
イネのアントラニル酸合成酵素遺伝子OASA2の新規改変遺伝子およびその利用
【課題】 トリプトファンによるフィードバック阻害に対する抵抗性を獲得した改変イネアントラニル酸合成酵素および当該改変酵素をコードする新規改変遺伝子を提供する。
【解決手段】 イネアントラニル酸合成酵素遺伝子OASA2の塩基配列に変異を導入し、特定の複数のアミノ酸に置換が生じたイネアントラニル酸合成酵素は、トリプトファンによるフィードバック阻害に対する抵抗性を獲得すると共に野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を維持している。
【解決手段】 イネアントラニル酸合成酵素遺伝子OASA2の塩基配列に変異を導入し、特定の複数のアミノ酸に置換が生じたイネアントラニル酸合成酵素は、トリプトファンによるフィードバック阻害に対する抵抗性を獲得すると共に野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を維持している。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イネのアントラニル酸合成酵素遺伝子OASA2の新規改変遺伝子およびその利用に関するものであり、特に野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有し、トリプトファンによるフィードバック阻害に対して抵抗性を有する改変イネアントラニル酸合成酵素をコードする新規改変遺伝子およびその利用に関するものである。
【背景技術】
【0002】
トリプトファンはタンパク質を構成するアミノ酸の1つであり、生物が生きていくうえで不可欠なものである。動物はトリプトファンを体内で合成できないため、食物から摂取しなければならない。イネ、トウモロコシ、コムギなどの穀物は、トリプトファンの含有量が非常に少ないため、通常穀物飼料には工業的に製造されたトリプトファンを添加する必要がある。
【0003】
トリプトファンの生合成経路において、コリスミン酸からアントラニル酸が生合成されるが、アントラニル酸の生成はアントラニル酸合成酵素の触媒作用が関与しており、これによってアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成することが知られている(非特許文献1参照)。
【0004】
発明者らは、既にイネアントラニル酸合成酵素アルファサブユニット遺伝子としてOASA1およびOASA2が存在することを見出し、これらを単離した(特許文献1参照)。そして、これらの特徴を詳細に検討した結果、遺伝子発現量が多く、イネにおいてアントラニル酸合成酵素アルファサブユニットとして主に機能するタンパク質をコードする遺伝子はOASA2であると考えられることを報告している(非特許文献2参照)。さらに、発明者らは、OASA2タンパク質は、極めてトリプトファン濃度に感受性が高く、細胞内のトリプトファン濃度が上昇するに伴い活性を低下させてしまうことを報告している(非特許文献3参照)。
【0005】
そこで、OASA2タンパク質の機能を改変することができれば、高濃度のトリプトファンが蓄積される新しい品種の植物を作出することができると考えられる。イネアントラニル酸合成酵素遺伝子の改変に関しては、例えばイネアントラニル酸合成酵素第1アイソザイムアルファサブユニットOASA1タンパク質の323番目のアスパラギン酸残基をアスパラギン残基に置換(D323N)することで得られたトリプトファンフィードバック阻害抵抗性変異体OASA1Dを形質転換すると、野生型よりもカルスで180倍、組換体イネで35倍、遊離のトリプトファン量が増加することが、発明者らにより報告されている(非特許文献2参照)。
【0006】
一方、遺伝子にランダムに変異を導入し、その変異遺伝子の産物である変異タンパク質の機能をスクリーニングすることは多大な時間と労力を必要とする。さらに、目的の位置に複数の変異を同時に導入することは、ランダムに変異を導入する方法では極めて困難であり、ほとんど不可能に近いと考えられている。
【特許文献1】国際公開第WO99/11800号
【非特許文献1】生化学実験講座、第11巻、東京化学同人、1976年、652頁〜653頁
【非特許文献2】Tozawa Y, Hasegawa H, Terakawa T and Wakasa K (2001) Characterization of rice anthranilate synthase alfa subunit genes OSASA1 and OSASA2: tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiology 126: 1493-1506
【非特許文献3】Takuya Kanno, Koji Kasai, Yasuko Ikejiri-Kanno, Kyo Wakasa and Yuzuru Tozawa (2004) In vitro reconstitution of rice anthranilate synthase: distinct functional properties of the alpha subunits OASA1 and OASA2. Plant Molecular Biology 54: 11-22
【発明の開示】
【発明が解決しようとする課題】
【0007】
上述のように、穀物飼料には工業的に製造されたトリプトファンを添加しているが、トリプトファンは他のアミノ酸に比べて高価であるため、トリプトファン含有量の高い穀物の作出が期待されている。そこで、イネアントラニル酸合成酵素遺伝子OASA2に変異を導入することにより、細胞内のトリプトファン濃度が上昇しても酵素活性が低下しないようにOASA2タンパク質の機能を改変することができれば、高濃度のトリプトファンが蓄積された新しい品種の植物を作出することが可能になる。
【0008】
本発明は、上記の問題点に鑑みてなされたものであり、その目的は、細胞内のトリプトファン濃度が上昇しても酵素活性が低下しないように機能改変されたイネアントラニル酸合成酵素および当該酵素をコードする新規改変遺伝子を提供し、高濃度のトリプトファンを含有する新しい品種の植物を実現することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記課題を解決するために鋭意検討した結果、イネアントラニル酸合成酵素遺伝子OASA2に変異を導入することにより、トリプトファンによるフィードバック阻害に対する抵抗性を有するタンパク質を創出した。さらに、発明者らは上記野生型イネアントラニル酸合成酵素遺伝子OASA2において複数の変異を導入し、これらを組み合わせることにより、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有するタンパク質を創出することに成功し、本発明を完成させるに至った。
【0010】
すなわち、本発明に係るポリペプチドは、アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴としている。上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることが好ましい。
【0011】
また、本発明に係るポリペプチドは、配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴としている。さらに、上記変異に加えて配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有することが好ましい。トリプトファンによるフィードバック阻害に対する抵抗性を有し、さらに野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有するポリペプチドは、細胞内のトリプトファン濃度が上昇してもトリプトファンを合成することができ、当該ポリペプチドを発現している植物は高濃度のトリプトファンを含有する栄養価の高い植物として有用となる。
【0012】
上記配列番号1に示されるアミノ酸配列の第126位の変異は、セリンをフェニルアラニンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第367位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第369位の変異は、アラニンをロイシンに置換する変異であることが好ましい。また、上記配列番号1に示されるアミノ酸配列の第351位の変異は、アスパラギンをアスパラギン酸に置換する変異であり、上記配列番号1に示されるアミノ酸配列の第522位の変異は、グリシンをチロシンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第530位の変異は、ロイシンをアラニンまたはアスパラギン酸に置換する変異であることが好ましい。上記に挙げたアミノ酸置換により、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異型イネアントラニル酸合成酵素、またはトリプトファンによるフィードバック阻害に対する抵抗性と野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性とを有する変異型イネアントラニル酸合成酵素が実現できる。
【0013】
上記変異型イネアントラニル酸合成酵素としては、配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドを挙げることができる。これらのアミノ酸配列を有することにより、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異型イネアントラニル酸合成酵素、またはトリプトファンによるフィードバック阻害に対する抵抗性と野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性とを有する変異型イネアントラニル酸合成酵素が実現できる。
【0014】
本発明に係るポリヌクレオチドは、上記本発明に係るポリペプチドをコードするポリヌクレオチドである。また、本発明に係るポリヌクレオチドは配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであることが好ましい。当該ポリヌクレオチドを細胞に導入することにより、細胞内で上記本発明に係るポリペプチドを発現できる形質転換体を作製することが可能となる。
【0015】
本発明に係る形質転換体選抜用マーカー遺伝子は、上記本発明に係るポリヌクレオチドからなるものである。本発明に係るポリヌクレオチドがコードするポリペプチドは、それが発現している細胞にトリプトファン類縁化合物に対する耐性を付与するので、当該トリプトファン類縁化合物に対する耐性を発現している形質転換細胞を選抜するためのマーカー遺伝子として利用することができる。
【0016】
本発明に係る組換え発現ベクターは、上記本発明に係るポリヌクレオチドからなるものである。当該組換え発現ベクターは、本発明に係るポリヌクレオチドを細胞に導入するための組換え発現ベクターとして利用できるだけでなく、本発明に係るポリヌクレオチドを選抜マーカーとして用いた場合には、他の遺伝子を細胞に導入するための組換え発現ベクターとしても利用できる。
【0017】
本発明に係る形質転換体は、上記本発明に係るポリヌクレオチドまたは上記本発明に係る組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体である。形質転換体は、植物細胞または植物体であることが好ましい。トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換植物は、高濃度のトリプトファンを含有する栄養価の高い植物として有用となる。また、本発明には上記形質転換植物から得られる種子も含まれる。
【0018】
本発明に係る形質転換細胞の選抜方法は、本発明に係るマーカー遺伝子または本発明に係る組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜することからなる選抜方法である。当該遺伝子を選択マーカーとして用いることで、イネ等の単子葉植物で利用できるマーカーの種類が限られるといった問題が解消される。また、これまで複数遺伝子を細胞に導入する際に使用できる選択マーカーの種類が制限されるといった問題に対しても、当該遺伝子を使用することで解決される。すなわち、通常選択マーカーとして使用されているハイグロマイシン等の抗生物質に対する耐性等の他に、トリプトファン類縁化合物、例えば5−メチルトリプトファンに対する抵抗性によって複数の目的DNAが導入された細胞の選抜が可能となる。さらに、当該マーカー遺伝子はイネ由来の遺伝子であるため、イネの形質転換体において本遺伝子がコードするタンパク質は低抗原性であることが期待される。
【0019】
本発明に係る形質転換キットは少なくとも本発明に係るポリヌクレオチド、あるいは本発明に係る組換え発現ベクターのいずれかを含むことを特徴としている。当該形質転換キットを用いれば、本発明に係るポリペプチドを発現する形質転換体を簡便かつ効率的に得ることができる。
【0020】
本発明に係るスクリーニング法は、本発明に係るポリペプチドおよび野生型イネアントラニル酸合成酵素のいずれか一方のみと結合する物質をスクリーニングする方法であって、本発明に係るポリペプチドと結合する物質をスクリーニングする工程と、野生型イネアントラニル酸合成酵素と結合する物質をスクリーニングする工程と、上記両工程の結果を比較する工程とを含むことを特徴としている。当該スクリーニング方法により、トリプトファン生合成経路におけるトリプトファンによるフィードバック阻害に関与する物質をスクリーニングすることができ、フィードバック阻害に対する抵抗性がより強力な変異型イネアントラニル酸合成酵素の作出に繋がる可能性がある。
【0021】
本発明に係るスクリーニングキットは上記本発明に係るスクリーニング方法を実現するためのキットであって、少なくとも本発明に係るポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むことを特徴としている。当該スクリーニングキットを用いれば、本発明に係るスクリーニング方法を簡便かつ効率的に実施することができる。
【発明の効果】
【0022】
本発明に係るポリペプチドは、トリプトファン合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有している。したがって、高濃度のトリプトファン存在下においてもトリプトファンを合成することができるという効果を奏する。
【0023】
本発明に係るポリヌクレオチドは上記本発明に係るポリペプチドをコードしている。したがって、当該ポリヌクレオチドを植物細胞に導入することにより、高濃度のトリプトファン存在下においてもトリプトファンを合成することができる形質転換植物を作出することができるという効果を奏する。
【0024】
さらに、本発明に係る形質転換植物はトリプトファン含量が高く栄養価に優れた食品や飼料として利用できるという効果を奏する。さらに、トリプトファン含量の高い飼料用イネを作出することにより、家畜用飼料のコストを低下できるという効果を奏すると共に、水田を有効利用し、我国の飼料の自給率を高めることができるという効果を奏する。
【発明を実施するための最良の形態】
【0025】
本発明の実施の一形態について説明すれば、以下の通りである。なお、本発明は、これに限定されるものではない。
【0026】
(1)本発明に係るポリペプチド
本発明に係るポリペプチドは、アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることが好ましい。
【0027】
図4Aにイネアントラニル酸合成酵素遺伝子OASA2(以下、適宜「OASA2遺伝子」と称する。)がコードするイネアントラニル酸合成酵素(以下、適宜「OASA2タンパク質」または「野生型OASA2タンパク質」と称する。)の模式図を示した。図4A中の数字はアミノ酸の位置を表す。cTPは葉緑体移行シグナル、I、IIおよびIIIはトリプトファン結合領域に位置するアミノ酸残基が存在するドメインを表す。また、図4Bに種々のトリプトファン合成系を有する生物のトリプトファン結合領域I、IIおよびIIIのアミノ酸配列の比較を示した。図4B中のOs1はイネOASA1 (Accession no. AB022602)、Os2はイネOASA2 (Accession no. AB022603)、At1はシロイヌナズナ(Arabidopsis)ASA1 (Accession no. M92353)、At2はシロイヌナズナ(Arabidopsis)ASA2 (Accession no. M92354)、Ssは好熱性古細菌(Sulfolobus solfataricus)TrpE (Accession no. 1QDL_A)、Stはサルモネラ菌(Salmonella tryphimurium)TrpE (Accession no. 1I1Q_A)、Smはセラチア菌(Serratia marcescens)TrpE (Accession no. 1I7Q_A)を表す。
【0028】
図4Aおよび図4Bから明らかなように、配列番号26に示されるアミノ酸配列(NPSPYM)は、トリプトファン結合領域IIにおいて、例示した全ての生物に保存されている。したがって、トリプトファン結合領域の中でも非常に重要な役割を果たしていると考えられている。このような保存されたアミノ酸配列を有する生物種としては、図4Bに例示した生物種以外にもニチニチソウ(Catharanthus roseus)α subunit (Accession no. CAC29060)、ヘンルーダ(Ruta graveolens)ASα1 (Accession no. L34343)、ヘンルーダ(Ruta graveolens)ASα2 (Accession no. L34344)、タバコ(tobacco)ASA2 (Accession no. T01990)、酵母(Saccharomyces cerevisiae)TRP2(Accession no. X68327)、大腸菌(Escherichia coli)TrpE (Accession no. V00368)、枯草菌(Bacillus subtilis)TrpE (Accession no. P03963)を挙げることができるが、これらに限定されるものではない。
【0029】
上記配列番号26に示されるアミノ酸配列は、例えば配列番号1に示されるイネアントラニル酸合成酵素遺伝子OASA2がコードする野生型OASA2タンパク質のアミノ酸配列では第363位〜第367位に該当する。そして、当該配列番号26に示される保存されたアミノ酸配列中のチロシンが他のアミノ酸、好ましくはアラニンまたはイソロイシンに置換されることにより、トリプトファンによるフィードバック阻害に対する抵抗性が獲得される。
【0030】
本発明に係るポリペプチドは、配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。上記配列番号1に示されるアミノ酸配列からなるポリペプチドは、イネアントラニル酸合成酵素遺伝子OASA2がコードする野生型イネアントラニル酸合成酵素である。すなわち、イネアントラニル酸合成酵素遺伝子OASA2(OASA2遺伝子)がコードする野生型イネアントラニル酸合成酵素(野生型OASA2タンパク質)の第126位、第367位または第369位の少なくとも1つに変異を有し、かつ、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。変異としては特に限定されるものではなく、例えばアミノ酸の置換、欠失、付加等を挙げることができる。なお、説明の便宜上、変異を有するOASA2タンパク質を、変異の位置、種類、数等を問わず、「変異型OASA2タンパク質」と称する。
【0031】
植物体中のトリプトファンの生合成経路においては、コリスミン酸からアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成する(非特許文献1参照)。アントラニル酸合成酵素は、上記経路中、コリスミン酸からアントラニル酸の生成を触媒する。また、アントラニル酸合成酵素は最終生成物であるトリプトファンによりフィードバック阻害を受け、細胞中のトリプトファン濃度が上昇するに伴い、酵素活性が低下する。したがって、トリプトファンがある一定量細胞に蓄積すると、それ以上トリプトファンが合成されなくなる。本発明に係るポリペプチドは、上述のようにトリプトファンによるフィードバック阻害に対して抵抗性を有するため、細胞中のトリプトファン濃度が上昇してもトリプトファンを合成することができ、トリプトファン含量の高い植物体を作出することが可能となる。
【0032】
トリプトファンによるフィードバック阻害に対する抵抗性は、例えばin vitro酵素活性測定系において、野生型OASA2タンパク質の酵素活性を測定した場合に、トリプトファン無添加の場合の酵素活性を100%としたとき、トリプトファンを添加した場合の酵素活性の比率(パーセンテージ)を基準として判断することができる。より具体的には、例えば図3に示すように、野生型OASA2タンパク質および変異型OASA2タンパク質のそれぞれのトリプトファン無添加時の酵素活性を100%とし、トリプトファン添加時の酵素活性の比率(パーセンテージ)を比較した場合に、野生型OASA2タンパク質のトリプトファン添加時の酵素活性の比率(パーセンテージ)を超えていれば、トリプトファンによるフィードバック阻害に対する抵抗性を有するといえる。野生型OASA2タンパク質のトリプトファン添加時の酵素活性の比率(パーセンテージ)をどの程度超えているかについては特に限定されるものではないが、少なくとも2倍以上であることが好ましく、少なくとも3倍以上であることがより好ましく、少なくとも4倍以上であることが特に好ましく、少なくとも5倍以上であることが最も好ましい。
【0033】
より具体的には、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系を用いて酵素活性を測定した場合において、トリプトファン無添加時の酵素活性(アントラニル酸合成量)を100%としたとき、100μMのトリプトファンを添加した場合の酵素活性(アントラニル酸合成量)が20%以上残存するものが好ましく、30%以上残存するものがより好ましく、40%以上残存するものが特に好ましく、50%以上残存するものが最も好ましい。
【0034】
OASA2タンパク質の第126位のセリンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、フェニルアラニンが好ましい。具体的には、配列番号29に示されるアミノ酸配列からなるポリペプチド(第126位のセリンをフェニルアラニンに置換)、または配列番号29に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0035】
OASA2タンパク質の第367位のチロシンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、アラニン、イソロイシン、フェニルアラニン、バリンが好ましく、アラニン、イソロイシンがより好ましい。具体的には、配列番号2に示されるアミノ酸配列からなるポリペプチド(第367位のチロシンをアラニンに置換)、および配列番号3に示されるアミノ酸配列からなるポリペプチド(第367位のチロシンをイソロイシンに置換)、または配列番号2もしくは3に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0036】
OASA2タンパク質の第369位のアラニンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、ロイシンが好ましい。具体的には、配列番号30に示されるアミノ酸配列からなるポリペプチド(第369位のアラニンをロイシンに置換)、または配列番号30に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0037】
本発明に係るポリペプチドとしては、上記OASA2タンパク質の第126位、第367位または第369位の少なくとも1つの変異に加えて、第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであることが好ましい。上記第351位、第522位、第530位のいずれか1つ、または2つ以上を組み合わせた変異を有する変異型OASA2タンパク質は、野生型OASA2タンパク質と比較して酵素活性が向上することが発明者らにより確認されている(図1参照)。これらの酵素活性が上昇する変異と上記トリプトファンによるフィードバック阻害に対する抵抗性を獲得する変異を組み合わせることにより、トリプトファンによるフィードバック阻害に対する抵抗性を有し、かつ、酵素活性が野生型OASA2タンパク質と同程度以上である変異型OASA2タンパク質を得ることができる。ここでいう酵素活性とは、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系を用いて測定したアントラニル酸合成量を挙げることができる。ただし、酵素活性を測定するための方法はこれに限定されるものではなく、アントラニル酸合成酵素活性を測定可能公知の酵素活性測定方法または、これらの改変方法であればよい。
【0038】
また、酵素活性が同程度以上とは、例えばin vitro酵素活性測定系(トリプトファン無添加)において、変異型OASA2タンパク質の酵素活性が野生型OASA2タンパク質の酵素活性の少なくとも0.7倍以上であることが好ましく、少なくとも0.8倍以上であることがより好ましく、少なくとも0.9倍以上であることが特に好ましく、少なくとも1倍以上であることが最も好ましい。より具体的には、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系(トリプトファン無添加)を用いて酵素活性を測定した場合に、変異型OASA2タンパク質のアントラニル酸合成量が野生型OASA2タンパク質のアントラニル酸合成量の少なくとも0.7倍以上であることが好ましく、少なくとも0.8倍以上であることがより好ましく、少なくとも0.9倍以上であることが特に好ましく、少なくとも1倍以上であることが最も好ましい。
【0039】
OASA2タンパク質の第351位はアスパラギンであり、第522位はグリシンであり、第530位はロイシンである。これらのアミノ酸とそれぞれ置換されるアミノ酸としては、それぞれの変異、またはこれらを組み合わせた変異と、さらに第126位、第367位または第369位の少なくとも1つの変異を組み合わせたときに、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するものであればよく、特に限定されるものではない。第351位のアスパラギンと置換されるアミノ酸としてはアスパラギン酸が好ましく、第522位のグリシンと置換されるアミノ酸としてはチロシンが好ましい。また、第530位のロイシンと置換されるアミノ酸としてはアラニン、アスパラギン酸が好ましく、アスパラギン酸がより好ましい。
【0040】
具体的には、以下に示す変異の組み合わせを有するポリペプチドが好ましい。
i)第367位のチロシンをアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号4)。
ii)第351位のアスパラギンをアスパラギン酸に置換し、第367位のチロシンをアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号5)。
iii)第367位のチロシンをアラニンに置換し、第522位のグリシンをチロシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号6)。
iv)第367位のチロシンをイソロイシンに置換し、第522位のグリシンをチロシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号7)。
v)第369位のアラニンをロイシンに置換したポリペプチド(配列番号30)。
vi)第126位のセリンをフェニルアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号31)。
vii)第369位のアラニンをロイシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号32)。
【0041】
すなわち、本発明に係るポリペプチドは配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するポリペプチドであることが好ましい。
【0042】
上記「1もしくは数個のアミノ酸が欠失、置換、もしくは付加された」とは、部位特異的突然変異誘発法等の公知の変異ポリペプチド作製法により欠失、置換、もしくは付加ができる程度の数(例えば20個以下、好ましくは10個以下、より好ましくは7個以下、さらに好ましくは5個以下、特に好ましくは3個以下)のアミノ酸が置換、欠失、もしくは付加されることを意味する。このような変異ポリペプチドは、公知の変異ポリペプチド作製法により人為的に導入された変異を有するポリペプチドに限定されるものではなく、天然に存在する同様の変異ポリペプチドを単離精製したものであってもよい。
【0043】
なお、本発明に係るポリペプチドは、アミノ酸がペプチド結合してなるポリペプチドであればよいが、これに限定されるものではなく、ポリペプチド以外の構造を含むものであってもよい。ここでいうポリペプチド以外の構造としては、糖鎖やイソプレノイド基等を挙げることができるが、特に限定されるものではない。
【0044】
また、本発明に係るポリペプチドは、付加的なポリペプチドを含むものであってもよい。このようなポリペプチドが付加される場合としては、例えば、HisやMyc、Flag等によってエピトープ標識されるような場合が挙げられる。
【0045】
また、本発明に係るポリペプチドは、後述する本発明に係るポリヌクレオチド(本発明に係るポリペプチドをコードするポリヌクレオチド)を宿主細胞に導入して、それがコードするポリペプチドを細胞内発現させた状態であってもよいし、細胞、組織などから単離精製された状態であってもよい。また、上記宿主細胞での発現条件によっては、本発明に係るポリペプチドは、他のポリペプチドとつながった融合タンパク質であってもよい。さらに本発明に係るポリペプチドは、化学合成されたものであってもよい。
【0046】
本発明に係るポリペプチドの取得方法(生産方法)は特に限定されるものではないが、本発明に係るポリペプチドは野生型OASA2タンパク質に1つ以上の変異を導入したものであるため、OASA2遺伝子の塩基配列に人工的に変異を導入した変異遺伝子を作製し、当該変異遺伝子にコードされたポリペプチドを発現させる方法により取得することが最適である。塩基配列に変異を導入する公知の方法としては、後述の実施例において発明者らが用いているKunkel法やPCRを利用して塩基配列に変異を導入する方法等を挙げることができる。また市販のキット(例えば、Mutan-K、Mutan-Super Express Km、LA-PCR in vitro mutagenesis Kit(いずれもタカラバイオ社製)等)を用いることもできる。このようにして人工的に変異を導入したOASA2遺伝子を適当な発現ベクターに挿入し、適当な宿主細胞に導入して宿主内で翻訳されて得られる産物(ポリペプチド)を生成することにより、本発明に係るポリペプチドを取得することができる。また、後述の実施例において発明者らが用いているようにコムギ胚芽無細胞システム等の公知の無細胞タンパク質合成系を用いて上記変異を導入したOASA2遺伝子の産物(ポリペプチド)を取得することもできる。本発明に係るポリペプチドの取得方法の1例は、後述の実施例において詳細に説明する。
【0047】
(2)本発明に係るポリヌクレオチド
本発明に係るポリヌクレオチドは、上記本発明に係るポリペプチドをコードするものであればよい。本発明に係るポリペプチドについては上記(1)で詳細に説明したので、ここでの説明は省略するが、例えば、具体的には、以下の〔1〕および〔2〕に記載のポリペプチドをコードするポリヌクレオチドであればよい。
〔1〕配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチド。
〔2〕上記〔1〕の変異に加えて、配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有するポリペプチド。
【0048】
例えば、配列番号2に示されるアミノ酸配列、すなわちOASA2タンパク質のアミノ酸配列の第367位チロシンがアラニンに置換されたポリペプチドをコードするポリヌクレオチドとしては、配列番号8に示される塩基配列(OASA2遺伝子のオープンリーディングフレーム領域の塩基配列)からなるポリヌクレオチドの1099、1100および1101位の塩基配列(tac:チロシン)がアラニンに対応するコドン、すなわちgct、gcc、gcaまたはgcgのいずれかに変異したポリヌクレオチドであればよい。また、1099〜1101位の塩基以外の塩基配列は配列番号8に示される塩基配列と同一でなくてもよく、例えばコードするアミノ酸が変異しない塩基置換を有するポリヌクレオチドでもよい。
【0049】
上記に例示した以外のアミノ酸変異を有するポリペプチドをコードするポリヌクレオチドについても同様であり、配列番号8に示される塩基配列のうち変異するアミノ酸に対応する位置の3つの塩基が、置換されるアミノ酸に対応するコドンの塩基に置換されたポリヌクレオチド、または当該塩基の変異とそれ以外の塩基配列におけるアミノ酸が変異しない塩基置換とを有するポリヌクレオチドであればよい。
【0050】
本発明に係るポリヌクレオチドとしては、以下の〔3〕および〔4〕に記載のポリペプチドをコードするポリヌクレオチドであることが好ましい。
〔3〕配列番号2、3、29または30に示されるアミノ酸配列からなるポリペプチド、または配列番号2、3、29または30に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチド。
〔4〕配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するポリペプチド。
【0051】
すなわち、配列番号2〜7および配列番号29〜32に示されるアミノ酸配列のいずれかをコードする塩基配列からなるポリヌクレオチドであればよく、具体的な塩基配列は特に限定されるものではない。ただし、配列番号8に示される塩基配列、すなわちOASA2遺伝子のオープンリーディングフレーム領域の塩基配列からなるポリヌクレオチドと相同性の高いポリヌクレオチドであることが好ましいことはいうまでもない。相同性としては、例えば、少なくとも90%の同一性、好ましくは少なくとも95%の同一性、最も好ましくは少なくとも97%の同一性を有していればよい。
【0052】
本発明に係るポリヌクレオチドとしては、配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチドを挙げることができる。ただし、これらに限定されるものではない。配列番号9ないし14および配列番号33ないし36に示される塩基配列からなるポリヌクレオチドは、それぞれ配列番号2ないし7および配列番号29ないし32に示されるアミノ酸配列からなるポリペプチドをコードする塩基配列からなるポリヌクレオチドである。配列番号9ないし14および配列番号33ないし36に示される塩基配列からなるポリヌクレオチドは、発明者らが、後述の実施例に記載の方法を用いて作製したポリヌクレオチドである。
【0053】
上記「ストリンジェントな条件」とは、少なくとも90%の同一性、好ましくは少なくとも95%の同一性、最も好ましくは少なくとも97%の同一性が配列間に存在するときにのみハイブリダイゼーションが起こることを意味し、例えば、60℃で2×SSC 洗浄条件下で結合することを意味する。上記ハイブリダイゼーションは、「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001) に記載されている方法等、従来公知の方法で行うことができる。通常、温度が高いほど、塩濃度が低いほどストリンジェンシーは高くなる。
【0054】
本発明に係るポリヌクレオチドにはDNAおよびRNAが含まれ、一本鎖でも二本鎖でもよい。また、非翻訳領域(UTR)の配列やベクター配列(発現ベクター配列を含む)などの配列を含むものであってもよい。
【0055】
本発明に係るポリヌクレオチドの取得方法(生産方法)は特に限定されるものではないが、例えば、OASA2遺伝子の塩基配列に人工的に変異を導入する方法を挙げることができる。塩基配列に変異を導入する公知の方法としては、Kunkel法やPCRを利用して塩基配列に変異を導入する方法等を挙げることができる。また市販のキット(例えば、Mutan-K、Mutan-Super Express Km、LA-PCR in vitro mutagenesis Kit(いずれもタカラバイオ社製)等)を用いることもできる。本発明に係るポリヌクレオチドの取得方法の1例は、後述の実施例において詳細に説明する。
【0056】
(3)形質転換用マーカー遺伝子および形質転換細胞の選抜方法
本発明に係るポリヌクレオチドは、形質転換用マーカー遺伝子として利用することができる。すなわち、本発明に係る形質転換体選抜用マーカー遺伝子は、上記(2)に記載した本発明に係るポリヌクレオチドからなるものであればよい。本発明に係るポリヌクレオチドを導入された細胞はトリプトファン耐性となるため、トリプトファン類縁化合物を培地に添加することにより、当該ポリヌクレオチドが導入された細胞のみを選抜することが可能となる。選抜用薬剤として用いることができる細胞の増殖を阻害するトリプトファン類縁化合物としては、例えば、5−メチルトリプトファン(5MT)、4−メチルトリプトファン(4MT)、6−メチルトリプトファン(6MT)、7−メチルトリプトファン(7MT)、6−メチルアントラニル酸(6MA)、5−メチルアントラニル酸(5MA)、3−メチルアントラニル酸(3MA)、5−フルオロアントラニル酸(5FA)、6−フルオロアントラニル酸(6FA)等を挙げることができる。
【0057】
具体的に、本発明に係るポリヌクレオチドを形質転換用マーカー遺伝子として利用するには、例えば、当該ポリヌクレオチドを組み込んだ発現ベクターを構築し、当該発現ベクターを目的の細胞に導入する。当該発現ベクターが導入され、本発明に係るポリヌクレオチドにコードされるポリペプチドが発現している細胞はトリプトファン耐性を獲得するため、上記例示した選抜用薬剤が添加された培地においても増殖ができ、一方、当該発現ベクターが導入されていない細胞や本発明に係るポリヌクレオチドにコードされるポリペプチドが発現していない細胞は増殖が阻害される。したがって、当該発現ベクターが導入され、本発明に係るポリヌクレオチドにコードされるポリペプチドが発現している形質転換細胞のみを選抜することが可能となる。
【0058】
上述の例では本発明に係るポリヌクレオチドを形質転換細胞に発現させる遺伝子とマーカー遺伝子との両方の目的で用いているが、本発明に係るポリヌクレオチドをマーカー遺伝子としてのみ用いることも可能である。また、例えば、植物のカルス細胞に特異的な転写プロモーターを使用することにより、本発明に係るポリヌクレオチドの選択マーカーとしての発現時期の制御も可能である。この場合は、さらに目的の細胞内で発現させたいタンパク質をコードする遺伝子を挿入した発現ベクターを構築し、当該発現ベクターを用いて形質転換すればよい。また、本発明に係るポリヌクレオチドを組み込んだ発現ベクターを構築せずに、本発明に係るポリヌクレオチドを単独で目的の細胞に導入することも可能である。
【0059】
本発明に係るポリヌクレオチドを選択マーカーとして用いることで、イネ等の単子葉植物で利用できるマーカーの種類が限られるといった問題が解消される。また、これまで複数遺伝子を細胞に導入する際に使用できる選択マーカーの種類が制限されるといった問題に対しても、当該遺伝子を使用することで解決される。すなわち、通常選択マーカーとして使用されているハイグロマイシン等の抗生物質に対する耐性等の他に、トリプトファン類縁化合物、例えば5−メチルトリプトファンに対する抵抗性によって複数の目的DNAが導入された細胞の選抜が可能となる。さらに、当該マーカー遺伝子はイネ由来の遺伝子であるため、イネの形質転換体において本遺伝子がコードするタンパク質は低抗原性であることが期待される。
【0060】
なお、本発明には上記本発明に係るマーカー遺伝子または以下に説明する組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜する形質転換細胞の選抜方法も含まれる。
【0061】
(4)組換え発現ベクターおよび形質転換キット
本発明に係る組換え発現ベクターは、上記(2)に記載した本発明に係るポリヌクレオチドを含むものであれば、特に限定されるものではない。例えば、cDNAが挿入された組換え発現ベクターが挙げられる。組換え発現ベクターの作製には、プラスミド、ファージ、またはコスミドなどを用いることができるが特に限定されるものではない。また、作製方法も公知の方法を用いて行えばよい。
【0062】
ベクターの具体的な種類は特に限定されるものではなく、ホスト細胞中で発現可能なベクターを適宜選択すればよい。すなわち、ホスト細胞の種類に応じて、確実に遺伝子を発現させるために適宜プロモーター配列を選択し、これと本発明に係るポリヌクレオチドを各種プラスミド等に組み込んだものを発現ベクターとして用いればよい。
【0063】
本組換え発現ベクターは、本発明に係るポリペプチドを発現させるために用いることができることはいうまでもないが、本発明に係るポリヌクレオチドをマーカー遺伝子として利用し、他の遺伝子を組み込んで当該他の遺伝子がコードするタンパク質を発現させるための組換え発現ベクターとしても利用できる。
【0064】
本発明に係るポリヌクレオチドがホスト細胞に導入されたか否か、さらにはホスト細胞中で確実に発現しているか否かを確認するために、各種マーカーを用いてもよい。例えば、ハイグロマイシンのような抗生物質に抵抗性を与える薬剤耐性遺伝子をマーカーとして用い、このマーカーと本発明に係るポリヌクレオチドとを含むプラスミド等を発現ベクターとしてホスト細胞に導入する。これによってマーカー遺伝子の発現から本発明の遺伝子の導入を確認することができる。
【0065】
上記ホスト細胞は、トリプトファン合成系を有する細胞、生物であれば、特に限定されるものではなく、従来公知の各種細胞を好適に用いることができる。具体的には、例えば、大腸菌(Escherichia coli)等の細菌、酵母(出芽酵母Saccharomyces cerevisiae、分裂酵母Schizosaccharomyces pombe)等を挙げることができるが、特に限定されるものではない。
【0066】
上記発現ベクターをホスト細胞に導入する方法、すなわち形質転換方法も特に限定されるものではなく、電気穿孔法(エレクトロポレーション法)、リン酸カルシウム法、プロトプラスト法、酢酸リチウム法等の従来公知の方法を好適に用いることができる。
【0067】
本発明に係る形質転換キットは、上記(2)に記載した本発明に係るポリヌクレオチドまたは当該本発明に係る組換え発現ベクターの少なくともいずれかを含むものであればよい。その他の具体的な構成については特に限定されるものではなく、必要な試薬や器具等を適宜選択してキットの構成とすればよい。当該形質転換期キットを用いることにより、簡便かつ効率的に形質転換細胞を得ることができる。
【0068】
(5)形質転換体
本発明に係る形質転換体は、上記(2)に記載した本発明に係るポリヌクレオチドまたは上記(4)に記載の組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体であれば、特に限定されるものではない。ここで「形質転換体」とは、細胞・組織・器官のみならず、生物個体を含む意味である。
【0069】
形質転換体の作製方法(生産方法)は特に限定されるものではないが、例えば、上述した組換え発現ベクターをホスト細胞に導入して形質転換する方法を挙げることができる。また、トリプトファン合成系を有する細胞、生物であれば、形質転換の対象となる生物も特に限定されるものではなく、上記(4)においてホスト細胞として例示した各種微生物等を挙げることができる。
【0070】
本発明に係る形質転換体は、植物細胞または植物体であることが好ましい。このような形質転換植物は、トリプトファンの含有量が高く、栄養価の高い食品原料や飼料として高い利用価値を有する。
【0071】
植物体の形質転換に用いられる組換え発現ベクターは、当該植物細胞内で挿入遺伝子を発現させることが可能なものであれば特に限定しない。特に、植物体へのベクターの導入法がアグロバクテリウムを用いる方法である場合には、pBI系等のバイナリーベクターを用いることが好ましい。バイナリーベクターとしては、具体的には、例えば、pBIG、pBIN19、pBI101、pBI121、pBI221等を挙げることができる。また、植物体内で遺伝子を発現させることが可能なプロモーターを有するベクターであることが好ましい。プロモーターとしては公知のプロモーターを好適に用いることができ、具体的には、例えば、カリフラワーモザイクウイルス35Sプロモーター(CaMV35S)、ユビキチンプロモーターやアクチンプロモーターを挙げることができる。なお、植物細胞には、種々の形態の植物細胞、例えば、懸濁培養細胞、プロトプラスト、葉の切片、カルスなどが含まれる。
【0072】
植物細胞への組み換え発現ベクターの導入には、ポリエチレングリコール法、電気穿孔法(エレクトロポレーション法)、アグロバクテリウムを介する方法、パーティクルガン法など、公知の種々の方法を用いることができる。また、形質転換細胞から植物体の再生は、植物細胞の種類に応じて公知の方法で行うことが可能である。
【0073】
ゲノム内に本発明に係るポリヌクレオチドが導入された形質転換植物体がいったん得られれば、当該植物体から得られる種子にも当該ポリヌクレオチドが導入されている。例えば、本発明に係るポリヌクレオチドで形質転換されたイネ等の穀物においては、トリプトファン含量の高い穀物を得ることができる。本発明には、形質転換植物から得られる種子も含まれる。
【0074】
(6)スクリーニング方法およびスクリーニングキット
本発明に係るスクリーニング方法は、(1)に記載した本発明に係るポリペプチド(変異型OASA2タンパク質)および野生型イネアントラニル酸合成酵素(OASA2タンパク質)のいずれか一方のみと結合する物質をスクリーニングする方法であって、変異型OASA2タンパク質と結合する物質をスクリーニングする工程と、野生型OASA2タンパク質結合する物質をスクリーニングする工程と、上記両工程の結果を比較する工程とを含むものであればよい。
【0075】
上記本発明に係るポリペプチド、すなわち変異型OASA2タンパク質はトリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するものであるため、上記変異型OASA2タンパク質および野生型OASA2タンパク質のいずれか一方のみと結合する物質は、トリプトファン生合成経路におけるフィードバック阻害に関与するシグナル物質である可能性が高いと考えられる。このような物質が、当該スクリーニング方法により見出されれば、フィードバック阻害のメカニズムの解明が進み、フィードバック阻害に対する抵抗性が一層強力な変異型OASA2タンパク質の開発に貢献できることが期待される。
【0076】
また、本発明に係るスクリーニングキットは、上記スクリーニング方法を実施するためのキットであって、少なくとも本発明に係るポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むものであればよい。その他の具体的な構成については特に限定されるものではなく、必要な試薬や器具等を適宜選択してキットの構成とすればよい。当該形質転換期キットを用いることにより、簡便かつ効率的に上記本発明に係るスクリーニング方法を実施することができる。
【0077】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0078】
〔実施例1:イネアントラニル酸合成酵素遺伝子OASA2への変異導入〕
<標的遺伝子のクローニングベクターへの挿入>
イネアントラニル酸合成酵素遺伝子OASA2(ACCESSION NO. AB022603)をクローニングベクターpBluescript SK+(Stratagene社製)のマルチクローニングサイトのEcoRIサイトに挿入し、pBS-OASA2を構築した。なお、挿入した遺伝子は、マルチクローニングサイトの制限酵素サイトKpnIからSacIの向きに挿入されている。
【0079】
<変異導入用プライマー>
Kunkel法によって目的タンパク質をコードする遺伝子に変異を導入する場合、変異を導入したい位置にセンス(あるいはアンチセンス)の変異導入用オリゴヌクレオチドを設定して用いる。目的タンパク質をコードする遺伝子にPCRを用いて変異を導入する場合、変異を導入したい位置に2つのプライマー、すなわちセンスおよびアンチセンスのオリゴヌクレオチドを設定して用いる。従って、当該変異導入用プライマーの一方は、上記Kunkel法に用いた変異導入用オリゴヌクレオチと同一のものを使用することができる。この際、遺伝子がコードするアミノ酸が変化しない適当な位置に制限酵素サイトを導入することで変異導入の有無の確認作業ができる。下記i)〜vi)の変異はKunkel法を用いて導入し、下記vii)およびviii)の変異はPCRを用いて導入した。
【0080】
<変異導入用プライマー>
i)351番目のアスパラギン残基をアスパラギン酸残基に置換(N351D)
351番目のアスパラギン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(BsiWI)を導入するように変異導入用プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
N351D-F:5'-GTTTGAGAGGCGAACGTACGCCgatCCATTTGAAGTCT-3'(配列番号15)
プライマーN351D-Fの23番目から25番目のgat(アスパラギン酸、D)および15番目から20番目のCGTACG(BsiWIサイト)は、それぞれ野生型OASA2のAAT(アスパラギン、N)およびCATACGの塩基配列よりデザインした。AATをGATにすることにより351番目のアスパラギン残基がアスパラギン酸残基へ置換される。また、CATACGをCGTACGとすることによりアミノ酸置換を伴わずに(ACA、ACGは共にスレオニン)制限酵素サイト(BsiWI: CGTACG)が導入され、変異導入後の選抜が容易となる。
【0081】
ii)367番目のチロシン残基をアラニン残基に置換(Y367A)
上記i)と同様に、367番目のチロシン残基をアラニン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(SnaBI:TACGTA)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
Y367A-F:5'-GTGAACCCAAGTCCAgccATGGCATACGTACAGGCAAGAGGC-3'(配列番号16)
iii)367番目のチロシン残基をイソロイシン残基に置換(Y367I)
上記i)と同様に、367番目のチロシン残基をイソロイシン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(SnaBI:TACGTA)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
Y367I-F:5'-GTGAACCCAAGTCCAatcATGGCATACGTACAGGCAAGAGGC-3'(配列番号17)
iv)530番目のロイシン残基をアラニン残基に置換(L530A)
上記i)と同様に、530番目のロイシン残基をアラニン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(NheI:GCTAGC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
L530A-F:5'-ACGGAGACATGgctATCGCGCTAGCACTCCGCACCATT-3'(配列番号18)
v)530番目のロイシン残基をアスパラギン酸残基に置換(L530D)
上記i)と同様に、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(NheI:GCTAGC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
L530D-F:5'-ACGGAGACATGgacATCGCGCTAGCACTCCGCACCATT-3'(配列番号19)
vi)522番目のグリシン残基をチロシン残基に、530番目のロイシン残基をアスパラギン酸残基に置換(G522Y+L530D)
上記i)と同様に、522番目のグリシン残基をチロシン残基に、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(BciVI:GTATCC/GGATAC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
G522Y+L530D-F:5'-AGTGGCGGCCTTGGAtacATATCATTTGACGGAGACATGgatATCGCTCTTGCACT-3'(配列番号20)
vii)126番目のセリン残基をフェニルアラニン残基に置換(S126F)
126番目のセリン残基をフェニルアラニン残基にアミノ酸置換するように変異導入用プライマーをデザインした。変異導入法にPCRを用いた手法で変異を導入したのでセンスおよびアンチセンスのオリゴヌクレオチドを作成した。変異導入用プライマーの塩基配列は下記の通りである。なお、小文字は変異導入コドンを示す。
S126F-F :5'-GCTTCCTCTTCGAGttcGTCGAGCAGGGGCC-3'(配列番号37)
S126F-R:5'-GGCCCCTGCTCGACgaaCTCGAAGAGGAAGC-3'(配列番号38)
プライマーS126F-Fの15番目から17番目のttc(フェニルアラニン、F)は、野生型OASA2のTCC(セリン、S)よりデザインした。TCCをTTCにすることにより126番目のセリン残基がフェニルアラニン残基へ置換される。プライマーS126F-RはプライマーS126F-Fの相補鎖である。
【0082】
viii)369番目のアラニン残基をロイシン残基に置換(A369L)
上記i)と同様に、369番目のアラニン残基をロイシン残基にアミノ酸置換するように変異導入用プライマーをデザインした。変異導入法にPCRを用いた手法で変異を導入したのでセンスおよびアンチセンスのオリゴヌクレオチドを作成した。変異導入用プライマーの塩基配列は下記の通りである。なお、小文字は変異導入コドンを示す。
A369L-F:5'-CAAGTCCATACATGctaTATGTACAGGCAA-3'(配列番号39)
A369L-R:5'-TTGCCTGTACATAtagCATGTATGGACTTG-3'(配列番号40)
プライマーA369L-RはプライマーA369L-Fの相補鎖である。
【0083】
<Kunkel法による変異導入DNAの調製>
Kunkel法は、下記の参考文献1−3に記載の方法にしたがって行った。
【0084】
参考文献1:Kunkel, T. A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proceedings of the National Academy of Science of the USA, 82: 488-492.
参考文献2:Kunkel, T. A., Bebenek, K. and McClary, J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods in Enzymology, 204: 125-139.
参考文献3:「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, (2001) Chapter 13.
例えば、351番目のアスパラギン残基をアスパラギン酸残基に置換する場合、前述のpBS-OASA2ベクターを鋳型とし、上記i)に記載した変異導入用プライマーを用いてKunkel法を行うことで変異を導入した。上記ii)、iv)、v)、vi)に記載の変異導入用プライマーを用いる場合も上記と同一の鋳型を用いてKunkel法を行った。以上の操作によりN351D、Y367A、L530A、L530D、G522Y+L530D変異DNAを調製した。
【0085】
N351D+L530Aの二重変異DNAは、変異導入工程を2回繰り返すことにより調製した。すなわち、最初に前述のpBS-OASA2ベクターを鋳型とし、上記i)に記載した変異導入用プライマーを用いてN351D変異DNAを調製した。次に、N351Dの変異が導入されたプラスミドDNA(pBS-OASA2(N351D)ベクター)を鋳型として、上記iv)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(N351D+L530A)を得た。同様に、N351Dの変異が導入されたプラスミドDNA(pBS-OASA2(N351D)ベクター)を鋳型として、上記v)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(N351D+L530D)を得、Y367Aの変異が導入されたプラスミドDNA(pBS-OASA2(Y367A)ベクター)を鋳型として、上記v)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(Y367A+L530D)を得た。
【0086】
三重変異DNAは、上記二重変異DNAを含むプラスミドDNAを鋳型としてKunkel法を行うことにより調製した。すなわち、N351D+Y367A+L530Aは、予めN351D+L530Aの変異が導入されたプラスミドDNA(pBS-OASA2(N351D+L530A)ベクター)を鋳型として、上記ii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(N351D+Y367A+L530A)を得た。同様に、G522Y+L530Dの変異が導入されたプラスミドDNA(pBS-OASA2(G522Y+L530D)ベクター)を鋳型として、上記ii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(Y367A+G522Y+L530A)を得た。同様に、G522Y+L530Dの変異が導入されたプラスミドDNA(pBS-OASA2(G522Y+L530D)ベクター)を鋳型として、上記iii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(Y367I+G522Y+L530A)を得た。
【0087】
<RCR法による変異導入用DNAの調製>
PCR法による変異導入法は下記の参考文献に記載の方法に従って行った。
【0088】
Higuchi R, Krummel B, Saiki RK (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351-7367
OASA2遺伝子が変異導入用のプライマー領域でオーバーラップするように5’側領域と3’側領域に分けてPCRを行った。例えば、上記vii)に記載した126番目のセリンをフェニルアラニンに置換するため変異導入用プライマーを用いる場合には、5’側領域はpBS-OASA2ベクターを鋳型とし、クローニング用センスプライマー(5'-AAAACTAGTATGGAGTCCATCGCCGCCGCCACG-3':配列番号41、下線は制限酵素SpeIサイトを示す)およびプライマーS126F-Rの組み合わせでPCRを行い、 3’領域はpBS-OASA2ベクターを鋳型とし、プライマーS126F-Fおよびクローニング用アンチセンスプライマー(5'-AAAGTCGACTGAGAGAGACTCTATTCCTTGTC-3':配列番号42、下線は制限酵素SalIサイトを示す)との組み合わせでPCRを行った。上記viii)に記載の変異導入用プライマーを用いる場合でも上記と同一の鋳型を用い、同様のプライマーの組み合わせによりPCRを行った。
【0089】
S126F+L530Dの二重変異DNAを調製する場合は、鋳型としてpBS-OASA2ベクターに前述のKunkel法でOASA2の530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する変異を既に導入したプラスミドDNAを用いた。従って、上記vii)に記載の変異導入用プライマー(S126F-FおよびS126F-R)を用いてPCRを行うことにより、OASA2の126番目のセリン残基をフェニルアラニン残基に置換し、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する2つの変異が導入された変異遺伝子が得られることになる。なお、プライマーの組み合わせは上記と同様である。A369L+L530Dの二重変異DNAの調製も上記と同様の鋳型と上記viii)に記載の変異導入用プライマー(A369L-FおよびA369L-R)を用いてPCRを行うことにより、OASA2の369番目のアラニン残基をロイシン残基に置換し、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する2つの変異が導入された変異遺伝子が得られる。
【0090】
PCR反応の組成は、1×Pyrobest buffer II(TaKaRa社)、0.2mM dNTP、0.5μM センスおよびアンチセンスプライマー、0.025 units/μl Pyrobest DNA polymerase(TaKaRa社)、10ng 鋳型DNAとし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分間保持した後4℃に冷却した。
【0091】
増幅したPCR産物(5’領域断片および3’領域断片)を20倍希釈し、PCR反応液[1×Pyrobest buffer II(TaKaRa社)、0.2mM dNTP、0.025 units/μl Pyrobest DNA polymerase(TaKaRa社)]に1μlずつ添加し、上記PCR反応装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を5サイクル繰り返し、伸長反応を行った。この反応によりクローニング用センスプライマーからクローニング用アンチセンスプライマーまで連結されたDNA断片が合成された。
【0092】
上記反応終了後すぐに、プライマーの濃度がそれぞれ0.2μMになるようにクローニング用センスプライマーおよびクローニング用アンチセンスプライマーを添加し、上記PCR装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を20サイクル繰り返し、72℃で10分間保持した後4℃に冷却した。以上の反応により、上記クローニング用センスプライマーからクローニング用アンチセンスプライマーまで連結されたDNA断片が増幅され、これをクローニングベクターpBluescript KS+(Stratagene社)のマルチクローニングサイトのSpeIおよびSalIに挿入し、pBS-OASA2(S126F)、pBS-OASA2(A369L) 、pBS-OASA2(S126F/L530D)、 pBS-OASA2(A369L/L530D)を構築した。
【0093】
〔実施例2:コムギ胚芽無細胞システムによるタンパク質合成〕
(1)Split-PCR法による転写鋳型DNAの合成
<Split-PCR用鋳型の調製>
OASA2遺伝子はイネの核ゲノム上にコードされている遺伝子であるが、合成されたタンパク質は葉緑体に移行して葉緑体内で機能していることが判っている。合成されたタンパク質のN末端領域には葉緑体へ移行するためのシグナル配列が存在し、その配列が除去されることで成熟型酵素となり、酵素活性を発揮する。したがって、OASA2タンパク質を成熟型酵素としてコムギ胚芽無細胞合成系で合成するため、N末端領域に存在する49残基からなるシグナル配列を除去した形で合成されるようにプライマーをデザインした。すなわち、コムギ胚芽無細胞合成システムでSplit-PCRを可能とするリンカー配列の下流にATG開始コドンを配し、OASA2遺伝子の148から164番目の塩基配列からなる全長36merのプライマーをデザインした。また、OASA2遺伝子が挿入されているベクター(pBluescript SK+)上に上記センスSplit-PCR用プライマーの相補鎖側の位置にSplit-PCR用アンチセンスプライマーを2種類設定した。挿入DNAの位置から遠い順にアンチセンスSplit-PCR用プライマー1およびアンチセンスSplit-PCR用プライマー2とした。Split-PCR用のプライマーの塩基配列は下記の通りである。なお、Split-PCRについては後述する。
センスSplit-PCR用プライマー:5'-CCTCTTCCAGGGCCCAATGTGCTCCGCGGGGAAGCC-3'(配列番号21、下線部分がリンカー配列)
アンチセンスSplit-PCR用プライマー1:5'-GGAGAAAGGCGGACAGGTAT-3'(配列番号22)
アンチセンスSplit-PCR用プライマー2:5'-GGGGAAACGCCTGGTATCTT-3'(配列番号23)
上記Kunkel法により変異が導入されたOASA2遺伝子を含むpBluescript SK+ベクターを鋳型として、上記センスSplit-PCR用プライマーおよびアンチセンスSplit-PCR用プライマー1を用いてPCR等の増幅反応を行い、増幅されたDNA断片を次に行うSplit-PCRの鋳型とすることができる。
【0094】
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センスSplit-PCR用プライマーおよびアンチセンスSplit-PCR用プライマー1、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA(各変異を導入したpBS-OASA2ベクター)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。以上の反応により、上記センスSplit-PCR用プライマーからアンチセンスSplit-PCR用プライマー1まで連結されたDNA断片が増幅され、これを次に行うSplit-PCRの鋳型とした。
【0095】
<転写鋳型DNAの合成>
上記Split-PCR用鋳型DNAを転写鋳型DNAとするためには、断片の5'末端上流にSP6 RNAポリメラーゼのプロモーターとタバコモザイクウイルス(TMV)由来の翻訳増強配列(オメガ配列)を付加する必要がある。そのため、Sawasakiらの方法(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)に従い、Split-PCRを行った。すなわち、上記Split-PCR用鋳型DNAを50倍希釈し、PCR反応液[1× EX Taq buffer (TaKaRa社)、0.2mM dNTP、0.025 units/μl TaKaRa EX Taq (TaKaRa社)、0.2μM SP6 promoterプライマー、1nM TMV-Omegaプライマー、0.2μM アンチセンスSplit-PCR用プライマー2]に1μl添加し、上記PCR反応装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を35サイクル繰り返し、72℃で10分保持した後4℃に冷却した。SP6 promoterプライマーおよびTMV-Omegaプライマーの塩基配列は下記の通りである。なお、下線はSP6プロモーター配列を示し、小文字は両プライマーの重複部分を示す。
SP6 promoterプライマー:5'-GCGTAGCATTTAggtgacact-3'(配列番号24)
TMV-Omegaプライマー:5'-ggtgacactATAGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACCTCTTCCAGGGCCCAATG-3'(配列番号25)
ここで、上記SP6 promoterプライマーとTMV-Omegaプライマーのように、プライマーを上流側プライマーと下流側プライマーとに分割し、上流側プライマーの3’末端と下流側プライマーの5’末端との配列が一部重複するように設計されたプライマーをSplit primerと称する。また、このようなプライマーを用いて行うPCR反応をSplit-PCRと称する。
【0096】
(2)コムギ胚芽無細胞システム用のmRNAの合成
上記(1)で得られたPCR産物を直接鋳型として用いてmRNAを合成(転写)した。すなわち、上記(1)で得られたPCR産物を、転写反応液[80mM HEPES-KOH (pH7.8)、16mM Mg(OAc)2、10mM spermidine、10mM DTT、3mM NTP、1 unit/μl RNasin RNase inhibitor (Promega社)、1 unit/μl SP6 RNA polymerase (Promega社)]に1/10量添加した。37℃で2時間反応後、エタノール沈殿、70% エタノール洗浄を行い、適当量の滅菌水に溶解した。260 nmの吸光度を測定してRNA量を算出した。
【0097】
(3)コムギ胚芽無細胞システムによるタンパク質合成
Sawasakiらの方法(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)に従って上記(3)で合成したmRNAを鋳型としてタンパク質を合成した。すなわち、50μlのコムギ胚芽無細胞タンパク質合成液を入れた透析カップに上記mRNA(約30-35μg)を添加し、1ウェルあたり1mlの基質液を入れた24穴プレートに上記透析カップを浸漬し、26℃で24時間インキュベートした。
【0098】
〔実施例3:変異型OASA2タンパク質の活性測定〕
(1)ウエスタンブロット法によるOASA2タンパク質の定量
OASA2タンパク質のアミノ酸配列(配列番号1)の第161位〜第175位の配列(MDHEKGKVTEQVVDD)のペプチド断片に基づいて作製したウサギ抗OASA2抗体とOASA2タンパク質精製標品を用いてコムギ胚芽無細胞システムにより合成されたOASA2タンパク質を定量した。ウエスタンブロット解析はTowbinらの方法(Towbin, H., Staehelin, T. and Gordon, J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354.)に従い行った。上記方法で見積もったOASA2タンパク質量で後述の酵素活性を補正した。
【0099】
(2)OASA2タンパク質の活性測定
イネアントラニル酸合成酵素のαサブユニットであるOASA2タンパク質は、βサブユニットが供給するグルタミンのアミド基由来のアンモニアを利用してコリスミン酸からアントラニル酸を生成する。生体内でOASA2タンパク質は、βサブユニットが供給するアンモニアを利用してアントラニル酸合成活性(以下、適宜「AS活性」と略記する。)を発揮するが、外部よりアンモニア、例えばNH4Clを添加することでも酵素活性を示す。
【0100】
OASA2タンパク質の活性は、周知の通りトリプトファンによってフィードバック抑制がかかるので、上記タンパク質合成反応液中からトリプトファンの除去は必須であり、また発現させたタンパク質の環境をAS活性測定用の緩衝液成分に変える必要がある。したがって、タンパク質合成反応液の成分をMicrospin G-25 columns(Amersham Biosciences社製)を用いてbuffer A(50mM Tris-HCl, pH7.6; 0.05mM EDTA; 2mM MgCl2; 0.05mM DTT; 5% グリセロール)に置換した。
【0101】
<酵素活性測定1>
90μlの反応液(20mM Tris-HCl, pH8.3; 100mM NH4Cl; 0.5mM コリスミン酸; 10mM MgCl2)にbuffer 置換した粗抽出液を2.5μl添加して、32℃で1時間反応させた。反応は10μlの1N HClを添加することで停止させ、300μlの酢酸エチルを添加して合成したアントラニル酸を抽出した。アントラニル酸の合成量は、Wallac 1420 ARVOsx-FL(パーキンエルマーライフサイエンスジャパン社製)を用いて励起波長355 nm/蛍光(emmision)460 nmで測定した。トリプトファンに対するフィードバック阻害活性を解析する場合、反応系に終濃度10μMまたは100μMになるようにトリプトファンを添加した。
【0102】
結果を図1および図2に示した。図1から明らかなように、6種類の変異型OASA2タンパク質(N351D、L530A、L530D、N351D/L530A、N351D/L530DおよびG522Y/L530D)は、野生型(wt)と比較して酵素活性が向上していた。また、図2から明らかなように、7種類の変異型OASA2タンパク質(A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、トリプトファンを添加していない場合の酵素活性が野生型と同程度以上であり、かつ、トリプトファンフィードバック阻害に対する抵抗性を獲得していた。
【0103】
<酵素活性測定2>
上記トリプトファンフィードバック阻害に対する抵抗性を獲得していた5種類の変異型OASA2タンパク質について、より詳細に酵素学的性質を検討するため、イネアントラニル酸合成酵素βサブユニットを用いた反応系において酵素活性およびトリプトファンフィードバック阻害を解析した。
【0104】
イネアントラニル酸合成酵素βサブユニットを過剰量添加した90μlの反応液(20mM Tris-HCl, pH8.3; 5mM グルタミン; 0.2〜0.8mM コリスミン酸; 10mM MgCl2;)にbuffer 置換した粗抽出液を2.5μl添加して、32℃で1時間反応させた。なお、イネアントラニル酸合成酵素βサブユニットの調製およびウエスタンブロット法による合成量の定量は、Kannoらの方法(Kanno et al.(2004) Plant Molecular Biology 54,11-22)に従って行った。反応は10μlの1N HClを添加することで停止させ、300μlの酢酸エチルを添加して合成したアントラニル酸を抽出した。アントラニル酸の合成量は、Wallac 1420 ARVOsx-FL(パーキンエルマーライフサイエンスジャパン社製)を用いて励起波長355 nm/蛍光(emmision)460 nmで測定した。トリプトファンに対するフィードバック阻害活性を解析する場合、反応系に終濃度10μM、25μMまたは50μMになるようにトリプトファンを添加した。
【0105】
結果を表1および図3に示した。表1から明らかなように、9種類の変異型OASA2タンパク質(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、S126F、Y367Aを除き、野生型(wt)と比較して同程度以上の酵素活性を有していた。より詳細には、野生型(wt)と比較してA369Lが約1倍、 S126F/L530Dが約2.5倍、Y367A/L530Dが約2.2倍、A369L/L530Dが約1.8倍、Y367A/G522Y/L530Dが約1.3倍、N351D/Y367A/L530Dが約1.2倍、Y367I/G522Y/L530Dが約0.8倍の酵素活性であった。
【0106】
【表1】

【0107】
また、図3はトリプトファン無添加時における野生型および各変異型OASA2タンパク質酵素活性を100%とし、各濃度のトリプトファンを添加した場合の酵素活性をトリプトファン無添加時の酵素活性と比較したものである。図3から明らかなように、野生型はトリプトファンを50μM以上添加すると酵素活性が無添加時の10%を下回っているが、9種類の変異型OASA2タンパク質(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、トリプトファンを50μM添加した場合においても無添加時の30%以上の酵素活性を有していた。
【0108】
〔実施例4:変異型OASA2遺伝子を発現させた酵母TRP2遺伝子欠損変異株(MATalpha his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 trp2::KANMX)の遊離トリプトファン含量解析〕
OASA2遺伝子に相当するTRP2遺伝子が欠損した酵母は生育のためにトリプトファンを要求する。従って、この欠損株にOASA2遺伝子を導入しトリプトファン非存在下で生育できれば導入したOASA2遺伝子はTRP2の機能を相補できることになる。また、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子をこの酵母で発現させ、生体内に蓄積する遊離トリプトファン含量を測定することで当該遺伝子の効果検定を行うことができる。
【0109】
発現ベクターを構築するために、トリプトファンフィードバック阻害に対する抵抗性を有する変異型OASA2遺伝子(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530D、Y367I/G522Y/L530D)および野生型(Wt)OASA2遺伝子が挿入されたpBluescriptベクターを鋳型として、下記のプライマーを用いてPCRを行った。前述の通りOASA2タンパク質のN末端領域には葉緑体移行シグナルが存在するので、上記(2)に記載の「Split-PCR用プライマー」のデザインと同様にシグナル配列を除去した形で発現するようにプライマーをデザインした。なお、酵母発現ベクターpYES2(Invitrogen社製)の制限酵素サイトKpnI/EcoRIに組み込めるようにセンスプライマーには制限酵素サイトKpnI(GGTACC)、アンチセンスプライマーには制限酵素サイトEcoRI(GAATTC)を導入した。これら制限酵素サイトは下線で示す。なお、pYES2ベクターはURA3遺伝子を有し、ガラクトースによってタンパク質の発現誘導が制御できる。
センスプライマー:5’-AAAGGTACCATGTGCTCCGCGGGGAAGCC-3’(配列番号27)
アンチセンスプライマー:5’-AAAGAATTCTGAGAGAGACTCTATTCCTTGTC-3’(配列番号28)
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センス及びアンチセンスプライマー、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA (pBS-OASA2ベクター)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。
【0110】
ベクターへのDNA断片のクローニングはSambrookらの方法(「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001))に従って行った。構築したベクターの酵母TRP2遺伝子欠損変異株への形質転換など基礎的手法はKaiserらの方法(Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual (Chris Kaiser, Susan Michaelis, Aaron Mitchell, Cold Spring Harbor Laboratory, 1994))に従って行った。
【0111】
酵母形質転換体は、2%グルコースを含む合成完全寒天培地(0.67%アミノ酸を含まないバクトイーストナイトロジェンベース、ウラシルを除いた0.2%ドロップアウト混合物、2%バクトアガー)でシングルコロニーを単離し、次いで菌体を2%ガラクトースを含む合成完全寒天培地(0.67%アミノ酸を含まないバクトイーストナイトロジェンベース、ウラシルとトリプトファンを除いた0.2%ドロップアウト混合物、2%バクトアガー)上に塗り、30℃で二日間培養した。寒天培地上から掻き取った菌体20mgを105μlの蒸留水で懸濁し、100℃で20分間処理した。次いで595μlのクロロホルム:メタノール混合液(5:12、v/v)を加え、よく混合した後、20,000×gの遠心加速度で10分間遠心分離した。得られた上清を新しいチューブに移し、175μlの蒸留水と263μlのクロロホルムを加え、激しく混合した後、20,000×gの遠心加速度で10分間遠心分離した。このようにして抽出された水層を新しいチューブに移し、減圧下で乾固させた後、200μlの10mM NaOHに溶解して、トリプトファン抽出液とした。これを高速液体クロマトグラフィー(HPLC)装置(Waters社製、Waters Alliance HPLC FLD System 2695)に供して遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、Xterra RP18 column (4.6 ×150 mm)(Waters社製)であり、トリプトファンは励起波長278nm/蛍光波長348nmで検出した。
【0112】
結果を表2に示した。表2から明らかなように、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530D、Y367I/G522Y/L530D)を発現させた酵母中で生成される遊離トリプトファン含量は、野生型(Wt)OASA2遺伝子を発現させた酵母のものより高濃度蓄積していることが確認された(野生型の1.9〜2.3倍)。
【0113】
【表2】
【0114】
〔実施例5:変異型OASA2遺伝子発現を発現させたイネカルス形質転換体の作製および当該形質転換カルスの遊離トリプトファン含量解析〕
<外来遺伝子導入用組換えベクターの構築>
外来遺伝子導入用組換えベクターの構築は、下記の参考文献に記載の方法に従って行った。
参考文献1:Urushibara S, Tozawa Y, Kawagishi-Kobayashi M and Wakasa K (2001) Efficient transformation of suspension-cultured rice cells mediated by Agrobacterium tumefaciens. Breeding Science 51: 33-38
参考文献2:Tozawa Y, Hasegawa H, Terakawa T and Wakasa K (2001) Characterization of rice anthranilate synthase alfa subunit genes OSASA1 and OSASA2: tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiology 126: 1493-1506
参考文献3:Hiei Y, Ohta S, Komari T and Kumashiro T (1994) Efficient transformation of rice (Oryza sativa) mediated by Agrobacterium and sequence analysis of boundaries of the T-DNA. Plant Journal 6: 271-282
外来遺伝子導入用組換えベクターpUB-Hm(Urushibara et al., Breeding Science 51: 33-38 (2001))は、トウモロコシのユビキチンプロモーターと、第一イントロンと、制限酵素Sse8387Iサイトと、NOSターミネーターと、ハイグロマイシン耐性遺伝子を有し、Sse8387Iサイトに目的外来遺伝子を挿入することでイネ形質転換用の組換えベクターを構築することができる。
【0115】
全長からなる野生型(Wt)OASA2遺伝子は、〔実施例1〕に記載のpBS-OASA2を鋳型とし、また下記に示すセンスプライマーおよびアンチセンスプライマーを用いてPCRで増幅した。センスプライマーには制限酵素サイトSpeI(ACTAGT)、アンチセンスプライマーには制限酵素サイトSalI(GTCGAC)を導入した。これら制限酵素サイトは下線で示す。
センスプライマー:5'-AAAACTAGTATGGAGTCCATCGCCGCCGCCACG-3'(配列番号43)
アンチセンスプライマー:5'-AAAGTCGACTGAGAGAGACTCTATTCCTTGTC-3'(配列番号44)
また、全長からなる変異型OASA2遺伝子(Y367A、Y367A/L530D)は、pBS-OASA2を鋳型とし、上記センスプライマー(配列番号43)およびアンチセンスプライマー(配列番号44)を用いて〔実施例1〕<RCR法による変異導入用DNAの調製>に記載の方法でDNA断片を調製した。以上のPCRにより増幅したDNA断片を制限酵素SpeIおよびSalIで消化して得られたDNA断片(1)は、Sawasakiらが構築した(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)コムギ胚芽無細胞合成システム用発現ベクターpEU3bを改良し、マルチクローニングサイトSpeI/SalIサイトの両端にSse8387Iサイトを導入したコムギ胚芽無細胞合成システム用発現ベクターpEU3sのSpeI/SalIサイトに挿入した。
【0116】
別途、上述の変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子を含有するpEU3sプラスミドベクターを制限酵素Sse8387Iで消化し、1.8kbサイズのDNA断片(2)を得た。
【0117】
上記の制限酵素Sse8387Iで消化したpUB-HmプラスミドベクターとDNA断片(2)をDNAライゲーション・キットver.2(タカラバイオ社製)で処理して、DNA連結反応を行った。これにより、環状の組換えベクターを構築した。この組換えベクターは、ユビキチンプロモーター領域の下流に第一イントロン領域を有し、第一イントロン領域とNOSターミネーター領域との間に変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子が挿入、連結されてあり、かつ植物中で発現するハイグロマイシン耐性遺伝子を含有する構造を有した。得られた外来遺伝子導入用組換えベクターを、それぞれpUB-OASA2(wt)-Hm(野生型OASA2遺伝子を有する)、pUB-OASA2(Y367A)-Hm(変異型OASA2遺伝子(Y367A)を有する)およびpUB-OASA2(Y367A/L530D)-Hm(変異型OASA2遺伝子(Y367A/L530D)を有する)と称する。
【0118】
図5にpUB-OASA2(wt)-Hm、pBU-OASA2(Y367A)-HmおよびpBU-OASA2(Y367A/L530D)-Hmの模式図を示した。図中PUbiはユビキチンプロモーター領域を表し、P35Sはカリフラワーモザイクウイルス35Sプロモーター領域を表し、nos3'はNOSターミネーター領域を表し、hptはハイグロマイシン耐性遺伝子領域を表し、Sse8387Iは制限酵素サイトを表し、RBは右側境界配列(Right Border)を表し、LBは左側境界配列(Left Border)を表す。
【0119】
<アグロバクテリウムの調製>
YEB培地(バクトペプトン5g/l、バクトビーフエクストラクト5g/l、バクトイーストエクストラクト1g/l、シュクロース5g/l、2mM MgCl2、pH7)の液体培地50mlに、アグロバクテリウム(Agrobacterium tumefaciens EHA101)を移植し、30℃で16時間振とう培養した後、培溶液を4℃で遠心分離により細菌の沈殿を得た。細菌の沈殿物を氷冷した10mM Tris-Hcl pH7.5で懸濁し、遠心分離で沈殿を得た。沈殿物を0.5mlのYEB培地に懸濁した。この菌懸濁液0.2mlに上記3種類の外来遺伝子導入用組換えベクターの溶解液(5μg)をそれぞれ添加し、よく混合した。これらを即座に凍結し、37℃で融解、再度凍結融解を計3回行った。16mlのYEB培地にこの懸濁液を加え、30℃で2時間振とう培養した。この培養液をL 培地(バクトトリプトン10g/l、バクトイーストエクストラクト5g/l、NaCl 5g/l、バクトアガー15g/l、pH7.5)にカナマイシン100mg/lおよびハイグロマイシン50mg/lを添加してなる寒天培地に塗布し、30℃で36時間培養して、生育してきたコロニーを組換えベクターが導入された形質転換アグロバクテリウムとして得た。
【0120】
<イネカルスの調製>
イネ(品種:日本晴)の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約4%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0121】
N6培地の無機塩組成にショ糖30g/l、2,4-D 2mg/l、カザミノ酸1g/lおよびゲルライト2g/l、およびN6ビタミンを添加してなる2N6固体培地(Urushibara et al., Breeding Science 51: 33-38 (2001))に、上記で殺菌したイネ種子を置床した。28℃で3週間インキュベートすることでカルスが形成された。それらカルスを胚乳部分から切り出し、そのカルスを上記と同じ組成の培地に移植して、7日間培養を行った。
【0122】
<イネカルスの形質転換>
上記のようにして得られた、遺伝子導入用の形質転換アグロバクテリウムをAA培地の組成(Hiei et al., Plant Journal 6: 271-282 (1994))にショ糖20g/l、2,4-D 2mg/l、カイネチン0.2mg/lおよびアセトシリンゴン10mg/lを添加してなる液体培地30mlに懸濁し菌液を得た。この懸濁液を9cmのシャーレに入れ、上記により得られたイネのカルスを100個入れた後、5分間浸せきした。浸せき後、ペーパータオルで余分な菌液を除去した後、N6培地の無機塩組成にショ糖30g/l、グルコース10g/l、2,4-D 2mg/l、カザミノ酸1g/l、ゲルライト2g/lおよびアセトシリンゴン10mg/lを添加してなる2N6CO固体培地に、上記の浸せきしたカルスを各シャーレ当たり20個ずつ置床した。24℃で3日間、暗黒下で培養し、イネのカルスへのアグロバクテリウムの感染を行った。
【0123】
<カルスの選抜>
組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩組成にショ糖30g/l、2,4-D 2mg/l、カルベニシリン500mg/l、ハイグロマイシン30mg/l、カザミノ酸1g/l、ゲルライト2mg/lを添加してなる2N6SE固体培地上に各シャーレ当たり20個ずつ移植した。28℃で3週間、暗黒下で培養して、ハイグロマイシン耐性の形質転換カルスを得た。さらに同じ組成の個体培地上へ増殖カルスを移植し、ハイグロマイシン耐性の形質転換カルスの培養を28℃で3週間行った。
【0124】
3週間後、増殖したカルスの一部からDNAを抽出し、下記に示すセンスプライマーおよびアンチセンスプライマーを用いたPCRにより形質転換カルスと同定した。
センスプライマー:5'-GGATGGCACCCGCAGCAGATCG-3' (配列番号45)
アンチセンスプライマー:5'-GTACTCATCACTTGTCATGGTTG-3'(配列番号46)
402塩基対に相当する形質転換用ベクター遺伝子に由来する断片の増幅により、形質転換体におけるOASA2変異遺伝子を確認した。元々含まれるゲノム上のOASA2遺伝子にはイントロン配列が含まれる、形質転換遺伝子にはイントロンが含まれないため、402塩基対のDNA増幅産物は、形質転換用遺伝子にのみ由来する。
【0125】
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センスプライマー(配列番号45)及びアンチセンスプライマー(配列番号46)、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA (カルスDNA)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。
【0126】
さらに、参考文献(Tozawa et al., Plant Physiology 126: 1493-1506 (2001))に記載された方法に従って形質転換カルスよりRNAを抽出し、同文献(Tozawa et al., Plant Physiology 126: 1493-1506 (2001))に記載されたジゴキシゲニン標識したOASA2のRNAプローブを用いる方法に従ってRNAブロットハイブリダイゼーションを行い、上記形質転換により導入したOASA2野生型遺伝子またはOASA2変異型遺伝子((Y367A)あるいは(Y367A/L530D))が選抜したカルスにRNAとして発現していることを確認した。
【0127】
<形質転換カルスの遊離トリプトファン含量解析>
変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子を発現するイネカルスそれぞれ2系統について、各イネカルスの100mgを液体窒素中で粉砕した。粉砕物を1.5ml容のチューブに移し替え、次いで500μlのクロロホルム:メタノール:水混合液(5:12:3の容積比)を加え、よく混合した後、5,000×rpmの遠心加速度で10分間遠心分離した。得られた上清を新しいチューブに移し、375μlの蒸留水と250μlのクロロホルムを加え、激しく混合した後、5,000×rpmの遠心加速度で10分間遠心分離した。このようにして抽出された水層を新しいチューブに移し、減圧下で乾固させた後、2mlの10mM NaOHに溶解して、トリプトファン抽出液とした。これを高速液体クロマトグラフィー(HPLC)装置(Waters社製、Waters Alliance HPLC FLD System 2695)に供して遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、Xterra RP18 column (4.6 ×150 mm)(Waters社製)であり、展開溶媒はアセトリトリル−0.1M H3PO4 水溶液(pH4.0) をアセトニトリルの5%〜45%濃度勾配で用い、流量は1ml/分として、励起波長278nmにおける蛍光波長348nmの測定を行いトリプトファン含量を評価した。
【0128】
対照として、形質転換されていない通常のイネ(品種:日本晴)のカルスを用いた。得られた測定結果を表3に示す。表3から明らかなように、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子(Y367A、Y367A/L530D)を発現させたイネカルス中に生成される遊離トリプトファン含量は、対照のイネカルスより高濃度蓄積していることが確認された(野生型の5.5〜38.8倍)。
【0129】
【表3】
【産業上の利用可能性】
【0130】
本発明により、高トリプトファン蓄積植物の作出が可能となる。したがって、本発明は広く農業に利用することができ、また高トリプトファン蓄積植物は家畜の飼料や食品原料に適しているため、本発明は畜産業や食品産業にも利用することができる。
【図面の簡単な説明】
【0131】
【図1】アミド基供与基質として100mM NH4Clを用いる測定系においてアントラニル酸合成活性を測定した結果を示すグラフであり、変異型OASA2タンパク質のうち、酵素活性が向上した変異体の結果を示すグラフである。
【図2】アミド基供与基質として100mM NH4Clを用いる測定系においてアントラニル酸合成活性を測定した結果を示すグラフであり、変異型OASA2タンパク質のうち、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異体の結果を示すグラフである。
【図3】トリプトファンによるフィードバック阻害に対する抵抗性を有する変異体および野生型のフィードバック阻害の程度を示すグラフである。
【図4】図4Aは野生型OASA2タンパク質の模式図であり、図4Bはトリプトファン合成系を有する生物のトリプトファン結合領域のアミノ酸配列を比較した図である。
【図5】アグロバクテリウム法によるイネカルスの形質転換に用いた変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(wt)OASA2遺伝子を含有する外来遺伝子導入用組換えベクターの模式図である。
【技術分野】
【0001】
本発明は、イネのアントラニル酸合成酵素遺伝子OASA2の新規改変遺伝子およびその利用に関するものであり、特に野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有し、トリプトファンによるフィードバック阻害に対して抵抗性を有する改変イネアントラニル酸合成酵素をコードする新規改変遺伝子およびその利用に関するものである。
【背景技術】
【0002】
トリプトファンはタンパク質を構成するアミノ酸の1つであり、生物が生きていくうえで不可欠なものである。動物はトリプトファンを体内で合成できないため、食物から摂取しなければならない。イネ、トウモロコシ、コムギなどの穀物は、トリプトファンの含有量が非常に少ないため、通常穀物飼料には工業的に製造されたトリプトファンを添加する必要がある。
【0003】
トリプトファンの生合成経路において、コリスミン酸からアントラニル酸が生合成されるが、アントラニル酸の生成はアントラニル酸合成酵素の触媒作用が関与しており、これによってアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成することが知られている(非特許文献1参照)。
【0004】
発明者らは、既にイネアントラニル酸合成酵素アルファサブユニット遺伝子としてOASA1およびOASA2が存在することを見出し、これらを単離した(特許文献1参照)。そして、これらの特徴を詳細に検討した結果、遺伝子発現量が多く、イネにおいてアントラニル酸合成酵素アルファサブユニットとして主に機能するタンパク質をコードする遺伝子はOASA2であると考えられることを報告している(非特許文献2参照)。さらに、発明者らは、OASA2タンパク質は、極めてトリプトファン濃度に感受性が高く、細胞内のトリプトファン濃度が上昇するに伴い活性を低下させてしまうことを報告している(非特許文献3参照)。
【0005】
そこで、OASA2タンパク質の機能を改変することができれば、高濃度のトリプトファンが蓄積される新しい品種の植物を作出することができると考えられる。イネアントラニル酸合成酵素遺伝子の改変に関しては、例えばイネアントラニル酸合成酵素第1アイソザイムアルファサブユニットOASA1タンパク質の323番目のアスパラギン酸残基をアスパラギン残基に置換(D323N)することで得られたトリプトファンフィードバック阻害抵抗性変異体OASA1Dを形質転換すると、野生型よりもカルスで180倍、組換体イネで35倍、遊離のトリプトファン量が増加することが、発明者らにより報告されている(非特許文献2参照)。
【0006】
一方、遺伝子にランダムに変異を導入し、その変異遺伝子の産物である変異タンパク質の機能をスクリーニングすることは多大な時間と労力を必要とする。さらに、目的の位置に複数の変異を同時に導入することは、ランダムに変異を導入する方法では極めて困難であり、ほとんど不可能に近いと考えられている。
【特許文献1】国際公開第WO99/11800号
【非特許文献1】生化学実験講座、第11巻、東京化学同人、1976年、652頁〜653頁
【非特許文献2】Tozawa Y, Hasegawa H, Terakawa T and Wakasa K (2001) Characterization of rice anthranilate synthase alfa subunit genes OSASA1 and OSASA2: tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiology 126: 1493-1506
【非特許文献3】Takuya Kanno, Koji Kasai, Yasuko Ikejiri-Kanno, Kyo Wakasa and Yuzuru Tozawa (2004) In vitro reconstitution of rice anthranilate synthase: distinct functional properties of the alpha subunits OASA1 and OASA2. Plant Molecular Biology 54: 11-22
【発明の開示】
【発明が解決しようとする課題】
【0007】
上述のように、穀物飼料には工業的に製造されたトリプトファンを添加しているが、トリプトファンは他のアミノ酸に比べて高価であるため、トリプトファン含有量の高い穀物の作出が期待されている。そこで、イネアントラニル酸合成酵素遺伝子OASA2に変異を導入することにより、細胞内のトリプトファン濃度が上昇しても酵素活性が低下しないようにOASA2タンパク質の機能を改変することができれば、高濃度のトリプトファンが蓄積された新しい品種の植物を作出することが可能になる。
【0008】
本発明は、上記の問題点に鑑みてなされたものであり、その目的は、細胞内のトリプトファン濃度が上昇しても酵素活性が低下しないように機能改変されたイネアントラニル酸合成酵素および当該酵素をコードする新規改変遺伝子を提供し、高濃度のトリプトファンを含有する新しい品種の植物を実現することにある。
【課題を解決するための手段】
【0009】
本発明者らは、上記課題を解決するために鋭意検討した結果、イネアントラニル酸合成酵素遺伝子OASA2に変異を導入することにより、トリプトファンによるフィードバック阻害に対する抵抗性を有するタンパク質を創出した。さらに、発明者らは上記野生型イネアントラニル酸合成酵素遺伝子OASA2において複数の変異を導入し、これらを組み合わせることにより、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有するタンパク質を創出することに成功し、本発明を完成させるに至った。
【0010】
すなわち、本発明に係るポリペプチドは、アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴としている。上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることが好ましい。
【0011】
また、本発明に係るポリペプチドは、配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴としている。さらに、上記変異に加えて配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有することが好ましい。トリプトファンによるフィードバック阻害に対する抵抗性を有し、さらに野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有するポリペプチドは、細胞内のトリプトファン濃度が上昇してもトリプトファンを合成することができ、当該ポリペプチドを発現している植物は高濃度のトリプトファンを含有する栄養価の高い植物として有用となる。
【0012】
上記配列番号1に示されるアミノ酸配列の第126位の変異は、セリンをフェニルアラニンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第367位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第369位の変異は、アラニンをロイシンに置換する変異であることが好ましい。また、上記配列番号1に示されるアミノ酸配列の第351位の変異は、アスパラギンをアスパラギン酸に置換する変異であり、上記配列番号1に示されるアミノ酸配列の第522位の変異は、グリシンをチロシンに置換する変異であり、上記配列番号1に示されるアミノ酸配列の第530位の変異は、ロイシンをアラニンまたはアスパラギン酸に置換する変異であることが好ましい。上記に挙げたアミノ酸置換により、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異型イネアントラニル酸合成酵素、またはトリプトファンによるフィードバック阻害に対する抵抗性と野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性とを有する変異型イネアントラニル酸合成酵素が実現できる。
【0013】
上記変異型イネアントラニル酸合成酵素としては、配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドを挙げることができる。これらのアミノ酸配列を有することにより、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異型イネアントラニル酸合成酵素、またはトリプトファンによるフィードバック阻害に対する抵抗性と野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性とを有する変異型イネアントラニル酸合成酵素が実現できる。
【0014】
本発明に係るポリヌクレオチドは、上記本発明に係るポリペプチドをコードするポリヌクレオチドである。また、本発明に係るポリヌクレオチドは配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチドであることが好ましい。当該ポリヌクレオチドを細胞に導入することにより、細胞内で上記本発明に係るポリペプチドを発現できる形質転換体を作製することが可能となる。
【0015】
本発明に係る形質転換体選抜用マーカー遺伝子は、上記本発明に係るポリヌクレオチドからなるものである。本発明に係るポリヌクレオチドがコードするポリペプチドは、それが発現している細胞にトリプトファン類縁化合物に対する耐性を付与するので、当該トリプトファン類縁化合物に対する耐性を発現している形質転換細胞を選抜するためのマーカー遺伝子として利用することができる。
【0016】
本発明に係る組換え発現ベクターは、上記本発明に係るポリヌクレオチドからなるものである。当該組換え発現ベクターは、本発明に係るポリヌクレオチドを細胞に導入するための組換え発現ベクターとして利用できるだけでなく、本発明に係るポリヌクレオチドを選抜マーカーとして用いた場合には、他の遺伝子を細胞に導入するための組換え発現ベクターとしても利用できる。
【0017】
本発明に係る形質転換体は、上記本発明に係るポリヌクレオチドまたは上記本発明に係る組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体である。形質転換体は、植物細胞または植物体であることが好ましい。トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換植物は、高濃度のトリプトファンを含有する栄養価の高い植物として有用となる。また、本発明には上記形質転換植物から得られる種子も含まれる。
【0018】
本発明に係る形質転換細胞の選抜方法は、本発明に係るマーカー遺伝子または本発明に係る組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜することからなる選抜方法である。当該遺伝子を選択マーカーとして用いることで、イネ等の単子葉植物で利用できるマーカーの種類が限られるといった問題が解消される。また、これまで複数遺伝子を細胞に導入する際に使用できる選択マーカーの種類が制限されるといった問題に対しても、当該遺伝子を使用することで解決される。すなわち、通常選択マーカーとして使用されているハイグロマイシン等の抗生物質に対する耐性等の他に、トリプトファン類縁化合物、例えば5−メチルトリプトファンに対する抵抗性によって複数の目的DNAが導入された細胞の選抜が可能となる。さらに、当該マーカー遺伝子はイネ由来の遺伝子であるため、イネの形質転換体において本遺伝子がコードするタンパク質は低抗原性であることが期待される。
【0019】
本発明に係る形質転換キットは少なくとも本発明に係るポリヌクレオチド、あるいは本発明に係る組換え発現ベクターのいずれかを含むことを特徴としている。当該形質転換キットを用いれば、本発明に係るポリペプチドを発現する形質転換体を簡便かつ効率的に得ることができる。
【0020】
本発明に係るスクリーニング法は、本発明に係るポリペプチドおよび野生型イネアントラニル酸合成酵素のいずれか一方のみと結合する物質をスクリーニングする方法であって、本発明に係るポリペプチドと結合する物質をスクリーニングする工程と、野生型イネアントラニル酸合成酵素と結合する物質をスクリーニングする工程と、上記両工程の結果を比較する工程とを含むことを特徴としている。当該スクリーニング方法により、トリプトファン生合成経路におけるトリプトファンによるフィードバック阻害に関与する物質をスクリーニングすることができ、フィードバック阻害に対する抵抗性がより強力な変異型イネアントラニル酸合成酵素の作出に繋がる可能性がある。
【0021】
本発明に係るスクリーニングキットは上記本発明に係るスクリーニング方法を実現するためのキットであって、少なくとも本発明に係るポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むことを特徴としている。当該スクリーニングキットを用いれば、本発明に係るスクリーニング方法を簡便かつ効率的に実施することができる。
【発明の効果】
【0022】
本発明に係るポリペプチドは、トリプトファン合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と同程度以上の酵素活性を有している。したがって、高濃度のトリプトファン存在下においてもトリプトファンを合成することができるという効果を奏する。
【0023】
本発明に係るポリヌクレオチドは上記本発明に係るポリペプチドをコードしている。したがって、当該ポリヌクレオチドを植物細胞に導入することにより、高濃度のトリプトファン存在下においてもトリプトファンを合成することができる形質転換植物を作出することができるという効果を奏する。
【0024】
さらに、本発明に係る形質転換植物はトリプトファン含量が高く栄養価に優れた食品や飼料として利用できるという効果を奏する。さらに、トリプトファン含量の高い飼料用イネを作出することにより、家畜用飼料のコストを低下できるという効果を奏すると共に、水田を有効利用し、我国の飼料の自給率を高めることができるという効果を奏する。
【発明を実施するための最良の形態】
【0025】
本発明の実施の一形態について説明すれば、以下の通りである。なお、本発明は、これに限定されるものではない。
【0026】
(1)本発明に係るポリペプチド
本発明に係るポリペプチドは、アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることが好ましい。
【0027】
図4Aにイネアントラニル酸合成酵素遺伝子OASA2(以下、適宜「OASA2遺伝子」と称する。)がコードするイネアントラニル酸合成酵素(以下、適宜「OASA2タンパク質」または「野生型OASA2タンパク質」と称する。)の模式図を示した。図4A中の数字はアミノ酸の位置を表す。cTPは葉緑体移行シグナル、I、IIおよびIIIはトリプトファン結合領域に位置するアミノ酸残基が存在するドメインを表す。また、図4Bに種々のトリプトファン合成系を有する生物のトリプトファン結合領域I、IIおよびIIIのアミノ酸配列の比較を示した。図4B中のOs1はイネOASA1 (Accession no. AB022602)、Os2はイネOASA2 (Accession no. AB022603)、At1はシロイヌナズナ(Arabidopsis)ASA1 (Accession no. M92353)、At2はシロイヌナズナ(Arabidopsis)ASA2 (Accession no. M92354)、Ssは好熱性古細菌(Sulfolobus solfataricus)TrpE (Accession no. 1QDL_A)、Stはサルモネラ菌(Salmonella tryphimurium)TrpE (Accession no. 1I1Q_A)、Smはセラチア菌(Serratia marcescens)TrpE (Accession no. 1I7Q_A)を表す。
【0028】
図4Aおよび図4Bから明らかなように、配列番号26に示されるアミノ酸配列(NPSPYM)は、トリプトファン結合領域IIにおいて、例示した全ての生物に保存されている。したがって、トリプトファン結合領域の中でも非常に重要な役割を果たしていると考えられている。このような保存されたアミノ酸配列を有する生物種としては、図4Bに例示した生物種以外にもニチニチソウ(Catharanthus roseus)α subunit (Accession no. CAC29060)、ヘンルーダ(Ruta graveolens)ASα1 (Accession no. L34343)、ヘンルーダ(Ruta graveolens)ASα2 (Accession no. L34344)、タバコ(tobacco)ASA2 (Accession no. T01990)、酵母(Saccharomyces cerevisiae)TRP2(Accession no. X68327)、大腸菌(Escherichia coli)TrpE (Accession no. V00368)、枯草菌(Bacillus subtilis)TrpE (Accession no. P03963)を挙げることができるが、これらに限定されるものではない。
【0029】
上記配列番号26に示されるアミノ酸配列は、例えば配列番号1に示されるイネアントラニル酸合成酵素遺伝子OASA2がコードする野生型OASA2タンパク質のアミノ酸配列では第363位〜第367位に該当する。そして、当該配列番号26に示される保存されたアミノ酸配列中のチロシンが他のアミノ酸、好ましくはアラニンまたはイソロイシンに置換されることにより、トリプトファンによるフィードバック阻害に対する抵抗性が獲得される。
【0030】
本発明に係るポリペプチドは、配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。上記配列番号1に示されるアミノ酸配列からなるポリペプチドは、イネアントラニル酸合成酵素遺伝子OASA2がコードする野生型イネアントラニル酸合成酵素である。すなわち、イネアントラニル酸合成酵素遺伝子OASA2(OASA2遺伝子)がコードする野生型イネアントラニル酸合成酵素(野生型OASA2タンパク質)の第126位、第367位または第369位の少なくとも1つに変異を有し、かつ、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであればよい。変異としては特に限定されるものではなく、例えばアミノ酸の置換、欠失、付加等を挙げることができる。なお、説明の便宜上、変異を有するOASA2タンパク質を、変異の位置、種類、数等を問わず、「変異型OASA2タンパク質」と称する。
【0031】
植物体中のトリプトファンの生合成経路においては、コリスミン酸からアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成する(非特許文献1参照)。アントラニル酸合成酵素は、上記経路中、コリスミン酸からアントラニル酸の生成を触媒する。また、アントラニル酸合成酵素は最終生成物であるトリプトファンによりフィードバック阻害を受け、細胞中のトリプトファン濃度が上昇するに伴い、酵素活性が低下する。したがって、トリプトファンがある一定量細胞に蓄積すると、それ以上トリプトファンが合成されなくなる。本発明に係るポリペプチドは、上述のようにトリプトファンによるフィードバック阻害に対して抵抗性を有するため、細胞中のトリプトファン濃度が上昇してもトリプトファンを合成することができ、トリプトファン含量の高い植物体を作出することが可能となる。
【0032】
トリプトファンによるフィードバック阻害に対する抵抗性は、例えばin vitro酵素活性測定系において、野生型OASA2タンパク質の酵素活性を測定した場合に、トリプトファン無添加の場合の酵素活性を100%としたとき、トリプトファンを添加した場合の酵素活性の比率(パーセンテージ)を基準として判断することができる。より具体的には、例えば図3に示すように、野生型OASA2タンパク質および変異型OASA2タンパク質のそれぞれのトリプトファン無添加時の酵素活性を100%とし、トリプトファン添加時の酵素活性の比率(パーセンテージ)を比較した場合に、野生型OASA2タンパク質のトリプトファン添加時の酵素活性の比率(パーセンテージ)を超えていれば、トリプトファンによるフィードバック阻害に対する抵抗性を有するといえる。野生型OASA2タンパク質のトリプトファン添加時の酵素活性の比率(パーセンテージ)をどの程度超えているかについては特に限定されるものではないが、少なくとも2倍以上であることが好ましく、少なくとも3倍以上であることがより好ましく、少なくとも4倍以上であることが特に好ましく、少なくとも5倍以上であることが最も好ましい。
【0033】
より具体的には、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系を用いて酵素活性を測定した場合において、トリプトファン無添加時の酵素活性(アントラニル酸合成量)を100%としたとき、100μMのトリプトファンを添加した場合の酵素活性(アントラニル酸合成量)が20%以上残存するものが好ましく、30%以上残存するものがより好ましく、40%以上残存するものが特に好ましく、50%以上残存するものが最も好ましい。
【0034】
OASA2タンパク質の第126位のセリンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、フェニルアラニンが好ましい。具体的には、配列番号29に示されるアミノ酸配列からなるポリペプチド(第126位のセリンをフェニルアラニンに置換)、または配列番号29に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0035】
OASA2タンパク質の第367位のチロシンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、アラニン、イソロイシン、フェニルアラニン、バリンが好ましく、アラニン、イソロイシンがより好ましい。具体的には、配列番号2に示されるアミノ酸配列からなるポリペプチド(第367位のチロシンをアラニンに置換)、および配列番号3に示されるアミノ酸配列からなるポリペプチド(第367位のチロシンをイソロイシンに置換)、または配列番号2もしくは3に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0036】
OASA2タンパク質の第369位のアラニンと置換されるアミノ酸は、上記フィードバック阻害に対する抵抗性の条件を満たすものであれば、特に限定されるものではないが、ロイシンが好ましい。具体的には、配列番号30に示されるアミノ酸配列からなるポリペプチド(第369位のアラニンをロイシンに置換)、または配列番号30に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドであることが好ましい。
【0037】
本発明に係るポリペプチドとしては、上記OASA2タンパク質の第126位、第367位または第369位の少なくとも1つの変異に加えて、第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであることが好ましい。上記第351位、第522位、第530位のいずれか1つ、または2つ以上を組み合わせた変異を有する変異型OASA2タンパク質は、野生型OASA2タンパク質と比較して酵素活性が向上することが発明者らにより確認されている(図1参照)。これらの酵素活性が上昇する変異と上記トリプトファンによるフィードバック阻害に対する抵抗性を獲得する変異を組み合わせることにより、トリプトファンによるフィードバック阻害に対する抵抗性を有し、かつ、酵素活性が野生型OASA2タンパク質と同程度以上である変異型OASA2タンパク質を得ることができる。ここでいう酵素活性とは、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系を用いて測定したアントラニル酸合成量を挙げることができる。ただし、酵素活性を測定するための方法はこれに限定されるものではなく、アントラニル酸合成酵素活性を測定可能公知の酵素活性測定方法または、これらの改変方法であればよい。
【0038】
また、酵素活性が同程度以上とは、例えばin vitro酵素活性測定系(トリプトファン無添加)において、変異型OASA2タンパク質の酵素活性が野生型OASA2タンパク質の酵素活性の少なくとも0.7倍以上であることが好ましく、少なくとも0.8倍以上であることがより好ましく、少なくとも0.9倍以上であることが特に好ましく、少なくとも1倍以上であることが最も好ましい。より具体的には、例えば、後述の実施例3(2)<酵素活性測定1>に示す活性測定系(トリプトファン無添加)を用いて酵素活性を測定した場合に、変異型OASA2タンパク質のアントラニル酸合成量が野生型OASA2タンパク質のアントラニル酸合成量の少なくとも0.7倍以上であることが好ましく、少なくとも0.8倍以上であることがより好ましく、少なくとも0.9倍以上であることが特に好ましく、少なくとも1倍以上であることが最も好ましい。
【0039】
OASA2タンパク質の第351位はアスパラギンであり、第522位はグリシンであり、第530位はロイシンである。これらのアミノ酸とそれぞれ置換されるアミノ酸としては、それぞれの変異、またはこれらを組み合わせた変異と、さらに第126位、第367位または第369位の少なくとも1つの変異を組み合わせたときに、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するものであればよく、特に限定されるものではない。第351位のアスパラギンと置換されるアミノ酸としてはアスパラギン酸が好ましく、第522位のグリシンと置換されるアミノ酸としてはチロシンが好ましい。また、第530位のロイシンと置換されるアミノ酸としてはアラニン、アスパラギン酸が好ましく、アスパラギン酸がより好ましい。
【0040】
具体的には、以下に示す変異の組み合わせを有するポリペプチドが好ましい。
i)第367位のチロシンをアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号4)。
ii)第351位のアスパラギンをアスパラギン酸に置換し、第367位のチロシンをアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号5)。
iii)第367位のチロシンをアラニンに置換し、第522位のグリシンをチロシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号6)。
iv)第367位のチロシンをイソロイシンに置換し、第522位のグリシンをチロシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号7)。
v)第369位のアラニンをロイシンに置換したポリペプチド(配列番号30)。
vi)第126位のセリンをフェニルアラニンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号31)。
vii)第369位のアラニンをロイシンに置換し、第530位のロイシンをアスパラギン酸に置換したポリペプチド(配列番号32)。
【0041】
すなわち、本発明に係るポリペプチドは配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するポリペプチドであることが好ましい。
【0042】
上記「1もしくは数個のアミノ酸が欠失、置換、もしくは付加された」とは、部位特異的突然変異誘発法等の公知の変異ポリペプチド作製法により欠失、置換、もしくは付加ができる程度の数(例えば20個以下、好ましくは10個以下、より好ましくは7個以下、さらに好ましくは5個以下、特に好ましくは3個以下)のアミノ酸が置換、欠失、もしくは付加されることを意味する。このような変異ポリペプチドは、公知の変異ポリペプチド作製法により人為的に導入された変異を有するポリペプチドに限定されるものではなく、天然に存在する同様の変異ポリペプチドを単離精製したものであってもよい。
【0043】
なお、本発明に係るポリペプチドは、アミノ酸がペプチド結合してなるポリペプチドであればよいが、これに限定されるものではなく、ポリペプチド以外の構造を含むものであってもよい。ここでいうポリペプチド以外の構造としては、糖鎖やイソプレノイド基等を挙げることができるが、特に限定されるものではない。
【0044】
また、本発明に係るポリペプチドは、付加的なポリペプチドを含むものであってもよい。このようなポリペプチドが付加される場合としては、例えば、HisやMyc、Flag等によってエピトープ標識されるような場合が挙げられる。
【0045】
また、本発明に係るポリペプチドは、後述する本発明に係るポリヌクレオチド(本発明に係るポリペプチドをコードするポリヌクレオチド)を宿主細胞に導入して、それがコードするポリペプチドを細胞内発現させた状態であってもよいし、細胞、組織などから単離精製された状態であってもよい。また、上記宿主細胞での発現条件によっては、本発明に係るポリペプチドは、他のポリペプチドとつながった融合タンパク質であってもよい。さらに本発明に係るポリペプチドは、化学合成されたものであってもよい。
【0046】
本発明に係るポリペプチドの取得方法(生産方法)は特に限定されるものではないが、本発明に係るポリペプチドは野生型OASA2タンパク質に1つ以上の変異を導入したものであるため、OASA2遺伝子の塩基配列に人工的に変異を導入した変異遺伝子を作製し、当該変異遺伝子にコードされたポリペプチドを発現させる方法により取得することが最適である。塩基配列に変異を導入する公知の方法としては、後述の実施例において発明者らが用いているKunkel法やPCRを利用して塩基配列に変異を導入する方法等を挙げることができる。また市販のキット(例えば、Mutan-K、Mutan-Super Express Km、LA-PCR in vitro mutagenesis Kit(いずれもタカラバイオ社製)等)を用いることもできる。このようにして人工的に変異を導入したOASA2遺伝子を適当な発現ベクターに挿入し、適当な宿主細胞に導入して宿主内で翻訳されて得られる産物(ポリペプチド)を生成することにより、本発明に係るポリペプチドを取得することができる。また、後述の実施例において発明者らが用いているようにコムギ胚芽無細胞システム等の公知の無細胞タンパク質合成系を用いて上記変異を導入したOASA2遺伝子の産物(ポリペプチド)を取得することもできる。本発明に係るポリペプチドの取得方法の1例は、後述の実施例において詳細に説明する。
【0047】
(2)本発明に係るポリヌクレオチド
本発明に係るポリヌクレオチドは、上記本発明に係るポリペプチドをコードするものであればよい。本発明に係るポリペプチドについては上記(1)で詳細に説明したので、ここでの説明は省略するが、例えば、具体的には、以下の〔1〕および〔2〕に記載のポリペプチドをコードするポリヌクレオチドであればよい。
〔1〕配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチド。
〔2〕上記〔1〕の変異に加えて、配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有すると共に、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有するポリペプチド。
【0048】
例えば、配列番号2に示されるアミノ酸配列、すなわちOASA2タンパク質のアミノ酸配列の第367位チロシンがアラニンに置換されたポリペプチドをコードするポリヌクレオチドとしては、配列番号8に示される塩基配列(OASA2遺伝子のオープンリーディングフレーム領域の塩基配列)からなるポリヌクレオチドの1099、1100および1101位の塩基配列(tac:チロシン)がアラニンに対応するコドン、すなわちgct、gcc、gcaまたはgcgのいずれかに変異したポリヌクレオチドであればよい。また、1099〜1101位の塩基以外の塩基配列は配列番号8に示される塩基配列と同一でなくてもよく、例えばコードするアミノ酸が変異しない塩基置換を有するポリヌクレオチドでもよい。
【0049】
上記に例示した以外のアミノ酸変異を有するポリペプチドをコードするポリヌクレオチドについても同様であり、配列番号8に示される塩基配列のうち変異するアミノ酸に対応する位置の3つの塩基が、置換されるアミノ酸に対応するコドンの塩基に置換されたポリヌクレオチド、または当該塩基の変異とそれ以外の塩基配列におけるアミノ酸が変異しない塩基置換とを有するポリヌクレオチドであればよい。
【0050】
本発明に係るポリヌクレオチドとしては、以下の〔3〕および〔4〕に記載のポリペプチドをコードするポリヌクレオチドであることが好ましい。
〔3〕配列番号2、3、29または30に示されるアミノ酸配列からなるポリペプチド、または配列番号2、3、29または30に示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチド。
〔4〕配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号4ないし7および配列番号30ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチドであって、トリプトファンによるフィードバック阻害に対する抵抗性を有すると共に野生型OASA2タンパク質の0.7倍以上の酵素活性を有するポリペプチド。
【0051】
すなわち、配列番号2〜7および配列番号29〜32に示されるアミノ酸配列のいずれかをコードする塩基配列からなるポリヌクレオチドであればよく、具体的な塩基配列は特に限定されるものではない。ただし、配列番号8に示される塩基配列、すなわちOASA2遺伝子のオープンリーディングフレーム領域の塩基配列からなるポリヌクレオチドと相同性の高いポリヌクレオチドであることが好ましいことはいうまでもない。相同性としては、例えば、少なくとも90%の同一性、好ましくは少なくとも95%の同一性、最も好ましくは少なくとも97%の同一性を有していればよい。
【0052】
本発明に係るポリヌクレオチドとしては、配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチドを挙げることができる。ただし、これらに限定されるものではない。配列番号9ないし14および配列番号33ないし36に示される塩基配列からなるポリヌクレオチドは、それぞれ配列番号2ないし7および配列番号29ないし32に示されるアミノ酸配列からなるポリペプチドをコードする塩基配列からなるポリヌクレオチドである。配列番号9ないし14および配列番号33ないし36に示される塩基配列からなるポリヌクレオチドは、発明者らが、後述の実施例に記載の方法を用いて作製したポリヌクレオチドである。
【0053】
上記「ストリンジェントな条件」とは、少なくとも90%の同一性、好ましくは少なくとも95%の同一性、最も好ましくは少なくとも97%の同一性が配列間に存在するときにのみハイブリダイゼーションが起こることを意味し、例えば、60℃で2×SSC 洗浄条件下で結合することを意味する。上記ハイブリダイゼーションは、「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001) に記載されている方法等、従来公知の方法で行うことができる。通常、温度が高いほど、塩濃度が低いほどストリンジェンシーは高くなる。
【0054】
本発明に係るポリヌクレオチドにはDNAおよびRNAが含まれ、一本鎖でも二本鎖でもよい。また、非翻訳領域(UTR)の配列やベクター配列(発現ベクター配列を含む)などの配列を含むものであってもよい。
【0055】
本発明に係るポリヌクレオチドの取得方法(生産方法)は特に限定されるものではないが、例えば、OASA2遺伝子の塩基配列に人工的に変異を導入する方法を挙げることができる。塩基配列に変異を導入する公知の方法としては、Kunkel法やPCRを利用して塩基配列に変異を導入する方法等を挙げることができる。また市販のキット(例えば、Mutan-K、Mutan-Super Express Km、LA-PCR in vitro mutagenesis Kit(いずれもタカラバイオ社製)等)を用いることもできる。本発明に係るポリヌクレオチドの取得方法の1例は、後述の実施例において詳細に説明する。
【0056】
(3)形質転換用マーカー遺伝子および形質転換細胞の選抜方法
本発明に係るポリヌクレオチドは、形質転換用マーカー遺伝子として利用することができる。すなわち、本発明に係る形質転換体選抜用マーカー遺伝子は、上記(2)に記載した本発明に係るポリヌクレオチドからなるものであればよい。本発明に係るポリヌクレオチドを導入された細胞はトリプトファン耐性となるため、トリプトファン類縁化合物を培地に添加することにより、当該ポリヌクレオチドが導入された細胞のみを選抜することが可能となる。選抜用薬剤として用いることができる細胞の増殖を阻害するトリプトファン類縁化合物としては、例えば、5−メチルトリプトファン(5MT)、4−メチルトリプトファン(4MT)、6−メチルトリプトファン(6MT)、7−メチルトリプトファン(7MT)、6−メチルアントラニル酸(6MA)、5−メチルアントラニル酸(5MA)、3−メチルアントラニル酸(3MA)、5−フルオロアントラニル酸(5FA)、6−フルオロアントラニル酸(6FA)等を挙げることができる。
【0057】
具体的に、本発明に係るポリヌクレオチドを形質転換用マーカー遺伝子として利用するには、例えば、当該ポリヌクレオチドを組み込んだ発現ベクターを構築し、当該発現ベクターを目的の細胞に導入する。当該発現ベクターが導入され、本発明に係るポリヌクレオチドにコードされるポリペプチドが発現している細胞はトリプトファン耐性を獲得するため、上記例示した選抜用薬剤が添加された培地においても増殖ができ、一方、当該発現ベクターが導入されていない細胞や本発明に係るポリヌクレオチドにコードされるポリペプチドが発現していない細胞は増殖が阻害される。したがって、当該発現ベクターが導入され、本発明に係るポリヌクレオチドにコードされるポリペプチドが発現している形質転換細胞のみを選抜することが可能となる。
【0058】
上述の例では本発明に係るポリヌクレオチドを形質転換細胞に発現させる遺伝子とマーカー遺伝子との両方の目的で用いているが、本発明に係るポリヌクレオチドをマーカー遺伝子としてのみ用いることも可能である。また、例えば、植物のカルス細胞に特異的な転写プロモーターを使用することにより、本発明に係るポリヌクレオチドの選択マーカーとしての発現時期の制御も可能である。この場合は、さらに目的の細胞内で発現させたいタンパク質をコードする遺伝子を挿入した発現ベクターを構築し、当該発現ベクターを用いて形質転換すればよい。また、本発明に係るポリヌクレオチドを組み込んだ発現ベクターを構築せずに、本発明に係るポリヌクレオチドを単独で目的の細胞に導入することも可能である。
【0059】
本発明に係るポリヌクレオチドを選択マーカーとして用いることで、イネ等の単子葉植物で利用できるマーカーの種類が限られるといった問題が解消される。また、これまで複数遺伝子を細胞に導入する際に使用できる選択マーカーの種類が制限されるといった問題に対しても、当該遺伝子を使用することで解決される。すなわち、通常選択マーカーとして使用されているハイグロマイシン等の抗生物質に対する耐性等の他に、トリプトファン類縁化合物、例えば5−メチルトリプトファンに対する抵抗性によって複数の目的DNAが導入された細胞の選抜が可能となる。さらに、当該マーカー遺伝子はイネ由来の遺伝子であるため、イネの形質転換体において本遺伝子がコードするタンパク質は低抗原性であることが期待される。
【0060】
なお、本発明には上記本発明に係るマーカー遺伝子または以下に説明する組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜する形質転換細胞の選抜方法も含まれる。
【0061】
(4)組換え発現ベクターおよび形質転換キット
本発明に係る組換え発現ベクターは、上記(2)に記載した本発明に係るポリヌクレオチドを含むものであれば、特に限定されるものではない。例えば、cDNAが挿入された組換え発現ベクターが挙げられる。組換え発現ベクターの作製には、プラスミド、ファージ、またはコスミドなどを用いることができるが特に限定されるものではない。また、作製方法も公知の方法を用いて行えばよい。
【0062】
ベクターの具体的な種類は特に限定されるものではなく、ホスト細胞中で発現可能なベクターを適宜選択すればよい。すなわち、ホスト細胞の種類に応じて、確実に遺伝子を発現させるために適宜プロモーター配列を選択し、これと本発明に係るポリヌクレオチドを各種プラスミド等に組み込んだものを発現ベクターとして用いればよい。
【0063】
本組換え発現ベクターは、本発明に係るポリペプチドを発現させるために用いることができることはいうまでもないが、本発明に係るポリヌクレオチドをマーカー遺伝子として利用し、他の遺伝子を組み込んで当該他の遺伝子がコードするタンパク質を発現させるための組換え発現ベクターとしても利用できる。
【0064】
本発明に係るポリヌクレオチドがホスト細胞に導入されたか否か、さらにはホスト細胞中で確実に発現しているか否かを確認するために、各種マーカーを用いてもよい。例えば、ハイグロマイシンのような抗生物質に抵抗性を与える薬剤耐性遺伝子をマーカーとして用い、このマーカーと本発明に係るポリヌクレオチドとを含むプラスミド等を発現ベクターとしてホスト細胞に導入する。これによってマーカー遺伝子の発現から本発明の遺伝子の導入を確認することができる。
【0065】
上記ホスト細胞は、トリプトファン合成系を有する細胞、生物であれば、特に限定されるものではなく、従来公知の各種細胞を好適に用いることができる。具体的には、例えば、大腸菌(Escherichia coli)等の細菌、酵母(出芽酵母Saccharomyces cerevisiae、分裂酵母Schizosaccharomyces pombe)等を挙げることができるが、特に限定されるものではない。
【0066】
上記発現ベクターをホスト細胞に導入する方法、すなわち形質転換方法も特に限定されるものではなく、電気穿孔法(エレクトロポレーション法)、リン酸カルシウム法、プロトプラスト法、酢酸リチウム法等の従来公知の方法を好適に用いることができる。
【0067】
本発明に係る形質転換キットは、上記(2)に記載した本発明に係るポリヌクレオチドまたは当該本発明に係る組換え発現ベクターの少なくともいずれかを含むものであればよい。その他の具体的な構成については特に限定されるものではなく、必要な試薬や器具等を適宜選択してキットの構成とすればよい。当該形質転換期キットを用いることにより、簡便かつ効率的に形質転換細胞を得ることができる。
【0068】
(5)形質転換体
本発明に係る形質転換体は、上記(2)に記載した本発明に係るポリヌクレオチドまたは上記(4)に記載の組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体であれば、特に限定されるものではない。ここで「形質転換体」とは、細胞・組織・器官のみならず、生物個体を含む意味である。
【0069】
形質転換体の作製方法(生産方法)は特に限定されるものではないが、例えば、上述した組換え発現ベクターをホスト細胞に導入して形質転換する方法を挙げることができる。また、トリプトファン合成系を有する細胞、生物であれば、形質転換の対象となる生物も特に限定されるものではなく、上記(4)においてホスト細胞として例示した各種微生物等を挙げることができる。
【0070】
本発明に係る形質転換体は、植物細胞または植物体であることが好ましい。このような形質転換植物は、トリプトファンの含有量が高く、栄養価の高い食品原料や飼料として高い利用価値を有する。
【0071】
植物体の形質転換に用いられる組換え発現ベクターは、当該植物細胞内で挿入遺伝子を発現させることが可能なものであれば特に限定しない。特に、植物体へのベクターの導入法がアグロバクテリウムを用いる方法である場合には、pBI系等のバイナリーベクターを用いることが好ましい。バイナリーベクターとしては、具体的には、例えば、pBIG、pBIN19、pBI101、pBI121、pBI221等を挙げることができる。また、植物体内で遺伝子を発現させることが可能なプロモーターを有するベクターであることが好ましい。プロモーターとしては公知のプロモーターを好適に用いることができ、具体的には、例えば、カリフラワーモザイクウイルス35Sプロモーター(CaMV35S)、ユビキチンプロモーターやアクチンプロモーターを挙げることができる。なお、植物細胞には、種々の形態の植物細胞、例えば、懸濁培養細胞、プロトプラスト、葉の切片、カルスなどが含まれる。
【0072】
植物細胞への組み換え発現ベクターの導入には、ポリエチレングリコール法、電気穿孔法(エレクトロポレーション法)、アグロバクテリウムを介する方法、パーティクルガン法など、公知の種々の方法を用いることができる。また、形質転換細胞から植物体の再生は、植物細胞の種類に応じて公知の方法で行うことが可能である。
【0073】
ゲノム内に本発明に係るポリヌクレオチドが導入された形質転換植物体がいったん得られれば、当該植物体から得られる種子にも当該ポリヌクレオチドが導入されている。例えば、本発明に係るポリヌクレオチドで形質転換されたイネ等の穀物においては、トリプトファン含量の高い穀物を得ることができる。本発明には、形質転換植物から得られる種子も含まれる。
【0074】
(6)スクリーニング方法およびスクリーニングキット
本発明に係るスクリーニング方法は、(1)に記載した本発明に係るポリペプチド(変異型OASA2タンパク質)および野生型イネアントラニル酸合成酵素(OASA2タンパク質)のいずれか一方のみと結合する物質をスクリーニングする方法であって、変異型OASA2タンパク質と結合する物質をスクリーニングする工程と、野生型OASA2タンパク質結合する物質をスクリーニングする工程と、上記両工程の結果を比較する工程とを含むものであればよい。
【0075】
上記本発明に係るポリペプチド、すなわち変異型OASA2タンパク質はトリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するものであるため、上記変異型OASA2タンパク質および野生型OASA2タンパク質のいずれか一方のみと結合する物質は、トリプトファン生合成経路におけるフィードバック阻害に関与するシグナル物質である可能性が高いと考えられる。このような物質が、当該スクリーニング方法により見出されれば、フィードバック阻害のメカニズムの解明が進み、フィードバック阻害に対する抵抗性が一層強力な変異型OASA2タンパク質の開発に貢献できることが期待される。
【0076】
また、本発明に係るスクリーニングキットは、上記スクリーニング方法を実施するためのキットであって、少なくとも本発明に係るポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むものであればよい。その他の具体的な構成については特に限定されるものではなく、必要な試薬や器具等を適宜選択してキットの構成とすればよい。当該形質転換期キットを用いることにより、簡便かつ効率的に上記本発明に係るスクリーニング方法を実施することができる。
【0077】
本発明は上述した各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0078】
〔実施例1:イネアントラニル酸合成酵素遺伝子OASA2への変異導入〕
<標的遺伝子のクローニングベクターへの挿入>
イネアントラニル酸合成酵素遺伝子OASA2(ACCESSION NO. AB022603)をクローニングベクターpBluescript SK+(Stratagene社製)のマルチクローニングサイトのEcoRIサイトに挿入し、pBS-OASA2を構築した。なお、挿入した遺伝子は、マルチクローニングサイトの制限酵素サイトKpnIからSacIの向きに挿入されている。
【0079】
<変異導入用プライマー>
Kunkel法によって目的タンパク質をコードする遺伝子に変異を導入する場合、変異を導入したい位置にセンス(あるいはアンチセンス)の変異導入用オリゴヌクレオチドを設定して用いる。目的タンパク質をコードする遺伝子にPCRを用いて変異を導入する場合、変異を導入したい位置に2つのプライマー、すなわちセンスおよびアンチセンスのオリゴヌクレオチドを設定して用いる。従って、当該変異導入用プライマーの一方は、上記Kunkel法に用いた変異導入用オリゴヌクレオチと同一のものを使用することができる。この際、遺伝子がコードするアミノ酸が変化しない適当な位置に制限酵素サイトを導入することで変異導入の有無の確認作業ができる。下記i)〜vi)の変異はKunkel法を用いて導入し、下記vii)およびviii)の変異はPCRを用いて導入した。
【0080】
<変異導入用プライマー>
i)351番目のアスパラギン残基をアスパラギン酸残基に置換(N351D)
351番目のアスパラギン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(BsiWI)を導入するように変異導入用プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
N351D-F:5'-GTTTGAGAGGCGAACGTACGCCgatCCATTTGAAGTCT-3'(配列番号15)
プライマーN351D-Fの23番目から25番目のgat(アスパラギン酸、D)および15番目から20番目のCGTACG(BsiWIサイト)は、それぞれ野生型OASA2のAAT(アスパラギン、N)およびCATACGの塩基配列よりデザインした。AATをGATにすることにより351番目のアスパラギン残基がアスパラギン酸残基へ置換される。また、CATACGをCGTACGとすることによりアミノ酸置換を伴わずに(ACA、ACGは共にスレオニン)制限酵素サイト(BsiWI: CGTACG)が導入され、変異導入後の選抜が容易となる。
【0081】
ii)367番目のチロシン残基をアラニン残基に置換(Y367A)
上記i)と同様に、367番目のチロシン残基をアラニン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(SnaBI:TACGTA)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
Y367A-F:5'-GTGAACCCAAGTCCAgccATGGCATACGTACAGGCAAGAGGC-3'(配列番号16)
iii)367番目のチロシン残基をイソロイシン残基に置換(Y367I)
上記i)と同様に、367番目のチロシン残基をイソロイシン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(SnaBI:TACGTA)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
Y367I-F:5'-GTGAACCCAAGTCCAatcATGGCATACGTACAGGCAAGAGGC-3'(配列番号17)
iv)530番目のロイシン残基をアラニン残基に置換(L530A)
上記i)と同様に、530番目のロイシン残基をアラニン残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(NheI:GCTAGC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
L530A-F:5'-ACGGAGACATGgctATCGCGCTAGCACTCCGCACCATT-3'(配列番号18)
v)530番目のロイシン残基をアスパラギン酸残基に置換(L530D)
上記i)と同様に、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(NheI:GCTAGC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
L530D-F:5'-ACGGAGACATGgacATCGCGCTAGCACTCCGCACCATT-3'(配列番号19)
vi)522番目のグリシン残基をチロシン残基に、530番目のロイシン残基をアスパラギン酸残基に置換(G522Y+L530D)
上記i)と同様に、522番目のグリシン残基をチロシン残基に、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換し、かつ、コードするアミノ酸が変化しない位置に制限酵素サイト(BciVI:GTATCC/GGATAC)を導入するように変異導入プライマーをデザインした。変異導入用プライマーの塩基配列は下記の通りである。なお、下線は制限酵素サイトを示し、小文字は変異導入コドンを示す。
G522Y+L530D-F:5'-AGTGGCGGCCTTGGAtacATATCATTTGACGGAGACATGgatATCGCTCTTGCACT-3'(配列番号20)
vii)126番目のセリン残基をフェニルアラニン残基に置換(S126F)
126番目のセリン残基をフェニルアラニン残基にアミノ酸置換するように変異導入用プライマーをデザインした。変異導入法にPCRを用いた手法で変異を導入したのでセンスおよびアンチセンスのオリゴヌクレオチドを作成した。変異導入用プライマーの塩基配列は下記の通りである。なお、小文字は変異導入コドンを示す。
S126F-F :5'-GCTTCCTCTTCGAGttcGTCGAGCAGGGGCC-3'(配列番号37)
S126F-R:5'-GGCCCCTGCTCGACgaaCTCGAAGAGGAAGC-3'(配列番号38)
プライマーS126F-Fの15番目から17番目のttc(フェニルアラニン、F)は、野生型OASA2のTCC(セリン、S)よりデザインした。TCCをTTCにすることにより126番目のセリン残基がフェニルアラニン残基へ置換される。プライマーS126F-RはプライマーS126F-Fの相補鎖である。
【0082】
viii)369番目のアラニン残基をロイシン残基に置換(A369L)
上記i)と同様に、369番目のアラニン残基をロイシン残基にアミノ酸置換するように変異導入用プライマーをデザインした。変異導入法にPCRを用いた手法で変異を導入したのでセンスおよびアンチセンスのオリゴヌクレオチドを作成した。変異導入用プライマーの塩基配列は下記の通りである。なお、小文字は変異導入コドンを示す。
A369L-F:5'-CAAGTCCATACATGctaTATGTACAGGCAA-3'(配列番号39)
A369L-R:5'-TTGCCTGTACATAtagCATGTATGGACTTG-3'(配列番号40)
プライマーA369L-RはプライマーA369L-Fの相補鎖である。
【0083】
<Kunkel法による変異導入DNAの調製>
Kunkel法は、下記の参考文献1−3に記載の方法にしたがって行った。
【0084】
参考文献1:Kunkel, T. A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proceedings of the National Academy of Science of the USA, 82: 488-492.
参考文献2:Kunkel, T. A., Bebenek, K. and McClary, J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods in Enzymology, 204: 125-139.
参考文献3:「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, (2001) Chapter 13.
例えば、351番目のアスパラギン残基をアスパラギン酸残基に置換する場合、前述のpBS-OASA2ベクターを鋳型とし、上記i)に記載した変異導入用プライマーを用いてKunkel法を行うことで変異を導入した。上記ii)、iv)、v)、vi)に記載の変異導入用プライマーを用いる場合も上記と同一の鋳型を用いてKunkel法を行った。以上の操作によりN351D、Y367A、L530A、L530D、G522Y+L530D変異DNAを調製した。
【0085】
N351D+L530Aの二重変異DNAは、変異導入工程を2回繰り返すことにより調製した。すなわち、最初に前述のpBS-OASA2ベクターを鋳型とし、上記i)に記載した変異導入用プライマーを用いてN351D変異DNAを調製した。次に、N351Dの変異が導入されたプラスミドDNA(pBS-OASA2(N351D)ベクター)を鋳型として、上記iv)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(N351D+L530A)を得た。同様に、N351Dの変異が導入されたプラスミドDNA(pBS-OASA2(N351D)ベクター)を鋳型として、上記v)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(N351D+L530D)を得、Y367Aの変異が導入されたプラスミドDNA(pBS-OASA2(Y367A)ベクター)を鋳型として、上記v)に記載した変異導入用プライマーを用いて2つの変異が導入された変異DNA(Y367A+L530D)を得た。
【0086】
三重変異DNAは、上記二重変異DNAを含むプラスミドDNAを鋳型としてKunkel法を行うことにより調製した。すなわち、N351D+Y367A+L530Aは、予めN351D+L530Aの変異が導入されたプラスミドDNA(pBS-OASA2(N351D+L530A)ベクター)を鋳型として、上記ii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(N351D+Y367A+L530A)を得た。同様に、G522Y+L530Dの変異が導入されたプラスミドDNA(pBS-OASA2(G522Y+L530D)ベクター)を鋳型として、上記ii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(Y367A+G522Y+L530A)を得た。同様に、G522Y+L530Dの変異が導入されたプラスミドDNA(pBS-OASA2(G522Y+L530D)ベクター)を鋳型として、上記iii)に記載した変異導入用プライマーを用いて3つの変異が導入された変異DNA(Y367I+G522Y+L530A)を得た。
【0087】
<RCR法による変異導入用DNAの調製>
PCR法による変異導入法は下記の参考文献に記載の方法に従って行った。
【0088】
Higuchi R, Krummel B, Saiki RK (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351-7367
OASA2遺伝子が変異導入用のプライマー領域でオーバーラップするように5’側領域と3’側領域に分けてPCRを行った。例えば、上記vii)に記載した126番目のセリンをフェニルアラニンに置換するため変異導入用プライマーを用いる場合には、5’側領域はpBS-OASA2ベクターを鋳型とし、クローニング用センスプライマー(5'-AAAACTAGTATGGAGTCCATCGCCGCCGCCACG-3':配列番号41、下線は制限酵素SpeIサイトを示す)およびプライマーS126F-Rの組み合わせでPCRを行い、 3’領域はpBS-OASA2ベクターを鋳型とし、プライマーS126F-Fおよびクローニング用アンチセンスプライマー(5'-AAAGTCGACTGAGAGAGACTCTATTCCTTGTC-3':配列番号42、下線は制限酵素SalIサイトを示す)との組み合わせでPCRを行った。上記viii)に記載の変異導入用プライマーを用いる場合でも上記と同一の鋳型を用い、同様のプライマーの組み合わせによりPCRを行った。
【0089】
S126F+L530Dの二重変異DNAを調製する場合は、鋳型としてpBS-OASA2ベクターに前述のKunkel法でOASA2の530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する変異を既に導入したプラスミドDNAを用いた。従って、上記vii)に記載の変異導入用プライマー(S126F-FおよびS126F-R)を用いてPCRを行うことにより、OASA2の126番目のセリン残基をフェニルアラニン残基に置換し、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する2つの変異が導入された変異遺伝子が得られることになる。なお、プライマーの組み合わせは上記と同様である。A369L+L530Dの二重変異DNAの調製も上記と同様の鋳型と上記viii)に記載の変異導入用プライマー(A369L-FおよびA369L-R)を用いてPCRを行うことにより、OASA2の369番目のアラニン残基をロイシン残基に置換し、530番目のロイシン残基をアスパラギン酸残基にアミノ酸置換する2つの変異が導入された変異遺伝子が得られる。
【0090】
PCR反応の組成は、1×Pyrobest buffer II(TaKaRa社)、0.2mM dNTP、0.5μM センスおよびアンチセンスプライマー、0.025 units/μl Pyrobest DNA polymerase(TaKaRa社)、10ng 鋳型DNAとし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分間保持した後4℃に冷却した。
【0091】
増幅したPCR産物(5’領域断片および3’領域断片)を20倍希釈し、PCR反応液[1×Pyrobest buffer II(TaKaRa社)、0.2mM dNTP、0.025 units/μl Pyrobest DNA polymerase(TaKaRa社)]に1μlずつ添加し、上記PCR反応装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を5サイクル繰り返し、伸長反応を行った。この反応によりクローニング用センスプライマーからクローニング用アンチセンスプライマーまで連結されたDNA断片が合成された。
【0092】
上記反応終了後すぐに、プライマーの濃度がそれぞれ0.2μMになるようにクローニング用センスプライマーおよびクローニング用アンチセンスプライマーを添加し、上記PCR装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を20サイクル繰り返し、72℃で10分間保持した後4℃に冷却した。以上の反応により、上記クローニング用センスプライマーからクローニング用アンチセンスプライマーまで連結されたDNA断片が増幅され、これをクローニングベクターpBluescript KS+(Stratagene社)のマルチクローニングサイトのSpeIおよびSalIに挿入し、pBS-OASA2(S126F)、pBS-OASA2(A369L) 、pBS-OASA2(S126F/L530D)、 pBS-OASA2(A369L/L530D)を構築した。
【0093】
〔実施例2:コムギ胚芽無細胞システムによるタンパク質合成〕
(1)Split-PCR法による転写鋳型DNAの合成
<Split-PCR用鋳型の調製>
OASA2遺伝子はイネの核ゲノム上にコードされている遺伝子であるが、合成されたタンパク質は葉緑体に移行して葉緑体内で機能していることが判っている。合成されたタンパク質のN末端領域には葉緑体へ移行するためのシグナル配列が存在し、その配列が除去されることで成熟型酵素となり、酵素活性を発揮する。したがって、OASA2タンパク質を成熟型酵素としてコムギ胚芽無細胞合成系で合成するため、N末端領域に存在する49残基からなるシグナル配列を除去した形で合成されるようにプライマーをデザインした。すなわち、コムギ胚芽無細胞合成システムでSplit-PCRを可能とするリンカー配列の下流にATG開始コドンを配し、OASA2遺伝子の148から164番目の塩基配列からなる全長36merのプライマーをデザインした。また、OASA2遺伝子が挿入されているベクター(pBluescript SK+)上に上記センスSplit-PCR用プライマーの相補鎖側の位置にSplit-PCR用アンチセンスプライマーを2種類設定した。挿入DNAの位置から遠い順にアンチセンスSplit-PCR用プライマー1およびアンチセンスSplit-PCR用プライマー2とした。Split-PCR用のプライマーの塩基配列は下記の通りである。なお、Split-PCRについては後述する。
センスSplit-PCR用プライマー:5'-CCTCTTCCAGGGCCCAATGTGCTCCGCGGGGAAGCC-3'(配列番号21、下線部分がリンカー配列)
アンチセンスSplit-PCR用プライマー1:5'-GGAGAAAGGCGGACAGGTAT-3'(配列番号22)
アンチセンスSplit-PCR用プライマー2:5'-GGGGAAACGCCTGGTATCTT-3'(配列番号23)
上記Kunkel法により変異が導入されたOASA2遺伝子を含むpBluescript SK+ベクターを鋳型として、上記センスSplit-PCR用プライマーおよびアンチセンスSplit-PCR用プライマー1を用いてPCR等の増幅反応を行い、増幅されたDNA断片を次に行うSplit-PCRの鋳型とすることができる。
【0094】
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センスSplit-PCR用プライマーおよびアンチセンスSplit-PCR用プライマー1、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA(各変異を導入したpBS-OASA2ベクター)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。以上の反応により、上記センスSplit-PCR用プライマーからアンチセンスSplit-PCR用プライマー1まで連結されたDNA断片が増幅され、これを次に行うSplit-PCRの鋳型とした。
【0095】
<転写鋳型DNAの合成>
上記Split-PCR用鋳型DNAを転写鋳型DNAとするためには、断片の5'末端上流にSP6 RNAポリメラーゼのプロモーターとタバコモザイクウイルス(TMV)由来の翻訳増強配列(オメガ配列)を付加する必要がある。そのため、Sawasakiらの方法(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)に従い、Split-PCRを行った。すなわち、上記Split-PCR用鋳型DNAを50倍希釈し、PCR反応液[1× EX Taq buffer (TaKaRa社)、0.2mM dNTP、0.025 units/μl TaKaRa EX Taq (TaKaRa社)、0.2μM SP6 promoterプライマー、1nM TMV-Omegaプライマー、0.2μM アンチセンスSplit-PCR用プライマー2]に1μl添加し、上記PCR反応装置を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を35サイクル繰り返し、72℃で10分保持した後4℃に冷却した。SP6 promoterプライマーおよびTMV-Omegaプライマーの塩基配列は下記の通りである。なお、下線はSP6プロモーター配列を示し、小文字は両プライマーの重複部分を示す。
SP6 promoterプライマー:5'-GCGTAGCATTTAggtgacact-3'(配列番号24)
TMV-Omegaプライマー:5'-ggtgacactATAGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACCTCTTCCAGGGCCCAATG-3'(配列番号25)
ここで、上記SP6 promoterプライマーとTMV-Omegaプライマーのように、プライマーを上流側プライマーと下流側プライマーとに分割し、上流側プライマーの3’末端と下流側プライマーの5’末端との配列が一部重複するように設計されたプライマーをSplit primerと称する。また、このようなプライマーを用いて行うPCR反応をSplit-PCRと称する。
【0096】
(2)コムギ胚芽無細胞システム用のmRNAの合成
上記(1)で得られたPCR産物を直接鋳型として用いてmRNAを合成(転写)した。すなわち、上記(1)で得られたPCR産物を、転写反応液[80mM HEPES-KOH (pH7.8)、16mM Mg(OAc)2、10mM spermidine、10mM DTT、3mM NTP、1 unit/μl RNasin RNase inhibitor (Promega社)、1 unit/μl SP6 RNA polymerase (Promega社)]に1/10量添加した。37℃で2時間反応後、エタノール沈殿、70% エタノール洗浄を行い、適当量の滅菌水に溶解した。260 nmの吸光度を測定してRNA量を算出した。
【0097】
(3)コムギ胚芽無細胞システムによるタンパク質合成
Sawasakiらの方法(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)に従って上記(3)で合成したmRNAを鋳型としてタンパク質を合成した。すなわち、50μlのコムギ胚芽無細胞タンパク質合成液を入れた透析カップに上記mRNA(約30-35μg)を添加し、1ウェルあたり1mlの基質液を入れた24穴プレートに上記透析カップを浸漬し、26℃で24時間インキュベートした。
【0098】
〔実施例3:変異型OASA2タンパク質の活性測定〕
(1)ウエスタンブロット法によるOASA2タンパク質の定量
OASA2タンパク質のアミノ酸配列(配列番号1)の第161位〜第175位の配列(MDHEKGKVTEQVVDD)のペプチド断片に基づいて作製したウサギ抗OASA2抗体とOASA2タンパク質精製標品を用いてコムギ胚芽無細胞システムにより合成されたOASA2タンパク質を定量した。ウエスタンブロット解析はTowbinらの方法(Towbin, H., Staehelin, T. and Gordon, J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354.)に従い行った。上記方法で見積もったOASA2タンパク質量で後述の酵素活性を補正した。
【0099】
(2)OASA2タンパク質の活性測定
イネアントラニル酸合成酵素のαサブユニットであるOASA2タンパク質は、βサブユニットが供給するグルタミンのアミド基由来のアンモニアを利用してコリスミン酸からアントラニル酸を生成する。生体内でOASA2タンパク質は、βサブユニットが供給するアンモニアを利用してアントラニル酸合成活性(以下、適宜「AS活性」と略記する。)を発揮するが、外部よりアンモニア、例えばNH4Clを添加することでも酵素活性を示す。
【0100】
OASA2タンパク質の活性は、周知の通りトリプトファンによってフィードバック抑制がかかるので、上記タンパク質合成反応液中からトリプトファンの除去は必須であり、また発現させたタンパク質の環境をAS活性測定用の緩衝液成分に変える必要がある。したがって、タンパク質合成反応液の成分をMicrospin G-25 columns(Amersham Biosciences社製)を用いてbuffer A(50mM Tris-HCl, pH7.6; 0.05mM EDTA; 2mM MgCl2; 0.05mM DTT; 5% グリセロール)に置換した。
【0101】
<酵素活性測定1>
90μlの反応液(20mM Tris-HCl, pH8.3; 100mM NH4Cl; 0.5mM コリスミン酸; 10mM MgCl2)にbuffer 置換した粗抽出液を2.5μl添加して、32℃で1時間反応させた。反応は10μlの1N HClを添加することで停止させ、300μlの酢酸エチルを添加して合成したアントラニル酸を抽出した。アントラニル酸の合成量は、Wallac 1420 ARVOsx-FL(パーキンエルマーライフサイエンスジャパン社製)を用いて励起波長355 nm/蛍光(emmision)460 nmで測定した。トリプトファンに対するフィードバック阻害活性を解析する場合、反応系に終濃度10μMまたは100μMになるようにトリプトファンを添加した。
【0102】
結果を図1および図2に示した。図1から明らかなように、6種類の変異型OASA2タンパク質(N351D、L530A、L530D、N351D/L530A、N351D/L530DおよびG522Y/L530D)は、野生型(wt)と比較して酵素活性が向上していた。また、図2から明らかなように、7種類の変異型OASA2タンパク質(A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、トリプトファンを添加していない場合の酵素活性が野生型と同程度以上であり、かつ、トリプトファンフィードバック阻害に対する抵抗性を獲得していた。
【0103】
<酵素活性測定2>
上記トリプトファンフィードバック阻害に対する抵抗性を獲得していた5種類の変異型OASA2タンパク質について、より詳細に酵素学的性質を検討するため、イネアントラニル酸合成酵素βサブユニットを用いた反応系において酵素活性およびトリプトファンフィードバック阻害を解析した。
【0104】
イネアントラニル酸合成酵素βサブユニットを過剰量添加した90μlの反応液(20mM Tris-HCl, pH8.3; 5mM グルタミン; 0.2〜0.8mM コリスミン酸; 10mM MgCl2;)にbuffer 置換した粗抽出液を2.5μl添加して、32℃で1時間反応させた。なお、イネアントラニル酸合成酵素βサブユニットの調製およびウエスタンブロット法による合成量の定量は、Kannoらの方法(Kanno et al.(2004) Plant Molecular Biology 54,11-22)に従って行った。反応は10μlの1N HClを添加することで停止させ、300μlの酢酸エチルを添加して合成したアントラニル酸を抽出した。アントラニル酸の合成量は、Wallac 1420 ARVOsx-FL(パーキンエルマーライフサイエンスジャパン社製)を用いて励起波長355 nm/蛍光(emmision)460 nmで測定した。トリプトファンに対するフィードバック阻害活性を解析する場合、反応系に終濃度10μM、25μMまたは50μMになるようにトリプトファンを添加した。
【0105】
結果を表1および図3に示した。表1から明らかなように、9種類の変異型OASA2タンパク質(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、S126F、Y367Aを除き、野生型(wt)と比較して同程度以上の酵素活性を有していた。より詳細には、野生型(wt)と比較してA369Lが約1倍、 S126F/L530Dが約2.5倍、Y367A/L530Dが約2.2倍、A369L/L530Dが約1.8倍、Y367A/G522Y/L530Dが約1.3倍、N351D/Y367A/L530Dが約1.2倍、Y367I/G522Y/L530Dが約0.8倍の酵素活性であった。
【0106】
【表1】
【0107】
また、図3はトリプトファン無添加時における野生型および各変異型OASA2タンパク質酵素活性を100%とし、各濃度のトリプトファンを添加した場合の酵素活性をトリプトファン無添加時の酵素活性と比較したものである。図3から明らかなように、野生型はトリプトファンを50μM以上添加すると酵素活性が無添加時の10%を下回っているが、9種類の変異型OASA2タンパク質(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530DおよびY367I/G522Y/L530D)は、トリプトファンを50μM添加した場合においても無添加時の30%以上の酵素活性を有していた。
【0108】
〔実施例4:変異型OASA2遺伝子を発現させた酵母TRP2遺伝子欠損変異株(MATalpha his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 trp2::KANMX)の遊離トリプトファン含量解析〕
OASA2遺伝子に相当するTRP2遺伝子が欠損した酵母は生育のためにトリプトファンを要求する。従って、この欠損株にOASA2遺伝子を導入しトリプトファン非存在下で生育できれば導入したOASA2遺伝子はTRP2の機能を相補できることになる。また、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子をこの酵母で発現させ、生体内に蓄積する遊離トリプトファン含量を測定することで当該遺伝子の効果検定を行うことができる。
【0109】
発現ベクターを構築するために、トリプトファンフィードバック阻害に対する抵抗性を有する変異型OASA2遺伝子(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530D、Y367I/G522Y/L530D)および野生型(Wt)OASA2遺伝子が挿入されたpBluescriptベクターを鋳型として、下記のプライマーを用いてPCRを行った。前述の通りOASA2タンパク質のN末端領域には葉緑体移行シグナルが存在するので、上記(2)に記載の「Split-PCR用プライマー」のデザインと同様にシグナル配列を除去した形で発現するようにプライマーをデザインした。なお、酵母発現ベクターpYES2(Invitrogen社製)の制限酵素サイトKpnI/EcoRIに組み込めるようにセンスプライマーには制限酵素サイトKpnI(GGTACC)、アンチセンスプライマーには制限酵素サイトEcoRI(GAATTC)を導入した。これら制限酵素サイトは下線で示す。なお、pYES2ベクターはURA3遺伝子を有し、ガラクトースによってタンパク質の発現誘導が制御できる。
センスプライマー:5’-AAAGGTACCATGTGCTCCGCGGGGAAGCC-3’(配列番号27)
アンチセンスプライマー:5’-AAAGAATTCTGAGAGAGACTCTATTCCTTGTC-3’(配列番号28)
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センス及びアンチセンスプライマー、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA (pBS-OASA2ベクター)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。
【0110】
ベクターへのDNA断片のクローニングはSambrookらの方法(「Molecular Cloning (Third Edition)」 (J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001))に従って行った。構築したベクターの酵母TRP2遺伝子欠損変異株への形質転換など基礎的手法はKaiserらの方法(Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual (Chris Kaiser, Susan Michaelis, Aaron Mitchell, Cold Spring Harbor Laboratory, 1994))に従って行った。
【0111】
酵母形質転換体は、2%グルコースを含む合成完全寒天培地(0.67%アミノ酸を含まないバクトイーストナイトロジェンベース、ウラシルを除いた0.2%ドロップアウト混合物、2%バクトアガー)でシングルコロニーを単離し、次いで菌体を2%ガラクトースを含む合成完全寒天培地(0.67%アミノ酸を含まないバクトイーストナイトロジェンベース、ウラシルとトリプトファンを除いた0.2%ドロップアウト混合物、2%バクトアガー)上に塗り、30℃で二日間培養した。寒天培地上から掻き取った菌体20mgを105μlの蒸留水で懸濁し、100℃で20分間処理した。次いで595μlのクロロホルム:メタノール混合液(5:12、v/v)を加え、よく混合した後、20,000×gの遠心加速度で10分間遠心分離した。得られた上清を新しいチューブに移し、175μlの蒸留水と263μlのクロロホルムを加え、激しく混合した後、20,000×gの遠心加速度で10分間遠心分離した。このようにして抽出された水層を新しいチューブに移し、減圧下で乾固させた後、200μlの10mM NaOHに溶解して、トリプトファン抽出液とした。これを高速液体クロマトグラフィー(HPLC)装置(Waters社製、Waters Alliance HPLC FLD System 2695)に供して遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、Xterra RP18 column (4.6 ×150 mm)(Waters社製)であり、トリプトファンは励起波長278nm/蛍光波長348nmで検出した。
【0112】
結果を表2に示した。表2から明らかなように、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子(S126F、Y367A、A369L、S126F/L530D、Y367A/L530D、A369L/L530D、N351D/Y367A/L530D、Y367A/G522Y/L530D、Y367I/G522Y/L530D)を発現させた酵母中で生成される遊離トリプトファン含量は、野生型(Wt)OASA2遺伝子を発現させた酵母のものより高濃度蓄積していることが確認された(野生型の1.9〜2.3倍)。
【0113】
【表2】
【0114】
〔実施例5:変異型OASA2遺伝子発現を発現させたイネカルス形質転換体の作製および当該形質転換カルスの遊離トリプトファン含量解析〕
<外来遺伝子導入用組換えベクターの構築>
外来遺伝子導入用組換えベクターの構築は、下記の参考文献に記載の方法に従って行った。
参考文献1:Urushibara S, Tozawa Y, Kawagishi-Kobayashi M and Wakasa K (2001) Efficient transformation of suspension-cultured rice cells mediated by Agrobacterium tumefaciens. Breeding Science 51: 33-38
参考文献2:Tozawa Y, Hasegawa H, Terakawa T and Wakasa K (2001) Characterization of rice anthranilate synthase alfa subunit genes OSASA1 and OSASA2: tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiology 126: 1493-1506
参考文献3:Hiei Y, Ohta S, Komari T and Kumashiro T (1994) Efficient transformation of rice (Oryza sativa) mediated by Agrobacterium and sequence analysis of boundaries of the T-DNA. Plant Journal 6: 271-282
外来遺伝子導入用組換えベクターpUB-Hm(Urushibara et al., Breeding Science 51: 33-38 (2001))は、トウモロコシのユビキチンプロモーターと、第一イントロンと、制限酵素Sse8387Iサイトと、NOSターミネーターと、ハイグロマイシン耐性遺伝子を有し、Sse8387Iサイトに目的外来遺伝子を挿入することでイネ形質転換用の組換えベクターを構築することができる。
【0115】
全長からなる野生型(Wt)OASA2遺伝子は、〔実施例1〕に記載のpBS-OASA2を鋳型とし、また下記に示すセンスプライマーおよびアンチセンスプライマーを用いてPCRで増幅した。センスプライマーには制限酵素サイトSpeI(ACTAGT)、アンチセンスプライマーには制限酵素サイトSalI(GTCGAC)を導入した。これら制限酵素サイトは下線で示す。
センスプライマー:5'-AAAACTAGTATGGAGTCCATCGCCGCCGCCACG-3'(配列番号43)
アンチセンスプライマー:5'-AAAGTCGACTGAGAGAGACTCTATTCCTTGTC-3'(配列番号44)
また、全長からなる変異型OASA2遺伝子(Y367A、Y367A/L530D)は、pBS-OASA2を鋳型とし、上記センスプライマー(配列番号43)およびアンチセンスプライマー(配列番号44)を用いて〔実施例1〕<RCR法による変異導入用DNAの調製>に記載の方法でDNA断片を調製した。以上のPCRにより増幅したDNA断片を制限酵素SpeIおよびSalIで消化して得られたDNA断片(1)は、Sawasakiらが構築した(Sawasaki et al. (2002) Proc Natl Acad Sci U S A 99, 14652-14657)コムギ胚芽無細胞合成システム用発現ベクターpEU3bを改良し、マルチクローニングサイトSpeI/SalIサイトの両端にSse8387Iサイトを導入したコムギ胚芽無細胞合成システム用発現ベクターpEU3sのSpeI/SalIサイトに挿入した。
【0116】
別途、上述の変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子を含有するpEU3sプラスミドベクターを制限酵素Sse8387Iで消化し、1.8kbサイズのDNA断片(2)を得た。
【0117】
上記の制限酵素Sse8387Iで消化したpUB-HmプラスミドベクターとDNA断片(2)をDNAライゲーション・キットver.2(タカラバイオ社製)で処理して、DNA連結反応を行った。これにより、環状の組換えベクターを構築した。この組換えベクターは、ユビキチンプロモーター領域の下流に第一イントロン領域を有し、第一イントロン領域とNOSターミネーター領域との間に変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子が挿入、連結されてあり、かつ植物中で発現するハイグロマイシン耐性遺伝子を含有する構造を有した。得られた外来遺伝子導入用組換えベクターを、それぞれpUB-OASA2(wt)-Hm(野生型OASA2遺伝子を有する)、pUB-OASA2(Y367A)-Hm(変異型OASA2遺伝子(Y367A)を有する)およびpUB-OASA2(Y367A/L530D)-Hm(変異型OASA2遺伝子(Y367A/L530D)を有する)と称する。
【0118】
図5にpUB-OASA2(wt)-Hm、pBU-OASA2(Y367A)-HmおよびpBU-OASA2(Y367A/L530D)-Hmの模式図を示した。図中PUbiはユビキチンプロモーター領域を表し、P35Sはカリフラワーモザイクウイルス35Sプロモーター領域を表し、nos3'はNOSターミネーター領域を表し、hptはハイグロマイシン耐性遺伝子領域を表し、Sse8387Iは制限酵素サイトを表し、RBは右側境界配列(Right Border)を表し、LBは左側境界配列(Left Border)を表す。
【0119】
<アグロバクテリウムの調製>
YEB培地(バクトペプトン5g/l、バクトビーフエクストラクト5g/l、バクトイーストエクストラクト1g/l、シュクロース5g/l、2mM MgCl2、pH7)の液体培地50mlに、アグロバクテリウム(Agrobacterium tumefaciens EHA101)を移植し、30℃で16時間振とう培養した後、培溶液を4℃で遠心分離により細菌の沈殿を得た。細菌の沈殿物を氷冷した10mM Tris-Hcl pH7.5で懸濁し、遠心分離で沈殿を得た。沈殿物を0.5mlのYEB培地に懸濁した。この菌懸濁液0.2mlに上記3種類の外来遺伝子導入用組換えベクターの溶解液(5μg)をそれぞれ添加し、よく混合した。これらを即座に凍結し、37℃で融解、再度凍結融解を計3回行った。16mlのYEB培地にこの懸濁液を加え、30℃で2時間振とう培養した。この培養液をL 培地(バクトトリプトン10g/l、バクトイーストエクストラクト5g/l、NaCl 5g/l、バクトアガー15g/l、pH7.5)にカナマイシン100mg/lおよびハイグロマイシン50mg/lを添加してなる寒天培地に塗布し、30℃で36時間培養して、生育してきたコロニーを組換えベクターが導入された形質転換アグロバクテリウムとして得た。
【0120】
<イネカルスの調製>
イネ(品種:日本晴)の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約4%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0121】
N6培地の無機塩組成にショ糖30g/l、2,4-D 2mg/l、カザミノ酸1g/lおよびゲルライト2g/l、およびN6ビタミンを添加してなる2N6固体培地(Urushibara et al., Breeding Science 51: 33-38 (2001))に、上記で殺菌したイネ種子を置床した。28℃で3週間インキュベートすることでカルスが形成された。それらカルスを胚乳部分から切り出し、そのカルスを上記と同じ組成の培地に移植して、7日間培養を行った。
【0122】
<イネカルスの形質転換>
上記のようにして得られた、遺伝子導入用の形質転換アグロバクテリウムをAA培地の組成(Hiei et al., Plant Journal 6: 271-282 (1994))にショ糖20g/l、2,4-D 2mg/l、カイネチン0.2mg/lおよびアセトシリンゴン10mg/lを添加してなる液体培地30mlに懸濁し菌液を得た。この懸濁液を9cmのシャーレに入れ、上記により得られたイネのカルスを100個入れた後、5分間浸せきした。浸せき後、ペーパータオルで余分な菌液を除去した後、N6培地の無機塩組成にショ糖30g/l、グルコース10g/l、2,4-D 2mg/l、カザミノ酸1g/l、ゲルライト2g/lおよびアセトシリンゴン10mg/lを添加してなる2N6CO固体培地に、上記の浸せきしたカルスを各シャーレ当たり20個ずつ置床した。24℃で3日間、暗黒下で培養し、イネのカルスへのアグロバクテリウムの感染を行った。
【0123】
<カルスの選抜>
組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩組成にショ糖30g/l、2,4-D 2mg/l、カルベニシリン500mg/l、ハイグロマイシン30mg/l、カザミノ酸1g/l、ゲルライト2mg/lを添加してなる2N6SE固体培地上に各シャーレ当たり20個ずつ移植した。28℃で3週間、暗黒下で培養して、ハイグロマイシン耐性の形質転換カルスを得た。さらに同じ組成の個体培地上へ増殖カルスを移植し、ハイグロマイシン耐性の形質転換カルスの培養を28℃で3週間行った。
【0124】
3週間後、増殖したカルスの一部からDNAを抽出し、下記に示すセンスプライマーおよびアンチセンスプライマーを用いたPCRにより形質転換カルスと同定した。
センスプライマー:5'-GGATGGCACCCGCAGCAGATCG-3' (配列番号45)
アンチセンスプライマー:5'-GTACTCATCACTTGTCATGGTTG-3'(配列番号46)
402塩基対に相当する形質転換用ベクター遺伝子に由来する断片の増幅により、形質転換体におけるOASA2変異遺伝子を確認した。元々含まれるゲノム上のOASA2遺伝子にはイントロン配列が含まれる、形質転換遺伝子にはイントロンが含まれないため、402塩基対のDNA増幅産物は、形質転換用遺伝子にのみ由来する。
【0125】
PCR反応液の組成は、1× Pyrobest buffer II (TaKaRa社)、0.2mM dNTP、0.5μM センスプライマー(配列番号45)及びアンチセンスプライマー(配列番号46)、0.025 units/μl Pyrobest DNA polymerase (TaKaRa社)、10ng 鋳型DNA (カルスDNA)とし、PCR反応装置(宝酒造社製、TaKaRa PCR Thermal Cycler MP TP3000型)を用いて、95℃で2分間保持した後、98℃で20秒間、60℃で35秒間、72℃で3分間の反応を25サイクル繰り返し、72℃で10分保持した後4℃に冷却した。
【0126】
さらに、参考文献(Tozawa et al., Plant Physiology 126: 1493-1506 (2001))に記載された方法に従って形質転換カルスよりRNAを抽出し、同文献(Tozawa et al., Plant Physiology 126: 1493-1506 (2001))に記載されたジゴキシゲニン標識したOASA2のRNAプローブを用いる方法に従ってRNAブロットハイブリダイゼーションを行い、上記形質転換により導入したOASA2野生型遺伝子またはOASA2変異型遺伝子((Y367A)あるいは(Y367A/L530D))が選抜したカルスにRNAとして発現していることを確認した。
【0127】
<形質転換カルスの遊離トリプトファン含量解析>
変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(Wt)OASA2遺伝子を発現するイネカルスそれぞれ2系統について、各イネカルスの100mgを液体窒素中で粉砕した。粉砕物を1.5ml容のチューブに移し替え、次いで500μlのクロロホルム:メタノール:水混合液(5:12:3の容積比)を加え、よく混合した後、5,000×rpmの遠心加速度で10分間遠心分離した。得られた上清を新しいチューブに移し、375μlの蒸留水と250μlのクロロホルムを加え、激しく混合した後、5,000×rpmの遠心加速度で10分間遠心分離した。このようにして抽出された水層を新しいチューブに移し、減圧下で乾固させた後、2mlの10mM NaOHに溶解して、トリプトファン抽出液とした。これを高速液体クロマトグラフィー(HPLC)装置(Waters社製、Waters Alliance HPLC FLD System 2695)に供して遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、Xterra RP18 column (4.6 ×150 mm)(Waters社製)であり、展開溶媒はアセトリトリル−0.1M H3PO4 水溶液(pH4.0) をアセトニトリルの5%〜45%濃度勾配で用い、流量は1ml/分として、励起波長278nmにおける蛍光波長348nmの測定を行いトリプトファン含量を評価した。
【0128】
対照として、形質転換されていない通常のイネ(品種:日本晴)のカルスを用いた。得られた測定結果を表3に示す。表3から明らかなように、トリプトファンフィードバック阻害に抵抗性を有する変異型OASA2遺伝子(Y367A、Y367A/L530D)を発現させたイネカルス中に生成される遊離トリプトファン含量は、対照のイネカルスより高濃度蓄積していることが確認された(野生型の5.5〜38.8倍)。
【0129】
【表3】
【産業上の利用可能性】
【0130】
本発明により、高トリプトファン蓄積植物の作出が可能となる。したがって、本発明は広く農業に利用することができ、また高トリプトファン蓄積植物は家畜の飼料や食品原料に適しているため、本発明は畜産業や食品産業にも利用することができる。
【図面の簡単な説明】
【0131】
【図1】アミド基供与基質として100mM NH4Clを用いる測定系においてアントラニル酸合成活性を測定した結果を示すグラフであり、変異型OASA2タンパク質のうち、酵素活性が向上した変異体の結果を示すグラフである。
【図2】アミド基供与基質として100mM NH4Clを用いる測定系においてアントラニル酸合成活性を測定した結果を示すグラフであり、変異型OASA2タンパク質のうち、トリプトファンによるフィードバック阻害に対する抵抗性を有する変異体の結果を示すグラフである。
【図3】トリプトファンによるフィードバック阻害に対する抵抗性を有する変異体および野生型のフィードバック阻害の程度を示すグラフである。
【図4】図4Aは野生型OASA2タンパク質の模式図であり、図4Bはトリプトファン合成系を有する生物のトリプトファン結合領域のアミノ酸配列を比較した図である。
【図5】アグロバクテリウム法によるイネカルスの形質転換に用いた変異型OASA2遺伝子(Y367A、Y367A/L530D)または野生型(wt)OASA2遺伝子を含有する外来遺伝子導入用組換えベクターの模式図である。
【特許請求の範囲】
【請求項1】
アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴とするポリペプチド。
【請求項2】
上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることを特徴とする請求項1に記載のポリペプチド。
【請求項3】
配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴とするポリペプチド。
【請求項4】
さらに、配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有することを特徴とする請求項3に記載のポリペプチド。
【請求項5】
上記配列番号1に示されるアミノ酸配列の第126位の変異は、セリンをフェニルアラニンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第367位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第369位の変異は、アラニンをロイシンに置換する変異であることを特徴とする請求項3または4に記載のポリペプチド。
【請求項6】
上記配列番号1に示されるアミノ酸配列の第351位の変異は、アスパラギンをアスパラギン酸に置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第522位の変異は、グリシンをチロシンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第530位の変異は、ロイシンをアラニンまたはアスパラギン酸に置換する変異であることを特徴とする請求項3ないし6のいずれか1項に記載のポリペプチド。
【請求項7】
請求項3ないし6のいずれか1項に記載のポリペプチドであって、
配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチド。
【請求項8】
請求項1ないし7のいずれか1項に記載のポリペプチドをコードするポリヌクレオチド。
【請求項9】
請求項8に記載のポリヌクレオチドであって、
配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチド。
【請求項10】
請求項8または9に記載のポリヌクレオチドからなる形質転換体選抜用マーカー遺伝子。
【請求項11】
請求項8または9に記載のポリヌクレオチドを含む組換え発現ベクター。
【請求項12】
請求項8または9に記載のポリヌクレオチドまたは請求項11に記載の組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体。
【請求項13】
上記形質転換体は、植物細胞または植物体であることを特徴とする請求項12に記載の形質転換体。
【請求項14】
請求項13に記載の植物体から得られる種子。
【請求項15】
請求項10に記載のマーカー遺伝子または請求項11に記載の組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜することからなる、形質転換細胞の選抜方法。
【請求項16】
少なくとも請求項8または9に記載のポリヌクレオチド、あるいは請求項11に記載の組換え発現ベクターのいずれかを含むことを特徴とする形質転換キット。
【請求項17】
請求項3ないし7のいずれか1項に記載のポリペプチドおよび野生型イネアントラニル酸合成酵素の、いずれか一方のみと結合する物質をスクリーニングする方法であって、
請求項3ないし7のいずれか1項に記載のポリペプチドと結合する物質をスクリーニングする工程と、
野生型イネアントラニル酸合成酵素と結合する物質をスクリーニングする工程と、
上記両工程の結果を比較する工程とを含むことを特徴とするスクリーニング方法。
【請求項18】
請求項17に記載のスクリーニング方法を実施するためのキットであって、
少なくとも請求項3ないし7のいずれか1項に記載のポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むことを特徴とするスクリーニングキット。
【請求項1】
アントラニル酸合成酵素のトリプトファン結合領域に変異を有するポリペプチドであって、配列番号26に示されるアミノ酸配列の第5位に変異を有し、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴とするポリペプチド。
【請求項2】
上記配列番号26に示されるアミノ酸配列の第5位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であることを特徴とする請求項1に記載のポリペプチド。
【請求項3】
配列番号1に示されるアミノ酸配列の第126位、第367位または第369位の少なくとも1つに変異を有するポリペプチドであって、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有することを特徴とするポリペプチド。
【請求項4】
さらに、配列番号1に示されるアミノ酸配列の第351位、第522位または第530位の少なくとも1つに変異を有するポリペプチドであって、野生型イネアントラニル酸合成酵素と比較して少なくとも0.7倍以上の酵素活性を有することを特徴とする請求項3に記載のポリペプチド。
【請求項5】
上記配列番号1に示されるアミノ酸配列の第126位の変異は、セリンをフェニルアラニンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第367位の変異は、チロシンをアラニンまたはイソロイシンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第369位の変異は、アラニンをロイシンに置換する変異であることを特徴とする請求項3または4に記載のポリペプチド。
【請求項6】
上記配列番号1に示されるアミノ酸配列の第351位の変異は、アスパラギンをアスパラギン酸に置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第522位の変異は、グリシンをチロシンに置換する変異であり、
上記配列番号1に示されるアミノ酸配列の第530位の変異は、ロイシンをアラニンまたはアスパラギン酸に置換する変異であることを特徴とする請求項3ないし6のいずれか1項に記載のポリペプチド。
【請求項7】
請求項3ないし6のいずれか1項に記載のポリペプチドであって、
配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列からなるポリペプチド、または配列番号2ないし7および配列番号29ないし32のいずれかに示されるアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換、もしくは付加されたアミノ酸配列からなるポリペプチド。
【請求項8】
請求項1ないし7のいずれか1項に記載のポリペプチドをコードするポリヌクレオチド。
【請求項9】
請求項8に記載のポリヌクレオチドであって、
配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチド、または配列番号9ないし14および配列番号33ないし36のいずれかに示される塩基配列からなるポリヌクレオチドと相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチド。
【請求項10】
請求項8または9に記載のポリヌクレオチドからなる形質転換体選抜用マーカー遺伝子。
【請求項11】
請求項8または9に記載のポリヌクレオチドを含む組換え発現ベクター。
【請求項12】
請求項8または9に記載のポリヌクレオチドまたは請求項11に記載の組換え発現ベクターが導入されており、かつ、トリプトファン生合成経路においてトリプトファンによるフィードバック阻害に対する抵抗性を有するポリペプチドが発現している形質転換体。
【請求項13】
上記形質転換体は、植物細胞または植物体であることを特徴とする請求項12に記載の形質転換体。
【請求項14】
請求項13に記載の植物体から得られる種子。
【請求項15】
請求項10に記載のマーカー遺伝子または請求項11に記載の組換え発現ベクターを細胞に導入することにより、細胞の増殖を阻害するトリプトファン類縁化合物に対する耐性を細胞に付与し、さらに当該トリプトファン類縁化合物に対する耐性を発現している細胞を選抜することからなる、形質転換細胞の選抜方法。
【請求項16】
少なくとも請求項8または9に記載のポリヌクレオチド、あるいは請求項11に記載の組換え発現ベクターのいずれかを含むことを特徴とする形質転換キット。
【請求項17】
請求項3ないし7のいずれか1項に記載のポリペプチドおよび野生型イネアントラニル酸合成酵素の、いずれか一方のみと結合する物質をスクリーニングする方法であって、
請求項3ないし7のいずれか1項に記載のポリペプチドと結合する物質をスクリーニングする工程と、
野生型イネアントラニル酸合成酵素と結合する物質をスクリーニングする工程と、
上記両工程の結果を比較する工程とを含むことを特徴とするスクリーニング方法。
【請求項18】
請求項17に記載のスクリーニング方法を実施するためのキットであって、
少なくとも請求項3ないし7のいずれか1項に記載のポリペプチドの1つと野生型イネアントラニル酸合成酵素とを含むことを特徴とするスクリーニングキット。
【図1】

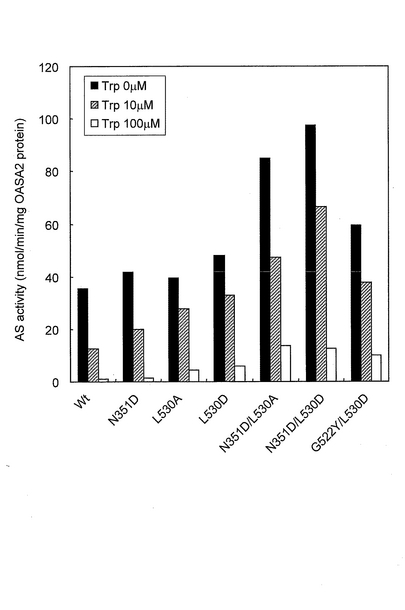
【図2】
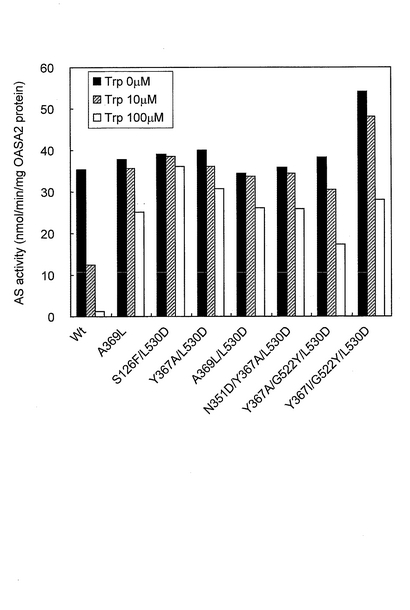
【図3】
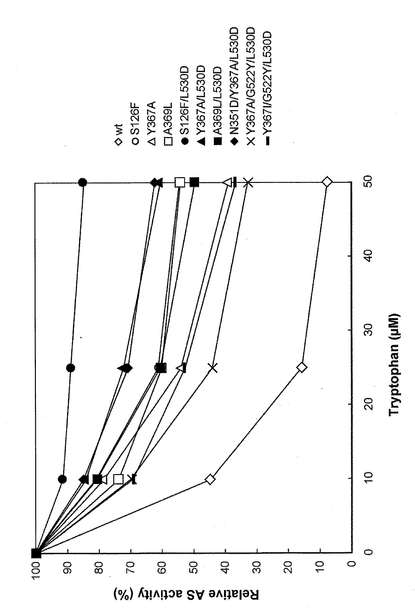
【図4】
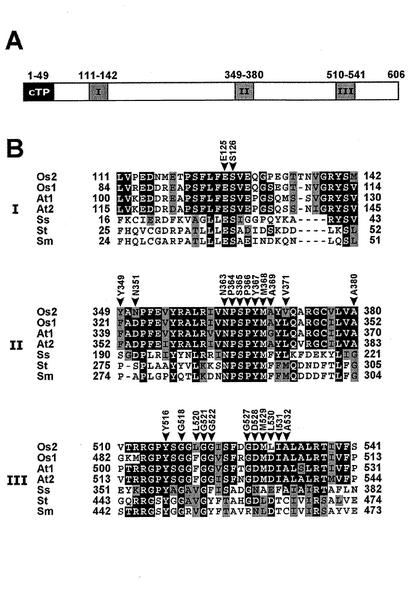
【図5】
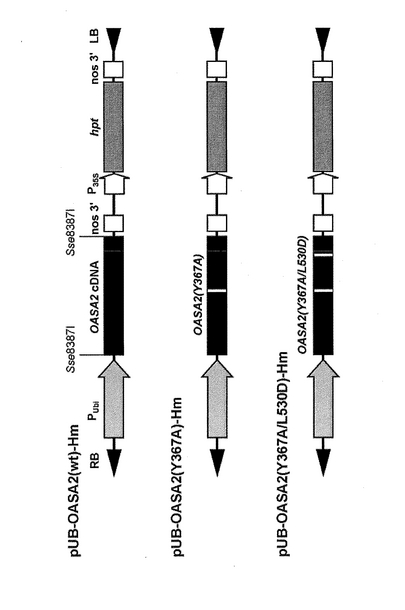
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2006−42801(P2006−42801A)
【公開日】平成18年2月16日(2006.2.16)
【国際特許分類】
【出願番号】特願2005−55165(P2005−55165)
【出願日】平成17年2月28日(2005.2.28)
【出願人】(504147254)国立大学法人愛媛大学 (214)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(501203344)独立行政法人農業・生物系特定産業技術研究機構 (827)
【Fターム(参考)】
【公開日】平成18年2月16日(2006.2.16)
【国際特許分類】
【出願日】平成17年2月28日(2005.2.28)
【出願人】(504147254)国立大学法人愛媛大学 (214)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(501203344)独立行政法人農業・生物系特定産業技術研究機構 (827)
【Fターム(参考)】
[ Back to top ]