
蛋白質/(ポリ)ペプチドライブラリー
【課題】1つまたは複数の相同蛋白質/(ポリ)ペプチドのコレクションをコードする合成DNA配列、ならびにこれらのDNA配列のライブラリーを作製および適用する方法を提供する。
【解決手段】ヒトのゲノム中にコードされる抗体の構造的レパートリーをカバーする合成コンセンサス配列を使用して、ヒト由来の抗体遺伝子ライブラリーを調製する。さらに高度に多様化した抗体ライブラリーの普遍的フレームワークとしての、単一のコンセンサス抗体遺伝子を使用する。
【解決手段】ヒトのゲノム中にコードされる抗体の構造的レパートリーをカバーする合成コンセンサス配列を使用して、ヒト由来の抗体遺伝子ライブラリーを調製する。さらに高度に多様化した抗体ライブラリーの普遍的フレームワークとしての、単一のコンセンサス抗体遺伝子を使用する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、1つまたは複数の相同蛋白質/(ポリ)ペプチドのコレクションをコードする合成DNA配列、ならびにこれらのDNA配列のライブラリーを作製および応用する方法に関する。特に、本発明はヒトのゲノム中にコードされる抗体の構造的レパートリーをカバーする合成コンセンサス配列を使用することによる、ヒト由来の抗体遺伝子ライブラリーの調製に関する。さらに本発明は、高度に多様化した抗体ライブラリーの普遍的フレームワークとしての、単一のコンセンサス抗体遺伝子の使用に関する。
【背景技術】
【0002】
発明の背景
例えば特定のリガンドに結合するというような、望ましい性質を有するメンバーをスクリーニングするために、例えば抗体などの蛋白質/(ポリ)ペプチドのライブラリーを用いる現行のすべての組み換え方法では、メンバーの望ましい性質を簡単かつ迅速に改良することはできない。通常、ライブラリーは、生物体からクローニングされた1つまたは複数のDNA配列に任意のオリゴヌクレオチド配列を挿入するか、またはDNA配列のファミリーをクローニングしライブラリーとして使用するかのいずれかによって作製される。その後、例えばファージのディスプレイなどを用いて、ライブラリーから所望の性質を示すメンバーをスクリーニングする。そして、この結果得られる1つまたは複数の分子の配列が決定される。これらの分子をさらに改良するために利用することのできる一般的な方法はない。
【0003】
Winter(欧州特許第0 368 684 B1号(特許文献1))により、抗体の可変領域遺伝子を増幅(PCRによる)し、クローニングし、発現させる方法が提供されている。これらの遺伝子から開始して、WinterはH鎖および/またはL鎖のCDR3を無作為化することにより、機能的な抗体断片のライブラリーを作製できた。この過程は、免疫系におけるB細胞の発生時に起こるVJおよびVDJの自然の組み換え過程と機能的には同等である。
【0004】
しかし、Winterの発明では、「親和性の成熟」という自然に起こる現象と機能的に同等であるような過程において、さらに抗体断片の結合親和性を最適化するための方法は提供されておらず、本発明によってこれは提供される。さらに、Winterの発明では、構造的に類似した天然の遺伝子ファミリー全体を表す、合成のDNAオリゴヌクレオチドから組み立てられるような、人工的可変領域遺伝子は提供されていない。また、Winterの発明では、抗体の可変領域の一部分を組み合わせて組み立てることはできないが、本発明によってこの機能が提供される。さらに、この方法では、PCRプライミング領域以外はライブラリーメンバーに関するさらなる配列情報は得られないため、スクリーニング過程で得られたすべての抗体遺伝子の配列を完全に決定する必要があるという欠点がある。これは時間と労力を要し、配列の誤りにもつながる可能性がある。
【0005】
Winterおよび他の方法の教示は、可能な限り多くの異なる抗原に対する抗体を見つけるために、相補性決定領域 (CDR) およびフレームワークにおいて、高度な多様性を有する大きな抗体ライブラリーを作製するよう試みられている。1つの普遍的なフレームワークが抗体ライブラリーを作製するために有用であることが示唆されているが、これはまだ成功を収めていない。
【0006】
もう1つの問題は、抗体に由来する試薬の生産である。小さな抗体断片は、治療薬、診断薬、および生化学研究に非常に有望である。したがって、これらは大量に必要とされ、例えば、大腸菌のペリプラズムにおけるFv、単鎖Fv (scFv)、またはFabのような抗体断片の発現(SkerraおよびPluckthun、1988(非特許文献1);Betterら、1988(非特許文献2))は、多くの研究室で日常的に使用されている。しかし、発現の収率には非常に差がある。振盪フラスコ培養の培養液1リットルおよびOD当たり機能的な可溶性蛋白質が数mgまで得られる断片もあるが(Carterら、1992(非特許文献3);Pluckthunら、1996(非特許文献4))、いわゆる封入体の中にしばしば見られる不溶性物質しかほとんど生成しない断片もある。後者からは、労力と時間をかけた再生過程を用いて、低い収量で機能的な蛋白質が得られる場合がある。抗体の発現量に影響を与える要素は、まだあまり分かっていない。抗体断片の折り畳み効率および安定性、プロテアーゼ感受性、ならびに発現された蛋白質の宿主細胞への毒性により実際の生産量がしばしばひどく制限されており、発現収率を上昇させるためのいくつかの試みが行われている。例えば、KnappikおよびPluckthun (1995)(非特許文献5)は、発現収率は抗体の配列に依存することを示した。彼等は、抗体のフレームワーク中で発現収率に大きな影響を与える鍵となる残基を同定した。同様に、Ullrichら(1995)(非特許文献6)は、CDR中の点変異がペリプラズムにおける抗体断片の発現の収量を上昇させ得ることを発見した。しかしながら、これらの戦略はごく一部の抗体にしか適用できない。Winterの発明は抗体の既存のレパートリーを使用するため、遺伝子の発現能力に影響を与えることはできない。
【0007】
さらに、KnappikおよびPluckthun、ならびにUllrichの結果は、抗体、特にその折り畳みと発現に関する知識がまだ増加中であることを示している。Winterの発明では、ライブラリーの設計にそのような改良を取り入れることはできない。
【0008】
大概の場合、スクリーニング手順はファージ表面における遺伝子産物のディスプレイに依存しており、効率の高いディスプレイには遺伝子が少なくともある程度は発現される必要があるため、遺伝子の発現能力は、ライブラリーの質にとっても重要である。
【0009】
本発明は、このような既存の方法の欠点を克服し、またすべての相同蛋白質のコレクションに適用できる。本発明は、以下に抗体を例として示される下記のような新規で有用な特徴を有する。
【0010】
相同抗体遺伝子のグループ全体の構造的性質を反映する既知の抗体配列に基づき、人工抗体およびその断片を作製することができる。したがって、構造的レパートリーを失うことなく、遺伝子の数を減らすことが可能である。この方法を用いることにより、制限された数の人工遺伝子が得られ、これを新規に合成することにより、切断部位を導入したり、不要な切断部位を除去したりすることができる。さらに、この方法によれば、(i) 遺伝子の使用コドンを、任意の所望の宿主細胞中で高度に発現する遺伝子の使用コドンに適合させることができ、また (ii) 抗体のL鎖とH鎖のすべての可能なペアについて、相互作用の選択、抗原の選択、または組み換え体の発現の力価に関して分析することができるが、これは生物体のすべての抗体遺伝子およびそのすべての組み合わせを用いた場合には、実質的に不可能である。
【0011】
限定された数の完全に合成された遺伝子のセットを使用すると、コードされる構造的サブ要素の境界部に切断部位を作製することが可能である。したがって、各遺伝子は、蛋白質/(ポリ)ペプチドレベルの構造的サブ要素を表すモジュールから築き上げられる。抗体の場合には、モジュールは「フレームワーク」と「CDR」から構成される。フレームワークとCDRのモジュールを別々に作製することにより、異なる組み合わせが可能になる。さらに、2つまたはそれ以上の人工遺伝子が、各々の遺伝子サブ要素の境界部に同一の切断部位を持っている場合には、特定の遺伝子配列に関する追加情報なしに、あらかじめ作製しておいたサブ要素のライブラリーを、同時にこれらの遺伝子中に挿入することができる。この戦略を用いると、遺伝子サブ要素のライブラリーをコードするDNAカセットを (i) あらかじめ作製し、保存、再使用でき、(ii) 実際の配列を知ったり各ライブラリーメンバーの配列決定をすることなくこれらの配列中の正しい位置に挿入することができるため、例えば抗体の親和性を迅速に最適化できる。
【0012】
さらに、既存のモジュールを新しい観察結果によって修飾したモジュールで置き換えることにより、結合、安定性、または可溶性、および発現に重要なアミノ酸残基に関する新しい情報を、ライブラリー設計に組み入れることができる。
【0013】
ライブラリー作製には限定された数のコンセンサス配列が使用されるため、スクリーニング後の、結合する抗体の同定が迅速化される。配列決定または制限部位のフィンガープリントを使用して、基礎となるコンセンサス遺伝子配列を同定した後に、無作為配列を含む部分のみを決定する。これにより配列の誤りおよび偽陽性の結果が生じる可能性が低下する。
【0014】
上記の切断部位は、人工遺伝子が挿入されるベクター系に1つだけ存在する場合に使用できる。このため、ベクターを、このような切断部位を含まないように修飾する必要がある。切断部位が除去され、抵抗性遺伝子や複製開始部位のような基本的な要素を含むベクターの構築は、クローニングのために広く興味を持たれている。さらに、これらのベクターは、上記の人工遺伝子およびあらかじめ作製してあるライブラリーを含むキットの一部ともなりうる。
【0015】
人工遺伝子のコレクションは、好ましくは齧歯類の抗体である非ヒト抗体を、迅速にヒト化するために使用できる。まず、好ましくは齧歯類の抗体である非ヒト抗体のアミノ酸配列を、人工遺伝子コレクションによってコードされるアミノ酸配列と比較して、最も相同性の高いL鎖とH鎖のフレームワーク領域を決定する。それから、これらの遺伝子を用いて、好ましくは齧歯類の抗体である非ヒト抗体のCDRをコードする遺伝子サブ要素を挿入する。
【0016】
驚くべきことに、1つのscFvフラグメントのL鎖とH鎖の各々のただ1つのコンセンサス配列を組み合わせることにより、実質的にすべての抗原に対する抗体を生成するような抗体レパートリーが作製できた。従って、本発明の1つの局面は、有用な(ポリ)ペプチドライブラリーを作製するための普遍的なフレームワークとしての、単一のコンセンサス配列の使用、およびそのために有用な抗体コンセンサス配列である。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】欧州特許第0 368 684 B1号
【非特許文献】
【0018】
【非特許文献1】SkerraおよびPluckthun、Science 240, 1038-1041(1988)
【非特許文献2】Betterら、Science 240, 1041-1043(1988)
【非特許文献3】Carterら、Bio/Technology 10, 163-167(1992)
【非特許文献4】Pluckthunら、A practical approach. Antibody Engineering (Ed. J. McCafferty). IRL Press. Oxford, pp. 203-252(1996)
【非特許文献5】KnappikおよびPluckthun、Protein Engineering 8, 81-89(1995)
【非特許文献6】Ullrichら、Proc. Natl. Acad. Sci. USA 92, 11907-11911(1995)
【発明の概要】
【0019】
発明の詳細な説明
本発明は、(ポリ)ペプチドの有用なライブラリーの作製を可能にする。第一の態様において、本発明は該ライブラリーの作製に適当な核酸配列の組み立て方法を提供する。第1の段階では、少なくとも3つの相同蛋白質を同定し分析する。したがって、蛋白質配列を整列させた蛋白質配列のデータベースが確立される。このデータベースを使って、配列と、情報がある場合には構造的配列との両方で、高度の類似性を示す蛋白質配列のサブグループを決定する。各サブグループにつき、サブグループのメンバーを表すため、少なくとも1つのコンセンサス配列を含む(ポリ)ペプチド配列を導き出す。したがって、(ポリ)ペプチド配列のコレクション全体は、相同蛋白質のコレクションの構造的レパートリー全体を代表することになる。その後、これらの人工(ポリ)ペプチド配列を、可能ならば構造的性質に基づいて分析し、該(ポリ)ペプチド配列内、または、たとえば多量体蛋白質において、該(ポリ)ペプチド配列間もしくは他の(ポリ)ペプチドとの間の好ましくない相互作用を同定する。該相互作用は、コンセンサス配列をしかるべく変更することにより除去する。それから、(ポリ)ペプチド配列を分析し、ドメイン、ループ、ヘリックス、またはCDRのようなサブ要素を同定する。アミノ酸配列は、該核酸配列を発現するために計画する宿主の使用コドンに適合させた核酸配列に逆翻訳する。上述のようにして同定されたサブ要素をコードする各サブ配列が2つの切断部位に挟まれるが、核酸配列内ではその2つの切断部位が2回以上存在しないように、1組の切断部位を組み立てる。これは、すでにサブ配列に隣接する切断部位を同定するか、または切断部位を作製するように1つまたは複数のヌクレオチドを変更し、さらに遺伝子の残りの部分からその部位を除去することにより達成できる。切断部位は対応するサブ要素またはサブ配列すべてに共通であるべきで、これにより核酸配列中のサブ配列および対応する(ポリ)ペプチド中のサブ要素が、完全にモジュール構造をとることになる。
【0020】
別の態様において、本発明は、上述の方法を実行する(ポリ)ペプチドの2つまたはそれ以上のセットで、切断部位が各セット内で唯一であるだけでなく任意の2つのセット間でも唯一であるようなセットを組み立てる方法を提供する。この方法は、新規のヘテロ結合ドメインをスクリーニングするために、例えば2つの異なる蛋白質由来の2つのαヘリックスドメインを含む(ポリ)ペプチドライブラリーを作製するために適用できる。
さらに別の態様において、上述のような少なくとも2つのセットが、同じ蛋白質のコレクションまたは少なくともその一部に由来する。これは、例えば、VHおよびVLのような抗体の2つのドメイン、または膜内外受容体の2つの細胞外ループを含むライブラリーであるが、これに限定されるわけではない。
さらに別の態様において、上述のように組み立てられる核酸配列が合成される。これは、例えば、遺伝子の全合成やPCRに基づく方法など、当業者に周知のいくつかの方法のいずれかにより達成できる。
【0021】
1つの態様において、核酸配列はベクターにクローニングされる。ベクターは当業者に周知の配列決定用ベクター、発現ベクター、またはディスプレイ(例、ファージディスプレイ)ベクターでありうる。すべてのベクターは、1つの核酸配列、または2つもしくはそれ以上の核酸配列を、異なるオペロンまたは同一のオペロン中に含み得る。後者の場合には、配列を別にクローニングするか、隣接する配列としてクローニングされるかのいずれかとなる。
1つの態様において、上述のように好ましくない相互作用を除去すると、修飾(ポリ)ペプチドの発現が増加する。
【0022】
好ましい態様において、核酸配列の1つまたは複数のサブ配列を、異なる配列によって置き換える。これは、例えば、当業者に周知の条件で制限酵素を用いて対応する切断部位で切断するように、サブ配列に隣接するまたはその末端にある制限部位を切断するために適当な条件を用いてサブ配列を切り出し、切断された核酸配列に適合する異なる配列によりそのサブ配列を置き換えることにより達成できる。さらに好ましい態様において、最初のサブ配列を置き換える異なる配列は、例えば、非ヒト抗体を迅速にヒト化するために非ヒト抗体のCDRをコンセンサス抗体配列に接合する場合のように、ゲノム配列または再配列したゲノム配列である。最も好ましい態様において、異なる配列は無作為配列であり、したがってそのサブ配列を配列のコレクションによって置換することにより、多様性を導入し、ライブラリーを作製できる。無作為配列は、例えばオリゴヌクレオチドの自動合成中に、モノヌクレオチド混合物、または好ましくはトリヌクレオチド混合物を使用したり(Virnekasら、1994)、誤りがちなPCRによって、または当業者に周知の他の方法など、様々な方法で作製することができる。無作為配列は、完全に無作為化してもよいし、または既知の蛋白質配列における特定の位置のアミノ酸分布にしたがって、特定のコドンを使うようにもしくは避けるように偏向してもよい。さらに、無作為サブ配列のコレクションには、異なる数のコドンが含まれ、サブ要素のコレクションが異なる長さを有するようにしてもよい。
【0023】
別の態様において、本発明は、当業者に周知の適当なベクターからの適当な条件下での核酸配列の発現を提供する。
さらに好ましい態様において、該核酸配列から発現される(ポリ)ペプチドをスクリーニングし、選択的に最適化する。スクリーニングは、ファージディスプレイ、選択的感染性を有するファージ、ポリソーム技術を用いた結合のスクリーニング、酵素活性のアッセイ系、または蛋白質の安定性など、当業者に周知の方法を用いて実行できる。望ましい性質を有する(ポリ)ペプチドは、対応する核酸配列の決定、またはアミノ酸の配列決定もしくは質量分析法によって同定できる。引き続き最適化する場合には、選択的に、最初に選択した(ポリ)ペプチドをコードする核酸配列を、配列決定せずに使用できる。最適化は異なる配列、好ましくは無作為配列によるサブ配列の置換と、スクリーニング段階を、1回または複数回繰り返すことにより実行する。
【0024】
(ポリ)ペプチドがスクリーニングされる望ましい性質は、好ましくは、標的分子に対する最適化した親和性または特異性、最適化した酵素活性、最適化した発現収量、最適化した安定性、および最適化した溶解度からなる群より選択されるが、これに限定されるわけではない。
【0025】
1つの態様において、サブ配列と隣接する切断部位は制限酵素によって認識および切断される部位であり、認識される配列と切断される配列は同一であるかまたは異なり、制限部位は平滑末端または付着末端のいずれかを有する。
サブ要素の長さは、好ましくは、例えば酵素の活性部位における1つの残基または1つの構造決定残基のように、1つのアミノ酸から、蛋白質ドメイン全体のように150アミノ酸にまでにわたるが、これに限定されるわけではない。最も好ましくは、抗体のCDRループ中に通常見られるように3アミノ酸から25アミノ酸までの長さにわたる。
核酸配列は、RNAでも、好ましくはDNAでもよい。
【0026】
1つの態様において、(ポリ)ペプチドは、特定の種に特徴的なアミノ酸パターンを有する。これは、例えば、1つの種のみの相同蛋白質のコレクション、最も好ましくはヒトの蛋白質コレクションから、コンセンサス配列を導き出すことにより達成できる。コンセンサス配列を含む(ポリ)ペプチドは人工的なものであるから、最も類似性の高い蛋白質配列と比較して、かかる特徴的なアミノ酸パターンを有することを確かめる必要がある。
【0027】
1つの態様において、本発明は免疫グロブリンスーパーファミリーのメンバーまたは誘導体の少なくとも一部、好ましくは免疫グロブリンのメンバーまたは誘導体の少なくとも一部を含む、(ポリ)ペプチドライブラリーの作製方法を提供する。最も好ましくは、本発明は、該(ポリ)ペプチドが、該構想的サブ要素がフレームワーク領域(FR)1、2、3、もしくは4、または相補性決定領域(CDR)1、2、もしくは3であるH鎖もしくはL鎖の可変領域であるかまたはそれに由来するもので、ヒトの抗体のライブラリーの作製方法を提供する。第1の段階において、ヒト由来の明らかにされている抗体配列を整列させたデータベースを作製する。このデータベースを用いて、配列とCDRループの共通の折り畳み(抗体構造の分析により決定)の両方において、高度の類似性を示す抗体配列のサブグループを決定する。各サブグループについて、そのサブグループのメンバーを代表するコンセンサス配列を導き出す。したがって、コンセンサス配列のコレクション全体は、ヒト抗体の構造的レパートリー全体を代表することになる。
【0028】
その後、例えば、遺伝子の全合成、または合成の遺伝子サブユニットの使用によって、これらの人工的な遺伝子を作製する。これらの遺伝子サブユニットは、(ポリ)ペプチドレベルでは構造的サブ要素に対応する。DNAレベルでは、これらの遺伝子サブユニットは、ベクター系に唯一である、各サブ要素の最初と最後にある切断部位が決定する。コンセンサス配列のコレクションのメンバーであるすべての遺伝子は、類似したパターンの対応する遺伝子サブ配列を含むように作製される。最も好ましくは、該(ポリ)ペプチドは、HuCALコンセンサス遺伝子Vκ1、 Vκ2、 Vκ3、 Vκ4、Vλ1、 Vλ2、 Vλ3、VH1A、 VH1B、 VH2、 VH3、 VH4、 VH5、 VH6、Cκ、Cλ、CH1もしくは該HuCALコンセンサス遺伝子の任意の組み合わせであるか、またはそれに由来する。
【0029】
その後、このDNA分子コレクションを使って、抗体または抗体断片、好ましくはFv、ジスルフィド結合したFv、単鎖Fv (scFv)、またはFabフラグメントのライブラリーを作製でき、これを新規の標的抗原に対する特異性の供給源として使用できる。さらに、あらかじめ作製してあるライブラリーカセットと一般的な手順を用いて、抗体の親和性を最適化できる。本発明は、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定するための方法を提供し、抗体断片を発現する段階、およびその後スクリーニングにより所定の標的分子に結合する1つまたは複数の抗体断片を単離する段階を含む。好ましくは、HuCAL VH3およびHuCAL Vλ2コンセンサス遺伝子の組み合わせ、および、H鎖CDR3サブ要素をコードする少なくとも1つの無作為サブ配列を含むscFvフラグメントライブラリーから、結合する抗体をスクリーニングする。必要ならば、その後、遺伝子のモジュラー型設計を用いて、抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、構造的サブ要素をコードする1つまたは複数の第2のサブ配列により置き換えることができる。その後、望ましい親和性を有する抗体が生成するまで、この発現およびスクリーニングの段階を繰り返すことができる。
【0030】
特に好ましいのは、該切断部位を使用して、1つまたは複数の遺伝子サブユニット(例、CDR)を、配列の無作為なコレクション(ライブラリー)によって置換する方法である。これらの切断部位は、(i) ベクター系で唯一であり、(ii) すべてのコンセンサス遺伝子に共通であるため、同じ(あらかじめ作製した)ライブラリーを、すべての人工抗体遺伝子に挿入できる。その結果得られるライブラリーを、任意の選択した抗原に対してスクリーニングする。結合する抗体を選択し、収集し、次のライブラリーの開始材料として使用する。ここでは、1つまたは複数の残りの遺伝子サブユニットを上述のようにして無作為化する。
【0031】
本発明のさらなる態様は、上述のような(ポリ)ペプチドと、付加部分の両方を含むDNAを提供することによる融合タンパク質に関する。特に好ましいのは、有用な治療機能を有する部分である。例えば、付加部分は細胞を殺すことのできる毒素分子であってもよい(Vitettaら、1993)。かかる毒素としては、シュードモナス外毒素Aおよびジフテリア毒素のような細菌毒素、ならびにリシン、アブリン、モデシン、サポリン、ゲロニンのような植物毒素など、数多くの例が当業者に周知である。かかる毒素を例えば抗体断片に融合すると、毒素は、例えば疾病を有する細胞を標的とし、有用な治療効果を有することができる。または、付加部分は、細胞のファミリーに特定の効果(この場合、T細胞の増殖効果)を有する
IL-2(RosenbergおよびLotze、1986)のようなサイトカインであってもよい。さらなる態様において、付加部分はその(ポリ)ペプチドパートナーに検出および/または精製の方法を与える場合がある。例えば、融合タンパク質は、修飾された抗体断片と、アルカリ性フォスファターゼ(Blakeら、1984)のような検出のために一般に用いられる酵素とを含むことができる。検出または精製のための標識として使用できる他の部分には、当業者に周知のものが数多くある。特に好ましいのは、金属イオンと結合することのできる、少なくとも5つのヒスチジン残基を含むペプチドで(Hochuliら、1988)、そのため、これはその融合した蛋白質の精製に使用できる(Lindnerら、1992)。また、本発明は、一般に使用されるC-mycおよびFLAG標識(Hoppら、1988;KnappikおよびPluckthun、1994)のような付加部分を提供する。
【0032】
1つまたは複数の付加ドメインを操作することにより、抗体断片または任意の他の(ポリ)ペプチドは、本発明の範囲内であるさらに大きな分子に組み立てることができる。例えば、ミニ抗体(Pack、1994)は、各々が自己会合性の二量化ドメインに融合した2つの抗体断片を含む二量体である。特に好ましい二量化ドメインには、ロイシンジッパー(PackおよびPluckthun、1992)またはαヘリックス・βターン・αヘリックスモチーフ(Packら、1993)がある。
【0033】
本発明の上記の態様はすべて、当業者に既知の分子生物学の標準的技法を使用して遂行できる。
さらなる態様において、サブ配列の無作為なコレクション(ライブラリー)を、1つの(ポリ)ペプチドをコードする単一の核酸配列に挿入し、1つの普遍的なフレームワークに基づく(ポリ)ペプチドライブラリーを作製する。好ましくは、CDRサブ配列の無作為なコレクションを、例えば、上述のH3κ2単鎖Fvフラグメントのような普遍的な抗体フレームワークに挿入する。
さらなる態様において、本発明は上述の方法にしたがって得られる核酸配列、核酸配列を含むベクター、ベクターを含む宿主細胞、および(ポリ)ペプチドを提供する。
さらに好ましい態様において、本発明は、(ポリ)ペプチドをコードするモジュール型の核酸配列と適合するモジュール型ベクターシステムを提供する。ベクターのモジュールは制限部位と隣接するが、この制限部位はベクター系中で唯一で、(ポリ)ペプチドをコードする核酸配列に組み込まれる制限部位については、例えば核酸配列をベクターにクローニングするために必要な制限部位を除いて本質的に唯一である。ベクターモジュールのリストには、一本鎖複製開始点、コピー数の多いおよび少ないプラスミドの二本鎖複製開始点、プロモーター/オペレーター、リプレッサーまたはターミネーター要素、抵抗性遺伝子、組み換え可能部位、線状ファージ上のディスプレイのための遺伝子III、シグナル配列、精製および検出用標識、および付加部分の配列を含む。
ベクターは、好ましくは発現ベクターまたは発現とライブラリーのスクリーニングに適当なベクターであるが、これに限定されるわけではない。
【0034】
他の態様において、本発明は、上述の方法にしたがった核酸配列、組み換え体ベクター、(ポリ)ペプチド、およびベクターのリスト、ならびに(ポリ)ペプチドを生産するために適当な宿主細胞のうちの1つまたは複数を含むキットを提供する。
好ましい態様において、本発明はヒトの抗体のライブラリーの作製方法を提供する。最初の段階では、ヒト由来の明らかにされている抗体配列のデータベースを確立する。このデータベースを用いて、配列と共通の折り畳み(抗体構造の分析により決定)の両方において、高度の類似性を示す抗体配列のサブグループを決定する。各サブグループについて、そのサブグループのメンバーを代表するコンセンサス配列を導き出す。したがって、コンセンサス配列のコレクション全体は、ヒト抗体の構造的レパートリー全体を代表することになる。
【0035】
その後、合成の遺伝子サブユニットを使用して、これらの人工的な遺伝子を作製する。これらの遺伝子サブユニットは、蛋白質レベルでは構造的サブ要素に対応する。DNAレベルでは、これらの遺伝子サブユニットは、ベクター系に唯一である、各サブ要素の最初と最後にある切断部位が決定する。コンセンサス配列のコレクションのメンバーであるすべての遺伝子は、該遺伝子サブユニットの類似したパターンを含むように作製される。
その後、DNA分子のこのコレクションを使って、抗体のライブラリーを作製でき、これを新規の標的抗原に対する特異性の供給源として使用できる。さらに、あらかじめ作製してあるライブラリーカセットと一般的な手順を用いて、抗体の親和性を最適化できる。本発明は、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定するための方法を提供し、抗体断片を発現する段階、およびその後スクリーニングにより所定の標的分子に結合する1つまたは複数の抗体断片を単離する段階を含む。必要ならば、その後、遺伝子のモジュラー型設計を用いて、抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、構造的サブ要素をコードする1つまたは複数の第2のサブ配列により置き換えることができる。その後、望ましい親和性を有する抗体が生成するまで、この発現およびスクリーニングの段階を繰り返すことができる。
【0036】
特に好ましいのは、該切断部位を使用して、1つまたは複数の遺伝子サブユニット(例、CDR)を、配列の無作為なコレクション(ライブラリー)によって置換する方法である。これらの切断部位は、(i) ベクター系に唯一で、(ii) すべてのコンセンサス遺伝子に共通であるため、同じ(あらかじめ作製した)ライブラリーを、すべての人工抗体遺伝子に挿入できる。その結果得られるライブラリーを、任意の選択した抗原に対してスクリーニングする。結合する抗体を溶出し、収集し、次のライブラリーの開始材料として使用する。ここでは、1つまたは複数の残りの遺伝子サブユニットを上述のようにして無作為化する。
【0037】
定義
蛋白質:
蛋白質という用語は、単量体のポリペプチド鎖、および2つまたはそれ以上のポリペプチド鎖が共有結合の相互作用(ジスルフィド結合のような結合)または非共有結合の相互作用(疎水性または静電的相互作用のような相互作用)のいずれかにより結合しているホモまたはヘテロ多量体複合体を含む。
相同蛋白質の分析:
3つまたはそれ以上の蛋白質のアミノ酸配列を、すべての位置で同一または類似したアミノ酸残基との一致が最大になるように、(ギャップの導入を可能にするよう)互いに整列させる。同一および/または類似した残基の合計の割合が一定の閾値を越える場合に、これらの整列させた配列は相同であると言う。当業者は一般に、整列させた遺伝子のアミノ酸の少なくとも15%が同一で、少なくとも30%が類似する場合、この閾値を越えていると見なす。相同蛋白質のファミリーの例には、免疫グロブリンスーパーファミリー、スカベンジャー受容体スーパーファミリー、フィブロネクチンスーパーファミリー(例、タイプIIおよびIII)、補体制御タンパク質スーパーファミリー、サイトカイン受容体スーパーファミリー、シスチンノット蛋白質、チロシンキナーゼ、および当業者に周知の数多くの他の例がある。
コンセンサス配列:
少なくとも3つの整列させたアミノ酸配列のマトリックスを用いて、並べる際にギャップを許すと、各位置で最も頻度の高いアミノ酸残基を決定することができる。コンセンサス配列は、各位置で最も出現頻度の高いアミノ酸を含む配列である。1つの位置で2つまたはそれ以上のアミノ酸が一つの部位で同じ出現頻度を有する場合は、コンセンサス配列は、これらのアミノ酸の両方またはすべてを含む。
好ましくない相互作用の除去:
コンセンサス配列自体は、大概、人工的なものであるため、結果として得られる分子が機能的な3次構造を取るのを妨げたり、または多量体複合体において他の(ポリ)ペプチド鎖との相互作用の障害となるアミノ酸残基を変更するために、分析する必要がある。これは、(i) 既知の関連構造を鋳型として用いて、コンセンサス配列の3次元モデルを構築し、モデル内で互いに好ましくない相互作用をする可能性があるアミノ酸残基を同定するか、または (ii) 整列させたアミノ酸配列のマトリックスを分析し、配列内で同時に1つの配列中に頻繁に出現し、したがって互いに相互作用をする可能性が高いアミノ酸残基の組み合わせを検出することにより行うことができる。これらの相互作用をすると思われるペアを表にし、この「相互作用地図」とコンセンサス配列を比較する。コンセンサス配列中で欠如する相互作用または不正な相互作用は、好ましくない相互作用を抑えるようにしかるべくアミノ酸配列を適当に変更することにより修復する。
構造的サブ要素の同定:
構造的サブ要素とは、分子の定義された構造的または機能的部分に対応する、蛋白質/(ポリ)ペプチド内のアミノ酸残基列である。これは、ループ(例、抗体のCDRループ)、または蛋白質/(ポリ)ペプチド内の任意の2次構造または機能的構造(ドメイン、αヘリックス、βシート、抗体のフレームワーク領域等)であり得る。構造的サブ要素は、類似または相同の(ポリ)ペプチドの既知の構造を用いて、または上述の整列したアミノ酸配列のマトリックスを用いて、同定できる。各位置での変動に基づき、構造的サブ要素(例、抗体の超可変領域)に属するアミノ酸残基列を決定する。
サブ配列:
サブ配列とは、唯一である切断部位によって隣接しており、少なくとも1つの構造的サブ要素をコードする遺伝子モジュールと定義される。これは構造的サブ要素と必ずしも同一ではない。
切断部位:
DNAを配列特異的に切断する試薬(例、制限エンドヌクレアーゼ)の特異的標的として使用される短いDNA配列。
適合する切断部位:
切断部位が修飾せずに、また好ましくはアダプター分子を加えずに、効果的に連結できる場合、これらの切断部位は互いに適合する。
唯一である切断部位:
興味のある遺伝子を少なくとも1つ含むベクター中で、切断部位が1度しか出現しない場合、または興味のある遺伝子を少なくとも1つ含むベクター中、その切断部位のうちのただ1つのみが切断試薬によって使用できるとして扱える場合、その切断部位は唯一であると定義する。
対応する(ポリ)ペプチド配列:
相同蛋白質の1グループの同じ部分から導き出される配列を、対応する(ポリ)ペプチド配列と呼ぶ。
共通の切断部位:
少なくとも2つの対応する配列中の切断部位で、同じ機能的位置(すなわち、定められたサブ配列と隣接する)に出現し、同一の切断ツールで加水分解され、同一の適合する末端を生成するような切断部位を、共通の切断部位と呼ぶ。
遺伝子サブ配列の切り出し:
唯一である切断部位が隣接する遺伝的サブ配列を単離、除去、または置換するために、唯一である切断部位と対応する切断試薬とを使用して、特定の位置で標的DNAを切断する方法。
遺伝的サブ配列の交換:
既存のサブ配列に隣接する切断部位を用いてこのサブ配列を除去し、このようにして作製した切断部位と適合する末端を含む新規のサブ配列またはサブ配列のコレクションを挿入する方法。
遺伝子の発現:
発現という用語は、遺伝子の情報がmRNAに転写され、それから蛋白質/(ポリ)ペプチドに翻訳されるインビボまたはインビトロの過程を示す。したがって、発現という用語は、遺伝子の情報がmRNAにそれから蛋白質に転写される、細胞内で発生する過程を示す。発現という用語はまた、(ポリ)ペプチドが機能を有するために必要な、翻訳後修飾と輸送のすべての事象も含む。
蛋白質/(ポリ)ペプチドライブラリーのスクリーニング:
望ましい性質を有する1つまたは複数の蛋白質/(ポリ)ペプチドを、ライブラリー内の他の蛋白質/(ポリ)ペプチドから単離することのできる任意の方法。
種に特徴的なアミノ酸パターン:
(ポリ)ペプチド配列は、1つの種のみに由来する相同蛋白質のコレクションから導き出される場合、その種に特徴的なアミノ酸パターンを示すと想定される。
免疫グロブリンスーパーファミリー (IgSF):
IgSFは免疫グロブリンの折り畳みを特徴とするドメインを含むタンパク質のファミリーである。IgSFには、例えばT細胞受容体および免疫グロブリン(抗体)が含まれる。
抗体のフレームワーク:
抗体の可変部のフレームワークは、可変部の一部でこの可変部の抗原結合ループの骨組みとして働く部分としてKabatら(1991)が定義したものである。
抗体のCDR:
抗体のCDR(相補性決定領域)は、 Kabatら(1991)が定義したように、抗原結合ループから構成される。抗体のFvフラグメントの2つの可変部には、各々3つのCDRが含まれる。
HuCAL:
ヒト組み合わせ抗体ライブラリー(Human Combinatorial Antibody Library)の頭字語。本発明に係るモジュール型のコンセンサス遺伝子に基づく抗体ライブラリー(実施例1参照)。
抗体断片:
例えば、抗原の結合のような、特定の機能を有する抗体の任意の部分。通常、抗体断片は抗体全体よりも小さい。例としては、Fv、ジスルフィド結合Fv、単鎖Fv (scFv)、またはFabフラグメントがある。さらに、抗体断片は、新規の機能または性質を含むように、しばしば操作される。
普遍的フレームワーク:
異なるフレームワークの大きなコレクションによって本来は維持されていた機能、特異性、または性質の完全な変動性を作製するために用いることのできる単一のフレームワークを、普遍的フレームワークと呼ぶ。
抗体の標的への結合:
抗体と、対応する分子またはリガンドとの間の強く特異的な会合に至る過程を、結合と呼ぶ。抗体によって認識される分子もしくはリガンド、または分子もしくはリガンドの任意の部分を、標的と呼ぶ。
遺伝子サブ配列の置換:
既存のサブ配列に隣接する切断部位を用いてこのサブ配列を除去し、このようにして作製した切断部位と適合する末端を含む新規のサブ配列またはサブ配列のコレクションを挿入する方法。
遺伝子配列の組み立て:
特定の方法で合成または天然の遺伝子配列を組み合わせ、使用された合成または天然の遺伝子配列の少なくとも一部を含む、さらに長い遺伝子配列を作製するために使用する任意の過程。
相同遺伝子の分析:
2つまたはそれ以上の遺伝子に対応するアミノ酸配列を、すべての位置で同一または類似したアミノ酸残基間の一致が最大になるように整列させる。同一および/または類似した残基の合計の割合が一定の閾値を越える場合に、これらの整列させた配列が相同であると言う。当業者は一般に、整列させた遺伝子のアミノ酸うちの少なくとも15%が同一で、少なくとも30%が類似する場合、この閾値を越えていると見なす。
【図面の簡単な説明】
【0038】
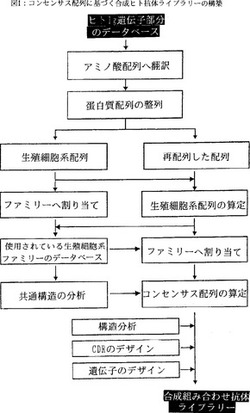
【図1】コンセンサス配列に基づく合成ヒト抗体ライブラリーの構築過程を示す流れ図。
【図2】各サブグループ(アミノ酸残基は標準的な1文字の略語で示されている)に設計されたコンセンサス配列を並べたもの。(A)κ配列、(B)λ配列、および(C)H鎖配列。位置はKabat (1991)に従って番号が付けられている。整列させた際の相同性を最大にするために、一定の位置の配列にギャップ(ミ)が挿入されている。
【図3】合成Vκコンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図4】合成Vλコンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図5】合成VH鎖コンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図6】コンセンサス遺伝子の作製に用いたオリゴヌクレオチド。例えば遺伝子Vκ1はO1K1〜O1K6の6つのオリゴヌクレオチドを用いて作製されたというように、オリゴは対応するコンセンサス遺伝子に従って命名されている。定常部ドメインCκ(OCLK1〜8)およびCH1(OCH1〜8)をコードする遺伝子の合成に用いられたオリゴヌクレオチドも示されている。
【図7AB】定常部ドメインCκ(A)およびCH1 (B)をコードする合成遺伝子の配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図7C】合成Cκ遺伝子部分(huCLλ)を含むモジュールM24の機能地図と配列。
【図7D】モジュールM24の合成に用いたオリゴヌクレオチド。
【図8】コンセンサス単鎖断片VH3-Vκ2をコードする合成遺伝子の配列と制限地図。シグナル配列(アミノ酸1〜21)は大腸菌phoA遺伝子(SkerraおよびPluckthun、1988)に由来する。 モノクローナル抗体M1 (KnappikおよびPluckthun、1994)を用いたウエスタンブロットまたはELISAで単鎖断片を検出するために、 phoAシグナル配列とVH3ドメインの間に、4つのアミノ酸残基(アミノ酸22〜25)をコードする短い配列が挿入された。配列の最後の6 塩基対は、クローニングを目的として導入された(EcoRI部位)。
【図9】H3κ2 scFvフラグメントのファージディスプレイに使用されたベクターpIG10.3のプラスミド地図。ベクターはpIG10由来で、lacオペロンリプレッサー遺伝子、lacI、lacプロモーターのコントロール下でH3κ2-gene3ssの融合をコードする人工オペロン、転写ターミネーターIpp、大腸菌ファージf1の一本鎖複製開始点(F1_ORI)、β-ラクタマーゼ (bla)をコードする遺伝子およびColEI由来の複製開始点を含む。
【図10】最初のライブラリーからの独立したクローンの配列結果を、対応するアミノ酸配列に翻訳したもの。(A) VH3コンセンサスH鎖CDR3(93〜102の位置、Kabatの番号付け)の佐見の酸配列。(B) 10量体ライブラリーの12のクローンのアミノ酸配列。(C) 15量体ライブラリーの11のクローンのアミノ酸配列、*:1塩基の欠失。
【図11】各ライブラリーメンバーの発現試験。(A) 10量体ライブラリーの独立した9クローンの発現。(B) 15量体ライブラリーの独立した9クローンの発現。Mと記したレーンはサイズマーカーが含まれている。gp3-scFv融合およびscFv単量体の両方が示されている。
【図12】FITC-BSAに対するパニングにおける特異的ファージ抗体の濃縮。最初およびその後のフルオレセイン特異的サブライブラリーをブロッキングバッファーに対してパニングし、各回のパニングでFITC-BSAコートしたウェルから溶出されたファージと、粉ミルクコートしたウェルから溶出されたファージの比率が「特異性係数」として示されている。
【図13】FITC-BSAへの結合を試験した3回のパニング後の24の独立したクローンのファージELISA。
【図14】選択されたFITC-BSA結合クローンの競合的ELISA。阻害なしの場合のscFv結合のELISAシグナル(OD405nm)を100%としている。
【図15】FITC-BSAに対する3回のパニング後の独立クローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図16】精製した抗フルオレセインscFvフラグメントのクーマシーブルー染色SDS-PAGE。M:分子量マーカー、A:誘導後の全可溶性細胞抽出物、B:フロースルー分画、C、D、E:それぞれ、精製scFvフラグメント1HA-3E4、1HA-3E5、1HA-3E10。
【図17】βエストラジオール-BSA、テストステロン-BSA、BSA、ESL-1、インターロイキン-2、リンホトキシン-β、LeY-BSAに対する3回のパニングによる特異的ファージ抗体の濃縮。
【図18】選択したESL-1およびβエストラジオール結合クローンのELISA。
【図19】HuCAL抗体の選択性と交差反応性:HuCAL抗体の特異的結合が対角線上に見られ、対角線以外のシグナルは非特異的交差反応を示す。
【図20】βエストラジオール-BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。1つのクローンは10量体ライブラリーに由来する。
【図21】テストステロン-BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図22】リンホトキシン-βに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。1つのクローンは、オリゴヌクレオチド合成中のトリヌクレオチド混合物の不完全なカップリングにより導入されたと思われる、14量体CDRを含む。
【図23】ESL-1に対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。2つのクローンは10量体ライブラリーに由来する。1つのクローンは、トリヌクレオチドを用いたオリゴヌクレオチド合成中の鎖の伸長により導入されたと思われる、16量体のCDRを含む。
【図24】BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図25】モジュール型pCALベクター系の概略図。
【図25a】モジュール型HuCAL遺伝子およびpCALベクター系にすでに使用されているか、または使用に適当な制限部位のリスト。
【図26】pCALベクターシリーズのモジュール型ベクター要素のリスト:モジュールシステムの一部となっている制限部位のみを示す。
【図27】多クローニング部位モジュール(MCS)の機能地図と配列。
【図28】pMCSクローニングベクターシリーズの機能地図と配列。
【図29】pCALモジュールM1(図26参照)の機能地図と配列。
【図30】pCALモジュールM7-III(図26参照)の機能地図と配列。
【図31】pCALモジュールM9-II(図26参照)の機能地図と配列。
【図32】pCALモジュールM11-II(図26参照)の機能地図と配列。
【図33】pCALモジュールM14-Ext2(図26参照)の機能地図と配列。
【図34】pCALモジュールM17(図26参照)の機能地図と配列。
【図35】モジュール型ベクターpCAL4の機能地図と配列。
【図35a】他のpCALモジュール(M2、M3、M7I、M7II、M8、M10II、M11II、M12、M13、M19、M20、M21、M41)および低コピー数プラスミドベクター(pCALO1〜pCALO3)の機能地図と配列。
【図35b】pCALベクターモジュールの合成に用いたオリゴヌクレオチドおよびプライマーのリスト。
【図36】CDRライブラリークローニングのための、CDRの置換用のβラクタマーゼカセットの機能地図と配列。
【図37】VκCDR3ライブラリーのためのオリゴとプライマーの設計。
【図38】VλCDR3ライブラリーのためのオリゴとプライマーの設計。
【図39】pBS13発現ベクターシリーズの機能地図。
【図40】7つのVH遺伝子の各々を7つのVL遺伝子の各々と組み合わせて得られる49のHuCAL scFvの発現(pBS13、30℃):可溶性物質と不溶性物資の割合、ならびにH3κ2の組み合わせを100%としてこれと比較した合計および可溶性の量の値が示されている。さらに、McPC603 scFvについて、対応する値も示されている。
【0039】
表1:再配列した配列の生殖細胞系メンバーを算定するために用いたヒト免疫グロブリンの生殖細胞系配列の概要。(A) κ配列、(B) λ配列、(C) H鎖配列。(1) 種々の計算に用いた生殖細胞の名称、(2) 対応する配列の参考文献番号(配列に関連する引用文献は付録を参照)、(3) 各配列が属するファミリー、 (4) 同一のアミノ酸配列を有する生殖細胞系遺伝子について文献に見られる種々の名称。
表2:コンセンサス配列の計算に用いた再配列したヒトの配列。(A) κ配列、(B) λ配列、(C) H鎖配列。配列の名称 (1)、アミノ酸にした配列の長さ (2)、生殖細胞系ファミリー (3)、および算定される生殖細胞系配列を表にまとめた。再配列した配列と生殖細胞系配列との間で交換されたアミノ酸数は(5)に、異なるアミノ酸の割合は(6)に記載されている。(7)は対応する配列(配列に関連する引用文献は付録を参照)の参考文献番号である。
表3:再配列したV配列の、生殖細胞系配列への割り当て。(A) κ配列、(B) λ配列、(C) H鎖配列。生殖細胞系遺伝子はファミリーごと(1)に記載され、各生殖細胞系遺伝子に見つかった再配列した遺伝子の数は(2)に記載されている。
表4:再配列したVκ配列のコンセンサス配列の算定。(A) Vκサブグループ1、(B) Vκサブグループ2、(C) Vκサブグループ3、(D) Vκサブグループ4。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。(1) アミノ酸は標準的な1文字の略称で記載されている(BはDまたはN、ZはEまたはQ、Xは任意のアミノ酸を表す)。統計学的分析では、各位置に見られる配列の数(2)、最も頻度の高いアミノ酸の出現回数(3)、その位置で最も出現頻度が高いアミノ酸残基(4)、最も出現頻度が高いアミノ酸の相対的な頻度(5)、各位置に見られる異なるアミノ酸の数(6)がまとめられている。
表5: 再配列したVλ配列のコンセンサス配列の算定。(A) Vλサブグループ1、(B) Vλサブグループ2、(C) Vλサブグループ3。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。略号は表4と同じである。
表6:再配列したH鎖配列のコンセンサス配列の算定。(A) VH鎖サブグループ1A、(B) V H鎖サブグループ1B、(C) V H鎖サブグループ2、(D) V H鎖サブグループ3、(E) V H鎖サブグループ4、(F) V H鎖サブグループ5、(G) V H鎖サブグループ6。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。略号は表4と同じである。
【実施例】
【0040】
実施例1:合成ヒト組み合わせ抗体ライブラリー(HuCAL)の設計
以下の実施例は、ヒト免疫グロブリンのレパートリーのコンセンサス配列と、コンセンサス遺伝子の合成に基づく、全合成ヒト組み合わせ抗体ライブラリー(HuCAL)の設計を説明する。全般的な手順は図1に略図が示されている。
1.1 配列データベース
1.1.1 ヒト免疫グロブリン配列の収集および整列
最初の段階では、ヒト免疫グロブリンの可変部ドメインの配列を収集して、3つのサブデータベース、VH鎖(VH)、Vκ、およびVλに分割した。それから、各配列につき、遺伝子配列を対応するアミノ酸配列に翻訳した。その後、すべてのアミノ酸配列を、Kabatら(1991)に従って整列させた。Vλ配列については、Chuchanaら(1990)の番号付けシステムを使用した。3つのメインデータベースを、さらに2つのサブデータベースに分割した。最初のサブデータベースには、配列の70以上の位置が既知である、再配列したV遺伝子に由来するすべての配列が含まれていた。第2のサブデータベースは、すべての生殖細胞系遺伝子部分が含まれていた(D-およびJ-ミニ遺伝子を含まない;内部に停止コドンを有する偽遺伝子も除去された)。すべてにおいて、同一のアミノ酸配列を持ちながら名称の異なる生殖細胞系配列が見つかった場合には、1つの配列のみが使用された(表1参照)。再配列した配列の最終的なデータベースには、Vκ、Vλ、VHがそれぞれ386、149、674含まれていた。生殖細胞系配列の最終的なデータベースには、 Vκ、Vλ、VHがそれぞれ48、26、141含まれていた。
【0041】
1.1.2 サブグループへの配列の割り当て
3つの生殖細胞系データベースの配列を、配列の相同性に基づいて分類した(Tomlinsonら、1992、WilliamsおよびWinter、1993、およびCoxら、1994も参照)。 Vκの場合、7つのファミリーが確立された。 Vλは8つのファミリー、VHは6つのファミリーに分類された。VH7ファミリー(Van Dijkら、1993)のVH生殖細胞系遺伝子は、VH1ファミリーの遺伝子と高い相同性を示すため、VH1ファミリーに分類された。各ファミリーに含まれる生殖細胞系遺伝子の数は、1(例えばVH6)から47(VH3)までにわたっていた。
【0042】
1.2 配列の分析
1.2.1 生殖細胞系メンバーの算定
次に、再配列した遺伝子のデータベース中の1209アミノ酸配列の各々について、最も近い生殖細胞系配列、すなわち、アミノ酸の違いが最も少ない生殖細胞系配列を計算した。生殖細胞系配列が見つかった後に、再配列した遺伝子中に起りアミノ酸を変更した体細胞変異の数を表にすることができた。140の場合には、同じ数のアミノ酸の変更を有する複数の生殖細胞系遺伝子が見つかったため、対応する生殖細胞系配列を正確に計算することができなかった。これらの再配列した配列は、データベースから除去された。いくつかの場合には、アミノ酸の変更の数は異常に大きく(VLについて>20、VHについて>25)、変異の大きな再配列遺伝子であるか、またはデータベースには存在しない生殖細胞系遺伝子に由来するかのいずれかであることを示していた。これらの2つの可能性を区別することはできないため、これらの配列も、データベースから除去した。最後に、非常に変わったCDRの長さおよび組成、または共通位置(以下参照)に異常なアミノ酸を有するため、12の再配列した配列をデータベースから除去した。要約すると、1209の再配列した配列のうち、1023 (85%)を、生殖細胞系配列に明確に割り当てることができた(表2参照)。
この計算の後、すべての再配列した遺伝子を、生殖細胞系遺伝子について確立したファミリーの1つに割り当てることができた。ここで、各生殖細胞系遺伝子の使用量、すなわち、各生殖細胞系遺伝子から生ずる再配列した遺伝子の数を計算することができる(表2参照)。使用量は生殖細胞系遺伝子の1つのサブセットに強く偏向しており、生殖細胞系遺伝子の大部分はデータベース中の再配列した遺伝子に存在せず、したがって免疫系では使用されていないと思われることが分かった(表3)。この観察結果は、Vκ(Coxら、1994)についてはすでに報告済みだった。割り当てられる再配列した配列がないまたは非常に少ないすべての生殖細胞系遺伝子ファミリーをデータベースから除去すると、4つのVκ、3つのVλ、6つのVHファミリーのみが残った。
【0043】
1.2.2 CDRの立体配座
抗体分子の抗原結合ループCDRの立体配座は、CDRの長さと、いわゆる共通位置(ChothiaおよびLesk、1987)に位置するアミノ酸残基の両方に、強く依存している。免疫グロブリンの可変部ドメインの構造的レパートリーを決定する共通構造は、わずか数種類しか存在しないことが分かった(Chothiaら、1989)。共通アミノ酸位置は、CDRおよびフレームワーク領域に見つかる。上記のように決定した使用されている13の生殖細胞系ファミリー(7つのVLおよび6つのVH)につき、これらのファミリーにコードされる構造的レパートリーを決定するため、共通構造に関して分析した。
4つのVκファミリーのうちの3つ( Vκ1、2および4)では、各ファミリーについてそれぞれ異なる1つのタイプのCDR1立体配座が決定された。 Vκ3は2つのタイプのCDR1立体配座を示した:1つはVκ1と同一で、1つはVκ3にのみ見られる。すべてのVκCDR2は、同じタイプの共通構造を使用していた。CDR3立体配座は、生殖細胞系遺伝子部分にはコードされていない。したがって、配列の相同性と使用量によって決定された4つのVκは、 Vκ生殖細胞系遺伝子中に見られる4つのタイプの共通構造にも対応する。
上記のように決定した3つのVλファミリーは、3つのタイプのCDR1立体配座を示し、各ファミリーがそれぞれ異なる1つのタイプを示した。 Vλ1ファミリーには2つの異なるCDR1の長さ(13および14アミノ酸)が含まれていたが、共通残基は同一で、両方とも同じ共通立体配座を取ると考えられた(ChothiaおよびLesk、1987)。使用されているVλ生殖細胞系のCDR2には1つの共通立体配座が存在するのみで、CDR3立体配座は生殖細胞系遺伝子部分にはコードされていない。したがって、配列の相同性と使用量によって決定された3つのVλは、 3つのタイプの共通構造にも対応する。
ヒトVH配列の構造的レパートリーは、Chothiaら、1992によって詳細に分析された。合わせて、CDR1の3つの立体配座(H1-1、H1-2、およびH1-3)およびCDR2の6つの立体配座(H2-1、 H2-2、 H2-3、 H2-4、 H2-5、およびH2-x)が決定された。CDR3はD-およびJ-ミニ遺伝子部分にコードされているため、このCDRには特に共通残基は決定されていない。
上記のように決定されたVH1ファミリーのすべてのメンバーは、CDR1立体配座H1-1を含んでいたが、CDR2立体配座は異なっていた:6の生殖細胞系遺伝子にはH2-2立体配座が見られたが、8つの生殖細胞系遺伝子にはH2-3が見られた。この2つのタイプのCDR2立体配座は、フレームワーク位置72にあるアミノ酸のタイプの違いにより定義されているため、VH1ファミリーは2つのサブグループに分類された。VH1AはH2-2のCDR2立体配座を、VH1BはH2-3の立体配座を有する。VH2ファミリーのメンバーはすべて、CDR1とCDR2にそれぞれH1-3とH2-1の立体配座を持っていた。VH3メンバーのCDR1の立体配座はすべての場合にH1-1だっ他が、CDR2には4つの異なるタイプが見られた(H2-1、H2-3、H2-4、およびH2-x)。これらのCDR2立体配座では、フレームワークの共通残基71は常にアルギニンに決められている。したがって、CDR2の4つのタイプの立体配座は、CDR2自身によってのみ決定されるため、VH3ファミリーをサブファミリーに分割する必要はなかった。VH4ファミリーに関しても同様だった。ここでは、3つのタイプのCDR1の立体配座がすべて見られたが、CDR1の立体配座はCDR自身によって決定されるため(フレームワークの共通残基26はすべての場合にグリシンだった)、さらに分割する必要はなかった。VH4メンバーのCDR2立体配座は、すべての場合にH2-1だった。VH5ファミリーのすべてのメンバーは、それぞれH1-1およびH2-2の立体配座を有することが分かった。VH6ファミリーの1つのみの生殖細胞系遺伝子は、CDR1とCDR2にそれぞれH1-3およびH2-5の立体配座を持っていた。
要約すると、VκおよびVλ遺伝子のすべての可能なCDR立体配座が、配列の比較により定義された7つのファミリーに存在していた。 VH1ファミリーを2つのサブファミリーに分け7つのVHファミリーを作製することにより、使用されているVH生殖細胞系遺伝子に見られる12の異なるCDR立体配座のうち、7つがカバーされた。残る5つのCDR立体配座(VH3ファミリーの3つおよびVH4ファミリーの2つ)は、CDR自身によって決定されており、CDRライブラリーの構築時に作製し得る。したがって、使用されているヒトV遺伝子の構造的レパートリーは、49 (7 x 7)の異なるフレームワークでカバーされ得る。
【0044】
1.2.3 コンセンサス配列の算定
再配列した配列の14のデータベース(4 Vκ、3 Vλ、および7 VH)を用いて、各サブグループのHuCALコンセンサス配列を算定した(4 HuCAL-Vκ、3 HuCAL-Vλ、7 HuCAL-VH、表4、5、6参照) 。これは、各位置において使用されるアミノ酸残基の数を数え(位置変動性)、その後、各位置において最も高頻度で使用されるアミノ酸残基を同定することにより行った。コンセンサスの算定に、使用されている生殖細胞系配列の代わりに再配列した配列を使用することで、コンセンサス配列は使用量の頻度にしたがって加重された。さらに、高頻度で変異したり、高度に保存される位置を同定できた。コンセンサス配列は、生殖細胞系ファミリーのコンセンサスと相互チェックし、再配列した配列が特定の位置で、収集した生殖細胞系配列には存在しないアミノ酸残基に偏向しているかどうかを確認したが、そのようなことはなかった。その後、14のコンセンサス配列の各々につき、各特定のファミリーに見られる生殖細胞系配列の各々に対する差異の数を計算した。最も相同性の高い生殖細胞系配列からの総合的な偏差は2.4アミノ酸残基(標準偏差=2.7)で、「人工的な」コンセンサス配列が、免疫原性に関しては正しくヒトの配列とみなすことができることが保証される。
【0045】
1.3 構造分析
ここまでは、コンセンサス配列の設計には、配列情報のみが使用されている。配列中では遠くに位置するが、3次元構造では相互に接触し、フレームワークを不安定にしたり、または誤った折り畳みを導くようなアミノ酸残基の特定の人工的な組み合わせが計算時に生成した可能性があるため、14のコンセンサス配列を、その構造的性質に従って分析した。
データベース中に存在する再配列した配列は、すべて機能的で、故に正しく折り畳まれた抗体分子に対応すると考えた。したがって、各コンセンサス配列について、最も相同性の高い再配列した配列を計算した。コンセンサス配列が、再配列した配列と異なる位置は、潜在的「人工残基」と同定し、検査した。
検査自体は2つの方向で行った。まず、各潜在的「人工残基」のまわりの局所的配列を、すべての再配列した配列の対応する列と比較した。この列が真に人工的ならば、すなわち、再配列した配列のいずれにも出現することがないならば、決定的な残基をその位置に2番目に高頻度で見られるアミノ酸に変換し、再度分析した。第2に、潜在的「人工残基」を長い範囲の相互作用について分析した。これは、対応するPDBファイルで利用できるヒト抗体可変部ドメインのすべての構造を収集し、すべての構造について、確立された各アミノ酸残基の各側鎖に対する相互作用の数とタイプを計算して行った。これらの「相互作用地図」を用いて、潜在的「人工残基」に考えられる側鎖/側鎖相互作用を分析した。この分析の結果、以下の残基を交換した(遺伝子の名称、Kabatの番号付けに従う位置、この位置で最も高頻度に見られるアミノ酸、およびその代わりに使用したアミノ酸が記載されている)。
VH2:S65T
Vκ1:N34A
Vκ3:G9A、D60A、R77S
Vλ3:V78T
【0046】
1.4. CDR配列の設計
上記の過程で、再配列した配列のデータベースのみに由来する完全なコンセンサス配列が得られる。再配列した配列および変異した配列のCDRは、特定の抗原に対して偏向しているため、CDR1およびCDR2領域は、使用されている生殖細胞系配列のデータベースから取るべきであると考えた。さらに、生殖細胞系CDR配列は、CDR3のみが異なる1次免疫応答において、様々な抗原への結合を可能にすることが知られている。したがって、VHおよびVκの場合には、上述の計算から得られるコンセンサスCDRを生殖細胞系CDRで置き換えた。Vλの場合は、プロテアーゼに切断される可能性のある部位、および構造的な制約の可能性を避けるため、選択された生殖細胞系CDRのいくつかで、少数のアミノ酸を変更した。
以下の生殖細胞系遺伝子のCDRが選択された:

CDR3の場合、これらは最初にオリゴヌクレオチドライブラリーにより置換する計画だったため、任意の配列を選択することができた。大腸菌においてコンセンサス配列の発現および折り畳みの振る舞いを調べるためには、すべての配列が同一のCDR3を有すると、折り畳みの振る舞いに対するCDR3の影響はすべての場合に同一になるため、これは有用である。VL鎖(κおよびλ)およびVH鎖に、それぞれQQHYTTPPおよびARWGGDGFYAMDYという架空配列を選択した。これらの配列は、大腸菌における抗体の折り畳みと適合することが既知である(Carterら、1992)。
【0047】
1.5 遺伝子設計
上述の過程により、最終的に、ヒトの免疫系に頻繁に使用されている構造的な抗体レパートリーを表す、14のHuCALアミノ酸配列のコレクションが得られた(図2参照)。これらの配列をDNA配列に逆翻訳した。最初の段階では、大腸菌で頻繁に使用されることが既知のコドンのみを使用して逆翻訳を行った。その後、これらの遺伝子配列を使って、対応するアミノ酸配列を変化させずに導入できる可能性のある制限エンドヌクレアーゼ部位のデータベースを作製した。このデータベースを用いて、遺伝子のすべてのサブ要素(CDRおよびフレームワーク領域)に隣接する領域に位置し、同じ位置で同時にすべてのHuCAL VH、VκまたはVλ遺伝子に導入できる切断部位を選択した。いくつかの場合には、1つのサブグループのすべての遺伝子の切断部位を見つけることができなかった。そのような場合は、利用できる配列および構造の情報に従ってアミノ酸配列を変更することが可能ならば、変更した。この変更は、再び上述のように分析した。この設計過程において、合計で以下の6つのアミノ酸残基を変更した(遺伝子の名称、Kabatの番号付けに従う位置、この位置で最も高頻度に見られるアミノ酸、およびその代わりに使用したアミノ酸が記載されている)。
VH2:T3Q
VH6:S42G
Vκ3:E1D、I58V
Vκ4:K24R
Vλ3:T22S
1つの場合には(VHフレームワーク3の5'末端)、7つのVH遺伝子すべてにある1つの切断部位を同定することができなかった。代わりに、2種類の切断部位を使用した:HuCAL VH1A、VH1B、VH4およびVH5にはBstEII、 そしてHuCAL VH2、VH3、VH4、およびVH6にはNspVを使用した。
サブ要素に隣接する領域には位置しないが、アミノ酸配列を変更することなく1つのグループのすべての遺伝子に導入することができる制限エンドヌクレアーゼ部位がいくつか同定された。これらの切断部位も、さらに改良するためにこのシステムの柔軟性を高めるために導入された。最後に、すべての遺伝子配列から、残りの制限エンドヌクレアーゼ部位のうち1つを除いてすべてを除去した。除去されなかった1つの切断部位は、1つのサブグループのすべての遺伝子で異なっており、制限酵素の消化により異なる遺伝子を簡単に同定するための「フィンガープリント」部位として使用できる。設計された遺伝子、それに対応するアミノ酸配列、およびグループに特異的な制限エンドヌクレアーゼ部位は、それぞれ図3、4、5に示されている。
【0048】
1.6 遺伝子合成およびクローニング
コンセンサス遺伝子は、図6に示されるオリゴヌクレオチドを用いてProdromouおよびPearl、1992によって記述された方法で合成された。Kabatら、1991(図6および図7参照)による配列情報に基づいて、ヒト定常部ドメインCκ、Cλ、およびCH1をコードする遺伝子部分も合成された。CDR3およびフレームワーク4遺伝子部分については、すべてのHuCAL Vκ、Vλ、およびVH遺伝子でそれぞれ同一の配列が選択されたため、定常部ドメインをコードする対応する遺伝子部分と共に、この部分は1度しか作製されなかった。PCR産物はpCR-Script KS(+)(Stratagene社)またはpZErO-1(Invitrogen社)にクローニングし、配列決定により確認した。
【0049】
実施例2:HuCALに基づく抗体ライブラリーのクローニングと試験
合成コンセンサス遺伝子の作製後、2つの合成コンセンサス遺伝子の組み合わせを選択し、これらの2つのコンセンサスフレームワークに基づくライブラリーから、結合する抗体断片が単離できるかどうかを試験した。2つの遺伝子は単鎖Fv (scFv)フラグメントとしてクローニングし、VH-CDR3ライブラリーを挿入した。ライブラリーに機能的な抗体分子が存在するかどうかを試験するため、抗原としてBSAに結合した小さなハプテンフルオレセイン (FITC-BSA)を用いた選択過程を実施した。
【0050】
2.1 HuCAL VH-3Vκ2 scFvフラグメントのクローニング
コンセンサス遺伝子の設計を試験するために、無作為に選択した合成のH鎖とH鎖遺伝子の組み合わせ(HuCAL Vκ2およびHuCAL VH-3)を用いて、単鎖抗体 (scFv)フラグメントを作製した。簡単に述べると、CDR3-フレームワーク4領域を含む、VH3コンセンサス遺伝子およびCH1遺伝子部分をコードする遺伝子部分、ならびに、CDR3-フレームワーク4領域を含む、Vκ2コンセンサス遺伝子およびCκ遺伝子部分を組み立て、それぞれVH3-CH1 Fd断片の遺伝子およびVκ2-CκL鎖をコードする遺伝子を生成した。その後CH1遺伝子部分を、AGGGSGGGGSGGGGSGGGGSという配列の20量体ペプチドリンカーをコードするオリゴヌクレオチドカセットによって置き換えた。このリンカーをコードする2つのオリゴヌクレオチドは、それぞれ5'- TCAGCGGGTGGCGGTTCTGGCGGCGGTGGGAGCGGTGGCGGTGGTTCTGGCGGTGGTGGTTCCGATATCGGTCCACGTACGG-3'およびAATTCCGTACGTGGACCGATATCGGAACCACCACCGCCAGAACCACCGCCACCGCTCCCACCGCCGCCAGAACCGCCACCCGC-3'である。最後に、HuCAL-Vκ2遺伝子をEcoRVおよびBSiWIを通してHuCAL-VH3-リンカー融合物に挿入し、単鎖形式のVH-リンカー-VLで2つのコンセンサス配列をコードする最終的な遺伝子、HuCAL-VH3-Vκ2を得た。全コーディング配列は図8に示されている。
【0051】
2.2 1価ファージディスプレイプラスミドベクターpIG10.3の作製
H3κ2 scFv遺伝子のファージディスプレイシステム(Winterら、1994)を作製するために、ファージミドpIG10.3(図9)を構築した。簡単に述べると、ファージミドベクターpIG10(Geら、1995)のEcoRI/HindIII制限断片を、アンバーコドン(アンバーサプレッサー株XL1 Blueでグルタミン酸、非サプレッサー株JM83で停止コドンをコードする)および遺伝子IIIの切形バージョン(コドン249の融合接合部、Lowmanら、1991参照)をPCR変異誘発を使って後につなげたc-mycによって置き換えた。
【0052】
2.3. H-CDR3ライブラリーの構築
トリヌクレオチドコドンを含むオリゴヌクレオチドを鋳型として用い、隣接領域に相補的なオリゴヌクレオチドをプライマーとして用いて、2つの長さ(10および15アミノ酸)のH鎖CDR3ライブラリーを構築した。機能的な抗体に最も高頻度で出現するCDR3構造にのみ集中するため、CDR3ループのRH94およびDH101のソルトブリッジを保存した。15量体のライブラリーに関しては、フェニルアラニンとメチオニンがこの長さのヒトCDR3に非常に頻繁に出現するため(データは示していない)、100の位置にはこの2つの残基の両方が導入された。同じ理由から、102の位置にはバリンとチロシンが導入された。無作為化したすべての他の位置には、トリヌクレオチド混合物には使用されていないシステイン以外のすべてのアミノ酸のコドンが含まれていた。
オリゴヌクレオチド鋳型O3HCDR103T (5'- GATACGGCCGTGTATTATTGCGCGCGT (TRI)6GATTATTGGGGCCAAGGCACCCTG-3') およびO3HCDR153T (5'- GATACGGCCGT GTATTATTGCGCGCGT(TRI)10(TTT/ATG)GAT(GTT/TAT)TGGGGCCAAGGCACCCTG-3') ならびにプライマーO3HCDR35 (5'-GATACGGCCGTGTATTATTGC-3')およびO3HCDR33 (5'-CAGGGTGCCTTGGCCCC-3') を用いたPCR断片から、長さが10および15のCDR3ライブラリーを作製した。ここで、TRIはシステイン以外のすべてのアミノ酸を含むトリヌクレオチド混合物、(TTT/ATG)および(GTT/TAT)はそれぞれ、アミノ酸のフェニルアラニン/メチオニン、およびバリン/チロシンをコードするトリヌクレオチド混合物である。これらのライブラリーの潜在的な多様性は、10量体と15量体につき、それぞれ4.7 x 107および3.4 x 1010である。ライブラリーカセットは、まず両方のプライマーの存在下でオリゴの鋳型をPCR増幅して合成した:総容量100 μl中にオリゴの鋳型O3HCDR103TまたはO3HCDR153Tが25 pmol、プライマーO3HCDR35およびO3HCDR33が各50 pmol、dNTPが20 nmol、10x緩衝液およびPfu DNAポリメラーゼ(Stratagene社)が2.5ユニットで、30サイクル(92℃で1分、62℃で1分、72℃で1分)。ホットスタート手順を使用した。この結果として得られた混合物をフェノール抽出し、エタノール沈殿し、さらにEagIおよびStyIにより一晩消化した。H-CDR3をコードするベクターpIG10.3-scH3κ2のEagI-StyI断片が、これらの2つの部位で挟まれているクロラムフェニコールアセチルトランスフェラーゼ遺伝子(cat)によって置き換えられたベクターpIG10.3-scH3κ2catを、同様に消化した。消化されたベクター(35 μg)をゲル精製し、100 μgのライブラリーカセットと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿物を100 μlのddH2Oに再溶解した。連結物は氷上で、新しく調製したエレクトロコンピテントXL1 Blue 1 mlと混合した。20回のエレクトロポレーションを行い、形質転換細胞をSOC培地で希釈し、37℃で30分間振盪し、37℃で6〜9時間大きなLBプレート(Amp/Tet/グルコース)に蒔いた。形質転換細胞の数(ライブラリーサイズ)は10量体と15量体につき、それぞれ3.2 x 107および2.3 x 107だった。コロニーは2xYT培地(Amp/Tet/グルコース)に懸濁し、グリセロール培養として保存した。
最初のライブラリーの質を試験するために、24の独立したコロニー(10量体から12、15量体から12)からファージミドを単離し、制限酵素による消化と配列決定を行って分析した。24のファージミドの分析では、すべてにおいて完全なベクターが存在していることが示された。これらのクローン(図10参照)の配列分析により、24のうちの22がH鎖CDR3領域の機能的な配列を含むことが示された。10量体ライブラリーの12クローンのうちの1つは、長さが10ではなく9のCDR3を持っていた。15量体ライブラリーの12のクローンのうちの2つは、オープンリーディングフレームを持たないため非機能的scFvを持っていた。これら2つのクローンのうちの1つは、2つの連続的インサートを含んでいたが、読み枠は、ずれていた(データは示していない)。導入されたすべてのコドンは、片寄りなく分布していた。
各ライブラリーメンバーの発現量も測定した。簡単に述べると、各ライブラリーから9クローンをAmp/Tet/0.5%グルコースを含む2xYT培地中で37℃で一晩培養した。次の日、培養液をAmp/Tetを含む新鮮な培地で希釈した。OD600nmが0.4の時点で、培養物を1 mMのIPTGで誘導し、室温で一晩振盪した。その後、細胞沈殿物を1 mM EDTAを添加したPBS緩衝液1 mlに懸濁した。懸濁液を超音波処理し、上清を還元条件のSDS-PAGEで分離し、ナイロン膜にブロッティングし、抗FLAG M1抗体で検出した(図11参照)。10量体のライブラリーの9つのクローンは、すべてscFvフラグメントを発現していた。さらに、すべてにおいて遺伝子III/scFv融合タンパク質が存在していた。15量体ライブラリーで分析した9つのクローンでは、6/9 (67%)がscFvと遺伝子III/scFv融合タンパク質の両方を発現していた。さらに重要なことに、 scFvと遺伝子III/scFv融合タンパク質を発現するすべてのクローンは、ほぼ同じ発現量を示した。
【0053】
2.4 バイオパニング
標準的なプロトコールを用いて、抗体ライブラリーをディスプレイするファージを調製した。10量体ライブラリー由来のファージを、15量体ライブラリーのファージと20:1の比率で混合した(1ウェル当たり10量体を1 x 1010 cfuおよび15量体を5 x 108)。続いて、ファージ溶液を使用して、PBS中100 μg/mlの濃度でFITC-BSA(Sigma社)によりコートしたELISAプレート(Maxisorp, Nunc社)で4℃で一晩のパニングを行った。抗原によりコートされたウェルをPBS中の3%粉ミルクでブロックし、各ウェルに1%粉ミルク中のファージ溶液を添加し、プレートを室温で1時間振盪した。その後ウェルをPBSTおよびPBSで洗った(室温で5分間の振盪を伴い各4回)。結合したファージを室温で0.1 Mトリエチルアミン(TEA)により10分間溶出した。溶出されたファージ溶液をただちに1/2の体積の1 M Tris-Cl、pH 7.6で中和した。溶出されたファージ溶液(約450 μl)を用いて37℃で30分間XL1 Blue細胞を感染した。その後、感染した培養液を大きなLBプレート(Amp/Tet/グルコース)上に蒔き、コロニーが見えるまで37℃で培養した。コロニーを2xYT培地に懸濁し、グリセロール培養を上述のようにして作製した。このパニング操作を2回繰り返し、3回目の溶出はPBS中に100 μg/mlの濃度でフルオレセインを添加して行った。特異的ファージ抗体の濃縮は、最初および後のフルオレセイン特異的サブライブラリーをブロッキング緩衝液に対してパニングしてモニターした(図12)。フルオレセインに対する特異性を有する抗体は、3回のパニング後に単離された。
【0054】
2.5 ELISA測定
バイオパニングの成功の重要な基準の1つは、標的抗原またはハプテンに結合する個々のファージクローンの単離である。我々は、抗FITCファージ抗体クローンを単離し、まずファージELISA形式で解析した。3回のバイオパニング(上記参照)の後、ファージミドを含む24のクローンを、ELISAプレート(Nunc社)中で100 μlの2xYT培地(Amp/Tet/グルコース)に接種し、その後37℃で5時間振盪した。100 μlの2xYT培地(Amp/Tet/1 mM IPTG)を添加し、振盪を30分間続けた。さらに、ヘルパーファージ(1 x 109 cfu/ウェル)を含む100 μlの2xYT培地(Amp/Tet)を加え、室温で3時間振盪した。ヘルパーファージの感染が成功したものを選択するためにカナマイシンを添加し、一晩振盪を継続した。その後プレートを遠心し、上清を100 μl FITC-BSA(100 μg/ml)でコートして粉ミルクでブロックしたELISAウェルに直接ピペットで添加した。パニング時と同様に洗い、可溶性POD基質(Boehringer-Mannheim社)を用いて、抗M13抗体-POD結合物(Pharmacia社)によって結合したファージを検出した。 FITC-BSAに対してスクリーニングした24のクローンのうち、22はELISAで活性を示した(図13)。同様の力価を有する最初のライブラリーでは、シグナルは検出されなかった。
フルオレセインに対する特異性は、競合的ELISAによって測定した。上述のようにして、FITCに特異的な5つのscFvのペリプラズム画分を調製した。ウエスタンブロッティングにより、すべてのクローンがほぼ同量のscFvフラグメントを発現することが示された(データは示していない)。上述のようにしてELISAを行ったが、さらに、ウェルに添加する前に、ペリプラズム画分を緩衝液(阻害なし)、10 mg/ml BSA(BSAによる阻害)、または10 mg/mlフルオレセイン(フルオレセインによる阻害)のいずれかで30分間インキュベートした。結合するscFvフラグメントを抗FLAG抗体M1を用いて検出した。可溶性フルオレセインを加えたときにのみELISAシグナルは阻害され、scFvの結合はフルオレセインに特異的であることが示された(図14)。
【0055】
2.6 配列分析
ライブラリー中のフルオレセイン結合抗体の配列の多様性を見積もるために、20クローンのH鎖CDR3領域の配列を分析した(図15)。合計では、20配列のうちの16(80%)が異なっており、構築したライブラリーにはフルオレセインと結合する非常に多様なレパートリーが含まれることが示された。CDR3は特定の配列の相同性を示さなかったが、平均4つのアルギニン残基を含んでいた。フルオレセイン結合抗体におけるアルギニンに対するこの偏向は、Barbasら、1992によってすでに記述されている。
【0056】
2.7 生産
選択された3つのクローンのファージミドDNAによって大腸菌JM83を形質転換し、0.5 L 2xYT培地中で培養した。OD600nm=0.4において1 mM IPTGにより誘導を行い、室温で一晩激しく振盪して増殖させた。細胞を収集し、沈殿物をPBS緩衝液中に懸濁し、超音波処理した。遠心により細胞破片から上清を分離し、BioLogicシステム(Bio-Rad社)により、POROS MC 20カラム(IMAC、PerSeptive Biosystems社)とイオン交換クロマトグラフィーを組み合わせて精製した。イオン交換カラムは、POROS HS、CM、もしくはHQ、またはPI 20(PerSeptive Biosystems社)のうちの1つで、精製するscFvの理論的pIに依存した。すべての緩衝液のpHは、常に精製するscFvのpIよりも1単位低いまたは高いように調製した。サンプルを最初のIMACカラムにかけ、カラム容積の7倍の20 mMリン酸ナトリウム、1 M NaCl、10 mMイミダゾールで洗った。洗浄後、カラム容積の7倍の20 mMリン酸ナトリウム、10 mMイミダゾールで洗った。その後カラム容積の3倍のイミダゾール勾配(10〜250 mM)をかけ、溶出物をイオン交換カラムに直接につなげた。カラム容積の9倍の250 mMイミダゾールによる無勾配洗浄後、カラム容積の15倍の250 mMから100 mM、およびカラム容積の7倍のイミダゾール/NaCl勾配(100〜10 mMイミダゾール、0〜1 M NaCl)で洗浄した。流速は5 ml/minだった。scFvフラグメントの純度は、SDS-PAGEクーマシー染色(図16)で確認した。フラグメントの濃度は、理論的に決定した吸光係数(Gillおよびvon Hippel、1989)を用いて、280 nmにおける吸光度により決定した。scFvフラグメントは均一に精製された(図16参照)。精製したフラグメントの収量は5〜10 mg/L/ODにわたっていた。
【0057】
実施例3:抗原のコレクションに対するHuCAL H3κ2ライブラリー
実施例2で使用したライブラリーをさらに試験するために、βエストラジオール 、テストステロン、ルイス-Yエピトープ(LeY)、インターロイキン-2(IL-2)、リンホトキシン-β(LT-β)、E-セレクチンリガンド-1(ESL-1)、およびBSAを含む様々な抗原を用いて、新規の選択手順を実行した。
【0058】
3.1 バイオパニング
ライブラリーおよびすべての手順は実施例2に記載されたものと同一である。ELISAプレートを、βエストラジオール-BSA(100 μg/ml)、テストステロン-BSA(100 μg/ml)、 LeY-BSA(20 μg/ml)、 IL-2(20 μg/ml)、ESL-1(20 μg/ml)、BSA(100 μg/ml)、LT-β(変性タンパク質、20 μg/ml)によってコートした。最初の2回には、結合したファージを0.1 Mトリエチルアミン(TEA)により室温で10分間溶出した。BSAの場合、3回のパニング後の溶出は、PBS中100 μg/mlの濃度のBSAをさらに添加して行った。他の抗原の場合は、3回目の溶出は0.1 Mトリエチルアミンによって行った。LeYを除くすべての場合に、結合するファージの濃縮が観察された(図17)。さらに、15量体ライブラリーのみを用いてバイオパニング実験を繰り返すと、LeY結合ファージも濃縮された(データは示されていない)。
【0059】
3.2 ELISA測定
FITCについて実施例2に記載された手順で、β-エストラジオール、テストステロン、LeY、LT-β、ESL-1およびBSAに結合するクローンをさらに分析および解析した。抗βエストラジオールおよび抗ESL-1抗体に関するELISAデータは図18に示されている。1つの実験では、scFvフラグメントの選択性と交差反応性を試験した。このために、ELISAプレートをFITC、テストステロン、βエストラジオール、BSA、およびESL-1の各抗原により5ウェルずつ5列に並べてコートし、各抗原につき1つ、合計5つの抗体を抗原についてスクリーニングした。図19は抗体が選択された抗原に特異的に結合し、他の4つの抗原には交差反応性が低いことを示す。
【0060】
3.3 配列分析
β-エストラジオール(34クローン)、テストステロン(12クローン)、LT-β(23クローン)、 ESL-1(34クローン)およびBSA(10クローン)に対するいくつかのクローンの配列データを図20〜24に示す。
【0061】
実施例4:ベクターの構築
コンセンサス遺伝子レパートリーがモジュール型であることを利用するためには、HuCALライブラリーのファージディスプレイスクリーニングとその後の最適化手順に使用できるベクター系を構築する必要がある。したがって、一本鎖または二本鎖の複製開始点、プロモーター/オペレーター、リプレッサーまたはターミネーター要素、抵抗性遺伝子、組み換え可能な部位、線状ファージ上のディスプレイのための遺伝子III、シグナル配列、または検出用標識のようなすべての必要なベクター要素が、モジュール型コンセンサス遺伝子の制限部位のパターンに適合するようにしなくてはならない。図25はpCALベクター系の概略図と、その中のベクターモジュールおよび制限部位の配置を示す。図25aは、すでにコンセンサス遺伝子またはベクター要素にモジュールシステムの一部として組み込まれているか、またはシステム全体にまだ存在しないすべての制限部位のリストを示す。後者は、後の段階で新規モジュールの導入のために、または新規モジュール中で使用することができる。
【0062】
4.1 ベクターモジュール
HuCAL遺伝子のサブ要素に隣接する制限部位を取り除き、ベクターモジュール自身が唯一である制限部位に隣接する、一連のベクターモジュールを作製した。これらのモジュールは、遺伝子合成または鋳型の変異誘発のいずれかによって作製した。変異誘発はアド・オンPCRにより、PCRに基づくアセンブリー方法を用いて部位特異的突然変異誘発法(Kunkelら、1991)または多部位のオリゴヌクレオチドを介する変異誘発(Sutherlandら、1995;Perlak、1990)によって行った。
図26は作製したモジュールのリストを含む。制限部位FseIを追加するために、ターミネーターモジュールM9 (HindIII-Ipp-PacI) の代わりに、より大きなカセットM9IIを調製した。M9IIはHindIII/BsrGIによりクローニングできる。
すべてのベクターモジュールは、制限分析と配列決定により解析した。モジュールM11-IIの場合は、モジュールの配列決定により、鋳型の配列データベースと比較すると164/65の位置の2つの塩基が異なることが分かった。これらの2つの塩基(CA→GC)により別のBanII部位が生成した。同じ2つの塩基の違いは、他のバクテリオファージのf1複製開始点中にも出現するため、この2つの塩基は鋳型に存在しておりクローニング中の変異誘発によって生成したものではないと考えられた。このBanII部位は部位特異的突然変異誘発法により除去され、モジュールM11-IIIが生成した。モジュールM14のBssSI部位は、初めはColE1複製開始点の機能に影響を与えずに除去することができなかったため、最初のpCALベクターシリーズのクローニングにはM14-Ext2が使用された。図29〜34はモジュール型ベクターpCAL4(以下参照)の組立てに使用したモジュールの機能地図と配列を示す。他のモジュールの機能地図と配列は図35aに記載されている。図35bにはモジュールの合成に使用したオリゴヌクレオチドおよびプライマーのリストが記載されている。
【0063】
4.2 クローニングベクターpMCS
各ベクターモジュールを組み立てるために、特異的な多クローニング部位モジュール(MCS)を含むクローニングベクターpMCSを作製した。まず、MCSカセット(図27)を遺伝子合成によって作製した。このカセットには、すべてのベクターモジュールを逐次導入するために必要なすべての制限部位が順番に含まれており、5'-HindIII部位および3'に突出したAatII部位と適合する4つの塩基を用いてクローニングできる。ベクターpMCS(図28)はpUC19をAatIIおよびHindIIIによって消化し、bla遺伝子とColE1複製開始点を含む2174塩基対の断片を単離し、MCSカセットを連結して作製した。
【0064】
4.3 モジュール型ベクターpCAL4のクローニング
これは、pMCSを制限酵素で消化し、引き続きモジュールM1(AatII/XbaIによる)、M7III(EcoRI/HindIIIによる)、M9II(HindIII/BsrGIによる)、M11-II(BsrGI/NheIによる)を連結して段階的に行った。最後に、bla遺伝子をcat遺伝子モジュールM17(AatII/BgIIIによる)によって、野生型のColE1複製開始点をモジュールM14-Ext2(BglII/NheI)によって置き換えた。図35はpCAL4の機能地図と配列を示す。
【0065】
4.4 低コピー数プラスミドベクターpCALOのクローニング
ColE1モジュールM14-Ext2の代わりにp15AモジュールM12を使用して、同様な方法で一連の低コピー数プラスミドベクターを作製した。図35aはベクターpCALO1〜pCALO3の機能地図と配列を示す。
実施例5:HuCAL scFvライブラリーの作製
【0066】
5.1 49のHuCAL scFvフラグメントすべてのクローニング
7つのHuCAL-VHおよび7つのHuCAL-VLコンセンサス遺伝子の49の組み合わせすべてを、実施例2でHuCAL VH3-Vκ2 scFvについて説明したようにして組み立て、抗体発現ベクターのpLiscシリーズの修飾バージョンであるベクターpBS12(Skerraら、1991)に挿入した。
【0067】
5.2 CDRクローニングカセットの構築
CDRの置換のために、5'末端および3'末端に複数のクローニング部位を有する普遍的βラクタマーゼクローニングカセットを作製した。5'の複数のクローニング部位には、 HuCAL VHおよびVLのCDRの5'末端に隣接するすべての制限部位が含まれており、3'の複数のクローニング部位には、 HuCAL VHおよびVLのCDRの3'末端に隣接するすべての制限部位が含まれている。5'および3'の複数のクローニング部位の両方とも、合成オリゴヌクレオチドを5'および3'プライマーとして使用し野生型βラクタマーゼ遺伝子を鋳型として使用したアド・オンPCRによってカセットとして調製した。図36はカセットbla-MCSの機能地図と配列を示す。
【0068】
5.3 VL-CDR3ライブラリーカセットの調製
7つの無作為位置を含むVL-CDR3ライブラリーは、Vκ遺伝子に関しては、オリゴヌクレオチド鋳型Vκ1およびVκ3、 Vκ2ならびにVκ4、プライマーO_K3L_5およびO_K3L_3(図37)、またVλ遺伝子に関しては、 VλおよびプライマーO_L3L_5 (5'-GCAGAAGGCGAACGTCC-3') およびO_L3LA_3(図38)を用いてPCR断片から作製した。カセットの構築は、実施例2.3に記述されるようにして行った。
【0069】
5.4 VL-CDR3ライブラリーを用いたHuCAL scFv遺伝子のクローニング
49の単鎖の各々をXbaI/EcoRIを用いてpCAL4にサブクローニングし、VL-CDR3をBbsI/MscIを用いてβラクタマーゼクローニングカセットにより置換し、その後これを上述のようにして合成した対応するVL-CDR3ライブラリーカセットによって置換した。このCDRの置換は、実施例2.3においてcat遺伝子を使用して説明されている。
【0070】
5.5 VH-CDR3ライブラリーカセットの調製
VH-CDR3ライブラリーは、実施例2.3に記載されるようにして設計および合成した。
【0071】
5.6 VL-およびVH-CDR3ライブラリーを用いたHuCAL scFv遺伝子のクローニング
VH-CDR3を置換するために、49の単鎖のVL-CDR3ライブラリーの各々をBssHII/StyIによって消化した。 BssHII/StyIによって消化された「ダミー(dummy)」カセットを挿入し、その後、これを上述のようにして合成した対応するVH-CDR3ライブラリーカセットによって置換した。
【0072】
実施例6:発現試験
scFv形式VH-リンカー-VLを用いて発現および毒性試験を行った。7つのHuCAL-VHおよび7つのHuCAL-VLコンセンサス遺伝子の49の組み合わせすべてを、実施例5で説明したようにして組み立て、抗体発現ベクターのpLiscシリーズの修飾バージョンであるベクターpBS13(Skerraら、1991)に挿入した。このベクターの地図は図39に示されている。
大腸菌JM83を各ベクターによって49回形質転換し、グリセロール保存株として保存した。4〜6クローンを同時に試験し、内部コントロールとして常にクローンH3κ2も試験に含んだ。別のコントロールとして、pBS13中のMcPC603 scFvフラグメント(KnappikおよびPluckthun、1995)も同一の条件下で発現させた。発現試験を行う2日前に、30 μg/mlクロラムフェニコールおよび60 mMグルコースを含むLBプレート上でクローンを培養した。このプレートを用いて、3 ml培養液(90 μgクロラムフェニコールおよび60 mMグルコースを含むLB培地)を37℃で一晩接種した。次の日、一晩培養した培養液を用いてクロラムフェニコール(30 μg/ml)を含む30 mlのLB培地を接種した。開始時のOD600nmを0.2に調製し、培養温度は30℃を使用した。8〜9時間の間、30分ごとに600 nmの光学密度を測定して細胞の生理を監視した。培養液のOD600nmが0.5に達したら、最終濃度が1 mMになるようにIPTGを添加して、抗体の発現を誘導した。2時間後に培養液から5 mlを採取し、抗体の発現を分析した。細胞を溶菌し、粗抽出物の可溶性画分および不溶性画分をKnappikおよびPluckthun、1995に記載されるようにして分離した。サンプルの光学密度を統一し、還元SDS-PAGEで画分を分析した。ブロッティングおよび1次抗体としてαFLAG抗体M1(Geら、1994参照)、2次抗体としてアルカリ性ホスファターゼ結合Fc特異的抗マウス抗血清を用いた免疫染色を行った後、レーンのスキャンを行い、期待されるサイズ(約30 kDa)のバンドの強度をデンシトメーターにより定量し、コントロールの抗体と比較して表にした(図40参照)。
【0073】
実施例7:フルオレセイン結合分子の最適化
7.1 L-CDR3およびH-CDR2ライブラリーカセットの構築
L-CDR3ライブラリーカセットは、オリゴヌクレオチド鋳型CDR3L (5'-TGGAAGCTGAAGACGTGGGCGTGTATTATTGCCAGCAG(TR5)(TRI)4CCG(TRI)TTTGGCCAGGGTACGAAAGTT-3') および相補鎖合成用プライマー5'-AACTTTCGTACCCTGGCC-3'を用いて調製した。ここで、(TRI)はCys以外のすべてのアミノ酸を含むトリヌクレオチド混合物で、(TR5)はAla、Arg、His、Ser、およびTyrの5つのコドンを含むトリヌクレオチドを含む。
H-CDR2ライブラリーカセットは、オリゴヌクレオチド鋳型CDRsH (5'-AGGGTCTCGAGTGGGTGAGC(TRI)ATT(TRI)2−3(6)2(TRI)ACC(TRI)TATGCGGATAGCGTGAAAGGCCGTTTTACCATTTCACGTGATAATTCGAAAAACACCA-3') および相補鎖合成用プライマー5'-TGGTGTTTTTCGAATTATCA-3'を用いて調製した。ここで、(TRI)はCys以外のすべてのアミノ酸を含むトリヌクレオチド混合物で、(6)は(A/G)(A/C/G)Tの混合を含みAla、Asn、Asp、Gly、Ser、およびThrの6つのコドンを形成し、長さの分布は、合成時に(TRI)混合物の準化学量論的カップリングを1度行いDNA合成に通常使用されるキャッピング段階を省略して得られた。
DNA合成を40 nmolスケールで行い、オリゴをTE緩衝液に溶解し、スピンカラム(S-200)を用いたゲル濾過により精製し、260 nmにおけるOD測定値によってDNA濃度を決定した(OD 1.0 = 40 μg/ml)。
オリゴヌクレオチド鋳型10 nmolおよび対応するプライマー12 nmolを混合し、80℃で1分間アニーリングさせ、20〜30分間かけて徐々に37℃に冷却した。クレノウポリメラーゼ(2.0 μl)および各dNTPを250 nmolずつ使用してフィルイン反応を37℃で2時間行った。過剰のdNTPをNick-Spinカラム(Pharmacia社)を用いたゲル濾過により除去し、BbsI/MscI(L-SCR3)またはXho/SfuI(H-CDR2)によって二本鎖DNAを37℃で一晩消化した。 Nick-Spinカラム(Pharmacia社)によりカセットを精製し、ODを測定して濃度を決定し、カセットを分注(15 pmol)して-80℃で保存した。
【0074】
7.2 ライブラリーのクローニング
実施例2で用いられたFITC結合クローンのコレクションからDNAを調製した(クローンに対し約104)。XbaI/EcoRI消化によりscFvフラグメントのコレクションを単離した。実施例4.3に説明されるベクターpCAL4(100 fmol、10 μg)を同様にXbaI/EcoRI消化し、ゲル濾過し、300 fmolのscFvフラグメントコレクションと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿物を100 μlのddH20に再溶解した。連結反応液を1 mlの新しく調製したエレクトロコンピテントなSCS 101細胞(L-CDR3の最適化用)またはXL1 Blue細胞(H-CDR2の最適化用)と氷上で混合した。エレクトロポレーションを1回行い、形質転換細胞をSOC培地で溶出し、37℃で30分間振盪し、一部をLBプレート(Amp/Tet/グルコース)上に37℃で6〜9時間蒔いた。形質転換細胞の数は5 x 104だった。
ベクターDNA(100 μg)を単離し、 L-CDR3の最適化にはBbsI/MscIで、 H-CDR2の最適化にはXhoI/NspVで消化した(scH3κ2の配列と制限地図は図8参照)。精製したベクター断片10μg (5 pmol)をL-CDR3またはH-CDR2ライブラリーカセット15 pmolと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿を100 μlのddH20に溶解した。連結反応液を新しく調製したエレクトロコンピテントなXL1 Blue細胞と氷上で混合した。エレクトロポレーションを行い、形質転換細胞をSOC培地で溶出し、37℃で30分間振盪し、一部をLBプレート(Amp/Tet/グルコース)上に37℃で6〜9時間蒔いた。形質転換細胞の数(ライブラリーサイズ)は両方のライブラリーとも108以上だった。ライブラリーはグリセロール培養として保存した。
【0075】
7.3 バイオパニング
これは実施例2.1において最初のH3κ2 H-CDR3ライブラリーについて記載したようにして実行した。FITCに結合する最適化したscFvは、実施例2.2および2.3に記載したようにして解析および分析したが、必要ならば、最適化をさらに繰り返すこともできる。
【0076】
参考文献
【技術分野】
【0001】
発明の分野
本発明は、1つまたは複数の相同蛋白質/(ポリ)ペプチドのコレクションをコードする合成DNA配列、ならびにこれらのDNA配列のライブラリーを作製および応用する方法に関する。特に、本発明はヒトのゲノム中にコードされる抗体の構造的レパートリーをカバーする合成コンセンサス配列を使用することによる、ヒト由来の抗体遺伝子ライブラリーの調製に関する。さらに本発明は、高度に多様化した抗体ライブラリーの普遍的フレームワークとしての、単一のコンセンサス抗体遺伝子の使用に関する。
【背景技術】
【0002】
発明の背景
例えば特定のリガンドに結合するというような、望ましい性質を有するメンバーをスクリーニングするために、例えば抗体などの蛋白質/(ポリ)ペプチドのライブラリーを用いる現行のすべての組み換え方法では、メンバーの望ましい性質を簡単かつ迅速に改良することはできない。通常、ライブラリーは、生物体からクローニングされた1つまたは複数のDNA配列に任意のオリゴヌクレオチド配列を挿入するか、またはDNA配列のファミリーをクローニングしライブラリーとして使用するかのいずれかによって作製される。その後、例えばファージのディスプレイなどを用いて、ライブラリーから所望の性質を示すメンバーをスクリーニングする。そして、この結果得られる1つまたは複数の分子の配列が決定される。これらの分子をさらに改良するために利用することのできる一般的な方法はない。
【0003】
Winter(欧州特許第0 368 684 B1号(特許文献1))により、抗体の可変領域遺伝子を増幅(PCRによる)し、クローニングし、発現させる方法が提供されている。これらの遺伝子から開始して、WinterはH鎖および/またはL鎖のCDR3を無作為化することにより、機能的な抗体断片のライブラリーを作製できた。この過程は、免疫系におけるB細胞の発生時に起こるVJおよびVDJの自然の組み換え過程と機能的には同等である。
【0004】
しかし、Winterの発明では、「親和性の成熟」という自然に起こる現象と機能的に同等であるような過程において、さらに抗体断片の結合親和性を最適化するための方法は提供されておらず、本発明によってこれは提供される。さらに、Winterの発明では、構造的に類似した天然の遺伝子ファミリー全体を表す、合成のDNAオリゴヌクレオチドから組み立てられるような、人工的可変領域遺伝子は提供されていない。また、Winterの発明では、抗体の可変領域の一部分を組み合わせて組み立てることはできないが、本発明によってこの機能が提供される。さらに、この方法では、PCRプライミング領域以外はライブラリーメンバーに関するさらなる配列情報は得られないため、スクリーニング過程で得られたすべての抗体遺伝子の配列を完全に決定する必要があるという欠点がある。これは時間と労力を要し、配列の誤りにもつながる可能性がある。
【0005】
Winterおよび他の方法の教示は、可能な限り多くの異なる抗原に対する抗体を見つけるために、相補性決定領域 (CDR) およびフレームワークにおいて、高度な多様性を有する大きな抗体ライブラリーを作製するよう試みられている。1つの普遍的なフレームワークが抗体ライブラリーを作製するために有用であることが示唆されているが、これはまだ成功を収めていない。
【0006】
もう1つの問題は、抗体に由来する試薬の生産である。小さな抗体断片は、治療薬、診断薬、および生化学研究に非常に有望である。したがって、これらは大量に必要とされ、例えば、大腸菌のペリプラズムにおけるFv、単鎖Fv (scFv)、またはFabのような抗体断片の発現(SkerraおよびPluckthun、1988(非特許文献1);Betterら、1988(非特許文献2))は、多くの研究室で日常的に使用されている。しかし、発現の収率には非常に差がある。振盪フラスコ培養の培養液1リットルおよびOD当たり機能的な可溶性蛋白質が数mgまで得られる断片もあるが(Carterら、1992(非特許文献3);Pluckthunら、1996(非特許文献4))、いわゆる封入体の中にしばしば見られる不溶性物質しかほとんど生成しない断片もある。後者からは、労力と時間をかけた再生過程を用いて、低い収量で機能的な蛋白質が得られる場合がある。抗体の発現量に影響を与える要素は、まだあまり分かっていない。抗体断片の折り畳み効率および安定性、プロテアーゼ感受性、ならびに発現された蛋白質の宿主細胞への毒性により実際の生産量がしばしばひどく制限されており、発現収率を上昇させるためのいくつかの試みが行われている。例えば、KnappikおよびPluckthun (1995)(非特許文献5)は、発現収率は抗体の配列に依存することを示した。彼等は、抗体のフレームワーク中で発現収率に大きな影響を与える鍵となる残基を同定した。同様に、Ullrichら(1995)(非特許文献6)は、CDR中の点変異がペリプラズムにおける抗体断片の発現の収量を上昇させ得ることを発見した。しかしながら、これらの戦略はごく一部の抗体にしか適用できない。Winterの発明は抗体の既存のレパートリーを使用するため、遺伝子の発現能力に影響を与えることはできない。
【0007】
さらに、KnappikおよびPluckthun、ならびにUllrichの結果は、抗体、特にその折り畳みと発現に関する知識がまだ増加中であることを示している。Winterの発明では、ライブラリーの設計にそのような改良を取り入れることはできない。
【0008】
大概の場合、スクリーニング手順はファージ表面における遺伝子産物のディスプレイに依存しており、効率の高いディスプレイには遺伝子が少なくともある程度は発現される必要があるため、遺伝子の発現能力は、ライブラリーの質にとっても重要である。
【0009】
本発明は、このような既存の方法の欠点を克服し、またすべての相同蛋白質のコレクションに適用できる。本発明は、以下に抗体を例として示される下記のような新規で有用な特徴を有する。
【0010】
相同抗体遺伝子のグループ全体の構造的性質を反映する既知の抗体配列に基づき、人工抗体およびその断片を作製することができる。したがって、構造的レパートリーを失うことなく、遺伝子の数を減らすことが可能である。この方法を用いることにより、制限された数の人工遺伝子が得られ、これを新規に合成することにより、切断部位を導入したり、不要な切断部位を除去したりすることができる。さらに、この方法によれば、(i) 遺伝子の使用コドンを、任意の所望の宿主細胞中で高度に発現する遺伝子の使用コドンに適合させることができ、また (ii) 抗体のL鎖とH鎖のすべての可能なペアについて、相互作用の選択、抗原の選択、または組み換え体の発現の力価に関して分析することができるが、これは生物体のすべての抗体遺伝子およびそのすべての組み合わせを用いた場合には、実質的に不可能である。
【0011】
限定された数の完全に合成された遺伝子のセットを使用すると、コードされる構造的サブ要素の境界部に切断部位を作製することが可能である。したがって、各遺伝子は、蛋白質/(ポリ)ペプチドレベルの構造的サブ要素を表すモジュールから築き上げられる。抗体の場合には、モジュールは「フレームワーク」と「CDR」から構成される。フレームワークとCDRのモジュールを別々に作製することにより、異なる組み合わせが可能になる。さらに、2つまたはそれ以上の人工遺伝子が、各々の遺伝子サブ要素の境界部に同一の切断部位を持っている場合には、特定の遺伝子配列に関する追加情報なしに、あらかじめ作製しておいたサブ要素のライブラリーを、同時にこれらの遺伝子中に挿入することができる。この戦略を用いると、遺伝子サブ要素のライブラリーをコードするDNAカセットを (i) あらかじめ作製し、保存、再使用でき、(ii) 実際の配列を知ったり各ライブラリーメンバーの配列決定をすることなくこれらの配列中の正しい位置に挿入することができるため、例えば抗体の親和性を迅速に最適化できる。
【0012】
さらに、既存のモジュールを新しい観察結果によって修飾したモジュールで置き換えることにより、結合、安定性、または可溶性、および発現に重要なアミノ酸残基に関する新しい情報を、ライブラリー設計に組み入れることができる。
【0013】
ライブラリー作製には限定された数のコンセンサス配列が使用されるため、スクリーニング後の、結合する抗体の同定が迅速化される。配列決定または制限部位のフィンガープリントを使用して、基礎となるコンセンサス遺伝子配列を同定した後に、無作為配列を含む部分のみを決定する。これにより配列の誤りおよび偽陽性の結果が生じる可能性が低下する。
【0014】
上記の切断部位は、人工遺伝子が挿入されるベクター系に1つだけ存在する場合に使用できる。このため、ベクターを、このような切断部位を含まないように修飾する必要がある。切断部位が除去され、抵抗性遺伝子や複製開始部位のような基本的な要素を含むベクターの構築は、クローニングのために広く興味を持たれている。さらに、これらのベクターは、上記の人工遺伝子およびあらかじめ作製してあるライブラリーを含むキットの一部ともなりうる。
【0015】
人工遺伝子のコレクションは、好ましくは齧歯類の抗体である非ヒト抗体を、迅速にヒト化するために使用できる。まず、好ましくは齧歯類の抗体である非ヒト抗体のアミノ酸配列を、人工遺伝子コレクションによってコードされるアミノ酸配列と比較して、最も相同性の高いL鎖とH鎖のフレームワーク領域を決定する。それから、これらの遺伝子を用いて、好ましくは齧歯類の抗体である非ヒト抗体のCDRをコードする遺伝子サブ要素を挿入する。
【0016】
驚くべきことに、1つのscFvフラグメントのL鎖とH鎖の各々のただ1つのコンセンサス配列を組み合わせることにより、実質的にすべての抗原に対する抗体を生成するような抗体レパートリーが作製できた。従って、本発明の1つの局面は、有用な(ポリ)ペプチドライブラリーを作製するための普遍的なフレームワークとしての、単一のコンセンサス配列の使用、およびそのために有用な抗体コンセンサス配列である。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】欧州特許第0 368 684 B1号
【非特許文献】
【0018】
【非特許文献1】SkerraおよびPluckthun、Science 240, 1038-1041(1988)
【非特許文献2】Betterら、Science 240, 1041-1043(1988)
【非特許文献3】Carterら、Bio/Technology 10, 163-167(1992)
【非特許文献4】Pluckthunら、A practical approach. Antibody Engineering (Ed. J. McCafferty). IRL Press. Oxford, pp. 203-252(1996)
【非特許文献5】KnappikおよびPluckthun、Protein Engineering 8, 81-89(1995)
【非特許文献6】Ullrichら、Proc. Natl. Acad. Sci. USA 92, 11907-11911(1995)
【発明の概要】
【0019】
発明の詳細な説明
本発明は、(ポリ)ペプチドの有用なライブラリーの作製を可能にする。第一の態様において、本発明は該ライブラリーの作製に適当な核酸配列の組み立て方法を提供する。第1の段階では、少なくとも3つの相同蛋白質を同定し分析する。したがって、蛋白質配列を整列させた蛋白質配列のデータベースが確立される。このデータベースを使って、配列と、情報がある場合には構造的配列との両方で、高度の類似性を示す蛋白質配列のサブグループを決定する。各サブグループにつき、サブグループのメンバーを表すため、少なくとも1つのコンセンサス配列を含む(ポリ)ペプチド配列を導き出す。したがって、(ポリ)ペプチド配列のコレクション全体は、相同蛋白質のコレクションの構造的レパートリー全体を代表することになる。その後、これらの人工(ポリ)ペプチド配列を、可能ならば構造的性質に基づいて分析し、該(ポリ)ペプチド配列内、または、たとえば多量体蛋白質において、該(ポリ)ペプチド配列間もしくは他の(ポリ)ペプチドとの間の好ましくない相互作用を同定する。該相互作用は、コンセンサス配列をしかるべく変更することにより除去する。それから、(ポリ)ペプチド配列を分析し、ドメイン、ループ、ヘリックス、またはCDRのようなサブ要素を同定する。アミノ酸配列は、該核酸配列を発現するために計画する宿主の使用コドンに適合させた核酸配列に逆翻訳する。上述のようにして同定されたサブ要素をコードする各サブ配列が2つの切断部位に挟まれるが、核酸配列内ではその2つの切断部位が2回以上存在しないように、1組の切断部位を組み立てる。これは、すでにサブ配列に隣接する切断部位を同定するか、または切断部位を作製するように1つまたは複数のヌクレオチドを変更し、さらに遺伝子の残りの部分からその部位を除去することにより達成できる。切断部位は対応するサブ要素またはサブ配列すべてに共通であるべきで、これにより核酸配列中のサブ配列および対応する(ポリ)ペプチド中のサブ要素が、完全にモジュール構造をとることになる。
【0020】
別の態様において、本発明は、上述の方法を実行する(ポリ)ペプチドの2つまたはそれ以上のセットで、切断部位が各セット内で唯一であるだけでなく任意の2つのセット間でも唯一であるようなセットを組み立てる方法を提供する。この方法は、新規のヘテロ結合ドメインをスクリーニングするために、例えば2つの異なる蛋白質由来の2つのαヘリックスドメインを含む(ポリ)ペプチドライブラリーを作製するために適用できる。
さらに別の態様において、上述のような少なくとも2つのセットが、同じ蛋白質のコレクションまたは少なくともその一部に由来する。これは、例えば、VHおよびVLのような抗体の2つのドメイン、または膜内外受容体の2つの細胞外ループを含むライブラリーであるが、これに限定されるわけではない。
さらに別の態様において、上述のように組み立てられる核酸配列が合成される。これは、例えば、遺伝子の全合成やPCRに基づく方法など、当業者に周知のいくつかの方法のいずれかにより達成できる。
【0021】
1つの態様において、核酸配列はベクターにクローニングされる。ベクターは当業者に周知の配列決定用ベクター、発現ベクター、またはディスプレイ(例、ファージディスプレイ)ベクターでありうる。すべてのベクターは、1つの核酸配列、または2つもしくはそれ以上の核酸配列を、異なるオペロンまたは同一のオペロン中に含み得る。後者の場合には、配列を別にクローニングするか、隣接する配列としてクローニングされるかのいずれかとなる。
1つの態様において、上述のように好ましくない相互作用を除去すると、修飾(ポリ)ペプチドの発現が増加する。
【0022】
好ましい態様において、核酸配列の1つまたは複数のサブ配列を、異なる配列によって置き換える。これは、例えば、当業者に周知の条件で制限酵素を用いて対応する切断部位で切断するように、サブ配列に隣接するまたはその末端にある制限部位を切断するために適当な条件を用いてサブ配列を切り出し、切断された核酸配列に適合する異なる配列によりそのサブ配列を置き換えることにより達成できる。さらに好ましい態様において、最初のサブ配列を置き換える異なる配列は、例えば、非ヒト抗体を迅速にヒト化するために非ヒト抗体のCDRをコンセンサス抗体配列に接合する場合のように、ゲノム配列または再配列したゲノム配列である。最も好ましい態様において、異なる配列は無作為配列であり、したがってそのサブ配列を配列のコレクションによって置換することにより、多様性を導入し、ライブラリーを作製できる。無作為配列は、例えばオリゴヌクレオチドの自動合成中に、モノヌクレオチド混合物、または好ましくはトリヌクレオチド混合物を使用したり(Virnekasら、1994)、誤りがちなPCRによって、または当業者に周知の他の方法など、様々な方法で作製することができる。無作為配列は、完全に無作為化してもよいし、または既知の蛋白質配列における特定の位置のアミノ酸分布にしたがって、特定のコドンを使うようにもしくは避けるように偏向してもよい。さらに、無作為サブ配列のコレクションには、異なる数のコドンが含まれ、サブ要素のコレクションが異なる長さを有するようにしてもよい。
【0023】
別の態様において、本発明は、当業者に周知の適当なベクターからの適当な条件下での核酸配列の発現を提供する。
さらに好ましい態様において、該核酸配列から発現される(ポリ)ペプチドをスクリーニングし、選択的に最適化する。スクリーニングは、ファージディスプレイ、選択的感染性を有するファージ、ポリソーム技術を用いた結合のスクリーニング、酵素活性のアッセイ系、または蛋白質の安定性など、当業者に周知の方法を用いて実行できる。望ましい性質を有する(ポリ)ペプチドは、対応する核酸配列の決定、またはアミノ酸の配列決定もしくは質量分析法によって同定できる。引き続き最適化する場合には、選択的に、最初に選択した(ポリ)ペプチドをコードする核酸配列を、配列決定せずに使用できる。最適化は異なる配列、好ましくは無作為配列によるサブ配列の置換と、スクリーニング段階を、1回または複数回繰り返すことにより実行する。
【0024】
(ポリ)ペプチドがスクリーニングされる望ましい性質は、好ましくは、標的分子に対する最適化した親和性または特異性、最適化した酵素活性、最適化した発現収量、最適化した安定性、および最適化した溶解度からなる群より選択されるが、これに限定されるわけではない。
【0025】
1つの態様において、サブ配列と隣接する切断部位は制限酵素によって認識および切断される部位であり、認識される配列と切断される配列は同一であるかまたは異なり、制限部位は平滑末端または付着末端のいずれかを有する。
サブ要素の長さは、好ましくは、例えば酵素の活性部位における1つの残基または1つの構造決定残基のように、1つのアミノ酸から、蛋白質ドメイン全体のように150アミノ酸にまでにわたるが、これに限定されるわけではない。最も好ましくは、抗体のCDRループ中に通常見られるように3アミノ酸から25アミノ酸までの長さにわたる。
核酸配列は、RNAでも、好ましくはDNAでもよい。
【0026】
1つの態様において、(ポリ)ペプチドは、特定の種に特徴的なアミノ酸パターンを有する。これは、例えば、1つの種のみの相同蛋白質のコレクション、最も好ましくはヒトの蛋白質コレクションから、コンセンサス配列を導き出すことにより達成できる。コンセンサス配列を含む(ポリ)ペプチドは人工的なものであるから、最も類似性の高い蛋白質配列と比較して、かかる特徴的なアミノ酸パターンを有することを確かめる必要がある。
【0027】
1つの態様において、本発明は免疫グロブリンスーパーファミリーのメンバーまたは誘導体の少なくとも一部、好ましくは免疫グロブリンのメンバーまたは誘導体の少なくとも一部を含む、(ポリ)ペプチドライブラリーの作製方法を提供する。最も好ましくは、本発明は、該(ポリ)ペプチドが、該構想的サブ要素がフレームワーク領域(FR)1、2、3、もしくは4、または相補性決定領域(CDR)1、2、もしくは3であるH鎖もしくはL鎖の可変領域であるかまたはそれに由来するもので、ヒトの抗体のライブラリーの作製方法を提供する。第1の段階において、ヒト由来の明らかにされている抗体配列を整列させたデータベースを作製する。このデータベースを用いて、配列とCDRループの共通の折り畳み(抗体構造の分析により決定)の両方において、高度の類似性を示す抗体配列のサブグループを決定する。各サブグループについて、そのサブグループのメンバーを代表するコンセンサス配列を導き出す。したがって、コンセンサス配列のコレクション全体は、ヒト抗体の構造的レパートリー全体を代表することになる。
【0028】
その後、例えば、遺伝子の全合成、または合成の遺伝子サブユニットの使用によって、これらの人工的な遺伝子を作製する。これらの遺伝子サブユニットは、(ポリ)ペプチドレベルでは構造的サブ要素に対応する。DNAレベルでは、これらの遺伝子サブユニットは、ベクター系に唯一である、各サブ要素の最初と最後にある切断部位が決定する。コンセンサス配列のコレクションのメンバーであるすべての遺伝子は、類似したパターンの対応する遺伝子サブ配列を含むように作製される。最も好ましくは、該(ポリ)ペプチドは、HuCALコンセンサス遺伝子Vκ1、 Vκ2、 Vκ3、 Vκ4、Vλ1、 Vλ2、 Vλ3、VH1A、 VH1B、 VH2、 VH3、 VH4、 VH5、 VH6、Cκ、Cλ、CH1もしくは該HuCALコンセンサス遺伝子の任意の組み合わせであるか、またはそれに由来する。
【0029】
その後、このDNA分子コレクションを使って、抗体または抗体断片、好ましくはFv、ジスルフィド結合したFv、単鎖Fv (scFv)、またはFabフラグメントのライブラリーを作製でき、これを新規の標的抗原に対する特異性の供給源として使用できる。さらに、あらかじめ作製してあるライブラリーカセットと一般的な手順を用いて、抗体の親和性を最適化できる。本発明は、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定するための方法を提供し、抗体断片を発現する段階、およびその後スクリーニングにより所定の標的分子に結合する1つまたは複数の抗体断片を単離する段階を含む。好ましくは、HuCAL VH3およびHuCAL Vλ2コンセンサス遺伝子の組み合わせ、および、H鎖CDR3サブ要素をコードする少なくとも1つの無作為サブ配列を含むscFvフラグメントライブラリーから、結合する抗体をスクリーニングする。必要ならば、その後、遺伝子のモジュラー型設計を用いて、抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、構造的サブ要素をコードする1つまたは複数の第2のサブ配列により置き換えることができる。その後、望ましい親和性を有する抗体が生成するまで、この発現およびスクリーニングの段階を繰り返すことができる。
【0030】
特に好ましいのは、該切断部位を使用して、1つまたは複数の遺伝子サブユニット(例、CDR)を、配列の無作為なコレクション(ライブラリー)によって置換する方法である。これらの切断部位は、(i) ベクター系で唯一であり、(ii) すべてのコンセンサス遺伝子に共通であるため、同じ(あらかじめ作製した)ライブラリーを、すべての人工抗体遺伝子に挿入できる。その結果得られるライブラリーを、任意の選択した抗原に対してスクリーニングする。結合する抗体を選択し、収集し、次のライブラリーの開始材料として使用する。ここでは、1つまたは複数の残りの遺伝子サブユニットを上述のようにして無作為化する。
【0031】
本発明のさらなる態様は、上述のような(ポリ)ペプチドと、付加部分の両方を含むDNAを提供することによる融合タンパク質に関する。特に好ましいのは、有用な治療機能を有する部分である。例えば、付加部分は細胞を殺すことのできる毒素分子であってもよい(Vitettaら、1993)。かかる毒素としては、シュードモナス外毒素Aおよびジフテリア毒素のような細菌毒素、ならびにリシン、アブリン、モデシン、サポリン、ゲロニンのような植物毒素など、数多くの例が当業者に周知である。かかる毒素を例えば抗体断片に融合すると、毒素は、例えば疾病を有する細胞を標的とし、有用な治療効果を有することができる。または、付加部分は、細胞のファミリーに特定の効果(この場合、T細胞の増殖効果)を有する
IL-2(RosenbergおよびLotze、1986)のようなサイトカインであってもよい。さらなる態様において、付加部分はその(ポリ)ペプチドパートナーに検出および/または精製の方法を与える場合がある。例えば、融合タンパク質は、修飾された抗体断片と、アルカリ性フォスファターゼ(Blakeら、1984)のような検出のために一般に用いられる酵素とを含むことができる。検出または精製のための標識として使用できる他の部分には、当業者に周知のものが数多くある。特に好ましいのは、金属イオンと結合することのできる、少なくとも5つのヒスチジン残基を含むペプチドで(Hochuliら、1988)、そのため、これはその融合した蛋白質の精製に使用できる(Lindnerら、1992)。また、本発明は、一般に使用されるC-mycおよびFLAG標識(Hoppら、1988;KnappikおよびPluckthun、1994)のような付加部分を提供する。
【0032】
1つまたは複数の付加ドメインを操作することにより、抗体断片または任意の他の(ポリ)ペプチドは、本発明の範囲内であるさらに大きな分子に組み立てることができる。例えば、ミニ抗体(Pack、1994)は、各々が自己会合性の二量化ドメインに融合した2つの抗体断片を含む二量体である。特に好ましい二量化ドメインには、ロイシンジッパー(PackおよびPluckthun、1992)またはαヘリックス・βターン・αヘリックスモチーフ(Packら、1993)がある。
【0033】
本発明の上記の態様はすべて、当業者に既知の分子生物学の標準的技法を使用して遂行できる。
さらなる態様において、サブ配列の無作為なコレクション(ライブラリー)を、1つの(ポリ)ペプチドをコードする単一の核酸配列に挿入し、1つの普遍的なフレームワークに基づく(ポリ)ペプチドライブラリーを作製する。好ましくは、CDRサブ配列の無作為なコレクションを、例えば、上述のH3κ2単鎖Fvフラグメントのような普遍的な抗体フレームワークに挿入する。
さらなる態様において、本発明は上述の方法にしたがって得られる核酸配列、核酸配列を含むベクター、ベクターを含む宿主細胞、および(ポリ)ペプチドを提供する。
さらに好ましい態様において、本発明は、(ポリ)ペプチドをコードするモジュール型の核酸配列と適合するモジュール型ベクターシステムを提供する。ベクターのモジュールは制限部位と隣接するが、この制限部位はベクター系中で唯一で、(ポリ)ペプチドをコードする核酸配列に組み込まれる制限部位については、例えば核酸配列をベクターにクローニングするために必要な制限部位を除いて本質的に唯一である。ベクターモジュールのリストには、一本鎖複製開始点、コピー数の多いおよび少ないプラスミドの二本鎖複製開始点、プロモーター/オペレーター、リプレッサーまたはターミネーター要素、抵抗性遺伝子、組み換え可能部位、線状ファージ上のディスプレイのための遺伝子III、シグナル配列、精製および検出用標識、および付加部分の配列を含む。
ベクターは、好ましくは発現ベクターまたは発現とライブラリーのスクリーニングに適当なベクターであるが、これに限定されるわけではない。
【0034】
他の態様において、本発明は、上述の方法にしたがった核酸配列、組み換え体ベクター、(ポリ)ペプチド、およびベクターのリスト、ならびに(ポリ)ペプチドを生産するために適当な宿主細胞のうちの1つまたは複数を含むキットを提供する。
好ましい態様において、本発明はヒトの抗体のライブラリーの作製方法を提供する。最初の段階では、ヒト由来の明らかにされている抗体配列のデータベースを確立する。このデータベースを用いて、配列と共通の折り畳み(抗体構造の分析により決定)の両方において、高度の類似性を示す抗体配列のサブグループを決定する。各サブグループについて、そのサブグループのメンバーを代表するコンセンサス配列を導き出す。したがって、コンセンサス配列のコレクション全体は、ヒト抗体の構造的レパートリー全体を代表することになる。
【0035】
その後、合成の遺伝子サブユニットを使用して、これらの人工的な遺伝子を作製する。これらの遺伝子サブユニットは、蛋白質レベルでは構造的サブ要素に対応する。DNAレベルでは、これらの遺伝子サブユニットは、ベクター系に唯一である、各サブ要素の最初と最後にある切断部位が決定する。コンセンサス配列のコレクションのメンバーであるすべての遺伝子は、該遺伝子サブユニットの類似したパターンを含むように作製される。
その後、DNA分子のこのコレクションを使って、抗体のライブラリーを作製でき、これを新規の標的抗原に対する特異性の供給源として使用できる。さらに、あらかじめ作製してあるライブラリーカセットと一般的な手順を用いて、抗体の親和性を最適化できる。本発明は、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定するための方法を提供し、抗体断片を発現する段階、およびその後スクリーニングにより所定の標的分子に結合する1つまたは複数の抗体断片を単離する段階を含む。必要ならば、その後、遺伝子のモジュラー型設計を用いて、抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、構造的サブ要素をコードする1つまたは複数の第2のサブ配列により置き換えることができる。その後、望ましい親和性を有する抗体が生成するまで、この発現およびスクリーニングの段階を繰り返すことができる。
【0036】
特に好ましいのは、該切断部位を使用して、1つまたは複数の遺伝子サブユニット(例、CDR)を、配列の無作為なコレクション(ライブラリー)によって置換する方法である。これらの切断部位は、(i) ベクター系に唯一で、(ii) すべてのコンセンサス遺伝子に共通であるため、同じ(あらかじめ作製した)ライブラリーを、すべての人工抗体遺伝子に挿入できる。その結果得られるライブラリーを、任意の選択した抗原に対してスクリーニングする。結合する抗体を溶出し、収集し、次のライブラリーの開始材料として使用する。ここでは、1つまたは複数の残りの遺伝子サブユニットを上述のようにして無作為化する。
【0037】
定義
蛋白質:
蛋白質という用語は、単量体のポリペプチド鎖、および2つまたはそれ以上のポリペプチド鎖が共有結合の相互作用(ジスルフィド結合のような結合)または非共有結合の相互作用(疎水性または静電的相互作用のような相互作用)のいずれかにより結合しているホモまたはヘテロ多量体複合体を含む。
相同蛋白質の分析:
3つまたはそれ以上の蛋白質のアミノ酸配列を、すべての位置で同一または類似したアミノ酸残基との一致が最大になるように、(ギャップの導入を可能にするよう)互いに整列させる。同一および/または類似した残基の合計の割合が一定の閾値を越える場合に、これらの整列させた配列は相同であると言う。当業者は一般に、整列させた遺伝子のアミノ酸の少なくとも15%が同一で、少なくとも30%が類似する場合、この閾値を越えていると見なす。相同蛋白質のファミリーの例には、免疫グロブリンスーパーファミリー、スカベンジャー受容体スーパーファミリー、フィブロネクチンスーパーファミリー(例、タイプIIおよびIII)、補体制御タンパク質スーパーファミリー、サイトカイン受容体スーパーファミリー、シスチンノット蛋白質、チロシンキナーゼ、および当業者に周知の数多くの他の例がある。
コンセンサス配列:
少なくとも3つの整列させたアミノ酸配列のマトリックスを用いて、並べる際にギャップを許すと、各位置で最も頻度の高いアミノ酸残基を決定することができる。コンセンサス配列は、各位置で最も出現頻度の高いアミノ酸を含む配列である。1つの位置で2つまたはそれ以上のアミノ酸が一つの部位で同じ出現頻度を有する場合は、コンセンサス配列は、これらのアミノ酸の両方またはすべてを含む。
好ましくない相互作用の除去:
コンセンサス配列自体は、大概、人工的なものであるため、結果として得られる分子が機能的な3次構造を取るのを妨げたり、または多量体複合体において他の(ポリ)ペプチド鎖との相互作用の障害となるアミノ酸残基を変更するために、分析する必要がある。これは、(i) 既知の関連構造を鋳型として用いて、コンセンサス配列の3次元モデルを構築し、モデル内で互いに好ましくない相互作用をする可能性があるアミノ酸残基を同定するか、または (ii) 整列させたアミノ酸配列のマトリックスを分析し、配列内で同時に1つの配列中に頻繁に出現し、したがって互いに相互作用をする可能性が高いアミノ酸残基の組み合わせを検出することにより行うことができる。これらの相互作用をすると思われるペアを表にし、この「相互作用地図」とコンセンサス配列を比較する。コンセンサス配列中で欠如する相互作用または不正な相互作用は、好ましくない相互作用を抑えるようにしかるべくアミノ酸配列を適当に変更することにより修復する。
構造的サブ要素の同定:
構造的サブ要素とは、分子の定義された構造的または機能的部分に対応する、蛋白質/(ポリ)ペプチド内のアミノ酸残基列である。これは、ループ(例、抗体のCDRループ)、または蛋白質/(ポリ)ペプチド内の任意の2次構造または機能的構造(ドメイン、αヘリックス、βシート、抗体のフレームワーク領域等)であり得る。構造的サブ要素は、類似または相同の(ポリ)ペプチドの既知の構造を用いて、または上述の整列したアミノ酸配列のマトリックスを用いて、同定できる。各位置での変動に基づき、構造的サブ要素(例、抗体の超可変領域)に属するアミノ酸残基列を決定する。
サブ配列:
サブ配列とは、唯一である切断部位によって隣接しており、少なくとも1つの構造的サブ要素をコードする遺伝子モジュールと定義される。これは構造的サブ要素と必ずしも同一ではない。
切断部位:
DNAを配列特異的に切断する試薬(例、制限エンドヌクレアーゼ)の特異的標的として使用される短いDNA配列。
適合する切断部位:
切断部位が修飾せずに、また好ましくはアダプター分子を加えずに、効果的に連結できる場合、これらの切断部位は互いに適合する。
唯一である切断部位:
興味のある遺伝子を少なくとも1つ含むベクター中で、切断部位が1度しか出現しない場合、または興味のある遺伝子を少なくとも1つ含むベクター中、その切断部位のうちのただ1つのみが切断試薬によって使用できるとして扱える場合、その切断部位は唯一であると定義する。
対応する(ポリ)ペプチド配列:
相同蛋白質の1グループの同じ部分から導き出される配列を、対応する(ポリ)ペプチド配列と呼ぶ。
共通の切断部位:
少なくとも2つの対応する配列中の切断部位で、同じ機能的位置(すなわち、定められたサブ配列と隣接する)に出現し、同一の切断ツールで加水分解され、同一の適合する末端を生成するような切断部位を、共通の切断部位と呼ぶ。
遺伝子サブ配列の切り出し:
唯一である切断部位が隣接する遺伝的サブ配列を単離、除去、または置換するために、唯一である切断部位と対応する切断試薬とを使用して、特定の位置で標的DNAを切断する方法。
遺伝的サブ配列の交換:
既存のサブ配列に隣接する切断部位を用いてこのサブ配列を除去し、このようにして作製した切断部位と適合する末端を含む新規のサブ配列またはサブ配列のコレクションを挿入する方法。
遺伝子の発現:
発現という用語は、遺伝子の情報がmRNAに転写され、それから蛋白質/(ポリ)ペプチドに翻訳されるインビボまたはインビトロの過程を示す。したがって、発現という用語は、遺伝子の情報がmRNAにそれから蛋白質に転写される、細胞内で発生する過程を示す。発現という用語はまた、(ポリ)ペプチドが機能を有するために必要な、翻訳後修飾と輸送のすべての事象も含む。
蛋白質/(ポリ)ペプチドライブラリーのスクリーニング:
望ましい性質を有する1つまたは複数の蛋白質/(ポリ)ペプチドを、ライブラリー内の他の蛋白質/(ポリ)ペプチドから単離することのできる任意の方法。
種に特徴的なアミノ酸パターン:
(ポリ)ペプチド配列は、1つの種のみに由来する相同蛋白質のコレクションから導き出される場合、その種に特徴的なアミノ酸パターンを示すと想定される。
免疫グロブリンスーパーファミリー (IgSF):
IgSFは免疫グロブリンの折り畳みを特徴とするドメインを含むタンパク質のファミリーである。IgSFには、例えばT細胞受容体および免疫グロブリン(抗体)が含まれる。
抗体のフレームワーク:
抗体の可変部のフレームワークは、可変部の一部でこの可変部の抗原結合ループの骨組みとして働く部分としてKabatら(1991)が定義したものである。
抗体のCDR:
抗体のCDR(相補性決定領域)は、 Kabatら(1991)が定義したように、抗原結合ループから構成される。抗体のFvフラグメントの2つの可変部には、各々3つのCDRが含まれる。
HuCAL:
ヒト組み合わせ抗体ライブラリー(Human Combinatorial Antibody Library)の頭字語。本発明に係るモジュール型のコンセンサス遺伝子に基づく抗体ライブラリー(実施例1参照)。
抗体断片:
例えば、抗原の結合のような、特定の機能を有する抗体の任意の部分。通常、抗体断片は抗体全体よりも小さい。例としては、Fv、ジスルフィド結合Fv、単鎖Fv (scFv)、またはFabフラグメントがある。さらに、抗体断片は、新規の機能または性質を含むように、しばしば操作される。
普遍的フレームワーク:
異なるフレームワークの大きなコレクションによって本来は維持されていた機能、特異性、または性質の完全な変動性を作製するために用いることのできる単一のフレームワークを、普遍的フレームワークと呼ぶ。
抗体の標的への結合:
抗体と、対応する分子またはリガンドとの間の強く特異的な会合に至る過程を、結合と呼ぶ。抗体によって認識される分子もしくはリガンド、または分子もしくはリガンドの任意の部分を、標的と呼ぶ。
遺伝子サブ配列の置換:
既存のサブ配列に隣接する切断部位を用いてこのサブ配列を除去し、このようにして作製した切断部位と適合する末端を含む新規のサブ配列またはサブ配列のコレクションを挿入する方法。
遺伝子配列の組み立て:
特定の方法で合成または天然の遺伝子配列を組み合わせ、使用された合成または天然の遺伝子配列の少なくとも一部を含む、さらに長い遺伝子配列を作製するために使用する任意の過程。
相同遺伝子の分析:
2つまたはそれ以上の遺伝子に対応するアミノ酸配列を、すべての位置で同一または類似したアミノ酸残基間の一致が最大になるように整列させる。同一および/または類似した残基の合計の割合が一定の閾値を越える場合に、これらの整列させた配列が相同であると言う。当業者は一般に、整列させた遺伝子のアミノ酸うちの少なくとも15%が同一で、少なくとも30%が類似する場合、この閾値を越えていると見なす。
【図面の簡単な説明】
【0038】
【図1】コンセンサス配列に基づく合成ヒト抗体ライブラリーの構築過程を示す流れ図。
【図2】各サブグループ(アミノ酸残基は標準的な1文字の略語で示されている)に設計されたコンセンサス配列を並べたもの。(A)κ配列、(B)λ配列、および(C)H鎖配列。位置はKabat (1991)に従って番号が付けられている。整列させた際の相同性を最大にするために、一定の位置の配列にギャップ(ミ)が挿入されている。
【図3】合成Vκコンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図4】合成Vλコンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図5】合成VH鎖コンセンサス遺伝子の遺伝子配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図6】コンセンサス遺伝子の作製に用いたオリゴヌクレオチド。例えば遺伝子Vκ1はO1K1〜O1K6の6つのオリゴヌクレオチドを用いて作製されたというように、オリゴは対応するコンセンサス遺伝子に従って命名されている。定常部ドメインCκ(OCLK1〜8)およびCH1(OCH1〜8)をコードする遺伝子の合成に用いられたオリゴヌクレオチドも示されている。
【図7AB】定常部ドメインCκ(A)およびCH1 (B)をコードする合成遺伝子の配列。対応するアミノ酸配列(図2参照)および唯一である切断部位も示されている。
【図7C】合成Cκ遺伝子部分(huCLλ)を含むモジュールM24の機能地図と配列。
【図7D】モジュールM24の合成に用いたオリゴヌクレオチド。
【図8】コンセンサス単鎖断片VH3-Vκ2をコードする合成遺伝子の配列と制限地図。シグナル配列(アミノ酸1〜21)は大腸菌phoA遺伝子(SkerraおよびPluckthun、1988)に由来する。 モノクローナル抗体M1 (KnappikおよびPluckthun、1994)を用いたウエスタンブロットまたはELISAで単鎖断片を検出するために、 phoAシグナル配列とVH3ドメインの間に、4つのアミノ酸残基(アミノ酸22〜25)をコードする短い配列が挿入された。配列の最後の6 塩基対は、クローニングを目的として導入された(EcoRI部位)。
【図9】H3κ2 scFvフラグメントのファージディスプレイに使用されたベクターpIG10.3のプラスミド地図。ベクターはpIG10由来で、lacオペロンリプレッサー遺伝子、lacI、lacプロモーターのコントロール下でH3κ2-gene3ssの融合をコードする人工オペロン、転写ターミネーターIpp、大腸菌ファージf1の一本鎖複製開始点(F1_ORI)、β-ラクタマーゼ (bla)をコードする遺伝子およびColEI由来の複製開始点を含む。
【図10】最初のライブラリーからの独立したクローンの配列結果を、対応するアミノ酸配列に翻訳したもの。(A) VH3コンセンサスH鎖CDR3(93〜102の位置、Kabatの番号付け)の佐見の酸配列。(B) 10量体ライブラリーの12のクローンのアミノ酸配列。(C) 15量体ライブラリーの11のクローンのアミノ酸配列、*:1塩基の欠失。
【図11】各ライブラリーメンバーの発現試験。(A) 10量体ライブラリーの独立した9クローンの発現。(B) 15量体ライブラリーの独立した9クローンの発現。Mと記したレーンはサイズマーカーが含まれている。gp3-scFv融合およびscFv単量体の両方が示されている。
【図12】FITC-BSAに対するパニングにおける特異的ファージ抗体の濃縮。最初およびその後のフルオレセイン特異的サブライブラリーをブロッキングバッファーに対してパニングし、各回のパニングでFITC-BSAコートしたウェルから溶出されたファージと、粉ミルクコートしたウェルから溶出されたファージの比率が「特異性係数」として示されている。
【図13】FITC-BSAへの結合を試験した3回のパニング後の24の独立したクローンのファージELISA。
【図14】選択されたFITC-BSA結合クローンの競合的ELISA。阻害なしの場合のscFv結合のELISAシグナル(OD405nm)を100%としている。
【図15】FITC-BSAに対する3回のパニング後の独立クローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図16】精製した抗フルオレセインscFvフラグメントのクーマシーブルー染色SDS-PAGE。M:分子量マーカー、A:誘導後の全可溶性細胞抽出物、B:フロースルー分画、C、D、E:それぞれ、精製scFvフラグメント1HA-3E4、1HA-3E5、1HA-3E10。
【図17】βエストラジオール-BSA、テストステロン-BSA、BSA、ESL-1、インターロイキン-2、リンホトキシン-β、LeY-BSAに対する3回のパニングによる特異的ファージ抗体の濃縮。
【図18】選択したESL-1およびβエストラジオール結合クローンのELISA。
【図19】HuCAL抗体の選択性と交差反応性:HuCAL抗体の特異的結合が対角線上に見られ、対角線以外のシグナルは非特異的交差反応を示す。
【図20】βエストラジオール-BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。1つのクローンは10量体ライブラリーに由来する。
【図21】テストステロン-BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図22】リンホトキシン-βに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。1つのクローンは、オリゴヌクレオチド合成中のトリヌクレオチド混合物の不完全なカップリングにより導入されたと思われる、14量体CDRを含む。
【図23】ESL-1に対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。2つのクローンは10量体ライブラリーに由来する。1つのクローンは、トリヌクレオチドを用いたオリゴヌクレオチド合成中の鎖の伸長により導入されたと思われる、16量体のCDRを含む。
【図24】BSAに対する3回のパニング後の独立したクローンのH鎖CDR3の配列分析結果を、対応するアミノ酸配列(93〜102、Kabatの番号付け)に翻訳したもの。
【図25】モジュール型pCALベクター系の概略図。
【図25a】モジュール型HuCAL遺伝子およびpCALベクター系にすでに使用されているか、または使用に適当な制限部位のリスト。
【図26】pCALベクターシリーズのモジュール型ベクター要素のリスト:モジュールシステムの一部となっている制限部位のみを示す。
【図27】多クローニング部位モジュール(MCS)の機能地図と配列。
【図28】pMCSクローニングベクターシリーズの機能地図と配列。
【図29】pCALモジュールM1(図26参照)の機能地図と配列。
【図30】pCALモジュールM7-III(図26参照)の機能地図と配列。
【図31】pCALモジュールM9-II(図26参照)の機能地図と配列。
【図32】pCALモジュールM11-II(図26参照)の機能地図と配列。
【図33】pCALモジュールM14-Ext2(図26参照)の機能地図と配列。
【図34】pCALモジュールM17(図26参照)の機能地図と配列。
【図35】モジュール型ベクターpCAL4の機能地図と配列。
【図35a】他のpCALモジュール(M2、M3、M7I、M7II、M8、M10II、M11II、M12、M13、M19、M20、M21、M41)および低コピー数プラスミドベクター(pCALO1〜pCALO3)の機能地図と配列。
【図35b】pCALベクターモジュールの合成に用いたオリゴヌクレオチドおよびプライマーのリスト。
【図36】CDRライブラリークローニングのための、CDRの置換用のβラクタマーゼカセットの機能地図と配列。
【図37】VκCDR3ライブラリーのためのオリゴとプライマーの設計。
【図38】VλCDR3ライブラリーのためのオリゴとプライマーの設計。
【図39】pBS13発現ベクターシリーズの機能地図。
【図40】7つのVH遺伝子の各々を7つのVL遺伝子の各々と組み合わせて得られる49のHuCAL scFvの発現(pBS13、30℃):可溶性物質と不溶性物資の割合、ならびにH3κ2の組み合わせを100%としてこれと比較した合計および可溶性の量の値が示されている。さらに、McPC603 scFvについて、対応する値も示されている。
【0039】
表1:再配列した配列の生殖細胞系メンバーを算定するために用いたヒト免疫グロブリンの生殖細胞系配列の概要。(A) κ配列、(B) λ配列、(C) H鎖配列。(1) 種々の計算に用いた生殖細胞の名称、(2) 対応する配列の参考文献番号(配列に関連する引用文献は付録を参照)、(3) 各配列が属するファミリー、 (4) 同一のアミノ酸配列を有する生殖細胞系遺伝子について文献に見られる種々の名称。
表2:コンセンサス配列の計算に用いた再配列したヒトの配列。(A) κ配列、(B) λ配列、(C) H鎖配列。配列の名称 (1)、アミノ酸にした配列の長さ (2)、生殖細胞系ファミリー (3)、および算定される生殖細胞系配列を表にまとめた。再配列した配列と生殖細胞系配列との間で交換されたアミノ酸数は(5)に、異なるアミノ酸の割合は(6)に記載されている。(7)は対応する配列(配列に関連する引用文献は付録を参照)の参考文献番号である。
表3:再配列したV配列の、生殖細胞系配列への割り当て。(A) κ配列、(B) λ配列、(C) H鎖配列。生殖細胞系遺伝子はファミリーごと(1)に記載され、各生殖細胞系遺伝子に見つかった再配列した遺伝子の数は(2)に記載されている。
表4:再配列したVκ配列のコンセンサス配列の算定。(A) Vκサブグループ1、(B) Vκサブグループ2、(C) Vκサブグループ3、(D) Vκサブグループ4。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。(1) アミノ酸は標準的な1文字の略称で記載されている(BはDまたはN、ZはEまたはQ、Xは任意のアミノ酸を表す)。統計学的分析では、各位置に見られる配列の数(2)、最も頻度の高いアミノ酸の出現回数(3)、その位置で最も出現頻度が高いアミノ酸残基(4)、最も出現頻度が高いアミノ酸の相対的な頻度(5)、各位置に見られる異なるアミノ酸の数(6)がまとめられている。
表5: 再配列したVλ配列のコンセンサス配列の算定。(A) Vλサブグループ1、(B) Vλサブグループ2、(C) Vλサブグループ3。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。略号は表4と同じである。
表6:再配列したH鎖配列のコンセンサス配列の算定。(A) VH鎖サブグループ1A、(B) V H鎖サブグループ1B、(C) V H鎖サブグループ2、(D) V H鎖サブグループ3、(E) V H鎖サブグループ4、(F) V H鎖サブグループ5、(G) V H鎖サブグループ6。各位置に見られるアミノ酸の数とデータの統計学的分析が表に記載されている。略号は表4と同じである。
【実施例】
【0040】
実施例1:合成ヒト組み合わせ抗体ライブラリー(HuCAL)の設計
以下の実施例は、ヒト免疫グロブリンのレパートリーのコンセンサス配列と、コンセンサス遺伝子の合成に基づく、全合成ヒト組み合わせ抗体ライブラリー(HuCAL)の設計を説明する。全般的な手順は図1に略図が示されている。
1.1 配列データベース
1.1.1 ヒト免疫グロブリン配列の収集および整列
最初の段階では、ヒト免疫グロブリンの可変部ドメインの配列を収集して、3つのサブデータベース、VH鎖(VH)、Vκ、およびVλに分割した。それから、各配列につき、遺伝子配列を対応するアミノ酸配列に翻訳した。その後、すべてのアミノ酸配列を、Kabatら(1991)に従って整列させた。Vλ配列については、Chuchanaら(1990)の番号付けシステムを使用した。3つのメインデータベースを、さらに2つのサブデータベースに分割した。最初のサブデータベースには、配列の70以上の位置が既知である、再配列したV遺伝子に由来するすべての配列が含まれていた。第2のサブデータベースは、すべての生殖細胞系遺伝子部分が含まれていた(D-およびJ-ミニ遺伝子を含まない;内部に停止コドンを有する偽遺伝子も除去された)。すべてにおいて、同一のアミノ酸配列を持ちながら名称の異なる生殖細胞系配列が見つかった場合には、1つの配列のみが使用された(表1参照)。再配列した配列の最終的なデータベースには、Vκ、Vλ、VHがそれぞれ386、149、674含まれていた。生殖細胞系配列の最終的なデータベースには、 Vκ、Vλ、VHがそれぞれ48、26、141含まれていた。
【0041】
1.1.2 サブグループへの配列の割り当て
3つの生殖細胞系データベースの配列を、配列の相同性に基づいて分類した(Tomlinsonら、1992、WilliamsおよびWinter、1993、およびCoxら、1994も参照)。 Vκの場合、7つのファミリーが確立された。 Vλは8つのファミリー、VHは6つのファミリーに分類された。VH7ファミリー(Van Dijkら、1993)のVH生殖細胞系遺伝子は、VH1ファミリーの遺伝子と高い相同性を示すため、VH1ファミリーに分類された。各ファミリーに含まれる生殖細胞系遺伝子の数は、1(例えばVH6)から47(VH3)までにわたっていた。
【0042】
1.2 配列の分析
1.2.1 生殖細胞系メンバーの算定
次に、再配列した遺伝子のデータベース中の1209アミノ酸配列の各々について、最も近い生殖細胞系配列、すなわち、アミノ酸の違いが最も少ない生殖細胞系配列を計算した。生殖細胞系配列が見つかった後に、再配列した遺伝子中に起りアミノ酸を変更した体細胞変異の数を表にすることができた。140の場合には、同じ数のアミノ酸の変更を有する複数の生殖細胞系遺伝子が見つかったため、対応する生殖細胞系配列を正確に計算することができなかった。これらの再配列した配列は、データベースから除去された。いくつかの場合には、アミノ酸の変更の数は異常に大きく(VLについて>20、VHについて>25)、変異の大きな再配列遺伝子であるか、またはデータベースには存在しない生殖細胞系遺伝子に由来するかのいずれかであることを示していた。これらの2つの可能性を区別することはできないため、これらの配列も、データベースから除去した。最後に、非常に変わったCDRの長さおよび組成、または共通位置(以下参照)に異常なアミノ酸を有するため、12の再配列した配列をデータベースから除去した。要約すると、1209の再配列した配列のうち、1023 (85%)を、生殖細胞系配列に明確に割り当てることができた(表2参照)。
この計算の後、すべての再配列した遺伝子を、生殖細胞系遺伝子について確立したファミリーの1つに割り当てることができた。ここで、各生殖細胞系遺伝子の使用量、すなわち、各生殖細胞系遺伝子から生ずる再配列した遺伝子の数を計算することができる(表2参照)。使用量は生殖細胞系遺伝子の1つのサブセットに強く偏向しており、生殖細胞系遺伝子の大部分はデータベース中の再配列した遺伝子に存在せず、したがって免疫系では使用されていないと思われることが分かった(表3)。この観察結果は、Vκ(Coxら、1994)についてはすでに報告済みだった。割り当てられる再配列した配列がないまたは非常に少ないすべての生殖細胞系遺伝子ファミリーをデータベースから除去すると、4つのVκ、3つのVλ、6つのVHファミリーのみが残った。
【0043】
1.2.2 CDRの立体配座
抗体分子の抗原結合ループCDRの立体配座は、CDRの長さと、いわゆる共通位置(ChothiaおよびLesk、1987)に位置するアミノ酸残基の両方に、強く依存している。免疫グロブリンの可変部ドメインの構造的レパートリーを決定する共通構造は、わずか数種類しか存在しないことが分かった(Chothiaら、1989)。共通アミノ酸位置は、CDRおよびフレームワーク領域に見つかる。上記のように決定した使用されている13の生殖細胞系ファミリー(7つのVLおよび6つのVH)につき、これらのファミリーにコードされる構造的レパートリーを決定するため、共通構造に関して分析した。
4つのVκファミリーのうちの3つ( Vκ1、2および4)では、各ファミリーについてそれぞれ異なる1つのタイプのCDR1立体配座が決定された。 Vκ3は2つのタイプのCDR1立体配座を示した:1つはVκ1と同一で、1つはVκ3にのみ見られる。すべてのVκCDR2は、同じタイプの共通構造を使用していた。CDR3立体配座は、生殖細胞系遺伝子部分にはコードされていない。したがって、配列の相同性と使用量によって決定された4つのVκは、 Vκ生殖細胞系遺伝子中に見られる4つのタイプの共通構造にも対応する。
上記のように決定した3つのVλファミリーは、3つのタイプのCDR1立体配座を示し、各ファミリーがそれぞれ異なる1つのタイプを示した。 Vλ1ファミリーには2つの異なるCDR1の長さ(13および14アミノ酸)が含まれていたが、共通残基は同一で、両方とも同じ共通立体配座を取ると考えられた(ChothiaおよびLesk、1987)。使用されているVλ生殖細胞系のCDR2には1つの共通立体配座が存在するのみで、CDR3立体配座は生殖細胞系遺伝子部分にはコードされていない。したがって、配列の相同性と使用量によって決定された3つのVλは、 3つのタイプの共通構造にも対応する。
ヒトVH配列の構造的レパートリーは、Chothiaら、1992によって詳細に分析された。合わせて、CDR1の3つの立体配座(H1-1、H1-2、およびH1-3)およびCDR2の6つの立体配座(H2-1、 H2-2、 H2-3、 H2-4、 H2-5、およびH2-x)が決定された。CDR3はD-およびJ-ミニ遺伝子部分にコードされているため、このCDRには特に共通残基は決定されていない。
上記のように決定されたVH1ファミリーのすべてのメンバーは、CDR1立体配座H1-1を含んでいたが、CDR2立体配座は異なっていた:6の生殖細胞系遺伝子にはH2-2立体配座が見られたが、8つの生殖細胞系遺伝子にはH2-3が見られた。この2つのタイプのCDR2立体配座は、フレームワーク位置72にあるアミノ酸のタイプの違いにより定義されているため、VH1ファミリーは2つのサブグループに分類された。VH1AはH2-2のCDR2立体配座を、VH1BはH2-3の立体配座を有する。VH2ファミリーのメンバーはすべて、CDR1とCDR2にそれぞれH1-3とH2-1の立体配座を持っていた。VH3メンバーのCDR1の立体配座はすべての場合にH1-1だっ他が、CDR2には4つの異なるタイプが見られた(H2-1、H2-3、H2-4、およびH2-x)。これらのCDR2立体配座では、フレームワークの共通残基71は常にアルギニンに決められている。したがって、CDR2の4つのタイプの立体配座は、CDR2自身によってのみ決定されるため、VH3ファミリーをサブファミリーに分割する必要はなかった。VH4ファミリーに関しても同様だった。ここでは、3つのタイプのCDR1の立体配座がすべて見られたが、CDR1の立体配座はCDR自身によって決定されるため(フレームワークの共通残基26はすべての場合にグリシンだった)、さらに分割する必要はなかった。VH4メンバーのCDR2立体配座は、すべての場合にH2-1だった。VH5ファミリーのすべてのメンバーは、それぞれH1-1およびH2-2の立体配座を有することが分かった。VH6ファミリーの1つのみの生殖細胞系遺伝子は、CDR1とCDR2にそれぞれH1-3およびH2-5の立体配座を持っていた。
要約すると、VκおよびVλ遺伝子のすべての可能なCDR立体配座が、配列の比較により定義された7つのファミリーに存在していた。 VH1ファミリーを2つのサブファミリーに分け7つのVHファミリーを作製することにより、使用されているVH生殖細胞系遺伝子に見られる12の異なるCDR立体配座のうち、7つがカバーされた。残る5つのCDR立体配座(VH3ファミリーの3つおよびVH4ファミリーの2つ)は、CDR自身によって決定されており、CDRライブラリーの構築時に作製し得る。したがって、使用されているヒトV遺伝子の構造的レパートリーは、49 (7 x 7)の異なるフレームワークでカバーされ得る。
【0044】
1.2.3 コンセンサス配列の算定
再配列した配列の14のデータベース(4 Vκ、3 Vλ、および7 VH)を用いて、各サブグループのHuCALコンセンサス配列を算定した(4 HuCAL-Vκ、3 HuCAL-Vλ、7 HuCAL-VH、表4、5、6参照) 。これは、各位置において使用されるアミノ酸残基の数を数え(位置変動性)、その後、各位置において最も高頻度で使用されるアミノ酸残基を同定することにより行った。コンセンサスの算定に、使用されている生殖細胞系配列の代わりに再配列した配列を使用することで、コンセンサス配列は使用量の頻度にしたがって加重された。さらに、高頻度で変異したり、高度に保存される位置を同定できた。コンセンサス配列は、生殖細胞系ファミリーのコンセンサスと相互チェックし、再配列した配列が特定の位置で、収集した生殖細胞系配列には存在しないアミノ酸残基に偏向しているかどうかを確認したが、そのようなことはなかった。その後、14のコンセンサス配列の各々につき、各特定のファミリーに見られる生殖細胞系配列の各々に対する差異の数を計算した。最も相同性の高い生殖細胞系配列からの総合的な偏差は2.4アミノ酸残基(標準偏差=2.7)で、「人工的な」コンセンサス配列が、免疫原性に関しては正しくヒトの配列とみなすことができることが保証される。
【0045】
1.3 構造分析
ここまでは、コンセンサス配列の設計には、配列情報のみが使用されている。配列中では遠くに位置するが、3次元構造では相互に接触し、フレームワークを不安定にしたり、または誤った折り畳みを導くようなアミノ酸残基の特定の人工的な組み合わせが計算時に生成した可能性があるため、14のコンセンサス配列を、その構造的性質に従って分析した。
データベース中に存在する再配列した配列は、すべて機能的で、故に正しく折り畳まれた抗体分子に対応すると考えた。したがって、各コンセンサス配列について、最も相同性の高い再配列した配列を計算した。コンセンサス配列が、再配列した配列と異なる位置は、潜在的「人工残基」と同定し、検査した。
検査自体は2つの方向で行った。まず、各潜在的「人工残基」のまわりの局所的配列を、すべての再配列した配列の対応する列と比較した。この列が真に人工的ならば、すなわち、再配列した配列のいずれにも出現することがないならば、決定的な残基をその位置に2番目に高頻度で見られるアミノ酸に変換し、再度分析した。第2に、潜在的「人工残基」を長い範囲の相互作用について分析した。これは、対応するPDBファイルで利用できるヒト抗体可変部ドメインのすべての構造を収集し、すべての構造について、確立された各アミノ酸残基の各側鎖に対する相互作用の数とタイプを計算して行った。これらの「相互作用地図」を用いて、潜在的「人工残基」に考えられる側鎖/側鎖相互作用を分析した。この分析の結果、以下の残基を交換した(遺伝子の名称、Kabatの番号付けに従う位置、この位置で最も高頻度に見られるアミノ酸、およびその代わりに使用したアミノ酸が記載されている)。
VH2:S65T
Vκ1:N34A
Vκ3:G9A、D60A、R77S
Vλ3:V78T
【0046】
1.4. CDR配列の設計
上記の過程で、再配列した配列のデータベースのみに由来する完全なコンセンサス配列が得られる。再配列した配列および変異した配列のCDRは、特定の抗原に対して偏向しているため、CDR1およびCDR2領域は、使用されている生殖細胞系配列のデータベースから取るべきであると考えた。さらに、生殖細胞系CDR配列は、CDR3のみが異なる1次免疫応答において、様々な抗原への結合を可能にすることが知られている。したがって、VHおよびVκの場合には、上述の計算から得られるコンセンサスCDRを生殖細胞系CDRで置き換えた。Vλの場合は、プロテアーゼに切断される可能性のある部位、および構造的な制約の可能性を避けるため、選択された生殖細胞系CDRのいくつかで、少数のアミノ酸を変更した。
以下の生殖細胞系遺伝子のCDRが選択された:
CDR3の場合、これらは最初にオリゴヌクレオチドライブラリーにより置換する計画だったため、任意の配列を選択することができた。大腸菌においてコンセンサス配列の発現および折り畳みの振る舞いを調べるためには、すべての配列が同一のCDR3を有すると、折り畳みの振る舞いに対するCDR3の影響はすべての場合に同一になるため、これは有用である。VL鎖(κおよびλ)およびVH鎖に、それぞれQQHYTTPPおよびARWGGDGFYAMDYという架空配列を選択した。これらの配列は、大腸菌における抗体の折り畳みと適合することが既知である(Carterら、1992)。
【0047】
1.5 遺伝子設計
上述の過程により、最終的に、ヒトの免疫系に頻繁に使用されている構造的な抗体レパートリーを表す、14のHuCALアミノ酸配列のコレクションが得られた(図2参照)。これらの配列をDNA配列に逆翻訳した。最初の段階では、大腸菌で頻繁に使用されることが既知のコドンのみを使用して逆翻訳を行った。その後、これらの遺伝子配列を使って、対応するアミノ酸配列を変化させずに導入できる可能性のある制限エンドヌクレアーゼ部位のデータベースを作製した。このデータベースを用いて、遺伝子のすべてのサブ要素(CDRおよびフレームワーク領域)に隣接する領域に位置し、同じ位置で同時にすべてのHuCAL VH、VκまたはVλ遺伝子に導入できる切断部位を選択した。いくつかの場合には、1つのサブグループのすべての遺伝子の切断部位を見つけることができなかった。そのような場合は、利用できる配列および構造の情報に従ってアミノ酸配列を変更することが可能ならば、変更した。この変更は、再び上述のように分析した。この設計過程において、合計で以下の6つのアミノ酸残基を変更した(遺伝子の名称、Kabatの番号付けに従う位置、この位置で最も高頻度に見られるアミノ酸、およびその代わりに使用したアミノ酸が記載されている)。
VH2:T3Q
VH6:S42G
Vκ3:E1D、I58V
Vκ4:K24R
Vλ3:T22S
1つの場合には(VHフレームワーク3の5'末端)、7つのVH遺伝子すべてにある1つの切断部位を同定することができなかった。代わりに、2種類の切断部位を使用した:HuCAL VH1A、VH1B、VH4およびVH5にはBstEII、 そしてHuCAL VH2、VH3、VH4、およびVH6にはNspVを使用した。
サブ要素に隣接する領域には位置しないが、アミノ酸配列を変更することなく1つのグループのすべての遺伝子に導入することができる制限エンドヌクレアーゼ部位がいくつか同定された。これらの切断部位も、さらに改良するためにこのシステムの柔軟性を高めるために導入された。最後に、すべての遺伝子配列から、残りの制限エンドヌクレアーゼ部位のうち1つを除いてすべてを除去した。除去されなかった1つの切断部位は、1つのサブグループのすべての遺伝子で異なっており、制限酵素の消化により異なる遺伝子を簡単に同定するための「フィンガープリント」部位として使用できる。設計された遺伝子、それに対応するアミノ酸配列、およびグループに特異的な制限エンドヌクレアーゼ部位は、それぞれ図3、4、5に示されている。
【0048】
1.6 遺伝子合成およびクローニング
コンセンサス遺伝子は、図6に示されるオリゴヌクレオチドを用いてProdromouおよびPearl、1992によって記述された方法で合成された。Kabatら、1991(図6および図7参照)による配列情報に基づいて、ヒト定常部ドメインCκ、Cλ、およびCH1をコードする遺伝子部分も合成された。CDR3およびフレームワーク4遺伝子部分については、すべてのHuCAL Vκ、Vλ、およびVH遺伝子でそれぞれ同一の配列が選択されたため、定常部ドメインをコードする対応する遺伝子部分と共に、この部分は1度しか作製されなかった。PCR産物はpCR-Script KS(+)(Stratagene社)またはpZErO-1(Invitrogen社)にクローニングし、配列決定により確認した。
【0049】
実施例2:HuCALに基づく抗体ライブラリーのクローニングと試験
合成コンセンサス遺伝子の作製後、2つの合成コンセンサス遺伝子の組み合わせを選択し、これらの2つのコンセンサスフレームワークに基づくライブラリーから、結合する抗体断片が単離できるかどうかを試験した。2つの遺伝子は単鎖Fv (scFv)フラグメントとしてクローニングし、VH-CDR3ライブラリーを挿入した。ライブラリーに機能的な抗体分子が存在するかどうかを試験するため、抗原としてBSAに結合した小さなハプテンフルオレセイン (FITC-BSA)を用いた選択過程を実施した。
【0050】
2.1 HuCAL VH-3Vκ2 scFvフラグメントのクローニング
コンセンサス遺伝子の設計を試験するために、無作為に選択した合成のH鎖とH鎖遺伝子の組み合わせ(HuCAL Vκ2およびHuCAL VH-3)を用いて、単鎖抗体 (scFv)フラグメントを作製した。簡単に述べると、CDR3-フレームワーク4領域を含む、VH3コンセンサス遺伝子およびCH1遺伝子部分をコードする遺伝子部分、ならびに、CDR3-フレームワーク4領域を含む、Vκ2コンセンサス遺伝子およびCκ遺伝子部分を組み立て、それぞれVH3-CH1 Fd断片の遺伝子およびVκ2-CκL鎖をコードする遺伝子を生成した。その後CH1遺伝子部分を、AGGGSGGGGSGGGGSGGGGSという配列の20量体ペプチドリンカーをコードするオリゴヌクレオチドカセットによって置き換えた。このリンカーをコードする2つのオリゴヌクレオチドは、それぞれ5'- TCAGCGGGTGGCGGTTCTGGCGGCGGTGGGAGCGGTGGCGGTGGTTCTGGCGGTGGTGGTTCCGATATCGGTCCACGTACGG-3'およびAATTCCGTACGTGGACCGATATCGGAACCACCACCGCCAGAACCACCGCCACCGCTCCCACCGCCGCCAGAACCGCCACCCGC-3'である。最後に、HuCAL-Vκ2遺伝子をEcoRVおよびBSiWIを通してHuCAL-VH3-リンカー融合物に挿入し、単鎖形式のVH-リンカー-VLで2つのコンセンサス配列をコードする最終的な遺伝子、HuCAL-VH3-Vκ2を得た。全コーディング配列は図8に示されている。
【0051】
2.2 1価ファージディスプレイプラスミドベクターpIG10.3の作製
H3κ2 scFv遺伝子のファージディスプレイシステム(Winterら、1994)を作製するために、ファージミドpIG10.3(図9)を構築した。簡単に述べると、ファージミドベクターpIG10(Geら、1995)のEcoRI/HindIII制限断片を、アンバーコドン(アンバーサプレッサー株XL1 Blueでグルタミン酸、非サプレッサー株JM83で停止コドンをコードする)および遺伝子IIIの切形バージョン(コドン249の融合接合部、Lowmanら、1991参照)をPCR変異誘発を使って後につなげたc-mycによって置き換えた。
【0052】
2.3. H-CDR3ライブラリーの構築
トリヌクレオチドコドンを含むオリゴヌクレオチドを鋳型として用い、隣接領域に相補的なオリゴヌクレオチドをプライマーとして用いて、2つの長さ(10および15アミノ酸)のH鎖CDR3ライブラリーを構築した。機能的な抗体に最も高頻度で出現するCDR3構造にのみ集中するため、CDR3ループのRH94およびDH101のソルトブリッジを保存した。15量体のライブラリーに関しては、フェニルアラニンとメチオニンがこの長さのヒトCDR3に非常に頻繁に出現するため(データは示していない)、100の位置にはこの2つの残基の両方が導入された。同じ理由から、102の位置にはバリンとチロシンが導入された。無作為化したすべての他の位置には、トリヌクレオチド混合物には使用されていないシステイン以外のすべてのアミノ酸のコドンが含まれていた。
オリゴヌクレオチド鋳型O3HCDR103T (5'- GATACGGCCGTGTATTATTGCGCGCGT (TRI)6GATTATTGGGGCCAAGGCACCCTG-3') およびO3HCDR153T (5'- GATACGGCCGT GTATTATTGCGCGCGT(TRI)10(TTT/ATG)GAT(GTT/TAT)TGGGGCCAAGGCACCCTG-3') ならびにプライマーO3HCDR35 (5'-GATACGGCCGTGTATTATTGC-3')およびO3HCDR33 (5'-CAGGGTGCCTTGGCCCC-3') を用いたPCR断片から、長さが10および15のCDR3ライブラリーを作製した。ここで、TRIはシステイン以外のすべてのアミノ酸を含むトリヌクレオチド混合物、(TTT/ATG)および(GTT/TAT)はそれぞれ、アミノ酸のフェニルアラニン/メチオニン、およびバリン/チロシンをコードするトリヌクレオチド混合物である。これらのライブラリーの潜在的な多様性は、10量体と15量体につき、それぞれ4.7 x 107および3.4 x 1010である。ライブラリーカセットは、まず両方のプライマーの存在下でオリゴの鋳型をPCR増幅して合成した:総容量100 μl中にオリゴの鋳型O3HCDR103TまたはO3HCDR153Tが25 pmol、プライマーO3HCDR35およびO3HCDR33が各50 pmol、dNTPが20 nmol、10x緩衝液およびPfu DNAポリメラーゼ(Stratagene社)が2.5ユニットで、30サイクル(92℃で1分、62℃で1分、72℃で1分)。ホットスタート手順を使用した。この結果として得られた混合物をフェノール抽出し、エタノール沈殿し、さらにEagIおよびStyIにより一晩消化した。H-CDR3をコードするベクターpIG10.3-scH3κ2のEagI-StyI断片が、これらの2つの部位で挟まれているクロラムフェニコールアセチルトランスフェラーゼ遺伝子(cat)によって置き換えられたベクターpIG10.3-scH3κ2catを、同様に消化した。消化されたベクター(35 μg)をゲル精製し、100 μgのライブラリーカセットと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿物を100 μlのddH2Oに再溶解した。連結物は氷上で、新しく調製したエレクトロコンピテントXL1 Blue 1 mlと混合した。20回のエレクトロポレーションを行い、形質転換細胞をSOC培地で希釈し、37℃で30分間振盪し、37℃で6〜9時間大きなLBプレート(Amp/Tet/グルコース)に蒔いた。形質転換細胞の数(ライブラリーサイズ)は10量体と15量体につき、それぞれ3.2 x 107および2.3 x 107だった。コロニーは2xYT培地(Amp/Tet/グルコース)に懸濁し、グリセロール培養として保存した。
最初のライブラリーの質を試験するために、24の独立したコロニー(10量体から12、15量体から12)からファージミドを単離し、制限酵素による消化と配列決定を行って分析した。24のファージミドの分析では、すべてにおいて完全なベクターが存在していることが示された。これらのクローン(図10参照)の配列分析により、24のうちの22がH鎖CDR3領域の機能的な配列を含むことが示された。10量体ライブラリーの12クローンのうちの1つは、長さが10ではなく9のCDR3を持っていた。15量体ライブラリーの12のクローンのうちの2つは、オープンリーディングフレームを持たないため非機能的scFvを持っていた。これら2つのクローンのうちの1つは、2つの連続的インサートを含んでいたが、読み枠は、ずれていた(データは示していない)。導入されたすべてのコドンは、片寄りなく分布していた。
各ライブラリーメンバーの発現量も測定した。簡単に述べると、各ライブラリーから9クローンをAmp/Tet/0.5%グルコースを含む2xYT培地中で37℃で一晩培養した。次の日、培養液をAmp/Tetを含む新鮮な培地で希釈した。OD600nmが0.4の時点で、培養物を1 mMのIPTGで誘導し、室温で一晩振盪した。その後、細胞沈殿物を1 mM EDTAを添加したPBS緩衝液1 mlに懸濁した。懸濁液を超音波処理し、上清を還元条件のSDS-PAGEで分離し、ナイロン膜にブロッティングし、抗FLAG M1抗体で検出した(図11参照)。10量体のライブラリーの9つのクローンは、すべてscFvフラグメントを発現していた。さらに、すべてにおいて遺伝子III/scFv融合タンパク質が存在していた。15量体ライブラリーで分析した9つのクローンでは、6/9 (67%)がscFvと遺伝子III/scFv融合タンパク質の両方を発現していた。さらに重要なことに、 scFvと遺伝子III/scFv融合タンパク質を発現するすべてのクローンは、ほぼ同じ発現量を示した。
【0053】
2.4 バイオパニング
標準的なプロトコールを用いて、抗体ライブラリーをディスプレイするファージを調製した。10量体ライブラリー由来のファージを、15量体ライブラリーのファージと20:1の比率で混合した(1ウェル当たり10量体を1 x 1010 cfuおよび15量体を5 x 108)。続いて、ファージ溶液を使用して、PBS中100 μg/mlの濃度でFITC-BSA(Sigma社)によりコートしたELISAプレート(Maxisorp, Nunc社)で4℃で一晩のパニングを行った。抗原によりコートされたウェルをPBS中の3%粉ミルクでブロックし、各ウェルに1%粉ミルク中のファージ溶液を添加し、プレートを室温で1時間振盪した。その後ウェルをPBSTおよびPBSで洗った(室温で5分間の振盪を伴い各4回)。結合したファージを室温で0.1 Mトリエチルアミン(TEA)により10分間溶出した。溶出されたファージ溶液をただちに1/2の体積の1 M Tris-Cl、pH 7.6で中和した。溶出されたファージ溶液(約450 μl)を用いて37℃で30分間XL1 Blue細胞を感染した。その後、感染した培養液を大きなLBプレート(Amp/Tet/グルコース)上に蒔き、コロニーが見えるまで37℃で培養した。コロニーを2xYT培地に懸濁し、グリセロール培養を上述のようにして作製した。このパニング操作を2回繰り返し、3回目の溶出はPBS中に100 μg/mlの濃度でフルオレセインを添加して行った。特異的ファージ抗体の濃縮は、最初および後のフルオレセイン特異的サブライブラリーをブロッキング緩衝液に対してパニングしてモニターした(図12)。フルオレセインに対する特異性を有する抗体は、3回のパニング後に単離された。
【0054】
2.5 ELISA測定
バイオパニングの成功の重要な基準の1つは、標的抗原またはハプテンに結合する個々のファージクローンの単離である。我々は、抗FITCファージ抗体クローンを単離し、まずファージELISA形式で解析した。3回のバイオパニング(上記参照)の後、ファージミドを含む24のクローンを、ELISAプレート(Nunc社)中で100 μlの2xYT培地(Amp/Tet/グルコース)に接種し、その後37℃で5時間振盪した。100 μlの2xYT培地(Amp/Tet/1 mM IPTG)を添加し、振盪を30分間続けた。さらに、ヘルパーファージ(1 x 109 cfu/ウェル)を含む100 μlの2xYT培地(Amp/Tet)を加え、室温で3時間振盪した。ヘルパーファージの感染が成功したものを選択するためにカナマイシンを添加し、一晩振盪を継続した。その後プレートを遠心し、上清を100 μl FITC-BSA(100 μg/ml)でコートして粉ミルクでブロックしたELISAウェルに直接ピペットで添加した。パニング時と同様に洗い、可溶性POD基質(Boehringer-Mannheim社)を用いて、抗M13抗体-POD結合物(Pharmacia社)によって結合したファージを検出した。 FITC-BSAに対してスクリーニングした24のクローンのうち、22はELISAで活性を示した(図13)。同様の力価を有する最初のライブラリーでは、シグナルは検出されなかった。
フルオレセインに対する特異性は、競合的ELISAによって測定した。上述のようにして、FITCに特異的な5つのscFvのペリプラズム画分を調製した。ウエスタンブロッティングにより、すべてのクローンがほぼ同量のscFvフラグメントを発現することが示された(データは示していない)。上述のようにしてELISAを行ったが、さらに、ウェルに添加する前に、ペリプラズム画分を緩衝液(阻害なし)、10 mg/ml BSA(BSAによる阻害)、または10 mg/mlフルオレセイン(フルオレセインによる阻害)のいずれかで30分間インキュベートした。結合するscFvフラグメントを抗FLAG抗体M1を用いて検出した。可溶性フルオレセインを加えたときにのみELISAシグナルは阻害され、scFvの結合はフルオレセインに特異的であることが示された(図14)。
【0055】
2.6 配列分析
ライブラリー中のフルオレセイン結合抗体の配列の多様性を見積もるために、20クローンのH鎖CDR3領域の配列を分析した(図15)。合計では、20配列のうちの16(80%)が異なっており、構築したライブラリーにはフルオレセインと結合する非常に多様なレパートリーが含まれることが示された。CDR3は特定の配列の相同性を示さなかったが、平均4つのアルギニン残基を含んでいた。フルオレセイン結合抗体におけるアルギニンに対するこの偏向は、Barbasら、1992によってすでに記述されている。
【0056】
2.7 生産
選択された3つのクローンのファージミドDNAによって大腸菌JM83を形質転換し、0.5 L 2xYT培地中で培養した。OD600nm=0.4において1 mM IPTGにより誘導を行い、室温で一晩激しく振盪して増殖させた。細胞を収集し、沈殿物をPBS緩衝液中に懸濁し、超音波処理した。遠心により細胞破片から上清を分離し、BioLogicシステム(Bio-Rad社)により、POROS MC 20カラム(IMAC、PerSeptive Biosystems社)とイオン交換クロマトグラフィーを組み合わせて精製した。イオン交換カラムは、POROS HS、CM、もしくはHQ、またはPI 20(PerSeptive Biosystems社)のうちの1つで、精製するscFvの理論的pIに依存した。すべての緩衝液のpHは、常に精製するscFvのpIよりも1単位低いまたは高いように調製した。サンプルを最初のIMACカラムにかけ、カラム容積の7倍の20 mMリン酸ナトリウム、1 M NaCl、10 mMイミダゾールで洗った。洗浄後、カラム容積の7倍の20 mMリン酸ナトリウム、10 mMイミダゾールで洗った。その後カラム容積の3倍のイミダゾール勾配(10〜250 mM)をかけ、溶出物をイオン交換カラムに直接につなげた。カラム容積の9倍の250 mMイミダゾールによる無勾配洗浄後、カラム容積の15倍の250 mMから100 mM、およびカラム容積の7倍のイミダゾール/NaCl勾配(100〜10 mMイミダゾール、0〜1 M NaCl)で洗浄した。流速は5 ml/minだった。scFvフラグメントの純度は、SDS-PAGEクーマシー染色(図16)で確認した。フラグメントの濃度は、理論的に決定した吸光係数(Gillおよびvon Hippel、1989)を用いて、280 nmにおける吸光度により決定した。scFvフラグメントは均一に精製された(図16参照)。精製したフラグメントの収量は5〜10 mg/L/ODにわたっていた。
【0057】
実施例3:抗原のコレクションに対するHuCAL H3κ2ライブラリー
実施例2で使用したライブラリーをさらに試験するために、βエストラジオール 、テストステロン、ルイス-Yエピトープ(LeY)、インターロイキン-2(IL-2)、リンホトキシン-β(LT-β)、E-セレクチンリガンド-1(ESL-1)、およびBSAを含む様々な抗原を用いて、新規の選択手順を実行した。
【0058】
3.1 バイオパニング
ライブラリーおよびすべての手順は実施例2に記載されたものと同一である。ELISAプレートを、βエストラジオール-BSA(100 μg/ml)、テストステロン-BSA(100 μg/ml)、 LeY-BSA(20 μg/ml)、 IL-2(20 μg/ml)、ESL-1(20 μg/ml)、BSA(100 μg/ml)、LT-β(変性タンパク質、20 μg/ml)によってコートした。最初の2回には、結合したファージを0.1 Mトリエチルアミン(TEA)により室温で10分間溶出した。BSAの場合、3回のパニング後の溶出は、PBS中100 μg/mlの濃度のBSAをさらに添加して行った。他の抗原の場合は、3回目の溶出は0.1 Mトリエチルアミンによって行った。LeYを除くすべての場合に、結合するファージの濃縮が観察された(図17)。さらに、15量体ライブラリーのみを用いてバイオパニング実験を繰り返すと、LeY結合ファージも濃縮された(データは示されていない)。
【0059】
3.2 ELISA測定
FITCについて実施例2に記載された手順で、β-エストラジオール、テストステロン、LeY、LT-β、ESL-1およびBSAに結合するクローンをさらに分析および解析した。抗βエストラジオールおよび抗ESL-1抗体に関するELISAデータは図18に示されている。1つの実験では、scFvフラグメントの選択性と交差反応性を試験した。このために、ELISAプレートをFITC、テストステロン、βエストラジオール、BSA、およびESL-1の各抗原により5ウェルずつ5列に並べてコートし、各抗原につき1つ、合計5つの抗体を抗原についてスクリーニングした。図19は抗体が選択された抗原に特異的に結合し、他の4つの抗原には交差反応性が低いことを示す。
【0060】
3.3 配列分析
β-エストラジオール(34クローン)、テストステロン(12クローン)、LT-β(23クローン)、 ESL-1(34クローン)およびBSA(10クローン)に対するいくつかのクローンの配列データを図20〜24に示す。
【0061】
実施例4:ベクターの構築
コンセンサス遺伝子レパートリーがモジュール型であることを利用するためには、HuCALライブラリーのファージディスプレイスクリーニングとその後の最適化手順に使用できるベクター系を構築する必要がある。したがって、一本鎖または二本鎖の複製開始点、プロモーター/オペレーター、リプレッサーまたはターミネーター要素、抵抗性遺伝子、組み換え可能な部位、線状ファージ上のディスプレイのための遺伝子III、シグナル配列、または検出用標識のようなすべての必要なベクター要素が、モジュール型コンセンサス遺伝子の制限部位のパターンに適合するようにしなくてはならない。図25はpCALベクター系の概略図と、その中のベクターモジュールおよび制限部位の配置を示す。図25aは、すでにコンセンサス遺伝子またはベクター要素にモジュールシステムの一部として組み込まれているか、またはシステム全体にまだ存在しないすべての制限部位のリストを示す。後者は、後の段階で新規モジュールの導入のために、または新規モジュール中で使用することができる。
【0062】
4.1 ベクターモジュール
HuCAL遺伝子のサブ要素に隣接する制限部位を取り除き、ベクターモジュール自身が唯一である制限部位に隣接する、一連のベクターモジュールを作製した。これらのモジュールは、遺伝子合成または鋳型の変異誘発のいずれかによって作製した。変異誘発はアド・オンPCRにより、PCRに基づくアセンブリー方法を用いて部位特異的突然変異誘発法(Kunkelら、1991)または多部位のオリゴヌクレオチドを介する変異誘発(Sutherlandら、1995;Perlak、1990)によって行った。
図26は作製したモジュールのリストを含む。制限部位FseIを追加するために、ターミネーターモジュールM9 (HindIII-Ipp-PacI) の代わりに、より大きなカセットM9IIを調製した。M9IIはHindIII/BsrGIによりクローニングできる。
すべてのベクターモジュールは、制限分析と配列決定により解析した。モジュールM11-IIの場合は、モジュールの配列決定により、鋳型の配列データベースと比較すると164/65の位置の2つの塩基が異なることが分かった。これらの2つの塩基(CA→GC)により別のBanII部位が生成した。同じ2つの塩基の違いは、他のバクテリオファージのf1複製開始点中にも出現するため、この2つの塩基は鋳型に存在しておりクローニング中の変異誘発によって生成したものではないと考えられた。このBanII部位は部位特異的突然変異誘発法により除去され、モジュールM11-IIIが生成した。モジュールM14のBssSI部位は、初めはColE1複製開始点の機能に影響を与えずに除去することができなかったため、最初のpCALベクターシリーズのクローニングにはM14-Ext2が使用された。図29〜34はモジュール型ベクターpCAL4(以下参照)の組立てに使用したモジュールの機能地図と配列を示す。他のモジュールの機能地図と配列は図35aに記載されている。図35bにはモジュールの合成に使用したオリゴヌクレオチドおよびプライマーのリストが記載されている。
【0063】
4.2 クローニングベクターpMCS
各ベクターモジュールを組み立てるために、特異的な多クローニング部位モジュール(MCS)を含むクローニングベクターpMCSを作製した。まず、MCSカセット(図27)を遺伝子合成によって作製した。このカセットには、すべてのベクターモジュールを逐次導入するために必要なすべての制限部位が順番に含まれており、5'-HindIII部位および3'に突出したAatII部位と適合する4つの塩基を用いてクローニングできる。ベクターpMCS(図28)はpUC19をAatIIおよびHindIIIによって消化し、bla遺伝子とColE1複製開始点を含む2174塩基対の断片を単離し、MCSカセットを連結して作製した。
【0064】
4.3 モジュール型ベクターpCAL4のクローニング
これは、pMCSを制限酵素で消化し、引き続きモジュールM1(AatII/XbaIによる)、M7III(EcoRI/HindIIIによる)、M9II(HindIII/BsrGIによる)、M11-II(BsrGI/NheIによる)を連結して段階的に行った。最後に、bla遺伝子をcat遺伝子モジュールM17(AatII/BgIIIによる)によって、野生型のColE1複製開始点をモジュールM14-Ext2(BglII/NheI)によって置き換えた。図35はpCAL4の機能地図と配列を示す。
【0065】
4.4 低コピー数プラスミドベクターpCALOのクローニング
ColE1モジュールM14-Ext2の代わりにp15AモジュールM12を使用して、同様な方法で一連の低コピー数プラスミドベクターを作製した。図35aはベクターpCALO1〜pCALO3の機能地図と配列を示す。
実施例5:HuCAL scFvライブラリーの作製
【0066】
5.1 49のHuCAL scFvフラグメントすべてのクローニング
7つのHuCAL-VHおよび7つのHuCAL-VLコンセンサス遺伝子の49の組み合わせすべてを、実施例2でHuCAL VH3-Vκ2 scFvについて説明したようにして組み立て、抗体発現ベクターのpLiscシリーズの修飾バージョンであるベクターpBS12(Skerraら、1991)に挿入した。
【0067】
5.2 CDRクローニングカセットの構築
CDRの置換のために、5'末端および3'末端に複数のクローニング部位を有する普遍的βラクタマーゼクローニングカセットを作製した。5'の複数のクローニング部位には、 HuCAL VHおよびVLのCDRの5'末端に隣接するすべての制限部位が含まれており、3'の複数のクローニング部位には、 HuCAL VHおよびVLのCDRの3'末端に隣接するすべての制限部位が含まれている。5'および3'の複数のクローニング部位の両方とも、合成オリゴヌクレオチドを5'および3'プライマーとして使用し野生型βラクタマーゼ遺伝子を鋳型として使用したアド・オンPCRによってカセットとして調製した。図36はカセットbla-MCSの機能地図と配列を示す。
【0068】
5.3 VL-CDR3ライブラリーカセットの調製
7つの無作為位置を含むVL-CDR3ライブラリーは、Vκ遺伝子に関しては、オリゴヌクレオチド鋳型Vκ1およびVκ3、 Vκ2ならびにVκ4、プライマーO_K3L_5およびO_K3L_3(図37)、またVλ遺伝子に関しては、 VλおよびプライマーO_L3L_5 (5'-GCAGAAGGCGAACGTCC-3') およびO_L3LA_3(図38)を用いてPCR断片から作製した。カセットの構築は、実施例2.3に記述されるようにして行った。
【0069】
5.4 VL-CDR3ライブラリーを用いたHuCAL scFv遺伝子のクローニング
49の単鎖の各々をXbaI/EcoRIを用いてpCAL4にサブクローニングし、VL-CDR3をBbsI/MscIを用いてβラクタマーゼクローニングカセットにより置換し、その後これを上述のようにして合成した対応するVL-CDR3ライブラリーカセットによって置換した。このCDRの置換は、実施例2.3においてcat遺伝子を使用して説明されている。
【0070】
5.5 VH-CDR3ライブラリーカセットの調製
VH-CDR3ライブラリーは、実施例2.3に記載されるようにして設計および合成した。
【0071】
5.6 VL-およびVH-CDR3ライブラリーを用いたHuCAL scFv遺伝子のクローニング
VH-CDR3を置換するために、49の単鎖のVL-CDR3ライブラリーの各々をBssHII/StyIによって消化した。 BssHII/StyIによって消化された「ダミー(dummy)」カセットを挿入し、その後、これを上述のようにして合成した対応するVH-CDR3ライブラリーカセットによって置換した。
【0072】
実施例6:発現試験
scFv形式VH-リンカー-VLを用いて発現および毒性試験を行った。7つのHuCAL-VHおよび7つのHuCAL-VLコンセンサス遺伝子の49の組み合わせすべてを、実施例5で説明したようにして組み立て、抗体発現ベクターのpLiscシリーズの修飾バージョンであるベクターpBS13(Skerraら、1991)に挿入した。このベクターの地図は図39に示されている。
大腸菌JM83を各ベクターによって49回形質転換し、グリセロール保存株として保存した。4〜6クローンを同時に試験し、内部コントロールとして常にクローンH3κ2も試験に含んだ。別のコントロールとして、pBS13中のMcPC603 scFvフラグメント(KnappikおよびPluckthun、1995)も同一の条件下で発現させた。発現試験を行う2日前に、30 μg/mlクロラムフェニコールおよび60 mMグルコースを含むLBプレート上でクローンを培養した。このプレートを用いて、3 ml培養液(90 μgクロラムフェニコールおよび60 mMグルコースを含むLB培地)を37℃で一晩接種した。次の日、一晩培養した培養液を用いてクロラムフェニコール(30 μg/ml)を含む30 mlのLB培地を接種した。開始時のOD600nmを0.2に調製し、培養温度は30℃を使用した。8〜9時間の間、30分ごとに600 nmの光学密度を測定して細胞の生理を監視した。培養液のOD600nmが0.5に達したら、最終濃度が1 mMになるようにIPTGを添加して、抗体の発現を誘導した。2時間後に培養液から5 mlを採取し、抗体の発現を分析した。細胞を溶菌し、粗抽出物の可溶性画分および不溶性画分をKnappikおよびPluckthun、1995に記載されるようにして分離した。サンプルの光学密度を統一し、還元SDS-PAGEで画分を分析した。ブロッティングおよび1次抗体としてαFLAG抗体M1(Geら、1994参照)、2次抗体としてアルカリ性ホスファターゼ結合Fc特異的抗マウス抗血清を用いた免疫染色を行った後、レーンのスキャンを行い、期待されるサイズ(約30 kDa)のバンドの強度をデンシトメーターにより定量し、コントロールの抗体と比較して表にした(図40参照)。
【0073】
実施例7:フルオレセイン結合分子の最適化
7.1 L-CDR3およびH-CDR2ライブラリーカセットの構築
L-CDR3ライブラリーカセットは、オリゴヌクレオチド鋳型CDR3L (5'-TGGAAGCTGAAGACGTGGGCGTGTATTATTGCCAGCAG(TR5)(TRI)4CCG(TRI)TTTGGCCAGGGTACGAAAGTT-3') および相補鎖合成用プライマー5'-AACTTTCGTACCCTGGCC-3'を用いて調製した。ここで、(TRI)はCys以外のすべてのアミノ酸を含むトリヌクレオチド混合物で、(TR5)はAla、Arg、His、Ser、およびTyrの5つのコドンを含むトリヌクレオチドを含む。
H-CDR2ライブラリーカセットは、オリゴヌクレオチド鋳型CDRsH (5'-AGGGTCTCGAGTGGGTGAGC(TRI)ATT(TRI)2−3(6)2(TRI)ACC(TRI)TATGCGGATAGCGTGAAAGGCCGTTTTACCATTTCACGTGATAATTCGAAAAACACCA-3') および相補鎖合成用プライマー5'-TGGTGTTTTTCGAATTATCA-3'を用いて調製した。ここで、(TRI)はCys以外のすべてのアミノ酸を含むトリヌクレオチド混合物で、(6)は(A/G)(A/C/G)Tの混合を含みAla、Asn、Asp、Gly、Ser、およびThrの6つのコドンを形成し、長さの分布は、合成時に(TRI)混合物の準化学量論的カップリングを1度行いDNA合成に通常使用されるキャッピング段階を省略して得られた。
DNA合成を40 nmolスケールで行い、オリゴをTE緩衝液に溶解し、スピンカラム(S-200)を用いたゲル濾過により精製し、260 nmにおけるOD測定値によってDNA濃度を決定した(OD 1.0 = 40 μg/ml)。
オリゴヌクレオチド鋳型10 nmolおよび対応するプライマー12 nmolを混合し、80℃で1分間アニーリングさせ、20〜30分間かけて徐々に37℃に冷却した。クレノウポリメラーゼ(2.0 μl)および各dNTPを250 nmolずつ使用してフィルイン反応を37℃で2時間行った。過剰のdNTPをNick-Spinカラム(Pharmacia社)を用いたゲル濾過により除去し、BbsI/MscI(L-SCR3)またはXho/SfuI(H-CDR2)によって二本鎖DNAを37℃で一晩消化した。 Nick-Spinカラム(Pharmacia社)によりカセットを精製し、ODを測定して濃度を決定し、カセットを分注(15 pmol)して-80℃で保存した。
【0074】
7.2 ライブラリーのクローニング
実施例2で用いられたFITC結合クローンのコレクションからDNAを調製した(クローンに対し約104)。XbaI/EcoRI消化によりscFvフラグメントのコレクションを単離した。実施例4.3に説明されるベクターpCAL4(100 fmol、10 μg)を同様にXbaI/EcoRI消化し、ゲル濾過し、300 fmolのscFvフラグメントコレクションと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿物を100 μlのddH20に再溶解した。連結反応液を1 mlの新しく調製したエレクトロコンピテントなSCS 101細胞(L-CDR3の最適化用)またはXL1 Blue細胞(H-CDR2の最適化用)と氷上で混合した。エレクトロポレーションを1回行い、形質転換細胞をSOC培地で溶出し、37℃で30分間振盪し、一部をLBプレート(Amp/Tet/グルコース)上に37℃で6〜9時間蒔いた。形質転換細胞の数は5 x 104だった。
ベクターDNA(100 μg)を単離し、 L-CDR3の最適化にはBbsI/MscIで、 H-CDR2の最適化にはXhoI/NspVで消化した(scH3κ2の配列と制限地図は図8参照)。精製したベクター断片10μg (5 pmol)をL-CDR3またはH-CDR2ライブラリーカセット15 pmolと16℃で一晩連結した。連結反応液をイソプロパノール沈殿し、空気乾燥し、沈殿を100 μlのddH20に溶解した。連結反応液を新しく調製したエレクトロコンピテントなXL1 Blue細胞と氷上で混合した。エレクトロポレーションを行い、形質転換細胞をSOC培地で溶出し、37℃で30分間振盪し、一部をLBプレート(Amp/Tet/グルコース)上に37℃で6〜9時間蒔いた。形質転換細胞の数(ライブラリーサイズ)は両方のライブラリーとも108以上だった。ライブラリーはグリセロール培養として保存した。
【0075】
7.3 バイオパニング
これは実施例2.1において最初のH3κ2 H-CDR3ライブラリーについて記載したようにして実行した。FITCに結合する最適化したscFvは、実施例2.2および2.3に記載したようにして解析および分析したが、必要ならば、最適化をさらに繰り返すこともできる。
【0076】
参考文献
【特許請求の範囲】
【請求項1】
(ポリ)ペプチドライブラリーの作製に適当な1つまたは複数の(ポリ)ペプチド配列をコードする1つまたは複数の核酸配列を組み立てる方法であって、該(ポリ)ペプチド配列がアミノ酸コンセンサス配列を含むものであり、以下の段階を含む方法:
(a) 少なくとも3つの相同蛋白質のコレクションから、少なくとも1つのアミノ酸コンセンサス配列を含む1つまたは複数の(ポリ)ペプチド配列を導き出す段階;
(b) 選択的に、該(ポリ)ペプチド配列内、または該(ポリ)ペプチドもしくは他の(ポリ)ペプチド配列の間においてアミノ酸の間の好ましくない相互作用を除去するため修飾される、該(ポリ)ペプチド配列内のアミノ酸を同定する段階;
(c) 各々の該(ポリ)ペプチド配列の中で、少なくとも1つの構造的サブ要素を同定する段階;
(d) 各々の該(ポリ)ペプチド配列を、対応する核酸コード配列に逆翻訳する段階;
(e) 該サブ要素をコードするサブ配列の末端に隣接する領域または末端の間にある領域に切断部位を組み立てる段階で、該切断部位の各々が、
(ea) 各々の該核酸コード配列内では唯一であり;
(eb) すべての該核酸コード配列の対応するサブ配列に共通である。
【請求項2】
切断部位はセットの間で唯一であるというさらなる条件で、各セットについて請求項1に記載される各段階の実行を含む、1つまたは複数の核酸配列の2つまたはそれ以上のセットを組み立てる方法。
【請求項3】
セットのうち少なくとも2つが、少なくとも3つの相同蛋白質の同じコレクションから導き出される、請求項2記載の方法。
【請求項4】
組立方法に核酸コード配列の合成がさらに含まれる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
核酸コード配列のベクターへのクローニングをさらに含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
好ましくない相互作用の除去により(ポリ)ペプチドの発現が増大される、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
以下の段階をさらに含む、請求項1〜6のいずれか一項に記載の方法:
(f) サブ配列の末端に隣接する領域または末端の間にある領域に位置する切断部位の少なくとも2つを切断する段階;および
(g) 該サブ配列を異なる配列と交換する段階;および
(h) 選択的に、段階(f)および(g)を1回またはそれ以上繰り返す段階。
【請求項8】
異なる配列が、同一または異なる(ポリ)ペプチドに由来する同一または異なるサブ要素をコードする異なるサブ配列からなる群より選択される、請求項7記載の方法。
【請求項9】
異なる配列が
(i) ゲノム配列またはゲノム配列に由来する配列、
(ii) 再配列したゲノム配列または再配列したゲノム配列に由来する配列、および
(iii) 無作為配列
からなる群より選択される、請求項7または8記載の方法。
【請求項10】
核酸コード配列の発現をさらに含む、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
以下の段階をさらに含む、請求項1〜10のいずれか一項に記載の方法:
(i) 発現後に、得られた(ポリ)ペプチドを所望の性質についてスクリーニングする段階、
(k) 選択的に、段階(i)で得られた1つまたは複数の(ポリ)ペプチドをコードする核酸配列について、段階(f)から(i)を1回またはそれ以上繰り返す段階。
【請求項12】
所望の性質が、標的分子に対して最適化した親和性または特異性、最適化した酵素活性、最適化した発現収量、最適化した安定性、および最適化した可溶性からなる群より選択される、請求項11記載の方法。
【請求項13】
切断部位が制限酵素によって切断される部位である、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
構造的サブ要素が1〜150アミノ酸を含む、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
構造的サブ要素が3〜25アミノ酸を含む、請求項14記載の方法。
【請求項16】
核酸がDNAである、請求項1〜15のいずれか一項に記載の方法。
【請求項17】
(ポリ)ペプチドが特定の種に特徴的なアミノ酸パターンを有する、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
種がヒトである、請求項17記載の方法。
【請求項19】
(ポリ)ペプチドが免疫グロブリンスーパーファミリーのメンバーまたは誘導体の少なくとも一部である、請求項1〜18のいずれか一項に記載の方法。
【請求項20】
免疫グロブリンスーパーファミリーのメンバーまたは誘導体が免疫グロブリンファミリーのメンバーまたは誘導体である、請求項19記載の方法。
【請求項21】
(ポリ)ペプチドが、構造的サブ要素がフレームワーク領域(FR) 1、2、3、もしくは4、または相補性決定領域(CDR) 1、2、もしくは3であるH鎖もしくはL鎖可変領域であるか、またはこれに由来するものである、請求項19または20記載の方法。
【請求項22】
(ポリ)ペプチドが、HuCALコンセンサス遺伝子:Vκ1、 Vκ2、 Vκ3、 Vκ4、 Vλ1、 Vλ2、 Vλ3、 VH1A、VH1B、VH2、VH3、VH4、VH5、VH6、Cκ、Cλ、CH1であるかもしくはこれに由来する、または該HuCALコンセンサス遺伝子の任意の組み合わせである、請求項20または21記載の方法。
【請求項23】
免疫グロブリンファミリーの誘導体または組み合わせが、Fv、ジスルフィド結合したFv、単鎖Fv (scFv)、またはFabフラグメントである、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
誘導体が、H鎖CDR3サブ要素をコードする任意のサブ配列を含む、HuCAL VH3コンセンサス遺伝子とHuCAL Vλ2コンセンサス遺伝子との組み合わせを有するscFvフラグメントである、請求項22〜23記載の方法。
【請求項25】
(ポリ)ペプチド配列または(ポリ)ペプチドの少なくとも一部が、それぞれ、少なくとも1つの付加的部分をコードする配列または少なくとも1つの付加的部分に接続する、請求項1〜24のいずれか一項に記載の方法。
【請求項26】
接続が、それぞれ、隣接する核酸配列またはアミノ酸配列を介して形成される、請求項25記載の方法。
【請求項27】
付加的部分が、毒素、サイトカイン、受容体酵素、金属イオンに結合することのできる部分、ペプチド、検出および/もしくは精製に適当な標識、またはホモもしくはヘテロ結合ドメインである、請求項25〜26に記載の方法。
【請求項28】
核酸配列の発現により、生物学的活性および/または特異性のレパートリーの生成、好ましくは普遍的フレームワークに基づくレパートリーの生成に至る、請求項10〜27のいずれか一項に記載の方法。
【請求項29】
請求項1〜28のいずれか一項に記載の方法によって得られる核酸配列。
【請求項30】
請求項1〜28のいずれか一項に記載の方法によって得られる核酸配列のコレクション。
【請求項31】
請求項5〜28のいずれか一項に記載の方法によって得られる組み換えベクター。
【請求項32】
請求項5〜30のいずれか一項に記載の方法によって得られる組み換えベクターのコレクション。
【請求項33】
請求項31記載の組み換えベクターで形質転換した宿主細胞。
【請求項34】
請求項32記載の組み換えベクターのコレクションで形質転換した宿主細胞のコレクション。
【請求項35】
請求項33記載の宿主細胞または請求項34記載の宿主細胞のコレクションを適当な条件下で培養する段階、および(ポリ)ペプチドまたは(ポリ)ペプチドのコレクションを単離する段階を含む、請求項1〜28のいずれかに定義される(ポリ)ペプチドまたは(ポリ)ペプチドのコレクションを生産する方法。
【請求項36】
請求項29記載の核酸配列によってコードされる、請求項1〜3のいずれか一項に記載の方法によって考案されるか、または請求項4〜28もしくは35のいずれか一項に記載の方法によって得られる、(ポリ)ペプチド。
【請求項37】
請求項30記載の核酸配列のコレクションによってコードされる、請求項1〜3のいずれか一項に記載の方法によって考案されるか、または請求項4〜28もしくは35のいずれか一項に記載の方法によって得られる、(ポリ)ペプチドのコレクション。
【請求項38】
請求項1(e)および2に定義される任意の切断部位を本質的に欠くという特徴を有する、請求項5〜28および35のいずれかに記載の方法において使用に適するベクター。
【請求項39】
発現ベクターである、請求項38記載のベクター。
【請求項40】
(a) 請求項29記載の核酸配列、
(b) 請求項30記載の核酸配列のコレクション;
(c) 請求項31記載の組み換えベクター;
(d) 請求項32記載の組み換えベクターのコレクション;
(e) 請求項36記載の(ポリ)ペプチド;
(f) 請求項37記載の(ポリ)ペプチドのコレクション;
(g) 請求項38または39記載のベクター;および選択的に、
(h) 請求項35記載の方法を実施するのに適当な宿主細胞
のうち少なくとも1つを含むキット。
【請求項41】
以下の段階を含む、2つまたはそれ以上の蛋白質のコレクションをコードする2つまたはそれ以上の遺伝子を設計する方法:
(a) (aa) 2つまたはそれ以上の相同遺伝子配列を同定するか、または
(ab) 少なくとも3つの相同配列を分析し、
それから2つまたはそれ以上のコンセンサス遺伝子配列を導き出す段階、
(b) 生成する蛋白質におけるアミノ酸の間の好ましくない相互作用を除去するため、選択的に、該コンセンサス遺伝子配列のコドンを修飾する段階、
(c) 該コンセンサス遺伝子配列において、構造的サブ要素をコードするサブ配列を同定する段階、
(d) 1つまたは複数の切断部位を画定するため、該サブ配列の末端に隣接する領域または末端の間にある領域において1つまたは複数の塩基を修飾する段階であって、その各々の切断部位は:
(da) 各コンセンサス遺伝子配列内では唯一であり、
(db) 単一のいずれのサブ配列に関しても適合する部位を形成せず、
(dc) すべての相同サブ配列に共通である。
【請求項42】
(a) 請求項41記載の遺伝子を設計する段階、および
(b) 該遺伝子を合成する段階
を含む、2つまたはそれ以上の蛋白質のコレクションをコードする2つまたはそれ以上の遺伝子を調製する方法。
【請求項43】
請求項42記載の方法に従って調製された遺伝子のコレクション。
【請求項44】
(a) 相同であるか、少なくとも3つの相同遺伝子に由来するコンセンサス遺伝子配列を表し、
(b) 各々の切断部位が、
(ba) 構造的サブ要素をコードする遺伝子サブ配列の末端にあるかまたは隣接しており、
(bb) 各遺伝子配列内では唯一であり、
(bc) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(bd) すべての相同サブ配列に共通である、切断部位を有する
遺伝子配列に由来する、2つまたはそれ以上の遺伝子のコレクション。
【請求項45】
各々の遺伝子配列が、特定の種に特徴的なヌクレオチド組成を有する、請求項43または44のいずれかに記載の遺伝子のコレクション。
【請求項46】
種がヒトである、請求項45記載の遺伝子のコレクション。
【請求項47】
1つまたは複数の遺伝子配列が、免疫グロブリンスーパーファミリー、好ましくは免疫グロブリンファミリーのメンバーの少なくとも一部をコードする、請求項43〜46のいずれかに記載の遺伝子のコレクション。
【請求項48】
構造的サブ要素が、抗体H鎖のフレームワーク領域1、2、3、および4、ならびに/またはCDR領域1、2、および3の任意の組み合わせに相当する、請求項47記載の遺伝子のコレクション。
【請求項49】
構造的サブ要素が、抗体L鎖のフレームワーク領域1、2、3、および4、ならびに/またはCDR領域1、2、および3の任意の組み合わせに相当する、請求項47記載の遺伝子のコレクション。
【請求項50】
請求項43〜49のいずれかに記載の遺伝子配列のコレクションを含むベクターのコレクション。
【請求項51】
ベクターが、請求項43〜49のいずれかに記載の遺伝子のコレクションに含まれる切断部位を含まないというさらなる特徴を含む、請求項50記載のベクターのコレクション。
【請求項52】
(a) 請求項50または51のいずれかに記載のベクターのコレクションから、蛋白質のコレクションを発現させる段階、
(b) 所望の性質を有する1つまたは複数の蛋白質を単離するため、該コレクションをスクリーニングする段階、
(c) 段階(b)で単離された蛋白質をコードする遺伝子を同定する段階、
(d) 請求項50または51のいずれかに記載の新規のベクターを生成するため、選択的に、段階(b)で単離された蛋白質をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、該サブ配列を、構造的サブ要素をコードする1つまたは複数の第2のサブ配列によって置換する段階、
(e) 選択的に、段階(a)〜(c)を繰り返す段階
を含む、所望の性質を有する1つまたは複数の蛋白質をコードする1つまたは複数の遺伝子を同定する方法。
【請求項53】
(a) 請求項50または51のいずれかに記載のベクターのコレクションから、蛋白質のコレクションを発現させる段階、
(b) 標的に結合する1つまたは複数の抗体断片を単離するため、該コレクションをスクリーニングする段階、
(c) 段階(b)で単離された蛋白質をコードする遺伝子を同定する段階、
(d) 請求項50または51のいずれかに記載の新規のベクターを生成するため、選択的に、段階(b)で単離された抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、該サブ配列を、構造的サブ要素をコードする1つまたは複数の第2のサブ配列によって置換する段階、
(e) 選択的に、段階(a)〜(c)を繰り返す段階
を含む、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定する方法。
【請求項54】
(a) 相同であるか、少なくとも3つの相同遺伝子に由来するコンセンサス遺伝子配列を表し、
(b) 各々の切断部位が
(ba) 構造的サブ要素をコードする遺伝子サブ配列の末端にあるかまたは隣接しており、
(bb) 各遺伝子配列内では唯一であり、
(bc) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(bd) すべての相同サブ配列に共通である、切断部位を有する
遺伝子配列に由来する、2つまたはそれ以上の遺伝子を含むキット。
【請求項55】
構造的サブ要素をコードし、組み立てて遺伝子を形成することができ、各々の切断部位が
(a) 該遺伝子サブ配列の末端にあるかまたは隣接しており、
(b) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(c) すべての相同サブ配列に共通である、
切断部位を有する、2つまたはそれ以上の遺伝子サブ配列を含むキット。
【請求項1】
(ポリ)ペプチドライブラリーの作製に適当な1つまたは複数の(ポリ)ペプチド配列をコードする1つまたは複数の核酸配列を組み立てる方法であって、該(ポリ)ペプチド配列がアミノ酸コンセンサス配列を含むものであり、以下の段階を含む方法:
(a) 少なくとも3つの相同蛋白質のコレクションから、少なくとも1つのアミノ酸コンセンサス配列を含む1つまたは複数の(ポリ)ペプチド配列を導き出す段階;
(b) 選択的に、該(ポリ)ペプチド配列内、または該(ポリ)ペプチドもしくは他の(ポリ)ペプチド配列の間においてアミノ酸の間の好ましくない相互作用を除去するため修飾される、該(ポリ)ペプチド配列内のアミノ酸を同定する段階;
(c) 各々の該(ポリ)ペプチド配列の中で、少なくとも1つの構造的サブ要素を同定する段階;
(d) 各々の該(ポリ)ペプチド配列を、対応する核酸コード配列に逆翻訳する段階;
(e) 該サブ要素をコードするサブ配列の末端に隣接する領域または末端の間にある領域に切断部位を組み立てる段階で、該切断部位の各々が、
(ea) 各々の該核酸コード配列内では唯一であり;
(eb) すべての該核酸コード配列の対応するサブ配列に共通である。
【請求項2】
切断部位はセットの間で唯一であるというさらなる条件で、各セットについて請求項1に記載される各段階の実行を含む、1つまたは複数の核酸配列の2つまたはそれ以上のセットを組み立てる方法。
【請求項3】
セットのうち少なくとも2つが、少なくとも3つの相同蛋白質の同じコレクションから導き出される、請求項2記載の方法。
【請求項4】
組立方法に核酸コード配列の合成がさらに含まれる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
核酸コード配列のベクターへのクローニングをさらに含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
好ましくない相互作用の除去により(ポリ)ペプチドの発現が増大される、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
以下の段階をさらに含む、請求項1〜6のいずれか一項に記載の方法:
(f) サブ配列の末端に隣接する領域または末端の間にある領域に位置する切断部位の少なくとも2つを切断する段階;および
(g) 該サブ配列を異なる配列と交換する段階;および
(h) 選択的に、段階(f)および(g)を1回またはそれ以上繰り返す段階。
【請求項8】
異なる配列が、同一または異なる(ポリ)ペプチドに由来する同一または異なるサブ要素をコードする異なるサブ配列からなる群より選択される、請求項7記載の方法。
【請求項9】
異なる配列が
(i) ゲノム配列またはゲノム配列に由来する配列、
(ii) 再配列したゲノム配列または再配列したゲノム配列に由来する配列、および
(iii) 無作為配列
からなる群より選択される、請求項7または8記載の方法。
【請求項10】
核酸コード配列の発現をさらに含む、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
以下の段階をさらに含む、請求項1〜10のいずれか一項に記載の方法:
(i) 発現後に、得られた(ポリ)ペプチドを所望の性質についてスクリーニングする段階、
(k) 選択的に、段階(i)で得られた1つまたは複数の(ポリ)ペプチドをコードする核酸配列について、段階(f)から(i)を1回またはそれ以上繰り返す段階。
【請求項12】
所望の性質が、標的分子に対して最適化した親和性または特異性、最適化した酵素活性、最適化した発現収量、最適化した安定性、および最適化した可溶性からなる群より選択される、請求項11記載の方法。
【請求項13】
切断部位が制限酵素によって切断される部位である、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
構造的サブ要素が1〜150アミノ酸を含む、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
構造的サブ要素が3〜25アミノ酸を含む、請求項14記載の方法。
【請求項16】
核酸がDNAである、請求項1〜15のいずれか一項に記載の方法。
【請求項17】
(ポリ)ペプチドが特定の種に特徴的なアミノ酸パターンを有する、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
種がヒトである、請求項17記載の方法。
【請求項19】
(ポリ)ペプチドが免疫グロブリンスーパーファミリーのメンバーまたは誘導体の少なくとも一部である、請求項1〜18のいずれか一項に記載の方法。
【請求項20】
免疫グロブリンスーパーファミリーのメンバーまたは誘導体が免疫グロブリンファミリーのメンバーまたは誘導体である、請求項19記載の方法。
【請求項21】
(ポリ)ペプチドが、構造的サブ要素がフレームワーク領域(FR) 1、2、3、もしくは4、または相補性決定領域(CDR) 1、2、もしくは3であるH鎖もしくはL鎖可変領域であるか、またはこれに由来するものである、請求項19または20記載の方法。
【請求項22】
(ポリ)ペプチドが、HuCALコンセンサス遺伝子:Vκ1、 Vκ2、 Vκ3、 Vκ4、 Vλ1、 Vλ2、 Vλ3、 VH1A、VH1B、VH2、VH3、VH4、VH5、VH6、Cκ、Cλ、CH1であるかもしくはこれに由来する、または該HuCALコンセンサス遺伝子の任意の組み合わせである、請求項20または21記載の方法。
【請求項23】
免疫グロブリンファミリーの誘導体または組み合わせが、Fv、ジスルフィド結合したFv、単鎖Fv (scFv)、またはFabフラグメントである、請求項20〜22のいずれか一項に記載の方法。
【請求項24】
誘導体が、H鎖CDR3サブ要素をコードする任意のサブ配列を含む、HuCAL VH3コンセンサス遺伝子とHuCAL Vλ2コンセンサス遺伝子との組み合わせを有するscFvフラグメントである、請求項22〜23記載の方法。
【請求項25】
(ポリ)ペプチド配列または(ポリ)ペプチドの少なくとも一部が、それぞれ、少なくとも1つの付加的部分をコードする配列または少なくとも1つの付加的部分に接続する、請求項1〜24のいずれか一項に記載の方法。
【請求項26】
接続が、それぞれ、隣接する核酸配列またはアミノ酸配列を介して形成される、請求項25記載の方法。
【請求項27】
付加的部分が、毒素、サイトカイン、受容体酵素、金属イオンに結合することのできる部分、ペプチド、検出および/もしくは精製に適当な標識、またはホモもしくはヘテロ結合ドメインである、請求項25〜26に記載の方法。
【請求項28】
核酸配列の発現により、生物学的活性および/または特異性のレパートリーの生成、好ましくは普遍的フレームワークに基づくレパートリーの生成に至る、請求項10〜27のいずれか一項に記載の方法。
【請求項29】
請求項1〜28のいずれか一項に記載の方法によって得られる核酸配列。
【請求項30】
請求項1〜28のいずれか一項に記載の方法によって得られる核酸配列のコレクション。
【請求項31】
請求項5〜28のいずれか一項に記載の方法によって得られる組み換えベクター。
【請求項32】
請求項5〜30のいずれか一項に記載の方法によって得られる組み換えベクターのコレクション。
【請求項33】
請求項31記載の組み換えベクターで形質転換した宿主細胞。
【請求項34】
請求項32記載の組み換えベクターのコレクションで形質転換した宿主細胞のコレクション。
【請求項35】
請求項33記載の宿主細胞または請求項34記載の宿主細胞のコレクションを適当な条件下で培養する段階、および(ポリ)ペプチドまたは(ポリ)ペプチドのコレクションを単離する段階を含む、請求項1〜28のいずれかに定義される(ポリ)ペプチドまたは(ポリ)ペプチドのコレクションを生産する方法。
【請求項36】
請求項29記載の核酸配列によってコードされる、請求項1〜3のいずれか一項に記載の方法によって考案されるか、または請求項4〜28もしくは35のいずれか一項に記載の方法によって得られる、(ポリ)ペプチド。
【請求項37】
請求項30記載の核酸配列のコレクションによってコードされる、請求項1〜3のいずれか一項に記載の方法によって考案されるか、または請求項4〜28もしくは35のいずれか一項に記載の方法によって得られる、(ポリ)ペプチドのコレクション。
【請求項38】
請求項1(e)および2に定義される任意の切断部位を本質的に欠くという特徴を有する、請求項5〜28および35のいずれかに記載の方法において使用に適するベクター。
【請求項39】
発現ベクターである、請求項38記載のベクター。
【請求項40】
(a) 請求項29記載の核酸配列、
(b) 請求項30記載の核酸配列のコレクション;
(c) 請求項31記載の組み換えベクター;
(d) 請求項32記載の組み換えベクターのコレクション;
(e) 請求項36記載の(ポリ)ペプチド;
(f) 請求項37記載の(ポリ)ペプチドのコレクション;
(g) 請求項38または39記載のベクター;および選択的に、
(h) 請求項35記載の方法を実施するのに適当な宿主細胞
のうち少なくとも1つを含むキット。
【請求項41】
以下の段階を含む、2つまたはそれ以上の蛋白質のコレクションをコードする2つまたはそれ以上の遺伝子を設計する方法:
(a) (aa) 2つまたはそれ以上の相同遺伝子配列を同定するか、または
(ab) 少なくとも3つの相同配列を分析し、
それから2つまたはそれ以上のコンセンサス遺伝子配列を導き出す段階、
(b) 生成する蛋白質におけるアミノ酸の間の好ましくない相互作用を除去するため、選択的に、該コンセンサス遺伝子配列のコドンを修飾する段階、
(c) 該コンセンサス遺伝子配列において、構造的サブ要素をコードするサブ配列を同定する段階、
(d) 1つまたは複数の切断部位を画定するため、該サブ配列の末端に隣接する領域または末端の間にある領域において1つまたは複数の塩基を修飾する段階であって、その各々の切断部位は:
(da) 各コンセンサス遺伝子配列内では唯一であり、
(db) 単一のいずれのサブ配列に関しても適合する部位を形成せず、
(dc) すべての相同サブ配列に共通である。
【請求項42】
(a) 請求項41記載の遺伝子を設計する段階、および
(b) 該遺伝子を合成する段階
を含む、2つまたはそれ以上の蛋白質のコレクションをコードする2つまたはそれ以上の遺伝子を調製する方法。
【請求項43】
請求項42記載の方法に従って調製された遺伝子のコレクション。
【請求項44】
(a) 相同であるか、少なくとも3つの相同遺伝子に由来するコンセンサス遺伝子配列を表し、
(b) 各々の切断部位が、
(ba) 構造的サブ要素をコードする遺伝子サブ配列の末端にあるかまたは隣接しており、
(bb) 各遺伝子配列内では唯一であり、
(bc) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(bd) すべての相同サブ配列に共通である、切断部位を有する
遺伝子配列に由来する、2つまたはそれ以上の遺伝子のコレクション。
【請求項45】
各々の遺伝子配列が、特定の種に特徴的なヌクレオチド組成を有する、請求項43または44のいずれかに記載の遺伝子のコレクション。
【請求項46】
種がヒトである、請求項45記載の遺伝子のコレクション。
【請求項47】
1つまたは複数の遺伝子配列が、免疫グロブリンスーパーファミリー、好ましくは免疫グロブリンファミリーのメンバーの少なくとも一部をコードする、請求項43〜46のいずれかに記載の遺伝子のコレクション。
【請求項48】
構造的サブ要素が、抗体H鎖のフレームワーク領域1、2、3、および4、ならびに/またはCDR領域1、2、および3の任意の組み合わせに相当する、請求項47記載の遺伝子のコレクション。
【請求項49】
構造的サブ要素が、抗体L鎖のフレームワーク領域1、2、3、および4、ならびに/またはCDR領域1、2、および3の任意の組み合わせに相当する、請求項47記載の遺伝子のコレクション。
【請求項50】
請求項43〜49のいずれかに記載の遺伝子配列のコレクションを含むベクターのコレクション。
【請求項51】
ベクターが、請求項43〜49のいずれかに記載の遺伝子のコレクションに含まれる切断部位を含まないというさらなる特徴を含む、請求項50記載のベクターのコレクション。
【請求項52】
(a) 請求項50または51のいずれかに記載のベクターのコレクションから、蛋白質のコレクションを発現させる段階、
(b) 所望の性質を有する1つまたは複数の蛋白質を単離するため、該コレクションをスクリーニングする段階、
(c) 段階(b)で単離された蛋白質をコードする遺伝子を同定する段階、
(d) 請求項50または51のいずれかに記載の新規のベクターを生成するため、選択的に、段階(b)で単離された蛋白質をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、該サブ配列を、構造的サブ要素をコードする1つまたは複数の第2のサブ配列によって置換する段階、
(e) 選択的に、段階(a)〜(c)を繰り返す段階
を含む、所望の性質を有する1つまたは複数の蛋白質をコードする1つまたは複数の遺伝子を同定する方法。
【請求項53】
(a) 請求項50または51のいずれかに記載のベクターのコレクションから、蛋白質のコレクションを発現させる段階、
(b) 標的に結合する1つまたは複数の抗体断片を単離するため、該コレクションをスクリーニングする段階、
(c) 段階(b)で単離された蛋白質をコードする遺伝子を同定する段階、
(d) 請求項50または51のいずれかに記載の新規のベクターを生成するため、選択的に、段階(b)で単離された抗体断片をコードする遺伝子から、構造的サブ要素をコードする1つまたは複数の遺伝子サブ配列を切り出し、該サブ配列を、構造的サブ要素をコードする1つまたは複数の第2のサブ配列によって置換する段階、
(e) 選択的に、段階(a)〜(c)を繰り返す段階
を含む、標的に結合する1つまたは複数の抗体断片をコードする1つまたは複数の遺伝子を同定する方法。
【請求項54】
(a) 相同であるか、少なくとも3つの相同遺伝子に由来するコンセンサス遺伝子配列を表し、
(b) 各々の切断部位が
(ba) 構造的サブ要素をコードする遺伝子サブ配列の末端にあるかまたは隣接しており、
(bb) 各遺伝子配列内では唯一であり、
(bc) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(bd) すべての相同サブ配列に共通である、切断部位を有する
遺伝子配列に由来する、2つまたはそれ以上の遺伝子を含むキット。
【請求項55】
構造的サブ要素をコードし、組み立てて遺伝子を形成することができ、各々の切断部位が
(a) 該遺伝子サブ配列の末端にあるかまたは隣接しており、
(b) 単一のいずれのサブ配列に関しても適合する部位を形成せず、かつ
(c) すべての相同サブ配列に共通である、
切断部位を有する、2つまたはそれ以上の遺伝子サブ配列を含むキット。
【図1】


【図2−1】

【図2−2】

【図2−3】

【図2−4】

【図2−5】

【図2−6】

【図3−1】

【図3−2】

【図3−3】

【図3−4】

【図3−5】

【図3−6】

【図3−7】

【図3−8】

【図4−1】

【図4−2】

【図4−3】

【図4−4】

【図4−5】

【図4−6】

【図5−1】

【図5−2】

【図5−3】

【図5−4】

【図5−5】

【図5−6】

【図5−7】

【図5−8】

【図5−9】

【図5−10】

【図5−11】

【図5−12】

【図5−13】

【図5−14】

【図6−1】

【図6−2】

【図6−3】

【図6−4】

【図6−5】

【図6−6】

【図6−7】

【図7−1】

【図7−2】

【図7−3】

【図7−4】

【図7−5】

【図7−6】

【図7−7】

【図7−8】

【図8−1】

【図8−2】

【図8−3】

【図8−4】

【図8−5】

【図9】

【図10−1】

【図10−2】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図24】

【図25−1】

【図25−2】

【図25−3】

【図26−1】

【図26−2】

【図26−3】

【図26−4】

【図27−1】

【図27−2】

【図28−1】

【図28−2】

【図28−3】

【図28−4】

【図28−5】

【図28−6】

【図28−7】

【図29−1】

【図29−2】

【図30−1】

【図30−2】

【図30−3】

【図31−1】

【図31−2】

【図32−1】

【図32−2】

【図32−3】

【図33−1】

【図33−2】

【図33−3】

【図33−4】

【図34−1】

【図34−2】

【図34−3】

【図34−4】

【図35−1】

【図35−2】

【図35−3】

【図35−4】

【図35−5】

【図35−6】

【図35−7】

【図35−8】

【図35−9】

【図35−10】

【図35−11】

【図35−12】

【図35−13】

【図35−14】

【図35−15】

【図35−16】

【図35−17】

【図35−18】

【図35−19】

【図35−20】

【図35−21】

【図35−22】

【図35−23】

【図35−24】

【図35−25】

【図35−26】

【図35−27】

【図35−28】

【図35−29】

【図35−30】

【図35−31】

【図35−32】

【図35−33】

【図35−34】

【図35−35】

【図35−36】

【図35−37】

【図35−38】

【図35−39】

【図35−40】

【図35−41】

【図35−42】

【図35−43】

【図35−44】

【図35−45】

【図35−46】

【図35−47】

【図35−48】

【図35−49】

【図35−50】

【図35−51】

【図35−52】

【図35−53】

【図35−54】

【図35−55】

【図35−56】

【図35−57】

【図35−58】

【図35−59】

【図35−60】

【図35−61】

【図35−62】

【図35−63】

【図35−64】

【図35−65】

【図35−66】

【図35−67】

【図35−68】

【図35−69】

【図35−70】

【図35−71】

【図35−72】

【図35−73】

【図35−74】

【図35−75】

【図35−76】

【図35−77】

【図35−78】

【図35−79】

【図35−80】

【図35−81】

【図36−1】

【図36−2】

【図36−3】

【図36−4】

【図36−5】

【図36−6】

【図37−1】

【図37−2】

【図37−3】

【図37−4】

【図38−1】

【図38−2】

【図38−3】

【図38−4】

【図39】

【図40−1】

【図40−2】

【図2−1】
【図2−2】
【図2−3】
【図2−4】
【図2−5】
【図2−6】
【図3−1】
【図3−2】
【図3−3】
【図3−4】
【図3−5】
【図3−6】
【図3−7】
【図3−8】
【図4−1】
【図4−2】
【図4−3】
【図4−4】
【図4−5】
【図4−6】
【図5−1】
【図5−2】
【図5−3】
【図5−4】
【図5−5】
【図5−6】
【図5−7】
【図5−8】
【図5−9】
【図5−10】
【図5−11】
【図5−12】
【図5−13】
【図5−14】
【図6−1】
【図6−2】
【図6−3】
【図6−4】
【図6−5】
【図6−6】
【図6−7】
【図7−1】
【図7−2】
【図7−3】
【図7−4】
【図7−5】
【図7−6】
【図7−7】
【図7−8】
【図8−1】
【図8−2】
【図8−3】
【図8−4】
【図8−5】
【図9】
【図10−1】
【図10−2】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25−1】
【図25−2】
【図25−3】
【図26−1】
【図26−2】
【図26−3】
【図26−4】
【図27−1】
【図27−2】
【図28−1】
【図28−2】
【図28−3】
【図28−4】
【図28−5】
【図28−6】
【図28−7】
【図29−1】
【図29−2】
【図30−1】
【図30−2】
【図30−3】
【図31−1】
【図31−2】
【図32−1】
【図32−2】
【図32−3】
【図33−1】
【図33−2】
【図33−3】
【図33−4】
【図34−1】
【図34−2】
【図34−3】
【図34−4】
【図35−1】
【図35−2】
【図35−3】
【図35−4】
【図35−5】
【図35−6】
【図35−7】
【図35−8】
【図35−9】
【図35−10】
【図35−11】
【図35−12】
【図35−13】
【図35−14】
【図35−15】
【図35−16】
【図35−17】
【図35−18】
【図35−19】
【図35−20】
【図35−21】
【図35−22】
【図35−23】
【図35−24】
【図35−25】
【図35−26】
【図35−27】
【図35−28】
【図35−29】
【図35−30】
【図35−31】
【図35−32】
【図35−33】
【図35−34】
【図35−35】
【図35−36】
【図35−37】
【図35−38】
【図35−39】
【図35−40】
【図35−41】
【図35−42】
【図35−43】
【図35−44】
【図35−45】
【図35−46】
【図35−47】
【図35−48】
【図35−49】
【図35−50】
【図35−51】
【図35−52】
【図35−53】
【図35−54】
【図35−55】
【図35−56】
【図35−57】
【図35−58】
【図35−59】
【図35−60】
【図35−61】
【図35−62】
【図35−63】
【図35−64】
【図35−65】
【図35−66】
【図35−67】
【図35−68】
【図35−69】
【図35−70】
【図35−71】
【図35−72】
【図35−73】
【図35−74】
【図35−75】
【図35−76】
【図35−77】
【図35−78】
【図35−79】
【図35−80】
【図35−81】
【図36−1】
【図36−2】
【図36−3】
【図36−4】
【図36−5】
【図36−6】
【図37−1】
【図37−2】
【図37−3】
【図37−4】
【図38−1】
【図38−2】
【図38−3】
【図38−4】
【図39】
【図40−1】
【図40−2】
【公開番号】特開2010−29198(P2010−29198A)
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願番号】特願2009−218390(P2009−218390)
【出願日】平成21年9月24日(2009.9.24)
【分割の表示】特願平9−509803の分割
【原出願日】平成8年8月19日(1996.8.19)
【出願人】(509265221)モルフォシス アイピー ゲーエムベーハー (2)
【Fターム(参考)】
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願日】平成21年9月24日(2009.9.24)
【分割の表示】特願平9−509803の分割
【原出願日】平成8年8月19日(1996.8.19)
【出願人】(509265221)モルフォシス アイピー ゲーエムベーハー (2)
【Fターム(参考)】
[ Back to top ]