
ステロイドホルモンとの性腺刺激ホルモン放出ホルモン類似体結合体
ホルモン成分又はその誘導体と結合した性腺刺激ホルモン放出ホルモン類似体を含んでなり、血漿ホルモン結合タンパク質に結合可能な化合物。この化合物は癌などのホルモン依存性疾患の治療、又は避妊薬として使用可能である。

【発明の詳細な説明】
【発明の開示】
【0001】
本発明は、結合体化合物、特に性腺刺激ホルモン(ゴナドトロピン)放出ホルモン結合体化合物に関する。
【0002】
性腺刺激ホルモン放出ホルモン(GnRH)は、生殖調節に関与する神経内分泌ホルモンであり、性腺刺激ホルモン、黄体形成ホルモン(LH)及び卵胞刺激ホルモン(FSH)の放出の誘因となる。
GnRH類似体は、視床下部−下垂体軸の検査においてもホルモン依存性症状の治療のための性腺刺激ホルモンの操作においても非常に有用な薬理学的作用剤である。GnRHアゴニスト及びアンタゴニストの多くはおよそ9又は10アミノ酸からなるペプチド分子で、一般的にレセプター結合親和性、レセプター活性を修飾するため、及びタンパク分解を減らすために天然でないアミノ酸を含む。
【0003】
GnRH類似体は、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維腫、月経前症候群、多嚢胞性卵巣症候群、多毛症、尋常性ざ瘡、性早熟症、急性間欠性ポルフィリン症、睾丸停留、思春期遅発症及び不妊治療の処置を含む臨床応用の範囲がある(Millar 2003)。
加えて、GnRH類似体は効果的な避妊薬になりうる。GnRHアンタゴニストはLHサージ期に服用すると排卵を抑制するように働く;しかしながら、用量のタイミングは重要であり、わずかな遅れ(時間)は如何なる効果をも十分無効にする2,4,6,7。あるいは、黄体期にGnRHアンタゴニスト処置を行うことによって黄体機能を抑制して初期妊娠の進行を抑制することができる。また、GnRHアゴニストは、より低い用量を必要とするレセプター脱感作によってではない性腺刺激ホルモンの放出を抑制することができる。性腺刺激ホルモンの放出を抑制するために、GnRH−レセプター(GnRH−R)で必要とされるレセプター占有率の高さに応じてGnRHアゴニストに比例してアンタゴニストを増やす必要がある。
【0004】
現在、女性ホルモン的な避妊は、性腺刺激ホルモンの分泌を抑制するために生理学的用量を超えてステロイドホルモン類似体が用いられている。周辺組織が同じレベルに曝されるので、結果としてさまざまな副作用が生じうる1。男性ホルモン的な避妊の開発は男性ホルモン置換と組み合わせた同様な原理に基づいているが、同じように副作用の問題に直面している。ゆえに、GnRHアンタゴニストは性腺ステロイドホルモン置換と組み合わせた男性及び女性の避妊の基本となる可能性がある1,3,7,12,18。
長期的GnRH類似体処置に関する主たる問題の一つは、性腺性ステロイドホルモンの減少である。したがって、ホルモン置換療法は女性の低エストロゲン性骨減少等の副作用の予防や男性の二次性徴の維持が必要である。
GnRH類似体処置に関する付加的問題は、経口投与したGnRH類似体の消化管での急速な分解である。更に、GnRH類似体は循環において比較的半減期が短く、しばしば初めの循環で腎臓を介して排出される(1〜7分、t1/2)。これらの問題により、GnRH類似体のインビボ有効濃度を維持するために徐放注射用貯蔵調製物が開発された。
【0005】
現在、GnRHペプチドアンタゴニストは注射により投与されている。非ペプチドアンタゴニスト及び経口活性型GnRHアンタゴニストの開発が試みられている。消化管内に活性的に取り込まれるビタミンB12などのハプテンとのGnRH類似体結合体はペプチドアンタゴニストに経口的活性を与える可能性がある15。GnRHアンタゴニストは既に修飾して付加的機能的部分を含んでいる。例えば、エモジン成分22とのGnRH結合体又は潜在的経口摂取15,20,21を亢進させるためのアンチドとのビタミンB12結合体が報告されている。ビタミンB12結合体の経口投与によりいくらか摂取の増加が見られるが21、これら成分の半減期が長くなっていないことが証明された。
多くのホルモンは循環内で血漿タンパク質と結合している。これは、腎クリアランス及び代謝分解からの保護による循環半減期の延長19など、多種の機能を司っていると考えられている。
【0006】
ヒト及び旧世界霊長類には二つの主な循環ステロイド結合タンパク質があり、それはコルチゾールとプロゲステロンの結合であるコルチゾール結合グロブリン(CBG)、及びテストステロンとエストラジオールの結合である性ホルモン結合グロブリン(SHBG)8である。また、モルモットなどのヤマアラシ類のげっ歯種はプロゲステロンに特異的に結合するプロゲステロン結合グロブリン(PBG)を持つ。
高分子量の血漿タンパク質に結合しているステロイドは腎及び代謝によりクリアランスされず、加えて細胞内への侵入が阻害されて核レセプターと相互作用する。ゆえに、循環内のステロイド有効濃度は、結合比率が平衡状態にあるときの非結合比率(ヒトではおよそ2%)により決定する16,19。
そこで我々は、ホルモン成分又はホルモン誘導体にGnRH類似体を結合させると血漿半減期が延長し、GnRH類似体が薬物動態学的にも薬力学的にも改善されることを示した。
【0007】
仮説にしばられることを望むものではないが、我々は、結合体化合物のホルモン又はホルモン誘導体成分はGnRH類似体の貯蔵として働く血漿ホルモン結合タンパク質に結合して、ゆっくりと持続してGnRH類似体を放出すると考える。血漿タンパク質への結合を介しての結合体の隔離は排泄及び不活性型への代謝から薬物を「保護」し、それによりGnRH類似体の半減期を延長するであろう。
我々はGnRH類似体−ホルモン結合体がそれ自身の半減期及び活性持続時間を延長し、生物学的効果に必要なGnRH類似体の用量を減らすことを示した。また、アンタゴニストが必要とされる前の重要な時期に結合体を投与することができたり、GnRH類似体の投与頻度及び用量を低くすることによって治療の副作用を潜在的に少なくすることができる。
【0008】
さらに、結合体はGnRH類似体と機能的ステロイド性ホルモンを単一分子に結合させたものであるため、ステロイド性ホルモンに結合したGnRH類似体での治療はホルモン置換療法の必要性を減らす又は軽減するであろう。
本発明の第一態様では、ホルモン成分又はその誘導体に結合したGnRH類似体を含んでなるものであり、血漿ホルモン結合タンパク質に結合可能な化合物を提供する。
GnRHは、デカペプチドpGlu−His−Trp−Ser−Tyr−Gly−Leu−Arg−Pro−Gly−NH2を意味し、pGluはピログルタミン酸である。
「GnRH類似体」はGnRHレセプターに結合するペプチド又は非ペプチド分子何れの分子をも含む。GnRHレセプターに対する分子の結合は当業者によって、例えば、レセプター結合アッセイ又は後述の実施例1などの全細胞結合アッセイを用いて迅速に決定することができる。
【0009】
一般的に、ペプチドGnRH類似体は6〜12のアミノ酸残基を持つペプチドである。より好ましくは、ペプチドGnRH類似体は7、8、9、10又は11のアミノ酸残基を持つ。さらにより好ましくは、ペプチドGnRH類似体は9又は、最も一般的には10アミノ酸残基を持つ。
一般的に、ペプチドGnRH類似体は少なくとも一つの修飾、すなわち非天然に生じたアミノ酸残基を含む。GnRH類似体は通常天然のGnRHデカペプチド構造の第6及び第10番目のアミノ酸を修飾して生成するのに対して、第1、2、3、5、6、8及び10番目の変更は通常は結果として拮抗作用が生じる(Thau 1984)。
Millar(2003)は、本発明の使用に適するかもしれないGnRH類似体だけでなくGnRH及びそのレセプターの構造について検討している。出典明記によりGnRH及びGnRH類似体に関するMillar(2003)の開示の全体をここに組み込む。
【0010】
一実施態様では、GnRH類似体はGnRHアンタゴニストである。一般的にGnRHアンタゴニストは、結合してGnRHレセプター(GnRH−R)活性又はシグナル伝達を遮断するようにGnRHの構造を修飾したペプチド分子である。
「GnRHアンタゴニスト」は、GnRHレセプターのシグナル伝達を阻害、減少又は防止するペプチド又は非ペプチドの如何なるGnRH類似体の意味をも含む。GnRH−Rシグナル伝達の阻害、減少又は防止は当業者によって、例えば、後述の実施例1などのイノシトールリン酸産生アッセイを用いて迅速に決定することができる。
Millar等(2000)は、本発明の使用に適するかもしれないGnRHアンタゴニストについて検討している。出典明記によりGnRHアンタゴニストに関するMillar等(2000)の開示の全体をここに組み込む。
【0011】
特定の位置に対してアミノ酸を明記していない場合、同じ位置に天然に生じたGnRHと同じアミノ酸残基が存在することを示す。
非天然に生じたアミノ酸に対して以下のような略語を用いる:AcD-Nal−アシルD-ナフチルアラニン;D-Cpa−D-クロロフェニルアラニン;D-Pal−D-ピリジルアラニン;D-Lys−D-リジン;D-Ala−D-アラニン;Ac-ΔPro−アシルΔ-プロリン;D-Fpa−D-フルオロフェニルアラニン;D-Trp−D-トリプトファン;Lys(Nic)−リジンニコチンアミド;及びiPr-Lys−イソプロピルリジン。
最も好ましいGnRHアンタゴニストは、セトロレリクス(Asta Medica AG)、ガニレリクス(Organon)、アバレリクス(Praecis Pharmaceuticals)、アンチド(Ares Serono SA)、テベレリクス(Ardana)、FE200486(Ferring)及びNal−Glu(NIH)である。これらGnRHアンタゴニストの構造を図8に示す。
【0012】
他の好適なGnRHアンタゴニストには、A−75998、A−76154及びA−84861(Abbott Laboratoriesにより最初に製造);D−26344及びD−63153(ASTA Medica AGにより最初に製造);ラモレリクス(Aventis AGにより最初に製造);デガレリクス(Ferring Research Institute (UK)により最初に製造)、NBI−42902(Neurocrine Biosciences Incにより最初に製造);Org−30850(Organonにより最初に製造)、デチレリクス(Roche Bioscienceにより最初に製造);イツレリクス(Serono SAにより最初に製造);TAK013及びTAK810(Takeda Chemical Industries Ltdにより最初に製造);AN 207(Tulane Universityにより最初に製造);Pfizer GnRHアンタゴニスト;Merck GnRHアンタゴニスト;及びWeizmann GnRHアンタゴニストが含まれる。また、以下も参照のこと:Goulet (1995) Ann. Reports Med. Chem. 30, 169-178;Nestor & Vickery (1988) Ann. Reports Med. Chem 23, 211-220;及びDutta & Barrington (1985) Ann. Reports Med. Chem. 20, 203-214これらはすべて出典明記によりここに組み込まれる。
他の好適なペプチドGnRHアンタゴニストには、
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
Ac−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;
D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;
[D−Pyr1, D−Phe2, D−Trp3,6]GnRH (Rahimipour等参照);
D−Lys6アンチド;Lys5アンチド;及びLys8アンチド
が含まれる。
【0013】
アンチド及びその誘導体は、Russel-Jones等(1995)及びWO94/28015(Biotech Australia Pty. Ltd)に記載されている。GnRHアンタゴニスト、GnRH類似体結合体化合物及びそれらの構造に関するRusselJones等及びWO94/28015の開示は出典明記によりその全体がここに含まれる。
本発明の使用に適するかもしれない非ペプチドGnRHアンタゴニストは、WO95/28405;WO96/24597;WO97/41126;WO99/33831;WO00/00493;WO00/56739及びWO01/78780 (Takeda Chemical Industries, Ltd)及びWO02/02533(Yamanouchi Pharmaceutical Co., Ltd)に記載されている。GnRHアンタゴニスト、その構造及び使用に関するこれら公報の開示は出典明記によりその全体がここに含まれる。
本発明に有用かもしれない更なるGnRH類似体には、米国特許第3,813,382;3,843,065;3,849,389;3,855,199;3,886,135;3,890,437;3,892,723;3,896,104;3,901,872;3,914,412;3,915,947,3,929,759;3,937,695;3,953,416;3,974,135;4,010,125,4,018,914、4,022,759;4,022,760;4,022,761;4,024,248;4,034,082;4,072,668;4,075,189;4,075,192;4,086,219;4,101,538;4,124,577;4,124,578、4,143,133;4,234,571、4,253,997;4,292,313;及び4,341,767号に記載のものが含まれる。GnRH類似体、その構造及び使用に関するこれら米国特許文献の開示は出典明記によりその全体がここに含まれる。
【0014】
本発明に有用かもしれないより更なるGnRH類似体には、米国特許4,504,414;4,677,193;4,705,778;5,064,939;5,371,070 5,413,990;5,502,035;5,633,248;5,756,497;6,156,731及び6,191,115;EP 0 081 877及びEP 0 192 492;以下の公開されているPCT出願WO93/03058、WO95/04541、WO95/28405、WO97/44321、WO97/44339、WO97/44041、W098/03632、WO98/55505、WO99/21557、WO99/41251、WO99/41252、WO99/46283及びWO00/53178;及びGB 2 310 660に記載のものが含まれる。アンタゴニスト及びアゴニストの両方、それらの構造及び使用を含むGnRH類似体に関するこれら公開の開示は出典明記によりその全体がここに含まれる。
一実施態様では、GnRH類似体はGnRHアゴニストである。
「GnRHアゴニスト」には、GnRHレセプターのシグナル伝達を刺激又は活性化するペプチド又は非ペプチドの如何なるGnRH類似体の意味をも含む。GnRHレセプターシグナル伝達の刺激又は活性化は、当業者によって、例えば実施例1に記載などのイノシトールリン酸産生アッセイを用いて迅速に決定することができる。
【0015】
特に6位でのDイソ型アミノ酸の取り込みは、GnRH類似体のアゴニストとしての可能性を高める。Rahimpour等(2001)は、3000以上のGnRH類似体を合成してその生物学的活性についての評価を報告している。上記のアゴニストのほとんどは、6位のGlyの代わりにD−アミノ酸を取り込み、多くはGly−NH2末端の代わりにN−エチルアミドを持つ。これら化学的修飾は、Gly−Leu結合でのGnRHの生物学的活性なβターン高次構造を亢進して、タンパク質分解に対するペプチドの感受性を低下させると報告されている。ゆえに、本発明の使用に好適なGnRHアゴニストには、これら修飾の何れか又は両方を有するGnRH類似体が含まれる。
一実施態様では、GnRH類似体の少なくとも一アミノ酸残基は、D−リシンである。一般的に、D−リシンは、類似体の6位にある;すなわち、GnRH類似体は[D−Lys6]GnRHである。
【0016】
最も好ましいGnRHは、ロイプロン(TAP)、ゾラデックス(Zeneca)、サプレリン(Roberts)、シナレル(Searle)、トリプトレリン(Ferring)、及びブセレリン(Hoechst)であり、各々は6位に非天然に生じる残基を持つ。GnRHアゴニストの構造を図8に示す。
他の好適なGnRHアゴニストには、デスロレリン(Balance Pharmaceuticals)、ProMaxx-100 (Epic Therapeutics);アボレリン(Mediolanum Farmaceutici SpA)、ヒストレリン(Ortho Pharmaceuticals)、及びナファレリン(Roche Bioscience)を含む。
【0017】
ペプチドGnRH類似体は当業者に知られた任意の方法で製造することができる。例えば、ペプチドは、Lu等(1981) J. Org. Chem. 46, 3433及びそこに記載の文献に開示されたような、固相ペプチド合成のFmoc-ポリアミド法によって合成することができる。一時的なN-アミノ基の保護は9-フルオレニルメチルオキシカルボニル(Fmoc)基によって得られる。この高度に塩基不安定保護基の繰り返しの開裂はN,N-ジメチルホルムアミド中20%ピペリジンを使用してなされる。側鎖官能基はそのブチルエステル(グルタミン酸及びアスパラギン酸の場合)、ブチルオキシカルボニル誘導体(リジン及びヒスチジンの場合)、トリチル誘導体(システインの場合)及び4-メトキシ-2,3,6-トリメチルベンゼンスルホニル誘導体(アルギニンの場合)として保護できる。グルタミン又はアスパラギンがC末端残基である場合、側鎖アミド官能基の保護には4,4'-ジメトキシベンズヒドリル基が使用される。固相支持体は、ジメチルアクリルアミド(骨格モノマー)、ビスアクリロイルエチレンジアミン(架橋剤)及びアクリロイルサルコシンメチルエステル(官能化剤)の三種のモノマーから構成されたポリジメチルアクリルアミドポリマーをベースとしている。使用されるペプチド対樹脂開裂架橋剤は酸不安定4-ヒドロキシメチル-フェノキシ酢酸誘導体である。全てのアミノ酸誘導体は、逆のN,N-ジシクロヘキシル-カルボジイミド/1-ヒドロキシベンゾトリアゾール媒介カップリング法を使用して添加されるアスパラギンとグルタミンを除いて、その予め形成された対称な無水物誘導体として添加される。全てのカップリング及び脱保護反応はニンヒドリン(ninhydrin)、トリニトロベンゼンスルホン酸又はイソチン(isotin)試験方法を使用してモニターされる。合成の完了時点で、ペプチドは50%のスカベンジャー混合物を含む95%トリフルオロ酢酸での処理によって側鎖保護基の同時の除去と共に樹脂支持体から開裂される。一般的に使用されるスカベンジャーはエタンジチオール、フェノール、アニソール及び水であり、正確な選択は合成されるペプチドの構成アミノ酸に依存する。トリフルオロ酢酸は真空蒸発によって除去され、続くジエチルエーテルでの倍散により粗ペプチドが得られる。存在する全てのスカベンジャーは、水相の凍結乾燥によりスカベンジャーのない粗ペプチドを生じる単純な抽出法によって除去される。ペプチド合成のための試薬は一般にCalbiochem-Novabiochem (UK)社, Nottingham NG7 2QJ, UKから入手できる。精製は、サイズ排除クロマトグラフィー、イオン交換クロマトグラフィー及び(主として)逆相高速液体クロマトグラフィーのような技術の何れか一つ又はその組み合わせによってなすことができる。ペプチドの分析は薄層クロマトグラフィー、逆相高速液体クロマトグラフィー、酸加水分解後のアミノ酸分析及び高速原子衝撃(FAB)質量分析法を使用して実施することができる。
【0018】
別法として、ペプチドGnRH類似体は、それらがDNA分子中でコード化されうるならば、標準的な分子生物学技術によって得ることができる。
本発明での使用に適したペプチド及び非ペプチドGnRH類似体はホルモン部分又はその誘導体への結合に適した原子又は官能基を有しているものである。
天然に生じるアミノ酸残基上に存在する好適な官能基には、Cys残基上のスルフヒドリル基、Ser、Thr又はTyr残基上のヒドロキシル基、Lys残基上のε-アミノ基、Asp及びGlu残基上のカルボキシル基、Arg残基上のグアニジノ基、Asn及びGln残基上のアミド基、His残基のイミダゾールNH基、Trp残基のインドールNH基、C末端カルボキシル基及びN末端アミノ基が含まれる。同じ官能基はこれらのアミノ酸残基のD-アイソフォーム上に存在する。
【0019】
修飾されたアミノ酸はまたホルモン部分への結合に好適な官能基を有しうる。これらには、ヒドロキシプロリン残基上のヒドロキシル基、O-ホスホセリン又はO-ホスホチロシン残基上のホスフェート基、γ-カルボキシグルタメート残基上の双方のカルボキシル基、iPr-Lys残基上のε-アミノ基及びシトルリン及びホモシトルリンの基が含まれる。
更に、ピリジルアラニン残基のピリジルN-原子及びN-アルキル化Arg残基の三級グアニジノN-原子がN-4級化によってホルモン部分に結合させるための好適な官能基である。再び、D-及びL-アイソフォームがGnRH類似体に存在しうる。
非ペプチドGnRH類似体上に存在するホルモン部分への結合のための好適な官能基にはケト、NH(アミノ、アミド又はウレイジル官能基の一部として)、ヒドロキシル、スルフヒドリル、カルボン酸及び三級アミノ基が含まれる。
【0020】
典型的には、GnRH類似体へ結合されるホルモン部分又はその誘導体は血漿ホルモン結合タンパク質にインビボで結合するものである。
典型的には、ホルモン結合タンパク質はグロブリンである。
一実施態様では、ホルモン部分又は誘導体は特異的血漿ホルモン結合タンパク質、例えばコルチゾール結合グロブリン(CBG)、性ホルモン結合グロブリン(SHBG)と、ある場合にはプロゲステロン結合グロブリン(PBG)に結合する。典型的には、ホルモン部分又はその誘導体はまた血清アルブミン(HSA)に結合する。疑義を避けるために、本発明においては、HSAは血漿ホルモン結合タンパク質である。
【0021】
ホルモン部分の「誘導体」には、我々は、誘導体が天然に見出されるホルモン部分の構造から改変されているという意味を含める。例えば、改変されて、GnRH類似体への新たな又は改良された結合部位を提供するか、又はその安定性又はその活性を改善する場合がある。しかし、ここで定義されるホルモン誘導体は血漿ホルモン結合タンパク質に結合するその能力を完全には失っていない。誘導体自体はホルモン活性を有しているか又は有していないことが理解される。
好適な実施態様では、ホルモン部分はステロイドホルモン部分である。
本発明での使用に適したステロイドホルモン部分及びその誘導体はGnRH類似体への結合のために適した原子又は官能基を有するものである。
【0022】
典型的には、ステロイドホルモンは3位にヒドロキシル基か又はケト基を有している。ステロイドホルモンの多くは17位にヒドロキシル基か又はケト基を有している。多くのステロイドホルモンは11位にヒドロキシル基を有している。幾つかのステロイドホルモンは21位にヒドロキシル基を有している。
好ましくは、ステロイドホルモン部分は、エストラジオール、プロゲステロン、コルチゾール、コルチコステロン、エストロン、テストステロン及びジヒドロキシテストステロン(DHT)である。
ステロイドホルモン誘導体には11、17又は21位にヒドロキシル基を付加することによって修飾されたものが含まれる。好適なプロゲステロン誘導体には11α-ヒドロキシプロゲステロン及び21-ヒドロキシプロゲステロンが含まれる。
ステロイドであるがもはやホルモン活性を有していないステロイドホルモン誘導体は、それらが血漿ホルモン結合タンパク質に結合する場合は使用することができることが理解される。
【0023】
血漿ホルモン結合タンパク質にステロイドホルモンが結合するのに必要とされる官能基は当業者に知られている。例えば、置換ステロイドを用いる構造調査において、SHBGと相互作用するためには、ステロイドは17β-ヒドロキシル基を含んでいなければならないことが証明されている(Burton及びWestphal (1972)及びCunningham等(1981))。幾つかの他の特徴、例えばC11でのヒドロキシル又はケト基の付加は結合親和性にマイナスの影響を及ぼす。ステロイド核中の炭素2、6、9及び11の修飾がまた結合親和性を低減させた(Cunningham等(1981))。
ヒトCBGへのステロイド結合に対しては、20-オキソ及び10β-メチル基が本質的であると報告されており、3-オキソ及び4ーエンがまた重要である。11β、17α-、及び21-ヒドロキシ基は比較的重要ではなく、ヒドロキシル基は11α、6α、6β、12α、14α、16α及び19位の結合を損なう(Mickelson等 1981)。
【0024】
しかして、血漿ホルモン結合タンパク質に結合するホルモン部分の能力を保持するようにGnRH類似体の特定の官能基にホルモン部分又はその誘導体を結合させるのは当業者の技量の十分な範囲内である。
リンカーは血漿ホルモン結合タンパク質とのホルモン又はGnRHレセプターとのGnRH類似体の相互作用を立体的に阻害しないのが好ましい。嵩がある、及び/又は親水性の基がホルモン部分又はGnRH類似体に近接したリンカー中に存在していないことが好ましい。
【0025】
本発明の幾つかの好適な実施態様では、ホルモン部分又はその誘導体は、GnRH類似体に結合した場合、全体的又は部分的にそのステロイド活性を保持している。あるいは、他の実施態様では、ホルモン部分又はその誘導体は、GnRH類似体に結合した場合、ステロイド活性は保持していないのが好ましい。
ホルモン活性が望まれる実施態様では、GnRH類似体への結合に使用される官能基は典型的にはその特定のホルモン又はホルモン誘導体の活性に必要とされないものである。逆に、ホルモン活性が望まれない場合は、GnRH類似体への結合に使用される官能基は典型的にはその特定のホルモン又はその誘導体の活性に必要とされるものである。
【0026】
ステロイドホルモンの活性に必要な官能基は当業者に知られている(例えば、ステロイドホルモン-レセプターの構造関係は、Duax等(1989) Advances in Drug Research 18,115-138;Aranyi (1982) Hung. Biologia (Budapest) 30,145-169;Raynaud及びOjasoo (1983) Nobel Symposium 57,141-170;Duax及びGriffin (1989) Alfred Benzon Symposium 28,62-77;Ojasoo等 (1992) Mol. Struct. Biol. Act. Steroids, pp 157-207, CRC, Boca Raton;及びDuax & Griffin (1998) NATO ASI series E : Applied Sciences 352,1-14にエストロゲン、グルココルチコイド、鉱質コルチコイド、アンドロゲン、及び代謝類似体及びアンタゴニストに対して記述されており、その全てが出典明示によりここに取り込まれる。従って、ホルモンの活性を保持し又は除去するようにGnRH類似体の特定の官能基にホルモン部分又はその誘導体を結合させるのは当業者の技量の範囲内にある。
【0027】
例えば、実施例1に示すように、21-ヒドロキシプロゲステロンの21位へのGnRH類似体の結合は結合化合物中でプロゲステロン活性を維持する。逆に、ステロイドホルモン活性が除去される場合には、GnRHは3位のケト基に結合させることができる。
「結合させる」という用語には、共有結合がGnRH類似体の原子とホルモン部分の原子の間に形成されている意味、又はGnRH類似体とホルモン部分が両方とも同じ結合基に共有結合的に結合している意味を含む。
好適な実施態様では、GnRH類似体とホルモン部分の間の結合は開裂可能であり、例えば結合には、エステラーゼにより開裂可能であるエステル結合又はアミダーゼにより開裂可能であるアミド結合が含まれる。
【0028】
一実施態様では、GnRH類似体及びホルモン部分は直接結合している。典型的にはこの場合、アミノ酸は既に結合しているホルモン部分と共に合成され、この修飾されたアミノ酸はペプチドGnRH類似体中に導入される。例えば、ホルモンのケト基とLysのε-アミノ基の間のイミンの形成を通して直接結合が生じうる。生じるイミンはインビボで加水分解的に不安定であり、よって短い半減期のコンジュゲートを生じる。半減期はイミンを還元してアミンを形成することにより増加させることができる。あるいは、直接結合は、ホルモン部分のα,β-不飽和ケトン官能基へのLys上のε-アミノ基のマイケル付加を通して(例えばプロゲステロン)、又はGnRH類似体の残基上の適切な求核基(例えばLys上のε-アミノ基又はSer上のβ-OH基)でOH基が離脱基(例えばハロゲン化物又はスルホン酸エステル)に転換されたホルモン部分の反応を通して生じうる
「結合基」はGnRH類似体にもホルモン部分にも内因的ではない一又は複数の原子によって形成された構造を意味する。
【0029】
結合基は一又は複数の原子を含み、GnRH類似体とホルモン部分又はその誘導体の間の最短経路は典型的には2又は3又は4又は5又は6又は7又は8又は9又は10又はそれ以上の結合を含む。
結合基は、場合によっては酸素、窒素及び/又は硫黄のような他の原子と共に、一又は複数の炭素原子をベースとした構造を含み得、GnRH類似体とホルモン部分又はその誘導体の間の最短経路は、場合によっては少なくとも一の酸素、窒素及び/又は硫黄原子と共に、典型的には2又は3又は4又は5又は6又は7又は8又は9又は10又はそれ以上の炭素原子を含む。
典型的には、結合基は、同時に又は任意の連続的な順序で、二官能性前駆体結合部分をGnRH類似体及びホルモン部分に反応させることによって導入される。あるいは、結合基はGnRH類似体及び/又はホルモン部分の誘導体化によって導入される構造的断片上に官能基をカップリングさせることによって形成される。
【0030】
好適な結合基には、式-A1-D-A2-によって表されるものが含まれ、ここで、Dは、例えば、
(a)アルキレン、アルケニレン又はアルキニレン(後者の3種の基は場合によってはNH、O又はS、炭素環又は複素環によって中断され及び/又は終端され、及び/又は場合によっては-S-S-によって中断されている)、
(b)炭素環、又は
(c)複素環
を表すか、
又はDは構造断片-D1-A3-D2-を表し、ここで、D1及びD2は独立して上の(a)から(c)に定義されたDを表し;
A1、A2及びA3は独立して例えば直接結合、-C(O)-、-C(S)-、-S(O)-、-S(O)2-又は-P(O)2-を表し、但し、A3は、D'及びD2が共に独立してA3への連結点でO又はNHによって終端する場合、直接結合を表さない。A1及びA2基はGnRH類似体又はホルモン部分に内因性の原子、例えばGnRH類似体又はホルモン部分のLys残基のN原子及びホルモン部分のヒドロキシル基のO原子に結合していることは明らかである。
【0031】
結合基はGnRH類似体及びホルモン部分に何れかの配向で結合されうる(つまり、結合基の基A1はGnRH類似体の原子かホルモン部分の原子の何れかに結合されうる)。
ここで定義されるアルキレン、アルケニレン又はアルキニレン基は1から12(例えば1から8、例えば1から6)のC原子を含み得、直鎖であり得、又は十分な数(つまり、アルキレン及びアルケニレンでは最小2つ、アルキニレンでは4)の炭素原子がある場合は、分枝状鎖であってもよい。
ここで使用される場合、「炭素環」という用語には、炭素骨格を有する環状構造であり、3又はそれ以上の数の一又は複数の環を含む基、例えば3から8員の単環環系、7から12員の二環環系及び12から18員の多環(例えば三又は四環)環系が含まれる。更に、各炭素環は完全に飽和しているか、部分的に飽和しているか、又は完全に又は部分的に芳香性でありうる。完全に飽和した炭素環の例にはシクロペンチル、シクロヘキシル及びシス-及びトランス-デカリニル等が含まれる。部分的に不飽和の炭素環の例にはシクロヘキセニル等が含まれる。部分的に芳香性の炭素環の例には、インデニル及び1,2,3,4-テトラヒドロナフチル等々が含まれる。完全に芳香性の炭素環の例にはフェニル、ナフチル等々が含まれる。
【0032】
ここで使用される場合、「複素環」という用語には、上述の炭素環基であり、環C原子の一又は複数(例えば1から3)が対応する数のヘテロ原子によって置換され、各ヘテロ原子が独立してO、S及びN(又は関連する場合はNH)から選択される基が含まれる。複素環基の例には、アゼチジニル、ベンゾジオキサニル、ベンゾジオキセパニル、ベンゾジオキソリル、ベンゾフラニル、ベンゾフラザニル、ベンズイミダゾリル、ベンゾモルホリニル、ベンゾチアゾリル、ベンゾチオフェニル、ベンゾキサゾリル、クロマニル、シンノリニル、クマリニル、ジオキサニル、フラニル、ヒダントイニル、イミダゾリル、イミダゾ[1,2-a]ピリジニル、インドリル、イソキノリニル、イソキサゾリル、マレイミド、モルホリニル、オキサゾリル、フタラジニル、ピペラジニル、ピペリジニル、プリニル、ピラニル、ピラジニル、ピラゾリル、ピリジニル、ピリミンジニル、ピロリジノニル、ピロリジニル、ピロリニル、ピロリル、キナゾリニル、キノリニル、3-スルホレニル、テトラヒドロピラニル、テトラヒドロフラニル、チアゾリル、チエニル、チオクロマニル、トリアゾリル等が含まれる。
【0033】
挙げることができる本発明の実施態様には、結合基が式-A1-D-A2-によって表されるものが含まれ、ここで、
DはC1−6アルキレン、C2−6アルケニレン又は-D1-A3-D2-を表し;
D1は-S-S-によって中断されていてもよいC2−6アルキレンを表し;
D2はC2−8アルキレンを表し;
A1及びA2は共にC(O)を表し;
A3はC(O)NHを表す。
挙げることができる特定の結合基には、C(O)-(CH2)2-C(O)(スクシニル)及びC(O)-(CH2)2-S-S-(CH2)2-C(O)NH-(CH2)6-NHC(O)が含まれる。
挙げることができる本発明の実施態様には、結合基がペプチドGnRH類似体中のLys残基のε-アミノ基をホルモン部分上のヒドロキシル基に連結するものが含まれる。
【0034】
GnRH類似体をホルモン部分又はその誘導体に結合させるために適した方法及び化学は当業者に知られており、実施例1に記載された方法が含まれる。リンカーを介して他の化学構造にGnRH類似体を結合させる方法はRahimipour等(2001)及びRussell Jones等(1995)に記載されている。本発明のコンジュゲートを形成する際に応用できるビタミンB12コンジュゲートを製造する方法はMcEwan等(1999)に記載されている。化学コンジュゲートの形成に関連しているこれらの3件の文献の開示全体は出典明示によりここに取り込まれる。
GnRH類似体は、例えばO'Sullivan等 Anal. Biochem. (1979) 100, 100-108に一般的に記載されたもののような、常套的な分子の架橋方法の何れかによってホルモン部分又はその誘導体に結合させることができる。例えば、一部分はチオール基に富んだものとされ得、他の部分はチオール基と反応可能な二官能性剤、例えばヨード酢酸のN-ヒドロキシスクシンイミドエステル(NHIA)又はN-ヒドロキシスクシンイミジル-3-(2-ピリジルジチオ)プロピオネート(SPDP)、結合種間にジスルフィド架橋を導入するヘテロ二官能性剤と反応させることができる。例えばm-マレイミドベンゾイル-N-ヒドロキシスクシンイミドエステルで達成されるアミド及びチオエーテル結合は、ジスルフィド結合よりもインビボで一般により安定である。
【0035】
更に有用な架橋剤には、第1級アミノ基を含む化合物に保護されたチオール官能基を導入するために使用される試薬であるS-アセチルチオグリコール酸N-ヒドロキシスクシンイミドエステル(SATA)が含まれる。アセチル化チオール基の脱保護は穏やかな条件で(Julian等(1983) Anal. Biochem. 132, 68)、つまりジメチルスベリミデート(dimethylsuberimidate)二塩酸塩及びN,N'-o-フェニレンジマレイミドとの反応によって達成される。
他の実施態様では、GnRH類似体はN末端アミン基を介してホルモン部分に結合させられる。
有利には、ここに記載された化合物は天然GnRHよりもインビボでの代謝又は腎臓クリアランスによる影響をあまり受けない、つまりインビボで長い半減期を有する。これは、以下の実施例1に記載されているように、当業者によって容易に決定することができる。
好ましくは、ここに記載の化合物はインビボにおいて天然GnRHよりも長い活性期間を有している。これはまた、例えば以下の実施例1に記載されているようにして、当業者によって容易に決定することができる。
好適な実施態様では、化合物は図1A又は図1Bに示される一般式を持ちうる。
【0036】
本発明には次の化合物が含まれる:
D-Lysの6位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Leu-Arg-Pro-D-Ala-NH2;
D-Lysの6位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAc-ΔPro-D-Fpa-D-Trp-Ser-Tyr-D-Lys-Leu-Arg-Pro-Gly-NH2;
Lysの7位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Lys-Leu-Arg-D-Ala-NH2;
N末端アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したD-Pal-Ser-Arg-D-Lys-Leu-Arg-Pro-D-Ala-NH2;
Lysの7位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Lys-Arg-Pro-D-Ala-NH2;
Lysの6位のε-アミン基で11α-ヒドロキシプロゲステロン11-スクシネートに結合した[DLys6]GnRH;
Lysの6位のε-アミン基で21α-ヒドロキシプロゲステロン21-スクシネートに結合した[DLys6]GnRH;
Lysの6位のε-アミン基でβ-エストラジオール17-スクシネートに結合した[DLys6]GnRH。
【0037】
更なる実施態様では、化合物は血漿ホルモン結合タンパク質に結合したホルモン部分又はその誘導体に結合したGnRH類似体を含む。結合タンパク質はCBG又はSHBGのようなホルモン特異的結合タンパク質であり得、又はHSAであり得る。
この実施態様に係る化合物は、投与時に直ぐに結合タンパク質の保護効果から恩恵を受け、第一回の通過で腎臓を経由するGnRH類似体の分泌を減少させ、よってGnRH類似体の半減期及び活性を更に延ばすために、有利である。
【0038】
本発明の第二の側面では、本発明の第一の側面に係る化合物と薬学的に許容可能な賦形剤、担体又は希釈剤を含有する薬学的組成物を提供する。
一実施態様では、薬学的組成物は経口投与に適している。
本発明の化合物は、無毒性の有機もしくは無機の酸もしくは塩基の付加塩の形態であってもよい、有効成分を含む薬学的製剤の形で、経口的又は任意の非経口経路により通常投与することができる。治療される疾患及び患者並びに投与経路に応じて、組成物を様々な用量で投与することができる。
ヒトの治療では、本発明の化合物は、単独で投与することができるが、一般には、目的とする投与経路及び標準的医薬規範を考慮して選択した適切な薬学的賦形剤、希釈剤又は担体と混合されて投与される。
【0039】
例えば、本発明の化合物は、即時、遅延又は徐放用途に対して、香味添加剤又は着色剤を含みうる錠剤、カプセル剤、卵剤、エリキシル剤、溶液又は懸濁液の形態で経口的、口腔内、又は舌下に投与することができる。また、本発明の化合物は、鼻腔内注入により投与してもよい。
そのような錠剤は賦形剤、例えばマイクロクリスタリンセルロース、ラクトース、クエン酸ナトリウム、炭酸カルシウム、二塩基性リン酸カルシウム及びグリシン、崩壊剤、例えばデンプン(好ましくはコーン、ポテト又はタピオカデンプン)、デンプングリコール酸ナトリウム、クロスカルメロース(croscarmellose)ナトリウム及びある種のケイ酸錯体、及び顆粒化バインダー、例えばポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ-プロピルセルロース(HPC)、スクロース、ゼラチン及びアカシアを含みうる。加えて、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリル、タルクなどの潤滑剤を含みうる。
同様なタイプの固形組成物はゼラチンカプセル中にフィラーとしても用いることができる。この点で好適な賦形剤には、ラクトース、デンプン、セルロース、ミルク糖又は高分子量ポリエチレングリコールが含まれる。水性懸濁液及び/又はエリキシルに対しては、本発明の化合物は様々な甘味料又は香味添加剤、着色物質又は染料、乳化及び/又は懸濁化剤、及び水、エタノール、プロピレングリコール及びグリセリンのような希釈剤、及びその組み合わせと組み合わせることができる。
【0040】
本発明の化合物は、非経口的に、例えば、静脈内、動脈内、腹腔内、腹腔内、くも膜下腔内、脳室内、胸骨内、頭蓋内、筋肉内又は皮下的に投与することもでき、あるいは点滴法により投与することができる。それらは、他の物質、例えば、溶液を血液と等張性にするのに十分な塩類又はブドウ糖を含んでいてよい無菌水溶液の形で用いるのが最もよい。水溶液は、必要な場合、適切に(好ましくは3〜9のpHとなるように)緩衝されなければならない。無菌条件下での適切な非経口製剤の調製は、当業者によく知られている標準的な製薬技術により容易に達成される。
一実施態様では、薬学的組成物又は製剤は、活性成分、つまり本発明の第一の側面で上述された化合物の毎日の用量又は単位、毎日の下位用量又はその適切なフラクションを含む単位投薬形態である。
他の実施態様では、薬学的組成物又は製剤は、注射可能なデポー剤のような徐放製剤である。
【0041】
非経口投与に適する製剤には、抗酸化剤、緩衝剤、静菌剤及び製剤を対象とする受容者の血液と等張性にする溶質を含みうる水性及び非水性無菌注射溶液;並びに懸濁剤及び増粘剤を含んでいてよい水性及び非水性無菌懸濁液が含まれる。製剤は、単位用量又は複数回用量容器、例えば、密閉アンプル及びバイアル入りで提供することができ、また、使用直前に無菌液体担体、例えば、注射用水の添加のみを必要とする凍結乾燥状態で貯蔵することができる。即時注射溶液及び懸濁液は、前述の製剤の既に記載された無菌散剤、顆粒剤及び錠剤から調製することができる。
現在、GnRHアゴニストは典型的には一日当たり約100μgで患者に投与される一方、GnRHアンタゴニストは典型的には一日当たり約1mgで患者に投与される。ヒトの患者への経口及び非経口投与に対しては、本発明の化合物の毎日の用量レベルは、通常、単一又は分割用量で投与される等価又は低レベルのGnRH類似体を含む。
よって、例えば、本発明の化合物の錠剤又はカプセルは、必要に応じて、一度に一回又は二回又はそれ以上の投与のために50μgから1mgの活性化合物を含みうる。とにかく、医師は各個々の患者に最も適した実際の用量を決定し、特定の患者の年齢、体重及び反応と共に変化する。上記用量は平均的な場合の例である。もちろん、高用量又は低用量範囲が有利な個々の例があり得、それも本発明の範囲内にある。
【0042】
本発明の化合物は、鼻腔内に、又は吸入で投与することもでき、適切な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、1,1,1,2-テトラフルオロエタン(HFA134ATM)又は1,1,1,2,3,3,3-ヘプタフルオロプロパン(HFA227EATM)等のヒドロフルオロアルカン、二酸化炭素又は他の適切なガスを用いた加圧容器、ポンプ、噴霧器もしくはネブライザーから、乾燥粉末吸入剤もしくはエアゾールスプレー提供形態で簡便に送達される。加圧エアゾールの場合には、計量された量を送達するためのバルブを設けることにより用量単位を決定することができる。加圧容器、ポンプ、噴霧器もしくはネブライザーは、潤滑剤、例えばトリオレイン酸ソルビタンをさらに含んでいてよい、例えば、溶媒としてのエタノールと噴射剤との混合物を用いた活性化合物の溶液又は懸濁液を含んでいてよい。吸入器又は注入器に用いるカプセル及びカートリッジ(例えば、ゼラチン製)は、本発明の化合物とラクトース又はデンプンのような適切な粉末基剤との粉末混合物を含むように製剤化することができる。
【0043】
エアゾール又は乾燥粉末製剤は、各計量用量すなわち「一吹き量」が患者に送達される本発明の化合物を少なくとも1mg含むように調製することが好ましい。エアゾールの総1日投与量は患者ごとに異なり、1日1回又は通常、分割して投与することができることは理解できよう。
あるいは、本発明の化合物は、座薬又はペッサリーの形態で投与することができ、あるいはローション、溶液、クリーム、軟膏又は粉剤の形態で局所適用してもよい。本発明の化合物は、例えば、皮膚パッチを用いて経皮投与することもできる。
皮膚に局所投与するために、本発明の化合物は、例えば、一又は複数の以下のもの:鉱油、流動ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックス及び水との混合物に、懸濁又は溶解した活性化合物を含む適切な軟膏剤として製剤することができる。あるいは、例えば、次の一又は複数の以下のもの:鉱油、モノステアリン酸ソルビタン、ポリエチレングリコール、流動パラフィン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2-オクチルドデカノール、ベンジルアルコール及び水との混合物に懸濁又は溶解した適切なローション又はクリームとしても製剤することができる。
【0044】
口中への局所適用に適した製剤には、通常はスクロース及びアカシア又はトラガカントである香味ベースに活性剤を含むトローチ剤;ゼラチン及びグリセリン、又はスクロース及びアカシアのような不活性な基剤中に活性成分を含む芳香錠(pastilles);及び適切な液体担体に活性成分を含む洗口剤が含まれる。
一般に、ヒトにおいて、本発明の化合物の経口又は局所投与が好ましい経路であり、最も簡便である。受容者が嚥下疾患又は経口投与後の薬物吸収障害を被っている状況では、薬剤は非経口的に、例えば舌下的に又は口腔的に投与することができる。
獣医学用途については、本発明の化合物は、通常の獣医学規範に従って適切に許容できる製剤として投与され、獣医は特定の動物に最も適する投与計画と投与経路を決定する。製剤は薬剤的製剤であることが都合がよい。有利には、獣医学用途については、製剤は獣医学的製剤である。
【0045】
本発明の第三の側面は、医薬品における使用のために、本発明の第一の側面による化合物又は本発明の第二の側面による薬学的組成物を提供する。
よって、化合物又は薬学的組成物は医薬品における使用のために包装され提供される。
本発明の第四の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物を個体に投与することを含んでなる、個体の繁殖性を減少させる方法を提供する。
GnRHアゴニスト及びGnRHアンタゴニストコンジュゲートの双方がゴナドトロピンの放出を阻害することによって個体において繁殖性を低減するために使用することができることが理解される。
本発明の第五の側面は、個体の繁殖性を低減させるための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
【0046】
典型的には、また好ましくは、治療される個体はヒトである。しかしながら、本発明の方法は、例えば次の種:ウシ、ウマ、ブタ、ヒツジ、ネコ及びイヌ、並びに他の霊長類、旧世界サル及び新世界サルを治療するために使用することができる。よって、本方法はヒトと獣医用医薬の両方に用途がある。特に、その獣医用途に対しては、コンジュゲートを用いて家畜、ウマ及び家庭内動物における去勢状態をつくり出すことができる。
雌における「繁殖性を低減する」は、受胎の可能性又は成功裏の妊娠を減じ、又は受胎又は成功裏の妊娠を防止するという意味を含む。よって、本発明は雌の避妊方法を含む。
雄における「繁殖性を低減する」は、テストステロンレベルを去勢レベルまで減じるという意味を含む。よって、本発明は雄の避妊方法を含む。
ここに記載された避妊方法は化合物、薬学的組成物又は医薬の投与の休止により可逆である。
【0047】
本発明の第六の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物をそれを必要とする個体に投与することを含んでなる、ホルモン依存性疾患又は症状と闘う方法を提供する。
疾患又は症状と「闘う」には、症状の徴候を軽減し(つまり対症用途)、又は疾患又は症状を治療し、又は疾患又は症状を防止する(つなり予防用途)意味が含まれる。
本発明の第七の側面は、必要とする個体におけるホルモン依存性疾患又は症状と闘うための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
本発明の方法、使用、化合物及び薬学的組成物によって闘うのに適したホルモン依存性疾患又は症状には、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維、月経前症候群、多嚢胞性卵巣症候群、多毛症、ざ瘡尋常性、思春期早発症、急性間欠ポルフィリン症、睾丸停留及び遅延思春期が含まれる。
本発明による治療に適したホルモン依存性癌はGnRHレセプターを発現し、乳癌、前立腺癌、子宮癌、子宮体癌、卵巣癌及び精巣癌を含む。
【0048】
本発明の第八の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物をそれを必要とする個体に投与することを含んでなる、不妊症と闘う方法を提供する。
本発明の第九の側面は、それを必要とする個体における不妊症と闘うための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
本発明の化合物は、特に補助生殖医療における排卵の誘導において、外因性ゴナドトロピンの制御された投与と共に内因性ゴナドトロピンの阻害によって不妊症と闘うために使用することができる。
よって、本発明の化合物は体外受精(IVF)技術において有用性を有している。
【0049】
本発明の第十の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物を個体に投与することを含んでなる、インビボでゴナドトロピン又は性ホルモンの産生を調節する方法を提供する。
「調節」には増大、減少又は阻害を含む。
本発明の第十一の側面は、インビボでゴナドトロピン又は性ホルモンの産生を調節するための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
例えば、受胎治療において、本発明の化合物を用いて完全に内因性のホルモン産生を阻害することができ、これがついでコンジュゲートの一部として又は別個に所望に応じて置き換えられる。受胎治療における使用のためのホルモン置換の量、頻度及び期間はこの分野の当業者にはよく知られており、とにかく医師によって決定される。
本発明のコンジュゲート化合物はまたGnRHレセプターを発現する細胞、例えば幹細胞、及びリンパ球のような免疫細胞の分化又は脱分化のためにインビトロで又はより少ない頻度でインビボで使用することもできる。
【0050】
本発明の第十二の側面は、GnRHと比較して増加したインビボ半減期を持つように、GnRH類似体を修飾する方法を提供し、該方法は、血漿ホルモン結合タンパク質に結合可能なホルモン部分又はその誘導体にGnRH類似体を結合させることを含んでなる。
本発明の第十三の側面は、GnRHと比較して増加したインビボ活性の期間を持つように、GnRH類似体を修飾する方法を提供し、該方法は、血漿ホルモン結合タンパク質に結合可能なホルモン部分又はその誘導体にGnRH類似体を結合させることを含んでなる。
好ましくは、GnRH類似体とホルモン部分又はその誘導体は結合基を介して結合される。
本発明の第十三及び第十四の側面の実施態様では、上記方法は、ホルモン部分に結合させられたGnRH類似体又はその誘導体を血漿ホルモン結合タンパク質に結合させることを含む。典型的には、この結合は、ホルモン部分に結合させられたGnRH類似体又はその誘導体を溶液中の血漿ホルモン結合タンパク質と接触させることによって実施される。
【0051】
GnRH類似体、ホルモン部分又はその誘導体、結合基、コンジュゲーションを実施する方法の好ましい例は実施例1及び本発明の第一側面に関して上に記載した通りである。
一実施態様では、本発明の第十三及び第十四の側面はまた結合されたGnRH類似体のインビボ半減期又は活性の期間を決定する工程を含む。典型的には、該方法は結合されたGnRH類似体の決定されたインビボ半減期又は活性の期間を、GnRHのインビボ半減期又は活性の期間と比較する工程を更に含む。
場合によっては、上記方法はGnRHの活性の期間又はインビボ半減期を定量する工程をまた含む。あるいは、過去に測定された値を使用することができる。
本発明を次の図面と実施例を参照しながら以下により詳細に説明する。
【0052】
図8で用いた略語を以下に示す:
DSer(tBu):D−Ser t−ブチルエーテル;
DHis(ImBzl):D−Hisベンジルイミダゾール;
NEt:N−エチルアミド;
DNal:D−ナフチルアラニン;
DCit:D−シトルリン;
DhCit:D−ホモシトルリン;
DhArg(Et)2:D−ジエチルホモアルギニン;
NMeTyr:N−メチルチロシン;
Aph(Hor):
DAph(Cba):
DGlu(AA):
【0053】
実施例1:GnRHホルモン結合体の合成及び性質
方法
GnRH−ステロイド結合体の合成
Mattox, Litwiller及びNelson9とRajkowski及びCittanova14からの結合方法を用いた。Amersham Pharmacia Biotech UK Limited (Little Chalfont, Buckinghamshire)から購入した放射性化学薬品を除き特に明記しない限りSigma-Aldrich Company Ltd. (Poole, Dorset)より全ての化学薬品を入手した。GnRHアゴニスト[D−Lys6]GnRH又は2つのGnRHアンタゴニストはJ. Rivier (Salk Institute, La Jolla, California)より快く提供していただいた。[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、アンタゴニスト1とする、及び[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、アンタゴニスト2とする、を0.1Mのリン酸緩衝液(pH7.0)に溶解して等量のDMFを加えた。20倍以上のステロイド([D−Lys6]GnRHの場合は11αヒドロキシプロゲステロン11−半コハク酸塩、GnRHアンタゴニスト結合体には21−ヒドロキシプロゲステロン21−半コハク酸塩)を等モルの1−ヒドロキシベンゾトリアゾール(HOBt)及びN,N’−ジシクロヘキシルカルボジイミド(DCC)を含む無水DMFに溶解した。混合液をボルテックスにかけ、1時間室温に放置した。溶液を含むステロイドを各ペプチド溶液50μlの一定分量に移した。トリブチルアミンでpH>7に調整した後、ペプチド−ステロイド混合液を20時間4℃で放置した。
【0054】
生成物の生成及び同定
HPLC
生成物のHPLC及び質量分析を行う前に、エチルアセテートの後ヘキサフルロプロパノール/DMF(70:30)をSep−Pak C18カートリッジ(Millipore UK Ltd., Harrow, Middlesex)に通して一次精製を行った。Beckman Coulter System Gold(登録商標)LC125ポンプ及びLC168ダイオードアレイ検出器に接続したNovapak C18カラム(4μmビーズ、3.9×150mm)で分析用RP−HPLCを行った。緩衝液系は、0.1%TFA/水をバッファーA、0.1%TFAを含むアセトニトリルをバッファーBとした。10%から100%のバッファーB勾配液を用いて流量1ml/分で30分以上カラムを展開した。
質量分析
α−シアノ−4−ヒドロキシ桂皮酸の基質を用いたTofspec 2E MALDI−TOF質量分析器(Micromass UK Ltd.)により質量分析を行った。
【0055】
細胞培養
10%胎仔ウシ血清、グルタミン及びペニシリン/ストレプトマイシン(通常培地)を含むDMEMにてCOS−7及びCOS−1細胞を維持した。全細胞レセプター結合アッセイのためにSuperfect形質移入試薬(Qiagen, Crawley, West Sussex)を用いて細胞にヒトGnRHレセプター(hGnRH−R)を形質移入して、好適培地(Invitrogen Life Technologies, Paisley, Scotland)に4時間置いた。更に通常培地(上記参照)に48時間置いた後に形質移入細胞を分析した。
hGnRHーR形質移入COS−1細胞にてGnRHアゴニストープロゲステロン結合体の膜結合アッセイを行った。100mmディッシュに入れたCOS−1細胞をHEPES修飾DMEMにて2度洗浄し、HEPES緩衝生理食塩水、ペニシリン/ストレプトマイシン及びDMEMを含むフィルター滅菌したDEAEデキストラン中で15μgのDNAを形質移入した。37度で5時間インキュベートした後、培養液を取り除き、ペニシリン/ストレプトマイシン、2%FCS及び2%クロロキン10mMを含むDMEMと置き換えて、プレートを更に1時間インキュベートした。培養液を吸引して、細胞を洗浄し、通常培地を加えて24時間後にアッセイを行った。
我々の研究室で開発したラットタイプIGnRHRを発現するHEK293安定細胞系を用いてイノシトールリン酸生成物アッセイを行った。この細胞系は、培養の間中G418を500μg/mlで添加した10%胎仔ウシ血清、グルタミン及びペニシリン/ストレプトマイシンを含むDMEMにて維持した。必要であれば、プレートをポリ−L−リジンでコートしてアッセイの間のプラスチック容器に対する吸着を亢進した。
【0056】
レセプター結合アッセイ
GnRHアゴニスト−プロゲステロン結合体の膜結合アッセイ:
形質移入したCOS−1細胞をPBSにて洗浄して、プレートから取り除き、細胞がペレット状になるように1500rpmにて5分間遠心分離を行った。PBSを吸引して、細胞をホモジネート緩衝液(20mMトリス、2mM MgCl2、pH7.2)に懸濁し、ボルテックスにかけた後氷上に10分放置した。細胞懸濁液を7mlホモジネータ(Jencons (Scientific) Ltd., Leighton Buzzard, Buckinghamshire)に移し、ゆるい吸引具(loose plunger)にて15回、きつい吸引具(tight plunger)にて15回突き刺した。そしてホモジナイズした細胞を最高速度、4度で10分間遠心分離を行って、上澄みを吸引ポンプにて除去した。膜に残ったペレットはアッセイ用の緩衝液(40mMトリス、2mM MgCl2、pH7.4)に懸濁して、氷上に置いた。事前に冷やした12mmガラスチューブを、200μlのアッセイ用緩衝液、50μlの細胞膜、アッセイ緩衝液(およそ1チューブ当たり120000CPM)中100μMの125I[His5,D−Tyr6]GnRH、及び漸増濃度の50μlのコールドリガンド(又はBoチューブのアッセイ緩衝液)で満たした。チューブを氷上にて2時間インキュベートした後、3mlの氷冷液体ポリエチレンイミン0.01%(PEE)を加えて、吸引下(1%PEEに予浸)にてWhatman GG/Cガラス繊維フィルター(Whatman International Ltd., Maidstone, Kent)で濾過した。そうしてすぐにガンマ計測器にてフィルターを計測した。
【0057】
GnRHアンタゴニスト−プロゲステロン結合体の全細胞結合アッセイ:
COS−7細胞を12ウェルプレートに播き、アッセイ前の24時間、37度でインキュベートした。細胞をPBSにて2度洗浄して、拮抗的リガンド及び125Iリガンド(125l[His5D−Tyr6]GnRH)を含むDMEM+0.1%BSA中に修飾したHEPES 500μlを添加した。プレートをPBSにて2度洗浄して、500μlの0.1M NaOHを添加して可溶性を高め、20分間振とうした。試料をガンマ計測器にて計測した。
【0058】
全イノシトールリン酸生成物の測定
ラットタイプIGnRH受容性発現HEK293細胞を12ウェルプレートにプレートアウトして、37度、5%CO2で24時間インキュベートし、その後、1%透析FCS(グルタミン及びペニシリン/ストレプトマイシンを含む)及び1μl/ウェルのミオ−[2−3H]イノシトールを含む特別DMEMにて更に48時間インキュベートした。培養液を吸引して培養緩衝液にて洗浄した後、10mM LiClを含む500μl緩衝液を更にプレートに加え、37度で30分間インキュベートした。1μMのアゴニストを最終濃度0.1μMになるように各ウェルに添加し、プレートを同じ条件下で更に1時間インキュベートした。500μlの10mMギ酸にて反応を終了させ、4度で30分間インキュベートした。ギ酸溶液を、500μlの50%AGl×樹脂懸濁液(Bio-Rad Laboratories Ltd, Hemel Hempsted, Hertfordshire)を含む12mmプラスチックチューブに移した。滅菌水(1ml)及び四ホウ酸ナトリウム、ギ酸ナトリウム(1ml、5mM、60mM)を徐々に加え、ボルテックスにて混合し、除去することによってイノシトールリン酸を溶出した。ギ酸、ギ酸アンモニウム(1ml、0.1M、1M)を添加してボルテックスした後、800μlの上澄みをシンチレーション流動にて計測した。
【0059】
血漿タンパク質結合アッセイ
[1,2,6,7−3H]プロゲステロン又は[3H]コルチゾール存在下にて、妊娠中のモルモット血漿又は妊娠中のヒト血清のそれぞれに対するステロイド結合体の拮抗的結合により血漿タンパク質結合を決定した。20μlの血清を2mlのデキストラン−コート木炭溶液(0.25gデキストランT−70、2.5g木炭脱色粉末、活性化酸性洗浄水[Merck Ltd., Lutterworth, Leicestershire]を含む500mlのPBS)で希釈して、室温にてインキュベートした。30分後、懸濁液を3000×gにて10分間遠心分離して、上澄みを取り除き、ペレットを破棄した。希釈した血清100μlを遠心チューブに等分して、その後に1pmol[3H]ステロイド/100μl PBSを入れた。100μl PBS(全結合)又は200pmol/100μl非標識拮抗リガンド(特異的結合)を2通りで希釈血清に添加した。チューブをボルテックスして、室温で1時間インキュベートしてから更に15分間氷上に置いた。更に、750μlのデキストラン−コート木炭懸濁液を添加し、氷上にて10分間インキュベーションした後、4度で5分間遠心分離した。上澄み液を計測した。
【0060】
雄及び雌共通マーモセット(Callithrix Jacchus)におけるインビボ研究
雌マーモセット研究:黄体機能を阻害するために必要な有効量を同定するために、中期黄体期に1.0、0.5又は0.25mgのGnRHアンタゴニスト−ステロイド結合体を1ml生理的食塩水にして皮下的ボーラス投与により2カ所に投与した。 プロゲステロン濃度をRIAによりモニターした。GnRHアンタゴニスト注入の前日に300μlの血液試料を回収した。更に注入1日目とその後3日間の0、4及び8時間後に同量の血液試料を回収した。次の排卵まで1週間に3回、血液試料を採取した。
雄マーモセット研究:GnRHレセプターでの活性持続時間を決定するために、0.5mgのGnRHアンタゴニスト−ステロイド結合体を1ml生理的食塩水にして皮下的ボーラス投与により6匹の成体雄マーモセットの2カ所に投与した。テストステロン濃度をRIAによりモニターした。GnRHアンタゴニスト(0.5mg)を1ml生理的食塩水にして皮下的ボーラス投与により3匹のマーモセットに投与した。テストステロン濃度をモニターした。GnRHアンタゴニスト注入の前日、注入1日目とその後3日間の0、4及び8時間後に300μlの血液試料を回収した。更に続く週に、3つの試料を回収した。
【0061】
雄ウサギにおけるインビボ研究
GnRHアゴニスト−プロゲステロン結合体の生物学的半減期を決定するために、およそ10000000cpmのヨウ化[D−Ala6]GnRH又は[D−Lys6]GnRH−プロゲステロン結合体を含む0.5mlの生理食塩水を雄ウサギの耳静脈内に静脈ボーラス投与により注入した。0.4mlのアセプロン10(Aceprom 10)(Milborrow Animal Health. Republic of South Africa)を実験開始3〜4分前にウサギに筋注して鎮静化した。2時間後に反復注入した。反対側の耳の静脈内に内設したカニューラからのヘパリン処理したチューブで1mlの血液試料を採取した。全血を直接計測して循環からの消失を決定した。Republic of South Africa 調整法に従って全実験を行った。
【0062】
結果
GnRHレセプター結合
GnRHアゴニスト−プロゲステロン結合体は、2.9±1.2×10−10M(標準誤差、n=1、データは示していない)のときED50でタイプIヒトGnRHレセプターに結合した。また、全細胞結合アッセイに示されるようにGnRHアンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体もレセプターに結合した(図2)。非修飾アンタゴニスト配列が1.6±0.4×10−8M(n=4)であるのに対して、アンタゴニストA−プロゲステロンのED50は1.1±0.2×10−7M(n=4)であった(p<0.01;STT)。アンタゴニストB及びアンタゴニストB−プロゲステロンのED50はそれぞれ、4.7±1.1×10−8M(n=4)及び1.1±0.3×10−7M(n=5)であった(p<0.05;STT)。
GnRH−刺激性イノシトールリン酸生成物の阻害
GnRHアゴニスト−プロゲステロン結合体は、5.2 ± 1.4×10−10M(n=2)のEC50でイノシトールリン酸生成を刺激しうる;これはペプチド単独の場合と有意な違いはなかった(STT、p>0.05)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体両方のGnRH拮抗作用を哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸生成物の阻害により確認した(図3)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体のIC50に有意な違いはなかった(p>0.05、STT)。
【0063】
GnRH刺激性イノシトールリン酸生成物の阻害
GnRHアゴニスト−プロゲステロン結合体は、5.2 ± 1.4×10−10M(n=2)のEC50でイノシトールリン酸生成を刺激しうる;これはペプチド単独の場合と有意な違いはなかった(STT、p>0.05)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体両方のGnRHR拮抗作用を哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸生成物の阻害により確認した(図3)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体のIC50はそれぞれ9.7±4.0×10−8M(n=6)及び8.6±2.6×10−8M(n=7)であり、有意な違いはなかった(p>0.05、STT)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体の何れによってもイノシトールリン酸生成物単独の活性化はみられず(データは示していない)、純粋な拮抗作用が確認された。
【0064】
血漿タンパク質結合部位に対する競合作用
モルモット種では高レベルのプロゲステロン結合グロブリン(PBG)が存在するため、妊娠中のモルモット血漿における血漿タンパク質結合を研究した。PBGは特定の形式で[3H]プロゲステロンへの結合がみられ、この結合は、IC50 9.6±1.8×10−8M(n=4)の非標識プロゲステロン、IC50 1.0±0.3×10−6M(n=6)のアンタゴニストA−プロゲステロン結合体、IC50 5.3±1.0×10−7M(n=4)のアンタゴニストB−プロゲステロン結合体により阻害された。PBGはプロゲステロンにしか結合しないので、このステロイド−血漿タンパク質相互作用の特異性は特異的[3H]プロゲステロン結合をコルチゾールが阻害しないことで証明された。また、アゴニスト−プロゲステロン結合体は、非標識のコルチゾール結合をIC50 6.7±2.4×10−9M(n=2)、及びプロゲステロン結合をIC50 7.3±1.5×10−8M(n=3)で阻害するのに対して、妊娠中のヒト血清(通常より高い濃度のCBGHを含む)への[3H]コルチゾール結合をIC50 1.2±0.3×10−6M(n=2)で阻害した(データは示していない)。
【0065】
プロゲステロンの活性化
CAT酵素活性のアッセイにより、すべてのGnRHアンタゴニスト21−ヒドロキシプロゲステロン21コハク酸塩結合体はT47D細胞のプロゲステロンレセプターに結合して活性化することができることを明らかにした。すべての結合体の有効性はプロゲステロンと同様であり、1nMで実質的に活性化されず、1μMに至るまで活性が増加する。試験した5つの結合体を以下の表1に示す:
【0066】
表1:GnRHアンタゴニスト結合体
哺乳動物のGnRHを対照として並べた試験したアンタゴニストのアミノ酸配列を示す。以下の略語を用いた:Glu;グルタミン酸、His;ヒスチジン、Tip;トリプトファン、Ser;セリン、Tyr;チロシン、Gly;グリシン、Leu;ロイシン、Arg;アルギニン、Pro;プロリン、AcD−Nal;アシルD−ナフチルアラニン、D−Cpa;D−クロロフェニルアラニン、D−Pal;D−ピリジルアラニン、D−Lys;D−リジン、D−Ala;D−アラニン、Ac−ΔPro;アシル デルタ−プロリン、D−Fpa;D−フルオロフェニルアラニン、D−Trp;D−トリプトファン。
【0067】
5つすべてのアンタゴニストは、アステリスクで示す異なる位置で同じステロイド、21−ヒドロキシプロゲステロン21−半コハク酸塩に結合する。結合体A及びBは6位のD−Lysのεアミンを介して結合する。結合体C及びEは7位のリジンのεアミンを介して結合する。結合体DのステロイドはD−Pal残基のN末端アミンに結合する。
【0068】
マーモセットのインビボ研究
成体雌マーモセット(cycling adult female marmosets)の研究により、1.0mgのアンタゴニストA−プロゲステロン結合体を摂取された個体では24.8±22.2(n=6)から8日間、0.5mgを投与した個体では21.0±1.2(n=7)から11日間の黄体期の持続期間の減少がみられた。血漿中ののプロゲステロン濃度が30ng/mlに達すると排卵が起こると考えられる。また、0.25mgのアンタゴニストA−プロゲステロン結合体を摂取した3匹目のマーモセットにも血漿中のプロゲステロン濃度の一時的な減少がみられたが、完全な黄体の退行及びそれに続く排卵はこのとき起こらなかった。
雄マーモセット(n=3)に対する0.5mgのペプチドA投与により、血漿中のテストステロン濃度の急激な低下が起こった(図5)。テストステロン濃度の低下は注入後8時間持続したが、24時間までに増加した。相対的に、0.5mgのペプチドA−プロゲステロン結合体(n=6)によってもテストステロン濃度が急速に低下したが、これは注入後少なくとも72時間後まで持続し(注入後24時間でp<0.05)、6日目までに回復した。既存内務省認可の制限があるため第4及び5日目の付加的な血液採取は行えなかった。
【0069】
雄ウサギにおけるインビボ研究
非修飾GnRHアゴニストと比較してヨウ化GnRHアゴニスト−プロゲステロンによる半減期を検討するためにインビボ雄ウサギの実験を設定した(図6)。ヨウ化化合物の消失により、分子の破壊に一致する初期の半減期及び代謝と腎クリアランスを表す第2期の半減期の両方を計測した。GnRHアゴニスト−プロゲステロン結合体の第2期半減期は53±13分(n=3)であった。GnRHアゴニスト[D−Ala6]GnRHの第2期半減期は21±3分(n=2)であった。2つの類似体間の半減期の違いは、統計学的に有意な差はなかったが(p>0.05、STT)、試料の数が多ければ有意になるかもしれない。
【0070】
検討
GnRHはGnRHレセプターに結合するときにコの字型高次構造をとると言われており10、それ故にホルモンは、側鎖又はN末端のアミン基を利用して、中央の6位又は7位又はN末端でD−Lysアミノ酸に結合した。
ステロイド分子の付加の結果生じる如何なる立体構造的障害をも最小限にすることが望まれた。全細胞結合アッセイによって決定されたアンタゴニストA−プロゲステロンに対するアンタゴニストAのED50での右方変動は有意であり(p<0.01、STT)、レセプターに対する親和性の減少が確認された。しかしながら、アンタゴニストB−プロゲステロンに対するアンタゴニストBでは観察されず、立体構造的障害がペプチド配列に依存していることが示唆された。イノシトールリン酸第二伝達系(second messenger system)の解析により、GnRH類似体の修飾がGnRHレセプターでの拮抗作用を潜在的に制限していたかどうかを同定した。これはアンタゴニストA−プロゲステロン又はアンタゴニストB−プロゲステロンの何れかについて観察されなかった。また、GnRHアゴニスト−プロゲステロン結合体のイノシトールリン酸生成物促進能力について検討した。やはり、ステロイド結合体の結果のようにこの系の促進の有意な減少はみられなかった。ゆえに、GnRH類似体−プロゲステロン結合体は重要な化学的修飾にもかかわらず、 GnRHR結合及び拮抗作用を保持していると結論づけた。
【0071】
検討した新規に結合したGnRHアンタゴニストはインビトロアッセイで血漿タンパク質に結合することが示唆され、これはインビボの場合においても同様である。これはウサギでの半減期実験でみられたように、結合したGnRH類似体の半減期の延長をもたらすであろう。結合タンパク質から持続的に放出される非結合の結合体に長く曝露すると、遊離成分が代謝されるにつれてGnRHRに結合するであろう。
この研究により、GnRH類似体−ステロイド分子がGnRH及びプロゲステロンレセプター結合と活性化に関して完全に二官能価であることが示唆される。これはエモディック酸(emodic acid)結合体のGnRH態様のみが官能性であるというRahimipour等22による以前の研究と大きく異なっている。また、これはステロイドへの他の分子の結合についての重要な情報であり、C21を介する化学的修飾がプロゲステロンとそのレセプター間の相互作用に有意な変化をもたらさないことが明らかである。
【0072】
ステロイド結合及び非結合のGnRHアンタゴニストを雄マーモセットに投与すると、CBGへ結合するという心配がなく、活性の持続時間の解析が可能であった。多くの新世界霊長類は糖質コルチコイドに対する見かけの耐性を共有し、全コルチゾールが高値でコルチゾール血漿受容能力が減少している8。ゆえに、コモンマーモセット(Callithrix jacchus)は官能性CBG受容能力をほとんどもたないインビボモデルであり(血漿タンパク質結合アッセイにより確認された、データは示していない)、血漿タンパク質相互作用のない生物活性の解析が可能である。それ故に、テストステロン産生の機能低下の延長はすべて、血漿タンパク質、膜及び脂肪内の蓄積効果との疎水性相互作用により半減期が延長している分子の疎水性増加に起因している可能性がある。この効果を認識することは、ヒトに類似したCBG生理機能を持つ他の霊長類モデルでみられる結果を理解する上で不可欠である。マーモセットの生理機能がヒトと類似しているとともにGnRHアンタゴニストの薬理がより複雑であるため、このデータは価値がある。
【0073】
CBGを産生する種である17雄ウサギで行ったインビボ試験により、プロゲステロン非結合のGnRHアゴニストと比較してGnRHアゴニスト−プロゲステロン結合体の半減期が延長することが示された。用いたGnRHアゴニストは、ここで試験したアンタゴニストの疎水性配列に比べて親水性であった。これは、ウサギの研究では、プロゲステロンへ結合した後に血漿輸送タンパク質に結合することによって半減期が延長した可能性がある。ゆえに、GnRH及びその類似体などの短いペプチド分子の結合体は循環中の半減期を亢進する機能があると考えられる。
【0074】
要約すれば、GnRH類似体−ステロイドホルモン結合体は血漿ステロイド結合タンパク質受容能力を誘導し、それによって、薬物動態学的及び薬力学的にGnRH類似体を修飾するように設定した。GnRH類似体、pGlu−His−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−GlyNH2、及び2つのGnRHアンタゴニスト、[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、及び[Ac−ΔPro1,D−Fpa2,D−Trp3,D−Lys6]GnRH(それぞれアンタゴニストA及びアンタゴニストBと呼称する)は、ジシクロヘキシルカルボジイミド(DCC)により、デカペプチドのD−リシン6位の側鎖を介してプロゲステロンのC11またはC21にある半コハク酸リンカーに結合した。生成物はダイオードアレイHPLCにより精製し、質量分析法にて定量した。全細胞結合アッセイでは、GnRHアゴニスト−プロゲステロン及びGnRHアンタゴニスト−プロゲステロン結合体はそれぞれ下垂体タイプIヒトGnRHレセプターに結合した。GnRH刺激性イノシトールリン酸産生が阻害されることにより、すべてのアゴニスト及びアンタゴニスト結合体がGnRHレセプターの純粋なアンタゴニストであることが明らかとなった。3つのペプチド−プロゲステロン結合体は、妊娠中のモルモット血清中の血漿タンパク質結合部位に対して[3H]プロゲステロンと拮抗することが示された。アンタゴニストペプチドA−プロゲステロンのIC50は8.5+2.8x10−7M(n=5)であり、アンタゴニストペプチドB−プロゲステロンのIC50は4.5±0.8x10−7M(n=3)、アゴニスト−プロゲステロン結合体のIC50は2.9+1.2x10−10M(n=1)であった。アンタゴニスト−プロゲステロン結合体のインビボ生物活性は、雄マーモセットに皮下注射した後に血漿中のテストステロン濃度が減少することで証明された。雄ウサギにGnRHアゴニスト−プロゲステロンを静注すると、アンタゴニストの半減期がプロゲステロン分子との結合により延長されることが示された。
【0075】
結論として、本発明に従って作製した特定の新規のGnRHハプテン分子を設定し、生成し、インビボ及びインビトロ検討を行った。GnRH類似体を化学的に修飾することにより、インビトロ活性に有意な影響を与えず、雄ウサギでのGnRHアゴニスト−プロゲステロン結合体の半減期は延長した。これら及び類似の分子はペプチド医薬に関する課題のいくつかを克服した。GnRHアンタゴニストをステロイドホルモンに加えて、摂取のための粒子内の担体分子や封入体へのGnRHアンタゴニスト結合体は経口生物学的利用能を亢進し、活性の持続時間を延長する可能性があるであろう。
【0076】
実施例2:GnRH結合体化合物を用いた乳癌の治療
乳癌に罹患した患者に、コハク酸塩連結基を介して21−ヒドロキシプロゲステロンに結合したテベレリクスを、治療部位の活性作用物質の治療的濃度が治療期間を通じて理想的にはEC90の濃度に維持され、好ましくはEC50以下にならないような用量及び頻度で投与する。この治療は、患者の容認性、副作用の回避及び全身的バイオアベイラビリティに応じて、経口的に、あるいはより局所的に行われる。
【0077】
実施例3:獣医的避妊薬としてのGnRH結合体化合物の使用
雌ウマに、コハク酸塩連結基を介して21−ヒドロキシプロゲステロンに結合したGnRHアンタゴニストを、受胎を防ぐような用量及び頻度で投与する。注射により投与する徐放剤として処置を行う。
【0078】
参考文献
1.Anderson RA (2000) Hormonal contraception in the male. Br Med Bull 56(3): 717-728.
2.Christin-Maitre S, Olivennes F, Dubourdieu S, Chabbert-Buffet N, Charbonnel B, Frydman R, and Bouchard P (2000) Effect of gonadotropin-releasing hormone (GnRH) antagonist during the LH surge in normal women and during controlled ovarian hyperstimulation. Clin Endocrinol (Oxf) 52(6): 721-726.
3.Danforth DR, Williams RF, Hsiu JG, Roh Sl, Hahn D., McGuire JL, Hodgen GD 1990 Intermittent GnRH antagonist plus progestin contraception conserving tonic ovarian estrogen secretion and reducing progestin exposure. Contraception 41(6).-6,13-631.
4.Ditkoff EC, Cassidenti DL, Paulson RJ, Saner MV, Paul WL, Rivier J, Yen SSC, Lobo RA (1991) The gonadotropin-releasing hormone antagonist (Nal-Glu ) acutely blocks the luteinizing hormone surge but allows for resumption of folliculogenesis in normal women. Am J Obs Gyne 165(6 Pt 1):1811-1817.
5.Fraser HM, Nestor JJ Jr, Vickery B H (1987) SSuppression of luteal function by a luteinizing hormone-releasing hormone antagonist during the early luteal phase in the stumptailed monkey and the effects of subsequent administration of human chorionic gonadotropin. Endocrinol 121(2):612-628.
6.Fraser HM, Lunn SF, Cowen GM, Smith KB, Conn PM, (1991) Effect of late follicular phase administration of amide on ovulation and inhibin secretion in macaques. Contraception 44(6):667-676.
7.Kenigsberg D, Hodgen GD, (1986) Ovulation inhibition by administration of weekly gonadotropin-releasing hormone antagonist. J Clin Endocrinol Metab 62(4):734-738.
8.Klosterman LL, Murai JT, Siiteri PK (1986) Cortisol levels, binding, and properties of corticosteroid-binding globulin in the serum of primates. Endocrinol 118(1):424-434.
9.Mattox VR, Litwiller RD, Nelson AN (1979) A comparison of procedures for attaching steroidal glucosiduronic acids to bovine serum albumin. J Steroid Biochem 10.167-172.
10.Millar RP, Assefa D, Ott T; Pawson A, Troskie B, Wakefield L, Katz A GnRH and GnRH analogues: Structure, actions and clinical applications. Horm Frontier Gynecol 5(4):77-83.
11.Nestor JJ, Tahilramani R, Ho TL, Goodpasture JC, Vickery B H, Ferrandon P (1992) Potent gonadotropin releasing hormone antagonists with low histamine-releasing activity. J Med Chem 35(21):3942-3948
12.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Vale WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
13.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Valc WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
14.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Vale WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
15.Russell-Jones GJ, Westwood SW, Farnworth PG, Findlay JK, Burger HG, (1995) Synthesis of LHRH antagonists suitable for oral administration via the Vitamin B12 uptake system. Bioconjugate Chem 6:34-42.
16.Siiteri PK, Murai JT, Hammond GL, Nisker JA, Raymoure WJ. Kuhn RW (1982) Tlie serum transport of steroid hormones Rec Prog Horm Res 38:457-510.
17.Seal US, Doe RP (1965) Vertebrate distribution of Cortlcosteroid-binding globulin and some endocrine effects on concentration. Steroids 5:827-841.
18.Swerdloff RS, Bagatell CJ, Wand C, Anawalt BD, Berman N, Steiner B, Bremner WJ (1998) Suppression of spermatogenesis in man induced by Nal-Glu gonadotropin releasing hormone antagonist and testosterone enanthate (TE) is maintained by TE alone. J Clin Endo Metab 83(10):3527-3533.
19.Westphal U (1993) Steroid-protein interaction: From past to present. J Steroid Biochem 19(1):1-15.
20.McEwan JF, Veitch ES, Russell-Jones GJ (1999) Synthesis and Biological Activity of Ribose-5'-Carbamate Derivatives of Vitamin B12. Bioconjugate Chem 20, 10, 1131-1136.
21.Alsenz J, Russell-Jones GJ, Westwood S, Levet-Traft B, de Smidt PC (2000) Oral Absorption of Peptides Through the Cobalamin (Vitamin B12) Pathway in the Rat Intestine. Pharmaceutical Research 21, Vol. 17, No. 7, 2000.
22.Rahimipour S, Ben-Aroya N, Fridkin M, Koch Y (2000) Design, Synthesis, and Evaluation of a Long-Acting, Potent Analogue of Gonadotropin-Releasing Hormone. J. Med. Chem. 22, 44, 3645- 3652.
23.Thau (1984) Luteinizing hormone-releasing honnone (LHRH) and its analogs for contraception in women: a review. Contraception, 29(2): 143-162.
24.Millar R.P. (2003) Gonadatropin-releasing hormones and their receptors, Chapter 1, Reproductive Medicine: Molecular, Cellular and Genetic Fundamentals, Eds. B.C.J.M. Fauser et al, The Parthenon Publishing Group, New York, USA. ISBN: 1-84214-019-1.
25.Millar R.P. 等 (2000) Progress towards the developmnent of nonpeptide orallyrnactive gonadatropin-releasing hormone (GnRH) antagonists: therapeutic implications. Br. Med. Bull. 56(3): 761-772.
26.Burton & Westphal (1972) Steroid hormone-binding proteins in blood plasma. Metabolism 21(3): 253-276.
27.Cunningham G.R. 等 (1981) Steroid structural requirements for high affinity binding to human sex steroid binding protein (SBP). Steroids 38(3) : 243-262.
28.Mickelson K.E. 等 (1981) Steroid-protein interactions. Human corticosteroid binding globulin: Some physiochemical properties and binding specificity. Biochemistry 20: 6211-6218.
【図面の簡単な説明】
【0079】
【図1】A:あるGnRHアンタゴニストステロイド結合体の構造を示す。結合体は、21−ヒドロキシプロゲステロン21−半コハク酸塩のカルボキシル基を有するペプチドの6位でD−リジン側鎖アミンの縮合によって生成された。ペプチドの残りの側鎖は1−5及び7−10の数で表す(21−ヒドロキシプロゲステロン21−半コハク酸塩も又、デオキシコルチコステロン21−半コハク酸塩及び21−ヒドロキシ−4−プレグネン−3,20−ジオン21−半コハク酸塩として知られている)。B:GnRHアゴニスト11−ヒドロキシプロゲステロン11−コハク酸塩の構造を示す。
【図2】GnRHレセプター結合親和性を示す。125I−GnRHアゴニストの置換はヒトGnRHレセプターを形質移入したCOS−7細胞全体に結合した。ペプチドAGnRHアンタゴニスト(○)、ペプチドAプロゲステロン結合体(●)、ペプチドBGnRHアンタゴニスト(□)、及びペプチドBプロゲステロン結合体(■)。
【図3】ラットI型GnRHRを安定して発現するHEK293細胞における哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸産生に対するペプチドAプロゲステロン結合体及びペプチドBプロゲステロン結合体の影響を示す。
【図4】妊娠中のモルモットに対する[1,2,6,7−3H]の結合に対するプロゲステロン(○)、ペプチドAプロゲステロン(●)及びペプチドBプロゲステロン(◆)の影響を、デキストランコート木炭懸濁液にて測定して示す。
【図5】0.5mgペプチドAプロゲステロン(○)又は0.5mgペプチドA(■)の何れかを皮下注射した雄マーモセットのテストステロン濃度を示す。
【図6】およそ15000000cpmの125I−GnRHアゴニスト−プロゲステロン結合体又は[125I−D−Ala6]GnRHを含む500μlの生理食塩水を耳静脈内に静脈内注入した2匹の雄ウサギの全血中に存在する放射性標識したGnRHアゴニスト−プロゲステロン(■)及び[D−Ala6]GnRH(▲)を示す。3.5時間を超えると全血から消失した。
【図7】T47D細胞におけるプロゲステロンレセプターに連結したCATリピート遺伝子の活性に対する結合体A、B、C、D及びEとプロゲステロンの影響を、CAT活性を測定することによって示す。
【図8】好ましいGnRH類似体の配列を示す。黒い円はGnRH類似体がGnRHと同じ位置に同じアミノ酸を持つことを示す。
【発明の開示】
【0001】
本発明は、結合体化合物、特に性腺刺激ホルモン(ゴナドトロピン)放出ホルモン結合体化合物に関する。
【0002】
性腺刺激ホルモン放出ホルモン(GnRH)は、生殖調節に関与する神経内分泌ホルモンであり、性腺刺激ホルモン、黄体形成ホルモン(LH)及び卵胞刺激ホルモン(FSH)の放出の誘因となる。
GnRH類似体は、視床下部−下垂体軸の検査においてもホルモン依存性症状の治療のための性腺刺激ホルモンの操作においても非常に有用な薬理学的作用剤である。GnRHアゴニスト及びアンタゴニストの多くはおよそ9又は10アミノ酸からなるペプチド分子で、一般的にレセプター結合親和性、レセプター活性を修飾するため、及びタンパク分解を減らすために天然でないアミノ酸を含む。
【0003】
GnRH類似体は、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維腫、月経前症候群、多嚢胞性卵巣症候群、多毛症、尋常性ざ瘡、性早熟症、急性間欠性ポルフィリン症、睾丸停留、思春期遅発症及び不妊治療の処置を含む臨床応用の範囲がある(Millar 2003)。
加えて、GnRH類似体は効果的な避妊薬になりうる。GnRHアンタゴニストはLHサージ期に服用すると排卵を抑制するように働く;しかしながら、用量のタイミングは重要であり、わずかな遅れ(時間)は如何なる効果をも十分無効にする2,4,6,7。あるいは、黄体期にGnRHアンタゴニスト処置を行うことによって黄体機能を抑制して初期妊娠の進行を抑制することができる。また、GnRHアゴニストは、より低い用量を必要とするレセプター脱感作によってではない性腺刺激ホルモンの放出を抑制することができる。性腺刺激ホルモンの放出を抑制するために、GnRH−レセプター(GnRH−R)で必要とされるレセプター占有率の高さに応じてGnRHアゴニストに比例してアンタゴニストを増やす必要がある。
【0004】
現在、女性ホルモン的な避妊は、性腺刺激ホルモンの分泌を抑制するために生理学的用量を超えてステロイドホルモン類似体が用いられている。周辺組織が同じレベルに曝されるので、結果としてさまざまな副作用が生じうる1。男性ホルモン的な避妊の開発は男性ホルモン置換と組み合わせた同様な原理に基づいているが、同じように副作用の問題に直面している。ゆえに、GnRHアンタゴニストは性腺ステロイドホルモン置換と組み合わせた男性及び女性の避妊の基本となる可能性がある1,3,7,12,18。
長期的GnRH類似体処置に関する主たる問題の一つは、性腺性ステロイドホルモンの減少である。したがって、ホルモン置換療法は女性の低エストロゲン性骨減少等の副作用の予防や男性の二次性徴の維持が必要である。
GnRH類似体処置に関する付加的問題は、経口投与したGnRH類似体の消化管での急速な分解である。更に、GnRH類似体は循環において比較的半減期が短く、しばしば初めの循環で腎臓を介して排出される(1〜7分、t1/2)。これらの問題により、GnRH類似体のインビボ有効濃度を維持するために徐放注射用貯蔵調製物が開発された。
【0005】
現在、GnRHペプチドアンタゴニストは注射により投与されている。非ペプチドアンタゴニスト及び経口活性型GnRHアンタゴニストの開発が試みられている。消化管内に活性的に取り込まれるビタミンB12などのハプテンとのGnRH類似体結合体はペプチドアンタゴニストに経口的活性を与える可能性がある15。GnRHアンタゴニストは既に修飾して付加的機能的部分を含んでいる。例えば、エモジン成分22とのGnRH結合体又は潜在的経口摂取15,20,21を亢進させるためのアンチドとのビタミンB12結合体が報告されている。ビタミンB12結合体の経口投与によりいくらか摂取の増加が見られるが21、これら成分の半減期が長くなっていないことが証明された。
多くのホルモンは循環内で血漿タンパク質と結合している。これは、腎クリアランス及び代謝分解からの保護による循環半減期の延長19など、多種の機能を司っていると考えられている。
【0006】
ヒト及び旧世界霊長類には二つの主な循環ステロイド結合タンパク質があり、それはコルチゾールとプロゲステロンの結合であるコルチゾール結合グロブリン(CBG)、及びテストステロンとエストラジオールの結合である性ホルモン結合グロブリン(SHBG)8である。また、モルモットなどのヤマアラシ類のげっ歯種はプロゲステロンに特異的に結合するプロゲステロン結合グロブリン(PBG)を持つ。
高分子量の血漿タンパク質に結合しているステロイドは腎及び代謝によりクリアランスされず、加えて細胞内への侵入が阻害されて核レセプターと相互作用する。ゆえに、循環内のステロイド有効濃度は、結合比率が平衡状態にあるときの非結合比率(ヒトではおよそ2%)により決定する16,19。
そこで我々は、ホルモン成分又はホルモン誘導体にGnRH類似体を結合させると血漿半減期が延長し、GnRH類似体が薬物動態学的にも薬力学的にも改善されることを示した。
【0007】
仮説にしばられることを望むものではないが、我々は、結合体化合物のホルモン又はホルモン誘導体成分はGnRH類似体の貯蔵として働く血漿ホルモン結合タンパク質に結合して、ゆっくりと持続してGnRH類似体を放出すると考える。血漿タンパク質への結合を介しての結合体の隔離は排泄及び不活性型への代謝から薬物を「保護」し、それによりGnRH類似体の半減期を延長するであろう。
我々はGnRH類似体−ホルモン結合体がそれ自身の半減期及び活性持続時間を延長し、生物学的効果に必要なGnRH類似体の用量を減らすことを示した。また、アンタゴニストが必要とされる前の重要な時期に結合体を投与することができたり、GnRH類似体の投与頻度及び用量を低くすることによって治療の副作用を潜在的に少なくすることができる。
【0008】
さらに、結合体はGnRH類似体と機能的ステロイド性ホルモンを単一分子に結合させたものであるため、ステロイド性ホルモンに結合したGnRH類似体での治療はホルモン置換療法の必要性を減らす又は軽減するであろう。
本発明の第一態様では、ホルモン成分又はその誘導体に結合したGnRH類似体を含んでなるものであり、血漿ホルモン結合タンパク質に結合可能な化合物を提供する。
GnRHは、デカペプチドpGlu−His−Trp−Ser−Tyr−Gly−Leu−Arg−Pro−Gly−NH2を意味し、pGluはピログルタミン酸である。
「GnRH類似体」はGnRHレセプターに結合するペプチド又は非ペプチド分子何れの分子をも含む。GnRHレセプターに対する分子の結合は当業者によって、例えば、レセプター結合アッセイ又は後述の実施例1などの全細胞結合アッセイを用いて迅速に決定することができる。
【0009】
一般的に、ペプチドGnRH類似体は6〜12のアミノ酸残基を持つペプチドである。より好ましくは、ペプチドGnRH類似体は7、8、9、10又は11のアミノ酸残基を持つ。さらにより好ましくは、ペプチドGnRH類似体は9又は、最も一般的には10アミノ酸残基を持つ。
一般的に、ペプチドGnRH類似体は少なくとも一つの修飾、すなわち非天然に生じたアミノ酸残基を含む。GnRH類似体は通常天然のGnRHデカペプチド構造の第6及び第10番目のアミノ酸を修飾して生成するのに対して、第1、2、3、5、6、8及び10番目の変更は通常は結果として拮抗作用が生じる(Thau 1984)。
Millar(2003)は、本発明の使用に適するかもしれないGnRH類似体だけでなくGnRH及びそのレセプターの構造について検討している。出典明記によりGnRH及びGnRH類似体に関するMillar(2003)の開示の全体をここに組み込む。
【0010】
一実施態様では、GnRH類似体はGnRHアンタゴニストである。一般的にGnRHアンタゴニストは、結合してGnRHレセプター(GnRH−R)活性又はシグナル伝達を遮断するようにGnRHの構造を修飾したペプチド分子である。
「GnRHアンタゴニスト」は、GnRHレセプターのシグナル伝達を阻害、減少又は防止するペプチド又は非ペプチドの如何なるGnRH類似体の意味をも含む。GnRH−Rシグナル伝達の阻害、減少又は防止は当業者によって、例えば、後述の実施例1などのイノシトールリン酸産生アッセイを用いて迅速に決定することができる。
Millar等(2000)は、本発明の使用に適するかもしれないGnRHアンタゴニストについて検討している。出典明記によりGnRHアンタゴニストに関するMillar等(2000)の開示の全体をここに組み込む。
【0011】
特定の位置に対してアミノ酸を明記していない場合、同じ位置に天然に生じたGnRHと同じアミノ酸残基が存在することを示す。
非天然に生じたアミノ酸に対して以下のような略語を用いる:AcD-Nal−アシルD-ナフチルアラニン;D-Cpa−D-クロロフェニルアラニン;D-Pal−D-ピリジルアラニン;D-Lys−D-リジン;D-Ala−D-アラニン;Ac-ΔPro−アシルΔ-プロリン;D-Fpa−D-フルオロフェニルアラニン;D-Trp−D-トリプトファン;Lys(Nic)−リジンニコチンアミド;及びiPr-Lys−イソプロピルリジン。
最も好ましいGnRHアンタゴニストは、セトロレリクス(Asta Medica AG)、ガニレリクス(Organon)、アバレリクス(Praecis Pharmaceuticals)、アンチド(Ares Serono SA)、テベレリクス(Ardana)、FE200486(Ferring)及びNal−Glu(NIH)である。これらGnRHアンタゴニストの構造を図8に示す。
【0012】
他の好適なGnRHアンタゴニストには、A−75998、A−76154及びA−84861(Abbott Laboratoriesにより最初に製造);D−26344及びD−63153(ASTA Medica AGにより最初に製造);ラモレリクス(Aventis AGにより最初に製造);デガレリクス(Ferring Research Institute (UK)により最初に製造)、NBI−42902(Neurocrine Biosciences Incにより最初に製造);Org−30850(Organonにより最初に製造)、デチレリクス(Roche Bioscienceにより最初に製造);イツレリクス(Serono SAにより最初に製造);TAK013及びTAK810(Takeda Chemical Industries Ltdにより最初に製造);AN 207(Tulane Universityにより最初に製造);Pfizer GnRHアンタゴニスト;Merck GnRHアンタゴニスト;及びWeizmann GnRHアンタゴニストが含まれる。また、以下も参照のこと:Goulet (1995) Ann. Reports Med. Chem. 30, 169-178;Nestor & Vickery (1988) Ann. Reports Med. Chem 23, 211-220;及びDutta & Barrington (1985) Ann. Reports Med. Chem. 20, 203-214これらはすべて出典明記によりここに組み込まれる。
他の好適なペプチドGnRHアンタゴニストには、
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
Ac−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;
D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;
[D−Pyr1, D−Phe2, D−Trp3,6]GnRH (Rahimipour等参照);
D−Lys6アンチド;Lys5アンチド;及びLys8アンチド
が含まれる。
【0013】
アンチド及びその誘導体は、Russel-Jones等(1995)及びWO94/28015(Biotech Australia Pty. Ltd)に記載されている。GnRHアンタゴニスト、GnRH類似体結合体化合物及びそれらの構造に関するRusselJones等及びWO94/28015の開示は出典明記によりその全体がここに含まれる。
本発明の使用に適するかもしれない非ペプチドGnRHアンタゴニストは、WO95/28405;WO96/24597;WO97/41126;WO99/33831;WO00/00493;WO00/56739及びWO01/78780 (Takeda Chemical Industries, Ltd)及びWO02/02533(Yamanouchi Pharmaceutical Co., Ltd)に記載されている。GnRHアンタゴニスト、その構造及び使用に関するこれら公報の開示は出典明記によりその全体がここに含まれる。
本発明に有用かもしれない更なるGnRH類似体には、米国特許第3,813,382;3,843,065;3,849,389;3,855,199;3,886,135;3,890,437;3,892,723;3,896,104;3,901,872;3,914,412;3,915,947,3,929,759;3,937,695;3,953,416;3,974,135;4,010,125,4,018,914、4,022,759;4,022,760;4,022,761;4,024,248;4,034,082;4,072,668;4,075,189;4,075,192;4,086,219;4,101,538;4,124,577;4,124,578、4,143,133;4,234,571、4,253,997;4,292,313;及び4,341,767号に記載のものが含まれる。GnRH類似体、その構造及び使用に関するこれら米国特許文献の開示は出典明記によりその全体がここに含まれる。
【0014】
本発明に有用かもしれないより更なるGnRH類似体には、米国特許4,504,414;4,677,193;4,705,778;5,064,939;5,371,070 5,413,990;5,502,035;5,633,248;5,756,497;6,156,731及び6,191,115;EP 0 081 877及びEP 0 192 492;以下の公開されているPCT出願WO93/03058、WO95/04541、WO95/28405、WO97/44321、WO97/44339、WO97/44041、W098/03632、WO98/55505、WO99/21557、WO99/41251、WO99/41252、WO99/46283及びWO00/53178;及びGB 2 310 660に記載のものが含まれる。アンタゴニスト及びアゴニストの両方、それらの構造及び使用を含むGnRH類似体に関するこれら公開の開示は出典明記によりその全体がここに含まれる。
一実施態様では、GnRH類似体はGnRHアゴニストである。
「GnRHアゴニスト」には、GnRHレセプターのシグナル伝達を刺激又は活性化するペプチド又は非ペプチドの如何なるGnRH類似体の意味をも含む。GnRHレセプターシグナル伝達の刺激又は活性化は、当業者によって、例えば実施例1に記載などのイノシトールリン酸産生アッセイを用いて迅速に決定することができる。
【0015】
特に6位でのDイソ型アミノ酸の取り込みは、GnRH類似体のアゴニストとしての可能性を高める。Rahimpour等(2001)は、3000以上のGnRH類似体を合成してその生物学的活性についての評価を報告している。上記のアゴニストのほとんどは、6位のGlyの代わりにD−アミノ酸を取り込み、多くはGly−NH2末端の代わりにN−エチルアミドを持つ。これら化学的修飾は、Gly−Leu結合でのGnRHの生物学的活性なβターン高次構造を亢進して、タンパク質分解に対するペプチドの感受性を低下させると報告されている。ゆえに、本発明の使用に好適なGnRHアゴニストには、これら修飾の何れか又は両方を有するGnRH類似体が含まれる。
一実施態様では、GnRH類似体の少なくとも一アミノ酸残基は、D−リシンである。一般的に、D−リシンは、類似体の6位にある;すなわち、GnRH類似体は[D−Lys6]GnRHである。
【0016】
最も好ましいGnRHは、ロイプロン(TAP)、ゾラデックス(Zeneca)、サプレリン(Roberts)、シナレル(Searle)、トリプトレリン(Ferring)、及びブセレリン(Hoechst)であり、各々は6位に非天然に生じる残基を持つ。GnRHアゴニストの構造を図8に示す。
他の好適なGnRHアゴニストには、デスロレリン(Balance Pharmaceuticals)、ProMaxx-100 (Epic Therapeutics);アボレリン(Mediolanum Farmaceutici SpA)、ヒストレリン(Ortho Pharmaceuticals)、及びナファレリン(Roche Bioscience)を含む。
【0017】
ペプチドGnRH類似体は当業者に知られた任意の方法で製造することができる。例えば、ペプチドは、Lu等(1981) J. Org. Chem. 46, 3433及びそこに記載の文献に開示されたような、固相ペプチド合成のFmoc-ポリアミド法によって合成することができる。一時的なN-アミノ基の保護は9-フルオレニルメチルオキシカルボニル(Fmoc)基によって得られる。この高度に塩基不安定保護基の繰り返しの開裂はN,N-ジメチルホルムアミド中20%ピペリジンを使用してなされる。側鎖官能基はそのブチルエステル(グルタミン酸及びアスパラギン酸の場合)、ブチルオキシカルボニル誘導体(リジン及びヒスチジンの場合)、トリチル誘導体(システインの場合)及び4-メトキシ-2,3,6-トリメチルベンゼンスルホニル誘導体(アルギニンの場合)として保護できる。グルタミン又はアスパラギンがC末端残基である場合、側鎖アミド官能基の保護には4,4'-ジメトキシベンズヒドリル基が使用される。固相支持体は、ジメチルアクリルアミド(骨格モノマー)、ビスアクリロイルエチレンジアミン(架橋剤)及びアクリロイルサルコシンメチルエステル(官能化剤)の三種のモノマーから構成されたポリジメチルアクリルアミドポリマーをベースとしている。使用されるペプチド対樹脂開裂架橋剤は酸不安定4-ヒドロキシメチル-フェノキシ酢酸誘導体である。全てのアミノ酸誘導体は、逆のN,N-ジシクロヘキシル-カルボジイミド/1-ヒドロキシベンゾトリアゾール媒介カップリング法を使用して添加されるアスパラギンとグルタミンを除いて、その予め形成された対称な無水物誘導体として添加される。全てのカップリング及び脱保護反応はニンヒドリン(ninhydrin)、トリニトロベンゼンスルホン酸又はイソチン(isotin)試験方法を使用してモニターされる。合成の完了時点で、ペプチドは50%のスカベンジャー混合物を含む95%トリフルオロ酢酸での処理によって側鎖保護基の同時の除去と共に樹脂支持体から開裂される。一般的に使用されるスカベンジャーはエタンジチオール、フェノール、アニソール及び水であり、正確な選択は合成されるペプチドの構成アミノ酸に依存する。トリフルオロ酢酸は真空蒸発によって除去され、続くジエチルエーテルでの倍散により粗ペプチドが得られる。存在する全てのスカベンジャーは、水相の凍結乾燥によりスカベンジャーのない粗ペプチドを生じる単純な抽出法によって除去される。ペプチド合成のための試薬は一般にCalbiochem-Novabiochem (UK)社, Nottingham NG7 2QJ, UKから入手できる。精製は、サイズ排除クロマトグラフィー、イオン交換クロマトグラフィー及び(主として)逆相高速液体クロマトグラフィーのような技術の何れか一つ又はその組み合わせによってなすことができる。ペプチドの分析は薄層クロマトグラフィー、逆相高速液体クロマトグラフィー、酸加水分解後のアミノ酸分析及び高速原子衝撃(FAB)質量分析法を使用して実施することができる。
【0018】
別法として、ペプチドGnRH類似体は、それらがDNA分子中でコード化されうるならば、標準的な分子生物学技術によって得ることができる。
本発明での使用に適したペプチド及び非ペプチドGnRH類似体はホルモン部分又はその誘導体への結合に適した原子又は官能基を有しているものである。
天然に生じるアミノ酸残基上に存在する好適な官能基には、Cys残基上のスルフヒドリル基、Ser、Thr又はTyr残基上のヒドロキシル基、Lys残基上のε-アミノ基、Asp及びGlu残基上のカルボキシル基、Arg残基上のグアニジノ基、Asn及びGln残基上のアミド基、His残基のイミダゾールNH基、Trp残基のインドールNH基、C末端カルボキシル基及びN末端アミノ基が含まれる。同じ官能基はこれらのアミノ酸残基のD-アイソフォーム上に存在する。
【0019】
修飾されたアミノ酸はまたホルモン部分への結合に好適な官能基を有しうる。これらには、ヒドロキシプロリン残基上のヒドロキシル基、O-ホスホセリン又はO-ホスホチロシン残基上のホスフェート基、γ-カルボキシグルタメート残基上の双方のカルボキシル基、iPr-Lys残基上のε-アミノ基及びシトルリン及びホモシトルリンの基が含まれる。
更に、ピリジルアラニン残基のピリジルN-原子及びN-アルキル化Arg残基の三級グアニジノN-原子がN-4級化によってホルモン部分に結合させるための好適な官能基である。再び、D-及びL-アイソフォームがGnRH類似体に存在しうる。
非ペプチドGnRH類似体上に存在するホルモン部分への結合のための好適な官能基にはケト、NH(アミノ、アミド又はウレイジル官能基の一部として)、ヒドロキシル、スルフヒドリル、カルボン酸及び三級アミノ基が含まれる。
【0020】
典型的には、GnRH類似体へ結合されるホルモン部分又はその誘導体は血漿ホルモン結合タンパク質にインビボで結合するものである。
典型的には、ホルモン結合タンパク質はグロブリンである。
一実施態様では、ホルモン部分又は誘導体は特異的血漿ホルモン結合タンパク質、例えばコルチゾール結合グロブリン(CBG)、性ホルモン結合グロブリン(SHBG)と、ある場合にはプロゲステロン結合グロブリン(PBG)に結合する。典型的には、ホルモン部分又はその誘導体はまた血清アルブミン(HSA)に結合する。疑義を避けるために、本発明においては、HSAは血漿ホルモン結合タンパク質である。
【0021】
ホルモン部分の「誘導体」には、我々は、誘導体が天然に見出されるホルモン部分の構造から改変されているという意味を含める。例えば、改変されて、GnRH類似体への新たな又は改良された結合部位を提供するか、又はその安定性又はその活性を改善する場合がある。しかし、ここで定義されるホルモン誘導体は血漿ホルモン結合タンパク質に結合するその能力を完全には失っていない。誘導体自体はホルモン活性を有しているか又は有していないことが理解される。
好適な実施態様では、ホルモン部分はステロイドホルモン部分である。
本発明での使用に適したステロイドホルモン部分及びその誘導体はGnRH類似体への結合のために適した原子又は官能基を有するものである。
【0022】
典型的には、ステロイドホルモンは3位にヒドロキシル基か又はケト基を有している。ステロイドホルモンの多くは17位にヒドロキシル基か又はケト基を有している。多くのステロイドホルモンは11位にヒドロキシル基を有している。幾つかのステロイドホルモンは21位にヒドロキシル基を有している。
好ましくは、ステロイドホルモン部分は、エストラジオール、プロゲステロン、コルチゾール、コルチコステロン、エストロン、テストステロン及びジヒドロキシテストステロン(DHT)である。
ステロイドホルモン誘導体には11、17又は21位にヒドロキシル基を付加することによって修飾されたものが含まれる。好適なプロゲステロン誘導体には11α-ヒドロキシプロゲステロン及び21-ヒドロキシプロゲステロンが含まれる。
ステロイドであるがもはやホルモン活性を有していないステロイドホルモン誘導体は、それらが血漿ホルモン結合タンパク質に結合する場合は使用することができることが理解される。
【0023】
血漿ホルモン結合タンパク質にステロイドホルモンが結合するのに必要とされる官能基は当業者に知られている。例えば、置換ステロイドを用いる構造調査において、SHBGと相互作用するためには、ステロイドは17β-ヒドロキシル基を含んでいなければならないことが証明されている(Burton及びWestphal (1972)及びCunningham等(1981))。幾つかの他の特徴、例えばC11でのヒドロキシル又はケト基の付加は結合親和性にマイナスの影響を及ぼす。ステロイド核中の炭素2、6、9及び11の修飾がまた結合親和性を低減させた(Cunningham等(1981))。
ヒトCBGへのステロイド結合に対しては、20-オキソ及び10β-メチル基が本質的であると報告されており、3-オキソ及び4ーエンがまた重要である。11β、17α-、及び21-ヒドロキシ基は比較的重要ではなく、ヒドロキシル基は11α、6α、6β、12α、14α、16α及び19位の結合を損なう(Mickelson等 1981)。
【0024】
しかして、血漿ホルモン結合タンパク質に結合するホルモン部分の能力を保持するようにGnRH類似体の特定の官能基にホルモン部分又はその誘導体を結合させるのは当業者の技量の十分な範囲内である。
リンカーは血漿ホルモン結合タンパク質とのホルモン又はGnRHレセプターとのGnRH類似体の相互作用を立体的に阻害しないのが好ましい。嵩がある、及び/又は親水性の基がホルモン部分又はGnRH類似体に近接したリンカー中に存在していないことが好ましい。
【0025】
本発明の幾つかの好適な実施態様では、ホルモン部分又はその誘導体は、GnRH類似体に結合した場合、全体的又は部分的にそのステロイド活性を保持している。あるいは、他の実施態様では、ホルモン部分又はその誘導体は、GnRH類似体に結合した場合、ステロイド活性は保持していないのが好ましい。
ホルモン活性が望まれる実施態様では、GnRH類似体への結合に使用される官能基は典型的にはその特定のホルモン又はホルモン誘導体の活性に必要とされないものである。逆に、ホルモン活性が望まれない場合は、GnRH類似体への結合に使用される官能基は典型的にはその特定のホルモン又はその誘導体の活性に必要とされるものである。
【0026】
ステロイドホルモンの活性に必要な官能基は当業者に知られている(例えば、ステロイドホルモン-レセプターの構造関係は、Duax等(1989) Advances in Drug Research 18,115-138;Aranyi (1982) Hung. Biologia (Budapest) 30,145-169;Raynaud及びOjasoo (1983) Nobel Symposium 57,141-170;Duax及びGriffin (1989) Alfred Benzon Symposium 28,62-77;Ojasoo等 (1992) Mol. Struct. Biol. Act. Steroids, pp 157-207, CRC, Boca Raton;及びDuax & Griffin (1998) NATO ASI series E : Applied Sciences 352,1-14にエストロゲン、グルココルチコイド、鉱質コルチコイド、アンドロゲン、及び代謝類似体及びアンタゴニストに対して記述されており、その全てが出典明示によりここに取り込まれる。従って、ホルモンの活性を保持し又は除去するようにGnRH類似体の特定の官能基にホルモン部分又はその誘導体を結合させるのは当業者の技量の範囲内にある。
【0027】
例えば、実施例1に示すように、21-ヒドロキシプロゲステロンの21位へのGnRH類似体の結合は結合化合物中でプロゲステロン活性を維持する。逆に、ステロイドホルモン活性が除去される場合には、GnRHは3位のケト基に結合させることができる。
「結合させる」という用語には、共有結合がGnRH類似体の原子とホルモン部分の原子の間に形成されている意味、又はGnRH類似体とホルモン部分が両方とも同じ結合基に共有結合的に結合している意味を含む。
好適な実施態様では、GnRH類似体とホルモン部分の間の結合は開裂可能であり、例えば結合には、エステラーゼにより開裂可能であるエステル結合又はアミダーゼにより開裂可能であるアミド結合が含まれる。
【0028】
一実施態様では、GnRH類似体及びホルモン部分は直接結合している。典型的にはこの場合、アミノ酸は既に結合しているホルモン部分と共に合成され、この修飾されたアミノ酸はペプチドGnRH類似体中に導入される。例えば、ホルモンのケト基とLysのε-アミノ基の間のイミンの形成を通して直接結合が生じうる。生じるイミンはインビボで加水分解的に不安定であり、よって短い半減期のコンジュゲートを生じる。半減期はイミンを還元してアミンを形成することにより増加させることができる。あるいは、直接結合は、ホルモン部分のα,β-不飽和ケトン官能基へのLys上のε-アミノ基のマイケル付加を通して(例えばプロゲステロン)、又はGnRH類似体の残基上の適切な求核基(例えばLys上のε-アミノ基又はSer上のβ-OH基)でOH基が離脱基(例えばハロゲン化物又はスルホン酸エステル)に転換されたホルモン部分の反応を通して生じうる
「結合基」はGnRH類似体にもホルモン部分にも内因的ではない一又は複数の原子によって形成された構造を意味する。
【0029】
結合基は一又は複数の原子を含み、GnRH類似体とホルモン部分又はその誘導体の間の最短経路は典型的には2又は3又は4又は5又は6又は7又は8又は9又は10又はそれ以上の結合を含む。
結合基は、場合によっては酸素、窒素及び/又は硫黄のような他の原子と共に、一又は複数の炭素原子をベースとした構造を含み得、GnRH類似体とホルモン部分又はその誘導体の間の最短経路は、場合によっては少なくとも一の酸素、窒素及び/又は硫黄原子と共に、典型的には2又は3又は4又は5又は6又は7又は8又は9又は10又はそれ以上の炭素原子を含む。
典型的には、結合基は、同時に又は任意の連続的な順序で、二官能性前駆体結合部分をGnRH類似体及びホルモン部分に反応させることによって導入される。あるいは、結合基はGnRH類似体及び/又はホルモン部分の誘導体化によって導入される構造的断片上に官能基をカップリングさせることによって形成される。
【0030】
好適な結合基には、式-A1-D-A2-によって表されるものが含まれ、ここで、Dは、例えば、
(a)アルキレン、アルケニレン又はアルキニレン(後者の3種の基は場合によってはNH、O又はS、炭素環又は複素環によって中断され及び/又は終端され、及び/又は場合によっては-S-S-によって中断されている)、
(b)炭素環、又は
(c)複素環
を表すか、
又はDは構造断片-D1-A3-D2-を表し、ここで、D1及びD2は独立して上の(a)から(c)に定義されたDを表し;
A1、A2及びA3は独立して例えば直接結合、-C(O)-、-C(S)-、-S(O)-、-S(O)2-又は-P(O)2-を表し、但し、A3は、D'及びD2が共に独立してA3への連結点でO又はNHによって終端する場合、直接結合を表さない。A1及びA2基はGnRH類似体又はホルモン部分に内因性の原子、例えばGnRH類似体又はホルモン部分のLys残基のN原子及びホルモン部分のヒドロキシル基のO原子に結合していることは明らかである。
【0031】
結合基はGnRH類似体及びホルモン部分に何れかの配向で結合されうる(つまり、結合基の基A1はGnRH類似体の原子かホルモン部分の原子の何れかに結合されうる)。
ここで定義されるアルキレン、アルケニレン又はアルキニレン基は1から12(例えば1から8、例えば1から6)のC原子を含み得、直鎖であり得、又は十分な数(つまり、アルキレン及びアルケニレンでは最小2つ、アルキニレンでは4)の炭素原子がある場合は、分枝状鎖であってもよい。
ここで使用される場合、「炭素環」という用語には、炭素骨格を有する環状構造であり、3又はそれ以上の数の一又は複数の環を含む基、例えば3から8員の単環環系、7から12員の二環環系及び12から18員の多環(例えば三又は四環)環系が含まれる。更に、各炭素環は完全に飽和しているか、部分的に飽和しているか、又は完全に又は部分的に芳香性でありうる。完全に飽和した炭素環の例にはシクロペンチル、シクロヘキシル及びシス-及びトランス-デカリニル等が含まれる。部分的に不飽和の炭素環の例にはシクロヘキセニル等が含まれる。部分的に芳香性の炭素環の例には、インデニル及び1,2,3,4-テトラヒドロナフチル等々が含まれる。完全に芳香性の炭素環の例にはフェニル、ナフチル等々が含まれる。
【0032】
ここで使用される場合、「複素環」という用語には、上述の炭素環基であり、環C原子の一又は複数(例えば1から3)が対応する数のヘテロ原子によって置換され、各ヘテロ原子が独立してO、S及びN(又は関連する場合はNH)から選択される基が含まれる。複素環基の例には、アゼチジニル、ベンゾジオキサニル、ベンゾジオキセパニル、ベンゾジオキソリル、ベンゾフラニル、ベンゾフラザニル、ベンズイミダゾリル、ベンゾモルホリニル、ベンゾチアゾリル、ベンゾチオフェニル、ベンゾキサゾリル、クロマニル、シンノリニル、クマリニル、ジオキサニル、フラニル、ヒダントイニル、イミダゾリル、イミダゾ[1,2-a]ピリジニル、インドリル、イソキノリニル、イソキサゾリル、マレイミド、モルホリニル、オキサゾリル、フタラジニル、ピペラジニル、ピペリジニル、プリニル、ピラニル、ピラジニル、ピラゾリル、ピリジニル、ピリミンジニル、ピロリジノニル、ピロリジニル、ピロリニル、ピロリル、キナゾリニル、キノリニル、3-スルホレニル、テトラヒドロピラニル、テトラヒドロフラニル、チアゾリル、チエニル、チオクロマニル、トリアゾリル等が含まれる。
【0033】
挙げることができる本発明の実施態様には、結合基が式-A1-D-A2-によって表されるものが含まれ、ここで、
DはC1−6アルキレン、C2−6アルケニレン又は-D1-A3-D2-を表し;
D1は-S-S-によって中断されていてもよいC2−6アルキレンを表し;
D2はC2−8アルキレンを表し;
A1及びA2は共にC(O)を表し;
A3はC(O)NHを表す。
挙げることができる特定の結合基には、C(O)-(CH2)2-C(O)(スクシニル)及びC(O)-(CH2)2-S-S-(CH2)2-C(O)NH-(CH2)6-NHC(O)が含まれる。
挙げることができる本発明の実施態様には、結合基がペプチドGnRH類似体中のLys残基のε-アミノ基をホルモン部分上のヒドロキシル基に連結するものが含まれる。
【0034】
GnRH類似体をホルモン部分又はその誘導体に結合させるために適した方法及び化学は当業者に知られており、実施例1に記載された方法が含まれる。リンカーを介して他の化学構造にGnRH類似体を結合させる方法はRahimipour等(2001)及びRussell Jones等(1995)に記載されている。本発明のコンジュゲートを形成する際に応用できるビタミンB12コンジュゲートを製造する方法はMcEwan等(1999)に記載されている。化学コンジュゲートの形成に関連しているこれらの3件の文献の開示全体は出典明示によりここに取り込まれる。
GnRH類似体は、例えばO'Sullivan等 Anal. Biochem. (1979) 100, 100-108に一般的に記載されたもののような、常套的な分子の架橋方法の何れかによってホルモン部分又はその誘導体に結合させることができる。例えば、一部分はチオール基に富んだものとされ得、他の部分はチオール基と反応可能な二官能性剤、例えばヨード酢酸のN-ヒドロキシスクシンイミドエステル(NHIA)又はN-ヒドロキシスクシンイミジル-3-(2-ピリジルジチオ)プロピオネート(SPDP)、結合種間にジスルフィド架橋を導入するヘテロ二官能性剤と反応させることができる。例えばm-マレイミドベンゾイル-N-ヒドロキシスクシンイミドエステルで達成されるアミド及びチオエーテル結合は、ジスルフィド結合よりもインビボで一般により安定である。
【0035】
更に有用な架橋剤には、第1級アミノ基を含む化合物に保護されたチオール官能基を導入するために使用される試薬であるS-アセチルチオグリコール酸N-ヒドロキシスクシンイミドエステル(SATA)が含まれる。アセチル化チオール基の脱保護は穏やかな条件で(Julian等(1983) Anal. Biochem. 132, 68)、つまりジメチルスベリミデート(dimethylsuberimidate)二塩酸塩及びN,N'-o-フェニレンジマレイミドとの反応によって達成される。
他の実施態様では、GnRH類似体はN末端アミン基を介してホルモン部分に結合させられる。
有利には、ここに記載された化合物は天然GnRHよりもインビボでの代謝又は腎臓クリアランスによる影響をあまり受けない、つまりインビボで長い半減期を有する。これは、以下の実施例1に記載されているように、当業者によって容易に決定することができる。
好ましくは、ここに記載の化合物はインビボにおいて天然GnRHよりも長い活性期間を有している。これはまた、例えば以下の実施例1に記載されているようにして、当業者によって容易に決定することができる。
好適な実施態様では、化合物は図1A又は図1Bに示される一般式を持ちうる。
【0036】
本発明には次の化合物が含まれる:
D-Lysの6位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Leu-Arg-Pro-D-Ala-NH2;
D-Lysの6位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAc-ΔPro-D-Fpa-D-Trp-Ser-Tyr-D-Lys-Leu-Arg-Pro-Gly-NH2;
Lysの7位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Lys-Leu-Arg-D-Ala-NH2;
N末端アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したD-Pal-Ser-Arg-D-Lys-Leu-Arg-Pro-D-Ala-NH2;
Lysの7位のε-アミンで21-ヒドロキシプロゲステロン21-スクシネートに結合したAcD-Nal-D-Cpa-D-Pal-Ser-Arg-D-Lys-Lys-Arg-Pro-D-Ala-NH2;
Lysの6位のε-アミン基で11α-ヒドロキシプロゲステロン11-スクシネートに結合した[DLys6]GnRH;
Lysの6位のε-アミン基で21α-ヒドロキシプロゲステロン21-スクシネートに結合した[DLys6]GnRH;
Lysの6位のε-アミン基でβ-エストラジオール17-スクシネートに結合した[DLys6]GnRH。
【0037】
更なる実施態様では、化合物は血漿ホルモン結合タンパク質に結合したホルモン部分又はその誘導体に結合したGnRH類似体を含む。結合タンパク質はCBG又はSHBGのようなホルモン特異的結合タンパク質であり得、又はHSAであり得る。
この実施態様に係る化合物は、投与時に直ぐに結合タンパク質の保護効果から恩恵を受け、第一回の通過で腎臓を経由するGnRH類似体の分泌を減少させ、よってGnRH類似体の半減期及び活性を更に延ばすために、有利である。
【0038】
本発明の第二の側面では、本発明の第一の側面に係る化合物と薬学的に許容可能な賦形剤、担体又は希釈剤を含有する薬学的組成物を提供する。
一実施態様では、薬学的組成物は経口投与に適している。
本発明の化合物は、無毒性の有機もしくは無機の酸もしくは塩基の付加塩の形態であってもよい、有効成分を含む薬学的製剤の形で、経口的又は任意の非経口経路により通常投与することができる。治療される疾患及び患者並びに投与経路に応じて、組成物を様々な用量で投与することができる。
ヒトの治療では、本発明の化合物は、単独で投与することができるが、一般には、目的とする投与経路及び標準的医薬規範を考慮して選択した適切な薬学的賦形剤、希釈剤又は担体と混合されて投与される。
【0039】
例えば、本発明の化合物は、即時、遅延又は徐放用途に対して、香味添加剤又は着色剤を含みうる錠剤、カプセル剤、卵剤、エリキシル剤、溶液又は懸濁液の形態で経口的、口腔内、又は舌下に投与することができる。また、本発明の化合物は、鼻腔内注入により投与してもよい。
そのような錠剤は賦形剤、例えばマイクロクリスタリンセルロース、ラクトース、クエン酸ナトリウム、炭酸カルシウム、二塩基性リン酸カルシウム及びグリシン、崩壊剤、例えばデンプン(好ましくはコーン、ポテト又はタピオカデンプン)、デンプングリコール酸ナトリウム、クロスカルメロース(croscarmellose)ナトリウム及びある種のケイ酸錯体、及び顆粒化バインダー、例えばポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ-プロピルセルロース(HPC)、スクロース、ゼラチン及びアカシアを含みうる。加えて、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリル、タルクなどの潤滑剤を含みうる。
同様なタイプの固形組成物はゼラチンカプセル中にフィラーとしても用いることができる。この点で好適な賦形剤には、ラクトース、デンプン、セルロース、ミルク糖又は高分子量ポリエチレングリコールが含まれる。水性懸濁液及び/又はエリキシルに対しては、本発明の化合物は様々な甘味料又は香味添加剤、着色物質又は染料、乳化及び/又は懸濁化剤、及び水、エタノール、プロピレングリコール及びグリセリンのような希釈剤、及びその組み合わせと組み合わせることができる。
【0040】
本発明の化合物は、非経口的に、例えば、静脈内、動脈内、腹腔内、腹腔内、くも膜下腔内、脳室内、胸骨内、頭蓋内、筋肉内又は皮下的に投与することもでき、あるいは点滴法により投与することができる。それらは、他の物質、例えば、溶液を血液と等張性にするのに十分な塩類又はブドウ糖を含んでいてよい無菌水溶液の形で用いるのが最もよい。水溶液は、必要な場合、適切に(好ましくは3〜9のpHとなるように)緩衝されなければならない。無菌条件下での適切な非経口製剤の調製は、当業者によく知られている標準的な製薬技術により容易に達成される。
一実施態様では、薬学的組成物又は製剤は、活性成分、つまり本発明の第一の側面で上述された化合物の毎日の用量又は単位、毎日の下位用量又はその適切なフラクションを含む単位投薬形態である。
他の実施態様では、薬学的組成物又は製剤は、注射可能なデポー剤のような徐放製剤である。
【0041】
非経口投与に適する製剤には、抗酸化剤、緩衝剤、静菌剤及び製剤を対象とする受容者の血液と等張性にする溶質を含みうる水性及び非水性無菌注射溶液;並びに懸濁剤及び増粘剤を含んでいてよい水性及び非水性無菌懸濁液が含まれる。製剤は、単位用量又は複数回用量容器、例えば、密閉アンプル及びバイアル入りで提供することができ、また、使用直前に無菌液体担体、例えば、注射用水の添加のみを必要とする凍結乾燥状態で貯蔵することができる。即時注射溶液及び懸濁液は、前述の製剤の既に記載された無菌散剤、顆粒剤及び錠剤から調製することができる。
現在、GnRHアゴニストは典型的には一日当たり約100μgで患者に投与される一方、GnRHアンタゴニストは典型的には一日当たり約1mgで患者に投与される。ヒトの患者への経口及び非経口投与に対しては、本発明の化合物の毎日の用量レベルは、通常、単一又は分割用量で投与される等価又は低レベルのGnRH類似体を含む。
よって、例えば、本発明の化合物の錠剤又はカプセルは、必要に応じて、一度に一回又は二回又はそれ以上の投与のために50μgから1mgの活性化合物を含みうる。とにかく、医師は各個々の患者に最も適した実際の用量を決定し、特定の患者の年齢、体重及び反応と共に変化する。上記用量は平均的な場合の例である。もちろん、高用量又は低用量範囲が有利な個々の例があり得、それも本発明の範囲内にある。
【0042】
本発明の化合物は、鼻腔内に、又は吸入で投与することもでき、適切な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、1,1,1,2-テトラフルオロエタン(HFA134ATM)又は1,1,1,2,3,3,3-ヘプタフルオロプロパン(HFA227EATM)等のヒドロフルオロアルカン、二酸化炭素又は他の適切なガスを用いた加圧容器、ポンプ、噴霧器もしくはネブライザーから、乾燥粉末吸入剤もしくはエアゾールスプレー提供形態で簡便に送達される。加圧エアゾールの場合には、計量された量を送達するためのバルブを設けることにより用量単位を決定することができる。加圧容器、ポンプ、噴霧器もしくはネブライザーは、潤滑剤、例えばトリオレイン酸ソルビタンをさらに含んでいてよい、例えば、溶媒としてのエタノールと噴射剤との混合物を用いた活性化合物の溶液又は懸濁液を含んでいてよい。吸入器又は注入器に用いるカプセル及びカートリッジ(例えば、ゼラチン製)は、本発明の化合物とラクトース又はデンプンのような適切な粉末基剤との粉末混合物を含むように製剤化することができる。
【0043】
エアゾール又は乾燥粉末製剤は、各計量用量すなわち「一吹き量」が患者に送達される本発明の化合物を少なくとも1mg含むように調製することが好ましい。エアゾールの総1日投与量は患者ごとに異なり、1日1回又は通常、分割して投与することができることは理解できよう。
あるいは、本発明の化合物は、座薬又はペッサリーの形態で投与することができ、あるいはローション、溶液、クリーム、軟膏又は粉剤の形態で局所適用してもよい。本発明の化合物は、例えば、皮膚パッチを用いて経皮投与することもできる。
皮膚に局所投与するために、本発明の化合物は、例えば、一又は複数の以下のもの:鉱油、流動ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックス及び水との混合物に、懸濁又は溶解した活性化合物を含む適切な軟膏剤として製剤することができる。あるいは、例えば、次の一又は複数の以下のもの:鉱油、モノステアリン酸ソルビタン、ポリエチレングリコール、流動パラフィン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2-オクチルドデカノール、ベンジルアルコール及び水との混合物に懸濁又は溶解した適切なローション又はクリームとしても製剤することができる。
【0044】
口中への局所適用に適した製剤には、通常はスクロース及びアカシア又はトラガカントである香味ベースに活性剤を含むトローチ剤;ゼラチン及びグリセリン、又はスクロース及びアカシアのような不活性な基剤中に活性成分を含む芳香錠(pastilles);及び適切な液体担体に活性成分を含む洗口剤が含まれる。
一般に、ヒトにおいて、本発明の化合物の経口又は局所投与が好ましい経路であり、最も簡便である。受容者が嚥下疾患又は経口投与後の薬物吸収障害を被っている状況では、薬剤は非経口的に、例えば舌下的に又は口腔的に投与することができる。
獣医学用途については、本発明の化合物は、通常の獣医学規範に従って適切に許容できる製剤として投与され、獣医は特定の動物に最も適する投与計画と投与経路を決定する。製剤は薬剤的製剤であることが都合がよい。有利には、獣医学用途については、製剤は獣医学的製剤である。
【0045】
本発明の第三の側面は、医薬品における使用のために、本発明の第一の側面による化合物又は本発明の第二の側面による薬学的組成物を提供する。
よって、化合物又は薬学的組成物は医薬品における使用のために包装され提供される。
本発明の第四の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物を個体に投与することを含んでなる、個体の繁殖性を減少させる方法を提供する。
GnRHアゴニスト及びGnRHアンタゴニストコンジュゲートの双方がゴナドトロピンの放出を阻害することによって個体において繁殖性を低減するために使用することができることが理解される。
本発明の第五の側面は、個体の繁殖性を低減させるための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
【0046】
典型的には、また好ましくは、治療される個体はヒトである。しかしながら、本発明の方法は、例えば次の種:ウシ、ウマ、ブタ、ヒツジ、ネコ及びイヌ、並びに他の霊長類、旧世界サル及び新世界サルを治療するために使用することができる。よって、本方法はヒトと獣医用医薬の両方に用途がある。特に、その獣医用途に対しては、コンジュゲートを用いて家畜、ウマ及び家庭内動物における去勢状態をつくり出すことができる。
雌における「繁殖性を低減する」は、受胎の可能性又は成功裏の妊娠を減じ、又は受胎又は成功裏の妊娠を防止するという意味を含む。よって、本発明は雌の避妊方法を含む。
雄における「繁殖性を低減する」は、テストステロンレベルを去勢レベルまで減じるという意味を含む。よって、本発明は雄の避妊方法を含む。
ここに記載された避妊方法は化合物、薬学的組成物又は医薬の投与の休止により可逆である。
【0047】
本発明の第六の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物をそれを必要とする個体に投与することを含んでなる、ホルモン依存性疾患又は症状と闘う方法を提供する。
疾患又は症状と「闘う」には、症状の徴候を軽減し(つまり対症用途)、又は疾患又は症状を治療し、又は疾患又は症状を防止する(つなり予防用途)意味が含まれる。
本発明の第七の側面は、必要とする個体におけるホルモン依存性疾患又は症状と闘うための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
本発明の方法、使用、化合物及び薬学的組成物によって闘うのに適したホルモン依存性疾患又は症状には、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維、月経前症候群、多嚢胞性卵巣症候群、多毛症、ざ瘡尋常性、思春期早発症、急性間欠ポルフィリン症、睾丸停留及び遅延思春期が含まれる。
本発明による治療に適したホルモン依存性癌はGnRHレセプターを発現し、乳癌、前立腺癌、子宮癌、子宮体癌、卵巣癌及び精巣癌を含む。
【0048】
本発明の第八の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物をそれを必要とする個体に投与することを含んでなる、不妊症と闘う方法を提供する。
本発明の第九の側面は、それを必要とする個体における不妊症と闘うための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
本発明の化合物は、特に補助生殖医療における排卵の誘導において、外因性ゴナドトロピンの制御された投与と共に内因性ゴナドトロピンの阻害によって不妊症と闘うために使用することができる。
よって、本発明の化合物は体外受精(IVF)技術において有用性を有している。
【0049】
本発明の第十の側面は、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物を個体に投与することを含んでなる、インビボでゴナドトロピン又は性ホルモンの産生を調節する方法を提供する。
「調節」には増大、減少又は阻害を含む。
本発明の第十一の側面は、インビボでゴナドトロピン又は性ホルモンの産生を調節するための医薬の調製における、本発明の第一の側面に係る化合物又は本発明の第二の側面に係る薬学的組成物の使用を提供する。
例えば、受胎治療において、本発明の化合物を用いて完全に内因性のホルモン産生を阻害することができ、これがついでコンジュゲートの一部として又は別個に所望に応じて置き換えられる。受胎治療における使用のためのホルモン置換の量、頻度及び期間はこの分野の当業者にはよく知られており、とにかく医師によって決定される。
本発明のコンジュゲート化合物はまたGnRHレセプターを発現する細胞、例えば幹細胞、及びリンパ球のような免疫細胞の分化又は脱分化のためにインビトロで又はより少ない頻度でインビボで使用することもできる。
【0050】
本発明の第十二の側面は、GnRHと比較して増加したインビボ半減期を持つように、GnRH類似体を修飾する方法を提供し、該方法は、血漿ホルモン結合タンパク質に結合可能なホルモン部分又はその誘導体にGnRH類似体を結合させることを含んでなる。
本発明の第十三の側面は、GnRHと比較して増加したインビボ活性の期間を持つように、GnRH類似体を修飾する方法を提供し、該方法は、血漿ホルモン結合タンパク質に結合可能なホルモン部分又はその誘導体にGnRH類似体を結合させることを含んでなる。
好ましくは、GnRH類似体とホルモン部分又はその誘導体は結合基を介して結合される。
本発明の第十三及び第十四の側面の実施態様では、上記方法は、ホルモン部分に結合させられたGnRH類似体又はその誘導体を血漿ホルモン結合タンパク質に結合させることを含む。典型的には、この結合は、ホルモン部分に結合させられたGnRH類似体又はその誘導体を溶液中の血漿ホルモン結合タンパク質と接触させることによって実施される。
【0051】
GnRH類似体、ホルモン部分又はその誘導体、結合基、コンジュゲーションを実施する方法の好ましい例は実施例1及び本発明の第一側面に関して上に記載した通りである。
一実施態様では、本発明の第十三及び第十四の側面はまた結合されたGnRH類似体のインビボ半減期又は活性の期間を決定する工程を含む。典型的には、該方法は結合されたGnRH類似体の決定されたインビボ半減期又は活性の期間を、GnRHのインビボ半減期又は活性の期間と比較する工程を更に含む。
場合によっては、上記方法はGnRHの活性の期間又はインビボ半減期を定量する工程をまた含む。あるいは、過去に測定された値を使用することができる。
本発明を次の図面と実施例を参照しながら以下により詳細に説明する。
【0052】
図8で用いた略語を以下に示す:
DSer(tBu):D−Ser t−ブチルエーテル;
DHis(ImBzl):D−Hisベンジルイミダゾール;
NEt:N−エチルアミド;
DNal:D−ナフチルアラニン;
DCit:D−シトルリン;
DhCit:D−ホモシトルリン;
DhArg(Et)2:D−ジエチルホモアルギニン;
NMeTyr:N−メチルチロシン;
Aph(Hor):
DAph(Cba):
DGlu(AA):
【0053】
実施例1:GnRHホルモン結合体の合成及び性質
方法
GnRH−ステロイド結合体の合成
Mattox, Litwiller及びNelson9とRajkowski及びCittanova14からの結合方法を用いた。Amersham Pharmacia Biotech UK Limited (Little Chalfont, Buckinghamshire)から購入した放射性化学薬品を除き特に明記しない限りSigma-Aldrich Company Ltd. (Poole, Dorset)より全ての化学薬品を入手した。GnRHアゴニスト[D−Lys6]GnRH又は2つのGnRHアンタゴニストはJ. Rivier (Salk Institute, La Jolla, California)より快く提供していただいた。[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、アンタゴニスト1とする、及び[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、アンタゴニスト2とする、を0.1Mのリン酸緩衝液(pH7.0)に溶解して等量のDMFを加えた。20倍以上のステロイド([D−Lys6]GnRHの場合は11αヒドロキシプロゲステロン11−半コハク酸塩、GnRHアンタゴニスト結合体には21−ヒドロキシプロゲステロン21−半コハク酸塩)を等モルの1−ヒドロキシベンゾトリアゾール(HOBt)及びN,N’−ジシクロヘキシルカルボジイミド(DCC)を含む無水DMFに溶解した。混合液をボルテックスにかけ、1時間室温に放置した。溶液を含むステロイドを各ペプチド溶液50μlの一定分量に移した。トリブチルアミンでpH>7に調整した後、ペプチド−ステロイド混合液を20時間4℃で放置した。
【0054】
生成物の生成及び同定
HPLC
生成物のHPLC及び質量分析を行う前に、エチルアセテートの後ヘキサフルロプロパノール/DMF(70:30)をSep−Pak C18カートリッジ(Millipore UK Ltd., Harrow, Middlesex)に通して一次精製を行った。Beckman Coulter System Gold(登録商標)LC125ポンプ及びLC168ダイオードアレイ検出器に接続したNovapak C18カラム(4μmビーズ、3.9×150mm)で分析用RP−HPLCを行った。緩衝液系は、0.1%TFA/水をバッファーA、0.1%TFAを含むアセトニトリルをバッファーBとした。10%から100%のバッファーB勾配液を用いて流量1ml/分で30分以上カラムを展開した。
質量分析
α−シアノ−4−ヒドロキシ桂皮酸の基質を用いたTofspec 2E MALDI−TOF質量分析器(Micromass UK Ltd.)により質量分析を行った。
【0055】
細胞培養
10%胎仔ウシ血清、グルタミン及びペニシリン/ストレプトマイシン(通常培地)を含むDMEMにてCOS−7及びCOS−1細胞を維持した。全細胞レセプター結合アッセイのためにSuperfect形質移入試薬(Qiagen, Crawley, West Sussex)を用いて細胞にヒトGnRHレセプター(hGnRH−R)を形質移入して、好適培地(Invitrogen Life Technologies, Paisley, Scotland)に4時間置いた。更に通常培地(上記参照)に48時間置いた後に形質移入細胞を分析した。
hGnRHーR形質移入COS−1細胞にてGnRHアゴニストープロゲステロン結合体の膜結合アッセイを行った。100mmディッシュに入れたCOS−1細胞をHEPES修飾DMEMにて2度洗浄し、HEPES緩衝生理食塩水、ペニシリン/ストレプトマイシン及びDMEMを含むフィルター滅菌したDEAEデキストラン中で15μgのDNAを形質移入した。37度で5時間インキュベートした後、培養液を取り除き、ペニシリン/ストレプトマイシン、2%FCS及び2%クロロキン10mMを含むDMEMと置き換えて、プレートを更に1時間インキュベートした。培養液を吸引して、細胞を洗浄し、通常培地を加えて24時間後にアッセイを行った。
我々の研究室で開発したラットタイプIGnRHRを発現するHEK293安定細胞系を用いてイノシトールリン酸生成物アッセイを行った。この細胞系は、培養の間中G418を500μg/mlで添加した10%胎仔ウシ血清、グルタミン及びペニシリン/ストレプトマイシンを含むDMEMにて維持した。必要であれば、プレートをポリ−L−リジンでコートしてアッセイの間のプラスチック容器に対する吸着を亢進した。
【0056】
レセプター結合アッセイ
GnRHアゴニスト−プロゲステロン結合体の膜結合アッセイ:
形質移入したCOS−1細胞をPBSにて洗浄して、プレートから取り除き、細胞がペレット状になるように1500rpmにて5分間遠心分離を行った。PBSを吸引して、細胞をホモジネート緩衝液(20mMトリス、2mM MgCl2、pH7.2)に懸濁し、ボルテックスにかけた後氷上に10分放置した。細胞懸濁液を7mlホモジネータ(Jencons (Scientific) Ltd., Leighton Buzzard, Buckinghamshire)に移し、ゆるい吸引具(loose plunger)にて15回、きつい吸引具(tight plunger)にて15回突き刺した。そしてホモジナイズした細胞を最高速度、4度で10分間遠心分離を行って、上澄みを吸引ポンプにて除去した。膜に残ったペレットはアッセイ用の緩衝液(40mMトリス、2mM MgCl2、pH7.4)に懸濁して、氷上に置いた。事前に冷やした12mmガラスチューブを、200μlのアッセイ用緩衝液、50μlの細胞膜、アッセイ緩衝液(およそ1チューブ当たり120000CPM)中100μMの125I[His5,D−Tyr6]GnRH、及び漸増濃度の50μlのコールドリガンド(又はBoチューブのアッセイ緩衝液)で満たした。チューブを氷上にて2時間インキュベートした後、3mlの氷冷液体ポリエチレンイミン0.01%(PEE)を加えて、吸引下(1%PEEに予浸)にてWhatman GG/Cガラス繊維フィルター(Whatman International Ltd., Maidstone, Kent)で濾過した。そうしてすぐにガンマ計測器にてフィルターを計測した。
【0057】
GnRHアンタゴニスト−プロゲステロン結合体の全細胞結合アッセイ:
COS−7細胞を12ウェルプレートに播き、アッセイ前の24時間、37度でインキュベートした。細胞をPBSにて2度洗浄して、拮抗的リガンド及び125Iリガンド(125l[His5D−Tyr6]GnRH)を含むDMEM+0.1%BSA中に修飾したHEPES 500μlを添加した。プレートをPBSにて2度洗浄して、500μlの0.1M NaOHを添加して可溶性を高め、20分間振とうした。試料をガンマ計測器にて計測した。
【0058】
全イノシトールリン酸生成物の測定
ラットタイプIGnRH受容性発現HEK293細胞を12ウェルプレートにプレートアウトして、37度、5%CO2で24時間インキュベートし、その後、1%透析FCS(グルタミン及びペニシリン/ストレプトマイシンを含む)及び1μl/ウェルのミオ−[2−3H]イノシトールを含む特別DMEMにて更に48時間インキュベートした。培養液を吸引して培養緩衝液にて洗浄した後、10mM LiClを含む500μl緩衝液を更にプレートに加え、37度で30分間インキュベートした。1μMのアゴニストを最終濃度0.1μMになるように各ウェルに添加し、プレートを同じ条件下で更に1時間インキュベートした。500μlの10mMギ酸にて反応を終了させ、4度で30分間インキュベートした。ギ酸溶液を、500μlの50%AGl×樹脂懸濁液(Bio-Rad Laboratories Ltd, Hemel Hempsted, Hertfordshire)を含む12mmプラスチックチューブに移した。滅菌水(1ml)及び四ホウ酸ナトリウム、ギ酸ナトリウム(1ml、5mM、60mM)を徐々に加え、ボルテックスにて混合し、除去することによってイノシトールリン酸を溶出した。ギ酸、ギ酸アンモニウム(1ml、0.1M、1M)を添加してボルテックスした後、800μlの上澄みをシンチレーション流動にて計測した。
【0059】
血漿タンパク質結合アッセイ
[1,2,6,7−3H]プロゲステロン又は[3H]コルチゾール存在下にて、妊娠中のモルモット血漿又は妊娠中のヒト血清のそれぞれに対するステロイド結合体の拮抗的結合により血漿タンパク質結合を決定した。20μlの血清を2mlのデキストラン−コート木炭溶液(0.25gデキストランT−70、2.5g木炭脱色粉末、活性化酸性洗浄水[Merck Ltd., Lutterworth, Leicestershire]を含む500mlのPBS)で希釈して、室温にてインキュベートした。30分後、懸濁液を3000×gにて10分間遠心分離して、上澄みを取り除き、ペレットを破棄した。希釈した血清100μlを遠心チューブに等分して、その後に1pmol[3H]ステロイド/100μl PBSを入れた。100μl PBS(全結合)又は200pmol/100μl非標識拮抗リガンド(特異的結合)を2通りで希釈血清に添加した。チューブをボルテックスして、室温で1時間インキュベートしてから更に15分間氷上に置いた。更に、750μlのデキストラン−コート木炭懸濁液を添加し、氷上にて10分間インキュベーションした後、4度で5分間遠心分離した。上澄み液を計測した。
【0060】
雄及び雌共通マーモセット(Callithrix Jacchus)におけるインビボ研究
雌マーモセット研究:黄体機能を阻害するために必要な有効量を同定するために、中期黄体期に1.0、0.5又は0.25mgのGnRHアンタゴニスト−ステロイド結合体を1ml生理的食塩水にして皮下的ボーラス投与により2カ所に投与した。 プロゲステロン濃度をRIAによりモニターした。GnRHアンタゴニスト注入の前日に300μlの血液試料を回収した。更に注入1日目とその後3日間の0、4及び8時間後に同量の血液試料を回収した。次の排卵まで1週間に3回、血液試料を採取した。
雄マーモセット研究:GnRHレセプターでの活性持続時間を決定するために、0.5mgのGnRHアンタゴニスト−ステロイド結合体を1ml生理的食塩水にして皮下的ボーラス投与により6匹の成体雄マーモセットの2カ所に投与した。テストステロン濃度をRIAによりモニターした。GnRHアンタゴニスト(0.5mg)を1ml生理的食塩水にして皮下的ボーラス投与により3匹のマーモセットに投与した。テストステロン濃度をモニターした。GnRHアンタゴニスト注入の前日、注入1日目とその後3日間の0、4及び8時間後に300μlの血液試料を回収した。更に続く週に、3つの試料を回収した。
【0061】
雄ウサギにおけるインビボ研究
GnRHアゴニスト−プロゲステロン結合体の生物学的半減期を決定するために、およそ10000000cpmのヨウ化[D−Ala6]GnRH又は[D−Lys6]GnRH−プロゲステロン結合体を含む0.5mlの生理食塩水を雄ウサギの耳静脈内に静脈ボーラス投与により注入した。0.4mlのアセプロン10(Aceprom 10)(Milborrow Animal Health. Republic of South Africa)を実験開始3〜4分前にウサギに筋注して鎮静化した。2時間後に反復注入した。反対側の耳の静脈内に内設したカニューラからのヘパリン処理したチューブで1mlの血液試料を採取した。全血を直接計測して循環からの消失を決定した。Republic of South Africa 調整法に従って全実験を行った。
【0062】
結果
GnRHレセプター結合
GnRHアゴニスト−プロゲステロン結合体は、2.9±1.2×10−10M(標準誤差、n=1、データは示していない)のときED50でタイプIヒトGnRHレセプターに結合した。また、全細胞結合アッセイに示されるようにGnRHアンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体もレセプターに結合した(図2)。非修飾アンタゴニスト配列が1.6±0.4×10−8M(n=4)であるのに対して、アンタゴニストA−プロゲステロンのED50は1.1±0.2×10−7M(n=4)であった(p<0.01;STT)。アンタゴニストB及びアンタゴニストB−プロゲステロンのED50はそれぞれ、4.7±1.1×10−8M(n=4)及び1.1±0.3×10−7M(n=5)であった(p<0.05;STT)。
GnRH−刺激性イノシトールリン酸生成物の阻害
GnRHアゴニスト−プロゲステロン結合体は、5.2 ± 1.4×10−10M(n=2)のEC50でイノシトールリン酸生成を刺激しうる;これはペプチド単独の場合と有意な違いはなかった(STT、p>0.05)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体両方のGnRH拮抗作用を哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸生成物の阻害により確認した(図3)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体のIC50に有意な違いはなかった(p>0.05、STT)。
【0063】
GnRH刺激性イノシトールリン酸生成物の阻害
GnRHアゴニスト−プロゲステロン結合体は、5.2 ± 1.4×10−10M(n=2)のEC50でイノシトールリン酸生成を刺激しうる;これはペプチド単独の場合と有意な違いはなかった(STT、p>0.05)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体両方のGnRHR拮抗作用を哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸生成物の阻害により確認した(図3)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体のIC50はそれぞれ9.7±4.0×10−8M(n=6)及び8.6±2.6×10−8M(n=7)であり、有意な違いはなかった(p>0.05、STT)。アンタゴニストA−プロゲステロン及びアンタゴニストB−プロゲステロン結合体の何れによってもイノシトールリン酸生成物単独の活性化はみられず(データは示していない)、純粋な拮抗作用が確認された。
【0064】
血漿タンパク質結合部位に対する競合作用
モルモット種では高レベルのプロゲステロン結合グロブリン(PBG)が存在するため、妊娠中のモルモット血漿における血漿タンパク質結合を研究した。PBGは特定の形式で[3H]プロゲステロンへの結合がみられ、この結合は、IC50 9.6±1.8×10−8M(n=4)の非標識プロゲステロン、IC50 1.0±0.3×10−6M(n=6)のアンタゴニストA−プロゲステロン結合体、IC50 5.3±1.0×10−7M(n=4)のアンタゴニストB−プロゲステロン結合体により阻害された。PBGはプロゲステロンにしか結合しないので、このステロイド−血漿タンパク質相互作用の特異性は特異的[3H]プロゲステロン結合をコルチゾールが阻害しないことで証明された。また、アゴニスト−プロゲステロン結合体は、非標識のコルチゾール結合をIC50 6.7±2.4×10−9M(n=2)、及びプロゲステロン結合をIC50 7.3±1.5×10−8M(n=3)で阻害するのに対して、妊娠中のヒト血清(通常より高い濃度のCBGHを含む)への[3H]コルチゾール結合をIC50 1.2±0.3×10−6M(n=2)で阻害した(データは示していない)。
【0065】
プロゲステロンの活性化
CAT酵素活性のアッセイにより、すべてのGnRHアンタゴニスト21−ヒドロキシプロゲステロン21コハク酸塩結合体はT47D細胞のプロゲステロンレセプターに結合して活性化することができることを明らかにした。すべての結合体の有効性はプロゲステロンと同様であり、1nMで実質的に活性化されず、1μMに至るまで活性が増加する。試験した5つの結合体を以下の表1に示す:
【0066】
表1:GnRHアンタゴニスト結合体
哺乳動物のGnRHを対照として並べた試験したアンタゴニストのアミノ酸配列を示す。以下の略語を用いた:Glu;グルタミン酸、His;ヒスチジン、Tip;トリプトファン、Ser;セリン、Tyr;チロシン、Gly;グリシン、Leu;ロイシン、Arg;アルギニン、Pro;プロリン、AcD−Nal;アシルD−ナフチルアラニン、D−Cpa;D−クロロフェニルアラニン、D−Pal;D−ピリジルアラニン、D−Lys;D−リジン、D−Ala;D−アラニン、Ac−ΔPro;アシル デルタ−プロリン、D−Fpa;D−フルオロフェニルアラニン、D−Trp;D−トリプトファン。
【0067】
5つすべてのアンタゴニストは、アステリスクで示す異なる位置で同じステロイド、21−ヒドロキシプロゲステロン21−半コハク酸塩に結合する。結合体A及びBは6位のD−Lysのεアミンを介して結合する。結合体C及びEは7位のリジンのεアミンを介して結合する。結合体DのステロイドはD−Pal残基のN末端アミンに結合する。
【0068】
マーモセットのインビボ研究
成体雌マーモセット(cycling adult female marmosets)の研究により、1.0mgのアンタゴニストA−プロゲステロン結合体を摂取された個体では24.8±22.2(n=6)から8日間、0.5mgを投与した個体では21.0±1.2(n=7)から11日間の黄体期の持続期間の減少がみられた。血漿中ののプロゲステロン濃度が30ng/mlに達すると排卵が起こると考えられる。また、0.25mgのアンタゴニストA−プロゲステロン結合体を摂取した3匹目のマーモセットにも血漿中のプロゲステロン濃度の一時的な減少がみられたが、完全な黄体の退行及びそれに続く排卵はこのとき起こらなかった。
雄マーモセット(n=3)に対する0.5mgのペプチドA投与により、血漿中のテストステロン濃度の急激な低下が起こった(図5)。テストステロン濃度の低下は注入後8時間持続したが、24時間までに増加した。相対的に、0.5mgのペプチドA−プロゲステロン結合体(n=6)によってもテストステロン濃度が急速に低下したが、これは注入後少なくとも72時間後まで持続し(注入後24時間でp<0.05)、6日目までに回復した。既存内務省認可の制限があるため第4及び5日目の付加的な血液採取は行えなかった。
【0069】
雄ウサギにおけるインビボ研究
非修飾GnRHアゴニストと比較してヨウ化GnRHアゴニスト−プロゲステロンによる半減期を検討するためにインビボ雄ウサギの実験を設定した(図6)。ヨウ化化合物の消失により、分子の破壊に一致する初期の半減期及び代謝と腎クリアランスを表す第2期の半減期の両方を計測した。GnRHアゴニスト−プロゲステロン結合体の第2期半減期は53±13分(n=3)であった。GnRHアゴニスト[D−Ala6]GnRHの第2期半減期は21±3分(n=2)であった。2つの類似体間の半減期の違いは、統計学的に有意な差はなかったが(p>0.05、STT)、試料の数が多ければ有意になるかもしれない。
【0070】
検討
GnRHはGnRHレセプターに結合するときにコの字型高次構造をとると言われており10、それ故にホルモンは、側鎖又はN末端のアミン基を利用して、中央の6位又は7位又はN末端でD−Lysアミノ酸に結合した。
ステロイド分子の付加の結果生じる如何なる立体構造的障害をも最小限にすることが望まれた。全細胞結合アッセイによって決定されたアンタゴニストA−プロゲステロンに対するアンタゴニストAのED50での右方変動は有意であり(p<0.01、STT)、レセプターに対する親和性の減少が確認された。しかしながら、アンタゴニストB−プロゲステロンに対するアンタゴニストBでは観察されず、立体構造的障害がペプチド配列に依存していることが示唆された。イノシトールリン酸第二伝達系(second messenger system)の解析により、GnRH類似体の修飾がGnRHレセプターでの拮抗作用を潜在的に制限していたかどうかを同定した。これはアンタゴニストA−プロゲステロン又はアンタゴニストB−プロゲステロンの何れかについて観察されなかった。また、GnRHアゴニスト−プロゲステロン結合体のイノシトールリン酸生成物促進能力について検討した。やはり、ステロイド結合体の結果のようにこの系の促進の有意な減少はみられなかった。ゆえに、GnRH類似体−プロゲステロン結合体は重要な化学的修飾にもかかわらず、 GnRHR結合及び拮抗作用を保持していると結論づけた。
【0071】
検討した新規に結合したGnRHアンタゴニストはインビトロアッセイで血漿タンパク質に結合することが示唆され、これはインビボの場合においても同様である。これはウサギでの半減期実験でみられたように、結合したGnRH類似体の半減期の延長をもたらすであろう。結合タンパク質から持続的に放出される非結合の結合体に長く曝露すると、遊離成分が代謝されるにつれてGnRHRに結合するであろう。
この研究により、GnRH類似体−ステロイド分子がGnRH及びプロゲステロンレセプター結合と活性化に関して完全に二官能価であることが示唆される。これはエモディック酸(emodic acid)結合体のGnRH態様のみが官能性であるというRahimipour等22による以前の研究と大きく異なっている。また、これはステロイドへの他の分子の結合についての重要な情報であり、C21を介する化学的修飾がプロゲステロンとそのレセプター間の相互作用に有意な変化をもたらさないことが明らかである。
【0072】
ステロイド結合及び非結合のGnRHアンタゴニストを雄マーモセットに投与すると、CBGへ結合するという心配がなく、活性の持続時間の解析が可能であった。多くの新世界霊長類は糖質コルチコイドに対する見かけの耐性を共有し、全コルチゾールが高値でコルチゾール血漿受容能力が減少している8。ゆえに、コモンマーモセット(Callithrix jacchus)は官能性CBG受容能力をほとんどもたないインビボモデルであり(血漿タンパク質結合アッセイにより確認された、データは示していない)、血漿タンパク質相互作用のない生物活性の解析が可能である。それ故に、テストステロン産生の機能低下の延長はすべて、血漿タンパク質、膜及び脂肪内の蓄積効果との疎水性相互作用により半減期が延長している分子の疎水性増加に起因している可能性がある。この効果を認識することは、ヒトに類似したCBG生理機能を持つ他の霊長類モデルでみられる結果を理解する上で不可欠である。マーモセットの生理機能がヒトと類似しているとともにGnRHアンタゴニストの薬理がより複雑であるため、このデータは価値がある。
【0073】
CBGを産生する種である17雄ウサギで行ったインビボ試験により、プロゲステロン非結合のGnRHアゴニストと比較してGnRHアゴニスト−プロゲステロン結合体の半減期が延長することが示された。用いたGnRHアゴニストは、ここで試験したアンタゴニストの疎水性配列に比べて親水性であった。これは、ウサギの研究では、プロゲステロンへ結合した後に血漿輸送タンパク質に結合することによって半減期が延長した可能性がある。ゆえに、GnRH及びその類似体などの短いペプチド分子の結合体は循環中の半減期を亢進する機能があると考えられる。
【0074】
要約すれば、GnRH類似体−ステロイドホルモン結合体は血漿ステロイド結合タンパク質受容能力を誘導し、それによって、薬物動態学的及び薬力学的にGnRH類似体を修飾するように設定した。GnRH類似体、pGlu−His−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−GlyNH2、及び2つのGnRHアンタゴニスト、[AcD−Nal1,D−Cpa2,D−Pal3,Arg5,D−Lys6,D−Ala10]GnRH、及び[Ac−ΔPro1,D−Fpa2,D−Trp3,D−Lys6]GnRH(それぞれアンタゴニストA及びアンタゴニストBと呼称する)は、ジシクロヘキシルカルボジイミド(DCC)により、デカペプチドのD−リシン6位の側鎖を介してプロゲステロンのC11またはC21にある半コハク酸リンカーに結合した。生成物はダイオードアレイHPLCにより精製し、質量分析法にて定量した。全細胞結合アッセイでは、GnRHアゴニスト−プロゲステロン及びGnRHアンタゴニスト−プロゲステロン結合体はそれぞれ下垂体タイプIヒトGnRHレセプターに結合した。GnRH刺激性イノシトールリン酸産生が阻害されることにより、すべてのアゴニスト及びアンタゴニスト結合体がGnRHレセプターの純粋なアンタゴニストであることが明らかとなった。3つのペプチド−プロゲステロン結合体は、妊娠中のモルモット血清中の血漿タンパク質結合部位に対して[3H]プロゲステロンと拮抗することが示された。アンタゴニストペプチドA−プロゲステロンのIC50は8.5+2.8x10−7M(n=5)であり、アンタゴニストペプチドB−プロゲステロンのIC50は4.5±0.8x10−7M(n=3)、アゴニスト−プロゲステロン結合体のIC50は2.9+1.2x10−10M(n=1)であった。アンタゴニスト−プロゲステロン結合体のインビボ生物活性は、雄マーモセットに皮下注射した後に血漿中のテストステロン濃度が減少することで証明された。雄ウサギにGnRHアゴニスト−プロゲステロンを静注すると、アンタゴニストの半減期がプロゲステロン分子との結合により延長されることが示された。
【0075】
結論として、本発明に従って作製した特定の新規のGnRHハプテン分子を設定し、生成し、インビボ及びインビトロ検討を行った。GnRH類似体を化学的に修飾することにより、インビトロ活性に有意な影響を与えず、雄ウサギでのGnRHアゴニスト−プロゲステロン結合体の半減期は延長した。これら及び類似の分子はペプチド医薬に関する課題のいくつかを克服した。GnRHアンタゴニストをステロイドホルモンに加えて、摂取のための粒子内の担体分子や封入体へのGnRHアンタゴニスト結合体は経口生物学的利用能を亢進し、活性の持続時間を延長する可能性があるであろう。
【0076】
実施例2:GnRH結合体化合物を用いた乳癌の治療
乳癌に罹患した患者に、コハク酸塩連結基を介して21−ヒドロキシプロゲステロンに結合したテベレリクスを、治療部位の活性作用物質の治療的濃度が治療期間を通じて理想的にはEC90の濃度に維持され、好ましくはEC50以下にならないような用量及び頻度で投与する。この治療は、患者の容認性、副作用の回避及び全身的バイオアベイラビリティに応じて、経口的に、あるいはより局所的に行われる。
【0077】
実施例3:獣医的避妊薬としてのGnRH結合体化合物の使用
雌ウマに、コハク酸塩連結基を介して21−ヒドロキシプロゲステロンに結合したGnRHアンタゴニストを、受胎を防ぐような用量及び頻度で投与する。注射により投与する徐放剤として処置を行う。
【0078】
参考文献
1.Anderson RA (2000) Hormonal contraception in the male. Br Med Bull 56(3): 717-728.
2.Christin-Maitre S, Olivennes F, Dubourdieu S, Chabbert-Buffet N, Charbonnel B, Frydman R, and Bouchard P (2000) Effect of gonadotropin-releasing hormone (GnRH) antagonist during the LH surge in normal women and during controlled ovarian hyperstimulation. Clin Endocrinol (Oxf) 52(6): 721-726.
3.Danforth DR, Williams RF, Hsiu JG, Roh Sl, Hahn D., McGuire JL, Hodgen GD 1990 Intermittent GnRH antagonist plus progestin contraception conserving tonic ovarian estrogen secretion and reducing progestin exposure. Contraception 41(6).-6,13-631.
4.Ditkoff EC, Cassidenti DL, Paulson RJ, Saner MV, Paul WL, Rivier J, Yen SSC, Lobo RA (1991) The gonadotropin-releasing hormone antagonist (Nal-Glu ) acutely blocks the luteinizing hormone surge but allows for resumption of folliculogenesis in normal women. Am J Obs Gyne 165(6 Pt 1):1811-1817.
5.Fraser HM, Nestor JJ Jr, Vickery B H (1987) SSuppression of luteal function by a luteinizing hormone-releasing hormone antagonist during the early luteal phase in the stumptailed monkey and the effects of subsequent administration of human chorionic gonadotropin. Endocrinol 121(2):612-628.
6.Fraser HM, Lunn SF, Cowen GM, Smith KB, Conn PM, (1991) Effect of late follicular phase administration of amide on ovulation and inhibin secretion in macaques. Contraception 44(6):667-676.
7.Kenigsberg D, Hodgen GD, (1986) Ovulation inhibition by administration of weekly gonadotropin-releasing hormone antagonist. J Clin Endocrinol Metab 62(4):734-738.
8.Klosterman LL, Murai JT, Siiteri PK (1986) Cortisol levels, binding, and properties of corticosteroid-binding globulin in the serum of primates. Endocrinol 118(1):424-434.
9.Mattox VR, Litwiller RD, Nelson AN (1979) A comparison of procedures for attaching steroidal glucosiduronic acids to bovine serum albumin. J Steroid Biochem 10.167-172.
10.Millar RP, Assefa D, Ott T; Pawson A, Troskie B, Wakefield L, Katz A GnRH and GnRH analogues: Structure, actions and clinical applications. Horm Frontier Gynecol 5(4):77-83.
11.Nestor JJ, Tahilramani R, Ho TL, Goodpasture JC, Vickery B H, Ferrandon P (1992) Potent gonadotropin releasing hormone antagonists with low histamine-releasing activity. J Med Chem 35(21):3942-3948
12.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Vale WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
13.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Valc WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
14.Pavlou SN, Brewer K, Farley G, Lindner J, Bastias M, Rogers J, Swift LL, Rivier JE, Vale WW, Conn PM, Herbert CM (1991) Combined administration of a gonadotropin-releasing hormone antagonist and testosterone in men induces reversible azoopermia without loss of libido. J Clin Endo Metab 73(6):1360-1369.
15.Russell-Jones GJ, Westwood SW, Farnworth PG, Findlay JK, Burger HG, (1995) Synthesis of LHRH antagonists suitable for oral administration via the Vitamin B12 uptake system. Bioconjugate Chem 6:34-42.
16.Siiteri PK, Murai JT, Hammond GL, Nisker JA, Raymoure WJ. Kuhn RW (1982) Tlie serum transport of steroid hormones Rec Prog Horm Res 38:457-510.
17.Seal US, Doe RP (1965) Vertebrate distribution of Cortlcosteroid-binding globulin and some endocrine effects on concentration. Steroids 5:827-841.
18.Swerdloff RS, Bagatell CJ, Wand C, Anawalt BD, Berman N, Steiner B, Bremner WJ (1998) Suppression of spermatogenesis in man induced by Nal-Glu gonadotropin releasing hormone antagonist and testosterone enanthate (TE) is maintained by TE alone. J Clin Endo Metab 83(10):3527-3533.
19.Westphal U (1993) Steroid-protein interaction: From past to present. J Steroid Biochem 19(1):1-15.
20.McEwan JF, Veitch ES, Russell-Jones GJ (1999) Synthesis and Biological Activity of Ribose-5'-Carbamate Derivatives of Vitamin B12. Bioconjugate Chem 20, 10, 1131-1136.
21.Alsenz J, Russell-Jones GJ, Westwood S, Levet-Traft B, de Smidt PC (2000) Oral Absorption of Peptides Through the Cobalamin (Vitamin B12) Pathway in the Rat Intestine. Pharmaceutical Research 21, Vol. 17, No. 7, 2000.
22.Rahimipour S, Ben-Aroya N, Fridkin M, Koch Y (2000) Design, Synthesis, and Evaluation of a Long-Acting, Potent Analogue of Gonadotropin-Releasing Hormone. J. Med. Chem. 22, 44, 3645- 3652.
23.Thau (1984) Luteinizing hormone-releasing honnone (LHRH) and its analogs for contraception in women: a review. Contraception, 29(2): 143-162.
24.Millar R.P. (2003) Gonadatropin-releasing hormones and their receptors, Chapter 1, Reproductive Medicine: Molecular, Cellular and Genetic Fundamentals, Eds. B.C.J.M. Fauser et al, The Parthenon Publishing Group, New York, USA. ISBN: 1-84214-019-1.
25.Millar R.P. 等 (2000) Progress towards the developmnent of nonpeptide orallyrnactive gonadatropin-releasing hormone (GnRH) antagonists: therapeutic implications. Br. Med. Bull. 56(3): 761-772.
26.Burton & Westphal (1972) Steroid hormone-binding proteins in blood plasma. Metabolism 21(3): 253-276.
27.Cunningham G.R. 等 (1981) Steroid structural requirements for high affinity binding to human sex steroid binding protein (SBP). Steroids 38(3) : 243-262.
28.Mickelson K.E. 等 (1981) Steroid-protein interactions. Human corticosteroid binding globulin: Some physiochemical properties and binding specificity. Biochemistry 20: 6211-6218.
【図面の簡単な説明】
【0079】
【図1】A:あるGnRHアンタゴニストステロイド結合体の構造を示す。結合体は、21−ヒドロキシプロゲステロン21−半コハク酸塩のカルボキシル基を有するペプチドの6位でD−リジン側鎖アミンの縮合によって生成された。ペプチドの残りの側鎖は1−5及び7−10の数で表す(21−ヒドロキシプロゲステロン21−半コハク酸塩も又、デオキシコルチコステロン21−半コハク酸塩及び21−ヒドロキシ−4−プレグネン−3,20−ジオン21−半コハク酸塩として知られている)。B:GnRHアゴニスト11−ヒドロキシプロゲステロン11−コハク酸塩の構造を示す。
【図2】GnRHレセプター結合親和性を示す。125I−GnRHアゴニストの置換はヒトGnRHレセプターを形質移入したCOS−7細胞全体に結合した。ペプチドAGnRHアンタゴニスト(○)、ペプチドAプロゲステロン結合体(●)、ペプチドBGnRHアンタゴニスト(□)、及びペプチドBプロゲステロン結合体(■)。
【図3】ラットI型GnRHRを安定して発現するHEK293細胞における哺乳動物GnRH(0.1μM)刺激性イノシトールリン酸産生に対するペプチドAプロゲステロン結合体及びペプチドBプロゲステロン結合体の影響を示す。
【図4】妊娠中のモルモットに対する[1,2,6,7−3H]の結合に対するプロゲステロン(○)、ペプチドAプロゲステロン(●)及びペプチドBプロゲステロン(◆)の影響を、デキストランコート木炭懸濁液にて測定して示す。
【図5】0.5mgペプチドAプロゲステロン(○)又は0.5mgペプチドA(■)の何れかを皮下注射した雄マーモセットのテストステロン濃度を示す。
【図6】およそ15000000cpmの125I−GnRHアゴニスト−プロゲステロン結合体又は[125I−D−Ala6]GnRHを含む500μlの生理食塩水を耳静脈内に静脈内注入した2匹の雄ウサギの全血中に存在する放射性標識したGnRHアゴニスト−プロゲステロン(■)及び[D−Ala6]GnRH(▲)を示す。3.5時間を超えると全血から消失した。
【図7】T47D細胞におけるプロゲステロンレセプターに連結したCATリピート遺伝子の活性に対する結合体A、B、C、D及びEとプロゲステロンの影響を、CAT活性を測定することによって示す。
【図8】好ましいGnRH類似体の配列を示す。黒い円はGnRH類似体がGnRHと同じ位置に同じアミノ酸を持つことを示す。
【特許請求の範囲】
【請求項1】
ホルモン成分に共役した性腺刺激ホルモン放出ホルモン(GnRH)類似体又はその誘導体を含んでなり、血漿ホルモン結合タンパク質に結合可能な化合物。
【請求項2】
GnRH類似体がペプチド類似体である、請求項1に記載の化合物。
【請求項3】
GnRH類似体がノナペプチド又はデカペプチドである、請求項2に記載の化合物。
【請求項4】
GnRH類似体のアミノ酸残基の一つがD−アミノ酸である、請求項1ないし3の何れか一に記載の化合物。
【請求項5】
D−アミノ酸がD−Lysである、請求項1ないし4の何れか一に記載の化合物。
【請求項6】
D−アミノ酸が6位である、請求項4又は5の何れかに記載の化合物。
【請求項7】
GnRH類似体がGnRHアンタゴニストである、請求項1ないし6の何れか一に記載の化合物。
【請求項8】
GnRHアンタゴニストが[AcD−Nal1,D−Cpa2, D−Pal3,Arg5, D−Lys6,D−Ala10]GnRH、又は[Ac−ΔPro1,D−Fpa2,D−Trp3,D−Lys6]GnRHである、請求項7に記載の化合物。
【請求項9】
GnRHアンタゴニストがセトロレリクス、ガニレリクス、アバレリクス、アンチド、テベレリクス、FE200486、Na−Glu、A−75998、A−76154、A−84861、D−26344、D−63153、D21775、ラモレリクス、デガレリクス、NBI−42902、Org−30850、デチレリクス、イツレリクス、TAK−013、TAK810、AN 207、AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;Ac−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;AcD−Nal−DCpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;[D−Pyr1,D−Phe2,D−Trp3−6]GnRH;D−Lys6アンチド;Lys5アンチド、又はLys8アンチドである、請求項7に記載の化合物。
【請求項10】
GnRH類似体がGnRHアゴニストである、請求項1ないし6の何れか一に記載の化合物。
【請求項11】
GnRHアゴニストがpGlu−His−Trp−Ser−Tyr−D−lys−Leu−Arg−Pro−GlyNH2、ロイプロン、ゾラデックス、サプレリン、シナレル、トリプトレリン、ブセレリン、ロイプロリド、ゴセレリン、デスロレリン、ProMaxx-100、アボレリン、ヒストレリン、ナファレリン、ロイプロレリン、又はトリプトレリンである、請求項10に記載の化合物。
【請求項12】
ホルモン成分がステロイドホルモン成分である、請求項1ないし12の何れか一に記載に記載の化合物。
【請求項13】
ステロイドホルモン成分がエストラジオール、プロゲステロン、コルチゾール、コルチコステロン、エストロン、テストステロン、又はジヒドロキシテストステロンである、請求項12に記載の化合物。
【請求項14】
プロゲステロン誘導体が11α−ヒドロキシプロゲステロン又は21−ヒドロキシプロゲステロンである、請求項13に記載の化合物。
【請求項15】
化合物がホルモン成分又はそれらの誘導体のインビボホルモン活性を維持している、請求項1ないし14の何れか一に記載の化合物。
【請求項16】
化合物がホルモン成分又はそれらの誘導体のインビボホルモン活性を持たない、請求項1ないし14の何れか一に記載の化合物。
【請求項17】
ホルモン成分がインビボで血漿ホルモン結合タンパク質に結合する、請求項1ないし16の何れか一に記載の化合物。
【請求項18】
ホルモン結合タンパク質がグロブリンである、請求項1ないし17の何れか一に記載の化合物。
【請求項19】
血漿ホルモン結合タンパク質がコルチゾール結合グロブリン(CBG)、性ホルモン結合グロブリン(SHBG)、又はプロゲステロン結合グロブリン(PBG)、又はアルブミンである、請求項18に記載の化合物。
【請求項20】
結合型GnRH類似体及びホルモン成分が切断可能である、請求項1ないし19の何れか一に記載の化合物。
【請求項21】
GnRH類似体及びホルモン成分が直接的に結合している、請求項1ないし19の何れか一に記載の化合物。
【請求項22】
GnRH類似体及びホルモン成分がリンカー基を介して結合している、請求項1ないし20の何れか一に記載の化合物。
【請求項23】
リンカーがコハク酸塩又はそれらの誘導体である、請求項22に記載の化合物。
【請求項24】
GnRH類似体がD−リシン残基であり、及び、GnRH類似体がD−リシンを介してホルモン成分に結合している、請求項1ないし23の何れか一に記載の化合物。
【請求項25】
天然GnRHよりインビボでの半減期が長い、請求項1ないし24の何れか一に記載の化合物。
【請求項26】
天然GnRHよりインビボでの活性持続時間が長い、請求項1ないし25の何れか一に記載の化合物。
【請求項27】
図1A又は1Bに示す化学式である、請求項1に記載の化合物。
【請求項28】
εアミンの6位のD−Lysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
εアミンの6位のD−Lysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAc−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;
εアミンの7位のLysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;
D−PalのN末端アミンで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているD−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
εアミンの7位のLysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;又は
εアミン族の6位のD−Lysで11α−ヒドロキシプロゲステロン11コハク酸塩に結合している[DLys6]GnRH
である、請求項1に記載の化合物。
【請求項29】
血漿ホルモン結合タンパク質に結合している、請求項1ないし28の何れか一に記載の化合物。
【請求項30】
血漿ホルモン結合タンパク質がCBG、SHBG、又はアルブミンである、請求項29に記載の化合物。
【請求項31】
請求項1ないし30の何れか一に記載の化合物と、薬剤的受容性のある賦形剤、担体又は希釈剤を含んでなる薬剤組成物。
【請求項32】
経口投与に適する、請求項31に記載の薬剤組成物。
【請求項33】
徐放製剤である、請求項31に記載の薬剤組成物。
【請求項34】
医薬への使用のための請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物。
【請求項35】
請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を個体に投与することを含んでなる個体の妊よう性減低方法。
【請求項36】
個体の妊よう性を減低するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項37】
必要とする個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、ホルモン依存性疾患又は症状の対処方法。
【請求項38】
必要とする個体のホルモン依存性疾患又は症状を対処するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項39】
ホルモン依存性疾患又は症状が、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維腫、月経前症候群、多嚢胞性卵巣症候群、多毛症、ざ瘡、性早熟症、急性間欠性ポルフィリン症、睾丸停留及び思春期遅発症から選択される、請求項37に記載の方法又は請求項38に記載の使用。
【請求項40】
ホルモン依存性癌が乳癌、前立腺癌、子宮癌、又は子宮内膜癌である、請求項39に記載の方法。
【請求項41】
必要とする個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、不妊の対処方法。
【請求項42】
必要とする個体の不妊に対処するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項43】
個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、インビボでの性腺刺激ホルモン又は性ホルモン産生の調節方法。
【請求項44】
インビボでの性腺刺激ホルモン又は性ホルモン産生を調節するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項45】
GnRH類似体を血漿ホルモン結合タンパク質に結合可能なホルモン成分又はその誘導体に結合させることを含んでなる、GnRHと比較してインビボでの半減期を延長させるためのGnRH類似体の修飾方法。
【請求項46】
GnRH類似体を血漿ホルモン結合タンパク質に結合可能なホルモン成分又はその誘導体に結合させることを含んでなる、GnRHと比較してインビボでの活性持続時間を延長させるためのGnRH類似体の修飾方法。
【請求項47】
結合工程がリンカー基を介してGnRH類似体とホルモン成分又はその誘導体を結合させることを含む、請求項45又は46に記載の方法。
【請求項48】
血漿ホルモン結合タンパク質にホルモン成分又はその誘導体を結合させることを含む、請求項45、46又は47に記載の方法。
【請求項49】
血漿ホルモン結合タンパク質がCBG、SHBG、又はアルブミンである、請求項48に記載の方法。
【請求項50】
結合型GnRH類似体のインビボでの半減期又は活性持続時間を検出することを含む、請求項46ないし59の何れか一に記載の方法。
【請求項51】
結合型GnRH類似体のインビボでの半減期又は活性持続時間をGnRHのインビボでの半減期又は活性持続時間と比較して、GnRHと比較してインビボでの半減期又は活性持続時間が延長しているGnRH類似体を同定することを含む、請求項50に記載の方法。
【請求項1】
ホルモン成分に共役した性腺刺激ホルモン放出ホルモン(GnRH)類似体又はその誘導体を含んでなり、血漿ホルモン結合タンパク質に結合可能な化合物。
【請求項2】
GnRH類似体がペプチド類似体である、請求項1に記載の化合物。
【請求項3】
GnRH類似体がノナペプチド又はデカペプチドである、請求項2に記載の化合物。
【請求項4】
GnRH類似体のアミノ酸残基の一つがD−アミノ酸である、請求項1ないし3の何れか一に記載の化合物。
【請求項5】
D−アミノ酸がD−Lysである、請求項1ないし4の何れか一に記載の化合物。
【請求項6】
D−アミノ酸が6位である、請求項4又は5の何れかに記載の化合物。
【請求項7】
GnRH類似体がGnRHアンタゴニストである、請求項1ないし6の何れか一に記載の化合物。
【請求項8】
GnRHアンタゴニストが[AcD−Nal1,D−Cpa2, D−Pal3,Arg5, D−Lys6,D−Ala10]GnRH、又は[Ac−ΔPro1,D−Fpa2,D−Trp3,D−Lys6]GnRHである、請求項7に記載の化合物。
【請求項9】
GnRHアンタゴニストがセトロレリクス、ガニレリクス、アバレリクス、アンチド、テベレリクス、FE200486、Na−Glu、A−75998、A−76154、A−84861、D−26344、D−63153、D21775、ラモレリクス、デガレリクス、NBI−42902、Org−30850、デチレリクス、イツレリクス、TAK−013、TAK810、AN 207、AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;Ac−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;AcD−Nal−DCpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;AcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;[D−Pyr1,D−Phe2,D−Trp3−6]GnRH;D−Lys6アンチド;Lys5アンチド、又はLys8アンチドである、請求項7に記載の化合物。
【請求項10】
GnRH類似体がGnRHアゴニストである、請求項1ないし6の何れか一に記載の化合物。
【請求項11】
GnRHアゴニストがpGlu−His−Trp−Ser−Tyr−D−lys−Leu−Arg−Pro−GlyNH2、ロイプロン、ゾラデックス、サプレリン、シナレル、トリプトレリン、ブセレリン、ロイプロリド、ゴセレリン、デスロレリン、ProMaxx-100、アボレリン、ヒストレリン、ナファレリン、ロイプロレリン、又はトリプトレリンである、請求項10に記載の化合物。
【請求項12】
ホルモン成分がステロイドホルモン成分である、請求項1ないし12の何れか一に記載に記載の化合物。
【請求項13】
ステロイドホルモン成分がエストラジオール、プロゲステロン、コルチゾール、コルチコステロン、エストロン、テストステロン、又はジヒドロキシテストステロンである、請求項12に記載の化合物。
【請求項14】
プロゲステロン誘導体が11α−ヒドロキシプロゲステロン又は21−ヒドロキシプロゲステロンである、請求項13に記載の化合物。
【請求項15】
化合物がホルモン成分又はそれらの誘導体のインビボホルモン活性を維持している、請求項1ないし14の何れか一に記載の化合物。
【請求項16】
化合物がホルモン成分又はそれらの誘導体のインビボホルモン活性を持たない、請求項1ないし14の何れか一に記載の化合物。
【請求項17】
ホルモン成分がインビボで血漿ホルモン結合タンパク質に結合する、請求項1ないし16の何れか一に記載の化合物。
【請求項18】
ホルモン結合タンパク質がグロブリンである、請求項1ないし17の何れか一に記載の化合物。
【請求項19】
血漿ホルモン結合タンパク質がコルチゾール結合グロブリン(CBG)、性ホルモン結合グロブリン(SHBG)、又はプロゲステロン結合グロブリン(PBG)、又はアルブミンである、請求項18に記載の化合物。
【請求項20】
結合型GnRH類似体及びホルモン成分が切断可能である、請求項1ないし19の何れか一に記載の化合物。
【請求項21】
GnRH類似体及びホルモン成分が直接的に結合している、請求項1ないし19の何れか一に記載の化合物。
【請求項22】
GnRH類似体及びホルモン成分がリンカー基を介して結合している、請求項1ないし20の何れか一に記載の化合物。
【請求項23】
リンカーがコハク酸塩又はそれらの誘導体である、請求項22に記載の化合物。
【請求項24】
GnRH類似体がD−リシン残基であり、及び、GnRH類似体がD−リシンを介してホルモン成分に結合している、請求項1ないし23の何れか一に記載の化合物。
【請求項25】
天然GnRHよりインビボでの半減期が長い、請求項1ないし24の何れか一に記載の化合物。
【請求項26】
天然GnRHよりインビボでの活性持続時間が長い、請求項1ないし25の何れか一に記載の化合物。
【請求項27】
図1A又は1Bに示す化学式である、請求項1に記載の化合物。
【請求項28】
εアミンの6位のD−Lysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
εアミンの6位のD−Lysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAc−ΔPro−D−Fpa−D−Trp−Ser−Tyr−D−Lys−Leu−Arg−Pro−Gly−NH2;
εアミンの7位のLysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Leu−Arg−D−Ala−NH2;
D−PalのN末端アミンで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているD−Pal−Ser−Arg−D−Lys−Leu−Arg−Pro−D−Ala−NH2;
εアミンの7位のLysで21−ヒドロキシプロゲステロン21コハク酸塩に結合しているAcD−Nal−D−Cpa−D−Pal−Ser−Arg−D−Lys−Lys−Arg−Pro−D−Ala−NH2;又は
εアミン族の6位のD−Lysで11α−ヒドロキシプロゲステロン11コハク酸塩に結合している[DLys6]GnRH
である、請求項1に記載の化合物。
【請求項29】
血漿ホルモン結合タンパク質に結合している、請求項1ないし28の何れか一に記載の化合物。
【請求項30】
血漿ホルモン結合タンパク質がCBG、SHBG、又はアルブミンである、請求項29に記載の化合物。
【請求項31】
請求項1ないし30の何れか一に記載の化合物と、薬剤的受容性のある賦形剤、担体又は希釈剤を含んでなる薬剤組成物。
【請求項32】
経口投与に適する、請求項31に記載の薬剤組成物。
【請求項33】
徐放製剤である、請求項31に記載の薬剤組成物。
【請求項34】
医薬への使用のための請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物。
【請求項35】
請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を個体に投与することを含んでなる個体の妊よう性減低方法。
【請求項36】
個体の妊よう性を減低するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項37】
必要とする個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、ホルモン依存性疾患又は症状の対処方法。
【請求項38】
必要とする個体のホルモン依存性疾患又は症状を対処するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項39】
ホルモン依存性疾患又は症状が、ホルモン依存性癌、良性前立腺肥大、子宮内膜症、子宮類線維腫、月経前症候群、多嚢胞性卵巣症候群、多毛症、ざ瘡、性早熟症、急性間欠性ポルフィリン症、睾丸停留及び思春期遅発症から選択される、請求項37に記載の方法又は請求項38に記載の使用。
【請求項40】
ホルモン依存性癌が乳癌、前立腺癌、子宮癌、又は子宮内膜癌である、請求項39に記載の方法。
【請求項41】
必要とする個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、不妊の対処方法。
【請求項42】
必要とする個体の不妊に対処するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項43】
個体に請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物を投与することを含んでなる、インビボでの性腺刺激ホルモン又は性ホルモン産生の調節方法。
【請求項44】
インビボでの性腺刺激ホルモン又は性ホルモン産生を調節するための医薬の調製における、請求項1ないし30の何れか一に記載の化合物、又は請求項31ないし33の何れか一に記載の薬剤組成物の使用。
【請求項45】
GnRH類似体を血漿ホルモン結合タンパク質に結合可能なホルモン成分又はその誘導体に結合させることを含んでなる、GnRHと比較してインビボでの半減期を延長させるためのGnRH類似体の修飾方法。
【請求項46】
GnRH類似体を血漿ホルモン結合タンパク質に結合可能なホルモン成分又はその誘導体に結合させることを含んでなる、GnRHと比較してインビボでの活性持続時間を延長させるためのGnRH類似体の修飾方法。
【請求項47】
結合工程がリンカー基を介してGnRH類似体とホルモン成分又はその誘導体を結合させることを含む、請求項45又は46に記載の方法。
【請求項48】
血漿ホルモン結合タンパク質にホルモン成分又はその誘導体を結合させることを含む、請求項45、46又は47に記載の方法。
【請求項49】
血漿ホルモン結合タンパク質がCBG、SHBG、又はアルブミンである、請求項48に記載の方法。
【請求項50】
結合型GnRH類似体のインビボでの半減期又は活性持続時間を検出することを含む、請求項46ないし59の何れか一に記載の方法。
【請求項51】
結合型GnRH類似体のインビボでの半減期又は活性持続時間をGnRHのインビボでの半減期又は活性持続時間と比較して、GnRHと比較してインビボでの半減期又は活性持続時間が延長しているGnRH類似体を同定することを含む、請求項50に記載の方法。
【図1】


【図2】

【図3】
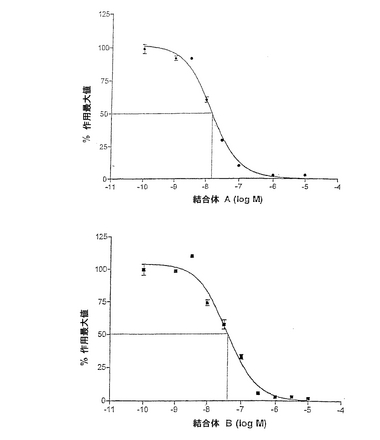
【図4】

【図5】

【図6】

【図7】

【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2006−522085(P2006−522085A)
【公表日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願番号】特願2006−506091(P2006−506091)
【出願日】平成16年4月5日(2004.4.5)
【国際出願番号】PCT/GB2004/001478
【国際公開番号】WO2004/087215
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(505103976)アーダナ バイオサイエンス リミテッド (11)
【Fターム(参考)】
【公表日】平成18年9月28日(2006.9.28)
【国際特許分類】
【出願日】平成16年4月5日(2004.4.5)
【国際出願番号】PCT/GB2004/001478
【国際公開番号】WO2004/087215
【国際公開日】平成16年10月14日(2004.10.14)
【出願人】(505103976)アーダナ バイオサイエンス リミテッド (11)
【Fターム(参考)】
[ Back to top ]