関連タンパクの免疫沈降)
PMAで活性化したII−23.D7細胞を表面ヨウ素化し、溶解してデタージェントで可溶化した。標識膜タンパク質の免疫沈降およびSDS−PAGE分析は、2つのタンパク質が抗rTNF−α抗血清により認識されることを示した。図4Aは、免疫前(PRE)あるいは免疫後(POST)抗rLT−α血清(ウサギ4由来)のいずれかで免疫沈降されたヨウ素化表面タンパク質のSDS−PAGE分析の結果を示す。
【0156】
図4Aに示されているように、LT−αの推定の大きさと相関する25〜26kD分子量形態(「LT−α」)を認め、そして、約33kDの別の形態(「LT−βあるいはp33」)を認めた。同じウサギ由来の免疫前血清(図4AのPREの欄)および抗rTNFウサギ血清のどちらもが、ヨウ素化PMA活性化II−23.D7細胞由来のどのバンドも免疫沈降させ得なかった。
【0157】
ヨウ素化バンドの1−D部分CNBrペプチドマッピングは、25〜26kD形態が、ヨウ素化rLT−αと同じパターンで切断されることを示した。従って、このバンドをLT−αと同定した。図4Bに示す実験で、パネルA由来の25〜26kDおよび33kDのバンドを切り出し、制限CNBr切断およびSDS−PAGEシステムでの電気泳動にかけた。比較のために、並行して行ったrTNFおよびrLT−αの両切断を図4Bに示す。ゲルをオートラジオグラフィーによって可視化した。レーン1はrTNFを示し、レーン2はrLTを示し、レーン3はLTを示し、そして、レーン4は、LT−βを示す。CNBrフラグメントの増大された大きさは、天然LT−βの炭水化物量の増加を反映する。ヨウ素化33kD形態はCNBrで切断されず(レーン4)、このことは、既知LT−α遺伝子生成物とは異なることを示す。rTNFは、このタンパク質中にメチオンが存在しないので、CNBrでは切断されなかった(レーン1)。
【0158】
免疫沈降と組み合わせて、35S−メチオニンおよび35S−システインでの代謝標識を行って、これらのLT−α関連表面形態のさらなる特徴付けを行った。TNF/LT−αペアの場合には、システインおよびメチオニン分布は、TNFとLT−αとの間、および、シグナル配列を伴う形態と伴わない形態との間の両方の区別を可能にし、これは、膜TNF形態の研究で活用された[M.Krieglerら、Cell,53,45〜53頁(1988)]。完全にプロセシングされたサイトカイン、すなわち、分泌形態の場合には、TNFはシステインを含有し、メチオニンを含有しないが、LT−αはメチオニンのみを含有し、システインを含有しない。しかし、LT−αは、シグナル配列ドメイン中に2つのシステイン残基を含有し、TNFは、N末端領域に1つのメチオニン残基を含有する。II−23.D7ハイブリドーマ細胞の分離培養物を、35S−メチオニンあるいは35S−システインのいずれかで標識し、免疫反応タンパク質を沈降させた。実験では(結果は図5に示す)、II−23.D7細胞を、10ng/ml PMAで活性化し、次に、8時間、35S−メチオニンあるいは35S−システインで標識した。培地および溶解細胞の両方を、免疫前(ウサギ4)(P)、抗rTNF(T)、および抗rLT−α(ウサギ4)(L)血清の順で、連続して免疫沈降を行った。図5は、分泌タンパク質を含有する上清あるいは洗浄細胞の、免疫沈降物のSDS−PAGEオートラジオグラフィー分析を示す。”s−TNF”は、35S−メチオニン標識の細胞由来の抗rTNF免疫沈降されたバンドを示し、TNFのプロセシングされていない26kD形態と推定される。”Met”および”Cys”は、使用した35S−標識アミノ酸のことである。35S−システイン含有のそれらのレーンを、35S−メチオニン含有レーンより長時間露光した。これらの細胞からの上清では(「分泌された」と記されたレーン)、25kD形態のLT(「LT−α」)は、PMA処理後に、35S−メチオニン標識された細胞により放出されるが、35S−システイン標識細胞によっては放出されない。このパターンは、完全にプロセシングされ、分泌されたLT−αと予測される。より長時間の露光は、上清に極少量のTNFを示し、標識の取り込みは、完全にプロセシングされた分泌TNFに対して予測された通りであった。mRNAレベルでもまた、低いTNFレベルでの優先的なLT−α発現を認めた(ShamanskyおよびWare、未公開情報)。洗浄細胞の分析(「細胞の」と記されたレーン)は、33 kD LT−βを伴って、25〜26kD LT−αの両方が存在することを示した。25〜26kDおよび33kD形態の相対的な量は、表面ヨウ素化により認められたものに相当した。25〜26kD表面LT−α形態はシステインを含まず、このことは、リーダー配列のプロセシングを示す。33kD形態は、35S−メチオニンおよび35S−システインの両方を取り込んだ。図5に示したフィルムのより長時間の露光(示されていない)は、約26〜27kDで細胞からの抗TNF免疫沈降されたバンドの存在を示した。バンドは、標識システインおよび標識メチオニンの両方の取り込みを示した。標識はシステインによって、より強かった。26kD−TNF形態ではcys:met比率は4:1であるので、この標識パターンは、このバンドの同一性を確実にする。
【0159】
細胞溶解物からの免疫沈降物中のLT−αを伴ったLT−βの存在は、LT−βが抗原的にLT−αに関連すること、あるいはLT−βがLT−αに結合されることのいずれかあるいは両方を示唆した。この論点に的を絞るために、35S−メチオニン標識細胞由来の25kDおよび33kDのバンドを、ウサギポリクローナル抗rLT−α血清で免疫沈降させ、切り出したゲル切断片から溶出して、抗rLT−αポリクローナル血清あるいはmAbで再度免疫沈降させた。LT−βではなくLT−αが、抗rLT−α抗体で免疫沈降され得、このことはLT−βが抗原的にLT−αに関連しないことを示唆する。これらの観察は、LT−βがLTαと物理的に結合することを示した。33kDのタンパク質は、抗原的にLT−α関連せず、単にLT−αと共に沈降されることを示した。
【0160】
表面ヨウ素化あるいは代謝標識のいずれかでは、LT−αあるいはTNFに結合した、既知の55あるいは80kD TNF/LT−αレセプター形態のいずれも検出され得なかった。おそらく、これは、レセプターが、T細胞の活性化の間に急速に費やされるからである。[C.Wareら、「腫瘍壊死因子によるCTL溶解経路の調節」Cellular Immunity And The Immunotherapy Of Cancer,UCLA Symposia on Molecular and Cell Biology,M.T.LotzeおよびO.J.Finn編 第135巻,121〜128頁(Wiley−Liss,Inc.New York)1990]。
【0161】
(実施例3:II−23.D7細胞上の表面LT−形態の生化学的特徴付け)
PMA処理したIl−23.D7細胞表面のLT−関連形態を、アフィニティークロマトグラフィーによって精製した。免疫沈降法によって、LT−βおよびLT−αがレンズマメレクチンセファロースに結合すること(このことは糖タンパク質構造を示す)に注目した。デタージェントで可溶化し、PMA処理したII−23.D7タンパク質を、レンズマメレクチンセファロースに結合し、アフィニティー精製の前にα−メチルマンノシドで溶出した。抗rLT−α血清によって特異的に認識されるこれらのタンパク質を、さらに正確に評価するために、コントロールIgGおよび抗IgGカラムの両方を調製した。カラムを低pHで溶出することにより、抗rLT−αカラムから約100〜200ngの2種のLT形態が放出された。
【0162】
図6Aは、免疫前(PRE)あるいは免疫後(POST)ウサギ血清から調製された抗rLT−αアフィニティーカラムから溶出されたタンパク質のSDS PAGE分析を示す。図6Bでは、パネルA中のゲルから33kDおよび20kDのバンドを切り出し、制限CNBr切断およびSDS−PAGEシステムでの電気泳動にかけた。比較のために、図6Bは、並行して行ったrTNFおよびrLT−α(CHO由来)のCNBr切断を示す。銀染色によってゲルを可視化した。溶出物のSDS−PAGEゲルは、免疫沈降された表面ヨウ素化PMA処理のII−23.D7細胞のゲルによく類似し、このことは類似のタンパク質が精製されたことを示す。
【0163】
アフィニティー精製の間に、25kDのLT−α形態は、19〜20kD形態に切断されるようである。すなわち、それは、未処理の組み換えCHO細胞由来のLT−αとともに移動した。RPMI 1788腫瘍株由来の天然LT−αの最初の単離もまた、N−末端切断された「des−20」LT−α形態を生じた。アフィニティー精製したタンパク質の一次元CNBr分解物は、切断された20kDのLT−α形態が、おそらくこのLT−α形態の切断型特性を反映するCNBr切断パターンを有することを示した。メチオニンの1つが、「des−20」LT−α形態では消失し、ゆえに、切断パターンは未処理のLT−α形態とは異なる。33kDのタンパク質(LT−β)は、CNBr切断で2つになるので、このことから、1つのメチオニンはCあるいはN末端から5〜20残基以内に存在しなければならないことが推定された。この切断パターンは、33kDのタンパク質が既知のLT形態と著しく異なることを示す。表面ヨウ素化33kDタンパク質のCNBr切断分析は、恐らく類似の結果を与えたが、しかし、ヨウ素化で達成され得る解像は、その2つを十分に現らわさなかった。ヨウ素化rLT−α、rTNF、およびII−23.D7 LT形態のStaphylococcusV8分解は、分解に対して耐性であるべきrLT−αおよび25〜26kDK II−23.D7 LT−α形態を示した。このことは、このタンパク質がLT−αであることを確実にした。33kDのタンパク質を、rTNFによく類似するパターンを有する、数個の小さなフラグメントに切断した。
【0164】
図7では、免疫沈降された表面ヨウ素化タンパク質を、SDS−PAGE分析で分離し、表面結合25〜26kDのタンパク質(「sLT−α」)および33kDのタンパク質(「LT−β」)のバンドを切り出した。切断片を、N−グリカナーゼ(N−Gly)、ノイラミニダーゼとO−グリカナーゼ(O−Gly)との混合物、あるいは3つ全ての酵素で分解した。分解した切断片を、再度SDS−PAGEにかけ、乾燥ゲルのオートラジオグラムを示す。図7に示したように、ヨウ素化表面LT形態の免疫沈降、その後の、N−グリカナーゼあるいはO−グリカナーゼのいずれか、またはその両方での分解で、25〜26kD LT−α形態が、N−連結オリゴ糖を含有することを示した。25−26kD LT−α形態は、N−グリカナーゼ分解での大きさの変化に関連する1つのN−連結部位のみを有する。同様に、33kD形態(LT−β)は、N−グリカナーゼ処理で約3kDの大きさを消失した。このことは、1つのN−連結オリゴ糖の存在を示唆する。25〜26kD LT−α形態に対して、O−グリカナーゼ処理は、LT−βの分子量に影響を与えなかった。しかし、グリカナーゼによる切断がないことは、炭水化物の存在を欠く明白な証拠ではない。
【0165】
(実施例4:LT−αの再免疫沈降)
LT−βのLT−αとの共沈降は、これらのタンパク質が抗原的に関連するか、あるいは物理的に結合されることを示唆した。この確認のために、SDS−PAGE分離されたLT−α(p25)およびLT−βタンパク質が免疫沈降され得るかどうかを試験した。125Iあるいは35S−Metで標識されたLT−αおよびLT−βを、免疫沈降によって部分精製し、SDS−PAGEで分離した。標識されたバンドを切り出し、緩衝液中で再度水和し、タンパク質を溶出した。次に、溶出タンパク質を、ポリクローナルあるいはモノクローナル抗rLT−α抗体を用いて、第二回目の免疫沈降を行った(図10)。ウサギ抗rLT−αは、LT−α(「p25」、レーン2)を再免疫沈降させたが、LT−βはさせなかった(レーン3)。抗rLT−α mAbは、LT−α(レーン5)および21kDのタンパク質(「p21」、レーン4)を沈降させた。これは、下記に示すように、LT−αの前駆体であるが、しかし、LT−βを沈降させなかった(レーン6)。結果は、LT−αおよびLT−βがSDS−PAGEにより分離された後に、ポリクローナルおよびモノクローナル抗rLT−α両抗体が、LT−αと反応し得るがLT−βとは反応し得ないことを示す。このデータは、LT−βが抗原的にLT−αと関連しない証拠を提供する。しかし、LT−αエピトープは完全なまま残存するが、推定のLT−β交差反応エピトープが、変性後に消失される可能性は除外され得ない。
【0166】
図8は、SDS−PAGEゲルから溶出された、125I標識および35S−Met標識のp25およびp33タンパク質の再免疫沈降の結果を示す。125I標識II−23.D7細胞(レーン2、3)および35S標識細胞(レーン4、5、6)由来のLT−αおよびLT−β種を、上記のようにゲル切断片から溶出した。溶出物を、抗rLT−α血清(レーン2、3)あるいは抗rLT−α mAb(レーン4、5、6)のいずれかで免疫沈降し、再沈降されたタンパク質をSDS−PAGEおよびオートラジオグラフィーで分析した。レーン1は、LT−αおよびLT−βの同定のためのコントロールレーンである。
【0167】
(実施例5:LT−αおよびLT−βの等電点電気泳動)
図9および10(各々は、オートラジオグラフ(9A、10A)、および移動距離対pHをグラフにした検量線を含む(9B、10B))は、変性(図9)および未変性(図10)条件下での等電点電気泳動分析を示す。II−23.D7細胞抽出物から免疫沈降された、125I標識LT−αおよびLT−βについて、上記のように、二次元ゲル分析を行った。2−Dゲル分析を、尿素が存在する変性条件下で実施した(図9A)。これに対し、未変性IEFを、尿素を含まない1% NP−40中で行った。125I標識II−23.D7細胞抽出物を、4℃でチューブゲルで泳動させた。泳動後に、チューブゲルを1cm切断片に切断し、泳動されたタンパク質をこれらの切断片から溶出し、免疫沈降して、SDS−PAGEで分析した(図10A)。ゲルレーン1〜12から免疫沈降された物質は、チューブゲル切断片の2〜13に対応する。1cmゲル増大量に基づいて変性および未変性チューブゲルに対してpH勾配を実施した。これらもまた、それぞれに9Bおよび10Bとして下にオートラジオグラムを示す。生化学的に、LT−βおよびLT−αは、非変性等電点電気泳動ゲル上で共に移動するが、複合体が尿素により解離されると、2つのタンパク質は別々に移動する。[図9A、10Aを参照]これらの観察は、LT−αおよびLT−βが細胞表面に複合体として存在するという結果を導く。
【0168】
(実施例6:LT−α発現の制御)
以下に示す表Iは、LT−αの表面形態発現についての、種々の細胞タイプのフローサイトフルオロメトリー分析による調査結果のまとめである。
【0169】
【表I】
これらの研究の際だった観察は、TおよびB細胞に対する表面LT−α発現の制限であった。leu−M3、単球マーカーおよびleu−4(CD3)抗体を、各細胞群を別々に観察するために、二色フローサイトフルオロメトリー分析に使用した。この分析で、表面TNFと表面LT−αと間に際だった相違が存在した。ここで、T細胞は表面LT−αのみを示したが、単球は表面TNFのみを発現した。この結果を図11に示す。
【0170】
図11に示されている実験では、PBLを、LPS(1μg/ml)、インターフェロン−γ(200U/ml)、およびOKT3(1ng/ml)の混合物で8時間処理し、次に、LT−α(ウサギ5由来の抗rLT−α血清)あるいはTNF(ウサギ7由来の抗rTNF血清)に対して染色し、その後、FITC抗ウサギ標識を行った。細胞を、フィコエリスリン−leu−4、pan T細胞マーカー、あるいはフィコエリスリン−leu−M3、単球マーカーで逆染色した。「T細胞パネル」をleu−4+に対してゲートし、一方、「単球」パネルをleu−M3+細胞に対してゲートした。細胞を、免疫前(破線)あるいは免疫後(実線)の血清で染色した。単球腫瘍株HL−60およびU937はLT−αを染色しなかった。二色フローサイトフルオロメトリー分析によって、活性化PBLのT4およびT8サブクラスが、同レベルの表面結合LT−αを示すことが認められた。一般的に、LT−αを発現し得る一次T細胞もまた表面LT−α形態を示し得るようである。
【0171】
3人の異なるヒトドナーの実験は、表面LT−α形態が用時単離された末梢T休止細胞に存在することを示した。PBLの場合には、細胞のOKT3活性化あるいは単なるIL−2処理が、発現の増大を導いた。蛍光チャネル数により、OKT3活性化の間の表面LT−αおよびIL−2レセプター(CD25)発現の両方の定量を試みた。OKT3による表面LT−αの最大誘導が、大量培養でIL−2レセプターのピーク発現(TAC発現)に先行するようであり、従って、表面結合LT形態(LT−α/LT−β複合体)は、初期T細胞活性化抗原のようである。抗T11および同種異系抗原の両方が、クローン化細胞障害性T細胞表面にLT−αを生じ得ることが見い出された。同様に、PMA刺激は、II−23.D7ハイブリドーマ表面でのLT−α出現の誘導に必要であった。T細胞活性化が表面LT−α形態を増加させるようである。II−23.D7ハイブリドーマに比較して、末梢リンパ球は、PMA処理後に急速に表面LT−α形態を下降制御する。同様に、OKT3活性化PBL群の二色分析では、活性化の前進段階でT細胞を含むはずであるDr+細胞は、表面LT−α形態を欠いた。
【0172】
新鮮なPBLの高レベルIL−2での活性化は、リンホカイン活性化キラー細胞(LAK細胞)を生じた。図12に示されているように、抗rLT−αおよびleu−19、NK/LAK細胞マーカーを用いた二色フローサイトフルオロメトリー分析は、表面LT−α形態のLAK細胞発現が、T細胞ハイブリドーマII−23.D7に類似することを示した。図12に示されている実験では、PBLを、20ng/ml IL−2とともに5日間培養し、次に二色分析のために、上記のように、フィコエリスリン標識leu−19および抗rLT(ウサギ5)で染色した。図12は、免疫前(破線)あるいは免疫後(実線)血清で染色した、leu−19+細胞上の表面LT−αレベルを示す。従って、LAK細胞は、あらゆる一次細胞形態中で最も高い表面LT−αレベルを有するようである。
【0173】
(実施例7:全TNFあるいはLT−αのT細胞活性化との機能関連性)
TNFおよびLT−αのT細胞活性化プロセスとの機能関連性を調べるために、混合リンパ球応答(MLR)およびOKT3活性化アッセイに、ウサギ抗rLT−αおよび抗rTNF血清を包含させた。MLRは、個人のT細胞が、他人のT細胞を外来物として認識し、増殖によるその存在に応答する能力を試験する、標準的な免疫学的アッセイである。以下の表IIは、種々の応答細胞/刺激細胞の組合せを使用した、MLR実験のデータを示す。
【表II】
【0174】
表IIに示したように、中和抗rLT−α血清(ウサギ4および5)は、5日目に評価されるように、増殖応答を阻害したが、免疫前血清あるいは非中和抗rLT−α(ウサギ6)血清は軽い刺激効果を有した。以前に報告されたように[M.Shalabyら、J.Immunol.141,499(1988)]、ポリクローナルおよびモノクローナル抗TNF調製物もまた阻害性であった。これらのアッセイを、過剰刺激細胞条件で行った。従って、阻害は最適化され得ない。表II中で使用した血清レベルは、その他の実験(データは示していない)よりかなり高く、1:1000までの抗体希釈物はまだ阻害性であった。PHAあるいはOKT3刺激T細胞増殖もまた、より低い程度で阻害された(データは示さない)。これらのデータは、T細胞表面のLTあるいはLT−関連エピトープが、T細胞活性化に関連し得ることを示した。
【0175】
MLRアッセイおよび中和モノクローナル抗体を用いた以前の研究は、T細胞活性化および次のこの系での増殖において、LT−αではなくTNFに関係した。[M.Shalabyら、J.Immunol.,141,499(1988)]。その研究では、モノクローナル抗rLT−α抗体は、MLRアッセイに効果がなかった。中和ポリクローナル抗CHO細胞誘導rLT−α血清が、部分的にMLRを阻害し得ることが、われわれの研究で示され、そのことは、この系におけるなんらかのLT形態に対する役割を示唆する。これらの抗体調製物の性質に相違が存在し得るが、この矛盾に対する理由は明確ではない。モノクローナル抗体を、グルタルアルデヒドに架橋結合された天然のLT−α(RPMI 1788分泌の)に対して産生し、一方、ポリクローナル抗rLT−α血清は、未変性r−LT−α(組み換えCHO細胞由来の)を、フロイントアジュバントともに直接リンパ節に注射して調製した。後者の系の成果に対する貯蔵効果は、免疫化マウスにおいて報告された問題を考慮するとおそらく重要であった[T.Bringmanら、「ヒト腫瘍壊死因子αおよびβに対するモノクローナル抗体:アフィニティー精製、イムノアッセイヘの適用、および構造プローブとしての適用」Hybridoma,6,489(1987)]。これらのデータは、本発明のポリクローナル抗rLT−α血清のMLRにおけるブロック効果が、従来の可溶性LT−α形態の認識ではなく、血清による表面LT形態の認識の結果であることを示唆する。
【0176】
(実施例8:LT−αおよびLT−βの精製およびの最初の配列決定)
上記のように、エドマン分解によって、LT−αおよびLT−βのN末端配列情報を得た。膜結合LT−αバンドの配列を以下のように認めた:Leu Pro Gly Val Gly Leu Thr Pro Ser。この配列は、分泌LT−αの既知配列に一致する。エドマン分解分析は、結合LT−βタンパク質のN末端部分が、2つの可能なアミノ酸配列を含むことを解明した:Gly Leu Glu Gly Arg Gly Gln Arg Leu GlnあるいはGly Leu Glu Gly Arg Leu Gln Arg Leu Gln。次のDNA分析は、7サイクル目でグリシンがグルタミンとして誤って同定されたこと以外は、前者の配列を確認した。
【0177】
(実施例9:LT−β配列決定およびクローニング)
上記のように、Abersoldらの方法によって、数個のペプチド配列の配列を得た。2つのアンチセンス17−merオリゴヌクレオチドプローブGTYTCNGGCTCYTCYTC[配列番号:9]およびGTYTCNGGTTCYTCYTC[配列番号:10]を、これらのペプチド、T−87/T−88の1つの配列部分と一致するように合成した。これらのプローブを、32Pで放射性標識した。ノーザン分析(J.Sambrookら、Molecular Cloning a Laboratory Manual,第2版(1989))は、プローブ1368(GTYTCNGGTCYTCYTC)[配列番号:10]が、先に記載のように、ホルボールエステルで前処理されたII−23.D7細胞中で強く誘導される、0.9〜1.1kb mRNAバンドに強くハイブリダイズしたことを示した。ベクターpCDM8(Invitrogen Corporation,San Diego,Californiaから入手可能)のcDNAライブラリーを、PMAで6時間誘導されたII−23.D7細胞から単離されたポリA+ mRNAから構築した[B.SeedおよびA.Aruffo,PNAS,84,3365〜3369(1987)を参照のこと]。ライブラリーを標識オリゴマー1368でスクリーニングし、ポジティブクローンを単離し、その後50℃の3M テトラメチルアンモニウムクロライドで洗浄した。[Jacobsら、Nucleic Acids Research,16,10,4637〜4649(1988)を参照のこと]。0.9kbの挿入物を含有するいくつかのクローンを、DNA配列分析にかけた。クローンpCDM8/LT−β−12(クローン12)が、コーディング配列を含むことが認められた。その他のクローンは、1〜30bpの種々の5’末端切断型以外は同定された。1つの可能なクローン(クローン4)はフレームシフトを含み、トランスフェクション実験のコントロールとして使用された。862位の終止配列AATAAAが、ポリA区域の直前に見いだされ、このことは、全3’末端が同定されたことを示す。同定されたタンパク質配列は、計算上の分子量25,390を有する少なくとも240アミノ酸、およびタイプII膜タンパク質の代表的なドメイン構造をコードする。このデータは、開始CTGが、gly 5(met=1)で始まるプロセシングされたN末端を導く、すなわち、CTGにコードされるロイシンが翻訳されないか、あるいは、ロイシン−4基がさらにプロセシングされて、アミノ酸配列決定で得られた成熟N末端を得ることを示唆する。
【0178】
LT−βの33kDaの大きさは、先に定義したようにN−結合グリコシル化に起因し、この結果は、CD40リガンドに認められる同様の部位に一致する位置の、アミノ酸配列中の1つの可能なN−結合炭水化物部位の存在により、裏付けられる。代表的なN−結合糖残基は、約3〜4kDa MWを付加し得、それゆえに、最終分子量は観察された30〜33kDaに近い。
【0179】
同定されていない配列が、データベースで認められた。細胞外ドメインに1つのシステイン残基、および最後のC末端17アミノ酸内に2つのメチオニンが存在し、このタンパク質で示される、非常に限定された臭化シアン切断パターンに一致する。
【0180】
(実施例10:LT−βの発現)
pCDM8/LT−βクローン12またはコントロールプラスミド、クローン4(非機能性cDNA挿入物を有するpCDM8)を、エレクトロポレーションによって、CHO dhfr−およびヒトLT−αで安定してトランスフェクトされたCHO細胞に導入した。3日後、細胞を、Ca/Mgを含まない、5mM EDTA含有のハンクス液で取り出し、上記のように、10μg/mlのコントロールIgG1、あるいは抗LTモノクローナル抗体(Boehringer−Mannheim)を使用するFACS分析のために染色し、その後、FITCあるいはフィコエリスリン標識ヤギ抗マウス調製物を用いる結合免疫グロブリンで標識した。
【0181】
別の実験で、COS細胞に、pCDM8中のクローン4あるいはクローン12 LT−β cDNAを、これもまたpCDM8ベクター中の等量のヒトLT−α cDNAの存在下あるいは非存在下で、エレクトロポレーションし、上記のように3日後に、FACS分析用に染色した。LT−αを発現するCOS細胞のみが、機能性LT−β DNA、すなわちクローン12でのトランスフェクションで、表面リンホトキシンを示した。
【0182】
クローン12は、開始ATGコドンを欠くが、いくつかのCTG開始コドンを有し、それゆえに、この発現実験は、1つあるいはいくつかの5’CTGコドンが翻訳を開始し得ることを示す。CTGコドンは、数種の真核タンパク質での翻訳開始部位として作用することが知られている。[M.Kozak,J.Cell.Biol.,115,4(1991)]。同様の結果が、LT−αおよびLT−β DNAの両方を有するCOS細胞のみが、FACS分析で実質的な表面LT−αを示すような二元トランスフェクション系を使用して観察された。
【0183】
II−23.D7細胞のノーザン分析は、LT−β cDNAの0.9〜1.0kb mRNAへのハイブリダイゼーションを示し、このことは、クローニングしたcDNAが転写される全遺伝子を本質的に表すことを示す。図15を参照のこと。LT−β遺伝子は、非処理II−23.D7ハイブリドーマ細胞において低レベルで発現されるが、しかし、ホルボールエステルによる細胞の活性化で、mRNAレベルが劇的に増大した。図16を参照のこと。
【0184】
(実施例11:LT−βとリンホカインのTNFフミリーのその他のメンバーとの間の相同性)
LT−βをコードするcDNAのクローニングは、LT−βが、TNF、LT−α、およびCD40レセプターのリガンドに著しい相同性を有するタイプII膜タンパク質であることを解明した。これらのタンパク質は、TNF/NGFレセプターファミリーのメンバーに結合することが知られている。LT−β、TNF、LT−αおよびCD−40レセプターのリガンドは、細胞外ドメインに4つの配列保存領域を共有する。図13を参照のこと。これらのドメインは、TNFおよびLT−α結晶構造の表面に存在し、サブユニット間相互作用に関連するようである。
【0185】
(実施例12:数個の可能な開始部位を明らかにするLT−βの5’末端の決定)
5’mRNA配列を、プライマー伸長分析によって決定した。プライマー伸長分析は、LT−βタンパク質の転写開始部位が、メチオニンATGの上流約7〜9塩基対であることを解明した。従って、mRNAは、少なくとも3つの可能な転写開始部位、Met−1,Leu−4、およびLeu−6コドンを有する。クローン12を用いる一時的な実験は、Leu−4あるいはLeu−6開始部位の1つあるいは両方が機能することを示した。5’mRNA配列は、上記のコスミドクローン031Aを使用したLT−βゲノム配列の決定、およびジデオキシ核酸配列決定によって明らかにされた。
【産業上の利用可能性】
【0186】
本発明は、リンホトキシン−β、リンパ球膜タイプタンパク質に関する。このタンパク質は、ホルボールエステル(PMA)刺激T細胞ハイブリドーマII−23.D7細胞を含む多数の細胞の表面で見られる。本発明はまた、リンホトキシン−βとリンホトキシン−αのような他のペプチドとの間で形成される複合体およびリンホトキシン−βの多数のサブユニットを包含する複合体に関する。これらのタンパク質および複合体は、細胞内で形成されるLT−αを細胞表面に保持するのに有用であり、LT−α/LT−β複合体が炎症調節剤、腫瘍増殖阻害剤、T細胞阻害剤、T細胞活性化剤、自己免疫疾患調節剤、またはHIV阻害剤として作用し得る。さらに、LT−α/LT−β複合体の抗腫瘍活性を、LT−βの遺伝子でトランスフェクトされた腫瘍湿潤リンパ球(TIL)によって腫瘍細胞に送達し得る。
【図面の簡単な説明】
【0187】
【図1】図1は、OKT3−刺激、IL−2拡張末梢血リンパ球(PBL)のフローサイトフルオロメトリー分析を示し、3つの、異なるウサギ抗rLT−α抗血清との反応を示し、そして本質的にウサギ抗rTNF抗血清との反応がなかったことを示す。
【図2】図2は、ヒトT細胞ハイブリドーマ、II−23.D7のフローサイトフルオロメトリー分析を示し、細胞表面上の(PMA処理後の)LT−α関連エピトープの存在を示す。
【図3】図3は、PMA活性化II−23.D7細胞の抗rLT−α抗体と結合する能力を示す。抗LT−α抗血清のサンプルと、PMA処理U937(非LT生成)細胞、(−〇−)、PMA活性化II−23.D7ハイブリドーマ細胞(108、−−;107、−・−)、および細胞なし(コントロール、−−)とをインキュベートした。インキュベーション後、無細胞抗血清の連続希釈をrLT−αに添加し、そしてL929(LT−αセンシティブ)細胞に対する細胞障害性アッセイに使用した。プロットは、rLT−α中和抗体が抗血清から活性化II−23.D7細胞によって除去されたことを示す。
【図4】図4は、PMA活性化II−23.D7細胞からの125I−標識表面タンパク質の免疫沈降を示す2つのオートラジオグラフ(AおよびB)を示す。図4Aは、免疫前ウサギ血清ではなくて免疫後ウサギ抗rLT−α抗血清によって、約25kD表面タンパク質(LT−α)および約33kD表面タンパク質(p33、すなわちLT−β)の免疫沈降を示す。図4Bは、図4Aからの25kDおよび33kDバンドの一次元CNBr切断マップを示し、E.coliで生産した組換えTNF(rTNF)、およびCHO細胞で生産される組換えリンホトキシン−α(rLT−α)[J.Browingら、J.Immunol.143、1859(1989)]と、CNBr切断の有り(+)およびなし(−)の両方で比較した。
【図5】図5は、35S−メチオニンまたは35S−システインで代謝的に標識したPMA刺激II−23.D7細胞からのTNF−およびLT−α−関連タンパク質の免疫沈降を示すオートラジオグラフである。この図は、約25kDメチオニン含有表面タンパク質(LT−α)および約33kDメチオニンおよびシステイン含有表面タンパク質(p33、すなわちLT−β)が、免疫前(P)または抗rTNF抗血清(T)ではない抗rLT−α抗血清(L)によって認識されることを示している。オートラジオグラフもまた、活性化II−23.D7細胞がまた26kD形態のTNFを生産し、そして可溶性リンホトキシン−αを分泌することを示す。
【図6】図6は、抗rLT−α血清によって認識されるPMA活性化II−2S.D7細胞からのこれらのタンパク質のアフィニティ精製1−D CNBrペプチドマッピングを示す。図6Aは、免疫前(PRE)または免疫後(POST)ウサギ血清のいずれかから調製した抗LT−αアフィニティーカラムから溶出されるタンパク質のSDS PAGE分析を表す。図6Aは、約33kDおよび約20kDタンパク質バンドは免疫前血清(PRE)を使用して調製したアフィニティーカラムに結合しないが、抗rLT−α抗血清(POST)を使用して調製したアフィニティーカラムに結合することを示す。図6Bは、POSTカラムから溶出される約33kDおよび約20kDタンパク質の部分的なCNBr切断を示し、並行して泳動したrTNFおよびrLT−αとの比較を示す。ゲルを、銀染色法で視覚化した。
【図7】図7は、約25kDおよび約33kD125I−標識タンパク質(それぞれLT−αおよびLT−βと命名した)のオートラジオグラフを示すが、これらは、N−グリカナーゼ(N−gly)、ノイラミニダーゼおよびO−グリカナーゼ(O−gly)の混合物で、または3つの酵素すべてで処理された活性化II−23.D7細胞から免疫沈降されたものである。
【図8】図8は、共沈降したp33(LT−β)およびp25(LT−α)タンパク質の再免疫沈降の結果を示し、さらにこれらが免疫学的に関連しているかどうか調査する。
【図9】図9(9Aおよび9B部分を含む)は、免疫沈降したp33(LT−β)およびp25(LT−α)タンパク質の変性条件下での等電点フォーカシングの結果を示す。
【図10】図10(10Aおよび10B部分を含む)は、免疫沈降したp33(LT−β)およびp25(LT−α)タンパク質の未変性条件下での等電点フォーカシングの結果を示す。図9および図10は共に、LT−βおよびLT−αが変性可能な複合体を形成することを示す。
【図11】図11は、LPS、IFN−γおよびOKT3の混合物で刺激した後、T細胞および単球上で分化的に発現した表面タンパク質をフローサイトフルオロメトリーを示す。刺激したPBLプールから分離したT細胞は、抗rLT−α抗血清(LT)によって認識される表面タンパク質を発現することが認められ、他方分離した単球は抗rTNF抗血清(TNF)によって認識される表面タンパク質を発現した。
【図12】図12は、IL−2で処理されたleu−19−およびleu−19+(すなわち天然キラー)細胞上の表面LT形態のフローサイトフローメトリー分析を示す。IL−2で処理したPBLを標識leu−19および抗rLT−αの両方で分析した結果、リンホカイン活性化キラー(LAK)細胞が表面LT形態を発現するのを確認した。
【図13】図13は、LT−βフラグメントのアミノ酸配列を示すが、これは、直接N−末端配列決定およびインサイチュトリプシン切断に続いて、切断されたペプチドの逆相HPLC分離によって得られた。
【図14】図14は、TNF/NGF−レセプターファミリーのメンバーと結合するリガンドのファミリーの4つのメンバーのアミノ酸配列比較を示す。ホモロジー領域を、太いタイプフェイスで、同一配列をドットで示し、そして保存配列を星印で示す。推定N−結合グルコシル化部位を四角で囲んでいる。
【図15】図15は、これはLT−αおよびLT−β発現のノーザン分析を示す。A)いくつかの細胞株のノーザンブロットでHut−78およびII−23.D7細胞中のみで両方のLT遺伝子を特異的に発現することを示す。B)II−23.D7細胞中のLT−αおよびLT−β mRNAのPMA誘導の時間経過 C)抗CD3またはIL−2単独のいずれかで活性化されたヒト末梢血リンパ球の同様の分析を示す。
【図16】図16は、CHO細胞中のLT−αおよびLT−βの発現を示す。A)LT−β cDNAで一時的にトランスフェクトされたCHO細胞のFACS分析。親dHFR−CHO株またはLT−αを発現するCHO細胞のいずれかを、無関係の挿入物(クローン4)を含有するpCDM8またはpCDM8/LT−βプラスミドのいずれかでエレクトロポレートし、そしてコントロールIgG(−−−)または抗LT−αモノクローナル抗体(___)で染色した。B)パネルBは、COS細胞中でLT−αおよびLT−βの発現を示す。細胞を、pCDM8/LTαと共に、または用いないで、コントロールDNAまたはpCDM8/LTβプラスミドでトランスフェクトし、そしてパネルAと同様に染色した。
【技術分野】
【0001】
(発明の技術分野)
本発明は、リンパ球膜タイプポリペプチドであるリンホトキシン−βに関する。リンホトキシン−β(p33と呼ばれる)は、Tリンパ球、T細胞株、B細胞株およびリンホカイン活性化キラー細胞の表面上で同定されている。
【0002】
本発明はまた、リンホトキシン−β(LT−β)およびリンホトキシン(ここでは「リンホトキシン−α(LT−α)」と称して、LT−βと区別している)のような他のリンホトキシンタイプペプチドとの間で形成される複合体ならびにLT−βの多数のサブユニットを含有する複合体に関する。本発明のLT−βポリペプチドは、細胞中で形成されたLT−αを細胞表面上に保持する点で有益であると考えられ、該表面で、LT−βまたはLT−α/LT−β複合体のいずれかが浸潤調節剤、腫瘍成長阻害剤、T細胞抑制剤、T細胞活性化剤、免疫モジュレ一ター剤、自己免疫疾患調節剤またはHIV調節剤として作用し得る。さらに、LT−α/LT−β複合体の抗腫瘍活性は、LT−βの遺伝子でトランスフェクトされた腫瘍浸潤リンパ球(TIL)によって腫瘍細胞へ送達され得る。
【0003】
ここに記述した発明は、国立衛生研究所(the National Institutes of Health)許可番号CA35638−07−10の下での一連の研究の中の一部でなされた。米国政府はこの発明に一定の権利を有している。
【背景技術】
【0004】
(発明の背景)
免疫応答の開始は、細胞間シグナルの複雑な配列を含む。これらのシグナルは、代表的に多数の細胞−細胞接触依存シグナルでカップルした可溶性サイトカインを含有する。接触依存事象(T細胞レセプターの最も有名な活性化)は、応答に特異性を与えるが、他方可溶性メディエーターは、細胞分化および細胞増殖の維持に対して一般的に応答可能である。腫瘍壊死因子(TNF)およびLT−αは、一般的に免疫応答の開始に関与すると認識されている2つのポリペプチドである。
【0005】
TNFおよびLT−αは、元来腫瘍の成長を阻害する能力が知られている可溶性タンパク質である。[L.Old、「腫瘍壊死因子」、Science、230、630(1985)]。さらなる研究により、両方のタンパク質は広範囲な活性を有することが示された。TNFは、造血細胞および非造血細胞の両方を含有する種々の細胞タイプによって、種々の炎症性の傷害に応答して合成されるが、対照的にLT−αはリンパ球によって特異的に作られる。2つの公知のTNFレセプターは、LT−αとTNFとをはっきりと区別することができない。[T.Schallら、「ヒト腫瘍壊死因子のレセプターの分子クローニングおよび発現」、Cell、61、361−370(1990);C.Smithら、「腫瘍壊死因子のレセプターが、細胞のおよびウイルス性タンパク質の異常なファミリーを定義する」Science、248、1019(1990)]。一般に、LT−αはしばしばほとんど強力ではないが、LT−αおよびTNFは、インビトロ系で類似の活性スペクトルを示す。[J.Browningら、「いくつかのヒト腫瘍系統の成長に対する腫瘍壊死因子およびリンホトキシンの異なる影響の研究」、J.Immunol.、143、1859(1989)]。
【0006】
TNFは、内毒素ショックヘの応答、および造血細胞発達の調節において、代謝制御の特別な局面で主要な役割をつとめるようである。[B.Beutlerら、「カケクチン(cachectin)の由来、性質、および生物学的効果」、Biochemistry、27、(1988);M.Akashiら、「リンホトキシン:線維芽細胞中のコロニー促進因子の促進および調節」、Blood、74、2383(1989);G.Roodmanら、「腫瘍壊死因子−αおよび造血前駆体:赤血球前駆体CFU−EおよびBFU−Eならびに造血幹系統k562、HL60、およびHEL細胞の成長に対する腫瘍壊死因子の効果」、Exp.Hematol.、15、928(1987)]。
【0007】
IL−1およびIL−6とともに、TNFもまた、炎症性応答の主要なメディエ一ターである。[D.Cavenderら、「腫瘍壊死因子およびリンホトキシンによって誘発される内皮細胞の活性化」、Amer.Jour.Path.、134、551(1989);R.Cotranら、「炎症性および免疫反応における内皮細胞活性化の役割」、Endothelial Cell Biology、(Plenum Press、SimonescuおよびSimonescu編、1988)335]。TNFはまた、ある条件下でT細胞活性化に関与しているようである。[M.Shalabyら、「混合リンパ球反応中のヒト腫瘍壊死因子−αおよび−βの関与」、J.Immunol.、141、499(1988);N.Damleら、「ヒトT細胞活性化のCD3依存およびCD3非依存開始の間のIL−4およびTNF−αの異なる調節効果」、Lymph.Res.、8、85(1989);G.Rangesら、「IL−2依存性T細胞株の増殖シグナルとしての腫瘍壊死因子−α:活性の絶対種特異性」、Amer.Assoc.Immunol、142、1203(1989);G.Rangesら、「腫瘍壊死因子α/カケクチンは胸腺細胞の成長因子である」、J.Exp.Med.、167、1472(1988);P.Scheurichら、「組換えヒト腫瘍壊死因子(TNF)−αの免疫調節活性:ヒトT細胞のTNFレセプターの誘発およびT細胞応答のTNF−α−仲介促進」、J.Immunol.、138、1786(1987)]。
【0008】
TNFは、単球、線維芽細胞、T細胞およびナチュラルキラー(NK)細胞を含む数種のタイプの細胞によって生産される。[D.Goeddelら、「腫瘍壊死因子:遺伝子構造および生物学的活性」、Cold Spring Harbor Symposium Quant.Biol.、51、597(1986);D.Spriggsら、「ヒト上皮腫瘍細胞株での腫瘍壊死因子の発現」、J.Clin.Invest.、81、455(1988);M.Turnerら、「自己免疫および正常個体からのヒトT細胞が腫瘍壊死因子を生産し得る」、Eur.J.Immunol.、17、1807(1987)]。研究者はまた、ネズミまたはヒト形態のTNFを検出し、それらがトランスメンブランタンパク質またはレセプター結合分子のいずれかとして種々の細胞の表面に結合していることを示した。[B.Luettigら、「膜腫瘍壊死因子の2形態の存在についての証拠:内在性タンパク質およびそのレセプターに付着した分子」、J.Immunol.、143、4034(1989);M.Krieglerら、「新規形態のTNF/カケクチンは細胞表面細胞傷害性トランスメンブランタンパク質である:TNFの複合生理学についての分枝」、Cell、53、45−53頁(1988)];およびM.Kinkhabwalaら、「T細胞レパートリーヘの新規追加」、J.Exp.Med.、171、941−946頁(1990)]。
【0009】
LT−αは、TNFの活性とは一般的に類似しているものの同一ではない多くの活性、例えば、腫瘍壊死、抗ウイルス状態の誘発、多形核白血球の活性化、内皮細胞上のクラスI主要組織適合性複合抗原の誘発、内皮細胞の接着分子の誘発および成長ホルモン刺激を含む。[N.RuddleおよびR.Homer、「炎症におけるリンホトキシンの役割」、Prog.Allergy、40、162−182頁(1988)]。LT−αおよびTNFの両方とも、神経成長因子(NGF)レセプターファミリーのメンバーのリガンドである。[S−MallettおよびA.N.Barclay、「神経成長因子レセプターに関連する細胞表面タンパク質の新しいスーパーファミリー」、Immunology Today、12、7、220−223(1991)]。
【0010】
TNFとは対照的に、LT−αの分泌は、活性化されたT細胞およびある種のB−リンパ芽球腫瘍のみの特異的な性質のようである。[N.Paulら、「リンホトキシン」、Ann.Rev.Immunol.、6、407(1988)]。ある研究者たちはまた、LT−αの膜結合形態は、ある環境下でリンパ球の表面上で発現され得ることを示した。[J.Hiserodtら、「インビトロでの異種の抗LT抗血清を使用するマイトジェン活性化ヒトリンパ球上の膜結合リンホトキシン(LT)の同定」、Cell.Immunol.、34、326−339頁(1977);C.wareら、「リンパ球仲介細胞障害性の機構」、J、Immunol.、126、1927−1933頁(1981);U.Andersonら、J.Immunol.Methods.、123、233(1989);Y.Abeら、Jpn.J.Canc.Res.、82、23(1991);Y.Abeら、「インビトロでのヒトリンホカイン活性化Tキラー細胞における膜結合および可溶性(分泌)リンホトキシンの研究」、Lymphokine and Cytokine Research、11、2、115−121(1992)]。
【0011】
近年、TNFおよびLT−αの両方の遺伝子が単離され、そしてクローン化されて、完全な特徴づけがなされ両方の組換え形態のタンパク質が入手可能となった。[P.Grayら、「ヒトリンホトキシンのcDNAのクローニングおよび発現、腫瘍壊死活性を有するリンホカイン」、Nature、312、121−124頁(1984);D.Pennicaら、「ヒト腫瘍壊死因子:前駆体の構造、発現およびリンホトキシンとのホモロジー」、Nature、312、724(1984)]。
【0012】
CD40タンパク質を含有する他の「サイトカイン様」細胞表面タンパク質は、TNFおよびLT−αとある種の類似性を共有していることが近年示された。TNFおよびLT−αと同様に、CD40タンパク質は、TNF/神経成長因子(NGF)レセプターファミリーのメンバーに対するリガンドである。[S.MallettおよびN.Barclay、Immunology Today、12、220−223頁(1991)]。CD40タンパク質は、B−リンパ球、上皮細胞、およびある種の癌腫細胞様の表面に発現する277アミノ酸のタンパク質である。[R.Armitageら、Nature、357、80−82頁(1992);T.FarrahおよびC.Smith、「サイトカインファミリーの発生」、Nature、358、p.26(1992)]。
【0013】
本発明者らは、新規な表面タンパク質、リンホトキシン−β(LT−β)すなわちp33を同定した。LT−βは、種々のタイプのリンパ球の表面で同定されたが、これらのリンパ球は、OKT3刺激一次T細胞、抗原特異的IL−2依存CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマII−23.D7を含む。LT−βは、細胞の中で生産され細胞膜に向かうLT−αを標的としており、細胞膜では、LT−βおよびLT−αは複合体(本開示を通して「LT−α/LT−β」と表示する)のようにみえる。LT−α/LT−βは、活性化T細胞によるLT−αの膜発現に対する新規機構であると考えられている。[Androlewiczら、「リンホトキシンは、活性化ヒトT細胞ハイブリドーマの表面上で別の33kDa糖タンパク質とヘテロマーな複合体として発現される」、Journal of Biological Chemistry、267、2542−2547頁(1992)]。LT−α/LT−β複合体は、可溶性LT−α、TNFおよびCD40タンパク質と類似の細胞溶解および細胞調節活性を示し得る。LT−αと複合した膜結合LT−βは、複合体としてT細胞が他の細胞と相互作用をする新規リガンドを表し得、そしてまた標的細胞の溶解に有用であり得る。
【発明の開示】
【課題を解決するための手段】
【0014】
(発明の要旨)
本発明の新規タンパク質はリンホトキシン−β(LT−β)と命名された。このタンパク質は、種々のタイプのリンパ球の表面で見い出されるが、これらのリンパ球はOKT3刺激一次T細胞、抗原特異的IL−2依存CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマII−23.D7を含む。それはLT−αとの新規複合体を形成し、そして他のLT−βサブユニットとの複合体を形成する(例えば、(LT−β)2LT−α複合体)。
【0015】
LT−βは、31−35kDの分子量を有するが、これは免疫沈降およびSDS−PAGEによって測定された。LT−βは、N−結合グルコシル化を有する。リンホトキシン−βのアミノ酸配列を配列番号2で示し、そしていくつかの可溶性リンホトキシン−βペプチドのアミノ酸配列を配列番号4、配列番号6および配列番号8で示す。リンホトキシン−βをコードするDNA配列を配列番号1で示し、数種の可溶性リンホトキシン−βペプチドをコードするDNA配列を、配列番号3、配列番号5および配列番号7で示す。
【0016】
細胞膜タンパク質としてのLT−βは、合成中のLT−αと結合する。従って、LT−αを細胞膜に「標的化」する。LT−βの非存在下で、LT−αは細胞外培地に分泌される。LT−α/LT−β複合体は、CHO細胞中で発現された組換えリンホトキシン−α(rLT−α)に対して生じたポリクローナル抗血清により、または天然LT−αに対して生じたモノクローナル抗体(mAbs)により認識される。さらに、LT−α/LT−β複合体および(LT−α)3を認識する抗血清は、混合リンパ球反応(MLR)をブロックする。MLRは同種間刺激に対するTリンパ球の予想される増殖応答であり、すなわち、他の個体からのTリンパ球を導入した際に「レスポンダー」リンパ球により異物(非自己)として認識される標準免疫学的アッセイである。[例えばM.Shalabyら、J.Immunl.、141、499(1988)を参照のこと]。
【0017】
LT−βタンパク質をアフィニティクロマトグラフィにより精製し、部分配列を決定し、そして特異的オリゴヌクレオチドプローブを設計した。LT−αをコードするcDNAを、活性化II−23.D7細胞(ホルボールエステル活性化により大量の表面リンホトキシンを表すヒトT細胞ハイブリドーマ)からのcDNAライブラリーをプローブすることによって単離した。同定したアミノ酸配列は、240〜244アミノ酸配列(約25130〜25390kDaの非修飾タンパク質の分子量)をコードする。配列番号2を参照のこと。アミノ酸配列およびトランスメンブラン領域位置はタイプII膜タンパク質の典型である。
【0018】
この配列は、短い14〜18アミノ酸N−末端「細胞質」ドメインを含有する。この細胞質ドメインの後ろに、おそらく膜固定ドメインとして働く30の疎水性アミノ酸の長い広がりがある。同一の配列は利用可能なデータベースには見られなかった。細胞外ドメインには1つのシステイン残基およびC−末端の最後の17アミノ酸に2つのメチオニン残基がある。これは、このタンパク質が示す非常に制限された臭化シアン切断パターンと一致する。
【0019】
LT−β配列とTNF/NGFレセプターファミリーのメンバーに結合することが知られている公知の他のタンパク質を比較すると、かなり構造の類似性がある。TNF/NGFレセプターファミリーのメンバーに対する4つのリガンド(TNF、LT−α、LT−βおよびCD40リガンド)は、タイプII膜タンパク質に類似し、そして図14に示されるような細胞外ドメイン中の保存配列の少なくとも4つの領域を共有している。LT−βと共有する、保存されたTNFおよびLT−αドメインは、サブユニット間相互作用およびβシート構造に関連しているようである。これらの保存領域は、LT−αおよびLT−βの間の結合を説明し得る。これらホモロジー領域の存在は、ポリペプチドを設計して、例えば、TNFまたはCD40リガンドと複合体を容易に形成させ得る。このような分子は、機能を混在し得、そして注文のデザインされた薬として使用され得る。[J.Fuhら、「ヒト成長ホルモンレセプターと拮抗するアンタゴニストの合理的なデザイン」、Science、256、1677(1992)参照のこと]。
【0020】
本発明のペプチド複合体はT細胞活性化の事象に重要であり、T細胞活性化あるいはT細胞抑制のための組成物および方法、ならびに炎症の治療剤として、あるいは腫瘍細胞または新生物の増殖阻害のような細胞障害活性を必要とする適用するための治療剤として有用であると考えられる。また、ポリペプチド複合体は細胞免疫療法に有用であり、腫瘍浸潤リンパ球(「TIL」)治療における腫瘍浸潤リンパ球の殺腫瘍能力を高めることが重要であると考えられる。TIL免疫療法は、遺伝子組換え技術により改善され得る。例えば、遺伝子を、体の免疫系を誘導する目的で腫瘍細胞に加えて、効果的な腫瘍特異的免疫応答を仲介し得る。[例えば、W.F.Anderson、「ヒト遺伝子療法」、Science、256、808−813(1992)参照のこと]。
【0021】
また、「Fas」として同定される分子とTNF/NGFレセプターファミリーのメンバーとの間の類似性に基づいて、本発明のポリペプチド複合体は、プログラムされた細胞死またはアポトーシスとして知られている細胞の内部プロセスに関与し得、それゆえ、自己免疫病の仲介に関与し得ると考えられる。[例えば、N.Itohら、「ヒト細胞表面抗体FasのcDNAによってコードされるポリペプチドはアポトーシスを仲介し得る」、Cell、66、233−243(1991);R.Watanabe−Fukunagaら、「アポトーシスを仲介するFas抗原の欠陥によって説明されるマウスにおけるリンパ増殖疾患」、Nature、356、341(1992)参照のこと]。
【0022】
LT−β、その関連ポリペプチド、LT−α/LT−β複合体または本発明の他のポリペプチド複合体に対する抗体はまた、特定のレセプターとの重要なLT相互作用を破壊する。すなわち、既知のTNFレセプター形態を介して仲介される事象以外の特異的に影響するLT仲介事象を形成し得る。同様に、TNF、LT−αまたはLT−α/LT−β、またはこれらの誘導体(例えば、可溶性レセプターおよびIgG/レセプター融合タンパク質)に対するレセプターも、本発明のポリペプチドおよび複合体の阻害に使用し得る。
【0023】
本発明は、以下を提供する:
(項目1) 配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−β。
【0024】
(項目2) 上記ポリペプチドが細胞表面に結合しているポリペプチドである、項目1に記載のポリペプチド。
【0025】
(項目3) 上記ポリペプチドが、OKT3刺激一次T細胞、抗原特異的IL−2依存性CTLクローン、およびPMA刺激ヒトT細胞ハイブリドーマであるII−23.D7に結合している、項目2に記載のポリペプチド。
【0026】
(項目4) 可溶性リンホトキシン−βペプチドであって、以下からなる群から選択されるアミノ酸配列:
(a)配列番号4;
(b)配列番号6;および
(c)以下の式で示されるアミノ酸配列:
X−配列番号6、
ここで、Xは配列番号8の3’末端から始まる1つまたはそれ以上のアミノ酸残基を含有する;
を包含するペプチド。
【0027】
(項目5) 前記ペプチドが、さらに5’末端でのリーダー配列を含有する、項目4に記載のペプチド。
【0028】
(項目6) ポリペプチドであって、以下からなる群からのDNA:
(a)配列番号1を含有するDNA配列;
(b)DNA配列であって、配列番号1によって定義されるDNAにハイブリダイズし、かつリンホトキシン−βと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(c)配列番号1によって定義されるDNA配列によってコードされるポリペプチドをコードする縮重ヌクレオチド配列を含有する、DNA;
によってコードされるアミノ酸配列を含有する、ポリペプチド。
【0029】
(項目7) ポリペプチドであって、以下からなる群からのDNAによってコードされるアミノ酸配列:
(a)配列番号3を含有するDNA配列;
(b)配列番号5を含有するDNA配列;
(c)以下の式で示されるDNA配列:
X−配列番号5、
ここで、Xは、配列番号7の3’末端から始まる1つまたはそれ以上のヌクレオシドトリプレットを含有する;
(d)DNA配列であって、配列番号3、配列番号5および上記(c)部のいずれか1つとハイブリダイズし、可溶性リンホトキシン−βペプチドと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(e)DNA配列であって、配列番号3、配列番号5および上記(c)部の配列のいずれか1つによってコードされるポリペプチドをコードしている、縮重ヌクレオチド配列を含有する、DNA配列;
を含有するポリペプチド。
【0030】
(項目8) 設計されたポリペプチドであって、配列番号2で定義されるアミノ酸配列を包含し、ここで配列Leu Gly Leuが上記配列の5’末端から切断され、かつ1つのMetまたは1っのLeu残基によって置換される、ポリペプチド。
【0031】
(項目9) 単離されたDNA配列であって、以下からなる群:
(a)配列番号1で定義されるヌクレオチド配列を含有するDNA配列;
(b)DNA配列であって、配列番号1によって定義されるDNA配列とハイブリダイズし、かつリンホトキシン−βと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;
(c)リンホトキシン−βをコードする縮重ヌクレオチド配列を含有するDNA配列;から選択される、DNA配列。
【0032】
(項目10) 単離されたDNA配列であって、以下からなる群:
(a)配列番号3で定義されるヌクレオチド配列を含有するDNA配列;
(b)配列番号5で定義されるヌクレオチド配列を含有するDNA配列;
(c)項目7(c)のヌクレオチド配列を含有するDNA配列;
(d)DNA配列であって、配列番号3、配列番号5または項目7(c)の配列のいずれか1つによって定義されるDNA配列にハイブリダイズし、かつ可溶性リンホトキシン−βペプチドと実質的に相同であるポリペプチドを発現するようにコードする、DNA配列;および
(e)可溶性リンホトキシン−βペプチドをコードする縮重ヌクレオチド配列を含有するDNA配列;
から選択される、DNA配列。
【0033】
(項目11) 設計されたDNA配列であって、配列番号1で定義されるヌクレオチド配列を含有し、ここでヌクレオチドCTGGGGCTGが上記配列の5’末端から切断され、かつ1つの開始コドンによって置換される、DNA配列。
【0034】
(項目12) 組換えDNA分子であって、以下からなる群:
(a)配列番号1で定義されるDNA配列;
(b)配列番号3で定義されるDNA配列;
(c)配列番号5で定義されるDNA配列;
(d)項目7(c)に記載のDNA配列;
(e)項目11に記載のDNA配列;
(f)DNA配列であって、配列番号1、配列番号3、配列番号5および項目7(c)に記載の配列のいずれか1つによって定義されるDNA配列とハイブリダイズし、かつリンホトキシン−βまたは可溶性リンホトキシン−βペプチドを発現するようにコードする、DNA配列;
(g)縮重ヌクレオチド配列を含有し、リンホトキシン−βに対してコードするDNA配列;および
(h)縮重ヌクレオチド配列を含有し、可溶性リンホトキシン−βペプチドに対してコードするDNA配列;
から選択されるDNA配列を含有する、組換えDNA分子。
【0035】
(項目13) 単細胞宿主、培養中の動物細胞および培養中のヒト細胞からなる群から選択される宿主であって、項目12に記載の組換えDNA分子でトランスフェクトされた、宿主。
【0036】
(項目14) 腫瘍浸潤リンパ球、リンホカイン活性化キラー細胞、キラー細胞および患者から取り出され、遺伝的に設計された腫瘍細胞からなる群から選択される項目13に記載の宿主。
【0037】
(項目15) 項目1〜8のいずれかに記載のポリペプチドを生産する方法であって、上記方法は項目13に記載の形質転換宿主を培養する工程、および該ポリペプチドを回収する工程を包含する、方法。
【0038】
(項目16) ポリペプチド複合体であって、配列番号2、配列番号4、配列番号6、項目8のポリペプチド、および項目4(c)の可溶性リンホトキシン−βペプチドからなる群から選択される第1のポリペプチド、および、リンホトキシン−α、天然ヒトまたは動物リンホトキシン、組換えリンホトキシン、可溶性リンホトキシン、分泌リンホトキシン、あるいはリンホトキシンまたは上記の任意のリンホトキシン活性フラグメントからなる群から選択される第2のポリペプチドを含有する、ポリペプチド複合体。
【0039】
(項目17) 複数のリンホトキシン−βポリペプチドユニットを含有するポリペプチド複合体。
【0040】
(項目18) 項目16に記載のポリペプチド複合体であって、上記複合体が細胞表面と結合している、ポリペプチド複合体。
【0041】
(項目19) 項目18に記載のポリペプチド複合体であって、上記第1のポリペプチドがOKT3刺激一次T細胞、抗原特異的IL−2依存性CTLクローン、およびPMA刺激非リンホトキシンヒトT細胞ハイブリドーマであるII−23.D7の表面と結合している、ポリペプチド複合体。
【0042】
(項目20) 細胞の表面上でリンホトキシンエピトープを生産する方法であって、項目12に記載の組換えDNA分子で該細胞をトランスフェクトする工程、および該細胞中でDNAを発現させる工程を包含する、方法。
【0043】
(項目21) 腫瘍浸潤リンパ球の標的殺腫瘍性活性を高める方法であって、項目12に記載の組換えDNA分子で上記リンパ球をトランスフェクトする工程、および上記形質転換されたリンパ球を患者に導入する工程を包含する、方法。
【0044】
(項目22) 項目21に記載の方法であって、上記形質転換されたリンパ球を項目12に記載の組換えDNAでトランスフェクトする前またはトランスフェクトした後に、リンホカインとともにインキュベートする、方法。
【0045】
(項目23) 項目22に記載の方法であって、前記リンホカインがIL−2である、方法。
【0046】
(項目24) HIV感染、新生物形成、炎症または炎症性の病気、ならびに自己免疫疾患の進行、重篤、または影響を、抑制、処理または軽減するための組成物であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを含有する、組成物。
【0047】
(項目25) HIV感染、新生物形成、炎症または炎症性の病気、ならびに自己免疫疾患の進行、重篤、または影響を、抑制、処理または軽減するための方法であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを投与することを含有する、方法。
【0048】
(項目26) 免疫系を抑制する組成物であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを含有する、組成物。
【0049】
(項目27) 免疫系を抑制する方法であって、項目1〜8のいずれかに記載のポリペプチド、項目16〜19のいずれかに記載のポリペプチド複合体、上記のいずれかに対する抗体、または上記のいずれかの組合せからなる群から選択される有効量のポリペプチド、および薬学的に受容可能なキャリアを投与することを包含する、方法。
【0050】
(項目28) リンホトキシン−βをコードするヌクレオチド配列であって、配列番号1により表される上記ヌクレオチド配列を含有し、さらに5’末端で設計されたヌクレオチド配列を含有し、上記設計されたヌクレオチド配列がATGまたはCTGである機能的な開始コドンを含有し、上記設計されたヌクレオチド配列中で、ロイシンをコードする他のコドンがCTGでない、ヌクレオチド配列。
【0051】
(発明の詳細な説明)
ここに記載された本発明を完全に理解するために、以下に詳細に説明する。
【0052】
本発明は、リンパ球膜タイプポリペプチドであるリンホトキシン−βに関する。リンホトキシン−βのアミノ酸配列を配列番号2に示す。
【0053】
ポリペプチド(またp33という)は、31〜35kDの分子量を有する。本発明のポリペプチドは、細胞表面に結合するか、またはそのような表面に結合しない。
【0054】
本発明はまた、リンホトキシン−βの可溶形態に関する。可溶性リンホトキシン−βペプチドは、リンホトキシン−βのアミノ酸配列で定義され、ここで、配列は、トランスメンブラン領域の端(すなわち、アミノ酸番号44付近)とアミノ酸番号95付近の最初のホモロジー領域との間の任意の位置で切断される。2つの可溶性リンホトキシン−βペプチドのアミノ酸配列は、配列番号4および6で定義されている。いくつかの付加的な可溶性リンホトキシン−βポリペプチドは、配列番号6によって定義されるアミノ酸配列の5’末端に付加的アミノ酸残基を有する配列を包含する。付加的な残基は、配列番号8によって定義される52アミノ酸残基を包含し得る。可溶性リンホトキシンはまた、配列番号6で定義される配列に3’末端から始まる1〜51のアミノ酸残基を含有する配列番号8の一部を加えたアミノ酸配列を包含する。このような可溶性ペプチドは、5’末端に周知のリーダー配列を任意の数包含し得る。このような配列により、真核系でペプチドが発現される。[例えば、Ernstら、米国特許番号第5,082,783(1992)]。
【0055】
本発明のポリペプチド複合体は、配列番号2、配列番号4、配列番号6、および配列番号6プラス上述したような配列番号8の全部または一部分によって定義される配列からなる群から選択されるアミノ酸配列を含有する第1のポリペプチドと、同一群から選択される第2のポリペプチド、および/またはリンホトキシン−α、天然ヒトまたは動物リンホトキシン−α、組換えリンホトキシン−α、可溶性リンホトキシン−α、分泌リンホトキシン−α、または上記の任意のリンホトキシン−α活性フラグメント等との第2のポリペプチドと複合した複合体に関する。
【0056】
新規LT−βペプチドはLT−αと複合体を形成し、そして他のLT−βサブユニットと複合体を形成する(例えば、(LT−β)2LT−α複合体)。これらの複合体は、細胞と結合し得、または結合し得ず、そして上述のように共通ホモロジー領域を共有する他のタイプII膜タンパク質との複合体であり得る。
【0057】
ポリペプチド複合体は、組換えLT−αを認識する多特異性抗血清によって認識され、この複合体はLT−αエピトープを示すことを示唆する。これらの抗血清は、市販の抗LT−αモノクローナル抗体(Boehring Mannheim)、およびトランスフェクトされたチャイニーズハムスター卵巣中(CHO)細胞で発現する組換えリンホトキシン−α(rLT−α)に対して生じる多特異性抗血清を包含する。これらの複合体を認識するポリクローナル抗血清はまた、混合リンパ球反応(MLR)をブロックするが、可溶性LT−αを認識するモノクローナル抗rLT−α抗体はMLRをブロックしない。従って、本発明の複合体は、T細胞活性化における重要な役割を行うようである。また、これらの複合体が可溶性LT−αまたはTNFの活性と同様にT細胞制御活性および細胞障害性を有することが予想される。
【0058】
本発明はまた、上記のアミノ酸配列を有するポリペプチドをコードするDNA配列を本質的に含有するDNA、このDNAによって特徴付けられる組換えDNA分子、本DNAでトランスフェクトされた培養物中の単細胞宿主または動物およびヒト細胞の群から選択される宿主、ならびにこのDNAおよびこれらの組換えDNA分子および宿主を使用しこれらによりコードされるポリペプチドを生産する組換え方法に関する。
【0059】
さらに詳しくは、本発明は、配列番号1によって定義されるヌクレオチド配列を含有する単離されたDNA配列に関する。
【0060】
本発明はさらに、上記配列、リンホトキシン−βと実質的に相同なポリペプチドをコードするDNAとハイブリダイズするDNA配列、およびリンホトキシン−βをコードするヌクレオチド配列を有する縮重DNA配列に関する。
【0061】
本発明はまた、可溶性リンホトキシン−βペプチドをコードするDNA配列に関する。これらのDNA配列は、配列番号3および配列番号5で定義される。本発明はまた、配列番号5のDNAの5’末端にいくつかの付加的なヌクレオチドトリプレットを有するDNAでコードされる数種の付加的な可溶性リンホトキシン−βペプチドに関する。付加的なヌクレオチドトリプレットは、配列番号7によって定義される52トリプレットを包含する。可溶性リンホトキシンペプチドはまた、配列番号5に3’末端から始まる1〜51ヌクレオチドトリプレットを含有する配列番号7の一部分が付加された配列にコードされ得る。
【0062】
本発明はまた、リンホトキシン−βまたは可溶性リンホトキシン−βペプチドを発現するようにコードする上記同定された任意の配列とハイブリダイズするDNA配列に関する。本発明はまた、リンホトキシン−βまたは可溶性リンホトキシン−βペプチドをコードする縮重ヌクレオチド配列、および可溶性リンホトキシン−βペプチドと実質的に相同なペプチドをコードするDNA配列に関する。
【0063】
リンホトキシン−βを同定し、単離しそして以下に記述する技術を使用して特徴付けた。
【0064】
(フローサイトフルオロメトリー分析)
まず、フローサイトフルオロメトリー分析を使用してT細胞の表面上のLT−αエピトープの発現を説明する。OKT3モノクローナル抗体で活性化されたヒト末梢血単核球細胞が、抗rLT−α抗血清と反応することによりLT−αエピトープの発現を示すことが判明した。抗rTNF抗血清ではなく、抗rLT−α抗血清のみが、OKT3刺激一次T細胞に結合した。
【0065】
また、ヒトT細胞ハイブリドーマ、II−23.D7[C.Wareら、「細胞障害性リンホトキシンを生産するヒトT細胞ハイブリドーマ:リンホトキシン放出の誘導および抗CD3モノクローナル抗体またはレクチンおよびホルボールエステルによるキラー細胞活性」、Lymph.Res.、5、313(1986)]が、PMA刺激によりLT−αを分泌すること、さらにまたPMA刺激により表面LT−α関連エピトープを発現することが観察された。また、PMA活性化II−23.D7細胞がウサギ抗rLT−α抗血清からLT中和抗体を除去できたが、一方コントロールU937細胞(表面LT−α形態全てが欠如している)は除去できなかったことが示された。さらに、ウサギ抗rLT−α抗血清がウサギLT−αと結合(複合体化)していた(生じた複合体が次にII−23.D7細胞上の細胞性LT−α/TNFレセプターと結合する)という可能性を排除したが、それは、細胞性レセプターを過剰の可溶性TNFまたはLT−αで飽和し、そしてこれが染色に関して影響を持たなかったことを観察したことによる。これらのアッセイの結果は、ハイブリドーマ上のLT−α関連エピトープが本当にLT−αと関係していることを示す。
【0066】
また、過剰のrLT−αによる抗血清の前処理は、抗血清がII−23.D7細胞を染色する能力を防ぐが、一方rTNFでの前処理は効果を持たなかったということが観察された。このアッセイは、LT−α関連エピトープに対する抗血清の特異性を示す。
【0067】
染色する前に刺激したII−23.D7細胞をトリプシン処理することにより、シグナルを失わせるが、これは、抗血清によって認識されるエピトープはタンパク質であったことを示した。
【0068】
また、CHO由来の混入物が活性化II−23.D7細胞の表面上の誘導タンパク質の抗血清による認識に寄与しなかったことが示されたが、それは可溶性LT−αのみを生産するLT−α遺伝子で安定にトランスフェクトされたCHO細胞が抗LT−α抗血清によって染色されなかったことによる。
【0069】
(免疫沈降)
さらに、これら表面LT−α関連タンパク質を、PMA活性化II−23.D7細胞の表面のヨウ素化(125I−標識化)または代謝標識化(35S−Metまたは35S−Cys)のいずれかにより標識し、続いてデタージェントで原形質膜を可溶化し、標識LT−α関連タンパク質の免疫沈降により特徴付けた。
【0070】
免疫沈降で結合した表面ヨウ素化によって、抗LT−α抗血清によって認識される2つのタンパク質:以降LT−αという25〜26kD形態、および以後、LT−βまたはp33という31〜35kD形態が明らかとなった。同じウサギからの免疫前抗血清も抗rTNF−ウサギ血清のいずれもヨウ素化されたPMA活性化II−23.D7細胞からのバンドを免疫沈降しなかったことを観察した。ヨウ素化し免疫沈降したバンドの一次元部分的CNBr処理ペプチドマップは、25〜26kD形態(LT−α)の切断によりヨウ素化した組換えLT−αのものと同一のパターンを示した。このことは、表面LT−αと可溶性LT−αとの相互関係を補強する。ヨウ素化された31〜35kD形態(LT−β、すなわちp33)はCNBrによって切断されず、公知のLT−α遺伝子生成物とは別であることが示された。
【0071】
さらに、PMA活性化II−23.D7細胞を35S−メチオニンまたは35S−システインで代謝標識し、LT−αおよびLT−βタンパク質を特徴付けた。システインおよびメチオニンの分布は、TNFとLT−αとの間、ならびに、シグナル配列を有する形態と有しない形態との間を区別するための方法が提供される。[M.Krieglerら、Cell、53、45(1988)]。分泌されたTNFはシステインを含有するが、メチオニンはなく、一方、分泌されたLT−αはメチオニンのみを含有し、システイン残基はない。しかし、LT−αはシグナル配列中に1つのシステイン残基を有するが、一方、TNFはシグナル配列中に2つのメチオニン残基を含有する。
【0072】
35S−メチオニンまたは35S−システインのいずれかでPMA処理II−23.D7細胞をそれぞれ培養して標識し、そして培地および細胞からの免疫反応性タンパク質を沈降した。次に、35S−メチオニンで標識した細胞の培地からの免疫沈降物をSDS−PAGE分析すると、LT−αの25kD形態が現れたが、一方35S−システインで標識した細胞の培地からの免疫沈降物では現れず、分泌LT−αに対して予測されるパターンではなかった。洗浄した細胞の分析では、25〜26kD LT−α形態および33kD LT−β形態の両方を示した。これらの結果は、表面ヨウ素化を用いて観察された膜結合形態と同じである。
【0073】
25〜26kD LT−αはシステインを欠如しており、これはリーディング配列のプロセッシングを示している。また、33kD LT−βは35S−メチオニンと35S−システインとの両方を取り込み、それ自身が25kD LT−α形態とは異なる形態であることが観察された。典型的に、そのレセプターと結合したLT−αは、BOSCOES(すなわち、ビス[2−[スクシンイミドオキシ−カルボニルオキシ]エチル]スルホン;Pierce、Rockford、IL)のような化学的架橋剤を用いてレセプターと架橋し得る。[J.S.Andrewsら、「ヒトTリンパ球上の腫瘍壊死因子(TNF)およびリンホトキシン(LT)に対するレセプターの特徴付け」、J.Immunol.、144、2582(1990)]。表面ヨウ素化II−23.D7細胞を架橋剤で処理した時、25〜26kD LT−αまたは33kD LT−β関連形態のいずれかが、他の付加的膜タンパク質と結合しなかったことが観察された。このアッセイは、レセプター結合はLT−αおよびLT−βが細胞膜と結合したままのメカニズムではないことを示した。LT−βの配列分析は、2つのTNFレセプター形態のいずれとも関係ないことを示した。[C.Smithら、Science、248、1019、(1990);T.J.Schallら、Cell、61、361(1990)]。
【0074】
(アフィニティークロマトグラフィー)
II−23.D7細胞の表面上のLT−βおよびLT−αタンパク質のさらなる特徴付けを、アフィニティークロマトグラフィーを用いて行った。PMA処理II−23.D7細胞の表面上のLT−α/LT−β複合体がレンチルレクチンと結合することが観察されたが、これは、それぞれの形態が糖タンパク質構造であることを示す。従って、精製工程としてレンチルレクチンクロマトグラフィー工程を、抗血清アフィニティークロマトグラフィーの前に使用した。デタージェントで可溶化したPMA処理II−23.D7タンパク質をレンチルレクチンセファロースに結合し、そしてα−メチルマンノシドで溶出した。コントロールIgGおよび抗LT−α−IgGアフィニティーカラムの両方を調製し、これらのタンパク質が抗LT−α抗血清によって特異的に認識されることを正確に評価した。次に、レンチルレクチンに結合したタンパク質をこれらのカラムに適用した。カラムを低pHで溶出したときに抗LT−αアフィニティーカラムからLT−βおよびLT−αタンパク質が放出されることを観察した。溶出液のSDS−PAGE分析結果は、表面ヨウ素化PMA処理II−23.D7細胞からの免疫沈降タンパク質のSDS−PAGE分析結果とかなり類似していた。この比較は、類似のタンパク質が2つの方法によって精製されたことを示した。
【0075】
アフィニティー精製の間、約25kD LT−α形態が切断されて19〜20kD形態、すなわち「デス(des)20」形態にとなるようであることを観察した。RPMI 1788腫瘍細胞株からの天然LT−αの最初の単離[B.Aggarwalら、「1788腫瘍細胞株由来のヒトリンホトキシンの一次構造」、J.Bio.Chem.、260、2334−2344頁(1985)]もまた、N−末端的に切断された20kD LT−α形態を与えていた。この「デス20」天然型LT−α中の1つのメチオニンが失われており、無傷の分子と比べると異なるCNBr切断パターンとなる。アフィニティー精製LT−αタンパク質の一次元CNBr切断は、短縮型の天然LT−α形態と一致する切断パターンを示し、そして、アフィニティー精製「デス20」LT−α形態はおそらく天然LT−α「デス20」で観測されたような類似の切断から生じたと結論した。
【0076】
さらに、LT−βが部分的CNBr切断でダブレットを生じることを観察した。LT−βタンパク質による切断パターンは、メチオニン残基が存在すること、および少なくとも1つのメチオニンがC−またはN−末端いずれかから5〜20残基の範囲内にある(このマッピング技術を使用して切断した時、いずれかの末端の1〜5残基の範囲内でメチオニン残基は検出されなかった)。このパターンは、LT−βは公知のLT−α配列の全部を含有していないことを示唆した。
【0077】
LT−βタンパク質はまた、抗原活性化一次細胞障害性Tリンパ球クローンによって発現されることが観察された。これらの細胞の代謝標識後の抗rLT−αを用いる免疫沈降の結果は、少量のLT−αと共にLT−βが現れた。これらの結果は、LT−βは一次T細胞およびII−23.D7ハイブリドーマによって作成され得ることを示した。
【0078】
(LT−βおよびLT−αタンパク質の初期精製)
以下の一般的な工程を使用してLT−αおよびLT−βタンパク質を精製した。まずホルボールミリスチンアセテート(PMA)をII−23.D7細胞に添加した。24時間後に、細胞を収集し、それらを無血清冷RPMI培地で洗浄した。冷たい細胞ペレットに、氷冷溶解緩衝液(HEPES、NP−40、EDTA、NaCl、およびアジ化ナトリウム)を加え、それにベンズアミジン、フェニルメチルスルホニルクロライド(PMSF)、ならびにN−エチルマレイミド(NEM)、ダイズトリプシンインヒビター、ペプスタチンおよびアプロチニンを新たに添加した。Dounceホモジナイザーで細胞を穏やかに均一化し、そして溶解液を遠心分離した。遠心分離し、そして上清を採取した。CaCl2およびMnCl2を添加した溶解緩衝液で予め平衡化したレンチルレクチンセファロースカラムに、上清をロードした。CaCl2およびMnCl2を有する溶解緩衝液でカラムを洗浄し、次にα−メチルマンノシドを含有する溶解緩衝液で溶出した。溶出画分をプールし、そしてウサギ非特異的IgGセファロースアフィニティーカラムの上にロードしたが、このカラムはウサギ抗rLT−αセファロースアフィニティーカラムに直接連結していた。EDTAを含有する同じ溶解緩衝液で、次にNP−40をMEGA−8(オクタノイル−N−メチルグルカミド、Boehringer−Mannheim)で置換した溶解緩衝液で両方のカラムを洗浄した。洗浄したカラムをそれぞれ別個に5mM MEGA−8、50mM グリシン、NaCl、ベンズアミジン、およびEDTA溶液で溶出したが、その方法は、Browningらの「リンホトキシンおよび関連33−kD糖タンパクは、活性化ヒトT細胞ハイブリドーマの表面上で発現される」、J.Immunol.147、1230−1237頁(1991)に記載された方法に従った。pHシフト後の第1回分をプールし、凍結乾燥し、SDSで水に再懸濁し、そしてHEPESおよびSDSの溶液に対して透析した。speed−vacで透析画分を乾燥し、そして水に再懸濁した。アリコートをLaemmliローディング緩衝液と混合し、そしてSDS−PAGE上で電気泳動した。銀染色でタンパク質を視覚化した。
【0079】
1つまたは複数のLT−αエピトープがPMA処理で生じるような細胞活性化によってのみ、II−23.D7T細胞ハイブリドーマの表面に存在することが観察された。反対に、一次T細胞上で存在する時、PMA処理により表面抗原が失われる。さらに、組換えLT−α(CHO細胞で生産される)あるいは天然LT−α(例えば、Genzyme、Boston、Mass.)のいずれかに対するウサギポリクローナル抗血清が1つまたは複数のLT−αエピトープを認識したことを見出した。
【0080】
また、LT−α/LT−β複合体を認識するこの抗血清はMLRをブロックするが、可溶性LT−αを認識する特定のモノクローナル抗体はブロックしないことを観察した[M.Shalabyら、J.Immunol.、141、499(1988)]。それゆえ、本発明のLT−α/LT−β複合体は、T細胞の活性化のメディエーターであり得る。
【0081】
細胞溶解物からの免疫沈降物中のLT−βがLT−αと共に存在することは、LT−βが免疫原性的にLT−αと関連するかあるいはLT−βがLT−αと結合されるか、または両方を示唆している。この問題を解決するために、35S−メチオニン標識細胞からの25kDおよび33kDバンドを、ウサギポリクローナル抗rLT−α血清で免疫沈降し、切り出したゲルスライスから溶出し、そして抗rLT−αポリクローナル抗血清または抗rLT−α mAbのいずれかで再免疫沈降した。LT−α(LT−βではなく)は、抗rLT−α抗体のいずれかで免疫沈降し、LT−βが免疫原性的にLT−αと関連しないことを示唆した。これらの観察は、LT−βが物理的にLT−αと結合していることを示した。
【0082】
表面LT−αおよびLT−βが複合体を形成するという仮説をさらに検証するために、変性および未変性条件下で等電点フォーカシング法(IEF)実験を行った。LT−αおよびLT−βが物理的に結合されれば、これらは未変性条件下では複合体としてフォーカスするはずであるが、変性条件下では分離した形でフォーカスするという理論的根拠に基づく。LT−αおよびLT−βに対する個々の等電点(pI)を、二次元ゲル分析(変性条件)によって決定した(図9A)。LT−αは、6.5〜7.3のpIの範囲で5つの荷電化したアイソマーを有するが、LT−βは5.5〜6.0のpIの範囲で4つの荷電化したアイソマーを有する。しかし、フォーカシング法を未変性条件下で行った時、LT−αおよびLT−βは共に6.3〜7.2のpIの範囲で広いバンド幅で集中した。それゆえ、LT−βの泳動は、未変性条件下で顕著に遅れた。
【0083】
(LT−αおよびLT−βのさらなる精製および同定)
LT−αおよびLT−βタンパク質を以下の一般的な工程を使用して精製した。ウシ胎児血清を含むRPMI培地でII−23.D7細胞を培養し、そして細胞を50lのRPMIから集め、そして培地に再懸濁し、そしてホルボールミリスチンアセテート(PMA)を添加した。6時間の活性化の後、遠心分離によって細胞を収集し、Dulbeccoのリン酸緩衝液生理食塩水で洗浄した。冷溶解緩衝液に最終細胞ペレットを懸濁し、そしてペレットをいったん窒素キャビテーターの中を通過させた。溶解した細胞を遠心分離し、そして上清を捨てた。一晩デタージェントと共に溶解緩衝液中でペレットを抽出し、次に再び遠心分離した。
【0084】
デタージェントで可溶化した膜を含有する上清を、Affi−gel(10)と結合したモノクローナル抗LT−αを含むアフィニティー樹脂に添加し、懸濁液を一晩振動した。樹脂を小さいカラム中に回収し、nonidet P40を含むHEPESで、次に1% w/v MEGA−8を含む同じ緩衝液でそれを洗浄し、MEGA−8を含むグリシン緩衝液で結合したタンパク質を溶出し、そしてその画分をトリス塩基で直ちに中和した。画分中にLT−βおよびLT−αの存在することをSDS−PAGE分析および銀染色法によって確認した。これらタンパク質を含有する画分をプールし、そしてSDSを添加し、そしてこのプールを0.l×laemmliサンプル緩衝液に対して透析した(MEGA−8デタージェントを除去するため何度も取り替えた)。透析した溶液を乾燥状態まで、凍結乾燥して、元の1/10量の水に再懸濁した。
【0085】
SDS−PAGEゲル上にサンプルを流し、ProBlotメンプラン上にプロットし、そしてクーマシーブルー染料で染色した。LT−βおよびLT−αバンドを切り出し、そしてタンパク質シーケンサーにロードした。エドマン分解によってN−末端配列を得た。膜に結合したLT−αバンドの配列は、分泌性のLT−αについて記載された配列と正確に一致することがわかった。すなわちLeu Pro Gly Val Gly Leu Thr Pro Ser(アミノ酸番号1〜9)[P.Grayら、Nature、312、121−124頁(1984)]と一致する。エドマン分解分析によって、結合LT−βタンパク質のN−末端部分のアミノ酸配列が2つのアミノ酸配列:Gly Leu Glu Gly Arg Gly Gln Arg Leu GlnまたはGly Leu Glu Gly Arg Leu Gln Arg Leu Glnを含む可能性があることを示した。引き続きより正確なcDNA技術を使用するDNA分析で、正しい配列はGly Leu Glu Gly Arg Gly Gly Arg Leu Glnであったことを確認した。
【0086】
LT−α形態が検出されたそれぞれの場合で、また、LT−βを検出し得た(すなわち、PMA活性化II−23.D7細胞、活性化CTLクローン、およびHut−78細胞の表面LT−α形態を構成的に発現する)。LT−αは表面LT−αの非存在下でトランスフェクトされたCHO細胞から分泌され、そしてLT−βの存在が表面結合LT−αと関連するので、LT−βはLT−αと複合体を形成し細胞表面でそれを標的化すると結論付けた。生化学的には、LT−βおよびLT−αは非変性等電点フォーカシングゲル上で共に泳動するが、複合体が尿素で解離すると、2つのタンパク質が分離して流れる。[図9A、10A参照のこと]。これらの観察は、LT−αおよびLT−βは細胞表面上で複合体として存在するという結論に導いた。
【0087】
(リンホトキシン−βおよび可溶性リンホトシン−βペプチドに対してコードするDNA配列の同定)
リンホトキシン−βを、上述したように免疫アフィニティークロマトグラフィーにより精製した。直接N−末端配列決定およびインサイチュトリプシン分解に続いて、分解ペプチドの逆相HPLC分離を行った。[Abersoldら、「ニトロセルロース上のインサイチュプロテアーゼ分解後の一次元または二次元ゲル電気泳動法によって分離されるタンパク質の内部アミノ酸配列分析」、PNAS、84、6970−6974(1987)参照のこと]。次に得られたN−末端および内部トリプシンフラグメントペプチドの配列を通常の方法を使用して決定した。N−末端およびT105、T87/88、T110およびT67と命名した内部ペプチドの配列決定を図13に示す。
【0088】
2つのアンチセンス17−merオリゴヌクレオチドプローブGTYTCNGGCTCYTCYTC[配列番号9]およびGTYTCNGGTTCYTCYTC[配列番号10](それぞれ1368および1369と命名した)を、ペプチドT−87/T−88の配列の一部分と一致するように合成し、そして32Pで放射能標識した。ノーザン分析の結果、1368と命名されたプローブが、0.9〜1.1kb mRNAバンドに強くハイブリダイズしたことを示した。このmRNAは、前述したようにホルボールエステルで前処理したII−23.D7細胞中で強く誘導された。
【0089】
ベクターpCDM8中のcDNAライブラリーを、PMAで6時間誘導したII−23.D7細胞から単離されたポリA+mRNAから構築した。ライブラリーを1368と命名された標識オリゴマーでスクリーニングし、そしてポジティブクローンを単離し、次に50℃で3M テトラメチルアンモニウムクロライドで洗浄した。0.8〜0.9kD挿入物を有するいくつかの(>16)クローンを、DNA配列分析にかけた。
【0090】
クローンpCDM8/LT−β−12は、配列番号1に示されるリンホトキシン−βをコードする配列を含んでいた。他のクローンは、5’末端で種々の短縮形であることを除いて、同一であった。クローン12 cDNAは、機能的なリンホトキシン−βをコードする。標準プライマー伸長法を用いて、アミノ酸残基−−MET GLY ALA−−をコードする付加的なコドンを同定した。862〜867位の終止コドン AATAAAは、3’ポリA領域のちょうど前に、あり、これは全3’末端領域が同定されたことを示す。タンパク質コード配列は、240アミノ酸をコードし非修飾分子量で約25,390kDaと計算される。
【0091】
クローン12 cDNAおよびプライマー伸長から同定されたLT−β DNA配列の5’末端、ATGGGGGCACTGGGGCTG[配列番号11]から、3つの開始コドン(下線)の可能性があることが明らかとなった。[例えば、M.Kozak、「脊椎動物mRNA配列の分析:翻訳調節の暗示」、J.Cell.Biol.、115、4、887−903(1991)]。設計されたLT−βポリペプチドおよびDNA配列は、この5’配列の全てまたは一部分を切断することならびに1つの開始コドンを置換することで誘導し得る。
【0092】
このアミノ酸配列プロフィルは、タイプII膜タンパク質に典型的である。短い(最大17)アミノ酸配列N−末端「細胞質」ドメインに続いて、膜固定ドメインとしておそらく働く30の疎水性アミノ酸が拡がる範囲がある。入手可能なデータベースでは同一の配列が発見されなかった。細胞外ドメイン中に1つのシステイン残基があり、そして最後のC−末端17アミノ酸中に2つのメチオニンがある。これは、このタンパク質で示される非常に制限された臭化シアン切断パターンと一致する。
【0093】
LT−βの配列を決定した後の比較では、LT−βは、タイプII膜タンパク質であり、TNF、LT−αおよびCD40タンパク質に対して有意に相同性を有することが明らかになった。これらのポリペプチドは、細胞外ドメイン中に4つの配列保存領域を共有している。図14参照のこと。このような保存領域は、これらのポリペプチドがお互いに複合体を形成するのを可能にするようである。[以下を参照のこと;例えば、M.EckおよびS.Sprang、「2.6Å分解能での腫瘍壊死因子−αの構造、レセプター結合との関係」、J.Bioloical Chemistry、264、29、17595−17605頁(1989);E.Jonesら、「腫瘍壊死因子の構造」、Nature、338、225−228頁(1989);M.Eckら、「1.9Å分解能でのヒトリンホトキシン(腫瘍壊死因子−β)の構造」、J.Bioloical Chemistry、267、4、2119−2112頁(1992);J.Tavernierら、「腫瘍壊死因子およびリンホトキシンの保存残基は、トリマー構造の骨組みを構成する」、Fed.Eur.Biochem.Soc.Lett.、257、2、(1989)]。
【0094】
(クローン化LT−βの発現)
pCDM8/LT−βクローン12*(*プラスミドpCDM/LT−βクローン12を有するE.coli K12は、BN1289(MC1061/P3/P33−クローン−12)と命名され、1992年11月13日にATCCに寄託された。)またはコントロールプラスミド、クローン4(非機能性LT−β cDNA挿入断片を有するpCDM8)を、エレクトロポレーションによって、安定にヒトLT−αでトランスフェクトしたCHO dhfr−細胞およびCHO細胞中に導入した。3日後、細胞を5mM EDTAを含むCa/Mgを含まないHank溶液に取り出し、そして上述したような10μg/mlコントロールIgG1または抗LT−αモノクローナル抗体(Boehringer−Mannheim)のいずれかを使用したFACS分析のために染色し、その後、FITCまたはフィコエリトリン標識ヤギ抗マウス調製物のいずれかで、結合免疫グロブリンを標識した。
【0095】
他の実験において、COS細胞を、pCDM8ベクター中でもまた等量のヒトLT−αcDNAの存在または非存在下で、pCDM8中のクローン4またはクローン12のLT−βcDNAのいずれかでエレクトポレートし、そして上記のように3日後FACS分析のために染色した。LT−αを発現するCHO細胞のみが、機能性LT−β DNA(すなわち、クローン12)でのトランスフェクションにおいて表面リンホトキシンを示した。
【0096】
クローン12は開始ATGコドンを欠損しているが、いくつかのCTG開始コドンを有しており、それゆえこの発現実験は1つまたはいくつかの5’CTGコドンが翻訳を開始しなければならないことを示す。CTGコドンは、いくつかの真核タンパク質の翻訳に対する開始部位となることは公知である。[M.Kozak、J.Cell.Biol、115、4、887−903(1991)]。LT−αおよびLT−β DNAの両方を受け入れるCOS細胞のみが、FACS分析中の実質的な表面LT−αを示すような同様の結果を、COS細胞中の2つのトランスフェクション系を使用して観測した。
【0097】
(LT−βおよびLT−αならびにLT−α/LT−βのポテンシャル使用)
上記のように、LT−β、LT−α、TNFおよびCD40リガンドの間にかなりの構造的類似性がある。LT−β、LT−α、TNFおよびCD40リガンドはタイプII膜タンパク質であり、そして細胞外ドメイン中に少なくとも4つの配列保存領域を共有している。
【0098】
構造的類似性の観点において、LT−αが、分泌される分子と同一の形態で活性化リンパ球の表面で見られるが、LT−αを表面におそらく固定する他の不可欠な膜タンパク質と複合されるという問題がある。発明者らは、この独特の複合体(すでにLT−α/LT−βであると決定された)が、LT−αのさらに関連している形態を示し、TNFと関連する特異性を与えると考えている。
【0099】
リンホカインのヘテロメトリックの複合体の存在は、免疫系に独特であるが、他の領域中のシグナル分子を暗示する(例えば、PDGFおよびインヒビン/アクチビンヘテロメトリック複合体)。LT−α/LT−β複合体の記述は、LT−αホモトリマーによって模倣できない複合体独特の免疫調節活性の可能性を提供する。複合体は、高い親和性相互作用および生化学的に関連したシグナルを導く独特のレセプターまたはレセプター鎖組合せに結合し得る。独特のレセプター複合体とのLTα−/LT−β相互作用の仮説は、多くの系でのTNF(2つの公知TNFレセプターの研究によって説明できない観測)と比較してLT−αホモトリマーの相対的に弱い活性を説明し得た。[T.Schallら、Cell、61、361−370(1990);C.Smithら、Science、248、1019(1990)]。
【0100】
LT−βとの複合体化を経て可溶性LT−αを細胞表面につなぐことは、LT−α/LT−βを介する細胞−細胞接触特異的シグナルが、免疫調節の重要な面であり得ることを示す。TNFおよび関連したLT−α形態は分泌されるので、LT−βもまた分泌され得ると考えられる。これは、抗LT−βモノクローナル抗体を使用する研究で実証し得る。このような抗体はまた、LT−βホモ−オリゴマーが自然に生じるかどうか決定するために使用され得る。
【0101】
一般的に、LT一およびTNFは、定性的に同じ活性スペクトルを示し、そしてLT−αおよびTNFは同じセットのレセプター(55および80kD TNFレセプターと呼ぶ)と相互作用すると考えられる。[C.Smithら、Science、248、1019(1990);T.Schallら、Cell、61、361(1990)]。それでもなお、LT−αおよびTNFによって示される生物学的能力の定量的パターンは劇的に異なり、LT−αはしばしばTNFよりさらに能力が少ない[例えば、Browningら、J.Immunol.、143、1859(1989)を参照のこと]。これらの観測は、存在するレセプター結合データと一致させることが難しい。LT−α/LT−β複合体は、他の現在まで不確定のレセプターと相互作用するような、独特の特性をLT−αに与える可能性がある。この場合、LT−α/LT−β複合体および本発明の他の複合体は、LT−αまたはTNFのいずれかと区別する独特の生物学的特性を有する。LT−α/LT−β複合体は、例えばLT−α/LT−βまたはLT−β特異的レセプターを同定し、そしてクローニングするために使用され得る。さらに、さらなる複合体の使用は、新規な生物学的活性を明らかにし得る。
【0102】
また、多数のT細胞およびマクロファージ細胞株がHIVウイルスによって感染可能であることは公知であるが、実際には、少数の細胞様が組織培養中のウイルスを増殖するのに有用である。例えば、H9株(Galloらによって最初に開発されたHut−78の誘導体[M.Popovicら、Science、224、497−500(1984)])、および他のヒトリンパ球株(C8166)は、HIV増殖に対して有用であった。[M.SomasundaranおよびH.Robinson、Science、242、1554−1557(1988)]。表面LT−α発現または表面LT−αの発現の能力は、所定の細胞をHIV増殖に対して良い標識にし得る。
【0103】
HIV増殖を高めることにおけるTNFの役割が提案された[L.Osbornら、Proc、Natl.Acad.Sci.USA、86、2336(1989);Z.RosenbergおよびA.Fauci、Immunol.Today、11、176(1990);C.Locardiら、J.Virology、64、5874(1990);G.Oliら、Proc.Natl.Acad、Sci.USA、87、782(1990)]。発明者らは、II−23.D7株がHIV株IIIBで感染可能であるが、PMA処理でこのウイルスによる感染が劇的に増加することを発見した。Hut−78細胞株が、表面LT形態を構成的に発現することが見出され、そしてC8166株は、その後のPMA処理で明らかとなる表面LT中でII−23.D7と似ている。[Wareら、J.Immunol.、印刷中(1992)]。
【0104】
これらのHIVによるII−23.D7の感染可能性の結果、および感染可能な細胞株と表面LT発現との関係を考慮すると、これらの株がHIV感染および複製に対し良い宿主であり得ることを提案する。なぜなら、LT−α/LT−β複合体、および本発明の他のポリペプチドおよび複合体が調節の役割を果たすからである。LT−α遺伝子がHIV転写活性化因子TATの発現によって誘導されることが証明され[K.Sastryら、J.Biol.Chem.、265、20091(1990)]、そして、さらに、HTLV−1感染もまたLT−α発現を誘導することが示された[N.Paulら、J.Virol.、64、5412(1990);E.Tschachlerら、Human Retrovirology(Raven Press 1990)、W.Blattner編、105頁]。従って、HIV感染および続いて起こるLT−α/LT−β複合体または他の複合体の発現によるLT−αの誘導、またはこれらのタンパク質を作る能力がある細胞株でのPMA処理によるLT−α/LT−βまたは他の複合体の発現の誘導は、ウイルス複製を高めるために用い得る。この理由のために、LT−α/LT−β複合体もしくは本発明の他のポリペプチド複合体、またはこれらの複合体の可溶性形態、またはLT−βおよび本発明の他のポリペプチドに対する抗体または特異的結合タンパク質(例えば、可溶性レセプター)は、HIV増殖を阻害し得、またはHIV誘導T細胞死をブロックし得る。
【0105】
LT−α/LT−βとCD40レセプターリガンドペアとの間の類似点が示され得、ここでT細胞表面CD40リガンドからのシグナルは、CD40レセプターを介してB細胞へ「ヘルプ」を与える。従って、表面LT−α/LT−βが、T細胞または他の造血系(LAKまたはNK細胞およびB−細胞など)のT細胞調節の成分であり得ると仮定し得る。さらに、この相互作用は、いくつかの自己免疫疾患中の機能不全であり得る。[例えば、R.Watanabe−Fukunaga、Nature、356、314−317頁(1992)を参照のこと]。
【0106】
さらに、Fas抗原と呼ばれる細胞表面タンパク質は、TNF、NGF、およびCD40タンパク質を含む多数の細胞表面レセプターとかなりの構造的相同性を有することを示した。Fas抗原は、アポトーシスを仲介することに関係しており、プロセスもまたプログラム細胞死として関連している[R.Watanabe−Fukunaga、Nature、356、314−317頁(1992);N.Itohら、Cell、66、233−243頁(1991)]。Fas抗原の欠損を説明するマウスの株は、全身性エリテマトーデス様自己免疫疾患を発生する。これは、構造的に類似しているLT−βまたはLT−α/LT−β複合体もまた、全身性エリテマトーデスを仲介する役割を行い得、それゆえ、この経路中の介入は、種々の自己免疫疾患を治療することに臨床的に有用であり得る。あるいは、LT−βまたはLT−α/LT−β複合体は、細胞−細胞接触依存機構を介するプログラム細胞死を誘導することに関連し得る。すでに大規模なTNF/NGFタイプレセプターのファミリーを補充するためのTNF関連リガンドのこのファミリーの出現は、免疫系中での重要な調節因子の追加配列の存在を示唆する。
【0107】
LT−βまたはLT−β/LT−α複合体は、免疫系を抑制する役割を同様に行い得、そしてアレルギーを治療し、耐性を誘発することに潜在的に有用であり得る。
【0108】
ゲノムのMHC領域のTNF/LT遺伝子座の位置は、研究者らに種々の自己免疫疾患、特にインスリン依存性真性糖尿病に対する関連を調査させた。[例えば、F.Pociotら、「モノカイン分泌とインスリン依存性真性糖尿病との関係における腫瘍壊死因子β遺伝子多形性」、Scand.J.Immunol.、33、37−49(1991);K.Badenhoopら、「(インスリン依存性)真性糖尿病タイプ1におけるTNF−α遺伝子多形性」、Diabetologia、32、445−448(1989)を参照のこと]。発明者らは、LT−β遺伝子がTNF/LT遺伝子座の次に位置することを発見したので、LT−β遺伝子またはそのレセプターがこの自己免疫条件に関連する可能性がある。従って、LT−βまたはそのレセプターまたはLT−βに対する抗体は、糖尿病のこの形態における置換療法を包含する。
【0109】
上述したように、LT−βポリペプチド、および本発明のポリペプチド複合体は、抗腫瘍、T細胞活性化、またはT細胞抑制適用を含む多数の可能性のある使用、全身性エリテマトーデスの治療を含む適用、および抗炎症性の組成物での使用および方法を有することが期待される。次に、LT−βポリペプチドをコードするDNA配列、同様なDNA配列を含む組換えDNA分子、ならびに同様の組換えDNA分子でトランスフェクトした培養物中の単細胞宿主および動物またはヒト細胞を用いて、組成物および上記の療法で使用するために、実質的に他のヒトタンパク質を含まない大量の本発明のポリペプチドを生成し得る。
【0110】
本発明のポリペプチド複合体の表面上で発現するリンパ球、および好ましくはLT−α/LT−β複合体は、腫瘍細胞を殺す能力を高め得たリンパ球のサブセットを示す。同様に、このサブセットは、LAK(リンホトキシン活性化キラー)細胞またはTIL(腫瘍浸潤リンパ球)細胞療法に有用であり得る。[H.Thomas、K.Sikora、「癌療法への生物学的アプローチ」、Jour.Int.Med.Res.、17、191(1989)]。TIL免疫療法は、遺伝子転移技法によって改善され得る。LT−βについての組換え遺伝子およびそれに基づく関連のポリペプチドは、例えばTIL療法で、治療上有用であり、ここでLT−β遺伝子は(LT−α遺伝子と共にまたはLT−α遺伝子なしのいずれかで)、腫瘍から単離したT細胞に導入し、そして患者に導入され得る。さらに好ましくは、細胞を患者から取り出し、本発明のポリペプチドを発現するようにコードするDNA配列でトランスフェクトし、トランスフェクションの前または後でリンホカイン、好ましくはIL−2でインキュベートし、そして患者に戻す。トランスフェクトしたT細胞(すでにLT−βを発現し、そしてまたその後LT−αと複合化している)は、取り出された腫瘍に戻り、そこでLT−αの殺腫瘍性作用を直接腫瘍に伝える。同様に、LAK細胞中に導入されるLT−β遺伝子が細胞上の表面複合体の数を増加させ、そしてそれらの活性を高めることが考えられる。あるいは、患者腫瘍細胞中へのLT−β遺伝子の導入は、LT−β改変腫瘍が、腫瘍自体に対する高められた免疫応答を誘発する腫瘍ワクチンを造り出すことに、有用であり得る。[例えば、W.F.Anderson、Science、256、808−813(1992)を参照のこと]。
【0111】
本発明のポリペプチドおよびポリペプチド複合体に対する抗体または抗体誘導体もまた、この細胞集団を富化するための通常の免疫学的方法(例えば、パンニングまたはフローサイトフルオロメトリー選別)に有用である。[L.J.WysockiおよびV.L.Sato、「リンパ球についてのパンニング:細胞選別の方法」、PNAS、75、2844(1978)]。
【0112】
本発明のポリペプチドおよびポリペプチド複合体、またはフラグメントまたはその誘導体は、リンホトキシン−αおよび腫瘍壊死因子が使用されるのと同様に、細胞調節または治療の適用に有用である。
【0113】
本発明の組成物は、特定の臨床状態に対して治療するために、有効な用量で投与され得る。所定の適用についての特定の用量の決定は、例えば、患者の状態および体重、所望の治療の程度および治療に対する患者の耐性を、当該技術分野では十分考慮に入れる。本発明の複合体およびポリペプチド、またはおそらくそれに由来するペプチド、またはそれからもしくはそれらのアミノ酸配列を使用して合成されるペプチド(ポリペプチドの単離および精製形態を包含する)、またはそれらの塩または薬学的に受容可能な誘導体の投与は、抗腫瘍、T細胞活性化、T細胞抑制または抗炎症活性を示す薬剤の任意の従来受容された態様を介するものであり得る。
【0114】
これらの療法で使用される組成物もまた、種々の形態であり得る。これらは、例えば、錠剤、丸剤、散剤、液体溶液または懸濁液、注入可能または浸出可能な溶液のような、固体、半固体、および液体の投与形態、および遺伝子療法を包含し得る。好ましい形態は、投与の意図した形式および療法の適用に依存する。投与の態様は、経口、非経口、皮下、静脈内、病変内、または局所投与を包含し得る。組成物はまた、好ましくは、通常の薬学的に受容可能なキャリアを包含し、そして他の医薬剤、キャリア、遺伝子キャリア、アジュバント、賦形剤など、例えば、ヒト血清アルブミンまたは血漿調製物を包含し得る。好ましくは、組成物は1単位用量の形態であり、そして通常1日1回またはそれ以上を投与される。
【0115】
以下は、本発明のLT−βおよびLT−α/LT−β複合体、およびこれらを特徴付けるために使用される方法を説明する実施例である。これらの実施例は限定して解釈されるべきものではなく:実施例は例証の目的のために含まれ、そして本発明は請求の範囲によってのみ限定される。
【実施例】
【0116】
実施例において以下の実験手順を使用した:
(抗血清)
組換えヒトLT−α(rLT−α)を、無血清条件培地中で安定にトランスフェクトしたチャイニーズハムスター卵巣(CHO)細胞株によって、発現しそして分泌した。無血清条件培地から、一連のセファロースS、レンズマメレクチンセファロースおよびFPLC MonoQカラムクロマトグラフィー工程によって、分泌rLT−αを精製した。CHO細胞由来のrLT−α調製物の特性は記載されている。[J.Browningら、J.Immunol.、143、1859(1989)]。リンパ腺手順[M.Sigelら、「リンパ腺中への接種による抗体の産生」、Met.Enz.、93、3(1983)]によって、完全フロイントアジュバンド中25μgの天然rLT−αで2匹のウサギ(4および5)を免疫した。3番目のウサギ(6)を、完全フロイントァジュバンド中25μgの変性rLT−αで同じ経路により免疫した。変性rLT−αをSDS−PAGEに続いて0.1%SDS−カーボネート緩衝液中で電気溶出によって調製した。
【0117】
上記の方法を使用して、3つの抗rLT−α抗血清を、2つを天然rLT−αに対して、3番目をSDS変性rLT−αに対して生成した。天然タンパク質(ウサギ4および5)での免疫によって生じた抗血清は、1:2000〜5000の希釈で50ユニット/ml溶液を中和し得た。変性rLT−α(ウサギ6)に対して生じた血清は、中和力価がないが、ウェスタンブロットではrLT−αと弱く反応した。抗血清は、r−ヒトTNFを中和し得ず、またウサギ6由来の抗血清中で非常に弱い力価であることを除いて、ELISAプレートに結合したr−ヒトTNFを認識し得なかった。ウサギ6由来の抗血清のみがウエスタン分析でrLTを認識可能であった。
【0118】
組換えヒトTNFで4番目のウサギを免疫した。完全フロイントアジュバント中で、続いて不完全フロイントアジュバント中のブーストにより、組換えヒトTNF(E.coli由来[D.Weirら、Handbook Of Experimental Immunology In Four Volumes、8章「実験動物の免疫」])を使用する古典的免疫スキームを経て、ポリクローナル抗rTNFウサギ血清を調製した。免疫によりrTNFに対して生じた血清は、良好な中和力価を有した。TNFに対する中和モノクローナル抗体が記述されている[Liangら、「組換えヒト腫瘍壊死因子/カケクチンに対するモノクローナル抗体の産生および特徴付け」、Biochem.Biophys.Res.Comm.、137、847(1986)]。予め免疫した血清を、コントロールとしての使用のために全ての動物から収集した。
【0119】
(細胞増殖およびT細胞活性化)
全ての細胞を、上述したLT−αトランスフェクトしたチャイニーズハムスター卵巣(CHO)細胞株を除いて、アメリカンタイプカルチャーコレクション(ATCC)から入手した[Browning、J.Immunol.、143、1859−1867頁(1989)]。
【0120】
細胞を、1%グルタミン、10mM HEPES緩衝液、pH7.5、ペニシリン/ストレプトマイシン、および10%ウシ胎児血清(Hyclone−defined)を補足したRPMI 1640(「完全RPMI」と呼ぶ)中で増殖した。ただし、上記のように補足したDulbecco改変Eagle培地中で増殖したトランスフェクトされたCHO細胞は除く。ヒトT細胞ハイブリドーマ、II−23は、活性化末梢リンパ球とヒトCEM腫瘍株との融合の結果であり、そしてさらにサブクローニングした(II−23.D7)[C.Wareら、「細胞障害性リンホカインを生産するヒトT細胞ハイブリドーマ:抗CD3モノクローナル抗体またはレクチンおよびホルボールエステルによるリンホトキシン放出およびキラー細胞活性の誘導」、Lymph.Res.、5、313(1986)]。ヒト末梢血リンパ球(PBL)をヘパリン添加ガラスチューブ中に入れ、Ficoll−Hypaque遠心分離によって単離し、洗浄し、そして完全RPMI培地で再懸濁した。1μg/ml インドメタシン存在下および、いくつかの実施例では10ng/ml rIL−2(Biogen,Inc.、Cambridge、MA)を用いて、OKT3条件培地の1:500希釈(最終的に約2ng/ml)を用いて、2×106細胞/mlで、PBLを処理した。ヒトCTL−クローンを、記述したように生成し[L.Greenら、「クローン化ヒト細胞障害性Tリンパ球によって生産される細胞障害性リンホカイン」、J、Immunol、135、4034(1985)]、そして照射刺激細胞(抗原)またはE.Reinherzによって提供された抗CD2モノクローナル抗体(Tl12+Tl13)の組合せのいずれかで活性化した。
【0121】
(フローサイトメトリー)
10%ウシ胎児血清(FBS)、0.1% アジ化ナトリウム、および0.1mg/ml ヒトIgGを含むRPMI 1640培地中で、0℃で、細胞を再懸濁した。ヒトIgGでのプレインキュベーションに続いて、所望の抗血清を含有する追加培地を添加した。代表的には、細胞を1:200の抗rLT−αおよび抗rTNF血清の最終希釈で60〜90分間インキュベートした。Dulbeccoのリン酸緩衝液生理食塩水(PBS)で細胞を2回洗浄し、次に、上記の培地中でフルオレセイン標識ヤギ抗ウサギIgG(Cappel Durham、N.C.)の1:500希釈で最低60分間これらをインキュベートした。次に細胞を1回洗浄し、そして直接に分析するか、またはいくつかの場合では0.5%パラホルムアルデヒドを用いて0℃にて10分間の固定後に分析するかのいずれかを行った。2番目の抗体段階でのフィコエリスリン標識leu−4、leu−2、leu−M3、またはleu−16もしくはleu−19(Becton−Dickinson、Mountain View、CA)を添加することを除いて、上記のように2つの色分析を行った。表面結合LT−αとIL−2レセプターレベルとの比較を、フルオレセイン標識抗IL−2レセプター(CD25)抗体(Becton−Dickinson、Mountain View、Ca.)を用いる分離単色分析で行った。分析を、FACStar機器(Becton−Dickinson)で行った。
【0122】
(活性化II−23.D7細胞による中和抗rLT−α抗体の吸着)
完全RPMI培地中で、1×106細胞/mlで8時間、10ng/mlのPMAと共に、II−23.D7およびU93前単球細胞を刺激した。3回培地で細胞(1×108個)を洗浄し、そして上清を吸引して、乾燥ペレットを得た。次に、細胞を、抗rLT−α血清(ウサギ4由来)の1:1000希釈を含有する1mlの培地で再懸濁し、そして混合しながら1.5時間氷上でインキュベートした。細胞を遠心分離によって抗血清から取り除いた。吸着した抗血清(免疫前および免疫後の両方)を、15U/mlのrLT−αを含有する等量(50μl)の培地と混合し、そして20分間室温でインキュベートした。この混合物を培地中に連続的に希釈し、そしてL929細胞(0.1ml中)に添加し、そしてさらに24時間インキュベートした。記載されたようなMTTアッセイ[L.Greenら、「細胞生存度に対する迅速な比色アッセイ:細胞障害性および増殖阻害リンホカインの定量への適用」、Jour.Immunol.Meth.、70、257−268(1984)]によって、細胞生存度を評価した。
【0123】
(T細胞の35S−メチオニンまたは35S−システインの代謝標識)
ペニシリン/ストレプトマイシン、グルタミン、10mM HEPES pH7.5、10% v/v 透析FBS、および2% v/v 通常のRPMI(冷キャリア添加)を補足したシステインを含まないまたはメチオニンを含まないRPMI 1640中に、細胞を移した。細胞濃度を2〜3×105細胞/mlに調節し、そして35S−メチオニンまたは35S−システインを100〜200μCi/mlのレベルにした適切な培地に添加した。新たに活性化したPBLの場合は、上清を穏やかに除去し、そして細胞を遠心分離し、標識培地中で再懸濁し、そして元の付着集団に戻して添加した。12〜18時間の標識期間の後、以下に記述するように細胞を洗浄し、そして溶解した。PBLを用いて、細胞をピペットにより除去し、そして付着集団をPBS中で5mMEDTAで処理することで部分的に除去した。
【0124】
(免疫沈降)
標識した細胞溶解物の0.2〜0.5mlに2〜4μlのウサギ血清を添加した。サンプルを1〜2時間、4℃で放置した。次に、洗浄したプロテインAセファロース(Pharmacia,Piscataway,N.J.)の60〜75%懸濁液の60μlアリコートを添加し、そして6〜18時間4℃でサンプルを振とうした。プロテインAセファロースペレットをカルシウム/マグネシウムを含まないPBS中 1%NP−40で3回洗浄し、それらを50μlのLaemmli SDSローディング緩衝液に再懸濁した。代表的には、1つの溶解物のサンプルを、連続した免疫沈降(免疫前抗rLT−α血清、抗rTNF抗血清、および最後に免疫後抗rLT−α抗血清)を介して循環した。1つのセットの実験で、5mM CaCl2およびMnCl2を溶解物に添加し、そして洗浄したレンズマメレクチン−セファロースの75%懸濁液の75μlと共に溶解物を一晩振とうした。このセファロースをNP−40/PBSで2回洗浄し、次に0.25M α−メチルマンノシドを含む75μlの1% NP−40/PBSの3回の連続的な添加により溶出した。プールした洗浄液を、免疫沈降プロトコールに用いた。
【0125】
(ウサギ抗rLTアフィニティーカラム)
プロテインAセファロースを用いて酸性pH溶出で抗rLT血清(ウサギ4由来)から免疫グロブリン画分を精製した。溶出したIgG含有面分をPBSで透析し、そしてアミコン濾過により濃縮した。抗rLT−α−IgG溶液(6mg/mlを15ml)を、使用説明書に従い8mlのAffi−gel 10樹脂(Biorad,Richmond,Ca.)に結合した。非特異的ウサギIgG(Cappel,Durham,N.C.)を用いて同一のアフィニティーカラムを調製した。両方のカラムを、PBS、1% NP40を含む1M 酢酸 pH3.0、そして最後にプロテアーゼインヒビターを含まない溶解緩衝液で洗浄した。
【0126】
(LT−βおよびLT−αの最初の精製)
II−2S.D7細胞(15L)を5×105細胞/mlの濃度になるまで増殖し、ホルボールミリスチンアセテート(PMA)を添加し、最終濃度25ng/mlにした。24時間後、細胞を採取し、そしてそれを冷無血清RPMI培地中で洗浄した。7×109細胞を含む凍結した細胞ペレットに、100mlの氷冷溶解緩衝液(50mM HEPES pH7.5、1% v/v NP−40、2mM EDTA、0.15M NaCl、および0.1% アジ化ナトリウム)を添加した。その溶解緩衝液には、5mM ベンズアミジン、1mM フェニルメチルスルホニルクロライド(PMSF)、および0.25mM N−エチルマレイミド(NEM)、10μg/ml大豆トリプシンインヒビター、0.7μg/ml ペプスタチンおよび10μg/ml アプロチニンが新たに添加されている。dounceホモジナイザーで細胞を穏やかにホモジナイズし、そして細胞溶解物を10,000×gで10分間遠心分離した。上清を60,000×gで90分間遠心分離し、そして上清を収集した。
【0127】
高速遠心分離からの上清に、5mM CaCl2および5mM MnCl2を添加した。次に、この上清を、CaCl2およびMnCl2を加えた溶解緩衝液で平衡化した20mlのレンズマメレクチンセファロースカラム(Pharmacia,Piscataway,N.J.)にロードした。溶解緩衝液(CaCl2およびMnCl2を含む)でカラムを洗浄し、そして次に0.25M α−メチルマンノシドを含有する溶解緩衝液でカラムを溶出した。
【0128】
レンズマメレクチン溶出画分を50mlの体積までプールし、そしてそれを2mlのウサギ非特異的IgGセファロースアフィニティーカラムに直接ロードした。このカラムを2mlのウサギ抗rLT−αセファロースアフィニティーカラムに直接連結した。両方のカラムをEDTAを含む同じ溶解緩衝液で洗浄し、次に1% NP−40が1% W/V
MEGA−8(オクタノイル−N−メチルグルカミド,Boehringer一Mannheim,Indianapolis,IN.)に置換されている溶解緩衝液で洗浄した。
【0129】
洗浄したカラムを、各々、1% MEGA−8、50mM グリシンpH2.5、0.05M NaCl、5mMベンズアミジン、および2mM EDTAで溶出した。pHが変わった後の最初の20mlをプールし、このプールを凍結乾燥し、0.05% SDSを含む水1mlに再懸濁し、そして10mM HEPES pH7.5、0.05% SDS、および0.1% MEGA−8で透析した。透析した画分をスピード真空(speed−vac)で乾燥し、そして0.15mlの水に再懸濁した。アリコートとLaemmliローディング緩衝液とを混合し、SDS−PAGEで電気泳動した。LT−βおよびLT−αタンパク質を銀染色により視覚化した。
【0130】
(II−23.D7細胞表面のヨウ素化)
コントロールまたはPMA誘導II−23.D7細胞のいずれかを、カルシウム/マグネシウムを含まないPBSで大量に洗浄し、1mM PMSFおよび0.25mM NEMで処理し、そして次に2回洗浄した。50μgのヨードジェン(iodogen)(Pierce)でコートした12×75mmガラスチューブに、0.3mlの細胞(1×107全量)および1〜2mCiの125ヨウ化ナトリウムを添加した。細胞を25分間、室温で定期的に撹拝しながら放置し、10%のFBSを含むPBSで3回洗浄し、そして上記の溶解緩衝液に再懸濁した。Eppendorf遠沈管での2分間の遠心分離により核を除去した。次に、この上清をさらに15分間遠心分離した。透明にした上清を免疫沈降のプロトコールに用いた。
【0131】
(一次元CNBrペプチドマッピング)
サンプルを、12%アクリルアミドSDS−PAGE Laemmliシステムゲル上にて短距離で電気泳動し、適切なゲル断片を切り出した。ゲルの薄片を、90%ギ酸中に新鮮な700mg/mlのCNBrを15μl含む1.0mlの0.1N HCl、0.2% 2−メルカプトエタノールに1時間浸漬した。次にこの薄片を取り出し、そして0.1M Tris−Cl pH8.0で5分間、25mM Tris−Cl pH8.0で5分間、そして最後に1×Laemmli SDS−PAGEローディング緩衝液で10分間洗浄した。12% アクリルアミドスタッキングゲルを有する15% SDS−PAGE Laemmliゲル上で薄片をロードした。ペプチドのバンドを銀染色または乾燥したゲルのオートラジオグラフィーにより視覚化した。
【0132】
(再免疫沈降)
SDS−PAGEで分離した抗原の再免疫沈降を、ゲルから標識したバンドを切り出し、TBS、0.2% SDSで10分間再水和化し、そして次にゲルの薄片を小さな切片に刻むことにより行った。タンパク質を、1ml TBS、0.2% SDS中で8時間、室温で回転しながらインキュベートすることにより溶出した。溶出後、ゲルの切片を遠心分離で取り除き、そしてNP−40を最終濃度が2%になるように上清に添加した。次に溶出されたタンパク質を上記の様に免疫沈降し、そしてSDS−PAGEで再分析した。
【0133】
(等電点電気泳動(IEF))
二次元IEFを、本質的にP.H.O’Farrell[J.Biol.Chem.,250,4007−4021(1975)]に記載されたように行った。125Iで標識した抗原を、II−23.D7細胞抽出物から免疫沈降し、そして免疫沈降したタンパク質を、9.5M尿素を含む100μlのO’Farrellサンプル緩衝液中で100℃、5分間の加熱により溶出した。次に、溶出したタンパク質を、最終濃度2%アンホリン(ampholines)(pH3〜10領域、Sigma)を有する14cm×3mmのチューブゲル上で室温で16時間定電圧(400V)で泳動した(第一次元)。第二次元は12%SDS−PAGEであった。
【0134】
天然(未変性)条件下でのIEFについて、125Iで標識した細胞抽出物を、尿素が存在することを除いて上記と同様にチューブゲルに直接電気泳動した。標識した抽出物(容積200μl)をチューブゲルでロードする前に、100,000×g(30psi、airfuge)で10分間遠心分離した。電気泳動を上記と同じ条件下で4℃にて行った。次にチューブゲルを取り出し、そして1cmの断片にスライスし、タンパク質を、1mlのTBS、2% NP−40、2mM PMSF中で、8時間室温でそれぞれの薄片を回転させながらインキュベートすることにより溶出した。次に溶出したタンパク質を含む上清を免疫沈降し、そしてSDS−PAGEにより分析した。変性および未変性のチューブゲルの両方についてのpH勾配を、並行で行ったゲルからの個々の薄片のpHの測定により決定した。
【0135】
(T細胞増殖アッセイ)
PBLを単離し、そして10%ヒト自己血清、1μg/ml インドメタシン、および50U/mlポリミキシンBを含むウシ胎児血清を除いた上記の完全RPMIに再懸濁した。MLR実験で、自己血清は応答動物の血清であった。異なるドナーからの刺激細胞を3000ラドで照射した。ウサギ血清を56℃で1時間前加熱し、そして増殖アッセイに用いる前に血清を希釈し、濾過滅菌した。丸底96ウェルプレートの0.2mlの細胞(全量1×105)を5μg/mlフィトヘマグルチニン、1〜2ng/ml OKT3または1.5〜2×105の照射した刺激細胞のどちらかで、種々の抗血清またはサイトカインの存在または非存在下で処理した。3日後(PHAまたはOKT3活性化)または5日後(MLR)、細胞を3H−チミジンでパルスし、採取しそして計数した。
【0136】
(LT−αおよびLT−βのさらなる精製)
II−23.D7細胞を10%ウシ胎児血清を含むRPMI培地で培養し、50LのRPMI培地から細胞を採取し、4×106細胞/mlの濃度になるよう培地に再懸濁し、そして50ng/ml ホルボールミリストイルアセテート(PMA)を添加した。6時間の活性化の後、遠心分離により細胞を採取し、そしてDulbeccoリン酸緩衝生理食塩水で洗浄した。200mlの冷溶解緩衝液(50mM HEPES緩衝液、pH7.0;0.1M NaCl、10mM EDTA、5mM ベンズアミジン、各10μg/mlの大豆トリプシンインヒビター(アプロチニン、キモスタチン、ロイペプチン、アンチパイン)、1μg/mlペプスタチン、および1mM フェニルメチルスルホニルフルオリド)中に最終的に4×1010細胞の細胞ペレットを懸濁し、このペレットを一回窒素キャビテ一ターに通した。溶解した細胞を、50.2 Tiローターで40,000rpm、60分間遠心分離し、上清を捨てた。ペレットを、1% w/v Nonidet
P40デタージェントを含む120mlの溶解緩衝液中で一晩抽出し、そして次に上記の様に遠心分離した。
【0137】
Affi−gel 10(BioRad)に結合しているモノクローナル抗リンホトキシン(Boehringer Mannheimから入手した抗腫瘍壊死因子−β)からなる2mlのアフィニティー樹脂に、デタージェントで可溶化した膜タンパク質を含む上清を添加し、そしてこの懸濁液を一晩振とうした。樹脂を小さなカラムに集め、1% Nonidet P40を含む50mM HEPES、pH7.0、そして次に1% w/v MEGA−8(Boehringer Mannheim)を含む同じ緩衝液で洗浄した。50mMグリシン緩衝液pH2.5中の1%MEGA−8で、結合したタンパク質を溶出し、そして画分を直ぐにTris塩基で中和した。SDS−PAGE分析および銀染色により、画分中のp33およびLTの存在を決定した。これらのタンパク質を含む画分をプールし、そしてSDSを最終濃度0.1% w/vになるまで添加し、そして0.1×Laemmliサンプル緩衝液(MEGA−8デタージェントを除去するために様々に変化している)でこのプールを透析した。透析した溶液を凍結乾燥し、そして元の水の1/10量に再懸濁した。SDS−PAGEゲル上にサンプルをロードし、ProBlotメンブラン(Applied Biosystems)にブロットし、そしてクマシーブルー染料で染色した。
【0138】
このスキームは、 ブロット上のバンドでLT−βを精製可能にする。当業者がイオン交換クロマトグラフィーによりアフィニティー樹脂から溶出したタンパク質を分離することは可能であるべきである。例えば、複合体を尿素で解離し得、そしてLT−αおよびLT−βタンパク質を、例えば1%非イオン性デタージェント(例えば、MEGA−8、Boehringer−Mannheim)および尿素を含むTris−Cl緩衝液pH8.0中で、MONO Q FPLC(Pharmacia)陰イオン交換クロマトグラフィーにより、塩勾配溶出を用いて分離し得る。このクロマトグラフィーの技術は、タンパク質の異なった電荷に基づいて分離する。2種類のタンパク質は、等電点電気泳動実験(上記参照)で電荷の差異に基づいて分離され得、このとき尿素を使用し、LT−α/LT−β複合体を解離する。このようなアフィニティークロマトグラフィー、尿素/非イオン性デタージェントでの解離、およびイオン交換クロマトグラフィーの組合せは、可溶性LT−βまたはLT−α/LT−β複合体の精製を可能にする。
【0139】
(ペプチド配列決定アッセイ)
ProblotからLT−βおよびLT−αバンドを切り出し、タンパク質シークエンサーにロードした。120A PTHアミノ酸分析機と連結した、Applied Biosystemsシークエンサーモデル470Aを用いて、Edman分解によりN末端の配列の情報を得た。LT−βを上記の免疫アフィニティークロマトグラフィーにより精製し、そしてトリプシン断片の配列を得た。[Abersoldら、「インサイチュ後の一次元または二次元ゲル電気泳動により分離したタンパク質の内部アミノ酸配列分析、ニトロセルロース上でのプロテアーゼ切断」PNAS,84,6970−6974(1987)を参照のこと]。すなわち、インサイチュでブロット上のタンパク質をトリプシンで切断し、切断したペプチドの逆相HPLC解析を行った。得られたN末端および内部のトリプシン断片ペプチドを、次にEdman分解により配列決定した。T105、T87/88、T100、およびT67と呼ばれるN末端および内部のペプチドの配列決定を図13に示す。
【0140】
(オリゴヌレオチドプローブの構築)
T87/88の配列から、以下のアンチセンスプローブを設計した:
1368 GTYTCNGGCTCYTCYTC[配列番号9]
1369 GTYTCNGGTTCYTCYTC[配列番号10]
そして標準方法により合成した。[例えば、J.Sambrookら、Molecular Cloning,A Laboratory Manual,第2版.(1989)を参照のこと]。
【0141】
(誘導されたII−23 cDNAライブラリーの調製)
cDNAサブライブラリーを以下のように調製した:
II−23.D7細胞を6時間、50ng/ml PMAで刺激し、LT−β mRNAの存在を確認した。これらの細胞からmRNAを単離し、そして当該技術分野で周知の技術を用いてそれを逆転写しcDNAにした。[B.SeedおよびA.Aruffo、「迅速免疫選択方法による、CD2抗原、T細胞赤血球レセプターの分子クローニング」PNAS、84,3365−3369(1987)]。標準手法を用いて、2本鎖cDNAを以下の配列を有するNotI−BstXIリンカー/アダプターに連結した:
【化1】
【0142】
次に4.2mlの5〜20%酢酸カリウム勾配、2mM EDTA、1μg/mlエチジウムブロマイド、Beckman(登録商標)SWローターで、50,000rpm、22℃にて3時間標準方法によりcDNAをサイズ選択した。500塩基対より大きなcDNA断片をプールした。次いで、ベクター、pCDM8(Brian Seed(Massachusetts General Hospital)から供与された)を調製した。このプラスミドをBstXIで切断した。400塩基対のスタッファー(stuffer)フラグメントを除去するために、この混合物を上記の様に酢酸カリウム勾配で遠心分離し、そしてより大きなフラグメントを単離した。このフラグメントをアガロースゲル電気泳動によりさらに精製し、そして次いでcDNAをベクターに連結した。この方法で、誘導したII−23.D7細胞で発現したmRNAに対するDNA配列を含む組換えDNA分子を作製した。これらのプラスミドを用いて、E.coli MC1061 P3を形質転換した。PMA誘導したII−23.D7 mRNAのcDNAライブラリーを有する、1×106以上の組換えクローンのコレクションを得た。
【0143】
(スリーニングおよびクローンのDNA配列決定)
pCDM8 II−23.D7ライブラリーを32P標識したオリゴマー1368でスクリーニングし、ポジティブなクローンを単離し、3M テトラメチルアンモニウムクロライドで50℃にて洗浄した。[J.Sambrookら、Molecular Cloning,A Laboratory Manual,(1989);Jacobsら、「テトラアルキルアンモニウム塩溶液中の配列非依存の二本鎖オリゴヌクレオチドの熱安定性:組換えDNAクローンの同定への適用」,Nucleic Acids Research,16,10,4637−4649(1988)]。0.9kbの挿入物を含むいくつかのクローンをジデオキシヌクレオチドDNA配列分析に用いた[同上]。
【0144】
(LT−β cDNAの発現)
pCDM8/LT−βクローン12またはコントロールプラスミドのクローン4(無関係なcDNA挿入物を含むpCDM8)を、エレクトロポレーションによりCHO dhfr−に導入し、CHO細胞をヒトLT−αで安定にトランスフェクトした。3日後、細胞を5mM EDTAを含みCa/Mgを含まないHank溶液に取り出し、10μg/ml コントロールIgG1または抗LTモノクローナル抗体(Boehringer−Mannheim)のどちらかを用いて上記の様にFACS分析のために染色し、次にFITCまたはフィコエリスリンのどちらかで標識したヤギ抗マウス調製物と結合した免疫グロブリンを標識した。その他の実験では、これもまたpCDM8ベクター中にある等量のヒトLT−α cDNAの存在または非存在下で、COS細胞を、pCDM8中のクローン4またはクローン12のいずれかのLT−β cDNAでエレクトロポレーションし、上記のように3日後FACS分析のために染色した。
【0145】
(LT−βの発現のノーザン分析)
ポリA+ RNAをII−23.D7細胞または末梢血単核細胞(PMBC)のどちらかでInvitrogenにより提供されたFast−TrackTMシステムを用いて単離した。J.Sambrookら、Molecular Cloning,A Laboratory Manual,(1989)に本質的には記載されているように、2μg/レーンのRNAおよびホルムアミドゲルでの電気泳動を用いてノーザンブロットを調製し、次にGene Screenナイロンメンブラン上へ転写し、そしてUV架橋した。ブロットを、ゲル精製したLT−β cDNAのランダムプライムBstEII/Xmn−Iフラグメント、あるいはヒトLT−αまたはアクチンのフラグメントでプローブした。II−23.D7細胞を、50ng/ml PMAで時間を変えて誘導し、そしてLT−αおよびLT−βの両方の発現が誘導されたことを見出した。PBMCを、RPMI培地のみまたは1000ユニット/mlのIL−2の存在下、あるいはT細胞を活性化するOKT3とともに培養した。
【0146】
(LT−βの5’末端の決定)
mRNAの5’の配列をプライマー伸長分析により決定した。[B.Wallnerら、Nature 320,77−81(1986).]オリゴヌクレオチドプライマー(プローブ360−121 5’GACAGTGATAGGCACCGCCAGCAACAA−3’)[配列番号13]を用いたプライマー伸長により、約128〜130bpの産物を得た。これは、MaxamおよびGilbert方法論[A.MaxamおよびW.Gilbert,「塩基特異的化学開裂を用いる末端標識DNAの配列決定」 Methods In Enzmology,65,499(1988)]を用いた配列決定により、メチオニンATGの7〜9bp上流にある転写開始部位を示した。一時的な実験でクローン12により示された発現は、1つまたは両方のLeu−4またはLeu−6の開始部位は機能的であることを示した。mRNAの5’配列を証明するために、コスミドクローン、031A[Spiesら、Science 243,214(1989)]をいくつかの制限酵素で切断し、電気泳動し、ブロッティングし、そしてLT−β cDNAのBST E2/Xmn−1フラグメントでプローブした。コスミドは、6kbのEcoRIフラグメント内にLT−β遺伝子を含有した。EcoRIフラグメントを、カナマイシン耐性遺伝子を有するpNN109と呼ぶpUC誘導体にサブクローニングした。ジデオキシ核酸配列決定により完全なゲノム配列を得た。
【0147】
(実施例1:T細胞がその表面にLT−関連エピトープを発現する)
上記の条件下で、ヒト末梢単核細胞(PMN)を、OKT3モノクローナル抗体で活性化し、培養の2日後に、フローサイトフルオロメトリー分析によって、LT−α/LT−β複合関連形態の発現について分析した。1つの実験(結果は図1に示す)で、新鮮なPBLをOKT3およびIL2とともに3日間培養し、それらを、未変性rLT−α(それぞれ、ウサギ4および5由来である、図1の「LT−4」および「LT−5」パネル)、変性rLT−α(ウサギ6由来である、図1の「LT−6」パネル)、および未変性rTNF(ウサギ7由来である、図1の「TNF」パネル)に対して1:200希釈の抗血清で染色した。細胞を、各動物からの免疫後の血清(図1のパネルの実線)あるいは免疫前の血清(図1のパネルの破線)で染色した。図1は、ウサギ4および5由来の抗rLT−α血清のみが、活性化末梢T細胞上のエピトープを認識したことを示す。
【0148】
図2に示す実験では、II−23.D7細胞を15時間、10ng/ml PMAで処理するかあるいは処理せずに、それらを、図1に記載の実験のように、ウサギ4抗rLT−α免疫後の血清(図2のパネルの実線)、あるいはウサギ4免疫前の血清(図2のパネルの破線)で染色した。図2に示すように、ホルボールエステル(PMA)刺激でLT−αを合成するT細胞ハイブリドーマII−23.D7が、PMA活性化で表面LT−関連エピトープを発現することを認めた。
【0149】
T細胞上のLT−α関連エピトープは、LTに関連し、組み換えLT−α調製物由来のCHO細胞中のなんらかの夾雑物に関連しないことを確認するために、PMA活性化し洗浄したII−23.D7あるいはU937細胞で、ウサギ4由来の抗血清の1:1000希釈物を処理した。図3に示す実験では、抗rLT−α血清(1:1000抗LT−4)の試料1mlを、細胞を含まない(−−)、1×108 U937細胞(−〇−)、1×108(−−)PMA活性化II−23.D7細胞、あるいは1×107(−−)PMA活性化II−23.D7細胞のいずれかで処理した。吸収抗血清の希釈物を、L929細胞障害性アッセイで限定量のrLT−αに加えて、最初のウエルに1:4000の最終希釈物が存在するようにした。このアッセイは、24時間以内に、マウス繊維芽細胞株L929を殺すLT−αの能力を測定する[L.Green,J.L.Reade,C.F.Ware,「細胞生存性についての迅速な比色アッセイ:細胞障害性および増殖阻害性リンホカインの定量への適用」J.Immunol.Methods,70,257(1984)]。24時間後に、細胞生存率をMTT読み取りによって評価した。図3のプロットは、吸収抗血清の希釈に対する吸光度(細胞生存率に比例する)である。データは、2つのウエルの平均であり、2つは概して、符号で示されている範囲内に存在する。図3に示されているように、標準L929細胞障害性アッセイでの吸収抗血清中和力価の分析は、U937細胞は効果がないが、活性化II−23.D7細胞が、LT−α中和抗体を除去することを実証した。これらのデータは、膜表面の抗原構造が実質的にLT−αに関連することを示す。
【0150】
ハイブリドーマII−23.D7を、T細胞表面のLT−α関連エピトープの明確な存在についての多くの平凡な説明を確かめるために、さらなる多くの処置を行った。まず最初に、抗血清中のLT−α:抗体複合体が、ハイブリドーマ上のTNF/LT−αレセプターに結合し得る可能性を除外した。TNFおよびLT−αの両方は、複合体中の抗体結合エピトープの存在を考慮し得る三量体構造を有する。しかし、細胞TNFレセプターの可溶性TNFあるいはLT−αでの前飽和は、表面染色には効果はなかった。このような飽和は、このような免疫複合体のこのようなレセプターヘの結合を妨害した。
【0151】
結合されたTNFをそのレセプターから解離し得る、pH3の乳酸処理は、シグナルには影響しなかった。このことは、LT−αがレセプター結合されないことを示唆する。しかし、II−23.D7細胞へ結合する125I−LT−αを使用する実験は、レセプター結合LT−αが、TNFよりも酸性pHではそのレセプターから解離させるのはより困難であることを示した。
【0152】
染色前の細胞のおだやかなトリプシン処理は、シグナルの消失を導き、このことは、エピトープがタンパク質であることを示す。表面結合LT−αがフォスファチジルイノシトール結合されたかどうかを決定するために、細胞をフォスファチジルイノシトール特異性フォスホリパーゼCで処理した。PI−結合抗原LFA−3が解離され得る条件下[A.Petersonら、T細胞赤血球レセプター(CD2)のモノクローナル抗体およびリガンド結合部位」Nature,329,842(1987)]で、LT−αエピトープには効果が認められなかった。
【0153】
LT−α遺伝子で安定にトランスフェクトされたCHO細胞を、(前PMA活性を伴うか、あるいは伴わない)染色し得なかった。このことは、ウサギを免疫するのに使用された、もともとのrLT−α中のCHO由来の夾雑物に対する抗体が、II−23.D7細胞の染色に寄与するのに十分な量が存在しなかったことを示す。同様に、任意のウシ胎児血清タンパク質を夾雑するLT−α調製物に対して誘起された抗体は、染色が10%ウシ胎児血清中で実施されたので、T細胞染色には影響しなかった。
【0154】
抗rLT−α血清のrLT−αでの前処理は、rTNFでの前処理では阻止しなかったが、II−23.D7細胞上のLT−α形態の染色を阻止した。
【0155】
(実施例2:T細胞ハイブリドーマII−23.D7上のLT−α関連タンパクの免疫沈降)
PMAで活性化したII−23.D7細胞を表面ヨウ素化し、溶解してデタージェントで可溶化した。標識膜タンパク質の免疫沈降およびSDS−PAGE分析は、2つのタンパク質が抗rTNF−α抗血清により認識されることを示した。図4Aは、免疫前(PRE)あるいは免疫後(POST)抗rLT−α血清(ウサギ4由来)のいずれかで免疫沈降されたヨウ素化表面タンパク質のSDS−PAGE分析の結果を示す。
【0156】
図4Aに示されているように、LT−αの推定の大きさと相関する25〜26kD分子量形態(「LT−α」)を認め、そして、約33kDの別の形態(「LT−βあるいはp33」)を認めた。同じウサギ由来の免疫前血清(図4AのPREの欄)および抗rTNFウサギ血清のどちらもが、ヨウ素化PMA活性化II−23.D7細胞由来のどのバンドも免疫沈降させ得なかった。
【0157】
ヨウ素化バンドの1−D部分CNBrペプチドマッピングは、25〜26kD形態が、ヨウ素化rLT−αと同じパターンで切断されることを示した。従って、このバンドをLT−αと同定した。図4Bに示す実験で、パネルA由来の25〜26kDおよび33kDのバンドを切り出し、制限CNBr切断およびSDS−PAGEシステムでの電気泳動にかけた。比較のために、並行して行ったrTNFおよびrLT−αの両切断を図4Bに示す。ゲルをオートラジオグラフィーによって可視化した。レーン1はrTNFを示し、レーン2はrLTを示し、レーン3はLTを示し、そして、レーン4は、LT−βを示す。CNBrフラグメントの増大された大きさは、天然LT−βの炭水化物量の増加を反映する。ヨウ素化33kD形態はCNBrで切断されず(レーン4)、このことは、既知LT−α遺伝子生成物とは異なることを示す。rTNFは、このタンパク質中にメチオンが存在しないので、CNBrでは切断されなかった(レーン1)。
【0158】
免疫沈降と組み合わせて、35S−メチオニンおよび35S−システインでの代謝標識を行って、これらのLT−α関連表面形態のさらなる特徴付けを行った。TNF/LT−αペアの場合には、システインおよびメチオニン分布は、TNFとLT−αとの間、および、シグナル配列を伴う形態と伴わない形態との間の両方の区別を可能にし、これは、膜TNF形態の研究で活用された[M.Krieglerら、Cell,53,45〜53頁(1988)]。完全にプロセシングされたサイトカイン、すなわち、分泌形態の場合には、TNFはシステインを含有し、メチオニンを含有しないが、LT−αはメチオニンのみを含有し、システインを含有しない。しかし、LT−αは、シグナル配列ドメイン中に2つのシステイン残基を含有し、TNFは、N末端領域に1つのメチオニン残基を含有する。II−23.D7ハイブリドーマ細胞の分離培養物を、35S−メチオニンあるいは35S−システインのいずれかで標識し、免疫反応タンパク質を沈降させた。実験では(結果は図5に示す)、II−23.D7細胞を、10ng/ml PMAで活性化し、次に、8時間、35S−メチオニンあるいは35S−システインで標識した。培地および溶解細胞の両方を、免疫前(ウサギ4)(P)、抗rTNF(T)、および抗rLT−α(ウサギ4)(L)血清の順で、連続して免疫沈降を行った。図5は、分泌タンパク質を含有する上清あるいは洗浄細胞の、免疫沈降物のSDS−PAGEオートラジオグラフィー分析を示す。”s−TNF”は、35S−メチオニン標識の細胞由来の抗rTNF免疫沈降されたバンドを示し、TNFのプロセシングされていない26kD形態と推定される。”Met”および”Cys”は、使用した35S−標識アミノ酸のことである。35S−システイン含有のそれらのレーンを、35S−メチオニン含有レーンより長時間露光した。これらの細胞からの上清では(「分泌された」と記されたレーン)、25kD形態のLT(「LT−α」)は、PMA処理後に、35S−メチオニン標識された細胞により放出されるが、35S−システイン標識細胞によっては放出されない。このパターンは、完全にプロセシングされ、分泌されたLT−αと予測される。より長時間の露光は、上清に極少量のTNFを示し、標識の取り込みは、完全にプロセシングされた分泌TNFに対して予測された通りであった。mRNAレベルでもまた、低いTNFレベルでの優先的なLT−α発現を認めた(ShamanskyおよびWare、未公開情報)。洗浄細胞の分析(「細胞の」と記されたレーン)は、33 kD LT−βを伴って、25〜26kD LT−αの両方が存在することを示した。25〜26kDおよび33kD形態の相対的な量は、表面ヨウ素化により認められたものに相当した。25〜26kD表面LT−α形態はシステインを含まず、このことは、リーダー配列のプロセシングを示す。33kD形態は、35S−メチオニンおよび35S−システインの両方を取り込んだ。図5に示したフィルムのより長時間の露光(示されていない)は、約26〜27kDで細胞からの抗TNF免疫沈降されたバンドの存在を示した。バンドは、標識システインおよび標識メチオニンの両方の取り込みを示した。標識はシステインによって、より強かった。26kD−TNF形態ではcys:met比率は4:1であるので、この標識パターンは、このバンドの同一性を確実にする。
【0159】
細胞溶解物からの免疫沈降物中のLT−αを伴ったLT−βの存在は、LT−βが抗原的にLT−αに関連すること、あるいはLT−βがLT−αに結合されることのいずれかあるいは両方を示唆した。この論点に的を絞るために、35S−メチオニン標識細胞由来の25kDおよび33kDのバンドを、ウサギポリクローナル抗rLT−α血清で免疫沈降させ、切り出したゲル切断片から溶出して、抗rLT−αポリクローナル血清あるいはmAbで再度免疫沈降させた。LT−βではなくLT−αが、抗rLT−α抗体で免疫沈降され得、このことはLT−βが抗原的にLT−αに関連しないことを示唆する。これらの観察は、LT−βがLTαと物理的に結合することを示した。33kDのタンパク質は、抗原的にLT−α関連せず、単にLT−αと共に沈降されることを示した。
【0160】
表面ヨウ素化あるいは代謝標識のいずれかでは、LT−αあるいはTNFに結合した、既知の55あるいは80kD TNF/LT−αレセプター形態のいずれも検出され得なかった。おそらく、これは、レセプターが、T細胞の活性化の間に急速に費やされるからである。[C.Wareら、「腫瘍壊死因子によるCTL溶解経路の調節」Cellular Immunity And The Immunotherapy Of Cancer,UCLA Symposia on Molecular and Cell Biology,M.T.LotzeおよびO.J.Finn編 第135巻,121〜128頁(Wiley−Liss,Inc.New York)1990]。
【0161】
(実施例3:II−23.D7細胞上の表面LT−形態の生化学的特徴付け)
PMA処理したIl−23.D7細胞表面のLT−関連形態を、アフィニティークロマトグラフィーによって精製した。免疫沈降法によって、LT−βおよびLT−αがレンズマメレクチンセファロースに結合すること(このことは糖タンパク質構造を示す)に注目した。デタージェントで可溶化し、PMA処理したII−23.D7タンパク質を、レンズマメレクチンセファロースに結合し、アフィニティー精製の前にα−メチルマンノシドで溶出した。抗rLT−α血清によって特異的に認識されるこれらのタンパク質を、さらに正確に評価するために、コントロールIgGおよび抗IgGカラムの両方を調製した。カラムを低pHで溶出することにより、抗rLT−αカラムから約100〜200ngの2種のLT形態が放出された。
【0162】
図6Aは、免疫前(PRE)あるいは免疫後(POST)ウサギ血清から調製された抗rLT−αアフィニティーカラムから溶出されたタンパク質のSDS PAGE分析を示す。図6Bでは、パネルA中のゲルから33kDおよび20kDのバンドを切り出し、制限CNBr切断およびSDS−PAGEシステムでの電気泳動にかけた。比較のために、図6Bは、並行して行ったrTNFおよびrLT−α(CHO由来)のCNBr切断を示す。銀染色によってゲルを可視化した。溶出物のSDS−PAGEゲルは、免疫沈降された表面ヨウ素化PMA処理のII−23.D7細胞のゲルによく類似し、このことは類似のタンパク質が精製されたことを示す。
【0163】
アフィニティー精製の間に、25kDのLT−α形態は、19〜20kD形態に切断されるようである。すなわち、それは、未処理の組み換えCHO細胞由来のLT−αとともに移動した。RPMI 1788腫瘍株由来の天然LT−αの最初の単離もまた、N−末端切断された「des−20」LT−α形態を生じた。アフィニティー精製したタンパク質の一次元CNBr分解物は、切断された20kDのLT−α形態が、おそらくこのLT−α形態の切断型特性を反映するCNBr切断パターンを有することを示した。メチオニンの1つが、「des−20」LT−α形態では消失し、ゆえに、切断パターンは未処理のLT−α形態とは異なる。33kDのタンパク質(LT−β)は、CNBr切断で2つになるので、このことから、1つのメチオニンはCあるいはN末端から5〜20残基以内に存在しなければならないことが推定された。この切断パターンは、33kDのタンパク質が既知のLT形態と著しく異なることを示す。表面ヨウ素化33kDタンパク質のCNBr切断分析は、恐らく類似の結果を与えたが、しかし、ヨウ素化で達成され得る解像は、その2つを十分に現らわさなかった。ヨウ素化rLT−α、rTNF、およびII−23.D7 LT形態のStaphylococcusV8分解は、分解に対して耐性であるべきrLT−αおよび25〜26kDK II−23.D7 LT−α形態を示した。このことは、このタンパク質がLT−αであることを確実にした。33kDのタンパク質を、rTNFによく類似するパターンを有する、数個の小さなフラグメントに切断した。
【0164】
図7では、免疫沈降された表面ヨウ素化タンパク質を、SDS−PAGE分析で分離し、表面結合25〜26kDのタンパク質(「sLT−α」)および33kDのタンパク質(「LT−β」)のバンドを切り出した。切断片を、N−グリカナーゼ(N−Gly)、ノイラミニダーゼとO−グリカナーゼ(O−Gly)との混合物、あるいは3つ全ての酵素で分解した。分解した切断片を、再度SDS−PAGEにかけ、乾燥ゲルのオートラジオグラムを示す。図7に示したように、ヨウ素化表面LT形態の免疫沈降、その後の、N−グリカナーゼあるいはO−グリカナーゼのいずれか、またはその両方での分解で、25〜26kD LT−α形態が、N−連結オリゴ糖を含有することを示した。25−26kD LT−α形態は、N−グリカナーゼ分解での大きさの変化に関連する1つのN−連結部位のみを有する。同様に、33kD形態(LT−β)は、N−グリカナーゼ処理で約3kDの大きさを消失した。このことは、1つのN−連結オリゴ糖の存在を示唆する。25〜26kD LT−α形態に対して、O−グリカナーゼ処理は、LT−βの分子量に影響を与えなかった。しかし、グリカナーゼによる切断がないことは、炭水化物の存在を欠く明白な証拠ではない。
【0165】
(実施例4:LT−αの再免疫沈降)
LT−βのLT−αとの共沈降は、これらのタンパク質が抗原的に関連するか、あるいは物理的に結合されることを示唆した。この確認のために、SDS−PAGE分離されたLT−α(p25)およびLT−βタンパク質が免疫沈降され得るかどうかを試験した。125Iあるいは35S−Metで標識されたLT−αおよびLT−βを、免疫沈降によって部分精製し、SDS−PAGEで分離した。標識されたバンドを切り出し、緩衝液中で再度水和し、タンパク質を溶出した。次に、溶出タンパク質を、ポリクローナルあるいはモノクローナル抗rLT−α抗体を用いて、第二回目の免疫沈降を行った(図10)。ウサギ抗rLT−αは、LT−α(「p25」、レーン2)を再免疫沈降させたが、LT−βはさせなかった(レーン3)。抗rLT−α mAbは、LT−α(レーン5)および21kDのタンパク質(「p21」、レーン4)を沈降させた。これは、下記に示すように、LT−αの前駆体であるが、しかし、LT−βを沈降させなかった(レーン6)。結果は、LT−αおよびLT−βがSDS−PAGEにより分離された後に、ポリクローナルおよびモノクローナル抗rLT−α両抗体が、LT−αと反応し得るがLT−βとは反応し得ないことを示す。このデータは、LT−βが抗原的にLT−αと関連しない証拠を提供する。しかし、LT−αエピトープは完全なまま残存するが、推定のLT−β交差反応エピトープが、変性後に消失される可能性は除外され得ない。
【0166】
図8は、SDS−PAGEゲルから溶出された、125I標識および35S−Met標識のp25およびp33タンパク質の再免疫沈降の結果を示す。125I標識II−23.D7細胞(レーン2、3)および35S標識細胞(レーン4、5、6)由来のLT−αおよびLT−β種を、上記のようにゲル切断片から溶出した。溶出物を、抗rLT−α血清(レーン2、3)あるいは抗rLT−α mAb(レーン4、5、6)のいずれかで免疫沈降し、再沈降されたタンパク質をSDS−PAGEおよびオートラジオグラフィーで分析した。レーン1は、LT−αおよびLT−βの同定のためのコントロールレーンである。
【0167】
(実施例5:LT−αおよびLT−βの等電点電気泳動)
図9および10(各々は、オートラジオグラフ(9A、10A)、および移動距離対pHをグラフにした検量線を含む(9B、10B))は、変性(図9)および未変性(図10)条件下での等電点電気泳動分析を示す。II−23.D7細胞抽出物から免疫沈降された、125I標識LT−αおよびLT−βについて、上記のように、二次元ゲル分析を行った。2−Dゲル分析を、尿素が存在する変性条件下で実施した(図9A)。これに対し、未変性IEFを、尿素を含まない1% NP−40中で行った。125I標識II−23.D7細胞抽出物を、4℃でチューブゲルで泳動させた。泳動後に、チューブゲルを1cm切断片に切断し、泳動されたタンパク質をこれらの切断片から溶出し、免疫沈降して、SDS−PAGEで分析した(図10A)。ゲルレーン1〜12から免疫沈降された物質は、チューブゲル切断片の2〜13に対応する。1cmゲル増大量に基づいて変性および未変性チューブゲルに対してpH勾配を実施した。これらもまた、それぞれに9Bおよび10Bとして下にオートラジオグラムを示す。生化学的に、LT−βおよびLT−αは、非変性等電点電気泳動ゲル上で共に移動するが、複合体が尿素により解離されると、2つのタンパク質は別々に移動する。[図9A、10Aを参照]これらの観察は、LT−αおよびLT−βが細胞表面に複合体として存在するという結果を導く。
【0168】
(実施例6:LT−α発現の制御)
以下に示す表Iは、LT−αの表面形態発現についての、種々の細胞タイプのフローサイトフルオロメトリー分析による調査結果のまとめである。
【0169】
【表I】
これらの研究の際だった観察は、TおよびB細胞に対する表面LT−α発現の制限であった。leu−M3、単球マーカーおよびleu−4(CD3)抗体を、各細胞群を別々に観察するために、二色フローサイトフルオロメトリー分析に使用した。この分析で、表面TNFと表面LT−αと間に際だった相違が存在した。ここで、T細胞は表面LT−αのみを示したが、単球は表面TNFのみを発現した。この結果を図11に示す。
【0170】
図11に示されている実験では、PBLを、LPS(1μg/ml)、インターフェロン−γ(200U/ml)、およびOKT3(1ng/ml)の混合物で8時間処理し、次に、LT−α(ウサギ5由来の抗rLT−α血清)あるいはTNF(ウサギ7由来の抗rTNF血清)に対して染色し、その後、FITC抗ウサギ標識を行った。細胞を、フィコエリスリン−leu−4、pan T細胞マーカー、あるいはフィコエリスリン−leu−M3、単球マーカーで逆染色した。「T細胞パネル」をleu−4+に対してゲートし、一方、「単球」パネルをleu−M3+細胞に対してゲートした。細胞を、免疫前(破線)あるいは免疫後(実線)の血清で染色した。単球腫瘍株HL−60およびU937はLT−αを染色しなかった。二色フローサイトフルオロメトリー分析によって、活性化PBLのT4およびT8サブクラスが、同レベルの表面結合LT−αを示すことが認められた。一般的に、LT−αを発現し得る一次T細胞もまた表面LT−α形態を示し得るようである。
【0171】
3人の異なるヒトドナーの実験は、表面LT−α形態が用時単離された末梢T休止細胞に存在することを示した。PBLの場合には、細胞のOKT3活性化あるいは単なるIL−2処理が、発現の増大を導いた。蛍光チャネル数により、OKT3活性化の間の表面LT−αおよびIL−2レセプター(CD25)発現の両方の定量を試みた。OKT3による表面LT−αの最大誘導が、大量培養でIL−2レセプターのピーク発現(TAC発現)に先行するようであり、従って、表面結合LT形態(LT−α/LT−β複合体)は、初期T細胞活性化抗原のようである。抗T11および同種異系抗原の両方が、クローン化細胞障害性T細胞表面にLT−αを生じ得ることが見い出された。同様に、PMA刺激は、II−23.D7ハイブリドーマ表面でのLT−α出現の誘導に必要であった。T細胞活性化が表面LT−α形態を増加させるようである。II−23.D7ハイブリドーマに比較して、末梢リンパ球は、PMA処理後に急速に表面LT−α形態を下降制御する。同様に、OKT3活性化PBL群の二色分析では、活性化の前進段階でT細胞を含むはずであるDr+細胞は、表面LT−α形態を欠いた。
【0172】
新鮮なPBLの高レベルIL−2での活性化は、リンホカイン活性化キラー細胞(LAK細胞)を生じた。図12に示されているように、抗rLT−αおよびleu−19、NK/LAK細胞マーカーを用いた二色フローサイトフルオロメトリー分析は、表面LT−α形態のLAK細胞発現が、T細胞ハイブリドーマII−23.D7に類似することを示した。図12に示されている実験では、PBLを、20ng/ml IL−2とともに5日間培養し、次に二色分析のために、上記のように、フィコエリスリン標識leu−19および抗rLT(ウサギ5)で染色した。図12は、免疫前(破線)あるいは免疫後(実線)血清で染色した、leu−19+細胞上の表面LT−αレベルを示す。従って、LAK細胞は、あらゆる一次細胞形態中で最も高い表面LT−αレベルを有するようである。
【0173】
(実施例7:全TNFあるいはLT−αのT細胞活性化との機能関連性)
TNFおよびLT−αのT細胞活性化プロセスとの機能関連性を調べるために、混合リンパ球応答(MLR)およびOKT3活性化アッセイに、ウサギ抗rLT−αおよび抗rTNF血清を包含させた。MLRは、個人のT細胞が、他人のT細胞を外来物として認識し、増殖によるその存在に応答する能力を試験する、標準的な免疫学的アッセイである。以下の表IIは、種々の応答細胞/刺激細胞の組合せを使用した、MLR実験のデータを示す。
【表II】
【0174】
表IIに示したように、中和抗rLT−α血清(ウサギ4および5)は、5日目に評価されるように、増殖応答を阻害したが、免疫前血清あるいは非中和抗rLT−α(ウサギ6)血清は軽い刺激効果を有した。以前に報告されたように[M.Shalabyら、J.Immunol.141,499(1988)]、ポリクローナルおよびモノクローナル抗TNF調製物もまた阻害性であった。これらのアッセイを、過剰刺激細胞条件で行った。従って、阻害は最適化され得ない。表II中で使用した血清レベルは、その他の実験(データは示していない)よりかなり高く、1:1000までの抗体希釈物はまだ阻害性であった。PHAあるいはOKT3刺激T細胞増殖もまた、より低い程度で阻害された(データは示さない)。これらのデータは、T細胞表面のLTあるいはLT−関連エピトープが、T細胞活性化に関連し得ることを示した。
【0175】
MLRアッセイおよび中和モノクローナル抗体を用いた以前の研究は、T細胞活性化および次のこの系での増殖において、LT−αではなくTNFに関係した。[M.Shalabyら、J.Immunol.,141,499(1988)]。その研究では、モノクローナル抗rLT−α抗体は、MLRアッセイに効果がなかった。中和ポリクローナル抗CHO細胞誘導rLT−α血清が、部分的にMLRを阻害し得ることが、われわれの研究で示され、そのことは、この系におけるなんらかのLT形態に対する役割を示唆する。これらの抗体調製物の性質に相違が存在し得るが、この矛盾に対する理由は明確ではない。モノクローナル抗体を、グルタルアルデヒドに架橋結合された天然のLT−α(RPMI 1788分泌の)に対して産生し、一方、ポリクローナル抗rLT−α血清は、未変性r−LT−α(組み換えCHO細胞由来の)を、フロイントアジュバントともに直接リンパ節に注射して調製した。後者の系の成果に対する貯蔵効果は、免疫化マウスにおいて報告された問題を考慮するとおそらく重要であった[T.Bringmanら、「ヒト腫瘍壊死因子αおよびβに対するモノクローナル抗体:アフィニティー精製、イムノアッセイヘの適用、および構造プローブとしての適用」Hybridoma,6,489(1987)]。これらのデータは、本発明のポリクローナル抗rLT−α血清のMLRにおけるブロック効果が、従来の可溶性LT−α形態の認識ではなく、血清による表面LT形態の認識の結果であることを示唆する。
【0176】
(実施例8:LT−αおよびLT−βの精製およびの最初の配列決定)
上記のように、エドマン分解によって、LT−αおよびLT−βのN末端配列情報を得た。膜結合LT−αバンドの配列を以下のように認めた:Leu Pro Gly Val Gly Leu Thr Pro Ser。この配列は、分泌LT−αの既知配列に一致する。エドマン分解分析は、結合LT−βタンパク質のN末端部分が、2つの可能なアミノ酸配列を含むことを解明した:Gly Leu Glu Gly Arg Gly Gln Arg Leu GlnあるいはGly Leu Glu Gly Arg Leu Gln Arg Leu Gln。次のDNA分析は、7サイクル目でグリシンがグルタミンとして誤って同定されたこと以外は、前者の配列を確認した。
【0177】
(実施例9:LT−β配列決定およびクローニング)
上記のように、Abersoldらの方法によって、数個のペプチド配列の配列を得た。2つのアンチセンス17−merオリゴヌクレオチドプローブGTYTCNGGCTCYTCYTC[配列番号:9]およびGTYTCNGGTTCYTCYTC[配列番号:10]を、これらのペプチド、T−87/T−88の1つの配列部分と一致するように合成した。これらのプローブを、32Pで放射性標識した。ノーザン分析(J.Sambrookら、Molecular Cloning a Laboratory Manual,第2版(1989))は、プローブ1368(GTYTCNGGTCYTCYTC)[配列番号:10]が、先に記載のように、ホルボールエステルで前処理されたII−23.D7細胞中で強く誘導される、0.9〜1.1kb mRNAバンドに強くハイブリダイズしたことを示した。ベクターpCDM8(Invitrogen Corporation,San Diego,Californiaから入手可能)のcDNAライブラリーを、PMAで6時間誘導されたII−23.D7細胞から単離されたポリA+ mRNAから構築した[B.SeedおよびA.Aruffo,PNAS,84,3365〜3369(1987)を参照のこと]。ライブラリーを標識オリゴマー1368でスクリーニングし、ポジティブクローンを単離し、その後50℃の3M テトラメチルアンモニウムクロライドで洗浄した。[Jacobsら、Nucleic Acids Research,16,10,4637〜4649(1988)を参照のこと]。0.9kbの挿入物を含有するいくつかのクローンを、DNA配列分析にかけた。クローンpCDM8/LT−β−12(クローン12)が、コーディング配列を含むことが認められた。その他のクローンは、1〜30bpの種々の5’末端切断型以外は同定された。1つの可能なクローン(クローン4)はフレームシフトを含み、トランスフェクション実験のコントロールとして使用された。862位の終止配列AATAAAが、ポリA区域の直前に見いだされ、このことは、全3’末端が同定されたことを示す。同定されたタンパク質配列は、計算上の分子量25,390を有する少なくとも240アミノ酸、およびタイプII膜タンパク質の代表的なドメイン構造をコードする。このデータは、開始CTGが、gly 5(met=1)で始まるプロセシングされたN末端を導く、すなわち、CTGにコードされるロイシンが翻訳されないか、あるいは、ロイシン−4基がさらにプロセシングされて、アミノ酸配列決定で得られた成熟N末端を得ることを示唆する。
【0178】
LT−βの33kDaの大きさは、先に定義したようにN−結合グリコシル化に起因し、この結果は、CD40リガンドに認められる同様の部位に一致する位置の、アミノ酸配列中の1つの可能なN−結合炭水化物部位の存在により、裏付けられる。代表的なN−結合糖残基は、約3〜4kDa MWを付加し得、それゆえに、最終分子量は観察された30〜33kDaに近い。
【0179】
同定されていない配列が、データベースで認められた。細胞外ドメインに1つのシステイン残基、および最後のC末端17アミノ酸内に2つのメチオニンが存在し、このタンパク質で示される、非常に限定された臭化シアン切断パターンに一致する。
【0180】
(実施例10:LT−βの発現)
pCDM8/LT−βクローン12またはコントロールプラスミド、クローン4(非機能性cDNA挿入物を有するpCDM8)を、エレクトロポレーションによって、CHO dhfr−およびヒトLT−αで安定してトランスフェクトされたCHO細胞に導入した。3日後、細胞を、Ca/Mgを含まない、5mM EDTA含有のハンクス液で取り出し、上記のように、10μg/mlのコントロールIgG1、あるいは抗LTモノクローナル抗体(Boehringer−Mannheim)を使用するFACS分析のために染色し、その後、FITCあるいはフィコエリスリン標識ヤギ抗マウス調製物を用いる結合免疫グロブリンで標識した。
【0181】
別の実験で、COS細胞に、pCDM8中のクローン4あるいはクローン12 LT−β cDNAを、これもまたpCDM8ベクター中の等量のヒトLT−α cDNAの存在下あるいは非存在下で、エレクトロポレーションし、上記のように3日後に、FACS分析用に染色した。LT−αを発現するCOS細胞のみが、機能性LT−β DNA、すなわちクローン12でのトランスフェクションで、表面リンホトキシンを示した。
【0182】
クローン12は、開始ATGコドンを欠くが、いくつかのCTG開始コドンを有し、それゆえに、この発現実験は、1つあるいはいくつかの5’CTGコドンが翻訳を開始し得ることを示す。CTGコドンは、数種の真核タンパク質での翻訳開始部位として作用することが知られている。[M.Kozak,J.Cell.Biol.,115,4(1991)]。同様の結果が、LT−αおよびLT−β DNAの両方を有するCOS細胞のみが、FACS分析で実質的な表面LT−αを示すような二元トランスフェクション系を使用して観察された。
【0183】
II−23.D7細胞のノーザン分析は、LT−β cDNAの0.9〜1.0kb mRNAへのハイブリダイゼーションを示し、このことは、クローニングしたcDNAが転写される全遺伝子を本質的に表すことを示す。図15を参照のこと。LT−β遺伝子は、非処理II−23.D7ハイブリドーマ細胞において低レベルで発現されるが、しかし、ホルボールエステルによる細胞の活性化で、mRNAレベルが劇的に増大した。図16を参照のこと。
【0184】
(実施例11:LT−βとリンホカインのTNFフミリーのその他のメンバーとの間の相同性)
LT−βをコードするcDNAのクローニングは、LT−βが、TNF、LT−α、およびCD40レセプターのリガンドに著しい相同性を有するタイプII膜タンパク質であることを解明した。これらのタンパク質は、TNF/NGFレセプターファミリーのメンバーに結合することが知られている。LT−β、TNF、LT−αおよびCD−40レセプターのリガンドは、細胞外ドメインに4つの配列保存領域を共有する。図13を参照のこと。これらのドメインは、TNFおよびLT−α結晶構造の表面に存在し、サブユニット間相互作用に関連するようである。
【0185】
(実施例12:数個の可能な開始部位を明らかにするLT−βの5’末端の決定)
5’mRNA配列を、プライマー伸長分析によって決定した。プライマー伸長分析は、LT−βタンパク質の転写開始部位が、メチオニンATGの上流約7〜9塩基対であることを解明した。従って、mRNAは、少なくとも3つの可能な転写開始部位、Met−1,Leu−4、およびLeu−6コドンを有する。クローン12を用いる一時的な実験は、Leu−4あるいはLeu−6開始部位の1つあるいは両方が機能することを示した。5’mRNA配列は、上記のコスミドクローン031Aを使用したLT−βゲノム配列の決定、およびジデオキシ核酸配列決定によって明らかにされた。
【産業上の利用可能性】
【0186】
本発明は、リンホトキシン−β、リンパ球膜タイプタンパク質に関する。このタンパク質は、ホルボールエステル(PMA)刺激T細胞ハイブリドーマII−23.D7細胞を含む多数の細胞の表面で見られる。本発明はまた、リンホトキシン−βとリンホトキシン−αのような他のペプチドとの間で形成される複合体およびリンホトキシン−βの多数のサブユニットを包含する複合体に関する。これらのタンパク質および複合体は、細胞内で形成されるLT−αを細胞表面に保持するのに有用であり、LT−α/LT−β複合体が炎症調節剤、腫瘍増殖阻害剤、T細胞阻害剤、T細胞活性化剤、自己免疫疾患調節剤、またはHIV阻害剤として作用し得る。さらに、LT−α/LT−β複合体の抗腫瘍活性を、LT−βの遺伝子でトランスフェクトされた腫瘍湿潤リンパ球(TIL)によって腫瘍細胞に送達し得る。
【図面の簡単な説明】
【0187】
【図1】図1は、OKT3−刺激、IL−2拡張末梢血リンパ球(PBL)のフローサイトフルオロメトリー分析を示し、3つの、異なるウサギ抗rLT−α抗血清との反応を示し、そして本質的にウサギ抗rTNF抗血清との反応がなかったことを示す。
【図2】図2は、ヒトT細胞ハイブリドーマ、II−23.D7のフローサイトフルオロメトリー分析を示し、細胞表面上の(PMA処理後の)LT−α関連エピトープの存在を示す。
【図3】図3は、PMA活性化II−23.D7細胞の抗rLT−α抗体と結合する能力を示す。抗LT−α抗血清のサンプルと、PMA処理U937(非LT生成)細胞、(−〇−)、PMA活性化II−23.D7ハイブリドーマ細胞(108、−−;107、−・−)、および細胞なし(コントロール、−−)とをインキュベートした。インキュベーション後、無細胞抗血清の連続希釈をrLT−αに添加し、そしてL929(LT−αセンシティブ)細胞に対する細胞障害性アッセイに使用した。プロットは、rLT−α中和抗体が抗血清から活性化II−23.D7細胞によって除去されたことを示す。
【図4】図4は、PMA活性化II−23.D7細胞からの125I−標識表面タンパク質の免疫沈降を示す2つのオートラジオグラフ(AおよびB)を示す。図4Aは、免疫前ウサギ血清ではなくて免疫後ウサギ抗rLT−α抗血清によって、約25kD表面タンパク質(LT−α)および約33kD表面タンパク質(p33、すなわちLT−β)の免疫沈降を示す。図4Bは、図4Aからの25kDおよび33kDバンドの一次元CNBr切断マップを示し、E.coliで生産した組換えTNF(rTNF)、およびCHO細胞で生産される組換えリンホトキシン−α(rLT−α)[J.Browingら、J.Immunol.143、1859(1989)]と、CNBr切断の有り(+)およびなし(−)の両方で比較した。
【図5】図5は、35S−メチオニンまたは35S−システインで代謝的に標識したPMA刺激II−23.D7細胞からのTNF−およびLT−α−関連タンパク質の免疫沈降を示すオートラジオグラフである。この図は、約25kDメチオニン含有表面タンパク質(LT−α)および約33kDメチオニンおよびシステイン含有表面タンパク質(p33、すなわちLT−β)が、免疫前(P)または抗rTNF抗血清(T)ではない抗rLT−α抗血清(L)によって認識されることを示している。オートラジオグラフもまた、活性化II−23.D7細胞がまた26kD形態のTNFを生産し、そして可溶性リンホトキシン−αを分泌することを示す。
【図6】図6は、抗rLT−α血清によって認識されるPMA活性化II−2S.D7細胞からのこれらのタンパク質のアフィニティ精製1−D CNBrペプチドマッピングを示す。図6Aは、免疫前(PRE)または免疫後(POST)ウサギ血清のいずれかから調製した抗LT−αアフィニティーカラムから溶出されるタンパク質のSDS PAGE分析を表す。図6Aは、約33kDおよび約20kDタンパク質バンドは免疫前血清(PRE)を使用して調製したアフィニティーカラムに結合しないが、抗rLT−α抗血清(POST)を使用して調製したアフィニティーカラムに結合することを示す。図6Bは、POSTカラムから溶出される約33kDおよび約20kDタンパク質の部分的なCNBr切断を示し、並行して泳動したrTNFおよびrLT−αとの比較を示す。ゲルを、銀染色法で視覚化した。
【図7】図7は、約25kDおよび約33kD125I−標識タンパク質(それぞれLT−αおよびLT−βと命名した)のオートラジオグラフを示すが、これらは、N−グリカナーゼ(N−gly)、ノイラミニダーゼおよびO−グリカナーゼ(O−gly)の混合物で、または3つの酵素すべてで処理された活性化II−23.D7細胞から免疫沈降されたものである。
【図8】図8は、共沈降したp33(LT−β)およびp25(LT−α)タンパク質の再免疫沈降の結果を示し、さらにこれらが免疫学的に関連しているかどうか調査する。
【図9】図9(9Aおよび9B部分を含む)は、免疫沈降したp33(LT−β)およびp25(LT−α)タンパク質の変性条件下での等電点フォーカシングの結果を示す。
【図10】図10(10Aおよび10B部分を含む)は、免疫沈降したp33(LT−β)およびp25(LT−α)タンパク質の未変性条件下での等電点フォーカシングの結果を示す。図9および図10は共に、LT−βおよびLT−αが変性可能な複合体を形成することを示す。
【図11】図11は、LPS、IFN−γおよびOKT3の混合物で刺激した後、T細胞および単球上で分化的に発現した表面タンパク質をフローサイトフルオロメトリーを示す。刺激したPBLプールから分離したT細胞は、抗rLT−α抗血清(LT)によって認識される表面タンパク質を発現することが認められ、他方分離した単球は抗rTNF抗血清(TNF)によって認識される表面タンパク質を発現した。
【図12】図12は、IL−2で処理されたleu−19−およびleu−19+(すなわち天然キラー)細胞上の表面LT形態のフローサイトフローメトリー分析を示す。IL−2で処理したPBLを標識leu−19および抗rLT−αの両方で分析した結果、リンホカイン活性化キラー(LAK)細胞が表面LT形態を発現するのを確認した。
【図13】図13は、LT−βフラグメントのアミノ酸配列を示すが、これは、直接N−末端配列決定およびインサイチュトリプシン切断に続いて、切断されたペプチドの逆相HPLC分離によって得られた。
【図14】図14は、TNF/NGF−レセプターファミリーのメンバーと結合するリガンドのファミリーの4つのメンバーのアミノ酸配列比較を示す。ホモロジー領域を、太いタイプフェイスで、同一配列をドットで示し、そして保存配列を星印で示す。推定N−結合グルコシル化部位を四角で囲んでいる。
【図15】図15は、これはLT−αおよびLT−β発現のノーザン分析を示す。A)いくつかの細胞株のノーザンブロットでHut−78およびII−23.D7細胞中のみで両方のLT遺伝子を特異的に発現することを示す。B)II−23.D7細胞中のLT−αおよびLT−β mRNAのPMA誘導の時間経過 C)抗CD3またはIL−2単独のいずれかで活性化されたヒト末梢血リンパ球の同様の分析を示す。
【図16】図16は、CHO細胞中のLT−αおよびLT−βの発現を示す。A)LT−β cDNAで一時的にトランスフェクトされたCHO細胞のFACS分析。親dHFR−CHO株またはLT−αを発現するCHO細胞のいずれかを、無関係の挿入物(クローン4)を含有するpCDM8またはpCDM8/LT−βプラスミドのいずれかでエレクトロポレートし、そしてコントロールIgG(−−−)または抗LT−αモノクローナル抗体(___)で染色した。B)パネルBは、COS細胞中でLT−αおよびLT−βの発現を示す。細胞を、pCDM8/LTαと共に、または用いないで、コントロールDNAまたはpCDM8/LTβプラスミドでトランスフェクトし、そしてパネルAと同様に染色した。
【特許請求の範囲】
【請求項1】
配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−βを使用する工程を包含する、新生物形成の進行を軽減するための方法。
【請求項1】
配列番号2の配列を含有する、リンパ球膜タイプポリペプチドであるリンホトキシン−βを使用する工程を包含する、新生物形成の進行を軽減するための方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】
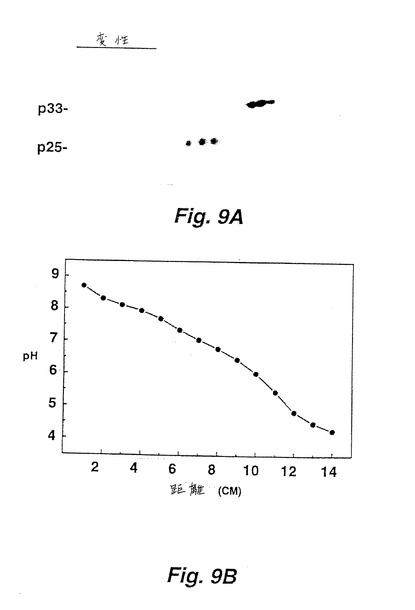
【図10】
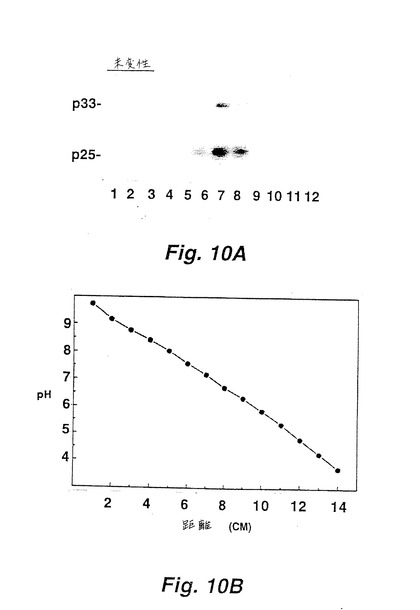
【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2007−39464(P2007−39464A)
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願番号】特願2006−253541(P2006−253541)
【出願日】平成18年9月19日(2006.9.19)
【分割の表示】特願2003−383099(P2003−383099)の分割
【原出願日】平成5年12月2日(1993.12.2)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【出願人】(500025503)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (26)
【Fターム(参考)】
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願日】平成18年9月19日(2006.9.19)
【分割の表示】特願2003−383099(P2003−383099)の分割
【原出願日】平成5年12月2日(1993.12.2)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【出願人】(500025503)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (26)
【Fターム(参考)】
[ Back to top ]