
無支持体の18Fで標識したアミノ酸O−(2−[18F]fluoroethyl)−L−Tyrosineの製造方法。
【課題】PET診断における腫瘍への集中度が高く、取扱いの容易なトレーサーとなる無支持体の18Fで標識したアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【解決手段】下記の化学式の前駆体を用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosine製造方法。
式中、R1カルボキシル基の保護基で、arylalkyl基、R2はアミノ基の保護基で、carboxyl基、R3は離脱基で、p-tosyloxy,methane sulfonyloxy基若しくはtrifluoromethanesulfonyloxy基又は臭素基である。
【化1】

【解決手段】下記の化学式の前駆体を用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosine製造方法。
式中、R1カルボキシル基の保護基で、arylalkyl基、R2はアミノ基の保護基で、carboxyl基、R3は離脱基で、p-tosyloxy,methane sulfonyloxy基若しくはtrifluoromethanesulfonyloxy基又は臭素基である。
【化1】
【発明の詳細な説明】
【技術分野】
【0001】
18Fで標識したアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineは、一種のアミノ酸トレーサー(追跡剤)で、人体実験によると腫瘍のポジトロンCT(PET)法診断に好適に適用することができる。
【背景技術】
【0002】
2-[18F]Fluoro-2-Deoxy-D-glucose([18F]FDG)は現在最も広く用いられているPET薬剤で、ブドウ糖により代謝するその生化学的な経路は腫瘍の診断や治療に対する評価に利用されている。しかし、 [18F]FDGの脳内での高度吸収量は脳のバックグランド放射線を増加して脳腫瘍の診断を困難にさせるだけでなく、炎症部位に関しても誤診され易い。一方、L-[methyl-11C]も脳腫瘍細胞に集中することができ、また上記のような高度吸収量による診断困難や誤診なども一切起こらない。しかしながら、このL-[methyl-11C]は、病院でサイクロトロンを備えていても半減期が20分にすぎないため、医療現場に至る途中での減衰が著しく、これらの欠点のためコストが高いものとなる。
【0003】
18Fで標識したアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineは、人体で吸収されるとアミノ酸により代謝する経路は大概C−11でラベルしたL-[methyl-11C] methionineと変わらず、[18F]FDGの脳内や炎症部位での高度吸収量のような影響で診断困難や誤診に至ることもない。[18F]FETの半減期は[18F]FDGと同じく109分間であるから病院間などの医療現場に至る間での所要時間からみて有利であって、C−11で標識したL-[methyl-11C] methionineよりもコストが低くできるという点は経済的である。
【0004】
O-(2-[18F]fluoroethyl)-L-Tyrosineの準備については、Westerら(J. Nucl. Med. 1999;40:205-212)及びHamacherら(Appl. Radiat. Isot. 2002;57:853-856)がその製造方法を明らかにしている。
【0005】
しかしながら、彼らの方法は、何れも高速液体クロマトグラフィー(high performance liquid chromatography, HPLC)によって混合物を分離、精製する方法によっているため、扱い難いだけでなく作業を自動化することも困難である。このHPLC自体は製造設備のコストが嵩むのみでなく、このHPLCによる精製・分離過程においてオペレーターは高純度のO-(2-[18F]fluoroethyl)-L-Tyrosine製品品質を維持するため、反応槽からの精製物質の収集過程を精密にコントロールする必要があり、これらのタイミングを失すれば収率の低下や純度の劣化など品質の低下や生産性の低下を招くため、品質管理を徹底することが困難であるなどの問題がある。
【非特許文献1】H.J.Wester, M.Herz, W.Weber, P.Heiss, R.Senekowitsch-Schmidtke, M.Schwaiger and G.Stocklin, Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-Tyrosine for tumor imaging. J. Nucl. Med. 40, 205-212 (1999).
【非特許文献2】K.Hamacher and H.H.Coenen, Efficient routine production of the 18F-labelled amino acid O-(2-[18F]fluoroethyl)-L-Tyrosine, Appl. Radiat. Isot. 57, 853-856 (2002).
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、O-(2-[18F]fluoroethyl)-L-Tyrosineの合成化合物の前駆体である、t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl(化1)を用いた新しい合成法により、上記問題を解決する。なお、該前駆体の製造方法に関する技術は、別途出願済である(特願2005−301071)。
【課題を解決するための手段】
【0007】
O-(2-[18F]fluoroethyl)-L-Tyrosineを合成するため、最初に標識した合成物(つまり前駆体)を用意する。このステップにおいては放射線被爆に懸念する必要はないので、放射線遮蔽エリアで行う必要はなく、一般的な化学実験室で行えば良い。
【0008】
O-(2-[18F]fluoroethyl)-L-Tyrosineの前駆体であるt-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlの合成方法
(1)Ethylene glycol-1,2-ditosylateの合成
ethylene glycol及びtoluenesulfonyl chlorideをpyridine溶液に溶解して低温で二〜三日間程度反応させ、反応溶液を常温に戻して固化させて、最後に再結晶させて精製することによりethylene glycol-1,2-ditosylate精製品が得られた。
【0009】
(2)t-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlの合成
t-Boc-L-Tyr-Obzlをethylene glycol-1,2-ditosylate並びにpotassium carbonateに加えてacrylonitrile溶液に溶解して攪拌しながら90℃まで加熱し、約4時間反応させる。反応終了後、溶剤を除去してChloroformで抽出することにより固体残留物を得て、column chromatographyに通して純粋のt-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlが得られる。
【0010】
本発明の次の反応過程である、O-(2-[18F]fluoroethyl)-L-Tyrosineの合成方法においては放射線遮蔽エリアや同様の機能を備えた区画など、放射線遮蔽という条件下で行う必要がある。このO-(2-[18F]fluoroethyl)-L-Tyrosineの合成にも二種類の方法がある:
(1)ryptofix 2.2.2を触媒とする合成方法。
t-Boc-(O-tosyloxyethyl)-L-Tyr-ObzlをKryptofix 2.2.2を触媒として、
acrylonitrile溶液で18Fイオンと交換反応し、OTsを18Fで置換えて、t-Boc-(O-[18F]fluoroethyl)-L-Tyr-Obzlとする。続いて、column chromatographyで精製して蒸発・乾燥させた後、更にアルコールで生成物を溶解反応させた後、1Nの HCLを加えて、100℃の液温で加水分解して1N NaOHを加えて反応精製物を中和する。最後に等張力を調整して、0.22μmの無菌フィルターで濾過して精製品を得た。
【0011】
(2)etrabutylammonium bicarbonate(TBAHCO3)を触媒とする合成方法。
t-Boc-(O-tosyloxyethyl)-L-Tyr-ObzlをTBAHCO3を触媒として、acrylonitrile溶液で18Fイオンと交換反応し、OTsを18Fで置換えてt-Boc-(O-[18F]fluoroethyl)-L-Tyr-Obzlとする。続いて、column chromatographyで精製して蒸発・乾燥させた後、更にアルコールで生成物を溶解反応させた後、1N HCLを加えて、100℃の液温で加水分解して1N NaOHを加えて生成物を中和する。最後に等張力を調整して、0.22 μmの無菌フィルターで濾過して精製品を得た。
【非特許文献3】H.J.Wester, M.Herz, W.Weber, P.Heiss, R.Senekowitsch-Schmidtke, M.Schwaiger and G.Stocklin, Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-Tyrosine for tumor imaging. J. Nucl. Med. 40, 205-212 (1999).
【非特許文献4】K.Hamacher and H.H.Coenen, Efficient routine production of the 18F-labelled amino acid O-(2-[18F]fluoroethyl)-L-Tyrosine, Appl. Radiat. Isot. 57, 853-856 (2002).
【発明の効果】
【0012】
本発明において使用する出発材料となる前駆体の合成過程は反応が容易であり、精製過程においては樹脂とシリカゲルコラムで分離することができ、現在最も広く使用されている[18F]FDGの製造方法と同様であるため作業の自動化に適している。制御の困難なHPLCに依存する必要もないことから、合成設備のコストを引下げることができる。本発明の設備であれば、PET−CT センターの[18F]FDG合成の一般的な技術者にも操作容易である。
【0013】
本発明が核子医学の応用において有する利点は:(1)半減期が109分である18F核種を放射性トレーサーとして、11Cより大幅に核子センターの臨床使用への供給過程の信用性を高めるため、中枢となる医療施設から他の一般病院に対しても供給することができるようになること。(2)樹脂とシリカゲルコラムを利用する精製化手法は自動化に適しているため、合成過程の容易さと反復再現性を高めて時間を短縮できるだけでなく人力をも節約できること。(3)HPLCを使わないため、製造費や製造工程をも簡略化できる上、合成の収率と製品品質の品質管理の適合率を更に向上すること、が挙げられる。
【実施例】
【0014】
本発明の実施例について、以下に具体的に説明する:
実施例1:18Fの前駆体t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する方法。
1. Ethylene glycol-1,2-ditosylateの製造:
(1)toluenesulfonyl chloride (TsCl) 17g (F.W.=190.65,0.089 mol)を採り、pyridine 20 mLの入った三角フラスコ(A)に加える。
(2)ethylene glycol 1.1 mL (F.W.=62.07,0.018 mol)を採り、pyridine 30 mLの入った三角フラスコ(B)に加える。
(3)ドライアイス−アセトン溶液の温度で(約−30℃)、三角フラスコ(A)の溶液を(B)に加えて、直ちに三角フラスコを−18℃として、2〜3日間反応させる。
(4)反応が終了した後、砕いた氷を加えた水の入った500 mLのビーカーに、三角フラスコ(B)の反応物を淹れて攪拌すると白い固体が析出する。
(5)適量の1N HClを上記のビーカーに淹れて、PHを6〜7に調整する。
(6)濾過して白い固体を分離して、methylene chlorideとnormal hexaneの混合溶液の中に再び結晶化させて精製すると収率80%でethylene glycol-1,2-ditosylate5.33 gが得られた。
【化2】
ethylene glycol-1,2-ditosylateの合成反応式。
2. t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlの製造:
t-BOC-L-Tyr-Obzlを原料として、t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する反応過程は以下の化3の通り:
【化3】
t-BOC-L-Tyr-Obzlを原料としてt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する反応式。
(1)N-tert-butyloxycarbonyl- L-tyrosine benzylester (t-BOC-L-Tyr-OBzl) 450mg ( F.W.=361, 1.24 mmol )を容量50 mLの丸底フラスコ中のpotassium carbonate 20 mgとethylene glycol-1,2- ditosylate (F.W.=370.35,3.73 mmol) 1.384 gに加え、更に25 mLの anhydrous acetonitrileを加えて90℃で3.5時間ほど攪拌する。
(2)反応終了後に、回転蒸発器(Rota vapor)で溶剤を除去し、更にchloroformで抽出を行う (5mL × 3)。Chloroform抽出物に対し、負圧の下で溶剤を除去する。
(3)最小量のmethylene chlorideで残留物を溶解して、シリカゲルコラムでクロマトグラフィーを行う;移動相に関する初期条件は100%のCH2Cl2で、ethylene glycol-1,2-ditosylateが流出した後、移動相をCH2Cl2/CHCl3=1/1に変更すると未精製の反応生成物が溶離され、更に減圧下で乾燥させて固体状の未精製品が得られた。
(4)この未精製品を最小量のCH2Cl2:CHCl3=8/2の溶剤でシリカゲルコラムのクロマトグラフィーを行う。移動相に関する初期条件はCH2Cl2/CHCl3=8/2(0.1% triethyl amineを加える)で、油状の淡黄色物質(398 mg)の純粋な
N-tert-butyloxycarbonyl-(O-tosyloxyethyl)-L-tyrosine benzylester
(t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl)が溶離され、2塩化メチルとそれ自身のアルキル溶液中で再結晶して精製すると融点85〜86℃で、収率60.1%で白色の固体を得た。
(5)Nuclear Magnetic Resonance (NMR):t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl 20 mgを採り、0.6mL CDCl3に溶解して1H-NMR spectrum(化学式3)を測定した結果は以下のとおりであった。
1H NMR (CDCl3) δ7.80 (d, 2H, J=8.4 Hz, Haryl), 7.31 (m, 7H, Haryl), 6.89 (d, 2H, J=8.4 Hz, Haryl), 6.62 (d, 2H, J=8.4 Hz, Haryl), 5.15 (d, 1H, 12.2 Hz, CH of benzyl), 5.08 (d, 1H, 12.2 Hz, CH of benzyl), 4.92 (d, 1H, J=8.0Hz, NH), 4.54 (m, 1H, CH), 4.33 (t, 2H, J=4.6 Hz, CH2), 4.07 (t, 2H, J=4.6 Hz, CH2), 2.99 (d, 2H, J=5.8 Hz, CH2 of Tyr), 2.43 (s, 3H, CH3 of toluene), 1.39 (s, 9H, CH3 of t-BOC)。
(6)元素分析:t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlの分子式はC30H35NO8Sで、元素分析計算値はC, 63.27; H, 6.15; N, 2.46、実測値は:C, 63.34; H, 5.62; N, 2.33であった。
実施例2:Kryptofix 2.2.2を触媒としてO-(2-[18F]fluoroethyl)-L-Tyrosineを製造する方法。
【化4】
Kryptofix 222でL-[18F]FETを製造する方法における反応式
(1)[18F]HF溶液(0.5-1.3 mL、放射活性1-500 mCi)を5ml先細形状のフラスコに入れたpotassium carbonate (4.6 mg)とkryptofix 222 (26 mg)に加え、130℃で加熱しながら窒素ガス(200 mL/min)を吹き込み、減圧吸引してほぼ液面を乾燥状態に保つ。
(2)3 mLの anhydrous acetonitrileを8分以内に緩やかに注いで、同じく130℃で加熱しながら窒素ガス(200 mL/min)を吹き込み、減圧吸引して水分とアセトニトリルを共沸蒸留させて乾燥する。
(3)5 mgのt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを0.8 mLのanhydrous acetonitrileで溶かし、更に上述の反応をしているボトルの中に加えて、110℃の温度を保って10分間程度反応させる。
(4) 反応終了後、温度110℃に維持した状態で、細い排気針を挿入して窒素(200 mL/min)をボトルの中が乾燥するまで吹込む(acetonitrileを取除くにはおよそ5分間を必要とする)。
(5)反応ボトルを室温まで冷却して1.5 ml のCHCl3を加えて、反応混合物を溶解し、放射能を決定する。溶液の一部分を取って薄層クロマトグラフを行う(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2);上記の溶液をpre-conditioned silica columnに通させて、低圧の窒素ガスで流速を上げる;1.5 mLの CHCl3で反応ボトルを洗浄して同じシリカゲルに通して流出した液体を廃棄瓶で収集する;また2.5 mLのetherをシリカゲルの溶離に使用して、キャップ付きの試験管で流出したether solutionを収集すると保護基のある生成物が得られる。収集した液体の一部で薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、収集した液体やシリカコラムや反応ボトルの放射能を測定する。
(6)キャップ付きの収集管を40℃の水浴で、窒素(200 mL/min)を吹込んで乾燥するまでゆっくり内部の気体を吸引する。固形物が0.3 mLのethanolに溶解後、0.3 mL 1Nのhydrochlorideを加えて、heating block(100℃)に載置して10分程度加水分解反応を行う。
(7)反応終了後、反応ボトルを取り出して0.35 mL 1Nの水酸化ナトリウムの水溶液で生成物を中和し、更に1.35 mLの純水を加えて体積を2mLまで引上げ、等張力の溶液にして室温まで冷却する。
(8)反応生成物を0.22 μmの無菌フィルターで濾過して無菌瓶に入れ、無菌のPH中性の無支持体L-[18F]FETの水溶液が得られる。続いて濾過後の生成物の放射能を測定し(収率30-40%,decay corrected)、薄層クロマトグラフで(reverse C18 plate, acetonitrile /10 mM ammonium acetate=7/3)生成物の放射化学純度分析を行う(>90%)。
実施例3、tetrabutylammonium bicarbonate (TBA+HCO3-)を触媒にしてO-(2-[18F]fluoroethyl)-L-Tyrosineを製造する方法。
【化5】
TBAHCO3でL-[18F]FETを製造する方法における化学反応式
(1)[18F]HFの溶液0.5〜1.3 mLを以て放射能を測定する。蠕動ポンプで[18F]HF溶液をpreconditioned QMA Sep-pak (1 mL/min)に通して、流出した廃水を [18O]の回収瓶に捨てて後に、流出液及びQMA Sep-pakの放射能を測定する。
(2)0.8 mL のTBAHCO3/CANでQMA Sep-pak (1 mL/min)を溶離して、TBAHCO3の流出液を先鋭基部のボトルに収納する;続きにボトルを100℃で加熱しながら窒素ガス(200 mL/min)を吹き込んで、真空吸引して液面をほぼ乾燥する。
(3)8分内にゆっくり2 mLの anhydrous acetonitrileを加えながら、100℃のもとで加熱して窒素を吹き込んで僅かに真空吸引することで水分とアセトニトリルを共沸蒸留してほぼ乾燥する。
(4)5 mg のt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを0.8 mLのanhydrous acetonitrileに溶解して上記の反応ボトルにおいて、90℃の下で10分程度反応させる。
(5)反応終了後、90℃を維持したまま、細い排気した針を挿入して窒素(200 mL/min)をボトル中が乾燥するまで吹込む(acetonitrileを取除くにはおよそ5分必要とする)。
(6)反応ボトルを室温まで冷却させる。1.5mlのCHCl3を加えて、振騰して反応混合物を溶解して放射能を測定する。この溶液の一部分を取って薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、上記の溶液をpre-conditioned silica columnに通して、低圧の窒素で流速を上げる。そして1.5 mLの CHCl3で反応ボトルを洗浄して同じシリカコラムに通してその流出液を廃棄液ボトルに排出する。続いて2.5 mLのetherをシリカコラムの溶離に使用し、流出した保護基を有する生成物のether solutionをキャップ付きの試験管中に集する。その中の一部の収集した液を取り出して薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、収集した液とシリカコラムと反応ボトルの放射能を測定する。
(7)キャップ付きの収集管を40℃の水浴に置いて、窒素(200 mL/min)を吹込んで乾燥するまでゆっくり内部の気体を排出する。0.3ml のethanolで固形物を溶解した後、さらに0.3ml の1N hydrochlorideを加えてheating block(100℃)に載置して10分程度加水分解反応させる。
(8)反応終了後、反応ボトルを取り出して0.35 mL 1N水酸化ナトリウム溶液を加えて生成物を中和する、そして1.35 mLの純水を加えて体積が2 mLの等張力溶液として室温までに冷却する。
(9)生成物を0.22 μmの無菌フィルターに通して無菌瓶に入れ、無菌で等張力の無支持体[18F]FET水溶液が得られた。この濾過後の産物放射能を測定したり(収率40−50%,decay corrected)、薄層クロマトグラフ(reverse C18 plate, acetonitrile /10 mM ammonium acetate=7/3)を行ったりその放射化学純度を測る。
実施例4. F98 glioma腫瘍細胞によるL-[18F]FETの評価。
(1)五つの6 wells plateの中に、三つのwellを1×106 のF98 glioma cells及び2 mL DMEM (1g glucose/L)に載置する。
(2)この実験には五つの実験時間点 (2、5、10、30分及び一時間),どの時間点とも一つの6 wells plateを使う。
(3)実験進行中に、全てのwellに固定放射活性のL-[18F]FET或[18F]FDG (10 μCi/well)を加える。そして細胞が摂取した放射活性を加えた放射活性で割算すると細胞摂取比率が得られる。
(4)L-[18F]FETに対してF98 glioma cellsの細胞摂取実験結果は化4に示したものとなった。F98 glioma cellsの L-[18F]FETに対する摂取は素早くて10分後で最大限蓄積量に至る(摂取比率0.47 %)。そしてL-[18F]FETを加えた10分後に、細胞はL-[18F]FETを代謝できないため、L-[18F]FETの細胞を出入りする速度がバランスした。更に一時間経過すると、L-[18F]FETが腫瘍細胞に蓄積量は僅かに下がっていた。(摂取比率は0.41 %)
実施例5. F98 gliomaのFischer 344 ratにおけるL-[18F]FETの生物分布。
(1)Fischer 344 male ratsを20匹用意してL-[18F]FETの生体内における生物分布評価に使う。この20匹のネズミが生後8〜10週間位に成長すると、脳部にあるbregmaとsagittal中線との交差点を原点として、左向き3mm並びに上向き5mmで深度5mmの周辺でF98 glioma cells (1 × 105cells /10 (L)を移植する。この脳腫瘍が植え付けられた11〜12日後に生物分布実験を開始する。
(2)異なった放射活性濃度の計算によってFischer 344 ratごとに体積は200〜300 μLで、活性は200〜250 μCiのL-[18F]FETを注射する。
(3)L-[18F]のFET薬物を尾静脈に注射してから計時して、15、30、60、90及び120分後ごとにネズミを犠牲する。
(4)ネズミが犠牲した直後ですぐ解剖する。まず生殖孔から上向きに顎まで胸部と腹部の毛皮を縦切る、続きに剣状突起の上方部の胸部を横切って肋骨中央部から垂直に上向きに剪んで心臓と肺臓を露出させる。肋骨を開いて0.5 mLの注射器で静脈洞から血液サンプルを抽出して、血液が消耗され尽くした後に注射針を引き出し、心臓と肺葉をも摘んで採集する。
(5)生殖孔から剣状突起の下部にかけて腹部の筋肉を縦に上向きに剪む。更に切口の上下際を横切って、筋肉層を左右に引っ繰り返して腹腔の器官を露出させる。それから下腹部で膀胱を探し、続いて胃・肝臓・脾臓・膵臓・小腸・大腸・腎臓・筋肉を順番に取って剪み切る。
(6)ネズミを裏返して、肩に毛皮がついている箇所を切り開いて皮下の腫瘍を露出させると剪み切る。それと大後頭孔上部の頭蓋下縁に沿って頭皮を上向きに剥がして頭蓋骨を現して、その大後頭孔で首をも横切って脊髄を現す。ハサミの尖った先を切り開かれた大後頭孔に刺し込んで頭蓋の内面に当てることで頭蓋腔の前縁に達して中央部から頭骨を剪み開き。頭骨が切り開かれた後、ハサミで頭骨を左右に引き開けて大小脳を露出させ、ハサミを頭蓋骨の底部に沿って前端に届く左脳・右脳・脳腫瘍をともに切り取る。
(7)全ての器官見本の重さを測った後に、γ-counterを放射活性測定に利用して各器官の%ID/g (% injection dose/g organ)に換算することでL-[18F]FETの生物分布の見積もりに用いる。生物分布実験からして見ればL-[18F]FETは差異あり持続時間を経てると、各組織のradiotracerに対する蓄積能力、並びに%ID/g (percent injected dose per gram of tissue)で表示する放射活性の蓄積量を共に観察できる。化学式1及び図2で示したような結果となる。文献によるとL-[18F]FETは非天然アミノ酸にも拘らず、system L amino acid transporterに依れば血液脳関門を通して脳部に入ることが出来る。それで本発明のL-[18F]FETの生物分布結果によると、L-[18F]FETの脳腫瘍と正常脳における蓄積値は90分辺りで最大限、1.49と0.48 %ID/gに至る。因みに脳腫瘍と正常脳における放射活性比(tumor-to-normal brain ratio)について、L-[18F]FET が15、30、60、90及び120分に当る数値は1.54、1.74、3.16、3.14及び2.34となっていた。この腫瘍の以外でも、L-[18F]FETはネズミの膵臓にも相当な蓄積量を示した(15、30、60、90及び120分に当る数値は0.98、1.76、2.55、2.28及び2.24 %ID/gである)。これは膵臓には各種の酵素とホルモンを合成するため大量なアミノ酸が必要とするため、L-[18F]FETの放射活性を増加させてしまった。ところで他の文献にも見られるのは、L-[18F]FETが人体の臨床実験に使われた時、PETには腫瘍患者の膵臓の活性蓄積は見かけないので、恐らく人間とネズミの代謝差異によって異なった結果を生じたと考えられる。またL-[18F]FETの生物分布結果によると、L-[18F]FETを注射してから二時間以内に腫瘍が蓄積した放射活性(%ID/gで表示する)は明らかに膵臓以外の他の正常器官(左脳、小腸、腎臓、肝臓、血液及び筋肉など)よりも高くなった。この結果によるとL-[18F]FETの他部位にての腫瘍ポジトロンCT(PET)の診断剤として使える可能性を示している。因みにL-[18F]FETは尿液により体外に排泄するので、尿液にはかなり高レベルの放射活性が検出される。
【表1】
実施例6: 脳腫瘍を移植したネズミで示されL-[18F]FETのポジトロンCT(PET)映像。
(1)脳腫瘍(F98 glioma)を移植した生後11〜12日後からL-[18F]FETの脳腫瘍ネズミ (Fischer 344 rats) におけるPETを進行する。
(2)L-[18F]FETの作り方は上記通り、放射濃度の割合によって異なった用量の薬物を注射する。
(3)最初に400 μCiのL-[18F]FETを尾静脈に注射してから計時する。0.5、1、1.5及び2時間の辺りにネズミをエーテルで麻酔して手足を伸張し透明なアクリルの板に固定した後にPETテーブルの上に配置する。そしてisofluraneの気体麻酔剤で持続的にネズミに麻酔をかけたり、頭部の脳に腫瘍が付いてる部分をPET のレーザーに合わせたり、レントゲン写真術のスタート位置を確実に固定する。
(4)PET をする前にblank scanを一回にして背景値を調整とする。
(5)Ga-68放射線源を利用してtransmission scanで組織衰弱の校正をする。
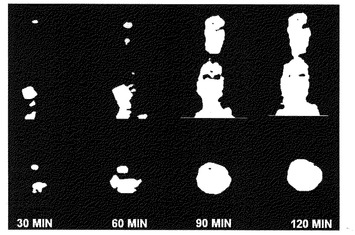
(6)PET及びmicroPETの進行を開始する。スキャナーによる映像資料を再編してから映像処理を行った冠状部位並びに横断面は図3と図4に示した。L-[18F]FETを注射したネズミの脳腫瘍の部位における放射活性の蓄積活動は明らかとなり、しかも注射してから90分の辺りに最大摂取量に至る。この脳腫瘍の部位と対照的に、正常の脳組織の必要するアミノ酸は割に少ないため、この部位におけるL-[18F]FETの放射活性の蓄積も相対的に少なくなった。L-[18F]FETのPET映像は脳腫瘍の位置を明確的に示している、これも生物分布実験結果に示したL-[18F]FETは腫瘍及び、腫瘍/脳の正常組織摂取量比率にはかなり高い蓄積量を持っているが、膵臓以外の正常器官には明らかな蓄積した様子はないことと一致している。
【図面の簡単な説明】
【0015】
【図1】L-[18F]FETに対してF98 glioma cellsが相異時間点で有していた摂取量。
【図2】ネズミにL-[18F]FETを注射してから、各器官が相異時間点で蓄積した放射活 性。
【図3】左脳にF98 rat gliomaを移植されたFischer344ネズミについてのmicroPET画像診断。それぞれL-[18F]FETを注射してから30、60、90、120分辺りの画像であって、上行は冠状断面で下行は横断面である。
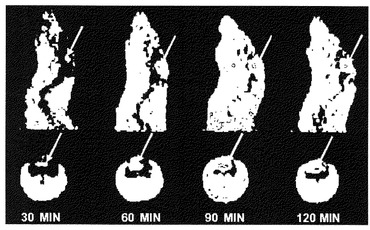
【図4】左脳にF98 rat glioma植え付けられたFischer344ネズミについてのPET画像診断。それぞれにはL-[18F]FETを注射してから30、60、90、120分辺りの画像であって、上行は冠状断面で下行は横断面である。
【技術分野】
【0001】
18Fで標識したアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineは、一種のアミノ酸トレーサー(追跡剤)で、人体実験によると腫瘍のポジトロンCT(PET)法診断に好適に適用することができる。
【背景技術】
【0002】
2-[18F]Fluoro-2-Deoxy-D-glucose([18F]FDG)は現在最も広く用いられているPET薬剤で、ブドウ糖により代謝するその生化学的な経路は腫瘍の診断や治療に対する評価に利用されている。しかし、 [18F]FDGの脳内での高度吸収量は脳のバックグランド放射線を増加して脳腫瘍の診断を困難にさせるだけでなく、炎症部位に関しても誤診され易い。一方、L-[methyl-11C]も脳腫瘍細胞に集中することができ、また上記のような高度吸収量による診断困難や誤診なども一切起こらない。しかしながら、このL-[methyl-11C]は、病院でサイクロトロンを備えていても半減期が20分にすぎないため、医療現場に至る途中での減衰が著しく、これらの欠点のためコストが高いものとなる。
【0003】
18Fで標識したアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineは、人体で吸収されるとアミノ酸により代謝する経路は大概C−11でラベルしたL-[methyl-11C] methionineと変わらず、[18F]FDGの脳内や炎症部位での高度吸収量のような影響で診断困難や誤診に至ることもない。[18F]FETの半減期は[18F]FDGと同じく109分間であるから病院間などの医療現場に至る間での所要時間からみて有利であって、C−11で標識したL-[methyl-11C] methionineよりもコストが低くできるという点は経済的である。
【0004】
O-(2-[18F]fluoroethyl)-L-Tyrosineの準備については、Westerら(J. Nucl. Med. 1999;40:205-212)及びHamacherら(Appl. Radiat. Isot. 2002;57:853-856)がその製造方法を明らかにしている。
【0005】
しかしながら、彼らの方法は、何れも高速液体クロマトグラフィー(high performance liquid chromatography, HPLC)によって混合物を分離、精製する方法によっているため、扱い難いだけでなく作業を自動化することも困難である。このHPLC自体は製造設備のコストが嵩むのみでなく、このHPLCによる精製・分離過程においてオペレーターは高純度のO-(2-[18F]fluoroethyl)-L-Tyrosine製品品質を維持するため、反応槽からの精製物質の収集過程を精密にコントロールする必要があり、これらのタイミングを失すれば収率の低下や純度の劣化など品質の低下や生産性の低下を招くため、品質管理を徹底することが困難であるなどの問題がある。
【非特許文献1】H.J.Wester, M.Herz, W.Weber, P.Heiss, R.Senekowitsch-Schmidtke, M.Schwaiger and G.Stocklin, Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-Tyrosine for tumor imaging. J. Nucl. Med. 40, 205-212 (1999).
【非特許文献2】K.Hamacher and H.H.Coenen, Efficient routine production of the 18F-labelled amino acid O-(2-[18F]fluoroethyl)-L-Tyrosine, Appl. Radiat. Isot. 57, 853-856 (2002).
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、O-(2-[18F]fluoroethyl)-L-Tyrosineの合成化合物の前駆体である、t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl(化1)を用いた新しい合成法により、上記問題を解決する。なお、該前駆体の製造方法に関する技術は、別途出願済である(特願2005−301071)。
【課題を解決するための手段】
【0007】
O-(2-[18F]fluoroethyl)-L-Tyrosineを合成するため、最初に標識した合成物(つまり前駆体)を用意する。このステップにおいては放射線被爆に懸念する必要はないので、放射線遮蔽エリアで行う必要はなく、一般的な化学実験室で行えば良い。
【0008】
O-(2-[18F]fluoroethyl)-L-Tyrosineの前駆体であるt-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlの合成方法
(1)Ethylene glycol-1,2-ditosylateの合成
ethylene glycol及びtoluenesulfonyl chlorideをpyridine溶液に溶解して低温で二〜三日間程度反応させ、反応溶液を常温に戻して固化させて、最後に再結晶させて精製することによりethylene glycol-1,2-ditosylate精製品が得られた。
【0009】
(2)t-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlの合成
t-Boc-L-Tyr-Obzlをethylene glycol-1,2-ditosylate並びにpotassium carbonateに加えてacrylonitrile溶液に溶解して攪拌しながら90℃まで加熱し、約4時間反応させる。反応終了後、溶剤を除去してChloroformで抽出することにより固体残留物を得て、column chromatographyに通して純粋のt-Boc-(O-tosyloxyethyl)-L-Tyr-Obzlが得られる。
【0010】
本発明の次の反応過程である、O-(2-[18F]fluoroethyl)-L-Tyrosineの合成方法においては放射線遮蔽エリアや同様の機能を備えた区画など、放射線遮蔽という条件下で行う必要がある。このO-(2-[18F]fluoroethyl)-L-Tyrosineの合成にも二種類の方法がある:
(1)ryptofix 2.2.2を触媒とする合成方法。
t-Boc-(O-tosyloxyethyl)-L-Tyr-ObzlをKryptofix 2.2.2を触媒として、
acrylonitrile溶液で18Fイオンと交換反応し、OTsを18Fで置換えて、t-Boc-(O-[18F]fluoroethyl)-L-Tyr-Obzlとする。続いて、column chromatographyで精製して蒸発・乾燥させた後、更にアルコールで生成物を溶解反応させた後、1Nの HCLを加えて、100℃の液温で加水分解して1N NaOHを加えて反応精製物を中和する。最後に等張力を調整して、0.22μmの無菌フィルターで濾過して精製品を得た。
【0011】
(2)etrabutylammonium bicarbonate(TBAHCO3)を触媒とする合成方法。
t-Boc-(O-tosyloxyethyl)-L-Tyr-ObzlをTBAHCO3を触媒として、acrylonitrile溶液で18Fイオンと交換反応し、OTsを18Fで置換えてt-Boc-(O-[18F]fluoroethyl)-L-Tyr-Obzlとする。続いて、column chromatographyで精製して蒸発・乾燥させた後、更にアルコールで生成物を溶解反応させた後、1N HCLを加えて、100℃の液温で加水分解して1N NaOHを加えて生成物を中和する。最後に等張力を調整して、0.22 μmの無菌フィルターで濾過して精製品を得た。
【非特許文献3】H.J.Wester, M.Herz, W.Weber, P.Heiss, R.Senekowitsch-Schmidtke, M.Schwaiger and G.Stocklin, Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-Tyrosine for tumor imaging. J. Nucl. Med. 40, 205-212 (1999).
【非特許文献4】K.Hamacher and H.H.Coenen, Efficient routine production of the 18F-labelled amino acid O-(2-[18F]fluoroethyl)-L-Tyrosine, Appl. Radiat. Isot. 57, 853-856 (2002).
【発明の効果】
【0012】
本発明において使用する出発材料となる前駆体の合成過程は反応が容易であり、精製過程においては樹脂とシリカゲルコラムで分離することができ、現在最も広く使用されている[18F]FDGの製造方法と同様であるため作業の自動化に適している。制御の困難なHPLCに依存する必要もないことから、合成設備のコストを引下げることができる。本発明の設備であれば、PET−CT センターの[18F]FDG合成の一般的な技術者にも操作容易である。
【0013】
本発明が核子医学の応用において有する利点は:(1)半減期が109分である18F核種を放射性トレーサーとして、11Cより大幅に核子センターの臨床使用への供給過程の信用性を高めるため、中枢となる医療施設から他の一般病院に対しても供給することができるようになること。(2)樹脂とシリカゲルコラムを利用する精製化手法は自動化に適しているため、合成過程の容易さと反復再現性を高めて時間を短縮できるだけでなく人力をも節約できること。(3)HPLCを使わないため、製造費や製造工程をも簡略化できる上、合成の収率と製品品質の品質管理の適合率を更に向上すること、が挙げられる。
【実施例】
【0014】
本発明の実施例について、以下に具体的に説明する:
実施例1:18Fの前駆体t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する方法。
1. Ethylene glycol-1,2-ditosylateの製造:
(1)toluenesulfonyl chloride (TsCl) 17g (F.W.=190.65,0.089 mol)を採り、pyridine 20 mLの入った三角フラスコ(A)に加える。
(2)ethylene glycol 1.1 mL (F.W.=62.07,0.018 mol)を採り、pyridine 30 mLの入った三角フラスコ(B)に加える。
(3)ドライアイス−アセトン溶液の温度で(約−30℃)、三角フラスコ(A)の溶液を(B)に加えて、直ちに三角フラスコを−18℃として、2〜3日間反応させる。
(4)反応が終了した後、砕いた氷を加えた水の入った500 mLのビーカーに、三角フラスコ(B)の反応物を淹れて攪拌すると白い固体が析出する。
(5)適量の1N HClを上記のビーカーに淹れて、PHを6〜7に調整する。
(6)濾過して白い固体を分離して、methylene chlorideとnormal hexaneの混合溶液の中に再び結晶化させて精製すると収率80%でethylene glycol-1,2-ditosylate5.33 gが得られた。
【化2】
ethylene glycol-1,2-ditosylateの合成反応式。
2. t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlの製造:
t-BOC-L-Tyr-Obzlを原料として、t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する反応過程は以下の化3の通り:
【化3】
t-BOC-L-Tyr-Obzlを原料としてt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを合成する反応式。
(1)N-tert-butyloxycarbonyl- L-tyrosine benzylester (t-BOC-L-Tyr-OBzl) 450mg ( F.W.=361, 1.24 mmol )を容量50 mLの丸底フラスコ中のpotassium carbonate 20 mgとethylene glycol-1,2- ditosylate (F.W.=370.35,3.73 mmol) 1.384 gに加え、更に25 mLの anhydrous acetonitrileを加えて90℃で3.5時間ほど攪拌する。
(2)反応終了後に、回転蒸発器(Rota vapor)で溶剤を除去し、更にchloroformで抽出を行う (5mL × 3)。Chloroform抽出物に対し、負圧の下で溶剤を除去する。
(3)最小量のmethylene chlorideで残留物を溶解して、シリカゲルコラムでクロマトグラフィーを行う;移動相に関する初期条件は100%のCH2Cl2で、ethylene glycol-1,2-ditosylateが流出した後、移動相をCH2Cl2/CHCl3=1/1に変更すると未精製の反応生成物が溶離され、更に減圧下で乾燥させて固体状の未精製品が得られた。
(4)この未精製品を最小量のCH2Cl2:CHCl3=8/2の溶剤でシリカゲルコラムのクロマトグラフィーを行う。移動相に関する初期条件はCH2Cl2/CHCl3=8/2(0.1% triethyl amineを加える)で、油状の淡黄色物質(398 mg)の純粋な
N-tert-butyloxycarbonyl-(O-tosyloxyethyl)-L-tyrosine benzylester
(t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl)が溶離され、2塩化メチルとそれ自身のアルキル溶液中で再結晶して精製すると融点85〜86℃で、収率60.1%で白色の固体を得た。
(5)Nuclear Magnetic Resonance (NMR):t-BOC-(O-tosyloxyethyl)-L-Tyr-OBzl 20 mgを採り、0.6mL CDCl3に溶解して1H-NMR spectrum(化学式3)を測定した結果は以下のとおりであった。
1H NMR (CDCl3) δ7.80 (d, 2H, J=8.4 Hz, Haryl), 7.31 (m, 7H, Haryl), 6.89 (d, 2H, J=8.4 Hz, Haryl), 6.62 (d, 2H, J=8.4 Hz, Haryl), 5.15 (d, 1H, 12.2 Hz, CH of benzyl), 5.08 (d, 1H, 12.2 Hz, CH of benzyl), 4.92 (d, 1H, J=8.0Hz, NH), 4.54 (m, 1H, CH), 4.33 (t, 2H, J=4.6 Hz, CH2), 4.07 (t, 2H, J=4.6 Hz, CH2), 2.99 (d, 2H, J=5.8 Hz, CH2 of Tyr), 2.43 (s, 3H, CH3 of toluene), 1.39 (s, 9H, CH3 of t-BOC)。
(6)元素分析:t-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlの分子式はC30H35NO8Sで、元素分析計算値はC, 63.27; H, 6.15; N, 2.46、実測値は:C, 63.34; H, 5.62; N, 2.33であった。
実施例2:Kryptofix 2.2.2を触媒としてO-(2-[18F]fluoroethyl)-L-Tyrosineを製造する方法。
【化4】
Kryptofix 222でL-[18F]FETを製造する方法における反応式
(1)[18F]HF溶液(0.5-1.3 mL、放射活性1-500 mCi)を5ml先細形状のフラスコに入れたpotassium carbonate (4.6 mg)とkryptofix 222 (26 mg)に加え、130℃で加熱しながら窒素ガス(200 mL/min)を吹き込み、減圧吸引してほぼ液面を乾燥状態に保つ。
(2)3 mLの anhydrous acetonitrileを8分以内に緩やかに注いで、同じく130℃で加熱しながら窒素ガス(200 mL/min)を吹き込み、減圧吸引して水分とアセトニトリルを共沸蒸留させて乾燥する。
(3)5 mgのt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを0.8 mLのanhydrous acetonitrileで溶かし、更に上述の反応をしているボトルの中に加えて、110℃の温度を保って10分間程度反応させる。
(4) 反応終了後、温度110℃に維持した状態で、細い排気針を挿入して窒素(200 mL/min)をボトルの中が乾燥するまで吹込む(acetonitrileを取除くにはおよそ5分間を必要とする)。
(5)反応ボトルを室温まで冷却して1.5 ml のCHCl3を加えて、反応混合物を溶解し、放射能を決定する。溶液の一部分を取って薄層クロマトグラフを行う(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2);上記の溶液をpre-conditioned silica columnに通させて、低圧の窒素ガスで流速を上げる;1.5 mLの CHCl3で反応ボトルを洗浄して同じシリカゲルに通して流出した液体を廃棄瓶で収集する;また2.5 mLのetherをシリカゲルの溶離に使用して、キャップ付きの試験管で流出したether solutionを収集すると保護基のある生成物が得られる。収集した液体の一部で薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、収集した液体やシリカコラムや反応ボトルの放射能を測定する。
(6)キャップ付きの収集管を40℃の水浴で、窒素(200 mL/min)を吹込んで乾燥するまでゆっくり内部の気体を吸引する。固形物が0.3 mLのethanolに溶解後、0.3 mL 1Nのhydrochlorideを加えて、heating block(100℃)に載置して10分程度加水分解反応を行う。
(7)反応終了後、反応ボトルを取り出して0.35 mL 1Nの水酸化ナトリウムの水溶液で生成物を中和し、更に1.35 mLの純水を加えて体積を2mLまで引上げ、等張力の溶液にして室温まで冷却する。
(8)反応生成物を0.22 μmの無菌フィルターで濾過して無菌瓶に入れ、無菌のPH中性の無支持体L-[18F]FETの水溶液が得られる。続いて濾過後の生成物の放射能を測定し(収率30-40%,decay corrected)、薄層クロマトグラフで(reverse C18 plate, acetonitrile /10 mM ammonium acetate=7/3)生成物の放射化学純度分析を行う(>90%)。
実施例3、tetrabutylammonium bicarbonate (TBA+HCO3-)を触媒にしてO-(2-[18F]fluoroethyl)-L-Tyrosineを製造する方法。
【化5】
TBAHCO3でL-[18F]FETを製造する方法における化学反応式
(1)[18F]HFの溶液0.5〜1.3 mLを以て放射能を測定する。蠕動ポンプで[18F]HF溶液をpreconditioned QMA Sep-pak (1 mL/min)に通して、流出した廃水を [18O]の回収瓶に捨てて後に、流出液及びQMA Sep-pakの放射能を測定する。
(2)0.8 mL のTBAHCO3/CANでQMA Sep-pak (1 mL/min)を溶離して、TBAHCO3の流出液を先鋭基部のボトルに収納する;続きにボトルを100℃で加熱しながら窒素ガス(200 mL/min)を吹き込んで、真空吸引して液面をほぼ乾燥する。
(3)8分内にゆっくり2 mLの anhydrous acetonitrileを加えながら、100℃のもとで加熱して窒素を吹き込んで僅かに真空吸引することで水分とアセトニトリルを共沸蒸留してほぼ乾燥する。
(4)5 mg のt-BOC-(O-tosyloxyethyl)-L-Tyr-Obzlを0.8 mLのanhydrous acetonitrileに溶解して上記の反応ボトルにおいて、90℃の下で10分程度反応させる。
(5)反応終了後、90℃を維持したまま、細い排気した針を挿入して窒素(200 mL/min)をボトル中が乾燥するまで吹込む(acetonitrileを取除くにはおよそ5分必要とする)。
(6)反応ボトルを室温まで冷却させる。1.5mlのCHCl3を加えて、振騰して反応混合物を溶解して放射能を測定する。この溶液の一部分を取って薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、上記の溶液をpre-conditioned silica columnに通して、低圧の窒素で流速を上げる。そして1.5 mLの CHCl3で反応ボトルを洗浄して同じシリカコラムに通してその流出液を廃棄液ボトルに排出する。続いて2.5 mLのetherをシリカコラムの溶離に使用し、流出した保護基を有する生成物のether solutionをキャップ付きの試験管中に集する。その中の一部の収集した液を取り出して薄層クロマトグラフを行って(silica gel plate, 現像剤CH2Cl2/CHCl3=8/2)、収集した液とシリカコラムと反応ボトルの放射能を測定する。
(7)キャップ付きの収集管を40℃の水浴に置いて、窒素(200 mL/min)を吹込んで乾燥するまでゆっくり内部の気体を排出する。0.3ml のethanolで固形物を溶解した後、さらに0.3ml の1N hydrochlorideを加えてheating block(100℃)に載置して10分程度加水分解反応させる。
(8)反応終了後、反応ボトルを取り出して0.35 mL 1N水酸化ナトリウム溶液を加えて生成物を中和する、そして1.35 mLの純水を加えて体積が2 mLの等張力溶液として室温までに冷却する。
(9)生成物を0.22 μmの無菌フィルターに通して無菌瓶に入れ、無菌で等張力の無支持体[18F]FET水溶液が得られた。この濾過後の産物放射能を測定したり(収率40−50%,decay corrected)、薄層クロマトグラフ(reverse C18 plate, acetonitrile /10 mM ammonium acetate=7/3)を行ったりその放射化学純度を測る。
実施例4. F98 glioma腫瘍細胞によるL-[18F]FETの評価。
(1)五つの6 wells plateの中に、三つのwellを1×106 のF98 glioma cells及び2 mL DMEM (1g glucose/L)に載置する。
(2)この実験には五つの実験時間点 (2、5、10、30分及び一時間),どの時間点とも一つの6 wells plateを使う。
(3)実験進行中に、全てのwellに固定放射活性のL-[18F]FET或[18F]FDG (10 μCi/well)を加える。そして細胞が摂取した放射活性を加えた放射活性で割算すると細胞摂取比率が得られる。
(4)L-[18F]FETに対してF98 glioma cellsの細胞摂取実験結果は化4に示したものとなった。F98 glioma cellsの L-[18F]FETに対する摂取は素早くて10分後で最大限蓄積量に至る(摂取比率0.47 %)。そしてL-[18F]FETを加えた10分後に、細胞はL-[18F]FETを代謝できないため、L-[18F]FETの細胞を出入りする速度がバランスした。更に一時間経過すると、L-[18F]FETが腫瘍細胞に蓄積量は僅かに下がっていた。(摂取比率は0.41 %)
実施例5. F98 gliomaのFischer 344 ratにおけるL-[18F]FETの生物分布。
(1)Fischer 344 male ratsを20匹用意してL-[18F]FETの生体内における生物分布評価に使う。この20匹のネズミが生後8〜10週間位に成長すると、脳部にあるbregmaとsagittal中線との交差点を原点として、左向き3mm並びに上向き5mmで深度5mmの周辺でF98 glioma cells (1 × 105cells /10 (L)を移植する。この脳腫瘍が植え付けられた11〜12日後に生物分布実験を開始する。
(2)異なった放射活性濃度の計算によってFischer 344 ratごとに体積は200〜300 μLで、活性は200〜250 μCiのL-[18F]FETを注射する。
(3)L-[18F]のFET薬物を尾静脈に注射してから計時して、15、30、60、90及び120分後ごとにネズミを犠牲する。
(4)ネズミが犠牲した直後ですぐ解剖する。まず生殖孔から上向きに顎まで胸部と腹部の毛皮を縦切る、続きに剣状突起の上方部の胸部を横切って肋骨中央部から垂直に上向きに剪んで心臓と肺臓を露出させる。肋骨を開いて0.5 mLの注射器で静脈洞から血液サンプルを抽出して、血液が消耗され尽くした後に注射針を引き出し、心臓と肺葉をも摘んで採集する。
(5)生殖孔から剣状突起の下部にかけて腹部の筋肉を縦に上向きに剪む。更に切口の上下際を横切って、筋肉層を左右に引っ繰り返して腹腔の器官を露出させる。それから下腹部で膀胱を探し、続いて胃・肝臓・脾臓・膵臓・小腸・大腸・腎臓・筋肉を順番に取って剪み切る。
(6)ネズミを裏返して、肩に毛皮がついている箇所を切り開いて皮下の腫瘍を露出させると剪み切る。それと大後頭孔上部の頭蓋下縁に沿って頭皮を上向きに剥がして頭蓋骨を現して、その大後頭孔で首をも横切って脊髄を現す。ハサミの尖った先を切り開かれた大後頭孔に刺し込んで頭蓋の内面に当てることで頭蓋腔の前縁に達して中央部から頭骨を剪み開き。頭骨が切り開かれた後、ハサミで頭骨を左右に引き開けて大小脳を露出させ、ハサミを頭蓋骨の底部に沿って前端に届く左脳・右脳・脳腫瘍をともに切り取る。
(7)全ての器官見本の重さを測った後に、γ-counterを放射活性測定に利用して各器官の%ID/g (% injection dose/g organ)に換算することでL-[18F]FETの生物分布の見積もりに用いる。生物分布実験からして見ればL-[18F]FETは差異あり持続時間を経てると、各組織のradiotracerに対する蓄積能力、並びに%ID/g (percent injected dose per gram of tissue)で表示する放射活性の蓄積量を共に観察できる。化学式1及び図2で示したような結果となる。文献によるとL-[18F]FETは非天然アミノ酸にも拘らず、system L amino acid transporterに依れば血液脳関門を通して脳部に入ることが出来る。それで本発明のL-[18F]FETの生物分布結果によると、L-[18F]FETの脳腫瘍と正常脳における蓄積値は90分辺りで最大限、1.49と0.48 %ID/gに至る。因みに脳腫瘍と正常脳における放射活性比(tumor-to-normal brain ratio)について、L-[18F]FET が15、30、60、90及び120分に当る数値は1.54、1.74、3.16、3.14及び2.34となっていた。この腫瘍の以外でも、L-[18F]FETはネズミの膵臓にも相当な蓄積量を示した(15、30、60、90及び120分に当る数値は0.98、1.76、2.55、2.28及び2.24 %ID/gである)。これは膵臓には各種の酵素とホルモンを合成するため大量なアミノ酸が必要とするため、L-[18F]FETの放射活性を増加させてしまった。ところで他の文献にも見られるのは、L-[18F]FETが人体の臨床実験に使われた時、PETには腫瘍患者の膵臓の活性蓄積は見かけないので、恐らく人間とネズミの代謝差異によって異なった結果を生じたと考えられる。またL-[18F]FETの生物分布結果によると、L-[18F]FETを注射してから二時間以内に腫瘍が蓄積した放射活性(%ID/gで表示する)は明らかに膵臓以外の他の正常器官(左脳、小腸、腎臓、肝臓、血液及び筋肉など)よりも高くなった。この結果によるとL-[18F]FETの他部位にての腫瘍ポジトロンCT(PET)の診断剤として使える可能性を示している。因みにL-[18F]FETは尿液により体外に排泄するので、尿液にはかなり高レベルの放射活性が検出される。
【表1】
実施例6: 脳腫瘍を移植したネズミで示されL-[18F]FETのポジトロンCT(PET)映像。
(1)脳腫瘍(F98 glioma)を移植した生後11〜12日後からL-[18F]FETの脳腫瘍ネズミ (Fischer 344 rats) におけるPETを進行する。
(2)L-[18F]FETの作り方は上記通り、放射濃度の割合によって異なった用量の薬物を注射する。
(3)最初に400 μCiのL-[18F]FETを尾静脈に注射してから計時する。0.5、1、1.5及び2時間の辺りにネズミをエーテルで麻酔して手足を伸張し透明なアクリルの板に固定した後にPETテーブルの上に配置する。そしてisofluraneの気体麻酔剤で持続的にネズミに麻酔をかけたり、頭部の脳に腫瘍が付いてる部分をPET のレーザーに合わせたり、レントゲン写真術のスタート位置を確実に固定する。
(4)PET をする前にblank scanを一回にして背景値を調整とする。
(5)Ga-68放射線源を利用してtransmission scanで組織衰弱の校正をする。
(6)PET及びmicroPETの進行を開始する。スキャナーによる映像資料を再編してから映像処理を行った冠状部位並びに横断面は図3と図4に示した。L-[18F]FETを注射したネズミの脳腫瘍の部位における放射活性の蓄積活動は明らかとなり、しかも注射してから90分の辺りに最大摂取量に至る。この脳腫瘍の部位と対照的に、正常の脳組織の必要するアミノ酸は割に少ないため、この部位におけるL-[18F]FETの放射活性の蓄積も相対的に少なくなった。L-[18F]FETのPET映像は脳腫瘍の位置を明確的に示している、これも生物分布実験結果に示したL-[18F]FETは腫瘍及び、腫瘍/脳の正常組織摂取量比率にはかなり高い蓄積量を持っているが、膵臓以外の正常器官には明らかな蓄積した様子はないことと一致している。
【図面の簡単な説明】
【0015】
【図1】L-[18F]FETに対してF98 glioma cellsが相異時間点で有していた摂取量。
【図2】ネズミにL-[18F]FETを注射してから、各器官が相異時間点で蓄積した放射活 性。
【図3】左脳にF98 rat gliomaを移植されたFischer344ネズミについてのmicroPET画像診断。それぞれL-[18F]FETを注射してから30、60、90、120分辺りの画像であって、上行は冠状断面で下行は横断面である。
【図4】左脳にF98 rat glioma植え付けられたFischer344ネズミについてのPET画像診断。それぞれにはL-[18F]FETを注射してから30、60、90、120分辺りの画像であって、上行は冠状断面で下行は横断面である。
【特許請求の範囲】
【請求項1】
化1の化学式の物質を前駆体として用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法において、
上記前駆体を原料として、Kryptofix 2.2.2を触媒として18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成する無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【化1】
R1カルボキシル基の保護基で、arylalkyl基であり;
R2はアミノ基の保護基で、carboxyl基であり;
R3は離脱基で、p-tosyloxy,methane sulfonyloxy基若しくはtrifluoromethanesulfonyloxy基又は臭素基である。
【請求項2】
化1の化学式の物質を前駆体として用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法において、
上記の前駆体を原料として、TBAHCO3を触媒として18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成する無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項3】
請求項1の製造方法において、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成後、シリカゲルコラムで精製することを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項4】
請求項1の製造方法において、触媒をTBAHCO3に替えて18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成することを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項5】
請求項1の製造方法により製造されたO-(2-[18F]fluoroethyl)-L-Tyrosineの注射液は、腫瘍の診断及び治療効果を追跡する核子医学レントゲン写真撮影法に用いられるものであることを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項6】
請求項2の製造方法により製造されたO-(2-[18F]fluoroethyl)-L-Tyrosineの注射液は、腫瘍の診断と治療効果を追跡する核子医学レントゲン写真撮影法に用いられるものであることを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項1】
化1の化学式の物質を前駆体として用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法において、
上記前駆体を原料として、Kryptofix 2.2.2を触媒として18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成する無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【化1】
R1カルボキシル基の保護基で、arylalkyl基であり;
R2はアミノ基の保護基で、carboxyl基であり;
R3は離脱基で、p-tosyloxy,methane sulfonyloxy基若しくはtrifluoromethanesulfonyloxy基又は臭素基である。
【請求項2】
化1の化学式の物質を前駆体として用いる18F-標識 O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法において、
上記の前駆体を原料として、TBAHCO3を触媒として18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成する無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項3】
請求項1の製造方法において、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成後、シリカゲルコラムで精製することを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項4】
請求項1の製造方法において、触媒をTBAHCO3に替えて18F-放射性同位元素と反応させ、O-(2-[18F]fluoroethyl)-L-Tyrosineを合成することを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項5】
請求項1の製造方法により製造されたO-(2-[18F]fluoroethyl)-L-Tyrosineの注射液は、腫瘍の診断及び治療効果を追跡する核子医学レントゲン写真撮影法に用いられるものであることを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【請求項6】
請求項2の製造方法により製造されたO-(2-[18F]fluoroethyl)-L-Tyrosineの注射液は、腫瘍の診断と治療効果を追跡する核子医学レントゲン写真撮影法に用いられるものであることを特徴とする無支持体の18Fアミノ酸O-(2-[18F]fluoroethyl)-L-Tyrosineの製造方法。
【図1】


【図2】

【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2007−112725(P2007−112725A)
【公開日】平成19年5月10日(2007.5.10)
【国際特許分類】
【出願番号】特願2005−303716(P2005−303716)
【出願日】平成17年10月18日(2005.10.18)
【出願人】(599171866)行政院原子能委員會核能研究所 (37)
【Fターム(参考)】
【公開日】平成19年5月10日(2007.5.10)
【国際特許分類】
【出願日】平成17年10月18日(2005.10.18)
【出願人】(599171866)行政院原子能委員會核能研究所 (37)
【Fターム(参考)】
[ Back to top ]