
気管支感染症の治療法
【課題】気管支上皮感染症の患者に対して、90〜130mgのアミノグリコシド系抗生物質を、ドライパウダーエアゾル組成物として患者の気管支系に、20〜36日間の第一治療期間中に1日1〜3回投与することによる治療法を提供すること。
【解決手段】本発明の態様によれば、第一治療期間の後に第二の治療休止期間を設けることができ、その間は患者の気管支系にはアミノグリコシド系抗生物質は投与されない。悪性の感染症の治療に対しては、患者の気管支系へのアミノグリコシド系抗生物質投与による第一の治療期およびその後のアミノグリコシド系抗生物質を投与しない第二の治療休止期のサイクルを、抗菌効果が現われるまで2回またはそれ以上繰返して行うことができる。CF患者が罹患する感染症のような慢性感染症の場合では、この第一および第二の治療期のサイクルを、患者治療中に複数回繰返すことができる。
【解決手段】本発明の態様によれば、第一治療期間の後に第二の治療休止期間を設けることができ、その間は患者の気管支系にはアミノグリコシド系抗生物質は投与されない。悪性の感染症の治療に対しては、患者の気管支系へのアミノグリコシド系抗生物質投与による第一の治療期およびその後のアミノグリコシド系抗生物質を投与しない第二の治療休止期のサイクルを、抗菌効果が現われるまで2回またはそれ以上繰返して行うことができる。CF患者が罹患する感染症のような慢性感染症の場合では、この第一および第二の治療期のサイクルを、患者治療中に複数回繰返すことができる。
【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本発明は、気管支感染症に抵抗性のない患者に対して、トブラマイシンのようなアミノグリコシド系抗生物質のドライパウダー製剤を用いる新規の改善された治療法に関する。
【背景技術】
【0002】
背景技術
嚢胞性線維症(CF)は、米国およびヨーロッパ北部において最も良く知られている寿命を縮める遺伝的疾患であって、米国では約30000人が罹患(Cunningham,J.C.ら、“An Introduction to Cystic Fibrosis for Patients and Families,”第5版、Bethesda:Cystic Fibrosis Foundation(2003))し、西ヨーロッパにおいても同様の患者数にのぼる。常染色体劣性であるこの疾患の遺伝的欠陥は、塩素イオンチャンネル蛋白質をコードするCF膜貫通コンダクタンス制御(CFTR)遺伝子(Collins,F.S.,“Cystic Fibrosis Molecular Biology and Therapeutic Implications,”Science 256:774−779(1992))におけるある突然変異である。一般的に慢性気管支感染症、副鼻腔炎、および膵臓の機能不全による消化器吸収不良を患うCF患者では、発汗による塩分損失、閉塞性肝胆嚢疾患および生殖能の低下が多く認められた(FitzSimmons,S.C.,“The Changing Epidemiology of Cystic Fibrosis,”J.Pediatr.122:1−9(1993))。CF患者では、呼吸器疾患が病状の主要な要因であり、それによる死亡率は90%にも達する(Cystic Fibrosis Foundation,Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report,Bethesda,MD:Cystic Fibrosis Foundation,(2004);Davis,P.B.ら、“Cystic fibrosis”,Amer.J.Respir.Crit.Care Med 154(5):1229−56(1996))。肺機能(努力呼気肺活1秒量(予測FEV1)として計測される)は、CFにおける生存率を予測する重要な因子の一つである。あるCF患者集団の2年生存率の減少率は、予測FEV1が10%減少すると2倍になり、患者のFEV1が予測値の30%未満になるとその2年生存率は50%未満となる(Kerem,E.ら、“Predictioin of Mortality in Patients with Cystic Fibrosis,”N.Engl.J.Med.326:1187−1191(1992))。肺機能の低下速度に関しては、患者によっても罹患期間もよっても異なる。遡及的な長期の分析から、肺機能低下速度は年間予測FEV1で2%未満から年間予測FEV1で9%超まであることがわかり、低下速度は全般に死亡年齢に関連しているとされる(Corey,M.ら、“Longitudinal Analysis of Pulmonary Function Decline in Patient with Cystic Fifrosis,”J.Pediatr.131(6):809−1(1997))。
【0003】
CF患者は、肺の宿主防御をそこなう上皮イオン輸送の混乱によって生じる粘液増粘症状を患っており、それによってStaphylococcus aureus,Haemophilus infuluenzaeおよびP.aeruginosaによる早期の気管支感染症に対する抵抗性が低下している。CF患者の大部分は、思春期までに喀痰中にP.aeruginosaが存在するようになる(Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report(2004))。慢性の気管支感染症、特にP.aeruginosaによる感染症によって気道の持続的な炎症反応が誘発され、それによって広汎性気管支拡張の所見を伴う進行性の閉塞性疾患が悪化する(Davis,P.B.ら、(1996),前記文献;Winnie,G.B.ら、“Respiratory Tract
Colonization with Pseudomonas aeruginosa in Cystic Fibrosis:Correlations Between Anti−Pseudomonas aeruginosa Antibody Levels And Pulmonary Function,”Pediatr.Pulmonol.10:92−100(1991);Ballmann,M.ら、“Long Term Follow Up of Changes in FEV1 and Treatment Intensity During Pseudomonas Aeruginosa Colinisation in Patients with Cystic Fibrosis”, Thorax 53:732−737(1998);Pamukcu,A.ら、”Effects of Pseudomonas aeruginosa Colonisation on Lung Function and Anthropometric Variables in Children with Cystic Fibrosis”,Pediatr.Pulmonol.19:10−15(1995))。P.aeruginosaによる慢性の気管支感染症への罹患から始まり、肺炎、肺機能の低下および最終的には死に至るまで関連性があることは、P.aeruginosaによる慢性感染症が伴うと生存率が優位に低下すること(Henry,R.L.ら、“Mucoid Pseudomonas aeruginosa is a Marker of Poor Survival in Cystic Fibrosis”,Pediatr.Pulmonol.12(3):158−61(1992))、ならびにP.aeruginosaによる慢性感染症への早期罹患と小児死亡率とに有意な関連性があること(Demko,C.A.ら、“Gender Differences in Cystic Fibrosis:Pseudomonas aeruginosa Infection”,J.Clin.Epidemiol.48:1041−1049(1995))によって示唆される。肺への細菌の侵入を抑える(MacLusky,I.B.ら、“Long−term Effects of Inhaled Tobramycin in Patients with Cystic Fibrosis Colonized with Pseudomonas aeruginosa”,Pediatr.Pulmonol.7(1):42−8(1989))か、またはその感染の結果生じる炎症を抑える(Konstan,M.W.ら、“Effect of high−dose Ibuprofen in Patients with Cystic Fibrosis”,N.Engl.J.Med.332(13):848−54(1995))ことのどちらかの長期持続的治療を行うと、感染症患者における肺機能低下速度が減少することがこれまでにわかっている。
【0004】
P.aeruginosaによる気管支上皮感染症に対する標準的な治療は、かつては一般にアミノグリコシドを代表とするシュードモナス属細菌に有効な抗生物質を、14〜21日間非経口的に投与して行なわれた。しかしながら、極性が高いアミノグリコシド系薬剤を非経口的に投与しても、気管支上皮内にはほとんど浸透しない。非経口投与によって感染部位での充分な薬物濃度を得るには、腎毒性、内耳前庭毒性および耳毒性に関連する血清中の薬物濃度が欠かせない(非特許文献1;非特許文献2)。
【0005】
アミノグリコシド系薬剤の吸入投与は、全身的な生物学的利用率を最小限にしながら、気管支上皮内の感染部位に高濃度の抗生物質を直接的に送達する、魅力的な代替法を提供する(非特許文献3;非特許文献4)。
【0006】
CF患者におけるP.aeruginosa感染症に対する現行の標準的な治療は、吸入用で保存剤不含、安定で使用しやすいトブラマイシン製剤であるTOBI(登録商標)トブラマイシン溶液(1/4生理食塩水5mL中に溶解したトブラマイシン60mg/mL溶液)をジェットネブライザーで投与して行なわれ、PathoGenesis社(米国、ワシントン州、シアトル市)(現在はChiron社(米国、カリフォルニア州、エメリービル市))で開発されたものである。5mlのTOBI(トブラマイシン300mg)の1日2回投与をPARI LC PLUS/PulmoAide(商品名)コンプレッサー送達システムと組合せは、CF患者におけるP.aeruginosa管理医療を目的とする長期断続的治療として、NDA50−753のもと、1997年12月にFDAにより承認され、そして、これが、この目的のための産業上の標準となっているままである。市販の用量300mgのTOBIの吸入法は、投与自体に20分間かかり、それ以外に投与の準備およびネブライザーの洗浄の時間を要する。患者の気管支上皮内のP.aeruginosaを抑制するための、1/4生理食塩水中に溶解したトブラマイシン300mg含有製剤を用量5mlでエアゾルで投与することに関しては、米国特許第5,508,269号においても開示されており、その開示については本明細書において参考文献としてそのまま掲載されている。
【0007】
トブラマイシンは、放線菌の一種であるStreptomyces tenebrariusによって生産されるアミノグリコシド系抗生物質である。トブラマイシンは低濃度(4μg/mL未満)でも多くのグラム陰性細菌の増殖を抑制するのに有効であり、条件しだいでは殺菌効果も示す(非特許文献5)。トブラマイシンは粘膜表面からの吸収性が悪く、通常は非経口的投与を要する。さらに、トブラマイシンの活性は化膿性喀痰によって抑制され、高濃度の二価性カチオン類、酸性条件、高いイオン強度およびトブラマイシン結合性高分子のいずれによっても、その環境中でトブラマイシンは抑制される。喀痰中のトブラマイシン濃度をさらに5〜10倍高めると、これらの抑制効果は克服できると考えられる(非特許文献6)。
【0008】
吸収性が悪い抗生物質トブラマイシンを、エアゾル投与経路によって嚢胞性線維症(CF)患者の気道に送達させる効率については、これまでに多くのデータが存在する。その研究の大半は、嚢胞性線維症(CF)患者におけるP.aeruginosaによる慢性の肺感染症の治療を目的として行われてきた。たとえば、71名のCF患者におけるP.aeruginosaによる気管支感染症に対する、エアゾル化トブラマイシン600mg、1日3回投与による多施設、二重盲検、プラセボ対照の交叉臨床試験では、治療群で病原体の喀痰中密度の有意な減少、ならびに肺活量の改善も認められた。トブラマイシンに対して高い抵抗性を有するP.aeruginosa株(MICが128μg/mL以上と規定されるもの)の出現は、プラセボ群と治療群とで同等であった。トブラマイシンに対して内在性の抵抗性を有するP.aeruginosa以外のグラム陰性細菌の喀痰中の存在は、トブラマイシンまたはプラセボの投与において同じ頻度で見られた(非特許文献7)。
【0009】
このレジメンでは安全性および有効性の両方が認められたが、費用がかさみ、使用性も悪い。1993年、Children’s Hospital CF Center(米国、ワシントン州、シアトル市)において、患者の初期喀痰を培養して単離したP.aeruginosaに関するMICの調査によって、単離菌の90%が16μg/mL以下のMICを有し、全単離菌の98%が128μg/mL以下のMICを有することが判明した。この調査から、喀痰中のトブラマイシン濃度128μg/mLが達成されると、CF患者における気管支感染症の治療に有効であることが示唆された(非特許文献6)。
【0010】
無作為化した交差試験では、エアゾル投与10分後に採取した喀痰試料中のトブラマイシンのピーク濃度を計測することによって種々のネブライザーのトブラマイシン送達能が比較された。この試験では、1/4生理食塩水5mL中にトブラマイシンが濃度60mg/mLで溶解したTOBI(登録商標)吸入用トブラマイシン溶液(PathoGenesis社(米国、ワシントン州、シアトル市)(現在はChiron社(米国、カリフォルニア州、エメリビル市)製)が、PALI(登録商標)LCジェットネブライザー(PALI Respiratory Equipment社(米国、バージニア州、リッチモンド市)製)の使用のもとで投与された。この薬物送達(標準偏差661.0μg/g)、ピーク濃度の中央値は433.0μg/gに達することが示された。患者のうち13%のみで喀痰中濃度が128μg/g以下であったが、患者の87%では128μg/g以上に達した(非特許文献8)。ごく最近になってPARI(登録商標)LCジェットネブライザーに一方向性流動バルブが追加されて改良され、PALI(登録商標)LC PLUSの名称のものが販売されている。そのPALI(登録商標)LC PLUSの中の一方向バルブは、PALI(登録商標)LCジェットネブライザーよりも多くの薬物送達を可能にするものと説明されてきたが、一方では不意のふきこぼれの可能性が減り、呼気用フィルターの使用もされるようになった。PALI LC PLUSジェットネブライザーを用いて得られた喀痰中のトブラマイシンのピーク濃度が、旧来のPALI(登録商標)LCジェットネブライザーを用いた場合よりも有意に高いことがEisenbergら(1997)の上述文献で明らかにされた。
【0011】
前述に挙げたことに加えて、P.aeruginosaに感染したCF患者において液体トブラマイシンエアゾル製剤を断続的に投与することによる、プラセボを対照とした多施設、無作為化二重盲検臨床試験の2件がRamsey,B.W.らによって報告された(非特許文献9)。これら2件の臨床試験では、520名の被験体に対して無作為にトブラマイシン300mgの吸入またはプラセボのいずれかを1日2回、28日間投与し、さらに投薬終了から28日間追跡調査を行った。被験体にはさらに3回の投薬−休薬サイクルでトブラマイシンまたはプラセボの投与を、合計で24週間継続した。有効性に関する変数には喀痰中のP.aeruginosa含量が含まれる。トブラマイシン治療群患者では、投与0〜20週間で喀痰中のP.aeruginosa含量が平均0.8log10減少したのに対し、プラセボ投与群の患者では逆に0.3log10増加した(P<0.001)。またトブラマイシン投与群の患者では、投与0〜4週間で喀痰中のP.aeruginosa含量が平均1.9log10減少したのに対し、プラセボ投与群では変化が見られなかった(P<0.001)。
【0012】
特許文献1および特許文献2では、気管支感染症に罹患している患者に対して、トブラマイシンのようなアミノグリコシド系抗生物質が生理学的適合性のある担体中に約60〜約200mg/ml含有する液体製剤を、ネブライザーによって4.0mlまたはそれ以下の用量で約10分間以下の時間をかけて吸入させて投与すると有効な治療ができることが開示されている。アミノグリコシド製剤のさらに効率的な投与がなされると、従来の投与レジームと比較して実質的に少ない量の液状アミノグリコシドで、および実質的に短い投与期間で投与が可能になり、その結果、投与コストおよび薬物の浪費が低減する。しかも、使用する製剤は比較的少量の生理学的適合性のある溶液中に処方されたアミノグリコシドを有効な最小量しか含有しないものであり、そのことによってアミノグリコシド製剤の吸入後の肺への刺激が低減した。
市場から購入可能なTOBI(登録商標)製品のような吸入用抗生物質のほかに、種々の長期療法が日常的に処方され、肺における閉塞、感染症および炎症の破壊サイクルが抑制される。積極的気道クリアランス療法(Aggressive Airway Clearance Therapy(非特許文献10))、吸入による気管支拡張薬療法(非特許文献11)、およびヒト組換え型α−ドルナーゼ(rhDNアーゼ;非特許文献12)のような粘液溶解療法については、いずれもCF患者に対する治療に明らかに役立つものである。それらの療法に関しては、CF患者にとって重大な問題があり(非特許文献13)、コンプライアンス(服薬遵守)の欠如で一定の治療にばらつきが出る(非特許文献14)ことが分かっている。
前述にように、市販されているTOBI(登録商標)吸入用液体エアゾルトブラマイシン溶液は、CF患者におけるP.aeruginosa感染症の治療に高い有効性を示すことがすでに証明されている。CF患者における肺機能の保持に伴う治療負荷および関連する免疫学的接種を行うと、治療に要する投与期間が短縮または患者に対する治療の利便性が増大する治療上の改善によって患者に便宜が図られ、その結果として治療効果も高まる。したがって、アミノグリコシド系抗生物質を患者に吸入させて送達し、投薬コストを下げ、患者のコンプライアンスを上げ、吸入療法の全般的有効性を増大させるための新規の改善された方法およびデバイス類の必要とされる。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】米国特許第6,890,907号明細書
【特許文献2】米国特許出願公開第2003/0143162号明細書
【非特許文献】
【0014】
【非特許文献1】“American Academy of Otolaryngology.Guide for the evaluation of hearing handicap”,JAMA(1979)241(19):2055−9
【非特許文献2】Brummett,R.E.,“Drug−induced ototoxicity”,Drugs(1980)19:412−28
【非特許文献3】Touw,D.J.ら、“Inhalation of Antibiotics in Cystic Fibrosis”,Eur.Respir.J.(1995)8:1594−604
【非特許文献4】Rosenfeld,M.ら、“Aerosolized Antibiotics for Bacterial Lower Airway Infections:Principle,Efficacy,and Pitfalls”,Clinical Pulmonary Medicine(1997)4(2):101−12
【非特許文献5】Neu,H.C.,“Tobramycin:an overview”,J.Infect.Dis.(1976)134:Suppl:S3−19
【非特許文献6】Levy,J.ら、“Bioactivity of Gentamicin in Purulent Sputum from Patients with Cystic Fibrosis or Bronchiectasis:Comparison with Activity in Serum”,J.Infect.Dis.(1983)148(6):1069−76
【非特許文献7】Ramsey,B.ら、“Response to Letter to the Editor:Aerosolised Tobramycin in Patients with Cystic Fibrosis”,N.Engl.J.Med.(1993)329:1660
【非特許文献8】Eisenberg,J.ら、“A Comparison of Peak Sputum Tobramycin Concentration in Patient With Cystic Fibrosis Using Jet and Ultrasonic Nebulizer System. Aerosolized Tobramycin Study Group”,Chest(1997)111(4):955−962
【非特許文献9】Ramsey,B.W.ら,“Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis.Cystic Fibrosis Inhaled Tobramycin Study Group.“N.Engl.J.Med.(1999)340(1):23−30
【非特許文献10】Reisman,J.J.ら、“Role of conventional physiology in cystic fibrosis”,J.Pediatr.(1988)113(4):632−6
【非特許文献11】Konig P.ら、“Short−term and Long−term Effects of Albuterol Aerozol Therapy in Cystic Fibrosis:A Preliminary Report”,Pediatr.Pulmol.(1995)20(4):205−14
【非特許文献12】Fuchs,H.J.ら、“Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symtoms and on Pulmonary Function in Patients with Cystic Fibrosis.the Pulmozyme Study Group”,N.Engl.J.Med.(1994)331(10):637−42
【非特許文献13】Conway,S.P.ら、“Compliance with treatment in adult patients with cystic fibrosis”,Thorax(1996)51(1):29−33
【非特許文献14】Abbot J.ら、“Treatment Compliance in Adults with Cystic Fibrosis”,Thorax(1994)49(2):115−20
【発明の概要】
【課題を解決するための手段】
【0015】
発明の要旨
本発明は、90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症患者の気管支系に対して、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む治療方法を提供する。本発明の態様によれば、第一治療期間の後に第二の治療休止期間を設けることができ、その間は患者の気管支系にはアミノグリコシド系抗生物質は投与されない。悪性の感染症の治療に対しては、患者の気管支系へのアミノグリコシド系抗生物質投与による第一の治療期およびその後のアミノグリコシド系抗生物質を投与しない第二の治療休止期のサイクルを、抗菌効果が現われるまで2回またはそれ以上繰返して行うことができる。CF患者が罹患する感染症のような慢性感染症の場合では、この第一および第二の治療期のサイクルを、患者治療中に複数回繰返すことができる。
本発明の方法は、CFに伴うシュードモナス属菌による気管支感染症のような、アミノグリコシド系抗生物質が有効ないずれの気管支感染症の治療にも有用である。
例えば、本発明は以下の項目を提供する。
(項目1)
90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症の患者の気管支系に対して、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む、気管支感染症の治療方法。
(項目2)
前記第一治療期間の後に、前記患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期間を設ける、項目1に記載の治療方法。
(項目3)
前記アミノグリコシド系抗生物質が、ゲンタマイシン、アミカシン、カナマイシン、ストレプトマイシン、ネオマイシン、ネチルミシンおよびトブラマイシン、あるいはそれらの薬学的に受容可能な塩からなる群から選択される、項目1に記載の治療方法。
(項目4)
前記アミノグリコシド系抗生物質が、トブラマイシンまたはその薬学的に受容可能な塩である、項目3に記載の治療方法。
(項目5)
前記エアゾルパウダー組成物が、100〜120mgのトブラマイシンを含有する、項目4に記載の治療方法。
(項目6)
前記エアゾルパウダー組成物が、110〜115mgのトブラマイシンを含有する、項目5に記載の治療方法。
(項目7)
前記患者に投与される前記エアゾルパウダー組成物が、2〜6個の用量単位に分割されている、項目1に記載の治療方法。
(項目8)
前記患者に投与される前記エアゾルパウダー組成物が、3〜5個の用量単位に分割されている、項目7に記載の治療方法。
(項目9)
前記エアゾルパウダー組成物が、4個の用量単位に分割されている、項目8に記載の治療方法。
(項目10)
前記第二の治療休止期間が、20〜36日間である、項目2に記載の治療方法。
(項目11)
前記第一の治療期間が、26〜30日間である、項目2に記載の治療方法。
(項目12)
前記第二の治療休止期間が、26〜30日間である、項目11に記載の治療方法。
(項目13)
前記第一の治療期間が、28日間である、項目2に記載の治療方法。
(項目14)
前記第二の治療休止期間が、26〜30日間である、項目13に記載の治療方法。
(項目15)
前記第一の治療期間の治療の後に、前記第二の治療休止期間を設ける治療レジメンを複数回繰返す、項目2に記載の治療方法。
(項目16)
前記パウダーが、空気力学的粒径域1〜5μmの粒子を少なくとも50%含有する、項目2に記載の治療方法。
(項目17)
前記患者が、嚢胞性線維症患者である、項目2に記載の治療方法。
(項目18)
前記嚢胞性線維症患者が、シュードモナス属菌による気管支感染症に罹患している、項目17に記載の治療方法。
(項目19)
前記エアゾルパウダーが、ドライパウダー吸入器を用いて前記患者に投与される、項目1に記載の治療方法。
(項目20)
前記エアゾルパウダーが、前記ドライパウダー吸入器内の単一容器に分配され、前記エアゾルパウダーが、前記容器から前記吸入器によってヒト被験体または動物被験体の肺内に送達される、項目1に記載の治療方法。
(項目21)
前記エアゾルパウダーが、前記ドライパウダー吸入器内の複数容器に分配され、前記エアゾルパウダーが、前記容器から前記吸入器によってヒト被験体または動物被験体の肺内に送達される、項目20に記載の治療方法。
(項目22)
110〜115mgのトブラマイシン抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症に罹患した嚢胞性線維症患者の気管支系に対して、28日間の第一治療期間に1日2回投与する工程、その後に前記患者の気管支系にトブラマイシン抗生物質が投与されない26〜30日間の第二の治療休止期間を設ける工程、および前記第一および前記第二の治療期間を繰返す工程を含む、前記患者の治療方法。
(項目23)
前記患者に投与される前記エアゾルパウダー組成物が、3〜5個の用量単位に分割されている、項目22に記載の治療方法。
(項目24)
前記エアゾルパウダー組成物が、4個の用量単位に分割されている、項目23に記載の治療方法。
(項目25)
前記嚢胞性線維症患者が、シュードモナス属菌による気管支感染症に罹患している、項目22に記載の治療方法。
(項目26)
前記第一および前記第二の治療期間が、複数回繰返される、項目22に記載の治療方法。
【図面の簡単な説明】
【0016】
本発明の前述の態様および付随する利点の多くについては、以下に述べる詳細な説明を参考とすると理解が深まると同時に、添付の図面と関連付けることでさらに容易に理解できるであろう。
【図1】図1は、規定用量のTPIおよびTOBI投与後の種々の時間における被験体の血清中トブラマイシンの平均濃度を示す。
【図2】図2は、曲線下面積(AUC)に対する吸入用トブラマイシン粉末製剤(TPI)および吸入用トブラマイシン溶液製剤(TOBI)の用量プロット(0,12)を示す。
【図3】図3は、AUCに対するTPIおよびTOBIの用量プロット(0,∞)を示す。
【図4】図4は、規定用量のTOBIまたはTPIが投与された被験体の喀痰中トブラマイシンの平均濃度を示す。
【発明を実施するための形態】
【0017】
好ましい実施形態の詳細な説明
本明細書において特別に規定されるものを除き、本明細書で使用される用語のすべてについては、当業者にとって公知と考えられるのと同じ意味を持つものとする。本明細書では以下の略語が用いられる。
【0018】
略語 意味
【0019】
AE 有害事象
ALT アラニン−アミノトランスフェラーゼ
AUC 曲線下面積
BID 1日2回
BUN 血中尿素体窒素
CaCl2 塩化カルシウム
CF 嚢胞性線維症
CFC クロロフルオロ炭素
Cmax 最高濃度
CFTR 嚢胞性線維症膜貫通コンダクタンス制御因子
DPI ドライパウダー吸入剤
DSPC 1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン
FDA 米国食品医薬品局
FEV1 1秒努力肺活量
FVC 努力肺活量
FEF25−75 25〜75%間の努力呼気流量
HPMC 2−ヒドロキシプロピルメチルセルロース
IRB 治験委員会
IVRS 対話型音声応答システム
MedDRA 医学規制用語集
MIC 最小抑制濃度
P.aeruginosa Pseudomonas aeruginosa(緑膿菌)
PFOB 臭化ペルフルオロオクチル
QPIT 定量的ピロカルピン電離療法検査
SAE 重篤な有害事象
tmax 最高濃度到達時間
TOBI(登録商標) 吸入用トブラマイシン300mg溶液(Chiron社(米国、カリフォルニア州、エメリビル市)製)
TPI 吸入用トブラマイシン粉末。
【0020】
一つの態様によれば、本発明は気管支感染症患者の気管支系に対して、90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む治療方法を提供する。本発明の実施によれば、第一治療期間の後に患者の気管支系にアミノグリコシド系抗生物質を投与しない第二治療休止期間を設けることができる。悪性感染症の治療には、アミノグリコシドで治療を行う第一治療期間およびそれに続く治療を行わない第二治療休止期間のサイクルを、所望の抗菌効果が得られるまで2回またはそれ以上繰返すこともできる。CF患者が罹患するような慢性感染症の場合では、第一および第二の治療期間はその患者の治療期間中に何回も繰返すことができる。
【0021】
別の態様によれば、本発明は気管支感染症患者の治療のための、アミノグリコシド系抗生物質90〜130mg含有ドライパウダーエアゾル組成物を20〜36日間の第一治療期間に1日1〜3回投与することによる治療用の医薬品製剤におけるアミノグリコシド系抗生物質の使用を提供する。本発明のこの態様の実施によれば、第一の治療期間の後に前述と同様に患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期間を設けることができ、本明細書に記載されているようにその第一および第二の治療期を繰返すことができる。
【0022】
本発明のこの態様の方法には、いずれにおいても20〜90重量%のアミノグリコシド系抗生物質および生理学的適合性のある担体を含有する治療上有効量のエアゾルパウダーを、必要に応じてヒト被験体または動物被験体に吸入させて投与する工程が含まれ、そのエアゾルパウダーは含有粉末粒子の少なくとも50%は空気力学的粒径1〜5μmの範囲に入るものである。
【0023】
「気管支感染症」という用語は、肺の気管支内に発生した細菌感染症を指す。本発明の方法を用いて治療することが可能な気管支感染症の例としては、Pseudomonas
aeruginosa、Staphylococcus aureus、Haemophilus influenzae、Burkholderia cepacia、Stenotrophomonas maltophilliaおよびAlcaligenesis xiloxidantsのようなグラム陰性微生物による感染症が挙げられる。本発明のこの態様の方法は、たとえば嚢胞性線維症に付随するPseudomonas aeruginosa感染症のような気管支感染症に罹患したヒトの治療に用いることができる。
【0024】
本発明の実施において有用なアミノグリコシド系抗生物質の例としては、ゲンタマイシン、アミカシン、カナマイシン、ストレプトマイシン、ネオマイシン、ネチルマイシン、パラメシンおよびトブラマイシンが挙げられる。本発明の実施において用いられる現時点で好ましいアミノグリコシド系抗生物質には、トブラマイシンがある。一般にアミノグリコシド系抗生物質は、その薬学的に受容可能な塩(たとえば硫酸塩、クエン酸塩、アスコルビン酸塩、グルコン酸塩、炭酸塩、酒石酸塩、コハク酸塩、酢酸塩またはリン酸塩)またはエステルの形で投与される。
【0025】
本発明の実施によれば、エアゾルパウダーはヒト被験体または動物被験体に吸入させることでその肺の中に到達する。そのエアゾルパウダーにはアミノグリコシド系抗生物質含有の粒子が含まれる。エアゾルパウダーは、その含有粒子の少なくとも50%が空気力学的粒径1〜5μmの範囲に入るもので、ヒト被験体または動物被験体の肺内に効果的に浸透し、その結果アミノグリコシド系抗生物質が効果的に肺内に送達されることが見出された。例を挙げると、本発明の実施において有用な(アミノグリコシド系抗生物質含有の)エアゾルパウダーのいくつかでは、その含有粒子の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%または少なくとも95%が空気力学的粒径1〜5μmの範囲にある。
【0026】
「空気力学的粒径」という用語は、対象の粒子と同じ限界沈降速度を有する単位密度球体の直径を指す(“Aerosol Measurement:Principles,Techniques and Applications”、Klaus WillekeおよびPaul A.Baron編、Van Nostrand Reinhold(ニューヨーク)社刊、1993などを参照のこと)。空気力学的粒径に関しては、たとえば粒子が気道内のどこに留まるのかを予測するのに用いられる。
【0027】
「空気力学的質量平均粒径」(略してMMAD)とは、分散した粒子の空気力学的大きさの計測値のことである。空気力学的大きさの分布は、吸入中におけるエアゾルの挙動を規定し、一般には空気中におけるその粒子と同じ限界沈降速度を有する単位密度球体の直径のことである。空気力学的粒径には粒子の形状、密度および物理学的大きさが含まれる。空気力学的大きさの分布が対数正規分布である場合には、空気力学的質量中央粒径(MMAD)で特性解析できる。本明細書において用いられるMMADは、Andersonのカスケード衝撃法によって求められるエアゾル化粉末の空気力学的粒子サイズ分布の中点または中央値を指す。
【0028】
カスケード衝撃装置には、簡単に述べると孔径サイズが小さくなる一連のスクリーンが含まれる。粒子は衝撃装置を通過する移動噴出流内で、一連のスクリーンにトラップされる。各々のスクリーン上にトラップされる粒子状物質(規定の大きさの範囲にある粒子)の量は、スクリーンを洗浄し溶離した物質の量を計測することで求めることができる。その際使用されるカスケード衝撃装置の例については、米国薬局方(第26版)の第601章(エアゾル)内に記載されており、その刊行物の該当部分は本明細書中に参考として援用されている。
【0029】
本発明の実施において有用な粉末化アミノグリコシド系抗生物質製剤は、水分含量が一般に15重量%未満、通常は約11重量%以下であり、好ましくは約8重量%以下である。
【0030】
本発明の実施によれば、アミノグリコシド系抗生物質含有のエアゾル粉末の治療有効量が、気管支感染症患者に吸入されて投与される。治療有効量のエアゾル粉末中には、患者の肺中に存在する薬物感受性細菌の増殖を完全にまたは部分的に抑制するのに充分なアミノグリコシド系抗生物質が含まれる。代表例として、アミノグリコシドであるトブラマイシンについては、その治療有効量は患者に1日1〜3回、本発明の好ましい態様では1日2回、トブラマイシンの用量(存在し得る対イオンの重量を除いた遊離塩基の重量として求められた用量)として約90〜約130mg、さらに好ましくは約100〜約120mg、および最も好ましくは約110〜115mgを含有するエアゾル粉末組成物を投与することで達成される。
【0031】
トブラマイシンのようなアミノグリコシドの投与量は、単回投与分の単一容器から投与でき、あるいはその抗生物質の送達に用いられる吸入デバイスしだいでは多回投与用容器内に、または連続投与用単位用量に分割して投与することもできる。アミノグリコシド系抗生物質の投与量は、たとえば2〜6単位用量、より好ましくは3〜5単位用量、およびさらに好ましくは4単位用量に分割できる。代表的な実施形態では、112mgのトブラマイシン投与量(存在し得る対イオンの重量を除いた遊離塩基の重量として求められた値)については、HPMC製2号カプセル4個に別々に、遊離塩基としてのトブラマイシンを27mgずつ分割充填できる。
【0032】
本発明のドライパウダーエアゾル組成物は、患者に対して20〜36日間、より好ましくは26〜30日間、およびさらに好ましくは約28日間の第一治療期間に投与される。この第一治療期間の後には、患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期を設ける。本発明の一つ態様によれば、第二治療休止期間は約20〜36日間、より好ましくは約26〜約30日間、および最も好ましくは約28日間継続される。
【0033】
代表的な実施形態では、本発明の方法はPseudomonas aeruginosaによる慢性感染症の療養管理を目的とするCF患者の治療に用いられる。この態様によれば、本発明は気管支感染症に罹患したCF患者の治療を企図するものであり、患者の気管支系に対し、抗生物質としてトブラマイシンを110〜115mg含有するドライパウダーエアゾル組成物を、28日間の第一治療期間で1日2回投与を行い、つづいて患者の気管支系にトブラマイシンを投与しない26〜30日間の第二治療休止期間を設け、さらに第一および第二の治療期を繰返す工程が含まれる。本発明のこの態様によれば、110〜115mgのトブラマイシン用量は、連続投与用に3〜5単位用量、好ましくは4単位用量に分割できる。CF患者はP.aeruginosaに慢性的に感染する傾向があるため、第一治療期間およびそれに続く第二治療休止期間のサイクルは、一般に複数回または多重回繰返して行われることになり、CF患者における気管支感染症の長期にわたる療養管理のために無期限継続することもできる。
【0034】
エアゾルパウダーには一般に20(重量)%〜90(重量)%のアミノグリコシド系抗生物質が含有する。したがって、本発明のいくつかの実施形態では、エアゾルパウダーには30(重量)%〜80(重量)%のアミノグリコシド系抗生物質が含有する。本発明のいくつかの実施形態では、エアゾルパウダーには40(重量)%〜70(重量)%のアミノグリコシド系抗生物質が含有する。この状況において、アミノグリコシド系抗生物質の(重量)%は、存在し得る対イオンの重量を除いた遊離型の抗生物質の量を指す。
【0035】
本発明のエアゾル粉末には、生理学上適合性のある少なくとも一種の担体が一般に含まれるが、必ずしも必須のものではない。エアゾル粉末にはたとえば一種または複数の賦形剤、および/またはアミノグリコシド系抗生物質の効力を高めるそのほかの成分も含有可能である。そのような賦形剤を用いると、患者に送達される粉末中の活性薬剤の含量を抑える必要があるときの簡便な希釈剤として役立つ。またその賦形剤を用いると、粉末分散デバイス中での粉末の分散性の改善にも役立ち、ひいては活性薬剤送達効率およびその再現性がさらに改善され、活性薬剤の取扱い易さ(たとえば流動性およびコンシステンシー)が改善されて製剤の製造および粉末充填が容易になる。特に賦形剤は、アミノグリコシドの物理的および化学的な安定性の改善、残存する水分含量の低減および吸湿防止、ならびに粒子サイズ、凝集度、表面特性(たとえばしわの度合)、吸入のしやすさおよび肺深部への発生粒子のターゲッティングの向上の機能が期待できる。
【0036】
本発明の実施において用いられるアミノグリコシド組成物で有用な医薬品用賦形剤および添加剤には、蛋白質、ペプチド、アミノ酸、脂質、ポリマー、および炭水化物(たとえば単糖類、二糖類、三糖類、四糖類およびオリゴ糖類を含めた糖類;アルジトール類、アルドン酸類、エステル化糖類のような糖誘導体;ならびに多糖類またはシュガーポリマー)が含まれ、それらは単独または組合わせて使用できるが、それらに限定されるものではない。蛋白質賦形剤の例としては、ヒト血清アルブミン(HSA)のような血清アルブミン、組換えヒトアルブミン(rHA)、ゼラチンおよびカゼインが挙げられる。代表的なアミノ酸/ポリペプチド成分であって緩衝機能も有するものには、アラニン、グリシン、アルギニン、ベタイン、ヒスチジン、グルタミン酸、アスパラギン酸、システイン、リシン、ロイシン、プロリン、イソロイシン、バリン、メチオニン、フェニルアラニンおよびアスパルテームが含まれるが、アルギニンについては好ましくない。ジロイシンおよびトリロイシンのような代表的なアミノ酸の重合体についても、本発明で用いるのに適している。
【0037】
本発明での使用に適した炭水化物賦形剤の例としては、フルクトース、マルトース、ガラクトース、グルコース、D−マンノースおよびソルボースのような単糖類;ラクトース、スクロース、トレハロース、セロビオースのような二糖類;ラフィノース、メレジトース、マルトデキストリン類、デキストラン類およびデンプンのような多糖類;ならびにマンニトール、キシリトール、マルチトール、ラクチトール、キシリトールソルビトール(グルシトール)およびミオイノシトールのようなアルジトール類が挙げられる。
【0038】
またアミノグリコシド組成物には緩衝剤またはpH調整剤も含有することが可能で、緩衝剤は一般に有機の酸または塩基から調製される塩である。代表的な緩衝剤には、クエン酸、アスコルビン酸、グルコン酸、カルボン酸、酒石酸、コハク酸、酢酸またはフタル酸の塩のような有機酸塩類;トリス、塩酸トロメタミン、またはリン酸塩緩衝剤が含まれる。
【0039】
本発明の実施において有用なアミノグリコシド組成物にはさらに、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース、メチルセルロース、エチルセルロース、フィコール類(糖ポリマー)、デキストラン、糖エステル類(たとえば2−ヒドロキシプロピル−β−シクロデキストリンのようなシクロデキストリン類、およびヒドロキシエチル澱粉)、ポリエチレングリコールおよびペクチンのような高分子性賦形剤/添加剤;塩類(たとえば塩化ナトリウム);酸化防止剤;帯電防止剤;界面活性剤(たとえば「TWEEN 20」および「TWEEN 80」のようなポリソルベート類、レシチン、オレイン酸、塩化ベンザルコニウム、およびソルビタンエステル類);脂質(たとえばリン脂質および脂肪酸);ステロイド(たとえばコレステロール);ならびにキレート剤(たとえばEDTA)が含有可能である。アミノグリコシド組成物中に使用するのに適したそのほかの医薬用賦形剤および/または添加剤の例については、“Remington:The Science & Practice of Pharmacy”,第19版、Williams & Williams社刊(1995)、および“Physician’s
Desk Reference”、第52版、Medical Economics社(米国、ニュージャージー州)刊(1998)に列挙されており、それらの開示箇所は本明細書中に参考として援用されている。
【0040】
現時点で好ましい賦形剤の組合わせとしては、レシチンと塩化カルシウムとの配合がある。レシチンは、哺乳類(ヒトを含めて)の肺内で界面活性剤としてはたらく、天然産リン脂質のうちのホスファチジルコリンのうちの一種である。
【0041】
本発明の実施において有用なアミノグリコシド組成物には、アミノグリコシド粉末固有の分散性を改善するための分散剤が添加できる。適した分散剤については、国際公開第95/31479号、国際公開第96/32096号および国際公開第96/32149号に開示されており、それらの公開特許はすべて本明細書中に参考として援用されている。それらの公開特許において記載されているように、適した分散剤には水溶性ポリペプチド、ならびにトリプトファン、ロイシン、フェニルアラニンおよびグリシンのような疎水性アミノ酸が含まれる。特にロイシンおよびトリロイシンは、本発明による使用に好適である。
【0042】
アミノグリコシドおよび賦形剤によって形成される固体状態のマトリックスによって、アミノグリコシドに安定した環境が与えられる。その安定性マトリックスについては、結晶性ガラス、無定形ガラス、または両者の混合物で形成できる。最適なものには、両者の混合物を含むドライパウダー製剤がある。実質的に無定形であるアミノグリコシドドライパウダー製剤が好ましいが、そのためには製剤のガラス転移温度(Tg)が約35℃超、好ましくは約45℃超、およびさらに好ましくは約55℃超とする。Tgについては保存温度よりも少なくとも20℃以上高い温度であるのが好ましい。好ましい実施形態によると、アミノグリコシド組成物は国際公開第99/16419号および国際公開第01/85136号において開示されたような固体状態のマトリックスとしてのリン脂質を含有し、それらの公開特許は本明細書においてすべて参考として援用されている。
【0043】
ドライパウダーアミノグリコシド組成物は、前述のような実質的に無定形のガラス質または実質的に結晶質の生物活性のある粉末が生成する条件下で噴霧乾燥して調製することができる。アミノグリコシド溶液の処方物の噴霧乾燥に関しては、たとえば“Spray
Drying Handbook”、第5版、K.Masters編、John Wiley & Sons社(米国ニューヨーク州、ニューヨーク市)刊(1991)、および国際公開第97/41833号に全般的に記載されているように実施される。尚、それに関係する記述箇所は、本明細書中に参考として援用されている。
【0044】
本発明の一つの実施形態による噴霧乾燥用アミノグリコシド溶液の調製のためには、一般に水のような生理学的適合性のある溶媒にアミノグリコシドを溶解する。噴霧乾燥用溶液のpH域は、一般には約3〜10、好ましくは5〜8、さらに好ましくは中性pH域になるように維持し、これによって噴霧乾燥後の粉末が肺内で再び溶解した時のpH域が生理学的に適合するように維持されるのに役立ち得る。この水性処方物には所望によりさらにアルコール、アセトン等のような水と混和する溶媒も添加できる。代表的なアルコールにはメタノール、エタノール、プロパノール、イソプロパノール等のような低級アルコールがある。アミノグリコシド溶液には、アミノグリコシドが一般に0.05%(w/v)−約20%(w/v)の濃度、通常は0.4〜5.0%(w/v)の濃度で溶解した状態で含有するものが考えられる。
【0045】
つづいてアミノグリコシド含有溶液は、Niro社(デンマーク)、Buchi社(スイス)などのような販売業者から購入可能なような従来からの噴霧乾燥機中で噴霧乾燥され、安定なアミノグリコシドドライパウダーが得られる。アミノグリコシド溶液の噴霧乾燥に関する最適な条件については、処方される成分しだいで変わると考えられ、一般には実験によって求められる。物質の噴霧乾燥に用いられる気体は空気が一般的であるが、窒素またはアルゴンのような不活性ガスも使用に適する。さらに、物質の噴霧乾燥に用いられる気体の給気および排気の温度も、噴霧乾燥中におけるアミノグリコシドの不活化が起きないように設定される。そのような温度域は一般に実験によって求められるが、一般的に給気温度は約50〜約200℃の範囲、排気温度は約30〜約150℃の範囲と考えられる。
【0046】
またアミノグリコシドドライパウダーについては凍結乾燥法、真空乾燥法、噴霧凍結乾燥法、超臨界流体プロセス法またはそれ以外の蒸発乾燥方法、あるいはドライパウダー処方中の製剤成分の混和法、粉砕法またはジェットミル法によっても調製できる。いくつかの場合では、取扱い/加工特性の改善、たとえば帯電性の低減化、流動性の向上、固化性の低下などが見込まれる処方でのアミノグリコシドドライパウダー製剤の提供が所望され、そのことは微粒子凝集体、すなわち前述のアミノグリコシドドライパウダー粒子の凝集体または塊から成る組成物を調製することによって達成され、その凝集体は肺内に送達されるとすぐに粉砕して微粉成分に戻るもので、このことについてはたとえば米国特許第5,654,007号に記載されており、本明細書においても参考文献として掲載されている。またアミノグリコシドドライパウダーは、粉末成分の凝集化、篩過による凝集体収集、球状化による球形に近い凝集体の形成、および分級による均一な大きさの製品化による代替製法によっても製造でき、これについてはたとえば国際特許第95/09616号に記載されており、本明細書にも参考文献として掲載されている。アミノグリコシドドライパウダーは、製造、加工および貯蔵中は乾燥条件下(すなわち相対湿度が低い条件下)に置かれるのが好ましい。
【0047】
一つの実施形態によると、本発明の実施において有用な粉末化トブラマイシン製剤の例に挙げられるものは、前述の国際特許第99/16419号および国際特許第01/85136号に開示された乳化/噴霧乾燥法によって製造できる。そのような実施形態による製剤は、少なくとも75%w/w、好ましくは少なくとも85%w/wのトブラマイシン、2〜25%w/w、好ましくは8〜18%w/wのリン脂質、および塩化カルシウムのような金属イオン0〜5%w/wを含有するドライパウダー粒子から構成されるように設計されている。この実施形態の粒子は、一般にMMAD(空気力学的質量平均粒径)が1〜5μmであり、かさ密度が0.08g/cm3超、好ましくは0.12g/cm3超である。
【0048】
本発明の実施において有用な粉末化トブラマイシン製剤の別の例に挙げられるものは、活性型トブラマイシン、1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)、CaCl2および臭化ペルフルオロオクチル(PFOB)を含有するエマルションを作製することによって製造できる。この供給原料としてのエマルションは、次にアトマイザーノズルを通過させて噴霧され、微細な液滴となる。その液滴が乾燥する際、水およびPFOBは蒸発し、多孔質構造の球形粒子を主体とするリン脂質が得られる。それらの球状物は低密度であるため、好ましい空気力学的特性を示す(たとえば空気力学的粒径が1〜5μmの範囲にある球形粒子)。またそれらの高い表面多孔性によって粒子同士の接触が少なくなり、エアゾル懸濁に要するエネルギーも少なくてすむようになる。
【0049】
エアゾル粉末(アミノグリコシド系抗生物質含有)は、たとえばヒト被験体または動物被験体の吸気を利用するドライパウダー吸入器を用いて投与でき、それによってその粉末化アミノグリコシド系抗生物質製剤は肺に送達される。有用なドライパウダー吸入器の例には、T−326型吸入器(Nektar Therapeutics社(米国、カリフォルニア州94070、サンカルロス市、インダストリアル・ロード150)製)がある。そのほかの有用なドライパウダー吸入デバイスについては、米国特許第5,458,135号、同第5,740,794号、同第5,775,320号および同第5,785,049号に開示されており、それらのいずれの特許も本明細書中に参考として援用されている。このタイプのデバイスを用いて投与する場合、粉末化医薬品は、穿刺用蓋またはそのほかのアクセス面を有する容器、好ましくはブリスター包装容器またはカートリッジに、1回投与単位分量または多回投与単位分量で充填される。計量用量分のドライパウダー医薬品で多数の空隙を充填する方法については、たとえば米国特許第5,826,633号に記載されており、その特許は本明細書中に参考として援用されている。
【0050】
本明細書において記載のアミノグリコシド粉末の送達に適したものには、たとえば米国特許第3,906,950号、同第4,013,075号、同第4,069,819号および同第4,995,385号に記載されているタイプのドライパウダー吸入器も含まれ、それらの特許はいずれも本明細書において参考として援用されており、患者に送達する分の予め計量された用量のアミノグリコシドドライパウダーが、ゼラチン硬カプセルのようなカプセルに充填されている。カプセルサイズは、たとえば00号、0号、1号または3号が挙げられるが、その粉末投与に使用される吸入デバイスやそれ以外の要因に依存して選ばれる。
【0051】
アミノグリコシドドライパウダーを肺に投与するためのそのほかのドライパウダー分散用デバイスについては、たとえば欧州特許出願公開第0129985号、同第0472598号、同第0467172号、ならびに米国特許第5,522,385号に記載されているものがあり、それらの特許はいずれもすべて本明細書中に参考として援用されている。また本発明のアミノグリコシドドライパウダーの送達に適するものとしては、Astra−Draco社製のTURBUHALER(商品名)のような吸入デバイスもある。このタイプのデバイスについては、米国特許第4,668,218号、同第4,667,668号および同第4,805,811号に詳しく記載されており、それらの特許のすべては本明細書中に参考として援用されている。
【0052】
また好ましいデバイスとしては、粉末化医薬品の浮遊化、担体スクリーンを通して空気を送ることによるスクリーンからの医薬品の浮揚化、または本明細書中に参考として援用されている米国特許第5,388,572号に記載されているようなマウスピースデバイスで粉末を患者に連続的に投入するための混合チャンバー内での空気と粉末医薬品とのミキシングのいずれかを目的とする空気供給用ピストンを使用するデバイスが挙げられる。
【0053】
前述事項を考慮すると、治療有効量のエアゾル粉末(アミノグリコシド系抗生物質含有)は、ドライパウダー吸入デバイス内に配置された単回投与用または複数回投与用の容器から投与できることが分かるであろう。たとえばドライパウダー吸入デバイスは、治療有効量のエアゾル粉末(アミノグリコシド系抗生物質含有)が入った1回投与用容器とともに装填でき、容器内容物がヒト被験体または動物被験体に吸入される。再び例を挙げると、ドライパウダー吸入器はHPMC製2号カプセルのような多重回投与分のユニット容器(たとえば2,3または4回投与分のユニット容器)とともに装填でき、それらのカプセルには別々に治療有効量未満のエアゾル粉末(アミノグリコシド系抗生物質含有)が入っており、それらのエアゾル粉末を合わせると治療有効量になる。ドライパウダー吸入器によって、その中に配置された容器内のすべての内容物が放出され、治療有効量のエアゾル粉末が患者に供給される。
【0054】
本発明のアミノグリコシド治療レジメンは単独でも使用でき、あるいは気管支感染症、特にP.aeruginosaによる感染症の治療用の一種または複数の追加薬剤と併用することもできる。本発明のこの態様によれば、気管支感染症治療用の一種または複数の追加薬剤は、アミノグリコシド系抗生物質による第一治療期間中、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間中、あるいは第一および第二の治療期間中の両方において投与することができる。本発明のこの態様の一つの実施形態では、気管支感染症の治療用の一種または複数の追加薬剤は、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間中に投与される。その気管支感染症の治療に適する追加薬剤の例としては、モノバクタム系、β−ラクタム系、マクロライド系、フルオロキノロン系および/またはグリコペプチド系の抗菌化合物のような非アミノグリコシド系の感染症治療薬剤が挙げられる。非アミノグリコシド系の感染症治療薬剤の例としては、アズトレオナムを挙げることができる。
【0055】
粉末化アミノグリコシド系抗生物質製剤の放出率(ED)は、一般には50%超である。本発明の実施において有用な粉末化アミノグリコシド系抗生物質製剤のEDは、さらに好ましくは70%超であり、しばしば80%を超える。本明細書において用いられる「放出用量(放出率)」または「ED」という用語は、パウダーユニット、カプセルまたはリザーバーから発射後または分散後における、適当な吸入デバイスからのドライパウダーの送達指標を指す。EDとは、吸入デバイスによって送達される用量の、正規の用量に対する比(すなわち、発射前の適当な吸入デバイス内に配置された単位用量に対する粉末質量の比)として定義される。EDは実験的に求められる量であって、一般には患者への投与をインビトロで模倣した構成のデバイスを用いて求められる。ED値を求めるには、乾燥粉末での正規用量(前述規定のような)を適当なドライパウダー吸入器内に入れ、吸入器を作動させて粉末を分散させる。生じたエアゾル浮遊物は次に減圧吸引によってデバイスから放出され、マウスピースデバイスに取付けられた風袋消去可能なフィルターの表面に捕捉される。そのフィルターに到達する粉末の量が送達用量とみなされる。たとえば、吸入デバイス内に配置された50mgのドライパウダー含有の2号カプセルに場合、粉末の分散によって、前述のように風袋消去可能なフィルター表面に40mgの粉末が回収されたとすると、そのドライパウダー組成物に関するEDは、40mg(搬送用量)/50mg(正規の用量)×100=80%となる。
【0056】
さらにほかの態様によれば、本発明は気管支感染症患者の治療に有用なキット類も提供し、キットには90〜130mgのドライパウダーのアミノグリコシド系抗生物質、ならびに患者の気管支系に一定の服用量を投与するための能書が含まれ、20〜36日間の第一治療期間に1日1〜3回、ドライパウダー吸入用のデバイスが用いられる。本発明のこの態様によれば、その能書によって第一治療期間の後に、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間を設け、本明細書において実質的に記載したように所望の抗菌効果が現われるまでさらに第一治療期間と第二治療休止期間とのサイクルを2回またはそれ以上繰返すことが指示され得る。本発明のキット類では、1回分または複数回分の用量を単一単位用量として単一容器内に入れるか、あるいは抗生物質の送達に使用する吸入用デバイスしだいでは多重容器または連続投与用単位用量に分割して入れることもできる。たとえばアミノグリコシド系抗生物質の投与用量は2〜6単位用量、好ましくは3〜5単位用量、およびさらに好ましくは4単位用量に分割することができる。一つの代表的な実施形態では、本発明のキット類には112mgのトブラマイシン(存在し得る対イオンの重量を除いた遊離塩基体としての重量で求められた値)が投与用量として含まれ、4個のHPMC製2号カプセルの各々に、遊離型塩基としてのトブラマイシンが27mgずつ別々に充填されている。
【0057】
以下の実施例は、本発明の実施を企図する最良の方式を説明するためのものにすぎず、本発明を限定する意味のものではない。
【実施例】
【0058】
(実施例1 吸入用トブラマイシン粉末(TPI)の調製)
硫酸トブラマイシンのドライパウダー組成物については、以下の手順によって調製した。ジステアロイルホスファチジルコリン(DSPC)がゲル状態から液晶状態に移行する温度(約80℃)以上になるように、潅注用滅菌水(SWFI)を加熱する。次にその熱水にDSPCおよび塩化カルシウム・二水和物を加える。得られた液体分散物を、UltraTurrax T−50(商品名)(IKA Labortechnik社製)中、8000rpmで5分間混合処理する。次にその液体分散物に混合しながら、臭化ペルフルオロオクチル(PFOB)を滴加(1分あたり15mlの滴加速度)する。滴加終了後に、得られた水中PFOB滴型エマルションをさらに10000rpmで10分間混合する。UltraTurraxでの乳化工程によってミクロンサイズの液滴が生成する。つづいてそのエマルションの連続相に硫酸トブラマイシンを溶解させ、得られた分散液を噴霧乾燥用の供給原料として用いる。次にその供給原料を噴霧乾燥して以下の表1に示した組成のドライパウダー製剤を得た。
【0059】
【表1】

その粉末を相対湿度10〜15%でカプセル充填機内にセットし、10分間平衡化させた後、50mg(トブラマイシン遊離塩基として27mg)ずつをHPMC製2号カプセルに充填する。
【0060】
(実施例2)
この実施例は、本発明のトブラマイシンドライパウダー組成物の単回投与が、トブラマイシンに関して同様の薬物動態学的挙動を示すトブラマイシン溶液での投与と比較して、トブラマイシンの送達効率が高いことを示す臨床試験の記述である。
【0061】
全般的な臨床試験デザインおよび試験計画
この試験は、無作為化、開放性、連続コホート、活性薬剤対照、単回投与および用量漸増による試験として設計した。連続コホートの各々において、被験体全体はT−326Inhaler(商品名)デバイス(Nektar Therapeutics社(米国、カリフォルニア州、サンカルロス市)製)を使用した以下に示す投与スケジュールに従うトブラマイシン粉末の単回投与、またはDeVilbiss PulmoAidesTMコンプレッサー装着のPALI LC PLUSTMジェットネブライザーによってエアゾル化させた吸入用300mgトブラマイシン溶液(TOBI)の単回投与のいずれかを、3:1に比率で無作為に分けて試験を行った。尚、患者は一つのコホートのみの参加とした。
【0062】
次の増量TPI(吸入用トブラマイシン粉末)治療コホートの実施については、試験が終了したコホートから得られた治療にまつわる有害事象(AE)およびそれ以外の安全性に関わる結果のすべてをデータ監視委員会(DMC)で検討してから進めた。DMCでは以下の判定基準、すなわちTPI治療コホート中の3名またはそれ以上が、投与終了後30分以内にFEV1が少なくとも20%低下することに該当せず、TPI投与被験体はいずれも試験薬に関係する重篤な有害事象(SAE)が認められないという場合に限り、次の増量投与試験段階に進むことが了承された。
【0063】
実施した治験
TPI(吸入用トブラマイシン粉末)の単回投与は、T−326Inhaler(商品名)デバイスを使用して行った。
コホート1:TPI(1カプセルあたりに遊離塩基としてのトブラマイシンが「活性用量」として14mg含有)カプセルを2個投与。
コホート2:TPI(活性用量14mg)カプセルを4個投与。
コホート3:TPI(活性用量28mg)カプセルを2個投与。
コホート4:TPI(活性用量28mg)カプセルを4個投与。
コホート5:TPI(活性用量28mg)カプセルを3個投与。
【0064】
対照の治験
300mg/5mL[保存剤不含、トブラマイシン濃度60mg/mL、溶媒として1/4生理食塩水5mL使用、pH6.0±0.5]溶液でのTOBIの単回投与は、DeVilbiss PulmoAideTMコンプレッサー装着のPARI LC PLUSTMジェットネブライザーによってエアゾル化して行った。
【0065】
本試験での被験体は、嚢胞性線維症(CF)と確定診断された6歳以上の80名までとし、無作為化して治験を行った。各コホートは16名で構成され、そのうちTPIが投与される12名とTOBIが投与される4名とに無作為に分けた。
【0066】
被験体については、投与試験の7〜9日前に適格かどうかのスクリーニングを行った。被験体に投与して、安全性、ならびに治験薬投与30分、1,2,4,8および12時間後の喀痰中および血清中のトブラマイシン濃度について監視下で評価を行った。被験体は、投与後7(±2)日に外来による追跡調査も行った。
【0067】
対照群の選択も含めた治験デザインの検討
本試験に関する一次結果の算定基準は、治験の全般的な安全性および忍容性とした。この結果をさらに評価するために、被験体を無作為に3:1に分けるスキームを選択して治験群への登録者数を最大にした。
【0068】
本試験で同時に行う活性対照は、DeVilbiss PulmoAideTMコンプレッサーによって駆動するPARI LC PLUSTMジェットネブライザーで送達される300mg/5mL TOBIとした。TOBIは、P.aeruginosa感染の嚢胞性線維症患者に対する予後管理用指標薬(対照薬)とした。
【0069】
試験集団の選定に関する採用基準
被験体については、以下の採用基準のすべてに該当する場合に、本試験の参加に適格であるとした。
・試験に関係するいかなる手続きを行う前でも、説明による同意(インフォームドコンセント)の書面およびHIPAA(健康保険計画実施協会)の認可が得られていること。
・スクリーニング時における年齢が6歳以上の男性および女性であること。
・定量的ピロカルピンイオン導入検査(QPIT)で記録された汗中の塩化物濃度が60meq/L以上であり、および/または嚢胞性線維症(CF)と整合性がある同定可能な二つの変異を有する遺伝子型を持ち、CFと一致する一種または複数の臨床的特徴を示すことからCFと診断されていること。
・女性の被験体の場合は11歳以上であるか、または初潮に達していて、しかも血清による妊娠検査で陰性であること。妊娠可能年齢の性的活動性の女性については、試験期間中の避妊に同意しなければならない。
・要求時に喀痰試料を排出できること。
・FEV1が予測値(性別、年齢および身長に基づいてKnudson式で算出される値)の40%以上であること。
・プロトコールの要求にすべて従うことができること。
・試験研究者の見解において臨床上、安定していること。
【0070】
試験対象者選定に関する除外基準
被験体については、以下の除外基準のいずれかに該当する場合、試験対象から除外される。
・試験薬投与前14日以内および試験期間中に、試験以外でアミノグリコシドを吸入または静脈内に投与した場合。
・試験薬投与前14日以内および試験期間中に、治療のためのいずれかの投薬が行われた場合。
・試験薬投与前7日以内および試験期間中に、ループ系利尿剤が投与された場合。
・試験薬投与前30日以内のいずれかにおいて、60cc超の喀血を生じた場合。
・アミノグリコシドまたは吸入による抗生物質に対して局所的または全身的な過敏症の既往歴がある場合。
・血清中クレアチニン濃度が2mg/dlまたはそれ以上、BUN(血液尿素窒素)が40mg/dlまたはそれ以上、あるいは尿検査で2+またはそれ以上の蛋白尿とされた場合。
【0071】
治験または評価から除外される被験体
被験体、その両親、または後見人は、試験のいずれの時期においても既得権を侵さずに参加への同意を撤回することができるものとする。試験研究者の側も、臨床的判断において最大の注意を払わなければならない被験体の場合、または試験プロトコールに応じることができないと考えられる被験体の場合にその被験体を除外できるものとする。可能な場合、試験終了時の外来による追跡調査で挙げられた検査および評価項目についても実施した。
【0072】
必要な再外来ができなかった患者では、できるだけその理由を調べるようにした。その情報については適切な症例報告書(CRF)に記録するようにした。
【0073】
投与が行われずに本試験から脱落した無作為抽出被験体は、代わりの被験体と入替えを行った。しかし、投与後に脱落した無作為抽出被験体については、いかなる場合でも入替えは行わなかった。脱落の理由および日時についても、CRFに記録するようにした。脱落の理由については、以下のように分類した。
【0074】
・有害事象;
・プロトコール違反;
・追跡調査の遺失;
・同意の撤回;
・死亡;
・不適切な登録;
・管理上の理由;
・そのほかの上記記載以外の事由。
【0075】
治験
80名までの被験体について、無作為化して治験を行った。被験体には対照薬剤または以下に挙げたような本試験薬のいずれかの単回投与が行われた。
【0076】
試験薬による治験:T−326Inhaler(商品名)デバイスを用いた投与
コホート1:TPI(活性用量14mg)カプセルを2個投与。
コホート2:TPI(活性用量14mg)カプセルを4個投与。
コホート3:TPI(活性用量28mg)カプセルを2個投与。
コホート4:TPI(活性用量28mg)カプセルを4個投与。
コホート5:TPI(活性用量28mg)カプセルを3個投与。
【0077】
対照の治験
TOBI 300mg/5mL[保存剤不含、トブラマイシン濃度60mg/mL(1/4生理食塩水5mL添加、pH6.0±0.5に調整]は、DeVilbiss PulmoAideコンプレッサー装着のPARI LC PLUSジェットネブライザーによってエアゾル化して投与した。
【0078】
試験薬製品の特徴
本試験で用いたTPIは、トブラマイシンならびに二種類の賦形剤、すなわち1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)および塩化カルシウム(CaCl2)から成るドライパウダー製剤である。TPIは25mgまたは50mgの粉末のいずれかを含有するように、対応するサイズの2−ヒドロキシプロピルメチルセルロース(HPMC)製カプセル内に充填して用いた。本試験ではトブラマイシン粉末は、二種類の活性用量、すなわち1カプセルあたりトブラマイシンとして14mgおよび28mgで用いた。
【0079】
吸入用トブラマイシン溶液(300mg/5mL)であるTOBI(登録商標)は、滅菌され、発熱物質が認められず、保存剤不含のエアゾル化用抗生物質製剤である。使用される製剤1mL当たりには、注射用滅菌水中に溶解したトブラマイシン60mgおよび塩化ナトリウム2.25mgが含まれ、pHは6.0±0.5に調整されている。
【0080】
投与に関する指示事項
無作為に対照薬治験群に割り当てられた被験体には、吸入用トブラマイシン60mg/mL溶液TOBI(登録商標)300mgを投与した。尚、吸入用トブラマイシン溶液は、PARI LC PLUSジェットネブライザーおよびDeVilbiss PulmoAideコンプレッサーを介して被験体に投与された。300mg用量のTOBI(登録商標)吸入用トブラマイシン溶液は、市販のTOBI(登録商標)吸入用トブラマイシン溶液アンプルを購入して準備した。5mLアンプル試験用薬2本は、ホイル袋に入れて用意した。この試験は単回投与で行ったが、試験薬の調製、ならびにネブライザーおよび送達系のセットアップの際に万一こぼれる場合には、2本の5mLアンプルの試験薬が入った前述のホイル袋を用いて患者に提供した。
【0081】
無作為に本試験薬の治療群に割り当てられた被験体には、トブラマイシン用量強度14mg含有のカプセル2個または4個によるTPI、あるいは用量強度28mg含有のカプセル2,3または4個によるTPIを単回投与した。TPIはT−326Inhaler(商品名)デバイスを介して患者に投与した。TPIカプセルはホイルで二重につつまれた防湿性の容器内に密閉し、容器を開けてから30分以内に投与するようにした。コホート1,2および3では、T−326Inhalerデバイスを1回だけ用いて単回用量の投与を完了させた。コホート4および5では、二個目のカプセルの投与後にT−326Inhalerデバイスを取替えるようにしたため、これらのコホートでは2個のT−326Inhalerデバイスを用いて単回用量の投与が完了した。
【0082】
対照薬および試験薬の調製および使用に関する詳細な指示事項は、本試験に関わる研究スタッフに予め伝えられた。
【0083】
治験群への被験体の割当て方法
適格被験体については、試験薬による治験群または対照薬による治験群のいずれかに3:1の比率で無作為に割り振った。被験体が適格基準に適合すると試験研究者または治験コーディネーターが確認すると、試験スタッフはその患者についての無作為化確認書の記入を完了し、対話型音声応答システム(IVRS)によって被験体番号および治験群割り当ての情報を得た。
【0084】
試験開始時には、5種のコホートにおける80名までの被験体が無作為化されるように準備し、試験薬治療群には60名(コホートごとに12名の被験体)および対照薬治験群には20名(コホートごとに4名の被験体)の割り振りを行った。
【0085】
試験で用いられる用量の選定
対照薬治験群での用量は、6歳またはそれ以上のCF患者におけるP.aeruginosaの予後管理用にFDAが認可した用量である300mgのTOBIに設定した。PK(薬物速度論)でのモデル解析を用いると、PARI LC PLUSジェットネブライザー/DeVilbiss PulmoAideコンプレッサーを介して300mg用量のTOBIを送達した場合の、全身における生物学的利用率は、噴霧された全投与用量の11.7%であると想定された。またTOBI300mg吸入1時間後における血清中トブラマイシン濃度の平均値および標準偏差は1.0±0.58μg/mLとなり、吸収されずに沈着する量が多いことが示唆された。
【0086】
試験薬治験群の用量については、トブラマイシン14mg入りTPIカプセル2個または4個、あるいはトブラマイシン28mg入りTPIカプセル2,3または4個とした。
【0087】
各被験体についての用量選定および投与時期の調整
被験体には、300mgTOBI(登録商標)トブラマイシン吸入用60mg/mL溶液の単回投与、あるいは用量強度14mgのカプセル2個または4個、または用量強度28mgのカプセル2,3または4個から成るTPIの単回投与のいずれかで投与を行った。尚、食事に関係する投与時期の制限は設けなかった。
【0088】
盲験性
本試験は、標識オープン式の臨床試験研究とした。2種類の異なる薬物送達系を用いるので、治験の同一性の盲験性は実現不能である。
【0089】
事前および同時に行われる治療
追加の支持療法については、試験現場ごとでの標準的な医療業務にしたがって投与が成された。除外基準の中に列挙された投薬は、被験体選定のスクリーニング時点から試験後の追跡調査での外来時に至るまで被験体には行わなかった。
【0090】
被験体には、試験薬投与前の気管支拡張剤の使用を認めた。但し、気管支拡張剤の投与は、臨床上日常的に気管支拡張剤が用いられている被験体に対してのみに行った。日常的使用とは、スクリーニング前の2週間において1日1回またはそれ以上の投与が行われていたことと規定する。短時間作用型の気管支拡張剤を被験体に投与する場合は、本試験薬投与開始の15〜60分前に行った。長時間作用型の気管支拡張剤を被験体に投与する場合では、24時間前に行った。
【0091】
以下の薬物の使用は禁じた。すなわち本試験薬投与14日以内および本試験期間中を通じてのアミノグリコシドのいかなる剤形およびいかなる療法;ならびに本試験薬投与7日以内および本試験期間中を通じてのループ系利尿薬の投薬。
【0092】
治験コンプライアンス
被験体は、研究者および治験コーディネーターの存在のもとで、自分で試験薬の単回投与を行った。本試験薬投与における早期の中止、中断または延期のいずれの場合でもその理由を原本およびCRFに記録した。試験薬投与期間のすべてについても、原本およびCRFに記録した。
【0093】
各被験体(または適切な場合では患者/法的後見人)は、試験研究者の管理下でHIPAAでの認可を含めたインフォームドコンセントの書面を提出し、(適切である場合は)試験に関係する手続きのいずれも、治験開始前に本試験への参加に同意を明確にした。試験研究者は、被験体について1回目の訪問時、および試験薬投与の7〜9日前(2回目の訪問時)にスクリーニングを行い、登録するのに適格性があるかの判断を下した。試験研究者は、患者に関する現疾患およびそれ以外の関連する呼吸器の病歴を含めた関連医療履歴、基礎的徴候および症状、スクリーニング前6ヶ月以内におけるドライパウダー吸入剤および抗生物質吸入剤の使用状況、ならびにスクリーニング中に実施された投薬および治療状況について精査し記録した。
【0094】
試験研究者は、被験体の身長、体重および生命徴候を含めた身体検査によるスクリーニングを実施した。生命徴候には心拍数、呼吸数、口内体温および座位動脈血圧を含めた。心拍数および呼吸数は、被験体を少なくとも10分間安静にしてから、1分間かけて計測した。血圧については、被験体を少なくとも10分間座らせてから計測した。
【0095】
スクリーニング前の2週間において、1日あたり少なくとも1回またはそれ以上の回数で短時間作用型の気管支拡張剤を治療のために日常的に使用したことが規則的投薬レジジメンに含まれるすべての患者に対しては、肺活量スクリーニング検査の15〜30分前および本試験薬投与の15〜60分前に気管支拡張剤の投与を行った。長時間作用型気管支拡張剤については、前述のように本試験薬投与の24時間前に被験体に投薬した。
【0096】
被験体は、1994年版の米国胸部学界指針に従ってルーチンの肺活量測定を済ませて1秒間努力呼気肺活量(FEV1)、努力肺活量(FVC)および中位努力肺活流速(FEF25−75)を計測し、信頼性の高い肺活量測定が実施できたかどうかを記録し、そのFEV1値が被験体の性別、年齢および身長に基づいたKnudson関係式を用いて計算された予測値の40%またはそれ以上である包含必要条件にあてはまることを保証した。
【0097】
被験体から血液試料を提供してもらって化学的および血液学的なスクリーニング検査を行い、尿試料については浸漬式試験スティックで蛋白尿の検査を行った。11歳またはそれ以上、あるいはすでに初潮に達した女性被験体については、尿試料での妊娠検査も行った。
【0098】
包含および除外の必要条件のすべてを満たした被験体については、本試験への参加資格があるものとし、プロトコールにおいて述べたように無作為化(無作為割付け)した。無作為化した被験体は、診察7〜9日後(2回目の訪問)の状況に復帰させて治験および関連する手続きを行った。
【0099】
訪問2日目、すなわち治験の投与1日前に、無作為化した被験体のすべては服薬前の手続きを完了した。その手続きには基礎的症状での変化、併用療法における変化および生命徴候の測定データも含めた。スクリーニングの際に被験体の身体系でなんらかの異常が見られ、それが臨床上明確であった場合、あるいは被験体の健康状態がスクリーニング時点から臨床上明らかに何らかの変化を示した場合に、試験研究者は被験体の身体検査を再度行った。スクリーニング時の蛋白尿検出用の尿浸漬式スティック検査の結果が微量検出または1+の結果であった場合は、同検査を投与前に再度行うようにした。またスクリーニング時の血清化学的および血液学的検査に異常が見られた場合も、同検査を再度行うようにした。試験研究者は、投与前の被験体の肺活量測定を実施し(通常の短時間作用型気管支拡張剤を使用した被験体の場合ではその15〜30分後に行う)、トブラマイシン分析用の血清および喀痰の試料も採取した。
【0100】
TPI投与群被験体のうち、その時点で短時間作用型気管支拡張剤を規則的または必要に応じて使用している被験体については、治験投与前に試験研究者の裁量の下で短時間作用型気管支拡張剤での予備治療もできるようにした。またその時点で短時間作用型気管支拡張剤を使用していない被験体についても、スクリーニング時および治験投与前の肺機能検査でのFEV1値の低下率が10%またはそれ以上に達した場合に、短時間作用型気管支拡張剤での予備治療を可能とした。スクリーニング時点からのFEV1の相対的変化%については、以下のように計算して求めた。
【0101】
・スクリーニング時からのFEV1の相対的変化%
・=[(予備投与時のFEV1−スクリーニング時のFEV1)/スクリーニング時のFEV1]×100。
【0102】
単回投与の治験は、気管支拡張剤の予備投薬、あるいは肺活量測定を行ってから60分以内に行い、以降の項で記述するようなプロトコールに従い、被験体へのエアゾル送達および安全性に関する客観的評価を行った。被験体は投与時に背筋を伸ばしていすに座って説明を受け、呼吸数を通常に整え、試験薬吸入中はノーズクリップを使用した。治験コーディネーターは、試験薬投与の開始および終了の時刻を記録した。被験体が長く咳込む場合(10秒間超)には、治験コーディネーターはタイマーを止め、被験体への投与が再開された時点でタイマーを再始動させた。TPI投与群の被験体の場合では、試験研究者および/または治験コーディネーターは、試験薬の二回目の吸入時に吸入器から出るガタガタ音(終了音)が聞こえるかどうかに注意をはらった。試験薬投与が完了した直後に、被験体は正規生理食塩水30mLで口をゆすぎ、5〜10秒間うがいを行い、喀出し、そのゆすぎ工程を3回実施した。
【0103】
被験体は試験薬投与開始後12時間、安全性の評価のために診療所に待機した。その後に被験体は診療所から退出し、その8(±2)日後に3回目の訪問のために診療所に戻るようにスケジュールを組んだ。追跡調査のための再訪問のスケジュール化については、患者によって柔軟に対処した(本実施例で許される限り、追跡調査のための再訪問は投与から「第8日目」と示す)。本プロトコールのスケジュールから幾分はずれても、客観的な試験に対する影響は最小限または無いと考えられた。
【0104】
追跡調査のための投与後第8日目の訪問において、治験コーディネーターは、被験体の同時発生疾患、新規または悪化性の有害事象、CFに付随する症状、現在の投薬およびその用量、ならびにOTC(店頭販売楽)の使用状況を含めての被験体の医学上の履歴におけるいかなる変化についても精査を行った。臨床上明らかな、試験薬に関係する有害事象(AE)が見られた被験体については、満足いくような解決がなされるまで繰返して追跡評価(電話によるかまたは診療所の訪問時のいずれかで)を行った。試験研究者は、被験体の身長、体重、生命徴候および肺活量の計測による身体検査を行い、ならびに血液および尿の試料を採取して化学的検査、血液学的検査および浸漬式スティックによる蛋白尿検査を行った。
【0105】
初回のエアゾル送達の変動要因
300mg/5mL TOBIに対するTPIの比較用量評価は、試験薬による治験と対照薬による治験とのエアゾル搬送特性の評価に基づくものである。試験薬による治験および対照薬による治験でのエアゾル特性に関しては、血清中および喀痰中のトブラマイシン濃度の時間推移、本実施例に記載されるような血清および喀痰に関するいわゆる薬物動態学的パラメーターの計算、治験薬の投与に要する時間の計測、ならびにT−326Inhalerデバイスとカプセルとのパーフォーマンスの評価に基づいて求めた。
【0106】
血清中のトブラマイシン濃度
血液試料は、投与前、ならびに治験での吸入における初回の吸入が開始されてから0.5,1,2,4,8および12時間後に採取した。試料の採取は可能な限り決められた時間に近づけて行い、たとえば試験薬投与の0.5時間後の採血ならば±2分以内に、またそれ以降の採血時間では設定時間から±10分以内の時間に行うこととした。それらの時間域からはずれて採取された血液については、プロトコール逸脱とみなした。
【0107】
血清は分離採取後、分析するまで−20℃またはそれ以下で保存した。血清中のトブラマイシン濃度は、Abott社製のTDx(登録商標)/TDxFLx(登録商標)システムを用いる改変型の蛍光偏光イムノアッセイ(FPIA)法で分析した。尚、試料はサンプルカートリッジの希釈ウェルに直接加えて行った。純偏光度についてはTDx/TDxFLx装置によって得た。トブラマイシン濃度の計算には、重み付けを行った4−パラメーター論理式を用いた。トブラマイシンの濃度は、その遊離塩基当量での記載で報告した。
【0108】
本試験の被験体試料のアッセイには、較正用標準(0.05μg/mL,0.10μg/mL,0.40μg/mL,0.80μg/mLおよび0.90μg/mL)ならびに品質管理用試料(0.10μg/mL,0.40μg/mLおよび0.80μg/mL)を準備した。アッセイは全6回での実行とした。
【0109】
定量性を保つ最小限界濃度は、0.05μg/mLであった。アッセイ精度については、品質管理用試料のCV(変動係数)によって反映されるが、0.10μg/mL,0.40μg/mLおよび0.80μg/mLでの品質管理用試料に対するCV値は各々、4.9%、5.7%および5.6%であった。このアッセイ法の正確度については、品質管理用試料のアッセイ間平均回収率によって反映されるが、0.10μg/mL,0.40μg/mLおよび0.80μg/mLでの品質管理用試料に対して回収率は各々、103%、103%および101%であった。全般的にみて、本アッセイ方法は薬物動態学的解析に適した正確度および精度を有することが認められた。
【0110】
血清中における薬物動態学的パラメーターの計算およびTPIの比較用量の推定については、本実施例に記載されている。
【0111】
喀痰中トブラマイシン濃度
喀痰試料は、治験第1日目の予備投与の前、ならびに試験薬吸入時における最初の吸気開始の0.5,1,2,4,8および12時間後に、被験体に深い咳をしてもらって喀出されたものを採取して得た。喀痰試料の採取は、可能な限り設定時間に近づけて行い、しかも血清採取と同じ時間帯で行った。これらの時間間隔をはずれて採取された試料については、プロトコール逸脱とみなした。
【0112】
喀痰試料(最低量100mg)は治験の単回投与前にも採取し、トブラマイシン濃度のベースラインを求めた。
【0113】
喀痰試料は、分析まで−70℃またはそれ以下で保存した。トブラマイシンの濃度は、紫外部検出器装着の妥当な逆相高速液体クロマトグラフィー(HPLC)法を用いる分析によって求めた。
【0114】
被験体の喀痰試料は、最初に0.2規定の水酸化ナトリウム溶液で液化させ、次にトリス緩衝液(トリズマ塩基濃度20.0g/L)で希釈した。喀痰標準試料は、治験前にCF被験体から予め採取したプール喀痰の希釈液に、最終濃度が0μg/g,20μg/g,40μg/g,100μg/g,200μg/g,400μg/gおよび1000μg/g(喀痰)になるようにトブラマイシンを添加して調製した。アッセイ品質管理用の試料は、希釈したプール喀痰にトブラマイシンを、40μg/g,300μg/gおよび800μg/g(喀痰)含有するように添加して調製した。内部標準用のシソマイシン(0.15mg/mL(トリス緩衝液)溶液、100μL)を、各標準試料、対照試料および被験体試料100μLに加えた後、アセトニトリル400μLおよび2,4−ジニトロフルオロベンゼン(0.17g/mL溶液)50μLを加えた。その試料の反応混合物を乾熱ブロックヒーター内、80℃で1時間加熱した。その反応液にアセトニトリル/水(60/40(v/v))混液600μLを加えた後、その50μLについてHPLCで分析した。
【0115】
試料は、Wters社製の600E型ポンプ、486型または2487型紫外部検出器(λmax=360nm)、および717PLUS型オートサンプラー装着のHPLC装置に接続したWaters社製Nova−Pak(登録商標)C−18カラム(3.9×150mm、充填材粒径4μm)上に注入した。移動相の組成は0.2%酢酸/アセトニトリル(39/61(v/v))混液とし、ポンプ流速は最初の5分間は1.5mL/分、その後9または10分間は2.0mL/分とし、運転時間によって加減した。生データ、データ処理および計算結果を得るために、Waters社製Millennium−32C/S LC Software(商品名)(ver.3.20)を用いてWaters社製HPLC装置を操作し、分析結果の報告を行った。内部標準シソマイシンに対するトブラマイシンのピーク高さ比(PHR)を計算した。そのアッセイの完了には全17回の運転を行った。
【0116】
HPLCにおけるトブラマイシンおよびシソマイシン(内部標準)の保持時間は各々、3.8〜4.1分および10.0〜10.6分の範囲に観測された。喀痰については、PHR(ピーク高さ比)と濃度との関係は20〜1000μg/gで線形性を有することが認められた。回帰式はPHR=Bx+A、重み付け1/x(xはトブラマイシン濃度)とした。定量に関する限界最低濃度は20μg/gであった。標準試料の濃度については、正規の濃度の97〜102%以内に入り、変動係数(CV)は5.7%以内であった。アッセイ値の精度は、品質管理用試料のCVによって反映され、濃度40μg/g,300μg/gおよび800μg/gの品質管理用試料ではCV値は各々、5.2%、3.9%および2.0%であった。本アッセイ法の正確度については、品質管理用試料のアッセイ間の回収率によって反映され、濃度40μg/g,300μg/gおよび800μg/gの品質管理用試料では回収率は各々、107%、99%および97%であった。全般的にみて、本アッセイ方法は薬物動態学的解析に適した正確度および精度を有することが認められた。
【0117】
治験投与時間
治験の投与時間は、被験体の最初の吸入の開始から投与完了までの時間と規定した。治験の投与は、T−326Inhalerデバイスがガタガタ音を発生し始めた時、または対照のPALI LC PLUSネブライザーがブツブツ音を発生し始めた時に終了させた。TPIカプセルの場合、被験体の二回目の呼吸の際にガタガタ音が聞こえるかどうかに試験研究者は注意し、その徴候があればカプセルはすでに空になっているとした。
【0118】
治験での投与が何らかの理由で中断された場合は、中断した時間、ならびに連続投与の開始および停止時間を記録した。投与の中断があった場合、全投与時間に中断中の時間は含めなかった。投与が中断し、その停止時間または投与再開時間のいずれかの記録が抜けてしまった場合は、治験での投与時間は計算不能とした。
【0119】
使用したT−326InhalerデバイスおよびTPIカプセルの検査および残存トブラマイシンの分析
本試験プロトコールには明記していないが、使用したT−326Inhalerデバイスおよび使用したTPIカプセルについては、使用後に分析してパーフォーマンスの評価を行った。
【0120】
有害事象
有害事象(AE)とは、医薬品の投与を受けた治験被験体において医学上不都合ないかなる出来事も規定し、用量にはかかわらず、また治験による投与と必ずしも明らかな因果関係が証明されない場合も含まれる。したがってAEは、医薬品の使用に一時的に付随する好ましくなく意図されない徴候(実験上の異常な見解も含む)、症状または疾患のいかなるものも、その医薬品との関連性の考慮の有無にかかわらず、含めることができよう。この定義には介在性の病気または傷害、およびさきに存在する病状の増悪も含まれる。予期しないAEとは、本質または重篤性が実用上の医薬品情報とは整合しない事象のことである。
【0121】
AEは、被検者による任意の申し出か、または試験研究者または治験コーディネーターによる一般的な問診の結果発見され得るものである。
【0122】
AEのすべては解明されるまでモニタリングするか、あるいはそのAEが慢性的なものと判定される場合は、原因をはっきりさせた。AEが試験終了時でも未解明な場合は、そのAEの追跡調査の継続が保証されるかどうかについて、試験研究者および医療監視者による臨床評価が成された。
【0123】
AE CRFに記録された有害事象の重篤性については、試験研究者が以下のような判定を下した。
軽度:通常生活に支障なし。
中程度:通常生活に何らかの支障あり。
重度:通常生活が不能。
【0124】
AEに対する本治験の関連性については、以下の規定に基づいて試験研究者が判定した。
すなわち、
関連性なし:治験薬を摂取しても起こらないか、またはAEの出現が治験薬の投与時間と合理的な関連性がみられないか、あるいはそのAEが治験医薬品とは関連性がないと考えられる場合。
関連する可能性あり:本試験医薬品の投与とAEとに合理的な時間関連性がみられ、そのAEについて試験医薬品の摂取以外の原因でも同様に説明し得る場合。
高い関連性あり:本治験とAEとに合理的な時間関連性がみられ、そのAEについて試験医薬品の摂取による出現説明の方がそれ以外の原因による説明よりも妥当である場合、またはそのAEの原因として試験医薬品が最も考えられる場合。
【0125】
重篤な有害事象(SAE)
本治験ではSAEは全く報告されなかった。
【0126】
研究室での臨床検査
血液学的基礎データ、血清化学、および浸漬式スティックによる蛋白尿検査の測定を対象とする研究室での検査は、スクリーニング時点、および治験薬投与後8日目の追跡調査時に実施した。治験第1日目の投与時には、投与開始から12時間後に血液尿素窒素(BUN)および血清クレアチニンを計測した。スクリーニング時における尿浸漬スティックによる蛋白尿検査の結果が微量陽性または1+であった場合は、さらに同検査を投与前に再度行い、また血清化学および血液学的検査についても、スクリーニング時点で異常値を示した場合は再度検査を行った。
【0127】
本治験で実施された血液学的検査には、全血球数計測(WBC)、赤血球数計測(RBC)、ヘモグロビン値、ヘマトクリット値、分化度および血小板数計測を含めた。
【0128】
実施された血清化学的検査には、血清中のナトリウム、カリウム、塩化物、炭酸水素塩、BUN、クレアチニン、グルコース、カルシウム、リン、γ−グルタミルトランスフェラーゼ(GGT)、アラニントランスアミナーゼ(ALTまたはSGPT)、アスパラギン酸トランスアミナーゼ(ASTまたはSGOT)、アルカリホスファターゼ、乳酸脱水素酵素(LDH)、総ビリルビン、直接ビリルビン、間接ビリルビン、総蛋白質、アルブミン、および血清中ヒト絨毛性腺刺激ホルモン(HCG)の定量を含めた。
【0129】
気管支痙攣および肺活量測定
肺活量測定検査(1秒間努力呼気肺活量[FEV1(L)]、努力肺活量[FVC(L)]、および努力肺活量の1/4〜3/4の中程度における努力性吐出流速[FEF25−75])については、本治験の単回投与の前日、および投与完了の30分後に実施して気道の反応を評価した。また投与後8日目の追跡調査時にもルーチンの肺活量測定を実施した。
【0130】
投与前から治験薬投与終了30後までの間に、FEV1が20%またはそれ以上低下した場合は、本試験プロトコールでは気管支痙攣であると予め規定した。
【0131】
投与30分後にFEV1が20%またはそれ以上低下した場合は、FEV1の低下率が10%未満になるまで、試験研究者によって定められたスケジュールにしたがって肺活量測定(FEV1、FVCおよびFEF25−75)を再度実施した(尚、この処置は被験体12/317集団では行わなかったが、1例のみでFEV1の減少率が20%またはそれ以上に達したことが予想された)。FEV1の低下率が20%またはそれ以上に達したことが予想されたケースのすべては、AEとして記録した(尚、この処置は被験体12/317集団では行われなかった)。適切な投薬およびその後の追跡調査を伴う治験は、試験研究者の裁量のもとで行われた。
【0132】
生命徴候
生命徴候は、投与前日、ならびに投与開始の30分、1,4,8および12時間後に計測し、その実施項目には10分間安静後の座位での動脈血圧、心拍数および呼吸数、ならびに体温の測定を含めた。また生命徴候の計測は、投与後8日目の追跡調査の訪問時にも行った。
【0133】
身体検査
被験体の身体検査は、投与後8日目の追跡調査の訪問時に行い、その実施項目は被験体の全身外観、皮膚、リンパ節、HEENT(頭部、眼部、耳部、鼻部および咽喉部)、両肺、心血管系、腹部、手足、筋骨格系、神経系、および(所望により)尿生殖器系の身体所見から構成されるようにした。
【0134】
計測値の適正さ
本試験において用いられた効力測定は標準的、すなわち広く用いられ、信頼性、正確さ、ならびに有効薬剤と無効薬剤との識別能を有すると一般に認識されるものとした。本試験において用いられる安全性の測定は、標準的な臨床上および研究室の手順で行った。
【0135】
薬物速度論
喀痰および血清中のトブラマイシンのアッセイから得られた濃度(C)対時間(t)のデータについては、モデル非依存的な方法によって解析して薬物動態学的パラメータ類を得た。最高濃度(Cmax)および最高濃度到達時間(tmax)に関しては視認によって求めた。最終速度定数(λz)に関しては、最終相の対数線形回帰によって求めた。半減期(t1/2)に関しては、式t1/2=ln(2)/λzで算出した。尚、計算は定量限界低濃度域をすべてゼロとして行った。0時間(投与前)から投与12時間後までの、喀痰および血清中の濃度−時間曲線下の面積、すなわちAUC(0,12)に関しては、台形則によって算出した。無限時間までのAUC,すなわちAUC(0、∞)に関しては、式AUC(0,12)+C(12)/λz;但し、C(12)は投与開始12時間後の濃度、で算出した。
【0136】
薬物動態学的パラメーターのすべては、平均値±SD(標準偏差)で表わした。調和半減期は、
【0137】
【数1】
で推定して求めた。
式中、
【0138】
【数2】
は用量ごとの最終速度定数の相加平均である。
調和平均半減期の標準偏差
【0139】
【数3】
は、
【0140】
【数4】
で求めた。
【0141】
式中、
【0142】
【数5】
は用量ごとの平均最終速度定数の標準誤差である。
【0143】
TOBIに対するTPIの相対用量の推定
線形回帰モデルは、全コホートから得られたTPIの血清中濃度データのlogAUC(0,12)対log(TPI用量)に当てはめ、TOBIに対するTPIの相対用量を推定した。その相対用量および95%信頼区間については、当てはめた回帰線の逆関数、ならびTOBIデータの平均logAUC(0,12)における上側および下側の95%信頼域を用いて求めた。
【0144】
モニタリング
本試験は、臨床試験実施に関する基準(GCP)に従い行われた。
【0145】
データの取扱い
症例報告書データは、Clintrial(登録商標)データベースに二重に入力した。
【0146】
データの品質管理は、手続き型言語/逐次クエリー言語(PL/SQL)およびSAS(登録商標)ソフトウエア第8.2版またはそれ以降の版(SAS Institute社(米国、ノースカロライナ州、キャリー市)製)を用いて実施した。データ解析は、予め規定した解析プランに基づき、SASソフトウエア第8.2版またはそれ以降の版を用いて実施した。全データベースを通じての誤り率は0.022%、すなわち上側95%信頼限界の0.157%(丸めた値)と推定された。この上側信頼限界は、選択した標準の0.5%未満である。
【0147】
統計処理方法および試料サイズの決定
この第1相試験の目標は、試験薬であるTPIおよび対照薬であるTOBIの治験の安全性の評価、およびTPI投与で得られる薬物動態学的プロフィールがTOBI300mg/5mL投与の場合に相当することの推断を行うことである。本試験で規定される目標には有効性の項目を含めなかった。設定された摘要事項および解析事項についてはすべて本質的な診査を行った。TPI治験群とTOBI治験群との間の統計的類似検定および両治験群間の差に関する統計的仮説検定は、いずれも本試験では企画しなかった。本試験では治験センターごとの登録可能な被験体が少数であるため、本試験の統計解析計画ではデータを全解析センターにプールするように前もって指定した。
【0148】
摘要事項および解析事項のすべては、6種の治験群のそれぞれに設けた。別に条件設定がない場合、頻度および百分率の値については絶対変数に対して算出し、非欠測値、平均値、標準偏差、最小値、中央値および最大値の数については連続変数に対して算出した。CRFに記録されたデータのすべては、被験体の個人データ一覧表中に存在するようにした。
【0149】
3種の被験体集団を以下のように規定した。
・すべてに登録された集団:本試験に登録され、無作為化された被験体。
・安全性評価が可能な集団:無作為化され、前述の試験薬のすべてまたは一部の投与を受けた被験体。
・薬物動態学的評価が可能な集団:無作為化され、前述の試験薬のすべての投与を受けた被験体。
【0150】
基礎的および個体群統計学的特性については、「すべてに登録された」被験体集団に対して摘要した。トブラマイシンの薬物速度論の評価については、「薬物動態学的評価が可能な」集団を用いて実施した。TOBI投与の場合と同様の全身的トブラマイシン摂取に相当するTPI用量の推定については、TPIの比較用量の評価が可能な集団で行なった。それ以外のすべての変数の解析については、「安全性評価が可能な」集団を用いて行った。
【0151】
すべての解析にSASソフトウエア(第8.2版)を用いた。試験結果のグラフ表示にはMicrosoft社製Excelソフトウエアを用いた。
【0152】
エアゾル搬送の分析および相対的TPI用量の推定
本単回投与治験を受けた被験体のすべては、喀痰および血清中のトブラマイシン濃度、血清に関して推定された薬物動態学的パラメータ類、ならびに治験の投与時間に基づき影響を受けるエアゾル送達特性の解析および評価にあてた。
【0153】
TOBIに相当するTPIの用量の推定には、血清に関するAUC(0,12)のデータを用いた。従属変数としてlogAUC(0,12)および独立変数としてlog(TPI用量)での線形回帰解析は、TPI投与群のすべてから得られたデータを用いて実施した。TPIの相当用量については、5種のTOBIコホートの組合わせから得られたTOBIにおける血清中トブラマイシン濃度の平均logAUC(0,12)値に当てはめた回帰線の逆関数を得て求めた。
【0154】
二次的なエアゾル送達の解析
本試験のプロトコールでは、エアゾル送達に関する二次変数を認定しなかった。
【0155】
安全性の解析
本治験の投薬を受けた被験体のすべてについては、AE、肺機能変化、臨床検査結果、生命徴候および身体検査に基づき、安全性の評価がなされた。
【0156】
有害事象の評価
基礎的症状、および治験の結果生じた有害事象(AE)に関しては、MedDRAシソーラスを用いて符号化した。個々の治験の結果生じたAEの全発生率(治験期間中または治験後に少なくとも1回の有害事象が認められた被験体の%)については、TPI治験とTOBI治験との間の顕著な差に関する記述的評価を行った。その統計的検定は企画しなかった。またAEについては、試験薬および対照薬での治験に関する重篤度(軽度、中程度、重度)および薬物との関連性(関連性なし、関連性高い)も摘要した。薬物との関連性を評価するための「関連性」分類には、個々のAEを本治験と「関連性あり」、「関連性高い」、および「関連性認めず」と試験研究者が判定を下して行った。
【0157】
肺機能の変化
被験体が肺疾患に罹患していない場合では、肺機能の正常値はFEV1、FVCおよびFEF25−75(肺活量計測値)で表されてきた。これらの標準値は、一般に肺疾患罹患被検者の試験において用いられている。肺活量計測値の生データは、以下に述べるようにKnudson式を用いて標準の予測%値に変換した。FEV1、FVCまたはFEF25−75に関するKnudson標準値とは、被験体ごとに以下の式を用いる年齢(歳)と身長(cm)との線形連結である。
Knudson標準値=C0+C1×身長+C2×年齢
式中、係数C0、C1およびC2は、被験体の性別および年齢群に基づき定められている。
【0158】
肺機能測定から得られる生データおよびKnudson標準値が得られると、予測%値は以下のようにして算出される。
%予測値=(生の観測値/Knudson標準値)×100
投与前から試験薬投与終了30分後までの予測されるFEV1%の変化(FEV1%)および相対変化については、以下の式を用いて算出した。
FEV1%の変化=投与後のFEV1%−投与前のFEV1%
相対変化=[(投与後のFEV1%−投与前のFEV1%)÷投与前のFEV1%]×100
基準値からの相対変化が20%以上の低下となった被験体については、気管支痙攣が起きたと規定した。
【0159】
FEV1%の変化および相対変化、ならびに気管支痙攣発生率に関しては、治験群間での記述的比較を行った。さらに以下の変数についても同様に導出し、摘要した。
【0160】
・投与前から投与終了30分後において予測されるFEV1%の相対低下が10%またはそれ以上である被験体の発生率
・投与前から投与終了30分後において予測されるFVC%およびFEF25−75%の変化および相対変化
・投与前から治験投与8日目の追跡時までに予測されるFEV1%、FVC%およびFEF25−75の変化および相対変化
安全性に関するほかの変数
臨床検査結果、生命徴候、併用薬、医療処置および身体検査を含むほかの安全性に関する変数に関しては、記述的な摘要を行った。さらに基底値から試験終了時の値の変化についても計算し、臨床検査データ、生命徴候および肺活量測定値に摘要した。尚、これらの計算では、試験薬投与のできるだけ近い時点に得られた評価値を基底値とした。各々の正常域よりも高いかまたは低い臨床検査結果の発生頻度の変化についても、評価を行った。
【0161】
試料サイズの設定
試料サイズおよび並列処理設計選択範囲については、統計力要件よりも臨床的および実地における要件に基づき設定した。すべての解析は診査によって行い、記述的方法を用いて取扱った。本試験では推論による統計解析は企画しなかった。
【0162】
暫定的解析
各コホートの試験終了時に、主要な安全性に関する変数の概要を作成し、治験データ監視委員会(DMC)に提出して用量増大の検討および決定がなされた。以下の結果は、被験体によって列挙され、治験完了後にコホートごとにまとめられたもので、DMCでの審議をサポートする。
【0163】
・被験体の個体群統計;
・投与前後で予測されたFEV1%および変化%;
・重篤な有害事象;
・治験の結果発生したAE;
・呼吸器系の、治験の結果発生したAE。
【0164】
この暫定的解析手順は、DMCによる安全性の検討および容量増大に関する議論をサポートするためにのみ設計された。尚、試験データの統計解析については企画せず、暫定的な安全性および用量増大の検討結果に、統計学上のα−水準の調整を行わなかった。
【0165】
被験体のスクリーニング、登録および無作為化
本試験には、15名の試験研究者によって全部で97名の被験体がスクリーニングされた。
【0166】
その97名の被験体うちの90名については、治験登録者基準に適合し、6種の治験群のうちの一つに無作為に登録(表2)した。97名のうちの残りの7名については、試験参加基準に適合せず、登録されなかった。
【0167】
【表2】
治験前の被験体の脱落
登録された90名の被験体のうちの3名については、治験薬投与の前にすでにAEが見られたため脱落させ、本治験の単回投与は行わなかった(表3)。90名のうちの残りの87名の被験体については、治験薬の投与が行われた。
【0168】
【表3】
試験の完遂
治験薬の投与が行われた87名の被験体のうちの86名が、試験を完了した。87名のうちの1名の被験体については、治験によって生じたAEによって試験から脱落した。
【0169】
試験を完了した86名の被験体のうちの1名については、投与は行われたが、試験に用いたカプセルがT−326Inhalerデバイスによって穿刺されなかったことが試験後に回収されたカプセルを評価して判明した。その被験体における血清中トブラマイシン濃度については、定量限界以下(BQL)であったため、その被験体には治験薬が投与されなかったことが確認された。
【0170】
エアゾル送達評価のために解析されたデータ集合
登録された90名の被験体のうちの86名は、無作為化し、治験薬の単回投与を行い、本プロトコールに関する安全性の評価対象とした。その90名のうちの3名については、AEのために治験薬投与前に試験から脱落したため、安全性の評価からは除外した。残りの1名については、投与時にカプセルがT−326Inhalerデバイスの不具合で穿刺されなかったために治験薬の投与が行われず、安全性の評価対象から除外した。
【0171】
【表4】
治験薬が投与された86名の被験体のうちの84名については、目的の薬物動態学的評価を行い、薬物速度論においてTOBI用量に相当すると考えられるTPIの用量を推定した。治験を受けた被験体のうちの2名については、薬物動態学的評価および相当用量の評価から除外した。
【0172】
【表5】
本治験を受けなかった被験体に対しては、薬物動態学的評価も行わなかった。
【0173】
個体群統計学的特性
本試験の登録被験体は、男性43名および女性47名、年齢は7〜50歳で嚢胞性線維症と診断された被験体であった。被験体の平均年齢はTOBI治験群での19.5歳からTPI2×14mg投与群での24.1歳までであり、治験群間でほぼ同等であった。被験体のうち15名が7〜12歳、22名が13〜17歳、および53名が18〜50歳であった。
【0174】
被験体のうち79名が白色人種系、5名がスペイン語系、3名がアフリカ系および3名がそのほかの系統であった。TPI治験群とTOBI治験群との間の性別および人種の分布はほぼ同等であったが、TPI4×14mg投与群とTOBI投与群とでは性別に関する不均衡が僅かに見られた(TPI4×14mg投与群の被験体は女性11名および男性3名;TOBI投与群の被験体は女性8名および男性12名)。試験結果に対するこの不均衡の影響は不明である。
【0175】
TPI投与群およびTOBI投与群の被験体は、スクリーニング時点で身長および体重の平均が同程度になるようにした。
【0176】
ほかの基本的特性
登録被験体については、臨床検査データ(定量的ピロカルピン−イオン導入検査(QPIT)時の汗中の塩化物濃度が60mEq/L以上であり、および/または二つの変異が確認された遺伝子型であること)の記録、および臨床的に安定型の嚢胞性線維症と診断しても臨床上明らかにに整合していることの記録がなされた。試験開始前に認められた医療履歴の所見、ならびに徴候および症状に関しては、治験群間で差はほとんどなかった。
【0177】
ほかの包含および除外基準については、いずれも満たしていた。但し、注目されることには、4名の被験体において、第1日目の投与後に軽度から中程度の持続性の喀血が見られたことである。被験体のすべては、スクリーニング時のFEV1が性別、年齢および身長に基づき予測される値の40%またはそれ以上という包含基準に適合した。被験体の予測されたFEV1%の中央値に関しては、6種の治験群間でほぼ同等であった(TPI2×14mg投与群での58.51%からTPI3×28mg投与群での82.40%まで、およびTOBI投与群での67.95%)。但し、合計3名の被験体では浸漬式スティックでの蛋白尿検査の結果が2+未満という治験参加必要条件を満たしていたが、スクリーニング時での尿試料の提供が行われなかった。
【0178】
登録被験体90名のうちの39名では、スクリーニング前の6ヶ月以内にドライパウダーの吸入投与を受けており、治験群によって25〜60%の被験体を占めた。被験体90名のうちの67名については、以前にTOBIの投与を受け(その割合は治験群によって40〜100%)、被験体のうちでほかのアミノグリコシドの吸入投与を受けたケースはなく、被験体90名のうちの10名では、過去6ヶ月以内にほかの抗生物質の吸入投与を受けた。
【0179】
登録被験体90名(すなわちTPI投与被験体およびTOBI投与被験体)のうちの36名では、治験薬投与前15〜60分以内に、それぞれに合った標準的CF治療の一部として短時間作用型気管支拡張剤の使用が行われた。そのうち二つのケースでは、治験薬投与の60分以上前に気管支拡張剤が使用された。
【0180】
治験コンプライアンスの評価
本プロトコールで要求される単回治験薬投与に関するコンプライアンスについては、90名のうちの86名の被験体が本治験を受けた(3名については基礎的症状で投与前に脱落し、1名については投与工程は行われたが、T−326Inhalerデバイスが治験薬カプセルを穿刺しなかったために実際には治験薬の投与は行われなかった)ことから、許容されるものであった。コンプライアンス不履行の被験体4名の事象については、表6にさらに列挙した。
【0181】
【表6】
血清中トブラマイシン濃度および薬物動態学的パラメータ類
図1に示すように、TPIおよびTOBI投与後のトブラマイシンの血清中平均濃度−時間プロフィールから、薬物が迅速に吸収されたことが認められた。すなわちtmax(最高濃度到達時間)の中央値が治験投与のすべてにおいて1時間であった。薬物の体内分布も極めて速いと考えられ、血清中からの消失は一相性の指数関数的減少挙動を示し、最終の平均半減期は2.8〜3.5時間の範囲であった。TOBI投与後のトブラマイシンの薬物動態学的パラメータ値については、過去に発表された試験結果と整合するものであった。
【0182】
TPIの投与量を上げると、AUC(0,∞)、AUC(0,12)およびCmaxの値が明らかに増加する(表8)ことから、トブラマイシンの体内摂取量が増大した。尚、これらのパラメータの増大は、用量比例的な増大よりは僅かに少ないものであった。
【0183】
4名の被験体では咳が生じたため、投与を一時中断した。その中断事例は、TPIの4×14mg、3×28mgおよび4×28mgの投与群で起こった。咳が見られた被験体のすべてに関しては、AUC(0,∞)、AUC(0,12)およびCmaxのいずれの値も、同じ投与群における一時中断なしで投与が終了したほかの被験体の値の範囲内にあった。
【0184】
AUCおよびCmaxの算出値から、4×14mgの手動充填カプセルの投与と2×28mgの自動充填カプセルの投与との間で被験体から検出されたトブラマイシンの体内摂取量の差は見られなかった(P>0.5)。したがって、手動充填および自動充填のカプセルのバイオアベイラビリティは同等であり、これら2群については統合した。
【0185】
デバイス内に残った粉末の%とAUC(0,∞)(r=−0.22、P=0.0771)、AUC(0,12)(r=−0.23、P=0.0707)およびCmax(r=−0.22、P=0.0838)との間には、全般に弱い負の相関性が見られた。体内摂取量が低いケースほど、デバイス内の残留TPI量が多い関係が見られた。4×28mgTPI投与群中の被験体のうちの1名(被験体No.04/409)では、粉末のデバイス内残留率が45%超であり、同一治験群内で最低のトブラマイシン体内摂取量となった。
【0186】
FEV1基底値の変化と血清でのAUCおよびCmaxとの間には有意な相関性は見られなかった[TPI投与の被験体:AUC(0,∞)(P=0.9838)、AUC(0,12)(P=0.9990)およびCmax(P=0.9110);TOBI投与の被験体:AUC(0,∞)(P=0.5261)、AUC(0,12)(P=0.4337)およびCmax(P=0.3878)]。
【0187】
【表7】
a中央値(領域)。最後の2列の被験体の数値を除き、それ以外の値は平均±標準偏差で表わす。
【0188】
TPIとTOBIとの比較用量解析
TOBI300mg投与後の体内摂取量に関しては、TPI4×28mgカプセル投与後の場合と極めて類似していた(図2および3)。全コホートから得られたAUC(0,12)データを用量の関数として調べてみると(図2)、その結果からTPIの115mg用量がTOBIの場合と同様な平均AUC(0,12)を示すと考えられることが分かる。AUC(0,∞)について考慮すると(図3)、TPIの112mg用量がTOBIの場合と同様なAUC(0,∞)を示すと考えられる。これらの結果に基づくと、TPIの28mgカプセル4個(総投与量112mg)で、TOBIの場合に極めて近い全身的体内摂取量になると考えられることがわかる。
【0189】
喀痰中トブラマイシン濃度および薬物動態学的パラメータ類
TPIおよびTOBI投与後では、喀痰中の最高濃度は平均で投与30分後に現われ(図4)、その後半減期0.8〜2.2時間で減衰した(表8)。
【0190】
薬物動態学的パラメータ類の変動性については、血清よりも喀痰の場合の方が大きい。喀痰から得られる薬物動態学的パラメータ類がもともと変動しやすいことに加えて、被験体のうちの数名では、必要な量の喀痰が得られなかった(いずれの時間でも喀痰試料が得られなかった被験体は治験群あたり3名以下であった)。このことは、体内摂取量、すなわちAUC(0,∞)、AUC(0,12)およびCmaxの計算に大きな影響を及ぼすと考えられる。結果として、用量を上げると喀痰中の量が上昇する傾向が見られる一方、喀痰中のレベルに基づいて用量の比例関係を確定することはできなかった。
【0191】
【表8】
a中央値(範囲)。最後の2列の被験体の数値を除き、それ以外の値は平均±標準偏差で表わす。
【0192】
bnはパラメータによって異なる場合がある。一回の解析のいずれで用いた被験体でもその最大数を挙げる。
【0193】
試験薬の投与時間
TOBIの被験体への投薬時間の平均は、約16分間であった(表9)。これに対して2カプセルのTPIの投薬では、2×14mgおよび2×28mgの投与の場合、各々平均で1.7分間および2.5分間であった。またTPI4×14mg、3×28mgおよび4×28mgの投薬の場合、各々平均で4.2分間,4.5分間および4.9分間であったが、それらのコホートでは2個目のデバイスは装填されておらず、各々3個目および4個目のカプセルの投与に用いた。したがってカプセル数が増えるとTPIの投与時間が増えるが、それによって用量強度も増す。
【0194】
【表9】
T−326InhalerデバイスおよびTPIカプセルの性能の評価
2個のカプセル以外のTPIカプセル投与のすべてケースでは、要求されるように投与を行った(但し、TPI4×28mg投与群中の被験体のうちの1名で、3個目および4個目のカプセルは、それからはずれた)。カプセル投与では85%またはそれ以上の頻度で2回目の呼吸時にガタガタ音(吸入終了の音)が聞こえた。
【0195】
Nektar解析によって治験施設および治験監視者が報告したデバイスの破損は存在しなかった。被験体の吸入器およびカプセル内の残存トブラマイシンの合計は、平均で正規用量の10.5%であった。その解析には、不適当な投与により残存粉末が45%超に成った被験体No.04/409も含んでいるので、デバイス中に実際に残存した粉末量はもっと少ないとも考えられる。
【0196】
脱落および遺失データの取り扱い
本試験では、無作為化した被験体のうちの3名が、投与前のAEによって治験薬投与の前に脱落した。無作為化した被験体のうちの別の1名については、本試験が完了したが、投与時に2個のTPIカプセルの両方とも穿刺されなかったために実際には吸入されなかったことが後になって判明した。これらの被験体については入替えがなされるまで、スクリーニングおよび登録の作業は継続し、入替え用の被験体には新たな無作為コードナンバーが割り振られた。
【0197】
1名の被験体は、早期に本試験から脱落した。その早期脱落理由の解析には、最終結果−繰越(LRCF)方略は用いなかった。
【0198】
定量限界以下の低レベルのトブラマイシン濃度に関しては、いずれの計算においてもゼロとして扱った。
【0199】
被験体の「エアゾル送達の部分集合」の適用
本プロトコールでは、無作為化された被験体および本治験の投与が完全に行われた被験体に関して、薬物速度論の評価が可能であるとした。試験が進行した時点で、予期されない投与およびTPIカプセルの状態の問題点によって、薬物速度論での評価の妥当性基準の見直しを行なった。6名の被験体の全員については、薬物速度論および用量比較の解析対象から除外した(4名の被験体については治験薬の投与が行われず、2名の被験体については治験薬投与が完全な用量で行われなかった)。それ以外の84名の被験体については、薬物速度論および用量比較の評価が可能とした。
【0200】
吸入による治験薬投与を受けた86名の被験体全員は、治験薬の投与時間の評価対象に含め、さらにカプセルおよびT−326Inhalerデバイス内の残存トブラマイシンの評価対象にも含めた。
【0201】
同等性の証明のための活性対照試験
本試験には、試験医薬品(TPI)と対照薬(TOBI)との臨床上および薬物動態学的な同等性を証明するようなデザインまたは検出力を設定しなかった。
【0202】
亜群の調査
以下の亜群解析は、TPIが正しい用量で投与された後の血清へのトブラマイシンの取込み(AUC(0,12)、AUC(0,∞)およびCmax)に関して行った。
【0203】
・12歳未満の被験体と12歳以上の被験体との間(P>0.4);
・以前にドライパウダーを使用した被験体と未使用の被験体との間(P>0.8);
・男性被験体と女性被験体との間(P>0.1);
・体重(P>0.2)
これらの変数の中でトブラマイシンの体内摂取に影響すると考えられるものはなかった。
【0204】
治験薬投与の15〜60分前に気管支拡張剤を使用したTPI投与被験体(N=27)においてAUCおよびCmaxの計算値としてのトブラマイシンの体内摂取量は、気管支拡張剤を使用しなかった被験体(N=37)の場合よりも僅かに高かった。AUC(0,∞)、AUC(0、12)およびCmaxの値は各々、19%(P=0.0685)、27%(P=0.0240)および38%(P=0.0038)高かった。気管支拡張剤の使用によって、結果的に全身血流循環に移行するトブラマイシン量が増す場合があり、このことはおそらく肺内に沈着するトブラマイシン量が増すためと考えられる。
【0205】
エアゾル送達のまとめ
TPIおよびTOBI投与後のトブラマイシンの血清中平均濃度−時間プロフィールから、薬物が迅速に吸収されることが分かる。すなわちtmaxはすべての治験例において1時間であった。薬物体内分布は極めて速いと考えられ、一相性の指数関数的な減衰が見られ、最終平均半減期は2.8〜3.5時間の範囲にある。TOBI投与後のトブラマイシンの薬物動態学的パラメータ値については、既存の報文と整合している。
【0206】
TPIの用量を上げると、トブラマイシンの体内摂取量が増大し、このことはAUC(0,∞)、AUC(0、12)およびCmaxの値の増加によって証明された。それらの増加は用量比例分と比較して僅かに少なかった。4×14mgの手動充填カプセル投与の被験体と2×28mgの自動充填カプセル投与の被験体との間で、トブラマイシン体内摂取量に関する差は検出されなかった。したがって、これらの二つの群は用量比較解析に関しては統合した。デバイス内の残存粉末の%とAUC(0,∞)(P=0.0707)、AUC(0,12)(P=0.0771)およびCmax(P=0.0838)との間には、全般に弱い負の相関性が見られた。FEV1の基底値からの変化とAUC(0,∞)(P=0.9838)、AUC(0,12)(P=0.9990)およびCmax(P=0.9110)との間には、有意な相関性は見られなかった。
【0207】
TOBI300mg投与後の体内摂取量は、TPI3×28mgカプセル投与後とTPI4×28mgカプセル投与後との体内摂取量の間にあった。その結果に基づくと、TPI4×28mgカプセル投与(総用量112mg)の場合が、TOBIでの全身体内摂取量に最も近いと考えられる。
【0208】
TPI投与亜群の解析から、年齢、以前のドライパウダー使用、性別および体重は、TPIにおけるトブラマイシンの体内摂取量に影響せず、トブラマイシンの薬物動態学的特徴はTPI亜群でほぼ同じであることが分かった。気管支拡張剤使用のTPI投与被験体(N=27)におけるトブラマイシンの体内摂取量は、気管支拡張剤を用いなかったTPI投与被験体(N=37)の場合と比較して平均で僅かに高かった。
【0209】
TPIおよびTOBI投与後の喀痰中のトブラマイシンの濃度は、平均30分で最高レベルに到達し、また平均半減期は0.8〜2.2時間の範囲にあることが推定された。
【0210】
薬物動態学的パラメータの変動性は、血清中の場合と比較して喀痰中の方が高かった。
【0211】
投薬時間は、TOBIでは平均約16分であった。これに対して2個のTPIカプセルの投与時間は、2×14mgおよび2×28mg投与では各々、平均1.7分および2.5分であった。またTPI4×14mg、3×28mgおよび4×28mgの場合の平均投与時間は各々、4.2分、4.5分および4.9分であった。したがって、TPIの投与時間は、第一にカプセル数が増えると増大し、第二に用量強度が上がると増大した。
【0212】
体内摂取量
90名の登録被験体のうちの86名について、以下に要約するように単回投与の治験を行った。
・TPI2×14mg、すなわち28mgまでの単回投与を受けた12名の被験体
・TPI4×14mg、すなわち56mgまでの単回投与を受けた13名の被験体
・TPI2×28mg、すなわち56mgまでの単回投与を受けた14名の被験体
・TPI3×28mg、すなわち84mgまでの単回投与を受けた15名の被験体
・TPI4×28mg、すなわち112mgまでの単回投与を受けた13名の被験体
・60mg/mLTOBI5mL、すなわち300mgまでの単回投与を受けた20名の被験体。
【0213】
有害事象の概要
治験薬の単回投与中または投与後における、治験の結果生じたAEの発生率に関しては、TOBI投与群の被験体の場合(6/20、30.0%)と比較してTPI投与群の被験体の方が高かった(40/66、60.6%)。TPI用量レベルが異なっても、いずれのAEについても被験体発生率は同様であった(TPI2×14mgでは45%、;4×14mgでは54%;2×28mgでは64%;3×28mgでは67%;4×28mgでは69%)が、試料サイズが小さすぎたため、TPI用量が増大したときにAE発生率が増大するという傾向があることは判断できなかった。治験で生じたAEの重篤度については、すべて軽度または中程度であった。
【0214】
TPI4×28mgの投与を受けた被験体の1名では、軽度の咳が認められ、投与後2日目まで喀痰の増加が続き、8日目に入院した。したがって、治験終了後の8日目におけるその被験体のAEについてはSAE(重度の有害事象)と判定した。しかし同一被験体で生じたSAEについては、いずれもTPI治験に関係しないと考えられた。TPI4×28mgの投与を受けた別の被験体1名に関しては、治験薬投与を行うと中程度で関連性が疑われる咳の悪化、味覚異常および流涙増加からなる重度ではないAEが認められため、投与を中断して最終的に投与中止とした。この被験体に関しては、治験の同意が撤回されたため、本試験から脱落した。4名の被験体(TPI4×14mg1名、4×28mg1名、3×28mg2名)では、TPI吸入中に重度ではないが咳から成るAEが認められたため、投薬の変更、中断または延期を行った。試験研究者の見解では、各々のAEについてはTPI治験と関連性が高いとされた。
【0215】
TPI投与群の被験体で最も多く見られたAEは、咳または咳の悪化(13/66=19.7%;治験開始前の基礎的症状として以前報告されたTPI投与被験体では12/13);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各々6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各々4.5%)であった。咳、咳の悪化および味覚異常の発生率は、TPIの用量が増すと僅かに増大したが、個々のAEの発生率に関しては、データに明確な傾向は認められなかった。これに対してTOBI投与の被験体のすべてでは、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化または味覚異常は全く見られなかった。しかしながら、TOBI投与の被験体のほとんど(17/20=85%)では、その通常療法の一部としてTOBIの投与が行われていた。
【0216】
試験研究者の見解では、咳および咳の悪化のほとんどのケース、味覚異常のすべてのケース、ならびに喀血および咽喉の炎症のほとんどのケースが、TPI治験と関連性があるかまたは高いとされた。これに対して胸部圧迫感、単純ヘルペス、好酸球数の増加、咽喉の渇き、咽頭炎、喀痰増加、および喀痰粘性上昇のそれぞれ単独のケースでは、TOBI治験に関係すると考えられた。
【0217】
治験の結果生じた有害事象:すべての原因
66名のTPI投与被験体のうちの40名、および20名のTOBI投与被験体のうちの6名では、治験中または治験後に治験に結果生じたAEが見られた。いずれのAEについてもその発生被験体%は、TPI用量レベル間で同等であった。治験により生じたAEのすべては、軽度または中程度のものであった。
【0218】
TPI投与被験体の最大数で見られたAEは、咳または咳の悪化(13/66=19.7%);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各4.5%)であった。
【0219】
TOBI投与の被験体は1名以外は、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化または味覚異常は見られなかった。
【0220】
治験に関係すると考えられ、治験の結果生じる有害事象
66名のTPI投与被験体のうちの24名(36.4%)、および20名のTOBI投与被験体のうちの2名(10%)では、それらケースで見られるAEが試験研究者の見解では治験と関連性があるかまたは高いものであった(表12)。試験研究者によると、咳および咳の悪化のほとんどのケース(10/66=15.2%)ならびに味覚異常のすべてのケース(16.7%)は、TPI治験と関連性があるかまたは高いとされた(表11)。喀血および咽喉の炎症(ともに4.5%)ならびに流涙増加(3.0%)についても、TPI治験に関係すると考えられた。これに対して、胸部圧迫感、単純ヘルペス、好酸球数増加、咽喉の渇き、咽頭炎、喀痰増加、および喀痰粘性上昇のそれぞれ単独のケースについては、TOBI治験に関係すると考えられた。
【0221】
ほかの重篤な有害事象(SAE)
TPI4×28mg投与の被験体のうちの1名では、投与後の2日目にSAE(中程度の咳および喀痰増加)が見られ、CFの肺疾患が悪化したため治験後の8日目に入院した。同一被験体で生じたそれらSAEについては、いずれもTPI治験に関係しないと考えられた。
【0222】
治験に関係する有害事象による試験からの被験体の脱落
TPI4×28mg投与被験体のうちの1名では、治験薬投与で中程度の咳の悪化、ならびに味覚異常および流涙増加が見られたため、投与を中断し、最終的に投与中止とし、その被験体を本試験から脱落させた。試験研究者の見解では、そのAEのいずれもTPI治験と関連性があるとされた。
【0223】
治験に関連した有害事象による投薬変更(dose intervention)
咳が見られた4名の被験体(TPI4×14mg被験体1名、4×28mg1名、3×28mg2名)については、投薬の変更、中断または延期を行った。試験研究者によると、そのAEはいずれもTPI治験と関連性があると考えられた。
【0224】
【表10】
死亡、ほかの重篤な有害事象、およびほかの明らかな有害事象に関する解析および考察
本試験では死亡例は報告されなかった。SAEについては、治験2日目に被験体のうちの1名にのみ中程度の咳および喀痰増加が報告され、その被験体は治験完了の8日目に入院したが、それらのAEは治験と関連性がないと考えられた。5名のTPI投与被験体では、TPI吸入の数分以内に中程度の咳または咳の悪化が生じ、そのうちの1名(味覚異常および流涙増加も併発)については、本試験から脱落させ、残りの4名については投薬を中断した。被験体のすべてで基礎的な呼吸器症状が報告され、またAEとしての咳も報告されたが、1名については基礎的症状としての咳が留意された。これらのAEについては、投薬の変更を最小限にするか、または変更しなくとも5〜35分以内に解消した。後者の4名の被験体には投薬を遅らせ、最初の中断から数分以内に投薬は無事に完了した。これらのAEは、嚢胞性線維症にしばしば伴う基礎的な気管支過敏症におそらく関係するものであったが、試験研究者の見解では、各AEは試験薬物の投与と関連性があるとされた。したがって、本試験では各用量のTPI、およびTOBI300mg/5mLの投与は良好な忍容性を示した。
【0225】
血液学
TOBIおよびTPI治験群では、試験前の基礎データから追跡調査時までの血液学的検査結果の平均値の変化には、いずれも注目するものはなかった。平均値の基底値からの変化については、TOBIおよびTPI治験群間で大きな差は見られず、いずれの血液学的検査においてもTPIの用量が上がっても平均値の増加パターンは見られなかった。
【0226】
TOBIおよびTPI治験群では、基底値から追跡調査時の値までの注目される増加、すなわち血液学的検査結果の高値または低値の発生頻度で増加は認められなかった。TOBI治験群とTPI治験群との間には大きな差は見られず、TPIの用量を上げても大きくはずれた血液学的検査結果の発生頻度の増加パターンは見られなかった。
【0227】
TOBI投与の被験体のうちの1名では、追跡調査時にAEとして記録された好酸球数の臨床上有意な変化が見られた。
【0228】
血清化学
TOBIおよびTPI治験群において、基底値から追跡調査時の値までの血清化学的検査結果の平均値の変化で注目されるものはなかった。いずれの血清化学的検査結果の平均値も、基底値からの変化に関してTOBI投与群とTPI投与群との間に大きな差は見られず、TPIの用量を上げても平均値の増加パターンは見られなかった。
【0229】
血清化学的検査結果において低値または高値の発生頻度に関する基底値から追跡調査時の値までの増加については、TOBIまたはTPI投与群で注目されるものはなかった。TOBI投与群とTPI投与群との間に大きな差は見られず、TPIの用量を上げても、大きくはずれた血清化学的検査結果の発生頻度の増加パターンは見られなかった。
【0230】
被験体の1名については、追跡調査時の血糖試験結果で臨床上有意な変化が見られたが、その結果はCFに関連した糖尿病に起因するものであり、AEとしては記録せず、TPI治験に起因しないものとした。
【0231】
浸漬式スティックによる蛋白尿検査
浸漬式スティックによる蛋白尿検査は定量的検査であるが、基底値から追跡調査時の値までの定量的変化は算出しなかった。TOBIまたはTPI投与群では浸漬式スティックによる尿蛋白質検査結果が2+またはそれ以上である頻度で、基底値から追跡調査時の値までの増加で注目されるものは見られなかった。
【0232】
TPI4×28mg投与の被験体のうち1名についてのみ、3+の陽性結果が注目された。
【0233】
個々の血液学的異常例
1名を除いて本試験では臨床上有意な血液学的検査結果は認められなかった。TOBI投与被験体No.12/405では、追跡調査時の好酸球数が9%という臨床上有意な結果(正常域0〜6%)が見られたため、AEとして記録し、それは試験研究者の見解ではTOBI治験と関連性があるとされた。その被験体の好酸球の基底値はもともと正常値の上限(5.9%)であったが、治験8日目にはその基底値から上昇した。この好酸球数9.0%という結果の臨床上の重要性に関しては、不確定である。
【0234】
血清化学
本試験においては1名を除き、臨床上有意な血清化学的結果は認められなかった。その1名の被験体(TPI3×28mg投与)では、追跡調査時の血糖値の結果が臨床上有意であったが、その結果はCFに関連した糖尿病に起因するものであった。
【0235】
浸漬式スティックによる尿蛋白検査
本試験では1例を除き、浸漬式スティックによる尿蛋白検査結果で臨床上有意なものは認められなかった。その1名の被験体(TPI4×28mg投与)では、治験後6日目に3+の陽性の結果となった。尚、その被験体では、スクリーニング時および投与当日の検査においては1+の陽性を示していた。この3+という陽性の尿蛋白検査結果については、治験2日目からのSAE(咳および喀痰の増加)と整合性があると治験コーディネーターが判断し、その被験体を入院させた。
【0236】
気管支痙攣および肺機能における急性疾患
TPI2×28mg単回吸入投与後、1例のみにおいて30分以内に起きたと考えられる無症候性の気管支痙攣(FEV1低下率20.9%)が見られた(表11)。尚、その所見は米国胸部学界(ATS)の診断基準によらずに行った肺活量測定で認められたものであった。ATS診断基準にしたがって検査を行えば、FEV1の低下率は少なくなる可能性があると考えられる(11%)。予想されるFEV1%の同様な無症候性の低下については、TOBI投与被験体でも認められた(19.1%の低下)。
【0237】
それ以外のTOBI投与被験体1名およびTPI投与被験体6名についても、予測されるFEV1の低下が10〜20%見られた(表11)。その6名のTPI投与被験体のうちの3名(TPI3×28mg投与の被験体No.02/507、ならびにTPI4×28mg投与の被験体No.01/415およびNo.08/406)では、投与後数分以内で咳または咳の悪化が見られた。
【0238】
【表11】
a予測FEV1%低下が10〜20%。
b予測FEV1%低下が20%以上。
【0239】
肺機能の定量的な変化
TOBIおよびTPI投与群において予測されたFEV1%、FVC%およびFEF25−75%の平均値の変化に関しては、スクリーニング時から投与1日前まで、ならびに投与前日から投与8日目の追跡調査時まで、比較的安定していた。また投与前から投与30分後までの予測されたFEV1、FVCまたはFEF25−75の%の平均値の変化に関しては、TOBI投与群とTPI投与群との間に明らかな差は見られず、異なる用量のTPI投与群間で用量に関連する差は見られなかった。
【0240】
生命徴候
本試験を通じての生命徴候またはその変化に関しては、TPI投与群とTOBI投与群との間に大きな差または整合する差は見られなかった。
【0241】
併用投薬
90名の被験体のうちの38名では、投与後に併用して投薬および医療を受けた。その併用の投薬および医療の頻度またはタイプによって、明らかな差は見られなかった。
【0242】
安全性の結論
治験中または治験後の、治験により生じたAEに関しては、TOBI投与の被験体の場合(30.0%)よりもTPI投与の被験体の場合(60.6%)の方が多く見られた。いずれのAEにおける被験体%も、各種TPI用量レベル間で同等(45〜69%)であった。これは試料サイズが小さすぎたため、いずれのAEも、その発生率がTPIの用量の増加で上昇する傾向があるか判定できなかったためである。
【0243】
TPI4×28mg投与の被験体の1名では、2種類のSAE(CFの肺疾患の悪化の指標となる中程度の咳および喀痰の増加)が見られたため、治験薬投与後8日目に入院させたが、それらのSAEはTPI治験には関係しないと考えられた。TPI4×28mg投与の別の1名の被験体では、中程度で、投薬と関連性がある咳の悪化、味覚異常および流涙増加が見られたため、投薬を中断して最終的に中止した。この被験体については、治験の同意が撤回され、本試験から脱落させた。4名の被験体(TPI4×14mg投与被験体1名、4×28mg被験体1名、および3×28mg被験体2名)では、TPIの吸入中に咳が見られたので、投薬の変更、中断または延期を行った。そのAEの各々に関しては、試験研究者の見解ではTPI治験と関連性があるとされた。しかしながら、この4例ではすべて、投薬を一時的に中断しただけであり、投薬は再開され、そつなく完了した。本試験の被験体の全員は、呼吸器に関する基礎的症状が報告され、各コホートごとに約90%に咳の報告が見られた。さらに中程度の咳の報告も、特に気道の炎症がすでに存在する場合に、ドライパウダー吸入器による最初の投与で予想されたことである。
【0244】
TPI投与被験体で見られた多くのAEは、咳または咳の悪化(13/66=19.7%);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各4.5%)であった。咳、咳の悪化および味覚異常の発生率は、TPIの用量を上げると僅かに増加した。TOBI投与の被験体では1名以外は、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化、または味覚異常が見られなかったが、TOBI投与群被験体の85%では日常的にTOBIが用いられていた。
【0245】
試験研究者によると、咳および咳の悪化のほとんどのケース、味覚異常のすべてのケース、ならびに喀血および咽喉の炎症のほとんどのケースは、TPI治験と関連性があるかまたは高いとされた。これに対して、胸部圧迫感、単純ヘルペス、好酸球数増加、咽喉の渇き、咽頭炎、喀痰増加および喀痰粘性上昇については、TOBI治験に関係すると考えられた。
【0246】
無症候性の気管支痙攣が見られ記録されたのは1例のみ(FEV1低下率20.9%)であって、TOBI投与被験体1例に見られた無症候性のFEV1の低下と同等(19.1%)であった。
【0247】
臨床検査結果における基底値からの留意される変化、TPI用量の増加に伴う増大パターン、低値または高値の検査結果の発生頻度の増大、ならびにTPIとTOBIとの明らかな差については、いずれも見られなかった。TOBI投与の被験体のうちの1名でのみ、好酸球数増加がAEとして記録された。
【0248】
考察および全体の結論
TPIの単回投与は、TOBI(登録商標)トブラマイシン吸入用溶液の投与の場合と比較して、トブラマイシンの薬物動態学的特徴は変わらないにもかかわらず、トブラマイシンの送達効率が高い結果となった。TOBI(登録商標)トブラマイシン吸入用溶液投与後の全身体内摂取量は、TPI4×28mgカプセル投与後に見られた量と極めて類似しており、相当する用量の計算値は115mgTPIであった。したがって、TPI28mgカプセル4個(総量112mg)で、300mgTOBI(登録商標)トブラマイシン吸入用溶液に相当する全身の体内摂取量が達成されると考えられる。
【0249】
TPIの最小用量での投与において、咳による中断(1例を除き、すべて一時的)が認められたが、TPI単回投与後の血清中トブラマイシンの薬物動態学的特性は変わらないと考えられた。
【0250】
TPI投与の被験体の約20%で咳が報告されたが、ほとんどのケースで軽度なものであり、時には短い投与中断を行うが、全般にすぐに落ち着いた。咳に関しては、ドライパウダー吸入療法で予想されるAEであり、大部分の被験体の基底症状でもある。AEの発生頻度については、TOBI治験群の被験体よりも、TPI治験群の被検者の方が高かったが、一方でほとんどのコホートの被験体の大部分では、日常的にTOBI(登録商標)トブラマイシン吸入用溶液が投薬されていた。
【0251】
若年の被験体1名では、通常の投薬(長時間作用型気管支拡張剤から成る)を忘れ、治験薬投与後に中程度の咳が見られたため、試験から脱落させた。通常行ってきた気管支保護療法が行われないと、この生体反応が起こると思われる。
【0252】
総投与量28〜112mgにおけるTPIの単回投与は、本試験でCF被験体に良好な忍容性を示した。112mgのTPI粉末における全身性体内摂取量は、TOBI(登録商標)トブラマイシン吸入用溶液の場合に近似している。
【0253】
本試験では、簡単な吸入用デバイスとともに本発明の方法を用いて吸入投与を行うと、TOBI(登録商標)トブラマイシン吸入用溶液の標準的用量での噴霧化による投与よりも著しく速く、効率的であることが示された。TPI吸入後に見られた咳増加および咽喉の炎症の副作用については、予想されたものであり、各用量に含まれる粉末量に依存した。しかしながら、ほとんどのケースで副作用は軽度であり、被験体の感想はTPIの投与がすばやく、簡単とのことであった。TOBI(登録商標)トブラマイシン吸入用溶液投与群では、本試験の前に85%で日常的にTOBIが使用されていたこともあって、咳および味の悪さの訴えがなかったといえる。これらの被験体では、トブラマイシンの湿式エアゾル剤形での効果に順化していた可能性がある。
【0254】
したがって、ドライパウダーでのトブラマイシンの単回投与では、TOBI(登録商標)トブラマイシン吸入用溶液の場合と比較して、トブラマイシンの薬物動態学的特徴が同様であるにもかかわらず、トブラマイシンの送達効率が高い結果となった。TOBI(登録商標)トブラマイシン吸入用溶液投与後の全身の体内摂取量は、TPIの4×28mgカプセルの投与後に見られる量にきわめて近く、計算上の相当用量は115mgTPIであった。したがって、TPI28mgカプセル4個(総量112mg)の全身の体内摂取量は、300mgTOBI(登録商標)トブラマイシン吸入用溶液の場合に相当すると考えられる。
【0255】
本発明の好ましい実施形態については、すでに説明および記載済みであるが、本発明の意図および視野からはずれなければ、種々の変更も可能であることが分かるであろう。
【技術分野】
【0001】
技術分野
本発明は、気管支感染症に抵抗性のない患者に対して、トブラマイシンのようなアミノグリコシド系抗生物質のドライパウダー製剤を用いる新規の改善された治療法に関する。
【背景技術】
【0002】
背景技術
嚢胞性線維症(CF)は、米国およびヨーロッパ北部において最も良く知られている寿命を縮める遺伝的疾患であって、米国では約30000人が罹患(Cunningham,J.C.ら、“An Introduction to Cystic Fibrosis for Patients and Families,”第5版、Bethesda:Cystic Fibrosis Foundation(2003))し、西ヨーロッパにおいても同様の患者数にのぼる。常染色体劣性であるこの疾患の遺伝的欠陥は、塩素イオンチャンネル蛋白質をコードするCF膜貫通コンダクタンス制御(CFTR)遺伝子(Collins,F.S.,“Cystic Fibrosis Molecular Biology and Therapeutic Implications,”Science 256:774−779(1992))におけるある突然変異である。一般的に慢性気管支感染症、副鼻腔炎、および膵臓の機能不全による消化器吸収不良を患うCF患者では、発汗による塩分損失、閉塞性肝胆嚢疾患および生殖能の低下が多く認められた(FitzSimmons,S.C.,“The Changing Epidemiology of Cystic Fibrosis,”J.Pediatr.122:1−9(1993))。CF患者では、呼吸器疾患が病状の主要な要因であり、それによる死亡率は90%にも達する(Cystic Fibrosis Foundation,Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report,Bethesda,MD:Cystic Fibrosis Foundation,(2004);Davis,P.B.ら、“Cystic fibrosis”,Amer.J.Respir.Crit.Care Med 154(5):1229−56(1996))。肺機能(努力呼気肺活1秒量(予測FEV1)として計測される)は、CFにおける生存率を予測する重要な因子の一つである。あるCF患者集団の2年生存率の減少率は、予測FEV1が10%減少すると2倍になり、患者のFEV1が予測値の30%未満になるとその2年生存率は50%未満となる(Kerem,E.ら、“Predictioin of Mortality in Patients with Cystic Fibrosis,”N.Engl.J.Med.326:1187−1191(1992))。肺機能の低下速度に関しては、患者によっても罹患期間もよっても異なる。遡及的な長期の分析から、肺機能低下速度は年間予測FEV1で2%未満から年間予測FEV1で9%超まであることがわかり、低下速度は全般に死亡年齢に関連しているとされる(Corey,M.ら、“Longitudinal Analysis of Pulmonary Function Decline in Patient with Cystic Fifrosis,”J.Pediatr.131(6):809−1(1997))。
【0003】
CF患者は、肺の宿主防御をそこなう上皮イオン輸送の混乱によって生じる粘液増粘症状を患っており、それによってStaphylococcus aureus,Haemophilus infuluenzaeおよびP.aeruginosaによる早期の気管支感染症に対する抵抗性が低下している。CF患者の大部分は、思春期までに喀痰中にP.aeruginosaが存在するようになる(Cystic Fibrosis Foundation Patient Registry 2003 Annual Data Report(2004))。慢性の気管支感染症、特にP.aeruginosaによる感染症によって気道の持続的な炎症反応が誘発され、それによって広汎性気管支拡張の所見を伴う進行性の閉塞性疾患が悪化する(Davis,P.B.ら、(1996),前記文献;Winnie,G.B.ら、“Respiratory Tract
Colonization with Pseudomonas aeruginosa in Cystic Fibrosis:Correlations Between Anti−Pseudomonas aeruginosa Antibody Levels And Pulmonary Function,”Pediatr.Pulmonol.10:92−100(1991);Ballmann,M.ら、“Long Term Follow Up of Changes in FEV1 and Treatment Intensity During Pseudomonas Aeruginosa Colinisation in Patients with Cystic Fibrosis”, Thorax 53:732−737(1998);Pamukcu,A.ら、”Effects of Pseudomonas aeruginosa Colonisation on Lung Function and Anthropometric Variables in Children with Cystic Fibrosis”,Pediatr.Pulmonol.19:10−15(1995))。P.aeruginosaによる慢性の気管支感染症への罹患から始まり、肺炎、肺機能の低下および最終的には死に至るまで関連性があることは、P.aeruginosaによる慢性感染症が伴うと生存率が優位に低下すること(Henry,R.L.ら、“Mucoid Pseudomonas aeruginosa is a Marker of Poor Survival in Cystic Fibrosis”,Pediatr.Pulmonol.12(3):158−61(1992))、ならびにP.aeruginosaによる慢性感染症への早期罹患と小児死亡率とに有意な関連性があること(Demko,C.A.ら、“Gender Differences in Cystic Fibrosis:Pseudomonas aeruginosa Infection”,J.Clin.Epidemiol.48:1041−1049(1995))によって示唆される。肺への細菌の侵入を抑える(MacLusky,I.B.ら、“Long−term Effects of Inhaled Tobramycin in Patients with Cystic Fibrosis Colonized with Pseudomonas aeruginosa”,Pediatr.Pulmonol.7(1):42−8(1989))か、またはその感染の結果生じる炎症を抑える(Konstan,M.W.ら、“Effect of high−dose Ibuprofen in Patients with Cystic Fibrosis”,N.Engl.J.Med.332(13):848−54(1995))ことのどちらかの長期持続的治療を行うと、感染症患者における肺機能低下速度が減少することがこれまでにわかっている。
【0004】
P.aeruginosaによる気管支上皮感染症に対する標準的な治療は、かつては一般にアミノグリコシドを代表とするシュードモナス属細菌に有効な抗生物質を、14〜21日間非経口的に投与して行なわれた。しかしながら、極性が高いアミノグリコシド系薬剤を非経口的に投与しても、気管支上皮内にはほとんど浸透しない。非経口投与によって感染部位での充分な薬物濃度を得るには、腎毒性、内耳前庭毒性および耳毒性に関連する血清中の薬物濃度が欠かせない(非特許文献1;非特許文献2)。
【0005】
アミノグリコシド系薬剤の吸入投与は、全身的な生物学的利用率を最小限にしながら、気管支上皮内の感染部位に高濃度の抗生物質を直接的に送達する、魅力的な代替法を提供する(非特許文献3;非特許文献4)。
【0006】
CF患者におけるP.aeruginosa感染症に対する現行の標準的な治療は、吸入用で保存剤不含、安定で使用しやすいトブラマイシン製剤であるTOBI(登録商標)トブラマイシン溶液(1/4生理食塩水5mL中に溶解したトブラマイシン60mg/mL溶液)をジェットネブライザーで投与して行なわれ、PathoGenesis社(米国、ワシントン州、シアトル市)(現在はChiron社(米国、カリフォルニア州、エメリービル市))で開発されたものである。5mlのTOBI(トブラマイシン300mg)の1日2回投与をPARI LC PLUS/PulmoAide(商品名)コンプレッサー送達システムと組合せは、CF患者におけるP.aeruginosa管理医療を目的とする長期断続的治療として、NDA50−753のもと、1997年12月にFDAにより承認され、そして、これが、この目的のための産業上の標準となっているままである。市販の用量300mgのTOBIの吸入法は、投与自体に20分間かかり、それ以外に投与の準備およびネブライザーの洗浄の時間を要する。患者の気管支上皮内のP.aeruginosaを抑制するための、1/4生理食塩水中に溶解したトブラマイシン300mg含有製剤を用量5mlでエアゾルで投与することに関しては、米国特許第5,508,269号においても開示されており、その開示については本明細書において参考文献としてそのまま掲載されている。
【0007】
トブラマイシンは、放線菌の一種であるStreptomyces tenebrariusによって生産されるアミノグリコシド系抗生物質である。トブラマイシンは低濃度(4μg/mL未満)でも多くのグラム陰性細菌の増殖を抑制するのに有効であり、条件しだいでは殺菌効果も示す(非特許文献5)。トブラマイシンは粘膜表面からの吸収性が悪く、通常は非経口的投与を要する。さらに、トブラマイシンの活性は化膿性喀痰によって抑制され、高濃度の二価性カチオン類、酸性条件、高いイオン強度およびトブラマイシン結合性高分子のいずれによっても、その環境中でトブラマイシンは抑制される。喀痰中のトブラマイシン濃度をさらに5〜10倍高めると、これらの抑制効果は克服できると考えられる(非特許文献6)。
【0008】
吸収性が悪い抗生物質トブラマイシンを、エアゾル投与経路によって嚢胞性線維症(CF)患者の気道に送達させる効率については、これまでに多くのデータが存在する。その研究の大半は、嚢胞性線維症(CF)患者におけるP.aeruginosaによる慢性の肺感染症の治療を目的として行われてきた。たとえば、71名のCF患者におけるP.aeruginosaによる気管支感染症に対する、エアゾル化トブラマイシン600mg、1日3回投与による多施設、二重盲検、プラセボ対照の交叉臨床試験では、治療群で病原体の喀痰中密度の有意な減少、ならびに肺活量の改善も認められた。トブラマイシンに対して高い抵抗性を有するP.aeruginosa株(MICが128μg/mL以上と規定されるもの)の出現は、プラセボ群と治療群とで同等であった。トブラマイシンに対して内在性の抵抗性を有するP.aeruginosa以外のグラム陰性細菌の喀痰中の存在は、トブラマイシンまたはプラセボの投与において同じ頻度で見られた(非特許文献7)。
【0009】
このレジメンでは安全性および有効性の両方が認められたが、費用がかさみ、使用性も悪い。1993年、Children’s Hospital CF Center(米国、ワシントン州、シアトル市)において、患者の初期喀痰を培養して単離したP.aeruginosaに関するMICの調査によって、単離菌の90%が16μg/mL以下のMICを有し、全単離菌の98%が128μg/mL以下のMICを有することが判明した。この調査から、喀痰中のトブラマイシン濃度128μg/mLが達成されると、CF患者における気管支感染症の治療に有効であることが示唆された(非特許文献6)。
【0010】
無作為化した交差試験では、エアゾル投与10分後に採取した喀痰試料中のトブラマイシンのピーク濃度を計測することによって種々のネブライザーのトブラマイシン送達能が比較された。この試験では、1/4生理食塩水5mL中にトブラマイシンが濃度60mg/mLで溶解したTOBI(登録商標)吸入用トブラマイシン溶液(PathoGenesis社(米国、ワシントン州、シアトル市)(現在はChiron社(米国、カリフォルニア州、エメリビル市)製)が、PALI(登録商標)LCジェットネブライザー(PALI Respiratory Equipment社(米国、バージニア州、リッチモンド市)製)の使用のもとで投与された。この薬物送達(標準偏差661.0μg/g)、ピーク濃度の中央値は433.0μg/gに達することが示された。患者のうち13%のみで喀痰中濃度が128μg/g以下であったが、患者の87%では128μg/g以上に達した(非特許文献8)。ごく最近になってPARI(登録商標)LCジェットネブライザーに一方向性流動バルブが追加されて改良され、PALI(登録商標)LC PLUSの名称のものが販売されている。そのPALI(登録商標)LC PLUSの中の一方向バルブは、PALI(登録商標)LCジェットネブライザーよりも多くの薬物送達を可能にするものと説明されてきたが、一方では不意のふきこぼれの可能性が減り、呼気用フィルターの使用もされるようになった。PALI LC PLUSジェットネブライザーを用いて得られた喀痰中のトブラマイシンのピーク濃度が、旧来のPALI(登録商標)LCジェットネブライザーを用いた場合よりも有意に高いことがEisenbergら(1997)の上述文献で明らかにされた。
【0011】
前述に挙げたことに加えて、P.aeruginosaに感染したCF患者において液体トブラマイシンエアゾル製剤を断続的に投与することによる、プラセボを対照とした多施設、無作為化二重盲検臨床試験の2件がRamsey,B.W.らによって報告された(非特許文献9)。これら2件の臨床試験では、520名の被験体に対して無作為にトブラマイシン300mgの吸入またはプラセボのいずれかを1日2回、28日間投与し、さらに投薬終了から28日間追跡調査を行った。被験体にはさらに3回の投薬−休薬サイクルでトブラマイシンまたはプラセボの投与を、合計で24週間継続した。有効性に関する変数には喀痰中のP.aeruginosa含量が含まれる。トブラマイシン治療群患者では、投与0〜20週間で喀痰中のP.aeruginosa含量が平均0.8log10減少したのに対し、プラセボ投与群の患者では逆に0.3log10増加した(P<0.001)。またトブラマイシン投与群の患者では、投与0〜4週間で喀痰中のP.aeruginosa含量が平均1.9log10減少したのに対し、プラセボ投与群では変化が見られなかった(P<0.001)。
【0012】
特許文献1および特許文献2では、気管支感染症に罹患している患者に対して、トブラマイシンのようなアミノグリコシド系抗生物質が生理学的適合性のある担体中に約60〜約200mg/ml含有する液体製剤を、ネブライザーによって4.0mlまたはそれ以下の用量で約10分間以下の時間をかけて吸入させて投与すると有効な治療ができることが開示されている。アミノグリコシド製剤のさらに効率的な投与がなされると、従来の投与レジームと比較して実質的に少ない量の液状アミノグリコシドで、および実質的に短い投与期間で投与が可能になり、その結果、投与コストおよび薬物の浪費が低減する。しかも、使用する製剤は比較的少量の生理学的適合性のある溶液中に処方されたアミノグリコシドを有効な最小量しか含有しないものであり、そのことによってアミノグリコシド製剤の吸入後の肺への刺激が低減した。
市場から購入可能なTOBI(登録商標)製品のような吸入用抗生物質のほかに、種々の長期療法が日常的に処方され、肺における閉塞、感染症および炎症の破壊サイクルが抑制される。積極的気道クリアランス療法(Aggressive Airway Clearance Therapy(非特許文献10))、吸入による気管支拡張薬療法(非特許文献11)、およびヒト組換え型α−ドルナーゼ(rhDNアーゼ;非特許文献12)のような粘液溶解療法については、いずれもCF患者に対する治療に明らかに役立つものである。それらの療法に関しては、CF患者にとって重大な問題があり(非特許文献13)、コンプライアンス(服薬遵守)の欠如で一定の治療にばらつきが出る(非特許文献14)ことが分かっている。
前述にように、市販されているTOBI(登録商標)吸入用液体エアゾルトブラマイシン溶液は、CF患者におけるP.aeruginosa感染症の治療に高い有効性を示すことがすでに証明されている。CF患者における肺機能の保持に伴う治療負荷および関連する免疫学的接種を行うと、治療に要する投与期間が短縮または患者に対する治療の利便性が増大する治療上の改善によって患者に便宜が図られ、その結果として治療効果も高まる。したがって、アミノグリコシド系抗生物質を患者に吸入させて送達し、投薬コストを下げ、患者のコンプライアンスを上げ、吸入療法の全般的有効性を増大させるための新規の改善された方法およびデバイス類の必要とされる。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】米国特許第6,890,907号明細書
【特許文献2】米国特許出願公開第2003/0143162号明細書
【非特許文献】
【0014】
【非特許文献1】“American Academy of Otolaryngology.Guide for the evaluation of hearing handicap”,JAMA(1979)241(19):2055−9
【非特許文献2】Brummett,R.E.,“Drug−induced ototoxicity”,Drugs(1980)19:412−28
【非特許文献3】Touw,D.J.ら、“Inhalation of Antibiotics in Cystic Fibrosis”,Eur.Respir.J.(1995)8:1594−604
【非特許文献4】Rosenfeld,M.ら、“Aerosolized Antibiotics for Bacterial Lower Airway Infections:Principle,Efficacy,and Pitfalls”,Clinical Pulmonary Medicine(1997)4(2):101−12
【非特許文献5】Neu,H.C.,“Tobramycin:an overview”,J.Infect.Dis.(1976)134:Suppl:S3−19
【非特許文献6】Levy,J.ら、“Bioactivity of Gentamicin in Purulent Sputum from Patients with Cystic Fibrosis or Bronchiectasis:Comparison with Activity in Serum”,J.Infect.Dis.(1983)148(6):1069−76
【非特許文献7】Ramsey,B.ら、“Response to Letter to the Editor:Aerosolised Tobramycin in Patients with Cystic Fibrosis”,N.Engl.J.Med.(1993)329:1660
【非特許文献8】Eisenberg,J.ら、“A Comparison of Peak Sputum Tobramycin Concentration in Patient With Cystic Fibrosis Using Jet and Ultrasonic Nebulizer System. Aerosolized Tobramycin Study Group”,Chest(1997)111(4):955−962
【非特許文献9】Ramsey,B.W.ら,“Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis.Cystic Fibrosis Inhaled Tobramycin Study Group.“N.Engl.J.Med.(1999)340(1):23−30
【非特許文献10】Reisman,J.J.ら、“Role of conventional physiology in cystic fibrosis”,J.Pediatr.(1988)113(4):632−6
【非特許文献11】Konig P.ら、“Short−term and Long−term Effects of Albuterol Aerozol Therapy in Cystic Fibrosis:A Preliminary Report”,Pediatr.Pulmol.(1995)20(4):205−14
【非特許文献12】Fuchs,H.J.ら、“Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symtoms and on Pulmonary Function in Patients with Cystic Fibrosis.the Pulmozyme Study Group”,N.Engl.J.Med.(1994)331(10):637−42
【非特許文献13】Conway,S.P.ら、“Compliance with treatment in adult patients with cystic fibrosis”,Thorax(1996)51(1):29−33
【非特許文献14】Abbot J.ら、“Treatment Compliance in Adults with Cystic Fibrosis”,Thorax(1994)49(2):115−20
【発明の概要】
【課題を解決するための手段】
【0015】
発明の要旨
本発明は、90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症患者の気管支系に対して、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む治療方法を提供する。本発明の態様によれば、第一治療期間の後に第二の治療休止期間を設けることができ、その間は患者の気管支系にはアミノグリコシド系抗生物質は投与されない。悪性の感染症の治療に対しては、患者の気管支系へのアミノグリコシド系抗生物質投与による第一の治療期およびその後のアミノグリコシド系抗生物質を投与しない第二の治療休止期のサイクルを、抗菌効果が現われるまで2回またはそれ以上繰返して行うことができる。CF患者が罹患する感染症のような慢性感染症の場合では、この第一および第二の治療期のサイクルを、患者治療中に複数回繰返すことができる。
本発明の方法は、CFに伴うシュードモナス属菌による気管支感染症のような、アミノグリコシド系抗生物質が有効ないずれの気管支感染症の治療にも有用である。
例えば、本発明は以下の項目を提供する。
(項目1)
90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症の患者の気管支系に対して、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む、気管支感染症の治療方法。
(項目2)
前記第一治療期間の後に、前記患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期間を設ける、項目1に記載の治療方法。
(項目3)
前記アミノグリコシド系抗生物質が、ゲンタマイシン、アミカシン、カナマイシン、ストレプトマイシン、ネオマイシン、ネチルミシンおよびトブラマイシン、あるいはそれらの薬学的に受容可能な塩からなる群から選択される、項目1に記載の治療方法。
(項目4)
前記アミノグリコシド系抗生物質が、トブラマイシンまたはその薬学的に受容可能な塩である、項目3に記載の治療方法。
(項目5)
前記エアゾルパウダー組成物が、100〜120mgのトブラマイシンを含有する、項目4に記載の治療方法。
(項目6)
前記エアゾルパウダー組成物が、110〜115mgのトブラマイシンを含有する、項目5に記載の治療方法。
(項目7)
前記患者に投与される前記エアゾルパウダー組成物が、2〜6個の用量単位に分割されている、項目1に記載の治療方法。
(項目8)
前記患者に投与される前記エアゾルパウダー組成物が、3〜5個の用量単位に分割されている、項目7に記載の治療方法。
(項目9)
前記エアゾルパウダー組成物が、4個の用量単位に分割されている、項目8に記載の治療方法。
(項目10)
前記第二の治療休止期間が、20〜36日間である、項目2に記載の治療方法。
(項目11)
前記第一の治療期間が、26〜30日間である、項目2に記載の治療方法。
(項目12)
前記第二の治療休止期間が、26〜30日間である、項目11に記載の治療方法。
(項目13)
前記第一の治療期間が、28日間である、項目2に記載の治療方法。
(項目14)
前記第二の治療休止期間が、26〜30日間である、項目13に記載の治療方法。
(項目15)
前記第一の治療期間の治療の後に、前記第二の治療休止期間を設ける治療レジメンを複数回繰返す、項目2に記載の治療方法。
(項目16)
前記パウダーが、空気力学的粒径域1〜5μmの粒子を少なくとも50%含有する、項目2に記載の治療方法。
(項目17)
前記患者が、嚢胞性線維症患者である、項目2に記載の治療方法。
(項目18)
前記嚢胞性線維症患者が、シュードモナス属菌による気管支感染症に罹患している、項目17に記載の治療方法。
(項目19)
前記エアゾルパウダーが、ドライパウダー吸入器を用いて前記患者に投与される、項目1に記載の治療方法。
(項目20)
前記エアゾルパウダーが、前記ドライパウダー吸入器内の単一容器に分配され、前記エアゾルパウダーが、前記容器から前記吸入器によってヒト被験体または動物被験体の肺内に送達される、項目1に記載の治療方法。
(項目21)
前記エアゾルパウダーが、前記ドライパウダー吸入器内の複数容器に分配され、前記エアゾルパウダーが、前記容器から前記吸入器によってヒト被験体または動物被験体の肺内に送達される、項目20に記載の治療方法。
(項目22)
110〜115mgのトブラマイシン抗生物質を含有するドライパウダーエアゾル組成物を、気管支感染症に罹患した嚢胞性線維症患者の気管支系に対して、28日間の第一治療期間に1日2回投与する工程、その後に前記患者の気管支系にトブラマイシン抗生物質が投与されない26〜30日間の第二の治療休止期間を設ける工程、および前記第一および前記第二の治療期間を繰返す工程を含む、前記患者の治療方法。
(項目23)
前記患者に投与される前記エアゾルパウダー組成物が、3〜5個の用量単位に分割されている、項目22に記載の治療方法。
(項目24)
前記エアゾルパウダー組成物が、4個の用量単位に分割されている、項目23に記載の治療方法。
(項目25)
前記嚢胞性線維症患者が、シュードモナス属菌による気管支感染症に罹患している、項目22に記載の治療方法。
(項目26)
前記第一および前記第二の治療期間が、複数回繰返される、項目22に記載の治療方法。
【図面の簡単な説明】
【0016】
本発明の前述の態様および付随する利点の多くについては、以下に述べる詳細な説明を参考とすると理解が深まると同時に、添付の図面と関連付けることでさらに容易に理解できるであろう。
【図1】図1は、規定用量のTPIおよびTOBI投与後の種々の時間における被験体の血清中トブラマイシンの平均濃度を示す。
【図2】図2は、曲線下面積(AUC)に対する吸入用トブラマイシン粉末製剤(TPI)および吸入用トブラマイシン溶液製剤(TOBI)の用量プロット(0,12)を示す。
【図3】図3は、AUCに対するTPIおよびTOBIの用量プロット(0,∞)を示す。
【図4】図4は、規定用量のTOBIまたはTPIが投与された被験体の喀痰中トブラマイシンの平均濃度を示す。
【発明を実施するための形態】
【0017】
好ましい実施形態の詳細な説明
本明細書において特別に規定されるものを除き、本明細書で使用される用語のすべてについては、当業者にとって公知と考えられるのと同じ意味を持つものとする。本明細書では以下の略語が用いられる。
【0018】
略語 意味
【0019】
AE 有害事象
ALT アラニン−アミノトランスフェラーゼ
AUC 曲線下面積
BID 1日2回
BUN 血中尿素体窒素
CaCl2 塩化カルシウム
CF 嚢胞性線維症
CFC クロロフルオロ炭素
Cmax 最高濃度
CFTR 嚢胞性線維症膜貫通コンダクタンス制御因子
DPI ドライパウダー吸入剤
DSPC 1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン
FDA 米国食品医薬品局
FEV1 1秒努力肺活量
FVC 努力肺活量
FEF25−75 25〜75%間の努力呼気流量
HPMC 2−ヒドロキシプロピルメチルセルロース
IRB 治験委員会
IVRS 対話型音声応答システム
MedDRA 医学規制用語集
MIC 最小抑制濃度
P.aeruginosa Pseudomonas aeruginosa(緑膿菌)
PFOB 臭化ペルフルオロオクチル
QPIT 定量的ピロカルピン電離療法検査
SAE 重篤な有害事象
tmax 最高濃度到達時間
TOBI(登録商標) 吸入用トブラマイシン300mg溶液(Chiron社(米国、カリフォルニア州、エメリビル市)製)
TPI 吸入用トブラマイシン粉末。
【0020】
一つの態様によれば、本発明は気管支感染症患者の気管支系に対して、90〜130mgのアミノグリコシド系抗生物質を含有するドライパウダーエアゾル組成物を、20〜36日間の第一治療期間に1日1〜3回投与する工程を含む治療方法を提供する。本発明の実施によれば、第一治療期間の後に患者の気管支系にアミノグリコシド系抗生物質を投与しない第二治療休止期間を設けることができる。悪性感染症の治療には、アミノグリコシドで治療を行う第一治療期間およびそれに続く治療を行わない第二治療休止期間のサイクルを、所望の抗菌効果が得られるまで2回またはそれ以上繰返すこともできる。CF患者が罹患するような慢性感染症の場合では、第一および第二の治療期間はその患者の治療期間中に何回も繰返すことができる。
【0021】
別の態様によれば、本発明は気管支感染症患者の治療のための、アミノグリコシド系抗生物質90〜130mg含有ドライパウダーエアゾル組成物を20〜36日間の第一治療期間に1日1〜3回投与することによる治療用の医薬品製剤におけるアミノグリコシド系抗生物質の使用を提供する。本発明のこの態様の実施によれば、第一の治療期間の後に前述と同様に患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期間を設けることができ、本明細書に記載されているようにその第一および第二の治療期を繰返すことができる。
【0022】
本発明のこの態様の方法には、いずれにおいても20〜90重量%のアミノグリコシド系抗生物質および生理学的適合性のある担体を含有する治療上有効量のエアゾルパウダーを、必要に応じてヒト被験体または動物被験体に吸入させて投与する工程が含まれ、そのエアゾルパウダーは含有粉末粒子の少なくとも50%は空気力学的粒径1〜5μmの範囲に入るものである。
【0023】
「気管支感染症」という用語は、肺の気管支内に発生した細菌感染症を指す。本発明の方法を用いて治療することが可能な気管支感染症の例としては、Pseudomonas
aeruginosa、Staphylococcus aureus、Haemophilus influenzae、Burkholderia cepacia、Stenotrophomonas maltophilliaおよびAlcaligenesis xiloxidantsのようなグラム陰性微生物による感染症が挙げられる。本発明のこの態様の方法は、たとえば嚢胞性線維症に付随するPseudomonas aeruginosa感染症のような気管支感染症に罹患したヒトの治療に用いることができる。
【0024】
本発明の実施において有用なアミノグリコシド系抗生物質の例としては、ゲンタマイシン、アミカシン、カナマイシン、ストレプトマイシン、ネオマイシン、ネチルマイシン、パラメシンおよびトブラマイシンが挙げられる。本発明の実施において用いられる現時点で好ましいアミノグリコシド系抗生物質には、トブラマイシンがある。一般にアミノグリコシド系抗生物質は、その薬学的に受容可能な塩(たとえば硫酸塩、クエン酸塩、アスコルビン酸塩、グルコン酸塩、炭酸塩、酒石酸塩、コハク酸塩、酢酸塩またはリン酸塩)またはエステルの形で投与される。
【0025】
本発明の実施によれば、エアゾルパウダーはヒト被験体または動物被験体に吸入させることでその肺の中に到達する。そのエアゾルパウダーにはアミノグリコシド系抗生物質含有の粒子が含まれる。エアゾルパウダーは、その含有粒子の少なくとも50%が空気力学的粒径1〜5μmの範囲に入るもので、ヒト被験体または動物被験体の肺内に効果的に浸透し、その結果アミノグリコシド系抗生物質が効果的に肺内に送達されることが見出された。例を挙げると、本発明の実施において有用な(アミノグリコシド系抗生物質含有の)エアゾルパウダーのいくつかでは、その含有粒子の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%または少なくとも95%が空気力学的粒径1〜5μmの範囲にある。
【0026】
「空気力学的粒径」という用語は、対象の粒子と同じ限界沈降速度を有する単位密度球体の直径を指す(“Aerosol Measurement:Principles,Techniques and Applications”、Klaus WillekeおよびPaul A.Baron編、Van Nostrand Reinhold(ニューヨーク)社刊、1993などを参照のこと)。空気力学的粒径に関しては、たとえば粒子が気道内のどこに留まるのかを予測するのに用いられる。
【0027】
「空気力学的質量平均粒径」(略してMMAD)とは、分散した粒子の空気力学的大きさの計測値のことである。空気力学的大きさの分布は、吸入中におけるエアゾルの挙動を規定し、一般には空気中におけるその粒子と同じ限界沈降速度を有する単位密度球体の直径のことである。空気力学的粒径には粒子の形状、密度および物理学的大きさが含まれる。空気力学的大きさの分布が対数正規分布である場合には、空気力学的質量中央粒径(MMAD)で特性解析できる。本明細書において用いられるMMADは、Andersonのカスケード衝撃法によって求められるエアゾル化粉末の空気力学的粒子サイズ分布の中点または中央値を指す。
【0028】
カスケード衝撃装置には、簡単に述べると孔径サイズが小さくなる一連のスクリーンが含まれる。粒子は衝撃装置を通過する移動噴出流内で、一連のスクリーンにトラップされる。各々のスクリーン上にトラップされる粒子状物質(規定の大きさの範囲にある粒子)の量は、スクリーンを洗浄し溶離した物質の量を計測することで求めることができる。その際使用されるカスケード衝撃装置の例については、米国薬局方(第26版)の第601章(エアゾル)内に記載されており、その刊行物の該当部分は本明細書中に参考として援用されている。
【0029】
本発明の実施において有用な粉末化アミノグリコシド系抗生物質製剤は、水分含量が一般に15重量%未満、通常は約11重量%以下であり、好ましくは約8重量%以下である。
【0030】
本発明の実施によれば、アミノグリコシド系抗生物質含有のエアゾル粉末の治療有効量が、気管支感染症患者に吸入されて投与される。治療有効量のエアゾル粉末中には、患者の肺中に存在する薬物感受性細菌の増殖を完全にまたは部分的に抑制するのに充分なアミノグリコシド系抗生物質が含まれる。代表例として、アミノグリコシドであるトブラマイシンについては、その治療有効量は患者に1日1〜3回、本発明の好ましい態様では1日2回、トブラマイシンの用量(存在し得る対イオンの重量を除いた遊離塩基の重量として求められた用量)として約90〜約130mg、さらに好ましくは約100〜約120mg、および最も好ましくは約110〜115mgを含有するエアゾル粉末組成物を投与することで達成される。
【0031】
トブラマイシンのようなアミノグリコシドの投与量は、単回投与分の単一容器から投与でき、あるいはその抗生物質の送達に用いられる吸入デバイスしだいでは多回投与用容器内に、または連続投与用単位用量に分割して投与することもできる。アミノグリコシド系抗生物質の投与量は、たとえば2〜6単位用量、より好ましくは3〜5単位用量、およびさらに好ましくは4単位用量に分割できる。代表的な実施形態では、112mgのトブラマイシン投与量(存在し得る対イオンの重量を除いた遊離塩基の重量として求められた値)については、HPMC製2号カプセル4個に別々に、遊離塩基としてのトブラマイシンを27mgずつ分割充填できる。
【0032】
本発明のドライパウダーエアゾル組成物は、患者に対して20〜36日間、より好ましくは26〜30日間、およびさらに好ましくは約28日間の第一治療期間に投与される。この第一治療期間の後には、患者の気管支系にアミノグリコシド系抗生物質を投与しない第二の治療休止期を設ける。本発明の一つ態様によれば、第二治療休止期間は約20〜36日間、より好ましくは約26〜約30日間、および最も好ましくは約28日間継続される。
【0033】
代表的な実施形態では、本発明の方法はPseudomonas aeruginosaによる慢性感染症の療養管理を目的とするCF患者の治療に用いられる。この態様によれば、本発明は気管支感染症に罹患したCF患者の治療を企図するものであり、患者の気管支系に対し、抗生物質としてトブラマイシンを110〜115mg含有するドライパウダーエアゾル組成物を、28日間の第一治療期間で1日2回投与を行い、つづいて患者の気管支系にトブラマイシンを投与しない26〜30日間の第二治療休止期間を設け、さらに第一および第二の治療期を繰返す工程が含まれる。本発明のこの態様によれば、110〜115mgのトブラマイシン用量は、連続投与用に3〜5単位用量、好ましくは4単位用量に分割できる。CF患者はP.aeruginosaに慢性的に感染する傾向があるため、第一治療期間およびそれに続く第二治療休止期間のサイクルは、一般に複数回または多重回繰返して行われることになり、CF患者における気管支感染症の長期にわたる療養管理のために無期限継続することもできる。
【0034】
エアゾルパウダーには一般に20(重量)%〜90(重量)%のアミノグリコシド系抗生物質が含有する。したがって、本発明のいくつかの実施形態では、エアゾルパウダーには30(重量)%〜80(重量)%のアミノグリコシド系抗生物質が含有する。本発明のいくつかの実施形態では、エアゾルパウダーには40(重量)%〜70(重量)%のアミノグリコシド系抗生物質が含有する。この状況において、アミノグリコシド系抗生物質の(重量)%は、存在し得る対イオンの重量を除いた遊離型の抗生物質の量を指す。
【0035】
本発明のエアゾル粉末には、生理学上適合性のある少なくとも一種の担体が一般に含まれるが、必ずしも必須のものではない。エアゾル粉末にはたとえば一種または複数の賦形剤、および/またはアミノグリコシド系抗生物質の効力を高めるそのほかの成分も含有可能である。そのような賦形剤を用いると、患者に送達される粉末中の活性薬剤の含量を抑える必要があるときの簡便な希釈剤として役立つ。またその賦形剤を用いると、粉末分散デバイス中での粉末の分散性の改善にも役立ち、ひいては活性薬剤送達効率およびその再現性がさらに改善され、活性薬剤の取扱い易さ(たとえば流動性およびコンシステンシー)が改善されて製剤の製造および粉末充填が容易になる。特に賦形剤は、アミノグリコシドの物理的および化学的な安定性の改善、残存する水分含量の低減および吸湿防止、ならびに粒子サイズ、凝集度、表面特性(たとえばしわの度合)、吸入のしやすさおよび肺深部への発生粒子のターゲッティングの向上の機能が期待できる。
【0036】
本発明の実施において用いられるアミノグリコシド組成物で有用な医薬品用賦形剤および添加剤には、蛋白質、ペプチド、アミノ酸、脂質、ポリマー、および炭水化物(たとえば単糖類、二糖類、三糖類、四糖類およびオリゴ糖類を含めた糖類;アルジトール類、アルドン酸類、エステル化糖類のような糖誘導体;ならびに多糖類またはシュガーポリマー)が含まれ、それらは単独または組合わせて使用できるが、それらに限定されるものではない。蛋白質賦形剤の例としては、ヒト血清アルブミン(HSA)のような血清アルブミン、組換えヒトアルブミン(rHA)、ゼラチンおよびカゼインが挙げられる。代表的なアミノ酸/ポリペプチド成分であって緩衝機能も有するものには、アラニン、グリシン、アルギニン、ベタイン、ヒスチジン、グルタミン酸、アスパラギン酸、システイン、リシン、ロイシン、プロリン、イソロイシン、バリン、メチオニン、フェニルアラニンおよびアスパルテームが含まれるが、アルギニンについては好ましくない。ジロイシンおよびトリロイシンのような代表的なアミノ酸の重合体についても、本発明で用いるのに適している。
【0037】
本発明での使用に適した炭水化物賦形剤の例としては、フルクトース、マルトース、ガラクトース、グルコース、D−マンノースおよびソルボースのような単糖類;ラクトース、スクロース、トレハロース、セロビオースのような二糖類;ラフィノース、メレジトース、マルトデキストリン類、デキストラン類およびデンプンのような多糖類;ならびにマンニトール、キシリトール、マルチトール、ラクチトール、キシリトールソルビトール(グルシトール)およびミオイノシトールのようなアルジトール類が挙げられる。
【0038】
またアミノグリコシド組成物には緩衝剤またはpH調整剤も含有することが可能で、緩衝剤は一般に有機の酸または塩基から調製される塩である。代表的な緩衝剤には、クエン酸、アスコルビン酸、グルコン酸、カルボン酸、酒石酸、コハク酸、酢酸またはフタル酸の塩のような有機酸塩類;トリス、塩酸トロメタミン、またはリン酸塩緩衝剤が含まれる。
【0039】
本発明の実施において有用なアミノグリコシド組成物にはさらに、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース、メチルセルロース、エチルセルロース、フィコール類(糖ポリマー)、デキストラン、糖エステル類(たとえば2−ヒドロキシプロピル−β−シクロデキストリンのようなシクロデキストリン類、およびヒドロキシエチル澱粉)、ポリエチレングリコールおよびペクチンのような高分子性賦形剤/添加剤;塩類(たとえば塩化ナトリウム);酸化防止剤;帯電防止剤;界面活性剤(たとえば「TWEEN 20」および「TWEEN 80」のようなポリソルベート類、レシチン、オレイン酸、塩化ベンザルコニウム、およびソルビタンエステル類);脂質(たとえばリン脂質および脂肪酸);ステロイド(たとえばコレステロール);ならびにキレート剤(たとえばEDTA)が含有可能である。アミノグリコシド組成物中に使用するのに適したそのほかの医薬用賦形剤および/または添加剤の例については、“Remington:The Science & Practice of Pharmacy”,第19版、Williams & Williams社刊(1995)、および“Physician’s
Desk Reference”、第52版、Medical Economics社(米国、ニュージャージー州)刊(1998)に列挙されており、それらの開示箇所は本明細書中に参考として援用されている。
【0040】
現時点で好ましい賦形剤の組合わせとしては、レシチンと塩化カルシウムとの配合がある。レシチンは、哺乳類(ヒトを含めて)の肺内で界面活性剤としてはたらく、天然産リン脂質のうちのホスファチジルコリンのうちの一種である。
【0041】
本発明の実施において有用なアミノグリコシド組成物には、アミノグリコシド粉末固有の分散性を改善するための分散剤が添加できる。適した分散剤については、国際公開第95/31479号、国際公開第96/32096号および国際公開第96/32149号に開示されており、それらの公開特許はすべて本明細書中に参考として援用されている。それらの公開特許において記載されているように、適した分散剤には水溶性ポリペプチド、ならびにトリプトファン、ロイシン、フェニルアラニンおよびグリシンのような疎水性アミノ酸が含まれる。特にロイシンおよびトリロイシンは、本発明による使用に好適である。
【0042】
アミノグリコシドおよび賦形剤によって形成される固体状態のマトリックスによって、アミノグリコシドに安定した環境が与えられる。その安定性マトリックスについては、結晶性ガラス、無定形ガラス、または両者の混合物で形成できる。最適なものには、両者の混合物を含むドライパウダー製剤がある。実質的に無定形であるアミノグリコシドドライパウダー製剤が好ましいが、そのためには製剤のガラス転移温度(Tg)が約35℃超、好ましくは約45℃超、およびさらに好ましくは約55℃超とする。Tgについては保存温度よりも少なくとも20℃以上高い温度であるのが好ましい。好ましい実施形態によると、アミノグリコシド組成物は国際公開第99/16419号および国際公開第01/85136号において開示されたような固体状態のマトリックスとしてのリン脂質を含有し、それらの公開特許は本明細書においてすべて参考として援用されている。
【0043】
ドライパウダーアミノグリコシド組成物は、前述のような実質的に無定形のガラス質または実質的に結晶質の生物活性のある粉末が生成する条件下で噴霧乾燥して調製することができる。アミノグリコシド溶液の処方物の噴霧乾燥に関しては、たとえば“Spray
Drying Handbook”、第5版、K.Masters編、John Wiley & Sons社(米国ニューヨーク州、ニューヨーク市)刊(1991)、および国際公開第97/41833号に全般的に記載されているように実施される。尚、それに関係する記述箇所は、本明細書中に参考として援用されている。
【0044】
本発明の一つの実施形態による噴霧乾燥用アミノグリコシド溶液の調製のためには、一般に水のような生理学的適合性のある溶媒にアミノグリコシドを溶解する。噴霧乾燥用溶液のpH域は、一般には約3〜10、好ましくは5〜8、さらに好ましくは中性pH域になるように維持し、これによって噴霧乾燥後の粉末が肺内で再び溶解した時のpH域が生理学的に適合するように維持されるのに役立ち得る。この水性処方物には所望によりさらにアルコール、アセトン等のような水と混和する溶媒も添加できる。代表的なアルコールにはメタノール、エタノール、プロパノール、イソプロパノール等のような低級アルコールがある。アミノグリコシド溶液には、アミノグリコシドが一般に0.05%(w/v)−約20%(w/v)の濃度、通常は0.4〜5.0%(w/v)の濃度で溶解した状態で含有するものが考えられる。
【0045】
つづいてアミノグリコシド含有溶液は、Niro社(デンマーク)、Buchi社(スイス)などのような販売業者から購入可能なような従来からの噴霧乾燥機中で噴霧乾燥され、安定なアミノグリコシドドライパウダーが得られる。アミノグリコシド溶液の噴霧乾燥に関する最適な条件については、処方される成分しだいで変わると考えられ、一般には実験によって求められる。物質の噴霧乾燥に用いられる気体は空気が一般的であるが、窒素またはアルゴンのような不活性ガスも使用に適する。さらに、物質の噴霧乾燥に用いられる気体の給気および排気の温度も、噴霧乾燥中におけるアミノグリコシドの不活化が起きないように設定される。そのような温度域は一般に実験によって求められるが、一般的に給気温度は約50〜約200℃の範囲、排気温度は約30〜約150℃の範囲と考えられる。
【0046】
またアミノグリコシドドライパウダーについては凍結乾燥法、真空乾燥法、噴霧凍結乾燥法、超臨界流体プロセス法またはそれ以外の蒸発乾燥方法、あるいはドライパウダー処方中の製剤成分の混和法、粉砕法またはジェットミル法によっても調製できる。いくつかの場合では、取扱い/加工特性の改善、たとえば帯電性の低減化、流動性の向上、固化性の低下などが見込まれる処方でのアミノグリコシドドライパウダー製剤の提供が所望され、そのことは微粒子凝集体、すなわち前述のアミノグリコシドドライパウダー粒子の凝集体または塊から成る組成物を調製することによって達成され、その凝集体は肺内に送達されるとすぐに粉砕して微粉成分に戻るもので、このことについてはたとえば米国特許第5,654,007号に記載されており、本明細書においても参考文献として掲載されている。またアミノグリコシドドライパウダーは、粉末成分の凝集化、篩過による凝集体収集、球状化による球形に近い凝集体の形成、および分級による均一な大きさの製品化による代替製法によっても製造でき、これについてはたとえば国際特許第95/09616号に記載されており、本明細書にも参考文献として掲載されている。アミノグリコシドドライパウダーは、製造、加工および貯蔵中は乾燥条件下(すなわち相対湿度が低い条件下)に置かれるのが好ましい。
【0047】
一つの実施形態によると、本発明の実施において有用な粉末化トブラマイシン製剤の例に挙げられるものは、前述の国際特許第99/16419号および国際特許第01/85136号に開示された乳化/噴霧乾燥法によって製造できる。そのような実施形態による製剤は、少なくとも75%w/w、好ましくは少なくとも85%w/wのトブラマイシン、2〜25%w/w、好ましくは8〜18%w/wのリン脂質、および塩化カルシウムのような金属イオン0〜5%w/wを含有するドライパウダー粒子から構成されるように設計されている。この実施形態の粒子は、一般にMMAD(空気力学的質量平均粒径)が1〜5μmであり、かさ密度が0.08g/cm3超、好ましくは0.12g/cm3超である。
【0048】
本発明の実施において有用な粉末化トブラマイシン製剤の別の例に挙げられるものは、活性型トブラマイシン、1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)、CaCl2および臭化ペルフルオロオクチル(PFOB)を含有するエマルションを作製することによって製造できる。この供給原料としてのエマルションは、次にアトマイザーノズルを通過させて噴霧され、微細な液滴となる。その液滴が乾燥する際、水およびPFOBは蒸発し、多孔質構造の球形粒子を主体とするリン脂質が得られる。それらの球状物は低密度であるため、好ましい空気力学的特性を示す(たとえば空気力学的粒径が1〜5μmの範囲にある球形粒子)。またそれらの高い表面多孔性によって粒子同士の接触が少なくなり、エアゾル懸濁に要するエネルギーも少なくてすむようになる。
【0049】
エアゾル粉末(アミノグリコシド系抗生物質含有)は、たとえばヒト被験体または動物被験体の吸気を利用するドライパウダー吸入器を用いて投与でき、それによってその粉末化アミノグリコシド系抗生物質製剤は肺に送達される。有用なドライパウダー吸入器の例には、T−326型吸入器(Nektar Therapeutics社(米国、カリフォルニア州94070、サンカルロス市、インダストリアル・ロード150)製)がある。そのほかの有用なドライパウダー吸入デバイスについては、米国特許第5,458,135号、同第5,740,794号、同第5,775,320号および同第5,785,049号に開示されており、それらのいずれの特許も本明細書中に参考として援用されている。このタイプのデバイスを用いて投与する場合、粉末化医薬品は、穿刺用蓋またはそのほかのアクセス面を有する容器、好ましくはブリスター包装容器またはカートリッジに、1回投与単位分量または多回投与単位分量で充填される。計量用量分のドライパウダー医薬品で多数の空隙を充填する方法については、たとえば米国特許第5,826,633号に記載されており、その特許は本明細書中に参考として援用されている。
【0050】
本明細書において記載のアミノグリコシド粉末の送達に適したものには、たとえば米国特許第3,906,950号、同第4,013,075号、同第4,069,819号および同第4,995,385号に記載されているタイプのドライパウダー吸入器も含まれ、それらの特許はいずれも本明細書において参考として援用されており、患者に送達する分の予め計量された用量のアミノグリコシドドライパウダーが、ゼラチン硬カプセルのようなカプセルに充填されている。カプセルサイズは、たとえば00号、0号、1号または3号が挙げられるが、その粉末投与に使用される吸入デバイスやそれ以外の要因に依存して選ばれる。
【0051】
アミノグリコシドドライパウダーを肺に投与するためのそのほかのドライパウダー分散用デバイスについては、たとえば欧州特許出願公開第0129985号、同第0472598号、同第0467172号、ならびに米国特許第5,522,385号に記載されているものがあり、それらの特許はいずれもすべて本明細書中に参考として援用されている。また本発明のアミノグリコシドドライパウダーの送達に適するものとしては、Astra−Draco社製のTURBUHALER(商品名)のような吸入デバイスもある。このタイプのデバイスについては、米国特許第4,668,218号、同第4,667,668号および同第4,805,811号に詳しく記載されており、それらの特許のすべては本明細書中に参考として援用されている。
【0052】
また好ましいデバイスとしては、粉末化医薬品の浮遊化、担体スクリーンを通して空気を送ることによるスクリーンからの医薬品の浮揚化、または本明細書中に参考として援用されている米国特許第5,388,572号に記載されているようなマウスピースデバイスで粉末を患者に連続的に投入するための混合チャンバー内での空気と粉末医薬品とのミキシングのいずれかを目的とする空気供給用ピストンを使用するデバイスが挙げられる。
【0053】
前述事項を考慮すると、治療有効量のエアゾル粉末(アミノグリコシド系抗生物質含有)は、ドライパウダー吸入デバイス内に配置された単回投与用または複数回投与用の容器から投与できることが分かるであろう。たとえばドライパウダー吸入デバイスは、治療有効量のエアゾル粉末(アミノグリコシド系抗生物質含有)が入った1回投与用容器とともに装填でき、容器内容物がヒト被験体または動物被験体に吸入される。再び例を挙げると、ドライパウダー吸入器はHPMC製2号カプセルのような多重回投与分のユニット容器(たとえば2,3または4回投与分のユニット容器)とともに装填でき、それらのカプセルには別々に治療有効量未満のエアゾル粉末(アミノグリコシド系抗生物質含有)が入っており、それらのエアゾル粉末を合わせると治療有効量になる。ドライパウダー吸入器によって、その中に配置された容器内のすべての内容物が放出され、治療有効量のエアゾル粉末が患者に供給される。
【0054】
本発明のアミノグリコシド治療レジメンは単独でも使用でき、あるいは気管支感染症、特にP.aeruginosaによる感染症の治療用の一種または複数の追加薬剤と併用することもできる。本発明のこの態様によれば、気管支感染症治療用の一種または複数の追加薬剤は、アミノグリコシド系抗生物質による第一治療期間中、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間中、あるいは第一および第二の治療期間中の両方において投与することができる。本発明のこの態様の一つの実施形態では、気管支感染症の治療用の一種または複数の追加薬剤は、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間中に投与される。その気管支感染症の治療に適する追加薬剤の例としては、モノバクタム系、β−ラクタム系、マクロライド系、フルオロキノロン系および/またはグリコペプチド系の抗菌化合物のような非アミノグリコシド系の感染症治療薬剤が挙げられる。非アミノグリコシド系の感染症治療薬剤の例としては、アズトレオナムを挙げることができる。
【0055】
粉末化アミノグリコシド系抗生物質製剤の放出率(ED)は、一般には50%超である。本発明の実施において有用な粉末化アミノグリコシド系抗生物質製剤のEDは、さらに好ましくは70%超であり、しばしば80%を超える。本明細書において用いられる「放出用量(放出率)」または「ED」という用語は、パウダーユニット、カプセルまたはリザーバーから発射後または分散後における、適当な吸入デバイスからのドライパウダーの送達指標を指す。EDとは、吸入デバイスによって送達される用量の、正規の用量に対する比(すなわち、発射前の適当な吸入デバイス内に配置された単位用量に対する粉末質量の比)として定義される。EDは実験的に求められる量であって、一般には患者への投与をインビトロで模倣した構成のデバイスを用いて求められる。ED値を求めるには、乾燥粉末での正規用量(前述規定のような)を適当なドライパウダー吸入器内に入れ、吸入器を作動させて粉末を分散させる。生じたエアゾル浮遊物は次に減圧吸引によってデバイスから放出され、マウスピースデバイスに取付けられた風袋消去可能なフィルターの表面に捕捉される。そのフィルターに到達する粉末の量が送達用量とみなされる。たとえば、吸入デバイス内に配置された50mgのドライパウダー含有の2号カプセルに場合、粉末の分散によって、前述のように風袋消去可能なフィルター表面に40mgの粉末が回収されたとすると、そのドライパウダー組成物に関するEDは、40mg(搬送用量)/50mg(正規の用量)×100=80%となる。
【0056】
さらにほかの態様によれば、本発明は気管支感染症患者の治療に有用なキット類も提供し、キットには90〜130mgのドライパウダーのアミノグリコシド系抗生物質、ならびに患者の気管支系に一定の服用量を投与するための能書が含まれ、20〜36日間の第一治療期間に1日1〜3回、ドライパウダー吸入用のデバイスが用いられる。本発明のこの態様によれば、その能書によって第一治療期間の後に、患者の気管支系にアミノグリコシド系抗生物質が投与されない第二治療休止期間を設け、本明細書において実質的に記載したように所望の抗菌効果が現われるまでさらに第一治療期間と第二治療休止期間とのサイクルを2回またはそれ以上繰返すことが指示され得る。本発明のキット類では、1回分または複数回分の用量を単一単位用量として単一容器内に入れるか、あるいは抗生物質の送達に使用する吸入用デバイスしだいでは多重容器または連続投与用単位用量に分割して入れることもできる。たとえばアミノグリコシド系抗生物質の投与用量は2〜6単位用量、好ましくは3〜5単位用量、およびさらに好ましくは4単位用量に分割することができる。一つの代表的な実施形態では、本発明のキット類には112mgのトブラマイシン(存在し得る対イオンの重量を除いた遊離塩基体としての重量で求められた値)が投与用量として含まれ、4個のHPMC製2号カプセルの各々に、遊離型塩基としてのトブラマイシンが27mgずつ別々に充填されている。
【0057】
以下の実施例は、本発明の実施を企図する最良の方式を説明するためのものにすぎず、本発明を限定する意味のものではない。
【実施例】
【0058】
(実施例1 吸入用トブラマイシン粉末(TPI)の調製)
硫酸トブラマイシンのドライパウダー組成物については、以下の手順によって調製した。ジステアロイルホスファチジルコリン(DSPC)がゲル状態から液晶状態に移行する温度(約80℃)以上になるように、潅注用滅菌水(SWFI)を加熱する。次にその熱水にDSPCおよび塩化カルシウム・二水和物を加える。得られた液体分散物を、UltraTurrax T−50(商品名)(IKA Labortechnik社製)中、8000rpmで5分間混合処理する。次にその液体分散物に混合しながら、臭化ペルフルオロオクチル(PFOB)を滴加(1分あたり15mlの滴加速度)する。滴加終了後に、得られた水中PFOB滴型エマルションをさらに10000rpmで10分間混合する。UltraTurraxでの乳化工程によってミクロンサイズの液滴が生成する。つづいてそのエマルションの連続相に硫酸トブラマイシンを溶解させ、得られた分散液を噴霧乾燥用の供給原料として用いる。次にその供給原料を噴霧乾燥して以下の表1に示した組成のドライパウダー製剤を得た。
【0059】
【表1】
その粉末を相対湿度10〜15%でカプセル充填機内にセットし、10分間平衡化させた後、50mg(トブラマイシン遊離塩基として27mg)ずつをHPMC製2号カプセルに充填する。
【0060】
(実施例2)
この実施例は、本発明のトブラマイシンドライパウダー組成物の単回投与が、トブラマイシンに関して同様の薬物動態学的挙動を示すトブラマイシン溶液での投与と比較して、トブラマイシンの送達効率が高いことを示す臨床試験の記述である。
【0061】
全般的な臨床試験デザインおよび試験計画
この試験は、無作為化、開放性、連続コホート、活性薬剤対照、単回投与および用量漸増による試験として設計した。連続コホートの各々において、被験体全体はT−326Inhaler(商品名)デバイス(Nektar Therapeutics社(米国、カリフォルニア州、サンカルロス市)製)を使用した以下に示す投与スケジュールに従うトブラマイシン粉末の単回投与、またはDeVilbiss PulmoAidesTMコンプレッサー装着のPALI LC PLUSTMジェットネブライザーによってエアゾル化させた吸入用300mgトブラマイシン溶液(TOBI)の単回投与のいずれかを、3:1に比率で無作為に分けて試験を行った。尚、患者は一つのコホートのみの参加とした。
【0062】
次の増量TPI(吸入用トブラマイシン粉末)治療コホートの実施については、試験が終了したコホートから得られた治療にまつわる有害事象(AE)およびそれ以外の安全性に関わる結果のすべてをデータ監視委員会(DMC)で検討してから進めた。DMCでは以下の判定基準、すなわちTPI治療コホート中の3名またはそれ以上が、投与終了後30分以内にFEV1が少なくとも20%低下することに該当せず、TPI投与被験体はいずれも試験薬に関係する重篤な有害事象(SAE)が認められないという場合に限り、次の増量投与試験段階に進むことが了承された。
【0063】
実施した治験
TPI(吸入用トブラマイシン粉末)の単回投与は、T−326Inhaler(商品名)デバイスを使用して行った。
コホート1:TPI(1カプセルあたりに遊離塩基としてのトブラマイシンが「活性用量」として14mg含有)カプセルを2個投与。
コホート2:TPI(活性用量14mg)カプセルを4個投与。
コホート3:TPI(活性用量28mg)カプセルを2個投与。
コホート4:TPI(活性用量28mg)カプセルを4個投与。
コホート5:TPI(活性用量28mg)カプセルを3個投与。
【0064】
対照の治験
300mg/5mL[保存剤不含、トブラマイシン濃度60mg/mL、溶媒として1/4生理食塩水5mL使用、pH6.0±0.5]溶液でのTOBIの単回投与は、DeVilbiss PulmoAideTMコンプレッサー装着のPARI LC PLUSTMジェットネブライザーによってエアゾル化して行った。
【0065】
本試験での被験体は、嚢胞性線維症(CF)と確定診断された6歳以上の80名までとし、無作為化して治験を行った。各コホートは16名で構成され、そのうちTPIが投与される12名とTOBIが投与される4名とに無作為に分けた。
【0066】
被験体については、投与試験の7〜9日前に適格かどうかのスクリーニングを行った。被験体に投与して、安全性、ならびに治験薬投与30分、1,2,4,8および12時間後の喀痰中および血清中のトブラマイシン濃度について監視下で評価を行った。被験体は、投与後7(±2)日に外来による追跡調査も行った。
【0067】
対照群の選択も含めた治験デザインの検討
本試験に関する一次結果の算定基準は、治験の全般的な安全性および忍容性とした。この結果をさらに評価するために、被験体を無作為に3:1に分けるスキームを選択して治験群への登録者数を最大にした。
【0068】
本試験で同時に行う活性対照は、DeVilbiss PulmoAideTMコンプレッサーによって駆動するPARI LC PLUSTMジェットネブライザーで送達される300mg/5mL TOBIとした。TOBIは、P.aeruginosa感染の嚢胞性線維症患者に対する予後管理用指標薬(対照薬)とした。
【0069】
試験集団の選定に関する採用基準
被験体については、以下の採用基準のすべてに該当する場合に、本試験の参加に適格であるとした。
・試験に関係するいかなる手続きを行う前でも、説明による同意(インフォームドコンセント)の書面およびHIPAA(健康保険計画実施協会)の認可が得られていること。
・スクリーニング時における年齢が6歳以上の男性および女性であること。
・定量的ピロカルピンイオン導入検査(QPIT)で記録された汗中の塩化物濃度が60meq/L以上であり、および/または嚢胞性線維症(CF)と整合性がある同定可能な二つの変異を有する遺伝子型を持ち、CFと一致する一種または複数の臨床的特徴を示すことからCFと診断されていること。
・女性の被験体の場合は11歳以上であるか、または初潮に達していて、しかも血清による妊娠検査で陰性であること。妊娠可能年齢の性的活動性の女性については、試験期間中の避妊に同意しなければならない。
・要求時に喀痰試料を排出できること。
・FEV1が予測値(性別、年齢および身長に基づいてKnudson式で算出される値)の40%以上であること。
・プロトコールの要求にすべて従うことができること。
・試験研究者の見解において臨床上、安定していること。
【0070】
試験対象者選定に関する除外基準
被験体については、以下の除外基準のいずれかに該当する場合、試験対象から除外される。
・試験薬投与前14日以内および試験期間中に、試験以外でアミノグリコシドを吸入または静脈内に投与した場合。
・試験薬投与前14日以内および試験期間中に、治療のためのいずれかの投薬が行われた場合。
・試験薬投与前7日以内および試験期間中に、ループ系利尿剤が投与された場合。
・試験薬投与前30日以内のいずれかにおいて、60cc超の喀血を生じた場合。
・アミノグリコシドまたは吸入による抗生物質に対して局所的または全身的な過敏症の既往歴がある場合。
・血清中クレアチニン濃度が2mg/dlまたはそれ以上、BUN(血液尿素窒素)が40mg/dlまたはそれ以上、あるいは尿検査で2+またはそれ以上の蛋白尿とされた場合。
【0071】
治験または評価から除外される被験体
被験体、その両親、または後見人は、試験のいずれの時期においても既得権を侵さずに参加への同意を撤回することができるものとする。試験研究者の側も、臨床的判断において最大の注意を払わなければならない被験体の場合、または試験プロトコールに応じることができないと考えられる被験体の場合にその被験体を除外できるものとする。可能な場合、試験終了時の外来による追跡調査で挙げられた検査および評価項目についても実施した。
【0072】
必要な再外来ができなかった患者では、できるだけその理由を調べるようにした。その情報については適切な症例報告書(CRF)に記録するようにした。
【0073】
投与が行われずに本試験から脱落した無作為抽出被験体は、代わりの被験体と入替えを行った。しかし、投与後に脱落した無作為抽出被験体については、いかなる場合でも入替えは行わなかった。脱落の理由および日時についても、CRFに記録するようにした。脱落の理由については、以下のように分類した。
【0074】
・有害事象;
・プロトコール違反;
・追跡調査の遺失;
・同意の撤回;
・死亡;
・不適切な登録;
・管理上の理由;
・そのほかの上記記載以外の事由。
【0075】
治験
80名までの被験体について、無作為化して治験を行った。被験体には対照薬剤または以下に挙げたような本試験薬のいずれかの単回投与が行われた。
【0076】
試験薬による治験:T−326Inhaler(商品名)デバイスを用いた投与
コホート1:TPI(活性用量14mg)カプセルを2個投与。
コホート2:TPI(活性用量14mg)カプセルを4個投与。
コホート3:TPI(活性用量28mg)カプセルを2個投与。
コホート4:TPI(活性用量28mg)カプセルを4個投与。
コホート5:TPI(活性用量28mg)カプセルを3個投与。
【0077】
対照の治験
TOBI 300mg/5mL[保存剤不含、トブラマイシン濃度60mg/mL(1/4生理食塩水5mL添加、pH6.0±0.5に調整]は、DeVilbiss PulmoAideコンプレッサー装着のPARI LC PLUSジェットネブライザーによってエアゾル化して投与した。
【0078】
試験薬製品の特徴
本試験で用いたTPIは、トブラマイシンならびに二種類の賦形剤、すなわち1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン(DSPC)および塩化カルシウム(CaCl2)から成るドライパウダー製剤である。TPIは25mgまたは50mgの粉末のいずれかを含有するように、対応するサイズの2−ヒドロキシプロピルメチルセルロース(HPMC)製カプセル内に充填して用いた。本試験ではトブラマイシン粉末は、二種類の活性用量、すなわち1カプセルあたりトブラマイシンとして14mgおよび28mgで用いた。
【0079】
吸入用トブラマイシン溶液(300mg/5mL)であるTOBI(登録商標)は、滅菌され、発熱物質が認められず、保存剤不含のエアゾル化用抗生物質製剤である。使用される製剤1mL当たりには、注射用滅菌水中に溶解したトブラマイシン60mgおよび塩化ナトリウム2.25mgが含まれ、pHは6.0±0.5に調整されている。
【0080】
投与に関する指示事項
無作為に対照薬治験群に割り当てられた被験体には、吸入用トブラマイシン60mg/mL溶液TOBI(登録商標)300mgを投与した。尚、吸入用トブラマイシン溶液は、PARI LC PLUSジェットネブライザーおよびDeVilbiss PulmoAideコンプレッサーを介して被験体に投与された。300mg用量のTOBI(登録商標)吸入用トブラマイシン溶液は、市販のTOBI(登録商標)吸入用トブラマイシン溶液アンプルを購入して準備した。5mLアンプル試験用薬2本は、ホイル袋に入れて用意した。この試験は単回投与で行ったが、試験薬の調製、ならびにネブライザーおよび送達系のセットアップの際に万一こぼれる場合には、2本の5mLアンプルの試験薬が入った前述のホイル袋を用いて患者に提供した。
【0081】
無作為に本試験薬の治療群に割り当てられた被験体には、トブラマイシン用量強度14mg含有のカプセル2個または4個によるTPI、あるいは用量強度28mg含有のカプセル2,3または4個によるTPIを単回投与した。TPIはT−326Inhaler(商品名)デバイスを介して患者に投与した。TPIカプセルはホイルで二重につつまれた防湿性の容器内に密閉し、容器を開けてから30分以内に投与するようにした。コホート1,2および3では、T−326Inhalerデバイスを1回だけ用いて単回用量の投与を完了させた。コホート4および5では、二個目のカプセルの投与後にT−326Inhalerデバイスを取替えるようにしたため、これらのコホートでは2個のT−326Inhalerデバイスを用いて単回用量の投与が完了した。
【0082】
対照薬および試験薬の調製および使用に関する詳細な指示事項は、本試験に関わる研究スタッフに予め伝えられた。
【0083】
治験群への被験体の割当て方法
適格被験体については、試験薬による治験群または対照薬による治験群のいずれかに3:1の比率で無作為に割り振った。被験体が適格基準に適合すると試験研究者または治験コーディネーターが確認すると、試験スタッフはその患者についての無作為化確認書の記入を完了し、対話型音声応答システム(IVRS)によって被験体番号および治験群割り当ての情報を得た。
【0084】
試験開始時には、5種のコホートにおける80名までの被験体が無作為化されるように準備し、試験薬治療群には60名(コホートごとに12名の被験体)および対照薬治験群には20名(コホートごとに4名の被験体)の割り振りを行った。
【0085】
試験で用いられる用量の選定
対照薬治験群での用量は、6歳またはそれ以上のCF患者におけるP.aeruginosaの予後管理用にFDAが認可した用量である300mgのTOBIに設定した。PK(薬物速度論)でのモデル解析を用いると、PARI LC PLUSジェットネブライザー/DeVilbiss PulmoAideコンプレッサーを介して300mg用量のTOBIを送達した場合の、全身における生物学的利用率は、噴霧された全投与用量の11.7%であると想定された。またTOBI300mg吸入1時間後における血清中トブラマイシン濃度の平均値および標準偏差は1.0±0.58μg/mLとなり、吸収されずに沈着する量が多いことが示唆された。
【0086】
試験薬治験群の用量については、トブラマイシン14mg入りTPIカプセル2個または4個、あるいはトブラマイシン28mg入りTPIカプセル2,3または4個とした。
【0087】
各被験体についての用量選定および投与時期の調整
被験体には、300mgTOBI(登録商標)トブラマイシン吸入用60mg/mL溶液の単回投与、あるいは用量強度14mgのカプセル2個または4個、または用量強度28mgのカプセル2,3または4個から成るTPIの単回投与のいずれかで投与を行った。尚、食事に関係する投与時期の制限は設けなかった。
【0088】
盲験性
本試験は、標識オープン式の臨床試験研究とした。2種類の異なる薬物送達系を用いるので、治験の同一性の盲験性は実現不能である。
【0089】
事前および同時に行われる治療
追加の支持療法については、試験現場ごとでの標準的な医療業務にしたがって投与が成された。除外基準の中に列挙された投薬は、被験体選定のスクリーニング時点から試験後の追跡調査での外来時に至るまで被験体には行わなかった。
【0090】
被験体には、試験薬投与前の気管支拡張剤の使用を認めた。但し、気管支拡張剤の投与は、臨床上日常的に気管支拡張剤が用いられている被験体に対してのみに行った。日常的使用とは、スクリーニング前の2週間において1日1回またはそれ以上の投与が行われていたことと規定する。短時間作用型の気管支拡張剤を被験体に投与する場合は、本試験薬投与開始の15〜60分前に行った。長時間作用型の気管支拡張剤を被験体に投与する場合では、24時間前に行った。
【0091】
以下の薬物の使用は禁じた。すなわち本試験薬投与14日以内および本試験期間中を通じてのアミノグリコシドのいかなる剤形およびいかなる療法;ならびに本試験薬投与7日以内および本試験期間中を通じてのループ系利尿薬の投薬。
【0092】
治験コンプライアンス
被験体は、研究者および治験コーディネーターの存在のもとで、自分で試験薬の単回投与を行った。本試験薬投与における早期の中止、中断または延期のいずれの場合でもその理由を原本およびCRFに記録した。試験薬投与期間のすべてについても、原本およびCRFに記録した。
【0093】
各被験体(または適切な場合では患者/法的後見人)は、試験研究者の管理下でHIPAAでの認可を含めたインフォームドコンセントの書面を提出し、(適切である場合は)試験に関係する手続きのいずれも、治験開始前に本試験への参加に同意を明確にした。試験研究者は、被験体について1回目の訪問時、および試験薬投与の7〜9日前(2回目の訪問時)にスクリーニングを行い、登録するのに適格性があるかの判断を下した。試験研究者は、患者に関する現疾患およびそれ以外の関連する呼吸器の病歴を含めた関連医療履歴、基礎的徴候および症状、スクリーニング前6ヶ月以内におけるドライパウダー吸入剤および抗生物質吸入剤の使用状況、ならびにスクリーニング中に実施された投薬および治療状況について精査し記録した。
【0094】
試験研究者は、被験体の身長、体重および生命徴候を含めた身体検査によるスクリーニングを実施した。生命徴候には心拍数、呼吸数、口内体温および座位動脈血圧を含めた。心拍数および呼吸数は、被験体を少なくとも10分間安静にしてから、1分間かけて計測した。血圧については、被験体を少なくとも10分間座らせてから計測した。
【0095】
スクリーニング前の2週間において、1日あたり少なくとも1回またはそれ以上の回数で短時間作用型の気管支拡張剤を治療のために日常的に使用したことが規則的投薬レジジメンに含まれるすべての患者に対しては、肺活量スクリーニング検査の15〜30分前および本試験薬投与の15〜60分前に気管支拡張剤の投与を行った。長時間作用型気管支拡張剤については、前述のように本試験薬投与の24時間前に被験体に投薬した。
【0096】
被験体は、1994年版の米国胸部学界指針に従ってルーチンの肺活量測定を済ませて1秒間努力呼気肺活量(FEV1)、努力肺活量(FVC)および中位努力肺活流速(FEF25−75)を計測し、信頼性の高い肺活量測定が実施できたかどうかを記録し、そのFEV1値が被験体の性別、年齢および身長に基づいたKnudson関係式を用いて計算された予測値の40%またはそれ以上である包含必要条件にあてはまることを保証した。
【0097】
被験体から血液試料を提供してもらって化学的および血液学的なスクリーニング検査を行い、尿試料については浸漬式試験スティックで蛋白尿の検査を行った。11歳またはそれ以上、あるいはすでに初潮に達した女性被験体については、尿試料での妊娠検査も行った。
【0098】
包含および除外の必要条件のすべてを満たした被験体については、本試験への参加資格があるものとし、プロトコールにおいて述べたように無作為化(無作為割付け)した。無作為化した被験体は、診察7〜9日後(2回目の訪問)の状況に復帰させて治験および関連する手続きを行った。
【0099】
訪問2日目、すなわち治験の投与1日前に、無作為化した被験体のすべては服薬前の手続きを完了した。その手続きには基礎的症状での変化、併用療法における変化および生命徴候の測定データも含めた。スクリーニングの際に被験体の身体系でなんらかの異常が見られ、それが臨床上明確であった場合、あるいは被験体の健康状態がスクリーニング時点から臨床上明らかに何らかの変化を示した場合に、試験研究者は被験体の身体検査を再度行った。スクリーニング時の蛋白尿検出用の尿浸漬式スティック検査の結果が微量検出または1+の結果であった場合は、同検査を投与前に再度行うようにした。またスクリーニング時の血清化学的および血液学的検査に異常が見られた場合も、同検査を再度行うようにした。試験研究者は、投与前の被験体の肺活量測定を実施し(通常の短時間作用型気管支拡張剤を使用した被験体の場合ではその15〜30分後に行う)、トブラマイシン分析用の血清および喀痰の試料も採取した。
【0100】
TPI投与群被験体のうち、その時点で短時間作用型気管支拡張剤を規則的または必要に応じて使用している被験体については、治験投与前に試験研究者の裁量の下で短時間作用型気管支拡張剤での予備治療もできるようにした。またその時点で短時間作用型気管支拡張剤を使用していない被験体についても、スクリーニング時および治験投与前の肺機能検査でのFEV1値の低下率が10%またはそれ以上に達した場合に、短時間作用型気管支拡張剤での予備治療を可能とした。スクリーニング時点からのFEV1の相対的変化%については、以下のように計算して求めた。
【0101】
・スクリーニング時からのFEV1の相対的変化%
・=[(予備投与時のFEV1−スクリーニング時のFEV1)/スクリーニング時のFEV1]×100。
【0102】
単回投与の治験は、気管支拡張剤の予備投薬、あるいは肺活量測定を行ってから60分以内に行い、以降の項で記述するようなプロトコールに従い、被験体へのエアゾル送達および安全性に関する客観的評価を行った。被験体は投与時に背筋を伸ばしていすに座って説明を受け、呼吸数を通常に整え、試験薬吸入中はノーズクリップを使用した。治験コーディネーターは、試験薬投与の開始および終了の時刻を記録した。被験体が長く咳込む場合(10秒間超)には、治験コーディネーターはタイマーを止め、被験体への投与が再開された時点でタイマーを再始動させた。TPI投与群の被験体の場合では、試験研究者および/または治験コーディネーターは、試験薬の二回目の吸入時に吸入器から出るガタガタ音(終了音)が聞こえるかどうかに注意をはらった。試験薬投与が完了した直後に、被験体は正規生理食塩水30mLで口をゆすぎ、5〜10秒間うがいを行い、喀出し、そのゆすぎ工程を3回実施した。
【0103】
被験体は試験薬投与開始後12時間、安全性の評価のために診療所に待機した。その後に被験体は診療所から退出し、その8(±2)日後に3回目の訪問のために診療所に戻るようにスケジュールを組んだ。追跡調査のための再訪問のスケジュール化については、患者によって柔軟に対処した(本実施例で許される限り、追跡調査のための再訪問は投与から「第8日目」と示す)。本プロトコールのスケジュールから幾分はずれても、客観的な試験に対する影響は最小限または無いと考えられた。
【0104】
追跡調査のための投与後第8日目の訪問において、治験コーディネーターは、被験体の同時発生疾患、新規または悪化性の有害事象、CFに付随する症状、現在の投薬およびその用量、ならびにOTC(店頭販売楽)の使用状況を含めての被験体の医学上の履歴におけるいかなる変化についても精査を行った。臨床上明らかな、試験薬に関係する有害事象(AE)が見られた被験体については、満足いくような解決がなされるまで繰返して追跡評価(電話によるかまたは診療所の訪問時のいずれかで)を行った。試験研究者は、被験体の身長、体重、生命徴候および肺活量の計測による身体検査を行い、ならびに血液および尿の試料を採取して化学的検査、血液学的検査および浸漬式スティックによる蛋白尿検査を行った。
【0105】
初回のエアゾル送達の変動要因
300mg/5mL TOBIに対するTPIの比較用量評価は、試験薬による治験と対照薬による治験とのエアゾル搬送特性の評価に基づくものである。試験薬による治験および対照薬による治験でのエアゾル特性に関しては、血清中および喀痰中のトブラマイシン濃度の時間推移、本実施例に記載されるような血清および喀痰に関するいわゆる薬物動態学的パラメーターの計算、治験薬の投与に要する時間の計測、ならびにT−326Inhalerデバイスとカプセルとのパーフォーマンスの評価に基づいて求めた。
【0106】
血清中のトブラマイシン濃度
血液試料は、投与前、ならびに治験での吸入における初回の吸入が開始されてから0.5,1,2,4,8および12時間後に採取した。試料の採取は可能な限り決められた時間に近づけて行い、たとえば試験薬投与の0.5時間後の採血ならば±2分以内に、またそれ以降の採血時間では設定時間から±10分以内の時間に行うこととした。それらの時間域からはずれて採取された血液については、プロトコール逸脱とみなした。
【0107】
血清は分離採取後、分析するまで−20℃またはそれ以下で保存した。血清中のトブラマイシン濃度は、Abott社製のTDx(登録商標)/TDxFLx(登録商標)システムを用いる改変型の蛍光偏光イムノアッセイ(FPIA)法で分析した。尚、試料はサンプルカートリッジの希釈ウェルに直接加えて行った。純偏光度についてはTDx/TDxFLx装置によって得た。トブラマイシン濃度の計算には、重み付けを行った4−パラメーター論理式を用いた。トブラマイシンの濃度は、その遊離塩基当量での記載で報告した。
【0108】
本試験の被験体試料のアッセイには、較正用標準(0.05μg/mL,0.10μg/mL,0.40μg/mL,0.80μg/mLおよび0.90μg/mL)ならびに品質管理用試料(0.10μg/mL,0.40μg/mLおよび0.80μg/mL)を準備した。アッセイは全6回での実行とした。
【0109】
定量性を保つ最小限界濃度は、0.05μg/mLであった。アッセイ精度については、品質管理用試料のCV(変動係数)によって反映されるが、0.10μg/mL,0.40μg/mLおよび0.80μg/mLでの品質管理用試料に対するCV値は各々、4.9%、5.7%および5.6%であった。このアッセイ法の正確度については、品質管理用試料のアッセイ間平均回収率によって反映されるが、0.10μg/mL,0.40μg/mLおよび0.80μg/mLでの品質管理用試料に対して回収率は各々、103%、103%および101%であった。全般的にみて、本アッセイ方法は薬物動態学的解析に適した正確度および精度を有することが認められた。
【0110】
血清中における薬物動態学的パラメーターの計算およびTPIの比較用量の推定については、本実施例に記載されている。
【0111】
喀痰中トブラマイシン濃度
喀痰試料は、治験第1日目の予備投与の前、ならびに試験薬吸入時における最初の吸気開始の0.5,1,2,4,8および12時間後に、被験体に深い咳をしてもらって喀出されたものを採取して得た。喀痰試料の採取は、可能な限り設定時間に近づけて行い、しかも血清採取と同じ時間帯で行った。これらの時間間隔をはずれて採取された試料については、プロトコール逸脱とみなした。
【0112】
喀痰試料(最低量100mg)は治験の単回投与前にも採取し、トブラマイシン濃度のベースラインを求めた。
【0113】
喀痰試料は、分析まで−70℃またはそれ以下で保存した。トブラマイシンの濃度は、紫外部検出器装着の妥当な逆相高速液体クロマトグラフィー(HPLC)法を用いる分析によって求めた。
【0114】
被験体の喀痰試料は、最初に0.2規定の水酸化ナトリウム溶液で液化させ、次にトリス緩衝液(トリズマ塩基濃度20.0g/L)で希釈した。喀痰標準試料は、治験前にCF被験体から予め採取したプール喀痰の希釈液に、最終濃度が0μg/g,20μg/g,40μg/g,100μg/g,200μg/g,400μg/gおよび1000μg/g(喀痰)になるようにトブラマイシンを添加して調製した。アッセイ品質管理用の試料は、希釈したプール喀痰にトブラマイシンを、40μg/g,300μg/gおよび800μg/g(喀痰)含有するように添加して調製した。内部標準用のシソマイシン(0.15mg/mL(トリス緩衝液)溶液、100μL)を、各標準試料、対照試料および被験体試料100μLに加えた後、アセトニトリル400μLおよび2,4−ジニトロフルオロベンゼン(0.17g/mL溶液)50μLを加えた。その試料の反応混合物を乾熱ブロックヒーター内、80℃で1時間加熱した。その反応液にアセトニトリル/水(60/40(v/v))混液600μLを加えた後、その50μLについてHPLCで分析した。
【0115】
試料は、Wters社製の600E型ポンプ、486型または2487型紫外部検出器(λmax=360nm)、および717PLUS型オートサンプラー装着のHPLC装置に接続したWaters社製Nova−Pak(登録商標)C−18カラム(3.9×150mm、充填材粒径4μm)上に注入した。移動相の組成は0.2%酢酸/アセトニトリル(39/61(v/v))混液とし、ポンプ流速は最初の5分間は1.5mL/分、その後9または10分間は2.0mL/分とし、運転時間によって加減した。生データ、データ処理および計算結果を得るために、Waters社製Millennium−32C/S LC Software(商品名)(ver.3.20)を用いてWaters社製HPLC装置を操作し、分析結果の報告を行った。内部標準シソマイシンに対するトブラマイシンのピーク高さ比(PHR)を計算した。そのアッセイの完了には全17回の運転を行った。
【0116】
HPLCにおけるトブラマイシンおよびシソマイシン(内部標準)の保持時間は各々、3.8〜4.1分および10.0〜10.6分の範囲に観測された。喀痰については、PHR(ピーク高さ比)と濃度との関係は20〜1000μg/gで線形性を有することが認められた。回帰式はPHR=Bx+A、重み付け1/x(xはトブラマイシン濃度)とした。定量に関する限界最低濃度は20μg/gであった。標準試料の濃度については、正規の濃度の97〜102%以内に入り、変動係数(CV)は5.7%以内であった。アッセイ値の精度は、品質管理用試料のCVによって反映され、濃度40μg/g,300μg/gおよび800μg/gの品質管理用試料ではCV値は各々、5.2%、3.9%および2.0%であった。本アッセイ法の正確度については、品質管理用試料のアッセイ間の回収率によって反映され、濃度40μg/g,300μg/gおよび800μg/gの品質管理用試料では回収率は各々、107%、99%および97%であった。全般的にみて、本アッセイ方法は薬物動態学的解析に適した正確度および精度を有することが認められた。
【0117】
治験投与時間
治験の投与時間は、被験体の最初の吸入の開始から投与完了までの時間と規定した。治験の投与は、T−326Inhalerデバイスがガタガタ音を発生し始めた時、または対照のPALI LC PLUSネブライザーがブツブツ音を発生し始めた時に終了させた。TPIカプセルの場合、被験体の二回目の呼吸の際にガタガタ音が聞こえるかどうかに試験研究者は注意し、その徴候があればカプセルはすでに空になっているとした。
【0118】
治験での投与が何らかの理由で中断された場合は、中断した時間、ならびに連続投与の開始および停止時間を記録した。投与の中断があった場合、全投与時間に中断中の時間は含めなかった。投与が中断し、その停止時間または投与再開時間のいずれかの記録が抜けてしまった場合は、治験での投与時間は計算不能とした。
【0119】
使用したT−326InhalerデバイスおよびTPIカプセルの検査および残存トブラマイシンの分析
本試験プロトコールには明記していないが、使用したT−326Inhalerデバイスおよび使用したTPIカプセルについては、使用後に分析してパーフォーマンスの評価を行った。
【0120】
有害事象
有害事象(AE)とは、医薬品の投与を受けた治験被験体において医学上不都合ないかなる出来事も規定し、用量にはかかわらず、また治験による投与と必ずしも明らかな因果関係が証明されない場合も含まれる。したがってAEは、医薬品の使用に一時的に付随する好ましくなく意図されない徴候(実験上の異常な見解も含む)、症状または疾患のいかなるものも、その医薬品との関連性の考慮の有無にかかわらず、含めることができよう。この定義には介在性の病気または傷害、およびさきに存在する病状の増悪も含まれる。予期しないAEとは、本質または重篤性が実用上の医薬品情報とは整合しない事象のことである。
【0121】
AEは、被検者による任意の申し出か、または試験研究者または治験コーディネーターによる一般的な問診の結果発見され得るものである。
【0122】
AEのすべては解明されるまでモニタリングするか、あるいはそのAEが慢性的なものと判定される場合は、原因をはっきりさせた。AEが試験終了時でも未解明な場合は、そのAEの追跡調査の継続が保証されるかどうかについて、試験研究者および医療監視者による臨床評価が成された。
【0123】
AE CRFに記録された有害事象の重篤性については、試験研究者が以下のような判定を下した。
軽度:通常生活に支障なし。
中程度:通常生活に何らかの支障あり。
重度:通常生活が不能。
【0124】
AEに対する本治験の関連性については、以下の規定に基づいて試験研究者が判定した。
すなわち、
関連性なし:治験薬を摂取しても起こらないか、またはAEの出現が治験薬の投与時間と合理的な関連性がみられないか、あるいはそのAEが治験医薬品とは関連性がないと考えられる場合。
関連する可能性あり:本試験医薬品の投与とAEとに合理的な時間関連性がみられ、そのAEについて試験医薬品の摂取以外の原因でも同様に説明し得る場合。
高い関連性あり:本治験とAEとに合理的な時間関連性がみられ、そのAEについて試験医薬品の摂取による出現説明の方がそれ以外の原因による説明よりも妥当である場合、またはそのAEの原因として試験医薬品が最も考えられる場合。
【0125】
重篤な有害事象(SAE)
本治験ではSAEは全く報告されなかった。
【0126】
研究室での臨床検査
血液学的基礎データ、血清化学、および浸漬式スティックによる蛋白尿検査の測定を対象とする研究室での検査は、スクリーニング時点、および治験薬投与後8日目の追跡調査時に実施した。治験第1日目の投与時には、投与開始から12時間後に血液尿素窒素(BUN)および血清クレアチニンを計測した。スクリーニング時における尿浸漬スティックによる蛋白尿検査の結果が微量陽性または1+であった場合は、さらに同検査を投与前に再度行い、また血清化学および血液学的検査についても、スクリーニング時点で異常値を示した場合は再度検査を行った。
【0127】
本治験で実施された血液学的検査には、全血球数計測(WBC)、赤血球数計測(RBC)、ヘモグロビン値、ヘマトクリット値、分化度および血小板数計測を含めた。
【0128】
実施された血清化学的検査には、血清中のナトリウム、カリウム、塩化物、炭酸水素塩、BUN、クレアチニン、グルコース、カルシウム、リン、γ−グルタミルトランスフェラーゼ(GGT)、アラニントランスアミナーゼ(ALTまたはSGPT)、アスパラギン酸トランスアミナーゼ(ASTまたはSGOT)、アルカリホスファターゼ、乳酸脱水素酵素(LDH)、総ビリルビン、直接ビリルビン、間接ビリルビン、総蛋白質、アルブミン、および血清中ヒト絨毛性腺刺激ホルモン(HCG)の定量を含めた。
【0129】
気管支痙攣および肺活量測定
肺活量測定検査(1秒間努力呼気肺活量[FEV1(L)]、努力肺活量[FVC(L)]、および努力肺活量の1/4〜3/4の中程度における努力性吐出流速[FEF25−75])については、本治験の単回投与の前日、および投与完了の30分後に実施して気道の反応を評価した。また投与後8日目の追跡調査時にもルーチンの肺活量測定を実施した。
【0130】
投与前から治験薬投与終了30後までの間に、FEV1が20%またはそれ以上低下した場合は、本試験プロトコールでは気管支痙攣であると予め規定した。
【0131】
投与30分後にFEV1が20%またはそれ以上低下した場合は、FEV1の低下率が10%未満になるまで、試験研究者によって定められたスケジュールにしたがって肺活量測定(FEV1、FVCおよびFEF25−75)を再度実施した(尚、この処置は被験体12/317集団では行わなかったが、1例のみでFEV1の減少率が20%またはそれ以上に達したことが予想された)。FEV1の低下率が20%またはそれ以上に達したことが予想されたケースのすべては、AEとして記録した(尚、この処置は被験体12/317集団では行われなかった)。適切な投薬およびその後の追跡調査を伴う治験は、試験研究者の裁量のもとで行われた。
【0132】
生命徴候
生命徴候は、投与前日、ならびに投与開始の30分、1,4,8および12時間後に計測し、その実施項目には10分間安静後の座位での動脈血圧、心拍数および呼吸数、ならびに体温の測定を含めた。また生命徴候の計測は、投与後8日目の追跡調査の訪問時にも行った。
【0133】
身体検査
被験体の身体検査は、投与後8日目の追跡調査の訪問時に行い、その実施項目は被験体の全身外観、皮膚、リンパ節、HEENT(頭部、眼部、耳部、鼻部および咽喉部)、両肺、心血管系、腹部、手足、筋骨格系、神経系、および(所望により)尿生殖器系の身体所見から構成されるようにした。
【0134】
計測値の適正さ
本試験において用いられた効力測定は標準的、すなわち広く用いられ、信頼性、正確さ、ならびに有効薬剤と無効薬剤との識別能を有すると一般に認識されるものとした。本試験において用いられる安全性の測定は、標準的な臨床上および研究室の手順で行った。
【0135】
薬物速度論
喀痰および血清中のトブラマイシンのアッセイから得られた濃度(C)対時間(t)のデータについては、モデル非依存的な方法によって解析して薬物動態学的パラメータ類を得た。最高濃度(Cmax)および最高濃度到達時間(tmax)に関しては視認によって求めた。最終速度定数(λz)に関しては、最終相の対数線形回帰によって求めた。半減期(t1/2)に関しては、式t1/2=ln(2)/λzで算出した。尚、計算は定量限界低濃度域をすべてゼロとして行った。0時間(投与前)から投与12時間後までの、喀痰および血清中の濃度−時間曲線下の面積、すなわちAUC(0,12)に関しては、台形則によって算出した。無限時間までのAUC,すなわちAUC(0、∞)に関しては、式AUC(0,12)+C(12)/λz;但し、C(12)は投与開始12時間後の濃度、で算出した。
【0136】
薬物動態学的パラメーターのすべては、平均値±SD(標準偏差)で表わした。調和半減期は、
【0137】
【数1】
で推定して求めた。
式中、
【0138】
【数2】
は用量ごとの最終速度定数の相加平均である。
調和平均半減期の標準偏差
【0139】
【数3】
は、
【0140】
【数4】
で求めた。
【0141】
式中、
【0142】
【数5】
は用量ごとの平均最終速度定数の標準誤差である。
【0143】
TOBIに対するTPIの相対用量の推定
線形回帰モデルは、全コホートから得られたTPIの血清中濃度データのlogAUC(0,12)対log(TPI用量)に当てはめ、TOBIに対するTPIの相対用量を推定した。その相対用量および95%信頼区間については、当てはめた回帰線の逆関数、ならびTOBIデータの平均logAUC(0,12)における上側および下側の95%信頼域を用いて求めた。
【0144】
モニタリング
本試験は、臨床試験実施に関する基準(GCP)に従い行われた。
【0145】
データの取扱い
症例報告書データは、Clintrial(登録商標)データベースに二重に入力した。
【0146】
データの品質管理は、手続き型言語/逐次クエリー言語(PL/SQL)およびSAS(登録商標)ソフトウエア第8.2版またはそれ以降の版(SAS Institute社(米国、ノースカロライナ州、キャリー市)製)を用いて実施した。データ解析は、予め規定した解析プランに基づき、SASソフトウエア第8.2版またはそれ以降の版を用いて実施した。全データベースを通じての誤り率は0.022%、すなわち上側95%信頼限界の0.157%(丸めた値)と推定された。この上側信頼限界は、選択した標準の0.5%未満である。
【0147】
統計処理方法および試料サイズの決定
この第1相試験の目標は、試験薬であるTPIおよび対照薬であるTOBIの治験の安全性の評価、およびTPI投与で得られる薬物動態学的プロフィールがTOBI300mg/5mL投与の場合に相当することの推断を行うことである。本試験で規定される目標には有効性の項目を含めなかった。設定された摘要事項および解析事項についてはすべて本質的な診査を行った。TPI治験群とTOBI治験群との間の統計的類似検定および両治験群間の差に関する統計的仮説検定は、いずれも本試験では企画しなかった。本試験では治験センターごとの登録可能な被験体が少数であるため、本試験の統計解析計画ではデータを全解析センターにプールするように前もって指定した。
【0148】
摘要事項および解析事項のすべては、6種の治験群のそれぞれに設けた。別に条件設定がない場合、頻度および百分率の値については絶対変数に対して算出し、非欠測値、平均値、標準偏差、最小値、中央値および最大値の数については連続変数に対して算出した。CRFに記録されたデータのすべては、被験体の個人データ一覧表中に存在するようにした。
【0149】
3種の被験体集団を以下のように規定した。
・すべてに登録された集団:本試験に登録され、無作為化された被験体。
・安全性評価が可能な集団:無作為化され、前述の試験薬のすべてまたは一部の投与を受けた被験体。
・薬物動態学的評価が可能な集団:無作為化され、前述の試験薬のすべての投与を受けた被験体。
【0150】
基礎的および個体群統計学的特性については、「すべてに登録された」被験体集団に対して摘要した。トブラマイシンの薬物速度論の評価については、「薬物動態学的評価が可能な」集団を用いて実施した。TOBI投与の場合と同様の全身的トブラマイシン摂取に相当するTPI用量の推定については、TPIの比較用量の評価が可能な集団で行なった。それ以外のすべての変数の解析については、「安全性評価が可能な」集団を用いて行った。
【0151】
すべての解析にSASソフトウエア(第8.2版)を用いた。試験結果のグラフ表示にはMicrosoft社製Excelソフトウエアを用いた。
【0152】
エアゾル搬送の分析および相対的TPI用量の推定
本単回投与治験を受けた被験体のすべては、喀痰および血清中のトブラマイシン濃度、血清に関して推定された薬物動態学的パラメータ類、ならびに治験の投与時間に基づき影響を受けるエアゾル送達特性の解析および評価にあてた。
【0153】
TOBIに相当するTPIの用量の推定には、血清に関するAUC(0,12)のデータを用いた。従属変数としてlogAUC(0,12)および独立変数としてlog(TPI用量)での線形回帰解析は、TPI投与群のすべてから得られたデータを用いて実施した。TPIの相当用量については、5種のTOBIコホートの組合わせから得られたTOBIにおける血清中トブラマイシン濃度の平均logAUC(0,12)値に当てはめた回帰線の逆関数を得て求めた。
【0154】
二次的なエアゾル送達の解析
本試験のプロトコールでは、エアゾル送達に関する二次変数を認定しなかった。
【0155】
安全性の解析
本治験の投薬を受けた被験体のすべてについては、AE、肺機能変化、臨床検査結果、生命徴候および身体検査に基づき、安全性の評価がなされた。
【0156】
有害事象の評価
基礎的症状、および治験の結果生じた有害事象(AE)に関しては、MedDRAシソーラスを用いて符号化した。個々の治験の結果生じたAEの全発生率(治験期間中または治験後に少なくとも1回の有害事象が認められた被験体の%)については、TPI治験とTOBI治験との間の顕著な差に関する記述的評価を行った。その統計的検定は企画しなかった。またAEについては、試験薬および対照薬での治験に関する重篤度(軽度、中程度、重度)および薬物との関連性(関連性なし、関連性高い)も摘要した。薬物との関連性を評価するための「関連性」分類には、個々のAEを本治験と「関連性あり」、「関連性高い」、および「関連性認めず」と試験研究者が判定を下して行った。
【0157】
肺機能の変化
被験体が肺疾患に罹患していない場合では、肺機能の正常値はFEV1、FVCおよびFEF25−75(肺活量計測値)で表されてきた。これらの標準値は、一般に肺疾患罹患被検者の試験において用いられている。肺活量計測値の生データは、以下に述べるようにKnudson式を用いて標準の予測%値に変換した。FEV1、FVCまたはFEF25−75に関するKnudson標準値とは、被験体ごとに以下の式を用いる年齢(歳)と身長(cm)との線形連結である。
Knudson標準値=C0+C1×身長+C2×年齢
式中、係数C0、C1およびC2は、被験体の性別および年齢群に基づき定められている。
【0158】
肺機能測定から得られる生データおよびKnudson標準値が得られると、予測%値は以下のようにして算出される。
%予測値=(生の観測値/Knudson標準値)×100
投与前から試験薬投与終了30分後までの予測されるFEV1%の変化(FEV1%)および相対変化については、以下の式を用いて算出した。
FEV1%の変化=投与後のFEV1%−投与前のFEV1%
相対変化=[(投与後のFEV1%−投与前のFEV1%)÷投与前のFEV1%]×100
基準値からの相対変化が20%以上の低下となった被験体については、気管支痙攣が起きたと規定した。
【0159】
FEV1%の変化および相対変化、ならびに気管支痙攣発生率に関しては、治験群間での記述的比較を行った。さらに以下の変数についても同様に導出し、摘要した。
【0160】
・投与前から投与終了30分後において予測されるFEV1%の相対低下が10%またはそれ以上である被験体の発生率
・投与前から投与終了30分後において予測されるFVC%およびFEF25−75%の変化および相対変化
・投与前から治験投与8日目の追跡時までに予測されるFEV1%、FVC%およびFEF25−75の変化および相対変化
安全性に関するほかの変数
臨床検査結果、生命徴候、併用薬、医療処置および身体検査を含むほかの安全性に関する変数に関しては、記述的な摘要を行った。さらに基底値から試験終了時の値の変化についても計算し、臨床検査データ、生命徴候および肺活量測定値に摘要した。尚、これらの計算では、試験薬投与のできるだけ近い時点に得られた評価値を基底値とした。各々の正常域よりも高いかまたは低い臨床検査結果の発生頻度の変化についても、評価を行った。
【0161】
試料サイズの設定
試料サイズおよび並列処理設計選択範囲については、統計力要件よりも臨床的および実地における要件に基づき設定した。すべての解析は診査によって行い、記述的方法を用いて取扱った。本試験では推論による統計解析は企画しなかった。
【0162】
暫定的解析
各コホートの試験終了時に、主要な安全性に関する変数の概要を作成し、治験データ監視委員会(DMC)に提出して用量増大の検討および決定がなされた。以下の結果は、被験体によって列挙され、治験完了後にコホートごとにまとめられたもので、DMCでの審議をサポートする。
【0163】
・被験体の個体群統計;
・投与前後で予測されたFEV1%および変化%;
・重篤な有害事象;
・治験の結果発生したAE;
・呼吸器系の、治験の結果発生したAE。
【0164】
この暫定的解析手順は、DMCによる安全性の検討および容量増大に関する議論をサポートするためにのみ設計された。尚、試験データの統計解析については企画せず、暫定的な安全性および用量増大の検討結果に、統計学上のα−水準の調整を行わなかった。
【0165】
被験体のスクリーニング、登録および無作為化
本試験には、15名の試験研究者によって全部で97名の被験体がスクリーニングされた。
【0166】
その97名の被験体うちの90名については、治験登録者基準に適合し、6種の治験群のうちの一つに無作為に登録(表2)した。97名のうちの残りの7名については、試験参加基準に適合せず、登録されなかった。
【0167】
【表2】
治験前の被験体の脱落
登録された90名の被験体のうちの3名については、治験薬投与の前にすでにAEが見られたため脱落させ、本治験の単回投与は行わなかった(表3)。90名のうちの残りの87名の被験体については、治験薬の投与が行われた。
【0168】
【表3】
試験の完遂
治験薬の投与が行われた87名の被験体のうちの86名が、試験を完了した。87名のうちの1名の被験体については、治験によって生じたAEによって試験から脱落した。
【0169】
試験を完了した86名の被験体のうちの1名については、投与は行われたが、試験に用いたカプセルがT−326Inhalerデバイスによって穿刺されなかったことが試験後に回収されたカプセルを評価して判明した。その被験体における血清中トブラマイシン濃度については、定量限界以下(BQL)であったため、その被験体には治験薬が投与されなかったことが確認された。
【0170】
エアゾル送達評価のために解析されたデータ集合
登録された90名の被験体のうちの86名は、無作為化し、治験薬の単回投与を行い、本プロトコールに関する安全性の評価対象とした。その90名のうちの3名については、AEのために治験薬投与前に試験から脱落したため、安全性の評価からは除外した。残りの1名については、投与時にカプセルがT−326Inhalerデバイスの不具合で穿刺されなかったために治験薬の投与が行われず、安全性の評価対象から除外した。
【0171】
【表4】
治験薬が投与された86名の被験体のうちの84名については、目的の薬物動態学的評価を行い、薬物速度論においてTOBI用量に相当すると考えられるTPIの用量を推定した。治験を受けた被験体のうちの2名については、薬物動態学的評価および相当用量の評価から除外した。
【0172】
【表5】
本治験を受けなかった被験体に対しては、薬物動態学的評価も行わなかった。
【0173】
個体群統計学的特性
本試験の登録被験体は、男性43名および女性47名、年齢は7〜50歳で嚢胞性線維症と診断された被験体であった。被験体の平均年齢はTOBI治験群での19.5歳からTPI2×14mg投与群での24.1歳までであり、治験群間でほぼ同等であった。被験体のうち15名が7〜12歳、22名が13〜17歳、および53名が18〜50歳であった。
【0174】
被験体のうち79名が白色人種系、5名がスペイン語系、3名がアフリカ系および3名がそのほかの系統であった。TPI治験群とTOBI治験群との間の性別および人種の分布はほぼ同等であったが、TPI4×14mg投与群とTOBI投与群とでは性別に関する不均衡が僅かに見られた(TPI4×14mg投与群の被験体は女性11名および男性3名;TOBI投与群の被験体は女性8名および男性12名)。試験結果に対するこの不均衡の影響は不明である。
【0175】
TPI投与群およびTOBI投与群の被験体は、スクリーニング時点で身長および体重の平均が同程度になるようにした。
【0176】
ほかの基本的特性
登録被験体については、臨床検査データ(定量的ピロカルピン−イオン導入検査(QPIT)時の汗中の塩化物濃度が60mEq/L以上であり、および/または二つの変異が確認された遺伝子型であること)の記録、および臨床的に安定型の嚢胞性線維症と診断しても臨床上明らかにに整合していることの記録がなされた。試験開始前に認められた医療履歴の所見、ならびに徴候および症状に関しては、治験群間で差はほとんどなかった。
【0177】
ほかの包含および除外基準については、いずれも満たしていた。但し、注目されることには、4名の被験体において、第1日目の投与後に軽度から中程度の持続性の喀血が見られたことである。被験体のすべては、スクリーニング時のFEV1が性別、年齢および身長に基づき予測される値の40%またはそれ以上という包含基準に適合した。被験体の予測されたFEV1%の中央値に関しては、6種の治験群間でほぼ同等であった(TPI2×14mg投与群での58.51%からTPI3×28mg投与群での82.40%まで、およびTOBI投与群での67.95%)。但し、合計3名の被験体では浸漬式スティックでの蛋白尿検査の結果が2+未満という治験参加必要条件を満たしていたが、スクリーニング時での尿試料の提供が行われなかった。
【0178】
登録被験体90名のうちの39名では、スクリーニング前の6ヶ月以内にドライパウダーの吸入投与を受けており、治験群によって25〜60%の被験体を占めた。被験体90名のうちの67名については、以前にTOBIの投与を受け(その割合は治験群によって40〜100%)、被験体のうちでほかのアミノグリコシドの吸入投与を受けたケースはなく、被験体90名のうちの10名では、過去6ヶ月以内にほかの抗生物質の吸入投与を受けた。
【0179】
登録被験体90名(すなわちTPI投与被験体およびTOBI投与被験体)のうちの36名では、治験薬投与前15〜60分以内に、それぞれに合った標準的CF治療の一部として短時間作用型気管支拡張剤の使用が行われた。そのうち二つのケースでは、治験薬投与の60分以上前に気管支拡張剤が使用された。
【0180】
治験コンプライアンスの評価
本プロトコールで要求される単回治験薬投与に関するコンプライアンスについては、90名のうちの86名の被験体が本治験を受けた(3名については基礎的症状で投与前に脱落し、1名については投与工程は行われたが、T−326Inhalerデバイスが治験薬カプセルを穿刺しなかったために実際には治験薬の投与は行われなかった)ことから、許容されるものであった。コンプライアンス不履行の被験体4名の事象については、表6にさらに列挙した。
【0181】
【表6】
血清中トブラマイシン濃度および薬物動態学的パラメータ類
図1に示すように、TPIおよびTOBI投与後のトブラマイシンの血清中平均濃度−時間プロフィールから、薬物が迅速に吸収されたことが認められた。すなわちtmax(最高濃度到達時間)の中央値が治験投与のすべてにおいて1時間であった。薬物の体内分布も極めて速いと考えられ、血清中からの消失は一相性の指数関数的減少挙動を示し、最終の平均半減期は2.8〜3.5時間の範囲であった。TOBI投与後のトブラマイシンの薬物動態学的パラメータ値については、過去に発表された試験結果と整合するものであった。
【0182】
TPIの投与量を上げると、AUC(0,∞)、AUC(0,12)およびCmaxの値が明らかに増加する(表8)ことから、トブラマイシンの体内摂取量が増大した。尚、これらのパラメータの増大は、用量比例的な増大よりは僅かに少ないものであった。
【0183】
4名の被験体では咳が生じたため、投与を一時中断した。その中断事例は、TPIの4×14mg、3×28mgおよび4×28mgの投与群で起こった。咳が見られた被験体のすべてに関しては、AUC(0,∞)、AUC(0,12)およびCmaxのいずれの値も、同じ投与群における一時中断なしで投与が終了したほかの被験体の値の範囲内にあった。
【0184】
AUCおよびCmaxの算出値から、4×14mgの手動充填カプセルの投与と2×28mgの自動充填カプセルの投与との間で被験体から検出されたトブラマイシンの体内摂取量の差は見られなかった(P>0.5)。したがって、手動充填および自動充填のカプセルのバイオアベイラビリティは同等であり、これら2群については統合した。
【0185】
デバイス内に残った粉末の%とAUC(0,∞)(r=−0.22、P=0.0771)、AUC(0,12)(r=−0.23、P=0.0707)およびCmax(r=−0.22、P=0.0838)との間には、全般に弱い負の相関性が見られた。体内摂取量が低いケースほど、デバイス内の残留TPI量が多い関係が見られた。4×28mgTPI投与群中の被験体のうちの1名(被験体No.04/409)では、粉末のデバイス内残留率が45%超であり、同一治験群内で最低のトブラマイシン体内摂取量となった。
【0186】
FEV1基底値の変化と血清でのAUCおよびCmaxとの間には有意な相関性は見られなかった[TPI投与の被験体:AUC(0,∞)(P=0.9838)、AUC(0,12)(P=0.9990)およびCmax(P=0.9110);TOBI投与の被験体:AUC(0,∞)(P=0.5261)、AUC(0,12)(P=0.4337)およびCmax(P=0.3878)]。
【0187】
【表7】
a中央値(領域)。最後の2列の被験体の数値を除き、それ以外の値は平均±標準偏差で表わす。
【0188】
TPIとTOBIとの比較用量解析
TOBI300mg投与後の体内摂取量に関しては、TPI4×28mgカプセル投与後の場合と極めて類似していた(図2および3)。全コホートから得られたAUC(0,12)データを用量の関数として調べてみると(図2)、その結果からTPIの115mg用量がTOBIの場合と同様な平均AUC(0,12)を示すと考えられることが分かる。AUC(0,∞)について考慮すると(図3)、TPIの112mg用量がTOBIの場合と同様なAUC(0,∞)を示すと考えられる。これらの結果に基づくと、TPIの28mgカプセル4個(総投与量112mg)で、TOBIの場合に極めて近い全身的体内摂取量になると考えられることがわかる。
【0189】
喀痰中トブラマイシン濃度および薬物動態学的パラメータ類
TPIおよびTOBI投与後では、喀痰中の最高濃度は平均で投与30分後に現われ(図4)、その後半減期0.8〜2.2時間で減衰した(表8)。
【0190】
薬物動態学的パラメータ類の変動性については、血清よりも喀痰の場合の方が大きい。喀痰から得られる薬物動態学的パラメータ類がもともと変動しやすいことに加えて、被験体のうちの数名では、必要な量の喀痰が得られなかった(いずれの時間でも喀痰試料が得られなかった被験体は治験群あたり3名以下であった)。このことは、体内摂取量、すなわちAUC(0,∞)、AUC(0,12)およびCmaxの計算に大きな影響を及ぼすと考えられる。結果として、用量を上げると喀痰中の量が上昇する傾向が見られる一方、喀痰中のレベルに基づいて用量の比例関係を確定することはできなかった。
【0191】
【表8】
a中央値(範囲)。最後の2列の被験体の数値を除き、それ以外の値は平均±標準偏差で表わす。
【0192】
bnはパラメータによって異なる場合がある。一回の解析のいずれで用いた被験体でもその最大数を挙げる。
【0193】
試験薬の投与時間
TOBIの被験体への投薬時間の平均は、約16分間であった(表9)。これに対して2カプセルのTPIの投薬では、2×14mgおよび2×28mgの投与の場合、各々平均で1.7分間および2.5分間であった。またTPI4×14mg、3×28mgおよび4×28mgの投薬の場合、各々平均で4.2分間,4.5分間および4.9分間であったが、それらのコホートでは2個目のデバイスは装填されておらず、各々3個目および4個目のカプセルの投与に用いた。したがってカプセル数が増えるとTPIの投与時間が増えるが、それによって用量強度も増す。
【0194】
【表9】
T−326InhalerデバイスおよびTPIカプセルの性能の評価
2個のカプセル以外のTPIカプセル投与のすべてケースでは、要求されるように投与を行った(但し、TPI4×28mg投与群中の被験体のうちの1名で、3個目および4個目のカプセルは、それからはずれた)。カプセル投与では85%またはそれ以上の頻度で2回目の呼吸時にガタガタ音(吸入終了の音)が聞こえた。
【0195】
Nektar解析によって治験施設および治験監視者が報告したデバイスの破損は存在しなかった。被験体の吸入器およびカプセル内の残存トブラマイシンの合計は、平均で正規用量の10.5%であった。その解析には、不適当な投与により残存粉末が45%超に成った被験体No.04/409も含んでいるので、デバイス中に実際に残存した粉末量はもっと少ないとも考えられる。
【0196】
脱落および遺失データの取り扱い
本試験では、無作為化した被験体のうちの3名が、投与前のAEによって治験薬投与の前に脱落した。無作為化した被験体のうちの別の1名については、本試験が完了したが、投与時に2個のTPIカプセルの両方とも穿刺されなかったために実際には吸入されなかったことが後になって判明した。これらの被験体については入替えがなされるまで、スクリーニングおよび登録の作業は継続し、入替え用の被験体には新たな無作為コードナンバーが割り振られた。
【0197】
1名の被験体は、早期に本試験から脱落した。その早期脱落理由の解析には、最終結果−繰越(LRCF)方略は用いなかった。
【0198】
定量限界以下の低レベルのトブラマイシン濃度に関しては、いずれの計算においてもゼロとして扱った。
【0199】
被験体の「エアゾル送達の部分集合」の適用
本プロトコールでは、無作為化された被験体および本治験の投与が完全に行われた被験体に関して、薬物速度論の評価が可能であるとした。試験が進行した時点で、予期されない投与およびTPIカプセルの状態の問題点によって、薬物速度論での評価の妥当性基準の見直しを行なった。6名の被験体の全員については、薬物速度論および用量比較の解析対象から除外した(4名の被験体については治験薬の投与が行われず、2名の被験体については治験薬投与が完全な用量で行われなかった)。それ以外の84名の被験体については、薬物速度論および用量比較の評価が可能とした。
【0200】
吸入による治験薬投与を受けた86名の被験体全員は、治験薬の投与時間の評価対象に含め、さらにカプセルおよびT−326Inhalerデバイス内の残存トブラマイシンの評価対象にも含めた。
【0201】
同等性の証明のための活性対照試験
本試験には、試験医薬品(TPI)と対照薬(TOBI)との臨床上および薬物動態学的な同等性を証明するようなデザインまたは検出力を設定しなかった。
【0202】
亜群の調査
以下の亜群解析は、TPIが正しい用量で投与された後の血清へのトブラマイシンの取込み(AUC(0,12)、AUC(0,∞)およびCmax)に関して行った。
【0203】
・12歳未満の被験体と12歳以上の被験体との間(P>0.4);
・以前にドライパウダーを使用した被験体と未使用の被験体との間(P>0.8);
・男性被験体と女性被験体との間(P>0.1);
・体重(P>0.2)
これらの変数の中でトブラマイシンの体内摂取に影響すると考えられるものはなかった。
【0204】
治験薬投与の15〜60分前に気管支拡張剤を使用したTPI投与被験体(N=27)においてAUCおよびCmaxの計算値としてのトブラマイシンの体内摂取量は、気管支拡張剤を使用しなかった被験体(N=37)の場合よりも僅かに高かった。AUC(0,∞)、AUC(0、12)およびCmaxの値は各々、19%(P=0.0685)、27%(P=0.0240)および38%(P=0.0038)高かった。気管支拡張剤の使用によって、結果的に全身血流循環に移行するトブラマイシン量が増す場合があり、このことはおそらく肺内に沈着するトブラマイシン量が増すためと考えられる。
【0205】
エアゾル送達のまとめ
TPIおよびTOBI投与後のトブラマイシンの血清中平均濃度−時間プロフィールから、薬物が迅速に吸収されることが分かる。すなわちtmaxはすべての治験例において1時間であった。薬物体内分布は極めて速いと考えられ、一相性の指数関数的な減衰が見られ、最終平均半減期は2.8〜3.5時間の範囲にある。TOBI投与後のトブラマイシンの薬物動態学的パラメータ値については、既存の報文と整合している。
【0206】
TPIの用量を上げると、トブラマイシンの体内摂取量が増大し、このことはAUC(0,∞)、AUC(0、12)およびCmaxの値の増加によって証明された。それらの増加は用量比例分と比較して僅かに少なかった。4×14mgの手動充填カプセル投与の被験体と2×28mgの自動充填カプセル投与の被験体との間で、トブラマイシン体内摂取量に関する差は検出されなかった。したがって、これらの二つの群は用量比較解析に関しては統合した。デバイス内の残存粉末の%とAUC(0,∞)(P=0.0707)、AUC(0,12)(P=0.0771)およびCmax(P=0.0838)との間には、全般に弱い負の相関性が見られた。FEV1の基底値からの変化とAUC(0,∞)(P=0.9838)、AUC(0,12)(P=0.9990)およびCmax(P=0.9110)との間には、有意な相関性は見られなかった。
【0207】
TOBI300mg投与後の体内摂取量は、TPI3×28mgカプセル投与後とTPI4×28mgカプセル投与後との体内摂取量の間にあった。その結果に基づくと、TPI4×28mgカプセル投与(総用量112mg)の場合が、TOBIでの全身体内摂取量に最も近いと考えられる。
【0208】
TPI投与亜群の解析から、年齢、以前のドライパウダー使用、性別および体重は、TPIにおけるトブラマイシンの体内摂取量に影響せず、トブラマイシンの薬物動態学的特徴はTPI亜群でほぼ同じであることが分かった。気管支拡張剤使用のTPI投与被験体(N=27)におけるトブラマイシンの体内摂取量は、気管支拡張剤を用いなかったTPI投与被験体(N=37)の場合と比較して平均で僅かに高かった。
【0209】
TPIおよびTOBI投与後の喀痰中のトブラマイシンの濃度は、平均30分で最高レベルに到達し、また平均半減期は0.8〜2.2時間の範囲にあることが推定された。
【0210】
薬物動態学的パラメータの変動性は、血清中の場合と比較して喀痰中の方が高かった。
【0211】
投薬時間は、TOBIでは平均約16分であった。これに対して2個のTPIカプセルの投与時間は、2×14mgおよび2×28mg投与では各々、平均1.7分および2.5分であった。またTPI4×14mg、3×28mgおよび4×28mgの場合の平均投与時間は各々、4.2分、4.5分および4.9分であった。したがって、TPIの投与時間は、第一にカプセル数が増えると増大し、第二に用量強度が上がると増大した。
【0212】
体内摂取量
90名の登録被験体のうちの86名について、以下に要約するように単回投与の治験を行った。
・TPI2×14mg、すなわち28mgまでの単回投与を受けた12名の被験体
・TPI4×14mg、すなわち56mgまでの単回投与を受けた13名の被験体
・TPI2×28mg、すなわち56mgまでの単回投与を受けた14名の被験体
・TPI3×28mg、すなわち84mgまでの単回投与を受けた15名の被験体
・TPI4×28mg、すなわち112mgまでの単回投与を受けた13名の被験体
・60mg/mLTOBI5mL、すなわち300mgまでの単回投与を受けた20名の被験体。
【0213】
有害事象の概要
治験薬の単回投与中または投与後における、治験の結果生じたAEの発生率に関しては、TOBI投与群の被験体の場合(6/20、30.0%)と比較してTPI投与群の被験体の方が高かった(40/66、60.6%)。TPI用量レベルが異なっても、いずれのAEについても被験体発生率は同様であった(TPI2×14mgでは45%、;4×14mgでは54%;2×28mgでは64%;3×28mgでは67%;4×28mgでは69%)が、試料サイズが小さすぎたため、TPI用量が増大したときにAE発生率が増大するという傾向があることは判断できなかった。治験で生じたAEの重篤度については、すべて軽度または中程度であった。
【0214】
TPI4×28mgの投与を受けた被験体の1名では、軽度の咳が認められ、投与後2日目まで喀痰の増加が続き、8日目に入院した。したがって、治験終了後の8日目におけるその被験体のAEについてはSAE(重度の有害事象)と判定した。しかし同一被験体で生じたSAEについては、いずれもTPI治験に関係しないと考えられた。TPI4×28mgの投与を受けた別の被験体1名に関しては、治験薬投与を行うと中程度で関連性が疑われる咳の悪化、味覚異常および流涙増加からなる重度ではないAEが認められため、投与を中断して最終的に投与中止とした。この被験体に関しては、治験の同意が撤回されたため、本試験から脱落した。4名の被験体(TPI4×14mg1名、4×28mg1名、3×28mg2名)では、TPI吸入中に重度ではないが咳から成るAEが認められたため、投薬の変更、中断または延期を行った。試験研究者の見解では、各々のAEについてはTPI治験と関連性が高いとされた。
【0215】
TPI投与群の被験体で最も多く見られたAEは、咳または咳の悪化(13/66=19.7%;治験開始前の基礎的症状として以前報告されたTPI投与被験体では12/13);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各々6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各々4.5%)であった。咳、咳の悪化および味覚異常の発生率は、TPIの用量が増すと僅かに増大したが、個々のAEの発生率に関しては、データに明確な傾向は認められなかった。これに対してTOBI投与の被験体のすべてでは、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化または味覚異常は全く見られなかった。しかしながら、TOBI投与の被験体のほとんど(17/20=85%)では、その通常療法の一部としてTOBIの投与が行われていた。
【0216】
試験研究者の見解では、咳および咳の悪化のほとんどのケース、味覚異常のすべてのケース、ならびに喀血および咽喉の炎症のほとんどのケースが、TPI治験と関連性があるかまたは高いとされた。これに対して胸部圧迫感、単純ヘルペス、好酸球数の増加、咽喉の渇き、咽頭炎、喀痰増加、および喀痰粘性上昇のそれぞれ単独のケースでは、TOBI治験に関係すると考えられた。
【0217】
治験の結果生じた有害事象:すべての原因
66名のTPI投与被験体のうちの40名、および20名のTOBI投与被験体のうちの6名では、治験中または治験後に治験に結果生じたAEが見られた。いずれのAEについてもその発生被験体%は、TPI用量レベル間で同等であった。治験により生じたAEのすべては、軽度または中程度のものであった。
【0218】
TPI投与被験体の最大数で見られたAEは、咳または咳の悪化(13/66=19.7%);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各4.5%)であった。
【0219】
TOBI投与の被験体は1名以外は、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化または味覚異常は見られなかった。
【0220】
治験に関係すると考えられ、治験の結果生じる有害事象
66名のTPI投与被験体のうちの24名(36.4%)、および20名のTOBI投与被験体のうちの2名(10%)では、それらケースで見られるAEが試験研究者の見解では治験と関連性があるかまたは高いものであった(表12)。試験研究者によると、咳および咳の悪化のほとんどのケース(10/66=15.2%)ならびに味覚異常のすべてのケース(16.7%)は、TPI治験と関連性があるかまたは高いとされた(表11)。喀血および咽喉の炎症(ともに4.5%)ならびに流涙増加(3.0%)についても、TPI治験に関係すると考えられた。これに対して、胸部圧迫感、単純ヘルペス、好酸球数増加、咽喉の渇き、咽頭炎、喀痰増加、および喀痰粘性上昇のそれぞれ単独のケースについては、TOBI治験に関係すると考えられた。
【0221】
ほかの重篤な有害事象(SAE)
TPI4×28mg投与の被験体のうちの1名では、投与後の2日目にSAE(中程度の咳および喀痰増加)が見られ、CFの肺疾患が悪化したため治験後の8日目に入院した。同一被験体で生じたそれらSAEについては、いずれもTPI治験に関係しないと考えられた。
【0222】
治験に関係する有害事象による試験からの被験体の脱落
TPI4×28mg投与被験体のうちの1名では、治験薬投与で中程度の咳の悪化、ならびに味覚異常および流涙増加が見られたため、投与を中断し、最終的に投与中止とし、その被験体を本試験から脱落させた。試験研究者の見解では、そのAEのいずれもTPI治験と関連性があるとされた。
【0223】
治験に関連した有害事象による投薬変更(dose intervention)
咳が見られた4名の被験体(TPI4×14mg被験体1名、4×28mg1名、3×28mg2名)については、投薬の変更、中断または延期を行った。試験研究者によると、そのAEはいずれもTPI治験と関連性があると考えられた。
【0224】
【表10】
死亡、ほかの重篤な有害事象、およびほかの明らかな有害事象に関する解析および考察
本試験では死亡例は報告されなかった。SAEについては、治験2日目に被験体のうちの1名にのみ中程度の咳および喀痰増加が報告され、その被験体は治験完了の8日目に入院したが、それらのAEは治験と関連性がないと考えられた。5名のTPI投与被験体では、TPI吸入の数分以内に中程度の咳または咳の悪化が生じ、そのうちの1名(味覚異常および流涙増加も併発)については、本試験から脱落させ、残りの4名については投薬を中断した。被験体のすべてで基礎的な呼吸器症状が報告され、またAEとしての咳も報告されたが、1名については基礎的症状としての咳が留意された。これらのAEについては、投薬の変更を最小限にするか、または変更しなくとも5〜35分以内に解消した。後者の4名の被験体には投薬を遅らせ、最初の中断から数分以内に投薬は無事に完了した。これらのAEは、嚢胞性線維症にしばしば伴う基礎的な気管支過敏症におそらく関係するものであったが、試験研究者の見解では、各AEは試験薬物の投与と関連性があるとされた。したがって、本試験では各用量のTPI、およびTOBI300mg/5mLの投与は良好な忍容性を示した。
【0225】
血液学
TOBIおよびTPI治験群では、試験前の基礎データから追跡調査時までの血液学的検査結果の平均値の変化には、いずれも注目するものはなかった。平均値の基底値からの変化については、TOBIおよびTPI治験群間で大きな差は見られず、いずれの血液学的検査においてもTPIの用量が上がっても平均値の増加パターンは見られなかった。
【0226】
TOBIおよびTPI治験群では、基底値から追跡調査時の値までの注目される増加、すなわち血液学的検査結果の高値または低値の発生頻度で増加は認められなかった。TOBI治験群とTPI治験群との間には大きな差は見られず、TPIの用量を上げても大きくはずれた血液学的検査結果の発生頻度の増加パターンは見られなかった。
【0227】
TOBI投与の被験体のうちの1名では、追跡調査時にAEとして記録された好酸球数の臨床上有意な変化が見られた。
【0228】
血清化学
TOBIおよびTPI治験群において、基底値から追跡調査時の値までの血清化学的検査結果の平均値の変化で注目されるものはなかった。いずれの血清化学的検査結果の平均値も、基底値からの変化に関してTOBI投与群とTPI投与群との間に大きな差は見られず、TPIの用量を上げても平均値の増加パターンは見られなかった。
【0229】
血清化学的検査結果において低値または高値の発生頻度に関する基底値から追跡調査時の値までの増加については、TOBIまたはTPI投与群で注目されるものはなかった。TOBI投与群とTPI投与群との間に大きな差は見られず、TPIの用量を上げても、大きくはずれた血清化学的検査結果の発生頻度の増加パターンは見られなかった。
【0230】
被験体の1名については、追跡調査時の血糖試験結果で臨床上有意な変化が見られたが、その結果はCFに関連した糖尿病に起因するものであり、AEとしては記録せず、TPI治験に起因しないものとした。
【0231】
浸漬式スティックによる蛋白尿検査
浸漬式スティックによる蛋白尿検査は定量的検査であるが、基底値から追跡調査時の値までの定量的変化は算出しなかった。TOBIまたはTPI投与群では浸漬式スティックによる尿蛋白質検査結果が2+またはそれ以上である頻度で、基底値から追跡調査時の値までの増加で注目されるものは見られなかった。
【0232】
TPI4×28mg投与の被験体のうち1名についてのみ、3+の陽性結果が注目された。
【0233】
個々の血液学的異常例
1名を除いて本試験では臨床上有意な血液学的検査結果は認められなかった。TOBI投与被験体No.12/405では、追跡調査時の好酸球数が9%という臨床上有意な結果(正常域0〜6%)が見られたため、AEとして記録し、それは試験研究者の見解ではTOBI治験と関連性があるとされた。その被験体の好酸球の基底値はもともと正常値の上限(5.9%)であったが、治験8日目にはその基底値から上昇した。この好酸球数9.0%という結果の臨床上の重要性に関しては、不確定である。
【0234】
血清化学
本試験においては1名を除き、臨床上有意な血清化学的結果は認められなかった。その1名の被験体(TPI3×28mg投与)では、追跡調査時の血糖値の結果が臨床上有意であったが、その結果はCFに関連した糖尿病に起因するものであった。
【0235】
浸漬式スティックによる尿蛋白検査
本試験では1例を除き、浸漬式スティックによる尿蛋白検査結果で臨床上有意なものは認められなかった。その1名の被験体(TPI4×28mg投与)では、治験後6日目に3+の陽性の結果となった。尚、その被験体では、スクリーニング時および投与当日の検査においては1+の陽性を示していた。この3+という陽性の尿蛋白検査結果については、治験2日目からのSAE(咳および喀痰の増加)と整合性があると治験コーディネーターが判断し、その被験体を入院させた。
【0236】
気管支痙攣および肺機能における急性疾患
TPI2×28mg単回吸入投与後、1例のみにおいて30分以内に起きたと考えられる無症候性の気管支痙攣(FEV1低下率20.9%)が見られた(表11)。尚、その所見は米国胸部学界(ATS)の診断基準によらずに行った肺活量測定で認められたものであった。ATS診断基準にしたがって検査を行えば、FEV1の低下率は少なくなる可能性があると考えられる(11%)。予想されるFEV1%の同様な無症候性の低下については、TOBI投与被験体でも認められた(19.1%の低下)。
【0237】
それ以外のTOBI投与被験体1名およびTPI投与被験体6名についても、予測されるFEV1の低下が10〜20%見られた(表11)。その6名のTPI投与被験体のうちの3名(TPI3×28mg投与の被験体No.02/507、ならびにTPI4×28mg投与の被験体No.01/415およびNo.08/406)では、投与後数分以内で咳または咳の悪化が見られた。
【0238】
【表11】
a予測FEV1%低下が10〜20%。
b予測FEV1%低下が20%以上。
【0239】
肺機能の定量的な変化
TOBIおよびTPI投与群において予測されたFEV1%、FVC%およびFEF25−75%の平均値の変化に関しては、スクリーニング時から投与1日前まで、ならびに投与前日から投与8日目の追跡調査時まで、比較的安定していた。また投与前から投与30分後までの予測されたFEV1、FVCまたはFEF25−75の%の平均値の変化に関しては、TOBI投与群とTPI投与群との間に明らかな差は見られず、異なる用量のTPI投与群間で用量に関連する差は見られなかった。
【0240】
生命徴候
本試験を通じての生命徴候またはその変化に関しては、TPI投与群とTOBI投与群との間に大きな差または整合する差は見られなかった。
【0241】
併用投薬
90名の被験体のうちの38名では、投与後に併用して投薬および医療を受けた。その併用の投薬および医療の頻度またはタイプによって、明らかな差は見られなかった。
【0242】
安全性の結論
治験中または治験後の、治験により生じたAEに関しては、TOBI投与の被験体の場合(30.0%)よりもTPI投与の被験体の場合(60.6%)の方が多く見られた。いずれのAEにおける被験体%も、各種TPI用量レベル間で同等(45〜69%)であった。これは試料サイズが小さすぎたため、いずれのAEも、その発生率がTPIの用量の増加で上昇する傾向があるか判定できなかったためである。
【0243】
TPI4×28mg投与の被験体の1名では、2種類のSAE(CFの肺疾患の悪化の指標となる中程度の咳および喀痰の増加)が見られたため、治験薬投与後8日目に入院させたが、それらのSAEはTPI治験には関係しないと考えられた。TPI4×28mg投与の別の1名の被験体では、中程度で、投薬と関連性がある咳の悪化、味覚異常および流涙増加が見られたため、投薬を中断して最終的に中止した。この被験体については、治験の同意が撤回され、本試験から脱落させた。4名の被験体(TPI4×14mg投与被験体1名、4×28mg被験体1名、および3×28mg被験体2名)では、TPIの吸入中に咳が見られたので、投薬の変更、中断または延期を行った。そのAEの各々に関しては、試験研究者の見解ではTPI治験と関連性があるとされた。しかしながら、この4例ではすべて、投薬を一時的に中断しただけであり、投薬は再開され、そつなく完了した。本試験の被験体の全員は、呼吸器に関する基礎的症状が報告され、各コホートごとに約90%に咳の報告が見られた。さらに中程度の咳の報告も、特に気道の炎症がすでに存在する場合に、ドライパウダー吸入器による最初の投与で予想されたことである。
【0244】
TPI投与被験体で見られた多くのAEは、咳または咳の悪化(13/66=19.7%);味覚異常(11名、16.7%);咽頭炎、喀血、および鼻漏(4名、各6.1%);喀痰増加、肺のクラックル音、流涙増加、上腹部痛、眩暈、頭痛(詳細不明)、および咽喉の炎症(3名、各4.5%)であった。咳、咳の悪化および味覚異常の発生率は、TPIの用量を上げると僅かに増加した。TOBI投与の被験体では1名以外は、いずれのAEも見られなかった。TOBI投与の被験体では咳、咳の悪化、または味覚異常が見られなかったが、TOBI投与群被験体の85%では日常的にTOBIが用いられていた。
【0245】
試験研究者によると、咳および咳の悪化のほとんどのケース、味覚異常のすべてのケース、ならびに喀血および咽喉の炎症のほとんどのケースは、TPI治験と関連性があるかまたは高いとされた。これに対して、胸部圧迫感、単純ヘルペス、好酸球数増加、咽喉の渇き、咽頭炎、喀痰増加および喀痰粘性上昇については、TOBI治験に関係すると考えられた。
【0246】
無症候性の気管支痙攣が見られ記録されたのは1例のみ(FEV1低下率20.9%)であって、TOBI投与被験体1例に見られた無症候性のFEV1の低下と同等(19.1%)であった。
【0247】
臨床検査結果における基底値からの留意される変化、TPI用量の増加に伴う増大パターン、低値または高値の検査結果の発生頻度の増大、ならびにTPIとTOBIとの明らかな差については、いずれも見られなかった。TOBI投与の被験体のうちの1名でのみ、好酸球数増加がAEとして記録された。
【0248】
考察および全体の結論
TPIの単回投与は、TOBI(登録商標)トブラマイシン吸入用溶液の投与の場合と比較して、トブラマイシンの薬物動態学的特徴は変わらないにもかかわらず、トブラマイシンの送達効率が高い結果となった。TOBI(登録商標)トブラマイシン吸入用溶液投与後の全身体内摂取量は、TPI4×28mgカプセル投与後に見られた量と極めて類似しており、相当する用量の計算値は115mgTPIであった。したがって、TPI28mgカプセル4個(総量112mg)で、300mgTOBI(登録商標)トブラマイシン吸入用溶液に相当する全身の体内摂取量が達成されると考えられる。
【0249】
TPIの最小用量での投与において、咳による中断(1例を除き、すべて一時的)が認められたが、TPI単回投与後の血清中トブラマイシンの薬物動態学的特性は変わらないと考えられた。
【0250】
TPI投与の被験体の約20%で咳が報告されたが、ほとんどのケースで軽度なものであり、時には短い投与中断を行うが、全般にすぐに落ち着いた。咳に関しては、ドライパウダー吸入療法で予想されるAEであり、大部分の被験体の基底症状でもある。AEの発生頻度については、TOBI治験群の被験体よりも、TPI治験群の被検者の方が高かったが、一方でほとんどのコホートの被験体の大部分では、日常的にTOBI(登録商標)トブラマイシン吸入用溶液が投薬されていた。
【0251】
若年の被験体1名では、通常の投薬(長時間作用型気管支拡張剤から成る)を忘れ、治験薬投与後に中程度の咳が見られたため、試験から脱落させた。通常行ってきた気管支保護療法が行われないと、この生体反応が起こると思われる。
【0252】
総投与量28〜112mgにおけるTPIの単回投与は、本試験でCF被験体に良好な忍容性を示した。112mgのTPI粉末における全身性体内摂取量は、TOBI(登録商標)トブラマイシン吸入用溶液の場合に近似している。
【0253】
本試験では、簡単な吸入用デバイスとともに本発明の方法を用いて吸入投与を行うと、TOBI(登録商標)トブラマイシン吸入用溶液の標準的用量での噴霧化による投与よりも著しく速く、効率的であることが示された。TPI吸入後に見られた咳増加および咽喉の炎症の副作用については、予想されたものであり、各用量に含まれる粉末量に依存した。しかしながら、ほとんどのケースで副作用は軽度であり、被験体の感想はTPIの投与がすばやく、簡単とのことであった。TOBI(登録商標)トブラマイシン吸入用溶液投与群では、本試験の前に85%で日常的にTOBIが使用されていたこともあって、咳および味の悪さの訴えがなかったといえる。これらの被験体では、トブラマイシンの湿式エアゾル剤形での効果に順化していた可能性がある。
【0254】
したがって、ドライパウダーでのトブラマイシンの単回投与では、TOBI(登録商標)トブラマイシン吸入用溶液の場合と比較して、トブラマイシンの薬物動態学的特徴が同様であるにもかかわらず、トブラマイシンの送達効率が高い結果となった。TOBI(登録商標)トブラマイシン吸入用溶液投与後の全身の体内摂取量は、TPIの4×28mgカプセルの投与後に見られる量にきわめて近く、計算上の相当用量は115mgTPIであった。したがって、TPI28mgカプセル4個(総量112mg)の全身の体内摂取量は、300mgTOBI(登録商標)トブラマイシン吸入用溶液の場合に相当すると考えられる。
【0255】
本発明の好ましい実施形態については、すでに説明および記載済みであるが、本発明の意図および視野からはずれなければ、種々の変更も可能であることが分かるであろう。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

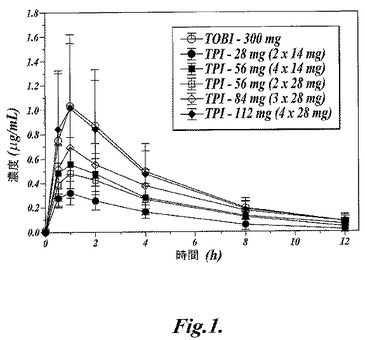
【図2】
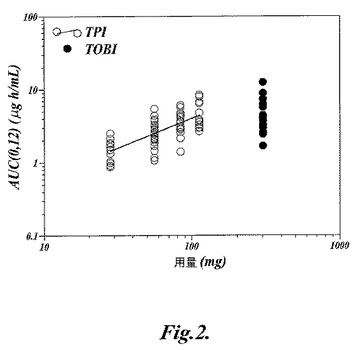
【図3】

【図4】

【図2】
【図3】
【図4】
【公開番号】特開2013−56947(P2013−56947A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−284937(P2012−284937)
【出願日】平成24年12月27日(2012.12.27)
【分割の表示】特願2012−36405(P2012−36405)の分割
【原出願日】平成17年6月20日(2005.6.20)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−284937(P2012−284937)
【出願日】平成24年12月27日(2012.12.27)
【分割の表示】特願2012−36405(P2012−36405)の分割
【原出願日】平成17年6月20日(2005.6.20)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
[ Back to top ]