
放射線増感剤を用いた放射線感受性腫瘍に対する方法
【解決手段】 本発明は、腫瘍の放射線増感剤としてPARP阻害剤を用いて癌を治療する方法に関するものである。特に、本発明は、化学式(I)の化合物或いはそれらの薬学的に許容可能な塩類を用いた、腫瘍の放射線増感の方法に関するものである。本発明はさらに、放射線感受性腫瘍に対するPARP阻害剤の薬学的組成物にも関するものである。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、腫瘍の放射線増感剤としてPARP阻害剤を用いて癌を治療する方法に関するものである。特に、本発明は、化学式(I)の化合物、
【0002】
【化1】

【0003】
或いはその薬学的に許容可能な塩形態を用いた、腫瘍の放射線増感方法に関するものである。本発明はさらに、放射線感受性腫瘍に対するPARP阻害剤の薬学的組成物に関するものである。
【背景技術】
【0004】
放射線は、DNA鎖破壊を作り出すことによって細胞障害を誘導する細胞障害性治療法である。ポリ(ADP−リボース)ポリメラーゼ1(PARP−1)は、核亜鉛フィンガーDNA結合タンパク質であり、DNA放射線誘導性障害及び修復によって活性化され、複製される。PARPは、DNA鎖破壊に結合し、ヌクレアーゼ攻撃或いは組換えからDNA鎖を保護するように働く。PARPはDNA修復を助けるように作用するので、阻害剤は細胞障害性薬剤の化学−及び放射線−感作を増強する潜在能力を有している(Curtin,2005)。
【0005】
治療上の失敗及び癌死亡率の最も重要な原因としては、放射線/化学−抵抗性である。細胞障害性薬剤に対する癌細胞抵抗性を克服するための薬剤は、癌治療を成功させる鍵となる因子となるであろう。化学及び放射線感作物質としての治療上のPARP阻害剤の潜在的な適用は、比較的最近まで、これら薬剤の力価、選択性及び薬学的特性によって制限されていた(Griffin et al.,1998;Bowman,et al.,1998;Bowman et al.,2001,Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Lui, et al.,1999)。最近、より強力で選択的なPARP阻害剤(ベンズイミダゾール−4−カルボキサミド及びキナゾリン−4−[3H]−オンズ(ones))が開発され、これらは、白血病、中枢神経系へのリンパ腫転移、大腸癌、肺癌及び乳癌のヒト及びマウス腫瘍モデルの両者を用いたin vitro及びin vivoにおける、放射線の効果、及びカンプトテシン(CPT)、トポテカン、イリノテカン、シスプラチン、エトポシド、ブレオマイシン、BCNU、及びテモゾロミド(TMZ)などの化学療法剤の効果を増強する能力を示した(Griffin et al.,1998;Bowman,et al.,1998;Bowman et al.,2001,Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Lui,et al.,1999,Tentori,et al.,2002)。異なるクラスの化学療法剤及び/若しくは放射線の作用に対して腫瘍細胞を増感させることができるPARP阻害剤は、確立された癌治療の成功率を上昇することができるであろう。
【0006】
PARP−1は、ヒストンポリメラーゼ、リガーゼ及びPARP自身(自己修飾)などの受容器タンパク質へとADP−リボースを移行するための基質としてNAD+を使用する116kD核亜鉛フィンガーDNA結合タンパク質である(Griffin et al.,1998;Tentori,et al.,2002;Baldwin et al.,2002)。PARP−1は、現在10種類のメンバーを含むタンパク質のファミリーに所属しており、その中のPARP−1及びPARP−2はDNA障害によって活性化される唯一の酵素である(Curtin,2005;Tentori,et al.,2002)。PARP−2の活性化はさらに、炎症誘発性活性を誘導し(Jagtop and Szabo,2005)、これは腫瘍細胞におけるPARP−2の阻害が付加的な治療利益となる可能性を示唆するものである。様々なPARPアイソフォームによって修飾された病態生理学的及び生理学的工程が大規模な研究の対象であるが(Ame et al.,2004)、一番特徴的なこのファミリーのメンバーであり、腫瘍学での治療上の標的薬剤を発見する試みの主な焦点は、PARP−1である。
【0007】
PARPは、転写レベルでのタンパク質発現、複製及び分化、テロメアーゼ活性、及び細胞骨格組織化を含む、たくさんの異なる生物学的工程の調節において活性である。しかしながら、化学/放射線増感剤としてPARP阻害剤を使用する目的であるDNA修復及びゲノム整合性の維持において、PARPは重要な役割を担っている(Smith,2001)。この役割は、イオン化放射線或いはアルキル化剤での治療に曝された場合の塩基切除修復の遅延及び高頻度の姉妹染色体交換を示す、PARP−1欠損細胞の使用を介して示される。更に、高レベルのイオン化放射線及びアルキル化剤は、野生型マウスと比較してPARP−1欠損マウスにおいてより高い死亡率を誘発する(Smith,2001;Virag&Szabo,2002)。
【0008】
PARPファミリーのメンバーの間で、PARP−1(及びPARP−2)は特に、イオン化放射線によって直接的に、或いはメチル化剤、トポイソメラーゼI阻害剤、及びシスプラチン及びブレオマイシンなどの他の化学療法剤によるDNA破壊の酵素的修復によって間接的に引き起こされたDNA破壊の修復によって活性化され複製される(Griffin et al.,1998;Delany et al.,2000;Tentori et al.,2002;de Murcia et al.,1997)。PARP−1は細胞生存及び亜致死性大量DNA損傷後の修復において重要な役割を担うことを示す実質的な生化学的及び遺伝学的証拠が存在する。さらに、PARP−1ノックアウトマウスによって例示さるように、DNA損傷が存在しないPARP−1機能は、細胞生存に対して重要ではなく、これにより、PARP−1の阻害が化学及び/若しくは放射線療法での使用に対して潜在的な実行可能な治療戦略となる(Delany et al.,2000;Burkle et al.,1993)。
【0009】
3−アミノベンズアミド、ニコチンアミド、及び関連誘導体などのPARP−1阻害剤の初期世代は、in vitro及びin vivoでのヒト及びマウス腫瘍モデルにおいて放射線、ブレオマイシン、CPT、シスプラチン及びTMZのin vitro及びin vivo細胞障害性活性の両者を増強した。疑いの余地のない抗腫瘍効果を与えることを阻止するような、これらの化合物の力価、選択性及び送達能力における本来の制限は、特にこれらの分子の非特異的活性に対するPARP−1の阻害に対してin vitro及びin vivoで観察された(Griffin et al.,1998;Griffin et al.,1995;Masuntani et al.,2000;Kato et al.,1988)。これらの課題は、様々なベンズイミダゾール−4−カルボキシアミド及びキナゾリン−4−[3H]−オン誘導体を含む、より強力で選択的なPARP−1阻害剤の構造クラスの開発において影響を及ぼした。in vitro及びin vivo解析によって、これらの化合物は、ヒト及びマウス腫瘍モデル両者を用いた化学療法剤の効果を増強可能であることが明らかになった(Griffin et al.,1998;Bowman, et al.,1998;Bowman et al.,2001;Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Liu,et al.,1999)。
【0010】
2001年11月15日付け公開のPCT公報第WO2001085686号では、PARP阻害活性を有するカルバゾール化合物が開示されている。
【0011】
PARPに対する高い選択性、高い力価、改善された送達能力、及び改善された耐用性特性を有する、腫瘍の治療に対する放射線増感薬剤としてのPARP阻害剤を発見し開発する必要性が存在する。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Curtin,NJ.(2005)PARP inhibitors for cancer therapy Expert Rev Molec Med 4:1−20.
【非特許文献2】Griffin,R et al.(1998)Resistance−modifying agents.5.Synthesis and biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(ADP−ribose)polymerase(PARP)J.Med.Chem.,42:5247−5256.
【非特許文献3】Bowman,KJ,et al.(1998)Potentiation of anti−cancer agent cytotoxicity by the potent poly(ADP−ribose)polymerase inhibitors NU 1025 and NU 1064 Br.J,Cancer 78:1269−1277.
【非特許文献4】Bowman,KJ,et al(2001)Differential effects of the poly(ADP−ribose)polymerase inhibitor NU 1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro Br.J.Cancer 84:106−112.
【非特許文献5】Chen&Pan(1988)Potentiation of antitumor activity of cisplatin in mice by 3−aminobenzamide and nicotinamide Cancer Chemoth.Pharmacol.22:303−307.
【非特許文献6】Delaney,CA et al.(2000)Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose)polymerase inhibitors in a panel of human tumor lines Clin.Cancer Res.6:2860−2867.
【非特許文献7】Griffin R et al.,(1995)The role of inhibitors of poly(ADP−ribose)polymerase as resistance−modifying agents in cancer therapy Biochimie 11:408−422.
【非特許文献8】Liu,L,et al.(1999)Pharmacologic disruption of base excision repair sensitizes mismatch repair−deficient and−proficient colon cancer cells to methylating agents Clin.Cancer Res.5:2908−2917.
【非特許文献9】Tentori,L.et al.(2002)Combined treatment with temozolomide and poly(ADP−ribose)polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site Blood 15:2241−2244.
【非特許文献10】Baldwin J.(2002)FDA evaluating oxaliplatin for advance colorectal cancer treatment J.Natl.Cancer Inst.94:1191−1193,2002.
【非特許文献11】Jagtap P and Szabo C(2005).Poly(ADP−Ribose)polymerase and the therapeutic effects of its inhibitors.Nature Rev Drug Disc 4:421−440.
【非特許文献12】Ame JC,et al(2004).The PARP superfamily.Bioessays 26:882−893.
【非特許文献13】Smith,S.(2001)The World According to PARP Trends Biochm.Sci.26:174−179.
【非特許文献14】Virag,L.and Szabo,C.(2002)The therapeutic potential of poly(ADP−ribose)polymerase inhibitors Pharmacol.Rev.54:375−429.
【非特許文献15】de Murcia,JM,et al.(1997)Requirement of poly(ADP−ribose)polymerase in recovery from DNA damage in mice and cells Proc Natl Acad Sci USA 94:7303−7307.
【非特許文献16】Masuntani,M,et al.(2000)The response of PARP knockout mice against DNA damaging agents Mutat.Res.462:159−166.
【非特許文献17】Kato,T et al.(1988)Enhancement of bleomycin activity by 3−aminobenzamide,a poly(ADP−ribose)synthesis inhibitor,in vitro and in vivo Anticancer Res.8:239−244.
【非特許文献18】Smith,L.et al.(2005)The novel poly(ADP−Ribose)polymerase inhibitor,AG14361,sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks Cl Cancer Res.11:8449−8457.
【発明の概要】
【課題を解決するための手段】
【0013】
本発明は、PARP−1のin vivo阻害によって腫瘍において放射線増感を引き起こすために4−メトキシ−カルバゾールを用いた方法を提供する。前記方法は、化学式(Ia)の4−メトキシ−カルバゾール、
【化2】
【0014】
そのプロドラッグ、好ましくはそのマンニッヒ塩基プロドラッグを有し、可溶性及び安定性を提供し、活性薬剤である7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン(trindene)−4,6−ジオンのin vivo送達を助けるものである。
【0015】
本発明はさらに、化学式(I)であって、
【0016】
【化3】
【0017】
式中、Xは、本明細書で定義されるように、H或いはプロドラッグ部位である、放射線増感剤或いは薬学的に許容可能なその塩形態を癌を患った哺乳動物へ投与する工程と、
前記哺乳動物組織へイオン化放射線を適用する工程と、によって癌を治療する方法を提供するものである。
【0018】
本発明の別の対象は、本発明の化合物を有する薬学的組成物を提供することであり、ここにおいて前記組成物は、1若しくはそれ以上の薬学的に許容可能な賦形剤及び治療上有効な量の少なくとも1つの本発明の化合物、或いはその薬学的に許容可能な塩或いはエステル形態を有するものである。
【0019】
本発明の別の対象は、化学式(II)の化合物、
【0020】
【化4】
【0021】
或いはその薬学的に許容可能な塩形態を提供するものである。
【0022】
別の実施形態において、本発明は、癌の治療に対する薬物を製造するための化学式(I)の化合物の使用を提供するものである。
【0023】
本発明のこれら及び他の対象、特性及び利点は、本開示の以下の詳細な説明において説明される。
【図面の簡単な説明】
【0024】
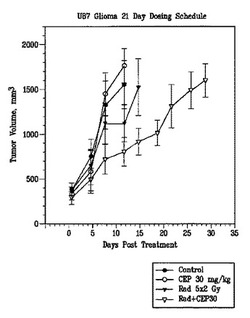
【図1】図1は、腫瘍の増殖遅延に対する、放射線耐性U87MG神経膠芽細胞腫異種移植片を用いた、放射線との組み合わせにおけるマンニッヒ塩基プロドラッグの効果を示したものである。
【図2】図2は、放射線療法或いはプロドラッグのみの比較投与計画によって達成された療法より強力な併用療法の効果の程度を示したものである。
【図3】図3は、ヌードマウスにおけるU87MGヒト神経膠芽細胞腫異所性移植片に対する実施例7の放射線増感効果を示したものである(非最適化スケジュール)。
【図4】図4は、本発明の観点内の化合物及びそれの前駆体を含む合成模式図を示したものである。
【図5】図5は、ヌードマウスにおけるU87MGヒト神経膠芽細胞腫異所性移植片に対する経口的に投与された実施例7の放射線増感効果を示したものである。
【発明を実施するための形態】
【0025】
第一の実施形態において、本発明は、化学式(I)であって、
【化5】
【0026】
式中、XはH或いはプロドラッグ部位である、放射線増感剤或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程、及び前記哺乳動物にイオン化放射線を適用させる工程によって癌を治療する方法を提供する。
【0027】
好ましい実施形態において、前記放射線増感剤は、前記組織内に或いは近接して存在するものであり、これは、局所的治療効果への前記適用イオン化放射線の転換効率を増強するものである。
【0028】
好ましい実施形態において、前記放射線増感剤は、放射線感受性癌細胞に対して有効な量で存在するものである。
【0029】
好ましい実施形態において、前記組織のイオン化放射線は、前記細胞を破壊するのに有効な放射線線量で実行されるものである。
【0030】
好ましい実施形態において、前記イオン化放射線は、既知の癌タイプに対して臨床的に許容可能な或いは推奨される放射線療法プロトコールで行われるものである。
【0031】
好ましい実施形態において、前記癌は悪性である。
【0032】
好ましい実施形態において、前記癌は良性である。
【0033】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、−C(=O)R4から成る群から選択されるものであり、ここにおいて、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【0034】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2であり、R1は、H或いはC1−4アルキルであり;R2は、H或いはC1−4アルキルであり、或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【0035】
好ましい実施形態において、前記プロドラッグ部位は、マンニッヒ塩基である。
【0036】
好ましい実施形態において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチル−、モルフォリン−4−イルメチル−、及び5−ジエチルアミノメチル−から選択されるものである。
【0037】
好ましい実施形態において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチルである。
【0038】
好ましい実施形態において、前記投与の経路は、静脈内、皮下、経口或いは腹腔内である。
【0039】
好ましい実施形態において、前記投与の経路は、静脈内である。
【0040】
好ましい実施形態において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌から選択されるものである。
【0041】
好ましい実施形態において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【0042】
好ましい実施形態において、本発明は、化学式7−メチル−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤を投与する工程によって癌を治療する方法を提供するものである。
【0043】
好ましい実施形態において、本発明は、化学式7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤を投与する工程によって癌を治療する方法を提供するものである。
【0044】
第二の実施形態において、本発明は、放射線感受性癌細胞に対して薬学的組成物を提供するものであり、この組成物は、放射線増感量の化学式(I)の化合物、
【化6】
【0045】
或いはその薬学的に許容可能な塩形態であって、ここにおいて、Xは、H或いはプロドラッグ部位である化合物或いは塩形態、
及び薬学的に許容可能な担体を有するものである。
【0046】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、−C(=O)R4から成る群から選択されるものであり、ここにおいて、
R1は、H或いはC1−4アルキル、
R2は、H或いはC1−4アルキル、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【0047】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【0048】
好ましい実施形態において、前記化合物は、
【化7】
【0049】
或いはその薬学的に許容可能な塩形態である。
【0050】
好ましい実施形態において、前記化合物は、
【化8】
【0051】
或いはその薬学的に許容可能な塩形態である。
【0052】
第三の実施形態において、本発明は、化学式(II)の化合物、
【化9】
【0053】
或いはその薬学的に許容可能な塩形態を提供するものである。
【0054】
第四の実施形態において、本発明は、癌を治療するための薬剤を製造するための化学式(I)の化合物の使用を提供するものである。
【0055】
好ましい実施形態において、本発明は、癌を治療するための薬剤を製造するための化学式(II)の化合物の使用を提供するものである。
【0056】
本発明の特定の特性は、明確にするために別々の実施形態の文脈において記載しているが、さらに単一の実施形態の組み合わせでも提供され得ることは理解される。反対に、本発明の様々な特性は、簡潔化するために単一の実施形態の文脈において記載しているが、別々に、或いはあらゆる適切なサブコンビネーションにおいても提供され得る。
【0057】
本明細書に含まれる以下の用語及び表現は、以下のように定義される。
【0058】
本明細書で用いられたように、「約」という用語は、特定値の±10%の範囲の値を意味する。例えば、「約50mg」という文章は、50の±10%、若しくは45から55mgを含む。
【0059】
本明細書で用いられる「アルキル」という用語は、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル及びtert−ブチルなどの、1から4個の炭素原子を持つ直鎖或いは分岐アルキル基を意味する。「C1−4アルキル」などの記号は、1から4個の炭素原子を含むアルキル遊離基を意味する。
【0060】
本明細書で用いられる「アミノ酸」という用語は、アミノ基及びカルボキシル基の両者を含む分子を意味する。それは、カルボキシル基に隣接する炭素上にアミノ機能基を生じるカルボキシル酸として本分野の当業者にはよく知られた「α−アミノ酸」を含む。アミノ酸は、天然由来或いは非天然由来であり得る。「天然由来アミノ酸」は、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン酸、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシン及びバリンを含む。
【0061】
本明細書で用いられる「ヘテロシクリル」という用語は、炭素原子、及び少なくともO、N或いはSから選択されたヘテロ原子を含む5或いは6員環基を意味し、ここにおいて前記ヘテロシクリル基は、飽和或いは不飽和されており、前記ヘテロシクリル基は、置換或いは非置換されているものである。窒素及び硫黄へテロ原子は任意に酸化されている。ヘテロシクリル基の例としては、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル、ピペラジニル、及びメチルピペラジニルが含まれる。
【0062】
本明細書で用いられたように、「哺乳動物」という用語は、本明細書で記載された1若しくはそれ以上の疾患及び症状に苦しめられている、若しくは苦しめられる可能性がある、マウス、ラット、ネコ、イヌ、サル或いはヒト、好ましくはヒト或いはヒト小児を含む温血動物を意味する。
【0063】
本明細書に用いられる「薬学的に許容可能な」化合物とは、適切な利益/リスク比に釣り合った、過度の有害な副作用(毒性、刺激及びアレルギー反応など)なしに、ヒト及び/若しくは動物に使用するのに適切なものである。
【0064】
本明細書で用いられる「安全且つ有効な量」という用語は、本発明のやり方で使用した場合、適切な利益/リスク比に釣り合った、過度の有害な副作用(毒性、刺激及びアレルギー反応など)なしに、望ましい治療反応を生むのに十分な成分の量を意味する。「治療上有効な量」は、望ましい治療反応を生むのに有効な本発明の化合物の量を意味している。例えば、癌、肉腫或いはリンパ腫の増殖或いはそれらを引き起こすのを遅延するのに有効な量、若しくは前記癌を収縮させる或いは転移を阻止するのに有効な量である。特異的に安全且つ有効な量、或いは治療上有効な量は、治療される特定の症状、患者の身体的状態、治療される哺乳動物或いは動物のタイプ、治療の持続期間、併用治療(もしあれば)の特性、及び使用された特異的処方、及び前記化合物或いはその誘導体の構造などの因子によって変わるであろう。
【0065】
本発明において、「イオン化放射線」という用語は、十分なエネルギーを持つ、若しくは核相互作用を介して十分なエネルギーを産生しイオン化(電子の獲得或いは損失)を産生できる粒子或いは光子を有する放射線を意味する。例示的で好ましいイオン化放射線は、X−放射線である。標的組織或いは細胞へX放射線を送達する手段は、本分野ではよく知られている。既知の細胞に必要とされるイオン化放射線の量は一般的に、その細胞の特性に依存している。放射線の有効な量を決定する手段は、本分野ではよく知られている。本明細書で使用されたように、イオン化放射線の「有効な線量」という用語は、本発明の化合物との組み合わせで与えられた場合、細胞障害或いは細胞死の増加を産生するイオン化放射線の線量を意味するものである。
【0066】
X線の投与量範囲は、長期間(3から4週間)の場合一日量で50から200レントゲンから、単一量で2000から6000レントゲンまでの範囲である。放射性同位元素に対する投与範囲は幅広く変わり、その同位元素の半減期、放射される放射線の強度及びタイプ、及び新生物細胞による取り込みに依存する。
【0067】
組織へ放射線を送達するあらゆる適切な手段は、本発明において使用される。組織へ放射線を送達する一般的な手段は、治療される体の外部にあるイオン化放射線源によるものである。組織へ放射線を送達する代替方法としては、例えばまず腫瘍の抗原に免疫反応を生じる放射線標識抗体をin vivoで送達する工程と、次に前記腫瘍へ有効な量の前記放射線標識抗体をin vivoで送達する工程とを含む。加えて、放射性同位元素は、組織或いは細胞へイオン化放射線を送達するために使用される。付加的に、前記放射線は、放射線類似薬剤の手段によって送達される。本明細書で用いられたように、「放射線類似薬剤」は、放射線の適用なしに放射線療法と同じタイプの細胞障害を生じる、例えばメルファランなどの化学療法剤である。
【0068】
本明細書で用いられれる「プロドラッグ部位」という用語は、前記プロドラッグが、生理学的条件下で、多数の化学的及び生物学的メカニズムによって生物学的活性薬剤へ変換され得るということを意味している。1実施形態において、生物学的活性薬剤への前記プロドラッグの変換は、前記プロドラッグ部位は化学的に或いは酵素学的に水で加水分解するという条件で、前記プロドラッグ部位の加水分解によって達成され得る。水との反応は一般的に、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。本発明の別の観点では、前記プロドラッグ部位の還元によって前記生物学的活性薬剤への前記プロドラッグの変換が提供される。この実施形態において一般的に、前記プロドラッグ部位は、生理学的条件下で、還元酵素工程の存在下において還元されるものである。前記還元は好ましくは、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。別の実施形態において、前記生物学的活性薬剤への前記プロドラッグの変換はさらに、前記プロドラッグ部位の酸化によっても達成され得る。この実施形態において一般的に、前記プロドラッグ部位は、生理学的条件下で、酸化酵素工程の存在下において酸化されるものである。前記酸化は好ましくは、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。本発明のさらなる態様では、前記プロドラッグ部位の除去によって前記生物学的活性薬剤への前記プロドラッグの変換が含まれる。一般的に言えば、この実施形態において、前記プロドラッグ部位は、生理学的条件下で、化学的或いは生物学的反応によって除去される。前記除去は、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。もちろん、本発明のあらゆるプロドラッグ化合物は、上述のメカニズムのあらゆる組み合わせを受け、前記プロドラッグを生物学的活性薬剤へと変換する。例えば、特定の化合物は、加水分解、酸化、除去、及び還元を受け、前記プロドラッグを生物学的活性化合物へと変換するであろう。同等に、特定の化合物は、これらメカニズムの1つのみを受け、前記プロドラッグを生物学的活性化合物へと変換するであろう。
【0069】
本明細書で用いられる「癌」とは、癌腫及び肉腫を含む、哺乳動物において見出された癌或いは新生物、若しくは悪性或いは良性腫瘍のあらゆるタイプを意味する。癌の例としては、脳の癌、乳癌、膵癌、子宮頸癌、大腸癌、頭頸部癌、腎癌、肺癌、肺非小細胞癌、メラノーマ、中皮腫、卵巣癌、肉腫、胃癌、子宮癌及び髄芽細胞腫である。
【0070】
「白血病」という用語は、造血器官の進行性、悪性疾患を広く意味し、一般的に血中及び骨髄中での白血球及びそれらの前駆体の歪んだ増殖及び発生によって特徴付けされている。白血病は一般的に、(1)疾患の持続期間及び特徴−急性或いは慢性、(2)関連した細胞の種類;骨髄球系(骨髄性)、リンパ球系(リンパ性)、或いは単球性、及び(3)血中の異常細胞の数が増加する或いは増加しない−白血病性或いは無白血病性(亜白血性)、という基礎に基づいて臨床的に分類されている。P388白血病モデルは、in vivo抗白血病活性を予想するものとして広く許容かせている。P388アッセイにおいてポジティブであった化合物は一般的に治療される白血病のタイプに関わらず、in vivoである程度の抗白血病活性を示すであろうと信じられている。従って、本発明は、白血病を治療する方法を含み、好ましくは急性非リンパ球性白血病、慢性リンパ球性白血病、急性顆粒球性白血病、慢性顆粒球性白血病、急性然骨髄球性白血病、成人T−細胞白血病、亜白血性白血病、白血性白血病(leukocythemic leukemia)、好塩基球性白血病、芽球細胞白血病、ウシ白血病、慢性骨髄球性白血病、皮膚白血病、胚性白血病、好酸球性白血病、グロス白血病、ヘアリー細胞白血病、血球母細胞白血病、血球芽性白血球、組織球性白血病、幹細胞白血病、急性単球性白血病、白血球減少性白血病、リンパ性白血病、リンパ芽球性白血病、リンパ球性白血病、リンパ向性白血病、リンパ様白血病、リンパ肉腫細胞白血病、マスト細胞白血病、巨核球性白血病、微小骨髄芽球性白血病、単球性白血病、骨髄芽球性白血病、骨髄球性白血病、骨髄性白血病、骨髄単球性白血病、ネーゲリ型白血病、形質細胞白血病、形質細胞性白血病、前骨髄球性白血病、リーダー細胞(Rieder cell)白血病、シリング白血病、幹細胞白血病、亜白血性白血病、及び未分化細胞白血病を治療する方法を含む。
【0071】
「肉腫」という用語は一般的に、胚性結合組織のような物質を作り上げる腫瘍を意味し、一般的に原線維或いは均一な物質に埋包された、密接に詰められた細胞から成る。4−メトキシ−カルバゾール及び放射線療法で治療され得る肉腫には、軟骨肉腫、胆管肉腫、線維肉腫、リンパ肉腫、メラノ肉腫、粘液肉腫、骨肉腫、アベメシー肉腫、脂肪(adipose)肉腫、脂肪肉腫(liposarcoma)、胞状軟部肉腫、エナメル芽細胞肉腫、ブドウ状肉腫、緑色肉腫、絨毛肉腫、胎児性肉腫、ウィルムス腫瘍肉腫、子宮内膜肉腫、間質性肉腫、ユーイング肉腫、筋膜肉腫、線維芽細胞肉腫、巨細胞肉腫、顆粒球性肉腫、ホジキン肉腫、特発性多発性色素性出血性肉腫、B細胞の免疫芽球性肉腫、リンパ腫、T細胞の免疫芽球性肉腫、イエンセン肉腫、カポジ肉腫、クッパー細胞肉腫、血管肉腫、白血肉腫、悪性間葉細胞肉腫、傍骨肉腫、細網肉腫、ラウス肉腫、漿液嚢胞性肉腫、軟部組織肉腫、滑膜肉腫、及び毛細血管拡張性肉腫が含まれる。
【0072】
「メラノーマ(黒色腫)」という用語は、皮膚及び他の器官のメラニン細胞システムから生じる腫瘍を意味する。4−メトキシ−カルバゾール及び放射線療法で治療され得るメラノーマには、例えば、末端性黒子性黒色腫、無色素性メラノーマ、良性若年性メラノーマ、クラウドマン黒色腫、S91メラノーマ、ハーディング−パッセー黒色腫、若年性黒色腫、悪性黒子型黒色腫、悪性黒色腫、結節性黒色腫、爪下黒色腫、及び表在性黒色腫が含まれる。
【0073】
「癌腫」という用語は、周囲組織へ浸潤し転移する傾向がある、上皮細胞から作られた悪性新生増植物を意味する。4−メトキシ−カルバゾール及び放射線療法で治療され得る癌腫の例としては、例えば、細葉細胞癌、腺房細胞癌、腺嚢癌、腺様嚢胞癌、乳癌、腺腫様癌、副腎皮質の癌腫、胞巣状癌、肺胞上皮細胞癌、基底細胞癌、好塩基球癌腫、類基底細胞癌、基底扁平上皮癌、膀胱癌、細気管支肺胞上皮癌、細気管支癌、気管支原性肺癌、脳回転様(cerebriform)癌、胆管細胞癌、絨毛がん、コロイド腺癌、結腸直腸癌、子宮頸癌、面疱癌、子宮体癌、篩状癌、鎧状癌、皮膚癌、円柱癌、円柱細胞癌、管癌、硬性癌、胎児性癌、脳様癌、類表皮癌、腺上皮癌、外方増殖性癌、潰瘍性癌、線維性癌、胃癌、膠様癌(gelatiniform carcinoma)、膠様癌(gelatinous carcinoma)、巨細胞癌、巨大細胞癌、腺癌、顆粒膜細胞癌、毛母癌、血様癌(hematoid carcinoma)、肝細胞癌、ハースル細胞癌、硝子状癌、hypemephroid癌、小児性胎児性癌、上皮内癌(carcinoma in situ)、表皮内癌、上皮内癌、Krompecher’s癌、クルチツキー(Kulchitzky)細胞癌、大細胞癌、レンズ状癌、レンズ状癌(carcinom lenticulare)、脂肪腫様癌、肺癌、リンパ上皮癌、髄様癌(carcinoma medullare)、髄様癌、黒色癌、軟性癌(carcinoma molle)、粘液癌、粘液性癌(carcinoma muciparum)、粘液細胞癌、粘表皮癌、粘膜癌(carcinoma mucosum)、粘膜癌(mucous carcinoma)、粘液腫状癌(carcinoma myxomatodes)、上咽頭癌、燕麦細胞癌、骨化性癌、類骨癌、卵巣癌、膵臓癌、前立腺癌、乳頭癌、門脈周囲癌、前浸潤癌、有棘細胞癌、糊性癌(pultaceous carcinoma)、腎臓の腎細胞癌、予備細胞癌(reserve cell carcinoma)、肉腫状癌(carcinoma sarcomatodes)、シュナイダー癌、スキルス癌、陰嚢癌(carcinoma scroti)、印環細胞癌、単純癌(carcinoma simplex)、小細胞癌、ソレノイド癌(solanoid carcinoma)、球状細胞癌、紡錘細胞癌、海綿細胞癌(carcinom spongiosum)、扁平上皮癌、扁平上皮細胞癌、ストリング癌(string carcinoma)、末梢血管拡張性癌、末梢血管拡張様癌(carcinoma telangiectodes)、精巣癌、移行細胞癌、甲状腺癌、結節性癌(carcunoma tuberosum)、結節性癌、疣状癌、及び絨毛様癌(carcunoma villosum)が含まれる。
【0074】
本発明に従った化合物で治療され得る好ましい癌としては、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌を含む。
【0075】
本発明に従った化合物で治療され得るより好ましい癌としては、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【0076】
本明細書で用いられる「4−メトキシ−カルバゾール」という用語は、以下の化学式、
【化10】
【0077】
を有する化学物、若しくはその薬学的に許容可能な塩形態を意味し、ここにおいて、XはH或いはプロドラッグ部位である。
【0078】
本発明の化合物は、プロドラッグ部位を含む。本発明によって考えられるプロドラッグ部位の例としては、リン酸エステル、アミノ酸エステル、アミノ酸アミド、アミノアルキルカルバミン酸、アルコキシアルキルカルバミン酸、ヒドロキシアルキルカルバミン酸、アルコキシアルキルエステル、ヒドロキシアルキルエステル、安息香酸エステル、ニコチンエステル、ピペラジン酢酸、モルフォリニル酢酸、及びマンニッヒ塩基から選択され得る。本発明によって考えられるプロドラッグ部位の例としては、
【化11】
【0079】
【化12】
【0080】
から選択され得る。好ましいプロドラッグ部位は、マンニッヒ塩基である。好ましいマンニッヒ塩基には、これに限定されるものではないが、4−メチル−ピペラジン−1−イルメチル−、モルフォリニル−4−イルメチル−、及びジエチルアミノメチル−が含まれる。
【0081】
本発明の化合物はさらに、薬理学的に許容可能な塩、水和物、溶媒和化合物、或いは代謝産物の形態もとる。薬理学的に許容可能な塩類には、これに限定されるものではないが、塩酸、臭化水素酸、硫酸、リン酸、メタンスルホン酸、エタンスルホン酸、リンゴ酸、酢酸、シュウ酸、酒石酸、クエン酸、乳酸、フマル酸、スクシニル酸、マレイン酸、サリチル酸、安息香酸、フェニル酢酸、マンデル酸、アスコルビン酸、グルコン酸、及びそれらと同等物を含む、無機及び有機酸の基礎塩類が含まれる。本発明の化合物がカルボキシ基などの酸性機能を含む場合、カルボキシ基に対して適切な薬学的に許容可能な陽イオン対は、本分野の当業者にはよく知られており、アルカリ、アルカリ土類、アンモニウム、四級アンモニウム陽イオン及びそれらと同等物を含む。本発明の化合物が薬理学的に許容可能な塩の形態をとる場合、前記塩形態はin situ或いは単離固形として生成されるということが本発明によって考えられる。
【0082】
本発明の化合物は、特に上記の塩類の形態において、薬学的に許容可能な担体として働き、例えば注射、懸濁液、乳剤、錠剤、カプセル及び軟膏などの形態において薬剤を投与可能にする、本分野ではよく知られている様々な賦形剤溶媒及び/若しくはアジュバントと組み合わされる。記載された化合物の放射線増感量を含むこれらの薬学的組成物は、低酸素腫瘍細胞の放射線増感を生じるあらゆる許容可能な手段によって投与される。温血動物に対して、及び特に放射線療法を受けるヒトにおいて、投与は経口、皮下、腹腔内、或いは静脈内に行われ得る。低酸素腫瘍細胞を破壊するために、放射線増感剤を含む前記薬学的組成物は、前記低酸素腫瘍細胞を放射線増感するのに有効な量で投与される。投与された特異的投与量は、患者の一般的健康状態及び体調、さらに年齢及び体重、患者疾患の段階、及びあらゆる併用療法の存在などの因子に依存するであろう。
【0083】
有効な量を投与する方法も、治療される疾患或いは病気に依存して変わる。適切な担体、付加的癌阻害化合物或いは化合物類、若しくは適用を容易にする希釈剤と処方される、4−メトキシ−カルバゾールの静脈内適用による治療は、温血動物へ前記化合物を投与する好ましい方法であろう。
【0084】
本明細書に記載された化合物は、純粋な形態で、他の有効成分との組み合わせで、若しくは薬学的に許容可能な非毒性賦形剤或いは担体との組み合わせで投与される。経口組成物は一般的に、不活性希釈担体或いは食用担体を含む。薬学的に適用性な結合剤、及び/若しくはアジュバント物質は、前記組成物の一部として含まれ得る。錠剤、ピル、カプセル、トローチ及びそれらと同等物は、あらゆる以下の成分或いは同様の性質の化合物:微結晶性セルロース、ガムトラガント或いはゼラチンなどの結合剤;スターチ或いはラクトースなどの賦形剤、アルギニン酸、プリモゲル(Primogel)或いはコーンスターチなどの分散剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド二酸化ケイ素などの流動促進剤;スクロース或いはサッカリンなどの甘味料;或いはペパーミント、サリチル酸メチル、或いはオレンジ香料などの香味料、を含む。前記投与ユニット形態がカプセルの場合、上記タイプの物質に加えて、脂肪油などの液体担体を含むことができる。加えて、投与ユニット形態は、例えば、糖のコーティング、セラック或いは腸溶剤などの、前記投薬ユニットの物理的形態を修飾する様々な他の物質を含むことができる。さらに、シロップは、前記有効化合物に加えて、甘味料としてのスクロース、及び特定の保存剤、色素、着色料及び香味料を含む。
【0085】
患者へ投与された化合物の量は、治療される悪性新生物を放射線増感するのに十分なものであるが、毒性作用を誘発する量以下である。この量は、腫瘍のタイプ、治療される患者の種類、意図された指標投与量、及び患者の体重或いは体表面量に依存するであろう。放射線は、様々な異なる分割計画でヒトへ投与される、すなわち総放射線線量が数日から数週間の期間で分割して与えられる。これらは、毎日(すなわち一週間で5回)線量から6週間まで、週1回線量から4から6週間までと変わる可能性が最も高い。
【0086】
患者へ投与された放射線増感化合物の量は、放射線治療前、放射線治療中、或いは放射線治療後に投与される。しかしながら、本発明の化合物は放射線治療前に投与されることが好ましい。
【0087】
低酸素腫瘍細胞への前記放射線増感組成物の投与、及び前記低酸素腫瘍細胞の放射線増感を増強するのに十分な時間間隔での継代後、前記低酸素腫瘍細胞は、前記低酸素腫瘍細胞を破壊するのに有効な放射線の線量で照射される。一般的に、患者は、1日当たり約2Gyの放射線線量を5日間受けるであろう。一般的に、前記患者は7から8週間で約70から約80Gyの総放射線線量を受け、それぞれ個々の放射線線量は、放射線増感剤の投与後約1から4時間以内に投与される。そのような一連の放射線増感治療及び照射は、寛解するのに、及び任意に悪性腫瘍の転移を減少或いは除去するのに必要な回数繰り返される。しかしながら、毎日の放射線線量及び総放射線線量は、患者の腫瘍タイプ、治療プロトコール、及び身体的状態に依存して変わるであろうことは本分野の当業者によって理解される。例えば、本発明の化合物の毎日の線量は、特別に限定されるものではないが、患者の年齢、癌、体重及び現在の治療プロトコール及び/若しくは薬物療法で変えることができる。付加的に、本発明の化合物は、放射線増感剤として有用であり、放射線に曝露される前に、1若しくはそれ以上の用量で、すなわち1から複数用量で投与され得る。
【0088】
ヌードマウスにおいてU87MG放射線耐性異種移植片を用いた初期放射線増感研究(非最適化用量スケジュール)
細胞の照射はチェックポイント停止を誘導し、これは、DNA障害の修復を促進する活性化PARPによる、細胞のDNA障害の修復を可能にする。仮定で、単一線量或いは分割放射線との組み合わせでのPARP阻害剤の投与は、DNA障害を修復するための照射細胞の能力を減少し、細胞殺傷を増加するであろう。従って、PARP阻害剤は、分割放射線と相乗的に作用し、腫瘍増殖遅延を増加しなくてはならない。実施例7/実施例6におけるこの仮説の初期テストは、ヌードマウスにおける放射線耐性U87MGヒト神経膠芽腫異種移植片で実行した。図3に示されたように、実施例7単独の投与、放射線単独、及び放射線(実施例6の100mg/kg用量当量、s.c.、放射線のqd2日前、及び3日間の7.5Gy放射線との組み合わせ)との組み合わせの実施例7は、確立された腫瘍を有するマウスにおいて実行した。単一剤として投与された実施例7は、腫瘍増殖に影響を及ぼさなかった。溶媒或いは実施例7で治療された腫瘍は、それぞれ10.0日或いは9.6日(コントロールに対して、p=0.798)で2000mm3の腫瘍容積に達した。放射線単独の投与は、2000mm3に達するための時間が16.1日と増加し、腫瘍増殖遅延(TGD)も6.1日(コントロールに対して、p=0.033)と増加した。対照的に、放射線療法との実施例7の投与は、2000mm3に達するための腫瘍に対する時間が24.8日と増加し、これは14.8日TGDに対応する。併用療法の効果の大きさは、実施例7単独(p=0.001)或いは放射線のみ(p=0.006)の比較計画によって見られたものより強く、これは、実施例7は真の放射線増感剤の特性を示すことを示している。有効性と関連したCmaxにおける実施例6の血漿レベル(100mg/kg実施例7での)は、23μMであり、これは化学−感作研究におけるこの用量で達成されるものに匹敵する。
【0089】
ヌードマウスにおいてU87MG放射線耐性異種移植片を用いた実施例7及び臨床的に関連した分割放射線療法投与スケジュールでの放射線増感研究
次の放射線増感研究は、臨床的に関連した分割放射線療法スケジュール(2Gyを5日間)との組み合わせで実施例7(30及び100mg/kg、s.c.)を評価することである。実施例7は、5日間の放射線後に0.5時間投与し、放射線計画後16日間続けた実施例7の投与を完了した。この投与計画に対する論理的根拠は、放射線障害からのDNA修復が照射後10から12日で起こり、従って、実施例7の連続投与及びPARP活性の修飾は、分割放射線と相乗的に作用し放射線増感及び腫瘍増殖遅延を増加するであろう、細胞周期停止及びDNA修復時間に影響を及ぼしている、という事実に基づいていた。図1及び2に示されたように、放射線単独の投与(2Gyを5日間)は、溶媒処理腫瘍と比較して2.5日のTGDを生じた。実施例7の投与(CEP 30;30mg/kg s.c.)は、TGDを15日まで増加し、放射線単独と比較して4倍増加し(p≦0.05);実施例7単独と比較して6倍増加した(p≦0.001)。放射線増感効果と関連したCmaxでの実施例6の血漿レベルは、5.5μMであった。分割放射線療法との実施例7の投与(100mg/kg、s.c.)は、有意な抗腫瘍効果を生じたが、80%死亡率は11日目で達成された。100mg/kg、s.c.でのCmaxにおける血漿レベルは21μMであり、これは化学−感作研究及び上述の初期放射線増感研究におけるこの用量で達成された照射レベルと一致している。
【0090】
これらのデータから、TGDのより大きな増加は、臨床的に関連した分割用量スケジュールを用いた、より低濃度の実施例7(CEP 30;実施例6の30mg/kg用量当量、s.c.、qdX21日間)で観察されたことが示された。加えて、実施例7(CEP 30;実施例6の30mg/kg用量当量、s.c.、qdX21日間)単独は、腫瘍増殖阻害に対して何も影響を及ぼさず、これは実施例7が「真の」放射線増感剤として作用することを示している。
【0091】
治療効果を評価するために、実施例7(実施例6の30及び100mg/kg用量当量、s.c.)プラス2Gy放射線X5日間は、骨髄及び空腸腺窩アッセイで評価し、実施例7が放射線誘導性正常組織(NT)毒性を増強するかどうか決定した。骨髄及び腸管粘膜の評価によって、実施例7(実施例6の30及び100mg/kg用量当量、s.c.)は、これらの組織において放射線毒性を増強しなかったことが明らかになった。この研究によって、CEP−9722は経口的に投与された場合、放射線増感効果を発揮することが示唆された。これらの組み合わせたデータによって、実施例7は、放射線耐性グリオーマモデルにおいて分割放射線療法の有効性を増加することによって、放射線増感剤としてより相加的に作用し、放射線誘導性NT毒性を増強しないことが示唆された。
【0092】
実施例
本発明の化合物は、これに限定されるのもではないが以下に記載された方法を含む本分野の当業者にはよく知られた多くの方法において、或いは標準技術を適用することによるこれら方法を修飾した方法を介して調合した。本発明に関連して開示された全ての工程は、ミリグラム、グラム、マルチグラム、キログラム、マルチキログラム或いは商工業スケールを含むあらゆるスケールで実行されると考えられる。
【0093】
本発明は、PARPの阻害剤として有用な、本明細書に記載された複環状化合物を調合する方法を扱う。前記方法は、4−メトキシインドールで開始する多段階合成から成る。特に、4−メトキシインドールAは、例えば、ブチルリチウム、二酸化炭素、t−ブチルリチウム、及びケトンBとともに連続的に処理し、2−置換4−メトキシインドール三級アルコールCを提供した。この三級アルコールは、例えば、塩酸或いはトルエンスルホン酸を用いた酸性条件下で除去し、置換2−ビニルインドールDを得た。限定されるものではないが、マレイミド(E)などの求ジエン体でのDのディールズ−アルダー環化付加反応によって、環化付加反応中間産物Fを得た。例えばパラジウム或いは白金などの触媒の存在下での酸素による、若しくはDDQ或いはテトラクロロキノンなどの酸化剤による前記環化付加反応中間産物の芳香環化によって、カルバゾールGを産生した。
アルキル化或いはアシル化剤でのGのさらなる処理によって、本発明のインドール−N−置換カルバゾール誘導体を得た。適切なプロドラッグ誘導体を選択及び調合するための従来の方法は、例えば、Prodrugs,Sloane,K.B.,Ed.;Marcel Dekker:New York,1992に記載されており、これはこの参照によってその全体が本明細書に組み込まれるものである。
【0094】
本発明の化合物はPARP阻害剤である。前記阻害剤の力価は、in vitro或いはin vivoでのPARP活性を測定することによってテストされ得る。好ましいアッセイは、[32P]NAD+からヒストン或いはPARP自身などのタンパク質受容体への放射線標識ADP−リボースユニットの転移をモニタリングすることである。PARPに対する日常的なアッセイは、Purnell and Whish,Biochem.J.1980,184,775に開示されており、これはこの参照によって本明細書に組み込まれるものである。
【実施例1】
【0095】
細胞株
U87MGヒト神経膠芽腫細胞は、1.5g/L炭酸水素ナトリウム、0.1nM非必須アミノ酸、10%ウシ胎児血清(FBS)を含む1.0nMピルビン酸ナトリウムと共に、商業的に利用可能な基礎培地(MEM)において培養した。
【実施例2】
【0096】
腫瘍細胞移植及び増殖
対数増殖細胞を播種し、商業的に利用可能な無胸腺NCRヌードマウスの右脇腹へ注入した((2X106)細胞/マウス)。200から400mm3の腫瘍を有する動物をサイズに基づいて適切な治療群へランダム化した(n=10)。腫瘍は、ノギスを用いて3から4日毎に測定した。腫瘍容積は、以下の公式:
V(mm3)=0.5236x長さ(mm)x幅(mm)[長さ(mm)+幅(mm)/2]
を用いて計算した。
【実施例3】
【0097】
方法:U87MGヒト神経膠芽腫細胞は、無胸腺NCR NUMマウスの右後肢へと皮下に(s.c.)注入し、平均腫瘍容積の200mm3へと増殖させた。放射線療法を受けたマウスは、100mg/kgケタミン+10mg/kgキシラジン(xylezine)、或いは37.5mg/kgケタミン+0.2mg/kgアセプロマジンをs.c.で照射する前に麻酔し、25から30分間鎮静させた。麻酔させたマウスは、過度の圧力なく、動物の体のサイズ及び形に合わせた可鍛性鉛遮蔽に位置付けした。その体は鉛によって遮蔽した。腫瘍を有する肢或いは曝露された腫瘍に適切な線量を照射した。腫瘍を照射した後、そのマウスは、麻酔から覚めるまでケージ或いは加温パッドへ戻した。実施例7は放射線(RT)後できるだけすぐ(30分以内)に与えた。図3:マウスは以下の治療群(n=10)へランダム化した:1)溶媒、2)放射線のみ(7.5Gyを3日間)、3)実施例7のみ(実施例6の100mg/kg用量当量、s.c.、5日間のQD)、及び4)実施例7プラス放射線。実施例7或いは溶媒は、1から5日目に、及び2、3及び4日目に放射線後30分にs.c.で投与した。データの解析は、時間及び治療の機能として腫瘍容積の10を底とする対数をモデルにした混合効果回帰を用いて行った。解析は、SAS8.3(SAS Institute Inc.,Cary,ノースカロライナ州)で実行した。図1及び2:マウスは、以下の治療群にランダム化し、投与した:1)溶媒、2)RT(5X2Gy)、3)RTプラス実施例7(30或いは100mg/kg、s.c.、実施例6の用量当量、qdx21日間)、或いは4)実施例7(30或いは100mg/kg、s.c.、実施例6の用量当量、qdx21日間)のみ。実施例7は1から21日目に投与し、RTは1から5日目に行った。全動物は、同日に測定した。個々の腫瘍容積測定は対数変換直線モデルをモデルにし、腫瘍が約2000mm3に到達する最適時間を測定した。一元ANOVA及び事後解析は、有意差を決定するために使用した。P値<0.05は有意差があると考えられた。
【0098】
結果:全群は、200mm3の同様のサイズの腫瘍の治療で開始した(P=0.83、0日目の比較群)。図3に示されたように、実施例7単独、放射線のみ、及び放射線との組み合わせでの実施例7(実施例6の100mg/kg用量当量s.c.放射線のqd2日前、及び7.5Gy放射線の組み合わせで3日間)の投与は、確立した腫瘍を有するマウスに行った。単剤として投与された実施例7は、腫瘍増殖に対して影響を及ぼさなかった。溶媒或いは実施例7のみで処理された腫瘍は、それぞれ10.0日或いは9.6日(P=0.798対コントロール)に2000mm3の腫瘍容積に達した。放射線単独の投与は、2000mm3に到達する時間が16.1日に延長され、腫瘍増殖遅延(TDG)も6.1日(P=0.033、対コントロール)に延長した。放射線療法との実施例7の併用療法は、腫瘍が2000mm3に到達する時間を24.8日に延長し、これは14.8日TGDに対応する。併用療法の有効性の程度は、実施例7のみ(P=0.001)或いは放射線のみ(P=0.006)の比較計画によって見られたものより強度であり、これは実施例7が真の放射線増感剤の特性を示すことを示唆するものである。図1及び2に示されたように、RTとの組み合わせでの実施例7の投与(CEP 30;実施例6の30mg/kg用量当量、s.c.)は、TGDを15日に延長(これは放射線のみ(P≦0.05)と比較して4倍増加);及び26日に延長(これは実施例7のみ(P≦0.001)と比較して6倍増加)した。分割放射線療法との実施例7(100mg/kg、s.c.)の投与は、有意な抗腫瘍効果を生じたが、80%死亡率は11日であった。これらのデータによって、TGDにおける有意な延長は、臨床的に関連する分割線量スケジュールを用いた実施例7(CEP 30;30mg/kg)の低濃度で観察されたことが示された。加えて、実施例7のみの投与は、腫瘍増殖阻害に対して影響を及ぼさず、これは実施例7が「真の」放射線増感剤として作用することを示すものである。
【実施例4】
【0099】
DNA障害の評価
抗体:一次抗体は、リン酸ヒストンH2AX(Cell Signaling,#2577,1:1000)及びGAPDH(Abcam,#9484,1:5000)に対して使用した。二次抗体は、ヤギ抗マウスIRDye800(Rockland,#610−132−121)及びヤギ抗ウサギAlexa fluor 700(Molecular Probe,#A21038)であった。
【0100】
U87MG細胞は、3Gy或いは5Gy放射線を照射し、その後実施例6(300nM及び1μM)で放射後0.5時間処理した。サンプルは次に、実施例6の添加後0.5、1及び4時間で回収した。次に前記細胞はRIPAバッファー(150mM NaCl、1% NP−40、0.5%デオキシコール酸ナトリウム、0.1% SDS、50mM トリス pH8.0)プラス阻害剤カクテル(プロテアーゼ阻害剤カクテルセットIII、Calbiochem)において氷上で溶解し、次に1mM Na3VO4はBCAタンパク質アッセイキット(Pierce#23225)を用いて定量化した。サンプルは、140ボルトで、MES SDSバッファー(Novex,#NP0002)での4−12%ビストリスゲル(Novex#NP0336)を用いて電気泳動(15μgタンパク質)で除去し、次に2Xトランスファーバッファー(Novex,#NP0006)を用いたセミドライトランスファー(18ボルトで35分)によって、ニトロセルロース膜(Biorad,#162−0145)へ移した。次に膜は、1XTBSでの1:1希釈のOdysseyブロッキングバッファー(Licor#927−40000)において室温で1時間ブロッキングし、次に1XTBS−T0.05%での1:1希釈のOdysseyブロッキングバッファーにおいて両一次抗体と共に4℃で一晩インキュベーションさせた。翌日、膜は1XTBS−T0.2%で各洗浄10分間を4回洗浄し、次に1:10,000(1XTBS−T0.05%での1:1希釈のOdysseyブロッキングバッファーにおいて)で両二次抗体と共に、遮光、室温で1.5時間インキュベーションした。ブロットは、1XTBS−T0.2%で各洗浄10分間(遮光)を4回洗浄し(遮光)、次にOdyssey Infrared Imagerで解析した。GAPDHは800nmシグナルを用いて可視化し、次にリン酸−H2AXは700nmで検出した。リン酸ヒストンH2Xの予想サイズは15kDaであり、GAPDHは36kDaであった。
【実施例5】
【0101】
細胞周期解析
U87MG細胞は、3Gy或いは5Gy放射線に照射し、次に照射後0.5時間で、実施例6(300nM及び1μM)で処理した。サンプルは、実施例6の添加後8、24及び48時間(或いは本分野の当業者によって決定されたあらゆる時間)で回収した。細胞は、4℃で一晩、100%エタノールにおいて固定させた。翌日細胞を、遮光、室温で1時間、細胞周期試薬(Guava Technologies#4500−0220)と共にインキュベーションした。染色された核はフローサイトメトリー(Guava EasyCyte;本分野の当業者に既知であるセッティングを用いる、例えば427X8;得られたデータ5,000イベント/サンプル)によって解析した。細胞周期の各相における細胞のパーセンテージは、細胞周期解析ソフトウェア(Guava Technologies)を用いて決定した。
【実施例6】
【0102】
7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化13】
【0103】
工程1:乾燥THF(20mL)における4−メトキシインドール(2.0g、13.1ミリモル)の冷却溶液(−78℃)へ、nBuLiヘキサン(2.5M、5.2mL、13.1ミリモル)をゆっくり添加した。その混合物を30分間−78℃でかき混ぜ、次にCO2ガスを15分間反応混合液へ泡立て、その後付加的に15分間かき混ぜた。過剰CO2及び半分のTHF容積を減圧下で除去した。付加的な乾燥THF(10mL)を、再び−78℃へ冷却した前記反応混合物へ添加した。1.7M t−BuLi(7.7mL、13.1ミリモル)を前記反応混合物へ30分かけてゆっくり添加した。−78℃で2時間かき混ぜ続け、その後乾燥THF(5mL)におけるシクロペンタノン(1.7g、20.4ミリモル)の溶液をゆっくり添加した。−78℃で1時間付加的にかき混ぜた後、前記反応混合物は水の液滴添加(5mL)、その後飽和NH4Cl溶液(20mL)によってクエンチした。エチルエーテル(50mL)を添加し、前記混合物を室温で10分間かき混ぜた。有機層を分離し、乾燥させ(MgSO4)、濃縮し、アルコール(1−(4−メトキシ−1H−インドール−2−イル)−シクロペンタノール)及びジエン(2−シクロペント−1−エニル−4−メトキシ−1H−インドール)の混合物を得た。アセトン(15mL)における前記混合物へ2N HCl(5mL)を添加した。前記混合物を10分間かき混ぜ、水(50mL)を添加し、前記ジエン産物2−シクロペント−1−エニル−4−メトキシ−1H−インドールを回収し、真空下で回収した。前記産物はシリカゲルクロマトグラフィー(EtOAC/ヘキサン9:1)で精製した。1H NMR(DMSO−d6)δ 1.9−2.1(m,3 H),2.6−2.75(m,3H),3.9(s,3H),6.1(s,IH),6.3(s,IH),6.4(m,IH),6.9−7.0(m,2H),11.1(s,IH)。この産物は次の工程に直接使用した。
【0104】
工程2:酢酸(5mL)における2−シクロペント−1−エニル−4−メトキシ−1H−インドール(0.1g、0.47ミリモル)及びマレイミド(0.09g、0.91ミリモル)の混合物を室温で1時間かき混ぜた。水を添加し、産物をEtOAcで抽出し、それは2N Na2CO3溶液、水、及び飽和NaCl溶液で洗浄し、乾燥させた(MgSO4)。乾燥剤を濾過によって除去し、溶液を濃縮し、0.13g MSを得た:m/z309(M−H)。
【0105】
工程3:トルエン(2mL)及び酢酸(3mL)における工程2からの産物(0.123g、0.4ミリモル)は、2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(DDQ、185mg、0.8ミリモル)へ添加した。0℃で30分かき混ぜた後、前記混合物を濃縮し、EtOAC及びアスコルビン酸で処理した。30分後前記混合物を2N Na2CO3で塩基性にした。EtOAc層を水で洗浄し、NaCl溶液で飽和させ、乾燥させ(MgSO4)、濃縮し、産物0.095mgを得た;MS:m/z305(M−H)+.1H NMR(DMSO−d6)δ 2.26−2.31(m,2H),3.1−3.2(m,2H),3.3−3.4(m,2H),3.9(s,3H),6.7(m,IH),7.1(m IH),6.4(m,IH),7.4(m,IH),10.6(s,IH),11.9(s,IH)。
【実施例7】
【0106】
7−メトキシ−5−(4−メチル−ピペラジン−1−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化14】
【0107】
エタノール(950mL)における実施例6(10.0g、30ミリモル)及びN−メチルピペラジン(12.4g、124ミリモル)のスラリーへパラホルムアルデヒド(5.60g、62.4ミリモル)を0.5時間で添加し、24時間かき混ぜた。前記スラリーは乾燥するまで蒸発させた。残留物へヘキサン(500mL)を添加し、15分間超音波処理し、1.5時間かき混ぜ、0℃で15分間冷却した。黄色の固形を回収し、冷却ヘキサンで洗浄した。この産物は温テトラヒドロフラン(THF)(250mL)に溶解し、濾過した。濾過物をヘキサン(3L)へ液滴添加し、15分間かき混ぜ、実施例7を沈殿物として回収し、ヘキサンで洗浄した(12.0g、96%収率)。1H NMR(DMSOd6)2.12(s,3H),2.35(m,8H),2.53(m,4H),3.18(m,2H),4.44(s,3H),6.70(d,lH),7.10(d,lH),7.40(t,lH),11.96(s,lH).MS m/z 419(M+H)。
【実施例8】
【0108】
7−メトキシ−5−(ジエチルアミノメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン(実施例8a)
7−メトキシ−5,11−(ビス−ジエチルアミノメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン(実施例8b)
【化15】
【0109】
DMF(5mL)における実施例6(50mg、0.16ミリモル)のスラリーへ、パラホルムアルデヒド(73mg、0.81ミリモル)、ジエチルアミン(84μL、0.81ミリモル)を添加し、室温で1日かき混ぜた。反応物を蒸発させ、残留物をヘキサンで粉砕し、蒸発させ、油として2つの産物を得た(比6−1、16b:16c)。1H−NMR(DMSOd6)0.98(t,3H),1.11(t,3H),2.27(m,2H),2.53(m,8H),2.57(m,15H),3.17(t,2H),3.50(m,lH),3.97(s,3H),4.14(d,2H),4.71(d,2H),6.82(t,2H),6.75(d,2H),7.13(d,2H),7.33(m,lH),7.46(t,3H),7.52(m,lH),11.95(s,lH).16b:MS m/z 392.16c MS m/z 476。
【実施例9】
【0110】
7−メトキシ−5,11−(ビス−モルフォリン−4−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化16】
【0111】
DMF(1mL)における実施例6(15mg、0.049ミリモル)のスラリーへ、パラホルムアルデヒド(42mg、0.05μL)、モルフォリン(160mg、1.9ミリモル)を添加し、70℃で18時間加熱した。混合物を蒸発させた。残留物をヘキサンで粉砕し、次にCH2Cl2に溶解し、濾過し蒸発させた。残留物をEt2Oで粉砕し、十知れを黄色の固形物として回収した(5mg、20%)。1HNMR(DMSO−J6)7.52(t,IH),7.39(d,IH),6.82(d,IH),5.0(s,2H),4.46(s,2H),3.98(s,3H),3.56(s,6H),3.49(s,4H),2.50(s,6H),2.49(s,4H),2.45(m,2H);MS m/z 505(M+H)。
【実施例10】
【0112】
7−メトキシ−5−(モルフォリン−4−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化17】
【0113】
エタノール(10mL)における実施例6(50mg、0.16ミリモル)のスラリーへ、パラホルムアルデヒド(72mg、0.8ミリモル)、モルフォリン(100mg、1.1ミリモル)を添加し、50℃で5時間加熱した。反応物を蒸発させ、水を添加し(15mL)、黄色の固形物を回収した(59mg)。1H NMR(DMSO−d6)11.98(s,IH),7.45(t,IH),7.13(d,IH),6.75(d,IH),4.44(s,2H),3.97(s,3H),3.56(s,4h),3.18(t,2h),2.29(t,2h).MS m/z 406(M+H)。
【実施例11】
【0114】
PARP酵素活性の測定
PARP活性は、[32P]NAD+からヒストン或いはPARP自身などのタンパク質受容体への放射線標識ADP−リボースユニットの転移によってモニタリングした。前記アッセイ混合物は、最終容積100μLにおいて100mMトリス(pH8.0)、2mM DTT、10mM MgCl2、20μg/ml DNA(超音波処理によってニックを入れた)、20mg/mlヒストンH1、5ng組換えヒトPARP、及び阻害剤或いはDMSO(<2.5%(v/v))を含む。反応は、2μCi[32P]NAD+/mL1を補充した00μM NAD+の添加で開始し、室温で12時間維持した。アッセイは100μMの50%TCAの添加によって終了し、放射線標識沈殿物を96ウェルフィルタープレート(Millipore,MADP NOB50)上で回収し、25% TCAで洗浄した。ポリADP−リボシル化タンパク質と一致する、酸不溶性放射線活性の量は、Wallac MicroBetaシンチレーションカウンターで定量化した。
【0115】
阻害剤に対するIC50の決定
一点阻害データは、DMSOのみ存在する場合の活性に対して、阻害剤の存在下でPARP、VEGFR2或いはMLK3活性と比較することによって計算した。化合物に対する阻害曲線は、パーセント阻害に対して化合物の濃度のlog10をプロットすることによって作成した。IC50値は、以下のGraphPad PrismにおけるS状用量反応性(可変傾き)方程式:
y=ボトム+(トップ−ボトム)/(1+10(logIC50−x)*斜面(Hillslope))
を用いた非直線回帰によって計算し、ここにおいて、yは化合物の既知濃度での%活性であり、xは化合物の濃度の対数であり、ボトムはテストした最も低い化合物濃度での%阻害であり、トップは試験した最も高い化合物濃度での%阻害である。ボトム及びトップに対する値は、それぞれ0及び100で固定した。IC50値は、少なくとも3回の別々の決定の平均として報告した。
【0116】
本明細書で開示されたアッセイを用いて、以下の表2から、PARP阻害に対する本発明の化合物の有用性が示された。本発明の化合物は、それらのIC50値が50μM以下の場合活性であると考えられた。PARP阻害に対する以下の表において、PARP阻害に対するIC50は、「+」となった本発明の化合物は10000nM以下であり;「++」となった本発明の化合物は1000nM以下であり;「+++」となった本発明の化合物は100nM以下である。IC50値が示されてない所は、まだデータを決定してない所である。
【0117】
【表1】
【実施例12】
【0118】
先行研究は、経口投与された実施例7の放射線増感能力を決定するために行った。
【0119】
腫瘍細胞移植及び増殖
指数関数的に増殖した細胞は播種し、商業的に利用可能な胸腺欠損NCR nu/nuヌードマウスの右側腹部へ注入した(2x106細胞/マウス)。200から400mm3の腫瘍を有する動物は、適切な処理群へサイズに従ってランダム化した(n=4)。腫瘍は、ノギスを用いて3から4日毎に測定した。腫瘍容積は、以下の公式を用いて計算した:
V=a2b/2、ここにおいてa及びbはそれぞれ、短寸法及び長寸法である。
【0120】
方法:U87MGヒト神経膠芽細胞腫細胞を、胸腺欠損NCR nu/nuヌードマウスの右後肢へ皮下的に(s.c.)注入し、平均腫瘍容積が200mm3になるように増殖させた。放射線療法を受けたマウスは、100mg/kgケタミン+10mg/kgキシラジン、若しくは37.5mg/kgケタミン+0.2mg/kgアセプロマジン、s.c.と共に照射を行う前に麻酔し、25から30分間鎮静させた。麻酔させたマウスは、過度の圧力なく、動物の体のサイズ及び形に合わせた可鍛性鉛遮蔽に位置付けした。その体は鉛によって遮蔽した。腫瘍を有する肢或いは曝露された腫瘍に適切な線量を照射した。腫瘍を照射した後、そのマウスは、麻酔から覚めるまでケージ或いは加温パッドへ戻した。実施例7は放射線(RT)後できるだけすぐ(30分以内)に与えた。マウスは以下の治療群へランダム化し、投薬した:1)溶媒、2)RT(2GyX5日)、3)RTプラス実施例7(200或いは300mg/kg p.o、実施例6の用量当量、qdX21日)、或いは4)実施例7(200或いは300mg/kg p.o、実施例6の用量当量、qdX21日)のみ。実施例7は、1から21日目に投与し、RTは1から5日目に行った。全動物は、同日に測定した。個々の腫瘍容積測定は対数変換直線モデルをモデルにし、腫瘍が約2000mm3に到達する最適時間を測定した。
【0121】
結果:図5に示したように、実施例7の投与(Cep 300;実施例6の300mg/kg用量当量、p.o.、qdx21日)プラスRT(2Gyx5日)は、8日目から腫瘍増殖静止状態となり、研究中(31日)続いた一方、実施例7(Cep 200;実施例6の200mg/kg用量当量、p.o.、qdx21日)プラスRT(2Gyx5日)及び実施例7のみ(実施例6の200及び300mg/kg用量当量、p.o.、qdx21日)の投与は、RTのみと比較して腫瘍増殖において影響を及ぼさなかった。得られた結果より、実施例7投与のみは腫瘍増殖に対して影響を及ぼさないということがs.c.投与から得られたデータによって確認されたことが示唆された。
【0122】
多数の変更及び修飾は本発明の好ましい実施形態になされ、そのような変更及び修飾は、本発明の観点から逸脱することなくなされ得るものであることは、本分野の当業者によって理解されるであろう。従って、添付された請求項は、本発明の観点及び範囲内にあるそのような同等なバリエーションの全てをカバーするものであることが意図される。
【0123】
本明細書に引用された全ての参考文献は、この参照によってその全てが本明細書に組み込まれる。
【技術分野】
【0001】
本発明は、腫瘍の放射線増感剤としてPARP阻害剤を用いて癌を治療する方法に関するものである。特に、本発明は、化学式(I)の化合物、
【0002】
【化1】
【0003】
或いはその薬学的に許容可能な塩形態を用いた、腫瘍の放射線増感方法に関するものである。本発明はさらに、放射線感受性腫瘍に対するPARP阻害剤の薬学的組成物に関するものである。
【背景技術】
【0004】
放射線は、DNA鎖破壊を作り出すことによって細胞障害を誘導する細胞障害性治療法である。ポリ(ADP−リボース)ポリメラーゼ1(PARP−1)は、核亜鉛フィンガーDNA結合タンパク質であり、DNA放射線誘導性障害及び修復によって活性化され、複製される。PARPは、DNA鎖破壊に結合し、ヌクレアーゼ攻撃或いは組換えからDNA鎖を保護するように働く。PARPはDNA修復を助けるように作用するので、阻害剤は細胞障害性薬剤の化学−及び放射線−感作を増強する潜在能力を有している(Curtin,2005)。
【0005】
治療上の失敗及び癌死亡率の最も重要な原因としては、放射線/化学−抵抗性である。細胞障害性薬剤に対する癌細胞抵抗性を克服するための薬剤は、癌治療を成功させる鍵となる因子となるであろう。化学及び放射線感作物質としての治療上のPARP阻害剤の潜在的な適用は、比較的最近まで、これら薬剤の力価、選択性及び薬学的特性によって制限されていた(Griffin et al.,1998;Bowman,et al.,1998;Bowman et al.,2001,Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Lui, et al.,1999)。最近、より強力で選択的なPARP阻害剤(ベンズイミダゾール−4−カルボキサミド及びキナゾリン−4−[3H]−オンズ(ones))が開発され、これらは、白血病、中枢神経系へのリンパ腫転移、大腸癌、肺癌及び乳癌のヒト及びマウス腫瘍モデルの両者を用いたin vitro及びin vivoにおける、放射線の効果、及びカンプトテシン(CPT)、トポテカン、イリノテカン、シスプラチン、エトポシド、ブレオマイシン、BCNU、及びテモゾロミド(TMZ)などの化学療法剤の効果を増強する能力を示した(Griffin et al.,1998;Bowman,et al.,1998;Bowman et al.,2001,Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Lui,et al.,1999,Tentori,et al.,2002)。異なるクラスの化学療法剤及び/若しくは放射線の作用に対して腫瘍細胞を増感させることができるPARP阻害剤は、確立された癌治療の成功率を上昇することができるであろう。
【0006】
PARP−1は、ヒストンポリメラーゼ、リガーゼ及びPARP自身(自己修飾)などの受容器タンパク質へとADP−リボースを移行するための基質としてNAD+を使用する116kD核亜鉛フィンガーDNA結合タンパク質である(Griffin et al.,1998;Tentori,et al.,2002;Baldwin et al.,2002)。PARP−1は、現在10種類のメンバーを含むタンパク質のファミリーに所属しており、その中のPARP−1及びPARP−2はDNA障害によって活性化される唯一の酵素である(Curtin,2005;Tentori,et al.,2002)。PARP−2の活性化はさらに、炎症誘発性活性を誘導し(Jagtop and Szabo,2005)、これは腫瘍細胞におけるPARP−2の阻害が付加的な治療利益となる可能性を示唆するものである。様々なPARPアイソフォームによって修飾された病態生理学的及び生理学的工程が大規模な研究の対象であるが(Ame et al.,2004)、一番特徴的なこのファミリーのメンバーであり、腫瘍学での治療上の標的薬剤を発見する試みの主な焦点は、PARP−1である。
【0007】
PARPは、転写レベルでのタンパク質発現、複製及び分化、テロメアーゼ活性、及び細胞骨格組織化を含む、たくさんの異なる生物学的工程の調節において活性である。しかしながら、化学/放射線増感剤としてPARP阻害剤を使用する目的であるDNA修復及びゲノム整合性の維持において、PARPは重要な役割を担っている(Smith,2001)。この役割は、イオン化放射線或いはアルキル化剤での治療に曝された場合の塩基切除修復の遅延及び高頻度の姉妹染色体交換を示す、PARP−1欠損細胞の使用を介して示される。更に、高レベルのイオン化放射線及びアルキル化剤は、野生型マウスと比較してPARP−1欠損マウスにおいてより高い死亡率を誘発する(Smith,2001;Virag&Szabo,2002)。
【0008】
PARPファミリーのメンバーの間で、PARP−1(及びPARP−2)は特に、イオン化放射線によって直接的に、或いはメチル化剤、トポイソメラーゼI阻害剤、及びシスプラチン及びブレオマイシンなどの他の化学療法剤によるDNA破壊の酵素的修復によって間接的に引き起こされたDNA破壊の修復によって活性化され複製される(Griffin et al.,1998;Delany et al.,2000;Tentori et al.,2002;de Murcia et al.,1997)。PARP−1は細胞生存及び亜致死性大量DNA損傷後の修復において重要な役割を担うことを示す実質的な生化学的及び遺伝学的証拠が存在する。さらに、PARP−1ノックアウトマウスによって例示さるように、DNA損傷が存在しないPARP−1機能は、細胞生存に対して重要ではなく、これにより、PARP−1の阻害が化学及び/若しくは放射線療法での使用に対して潜在的な実行可能な治療戦略となる(Delany et al.,2000;Burkle et al.,1993)。
【0009】
3−アミノベンズアミド、ニコチンアミド、及び関連誘導体などのPARP−1阻害剤の初期世代は、in vitro及びin vivoでのヒト及びマウス腫瘍モデルにおいて放射線、ブレオマイシン、CPT、シスプラチン及びTMZのin vitro及びin vivo細胞障害性活性の両者を増強した。疑いの余地のない抗腫瘍効果を与えることを阻止するような、これらの化合物の力価、選択性及び送達能力における本来の制限は、特にこれらの分子の非特異的活性に対するPARP−1の阻害に対してin vitro及びin vivoで観察された(Griffin et al.,1998;Griffin et al.,1995;Masuntani et al.,2000;Kato et al.,1988)。これらの課題は、様々なベンズイミダゾール−4−カルボキシアミド及びキナゾリン−4−[3H]−オン誘導体を含む、より強力で選択的なPARP−1阻害剤の構造クラスの開発において影響を及ぼした。in vitro及びin vivo解析によって、これらの化合物は、ヒト及びマウス腫瘍モデル両者を用いた化学療法剤の効果を増強可能であることが明らかになった(Griffin et al.,1998;Bowman, et al.,1998;Bowman et al.,2001;Chen&Pan,1998;Delany et al.,2000;Griffin et al.,1995;Liu,et al.,1999)。
【0010】
2001年11月15日付け公開のPCT公報第WO2001085686号では、PARP阻害活性を有するカルバゾール化合物が開示されている。
【0011】
PARPに対する高い選択性、高い力価、改善された送達能力、及び改善された耐用性特性を有する、腫瘍の治療に対する放射線増感薬剤としてのPARP阻害剤を発見し開発する必要性が存在する。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Curtin,NJ.(2005)PARP inhibitors for cancer therapy Expert Rev Molec Med 4:1−20.
【非特許文献2】Griffin,R et al.(1998)Resistance−modifying agents.5.Synthesis and biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(ADP−ribose)polymerase(PARP)J.Med.Chem.,42:5247−5256.
【非特許文献3】Bowman,KJ,et al.(1998)Potentiation of anti−cancer agent cytotoxicity by the potent poly(ADP−ribose)polymerase inhibitors NU 1025 and NU 1064 Br.J,Cancer 78:1269−1277.
【非特許文献4】Bowman,KJ,et al(2001)Differential effects of the poly(ADP−ribose)polymerase inhibitor NU 1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro Br.J.Cancer 84:106−112.
【非特許文献5】Chen&Pan(1988)Potentiation of antitumor activity of cisplatin in mice by 3−aminobenzamide and nicotinamide Cancer Chemoth.Pharmacol.22:303−307.
【非特許文献6】Delaney,CA et al.(2000)Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose)polymerase inhibitors in a panel of human tumor lines Clin.Cancer Res.6:2860−2867.
【非特許文献7】Griffin R et al.,(1995)The role of inhibitors of poly(ADP−ribose)polymerase as resistance−modifying agents in cancer therapy Biochimie 11:408−422.
【非特許文献8】Liu,L,et al.(1999)Pharmacologic disruption of base excision repair sensitizes mismatch repair−deficient and−proficient colon cancer cells to methylating agents Clin.Cancer Res.5:2908−2917.
【非特許文献9】Tentori,L.et al.(2002)Combined treatment with temozolomide and poly(ADP−ribose)polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site Blood 15:2241−2244.
【非特許文献10】Baldwin J.(2002)FDA evaluating oxaliplatin for advance colorectal cancer treatment J.Natl.Cancer Inst.94:1191−1193,2002.
【非特許文献11】Jagtap P and Szabo C(2005).Poly(ADP−Ribose)polymerase and the therapeutic effects of its inhibitors.Nature Rev Drug Disc 4:421−440.
【非特許文献12】Ame JC,et al(2004).The PARP superfamily.Bioessays 26:882−893.
【非特許文献13】Smith,S.(2001)The World According to PARP Trends Biochm.Sci.26:174−179.
【非特許文献14】Virag,L.and Szabo,C.(2002)The therapeutic potential of poly(ADP−ribose)polymerase inhibitors Pharmacol.Rev.54:375−429.
【非特許文献15】de Murcia,JM,et al.(1997)Requirement of poly(ADP−ribose)polymerase in recovery from DNA damage in mice and cells Proc Natl Acad Sci USA 94:7303−7307.
【非特許文献16】Masuntani,M,et al.(2000)The response of PARP knockout mice against DNA damaging agents Mutat.Res.462:159−166.
【非特許文献17】Kato,T et al.(1988)Enhancement of bleomycin activity by 3−aminobenzamide,a poly(ADP−ribose)synthesis inhibitor,in vitro and in vivo Anticancer Res.8:239−244.
【非特許文献18】Smith,L.et al.(2005)The novel poly(ADP−Ribose)polymerase inhibitor,AG14361,sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks Cl Cancer Res.11:8449−8457.
【発明の概要】
【課題を解決するための手段】
【0013】
本発明は、PARP−1のin vivo阻害によって腫瘍において放射線増感を引き起こすために4−メトキシ−カルバゾールを用いた方法を提供する。前記方法は、化学式(Ia)の4−メトキシ−カルバゾール、
【化2】
【0014】
そのプロドラッグ、好ましくはそのマンニッヒ塩基プロドラッグを有し、可溶性及び安定性を提供し、活性薬剤である7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン(trindene)−4,6−ジオンのin vivo送達を助けるものである。
【0015】
本発明はさらに、化学式(I)であって、
【0016】
【化3】
【0017】
式中、Xは、本明細書で定義されるように、H或いはプロドラッグ部位である、放射線増感剤或いは薬学的に許容可能なその塩形態を癌を患った哺乳動物へ投与する工程と、
前記哺乳動物組織へイオン化放射線を適用する工程と、によって癌を治療する方法を提供するものである。
【0018】
本発明の別の対象は、本発明の化合物を有する薬学的組成物を提供することであり、ここにおいて前記組成物は、1若しくはそれ以上の薬学的に許容可能な賦形剤及び治療上有効な量の少なくとも1つの本発明の化合物、或いはその薬学的に許容可能な塩或いはエステル形態を有するものである。
【0019】
本発明の別の対象は、化学式(II)の化合物、
【0020】
【化4】
【0021】
或いはその薬学的に許容可能な塩形態を提供するものである。
【0022】
別の実施形態において、本発明は、癌の治療に対する薬物を製造するための化学式(I)の化合物の使用を提供するものである。
【0023】
本発明のこれら及び他の対象、特性及び利点は、本開示の以下の詳細な説明において説明される。
【図面の簡単な説明】
【0024】
【図1】図1は、腫瘍の増殖遅延に対する、放射線耐性U87MG神経膠芽細胞腫異種移植片を用いた、放射線との組み合わせにおけるマンニッヒ塩基プロドラッグの効果を示したものである。
【図2】図2は、放射線療法或いはプロドラッグのみの比較投与計画によって達成された療法より強力な併用療法の効果の程度を示したものである。
【図3】図3は、ヌードマウスにおけるU87MGヒト神経膠芽細胞腫異所性移植片に対する実施例7の放射線増感効果を示したものである(非最適化スケジュール)。
【図4】図4は、本発明の観点内の化合物及びそれの前駆体を含む合成模式図を示したものである。
【図5】図5は、ヌードマウスにおけるU87MGヒト神経膠芽細胞腫異所性移植片に対する経口的に投与された実施例7の放射線増感効果を示したものである。
【発明を実施するための形態】
【0025】
第一の実施形態において、本発明は、化学式(I)であって、
【化5】
【0026】
式中、XはH或いはプロドラッグ部位である、放射線増感剤或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程、及び前記哺乳動物にイオン化放射線を適用させる工程によって癌を治療する方法を提供する。
【0027】
好ましい実施形態において、前記放射線増感剤は、前記組織内に或いは近接して存在するものであり、これは、局所的治療効果への前記適用イオン化放射線の転換効率を増強するものである。
【0028】
好ましい実施形態において、前記放射線増感剤は、放射線感受性癌細胞に対して有効な量で存在するものである。
【0029】
好ましい実施形態において、前記組織のイオン化放射線は、前記細胞を破壊するのに有効な放射線線量で実行されるものである。
【0030】
好ましい実施形態において、前記イオン化放射線は、既知の癌タイプに対して臨床的に許容可能な或いは推奨される放射線療法プロトコールで行われるものである。
【0031】
好ましい実施形態において、前記癌は悪性である。
【0032】
好ましい実施形態において、前記癌は良性である。
【0033】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、−C(=O)R4から成る群から選択されるものであり、ここにおいて、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【0034】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2であり、R1は、H或いはC1−4アルキルであり;R2は、H或いはC1−4アルキルであり、或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【0035】
好ましい実施形態において、前記プロドラッグ部位は、マンニッヒ塩基である。
【0036】
好ましい実施形態において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチル−、モルフォリン−4−イルメチル−、及び5−ジエチルアミノメチル−から選択されるものである。
【0037】
好ましい実施形態において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチルである。
【0038】
好ましい実施形態において、前記投与の経路は、静脈内、皮下、経口或いは腹腔内である。
【0039】
好ましい実施形態において、前記投与の経路は、静脈内である。
【0040】
好ましい実施形態において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌から選択されるものである。
【0041】
好ましい実施形態において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【0042】
好ましい実施形態において、本発明は、化学式7−メチル−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤を投与する工程によって癌を治療する方法を提供するものである。
【0043】
好ましい実施形態において、本発明は、化学式7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤を投与する工程によって癌を治療する方法を提供するものである。
【0044】
第二の実施形態において、本発明は、放射線感受性癌細胞に対して薬学的組成物を提供するものであり、この組成物は、放射線増感量の化学式(I)の化合物、
【化6】
【0045】
或いはその薬学的に許容可能な塩形態であって、ここにおいて、Xは、H或いはプロドラッグ部位である化合物或いは塩形態、
及び薬学的に許容可能な担体を有するものである。
【0046】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、−C(=O)R4から成る群から選択されるものであり、ここにおいて、
R1は、H或いはC1−4アルキル、
R2は、H或いはC1−4アルキル、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【0047】
好ましい実施形態において、前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【0048】
好ましい実施形態において、前記化合物は、
【化7】
【0049】
或いはその薬学的に許容可能な塩形態である。
【0050】
好ましい実施形態において、前記化合物は、
【化8】
【0051】
或いはその薬学的に許容可能な塩形態である。
【0052】
第三の実施形態において、本発明は、化学式(II)の化合物、
【化9】
【0053】
或いはその薬学的に許容可能な塩形態を提供するものである。
【0054】
第四の実施形態において、本発明は、癌を治療するための薬剤を製造するための化学式(I)の化合物の使用を提供するものである。
【0055】
好ましい実施形態において、本発明は、癌を治療するための薬剤を製造するための化学式(II)の化合物の使用を提供するものである。
【0056】
本発明の特定の特性は、明確にするために別々の実施形態の文脈において記載しているが、さらに単一の実施形態の組み合わせでも提供され得ることは理解される。反対に、本発明の様々な特性は、簡潔化するために単一の実施形態の文脈において記載しているが、別々に、或いはあらゆる適切なサブコンビネーションにおいても提供され得る。
【0057】
本明細書に含まれる以下の用語及び表現は、以下のように定義される。
【0058】
本明細書で用いられたように、「約」という用語は、特定値の±10%の範囲の値を意味する。例えば、「約50mg」という文章は、50の±10%、若しくは45から55mgを含む。
【0059】
本明細書で用いられる「アルキル」という用語は、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル及びtert−ブチルなどの、1から4個の炭素原子を持つ直鎖或いは分岐アルキル基を意味する。「C1−4アルキル」などの記号は、1から4個の炭素原子を含むアルキル遊離基を意味する。
【0060】
本明細書で用いられる「アミノ酸」という用語は、アミノ基及びカルボキシル基の両者を含む分子を意味する。それは、カルボキシル基に隣接する炭素上にアミノ機能基を生じるカルボキシル酸として本分野の当業者にはよく知られた「α−アミノ酸」を含む。アミノ酸は、天然由来或いは非天然由来であり得る。「天然由来アミノ酸」は、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン酸、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシン及びバリンを含む。
【0061】
本明細書で用いられる「ヘテロシクリル」という用語は、炭素原子、及び少なくともO、N或いはSから選択されたヘテロ原子を含む5或いは6員環基を意味し、ここにおいて前記ヘテロシクリル基は、飽和或いは不飽和されており、前記ヘテロシクリル基は、置換或いは非置換されているものである。窒素及び硫黄へテロ原子は任意に酸化されている。ヘテロシクリル基の例としては、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル、ピペラジニル、及びメチルピペラジニルが含まれる。
【0062】
本明細書で用いられたように、「哺乳動物」という用語は、本明細書で記載された1若しくはそれ以上の疾患及び症状に苦しめられている、若しくは苦しめられる可能性がある、マウス、ラット、ネコ、イヌ、サル或いはヒト、好ましくはヒト或いはヒト小児を含む温血動物を意味する。
【0063】
本明細書に用いられる「薬学的に許容可能な」化合物とは、適切な利益/リスク比に釣り合った、過度の有害な副作用(毒性、刺激及びアレルギー反応など)なしに、ヒト及び/若しくは動物に使用するのに適切なものである。
【0064】
本明細書で用いられる「安全且つ有効な量」という用語は、本発明のやり方で使用した場合、適切な利益/リスク比に釣り合った、過度の有害な副作用(毒性、刺激及びアレルギー反応など)なしに、望ましい治療反応を生むのに十分な成分の量を意味する。「治療上有効な量」は、望ましい治療反応を生むのに有効な本発明の化合物の量を意味している。例えば、癌、肉腫或いはリンパ腫の増殖或いはそれらを引き起こすのを遅延するのに有効な量、若しくは前記癌を収縮させる或いは転移を阻止するのに有効な量である。特異的に安全且つ有効な量、或いは治療上有効な量は、治療される特定の症状、患者の身体的状態、治療される哺乳動物或いは動物のタイプ、治療の持続期間、併用治療(もしあれば)の特性、及び使用された特異的処方、及び前記化合物或いはその誘導体の構造などの因子によって変わるであろう。
【0065】
本発明において、「イオン化放射線」という用語は、十分なエネルギーを持つ、若しくは核相互作用を介して十分なエネルギーを産生しイオン化(電子の獲得或いは損失)を産生できる粒子或いは光子を有する放射線を意味する。例示的で好ましいイオン化放射線は、X−放射線である。標的組織或いは細胞へX放射線を送達する手段は、本分野ではよく知られている。既知の細胞に必要とされるイオン化放射線の量は一般的に、その細胞の特性に依存している。放射線の有効な量を決定する手段は、本分野ではよく知られている。本明細書で使用されたように、イオン化放射線の「有効な線量」という用語は、本発明の化合物との組み合わせで与えられた場合、細胞障害或いは細胞死の増加を産生するイオン化放射線の線量を意味するものである。
【0066】
X線の投与量範囲は、長期間(3から4週間)の場合一日量で50から200レントゲンから、単一量で2000から6000レントゲンまでの範囲である。放射性同位元素に対する投与範囲は幅広く変わり、その同位元素の半減期、放射される放射線の強度及びタイプ、及び新生物細胞による取り込みに依存する。
【0067】
組織へ放射線を送達するあらゆる適切な手段は、本発明において使用される。組織へ放射線を送達する一般的な手段は、治療される体の外部にあるイオン化放射線源によるものである。組織へ放射線を送達する代替方法としては、例えばまず腫瘍の抗原に免疫反応を生じる放射線標識抗体をin vivoで送達する工程と、次に前記腫瘍へ有効な量の前記放射線標識抗体をin vivoで送達する工程とを含む。加えて、放射性同位元素は、組織或いは細胞へイオン化放射線を送達するために使用される。付加的に、前記放射線は、放射線類似薬剤の手段によって送達される。本明細書で用いられたように、「放射線類似薬剤」は、放射線の適用なしに放射線療法と同じタイプの細胞障害を生じる、例えばメルファランなどの化学療法剤である。
【0068】
本明細書で用いられれる「プロドラッグ部位」という用語は、前記プロドラッグが、生理学的条件下で、多数の化学的及び生物学的メカニズムによって生物学的活性薬剤へ変換され得るということを意味している。1実施形態において、生物学的活性薬剤への前記プロドラッグの変換は、前記プロドラッグ部位は化学的に或いは酵素学的に水で加水分解するという条件で、前記プロドラッグ部位の加水分解によって達成され得る。水との反応は一般的に、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。本発明の別の観点では、前記プロドラッグ部位の還元によって前記生物学的活性薬剤への前記プロドラッグの変換が提供される。この実施形態において一般的に、前記プロドラッグ部位は、生理学的条件下で、還元酵素工程の存在下において還元されるものである。前記還元は好ましくは、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。別の実施形態において、前記生物学的活性薬剤への前記プロドラッグの変換はさらに、前記プロドラッグ部位の酸化によっても達成され得る。この実施形態において一般的に、前記プロドラッグ部位は、生理学的条件下で、酸化酵素工程の存在下において酸化されるものである。前記酸化は好ましくは、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。本発明のさらなる態様では、前記プロドラッグ部位の除去によって前記生物学的活性薬剤への前記プロドラッグの変換が含まれる。一般的に言えば、この実施形態において、前記プロドラッグ部位は、生理学的条件下で、化学的或いは生物学的反応によって除去される。前記除去は、前記プロドラッグ部位の除去、及び前記生物学的活性薬剤の遊離を生じる。もちろん、本発明のあらゆるプロドラッグ化合物は、上述のメカニズムのあらゆる組み合わせを受け、前記プロドラッグを生物学的活性薬剤へと変換する。例えば、特定の化合物は、加水分解、酸化、除去、及び還元を受け、前記プロドラッグを生物学的活性化合物へと変換するであろう。同等に、特定の化合物は、これらメカニズムの1つのみを受け、前記プロドラッグを生物学的活性化合物へと変換するであろう。
【0069】
本明細書で用いられる「癌」とは、癌腫及び肉腫を含む、哺乳動物において見出された癌或いは新生物、若しくは悪性或いは良性腫瘍のあらゆるタイプを意味する。癌の例としては、脳の癌、乳癌、膵癌、子宮頸癌、大腸癌、頭頸部癌、腎癌、肺癌、肺非小細胞癌、メラノーマ、中皮腫、卵巣癌、肉腫、胃癌、子宮癌及び髄芽細胞腫である。
【0070】
「白血病」という用語は、造血器官の進行性、悪性疾患を広く意味し、一般的に血中及び骨髄中での白血球及びそれらの前駆体の歪んだ増殖及び発生によって特徴付けされている。白血病は一般的に、(1)疾患の持続期間及び特徴−急性或いは慢性、(2)関連した細胞の種類;骨髄球系(骨髄性)、リンパ球系(リンパ性)、或いは単球性、及び(3)血中の異常細胞の数が増加する或いは増加しない−白血病性或いは無白血病性(亜白血性)、という基礎に基づいて臨床的に分類されている。P388白血病モデルは、in vivo抗白血病活性を予想するものとして広く許容かせている。P388アッセイにおいてポジティブであった化合物は一般的に治療される白血病のタイプに関わらず、in vivoである程度の抗白血病活性を示すであろうと信じられている。従って、本発明は、白血病を治療する方法を含み、好ましくは急性非リンパ球性白血病、慢性リンパ球性白血病、急性顆粒球性白血病、慢性顆粒球性白血病、急性然骨髄球性白血病、成人T−細胞白血病、亜白血性白血病、白血性白血病(leukocythemic leukemia)、好塩基球性白血病、芽球細胞白血病、ウシ白血病、慢性骨髄球性白血病、皮膚白血病、胚性白血病、好酸球性白血病、グロス白血病、ヘアリー細胞白血病、血球母細胞白血病、血球芽性白血球、組織球性白血病、幹細胞白血病、急性単球性白血病、白血球減少性白血病、リンパ性白血病、リンパ芽球性白血病、リンパ球性白血病、リンパ向性白血病、リンパ様白血病、リンパ肉腫細胞白血病、マスト細胞白血病、巨核球性白血病、微小骨髄芽球性白血病、単球性白血病、骨髄芽球性白血病、骨髄球性白血病、骨髄性白血病、骨髄単球性白血病、ネーゲリ型白血病、形質細胞白血病、形質細胞性白血病、前骨髄球性白血病、リーダー細胞(Rieder cell)白血病、シリング白血病、幹細胞白血病、亜白血性白血病、及び未分化細胞白血病を治療する方法を含む。
【0071】
「肉腫」という用語は一般的に、胚性結合組織のような物質を作り上げる腫瘍を意味し、一般的に原線維或いは均一な物質に埋包された、密接に詰められた細胞から成る。4−メトキシ−カルバゾール及び放射線療法で治療され得る肉腫には、軟骨肉腫、胆管肉腫、線維肉腫、リンパ肉腫、メラノ肉腫、粘液肉腫、骨肉腫、アベメシー肉腫、脂肪(adipose)肉腫、脂肪肉腫(liposarcoma)、胞状軟部肉腫、エナメル芽細胞肉腫、ブドウ状肉腫、緑色肉腫、絨毛肉腫、胎児性肉腫、ウィルムス腫瘍肉腫、子宮内膜肉腫、間質性肉腫、ユーイング肉腫、筋膜肉腫、線維芽細胞肉腫、巨細胞肉腫、顆粒球性肉腫、ホジキン肉腫、特発性多発性色素性出血性肉腫、B細胞の免疫芽球性肉腫、リンパ腫、T細胞の免疫芽球性肉腫、イエンセン肉腫、カポジ肉腫、クッパー細胞肉腫、血管肉腫、白血肉腫、悪性間葉細胞肉腫、傍骨肉腫、細網肉腫、ラウス肉腫、漿液嚢胞性肉腫、軟部組織肉腫、滑膜肉腫、及び毛細血管拡張性肉腫が含まれる。
【0072】
「メラノーマ(黒色腫)」という用語は、皮膚及び他の器官のメラニン細胞システムから生じる腫瘍を意味する。4−メトキシ−カルバゾール及び放射線療法で治療され得るメラノーマには、例えば、末端性黒子性黒色腫、無色素性メラノーマ、良性若年性メラノーマ、クラウドマン黒色腫、S91メラノーマ、ハーディング−パッセー黒色腫、若年性黒色腫、悪性黒子型黒色腫、悪性黒色腫、結節性黒色腫、爪下黒色腫、及び表在性黒色腫が含まれる。
【0073】
「癌腫」という用語は、周囲組織へ浸潤し転移する傾向がある、上皮細胞から作られた悪性新生増植物を意味する。4−メトキシ−カルバゾール及び放射線療法で治療され得る癌腫の例としては、例えば、細葉細胞癌、腺房細胞癌、腺嚢癌、腺様嚢胞癌、乳癌、腺腫様癌、副腎皮質の癌腫、胞巣状癌、肺胞上皮細胞癌、基底細胞癌、好塩基球癌腫、類基底細胞癌、基底扁平上皮癌、膀胱癌、細気管支肺胞上皮癌、細気管支癌、気管支原性肺癌、脳回転様(cerebriform)癌、胆管細胞癌、絨毛がん、コロイド腺癌、結腸直腸癌、子宮頸癌、面疱癌、子宮体癌、篩状癌、鎧状癌、皮膚癌、円柱癌、円柱細胞癌、管癌、硬性癌、胎児性癌、脳様癌、類表皮癌、腺上皮癌、外方増殖性癌、潰瘍性癌、線維性癌、胃癌、膠様癌(gelatiniform carcinoma)、膠様癌(gelatinous carcinoma)、巨細胞癌、巨大細胞癌、腺癌、顆粒膜細胞癌、毛母癌、血様癌(hematoid carcinoma)、肝細胞癌、ハースル細胞癌、硝子状癌、hypemephroid癌、小児性胎児性癌、上皮内癌(carcinoma in situ)、表皮内癌、上皮内癌、Krompecher’s癌、クルチツキー(Kulchitzky)細胞癌、大細胞癌、レンズ状癌、レンズ状癌(carcinom lenticulare)、脂肪腫様癌、肺癌、リンパ上皮癌、髄様癌(carcinoma medullare)、髄様癌、黒色癌、軟性癌(carcinoma molle)、粘液癌、粘液性癌(carcinoma muciparum)、粘液細胞癌、粘表皮癌、粘膜癌(carcinoma mucosum)、粘膜癌(mucous carcinoma)、粘液腫状癌(carcinoma myxomatodes)、上咽頭癌、燕麦細胞癌、骨化性癌、類骨癌、卵巣癌、膵臓癌、前立腺癌、乳頭癌、門脈周囲癌、前浸潤癌、有棘細胞癌、糊性癌(pultaceous carcinoma)、腎臓の腎細胞癌、予備細胞癌(reserve cell carcinoma)、肉腫状癌(carcinoma sarcomatodes)、シュナイダー癌、スキルス癌、陰嚢癌(carcinoma scroti)、印環細胞癌、単純癌(carcinoma simplex)、小細胞癌、ソレノイド癌(solanoid carcinoma)、球状細胞癌、紡錘細胞癌、海綿細胞癌(carcinom spongiosum)、扁平上皮癌、扁平上皮細胞癌、ストリング癌(string carcinoma)、末梢血管拡張性癌、末梢血管拡張様癌(carcinoma telangiectodes)、精巣癌、移行細胞癌、甲状腺癌、結節性癌(carcunoma tuberosum)、結節性癌、疣状癌、及び絨毛様癌(carcunoma villosum)が含まれる。
【0074】
本発明に従った化合物で治療され得る好ましい癌としては、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌を含む。
【0075】
本発明に従った化合物で治療され得るより好ましい癌としては、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【0076】
本明細書で用いられる「4−メトキシ−カルバゾール」という用語は、以下の化学式、
【化10】
【0077】
を有する化学物、若しくはその薬学的に許容可能な塩形態を意味し、ここにおいて、XはH或いはプロドラッグ部位である。
【0078】
本発明の化合物は、プロドラッグ部位を含む。本発明によって考えられるプロドラッグ部位の例としては、リン酸エステル、アミノ酸エステル、アミノ酸アミド、アミノアルキルカルバミン酸、アルコキシアルキルカルバミン酸、ヒドロキシアルキルカルバミン酸、アルコキシアルキルエステル、ヒドロキシアルキルエステル、安息香酸エステル、ニコチンエステル、ピペラジン酢酸、モルフォリニル酢酸、及びマンニッヒ塩基から選択され得る。本発明によって考えられるプロドラッグ部位の例としては、
【化11】
【0079】
【化12】
【0080】
から選択され得る。好ましいプロドラッグ部位は、マンニッヒ塩基である。好ましいマンニッヒ塩基には、これに限定されるものではないが、4−メチル−ピペラジン−1−イルメチル−、モルフォリニル−4−イルメチル−、及びジエチルアミノメチル−が含まれる。
【0081】
本発明の化合物はさらに、薬理学的に許容可能な塩、水和物、溶媒和化合物、或いは代謝産物の形態もとる。薬理学的に許容可能な塩類には、これに限定されるものではないが、塩酸、臭化水素酸、硫酸、リン酸、メタンスルホン酸、エタンスルホン酸、リンゴ酸、酢酸、シュウ酸、酒石酸、クエン酸、乳酸、フマル酸、スクシニル酸、マレイン酸、サリチル酸、安息香酸、フェニル酢酸、マンデル酸、アスコルビン酸、グルコン酸、及びそれらと同等物を含む、無機及び有機酸の基礎塩類が含まれる。本発明の化合物がカルボキシ基などの酸性機能を含む場合、カルボキシ基に対して適切な薬学的に許容可能な陽イオン対は、本分野の当業者にはよく知られており、アルカリ、アルカリ土類、アンモニウム、四級アンモニウム陽イオン及びそれらと同等物を含む。本発明の化合物が薬理学的に許容可能な塩の形態をとる場合、前記塩形態はin situ或いは単離固形として生成されるということが本発明によって考えられる。
【0082】
本発明の化合物は、特に上記の塩類の形態において、薬学的に許容可能な担体として働き、例えば注射、懸濁液、乳剤、錠剤、カプセル及び軟膏などの形態において薬剤を投与可能にする、本分野ではよく知られている様々な賦形剤溶媒及び/若しくはアジュバントと組み合わされる。記載された化合物の放射線増感量を含むこれらの薬学的組成物は、低酸素腫瘍細胞の放射線増感を生じるあらゆる許容可能な手段によって投与される。温血動物に対して、及び特に放射線療法を受けるヒトにおいて、投与は経口、皮下、腹腔内、或いは静脈内に行われ得る。低酸素腫瘍細胞を破壊するために、放射線増感剤を含む前記薬学的組成物は、前記低酸素腫瘍細胞を放射線増感するのに有効な量で投与される。投与された特異的投与量は、患者の一般的健康状態及び体調、さらに年齢及び体重、患者疾患の段階、及びあらゆる併用療法の存在などの因子に依存するであろう。
【0083】
有効な量を投与する方法も、治療される疾患或いは病気に依存して変わる。適切な担体、付加的癌阻害化合物或いは化合物類、若しくは適用を容易にする希釈剤と処方される、4−メトキシ−カルバゾールの静脈内適用による治療は、温血動物へ前記化合物を投与する好ましい方法であろう。
【0084】
本明細書に記載された化合物は、純粋な形態で、他の有効成分との組み合わせで、若しくは薬学的に許容可能な非毒性賦形剤或いは担体との組み合わせで投与される。経口組成物は一般的に、不活性希釈担体或いは食用担体を含む。薬学的に適用性な結合剤、及び/若しくはアジュバント物質は、前記組成物の一部として含まれ得る。錠剤、ピル、カプセル、トローチ及びそれらと同等物は、あらゆる以下の成分或いは同様の性質の化合物:微結晶性セルロース、ガムトラガント或いはゼラチンなどの結合剤;スターチ或いはラクトースなどの賦形剤、アルギニン酸、プリモゲル(Primogel)或いはコーンスターチなどの分散剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド二酸化ケイ素などの流動促進剤;スクロース或いはサッカリンなどの甘味料;或いはペパーミント、サリチル酸メチル、或いはオレンジ香料などの香味料、を含む。前記投与ユニット形態がカプセルの場合、上記タイプの物質に加えて、脂肪油などの液体担体を含むことができる。加えて、投与ユニット形態は、例えば、糖のコーティング、セラック或いは腸溶剤などの、前記投薬ユニットの物理的形態を修飾する様々な他の物質を含むことができる。さらに、シロップは、前記有効化合物に加えて、甘味料としてのスクロース、及び特定の保存剤、色素、着色料及び香味料を含む。
【0085】
患者へ投与された化合物の量は、治療される悪性新生物を放射線増感するのに十分なものであるが、毒性作用を誘発する量以下である。この量は、腫瘍のタイプ、治療される患者の種類、意図された指標投与量、及び患者の体重或いは体表面量に依存するであろう。放射線は、様々な異なる分割計画でヒトへ投与される、すなわち総放射線線量が数日から数週間の期間で分割して与えられる。これらは、毎日(すなわち一週間で5回)線量から6週間まで、週1回線量から4から6週間までと変わる可能性が最も高い。
【0086】
患者へ投与された放射線増感化合物の量は、放射線治療前、放射線治療中、或いは放射線治療後に投与される。しかしながら、本発明の化合物は放射線治療前に投与されることが好ましい。
【0087】
低酸素腫瘍細胞への前記放射線増感組成物の投与、及び前記低酸素腫瘍細胞の放射線増感を増強するのに十分な時間間隔での継代後、前記低酸素腫瘍細胞は、前記低酸素腫瘍細胞を破壊するのに有効な放射線の線量で照射される。一般的に、患者は、1日当たり約2Gyの放射線線量を5日間受けるであろう。一般的に、前記患者は7から8週間で約70から約80Gyの総放射線線量を受け、それぞれ個々の放射線線量は、放射線増感剤の投与後約1から4時間以内に投与される。そのような一連の放射線増感治療及び照射は、寛解するのに、及び任意に悪性腫瘍の転移を減少或いは除去するのに必要な回数繰り返される。しかしながら、毎日の放射線線量及び総放射線線量は、患者の腫瘍タイプ、治療プロトコール、及び身体的状態に依存して変わるであろうことは本分野の当業者によって理解される。例えば、本発明の化合物の毎日の線量は、特別に限定されるものではないが、患者の年齢、癌、体重及び現在の治療プロトコール及び/若しくは薬物療法で変えることができる。付加的に、本発明の化合物は、放射線増感剤として有用であり、放射線に曝露される前に、1若しくはそれ以上の用量で、すなわち1から複数用量で投与され得る。
【0088】
ヌードマウスにおいてU87MG放射線耐性異種移植片を用いた初期放射線増感研究(非最適化用量スケジュール)
細胞の照射はチェックポイント停止を誘導し、これは、DNA障害の修復を促進する活性化PARPによる、細胞のDNA障害の修復を可能にする。仮定で、単一線量或いは分割放射線との組み合わせでのPARP阻害剤の投与は、DNA障害を修復するための照射細胞の能力を減少し、細胞殺傷を増加するであろう。従って、PARP阻害剤は、分割放射線と相乗的に作用し、腫瘍増殖遅延を増加しなくてはならない。実施例7/実施例6におけるこの仮説の初期テストは、ヌードマウスにおける放射線耐性U87MGヒト神経膠芽腫異種移植片で実行した。図3に示されたように、実施例7単独の投与、放射線単独、及び放射線(実施例6の100mg/kg用量当量、s.c.、放射線のqd2日前、及び3日間の7.5Gy放射線との組み合わせ)との組み合わせの実施例7は、確立された腫瘍を有するマウスにおいて実行した。単一剤として投与された実施例7は、腫瘍増殖に影響を及ぼさなかった。溶媒或いは実施例7で治療された腫瘍は、それぞれ10.0日或いは9.6日(コントロールに対して、p=0.798)で2000mm3の腫瘍容積に達した。放射線単独の投与は、2000mm3に達するための時間が16.1日と増加し、腫瘍増殖遅延(TGD)も6.1日(コントロールに対して、p=0.033)と増加した。対照的に、放射線療法との実施例7の投与は、2000mm3に達するための腫瘍に対する時間が24.8日と増加し、これは14.8日TGDに対応する。併用療法の効果の大きさは、実施例7単独(p=0.001)或いは放射線のみ(p=0.006)の比較計画によって見られたものより強く、これは、実施例7は真の放射線増感剤の特性を示すことを示している。有効性と関連したCmaxにおける実施例6の血漿レベル(100mg/kg実施例7での)は、23μMであり、これは化学−感作研究におけるこの用量で達成されるものに匹敵する。
【0089】
ヌードマウスにおいてU87MG放射線耐性異種移植片を用いた実施例7及び臨床的に関連した分割放射線療法投与スケジュールでの放射線増感研究
次の放射線増感研究は、臨床的に関連した分割放射線療法スケジュール(2Gyを5日間)との組み合わせで実施例7(30及び100mg/kg、s.c.)を評価することである。実施例7は、5日間の放射線後に0.5時間投与し、放射線計画後16日間続けた実施例7の投与を完了した。この投与計画に対する論理的根拠は、放射線障害からのDNA修復が照射後10から12日で起こり、従って、実施例7の連続投与及びPARP活性の修飾は、分割放射線と相乗的に作用し放射線増感及び腫瘍増殖遅延を増加するであろう、細胞周期停止及びDNA修復時間に影響を及ぼしている、という事実に基づいていた。図1及び2に示されたように、放射線単独の投与(2Gyを5日間)は、溶媒処理腫瘍と比較して2.5日のTGDを生じた。実施例7の投与(CEP 30;30mg/kg s.c.)は、TGDを15日まで増加し、放射線単独と比較して4倍増加し(p≦0.05);実施例7単独と比較して6倍増加した(p≦0.001)。放射線増感効果と関連したCmaxでの実施例6の血漿レベルは、5.5μMであった。分割放射線療法との実施例7の投与(100mg/kg、s.c.)は、有意な抗腫瘍効果を生じたが、80%死亡率は11日目で達成された。100mg/kg、s.c.でのCmaxにおける血漿レベルは21μMであり、これは化学−感作研究及び上述の初期放射線増感研究におけるこの用量で達成された照射レベルと一致している。
【0090】
これらのデータから、TGDのより大きな増加は、臨床的に関連した分割用量スケジュールを用いた、より低濃度の実施例7(CEP 30;実施例6の30mg/kg用量当量、s.c.、qdX21日間)で観察されたことが示された。加えて、実施例7(CEP 30;実施例6の30mg/kg用量当量、s.c.、qdX21日間)単独は、腫瘍増殖阻害に対して何も影響を及ぼさず、これは実施例7が「真の」放射線増感剤として作用することを示している。
【0091】
治療効果を評価するために、実施例7(実施例6の30及び100mg/kg用量当量、s.c.)プラス2Gy放射線X5日間は、骨髄及び空腸腺窩アッセイで評価し、実施例7が放射線誘導性正常組織(NT)毒性を増強するかどうか決定した。骨髄及び腸管粘膜の評価によって、実施例7(実施例6の30及び100mg/kg用量当量、s.c.)は、これらの組織において放射線毒性を増強しなかったことが明らかになった。この研究によって、CEP−9722は経口的に投与された場合、放射線増感効果を発揮することが示唆された。これらの組み合わせたデータによって、実施例7は、放射線耐性グリオーマモデルにおいて分割放射線療法の有効性を増加することによって、放射線増感剤としてより相加的に作用し、放射線誘導性NT毒性を増強しないことが示唆された。
【0092】
実施例
本発明の化合物は、これに限定されるのもではないが以下に記載された方法を含む本分野の当業者にはよく知られた多くの方法において、或いは標準技術を適用することによるこれら方法を修飾した方法を介して調合した。本発明に関連して開示された全ての工程は、ミリグラム、グラム、マルチグラム、キログラム、マルチキログラム或いは商工業スケールを含むあらゆるスケールで実行されると考えられる。
【0093】
本発明は、PARPの阻害剤として有用な、本明細書に記載された複環状化合物を調合する方法を扱う。前記方法は、4−メトキシインドールで開始する多段階合成から成る。特に、4−メトキシインドールAは、例えば、ブチルリチウム、二酸化炭素、t−ブチルリチウム、及びケトンBとともに連続的に処理し、2−置換4−メトキシインドール三級アルコールCを提供した。この三級アルコールは、例えば、塩酸或いはトルエンスルホン酸を用いた酸性条件下で除去し、置換2−ビニルインドールDを得た。限定されるものではないが、マレイミド(E)などの求ジエン体でのDのディールズ−アルダー環化付加反応によって、環化付加反応中間産物Fを得た。例えばパラジウム或いは白金などの触媒の存在下での酸素による、若しくはDDQ或いはテトラクロロキノンなどの酸化剤による前記環化付加反応中間産物の芳香環化によって、カルバゾールGを産生した。
アルキル化或いはアシル化剤でのGのさらなる処理によって、本発明のインドール−N−置換カルバゾール誘導体を得た。適切なプロドラッグ誘導体を選択及び調合するための従来の方法は、例えば、Prodrugs,Sloane,K.B.,Ed.;Marcel Dekker:New York,1992に記載されており、これはこの参照によってその全体が本明細書に組み込まれるものである。
【0094】
本発明の化合物はPARP阻害剤である。前記阻害剤の力価は、in vitro或いはin vivoでのPARP活性を測定することによってテストされ得る。好ましいアッセイは、[32P]NAD+からヒストン或いはPARP自身などのタンパク質受容体への放射線標識ADP−リボースユニットの転移をモニタリングすることである。PARPに対する日常的なアッセイは、Purnell and Whish,Biochem.J.1980,184,775に開示されており、これはこの参照によって本明細書に組み込まれるものである。
【実施例1】
【0095】
細胞株
U87MGヒト神経膠芽腫細胞は、1.5g/L炭酸水素ナトリウム、0.1nM非必須アミノ酸、10%ウシ胎児血清(FBS)を含む1.0nMピルビン酸ナトリウムと共に、商業的に利用可能な基礎培地(MEM)において培養した。
【実施例2】
【0096】
腫瘍細胞移植及び増殖
対数増殖細胞を播種し、商業的に利用可能な無胸腺NCRヌードマウスの右脇腹へ注入した((2X106)細胞/マウス)。200から400mm3の腫瘍を有する動物をサイズに基づいて適切な治療群へランダム化した(n=10)。腫瘍は、ノギスを用いて3から4日毎に測定した。腫瘍容積は、以下の公式:
V(mm3)=0.5236x長さ(mm)x幅(mm)[長さ(mm)+幅(mm)/2]
を用いて計算した。
【実施例3】
【0097】
方法:U87MGヒト神経膠芽腫細胞は、無胸腺NCR NUMマウスの右後肢へと皮下に(s.c.)注入し、平均腫瘍容積の200mm3へと増殖させた。放射線療法を受けたマウスは、100mg/kgケタミン+10mg/kgキシラジン(xylezine)、或いは37.5mg/kgケタミン+0.2mg/kgアセプロマジンをs.c.で照射する前に麻酔し、25から30分間鎮静させた。麻酔させたマウスは、過度の圧力なく、動物の体のサイズ及び形に合わせた可鍛性鉛遮蔽に位置付けした。その体は鉛によって遮蔽した。腫瘍を有する肢或いは曝露された腫瘍に適切な線量を照射した。腫瘍を照射した後、そのマウスは、麻酔から覚めるまでケージ或いは加温パッドへ戻した。実施例7は放射線(RT)後できるだけすぐ(30分以内)に与えた。図3:マウスは以下の治療群(n=10)へランダム化した:1)溶媒、2)放射線のみ(7.5Gyを3日間)、3)実施例7のみ(実施例6の100mg/kg用量当量、s.c.、5日間のQD)、及び4)実施例7プラス放射線。実施例7或いは溶媒は、1から5日目に、及び2、3及び4日目に放射線後30分にs.c.で投与した。データの解析は、時間及び治療の機能として腫瘍容積の10を底とする対数をモデルにした混合効果回帰を用いて行った。解析は、SAS8.3(SAS Institute Inc.,Cary,ノースカロライナ州)で実行した。図1及び2:マウスは、以下の治療群にランダム化し、投与した:1)溶媒、2)RT(5X2Gy)、3)RTプラス実施例7(30或いは100mg/kg、s.c.、実施例6の用量当量、qdx21日間)、或いは4)実施例7(30或いは100mg/kg、s.c.、実施例6の用量当量、qdx21日間)のみ。実施例7は1から21日目に投与し、RTは1から5日目に行った。全動物は、同日に測定した。個々の腫瘍容積測定は対数変換直線モデルをモデルにし、腫瘍が約2000mm3に到達する最適時間を測定した。一元ANOVA及び事後解析は、有意差を決定するために使用した。P値<0.05は有意差があると考えられた。
【0098】
結果:全群は、200mm3の同様のサイズの腫瘍の治療で開始した(P=0.83、0日目の比較群)。図3に示されたように、実施例7単独、放射線のみ、及び放射線との組み合わせでの実施例7(実施例6の100mg/kg用量当量s.c.放射線のqd2日前、及び7.5Gy放射線の組み合わせで3日間)の投与は、確立した腫瘍を有するマウスに行った。単剤として投与された実施例7は、腫瘍増殖に対して影響を及ぼさなかった。溶媒或いは実施例7のみで処理された腫瘍は、それぞれ10.0日或いは9.6日(P=0.798対コントロール)に2000mm3の腫瘍容積に達した。放射線単独の投与は、2000mm3に到達する時間が16.1日に延長され、腫瘍増殖遅延(TDG)も6.1日(P=0.033、対コントロール)に延長した。放射線療法との実施例7の併用療法は、腫瘍が2000mm3に到達する時間を24.8日に延長し、これは14.8日TGDに対応する。併用療法の有効性の程度は、実施例7のみ(P=0.001)或いは放射線のみ(P=0.006)の比較計画によって見られたものより強度であり、これは実施例7が真の放射線増感剤の特性を示すことを示唆するものである。図1及び2に示されたように、RTとの組み合わせでの実施例7の投与(CEP 30;実施例6の30mg/kg用量当量、s.c.)は、TGDを15日に延長(これは放射線のみ(P≦0.05)と比較して4倍増加);及び26日に延長(これは実施例7のみ(P≦0.001)と比較して6倍増加)した。分割放射線療法との実施例7(100mg/kg、s.c.)の投与は、有意な抗腫瘍効果を生じたが、80%死亡率は11日であった。これらのデータによって、TGDにおける有意な延長は、臨床的に関連する分割線量スケジュールを用いた実施例7(CEP 30;30mg/kg)の低濃度で観察されたことが示された。加えて、実施例7のみの投与は、腫瘍増殖阻害に対して影響を及ぼさず、これは実施例7が「真の」放射線増感剤として作用することを示すものである。
【実施例4】
【0099】
DNA障害の評価
抗体:一次抗体は、リン酸ヒストンH2AX(Cell Signaling,#2577,1:1000)及びGAPDH(Abcam,#9484,1:5000)に対して使用した。二次抗体は、ヤギ抗マウスIRDye800(Rockland,#610−132−121)及びヤギ抗ウサギAlexa fluor 700(Molecular Probe,#A21038)であった。
【0100】
U87MG細胞は、3Gy或いは5Gy放射線を照射し、その後実施例6(300nM及び1μM)で放射後0.5時間処理した。サンプルは次に、実施例6の添加後0.5、1及び4時間で回収した。次に前記細胞はRIPAバッファー(150mM NaCl、1% NP−40、0.5%デオキシコール酸ナトリウム、0.1% SDS、50mM トリス pH8.0)プラス阻害剤カクテル(プロテアーゼ阻害剤カクテルセットIII、Calbiochem)において氷上で溶解し、次に1mM Na3VO4はBCAタンパク質アッセイキット(Pierce#23225)を用いて定量化した。サンプルは、140ボルトで、MES SDSバッファー(Novex,#NP0002)での4−12%ビストリスゲル(Novex#NP0336)を用いて電気泳動(15μgタンパク質)で除去し、次に2Xトランスファーバッファー(Novex,#NP0006)を用いたセミドライトランスファー(18ボルトで35分)によって、ニトロセルロース膜(Biorad,#162−0145)へ移した。次に膜は、1XTBSでの1:1希釈のOdysseyブロッキングバッファー(Licor#927−40000)において室温で1時間ブロッキングし、次に1XTBS−T0.05%での1:1希釈のOdysseyブロッキングバッファーにおいて両一次抗体と共に4℃で一晩インキュベーションさせた。翌日、膜は1XTBS−T0.2%で各洗浄10分間を4回洗浄し、次に1:10,000(1XTBS−T0.05%での1:1希釈のOdysseyブロッキングバッファーにおいて)で両二次抗体と共に、遮光、室温で1.5時間インキュベーションした。ブロットは、1XTBS−T0.2%で各洗浄10分間(遮光)を4回洗浄し(遮光)、次にOdyssey Infrared Imagerで解析した。GAPDHは800nmシグナルを用いて可視化し、次にリン酸−H2AXは700nmで検出した。リン酸ヒストンH2Xの予想サイズは15kDaであり、GAPDHは36kDaであった。
【実施例5】
【0101】
細胞周期解析
U87MG細胞は、3Gy或いは5Gy放射線に照射し、次に照射後0.5時間で、実施例6(300nM及び1μM)で処理した。サンプルは、実施例6の添加後8、24及び48時間(或いは本分野の当業者によって決定されたあらゆる時間)で回収した。細胞は、4℃で一晩、100%エタノールにおいて固定させた。翌日細胞を、遮光、室温で1時間、細胞周期試薬(Guava Technologies#4500−0220)と共にインキュベーションした。染色された核はフローサイトメトリー(Guava EasyCyte;本分野の当業者に既知であるセッティングを用いる、例えば427X8;得られたデータ5,000イベント/サンプル)によって解析した。細胞周期の各相における細胞のパーセンテージは、細胞周期解析ソフトウェア(Guava Technologies)を用いて決定した。
【実施例6】
【0102】
7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化13】
【0103】
工程1:乾燥THF(20mL)における4−メトキシインドール(2.0g、13.1ミリモル)の冷却溶液(−78℃)へ、nBuLiヘキサン(2.5M、5.2mL、13.1ミリモル)をゆっくり添加した。その混合物を30分間−78℃でかき混ぜ、次にCO2ガスを15分間反応混合液へ泡立て、その後付加的に15分間かき混ぜた。過剰CO2及び半分のTHF容積を減圧下で除去した。付加的な乾燥THF(10mL)を、再び−78℃へ冷却した前記反応混合物へ添加した。1.7M t−BuLi(7.7mL、13.1ミリモル)を前記反応混合物へ30分かけてゆっくり添加した。−78℃で2時間かき混ぜ続け、その後乾燥THF(5mL)におけるシクロペンタノン(1.7g、20.4ミリモル)の溶液をゆっくり添加した。−78℃で1時間付加的にかき混ぜた後、前記反応混合物は水の液滴添加(5mL)、その後飽和NH4Cl溶液(20mL)によってクエンチした。エチルエーテル(50mL)を添加し、前記混合物を室温で10分間かき混ぜた。有機層を分離し、乾燥させ(MgSO4)、濃縮し、アルコール(1−(4−メトキシ−1H−インドール−2−イル)−シクロペンタノール)及びジエン(2−シクロペント−1−エニル−4−メトキシ−1H−インドール)の混合物を得た。アセトン(15mL)における前記混合物へ2N HCl(5mL)を添加した。前記混合物を10分間かき混ぜ、水(50mL)を添加し、前記ジエン産物2−シクロペント−1−エニル−4−メトキシ−1H−インドールを回収し、真空下で回収した。前記産物はシリカゲルクロマトグラフィー(EtOAC/ヘキサン9:1)で精製した。1H NMR(DMSO−d6)δ 1.9−2.1(m,3 H),2.6−2.75(m,3H),3.9(s,3H),6.1(s,IH),6.3(s,IH),6.4(m,IH),6.9−7.0(m,2H),11.1(s,IH)。この産物は次の工程に直接使用した。
【0104】
工程2:酢酸(5mL)における2−シクロペント−1−エニル−4−メトキシ−1H−インドール(0.1g、0.47ミリモル)及びマレイミド(0.09g、0.91ミリモル)の混合物を室温で1時間かき混ぜた。水を添加し、産物をEtOAcで抽出し、それは2N Na2CO3溶液、水、及び飽和NaCl溶液で洗浄し、乾燥させた(MgSO4)。乾燥剤を濾過によって除去し、溶液を濃縮し、0.13g MSを得た:m/z309(M−H)。
【0105】
工程3:トルエン(2mL)及び酢酸(3mL)における工程2からの産物(0.123g、0.4ミリモル)は、2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(DDQ、185mg、0.8ミリモル)へ添加した。0℃で30分かき混ぜた後、前記混合物を濃縮し、EtOAC及びアスコルビン酸で処理した。30分後前記混合物を2N Na2CO3で塩基性にした。EtOAc層を水で洗浄し、NaCl溶液で飽和させ、乾燥させ(MgSO4)、濃縮し、産物0.095mgを得た;MS:m/z305(M−H)+.1H NMR(DMSO−d6)δ 2.26−2.31(m,2H),3.1−3.2(m,2H),3.3−3.4(m,2H),3.9(s,3H),6.7(m,IH),7.1(m IH),6.4(m,IH),7.4(m,IH),10.6(s,IH),11.9(s,IH)。
【実施例7】
【0106】
7−メトキシ−5−(4−メチル−ピペラジン−1−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化14】
【0107】
エタノール(950mL)における実施例6(10.0g、30ミリモル)及びN−メチルピペラジン(12.4g、124ミリモル)のスラリーへパラホルムアルデヒド(5.60g、62.4ミリモル)を0.5時間で添加し、24時間かき混ぜた。前記スラリーは乾燥するまで蒸発させた。残留物へヘキサン(500mL)を添加し、15分間超音波処理し、1.5時間かき混ぜ、0℃で15分間冷却した。黄色の固形を回収し、冷却ヘキサンで洗浄した。この産物は温テトラヒドロフラン(THF)(250mL)に溶解し、濾過した。濾過物をヘキサン(3L)へ液滴添加し、15分間かき混ぜ、実施例7を沈殿物として回収し、ヘキサンで洗浄した(12.0g、96%収率)。1H NMR(DMSOd6)2.12(s,3H),2.35(m,8H),2.53(m,4H),3.18(m,2H),4.44(s,3H),6.70(d,lH),7.10(d,lH),7.40(t,lH),11.96(s,lH).MS m/z 419(M+H)。
【実施例8】
【0108】
7−メトキシ−5−(ジエチルアミノメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン(実施例8a)
7−メトキシ−5,11−(ビス−ジエチルアミノメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン(実施例8b)
【化15】
【0109】
DMF(5mL)における実施例6(50mg、0.16ミリモル)のスラリーへ、パラホルムアルデヒド(73mg、0.81ミリモル)、ジエチルアミン(84μL、0.81ミリモル)を添加し、室温で1日かき混ぜた。反応物を蒸発させ、残留物をヘキサンで粉砕し、蒸発させ、油として2つの産物を得た(比6−1、16b:16c)。1H−NMR(DMSOd6)0.98(t,3H),1.11(t,3H),2.27(m,2H),2.53(m,8H),2.57(m,15H),3.17(t,2H),3.50(m,lH),3.97(s,3H),4.14(d,2H),4.71(d,2H),6.82(t,2H),6.75(d,2H),7.13(d,2H),7.33(m,lH),7.46(t,3H),7.52(m,lH),11.95(s,lH).16b:MS m/z 392.16c MS m/z 476。
【実施例9】
【0110】
7−メトキシ−5,11−(ビス−モルフォリン−4−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化16】
【0111】
DMF(1mL)における実施例6(15mg、0.049ミリモル)のスラリーへ、パラホルムアルデヒド(42mg、0.05μL)、モルフォリン(160mg、1.9ミリモル)を添加し、70℃で18時間加熱した。混合物を蒸発させた。残留物をヘキサンで粉砕し、次にCH2Cl2に溶解し、濾過し蒸発させた。残留物をEt2Oで粉砕し、十知れを黄色の固形物として回収した(5mg、20%)。1HNMR(DMSO−J6)7.52(t,IH),7.39(d,IH),6.82(d,IH),5.0(s,2H),4.46(s,2H),3.98(s,3H),3.56(s,6H),3.49(s,4H),2.50(s,6H),2.49(s,4H),2.45(m,2H);MS m/z 505(M+H)。
【実施例10】
【0112】
7−メトキシ−5−(モルフォリン−4−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオン
【化17】
【0113】
エタノール(10mL)における実施例6(50mg、0.16ミリモル)のスラリーへ、パラホルムアルデヒド(72mg、0.8ミリモル)、モルフォリン(100mg、1.1ミリモル)を添加し、50℃で5時間加熱した。反応物を蒸発させ、水を添加し(15mL)、黄色の固形物を回収した(59mg)。1H NMR(DMSO−d6)11.98(s,IH),7.45(t,IH),7.13(d,IH),6.75(d,IH),4.44(s,2H),3.97(s,3H),3.56(s,4h),3.18(t,2h),2.29(t,2h).MS m/z 406(M+H)。
【実施例11】
【0114】
PARP酵素活性の測定
PARP活性は、[32P]NAD+からヒストン或いはPARP自身などのタンパク質受容体への放射線標識ADP−リボースユニットの転移によってモニタリングした。前記アッセイ混合物は、最終容積100μLにおいて100mMトリス(pH8.0)、2mM DTT、10mM MgCl2、20μg/ml DNA(超音波処理によってニックを入れた)、20mg/mlヒストンH1、5ng組換えヒトPARP、及び阻害剤或いはDMSO(<2.5%(v/v))を含む。反応は、2μCi[32P]NAD+/mL1を補充した00μM NAD+の添加で開始し、室温で12時間維持した。アッセイは100μMの50%TCAの添加によって終了し、放射線標識沈殿物を96ウェルフィルタープレート(Millipore,MADP NOB50)上で回収し、25% TCAで洗浄した。ポリADP−リボシル化タンパク質と一致する、酸不溶性放射線活性の量は、Wallac MicroBetaシンチレーションカウンターで定量化した。
【0115】
阻害剤に対するIC50の決定
一点阻害データは、DMSOのみ存在する場合の活性に対して、阻害剤の存在下でPARP、VEGFR2或いはMLK3活性と比較することによって計算した。化合物に対する阻害曲線は、パーセント阻害に対して化合物の濃度のlog10をプロットすることによって作成した。IC50値は、以下のGraphPad PrismにおけるS状用量反応性(可変傾き)方程式:
y=ボトム+(トップ−ボトム)/(1+10(logIC50−x)*斜面(Hillslope))
を用いた非直線回帰によって計算し、ここにおいて、yは化合物の既知濃度での%活性であり、xは化合物の濃度の対数であり、ボトムはテストした最も低い化合物濃度での%阻害であり、トップは試験した最も高い化合物濃度での%阻害である。ボトム及びトップに対する値は、それぞれ0及び100で固定した。IC50値は、少なくとも3回の別々の決定の平均として報告した。
【0116】
本明細書で開示されたアッセイを用いて、以下の表2から、PARP阻害に対する本発明の化合物の有用性が示された。本発明の化合物は、それらのIC50値が50μM以下の場合活性であると考えられた。PARP阻害に対する以下の表において、PARP阻害に対するIC50は、「+」となった本発明の化合物は10000nM以下であり;「++」となった本発明の化合物は1000nM以下であり;「+++」となった本発明の化合物は100nM以下である。IC50値が示されてない所は、まだデータを決定してない所である。
【0117】
【表1】
【実施例12】
【0118】
先行研究は、経口投与された実施例7の放射線増感能力を決定するために行った。
【0119】
腫瘍細胞移植及び増殖
指数関数的に増殖した細胞は播種し、商業的に利用可能な胸腺欠損NCR nu/nuヌードマウスの右側腹部へ注入した(2x106細胞/マウス)。200から400mm3の腫瘍を有する動物は、適切な処理群へサイズに従ってランダム化した(n=4)。腫瘍は、ノギスを用いて3から4日毎に測定した。腫瘍容積は、以下の公式を用いて計算した:
V=a2b/2、ここにおいてa及びbはそれぞれ、短寸法及び長寸法である。
【0120】
方法:U87MGヒト神経膠芽細胞腫細胞を、胸腺欠損NCR nu/nuヌードマウスの右後肢へ皮下的に(s.c.)注入し、平均腫瘍容積が200mm3になるように増殖させた。放射線療法を受けたマウスは、100mg/kgケタミン+10mg/kgキシラジン、若しくは37.5mg/kgケタミン+0.2mg/kgアセプロマジン、s.c.と共に照射を行う前に麻酔し、25から30分間鎮静させた。麻酔させたマウスは、過度の圧力なく、動物の体のサイズ及び形に合わせた可鍛性鉛遮蔽に位置付けした。その体は鉛によって遮蔽した。腫瘍を有する肢或いは曝露された腫瘍に適切な線量を照射した。腫瘍を照射した後、そのマウスは、麻酔から覚めるまでケージ或いは加温パッドへ戻した。実施例7は放射線(RT)後できるだけすぐ(30分以内)に与えた。マウスは以下の治療群へランダム化し、投薬した:1)溶媒、2)RT(2GyX5日)、3)RTプラス実施例7(200或いは300mg/kg p.o、実施例6の用量当量、qdX21日)、或いは4)実施例7(200或いは300mg/kg p.o、実施例6の用量当量、qdX21日)のみ。実施例7は、1から21日目に投与し、RTは1から5日目に行った。全動物は、同日に測定した。個々の腫瘍容積測定は対数変換直線モデルをモデルにし、腫瘍が約2000mm3に到達する最適時間を測定した。
【0121】
結果:図5に示したように、実施例7の投与(Cep 300;実施例6の300mg/kg用量当量、p.o.、qdx21日)プラスRT(2Gyx5日)は、8日目から腫瘍増殖静止状態となり、研究中(31日)続いた一方、実施例7(Cep 200;実施例6の200mg/kg用量当量、p.o.、qdx21日)プラスRT(2Gyx5日)及び実施例7のみ(実施例6の200及び300mg/kg用量当量、p.o.、qdx21日)の投与は、RTのみと比較して腫瘍増殖において影響を及ぼさなかった。得られた結果より、実施例7投与のみは腫瘍増殖に対して影響を及ぼさないということがs.c.投与から得られたデータによって確認されたことが示唆された。
【0122】
多数の変更及び修飾は本発明の好ましい実施形態になされ、そのような変更及び修飾は、本発明の観点から逸脱することなくなされ得るものであることは、本分野の当業者によって理解されるであろう。従って、添付された請求項は、本発明の観点及び範囲内にあるそのような同等なバリエーションの全てをカバーするものであることが意図される。
【0123】
本明細書に引用された全ての参考文献は、この参照によってその全てが本明細書に組み込まれる。
【特許請求の範囲】
【請求項1】
化学式(I)の放射線増感剤、
【化18】
或いはその薬学的に許容可能な塩形態(XはH或いはプロドラッグ部位である)を、癌を患った哺乳動物に投与する工程と
イオン化放射線を前記哺乳動物に適用させる工程と
により、癌を治療する方法。
【請求項2】
請求項1記載の方法において、前記放射線増感剤は、前記組織内或いは前記組織に近接して存在し、前記適用イオン化放射線を局所的治療効果に変換する効率を増強させるものである。
【請求項3】
請求項2記載の方法において、前記放射線増感剤は、癌細胞を放射線増感させるのに有効な量で存在するものである。
【請求項4】
請求項3記載の方法において、前記組織のイオン化放射線は、前記細胞を破壊するのに有効な放射線線量で実行されるものである。
【請求項5】
請求項4記載の方法において、前記組織のイオン化放射線は、所定の癌タイプに対して臨床的に許容可能或いは推奨される放射線療法プロトコールである。
【請求項6】
請求項4記載の方法において、前記癌は、悪性である。
【請求項7】
請求項4記載の方法において、前記癌は、良性である。
【請求項8】
請求項1の方法において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、及び−C(=O)R4から成る群から選択されるものであり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素原子とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【請求項9】
請求項1記載の方法において、前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【請求項10】
請求項1の方法において、前記プロドラッグ部位は、マンニッヒ塩基である。
【請求項11】
請求項8記載の方法において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチル−、モルフォリン−4−イルメチル−、及び5−ジエチルアミノメチル−から選択されるものである。
【請求項12】
請求項8記載の方法において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチルである。
【請求項13】
請求項1記載の方法において、投与の経路は、静脈内、皮下、経口或いは腹腔内である。
【請求項14】
請求項1の方法において、投与の経路は、静脈内である。
【請求項15】
請求項1記載の方法において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌から選択されるものである。
【請求項16】
請求項1記載の方法において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【請求項17】
癌を治療する方法であって、
化学式7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤、或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程と、
イオン化放射線を前記哺乳動物に適用させる工程と
により、癌を治療する方法。
【請求項18】
癌を治療する方法であって、
化学式7−メトキシ−5−(4−メチル−ピペラジン−1−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤、或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程と、
イオン化放射線を前記哺乳動物に適用させる工程と、
により、癌を治療する方法。
【請求項19】
癌細胞を放射線増感するための薬学的組成物であって、
化学式(I)の化合物、
【化19】
或いはその薬学的に許容可能な塩形態(XはH或いはプロドラッグ部位である)の放射線増感量を有する、薬学的組成物。
【請求項20】
請求項19記載の薬学的組成物において、
前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、及び−C(=O)R4から成る群から選択されるものであり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択さるヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【請求項21】
請求項19記載の薬学的組成物において、
前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【請求項22】
請求項19記載の薬学的組成物において、前記化合物は、
【化20】
或いはその薬学的に許容可能な塩形態である。
【請求項23】
請求項19記載の薬学的組成物において、前記化合物は、
【化21】
或いはその薬学的に許容可能な塩形態である。
【請求項24】
化学式(II)の化合物、
【化22】
或いはその薬学的に許容可能な塩形態。
【請求項1】
化学式(I)の放射線増感剤、
【化18】
或いはその薬学的に許容可能な塩形態(XはH或いはプロドラッグ部位である)を、癌を患った哺乳動物に投与する工程と
イオン化放射線を前記哺乳動物に適用させる工程と
により、癌を治療する方法。
【請求項2】
請求項1記載の方法において、前記放射線増感剤は、前記組織内或いは前記組織に近接して存在し、前記適用イオン化放射線を局所的治療効果に変換する効率を増強させるものである。
【請求項3】
請求項2記載の方法において、前記放射線増感剤は、癌細胞を放射線増感させるのに有効な量で存在するものである。
【請求項4】
請求項3記載の方法において、前記組織のイオン化放射線は、前記細胞を破壊するのに有効な放射線線量で実行されるものである。
【請求項5】
請求項4記載の方法において、前記組織のイオン化放射線は、所定の癌タイプに対して臨床的に許容可能或いは推奨される放射線療法プロトコールである。
【請求項6】
請求項4記載の方法において、前記癌は、悪性である。
【請求項7】
請求項4記載の方法において、前記癌は、良性である。
【請求項8】
請求項1の方法において、前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、及び−C(=O)R4から成る群から選択されるものであり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素原子とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【請求項9】
請求項1記載の方法において、前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【請求項10】
請求項1の方法において、前記プロドラッグ部位は、マンニッヒ塩基である。
【請求項11】
請求項8記載の方法において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチル−、モルフォリン−4−イルメチル−、及び5−ジエチルアミノメチル−から選択されるものである。
【請求項12】
請求項8記載の方法において、前記マンニッヒ塩基は、4−メチル−ピペラジン−1−イルメチルである。
【請求項13】
請求項1記載の方法において、投与の経路は、静脈内、皮下、経口或いは腹腔内である。
【請求項14】
請求項1の方法において、投与の経路は、静脈内である。
【請求項15】
請求項1記載の方法において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、星状細胞腫、乏突起膠細胞腫、オリゴ星状細胞腫、髄膜腫、神経芽細胞腫、横紋筋肉腫、軟部組織肉腫、骨肉腫、中枢神経系部位での造血器腫瘍、乳癌(腺管及び上皮内癌)、甲状腺癌(乳頭状及び瀘胞状)、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、肝細胞癌、大腸−結腸癌、子宮頸癌、卵巣癌、前立腺癌、精巣癌、胃癌、膵臓癌、胆管肉腫、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、白血病(骨髄及びリンパ由来の急性及び慢性白血病)、及び膀胱癌から選択されるものである。
【請求項16】
請求項1記載の方法において、前記癌は、頭頸部扁平上皮癌(目、唇、口腔、咽頭、喉頭、舌の癌腫、及び食道癌腫)、メラノーマ、扁平上皮細胞癌(表皮)、神経膠芽細胞腫、神経芽細胞腫、横紋筋肉腫、肺癌(細気管支肺胞癌、肺小細胞癌、小細胞/大細胞混合肺癌、混合肺小細胞癌、非小細胞肺癌、扁平上皮細胞癌、大細胞癌、及び肺の腺癌)、リンパ腫(T細胞及びB細胞由来のホジキン及び非ホジキン型)、及び白血病(骨髄及びリンパ由来の急性及び慢性白血病)から選択されるものである。
【請求項17】
癌を治療する方法であって、
化学式7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤、或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程と、
イオン化放射線を前記哺乳動物に適用させる工程と
により、癌を治療する方法。
【請求項18】
癌を治療する方法であって、
化学式7−メトキシ−5−(4−メチル−ピペラジン−1−イルメチル)−1,2,3,11−テトラヒドロ−5,11−ジアザ−ベンゾ[a]トリンデン−4,6−ジオンの放射線増感剤、或いはその薬学的に許容可能な塩形態を、癌を患った哺乳動物に投与する工程と、
イオン化放射線を前記哺乳動物に適用させる工程と、
により、癌を治療する方法。
【請求項19】
癌細胞を放射線増感するための薬学的組成物であって、
化学式(I)の化合物、
【化19】
或いはその薬学的に許容可能な塩形態(XはH或いはプロドラッグ部位である)の放射線増感量を有する、薬学的組成物。
【請求項20】
請求項19記載の薬学的組成物において、
前記プロドラッグ部位は、−CH2NR1R2、−CH2OC(=O)R3、−CH2OP(=O)(OH)2、及び−C(=O)R4から成る群から選択されるものであり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択さるヘテロシクリル基を形成するものであり、前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものであり、
R3は、−C1−4アルキル−NR1R2、−C1−4アルキル−OR5、ピリジニル、−フェニル(CH2NR1R2)、及び−CH(R6)NH2から成る群から選択されるものであり、
R4は、−O−(C1−4アルキル)−NR1R2、−O−(C1−4アルキル)−OR5、及び−CH(R6)NH2から成る群から選択されるものであり、
R5は、H或いはC1−4アルキルであり、
R6は、天然由来アミノ酸の側鎖である。
【請求項21】
請求項19記載の薬学的組成物において、
前記プロドラッグ部位は、−CH2NR1R2であり、
R1は、H或いはC1−4アルキルであり、
R2は、H或いはC1−4アルキルであるか、
或いは、R1及びR2は、それらが結合する窒素元素とともに、ピロリル、ピロリジニル、ピペリジニル、モルフォリニル、チオモルフォリニル及びピペラジニルから選択されたヘテロシクリル基を形成するものであり、ここにおいて前記ヘテロシクリル基は任意にC1−4アルキルで置換されるものである。
【請求項22】
請求項19記載の薬学的組成物において、前記化合物は、
【化20】
或いはその薬学的に許容可能な塩形態である。
【請求項23】
請求項19記載の薬学的組成物において、前記化合物は、
【化21】
或いはその薬学的に許容可能な塩形態である。
【請求項24】
化学式(II)の化合物、
【化22】
或いはその薬学的に許容可能な塩形態。
【図1】


【図2】

【図3】

【図4】

【図5】

【図2】
【図3】
【図4】
【図5】
【公表番号】特表2010−510312(P2010−510312A)
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願番号】特願2009−538405(P2009−538405)
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/US2007/024271
【国際公開番号】WO2008/063644
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/US2007/024271
【国際公開番号】WO2008/063644
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
[ Back to top ]