
微水溶性活性物質の経口製薬学的組成物
本発明は、ベンズアゼピン−1−酢酸誘導体の経口組成物であって、a)調剤の合計重量の10〜65%の間の量の該活性化合物、b)少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物を含んでなり、c)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなる、組成物に関する。本発明はさらに、アルカリ性化合物として特定の粒子寸法および/または表面積を有する炭酸ナトリウムを含んでなる以上で定義された経口組成物にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、一般式
【0002】
【化1】

【0003】
[式中、
R1は
(1)場合により(C1−C6)アルコキシにより置換されていてもよい、(C1−C6
)アルコキシ(C1−C6)アルキル、
(2)フェニル基が場合により(C1−C6)アルキル、(C1−C6)アルコキシまた
はハロゲンで置換されていてもよい、フェニル−(C1−C6)−アルキルおよびフェ
ニルオキシ−(C1−C6)−アルキル、並びに
(3)ナフチル−(C1−C6)−アルキル
から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口調剤に関する。
【背景技術】
【0004】
これらの化合物並びにそれらの塩類および生不安定性エステル類は本発明の保護範囲下に入りそして有効なECE/NEP阻害剤でありそしてWaldeck他の特許文献1および特許文献2に記載されている。本発明で使用されるベンズアゼピン−N−酢酸化合物は特許文献2、特許文献3、特許文献4および特許文献5から既知であり、そして該特許文献1および特許文献2に記載された方法により製造できる。これらの特許は該化合物およびそれらの生理学的に許容可能な塩類自体並びに心不全における化合物の使用に関する。特許文献6はこれらの化合物の特定の塩類、特にカルシウム塩に関する。特許文献3、特許文献4および特許文献5は、胃腸管流の改良における、高血圧症の処置における、そしてそれぞれアドリアマイシン(adriamycin)および匹敵する抗癌薬により誘発される心臓損傷の処置および予防における、上記化合物の使用に関する。
【0005】
上記の式(I)の化合物を包含する種々の活性化合物は水中で非常に劣悪な溶解度を有する。これらの化合物を身体に投与する時には、それらは消化液中の劣悪な溶解度のために劣悪なバイオ−アベイラビリティーをしばしば有する。この問題を解決するために、数種の方法、例えば微細化、シクロデキストリン類中への包含、不活性な水溶性担体の使用、固体分散液(特許文献7)もしくは固体溶液または活性化合物のナノ結晶もしくは非晶質形態の使用が開発された。
【0006】
特許文献8は、従来法で調合された形態の該活性化合物と比べて増加したバイオ−アベイラビリティーを有する式(I)の経口固体溶液調剤を記載している。この調剤は良好なバイオアベイラビリティー性質を有するが、それを溶融−押し出し技術によりカプセル剤または錠剤のいずれかに調合しなければならないという幾つかの制限をもたらす溶融混合物を介してそれが製造されるという欠点を有する。さらに、比較的高い薬用量にとっては調剤の寸法が大きすぎるであろう。
【0007】
特許文献9(予備−公開されていない)は、調剤の合計重量の60%までの量の活性化合物、少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物、0.1〜10%w/wの間の1種もしくはそれ以上の界面活性剤および場合により調剤の合計重量の1%〜45%の間の量の助剤物質を含んでなる式(I)の化合物の経口即時放出調剤を記載している。特にドキュセート(docusate)ナトリウムが界面活性剤として使用される時には活性化合物の良好なバイオアベイラビリティーが得られる。
【特許文献1】米国特許第5,677,297号明細書
【特許文献2】欧州特許第0733642号明細書
【特許文献3】欧州特許第0830863号明細書
【特許文献4】国際公開第00/48601号パンフレット
【特許文献5】国際公開第01/03699号パンフレット
【特許文献6】国際公開第03/059939号パンフレット
【特許文献7】国際公開第00/00179号パンフレット
【特許文献8】国際公開第03/068266号パンフレット
【特許文献9】国際公開第2006/067150号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0008】
商業的使用にとって不充分に安定でありそして妥当な寸法を有する高い含有量の活性化合物を有する調剤を製造するためにも使用できる従来法で調合される形態の活性化合物と比べて、バイオ−アベイラビリティーにおける有意な増加を有する以上で定義された式Iの化合物用の代替経口調剤を提供することが本発明の目的である。通常の調合工程および装置を用いて製造できるので大資本が必要でない調剤を提供することが本発明の別の目的である。
【課題を解決するための手段】
【0009】
この目的は、本発明に従い、一般式
【0010】
【化2】
【0011】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ
(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフチル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口調剤であって、
a)調剤の合計重量の10%〜80%の間の量の該活性化合物、
b)少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物を含ん
でなり、
c)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなり、
但し調剤が界面活性剤を含有しない調剤により達成することができる。
【0012】
Mは好ましくはLi+、Ca2+、Mg2+およびZn2+よりなる群から選択され、そして最も好ましくはCa2+である。(C1−C6)−アルキルは1〜6個の間の炭素原子よりなる直鎖状もしくは分枝鎖状のアルキル基として定義される。(C1−C6)−アルコキシは1〜6個の間の炭素原子よりなる直鎖状もしくは分枝鎖状のアルコキシ基として定義される。R1は好ましくはフェニルエチルであり、R2およびR3は好ましくは水素でありそしてR4は好ましくはエチルである。
【0013】
本発明の枠内では、生不安定性エステル類を生成しうる適当なR4基は低級アルキル基、場合によりフェニル環中で2個の隣接炭素原子に結合された低級アルキルもしくは低級アルキレン鎖により置換されていてもよいフェニルもしくはフェニル−低級−アルキル基、場合によりジオキサン環中で低級アルキルにより置換されていてもよいジオキソラニルメチル基、または場合によりオキシメチル基上で低級アルキルにより置換されていてもよいC2−C6−アルカノイルオキシメチル基を包含する。生不安定性エステルを生成する基R4が低級アルキルである場合には、これは好ましくは炭素数1〜4、好ましくは2の非分枝鎖状アルキル基でありうる。生不安定性エステルを生成する基が場合により置換されていてもよいフェニル−低級アルキルである場合には、そのアルキレン鎖は1〜3個の、好ましくは1個の炭素原子を含有しうる。フェニル環が低級アルキレン鎖により置換されている場合には、これは3〜4個の、好ましくは3個の炭素原子を含有しうる。特に適するフェニル−含有置換基R4はフェニル、ベンジルまたはインダニルである。R4が場合により置換されていてもよいアルカノイルオキシメチル基である場合には、そのアルカノイルオキシ基は2〜6個の、好ましくは3〜5個の炭素原子を含有することができ、好ましくは分岐しており、そして、例えば、ピバロイルオキシメチル基(tert−ブチルカルボニルオキシメチル基)でありうる。
【0014】
好ましい化合物は酸である3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]シクロペンチル]カルボニル]アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンズアゼピン−1−酢酸のカルシウム塩である。最も好ましい化合物は、ダグルトリル(daglutril)カルシウムまたはSLV 306カルシウムとしても知られるその3S,2’R形態での該化合物である。この化合物は化合物S−Caと称され、ダグルトリルまたはSLV 306としても知られる対応する酸(3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]−シクロペンチル]−カルボニルアミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンズアゼピン−1−酢酸)は化合物S−Hと称される。
【0015】
式(I)の活性化合物は通常は約10〜80重量%の間の量で、より好ましくは約15
〜75重量%の間の量で、さらにより好ましくは約20〜65重量%の間の量で、そして最も好ましくは約45〜65重量%の間の量で使用される。活性化合物は微細化された形態で使用されるかまたは場合により使用されうる。
【0016】
本発明の枠内で使用されるある種の用語の理解を助けるために以下の定義が提供される。
【0017】
商業的使用にとって充分な安定性は、周囲条件における少なくとも1年間、好ましくは少なくとも2年間、さらにより好ましくは少なくとも3年間、そして最も好ましくは少なくとも5年間の貯蔵期間中の許容可能な化学的および物理的安定性を意味する。許容可能な化学的安定性は、貯蔵期間中の活性化合物の5%より多くない好ましくは3%より多くない、そして最も好ましくは1%より多くない活性物質の崩壊を意味する。許容可能な物理的安定性は、外観における有意な変化なし、貯蔵期間の終了時に脱ブリスター化(deblistering)中の破壊なし、および20%より多くない崩壊時間の変化を意味する。用語「微細化された」は、容量基準で粒子の95%より多くが75ミクロンより小さい粒子寸法をさす。
【0018】
界面活性剤は、分子を溶液中で集積させてミセルを形成させうる良く規定された極性および非−極性領域を有する分子として定義される。極性領域の性質により、界面活性剤は非−イオン性、アニオン性、カチオン性および両性でありうる。非−イオン性の親水性界面活性剤の例はポリオキシエチレンソルビタンエステル類、クレモフォア類(cremophores)、およびポロキサマー類である。アニオン性界面活性剤の例はナトリウムラウリルサルコシネート(sodium lauryl sarcosinate)、ドキュセート(docusate)、並びに製薬学的に許容可能なドキュセート塩類、例えばドキュセートカルシウム、ドキュセートナトリウムおよびドキュセートカリウムである。
【0019】
アルカリ性化合物は無機および有機アルカリ性化合物、例えば炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、クエン酸ナトリウム、トリス緩衝液、トリエタノールアミン、アルカリ性水酸化物、例えば水酸化ナトリウム、水酸化カリウムまたは水酸化マグネシウム、アルカリ性燐酸塩、例えば燐酸水素二カリウム、並びにメグルミンよりなる群から選択される。これらのアルカリ性化合物の混合物も使用することができる。好ましいアルカリ性化合物は炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウムおよび炭酸カルシウムである。さらにより好ましいアルカリ性化合物は炭酸ナトリウムである。
【0020】
好ましくは、アルカリ系は未コーティング組成物の10%w/wより多い、より好ましくは20%w/wより多い量で存在し、または未コーティング組成物の25%、30%w/w、40%w/w、45%w/w、50%w/w、55%w/wもしくは60%w/wより多い量で存在する。用語「未コーティング調剤」は1種もしくは複数の場合により存在するコーティング物質の適用前の調剤を意味する。
【0021】
炭酸塩が使用される場合には、それは好ましくは未コーティング調剤の合計重量の25%の量で使用され、或いはそれは未コーティング調剤は少なくとも45%w/wの量で存在し、または未コーティング調剤の50%w/w、55%w/wもしくは60%w/wより多い量で存在する。
【0022】
さらに好ましい態様では、本発明は一般式
【0023】
【化3】
【0024】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフフル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wの、粒子の97%より多くが500μmより小さく、粒子の
40%より多くが160μmより小さくそして粒子の10%より多くが63μmより
小さい粒子寸法分布を有する炭酸ナトリウムを含んでなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなる組成物
に関する。
【0025】
この好ましい態様では、アルカリ系は通常の炭酸ナトリウムより小さい粒子寸法を有する炭酸ナトリウムを含んでなり、それは粒子の多くとも25%が160μmより小さい粒子寸法分布(ふるい分析により測定されそして重量基準である)を有する。この炭酸ナトリウムは通常の炭酸ナトリウムより高い比表面積を有し、それは約0.2m2/gの比表面積(標準的なBET面積測定に従い測定される)を有する。以上で示されたように、好ましい態様で使用される炭酸ナトリウムは粒子の97%より多くが500μmより小さい、粒子の40%より多くが160μmより小さいそして粒子の10%より多くが63μmより小さい粒子寸法分布(ふるい分析により測定されそして重量基準である)を有する。さらにより好ましい態様では、粒子の98%より多くは500μmより小さく、粒子の約60%より多くは160μmより小さくそして粒子の30%より多くは63μmより小さい。比表面積は好ましくは1m2/gより高く、そしてより好ましくは1.5m2/gより高い。最も好ましい炭酸ナトリウムはソルベイ・SA(Solvay SA)によりソーダ・アッシュ(Soda Ash)IPHとして販売されている特定の炭酸ナトリウムである。このタイプの炭酸ナトリウムでは、典型的には粒子の99.8%が500μmより小さく、粒子の80%が160μmより小さくそして粒子の40%が63μmより小さく、そしてこのタイプの炭酸ナトリウムは2m2/gの比表面積を有する。
【0026】
驚くべきことに、本発明の発明者はアルカリ性化合物を調剤中で、単独でまたは混合して、使用すると、組成物中に界面活性剤を含まなくても、ゲルを酸性胃液の中に溶解させる際の難点の発生を防止し、それによりインビボ溶解度における改良並びにバイオアベイ
ラビリティーにおける改良を指示する二相溶解モデル(実施例1a参照)におけるインビトロ溶解中に特に証明されているようなSLV−306の溶解度を高めることを見出した。特に(ソーダ・アッシュIPH)の如き100μmの平均粒子寸法および2m2/gの比表面積を有する炭酸ナトリウムを用いると、良好なインビボ溶解度および良好なバイオアベイラビリティーを有する調剤が得られる。さらに、組成物は貯蔵時に良好な安定性を有することが予測される。
【0027】
特定の粒子寸法および比表面積を有する上記の炭酸塩が使用される場合には、それは好ましくは未コーティング調剤の合計重量の少なくとも15%の量で、より好ましくは少なくとも18%の量で、さらにより好ましくは少なくとも20%の量で使用され、またはそれは未コーティングの25%w/w、30%w/w、40%w/w、50%w/wもしくは60%w/wより多い量で存在する。
【0028】
以上で示された通りの炭酸水素塩および炭酸塩のような特定の固体アルカリ性化合物は発泡性組成物中でしばしば固体の酸性化合物(例えばクエン酸、酒石酸、アジピン酸、フマル酸、琥珀酸、アスコルビン酸、ニコチン酸、サッカリン、アスピリン、リンゴ酸、燐酸二水素ナトリウム、ピロ燐酸二水素二ナトリウム、クエン酸二水素ナトリウムおよびクエン酸水素二ナトリウム)と組み合わせて使用される。本発明では、組成物は好ましくは酸性化合物を含有しない。
【0029】
調剤は場合により補助物質を調剤の合計重量の45%までのそして好ましくは調剤の合計重量の1%〜45%の間の量で含んでなる。これらの補助物質の例は以下のものを包含するが、それらに限定されない:
a)アラビアゴム、アルギン酸およびその塩類、セルロース誘導体、メチルセルロース、
ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ポリエチレングリ
コール、ゴム類、多糖酸類、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリ
ビニルピロリドン、ポリビニルピロリドン/酢酸ビニル共重合体、ポリメタクリレー
ト類、ヒドロキシプロピルメチルセルロース、澱粉、予備ゼラチン化された澱粉、エ
チルセルロース、トラガカント、デキストリン、微結晶性セルロース、スクロース、
またはグルコースなどを包含するがそれらに限定されない結合剤。
b)澱粉類、予備ゼラチン化されたコーンスターチ、予備ゼラチン化された澱粉、セル
ロース、架橋−結合されたカルボキシメチルセルロース、クロスポビドン、架橋−結
合されたポリビニルピロリドン、アルギン酸カルシウムもしくはナトリウム複合体、
クレイ類、アルギネート類、ゴム類、またはグリコール酸ナトリウム澱粉、および錠
剤製造において使用されるいずれかの崩壊剤を包含するがそれらに限定されない崩壊
剤。
c)ラクトース、炭酸カルシウム、燐酸カルシウム、二塩基性燐酸カルシウム、硫酸カル
シウム、微結晶性セルロース、セルロース粉末、デキストロース、デキシトラート
類、デキストラン、澱粉類、予備ゼラチン化された澱粉、スクロース、キシリトー
ル、ラクチトール、マンニトール、ソルビトールなどを包含するがそれらに限定され
ない充填剤。
d)いずれかの酸化防止剤、緩衝液、または酸類などを包含するがそれらに限定されない
安定剤。
e)ステアリン酸マグネシウム、水酸化カルシウム、タルク、コロイド状二酸化珪素、ス
テアリルフマル酸ナトリウム、水素化された植物油、ステアリン酸、ベヘン酸グリセ
リル、ステアリン酸マグネシウム、カルシウムおよびナトリウム、ワックス類、ステ
アロウェット(Stearowet)、ホウ酸、安息香酸ナトリウム、酢酸ナトリウ
ム、DL−ロイシン、ポリエチレングリコール類、オレイン酸ナトリウム、またはラ
ウリル硫酸ナトリウムなどを包含するがそれらに限定されない潤滑剤。
f)オレイン酸、モノステアリン酸グリセリル、モノオレイン酸ソルビタン、モノラウリ
ン酸ソルビタン、オレイン酸トリエタノールアミン、モノオレイン酸ポリオキシエチ
レンソルビタン、モノラウリン酸ポリオキシエチレンソルビタン、オレイン酸ナトリ
ウム、またはラウリル硫酸ナトリウムなどを包含するがそれらに限定されない湿潤剤
。
g)ラクトース、澱粉、マンニトール、ソルビトール、デキストロース、微結晶性セルロ
ース、二塩基性燐酸カルシウム、スクロースをベースとした希釈剤、菓子用糖、一塩
基性硫酸カルシウム一水和物、硫酸カルシウム二水和物、乳酸カルシウム三水和物、
デキストレート類、イノシトール、加水分解された穀類固体、アミロース、粉末状セ
ルロース、炭酸カルシウム、グリシン、またはベントナイトなどの如き希釈剤。
h)コロイド状シリカ、タルク、コーンスターチ、DL−ロイシン、ラウリル硫酸ナトリ
ウム、およびステアリン酸マグネシウム、カルシウムまたはナトリウムなどを包含す
るがそれらに限定されない抗−付着剤または滑り剤。
i)アラビアゴム、ゼラチン、コロイド状二酸化珪素、グリセロ燐酸カルシウム、乳酸カ
ルシウム、マルトデキストリン、グリセリン、珪酸マグネシウム、カゼインナトリウ
ム、大豆レシチン、塩化ナトリウム、燐酸三カルシウム、燐酸二カリウム、ナトリウ
ムステアロイルラクチレート、カラゲナン、モノグリセリド、ジグリセリド、または
予備ゼラチン化された澱粉などを包含するがそれらに限定されない製薬学的に相容性
の担体。
【0030】
調剤が上記の通りの特定の粒子寸法分布および/または表面積を有する炭酸ナトリウムを少なくとも10%w/wを含有する好ましい態様の場合には、調剤は界面活性剤を補助物質として含有することもできる。
【0031】
最終的な調剤は好ましくは粒剤、圧縮錠剤、またはカプセル剤の形態である。
【0032】
上記の調剤は普遍的な調合工程および装置を用いて製造することができる。従って、以下の段階:
a)式Iの活性化合物をアルカリ性化合物またはアルカリ性化合物の混合物とそして場合
により1種もしくはそれ以上の補助物質と混合し、
b)混合物を圧縮し、
c)該圧縮から得られた粒子を粉砕しそしてふるいにかけ、そして場合により該ふるいに
かけられた粒子を1種もしくはそれ以上の補助物質と混合し、そして
d)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる、上記のような調剤の製造方法を提供することが本発明の別の面である。
【0033】
本発明の別の態様では、調剤は以下の段階:
a)活性化合物を1種もしくはそれ以上の補助物質と混合し、
b)有機溶媒を用いて該混合物を造粒し、
c)有機溶媒を除去して粒子を得、
d)粒子を粉砕しそしてふるいにかけ、そしてふるいにかけられた粒子を補助物質の残り
の部分と混合し、
e)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる有機造粒方法で製造される。
【0034】
有機造粒方法では、数種の有機溶媒を使用することができる。例は、メチルt−ブチルエーテル(MTBE)、ジクロロメタンおよび酢酸エチルである。使用される好ましい溶媒は酢酸エチルである。
【0035】
本発明の調剤が錠剤の形態で提供される時には、これらの錠剤は5分〜90分の間の崩壊時間を有する。好ましくは、崩壊時間は60分以内でありそして最も好ましくはそれらは45分以内である。短い崩壊時間を有する調剤は、ソーダ・アッシュIPH中で入手可能な多孔性炭酸ナトリウムを使用することにより、製造することができる。
【0036】
種々の追加段階、例えば乾燥、破壊、ふるいかけ、混合および包装、もこの方法の一部であることができるが、これらの段階は本発明に従う調剤を得る際の必須特徴ではない。
【0037】
別の面で、本発明は好ましい放出特徴を有する組成物を提供する。従って、本発明は組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%の溶解度を有する上記の経口製薬学的組成物にも関する。5分後の溶解は好ましくは少なくとも55%そしてさらにより好ましくは60%である。15分後に、溶解は少なくとも65%、好ましくは少なくとも70%そしてより好ましくは75%である。30分後に、溶解は少なくとも75%、好ましくは少なくとも80%そしてより好ましくは少なくとも85%である。
【0038】
2.0のpHにおいて、本発明に従う製薬学的組成物は活性化合物を有意に放出しない。5%より少ない活性化合物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH2.0において測定して30分以内に放出される。好ましくは2%より少ないそして好ましくは1%より少ない量が放出される。
【0039】
以下の実施例は本発明をより詳細にさらに説明することが意図され、そしてそのためにこれらの実施例は本発明の範囲をいずれかの方法で制限するものとは考えられない。
【実施例】
【0040】
実施例1.物質、装置および方法
【0041】
物質。
S−Caは、国際公開第03/059939号パンフレットの実施例2および3に示された指示に従い、欧州特許第0733642号明細書の実施例2に従い製造された酸で出発して製造された。
【0042】
ソーダ・アッシュIPH(ソーダ、多孔性または多孔性ソーダとも称する)はベルギー、ブリュッセルのソルベイ・SAから得られた。
【0043】
全ての他の補助物質は容易に商業的に入手可能である。
【0044】
装置
ローラー圧縮用には、フィッツミル(Fitzmill)L1Aが装着されたフィッツパトリック(Fitzpatrick)タイプIR200ローラー圧縮機が使用された。
【0045】
ローラー圧縮の設定は、
回転速度水平供給スクリュー(HFS、rpm)
回転速度垂直供給スクリュー(VFS、rpm)
回転速度ローラー(N1、rpm)
間隙(d、mm)
圧縮力(F、KN/cm)
であった。
【0046】
フィッツミルの設定は、
回転速度ハンマーナイフ(N2、rpm)
スクリーン寸法(mm)
であった。
【0047】
方法。
a)二相インビトロ溶解方法の記述。
二相溶解はUSP装置2配置を用いて行われた。パドル速度は50rpmでありそして容器(および溶解媒体)の温度はバンケル(Vankel)VK7010を用いて37.0℃に保たれた。
【0048】
調剤の溶解は500mlの水中0.1M塩酸(500mlの4.2mlの濃塩酸(HCl))中で開始された(相1)。0、5、15、および30分後に、試料を採取した。30分後に、500mlの1M燐酸塩緩衝液(1000mlの水中の32.4グラムの燐酸二水素塩NaH2PO4および124.8グラムの燐酸水素二ナトリウム(Na2HPO4)を相1に加えた。燐酸水素塩緩衝液の添加が溶解媒体のpHを相1におけるpH1から相2におけるpH6.8に変えた。溶解試験中に、両方の相のpHは未変化のままであった。試料を35、45、および60分後に採取した。
【0049】
全ての試料をポール・ジマーク・アクロディスク(Pall Zymark Acrodisc)PSF、GxF/GHP、0.45μmまたはミリポア・ミレックス(Millipore Millex)−FH(疎水性PTFE 0.45μm)フィルターを通して濾過した。
【0050】
濾過された試料中に溶解したダグルトリルの量を外部基準を用いる240nmにおけるオフ−ラインUV測定により分析した。
【0051】
化合物SLV306のカルシウム塩(S−Ca)を用いるこれまでの比較試験では、この二相インビトロ溶解方法はインビボ結果と良好な相関性を有した。
【0052】
b)多孔性炭酸ナトリウムを同定する方法の記述。
多孔性炭酸ナトリウムの粒子寸法分布を機械的システムにより測定した。約70gの生成物の試料を重量測定しそして0.5、0.25、0.16、0.125、0.1、0.063、および0.045mmのスクリーンを有するふるいを含有する移行機械(ふるいのセットへの水平運動および垂直反射運動の組み合わせを動かしうる自動装置、例えばROTAPまたはRETSCHシフト機)の上方ふるいの中に入れた。ふるいかけ工程は約15分間にわたり行われた。各ふるいの内容物を重量測定しそして500μmより小さい粒子寸法を有する粒子の質量、160μmより小さい粒子寸法を有する粒子の質量、および63μmより小さい粒子寸法を有する粒子の質量を計算した。多孔性炭酸ナトリウムの比表面積を標準的BET面積方法に従い測定した。
【0053】
c)他の物理的方法の記述。
粉末流を径8mmの出口を有する黄銅漏斗中で測定した。粉末流は秒/100グラムで表示された。
− ローラー−圧縮された物質の粒子寸法分布は0.25mm、0.50mm、0.71mm、0.85mm、1.0mm、および2.0mmスクリーンを用いる手動移行から得られた。<0.25mmの量(%)、>1.0mmの量(%)およびd(50%)が計算された。
【0054】
混合された粒子のかさおよび叩き密度を以下の通りにして測定した:
− 100〜150グラムの量の粒状物を造粒シリンダー中に充填した。
− 占有された量を測定した。
− 1200叩き後に、占有された量を再び測定した。カル(Carr)−指数をかさおよび叩き密度から計算した。
【0055】
5個の錠剤をシュレウニガー(Schleuniger)硬度試験機中で粉砕すること
により錠剤の粉砕強度を測定した。平均値が報告された。
【0056】
Erwekaもろさ試験機を用いて20個の錠剤についてもろさを測定した。試験条件は40rpmにおいて10分であった。もろさは%錠剤重量損失で表した。
【0057】
溶解媒体として水を用いて、1個の錠剤上で崩壊を試験した。
【0058】
実施例2:S−Caの従来法で調合されたコーティング錠剤の製造。
【表1】
【0059】
工程:
i)S−Caを圧縮しそして1.0mmふるい中に通した。
ii)段階(i)の物質を微結晶性セルロースPH301、架橋−結合されたポリビニル
ピロリドンおよびステアリル蟻酸ナトリウムと混合して均一混合物を得た。
iii)段階(ii)の物質を錠剤圧縮機械を用いて圧縮した。
iv)段階(iii)からの錠剤を適当なコーティング装置中でコーティングした。
【0060】
実施例3:ソーダ・アッシュIPHを含有するS−Caの未コーティング錠剤の製造
【表2】
【0061】
工程:
i)S−Ca、ソーダ・アッシュIPH、ステアリン酸マグネシウムおよびグリコール酸
ナトリウム澱粉(プリモジェル)を#40メッシュふるいを通して移した。
ii)以上で移されたS−Ca、ソーダ・アッシュIPH(内部として示される)および
ステアリン酸マグネシウムの一部(内部として示される)および場合によりグリコ
ール酸ナトリウム澱粉(プリモジェル(R))(内部として示される)および/ま
たはラクトースを混合して均一混合物を得た。
iii)段階(ii)の物質をローラー圧縮機で指示された設定で混合した。
iv)混合された物質を粉砕しそして2.5mmのスクリーン寸法上でふるいにかけた。v)粉末流および粒子寸法分布を測定した。
【0062】
【表3】
【0063】
工程:
vi)段階(iv)の物質を残りの量のステアリン酸マグネシウム(外部)、グリコール
酸ナトリウム澱粉(プリモジェル(R))(外部)、ソーダ・アッシュIPH(外
部)または微結晶性セルロース(外部)と混合した。
vii)かさおよび叩き密度を測定しそしてカル−指数を計算した。
viii)段階(vi)の物質を錠剤圧縮機械を用いて指示された圧縮力で圧縮した。
ix)脆さ、粉砕強度、および崩壊時間を測定した。
【0064】
実施例4.ソーダ・アッシュIPHを有するSLV306調剤および従来法で調合された錠剤に関する比較溶解試験
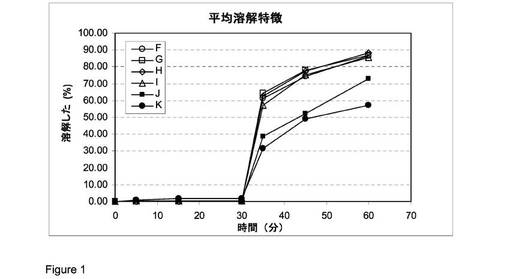
実施例1(方法a)に記載された方法に従う比較溶解試験を従来法で調合された錠剤(錠剤K、実施例2に記載された通りにして製造された)の1つのバッチおよび実施例3に従い製造されたSLV−306(S−Ca)のカルシウム塩の5つの600/965バッチ(表中のバッチ指示を参照のこと)(9kNで圧縮された錠剤F、12kNで圧縮された錠剤G、6kNで圧縮された錠剤H、11kNで圧縮された錠剤Iおよび10kNで圧縮された錠剤J)に対して行った。
【0065】
これらの調剤の放出特徴を以下の表に示しそして図1に示す(○=調剤F;□=調剤G;◇=調剤H;Δ=調剤I;■=調剤J;●=調剤K)
【0066】
【表4】
【0067】
この試験から、妥当な寸法および好ましい放出特徴を有する高い薬品負荷のあるS−Caの調剤が製造されたことが結論づけられた。
【図面の簡単な説明】
【0068】
【図1】図1は調剤の放出特徴を示す。
【技術分野】
【0001】
本発明は、一般式
【0002】
【化1】
【0003】
[式中、
R1は
(1)場合により(C1−C6)アルコキシにより置換されていてもよい、(C1−C6
)アルコキシ(C1−C6)アルキル、
(2)フェニル基が場合により(C1−C6)アルキル、(C1−C6)アルコキシまた
はハロゲンで置換されていてもよい、フェニル−(C1−C6)−アルキルおよびフェ
ニルオキシ−(C1−C6)−アルキル、並びに
(3)ナフチル−(C1−C6)−アルキル
から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口調剤に関する。
【背景技術】
【0004】
これらの化合物並びにそれらの塩類および生不安定性エステル類は本発明の保護範囲下に入りそして有効なECE/NEP阻害剤でありそしてWaldeck他の特許文献1および特許文献2に記載されている。本発明で使用されるベンズアゼピン−N−酢酸化合物は特許文献2、特許文献3、特許文献4および特許文献5から既知であり、そして該特許文献1および特許文献2に記載された方法により製造できる。これらの特許は該化合物およびそれらの生理学的に許容可能な塩類自体並びに心不全における化合物の使用に関する。特許文献6はこれらの化合物の特定の塩類、特にカルシウム塩に関する。特許文献3、特許文献4および特許文献5は、胃腸管流の改良における、高血圧症の処置における、そしてそれぞれアドリアマイシン(adriamycin)および匹敵する抗癌薬により誘発される心臓損傷の処置および予防における、上記化合物の使用に関する。
【0005】
上記の式(I)の化合物を包含する種々の活性化合物は水中で非常に劣悪な溶解度を有する。これらの化合物を身体に投与する時には、それらは消化液中の劣悪な溶解度のために劣悪なバイオ−アベイラビリティーをしばしば有する。この問題を解決するために、数種の方法、例えば微細化、シクロデキストリン類中への包含、不活性な水溶性担体の使用、固体分散液(特許文献7)もしくは固体溶液または活性化合物のナノ結晶もしくは非晶質形態の使用が開発された。
【0006】
特許文献8は、従来法で調合された形態の該活性化合物と比べて増加したバイオ−アベイラビリティーを有する式(I)の経口固体溶液調剤を記載している。この調剤は良好なバイオアベイラビリティー性質を有するが、それを溶融−押し出し技術によりカプセル剤または錠剤のいずれかに調合しなければならないという幾つかの制限をもたらす溶融混合物を介してそれが製造されるという欠点を有する。さらに、比較的高い薬用量にとっては調剤の寸法が大きすぎるであろう。
【0007】
特許文献9(予備−公開されていない)は、調剤の合計重量の60%までの量の活性化合物、少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物、0.1〜10%w/wの間の1種もしくはそれ以上の界面活性剤および場合により調剤の合計重量の1%〜45%の間の量の助剤物質を含んでなる式(I)の化合物の経口即時放出調剤を記載している。特にドキュセート(docusate)ナトリウムが界面活性剤として使用される時には活性化合物の良好なバイオアベイラビリティーが得られる。
【特許文献1】米国特許第5,677,297号明細書
【特許文献2】欧州特許第0733642号明細書
【特許文献3】欧州特許第0830863号明細書
【特許文献4】国際公開第00/48601号パンフレット
【特許文献5】国際公開第01/03699号パンフレット
【特許文献6】国際公開第03/059939号パンフレット
【特許文献7】国際公開第00/00179号パンフレット
【特許文献8】国際公開第03/068266号パンフレット
【特許文献9】国際公開第2006/067150号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0008】
商業的使用にとって不充分に安定でありそして妥当な寸法を有する高い含有量の活性化合物を有する調剤を製造するためにも使用できる従来法で調合される形態の活性化合物と比べて、バイオ−アベイラビリティーにおける有意な増加を有する以上で定義された式Iの化合物用の代替経口調剤を提供することが本発明の目的である。通常の調合工程および装置を用いて製造できるので大資本が必要でない調剤を提供することが本発明の別の目的である。
【課題を解決するための手段】
【0009】
この目的は、本発明に従い、一般式
【0010】
【化2】
【0011】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ
(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフチル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口調剤であって、
a)調剤の合計重量の10%〜80%の間の量の該活性化合物、
b)少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物を含ん
でなり、
c)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなり、
但し調剤が界面活性剤を含有しない調剤により達成することができる。
【0012】
Mは好ましくはLi+、Ca2+、Mg2+およびZn2+よりなる群から選択され、そして最も好ましくはCa2+である。(C1−C6)−アルキルは1〜6個の間の炭素原子よりなる直鎖状もしくは分枝鎖状のアルキル基として定義される。(C1−C6)−アルコキシは1〜6個の間の炭素原子よりなる直鎖状もしくは分枝鎖状のアルコキシ基として定義される。R1は好ましくはフェニルエチルであり、R2およびR3は好ましくは水素でありそしてR4は好ましくはエチルである。
【0013】
本発明の枠内では、生不安定性エステル類を生成しうる適当なR4基は低級アルキル基、場合によりフェニル環中で2個の隣接炭素原子に結合された低級アルキルもしくは低級アルキレン鎖により置換されていてもよいフェニルもしくはフェニル−低級−アルキル基、場合によりジオキサン環中で低級アルキルにより置換されていてもよいジオキソラニルメチル基、または場合によりオキシメチル基上で低級アルキルにより置換されていてもよいC2−C6−アルカノイルオキシメチル基を包含する。生不安定性エステルを生成する基R4が低級アルキルである場合には、これは好ましくは炭素数1〜4、好ましくは2の非分枝鎖状アルキル基でありうる。生不安定性エステルを生成する基が場合により置換されていてもよいフェニル−低級アルキルである場合には、そのアルキレン鎖は1〜3個の、好ましくは1個の炭素原子を含有しうる。フェニル環が低級アルキレン鎖により置換されている場合には、これは3〜4個の、好ましくは3個の炭素原子を含有しうる。特に適するフェニル−含有置換基R4はフェニル、ベンジルまたはインダニルである。R4が場合により置換されていてもよいアルカノイルオキシメチル基である場合には、そのアルカノイルオキシ基は2〜6個の、好ましくは3〜5個の炭素原子を含有することができ、好ましくは分岐しており、そして、例えば、ピバロイルオキシメチル基(tert−ブチルカルボニルオキシメチル基)でありうる。
【0014】
好ましい化合物は酸である3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]シクロペンチル]カルボニル]アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンズアゼピン−1−酢酸のカルシウム塩である。最も好ましい化合物は、ダグルトリル(daglutril)カルシウムまたはSLV 306カルシウムとしても知られるその3S,2’R形態での該化合物である。この化合物は化合物S−Caと称され、ダグルトリルまたはSLV 306としても知られる対応する酸(3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]−シクロペンチル]−カルボニルアミノ]−2,3,4,5−テトラヒドロ−2−オキソ−1H−1−ベンズアゼピン−1−酢酸)は化合物S−Hと称される。
【0015】
式(I)の活性化合物は通常は約10〜80重量%の間の量で、より好ましくは約15
〜75重量%の間の量で、さらにより好ましくは約20〜65重量%の間の量で、そして最も好ましくは約45〜65重量%の間の量で使用される。活性化合物は微細化された形態で使用されるかまたは場合により使用されうる。
【0016】
本発明の枠内で使用されるある種の用語の理解を助けるために以下の定義が提供される。
【0017】
商業的使用にとって充分な安定性は、周囲条件における少なくとも1年間、好ましくは少なくとも2年間、さらにより好ましくは少なくとも3年間、そして最も好ましくは少なくとも5年間の貯蔵期間中の許容可能な化学的および物理的安定性を意味する。許容可能な化学的安定性は、貯蔵期間中の活性化合物の5%より多くない好ましくは3%より多くない、そして最も好ましくは1%より多くない活性物質の崩壊を意味する。許容可能な物理的安定性は、外観における有意な変化なし、貯蔵期間の終了時に脱ブリスター化(deblistering)中の破壊なし、および20%より多くない崩壊時間の変化を意味する。用語「微細化された」は、容量基準で粒子の95%より多くが75ミクロンより小さい粒子寸法をさす。
【0018】
界面活性剤は、分子を溶液中で集積させてミセルを形成させうる良く規定された極性および非−極性領域を有する分子として定義される。極性領域の性質により、界面活性剤は非−イオン性、アニオン性、カチオン性および両性でありうる。非−イオン性の親水性界面活性剤の例はポリオキシエチレンソルビタンエステル類、クレモフォア類(cremophores)、およびポロキサマー類である。アニオン性界面活性剤の例はナトリウムラウリルサルコシネート(sodium lauryl sarcosinate)、ドキュセート(docusate)、並びに製薬学的に許容可能なドキュセート塩類、例えばドキュセートカルシウム、ドキュセートナトリウムおよびドキュセートカリウムである。
【0019】
アルカリ性化合物は無機および有機アルカリ性化合物、例えば炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、クエン酸ナトリウム、トリス緩衝液、トリエタノールアミン、アルカリ性水酸化物、例えば水酸化ナトリウム、水酸化カリウムまたは水酸化マグネシウム、アルカリ性燐酸塩、例えば燐酸水素二カリウム、並びにメグルミンよりなる群から選択される。これらのアルカリ性化合物の混合物も使用することができる。好ましいアルカリ性化合物は炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウムおよび炭酸カルシウムである。さらにより好ましいアルカリ性化合物は炭酸ナトリウムである。
【0020】
好ましくは、アルカリ系は未コーティング組成物の10%w/wより多い、より好ましくは20%w/wより多い量で存在し、または未コーティング組成物の25%、30%w/w、40%w/w、45%w/w、50%w/w、55%w/wもしくは60%w/wより多い量で存在する。用語「未コーティング調剤」は1種もしくは複数の場合により存在するコーティング物質の適用前の調剤を意味する。
【0021】
炭酸塩が使用される場合には、それは好ましくは未コーティング調剤の合計重量の25%の量で使用され、或いはそれは未コーティング調剤は少なくとも45%w/wの量で存在し、または未コーティング調剤の50%w/w、55%w/wもしくは60%w/wより多い量で存在する。
【0022】
さらに好ましい態様では、本発明は一般式
【0023】
【化3】
【0024】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフフル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wの、粒子の97%より多くが500μmより小さく、粒子の
40%より多くが160μmより小さくそして粒子の10%より多くが63μmより
小さい粒子寸法分布を有する炭酸ナトリウムを含んでなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなる組成物
に関する。
【0025】
この好ましい態様では、アルカリ系は通常の炭酸ナトリウムより小さい粒子寸法を有する炭酸ナトリウムを含んでなり、それは粒子の多くとも25%が160μmより小さい粒子寸法分布(ふるい分析により測定されそして重量基準である)を有する。この炭酸ナトリウムは通常の炭酸ナトリウムより高い比表面積を有し、それは約0.2m2/gの比表面積(標準的なBET面積測定に従い測定される)を有する。以上で示されたように、好ましい態様で使用される炭酸ナトリウムは粒子の97%より多くが500μmより小さい、粒子の40%より多くが160μmより小さいそして粒子の10%より多くが63μmより小さい粒子寸法分布(ふるい分析により測定されそして重量基準である)を有する。さらにより好ましい態様では、粒子の98%より多くは500μmより小さく、粒子の約60%より多くは160μmより小さくそして粒子の30%より多くは63μmより小さい。比表面積は好ましくは1m2/gより高く、そしてより好ましくは1.5m2/gより高い。最も好ましい炭酸ナトリウムはソルベイ・SA(Solvay SA)によりソーダ・アッシュ(Soda Ash)IPHとして販売されている特定の炭酸ナトリウムである。このタイプの炭酸ナトリウムでは、典型的には粒子の99.8%が500μmより小さく、粒子の80%が160μmより小さくそして粒子の40%が63μmより小さく、そしてこのタイプの炭酸ナトリウムは2m2/gの比表面積を有する。
【0026】
驚くべきことに、本発明の発明者はアルカリ性化合物を調剤中で、単独でまたは混合して、使用すると、組成物中に界面活性剤を含まなくても、ゲルを酸性胃液の中に溶解させる際の難点の発生を防止し、それによりインビボ溶解度における改良並びにバイオアベイ
ラビリティーにおける改良を指示する二相溶解モデル(実施例1a参照)におけるインビトロ溶解中に特に証明されているようなSLV−306の溶解度を高めることを見出した。特に(ソーダ・アッシュIPH)の如き100μmの平均粒子寸法および2m2/gの比表面積を有する炭酸ナトリウムを用いると、良好なインビボ溶解度および良好なバイオアベイラビリティーを有する調剤が得られる。さらに、組成物は貯蔵時に良好な安定性を有することが予測される。
【0027】
特定の粒子寸法および比表面積を有する上記の炭酸塩が使用される場合には、それは好ましくは未コーティング調剤の合計重量の少なくとも15%の量で、より好ましくは少なくとも18%の量で、さらにより好ましくは少なくとも20%の量で使用され、またはそれは未コーティングの25%w/w、30%w/w、40%w/w、50%w/wもしくは60%w/wより多い量で存在する。
【0028】
以上で示された通りの炭酸水素塩および炭酸塩のような特定の固体アルカリ性化合物は発泡性組成物中でしばしば固体の酸性化合物(例えばクエン酸、酒石酸、アジピン酸、フマル酸、琥珀酸、アスコルビン酸、ニコチン酸、サッカリン、アスピリン、リンゴ酸、燐酸二水素ナトリウム、ピロ燐酸二水素二ナトリウム、クエン酸二水素ナトリウムおよびクエン酸水素二ナトリウム)と組み合わせて使用される。本発明では、組成物は好ましくは酸性化合物を含有しない。
【0029】
調剤は場合により補助物質を調剤の合計重量の45%までのそして好ましくは調剤の合計重量の1%〜45%の間の量で含んでなる。これらの補助物質の例は以下のものを包含するが、それらに限定されない:
a)アラビアゴム、アルギン酸およびその塩類、セルロース誘導体、メチルセルロース、
ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ポリエチレングリ
コール、ゴム類、多糖酸類、ヒドロキシプロピルメチルセルロース、ゼラチン、ポリ
ビニルピロリドン、ポリビニルピロリドン/酢酸ビニル共重合体、ポリメタクリレー
ト類、ヒドロキシプロピルメチルセルロース、澱粉、予備ゼラチン化された澱粉、エ
チルセルロース、トラガカント、デキストリン、微結晶性セルロース、スクロース、
またはグルコースなどを包含するがそれらに限定されない結合剤。
b)澱粉類、予備ゼラチン化されたコーンスターチ、予備ゼラチン化された澱粉、セル
ロース、架橋−結合されたカルボキシメチルセルロース、クロスポビドン、架橋−結
合されたポリビニルピロリドン、アルギン酸カルシウムもしくはナトリウム複合体、
クレイ類、アルギネート類、ゴム類、またはグリコール酸ナトリウム澱粉、および錠
剤製造において使用されるいずれかの崩壊剤を包含するがそれらに限定されない崩壊
剤。
c)ラクトース、炭酸カルシウム、燐酸カルシウム、二塩基性燐酸カルシウム、硫酸カル
シウム、微結晶性セルロース、セルロース粉末、デキストロース、デキシトラート
類、デキストラン、澱粉類、予備ゼラチン化された澱粉、スクロース、キシリトー
ル、ラクチトール、マンニトール、ソルビトールなどを包含するがそれらに限定され
ない充填剤。
d)いずれかの酸化防止剤、緩衝液、または酸類などを包含するがそれらに限定されない
安定剤。
e)ステアリン酸マグネシウム、水酸化カルシウム、タルク、コロイド状二酸化珪素、ス
テアリルフマル酸ナトリウム、水素化された植物油、ステアリン酸、ベヘン酸グリセ
リル、ステアリン酸マグネシウム、カルシウムおよびナトリウム、ワックス類、ステ
アロウェット(Stearowet)、ホウ酸、安息香酸ナトリウム、酢酸ナトリウ
ム、DL−ロイシン、ポリエチレングリコール類、オレイン酸ナトリウム、またはラ
ウリル硫酸ナトリウムなどを包含するがそれらに限定されない潤滑剤。
f)オレイン酸、モノステアリン酸グリセリル、モノオレイン酸ソルビタン、モノラウリ
ン酸ソルビタン、オレイン酸トリエタノールアミン、モノオレイン酸ポリオキシエチ
レンソルビタン、モノラウリン酸ポリオキシエチレンソルビタン、オレイン酸ナトリ
ウム、またはラウリル硫酸ナトリウムなどを包含するがそれらに限定されない湿潤剤
。
g)ラクトース、澱粉、マンニトール、ソルビトール、デキストロース、微結晶性セルロ
ース、二塩基性燐酸カルシウム、スクロースをベースとした希釈剤、菓子用糖、一塩
基性硫酸カルシウム一水和物、硫酸カルシウム二水和物、乳酸カルシウム三水和物、
デキストレート類、イノシトール、加水分解された穀類固体、アミロース、粉末状セ
ルロース、炭酸カルシウム、グリシン、またはベントナイトなどの如き希釈剤。
h)コロイド状シリカ、タルク、コーンスターチ、DL−ロイシン、ラウリル硫酸ナトリ
ウム、およびステアリン酸マグネシウム、カルシウムまたはナトリウムなどを包含す
るがそれらに限定されない抗−付着剤または滑り剤。
i)アラビアゴム、ゼラチン、コロイド状二酸化珪素、グリセロ燐酸カルシウム、乳酸カ
ルシウム、マルトデキストリン、グリセリン、珪酸マグネシウム、カゼインナトリウ
ム、大豆レシチン、塩化ナトリウム、燐酸三カルシウム、燐酸二カリウム、ナトリウ
ムステアロイルラクチレート、カラゲナン、モノグリセリド、ジグリセリド、または
予備ゼラチン化された澱粉などを包含するがそれらに限定されない製薬学的に相容性
の担体。
【0030】
調剤が上記の通りの特定の粒子寸法分布および/または表面積を有する炭酸ナトリウムを少なくとも10%w/wを含有する好ましい態様の場合には、調剤は界面活性剤を補助物質として含有することもできる。
【0031】
最終的な調剤は好ましくは粒剤、圧縮錠剤、またはカプセル剤の形態である。
【0032】
上記の調剤は普遍的な調合工程および装置を用いて製造することができる。従って、以下の段階:
a)式Iの活性化合物をアルカリ性化合物またはアルカリ性化合物の混合物とそして場合
により1種もしくはそれ以上の補助物質と混合し、
b)混合物を圧縮し、
c)該圧縮から得られた粒子を粉砕しそしてふるいにかけ、そして場合により該ふるいに
かけられた粒子を1種もしくはそれ以上の補助物質と混合し、そして
d)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる、上記のような調剤の製造方法を提供することが本発明の別の面である。
【0033】
本発明の別の態様では、調剤は以下の段階:
a)活性化合物を1種もしくはそれ以上の補助物質と混合し、
b)有機溶媒を用いて該混合物を造粒し、
c)有機溶媒を除去して粒子を得、
d)粒子を粉砕しそしてふるいにかけ、そしてふるいにかけられた粒子を補助物質の残り
の部分と混合し、
e)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる有機造粒方法で製造される。
【0034】
有機造粒方法では、数種の有機溶媒を使用することができる。例は、メチルt−ブチルエーテル(MTBE)、ジクロロメタンおよび酢酸エチルである。使用される好ましい溶媒は酢酸エチルである。
【0035】
本発明の調剤が錠剤の形態で提供される時には、これらの錠剤は5分〜90分の間の崩壊時間を有する。好ましくは、崩壊時間は60分以内でありそして最も好ましくはそれらは45分以内である。短い崩壊時間を有する調剤は、ソーダ・アッシュIPH中で入手可能な多孔性炭酸ナトリウムを使用することにより、製造することができる。
【0036】
種々の追加段階、例えば乾燥、破壊、ふるいかけ、混合および包装、もこの方法の一部であることができるが、これらの段階は本発明に従う調剤を得る際の必須特徴ではない。
【0037】
別の面で、本発明は好ましい放出特徴を有する組成物を提供する。従って、本発明は組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%の溶解度を有する上記の経口製薬学的組成物にも関する。5分後の溶解は好ましくは少なくとも55%そしてさらにより好ましくは60%である。15分後に、溶解は少なくとも65%、好ましくは少なくとも70%そしてより好ましくは75%である。30分後に、溶解は少なくとも75%、好ましくは少なくとも80%そしてより好ましくは少なくとも85%である。
【0038】
2.0のpHにおいて、本発明に従う製薬学的組成物は活性化合物を有意に放出しない。5%より少ない活性化合物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH2.0において測定して30分以内に放出される。好ましくは2%より少ないそして好ましくは1%より少ない量が放出される。
【0039】
以下の実施例は本発明をより詳細にさらに説明することが意図され、そしてそのためにこれらの実施例は本発明の範囲をいずれかの方法で制限するものとは考えられない。
【実施例】
【0040】
実施例1.物質、装置および方法
【0041】
物質。
S−Caは、国際公開第03/059939号パンフレットの実施例2および3に示された指示に従い、欧州特許第0733642号明細書の実施例2に従い製造された酸で出発して製造された。
【0042】
ソーダ・アッシュIPH(ソーダ、多孔性または多孔性ソーダとも称する)はベルギー、ブリュッセルのソルベイ・SAから得られた。
【0043】
全ての他の補助物質は容易に商業的に入手可能である。
【0044】
装置
ローラー圧縮用には、フィッツミル(Fitzmill)L1Aが装着されたフィッツパトリック(Fitzpatrick)タイプIR200ローラー圧縮機が使用された。
【0045】
ローラー圧縮の設定は、
回転速度水平供給スクリュー(HFS、rpm)
回転速度垂直供給スクリュー(VFS、rpm)
回転速度ローラー(N1、rpm)
間隙(d、mm)
圧縮力(F、KN/cm)
であった。
【0046】
フィッツミルの設定は、
回転速度ハンマーナイフ(N2、rpm)
スクリーン寸法(mm)
であった。
【0047】
方法。
a)二相インビトロ溶解方法の記述。
二相溶解はUSP装置2配置を用いて行われた。パドル速度は50rpmでありそして容器(および溶解媒体)の温度はバンケル(Vankel)VK7010を用いて37.0℃に保たれた。
【0048】
調剤の溶解は500mlの水中0.1M塩酸(500mlの4.2mlの濃塩酸(HCl))中で開始された(相1)。0、5、15、および30分後に、試料を採取した。30分後に、500mlの1M燐酸塩緩衝液(1000mlの水中の32.4グラムの燐酸二水素塩NaH2PO4および124.8グラムの燐酸水素二ナトリウム(Na2HPO4)を相1に加えた。燐酸水素塩緩衝液の添加が溶解媒体のpHを相1におけるpH1から相2におけるpH6.8に変えた。溶解試験中に、両方の相のpHは未変化のままであった。試料を35、45、および60分後に採取した。
【0049】
全ての試料をポール・ジマーク・アクロディスク(Pall Zymark Acrodisc)PSF、GxF/GHP、0.45μmまたはミリポア・ミレックス(Millipore Millex)−FH(疎水性PTFE 0.45μm)フィルターを通して濾過した。
【0050】
濾過された試料中に溶解したダグルトリルの量を外部基準を用いる240nmにおけるオフ−ラインUV測定により分析した。
【0051】
化合物SLV306のカルシウム塩(S−Ca)を用いるこれまでの比較試験では、この二相インビトロ溶解方法はインビボ結果と良好な相関性を有した。
【0052】
b)多孔性炭酸ナトリウムを同定する方法の記述。
多孔性炭酸ナトリウムの粒子寸法分布を機械的システムにより測定した。約70gの生成物の試料を重量測定しそして0.5、0.25、0.16、0.125、0.1、0.063、および0.045mmのスクリーンを有するふるいを含有する移行機械(ふるいのセットへの水平運動および垂直反射運動の組み合わせを動かしうる自動装置、例えばROTAPまたはRETSCHシフト機)の上方ふるいの中に入れた。ふるいかけ工程は約15分間にわたり行われた。各ふるいの内容物を重量測定しそして500μmより小さい粒子寸法を有する粒子の質量、160μmより小さい粒子寸法を有する粒子の質量、および63μmより小さい粒子寸法を有する粒子の質量を計算した。多孔性炭酸ナトリウムの比表面積を標準的BET面積方法に従い測定した。
【0053】
c)他の物理的方法の記述。
粉末流を径8mmの出口を有する黄銅漏斗中で測定した。粉末流は秒/100グラムで表示された。
− ローラー−圧縮された物質の粒子寸法分布は0.25mm、0.50mm、0.71mm、0.85mm、1.0mm、および2.0mmスクリーンを用いる手動移行から得られた。<0.25mmの量(%)、>1.0mmの量(%)およびd(50%)が計算された。
【0054】
混合された粒子のかさおよび叩き密度を以下の通りにして測定した:
− 100〜150グラムの量の粒状物を造粒シリンダー中に充填した。
− 占有された量を測定した。
− 1200叩き後に、占有された量を再び測定した。カル(Carr)−指数をかさおよび叩き密度から計算した。
【0055】
5個の錠剤をシュレウニガー(Schleuniger)硬度試験機中で粉砕すること
により錠剤の粉砕強度を測定した。平均値が報告された。
【0056】
Erwekaもろさ試験機を用いて20個の錠剤についてもろさを測定した。試験条件は40rpmにおいて10分であった。もろさは%錠剤重量損失で表した。
【0057】
溶解媒体として水を用いて、1個の錠剤上で崩壊を試験した。
【0058】
実施例2:S−Caの従来法で調合されたコーティング錠剤の製造。
【表1】
【0059】
工程:
i)S−Caを圧縮しそして1.0mmふるい中に通した。
ii)段階(i)の物質を微結晶性セルロースPH301、架橋−結合されたポリビニル
ピロリドンおよびステアリル蟻酸ナトリウムと混合して均一混合物を得た。
iii)段階(ii)の物質を錠剤圧縮機械を用いて圧縮した。
iv)段階(iii)からの錠剤を適当なコーティング装置中でコーティングした。
【0060】
実施例3:ソーダ・アッシュIPHを含有するS−Caの未コーティング錠剤の製造
【表2】
【0061】
工程:
i)S−Ca、ソーダ・アッシュIPH、ステアリン酸マグネシウムおよびグリコール酸
ナトリウム澱粉(プリモジェル)を#40メッシュふるいを通して移した。
ii)以上で移されたS−Ca、ソーダ・アッシュIPH(内部として示される)および
ステアリン酸マグネシウムの一部(内部として示される)および場合によりグリコ
ール酸ナトリウム澱粉(プリモジェル(R))(内部として示される)および/ま
たはラクトースを混合して均一混合物を得た。
iii)段階(ii)の物質をローラー圧縮機で指示された設定で混合した。
iv)混合された物質を粉砕しそして2.5mmのスクリーン寸法上でふるいにかけた。v)粉末流および粒子寸法分布を測定した。
【0062】
【表3】
【0063】
工程:
vi)段階(iv)の物質を残りの量のステアリン酸マグネシウム(外部)、グリコール
酸ナトリウム澱粉(プリモジェル(R))(外部)、ソーダ・アッシュIPH(外
部)または微結晶性セルロース(外部)と混合した。
vii)かさおよび叩き密度を測定しそしてカル−指数を計算した。
viii)段階(vi)の物質を錠剤圧縮機械を用いて指示された圧縮力で圧縮した。
ix)脆さ、粉砕強度、および崩壊時間を測定した。
【0064】
実施例4.ソーダ・アッシュIPHを有するSLV306調剤および従来法で調合された錠剤に関する比較溶解試験
実施例1(方法a)に記載された方法に従う比較溶解試験を従来法で調合された錠剤(錠剤K、実施例2に記載された通りにして製造された)の1つのバッチおよび実施例3に従い製造されたSLV−306(S−Ca)のカルシウム塩の5つの600/965バッチ(表中のバッチ指示を参照のこと)(9kNで圧縮された錠剤F、12kNで圧縮された錠剤G、6kNで圧縮された錠剤H、11kNで圧縮された錠剤Iおよび10kNで圧縮された錠剤J)に対して行った。
【0065】
これらの調剤の放出特徴を以下の表に示しそして図1に示す(○=調剤F;□=調剤G;◇=調剤H;Δ=調剤I;■=調剤J;●=調剤K)
【0066】
【表4】
【0067】
この試験から、妥当な寸法および好ましい放出特徴を有する高い薬品負荷のあるS−Caの調剤が製造されたことが結論づけられた。
【図面の簡単な説明】
【0068】
【図1】図1は調剤の放出特徴を示す。
【特許請求の範囲】
【請求項1】
一般式
【化1】
[式中、
R1は
(1)場合により(C1−C6)アルコキシにより置換されていてもよい、(C1−C6
)アルコキシ(C1−C6)アルキル、
(2)フェニル基が場合により(C1−C6)アルキル、(C1−C6)アルコキシまた
はハロゲンで置換されていてもよい、フェニル−(C1−C6)−アルキルおよびフェ
ニルオキシ−(C1−C6)−アルキル、並びに
(3)ナフチル−(C1−C6)−アルキル
から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物を含ん
でなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなり、
但し調剤が界面活性剤を含有しない組成物。
【請求項2】
アルカリ性化合物が無機および有機アルカリ性化合物、例えば炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、クエン酸ナトリウム、トリス緩衝液、トリエタノールアミン、アルカリ性水酸化物、例えば水酸化ナトリウム、水酸化カリウムもしくは水酸化マグネシウム、アルカリ性燐酸塩、例えば燐酸水素二カリウム、およびメグルミン、またはこれらのアルカリ性化合物の混合物よりなる群から選択される請求項1のいずれかに記載の組成物。
【請求項3】
一般式
【化2】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフチル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wの、粒子の97%より多くが500μmより小さく、粒子の
40%より多くが160μmより小さくそして粒子の10%より多くが63μmより
小さい粒子寸法分布を有する炭酸ナトリウムを含んでなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなる組成
物。
【請求項4】
該炭酸ナトリウムが、粒子の98%より多くが500μmより小さく、粒子の60%より多くが160μmより小さくそして粒子の30%より多くが63μmより小さい粒子寸法分布を有する請求項3に記載の組成物。
【請求項5】
該炭酸ナトリウムが、粒子の約99.8%が500μmより小さく、粒子の約80%が160μmより小さくそして粒子の約40%が63μmより小さい粒子寸法分布を有する請求項4に記載の組成物。
【請求項6】
該炭酸ナトリウムが1.0m2/gより大きい比表面積を有する請求項3−5に記載の組成物。
【請求項7】
該炭酸ナトリウムが1.5m2/gより大きい比表面積を有する請求項6の組成物。
【請求項8】
該炭酸ナトリウムが約2.0m2/gの比表面積を有する請求項7の組成物。
【請求項9】
該炭酸ナトリウムが組成物の少なくとも20%w/wの量で存在する請求項3〜8のいずれかに記載の組成物。
【請求項10】
Mがその2+形態のカルシウムである請求項1〜9のいずれかに記載の組成物。
【請求項11】
アルカリ性化合物の量が55%w/wより多い、好ましくは60%w/wより多いこと
を特徴とする請求項1〜10に記載の組成物。
【請求項12】
該活性化合物が1H−1−ベンズアゼピン−1−酢酸、3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]−シクロペンチル]カルボニル]−アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−のカルシウム塩、好ましくはその3S,2’R形態であることを特徴とする請求項1〜11に記載の組成物。
【請求項13】
粒剤、圧縮錠剤またはカプセル剤の形態の請求項1〜12に記載の組成物。
【請求項14】
以下の段階:
e)式Iの活性化合物をアルカリ性化合物またはアルカリ性化合物の混合物とそして場合
により1種もしくはそれ以上の補助物質と混合し、
f)混合物を圧縮し、
g)該圧縮から得られた粒子を粉砕しそしてふるいにかけ、そして場合により該ふるいに
かけられた粒子を1種もしくはそれ以上の補助物質と混合し、そして
h)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる請求項1〜13に記載の組成物の製造方法。
【請求項15】
以下の段階:
a)活性化合物を1種もしくはそれ以上の補助物質と混合し、
b)有機溶媒を用いて該混合物を造粒し、
c)有機溶媒を除去して粒子を得、
d)粒子を粉砕しそしてふるいにかけ、そしてふるいにかけられた粒子を補助物質の残り
の部分と混合し、
e)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる請求項1〜14に記載の組成物の製造方法。
【請求項16】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%の溶解度を有する請求項1〜14に記載の製薬学的組成物。
【請求項17】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して15分以内で少なくとも65%の溶解度を有する請求項17に記載の製薬学的組成物。
【請求項18】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して30分以内で少なくとも75%の溶解度を有する請求項17に記載の経口製薬学的組成物。
【請求項19】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%、15分以内で少なくとも65%そして30分以内で少なくとも75%の溶解度を有する請求項17に記載の経口製薬学的組成物。
【請求項20】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH2.0において測定して30分以内で5%より少ない溶解度を有する請求項17〜20に記載の経口製薬学的組成物。
【請求項1】
一般式
【化1】
[式中、
R1は
(1)場合により(C1−C6)アルコキシにより置換されていてもよい、(C1−C6
)アルコキシ(C1−C6)アルキル、
(2)フェニル基が場合により(C1−C6)アルキル、(C1−C6)アルコキシまた
はハロゲンで置換されていてもよい、フェニル−(C1−C6)−アルキルおよびフェ
ニルオキシ−(C1−C6)−アルキル、並びに
(3)ナフチル−(C1−C6)−アルキル
から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wのアルカリ性化合物またはアルカリ性化合物の混合物を含ん
でなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなり、
但し調剤が界面活性剤を含有しない組成物。
【請求項2】
アルカリ性化合物が無機および有機アルカリ性化合物、例えば炭酸水素ナトリウム、炭酸水素カリウム、炭酸ナトリウム、炭酸カリウム、クエン酸ナトリウム、トリス緩衝液、トリエタノールアミン、アルカリ性水酸化物、例えば水酸化ナトリウム、水酸化カリウムもしくは水酸化マグネシウム、アルカリ性燐酸塩、例えば燐酸水素二カリウム、およびメグルミン、またはこれらのアルカリ性化合物の混合物よりなる群から選択される請求項1のいずれかに記載の組成物。
【請求項3】
一般式
【化2】
[式中、
R1は(C1−C6)アルコキシにより置換されていてもよい(C1−C6)アルコキシ(C1−C6)アルキル、フェニル基が(C1−C6)アルキル、(C1−C6)アルコキシまたはハロゲンで置換されていてもよいフェニル−(C1−C6)−アルキルおよびフェニルオキシ−(C1−C6)−アルキル、並びにナフチル−(C1−C6)−アルキルよりなる群から選択され、
R2およびR3は両者とも独立して水素またはハロゲンであり、
R4は生不安定性エステル生成基であり、
Mは水素または金属イオン、好ましくは2価金属イオンであり、そして
nは1、2または3である]
の活性化合物の改良された経口製薬学的組成物であって、
a)調剤の合計重量の10〜65%の量の該活性化合物、
b)少なくとも10%w/wの、粒子の97%より多くが500μmより小さく、粒子の
40%より多くが160μmより小さくそして粒子の10%より多くが63μmより
小さい粒子寸法分布を有する炭酸ナトリウムを含んでなり、
d)場合により、調剤の合計重量の1%〜45%の間の量の補助物質を含んでなる組成
物。
【請求項4】
該炭酸ナトリウムが、粒子の98%より多くが500μmより小さく、粒子の60%より多くが160μmより小さくそして粒子の30%より多くが63μmより小さい粒子寸法分布を有する請求項3に記載の組成物。
【請求項5】
該炭酸ナトリウムが、粒子の約99.8%が500μmより小さく、粒子の約80%が160μmより小さくそして粒子の約40%が63μmより小さい粒子寸法分布を有する請求項4に記載の組成物。
【請求項6】
該炭酸ナトリウムが1.0m2/gより大きい比表面積を有する請求項3−5に記載の組成物。
【請求項7】
該炭酸ナトリウムが1.5m2/gより大きい比表面積を有する請求項6の組成物。
【請求項8】
該炭酸ナトリウムが約2.0m2/gの比表面積を有する請求項7の組成物。
【請求項9】
該炭酸ナトリウムが組成物の少なくとも20%w/wの量で存在する請求項3〜8のいずれかに記載の組成物。
【請求項10】
Mがその2+形態のカルシウムである請求項1〜9のいずれかに記載の組成物。
【請求項11】
アルカリ性化合物の量が55%w/wより多い、好ましくは60%w/wより多いこと
を特徴とする請求項1〜10に記載の組成物。
【請求項12】
該活性化合物が1H−1−ベンズアゼピン−1−酢酸、3−[[[1−[2−(エトキシカルボニル)−4−フェニルブチル]−シクロペンチル]カルボニル]−アミノ]−2,3,4,5−テトラヒドロ−2−オキソ−のカルシウム塩、好ましくはその3S,2’R形態であることを特徴とする請求項1〜11に記載の組成物。
【請求項13】
粒剤、圧縮錠剤またはカプセル剤の形態の請求項1〜12に記載の組成物。
【請求項14】
以下の段階:
e)式Iの活性化合物をアルカリ性化合物またはアルカリ性化合物の混合物とそして場合
により1種もしくはそれ以上の補助物質と混合し、
f)混合物を圧縮し、
g)該圧縮から得られた粒子を粉砕しそしてふるいにかけ、そして場合により該ふるいに
かけられた粒子を1種もしくはそれ以上の補助物質と混合し、そして
h)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる請求項1〜13に記載の組成物の製造方法。
【請求項15】
以下の段階:
a)活性化合物を1種もしくはそれ以上の補助物質と混合し、
b)有機溶媒を用いて該混合物を造粒し、
c)有機溶媒を除去して粒子を得、
d)粒子を粉砕しそしてふるいにかけ、そしてふるいにかけられた粒子を補助物質の残り
の部分と混合し、
e)場合により混合物を錠剤に圧縮し、場合により次いでコーティングし、および/また
は場合により混合物をカプセル剤中に充填すること
を含んでなる請求項1〜14に記載の組成物の製造方法。
【請求項16】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%の溶解度を有する請求項1〜14に記載の製薬学的組成物。
【請求項17】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して15分以内で少なくとも65%の溶解度を有する請求項17に記載の製薬学的組成物。
【請求項18】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して30分以内で少なくとも75%の溶解度を有する請求項17に記載の経口製薬学的組成物。
【請求項19】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH6.8において測定して5分以内で少なくとも50%、15分以内で少なくとも65%そして30分以内で少なくとも75%の溶解度を有する請求項17に記載の経口製薬学的組成物。
【請求項20】
組成物がUSP装置2配置を用いて50rpmのパドル速度で37.0℃においてそしてpH2.0において測定して30分以内で5%より少ない溶解度を有する請求項17〜20に記載の経口製薬学的組成物。
【図1】

【公表番号】特表2009−539940(P2009−539940A)
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願番号】特願2009−514807(P2009−514807)
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/EP2007/055937
【国際公開番号】WO2007/144418
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【Fターム(参考)】
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/EP2007/055937
【国際公開番号】WO2007/144418
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【Fターム(参考)】
[ Back to top ]