
ポリマー−フォンビルブラント因子結合体
【課題】哺乳動物血液中のVWFおよび/またはFVIIIの生体内半減期を延長する新規の系を提供すること。
【解決手段】本発明は、血漿のおよび/または組換えフォンビルブラント因子(VWF)を含むタンパク質性構築物に関し、上記VWFが少なくとも1つの生理学的に許容し得るポリマー分子に結合するとともに、上記タンパク質性構築物と少なくとも1つの第VIII因子(FVIII)タンパク質との複合体と結合する。さらに、本発明は、FVIIIまたはVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法に関する。
【解決手段】本発明は、血漿のおよび/または組換えフォンビルブラント因子(VWF)を含むタンパク質性構築物に関し、上記VWFが少なくとも1つの生理学的に許容し得るポリマー分子に結合するとともに、上記タンパク質性構築物と少なくとも1つの第VIII因子(FVIII)タンパク質との複合体と結合する。さらに、本発明は、FVIIIまたはVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、血漿のおよび/または組換えフォンビルブラント因子(VWF)を含むタンパク質性構築物に関し、上記VWFが少なくとも1つの生理学的に許容し得るポリマー分子に結合するとともに、上記タンパク質性構築物と少なくとも1つの第VIII因子(FVIII)タンパク質との複合体と結合する。さらに、本発明は、FVIIIまたはVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法に関する。
【背景技術】
【0002】
(発明の背景)
VWFは、哺乳動物の血漿で存在する多量体付着性糖タンパク質であり、複数の生理機能を持つ。一次止血の間、VWFは血小板表面上の特異的な受容体とコラーゲン等の細胞外マトリックス成分と間にメディエーターとして働く。さらに、VWFはプロコアギュラントFVIII用担体を安定させるタンパク質として働く。VWFは、2813個のアミノ酸からなる前駆体分子として血管内皮細胞および巨核球で合成される。前駆体ポリペチド(プレプロVWF)は、成熟血漿VWFで見つかる22残基シグナルペプチド、741残基プロペプチド、および2050残基ポリペチドからなる(非特許文献1)。血漿へ分泌されると、VWFは異なる分子の大きさで多用な種類の形態で循環する。これらのVWF分子は、2050のアミノ酸残基の成熟サブユニットのオリゴマーおよびマルチマーからなる。VWFは、通常、1つのダイマーから最大で50〜100のダイマーからなるマルチマーまでのダイマーとして、血漿で見いだされる(非特許文献2)。ヒト血行内のヒトVWFの生体内半減期は、約12〜20時間である。
【0003】
ヒトで最も頻繁に起こる遺伝性出血障害はフォンビルブラント病(VWD)であり、血漿または組換え起源のコンセントレートを含むVWFによる補充療法によって処置することができる。血液中のVWFの半減期が短いため、VWFの生体内半減期を長期にしたVWFコンセントレートを発現することが強く求められている。同じことがFVIIIにもあてはまり、FVIIIも約8〜12時間の比較的短い生体内半減期を持つことから、FVIIIおよびVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害の患者を処置するために頻繁に投薬を行うことを必要とする。
【0004】
先行技術では、真核細胞培養で産生される組換えVWF(rVWF)がより完全であり、血漿由来VWFよりも蛋白質加水分解による分解が少ないことが記載されている(非特許文献3)。特許文献1は、陰イオン交換クロマトグラフィーを使用して組換え型VWFを精製することによって高純度のVWFを単離する方法を記載する。相同かつ構造的に完全なVWFの大量産生のための方法は、当該技術分野でも知られている(非特許文献4;非特許文献5)。組換え型VWFは、フォンビルブラント病(VWD)のイヌ、マウス、およびブタモデルを用いたことを特徴とする(非特許文献6;非特許文献7;非特許文献8;非特許文献9)。特許文献2は、プロVWFをトロンビンで処置することによってVWF調製を生産する方法を記載する。担体の上で固定されるrVWFを用いてVWFと結合するタンパク質を精製する方法は、特許文献3に開示される。血液凝固障害を処置するための血漿由来および組換えVWFプロペプチド(pro−VWF)の製薬的使用は、特許文献4に記載される。特許文献5は、リンカーまたはカップリング剤を介したポリ(酸化アルキレン)に対するFVIIIおよびIX因子(FIX)の結合について記載する。特許文献6では、長期間にわたる生物学的生体内半減期を持つ組換え型VWFの使用が哺乳動物血液でFVIIIを安定化させ、内因性FVIIIの産生を誘導することが示されている。にもかかわらず、VWF−またはFVIIIベースの出血障害がある患者にとっては、さらにVWFおよびFVIIIの生体内半減期を増大させる必要性が存在する。
【0005】
VWFは生体内でFVIIIを安定化させることで、FVIIIの血漿レベルを調節する重要な役割を担うことで、結果として一次および二次止血を制御するための中心因子となることが知られている。VWFを含む治療的産物を使用した後、24時間内に1mlあたり1〜3単位まで内因性FVMI:Cが増大することが観察され、このことがFVIII上でのVWFの生体内安定効果を示していることも知られている
凝固FVIII欠乏(別名血友病A)および/またはVWFの質的および定量的欠乏(別名VWD)に対する処置の範囲を広げるための新規の物質が強く求められている。機能性VWFが欠如していることから、VWDを有する患者は、正常値によりも低いFVIII血漿レベルによって示されるFVIIIの二次欠乏を呈する。VWDの型と疾患の重症度とに応じて、これらのFVIIIレベルが変化し得るが、通常、健常なヒトで見出されるFVIII血漿レベルよりも多少低い。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許出願公開第0 784 632号明細書
【特許文献2】国際公開第00/49047号パンフレット
【特許文献3】国際公開第98/25969号パンフレット
【特許文献4】欧州特許出願公開第0 977 584号明細書
【特許文献5】米国特許第6,037,452号明細書
【特許文献6】欧州特許出願公開第0 774 261号明細書
【非特許文献】
【0007】
【非特許文献1】Fischer et al.,FEBS Lett.351:345−348,1994
【非特許文献2】Ruggeri et al.Thromb.Haemost.82:576−584,1999
【非特許文献3】Fischer et al.,FEBS Lett.375:259−262,1995
【非特許文献4】Schlokat et al.,Biotechnol.Appl.Biochem.24:257−267,1996
【非特許文献5】Fischer et al.,CMLS 53:943−950,1997
【非特許文献6】Turecek et al.,Blood 90:3555−3567,1997
【非特許文献7】Roussi et al.,Blood Coag.Fibrinol.9:361−372,1998
【非特許文献8】Schwarz et al.,Haemophilia 4:53−62,1998
【非特許文献9】Schwarz et al.,Semin.Thromb.Hemost.28:215−225,2002
【発明の概要】
【発明が解決しようとする課題】
【0008】
このように、本発明は哺乳動物血液中のVWFおよび/またはFVIIIの生体内半減期を延長する新規の系を提供する。本発明のさらなる目的は、FVIIIおよびVWFの一方または両方の機能欠陥または欠乏を伴う出血障害に対する改善された処置のための方法を提供することである。
【課題を解決するための手段】
【0009】
(発明の要旨)
本発明は、血漿および/または組換えフォンビルブラント因子(VWF)またはその生物学的活性誘導体を含むタンパク質性構築物に関し、上記VWFまたは上記その生物活性誘導体が一つ以上の生理学的に許容し得るポリマー分子に結合しており、タンパク質性構築物の生体内半減期が哺乳動物(特にヒト)の血液で長くなる。さらに、本発明は上記タンパク質性構築物と少なくとも1つのFVIII因子(FVIII)タンパク質またはその生物学的活性誘導体との複合体に関し、上記FVIIIタンパク質または上記その上記生物学的活性誘導体の半減期が哺乳動物の血液でも長くなる。また、本発明により、上記タンパク質性構築物または上記複合体を用いたFVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法と同様に、上記タンパク質性構築物または上記複合体を含む医薬品組成物を提供する。蛋白質様構築物を作る方法も、提供する。
本発明は、例えば、以下を提供する:
(項目1)
タンパク質性構築物であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(b)該VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子と、を含み、
該構築物が少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体に対する結合能を有し、該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大している、タンパク質性構築物。
(項目2)
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目1に記載のタンパク質性構築物。
(項目3)
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して、少なくとも約2倍増大している、項目1に記載のタンパク質性構築物。
(項目4)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体の糖質残基に結合している、項目1に記載のタンパク質性構築物。
(項目5)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体のリジン残基に結合している、項目1に記載のタンパク質性構築物。
(項目6)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目1に記載のタンパク質性構築物。
(項目7)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目1に記載のタンパク質性構築物。
(項目8)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目1に記載のタンパク質性構築物。
(項目9)
上記構築物に含まれる上記VWFが、VWFの一次止血における生物学的活性を保持し、上記生物学的活性が、血小板上の受容体に対する結合および細胞間マトリックス成分上の受容体に対する結合を含み、上記成分がコラーゲンを含む、項目1に記載のタンパク質性構築物。
(項目10)
タンパク質性構築物であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(b)上記VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子と、を含み、
該構築物が少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に対する結合能を有し、該構築物に結合した該FVIII分子の生体内半減期が、該構築物とは結合していないFVIII分子の生体内半減期と比較して増大している、タンパク質性構築物。
(項目11)
上記構築物に結合した場合、上記FVIII分子の生体内半減期が、上記構築物とは結合していないFVIII分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目10に記載のタンパク質性構築物。
(項目12)
上記構築物に結合した場合、上記FVIII分子の生体内半減期が、上記構築物とは結合していないFVIII分子の生体内半減期と比較して、少なくとも約2倍増大している、項目10に記載のタンパク質性構築物。
(項目13)
上記VWFまたは上記その生物活性誘導体が組換え産物である、項目1または10に記載のタンパク質性構築物。
(項目14)
項目1または10に記載のタンパク質性構築物および少なくとも1つのFVIII分子またはその生物活性誘導体を含む、複合体。
(項目15)
上記FVIII分子またはその生物活性誘導体が組換え産物である、項目14に記載の複合体。
(項目16)
FVIIIの機能的な欠陥または欠乏に関連した出血性障害を持つ哺乳動物血液中でのFVIIIまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物の第1の用量を該哺乳動物に対して投与するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体の第1の用量を該哺乳動物に対して投与するステップと、
を含む、方法。
(項目17)
FVIIIおよびVWFのうちの少なくとも1つの機能的な欠陥に関連した出血性障害を持つ哺乳動物血液中でのFVIIIまたはその生物活性誘導体の生体内半減期およびVWFまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物の第1の用量を該哺乳動物に対して投与するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体の第1の用量を該哺乳動物に対して投与するステップと、
を含む、方法。
(項目18)
FVIIIおよびVWFのうちの少なくとも1つの機能的な欠陥または欠乏に関連した出血性障害を持つ哺乳動物血液中での第VIII因子(FVIII)またはその生物活性誘導体およびVWFまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10の少なくとも1つのタンパク質性構築物を提供するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体を提供するステップと、
(c)上記タンパク質性構築物と上記FVIII分子またはその生物活性誘導体との間で複合体を形成するステップと、
を含む、方法。
(項目19)
FVIIIの機能的な欠陥に関連した出血性障害またはFVIIIの機能的な欠陥もしくは欠乏に関連した障害を持つ哺乳動物血液中での第VIII因子(FVIII)またはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物を提供するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体を提供するステップと、
(c)該タンパク質性構築物と該FVIII分子またはその生物活性誘導体との間で複合体を形成するステップと、
を含む、方法。
(項目20)
ステップ(c)の複合体を上記哺乳動物に投与する、項目18または19に記載の方法。
(項目21)
上記少なくとも1つのFVIII分子またはその生物活性誘導体を、上記タンパク質性構築物と同時に投与する、項目16または17に記載の方法。
(項目22)
上記少なくとも1つのFVIII分子またはその生物活性誘導体を、上記タンパク質性構築物の投与前または投与後に、逐次投与する、項目16または17に記載の方法。
(項目23)
医薬品組成物であって、
項目1、2、3、または6に記載のタンパク質性構築物の有効量と、
薬学的に許容可能な担体、希釈剤、塩、緩衝液および賦形剤からなる群から選択される一つ以上の化合物と、
を含む、医薬品組成物。
(項目24)
医薬品組成物であって、
項目10、11、または12に記載のタンパク質性構築物の有効量と、
薬学的に許容可能な担体、希釈剤、塩、緩衝液および賦形剤からなる群から選択される一つ以上の化合物と、
を含む、医薬品組成物。
(項目25)
複合体であって、
(a)タンパク質性構築物であって、
(i)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(ii)該VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子とを含む、タンパク質性構築物と、
(b)該タンパク質性構築物に結合した少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と、
を含み、
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、増大している、複合体。
(項目26)
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、少なくとも約1.5倍増大している、項目25に記載の複合体。
(項目27)
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、少なくとも約2倍増大している、項目25に記載の複合体。
(項目28)
VWF分子と、該VWF分子上の少なくとも1つの糖質残基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質残基を酸化するステップと、
(c)該糖質残基を、ヒドラジド基含有PEG試薬と接触させるステップと、
(d)該試薬PEGヒドラジドを該VWF上の少なくとも1つの糖質残基と共有結合させることにより、上記構築物を形成させるステップとを含み、
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
上記構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に結合することが可能である、方法。
(項目29)
上記酸化ステップ(b)が化学的酸化剤を使用して実行される、項目28に記載の方法。
(項目30)
上記化学的酸化剤がNaIO4である、項目29に記載の方法。
(項目31)
上記酸化ステップ(b)が酵素的酸化剤を使用して実行される、項目28の方法。
(項目32)
上記酵素的酸化剤がガラクトースオキシダーゼである、項目31に記載の方法。
(項目33)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目28に記載の方法。
(項目34)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目33に記載の方法。
(項目35)
項目28に記載の方法によって調製されたタンパク質性構築物。
(項目36)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目35に記載のタンパク質性構築物。
(項目37)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも約2倍増大している、項目35に記載のタンパク質性構築物。
(項目38)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも約1.5倍増大している、項目35に記載のタンパク質性構築物。
(項目39)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも約2倍増大している、項目35に記載のタンパク質性構築物。
(項目40)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目28に記載の方法。
(項目41)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目28に記載の方法。
(項目42)
VWF分子と、該VWF分子上の少なくとも1つの一級アミノ基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の該一級アミノ基をPEG試薬と接触させるステップと、
(c)該PEG試薬を該VWF上の該一級アミノ基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に結合することが可能である、方法。
(項目43)
上記一級アミノ基が、上記VWF分子のリジン残基上に含まれる、項目42に記載の方法。
(項目44)
上記一級アミノ基が、上記VWF分子のN末端上に含まれる、項目42に記載の方法。
(項目45)
上記PEG試薬が、N−ヒドロキシスクシンイミド−エステル、PEGカーボネート、カルボキシル基含有PEG誘導体、およびアルデヒド基含有PEG誘導体からなる群から選択される、項目42に記載の方法。
(項目46)
上記試薬が、PEG−スクシンイミジルスクシネート、PEG−スクシンイミジルグルタレート、PEG−スクシンイミジルブタノエート、PEG−スクシンイミジルヘキサノエート、分解性PEG試薬、および加水分解性PEG試薬からなる群から選択されるN−ヒドロキシスクシンイミド−エステルである、項目45に記載の方法。
(項目47)
上記試薬が、p−ニトロフェニルカーボネートまたはスクシンイミジルカーボネートであるPEGカーボネートである、項目45に記載の方法。
(項目48)
上記試薬が、水溶性カルボジイミドの使用によって一級アミノ基に連結することができるカルボキシル基を含むPEG誘導体の群から選択される、項目45に記載の方法。
(項目49)
上記試薬がPEGプロピオンアルデヒドであり、還元性アミノ化によって一級アミノ基に連結することができる、項目45に記載の方法。
(項目50)
VWF分子と、該VWF分子上の少なくとも1つの一級アミノ基に、還元的アミノ化によって共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で該VWF分子上のリジン残基をアルデヒド基含有PEG試薬と接触させてシッフ塩基を形成するステップと、
(c)該溶液を還元剤と接触させて二級アミド結合を形成するステップと、
(d)該PEG試薬を上記VWF分子に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目51)
上記還元剤がNaCNBH3である、項目50に記載の方法。
(項目52)
上記一級アミノ基が上記VWF分子上のN末端アミノ基であり、該N末端アミノ基が選択的にPEG付加されている、項目50に記載の方法。
(項目53)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目50に記載の方法。
(項目54)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目53に記載の方法。
(項目55)
項目50に記載の方法によって調製されたタンパク質性構築物。
(項目56)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目55に記載のタンパク質性構築物。
(項目57)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目55に記載のタンパク質性構築物。
(項目58)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目55に記載のタンパク質性構築物。
(項目59)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目55に記載のタンパク質性構築物。
(項目60)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目50に記載の方法。
(項目61)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目50に記載の方法。
(項目62)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で該VWF分子上のリジン残基を試薬PEGアルデヒドと接触させてシッフ塩基を形成するステップと、
(c)該溶液を反応性試薬と接触させて二級アミド結合を形成するステップと、
(d)該PEG試薬を該VWF分子に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目63)
VWF分子と、該VWF分子上の少なくとも1つの遊離または生成スルフヒドリル基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の該スルフヒドリル基を、マレイミド基含有PEG試薬と接触させるステップと、
(c)該PEG試薬を該VWF分子上の少なくとも1つのスルフヒドリル基に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目64)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目63に記載の方法。
(項目65)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目64に記載の方法。
(項目66)
項目63に記載の方法によって調製されたタンパク質性構築物。
(項目67)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目66に記載のタンパク質性構築物。
(項目68)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目66に記載のタンパク質性構築物。
(項目69)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目66に記載のタンパク質性構築物。
(項目70)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目66に記載のタンパク質性構築物。
(項目71)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目63に記載の方法。
(項目72)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目71に記載の方法。
(項目73)
VWF分子と、該VWF分子上の少なくとも1つのカルボキシル基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上のカルボキシル基を、アミノ基および水溶性カルボジイミドを含むPEG試薬と接触させてアミド結合を形成するステップと、
(c)該PEG部分を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目74)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目73に記載の方法。
(項目75)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目74に記載の方法。
(項目76)
項目73に記載の方法によって調製されたタンパク質性構築物。
(項目77)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目76に記載の構築物。
(項目78)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目76に記載の構築物。
(項目79)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目76に記載の構築物。
(項目80)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目76に記載の構築物。
(項目81)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目73に記載の方法。
(項目82)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目81に記載の方法。
(項目83)
VWF分子と、該VWF分子上に共有結合しており、該VWF分子のFVIII結合部位に対しては結合していないPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上のFVIII結合部位を第VIII因子結合部位保護剤と接触させることにより、保護されたFVIII結合部位を持つVWF分子を形成させるステップと、
(c)ステップ(b)の該VWF分子上の反応部位をPEG試薬と接触させるステップと、
(d)該PEG試薬を該VWF分子と共有結合させるステップと、
(e)該VWF分子から該FVIII結合部位保護剤を分離することにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目84)
上記第VIII因子結合部位保護剤が親和性カラムに含まれる、項目83に記載の方法。
(項目85)
上記第VIII因子結合部位保護剤が、第VIII因子、FVIIIの誘導体、ヘパリン、およびヘパリンの誘導体からなる群から選択される、項目83の方法。
(項目86)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目83に記載の方法。
(項目87)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目86に記載の方法。
(項目88)
項目83に記載の方法によって調製されたタンパク質性構築物。
(項目89)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目88に記載の構築物。
(項目90)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目88に記載の構築物。
(項目91)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目88に記載の構築物。
(項目92)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目88に記載の構築物。
(項目93)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目83に記載の方法。
(項目94)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目93に記載の方法。
(項目95)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基と共有結合したポリシアル酸(PSA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で、PSAを、アルデヒド含有PEG試薬と接触させてシッフ塩基を形成するステップと、
(c)該溶液を還元剤と接触させて二級アミド結合を形成するステップと、
(d)該PSAを該VWF上の少なくとも1つのリジン残基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目96)
上記還元剤がNaCNBH3である、項目95に記載の方法。
(項目97)
項目95に記載の方法によって調製されたタンパク質性構築物。
(項目98)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目97に記載の構築物。
(項目99)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目97に記載の構築物。
(項目100)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目97に記載のタンパク質性構築物。
(項目101)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目97に記載のタンパク質性構築物。
(項目102)
VWF分子と、該VWF分子と共有結合により架橋したポリシアル酸(PSA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子を、PSAとグルタルアルデヒドとを含む溶液と接触させるステップと、
(c)該PSAを該VWFへと共有結合により架橋させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目103)
項目102に記載の方法によって調製されたタンパク質性構築物。
(項目104)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目103に記載の構築物。
(項目105)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目103に記載の構築物。
(項目106)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目103に記載のタンパク質性構築物。
(項目107)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目103に記載のタンパク質性構築物。
(項目108)
VWF分子と、該VWF分子上の少なくとも1つの糖質残基に共有結合したPSA部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質残基を酸化するステップと
(c)該糖質残基を、ヒドラジド基含有PSA試薬と接触させるステップと、
(d)該PSA試薬を該VWF上の少なくとも1つの糖質残基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目109)
上記酸化ステップ(b)が酵素的酸化剤を用いて実行される、項目108に記載の方法。
(項目110)
上記酵素的酸化剤がガラクトースオキシダーゼである、項目109に記載の方法。
(項目111)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基に共有結合したヒアルロン酸(HA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)HAの溶液を酸化剤と接触させて活性化HAを形成させるステップと、
(c)該VWF分子を該活性化HAと接触させることにより、該構築物を形成させるステップとを含み、
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して増大している、上記構築物が、少なくとも1つのFVIII因子分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目112)
項目111に記載の方法によって調製されたタンパク質性構築物。
(項目113)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目112に記載の構築物。
(項目114)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目112に記載のタンパク質性構築物。
(項目115)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目112に記載のタンパク質性構築物。
(項目116)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目112に記載のタンパク質性構築物。
(項目117)
VWF分子と、上記VWF分子上の少なくとも1つの糖質基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質基を酸化酵素と接触させて、該VWF上で酸化型の糖質部分を形成させるステップと、
(c)該VWF上の該酸化型の糖質部分をヒドラジド基含有PEG試薬と接触させるステップと、
(d)該PEG部分を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目118)
項目117に記載の方法によって調製されたタンパク質性構築物。
(項目119)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目118に記載の構築物。
(項目120)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目118に記載の構築物。
(項目121)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目118に記載のタンパク質性構築物。
(項目122)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目118に記載のタンパク質性構築物。
(項目123)
VWF分子と、該VWF分子に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子をPEG試薬に接触させて溶液を形成するステップと、
(c)該溶液を灌流チェンバーに入れ、蠕動ポンプの使用により上記VWF分子上に剪断応力を発生させるステップと、
(c)該PEG試薬を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目124)
上記PEG試薬が、N−ヒドロキシスクシンイミド−エステル、PEGカーボネート、カルボキシル基含有PEG誘導体、およびアミノ基含有PEG誘導体からなる群から選択される、項目123に記載の方法。
(項目125)
上記PEG試薬がPEG−スクシンイミジルスクシネート、PEG−スクシンイミジルグルタレート、PEG−スクシンイミジルブタノエート、PEG−スクシンイミジルヘキサノエート、加水分解感受性のエステルまたはアミド結合によって結合される加水分解性PEG、mPEG−(カルボキシメチル)−3−ヒドロキシ−ブタン酸N−ヒドロキシスクシンイミドエステル、PEG−p−ニトロフェニルカーボネート、および、PEG−スクシンイミジルカーボネートからなる群から選択される、項目123に記載の方法。
(項目126)
項目123に記載の方法によって調製されたタンパク質性構築物。
(項目127)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目126に記載の構築物。
(項目128)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目126に記載の構築物。
(項目129)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目126に記載の構築物。
(項目130)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目126に記載の構築物。
(項目131)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体の糖質残基に結合している、項目10に記載のタンパク質性構築物。
(項目132)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体のリジン残基に結合している、項目10に記載のタンパク質性構築物。
(項目133)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目10に記載のタンパク質性構築物。
(項目134)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目10に記載のタンパク質性構築物。
(項目135)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目10に記載のタンパク質性構築物。
(項目136)
上記構築物に含まれる上記VWFが、VWFの一次止血における生物学的活性を保持し、上記生物学的活性が、血小板上の受容体に対する結合および細胞間マトリックス成分上の受容体に対する結合を含み、上記成分がコラーゲンを含む、項目10に記載のタンパク質性構築物。
(項目137)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目10に記載のタンパク質性構築物。
(項目138)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目25に記載の複合体。
(項目139)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目26に記載の複合体。
(項目140)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目26に記載の複合体。
【図面の簡単な説明】
【0010】
【図1】図1Aおよび図1Bは、結合標的部位に関する実施例によるVWFの模式的構造を示す。図1Aの灰色の点は少なくとも1つのポリマー分子によって結合し得るVWFのリジン残基を示し、図1Bは少なくとも1つのポリマー分子によって結合し得るVWFの糖質残基を示す。
【図2】図2は、VWDを有するマウスでの非結合体化VWFと比較したポリマーVWF結合体の薬物動態を示す。
【図3】図3は、血友病マウスでの非結合体化VWFと比較したポリマーVWF結合体の薬物動態を示す。
【図4】図4は、rVWFまたはPEG−rVWFで処置されるVWDマウスでのFVIIIの薬物動態を示す。
【図5】図5は、ポリマーVWF結合体と複合体を形成したrFVIIIおよびVWFと複合体を形成したrFVIIIを適用した後の血友病マウスにおけるrFVIIIの回収率を示す。
【図6】図6は、ポリマーVWF結合体と複合体を形成したrFVIIIおよびVWFと複合体を形成したrFVIIIを適用した後の血友病マウスにおけるVWFの回収率を示す。
【図7】図7は、FVIII×VWFダブルノックアウトマウスにおけるrFVIIIおよびPEG−rVWFの薬物動態を示す。
【図8】図8は、雑種−マウスにおけるrFVIIIとPEG−rVWFの薬物動態を示す。
【図9】図9は、マウス血漿におけるVWF:Ag増加(リジンおよび糖質結合)を示す。
【図10】図10は、非結合体化rVWFと比較した場合のPEGに結合したrVWFの生体分子相互作用検討(左側図)とFVIII(右側図)の結合能の比較とを示す。
【図11】図11は、SDS−PAGEによって計測されるポリマー結合の後のVWFの大量の増加を示す。
【図12】図12は、VWFマルチマーを分析するためにアガロース電気泳動で計測されるポリマー結合後のVWFの大量の増加を示す。
【図13】図13は、ポリマーVWF結合体(PEG付加リジン残基を有するrVWF)と複合体を形成したrFVIIIと、rVWFと複合体を形成したrFVIIIの適用後のFVIII−K.O.マウスにおけるrFVIIIの薬物動態を示す。
【図14】図14は、ポリマーVWF結合体(PEG付加リジン残基によるrVWF)と複合体を形成したrFVIIIおよびrVWFと複合体を形成したrFVIIIの適用後のFVIII−K.O.マウスにおけるrVWFの薬物動態を示す。
【図15】図15は、組み合わされたELISAおよび色素アッセイ(ECA)を使用して、非結合体化rVWFと比較した場合のPEGと結合したrVWFのFVIII結合能の測定を示す。
【図16】図16は、表面プラズモン共鳴技術を使用したPEGと結合したrVWFのFVIII結合能の側定を示す。
【図17】図17は、実施例13に記載のように、SDS−PAGEで計測されるポリマー結合後のVWFの大量増加を示す。
【図18】図18は、組み合わされたELISAおよび色素アッセイ(ECA)を用いたpdVWFおよび無処置rVWFと比較した場合のフリン成熟rVWFのFVIII結合能の測定を示す。
【図19】図19は、表面プラズモン共鳴技術を使用してpdVWFおよび無処置rVWFと比較した場合のフリン成熟rVWFのFVIII結合能の測定を示す。
【図20】図20は、還元条件下でのrVWF−ポリシアル酸結合体およびrVWFの等電点電気泳動パターンを示す。
【図21】図21は、VWF欠乏マウスにおけるrVWF−ポリシアル酸結合体およびrVWFの薬物動態を示す。
【図22】図22は、VWF欠乏マウスにおけるrVWF−ポリシアル酸結合体またはrVWFの使用後、マウスFVIII活性の時間経過を示す。
【図23】図23は、FVIII欠乏マウスにおけるPEG−rVWF(分枝PEG20K SG)およびrVWFの薬物動態を示す。
【図24】図24は、FVIII欠乏マウスでのPEG−rVWF(分枝PEG20K SG)またはrVWFを共に注入したrFVIIの薬物動態を示す。
【図25】図25は、FVIII欠乏マウスで種々の濃度のPEG−rVWF(分枝PEG20K SG)を注入したrFVIIの薬物動態を示す。
【図26】図26は、FVIII欠乏マウスにおけるPEG−rVWF#A(5mgPEG/mgタンパク質)、PEG−rVWF#B(20mgPEG/mgタンパク質)とネイティブrVWFの薬物動態を示す。
【図27】図27は、PEG−rVWF#A(5mgPEG/mgタンパク質)、PEG−rVWF #B(20mgPEG/mgタンパク質)またはネイティブrVWFを同時注入した場合のrFVIIIの薬物動態を示す。
【発明を実施するための形態】
【0011】
(発明の詳細な説明)
本発明の一態様は、血漿および/または組換えVWFあるいはその生物活性誘導体を含むタンパク質性構築物(また、以下で「ポリマーVWF結合体」と称される)に関し、上記VWFまたは上記その生物活性誘導体が一種類以上の生理学的に許容可能なポリマー分子に結合しており、上記VWFまたは上記その生物活性誘導体の生体内半減期が哺乳動物血液で長くなる。
【0012】
本発明のさらなる態様は、ポリマーVWF結合体を提供することであり、該ポリマーVWF結合体は、2つの形態の中間にある特徴を有する上記ポリマーVWF結合体の形態をなす。1つの形態は、VWFと結合体を形成するポリマーを安定して担持し、哺乳動物への適用の後、時間が経つにつれて内在性分子として除去される。他の形態は、VWFに結合体を形成するポリマーの可逆性を特徴とする。哺乳動物に対する投与の後、VWFに結合したポリマー分子はVWFから段階的に遊離され、非結合VWFは薬理的機能因子として利用可能となる。剥離性は、共役化学に依存するとともに、VWFに結合したポリマー分子の組成および構造に依存する。
【0013】
ポリマーVWF結合体は、VWDの処置に対して単独で、または半減期の増大に関してFVIIIを安定化させるためにFVIIIと結合して、あるいは両方で、有用である。VWDの処置に対して単独使用される場合、結合体は2つの形態のうちの一方を取ると考えらえる。第1の形態は、ポリマーが遊離自在にVWFに結合するものである。このようにして、VWFは、ポリマーが遊離または分解するにつれて活性化される。第2の形態は、VWFに結合したポリマー濃度がVWF活性と干渉しない程度のものである。結合体がFVIIIを結合して安定させるために調製される場合、VWFに結合したポリマーの度合いまたはレベルは、VWFの結合領域と干渉しないように提供される。実施例で示されるように、VWFに対する満足のいくポリマーの結合は、VWFおよびFVIIIの結合能に干渉することなく達成し得る。ポリマー結合の度合いもまた、制御または修飾してVWF活性を保つ一方で、FVIIIに対して結合するVWFの能力も保たれる。この形態で、ポリマーVWF結合体は治療的活性VWFを提供する一方で、半減期増大に関してFVIIIを安定化させる。
【0014】
本発明に有用なVWFおよびFVIII分子として、完全長タンパク質、タンパク質の前駆体、タンパク質のサブユニットまたはフラグメント、ならびにその機能的誘導体が挙げられる。VWFおおびFVIIIまたはFVIIIへの言及は、このようなタンパク質のすべての潜在的形態を含むものと考える。
【0015】
本明細書中で用いられる場合「生物活性誘導体」は、分子の任意の誘導体を含み、該誘導体は、上記分子の同じ機能的なおよび/または生物学的な性質(例えば結合性を持つ)および/または同じ構造的基礎(例えばペプチドの主鎖または基本的高分子単位)を、実質的に持つ。
【0016】
本発明に有用なVWFは、単一および多量体形態を含むすべての潜在的形態を含む。VWFの1つの特に有用な形態は、少なくとも2つのVWFからなるホモマルチマーである。VWFタンパク質は、いずれの生物活性誘導体であってもよく、あるいはFVIIIの安定剤として単独で使用される場合、VWFが生物学的に不活性である形態であってもよい。併用使用されるVWFの異なる形態を本発明が含むことも理解すべきである。例えば、本発明に有用な組成物は、異なるマルチマー、異なる誘導体、ならびに生物活性誘導体と生物活性のない誘導体とを含むものであってもよい。一次止血において、VWFは血小板と特異的な細胞外マトリックス成分(例えばコラーゲン)間の架橋として働く。このプロセスでのVWFの生物学的活性を、2つの異なる生体外アッセイで計測することができる(Turecek et al.,Semin.Thromb.Hemost.28:149−160,2002)。リストセチンコファクターアッセイは、VWF存在下で抗生物質のリストセチンによって誘発される新鮮またはホルマリン固定血小板の凝集に基づく。血小板凝集反応の程度は、VWF濃度次第であって、比濁法によって、例えば血小板凝集計を使用することで計測することができる(Weiss et al.,J.Clin.Invest.52:2708−2716,1973;Macfarlane et al.,Thromb.Diath.Haemorrh.34:306−308,1975)。第2の方法はコラーゲン結合実験であり、これはELISA技術に基づく(Brown et Bosak,Thromb.Res.43:303−311,1986;Favaloro,Thromb.Haemost.83:127−135,2000)。マイクロタイタープレートは、I型またはIII型のコラーゲンで被覆されている。さらに、VWFはコラーゲン面に結合し、続いて、酵素で標識されたポリクローナル抗体で検出される。最後のステップは、基質反応であり、ELISAリーダーで光度分析的にモニターすることができる。
【0017】
本明細書中で用いられる場合、「血漿由来VWF(pdVWF)」は血液中に見いだされるタンパク質のすべての形態を含むもので、該形態として、少なくとも1つのFVIII分子を生体内安定化(例えば結合)させる性質がある哺乳動物から得られる成熟VWFが挙げられる。しかし、本発明は成熟VWFに限らない。1つに、上記pVWFの生物活性誘導体は、プロペプチドを含むプロVWFである。本発明に有用な他の型のVWFとして、タンパク質性構築物が挙げられる。このタンパク質性構築物は未熟なVWFを含み、該未熟VWFとして、血管内皮細胞および巨核球によって合成される前駆体VWF分子(プレプロVWF)および/またはシグナルペプチドおよびプロペプチドの開裂で得られるVWFプロペプチド(プロVWF)および/または成熟pdVWF(それぞれ前駆体分子のもの)が挙げられる。pdVWFの生物活性誘導体のさらなる例として、プロドラッグが挙げられ、該プロドラッグは、処理または変換され、生物活性形態となる。あるいは、プロドラッグは、天然型と比べて、生物学的に活性があることから、切断形態、欠失を持つ形態、置換を持つ形態、プロ形態以外の付加を有する形態、成熟形態のフラグメント、キメラ形態、および後翻訳修飾を持つ形態である。本発明に有用なPdVWFも、生物学的活性のないそれらの形態を含む。このことは、血液中見いだされる成熟VWFまたは他の天然形態の修飾によって達成し得る。本発明に有用なVWFの供給源は、ブタおよびヒトバージョンを含む哺乳動物である。
【0018】
本明細書中で用いられる場合、「組換え型VWF(rVWF)」は組換えDNA技術を経て得られるVWFを含む。有用なrVWFの1つの形態は、少なくとも1つのFVIII分子の少なくとも生体内安定化(例えば結合)の性質を持ち、薬理学的に許容可能なグリコシル化パターンを任意に有する。その具体例として、A2領域を持たないことからタンパク質分解に対して耐性を持つVWF(Lankhof et al.,Thromb.Haemost.77:1008−1013,1997)、糖タンパク質Ib結合領域とコラーゲンおよびヘパリンの結合部位とをVal449からAsn730までのVWFフラグメント(Pietu et al.,Biochem.Biophys.Res.Commun.164:1339−1347,1989)が挙げられる。少なくとも1つのFVIII分子の安定化の側定は、当該技術分野における状態で公知の方法によって、VWF欠乏哺乳動物で行うことができる。例えば、下記の実施例8に記載のように、VWF欠乏マウスに対してVWFを経尾静脈静注することにより処置すると、その血漿のFVIII活性のレベルが時間とともに変化する。例えば、FVIII活性のレベルは、色素アッセイ(例えば、European Pharmacopoeiaで発表されているもの)で計測することができる(Ph.Eur.,3rd Ed.1997:2.7.4)。
【0019】
FVIII(FVIlI:C)含有試料を、カルシウム含有緩衝液中でトロンビン、活性化第IX因子(FIXa)、リン脂質、および第X因子(FX)と混合する。FVIIIはトロンビンにより活性化され、続いてリン脂質、FIXaおよびカルシウムイオンと複合体を形成する。この複合体は、FXおよびFXaを活性化し、順次に色素基質(例えば、AcOH*CH3OCO−D−CHA−Gly−Arg−pNA)を切断する。放出されたp−ニトロアニリン(pNA)の時間経過を、405nmで計測する。反応の傾斜は、試料中のFVIII濃度に比例している。
【0020】
本発明のrVWFは、当該技術分野で知られている任意の方法によって生産可能である。一具体例が、WO86/06096(1986年10月23日発行)および米国特許出願第07/559509号(Ginsburg et al.名義で1990年7月23日出願)に開示されており、組換え型VWFを生産する方法に関連して本明細書中に援用する。これは、当該技術分野で公知の任意の方法を含むものであってよく、該方法は、(i)遺伝子工学による組換えDNAの産生(例えばRNAの逆転写および/またはDNAの増幅を介する)、(ii)トランスフェクションによって組換えDNAを原核または真核細胞に導入すること(例えば電気穿孔法またはマイクロインジェクションを介する)、(iii)上記形質転換細胞を培養すること(例えば連続またはバッチ方式によるもの)、(iv)VWFを発現すること(例えば構成的または誘導的)、ならびに(v)上記VWFを単離すること(例えば、培地から、または形質転換細胞を収集することによって)、そのために(vi)精製されたrVWF(例えば陰イオン交換クロマトグラフィーまたはアフィニティークロマトグラフィーを経て)を得ることのためのものである。
【0021】
薬理学的に許容可能なVWF分子を生産することを特徴とする適当な原核生物または真核生物宿主系での発現によって、rVWFを生産することができる。真核細胞の例は、哺乳動物細胞(例えばCHO、COS、HEK 293、BHK、SK−Hep、およびHepG2)である。本発明によるVWFを生産または単離するための試薬または条件に対しては何ら特定の制限はなく、当該技術分野で公知または市販されている任意の系を用いることができる。本発明の好ましい実施形態では、先端技術に記載の方法でrVWFが得られる。
【0022】
多種多様なベクターをrVWFの調製に使用することができ、該ベクターを真核生物および原核生物発現ベクターから選択することができる。原核生物発現のためのベクターの例として、プラスミド(例えばpRSET、pET、pBAD、その他)が挙げられ、原核生物発現ベクターで使用されるプロモーターとして、lac、trc、trp、recA、araBAD、その他が挙げられる。真核生物発現のためのベクターの例として、以下のものが挙げられる。すなわち、(i)酵母での発現用として、プロモーター(例えば、AOX1、GAP、GAL1、AUG1、その他)を用いるベクター(例えば、pAO、pPIC、pYES、pMET)、(ii)昆虫細胞での発現用として、プロモーター(例えば、PH、p10、MT、Ac5、OplE2、gp64、polh、その他)を用いるベクター(例えば、pMT、pAc5、plB、pMIB、pBAC,その他)、ならびに(iii)哺乳動物細胞での発現として、ベクター(例えば、pSVL、pCMV、pRc/RSV、pcDNA3、pBPV、その他)、およびプロモーター(例えばCMV、SV40、EF−1、UbC、RSV、ADV、BPV、β−actin)を用いたこのようなウイルス系に由来するベクター(例えば、ワクシニアウイルス、アデノ関連性ウイルス、ヘルペスウイルス、レトロウイルス)が挙げられる。
【0023】
本発明に有用なFVIIIは、それらの形態を含み、該形態は、生物学的に活性があり、完全長FVIII、凝固FIXの活性化でコファクターとして働く任意の派生的な能力、およびVWFで複合体を形成する能力が含まれる。本発明によって使用されるFVIIIは、血漿由来FVIII(pdFVIII)または組換え型FVIII(rFVIII)またはその生物活性誘導体であってもよい。pdFVIIIおよびrFVIIIは、当該技術分野で知られている任意の方法によって生産することが可能である。PdFVIIIは、任意の適当な手段で精製可能である。1つの有用な方法は米国特許第5,470,954号に記載されており、該特許文献を本明細書中に援用する。RFVIIIタンパク質は、任意の適当な手段で調製可能である。このようなrFVIIIの例として、Baxter Healthcare社によって製造販売されているRECOMBINATEとADVATE、Wyeth社によって製造販売されているREFACTO(FVIIIのB−領域が欠失した形態)、およびBayer社によって製造販売されているKOGENATEが挙げられる。rFVIIIの方法および例は、米国特許第4,757,006号、第4,965,199号、および第5,618,788号(いずれも本明細書中に援用)に記載されている。
【0024】
本明細書中で用いられる場合、「生理学的に許容し得るポリマー」は、水溶液または懸濁液に可溶であり、医薬的に有効な量のポリマーVWF結合体の投与に際して哺乳動物に対し陰性影響(例えば副作用)がないポリマーを含む。本発明によって使用される生理学的に許容し得るポリマーは、特に制限されるものではない。ポリマーは、反復単位を好ましくは2ないし約300持つことによって一般に特徴づけられる。このようなポリマーの例として、限定されるものではないが、ポリ(アルキレングリコール)、例えばポリエチレングリコール(PEG)、ポリ(プロピレングリコール)(PPG)、エチレングリコールとプロピレングリコールとの共重合体、その他、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリル酸塩)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファゼン、ポリオキサゾリン、ポリ(N−重合開始剤)、ならびに前述のいずれかの組合せが挙げられる。
【0025】
生理学的に許容し得るポリマーは特定の構造に限定されず、直鎖状(例えばアルコキシPEGまたは二官能性PEG)、分岐状、またはマルチアーム(例えばフォーク状PEGまたはポリオール核に結合したPEG)、樹状、または分解可能な結合を有することができる。さらに、ポリマーの内部構造は、任意の数の異なるパターンで系統化され、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される。
【0026】
これらのポリマーもまた、ポリ(酸化アルキレン)ポリマー、ポリ(マレイン酸)、ポリ(DL−アラニン)、例えばカルボキシメチルセルロース、デキストラン、ヒアルロン酸およびキチン、ならびにポリ(メセドリン)アクリレート系が挙げられる。
【0027】
本発明の実施形態において、生理学的に許容し得るポリマーはそのPEGと誘導体である。PEG側鎖は直鎖状、分岐状、フォーク状、マルチアームからなることができる。本発明によって使用されるPEGは、特に限定されない。特に有用なPEGは、3,000〜20,000の範囲に、分子量を持つ。パブリックドメイン(すなわち、決して特許権を得なかったか、現在は特許期限切れ.)にあるいくつかの有用なPEG分子がある。他の有用なPEG分子は、WO03/040211、US6,566,506、US6,864,350、およびUS6,455,639に開示されている。
【0028】
本発明の別の実施形態において、生理学的に許容し得るポリマーはポリシアル酸(PSA)とその誘導体である。PSAはUS4,356,170(本明細書中に援用)に記載の方法によってVWFに結合することができる。本発明の一実施形態において、多糖化合物を、天然に存在する多糖類、天然に存在する多糖類の誘導体または天然に存在する多糖誘導体することが可能である。化合物の多糖部分は、5以上(概して少なくとも10)のシアル酸残基をポリマー鎖に有し、別の実施形態では少なくとも20〜50のシアル酸残基をポリマー鎖に有する。すぐに利用できる多糖化合物は全体で最高500のサッカライド残基を持つと考えられ、通常ポリマー鎖で300未満の残基を持つものであってもよい。通常、化合物のサッカライド残基の全てが、シアル酸残基である。
【0029】
多糖化合物で少なくともポリシアル酸部分、また一実施形態では全ての化合物で、高度に親水性である。親水性は、水酸基と同様に、シアル酸単位のペンダントカルボキシル基によって主に与えられる。サッカライド単位は、他の官能基(例えばアミン、ヒドロキシル基または硫酸塩基)またはその組合せを含むものであってもよい。これらの基は天然のサッカライド化合物上に存在可能であり、あるいは派生的な多糖化合物に導入されるものであってもよい。
【0030】
本発明の特定用途の多糖化合物は、細菌によって産生されるものである。これらの天然の多糖の一部は、グリコリピドとして知られている。特に有利なことは、多糖化合物が末端ガラクトース単位を実質的に有しない場合であり、このことは肝細胞およびクッパー細胞のガラクトース受容体によって認識される傾向にある。
【0031】
VWFマルチマーは、当業者に知られている種々の技術のいずれかによって、多糖化合物に共有結合可能である。実施例は、VWFまたは多糖のいずれか一方の上にあるカルボキシル基と他方のアミン基との間のペプチド結合、あるいは一方のカルボキシ基と他方の水酸基との間のエステル結合を介した結合を含む。あるいは、シッフ塩基は一方のアミノ基と他方のアルデヒド基との間に形成することができる。結合の他の機構は、当該技術分野の通常の技量内である。種々の例が米国特許第5,846,951号の第7欄15合目から第8欄第5行目までに同定される。
【0032】
本明細書中で用いられる場合、一つ以上の生理学的に許容し得るポリマー分子に結合しているVWFへの言及は任意の適当な化学的結合、例えば共有結合または非共有結合(例えばイオン性、疎水性、親和性、生物親和性相互作用)を含む。ポリマーを、二価性試薬の使用によって、またスペーサーアームを経てタンパク質に連結させることもできる。加えて、ポリマー分子は親和性相互作用によってVWFに連結することができる。例えば、VWFをビオチン標識させることができ、アビジンまたはストレプトアビジン結合ポリマーをVWFに結合させることができる。また、ポリクローナルまたはモノクローナル抗VWF抗体およびそのフラグメントをポリマーに結合することができ、この複合体をVWFに結合させることができる。ポリマーを酵素的方法、例えばUS6,379,933に教示されるポリグリコシルトランスフェラーゼによるサッカライドの転移またはUS20040132640A1に教示されるものとしてのグリコペジレーション(glycopegylation)の転移によってもVWFに結合させることができ、それらの特許文献を本明細書中に援用する。もう一つのアプローチは、VWFのA1およびA3領域に対するPEG付加コラーゲンまたはコラーゲンフラグメントの結合のようなそれらの生体機能を基礎としたVWFに対するポリマーの結合である。この目的のために、VWFで強い相互作用を示すIおよびIII型(例えばヒト胎盤由来)由来コラーゲンを使うことができる。ポリマーの結合は、タンパク質性構築物の生体内使用の後、安定または可逆的であると思われる。
【0033】
本明細書中で用いられる場合、「PEG付加VWF」は一つ以上のPEGに結合するVWFを含み、また本明細書中で用いられる場合、「PEG付加(PEGylation)」はVWFに一つ以上のPEGを結合させるプロセスを含む。PEG付加の適当な方法は、米国特許第5,122,614号および第5,539,063号(PEG付加方法の全てを本明細書中に援用)に開示されている。
【0034】
本発明の実施形態によれば、生理学的に許容し得るポリマーはPEGまたはPEG誘導体であり、当該技術分野で知られている任意の戦略と方法によってVWFに共有結合する。最も頻度が高い修飾戦略は、リジン残基のアミノ基を経た少なくとも1つのポリマー分子の結合、糖質側鎖を経た少なくとも1つのポリマー分子の結合、スルフヒドリル基を経た少なくとも1つのポリマー分子の結合、水酸基の少なくとも1つのポリマー分子の結合と同様にアスパラギン酸およびグルタミン酸のカルボキシル基を経た少なくとも1つのポリマー分子の結合、ならびにN末端の少なくとも1つのポリマー分子の結合である。
【0035】
本発明の一実施形態において、VWFに対する少なくとも1つのポリマー分子の結合は、上記ポリマー分子をVWFのリジン側鎖のアミノ基に連結される共有結合によって行うことができる。ヒトVWFは側鎖でNH2基とともに遊離の108リジン残基を含み、少なくとも1つのポリマー分子の結合の影響を受けやすい。ポリマー分子が本発明によって共有結合であるVWFのリジン残基の実施例を図1Aに示す。VWFのリジン残基に共有結合である可能性がある適当なPEG誘導体は、例えば、活性N−ヒドロキシスクシンイミドエステル(NHS)(例えばスクシンイミジル琥珀酸エステル、スクシンイミジルグルタラートまたはスクシンイミジルプロピオン酸)を有するポリエチレングリコールであり、それらはアミド結合を形成することによって、温和な条件下でリジン残基と反応する。活性PEGの他の例として、活性カーボネート(例えばスクシンイミジルカーボネート(SC−PEG)、およびベンゾトリアゾールカーボネート(BTC−PEG))を持つものが挙げられる(Roberts et al.,Advanced Drug Delivery Reviews 54:459−476,2002の463ページを参照)。SC−PEGおよびBTC−PEGは、カルバミン酸塩結合を形成するためにリジン残基に優先して反応するが、ヒスチジンおよびチロシン残基と反応することも知られている。別の方法は、ウレタン結合を形成するPEGカーボネートまたは第二級アミンを形成するアルデヒドまたはケトンと、例えばナトリウムシアノホウ化水素による還元の後、少なくとも1つのポリマー分子との結合である。ウレタンを生産する他の試薬をアシル化しているPEGは、タンパク質を連結したp−ニトロフェニルカーボネート、トリクロロフェニルカーボネート、およびカルボニルイミダゾールを含む。これらの試薬は、モノメトキシPEG(mPEG)(前述のRoberts et al.の464ページを参照)上で、末期の水酸基とクロロギ酸エステルまたはカルボニルイミダゾールとを反応させることによって調製される。もう一つの実施例は、所謂「第二世代」PEG付加化学に関するもので、それによって、酸性条件下のmPEG−プロピオンアルデヒドが、N末端a−アミンに対して選択的である(上記のRoberts et al.の464ページを参照)。遊離可能なPEG試薬(例えばPEG無水マレイン酸、mPEGフェニルエーテルスクシンイミジルカーボネート、およびmPEGベンズアミドスクシンイミジルカーボネート)は、結合体の生産に用いることが可能である。上記のRoberts et al.の469ページに記載のように、結合体は、生理学的条件下において「非標識(ノータグ)」で治療的タンパク質を遊離する。
【0036】
本発明のさらなる実施形態において、VWFはその糖質残基を経て少なくとも1つのポリマー分子に結合することができる。このことは、例えば糖質のマイルド酸化(例えば、NaIO4による)によって、実施することができ、アルデヒド機能を形成するとともに、その後PEG(例えばPEG−ヒドラジド)に対して連結する。この手法の長所は、以下の事実に基づく。すなわち、VWFのA1およびA3ループは、コラーゲン結合部位を含むことから、それらはVWFの生物学的活性について重要な領域であり、また糖質残基を含まずに、図1Bで分かるように、この手法によって修飾されない場合もある。この図は、アスパラギンN結合型GIcNAcとスレオニンまたはセリンO結合型GaINAc残基の実施例を表し、それらは酸化型であり、その後少なくとも1つのポリマー分子によって結合し得る。VWFの糖質が特定の領域に集まるという事実のため、VWFはこの修飾手法の使用によって少なくとも1つのポリマー分子と直接結合する部位であると考えられる。VWFの治療的活性形態が求められる場合、糖質残基に対するポリマーの結合が特に有利である。これは、既知の方法によってN結合型残基またはO結合型残基のいずれかに対してポリマーが選択的に反応することによって高められる。例えば、酵素グルコースオキシダーゼは複数の反応性アルデヒド基を生成するためにVWF上の糖質残基を酸化する際に用いることができ、PEG−ヒドラジドと反応してヒドラゾン結合を生産することができ、またはPEG−アミンと反応して可逆性シッフ塩基を生産することができる(前述のRoberts et al.の467ページを参照)。特定の状況下では、VWFのN末端セリンまたはスレオニンを、過沃素酸酸化によってグリオキシル誘導体に変換することによって、部位特異的結合に用いることができる(前述のRoberts et al.の467ページを参照)。
【0037】
本発明の別の実施形態は、スルフヒドリル基を経たVWFに対する少なくとも1つのポリマー分子の結合である。ヒトVWFは、例えばPEGマレイミドによって、177個の遊離SH基(修飾し得る)を持ち、安定なスルフィドを形成している。システイン残基のPEG付加は、例えば、PEG−ビニルスルホン、PEG−ヨードアセトアミドまたはPEG−オルソピリジルジスルフィドを使用して、行うことが可能である(前述のRoberts et al.の466ページを参照)。
【0038】
その複数の機能によって、VWF分子は、特異的な受容体またはリガンドに対するいくつかの結合部位を有する(Girma et al.,Thromb Haemost.74:156−60,1995)。1つの重要な結合部位はFVIII結合領域であり、成熟サブユニット(アミノ酸1〜272)のN末端に位置する。このエピトープを、遊離FVIIIによるインキュベーションと複雑なFVIII/VWFの形成にとよって保護することができる。その後、複合体を化学的に修飾(PEG付加またはポリシアル酸付加)し、遊離FVIII結合部位によるポリマー結合VWFを、FVIIIから分離する(例えば0.3MCaCl2または2MNaClによるサイズ排除クロマトグラフィーによる)。同様に、VWFを固定FVIIIを持つ親和性樹脂に結合させることができる。その後、VWFとポリマー(例えばポリエチレングリコールまたはポリシアル酸誘導体)とから化学的に結合させ、バッチモードまたはクロマトグラフィーカラムの使用により、このマトリックス(例えば高塩条件下、例えば0.3MCaCl2または2MNaCl)から溶出させる。
【0039】
VWFのFVIII結合エピトープは、ヘパリン結合部位位とほとんど同一である。このように、FVIII結合部位はヘパリンに対するVWFの結合または固定化ヘパリンを有する親和性樹脂に対するVWFの結合によって、化学修飾法の過程でブロックおよび保護される。
【0040】
本明細書中で用いられる場合、用語「第VIII因子結合部位保護剤」とは、VWF分子上でFVIII結合領域またはエピトープと結合する任意の薬剤のことをいう。第VIII因子結合部位保護剤は、第VIII因子、FVIIIの誘導体、ヘパリン、およびヘパリン誘導体から選択可能である。
【0041】
本発明は、少なくとも1つの生理学的に許容可能なポリマー分子に連結されないVWFの生体内半減期と比較して、VWFまたはその生物活性誘導体の生体内半減期の増大に関する。本発明の一実施形態において、別の実施形態で生体内半減期が少なくとも3倍増大している一方で、VWFの生体内半減期は少なくとも2倍長くなる。さらに別の実施例において、生体内半減期は少なくとも1つの生理学的に許容可能なポリマー分子の結合によって、5倍に増大する。下記の実施例7に記載のように、VWF半減期の増大または延長を、FVIII欠乏マウスでVWFの薬物動態学を計測することによって評価することができる。手短に言うと、FVIII欠乏マウスは尾静脈を経てFVIIIで予備混合されるVWFの大量瞬時投与で処置され、VWF抗原量は種々の時点で、血漿サンプルで計測される。VWF抗原(FVIII抗原と同様に)を、ELISAアッセイを介して計測することができる。
【0042】
本発明のさらなる態様は、FVIIIの生体内半減期がポリマーVWF結合体によって哺乳動物血液中で長くなる、少なくとも1つのポリマーVWF結合体と少なくとも1つのFVIII分子との間に形成される複合体に関する。
【0043】
ポリマーVWF結合体とFVIIIとの結合は、FVIIIの生体内半減期と比較すると、上記FVIIIの生体内半減期を延長または増加させ、VWFを少なくとも1つの生理学的に許容可能なポリマー分子に連結していない複合体を形成する。本発明の一実施形態において、FVIIIの生体内半減期は、少なくとも1.5倍であり、別の実施形態では少なくとも2倍、別の実施形態では少なくとも3倍、さらに別の実施形態では少なくとも5倍である。
【0044】
本発明のポリマーVWF結合体を、血友病Aおよび/またはVWDの処置またはこれらの2つの疾患の亜型の処置に使用することができる。VWDの場合、ポリマーVWF結合体の使用は、予防プロトコルまたはオンデマンド処置とも各々呼ばれる規則的または不規則な処置によってコンセントレートを含むVWFによる現在の補充療法と類似している。ポリマーVWF結合体を、血友病A予防のアジュバント処置として用いることもできる。これらの処置状況下で、ポリマーVWF結合体は、血漿由来または組換え型のFVIIIコンセントレートとは別個に時間間隔で投与され、血友病Aの規則的な処置のために現在使われる同じことが通常通り与えられるが、ポリマーVWF結合体の半減期延長能力による長期間の処置間隔による。
【0045】
血友病AおよびVWDの予防と血友病AおよびVWDの急性出血の処置とに関する本発明の一実施形態では、血友病AまたはVWDを有する患者に、ポリマーVWF結合体をFVIIIと共に投与するか、あるいはFVIIIで複合体の形で投与する。このような場合、体内または外部の出血の停止が、治療的に有効なレベルにVWFおよび/またはFVIIIのいずれかのさもなければ低い血漿レベルを上げることで、直ちに達成することが必要である。ポリマーVWF結合体を免疫寛容療法に用いて、阻害抗体を根絶することもでき、該阻害抗体はFVIII(凝血抑制因子増加性血友病としても公知の臨床状況、またはVWFに対して)に対して発現した。このような状況下で、FVIIIの超生理学的および超薬理学レベルを、FVIIIまたはVWFに対する発現した阻害因子を有する患者に投与する。治療のこの形態は、上記製剤を投与された患者の血行で通常より高い回復とVWFより長い持続性とを持っているポリマーVWF結合体の使用によって促進される。
【0046】
治療の技術水準によれば、また国際的なガイドラインと規則に従って、注入されたFVIIIの薬物動態学が認識され、有効性のための妥当な代用のマーカーと認められる。このことは、機能的な活性のために標準検査によって特徴づけられる注入されたFVIII産物が血流量で見いだされること、またテナーゼ複合体(FIXaおよびリン脂質に結合することによる第X因子の活性化複合体)のコファクターとして予想されたようにそこで作用するという、確証された仮定に基づく(Elodi et al.,Thromb.Res.21:695−700,1981)。したがって、動物モデルの任意の薬動力学の分析は、FVIII産物で処置される患者で、期待される有効性を予測する。
【0047】
FIXコファクター活性を判定するために、FVIIIまたはFVIIIa試料(トロンビンで完全に活性化されるFVIII)を、FIXa、FX、燐脂質、およびCaCl2からなる調製された混合物に加える。この反応混合物を37℃でインキュベートして複合体形成を可能にするとともに、その後のFXa生成を可能にする。副試料を最高20分の間隔で採血し、FXaによって選択的に分けられる色素基質に加える。15分のインキュベーションの後、酢酸を添加することで反応を停止させる。吸光度(A405)値(それはFXa濃度に比例)をELISAリーダーで計測し、反応混合物のインキュベーション時間に対してプロットする。
【0048】
本発明のさらなる態様は、FVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害がある哺乳動物血液でFVIIIの生体内半減期を延長する方法の提供であり、該方法は、
(a)上記に定義された少なくとも1種類のタンパク質性構築物を提供するステップと、
(b)上記に定義した少なくとも1種類のFVIIIを提供するステップと、
(c)上記タンパク質性構築物と上記FVIIIとの間の複合体を形成するステップと、を有する。
【0049】
上記の方法の一実施形態では、ステップ(c)の複合体は、例えばタンパク質性構築物と上記FVIIIとを混合することによって「体外」(すなわち、哺乳動物の体外)で形成され、このように形成された複合体を上記出血障害がある哺乳動物に対して有効量投与する。
【0050】
上記の方法のさらなる実施形態では、ステップ(c)の複合体は、上記哺乳動物に有効な量でタンパク質性構築物を投与すると即座に、上記出血障害がある哺乳動物血液中に存在するタンパク質性構築物と内因性FVIIIとの間に体内(すなわち哺乳動物の体内)で形成される。
【0051】
上記の方法のさらに別の実施例では、ステップ(c)の複合体は、上記哺乳動物に有効な量でタンパク質性構築物を投与すると即座に、上記出血障害がある哺乳動物血液で存在する外因性FVIIIとタンパク質性構築物との間に「体内で」形成される。外因性FVIIIを有効量で上記タンパク質性構築物と同時に、または経時的に、例えば上記タンパク質性構築物の前または後に、投与することが可能である。
【0052】
本明細書中で用いられる場合、「内因性FVIII」は上記哺乳動物から生じるFVIIIを含む。上記哺乳動物に存在する導入遺伝子または任意の他の外来性DNAから転写されるFVIIIも含む。本明細書中で用いられる場合、「外因性FVIII」は既に概説されたpdFVIIIおよびrFVIIIを含むFVIII(上記哺乳動物からは生じない)を有するとともに、pdFVIII(上記の定義された複合体を形成した後に単離された哺乳動物に再投与される)、さらにrFVIII(DNAが上記rFVIIIの産生で使われた哺乳動物に投与される)が挙げられる。
【0053】
本明細書中で用いられる場合、上で概説されるように、「有効量」は出血障害がある哺乳動物を処置するための適当な用量を含み、例えば、ヒトに関して、好ましくは1回の注入あたり5〜1,000IU、より好ましくは1回の注入あたり10〜250IUの範囲内である。
【0054】
投与経路は特に制限されるものではなく、一実施形態において、タンパク質性構築物または本発明の複合体を注射(例えば静脈内、筋内、または腹腔内注射)によって投与することも可能である。
【0055】
本発明も、本明細書中で用いられるFVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害を処置することに関し、出血障害の原因が、FVIIIおよび/またはVWFの短くなる生体内半減期、FVIIIおよび/またはVWFの改変された結着性、FVIIIおよび/またはVWFの遺伝子欠損とFVIIIおよび/またはVWFの減少した発現からなる群から選択されるものであってもよい出血障害が含まれる。本発明の一実施形態において、出血障害は血友病A、VWDまたはVWFの障害のある機能または他の分子によるVWFの障害のある相互作用に関連した他の疾患からなる群から選択される。
【0056】
さらに、本発明は上記に定義されたタンパク質性構築物の有効量または上記に定義された複合体の有効量を含む医薬品組成物に関する。医薬品組成物は、薬学的に許容可能な担体、希釈剤、塩、緩衝液、または賦形剤をさらに含む可能性がある。医薬品組成物が、上記に定義された出血障害を処置するために使用し得る。本発明の医薬品組成物は、溶液または凍結乾燥された産物である可能性がある。タンパク質と特にVWFとFVIIIの安定溶液を形成する多くの既知の方法がある。例えば、米国特許第6,586,573号、第5,565,427号、第5,763,401号、第5,733,873号、第4,877,608号、第5,605,884号、および第5,328,694号が挙げられる。これらの溶液は任意の適当な凍結乾燥プロセス(例えばUS 6,586,573に記載のプロセス)にかけることができ、そのことは本明細書中に援用する。本発明は、単独ポリマーVWF結合体単独またはFVIIIと組み合わせた他の適当な形態を含む。
【0057】
本発明は、以下の実施例でさらに詳しく説明されるが、それらになんら限定されるものではない。
【実施例】
【0058】
(実施例1:糖質残基の修飾によるポリマーVWF結合体の調製)
糖質残基(図1B)を介してポリマーVWF結合体を調製するために、rVWF(最終濃度:500μg/ml)の溶液を20mM酢酸ナトリウム緩衝液(pH6.0)で調製し、NaIO4を糖質残基の酸化のために添加した(5mM最終濃度)。酸化を4℃で20分間行い、亜硫酸水素ナトリウム(5mM最終濃度)を添加して反応を停止させた。その後、mPEGヒドラジド(鎖長:3kD)を添加し(10mM最終濃度)、さらにVWFのPEG付加を室温で1時間行った。次に、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラム(寸法26mm×840mm)に加え、PEG付加VWFを20mM HEPES緩衝液(150mM NaCl、pH7.4、5%トレハロース含有)を用いて、試薬から分離した。280ナノメートルVWF抗原量およびODの計測によって示されるように、修飾VWFをボイド容量で溶出させた。VWF含有分画を、さらに精製するために、EMD TMAE 650M(Merck)充填陰イオン交換カラムに直接載せた(寸法:10mm×108mm)。次に、PEG付加VWFを20mM HEPES緩衝液(5%トレハロースおよび1000mM NaCl含有)で溶出させた。
【0059】
(実施例2:mPEGスクシンイミジルスクシネートによるVWFのリジン残基のPEG付加)
リジン残基(図1A)を介したVWFのPEG付加のために、rVWF(最終濃度:500μg/ml)溶液を20mM HEPES緩衝液(150mM NaCl、pH7.4、5%ショ糖含有)で調製し、mPEGスクシンイミジルスクシネート(鎖長:5kD)を添加した(10mM最終濃度)。VWFを室温で1時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、修飾VWFをボイド容量で溶出させた。VWF含有分画を、さらに精製するために、EMD TMAE650M(Merck)充填陰イオン交換カラムに直接載せた(寸法:26mm×840mm)。次に、PEG付加VWFを20mM HEPES緩衝液(5%ショ糖および1000mM NaCl含有)で溶出させた。
【0060】
(実施例3:mPEGp−ニトロフェニルカーボネートによるVWFのリジン残基のPEG付加)
mPEG p−ニトロフェニルカーボネートによるVWFのPEG付加のために、血漿由来VWF(最終濃度:500μg/ml)を20mM HEPES緩衝液(150mM NaCl、pH7.6、5%ショ糖含有)で調製し、mPEG p−ニトロフェニルカーボネート(鎖長:2kD)を添加した(最終濃度:10mM)。VWFを室温で2時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、VWFをボイド容量で溶出させた。
【0061】
(実施例4:mPEGマレイミドによるVWFのスルフヒドリル残基のPEG付加)
mPEGマレイミドによる遊離SH残基を介したVWFのPEG付加のために、rVWF溶液(最終濃度:500μg/m)を20mM HEPES緩衝液(150mM NaCl、pH7.6、4%マンノースおよび1%トレハロース含有)で調製し、mPEG マレイミド(鎖長:10kD)を添加した(最終濃度:10mM)。VWFを室温で2時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、修飾VWFをボイド容量で溶出させた。
【0062】
(実施例5:デキストランとVWFとの結合)
6mg/mlのデキストラン(MW40kD)溶液を20mM酢酸ナトリウム緩衝液(pH6.0)で調製し、NaIO4を添加した(最終濃度10mM)を添加して遊離アルデヒド基を生成した。酸化を暗所で4℃、1時間実施し、亜硫酸水素ナトリウム(最終濃度5mM)を添加して反応を停止させた。活性化デキストランを0.15MNaCl(PBS緩衝液)含有0.1Mリン酸ナトリウム緩衝液(pH7.2)で透析した。次に、この活性化デキストラン溶液2.4mlを10mlのrVWF溶液(濃度:PBS緩衝液中0.6mg/ml)に添加した。この混合物に、5mlのシクラミン酸ナトリウム溶液(PBS緩衝液中64mg/ml)を添加し、暗所で室温、一晩にわたってインキュベートした。次に、3mlの1.0MTRIS−HCl溶液(pH7.2)を添加して残留アルデヒド基をブロックし、室温で1時間インキュベートし、さらに20mM HEPES緩衝液(pH7.4、5%ショ糖含有)でインキュベートした。次に、rVWF誘導体に結合したデキストランをさらに、セファクリルS−300HR(緩衝液:20mM HEPES、5%ショ糖、pH7.4)を充填したクロマトグラフィーカラム(寸法:50mm×860mm)に混合物を載せることで、サイズ排除クロマトグラフィーにより精製した。rVWF誘導体を、VWF抗原量およびOD280nmの計測によって示されるように、VWFをボイド容量で溶出させた。これらの分画を回収し、100kDの再生セルロース膜(Millipore)を用いて、限界濾過法によって濃縮した。
【0063】
(実施例6:VWDマウスにおける薬物動態)
Denis et al.(PNAS 95:9524−9529,1998)に詳述されているVWF欠乏マウスを、重度III型VWD類似ヒトVWDモデルとして使用した。マウス5匹からなる群に対して、PEG−rVWF(鎖長3kD、実施例1によるrVWFのPEG付加)または対照としてのネイティブrVWFを、PEG付加後、検出可能なVWF(ELISA)に基づいて、40単位(U)VWF:Ag/kg体重の用量で、経尾静脈ボーラス注入法により投与した。PEG−rVWF群を、注射後5分、30分、1時間、2時間、6時間、10時間、および24時間(対照群の場合、5分、15分、30分、1時間、2時間、4時間、6時間、10時間、および24時間)麻酔し、クエン酸血漿を心臓穿刺から調製した。血漿でVWF抗原量を追跡した。この実験の結果を図2にまとめる。ネイティブrVWFを、文献(Lenting et al.,J.Biol.Chem.279:12102−12109,2004)に記載されているように、典型的な二相法で血行から除去することで、600分と1440分(10時間および24時間に等しい)との間の検出限界以下に落とす。対照的に、注射時の0から注射後10時間の約0.6U/ml血漿までの初期増加後のPEG付加rVWFは、依然として注射後24時間であっても約0.4U/mlの実質的に高いレベルに存在し、10時間ないし24時間の範囲では平坦な勾配となったことから、PEG付加rVWFがより長期にわたって持続することが示される。経時的な計測可能VWFの漸増は、VWFによって結合体を形成したポリマーPEGの結合の可逆性を示すもので、ポリマーからの放出後、計測のためにアクセス可能になる。このモデルでのPEG−VWFの長い循環時間は、この調製物がVWDの予防療法に使用し得ることを示す。
【0064】
(実施例7:FVIII−K.O.マウスにおける薬物動態)
Bi et al.(Nat.Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々をrFVIIIと事前に混合して3UFVIII/mlおよび3UVWF:Ag/mlを達成するためにPEG−rVWF(鎖長3kD、実施例1によるrVWFのPEG付加)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(13ml/kg)を行った。麻酔後、生成物を注射してから5分および6時間後に、心臓穿刺によってクエン酸血漿を各々の群から調製した。対照群に対しては緩衝液を注射し、注射5分後に出血させた。VWF抗原量を血漿サンプルで計測した。この実験の結果を図3にまとめた。曲線は、VWFのベースレベルの近くまで落ち込んだ、FVIII欠乏マウスに存在するrVWFの典型的除去を示し、その一方でPEG付加rVWFの使用後、レベルが6時間の観察期間中に増加したことを示す。このことは再び、ポリマーPEGの結合の可逆性がVWFと結合体を形成したことを示し、ポリマーからの放出の後、計測のためにアクセス可能になり、適用後360分(6時間)経過しても増加を持続することを示している。
【0065】
(実施例8:VWDマウスでのFVIII増加)
Denis et al.(PNAS 95:9524−9529,1998)によって詳述されるフォンビルブラント欠乏マウスを、重度III型VWD類似ヒトVWDモデルとして使用した。4〜5匹のマウスからなる群に対して、PEG−rVWF(鎖長5kD、実施例2によるrVWFのPEG付加)含有20mM HEPES(150mM NaCl、5%ショ糖、pH7.4)またはネイティブrVWFを、尾静脈を経る静脈内注射した。対照群を緩衝液で処置した。(ELISA換算PEG−rVWF−用量18UVWF:Ag/kg、2700μg/kg、rVWFネイティブ用量:2400μg/kg)。
【0066】
各マウスに対して、10ml/kg体積用量を投与した。PEG−rVWF投与後の時点(5分、1時間、3時間、6時間、10時間、24時間、および48時間)またはネイティブrVWF投与後の時点(5分、15分、30分、1時間、2時間、6時間、24時間、および32時間)の注射の後の時点で、マウス4〜5匹からなる群を麻酔し、クエン酸血漿を心臓穿刺から調製し、FVIII活性(ハウスアッセイにおける発色)のレベルを血漿で追跡した。対照群を注射15分後に出血させた。この実験の結果を図4にまとめた。
【0067】
マウスにおける内因性FVIIIのレベルは、rVWF注入の結果として増加する。PEG付加rVWF適用後の血中濃度時間曲線下面積(AUC)は、rVWF使用後のたったの3.3U*h/mlと比較して、8.0U*h/mlであった。これは、PEG付加rVWFのための実質的により長い循環時間を示す。結果は、PEG付加VWFがVWDにおける二次FVIII欠乏の予防療法に使用し得ることを示す。
【0068】
(実施例9:FVIII−K.O.マウスでのrFVIIIおよびVWFの回収)
Bi et al.(Nat Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々をrFVIIIと事前に混合して3UFVIII/mlを達成するためにPEG−rVWF(HZ−PEG、3K、糖質を介して結合)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(13ml/kg)を行った。麻酔後、注射してから5分および6時間後に、心臓穿刺によってクエン酸血漿を各々の群から調製した。FVIII活性およびVWF抗原回収量を血漿サンプルで計測した。この実験の結果を図5および図6にまとめた。
【0069】
無処置のrVWFと比較して、両調製物のPEG付加rVWFは、同時注射されたrFVIllのより高度な回収をもたらした。経時的にPEG付加rVWFに対するVWFレベルが上昇し、その一方で正常rVWFが360分以内に完全に除去された。結果は、FVIIIと複合体を形成したPEG付加VWFを、FVIII循環時間の増大という利点をもって血友病Aの急性処置に用いることができることを示している。
【0070】
(実施例10:FVIII×VWFダブルノックアウトマウスにおけるFVIII半減期の増大)
FVIII×VWFダブルノックアウトマウスは、FVIII欠乏およびVWF欠乏マウスの交雑育種によって得られた。それらのマウスはVWF欠乏と同様にFVIII欠乏を患う。このように、動物モデルにおけるFVIII−VWF相互作用を研究するための理想的なモデルを提供する。
【0071】
FVIII×VWFダブルノックアウトマウス(FVIII欠乏マウスをVWF欠乏マウスと交雑育種した)5匹からなる群に対して、PEG−rVWF(鎖長5kD、rVWFのPEG付加を、mPEGスクシンイミジルスクシネートのリジン残基の修飾によって、実施例2により行った)含有20mM HEPES(150mM NaCl、5%ショ糖、pH7.4(またはネイティブrVWFSS−PEGまたはネイティブrVWF(各々をrFVIIIと事前に混ぜ合わせ、9UFVIII/mlおよび9UVWF抗原/mlおよび0.67UVWF:RCo/mlを達成)を、経尾静脈によりボーラス注入(11ml/kg)を行った。公開(Ingerslev(Scand.J.Clin).Invest.47:143−149(1987))されているようにして、VWF−抗原値をELISA法で計測した。一次止血のプロセスにおけるVWFの血小板結着性を反映している機能的なVWF:RCo活性をMacfarlane et al.(Thromb.Diath.Haemorrh.34:306−308,1975)により、計測した。注射後5分、3時間、6時間、10時間、および24時間、麻酔後の心臓穿刺によるクエン酸血漿を、それぞれの群から調製した。FVIII活性およびVWF抗原量を、血漿サンプルで計測した。
【0072】
FVIIIおよびVWFの半減期を、薬物動態学ライブラリーの1つの区画をモデルとして使用するMicroMath科学者プログラム(Micromath Research,Saint Luis,MO,US)を用いて算出した。rVWFまたはPEG付加rVWFのいずれかと同時注入されたFVIIIの半減期は、1.88時間から2.58時間へと増大し、血中濃度時間曲線下面積(AUC)は4.3から7.3U*h/mlへと増大した。VWFの半減期は、3.1から10.4へと増大し、血中濃度時間曲線下面積は、5.7から22.8に増やされる。結果を図7および図8にまとめた。データによれば、PEG−VWFを、VWDおよびFVIIIの循環時間を長くするという利点とともに、血友病AおよびVWDの急性処置および予防処置のために使用し得ることが示された。
【0073】
(実施例11:マウス血漿におけるPEG付加の可逆性の実証)
PEG付加の可逆性を、VWF欠乏血漿による試験管内実験によって示した。クエン酸血漿を、4℃、15分間、1100xgの遠心によって、VWF欠乏マウスから得た(Denis et al.PNAS 95:9524−9529,1998)。4容量のマウス血漿を、実施例1(糖質を経たPEGカップリング)または実施例2(リジン残基を経たPEGカップリング)によって調製し、48時間にわたり37℃に保ち、1容量のPEG付加rVWFと混ぜ合わせた。非PEG付加rVWFが、両方の実験における対照として用いた。副試料を混合直後、ならびに1時間、5.5時間、24時間、および48時間後に回収し、VWF抗原量を、サンドイッチELISAシステムを用いて凍結試料からアッセイした。ポリクローナル抗VWF抗体(DAKO)を用いて、96穴ELISAプレートを被覆し、ヤギ−抗ウサギ−lgG−HRP−結合体(AXELL)を結合VWF因子検出のためにアッセイした。経時的にVWF抗原の増加性量を計測し、生体外血漿サンプルにおける場合も含めVWFに対するポリエチレングリコールの結合の可逆性が示された(図9)。
【0074】
(実施例12:PEG付加VWF調製試料のFVIII結合能の側定)
異なるPEG付加rVWF調製試料のFVIII結合能を、BIACORE(登録商標)3000装置(BIACORE、Uppsala、Sweden)を使用して表面プラズモン共鳴実験によって比較した(Karlsson et Falt,J.Immunol.Methods 200:121−33,1997)。通常、リガンドをセンサチップに固定し、該リガンドに対する他の構成要素の結合を表面プラズモン共鳴によって測定する。この技術の使用によって、チップ表面の近くの溶液の屈折率の変化が計測される。チップの表面の結合した構成要素の濃度変化を信号として検出し、それを任意の共鳴音単位(RU)で表す。固定リガンドに結合したタンパク質量と観察されるRUとのあいだに、比例関係が存在する。PEG付加VWF調製試料を、7000〜9000RUおよび25℃で、NHS/EDC化学を用いて、BIACORE(商標)センサチップのデキストラン表面に25℃で固定した。150mM NaCl、3mM EDTA、および0.005%界面活性剤P20(HBS緩衝液、BIACORE)を含む10mM HEPES緩衝液(pH7.4)を流速15μl/分で使用した。図10に例示されるように、市販のFVIII産物(ADVATE、Baxter AG、Vienna、Austria)の異なる量の結合が計測された。この図は、mPEGマレイミド5000で修飾され、かつ実施例4によって調製されるPEG付加rVWF調製試料のFVIII結合能を示す。異なるPEG付加rVWF調製試料のBIACORE実験の結果を表1にまとめている。この表では、PEG付加rVWF調製試料の異なるFVIII結合能は、10〜20IUFVIII/ml(色素アッセイ)の範囲にある参照の最大レベルでの非PEG付加参照調製試料のRU値の割合として与えられる。
【0075】
【表1】

(実施例13:ポリマー結合後のVWFの質量増加)
rVWFを、種々の濃度(1mM、2.5mM、5mM、7.5mM、および10mM)でmPEGスクシンイミジルスクシネート(鎖長:5kD)を使用して、実施例2により、PEG付加した。PEG付加VWF種を2つの異なる方法で分析した。すなわち、SDSポリアクリルアミドゲル電気泳動およびVWFマルチマー分析である。SDSゲル電気泳動を、3〜8%勾配ゲルを使用して還元条件下で行った(Tris Acetat Gel/Bio−Rad)。VWFマルチマー分析を、1.6%のアガロースゲルを使用してRuggeri et Zimmerman(Blood 57:1140−43,1981)により行った。VWFマルチマーの明視化を、Aihara et al.(Thromb.Haemost.55:263−67,1986)によって実施した。
【0076】
SDSゲル電気泳動(図11)は、成熟VWF(下部のバンド)とプロVWF(上層ブライトバンド)とからなるrVWF調製試料を示すとともに、異なる試薬濃度の使用によってPEG付加後の分子量が増加することを示す。PEG付加の後、ヒト血清アルブミン(HSA)調製試料(PEG付加は、実施例2によって実施)からの分子量の推移は、参照調製試料として示される。HSAの分子量が66,000のダルトンから190,000のダルトンまでシフトしていることで、PEG付加手法が有効であることが示されることが明らかになった。
【0077】
図12は、異なる試薬濃度でのPEG付加前後におけるrVWFの多量体パターンを示す。試薬濃度の上昇に伴って異なるマルチマーの分子量がより高いほうへ移行および広幅化することが明らかに示される。
【0078】
(実施例14:mPEGスクシンイミジルグルタレートによるVWFのリジン残基のPEG付加)
リジン残基(図1A)を介したVWFのPEG付加のために、rVWF(最終濃度:500μg/ml)を、20mM HEPES緩衝液(150mM NaCl、pH7.4、5%スクロース含有)で調製し、mPEGスクシンイミジルグルタレート(鎖長:5kD)を添加した(最終濃度:200mgPEGスクシンイミジルグルタレート/mgタンパク質)。つぎに、pH値を0.1MNaOHで7.4に合わせた。VWFを室温で1時間、PEG付加し、実施例2の記載に従って精製した。
【0079】
(実施例15:FVIII−K.O.マウスでのFVIII半減期の増大)
Bi et al.(Nat.Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々を組換え型FVIIIと事前に混合して10UFVIII/mlおよび10UVWF/mlを達成するために、実施例2により調製したPEG−rVWF(SS−PEG、5K)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(10ml/kg)を行った。麻酔後、生成物を注射してから5分、3時間、9時間、および24時間後に、心臓穿刺によるクエン酸血漿を調製した。FVIII活性およびVWF抗原回収量を血漿サンプルで計測した。この実験の結果を図13および図14にまとめた。
【0080】
FVIIIのための半減期は1.8時間(ネイティブrVWF存在下)から3.9時間(PEG−rVWFと併用)へと増大し、血中濃度時間曲線下面積(AUC)が4.1から7.8U*hへと増大した。VWF半減期は3.2時間から13.6時間へと増大し、VWFのAUCは7.7から32.1U*hへとほぼ4倍になった。
【0081】
(実施例16:異なる方法によるSS−PEG付加VWF調製試料のFVIII結合能の測定)
異なるPEG付加rVWF調製試料のFVIII結合能を、ELISAと色素アッセイシステム(ECA)とを組み合わせ、Bendetowicz et al.(Blood 92:529−538,1998)の方法の変法によって計測した。マイクロタイタープレートを、200μlの2.6μg/mL抗vWFポリクローナル抗体含有50mmol/lNa2CO3/NaHCO3(pH9.6)で覆った。プレートを、PBS−Tween緩衝液(100mM Na2HPO4/KH2PO4、150mM NaCl、pH7.6と0.05%Tween20)で、各ステップの後に洗った。プレートを、0.1%粉ミルク/2mMベンズアミジン含有PBS−Tweenで37℃、1時間にわたりブロックした。VWFの増加量を37℃、25分間にわたり、0.2U/mlrFVIII(ADVATE,Baxter AG,Vienna,Austria)とともにプレインキュベートし、これらの混合物の100μlをプレートに添加した。インキュベーション後、捕獲されたVWFに結合したFVIIIの量を、FVIII色素アッセイ(Technoclone,Vienna,Austria)で計測した。FVIII結合能を、1分内で405nm(dA405)で計測される吸光度の変化として表した。図15は、2つのPEG付加VWF調製試料(両方ともmPEG−スクシンイミジルスクシネート(SS)で修飾)のVWF用量依存性FVIII結合を示す。修飾VWF調製試料のFVIII結合能は、非修飾開始VWF調製試料の%として算出され、PEG−SS−rVWF−1では20%およびPEG−SS−rVWF−2では50%であると算出された。
【0082】
図16は、実施例12に記載の表面プラズモン共鳴方法で計測される2つのPEG−SS−rVWF調製試料のFVIII結合能を示す。算出結合能は、それぞれ25および45%であった。
【0083】
SDS−PAGEで計測されるPEG−SS結合後のrVWF分子の分子質量における適当な増加を、図17に示す。
【0084】
(実施例17:分枝PEGマレイミドによるVWFにおけるスルフヒドリル基のPEG付加)
分枝PEGマレイミドによる遊離SH残基を経たVWFのPEG付加のために組換え型VWF(最終濃度:500μg/ml)の溶液を、20mM HEPES緩衝液(150mM NaCl、pH7.6、3%トレハロース含有)によって調製する。次に、NOF社(NOF Europe,Grobbendonk,Belgium)によって供給される分岐mPEGマレイミド(鎖長:20kD)を添加した(最終濃度10mM)。VWFを静かに撹拌しながら室温で2時間にわたり、PEG付加した。その後、PEG付加rVWFを、再生セルロース(Millipore)からなる100kD膜を使用して、限界濾過法/ダイアフィルトレーション(UF/DF)によって、試薬から分離する。
【0085】
(実施例18:mPEGヒドラジド/EDCによるVWFのカルボキシル基のPEG付加)
実施例23により、フリン成熟rVWFを調製および精製した。調製試料を50mMリン酸緩衝液(pH6.2)に対して透析し、400μg/mlの濃度まで希釈した。次に、鎖長5kDのmPEGヒドラジド(mPEG Hz)を添加した(濃度:60mg mPEG Hz/mgVWF)。500mM 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC)の新しく調製された溶液30μlを、1mlのVWF含有混合物の1mlに添加し、5時間にわたって静かに振とうさせながら室温でインキュベートした。100kD膜(再生セルロース/Millipore)を用いて、20mM HEPES緩衝液(150mM NaCl、pH7.4)に対して、UF/DFによりPEG付加rVWFから試薬を分離する。
【0086】
(実施例19:ポリシアル酸によるVWFのリジン残基の修飾)
ポリシアル酸(コロミン酸、CA)によるリジン残基の修飾を、Fernandes et Gregoriadis(Biochim.Biophys.Acta 1341:26−34,1997)およびJennings et Lugowski(J.Immunol.127:1011−1018,1981)に記載のようにして、実施した。0.1MNaIO4含有コロミン酸(濃度:20mg/ml)溶液を暗所、室温で15分間撹拌させて、CAを酸化した。活性化CA溶液1mlあたり2mlのエチレングリコールを添加し、暗所、室温でさらに30分間撹拌した。溶液を一晩にわたって暗所で0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して透析した。その後、この溶液のアリコートをrVWF溶液(400μg/ml)含有0.05Mリン酸ナトリウムに添加し、1mgVWFあたり50mg活性化CAの最終濃度を得た。この混合物を暗所、室温で30分間撹拌した。NaCNBH3を添加し(1mg/mgrVWF)、混合物を暗所、室温で18時間、静かに撹拌しながらインキュベートした。1MTRIS水溶液(pH7.2)を添加し(50μl/mgNaCNBH3)、1時間にわたって撹拌することで反応を停止させた。遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して、UF/DFによりrVWF−ポリシアル酸結合体から分離した。
【0087】
この調製試料のFVIII結合能を実施例16の記載通りに測定した。
【0088】
ポリシアル酸によるrVWFの結合を、還元条件の下で等電点電気泳動(IEF)によって検出される等電点(PI)の移動によって示した。図20は、この実施例によりポリシアル酸による結合の前後におけるVWF調製試料の比較を示す。製造元(Amersham Bioscience)の指示により、Ampholine PAGplateシステム(pH3.5ないし9.5)を用いた。ポリシアル酸との結合による酸性基の導入によって、修飾rVWFの1つの酸性バンドが生ずる。
【0089】
実施例16に記載のように、54%のFVIII結合能がECA試験を用いて側定された。70%のFVIII結合能を表面プラズモン共鳴方法(実施例12)を用いて側定した。
【0090】
(実施例20:剪断応力条件下のPEG付加)
Sakariassen et al.(J.Lab.Clin.Med.102:522−535,1983)に記載のように、灌流チェンバーを作製し、剪断応力条件下でのrVWFのPEG付加に使用した。rVWF(500μg/ml)含有20mM HEPES(pH7.4、3%トレハロース含有)の溶液を調製した。次に、mPEGスクシンイミジルスクシネート(最終濃度:5mM)を添加し、0.1MNaOHを添加してpHを7.4に調製し、直ちに灌流システムに充填した。次に、PEG付加はSakariassen et al.(J.Lab.Clin.Med.102:522−535,1983に記載のように、灌流条件下で、蠕動ポンプを使用することで、2500秒−1の剪断速度で、室温で行った。
【0091】
(実施例21:pdVWFの調製)
dVWFの調製を、改良を加えたThorell et Blomback(Thromb.Res.35:431−450,1984)により実施した。pdVWFの調製のために、1.5kgのクリオプレシピテートを20ないし30℃で6リットルの水に溶解した。1時間撹拌させた後に、フィブロネクチン沈殿物を遠心によって除去した。上清1リットルあたり8gNaClを添加し、溶液を室温まで温め、ウイルスを失活させるためにSDストック試薬(1%Tween80+0.18%アセチルトリエチルシトレート最終濃度)を添加した。
【0092】
溶液を、0.2MNaCl、0.02M酢酸ナトリウム(pH6.5)(洗浄用緩衝液)で事前に平衡化されたEMD−TMAEフラクトゲル650Mカラム(XK50/180)上で精製した。洗浄用緩衝液20カラム容量で洗浄した後、VWFを0.5MNaCl、0.02MNaクエン酸塩(pH6.9)で溶出させた。VWFを沈降させるために、グリシン(1M最終濃度)およびNaCl(3M最終濃度)を添加した。沈殿物を緩衝液に溶解し、最終濃度に対して0.3MCaCl2、0.15MNaCl、20mM HEPES(pH7.4)で平衡化したカラムをセファクリルS−400HR(Amersham)上に載せた。
【0093】
【表2】
(実施例22:PEGSSによるpdVWFのリジン残基のPEG付加)
PdVWFを実施例21により調製し、20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含む)で希釈し、最終濃度を400μg/mlとする。次に、mPEGスクシイミジルスクシネート(鎖長:5kD)を添加し(濃度:10mgPEGSS5000/mg VWF)、pdVWFを室温で、1時間にわたりPEG付加した。次に、再生セルロース(Millipore)からなる100kD膜を使用して、UF/DFによって、PEG付加rVWFから試薬を分離した。
【0094】
(実施例23:フリン成熟rVWFのフリン成熟および精製)
rFVIII発酵および精製プロセス由来の抗FVIII抗体カラムのフロースルー分画の143kgを、Schlokat et al.(Biotechnol.Appl.Biochem.24:257−267,1996)に記載のように、プロペプチド除去のためにフリンで処理し、滅菌濾過した。このプロセスは、初期の研究に基づいた(Fischer et al.,FEBS Lett.375:259−262,1995;Fischer et al.PCT/AT98/00034[WO98/38219],1−33,1998,18−2−1998 and Kaersgaard et Barington,J.Chromatogr.B 715:357−367,1998)。
【0095】
水で4倍希釈して16mS/cmにした後、633kgの希釈溶液を、10mM Tris、100mM NaAc、86mM NaCl、pH6.5、mS/cm(平衡緩衝液)により平衡化させたXK50/15EMD−TMAEフラクトゲル650Mカラム(300mlゲル;Merck;#K14540281)に載せた。カラムを平衡化緩衝液で洗浄し、100mM NaAc、250mM NaCl、100mMグリシン、3mM CaCl2で溶出した。
【0096】
4506gのTMAE溶出液をSartoclean GF(0.8及び0.65μ)およびSartobran P(0.45及び0.2μ)上で濾過し、1.5倍希釈して29mS/cmとし、Mustang Qフィルター(#IH18770932)を介してポンピングする。22+/−2℃で60分間にわたりSD処置し、かつ水で2倍に希釈して16mS/cmにした後、溶液を、TMAEステップの平衡用緩衝液で平衡化させたAmicon 70/29 UNOsphere Sカラム(600mlゲル;Bio−Rad,#78960C)に載せた。カラムを平衡用緩衝液で洗浄し、そしてTMAEステップの溶離緩衝液で溶出させた。
【0097】
3223gのUNO−S溶出液を、再生セルロースからなる30kDaの0.1m2膜(Hydrosart #01080217、Sartorius)を使用して限界濾過法により15倍濃縮した。コンセントレートの201gを、100mM NaAc、500mM NaCl、pH7.0(1698mlのゲル;GE Healthcare #17−0489−01)で平衡化させたXK50/86.5Superose 6 Prep Gradeカラム上で、サイズ排除クロマトグラフィーにより最終的に精製された。
【0098】
【表3】
(実施例24:フリン成熟rVWFのPEG付加)
フリン成熟VWFを実施例23により調製し、20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含有)に対して透析した。次に、溶液を20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含有)により最終濃度を300μg/mlに希釈した。続いて、mPEGスクシミジルスクシネート(鎖長:5kD)を添加し(濃度:25mgPEGSS5000/mgVWF)、フリン成熟VWFを室温で1時間にわたってPEG付加した。次に、再生セルロース(Millipore)からなる100kD膜を使用して、UF/DFによって、PEG付加VWFから試薬を分離した。
【0099】
(実施例25:フリン成熟rVWFの生体外特徴付け)
実施例12および図15に記載のように、フリン成熟rVWFのFVIII結合能を表面プラズモン共鳴技術およびECAによって測定した。結果を、実施例21によって調製されるアルブミンを含まない血漿由来VWF調製試料と比較した。
【0100】
図18および図19に示されるように、フリン成熟rVWFのFVIII結合能は血漿由来参照調製試料と同等だった。
【0101】
(実施例26:グルタルアルデヒドで架橋させることによるポリシアル酸とVWFとの結合)
グルタルアルデヒドを架橋試薬(Migneault et al.,Biotechniques 37:790−796,2004)として用いたrVWFとポリシアル酸(コロミン酸)との結合のために、コロミン酸(濃度:20mg/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4)の溶液4mlを調製し、0.1MNaOHを添加してpHを7.4に合わせた。グルタルアルデヒドを添加して最終濃度を0.01%にした。続いて、rVWF(400μg/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4)の溶液1mlを100μlのアリコートに添加し、混合物を静かに振とうさせながら1時間インキュベートした。次に、混合物を透析し、トレハロースを添加し(最終濃度:3%)、rVWFポリシアル酸結合体を限界濾過法によって濃縮した。
【0102】
(実施例27:VWF欠乏マウスでのVWF半減期の増大)
Denis et al.(PNAS 95:9524−9529,1998)により記載されたVWF欠乏マウスを、ヒトVWDの動物モデルとして使用した。マウス5匹からなる群に対して、100UVWF:Ag/kgを達成するために、実施例19により調製したrVWFポリシアル酸結合体またはネイティブrVWFのいずれかをボーラス注入法(10ml/kg)によって経尾静脈により投与した。麻酔後の心臓穿刺によるクエン酸血漿は、注射後5分、1、3、6、9、および21時間で、それぞれの群から調製した。VWF抗原回収および内因性マウスFVIII活性レベルを血漿サンプルで計測した。この実験の結果は、図21および図22にまとめられている。
【0103】
VWFの半減期を、薬物動態学ライブラリーの1つの区画をモデルとして使用するMicroMath科学者プログラム(Micromath Research,Saint Luis,MO,US)を用いて算出した。FVIII活性の曲線下面積は、ベースラインサブトラクションによる台形モデルで算出した。
【0104】
VWFの半減期は1.3時間(ネイティブrVWF)から2.4時間(rVWF−ポリシアル酸結合体)へと増大し、FVIIIのAUCがそれぞれ3.3U*hr/mlから5.3U*hr/mlへと増大した。
【0105】
(実施例28:還元性アミノ化によるリジン基に対するmPEGプロピオンアルデヒドの結合)
rVWF溶液(400μg/ml)を0.05Mリン酸ナトリウム緩衝液(pH7.2)に調製し、mPEGプロピオンアルデヒド(5kDa鎖長)を添加して最終濃度を1mgVWFあたり10mgのmPEGプロピオンアルデヒドとする。この混合物を30分間撹拌する。つぎに、NaCNBH3を添加し(1mg/mgのrVWF)、混合物を静かに撹拌させながら室温で15時間にわたりインキュベートする。1MのTRIS水溶液(pH7.2)を添加し(1mgのNaCNBH3あたり50μl)、1時間にわたって撹拌することで反応を停止させる。その後、PEG付加rVWFは、100kD膜(再生セルロース/Millipore)を使用している限界濾過法/ダイアフィルトレーションによって、試薬から分離される。
【0106】
(実施例29:VWFのN末端PEG付加)
rVWFのN末端PEG付加をLee et al.(Pharm.Res.20:818−825,2003)の記載のように行う。rVWF溶液(最終濃度:500のμg/ml)を50mMの酢酸ナトリウム緩衝液(pH5.5)に調製し、mPEGプロピオンアルデヒド(鎖長:5kD)を添加した(濃度:10mg mPEGプロピオンアルデヒド/mgVWF)。PEG付加を、還元剤として2mMのNaCNBH3の存在下、室温で24時間にわたって行う。その後、PEG付加rVWFを、100kD膜(再生セルロース/Millipore)を使用して限界濾過法/ダイアフィルトレーションにより試薬から分離される。
【0107】
(実施例30:リジン残基およびrVWFのSH残基の逐次的なPEG付加)
RVWFを、実施例2によりmPEGスクシンイミジルスクシネート(鎖長:5kD)で、リジン残基を経てPEG付加する。PEG付加を1時間にわたって室温で行い、遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して20mM HEPES緩衝液(pH7.4、5%サッカロース)に対してUF/DFによりrVWFPEG結合体から分離する。次に、溶液のpH値を、0.1MNaOHで7.6に合わせ、mPEGマレイミド(10mM鎖長5kD/最終濃度)を加えて、遊離SH基のPEG付加を行う。静かに振とうしながら、室温で2時間にわたってPEG付加を行う。次に、試薬を再び、100kD膜(再生セルロース/Millipore)を使用して反応混合物から分離する。
【0108】
(実施例31:糖質残基の酵素酸化とPEG−Hzによるその後のPEG付加)
Avigad et al.(J.Biol.Chem.237:2736−43,1962)に記載のように、アルデヒド基を生じさせるrVWFの糖質残基(Wilchek et Bayer,Meth.Enzymol.138;429−442,1987)の酵素酸化を、Dactylium dendroides(Sigma)由来のガラクトースオキシダーゼを使用して行う。得られた溶液を、50mMリン酸緩衝液(pH7.2)に対して透析し、希釈してVWF濃度を400μg/mlにする。次に、鎖長5kDのmPEGヒドラジド(mPEG Hz)(最終濃度:40mg mPEG−Hz/mg VWF)を添加する。混合物を静かに振とうさせながら室温で3時間インキュベートする。試薬を、100kD膜(再生セルロース/Millipore)を使用して、5%のサッカロース含有20mM HEPES緩衝液(150mM NaCl、pH7.4)に対して、UF/DFによりPEG付加rVWFから分離する。
【0109】
(実施例32:rFVIIIによるFVIII結合部位のブロッキングによるrVWFのPEG付加)
非修飾FVIII結合部位を持つPEG付加rVWFを調製するために、rFVIII(200U/ml)およびrVWF(40U VWF:Ag/ml)を含む50mM HEPES緩衝液(50mM HEPES、150mM NaCl、2%トレハロース、pH7.4)の溶液3mlを調製して、37℃で1時間インキュベートする。混合物を、室温まで冷やし、PEGスクシンイミジルスクシネート(PEG−SS/鎖長:5kD)を添加し(最終濃度:1mgPEG−SS/UVWF:Ag)、静かに振とうさせながら1時間インキュベートする。次に、CaCl2を静かに振とうさせながら添加することで、最終濃度で400mMを得る。この溶液を、セファクリルS−400HR(Amersham)充填クロマトグラフィーカラム(2.6×80cm)に載せ、FVIIIの遊離結合部位を持つPEG付加rVWFを、サイズ排除クロマトグラフィー(溶離緩衝液:50mM HEPES緩衝液、400mM CaCI2、pH7.4)によって、rFVIIIから分離する。
【0110】
(実施例33:ヘパリンによるFVIII結合部位のブロッキングによるrVWFのPEG付加)
rVWF(300μg/ml)含有50mM HEPES緩衝液(pH7.4)の溶液5mlを調製し、同一緩衝液のヘパリンセファロースCL−6B懸濁液(Amersham Bioscience)2mlに添加する。この混合物を静かに振とうさせながら2時間にわたってインキュベートし、VWFをゲルに結合させる(de Romeuf et Mazurier,Thromb.Hamost.69:436−440,1993)。その後、mPEGスクシンイミジルスクシネート(200mg/mgのVWF)を混合物に加え、静かに振とうさせなら、PEG付加を室温で1時間行う。混合物を等量の2MNaCl含有HEPES緩衝液(pH7.4)で希釈する。ゲルを濾過によって上清から分離する。次に、ゲルは2mlHEPES緩衝液(pH7.4)(20mM HEPES、1MNaCl)で3回洗浄し、上清および洗い溶液を組み合わせる。続いて、PEG付加VWFを含む溶液を限界濾過法によって濃縮し、再生セルロース(Millipore)からなる100kD膜を用いて20mM HEPES緩衝液(pH7.4)(150mM NaCl、3%のサッカロース)に対して透析する。得られた誘導体は、バイアコア技術またはECA試験(試験システムは、米国仮特許出願第60/668,378号(2005年4月4日出願)に記載されている)の使用によって、完全なFVIII結合能を示す。
【0111】
(実施例34:ヒアルロン酸とVWFとの結合)
ヒアルロン酸(HA)によるリジン残基の修飾を、Sigma(C53747)から得られるヒアルロン酸を使用して還元性アミノ化によって行った。HAを、新たに調製された0.1MのNaIO4溶液に溶解して最終濃度を5mgHA/mlにした。次に、静かに撹拌させながら15分間にわたり酸化を行った。1mlの酸化HA溶液に対して2mlのエチレングリコールを添加し、室温、暗所で30分間さらに撹拌することで、反応を停止させた。溶液を、0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して、暗所4℃で一晩透析を行った。続いて、この溶液のアリコートを、0.05Mリン酸ナトリウム緩衝液(pH7.2))からなるrVWF溶液(40UVWF:Ag/ml)に添加して、最終濃度50mgの活性化HA/mgタンパク質を得た。この混合物を、暗所、室温で120分間にわたり撹拌した。NaCNBH3を添加し(1mg/mgタンパク質)、混合物を静かに振とうさせながら、18時間、室温でインキュベートした。つぎに、100μlの1MTris緩衝液(pH7.2)をこの混合物1mlに対して添加し、1時間にわたり撹拌することで、反応を停止させた。遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して、UF/DFによりrVWFHA結合体から分離した。
【0112】
(実施例35:PSA結合VWFの生体外生化学的特徴づけ)
実施例19によりRVWFをポリシアル化した。この実施例に記載のパラメーター、例えば等電点電気泳動(図20)、実施例16によるECA試験(54%)および実施例12による表面プラズモン共鳴(70%)によってFVIII結合能の測定に加えて、比率VWF:RCo/VWF:Agを算出した。VWF抗原量を、市販のアッセイシステム(Asserachrom vWF,Roche,Basel,Switzerland)を用いて側定した。Macfarlane et al.(Thromb.Diath.Haemorrh 34:306−308,1975)に記載のように、調製試料の機能的な活性をリストセチンコファクターアッセイで測定した。rVWF出発原料に関して算出される0.39という比率の値は、ポリシアル酸付加後に減少して0.13となる。
【0113】
(実施例36:リジン残基を介した分岐PEGによるrVWFの結合)
成熟rVWF(35UVWF:Ag/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)の溶液を実施例23により調製した。次に、静かに撹拌させながら、NOF会社(NOF Europe,Grobbendonk,Belgium)によって供給される分枝mPEGスクシンイミジルグルタレート(PE−SG/鎖長:20kD)を、この溶液に添加し(5mgPEG−SG/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。次に、室温で1時間、静かに撹拌させながらPEG付加を行った。続いて、反応混合物を平衡イオン交換クロマトグラフィー樹脂(Fractogel EMD TMAE 650M)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)に載せた。次に、このカラムを20CV平衡化緩衝液で洗い、PEG付加rVWFを溶離緩衝液(20mM HEPES、0.5MNaCl、0.5%ショ糖、pH7.4)で溶出した。溶出液を、20mMのHEPES、150mMのNaCl、0.5%ショ糖、pH7.4からなる緩衝系を用い、再生セルロースからなる膜および100kD分子量カットオフで、限界濾過法/ダイアフィルトレーションにより濃縮した。得られたPEG付加誘導体のVWF:RCo/VWF:Ag 比は、rVWF出発材料(VWF:Rco/VWF:Ag比:0.89)と比較して、わずかに減少して0.79であった。また、PEG付加rVWF出発原料のFVIII結合能は、ECA試験(実施例16)で計測されたように、83%であった。
【0114】
(実施例37:FVIII−K.O.マウスでの分岐PEGと結合体を形成したVWFの薬物動態)
FVIII欠乏マウス(Bi et al.,Nat.Genet.10:119−121,1995)を、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、PEG−rVWF(分岐PEG,SG)およびrFVIIIの混合物またはネイティブrVWFおよびrFVIIIの混合物を経尾静脈によるボーラス注入法(10ml/kg)によって投与することで、30UFVIII/mlおよび25UVWF/mlを達成した。注射後5分、3、9、24、および32時間、麻酔後の心臓穿刺によってそれぞれの群からクエン酸血漿を調製した。FVIII活性およびVWF抗原回収濃度を血漿サンプルで計測した。VWFおよびFVIIIの抽出曲線を図23および図24に示す。VWF半減期は1.4から9.7時間へと増大し、VWFのAUCは11.8から49.2U*h/mlに増加した。FVIIIの半減期は、1.2時間(ネイティブrVWF存在下)から4.4時間(PEG−rVWFとともに適用)へと増大し、血中濃度時間曲線下面積(AUC)が12.1から30.5U*h/mlへと増大した。
【0115】
(実施例38:FVIII−K.O.マウスにおける異なる量のPEG−VWFと混合されたFVIIIの比較薬物動態)
FVIII欠乏K.O.マウス5匹からなる群に対して、PEG−rVWF(25mg分岐PEG−SG20000/mgタンパク質)とrFVIII(A:20IUPEG−rVWF/ml+20IUFVIII/ml、B:10IUPEG−rVWF/ml+20IUFVIII/ml、C:3IUPEG−rVWF/ml+20IUFVIII/ml)の種々の混合物を経尾静脈によるボーラス注入法(10ml/kg)によって投与した。PEG付加rVWFについて、算出された比率は3モルPEG/moleリジンであった。異なるPEG−rVWF/rFVIII混合物において等量(単位換算)のPEG−rVWFとrFVIIIとを比較すると、以下のPEG/FVIII比が算出された。すなわち、3:1(A)、1.5:1(B)、0.45:1(C)である。麻酔後、クエン酸血漿を注射後5分、1、3、9、および24時間で、それぞれの群から心臓穿刺によって調製した。FVIII半減期(A:2.1時間(B):2.0時間、および(C):2.5時間)とAUC(A:18.9(B):14.5、およびC:13.2U*hr/ml)との間に関連した違いは見出されなかった。FVIIIの抽出曲線を図25に示す。
【0116】
(実施例39:PEG付加の度合いが異なるPEG−VWF調製試料を用いた比較薬物動態)
FVIII×VWFダブルノックアウトマウスを、FVIII欠乏マウスとVWF欠乏マウスとの交雑育種によって得た。それらのマウスは、VWF欠乏と同様にFVIII欠乏を患う。FVIII×VWFダブルノックアウトマウス5匹からなる群に対して、ネイティブrVWF/rFVIII(100/150IU/kg)混合物、PEG付加rVWF#AとrFVIIIとの混合物(100/150IU/kg)、あるいはPEG付加rVWF#BとrFVIIIとの混合物(150/150IU/kg)を経尾静脈により注入した。PEG−rVWF#A(5mg PEG−SS 5000/mgタンパク質)およびPEG−rVWF#B(20mg PEG−SS 5000/mgタンパク質)を実施例24により調製した。調製試料#Aに関しては、算出された比率は2.5molePEG/moleリジンであった。調製試料#Bに関しては、算出された比率は10molePEG/moleリジンであった。このクエン酸血漿に関して、試料適用後5分、1、3、9、および24時間、試料を調製した。VWF:Agの血漿レベルおよびFVIII活性を計測し、最大血漿レベルのパーセントに概ね注射後5分で達する。VWFおよびFVIIIの抽出曲線を、それぞれ図26および図27に示す。半減期は、PEG−rVWF #Aおよび#Bに関して、それぞれ6.3時間および8.1時間であった。ネイティブrVWFに関しては、算出された半減期は2.0時間であった。PEG付加rVWF両方の正規化AUC(%最大×h)は、360%*h(ネイティブrVWF)から901%*h(#A)および1064%*h(#B)へと増大した。同時注入されたrFVIIIの循環時間は、ネイティブVWFと比較してPEG付加rVWFによって改善された。FVIIIの半減期は、ネイティブrVWFの存在下で0.8時間であって、それぞれPEG付加rVWF#Aおよび#Bが注入される場合、1.5および1.8時間へと増大した。FVIIIのAUCは、214、370、および358%*hであった。
【0117】
(実施例40:VWFダイマーのPEG付加)
VWFダイマー(58IUのVWF:Ag/ml)(組換えCHO細胞株の条件培地から精製(Baxter BioScience))を20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖)で調製した。NOF社から供給された分岐mPEGスクシンイミジルグルタレート(PE−SG/鎖長:20kD)を、静かに撹拌しながらこの溶液に添加し(5mgPEG−SG/mgタンパク質)、0.5MNaOHを滴加してpH値を7.4に合わせた。PEG付加を、室温で1時間、静かに撹拌させながら行った。続いて、反応混合物を、反応混合物を平衡イオン交換クロマトグラフィー樹脂(Fractogel EMD TMAE 650M)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)に載せた。次に、カラムを20CV平衡化緩衝液で洗い、過剰の試薬を取り除いた。PEG付加rVWFダイマーを溶離緩衝液(20mM HEPES、0.5MNaCl、0.5%ショ糖、pH7.4)で溶離させた。溶出液を、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を使用し、再生セルロース(Millipore)からなる膜および100kD分子量カットオフで、限界濾過法/ダイアフィルトレーションによって濃縮した。
【0118】
(実施例41:低マルチマーrVWFのPEG付加および生体外特徴付け)
成熟rVWFを、実施例23により精製した。精製手法は、イオン交換クロマトグラフィーステップおよびスペローズ6上での最終ゲル濾過ステップを含むもので、該手法を20mM HEPES、150mM NaCl、pH7.4で実行し、高マルチマーrVWF(17のマルチマー)がボイド容量で溶出した。VWFマルチマー分析を、1.0%のアガロースゲルを使用してRuggeri and Zimmerman(Blood 57:1140−43,1981)により行った。低マルチマーrVWF調製試料(6マルチマー)がサイド分画から得られ、より高い持続時間で溶離した。0.5%のショ糖pH7.4を加えることにより、この分画を安定化させた。次に、低マルチマーrVWFを、mPEGスクシンイミジルスクシネート(PEG−SS)を使用してPEG付加した。PEG−SSを、静かに撹拌させながらこの溶液に添加し(5mgPEG−SS/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。PEG付加を、室温で1時間、静かに撹拌させながら行った。その後、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を用いて過剰試薬を、再生セルロースからなる膜および100kDの分子量カットオフで、限界濾過法/ダイアフィルトレーションによって除去した。実施例16によるECAアッセイによって測定されるFVIII結合能は、出発原料の49%からPEG付加調製試料の34%まで、わずかに減少した。低マルチマーrVWFについて計測されたVWF:RCo/VWF:Ag比率の値は、0.02であり、PEG付加手法の影響を受けなかった。
【0119】
(実施例42:可逆的にブロックされたFVIII結合エピトープ(FVIIIおよびヘパリンでブロック)によるVWFの誘導体化)
クロマトグラフィーカラム(15mm×148mm)をヘパリンハイパーD(Bio−Sepra)で充填し、20mM HEPES、68mM NaCl、0.5%ショ糖、pH7.4からなる平衡化緩衝液で平衡させた。次に、成熟rVWF(48IUVWF:Ag/ml)含有20mM HEPES(150mM NaCl、0.5ショ糖)の溶液をH2Oで希釈することで伝導率を7〜8mS/cmとし、線流速1.5cm/分を使用してカラム上に載せた。その後、NOF社(NOF Europe,Grobbendonk,Belgium)によって供給された分枝mPEGスクシンイミジルグルタレート(鎖長:20kD)を新たに15mlの平衡化緩衝液に溶かし、最終濃度を5mgPEG−SG/mg結合タンパク質とした。次に、この試薬液をカラム上にポンプで送り、PEG付加を静的条件下で2時間行った。次に、0.05%リジンを含む10CV平衡化緩衝液でカラムを洗った。次に、保護されたFVIII結合エピトープによるPEG付加rVWFを20mM HEPES、1MNaCl、0.5%ショ糖、pH7.4からなる緩衝液で溶出させた。最終的に、再生セルロース(Millipore)からなる100kD膜を使用して、20mM HEPES緩衝液(pH7.4、150mM NaCl、0.5%ショ糖)に対する限界濾過法/ダイアフィルトレーションによって、この溶液を濃縮した。得られた誘導体のVWF:RCo/VWF:Ag比率は0.48であり、rVWF出発原料(比率0.47)と同一であった。実施例36に記載の分枝PEG−SG20000によるrVWFのPEG付加手法と対照的にFVIII結合能は、ECA試験(実施例16)によって計測されるように、FVIIIエピトープキャッピングでこのPEG付加手法に影響を受けなかった。
【0120】
(実施例43:リジン残基を介した分解可能PEGによるVWFの結合)
成熟rVWFを、実施例23によって精製する。次に、このrVWF(40UVWF:Ag/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)の溶液を、調製する。続いて、PEG付加は、静かに撹拌させながら、mPEG−(カルボキシメチル)−3−ヒドロキシ−ブタン酸N−ヒドロキシスクシンイミドエステル(鎖長:5kD)をこの溶液に加えることによって行い(5mgのPEG試薬/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。次に、PEG付加反応を、室温で1時間、静かに撹拌させながら行った。その後、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を用いて過剰試薬を、再生セルロースからなる膜および100kDの分子量カットオフで、限界濾過法/ダイアフィルトレーションによって、PEG付加rVWFから分離した。
【技術分野】
【0001】
(発明の分野)
本発明は、血漿のおよび/または組換えフォンビルブラント因子(VWF)を含むタンパク質性構築物に関し、上記VWFが少なくとも1つの生理学的に許容し得るポリマー分子に結合するとともに、上記タンパク質性構築物と少なくとも1つの第VIII因子(FVIII)タンパク質との複合体と結合する。さらに、本発明は、FVIIIまたはVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法に関する。
【背景技術】
【0002】
(発明の背景)
VWFは、哺乳動物の血漿で存在する多量体付着性糖タンパク質であり、複数の生理機能を持つ。一次止血の間、VWFは血小板表面上の特異的な受容体とコラーゲン等の細胞外マトリックス成分と間にメディエーターとして働く。さらに、VWFはプロコアギュラントFVIII用担体を安定させるタンパク質として働く。VWFは、2813個のアミノ酸からなる前駆体分子として血管内皮細胞および巨核球で合成される。前駆体ポリペチド(プレプロVWF)は、成熟血漿VWFで見つかる22残基シグナルペプチド、741残基プロペプチド、および2050残基ポリペチドからなる(非特許文献1)。血漿へ分泌されると、VWFは異なる分子の大きさで多用な種類の形態で循環する。これらのVWF分子は、2050のアミノ酸残基の成熟サブユニットのオリゴマーおよびマルチマーからなる。VWFは、通常、1つのダイマーから最大で50〜100のダイマーからなるマルチマーまでのダイマーとして、血漿で見いだされる(非特許文献2)。ヒト血行内のヒトVWFの生体内半減期は、約12〜20時間である。
【0003】
ヒトで最も頻繁に起こる遺伝性出血障害はフォンビルブラント病(VWD)であり、血漿または組換え起源のコンセントレートを含むVWFによる補充療法によって処置することができる。血液中のVWFの半減期が短いため、VWFの生体内半減期を長期にしたVWFコンセントレートを発現することが強く求められている。同じことがFVIIIにもあてはまり、FVIIIも約8〜12時間の比較的短い生体内半減期を持つことから、FVIIIおよびVWFの少なくとも1つの機能的な欠陥または不足を伴う出血障害の患者を処置するために頻繁に投薬を行うことを必要とする。
【0004】
先行技術では、真核細胞培養で産生される組換えVWF(rVWF)がより完全であり、血漿由来VWFよりも蛋白質加水分解による分解が少ないことが記載されている(非特許文献3)。特許文献1は、陰イオン交換クロマトグラフィーを使用して組換え型VWFを精製することによって高純度のVWFを単離する方法を記載する。相同かつ構造的に完全なVWFの大量産生のための方法は、当該技術分野でも知られている(非特許文献4;非特許文献5)。組換え型VWFは、フォンビルブラント病(VWD)のイヌ、マウス、およびブタモデルを用いたことを特徴とする(非特許文献6;非特許文献7;非特許文献8;非特許文献9)。特許文献2は、プロVWFをトロンビンで処置することによってVWF調製を生産する方法を記載する。担体の上で固定されるrVWFを用いてVWFと結合するタンパク質を精製する方法は、特許文献3に開示される。血液凝固障害を処置するための血漿由来および組換えVWFプロペプチド(pro−VWF)の製薬的使用は、特許文献4に記載される。特許文献5は、リンカーまたはカップリング剤を介したポリ(酸化アルキレン)に対するFVIIIおよびIX因子(FIX)の結合について記載する。特許文献6では、長期間にわたる生物学的生体内半減期を持つ組換え型VWFの使用が哺乳動物血液でFVIIIを安定化させ、内因性FVIIIの産生を誘導することが示されている。にもかかわらず、VWF−またはFVIIIベースの出血障害がある患者にとっては、さらにVWFおよびFVIIIの生体内半減期を増大させる必要性が存在する。
【0005】
VWFは生体内でFVIIIを安定化させることで、FVIIIの血漿レベルを調節する重要な役割を担うことで、結果として一次および二次止血を制御するための中心因子となることが知られている。VWFを含む治療的産物を使用した後、24時間内に1mlあたり1〜3単位まで内因性FVMI:Cが増大することが観察され、このことがFVIII上でのVWFの生体内安定効果を示していることも知られている
凝固FVIII欠乏(別名血友病A)および/またはVWFの質的および定量的欠乏(別名VWD)に対する処置の範囲を広げるための新規の物質が強く求められている。機能性VWFが欠如していることから、VWDを有する患者は、正常値によりも低いFVIII血漿レベルによって示されるFVIIIの二次欠乏を呈する。VWDの型と疾患の重症度とに応じて、これらのFVIIIレベルが変化し得るが、通常、健常なヒトで見出されるFVIII血漿レベルよりも多少低い。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許出願公開第0 784 632号明細書
【特許文献2】国際公開第00/49047号パンフレット
【特許文献3】国際公開第98/25969号パンフレット
【特許文献4】欧州特許出願公開第0 977 584号明細書
【特許文献5】米国特許第6,037,452号明細書
【特許文献6】欧州特許出願公開第0 774 261号明細書
【非特許文献】
【0007】
【非特許文献1】Fischer et al.,FEBS Lett.351:345−348,1994
【非特許文献2】Ruggeri et al.Thromb.Haemost.82:576−584,1999
【非特許文献3】Fischer et al.,FEBS Lett.375:259−262,1995
【非特許文献4】Schlokat et al.,Biotechnol.Appl.Biochem.24:257−267,1996
【非特許文献5】Fischer et al.,CMLS 53:943−950,1997
【非特許文献6】Turecek et al.,Blood 90:3555−3567,1997
【非特許文献7】Roussi et al.,Blood Coag.Fibrinol.9:361−372,1998
【非特許文献8】Schwarz et al.,Haemophilia 4:53−62,1998
【非特許文献9】Schwarz et al.,Semin.Thromb.Hemost.28:215−225,2002
【発明の概要】
【発明が解決しようとする課題】
【0008】
このように、本発明は哺乳動物血液中のVWFおよび/またはFVIIIの生体内半減期を延長する新規の系を提供する。本発明のさらなる目的は、FVIIIおよびVWFの一方または両方の機能欠陥または欠乏を伴う出血障害に対する改善された処置のための方法を提供することである。
【課題を解決するための手段】
【0009】
(発明の要旨)
本発明は、血漿および/または組換えフォンビルブラント因子(VWF)またはその生物学的活性誘導体を含むタンパク質性構築物に関し、上記VWFまたは上記その生物活性誘導体が一つ以上の生理学的に許容し得るポリマー分子に結合しており、タンパク質性構築物の生体内半減期が哺乳動物(特にヒト)の血液で長くなる。さらに、本発明は上記タンパク質性構築物と少なくとも1つのFVIII因子(FVIII)タンパク質またはその生物学的活性誘導体との複合体に関し、上記FVIIIタンパク質または上記その上記生物学的活性誘導体の半減期が哺乳動物の血液でも長くなる。また、本発明により、上記タンパク質性構築物または上記複合体を用いたFVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害がある哺乳動物の血液でVWFまたはFVIIIの生体内半減期を延長する方法と同様に、上記タンパク質性構築物または上記複合体を含む医薬品組成物を提供する。蛋白質様構築物を作る方法も、提供する。
本発明は、例えば、以下を提供する:
(項目1)
タンパク質性構築物であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(b)該VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子と、を含み、
該構築物が少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体に対する結合能を有し、該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大している、タンパク質性構築物。
(項目2)
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目1に記載のタンパク質性構築物。
(項目3)
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して、少なくとも約2倍増大している、項目1に記載のタンパク質性構築物。
(項目4)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体の糖質残基に結合している、項目1に記載のタンパク質性構築物。
(項目5)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体のリジン残基に結合している、項目1に記載のタンパク質性構築物。
(項目6)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目1に記載のタンパク質性構築物。
(項目7)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目1に記載のタンパク質性構築物。
(項目8)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目1に記載のタンパク質性構築物。
(項目9)
上記構築物に含まれる上記VWFが、VWFの一次止血における生物学的活性を保持し、上記生物学的活性が、血小板上の受容体に対する結合および細胞間マトリックス成分上の受容体に対する結合を含み、上記成分がコラーゲンを含む、項目1に記載のタンパク質性構築物。
(項目10)
タンパク質性構築物であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(b)上記VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子と、を含み、
該構築物が少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に対する結合能を有し、該構築物に結合した該FVIII分子の生体内半減期が、該構築物とは結合していないFVIII分子の生体内半減期と比較して増大している、タンパク質性構築物。
(項目11)
上記構築物に結合した場合、上記FVIII分子の生体内半減期が、上記構築物とは結合していないFVIII分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目10に記載のタンパク質性構築物。
(項目12)
上記構築物に結合した場合、上記FVIII分子の生体内半減期が、上記構築物とは結合していないFVIII分子の生体内半減期と比較して、少なくとも約2倍増大している、項目10に記載のタンパク質性構築物。
(項目13)
上記VWFまたは上記その生物活性誘導体が組換え産物である、項目1または10に記載のタンパク質性構築物。
(項目14)
項目1または10に記載のタンパク質性構築物および少なくとも1つのFVIII分子またはその生物活性誘導体を含む、複合体。
(項目15)
上記FVIII分子またはその生物活性誘導体が組換え産物である、項目14に記載の複合体。
(項目16)
FVIIIの機能的な欠陥または欠乏に関連した出血性障害を持つ哺乳動物血液中でのFVIIIまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物の第1の用量を該哺乳動物に対して投与するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体の第1の用量を該哺乳動物に対して投与するステップと、
を含む、方法。
(項目17)
FVIIIおよびVWFのうちの少なくとも1つの機能的な欠陥に関連した出血性障害を持つ哺乳動物血液中でのFVIIIまたはその生物活性誘導体の生体内半減期およびVWFまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物の第1の用量を該哺乳動物に対して投与するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体の第1の用量を該哺乳動物に対して投与するステップと、
を含む、方法。
(項目18)
FVIIIおよびVWFのうちの少なくとも1つの機能的な欠陥または欠乏に関連した出血性障害を持つ哺乳動物血液中での第VIII因子(FVIII)またはその生物活性誘導体およびVWFまたはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10の少なくとも1つのタンパク質性構築物を提供するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体を提供するステップと、
(c)上記タンパク質性構築物と上記FVIII分子またはその生物活性誘導体との間で複合体を形成するステップと、
を含む、方法。
(項目19)
FVIIIの機能的な欠陥に関連した出血性障害またはFVIIIの機能的な欠陥もしくは欠乏に関連した障害を持つ哺乳動物血液中での第VIII因子(FVIII)またはその生物活性誘導体の生体内半減期を延長させるための方法であって、
(a)項目1または10に記載の少なくとも1つのタンパク質性構築物を提供するステップと、
(b)少なくとも1つのFVIII分子またはその生物活性誘導体を提供するステップと、
(c)該タンパク質性構築物と該FVIII分子またはその生物活性誘導体との間で複合体を形成するステップと、
を含む、方法。
(項目20)
ステップ(c)の複合体を上記哺乳動物に投与する、項目18または19に記載の方法。
(項目21)
上記少なくとも1つのFVIII分子またはその生物活性誘導体を、上記タンパク質性構築物と同時に投与する、項目16または17に記載の方法。
(項目22)
上記少なくとも1つのFVIII分子またはその生物活性誘導体を、上記タンパク質性構築物の投与前または投与後に、逐次投与する、項目16または17に記載の方法。
(項目23)
医薬品組成物であって、
項目1、2、3、または6に記載のタンパク質性構築物の有効量と、
薬学的に許容可能な担体、希釈剤、塩、緩衝液および賦形剤からなる群から選択される一つ以上の化合物と、
を含む、医薬品組成物。
(項目24)
医薬品組成物であって、
項目10、11、または12に記載のタンパク質性構築物の有効量と、
薬学的に許容可能な担体、希釈剤、塩、緩衝液および賦形剤からなる群から選択される一つ以上の化合物と、
を含む、医薬品組成物。
(項目25)
複合体であって、
(a)タンパク質性構築物であって、
(i)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子と、
(ii)該VWF分子に結合した少なくとも1つの生理学的に許容可能なポリマー分子とを含む、タンパク質性構築物と、
(b)該タンパク質性構築物に結合した少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と、
を含み、
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、増大している、複合体。
(項目26)
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、少なくとも約1.5倍増大している、項目25に記載の複合体。
(項目27)
上記複合体の生体内半減期が、VWFに結合したFVIIIの生体内半減期と比較して、少なくとも約2倍増大している、項目25に記載の複合体。
(項目28)
VWF分子と、該VWF分子上の少なくとも1つの糖質残基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質残基を酸化するステップと、
(c)該糖質残基を、ヒドラジド基含有PEG試薬と接触させるステップと、
(d)該試薬PEGヒドラジドを該VWF上の少なくとも1つの糖質残基と共有結合させることにより、上記構築物を形成させるステップとを含み、
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
上記構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に結合することが可能である、方法。
(項目29)
上記酸化ステップ(b)が化学的酸化剤を使用して実行される、項目28に記載の方法。
(項目30)
上記化学的酸化剤がNaIO4である、項目29に記載の方法。
(項目31)
上記酸化ステップ(b)が酵素的酸化剤を使用して実行される、項目28の方法。
(項目32)
上記酵素的酸化剤がガラクトースオキシダーゼである、項目31に記載の方法。
(項目33)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目28に記載の方法。
(項目34)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目33に記載の方法。
(項目35)
項目28に記載の方法によって調製されたタンパク質性構築物。
(項目36)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも約1.5倍増大している、項目35に記載のタンパク質性構築物。
(項目37)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも約2倍増大している、項目35に記載のタンパク質性構築物。
(項目38)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも約1.5倍増大している、項目35に記載のタンパク質性構築物。
(項目39)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも約2倍増大している、項目35に記載のタンパク質性構築物。
(項目40)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目28に記載の方法。
(項目41)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目28に記載の方法。
(項目42)
VWF分子と、該VWF分子上の少なくとも1つの一級アミノ基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の該一級アミノ基をPEG試薬と接触させるステップと、
(c)該PEG試薬を該VWF上の該一級アミノ基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体に結合することが可能である、方法。
(項目43)
上記一級アミノ基が、上記VWF分子のリジン残基上に含まれる、項目42に記載の方法。
(項目44)
上記一級アミノ基が、上記VWF分子のN末端上に含まれる、項目42に記載の方法。
(項目45)
上記PEG試薬が、N−ヒドロキシスクシンイミド−エステル、PEGカーボネート、カルボキシル基含有PEG誘導体、およびアルデヒド基含有PEG誘導体からなる群から選択される、項目42に記載の方法。
(項目46)
上記試薬が、PEG−スクシンイミジルスクシネート、PEG−スクシンイミジルグルタレート、PEG−スクシンイミジルブタノエート、PEG−スクシンイミジルヘキサノエート、分解性PEG試薬、および加水分解性PEG試薬からなる群から選択されるN−ヒドロキシスクシンイミド−エステルである、項目45に記載の方法。
(項目47)
上記試薬が、p−ニトロフェニルカーボネートまたはスクシンイミジルカーボネートであるPEGカーボネートである、項目45に記載の方法。
(項目48)
上記試薬が、水溶性カルボジイミドの使用によって一級アミノ基に連結することができるカルボキシル基を含むPEG誘導体の群から選択される、項目45に記載の方法。
(項目49)
上記試薬がPEGプロピオンアルデヒドであり、還元性アミノ化によって一級アミノ基に連結することができる、項目45に記載の方法。
(項目50)
VWF分子と、該VWF分子上の少なくとも1つの一級アミノ基に、還元的アミノ化によって共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で該VWF分子上のリジン残基をアルデヒド基含有PEG試薬と接触させてシッフ塩基を形成するステップと、
(c)該溶液を還元剤と接触させて二級アミド結合を形成するステップと、
(d)該PEG試薬を上記VWF分子に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目51)
上記還元剤がNaCNBH3である、項目50に記載の方法。
(項目52)
上記一級アミノ基が上記VWF分子上のN末端アミノ基であり、該N末端アミノ基が選択的にPEG付加されている、項目50に記載の方法。
(項目53)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目50に記載の方法。
(項目54)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目53に記載の方法。
(項目55)
項目50に記載の方法によって調製されたタンパク質性構築物。
(項目56)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目55に記載のタンパク質性構築物。
(項目57)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目55に記載のタンパク質性構築物。
(項目58)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目55に記載のタンパク質性構築物。
(項目59)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目55に記載のタンパク質性構築物。
(項目60)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目50に記載の方法。
(項目61)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目50に記載の方法。
(項目62)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で該VWF分子上のリジン残基を試薬PEGアルデヒドと接触させてシッフ塩基を形成するステップと、
(c)該溶液を反応性試薬と接触させて二級アミド結合を形成するステップと、
(d)該PEG試薬を該VWF分子に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目63)
VWF分子と、該VWF分子上の少なくとも1つの遊離または生成スルフヒドリル基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の該スルフヒドリル基を、マレイミド基含有PEG試薬と接触させるステップと、
(c)該PEG試薬を該VWF分子上の少なくとも1つのスルフヒドリル基に共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目64)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目63に記載の方法。
(項目65)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目64に記載の方法。
(項目66)
項目63に記載の方法によって調製されたタンパク質性構築物。
(項目67)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目66に記載のタンパク質性構築物。
(項目68)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目66に記載のタンパク質性構築物。
(項目69)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目66に記載のタンパク質性構築物。
(項目70)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目66に記載のタンパク質性構築物。
(項目71)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目63に記載の方法。
(項目72)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目71に記載の方法。
(項目73)
VWF分子と、該VWF分子上の少なくとも1つのカルボキシル基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上のカルボキシル基を、アミノ基および水溶性カルボジイミドを含むPEG試薬と接触させてアミド結合を形成するステップと、
(c)該PEG部分を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目74)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目73に記載の方法。
(項目75)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目74に記載の方法。
(項目76)
項目73に記載の方法によって調製されたタンパク質性構築物。
(項目77)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目76に記載の構築物。
(項目78)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目76に記載の構築物。
(項目79)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目76に記載の構築物。
(項目80)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目76に記載の構築物。
(項目81)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目73に記載の方法。
(項目82)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目81に記載の方法。
(項目83)
VWF分子と、該VWF分子上に共有結合しており、該VWF分子のFVIII結合部位に対しては結合していないPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上のFVIII結合部位を第VIII因子結合部位保護剤と接触させることにより、保護されたFVIII結合部位を持つVWF分子を形成させるステップと、
(c)ステップ(b)の該VWF分子上の反応部位をPEG試薬と接触させるステップと、
(d)該PEG試薬を該VWF分子と共有結合させるステップと、
(e)該VWF分子から該FVIII結合部位保護剤を分離することにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つの第VIII因子(FVIII)分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目84)
上記第VIII因子結合部位保護剤が親和性カラムに含まれる、項目83に記載の方法。
(項目85)
上記第VIII因子結合部位保護剤が、第VIII因子、FVIIIの誘導体、ヘパリン、およびヘパリンの誘導体からなる群から選択される、項目83の方法。
(項目86)
上記PEG試薬が、直鎖状アルコキシPEG、直鎖状二官能性PEG、分岐状PEG、マルチアームPEG、フォーク状PEG、ポリオール核に結合したPEG、樹状PEG、安定な結合を有するPEG、分解可能な結合を有するPEG、および加水分解可能な結合を有するPEGからなる群から選択される、項目83に記載の方法。
(項目87)
上記PEG試薬が、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される内部構造を有する、項目86に記載の方法。
(項目88)
項目83に記載の方法によって調製されたタンパク質性構築物。
(項目89)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目88に記載の構築物。
(項目90)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目88に記載の構築物。
(項目91)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目88に記載の構築物。
(項目92)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目88に記載の構築物。
(項目93)
上記PEG試薬が約0.1〜約200mg PEG/mg VWFタンパク質の比率で提供される、項目83に記載の方法。
(項目94)
上記PEG試薬が約1.0〜約25mg PEG/mg VWFタンパク質の比率で提供される、項目93に記載の方法。
(項目95)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基と共有結合したポリシアル酸(PSA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)溶液中で、PSAを、アルデヒド含有PEG試薬と接触させてシッフ塩基を形成するステップと、
(c)該溶液を還元剤と接触させて二級アミド結合を形成するステップと、
(d)該PSAを該VWF上の少なくとも1つのリジン残基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目96)
上記還元剤がNaCNBH3である、項目95に記載の方法。
(項目97)
項目95に記載の方法によって調製されたタンパク質性構築物。
(項目98)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目97に記載の構築物。
(項目99)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目97に記載の構築物。
(項目100)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目97に記載のタンパク質性構築物。
(項目101)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目97に記載のタンパク質性構築物。
(項目102)
VWF分子と、該VWF分子と共有結合により架橋したポリシアル酸(PSA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子を、PSAとグルタルアルデヒドとを含む溶液と接触させるステップと、
(c)該PSAを該VWFへと共有結合により架橋させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期が、VWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目103)
項目102に記載の方法によって調製されたタンパク質性構築物。
(項目104)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目103に記載の構築物。
(項目105)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目103に記載の構築物。
(項目106)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目103に記載のタンパク質性構築物。
(項目107)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目103に記載のタンパク質性構築物。
(項目108)
VWF分子と、該VWF分子上の少なくとも1つの糖質残基に共有結合したPSA部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質残基を酸化するステップと
(c)該糖質残基を、ヒドラジド基含有PSA試薬と接触させるステップと、
(d)該PSA試薬を該VWF上の少なくとも1つの糖質残基と共有結合させることにより、該構築物を形成させるステップとを含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目109)
上記酸化ステップ(b)が酵素的酸化剤を用いて実行される、項目108に記載の方法。
(項目110)
上記酵素的酸化剤がガラクトースオキシダーゼである、項目109に記載の方法。
(項目111)
VWF分子と、該VWF分子上の少なくとも1つのリジン残基に共有結合したヒアルロン酸(HA)部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)HAの溶液を酸化剤と接触させて活性化HAを形成させるステップと、
(c)該VWF分子を該活性化HAと接触させることにより、該構築物を形成させるステップとを含み、
上記構築物の生体内半減期がVWF分子の生体内半減期と比較して増大している、上記構築物が、少なくとも1つのFVIII因子分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目112)
項目111に記載の方法によって調製されたタンパク質性構築物。
(項目113)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目112に記載の構築物。
(項目114)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目112に記載のタンパク質性構築物。
(項目115)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目112に記載のタンパク質性構築物。
(項目116)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目112に記載のタンパク質性構築物。
(項目117)
VWF分子と、上記VWF分子上の少なくとも1つの糖質基に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子上の糖質基を酸化酵素と接触させて、該VWF上で酸化型の糖質部分を形成させるステップと、
(c)該VWF上の該酸化型の糖質部分をヒドラジド基含有PEG試薬と接触させるステップと、
(d)該PEG部分を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合できる、方法。
(項目118)
項目117に記載の方法によって調製されたタンパク質性構築物。
(項目119)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目118に記載の構築物。
(項目120)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目118に記載の構築物。
(項目121)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目118に記載のタンパク質性構築物。
(項目122)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目118に記載のタンパク質性構築物。
(項目123)
VWF分子と、該VWF分子に共有結合したPEG部分とを含むタンパク質性構築物を形成するための方法であって、
(a)血漿フォンビルブラント因子(VWF)、組換え型VWF、VWFの生物活性誘導体、ならびにそれらのダイマーおよびマルチマーからなる群から選択されるVWF分子を提供するステップと、
(b)該VWF分子をPEG試薬に接触させて溶液を形成するステップと、
(c)該溶液を灌流チェンバーに入れ、蠕動ポンプの使用により上記VWF分子上に剪断応力を発生させるステップと、
(c)該PEG試薬を該VWFに共有結合させることにより、該構築物を形成させるステップと、
を含み、
該構築物の生体内半減期がVWF分子の生体内半減期と比較して増大しており、
該構築物が、少なくとも1つのFVIII分子またはFVIIIの生物活性誘導体と結合することができる、方法。
(項目124)
上記PEG試薬が、N−ヒドロキシスクシンイミド−エステル、PEGカーボネート、カルボキシル基含有PEG誘導体、およびアミノ基含有PEG誘導体からなる群から選択される、項目123に記載の方法。
(項目125)
上記PEG試薬がPEG−スクシンイミジルスクシネート、PEG−スクシンイミジルグルタレート、PEG−スクシンイミジルブタノエート、PEG−スクシンイミジルヘキサノエート、加水分解感受性のエステルまたはアミド結合によって結合される加水分解性PEG、mPEG−(カルボキシメチル)−3−ヒドロキシ−ブタン酸N−ヒドロキシスクシンイミドエステル、PEG−p−ニトロフェニルカーボネート、および、PEG−スクシンイミジルカーボネートからなる群から選択される、項目123に記載の方法。
(項目126)
項目123に記載の方法によって調製されたタンパク質性構築物。
(項目127)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも1.5倍増大している、項目126に記載の構築物。
(項目128)
上記構築物の生体内半減期が、VWF分子の生体内半減期と比較して、少なくとも2倍増大している、項目126に記載の構築物。
(項目129)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも1.5倍増大している、項目126に記載の構築物。
(項目130)
上記構築物に結合した場合、FVIII分子またはFVIIIの生物活性誘導体の生体内半減期が、該構築物に結合していない該FVIII分子または該FVIIIの生物活性誘導体の生体内半減期と比較して、少なくとも2倍増大している、項目126に記載の構築物。
(項目131)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体の糖質残基に結合している、項目10に記載のタンパク質性構築物。
(項目132)
上記少なくとも1つの生理学的に許容可能なポリマー分子が上記VWFまたは上記VWFの生物活性誘導体のリジン残基に結合している、項目10に記載のタンパク質性構築物。
(項目133)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目10に記載のタンパク質性構築物。
(項目134)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目10に記載のタンパク質性構築物。
(項目135)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目10に記載のタンパク質性構築物。
(項目136)
上記構築物に含まれる上記VWFが、VWFの一次止血における生物学的活性を保持し、上記生物学的活性が、血小板上の受容体に対する結合および細胞間マトリックス成分上の受容体に対する結合を含み、上記成分がコラーゲンを含む、項目10に記載のタンパク質性構築物。
(項目137)
上記生理学的に許容可能なポリマー分子は、ポリ(アルキレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールとの共重合体、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリレート)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファスファゼン、ポリオキサゾリン、およびポリ(N−アクリロイルモルホリン)からなる群から選択される、項目10に記載のタンパク質性構築物。
(項目138)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目25に記載の複合体。
(項目139)
上記生理学的に許容可能なポリマー分子がポリエチレングリコール(PEG)またはその誘導体である、項目26に記載の複合体。
(項目140)
上記生理学的に許容可能なポリマー分子がポリシアル酸(PSA)またはその誘導体である、項目26に記載の複合体。
【図面の簡単な説明】
【0010】
【図1】図1Aおよび図1Bは、結合標的部位に関する実施例によるVWFの模式的構造を示す。図1Aの灰色の点は少なくとも1つのポリマー分子によって結合し得るVWFのリジン残基を示し、図1Bは少なくとも1つのポリマー分子によって結合し得るVWFの糖質残基を示す。
【図2】図2は、VWDを有するマウスでの非結合体化VWFと比較したポリマーVWF結合体の薬物動態を示す。
【図3】図3は、血友病マウスでの非結合体化VWFと比較したポリマーVWF結合体の薬物動態を示す。
【図4】図4は、rVWFまたはPEG−rVWFで処置されるVWDマウスでのFVIIIの薬物動態を示す。
【図5】図5は、ポリマーVWF結合体と複合体を形成したrFVIIIおよびVWFと複合体を形成したrFVIIIを適用した後の血友病マウスにおけるrFVIIIの回収率を示す。
【図6】図6は、ポリマーVWF結合体と複合体を形成したrFVIIIおよびVWFと複合体を形成したrFVIIIを適用した後の血友病マウスにおけるVWFの回収率を示す。
【図7】図7は、FVIII×VWFダブルノックアウトマウスにおけるrFVIIIおよびPEG−rVWFの薬物動態を示す。
【図8】図8は、雑種−マウスにおけるrFVIIIとPEG−rVWFの薬物動態を示す。
【図9】図9は、マウス血漿におけるVWF:Ag増加(リジンおよび糖質結合)を示す。
【図10】図10は、非結合体化rVWFと比較した場合のPEGに結合したrVWFの生体分子相互作用検討(左側図)とFVIII(右側図)の結合能の比較とを示す。
【図11】図11は、SDS−PAGEによって計測されるポリマー結合の後のVWFの大量の増加を示す。
【図12】図12は、VWFマルチマーを分析するためにアガロース電気泳動で計測されるポリマー結合後のVWFの大量の増加を示す。
【図13】図13は、ポリマーVWF結合体(PEG付加リジン残基を有するrVWF)と複合体を形成したrFVIIIと、rVWFと複合体を形成したrFVIIIの適用後のFVIII−K.O.マウスにおけるrFVIIIの薬物動態を示す。
【図14】図14は、ポリマーVWF結合体(PEG付加リジン残基によるrVWF)と複合体を形成したrFVIIIおよびrVWFと複合体を形成したrFVIIIの適用後のFVIII−K.O.マウスにおけるrVWFの薬物動態を示す。
【図15】図15は、組み合わされたELISAおよび色素アッセイ(ECA)を使用して、非結合体化rVWFと比較した場合のPEGと結合したrVWFのFVIII結合能の測定を示す。
【図16】図16は、表面プラズモン共鳴技術を使用したPEGと結合したrVWFのFVIII結合能の側定を示す。
【図17】図17は、実施例13に記載のように、SDS−PAGEで計測されるポリマー結合後のVWFの大量増加を示す。
【図18】図18は、組み合わされたELISAおよび色素アッセイ(ECA)を用いたpdVWFおよび無処置rVWFと比較した場合のフリン成熟rVWFのFVIII結合能の測定を示す。
【図19】図19は、表面プラズモン共鳴技術を使用してpdVWFおよび無処置rVWFと比較した場合のフリン成熟rVWFのFVIII結合能の測定を示す。
【図20】図20は、還元条件下でのrVWF−ポリシアル酸結合体およびrVWFの等電点電気泳動パターンを示す。
【図21】図21は、VWF欠乏マウスにおけるrVWF−ポリシアル酸結合体およびrVWFの薬物動態を示す。
【図22】図22は、VWF欠乏マウスにおけるrVWF−ポリシアル酸結合体またはrVWFの使用後、マウスFVIII活性の時間経過を示す。
【図23】図23は、FVIII欠乏マウスにおけるPEG−rVWF(分枝PEG20K SG)およびrVWFの薬物動態を示す。
【図24】図24は、FVIII欠乏マウスでのPEG−rVWF(分枝PEG20K SG)またはrVWFを共に注入したrFVIIの薬物動態を示す。
【図25】図25は、FVIII欠乏マウスで種々の濃度のPEG−rVWF(分枝PEG20K SG)を注入したrFVIIの薬物動態を示す。
【図26】図26は、FVIII欠乏マウスにおけるPEG−rVWF#A(5mgPEG/mgタンパク質)、PEG−rVWF#B(20mgPEG/mgタンパク質)とネイティブrVWFの薬物動態を示す。
【図27】図27は、PEG−rVWF#A(5mgPEG/mgタンパク質)、PEG−rVWF #B(20mgPEG/mgタンパク質)またはネイティブrVWFを同時注入した場合のrFVIIIの薬物動態を示す。
【発明を実施するための形態】
【0011】
(発明の詳細な説明)
本発明の一態様は、血漿および/または組換えVWFあるいはその生物活性誘導体を含むタンパク質性構築物(また、以下で「ポリマーVWF結合体」と称される)に関し、上記VWFまたは上記その生物活性誘導体が一種類以上の生理学的に許容可能なポリマー分子に結合しており、上記VWFまたは上記その生物活性誘導体の生体内半減期が哺乳動物血液で長くなる。
【0012】
本発明のさらなる態様は、ポリマーVWF結合体を提供することであり、該ポリマーVWF結合体は、2つの形態の中間にある特徴を有する上記ポリマーVWF結合体の形態をなす。1つの形態は、VWFと結合体を形成するポリマーを安定して担持し、哺乳動物への適用の後、時間が経つにつれて内在性分子として除去される。他の形態は、VWFに結合体を形成するポリマーの可逆性を特徴とする。哺乳動物に対する投与の後、VWFに結合したポリマー分子はVWFから段階的に遊離され、非結合VWFは薬理的機能因子として利用可能となる。剥離性は、共役化学に依存するとともに、VWFに結合したポリマー分子の組成および構造に依存する。
【0013】
ポリマーVWF結合体は、VWDの処置に対して単独で、または半減期の増大に関してFVIIIを安定化させるためにFVIIIと結合して、あるいは両方で、有用である。VWDの処置に対して単独使用される場合、結合体は2つの形態のうちの一方を取ると考えらえる。第1の形態は、ポリマーが遊離自在にVWFに結合するものである。このようにして、VWFは、ポリマーが遊離または分解するにつれて活性化される。第2の形態は、VWFに結合したポリマー濃度がVWF活性と干渉しない程度のものである。結合体がFVIIIを結合して安定させるために調製される場合、VWFに結合したポリマーの度合いまたはレベルは、VWFの結合領域と干渉しないように提供される。実施例で示されるように、VWFに対する満足のいくポリマーの結合は、VWFおよびFVIIIの結合能に干渉することなく達成し得る。ポリマー結合の度合いもまた、制御または修飾してVWF活性を保つ一方で、FVIIIに対して結合するVWFの能力も保たれる。この形態で、ポリマーVWF結合体は治療的活性VWFを提供する一方で、半減期増大に関してFVIIIを安定化させる。
【0014】
本発明に有用なVWFおよびFVIII分子として、完全長タンパク質、タンパク質の前駆体、タンパク質のサブユニットまたはフラグメント、ならびにその機能的誘導体が挙げられる。VWFおおびFVIIIまたはFVIIIへの言及は、このようなタンパク質のすべての潜在的形態を含むものと考える。
【0015】
本明細書中で用いられる場合「生物活性誘導体」は、分子の任意の誘導体を含み、該誘導体は、上記分子の同じ機能的なおよび/または生物学的な性質(例えば結合性を持つ)および/または同じ構造的基礎(例えばペプチドの主鎖または基本的高分子単位)を、実質的に持つ。
【0016】
本発明に有用なVWFは、単一および多量体形態を含むすべての潜在的形態を含む。VWFの1つの特に有用な形態は、少なくとも2つのVWFからなるホモマルチマーである。VWFタンパク質は、いずれの生物活性誘導体であってもよく、あるいはFVIIIの安定剤として単独で使用される場合、VWFが生物学的に不活性である形態であってもよい。併用使用されるVWFの異なる形態を本発明が含むことも理解すべきである。例えば、本発明に有用な組成物は、異なるマルチマー、異なる誘導体、ならびに生物活性誘導体と生物活性のない誘導体とを含むものであってもよい。一次止血において、VWFは血小板と特異的な細胞外マトリックス成分(例えばコラーゲン)間の架橋として働く。このプロセスでのVWFの生物学的活性を、2つの異なる生体外アッセイで計測することができる(Turecek et al.,Semin.Thromb.Hemost.28:149−160,2002)。リストセチンコファクターアッセイは、VWF存在下で抗生物質のリストセチンによって誘発される新鮮またはホルマリン固定血小板の凝集に基づく。血小板凝集反応の程度は、VWF濃度次第であって、比濁法によって、例えば血小板凝集計を使用することで計測することができる(Weiss et al.,J.Clin.Invest.52:2708−2716,1973;Macfarlane et al.,Thromb.Diath.Haemorrh.34:306−308,1975)。第2の方法はコラーゲン結合実験であり、これはELISA技術に基づく(Brown et Bosak,Thromb.Res.43:303−311,1986;Favaloro,Thromb.Haemost.83:127−135,2000)。マイクロタイタープレートは、I型またはIII型のコラーゲンで被覆されている。さらに、VWFはコラーゲン面に結合し、続いて、酵素で標識されたポリクローナル抗体で検出される。最後のステップは、基質反応であり、ELISAリーダーで光度分析的にモニターすることができる。
【0017】
本明細書中で用いられる場合、「血漿由来VWF(pdVWF)」は血液中に見いだされるタンパク質のすべての形態を含むもので、該形態として、少なくとも1つのFVIII分子を生体内安定化(例えば結合)させる性質がある哺乳動物から得られる成熟VWFが挙げられる。しかし、本発明は成熟VWFに限らない。1つに、上記pVWFの生物活性誘導体は、プロペプチドを含むプロVWFである。本発明に有用な他の型のVWFとして、タンパク質性構築物が挙げられる。このタンパク質性構築物は未熟なVWFを含み、該未熟VWFとして、血管内皮細胞および巨核球によって合成される前駆体VWF分子(プレプロVWF)および/またはシグナルペプチドおよびプロペプチドの開裂で得られるVWFプロペプチド(プロVWF)および/または成熟pdVWF(それぞれ前駆体分子のもの)が挙げられる。pdVWFの生物活性誘導体のさらなる例として、プロドラッグが挙げられ、該プロドラッグは、処理または変換され、生物活性形態となる。あるいは、プロドラッグは、天然型と比べて、生物学的に活性があることから、切断形態、欠失を持つ形態、置換を持つ形態、プロ形態以外の付加を有する形態、成熟形態のフラグメント、キメラ形態、および後翻訳修飾を持つ形態である。本発明に有用なPdVWFも、生物学的活性のないそれらの形態を含む。このことは、血液中見いだされる成熟VWFまたは他の天然形態の修飾によって達成し得る。本発明に有用なVWFの供給源は、ブタおよびヒトバージョンを含む哺乳動物である。
【0018】
本明細書中で用いられる場合、「組換え型VWF(rVWF)」は組換えDNA技術を経て得られるVWFを含む。有用なrVWFの1つの形態は、少なくとも1つのFVIII分子の少なくとも生体内安定化(例えば結合)の性質を持ち、薬理学的に許容可能なグリコシル化パターンを任意に有する。その具体例として、A2領域を持たないことからタンパク質分解に対して耐性を持つVWF(Lankhof et al.,Thromb.Haemost.77:1008−1013,1997)、糖タンパク質Ib結合領域とコラーゲンおよびヘパリンの結合部位とをVal449からAsn730までのVWFフラグメント(Pietu et al.,Biochem.Biophys.Res.Commun.164:1339−1347,1989)が挙げられる。少なくとも1つのFVIII分子の安定化の側定は、当該技術分野における状態で公知の方法によって、VWF欠乏哺乳動物で行うことができる。例えば、下記の実施例8に記載のように、VWF欠乏マウスに対してVWFを経尾静脈静注することにより処置すると、その血漿のFVIII活性のレベルが時間とともに変化する。例えば、FVIII活性のレベルは、色素アッセイ(例えば、European Pharmacopoeiaで発表されているもの)で計測することができる(Ph.Eur.,3rd Ed.1997:2.7.4)。
【0019】
FVIII(FVIlI:C)含有試料を、カルシウム含有緩衝液中でトロンビン、活性化第IX因子(FIXa)、リン脂質、および第X因子(FX)と混合する。FVIIIはトロンビンにより活性化され、続いてリン脂質、FIXaおよびカルシウムイオンと複合体を形成する。この複合体は、FXおよびFXaを活性化し、順次に色素基質(例えば、AcOH*CH3OCO−D−CHA−Gly−Arg−pNA)を切断する。放出されたp−ニトロアニリン(pNA)の時間経過を、405nmで計測する。反応の傾斜は、試料中のFVIII濃度に比例している。
【0020】
本発明のrVWFは、当該技術分野で知られている任意の方法によって生産可能である。一具体例が、WO86/06096(1986年10月23日発行)および米国特許出願第07/559509号(Ginsburg et al.名義で1990年7月23日出願)に開示されており、組換え型VWFを生産する方法に関連して本明細書中に援用する。これは、当該技術分野で公知の任意の方法を含むものであってよく、該方法は、(i)遺伝子工学による組換えDNAの産生(例えばRNAの逆転写および/またはDNAの増幅を介する)、(ii)トランスフェクションによって組換えDNAを原核または真核細胞に導入すること(例えば電気穿孔法またはマイクロインジェクションを介する)、(iii)上記形質転換細胞を培養すること(例えば連続またはバッチ方式によるもの)、(iv)VWFを発現すること(例えば構成的または誘導的)、ならびに(v)上記VWFを単離すること(例えば、培地から、または形質転換細胞を収集することによって)、そのために(vi)精製されたrVWF(例えば陰イオン交換クロマトグラフィーまたはアフィニティークロマトグラフィーを経て)を得ることのためのものである。
【0021】
薬理学的に許容可能なVWF分子を生産することを特徴とする適当な原核生物または真核生物宿主系での発現によって、rVWFを生産することができる。真核細胞の例は、哺乳動物細胞(例えばCHO、COS、HEK 293、BHK、SK−Hep、およびHepG2)である。本発明によるVWFを生産または単離するための試薬または条件に対しては何ら特定の制限はなく、当該技術分野で公知または市販されている任意の系を用いることができる。本発明の好ましい実施形態では、先端技術に記載の方法でrVWFが得られる。
【0022】
多種多様なベクターをrVWFの調製に使用することができ、該ベクターを真核生物および原核生物発現ベクターから選択することができる。原核生物発現のためのベクターの例として、プラスミド(例えばpRSET、pET、pBAD、その他)が挙げられ、原核生物発現ベクターで使用されるプロモーターとして、lac、trc、trp、recA、araBAD、その他が挙げられる。真核生物発現のためのベクターの例として、以下のものが挙げられる。すなわち、(i)酵母での発現用として、プロモーター(例えば、AOX1、GAP、GAL1、AUG1、その他)を用いるベクター(例えば、pAO、pPIC、pYES、pMET)、(ii)昆虫細胞での発現用として、プロモーター(例えば、PH、p10、MT、Ac5、OplE2、gp64、polh、その他)を用いるベクター(例えば、pMT、pAc5、plB、pMIB、pBAC,その他)、ならびに(iii)哺乳動物細胞での発現として、ベクター(例えば、pSVL、pCMV、pRc/RSV、pcDNA3、pBPV、その他)、およびプロモーター(例えばCMV、SV40、EF−1、UbC、RSV、ADV、BPV、β−actin)を用いたこのようなウイルス系に由来するベクター(例えば、ワクシニアウイルス、アデノ関連性ウイルス、ヘルペスウイルス、レトロウイルス)が挙げられる。
【0023】
本発明に有用なFVIIIは、それらの形態を含み、該形態は、生物学的に活性があり、完全長FVIII、凝固FIXの活性化でコファクターとして働く任意の派生的な能力、およびVWFで複合体を形成する能力が含まれる。本発明によって使用されるFVIIIは、血漿由来FVIII(pdFVIII)または組換え型FVIII(rFVIII)またはその生物活性誘導体であってもよい。pdFVIIIおよびrFVIIIは、当該技術分野で知られている任意の方法によって生産することが可能である。PdFVIIIは、任意の適当な手段で精製可能である。1つの有用な方法は米国特許第5,470,954号に記載されており、該特許文献を本明細書中に援用する。RFVIIIタンパク質は、任意の適当な手段で調製可能である。このようなrFVIIIの例として、Baxter Healthcare社によって製造販売されているRECOMBINATEとADVATE、Wyeth社によって製造販売されているREFACTO(FVIIIのB−領域が欠失した形態)、およびBayer社によって製造販売されているKOGENATEが挙げられる。rFVIIIの方法および例は、米国特許第4,757,006号、第4,965,199号、および第5,618,788号(いずれも本明細書中に援用)に記載されている。
【0024】
本明細書中で用いられる場合、「生理学的に許容し得るポリマー」は、水溶液または懸濁液に可溶であり、医薬的に有効な量のポリマーVWF結合体の投与に際して哺乳動物に対し陰性影響(例えば副作用)がないポリマーを含む。本発明によって使用される生理学的に許容し得るポリマーは、特に制限されるものではない。ポリマーは、反復単位を好ましくは2ないし約300持つことによって一般に特徴づけられる。このようなポリマーの例として、限定されるものではないが、ポリ(アルキレングリコール)、例えばポリエチレングリコール(PEG)、ポリ(プロピレングリコール)(PPG)、エチレングリコールとプロピレングリコールとの共重合体、その他、ポリ(オキシエチル化ポリオール)、ポリ(オレフィンアルコール)、ポリ(ビニルピロリドン)、ポリ(ヒドロキシアルキルメタクリルアミド)、ポリ(ヒドロキシアルキルメタクリル酸塩)、ポリ(サッカライド)、ポリ(α−ヒドロキシ酸)、ポリ(ビニルアルコール)、ポリホスファゼン、ポリオキサゾリン、ポリ(N−重合開始剤)、ならびに前述のいずれかの組合せが挙げられる。
【0025】
生理学的に許容し得るポリマーは特定の構造に限定されず、直鎖状(例えばアルコキシPEGまたは二官能性PEG)、分岐状、またはマルチアーム(例えばフォーク状PEGまたはポリオール核に結合したPEG)、樹状、または分解可能な結合を有することができる。さらに、ポリマーの内部構造は、任意の数の異なるパターンで系統化され、ホモポリマー、交互共重合体、ランダム共重合体、ブロック共重合体、交互トリポリマー、ランダムトリポリマー、およびブロックトリポリマーからなる群から選択される。
【0026】
これらのポリマーもまた、ポリ(酸化アルキレン)ポリマー、ポリ(マレイン酸)、ポリ(DL−アラニン)、例えばカルボキシメチルセルロース、デキストラン、ヒアルロン酸およびキチン、ならびにポリ(メセドリン)アクリレート系が挙げられる。
【0027】
本発明の実施形態において、生理学的に許容し得るポリマーはそのPEGと誘導体である。PEG側鎖は直鎖状、分岐状、フォーク状、マルチアームからなることができる。本発明によって使用されるPEGは、特に限定されない。特に有用なPEGは、3,000〜20,000の範囲に、分子量を持つ。パブリックドメイン(すなわち、決して特許権を得なかったか、現在は特許期限切れ.)にあるいくつかの有用なPEG分子がある。他の有用なPEG分子は、WO03/040211、US6,566,506、US6,864,350、およびUS6,455,639に開示されている。
【0028】
本発明の別の実施形態において、生理学的に許容し得るポリマーはポリシアル酸(PSA)とその誘導体である。PSAはUS4,356,170(本明細書中に援用)に記載の方法によってVWFに結合することができる。本発明の一実施形態において、多糖化合物を、天然に存在する多糖類、天然に存在する多糖類の誘導体または天然に存在する多糖誘導体することが可能である。化合物の多糖部分は、5以上(概して少なくとも10)のシアル酸残基をポリマー鎖に有し、別の実施形態では少なくとも20〜50のシアル酸残基をポリマー鎖に有する。すぐに利用できる多糖化合物は全体で最高500のサッカライド残基を持つと考えられ、通常ポリマー鎖で300未満の残基を持つものであってもよい。通常、化合物のサッカライド残基の全てが、シアル酸残基である。
【0029】
多糖化合物で少なくともポリシアル酸部分、また一実施形態では全ての化合物で、高度に親水性である。親水性は、水酸基と同様に、シアル酸単位のペンダントカルボキシル基によって主に与えられる。サッカライド単位は、他の官能基(例えばアミン、ヒドロキシル基または硫酸塩基)またはその組合せを含むものであってもよい。これらの基は天然のサッカライド化合物上に存在可能であり、あるいは派生的な多糖化合物に導入されるものであってもよい。
【0030】
本発明の特定用途の多糖化合物は、細菌によって産生されるものである。これらの天然の多糖の一部は、グリコリピドとして知られている。特に有利なことは、多糖化合物が末端ガラクトース単位を実質的に有しない場合であり、このことは肝細胞およびクッパー細胞のガラクトース受容体によって認識される傾向にある。
【0031】
VWFマルチマーは、当業者に知られている種々の技術のいずれかによって、多糖化合物に共有結合可能である。実施例は、VWFまたは多糖のいずれか一方の上にあるカルボキシル基と他方のアミン基との間のペプチド結合、あるいは一方のカルボキシ基と他方の水酸基との間のエステル結合を介した結合を含む。あるいは、シッフ塩基は一方のアミノ基と他方のアルデヒド基との間に形成することができる。結合の他の機構は、当該技術分野の通常の技量内である。種々の例が米国特許第5,846,951号の第7欄15合目から第8欄第5行目までに同定される。
【0032】
本明細書中で用いられる場合、一つ以上の生理学的に許容し得るポリマー分子に結合しているVWFへの言及は任意の適当な化学的結合、例えば共有結合または非共有結合(例えばイオン性、疎水性、親和性、生物親和性相互作用)を含む。ポリマーを、二価性試薬の使用によって、またスペーサーアームを経てタンパク質に連結させることもできる。加えて、ポリマー分子は親和性相互作用によってVWFに連結することができる。例えば、VWFをビオチン標識させることができ、アビジンまたはストレプトアビジン結合ポリマーをVWFに結合させることができる。また、ポリクローナルまたはモノクローナル抗VWF抗体およびそのフラグメントをポリマーに結合することができ、この複合体をVWFに結合させることができる。ポリマーを酵素的方法、例えばUS6,379,933に教示されるポリグリコシルトランスフェラーゼによるサッカライドの転移またはUS20040132640A1に教示されるものとしてのグリコペジレーション(glycopegylation)の転移によってもVWFに結合させることができ、それらの特許文献を本明細書中に援用する。もう一つのアプローチは、VWFのA1およびA3領域に対するPEG付加コラーゲンまたはコラーゲンフラグメントの結合のようなそれらの生体機能を基礎としたVWFに対するポリマーの結合である。この目的のために、VWFで強い相互作用を示すIおよびIII型(例えばヒト胎盤由来)由来コラーゲンを使うことができる。ポリマーの結合は、タンパク質性構築物の生体内使用の後、安定または可逆的であると思われる。
【0033】
本明細書中で用いられる場合、「PEG付加VWF」は一つ以上のPEGに結合するVWFを含み、また本明細書中で用いられる場合、「PEG付加(PEGylation)」はVWFに一つ以上のPEGを結合させるプロセスを含む。PEG付加の適当な方法は、米国特許第5,122,614号および第5,539,063号(PEG付加方法の全てを本明細書中に援用)に開示されている。
【0034】
本発明の実施形態によれば、生理学的に許容し得るポリマーはPEGまたはPEG誘導体であり、当該技術分野で知られている任意の戦略と方法によってVWFに共有結合する。最も頻度が高い修飾戦略は、リジン残基のアミノ基を経た少なくとも1つのポリマー分子の結合、糖質側鎖を経た少なくとも1つのポリマー分子の結合、スルフヒドリル基を経た少なくとも1つのポリマー分子の結合、水酸基の少なくとも1つのポリマー分子の結合と同様にアスパラギン酸およびグルタミン酸のカルボキシル基を経た少なくとも1つのポリマー分子の結合、ならびにN末端の少なくとも1つのポリマー分子の結合である。
【0035】
本発明の一実施形態において、VWFに対する少なくとも1つのポリマー分子の結合は、上記ポリマー分子をVWFのリジン側鎖のアミノ基に連結される共有結合によって行うことができる。ヒトVWFは側鎖でNH2基とともに遊離の108リジン残基を含み、少なくとも1つのポリマー分子の結合の影響を受けやすい。ポリマー分子が本発明によって共有結合であるVWFのリジン残基の実施例を図1Aに示す。VWFのリジン残基に共有結合である可能性がある適当なPEG誘導体は、例えば、活性N−ヒドロキシスクシンイミドエステル(NHS)(例えばスクシンイミジル琥珀酸エステル、スクシンイミジルグルタラートまたはスクシンイミジルプロピオン酸)を有するポリエチレングリコールであり、それらはアミド結合を形成することによって、温和な条件下でリジン残基と反応する。活性PEGの他の例として、活性カーボネート(例えばスクシンイミジルカーボネート(SC−PEG)、およびベンゾトリアゾールカーボネート(BTC−PEG))を持つものが挙げられる(Roberts et al.,Advanced Drug Delivery Reviews 54:459−476,2002の463ページを参照)。SC−PEGおよびBTC−PEGは、カルバミン酸塩結合を形成するためにリジン残基に優先して反応するが、ヒスチジンおよびチロシン残基と反応することも知られている。別の方法は、ウレタン結合を形成するPEGカーボネートまたは第二級アミンを形成するアルデヒドまたはケトンと、例えばナトリウムシアノホウ化水素による還元の後、少なくとも1つのポリマー分子との結合である。ウレタンを生産する他の試薬をアシル化しているPEGは、タンパク質を連結したp−ニトロフェニルカーボネート、トリクロロフェニルカーボネート、およびカルボニルイミダゾールを含む。これらの試薬は、モノメトキシPEG(mPEG)(前述のRoberts et al.の464ページを参照)上で、末期の水酸基とクロロギ酸エステルまたはカルボニルイミダゾールとを反応させることによって調製される。もう一つの実施例は、所謂「第二世代」PEG付加化学に関するもので、それによって、酸性条件下のmPEG−プロピオンアルデヒドが、N末端a−アミンに対して選択的である(上記のRoberts et al.の464ページを参照)。遊離可能なPEG試薬(例えばPEG無水マレイン酸、mPEGフェニルエーテルスクシンイミジルカーボネート、およびmPEGベンズアミドスクシンイミジルカーボネート)は、結合体の生産に用いることが可能である。上記のRoberts et al.の469ページに記載のように、結合体は、生理学的条件下において「非標識(ノータグ)」で治療的タンパク質を遊離する。
【0036】
本発明のさらなる実施形態において、VWFはその糖質残基を経て少なくとも1つのポリマー分子に結合することができる。このことは、例えば糖質のマイルド酸化(例えば、NaIO4による)によって、実施することができ、アルデヒド機能を形成するとともに、その後PEG(例えばPEG−ヒドラジド)に対して連結する。この手法の長所は、以下の事実に基づく。すなわち、VWFのA1およびA3ループは、コラーゲン結合部位を含むことから、それらはVWFの生物学的活性について重要な領域であり、また糖質残基を含まずに、図1Bで分かるように、この手法によって修飾されない場合もある。この図は、アスパラギンN結合型GIcNAcとスレオニンまたはセリンO結合型GaINAc残基の実施例を表し、それらは酸化型であり、その後少なくとも1つのポリマー分子によって結合し得る。VWFの糖質が特定の領域に集まるという事実のため、VWFはこの修飾手法の使用によって少なくとも1つのポリマー分子と直接結合する部位であると考えられる。VWFの治療的活性形態が求められる場合、糖質残基に対するポリマーの結合が特に有利である。これは、既知の方法によってN結合型残基またはO結合型残基のいずれかに対してポリマーが選択的に反応することによって高められる。例えば、酵素グルコースオキシダーゼは複数の反応性アルデヒド基を生成するためにVWF上の糖質残基を酸化する際に用いることができ、PEG−ヒドラジドと反応してヒドラゾン結合を生産することができ、またはPEG−アミンと反応して可逆性シッフ塩基を生産することができる(前述のRoberts et al.の467ページを参照)。特定の状況下では、VWFのN末端セリンまたはスレオニンを、過沃素酸酸化によってグリオキシル誘導体に変換することによって、部位特異的結合に用いることができる(前述のRoberts et al.の467ページを参照)。
【0037】
本発明の別の実施形態は、スルフヒドリル基を経たVWFに対する少なくとも1つのポリマー分子の結合である。ヒトVWFは、例えばPEGマレイミドによって、177個の遊離SH基(修飾し得る)を持ち、安定なスルフィドを形成している。システイン残基のPEG付加は、例えば、PEG−ビニルスルホン、PEG−ヨードアセトアミドまたはPEG−オルソピリジルジスルフィドを使用して、行うことが可能である(前述のRoberts et al.の466ページを参照)。
【0038】
その複数の機能によって、VWF分子は、特異的な受容体またはリガンドに対するいくつかの結合部位を有する(Girma et al.,Thromb Haemost.74:156−60,1995)。1つの重要な結合部位はFVIII結合領域であり、成熟サブユニット(アミノ酸1〜272)のN末端に位置する。このエピトープを、遊離FVIIIによるインキュベーションと複雑なFVIII/VWFの形成にとよって保護することができる。その後、複合体を化学的に修飾(PEG付加またはポリシアル酸付加)し、遊離FVIII結合部位によるポリマー結合VWFを、FVIIIから分離する(例えば0.3MCaCl2または2MNaClによるサイズ排除クロマトグラフィーによる)。同様に、VWFを固定FVIIIを持つ親和性樹脂に結合させることができる。その後、VWFとポリマー(例えばポリエチレングリコールまたはポリシアル酸誘導体)とから化学的に結合させ、バッチモードまたはクロマトグラフィーカラムの使用により、このマトリックス(例えば高塩条件下、例えば0.3MCaCl2または2MNaCl)から溶出させる。
【0039】
VWFのFVIII結合エピトープは、ヘパリン結合部位位とほとんど同一である。このように、FVIII結合部位はヘパリンに対するVWFの結合または固定化ヘパリンを有する親和性樹脂に対するVWFの結合によって、化学修飾法の過程でブロックおよび保護される。
【0040】
本明細書中で用いられる場合、用語「第VIII因子結合部位保護剤」とは、VWF分子上でFVIII結合領域またはエピトープと結合する任意の薬剤のことをいう。第VIII因子結合部位保護剤は、第VIII因子、FVIIIの誘導体、ヘパリン、およびヘパリン誘導体から選択可能である。
【0041】
本発明は、少なくとも1つの生理学的に許容可能なポリマー分子に連結されないVWFの生体内半減期と比較して、VWFまたはその生物活性誘導体の生体内半減期の増大に関する。本発明の一実施形態において、別の実施形態で生体内半減期が少なくとも3倍増大している一方で、VWFの生体内半減期は少なくとも2倍長くなる。さらに別の実施例において、生体内半減期は少なくとも1つの生理学的に許容可能なポリマー分子の結合によって、5倍に増大する。下記の実施例7に記載のように、VWF半減期の増大または延長を、FVIII欠乏マウスでVWFの薬物動態学を計測することによって評価することができる。手短に言うと、FVIII欠乏マウスは尾静脈を経てFVIIIで予備混合されるVWFの大量瞬時投与で処置され、VWF抗原量は種々の時点で、血漿サンプルで計測される。VWF抗原(FVIII抗原と同様に)を、ELISAアッセイを介して計測することができる。
【0042】
本発明のさらなる態様は、FVIIIの生体内半減期がポリマーVWF結合体によって哺乳動物血液中で長くなる、少なくとも1つのポリマーVWF結合体と少なくとも1つのFVIII分子との間に形成される複合体に関する。
【0043】
ポリマーVWF結合体とFVIIIとの結合は、FVIIIの生体内半減期と比較すると、上記FVIIIの生体内半減期を延長または増加させ、VWFを少なくとも1つの生理学的に許容可能なポリマー分子に連結していない複合体を形成する。本発明の一実施形態において、FVIIIの生体内半減期は、少なくとも1.5倍であり、別の実施形態では少なくとも2倍、別の実施形態では少なくとも3倍、さらに別の実施形態では少なくとも5倍である。
【0044】
本発明のポリマーVWF結合体を、血友病Aおよび/またはVWDの処置またはこれらの2つの疾患の亜型の処置に使用することができる。VWDの場合、ポリマーVWF結合体の使用は、予防プロトコルまたはオンデマンド処置とも各々呼ばれる規則的または不規則な処置によってコンセントレートを含むVWFによる現在の補充療法と類似している。ポリマーVWF結合体を、血友病A予防のアジュバント処置として用いることもできる。これらの処置状況下で、ポリマーVWF結合体は、血漿由来または組換え型のFVIIIコンセントレートとは別個に時間間隔で投与され、血友病Aの規則的な処置のために現在使われる同じことが通常通り与えられるが、ポリマーVWF結合体の半減期延長能力による長期間の処置間隔による。
【0045】
血友病AおよびVWDの予防と血友病AおよびVWDの急性出血の処置とに関する本発明の一実施形態では、血友病AまたはVWDを有する患者に、ポリマーVWF結合体をFVIIIと共に投与するか、あるいはFVIIIで複合体の形で投与する。このような場合、体内または外部の出血の停止が、治療的に有効なレベルにVWFおよび/またはFVIIIのいずれかのさもなければ低い血漿レベルを上げることで、直ちに達成することが必要である。ポリマーVWF結合体を免疫寛容療法に用いて、阻害抗体を根絶することもでき、該阻害抗体はFVIII(凝血抑制因子増加性血友病としても公知の臨床状況、またはVWFに対して)に対して発現した。このような状況下で、FVIIIの超生理学的および超薬理学レベルを、FVIIIまたはVWFに対する発現した阻害因子を有する患者に投与する。治療のこの形態は、上記製剤を投与された患者の血行で通常より高い回復とVWFより長い持続性とを持っているポリマーVWF結合体の使用によって促進される。
【0046】
治療の技術水準によれば、また国際的なガイドラインと規則に従って、注入されたFVIIIの薬物動態学が認識され、有効性のための妥当な代用のマーカーと認められる。このことは、機能的な活性のために標準検査によって特徴づけられる注入されたFVIII産物が血流量で見いだされること、またテナーゼ複合体(FIXaおよびリン脂質に結合することによる第X因子の活性化複合体)のコファクターとして予想されたようにそこで作用するという、確証された仮定に基づく(Elodi et al.,Thromb.Res.21:695−700,1981)。したがって、動物モデルの任意の薬動力学の分析は、FVIII産物で処置される患者で、期待される有効性を予測する。
【0047】
FIXコファクター活性を判定するために、FVIIIまたはFVIIIa試料(トロンビンで完全に活性化されるFVIII)を、FIXa、FX、燐脂質、およびCaCl2からなる調製された混合物に加える。この反応混合物を37℃でインキュベートして複合体形成を可能にするとともに、その後のFXa生成を可能にする。副試料を最高20分の間隔で採血し、FXaによって選択的に分けられる色素基質に加える。15分のインキュベーションの後、酢酸を添加することで反応を停止させる。吸光度(A405)値(それはFXa濃度に比例)をELISAリーダーで計測し、反応混合物のインキュベーション時間に対してプロットする。
【0048】
本発明のさらなる態様は、FVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害がある哺乳動物血液でFVIIIの生体内半減期を延長する方法の提供であり、該方法は、
(a)上記に定義された少なくとも1種類のタンパク質性構築物を提供するステップと、
(b)上記に定義した少なくとも1種類のFVIIIを提供するステップと、
(c)上記タンパク質性構築物と上記FVIIIとの間の複合体を形成するステップと、を有する。
【0049】
上記の方法の一実施形態では、ステップ(c)の複合体は、例えばタンパク質性構築物と上記FVIIIとを混合することによって「体外」(すなわち、哺乳動物の体外)で形成され、このように形成された複合体を上記出血障害がある哺乳動物に対して有効量投与する。
【0050】
上記の方法のさらなる実施形態では、ステップ(c)の複合体は、上記哺乳動物に有効な量でタンパク質性構築物を投与すると即座に、上記出血障害がある哺乳動物血液中に存在するタンパク質性構築物と内因性FVIIIとの間に体内(すなわち哺乳動物の体内)で形成される。
【0051】
上記の方法のさらに別の実施例では、ステップ(c)の複合体は、上記哺乳動物に有効な量でタンパク質性構築物を投与すると即座に、上記出血障害がある哺乳動物血液で存在する外因性FVIIIとタンパク質性構築物との間に「体内で」形成される。外因性FVIIIを有効量で上記タンパク質性構築物と同時に、または経時的に、例えば上記タンパク質性構築物の前または後に、投与することが可能である。
【0052】
本明細書中で用いられる場合、「内因性FVIII」は上記哺乳動物から生じるFVIIIを含む。上記哺乳動物に存在する導入遺伝子または任意の他の外来性DNAから転写されるFVIIIも含む。本明細書中で用いられる場合、「外因性FVIII」は既に概説されたpdFVIIIおよびrFVIIIを含むFVIII(上記哺乳動物からは生じない)を有するとともに、pdFVIII(上記の定義された複合体を形成した後に単離された哺乳動物に再投与される)、さらにrFVIII(DNAが上記rFVIIIの産生で使われた哺乳動物に投与される)が挙げられる。
【0053】
本明細書中で用いられる場合、上で概説されるように、「有効量」は出血障害がある哺乳動物を処置するための適当な用量を含み、例えば、ヒトに関して、好ましくは1回の注入あたり5〜1,000IU、より好ましくは1回の注入あたり10〜250IUの範囲内である。
【0054】
投与経路は特に制限されるものではなく、一実施形態において、タンパク質性構築物または本発明の複合体を注射(例えば静脈内、筋内、または腹腔内注射)によって投与することも可能である。
【0055】
本発明も、本明細書中で用いられるFVIIIおよびVWFの少なくとも1つの機能的な欠陥または欠乏を伴う出血障害を処置することに関し、出血障害の原因が、FVIIIおよび/またはVWFの短くなる生体内半減期、FVIIIおよび/またはVWFの改変された結着性、FVIIIおよび/またはVWFの遺伝子欠損とFVIIIおよび/またはVWFの減少した発現からなる群から選択されるものであってもよい出血障害が含まれる。本発明の一実施形態において、出血障害は血友病A、VWDまたはVWFの障害のある機能または他の分子によるVWFの障害のある相互作用に関連した他の疾患からなる群から選択される。
【0056】
さらに、本発明は上記に定義されたタンパク質性構築物の有効量または上記に定義された複合体の有効量を含む医薬品組成物に関する。医薬品組成物は、薬学的に許容可能な担体、希釈剤、塩、緩衝液、または賦形剤をさらに含む可能性がある。医薬品組成物が、上記に定義された出血障害を処置するために使用し得る。本発明の医薬品組成物は、溶液または凍結乾燥された産物である可能性がある。タンパク質と特にVWFとFVIIIの安定溶液を形成する多くの既知の方法がある。例えば、米国特許第6,586,573号、第5,565,427号、第5,763,401号、第5,733,873号、第4,877,608号、第5,605,884号、および第5,328,694号が挙げられる。これらの溶液は任意の適当な凍結乾燥プロセス(例えばUS 6,586,573に記載のプロセス)にかけることができ、そのことは本明細書中に援用する。本発明は、単独ポリマーVWF結合体単独またはFVIIIと組み合わせた他の適当な形態を含む。
【0057】
本発明は、以下の実施例でさらに詳しく説明されるが、それらになんら限定されるものではない。
【実施例】
【0058】
(実施例1:糖質残基の修飾によるポリマーVWF結合体の調製)
糖質残基(図1B)を介してポリマーVWF結合体を調製するために、rVWF(最終濃度:500μg/ml)の溶液を20mM酢酸ナトリウム緩衝液(pH6.0)で調製し、NaIO4を糖質残基の酸化のために添加した(5mM最終濃度)。酸化を4℃で20分間行い、亜硫酸水素ナトリウム(5mM最終濃度)を添加して反応を停止させた。その後、mPEGヒドラジド(鎖長:3kD)を添加し(10mM最終濃度)、さらにVWFのPEG付加を室温で1時間行った。次に、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラム(寸法26mm×840mm)に加え、PEG付加VWFを20mM HEPES緩衝液(150mM NaCl、pH7.4、5%トレハロース含有)を用いて、試薬から分離した。280ナノメートルVWF抗原量およびODの計測によって示されるように、修飾VWFをボイド容量で溶出させた。VWF含有分画を、さらに精製するために、EMD TMAE 650M(Merck)充填陰イオン交換カラムに直接載せた(寸法:10mm×108mm)。次に、PEG付加VWFを20mM HEPES緩衝液(5%トレハロースおよび1000mM NaCl含有)で溶出させた。
【0059】
(実施例2:mPEGスクシンイミジルスクシネートによるVWFのリジン残基のPEG付加)
リジン残基(図1A)を介したVWFのPEG付加のために、rVWF(最終濃度:500μg/ml)溶液を20mM HEPES緩衝液(150mM NaCl、pH7.4、5%ショ糖含有)で調製し、mPEGスクシンイミジルスクシネート(鎖長:5kD)を添加した(10mM最終濃度)。VWFを室温で1時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、修飾VWFをボイド容量で溶出させた。VWF含有分画を、さらに精製するために、EMD TMAE650M(Merck)充填陰イオン交換カラムに直接載せた(寸法:26mm×840mm)。次に、PEG付加VWFを20mM HEPES緩衝液(5%ショ糖および1000mM NaCl含有)で溶出させた。
【0060】
(実施例3:mPEGp−ニトロフェニルカーボネートによるVWFのリジン残基のPEG付加)
mPEG p−ニトロフェニルカーボネートによるVWFのPEG付加のために、血漿由来VWF(最終濃度:500μg/ml)を20mM HEPES緩衝液(150mM NaCl、pH7.6、5%ショ糖含有)で調製し、mPEG p−ニトロフェニルカーボネート(鎖長:2kD)を添加した(最終濃度:10mM)。VWFを室温で2時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、VWFをボイド容量で溶出させた。
【0061】
(実施例4:mPEGマレイミドによるVWFのスルフヒドリル残基のPEG付加)
mPEGマレイミドによる遊離SH残基を介したVWFのPEG付加のために、rVWF溶液(最終濃度:500μg/m)を20mM HEPES緩衝液(150mM NaCl、pH7.6、4%マンノースおよび1%トレハロース含有)で調製し、mPEG マレイミド(鎖長:10kD)を添加した(最終濃度:10mM)。VWFを室温で2時間、PEG付加した。その後、PEG付加VWFをサイズ排除クロマトグラフィーによって精製した。反応混合物をセファクリルS−300HR(Amersham)充填クロマトグラフィーカラムに載せ、PEG付加VWFを、PEG付加反応に使用されるのと同じ緩衝系によって分離した。VWF抗原量およびOD280nmの計測によって示されるように、修飾VWFをボイド容量で溶出させた。
【0062】
(実施例5:デキストランとVWFとの結合)
6mg/mlのデキストラン(MW40kD)溶液を20mM酢酸ナトリウム緩衝液(pH6.0)で調製し、NaIO4を添加した(最終濃度10mM)を添加して遊離アルデヒド基を生成した。酸化を暗所で4℃、1時間実施し、亜硫酸水素ナトリウム(最終濃度5mM)を添加して反応を停止させた。活性化デキストランを0.15MNaCl(PBS緩衝液)含有0.1Mリン酸ナトリウム緩衝液(pH7.2)で透析した。次に、この活性化デキストラン溶液2.4mlを10mlのrVWF溶液(濃度:PBS緩衝液中0.6mg/ml)に添加した。この混合物に、5mlのシクラミン酸ナトリウム溶液(PBS緩衝液中64mg/ml)を添加し、暗所で室温、一晩にわたってインキュベートした。次に、3mlの1.0MTRIS−HCl溶液(pH7.2)を添加して残留アルデヒド基をブロックし、室温で1時間インキュベートし、さらに20mM HEPES緩衝液(pH7.4、5%ショ糖含有)でインキュベートした。次に、rVWF誘導体に結合したデキストランをさらに、セファクリルS−300HR(緩衝液:20mM HEPES、5%ショ糖、pH7.4)を充填したクロマトグラフィーカラム(寸法:50mm×860mm)に混合物を載せることで、サイズ排除クロマトグラフィーにより精製した。rVWF誘導体を、VWF抗原量およびOD280nmの計測によって示されるように、VWFをボイド容量で溶出させた。これらの分画を回収し、100kDの再生セルロース膜(Millipore)を用いて、限界濾過法によって濃縮した。
【0063】
(実施例6:VWDマウスにおける薬物動態)
Denis et al.(PNAS 95:9524−9529,1998)に詳述されているVWF欠乏マウスを、重度III型VWD類似ヒトVWDモデルとして使用した。マウス5匹からなる群に対して、PEG−rVWF(鎖長3kD、実施例1によるrVWFのPEG付加)または対照としてのネイティブrVWFを、PEG付加後、検出可能なVWF(ELISA)に基づいて、40単位(U)VWF:Ag/kg体重の用量で、経尾静脈ボーラス注入法により投与した。PEG−rVWF群を、注射後5分、30分、1時間、2時間、6時間、10時間、および24時間(対照群の場合、5分、15分、30分、1時間、2時間、4時間、6時間、10時間、および24時間)麻酔し、クエン酸血漿を心臓穿刺から調製した。血漿でVWF抗原量を追跡した。この実験の結果を図2にまとめる。ネイティブrVWFを、文献(Lenting et al.,J.Biol.Chem.279:12102−12109,2004)に記載されているように、典型的な二相法で血行から除去することで、600分と1440分(10時間および24時間に等しい)との間の検出限界以下に落とす。対照的に、注射時の0から注射後10時間の約0.6U/ml血漿までの初期増加後のPEG付加rVWFは、依然として注射後24時間であっても約0.4U/mlの実質的に高いレベルに存在し、10時間ないし24時間の範囲では平坦な勾配となったことから、PEG付加rVWFがより長期にわたって持続することが示される。経時的な計測可能VWFの漸増は、VWFによって結合体を形成したポリマーPEGの結合の可逆性を示すもので、ポリマーからの放出後、計測のためにアクセス可能になる。このモデルでのPEG−VWFの長い循環時間は、この調製物がVWDの予防療法に使用し得ることを示す。
【0064】
(実施例7:FVIII−K.O.マウスにおける薬物動態)
Bi et al.(Nat.Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々をrFVIIIと事前に混合して3UFVIII/mlおよび3UVWF:Ag/mlを達成するためにPEG−rVWF(鎖長3kD、実施例1によるrVWFのPEG付加)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(13ml/kg)を行った。麻酔後、生成物を注射してから5分および6時間後に、心臓穿刺によってクエン酸血漿を各々の群から調製した。対照群に対しては緩衝液を注射し、注射5分後に出血させた。VWF抗原量を血漿サンプルで計測した。この実験の結果を図3にまとめた。曲線は、VWFのベースレベルの近くまで落ち込んだ、FVIII欠乏マウスに存在するrVWFの典型的除去を示し、その一方でPEG付加rVWFの使用後、レベルが6時間の観察期間中に増加したことを示す。このことは再び、ポリマーPEGの結合の可逆性がVWFと結合体を形成したことを示し、ポリマーからの放出の後、計測のためにアクセス可能になり、適用後360分(6時間)経過しても増加を持続することを示している。
【0065】
(実施例8:VWDマウスでのFVIII増加)
Denis et al.(PNAS 95:9524−9529,1998)によって詳述されるフォンビルブラント欠乏マウスを、重度III型VWD類似ヒトVWDモデルとして使用した。4〜5匹のマウスからなる群に対して、PEG−rVWF(鎖長5kD、実施例2によるrVWFのPEG付加)含有20mM HEPES(150mM NaCl、5%ショ糖、pH7.4)またはネイティブrVWFを、尾静脈を経る静脈内注射した。対照群を緩衝液で処置した。(ELISA換算PEG−rVWF−用量18UVWF:Ag/kg、2700μg/kg、rVWFネイティブ用量:2400μg/kg)。
【0066】
各マウスに対して、10ml/kg体積用量を投与した。PEG−rVWF投与後の時点(5分、1時間、3時間、6時間、10時間、24時間、および48時間)またはネイティブrVWF投与後の時点(5分、15分、30分、1時間、2時間、6時間、24時間、および32時間)の注射の後の時点で、マウス4〜5匹からなる群を麻酔し、クエン酸血漿を心臓穿刺から調製し、FVIII活性(ハウスアッセイにおける発色)のレベルを血漿で追跡した。対照群を注射15分後に出血させた。この実験の結果を図4にまとめた。
【0067】
マウスにおける内因性FVIIIのレベルは、rVWF注入の結果として増加する。PEG付加rVWF適用後の血中濃度時間曲線下面積(AUC)は、rVWF使用後のたったの3.3U*h/mlと比較して、8.0U*h/mlであった。これは、PEG付加rVWFのための実質的により長い循環時間を示す。結果は、PEG付加VWFがVWDにおける二次FVIII欠乏の予防療法に使用し得ることを示す。
【0068】
(実施例9:FVIII−K.O.マウスでのrFVIIIおよびVWFの回収)
Bi et al.(Nat Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々をrFVIIIと事前に混合して3UFVIII/mlを達成するためにPEG−rVWF(HZ−PEG、3K、糖質を介して結合)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(13ml/kg)を行った。麻酔後、注射してから5分および6時間後に、心臓穿刺によってクエン酸血漿を各々の群から調製した。FVIII活性およびVWF抗原回収量を血漿サンプルで計測した。この実験の結果を図5および図6にまとめた。
【0069】
無処置のrVWFと比較して、両調製物のPEG付加rVWFは、同時注射されたrFVIllのより高度な回収をもたらした。経時的にPEG付加rVWFに対するVWFレベルが上昇し、その一方で正常rVWFが360分以内に完全に除去された。結果は、FVIIIと複合体を形成したPEG付加VWFを、FVIII循環時間の増大という利点をもって血友病Aの急性処置に用いることができることを示している。
【0070】
(実施例10:FVIII×VWFダブルノックアウトマウスにおけるFVIII半減期の増大)
FVIII×VWFダブルノックアウトマウスは、FVIII欠乏およびVWF欠乏マウスの交雑育種によって得られた。それらのマウスはVWF欠乏と同様にFVIII欠乏を患う。このように、動物モデルにおけるFVIII−VWF相互作用を研究するための理想的なモデルを提供する。
【0071】
FVIII×VWFダブルノックアウトマウス(FVIII欠乏マウスをVWF欠乏マウスと交雑育種した)5匹からなる群に対して、PEG−rVWF(鎖長5kD、rVWFのPEG付加を、mPEGスクシンイミジルスクシネートのリジン残基の修飾によって、実施例2により行った)含有20mM HEPES(150mM NaCl、5%ショ糖、pH7.4(またはネイティブrVWFSS−PEGまたはネイティブrVWF(各々をrFVIIIと事前に混ぜ合わせ、9UFVIII/mlおよび9UVWF抗原/mlおよび0.67UVWF:RCo/mlを達成)を、経尾静脈によりボーラス注入(11ml/kg)を行った。公開(Ingerslev(Scand.J.Clin).Invest.47:143−149(1987))されているようにして、VWF−抗原値をELISA法で計測した。一次止血のプロセスにおけるVWFの血小板結着性を反映している機能的なVWF:RCo活性をMacfarlane et al.(Thromb.Diath.Haemorrh.34:306−308,1975)により、計測した。注射後5分、3時間、6時間、10時間、および24時間、麻酔後の心臓穿刺によるクエン酸血漿を、それぞれの群から調製した。FVIII活性およびVWF抗原量を、血漿サンプルで計測した。
【0072】
FVIIIおよびVWFの半減期を、薬物動態学ライブラリーの1つの区画をモデルとして使用するMicroMath科学者プログラム(Micromath Research,Saint Luis,MO,US)を用いて算出した。rVWFまたはPEG付加rVWFのいずれかと同時注入されたFVIIIの半減期は、1.88時間から2.58時間へと増大し、血中濃度時間曲線下面積(AUC)は4.3から7.3U*h/mlへと増大した。VWFの半減期は、3.1から10.4へと増大し、血中濃度時間曲線下面積は、5.7から22.8に増やされる。結果を図7および図8にまとめた。データによれば、PEG−VWFを、VWDおよびFVIIIの循環時間を長くするという利点とともに、血友病AおよびVWDの急性処置および予防処置のために使用し得ることが示された。
【0073】
(実施例11:マウス血漿におけるPEG付加の可逆性の実証)
PEG付加の可逆性を、VWF欠乏血漿による試験管内実験によって示した。クエン酸血漿を、4℃、15分間、1100xgの遠心によって、VWF欠乏マウスから得た(Denis et al.PNAS 95:9524−9529,1998)。4容量のマウス血漿を、実施例1(糖質を経たPEGカップリング)または実施例2(リジン残基を経たPEGカップリング)によって調製し、48時間にわたり37℃に保ち、1容量のPEG付加rVWFと混ぜ合わせた。非PEG付加rVWFが、両方の実験における対照として用いた。副試料を混合直後、ならびに1時間、5.5時間、24時間、および48時間後に回収し、VWF抗原量を、サンドイッチELISAシステムを用いて凍結試料からアッセイした。ポリクローナル抗VWF抗体(DAKO)を用いて、96穴ELISAプレートを被覆し、ヤギ−抗ウサギ−lgG−HRP−結合体(AXELL)を結合VWF因子検出のためにアッセイした。経時的にVWF抗原の増加性量を計測し、生体外血漿サンプルにおける場合も含めVWFに対するポリエチレングリコールの結合の可逆性が示された(図9)。
【0074】
(実施例12:PEG付加VWF調製試料のFVIII結合能の側定)
異なるPEG付加rVWF調製試料のFVIII結合能を、BIACORE(登録商標)3000装置(BIACORE、Uppsala、Sweden)を使用して表面プラズモン共鳴実験によって比較した(Karlsson et Falt,J.Immunol.Methods 200:121−33,1997)。通常、リガンドをセンサチップに固定し、該リガンドに対する他の構成要素の結合を表面プラズモン共鳴によって測定する。この技術の使用によって、チップ表面の近くの溶液の屈折率の変化が計測される。チップの表面の結合した構成要素の濃度変化を信号として検出し、それを任意の共鳴音単位(RU)で表す。固定リガンドに結合したタンパク質量と観察されるRUとのあいだに、比例関係が存在する。PEG付加VWF調製試料を、7000〜9000RUおよび25℃で、NHS/EDC化学を用いて、BIACORE(商標)センサチップのデキストラン表面に25℃で固定した。150mM NaCl、3mM EDTA、および0.005%界面活性剤P20(HBS緩衝液、BIACORE)を含む10mM HEPES緩衝液(pH7.4)を流速15μl/分で使用した。図10に例示されるように、市販のFVIII産物(ADVATE、Baxter AG、Vienna、Austria)の異なる量の結合が計測された。この図は、mPEGマレイミド5000で修飾され、かつ実施例4によって調製されるPEG付加rVWF調製試料のFVIII結合能を示す。異なるPEG付加rVWF調製試料のBIACORE実験の結果を表1にまとめている。この表では、PEG付加rVWF調製試料の異なるFVIII結合能は、10〜20IUFVIII/ml(色素アッセイ)の範囲にある参照の最大レベルでの非PEG付加参照調製試料のRU値の割合として与えられる。
【0075】
【表1】
(実施例13:ポリマー結合後のVWFの質量増加)
rVWFを、種々の濃度(1mM、2.5mM、5mM、7.5mM、および10mM)でmPEGスクシンイミジルスクシネート(鎖長:5kD)を使用して、実施例2により、PEG付加した。PEG付加VWF種を2つの異なる方法で分析した。すなわち、SDSポリアクリルアミドゲル電気泳動およびVWFマルチマー分析である。SDSゲル電気泳動を、3〜8%勾配ゲルを使用して還元条件下で行った(Tris Acetat Gel/Bio−Rad)。VWFマルチマー分析を、1.6%のアガロースゲルを使用してRuggeri et Zimmerman(Blood 57:1140−43,1981)により行った。VWFマルチマーの明視化を、Aihara et al.(Thromb.Haemost.55:263−67,1986)によって実施した。
【0076】
SDSゲル電気泳動(図11)は、成熟VWF(下部のバンド)とプロVWF(上層ブライトバンド)とからなるrVWF調製試料を示すとともに、異なる試薬濃度の使用によってPEG付加後の分子量が増加することを示す。PEG付加の後、ヒト血清アルブミン(HSA)調製試料(PEG付加は、実施例2によって実施)からの分子量の推移は、参照調製試料として示される。HSAの分子量が66,000のダルトンから190,000のダルトンまでシフトしていることで、PEG付加手法が有効であることが示されることが明らかになった。
【0077】
図12は、異なる試薬濃度でのPEG付加前後におけるrVWFの多量体パターンを示す。試薬濃度の上昇に伴って異なるマルチマーの分子量がより高いほうへ移行および広幅化することが明らかに示される。
【0078】
(実施例14:mPEGスクシンイミジルグルタレートによるVWFのリジン残基のPEG付加)
リジン残基(図1A)を介したVWFのPEG付加のために、rVWF(最終濃度:500μg/ml)を、20mM HEPES緩衝液(150mM NaCl、pH7.4、5%スクロース含有)で調製し、mPEGスクシンイミジルグルタレート(鎖長:5kD)を添加した(最終濃度:200mgPEGスクシンイミジルグルタレート/mgタンパク質)。つぎに、pH値を0.1MNaOHで7.4に合わせた。VWFを室温で1時間、PEG付加し、実施例2の記載に従って精製した。
【0079】
(実施例15:FVIII−K.O.マウスでのFVIII半減期の増大)
Bi et al.(Nat.Genet.10:119−121,1995)に詳しく記載されているFVIII欠乏マウスを、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、各々を組換え型FVIIIと事前に混合して10UFVIII/mlおよび10UVWF/mlを達成するために、実施例2により調製したPEG−rVWF(SS−PEG、5K)またはネイティブrVWFのいずれかによる経尾静脈ボーラス注入法による投与(10ml/kg)を行った。麻酔後、生成物を注射してから5分、3時間、9時間、および24時間後に、心臓穿刺によるクエン酸血漿を調製した。FVIII活性およびVWF抗原回収量を血漿サンプルで計測した。この実験の結果を図13および図14にまとめた。
【0080】
FVIIIのための半減期は1.8時間(ネイティブrVWF存在下)から3.9時間(PEG−rVWFと併用)へと増大し、血中濃度時間曲線下面積(AUC)が4.1から7.8U*hへと増大した。VWF半減期は3.2時間から13.6時間へと増大し、VWFのAUCは7.7から32.1U*hへとほぼ4倍になった。
【0081】
(実施例16:異なる方法によるSS−PEG付加VWF調製試料のFVIII結合能の測定)
異なるPEG付加rVWF調製試料のFVIII結合能を、ELISAと色素アッセイシステム(ECA)とを組み合わせ、Bendetowicz et al.(Blood 92:529−538,1998)の方法の変法によって計測した。マイクロタイタープレートを、200μlの2.6μg/mL抗vWFポリクローナル抗体含有50mmol/lNa2CO3/NaHCO3(pH9.6)で覆った。プレートを、PBS−Tween緩衝液(100mM Na2HPO4/KH2PO4、150mM NaCl、pH7.6と0.05%Tween20)で、各ステップの後に洗った。プレートを、0.1%粉ミルク/2mMベンズアミジン含有PBS−Tweenで37℃、1時間にわたりブロックした。VWFの増加量を37℃、25分間にわたり、0.2U/mlrFVIII(ADVATE,Baxter AG,Vienna,Austria)とともにプレインキュベートし、これらの混合物の100μlをプレートに添加した。インキュベーション後、捕獲されたVWFに結合したFVIIIの量を、FVIII色素アッセイ(Technoclone,Vienna,Austria)で計測した。FVIII結合能を、1分内で405nm(dA405)で計測される吸光度の変化として表した。図15は、2つのPEG付加VWF調製試料(両方ともmPEG−スクシンイミジルスクシネート(SS)で修飾)のVWF用量依存性FVIII結合を示す。修飾VWF調製試料のFVIII結合能は、非修飾開始VWF調製試料の%として算出され、PEG−SS−rVWF−1では20%およびPEG−SS−rVWF−2では50%であると算出された。
【0082】
図16は、実施例12に記載の表面プラズモン共鳴方法で計測される2つのPEG−SS−rVWF調製試料のFVIII結合能を示す。算出結合能は、それぞれ25および45%であった。
【0083】
SDS−PAGEで計測されるPEG−SS結合後のrVWF分子の分子質量における適当な増加を、図17に示す。
【0084】
(実施例17:分枝PEGマレイミドによるVWFにおけるスルフヒドリル基のPEG付加)
分枝PEGマレイミドによる遊離SH残基を経たVWFのPEG付加のために組換え型VWF(最終濃度:500μg/ml)の溶液を、20mM HEPES緩衝液(150mM NaCl、pH7.6、3%トレハロース含有)によって調製する。次に、NOF社(NOF Europe,Grobbendonk,Belgium)によって供給される分岐mPEGマレイミド(鎖長:20kD)を添加した(最終濃度10mM)。VWFを静かに撹拌しながら室温で2時間にわたり、PEG付加した。その後、PEG付加rVWFを、再生セルロース(Millipore)からなる100kD膜を使用して、限界濾過法/ダイアフィルトレーション(UF/DF)によって、試薬から分離する。
【0085】
(実施例18:mPEGヒドラジド/EDCによるVWFのカルボキシル基のPEG付加)
実施例23により、フリン成熟rVWFを調製および精製した。調製試料を50mMリン酸緩衝液(pH6.2)に対して透析し、400μg/mlの濃度まで希釈した。次に、鎖長5kDのmPEGヒドラジド(mPEG Hz)を添加した(濃度:60mg mPEG Hz/mgVWF)。500mM 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC)の新しく調製された溶液30μlを、1mlのVWF含有混合物の1mlに添加し、5時間にわたって静かに振とうさせながら室温でインキュベートした。100kD膜(再生セルロース/Millipore)を用いて、20mM HEPES緩衝液(150mM NaCl、pH7.4)に対して、UF/DFによりPEG付加rVWFから試薬を分離する。
【0086】
(実施例19:ポリシアル酸によるVWFのリジン残基の修飾)
ポリシアル酸(コロミン酸、CA)によるリジン残基の修飾を、Fernandes et Gregoriadis(Biochim.Biophys.Acta 1341:26−34,1997)およびJennings et Lugowski(J.Immunol.127:1011−1018,1981)に記載のようにして、実施した。0.1MNaIO4含有コロミン酸(濃度:20mg/ml)溶液を暗所、室温で15分間撹拌させて、CAを酸化した。活性化CA溶液1mlあたり2mlのエチレングリコールを添加し、暗所、室温でさらに30分間撹拌した。溶液を一晩にわたって暗所で0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して透析した。その後、この溶液のアリコートをrVWF溶液(400μg/ml)含有0.05Mリン酸ナトリウムに添加し、1mgVWFあたり50mg活性化CAの最終濃度を得た。この混合物を暗所、室温で30分間撹拌した。NaCNBH3を添加し(1mg/mgrVWF)、混合物を暗所、室温で18時間、静かに撹拌しながらインキュベートした。1MTRIS水溶液(pH7.2)を添加し(50μl/mgNaCNBH3)、1時間にわたって撹拌することで反応を停止させた。遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して、UF/DFによりrVWF−ポリシアル酸結合体から分離した。
【0087】
この調製試料のFVIII結合能を実施例16の記載通りに測定した。
【0088】
ポリシアル酸によるrVWFの結合を、還元条件の下で等電点電気泳動(IEF)によって検出される等電点(PI)の移動によって示した。図20は、この実施例によりポリシアル酸による結合の前後におけるVWF調製試料の比較を示す。製造元(Amersham Bioscience)の指示により、Ampholine PAGplateシステム(pH3.5ないし9.5)を用いた。ポリシアル酸との結合による酸性基の導入によって、修飾rVWFの1つの酸性バンドが生ずる。
【0089】
実施例16に記載のように、54%のFVIII結合能がECA試験を用いて側定された。70%のFVIII結合能を表面プラズモン共鳴方法(実施例12)を用いて側定した。
【0090】
(実施例20:剪断応力条件下のPEG付加)
Sakariassen et al.(J.Lab.Clin.Med.102:522−535,1983)に記載のように、灌流チェンバーを作製し、剪断応力条件下でのrVWFのPEG付加に使用した。rVWF(500μg/ml)含有20mM HEPES(pH7.4、3%トレハロース含有)の溶液を調製した。次に、mPEGスクシンイミジルスクシネート(最終濃度:5mM)を添加し、0.1MNaOHを添加してpHを7.4に調製し、直ちに灌流システムに充填した。次に、PEG付加はSakariassen et al.(J.Lab.Clin.Med.102:522−535,1983に記載のように、灌流条件下で、蠕動ポンプを使用することで、2500秒−1の剪断速度で、室温で行った。
【0091】
(実施例21:pdVWFの調製)
dVWFの調製を、改良を加えたThorell et Blomback(Thromb.Res.35:431−450,1984)により実施した。pdVWFの調製のために、1.5kgのクリオプレシピテートを20ないし30℃で6リットルの水に溶解した。1時間撹拌させた後に、フィブロネクチン沈殿物を遠心によって除去した。上清1リットルあたり8gNaClを添加し、溶液を室温まで温め、ウイルスを失活させるためにSDストック試薬(1%Tween80+0.18%アセチルトリエチルシトレート最終濃度)を添加した。
【0092】
溶液を、0.2MNaCl、0.02M酢酸ナトリウム(pH6.5)(洗浄用緩衝液)で事前に平衡化されたEMD−TMAEフラクトゲル650Mカラム(XK50/180)上で精製した。洗浄用緩衝液20カラム容量で洗浄した後、VWFを0.5MNaCl、0.02MNaクエン酸塩(pH6.9)で溶出させた。VWFを沈降させるために、グリシン(1M最終濃度)およびNaCl(3M最終濃度)を添加した。沈殿物を緩衝液に溶解し、最終濃度に対して0.3MCaCl2、0.15MNaCl、20mM HEPES(pH7.4)で平衡化したカラムをセファクリルS−400HR(Amersham)上に載せた。
【0093】
【表2】
(実施例22:PEGSSによるpdVWFのリジン残基のPEG付加)
PdVWFを実施例21により調製し、20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含む)で希釈し、最終濃度を400μg/mlとする。次に、mPEGスクシイミジルスクシネート(鎖長:5kD)を添加し(濃度:10mgPEGSS5000/mg VWF)、pdVWFを室温で、1時間にわたりPEG付加した。次に、再生セルロース(Millipore)からなる100kD膜を使用して、UF/DFによって、PEG付加rVWFから試薬を分離した。
【0094】
(実施例23:フリン成熟rVWFのフリン成熟および精製)
rFVIII発酵および精製プロセス由来の抗FVIII抗体カラムのフロースルー分画の143kgを、Schlokat et al.(Biotechnol.Appl.Biochem.24:257−267,1996)に記載のように、プロペプチド除去のためにフリンで処理し、滅菌濾過した。このプロセスは、初期の研究に基づいた(Fischer et al.,FEBS Lett.375:259−262,1995;Fischer et al.PCT/AT98/00034[WO98/38219],1−33,1998,18−2−1998 and Kaersgaard et Barington,J.Chromatogr.B 715:357−367,1998)。
【0095】
水で4倍希釈して16mS/cmにした後、633kgの希釈溶液を、10mM Tris、100mM NaAc、86mM NaCl、pH6.5、mS/cm(平衡緩衝液)により平衡化させたXK50/15EMD−TMAEフラクトゲル650Mカラム(300mlゲル;Merck;#K14540281)に載せた。カラムを平衡化緩衝液で洗浄し、100mM NaAc、250mM NaCl、100mMグリシン、3mM CaCl2で溶出した。
【0096】
4506gのTMAE溶出液をSartoclean GF(0.8及び0.65μ)およびSartobran P(0.45及び0.2μ)上で濾過し、1.5倍希釈して29mS/cmとし、Mustang Qフィルター(#IH18770932)を介してポンピングする。22+/−2℃で60分間にわたりSD処置し、かつ水で2倍に希釈して16mS/cmにした後、溶液を、TMAEステップの平衡用緩衝液で平衡化させたAmicon 70/29 UNOsphere Sカラム(600mlゲル;Bio−Rad,#78960C)に載せた。カラムを平衡用緩衝液で洗浄し、そしてTMAEステップの溶離緩衝液で溶出させた。
【0097】
3223gのUNO−S溶出液を、再生セルロースからなる30kDaの0.1m2膜(Hydrosart #01080217、Sartorius)を使用して限界濾過法により15倍濃縮した。コンセントレートの201gを、100mM NaAc、500mM NaCl、pH7.0(1698mlのゲル;GE Healthcare #17−0489−01)で平衡化させたXK50/86.5Superose 6 Prep Gradeカラム上で、サイズ排除クロマトグラフィーにより最終的に精製された。
【0098】
【表3】
(実施例24:フリン成熟rVWFのPEG付加)
フリン成熟VWFを実施例23により調製し、20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含有)に対して透析した。次に、溶液を20mM HEPES緩衝液(pH7.4)(150mM NaClおよび3%ショ糖を含有)により最終濃度を300μg/mlに希釈した。続いて、mPEGスクシミジルスクシネート(鎖長:5kD)を添加し(濃度:25mgPEGSS5000/mgVWF)、フリン成熟VWFを室温で1時間にわたってPEG付加した。次に、再生セルロース(Millipore)からなる100kD膜を使用して、UF/DFによって、PEG付加VWFから試薬を分離した。
【0099】
(実施例25:フリン成熟rVWFの生体外特徴付け)
実施例12および図15に記載のように、フリン成熟rVWFのFVIII結合能を表面プラズモン共鳴技術およびECAによって測定した。結果を、実施例21によって調製されるアルブミンを含まない血漿由来VWF調製試料と比較した。
【0100】
図18および図19に示されるように、フリン成熟rVWFのFVIII結合能は血漿由来参照調製試料と同等だった。
【0101】
(実施例26:グルタルアルデヒドで架橋させることによるポリシアル酸とVWFとの結合)
グルタルアルデヒドを架橋試薬(Migneault et al.,Biotechniques 37:790−796,2004)として用いたrVWFとポリシアル酸(コロミン酸)との結合のために、コロミン酸(濃度:20mg/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4)の溶液4mlを調製し、0.1MNaOHを添加してpHを7.4に合わせた。グルタルアルデヒドを添加して最終濃度を0.01%にした。続いて、rVWF(400μg/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4)の溶液1mlを100μlのアリコートに添加し、混合物を静かに振とうさせながら1時間インキュベートした。次に、混合物を透析し、トレハロースを添加し(最終濃度:3%)、rVWFポリシアル酸結合体を限界濾過法によって濃縮した。
【0102】
(実施例27:VWF欠乏マウスでのVWF半減期の増大)
Denis et al.(PNAS 95:9524−9529,1998)により記載されたVWF欠乏マウスを、ヒトVWDの動物モデルとして使用した。マウス5匹からなる群に対して、100UVWF:Ag/kgを達成するために、実施例19により調製したrVWFポリシアル酸結合体またはネイティブrVWFのいずれかをボーラス注入法(10ml/kg)によって経尾静脈により投与した。麻酔後の心臓穿刺によるクエン酸血漿は、注射後5分、1、3、6、9、および21時間で、それぞれの群から調製した。VWF抗原回収および内因性マウスFVIII活性レベルを血漿サンプルで計測した。この実験の結果は、図21および図22にまとめられている。
【0103】
VWFの半減期を、薬物動態学ライブラリーの1つの区画をモデルとして使用するMicroMath科学者プログラム(Micromath Research,Saint Luis,MO,US)を用いて算出した。FVIII活性の曲線下面積は、ベースラインサブトラクションによる台形モデルで算出した。
【0104】
VWFの半減期は1.3時間(ネイティブrVWF)から2.4時間(rVWF−ポリシアル酸結合体)へと増大し、FVIIIのAUCがそれぞれ3.3U*hr/mlから5.3U*hr/mlへと増大した。
【0105】
(実施例28:還元性アミノ化によるリジン基に対するmPEGプロピオンアルデヒドの結合)
rVWF溶液(400μg/ml)を0.05Mリン酸ナトリウム緩衝液(pH7.2)に調製し、mPEGプロピオンアルデヒド(5kDa鎖長)を添加して最終濃度を1mgVWFあたり10mgのmPEGプロピオンアルデヒドとする。この混合物を30分間撹拌する。つぎに、NaCNBH3を添加し(1mg/mgのrVWF)、混合物を静かに撹拌させながら室温で15時間にわたりインキュベートする。1MのTRIS水溶液(pH7.2)を添加し(1mgのNaCNBH3あたり50μl)、1時間にわたって撹拌することで反応を停止させる。その後、PEG付加rVWFは、100kD膜(再生セルロース/Millipore)を使用している限界濾過法/ダイアフィルトレーションによって、試薬から分離される。
【0106】
(実施例29:VWFのN末端PEG付加)
rVWFのN末端PEG付加をLee et al.(Pharm.Res.20:818−825,2003)の記載のように行う。rVWF溶液(最終濃度:500のμg/ml)を50mMの酢酸ナトリウム緩衝液(pH5.5)に調製し、mPEGプロピオンアルデヒド(鎖長:5kD)を添加した(濃度:10mg mPEGプロピオンアルデヒド/mgVWF)。PEG付加を、還元剤として2mMのNaCNBH3の存在下、室温で24時間にわたって行う。その後、PEG付加rVWFを、100kD膜(再生セルロース/Millipore)を使用して限界濾過法/ダイアフィルトレーションにより試薬から分離される。
【0107】
(実施例30:リジン残基およびrVWFのSH残基の逐次的なPEG付加)
RVWFを、実施例2によりmPEGスクシンイミジルスクシネート(鎖長:5kD)で、リジン残基を経てPEG付加する。PEG付加を1時間にわたって室温で行い、遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して20mM HEPES緩衝液(pH7.4、5%サッカロース)に対してUF/DFによりrVWFPEG結合体から分離する。次に、溶液のpH値を、0.1MNaOHで7.6に合わせ、mPEGマレイミド(10mM鎖長5kD/最終濃度)を加えて、遊離SH基のPEG付加を行う。静かに振とうしながら、室温で2時間にわたってPEG付加を行う。次に、試薬を再び、100kD膜(再生セルロース/Millipore)を使用して反応混合物から分離する。
【0108】
(実施例31:糖質残基の酵素酸化とPEG−Hzによるその後のPEG付加)
Avigad et al.(J.Biol.Chem.237:2736−43,1962)に記載のように、アルデヒド基を生じさせるrVWFの糖質残基(Wilchek et Bayer,Meth.Enzymol.138;429−442,1987)の酵素酸化を、Dactylium dendroides(Sigma)由来のガラクトースオキシダーゼを使用して行う。得られた溶液を、50mMリン酸緩衝液(pH7.2)に対して透析し、希釈してVWF濃度を400μg/mlにする。次に、鎖長5kDのmPEGヒドラジド(mPEG Hz)(最終濃度:40mg mPEG−Hz/mg VWF)を添加する。混合物を静かに振とうさせながら室温で3時間インキュベートする。試薬を、100kD膜(再生セルロース/Millipore)を使用して、5%のサッカロース含有20mM HEPES緩衝液(150mM NaCl、pH7.4)に対して、UF/DFによりPEG付加rVWFから分離する。
【0109】
(実施例32:rFVIIIによるFVIII結合部位のブロッキングによるrVWFのPEG付加)
非修飾FVIII結合部位を持つPEG付加rVWFを調製するために、rFVIII(200U/ml)およびrVWF(40U VWF:Ag/ml)を含む50mM HEPES緩衝液(50mM HEPES、150mM NaCl、2%トレハロース、pH7.4)の溶液3mlを調製して、37℃で1時間インキュベートする。混合物を、室温まで冷やし、PEGスクシンイミジルスクシネート(PEG−SS/鎖長:5kD)を添加し(最終濃度:1mgPEG−SS/UVWF:Ag)、静かに振とうさせながら1時間インキュベートする。次に、CaCl2を静かに振とうさせながら添加することで、最終濃度で400mMを得る。この溶液を、セファクリルS−400HR(Amersham)充填クロマトグラフィーカラム(2.6×80cm)に載せ、FVIIIの遊離結合部位を持つPEG付加rVWFを、サイズ排除クロマトグラフィー(溶離緩衝液:50mM HEPES緩衝液、400mM CaCI2、pH7.4)によって、rFVIIIから分離する。
【0110】
(実施例33:ヘパリンによるFVIII結合部位のブロッキングによるrVWFのPEG付加)
rVWF(300μg/ml)含有50mM HEPES緩衝液(pH7.4)の溶液5mlを調製し、同一緩衝液のヘパリンセファロースCL−6B懸濁液(Amersham Bioscience)2mlに添加する。この混合物を静かに振とうさせながら2時間にわたってインキュベートし、VWFをゲルに結合させる(de Romeuf et Mazurier,Thromb.Hamost.69:436−440,1993)。その後、mPEGスクシンイミジルスクシネート(200mg/mgのVWF)を混合物に加え、静かに振とうさせなら、PEG付加を室温で1時間行う。混合物を等量の2MNaCl含有HEPES緩衝液(pH7.4)で希釈する。ゲルを濾過によって上清から分離する。次に、ゲルは2mlHEPES緩衝液(pH7.4)(20mM HEPES、1MNaCl)で3回洗浄し、上清および洗い溶液を組み合わせる。続いて、PEG付加VWFを含む溶液を限界濾過法によって濃縮し、再生セルロース(Millipore)からなる100kD膜を用いて20mM HEPES緩衝液(pH7.4)(150mM NaCl、3%のサッカロース)に対して透析する。得られた誘導体は、バイアコア技術またはECA試験(試験システムは、米国仮特許出願第60/668,378号(2005年4月4日出願)に記載されている)の使用によって、完全なFVIII結合能を示す。
【0111】
(実施例34:ヒアルロン酸とVWFとの結合)
ヒアルロン酸(HA)によるリジン残基の修飾を、Sigma(C53747)から得られるヒアルロン酸を使用して還元性アミノ化によって行った。HAを、新たに調製された0.1MのNaIO4溶液に溶解して最終濃度を5mgHA/mlにした。次に、静かに撹拌させながら15分間にわたり酸化を行った。1mlの酸化HA溶液に対して2mlのエチレングリコールを添加し、室温、暗所で30分間さらに撹拌することで、反応を停止させた。溶液を、0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して、暗所4℃で一晩透析を行った。続いて、この溶液のアリコートを、0.05Mリン酸ナトリウム緩衝液(pH7.2))からなるrVWF溶液(40UVWF:Ag/ml)に添加して、最終濃度50mgの活性化HA/mgタンパク質を得た。この混合物を、暗所、室温で120分間にわたり撹拌した。NaCNBH3を添加し(1mg/mgタンパク質)、混合物を静かに振とうさせながら、18時間、室温でインキュベートした。つぎに、100μlの1MTris緩衝液(pH7.2)をこの混合物1mlに対して添加し、1時間にわたり撹拌することで、反応を停止させた。遊離試薬を、100kD膜(再生セルロース/Millipore)を使用して、UF/DFによりrVWFHA結合体から分離した。
【0112】
(実施例35:PSA結合VWFの生体外生化学的特徴づけ)
実施例19によりRVWFをポリシアル化した。この実施例に記載のパラメーター、例えば等電点電気泳動(図20)、実施例16によるECA試験(54%)および実施例12による表面プラズモン共鳴(70%)によってFVIII結合能の測定に加えて、比率VWF:RCo/VWF:Agを算出した。VWF抗原量を、市販のアッセイシステム(Asserachrom vWF,Roche,Basel,Switzerland)を用いて側定した。Macfarlane et al.(Thromb.Diath.Haemorrh 34:306−308,1975)に記載のように、調製試料の機能的な活性をリストセチンコファクターアッセイで測定した。rVWF出発原料に関して算出される0.39という比率の値は、ポリシアル酸付加後に減少して0.13となる。
【0113】
(実施例36:リジン残基を介した分岐PEGによるrVWFの結合)
成熟rVWF(35UVWF:Ag/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)の溶液を実施例23により調製した。次に、静かに撹拌させながら、NOF会社(NOF Europe,Grobbendonk,Belgium)によって供給される分枝mPEGスクシンイミジルグルタレート(PE−SG/鎖長:20kD)を、この溶液に添加し(5mgPEG−SG/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。次に、室温で1時間、静かに撹拌させながらPEG付加を行った。続いて、反応混合物を平衡イオン交換クロマトグラフィー樹脂(Fractogel EMD TMAE 650M)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)に載せた。次に、このカラムを20CV平衡化緩衝液で洗い、PEG付加rVWFを溶離緩衝液(20mM HEPES、0.5MNaCl、0.5%ショ糖、pH7.4)で溶出した。溶出液を、20mMのHEPES、150mMのNaCl、0.5%ショ糖、pH7.4からなる緩衝系を用い、再生セルロースからなる膜および100kD分子量カットオフで、限界濾過法/ダイアフィルトレーションにより濃縮した。得られたPEG付加誘導体のVWF:RCo/VWF:Ag 比は、rVWF出発材料(VWF:Rco/VWF:Ag比:0.89)と比較して、わずかに減少して0.79であった。また、PEG付加rVWF出発原料のFVIII結合能は、ECA試験(実施例16)で計測されたように、83%であった。
【0114】
(実施例37:FVIII−K.O.マウスでの分岐PEGと結合体を形成したVWFの薬物動態)
FVIII欠乏マウス(Bi et al.,Nat.Genet.10:119−121,1995)を、重度ヒト血友病Aのモデルとして使用した。マウス5匹からなる群に対して、PEG−rVWF(分岐PEG,SG)およびrFVIIIの混合物またはネイティブrVWFおよびrFVIIIの混合物を経尾静脈によるボーラス注入法(10ml/kg)によって投与することで、30UFVIII/mlおよび25UVWF/mlを達成した。注射後5分、3、9、24、および32時間、麻酔後の心臓穿刺によってそれぞれの群からクエン酸血漿を調製した。FVIII活性およびVWF抗原回収濃度を血漿サンプルで計測した。VWFおよびFVIIIの抽出曲線を図23および図24に示す。VWF半減期は1.4から9.7時間へと増大し、VWFのAUCは11.8から49.2U*h/mlに増加した。FVIIIの半減期は、1.2時間(ネイティブrVWF存在下)から4.4時間(PEG−rVWFとともに適用)へと増大し、血中濃度時間曲線下面積(AUC)が12.1から30.5U*h/mlへと増大した。
【0115】
(実施例38:FVIII−K.O.マウスにおける異なる量のPEG−VWFと混合されたFVIIIの比較薬物動態)
FVIII欠乏K.O.マウス5匹からなる群に対して、PEG−rVWF(25mg分岐PEG−SG20000/mgタンパク質)とrFVIII(A:20IUPEG−rVWF/ml+20IUFVIII/ml、B:10IUPEG−rVWF/ml+20IUFVIII/ml、C:3IUPEG−rVWF/ml+20IUFVIII/ml)の種々の混合物を経尾静脈によるボーラス注入法(10ml/kg)によって投与した。PEG付加rVWFについて、算出された比率は3モルPEG/moleリジンであった。異なるPEG−rVWF/rFVIII混合物において等量(単位換算)のPEG−rVWFとrFVIIIとを比較すると、以下のPEG/FVIII比が算出された。すなわち、3:1(A)、1.5:1(B)、0.45:1(C)である。麻酔後、クエン酸血漿を注射後5分、1、3、9、および24時間で、それぞれの群から心臓穿刺によって調製した。FVIII半減期(A:2.1時間(B):2.0時間、および(C):2.5時間)とAUC(A:18.9(B):14.5、およびC:13.2U*hr/ml)との間に関連した違いは見出されなかった。FVIIIの抽出曲線を図25に示す。
【0116】
(実施例39:PEG付加の度合いが異なるPEG−VWF調製試料を用いた比較薬物動態)
FVIII×VWFダブルノックアウトマウスを、FVIII欠乏マウスとVWF欠乏マウスとの交雑育種によって得た。それらのマウスは、VWF欠乏と同様にFVIII欠乏を患う。FVIII×VWFダブルノックアウトマウス5匹からなる群に対して、ネイティブrVWF/rFVIII(100/150IU/kg)混合物、PEG付加rVWF#AとrFVIIIとの混合物(100/150IU/kg)、あるいはPEG付加rVWF#BとrFVIIIとの混合物(150/150IU/kg)を経尾静脈により注入した。PEG−rVWF#A(5mg PEG−SS 5000/mgタンパク質)およびPEG−rVWF#B(20mg PEG−SS 5000/mgタンパク質)を実施例24により調製した。調製試料#Aに関しては、算出された比率は2.5molePEG/moleリジンであった。調製試料#Bに関しては、算出された比率は10molePEG/moleリジンであった。このクエン酸血漿に関して、試料適用後5分、1、3、9、および24時間、試料を調製した。VWF:Agの血漿レベルおよびFVIII活性を計測し、最大血漿レベルのパーセントに概ね注射後5分で達する。VWFおよびFVIIIの抽出曲線を、それぞれ図26および図27に示す。半減期は、PEG−rVWF #Aおよび#Bに関して、それぞれ6.3時間および8.1時間であった。ネイティブrVWFに関しては、算出された半減期は2.0時間であった。PEG付加rVWF両方の正規化AUC(%最大×h)は、360%*h(ネイティブrVWF)から901%*h(#A)および1064%*h(#B)へと増大した。同時注入されたrFVIIIの循環時間は、ネイティブVWFと比較してPEG付加rVWFによって改善された。FVIIIの半減期は、ネイティブrVWFの存在下で0.8時間であって、それぞれPEG付加rVWF#Aおよび#Bが注入される場合、1.5および1.8時間へと増大した。FVIIIのAUCは、214、370、および358%*hであった。
【0117】
(実施例40:VWFダイマーのPEG付加)
VWFダイマー(58IUのVWF:Ag/ml)(組換えCHO細胞株の条件培地から精製(Baxter BioScience))を20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖)で調製した。NOF社から供給された分岐mPEGスクシンイミジルグルタレート(PE−SG/鎖長:20kD)を、静かに撹拌しながらこの溶液に添加し(5mgPEG−SG/mgタンパク質)、0.5MNaOHを滴加してpH値を7.4に合わせた。PEG付加を、室温で1時間、静かに撹拌させながら行った。続いて、反応混合物を、反応混合物を平衡イオン交換クロマトグラフィー樹脂(Fractogel EMD TMAE 650M)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)に載せた。次に、カラムを20CV平衡化緩衝液で洗い、過剰の試薬を取り除いた。PEG付加rVWFダイマーを溶離緩衝液(20mM HEPES、0.5MNaCl、0.5%ショ糖、pH7.4)で溶離させた。溶出液を、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を使用し、再生セルロース(Millipore)からなる膜および100kD分子量カットオフで、限界濾過法/ダイアフィルトレーションによって濃縮した。
【0118】
(実施例41:低マルチマーrVWFのPEG付加および生体外特徴付け)
成熟rVWFを、実施例23により精製した。精製手法は、イオン交換クロマトグラフィーステップおよびスペローズ6上での最終ゲル濾過ステップを含むもので、該手法を20mM HEPES、150mM NaCl、pH7.4で実行し、高マルチマーrVWF(17のマルチマー)がボイド容量で溶出した。VWFマルチマー分析を、1.0%のアガロースゲルを使用してRuggeri and Zimmerman(Blood 57:1140−43,1981)により行った。低マルチマーrVWF調製試料(6マルチマー)がサイド分画から得られ、より高い持続時間で溶離した。0.5%のショ糖pH7.4を加えることにより、この分画を安定化させた。次に、低マルチマーrVWFを、mPEGスクシンイミジルスクシネート(PEG−SS)を使用してPEG付加した。PEG−SSを、静かに撹拌させながらこの溶液に添加し(5mgPEG−SS/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。PEG付加を、室温で1時間、静かに撹拌させながら行った。その後、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を用いて過剰試薬を、再生セルロースからなる膜および100kDの分子量カットオフで、限界濾過法/ダイアフィルトレーションによって除去した。実施例16によるECAアッセイによって測定されるFVIII結合能は、出発原料の49%からPEG付加調製試料の34%まで、わずかに減少した。低マルチマーrVWFについて計測されたVWF:RCo/VWF:Ag比率の値は、0.02であり、PEG付加手法の影響を受けなかった。
【0119】
(実施例42:可逆的にブロックされたFVIII結合エピトープ(FVIIIおよびヘパリンでブロック)によるVWFの誘導体化)
クロマトグラフィーカラム(15mm×148mm)をヘパリンハイパーD(Bio−Sepra)で充填し、20mM HEPES、68mM NaCl、0.5%ショ糖、pH7.4からなる平衡化緩衝液で平衡させた。次に、成熟rVWF(48IUVWF:Ag/ml)含有20mM HEPES(150mM NaCl、0.5ショ糖)の溶液をH2Oで希釈することで伝導率を7〜8mS/cmとし、線流速1.5cm/分を使用してカラム上に載せた。その後、NOF社(NOF Europe,Grobbendonk,Belgium)によって供給された分枝mPEGスクシンイミジルグルタレート(鎖長:20kD)を新たに15mlの平衡化緩衝液に溶かし、最終濃度を5mgPEG−SG/mg結合タンパク質とした。次に、この試薬液をカラム上にポンプで送り、PEG付加を静的条件下で2時間行った。次に、0.05%リジンを含む10CV平衡化緩衝液でカラムを洗った。次に、保護されたFVIII結合エピトープによるPEG付加rVWFを20mM HEPES、1MNaCl、0.5%ショ糖、pH7.4からなる緩衝液で溶出させた。最終的に、再生セルロース(Millipore)からなる100kD膜を使用して、20mM HEPES緩衝液(pH7.4、150mM NaCl、0.5%ショ糖)に対する限界濾過法/ダイアフィルトレーションによって、この溶液を濃縮した。得られた誘導体のVWF:RCo/VWF:Ag比率は0.48であり、rVWF出発原料(比率0.47)と同一であった。実施例36に記載の分枝PEG−SG20000によるrVWFのPEG付加手法と対照的にFVIII結合能は、ECA試験(実施例16)によって計測されるように、FVIIIエピトープキャッピングでこのPEG付加手法に影響を受けなかった。
【0120】
(実施例43:リジン残基を介した分解可能PEGによるVWFの結合)
成熟rVWFを、実施例23によって精製する。次に、このrVWF(40UVWF:Ag/ml)含有20mM HEPES緩衝液(150mM NaCl、pH7.4、0.5%ショ糖含有)の溶液を、調製する。続いて、PEG付加は、静かに撹拌させながら、mPEG−(カルボキシメチル)−3−ヒドロキシ−ブタン酸N−ヒドロキシスクシンイミドエステル(鎖長:5kD)をこの溶液に加えることによって行い(5mgのPEG試薬/mgタンパク質)、0.5MNaOHを滴加することでpH値を7.4に合わせた。次に、PEG付加反応を、室温で1時間、静かに撹拌させながら行った。その後、20mM HEPES、150mM NaCl、0.5%ショ糖、pH7.4からなる緩衝系を用いて過剰試薬を、再生セルロースからなる膜および100kDの分子量カットオフで、限界濾過法/ダイアフィルトレーションによって、PEG付加rVWFから分離した。
【特許請求の範囲】
【請求項1】
本明細書中に記載される発明。
【請求項1】
本明細書中に記載される発明。
【図1】


【図2】
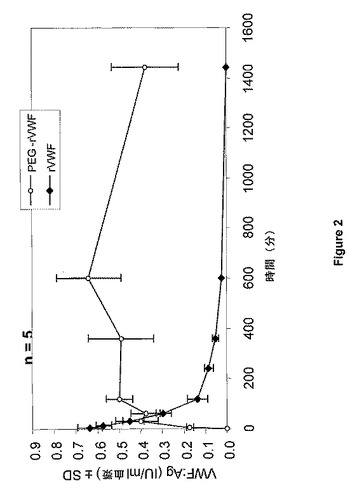
【図3】
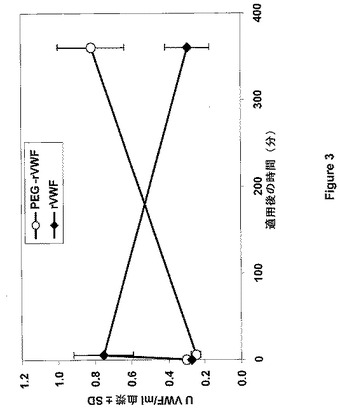
【図4】
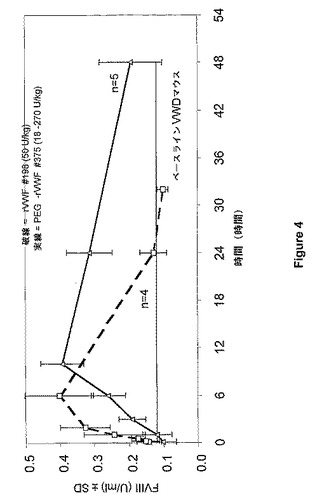
【図5】

【図6】
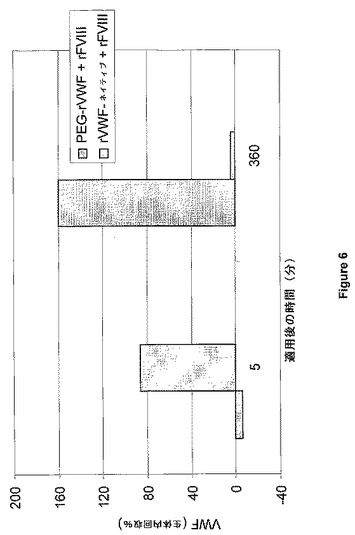
【図7】
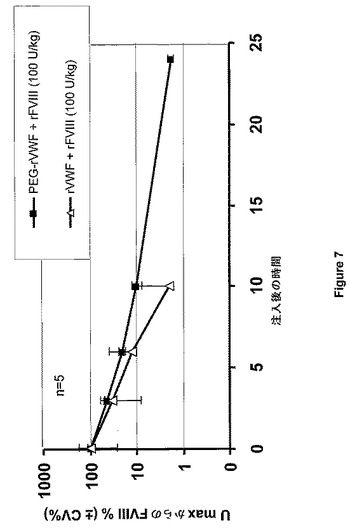
【図8】

【図9】
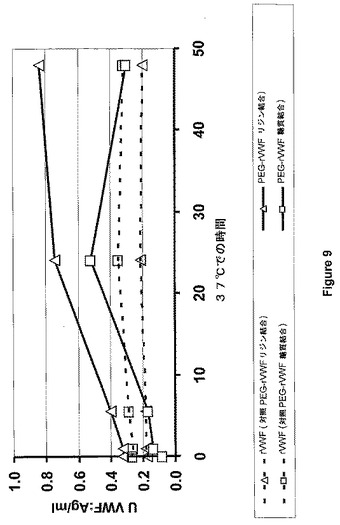
【図10】
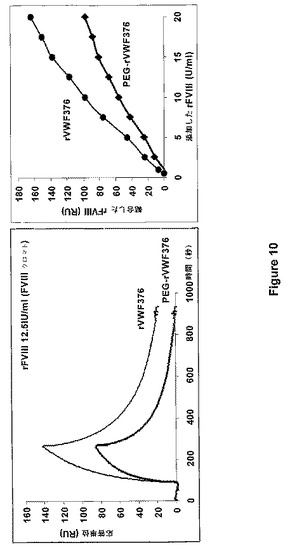
【図11】
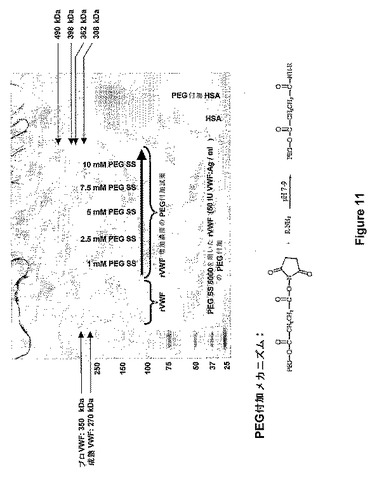
【図12】

【図13】

【図14】
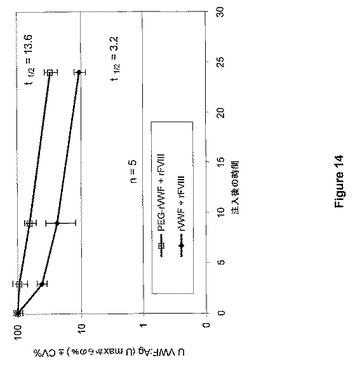
【図15】
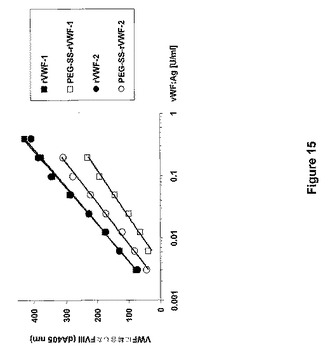
【図16】
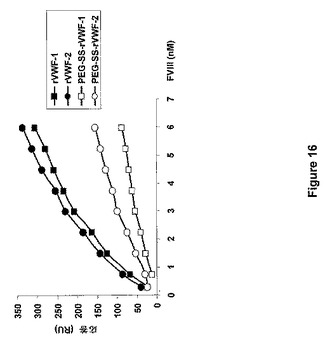
【図17】
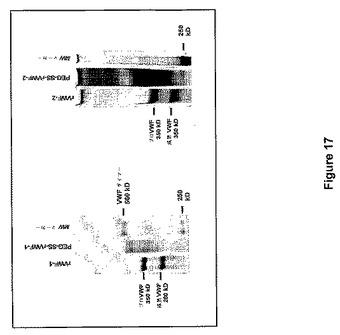
【図18】
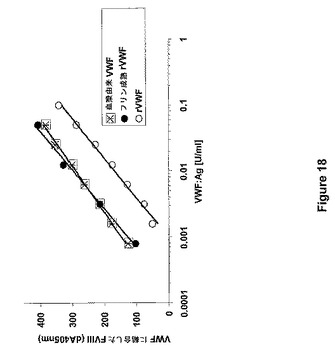
【図19】
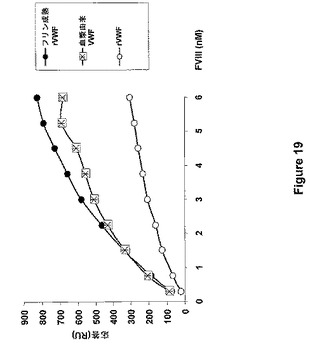
【図20】

【図21】
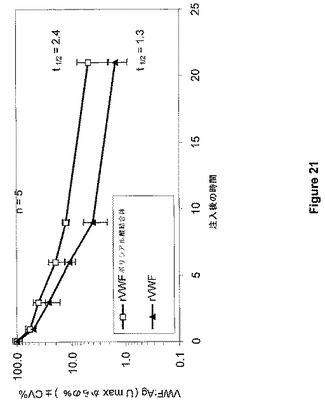
【図22】
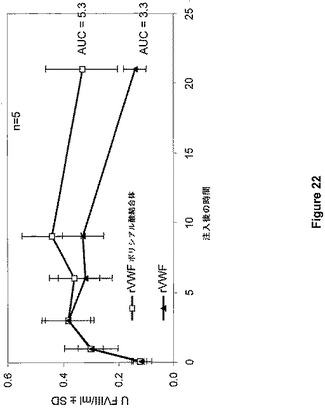
【図23】

【図24】

【図25】

【図26】
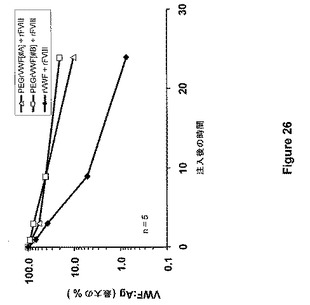
【図27】
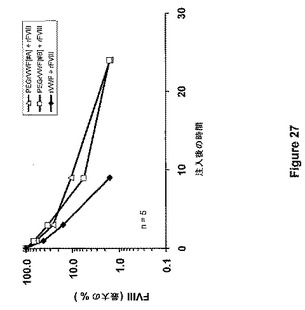
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2012−126750(P2012−126750A)
【公開日】平成24年7月5日(2012.7.5)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−81700(P2012−81700)
【出願日】平成24年3月30日(2012.3.30)
【分割の表示】特願2007−548552(P2007−548552)の分割
【原出願日】平成17年12月23日(2005.12.23)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
【公開日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願番号】特願2012−81700(P2012−81700)
【出願日】平成24年3月30日(2012.3.30)
【分割の表示】特願2007−548552(P2007−548552)の分割
【原出願日】平成17年12月23日(2005.12.23)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
[ Back to top ]