
初代細胞の形質導入
造血系の初代細胞および/または造血幹細胞の安定形質導入に関する方法、組成物、およびシステム。造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法。対象において腫瘍または感染症を治療、診断、緩和または予防するための形質導入した初代細胞の使用も開示する。初代細胞を成長させるための容器またはフラスコを含む系も記載する。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本出願は、2005年5月20日に出願された米国仮特許出願第60/683,527号に関し、その全体が本明細書中に参考として援用される。
【0002】
これらの文書の内容は、本明細書中に参考として援用される。
【0003】
(技術分野)
本発明は、一般にウイルス学、細胞生物学および生命工学に関する。具体的には、本発明は、初代細胞の作製、初代細胞の形質導入、および初代細胞集団の増殖を行うための新規プロセスを提供する。
【0004】
本発明は、ウイルスベクターを用いて細胞の効率的かつ安定した形質導入を行うための方法、ならびにそれに関連する組成物を対象とする。本方法により、形質導入した細胞数の増加がもたらされる。形質導入した細胞は、実験室および臨床応用のどちらにも用いることができる。
【0005】
本発明は、ウイルスベクターを用いて細胞の効率的かつ安定した形質導入を行うための方法、ならびにそれに関連する組成物およびシステムを対象とする。本方法は、たとえば、形質導入する細胞と細胞表面に結合する1つまたは複数の分子とを接触させることによって、形質導入の効率を増加させる。接触させるステップは、細胞へのウイルスベクターの導入の前、後、またはそれと同時に起こり得る。また、本方法では、単層の容器またはフラスコよりも多数の細胞を収容することができる多層の容器またはフラスコ中で細胞を培養することによって、培養および/または成長させている初代細胞がより多くの数もたらされる。また、本発明は、ベクターによって運ばれる核酸の発現または生きた生物の治療を含めた他の応用における、安定形質導入した細胞の使用にも関する。
【背景技術】
【0006】
(背景)
Barry, S.C.ら、2000年、「Lentiviral and murine retroviral transduction of T cells for expression of human CD40 ligand」、Human Gene Therapy、11巻:323〜332頁。Costello, E.ら、2000年、「Gene transfer into stimulated and unstimulated T lymphocytes by HIV−1−derived lentiviral vectors」、Gene Therapy、7巻:596〜604頁。Douglas, J.ら、1999年、「Efficient transduction of human lymphocytes and CD34+ cells via human immunodeficiency virus−based gene transfer vectors」、Human Gene Therapy、10巻:935〜945頁。Follenzi, A.ら、2000年、「Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV−1 pol sequences」、Nature Genetics、25巻:217〜222頁。Han, W.ら、2000年、「A soluble form of human Delta−like−1 inhibits differentiation of hematopoietic progenitor cells」、Blood、95巻:1616〜1625頁。Haas, D.L.ら、2000年、「Critical factors influencing stable transduction of human CD34+ cells with HIV−1−derived lentiviral vectors」、Molecular Therapy、2巻:71〜80頁。Hooijberg E.ら、2000年、「NFAT−controlled expression of GFP permits visualization and isolation of antigen−stimulated primary human T cells」、Blood、96巻:459〜466頁。Kishimoto, T.編、Leucocyte Typing VI: White Cell Differentiation Antigens: Proceedings of the Sixth International Workshop and Conference Held in Kobe, Japan、1996年11月10〜14日、Garland Publishing、New York、1998年。Klebba, C.ら、2000年、「Retrovirally expressed anti−HIV ribozymes confer a selective survival advantage on CD4+ T cells in vitro」、Gene Therapy、7巻:408〜416頁。Koc, O.N.ら、1999年、「Transfer of drug resistance genes into hematopoietic progenitors」、第11章、Gene Therapy of Cancer、Academic Press、San Diego、177〜195頁。Movassagh, M.ら、2000年、「Retrovirus−mediated gene transfer into T cells: 95% transduction efficiency without further in vitro selection」、Human Gene Therapy、11巻:1189〜1200頁。Onodera, M.ら、1998年、「Successful peripheral T−lymphocyte−directed gene transfer for a subject with severe combined immune deficiency caused by adenosine deaminase deficiency」、Blood、91巻:30〜36頁。St. Croix, B.ら、2000年、「Genes expressed in human tumor endothelium」、Science、289巻:1197〜1202頁。Unutmaz, D.ら、1999年、「Cytokine signals are sufficient for HIV−1 infection of resting human T lymphocytes」、J. Exp. Med.、11巻:1735〜1746頁。Zennou, V.ら、2000年、「HIV−1 genome Nuclear import is mediated by a central DNA flap」、Cell、101巻:173〜185頁。
【0007】
一般に遺伝物質を細胞内に導入するための技術をいう「トランスフェクション(transufection)」は、生物学において分子および組換えの革命に大きく貢献した。高等真核細胞で用いるためのトランスフェクション技術の例には、リン酸カルシウム沈殿、DEAE−デキストラン処理、電気穿孔、微量注入、リポフェクチン、ウイルス感染症、ならびに数々の科学の教科書および学術誌に見られる他の方法が含まれる。
【0008】
形質導入(transduction)技術のうち、ウイルス感染の使用は、目的の核酸分子を細胞内に移すために、ウイルスに天然に存在する、その遺伝物質を細胞内に導入する手段を利用するという点で独特である。そのような技術のために改変および応用されたウイルスの例には、アデノウイルス、アデノ関連ウイルス、単純ヘルペスウイルス、およびレトロウイルスが含まれる。一般に、目的の核酸分子は、ウイルスゲノム中にクローニングし得る。ウイルスゲノムの複製およびパッケージング後、生じたウイルス粒子は、ウイルスの侵入機構を介して目的の核酸を細胞内に送達することができる。
【0009】
一般的に、目的の核酸を付加する前に、最初に核酸操作によってウイルスゲノムを複製欠損にする。生じたウイルスゲノム、またはウイルスベクターは、ウイルス粒子の組立ての完了および細胞からの放出にヘルパーウイルスまたはパッケージング系の使用を必要とする。ウイルスベクターまたはウイルス粒子を用いて目的の遺伝物質を細胞内に導入する場合、この技術は「形質導入」と呼ばれる。したがって、一般に、細胞の「形質導入」を行うこととは、ウイルスベクターまたはウイルス粒子を用いて遺伝物質を細胞内に導入することである。
【0010】
形質導入技術では、とりわけ、レトロウイルスの使用が哺乳動物細胞の遺伝組換えの非常な関心の対象となっている。特に関心が持たれているのは、遺伝的欠陥および他の疾患を治療するために遺伝物質を細胞内に導入するための、改変レトロウイルスの使用である。この手法の一例は、レトロウイルスおよびレンチウイルスベクターが熱心な研究の対象である造血系の細胞の事例で見られる。
【0011】
たとえば、Movassaghらは、活性T細胞の細胞周期の研究からの結果を含めることによって、レトロウイルスに媒介される形質導入の効率を上昇させる彼らの試みに関する研究を記述している。それ自体として、その結果は形質導入中の活性細胞分裂に依存する。また、この研究は、ネズミオンコレトロウイルスの使用および形質導入前に細胞の有意な予備刺激を必要とすることに制限されている。
【0012】
Juneら(国際公開公報WO96/34970号)は、T細胞のトランスフェクションを増加させる手段としてのT細胞刺激の使用を記載している。活性化または刺激した細胞を用いたT細胞の形質導入に関する他の研究には、Douglasら、Hooijbergら、Onoderaら、Klebbaら、Barryら、およびUnutmazらの研究が含まれる。残念ながら、この研究のいずれにおいても、約65%を超える形質導入効率が実証されなかった。
【0013】
Costelloらは、ヒト免疫不全ウイルス−1(HIV−1)レンチウイルスベクターを用いた、刺激および非刺激のT細胞の両方の形質導入を記載している。ここでは、刺激した初代T細胞で最大約17%の効率、非刺激のT細胞で19%未満の効率しか観察されなかった。彼らはまた、HIV−1アクセサリータンパク質の存在を含めることによって刺激したT細胞で効率を36%以下まで増加させるという、限定された能力に注目している。
【0014】
また、Chinnasamyらは、非刺激およびマイトジェン刺激したT細胞における、HIV−1アクセサリータンパク質の存在下での形質導入の効率の増加を記載している。Movassaghらと同様、Chinnasamyらは、レンチウイルスベクターを用いて形質導入する前に、血液リンパ球を有意な期間、予備刺激した。Chinnasamyらは、最初、形質導入の3日後に96%を超える形質導入効率を観察したが、安定形質導入した細胞の割合は形質導入の2週間後に71.2%まで低下した。また、Haasらは、マーカー遺伝子(緑色蛍光タンパク質)を発現する能力を有するレンチウイルスベクターを用いて形質導入した細胞において一過性形質導入および「偽形質導入」を観察した。形質導入の3日後でも、形質導入した初代CD34+臍帯血細胞におけるマーカー遺伝子の非組込み発現に基づいて、有意な(10%を超える)一過性形質導入が検出された。一過性形質導入からのこのような発現は、形質導入の7日後でも約5%で検出可能に保たれていた。形質導入の約10日後になってやっと、一過性形質導入からの発現はマーカーなしのベクターを用いて形質導入した細胞における発現の反映となった。
【0015】
したがって、Chinnasamyらは、2週間後の効率によって反映されるように、ウイルスベクターの組込み型を形質導入した細胞の染色体DNA内に挿入した場合、71.2%を超える初代リンパ球の安定形質導入を達成することができなかった。これは、細胞を予備刺激するためにサイトカインを使用したにもかかわらず、そうであった。さらに、Chinnasamyは、形質導入後に細胞をPHAマイトジェンおよびIL−2サイトカインで後に刺激したにもかかわらず、アクセサリータンパク質(Vif、Vpr、VpuおよびNef)を発現しなかったHIVベクターを用いて非刺激のリンパ球を有意に形質導入できなかったことを記載している(形質導入の14日後に3.6%のみ)。結果は非刺激の細胞およびアクセサリータンパク質を含むベクターの使用である程度改善されていたが、ベクターと共に刺激プロトコルを用いたにもかかわらず、どの場合でも、刺激または非刺激の細胞の安定形質導入の効率は、形質導入後14日目に75%を超えていなかった。
【0016】
また、Hassらによってもレンチウイルスベクターを用いた低頻度の安定形質導入が観察され、彼らは、初代CD34陽性臍帯血細胞を用いた形質導入の7日後に25%未満の最大安定形質導入効率しか達成することができなかった。顕著なことに、この25%の形質導入の上限は、極めて高い感染多重度またはベクター濃度、たとえば9000までの感染多重度(MOI)および108感染単位/ミリリットルまでのベクター濃度などの後にも、向上させることができなかった。
【0017】
Follenziらも、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)および幹細胞因子(SCF)を含む3つのサイトカインカクテルの存在下で細胞を形質導入するために、500という非常に高いMOIを用いた。興味深いことに、カクテルを使用することにより、細胞がヒトへの臨床移植に不適切となる。
【特許文献1】国際公開第96/34970号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0018】
したがって、高頻度でベクターを用いた細胞の安定形質導入を行う、より効率的な手段を提供する必要性が、依然として存在する。さらに、研究ツールおよび治療剤のどちらのために使用するとしても、非刺激の細胞を形質導入するためのより効率的な手段の必要性が存在する。
【0019】
さらに、実験室および/または臨床応用において使用できる、多数の初代細胞を同時に形質導入ならびに培養および/または成長させる必要性が、依然として存在する。たとえば、形質導入から1「バッチ」の細胞を得て対象に投与し、様々な形質導入効率を有する複数バッチの細胞を合わせなくてもよいことが望ましい。
【課題を解決するための手段】
【0020】
(発明の開示)
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法である。
【0021】
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行い;細胞とレンチウイルスベクターとを約24時間接触させ、任意選択で少なくとも1回繰り返す方法である。
【0022】
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;約7〜10日後、もしくは約14日目に細胞の少なくとも約50%が安定に形質導入されており;任意選択で約14日後に細胞の少なくとも50%が安定に形質導入されたまま保たれているか;または約7〜10日後、もしくは約14日目に細胞の少なくとも約75%が安定に形質導入されており、かつ任意選択で約14日後に細胞の少なくとも75%が安定に形質導入されたまま保たれているか;または約14日後に80%、85%、89%、90%、91%、92%、93%、94%もしくは95%を超える細胞が安定に形質導入されているか;または約2〜約50、もしくは約10〜約30、もしくは10、もしくは約20、もしくは約30、もしくは約40、もしくは約50、もしくは約1〜約400、もしくは500未満の感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または形質導入した細胞1個あたりのレンチウイルスベクターのコピーが約1〜約100個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または細胞とレンチウイルスベクターとを約24時間接触させ、任意選択で少なくとも1回繰り返し;細胞表面分子はアポトーシスを誘導せず、細胞表面結合分子により、ウイルスレンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなることをもたらす方法である。
【0023】
初代細胞は、単離するか、または対象から導くことができる。初代細胞は、以下の手順の1つまたは複数によって、すなわち、(a)対象の血液のアフェレーシスによって;または(b)対象の骨由来の骨髄から;または(c)同種の対象の血液のアフェレーシスによって;または(d)同種の対象の骨由来の骨髄から、単離することができる。初代細胞はまた、当業者に知られている代替技術を用いて単離することもできる。
【0024】
アフェレーシスとは、ドナーまたは対象の血液を、1つの特定の構成成分を分離して残りを循環に戻す装置に通す医療技術である。
【0025】
除去する物質に応じて、異なるプロセスをアフェレーシスで用いる。たとえば、重量による分離が必要な場合は、遠心分離が選択する方法となる。本発明で用いることができる他の例示的な方法は、吸収材料でコーティングしたビーズ上への吸収を含む。
【0026】
多種類のアフェレーシスが存在する。たとえば、血漿アフェレーシス−血漿;血小板アフェレーシス(plateletpheresis、thrombapheresis、thrombocytapheresis)−血小板;白血球アフェレーシス−白血球(leukocytes、white blood cells);幹細胞収集−骨髄移植で用いるために循環骨髄細胞を収集する;およびLDLアフェレーシス−家族性高コレステロール血症に罹患している対象における低密度リポタンパク質の除去が存在する。これらのアフェレーシスの種類は単なる例示である。当業者には他の種類が知られ得る。
【0027】
血液構成成分は、たとえば、血液バッグに採取する前に採取した全血のバッグまたはドナーの血流から分離することができる。様々な種類の血液構成成分をドナーからアフェレーシスによって得ることができる。これには、たとえば血小板および血漿が含まれる。
【0028】
様々なアフェレーシス技術は、たとえば、除去した構成成分が疾患の重篤な症状を引き起こしている場合にいつでも用い得る。一般に、アフェレーシスは相当頻繁に行う必要があり、侵襲性のプロセスである。したがって、通常は、特定の疾患を制御するための他の手段が失敗した場合、または症状の性質が、医薬品が効果的になるまで待つことで苦痛もしくは合併症の危険性を引き起こすものである場合にのみ用いる。
【0029】
骨髄は、当業者に知られている方法によって得る、保存、および操作することができる。たとえば、米国特許第6,991,787号は、骨髄間質細胞を得る方法を記載しており、米国特許第6,110,176号は、遺伝的に適合性のある骨髄を採取および保存する方法を記載しており、米国特許第4,366,822号は、骨髄細胞を分離および分析する方法および装置を記載している。
【0030】
対象はヒト免疫不全ウイルス(HIV)に感染していてよく、任意選択でHIVはHIV−1またはHIV−2である。対象は癌に罹患していてよく、任意選択で癌は乳癌である。対象はヒトまたは動物のどちらかであることができる。
【0031】
初代細胞は、細胞を密度勾配緩衝液に通すことによって、および/または磁場上で免疫精製を行うことによって、レンチウイルスベクターまたは細胞表面結合分子と接触させる前に富化することができる(磁気細胞分別)。初代細胞は、細胞の培養および/または成長の前またはその間に富化することもできる。当業者によって知られている、細胞集団を富化するための他の知られている手段も、本発明の方法で用いることができる。たとえば、細胞に特異的な標的構造体を同定および富化するプロセスを記載している米国特許第6,974,675号を参照されたい。たとえば、サイトカインを組織培養培地に加えることにより、特定の細胞種の富化がもたらされる。具体的には、培地中に特定のサイトカインが存在すると、特定の細胞集団が死滅する。
【0032】
初代細胞と(a)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触の前に起こるか;または(b)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触と同時に起こるか;または(c)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触の後に起こるか;または(d)レンチウイルスベクターとの接触は、複数回起こるか;または(e)レンチウイルスベクターとの接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または(f)細胞表面結合分子との接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または(g)レンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との最初の同時接触の後、連続的に起こるか;または(h)(a)〜(g)の任意のものは、約24〜36時間の期間中に少なくとも1回起こる。
【0033】
初代細胞は、少なくとも1つの細胞表面結合分子で予備刺激することができ、任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約24時間以内に、細胞を少なくとも1つの細胞表面結合分子で予備刺激するか、または任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約12〜96時間以内に、細胞を少なくとも1つの細胞表面結合分子で予備刺激する。
【0034】
レンチウイルスベクターは、gag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来する少なくとも1つのシス作用性ヌクレオチド配列を含むことができ、任意選択で、配列は発現されない、またはgag、pol、env、vif、vpr、vpu、tatもしくはrev遺伝子の断片もしくは突然変異体である。
【0035】
レンチウイルスベクターは、(a)偽型であり、任意選択で偽型ベクターは水疱性口内炎ウイルスGエンベロープタンパク質を含むことができるか;または(b)偽型であり、偽型化は、レンチウイルスベクター遺伝物質と別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質もしくは細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションもしくは同時感染を行うことを含むことができるか;または(c)ラブドウイルスを用いた偽型であり、任意選択でラブドウイルスは水疱性口内炎ウイルスエンベロープG(VSV−G)タンパク質であることができる。
【0036】
初代細胞は、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体であることができる。
【0037】
少なくとも1つの細胞表面結合分子は、(a)ポリペプチド、脂質、核酸、炭水化物またはイオンを含むか;あるいは(b)抗体、抗原結合断片、リガンド、または細胞表面分子を含むか;あるいは(c)FLT−3リガンド、TPOリガンド、もしくはKitリガンド、またはFLT−3リガンド、TPOリガンド、もしくはKitリガンドの細胞表面結合類似体であるポリペプチドもしくは他の結合分子を含むか;あるいは(d)CD34、CD3リガンド、CD28リガンド、CD25リガンド、CD71リガンド、もしくはCD69リガンド、またはCD34、CD3、CD25、CD28、CD69もしくはCD71リガンドと同じ細胞表面結合特異性を有するポリペプチドもしくは他の結合分子を含むか;あるいは(e)GM−CSF、IL−4、およびTNF−α;GM−CSFおよびインターフェロン−α;またはGM−CSF、IL−4、およびTNF−αの細胞表面結合類似体であるポリペプチドもしくは他の結合分子;GM−CSFもしくはインターフェロン−αを含む組成物を含むか;あるいは(f)CD3抗体またはその細胞表面結合断片、CD28抗体またはその細胞表面結合断片、抗体とその細胞表面結合断片との組合せ、および抗体と同じ細胞表面結合特異性を有する結合分子を含むか;あるいは(g)ビーズまたは表面上に固定したCD3およびCD28抗体の組合せを含み、任意選択で、ビーズまたは表面はコーティングしたビーズを含むか;あるいは(h)(a)〜(g)の任意のものから選択された2つ以上の細胞表面結合分子を含むか;あるいは(i)分子が細胞の表面に結合する能力を増加または増強するために用いる別の分子を含むか;あるいは(j)別の分子と複合しているか;あるいは(k)初代細胞の表面上に見つかり、別の細胞の表面に結合する。
【0038】
細胞培養条件は、(a)細胞表面結合分子もしくはサイトカインと共にさらなるインキュベーションを行うこと;または(b)インターロイキン−2と共にさらなるインキュベーションを行うこと;または(c)細胞を約7日間培養すること;または(d)細胞を約14日間培養することを含むことができる。
【0039】
レンチウイルスベクターは、(a)ヒト免疫不全ウイルス(HIV)由来であるか;または(b)HIV−1、HIV−2、もしくはそれらの組合せ由来であるか;または(c)HIV配列を含むキメラベクターであり、任意選択で、HIV配列はHIV−1およびHIV−2配列を含むか;または(d)VRX496、もしくはVRX496の誘導体であることができる。
【0040】
細胞とレンチウイルスベクターまたは細胞表面結合分子との接触は、ex vivoで、混合もしくは純粋細胞培養物、組織または器官系中で起こることができる。
【0041】
本発明の別の態様は、本明細書中に記載した任意の方法によって形質導入した細胞を生きた対象、組織、器官、胚盤胞または胚性幹細胞内にex vivo導入することを含む、遺伝物質を細胞内に導入する方法である。
【0042】
本発明の別の態様は、薬剤組成物を調製するための、本明細書中に記載した任意の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用である。薬剤組成物は、対象におけるウイルス感染症の治療もしくは予防、対象におけるHIV感染症の治療もしくは予防、または癌の治療もしくは予防に用いることができる。癌は、任意の種類の癌、たとえば乳癌または内皮細胞の任意の癌であることができる。
【0043】
本発明の別の態様は、本明細書中に記載した任意の方法によって生成した、遺伝的欠陥によって引き起こされた異常を治療もしくは予防する、または腫瘍もしくは癌を治療、診断、緩和もしくは予防するための遺伝子治療用の薬剤組成物であり、任意選択で、遺伝的欠陥または腫瘍もしくは癌によって引き起こされた異常は乳癌腫瘍である。
【0044】
本発明の別の態様は、本明細書中に記載した任意の方法によって生成した、感染症によって引き起こされた異常を治療または予防するための遺伝子治療用の薬剤組成物。感染症はウイルス感染症であることができ、任意選択で、ウイルス感染症はヒト免疫不全ウイルス(HIV)感染症である。薬剤組成物は、ex vivoで使用するために配合する。
【0045】
本発明の別の態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、前記初代細胞と細胞表面分子との接触により、レンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなる方法である。
【0046】
初代細胞の表面における細胞表面分子の存在により、(a)細胞クロマチンのDNA組込みに対する受容性がより高くなること;または(b)レンチウイルスベクターからの遺伝子の発現に有利な細胞部位内へのレンチウイルスベクターの組込み;または(c)カプシドを含む核酸の、細胞の細胞質内へのより効率的な侵入;または(d)ウイルスの、細胞膜を横断するもしくは細胞の内部膜構造体を横断するより効率的な侵入;または(e)ウイルスベクター内に含まれる遺伝物質の核内移行に対する初代細胞の許容性がより高くなることが、もたらされることができる。
【0047】
細胞表面結合分子、抗体、抗原結合断片、リガンドまたは細胞表面分子は、細胞に結合してベクター形質導入に対するその受容性をより高くする抗CD3または抗CD28抗体;細胞に結合してベクター形質導入に対するその受容性をより高くする、FLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンド;樹状細胞もしくはその前駆体、単球、CD34陽性幹細胞、または樹状細胞系列上の分化したその前駆細胞に結合してベクター形質導入に対するその受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンド;細胞上の
【0048】
【化2】
に結合してベクター形質導入に対するその受容性をより高くする、ポリペプチド、核酸、炭水化物、脂質もしくはイオン、または別の物質と複合したポリペプチド、核酸、炭水化物、脂質もしくはイオンを含む。
【0049】
本発明の別の態様は、HIVに感染した対象から単離した造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、(a)HIVに感染した対象から造血系細胞の初代細胞または造血幹細胞を単離するステップと;(b)任意選択で、初代細胞または造血幹細胞を少なくとも1つの細胞表面結合分子で予備刺激するステップと;(c)in vitroまたはex vivoで、造血系細胞または造血幹細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させるステップと;(d)2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養するステップとを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法である。
【0050】
本発明の別の態様は、(a)2つ以上の層を含む換気容器;ならびに(b)単離した非接着性の造血系の初代細胞および/または造血幹細胞を含むシステムである。システムの初代培養は、本発明の方法について上述した任意の細胞であることができる。
【0051】
多層容器は、細胞をその中で培養および/または成長させるために実用的な任意の形状であることができる。たとえば、容器は、四角形の形状、正方形の形状、または曲線縁の四角形の形状、もしくは曲線縁の正方形の形状であることができる。
【0052】
組織培養フラスコまたは容器が、細胞を成長させるために実験室で幅広く使用されている。典型的には、これらのフラスコを用いて細胞を培地中で培養し、細胞がフラスコの内部表面に接着する。細胞は、開口部からフラスコまたは容器内に導入する。フラスコまたは容器を閉じ(ただし換気を許容する)、培地中の細胞の成長を促進するために、オーブンなどのスタッキング施設またはチャンバ内に挿入する。
【0053】
造血系の初代細胞および/または造血幹細胞は、培養バッグ、または他の単層の容器もしくはフラスコ中で成長させる。これにより、一度に成長させることができる細胞の数が制限される。平坦な表面を有する多層のフラスコまたは容器により、一度に多数の細胞を培養することが可能となるが、これは平坦な表面に接着する細胞に用いられている。造血系の初代細胞および/または造血幹細胞は非接着性細胞である。しかし、これらの細胞は多層のフラスコまたは容器中で成長させることができ、これにより、一度に多数の細胞を成長させることが可能となる。多層容器の一例は細胞工場である。
【0054】
本発明の方法および組成物は、多層の組織培養フラスコまたは容器中における、非接着性の造血系の初代細胞および/または造血幹細胞の大スケールの培養および/または成長を可能にし、少なくとも約1億個の細胞の成長をもたらす。
【0055】
細胞工場、または細胞工場の概念のその変型を、たとえば、ワクチン、モノクローナル抗体または医薬品の大スケール産生に用いることができる。これらは接着性細胞に理想的であるが、懸濁培養にも用いることができる。細胞の成長の速度論は、実験室スケールの培養から変化せずに保たれる。細胞工場は、容易なスケールアップのために、たとえば、1、2、4、10および40個のトレイの型で入手可能である。これらの汚染危険性は低く、コンパクトな設計である。
【発明を実施するための最良の形態】
【0056】
上記の文書の引用は、前述の任意のものが関連する従来技術であると承認することを意図しない。これらの文書の内容に関する日付または表示に関するすべての記述は、出願人に利用可能な情報に基づいており、これらの文書の日付または内容の正確性に関していかなる承認も構成しない。
【0057】
本明細書中に引用するすべての参考文献、出版物、特許出願および特許は、具体的に組み込んでいるかいないかにかかわらず、その全体で参考として本明細書中に組み込まれている。
【0058】
以下の記述は、本発明の方法で用いることができる容器の種類の例である。これらは単なる例示であり、本発明の範囲を限定することを意図しない。
【0059】
細胞をその中で成長させることができる容器の種類の例を図1に示す。容器は、細胞を中で成長させるために実用的な任意の形状、たとえば、正方形、四角形、円形、楕円形、または成形端部を有する正方形もしくは四角形であることができる。容器は複数層でなければならないが、容器が含むことができる層の数の上限は、細胞を中で培養および/または成長させるチャンバ、オーブン、または容器の大きさにのみ制限される。容器は、たとえばプラスチック、または細胞をその中で培養および/もしくは成長させるために適した任意の他の材料から作製することができる。
【0060】
細胞工場とは、単独のユニットに密着させ、共通の通気口および注入口を共有する、積み重ねたチャンバである。それぞれのチャンバは、たとえば、632cm2の平坦な成長表面を有する。細胞工場は、大スケール細胞培養ならびにワクチン、モノクローナル抗体およびインターフェロンなどの生体材料の産生に用いる。細胞工場は、小さな面積で大量の成長表面を提供し、取扱いが容易で汚染の危険性が低い。成長面積が25,280cmの、チャンバが40個のユニットは、14個の大きなローラーボトル(それぞれ1,750cm)に対応する。細胞工場では1回の充填および排出作業が必要であることに対して、ローラーボトルでは14回必要である。細胞工場は無菌的である。細胞工場は、限られた空間の面積で大きな成長表面を提供する。
【0061】
細胞工場の他の態様は、たとえば、細胞懸濁液を保つ通気ストッパを備えたアスピレーターボトルからなる、培地リザーバである。細胞工場を用いた細胞の導入、培養および収集は当分野で知られている。本発明で用いることができるインキュベーターおよび細胞培養装置の種類の変型は、たとえば、公開された特許出願第20060057713号および第20060057712号、ならびに米国特許第6,114,165号に記載されている。
【0062】
チャンバが10個および40個の細胞工場の高さが原因で、細胞を一般的な顕微鏡で見ることができない。ほとんどの場合、対照として細胞をチャンバが1個またはチャンバが2個の細胞工場に播種する。これらの製品のそれぞれで成長させた細胞は、顕微鏡下で見ることができる。見る側に強力な光源を備え、細胞工場の高さに調節した倒立双眼実体顕微鏡を用いて、最初の数層を見た。
【0063】
例示的な細胞工場の寸法および培養面積:
【0064】
【化3】
本発明の方法では、新規組成物、すなわち、高い形質導入レベルを有する細胞の試料、「集団」または「バッチ」であり、少なくとも約1億個の細胞を含む組成物を生成する。
【0065】
本発明は、ウイルスベクターおよびウイルス粒子を用いて細胞の安定形質導入を行うための、効率の高い方法、およびそれに関連する組成物を提供する。「安定形質導入」とは、ウイルスベクターの組込み型を形質導入した細胞の染色体DNA内に挿入したことを意味する。この方法は、細胞表面に結合する少なくとも1つの分子と接触するように形質導入する細胞を曝露することを含む。この接触させるステップは、ウイルスベクターまたはウイルス粒子に細胞を曝露する前、その間、またはその後に起こり得る。本明細書中では以降、用語「ウイルスベクター」とは、ウイルスに由来し、形質導入を介して遺伝物質を細胞内に導入するために用いる、任意の形態の核酸を表すために用いる。この用語には、DNAおよびRNAなどのウイルスベクター核酸、これらの核酸のカプセル封入した形態、ならびにウイルスベクター核酸をパッケージングしたウイルス粒子が包含される。
【0066】
また、本発明には、ベクター中に存在する核酸の発現による有用な遺伝子産物およびタンパク質の産生、または疾患に罹患している、もしくは罹患する危険性のある生きた対象の治療を含めた、他の応用における形質導入した細胞の使用も含まれる。たとえば、対象はヒトである。
【0067】
形質導入する細胞の表面に結合する少なくとも1つの分子には、細胞の表面上の受容体、マーカー、または他の認識可能な部分と物理的に相互作用する任意の分子が含まれる。原理上は、任意の細胞表面結合分子を細胞の高効率形質導入に用い得る。本発明を理論に束縛せずに、細胞表面結合分子は、宿主細胞のクロマチンのDNA組込みに対する受容性がより高くなること;ベクターの遺伝子発現に有利な部位内へのウイルスベクターの優先的な組込み;カプシドを含む核酸の細胞質内へのより効率的な侵入;細胞膜もしくはエンドソームなどの内部膜構造体を横切るウイルスのより効率的な侵入;またはウイルスベクターの遺伝物質核内移行に対する細胞の許容性をより高くさせることをもたらし得る。本発明の方法はまた、上記の可能性の複数に関与し得る。また、上記の可能性の数および多様性から明らかなように、本発明を任意の1つの理論に限定することはできない。そうではなく、ヒト治療における細胞の使用可能性に負の影響を与えずに処理した細胞の最高100%の安定形質導入を行うという本発明の驚くべき発見を考慮すると、本発明は、ヒト細胞治療の分野において新しい手法を開くとみなすべきである。
【0068】
しかし、すべての細胞表面結合分子でウイルスベクターによる効率的かつ安定した形質導入がもたらされるわけではない。たとえば、アポトーシスを誘導する細胞表面分子との結合では、細胞の効率的な形質導入はもたらされず、むしろ細胞死がもたらされる。細胞死は細胞を死滅させるため(たとえば腫瘍細胞)には好ましい場合もあるが、ペイロード(payroad)遺伝子または核酸配列を含むベクターを用いた細胞の安定形質導入には好ましくない。好ましい細胞表面結合分子により、細胞は、ウイルスベクターによる形質導入に対して受容性がより高くなる。そのような分子の例には、特異的細胞表面受容体またはその一部分に対する抗体、およびそのような受容体に対するリガンドまたは結合ドメインが含まれる。さらに、FabおよびFv断片などの抗体の抗原結合断片が、本発明における使用に企図される。特異的な細胞表面受容体に対する結合ドメインは、単一のエピトープまたは2つ以上のエピトープを含んでいることができる。
【0069】
本発明において使用する細胞表面結合分子の例は、T細胞に結合してベクター形質導入に対するその受容性をより高くする、抗CD3および抗CD28抗体である。他の細胞表面結合分子は、造血幹細胞などの受容体を発現する細胞の、ベクター形質導入に対する受容性をより高くするFLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンドである。さらなる細胞表面結合分子は、単球などの樹状細胞もしくはその前駆体、CD34陽性幹細胞、または樹状細胞系列上にあるその分化した前駆細胞の、ベクター形質導入に対する受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンドである。他の細胞表面結合分子には、別の細胞の表面に結合する、細胞表面上に見つかる分子が含まれる。
【0070】
細胞表面結合分子のさらなる例には、すべて任意選択で他の物質と複合したポリペプチド、核酸、炭水化物、脂質、およびイオンが含まれる。これらの分子は、血液細胞の表面上に見つかる因子、たとえば、
【0071】
【化4−1】
などに結合することができる。小文字(たとえば「a」または「b」)は、複数の遺伝子産物からなる、または構造的に関連するタンパク質のファミリーに属する複合CD分子を示す。記号「w」は、完全に確認されてない推定上のCD分子を指す。
【0072】
リンパ球、T細胞および白血球の表面上に見つかる因子に結合するさらなる分子は、
【0073】
【化4−2】
である。
【0074】
細胞の表面に結合し、本発明における使用に適したさらなる抗体および分子は、完全に記載したかのように本明細書中に参考として組み込まれているLinscott’s Directory of Immunological and Biological Reagents、第11版、2000年1月、出版社:W.D. Linscott、カリフォルニア州Petalumaに開示されている。しかし、本発明の一部の実施形態では、細胞表面結合分子はサイトカインではない。
【0075】
本発明は、細胞のベクター形質導入を促進する可溶性細胞表面結合分子を使用することによって実施し得るが、他の実施形態には、固定した細胞表面結合分子の使用が含まれる。たとえば、固定した分子は抗体である。あるいは、固定は、細胞表面結合分子を発現する他の細胞の使用を介し得る。造血幹細胞の効率的な形質導入の例示的な方法は、分化させずに幹細胞の維持を容易にするリガンドをその表面上に発現する骨髄間質細胞を、形質導入中に含めることである。刺激性細胞はネイティブ細胞に限定されず、任意の細胞を、形質導入のために正しい刺激を提供するために適切な細胞表面結合分子を発現するように操作することができる。
【0076】
少なくとも1つの分子が細胞表面に結合する能力を増加または増強させるさらなる分子も含め得る。たとえば、特異的細胞表面受容体に対する(一次)抗体の可溶形を、既に細胞表面と結合している一次抗体に架橋結合することができる二次抗体と組み合わせて用い得る。
【0077】
もちろん、任意の細胞を本発明の実施に用いることができる。たとえば、形質導入する細胞は真核細胞である。たとえば、細胞は初代細胞である。しかし、細胞系は、本発明の方法を用いて形質導入してもよく、多くの場合、より容易に形質導入される。一実施形態では、形質導入する細胞は、初代リンパ球(Tリンパ球など)もしくはマクロファージ(単球マクロファージなど)であるか、またはこれらの細胞のいずれかの前駆体、たとえば造血幹細胞などである。一般に、形質導入させる他の例示的な細胞は造血系の細胞であるか、または、より一般的には、造血によって形成される細胞ならびにそれらを形成する幹細胞および血液細胞機能に関連する細胞である。そのような細胞には、造血によって形成される顆粒球およびリンパ球、ならびに前駆体分化多能性幹細胞、リンパ球幹細胞、および骨髄幹細胞が含まれる。血液細胞機能に関連する細胞には、樹状細胞等の抗原提示細胞、内皮細胞、単球、およびランゲルハンス細胞などの、免疫系細胞の機能を補助する細胞が含まれる。一実施形態では、細胞は、CD4およびCD8マーカーを発現するものなどのTリンパ球(またはT細胞)である。
【0078】
別の実施形態では、細胞は初代CD4+Tリンパ球または初代CD34+造血幹細胞である。しかし、本発明で用いるためのウイルスベクターは、水疱性口内炎ウイルスエンベロープGタンパク質を用いて偽型とし得ることを考慮すると(以下に記述)、任意の細胞を本発明の方法で形質導入することができる。そのような細胞には、それだけには限定されないが、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、リンパ球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞または精母細胞(たとえばトランスジェニック動物を作製するため)、生殖系列に寄与する細胞、胚性分化多能性幹細胞またはその前駆体、血小板および赤血球等の無核細胞を含めた血液細胞などが含まれる。たとえば、細胞は、真核の多細胞種(たとえば単細胞の酵母細胞に対抗して)のものであるか、または哺乳動物由来のもの、たとえばヒト細胞である。
【0079】
形質導入する細胞は、単独の実態として存在するか、またはより大きな細胞コレクションの一部であることができる。そのような「細胞のより大きなコレクション」は、たとえば、細胞培養物(混合もしくは純粋のどちらか)、組織(たとえば、上皮、間質もしくは他の組織)、器官(たとえば、心臓、肺、肝臓、胆嚢、膀胱、眼、および他の器官)、器官系(たとえば、循環系、呼吸器系、胃腸管系、泌尿器系、神経系、外皮系もしくは他の器官系)、胚盤胞、胚性幹細胞、胎児由来の細胞(たとえば、遺伝的障害/疾患を治療するため、もしくはトランスジェニック動物を作製するため)、腫瘍もしくは感染部位などの患部組織、または生物(たとえば、鳥、哺乳動物、海洋生物、魚、植物など)を含むことができる。標的とする器官/組織/細胞は、循環系(たとえば、それだけには限定されないが、心臓、血管、および血液を含む)、呼吸器系(たとえば、鼻、咽頭、喉頭、気管、気管支、細気管支、肺など)、胃腸管系(たとえば、口および口腔組織、咽頭、食道、胃、腸管、唾液腺、膵臓、肝臓、胆嚢などを含む)、乳腺系(乳房上皮細胞および組織中の支持細胞など)、泌尿器系(腎臓、子宮、膀胱、尿道など)、神経系(それだけには限定されないが、脳および脊髄、ならびに眼などの特殊感覚器を含む)、外皮系(たとえば皮膚)のものであることができる。
【0080】
形質導入する細胞は、心臓、腫瘍血管および感染組織または患部組織に関連する血管を含めた血管、骨髄、血液、脳、リンパ組織、リンパ節、脾臓、肺、肝臓、胆嚢、膀胱、ならびに眼細胞からなる群から選択することができる。本発明の特定の一実施形態では、細胞は使用を意図する宿主の自己由来であるが、同種、部分ミスマッチ、完全ミスマッチ、またはさらには宿主に異種の細胞も用い得る。さらに、人間など生物または種の関連した群である任意の与えられた宿主生物での使用に適したユニバーサルドナー細胞を形質導入してよい。本発明のこの後者の実施形態は、形質導入した細胞の供給源が何であるかが移植の結果に決定的であり得る、細胞、組織または器官における移植に特に重要である。
【0081】
本発明の方法による形質導入のための別の細胞種は、腫瘍細胞、患部細胞、またはその遺伝子構造もしくは同一生物中に存在する他の細胞の遺伝子構造が原因でいずれ異常となる危険性にある細胞である。後者の実施形態により、本発明の形質導入した細胞を予防に用いることが可能となる。乳癌は、予後指標により、疾患が引き起こされる前の初期遺伝的介入としての本発明の形質導入した細胞を用いた治療が可能となる、疾患プロセスの一例である。しかし、本発明の方法は、疾患が検出された後の乳癌の治療処置にも用い得る。癌治療における本発明のさらなる用途は多数存在し、当業者は、必要以上の実験を行わずに、多種類の癌を治療するために本明細書中に記載した本発明を用いることができるであろう。
【0082】
例として、かつ本発明を限定せずに、一用途はエストロゲン依存性の乳癌にある。エストロゲン依存性乳癌中の癌細胞は、たとえば、エストロゲン受容体に結合する抗体またはリガンドを治療ウイルスベクターと組み合わせて用いることによって形質導入する。ベクターは、たとえば、ヘルペスウイルスチミジンキナーゼ遺伝子などの腫瘍阻害遺伝子を含み得る。したがって、ヘルペスチミジンキナーゼによって活性化することができるプロドラッグであるガンシクロビルを加えることによって、形質導入した細胞を選択的に死滅させることができる。腫瘍阻害遺伝子および対応するプロドラッグのさらなる例は多数存在し、当分野で周知であり、必要以上の実験を行わずに当業者によって選択し得る。本発明の形質導入方法の用途と組み合わせた活性化可能なプロドラッグの使用は、他の腫瘍種に幅広く応用してよく、上記の例は、本発明をホルモン依存性または成長もしくは増殖を何らかの可溶性因子に依存する腫瘍に限定しない。
【0083】
たとえば、Her−2/neu陽性腫瘍細胞はエストロゲン依存性ではなく、そのような細胞を含む非エストロゲン依存性腫瘍は、エストロゲン拮抗剤であるタキソールなどの薬物を用いた治療に高度に耐性を有するので、これは乏しい予後指標である。本発明の別の実施形態は、骨髄移植プロトコル中などの腫瘍が混入した細胞の形質導入中に、Her−2/neuまたはヘレグリンに結合する抗体または他の分子をウイルスベクター調製物と共に含めることである。あるいは、形質導入を、腫瘍部位に直接、またはin vivoで腫瘍形成を調節するベクターと共に血管内に行い得る。
【0084】
本発明のさらに別の実施形態は、腫瘍血管構造を、単独でまたは腫瘍細胞の標的化と組み合わせて標的とすることである。完全に記載したかのように本明細書中に参考として組み込まれているSt Croixらは、SAGE分析によって、正常内皮と比較して腫瘍内皮細胞中で特異的に過剰発現されている遺伝子を同定している。これらの遺伝子の多くは、Thy−1細胞表面抗原またはEndo180レクチンなどの細胞表面分子をコードしている。アップレギュレーションされた細胞表面因子のすべては、効率的な安定遺伝子形質導入を行うための刺激を提供するために、本発明の細胞表面結合分子と結合し得る。したがって、腫瘍を治療するための一手法は、腫瘍血管構造と選択的に結合し、正常内皮細胞とは結合しない細胞表面結合分子の存在下で治療ウイルスベクターを用いて形質導入した後で、腫瘍内皮細胞を死滅させることによって腫瘍血管構造を破壊することである。
【0085】
本発明のさらに別の実施形態は、腫瘍中で抗腫瘍遺伝子の発現を選択的に促進するが、正常な血管内皮では促進しないウイルスベクター中に、要素(たとえば、mRNAのプロモーターまたはシス作用性の安定化/分解要素)を組み込むことによって、腫瘍血管構造中で抗腫瘍遺伝子を選択的に発現させることである。このような方法は、ex vivo、in vitroまたはin vivoで起こることができる。腫瘍血管内皮を標的とする場合はin vivoが治療の一方法である。あるいは、たとえば目的が骨髄移植のために骨髄から混入した腫瘍細胞を追放することである場合は、治療方法はex vivoまたはin vitroで起こることができる。
【0086】
さらに、in vivoでの使用は病状に限定されず、正常細胞に形質導入に用いることができる。たとえば、本発明は、骨髄中で、in vivoで造血幹細胞を形質導入するために用い得る。高効率の骨髄形質導入のために、抗体またはFLT−3リガンド、TPOおよびKitリガンド、もしくはその機能的類似体などの他の細胞表面結合分子、あるいは細胞表面結合分子を発現する間質細胞の任意の組合せを、骨髄内への直接注射を行う際にベクターと共に加えることができる。用語「機能的類似体」とは、本発明の細胞表面結合分子の細胞表面結合活性を保持する任意の分子をいう。そのような機能的類似体には、FLT−3リガンド、TPOおよびKitリガンド;1つまたは複数のアミノ酸の置換、付加または欠失を含むFLT−3リガンド、TPOおよびKitリガンド分子;ならびに細胞表面結合分子の細胞表面結合活性を模倣する抗体の断片が含まれる。
【0087】
上記の代替手法は、骨髄間質細胞をウイルスベクターの産生細胞として使用し、したがって、ベクター調製物としてではなく細胞治療を介してベクターおよび細胞表面結合分子を提供することである。別の例は、CD3およびCD28抗体またはGM−CSFおよびIL−4の機能的類似体を、それぞれベクターと共に皮下注射中に加えることによる、T細胞または樹状細胞の形質導入である。皮下組織中のリンパ液が、標的細胞の効率的な形質導入のためにベクターおよび刺激因子をリンパ節内に流す。
【0088】
本発明には、任意選択で、形質導入する細胞の精製が必須ではないという利点が含まれる。主に目的の細胞種の形質導入は、結合させる細胞表面部分の選択によって達成することができる。したがって、血液細胞の混合集団中では、たとえば、特定のT細胞などのCD3を発現する細胞の形質導入は、細胞との相互作用にCD3に特異的な抗体を用いた場合に増強される。これは、CD3を発現しない顆粒球および単球などの集団中の他の細胞種より優先的に起こる。
【0089】
また、本発明は、所望する場合は、精製または単離した細胞種の形質導入も包含する。精製または単離した細胞種の使用により、形質導入する細胞よりも高いベクター濃度に起因するより高い形質導入効率などの、さらなる利点が提供される。
【0090】
精製したT細胞を形質導入する場合は、少なくとも1つの分子はT細胞上に見られる細胞表面分子に結合する。そのような細胞表面分子の例には、CD3、CD28、CD25、CD71、およびCD69が含まれる。これらの細胞表面分子に結合する分子の例には、それらを認識する抗体およびモノクローナル抗体が含まれ、それらの多くが市販されているか、または必要以上の実験を行わずに標準の技術を用いて容易かつ日常的に調製される。CD4+またはCD8+細胞の形質導入の例示的な実施形態では、CD3および/またはCD28を認識するモノクローナル抗体を用い得る。そのような抗体の市販されている例には、CD3用のOKT3およびCD28用のCD28.2が含まれる。これらの抗体分子は、可溶形で用いてよく、任意選択で後に他の分子に架橋結合して、またはビーズもしくは他の固体表面上などの固定した形態で用いてよい。本発明の一実施形態では、抗体を、ウイルスベクターに媒介される形質導入に用いる組織培養ウェル、プレート、またはバッグの壁などの容器の表面上に固定する。理論に束縛されずに、細胞が接着または接触する表面上の固定した抗体を使用することにより、細胞表面上の細胞表面相互作用の局所濃度が増加し得る。
【0091】
形質導入を造血幹細胞する場合は、FLT−3リガンド、TPO(トロンボポエチンもしくは巨核球成長および発達因子)、またはKitリガンドの造血幹細胞受容体に特異的な抗体を細胞表面結合分子として用い得る。あるいは、それだけには限定されないが、CD34またはAC133を含めた幹細胞陽性細胞マーカーに対する抗体を用い得る。化合物または組成物を含むリガンドを細胞表面結合分子として用いる場合は、陰性リガンド含有タンパク質、リガンドまたは異種タンパク質に結合したリガンドの全体を、可溶性または固定した形態のどちらかで用いることができる。固定した形態には、たとえばアビジン/ビオチンを用いたマイクロビーズへの直接または間接的な付着が含まれる。
【0092】
あるいは、リガンドは、ウイルスベクターのウイルスエンベロープ中、任意選択でキメラもしくは融合タンパク質の形態で、および/または1つもしくは複数の他のタンパク質と(共有もしくは非共有)結合して、発現され得る。そのような実施形態では、細胞表面結合分子は、細胞を形質導入するための単一の組成物として、ウイルスベクターと組み合わせて提供する。ウイルスエンベロープ中で発現させ得る細胞表面結合分子のさらなる例には、上記の多数の表面因子が含まれる。
【0093】
抗体またはその断片などの他の細胞表面結合分子は、ノッチもしくはデルタの造血幹細胞受容体に結合するもの、またはノッチもしくはデルタタンパク質自体、または異種タンパク質に結合するノッチもしくはデルタのリガンドである。デルタおよびノッチは、ショウジョウバエの発達において広範な細胞運命の決定に影響を与える細胞表面タンパク質をコードしている。デルタおよびノッチの脊椎動物相同体は、正常な胚発達に必須である。デルタ相同体は、造血の制御に重要に関与している。相同体の可溶形であるデルタ−Serrate−lag2(DSL)は、原始造血前駆体の増殖を増強する。インターロイキン−3(IL−3)、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)を含めた造血成長因子と合わせた場合、DSLは原始造血前駆体の増殖を促進し、同時に、原始前駆体が単独でIL−3に応答性のあるより成熟な前駆細胞へと分化することを阻害した(Hanら参照)。DSLは、造血細胞中で発現されるノッチ受容体を活性化し、細胞分化を選択的に遮断するが増殖シグナルを遮断しないことで慣用の造血成長因子に応答する細胞の能力を調節することによって作用する可能性が最も高い(HanおよびMoore、Blood、1999年参照)。したがって、相同体の機能的類似体の抗体であるデルタおよびノッチ相同体は、細胞、特に造血幹細胞の、75%を超える効率のベクター形質導入を達成するのに使用する細胞表面結合分子の例である。
【0094】
本発明には、開示した方法で使用するためのウイルスベクターおよびそれらを含む組成物が含まれる。ベクターは、レトロウイルス(レトロウイルス科)ベクター、たとえばレンチウイルスベクターであることができる。オンコウイルスまたはネズミレトロウイルスベクターなどの他のレトロウイルスベクターも用い得る。さらなるベクターは、他のDNAウイルス、またはその生活環のある時点でそのゲノムをDNA内に転換することができるウイルスに由来し得る。ウイルスは、たとえば、アデノウイルス科、パルボウイルス科、ヘパンダウイルス科(Hepandaviridae)(通常はヘパンダウイルス科に分類されないδ型肝炎ウイルスおよびE型肝炎ウイルスを含む)、パポウイルス科(Papoviridae)(ポリオーマウイルス亜科およびパピローマウイルス亜科を含む)、ヘルペスウイルス科、ならびにポックスウイルス科由来であることができる。
【0095】
レトロウイルス科(すなわちレトロウイルス)のさらなるウイルスは、オンコウイルス亜科、スプマウイルス亜科、スプマウイルス、レンチウイルス亜科、およびレンチウイルスの属または亜科のものである。亜科であるオンコウイルス亜科のRNAウイルスは、望ましくは、ヒトT−リンパ球向性ウイルス1型もしくは2型(すなわちHTLV−1もしくはHTLV−2)またはウシ白血病ウイルス(BLV)、トリ白血症肉腫ウイルス(たとえば、ラウス肉腫ウイルス(RSV)、トリ骨髄芽球症ウイルス(AMV)、トリ赤芽球症ウイルス(AEV)、およびラウス関連ウイルス(RAV;RAV−0からRAV−50)、哺乳動物C型ウイルス(たとえば、モロニーマウス白血病ウイルス(MuLV)、ハーベイマウス肉腫ウイルス(HaMSV)、アベルソンマウス白血病ウイルス(A−MuLV)、AKR−MuLV、ネコ白血病ウイルス(FeLV)、サル肉腫ウイルス、細網内皮症ウイルス(REV)、脾臓壊死ウイルス(SNV))、B型ウイルス(たとえばマウス乳癌ウイルス(MMTV))、ならびにD型ウイルス(たとえば、メーソン−ファイザーサルウイルス(MPMV)および「SAIDS」ウイルス)である。
【0096】
亜科レンチウイルスのRNAウイルスは、望ましくは、ヒト免疫不全ウイルス1型もしくは2型(すなわちHIV−1もしくはHIV−2であり、HIV−1は、以前はリンパ節腫脹関連ウイルス3(HTLV−III)および後天性免疫不全症候群(AIDS)関連ウイルス(ARV)と呼ばれていた)、または、AIDSもしくはAIDS様の疾患に関連付けられた同定されたHIV−1もしくはHIV−2に関連する別のウイルスである。頭字語「HIV」または用語「AIDSウイルス」もしくは「ヒト免疫不全ウイルス」は、本明細書中で、これらのHIVウイルス、ならびにHIV関係およびHIV関連ウイルスを総称的に指すために使用する。さらに、亜科レンチウイルスのRNAウイルスは、たとえば、ビスナ/マエディウイルス(たとえばヒツジに感染)、ネコ免疫不全ウイルス(FIV)、ウシレンチウイルス、サル免疫不全ウイルス(SIV)、ウマ伝染性貧血ウイルス(EIAV)、もしくはヤギ関節炎−脳炎ウイルス(CAEV)、またはそれらの組合せであることができる。
【0097】
例示的なレンチウイルスベクターは、HIVに由来するもの、たとえば、HIV−1、HIV−2、またはそれらの組合せキメラである。もちろん、レトロウイルスの異なる血清型、特にHIVを単独または任意の組合せで用いて、本発明で用いるためのベクターを調製し得る。本発明のベクターは、たとえば、野生型ウイルス中には存在するが、「基本」レンチウイルスベクター中には存在しないシス作用性要素を含むことができる。「基本」レンチウイルスベクターは、最小限で5’リーダー配列およびgagコード配列中にLTRおよびパッケージング配列しか含まないが、任意選択で、Rev依存性の様式でベクターRNAの核外移行を容易にするために、RRE要素を含むこともできる。ベクターは、細胞内への形質導入の効率を増強するヌクレオチド配列をさらに含むことができる。
【0098】
そのようなベクターの例は、gagおよびpolの完全な配列を含むベクターである、pN2cGFPである。別の例は、pol中の位置が約4551〜約5096の配列を含むベクターである(参照位置は、pNL4−3配列、寄託番号M19921、HIVNL43 9709bp、C.E. Buckler、NIAID、NIH、メリーランド州Bethesdaから提供)。しかし、ベクターの形質導入効率を向上することができる、wt−HIVからの任意のシス作用性配列を用い得る。本発明による効率的な形質導入が可能なベクターの他の例は、米国特許第5,885,806号に記載のcr2HIV構築体である。
【0099】
Zennouら、2000年が中心DNAフラップ(pNL4−3上の位置4757〜4935に対応する、pLAI3上の位置4793〜4971の178塩基対の断片)として記載した、形質導入効率の有意に増加させるために不十分であったと以前に同定された配列は、形質導入効率を増加させることができる。本発明には、この小断片は形質導入効率を増加させるために不十分であるが、より大きな545塩基対の断片(pNL4−3中の位置4551〜5096)、またはそれを含むさらに大きな断片が、本発明の一部として形質導入を増加させることができたという発見が含まれる。
【0100】
本発明で用い得るウイルスベクター構築体のさらなる例は、完全に記載したかのように本明細書中に参考として組み込まれている米国特許第5,885,806号に見られる。特許5,885,806号の構築体は単なる例であり、細胞を効率的に形質導入するベクターの範囲を限定しない。むしろ、これらの構築体は、当業者に、本発明で用いるウイルスベクターが野生型ウイルスからの最小限の配列、または野生型ウイルスのゲノムのほぼ全体までの配列を含んでいても、疾患の複製および/または生産に必要な必須の核酸配列を排除し得るというさらなる指針を提供する。細胞の効率的な形質導入に必要な配列を正確に決定する方法は日常的であり、当分野で周知である。たとえば、ウイルス配列を「基本」ベクター内に戻す体系的な組み込ませること、またはcr2HIVなどの実質的にHIVゲノムの全体を含むベクターから配列を欠失させることは日常的であり、当分野で周知である。
【0101】
さらに、他のウイルスの主鎖からの配列を、サイトメガロウイルス(CMV)などの目的のウイルスベクター内に配置することも、当分野で周知である。実際に用いるウイルスベクターが何であるかにかかわらず、ウイルス遺伝物質によってコードされている様々なアクセサリータンパク質、およびウイルス遺伝物質中に存在する配列は、これらのタンパク質または配列が特定の細胞種において形質導入効率を増加させる場合は、ベクターまたはヘルパーゲノム中に残してもよい。特定の遺伝物質が、ベクターまたはヘルパーゲノムのどちらかに配列を組み込ませることによって形質導入効率を増加させるかどうかを決定するために、多数の日常的なスクリーニングが利用可能である。本発明の一実施形態は、ベクターまたはヘルパーゲノムのどちらかにアクセサリータンパク質を含めないことである。この実施形態は、形質導入効率を増加させるためにアクセサリータンパク質および他の配列をベクターまたはヘルパーゲノムのどちらかに残す本発明の実施形態を排除しない。
【0102】
本発明で用いるウイルスベクターは、「偽型」の形成の結果であってもよく、ここでは、異なるウイルスによる細胞の同時感染により、1つのウイルスのゲノムが別のウイルスの1つまたは複数のエンベロープタンパク質を含む外層内にカプセル封入されて含まれる、子孫ビリオンが生じる。この現象は、目的のウイルスベクターと別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質または細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションまたは同時感染を行うことによって、目的のウイルスベクターを「偽型」ビリオン内にパッケージングするために用いられている。米国特許第5,512,421号を参照されたい。このような混合ウイルスは、用いる1つまたは複数の異種エンベロープタンパク質に対する抗血清によって中和することができる。偽型の形成に一般的に用いられる1つのウイルスは、ラブドウイルスの水疱性口内炎ウイルス(VSV)である。偽型化を用いることで、用いる異種ウイルスの侵入機構の要素を含めることによって、ウイルスの宿主細胞範囲が広がる。
【0103】
本発明で用いるためのウイルスベクターおよびVSVの偽型化により、VSV Gタンパク質を含む膜に囲まれたヌクレオカプシド中にカプセル封入されたウイルスベクター核酸を含むウイルス粒子がもたらされる。ヌクレオカプシドは、通常はウイルスベクターに関連しているタンパク質を含むことができる。膜を含む周囲のVSV Gタンパク質は、ウイルスベクターのパッケージングに用いる細胞から放出された際にウイルス粒子の一部を形成する。パッケージング細胞の例は米国特許第5,739,018号に記載されている。本発明の別の実施形態では、ウイルス粒子はHIVに由来し、VSV Gタンパク質を用いて偽型にする。VSV Gタンパク質を含む偽型ウイルス粒子は、広宿主性のウイルスベクターよりも高い効率で多様なアレイの細胞種に感染することができる。宿主細胞の範囲には、ヒト、げっ歯類、魚、両生類および昆虫などの哺乳動物および非哺乳動物種がどちらも含まれる。
【0104】
また、本発明の形質導入方法で用いるためのウイルスベクターは、1つまたは複数の核酸配列を含み、ウイルス中に存在するプロモーターの制御下またはベクター内に導入した異種プロモーターの制御下でそれを発現することもできる。プロモーターは、厳しく制御された遺伝子発現のためにオペロンに隣接するように、赤血球DNAse過敏部位などの遮蔽要素をさらに含み得る。プロモーターの例には、HIV−LTR、CMVプロモーター、PGK、U1、エプスタインバーウイルス由来のEBER転写ユニット、tRNA、U6およびU7が含まれる。Pol IIプロモーターが有用であり、Pol IIIプロモーターも用い得る。組織特異的プロモーターの使用も、実施形態の1つである。たとえば、βグロビン遺伝子座調節領域エンハンサーならびにαおよびβグロビンプロモーターは、赤血球および赤血球細胞中で組織特異的発現をもたらすことができる。別の実施形態は、プロモーターに関連するシス作用性配列を用いることである。たとえば、本明細書中に参考として組み込まれている米国特許第5,814,500号に記載のように、U1遺伝子を用いてアンチセンス遺伝子発現を増強させ得る一方で、非プロモーター配列を用いて、標的のスプライシングされたRNAをアンチセンスまたはリボザイム分子の標的とする。
【0105】
もちろん、ウイルス由来の任意のシス作用性ヌクレオチド配列を本発明のウイルスベクター内に組み込ませ得る。たとえば、レトロウイルスゲノム中に見られるシス作用性配列を用いることができる。たとえば、形質導入効率をさらに増加させるために、gag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来するシス作用性ヌクレオチド配列を本発明のウイルスベクター内に組み込ませ得る。シス作用性配列は、発現されたポリペプチドをコードしている必要はなく;翻訳開始部位の欠失などの遺伝子変化によってポリペプチドもしくはその一部として発現される必要はなく;より大きなポリペプチドの一部分もしくは断片のみをコードしていることができ;またはネイティブ配列から1つもしくは複数の置換、付加、もしくは欠失を含む突然変異体配列であることができる。シス作用性配列の例は、HIV pol遺伝子内で同定されたcPPT(中央ポリプリントラクト)配列である。
【0106】
本発明のウイルスベクター中の1つまたは複数の核酸配列は、ベクターが由来するウイルス中に見つかるか、または異種配列であってもよい。これは、目的の遺伝子産物であるか、それをコードしている完全長配列または部分配列であることができる。このような配列および遺伝子産物は、細胞中で生物学的効果を生じる能力を有する、生物活性のある因子であることができる。そのような因子の例には、タンパク質、リボ核酸、酵素、トランスポーターまたは他の生物活性のある分子が含まれる。
【0107】
一実施形態では、因子は、毒素、転写因子、成長因子もしくはサイトカインなどのタンパク質、構造タンパク質、または細胞表面分子である。タンパク質は、機能が同定されていない1つまたは複数のドメインを含んでいてよく、形質導入した細胞に相同的であり得る。さらに、タンパク質は、形質導入する細胞中に存在しない、またはその中で欠損もしくは改変されていてもよい。あるいは、タンパク質は、形質導入した細胞中で天然タンパク質が正常の活性を実行することを妨げるための、トランス優勢の陰性突然変異体またはデコイであり得る。
【0108】
たとえば、核酸配列は、形質導入した細胞中で発現された、またはこれから発現されるRNAに結合し、切断し、破壊するリボザイムをコードし得る。あるいは、核酸配列は、特定の核酸配列を標的として、その分解をもたらすように設計されたアンチセンス分子をコードし得る。ベクターに含まれる配列は、形質導入した細胞中で過剰発現されるか、誘導発現されるか、または細胞もしくはウイルスの制御的転写調節下にあり得る。意図する使用に応じて、異種配列は、形質導入した細胞のマーカーを含めた任意の所望のタンパク質をコードし得る。そのようなマーカーには、ネオマイシン等の特定の耐性の表現型、MDR−1(P−糖タンパク質)、O6−メチルグアニン−DNA−メチルトランスフェラーゼ(MGMT)、ジヒドロ葉酸還元酵素(DHFR)、アルデヒドデヒドロゲナーゼ(ALDH)、グルタチオン−S−トランスフェラーゼ(GST)、スーパーオキシドジスムターゼ(SOD)およびシトシンデアミナーゼなどの選択マーカーが含まれる。総説には、本明細書中に参考として組み込まれているKocらを参照されたい。
【0109】
本発明の方法では、形質導入する細胞を、ウイルスベクターの施用の前、後、またはそれと同時に、細胞表面に結合する少なくとも1つの分子に接触曝露させる。たとえば、細胞は、形質導入するウイルスベクターの前、後、またはその存在下で、CD3およびCD28抗体(培養皿の表面上にコーティングまたは培養物中に存在させるビーズに固定)を有する培地中で培養することができる。細胞は、ウイルスベクターとの最初の接触後または接触時にしか、固定したCD3および/またはCD28に曝すことができない。このような条件下では、細胞は、ウイルスベクターを用いた実際の形質導入の前には細胞表面結合分子(または複数の細胞表面結合分子)に曝されない。細胞をウイルスベクターに曝した(形質導入)後に細胞表面結合分子との接触が起こる実施形態では、接触は形質導入後3日以内、あるいは形質導入後1〜2日以内に起こることができる。
【0110】
インキュベーションまたは細胞とウイルスベクターとの接触は、用いる条件および材料に応じて様々な長さの時間であり得る。インキュベーション時間に影響を与える要素には、用いる細胞、ベクターおよびMOI(感染多重度)、細胞表面の結合に用いる分子(または複数の分子)および量、前記分子(または複数の分子)が固定または可溶化されているかおよびどのように固定または可溶化されているか、ならびに所望する形質導入効率のレベルが含まれる。たとえば、インキュベーションは約8〜約72時間、または約12〜約48時間である。別の実施形態では、インキュベーションまたは接触は約24時間であり、任意選択で1回繰り返す。別の実施形態では、インキュベーションまたは接触は約24時間〜約36時間である。
【0111】
形質導入する細胞とウイルスベクターとの接触少なくとも1回起こるが、細胞種に応じて複数回起こり得る。たとえば、CD34陽性幹細胞の高効率形質導入が、ベクターを用いた複数の形質導入で達成されている。本発明の別の方法は、ウイルスベクターを細胞表面結合分子(たとえば、CD3および/もしくはCD28抗体またはFLT−3リガンド、TPOもしくはKitリガンド)と組み合わせて同時導入し、形質導入後の間の約1〜約8日間、培地の交換を避けることである。あるいは、形質導入後3日間、培地を交換しない。形質導入は、プロセスが細胞またはそれを含む生物に対して顕著に有害とならない限り、条件が許容する間、進行させることができる。このような使用のための細胞表面結合タンパク質のさらなる例には、本明細書中に上述したものが含まれる。
【0112】
使用するMOIは約1〜約400、または500未満であることができる。MOIは約2〜約50であることができる。あるいは、MOIは約10〜約30であるが、約1〜約10、約20、約30、または約40の範囲も企図される。あるいは、約20または約0.5〜10のMOI。さらに、細胞1個あたりのウイルスベクターのコピー数は少なくとも1であるべきである。しかし、細胞1個あたり多数のコピーのベクターも、上述の方法で用い得る。例示的な細胞1個あたりのコピー数は約1〜約100である。所望のコピー数は、ベクター形質導入の結果として治療的、予防的もしくは生物学的影響をもたらす、または最も効率的な形質導入をもたらす最小コピー数である。
【0113】
治療的または予防的応用には、所望のコピー数は、細胞またはそれを含む生物に対して顕著に有害とならずに細胞によって許容される最大コピー数である。細胞1個あたりの最小および最大コピー数はどちらも、形質導入する細胞および存在し得る他の細胞に応じて変動する。最適コピー数は当業者によって日常的な方法を用いて容易に決定される。たとえば、細胞を漸進的に増加する濃度または感染多重度で形質導入する。その後、細胞をコピー数、治療的または生物学的影響ならびに形質導入した細胞またはそれを含む宿主に対する有害効果(たとえば、安全性および毒性)について分析する。
【0114】
ウイルスベクターとin vitroでインキュベーションを行った後、細胞を細胞表面結合分子(または複数の細胞表面結合分子)の存在下で様々な期間培養し、その後、形質導入の効率について分析するか、または他の方法で用い得る。あるいは、細胞を細胞の成長および増殖をもたらす任意の条件下、たとえば、インターロイキン−2(IL−2)と共にインキュベーションまたは細胞表面結合分子(もしくは複数の細胞表面結合分子)と共に、次いでIL−2と共にインキュベーションなどで培養し得る。形質導入後のインキュベーションは任意の期間、たとえば約1〜約7〜10日間であり得る。約14日間などのより長い期間を用いてもよいが、細胞成長に有害な期間は望ましくない。ウイルスベクターとのインキュベーションの前に細胞を細胞表面結合分子(または複数の細胞表面結合分子)と共に培養する本発明の実施形態では、培養時間は約24〜約72時間の範囲、または約24時間であり得る。
【0115】
このような形質導入前の培養は、たとえば当分野で教示されている、形質導入前のサイトカインおよび/またはマイトジェンを用いた細胞の刺激と比較し得る。本発明には、そのような刺激の回避から生じる利点が含まれる。たとえば、刺激により、細胞の数が増殖によって増加して、刺激前よりも刺激後の細胞の方がはるかに多くなる。この増加した細胞セットの形質導入には、はるかに多数のウイルスベクターおよび関連する形質導入の材料(たとえば、容器、培地、サイトカインなど)が必要であり、関連するコストが増加する。さらに、細胞の刺激により、さらなる応用のためのその品質に影響が与えられる。Movassaghらは、3日間の形質導入前刺激の使用を記載しているが、これは形質導入およびさらなる培養後にT細胞レパートリー多様性の劣化をもたらす。さらに、形質導入前刺激により、活動的に分裂していない細胞の形質導入から得られる利点が取り除かれる。
【0116】
本発明で観察された形質導入の効率は約50〜100%である。たとえば、効率は少なくとも約50〜75%または約75〜90%である。本発明の他の実施形態は、形質導入効率が少なくとも約90〜100%である実施形態である。さらなる実施形態は、少なくとも91、92、93、94、95、96、97、98、99および100%の形質導入効率を有する。
【0117】
上記に加えて、形質導入した細胞は、研究または生きた対象における病状の治療もしくは予防に用い得る。研究使用の例は、Unutmazらによって記載されている構造−機能研究である。形質導入した細胞の治療的使用には、生きた生物内への細胞の導入が含まれる。たとえば、HIVに感染している、または感染している危険性にある個体から単離した未刺激の初代T細胞を、最初に、本発明の方法を用いて米国特許第5,885,806に記載のものなどのベクターによって形質導入し、続いて形質導入した細胞を個体に注射して戻し得る。あるいは、細胞をウイルスベクター中に存在する異種配列の発現に直接用い得る。
【0118】
HIVの治療または予防の一部として用いる場合は、ベクターは抗HIV用途に適用された毒素または他の抗ウイルス剤をコードしていてよい。あるいは、ベクターは、tat、rev、nef、vpu、またはvpr遺伝子のトランス優勢の陰性突然変異体など、HIVを標的とするよう設計された薬剤をコードしていてよい。他の応用では、形質導入した細胞は、鎌状赤血球貧血または地中海貧血を矯正するための適切なグロビン遺伝子を発現するように矯正し得る。また、免疫細胞も、その免疫機能、抗原に対するその応答、または他の細胞とのその相互作用を調節するために形質導入し得る。当業者は、本発明の形質導入方法ならびに当分野で知られている数々の他の使用および用途の上記の使用を認識しているであろう。
【0119】
本発明は、自己T細胞を製造し、およびT細胞を形質導入するプロセスを提供する。本発明の方法を用いることで、細胞をプラスチックバッグに対して中実のプラスチックフラスコ中で培養した場合に高い形質導入レベルが達成される。一態様では、大スケールの形質導入には、10層の細胞工場を用いる。一態様では、臨床的スケールでのT細胞の効率的な形質導入にはベクター体積の2倍縮小が必要である。一態様では、形質導入をさらに増加させるために、ベクター(T細胞の形質導入用)を24時間間隔で2回加える。
【0120】
一態様では、T細胞の増殖には、通常の培地よりも低い酸素濃度および僅かに低いpH。本発明では、通常の培地よりも低い酸素濃度および僅かに低いpHの存在下でT細胞がより良好に増殖することが判明した。一態様では、通常の空気(約20%のO2)と対比して、N2/O290%/10%を用いてT細胞を培養する。一態様では、pHを低下するために、混合空気中のCO2濃度を通常の5%から10%まで増加する。この混合気体の変更により、より高い増殖率が可能となった。
【0121】
一態様では、増殖の終わりに約1000億個の細胞、培地の灌流を用いる。本発明者らは、増殖の終わりに約1000億個の細胞を保有するためには、培地の灌流を用いなければならないことを発見した。一態様では、以前に用いられていた20Lバッグでは十分な細胞を支持することができなかったので、50Lの灌流バッグを用いる。一態様では、細胞濃度が0.5×106個の細胞/mlを超えたらすぐに、約3L/日の速度で灌流を開始する。一態様では、翌日、揺動速度および角度を1単位同時に増加して、速度を約2倍増加する。
【0122】
一態様では、収集中、続く凍結および解凍後のその生存度を増加するためにT細胞を冷却する。一部の用途では、大量の細胞の収集は長時間かかり、細胞はより低い温度でより良好に生存するので、これを行う必要がある。一態様では、細胞をより小さな10Lバッグに移し、冷却のために冷蔵庫に入れる。細胞の洗浄には冷蔵した緩衝液を用いるべきである。
【0123】
本発明は、約75%を超える効率までのウイルスベクターを用いた細胞の安定形質導入を行うための、方法およびそれに関連する組成物を対象とする。安定形質導入した細胞は、形質導入の約7〜10日後、または任意選択で約14日後に、一過的に形質導入したまたは偽形質導入した細胞から区別し得る。この方法は、形質導入する細胞と細胞表面に結合する少なくとも1つの分子との接触により安定形質導入の効率が増加するという事実に関連する。驚くべきことに、接触ステップは形質導入ステップの後に起こってもよい。さらに驚くべきことに、最も高い安定形質導入レベルは、形質導入が最初に起こり、続いて固定した細胞表面結合分子との接触が起こった場合に見られた。
【0124】
本発明の方法は、細胞表面結合分子との接触と組み合わせた、ウイルスベクターを用いた形質導入のステップを含む。上に注記したように、接触は、ベクターの形質導入の前、後、またはそれと同時に起こり得る。本発明は、任意の細胞および任意の細胞表面結合分子の使用に広く適用可能である。本方法で用いる細胞には、in vivo供給源から新しく単離した未刺激の初代細胞、および細胞を増殖状態に保つ因子の存在下で様々な期間事前に培養していてもよい細胞系が含まれる。細胞系を用いる場合は、本方法を用いて形質導入する前に、最初に刺激因子を存在させずに培養してもよい。
【0125】
初代細胞の場合は、最初にこれらをin vivo供給源から得て、続いて任意選択で特定の細胞種について選択を行う。たとえば、初代CD4+および/またはCD8+T細胞を用いる場合は、末梢血(PB)または臍帯血(臍源由来の「CB」)試料を最初に得て、続いてCD4+および/またはCD8+細胞種について富化を行う。標準の磁気ビーズ陽性選択、プラスチック接着性陰性選択、および/または当分野で認められている他の標準技術を用いて、CD4+および/またはCD8+細胞を混入PB細胞から離して単離し得る。単離した細胞種の純度は、標準技術を用いた免疫表現型決定およびフローサイトメトリーによって決定し得る。
【0126】
単離後、50%を超える、または75%を超える効率でウイルスベクターを用いて形質導入するために、初代細胞を本方法で用い得る。本発明は、T細胞などの初代リンパ球、目的の異種遺伝物質を発現する能力を有するHIV−1系ベクターで形質導入したにおいて、最も有利に用いられる。別の使用は、CD34陽性細胞などの初代造血幹細胞を用いた使用である。異種遺伝物質が疾患を治療または予防するためにin vivoで使用する治療的または予防的生成物であるか、それをコードしている場合は、形質導入した初代細胞を対象などのin vivo環境に導入して戻すことができる。したがって、本発明は、遺伝的欠陥と戦うことまたはウイルス感染を標的とすることによる、疾患を治療または予防する遺伝子治療における形質導入した細胞の使用を企図する。
【0127】
また、本発明は、遺伝子の機能を決定するため、哺乳動物細胞中で遺伝子を効率的に発現するため、目的の遺伝子の機能的スクリーニングのために遺伝子ライブラリ(cDNAライブラリおよび遺伝子のアンチセンスもしくはリボザイムライブラリ)を発現するため、タンパク質−タンパク質またはタンパク質−核酸の二重ハイブリッド様の検出戦略、遺伝子捕捉手法、マイクロアレイもしくはタンパク質アレイを用いた高スループット遺伝子スクリーニング分析で使用するため、またはSAGE、プロテオミクスおよび他の機能的分析方法を用いて研究を行うために、細胞を効率的に形質導入するための使用も企図される。
【0128】
混合集団における初代細胞の形質導入には、上記の単離/精製ステップは使用しない。その代わりに、形質導入する細胞は、少なくとも1つの適切な細胞表面分子またはその細胞種上に見つかる部分の選択および前記部分に結合する能力を有する1つまたは複数の分子の調製によって標的とする。細胞表面部分は、標的とした細胞の表面上にある受容体、マーカー、または他の認識可能なエピトープであり得る。選択後、本発明で用いるために、特異的抗体などの部分と相互作用する分子を調製し得る。
【0129】
たとえば、CD4+および/またはCD8+細胞は、最初に精製し、その後、固定したCD3およびCD28抗体を用いて本発明の方法によって形質導入するか、または末梢血細胞(PBC)もしくは末梢血単核細胞(PBMNC)などの混合集団の一部として、同じ抗体を用いることによって形質導入することができる。精製または単離が困難であり得る全白血球集団中の造血幹細胞は、固定したCD34抗体を用いることによって混合集団中で形質導入し得る。
【0130】
本発明の細胞表面結合分子は、形質導入する細胞の表面上に見られる任意の部分を標的としてそれに結合し得る。部分は、細胞表面上の受容体、マーカー、または他のタンパク質性もしくは非タンパク質性因子の一部として見つかる。部分には、細胞表面結合分子によって認識されるエピトープが含まれる。これらのエピトープには、ポリペプチド配列、炭水化物、脂質、核酸、またはイオンおよびそれらの組合せを含むものが含まれる。
【0131】
細胞表面結合分子の例には、抗体またはその抗原結合断片およびリガンドまたは細胞表面受容体の結合ドメインが含まれる。細胞表面結合分子自体がポリペプチド、核酸、炭水化物、脂質、またはイオンであり得る。分子は、抗体またはFabもしくはFv断片などのその断片であることができる。あるいは、分子は可溶形で用いるのではなく、形質導入する細胞をそれと共に培養し得るビーズなどの固体培地上、または形質導入する細胞をそれ上で培養し得る組織培養皿、バッグもしくはプレートの表面に固定されている。一実施形態では、CD4+またはCD8+細胞の形質導入には、CD3および/またはCD28を認識するモノクローナル抗体は、ウイルスベクターの存在下で、細胞培養バッグ中で用い得る。
【0132】
本発明には、開示した方法の一部として用いるための細胞表面結合分子を含む組成物が含まれる。例示的な組成物は、形質導入する分子およびウイルスベクターを、任意選択で形質導入する細胞の存在下で含む。ウイルスベクターは、任意の源、たとえばレトロウイルスベクター由来であり得る。たとえば、これらはレンチウイルスベクターであることができる。例示的なレンチウイルスベクターは、ヒト免疫不全ウイルス(HIV)、たとえば、HIV−1、HIV−2、またはそれらのキメラの組合せに由来するものである。もちろん、本方法を用いることによって異なるウイルスベクターを同一細胞内に同時に形質導入し得る。たとえば、1つのベクターが複製欠損性または条件的に複製するレトロウイルスベクターであることができ、一方で第2のベクターは第1のベクターの複製/パッケージングおよび増殖を可能にするパッケージング構築体であることができる。様々なウイルスアクセサリータンパク質をウイルスベクターによってコードさせる場合は、細胞内に形質導入するベクターの任意の1つ中に存在し得る。あるいは、ウイルスアクセサリータンパク質は、形質導入に用いるウイルス粒子中のその存在を介して、形質導入プロセスに存在し得る。このようなウイルス粒子は、形質導入効率の増加をもたらすために有効な量の同時パッケージングされたアクセサリータンパク質を有し得る。一実施形態では、ウイルスベクターは、アクセサリータンパク質の1つまたは複数をコードしない。
【0133】
本発明の形質導入方法で用いるためのウイルスベクターはまた、1つまたは複数の核酸配列を含むことができ、プロモーターの制御下でそれを発現することができる。本発明の一実施形態では、核酸配列は、発現の際に形質導入する細胞において遺伝的欠陥を緩和または矯正する遺伝子産物をコードしている。別の実施形態では、核酸配列は、ウイルス感染症を予防または治療することができる遺伝的抗ウイルス剤をコードまたは構成している。「遺伝的抗ウイルス剤」とは、遺伝物質によってコードまたは構成されている任意の物質を意味する。そのような薬剤の例は米国特許第5,885,806号に提供されている。これらには、逆転写酵素もしくはプロテアーゼなどのウイルスタンパク質を阻害すること;結合部位もしくは標的部位についてウイルス因子と競合すること;またはリボザイムおよびアンチセンス構築体の場合などのようにウイルス標的を直接標的として分解することによって機能する薬剤が含まれる。遺伝的抗ウイルス剤の他の例には、アンチセンス、RNA囮、トランス優勢の突然変異体、インターフェロン、毒素、RNAスプライシングを調節または改変する核酸、免疫原、および「ハンマーヘッド」などのリボザイム、ならびにそれらの外部ガイド配列(EGS)に媒介された形態が含まれる。
【0134】
あるいは、ウイルスベクターは、形質導入した細胞のマーカーをコードすることができる。図および以下に示す例では、CD4+細胞内に形質導入したウイルスベクターによってコードされているマーカーは緑色蛍光タンパク質(GFP)である。他のマーカーには、上述のものが含まれる。GFPの検出は、機能的に形質導入された細胞の数を同定する役割を果たし得、これらは、ベクターで形質導入されただけでなく、FACS分析によって検出されるレベルでGFPを機能的に発現することができた。一部の細胞は、ベクターで形質導入されているが、FACS検出で用いる限界未満のレベルでGFPを発現している可能性があるので、検出は形質導入した細胞の実際の数を表さない可能性があることに注意されたい。
【0135】
トランスフェクション効率を検出する代替手法は、ポリメラーゼ連鎖反応(PCR)を用いる方法である。たとえば、TaqMan PCRを用いて、形質導入した細胞中の安定に組み込まれたウイルスベクターの実際のコピー数を決定することができる。
【0136】
形質導入する細胞は、細胞表面結合分子との接触の前、後、またはそれと同時に、ウイルスベクターと接触曝露させ得る。したがって、最初に細胞をベクターに一定期間曝し、続いて細胞表面結合分子の導入を行うことができる。このような細胞は、細胞周期に入るように意図的に刺激していない、新しく単離したまたは調製した初代細胞であり得る。あるいは、最初に細胞を細胞表面結合分子に一定期間曝し、続いてウイルスベクターと接触させることができる。ベクターとの接触後、過剰のベクターを除去する必要はなく、細胞の成長および/または増殖に貢献する条件下で細胞を培養することができる。そのような条件は、細胞表面結合分子またはT細胞の場合はサイトカインおよびリンホカインなどの他の刺激/活性化因子の存在下であり得る。あるいは、細胞との接触後かつさらなる培養の前に過剰のベクターを除去し得る。
【0137】
本発明の別の実施形態は、ウイルスベクターおよび細胞表面結合分子をどちらも同時に存在させて細胞を培養することである。このような細胞は、事前に刺激する必要がない。一定期間後、連続的に存在する細胞表面結合分子または他の刺激/活性化因子などの成長または増殖誘導条件下で細胞を培養する。あるいは、さらなる培養の前に過剰のベクターを除去し得る。
【0138】
ウイルスベクターおよび細胞表面結合分子の投与の上記の任意の組合せにおいて、ベクターとのインキュベーションを任意選択で少なくとも1回繰り返すことができる。ベクターとの接触は、複数回、たとえば2回、3回、4回、またはそれ以上繰り返すこともできる。
【0139】
ウイルスベクターを用いて形質導入する細胞のインキュベーションは、用いる条件および材料に応じて様々な長さの時間であり得る。インキュベーション時間に影響を与える要素には、用いる細胞、ベクターおよびMOI(感染多重度)、細胞表面の結合に用いる分子(または複数の分子)および量、前記分子(または複数の分子)が固定されているかおよびどのように固定されているか、ならびに所望する形質導入効率のレベルが含まれる。本発明の一実施形態では、細胞はTリンパ球であり、ベクターはHIV系であり、MOIは約20であり、細胞表面結合分子はビーズ上に固定したCD3およびCD28抗体であり、生じる効率は少なくとも93%である。当業者には明らかなように、上記要素の一部は直接相関しており、一方で他の要素は逆相関している。たとえば、MOIの低下は効率のレベルを低下させる可能性が高く、一方で用いる細胞表面結合分子の量を増加した場合は効率が維持される可能性が高い。
【0140】
ウイルスベクターおよび形質転換させる細胞のインキュベーションの長さは、たとえば24時間であることができ、任意選択で、リンパ球では1回、造血幹細胞では4回まで繰り返す。同様に、ウイルスベクターを導入する前に細胞を細胞表面結合分子と共にインキュベーションする実施形態では、インキュベーションは約12時間〜約96時間であり得る。細胞表面結合分子とのインキュベーションは、細胞とウイルスベクターとの接触と同時に起こることができる。このような状況下では、ベクターを導入する際に細胞表面結合分子を細胞と接触させたままであってもよい。あるいは、ベクターを細胞に導入する前に、最初に過剰の細胞表面結合分子を培養から除去してもよい。
【0141】
ベクターとの接触後、細胞を、その成長または増殖に貢献する条件下で培養する。たとえば、条件は、細胞表面結合分子の存在下における連続培養である。あるいは、細胞を最初に細胞表面結合分子と共に培養し、続いてインターロイキン−2などの細胞成長に貢献する別の因子を含む培地に置き換える。さらに別の実施形態は、過剰の細胞表面結合分子および過剰のベクターをどちらも除去し、続いて成長または増殖に貢献する因子の存在下での培養およびさらなるベクター形質導入を増強することであろう。そのような因子には、フィトヘマグルチニン(PHA)およびサイトカインなどのマイトジェン、成長因子、活性化剤、細胞表面受容体、細胞表面分子、可溶性因子、あるいはそれらの組合せ、ならびに単独でまたは別のタンパク質もしくは因子と組み合わせたそのような分子の活性断片、あるいはそれらの組合せが含まれる。
【0142】
さらなる因子の例には、表皮成長因子(EGF)、トランスフォーミング成長因子α(TGF−α)、アンジオテンシン、トランスフォーミング成長因子β(TGF−β)、GDF、骨形態形成タンパク質(BMP)、線維芽細胞成長因子(FGF酸性および塩基性)、血管内皮成長因子(VEGF)、PIGF、ヒト成長ホルモン(HGH)、ウシ成長ホルモン(BGH)、ヘレグリン、アンフィレギュリン、Ach受容体誘導活性(ARIA)、RANTES(活性化の際に制御、正常T発現および分泌)、アンジオゲニン、肝細胞成長因子、腫瘍壊死因子β(TNF−β)、腫瘍壊死因子α(TNF−α)、アンジオポイエチン1もしくは2、インスリン、インスリン成長因子IもしくはII(IGF−IもしくはIGF−2)、エフリン、レプチン、インターロイキン1、2、3、4、5、6、7、8、9、10、11、12、13、14、もしくは15(IL−1、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、IL−12、IL−13、IL−14、もしくはIL−15)、G−CSF(顆粒球コロニー刺激因子)、GM−CSF(顆粒球−マクロファージコロニー刺激因子)、M−CSF(マクロファージコロニー刺激因子)、LIF(白血病阻害因子)、アンジオスタチン、オンコスタチン、エリスロポイエチン(EPO)、インターフェロンα(亜型を含む)、インターフェロンβ、γ、およびω、ケモカイン、マクロファージ炎症性タンパク質−1αもしくはβ(MIP−1αもしくはβ)、単球化学走性タンパク質−1もしくは−2(MCP−1もしくは2)、GROβ、MIF(マクロファージ遊走阻害因子)、MGSA(黒色腫成長刺激活性)、αインヒビンHGF、PD−ECGF、bFGF、リンフォトキシン、ミュラー阻害物質、FASリガンド、骨原性タンパク質、プレイオトロフィン/ミッドカイン、毛様体神経栄養因子、アンドロゲン誘導性成長因子、自己分泌運動性因子、ヘッジホッグタンパク質、エストロゲン、プロゲステロン、アンドロゲン、糖質コルチコイド受容体、RAR/RXR、甲状腺受容体、TRAP/CD40、EDF(赤血球分化因子)、Fic(成長因子誘導性ケモカイン)、IL−1RA、SDF、NGRもしくはRGDリガンド、NGF、チモシン−α1、OSM、ケモカイン受容体、幹細胞因子(SCF)、またはそれらの組合せが含まれる。当業者には明らかなように、培養条件の選択は、形質導入する細胞に関する知識、および続く細胞の使用意図に依存する。たとえば、IL−3、IL−6および幹細胞因子の組合せは、ヒト移植に用いる形質導入細胞の選択肢ではない。同様に、培養条件の選択は、細胞生存度または形質導入効率に有害でないことが望ましい。
【0143】
形質導入後のインキュベーションは、たとえば、約4時間、または約1日間から約7〜10日間の期間である。形質導入後のインキュベーションは、たとえば、約16〜約20時間、または約4日間、約5日間もしくは約6日間であることもできる。約14日間の形質導入後インキュベーションも企図される。
【0144】
本発明で観察される形質導入の効率は約50〜100%である。効率は、少なくとも約50〜75%、または少なくとも約75〜90%であることができる。本発明の他の実施形態は、形質導入効率が少なくとも約90〜95%である実施形態である。さらなる実施形態は、少なくとも91、92、93、94、95、96、97、98、99および100%の形質導入効率を有する。
【0145】
上記に加えて、形質導入した細胞は、研究または生きた対象における疾患状態の治療に用い得る。本発明の一部として、目的の遺伝子産物を産生するための形質導入した細胞の治療的使用または遺伝子治療の一部としての生きた生物への直接導入が存在する。たとえば、以下に例示するように、初代T細胞を単離して、ウイルスベクターを用いて形質導入することができる。形質導入の成功は、ベクターによってコードされている遺伝子産物の産生もしくは過剰産生、またはベクターによって与えられた表現型の発生によって示される。したがって、最初に初代T細胞を、望ましいまたは有用な核酸配列を含み、それを発現する能力を有するベクターを用いて形質導入し、その後、生きた対象などのin vivo環境に戻すことができる。たとえば、生きた対象は、HIV−1に感染している、または感染している危険性にある個体である。
【0146】
別の実施形態では、T細胞を、宿主生物内に導入された際にT細胞を条件的に死滅させる能力を有する遺伝子または核酸を用いて形質導入する。これは、同種骨髄移植において、プロドラッグ手法を用いてT細胞を死滅させることによる移植片対宿主病の予防に応用がある。
【0147】
あるいは、初代細胞は遺伝子産物に欠陥を有することができ、血管は形質導入したウイルスベクターによって矯正可能である。このような細胞は、ベクターを用いて形質導入した後に、生きた対象内に再導入する。
【0148】
したがって、本発明のin vitroおよびex vivoの応用がどちらも企図される。生きた対象内への導入には、形質導入した細胞を、たとえば、生物学的に許容される溶液または製薬上許容される配合物中に入れる。そのような導入は、静脈内、腹腔内または当分野で知られている他の注射および非注射方法によって行い得る。投与する用量は様々な要素に応じて変動するが、当分野の従事者によって容易に決定され得る。本発明には、ウイルスベクター中に既知またはよく設計されたペイロードを用いた数々の応用があり、形質導入した遺伝物質によって与えられる利点が負の効果のすべてのリスクに勝る。
【0149】
最初は、導入する形質導入した細胞の合計数約104〜約1010個である。したがって、105、106、107、108、または109個の細胞を用い得る。実際の数は形質導入する細胞に応じて変動する。必要な場合は、形質導入した細胞の複数の導入が別の実施形態である。さらに、必要な場合は、形質導入した細胞を導入する前の宿主の調節が別の実施形態である。調節レジメンは当分野で知られており、レジメンの例は骨髄移植のレジメンである。
【0150】
多層の容器またはフラスコ中で成長させることができる細胞の量は、少なくとも約1億個の細胞、または少なくとも約7千万個の細胞、または少なくとも約8千万個の細胞、または少なくとも約9千万個の細胞である。
【0151】
本発明は、ウイルスベクターおよびウイルス粒子を用いて細胞の安定形質導入を行うための、効率の高い方法およびそれに関連する組成物を提供する。本発明は、形質導入した細胞、たとえばレンチウイルスベクターで改変した細胞、たとえば自己CD4+T細胞、たとえば本明細書中に記載の例示的なVRX496で形質導入したCD4+T細胞の細胞処理を行うための、新規の製造施設および製造プロセスを提供する。
【0152】
一態様では、自己T細胞を製造するための本発明のプロセスは、以下に記載のように、たとえば血液の凍結アフェレーシスによるリンパ球の単離、たとえばCytoMate(商標)中での洗浄、その後、CD4+富化、次いでCD8の枯渇、次いでウイルス、たとえばレンチウイルスを用いた形質導入を含む。さらなる処理を以下に記載し、本明細書中の図に例示する。
【0153】
一態様では、自己VRX496で形質導入したCD4+T細胞を産生するための出発物質は末梢血単核細胞(PBMC)である。PBMCは、HIVに感染した対象から白血球アフェレーシス中に得る。白血球アフェレーシス手順は、自動細胞分離器を用いて血液採取施設で起こることができる。
【0154】
一態様では、血漿を除去するために細胞を洗浄し、CD4抗原の発現に基づいたヒト細胞の分離のために開発されたCD4マイクロビーズ(Miltenyi Biotech、ドイツ)で磁気標識した(インキュベーション)。第I相臨床研究中、血漿を除去するために出発物質を低速遠心分離によるフィコール密度勾配分離を行い、その後、COBE(Baxter)洗浄を行い、標準緩衝液中に再懸濁させた。その後、洗浄した細胞材料を、CD8高密度微粒子(CD8−HDMニッケルビーズ)(Biotransport)と共にインキュベーションし、続いてEligix Magnetic Cell Separation Systemを用いて磁気分離を行った。
【0155】
第II相臨床研究では、CYTOMATE(商標)Cell Processing System(Miltinyi Biotech、ドイツ)を用いて血漿を除去するための細胞洗浄を行うことができる。CYTOMATE(商標)Cell Processing Systemは、細胞生成物を洗浄および濃縮するため、および液体移送用途のための、独立型の閉鎖された自動装置である。これは、低い細胞損失および高い生存度の効率的な細胞洗浄を可能にする。この系は、cGMP環境における細胞処理のための閉鎖系の液体経路を生み出す使い捨てチューブセットを特徴とする。また、これは液体移送を柔軟、急速かつ正確にする。液体を、すべて1つの閉鎖系の液体経路の内部で、単一または複数の容器から中および外に移送することができる。
【0156】
一態様では、加えたCD4+マイクロビーズ(Miltinyi Biotech)のインキュベーション中の非特異的な細胞結合を防ぐために、免疫グロブリン溶液(Immune Globubin Intravenous、USP、Grifols)を加える。一態様では、最終産物のバッグ(CD4マイクロビーズのインキュベーションした細胞懸濁液)を熱密封し、バッグを取り出して生物学的安全フッド下に置く。
【0157】
一態様では、Eligix(商標)Cell Separation SystemをCD8の枯渇に用いる。第II相臨床研究では、CD4+陽性選択は、CliniMACS(商標)磁気細胞分離系介して行うことができる。この系では、(1)移送パック容器と、(2)緩衝液バッグおよび細胞懸濁液バッグに接続するためのメスルアーアダプターを備えた血漿移送セットと(3)陽性選択バッグおよび廃棄物採取バッグに接続するためのメスルアーアダプターを備えた血漿移送セットとからなる滅菌CliniMACS(商標)使い捨てセットを用いる。
【0158】
一態様では、滅菌使い捨てセットの緩衝液および細胞懸濁液系をそれぞれのバッグに加えることは、滅菌を保つために生物学的安全フッド下で行うことができる。リン酸緩衝生理食塩水(PBS)緩衝液および細胞懸濁液系を加えた後、使い捨てセットをCliniMACS(商標)に取り付けることができ、CD4磁気標識細胞懸濁液をCliniMACS(商標)に通すことができる。採取した陽性画分を用いてプロセスを続けることができる。
【0159】
一態様では、PBSからX−VIVO−15培地(Cambrex;メリーランド州Walkersville)への交換は、CytoMate(商標)を介して達成される。一態様では、最終産物のバッグを取り外し、熱密封し、生物学的安全フッド下に置く。メスルアーアダプターを備えた移送セットを生成物のバッグに取り付けることができ、以下についてのQC試験のために、シリンジによって5ccの試料を得る:CD3+CD8+細胞およびCD3+CD4+細胞の割合、細胞生存度、細胞数、および増殖前HIV gagの測定値。一態様では、QCの結果が得られるまで細胞の産生を停止する。細胞が規格に合っていれば、細胞の形質導入で産生を続ける。
【0160】
一態様では、CD3/CD28同時刺激ビーズ(Dynal beads、ノルウェー、Oslo、抗CD3(OKT3)および抗CD28(UPennモノクローナル抗体9.3)でコーティングをCD4+T細胞懸濁液に加え、続いてVRX496ウイルスベクター産物を加える。一態様では、CD4+T細胞、X−VIVO+5%ヒト血清アルブミン、IL2、NAC、CD3/CD28マイクロビーズおよびVRX496ベクター懸濁液(5%W/V)の混合物全体を、RetroNectin(Takara Bio、日本)でコーティングしたNunc(商標)細胞工場に加え、細胞工場を加湿した37℃、5%CO2インキュベーターに入れる。翌日、VRX496ベクター懸濁液(5%W/V)を再度加える。一態様では、細胞をベクターと共に3日間インキュベーションし、その後、WAVE(商標)細胞バッグに移し、Wave(商標)バイオリアクター(WAVE(商標)Biotech LLC、ニュージャージー州Bridgewater)に入れる。
【0161】
WAVEバイオリアクターは、特別な揺動プラットホームを有する。このプラットホームの揺動動作が、培養溶液に波を誘起する。これらの波が混合および酸素の伝達を提供し、20×106を超える細胞/mlを容易に支持することができる、細胞成長に完璧な環境をもたらす。バッグおよび様々な接続装置上のチューブ導線により(接続はスパイクコネクターを介し、溶接はTerumo Sterile Connecting Deviceを介して行う)、汚染の危険性を最小限にして細胞を閉鎖系で成長させることが可能となる。
【0162】
ベクターを除去するために、4日目に、CytoMate(商標)細胞洗浄器を用いて細胞をX−VIVO15で2回洗浄することができる。
【0163】
収集時まで培養を7〜12日間保つ。少なくとも1日おきに細胞を計数し、細胞を約0.5〜1.5×106個の細胞/mlの密度に保つために新鮮な培地を加える。細胞の培養中にHIVの複製を阻害するために、抗レトロウイルス薬(Norvir、Abbot Laboratories、およびRetrovir、GlaxoSmithKline)(1μmol/L)を加える。他の種類の抗レトロウイルス薬を用いることができる。当業者は、適切な抗レトロウイルス薬の選択および投与方法を知っているであろう。約10日目に、細胞は収集の準備が整う。増殖後のHIVコピー数が増殖前のHIVコピー数を超えていないことを確認するために、増殖後HIV gag測定を行う。収集前の細胞から、マイコプラズマについて試験するために試料を採取する。
【0164】
一態様では、CD3/CD28マイクロビーズは、培養バッグをMaxSep(商標)磁石(Baxter)上に通すことによって除去する。ビーズは磁石上に保持され、細胞は別のバッグ中に注がれる。細胞は、残留ビーズについてアッセイする。
【0165】
別段に定義しない限りは、本明細書中で用いるすべての専門用語および科学用語は、本発明が属する分野の技術者に一般的に理解される意味と同じ意味を有する。本明細書中で引用したすべての特許、特許出願、公開特許出願および他の出版物は、その全体で参考として組み込まれている。本セクションに記載した定義が、本明細書中に参考として組み込まれている特許、特許出願、公開特許出願および他の出版物に記載されている定義に反するまたは他の様式で矛盾している場合は、本セクションに記載した定義が、本明細書中に参考として組み込まれている定義に勝る。
【0166】
本発明の1つまたは複数の実施形態の詳細を、添付の図および以下の説明に記載する。本発明の他の特徴、目的、および利点は、説明および図、ならびに特許請求の範囲から明らかであろう。
【0167】
本発明を一般に説明したが、本発明は、例示として提供する、指定のない限りは本発明を限定することを意図しない以下の実施例を参照することでより容易に理解されるであろう。
【0168】
(実施例1)
初代CD4+T細胞の調製
CD4+T細胞は、軽微な改変をもつ標準のプロトコルを用いて末梢血から単離した。より詳細には、汚染単球を付着によって枯渇させた。CD4+細胞の陽性選択を行うために、非接着性細胞を抗CD4抗体でコーティングした磁気ビーズの存在下に置いた。磁気ビーズを除去し、CD4+細胞を単離した。
【0169】
高度に精製されたCD4+細胞は、フローサイトメトリーによって90%を超えることが確認された。
【0170】
(実施例2)
様々な時間の細胞表面結合分子との接触を用いた初代CD4+T細胞の形質導入
細胞表面結合前の形質導入
初代CD4+細胞(約500,000個)をpN2cGFPと共に、20のMOIで24時間培養し、次いで、さらに7日間、培養物にαCD3およびαCD28でコーティングしたビーズを加えた。図2はpN2cGFPのマップを含む。
【0171】
細胞表面結合後の形質導入
初代CD4+細胞(約500,000個)を24時間、αCD3およびαCD28でコーティングしたビーズと共に24時間培養し、次いで、さらに24時間、pN2cGFPを20のMOIで培養物に導入した。過剰のベクターを除去するために細胞を洗浄し、次いで、ビーズを含む、ベクターを含まない培地中でさらに7日間インキュベーションした。
【0172】
同時の形質導入および細胞表面結合
初代CD4+細胞(約500,000個)をpN2cGFPと共に、20のMOIで24時間、αCD3およびαCD28でコーティングしたビーズの存在下で培養した。過剰のベクターを除去するために細胞を洗浄し、次いで、ビーズを含む、ベクターを含まない培地中でさらに7日間インキュベーションした。
【0173】
任意選択のプロトコルの置換
他のウイルスベクターでpN2cGFPを置換し得る。さらに、過剰のベクターを除去する前に形質導入を合計2回繰り返し得る。さらに、形質導入および過剰のベクターの除去後に、αCD3およびαCD28でコーティングしたビーズをインターロイキン−2(10ng/ml)およびPHA−P(3mg/ml)によって置換し得る。7日後、培地をインターロイキン−2(10ng/ml)を含むPHA−Pを含まない培地で置き換え、インキュベーションをさらに7日間続ける。
【0174】
あるいは、αCD3およびαCD28でコーティングしたビーズと共に形質導入後のインキュベーションを7日間行った後、細胞を洗浄し、インターロイキン−2(10ng/ml)の存在下でインキュベーションを続ける。
【0175】
(実施例3)
形質導入後の分析
形質導入後およびインキュベーションの7または14日後、細胞を、CD4+および/または緑色蛍光タンパク質(GFP)についてフローサイトメトリーによって分析した。
【0176】
上記3つの形質導入プロトコルの比較を図3に示す。pN2cGFPを用いた20のMOIでの形質導入後の、ビーズに固定したCD3およびCD28抗体との接触により、約91%の効率がもたらされた。形質導入前でのビーズとの接触では約89%の効率がもたらされ、同時のビーズ接触および形質導入では約80%の効率がもたらされた。この実験では、CD4+T細胞は、接着性単球枯渇、CD14MACS枯渇およびCD4MACS富化によって選択した。抗体は、以下に記載のように固定した。ベクターとの接触は37℃、5%のCO2で行った。培養条件は、2%のヒト血清アルブミンを添加したアイセル培地中に500,000個のCD4+T細胞/mlであった。FACS分析は選択後7日目に行った。MFとは、平均蛍光をいう。
【0177】
様々な刺激条件を比較する、7日後の実験結果を図4に示す。CD4+細胞をIL−2およびPHA−Pのどちらかまたはビーズに固定したCD3およびCD28抗体で24時間処理し、次いでpN2cGFPを20のMOIで用いた形質導入を1ラウンド行った。隣り合わせの比較では、固定した抗体を使用することにより、毎回95%を超える形質導入効率がもたらされた(CD4およびGFPのどちらに対しても陽性な細胞によって示された)。それに比較して、IL−2およびPHA刺激を用いた結果では、70.2〜84.5%の効率しかもたらされなかった。FACS分析は選択後7日目に行った。
【0178】
図5は、選択後15日での類似の実験の結果を示す。ここでも、細胞をIL−2およびPHA−Pのどちらかまたはビーズに固定したCD3およびCD28抗体で24時間処理し、次いでpN2cGFPを20のMOIで用いた形質導入を1ラウンド行った。PHA−Pおよびビーズは形質導入後7日目に除去し、細胞は選択後15日目までIL−2のみと共に500,000個の細胞/mlで培養した。固定した抗体を使用した後、細胞の約93%がCD4およびGFPのどちらに対しても陽性であった。IL−2およびPHAで処理した細胞の約75%のみが、CD4およびGFPのどちらに対しても陽性のまま保たれた。これらの結果は、7日後に陽性として検出された細胞の小さな画分(図4)は、「偽型トランスフェクション」が原因であった可能性を示している。
【0179】
(実施例4)
様々なベクターが細胞を高効率で安定に形質導入する:
本例は、形質導入に用いたベクターの比較である。pN2cGFPはgagおよびpolコード配列の全体を含む一方で、pN1(cpt)cGFPは4551〜5096の部分的(非コード)pol配列を含む。図6に示す結果から見ることができるように、どちらのベクターも、ビーズに固定したCD3およびCD28抗体ならびに20のMOIのベクターで同時刺激を行った後、初代CD4細胞の非常に効率的な形質導入を示す。FACS分析は選択後10日目に行った。
【0180】
(実施例5)
トランスフェクション効率に対するMOIの効果
様々なMOIの効果を図7に示し、2〜20のMOIを使用することにより、72.7〜83.8%の形質導入効率がもたらされた。様々なMOIでのpN1(cpt)CGFPを用いた形質導入の前に、細胞をビーズに固定したCD3およびCD28抗体と24時間接触させた。
【0181】
(実施例6)
CD34陽性細胞の形質導入
CD34陽性細胞を臍帯血から調製し、FLT−3リガンド、TPOおよびKitリガンド(それぞれ100ng/ml)の存在下で、pN1cptGFPを用いて4回同時に形質導入した。細胞を長期培養(LTC−IC)で5週間培養し、その後、分析前に細胞をメチルセルロース中で10日間培養した(結果は、6週間の培養にわたる経過時間からのものである)。図8の結果は、CD34未熟細胞から生じる成熟CD45陽性細胞を分析している。対照細胞は有意な形質導入を示さなかったが、ベクターで形質導入した細胞は、88%を超える細胞がCD45およびGFPに陽性であることを示す。
【0182】
(実施例7)
CD34陽性細胞の長期形質導入
上述のように、CD34陽性細胞をpN1(cpt)GFPで形質導入し、部分的に照射したSCIDマウスの骨髄に移植した。8週間後、細胞を単離し、FACSによってCD45を保有する成熟ヒト細胞およびGFP発現について分析した。結果を図9、パネルA〜Dに示す。
【0183】
パネルAは、ベクターで形質導入していないヒト細胞を移植した対照マウスを用いた結果を示す。
【0184】
パネルBは、50のMOIで連続した4日間、100ng/mlのFLT−3リガンド、TPOおよびKitリガンドの存在下で、pN1(cpt)GFPベクターで形質導入した細胞で形質導入した細胞を移植したマウスを用いた結果を示す。このマウスは、形質導入の8週間後に、形質導入したヒト細胞(CD45陽性細胞)が96.3%という印象的な形質導入効率を示す。このマウスにおけるヒト細胞移植のレベルは11.1%であり、これは以前に報告された結果と矛盾しない。
【0185】
パネルCおよびDは、パネルBと同様に治療した2匹の他のマウスを用いた結果を示す。この結果により、87.8%および89.6%のCD45陽性細胞という高効率形質導入、ならびにGFP陽性であることの再現性が確認された。
【0186】
平均効率は91.2%であり、これは長期安定形質導入を反映している。
【0187】
(実施例8)
細胞表面結合分子の固定
この例は、以下の実施例で用いるための、エポキシdynalビーズに対するCD3(B−B11)抗体およびCD28(B−T3)抗体の直接結合を記載している。
【0188】
1.0.618gのホウ酸を95mlの組織培養グレードの水に溶かすことによって0.1ホウ酸溶液を調製する。よく混合し、最高品質のNaOHを用いてpHを9.5に調節する。最終体積を100mlにし、0.2μmのフィルターを介して滅菌する。容器を密閉し、4℃で保存する。
【0189】
2.抗体を上記ホウ酸溶液に150μg/mlの濃度で加える。B−B11およびB−T3抗体のどちらについても、ホウ酸溶液1mlあたりそれぞれ75μgを加える。体積を合計1mlにする。ホウ酸濃度は、抗体を加えた後に0.05M以上であるべきである。1mlのホウ酸/抗体溶液のそれぞれに、4×108個のエポキシビーズを加える。
【0190】
3.24時間、37℃で、回転輪上でインキュベーションを行う。
【0191】
4.ビーズを3回、それぞれ10分間、22℃で、ビーズ洗浄媒体(カルシウムおよびマグネシウムを含まないリン酸緩衝生理食塩水、3%のヒト血清アルブミン、5mMのEDTA、ならびに0.1アジ化ナトリウム)を用いて洗浄する。
【0192】
5.ビーズを1回、30分間、22℃で洗浄する。
【0193】
6.4℃で終夜洗浄する。
【0194】
7.新鮮なビーズ洗浄媒体に交換し、ビーズを2×108個のビーズ/mlで再懸濁させる。IgGでコーティングしたビーズは、4℃で少なくとも6カ月安定である。
【0195】
(実施例9)
樹状細胞の形質導入
末梢血由来の単球を単離し、その後、50のMOIで連続した3日間、2つの同時のサイトカイン条件、すなわち、GM−CSF(800単位/ml)、IL−4(500単位/ml)およびTNF−α(100単位/ml)またはGM−CSF(500単位/ml)およびインターフェロン−α(800単位/ml)を用いて、pN2cGFPで形質導入した。図10、パネルAは、形質導入の7日後の結果を示し、第1のサイトカイン条件では90.2%の効率がもたらされた。第2のサイトカイン条件下でベクターを用いて形質導入した細胞は、7日後に92.9%の効率を示した(パネルB)。CD86が樹状細胞の唯一の可能なマーカーであり、CD86陰性細胞は樹状細胞であり得ることに注意されたい。
【0196】
(実施例10)
製造施設
この要約では、レンチウイルスベクターで改変した細胞、具体的には自己VRX496で形質導入したCD4+T細胞の細胞を処理するための新しい製造施設および製造プロセスを説明する情報を提供する。
【0197】
第I相臨床研究中、自己VRX496で形質導入したCD4+T細胞は、ペンシルバニア大学(UPenn)、細胞およびワクチン産生施設(Cell and Vaccine Production Facility、CVPF)で製造した(Levineら、2002年)。
【0198】
第II相臨床研究では、自己VRX496で形質導入したCD4+T細胞生成物を製造する。このプロセスにより、レンチウイルスベクターによって改変したHIVに感染したCD4T細胞の、前例のない100倍を超える増殖が可能となる。
【0199】
本要約には、以下を提示する:
全体的な施設配置図を含めた製造施設の説明、
出発物質、プロセス中およびリリースの品質管理試験、安定性を含めた新しい製造プロセスの説明、ならびに
製造の一貫性およびUPennのCVPFのプロセスのデータとの比較を実証するデータ。
【0200】
(実施例11)
全体的な施設の説明
GMP製造面積および品質管理(QC)試験面積は封じ込められており、物理的な仕切りによって他の設立面積および互いから隔離されている。また、製造面積およびQC試験面積はカードキーの鍵によって制限および制御されている。
【0201】
2つのクリーンルームの使用意図は、VRX496レンチウイルスベクターの臨床的cGMP産生用およびこのベクターを用いた対象細胞の対応するcGMP ex vivo形質導入用である。ベクター産生用のクリーンルームは複数の生成物産生施設であり、細胞処理用のクリーンルームは、現時点では自己VRX496で形質導入したCD4+T細胞の産生のみを目的とする。ベクター産生と対象細胞の形質転換との間には適切な切替え手順が配置されている。すべての対象細胞性生物、バーコードシステムによって産生プロセス全体にわたって追跡する(以下を参照)。
【0202】
相互汚染のすべての可能性を最小限にするために、ベクター産生および細胞処理操作では人員を共有せず、領域は位置が物理的に隔離されている。書面による標準操作手順(SOP)およびこれらの手順で訓練した人員によってさらなる制御を維持する。
【0203】
ベクター産生用のクリーンルームスイートおよび細胞処理用のクリーンルームスイートは、どちらもcGMP産生および生物学的安全性の封じ込めのために設計されている。どちらのクリーンルームも、クラス10,000(ISOクラス7)および生物学的安全性レベル2(BSL−2)、大スケールとして設計されている。
【0204】
クリーンルームは、個別の空調ユニット(AHU)を有する。ベクタークリーンルームのAHUは、供給および戻りファン;45%前置フィルターおよび95%最終フィルター;冷却コイル、ならびにDX凝縮ユニットを含む、一定体積の再循環ユニットである。細胞処理用のクリーンルームのAHUは、100%外気を供給する。クリーンルームの仕上げは、滑らかで硬い清掃可能な防水かつ化学薬品耐性の表面、および一体カバーを備えた継ぎ目のないビニル製の床材から構成される。扉は安全ガラスの覗きパネルを備えた亜鉛めっき鋼から構成される。扉の金具の特徴には、蹴板、モップ板および扉閉鎖が含まれる。
【0205】
産生領域内の移動と同様、品質管理(QC)研究室間の移動はSOPによって制御されている。また、汚染のすべての危険性を最小限にするために、DNA抽出研究室(Q2)はPCR研究室(Q5およびQ6)から物理的に隔離されている。
【0206】
(実施例12)
製造された細胞生成物
本補正書中で製造および記載した例示的な細胞生成物の名称は、自己VRX496で形質導入したCD4+T細胞である。
【0207】
VRX496は、ヒト免疫不全ウイルス(HIV)エンベロープ遺伝子を標的とした937個のヌクレオチドのアンチセンス配列を含む。
【0208】
自己VRX496で形質導入した細胞を輸液バッグに分割した(90mlのバッグ1個あたり5×109〜1010個の形質導入した細胞)。細胞を以下からなる輸液用低温培地に懸濁させた:
31.25%のPlasmalyte−A、
31.25%のデキストロース(5%)、
0.45%の塩化ナトリウム、
7.5%のジメチルスルホキシド(DMSO)、
1%のデキストラン40、および
5%のヒト血清アルブミン。
【0209】
(実施例13)
出発物質
以下の表に、自己VRX496で形質導入したCD4+T細胞の産生に用いた出発物質を例示的に記載する。
【0210】
【化5】
【0211】
【化6】
すべての出発物質は、材料管理の人員によって受取りおよび検査を行った。検査には、認可された「原材料規格書および受取りシート」の完全記入が含まれる。ロット番号を割り当て、梱包ラベルを調査し、認可された「原材料規格書および受取りシート」の適合性について分析証明書(C of A)を再調査した。
【0212】
品質保証(QA)では、材料管理によって完全記入された「原材料規格書および受取りシート」を再調査し、材料を認可または却下する。材料がQAによって「認可」された場合は、「認可」と標識され、材料管理の人員によって倉庫領域内の適切な認可された保存場所に移送される。
【0213】
出発物質がQAによって「却下」された場合は、「却下」と標識され、最終的な処理、すなわち、廃棄するか、販売者に返却するか、またはR&Dへ移送するかが決定されるまで、認可された材料から隔離される。
【0214】
すべての出発物質はQAによって目録ログに入力される。このログには、割り当てられたロット番号、受け取った量、処理、および使用期限が含まれる。この目録には、製造プロセスまたはQC試験手順で用いる緩衝液などの企業内用配合物が含まれる。QAによって、月末に期限の切れる材料の月ごとの表が材料管理および/または産生用に作成されて、倉庫領域からのその除去が確実となり、不注意による使用が防がれる。
【0215】
(実施例14)
産生および日常的な制御
プロセスの流れ図
細胞の処理精製(図11)および製造手順(図12)の図を添付する。
【0216】
(実施例15)
プロセスの説明
細胞の採取方法
自己VRX496で形質導入したCD4+T細胞を産生するための出発物質は末梢血単核細胞(PBMC)である。PBMCは、白血球アフェレーシス中にHIVに感染した対象から得られる。白血球アフェレーシス手順は、自動細胞分離器(Cobe Spectra CS−3000、Baxter;コロラド州、Lakewood)を用いて血液採取施設で起こる。
【0217】
第II相臨床研究中にそれぞれ約5×109から10〜1010個の自己VRX496で形質導入したCD4+T細胞の8つまでの細胞輸液を対象に与え得るので、約3〜4倍血液量(15L)の血液をCobe Spectraを通して処理し、十分なPBMC(約100〜200億個)を採取し、細胞の洗浄および選択手順(すなわち精製)を受けさせ、細胞の形質導入および増殖プロセスを開始するために必要な数のCD4T細胞(すなわち約10〜20億個)をもたらす必要がある。1回の白血球アフェレーシス手順は完了するまで約3時間かかる。
【0218】
対照的に、第I相臨床研究中では、それぞれの対象は約1×1010個の自己VRX496で形質導入したCD4+T細胞の輸液を1回のみを受けたので、採取された白血球アフェレーシス生成物はより少なく、約70mL中に約50億個のPBMCからなる。
【0219】
白血球アフェレーシス生成物は、採取した日に、周囲温度で、それぞれの血液センターからその生成物がさらに処理される場所へと、IATAおよびDODの規制に従って空輸または陸輸配達によって発送される。産生の人員が採取後24時間のうちに白血球アフェレーシス生成物を受け取って処理することを確実にするために、移送時間の計画を立てる。
【0220】
生成物は自己かつ感染性であるので、生成物の追跡制御を確実にし、すべての生成物の混同の可能性を減らすために、それぞれの白血球アフェレーシスのバッグを以下で標識する:
固有のロット番号、
含まれる細胞体積、
固有のバーコードラベル、
対象の研究ID(研究の現場の識別を含む)、
対象のイニシャル、および
対象の生年月日。
【0221】
混同の可能性を減らすために採るさらなる予防措置は、以下である:
細胞処理用のクリーンルーム内では任意の時点で2つの個別の細胞生成物だけを操作することを許可すること、および
産生操作は、産生プロセス中の様々な段階(たとえば、CD4+T細胞の選択、ベクターの除去または細胞の収集)を含まなければならないことを記載した書面による手順。
【0222】
この方針の例外はインキュベーションおよび保存ステップ中であり、任意の時点で80個もの個別の生成物のインキュベーションをWAVE(商標)バイオリアクター中で行い、30個までを冷凍庫内で保存し得る。
【0223】
バーコードシステムによる支援の下、製造プロセス全体にわたって細胞生成物を追跡する。
【0224】
(実施例16)
白血球アフェレーシス生成物の受取り
白血球アフェレーシス生成物が細胞処理施設で受け取られた後、品質保証(QA)は受取室でバーコードの読取りを行い、同時に生成物のバッグのラベルおよび記録を確認する:
受け取った総体積、
バッグに記載のロット番号、
バッグを受け取った時間、および
白血球アフェレーシス時から受取時までの時間数。
【0225】
QAが白血球アフェレーシス生成物をリリースした後、材料管理の人員によって細胞処理用のクリーンルーム(クラス10,000)(生物学的安全性レベル2、大スケール)に届けられる。産生の人員が約5ccの細胞生成物の試料をシリンジで採り、QC試験のためにバイアルに加える。全CD4+生細胞のQC試験は≧6×108個の細胞であるべきである。
【0226】
(実施例17)
血漿の洗浄およびMACS CD4インキュベーション
細胞を洗浄して血漿を除去し、CD4抗原の発現に基づいたヒト細胞の分離のために開発されたCD4マイクロビーズ(Miltenyi Biotech、ドイツ)で磁気標識した(インキュベーション)。
【0227】
第I相臨床研究中、血漿を除去するために出発物質を低速遠心分離によるフィコール密度勾配分離を行い、その後、COBE(Baxter)洗浄を行い、標準緩衝液中に再懸濁させた。その後、洗浄した細胞材料を、CD8高密度微粒子(CD8−HDMニッケルビーズ)(Biotransport)と共にインキュベーションし、続いてEligix Magnetic Cell Separation Systemを用いて磁気分離を行った。
【0228】
第II相臨床研究では、CYTOMATE Cell Processing System(Miltinyi Biotech、ドイツ)を用いて血漿を除去するための細胞洗浄を行う。CYTOMATE Cell Processing Systemは、細胞生成物を洗浄および濃縮するため、および液体移送用途のための、独立型の閉鎖された自動装置である。これは、低い細胞損失および高い生存度の効率的な細胞洗浄を可能にする。この系は、cGMP環境における細胞処理のための閉鎖系の液体経路を生み出す使い捨てチューブセットを特徴とする。また、これは液体移送を柔軟、急速かつ正確にする。液体を、すべて1つの閉鎖系の液体経路の内部で、単一または複数の容器から中および外に移送することができる。
【0229】
さらに、加えたCD4+マイクロビーズ(Miltinyi Biotech)のインキュベーション中の非特異的な細胞結合を防ぐために、免疫グロブリン溶液(Immune Globubin Intravenous、USP、Grifols)を加える。
【0230】
最終産物のバッグ(CD4マイクロビーズのインキュベーションした細胞懸濁液)を熱密封し、バッグを取り出して生物学的安全フッド下に置いた。以下を行うために約5ccのQC試料を採取した:
細胞濃度および
CD3+CD8+の割合およびCD3+CD4+の割合を決定するためのFACS分析。
【0231】
QCの結果を受け取るまで産生を停止する(約30分間)。CytoMate最終産物体積の体積を計算する。
【0232】
(実施例18)
CD4+の選択
上に注記したように、第I相臨床研究中、Eligix(商標)Cell Separation SystemをCD8の枯渇に用いた。第II相臨床研究では、CD4+陽性選択はCliniMACS磁気細胞分離系を介して行う。この系では、(1)移送パック容器と、(2)緩衝液バッグおよび細胞懸濁液バッグに接続するためのメスルアーアダプターを備えた血漿移送セットと(3)陽性選択バッグおよび廃棄物採取バッグに接続するためのメスルアーアダプターを備えた血漿移送セットとからなる滅菌CliniMACS使い捨てセットを用いる。
【0233】
滅菌使い捨てセットの緩衝液および細胞懸濁液系をそれぞれのバッグに加えることは、滅菌を保つために生物学的安全フッド下で行った。
【0234】
リン酸緩衝生理食塩水(PBS)緩衝液および細胞懸濁液系を加えた後、使い捨てセットをCliniMACSに取り付け、CD4磁気標識細胞懸濁液をCliniMACSに通す。採取した陽性画分を用いてプロセスを続ける。
【0235】
第I相臨床研究中の2ステップのプロセス、すなわち、CD8の枯渇およびCD3の選択から第II相臨床研究中の1ステップのプロセス、すなわち、CD4の選択へと変更した理論的根拠は、細胞処理効率およびより純粋な細胞生成物を得るためである。
【0236】
(実施例19)
緩衝液から培地への交換
PBSからX−VIVO−15培地(Cambrex;メリーランド州、Walkersville)への交換は、CytoMateを介して達成される。最終産物のバッグを取り外し、熱密封し、生物学的安全フッド下に置いた。メスルアーアダプターを備えた移送セットを生成物のバッグに取り付け、以下についてのQC試験のために、シリンジによって5ccの試料を得る:
CD3+CD8+細胞およびCD3+CD4+細胞の割合、
細胞生存度、
細胞数、および
増殖前GIV Gag測定。
【0237】
QCの結果が得られるまで細胞の産生を停止する。細胞が規格に合っていれば、細胞の形質導入で産生を続ける。
【0238】
(実施例20)
CD4+T細胞の形質導入
CD3/CD28同時刺激ビーズ(Dynal beads、ノルウェー、Oslo、抗CD3(OKT3)および抗CD28(UPennモノクローナル抗体9.3)でコーティングをCD4+T細胞懸濁液に加え、続いてVRX496ウイルスベクター産物を加える。CD4+T細胞、X−VIVO+5%ヒト血清アルブミン、IL2、NAC、CD3/CD28マイクロビーズおよびVRX496ベクター懸濁液(5%W/V)の混合物全体を、RetroNectin(Takara Bio、日本)でコーティングしたNunc(商標)細胞工場に加え、細胞工場を加湿した37℃、5%CO2インキュベーターに入れた。翌日、VRX496ベクター懸濁液(5%W/V)を再度加える。細胞をベクターと共に3日間インキュベーションし、その後、WAVE(商標)細胞バッグに移し、Wave(商標)バイオリアクター(WAVE(商標)Biotech LLC、ニュージャージー州、Bridgewater)に入れる。
【0239】
WAVEバイオリアクターは、特別な揺動プラットホームを有する。このプラットホームの揺動動作が、培養溶液に波を誘起する。これらの波が混合および酸素の伝達を提供し、20×106個を超える細胞/mlを容易に支持することができる、細胞成長に完璧な環境をもたらす。バッグおよび様々な接続装置上のチューブ導線により(接続はスパイクコネクターを介し、溶接はTerumo Sterile Connecting Deviceを介して行う)、汚染の危険性を最小限にして細胞を閉鎖系で成長させることが可能となる。
【0240】
(実施例21)
ベクターを除去するための洗浄
4日目に、CytoMate細胞洗浄器を用いて細胞をX−VIVO15で2回洗浄する。
【0241】
(実施例22)
細胞の増殖
収集時まで培養を7〜12日間保つ。少なくとも1日おきに細胞を計数し、細胞を約0.5〜1.5×106個の細胞/mlの密度に保つために新鮮な培地を加えた。細胞の培養中にHIVの複製を阻害するために、抗レトロウイルス薬(Norvir、Abbot Laboratories、およびRetrovir、GlaxoSmithKline)(1μmol/L)を加える。約10日目に、細胞は収集の準備が整う。増殖後のHIVコピー数が増殖前HIVコピー数を超えていないことを確認するために、増殖後のHIV gag測定を行う。収集前の細胞から、マイコプラズマについて試験するために試料を採取する。
【0242】
(実施例23)
洗浄、体積縮小および配合
細胞のバッグをCytoMateに載せ、細胞を栄養素培地から洗浄して出し、以下からなる輸液用低温培地溶液に入れる:
31.25%のPlasmaLyte A、
31.25%のデキストロース5%、
0.45%のNACl、
5%のヒト血清アルブミン(HSA)、
1%のデキストラン40、および
7.5%のDMSO。
【0243】
(実施例24)
CD3/CD28マイクロビーズの枯渇
CD3/CD28マイクロビーズは、培養バッグをMaxSep(商標)磁石(Baxter)上に通すことによって除去する。ビーズは磁石上に保持され、細胞は別のバッグ中に注がれる。細胞は、残留ビーズについてアッセイする。
【0244】
(実施例25)
冷凍保存
VRX496で形質導入したCD4+T細胞は、速度を制御して凍結した。生成物を冷却する細胞は生成物が相転移点に達するまで1℃/分で冷却し、その後、温度が−90℃に達するまで凍結速度を増加する。
【0245】
(実施例26)
品質管理(QC)リリース試験
QCリリース試験用に試料を採取する。細胞生成物は、QC試験の完了まで液体窒素冷凍庫の蒸気相中で保存する(設定値<−130℃)。
【0246】
(実施例27)
品質保証(QA)リリース
QC試験の完了後、QAはすべての試験の再調査を行い、リリース規格書が満たされている場合は、臨床治験で用いるための細胞生成物のリリースを認可する。
【0247】
(実施例28)
保存
QAからリリースされた細胞生成物は、臨床施設の現場へ運送する準備が整うまで液体窒素中で保存する。
【0248】
(実施例29)
臨床施設の現場への運送
細胞生成物は、液体窒素蒸気運送容器(Chart Inc.、ジョージア州、Marietta、以前はMVE Cryogenics)中、≦140℃の温度で、契約輸送業者(Cavalier Logistics Management,Inc.、バージニア州、Dulles)によって、その自社貨物トラックまたは民間航空会社によって臨床施設の現場に輸送される。これらの低温運送容器は、請け負わせたその内容物を8日間維持することが確認されている。
【0249】
(実施例30)
品質管理(QC)試験
QCのプロセス中の試験
プロセス中のQC試験を以下のように行う。開発のこの段階では、これらの試験は情報としてのみ行う。
【0250】
【化7】
(実施例31)
QCのリリース試験
最終的な細胞生成物のリリース試験および規格書を、以下の分析の証明書に示す。リリース試験はプロセスステップで、および以下に示す被験物質に対して行う。
【0251】
【化8】
【0252】
【化9】
(実施例32)
産生プロセスの認定
第I相および第II相の間で行った主要な製造の変更の要約
表1は、第I相および第II相の間で行った主要な製造の変更の要約を示す。
【0253】
【化10】
(実施例33)
アフェレーシスの細胞生成物および陽性選択した後の細胞生成物の品質:
第I相対第II相
表2は、第I相および第II相の出発物質(すなわち、洗浄後のアフェレーシス生成物)と陽性選択した後の細胞生成物(すなわち、CD4+T細胞の形質導入および細胞の増殖を開始するために用いた細胞生成物)とのCD4+T細胞の純度の比較を提供する。
【0254】
これらのデータから見られるように、第II相細胞産生プロセスは、より純粋なCD4+出発物質(平均28.04%のCD4対第I相で14.58%のCD4)ならびにVRX496形質導入および細胞の増殖を開始するためのより純粋なCD4+細胞生成物(平均95.60%のCD4対第I相で36.82%のCD4をもたらす。
【0255】
さらに、データは、第II相産生プロセスは、第I相臨床研究で用いた生成物よりも純度が一貫した陽性選択後の細胞生成物をもたらすことを示す。これは、単一のCD4+陽性選択ステップに寄与することができ、一方で、第I相プロセスでは2ステップのプロセス、すなわちCD8+枯渇およびCD3+/CD4+陽性選択を用いた。
【0256】
【化11】
(実施例34)
VRX496形質導入効率の比較:第I相対第II相
表4は、第I相細胞生成物および第II相開発ロットの間のVRX496平均ベクターコピー数/細胞の比較を示す。これから見ることができるように、平均ベクターコピー数は本質的に第I相から変化なしに保たれるが、第II相開発ロットで、平均ベクターコピー数は第I相のそれよりも一貫性がある。
【0257】
【化12】
(実施例35)
第II相開発ロットのリリース試験の結果の要約
表5は、第II相開発ロット1、2および3のリリース試験の結果の要約を示す。3つの開発ロットすべてがロットリリースの規格を満たしていた。
【0258】
【化13】
(実施例36)
細胞生成物の安定性
第I相臨床治験のために製造したVRX496で形質導入したCD4+T細胞生成物を凍結保存し、対象に輸液を行う予定まで≦−80℃で保存した。輸液の前日、細胞生成物のセンチネルバイアル試料を解凍し、細胞生成物リリース基準の一部として細胞生存度を測定した。第I相の製造した生成物のそれぞれの細胞生存度は≧70%であった。<−80℃保存の最長期間は6カ月であった。これらのデータは、≦−80℃で6カ月間まで保存した場合の自己VRX496で形質導入した細胞生成物の安定性を支持している。
【0259】
安定性を評価するために、自己VRX496で形質導入したCD4+T細胞の24カ月間の安定性研究を6つの自己VRX496で形質導入したCD4+T細胞のロットで行う。これらのロットは、既存の製造プランに従って製造した2つの異なるVRX496ベクターのロットを用いて形質導入する(すなわち、3つのVRX496で形質導入した細胞生成物のロット/ベクターロット)。保存条件は液体窒素である。形質導入した細胞生成物の試料(15ml)を3、6、12、18および24カ月にアッセイする。0時でのデータを、形質導入した細胞生成物のロットのリリースデータとする。それぞれの時点でのアッセイに用いるために、十分な試料(20個のバッグ/ロット)を細胞処理終了時に採取する。アッセイには、外観、Gtagコピー数、細胞生存度、回収、細胞内サイトカイン染色、滅菌性、および細胞外DNA濃度が含まれる。それぞれの時間間隔の試験の完了時に暫定的な報告書を作成する。最終報告書は研究の最後に作成する。QA部門が、生じたデータの完全性を保証すること、およびcGMPとのコンプライアンスを確証する責任を持つ。作成されたすべての生データ、記録および報告書は、企業内で保有する。保有する記録には、保存条件、保存ユニットの妥当性確認および維持、試料の調製ならびに生のアッセイデータが含まれる。
【0260】
(実施例37)
自己細胞生成物の追跡手順
4人の異なる対象の自己CD4+T細胞を同時に処理し得る。この同時製造中にこれらの細胞生成物を潜在的な混同および汚染から保護するために、これら4つの細胞生成物を産生の異なる段階中に処理する。最新の適正製造基準に従う。認可された書面の細胞処理手順が存在し、すべての産生人員はこれらの手順の訓練を受ける。産生ロットを切り替える手順では専用の産生装置を用いる。重要な装置(インキュベーター、冷凍庫、HVAC)は妥当性が確認されている。使用する処理用水およびすべての産生用材料は認可された販売者から、確立された規格書に従って得る。細胞処理手順全体にわたって対象細胞生成物を追跡するための以下の特別な制御も実行されている:
(実施例38)
バーコードシステム
特注デザインのバーコードシステムにより、細胞生成物およびQC試験プロセスの全体、すなわち、受取り、細胞の形質導入、増殖、冷凍保存、保存、梱包および運送にわたって対象細胞を追跡する。
【0261】
バーコードシステムは、監査追跡、ユーザレベルアクセスおよび完全な報告能力を提供する。
【0262】
対象細胞生成物を処理または試験する前に、産生人員が処理または試験する材料およびバッチ産生記録または品質管理(QC)試験書類に添付されているバーコードの両方を、完全に一致するかどうかスキャンする。これらが一致しない場合は、コンピュータスクリーン上に警告が表示される。その場合、材料をスキャンする人は、照合を行ったことを証明しなければならず、バッチ産生記録またはQC試験書類にイニシャルおよび日付を記入する。
【0263】
(実施例39)
書類
それぞれの対象細胞生成物ロットの書類には、処理中に対象細胞生成物を視覚的に区分するために異なる色を割り当てる(すなわち、バッチ産生記録およびQC試験書類に固有の色)。
【0264】
(実施例40)
処理中の細胞生成物の隔離および制御
任意の時点において1人の産生人員だけが1つの対象細胞生成物で作業することが許可され、次の対象細胞生成物の処理を行うことができる前に、この細胞生成物に関与するすべての操作を終えなければならない。
【0265】
すべての開放された場での細胞生成物の操作は、クラス100生物学的安全フッド内で行う。任意の時点で1人の対象由来の細胞のみを操作する。
【0266】
対象細胞生成物はWAVE(商標)バッグ内でインキュベーションし、それぞれの対象細胞生成物ロットはそれ専用のWAVE(商標)インキュベーターを有する。
【0267】
フッド内に入れる緩衝液および試薬などの原材料は1つの対象細胞生成物ロットに専用であり、処理の最後に廃棄する。
【0268】
(実施例41)
保存中の細胞生成物の隔離および責任
一度に1つの対象細胞生成物のみを凍結保存する。
【0269】
冷凍保存中、それぞれの対象細胞生成物バッグは金属カセット内で保護される。冷凍保存後、これらのカセットをケーブル線で接続し、冷凍庫のラック内に隔離して保存する。
【0270】
保存されているすべての生成物の目録をバーコードシステムおよびハードコピーの書類によって維持する。
【0271】
(実施例42)
現在のプロセスの概観
間もなく行われる施設での第I相/第II相臨床治験の提案された細胞処理手順を以下に要約する。手短に述べると、最初に、COBE2991細胞プロセッサー(Gambro BCT)を用いて、アフェレーシス生成物に赤血球(RBC)枯渇を行う。その後、生じた生成物をMiltenyi抗CD4MACSと共にインキュベーションし、COBE2991細胞プロセッサーで洗浄する。抗CD4とインキュベーションした生成物を、恐らくは最大収率のために2回、CliniMACS装置で処理する。
【0272】
CD4で選択された生成物は、刺激ビーズの存在下、RetroNectinでコーティングしたバッグ中で、ベクターを用いてすぐに形質導入する。形質導入は3日間、37℃、5%のCO2インキュベーターで実施する。形質導入後の細胞は、Cytomate装置(Baxter Oncology)を用いて洗浄した後、8〜10日間の期間、Waveバイオリアクター中で増殖する。増殖後、Isolex 300iまたはMaxSep(どちらもBaxter Oncology)を用いて刺激ビーズを除去し、細胞培養物の体積を縮小し、再度Cytomateを用いて細胞を洗浄し、冷凍保存用に調製する(配合)。冷凍保存はCryo−Med速度制御冷凍庫を用いて行い、細胞は蒸気相液体窒素MVEタンク中に保存する。全体的に、プロセスは11〜13日間かかるはずである。
【0273】
提案のとおり、現在の細胞処理手順は時間がかかり、かつ高価であるが、迅速に実行することができる。このプロセスで確認された主なコストは抗体選択ステップであり、多数の対象を処理するための主な制限は8〜10日間の増殖ステップである。
【0274】
以下に、現在の細胞処理手順を簡素化することを目的としたいくつかの技術的な代替方法を示し、実行が容易なものから始める。
【0275】
第1の技術的代替方法は増殖ステップの長さに関し、8〜10日間から0日間へと短縮する。手短に述べると、3日間形質導入した細胞を直接冷凍保存処理する(ビーズ枯渇、洗浄、および配合)。生成物の調製時間を11〜13日間から3日間へと短縮することによって、同じ期間内により多くの生成物を処理することが可能となり(4個対1個)、また、増殖に関連するコストも削減される(Waveバイオリアクターおよび培地)。
【0276】
この第1の代替方法に関連して、実行できる精製ステップが制限されるまたは実行できず、したがって関連する精製コストが削減される。
【0277】
第2の代替方法は、過剰かつ時間のかかる操作手順なしに臨床施設の現場で用いられるほど単純な形質導入キットの作製である。手短に述べると、生アフェレーシス生成物を、CD8+およびCD19+リンパ球(Stemcell Technologiesの製品RosetteSep)などの、RBCを所望しない細胞と結合させる抗体カクテルと共に直接インキュベーションする。コンパクトな自動かつ密閉型のSepax装置(Biosafe)を用いて、フィコール層上での遠心分離中に所望しない細胞をRBCと共に沈殿させる。単核細胞を採取し、同じ装置を用いてフィコールを洗浄して除去し、ベクターおよび生分解性刺激ナノビーズを既に含むテフロン(登録商標)バッグに移す。形質導入を3日間、37℃、5%のCO2インキュベーターで実施した後、再度Sepax装置を用いて洗浄し、すぐに対象に再注射する。
【0278】
第3の代替方法を図15に要約する。この技術的代替方法では、1つの処理ステップ、すなわちアフェレーシス手順のみを用い、臨床施設の現場で数時間内に行われる。手短に述べると、対象は、現在および他の提案された代替方法と同じ方法でアフェレーシス手順を受ける。RBCおよび血漿を対象に連続的に再輸液しながら、濃縮白血球は通常バッグ内に採取する。その後、採取した白血球は、対象に再輸液する前に採取バッグ中で形質導入させる。ex vivo操作は必要ない。
【0279】
(実施例43)
単離
RBC枯渇のみを行うことが最も安価な代替方法であり、臨床グレードの抗体は必要ない。RBC枯渇は時間がかからず、装置は1点しか必要としない。しかし、CD4含有量に関しては制御がない。
【0280】
Sepax装置を用いた制限のあるCD8CD19枯渇の代替方法は、1点の装置のみを必要とするが、枯渇手順(3)に必要な臨床的抗体の数およびStemcell Technologies IPの技術許可が原因でより拡張的な可能性がある。
【0281】
現在の単離手順、すなわちCD4陽性選択は、実行するために2点の装置および1つの臨床グレードの抗体(間もなく市販される)を必要とする。コストは制限された選択と同様の可能性があるが、この手順はより時間がかかる。しかし、CD4含有量の制御は良好である。
【0282】
1年前に、CD14枯渇のみ、またはCD14およびCD8の枯渇、またはCD14枯渇およびCD4精製を受けた細胞の、フローサイトメトリーによってアッセイした形質導入レベルが同様であったことが実証されている。しかし、その差異は7日間の培養期間後の増殖のレベルにあった。
【0283】
(実施例44)
増殖
現在のプロセスの培養期間は8〜10日間である。
【0284】
提案された代替方法は、増殖時間を形質導入に必要な最小限の時間に短縮することである。しかし、3日間操作したT細胞のin vivo増殖の潜在性を評価するために現在利用可能なデータは、存在していたとしても僅かである。さらに、T細胞の再構成を評価するための適切な小動物モデルを欠くことが主要な制限である。
【0285】
形質導入したT細胞のin vivo増殖を達成するための、提案された一方法は、MGMT手法を用いてそれらを選択することである。Brian Davisらは、2年前に、形質導入した初代CD4T細胞をin vitroで、BG/BCNU薬物処置を用いて5%〜80%を超えて選択することが可能であることを実証した。ここでも、正確なin vivo薬物投薬を評価するための適切な動物モデルを欠くことが主要な制限である。
【0286】
別の選択肢は、対象を、操作細胞を再輸液する前に抗CD3抗体で(in vivo)プレコンディショニングすることである。形質導入した細胞の再輸液前のin vivoT細胞枯渇は、再注射した細胞を用いたT細胞サブセットの迅速な再構成をもたらす可能性がある。大動物モデルまたは第I相臨床治験での直接評価が、最も適切な方法であると見受けられる。
【0287】
(実施例45)
刺激
現在の刺激手順では、Dynalエポキシビーズにコーティングした抗ヒトCD3および抗ヒトCD28ネズミ抗体を用いる。CD3/CD28刺激は、細胞形質導入プロトコルの特徴である。
【0288】
以下に4つの代替実施形態を記載する:
生分解性ナノビーズ上に結合したCD3およびCD28抗体の使用。この手法では、関連する抗体刺激コストが削減されず、生成物の操作性も低下する(ビーズの除去なし)。
【0289】
過作用性抗ヒトCD28の使用。この抗体は、抗CD3抗体を必要とせずにT細胞の増殖を効率的に刺激することが示されている。
【0290】
ベクター自体をT細胞刺激タンパク質担体として使用。この手法では、抗体産生を必要としないが、パッケージング細胞系の改変を必要とする。
【0291】
Stemcell Technologiesによって開発されたTetralinkシステムの使用。このシステムでは、ビーズ支持体の必要性が回避され、したがってビーズ枯渇ステップが回避される。このシステムでは、ネズミIgG1モノクローナル抗体の使用が機能的であることを必要とする。
【0292】
(実施例46)
キット
本発明の一実施形態は、生分解性ナノビーズを含まない刺激系を用いた、改変したパッケージング細胞系から産生した臨床グレードのベクターを含めたキット、ならびに3日間の培養期間について大動物モデルからの安全性/有効性およびin vivoT細胞再構成である。
【0293】
本発明を完全に説明したので、当業者には、本発明の精神および範囲から逸脱せず、かつ必要以上の実験を行わずに、広範囲の均等なパラメータ、濃度、および条件内で同発明を行うことができることを理解されよう。
【0294】
本発明の基本的な態様から逸脱せずに前述の改変を行い得る。本発明は、1つまたは複数の具体的な実施形態を参照しながら相当に詳細に記載したが、当業者は、本出願中に具体的に開示した実施形態に変更を行ってもよく、それでもこれらの改変および改良が本発明の範囲および精神の範囲内にあることを理解されよう。本明細書中に例示的に記載した本発明は、具体的に開示していない任意の要素(または複数の要素)が存在しない場合に適切に実施し得る。したがって、たとえば、本明細書中のそれぞれの例において、「含む」、「本質的に〜からなる」、および「からなる」のうちの任意の用語を、他の2つの用語のどちらかと置き換え得る。したがって、用いた用語および表現は、限定ではなく説明の用語として用い、表示かつ記載した特徴の均等物またはその部分は排除されず、様々な改変が本発明の範囲内で可能であることを理解されたい。本発明の実施形態は、以下の特許請求の範囲に記載する。
【0295】
本明細書中上記および以下の特許請求の範囲では、用語「a」または「an」の使用は、単数形の定義に限定されない。そうではなく、これらの用語の使用は複数形を包含する。たとえば、用語「an抗体」は、1つの単一の抗体分子の単数形には限定されず、むしろ、参照する抗体の同一コピーである限りは複数の抗体分子の存在を包含する。同様に、「aウイルスベクター」は1つの単一のウイルスベクター分子または1つの単一のウイルス粒子に限定されない。用語「or」は、それが指す用語の1つに排他的であることを意図しない。たとえば、構造「A or B」の語句で用いられる場合は、これは、A単独、B単独、またはAおよびB両方を示し得る。
【0296】
以下の図は、本発明の態様の例示であり、特許請求の範囲によって包含される本発明の範囲を限定することを意図しない。
【0297】
特許または特許出願のファイルは、少なくとも1つのカラーで作成した図を含む。カラーの図(または複数の図)を含む本特許または特許出願公報のコピーは、請求に応じて必要経費の支払い後に特許局(Office)により提供される。
【0298】
図を添付する。異なる図中の同様の参照記号は同様の要素を示す。
【図面の簡単な説明】
【0299】
【図1】図1は、本発明の方法およびシステムで用いることができる例示的な容器またはフラスコを示す図である。
【図2】図2Aおよび2Bは、それぞれpN2cGFPおよびpN1GFP(cPT)のマップを示す図である。様々な制限酵素部位およびHIVに由来する構成成分を示す。pN2cGFP構築体は、CMV(サイトメガロウイルス)プロモーターに作動可能に連結し、したがってGFPの発現を制御するGFPコード配列を含む。pN1GFP(cPT)構築体は、以下でpN1(cpt)CGFPとも呼ばれ、HIV pol遺伝子由来のcPPTを含む。これらの構築体は、以下に記載の実施例で用いる。
【図3】図3は、固定したCD3およびCD28抗体でコーティングしたビーズを用いた初代T細胞の形質導入の結果を示す図である。細胞は、ビーズと接触させる前にベクターと接触させたか(パネルA)、ベクターと接触させる前にビーズと接触させたか(パネルB)、またはベクターおよびビーズをどちらも同時に接触させた(パネルC)。形質導入したベクターによってコードされているGFPからの蛍光に基づいたフローサイトメトリーの結果は、パネルA〜Cの細胞はそれぞれ90.70、87.19、および79.14%形質導入されていたことを示している。
【図4】図4は、ウイルスベクターと接触させる前にCD4+細胞を刺激するためにIL−2およびPHA−Pまたはビーズに固定したCD3およびCD28抗体のどちらかを用いた、形質導入の比較を示す図である。固定した抗体を使用することにより、毎回95%を超える形質導入効率がもたらされた。IL−2およびPHAの使用では、70.2〜84.5%の効率しかもたらされなかった。
【図5】図5は、本方法を用いたヒトCD4+T細胞の形質導入の頻度を示す図である。形質導入の15日後、対照細胞対緑色蛍光タンパク質(GFP)を発現する能力を有するベクターを用いて20のMOIで形質導入した細胞のフローサイトメトリー分析の比較により、形質導入した細胞の約93%も緑色蛍光を示すことが示されている。
【技術分野】
【0001】
(関連出願)
本出願は、2005年5月20日に出願された米国仮特許出願第60/683,527号に関し、その全体が本明細書中に参考として援用される。
【0002】
これらの文書の内容は、本明細書中に参考として援用される。
【0003】
(技術分野)
本発明は、一般にウイルス学、細胞生物学および生命工学に関する。具体的には、本発明は、初代細胞の作製、初代細胞の形質導入、および初代細胞集団の増殖を行うための新規プロセスを提供する。
【0004】
本発明は、ウイルスベクターを用いて細胞の効率的かつ安定した形質導入を行うための方法、ならびにそれに関連する組成物を対象とする。本方法により、形質導入した細胞数の増加がもたらされる。形質導入した細胞は、実験室および臨床応用のどちらにも用いることができる。
【0005】
本発明は、ウイルスベクターを用いて細胞の効率的かつ安定した形質導入を行うための方法、ならびにそれに関連する組成物およびシステムを対象とする。本方法は、たとえば、形質導入する細胞と細胞表面に結合する1つまたは複数の分子とを接触させることによって、形質導入の効率を増加させる。接触させるステップは、細胞へのウイルスベクターの導入の前、後、またはそれと同時に起こり得る。また、本方法では、単層の容器またはフラスコよりも多数の細胞を収容することができる多層の容器またはフラスコ中で細胞を培養することによって、培養および/または成長させている初代細胞がより多くの数もたらされる。また、本発明は、ベクターによって運ばれる核酸の発現または生きた生物の治療を含めた他の応用における、安定形質導入した細胞の使用にも関する。
【背景技術】
【0006】
(背景)
Barry, S.C.ら、2000年、「Lentiviral and murine retroviral transduction of T cells for expression of human CD40 ligand」、Human Gene Therapy、11巻:323〜332頁。Costello, E.ら、2000年、「Gene transfer into stimulated and unstimulated T lymphocytes by HIV−1−derived lentiviral vectors」、Gene Therapy、7巻:596〜604頁。Douglas, J.ら、1999年、「Efficient transduction of human lymphocytes and CD34+ cells via human immunodeficiency virus−based gene transfer vectors」、Human Gene Therapy、10巻:935〜945頁。Follenzi, A.ら、2000年、「Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV−1 pol sequences」、Nature Genetics、25巻:217〜222頁。Han, W.ら、2000年、「A soluble form of human Delta−like−1 inhibits differentiation of hematopoietic progenitor cells」、Blood、95巻:1616〜1625頁。Haas, D.L.ら、2000年、「Critical factors influencing stable transduction of human CD34+ cells with HIV−1−derived lentiviral vectors」、Molecular Therapy、2巻:71〜80頁。Hooijberg E.ら、2000年、「NFAT−controlled expression of GFP permits visualization and isolation of antigen−stimulated primary human T cells」、Blood、96巻:459〜466頁。Kishimoto, T.編、Leucocyte Typing VI: White Cell Differentiation Antigens: Proceedings of the Sixth International Workshop and Conference Held in Kobe, Japan、1996年11月10〜14日、Garland Publishing、New York、1998年。Klebba, C.ら、2000年、「Retrovirally expressed anti−HIV ribozymes confer a selective survival advantage on CD4+ T cells in vitro」、Gene Therapy、7巻:408〜416頁。Koc, O.N.ら、1999年、「Transfer of drug resistance genes into hematopoietic progenitors」、第11章、Gene Therapy of Cancer、Academic Press、San Diego、177〜195頁。Movassagh, M.ら、2000年、「Retrovirus−mediated gene transfer into T cells: 95% transduction efficiency without further in vitro selection」、Human Gene Therapy、11巻:1189〜1200頁。Onodera, M.ら、1998年、「Successful peripheral T−lymphocyte−directed gene transfer for a subject with severe combined immune deficiency caused by adenosine deaminase deficiency」、Blood、91巻:30〜36頁。St. Croix, B.ら、2000年、「Genes expressed in human tumor endothelium」、Science、289巻:1197〜1202頁。Unutmaz, D.ら、1999年、「Cytokine signals are sufficient for HIV−1 infection of resting human T lymphocytes」、J. Exp. Med.、11巻:1735〜1746頁。Zennou, V.ら、2000年、「HIV−1 genome Nuclear import is mediated by a central DNA flap」、Cell、101巻:173〜185頁。
【0007】
一般に遺伝物質を細胞内に導入するための技術をいう「トランスフェクション(transufection)」は、生物学において分子および組換えの革命に大きく貢献した。高等真核細胞で用いるためのトランスフェクション技術の例には、リン酸カルシウム沈殿、DEAE−デキストラン処理、電気穿孔、微量注入、リポフェクチン、ウイルス感染症、ならびに数々の科学の教科書および学術誌に見られる他の方法が含まれる。
【0008】
形質導入(transduction)技術のうち、ウイルス感染の使用は、目的の核酸分子を細胞内に移すために、ウイルスに天然に存在する、その遺伝物質を細胞内に導入する手段を利用するという点で独特である。そのような技術のために改変および応用されたウイルスの例には、アデノウイルス、アデノ関連ウイルス、単純ヘルペスウイルス、およびレトロウイルスが含まれる。一般に、目的の核酸分子は、ウイルスゲノム中にクローニングし得る。ウイルスゲノムの複製およびパッケージング後、生じたウイルス粒子は、ウイルスの侵入機構を介して目的の核酸を細胞内に送達することができる。
【0009】
一般的に、目的の核酸を付加する前に、最初に核酸操作によってウイルスゲノムを複製欠損にする。生じたウイルスゲノム、またはウイルスベクターは、ウイルス粒子の組立ての完了および細胞からの放出にヘルパーウイルスまたはパッケージング系の使用を必要とする。ウイルスベクターまたはウイルス粒子を用いて目的の遺伝物質を細胞内に導入する場合、この技術は「形質導入」と呼ばれる。したがって、一般に、細胞の「形質導入」を行うこととは、ウイルスベクターまたはウイルス粒子を用いて遺伝物質を細胞内に導入することである。
【0010】
形質導入技術では、とりわけ、レトロウイルスの使用が哺乳動物細胞の遺伝組換えの非常な関心の対象となっている。特に関心が持たれているのは、遺伝的欠陥および他の疾患を治療するために遺伝物質を細胞内に導入するための、改変レトロウイルスの使用である。この手法の一例は、レトロウイルスおよびレンチウイルスベクターが熱心な研究の対象である造血系の細胞の事例で見られる。
【0011】
たとえば、Movassaghらは、活性T細胞の細胞周期の研究からの結果を含めることによって、レトロウイルスに媒介される形質導入の効率を上昇させる彼らの試みに関する研究を記述している。それ自体として、その結果は形質導入中の活性細胞分裂に依存する。また、この研究は、ネズミオンコレトロウイルスの使用および形質導入前に細胞の有意な予備刺激を必要とすることに制限されている。
【0012】
Juneら(国際公開公報WO96/34970号)は、T細胞のトランスフェクションを増加させる手段としてのT細胞刺激の使用を記載している。活性化または刺激した細胞を用いたT細胞の形質導入に関する他の研究には、Douglasら、Hooijbergら、Onoderaら、Klebbaら、Barryら、およびUnutmazらの研究が含まれる。残念ながら、この研究のいずれにおいても、約65%を超える形質導入効率が実証されなかった。
【0013】
Costelloらは、ヒト免疫不全ウイルス−1(HIV−1)レンチウイルスベクターを用いた、刺激および非刺激のT細胞の両方の形質導入を記載している。ここでは、刺激した初代T細胞で最大約17%の効率、非刺激のT細胞で19%未満の効率しか観察されなかった。彼らはまた、HIV−1アクセサリータンパク質の存在を含めることによって刺激したT細胞で効率を36%以下まで増加させるという、限定された能力に注目している。
【0014】
また、Chinnasamyらは、非刺激およびマイトジェン刺激したT細胞における、HIV−1アクセサリータンパク質の存在下での形質導入の効率の増加を記載している。Movassaghらと同様、Chinnasamyらは、レンチウイルスベクターを用いて形質導入する前に、血液リンパ球を有意な期間、予備刺激した。Chinnasamyらは、最初、形質導入の3日後に96%を超える形質導入効率を観察したが、安定形質導入した細胞の割合は形質導入の2週間後に71.2%まで低下した。また、Haasらは、マーカー遺伝子(緑色蛍光タンパク質)を発現する能力を有するレンチウイルスベクターを用いて形質導入した細胞において一過性形質導入および「偽形質導入」を観察した。形質導入の3日後でも、形質導入した初代CD34+臍帯血細胞におけるマーカー遺伝子の非組込み発現に基づいて、有意な(10%を超える)一過性形質導入が検出された。一過性形質導入からのこのような発現は、形質導入の7日後でも約5%で検出可能に保たれていた。形質導入の約10日後になってやっと、一過性形質導入からの発現はマーカーなしのベクターを用いて形質導入した細胞における発現の反映となった。
【0015】
したがって、Chinnasamyらは、2週間後の効率によって反映されるように、ウイルスベクターの組込み型を形質導入した細胞の染色体DNA内に挿入した場合、71.2%を超える初代リンパ球の安定形質導入を達成することができなかった。これは、細胞を予備刺激するためにサイトカインを使用したにもかかわらず、そうであった。さらに、Chinnasamyは、形質導入後に細胞をPHAマイトジェンおよびIL−2サイトカインで後に刺激したにもかかわらず、アクセサリータンパク質(Vif、Vpr、VpuおよびNef)を発現しなかったHIVベクターを用いて非刺激のリンパ球を有意に形質導入できなかったことを記載している(形質導入の14日後に3.6%のみ)。結果は非刺激の細胞およびアクセサリータンパク質を含むベクターの使用である程度改善されていたが、ベクターと共に刺激プロトコルを用いたにもかかわらず、どの場合でも、刺激または非刺激の細胞の安定形質導入の効率は、形質導入後14日目に75%を超えていなかった。
【0016】
また、Hassらによってもレンチウイルスベクターを用いた低頻度の安定形質導入が観察され、彼らは、初代CD34陽性臍帯血細胞を用いた形質導入の7日後に25%未満の最大安定形質導入効率しか達成することができなかった。顕著なことに、この25%の形質導入の上限は、極めて高い感染多重度またはベクター濃度、たとえば9000までの感染多重度(MOI)および108感染単位/ミリリットルまでのベクター濃度などの後にも、向上させることができなかった。
【0017】
Follenziらも、インターロイキン−3(IL−3)、インターロイキン−6(IL−6)および幹細胞因子(SCF)を含む3つのサイトカインカクテルの存在下で細胞を形質導入するために、500という非常に高いMOIを用いた。興味深いことに、カクテルを使用することにより、細胞がヒトへの臨床移植に不適切となる。
【特許文献1】国際公開第96/34970号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0018】
したがって、高頻度でベクターを用いた細胞の安定形質導入を行う、より効率的な手段を提供する必要性が、依然として存在する。さらに、研究ツールおよび治療剤のどちらのために使用するとしても、非刺激の細胞を形質導入するためのより効率的な手段の必要性が存在する。
【0019】
さらに、実験室および/または臨床応用において使用できる、多数の初代細胞を同時に形質導入ならびに培養および/または成長させる必要性が、依然として存在する。たとえば、形質導入から1「バッチ」の細胞を得て対象に投与し、様々な形質導入効率を有する複数バッチの細胞を合わせなくてもよいことが望ましい。
【課題を解決するための手段】
【0020】
(発明の開示)
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法である。
【0021】
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行い;細胞とレンチウイルスベクターとを約24時間接触させ、任意選択で少なくとも1回繰り返す方法である。
【0022】
本発明の一態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;約7〜10日後、もしくは約14日目に細胞の少なくとも約50%が安定に形質導入されており;任意選択で約14日後に細胞の少なくとも50%が安定に形質導入されたまま保たれているか;または約7〜10日後、もしくは約14日目に細胞の少なくとも約75%が安定に形質導入されており、かつ任意選択で約14日後に細胞の少なくとも75%が安定に形質導入されたまま保たれているか;または約14日後に80%、85%、89%、90%、91%、92%、93%、94%もしくは95%を超える細胞が安定に形質導入されているか;または約2〜約50、もしくは約10〜約30、もしくは10、もしくは約20、もしくは約30、もしくは約40、もしくは約50、もしくは約1〜約400、もしくは500未満の感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または形質導入した細胞1個あたりのレンチウイルスベクターのコピーが約1〜約100個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて細胞の形質導入を行うか;または細胞とレンチウイルスベクターとを約24時間接触させ、任意選択で少なくとも1回繰り返し;細胞表面分子はアポトーシスを誘導せず、細胞表面結合分子により、ウイルスレンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなることをもたらす方法である。
【0023】
初代細胞は、単離するか、または対象から導くことができる。初代細胞は、以下の手順の1つまたは複数によって、すなわち、(a)対象の血液のアフェレーシスによって;または(b)対象の骨由来の骨髄から;または(c)同種の対象の血液のアフェレーシスによって;または(d)同種の対象の骨由来の骨髄から、単離することができる。初代細胞はまた、当業者に知られている代替技術を用いて単離することもできる。
【0024】
アフェレーシスとは、ドナーまたは対象の血液を、1つの特定の構成成分を分離して残りを循環に戻す装置に通す医療技術である。
【0025】
除去する物質に応じて、異なるプロセスをアフェレーシスで用いる。たとえば、重量による分離が必要な場合は、遠心分離が選択する方法となる。本発明で用いることができる他の例示的な方法は、吸収材料でコーティングしたビーズ上への吸収を含む。
【0026】
多種類のアフェレーシスが存在する。たとえば、血漿アフェレーシス−血漿;血小板アフェレーシス(plateletpheresis、thrombapheresis、thrombocytapheresis)−血小板;白血球アフェレーシス−白血球(leukocytes、white blood cells);幹細胞収集−骨髄移植で用いるために循環骨髄細胞を収集する;およびLDLアフェレーシス−家族性高コレステロール血症に罹患している対象における低密度リポタンパク質の除去が存在する。これらのアフェレーシスの種類は単なる例示である。当業者には他の種類が知られ得る。
【0027】
血液構成成分は、たとえば、血液バッグに採取する前に採取した全血のバッグまたはドナーの血流から分離することができる。様々な種類の血液構成成分をドナーからアフェレーシスによって得ることができる。これには、たとえば血小板および血漿が含まれる。
【0028】
様々なアフェレーシス技術は、たとえば、除去した構成成分が疾患の重篤な症状を引き起こしている場合にいつでも用い得る。一般に、アフェレーシスは相当頻繁に行う必要があり、侵襲性のプロセスである。したがって、通常は、特定の疾患を制御するための他の手段が失敗した場合、または症状の性質が、医薬品が効果的になるまで待つことで苦痛もしくは合併症の危険性を引き起こすものである場合にのみ用いる。
【0029】
骨髄は、当業者に知られている方法によって得る、保存、および操作することができる。たとえば、米国特許第6,991,787号は、骨髄間質細胞を得る方法を記載しており、米国特許第6,110,176号は、遺伝的に適合性のある骨髄を採取および保存する方法を記載しており、米国特許第4,366,822号は、骨髄細胞を分離および分析する方法および装置を記載している。
【0030】
対象はヒト免疫不全ウイルス(HIV)に感染していてよく、任意選択でHIVはHIV−1またはHIV−2である。対象は癌に罹患していてよく、任意選択で癌は乳癌である。対象はヒトまたは動物のどちらかであることができる。
【0031】
初代細胞は、細胞を密度勾配緩衝液に通すことによって、および/または磁場上で免疫精製を行うことによって、レンチウイルスベクターまたは細胞表面結合分子と接触させる前に富化することができる(磁気細胞分別)。初代細胞は、細胞の培養および/または成長の前またはその間に富化することもできる。当業者によって知られている、細胞集団を富化するための他の知られている手段も、本発明の方法で用いることができる。たとえば、細胞に特異的な標的構造体を同定および富化するプロセスを記載している米国特許第6,974,675号を参照されたい。たとえば、サイトカインを組織培養培地に加えることにより、特定の細胞種の富化がもたらされる。具体的には、培地中に特定のサイトカインが存在すると、特定の細胞集団が死滅する。
【0032】
初代細胞と(a)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触の前に起こるか;または(b)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触と同時に起こるか;または(c)レンチウイルスベクターとの接触は、細胞と少なくとも1つの細胞表面結合分子との接触の後に起こるか;または(d)レンチウイルスベクターとの接触は、複数回起こるか;または(e)レンチウイルスベクターとの接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または(f)細胞表面結合分子との接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または(g)レンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との接触は、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との最初の同時接触の後、連続的に起こるか;または(h)(a)〜(g)の任意のものは、約24〜36時間の期間中に少なくとも1回起こる。
【0033】
初代細胞は、少なくとも1つの細胞表面結合分子で予備刺激することができ、任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約24時間以内に、細胞を少なくとも1つの細胞表面結合分子で予備刺激するか、または任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約12〜96時間以内に、細胞を少なくとも1つの細胞表面結合分子で予備刺激する。
【0034】
レンチウイルスベクターは、gag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来する少なくとも1つのシス作用性ヌクレオチド配列を含むことができ、任意選択で、配列は発現されない、またはgag、pol、env、vif、vpr、vpu、tatもしくはrev遺伝子の断片もしくは突然変異体である。
【0035】
レンチウイルスベクターは、(a)偽型であり、任意選択で偽型ベクターは水疱性口内炎ウイルスGエンベロープタンパク質を含むことができるか;または(b)偽型であり、偽型化は、レンチウイルスベクター遺伝物質と別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質もしくは細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションもしくは同時感染を行うことを含むことができるか;または(c)ラブドウイルスを用いた偽型であり、任意選択でラブドウイルスは水疱性口内炎ウイルスエンベロープG(VSV−G)タンパク質であることができる。
【0036】
初代細胞は、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体であることができる。
【0037】
少なくとも1つの細胞表面結合分子は、(a)ポリペプチド、脂質、核酸、炭水化物またはイオンを含むか;あるいは(b)抗体、抗原結合断片、リガンド、または細胞表面分子を含むか;あるいは(c)FLT−3リガンド、TPOリガンド、もしくはKitリガンド、またはFLT−3リガンド、TPOリガンド、もしくはKitリガンドの細胞表面結合類似体であるポリペプチドもしくは他の結合分子を含むか;あるいは(d)CD34、CD3リガンド、CD28リガンド、CD25リガンド、CD71リガンド、もしくはCD69リガンド、またはCD34、CD3、CD25、CD28、CD69もしくはCD71リガンドと同じ細胞表面結合特異性を有するポリペプチドもしくは他の結合分子を含むか;あるいは(e)GM−CSF、IL−4、およびTNF−α;GM−CSFおよびインターフェロン−α;またはGM−CSF、IL−4、およびTNF−αの細胞表面結合類似体であるポリペプチドもしくは他の結合分子;GM−CSFもしくはインターフェロン−αを含む組成物を含むか;あるいは(f)CD3抗体またはその細胞表面結合断片、CD28抗体またはその細胞表面結合断片、抗体とその細胞表面結合断片との組合せ、および抗体と同じ細胞表面結合特異性を有する結合分子を含むか;あるいは(g)ビーズまたは表面上に固定したCD3およびCD28抗体の組合せを含み、任意選択で、ビーズまたは表面はコーティングしたビーズを含むか;あるいは(h)(a)〜(g)の任意のものから選択された2つ以上の細胞表面結合分子を含むか;あるいは(i)分子が細胞の表面に結合する能力を増加または増強するために用いる別の分子を含むか;あるいは(j)別の分子と複合しているか;あるいは(k)初代細胞の表面上に見つかり、別の細胞の表面に結合する。
【0038】
細胞培養条件は、(a)細胞表面結合分子もしくはサイトカインと共にさらなるインキュベーションを行うこと;または(b)インターロイキン−2と共にさらなるインキュベーションを行うこと;または(c)細胞を約7日間培養すること;または(d)細胞を約14日間培養することを含むことができる。
【0039】
レンチウイルスベクターは、(a)ヒト免疫不全ウイルス(HIV)由来であるか;または(b)HIV−1、HIV−2、もしくはそれらの組合せ由来であるか;または(c)HIV配列を含むキメラベクターであり、任意選択で、HIV配列はHIV−1およびHIV−2配列を含むか;または(d)VRX496、もしくはVRX496の誘導体であることができる。
【0040】
細胞とレンチウイルスベクターまたは細胞表面結合分子との接触は、ex vivoで、混合もしくは純粋細胞培養物、組織または器官系中で起こることができる。
【0041】
本発明の別の態様は、本明細書中に記載した任意の方法によって形質導入した細胞を生きた対象、組織、器官、胚盤胞または胚性幹細胞内にex vivo導入することを含む、遺伝物質を細胞内に導入する方法である。
【0042】
本発明の別の態様は、薬剤組成物を調製するための、本明細書中に記載した任意の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用である。薬剤組成物は、対象におけるウイルス感染症の治療もしくは予防、対象におけるHIV感染症の治療もしくは予防、または癌の治療もしくは予防に用いることができる。癌は、任意の種類の癌、たとえば乳癌または内皮細胞の任意の癌であることができる。
【0043】
本発明の別の態様は、本明細書中に記載した任意の方法によって生成した、遺伝的欠陥によって引き起こされた異常を治療もしくは予防する、または腫瘍もしくは癌を治療、診断、緩和もしくは予防するための遺伝子治療用の薬剤組成物であり、任意選択で、遺伝的欠陥または腫瘍もしくは癌によって引き起こされた異常は乳癌腫瘍である。
【0044】
本発明の別の態様は、本明細書中に記載した任意の方法によって生成した、感染症によって引き起こされた異常を治療または予防するための遺伝子治療用の薬剤組成物。感染症はウイルス感染症であることができ、任意選択で、ウイルス感染症はヒト免疫不全ウイルス(HIV)感染症である。薬剤組成物は、ex vivoで使用するために配合する。
【0045】
本発明の別の態様は、造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、前記初代細胞と細胞表面分子との接触により、レンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなる方法である。
【0046】
初代細胞の表面における細胞表面分子の存在により、(a)細胞クロマチンのDNA組込みに対する受容性がより高くなること;または(b)レンチウイルスベクターからの遺伝子の発現に有利な細胞部位内へのレンチウイルスベクターの組込み;または(c)カプシドを含む核酸の、細胞の細胞質内へのより効率的な侵入;または(d)ウイルスの、細胞膜を横断するもしくは細胞の内部膜構造体を横断するより効率的な侵入;または(e)ウイルスベクター内に含まれる遺伝物質の核内移行に対する初代細胞の許容性がより高くなることが、もたらされることができる。
【0047】
細胞表面結合分子、抗体、抗原結合断片、リガンドまたは細胞表面分子は、細胞に結合してベクター形質導入に対するその受容性をより高くする抗CD3または抗CD28抗体;細胞に結合してベクター形質導入に対するその受容性をより高くする、FLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンド;樹状細胞もしくはその前駆体、単球、CD34陽性幹細胞、または樹状細胞系列上の分化したその前駆細胞に結合してベクター形質導入に対するその受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンド;細胞上の
【0048】
【化2】
に結合してベクター形質導入に対するその受容性をより高くする、ポリペプチド、核酸、炭水化物、脂質もしくはイオン、または別の物質と複合したポリペプチド、核酸、炭水化物、脂質もしくはイオンを含む。
【0049】
本発明の別の態様は、HIVに感染した対象から単離した造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、(a)HIVに感染した対象から造血系細胞の初代細胞または造血幹細胞を単離するステップと;(b)任意選択で、初代細胞または造血幹細胞を少なくとも1つの細胞表面結合分子で予備刺激するステップと;(c)in vitroまたはex vivoで、造血系細胞または造血幹細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させるステップと;(d)2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養するステップとを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法である。
【0050】
本発明の別の態様は、(a)2つ以上の層を含む換気容器;ならびに(b)単離した非接着性の造血系の初代細胞および/または造血幹細胞を含むシステムである。システムの初代培養は、本発明の方法について上述した任意の細胞であることができる。
【0051】
多層容器は、細胞をその中で培養および/または成長させるために実用的な任意の形状であることができる。たとえば、容器は、四角形の形状、正方形の形状、または曲線縁の四角形の形状、もしくは曲線縁の正方形の形状であることができる。
【0052】
組織培養フラスコまたは容器が、細胞を成長させるために実験室で幅広く使用されている。典型的には、これらのフラスコを用いて細胞を培地中で培養し、細胞がフラスコの内部表面に接着する。細胞は、開口部からフラスコまたは容器内に導入する。フラスコまたは容器を閉じ(ただし換気を許容する)、培地中の細胞の成長を促進するために、オーブンなどのスタッキング施設またはチャンバ内に挿入する。
【0053】
造血系の初代細胞および/または造血幹細胞は、培養バッグ、または他の単層の容器もしくはフラスコ中で成長させる。これにより、一度に成長させることができる細胞の数が制限される。平坦な表面を有する多層のフラスコまたは容器により、一度に多数の細胞を培養することが可能となるが、これは平坦な表面に接着する細胞に用いられている。造血系の初代細胞および/または造血幹細胞は非接着性細胞である。しかし、これらの細胞は多層のフラスコまたは容器中で成長させることができ、これにより、一度に多数の細胞を成長させることが可能となる。多層容器の一例は細胞工場である。
【0054】
本発明の方法および組成物は、多層の組織培養フラスコまたは容器中における、非接着性の造血系の初代細胞および/または造血幹細胞の大スケールの培養および/または成長を可能にし、少なくとも約1億個の細胞の成長をもたらす。
【0055】
細胞工場、または細胞工場の概念のその変型を、たとえば、ワクチン、モノクローナル抗体または医薬品の大スケール産生に用いることができる。これらは接着性細胞に理想的であるが、懸濁培養にも用いることができる。細胞の成長の速度論は、実験室スケールの培養から変化せずに保たれる。細胞工場は、容易なスケールアップのために、たとえば、1、2、4、10および40個のトレイの型で入手可能である。これらの汚染危険性は低く、コンパクトな設計である。
【発明を実施するための最良の形態】
【0056】
上記の文書の引用は、前述の任意のものが関連する従来技術であると承認することを意図しない。これらの文書の内容に関する日付または表示に関するすべての記述は、出願人に利用可能な情報に基づいており、これらの文書の日付または内容の正確性に関していかなる承認も構成しない。
【0057】
本明細書中に引用するすべての参考文献、出版物、特許出願および特許は、具体的に組み込んでいるかいないかにかかわらず、その全体で参考として本明細書中に組み込まれている。
【0058】
以下の記述は、本発明の方法で用いることができる容器の種類の例である。これらは単なる例示であり、本発明の範囲を限定することを意図しない。
【0059】
細胞をその中で成長させることができる容器の種類の例を図1に示す。容器は、細胞を中で成長させるために実用的な任意の形状、たとえば、正方形、四角形、円形、楕円形、または成形端部を有する正方形もしくは四角形であることができる。容器は複数層でなければならないが、容器が含むことができる層の数の上限は、細胞を中で培養および/または成長させるチャンバ、オーブン、または容器の大きさにのみ制限される。容器は、たとえばプラスチック、または細胞をその中で培養および/もしくは成長させるために適した任意の他の材料から作製することができる。
【0060】
細胞工場とは、単独のユニットに密着させ、共通の通気口および注入口を共有する、積み重ねたチャンバである。それぞれのチャンバは、たとえば、632cm2の平坦な成長表面を有する。細胞工場は、大スケール細胞培養ならびにワクチン、モノクローナル抗体およびインターフェロンなどの生体材料の産生に用いる。細胞工場は、小さな面積で大量の成長表面を提供し、取扱いが容易で汚染の危険性が低い。成長面積が25,280cmの、チャンバが40個のユニットは、14個の大きなローラーボトル(それぞれ1,750cm)に対応する。細胞工場では1回の充填および排出作業が必要であることに対して、ローラーボトルでは14回必要である。細胞工場は無菌的である。細胞工場は、限られた空間の面積で大きな成長表面を提供する。
【0061】
細胞工場の他の態様は、たとえば、細胞懸濁液を保つ通気ストッパを備えたアスピレーターボトルからなる、培地リザーバである。細胞工場を用いた細胞の導入、培養および収集は当分野で知られている。本発明で用いることができるインキュベーターおよび細胞培養装置の種類の変型は、たとえば、公開された特許出願第20060057713号および第20060057712号、ならびに米国特許第6,114,165号に記載されている。
【0062】
チャンバが10個および40個の細胞工場の高さが原因で、細胞を一般的な顕微鏡で見ることができない。ほとんどの場合、対照として細胞をチャンバが1個またはチャンバが2個の細胞工場に播種する。これらの製品のそれぞれで成長させた細胞は、顕微鏡下で見ることができる。見る側に強力な光源を備え、細胞工場の高さに調節した倒立双眼実体顕微鏡を用いて、最初の数層を見た。
【0063】
例示的な細胞工場の寸法および培養面積:
【0064】
【化3】
本発明の方法では、新規組成物、すなわち、高い形質導入レベルを有する細胞の試料、「集団」または「バッチ」であり、少なくとも約1億個の細胞を含む組成物を生成する。
【0065】
本発明は、ウイルスベクターおよびウイルス粒子を用いて細胞の安定形質導入を行うための、効率の高い方法、およびそれに関連する組成物を提供する。「安定形質導入」とは、ウイルスベクターの組込み型を形質導入した細胞の染色体DNA内に挿入したことを意味する。この方法は、細胞表面に結合する少なくとも1つの分子と接触するように形質導入する細胞を曝露することを含む。この接触させるステップは、ウイルスベクターまたはウイルス粒子に細胞を曝露する前、その間、またはその後に起こり得る。本明細書中では以降、用語「ウイルスベクター」とは、ウイルスに由来し、形質導入を介して遺伝物質を細胞内に導入するために用いる、任意の形態の核酸を表すために用いる。この用語には、DNAおよびRNAなどのウイルスベクター核酸、これらの核酸のカプセル封入した形態、ならびにウイルスベクター核酸をパッケージングしたウイルス粒子が包含される。
【0066】
また、本発明には、ベクター中に存在する核酸の発現による有用な遺伝子産物およびタンパク質の産生、または疾患に罹患している、もしくは罹患する危険性のある生きた対象の治療を含めた、他の応用における形質導入した細胞の使用も含まれる。たとえば、対象はヒトである。
【0067】
形質導入する細胞の表面に結合する少なくとも1つの分子には、細胞の表面上の受容体、マーカー、または他の認識可能な部分と物理的に相互作用する任意の分子が含まれる。原理上は、任意の細胞表面結合分子を細胞の高効率形質導入に用い得る。本発明を理論に束縛せずに、細胞表面結合分子は、宿主細胞のクロマチンのDNA組込みに対する受容性がより高くなること;ベクターの遺伝子発現に有利な部位内へのウイルスベクターの優先的な組込み;カプシドを含む核酸の細胞質内へのより効率的な侵入;細胞膜もしくはエンドソームなどの内部膜構造体を横切るウイルスのより効率的な侵入;またはウイルスベクターの遺伝物質核内移行に対する細胞の許容性をより高くさせることをもたらし得る。本発明の方法はまた、上記の可能性の複数に関与し得る。また、上記の可能性の数および多様性から明らかなように、本発明を任意の1つの理論に限定することはできない。そうではなく、ヒト治療における細胞の使用可能性に負の影響を与えずに処理した細胞の最高100%の安定形質導入を行うという本発明の驚くべき発見を考慮すると、本発明は、ヒト細胞治療の分野において新しい手法を開くとみなすべきである。
【0068】
しかし、すべての細胞表面結合分子でウイルスベクターによる効率的かつ安定した形質導入がもたらされるわけではない。たとえば、アポトーシスを誘導する細胞表面分子との結合では、細胞の効率的な形質導入はもたらされず、むしろ細胞死がもたらされる。細胞死は細胞を死滅させるため(たとえば腫瘍細胞)には好ましい場合もあるが、ペイロード(payroad)遺伝子または核酸配列を含むベクターを用いた細胞の安定形質導入には好ましくない。好ましい細胞表面結合分子により、細胞は、ウイルスベクターによる形質導入に対して受容性がより高くなる。そのような分子の例には、特異的細胞表面受容体またはその一部分に対する抗体、およびそのような受容体に対するリガンドまたは結合ドメインが含まれる。さらに、FabおよびFv断片などの抗体の抗原結合断片が、本発明における使用に企図される。特異的な細胞表面受容体に対する結合ドメインは、単一のエピトープまたは2つ以上のエピトープを含んでいることができる。
【0069】
本発明において使用する細胞表面結合分子の例は、T細胞に結合してベクター形質導入に対するその受容性をより高くする、抗CD3および抗CD28抗体である。他の細胞表面結合分子は、造血幹細胞などの受容体を発現する細胞の、ベクター形質導入に対する受容性をより高くするFLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンドである。さらなる細胞表面結合分子は、単球などの樹状細胞もしくはその前駆体、CD34陽性幹細胞、または樹状細胞系列上にあるその分化した前駆細胞の、ベクター形質導入に対する受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンドである。他の細胞表面結合分子には、別の細胞の表面に結合する、細胞表面上に見つかる分子が含まれる。
【0070】
細胞表面結合分子のさらなる例には、すべて任意選択で他の物質と複合したポリペプチド、核酸、炭水化物、脂質、およびイオンが含まれる。これらの分子は、血液細胞の表面上に見つかる因子、たとえば、
【0071】
【化4−1】
などに結合することができる。小文字(たとえば「a」または「b」)は、複数の遺伝子産物からなる、または構造的に関連するタンパク質のファミリーに属する複合CD分子を示す。記号「w」は、完全に確認されてない推定上のCD分子を指す。
【0072】
リンパ球、T細胞および白血球の表面上に見つかる因子に結合するさらなる分子は、
【0073】
【化4−2】
である。
【0074】
細胞の表面に結合し、本発明における使用に適したさらなる抗体および分子は、完全に記載したかのように本明細書中に参考として組み込まれているLinscott’s Directory of Immunological and Biological Reagents、第11版、2000年1月、出版社:W.D. Linscott、カリフォルニア州Petalumaに開示されている。しかし、本発明の一部の実施形態では、細胞表面結合分子はサイトカインではない。
【0075】
本発明は、細胞のベクター形質導入を促進する可溶性細胞表面結合分子を使用することによって実施し得るが、他の実施形態には、固定した細胞表面結合分子の使用が含まれる。たとえば、固定した分子は抗体である。あるいは、固定は、細胞表面結合分子を発現する他の細胞の使用を介し得る。造血幹細胞の効率的な形質導入の例示的な方法は、分化させずに幹細胞の維持を容易にするリガンドをその表面上に発現する骨髄間質細胞を、形質導入中に含めることである。刺激性細胞はネイティブ細胞に限定されず、任意の細胞を、形質導入のために正しい刺激を提供するために適切な細胞表面結合分子を発現するように操作することができる。
【0076】
少なくとも1つの分子が細胞表面に結合する能力を増加または増強させるさらなる分子も含め得る。たとえば、特異的細胞表面受容体に対する(一次)抗体の可溶形を、既に細胞表面と結合している一次抗体に架橋結合することができる二次抗体と組み合わせて用い得る。
【0077】
もちろん、任意の細胞を本発明の実施に用いることができる。たとえば、形質導入する細胞は真核細胞である。たとえば、細胞は初代細胞である。しかし、細胞系は、本発明の方法を用いて形質導入してもよく、多くの場合、より容易に形質導入される。一実施形態では、形質導入する細胞は、初代リンパ球(Tリンパ球など)もしくはマクロファージ(単球マクロファージなど)であるか、またはこれらの細胞のいずれかの前駆体、たとえば造血幹細胞などである。一般に、形質導入させる他の例示的な細胞は造血系の細胞であるか、または、より一般的には、造血によって形成される細胞ならびにそれらを形成する幹細胞および血液細胞機能に関連する細胞である。そのような細胞には、造血によって形成される顆粒球およびリンパ球、ならびに前駆体分化多能性幹細胞、リンパ球幹細胞、および骨髄幹細胞が含まれる。血液細胞機能に関連する細胞には、樹状細胞等の抗原提示細胞、内皮細胞、単球、およびランゲルハンス細胞などの、免疫系細胞の機能を補助する細胞が含まれる。一実施形態では、細胞は、CD4およびCD8マーカーを発現するものなどのTリンパ球(またはT細胞)である。
【0078】
別の実施形態では、細胞は初代CD4+Tリンパ球または初代CD34+造血幹細胞である。しかし、本発明で用いるためのウイルスベクターは、水疱性口内炎ウイルスエンベロープGタンパク質を用いて偽型とし得ることを考慮すると(以下に記述)、任意の細胞を本発明の方法で形質導入することができる。そのような細胞には、それだけには限定されないが、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、リンパ球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞または精母細胞(たとえばトランスジェニック動物を作製するため)、生殖系列に寄与する細胞、胚性分化多能性幹細胞またはその前駆体、血小板および赤血球等の無核細胞を含めた血液細胞などが含まれる。たとえば、細胞は、真核の多細胞種(たとえば単細胞の酵母細胞に対抗して)のものであるか、または哺乳動物由来のもの、たとえばヒト細胞である。
【0079】
形質導入する細胞は、単独の実態として存在するか、またはより大きな細胞コレクションの一部であることができる。そのような「細胞のより大きなコレクション」は、たとえば、細胞培養物(混合もしくは純粋のどちらか)、組織(たとえば、上皮、間質もしくは他の組織)、器官(たとえば、心臓、肺、肝臓、胆嚢、膀胱、眼、および他の器官)、器官系(たとえば、循環系、呼吸器系、胃腸管系、泌尿器系、神経系、外皮系もしくは他の器官系)、胚盤胞、胚性幹細胞、胎児由来の細胞(たとえば、遺伝的障害/疾患を治療するため、もしくはトランスジェニック動物を作製するため)、腫瘍もしくは感染部位などの患部組織、または生物(たとえば、鳥、哺乳動物、海洋生物、魚、植物など)を含むことができる。標的とする器官/組織/細胞は、循環系(たとえば、それだけには限定されないが、心臓、血管、および血液を含む)、呼吸器系(たとえば、鼻、咽頭、喉頭、気管、気管支、細気管支、肺など)、胃腸管系(たとえば、口および口腔組織、咽頭、食道、胃、腸管、唾液腺、膵臓、肝臓、胆嚢などを含む)、乳腺系(乳房上皮細胞および組織中の支持細胞など)、泌尿器系(腎臓、子宮、膀胱、尿道など)、神経系(それだけには限定されないが、脳および脊髄、ならびに眼などの特殊感覚器を含む)、外皮系(たとえば皮膚)のものであることができる。
【0080】
形質導入する細胞は、心臓、腫瘍血管および感染組織または患部組織に関連する血管を含めた血管、骨髄、血液、脳、リンパ組織、リンパ節、脾臓、肺、肝臓、胆嚢、膀胱、ならびに眼細胞からなる群から選択することができる。本発明の特定の一実施形態では、細胞は使用を意図する宿主の自己由来であるが、同種、部分ミスマッチ、完全ミスマッチ、またはさらには宿主に異種の細胞も用い得る。さらに、人間など生物または種の関連した群である任意の与えられた宿主生物での使用に適したユニバーサルドナー細胞を形質導入してよい。本発明のこの後者の実施形態は、形質導入した細胞の供給源が何であるかが移植の結果に決定的であり得る、細胞、組織または器官における移植に特に重要である。
【0081】
本発明の方法による形質導入のための別の細胞種は、腫瘍細胞、患部細胞、またはその遺伝子構造もしくは同一生物中に存在する他の細胞の遺伝子構造が原因でいずれ異常となる危険性にある細胞である。後者の実施形態により、本発明の形質導入した細胞を予防に用いることが可能となる。乳癌は、予後指標により、疾患が引き起こされる前の初期遺伝的介入としての本発明の形質導入した細胞を用いた治療が可能となる、疾患プロセスの一例である。しかし、本発明の方法は、疾患が検出された後の乳癌の治療処置にも用い得る。癌治療における本発明のさらなる用途は多数存在し、当業者は、必要以上の実験を行わずに、多種類の癌を治療するために本明細書中に記載した本発明を用いることができるであろう。
【0082】
例として、かつ本発明を限定せずに、一用途はエストロゲン依存性の乳癌にある。エストロゲン依存性乳癌中の癌細胞は、たとえば、エストロゲン受容体に結合する抗体またはリガンドを治療ウイルスベクターと組み合わせて用いることによって形質導入する。ベクターは、たとえば、ヘルペスウイルスチミジンキナーゼ遺伝子などの腫瘍阻害遺伝子を含み得る。したがって、ヘルペスチミジンキナーゼによって活性化することができるプロドラッグであるガンシクロビルを加えることによって、形質導入した細胞を選択的に死滅させることができる。腫瘍阻害遺伝子および対応するプロドラッグのさらなる例は多数存在し、当分野で周知であり、必要以上の実験を行わずに当業者によって選択し得る。本発明の形質導入方法の用途と組み合わせた活性化可能なプロドラッグの使用は、他の腫瘍種に幅広く応用してよく、上記の例は、本発明をホルモン依存性または成長もしくは増殖を何らかの可溶性因子に依存する腫瘍に限定しない。
【0083】
たとえば、Her−2/neu陽性腫瘍細胞はエストロゲン依存性ではなく、そのような細胞を含む非エストロゲン依存性腫瘍は、エストロゲン拮抗剤であるタキソールなどの薬物を用いた治療に高度に耐性を有するので、これは乏しい予後指標である。本発明の別の実施形態は、骨髄移植プロトコル中などの腫瘍が混入した細胞の形質導入中に、Her−2/neuまたはヘレグリンに結合する抗体または他の分子をウイルスベクター調製物と共に含めることである。あるいは、形質導入を、腫瘍部位に直接、またはin vivoで腫瘍形成を調節するベクターと共に血管内に行い得る。
【0084】
本発明のさらに別の実施形態は、腫瘍血管構造を、単独でまたは腫瘍細胞の標的化と組み合わせて標的とすることである。完全に記載したかのように本明細書中に参考として組み込まれているSt Croixらは、SAGE分析によって、正常内皮と比較して腫瘍内皮細胞中で特異的に過剰発現されている遺伝子を同定している。これらの遺伝子の多くは、Thy−1細胞表面抗原またはEndo180レクチンなどの細胞表面分子をコードしている。アップレギュレーションされた細胞表面因子のすべては、効率的な安定遺伝子形質導入を行うための刺激を提供するために、本発明の細胞表面結合分子と結合し得る。したがって、腫瘍を治療するための一手法は、腫瘍血管構造と選択的に結合し、正常内皮細胞とは結合しない細胞表面結合分子の存在下で治療ウイルスベクターを用いて形質導入した後で、腫瘍内皮細胞を死滅させることによって腫瘍血管構造を破壊することである。
【0085】
本発明のさらに別の実施形態は、腫瘍中で抗腫瘍遺伝子の発現を選択的に促進するが、正常な血管内皮では促進しないウイルスベクター中に、要素(たとえば、mRNAのプロモーターまたはシス作用性の安定化/分解要素)を組み込むことによって、腫瘍血管構造中で抗腫瘍遺伝子を選択的に発現させることである。このような方法は、ex vivo、in vitroまたはin vivoで起こることができる。腫瘍血管内皮を標的とする場合はin vivoが治療の一方法である。あるいは、たとえば目的が骨髄移植のために骨髄から混入した腫瘍細胞を追放することである場合は、治療方法はex vivoまたはin vitroで起こることができる。
【0086】
さらに、in vivoでの使用は病状に限定されず、正常細胞に形質導入に用いることができる。たとえば、本発明は、骨髄中で、in vivoで造血幹細胞を形質導入するために用い得る。高効率の骨髄形質導入のために、抗体またはFLT−3リガンド、TPOおよびKitリガンド、もしくはその機能的類似体などの他の細胞表面結合分子、あるいは細胞表面結合分子を発現する間質細胞の任意の組合せを、骨髄内への直接注射を行う際にベクターと共に加えることができる。用語「機能的類似体」とは、本発明の細胞表面結合分子の細胞表面結合活性を保持する任意の分子をいう。そのような機能的類似体には、FLT−3リガンド、TPOおよびKitリガンド;1つまたは複数のアミノ酸の置換、付加または欠失を含むFLT−3リガンド、TPOおよびKitリガンド分子;ならびに細胞表面結合分子の細胞表面結合活性を模倣する抗体の断片が含まれる。
【0087】
上記の代替手法は、骨髄間質細胞をウイルスベクターの産生細胞として使用し、したがって、ベクター調製物としてではなく細胞治療を介してベクターおよび細胞表面結合分子を提供することである。別の例は、CD3およびCD28抗体またはGM−CSFおよびIL−4の機能的類似体を、それぞれベクターと共に皮下注射中に加えることによる、T細胞または樹状細胞の形質導入である。皮下組織中のリンパ液が、標的細胞の効率的な形質導入のためにベクターおよび刺激因子をリンパ節内に流す。
【0088】
本発明には、任意選択で、形質導入する細胞の精製が必須ではないという利点が含まれる。主に目的の細胞種の形質導入は、結合させる細胞表面部分の選択によって達成することができる。したがって、血液細胞の混合集団中では、たとえば、特定のT細胞などのCD3を発現する細胞の形質導入は、細胞との相互作用にCD3に特異的な抗体を用いた場合に増強される。これは、CD3を発現しない顆粒球および単球などの集団中の他の細胞種より優先的に起こる。
【0089】
また、本発明は、所望する場合は、精製または単離した細胞種の形質導入も包含する。精製または単離した細胞種の使用により、形質導入する細胞よりも高いベクター濃度に起因するより高い形質導入効率などの、さらなる利点が提供される。
【0090】
精製したT細胞を形質導入する場合は、少なくとも1つの分子はT細胞上に見られる細胞表面分子に結合する。そのような細胞表面分子の例には、CD3、CD28、CD25、CD71、およびCD69が含まれる。これらの細胞表面分子に結合する分子の例には、それらを認識する抗体およびモノクローナル抗体が含まれ、それらの多くが市販されているか、または必要以上の実験を行わずに標準の技術を用いて容易かつ日常的に調製される。CD4+またはCD8+細胞の形質導入の例示的な実施形態では、CD3および/またはCD28を認識するモノクローナル抗体を用い得る。そのような抗体の市販されている例には、CD3用のOKT3およびCD28用のCD28.2が含まれる。これらの抗体分子は、可溶形で用いてよく、任意選択で後に他の分子に架橋結合して、またはビーズもしくは他の固体表面上などの固定した形態で用いてよい。本発明の一実施形態では、抗体を、ウイルスベクターに媒介される形質導入に用いる組織培養ウェル、プレート、またはバッグの壁などの容器の表面上に固定する。理論に束縛されずに、細胞が接着または接触する表面上の固定した抗体を使用することにより、細胞表面上の細胞表面相互作用の局所濃度が増加し得る。
【0091】
形質導入を造血幹細胞する場合は、FLT−3リガンド、TPO(トロンボポエチンもしくは巨核球成長および発達因子)、またはKitリガンドの造血幹細胞受容体に特異的な抗体を細胞表面結合分子として用い得る。あるいは、それだけには限定されないが、CD34またはAC133を含めた幹細胞陽性細胞マーカーに対する抗体を用い得る。化合物または組成物を含むリガンドを細胞表面結合分子として用いる場合は、陰性リガンド含有タンパク質、リガンドまたは異種タンパク質に結合したリガンドの全体を、可溶性または固定した形態のどちらかで用いることができる。固定した形態には、たとえばアビジン/ビオチンを用いたマイクロビーズへの直接または間接的な付着が含まれる。
【0092】
あるいは、リガンドは、ウイルスベクターのウイルスエンベロープ中、任意選択でキメラもしくは融合タンパク質の形態で、および/または1つもしくは複数の他のタンパク質と(共有もしくは非共有)結合して、発現され得る。そのような実施形態では、細胞表面結合分子は、細胞を形質導入するための単一の組成物として、ウイルスベクターと組み合わせて提供する。ウイルスエンベロープ中で発現させ得る細胞表面結合分子のさらなる例には、上記の多数の表面因子が含まれる。
【0093】
抗体またはその断片などの他の細胞表面結合分子は、ノッチもしくはデルタの造血幹細胞受容体に結合するもの、またはノッチもしくはデルタタンパク質自体、または異種タンパク質に結合するノッチもしくはデルタのリガンドである。デルタおよびノッチは、ショウジョウバエの発達において広範な細胞運命の決定に影響を与える細胞表面タンパク質をコードしている。デルタおよびノッチの脊椎動物相同体は、正常な胚発達に必須である。デルタ相同体は、造血の制御に重要に関与している。相同体の可溶形であるデルタ−Serrate−lag2(DSL)は、原始造血前駆体の増殖を増強する。インターロイキン−3(IL−3)、顆粒球コロニー刺激因子(G−CSF)または顆粒球マクロファージコロニー刺激因子(GM−CSF)を含めた造血成長因子と合わせた場合、DSLは原始造血前駆体の増殖を促進し、同時に、原始前駆体が単独でIL−3に応答性のあるより成熟な前駆細胞へと分化することを阻害した(Hanら参照)。DSLは、造血細胞中で発現されるノッチ受容体を活性化し、細胞分化を選択的に遮断するが増殖シグナルを遮断しないことで慣用の造血成長因子に応答する細胞の能力を調節することによって作用する可能性が最も高い(HanおよびMoore、Blood、1999年参照)。したがって、相同体の機能的類似体の抗体であるデルタおよびノッチ相同体は、細胞、特に造血幹細胞の、75%を超える効率のベクター形質導入を達成するのに使用する細胞表面結合分子の例である。
【0094】
本発明には、開示した方法で使用するためのウイルスベクターおよびそれらを含む組成物が含まれる。ベクターは、レトロウイルス(レトロウイルス科)ベクター、たとえばレンチウイルスベクターであることができる。オンコウイルスまたはネズミレトロウイルスベクターなどの他のレトロウイルスベクターも用い得る。さらなるベクターは、他のDNAウイルス、またはその生活環のある時点でそのゲノムをDNA内に転換することができるウイルスに由来し得る。ウイルスは、たとえば、アデノウイルス科、パルボウイルス科、ヘパンダウイルス科(Hepandaviridae)(通常はヘパンダウイルス科に分類されないδ型肝炎ウイルスおよびE型肝炎ウイルスを含む)、パポウイルス科(Papoviridae)(ポリオーマウイルス亜科およびパピローマウイルス亜科を含む)、ヘルペスウイルス科、ならびにポックスウイルス科由来であることができる。
【0095】
レトロウイルス科(すなわちレトロウイルス)のさらなるウイルスは、オンコウイルス亜科、スプマウイルス亜科、スプマウイルス、レンチウイルス亜科、およびレンチウイルスの属または亜科のものである。亜科であるオンコウイルス亜科のRNAウイルスは、望ましくは、ヒトT−リンパ球向性ウイルス1型もしくは2型(すなわちHTLV−1もしくはHTLV−2)またはウシ白血病ウイルス(BLV)、トリ白血症肉腫ウイルス(たとえば、ラウス肉腫ウイルス(RSV)、トリ骨髄芽球症ウイルス(AMV)、トリ赤芽球症ウイルス(AEV)、およびラウス関連ウイルス(RAV;RAV−0からRAV−50)、哺乳動物C型ウイルス(たとえば、モロニーマウス白血病ウイルス(MuLV)、ハーベイマウス肉腫ウイルス(HaMSV)、アベルソンマウス白血病ウイルス(A−MuLV)、AKR−MuLV、ネコ白血病ウイルス(FeLV)、サル肉腫ウイルス、細網内皮症ウイルス(REV)、脾臓壊死ウイルス(SNV))、B型ウイルス(たとえばマウス乳癌ウイルス(MMTV))、ならびにD型ウイルス(たとえば、メーソン−ファイザーサルウイルス(MPMV)および「SAIDS」ウイルス)である。
【0096】
亜科レンチウイルスのRNAウイルスは、望ましくは、ヒト免疫不全ウイルス1型もしくは2型(すなわちHIV−1もしくはHIV−2であり、HIV−1は、以前はリンパ節腫脹関連ウイルス3(HTLV−III)および後天性免疫不全症候群(AIDS)関連ウイルス(ARV)と呼ばれていた)、または、AIDSもしくはAIDS様の疾患に関連付けられた同定されたHIV−1もしくはHIV−2に関連する別のウイルスである。頭字語「HIV」または用語「AIDSウイルス」もしくは「ヒト免疫不全ウイルス」は、本明細書中で、これらのHIVウイルス、ならびにHIV関係およびHIV関連ウイルスを総称的に指すために使用する。さらに、亜科レンチウイルスのRNAウイルスは、たとえば、ビスナ/マエディウイルス(たとえばヒツジに感染)、ネコ免疫不全ウイルス(FIV)、ウシレンチウイルス、サル免疫不全ウイルス(SIV)、ウマ伝染性貧血ウイルス(EIAV)、もしくはヤギ関節炎−脳炎ウイルス(CAEV)、またはそれらの組合せであることができる。
【0097】
例示的なレンチウイルスベクターは、HIVに由来するもの、たとえば、HIV−1、HIV−2、またはそれらの組合せキメラである。もちろん、レトロウイルスの異なる血清型、特にHIVを単独または任意の組合せで用いて、本発明で用いるためのベクターを調製し得る。本発明のベクターは、たとえば、野生型ウイルス中には存在するが、「基本」レンチウイルスベクター中には存在しないシス作用性要素を含むことができる。「基本」レンチウイルスベクターは、最小限で5’リーダー配列およびgagコード配列中にLTRおよびパッケージング配列しか含まないが、任意選択で、Rev依存性の様式でベクターRNAの核外移行を容易にするために、RRE要素を含むこともできる。ベクターは、細胞内への形質導入の効率を増強するヌクレオチド配列をさらに含むことができる。
【0098】
そのようなベクターの例は、gagおよびpolの完全な配列を含むベクターである、pN2cGFPである。別の例は、pol中の位置が約4551〜約5096の配列を含むベクターである(参照位置は、pNL4−3配列、寄託番号M19921、HIVNL43 9709bp、C.E. Buckler、NIAID、NIH、メリーランド州Bethesdaから提供)。しかし、ベクターの形質導入効率を向上することができる、wt−HIVからの任意のシス作用性配列を用い得る。本発明による効率的な形質導入が可能なベクターの他の例は、米国特許第5,885,806号に記載のcr2HIV構築体である。
【0099】
Zennouら、2000年が中心DNAフラップ(pNL4−3上の位置4757〜4935に対応する、pLAI3上の位置4793〜4971の178塩基対の断片)として記載した、形質導入効率の有意に増加させるために不十分であったと以前に同定された配列は、形質導入効率を増加させることができる。本発明には、この小断片は形質導入効率を増加させるために不十分であるが、より大きな545塩基対の断片(pNL4−3中の位置4551〜5096)、またはそれを含むさらに大きな断片が、本発明の一部として形質導入を増加させることができたという発見が含まれる。
【0100】
本発明で用い得るウイルスベクター構築体のさらなる例は、完全に記載したかのように本明細書中に参考として組み込まれている米国特許第5,885,806号に見られる。特許5,885,806号の構築体は単なる例であり、細胞を効率的に形質導入するベクターの範囲を限定しない。むしろ、これらの構築体は、当業者に、本発明で用いるウイルスベクターが野生型ウイルスからの最小限の配列、または野生型ウイルスのゲノムのほぼ全体までの配列を含んでいても、疾患の複製および/または生産に必要な必須の核酸配列を排除し得るというさらなる指針を提供する。細胞の効率的な形質導入に必要な配列を正確に決定する方法は日常的であり、当分野で周知である。たとえば、ウイルス配列を「基本」ベクター内に戻す体系的な組み込ませること、またはcr2HIVなどの実質的にHIVゲノムの全体を含むベクターから配列を欠失させることは日常的であり、当分野で周知である。
【0101】
さらに、他のウイルスの主鎖からの配列を、サイトメガロウイルス(CMV)などの目的のウイルスベクター内に配置することも、当分野で周知である。実際に用いるウイルスベクターが何であるかにかかわらず、ウイルス遺伝物質によってコードされている様々なアクセサリータンパク質、およびウイルス遺伝物質中に存在する配列は、これらのタンパク質または配列が特定の細胞種において形質導入効率を増加させる場合は、ベクターまたはヘルパーゲノム中に残してもよい。特定の遺伝物質が、ベクターまたはヘルパーゲノムのどちらかに配列を組み込ませることによって形質導入効率を増加させるかどうかを決定するために、多数の日常的なスクリーニングが利用可能である。本発明の一実施形態は、ベクターまたはヘルパーゲノムのどちらかにアクセサリータンパク質を含めないことである。この実施形態は、形質導入効率を増加させるためにアクセサリータンパク質および他の配列をベクターまたはヘルパーゲノムのどちらかに残す本発明の実施形態を排除しない。
【0102】
本発明で用いるウイルスベクターは、「偽型」の形成の結果であってもよく、ここでは、異なるウイルスによる細胞の同時感染により、1つのウイルスのゲノムが別のウイルスの1つまたは複数のエンベロープタンパク質を含む外層内にカプセル封入されて含まれる、子孫ビリオンが生じる。この現象は、目的のウイルスベクターと別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質または細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションまたは同時感染を行うことによって、目的のウイルスベクターを「偽型」ビリオン内にパッケージングするために用いられている。米国特許第5,512,421号を参照されたい。このような混合ウイルスは、用いる1つまたは複数の異種エンベロープタンパク質に対する抗血清によって中和することができる。偽型の形成に一般的に用いられる1つのウイルスは、ラブドウイルスの水疱性口内炎ウイルス(VSV)である。偽型化を用いることで、用いる異種ウイルスの侵入機構の要素を含めることによって、ウイルスの宿主細胞範囲が広がる。
【0103】
本発明で用いるためのウイルスベクターおよびVSVの偽型化により、VSV Gタンパク質を含む膜に囲まれたヌクレオカプシド中にカプセル封入されたウイルスベクター核酸を含むウイルス粒子がもたらされる。ヌクレオカプシドは、通常はウイルスベクターに関連しているタンパク質を含むことができる。膜を含む周囲のVSV Gタンパク質は、ウイルスベクターのパッケージングに用いる細胞から放出された際にウイルス粒子の一部を形成する。パッケージング細胞の例は米国特許第5,739,018号に記載されている。本発明の別の実施形態では、ウイルス粒子はHIVに由来し、VSV Gタンパク質を用いて偽型にする。VSV Gタンパク質を含む偽型ウイルス粒子は、広宿主性のウイルスベクターよりも高い効率で多様なアレイの細胞種に感染することができる。宿主細胞の範囲には、ヒト、げっ歯類、魚、両生類および昆虫などの哺乳動物および非哺乳動物種がどちらも含まれる。
【0104】
また、本発明の形質導入方法で用いるためのウイルスベクターは、1つまたは複数の核酸配列を含み、ウイルス中に存在するプロモーターの制御下またはベクター内に導入した異種プロモーターの制御下でそれを発現することもできる。プロモーターは、厳しく制御された遺伝子発現のためにオペロンに隣接するように、赤血球DNAse過敏部位などの遮蔽要素をさらに含み得る。プロモーターの例には、HIV−LTR、CMVプロモーター、PGK、U1、エプスタインバーウイルス由来のEBER転写ユニット、tRNA、U6およびU7が含まれる。Pol IIプロモーターが有用であり、Pol IIIプロモーターも用い得る。組織特異的プロモーターの使用も、実施形態の1つである。たとえば、βグロビン遺伝子座調節領域エンハンサーならびにαおよびβグロビンプロモーターは、赤血球および赤血球細胞中で組織特異的発現をもたらすことができる。別の実施形態は、プロモーターに関連するシス作用性配列を用いることである。たとえば、本明細書中に参考として組み込まれている米国特許第5,814,500号に記載のように、U1遺伝子を用いてアンチセンス遺伝子発現を増強させ得る一方で、非プロモーター配列を用いて、標的のスプライシングされたRNAをアンチセンスまたはリボザイム分子の標的とする。
【0105】
もちろん、ウイルス由来の任意のシス作用性ヌクレオチド配列を本発明のウイルスベクター内に組み込ませ得る。たとえば、レトロウイルスゲノム中に見られるシス作用性配列を用いることができる。たとえば、形質導入効率をさらに増加させるために、gag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来するシス作用性ヌクレオチド配列を本発明のウイルスベクター内に組み込ませ得る。シス作用性配列は、発現されたポリペプチドをコードしている必要はなく;翻訳開始部位の欠失などの遺伝子変化によってポリペプチドもしくはその一部として発現される必要はなく;より大きなポリペプチドの一部分もしくは断片のみをコードしていることができ;またはネイティブ配列から1つもしくは複数の置換、付加、もしくは欠失を含む突然変異体配列であることができる。シス作用性配列の例は、HIV pol遺伝子内で同定されたcPPT(中央ポリプリントラクト)配列である。
【0106】
本発明のウイルスベクター中の1つまたは複数の核酸配列は、ベクターが由来するウイルス中に見つかるか、または異種配列であってもよい。これは、目的の遺伝子産物であるか、それをコードしている完全長配列または部分配列であることができる。このような配列および遺伝子産物は、細胞中で生物学的効果を生じる能力を有する、生物活性のある因子であることができる。そのような因子の例には、タンパク質、リボ核酸、酵素、トランスポーターまたは他の生物活性のある分子が含まれる。
【0107】
一実施形態では、因子は、毒素、転写因子、成長因子もしくはサイトカインなどのタンパク質、構造タンパク質、または細胞表面分子である。タンパク質は、機能が同定されていない1つまたは複数のドメインを含んでいてよく、形質導入した細胞に相同的であり得る。さらに、タンパク質は、形質導入する細胞中に存在しない、またはその中で欠損もしくは改変されていてもよい。あるいは、タンパク質は、形質導入した細胞中で天然タンパク質が正常の活性を実行することを妨げるための、トランス優勢の陰性突然変異体またはデコイであり得る。
【0108】
たとえば、核酸配列は、形質導入した細胞中で発現された、またはこれから発現されるRNAに結合し、切断し、破壊するリボザイムをコードし得る。あるいは、核酸配列は、特定の核酸配列を標的として、その分解をもたらすように設計されたアンチセンス分子をコードし得る。ベクターに含まれる配列は、形質導入した細胞中で過剰発現されるか、誘導発現されるか、または細胞もしくはウイルスの制御的転写調節下にあり得る。意図する使用に応じて、異種配列は、形質導入した細胞のマーカーを含めた任意の所望のタンパク質をコードし得る。そのようなマーカーには、ネオマイシン等の特定の耐性の表現型、MDR−1(P−糖タンパク質)、O6−メチルグアニン−DNA−メチルトランスフェラーゼ(MGMT)、ジヒドロ葉酸還元酵素(DHFR)、アルデヒドデヒドロゲナーゼ(ALDH)、グルタチオン−S−トランスフェラーゼ(GST)、スーパーオキシドジスムターゼ(SOD)およびシトシンデアミナーゼなどの選択マーカーが含まれる。総説には、本明細書中に参考として組み込まれているKocらを参照されたい。
【0109】
本発明の方法では、形質導入する細胞を、ウイルスベクターの施用の前、後、またはそれと同時に、細胞表面に結合する少なくとも1つの分子に接触曝露させる。たとえば、細胞は、形質導入するウイルスベクターの前、後、またはその存在下で、CD3およびCD28抗体(培養皿の表面上にコーティングまたは培養物中に存在させるビーズに固定)を有する培地中で培養することができる。細胞は、ウイルスベクターとの最初の接触後または接触時にしか、固定したCD3および/またはCD28に曝すことができない。このような条件下では、細胞は、ウイルスベクターを用いた実際の形質導入の前には細胞表面結合分子(または複数の細胞表面結合分子)に曝されない。細胞をウイルスベクターに曝した(形質導入)後に細胞表面結合分子との接触が起こる実施形態では、接触は形質導入後3日以内、あるいは形質導入後1〜2日以内に起こることができる。
【0110】
インキュベーションまたは細胞とウイルスベクターとの接触は、用いる条件および材料に応じて様々な長さの時間であり得る。インキュベーション時間に影響を与える要素には、用いる細胞、ベクターおよびMOI(感染多重度)、細胞表面の結合に用いる分子(または複数の分子)および量、前記分子(または複数の分子)が固定または可溶化されているかおよびどのように固定または可溶化されているか、ならびに所望する形質導入効率のレベルが含まれる。たとえば、インキュベーションは約8〜約72時間、または約12〜約48時間である。別の実施形態では、インキュベーションまたは接触は約24時間であり、任意選択で1回繰り返す。別の実施形態では、インキュベーションまたは接触は約24時間〜約36時間である。
【0111】
形質導入する細胞とウイルスベクターとの接触少なくとも1回起こるが、細胞種に応じて複数回起こり得る。たとえば、CD34陽性幹細胞の高効率形質導入が、ベクターを用いた複数の形質導入で達成されている。本発明の別の方法は、ウイルスベクターを細胞表面結合分子(たとえば、CD3および/もしくはCD28抗体またはFLT−3リガンド、TPOもしくはKitリガンド)と組み合わせて同時導入し、形質導入後の間の約1〜約8日間、培地の交換を避けることである。あるいは、形質導入後3日間、培地を交換しない。形質導入は、プロセスが細胞またはそれを含む生物に対して顕著に有害とならない限り、条件が許容する間、進行させることができる。このような使用のための細胞表面結合タンパク質のさらなる例には、本明細書中に上述したものが含まれる。
【0112】
使用するMOIは約1〜約400、または500未満であることができる。MOIは約2〜約50であることができる。あるいは、MOIは約10〜約30であるが、約1〜約10、約20、約30、または約40の範囲も企図される。あるいは、約20または約0.5〜10のMOI。さらに、細胞1個あたりのウイルスベクターのコピー数は少なくとも1であるべきである。しかし、細胞1個あたり多数のコピーのベクターも、上述の方法で用い得る。例示的な細胞1個あたりのコピー数は約1〜約100である。所望のコピー数は、ベクター形質導入の結果として治療的、予防的もしくは生物学的影響をもたらす、または最も効率的な形質導入をもたらす最小コピー数である。
【0113】
治療的または予防的応用には、所望のコピー数は、細胞またはそれを含む生物に対して顕著に有害とならずに細胞によって許容される最大コピー数である。細胞1個あたりの最小および最大コピー数はどちらも、形質導入する細胞および存在し得る他の細胞に応じて変動する。最適コピー数は当業者によって日常的な方法を用いて容易に決定される。たとえば、細胞を漸進的に増加する濃度または感染多重度で形質導入する。その後、細胞をコピー数、治療的または生物学的影響ならびに形質導入した細胞またはそれを含む宿主に対する有害効果(たとえば、安全性および毒性)について分析する。
【0114】
ウイルスベクターとin vitroでインキュベーションを行った後、細胞を細胞表面結合分子(または複数の細胞表面結合分子)の存在下で様々な期間培養し、その後、形質導入の効率について分析するか、または他の方法で用い得る。あるいは、細胞を細胞の成長および増殖をもたらす任意の条件下、たとえば、インターロイキン−2(IL−2)と共にインキュベーションまたは細胞表面結合分子(もしくは複数の細胞表面結合分子)と共に、次いでIL−2と共にインキュベーションなどで培養し得る。形質導入後のインキュベーションは任意の期間、たとえば約1〜約7〜10日間であり得る。約14日間などのより長い期間を用いてもよいが、細胞成長に有害な期間は望ましくない。ウイルスベクターとのインキュベーションの前に細胞を細胞表面結合分子(または複数の細胞表面結合分子)と共に培養する本発明の実施形態では、培養時間は約24〜約72時間の範囲、または約24時間であり得る。
【0115】
このような形質導入前の培養は、たとえば当分野で教示されている、形質導入前のサイトカインおよび/またはマイトジェンを用いた細胞の刺激と比較し得る。本発明には、そのような刺激の回避から生じる利点が含まれる。たとえば、刺激により、細胞の数が増殖によって増加して、刺激前よりも刺激後の細胞の方がはるかに多くなる。この増加した細胞セットの形質導入には、はるかに多数のウイルスベクターおよび関連する形質導入の材料(たとえば、容器、培地、サイトカインなど)が必要であり、関連するコストが増加する。さらに、細胞の刺激により、さらなる応用のためのその品質に影響が与えられる。Movassaghらは、3日間の形質導入前刺激の使用を記載しているが、これは形質導入およびさらなる培養後にT細胞レパートリー多様性の劣化をもたらす。さらに、形質導入前刺激により、活動的に分裂していない細胞の形質導入から得られる利点が取り除かれる。
【0116】
本発明で観察された形質導入の効率は約50〜100%である。たとえば、効率は少なくとも約50〜75%または約75〜90%である。本発明の他の実施形態は、形質導入効率が少なくとも約90〜100%である実施形態である。さらなる実施形態は、少なくとも91、92、93、94、95、96、97、98、99および100%の形質導入効率を有する。
【0117】
上記に加えて、形質導入した細胞は、研究または生きた対象における病状の治療もしくは予防に用い得る。研究使用の例は、Unutmazらによって記載されている構造−機能研究である。形質導入した細胞の治療的使用には、生きた生物内への細胞の導入が含まれる。たとえば、HIVに感染している、または感染している危険性にある個体から単離した未刺激の初代T細胞を、最初に、本発明の方法を用いて米国特許第5,885,806に記載のものなどのベクターによって形質導入し、続いて形質導入した細胞を個体に注射して戻し得る。あるいは、細胞をウイルスベクター中に存在する異種配列の発現に直接用い得る。
【0118】
HIVの治療または予防の一部として用いる場合は、ベクターは抗HIV用途に適用された毒素または他の抗ウイルス剤をコードしていてよい。あるいは、ベクターは、tat、rev、nef、vpu、またはvpr遺伝子のトランス優勢の陰性突然変異体など、HIVを標的とするよう設計された薬剤をコードしていてよい。他の応用では、形質導入した細胞は、鎌状赤血球貧血または地中海貧血を矯正するための適切なグロビン遺伝子を発現するように矯正し得る。また、免疫細胞も、その免疫機能、抗原に対するその応答、または他の細胞とのその相互作用を調節するために形質導入し得る。当業者は、本発明の形質導入方法ならびに当分野で知られている数々の他の使用および用途の上記の使用を認識しているであろう。
【0119】
本発明は、自己T細胞を製造し、およびT細胞を形質導入するプロセスを提供する。本発明の方法を用いることで、細胞をプラスチックバッグに対して中実のプラスチックフラスコ中で培養した場合に高い形質導入レベルが達成される。一態様では、大スケールの形質導入には、10層の細胞工場を用いる。一態様では、臨床的スケールでのT細胞の効率的な形質導入にはベクター体積の2倍縮小が必要である。一態様では、形質導入をさらに増加させるために、ベクター(T細胞の形質導入用)を24時間間隔で2回加える。
【0120】
一態様では、T細胞の増殖には、通常の培地よりも低い酸素濃度および僅かに低いpH。本発明では、通常の培地よりも低い酸素濃度および僅かに低いpHの存在下でT細胞がより良好に増殖することが判明した。一態様では、通常の空気(約20%のO2)と対比して、N2/O290%/10%を用いてT細胞を培養する。一態様では、pHを低下するために、混合空気中のCO2濃度を通常の5%から10%まで増加する。この混合気体の変更により、より高い増殖率が可能となった。
【0121】
一態様では、増殖の終わりに約1000億個の細胞、培地の灌流を用いる。本発明者らは、増殖の終わりに約1000億個の細胞を保有するためには、培地の灌流を用いなければならないことを発見した。一態様では、以前に用いられていた20Lバッグでは十分な細胞を支持することができなかったので、50Lの灌流バッグを用いる。一態様では、細胞濃度が0.5×106個の細胞/mlを超えたらすぐに、約3L/日の速度で灌流を開始する。一態様では、翌日、揺動速度および角度を1単位同時に増加して、速度を約2倍増加する。
【0122】
一態様では、収集中、続く凍結および解凍後のその生存度を増加するためにT細胞を冷却する。一部の用途では、大量の細胞の収集は長時間かかり、細胞はより低い温度でより良好に生存するので、これを行う必要がある。一態様では、細胞をより小さな10Lバッグに移し、冷却のために冷蔵庫に入れる。細胞の洗浄には冷蔵した緩衝液を用いるべきである。
【0123】
本発明は、約75%を超える効率までのウイルスベクターを用いた細胞の安定形質導入を行うための、方法およびそれに関連する組成物を対象とする。安定形質導入した細胞は、形質導入の約7〜10日後、または任意選択で約14日後に、一過的に形質導入したまたは偽形質導入した細胞から区別し得る。この方法は、形質導入する細胞と細胞表面に結合する少なくとも1つの分子との接触により安定形質導入の効率が増加するという事実に関連する。驚くべきことに、接触ステップは形質導入ステップの後に起こってもよい。さらに驚くべきことに、最も高い安定形質導入レベルは、形質導入が最初に起こり、続いて固定した細胞表面結合分子との接触が起こった場合に見られた。
【0124】
本発明の方法は、細胞表面結合分子との接触と組み合わせた、ウイルスベクターを用いた形質導入のステップを含む。上に注記したように、接触は、ベクターの形質導入の前、後、またはそれと同時に起こり得る。本発明は、任意の細胞および任意の細胞表面結合分子の使用に広く適用可能である。本方法で用いる細胞には、in vivo供給源から新しく単離した未刺激の初代細胞、および細胞を増殖状態に保つ因子の存在下で様々な期間事前に培養していてもよい細胞系が含まれる。細胞系を用いる場合は、本方法を用いて形質導入する前に、最初に刺激因子を存在させずに培養してもよい。
【0125】
初代細胞の場合は、最初にこれらをin vivo供給源から得て、続いて任意選択で特定の細胞種について選択を行う。たとえば、初代CD4+および/またはCD8+T細胞を用いる場合は、末梢血(PB)または臍帯血(臍源由来の「CB」)試料を最初に得て、続いてCD4+および/またはCD8+細胞種について富化を行う。標準の磁気ビーズ陽性選択、プラスチック接着性陰性選択、および/または当分野で認められている他の標準技術を用いて、CD4+および/またはCD8+細胞を混入PB細胞から離して単離し得る。単離した細胞種の純度は、標準技術を用いた免疫表現型決定およびフローサイトメトリーによって決定し得る。
【0126】
単離後、50%を超える、または75%を超える効率でウイルスベクターを用いて形質導入するために、初代細胞を本方法で用い得る。本発明は、T細胞などの初代リンパ球、目的の異種遺伝物質を発現する能力を有するHIV−1系ベクターで形質導入したにおいて、最も有利に用いられる。別の使用は、CD34陽性細胞などの初代造血幹細胞を用いた使用である。異種遺伝物質が疾患を治療または予防するためにin vivoで使用する治療的または予防的生成物であるか、それをコードしている場合は、形質導入した初代細胞を対象などのin vivo環境に導入して戻すことができる。したがって、本発明は、遺伝的欠陥と戦うことまたはウイルス感染を標的とすることによる、疾患を治療または予防する遺伝子治療における形質導入した細胞の使用を企図する。
【0127】
また、本発明は、遺伝子の機能を決定するため、哺乳動物細胞中で遺伝子を効率的に発現するため、目的の遺伝子の機能的スクリーニングのために遺伝子ライブラリ(cDNAライブラリおよび遺伝子のアンチセンスもしくはリボザイムライブラリ)を発現するため、タンパク質−タンパク質またはタンパク質−核酸の二重ハイブリッド様の検出戦略、遺伝子捕捉手法、マイクロアレイもしくはタンパク質アレイを用いた高スループット遺伝子スクリーニング分析で使用するため、またはSAGE、プロテオミクスおよび他の機能的分析方法を用いて研究を行うために、細胞を効率的に形質導入するための使用も企図される。
【0128】
混合集団における初代細胞の形質導入には、上記の単離/精製ステップは使用しない。その代わりに、形質導入する細胞は、少なくとも1つの適切な細胞表面分子またはその細胞種上に見つかる部分の選択および前記部分に結合する能力を有する1つまたは複数の分子の調製によって標的とする。細胞表面部分は、標的とした細胞の表面上にある受容体、マーカー、または他の認識可能なエピトープであり得る。選択後、本発明で用いるために、特異的抗体などの部分と相互作用する分子を調製し得る。
【0129】
たとえば、CD4+および/またはCD8+細胞は、最初に精製し、その後、固定したCD3およびCD28抗体を用いて本発明の方法によって形質導入するか、または末梢血細胞(PBC)もしくは末梢血単核細胞(PBMNC)などの混合集団の一部として、同じ抗体を用いることによって形質導入することができる。精製または単離が困難であり得る全白血球集団中の造血幹細胞は、固定したCD34抗体を用いることによって混合集団中で形質導入し得る。
【0130】
本発明の細胞表面結合分子は、形質導入する細胞の表面上に見られる任意の部分を標的としてそれに結合し得る。部分は、細胞表面上の受容体、マーカー、または他のタンパク質性もしくは非タンパク質性因子の一部として見つかる。部分には、細胞表面結合分子によって認識されるエピトープが含まれる。これらのエピトープには、ポリペプチド配列、炭水化物、脂質、核酸、またはイオンおよびそれらの組合せを含むものが含まれる。
【0131】
細胞表面結合分子の例には、抗体またはその抗原結合断片およびリガンドまたは細胞表面受容体の結合ドメインが含まれる。細胞表面結合分子自体がポリペプチド、核酸、炭水化物、脂質、またはイオンであり得る。分子は、抗体またはFabもしくはFv断片などのその断片であることができる。あるいは、分子は可溶形で用いるのではなく、形質導入する細胞をそれと共に培養し得るビーズなどの固体培地上、または形質導入する細胞をそれ上で培養し得る組織培養皿、バッグもしくはプレートの表面に固定されている。一実施形態では、CD4+またはCD8+細胞の形質導入には、CD3および/またはCD28を認識するモノクローナル抗体は、ウイルスベクターの存在下で、細胞培養バッグ中で用い得る。
【0132】
本発明には、開示した方法の一部として用いるための細胞表面結合分子を含む組成物が含まれる。例示的な組成物は、形質導入する分子およびウイルスベクターを、任意選択で形質導入する細胞の存在下で含む。ウイルスベクターは、任意の源、たとえばレトロウイルスベクター由来であり得る。たとえば、これらはレンチウイルスベクターであることができる。例示的なレンチウイルスベクターは、ヒト免疫不全ウイルス(HIV)、たとえば、HIV−1、HIV−2、またはそれらのキメラの組合せに由来するものである。もちろん、本方法を用いることによって異なるウイルスベクターを同一細胞内に同時に形質導入し得る。たとえば、1つのベクターが複製欠損性または条件的に複製するレトロウイルスベクターであることができ、一方で第2のベクターは第1のベクターの複製/パッケージングおよび増殖を可能にするパッケージング構築体であることができる。様々なウイルスアクセサリータンパク質をウイルスベクターによってコードさせる場合は、細胞内に形質導入するベクターの任意の1つ中に存在し得る。あるいは、ウイルスアクセサリータンパク質は、形質導入に用いるウイルス粒子中のその存在を介して、形質導入プロセスに存在し得る。このようなウイルス粒子は、形質導入効率の増加をもたらすために有効な量の同時パッケージングされたアクセサリータンパク質を有し得る。一実施形態では、ウイルスベクターは、アクセサリータンパク質の1つまたは複数をコードしない。
【0133】
本発明の形質導入方法で用いるためのウイルスベクターはまた、1つまたは複数の核酸配列を含むことができ、プロモーターの制御下でそれを発現することができる。本発明の一実施形態では、核酸配列は、発現の際に形質導入する細胞において遺伝的欠陥を緩和または矯正する遺伝子産物をコードしている。別の実施形態では、核酸配列は、ウイルス感染症を予防または治療することができる遺伝的抗ウイルス剤をコードまたは構成している。「遺伝的抗ウイルス剤」とは、遺伝物質によってコードまたは構成されている任意の物質を意味する。そのような薬剤の例は米国特許第5,885,806号に提供されている。これらには、逆転写酵素もしくはプロテアーゼなどのウイルスタンパク質を阻害すること;結合部位もしくは標的部位についてウイルス因子と競合すること;またはリボザイムおよびアンチセンス構築体の場合などのようにウイルス標的を直接標的として分解することによって機能する薬剤が含まれる。遺伝的抗ウイルス剤の他の例には、アンチセンス、RNA囮、トランス優勢の突然変異体、インターフェロン、毒素、RNAスプライシングを調節または改変する核酸、免疫原、および「ハンマーヘッド」などのリボザイム、ならびにそれらの外部ガイド配列(EGS)に媒介された形態が含まれる。
【0134】
あるいは、ウイルスベクターは、形質導入した細胞のマーカーをコードすることができる。図および以下に示す例では、CD4+細胞内に形質導入したウイルスベクターによってコードされているマーカーは緑色蛍光タンパク質(GFP)である。他のマーカーには、上述のものが含まれる。GFPの検出は、機能的に形質導入された細胞の数を同定する役割を果たし得、これらは、ベクターで形質導入されただけでなく、FACS分析によって検出されるレベルでGFPを機能的に発現することができた。一部の細胞は、ベクターで形質導入されているが、FACS検出で用いる限界未満のレベルでGFPを発現している可能性があるので、検出は形質導入した細胞の実際の数を表さない可能性があることに注意されたい。
【0135】
トランスフェクション効率を検出する代替手法は、ポリメラーゼ連鎖反応(PCR)を用いる方法である。たとえば、TaqMan PCRを用いて、形質導入した細胞中の安定に組み込まれたウイルスベクターの実際のコピー数を決定することができる。
【0136】
形質導入する細胞は、細胞表面結合分子との接触の前、後、またはそれと同時に、ウイルスベクターと接触曝露させ得る。したがって、最初に細胞をベクターに一定期間曝し、続いて細胞表面結合分子の導入を行うことができる。このような細胞は、細胞周期に入るように意図的に刺激していない、新しく単離したまたは調製した初代細胞であり得る。あるいは、最初に細胞を細胞表面結合分子に一定期間曝し、続いてウイルスベクターと接触させることができる。ベクターとの接触後、過剰のベクターを除去する必要はなく、細胞の成長および/または増殖に貢献する条件下で細胞を培養することができる。そのような条件は、細胞表面結合分子またはT細胞の場合はサイトカインおよびリンホカインなどの他の刺激/活性化因子の存在下であり得る。あるいは、細胞との接触後かつさらなる培養の前に過剰のベクターを除去し得る。
【0137】
本発明の別の実施形態は、ウイルスベクターおよび細胞表面結合分子をどちらも同時に存在させて細胞を培養することである。このような細胞は、事前に刺激する必要がない。一定期間後、連続的に存在する細胞表面結合分子または他の刺激/活性化因子などの成長または増殖誘導条件下で細胞を培養する。あるいは、さらなる培養の前に過剰のベクターを除去し得る。
【0138】
ウイルスベクターおよび細胞表面結合分子の投与の上記の任意の組合せにおいて、ベクターとのインキュベーションを任意選択で少なくとも1回繰り返すことができる。ベクターとの接触は、複数回、たとえば2回、3回、4回、またはそれ以上繰り返すこともできる。
【0139】
ウイルスベクターを用いて形質導入する細胞のインキュベーションは、用いる条件および材料に応じて様々な長さの時間であり得る。インキュベーション時間に影響を与える要素には、用いる細胞、ベクターおよびMOI(感染多重度)、細胞表面の結合に用いる分子(または複数の分子)および量、前記分子(または複数の分子)が固定されているかおよびどのように固定されているか、ならびに所望する形質導入効率のレベルが含まれる。本発明の一実施形態では、細胞はTリンパ球であり、ベクターはHIV系であり、MOIは約20であり、細胞表面結合分子はビーズ上に固定したCD3およびCD28抗体であり、生じる効率は少なくとも93%である。当業者には明らかなように、上記要素の一部は直接相関しており、一方で他の要素は逆相関している。たとえば、MOIの低下は効率のレベルを低下させる可能性が高く、一方で用いる細胞表面結合分子の量を増加した場合は効率が維持される可能性が高い。
【0140】
ウイルスベクターおよび形質転換させる細胞のインキュベーションの長さは、たとえば24時間であることができ、任意選択で、リンパ球では1回、造血幹細胞では4回まで繰り返す。同様に、ウイルスベクターを導入する前に細胞を細胞表面結合分子と共にインキュベーションする実施形態では、インキュベーションは約12時間〜約96時間であり得る。細胞表面結合分子とのインキュベーションは、細胞とウイルスベクターとの接触と同時に起こることができる。このような状況下では、ベクターを導入する際に細胞表面結合分子を細胞と接触させたままであってもよい。あるいは、ベクターを細胞に導入する前に、最初に過剰の細胞表面結合分子を培養から除去してもよい。
【0141】
ベクターとの接触後、細胞を、その成長または増殖に貢献する条件下で培養する。たとえば、条件は、細胞表面結合分子の存在下における連続培養である。あるいは、細胞を最初に細胞表面結合分子と共に培養し、続いてインターロイキン−2などの細胞成長に貢献する別の因子を含む培地に置き換える。さらに別の実施形態は、過剰の細胞表面結合分子および過剰のベクターをどちらも除去し、続いて成長または増殖に貢献する因子の存在下での培養およびさらなるベクター形質導入を増強することであろう。そのような因子には、フィトヘマグルチニン(PHA)およびサイトカインなどのマイトジェン、成長因子、活性化剤、細胞表面受容体、細胞表面分子、可溶性因子、あるいはそれらの組合せ、ならびに単独でまたは別のタンパク質もしくは因子と組み合わせたそのような分子の活性断片、あるいはそれらの組合せが含まれる。
【0142】
さらなる因子の例には、表皮成長因子(EGF)、トランスフォーミング成長因子α(TGF−α)、アンジオテンシン、トランスフォーミング成長因子β(TGF−β)、GDF、骨形態形成タンパク質(BMP)、線維芽細胞成長因子(FGF酸性および塩基性)、血管内皮成長因子(VEGF)、PIGF、ヒト成長ホルモン(HGH)、ウシ成長ホルモン(BGH)、ヘレグリン、アンフィレギュリン、Ach受容体誘導活性(ARIA)、RANTES(活性化の際に制御、正常T発現および分泌)、アンジオゲニン、肝細胞成長因子、腫瘍壊死因子β(TNF−β)、腫瘍壊死因子α(TNF−α)、アンジオポイエチン1もしくは2、インスリン、インスリン成長因子IもしくはII(IGF−IもしくはIGF−2)、エフリン、レプチン、インターロイキン1、2、3、4、5、6、7、8、9、10、11、12、13、14、もしくは15(IL−1、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、IL−12、IL−13、IL−14、もしくはIL−15)、G−CSF(顆粒球コロニー刺激因子)、GM−CSF(顆粒球−マクロファージコロニー刺激因子)、M−CSF(マクロファージコロニー刺激因子)、LIF(白血病阻害因子)、アンジオスタチン、オンコスタチン、エリスロポイエチン(EPO)、インターフェロンα(亜型を含む)、インターフェロンβ、γ、およびω、ケモカイン、マクロファージ炎症性タンパク質−1αもしくはβ(MIP−1αもしくはβ)、単球化学走性タンパク質−1もしくは−2(MCP−1もしくは2)、GROβ、MIF(マクロファージ遊走阻害因子)、MGSA(黒色腫成長刺激活性)、αインヒビンHGF、PD−ECGF、bFGF、リンフォトキシン、ミュラー阻害物質、FASリガンド、骨原性タンパク質、プレイオトロフィン/ミッドカイン、毛様体神経栄養因子、アンドロゲン誘導性成長因子、自己分泌運動性因子、ヘッジホッグタンパク質、エストロゲン、プロゲステロン、アンドロゲン、糖質コルチコイド受容体、RAR/RXR、甲状腺受容体、TRAP/CD40、EDF(赤血球分化因子)、Fic(成長因子誘導性ケモカイン)、IL−1RA、SDF、NGRもしくはRGDリガンド、NGF、チモシン−α1、OSM、ケモカイン受容体、幹細胞因子(SCF)、またはそれらの組合せが含まれる。当業者には明らかなように、培養条件の選択は、形質導入する細胞に関する知識、および続く細胞の使用意図に依存する。たとえば、IL−3、IL−6および幹細胞因子の組合せは、ヒト移植に用いる形質導入細胞の選択肢ではない。同様に、培養条件の選択は、細胞生存度または形質導入効率に有害でないことが望ましい。
【0143】
形質導入後のインキュベーションは、たとえば、約4時間、または約1日間から約7〜10日間の期間である。形質導入後のインキュベーションは、たとえば、約16〜約20時間、または約4日間、約5日間もしくは約6日間であることもできる。約14日間の形質導入後インキュベーションも企図される。
【0144】
本発明で観察される形質導入の効率は約50〜100%である。効率は、少なくとも約50〜75%、または少なくとも約75〜90%であることができる。本発明の他の実施形態は、形質導入効率が少なくとも約90〜95%である実施形態である。さらなる実施形態は、少なくとも91、92、93、94、95、96、97、98、99および100%の形質導入効率を有する。
【0145】
上記に加えて、形質導入した細胞は、研究または生きた対象における疾患状態の治療に用い得る。本発明の一部として、目的の遺伝子産物を産生するための形質導入した細胞の治療的使用または遺伝子治療の一部としての生きた生物への直接導入が存在する。たとえば、以下に例示するように、初代T細胞を単離して、ウイルスベクターを用いて形質導入することができる。形質導入の成功は、ベクターによってコードされている遺伝子産物の産生もしくは過剰産生、またはベクターによって与えられた表現型の発生によって示される。したがって、最初に初代T細胞を、望ましいまたは有用な核酸配列を含み、それを発現する能力を有するベクターを用いて形質導入し、その後、生きた対象などのin vivo環境に戻すことができる。たとえば、生きた対象は、HIV−1に感染している、または感染している危険性にある個体である。
【0146】
別の実施形態では、T細胞を、宿主生物内に導入された際にT細胞を条件的に死滅させる能力を有する遺伝子または核酸を用いて形質導入する。これは、同種骨髄移植において、プロドラッグ手法を用いてT細胞を死滅させることによる移植片対宿主病の予防に応用がある。
【0147】
あるいは、初代細胞は遺伝子産物に欠陥を有することができ、血管は形質導入したウイルスベクターによって矯正可能である。このような細胞は、ベクターを用いて形質導入した後に、生きた対象内に再導入する。
【0148】
したがって、本発明のin vitroおよびex vivoの応用がどちらも企図される。生きた対象内への導入には、形質導入した細胞を、たとえば、生物学的に許容される溶液または製薬上許容される配合物中に入れる。そのような導入は、静脈内、腹腔内または当分野で知られている他の注射および非注射方法によって行い得る。投与する用量は様々な要素に応じて変動するが、当分野の従事者によって容易に決定され得る。本発明には、ウイルスベクター中に既知またはよく設計されたペイロードを用いた数々の応用があり、形質導入した遺伝物質によって与えられる利点が負の効果のすべてのリスクに勝る。
【0149】
最初は、導入する形質導入した細胞の合計数約104〜約1010個である。したがって、105、106、107、108、または109個の細胞を用い得る。実際の数は形質導入する細胞に応じて変動する。必要な場合は、形質導入した細胞の複数の導入が別の実施形態である。さらに、必要な場合は、形質導入した細胞を導入する前の宿主の調節が別の実施形態である。調節レジメンは当分野で知られており、レジメンの例は骨髄移植のレジメンである。
【0150】
多層の容器またはフラスコ中で成長させることができる細胞の量は、少なくとも約1億個の細胞、または少なくとも約7千万個の細胞、または少なくとも約8千万個の細胞、または少なくとも約9千万個の細胞である。
【0151】
本発明は、ウイルスベクターおよびウイルス粒子を用いて細胞の安定形質導入を行うための、効率の高い方法およびそれに関連する組成物を提供する。本発明は、形質導入した細胞、たとえばレンチウイルスベクターで改変した細胞、たとえば自己CD4+T細胞、たとえば本明細書中に記載の例示的なVRX496で形質導入したCD4+T細胞の細胞処理を行うための、新規の製造施設および製造プロセスを提供する。
【0152】
一態様では、自己T細胞を製造するための本発明のプロセスは、以下に記載のように、たとえば血液の凍結アフェレーシスによるリンパ球の単離、たとえばCytoMate(商標)中での洗浄、その後、CD4+富化、次いでCD8の枯渇、次いでウイルス、たとえばレンチウイルスを用いた形質導入を含む。さらなる処理を以下に記載し、本明細書中の図に例示する。
【0153】
一態様では、自己VRX496で形質導入したCD4+T細胞を産生するための出発物質は末梢血単核細胞(PBMC)である。PBMCは、HIVに感染した対象から白血球アフェレーシス中に得る。白血球アフェレーシス手順は、自動細胞分離器を用いて血液採取施設で起こることができる。
【0154】
一態様では、血漿を除去するために細胞を洗浄し、CD4抗原の発現に基づいたヒト細胞の分離のために開発されたCD4マイクロビーズ(Miltenyi Biotech、ドイツ)で磁気標識した(インキュベーション)。第I相臨床研究中、血漿を除去するために出発物質を低速遠心分離によるフィコール密度勾配分離を行い、その後、COBE(Baxter)洗浄を行い、標準緩衝液中に再懸濁させた。その後、洗浄した細胞材料を、CD8高密度微粒子(CD8−HDMニッケルビーズ)(Biotransport)と共にインキュベーションし、続いてEligix Magnetic Cell Separation Systemを用いて磁気分離を行った。
【0155】
第II相臨床研究では、CYTOMATE(商標)Cell Processing System(Miltinyi Biotech、ドイツ)を用いて血漿を除去するための細胞洗浄を行うことができる。CYTOMATE(商標)Cell Processing Systemは、細胞生成物を洗浄および濃縮するため、および液体移送用途のための、独立型の閉鎖された自動装置である。これは、低い細胞損失および高い生存度の効率的な細胞洗浄を可能にする。この系は、cGMP環境における細胞処理のための閉鎖系の液体経路を生み出す使い捨てチューブセットを特徴とする。また、これは液体移送を柔軟、急速かつ正確にする。液体を、すべて1つの閉鎖系の液体経路の内部で、単一または複数の容器から中および外に移送することができる。
【0156】
一態様では、加えたCD4+マイクロビーズ(Miltinyi Biotech)のインキュベーション中の非特異的な細胞結合を防ぐために、免疫グロブリン溶液(Immune Globubin Intravenous、USP、Grifols)を加える。一態様では、最終産物のバッグ(CD4マイクロビーズのインキュベーションした細胞懸濁液)を熱密封し、バッグを取り出して生物学的安全フッド下に置く。
【0157】
一態様では、Eligix(商標)Cell Separation SystemをCD8の枯渇に用いる。第II相臨床研究では、CD4+陽性選択は、CliniMACS(商標)磁気細胞分離系介して行うことができる。この系では、(1)移送パック容器と、(2)緩衝液バッグおよび細胞懸濁液バッグに接続するためのメスルアーアダプターを備えた血漿移送セットと(3)陽性選択バッグおよび廃棄物採取バッグに接続するためのメスルアーアダプターを備えた血漿移送セットとからなる滅菌CliniMACS(商標)使い捨てセットを用いる。
【0158】
一態様では、滅菌使い捨てセットの緩衝液および細胞懸濁液系をそれぞれのバッグに加えることは、滅菌を保つために生物学的安全フッド下で行うことができる。リン酸緩衝生理食塩水(PBS)緩衝液および細胞懸濁液系を加えた後、使い捨てセットをCliniMACS(商標)に取り付けることができ、CD4磁気標識細胞懸濁液をCliniMACS(商標)に通すことができる。採取した陽性画分を用いてプロセスを続けることができる。
【0159】
一態様では、PBSからX−VIVO−15培地(Cambrex;メリーランド州Walkersville)への交換は、CytoMate(商標)を介して達成される。一態様では、最終産物のバッグを取り外し、熱密封し、生物学的安全フッド下に置く。メスルアーアダプターを備えた移送セットを生成物のバッグに取り付けることができ、以下についてのQC試験のために、シリンジによって5ccの試料を得る:CD3+CD8+細胞およびCD3+CD4+細胞の割合、細胞生存度、細胞数、および増殖前HIV gagの測定値。一態様では、QCの結果が得られるまで細胞の産生を停止する。細胞が規格に合っていれば、細胞の形質導入で産生を続ける。
【0160】
一態様では、CD3/CD28同時刺激ビーズ(Dynal beads、ノルウェー、Oslo、抗CD3(OKT3)および抗CD28(UPennモノクローナル抗体9.3)でコーティングをCD4+T細胞懸濁液に加え、続いてVRX496ウイルスベクター産物を加える。一態様では、CD4+T細胞、X−VIVO+5%ヒト血清アルブミン、IL2、NAC、CD3/CD28マイクロビーズおよびVRX496ベクター懸濁液(5%W/V)の混合物全体を、RetroNectin(Takara Bio、日本)でコーティングしたNunc(商標)細胞工場に加え、細胞工場を加湿した37℃、5%CO2インキュベーターに入れる。翌日、VRX496ベクター懸濁液(5%W/V)を再度加える。一態様では、細胞をベクターと共に3日間インキュベーションし、その後、WAVE(商標)細胞バッグに移し、Wave(商標)バイオリアクター(WAVE(商標)Biotech LLC、ニュージャージー州Bridgewater)に入れる。
【0161】
WAVEバイオリアクターは、特別な揺動プラットホームを有する。このプラットホームの揺動動作が、培養溶液に波を誘起する。これらの波が混合および酸素の伝達を提供し、20×106を超える細胞/mlを容易に支持することができる、細胞成長に完璧な環境をもたらす。バッグおよび様々な接続装置上のチューブ導線により(接続はスパイクコネクターを介し、溶接はTerumo Sterile Connecting Deviceを介して行う)、汚染の危険性を最小限にして細胞を閉鎖系で成長させることが可能となる。
【0162】
ベクターを除去するために、4日目に、CytoMate(商標)細胞洗浄器を用いて細胞をX−VIVO15で2回洗浄することができる。
【0163】
収集時まで培養を7〜12日間保つ。少なくとも1日おきに細胞を計数し、細胞を約0.5〜1.5×106個の細胞/mlの密度に保つために新鮮な培地を加える。細胞の培養中にHIVの複製を阻害するために、抗レトロウイルス薬(Norvir、Abbot Laboratories、およびRetrovir、GlaxoSmithKline)(1μmol/L)を加える。他の種類の抗レトロウイルス薬を用いることができる。当業者は、適切な抗レトロウイルス薬の選択および投与方法を知っているであろう。約10日目に、細胞は収集の準備が整う。増殖後のHIVコピー数が増殖前のHIVコピー数を超えていないことを確認するために、増殖後HIV gag測定を行う。収集前の細胞から、マイコプラズマについて試験するために試料を採取する。
【0164】
一態様では、CD3/CD28マイクロビーズは、培養バッグをMaxSep(商標)磁石(Baxter)上に通すことによって除去する。ビーズは磁石上に保持され、細胞は別のバッグ中に注がれる。細胞は、残留ビーズについてアッセイする。
【0165】
別段に定義しない限りは、本明細書中で用いるすべての専門用語および科学用語は、本発明が属する分野の技術者に一般的に理解される意味と同じ意味を有する。本明細書中で引用したすべての特許、特許出願、公開特許出願および他の出版物は、その全体で参考として組み込まれている。本セクションに記載した定義が、本明細書中に参考として組み込まれている特許、特許出願、公開特許出願および他の出版物に記載されている定義に反するまたは他の様式で矛盾している場合は、本セクションに記載した定義が、本明細書中に参考として組み込まれている定義に勝る。
【0166】
本発明の1つまたは複数の実施形態の詳細を、添付の図および以下の説明に記載する。本発明の他の特徴、目的、および利点は、説明および図、ならびに特許請求の範囲から明らかであろう。
【0167】
本発明を一般に説明したが、本発明は、例示として提供する、指定のない限りは本発明を限定することを意図しない以下の実施例を参照することでより容易に理解されるであろう。
【0168】
(実施例1)
初代CD4+T細胞の調製
CD4+T細胞は、軽微な改変をもつ標準のプロトコルを用いて末梢血から単離した。より詳細には、汚染単球を付着によって枯渇させた。CD4+細胞の陽性選択を行うために、非接着性細胞を抗CD4抗体でコーティングした磁気ビーズの存在下に置いた。磁気ビーズを除去し、CD4+細胞を単離した。
【0169】
高度に精製されたCD4+細胞は、フローサイトメトリーによって90%を超えることが確認された。
【0170】
(実施例2)
様々な時間の細胞表面結合分子との接触を用いた初代CD4+T細胞の形質導入
細胞表面結合前の形質導入
初代CD4+細胞(約500,000個)をpN2cGFPと共に、20のMOIで24時間培養し、次いで、さらに7日間、培養物にαCD3およびαCD28でコーティングしたビーズを加えた。図2はpN2cGFPのマップを含む。
【0171】
細胞表面結合後の形質導入
初代CD4+細胞(約500,000個)を24時間、αCD3およびαCD28でコーティングしたビーズと共に24時間培養し、次いで、さらに24時間、pN2cGFPを20のMOIで培養物に導入した。過剰のベクターを除去するために細胞を洗浄し、次いで、ビーズを含む、ベクターを含まない培地中でさらに7日間インキュベーションした。
【0172】
同時の形質導入および細胞表面結合
初代CD4+細胞(約500,000個)をpN2cGFPと共に、20のMOIで24時間、αCD3およびαCD28でコーティングしたビーズの存在下で培養した。過剰のベクターを除去するために細胞を洗浄し、次いで、ビーズを含む、ベクターを含まない培地中でさらに7日間インキュベーションした。
【0173】
任意選択のプロトコルの置換
他のウイルスベクターでpN2cGFPを置換し得る。さらに、過剰のベクターを除去する前に形質導入を合計2回繰り返し得る。さらに、形質導入および過剰のベクターの除去後に、αCD3およびαCD28でコーティングしたビーズをインターロイキン−2(10ng/ml)およびPHA−P(3mg/ml)によって置換し得る。7日後、培地をインターロイキン−2(10ng/ml)を含むPHA−Pを含まない培地で置き換え、インキュベーションをさらに7日間続ける。
【0174】
あるいは、αCD3およびαCD28でコーティングしたビーズと共に形質導入後のインキュベーションを7日間行った後、細胞を洗浄し、インターロイキン−2(10ng/ml)の存在下でインキュベーションを続ける。
【0175】
(実施例3)
形質導入後の分析
形質導入後およびインキュベーションの7または14日後、細胞を、CD4+および/または緑色蛍光タンパク質(GFP)についてフローサイトメトリーによって分析した。
【0176】
上記3つの形質導入プロトコルの比較を図3に示す。pN2cGFPを用いた20のMOIでの形質導入後の、ビーズに固定したCD3およびCD28抗体との接触により、約91%の効率がもたらされた。形質導入前でのビーズとの接触では約89%の効率がもたらされ、同時のビーズ接触および形質導入では約80%の効率がもたらされた。この実験では、CD4+T細胞は、接着性単球枯渇、CD14MACS枯渇およびCD4MACS富化によって選択した。抗体は、以下に記載のように固定した。ベクターとの接触は37℃、5%のCO2で行った。培養条件は、2%のヒト血清アルブミンを添加したアイセル培地中に500,000個のCD4+T細胞/mlであった。FACS分析は選択後7日目に行った。MFとは、平均蛍光をいう。
【0177】
様々な刺激条件を比較する、7日後の実験結果を図4に示す。CD4+細胞をIL−2およびPHA−Pのどちらかまたはビーズに固定したCD3およびCD28抗体で24時間処理し、次いでpN2cGFPを20のMOIで用いた形質導入を1ラウンド行った。隣り合わせの比較では、固定した抗体を使用することにより、毎回95%を超える形質導入効率がもたらされた(CD4およびGFPのどちらに対しても陽性な細胞によって示された)。それに比較して、IL−2およびPHA刺激を用いた結果では、70.2〜84.5%の効率しかもたらされなかった。FACS分析は選択後7日目に行った。
【0178】
図5は、選択後15日での類似の実験の結果を示す。ここでも、細胞をIL−2およびPHA−Pのどちらかまたはビーズに固定したCD3およびCD28抗体で24時間処理し、次いでpN2cGFPを20のMOIで用いた形質導入を1ラウンド行った。PHA−Pおよびビーズは形質導入後7日目に除去し、細胞は選択後15日目までIL−2のみと共に500,000個の細胞/mlで培養した。固定した抗体を使用した後、細胞の約93%がCD4およびGFPのどちらに対しても陽性であった。IL−2およびPHAで処理した細胞の約75%のみが、CD4およびGFPのどちらに対しても陽性のまま保たれた。これらの結果は、7日後に陽性として検出された細胞の小さな画分(図4)は、「偽型トランスフェクション」が原因であった可能性を示している。
【0179】
(実施例4)
様々なベクターが細胞を高効率で安定に形質導入する:
本例は、形質導入に用いたベクターの比較である。pN2cGFPはgagおよびpolコード配列の全体を含む一方で、pN1(cpt)cGFPは4551〜5096の部分的(非コード)pol配列を含む。図6に示す結果から見ることができるように、どちらのベクターも、ビーズに固定したCD3およびCD28抗体ならびに20のMOIのベクターで同時刺激を行った後、初代CD4細胞の非常に効率的な形質導入を示す。FACS分析は選択後10日目に行った。
【0180】
(実施例5)
トランスフェクション効率に対するMOIの効果
様々なMOIの効果を図7に示し、2〜20のMOIを使用することにより、72.7〜83.8%の形質導入効率がもたらされた。様々なMOIでのpN1(cpt)CGFPを用いた形質導入の前に、細胞をビーズに固定したCD3およびCD28抗体と24時間接触させた。
【0181】
(実施例6)
CD34陽性細胞の形質導入
CD34陽性細胞を臍帯血から調製し、FLT−3リガンド、TPOおよびKitリガンド(それぞれ100ng/ml)の存在下で、pN1cptGFPを用いて4回同時に形質導入した。細胞を長期培養(LTC−IC)で5週間培養し、その後、分析前に細胞をメチルセルロース中で10日間培養した(結果は、6週間の培養にわたる経過時間からのものである)。図8の結果は、CD34未熟細胞から生じる成熟CD45陽性細胞を分析している。対照細胞は有意な形質導入を示さなかったが、ベクターで形質導入した細胞は、88%を超える細胞がCD45およびGFPに陽性であることを示す。
【0182】
(実施例7)
CD34陽性細胞の長期形質導入
上述のように、CD34陽性細胞をpN1(cpt)GFPで形質導入し、部分的に照射したSCIDマウスの骨髄に移植した。8週間後、細胞を単離し、FACSによってCD45を保有する成熟ヒト細胞およびGFP発現について分析した。結果を図9、パネルA〜Dに示す。
【0183】
パネルAは、ベクターで形質導入していないヒト細胞を移植した対照マウスを用いた結果を示す。
【0184】
パネルBは、50のMOIで連続した4日間、100ng/mlのFLT−3リガンド、TPOおよびKitリガンドの存在下で、pN1(cpt)GFPベクターで形質導入した細胞で形質導入した細胞を移植したマウスを用いた結果を示す。このマウスは、形質導入の8週間後に、形質導入したヒト細胞(CD45陽性細胞)が96.3%という印象的な形質導入効率を示す。このマウスにおけるヒト細胞移植のレベルは11.1%であり、これは以前に報告された結果と矛盾しない。
【0185】
パネルCおよびDは、パネルBと同様に治療した2匹の他のマウスを用いた結果を示す。この結果により、87.8%および89.6%のCD45陽性細胞という高効率形質導入、ならびにGFP陽性であることの再現性が確認された。
【0186】
平均効率は91.2%であり、これは長期安定形質導入を反映している。
【0187】
(実施例8)
細胞表面結合分子の固定
この例は、以下の実施例で用いるための、エポキシdynalビーズに対するCD3(B−B11)抗体およびCD28(B−T3)抗体の直接結合を記載している。
【0188】
1.0.618gのホウ酸を95mlの組織培養グレードの水に溶かすことによって0.1ホウ酸溶液を調製する。よく混合し、最高品質のNaOHを用いてpHを9.5に調節する。最終体積を100mlにし、0.2μmのフィルターを介して滅菌する。容器を密閉し、4℃で保存する。
【0189】
2.抗体を上記ホウ酸溶液に150μg/mlの濃度で加える。B−B11およびB−T3抗体のどちらについても、ホウ酸溶液1mlあたりそれぞれ75μgを加える。体積を合計1mlにする。ホウ酸濃度は、抗体を加えた後に0.05M以上であるべきである。1mlのホウ酸/抗体溶液のそれぞれに、4×108個のエポキシビーズを加える。
【0190】
3.24時間、37℃で、回転輪上でインキュベーションを行う。
【0191】
4.ビーズを3回、それぞれ10分間、22℃で、ビーズ洗浄媒体(カルシウムおよびマグネシウムを含まないリン酸緩衝生理食塩水、3%のヒト血清アルブミン、5mMのEDTA、ならびに0.1アジ化ナトリウム)を用いて洗浄する。
【0192】
5.ビーズを1回、30分間、22℃で洗浄する。
【0193】
6.4℃で終夜洗浄する。
【0194】
7.新鮮なビーズ洗浄媒体に交換し、ビーズを2×108個のビーズ/mlで再懸濁させる。IgGでコーティングしたビーズは、4℃で少なくとも6カ月安定である。
【0195】
(実施例9)
樹状細胞の形質導入
末梢血由来の単球を単離し、その後、50のMOIで連続した3日間、2つの同時のサイトカイン条件、すなわち、GM−CSF(800単位/ml)、IL−4(500単位/ml)およびTNF−α(100単位/ml)またはGM−CSF(500単位/ml)およびインターフェロン−α(800単位/ml)を用いて、pN2cGFPで形質導入した。図10、パネルAは、形質導入の7日後の結果を示し、第1のサイトカイン条件では90.2%の効率がもたらされた。第2のサイトカイン条件下でベクターを用いて形質導入した細胞は、7日後に92.9%の効率を示した(パネルB)。CD86が樹状細胞の唯一の可能なマーカーであり、CD86陰性細胞は樹状細胞であり得ることに注意されたい。
【0196】
(実施例10)
製造施設
この要約では、レンチウイルスベクターで改変した細胞、具体的には自己VRX496で形質導入したCD4+T細胞の細胞を処理するための新しい製造施設および製造プロセスを説明する情報を提供する。
【0197】
第I相臨床研究中、自己VRX496で形質導入したCD4+T細胞は、ペンシルバニア大学(UPenn)、細胞およびワクチン産生施設(Cell and Vaccine Production Facility、CVPF)で製造した(Levineら、2002年)。
【0198】
第II相臨床研究では、自己VRX496で形質導入したCD4+T細胞生成物を製造する。このプロセスにより、レンチウイルスベクターによって改変したHIVに感染したCD4T細胞の、前例のない100倍を超える増殖が可能となる。
【0199】
本要約には、以下を提示する:
全体的な施設配置図を含めた製造施設の説明、
出発物質、プロセス中およびリリースの品質管理試験、安定性を含めた新しい製造プロセスの説明、ならびに
製造の一貫性およびUPennのCVPFのプロセスのデータとの比較を実証するデータ。
【0200】
(実施例11)
全体的な施設の説明
GMP製造面積および品質管理(QC)試験面積は封じ込められており、物理的な仕切りによって他の設立面積および互いから隔離されている。また、製造面積およびQC試験面積はカードキーの鍵によって制限および制御されている。
【0201】
2つのクリーンルームの使用意図は、VRX496レンチウイルスベクターの臨床的cGMP産生用およびこのベクターを用いた対象細胞の対応するcGMP ex vivo形質導入用である。ベクター産生用のクリーンルームは複数の生成物産生施設であり、細胞処理用のクリーンルームは、現時点では自己VRX496で形質導入したCD4+T細胞の産生のみを目的とする。ベクター産生と対象細胞の形質転換との間には適切な切替え手順が配置されている。すべての対象細胞性生物、バーコードシステムによって産生プロセス全体にわたって追跡する(以下を参照)。
【0202】
相互汚染のすべての可能性を最小限にするために、ベクター産生および細胞処理操作では人員を共有せず、領域は位置が物理的に隔離されている。書面による標準操作手順(SOP)およびこれらの手順で訓練した人員によってさらなる制御を維持する。
【0203】
ベクター産生用のクリーンルームスイートおよび細胞処理用のクリーンルームスイートは、どちらもcGMP産生および生物学的安全性の封じ込めのために設計されている。どちらのクリーンルームも、クラス10,000(ISOクラス7)および生物学的安全性レベル2(BSL−2)、大スケールとして設計されている。
【0204】
クリーンルームは、個別の空調ユニット(AHU)を有する。ベクタークリーンルームのAHUは、供給および戻りファン;45%前置フィルターおよび95%最終フィルター;冷却コイル、ならびにDX凝縮ユニットを含む、一定体積の再循環ユニットである。細胞処理用のクリーンルームのAHUは、100%外気を供給する。クリーンルームの仕上げは、滑らかで硬い清掃可能な防水かつ化学薬品耐性の表面、および一体カバーを備えた継ぎ目のないビニル製の床材から構成される。扉は安全ガラスの覗きパネルを備えた亜鉛めっき鋼から構成される。扉の金具の特徴には、蹴板、モップ板および扉閉鎖が含まれる。
【0205】
産生領域内の移動と同様、品質管理(QC)研究室間の移動はSOPによって制御されている。また、汚染のすべての危険性を最小限にするために、DNA抽出研究室(Q2)はPCR研究室(Q5およびQ6)から物理的に隔離されている。
【0206】
(実施例12)
製造された細胞生成物
本補正書中で製造および記載した例示的な細胞生成物の名称は、自己VRX496で形質導入したCD4+T細胞である。
【0207】
VRX496は、ヒト免疫不全ウイルス(HIV)エンベロープ遺伝子を標的とした937個のヌクレオチドのアンチセンス配列を含む。
【0208】
自己VRX496で形質導入した細胞を輸液バッグに分割した(90mlのバッグ1個あたり5×109〜1010個の形質導入した細胞)。細胞を以下からなる輸液用低温培地に懸濁させた:
31.25%のPlasmalyte−A、
31.25%のデキストロース(5%)、
0.45%の塩化ナトリウム、
7.5%のジメチルスルホキシド(DMSO)、
1%のデキストラン40、および
5%のヒト血清アルブミン。
【0209】
(実施例13)
出発物質
以下の表に、自己VRX496で形質導入したCD4+T細胞の産生に用いた出発物質を例示的に記載する。
【0210】
【化5】
【0211】
【化6】
すべての出発物質は、材料管理の人員によって受取りおよび検査を行った。検査には、認可された「原材料規格書および受取りシート」の完全記入が含まれる。ロット番号を割り当て、梱包ラベルを調査し、認可された「原材料規格書および受取りシート」の適合性について分析証明書(C of A)を再調査した。
【0212】
品質保証(QA)では、材料管理によって完全記入された「原材料規格書および受取りシート」を再調査し、材料を認可または却下する。材料がQAによって「認可」された場合は、「認可」と標識され、材料管理の人員によって倉庫領域内の適切な認可された保存場所に移送される。
【0213】
出発物質がQAによって「却下」された場合は、「却下」と標識され、最終的な処理、すなわち、廃棄するか、販売者に返却するか、またはR&Dへ移送するかが決定されるまで、認可された材料から隔離される。
【0214】
すべての出発物質はQAによって目録ログに入力される。このログには、割り当てられたロット番号、受け取った量、処理、および使用期限が含まれる。この目録には、製造プロセスまたはQC試験手順で用いる緩衝液などの企業内用配合物が含まれる。QAによって、月末に期限の切れる材料の月ごとの表が材料管理および/または産生用に作成されて、倉庫領域からのその除去が確実となり、不注意による使用が防がれる。
【0215】
(実施例14)
産生および日常的な制御
プロセスの流れ図
細胞の処理精製(図11)および製造手順(図12)の図を添付する。
【0216】
(実施例15)
プロセスの説明
細胞の採取方法
自己VRX496で形質導入したCD4+T細胞を産生するための出発物質は末梢血単核細胞(PBMC)である。PBMCは、白血球アフェレーシス中にHIVに感染した対象から得られる。白血球アフェレーシス手順は、自動細胞分離器(Cobe Spectra CS−3000、Baxter;コロラド州、Lakewood)を用いて血液採取施設で起こる。
【0217】
第II相臨床研究中にそれぞれ約5×109から10〜1010個の自己VRX496で形質導入したCD4+T細胞の8つまでの細胞輸液を対象に与え得るので、約3〜4倍血液量(15L)の血液をCobe Spectraを通して処理し、十分なPBMC(約100〜200億個)を採取し、細胞の洗浄および選択手順(すなわち精製)を受けさせ、細胞の形質導入および増殖プロセスを開始するために必要な数のCD4T細胞(すなわち約10〜20億個)をもたらす必要がある。1回の白血球アフェレーシス手順は完了するまで約3時間かかる。
【0218】
対照的に、第I相臨床研究中では、それぞれの対象は約1×1010個の自己VRX496で形質導入したCD4+T細胞の輸液を1回のみを受けたので、採取された白血球アフェレーシス生成物はより少なく、約70mL中に約50億個のPBMCからなる。
【0219】
白血球アフェレーシス生成物は、採取した日に、周囲温度で、それぞれの血液センターからその生成物がさらに処理される場所へと、IATAおよびDODの規制に従って空輸または陸輸配達によって発送される。産生の人員が採取後24時間のうちに白血球アフェレーシス生成物を受け取って処理することを確実にするために、移送時間の計画を立てる。
【0220】
生成物は自己かつ感染性であるので、生成物の追跡制御を確実にし、すべての生成物の混同の可能性を減らすために、それぞれの白血球アフェレーシスのバッグを以下で標識する:
固有のロット番号、
含まれる細胞体積、
固有のバーコードラベル、
対象の研究ID(研究の現場の識別を含む)、
対象のイニシャル、および
対象の生年月日。
【0221】
混同の可能性を減らすために採るさらなる予防措置は、以下である:
細胞処理用のクリーンルーム内では任意の時点で2つの個別の細胞生成物だけを操作することを許可すること、および
産生操作は、産生プロセス中の様々な段階(たとえば、CD4+T細胞の選択、ベクターの除去または細胞の収集)を含まなければならないことを記載した書面による手順。
【0222】
この方針の例外はインキュベーションおよび保存ステップ中であり、任意の時点で80個もの個別の生成物のインキュベーションをWAVE(商標)バイオリアクター中で行い、30個までを冷凍庫内で保存し得る。
【0223】
バーコードシステムによる支援の下、製造プロセス全体にわたって細胞生成物を追跡する。
【0224】
(実施例16)
白血球アフェレーシス生成物の受取り
白血球アフェレーシス生成物が細胞処理施設で受け取られた後、品質保証(QA)は受取室でバーコードの読取りを行い、同時に生成物のバッグのラベルおよび記録を確認する:
受け取った総体積、
バッグに記載のロット番号、
バッグを受け取った時間、および
白血球アフェレーシス時から受取時までの時間数。
【0225】
QAが白血球アフェレーシス生成物をリリースした後、材料管理の人員によって細胞処理用のクリーンルーム(クラス10,000)(生物学的安全性レベル2、大スケール)に届けられる。産生の人員が約5ccの細胞生成物の試料をシリンジで採り、QC試験のためにバイアルに加える。全CD4+生細胞のQC試験は≧6×108個の細胞であるべきである。
【0226】
(実施例17)
血漿の洗浄およびMACS CD4インキュベーション
細胞を洗浄して血漿を除去し、CD4抗原の発現に基づいたヒト細胞の分離のために開発されたCD4マイクロビーズ(Miltenyi Biotech、ドイツ)で磁気標識した(インキュベーション)。
【0227】
第I相臨床研究中、血漿を除去するために出発物質を低速遠心分離によるフィコール密度勾配分離を行い、その後、COBE(Baxter)洗浄を行い、標準緩衝液中に再懸濁させた。その後、洗浄した細胞材料を、CD8高密度微粒子(CD8−HDMニッケルビーズ)(Biotransport)と共にインキュベーションし、続いてEligix Magnetic Cell Separation Systemを用いて磁気分離を行った。
【0228】
第II相臨床研究では、CYTOMATE Cell Processing System(Miltinyi Biotech、ドイツ)を用いて血漿を除去するための細胞洗浄を行う。CYTOMATE Cell Processing Systemは、細胞生成物を洗浄および濃縮するため、および液体移送用途のための、独立型の閉鎖された自動装置である。これは、低い細胞損失および高い生存度の効率的な細胞洗浄を可能にする。この系は、cGMP環境における細胞処理のための閉鎖系の液体経路を生み出す使い捨てチューブセットを特徴とする。また、これは液体移送を柔軟、急速かつ正確にする。液体を、すべて1つの閉鎖系の液体経路の内部で、単一または複数の容器から中および外に移送することができる。
【0229】
さらに、加えたCD4+マイクロビーズ(Miltinyi Biotech)のインキュベーション中の非特異的な細胞結合を防ぐために、免疫グロブリン溶液(Immune Globubin Intravenous、USP、Grifols)を加える。
【0230】
最終産物のバッグ(CD4マイクロビーズのインキュベーションした細胞懸濁液)を熱密封し、バッグを取り出して生物学的安全フッド下に置いた。以下を行うために約5ccのQC試料を採取した:
細胞濃度および
CD3+CD8+の割合およびCD3+CD4+の割合を決定するためのFACS分析。
【0231】
QCの結果を受け取るまで産生を停止する(約30分間)。CytoMate最終産物体積の体積を計算する。
【0232】
(実施例18)
CD4+の選択
上に注記したように、第I相臨床研究中、Eligix(商標)Cell Separation SystemをCD8の枯渇に用いた。第II相臨床研究では、CD4+陽性選択はCliniMACS磁気細胞分離系を介して行う。この系では、(1)移送パック容器と、(2)緩衝液バッグおよび細胞懸濁液バッグに接続するためのメスルアーアダプターを備えた血漿移送セットと(3)陽性選択バッグおよび廃棄物採取バッグに接続するためのメスルアーアダプターを備えた血漿移送セットとからなる滅菌CliniMACS使い捨てセットを用いる。
【0233】
滅菌使い捨てセットの緩衝液および細胞懸濁液系をそれぞれのバッグに加えることは、滅菌を保つために生物学的安全フッド下で行った。
【0234】
リン酸緩衝生理食塩水(PBS)緩衝液および細胞懸濁液系を加えた後、使い捨てセットをCliniMACSに取り付け、CD4磁気標識細胞懸濁液をCliniMACSに通す。採取した陽性画分を用いてプロセスを続ける。
【0235】
第I相臨床研究中の2ステップのプロセス、すなわち、CD8の枯渇およびCD3の選択から第II相臨床研究中の1ステップのプロセス、すなわち、CD4の選択へと変更した理論的根拠は、細胞処理効率およびより純粋な細胞生成物を得るためである。
【0236】
(実施例19)
緩衝液から培地への交換
PBSからX−VIVO−15培地(Cambrex;メリーランド州、Walkersville)への交換は、CytoMateを介して達成される。最終産物のバッグを取り外し、熱密封し、生物学的安全フッド下に置いた。メスルアーアダプターを備えた移送セットを生成物のバッグに取り付け、以下についてのQC試験のために、シリンジによって5ccの試料を得る:
CD3+CD8+細胞およびCD3+CD4+細胞の割合、
細胞生存度、
細胞数、および
増殖前GIV Gag測定。
【0237】
QCの結果が得られるまで細胞の産生を停止する。細胞が規格に合っていれば、細胞の形質導入で産生を続ける。
【0238】
(実施例20)
CD4+T細胞の形質導入
CD3/CD28同時刺激ビーズ(Dynal beads、ノルウェー、Oslo、抗CD3(OKT3)および抗CD28(UPennモノクローナル抗体9.3)でコーティングをCD4+T細胞懸濁液に加え、続いてVRX496ウイルスベクター産物を加える。CD4+T細胞、X−VIVO+5%ヒト血清アルブミン、IL2、NAC、CD3/CD28マイクロビーズおよびVRX496ベクター懸濁液(5%W/V)の混合物全体を、RetroNectin(Takara Bio、日本)でコーティングしたNunc(商標)細胞工場に加え、細胞工場を加湿した37℃、5%CO2インキュベーターに入れた。翌日、VRX496ベクター懸濁液(5%W/V)を再度加える。細胞をベクターと共に3日間インキュベーションし、その後、WAVE(商標)細胞バッグに移し、Wave(商標)バイオリアクター(WAVE(商標)Biotech LLC、ニュージャージー州、Bridgewater)に入れる。
【0239】
WAVEバイオリアクターは、特別な揺動プラットホームを有する。このプラットホームの揺動動作が、培養溶液に波を誘起する。これらの波が混合および酸素の伝達を提供し、20×106個を超える細胞/mlを容易に支持することができる、細胞成長に完璧な環境をもたらす。バッグおよび様々な接続装置上のチューブ導線により(接続はスパイクコネクターを介し、溶接はTerumo Sterile Connecting Deviceを介して行う)、汚染の危険性を最小限にして細胞を閉鎖系で成長させることが可能となる。
【0240】
(実施例21)
ベクターを除去するための洗浄
4日目に、CytoMate細胞洗浄器を用いて細胞をX−VIVO15で2回洗浄する。
【0241】
(実施例22)
細胞の増殖
収集時まで培養を7〜12日間保つ。少なくとも1日おきに細胞を計数し、細胞を約0.5〜1.5×106個の細胞/mlの密度に保つために新鮮な培地を加えた。細胞の培養中にHIVの複製を阻害するために、抗レトロウイルス薬(Norvir、Abbot Laboratories、およびRetrovir、GlaxoSmithKline)(1μmol/L)を加える。約10日目に、細胞は収集の準備が整う。増殖後のHIVコピー数が増殖前HIVコピー数を超えていないことを確認するために、増殖後のHIV gag測定を行う。収集前の細胞から、マイコプラズマについて試験するために試料を採取する。
【0242】
(実施例23)
洗浄、体積縮小および配合
細胞のバッグをCytoMateに載せ、細胞を栄養素培地から洗浄して出し、以下からなる輸液用低温培地溶液に入れる:
31.25%のPlasmaLyte A、
31.25%のデキストロース5%、
0.45%のNACl、
5%のヒト血清アルブミン(HSA)、
1%のデキストラン40、および
7.5%のDMSO。
【0243】
(実施例24)
CD3/CD28マイクロビーズの枯渇
CD3/CD28マイクロビーズは、培養バッグをMaxSep(商標)磁石(Baxter)上に通すことによって除去する。ビーズは磁石上に保持され、細胞は別のバッグ中に注がれる。細胞は、残留ビーズについてアッセイする。
【0244】
(実施例25)
冷凍保存
VRX496で形質導入したCD4+T細胞は、速度を制御して凍結した。生成物を冷却する細胞は生成物が相転移点に達するまで1℃/分で冷却し、その後、温度が−90℃に達するまで凍結速度を増加する。
【0245】
(実施例26)
品質管理(QC)リリース試験
QCリリース試験用に試料を採取する。細胞生成物は、QC試験の完了まで液体窒素冷凍庫の蒸気相中で保存する(設定値<−130℃)。
【0246】
(実施例27)
品質保証(QA)リリース
QC試験の完了後、QAはすべての試験の再調査を行い、リリース規格書が満たされている場合は、臨床治験で用いるための細胞生成物のリリースを認可する。
【0247】
(実施例28)
保存
QAからリリースされた細胞生成物は、臨床施設の現場へ運送する準備が整うまで液体窒素中で保存する。
【0248】
(実施例29)
臨床施設の現場への運送
細胞生成物は、液体窒素蒸気運送容器(Chart Inc.、ジョージア州、Marietta、以前はMVE Cryogenics)中、≦140℃の温度で、契約輸送業者(Cavalier Logistics Management,Inc.、バージニア州、Dulles)によって、その自社貨物トラックまたは民間航空会社によって臨床施設の現場に輸送される。これらの低温運送容器は、請け負わせたその内容物を8日間維持することが確認されている。
【0249】
(実施例30)
品質管理(QC)試験
QCのプロセス中の試験
プロセス中のQC試験を以下のように行う。開発のこの段階では、これらの試験は情報としてのみ行う。
【0250】
【化7】
(実施例31)
QCのリリース試験
最終的な細胞生成物のリリース試験および規格書を、以下の分析の証明書に示す。リリース試験はプロセスステップで、および以下に示す被験物質に対して行う。
【0251】
【化8】
【0252】
【化9】
(実施例32)
産生プロセスの認定
第I相および第II相の間で行った主要な製造の変更の要約
表1は、第I相および第II相の間で行った主要な製造の変更の要約を示す。
【0253】
【化10】
(実施例33)
アフェレーシスの細胞生成物および陽性選択した後の細胞生成物の品質:
第I相対第II相
表2は、第I相および第II相の出発物質(すなわち、洗浄後のアフェレーシス生成物)と陽性選択した後の細胞生成物(すなわち、CD4+T細胞の形質導入および細胞の増殖を開始するために用いた細胞生成物)とのCD4+T細胞の純度の比較を提供する。
【0254】
これらのデータから見られるように、第II相細胞産生プロセスは、より純粋なCD4+出発物質(平均28.04%のCD4対第I相で14.58%のCD4)ならびにVRX496形質導入および細胞の増殖を開始するためのより純粋なCD4+細胞生成物(平均95.60%のCD4対第I相で36.82%のCD4をもたらす。
【0255】
さらに、データは、第II相産生プロセスは、第I相臨床研究で用いた生成物よりも純度が一貫した陽性選択後の細胞生成物をもたらすことを示す。これは、単一のCD4+陽性選択ステップに寄与することができ、一方で、第I相プロセスでは2ステップのプロセス、すなわちCD8+枯渇およびCD3+/CD4+陽性選択を用いた。
【0256】
【化11】
(実施例34)
VRX496形質導入効率の比較:第I相対第II相
表4は、第I相細胞生成物および第II相開発ロットの間のVRX496平均ベクターコピー数/細胞の比較を示す。これから見ることができるように、平均ベクターコピー数は本質的に第I相から変化なしに保たれるが、第II相開発ロットで、平均ベクターコピー数は第I相のそれよりも一貫性がある。
【0257】
【化12】
(実施例35)
第II相開発ロットのリリース試験の結果の要約
表5は、第II相開発ロット1、2および3のリリース試験の結果の要約を示す。3つの開発ロットすべてがロットリリースの規格を満たしていた。
【0258】
【化13】
(実施例36)
細胞生成物の安定性
第I相臨床治験のために製造したVRX496で形質導入したCD4+T細胞生成物を凍結保存し、対象に輸液を行う予定まで≦−80℃で保存した。輸液の前日、細胞生成物のセンチネルバイアル試料を解凍し、細胞生成物リリース基準の一部として細胞生存度を測定した。第I相の製造した生成物のそれぞれの細胞生存度は≧70%であった。<−80℃保存の最長期間は6カ月であった。これらのデータは、≦−80℃で6カ月間まで保存した場合の自己VRX496で形質導入した細胞生成物の安定性を支持している。
【0259】
安定性を評価するために、自己VRX496で形質導入したCD4+T細胞の24カ月間の安定性研究を6つの自己VRX496で形質導入したCD4+T細胞のロットで行う。これらのロットは、既存の製造プランに従って製造した2つの異なるVRX496ベクターのロットを用いて形質導入する(すなわち、3つのVRX496で形質導入した細胞生成物のロット/ベクターロット)。保存条件は液体窒素である。形質導入した細胞生成物の試料(15ml)を3、6、12、18および24カ月にアッセイする。0時でのデータを、形質導入した細胞生成物のロットのリリースデータとする。それぞれの時点でのアッセイに用いるために、十分な試料(20個のバッグ/ロット)を細胞処理終了時に採取する。アッセイには、外観、Gtagコピー数、細胞生存度、回収、細胞内サイトカイン染色、滅菌性、および細胞外DNA濃度が含まれる。それぞれの時間間隔の試験の完了時に暫定的な報告書を作成する。最終報告書は研究の最後に作成する。QA部門が、生じたデータの完全性を保証すること、およびcGMPとのコンプライアンスを確証する責任を持つ。作成されたすべての生データ、記録および報告書は、企業内で保有する。保有する記録には、保存条件、保存ユニットの妥当性確認および維持、試料の調製ならびに生のアッセイデータが含まれる。
【0260】
(実施例37)
自己細胞生成物の追跡手順
4人の異なる対象の自己CD4+T細胞を同時に処理し得る。この同時製造中にこれらの細胞生成物を潜在的な混同および汚染から保護するために、これら4つの細胞生成物を産生の異なる段階中に処理する。最新の適正製造基準に従う。認可された書面の細胞処理手順が存在し、すべての産生人員はこれらの手順の訓練を受ける。産生ロットを切り替える手順では専用の産生装置を用いる。重要な装置(インキュベーター、冷凍庫、HVAC)は妥当性が確認されている。使用する処理用水およびすべての産生用材料は認可された販売者から、確立された規格書に従って得る。細胞処理手順全体にわたって対象細胞生成物を追跡するための以下の特別な制御も実行されている:
(実施例38)
バーコードシステム
特注デザインのバーコードシステムにより、細胞生成物およびQC試験プロセスの全体、すなわち、受取り、細胞の形質導入、増殖、冷凍保存、保存、梱包および運送にわたって対象細胞を追跡する。
【0261】
バーコードシステムは、監査追跡、ユーザレベルアクセスおよび完全な報告能力を提供する。
【0262】
対象細胞生成物を処理または試験する前に、産生人員が処理または試験する材料およびバッチ産生記録または品質管理(QC)試験書類に添付されているバーコードの両方を、完全に一致するかどうかスキャンする。これらが一致しない場合は、コンピュータスクリーン上に警告が表示される。その場合、材料をスキャンする人は、照合を行ったことを証明しなければならず、バッチ産生記録またはQC試験書類にイニシャルおよび日付を記入する。
【0263】
(実施例39)
書類
それぞれの対象細胞生成物ロットの書類には、処理中に対象細胞生成物を視覚的に区分するために異なる色を割り当てる(すなわち、バッチ産生記録およびQC試験書類に固有の色)。
【0264】
(実施例40)
処理中の細胞生成物の隔離および制御
任意の時点において1人の産生人員だけが1つの対象細胞生成物で作業することが許可され、次の対象細胞生成物の処理を行うことができる前に、この細胞生成物に関与するすべての操作を終えなければならない。
【0265】
すべての開放された場での細胞生成物の操作は、クラス100生物学的安全フッド内で行う。任意の時点で1人の対象由来の細胞のみを操作する。
【0266】
対象細胞生成物はWAVE(商標)バッグ内でインキュベーションし、それぞれの対象細胞生成物ロットはそれ専用のWAVE(商標)インキュベーターを有する。
【0267】
フッド内に入れる緩衝液および試薬などの原材料は1つの対象細胞生成物ロットに専用であり、処理の最後に廃棄する。
【0268】
(実施例41)
保存中の細胞生成物の隔離および責任
一度に1つの対象細胞生成物のみを凍結保存する。
【0269】
冷凍保存中、それぞれの対象細胞生成物バッグは金属カセット内で保護される。冷凍保存後、これらのカセットをケーブル線で接続し、冷凍庫のラック内に隔離して保存する。
【0270】
保存されているすべての生成物の目録をバーコードシステムおよびハードコピーの書類によって維持する。
【0271】
(実施例42)
現在のプロセスの概観
間もなく行われる施設での第I相/第II相臨床治験の提案された細胞処理手順を以下に要約する。手短に述べると、最初に、COBE2991細胞プロセッサー(Gambro BCT)を用いて、アフェレーシス生成物に赤血球(RBC)枯渇を行う。その後、生じた生成物をMiltenyi抗CD4MACSと共にインキュベーションし、COBE2991細胞プロセッサーで洗浄する。抗CD4とインキュベーションした生成物を、恐らくは最大収率のために2回、CliniMACS装置で処理する。
【0272】
CD4で選択された生成物は、刺激ビーズの存在下、RetroNectinでコーティングしたバッグ中で、ベクターを用いてすぐに形質導入する。形質導入は3日間、37℃、5%のCO2インキュベーターで実施する。形質導入後の細胞は、Cytomate装置(Baxter Oncology)を用いて洗浄した後、8〜10日間の期間、Waveバイオリアクター中で増殖する。増殖後、Isolex 300iまたはMaxSep(どちらもBaxter Oncology)を用いて刺激ビーズを除去し、細胞培養物の体積を縮小し、再度Cytomateを用いて細胞を洗浄し、冷凍保存用に調製する(配合)。冷凍保存はCryo−Med速度制御冷凍庫を用いて行い、細胞は蒸気相液体窒素MVEタンク中に保存する。全体的に、プロセスは11〜13日間かかるはずである。
【0273】
提案のとおり、現在の細胞処理手順は時間がかかり、かつ高価であるが、迅速に実行することができる。このプロセスで確認された主なコストは抗体選択ステップであり、多数の対象を処理するための主な制限は8〜10日間の増殖ステップである。
【0274】
以下に、現在の細胞処理手順を簡素化することを目的としたいくつかの技術的な代替方法を示し、実行が容易なものから始める。
【0275】
第1の技術的代替方法は増殖ステップの長さに関し、8〜10日間から0日間へと短縮する。手短に述べると、3日間形質導入した細胞を直接冷凍保存処理する(ビーズ枯渇、洗浄、および配合)。生成物の調製時間を11〜13日間から3日間へと短縮することによって、同じ期間内により多くの生成物を処理することが可能となり(4個対1個)、また、増殖に関連するコストも削減される(Waveバイオリアクターおよび培地)。
【0276】
この第1の代替方法に関連して、実行できる精製ステップが制限されるまたは実行できず、したがって関連する精製コストが削減される。
【0277】
第2の代替方法は、過剰かつ時間のかかる操作手順なしに臨床施設の現場で用いられるほど単純な形質導入キットの作製である。手短に述べると、生アフェレーシス生成物を、CD8+およびCD19+リンパ球(Stemcell Technologiesの製品RosetteSep)などの、RBCを所望しない細胞と結合させる抗体カクテルと共に直接インキュベーションする。コンパクトな自動かつ密閉型のSepax装置(Biosafe)を用いて、フィコール層上での遠心分離中に所望しない細胞をRBCと共に沈殿させる。単核細胞を採取し、同じ装置を用いてフィコールを洗浄して除去し、ベクターおよび生分解性刺激ナノビーズを既に含むテフロン(登録商標)バッグに移す。形質導入を3日間、37℃、5%のCO2インキュベーターで実施した後、再度Sepax装置を用いて洗浄し、すぐに対象に再注射する。
【0278】
第3の代替方法を図15に要約する。この技術的代替方法では、1つの処理ステップ、すなわちアフェレーシス手順のみを用い、臨床施設の現場で数時間内に行われる。手短に述べると、対象は、現在および他の提案された代替方法と同じ方法でアフェレーシス手順を受ける。RBCおよび血漿を対象に連続的に再輸液しながら、濃縮白血球は通常バッグ内に採取する。その後、採取した白血球は、対象に再輸液する前に採取バッグ中で形質導入させる。ex vivo操作は必要ない。
【0279】
(実施例43)
単離
RBC枯渇のみを行うことが最も安価な代替方法であり、臨床グレードの抗体は必要ない。RBC枯渇は時間がかからず、装置は1点しか必要としない。しかし、CD4含有量に関しては制御がない。
【0280】
Sepax装置を用いた制限のあるCD8CD19枯渇の代替方法は、1点の装置のみを必要とするが、枯渇手順(3)に必要な臨床的抗体の数およびStemcell Technologies IPの技術許可が原因でより拡張的な可能性がある。
【0281】
現在の単離手順、すなわちCD4陽性選択は、実行するために2点の装置および1つの臨床グレードの抗体(間もなく市販される)を必要とする。コストは制限された選択と同様の可能性があるが、この手順はより時間がかかる。しかし、CD4含有量の制御は良好である。
【0282】
1年前に、CD14枯渇のみ、またはCD14およびCD8の枯渇、またはCD14枯渇およびCD4精製を受けた細胞の、フローサイトメトリーによってアッセイした形質導入レベルが同様であったことが実証されている。しかし、その差異は7日間の培養期間後の増殖のレベルにあった。
【0283】
(実施例44)
増殖
現在のプロセスの培養期間は8〜10日間である。
【0284】
提案された代替方法は、増殖時間を形質導入に必要な最小限の時間に短縮することである。しかし、3日間操作したT細胞のin vivo増殖の潜在性を評価するために現在利用可能なデータは、存在していたとしても僅かである。さらに、T細胞の再構成を評価するための適切な小動物モデルを欠くことが主要な制限である。
【0285】
形質導入したT細胞のin vivo増殖を達成するための、提案された一方法は、MGMT手法を用いてそれらを選択することである。Brian Davisらは、2年前に、形質導入した初代CD4T細胞をin vitroで、BG/BCNU薬物処置を用いて5%〜80%を超えて選択することが可能であることを実証した。ここでも、正確なin vivo薬物投薬を評価するための適切な動物モデルを欠くことが主要な制限である。
【0286】
別の選択肢は、対象を、操作細胞を再輸液する前に抗CD3抗体で(in vivo)プレコンディショニングすることである。形質導入した細胞の再輸液前のin vivoT細胞枯渇は、再注射した細胞を用いたT細胞サブセットの迅速な再構成をもたらす可能性がある。大動物モデルまたは第I相臨床治験での直接評価が、最も適切な方法であると見受けられる。
【0287】
(実施例45)
刺激
現在の刺激手順では、Dynalエポキシビーズにコーティングした抗ヒトCD3および抗ヒトCD28ネズミ抗体を用いる。CD3/CD28刺激は、細胞形質導入プロトコルの特徴である。
【0288】
以下に4つの代替実施形態を記載する:
生分解性ナノビーズ上に結合したCD3およびCD28抗体の使用。この手法では、関連する抗体刺激コストが削減されず、生成物の操作性も低下する(ビーズの除去なし)。
【0289】
過作用性抗ヒトCD28の使用。この抗体は、抗CD3抗体を必要とせずにT細胞の増殖を効率的に刺激することが示されている。
【0290】
ベクター自体をT細胞刺激タンパク質担体として使用。この手法では、抗体産生を必要としないが、パッケージング細胞系の改変を必要とする。
【0291】
Stemcell Technologiesによって開発されたTetralinkシステムの使用。このシステムでは、ビーズ支持体の必要性が回避され、したがってビーズ枯渇ステップが回避される。このシステムでは、ネズミIgG1モノクローナル抗体の使用が機能的であることを必要とする。
【0292】
(実施例46)
キット
本発明の一実施形態は、生分解性ナノビーズを含まない刺激系を用いた、改変したパッケージング細胞系から産生した臨床グレードのベクターを含めたキット、ならびに3日間の培養期間について大動物モデルからの安全性/有効性およびin vivoT細胞再構成である。
【0293】
本発明を完全に説明したので、当業者には、本発明の精神および範囲から逸脱せず、かつ必要以上の実験を行わずに、広範囲の均等なパラメータ、濃度、および条件内で同発明を行うことができることを理解されよう。
【0294】
本発明の基本的な態様から逸脱せずに前述の改変を行い得る。本発明は、1つまたは複数の具体的な実施形態を参照しながら相当に詳細に記載したが、当業者は、本出願中に具体的に開示した実施形態に変更を行ってもよく、それでもこれらの改変および改良が本発明の範囲および精神の範囲内にあることを理解されよう。本明細書中に例示的に記載した本発明は、具体的に開示していない任意の要素(または複数の要素)が存在しない場合に適切に実施し得る。したがって、たとえば、本明細書中のそれぞれの例において、「含む」、「本質的に〜からなる」、および「からなる」のうちの任意の用語を、他の2つの用語のどちらかと置き換え得る。したがって、用いた用語および表現は、限定ではなく説明の用語として用い、表示かつ記載した特徴の均等物またはその部分は排除されず、様々な改変が本発明の範囲内で可能であることを理解されたい。本発明の実施形態は、以下の特許請求の範囲に記載する。
【0295】
本明細書中上記および以下の特許請求の範囲では、用語「a」または「an」の使用は、単数形の定義に限定されない。そうではなく、これらの用語の使用は複数形を包含する。たとえば、用語「an抗体」は、1つの単一の抗体分子の単数形には限定されず、むしろ、参照する抗体の同一コピーである限りは複数の抗体分子の存在を包含する。同様に、「aウイルスベクター」は1つの単一のウイルスベクター分子または1つの単一のウイルス粒子に限定されない。用語「or」は、それが指す用語の1つに排他的であることを意図しない。たとえば、構造「A or B」の語句で用いられる場合は、これは、A単独、B単独、またはAおよびB両方を示し得る。
【0296】
以下の図は、本発明の態様の例示であり、特許請求の範囲によって包含される本発明の範囲を限定することを意図しない。
【0297】
特許または特許出願のファイルは、少なくとも1つのカラーで作成した図を含む。カラーの図(または複数の図)を含む本特許または特許出願公報のコピーは、請求に応じて必要経費の支払い後に特許局(Office)により提供される。
【0298】
図を添付する。異なる図中の同様の参照記号は同様の要素を示す。
【図面の簡単な説明】
【0299】
【図1】図1は、本発明の方法およびシステムで用いることができる例示的な容器またはフラスコを示す図である。
【図2】図2Aおよび2Bは、それぞれpN2cGFPおよびpN1GFP(cPT)のマップを示す図である。様々な制限酵素部位およびHIVに由来する構成成分を示す。pN2cGFP構築体は、CMV(サイトメガロウイルス)プロモーターに作動可能に連結し、したがってGFPの発現を制御するGFPコード配列を含む。pN1GFP(cPT)構築体は、以下でpN1(cpt)CGFPとも呼ばれ、HIV pol遺伝子由来のcPPTを含む。これらの構築体は、以下に記載の実施例で用いる。
【図3】図3は、固定したCD3およびCD28抗体でコーティングしたビーズを用いた初代T細胞の形質導入の結果を示す図である。細胞は、ビーズと接触させる前にベクターと接触させたか(パネルA)、ベクターと接触させる前にビーズと接触させたか(パネルB)、またはベクターおよびビーズをどちらも同時に接触させた(パネルC)。形質導入したベクターによってコードされているGFPからの蛍光に基づいたフローサイトメトリーの結果は、パネルA〜Cの細胞はそれぞれ90.70、87.19、および79.14%形質導入されていたことを示している。
【図4】図4は、ウイルスベクターと接触させる前にCD4+細胞を刺激するためにIL−2およびPHA−Pまたはビーズに固定したCD3およびCD28抗体のどちらかを用いた、形質導入の比較を示す図である。固定した抗体を使用することにより、毎回95%を超える形質導入効率がもたらされた。IL−2およびPHAの使用では、70.2〜84.5%の効率しかもたらされなかった。
【図5】図5は、本方法を用いたヒトCD4+T細胞の形質導入の頻度を示す図である。形質導入の15日後、対照細胞対緑色蛍光タンパク質(GFP)を発現する能力を有するベクターを用いて20のMOIで形質導入した細胞のフローサイトメトリー分析の比較により、形質導入した細胞の約93%も緑色蛍光を示すことが示されている。
【特許請求の範囲】
【請求項1】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法。
【請求項2】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;
形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入され;
該細胞とレンチウイルスベクターとの接触は約24時間にわたり、そして任意選択で少なくとも1回繰り返される、方法。
【請求項3】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;
約7〜10日後、もしくは約14日目に細胞の少なくとも約50%が安定に形質導入されており;かつ任意選択で約14日後に細胞の少なくとも50%が安定に形質導入されたまま保たれているか;または
約7〜10日後、もしくは約14日目に細胞の少なくとも約75%が安定に形質導入されており、かつ任意選択で約14日後に細胞の少なくとも75%が安定に形質導入されたまま保たれているか;または
約14日後に80%、85%、89%、90%、91%、92%、93%、94%もしくは95%を超える細胞が安定に形質導入されているか;または
約2〜約50、もしくは約10〜約30、もしくは10、もしくは約20、もしくは約30、もしくは約40、もしくは約50、もしくは約1〜約400、もしくは500未満の感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
形質導入した細胞1個あたりのレンチウイルスベクターのコピーが約1〜約100個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
細胞とレンチウイルスベクターとをの接触が約24時間にわたり、任意選択で少なくとも1回繰り返され;
細胞表面分子はアポトーシスを誘導せず、細胞表面結合分子により、ウイルスレンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなることをもたらす方法。
【請求項4】
初代細胞は、単離されるか、または対象から誘導される、請求項1に記載の方法。
【請求項5】
以下の手順:
(a)対象の血液のアフェレーシスによって;または
(b)対象の骨由来の骨髄から;または
(c)同種の対象の血液のアフェレーシスによって;または
(d)同種の対象の骨由来の骨髄から
1つまたは複数によって、初代細胞が単離される、請求項4に記載の方法。
【請求項6】
対象がヒト免疫不全ウイルス(HIV)に感染しており、任意選択でHIVがHIV−1またはHIV−2である、請求項4に記載の方法。
【請求項7】
対象が癌に罹患しており、任意選択で癌が乳癌である、請求項4に記載の方法。
【請求項8】
対象がヒトまたは動物である、請求項4に記載の方法。
【請求項9】
初代細胞が、細胞を密度勾配緩衝液に通すことによって、および/または磁場上で免疫精製を行うことによって、レンチウイルスベクターまたは細胞表面結合分子と接触させる前に富化される、請求項1に記載の方法。
【請求項10】
初代細胞と、以下:
(a)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触の前に起こるか;または
(b)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触と同時に起こるか;または
(c)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触の後に起こるか;または
(d)レンチウイルスベクターとの接触が、複数回起こるか;または
(e)レンチウイルスベクターとの接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または
(f)細胞表面結合分子との接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または
(g)レンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との最初の同時接触の後、連続的に起こるか;または
(h)(a)〜(g)の任意のものが、約24〜36時間の期間中に少なくとも1回起こる、請求項1に記載の方法。
【請求項11】
初代細胞は、少なくとも1つの細胞表面結合分子で予備刺激され、任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約24時間以内に、細胞が少なくとも1つの細胞表面結合分子で予備刺激されるか、または任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約12〜96時間以内に、細胞が少なくとも1つの細胞表面結合分子で予備刺激される、請求項1に記載の方法。
【請求項12】
レンチウイルスベクターがgag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来する少なくとも1つのシス作用性ヌクレオチド配列を含み、任意選択で、配列が発現されないか、またはgag、pol、env、vif、vpr、vpu、tatもしくはrev遺伝子の断片もしくは突然変異体である、請求項1に記載の方法。
【請求項13】
レンチウイルスベクターが:
(a)偽型であり、任意選択で偽型ベクターは水疱性口内炎ウイルスGエンベロープタンパク質を含むか;または
(b)偽型であり、偽型化は、レンチウイルスベクター遺伝物質と別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質もしくは細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションもしくは同時感染を行うことを含むか;または
(c)ラブドウイルスを用いた偽型であり、任意選択でラブドウイルスは水疱性口内炎ウイルスエンベロープG(VSV−G)タンパク質である、
請求項1に記載の方法。
【請求項14】
初代細胞が、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体である、請求項1に記載の方法。
【請求項15】
少なくとも1つの細胞表面結合分子が:
(a)ポリペプチド、脂質、核酸、炭水化物またはイオンを含むか;あるいは
(b)抗体、抗原結合断片、リガンド、または細胞表面分子を含むか;あるいは
(c)FLT−3リガンド、TPOリガンド、もしくはKitリガンド、またはFLT−3リガンド、TPOリガンド、もしくはKitリガンドの細胞表面結合類似体であるポリペプチドもしくは他の結合分子を含むか;あるいは
(d)CD34、CD3リガンド、CD28リガンド、CD25リガンド、CD71リガンド、もしくはCD69リガンド、またはCD34、CD3、CD25、CD28、CD69もしくはCD71リガンドと同じ細胞表面結合特異性を有するポリペプチドもしくは他の結合分子を含むか;あるいは
(e)GM−CSF、IL−4、およびTNF−α;GM−CSFおよびインターフェロン−α;またはGM−CSF、IL−4、およびTNF−αの細胞表面結合類似体であるポリペプチドもしくは他の結合分子;GM−CSFもしくはインターフェロン−αを含む組成物を含むか;あるいは
(f)CD3抗体またはその細胞表面結合断片、CD28抗体またはその細胞表面結合断片、抗体とその細胞表面結合断片との組合せ、および抗体と同じ細胞表面結合特異性を有する結合分子を含むか;あるいは
(g)ビーズまたは表面上に固定したCD3およびCD28抗体の組合せを含み、任意選択で、ビーズまたは表面はコーティングしたビーズを含むか;あるいは
(h)(a)〜(g)の任意のものから選択された2つ以上の細胞表面結合分子を含むか;あるいは
(i)分子が細胞の表面に結合する能力を増加または増強するために用いる別の分子を含むか;あるいは
(j)別の分子と複合体化しているか;あるいは
(k)初代細胞の表面上に見られ、別の細胞の表面に結合する、
請求項1に記載の方法。
【請求項16】
条件が:
(a)細胞表面結合分子もしくはサイトカインと共にさらなるインキュベーションを行うこと;または
(b)インターロイキン−2と共にさらなるインキュベーションを行うこと;または
(c)細胞を約7日間培養すること;または
(d)細胞を約14日間培養すること
を含む、請求項1に記載の方法。
【請求項17】
レンチウイルスベクターが:
(a)ヒト免疫不全ウイルス(HIV)由来であるか;または
(b)HIV−1、HIV−2、もしくはそれらの組合せ由来であるか;または
(c)HIV配列を含むキメラベクターであり、任意選択で、HIV配列はHIV−1およびHIV−2配列を含むか;または
(d)VRX496もしくはその誘導体である、
請求項1に記載の方法。
【請求項18】
前記接触がex vivoで、混合もしくは純粋細胞培養物、組織または器官系中で起こる、請求項1に記載の方法。
【請求項19】
請求項1に記載の方法によって形質導入した細胞を、生存する対象、組織、器官、胚盤胞または胚性幹細胞内にex vivo導入することを含む、遺伝物質を細胞内に導入する方法。
【請求項20】
薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項21】
対象におけるウイルス感染症を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項22】
対象におけるHIV感染症を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項23】
癌を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項24】
癌が乳癌または内皮細胞癌である、請求項23に記載の使用。
【請求項25】
請求項1から18のいずれか一項に記載の方法によって生成した、遺伝的欠陥によって引き起こされた異常を治療もしくは予防する、または腫瘍もしくは癌を治療、診断、緩和もしくは予防するための遺伝子治療用の薬剤組成物であって、任意選択で遺伝的欠陥または腫瘍もしくは癌によって引き起こされた異常が乳癌腫瘍である、薬剤組成物。
【請求項26】
請求項1から18のいずれか一項に記載の方法によって生成した、感染症によって引き起こされた異常を治療または予防するための遺伝子治療用の薬剤組成物。
【請求項27】
感染症がウイルス感染症であり、任意選択で、ウイルス感染症がヒト免疫不全ウイルス(HIV)感染症である、請求項26に記載の薬剤組成物。
【請求項28】
ex vivoで使用するために配合された、請求項25に記載の薬剤組成物。
【請求項29】
ex vivoで使用するために配合された、請求項26に記載の薬剤組成物。
【請求項30】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、前記初代細胞と細胞表面分子との接触により、レンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなる方法。
【請求項31】
細胞表面分子の存在により:
(a)細胞クロマチンのDNA組込みに対する受容性がより高くなること;または
(b)レンチウイルスベクターからの遺伝子の発現に有利な細胞部位内へのレンチウイルスベクターの組込み;または
(c)カプシドを含む核酸の、細胞の細胞質内へのより効率的な侵入;または
(d)ウイルスの、細胞膜を横断するもしくは細胞の内部膜構造体を横断するより効率的な侵入;または
(e)ウイルスベクター内に含まれる遺伝物質の核内移行に対する初代細胞の許容性がより高くなること
がもたらされる、請求項30に記載の方法。
【請求項32】
細胞表面結合分子、抗体、抗原結合断片、リガンドまたは細胞表面分子が:細胞に結合してベクター形質導入に対するその受容性をより高くする抗CD3または抗CD28抗体;細胞に結合してベクター形質導入に対するその受容性をより高くする、FLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンド;樹状細胞もしくはその前駆体、単球、CD34陽性幹細胞、または樹状細胞系列上の分化したその前駆細胞に結合してベクター形質導入に対するその受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンド;細胞上の
【化1】
に結合してベクター形質導入に対するその受容性をより高くする、ポリペプチド、核酸、炭水化物、脂質もしくはイオン、または別の物質と複合体化したポリペプチド、核酸、炭水化物、脂質もしくはイオンを含む、請求項1または請求項31に記載の方法。
【請求項33】
HIVに感染した対象から単離した造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、
(a)HIVに感染した対象から造血系細胞の初代細胞または造血幹細胞を単離するステップと;
(b)任意選択で、初代細胞または造血幹細胞を少なくとも1つの細胞表面結合分子で予備刺激するステップと;
(c)in vitroまたはex vivoで、造血系細胞または造血幹細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させるステップと;
(d)2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養するステップとを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法。
【請求項34】
(a)2つ以上の層を含む換気容器;ならびに
(b)単離した非接着性の造血系の初代細胞および/または造血幹細胞
を含むシステム。
【請求項35】
初代細胞が、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体である、請求項34に記載のシステム。
【請求項36】
多層容器が、四角形の形状、正方形の形状、または曲線縁の四角形の形状、もしくは曲線縁の正方形の形状である、請求項34に記載のシステム。
【請求項37】
容器が少なくとも約1億個の細胞の培養に適している、請求項34に記載のシステム。
【請求項1】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法。
【請求項2】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;
形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入され;
該細胞とレンチウイルスベクターとの接触は約24時間にわたり、そして任意選択で少なくとも1回繰り返される、方法。
【請求項3】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており;
約7〜10日後、もしくは約14日目に細胞の少なくとも約50%が安定に形質導入されており;かつ任意選択で約14日後に細胞の少なくとも50%が安定に形質導入されたまま保たれているか;または
約7〜10日後、もしくは約14日目に細胞の少なくとも約75%が安定に形質導入されており、かつ任意選択で約14日後に細胞の少なくとも75%が安定に形質導入されたまま保たれているか;または
約14日後に80%、85%、89%、90%、91%、92%、93%、94%もしくは95%を超える細胞が安定に形質導入されているか;または
約2〜約50、もしくは約10〜約30、もしくは10、もしくは約20、もしくは約30、もしくは約40、もしくは約50、もしくは約1〜約400、もしくは500未満の感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
形質導入した細胞1個あたりのレンチウイルスベクターのコピーが約1〜約100個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
形質導入した細胞1個あたりのレンチウイルスベクターのコピー数が約0.5〜約10個となるような感染多重度(MOI)で、レンチウイルスベクターを用いて、細胞が形質導入されるか;または
細胞とレンチウイルスベクターとをの接触が約24時間にわたり、任意選択で少なくとも1回繰り返され;
細胞表面分子はアポトーシスを誘導せず、細胞表面結合分子により、ウイルスレンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなることをもたらす方法。
【請求項4】
初代細胞は、単離されるか、または対象から誘導される、請求項1に記載の方法。
【請求項5】
以下の手順:
(a)対象の血液のアフェレーシスによって;または
(b)対象の骨由来の骨髄から;または
(c)同種の対象の血液のアフェレーシスによって;または
(d)同種の対象の骨由来の骨髄から
1つまたは複数によって、初代細胞が単離される、請求項4に記載の方法。
【請求項6】
対象がヒト免疫不全ウイルス(HIV)に感染しており、任意選択でHIVがHIV−1またはHIV−2である、請求項4に記載の方法。
【請求項7】
対象が癌に罹患しており、任意選択で癌が乳癌である、請求項4に記載の方法。
【請求項8】
対象がヒトまたは動物である、請求項4に記載の方法。
【請求項9】
初代細胞が、細胞を密度勾配緩衝液に通すことによって、および/または磁場上で免疫精製を行うことによって、レンチウイルスベクターまたは細胞表面結合分子と接触させる前に富化される、請求項1に記載の方法。
【請求項10】
初代細胞と、以下:
(a)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触の前に起こるか;または
(b)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触と同時に起こるか;または
(c)レンチウイルスベクターとの接触が、細胞と少なくとも1つの細胞表面結合分子との接触の後に起こるか;または
(d)レンチウイルスベクターとの接触が、複数回起こるか;または
(e)レンチウイルスベクターとの接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または
(f)細胞表面結合分子との接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との同時接触の後、連続的に起こるか;または
(g)レンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との接触が、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子との最初の同時接触の後、連続的に起こるか;または
(h)(a)〜(g)の任意のものが、約24〜36時間の期間中に少なくとも1回起こる、請求項1に記載の方法。
【請求項11】
初代細胞は、少なくとも1つの細胞表面結合分子で予備刺激され、任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約24時間以内に、細胞が少なくとも1つの細胞表面結合分子で予備刺激されるか、または任意選択で、細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させる前の約12〜96時間以内に、細胞が少なくとも1つの細胞表面結合分子で予備刺激される、請求項1に記載の方法。
【請求項12】
レンチウイルスベクターがgag、pol、env、vif、vpr、vpu、tatまたはrev遺伝子に由来する少なくとも1つのシス作用性ヌクレオチド配列を含み、任意選択で、配列が発現されないか、またはgag、pol、env、vif、vpr、vpu、tatもしくはrev遺伝子の断片もしくは突然変異体である、請求項1に記載の方法。
【請求項13】
レンチウイルスベクターが:
(a)偽型であり、任意選択で偽型ベクターは水疱性口内炎ウイルスGエンベロープタンパク質を含むか;または
(b)偽型であり、偽型化は、レンチウイルスベクター遺伝物質と別のウイルスの少なくとも1つのエンベロープタンパク質をコードしている遺伝物質もしくは細胞表面分子をコードしている遺伝物質とをどちらも用いて、パッケージング細胞の同時トランスフェクションもしくは同時感染を行うことを含むか;または
(c)ラブドウイルスを用いた偽型であり、任意選択でラブドウイルスは水疱性口内炎ウイルスエンベロープG(VSV−G)タンパク質である、
請求項1に記載の方法。
【請求項14】
初代細胞が、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体である、請求項1に記載の方法。
【請求項15】
少なくとも1つの細胞表面結合分子が:
(a)ポリペプチド、脂質、核酸、炭水化物またはイオンを含むか;あるいは
(b)抗体、抗原結合断片、リガンド、または細胞表面分子を含むか;あるいは
(c)FLT−3リガンド、TPOリガンド、もしくはKitリガンド、またはFLT−3リガンド、TPOリガンド、もしくはKitリガンドの細胞表面結合類似体であるポリペプチドもしくは他の結合分子を含むか;あるいは
(d)CD34、CD3リガンド、CD28リガンド、CD25リガンド、CD71リガンド、もしくはCD69リガンド、またはCD34、CD3、CD25、CD28、CD69もしくはCD71リガンドと同じ細胞表面結合特異性を有するポリペプチドもしくは他の結合分子を含むか;あるいは
(e)GM−CSF、IL−4、およびTNF−α;GM−CSFおよびインターフェロン−α;またはGM−CSF、IL−4、およびTNF−αの細胞表面結合類似体であるポリペプチドもしくは他の結合分子;GM−CSFもしくはインターフェロン−αを含む組成物を含むか;あるいは
(f)CD3抗体またはその細胞表面結合断片、CD28抗体またはその細胞表面結合断片、抗体とその細胞表面結合断片との組合せ、および抗体と同じ細胞表面結合特異性を有する結合分子を含むか;あるいは
(g)ビーズまたは表面上に固定したCD3およびCD28抗体の組合せを含み、任意選択で、ビーズまたは表面はコーティングしたビーズを含むか;あるいは
(h)(a)〜(g)の任意のものから選択された2つ以上の細胞表面結合分子を含むか;あるいは
(i)分子が細胞の表面に結合する能力を増加または増強するために用いる別の分子を含むか;あるいは
(j)別の分子と複合体化しているか;あるいは
(k)初代細胞の表面上に見られ、別の細胞の表面に結合する、
請求項1に記載の方法。
【請求項16】
条件が:
(a)細胞表面結合分子もしくはサイトカインと共にさらなるインキュベーションを行うこと;または
(b)インターロイキン−2と共にさらなるインキュベーションを行うこと;または
(c)細胞を約7日間培養すること;または
(d)細胞を約14日間培養すること
を含む、請求項1に記載の方法。
【請求項17】
レンチウイルスベクターが:
(a)ヒト免疫不全ウイルス(HIV)由来であるか;または
(b)HIV−1、HIV−2、もしくはそれらの組合せ由来であるか;または
(c)HIV配列を含むキメラベクターであり、任意選択で、HIV配列はHIV−1およびHIV−2配列を含むか;または
(d)VRX496もしくはその誘導体である、
請求項1に記載の方法。
【請求項18】
前記接触がex vivoで、混合もしくは純粋細胞培養物、組織または器官系中で起こる、請求項1に記載の方法。
【請求項19】
請求項1に記載の方法によって形質導入した細胞を、生存する対象、組織、器官、胚盤胞または胚性幹細胞内にex vivo導入することを含む、遺伝物質を細胞内に導入する方法。
【請求項20】
薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項21】
対象におけるウイルス感染症を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項22】
対象におけるHIV感染症を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項23】
癌を治療または予防するための薬剤組成物を調製するための、請求項1から18のいずれか一項に記載の方法によって形質導入した造血系の初代細胞または造血幹細胞の使用。
【請求項24】
癌が乳癌または内皮細胞癌である、請求項23に記載の使用。
【請求項25】
請求項1から18のいずれか一項に記載の方法によって生成した、遺伝的欠陥によって引き起こされた異常を治療もしくは予防する、または腫瘍もしくは癌を治療、診断、緩和もしくは予防するための遺伝子治療用の薬剤組成物であって、任意選択で遺伝的欠陥または腫瘍もしくは癌によって引き起こされた異常が乳癌腫瘍である、薬剤組成物。
【請求項26】
請求項1から18のいずれか一項に記載の方法によって生成した、感染症によって引き起こされた異常を治療または予防するための遺伝子治療用の薬剤組成物。
【請求項27】
感染症がウイルス感染症であり、任意選択で、ウイルス感染症がヒト免疫不全ウイルス(HIV)感染症である、請求項26に記載の薬剤組成物。
【請求項28】
ex vivoで使用するために配合された、請求項25に記載の薬剤組成物。
【請求項29】
ex vivoで使用するために配合された、請求項26に記載の薬剤組成物。
【請求項30】
造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、細胞の表面とレンチウイルスベクターおよび細胞表面に結合する少なくとも1つの分子の両方とをin vitroまたはex vivoで接触させること、および2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養することを含み、前記容器は少なくとも約1億個の細胞の培養に適しており、前記初代細胞と細胞表面分子との接触により、レンチウイルスベクターによる形質導入に対する細胞の受容性がより高くなる方法。
【請求項31】
細胞表面分子の存在により:
(a)細胞クロマチンのDNA組込みに対する受容性がより高くなること;または
(b)レンチウイルスベクターからの遺伝子の発現に有利な細胞部位内へのレンチウイルスベクターの組込み;または
(c)カプシドを含む核酸の、細胞の細胞質内へのより効率的な侵入;または
(d)ウイルスの、細胞膜を横断するもしくは細胞の内部膜構造体を横断するより効率的な侵入;または
(e)ウイルスベクター内に含まれる遺伝物質の核内移行に対する初代細胞の許容性がより高くなること
がもたらされる、請求項30に記載の方法。
【請求項32】
細胞表面結合分子、抗体、抗原結合断片、リガンドまたは細胞表面分子が:細胞に結合してベクター形質導入に対するその受容性をより高くする抗CD3または抗CD28抗体;細胞に結合してベクター形質導入に対するその受容性をより高くする、FLT−3リガンド、TPO、およびKitリガンド受容体に対する抗体またはリガンド;樹状細胞もしくはその前駆体、単球、CD34陽性幹細胞、または樹状細胞系列上の分化したその前駆細胞に結合してベクター形質導入に対するその受容性をより高くする、GM−CSFおよびIL−4受容体に対する抗体またはリガンド;細胞上の
【化1】
に結合してベクター形質導入に対するその受容性をより高くする、ポリペプチド、核酸、炭水化物、脂質もしくはイオン、または別の物質と複合体化したポリペプチド、核酸、炭水化物、脂質もしくはイオンを含む、請求項1または請求項31に記載の方法。
【請求項33】
HIVに感染した対象から単離した造血系の初代細胞および/または造血幹細胞の安定な形質導入のための方法であって、
(a)HIVに感染した対象から造血系細胞の初代細胞または造血幹細胞を単離するステップと;
(b)任意選択で、初代細胞または造血幹細胞を少なくとも1つの細胞表面結合分子で予備刺激するステップと;
(c)in vitroまたはex vivoで、造血系細胞または造血幹細胞とレンチウイルスベクターおよび少なくとも1つの細胞表面結合分子とを同時に接触させるステップと;
(d)2つ以上の層を含む換気容器内にて、成長および/または増殖を助ける条件下で細胞を培養するステップとを含み、前記容器は少なくとも約1億個の細胞の培養に適している方法。
【請求項34】
(a)2つ以上の層を含む換気容器;ならびに
(b)単離した非接着性の造血系の初代細胞および/または造血幹細胞
を含むシステム。
【請求項35】
初代細胞が、リンパ球、リンパ球の前駆体、CD4陽性細胞、CD4陽性細胞の造血幹細胞、CD8陽性細胞、CD8陽性細胞の造血幹細胞、CD34陽性細胞、CD34陽性細胞の造血幹細胞、樹状細胞、樹状細胞に分化する能力を有する細胞、ヒト造血系の初代細胞および/もしくはヒト造血幹細胞、ヒト造血幹細胞の前駆体、星細胞、皮膚線維芽細胞、上皮細胞、ニューロン、樹状細胞、白血球、免疫応答に関連する細胞、血管内皮細胞、腫瘍細胞、腫瘍血管内皮細胞、肝細胞、肺細胞、骨髄細胞、抗原提示細胞、間質細胞、脂肪細胞、筋肉細胞、膵臓細胞、腎細胞、卵細胞、精母細胞、生殖系列に寄与する細胞、胚性分化多能性幹細胞もしくは前駆細胞、血液細胞、無核細胞、血小板細胞、または赤血球、あるいはその誘導体である、請求項34に記載のシステム。
【請求項36】
多層容器が、四角形の形状、正方形の形状、または曲線縁の四角形の形状、もしくは曲線縁の正方形の形状である、請求項34に記載のシステム。
【請求項37】
容器が少なくとも約1億個の細胞の培養に適している、請求項34に記載のシステム。
【図5】図5は、IL−2およびPHA−Pまたはビーズに固定したCD3およびCD28抗体のどちらかを用いた形質導入の14日後の、CD4+およびGFP+細胞のFACS分析の結果を示す図である。抗体で処理した細胞の約93%が、14日後に安定した形質導入が保たれていた。IL−2およびPHAで処理した細胞の約75%のみが、同じ時点で安定した形質導入が保たれていた。
【図6A】図6Aは、様々なウイルスベクターを用いて形質導入した細胞の結果を示す図である。
【図6B】図6Bは、様々なウイルスベクターを用いて形質導入した細胞の結果を示す図である。
【図7】図7は、トランスフェクション効率に対する様々なMOIを使用した効果を示す図である。
【図8】図8は、細胞表面結合分子の存在下でウイルスベクターを用いた複数の形質導入後の、臍帯血から調製したCD34+細胞の安定形質導入を実証する図である。88%を超える細胞が、形質導入後6週間を超えて陽性に保たれていた。
【図9】図9、パネルA〜Dは、SCID(重症複合型免疫不全)マウスへ移植した後の長期形質導入の効率を示す図である。約8週間後、平均して91%を超える形質導入した細胞が、形質導入したGFPマーカーの発現について陽性が保たれており、これらは成熟を続けていた。
【図10】図10、パネルAおよびBは、形質導入の7日後の樹状細胞の効率を示す図である。
【図11】図11は、CD8の枯渇(CD4富化)またはCD4陽性選択(CD4単離)のどちらかを用いた、対象のアフェレーシス生成物からCD4Tリンパ球を単離または富化するための精製プロセスおよび関連する装置を示す模式図である。
【図12】図12は、形質導入、増殖および冷凍保存を含めた細胞処理の製造、および関連する装置を示す模式図である。この図で用いた生成物は、上述した図11の最初の単離手順からものである。
【図13】図13は、「増殖なし」の細胞処理手順および関連する装置を示す模式図の概要である。ここではCD4陽性選択を示すが、CD4陽性選択、または非CD4細胞の枯渇を介したCD4富化を用いることもできる。「スマートベクター」とは、そのエンベロープに組み込まれているタンパク質により、細胞を結合、刺激、および形質導入する能力が増強されている、米国仮特許第60/585,464号に記載の特別にパッケージングされたレンチウイルスベクターをいう。
【図14】図14は、細胞処理用の「増殖なし」の形質導入キットおよび関連する装置を示す模式図の概要である。この手順により、分散した分布のための閉鎖系での細胞の単離および形質導入可能となる。このプロセスは増殖ステップを含まない。CD4細胞の富化は、非CD4細胞の枯渇方法であるRosette−Sepを用いて行う。
【図15】図15は、「インライン」形質導入プロセスを示す模式図の概要である。このプロセスでは、任意の精製(これは用いても用いなくてもよい)、および対象に直接自己輸血するための形質導入を行うための、閉鎖系のアフェレーシス機構を使用する。
【図16A】図16Aは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図16B】図16Bは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図16C】図16Cは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図17】図17は、例示的なレンチウイルスベクターを示す図である。具体的には、WT HIVに由来するHIV系ベクターである。
【図18】図18は、いくつかのレトロウイルスベクター:VRX496、VRX494、およびVRX577を示す図である。(上部)この図は、pN1cptASenv、(VRX496)の模式図であり、それが由来するWT HIVの要素および領域を示す。ベクター中の数字は、遺伝子要素の大きさを指す。このベクターは、HIVエンベロップ遺伝子(ASenv)に対して標的化されている937bpのアンチセンスセグメントを発現する。アンチセンスペイロードはTatであり、Rev依存性であり、したがって、HIVがベクター含有細胞に感染した後にのみ発現される。HIV由来の要素には、5’および3’末端反復配列(LTR)、パッケージングシグナル(Ψ)、tRNAプライマー結合部位(PBS)、中央ポリプリントラクトおよび中心終止配列(cPPTおよびCTS)、スプライスアクセプターおよびドナー部位(SA/SD)、Tat依存性HIVプロモーター(P)、Gag遺伝子、rev応答要素(RRE)、ならびに3’ポリプリントラクト(PPT)が含まれる。操作した要素には、gag中のストップコドンが含まれる。Gtagは、GFPからの非コードマーカー配列である。
【0300】
(下部)これは、ヘルパーパッケージング構築体であるpVRX577(VIRPAC)の模式図である。VRX496は、水疱性口内炎ウイルスタンパク質G(VSV−G)エンベロープを用いて偽型化されている。GagおよびPolはサイトメガロウイルス(CMV)プロモーターの制御下、Revは、VRX496とVIRPACとの相同性を低下させるために用いるHIV−2に由来するrev応答要素(RRE−2)の制御下、Tatは内部リボソーム侵入部位(IRES)の制御下で発現され、VSV−Gは伸長因子1α(EF−1α)/ヒトT細胞リンパ増殖性ウイルス(HTLV)キメラプロモーターによって発現される。安全性のために、VSV−Gは、数個のポーズシグナル、および任意の読み過ごしRNAを切断するタバコモザイクリングスポットウイルス(sTobRV+Rz)由来のシス作用性リボザイムによって、他のパッケージング遺伝子から離す。ベクターとの相同性を低下させるために、revおよびtat遺伝子の配列を部分的に縮退させた。
【図19】図19は、どのようにしてVRX496アンチセンスDNAが対象のT細胞のDNA内に組み込まれるかを示す図である。図19は、レンチウイルスベクターVRX496およびそのアンチセンスペイロードを利用したその作用機構の説明である。
【図20】図20は、どのようにしてVRX496がHIVのRNA産生を破壊するかを示す図である。図20は、レンチウイルスベクターVRX496およびそのアンチセンスペイロードを利用したその作用機構の説明である。
【図21】図21は、どのようにしてVRX496を使用することによりHIV耐性の問題が解決されるかを示す図である。このグラフは、抗HIV薬に対する耐性が発生するために必要な突然変異の数と、VRX496に対する耐性が発生するために必要な突然変異の数とを比較する。HIVは、アンチセンスペイロードによって破壊されるか、またはウイルスが疾患を引き起こすために不適合なレベルまで変異する。
【図22】図22は、緑色蛍光タンパク質を用いてタグ付けしたウイルスベクターを用いた遺伝子導入の効率が優れていることを示す図である。図22は、フローサイトメトリーによって初代T細胞内への遺伝子形質導入のレベルを示す。
【図23】図23は、HIV(+)患者から採取した初代リンパ球において、どのようにしてVRX496がWT HIVの複製を阻害するかを示す図である。
【図24】図24は、HIV感染の21日後における、未保護の細胞と比較した、VRX496で形質導入した細胞の生存の優位を示す図である。
【図25】図25は、HIVを治療するために自己細胞治療を用いた、例示的な臨床的プロセスの概要を示す図である。図25は、VIRxSYS第I相臨床治験の模式図である。
【図26】図26は、研究対象の基礎特徴を示す表である。
【図27】図27は、VRX496で輸液を行った対象の第I相臨床治験のCD4細胞数を示す図である。
【図28】図28は、VRX496で輸液を行った対象の第I相臨床治験のHIVウイルス量を示す図である。
【図29】図29は、いくつかのレトロウイルス薬、すなわちAZT、ddC/サキナビル、D4T/3Te/クリキシバン、Norvir/アムプレニビル/ddl/アデフォビル、Sustiva/Ziagen/Kaletra/3Te/Vireadの使用を示す図である。図29は、VIRxSYS第I相臨床治験の対象第2号のウイルス量履歴を示す。
【図30】図30は、VRX496で改変したCD4T細胞の持続生着および残留性を示す図である。図30は、VRX496で改変したCD4+細胞の輸液の20分後から開始して、その後、72時間後、1、2、3、および6週間後、ならびに3、6、9、および12カ月後に評価した、VRX496ベクターの残留性を示す折れ線グラフである。示した時点でPBMCを採取し、実時間PCRを用いてVRX496検出についてDNA分析を行った。定量の限界は100個のベクターコピー数/106個のPBMCである。1年後の時点で、患者第4号は、9カ月の時点で検出不可能であった後に生着の頻度が0.04%(400個のコピー)であった。
【図31】図31は、免疫機能分析:IFN−g ELISPOT Envを示す図である。図31は、HIV−1 env特異的エフェクター細胞IFN−γ分泌を示す棒グラフである。血液試料を基底および遺伝子導入治療の3および6カ月後に得た。標準のフィコール分離技術によってPBMCを対象およびHIV−1陽性対照対象(n=25)から単離した。HIV−1 envのPBMCのin vitro刺激後のIFN−γの産生を、標準のELISPOTによって、env抗原特異的応答について評価した。対照対象の平均±95%の信頼区間をプロットした。
【図32】図32は、免疫機能分析:IFN−g ELISPOT−Gagを示す図である。図32は、HIV−1 gag特異的エフェクター細胞IFN−γ分泌を示す棒グラフである。遺伝子導入治療の前ならびに3および6カ月後に血液試料を採取した。標準のフィコール分離技術によってPBMCを対象およびHIV−1陽性対照対象(n=25)から単離した。HIV−1 gagのPBMCのin vitro刺激後のIFN−γ産生を、標準のELISPOTによって、gag抗原特異的応答について評価した。対照対象の平均±95%の信頼区間をプロットした。
【図33】図33は、第I相および第II相の産生で行った、増加したベクター産生および収率の調節を示す表である。
【図34】図34は、例示的な自己T細胞製造プロセスを示す図である。図34は、図16A、BおよびCに関連する臨床グレード、大スケールの細胞処理の模式図である。図39に記載のプロセスで生または凍結アフェレーシス生成物を用いることができる。
【図35】図35は、第I相CD4+生成物の純度とVIRxSYS第II相開発プロセスとの比較を示す図である。
【図36】図36は、第I相VRX496のコピー数とVIRxSYS第II相VRX496ウイルスのコピー数との比較を示す図である。
【図37】図37は、第I相細胞生成物の増殖とVIRxSYS第II相細胞生成物の増殖との比較を示す図である。
【図1】


【図2A】

【図2B】

【図3】

【図4A】

【図4B】

【図4C】

【図4D】

【図4E】

【図4F】

【図5】

【図6A】

【図6B】

【図7A】

【図7B】

【図7C】

【図7D】

【図7E】

【図8】

【図9A】

【図9B】

【図9C】

【図9D】

【図10A】

【図10B】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16A】

【図16B】

【図16C】

【図17】

【図18】

【図19】
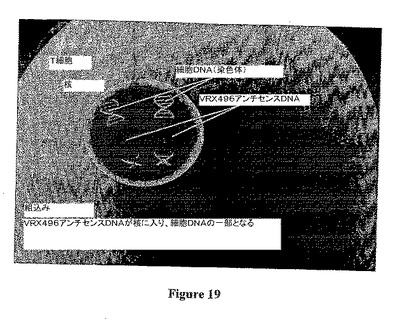
【図20】

【図21】

【図22】

【図23】

【図24】

【図25】

【図26】

【図27】

【図28】

【図29】

【図30】

【図31】

【図32】

【図33】

【図34】

【図35】

【図36】

【図37】

【図6A】図6Aは、様々なウイルスベクターを用いて形質導入した細胞の結果を示す図である。
【図6B】図6Bは、様々なウイルスベクターを用いて形質導入した細胞の結果を示す図である。
【図7】図7は、トランスフェクション効率に対する様々なMOIを使用した効果を示す図である。
【図8】図8は、細胞表面結合分子の存在下でウイルスベクターを用いた複数の形質導入後の、臍帯血から調製したCD34+細胞の安定形質導入を実証する図である。88%を超える細胞が、形質導入後6週間を超えて陽性に保たれていた。
【図9】図9、パネルA〜Dは、SCID(重症複合型免疫不全)マウスへ移植した後の長期形質導入の効率を示す図である。約8週間後、平均して91%を超える形質導入した細胞が、形質導入したGFPマーカーの発現について陽性が保たれており、これらは成熟を続けていた。
【図10】図10、パネルAおよびBは、形質導入の7日後の樹状細胞の効率を示す図である。
【図11】図11は、CD8の枯渇(CD4富化)またはCD4陽性選択(CD4単離)のどちらかを用いた、対象のアフェレーシス生成物からCD4Tリンパ球を単離または富化するための精製プロセスおよび関連する装置を示す模式図である。
【図12】図12は、形質導入、増殖および冷凍保存を含めた細胞処理の製造、および関連する装置を示す模式図である。この図で用いた生成物は、上述した図11の最初の単離手順からものである。
【図13】図13は、「増殖なし」の細胞処理手順および関連する装置を示す模式図の概要である。ここではCD4陽性選択を示すが、CD4陽性選択、または非CD4細胞の枯渇を介したCD4富化を用いることもできる。「スマートベクター」とは、そのエンベロープに組み込まれているタンパク質により、細胞を結合、刺激、および形質導入する能力が増強されている、米国仮特許第60/585,464号に記載の特別にパッケージングされたレンチウイルスベクターをいう。
【図14】図14は、細胞処理用の「増殖なし」の形質導入キットおよび関連する装置を示す模式図の概要である。この手順により、分散した分布のための閉鎖系での細胞の単離および形質導入可能となる。このプロセスは増殖ステップを含まない。CD4細胞の富化は、非CD4細胞の枯渇方法であるRosette−Sepを用いて行う。
【図15】図15は、「インライン」形質導入プロセスを示す模式図の概要である。このプロセスでは、任意の精製(これは用いても用いなくてもよい)、および対象に直接自己輸血するための形質導入を行うための、閉鎖系のアフェレーシス機構を使用する。
【図16A】図16Aは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図16B】図16Bは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図16C】図16Cは、初代細胞のアフェレーシス、形質導入および増殖を説明する流れ図であり、多くの場合、これはその後対象に再輸液する。さらに、細胞処理ステップおよび関連する品質管理手段を示す。
【図17】図17は、例示的なレンチウイルスベクターを示す図である。具体的には、WT HIVに由来するHIV系ベクターである。
【図18】図18は、いくつかのレトロウイルスベクター:VRX496、VRX494、およびVRX577を示す図である。(上部)この図は、pN1cptASenv、(VRX496)の模式図であり、それが由来するWT HIVの要素および領域を示す。ベクター中の数字は、遺伝子要素の大きさを指す。このベクターは、HIVエンベロップ遺伝子(ASenv)に対して標的化されている937bpのアンチセンスセグメントを発現する。アンチセンスペイロードはTatであり、Rev依存性であり、したがって、HIVがベクター含有細胞に感染した後にのみ発現される。HIV由来の要素には、5’および3’末端反復配列(LTR)、パッケージングシグナル(Ψ)、tRNAプライマー結合部位(PBS)、中央ポリプリントラクトおよび中心終止配列(cPPTおよびCTS)、スプライスアクセプターおよびドナー部位(SA/SD)、Tat依存性HIVプロモーター(P)、Gag遺伝子、rev応答要素(RRE)、ならびに3’ポリプリントラクト(PPT)が含まれる。操作した要素には、gag中のストップコドンが含まれる。Gtagは、GFPからの非コードマーカー配列である。
【0300】
(下部)これは、ヘルパーパッケージング構築体であるpVRX577(VIRPAC)の模式図である。VRX496は、水疱性口内炎ウイルスタンパク質G(VSV−G)エンベロープを用いて偽型化されている。GagおよびPolはサイトメガロウイルス(CMV)プロモーターの制御下、Revは、VRX496とVIRPACとの相同性を低下させるために用いるHIV−2に由来するrev応答要素(RRE−2)の制御下、Tatは内部リボソーム侵入部位(IRES)の制御下で発現され、VSV−Gは伸長因子1α(EF−1α)/ヒトT細胞リンパ増殖性ウイルス(HTLV)キメラプロモーターによって発現される。安全性のために、VSV−Gは、数個のポーズシグナル、および任意の読み過ごしRNAを切断するタバコモザイクリングスポットウイルス(sTobRV+Rz)由来のシス作用性リボザイムによって、他のパッケージング遺伝子から離す。ベクターとの相同性を低下させるために、revおよびtat遺伝子の配列を部分的に縮退させた。
【図19】図19は、どのようにしてVRX496アンチセンスDNAが対象のT細胞のDNA内に組み込まれるかを示す図である。図19は、レンチウイルスベクターVRX496およびそのアンチセンスペイロードを利用したその作用機構の説明である。
【図20】図20は、どのようにしてVRX496がHIVのRNA産生を破壊するかを示す図である。図20は、レンチウイルスベクターVRX496およびそのアンチセンスペイロードを利用したその作用機構の説明である。
【図21】図21は、どのようにしてVRX496を使用することによりHIV耐性の問題が解決されるかを示す図である。このグラフは、抗HIV薬に対する耐性が発生するために必要な突然変異の数と、VRX496に対する耐性が発生するために必要な突然変異の数とを比較する。HIVは、アンチセンスペイロードによって破壊されるか、またはウイルスが疾患を引き起こすために不適合なレベルまで変異する。
【図22】図22は、緑色蛍光タンパク質を用いてタグ付けしたウイルスベクターを用いた遺伝子導入の効率が優れていることを示す図である。図22は、フローサイトメトリーによって初代T細胞内への遺伝子形質導入のレベルを示す。
【図23】図23は、HIV(+)患者から採取した初代リンパ球において、どのようにしてVRX496がWT HIVの複製を阻害するかを示す図である。
【図24】図24は、HIV感染の21日後における、未保護の細胞と比較した、VRX496で形質導入した細胞の生存の優位を示す図である。
【図25】図25は、HIVを治療するために自己細胞治療を用いた、例示的な臨床的プロセスの概要を示す図である。図25は、VIRxSYS第I相臨床治験の模式図である。
【図26】図26は、研究対象の基礎特徴を示す表である。
【図27】図27は、VRX496で輸液を行った対象の第I相臨床治験のCD4細胞数を示す図である。
【図28】図28は、VRX496で輸液を行った対象の第I相臨床治験のHIVウイルス量を示す図である。
【図29】図29は、いくつかのレトロウイルス薬、すなわちAZT、ddC/サキナビル、D4T/3Te/クリキシバン、Norvir/アムプレニビル/ddl/アデフォビル、Sustiva/Ziagen/Kaletra/3Te/Vireadの使用を示す図である。図29は、VIRxSYS第I相臨床治験の対象第2号のウイルス量履歴を示す。
【図30】図30は、VRX496で改変したCD4T細胞の持続生着および残留性を示す図である。図30は、VRX496で改変したCD4+細胞の輸液の20分後から開始して、その後、72時間後、1、2、3、および6週間後、ならびに3、6、9、および12カ月後に評価した、VRX496ベクターの残留性を示す折れ線グラフである。示した時点でPBMCを採取し、実時間PCRを用いてVRX496検出についてDNA分析を行った。定量の限界は100個のベクターコピー数/106個のPBMCである。1年後の時点で、患者第4号は、9カ月の時点で検出不可能であった後に生着の頻度が0.04%(400個のコピー)であった。
【図31】図31は、免疫機能分析:IFN−g ELISPOT Envを示す図である。図31は、HIV−1 env特異的エフェクター細胞IFN−γ分泌を示す棒グラフである。血液試料を基底および遺伝子導入治療の3および6カ月後に得た。標準のフィコール分離技術によってPBMCを対象およびHIV−1陽性対照対象(n=25)から単離した。HIV−1 envのPBMCのin vitro刺激後のIFN−γの産生を、標準のELISPOTによって、env抗原特異的応答について評価した。対照対象の平均±95%の信頼区間をプロットした。
【図32】図32は、免疫機能分析:IFN−g ELISPOT−Gagを示す図である。図32は、HIV−1 gag特異的エフェクター細胞IFN−γ分泌を示す棒グラフである。遺伝子導入治療の前ならびに3および6カ月後に血液試料を採取した。標準のフィコール分離技術によってPBMCを対象およびHIV−1陽性対照対象(n=25)から単離した。HIV−1 gagのPBMCのin vitro刺激後のIFN−γ産生を、標準のELISPOTによって、gag抗原特異的応答について評価した。対照対象の平均±95%の信頼区間をプロットした。
【図33】図33は、第I相および第II相の産生で行った、増加したベクター産生および収率の調節を示す表である。
【図34】図34は、例示的な自己T細胞製造プロセスを示す図である。図34は、図16A、BおよびCに関連する臨床グレード、大スケールの細胞処理の模式図である。図39に記載のプロセスで生または凍結アフェレーシス生成物を用いることができる。
【図35】図35は、第I相CD4+生成物の純度とVIRxSYS第II相開発プロセスとの比較を示す図である。
【図36】図36は、第I相VRX496のコピー数とVIRxSYS第II相VRX496ウイルスのコピー数との比較を示す図である。
【図37】図37は、第I相細胞生成物の増殖とVIRxSYS第II相細胞生成物の増殖との比較を示す図である。
【図1】
【図2A】
【図2B】
【図3】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図4F】
【図5】
【図6A】
【図6B】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図8】
【図9A】
【図9B】
【図9C】
【図9D】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A】
【図16B】
【図16C】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【公表番号】特表2008−539796(P2008−539796A)
【公表日】平成20年11月20日(2008.11.20)
【国際特許分類】
【出願番号】特願2008−512590(P2008−512590)
【出願日】平成18年5月22日(2006.5.22)
【国際出願番号】PCT/US2006/019709
【国際公開番号】WO2006/127585
【国際公開日】平成18年11月30日(2006.11.30)
【出願人】(503081933)バイレクシス コーポレイション (6)
【Fターム(参考)】
【公表日】平成20年11月20日(2008.11.20)
【国際特許分類】
【出願日】平成18年5月22日(2006.5.22)
【国際出願番号】PCT/US2006/019709
【国際公開番号】WO2006/127585
【国際公開日】平成18年11月30日(2006.11.30)
【出願人】(503081933)バイレクシス コーポレイション (6)
【Fターム(参考)】
[ Back to top ]