
低紅潮ナイアシン製剤
本発明は、ナイアシン、放出遅延剤及びその他の賦形剤を含む錠剤へと直接圧縮することができる徐放マトリクス製剤に関する。得られる本発明の錠剤は、好都合な放出特性及びナイアシン療法に一般に付随する皮膚の紅潮の重症度、持続時間及び発生率の低下を示す。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ナイアシン、放出遅延剤及びその他の賦形剤を含む錠剤へと直接圧縮することができる徐放マトリクス製剤に関する。本発明の得られる錠剤は、製造特性が向上しており、好都合な放出特性を示し、ナイアシン療法に一般に付随する皮膚紅潮の持続時間が短く、重症度及び発生率が低い。
【背景技術】
【0002】
ナイアシン(ニコチン酸、3−ピリジンカルボン酸としても知られる。化学式C6H5NO2)は、高比重リポタンパク質(HDL)のレベルを上昇させ、総血清コレステロール低比重リポタンパク質(LDL)及びトリグリセリドのレベルを低下させるので、高コレステロール血症の治療に関連する長所を有することが知られている。
【0003】
ナイアシンは血液脂質において非常に有益な効果をもたらすことが知られているが、NIASPAN(R)(Kos Pharmaceuticals、Inc.、Cranbury、NJ)を除き、有効な脂質治療に必要とされる高用量のナイアシンでよく起こる「紅潮」の発生率が高いために、ナイアシンを広範には使用しにくい。紅潮は、ナイアシン誘発性血管拡張を述べるために一般に使用される用語である。結果として、ナイアシン投与時に、紅潮が起こっている個体では、目に見える、不快なのぼせ又は紅潮感が発現し得る。ある種の物質及び/又は製剤が、皮膚紅潮を回避するか又は軽減することが示唆されている一方(米国特許第4,956,252号、同第5,023,245号及び同第5,126,145号参照)、この望ましくない副作用はナイアシン製品を広く利用する上で依然として問題となっている。
【0004】
さらに、市販のNIASPAN(R)製剤中の現在の放出遅延剤(一般に「膨張剤」とも呼ばれる。)は質的に非常にばらつきがあり、それにより、内部の仕様書に合わせるために市販業者から特定のバッチ製品を得る必要がある。
【0005】
したがって、医薬の技術分野において既存のナイアシン製剤よりも皮膚紅潮のレベルが低く、一方で、物理的、化学的及び工学的特性が向上していることを特徴とする、ロバストな製造プロセスも可能となる、徐放ニコチン酸製剤が必要とされている。
【発明の開示】
【発明が解決しようとする課題】
【0006】
(発明の要旨)
本発明は、ナイアシン及び放出遅延剤を含む徐放(ER)錠剤製剤を提供する。ある実施形態において、本発明は、既存の1000mg処方ナイアシン製剤よりも流動性、圧縮性、充填性及び硬度が改善された1000mgERナイアシン錠剤製剤を提供する。さらに、本発明の1000mgERナイアシン錠剤は、製造ロバスト性(ロバストなプロセスは、原材料又は製造プロセスの小さな変化など、様々な環境又は条件下で目標とする最終物を再現する能力を有するものである。)又は工業的に望まれる条件(例えばサイズ)を何ら低下させることなく、市販の500mg NIASPAN(R)錠剤の放出速度及び/又は吸収速度を再現する能力を示す。2個の500mg NIASPAN(R)錠剤は1個の1000mg NIASPAN(R)錠剤よりも紅潮が少ないことが特徴であると考えられているので、本発明のある目的は、2個の500mg NIASPAN(R)錠剤と生物学的に同等である1000mg ERナイアシン錠剤製剤を提供することである。
【課題を解決するための手段】
【0007】
特に、本発明は、
(a)ナイアシン約70%から約92%w/wと、
(b)放出遅延剤約7%から約25%w/wと、
(c)結合剤約0.1%から約4.3%w/wと、
(d)潤滑剤約0.5%から約1.5%w/wと、を含む医薬組成物を提供する。
【0008】
ある実施形態において、医薬錠剤は直接圧縮錠剤である。
【0009】
さらに、本発明は、(a)ナイアシン約70%から約92%w/wと、放出遅延剤約7%から約25%w/wと、結合剤約0.1%から約4.3%w/wと、潤滑剤約1.3%から約4.3%w/wと、の混合物を混合し;
(b)段階(a)の混合物を錠剤へと圧縮する
段階を含む、徐放ナイアシン錠剤を調製する方法を提供する。
【0010】
好ましい実施形態において、徐放ナイアシン錠剤は、粒状ナイアシンを混合することにより調製される。
【0011】
患者においてナイアシン処置療法に伴う紅潮を軽減する方法も提供されるが、この方法は、ナイアシン治療を必要とする患者に本発明の徐放ナイアシン錠剤形態を投与することを含む。好ましい実施形態において、本発明によるナイアシン製剤は、夕方又は夜間に1日1回投与される。
【0012】
本発明のある実施形態は、4個の500mg NIASPAN(R)錠剤の単回投与を改質1000mg徐放ナイアシン組成物の単回投与と比較する生物学的同等性試験において対象に投与される場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、改質1000mg徐放ナイアシン医薬組成物を含む。
【0013】
本発明によると、非ステロイド抗炎症薬(NSAID)と組み合わせて本発明の徐放ナイアシン製剤を投与することによって、紅潮をさらに軽減することができる。好ましい実施形態において、NSAIDはアスピリンである。
【0014】
本発明による医薬組成物は、即放型紅潮抑制剤成分及び遅延放出ナイアシン成分を含み得、このナイアシンは遅延放出される(即ち、ナイアシンが遅延時間後に放出される。)。好ましい実施形態において、ナイアシンは紅潮抑制剤の放出から少なくとも約30分から約40分後に放出される。
【0015】
(詳細な説明)
本発明の徐放マトリクス錠剤製剤は、(1)活性成分としてのナイアシン及び(2)活性成分の徐放を達成するための親水性ポリマーマトリクス、即ち、放出遅延剤を含む。本明細書中で使用される場合、「徐放」製剤は、1日1回投与で患者の脂質異常症に対して有効な治療を提供する製剤を意味する。
【0016】
本発明の徐放ナイアシン製剤によって、結果として、患者において脂質プロファイルが改善し得る。例えば、患者への本発明の徐放ナイアシン製剤の投与は、患者の血流中の、総コレステロール、低比重リポタンパク質(LDL)、トリグリセリド及びリポタンパク質A(Lp(a))を低下させ、高比重リポタンパク質(HDL)を向上させ得る。患者の血流中の、総コレステロール、LDL、トリグリセリド及び/又はリポタンパク質A(Lp(a))を低下させ、及び/又はHDLを向上させるための治療を必要とする状態は、本明細書中で「脂質異常症」と呼ぶ。したがって、本発明は、脂質異常症の治療を必要とする患者に本発明の徐放ナイアシン製剤を投与することにによる、脂質異常症の治療を包含する。
【0017】
生物学的同等性は、適切に計画された試験において同様の条件下で同じモル濃度用量で投与された場合に、医薬的同等物又は医薬的代替物中の活性成分又は活性部分が薬物作用部位で利用可能になる速度及び程度について顕著な差がないことである。通常、2つの製剤が生物学的同等であると結論するためには、自然対数変換されたCmax及びAUC又はこれらの生物学的同等性パラメータ計算値に対する何らかの適切な代替物のTest/参照治療比に対する90%信頼区間が80%から125%の間(両端を含む。)に入ることを示すことで十分である。
【0018】
本発明の範囲内の製剤は、自然対数変換生物学的利用率パラメータの試験/参照治療比に対する90%CIが標準的な80%から125%の間隔内に入る場合、本発明の製剤と生物学的に同等であるとみなされるものである(例えば、Guidance for Industry:Bioavailability and Bioequivalence Studies for Orally Administered Drug Products−General Considerations、U.S.Department of Health & Human Services、Food and Drug Administration、CDER、2003年3月;Guidance for Industry Food−Effect Bioavailability and Fed Bioequivalence Studies、2002年12月を参照のこと;両刊行物の内容は参照により本明明細書中に組み込まれる。)。当業者にとって公知のように、関連性のある生物学的同等パラメータを用いて(参照製剤が対照として使用される。)同じ分析条件下(例えば、分析的及び技術的条件分析)でこのような製剤を参照製剤(本明細書中に記載のもの又は本明細書中に記載の本発明の実施形態など)と比較する。
【0019】
ナイアシン
水溶性薬剤であるナイアシンは、微粉白色結晶、顆粒又は白色結晶粉末として市販されている。本発明の医薬組成物は、ナイアシン結晶、顆粒又は粉末を用いて製造することができる。好ましい実施形態において、本医薬組成物は、粒状ナイアシン(これはナイアシン粉末と比較して流動性が大きい。)を用いて製造される。流動性は、錠剤製造にのための重要な加工パラメータである。本発明による粒状ナイアシンの使用により、流動性が向上し、製造スケールで実行可能なナイアシン錠剤の直接圧縮が可能となる。何の粒状ナイアシン粒子サイズも本発明によるナイアシン錠剤を調製するのに適している。粒状ナイアシンに対する好ましい粒子サイズは、顆粒が100−425μmの範囲となるようにふるい分画に対してNLT85%(w/w)であり、ダスト<100μmに対してNMT10%(w/w)である。乾式造粒又は湿式造粒プロセスを用いて、ナイアシン粉末の流動性を向上させることができる。
【0020】
ナイアシンは通常、約70%から約95%w/w、好ましくは約76%から約90%w/w、より好ましくは約78%から約82%w/wの濃度で本発明の錠剤中に存在する。ナイアシンは、約100mgから3000mgの量で本発明の徐放製剤中に存在し得る。ある実施形態において、本発明の製剤は、ナイアシン約500mg、約750mg又は約1000mgを含む。ナイアシンの好ましい1日投与量は、約1000mg、約1500mg又は約2000mgである。したがって、1日1回、2個の1000mg錠剤を患者に投与することにより、例えば、ナイアシンの1日投与量を患者に与えることができる。
【0021】
放出遅延剤
ポリマーマトリクス系からの徐放は通常、ポリマー湿潤、ポリマー水和、ゲル形成、膨潤及びポリマー溶解に関与する。可溶性薬物の場合、これらの薬物は濡れ、溶解し、ポリマーマトリクスにより形成されたゲル層から拡散する。可溶性薬物がマトリクス錠剤中で放出される機構は多くの可変要素に依存するが、一般的原理は、錠剤全体に存在する水溶性ポリマーが錠剤の外表面で水和しゲル層を形成するということである。水が錠剤に浸透するにつれて、ゲル層の厚みが増し、可溶性薬物がゲル層を通じて拡散する。摂取された錠剤の寿命中、薬物放出速度は、ゲルを通じた可溶性薬物の拡散及び錠剤浸食の速度により決定される。
【0022】
本発明の放出遅延成分は、好都合な膨潤及びゲル化特性を示す当業者にとって公知の何れかの薬物であり得る。適切な放出遅延剤の例には、以下に限定されないが、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(一般にHPMC又はヒプロメロースとも呼ばれる。)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)及びポリビニルピロリドン(PVP)、キサンタンガム及びトリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))ならびにこれらの放出遅延剤の混合物が含まれる。ある実施形態において、放出遅延剤は親水性の水溶性ポリマーである。好ましい親水性ポリマーは中程度の粘度のヒドロキシプロピルメチルセルロース及び中程度の粘度のポリビニルアルコールである。
【0023】
放出遅延剤は通常、約7.0%から約25.0%w/w(製剤の総重量に対する%重量)、好ましくは約11.0%から約20.0%w/w、より好ましくは約14%から約18%w/wの濃度で本発明の錠剤中に存在する。
【0024】
ある実施形態において、放出遅延剤はヒドロキシプロピルメチルセルロースである。HPMCは、セルロース(無水グルコース単位の基本的繰り返し構造を含有する天然の炭水化物)のポリマー性骨格を有する。溶解度(例えば水和率)及びHPMCにより形成されるゲル層の強度は、HPMCのセルロース骨格(セルロースは無水グルコース単位の基本的繰り返し構造を含有する天然の炭水化物である。)に結合した、2種類の化学置換基、ヒドロキシプロポキシル(ヒドロキシプロピルと呼ばれることがある。)及びメトキシル(メチルと呼ばれることがある。)置換の割合により影響を受ける。ヒドロキシプロポキシル置換は本来、比較的親水性であり、水和率に大きく関与するが、一方メトキシル置換は本来、比較的疎水性である。セルロースの無水グルコース単位における置換基の量は、1個の無水グルコース環に結合する平均置換基数により示すことができ、これは、当業者により「置換度」として一般に知られている概念である。METHOCEL(R)Cellulose Eithers Technical Handbook、Dow Chemical Company(2002年9月刊行、Form番号192−01062−0902 AMS)を参照のこと;及びMETHOCEL(R)セルロース Eithers for Controlled Release of Drugs in Hydrophilic matrix Systems(2002年7月刊行、Form番号198−02075−0702 AMS)を用いる。本発明のある実施形態において、HPMC放出遅延剤は、約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度、好ましくは約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度、より好ましくは約1.39から約1.41のメトキシル置換度及び約0.20から約0.22のヒドロキシプロポキシルモル置換度、より好ましくは約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する。METHOCEL(R)K−15M(Dow Chemical Companyより入手可能、K−15M Premium及びK−15M Premium CRなどの特定のK−15サブブランドを含む。)は好ましい放出遅延剤である。
【0025】
さらに、ヒドロキシプロピルメチルセルロースポリマーは様々な粘度グレードのものが市販されている。これらには、例えば、METHOCEL(R)K、即ちMETHOCEL(R)K4M及びMETHOCEL(R)K15M(Dow Chemical Co、USAより入手可能)の4000及び15000mPas(1センチポアズ(cps)=1mPas(ミリパスカル秒))粘度グレード;及びMetalose90 SH(Shin Etsu Ltd、Japanより入手可能)の4000、15,000及び39000mPas粘度グレードが含まれる。本発明の実施形態において、HPMC粘度(20℃にて水中2%濃度で測定、例えばASTM D2363)は約11,000から約22,000mPas、好ましくは約13,000から約18,000mPasである。
【0026】
HPMC以外の、適切なポリマーの置換に必要な具体的な特徴を調べるために、当業者はポリマー(例えばヒドロキシプロピルセルロース)の置換度を変化させ、本発明によるHPMCを使用する製剤(例えば実施例1又は2による製剤)の溶出プロファイルに合致する置換を同定することができる。
【0027】
賦形剤
本発明の錠剤は結合剤をさらに含む。結合剤は、何れかの従来から知られている医薬的に許容可能な結合剤、例えばポリビニルピロリドン(PVP、ポビドン、ポリビドンとしても知られる。)、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート、ワックスなどであり得る。上述の結合剤の混合物も使用し得る。本発明の実施形態において、結合剤は、錠剤の総重量の約0.1%から4.3%w/w、好ましくは約0.2%から3.25%w/w、より好ましくは約2.5%から3.0%w/wを構成する。
【0028】
さらに、本発明の錠剤は潤滑剤を含む。潤滑剤は疎水性又は親水性であり得、当業者にとって公知の潤滑剤(以下に限定されないが、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、硬化植物油など)を含み得る。好ましくは、潤滑剤はステアリン酸である。製剤への潤滑剤の添加によって、圧縮中のダイ壁と錠剤製剤との間の摩擦が減少し、粉末の流れが促進され(即ち混合された製剤のホッパー及びダイへの流れ)、加工装置に錠剤材料が接着するのを防ぐのに役立つ。ある実施形態において、本発明の錠剤製剤は、潤滑剤約0.5%から1.5%w/w、好ましくは約0.75%から1.25%w/w、より好ましくは約0.85%から1.15%w/w、より好ましくは約0.95%から1.05%w/wを含む。
【0029】
コーティング
本発明の徐放錠剤製剤は、色塗を与え、視覚的特性を促進し、湿気又は臭気を防御するものとして作用し、日光、温度変化などの環境因子による劣化を防ぐか、又は錠剤の味を隠すことが医薬固形剤形の分野で知られているようなコーティングをさらに含み得る。当業者にとって公知であるように、このようなコーティングは、ポリマー、可塑剤及び/又は着色顔料を含有し得る。例としては、OPADRY(R)コーティングが挙げられる。このコーティングは、流動床塗布装置(例えばWursterコーティング)又はパンコーティングシステムなどの何らかの公知の手段を使用して、溶液(例えば水性)、溶媒又は懸濁液から塗布することができる。本発明のある実施形態において、コーティングは色彩塗料、特にOPADRY(R)コーティングである。さらなる実施形態において、約1.5から約8.0%の重量増加、好ましくは約1.75から約5.0%重量増加で色彩塗料を錠剤に塗布する。
【0030】
NIASPAN(R)500mg錠剤に対する同等性
以前の臨床試験の検討から、2個の(2)NIASPAN(R)1000mg錠剤(錠剤重量1203.6mg)が4個の(4)NIASPAN(R)500mg錠剤と生物学的に同等ではなく、NIASPAN(R)500mg錠剤からよりもNIASPAN(R)1000mg錠剤からナイアシンが速く放出されることが明らかになった。さらなる研究から、NIASPAN(R)500mg錠剤中の成分量の2倍を有するナイアシンER1000mg錠剤(錠剤重量1419.0mg)も生物学的に同等でないことが示された。後者の事例では、ナイアシン溶出は、インビトロで、NIASPAN(R)500mg錠剤からよりも1000mg錠剤からの方が遅く、ナイアシンは、インビボで参照製品(500mg)よりもナイアシンER1000mg錠剤からの方が吸収が遅かった。さらなる研究から、1300.0mg及び1280.0mgの錠剤重量の改質ナイアシンER1000mg錠剤が、放出速度が遅いため、NIASPAN(R)500mg錠剤と生物学的に同等でないことも分かった。
【0031】
2個のNIASPAN(R)500mg錠剤と生物学的に同等なナイアシンER1000mg錠剤を処方するために、発明者らは、インビボでの放出及び吸収特性を予想するため、複数のナイアシンER1000mg製剤を調製し、インビトロで試験した。錠剤中のポリマー(放出遅延剤)レベル(w/w)が上昇するに従い溶解度が低下するという事実に基づき、試験ナイアシンER1000mg錠剤をさらに改質した。したがって、評価には、新しい成分(様々なタイプのポリマーなど)及び代替的製造技術(直接圧縮又はローラー圧縮など)の分析が含まれた。
【0032】
表1は、総錠剤重量が様々である1000mg錠剤に対する様々な試験製剤を示す。
【0033】
【表1】
【0034】
複数の可変要素の最初の評価後、下記の4種類の製剤をさらなる評価のために選択した。全時間点に対して、37℃で1.2のpHに維持した擬似胃液250mL中で60分間、次いで37℃で6.8のpHに維持した250mLの擬似腸液中で、参照として500mg NIASPAN(R)を用い、USPタイプ3装置を採用して、溶出プロファイルに基づき、下記製剤に対する変化を分析した。
【0035】
(i)湿式造粒(WG)を用いて調製したMETHOCEL(R)E10M
1240mg、1260mg、1280mg及び1300mg製剤に対して指定された製法に従い、粒状ナイアシン、METHOCEL(R)E10M、ポビドンK90及びステアリン酸の重量を測定し、次いで、造粒溶液として脱イオン水を用いて高せん断造粒機中で造粒した。湿った顆粒を乾燥させ、粉砕し、次いで追加の粒状METHOCEL(R)E10M及びステアリン酸と混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった最終的な混合物を錠剤へと圧縮した。
【0036】
(ii)直接圧縮(DC)法を用いて調製したMETHOCEL(R)E10M
表1で概説した指定された製法に従い、粒状ナイアシン、METHOCEL(R)E10M、ポビドンK90及びステアリン酸の重量を測定し、次いで、8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0037】
(iii)WG法を用いて調製したMETHOCEL(R)K15M
表1で概説した指定された製法に従い、ナイアシンUSP、METHOCEL(R)K15M及びポビドンK90の重量を測定し、次いで造粒溶液として脱イオン水を用いて高せん断造粒機中で造粒した。湿った顆粒を乾燥させ、粉砕し、次いで追加の粒状METHOCEL(R)K15M及びステアリン酸と混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、最終的なよく混ざった混合物を錠剤へと圧縮した。
【0038】
(iv)DC法を用いて調製したMETHOCEL(R)K15M
表1で概説した指定された製法に従い、粒状ナイアシン、METHOCEL(R)K15M、ポビドンK90及びステアリン酸の重量を測定し、次いで、8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0039】
分析には、加工機器ツールの変化;ポリマーレベルの変動(表1参照);湿式造粒、直接圧縮及びローラー圧縮法の置き換え;PVPレベルの変動;錠剤硬度の変化;重量変動(+/−5%);再現性;錠剤化スピードの変動及び錠剤安定性(保管後の放出速度、吸湿など)が含まれた。次の3種類の製剤に対して目標とする薬物放出プロファイルを達成した:(i)METHOCEL(R)E−10M湿式造粒、(iii)METHOCEL(R)K−15M湿式造粒及び(iv)METHOCEL(R)K15M直接圧縮。この3種類の製剤は、3ヶ月間の安定性試験後、好都合な安定性の結果をさらに示した。
【0040】
上述の分析で分かった経済的及び安定性に関する長所により、さらなる分析のための好ましい実施形態として、METHOCEL(R)K−15を用いた直接圧縮錠剤を選択した。したがって、改質1000mgナイアシンDC錠剤に関して、造粒サイズ;各成分の粒子サイズ分布、バルク及び各成分のタップ密度;各成分の様々なロット;含量均一性;Hauser及びCarr指標;流動性;圧縮率及びもろさの影響についてさらに評価を行った。表2は、様々な実験製剤で使用される具体的な主要材料を概説する。DMFはドラッグマスターファイルである。
【0041】
【表2】
【0042】
表3は、賦形剤の様々なレベルを含有する改質試験1000mgDC錠剤及び関連する物理的品質を示す。表3に記述の場合各成分のw/w%で、上述のようにこれらの製剤を調製した。
【0043】
【表3】
【0044】
図1は、表3で示した製剤に対する溶出プロファイルの比較例を与える。
【0045】
表4は、臨床試験での500mgのNiaspan(R)に対する、複数の1000mg実験ナイアシン製剤の溶出及び生物学的利用率データを示す。USP装置1を用い、37℃にて100rpm(バスケット法)で900mL脱イオン水を用いて、溶出を計算した。
【0046】
【表4】
【0047】
次のパラメータを変化させることにより、改質1000mgナイアシンER直接圧縮錠剤の再現性を調べた:
METHOCEL(R)K−15MP CRの粘度及びヒドロキシプロポキシル含量
METHOCEL(R)K−15Mの粒子サイズ
粒状ナイアシンの粒子サイズ
ステアリン酸含量
PVP K−90のふるい
加工パラメータ
混合順序及び時間
錠剤硬度
錠剤化スピード
下記表5及び図2−4は、上述の再現性試験中に得られたデータを示す。
【0048】
【表5】
【0049】
上述の可変要素の分析完了後、出願者らは、次の可変要素を変化させた場合に作られた錠剤からのナイアシン溶出に顕著な差がないことを見出した:METHOCEL(R)K−15MPremium(CR)の粘度及びヒドロキシプロポキシル置換、様々な粒子サイズの粒状ナイアシン、40メッシュスクリーンを通したPVP K−90のふるい、0.5%から2.0%のステアリン酸含量、混合段階及び混合時間。METHOCEL(R)K−15M Premium(CR)の粒子サイズ及び錠剤の硬度(特に8kp未満)を向上させると、ナイアシン溶出度が上昇した。粒状ナイアシン及びMETHOCEL(R)K−15M premium CRの粒子サイズが小さいほど、圧縮率が大きくなった。製剤中のステアリン酸含量が増加するにつれて、突出力が著しく低下した。錠剤硬度を高くするには、高い圧縮力及び突出力が必要であり、錠剤化スピードを上昇させる場合、目標錠剤硬度(18Kp)を得るには、より大きい圧縮力が必要であった。
【0050】
上記によると、本発明は、
(a)ナイアシン約70%から約92%w/wと、
(b)約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する放出遅延剤約7%から約25%w/wと、
(c)結合剤約0.1%から約4.3%w/wと、
(d)潤滑剤約0.5%から約1.5%w/wと、を含む、湿式造粒又は直接圧縮1000mgナイアシン徐放(ER)錠剤製剤を包含する。
【0051】
好ましい実施形態において、本製剤は、直接圧縮法を使用して調製される。
【0052】
本発明の1000mg徐放ナイアシン製剤は2個の500mg NIASPAN(R)錠剤と生物学的に同等なので、これらは、効力及び毒性プロファイルの両方が同じであると予想される。したがって本発明の1000mg徐放ナイアシン製剤の投与により、本発明の製剤の使用を中止することが求められるレベルの、治療を制限する肝毒性又は治療を制限する尿酸又はグルコースレベル上昇を生じることなく、2個の500mg NIASPAN(R)の治療効果と同様の治療効果が与えられ得る。持続放出ナイアシン製剤に関連する毒性の問題は当業者にとって周知である。例えば「A comparison of the Efficacy and Toxic Effects of Sustained−v.Immediate−Release Nyacin Hypercholesterolemic Patients」、McKenneyら、JAMA Vol.271、No.9、1994年3月2日;及び「Hepatic Toxicity of Unmodified and Time Release Preparations of Nyacine」、Raderら、The Am.Jour.Of Med.、Vol.92、1992年1月、77頁を参照のこと。
【0053】
したがって、本発明のある態様は、それを必要とする患者を治療するための本発明の医薬組成物の投与を含み、この治療によって、1日1回患者によりこの組成物が摂取される場合、患者への投与後に、このような治療を中止することが必要となる、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を一般に生じさせることなく、血清脂質を低下させることができる。さらなる実施形態において、投与は、夕方又は夜間に1日1回である(例えば夕食後又は就寝前)。
【0054】
併用療法
本発明の1日1回のナイアシン製剤をHMG−CoA還元酵素阻害剤と組み合わせることができる。本明細書中で使用される場合、「併用療法(combination therapy)」及び「併用療法(combination treatment)」は、同じ又は個別の医薬剤形での、本発明のナイアシン製剤と少なくとも1つのさらなる活性成分との投与を包含する。併用療法は、本明細書中で使用される場合、治療計画の一部としての、活性薬剤の同時投与及び活性薬剤の連続投与を含む。
【0055】
HMG−CoA還元酵素阻害剤の例には、以下に限定されないが、ロバスタチン及び米国特許第4,231,938号で開示されるような関連化合物、プラバスタチン及び米国特許第4,346,227号及び同第4,448,979号で報告されるような関連化合物、メバスタチン及び米国特許第3,983,140号で開示されるような関連化合物、ベロスタチン及シンバスタチン及び米国特許第4,448,784号及び同第4,450,171号で考察されるような関連化合物、フルバスタチン、アトルバスタチン、リバスタチン及びフルインドスタチン(Sandoz XU−62−320)が含まれる。その他のHMG−CoA還元性阻害剤には、以下に限定されないが、米国特許第4,613,610号で開示されるようなメバロノラクトン誘導体のピラゾール類似体、PCT出願WO86/03488で開示されるようなメバロノラクトン誘導体のインデント類似体(indent analogs)、米国特許第4,647,576号で開示されるような、6−[2−(置換ピロール−1−イル)アルキル]ピラン−2−オン及びその誘導体、SearleのSC45355(3−置換ペンタンジオン酸誘導体)ジクロロ酢酸、PCR出願WO86/07054で開示されるようなメバロノラクトンのイミダゾール類似体、仏国特許第2,596,393号で開示されるような3−カルボキシ−2−ヒドロキシ−プロパン−リン酸誘導体、欧州特許出願0221025 A14で開示されるような2,3−ジ−置換ピロール、フラン及びチオフェン誘導体、米国特許第4,686,237号で開示されるようなメバロノラクトンのナフチル類似体、米国特許第4,499,289号で開示されるようなオクタヒドロ−ナフテレン、欧州特許出願0142146 A2で開示されるようなロバスタチンのケト類似体ならびにその他の公知のHMG−CoA還元酵素阻害剤、例えば英国特許第2,205,837号及び同第2,205,838号;及び米国特許第5,217,992号;同第5,196,440号;同第5,189,180号;同第5,166,364号;同第5,157,134号;同第5,110,940号;同第5,106,992号;同第5,099,035号;同第5,081,136号;同第5,049,696号;同第5,049,577号;同第5,025,017号;同第5,011,947号;同第5,010,105号;同第4,970,221号;同第4,940,800号;同第4,866,058号;同第4,686,237号で開示されているものなどである。
【0056】
場合によっては、本発明の製剤処方をその他の抗高脂血剤と組み合わせて投与することもできる。抗高脂血剤の具体例には、以下に限定されないが、胆汁酸抑制剤、例えば、コレスチラミン、コレスチポールDEAESephadex(Secholex.RTM.及びPolidexide.RTM.)、プロブコール及び米国特許第3,674,836号で開示されているような関連化合物、リポスタビル(Rhone−Poulanc)、エイザイE5050(N−置換エタノールアミン誘導体)、イマニキシル(HOE−402)テトラヒドロリプスタチン(THL)、イシチグマスタニルホスホリルコリン(SPC Roche)、アミノシクロデキストリン(田辺製薬)、味の素A J−814(アズレン誘導体)、メリナミド(住友)、Sandoz 58−035、American Cyanimid CL−277,082及びCL−283,546(二置換尿素誘導体)、ネオマイシン、p−アミノサリチル酸、アスピリン、四級アミンポリ(ジアリルジメチルアンモニウムクロリド)及び米国特許第4,027,009号で開示されるようなイオネン、米国特許第4,759,923号で開示されるようなポリ(ジアリルメチルアミン)誘導体、様々な魚油栄養剤で見出されるオメガ−3−脂肪酸、フィブリン酸誘導体、例えば、ゲムフィブロジル、クロフィブレート、ベザフィブレート、フェノフィブレート、シプロフィブレート及びクリノフィブレート及び米国特許第5,200,424号;欧州特許出願0065835 A1、欧州特許164−698−A、英国特許第1,586,152号及び英国特許出願2162−179−Aで開示されるものなどのその他の公知のコレステロール低下剤が含まれる。
【0057】
さらに、紅潮抑制剤と組み合わせて本発明の製剤処方を投与することができる。紅潮抑制剤には、以下に限定されないが、アスピリン及びサリチル酸塩などの非ステロイド抗炎症薬;イブプロフェン、フルルビプロフェン、フェノプロフェン、ケトプロフェン、ナプロキセン、ナプロキセンナトリウム、カルプロフェン及びスプロフェンなどのプロピオン酸;インドメタシン、エトドラク及びスリンダクなどのインドール酢酸誘導体;アクロフェナク、ジクロフェナク及びフェンクロフェナクなどのベンゼン酢酸;ゾメピラク及びトルメクチンなどのピロール酢酸;フェニルブタゾン及びオキシフェンブタゾンなどのピラゾール;ピロキシカムなどのオキシカム;及びメクロフェナメート及びメフェナム酸などのアントラニル酸が含まれる。
【0058】
紅潮抑制剤は、以下に限定されないが、米国特許公開第2004/0229844号及び同第2005/0154044号で開示されている化合物を含むプロスタグランジンD2受容体アンタゴニストでもあり得る。好ましいプロスタグランジンD2受容体アンタゴニストはMK−0524(Merck & Co.)である。
【0059】
遅延放出
本発明は遅延放出剤形を包含する。本明細書中で使用される場合、「遅延放出」とは、患者への投与後ある一定時間(即ち遅延時間)殆ど又は全く放出が起こらないことを意味する。医薬組成物中の唯一の活性薬剤として又は医薬剤形中の複数の活性薬剤の1つとして(その他の活性薬剤は遅延放出されてもよいし又はされなくてもよい。)、遅延放出形態で本発明のナイアシン製剤を提供することができる。したがって、例えば、医薬組成物は、遅延放出ナイアシン成分と組み合わせて即放型紅潮抑制剤成分に迎合することができる。例えば、患者への本発明の医薬組成物の投与時に、即放型紅潮抑制剤がすぐに放出され、遅延放出ナイアシン成分が遅延時間(例えば少なくとも約30分から約40分)後に放出される。
【0060】
当技術分野で周知の材料及び方法を用いて、遅延放出を行うことができる。これらの材料及び方法には次のものが含まれる。単一ユニットの、カプセル薬物送達系(これには、薬物及びプラグを収容する不溶性カプセルが含まれる。)。膨潤、浸食又は溶解の結果、予め定められた遅延時間後にこのプラグが除去される。Pulsincap(R)システム(Scherer DDS、Ltd)はこのような系の一例であり、この場合、本体の開口端が膨潤可能なヒドロゲルプラグで閉じられている。溶解液又は胃腸液と接触すると、プラグが膨潤し、遅延時間後にそれ自身をカプセルから押し出す。この後、薬物が急速に放出される。プラグの直径及び位置を操作することにより遅延時間を調節することができる。例えば、WO90/09168;Wildingら、Pharm Res.1992;9:654−657を参照のこと。プラグの材料を、不溶性であるが浸透性で膨潤性のポリマー(例えば、ポリメタクリレート)(Krogel I、Bodmeier R、Pharm Res.1998;15(3):474−481;Krogel I、Bodmeier R、Pharm Res.1999;16(9):1424−1429)浸食性圧縮ポリマー(例えば、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリエチレンオキシド)、凝結融解ポリマー(例えば飽和ポリグリコール化グリセリド、グリセリルモノオレエート)及び酵素的に調節された侵食性ポリマー(例えばペクチン)製にすることができる。小腸のより高いpH領域でのみ溶解が起こるように系を腸溶性被覆することによって、胃での滞留時間が変動するという潜在的問題を解決することができる。Saeger H、Virley P.Pulsincap & Mac226:パルス放出剤形。Scherer DDS、Ltd;2004からの製品情報。
【0061】
Port(R)システム(Port Systems、LLC)は、浸透に基づくカプセル系であり、製剤とともに。不溶性プラグ(例えば脂質性のもの)を収容する半透性の膜(例えば酢酸セルロース)で被覆されたゼラチンカプセル及び浸透圧的に活性のある物質からなる。Crisonら、Proceed Intern Symp Control Rel Bioact Mater.1995;22:278−279。水性溶媒と接触すると、水が半透性の膜を横切って拡散し、結果として、内圧が上昇し、遅延時間後にプラグが噴出される。コーティングの厚さによって遅延時間を調節する。
【0062】
薬物を液体の形態で送達するために、浸透圧によって駆動されるカプセル系を使用することができ、この場合、液体薬物は非常に多孔性である粒子に吸収されており、この粒子は、障壁層が溶解した後に拡大する浸透性の層により支持された半透性カプセルのオリフィスを通じて薬物を放出する。米国特許第5,318,558号を参照のこと。このカプセル系は、身体からの水分が浸透圧によって導入されることにより薬物を送達する。カプセルの壁は弾性材料からなり、オリフィスを有する。浸透が進行するにつれて、カプセル内の圧力が上昇し、壁が伸張する。オリフィスは弾性壁が弛緩しているときはオリフィスを通じた薬物の流れが基本的に停止するが、弾性壁が閾値を超えて広がった場合はオリフィスが十分に広がり、望まれる速度で薬物が放出されるよう、十分に小さい。スチレン−ブタンジエンコポリマーなどのエラストマーを使用することができる。米国特許第5,221,278号;米国特許第5209746号を参照のこと。
【0063】
Time Clock(R)システム(West Pharmaceutical Services Drug Delivery & Clinical Research Centre)は、ポリオキシエチレンソルビタンモノオレエートなど、界面活性剤とともにカルナバワックス及び蜜蝋を含有する脂質性の障壁で被覆された固形の剤形である。Wildingら、Int J Pharm.1994;111:99−102;Niwaら、J Drug Target.1995;3:83−89。このコーティングはフィルムの厚さに比例した時間で水性環境において浸食又は乳化し、次にコアが分散に利用可能になる。ヒトのボランティアの試験において、遅延時間が胃での滞留時間とは独立であることが分かり、疎水性フィルム再分散は腸の酵素の有無又は胃の機械的作用又は胃腸のpHにより影響を受けないと思われる。Gazzanigaら、Int J Pharm.1994;2(108):77−83。コーティングが厚くなるにつれ遅延時間が長くなった。
【0064】
Chronotropic(R)システムは親水性の膨潤性ヒドロキシプロピルメチルセルロース(HPMC)で被覆された薬物含有コアであり、これは、放出前の遅延期間に関与する。Gazzanigaら、Eur J Biopharm.1994;40(4):246−250;Gazzanigaら、Proceed Intern Symp Control Rel Bioact Mater.1995;22:242−243;EP 0 572 942。胃に耐性のある外側の腸溶性フィルムによって、胃が空である時間の変動に関する問題を解決することができる。Sanagalliら、J Contr Rel.2001;73:103−110。HPMCの厚さ及び粘性グレードによって遅延時間が調節される。この系は錠剤及びカプセルの両方に適している。Conteら、Drug Dev Ind Pharm.1989;15(14−16):2583−2596。
【0065】
薬物不含のゲル化可能ポリマー性障壁層により分離されている2つの活性薬剤含有層を含む3層の錠剤構造から2種類の活性薬剤を含有する多層錠剤を形成することができる。米国特許第4,865,849号;Conteら、Eur J Pharm.1992;38(6):209−212;Krogel I、Bodmeier R、Int J Pharm.1999;187:175−184。この3層性錠剤の3つの側面を不浸透性のエチルセルロースで被覆し、上の部分は被覆しない。溶解液と接触すると、一番上の層に組み込まれた用量が非被覆面から急速に放出される。第二の用量は、HPMCのゲル化した障壁層が浸食し溶解した後に最下層から放出される。障壁層のゲル化及び/又は溶解の速度によって第二の要領の出現が調節される。ゲル化ポリマーには、セルロース誘導体、例えばHPMC、メチルセルロース又は様々な分子量のポリビニルアルコールなどが含まれ得、コーティング材料には、エチルセルロース、酢酸−プロピオン酸セルロース、メタクリルポリマー、アクリル及びメタクリルコポリマー及び多価アルコールが含まれる。
【0066】
破裂可能なコーティング付きのパルス系は、薬物の放出のためのコーティングの崩壊に依存する。発泡性賦形剤、膨潤剤又は浸透圧により、コーティングの破裂に必要な圧力を得ることができる。クエン酸及び重炭酸ナトリウムの発泡性混合物をエチルセルロースで被覆された錠剤コア中に組み込むことができる。コアへの水の浸透後に生成する二酸化炭素によって、コーティングの破裂後、薬物が放出される。Bussemer T、Bodmeier R、AAPS Pharm Sci.1999;1(4 suppl):434(1999)。コーティングが厚くなり、コア錠剤の硬度が向上するにつれて遅延時間が長くなる。
【0067】
薬物、膨潤剤及び破裂可能なポリマー層を含むカプセルに基づく系を設計するために、超崩壊剤(superdisintegrant)とも呼ばれる高度膨潤性物質を使用することもできる。米国特許第5,229,131号。超崩壊剤の例には、クロスカメロース、デンプングリコール酸ナトリウム及び低置換度ヒドロキシプロピルセルロースが含まれる。これらの材料が膨潤する結果、フィルムが完全に破裂し、次いで薬物が放出される。遅延時間は、外側のポリマー層の組成の関数である。HPMCなどの親水性ポリマーが存在すると遅延時間は短くなる。固形及び液体製剤両方の送達のためにこの系を使用することができる。
【0068】
ある活性薬剤の遅延放出を行い、第二の活性薬剤の遅延又はその他のタイプ(例えば即放性)の放出を行うために、多粒子系(例えばビーズ又はカプセル中ペレット)を使用することができる。例えば、米国特許第4,871,549号を参照のこと。
【0069】
時間制御型破裂系(Time−Controlled Explosion System)(藤沢薬品工業)は多粒子系であり、ここでは薬物がノンパレイユシュガーシード(non−pareil sugar seed)、次いで膨潤性の層及び不溶性の一番外側の層で被覆される。Uedaら、J Drug Targeting 1994;2:35−44;Uedaら、Chem Pharm Bull.1994;42(2):359−363;Uedaら、Chem Pharm Bull.1994;42(2):364−367;Hataら、Int J Pharm.1994;110:1−7。膨潤剤には、カルボキシメチルセルロースナトリウム、デンプングリコール酸ナトリウム、L−ヒドロキシプロピルセルロースのような超崩壊剤、酢酸ポリビニル、ポリアクリル酸、ポリエチレングリコールのようなポリマーなどが含まれ得る。あるいは、酒石酸及び重炭酸ナトリウムの混合物を含む発泡系を使用することができる。水が浸入すると、膨潤性の層が拡張し、その結果、フィルムが破裂し、続いて薬物が急速に放出される。この放出は、pHなどの環境要因及び薬物の溶解度とは独立である。コーティングの厚さを変化させるか又は多量の脂溶性の可塑剤を最外層に添加することによって、遅延時間を変化させることができる。米国特許第5,508,040号。
【0070】
浸透性制御系(Permeability Controlled System)は、浸透及び膨潤効果の組み合わせに基づく。コアは、薬物、低かさ密度の固形及び/又は液体脂質性物質(例えば鉱物油)及び崩壊剤を含有する。次に、このコアは酢酸セルロースで被覆されている。水性溶媒中での浸漬の際、水がコアに浸透し、脂質性物質と置き換わる。脂質性物質がなくなった後、臨界応力に到達するまで内圧が上昇し、その結果、コーティングが破裂する。米国特許第5,229,131号。
【0071】
別の系は、2以上のペレット又は部分からなる多数のペレット(即ち集団)でできているカプセル又は錠剤に基づく。Schultz P、Kleinebudde P.J Contr Rel.1997;47:181−189。各ペレットは、治療薬物及び水溶性浸透物質を含有するコアを有する。透水性で不水溶性のポリマーフィルムは各コアを封入する。浸透性を変化させる疎水性の不水溶性の物質(例えば、脂肪酸、ワックス又は脂肪酸の塩)をポリマーフィルムに組み込む。水の流入及び薬物の流出の速度によって、各集団のフィルムコーティングが剤形中の何れのその他のペレットコーティングとも異なるようになる。浸透物質が水中で溶解してペレットが膨潤するが、それにより薬物拡散速度が制御される。その薬物含量を連続的に放出する各ペレット集団の効果によって、1つの剤形から一連の薬物放出が起こる。ペレット間でコーティングの厚さを変化させることができる。
【0072】
遅延放出を行うために、膨潤しない浸透圧的に活性のある物質を使用することもできる。Schultzら、J Contr Rel.1997; 47:191−199;米国特許第5,260,069号。ペレットコアは薬物及び塩化ナトリウムからなる。このコアは半透性酢酸セルロースポリマーで被覆されている。このポリマーは、選択的に水を浸透させ、薬物に対して非浸透性である。遅延時間は、コーティングが厚くなるに従い、及びコーティング中のタルク又は脂溶性可塑剤の量が増えるに従い、長くなる。塩化ナトリウムは薬物の高速放出を促進する。塩化ナトリウム非存在下では、コア膨潤の程度が低くなり、その結果小さな亀裂が生じるために、遅延時間後、徐放を行うことができる。
【0073】
遅延放出を行うために、不溶性の浸透性膜で被覆された、薬物及び浸透圧的に活性のある物質(塩化ナトリウム)のコアを含有する系を使用することができる。米国特許第5,260,068号。コーティング材料には、様々なタイプのポリ(アクリレート−メタクリレート)コポリマー及びステアリン酸マグネシウムが含まれるが、これは、膜の透水性を低下させ、従ってより薄い膜の使用が可能となる。厚いフィルムは完全に破裂し得ないので、避けるべきである。コーティング材料としてエチルセルロースを使用して、予め決定された時間後に破裂させるために、腸溶性ポリマーに対する遅延時間に影響を与えることができる。Bodmeierら、Pharm Res.1996;13(1):52−56。
【0074】
溶媒中の様々な対イオンの存在によって、四級アンモニウム基を有するアクリル性ポリマーの浸透性及び水取り込みに影響を及ぼし得る。Beckertら、Proceed Int’l Symp Control Rel Bioact Mater.1999;26:533−534。このイオン交換に基づくいくつかの送達系が開発されてきた。Eudragit RS 30Dは、ポリマー側鎖中に正に分極した四級アンモニウム基を含有し、これは負の塩酸対イオンを付随するので、この目的のための好ましいポリマーである。アンモニウム基は親水性であり、ポリマーの水との相互作用を促進し、それによってその浸透性が変化し、制御下で水が活性コアに浸透できるようになる。4種類の異なる層厚さのEUDRAGIT RS30D(R)(10%から40%重量増加)でペレットを被覆することができる。遅延時間は、フィルムの厚さに相関する。EUDRAGITフィルムの薬物浸透性はペレットコア中の酢酸ナトリウム量に依存する。遅延時間後、酢酸塩とポリマーとの間の相互作用によりコーティングの浸透性が向上し、活性用量全体が数分以内に遊離する。Guo X.Physicochemical and Mechanical Properties Influencing the Drug Release From Coated Dosage Forms.Doctoral Thesis.The University of Texas at Austin;1996。
【0075】
シグモイド放出系は、アンモニオ−メタクリレートコポリマー USP/NFタイプBで被覆された薬物及びコハク酸を含有するペレットコアを含む。Narisawaら、Pharm Res.1994;11(1):111−116。遅延時間は、ポリマー膜を通じた水流入の速度により制御される。水はコア中のコハク酸及び薬物を溶解する。この酸溶液が次に、水和ポリマーフィルムの浸透性を向上させる。コハク酸に加えて、酢酸、グルタル酸、酒石酸、リンゴ酸又はクエン酸を使用することができる。浸透性の向上は、フィルムの水和が向上することにより説明できるが、これによって自由体積が増加する。酸含有コアを有する被覆送達系を設計するために、これらの知見を使用した。Narisawaら、Pharm Res.1994;11(1):111−116;Narisawaら、J Contr Rel.1995;33:253−260。インビトロ遅延時間は、ビーグル犬で試験した際のインビボデータとよく相関した。Narisawaら、J Contr Rel.1995;33:253−260。
【0076】
本発明は、紅潮抑制剤と組み合わせた遅延放出型の本発明のナイアシン製剤を含む医薬組成物を包含する。1つの剤形又は別個の剤形で遅延放出ナイアシン及び紅潮抑制剤を与えることができる。したがって、例えば、本医薬組成物は、外側に即放型紅潮抑制剤成分及び内側に遅延放出ナイアシンを有する固形剤形を含み得る。好ましい実施形態において、紅潮抑制剤は、ナイアシンが放出される約30分から約40分前に放出される。
【0077】
次の実施例は、本発明の複数の実施形態をより詳細に説明するためのものである(これらに限定されない。)。
【実施例1】
【0078】
この実施例において次の処方を使用した:
【0079】
【表6】
【0080】
好ましくは、METHOCEL(R)K−15M Premiumを使用する場合、METHOCEL(R)K−15M Premiumに対する粒子サイズ規格は、最低90%が100メッシュUS標準ふるいを通過することである。METHOCEL(R)K−15M Premium CRの場合、好ましくは最低99%が40メッシュUS標準ふるいを通過し、最低90%が100メッシュUS標準ふるいを通過する。
【0081】
20kgのバッチサイズの場合、上記処方に従い、砕いた(delumped)粒状ナイアシン及び賦形剤の重量を測定し、次いで8−quartブレンダーに添加し、24rpmで10分間混合した。特に、METHOCEL(R)K−15M及びステアリン酸を分散させるために12メッシュ(1.68mm)スクリーンを選択し、粒状ナイアシン及びポビドンK−90(場合によってはふるいにかける、粉砕する、又は両方)を分散させるために16メッシュ(1.19mm)スクリーンを選択した。30kNで19mm長の楕円形のツーリングを有するBWI Manesty Beta Pressを用いて、得られた粒状成分を直接錠剤へと圧縮した。目標とする18kPの錠剤硬度に対して、標準的な錠剤硬度テスターを用いて、錠剤の硬度(即ち、当業者にとって公知の標準的圧縮試験方法によって測定した場合の錠剤の圧縮強度)を16kP(キロポンド)から22kPの範囲内に調節した。場合によっては、40メッシュスクリーンなど、ステアリン酸又はポビドンをメッシュスクリーンに通してふるいにかけることができ、代替的実施形態において、混合段階(1又は2)及び混合時間(10、15又は20)を変化させることができる。
【0082】
OPADRY(R)Orange 03B93199の2%増量の色彩塗料で、得られた圧縮錠剤を被覆した。コーティング条件は次のとおりであった。
【0083】
【表7】
【0084】
安定性試験前後での、ナイアシンアッセイ、ナイアシン溶解、錠剤の水分及び被覆錠剤の外見を比較することによって、被覆したナイアシン1000mg直接圧縮錠剤が40℃/75%相対湿度(RH)及び25℃/60%相対湿度で3ヶ月間安定であることが分かった。
【0085】
図5は、本発明の実施形態による錠剤製剤を調製するための直接圧縮製造プロセスの流れ図を示す。
【0086】
別段の断りがない限り、次の実施例に記載の本発明の1000mg徐放ナイアシン製剤は実施例1に従い調製した。
【実施例2】
【0087】
本明細書中に記載のプロセスを使用して、下記表8及び9で述べる含量濃度を有する、500mg及び750mg徐放直接圧縮錠剤(被覆又は被覆なし)を調製することができる。
【0088】
【表8】
【0089】
【表9】
【0090】
500mg及び750mg錠剤の場合、表8及び表9で示す成分濃度に従い、砕いた(delumped)粒状ナイアシン及び賦形剤の重量を測定し、次いで成分を十分に混合するために適切なブレンダー又はミキサー中で適切な時間、混合する。次に、上述のBWI Manesty Beta Pressなどの適切なプレスを用いて、得られた粒状成分を錠剤へと直接圧縮し、所望のような500mg又は750mg錠剤強度を形成することができる。場合によっては、500mg又は750mg錠剤強度を当技術分野で公知のように、色彩塗料などで被覆することができる。
【実施例3】
【0091】
被覆徐放1000mgナイアシン直接圧縮マトリクス錠剤と1000mg NIASPAN(R)との間の紅潮の相対的発生率
方法
本試験は、単一施設で行われた、無作為の二重盲検、ダブルダミー、単回投与、プラセボを対照とした、3期クロスオーバー、紅潮誘発試験である。試験中にアスピリン又はNSAIDを対象に使用させなかった。
【0092】
本試験には、ボディーマスインデックス(BMI)が22から31の、18歳から70歳の健常な非喫煙の男性ボランティアが含まれた。人間ドック、病歴、心電図及びスクリーニングの来院時又は最初の試験期間の入院の来院時に行った臨床検査によって、対象が健常であることを確認した。ナイアシン又は関連誘導体に対するアレルギー又は過敏性がある場合;過去3年以内に薬物乱用又は依存症があった場合;偏頭痛、糖尿病、胆嚢疾患、肝臓疾患、重度の高血圧又は低血圧、心臓の異常、腎臓疾患又は薬物誘発性ミオパシーの病歴がある場合は対象を除外した。対象は、試験開始の21日以内のいかなる処方投薬も、又は、試験開始の10日前以内の市販薬、ビタミン又はハーブも摂取してはならなかった。
【0093】
試験期間1への入院前21日以内でスクリーニング手順を完了した(図6)。3回の各試験期間中、第1日の午前7:00前後から第2日の午前の全ての試験手順が完了するまで対象を隔離した(午前7:00から午前10:00の間)。食事構成及び開始時間は各試験期間で同じであった。各試験期間中、対象は、ナイアシン及び脂肪含量を調節した特別メニューに従う食事を取った。併用薬、ビタミン又はハーブ及び/又は栄養剤は試験中一切禁止した。
【0094】
試験治療
3回の試験期間中に投与された製剤を図6で記す。試験治療は、本発明の2個のフィルム被覆1000mg錠剤製剤を使用し(実施例1参照)(Test−改質ナイアシンER錠剤)、一方、参照治療は、2個の非被覆1000mg市販ナイアシンER錠剤(Reference−NIASPAN(R))を使用した。対照治療は、2個の非被覆プラセボ錠剤(対照)を使用した。この試験は対象が報告する紅潮に焦点を当てたので、対象及び試験者が治療において投与される製剤がどれであるかについて完全に分からないことが重要であった。いくつかの方法によって分からないようにした。各活性治療において、活性錠剤に似ている2個のフィルム被覆プラセボ又は非被覆プラセボ錠剤を活性錠剤とともに同時投与し、全対象が治療にかかわらず2個のフィルム被覆及び2個の非被覆錠剤を摂取するようにした。また、不透明な投与カップから対象に試験薬を投与し、試験薬投与中、対象に目隠しを施した。予想されるプラセボ反応に対して紅潮の結果を補正するために、試験にプラセボ−対照治療を入れた。
【0095】
各試験期間、第1日の午後11:00前後に、無作為計画に従うクロスオーバー方式で試験薬の単回投与を行った。各試験期間の間に7日以上のウォッシュアウト期間を置いた。試験者及び現場人員に対して治療割り当て方式が分からないようにし、治療割り当て調製及び/又は投与に関わった何れの現場人員も、治療中に発生した有害事象を収集又は評価することを禁じた。
【0096】
低脂肪スナックの後に、水240mLとともに、各用量を経口投与した。このスナックを全て、試験薬投与前15分以内に摂取させた。錠剤を1度に一緒に飲ませるか、又は1個を飲んだ後すぐに他方を飲ませ、投与を完了するのに1分を超えないよう各対象に指導した。錠剤を咀嚼又は噛むことは禁じた。錠剤を飲み込むために対象がさらなる水を所望した場合、120mL単位でさらなる水を与えた。用量を摂取したことを確認するために、試験用量の投与後、各対象の口腔を調べた。
【0097】
紅潮可変要素
第一の紅潮可変要素は、対象が報告する紅潮事象又は発現の発生率であった。紅潮事象又は発現を次の共通の紅潮症状の1以上として記した:発赤、熱感、刺痛及び掻痒。各試験期間中、試験薬物投与後、最長で8時間、1時間ごとに、紅潮症状の有無を対象に評価させた。紅潮症状の開始及び停止時間を対象に記録させ、水平の10cm視覚的アナログ尺度(VAS)上で垂直線を作ることによって(「なし」(0)を左端に「容認できない。」(100)を右端にした。)各症状の強度(重症度)のランク付けを行わせた。コンピュータの紅潮日誌に情報を記録した。
【0098】
第二の紅潮可変要素には、紅潮発現の回数、強度及び全体的紅潮事象及び紅潮の個々の症状(発赤、熱感、刺痛及び掻痒)の両方に対する紅潮の持続時間が含まれた。各対象が、試験期間中に発生した1以上の共通紅潮症状の最初のものの開始時間に始まるものとして定義される、最初の紅潮事象又は発現の全体的強度をランク付けした。紅潮発現の終了時間は、その発現において起こった1以上の共通の紅潮症状の最後の停止時間(また、この後、無症状期間が30分以上続く。)として定義した。
【0099】
統計分析
マクネマー検定(nQuery Advisor(R)、バージョン5.0)を用いて5%のアルファ(α)で治療間の紅潮の発生率の統計的有意差を示すためには144名の対象のサンプルサイズが必要であると決定した。対象の適切な人数が試験を完遂し、少なくとも2つの治療から評価可能なデータを得ることを確実にするために、早期に撤退した患者を入れ替えた。
【0100】
対応のある比率の同等性のマクネマー検定を用いて治療群間で最初の有効性評価(紅潮の発生率)を比較した。少なくとも2回の試験期間で少なくとも1回試験薬投与を受けた対象において、ナイアシンERのTest製剤と参照製剤との間で最初の比較を行った。ナイアシンとプラセボとの間の比較も行った。マクネマー検定(カテゴリ変数に対して)又はマッチドペアt検定の何れかを用いて第二の評価を比較した。比較は全て両側検定であり、アルファ(α)=0.05で行った。
【0101】
結果
全部で156名の対象をこの試験に登録し、少なくとも1回の試験薬投与を受けさせた。これらの平均年齢は33.5歳であり、これらの平均BMIは26.2であった。対象デモグラフィックの要約を下記表10で示す。
【0102】
【表10】
【0103】
ピリオド1において156名の対象全員が試験薬の投与を受け、143名の対象(92%)がピリオド2で試験薬の投与を受け、131名の対象(84%)がピリオド3で試験薬の投与を受けた。全部で130名の対象(83%)が全3期間での投与を完遂した。26名の対象(17%)は試験から途中で撤退し:8名(5%)が同意を取り消し、3名(2%)がフォローアップできず、2名(1%)で有害事象が起こり、2名(1%)がプロトコール違反し、1名(1%)が薬物スクリーニング検査で陽性となり、残り10名(6%)が「その他」の理由で撤退した。十分な能力を保証するために、途中で試験から撤退した対象のうち11名を置き換えた。
【0104】
紅潮
目的どおり、活性治療における紅潮発生が対照治療のものよりもおよそ4倍高かったので、紅潮誘発を達成した。表11は、少なくとも1回の試験薬投与を受け、少なくとも1回の試験期間を完遂した対象として定義される包括解析(ITT)集団における、最初の紅潮事象の、発生率、強度及び持続時間を示すが、入れ替えた対象は含まなかった。この試験で見られたプラセボ反応は、一般にプラセボ反応に特有である。
【0105】
【表11】
【0106】
参照(市販ナイアシンER)(参照)に対するTest(改質ナイアシンER)に関する、少なくとも2回の試験期間で少なくとも1回の試験薬投与を受けた対象における最初の紅潮事象の発生率及び全体的強度及び持続時間:A)最初の紅潮事象の発生率(p=0.0027);B)VASに基づく最初の紅潮事象の強度;中央値は、[平均値が、Testで35.6±22.78(Min、Max:0.0、99.0)であり、参照で52.8±23.86(Min、Max:0.0、95.0)、p<0.001であった。]ことを示し;C)最初の紅潮事象の持続時間(分);中央値は、[平均値が、Testで130.3±95.01(Min、Max:9.0、473.0)であり、参照で195.7±136.32[Min、Max:5.0、984.0]、p<0.0001であった。]ことを示した。
【0107】
図7で示されるように、最初の有効性評価に対して、少なくとも2回の試験期間で少なくとも1回の試験薬投与を受けた対象中で、Test製剤での治療中に118名(89%)の対象で紅潮が起こり、参照製剤によって130名(98%)の対象で治療中に紅潮が起こった。0.0027のp値でこの差は統計的に有意であった。
【0108】
図8及び9は、最初の紅潮事象の中央値強度及び持続時間を示す。これらの個々の中央値との強度及び持続時間に対する平均値の比較から、これらのデータの基礎をなす分布が非対称であったことが示唆された。Test治療の結果、参照治療と比較して、紅潮強度中央値が42%低下し(平均紅潮強度は33%低下)、紅潮持続時間中央値が43%低下(平均紅潮持続時間は33%低下)した。対応のあるt検定によって、最初の紅潮事象の平均強度(p<0.0001)及び平均持続時間(p<0.0001)の両方に関して、Test治療で統計的に有意な改善が示された。
【0109】
参照製剤に対するTest製剤で、4種類の紅潮症状(発赤、熱感、刺痛及び掻痒)のそれぞれの発生率が低下した(図10)。この2種類の製剤の比較は有意差があり、マクネマー検定を用いて、最初の紅潮事象に対する4種類の個々の紅潮症状のそれぞれに対してTest製剤が選択される。発赤は、Test製剤によって71%で発生したのに対して参照製剤では86%で発生し(p=0.0016);熱感は、Test製剤によって68%で発生したのに対して参照製剤では80%で発生し(p=0.0163);刺痛は、Test製剤によって47%で発生したのに対して参照製剤では62%で発生し(p=0.0039);掻痒は、Test製剤によって48%で発生したのに対して参照製剤では65%で発生した(p=0.0015)。
【0110】
このデータから、市販の製剤と比較して、本発明の製剤によって、紅潮の発生率、強度(重症度)及び持続時間が低下することが示される。ナイアシン治療を受けたことがなかった対象に単回大量(2000mg)用量を投与することによって紅潮を誘発するようにこの試験が計画されたにもかかわらず、全体的に市販ナイアシンER製剤NIASPAN(R)(98%)と比較して、本発明の製剤で(89%)紅潮の発生率が統計的に有意に9%低下した。この紅潮誘発試験での本発明の製剤の投与の結果、また、紅潮強度及び持続時間が非常に統計的に有意に低下した。市販ナイアシンER治療と比較して、中央値紅潮強度及び持続時間はそれぞれ42%及び43%低下した。また、最初の紅潮事象の持続時間は本発明の製剤によって1時間を超えて短くなった。
【実施例4】
【0111】
この試験の目的は、2000mgの単回投与で投与した際の、市販の1000mgNIASPAN(R)錠剤(REF)との、本発明の1000mg徐放ナイアシン錠剤(本明細書中で以後「改質」錠剤と呼ぶ。)(Test)の生物学的同等性(BE)を調べることであった。
【0112】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙男性及び女性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、2期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間に少なくとも10日間のウォッシュアウト期間を置いて、2回の別個の時に、2000mgの同じ単回投与で、2つのナイアシン製剤、Test及びREFの投与を受けた。試験製品は改質1000mg徐放ナイアシン錠剤であり、参照製品(REF)は1000mg NIASPAN(R)錠剤であった。各期間の第1日の22:00前後から開始する低脂肪スナックの後に水240mLとともに各用量を投与した。各試験期間中、試験施設で患者を隔離し(ピリオド1に対して5日間、ピリオド2に対して6日間)、スポンサーが提供するメニューに従い食事を与えた。他の薬、ビタミン、ハーブ又は栄養剤を試験中禁じた。
【0113】
−30分(投与前)、1、2、3、4、4.5、5、6、7、8、10、12、14、16及び24時間(投与後)の間隔で、投与前30分以内から、投与後24時間まで、連続血液試料を回収した。−24から−18、−18から−12、−12から−6及び−6から0時間(投与前);0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)の間隔で、投与前24時間から投与後96時間まで尿を回収した。ナイアシン及びニコチン尿酸(NUA)について血漿を分析した。ナイアシン及びその代謝産物:NUA、N−メチルニコチンアミド(MNA)及び2−PY(N−メチル−2−ピリドン−5−カルボキサミド)について尿を分析した。
【0114】
ナイアシンは十分に代謝され、血漿濃度から、その主要な代謝産物の1つであるNUAと比較して非常に変動性が高いことが示される。したがって、NUAに対する最大血漿濃度(Cmax)がナイアシン吸収速度を決定するために使用されてきた。AUCは非線形的な薬物動態の影響をより受けやすいので、NIASPAN(R)NDAで示されるように、総尿回収は、AUCよりも、吸収の程度のより正確な目安である。したがって、尿中でナイアシン及びその代謝産物のうち3種類、NUA、MNA及び2PYとして排出されるナイアシンの総量は、ナイアシン吸収の程度の目安として役立つ。本プロトコールにおいて定義される、NUA生物学的同等性を評価するための主要な可変要素は、ゆえに、NUAに対するCmax及びナイアシン及び3種類の代謝産物(NUA、MNA及び2PY)の総尿回収である。
【0115】
Test薬は、本発明の改質1000mg徐放錠剤の2個の錠剤からなる。REF薬は、1000mg NIASPAN(R)錠剤の2個の錠剤からなる。治療を少なくとも10日間離した。
【0116】
対象は、各期間中、病院に隔離されているときは、各日の同じ時間に食事を開始した。食事は各期間に対して同じに維持し、各食事を全量摂取するよう求めた。朝食、昼食、夕食及び夜食はそれぞれ、07:00、12:00、18:00及び21:45前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに水240mLを与えたのに加え、第−1日に水を最低720mL飲み、第1日から5日に1440mL飲むよう求めた。
【0117】
第−1日に、夕食及び夜食を供した。第1日から5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では、投与直前15分以内に取らせた。ピリオド2の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0118】
薬物動態の評価
a.血漿回収及び分析
各期間において、投与前30分以内から、投与後24時間まで、連続血液試料を回収した(治療あたり15試料)。ヘパリンナトリウムを含有する1本の10mL真空採血システムに各血液試料を回収し、氷片及び水浴中で回収後最低5分間、試料を冷却した。4℃にておよそ3000rpmで15分間試料を遠心して血漿を分離した。各血漿試料を2つ、アリコートA及びB、に分け、予め冷却し適切にラベルを貼った2本のポリプロピレンチューブに移した。次に、試料をおよそ−20℃で凍結保存した。
【0119】
有効な液体クロマトグラフィータンデム質量分析(LC/MS/MS)によって、ナイアシン及びNUA濃度を分析した。ナイアシン及びNUA濃度を同じ注入から得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0120】
b.採尿及び分析
次の間隔で採尿した:−24から−18、−18から−12、−12から−6及び−6から0時間(投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(全部で11回の回収)。
【0121】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、採尿した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。空の容器の重量を測定し、グラムの小数点以下1桁に四捨五入して(例えば100.1g)、これを容器に記入し、実験室のソースドキュメントワークシートに記録した。各間隔の終わりに、容器及び回収した尿の総重量を測定し、グラムの小数点以下1桁に四捨五入して記録した。尿重量は、容器+尿の総重量から空の容器の重量を差し引くことにより得られた。場合によっては、ある一定の回収間隔中の尿体積が1個の容器の容量を超え、従って採尿を完遂するために第二の容器を必要とした。各採尿間隔の開始及び終了日時も記録した。各回収間隔からの2つのアリコート(各およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。特定の回収間隔中に複数の容器が必要であった場合、両方の容器からの尿を一緒にして混合し、その後アリコートを取った。分析までおよそ−20℃で試料を凍結保存した。
【0122】
有効なLC/MS/MSにより、ナイアシン、NUA、NMA及び2−PYの濃度について尿試料を分析した。同じ注入から尿ナイアシン及びNUA濃度を得て、一方、NMA及び2−PY濃度を同じ注入から得た。尿において、LLQ値は、ナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2PYのLLQ値はそれぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0123】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対するPKパラメータを計算するために十分な情報を提供する対象からのデータが、PK分析に含まれた。各治療の投与後、各対象に対して次のPKパラメータを計算した:
・Cmax:観察される最大濃度
・Tmax:最大観察濃度の時間
・AUClast:直線的台形規則による、時間0から最後の測定可能(0ではない)濃度までの濃度−時間プロファイル下面積
・AUCinf:時間0から無限大の血漿濃度−時間プロファイル下面積;AUClast及びCl/λの合計として計算(Clは、最後に観察された濃度であり、λは、時間プロットに対する自然対数濃度のプロットから得られた最終消失速度定数である。)
・T1/2:見かけの最終半減期;0.693/λの比として計算
ナイアシン及びその代謝産物(NUA、MNA及び2−PY)の尿データから、次のパラメータを計算した:
・CumXu:投与後0から96時間の尿から回収した各代謝産物の累積量。
【0124】
・%Fe:投与後96時間での、ベースライン回収及び分子量に対する補正後の、ナイアシン投与に対する、尿中で排出された各代謝産物の分画。
【0125】
・総%Fe:投与後96時間における4種類の代謝産物の総分画。
【0126】
尿中の各被分析物に対する%Feを以下のように計算した:
【数1】
【0127】
定量限界未満の濃度をゼロとして処理した。血漿分析に対して、実際の試料回収時間を使用してPKパラメータを計算した。各代謝産物濃度に各間隔で回収された尿の体積を掛けることによって、尿中で回収されたナイアシン及びその代謝産物の量を決定した。24時間投与前間隔で回収された量を差し引くことによって、投与後各24時間間隔に対して尿中で回収された総量をベースラインに対して調整した。投与後測定値がベースライン未満となった場合、その量を0とした。ナイアシン及びその代謝産物の分子量は、ナイアシン、NUA、MNA及び2−PYに対してそれぞれ、123.1、180.2、137.1及び153.1であった。4つの尿被分析物からの%Feの合計を計算し、総%Feとした。
【0128】
WinNonlin Linear Mixed Effects Modeling/bioequivalence、バージョン5.0.1(2005年7月26日)を用いて、(上述のような)生物学的利用率パラメータを計算した。
【0129】
統計分析
WindowsTM用SAS(R)Systemバージョン8.2を用いて、上記で計算された生物学的利用率パラメータの統計分析を行った。
【0130】
治療及び期間によって、血漿薬物動態パラメータ(Cmax、Tmax、T1/2、AUClast及びAUCinf)、それらの自然対数変換値(Tmax及びT1/2を除く。)及び要約統計量(n、平均、std、中央値、min、max、CV%)を計算した。ナイアシン及びNUAの血漿濃度を時間及び治療によってまとめた。
【0131】
ナイアシン及びNUA PK分析の場合、自然対数変換したCmax及びAUClastのデータが正規分布に従い、2つの治療間で独立であると仮定する。固定効果として治療、期間及び順序を、変量効果として順序内の対象を用いて、SAS PROC MIXEDを使用して混合効果によってこのデータをANOVAモデルに適合させた。Cmax及びAUClastのTest/REF比及びそれらの対応する90%信頼区間をこのモデルに基づき評価した。
【0132】
尿からのナイアシン及びその代謝産物の平均回収を計算し、治療により及び間隔によりまとめた。個々の成分のCumXu及び%Fe及び投与後96時間での合計を計算し、治療によりまとめた。
【0133】
血漿PK分析に対して用いたものと同じANOVAモデルに適合させることにより、総%FeのTest/REF平均比に対する90%信頼区間(CI)を計算した。
【0134】
対象のデモグラフィック可変要素(年齢、性別、人種、体重、身長及び肘幅)を性別によりまとめた。連続的デモグラフィック可変要素の、平均、標準偏差(SD)、中央値、最小及び最大を計算した。
【0135】
結果
対象の動態を表12でまとめる。プロトコールの包除基準に合致した後に全部で44名の対象を試験に登録した。対象者全てが、少なくとも1回の試験薬投与を受け、このうち41名がこの試験を完遂した。44名の対象がプロトコールの無作為治療割り当てに従いピリオド1で試験薬投与を受け;41名の対象がピリオド2で試験薬投与を受けた。全部で3名の対象が試験から撤退した。対象0012及び0039がピリオド2で撤退した。対象0038はピリオド2で同意を撤回した。試験から撤退した対象数は、前もって容認するとした10%脱落の範囲であり、この試験の結果又は結論に影響しないと考えられた。
【0136】
【表12】
【0137】
登録した44名の対象のうち、25名の対象が男性で19名が女性であった。平均年齢は54.5歳;平均体重は169.8ポンド;平均身長は68.0インチ;平均肘幅は2.6インチであった。対象のうち37名が白人、6名が黒人、1名がアメリカインディアンであった。表13で詳細なデモグラフィックをまとめる。
【0138】
【表13】
【0139】
a.生物学的同等性の評価
尿の分析のために、1g/mLの比重を使用して、尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究において、962個の試料で測定した平均比重が1.009g/mLであり、962個の試料で測定した最大比重が1.025g/mLであった。)。
【0140】
治療によるナイアシン及びNUAの平均血漿濃度のプロットを図11及び12でそれぞれ示す。平均尿中回収データを図13で示す。
【0141】
b.血漿NUA及び尿中で排出される総量
表14は、2種類の主要な可変要素(NUAに対するCmax及びナイアシン及び3種類の代謝産物の総尿中回収)に対する、及びNUA AUClastに対する、平均(SD)及び統計結果を示す。この表は、投与後に嘔吐の発現があった対象0001、0003及び0014に対する参照治療あり及びなしでのBE分析の結果を与える。
【0142】
対象0001は、ピリオド2でのREF製品投与後7時間20分で嘔吐があった。対象0003は、ピリオド2でのREF製品投与後それぞれ8時間34分及び9時間及び20分で2回の嘔吐があった。対象0014は、ピリオド1でのREF製品投与後11時間20分で嘔吐があった。この3名の対象全員の嘔吐発現時間は、投与から少なくとも7時間20分後であった。NUA及びナイアシンの両方に対するTmaxは、投与後6時間以内であった。したがって、嘔吐はこれらの対象のPKパラメータに影響しないと思われた。
【0143】
【表14】
【0144】
上記の表で示されるように、NUA Cmaxの平均Test/REF比に対する90%CIは、80−125%の生物学的同等性の範囲外であったが、尿から回収されたナイアシン及び代謝産物のTest/REFの平均比に対する90%CIは、80−125%以内であった。この結果は、対象0001、0003及び0014に対するREF治療あり及びなしで同様であった。
【0145】
治療によって、各対象に対して最終消失速度を計算した。それぞれTest及びREFに対して、平均NUA T1/2は3.16及び3.47時間であり、平均NUA Tmaxは5.55及び5.80時間であり、平均NUA AUCinfは12510.8及び18980.8ng*hr/mLであった。
【0146】
c.血漿ナイアシン
統計分析付きで血漿ナイアシンに対する平均PKパラメータを表15で示す。この表は、対象0001、0003及び0014に対するREF治療あり及びなしでのBE分析の結果を与える。ナイアシンCmax及びAUClastのTest/REFの平均比は100%未満であった。この比に対する対応する90%CIは、変動が大きかったため、80−125%間隔の外であった。この結果は、対象0001、0003及び0014に対するREF治療あり及びなしで同様であった。
【0147】
【表15】
【0148】
それぞれTest及びREFに対して、ナイアシンの平均T1/2は5.46及び4.42時間であり、平均Tmaxは5.56及び5.55時間であり、平均AUCinfは13987.8及び35296.6ng*hr/mLであった。
【0149】
d.個々の被分析物の尿中回収
個々の被分析物の平均尿中回収を表16で与える。
【0150】
【表16】
【0151】
上記の表で示されるように、平均尿中回収は2PYに対して最大であり、続いてMNA、NUA及びナイアシンであった。
【0152】
e.生物学的同等性評価の結論
NUA Cmax及びナイアシン及びその代謝産物の尿中回収(総%Fe)の平均Test/REF比に対する90%CIに基づき、生物学的同等性を評価した。総%Feに対するTest/REF平均比の90%CIは80−125%の求められるBE範囲内であったが、NUA Cmaxについては生物学的同等性の範囲外であった。NUA AUClastを含む補助的測定に対するTest/REF平均比の90%CIもまた、80−125%の範囲から外れた。したがって、改質1000mgERナイアシン錠剤(Test)は、NIASPAN(R)1000mg錠剤(REF)と比較した場合、吸収速度が低く、吸収が同程度である。Test治療はREF治療と生物学的に同等ではない。
【実施例5】
【0153】
2000mgナイアシン単回投与後の、市販のNIASPAN(R)500mg錠剤に対する本発明の1000mg徐放ナイアシン錠剤の3種類の製剤(本明細書中で以後「改質」錠剤と呼ぶ。)の生物学的同等性を調べるために、この試験を計画した。
【0154】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙女性及び男性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、4期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間で10日以上のウォッシュアウト期間を設けて4回、経口試験薬、2000mgナイアシン、の同じ用量の投与を受けた。各対象は、1000mgERナイアシン製剤の2個の錠剤(ERN−1、ERN−2、ERN−3)及び4個の500mg NIASPAN(R)錠剤の投与を受けた。
【0155】
低脂肪スナック(22時前後に開始)の後に、水300mLとともに各用量を投与した。対象は、各治療期間中、スポンサーが提供するメニューに従い食事を摂取した。試験中、他の薬、ビタミン、ハーブ又は栄養剤を禁じた。投与前及び投与後24時間まで頻繁に血液試料を回収し、投与前24時間及び投与後96時間、尿を回収した。NUA及びナイアシンに対して血漿を分析した。ナイアシン及びその3種類の主要な代謝産物、NUA、MNA及び2PYについて尿を分析した。各治療の5日間の治療期間中、対象を隔離した。
【0156】
各治療期間中、ナイアシン含量を調整した食事(朝食、昼食、夕食及び夜食)を提供した。
【0157】
参照治療は、高力価の造粒物(ナイアシン、ポビドン及びヒドロキシプロピルメチルセルロース[HPMC])(次いで錠剤への圧縮前にステアリン酸及びさらなるHPMCと混合される。)からなる市販の500mg NIASPAN(R)(NSP)製剤であった。
【0158】
試験治療は、下記表17に従い調製された3種類の様々な改質1000mg NIASPAN(R)製剤(ERN−1、ERN−2及びERN−3)であった。
【0159】
【表17】
【0160】
上記表17で指定された処方に従い、粒状ナイアシン、METHOCEL(R)K15M、ポビドンK90及びステアリン酸の重量を測定し、次いで8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0161】
全ての食事及び飲料はアルコール及びキサンチン不含であった。ナイアシンは通常の食事で摂取可能なので、およそ25mg/日のナイアシン摂取を維持するためにこの試験の食事を調整し、一方、対象を病院に拘束した。低脂肪スナックの直後、22:00前後に各用量を投与した。
【0162】
対象は、各期間中、毎日、同じ時間に食事を開始し、病院に拘束された。食事は各期間で同じであり、各食事の全量を摂取させた。朝食、昼食、夕食及び夜食は、それぞれ0700、1200、1700及び2145前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに与えた300mLの水に加え、第−1日に水を720mL以上飲み、第1、2、3、4及び5日に水を1440mL以上飲むよう求めた。
【0163】
第−1日に、夕食及び夜食を供した。第1、2、3、4及び5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では15分以内に取らせた。各期間の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0164】
薬物動態の評価
a.血漿回収及び分析
各期間、投与前30分以内(即ち投与前)及び投与後1、2、3、4、4.5、5、6、7、8、9、10、11、12、14、16及び24時間で血液試料を得た。対象をまっすぐに椅子に座らせ、試験区域で試料を採取した。ヘパリンナトリウムを含有する7mL真空採血システムに採血し、氷片及び水浴中で回収後少なくとも5分間、試料を冷却した。4℃にておよそ3000rpmで15分間試料を遠心して血漿を分離した。冷却し予めラベルした2本のポリプロピレンチューブに血漿分画を移した。分析までおよそ−70℃で試料を凍結保存した。
【0165】
MS/MS検出付きのHPLCクロマトグラフィーによって血漿ナイアシン及びNUA濃度の生物分析を行った。同じ注入からナイアシン及びNUA濃度を得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0166】
b.採尿及び分析
次の間隔で採尿した:−24から−18、−18から−12、−12から−6及び−6から0時間(即ち投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(治療あたり全部で11回の回収)。
【0167】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、回収した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。各間隔中に回収した尿の総重量を測定してグラムの小数点以下1桁に四捨五入し(例えば100.1g)、記録した。各採尿間隔の開始及び終了の日時も記録した。各回収間隔からの2つのアリコート(およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。Kos、プロトコール番号、対象番号、日付、回収間隔、試験日及び期間を識別するために、分析用検体にラベルを付した。発送の準備ができるまで、およそ−20℃でアリコートを保存した。病院側は、比重測定のためにさらなるアリコートを得た。
【0168】
ナイアシン、NUA、MNA及び2−PYについて尿試料を分析した。MS/MS検出付きのHPLCクロマトグラフィーによって、尿中ナイアシン、NUA、MNA及び2−PY濃度の生物分析を行った。ナイアシン及びNUA濃度を同じ注入から得た。
【0169】
尿中で、LLQ値はナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2−PYのLLQ値は、それぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0170】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対する薬物動態パラメータを計算するために十分な情報を提供する対象からのデータが薬物動態分析に含まれた。各治療の投与後、各対象に対して次の薬物動態パラメータを計算した:
血漿ナイアシン及びNUAデータから:
・Cmax:観察される最大血漿濃度
・Tmax:最大観察濃度の時間
・AUC0−last:血漿濃度−時間プロファイル下面積;直線的台形規則によって時間0から最後の測定可能濃度まで計算
NUAデータから、さらなるPKパラメータも計算した:
・AUC0−inf:血漿濃度−時間プロファイル下面積;AUC0−last+Ct/Kelとして時間0から無限大まで計算(Ctは最後に観察された定量可能濃度であり、Kelは最終消失速度定数である。)
・T1/2:0.693/Kelとして計算された最終半減期
尿中のナイアシン、NUA、MNA及び2PYデータから:
・CumXu:各被分析物個々に対する尿中回収(即ち尿中で回収された各被分析物の量)
・Fe:
【数2】
として計算された尿中で排出された分画
・総%Fe:ナイアシン、NUA、MNA及び2PYの総回収。
【0171】
LLQ未満の濃度をゼロとして処理し、分析において実際の試料回収時間を使用した。WinNonlin Professional Network Edition、バージョン4.1を用いて、血漿ナイアシン及びNUA濃度の個々のプロットを生成させた。血漿ナイアシン及びNUA濃度及び尿中回収データの平均プロットはWinNonlin4.1及びMicrosoft(R)Excel 2000により生成させた。WinNonlinによって各プロファイルから血漿薬物動態パラメータを決定した。各血漿プロファイルでの定量可能なナイアシン濃度が少数であったこと及びはっきりと定義される最終相がないことから、血漿ナイアシンデータに対して最終相勾配及び見かけの半減期を計算しなかった。
【0172】
WinNonlin4.1及びExcel 2000を用いて全ての尿中薬物動態パラメータを決定した。被分析物濃度に各間隔に対して回収された尿の体積を掛けることによって、尿中で回収された各被分析物の量を調べ;次に、24時間投与前間隔で見出された量を差し引くことによって、投与後24時間間隔に対する尿中回収量をベースライン回収に対して補正した。
【0173】
被分析物の分子量は、ナイアシン、123.1;NUA、180.2;MNA、137.1;2PY、153.1である。個々の被分析物の%回収を合計し、回収した用量の総%を計算した。
【0174】
統計分析
WindowsTM、バージョン8.02用のSAS Systemを用いてデモグラフィックをまとめた。平均、標準偏差(SD)、中央値、最小及び最大値により、連続的デモグラフィックデータをまとめた。
【0175】
自然対数変換データを用いて、WinNonlin4.1で構築された生物学的同等性ウィザードを使用することによって、生物学的同等性パラメータを評価した。このモデルには、順序、順序内の対象、期間及び治療が含まれた。
【0176】
自然対数変換したデータに基づき、最小二乗平均の参照に対するTestの比(ERN−1/NSP、ERN−2/NSP、ERN−3/NSP)についての古典的な90%信頼区間(CI)推定により生物学的同等性を評価した。90%CIが80から125%内であった場合、治療が生物学的に同等であるとみなした。生物学的同等性決定に対して、使用したパラメータは、血漿中のNUAに対するCmax及び尿中で排出されるナイアシン及び代謝産物の総量であった。ナイアシン血漿(Cmax及びAUC0−last)及び尿データに対する個々の%Fe(ナイアシン、NUA、MNA、2PY)に対しても信頼区間を調べた。
【0177】
結果
プロトコールの包除基準に合致した40歳から70歳(両端を含む。)の44名の健常な非喫煙女性及び男性をこの試験に登録した。試験薬の最初の投与を受ける少なくとも120日前から喫煙せず、病歴、健康診断、心電図(ECG)及び臨床検査評価から臨床的に意味のある知見がないことを基として、対象を選択した。
【0178】
登録した44名の対象のうち、41名の対象がこの試験を完遂した。28名の対象が男性であり、16名の対象が女性であった。平均年齢は51歳、平均体重は171ポンド、平均身長は68インチであった。36名の対象が白人、6名が黒人、2名がヒスパニックであった。詳細なデモグラフィックを下記表18で示す。
【0179】
【表18】
【0180】
a.生物学的同等性の評価
参照(NSP)治療に対して43名の対象が血漿及び尿データを提供した。42名の対象が、ERN−1及びERN−2試験治療に対して血漿及び尿データを提供した。41名の対象が、ERN−3試験治療に対して血漿及び尿データを提供した。平均の表、平均プロット、個々のプロット及び濃度リストに対して公称の時間を使用した。PK分析に対して次の規則を使用した:
1−10時間(両端を含む。)の試料採取時間の場合:5分以上ずれる場合は実際の時間を使用した。5分未満のずれの場合は公称の時間を使用する。
【0181】
10時間を超える試料採取時間の場合:10分以上ずれる場合は実際の時間を使用した。10分未満のずれの場合は公称の時間を使用する。
【0182】
血漿ナイアシン及びNUA薬物動態パラメータに対する要約統計量を表18Aで示す。
【0183】
【表19】
【0184】
ナイアシン及びNUAに対する平均血漿プロファイルを図14及び15で示す。
【0185】
b.血漿データ
血漿ナイアシン
全対象の投与前値がLLQ未満であった。各治療後、投与後4.5から12時間で、全対象のナイアシン濃度が測定可能な濃度であった。
【0186】
平均ナイアシンCmaxは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、5288、4223、5671及び4707ng/mLであった。平均ナイアシンAUC0−lastは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13896、10207、13507及び12315ng*hr/mLであった。ナイアシンに対するTmax中央値は、ERN−1及びERN−3については6.0時間であり、ERN−2及びNSPについては5時間であった。
【0187】
NSPと比較した場合、全ての3種類の試験治療について、自然対数変換されたCmax及びAUC0−lastの比は100%より大きかった。ナイアシンCmaxの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、139%、116%及び166%であった。ナイアシンAUC0−lastの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、137%、112%及び151%であった。自然対数変換したナイアシンCmaxに対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、113から170%、94から142%及び135から203%であった。自然対数変換したナイアシンAUC0−lastに対して、90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、114から164%、94から134%及び126から181%であった。Cmax及びAUC0−lastの両方に対する90%CIは、80−125%の同等範囲外であった。
【0188】
4種類全ての治療についてのCmax及びAUC0−lastに関して、ナイアシンデータは、76から171%の範囲のCVで非常に大きく変動した。
【0189】
血漿ニコチン尿酸
3名の対象の投与前NUA濃度が陽性であった。これらは、対象0028(ピリオド2、ERN−1、濃度4.47ng/mL)、対象30(ピリオド2、ERN−3、濃度2.75ng/mL)及び対象33(ピリオド2、ERN−2、濃度3.26ng/mL)であった。投与前濃度が、対象0028、0030及び0033に対してそれぞれCmaxのわずか約0.24%、0.06%及び0.53%であったので、これらの血漿プロファイルに対して補正を行わなかった。全対象のNUA濃度が各治療後の投与後3から16時間で測定可能な濃度であった。
【0190】
平均NUA Cmaxは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、2822、2616、3058及び2540ng/mLであった。平均NUA AUC0−lastは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13664、12069、13960及び13070ng*hr/mLであった。NUA Tmax中央値は、ERN−1及びERN−3については6.0時間、ERN−2については5.5時間、NSPについては5.0時間であった。
【0191】
各対象及び治療について、可能な場合は最終消失速度を計算した。平均t1/2は、ERN−1、ERN−2及びERN−3についてそれぞれ3.4時間であり、NSPについて3.1時間であった。平均AUCinfは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13602、11913、14136及び13009ng*hr/mLであった。
【0192】
自然対数変換したCmax及びAUC0−lastの比は、NSPと比較した場合、治療ERN−1及びERN−3については100%より大きかった。ERN−2の場合、Cmax比は100%より大きかったが、一方、AUC0−last比は100%未満であった。NUA Cmaxの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、111%、105%及び123%であった。NUA AUC0−lastの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、104、95%及び109%であった。自然対数変換したNUA Cmaxに対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、101から121%、96から115%及び113から135%であった。自然対数変換したNUA AUC0−lastの場合、90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、96から111%、88から102%及び102から118%であった。Cmax及びAUC0−lastの両方に対する90%CIは、ERN−1及びERN−2については80−125%の生物学的同等性の範囲内であった。ERN−3については、Cmaxに対する90%CIは80−125%の範囲外であったが、AUC0−lastに対しては80−125%の範囲内であった。
【0193】
NUAデータは、4種類の治療全てについて、Cmax及びAUC0−lastに対してCV約48−58%で非常に大きく変動した。
【0194】
c.ナイアシン及び代謝産物の尿中回収
1の比重を使用して尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究では、962個の試料で測定した平均比重は1.009g/mLであり、962個の試料で測定した最大比重は1.025g/mLであった。)。
【0195】
平均尿回収データを表19で示し、図16で述べる。
【0196】
【表20】
【0197】
総回収
ナイアシン、NUA、MNA及び2PYとしての尿中のナイアシンの総回収は、NSPの場合67.14%、ERN−1、ERN−2及びERN−3の場合それぞれ63.91%、63.44%及び66.16%であった。総回収に対する、loge変換された%Feの最小二乗平均比は、ERN−1、ERN−2及びERN−3についてそれぞれ95%、94%及び98%であった。この比に対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ91から99%、90から98%及び94から102%であり、このことから、3種類の試験製剤によって尿中で排出される総量は、80−125%信頼区間に基づきNSPと同等であることが示された。
【0198】
d.生物同等性評価の結論
NUAデータの薬物動態分析から、Cmaxにより測定されたピーク曝露が、試験製剤NSPと比較した場合、3種類全ての試験製剤(ERN−1、ERN−2、ERN−3)について5から23%高いことが示された。loge変換されたCmaxの場合の最小二乗平均比に対する90%CIは、ERN−1及びERN−2については80−125%の範囲内であり、このことから、これらの製剤がNUAに関してNSPと生物学的に同等であることが示された。ERN−3の場合、90%CIはCmaxに対して80−125%の範囲外であり、このことから、製剤ERN−3がNSPと生物学的に同等でないことが示された。
【0199】
尿中で排出されるナイアシン及び代謝産物の平均総量は、3種類の試験製剤では63から66%であり、NSPの場合は67%であった。排出された分画は親ナイアシンに対して最小であり、続いてNUA、MNA及び2PYであった(37.2−40.0%)。測定した総回収は、NSPと比較した場合、試験製剤では2から6%低かった。loge変換した総回収の最小二乗平均比に対する90%CIは80−125%の範囲内であり、したがって、NSPと比較した場合、3種類の試験製剤は同等であった。
【0200】
したがって、本発明のある実施形態は、4個の500mg NIASPAN(R)錠剤の単回投与を改質1000mg徐放ナイアシン組成物の単回投与と比較する生物学的同等性試験において対象に投与した場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、改質1000mg徐放ナイアシン医薬組成物を含む。
【0201】
好ましい実施形態において、生物学的利用率パラメータは、NUA Cmax(ng/mL)及び総回収又はナイアシンCmax(ng/mL)及びナイアシンAUCである。
【実施例6】
【0202】
この試験の目的は、2000mgの単回投与として投与した場合の、非被覆に対する被覆の本発明1000mg徐放ナイアシン錠剤(本明細書中で以後「改質」錠剤と呼ぶ。)の生物学的同等性(BE)を調べることである。
【0203】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙男性及び女性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、2期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間に10日以上のウォッシュアウト期間を設け、2回の個別の日時において、同じ2000mgの単回投与で、2種類のナイアシン製剤、Test及びREFの投与を受けた。Testは、被覆改質1000mg徐放ナイアシンの2個の錠剤から構成され、REFは非被覆改質1000mg徐放ナイアシンの2個の錠剤から構成された。各期間の第1日の22:00前後から開始する低脂肪スナックの後に水240mLとともに各用量を投与した。各治療の6日間の試験期間(第−1から第5日)の間、患者を隔離し、各治療期間中、スポンサーが提供するメニューに従い食事を与えた。他の薬、ビタミン、ハーブ又は栄養剤を試験中禁じた。
【0204】
−30分(投与前)、1、2、3、4、4.5、5、6、7、8、10、12、14、16及び24時間(投与後)の間隔で、投与前30分以内から、投与後24時間まで、連続血液試料を回収した。−24から−18、−18から−12、−12から−6及び−6から0時間(投与前);0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)の間隔で、投与前24時間から投与後96時間まで採尿した。ナイアシン及びNUAについて血漿を分析した。ナイアシン及びその代謝産物:NUA、MNA及び2−PYについて尿を分析した。
【0205】
対象は、各期間中、病院に隔離されているときは、毎日同じ時間に食事を開始した。食事は各期間で同じとなるように維持し、各食事の全量を摂取するよう求めた。朝食、昼食、夕食及び夜食はそれぞれ、07:00、12:00、18:00及び21:45前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに与える水240mLに加え、第−1日に水を720mL以上飲み、第1日から5日に水を1440mL以上飲むよう求めた。
【0206】
第−1日に、夕食及び夜食を供した。第1日から5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では、投与直前15分以内に取らせた。ピリオド2の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0207】
薬物動態の評価
a.血漿回収及び分析
各期間、投与前30分以内から、投与後24時間まで、連続血液試料を回収した(治療あたり15試料)。ヘパリンナトリウムを含有する1本の17−mL真空採血システムに各血液試料を回収し、回収後、氷片及び水浴中で5分以上、試料を冷却した。試料を4℃にておよそ3000rpmで15分間遠心して血漿を分離した。各血漿試料を2つのアリコート、アリコートA及びBに分け、予め冷却し適切にラベルを貼った2本のポリプロピレンチューブに移した。次に、分析まで試料をおよそ−20℃で凍結保存した。
【0208】
有効な液体クロマトグラフィータンデム質量分析(LC/MS/MS)によって、ナイアシン及びNUA濃度を分析した。ナイアシン及びNUA濃度を同じ注入から得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0209】
b.採尿及び分析
次の間隔で尿を回収した:−24から−18、−18から−12、−12から−6及び−6から0時間(投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)(全部で11回の回収)。
【0210】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、回収した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。空の容器の重量を測定し、グラムの小数点以下1桁に四捨五入し(例えば100.1g)、これを容器に記入し、実験室のソースドキュメントワークシートに記録した。各間隔の終わりに、容器及び回収した尿の総重量を測定し、グラムの小数点以下1桁に四捨五入し、記録した。尿重量は、容器+尿の総重量から空の容器の重量を差し引くことにより得られた。場合によっては、ある回収間隔中の尿体積が1個の容器の容量を超え、従って採尿を完遂するために第二の容器を必要とした。各採尿間隔の開始及び終了日時も記録した。各回収間隔からの2つのアリコート(各およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。特定の回収間隔中に複数の容器を必要とした場合、両方の容器からの尿を一緒にして混合し、その後アリコートを取った。分析まで、およそ−20℃で試料を凍結保存した。
【0211】
有効なLC/MS/MSにより、ナイアシン、NUA、NMA及び2−PYの濃度について尿試料を分析した。同じ注入から尿ナイアシン及びNUA濃度を得て、一方、NMA及び2−PY濃度を同じ注入から得た。尿において、LLQ値はナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2PYのLLQ値はそれぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0212】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対するPKパラメータを計算するために十分な情報を提供する対象からのデータがPK分析に含まれた。血漿中のナイアシン及びNUAについて、各治療の投与後、各対象に対して次のPKパラメータを計算した:
・Cmax:観察される最大濃度
・Tmax:最大観察濃度の時間
・AUClast:直線的台形規則による、時間0から最後の測定可能(0ではない)濃度までの濃度−時間プロファイル下面積
・AUCinf:時間0から無限大の血漿濃度−時間プロファイル下面積;AUClast及びCt/λzの合計として計算(Ctは、最後に観察された濃度であり、λzは、時間プロットに対する自然対数濃度のプロットから得られた最終消失速度定数である。)
・T1/2:見かけの最終半減期;0.693/λzの比として計算
ナイアシン及びその代謝産物(NUA、MNA及び2−PY)の尿データから、次のパラメータを計算した:
・CumXu:投与後0から96時間の尿から回収した各代謝産物の累積量。
【0213】
・%Fe:投与後96時間での、ベースライン回収及び分子量に対する補正後のナイアシンの用量に対する尿中で排出された各代謝産物の分画
・総%Fe:投与後96時間における4種類の代謝産物の総分画。尿中の各被分析物に対して、%Feは以下のように計算:
【数3】
【0214】
定量限界未満の濃度をゼロとして処理した。各代謝産物濃度に各間隔で回収された尿の体積を掛けることによって、尿中で回収されたナイアシン及びその代謝産物の量を決定した。24時間投与前間隔において回収された量を差し引くことによって、投与後各24時間間隔に対して尿中で回収された総量をベースラインに対して調整した。投与後測定値がベースライン未満となった場合、その量を0とした。ナイアシン及びその代謝産物の分子量は、ナイアシン、NUA、MNA及び2−PYに対してそれぞれ、123.1、180.2、137.1及び153.1であった。4種類の尿被分析物からの%Feの合計を計算し、総%Feとした。
【0215】
WinNonlin Linear Mixed Effects Modeling/bioequivalence、バージョン5.0.1(2005年7月26日)を用いて、(上述のような)生物学的利用率パラメータを計算した。
【0216】
統計分析
WindowsTM用SAS(R)Systemバージョン8.2を用いて、上記で計算された生物学的利用率パラメータの統計分析を行い、データ分析に用いた。
【0217】
治療及び期間によって、血漿薬物動態パラメータ(Cmax、Tmax、T1/2、AUClast及びAUCinf)、それらの自然対数変換値(Tmax及びT1/2を除く。)及び要約統計量(n、平均、std、中央値、min、max、CV%)を計算した。ナイアシン及びNUAの血漿濃度を時間及び治療によってまとめる。
【0218】
ナイアシン及びNUA PK分析の場合、自然対数変換したCmax及びAUClastのデータが正規分布に従い、2つの治療間で独立であると仮定する。固定効果として治療、期間及び順序を、変量効果として順序内の対象を用いて、SAS PROC MIXEDを使用して混合効果によってこのデータをANOVAモデルに適合させた。Cmax及びAUClastのTest/REF比及びそれらの対応する90%信頼区間をこのモデルに基づき評価した。
【0219】
尿からのナイアシン及びその代謝産物の平均回収を計算し、治療により及び間隔によりまとめた。個々の成分のCumXu及び%Fe及び投与後96時間における合計を計算し、治療によりまとめた。
【0220】
血漿PK分析に対して用いたものと同じANOVAモデルに適合させることにより、総%FeのTest/REF平均比に対する90%信頼区間(CI)を計算した。
【0221】
対象のデモグラフィック可変要素(年齢、性別、人種、体重、身長、女性サイズ、肘幅及びBMI)を性別によりまとめた。連続的デモグラフィック可変要素の、平均、標準偏差(SD)、中央値、最小及び最大を計算した。
【0222】
結果
対象の動態を表20でまとめる。プロトコールの包除基準に合致した後に全部で44名の対象を試験に登録した。対象者全てが、試験薬の少なくとも1回の投与を受け、このうち42名がこの試験を完遂した。44名の対象がプロトコールの無作為治療割り当てに従いピリオド1で試験薬の投与を受け;42名の対象がピリオド2で試験薬の投与を受けた。全部で2名の対象が試験から撤退した。試験から撤退した対象の数は、前もって容認するとした10%脱落の範囲内であり、この試験の結果又は結論に影響しないと考えられた。
【0223】
【表21】
【0224】
登録した44名の対象のうち、20名の対象が男性で24名が女性であった。平均年齢は53.1歳;平均体重は161.5ポンド;平均身長は65.6インチ;平均肘幅は2.7インチ;平均BMIは26.3kg/m2であった。女性サイズを小、中及び大に分類した。9名の対象が小の女性サイズであり、20名の対象が中、15名の対象が大の女性サイズであった。対象のうち38名がヒスパニック、4名が白人、2名が黒人であった。表21で詳細なデモグラフィックをまとめる。
【0225】
【表22】
【0226】
a.生物学的同等性の評価
生物学的同等性を調べるために、Test治療で42名の対象から、REF治療で44名の対象から得たデータを分析した。全分析に対し、投与時間に対して実際の時間を使用した。
【0227】
尿の分析のために、1g/mLの比重を使用して、尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究において、962個の試料で測定した平均比重は1.009g/mLであり、962個の試料で測定した最大比重は1.025g/mLであった。)。
【0228】
治療によるナイアシン及びNUAの平均血漿濃度のプロットを図17及び18でそれぞれ示す。平均尿中回収データを図19で示す。
【0229】
b.血漿NUA及び尿中で排出される総量
ナイアシン生物学的同等性を評価するための主要な可変要素は、NUAに対するCmax及びナイアシン及び3種類の代謝産物(NUA、MNA及び2PY)の総尿中回収として定義される。
【0230】
表22は、これら2種類の可変要素に対する平均(SD)及び統計結果を示す。表22はまた、NUA AUClastに対する平均(SD)値及び統計解析も示す。
【0231】
【表23】
【0232】
上記の表で示されるように、主要なBE可変要素である、自然対数変換した、NUA Cmax及びナイアシン及び代謝産物の総回収の、参照に対する試験比に対する90%CIは、80から25%内であった。自然対数変換したNUA AUClastの参照に対する試験比も80から125%内であった。
【0233】
治療によって、各対象について最終消失速度を計算した。Test及びREFに対してそれぞれ、平均NUA T1/2は3.16及び3.04時間、平均NUA Tmaxは4.90及び4.80時間、平均NUA AUCinfは、10914.7及び11770.6ng*hr/mLであった。
【0234】
c.血漿ナイアシン
統計解析付きの血漿ナイアシンに対する平均PKパラメータを表23で示す。自然対数変換したナイアシンCmax及びAUClastの参照に対する試験比は100%未満であった。自然対数変換したナイアシンCmax及びAUClastの比に対する90%CIは、変動が大きかったため、80から125%の間隔外であった。
【0235】
【表24】
【0236】
Test及びREFに対してそれぞれ、ナイアシンの平均T1/2は4.73及び2.94時間であり、平均Tmaxは4.68及び4.64時間であり、平均AUCinfは11553.1及び16134.3ng*hr/mLであった。
【0237】
d.個々の被分析物の尿中回収
個々の被分析物の平均尿中回収を表24で与える。
【0238】
【表25】
【0239】
平均尿中回収は2PYに対して最大であり、続いてMNA、NUA及びナイアシンであった。
【0240】
e.生物学的同等性評価の結論
NUA Cmaxの平均Test/REF比及びナイアシン及びその代謝産物の尿中回収(総%Fe)に対する90%CTに基づき、生物学的同等性を評価した。自然対数変換したナイアシン吸収の速度(NUA Cmax)及び程度(尿中総%Fe)のTest/REFの平均比に対する90%CIは80から125%の求められるBE範囲内であり、このことから、Test及びREF製剤が生物学的に同等であることが示される。NUA AUClastに対する90%CIも80から125%の範囲内であり、BEの結論を支持する。
【0241】
ナイアシンCmax及びAUClastについて、ナイアシンCmax及びAUClastの両方に対するTest/REFの平均比の90%CIの上限は生物学的同等性範囲内であり、下限は、生物学的同等性範囲の下限である80%に両方とも非常に近かった。
【実施例7】
【0242】
実施例4、5及び6で分析した本発明の1000mg製剤について、NUAに対するCmax(ng/mL)、総尿中回収(%)、ナイアシンCmax(ng/mL)及びナイアシンAUCに対する平均を下記表25で示す(ERN−3を除く。)。
【0243】
【表26】
【0244】
したがって、本発明のある実施形態は、それを必要とする患者に2個の1000mg錠剤の単回投与として投与した場合、次の生物学的利用率パラメータの少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%内となるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物を含む:
(a)2601.8ng/mLのNUA Cmax;
(b)60.5%の尿中ナイアシン総回収;
(c)4958.9ng/mLのナイアシンCmax;及び
(d)12414.5ng/mLのナイアシンAUC。
【0245】
下記表25aは、標準誤差(括弧で示す。)を考慮に入れた、表25から選択された生物学的利用率パラメータの上限及び下限を示す。特に、上記表25で割り出された各パラメータに対して上記実施例4、5及び6からの最低平均を割り出し、次いで2つの標準誤差をその平均から差し引いて下限を出すことによって、下限を計算した。標準偏差を試料サイズの平方根で割ることによって(例えば、1430/V44=326)、標準誤差を計算した。同様に、上限は、各パラメータ+2つの標準誤差に対する実施例4、5及び6からの最大平均を表す。
【0246】
【表27】
【0247】
したがって、本発明のさらなる実施形態は、それを必要とする患者に2個の1000mg錠剤の単回投与として投与した場合、次の生物学的利用率パラメータの少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%間隔内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物を含む:
(a)約2111.0ng/mLから約3253ng/mLのNUA Cmax;
(b)約49.24%から約70.23%の尿中ナイアシンの総回収;
(c)約3096ng/mLから約6750ng/mLのナイアシンCmax;及び
(d)約6723ng/mLから約18643ng/mLのナイアシンAUC。
【実施例8】
【0248】
アスピリンで前治療されるか又はアスピリンと同時投与される場合の、徐放ナイアシンの2000mg用量により誘発される紅潮の相対的発生率
この試験は、単一施設で行われ、本発明の徐放ナイアシン錠剤の経口投与の結果として起こる紅潮反応におけるアスピリン前治療及びアスピリン同時投与の影響を調べるために計画された、無作為二重盲検、ダブルダミー、単回投与、3期クロスオーバー試験である。この試験計画及び治療を図20で示す。対象にはまた、試験中如何なる時点でも、非研究関連アスピリン又はその他のNSAIDを用いることもまた禁止した。この試験は、病院のInstitutional Review Boardから承認を得、各対象は参加前に書面でインフォームドコンセントを提出した。
【0249】
この試験には、ボディーマスインデックス(BMI)が22から31kg/m2である、19歳から70歳の健常成人男性が含まれた。ナイアシン誘発性の紅潮事象と更年期の紅潮との混同を避けるため、この試験から女性を除外した。人間ドック、病歴、心電図及びスクリーニングのための来院時及び最初の試験期間のための入院時に行った臨床検査の結果によって、対象が健常であることを確認した。試験に入る前の4ヶ月以内に煙草又はニコチン製品を使用した場合、ナイアシン、アスピリンもしくは関連誘導体に対するアレルギー又は過敏性がある場合;過去3年間で薬物乱用又は依存症があった場合;又は偏頭痛、糖尿病、胆嚢疾患、肝臓疾患、重度の高血圧もしくは低血圧、心臓の異常、腎臓疾患もしくは薬物誘発性ミオパシーの病歴がある場合は対象を除外した。対象に対して、試験に入る前の21日以内及び試験中、処方薬を禁じ、試験に入る前の10日以内及び試験中、市販薬、ビタミン又はハーブ調合剤を禁じた。
【0250】
ピリオド1のための入院前21日以内でスクリーニング手順を完了した。3回の試験期間それぞれに対して、対象をおよそ24時間隔離し、少なくとも7日間あけて治療薬を投与し、対象は、ナイアシン及び脂肪含量が調節された特別メニューに従う食事を取った。食事構成及び開始時間は各試験期間で同じであった。
【0251】
試験治療
無作為化スケジュールに従い、クロスオーバー方式で、試験薬を経口投与した。アスピリン(ASA)及びプラセボ投与は各試験期間で異なったが、本発明の被覆徐放ナイアシン錠剤(本明細書中で「改質ナイアシンER錠剤」又は「rNER」とも呼ばれる。)(2個の1000mg錠剤)の投与は同じであった。ある期間において、対象は、2個のプラセボ錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のアスピリン325mg錠剤の投与を受けた(「ASA前治療」)。別の期間において、対象は、2個のアスピリン325mg錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のプラセボ錠剤の投与を受けた(「同時ASA」)。第三の期間において、対象は、2個のプラセボ錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のプラセボ錠剤からなる対照治療の投与を受けた(「R−ナイアシンER単独」)。
【0252】
紅潮事象を評価することが目的であるので、いくつかの方法によって試験者及び対象が投与薬物を識別できないようにした。各投与期間において、対象は、各投与に対して同じ数の錠剤の投与を受けた(図21参照)。プラセボ及びアスピリン錠剤の外観は同様であり、これらは識別されず、一方、それとともに、不透明な投与カップから試験薬を投与し、試験薬投与の間、対象に目隠しをした。アスピリンなしでの紅潮反応を評価するために、対照治療、R−ナイアシンER単独、がこの試験に含まれた。試験スポンサー及び各期間について投与薬を調製した試験施設の人員のみが、試験中の治療の無作為割り当てについて知っていた。治験医師、試験施設の人員及び試験モニターは治療割り当てスキームを知らず、治療薬調製又は投与に関与する試験施設の何れの者も、紅潮事象又は治療中に発生した有害事象を回収又は評価することを禁じた。
【0253】
各対象は、改質ナイアシンER投与前に、前治療薬及びスナックを摂取した。対象は、21:30前後に水180mLとともに、割り当てられた前治療薬(アスピリン又はプラセボ)の経口投与を受け、次いで、21:45前後に低脂肪スナックを食べ始めた。水240mLとともに22:00前後に対象が割り当てられた治療薬の残りの投与を対象が受ける前に、このスナックを全て摂取させた。各薬の投与には複数の錠剤が必要であり、錠剤を一度に一緒に摂取するか又は1個を飲んだ後すぐに続いて別のものを摂取するというように、これを1分以内に摂取させた。錠剤を飲み込むために必要である場合、120mL単位でさらなる水を与え;錠剤を咀嚼するか又は噛むことは禁じた。用量全てを摂取したことを確認するために、試験用量の投与後、各対象の口腔を調べた。
【0254】
紅潮評価
対象が次の紅潮症状の1以上:発赤、熱感、刺痛及び掻痒(これらの症状は個々に又は同時に起こり得た。)を報告することとして紅潮事象を定義した。各試験期間中、改質ナイアシンER投与から8時間後まで、1時間間隔で紅潮症状の有無を評価するよう、対象に促した。電子日誌において各紅潮症状についての開始時間、停止時間及び強度を記録するよう、対象に促した。
【0255】
各対象は、連続及び分類別評価基準の両方によって、各対象が感じる症状強度をランク付けした。対象は、コンピュータの水平視覚的アナログスケール(VAS)(「なし」(0)を左端に「容認できない。」(100)を右端にした。)上に垂直線で強度に対して印を付け、軽度、中度又は重度としても症状をランク付けした。容易に耐えられるか又は活動を制限しなかった症状を軽度と定義し;活動を行う上で困難を生じる症状を重度とした。
【0256】
各対象は、ナイアシンER投与後に起こる1以上の平行する紅潮症状の最初として定義される最初の全体的紅潮事象を同様にランク付けした。最初の症状の開始時間はまた、最初の全体的紅潮事象の開始時間でもあり;その事象の最後の症状が消散し、さらなる紅潮症状が起こることなく少なくとも30分間経過した場合、事象全体の終了とした。
【0257】
統計分析
少なくとも144名の対象が3つの治療全てを完遂することを確実にするために、全部で164名の対象を登録するよう計画した。早期に撤退した対象を入れ替えなかった。
【0258】
アルファ(α)=0.05の両側として全ての比較を行った。主要評価項目は、試験中に少なくとも1回紅潮事象が起こった対象の人数であった。マクネマー検定を用いて、「ASA前治療」及び対象治療、「R−ナイアシンER単独」の間で、紅潮発生率を比較した。この試験には、比較に含まれるべき両治療後に対象が反応(この場合、紅潮)することが必要である。「同時ASA」と「R−ナイアシンER単独」治療との間及び「ASA前治療」と「同時ASA」治療との間で、紅潮発生率の比較を同様に行った。
【0259】
副次的評価項目には、紅潮事象数及び最初の全体的紅潮事象ならびに個々の紅潮症状のの、強度、開始時間及び持続時間が含まれた。頻度数により事象の数をまとめ、マクネマー検定を用いて比較した。VAS線の左端からの距離として対象の垂直マークを表すことにより(100mmに規格化)、図式データから数字データにVAS強度評価を変換した。平均値に対して対応のあるt検定及び中央値に対してWilcoxon符号付き検定を用いて治療間でVASにより測定された強度及び持続時間を比較し、一方、Bowkerの対称性の検定(対象が比較において両治療に対するデータを有することも必要とするマクネマー検定の一般化)を用いて、分類別スケールにより測定された強度を比較した。主要評価項目と同じ治療のペアについて、副次的評価項目に対する治療間の比較を行った。
【0260】
Medical Dictionary for Regulatory Activities(MedDRA、バージョン7.0)を用いて有害事象(紅潮を除く。)を符号化した。有害事象は治療間で比較しなかった。
【0261】
結果
平均年齢29歳で平均BMIが26.5kg/m2の全部で164名の男性を登録し、少なくとも1回の試験薬投与を受けさせた。対象デモグラフィックを表26でまとめる。164名の対象のうち、148名(90%)が3回全ての治療を受け、紅潮反応について評価可能であった。16名の対象(10%)が早期に撤退し:4名(2%)が同意を取り消し、4名(2%)がフォローアップできず、1名(1%)で有害事象が起こり、3名(2%)がプロトコール違反し、3名(2%)が薬物スクリーニング検査で陽性となり、1名(1%)が投与ミスにより撤退した。
【0262】
【表28】
【0263】
紅潮
3回全ての治療を受けた148名の対象において、「同時ASA」(61%、p0.001)又は「ASA前治療」(53%、p0.001;表27)の後よりも、「R−ナイアシンER単独」(77%)の後に紅潮発生率が有意に高かった。
【0264】
【表29】
【0265】
両アスピリン含有治療は紅潮発生率において互いに有意な差はなかった。図21で示されるように、最初の全体的紅潮事象における個々の症状(発赤、熱感、刺痛及び掻痒)の発生率は、「R−ナイアシンER単独」と比較して、「ASA前治療」後では30%から50%低かった。4種類全ての症状を報告した対象数が最低であったのは「ASA前治療」後であり、対象数が最大であったのは「R−ナイアシンER単独」後であった。治療間で個々の症状は比較しなかった。紅潮事象の数は紅潮発生率と同じ傾向であり、「R−ナイアシンER単独」の後に報告した人数が最多であり、「ASA前治療」後に報告した人数が最少であった(データは示さない。)。
【0266】
「ASA前治療」及び「R−ナイアシンER単独」治療両方の後に紅潮した対象において(下記表28)、「ASA前治療」では、分類別評価又はVASの何れかにより測定された最初の全体的紅潮事象の強度が有意に低下していた(各p<0.001)。
【0267】
【表30】
【0268】
両治療で、紅潮事象の殆どが軽度にランク付けされ、1例のみが(「R−ナイアシンER単独」の後に)重度であった。軽度にランク付けされた事象があった対象の人数は、「R−ナイアシンER単独」と比較して、「ASA前治療」後では36%多く;同様に、中度又は重度にランク付けされた紅潮があった対象の人数は62%少なかった。VASのランク付けは、「R−ナイアシンER単独」の後よりも、「ASA前治療」の後で30%を超えて低かった。最初の全体的紅潮事象の持続時間について、平均及び中央値データは一貫しなかったが、このことから、非正規分布が示唆される。「ASA前治療」の場合の持続時間中央値は、「R−ナイアシンER単独」の場合よりも43%短かった(p=0.008)。個々の症状について、発赤、熱感及び刺痛は、「ASA前治療」後有意に強度が低く(p≦0.025、データは示さない。);何れの症状の持続時間についても治療間で有意差はなかった。
【0269】
「同時ASA」及び「R−ナイアシンER単独」両治療後に紅潮があった対象において(下記表29)、最初の全体的紅潮事象の強度は、分類別データについては有意差があったが(p=0.028)、VASデータについては有意差はなかった。
【0270】
【表31】
【0271】
ここで、「同時ASA」後に軽度紅潮事象のあった対象数は、「R−ナイアシンER単独」の後よりも22%高く、中度又は重度事象は38%少なかった。最初の全体的紅潮事象の持続時間の差は有意ではなかった。個々の症状について、発赤及び熱感の強度は、「同時ASA」治療後、有意に低く(p≦0.024、データは示さない。);何れの症状の持続時間も有意差はなかった。
【0272】
「ASA前治療」及び「同時ASA」両治療後に紅潮があった対象において(下記表30参照)、最初の全体的紅潮事象の強度の差は、分類別の尺度では有意ではなかったが、「ASA前治療」に対するVASスコアは20%低く、統計学的に有意であった。
【0273】
【表32】
【0274】
最初の全体的紅潮事象の持続時間は、これらの治療間で有意差はなかった。個々の症状について、強度及び持続時間のどちらもこの2つの治療間で有意差がなかった。
【0275】
上記の結果から、本発明の徐放錠剤の30分前に摂取されるアスピリンの650mg(2x325mg錠剤)によって、本発明の錠剤単独の使用と比較して、対象が報告する紅潮の発生率、強度及び持続時間が有意に低下した。アスピリン650mg及び本発明の錠剤の同時投与により、それほどではないにせよ、紅潮発生率、強度及び持続時間が低下した。
【0276】
実施例3及び実施例8からの紅潮発生率及び強度の結果をまとめ、図22及び23で一緒に示す。これらの図面から、本発明の徐放医薬組成物が、元の1000mg錠剤(Nisapan(R))と比較して、紅潮発生率の低下は小さいが、紅潮強度及び持続時間(〜40%)を低下させることが示される(実施例3参照)。実施例8から、アスピリンを本発明の徐放医薬組成物の30分前又はこれと同時に摂取することによって紅潮の発生率が低下し、さらに、紅潮強度及び持続時間が低下することが分かる。実施例3において、患者のほぼ全員(98%)が、元の1000mg錠剤の単回の2000mg投与で(2錠剤投与)紅潮(発生)を報告した。実施例8において、本発明の徐放錠剤の単回2000mg投与(2錠剤投与)+アスピリンで紅潮したのは対象の50−60%のみであった。先行試験における元の1000mg錠剤での強度中央値はVASで54mmであった。今回の試験において、本発明の徐放錠剤+アスピリンでは、強度中央値がわずか19−23mmであり、大多数(約80%以上)は紅潮が「軽度」であると報告した。
【0277】
本発明の具体的な実施形態を参照して本発明を上記で説明してきたが、本明細書中で開示される本発明の概念から逸脱することなく、多くの変化、修飾及び変更がなされ得ることは明らかである。したがって、添付の特許請求の範囲の精神及び幅広い範囲内に入る全てのかかる変化、修飾及び変更を包含するものとする。本明細書中で引用される全ての特許出願、特許及びその他の刊行物は、その全体が参照により本明細書中に組み込まれる。
【図面の簡単な説明】
【0278】
【図1】図1は、METHOCEL(R)K−15M Premiumの様々なレベルを含有する1000mgナイアシン徐放錠剤からの平均ナイアシン溶出を示すグラフである。
【図2】図2は、1000mgナイアシン徐放錠剤(1240mg総重量)からのナイアシン溶出における、METHOCEL(R)K−15MPCRの粘度の変化の影響を示すグラフである。
【図3】図3は、バルク及び40メッシュPVP K−90を用いて製造された1000mgナイアシン徐放錠剤からのナイアシン溶出プロファイルを示すグラフである。
【図4】図4は、様々な混合段階を用いて製造された1000mgナイアシン徐放錠剤(1240mg総重量)からのナイアシン溶出プロファイルを示すグラフである。
【図5】図5は、直接圧縮製造プロセスを示す流れ図である。
【図6】図6は、実施例3に記載の臨床試験の流れ図である。
【図7】図7は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mgNIASPAN(R)錠剤(参照)投与後の紅潮の発生率を示す棒グラフである。
【図8】図8は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mgNIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象の中央値強度を示す棒グラフである。
【図9】図9は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mg NIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象の中央値持続時間を示す棒グラフである。
【図10】図10は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mg NIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象における個々の紅潮症状の発生率を示す棒グラフである。
【図11】図11は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の1000mg NIASPAN(R)錠剤(「Ref」)投与後の、ナイアシンの平均血漿濃度を示すグラフである。
【図12】図12は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の1000mg NIASPAN(R)錠剤(「Ref」)投与後の、NUAの平均血漿濃度を示すグラフである。
【図13】図13は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の非被覆1000mg NIASPAN(R)錠剤(「Ref」)の投与から96時間後の、ナイアシン及びその代謝産物(ナイアシン用量の%として)の平均尿中回収を示す棒グラフである。
【図14】図14aは、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する線形平均血漿ナイアシンプロファイル示すグラフであり;14bは、3種類の試験及び1種類の参照製剤に対する、片対数平均血漿ナイアシンプロファイルを示すグラフである。
【図15】図15aは、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する、線形平均血漿NUAプロファイルを示すグラフであり;15bは、3種類の試験及び1種類の参照製剤に対する、片対数平均血漿ナイアシンプロファイルを示すグラフである。
【図16】図16は、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する、ナイアシン及びその代謝産物(ナイアシン用量の%として)の平均尿中回収を示す棒グラフである。
【図17】図17aは、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)に対する、線形平均血漿ナイアシンプロファイルを示すグラフであり;17bは、試験及び参照製剤に対する、対数変換平均血漿ナイアシンプロファイルを示すグラフである。
【図18】図18aは、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)に対する、線形平均血漿NUAプロファイルを示すグラフであり;18bは、試験及び参照製剤に対する、対数変換平均血漿NUAプロファイルを示すグラフである。
【図19】図19は、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)の投与から96時間後の、ナイアシン及びその代謝産物の平均尿中回収を示す棒グラフである。
【図20】図20は、実施例6の試験計画を示す流れ図である。
【図21】図21は、(1)対象をアスピリン(ASA)で予め処置した場合、(2)ナイアシン製剤とともにASAを投与した場合及び(3)ナイアシン製剤のみを投与した場合の、本発明の2個の1000mg徐放ナイアシン製剤(「NIASPAN(R)CF」)の投与後の、最初の紅潮事象に対する個々の紅潮症状の発生率を示す棒グラフである。
【図22】図22は、実施例3及び実施例8の両方に対する、紅潮事象の発生率を示す棒グラフである。
【図23】図23は、実施例3及び実施例8の両方に対する、紅潮事象の強度を示す棒グラフである。
【技術分野】
【0001】
本発明は、ナイアシン、放出遅延剤及びその他の賦形剤を含む錠剤へと直接圧縮することができる徐放マトリクス製剤に関する。本発明の得られる錠剤は、製造特性が向上しており、好都合な放出特性を示し、ナイアシン療法に一般に付随する皮膚紅潮の持続時間が短く、重症度及び発生率が低い。
【背景技術】
【0002】
ナイアシン(ニコチン酸、3−ピリジンカルボン酸としても知られる。化学式C6H5NO2)は、高比重リポタンパク質(HDL)のレベルを上昇させ、総血清コレステロール低比重リポタンパク質(LDL)及びトリグリセリドのレベルを低下させるので、高コレステロール血症の治療に関連する長所を有することが知られている。
【0003】
ナイアシンは血液脂質において非常に有益な効果をもたらすことが知られているが、NIASPAN(R)(Kos Pharmaceuticals、Inc.、Cranbury、NJ)を除き、有効な脂質治療に必要とされる高用量のナイアシンでよく起こる「紅潮」の発生率が高いために、ナイアシンを広範には使用しにくい。紅潮は、ナイアシン誘発性血管拡張を述べるために一般に使用される用語である。結果として、ナイアシン投与時に、紅潮が起こっている個体では、目に見える、不快なのぼせ又は紅潮感が発現し得る。ある種の物質及び/又は製剤が、皮膚紅潮を回避するか又は軽減することが示唆されている一方(米国特許第4,956,252号、同第5,023,245号及び同第5,126,145号参照)、この望ましくない副作用はナイアシン製品を広く利用する上で依然として問題となっている。
【0004】
さらに、市販のNIASPAN(R)製剤中の現在の放出遅延剤(一般に「膨張剤」とも呼ばれる。)は質的に非常にばらつきがあり、それにより、内部の仕様書に合わせるために市販業者から特定のバッチ製品を得る必要がある。
【0005】
したがって、医薬の技術分野において既存のナイアシン製剤よりも皮膚紅潮のレベルが低く、一方で、物理的、化学的及び工学的特性が向上していることを特徴とする、ロバストな製造プロセスも可能となる、徐放ニコチン酸製剤が必要とされている。
【発明の開示】
【発明が解決しようとする課題】
【0006】
(発明の要旨)
本発明は、ナイアシン及び放出遅延剤を含む徐放(ER)錠剤製剤を提供する。ある実施形態において、本発明は、既存の1000mg処方ナイアシン製剤よりも流動性、圧縮性、充填性及び硬度が改善された1000mgERナイアシン錠剤製剤を提供する。さらに、本発明の1000mgERナイアシン錠剤は、製造ロバスト性(ロバストなプロセスは、原材料又は製造プロセスの小さな変化など、様々な環境又は条件下で目標とする最終物を再現する能力を有するものである。)又は工業的に望まれる条件(例えばサイズ)を何ら低下させることなく、市販の500mg NIASPAN(R)錠剤の放出速度及び/又は吸収速度を再現する能力を示す。2個の500mg NIASPAN(R)錠剤は1個の1000mg NIASPAN(R)錠剤よりも紅潮が少ないことが特徴であると考えられているので、本発明のある目的は、2個の500mg NIASPAN(R)錠剤と生物学的に同等である1000mg ERナイアシン錠剤製剤を提供することである。
【課題を解決するための手段】
【0007】
特に、本発明は、
(a)ナイアシン約70%から約92%w/wと、
(b)放出遅延剤約7%から約25%w/wと、
(c)結合剤約0.1%から約4.3%w/wと、
(d)潤滑剤約0.5%から約1.5%w/wと、を含む医薬組成物を提供する。
【0008】
ある実施形態において、医薬錠剤は直接圧縮錠剤である。
【0009】
さらに、本発明は、(a)ナイアシン約70%から約92%w/wと、放出遅延剤約7%から約25%w/wと、結合剤約0.1%から約4.3%w/wと、潤滑剤約1.3%から約4.3%w/wと、の混合物を混合し;
(b)段階(a)の混合物を錠剤へと圧縮する
段階を含む、徐放ナイアシン錠剤を調製する方法を提供する。
【0010】
好ましい実施形態において、徐放ナイアシン錠剤は、粒状ナイアシンを混合することにより調製される。
【0011】
患者においてナイアシン処置療法に伴う紅潮を軽減する方法も提供されるが、この方法は、ナイアシン治療を必要とする患者に本発明の徐放ナイアシン錠剤形態を投与することを含む。好ましい実施形態において、本発明によるナイアシン製剤は、夕方又は夜間に1日1回投与される。
【0012】
本発明のある実施形態は、4個の500mg NIASPAN(R)錠剤の単回投与を改質1000mg徐放ナイアシン組成物の単回投与と比較する生物学的同等性試験において対象に投与される場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、改質1000mg徐放ナイアシン医薬組成物を含む。
【0013】
本発明によると、非ステロイド抗炎症薬(NSAID)と組み合わせて本発明の徐放ナイアシン製剤を投与することによって、紅潮をさらに軽減することができる。好ましい実施形態において、NSAIDはアスピリンである。
【0014】
本発明による医薬組成物は、即放型紅潮抑制剤成分及び遅延放出ナイアシン成分を含み得、このナイアシンは遅延放出される(即ち、ナイアシンが遅延時間後に放出される。)。好ましい実施形態において、ナイアシンは紅潮抑制剤の放出から少なくとも約30分から約40分後に放出される。
【0015】
(詳細な説明)
本発明の徐放マトリクス錠剤製剤は、(1)活性成分としてのナイアシン及び(2)活性成分の徐放を達成するための親水性ポリマーマトリクス、即ち、放出遅延剤を含む。本明細書中で使用される場合、「徐放」製剤は、1日1回投与で患者の脂質異常症に対して有効な治療を提供する製剤を意味する。
【0016】
本発明の徐放ナイアシン製剤によって、結果として、患者において脂質プロファイルが改善し得る。例えば、患者への本発明の徐放ナイアシン製剤の投与は、患者の血流中の、総コレステロール、低比重リポタンパク質(LDL)、トリグリセリド及びリポタンパク質A(Lp(a))を低下させ、高比重リポタンパク質(HDL)を向上させ得る。患者の血流中の、総コレステロール、LDL、トリグリセリド及び/又はリポタンパク質A(Lp(a))を低下させ、及び/又はHDLを向上させるための治療を必要とする状態は、本明細書中で「脂質異常症」と呼ぶ。したがって、本発明は、脂質異常症の治療を必要とする患者に本発明の徐放ナイアシン製剤を投与することにによる、脂質異常症の治療を包含する。
【0017】
生物学的同等性は、適切に計画された試験において同様の条件下で同じモル濃度用量で投与された場合に、医薬的同等物又は医薬的代替物中の活性成分又は活性部分が薬物作用部位で利用可能になる速度及び程度について顕著な差がないことである。通常、2つの製剤が生物学的同等であると結論するためには、自然対数変換されたCmax及びAUC又はこれらの生物学的同等性パラメータ計算値に対する何らかの適切な代替物のTest/参照治療比に対する90%信頼区間が80%から125%の間(両端を含む。)に入ることを示すことで十分である。
【0018】
本発明の範囲内の製剤は、自然対数変換生物学的利用率パラメータの試験/参照治療比に対する90%CIが標準的な80%から125%の間隔内に入る場合、本発明の製剤と生物学的に同等であるとみなされるものである(例えば、Guidance for Industry:Bioavailability and Bioequivalence Studies for Orally Administered Drug Products−General Considerations、U.S.Department of Health & Human Services、Food and Drug Administration、CDER、2003年3月;Guidance for Industry Food−Effect Bioavailability and Fed Bioequivalence Studies、2002年12月を参照のこと;両刊行物の内容は参照により本明明細書中に組み込まれる。)。当業者にとって公知のように、関連性のある生物学的同等パラメータを用いて(参照製剤が対照として使用される。)同じ分析条件下(例えば、分析的及び技術的条件分析)でこのような製剤を参照製剤(本明細書中に記載のもの又は本明細書中に記載の本発明の実施形態など)と比較する。
【0019】
ナイアシン
水溶性薬剤であるナイアシンは、微粉白色結晶、顆粒又は白色結晶粉末として市販されている。本発明の医薬組成物は、ナイアシン結晶、顆粒又は粉末を用いて製造することができる。好ましい実施形態において、本医薬組成物は、粒状ナイアシン(これはナイアシン粉末と比較して流動性が大きい。)を用いて製造される。流動性は、錠剤製造にのための重要な加工パラメータである。本発明による粒状ナイアシンの使用により、流動性が向上し、製造スケールで実行可能なナイアシン錠剤の直接圧縮が可能となる。何の粒状ナイアシン粒子サイズも本発明によるナイアシン錠剤を調製するのに適している。粒状ナイアシンに対する好ましい粒子サイズは、顆粒が100−425μmの範囲となるようにふるい分画に対してNLT85%(w/w)であり、ダスト<100μmに対してNMT10%(w/w)である。乾式造粒又は湿式造粒プロセスを用いて、ナイアシン粉末の流動性を向上させることができる。
【0020】
ナイアシンは通常、約70%から約95%w/w、好ましくは約76%から約90%w/w、より好ましくは約78%から約82%w/wの濃度で本発明の錠剤中に存在する。ナイアシンは、約100mgから3000mgの量で本発明の徐放製剤中に存在し得る。ある実施形態において、本発明の製剤は、ナイアシン約500mg、約750mg又は約1000mgを含む。ナイアシンの好ましい1日投与量は、約1000mg、約1500mg又は約2000mgである。したがって、1日1回、2個の1000mg錠剤を患者に投与することにより、例えば、ナイアシンの1日投与量を患者に与えることができる。
【0021】
放出遅延剤
ポリマーマトリクス系からの徐放は通常、ポリマー湿潤、ポリマー水和、ゲル形成、膨潤及びポリマー溶解に関与する。可溶性薬物の場合、これらの薬物は濡れ、溶解し、ポリマーマトリクスにより形成されたゲル層から拡散する。可溶性薬物がマトリクス錠剤中で放出される機構は多くの可変要素に依存するが、一般的原理は、錠剤全体に存在する水溶性ポリマーが錠剤の外表面で水和しゲル層を形成するということである。水が錠剤に浸透するにつれて、ゲル層の厚みが増し、可溶性薬物がゲル層を通じて拡散する。摂取された錠剤の寿命中、薬物放出速度は、ゲルを通じた可溶性薬物の拡散及び錠剤浸食の速度により決定される。
【0022】
本発明の放出遅延成分は、好都合な膨潤及びゲル化特性を示す当業者にとって公知の何れかの薬物であり得る。適切な放出遅延剤の例には、以下に限定されないが、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(一般にHPMC又はヒプロメロースとも呼ばれる。)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)及びポリビニルピロリドン(PVP)、キサンタンガム及びトリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))ならびにこれらの放出遅延剤の混合物が含まれる。ある実施形態において、放出遅延剤は親水性の水溶性ポリマーである。好ましい親水性ポリマーは中程度の粘度のヒドロキシプロピルメチルセルロース及び中程度の粘度のポリビニルアルコールである。
【0023】
放出遅延剤は通常、約7.0%から約25.0%w/w(製剤の総重量に対する%重量)、好ましくは約11.0%から約20.0%w/w、より好ましくは約14%から約18%w/wの濃度で本発明の錠剤中に存在する。
【0024】
ある実施形態において、放出遅延剤はヒドロキシプロピルメチルセルロースである。HPMCは、セルロース(無水グルコース単位の基本的繰り返し構造を含有する天然の炭水化物)のポリマー性骨格を有する。溶解度(例えば水和率)及びHPMCにより形成されるゲル層の強度は、HPMCのセルロース骨格(セルロースは無水グルコース単位の基本的繰り返し構造を含有する天然の炭水化物である。)に結合した、2種類の化学置換基、ヒドロキシプロポキシル(ヒドロキシプロピルと呼ばれることがある。)及びメトキシル(メチルと呼ばれることがある。)置換の割合により影響を受ける。ヒドロキシプロポキシル置換は本来、比較的親水性であり、水和率に大きく関与するが、一方メトキシル置換は本来、比較的疎水性である。セルロースの無水グルコース単位における置換基の量は、1個の無水グルコース環に結合する平均置換基数により示すことができ、これは、当業者により「置換度」として一般に知られている概念である。METHOCEL(R)Cellulose Eithers Technical Handbook、Dow Chemical Company(2002年9月刊行、Form番号192−01062−0902 AMS)を参照のこと;及びMETHOCEL(R)セルロース Eithers for Controlled Release of Drugs in Hydrophilic matrix Systems(2002年7月刊行、Form番号198−02075−0702 AMS)を用いる。本発明のある実施形態において、HPMC放出遅延剤は、約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度、好ましくは約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度、より好ましくは約1.39から約1.41のメトキシル置換度及び約0.20から約0.22のヒドロキシプロポキシルモル置換度、より好ましくは約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する。METHOCEL(R)K−15M(Dow Chemical Companyより入手可能、K−15M Premium及びK−15M Premium CRなどの特定のK−15サブブランドを含む。)は好ましい放出遅延剤である。
【0025】
さらに、ヒドロキシプロピルメチルセルロースポリマーは様々な粘度グレードのものが市販されている。これらには、例えば、METHOCEL(R)K、即ちMETHOCEL(R)K4M及びMETHOCEL(R)K15M(Dow Chemical Co、USAより入手可能)の4000及び15000mPas(1センチポアズ(cps)=1mPas(ミリパスカル秒))粘度グレード;及びMetalose90 SH(Shin Etsu Ltd、Japanより入手可能)の4000、15,000及び39000mPas粘度グレードが含まれる。本発明の実施形態において、HPMC粘度(20℃にて水中2%濃度で測定、例えばASTM D2363)は約11,000から約22,000mPas、好ましくは約13,000から約18,000mPasである。
【0026】
HPMC以外の、適切なポリマーの置換に必要な具体的な特徴を調べるために、当業者はポリマー(例えばヒドロキシプロピルセルロース)の置換度を変化させ、本発明によるHPMCを使用する製剤(例えば実施例1又は2による製剤)の溶出プロファイルに合致する置換を同定することができる。
【0027】
賦形剤
本発明の錠剤は結合剤をさらに含む。結合剤は、何れかの従来から知られている医薬的に許容可能な結合剤、例えばポリビニルピロリドン(PVP、ポビドン、ポリビドンとしても知られる。)、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート、ワックスなどであり得る。上述の結合剤の混合物も使用し得る。本発明の実施形態において、結合剤は、錠剤の総重量の約0.1%から4.3%w/w、好ましくは約0.2%から3.25%w/w、より好ましくは約2.5%から3.0%w/wを構成する。
【0028】
さらに、本発明の錠剤は潤滑剤を含む。潤滑剤は疎水性又は親水性であり得、当業者にとって公知の潤滑剤(以下に限定されないが、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、硬化植物油など)を含み得る。好ましくは、潤滑剤はステアリン酸である。製剤への潤滑剤の添加によって、圧縮中のダイ壁と錠剤製剤との間の摩擦が減少し、粉末の流れが促進され(即ち混合された製剤のホッパー及びダイへの流れ)、加工装置に錠剤材料が接着するのを防ぐのに役立つ。ある実施形態において、本発明の錠剤製剤は、潤滑剤約0.5%から1.5%w/w、好ましくは約0.75%から1.25%w/w、より好ましくは約0.85%から1.15%w/w、より好ましくは約0.95%から1.05%w/wを含む。
【0029】
コーティング
本発明の徐放錠剤製剤は、色塗を与え、視覚的特性を促進し、湿気又は臭気を防御するものとして作用し、日光、温度変化などの環境因子による劣化を防ぐか、又は錠剤の味を隠すことが医薬固形剤形の分野で知られているようなコーティングをさらに含み得る。当業者にとって公知であるように、このようなコーティングは、ポリマー、可塑剤及び/又は着色顔料を含有し得る。例としては、OPADRY(R)コーティングが挙げられる。このコーティングは、流動床塗布装置(例えばWursterコーティング)又はパンコーティングシステムなどの何らかの公知の手段を使用して、溶液(例えば水性)、溶媒又は懸濁液から塗布することができる。本発明のある実施形態において、コーティングは色彩塗料、特にOPADRY(R)コーティングである。さらなる実施形態において、約1.5から約8.0%の重量増加、好ましくは約1.75から約5.0%重量増加で色彩塗料を錠剤に塗布する。
【0030】
NIASPAN(R)500mg錠剤に対する同等性
以前の臨床試験の検討から、2個の(2)NIASPAN(R)1000mg錠剤(錠剤重量1203.6mg)が4個の(4)NIASPAN(R)500mg錠剤と生物学的に同等ではなく、NIASPAN(R)500mg錠剤からよりもNIASPAN(R)1000mg錠剤からナイアシンが速く放出されることが明らかになった。さらなる研究から、NIASPAN(R)500mg錠剤中の成分量の2倍を有するナイアシンER1000mg錠剤(錠剤重量1419.0mg)も生物学的に同等でないことが示された。後者の事例では、ナイアシン溶出は、インビトロで、NIASPAN(R)500mg錠剤からよりも1000mg錠剤からの方が遅く、ナイアシンは、インビボで参照製品(500mg)よりもナイアシンER1000mg錠剤からの方が吸収が遅かった。さらなる研究から、1300.0mg及び1280.0mgの錠剤重量の改質ナイアシンER1000mg錠剤が、放出速度が遅いため、NIASPAN(R)500mg錠剤と生物学的に同等でないことも分かった。
【0031】
2個のNIASPAN(R)500mg錠剤と生物学的に同等なナイアシンER1000mg錠剤を処方するために、発明者らは、インビボでの放出及び吸収特性を予想するため、複数のナイアシンER1000mg製剤を調製し、インビトロで試験した。錠剤中のポリマー(放出遅延剤)レベル(w/w)が上昇するに従い溶解度が低下するという事実に基づき、試験ナイアシンER1000mg錠剤をさらに改質した。したがって、評価には、新しい成分(様々なタイプのポリマーなど)及び代替的製造技術(直接圧縮又はローラー圧縮など)の分析が含まれた。
【0032】
表1は、総錠剤重量が様々である1000mg錠剤に対する様々な試験製剤を示す。
【0033】
【表1】
【0034】
複数の可変要素の最初の評価後、下記の4種類の製剤をさらなる評価のために選択した。全時間点に対して、37℃で1.2のpHに維持した擬似胃液250mL中で60分間、次いで37℃で6.8のpHに維持した250mLの擬似腸液中で、参照として500mg NIASPAN(R)を用い、USPタイプ3装置を採用して、溶出プロファイルに基づき、下記製剤に対する変化を分析した。
【0035】
(i)湿式造粒(WG)を用いて調製したMETHOCEL(R)E10M
1240mg、1260mg、1280mg及び1300mg製剤に対して指定された製法に従い、粒状ナイアシン、METHOCEL(R)E10M、ポビドンK90及びステアリン酸の重量を測定し、次いで、造粒溶液として脱イオン水を用いて高せん断造粒機中で造粒した。湿った顆粒を乾燥させ、粉砕し、次いで追加の粒状METHOCEL(R)E10M及びステアリン酸と混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった最終的な混合物を錠剤へと圧縮した。
【0036】
(ii)直接圧縮(DC)法を用いて調製したMETHOCEL(R)E10M
表1で概説した指定された製法に従い、粒状ナイアシン、METHOCEL(R)E10M、ポビドンK90及びステアリン酸の重量を測定し、次いで、8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0037】
(iii)WG法を用いて調製したMETHOCEL(R)K15M
表1で概説した指定された製法に従い、ナイアシンUSP、METHOCEL(R)K15M及びポビドンK90の重量を測定し、次いで造粒溶液として脱イオン水を用いて高せん断造粒機中で造粒した。湿った顆粒を乾燥させ、粉砕し、次いで追加の粒状METHOCEL(R)K15M及びステアリン酸と混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、最終的なよく混ざった混合物を錠剤へと圧縮した。
【0038】
(iv)DC法を用いて調製したMETHOCEL(R)K15M
表1で概説した指定された製法に従い、粒状ナイアシン、METHOCEL(R)K15M、ポビドンK90及びステアリン酸の重量を測定し、次いで、8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0039】
分析には、加工機器ツールの変化;ポリマーレベルの変動(表1参照);湿式造粒、直接圧縮及びローラー圧縮法の置き換え;PVPレベルの変動;錠剤硬度の変化;重量変動(+/−5%);再現性;錠剤化スピードの変動及び錠剤安定性(保管後の放出速度、吸湿など)が含まれた。次の3種類の製剤に対して目標とする薬物放出プロファイルを達成した:(i)METHOCEL(R)E−10M湿式造粒、(iii)METHOCEL(R)K−15M湿式造粒及び(iv)METHOCEL(R)K15M直接圧縮。この3種類の製剤は、3ヶ月間の安定性試験後、好都合な安定性の結果をさらに示した。
【0040】
上述の分析で分かった経済的及び安定性に関する長所により、さらなる分析のための好ましい実施形態として、METHOCEL(R)K−15を用いた直接圧縮錠剤を選択した。したがって、改質1000mgナイアシンDC錠剤に関して、造粒サイズ;各成分の粒子サイズ分布、バルク及び各成分のタップ密度;各成分の様々なロット;含量均一性;Hauser及びCarr指標;流動性;圧縮率及びもろさの影響についてさらに評価を行った。表2は、様々な実験製剤で使用される具体的な主要材料を概説する。DMFはドラッグマスターファイルである。
【0041】
【表2】
【0042】
表3は、賦形剤の様々なレベルを含有する改質試験1000mgDC錠剤及び関連する物理的品質を示す。表3に記述の場合各成分のw/w%で、上述のようにこれらの製剤を調製した。
【0043】
【表3】
【0044】
図1は、表3で示した製剤に対する溶出プロファイルの比較例を与える。
【0045】
表4は、臨床試験での500mgのNiaspan(R)に対する、複数の1000mg実験ナイアシン製剤の溶出及び生物学的利用率データを示す。USP装置1を用い、37℃にて100rpm(バスケット法)で900mL脱イオン水を用いて、溶出を計算した。
【0046】
【表4】
【0047】
次のパラメータを変化させることにより、改質1000mgナイアシンER直接圧縮錠剤の再現性を調べた:
METHOCEL(R)K−15MP CRの粘度及びヒドロキシプロポキシル含量
METHOCEL(R)K−15Mの粒子サイズ
粒状ナイアシンの粒子サイズ
ステアリン酸含量
PVP K−90のふるい
加工パラメータ
混合順序及び時間
錠剤硬度
錠剤化スピード
下記表5及び図2−4は、上述の再現性試験中に得られたデータを示す。
【0048】
【表5】
【0049】
上述の可変要素の分析完了後、出願者らは、次の可変要素を変化させた場合に作られた錠剤からのナイアシン溶出に顕著な差がないことを見出した:METHOCEL(R)K−15MPremium(CR)の粘度及びヒドロキシプロポキシル置換、様々な粒子サイズの粒状ナイアシン、40メッシュスクリーンを通したPVP K−90のふるい、0.5%から2.0%のステアリン酸含量、混合段階及び混合時間。METHOCEL(R)K−15M Premium(CR)の粒子サイズ及び錠剤の硬度(特に8kp未満)を向上させると、ナイアシン溶出度が上昇した。粒状ナイアシン及びMETHOCEL(R)K−15M premium CRの粒子サイズが小さいほど、圧縮率が大きくなった。製剤中のステアリン酸含量が増加するにつれて、突出力が著しく低下した。錠剤硬度を高くするには、高い圧縮力及び突出力が必要であり、錠剤化スピードを上昇させる場合、目標錠剤硬度(18Kp)を得るには、より大きい圧縮力が必要であった。
【0050】
上記によると、本発明は、
(a)ナイアシン約70%から約92%w/wと、
(b)約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する放出遅延剤約7%から約25%w/wと、
(c)結合剤約0.1%から約4.3%w/wと、
(d)潤滑剤約0.5%から約1.5%w/wと、を含む、湿式造粒又は直接圧縮1000mgナイアシン徐放(ER)錠剤製剤を包含する。
【0051】
好ましい実施形態において、本製剤は、直接圧縮法を使用して調製される。
【0052】
本発明の1000mg徐放ナイアシン製剤は2個の500mg NIASPAN(R)錠剤と生物学的に同等なので、これらは、効力及び毒性プロファイルの両方が同じであると予想される。したがって本発明の1000mg徐放ナイアシン製剤の投与により、本発明の製剤の使用を中止することが求められるレベルの、治療を制限する肝毒性又は治療を制限する尿酸又はグルコースレベル上昇を生じることなく、2個の500mg NIASPAN(R)の治療効果と同様の治療効果が与えられ得る。持続放出ナイアシン製剤に関連する毒性の問題は当業者にとって周知である。例えば「A comparison of the Efficacy and Toxic Effects of Sustained−v.Immediate−Release Nyacin Hypercholesterolemic Patients」、McKenneyら、JAMA Vol.271、No.9、1994年3月2日;及び「Hepatic Toxicity of Unmodified and Time Release Preparations of Nyacine」、Raderら、The Am.Jour.Of Med.、Vol.92、1992年1月、77頁を参照のこと。
【0053】
したがって、本発明のある態様は、それを必要とする患者を治療するための本発明の医薬組成物の投与を含み、この治療によって、1日1回患者によりこの組成物が摂取される場合、患者への投与後に、このような治療を中止することが必要となる、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を一般に生じさせることなく、血清脂質を低下させることができる。さらなる実施形態において、投与は、夕方又は夜間に1日1回である(例えば夕食後又は就寝前)。
【0054】
併用療法
本発明の1日1回のナイアシン製剤をHMG−CoA還元酵素阻害剤と組み合わせることができる。本明細書中で使用される場合、「併用療法(combination therapy)」及び「併用療法(combination treatment)」は、同じ又は個別の医薬剤形での、本発明のナイアシン製剤と少なくとも1つのさらなる活性成分との投与を包含する。併用療法は、本明細書中で使用される場合、治療計画の一部としての、活性薬剤の同時投与及び活性薬剤の連続投与を含む。
【0055】
HMG−CoA還元酵素阻害剤の例には、以下に限定されないが、ロバスタチン及び米国特許第4,231,938号で開示されるような関連化合物、プラバスタチン及び米国特許第4,346,227号及び同第4,448,979号で報告されるような関連化合物、メバスタチン及び米国特許第3,983,140号で開示されるような関連化合物、ベロスタチン及シンバスタチン及び米国特許第4,448,784号及び同第4,450,171号で考察されるような関連化合物、フルバスタチン、アトルバスタチン、リバスタチン及びフルインドスタチン(Sandoz XU−62−320)が含まれる。その他のHMG−CoA還元性阻害剤には、以下に限定されないが、米国特許第4,613,610号で開示されるようなメバロノラクトン誘導体のピラゾール類似体、PCT出願WO86/03488で開示されるようなメバロノラクトン誘導体のインデント類似体(indent analogs)、米国特許第4,647,576号で開示されるような、6−[2−(置換ピロール−1−イル)アルキル]ピラン−2−オン及びその誘導体、SearleのSC45355(3−置換ペンタンジオン酸誘導体)ジクロロ酢酸、PCR出願WO86/07054で開示されるようなメバロノラクトンのイミダゾール類似体、仏国特許第2,596,393号で開示されるような3−カルボキシ−2−ヒドロキシ−プロパン−リン酸誘導体、欧州特許出願0221025 A14で開示されるような2,3−ジ−置換ピロール、フラン及びチオフェン誘導体、米国特許第4,686,237号で開示されるようなメバロノラクトンのナフチル類似体、米国特許第4,499,289号で開示されるようなオクタヒドロ−ナフテレン、欧州特許出願0142146 A2で開示されるようなロバスタチンのケト類似体ならびにその他の公知のHMG−CoA還元酵素阻害剤、例えば英国特許第2,205,837号及び同第2,205,838号;及び米国特許第5,217,992号;同第5,196,440号;同第5,189,180号;同第5,166,364号;同第5,157,134号;同第5,110,940号;同第5,106,992号;同第5,099,035号;同第5,081,136号;同第5,049,696号;同第5,049,577号;同第5,025,017号;同第5,011,947号;同第5,010,105号;同第4,970,221号;同第4,940,800号;同第4,866,058号;同第4,686,237号で開示されているものなどである。
【0056】
場合によっては、本発明の製剤処方をその他の抗高脂血剤と組み合わせて投与することもできる。抗高脂血剤の具体例には、以下に限定されないが、胆汁酸抑制剤、例えば、コレスチラミン、コレスチポールDEAESephadex(Secholex.RTM.及びPolidexide.RTM.)、プロブコール及び米国特許第3,674,836号で開示されているような関連化合物、リポスタビル(Rhone−Poulanc)、エイザイE5050(N−置換エタノールアミン誘導体)、イマニキシル(HOE−402)テトラヒドロリプスタチン(THL)、イシチグマスタニルホスホリルコリン(SPC Roche)、アミノシクロデキストリン(田辺製薬)、味の素A J−814(アズレン誘導体)、メリナミド(住友)、Sandoz 58−035、American Cyanimid CL−277,082及びCL−283,546(二置換尿素誘導体)、ネオマイシン、p−アミノサリチル酸、アスピリン、四級アミンポリ(ジアリルジメチルアンモニウムクロリド)及び米国特許第4,027,009号で開示されるようなイオネン、米国特許第4,759,923号で開示されるようなポリ(ジアリルメチルアミン)誘導体、様々な魚油栄養剤で見出されるオメガ−3−脂肪酸、フィブリン酸誘導体、例えば、ゲムフィブロジル、クロフィブレート、ベザフィブレート、フェノフィブレート、シプロフィブレート及びクリノフィブレート及び米国特許第5,200,424号;欧州特許出願0065835 A1、欧州特許164−698−A、英国特許第1,586,152号及び英国特許出願2162−179−Aで開示されるものなどのその他の公知のコレステロール低下剤が含まれる。
【0057】
さらに、紅潮抑制剤と組み合わせて本発明の製剤処方を投与することができる。紅潮抑制剤には、以下に限定されないが、アスピリン及びサリチル酸塩などの非ステロイド抗炎症薬;イブプロフェン、フルルビプロフェン、フェノプロフェン、ケトプロフェン、ナプロキセン、ナプロキセンナトリウム、カルプロフェン及びスプロフェンなどのプロピオン酸;インドメタシン、エトドラク及びスリンダクなどのインドール酢酸誘導体;アクロフェナク、ジクロフェナク及びフェンクロフェナクなどのベンゼン酢酸;ゾメピラク及びトルメクチンなどのピロール酢酸;フェニルブタゾン及びオキシフェンブタゾンなどのピラゾール;ピロキシカムなどのオキシカム;及びメクロフェナメート及びメフェナム酸などのアントラニル酸が含まれる。
【0058】
紅潮抑制剤は、以下に限定されないが、米国特許公開第2004/0229844号及び同第2005/0154044号で開示されている化合物を含むプロスタグランジンD2受容体アンタゴニストでもあり得る。好ましいプロスタグランジンD2受容体アンタゴニストはMK−0524(Merck & Co.)である。
【0059】
遅延放出
本発明は遅延放出剤形を包含する。本明細書中で使用される場合、「遅延放出」とは、患者への投与後ある一定時間(即ち遅延時間)殆ど又は全く放出が起こらないことを意味する。医薬組成物中の唯一の活性薬剤として又は医薬剤形中の複数の活性薬剤の1つとして(その他の活性薬剤は遅延放出されてもよいし又はされなくてもよい。)、遅延放出形態で本発明のナイアシン製剤を提供することができる。したがって、例えば、医薬組成物は、遅延放出ナイアシン成分と組み合わせて即放型紅潮抑制剤成分に迎合することができる。例えば、患者への本発明の医薬組成物の投与時に、即放型紅潮抑制剤がすぐに放出され、遅延放出ナイアシン成分が遅延時間(例えば少なくとも約30分から約40分)後に放出される。
【0060】
当技術分野で周知の材料及び方法を用いて、遅延放出を行うことができる。これらの材料及び方法には次のものが含まれる。単一ユニットの、カプセル薬物送達系(これには、薬物及びプラグを収容する不溶性カプセルが含まれる。)。膨潤、浸食又は溶解の結果、予め定められた遅延時間後にこのプラグが除去される。Pulsincap(R)システム(Scherer DDS、Ltd)はこのような系の一例であり、この場合、本体の開口端が膨潤可能なヒドロゲルプラグで閉じられている。溶解液又は胃腸液と接触すると、プラグが膨潤し、遅延時間後にそれ自身をカプセルから押し出す。この後、薬物が急速に放出される。プラグの直径及び位置を操作することにより遅延時間を調節することができる。例えば、WO90/09168;Wildingら、Pharm Res.1992;9:654−657を参照のこと。プラグの材料を、不溶性であるが浸透性で膨潤性のポリマー(例えば、ポリメタクリレート)(Krogel I、Bodmeier R、Pharm Res.1998;15(3):474−481;Krogel I、Bodmeier R、Pharm Res.1999;16(9):1424−1429)浸食性圧縮ポリマー(例えば、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリエチレンオキシド)、凝結融解ポリマー(例えば飽和ポリグリコール化グリセリド、グリセリルモノオレエート)及び酵素的に調節された侵食性ポリマー(例えばペクチン)製にすることができる。小腸のより高いpH領域でのみ溶解が起こるように系を腸溶性被覆することによって、胃での滞留時間が変動するという潜在的問題を解決することができる。Saeger H、Virley P.Pulsincap & Mac226:パルス放出剤形。Scherer DDS、Ltd;2004からの製品情報。
【0061】
Port(R)システム(Port Systems、LLC)は、浸透に基づくカプセル系であり、製剤とともに。不溶性プラグ(例えば脂質性のもの)を収容する半透性の膜(例えば酢酸セルロース)で被覆されたゼラチンカプセル及び浸透圧的に活性のある物質からなる。Crisonら、Proceed Intern Symp Control Rel Bioact Mater.1995;22:278−279。水性溶媒と接触すると、水が半透性の膜を横切って拡散し、結果として、内圧が上昇し、遅延時間後にプラグが噴出される。コーティングの厚さによって遅延時間を調節する。
【0062】
薬物を液体の形態で送達するために、浸透圧によって駆動されるカプセル系を使用することができ、この場合、液体薬物は非常に多孔性である粒子に吸収されており、この粒子は、障壁層が溶解した後に拡大する浸透性の層により支持された半透性カプセルのオリフィスを通じて薬物を放出する。米国特許第5,318,558号を参照のこと。このカプセル系は、身体からの水分が浸透圧によって導入されることにより薬物を送達する。カプセルの壁は弾性材料からなり、オリフィスを有する。浸透が進行するにつれて、カプセル内の圧力が上昇し、壁が伸張する。オリフィスは弾性壁が弛緩しているときはオリフィスを通じた薬物の流れが基本的に停止するが、弾性壁が閾値を超えて広がった場合はオリフィスが十分に広がり、望まれる速度で薬物が放出されるよう、十分に小さい。スチレン−ブタンジエンコポリマーなどのエラストマーを使用することができる。米国特許第5,221,278号;米国特許第5209746号を参照のこと。
【0063】
Time Clock(R)システム(West Pharmaceutical Services Drug Delivery & Clinical Research Centre)は、ポリオキシエチレンソルビタンモノオレエートなど、界面活性剤とともにカルナバワックス及び蜜蝋を含有する脂質性の障壁で被覆された固形の剤形である。Wildingら、Int J Pharm.1994;111:99−102;Niwaら、J Drug Target.1995;3:83−89。このコーティングはフィルムの厚さに比例した時間で水性環境において浸食又は乳化し、次にコアが分散に利用可能になる。ヒトのボランティアの試験において、遅延時間が胃での滞留時間とは独立であることが分かり、疎水性フィルム再分散は腸の酵素の有無又は胃の機械的作用又は胃腸のpHにより影響を受けないと思われる。Gazzanigaら、Int J Pharm.1994;2(108):77−83。コーティングが厚くなるにつれ遅延時間が長くなった。
【0064】
Chronotropic(R)システムは親水性の膨潤性ヒドロキシプロピルメチルセルロース(HPMC)で被覆された薬物含有コアであり、これは、放出前の遅延期間に関与する。Gazzanigaら、Eur J Biopharm.1994;40(4):246−250;Gazzanigaら、Proceed Intern Symp Control Rel Bioact Mater.1995;22:242−243;EP 0 572 942。胃に耐性のある外側の腸溶性フィルムによって、胃が空である時間の変動に関する問題を解決することができる。Sanagalliら、J Contr Rel.2001;73:103−110。HPMCの厚さ及び粘性グレードによって遅延時間が調節される。この系は錠剤及びカプセルの両方に適している。Conteら、Drug Dev Ind Pharm.1989;15(14−16):2583−2596。
【0065】
薬物不含のゲル化可能ポリマー性障壁層により分離されている2つの活性薬剤含有層を含む3層の錠剤構造から2種類の活性薬剤を含有する多層錠剤を形成することができる。米国特許第4,865,849号;Conteら、Eur J Pharm.1992;38(6):209−212;Krogel I、Bodmeier R、Int J Pharm.1999;187:175−184。この3層性錠剤の3つの側面を不浸透性のエチルセルロースで被覆し、上の部分は被覆しない。溶解液と接触すると、一番上の層に組み込まれた用量が非被覆面から急速に放出される。第二の用量は、HPMCのゲル化した障壁層が浸食し溶解した後に最下層から放出される。障壁層のゲル化及び/又は溶解の速度によって第二の要領の出現が調節される。ゲル化ポリマーには、セルロース誘導体、例えばHPMC、メチルセルロース又は様々な分子量のポリビニルアルコールなどが含まれ得、コーティング材料には、エチルセルロース、酢酸−プロピオン酸セルロース、メタクリルポリマー、アクリル及びメタクリルコポリマー及び多価アルコールが含まれる。
【0066】
破裂可能なコーティング付きのパルス系は、薬物の放出のためのコーティングの崩壊に依存する。発泡性賦形剤、膨潤剤又は浸透圧により、コーティングの破裂に必要な圧力を得ることができる。クエン酸及び重炭酸ナトリウムの発泡性混合物をエチルセルロースで被覆された錠剤コア中に組み込むことができる。コアへの水の浸透後に生成する二酸化炭素によって、コーティングの破裂後、薬物が放出される。Bussemer T、Bodmeier R、AAPS Pharm Sci.1999;1(4 suppl):434(1999)。コーティングが厚くなり、コア錠剤の硬度が向上するにつれて遅延時間が長くなる。
【0067】
薬物、膨潤剤及び破裂可能なポリマー層を含むカプセルに基づく系を設計するために、超崩壊剤(superdisintegrant)とも呼ばれる高度膨潤性物質を使用することもできる。米国特許第5,229,131号。超崩壊剤の例には、クロスカメロース、デンプングリコール酸ナトリウム及び低置換度ヒドロキシプロピルセルロースが含まれる。これらの材料が膨潤する結果、フィルムが完全に破裂し、次いで薬物が放出される。遅延時間は、外側のポリマー層の組成の関数である。HPMCなどの親水性ポリマーが存在すると遅延時間は短くなる。固形及び液体製剤両方の送達のためにこの系を使用することができる。
【0068】
ある活性薬剤の遅延放出を行い、第二の活性薬剤の遅延又はその他のタイプ(例えば即放性)の放出を行うために、多粒子系(例えばビーズ又はカプセル中ペレット)を使用することができる。例えば、米国特許第4,871,549号を参照のこと。
【0069】
時間制御型破裂系(Time−Controlled Explosion System)(藤沢薬品工業)は多粒子系であり、ここでは薬物がノンパレイユシュガーシード(non−pareil sugar seed)、次いで膨潤性の層及び不溶性の一番外側の層で被覆される。Uedaら、J Drug Targeting 1994;2:35−44;Uedaら、Chem Pharm Bull.1994;42(2):359−363;Uedaら、Chem Pharm Bull.1994;42(2):364−367;Hataら、Int J Pharm.1994;110:1−7。膨潤剤には、カルボキシメチルセルロースナトリウム、デンプングリコール酸ナトリウム、L−ヒドロキシプロピルセルロースのような超崩壊剤、酢酸ポリビニル、ポリアクリル酸、ポリエチレングリコールのようなポリマーなどが含まれ得る。あるいは、酒石酸及び重炭酸ナトリウムの混合物を含む発泡系を使用することができる。水が浸入すると、膨潤性の層が拡張し、その結果、フィルムが破裂し、続いて薬物が急速に放出される。この放出は、pHなどの環境要因及び薬物の溶解度とは独立である。コーティングの厚さを変化させるか又は多量の脂溶性の可塑剤を最外層に添加することによって、遅延時間を変化させることができる。米国特許第5,508,040号。
【0070】
浸透性制御系(Permeability Controlled System)は、浸透及び膨潤効果の組み合わせに基づく。コアは、薬物、低かさ密度の固形及び/又は液体脂質性物質(例えば鉱物油)及び崩壊剤を含有する。次に、このコアは酢酸セルロースで被覆されている。水性溶媒中での浸漬の際、水がコアに浸透し、脂質性物質と置き換わる。脂質性物質がなくなった後、臨界応力に到達するまで内圧が上昇し、その結果、コーティングが破裂する。米国特許第5,229,131号。
【0071】
別の系は、2以上のペレット又は部分からなる多数のペレット(即ち集団)でできているカプセル又は錠剤に基づく。Schultz P、Kleinebudde P.J Contr Rel.1997;47:181−189。各ペレットは、治療薬物及び水溶性浸透物質を含有するコアを有する。透水性で不水溶性のポリマーフィルムは各コアを封入する。浸透性を変化させる疎水性の不水溶性の物質(例えば、脂肪酸、ワックス又は脂肪酸の塩)をポリマーフィルムに組み込む。水の流入及び薬物の流出の速度によって、各集団のフィルムコーティングが剤形中の何れのその他のペレットコーティングとも異なるようになる。浸透物質が水中で溶解してペレットが膨潤するが、それにより薬物拡散速度が制御される。その薬物含量を連続的に放出する各ペレット集団の効果によって、1つの剤形から一連の薬物放出が起こる。ペレット間でコーティングの厚さを変化させることができる。
【0072】
遅延放出を行うために、膨潤しない浸透圧的に活性のある物質を使用することもできる。Schultzら、J Contr Rel.1997; 47:191−199;米国特許第5,260,069号。ペレットコアは薬物及び塩化ナトリウムからなる。このコアは半透性酢酸セルロースポリマーで被覆されている。このポリマーは、選択的に水を浸透させ、薬物に対して非浸透性である。遅延時間は、コーティングが厚くなるに従い、及びコーティング中のタルク又は脂溶性可塑剤の量が増えるに従い、長くなる。塩化ナトリウムは薬物の高速放出を促進する。塩化ナトリウム非存在下では、コア膨潤の程度が低くなり、その結果小さな亀裂が生じるために、遅延時間後、徐放を行うことができる。
【0073】
遅延放出を行うために、不溶性の浸透性膜で被覆された、薬物及び浸透圧的に活性のある物質(塩化ナトリウム)のコアを含有する系を使用することができる。米国特許第5,260,068号。コーティング材料には、様々なタイプのポリ(アクリレート−メタクリレート)コポリマー及びステアリン酸マグネシウムが含まれるが、これは、膜の透水性を低下させ、従ってより薄い膜の使用が可能となる。厚いフィルムは完全に破裂し得ないので、避けるべきである。コーティング材料としてエチルセルロースを使用して、予め決定された時間後に破裂させるために、腸溶性ポリマーに対する遅延時間に影響を与えることができる。Bodmeierら、Pharm Res.1996;13(1):52−56。
【0074】
溶媒中の様々な対イオンの存在によって、四級アンモニウム基を有するアクリル性ポリマーの浸透性及び水取り込みに影響を及ぼし得る。Beckertら、Proceed Int’l Symp Control Rel Bioact Mater.1999;26:533−534。このイオン交換に基づくいくつかの送達系が開発されてきた。Eudragit RS 30Dは、ポリマー側鎖中に正に分極した四級アンモニウム基を含有し、これは負の塩酸対イオンを付随するので、この目的のための好ましいポリマーである。アンモニウム基は親水性であり、ポリマーの水との相互作用を促進し、それによってその浸透性が変化し、制御下で水が活性コアに浸透できるようになる。4種類の異なる層厚さのEUDRAGIT RS30D(R)(10%から40%重量増加)でペレットを被覆することができる。遅延時間は、フィルムの厚さに相関する。EUDRAGITフィルムの薬物浸透性はペレットコア中の酢酸ナトリウム量に依存する。遅延時間後、酢酸塩とポリマーとの間の相互作用によりコーティングの浸透性が向上し、活性用量全体が数分以内に遊離する。Guo X.Physicochemical and Mechanical Properties Influencing the Drug Release From Coated Dosage Forms.Doctoral Thesis.The University of Texas at Austin;1996。
【0075】
シグモイド放出系は、アンモニオ−メタクリレートコポリマー USP/NFタイプBで被覆された薬物及びコハク酸を含有するペレットコアを含む。Narisawaら、Pharm Res.1994;11(1):111−116。遅延時間は、ポリマー膜を通じた水流入の速度により制御される。水はコア中のコハク酸及び薬物を溶解する。この酸溶液が次に、水和ポリマーフィルムの浸透性を向上させる。コハク酸に加えて、酢酸、グルタル酸、酒石酸、リンゴ酸又はクエン酸を使用することができる。浸透性の向上は、フィルムの水和が向上することにより説明できるが、これによって自由体積が増加する。酸含有コアを有する被覆送達系を設計するために、これらの知見を使用した。Narisawaら、Pharm Res.1994;11(1):111−116;Narisawaら、J Contr Rel.1995;33:253−260。インビトロ遅延時間は、ビーグル犬で試験した際のインビボデータとよく相関した。Narisawaら、J Contr Rel.1995;33:253−260。
【0076】
本発明は、紅潮抑制剤と組み合わせた遅延放出型の本発明のナイアシン製剤を含む医薬組成物を包含する。1つの剤形又は別個の剤形で遅延放出ナイアシン及び紅潮抑制剤を与えることができる。したがって、例えば、本医薬組成物は、外側に即放型紅潮抑制剤成分及び内側に遅延放出ナイアシンを有する固形剤形を含み得る。好ましい実施形態において、紅潮抑制剤は、ナイアシンが放出される約30分から約40分前に放出される。
【0077】
次の実施例は、本発明の複数の実施形態をより詳細に説明するためのものである(これらに限定されない。)。
【実施例1】
【0078】
この実施例において次の処方を使用した:
【0079】
【表6】
【0080】
好ましくは、METHOCEL(R)K−15M Premiumを使用する場合、METHOCEL(R)K−15M Premiumに対する粒子サイズ規格は、最低90%が100メッシュUS標準ふるいを通過することである。METHOCEL(R)K−15M Premium CRの場合、好ましくは最低99%が40メッシュUS標準ふるいを通過し、最低90%が100メッシュUS標準ふるいを通過する。
【0081】
20kgのバッチサイズの場合、上記処方に従い、砕いた(delumped)粒状ナイアシン及び賦形剤の重量を測定し、次いで8−quartブレンダーに添加し、24rpmで10分間混合した。特に、METHOCEL(R)K−15M及びステアリン酸を分散させるために12メッシュ(1.68mm)スクリーンを選択し、粒状ナイアシン及びポビドンK−90(場合によってはふるいにかける、粉砕する、又は両方)を分散させるために16メッシュ(1.19mm)スクリーンを選択した。30kNで19mm長の楕円形のツーリングを有するBWI Manesty Beta Pressを用いて、得られた粒状成分を直接錠剤へと圧縮した。目標とする18kPの錠剤硬度に対して、標準的な錠剤硬度テスターを用いて、錠剤の硬度(即ち、当業者にとって公知の標準的圧縮試験方法によって測定した場合の錠剤の圧縮強度)を16kP(キロポンド)から22kPの範囲内に調節した。場合によっては、40メッシュスクリーンなど、ステアリン酸又はポビドンをメッシュスクリーンに通してふるいにかけることができ、代替的実施形態において、混合段階(1又は2)及び混合時間(10、15又は20)を変化させることができる。
【0082】
OPADRY(R)Orange 03B93199の2%増量の色彩塗料で、得られた圧縮錠剤を被覆した。コーティング条件は次のとおりであった。
【0083】
【表7】
【0084】
安定性試験前後での、ナイアシンアッセイ、ナイアシン溶解、錠剤の水分及び被覆錠剤の外見を比較することによって、被覆したナイアシン1000mg直接圧縮錠剤が40℃/75%相対湿度(RH)及び25℃/60%相対湿度で3ヶ月間安定であることが分かった。
【0085】
図5は、本発明の実施形態による錠剤製剤を調製するための直接圧縮製造プロセスの流れ図を示す。
【0086】
別段の断りがない限り、次の実施例に記載の本発明の1000mg徐放ナイアシン製剤は実施例1に従い調製した。
【実施例2】
【0087】
本明細書中に記載のプロセスを使用して、下記表8及び9で述べる含量濃度を有する、500mg及び750mg徐放直接圧縮錠剤(被覆又は被覆なし)を調製することができる。
【0088】
【表8】
【0089】
【表9】
【0090】
500mg及び750mg錠剤の場合、表8及び表9で示す成分濃度に従い、砕いた(delumped)粒状ナイアシン及び賦形剤の重量を測定し、次いで成分を十分に混合するために適切なブレンダー又はミキサー中で適切な時間、混合する。次に、上述のBWI Manesty Beta Pressなどの適切なプレスを用いて、得られた粒状成分を錠剤へと直接圧縮し、所望のような500mg又は750mg錠剤強度を形成することができる。場合によっては、500mg又は750mg錠剤強度を当技術分野で公知のように、色彩塗料などで被覆することができる。
【実施例3】
【0091】
被覆徐放1000mgナイアシン直接圧縮マトリクス錠剤と1000mg NIASPAN(R)との間の紅潮の相対的発生率
方法
本試験は、単一施設で行われた、無作為の二重盲検、ダブルダミー、単回投与、プラセボを対照とした、3期クロスオーバー、紅潮誘発試験である。試験中にアスピリン又はNSAIDを対象に使用させなかった。
【0092】
本試験には、ボディーマスインデックス(BMI)が22から31の、18歳から70歳の健常な非喫煙の男性ボランティアが含まれた。人間ドック、病歴、心電図及びスクリーニングの来院時又は最初の試験期間の入院の来院時に行った臨床検査によって、対象が健常であることを確認した。ナイアシン又は関連誘導体に対するアレルギー又は過敏性がある場合;過去3年以内に薬物乱用又は依存症があった場合;偏頭痛、糖尿病、胆嚢疾患、肝臓疾患、重度の高血圧又は低血圧、心臓の異常、腎臓疾患又は薬物誘発性ミオパシーの病歴がある場合は対象を除外した。対象は、試験開始の21日以内のいかなる処方投薬も、又は、試験開始の10日前以内の市販薬、ビタミン又はハーブも摂取してはならなかった。
【0093】
試験期間1への入院前21日以内でスクリーニング手順を完了した(図6)。3回の各試験期間中、第1日の午前7:00前後から第2日の午前の全ての試験手順が完了するまで対象を隔離した(午前7:00から午前10:00の間)。食事構成及び開始時間は各試験期間で同じであった。各試験期間中、対象は、ナイアシン及び脂肪含量を調節した特別メニューに従う食事を取った。併用薬、ビタミン又はハーブ及び/又は栄養剤は試験中一切禁止した。
【0094】
試験治療
3回の試験期間中に投与された製剤を図6で記す。試験治療は、本発明の2個のフィルム被覆1000mg錠剤製剤を使用し(実施例1参照)(Test−改質ナイアシンER錠剤)、一方、参照治療は、2個の非被覆1000mg市販ナイアシンER錠剤(Reference−NIASPAN(R))を使用した。対照治療は、2個の非被覆プラセボ錠剤(対照)を使用した。この試験は対象が報告する紅潮に焦点を当てたので、対象及び試験者が治療において投与される製剤がどれであるかについて完全に分からないことが重要であった。いくつかの方法によって分からないようにした。各活性治療において、活性錠剤に似ている2個のフィルム被覆プラセボ又は非被覆プラセボ錠剤を活性錠剤とともに同時投与し、全対象が治療にかかわらず2個のフィルム被覆及び2個の非被覆錠剤を摂取するようにした。また、不透明な投与カップから対象に試験薬を投与し、試験薬投与中、対象に目隠しを施した。予想されるプラセボ反応に対して紅潮の結果を補正するために、試験にプラセボ−対照治療を入れた。
【0095】
各試験期間、第1日の午後11:00前後に、無作為計画に従うクロスオーバー方式で試験薬の単回投与を行った。各試験期間の間に7日以上のウォッシュアウト期間を置いた。試験者及び現場人員に対して治療割り当て方式が分からないようにし、治療割り当て調製及び/又は投与に関わった何れの現場人員も、治療中に発生した有害事象を収集又は評価することを禁じた。
【0096】
低脂肪スナックの後に、水240mLとともに、各用量を経口投与した。このスナックを全て、試験薬投与前15分以内に摂取させた。錠剤を1度に一緒に飲ませるか、又は1個を飲んだ後すぐに他方を飲ませ、投与を完了するのに1分を超えないよう各対象に指導した。錠剤を咀嚼又は噛むことは禁じた。錠剤を飲み込むために対象がさらなる水を所望した場合、120mL単位でさらなる水を与えた。用量を摂取したことを確認するために、試験用量の投与後、各対象の口腔を調べた。
【0097】
紅潮可変要素
第一の紅潮可変要素は、対象が報告する紅潮事象又は発現の発生率であった。紅潮事象又は発現を次の共通の紅潮症状の1以上として記した:発赤、熱感、刺痛及び掻痒。各試験期間中、試験薬物投与後、最長で8時間、1時間ごとに、紅潮症状の有無を対象に評価させた。紅潮症状の開始及び停止時間を対象に記録させ、水平の10cm視覚的アナログ尺度(VAS)上で垂直線を作ることによって(「なし」(0)を左端に「容認できない。」(100)を右端にした。)各症状の強度(重症度)のランク付けを行わせた。コンピュータの紅潮日誌に情報を記録した。
【0098】
第二の紅潮可変要素には、紅潮発現の回数、強度及び全体的紅潮事象及び紅潮の個々の症状(発赤、熱感、刺痛及び掻痒)の両方に対する紅潮の持続時間が含まれた。各対象が、試験期間中に発生した1以上の共通紅潮症状の最初のものの開始時間に始まるものとして定義される、最初の紅潮事象又は発現の全体的強度をランク付けした。紅潮発現の終了時間は、その発現において起こった1以上の共通の紅潮症状の最後の停止時間(また、この後、無症状期間が30分以上続く。)として定義した。
【0099】
統計分析
マクネマー検定(nQuery Advisor(R)、バージョン5.0)を用いて5%のアルファ(α)で治療間の紅潮の発生率の統計的有意差を示すためには144名の対象のサンプルサイズが必要であると決定した。対象の適切な人数が試験を完遂し、少なくとも2つの治療から評価可能なデータを得ることを確実にするために、早期に撤退した患者を入れ替えた。
【0100】
対応のある比率の同等性のマクネマー検定を用いて治療群間で最初の有効性評価(紅潮の発生率)を比較した。少なくとも2回の試験期間で少なくとも1回試験薬投与を受けた対象において、ナイアシンERのTest製剤と参照製剤との間で最初の比較を行った。ナイアシンとプラセボとの間の比較も行った。マクネマー検定(カテゴリ変数に対して)又はマッチドペアt検定の何れかを用いて第二の評価を比較した。比較は全て両側検定であり、アルファ(α)=0.05で行った。
【0101】
結果
全部で156名の対象をこの試験に登録し、少なくとも1回の試験薬投与を受けさせた。これらの平均年齢は33.5歳であり、これらの平均BMIは26.2であった。対象デモグラフィックの要約を下記表10で示す。
【0102】
【表10】
【0103】
ピリオド1において156名の対象全員が試験薬の投与を受け、143名の対象(92%)がピリオド2で試験薬の投与を受け、131名の対象(84%)がピリオド3で試験薬の投与を受けた。全部で130名の対象(83%)が全3期間での投与を完遂した。26名の対象(17%)は試験から途中で撤退し:8名(5%)が同意を取り消し、3名(2%)がフォローアップできず、2名(1%)で有害事象が起こり、2名(1%)がプロトコール違反し、1名(1%)が薬物スクリーニング検査で陽性となり、残り10名(6%)が「その他」の理由で撤退した。十分な能力を保証するために、途中で試験から撤退した対象のうち11名を置き換えた。
【0104】
紅潮
目的どおり、活性治療における紅潮発生が対照治療のものよりもおよそ4倍高かったので、紅潮誘発を達成した。表11は、少なくとも1回の試験薬投与を受け、少なくとも1回の試験期間を完遂した対象として定義される包括解析(ITT)集団における、最初の紅潮事象の、発生率、強度及び持続時間を示すが、入れ替えた対象は含まなかった。この試験で見られたプラセボ反応は、一般にプラセボ反応に特有である。
【0105】
【表11】
【0106】
参照(市販ナイアシンER)(参照)に対するTest(改質ナイアシンER)に関する、少なくとも2回の試験期間で少なくとも1回の試験薬投与を受けた対象における最初の紅潮事象の発生率及び全体的強度及び持続時間:A)最初の紅潮事象の発生率(p=0.0027);B)VASに基づく最初の紅潮事象の強度;中央値は、[平均値が、Testで35.6±22.78(Min、Max:0.0、99.0)であり、参照で52.8±23.86(Min、Max:0.0、95.0)、p<0.001であった。]ことを示し;C)最初の紅潮事象の持続時間(分);中央値は、[平均値が、Testで130.3±95.01(Min、Max:9.0、473.0)であり、参照で195.7±136.32[Min、Max:5.0、984.0]、p<0.0001であった。]ことを示した。
【0107】
図7で示されるように、最初の有効性評価に対して、少なくとも2回の試験期間で少なくとも1回の試験薬投与を受けた対象中で、Test製剤での治療中に118名(89%)の対象で紅潮が起こり、参照製剤によって130名(98%)の対象で治療中に紅潮が起こった。0.0027のp値でこの差は統計的に有意であった。
【0108】
図8及び9は、最初の紅潮事象の中央値強度及び持続時間を示す。これらの個々の中央値との強度及び持続時間に対する平均値の比較から、これらのデータの基礎をなす分布が非対称であったことが示唆された。Test治療の結果、参照治療と比較して、紅潮強度中央値が42%低下し(平均紅潮強度は33%低下)、紅潮持続時間中央値が43%低下(平均紅潮持続時間は33%低下)した。対応のあるt検定によって、最初の紅潮事象の平均強度(p<0.0001)及び平均持続時間(p<0.0001)の両方に関して、Test治療で統計的に有意な改善が示された。
【0109】
参照製剤に対するTest製剤で、4種類の紅潮症状(発赤、熱感、刺痛及び掻痒)のそれぞれの発生率が低下した(図10)。この2種類の製剤の比較は有意差があり、マクネマー検定を用いて、最初の紅潮事象に対する4種類の個々の紅潮症状のそれぞれに対してTest製剤が選択される。発赤は、Test製剤によって71%で発生したのに対して参照製剤では86%で発生し(p=0.0016);熱感は、Test製剤によって68%で発生したのに対して参照製剤では80%で発生し(p=0.0163);刺痛は、Test製剤によって47%で発生したのに対して参照製剤では62%で発生し(p=0.0039);掻痒は、Test製剤によって48%で発生したのに対して参照製剤では65%で発生した(p=0.0015)。
【0110】
このデータから、市販の製剤と比較して、本発明の製剤によって、紅潮の発生率、強度(重症度)及び持続時間が低下することが示される。ナイアシン治療を受けたことがなかった対象に単回大量(2000mg)用量を投与することによって紅潮を誘発するようにこの試験が計画されたにもかかわらず、全体的に市販ナイアシンER製剤NIASPAN(R)(98%)と比較して、本発明の製剤で(89%)紅潮の発生率が統計的に有意に9%低下した。この紅潮誘発試験での本発明の製剤の投与の結果、また、紅潮強度及び持続時間が非常に統計的に有意に低下した。市販ナイアシンER治療と比較して、中央値紅潮強度及び持続時間はそれぞれ42%及び43%低下した。また、最初の紅潮事象の持続時間は本発明の製剤によって1時間を超えて短くなった。
【実施例4】
【0111】
この試験の目的は、2000mgの単回投与で投与した際の、市販の1000mgNIASPAN(R)錠剤(REF)との、本発明の1000mg徐放ナイアシン錠剤(本明細書中で以後「改質」錠剤と呼ぶ。)(Test)の生物学的同等性(BE)を調べることであった。
【0112】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙男性及び女性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、2期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間に少なくとも10日間のウォッシュアウト期間を置いて、2回の別個の時に、2000mgの同じ単回投与で、2つのナイアシン製剤、Test及びREFの投与を受けた。試験製品は改質1000mg徐放ナイアシン錠剤であり、参照製品(REF)は1000mg NIASPAN(R)錠剤であった。各期間の第1日の22:00前後から開始する低脂肪スナックの後に水240mLとともに各用量を投与した。各試験期間中、試験施設で患者を隔離し(ピリオド1に対して5日間、ピリオド2に対して6日間)、スポンサーが提供するメニューに従い食事を与えた。他の薬、ビタミン、ハーブ又は栄養剤を試験中禁じた。
【0113】
−30分(投与前)、1、2、3、4、4.5、5、6、7、8、10、12、14、16及び24時間(投与後)の間隔で、投与前30分以内から、投与後24時間まで、連続血液試料を回収した。−24から−18、−18から−12、−12から−6及び−6から0時間(投与前);0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)の間隔で、投与前24時間から投与後96時間まで尿を回収した。ナイアシン及びニコチン尿酸(NUA)について血漿を分析した。ナイアシン及びその代謝産物:NUA、N−メチルニコチンアミド(MNA)及び2−PY(N−メチル−2−ピリドン−5−カルボキサミド)について尿を分析した。
【0114】
ナイアシンは十分に代謝され、血漿濃度から、その主要な代謝産物の1つであるNUAと比較して非常に変動性が高いことが示される。したがって、NUAに対する最大血漿濃度(Cmax)がナイアシン吸収速度を決定するために使用されてきた。AUCは非線形的な薬物動態の影響をより受けやすいので、NIASPAN(R)NDAで示されるように、総尿回収は、AUCよりも、吸収の程度のより正確な目安である。したがって、尿中でナイアシン及びその代謝産物のうち3種類、NUA、MNA及び2PYとして排出されるナイアシンの総量は、ナイアシン吸収の程度の目安として役立つ。本プロトコールにおいて定義される、NUA生物学的同等性を評価するための主要な可変要素は、ゆえに、NUAに対するCmax及びナイアシン及び3種類の代謝産物(NUA、MNA及び2PY)の総尿回収である。
【0115】
Test薬は、本発明の改質1000mg徐放錠剤の2個の錠剤からなる。REF薬は、1000mg NIASPAN(R)錠剤の2個の錠剤からなる。治療を少なくとも10日間離した。
【0116】
対象は、各期間中、病院に隔離されているときは、各日の同じ時間に食事を開始した。食事は各期間に対して同じに維持し、各食事を全量摂取するよう求めた。朝食、昼食、夕食及び夜食はそれぞれ、07:00、12:00、18:00及び21:45前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに水240mLを与えたのに加え、第−1日に水を最低720mL飲み、第1日から5日に1440mL飲むよう求めた。
【0117】
第−1日に、夕食及び夜食を供した。第1日から5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では、投与直前15分以内に取らせた。ピリオド2の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0118】
薬物動態の評価
a.血漿回収及び分析
各期間において、投与前30分以内から、投与後24時間まで、連続血液試料を回収した(治療あたり15試料)。ヘパリンナトリウムを含有する1本の10mL真空採血システムに各血液試料を回収し、氷片及び水浴中で回収後最低5分間、試料を冷却した。4℃にておよそ3000rpmで15分間試料を遠心して血漿を分離した。各血漿試料を2つ、アリコートA及びB、に分け、予め冷却し適切にラベルを貼った2本のポリプロピレンチューブに移した。次に、試料をおよそ−20℃で凍結保存した。
【0119】
有効な液体クロマトグラフィータンデム質量分析(LC/MS/MS)によって、ナイアシン及びNUA濃度を分析した。ナイアシン及びNUA濃度を同じ注入から得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0120】
b.採尿及び分析
次の間隔で採尿した:−24から−18、−18から−12、−12から−6及び−6から0時間(投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(全部で11回の回収)。
【0121】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、採尿した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。空の容器の重量を測定し、グラムの小数点以下1桁に四捨五入して(例えば100.1g)、これを容器に記入し、実験室のソースドキュメントワークシートに記録した。各間隔の終わりに、容器及び回収した尿の総重量を測定し、グラムの小数点以下1桁に四捨五入して記録した。尿重量は、容器+尿の総重量から空の容器の重量を差し引くことにより得られた。場合によっては、ある一定の回収間隔中の尿体積が1個の容器の容量を超え、従って採尿を完遂するために第二の容器を必要とした。各採尿間隔の開始及び終了日時も記録した。各回収間隔からの2つのアリコート(各およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。特定の回収間隔中に複数の容器が必要であった場合、両方の容器からの尿を一緒にして混合し、その後アリコートを取った。分析までおよそ−20℃で試料を凍結保存した。
【0122】
有効なLC/MS/MSにより、ナイアシン、NUA、NMA及び2−PYの濃度について尿試料を分析した。同じ注入から尿ナイアシン及びNUA濃度を得て、一方、NMA及び2−PY濃度を同じ注入から得た。尿において、LLQ値は、ナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2PYのLLQ値はそれぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0123】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対するPKパラメータを計算するために十分な情報を提供する対象からのデータが、PK分析に含まれた。各治療の投与後、各対象に対して次のPKパラメータを計算した:
・Cmax:観察される最大濃度
・Tmax:最大観察濃度の時間
・AUClast:直線的台形規則による、時間0から最後の測定可能(0ではない)濃度までの濃度−時間プロファイル下面積
・AUCinf:時間0から無限大の血漿濃度−時間プロファイル下面積;AUClast及びCl/λの合計として計算(Clは、最後に観察された濃度であり、λは、時間プロットに対する自然対数濃度のプロットから得られた最終消失速度定数である。)
・T1/2:見かけの最終半減期;0.693/λの比として計算
ナイアシン及びその代謝産物(NUA、MNA及び2−PY)の尿データから、次のパラメータを計算した:
・CumXu:投与後0から96時間の尿から回収した各代謝産物の累積量。
【0124】
・%Fe:投与後96時間での、ベースライン回収及び分子量に対する補正後の、ナイアシン投与に対する、尿中で排出された各代謝産物の分画。
【0125】
・総%Fe:投与後96時間における4種類の代謝産物の総分画。
【0126】
尿中の各被分析物に対する%Feを以下のように計算した:
【数1】
【0127】
定量限界未満の濃度をゼロとして処理した。血漿分析に対して、実際の試料回収時間を使用してPKパラメータを計算した。各代謝産物濃度に各間隔で回収された尿の体積を掛けることによって、尿中で回収されたナイアシン及びその代謝産物の量を決定した。24時間投与前間隔で回収された量を差し引くことによって、投与後各24時間間隔に対して尿中で回収された総量をベースラインに対して調整した。投与後測定値がベースライン未満となった場合、その量を0とした。ナイアシン及びその代謝産物の分子量は、ナイアシン、NUA、MNA及び2−PYに対してそれぞれ、123.1、180.2、137.1及び153.1であった。4つの尿被分析物からの%Feの合計を計算し、総%Feとした。
【0128】
WinNonlin Linear Mixed Effects Modeling/bioequivalence、バージョン5.0.1(2005年7月26日)を用いて、(上述のような)生物学的利用率パラメータを計算した。
【0129】
統計分析
WindowsTM用SAS(R)Systemバージョン8.2を用いて、上記で計算された生物学的利用率パラメータの統計分析を行った。
【0130】
治療及び期間によって、血漿薬物動態パラメータ(Cmax、Tmax、T1/2、AUClast及びAUCinf)、それらの自然対数変換値(Tmax及びT1/2を除く。)及び要約統計量(n、平均、std、中央値、min、max、CV%)を計算した。ナイアシン及びNUAの血漿濃度を時間及び治療によってまとめた。
【0131】
ナイアシン及びNUA PK分析の場合、自然対数変換したCmax及びAUClastのデータが正規分布に従い、2つの治療間で独立であると仮定する。固定効果として治療、期間及び順序を、変量効果として順序内の対象を用いて、SAS PROC MIXEDを使用して混合効果によってこのデータをANOVAモデルに適合させた。Cmax及びAUClastのTest/REF比及びそれらの対応する90%信頼区間をこのモデルに基づき評価した。
【0132】
尿からのナイアシン及びその代謝産物の平均回収を計算し、治療により及び間隔によりまとめた。個々の成分のCumXu及び%Fe及び投与後96時間での合計を計算し、治療によりまとめた。
【0133】
血漿PK分析に対して用いたものと同じANOVAモデルに適合させることにより、総%FeのTest/REF平均比に対する90%信頼区間(CI)を計算した。
【0134】
対象のデモグラフィック可変要素(年齢、性別、人種、体重、身長及び肘幅)を性別によりまとめた。連続的デモグラフィック可変要素の、平均、標準偏差(SD)、中央値、最小及び最大を計算した。
【0135】
結果
対象の動態を表12でまとめる。プロトコールの包除基準に合致した後に全部で44名の対象を試験に登録した。対象者全てが、少なくとも1回の試験薬投与を受け、このうち41名がこの試験を完遂した。44名の対象がプロトコールの無作為治療割り当てに従いピリオド1で試験薬投与を受け;41名の対象がピリオド2で試験薬投与を受けた。全部で3名の対象が試験から撤退した。対象0012及び0039がピリオド2で撤退した。対象0038はピリオド2で同意を撤回した。試験から撤退した対象数は、前もって容認するとした10%脱落の範囲であり、この試験の結果又は結論に影響しないと考えられた。
【0136】
【表12】
【0137】
登録した44名の対象のうち、25名の対象が男性で19名が女性であった。平均年齢は54.5歳;平均体重は169.8ポンド;平均身長は68.0インチ;平均肘幅は2.6インチであった。対象のうち37名が白人、6名が黒人、1名がアメリカインディアンであった。表13で詳細なデモグラフィックをまとめる。
【0138】
【表13】
【0139】
a.生物学的同等性の評価
尿の分析のために、1g/mLの比重を使用して、尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究において、962個の試料で測定した平均比重が1.009g/mLであり、962個の試料で測定した最大比重が1.025g/mLであった。)。
【0140】
治療によるナイアシン及びNUAの平均血漿濃度のプロットを図11及び12でそれぞれ示す。平均尿中回収データを図13で示す。
【0141】
b.血漿NUA及び尿中で排出される総量
表14は、2種類の主要な可変要素(NUAに対するCmax及びナイアシン及び3種類の代謝産物の総尿中回収)に対する、及びNUA AUClastに対する、平均(SD)及び統計結果を示す。この表は、投与後に嘔吐の発現があった対象0001、0003及び0014に対する参照治療あり及びなしでのBE分析の結果を与える。
【0142】
対象0001は、ピリオド2でのREF製品投与後7時間20分で嘔吐があった。対象0003は、ピリオド2でのREF製品投与後それぞれ8時間34分及び9時間及び20分で2回の嘔吐があった。対象0014は、ピリオド1でのREF製品投与後11時間20分で嘔吐があった。この3名の対象全員の嘔吐発現時間は、投与から少なくとも7時間20分後であった。NUA及びナイアシンの両方に対するTmaxは、投与後6時間以内であった。したがって、嘔吐はこれらの対象のPKパラメータに影響しないと思われた。
【0143】
【表14】
【0144】
上記の表で示されるように、NUA Cmaxの平均Test/REF比に対する90%CIは、80−125%の生物学的同等性の範囲外であったが、尿から回収されたナイアシン及び代謝産物のTest/REFの平均比に対する90%CIは、80−125%以内であった。この結果は、対象0001、0003及び0014に対するREF治療あり及びなしで同様であった。
【0145】
治療によって、各対象に対して最終消失速度を計算した。それぞれTest及びREFに対して、平均NUA T1/2は3.16及び3.47時間であり、平均NUA Tmaxは5.55及び5.80時間であり、平均NUA AUCinfは12510.8及び18980.8ng*hr/mLであった。
【0146】
c.血漿ナイアシン
統計分析付きで血漿ナイアシンに対する平均PKパラメータを表15で示す。この表は、対象0001、0003及び0014に対するREF治療あり及びなしでのBE分析の結果を与える。ナイアシンCmax及びAUClastのTest/REFの平均比は100%未満であった。この比に対する対応する90%CIは、変動が大きかったため、80−125%間隔の外であった。この結果は、対象0001、0003及び0014に対するREF治療あり及びなしで同様であった。
【0147】
【表15】
【0148】
それぞれTest及びREFに対して、ナイアシンの平均T1/2は5.46及び4.42時間であり、平均Tmaxは5.56及び5.55時間であり、平均AUCinfは13987.8及び35296.6ng*hr/mLであった。
【0149】
d.個々の被分析物の尿中回収
個々の被分析物の平均尿中回収を表16で与える。
【0150】
【表16】
【0151】
上記の表で示されるように、平均尿中回収は2PYに対して最大であり、続いてMNA、NUA及びナイアシンであった。
【0152】
e.生物学的同等性評価の結論
NUA Cmax及びナイアシン及びその代謝産物の尿中回収(総%Fe)の平均Test/REF比に対する90%CIに基づき、生物学的同等性を評価した。総%Feに対するTest/REF平均比の90%CIは80−125%の求められるBE範囲内であったが、NUA Cmaxについては生物学的同等性の範囲外であった。NUA AUClastを含む補助的測定に対するTest/REF平均比の90%CIもまた、80−125%の範囲から外れた。したがって、改質1000mgERナイアシン錠剤(Test)は、NIASPAN(R)1000mg錠剤(REF)と比較した場合、吸収速度が低く、吸収が同程度である。Test治療はREF治療と生物学的に同等ではない。
【実施例5】
【0153】
2000mgナイアシン単回投与後の、市販のNIASPAN(R)500mg錠剤に対する本発明の1000mg徐放ナイアシン錠剤の3種類の製剤(本明細書中で以後「改質」錠剤と呼ぶ。)の生物学的同等性を調べるために、この試験を計画した。
【0154】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙女性及び男性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、4期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間で10日以上のウォッシュアウト期間を設けて4回、経口試験薬、2000mgナイアシン、の同じ用量の投与を受けた。各対象は、1000mgERナイアシン製剤の2個の錠剤(ERN−1、ERN−2、ERN−3)及び4個の500mg NIASPAN(R)錠剤の投与を受けた。
【0155】
低脂肪スナック(22時前後に開始)の後に、水300mLとともに各用量を投与した。対象は、各治療期間中、スポンサーが提供するメニューに従い食事を摂取した。試験中、他の薬、ビタミン、ハーブ又は栄養剤を禁じた。投与前及び投与後24時間まで頻繁に血液試料を回収し、投与前24時間及び投与後96時間、尿を回収した。NUA及びナイアシンに対して血漿を分析した。ナイアシン及びその3種類の主要な代謝産物、NUA、MNA及び2PYについて尿を分析した。各治療の5日間の治療期間中、対象を隔離した。
【0156】
各治療期間中、ナイアシン含量を調整した食事(朝食、昼食、夕食及び夜食)を提供した。
【0157】
参照治療は、高力価の造粒物(ナイアシン、ポビドン及びヒドロキシプロピルメチルセルロース[HPMC])(次いで錠剤への圧縮前にステアリン酸及びさらなるHPMCと混合される。)からなる市販の500mg NIASPAN(R)(NSP)製剤であった。
【0158】
試験治療は、下記表17に従い調製された3種類の様々な改質1000mg NIASPAN(R)製剤(ERN−1、ERN−2及びERN−3)であった。
【0159】
【表17】
【0160】
上記表17で指定された処方に従い、粒状ナイアシン、METHOCEL(R)K15M、ポビドンK90及びステアリン酸の重量を測定し、次いで8qtブレンダー(LB−9322、Petterson Kelly、East Stroudsburg、PA)に添加し、10分間混合した。16から18Kpの目標錠剤硬度になるように、500錠剤/分のスピードで、BWI Manesty Beta Press(Thomas Eng、Hoffman Estate、IL)を用いて、よく混ざった混合物を錠剤へと圧縮した。
【0161】
全ての食事及び飲料はアルコール及びキサンチン不含であった。ナイアシンは通常の食事で摂取可能なので、およそ25mg/日のナイアシン摂取を維持するためにこの試験の食事を調整し、一方、対象を病院に拘束した。低脂肪スナックの直後、22:00前後に各用量を投与した。
【0162】
対象は、各期間中、毎日、同じ時間に食事を開始し、病院に拘束された。食事は各期間で同じであり、各食事の全量を摂取させた。朝食、昼食、夕食及び夜食は、それぞれ0700、1200、1700及び2145前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに与えた300mLの水に加え、第−1日に水を720mL以上飲み、第1、2、3、4及び5日に水を1440mL以上飲むよう求めた。
【0163】
第−1日に、夕食及び夜食を供した。第1、2、3、4及び5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では15分以内に取らせた。各期間の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0164】
薬物動態の評価
a.血漿回収及び分析
各期間、投与前30分以内(即ち投与前)及び投与後1、2、3、4、4.5、5、6、7、8、9、10、11、12、14、16及び24時間で血液試料を得た。対象をまっすぐに椅子に座らせ、試験区域で試料を採取した。ヘパリンナトリウムを含有する7mL真空採血システムに採血し、氷片及び水浴中で回収後少なくとも5分間、試料を冷却した。4℃にておよそ3000rpmで15分間試料を遠心して血漿を分離した。冷却し予めラベルした2本のポリプロピレンチューブに血漿分画を移した。分析までおよそ−70℃で試料を凍結保存した。
【0165】
MS/MS検出付きのHPLCクロマトグラフィーによって血漿ナイアシン及びNUA濃度の生物分析を行った。同じ注入からナイアシン及びNUA濃度を得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0166】
b.採尿及び分析
次の間隔で採尿した:−24から−18、−18から−12、−12から−6及び−6から0時間(即ち投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(治療あたり全部で11回の回収)。
【0167】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、回収した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。各間隔中に回収した尿の総重量を測定してグラムの小数点以下1桁に四捨五入し(例えば100.1g)、記録した。各採尿間隔の開始及び終了の日時も記録した。各回収間隔からの2つのアリコート(およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。Kos、プロトコール番号、対象番号、日付、回収間隔、試験日及び期間を識別するために、分析用検体にラベルを付した。発送の準備ができるまで、およそ−20℃でアリコートを保存した。病院側は、比重測定のためにさらなるアリコートを得た。
【0168】
ナイアシン、NUA、MNA及び2−PYについて尿試料を分析した。MS/MS検出付きのHPLCクロマトグラフィーによって、尿中ナイアシン、NUA、MNA及び2−PY濃度の生物分析を行った。ナイアシン及びNUA濃度を同じ注入から得た。
【0169】
尿中で、LLQ値はナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2−PYのLLQ値は、それぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0170】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対する薬物動態パラメータを計算するために十分な情報を提供する対象からのデータが薬物動態分析に含まれた。各治療の投与後、各対象に対して次の薬物動態パラメータを計算した:
血漿ナイアシン及びNUAデータから:
・Cmax:観察される最大血漿濃度
・Tmax:最大観察濃度の時間
・AUC0−last:血漿濃度−時間プロファイル下面積;直線的台形規則によって時間0から最後の測定可能濃度まで計算
NUAデータから、さらなるPKパラメータも計算した:
・AUC0−inf:血漿濃度−時間プロファイル下面積;AUC0−last+Ct/Kelとして時間0から無限大まで計算(Ctは最後に観察された定量可能濃度であり、Kelは最終消失速度定数である。)
・T1/2:0.693/Kelとして計算された最終半減期
尿中のナイアシン、NUA、MNA及び2PYデータから:
・CumXu:各被分析物個々に対する尿中回収(即ち尿中で回収された各被分析物の量)
・Fe:
【数2】
として計算された尿中で排出された分画
・総%Fe:ナイアシン、NUA、MNA及び2PYの総回収。
【0171】
LLQ未満の濃度をゼロとして処理し、分析において実際の試料回収時間を使用した。WinNonlin Professional Network Edition、バージョン4.1を用いて、血漿ナイアシン及びNUA濃度の個々のプロットを生成させた。血漿ナイアシン及びNUA濃度及び尿中回収データの平均プロットはWinNonlin4.1及びMicrosoft(R)Excel 2000により生成させた。WinNonlinによって各プロファイルから血漿薬物動態パラメータを決定した。各血漿プロファイルでの定量可能なナイアシン濃度が少数であったこと及びはっきりと定義される最終相がないことから、血漿ナイアシンデータに対して最終相勾配及び見かけの半減期を計算しなかった。
【0172】
WinNonlin4.1及びExcel 2000を用いて全ての尿中薬物動態パラメータを決定した。被分析物濃度に各間隔に対して回収された尿の体積を掛けることによって、尿中で回収された各被分析物の量を調べ;次に、24時間投与前間隔で見出された量を差し引くことによって、投与後24時間間隔に対する尿中回収量をベースライン回収に対して補正した。
【0173】
被分析物の分子量は、ナイアシン、123.1;NUA、180.2;MNA、137.1;2PY、153.1である。個々の被分析物の%回収を合計し、回収した用量の総%を計算した。
【0174】
統計分析
WindowsTM、バージョン8.02用のSAS Systemを用いてデモグラフィックをまとめた。平均、標準偏差(SD)、中央値、最小及び最大値により、連続的デモグラフィックデータをまとめた。
【0175】
自然対数変換データを用いて、WinNonlin4.1で構築された生物学的同等性ウィザードを使用することによって、生物学的同等性パラメータを評価した。このモデルには、順序、順序内の対象、期間及び治療が含まれた。
【0176】
自然対数変換したデータに基づき、最小二乗平均の参照に対するTestの比(ERN−1/NSP、ERN−2/NSP、ERN−3/NSP)についての古典的な90%信頼区間(CI)推定により生物学的同等性を評価した。90%CIが80から125%内であった場合、治療が生物学的に同等であるとみなした。生物学的同等性決定に対して、使用したパラメータは、血漿中のNUAに対するCmax及び尿中で排出されるナイアシン及び代謝産物の総量であった。ナイアシン血漿(Cmax及びAUC0−last)及び尿データに対する個々の%Fe(ナイアシン、NUA、MNA、2PY)に対しても信頼区間を調べた。
【0177】
結果
プロトコールの包除基準に合致した40歳から70歳(両端を含む。)の44名の健常な非喫煙女性及び男性をこの試験に登録した。試験薬の最初の投与を受ける少なくとも120日前から喫煙せず、病歴、健康診断、心電図(ECG)及び臨床検査評価から臨床的に意味のある知見がないことを基として、対象を選択した。
【0178】
登録した44名の対象のうち、41名の対象がこの試験を完遂した。28名の対象が男性であり、16名の対象が女性であった。平均年齢は51歳、平均体重は171ポンド、平均身長は68インチであった。36名の対象が白人、6名が黒人、2名がヒスパニックであった。詳細なデモグラフィックを下記表18で示す。
【0179】
【表18】
【0180】
a.生物学的同等性の評価
参照(NSP)治療に対して43名の対象が血漿及び尿データを提供した。42名の対象が、ERN−1及びERN−2試験治療に対して血漿及び尿データを提供した。41名の対象が、ERN−3試験治療に対して血漿及び尿データを提供した。平均の表、平均プロット、個々のプロット及び濃度リストに対して公称の時間を使用した。PK分析に対して次の規則を使用した:
1−10時間(両端を含む。)の試料採取時間の場合:5分以上ずれる場合は実際の時間を使用した。5分未満のずれの場合は公称の時間を使用する。
【0181】
10時間を超える試料採取時間の場合:10分以上ずれる場合は実際の時間を使用した。10分未満のずれの場合は公称の時間を使用する。
【0182】
血漿ナイアシン及びNUA薬物動態パラメータに対する要約統計量を表18Aで示す。
【0183】
【表19】
【0184】
ナイアシン及びNUAに対する平均血漿プロファイルを図14及び15で示す。
【0185】
b.血漿データ
血漿ナイアシン
全対象の投与前値がLLQ未満であった。各治療後、投与後4.5から12時間で、全対象のナイアシン濃度が測定可能な濃度であった。
【0186】
平均ナイアシンCmaxは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、5288、4223、5671及び4707ng/mLであった。平均ナイアシンAUC0−lastは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13896、10207、13507及び12315ng*hr/mLであった。ナイアシンに対するTmax中央値は、ERN−1及びERN−3については6.0時間であり、ERN−2及びNSPについては5時間であった。
【0187】
NSPと比較した場合、全ての3種類の試験治療について、自然対数変換されたCmax及びAUC0−lastの比は100%より大きかった。ナイアシンCmaxの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、139%、116%及び166%であった。ナイアシンAUC0−lastの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、137%、112%及び151%であった。自然対数変換したナイアシンCmaxに対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、113から170%、94から142%及び135から203%であった。自然対数変換したナイアシンAUC0−lastに対して、90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、114から164%、94から134%及び126から181%であった。Cmax及びAUC0−lastの両方に対する90%CIは、80−125%の同等範囲外であった。
【0188】
4種類全ての治療についてのCmax及びAUC0−lastに関して、ナイアシンデータは、76から171%の範囲のCVで非常に大きく変動した。
【0189】
血漿ニコチン尿酸
3名の対象の投与前NUA濃度が陽性であった。これらは、対象0028(ピリオド2、ERN−1、濃度4.47ng/mL)、対象30(ピリオド2、ERN−3、濃度2.75ng/mL)及び対象33(ピリオド2、ERN−2、濃度3.26ng/mL)であった。投与前濃度が、対象0028、0030及び0033に対してそれぞれCmaxのわずか約0.24%、0.06%及び0.53%であったので、これらの血漿プロファイルに対して補正を行わなかった。全対象のNUA濃度が各治療後の投与後3から16時間で測定可能な濃度であった。
【0190】
平均NUA Cmaxは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、2822、2616、3058及び2540ng/mLであった。平均NUA AUC0−lastは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13664、12069、13960及び13070ng*hr/mLであった。NUA Tmax中央値は、ERN−1及びERN−3については6.0時間、ERN−2については5.5時間、NSPについては5.0時間であった。
【0191】
各対象及び治療について、可能な場合は最終消失速度を計算した。平均t1/2は、ERN−1、ERN−2及びERN−3についてそれぞれ3.4時間であり、NSPについて3.1時間であった。平均AUCinfは、ERN−1、ERN−2、ERN−3及びNSPについてそれぞれ、13602、11913、14136及び13009ng*hr/mLであった。
【0192】
自然対数変換したCmax及びAUC0−lastの比は、NSPと比較した場合、治療ERN−1及びERN−3については100%より大きかった。ERN−2の場合、Cmax比は100%より大きかったが、一方、AUC0−last比は100%未満であった。NUA Cmaxの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、111%、105%及び123%であった。NUA AUC0−lastの比は、ERN−1、ERN−2及びERN−3についてそれぞれ、104、95%及び109%であった。自然対数変換したNUA Cmaxに対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、101から121%、96から115%及び113から135%であった。自然対数変換したNUA AUC0−lastの場合、90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ、96から111%、88から102%及び102から118%であった。Cmax及びAUC0−lastの両方に対する90%CIは、ERN−1及びERN−2については80−125%の生物学的同等性の範囲内であった。ERN−3については、Cmaxに対する90%CIは80−125%の範囲外であったが、AUC0−lastに対しては80−125%の範囲内であった。
【0193】
NUAデータは、4種類の治療全てについて、Cmax及びAUC0−lastに対してCV約48−58%で非常に大きく変動した。
【0194】
c.ナイアシン及び代謝産物の尿中回収
1の比重を使用して尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究では、962個の試料で測定した平均比重は1.009g/mLであり、962個の試料で測定した最大比重は1.025g/mLであった。)。
【0195】
平均尿回収データを表19で示し、図16で述べる。
【0196】
【表20】
【0197】
総回収
ナイアシン、NUA、MNA及び2PYとしての尿中のナイアシンの総回収は、NSPの場合67.14%、ERN−1、ERN−2及びERN−3の場合それぞれ63.91%、63.44%及び66.16%であった。総回収に対する、loge変換された%Feの最小二乗平均比は、ERN−1、ERN−2及びERN−3についてそれぞれ95%、94%及び98%であった。この比に対する90%CIは、ERN−1、ERN−2及びERN−3についてそれぞれ91から99%、90から98%及び94から102%であり、このことから、3種類の試験製剤によって尿中で排出される総量は、80−125%信頼区間に基づきNSPと同等であることが示された。
【0198】
d.生物同等性評価の結論
NUAデータの薬物動態分析から、Cmaxにより測定されたピーク曝露が、試験製剤NSPと比較した場合、3種類全ての試験製剤(ERN−1、ERN−2、ERN−3)について5から23%高いことが示された。loge変換されたCmaxの場合の最小二乗平均比に対する90%CIは、ERN−1及びERN−2については80−125%の範囲内であり、このことから、これらの製剤がNUAに関してNSPと生物学的に同等であることが示された。ERN−3の場合、90%CIはCmaxに対して80−125%の範囲外であり、このことから、製剤ERN−3がNSPと生物学的に同等でないことが示された。
【0199】
尿中で排出されるナイアシン及び代謝産物の平均総量は、3種類の試験製剤では63から66%であり、NSPの場合は67%であった。排出された分画は親ナイアシンに対して最小であり、続いてNUA、MNA及び2PYであった(37.2−40.0%)。測定した総回収は、NSPと比較した場合、試験製剤では2から6%低かった。loge変換した総回収の最小二乗平均比に対する90%CIは80−125%の範囲内であり、したがって、NSPと比較した場合、3種類の試験製剤は同等であった。
【0200】
したがって、本発明のある実施形態は、4個の500mg NIASPAN(R)錠剤の単回投与を改質1000mg徐放ナイアシン組成物の単回投与と比較する生物学的同等性試験において対象に投与した場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、改質1000mg徐放ナイアシン医薬組成物を含む。
【0201】
好ましい実施形態において、生物学的利用率パラメータは、NUA Cmax(ng/mL)及び総回収又はナイアシンCmax(ng/mL)及びナイアシンAUCである。
【実施例6】
【0202】
この試験の目的は、2000mgの単回投与として投与した場合の、非被覆に対する被覆の本発明1000mg徐放ナイアシン錠剤(本明細書中で以後「改質」錠剤と呼ぶ。)の生物学的同等性(BE)を調べることである。
【0203】
試験計画
この試験は、40歳から70歳(両端を含む。)の44名の健常な非喫煙男性及び女性ボランティア対象における、無作為の、単一施設で行われた、非盲検、単回投与、2期クロスオーバー試験であった。脱落者の置き換えは行わなかった。各対象は、投与間に10日以上のウォッシュアウト期間を設け、2回の個別の日時において、同じ2000mgの単回投与で、2種類のナイアシン製剤、Test及びREFの投与を受けた。Testは、被覆改質1000mg徐放ナイアシンの2個の錠剤から構成され、REFは非被覆改質1000mg徐放ナイアシンの2個の錠剤から構成された。各期間の第1日の22:00前後から開始する低脂肪スナックの後に水240mLとともに各用量を投与した。各治療の6日間の試験期間(第−1から第5日)の間、患者を隔離し、各治療期間中、スポンサーが提供するメニューに従い食事を与えた。他の薬、ビタミン、ハーブ又は栄養剤を試験中禁じた。
【0204】
−30分(投与前)、1、2、3、4、4.5、5、6、7、8、10、12、14、16及び24時間(投与後)の間隔で、投与前30分以内から、投与後24時間まで、連続血液試料を回収した。−24から−18、−18から−12、−12から−6及び−6から0時間(投与前);0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)の間隔で、投与前24時間から投与後96時間まで採尿した。ナイアシン及びNUAについて血漿を分析した。ナイアシン及びその代謝産物:NUA、MNA及び2−PYについて尿を分析した。
【0205】
対象は、各期間中、病院に隔離されているときは、毎日同じ時間に食事を開始した。食事は各期間で同じとなるように維持し、各食事の全量を摂取するよう求めた。朝食、昼食、夕食及び夜食はそれぞれ、07:00、12:00、18:00及び21:45前後に開始した。各対象に対する実際の食事又はスナックの時間は実際の投与時間に対して計画した。対象に対して、第1日に試験薬とともに与える水240mLに加え、第−1日に水を720mL以上飲み、第1日から5日に水を1440mL以上飲むよう求めた。
【0206】
第−1日に、夕食及び夜食を供した。第1日から5日に、朝食、昼食、夕食及び夜食を供した。夜食は、各期間の第1日では、投与直前15分以内に取らせた。ピリオド2の第6日には、全ての治療手順終了後に対象が病院から退院するので、食事は供さなかった。
【0207】
薬物動態の評価
a.血漿回収及び分析
各期間、投与前30分以内から、投与後24時間まで、連続血液試料を回収した(治療あたり15試料)。ヘパリンナトリウムを含有する1本の17−mL真空採血システムに各血液試料を回収し、回収後、氷片及び水浴中で5分以上、試料を冷却した。試料を4℃にておよそ3000rpmで15分間遠心して血漿を分離した。各血漿試料を2つのアリコート、アリコートA及びBに分け、予め冷却し適切にラベルを貼った2本のポリプロピレンチューブに移した。次に、分析まで試料をおよそ−20℃で凍結保存した。
【0208】
有効な液体クロマトグラフィータンデム質量分析(LC/MS/MS)によって、ナイアシン及びNUA濃度を分析した。ナイアシン及びNUA濃度を同じ注入から得た。ナイアシン及びNUAの両方に対する定量の下限(LLQ)は血漿中2ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0209】
b.採尿及び分析
次の間隔で尿を回収した:−24から−18、−18から−12、−12から−6及び−6から0時間(投与前)及び投与後0から6、6から12、12から18、18から24、24から48、48から72及び72から96時間(投与後)(全部で11回の回収)。
【0210】
採尿し、ぴったりと合う蓋を備えたプラスチック容器に移した。回収間隔中、回収した尿を冷蔵庫又は氷−水浴で保存した。対象番号及びイニシャル、回収間隔及びプロトコール番号を識別するために回収容器にラベルを付した。空の容器の重量を測定し、グラムの小数点以下1桁に四捨五入し(例えば100.1g)、これを容器に記入し、実験室のソースドキュメントワークシートに記録した。各間隔の終わりに、容器及び回収した尿の総重量を測定し、グラムの小数点以下1桁に四捨五入し、記録した。尿重量は、容器+尿の総重量から空の容器の重量を差し引くことにより得られた。場合によっては、ある回収間隔中の尿体積が1個の容器の容量を超え、従って採尿を完遂するために第二の容器を必要とした。各採尿間隔の開始及び終了日時も記録した。各回収間隔からの2つのアリコート(各およそ2.5mL)を適切にラベルした2本のポリプロピレンチューブに移した。特定の回収間隔中に複数の容器を必要とした場合、両方の容器からの尿を一緒にして混合し、その後アリコートを取った。分析まで、およそ−20℃で試料を凍結保存した。
【0211】
有効なLC/MS/MSにより、ナイアシン、NUA、NMA及び2−PYの濃度について尿試料を分析した。同じ注入から尿ナイアシン及びNUA濃度を得て、一方、NMA及び2−PY濃度を同じ注入から得た。尿において、LLQ値はナイアシンに対して20ng/mLであり、NUAに対して200ng/mLであった。MNA及び2PYのLLQ値はそれぞれ500ng/mL及び2500ng/mLであった。各分析操作とともに品質管理試料を評価した。
【0212】
c.血漿薬物動態パラメータ及び尿中回収
少なくとも1つの治療に対するPKパラメータを計算するために十分な情報を提供する対象からのデータがPK分析に含まれた。血漿中のナイアシン及びNUAについて、各治療の投与後、各対象に対して次のPKパラメータを計算した:
・Cmax:観察される最大濃度
・Tmax:最大観察濃度の時間
・AUClast:直線的台形規則による、時間0から最後の測定可能(0ではない)濃度までの濃度−時間プロファイル下面積
・AUCinf:時間0から無限大の血漿濃度−時間プロファイル下面積;AUClast及びCt/λzの合計として計算(Ctは、最後に観察された濃度であり、λzは、時間プロットに対する自然対数濃度のプロットから得られた最終消失速度定数である。)
・T1/2:見かけの最終半減期;0.693/λzの比として計算
ナイアシン及びその代謝産物(NUA、MNA及び2−PY)の尿データから、次のパラメータを計算した:
・CumXu:投与後0から96時間の尿から回収した各代謝産物の累積量。
【0213】
・%Fe:投与後96時間での、ベースライン回収及び分子量に対する補正後のナイアシンの用量に対する尿中で排出された各代謝産物の分画
・総%Fe:投与後96時間における4種類の代謝産物の総分画。尿中の各被分析物に対して、%Feは以下のように計算:
【数3】
【0214】
定量限界未満の濃度をゼロとして処理した。各代謝産物濃度に各間隔で回収された尿の体積を掛けることによって、尿中で回収されたナイアシン及びその代謝産物の量を決定した。24時間投与前間隔において回収された量を差し引くことによって、投与後各24時間間隔に対して尿中で回収された総量をベースラインに対して調整した。投与後測定値がベースライン未満となった場合、その量を0とした。ナイアシン及びその代謝産物の分子量は、ナイアシン、NUA、MNA及び2−PYに対してそれぞれ、123.1、180.2、137.1及び153.1であった。4種類の尿被分析物からの%Feの合計を計算し、総%Feとした。
【0215】
WinNonlin Linear Mixed Effects Modeling/bioequivalence、バージョン5.0.1(2005年7月26日)を用いて、(上述のような)生物学的利用率パラメータを計算した。
【0216】
統計分析
WindowsTM用SAS(R)Systemバージョン8.2を用いて、上記で計算された生物学的利用率パラメータの統計分析を行い、データ分析に用いた。
【0217】
治療及び期間によって、血漿薬物動態パラメータ(Cmax、Tmax、T1/2、AUClast及びAUCinf)、それらの自然対数変換値(Tmax及びT1/2を除く。)及び要約統計量(n、平均、std、中央値、min、max、CV%)を計算した。ナイアシン及びNUAの血漿濃度を時間及び治療によってまとめる。
【0218】
ナイアシン及びNUA PK分析の場合、自然対数変換したCmax及びAUClastのデータが正規分布に従い、2つの治療間で独立であると仮定する。固定効果として治療、期間及び順序を、変量効果として順序内の対象を用いて、SAS PROC MIXEDを使用して混合効果によってこのデータをANOVAモデルに適合させた。Cmax及びAUClastのTest/REF比及びそれらの対応する90%信頼区間をこのモデルに基づき評価した。
【0219】
尿からのナイアシン及びその代謝産物の平均回収を計算し、治療により及び間隔によりまとめた。個々の成分のCumXu及び%Fe及び投与後96時間における合計を計算し、治療によりまとめた。
【0220】
血漿PK分析に対して用いたものと同じANOVAモデルに適合させることにより、総%FeのTest/REF平均比に対する90%信頼区間(CI)を計算した。
【0221】
対象のデモグラフィック可変要素(年齢、性別、人種、体重、身長、女性サイズ、肘幅及びBMI)を性別によりまとめた。連続的デモグラフィック可変要素の、平均、標準偏差(SD)、中央値、最小及び最大を計算した。
【0222】
結果
対象の動態を表20でまとめる。プロトコールの包除基準に合致した後に全部で44名の対象を試験に登録した。対象者全てが、試験薬の少なくとも1回の投与を受け、このうち42名がこの試験を完遂した。44名の対象がプロトコールの無作為治療割り当てに従いピリオド1で試験薬の投与を受け;42名の対象がピリオド2で試験薬の投与を受けた。全部で2名の対象が試験から撤退した。試験から撤退した対象の数は、前もって容認するとした10%脱落の範囲内であり、この試験の結果又は結論に影響しないと考えられた。
【0223】
【表21】
【0224】
登録した44名の対象のうち、20名の対象が男性で24名が女性であった。平均年齢は53.1歳;平均体重は161.5ポンド;平均身長は65.6インチ;平均肘幅は2.7インチ;平均BMIは26.3kg/m2であった。女性サイズを小、中及び大に分類した。9名の対象が小の女性サイズであり、20名の対象が中、15名の対象が大の女性サイズであった。対象のうち38名がヒスパニック、4名が白人、2名が黒人であった。表21で詳細なデモグラフィックをまとめる。
【0225】
【表22】
【0226】
a.生物学的同等性の評価
生物学的同等性を調べるために、Test治療で42名の対象から、REF治療で44名の対象から得たデータを分析した。全分析に対し、投与時間に対して実際の時間を使用した。
【0227】
尿の分析のために、1g/mLの比重を使用して、尿重量を体積に変換した。これは、NIASPAN(R)を用いた先行研究に基づくものであった(この先行研究において、962個の試料で測定した平均比重は1.009g/mLであり、962個の試料で測定した最大比重は1.025g/mLであった。)。
【0228】
治療によるナイアシン及びNUAの平均血漿濃度のプロットを図17及び18でそれぞれ示す。平均尿中回収データを図19で示す。
【0229】
b.血漿NUA及び尿中で排出される総量
ナイアシン生物学的同等性を評価するための主要な可変要素は、NUAに対するCmax及びナイアシン及び3種類の代謝産物(NUA、MNA及び2PY)の総尿中回収として定義される。
【0230】
表22は、これら2種類の可変要素に対する平均(SD)及び統計結果を示す。表22はまた、NUA AUClastに対する平均(SD)値及び統計解析も示す。
【0231】
【表23】
【0232】
上記の表で示されるように、主要なBE可変要素である、自然対数変換した、NUA Cmax及びナイアシン及び代謝産物の総回収の、参照に対する試験比に対する90%CIは、80から25%内であった。自然対数変換したNUA AUClastの参照に対する試験比も80から125%内であった。
【0233】
治療によって、各対象について最終消失速度を計算した。Test及びREFに対してそれぞれ、平均NUA T1/2は3.16及び3.04時間、平均NUA Tmaxは4.90及び4.80時間、平均NUA AUCinfは、10914.7及び11770.6ng*hr/mLであった。
【0234】
c.血漿ナイアシン
統計解析付きの血漿ナイアシンに対する平均PKパラメータを表23で示す。自然対数変換したナイアシンCmax及びAUClastの参照に対する試験比は100%未満であった。自然対数変換したナイアシンCmax及びAUClastの比に対する90%CIは、変動が大きかったため、80から125%の間隔外であった。
【0235】
【表24】
【0236】
Test及びREFに対してそれぞれ、ナイアシンの平均T1/2は4.73及び2.94時間であり、平均Tmaxは4.68及び4.64時間であり、平均AUCinfは11553.1及び16134.3ng*hr/mLであった。
【0237】
d.個々の被分析物の尿中回収
個々の被分析物の平均尿中回収を表24で与える。
【0238】
【表25】
【0239】
平均尿中回収は2PYに対して最大であり、続いてMNA、NUA及びナイアシンであった。
【0240】
e.生物学的同等性評価の結論
NUA Cmaxの平均Test/REF比及びナイアシン及びその代謝産物の尿中回収(総%Fe)に対する90%CTに基づき、生物学的同等性を評価した。自然対数変換したナイアシン吸収の速度(NUA Cmax)及び程度(尿中総%Fe)のTest/REFの平均比に対する90%CIは80から125%の求められるBE範囲内であり、このことから、Test及びREF製剤が生物学的に同等であることが示される。NUA AUClastに対する90%CIも80から125%の範囲内であり、BEの結論を支持する。
【0241】
ナイアシンCmax及びAUClastについて、ナイアシンCmax及びAUClastの両方に対するTest/REFの平均比の90%CIの上限は生物学的同等性範囲内であり、下限は、生物学的同等性範囲の下限である80%に両方とも非常に近かった。
【実施例7】
【0242】
実施例4、5及び6で分析した本発明の1000mg製剤について、NUAに対するCmax(ng/mL)、総尿中回収(%)、ナイアシンCmax(ng/mL)及びナイアシンAUCに対する平均を下記表25で示す(ERN−3を除く。)。
【0243】
【表26】
【0244】
したがって、本発明のある実施形態は、それを必要とする患者に2個の1000mg錠剤の単回投与として投与した場合、次の生物学的利用率パラメータの少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%内となるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物を含む:
(a)2601.8ng/mLのNUA Cmax;
(b)60.5%の尿中ナイアシン総回収;
(c)4958.9ng/mLのナイアシンCmax;及び
(d)12414.5ng/mLのナイアシンAUC。
【0245】
下記表25aは、標準誤差(括弧で示す。)を考慮に入れた、表25から選択された生物学的利用率パラメータの上限及び下限を示す。特に、上記表25で割り出された各パラメータに対して上記実施例4、5及び6からの最低平均を割り出し、次いで2つの標準誤差をその平均から差し引いて下限を出すことによって、下限を計算した。標準偏差を試料サイズの平方根で割ることによって(例えば、1430/V44=326)、標準誤差を計算した。同様に、上限は、各パラメータ+2つの標準誤差に対する実施例4、5及び6からの最大平均を表す。
【0246】
【表27】
【0247】
したがって、本発明のさらなる実施形態は、それを必要とする患者に2個の1000mg錠剤の単回投与として投与した場合、次の生物学的利用率パラメータの少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%間隔内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物を含む:
(a)約2111.0ng/mLから約3253ng/mLのNUA Cmax;
(b)約49.24%から約70.23%の尿中ナイアシンの総回収;
(c)約3096ng/mLから約6750ng/mLのナイアシンCmax;及び
(d)約6723ng/mLから約18643ng/mLのナイアシンAUC。
【実施例8】
【0248】
アスピリンで前治療されるか又はアスピリンと同時投与される場合の、徐放ナイアシンの2000mg用量により誘発される紅潮の相対的発生率
この試験は、単一施設で行われ、本発明の徐放ナイアシン錠剤の経口投与の結果として起こる紅潮反応におけるアスピリン前治療及びアスピリン同時投与の影響を調べるために計画された、無作為二重盲検、ダブルダミー、単回投与、3期クロスオーバー試験である。この試験計画及び治療を図20で示す。対象にはまた、試験中如何なる時点でも、非研究関連アスピリン又はその他のNSAIDを用いることもまた禁止した。この試験は、病院のInstitutional Review Boardから承認を得、各対象は参加前に書面でインフォームドコンセントを提出した。
【0249】
この試験には、ボディーマスインデックス(BMI)が22から31kg/m2である、19歳から70歳の健常成人男性が含まれた。ナイアシン誘発性の紅潮事象と更年期の紅潮との混同を避けるため、この試験から女性を除外した。人間ドック、病歴、心電図及びスクリーニングのための来院時及び最初の試験期間のための入院時に行った臨床検査の結果によって、対象が健常であることを確認した。試験に入る前の4ヶ月以内に煙草又はニコチン製品を使用した場合、ナイアシン、アスピリンもしくは関連誘導体に対するアレルギー又は過敏性がある場合;過去3年間で薬物乱用又は依存症があった場合;又は偏頭痛、糖尿病、胆嚢疾患、肝臓疾患、重度の高血圧もしくは低血圧、心臓の異常、腎臓疾患もしくは薬物誘発性ミオパシーの病歴がある場合は対象を除外した。対象に対して、試験に入る前の21日以内及び試験中、処方薬を禁じ、試験に入る前の10日以内及び試験中、市販薬、ビタミン又はハーブ調合剤を禁じた。
【0250】
ピリオド1のための入院前21日以内でスクリーニング手順を完了した。3回の試験期間それぞれに対して、対象をおよそ24時間隔離し、少なくとも7日間あけて治療薬を投与し、対象は、ナイアシン及び脂肪含量が調節された特別メニューに従う食事を取った。食事構成及び開始時間は各試験期間で同じであった。
【0251】
試験治療
無作為化スケジュールに従い、クロスオーバー方式で、試験薬を経口投与した。アスピリン(ASA)及びプラセボ投与は各試験期間で異なったが、本発明の被覆徐放ナイアシン錠剤(本明細書中で「改質ナイアシンER錠剤」又は「rNER」とも呼ばれる。)(2個の1000mg錠剤)の投与は同じであった。ある期間において、対象は、2個のプラセボ錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のアスピリン325mg錠剤の投与を受けた(「ASA前治療」)。別の期間において、対象は、2個のアスピリン325mg錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のプラセボ錠剤の投与を受けた(「同時ASA」)。第三の期間において、対象は、2個のプラセボ錠剤と改質ナイアシンER2000mgを同時投与する30分前に、2個のプラセボ錠剤からなる対照治療の投与を受けた(「R−ナイアシンER単独」)。
【0252】
紅潮事象を評価することが目的であるので、いくつかの方法によって試験者及び対象が投与薬物を識別できないようにした。各投与期間において、対象は、各投与に対して同じ数の錠剤の投与を受けた(図21参照)。プラセボ及びアスピリン錠剤の外観は同様であり、これらは識別されず、一方、それとともに、不透明な投与カップから試験薬を投与し、試験薬投与の間、対象に目隠しをした。アスピリンなしでの紅潮反応を評価するために、対照治療、R−ナイアシンER単独、がこの試験に含まれた。試験スポンサー及び各期間について投与薬を調製した試験施設の人員のみが、試験中の治療の無作為割り当てについて知っていた。治験医師、試験施設の人員及び試験モニターは治療割り当てスキームを知らず、治療薬調製又は投与に関与する試験施設の何れの者も、紅潮事象又は治療中に発生した有害事象を回収又は評価することを禁じた。
【0253】
各対象は、改質ナイアシンER投与前に、前治療薬及びスナックを摂取した。対象は、21:30前後に水180mLとともに、割り当てられた前治療薬(アスピリン又はプラセボ)の経口投与を受け、次いで、21:45前後に低脂肪スナックを食べ始めた。水240mLとともに22:00前後に対象が割り当てられた治療薬の残りの投与を対象が受ける前に、このスナックを全て摂取させた。各薬の投与には複数の錠剤が必要であり、錠剤を一度に一緒に摂取するか又は1個を飲んだ後すぐに続いて別のものを摂取するというように、これを1分以内に摂取させた。錠剤を飲み込むために必要である場合、120mL単位でさらなる水を与え;錠剤を咀嚼するか又は噛むことは禁じた。用量全てを摂取したことを確認するために、試験用量の投与後、各対象の口腔を調べた。
【0254】
紅潮評価
対象が次の紅潮症状の1以上:発赤、熱感、刺痛及び掻痒(これらの症状は個々に又は同時に起こり得た。)を報告することとして紅潮事象を定義した。各試験期間中、改質ナイアシンER投与から8時間後まで、1時間間隔で紅潮症状の有無を評価するよう、対象に促した。電子日誌において各紅潮症状についての開始時間、停止時間及び強度を記録するよう、対象に促した。
【0255】
各対象は、連続及び分類別評価基準の両方によって、各対象が感じる症状強度をランク付けした。対象は、コンピュータの水平視覚的アナログスケール(VAS)(「なし」(0)を左端に「容認できない。」(100)を右端にした。)上に垂直線で強度に対して印を付け、軽度、中度又は重度としても症状をランク付けした。容易に耐えられるか又は活動を制限しなかった症状を軽度と定義し;活動を行う上で困難を生じる症状を重度とした。
【0256】
各対象は、ナイアシンER投与後に起こる1以上の平行する紅潮症状の最初として定義される最初の全体的紅潮事象を同様にランク付けした。最初の症状の開始時間はまた、最初の全体的紅潮事象の開始時間でもあり;その事象の最後の症状が消散し、さらなる紅潮症状が起こることなく少なくとも30分間経過した場合、事象全体の終了とした。
【0257】
統計分析
少なくとも144名の対象が3つの治療全てを完遂することを確実にするために、全部で164名の対象を登録するよう計画した。早期に撤退した対象を入れ替えなかった。
【0258】
アルファ(α)=0.05の両側として全ての比較を行った。主要評価項目は、試験中に少なくとも1回紅潮事象が起こった対象の人数であった。マクネマー検定を用いて、「ASA前治療」及び対象治療、「R−ナイアシンER単独」の間で、紅潮発生率を比較した。この試験には、比較に含まれるべき両治療後に対象が反応(この場合、紅潮)することが必要である。「同時ASA」と「R−ナイアシンER単独」治療との間及び「ASA前治療」と「同時ASA」治療との間で、紅潮発生率の比較を同様に行った。
【0259】
副次的評価項目には、紅潮事象数及び最初の全体的紅潮事象ならびに個々の紅潮症状のの、強度、開始時間及び持続時間が含まれた。頻度数により事象の数をまとめ、マクネマー検定を用いて比較した。VAS線の左端からの距離として対象の垂直マークを表すことにより(100mmに規格化)、図式データから数字データにVAS強度評価を変換した。平均値に対して対応のあるt検定及び中央値に対してWilcoxon符号付き検定を用いて治療間でVASにより測定された強度及び持続時間を比較し、一方、Bowkerの対称性の検定(対象が比較において両治療に対するデータを有することも必要とするマクネマー検定の一般化)を用いて、分類別スケールにより測定された強度を比較した。主要評価項目と同じ治療のペアについて、副次的評価項目に対する治療間の比較を行った。
【0260】
Medical Dictionary for Regulatory Activities(MedDRA、バージョン7.0)を用いて有害事象(紅潮を除く。)を符号化した。有害事象は治療間で比較しなかった。
【0261】
結果
平均年齢29歳で平均BMIが26.5kg/m2の全部で164名の男性を登録し、少なくとも1回の試験薬投与を受けさせた。対象デモグラフィックを表26でまとめる。164名の対象のうち、148名(90%)が3回全ての治療を受け、紅潮反応について評価可能であった。16名の対象(10%)が早期に撤退し:4名(2%)が同意を取り消し、4名(2%)がフォローアップできず、1名(1%)で有害事象が起こり、3名(2%)がプロトコール違反し、3名(2%)が薬物スクリーニング検査で陽性となり、1名(1%)が投与ミスにより撤退した。
【0262】
【表28】
【0263】
紅潮
3回全ての治療を受けた148名の対象において、「同時ASA」(61%、p0.001)又は「ASA前治療」(53%、p0.001;表27)の後よりも、「R−ナイアシンER単独」(77%)の後に紅潮発生率が有意に高かった。
【0264】
【表29】
【0265】
両アスピリン含有治療は紅潮発生率において互いに有意な差はなかった。図21で示されるように、最初の全体的紅潮事象における個々の症状(発赤、熱感、刺痛及び掻痒)の発生率は、「R−ナイアシンER単独」と比較して、「ASA前治療」後では30%から50%低かった。4種類全ての症状を報告した対象数が最低であったのは「ASA前治療」後であり、対象数が最大であったのは「R−ナイアシンER単独」後であった。治療間で個々の症状は比較しなかった。紅潮事象の数は紅潮発生率と同じ傾向であり、「R−ナイアシンER単独」の後に報告した人数が最多であり、「ASA前治療」後に報告した人数が最少であった(データは示さない。)。
【0266】
「ASA前治療」及び「R−ナイアシンER単独」治療両方の後に紅潮した対象において(下記表28)、「ASA前治療」では、分類別評価又はVASの何れかにより測定された最初の全体的紅潮事象の強度が有意に低下していた(各p<0.001)。
【0267】
【表30】
【0268】
両治療で、紅潮事象の殆どが軽度にランク付けされ、1例のみが(「R−ナイアシンER単独」の後に)重度であった。軽度にランク付けされた事象があった対象の人数は、「R−ナイアシンER単独」と比較して、「ASA前治療」後では36%多く;同様に、中度又は重度にランク付けされた紅潮があった対象の人数は62%少なかった。VASのランク付けは、「R−ナイアシンER単独」の後よりも、「ASA前治療」の後で30%を超えて低かった。最初の全体的紅潮事象の持続時間について、平均及び中央値データは一貫しなかったが、このことから、非正規分布が示唆される。「ASA前治療」の場合の持続時間中央値は、「R−ナイアシンER単独」の場合よりも43%短かった(p=0.008)。個々の症状について、発赤、熱感及び刺痛は、「ASA前治療」後有意に強度が低く(p≦0.025、データは示さない。);何れの症状の持続時間についても治療間で有意差はなかった。
【0269】
「同時ASA」及び「R−ナイアシンER単独」両治療後に紅潮があった対象において(下記表29)、最初の全体的紅潮事象の強度は、分類別データについては有意差があったが(p=0.028)、VASデータについては有意差はなかった。
【0270】
【表31】
【0271】
ここで、「同時ASA」後に軽度紅潮事象のあった対象数は、「R−ナイアシンER単独」の後よりも22%高く、中度又は重度事象は38%少なかった。最初の全体的紅潮事象の持続時間の差は有意ではなかった。個々の症状について、発赤及び熱感の強度は、「同時ASA」治療後、有意に低く(p≦0.024、データは示さない。);何れの症状の持続時間も有意差はなかった。
【0272】
「ASA前治療」及び「同時ASA」両治療後に紅潮があった対象において(下記表30参照)、最初の全体的紅潮事象の強度の差は、分類別の尺度では有意ではなかったが、「ASA前治療」に対するVASスコアは20%低く、統計学的に有意であった。
【0273】
【表32】
【0274】
最初の全体的紅潮事象の持続時間は、これらの治療間で有意差はなかった。個々の症状について、強度及び持続時間のどちらもこの2つの治療間で有意差がなかった。
【0275】
上記の結果から、本発明の徐放錠剤の30分前に摂取されるアスピリンの650mg(2x325mg錠剤)によって、本発明の錠剤単独の使用と比較して、対象が報告する紅潮の発生率、強度及び持続時間が有意に低下した。アスピリン650mg及び本発明の錠剤の同時投与により、それほどではないにせよ、紅潮発生率、強度及び持続時間が低下した。
【0276】
実施例3及び実施例8からの紅潮発生率及び強度の結果をまとめ、図22及び23で一緒に示す。これらの図面から、本発明の徐放医薬組成物が、元の1000mg錠剤(Nisapan(R))と比較して、紅潮発生率の低下は小さいが、紅潮強度及び持続時間(〜40%)を低下させることが示される(実施例3参照)。実施例8から、アスピリンを本発明の徐放医薬組成物の30分前又はこれと同時に摂取することによって紅潮の発生率が低下し、さらに、紅潮強度及び持続時間が低下することが分かる。実施例3において、患者のほぼ全員(98%)が、元の1000mg錠剤の単回の2000mg投与で(2錠剤投与)紅潮(発生)を報告した。実施例8において、本発明の徐放錠剤の単回2000mg投与(2錠剤投与)+アスピリンで紅潮したのは対象の50−60%のみであった。先行試験における元の1000mg錠剤での強度中央値はVASで54mmであった。今回の試験において、本発明の徐放錠剤+アスピリンでは、強度中央値がわずか19−23mmであり、大多数(約80%以上)は紅潮が「軽度」であると報告した。
【0277】
本発明の具体的な実施形態を参照して本発明を上記で説明してきたが、本明細書中で開示される本発明の概念から逸脱することなく、多くの変化、修飾及び変更がなされ得ることは明らかである。したがって、添付の特許請求の範囲の精神及び幅広い範囲内に入る全てのかかる変化、修飾及び変更を包含するものとする。本明細書中で引用される全ての特許出願、特許及びその他の刊行物は、その全体が参照により本明細書中に組み込まれる。
【図面の簡単な説明】
【0278】
【図1】図1は、METHOCEL(R)K−15M Premiumの様々なレベルを含有する1000mgナイアシン徐放錠剤からの平均ナイアシン溶出を示すグラフである。
【図2】図2は、1000mgナイアシン徐放錠剤(1240mg総重量)からのナイアシン溶出における、METHOCEL(R)K−15MPCRの粘度の変化の影響を示すグラフである。
【図3】図3は、バルク及び40メッシュPVP K−90を用いて製造された1000mgナイアシン徐放錠剤からのナイアシン溶出プロファイルを示すグラフである。
【図4】図4は、様々な混合段階を用いて製造された1000mgナイアシン徐放錠剤(1240mg総重量)からのナイアシン溶出プロファイルを示すグラフである。
【図5】図5は、直接圧縮製造プロセスを示す流れ図である。
【図6】図6は、実施例3に記載の臨床試験の流れ図である。
【図7】図7は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mgNIASPAN(R)錠剤(参照)投与後の紅潮の発生率を示す棒グラフである。
【図8】図8は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mgNIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象の中央値強度を示す棒グラフである。
【図9】図9は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mg NIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象の中央値持続時間を示す棒グラフである。
【図10】図10は、本発明の2個のフィルム被覆1000mg徐放ナイアシン製剤(Test)及び2個の非被覆1000mg NIASPAN(R)錠剤(参照)投与後の、最初の紅潮事象における個々の紅潮症状の発生率を示す棒グラフである。
【図11】図11は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の1000mg NIASPAN(R)錠剤(「Ref」)投与後の、ナイアシンの平均血漿濃度を示すグラフである。
【図12】図12は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の1000mg NIASPAN(R)錠剤(「Ref」)投与後の、NUAの平均血漿濃度を示すグラフである。
【図13】図13は、本発明の2個の1000mg徐放製剤(「Test」又は「改質」)及び2個の非被覆1000mg NIASPAN(R)錠剤(「Ref」)の投与から96時間後の、ナイアシン及びその代謝産物(ナイアシン用量の%として)の平均尿中回収を示す棒グラフである。
【図14】図14aは、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する線形平均血漿ナイアシンプロファイル示すグラフであり;14bは、3種類の試験及び1種類の参照製剤に対する、片対数平均血漿ナイアシンプロファイルを示すグラフである。
【図15】図15aは、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する、線形平均血漿NUAプロファイルを示すグラフであり;15bは、3種類の試験及び1種類の参照製剤に対する、片対数平均血漿ナイアシンプロファイルを示すグラフである。
【図16】図16は、3種類の試験徐放ナイアシン製剤(ERN−1、ERN−2、ERN−3)及び参照徐放ナイアシン製剤(NSP)に対する、ナイアシン及びその代謝産物(ナイアシン用量の%として)の平均尿中回収を示す棒グラフである。
【図17】図17aは、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)に対する、線形平均血漿ナイアシンプロファイルを示すグラフであり;17bは、試験及び参照製剤に対する、対数変換平均血漿ナイアシンプロファイルを示すグラフである。
【図18】図18aは、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)に対する、線形平均血漿NUAプロファイルを示すグラフであり;18bは、試験及び参照製剤に対する、対数変換平均血漿NUAプロファイルを示すグラフである。
【図19】図19は、本発明の2個の被覆1000mg徐放ナイアシン製剤(Test)及び本発明の2個の非被覆1000mg徐放ナイアシン製剤(Ref)の投与から96時間後の、ナイアシン及びその代謝産物の平均尿中回収を示す棒グラフである。
【図20】図20は、実施例6の試験計画を示す流れ図である。
【図21】図21は、(1)対象をアスピリン(ASA)で予め処置した場合、(2)ナイアシン製剤とともにASAを投与した場合及び(3)ナイアシン製剤のみを投与した場合の、本発明の2個の1000mg徐放ナイアシン製剤(「NIASPAN(R)CF」)の投与後の、最初の紅潮事象に対する個々の紅潮症状の発生率を示す棒グラフである。
【図22】図22は、実施例3及び実施例8の両方に対する、紅潮事象の発生率を示す棒グラフである。
【図23】図23は、実施例3及び実施例8の両方に対する、紅潮事象の強度を示す棒グラフである。
【特許請求の範囲】
【請求項1】
(a)ナイアシン約78%から約82%w/w、
(b)約1.39から約1.41のメトキシル置換度及び約0.20から約0.22のヒドロキシプロポキシルモル置換度を有するヒドロキシプロピルメチルセルロース約14%から約18%、
(c)約2.5%から約3.0%w/wのポリビニルピロリドン、及び
(d)約0.95%から約1.05%w/wのステアリン酸、
を含む、1000mgナイアシン医薬組成物。
【請求項2】
(a)ナイアシン約70%から約92%w/w、
(b)放出遅延剤約7%から約25%w/w、
(c)結合剤約0.1%から約4.3%w/w、及び
(d)潤滑剤約0.5%から約1.5%w/w、
を含む医薬組成物であり、患者への投与後、NIASPAN(R)錠剤の同等用量の投与と比較して、紅潮を軽減させる結果をもたらす、医薬組成物。
【請求項3】
組成物が1000mg徐放ナイアシン錠剤製剤である、請求項2の医薬組成物。
【請求項4】
請求項3の医薬組成物であり、患者により1日1回前記組成物が摂取される場合、前記患者への投与後、このような治療を中断する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項5】
前記患者への投与が、夕方又は夜間に1日1回である、請求項4の医薬組成物。
【請求項6】
放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)及びキサンタンガム及びそれらの混合物からなる群から選択される、請求項2の医薬組成物。
【請求項7】
放出遅延剤がヒドロキシプロピルメチルセルロースである、請求項6の医薬組成物。
【請求項8】
ヒドロキシプロピルメチルセルロースが、約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項7の医薬組成物。
【請求項9】
ヒドロキシプロピルメチルセルロースが、約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度を有する、請求項8の医薬組成物。
【請求項10】
ヒドロキシプロピルメチルセルロースが、約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する、請求項8の医薬組成物。
【請求項11】
ヒドロキシプロピルメチルセルロースが約11,000から約22,000mPasの粘度を有する、請求項8の医薬組成物。
【請求項12】
ヒドロキシプロピルメチルセルロースが約13,000から約18,000mPasの粘度を有する、請求項11の医薬組成物。
【請求項13】
コーティングをさらに含む、請求項2の医薬組成物。
【請求項14】
前記コーティングが、約1.5から約8.0%の重量増加となる色彩塗料である、請求項13の医薬組成物。
【請求項15】
前記コーティングが、錠剤の重量を約1.75から約5.0%増加させるために塗布される色彩塗料である、請求項14の医薬組成物。
【請求項16】
前記結合剤が、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート及びワックス又はそれらの混合物からなる群から選択される、請求項7の医薬組成物。
【請求項17】
前記結合剤がポリビニルピロリドンである、請求項16の医薬組成物。
【請求項18】
前記潤滑剤が、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及び硬化植物油及びそれらの混合物からなる群から選択される、請求項7の医薬組成物。
【請求項19】
前記潤滑剤がステアリン酸である、請求項18の医薬組成物。
【請求項20】
(a)ナイアシン約76%から約88%w/w、
(b)放出遅延剤約11.0%から約20.0%w/w、
(c)結合剤約0.2%から約3.25%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、請求項2の医薬組成物。
【請求項21】
(a)ナイアシン約78%から約82%w/w、
(b)放出遅延剤約14%から約18%w/w、
(c)結合剤約2.5%から約3.0%w/w、及び
(d)潤滑剤約0.85%から約1.05%w/w、
を含む、請求項20の医薬組成物。
【請求項22】
潤滑剤約0.95%から約1.05%w/wを含む、請求項21の医薬組成物。
【請求項23】
ナイアシン処置療法に付随する紅潮を軽減する方法であり、
(a)ナイアシン約70%から約92%w/w、
(b)放出遅延剤約7%から約25%w/w、
(c)結合剤約0.1%から約4.3%w/w、及び
(d)潤滑剤約0.5%から約1.5%w/w、
を含む医薬剤形を1日1回投与することを含む、前記方法。
【請求項24】
前記1日1回剤形が、2個の1000mg錠剤を含む、請求項23の方法。
【請求項25】
前記錠剤が、
(a)ナイアシン約76%から約88%w/w、
(b)放出遅延剤約11.0%から約20.0%w/w、
(c)結合剤約0.2%から約3.25%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/wと、
を含む1000mg錠剤である、請求項23の方法。
【請求項26】
前記1000mg錠剤が、
(a)ナイアシン約78%から約82%w/w、
(b)放出遅延剤約14%から約18%w/w、
(c)結合剤約2.5%から約3.0%w/w、及び
(d)潤滑剤約0.85%から約1.05%w/w、
を含む、請求項25の方法。
【請求項27】
前記1000mg錠剤が、潤滑剤約0.95%から約1.05%w/wを含む、請求項26の方法。
【請求項28】
放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)、トリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))及びキサンタンガム及びそれらの混合物からなる群から選択される、請求項23の方法。
【請求項29】
放出遅延剤がヒドロキシプロピルメチルセルロースであり、並びにヒドロキシプロピルメチルセルロースが約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項28の方法。
【請求項30】
ヒドロキシプロピルメチルセルロースが約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度を有する、請求項29の方法。
【請求項31】
ヒドロキシプロピルメチルセルロースが、約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する、請求項30の方法。
【請求項32】
ヒドロキシプロピルメチルセルロースが約11,000から約22,000mPasの粘度を有する、請求項29の方法。
【請求項33】
ヒドロキシプロピルメチルセルロースが約13,000から約18,000mPasの粘度を有する、請求項32の方法。
【請求項34】
医薬剤形がコーティングをさらに含む、請求項23の方法。
【請求項35】
前記コーティングが、医薬剤形の重量を約1.5から約8.0%増加させるために塗布される色彩塗料である、請求項34の医薬組成物。
【請求項36】
前記コーティングが、医薬剤形の重量を約1.75から約5.0%増加させるために適用される色彩塗料である、請求項35の医薬組成物。
【請求項37】
直接圧縮ナイアシン錠剤を調製する方法であり
(a)ナイアシン約70%から約92%w/w、放出遅延剤約7%から約25%w/w、結合剤約0.1%から約4.3%w/w、及び潤滑剤約0.5%から約1.5%w/wの混合物を混合する段階、
(b)段階(a)の混合物を錠剤へと圧縮する段階
を含む、方法。
【請求項38】
前記ナイアシン錠剤が1000mgナイアシン投与製剤である、請求項37の方法。
【請求項39】
錠剤を被覆することをさらに含む、請求項37の方法。
【請求項40】
錠剤の重量を約1.5から約8.0%増加させるために色彩塗料で錠剤を被覆することを含む、請求項37の方法。
【請求項41】
前記色彩塗料が約1.75から5.0%重量増加となる、請求項40の方法。
【請求項42】
前記放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)、トリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))及びキサンタンガム又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項43】
前記結合剤が、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート及びワックス又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項44】
前記潤滑剤が、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及び硬化植物油又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項45】
前記錠剤が、ナイアシン約76%から約88%w/w、放出遅延剤約11.0%から約20%w/w、結合剤約0.2%から約3.25%w/w、及び潤滑剤約0.75%から約1.25%w/wを含む、請求項40の方法。
【請求項46】
前記錠剤が、ナイアシン約78%から約82%w/w、放出遅延剤約14%から約18%w/w、結合剤約2.5%から約3.0%w/w、及び潤滑剤約0.95%から約1.05%w/wと、を含む、請求項45の方法。
【請求項47】
前記放出遅延剤がヒドロキシプロピルメチルセルロースであり、前記結合剤がポリビニルピロリドンであり、前記潤滑剤がステアリン酸であり、並びにヒドロキシプロピルメチルセルロースが約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項37の方法。
【請求項48】
(a)ナイアシン約65%から約85%w/w、
(b)放出遅延剤約20%から約32%w/w、
(c)結合剤約2%から約3%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項49】
(a)ナイアシン約68%から約75%w/w、
(b)放出遅延剤約24%から約29%w/w、
(c)結合剤約2.25%から約2.75%w/w、及び
(d)潤滑剤約0.95%から約1.05%w/w、
を含む、請求項48に記載の直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項50】
コーティングをさらに含み、該コーティングが約1.5から約8.0%重量増加となる、請求項48に記載の直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項51】
(a)ナイアシン約74%から約80%w/w、
(b)放出遅延剤約16%から約22%w/w、
(c)結合剤約2.5%から約2.75%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項52】
(a)ナイアシン約76%から約79%w/w、
(b)放出遅延剤約18%から約21%w/w、
(c)結合剤約2.5%から約2.7%w/w、及び
(d)潤滑剤約0.95%から約1.05%w/w、
を含む、請求項51に記載の直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項53】
コーティングをさらに含み、該コーティングが約1.5から約8.0%の重量増加となる、請求項52に記載の直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項54】
抗高脂血剤をさらに含む、請求項2の医薬組成物。
【請求項55】
抗高脂血剤がHMG−CoA還元酵素阻害剤である、請求項54の医薬組成物。
【請求項56】
紅潮抑制剤をさらに含む、請求項55の医薬組成物。
【請求項57】
紅潮抑制剤をさらに含む、請求項2の医薬組成物。
【請求項58】
紅潮抑制剤が非ステロイド抗炎症薬(NSAID)である、請求項57の医薬組成物。
【請求項59】
紅潮抑制剤がアスピリン(ASA)である、請求項58の医薬組成物。
【請求項60】
紅潮抑制剤がプロスタグランジンD2受容体アンタゴニストである、請求項57の医薬組成物。
【請求項61】
プロスタグランジンD2受容体アンタゴニストがMK−0524である、請求項60の医薬組成物。
【請求項62】
ナイアシンが粒状ナイアシンである、請求項23、37、48又は51の何れか一項の方法。
【請求項63】
粒状ナイアシン粒子サイズが、ふるい分画100−425μmに対してNLT85%(w/w)であり、及びダスト<100μmに対してNMT10%(w/w)である、請求項62の方法。
【請求項64】
1000mg徐放ナイアシン医薬組成物であり、これを必要とする患者に2個の1000mg錠剤を単回投与として投与する場合、次の生物学的利用率パラメータ
(a)2601.8ng/mLのNUA Cmax;
(b)60.5%の尿中ナイアシン総回収;
(c)4958.9ng/mLのナイアシンCmax;及び
(d)12414.5ng/mLのナイアシンAUC
の少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物。
【請求項65】
自然対数変換比が90%から115%内である、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項66】
自然対数変換比が95%から110%内である、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項67】
紅潮抑制剤及び抗高脂血剤からなる群から選択される少なくとも1つのさらなる治療剤をさらに含む、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項68】
請求項77の1000mg徐放ナイアシン医薬組成物であり、前記組成物が前記患者に1日1回摂取される場合、このような治療を中止する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項69】
1000mg徐放ナイアシン医薬組成物であり、4個の500mg NIASPAN(R)錠剤の単回投与を前記1000mg徐放ナイアシン組成物2個の単回投与と比較する生物学的同等性試験において対象に投与される場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、医薬組成物。
【請求項70】
前記生物学的利用率パラメータが、NUA Cmax(ng/mL)及び総回収又はナイアシンCmax(ng/mL)及びナイアシンAUCである、請求項69に記載の1000mg徐放ナイアシン医薬組成物。
【請求項71】
1000mg徐放ナイアシン医薬組成物であり、これを必要とする患者に2個の1000mg錠剤の単回投与として投与される場合、次の生物学的利用率パラメータ
(a)約2111.0ng/mLから約3253ng/mL NUA Cmax;
(b)約49.24%から約70.23%の尿中ナイアシンの総回収;
(c)約3096ng/mLから約6750ng/mLのナイアシンCmax;及び
(d)約6723ng/mLから約18643ng/mLのナイアシンAUC
の少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%間隔内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物。
【請求項72】
請求項71の1000mg徐放ナイアシン医薬組成物であり、前記組成物が前記患者に1日1回摂取される場合、治療を中止する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項73】
ナイアシン放出が遅延される、請求項3の医薬組成物。
【請求項74】
即放型紅潮抑制剤をさらに含む、請求項73の医薬組成物。
【請求項75】
紅潮抑制剤がプロスタグランジンD2受容体である、請求項74の医薬組成物。
【請求項76】
プロスタグランジンD2受容体がMK−0524である、請求項75の医薬組成物。
【請求項77】
紅潮抑制剤が非ステロイド抗炎症薬(NSAID)である、請求項74の医薬組成物。
【請求項1】
(a)ナイアシン約78%から約82%w/w、
(b)約1.39から約1.41のメトキシル置換度及び約0.20から約0.22のヒドロキシプロポキシルモル置換度を有するヒドロキシプロピルメチルセルロース約14%から約18%、
(c)約2.5%から約3.0%w/wのポリビニルピロリドン、及び
(d)約0.95%から約1.05%w/wのステアリン酸、
を含む、1000mgナイアシン医薬組成物。
【請求項2】
(a)ナイアシン約70%から約92%w/w、
(b)放出遅延剤約7%から約25%w/w、
(c)結合剤約0.1%から約4.3%w/w、及び
(d)潤滑剤約0.5%から約1.5%w/w、
を含む医薬組成物であり、患者への投与後、NIASPAN(R)錠剤の同等用量の投与と比較して、紅潮を軽減させる結果をもたらす、医薬組成物。
【請求項3】
組成物が1000mg徐放ナイアシン錠剤製剤である、請求項2の医薬組成物。
【請求項4】
請求項3の医薬組成物であり、患者により1日1回前記組成物が摂取される場合、前記患者への投与後、このような治療を中断する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項5】
前記患者への投与が、夕方又は夜間に1日1回である、請求項4の医薬組成物。
【請求項6】
放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)及びキサンタンガム及びそれらの混合物からなる群から選択される、請求項2の医薬組成物。
【請求項7】
放出遅延剤がヒドロキシプロピルメチルセルロースである、請求項6の医薬組成物。
【請求項8】
ヒドロキシプロピルメチルセルロースが、約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項7の医薬組成物。
【請求項9】
ヒドロキシプロピルメチルセルロースが、約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度を有する、請求項8の医薬組成物。
【請求項10】
ヒドロキシプロピルメチルセルロースが、約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する、請求項8の医薬組成物。
【請求項11】
ヒドロキシプロピルメチルセルロースが約11,000から約22,000mPasの粘度を有する、請求項8の医薬組成物。
【請求項12】
ヒドロキシプロピルメチルセルロースが約13,000から約18,000mPasの粘度を有する、請求項11の医薬組成物。
【請求項13】
コーティングをさらに含む、請求項2の医薬組成物。
【請求項14】
前記コーティングが、約1.5から約8.0%の重量増加となる色彩塗料である、請求項13の医薬組成物。
【請求項15】
前記コーティングが、錠剤の重量を約1.75から約5.0%増加させるために塗布される色彩塗料である、請求項14の医薬組成物。
【請求項16】
前記結合剤が、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート及びワックス又はそれらの混合物からなる群から選択される、請求項7の医薬組成物。
【請求項17】
前記結合剤がポリビニルピロリドンである、請求項16の医薬組成物。
【請求項18】
前記潤滑剤が、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及び硬化植物油及びそれらの混合物からなる群から選択される、請求項7の医薬組成物。
【請求項19】
前記潤滑剤がステアリン酸である、請求項18の医薬組成物。
【請求項20】
(a)ナイアシン約76%から約88%w/w、
(b)放出遅延剤約11.0%から約20.0%w/w、
(c)結合剤約0.2%から約3.25%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、請求項2の医薬組成物。
【請求項21】
(a)ナイアシン約78%から約82%w/w、
(b)放出遅延剤約14%から約18%w/w、
(c)結合剤約2.5%から約3.0%w/w、及び
(d)潤滑剤約0.85%から約1.05%w/w、
を含む、請求項20の医薬組成物。
【請求項22】
潤滑剤約0.95%から約1.05%w/wを含む、請求項21の医薬組成物。
【請求項23】
ナイアシン処置療法に付随する紅潮を軽減する方法であり、
(a)ナイアシン約70%から約92%w/w、
(b)放出遅延剤約7%から約25%w/w、
(c)結合剤約0.1%から約4.3%w/w、及び
(d)潤滑剤約0.5%から約1.5%w/w、
を含む医薬剤形を1日1回投与することを含む、前記方法。
【請求項24】
前記1日1回剤形が、2個の1000mg錠剤を含む、請求項23の方法。
【請求項25】
前記錠剤が、
(a)ナイアシン約76%から約88%w/w、
(b)放出遅延剤約11.0%から約20.0%w/w、
(c)結合剤約0.2%から約3.25%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/wと、
を含む1000mg錠剤である、請求項23の方法。
【請求項26】
前記1000mg錠剤が、
(a)ナイアシン約78%から約82%w/w、
(b)放出遅延剤約14%から約18%w/w、
(c)結合剤約2.5%から約3.0%w/w、及び
(d)潤滑剤約0.85%から約1.05%w/w、
を含む、請求項25の方法。
【請求項27】
前記1000mg錠剤が、潤滑剤約0.95%から約1.05%w/wを含む、請求項26の方法。
【請求項28】
放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)、トリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))及びキサンタンガム及びそれらの混合物からなる群から選択される、請求項23の方法。
【請求項29】
放出遅延剤がヒドロキシプロピルメチルセルロースであり、並びにヒドロキシプロピルメチルセルロースが約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項28の方法。
【請求項30】
ヒドロキシプロピルメチルセルロースが約1.4から約1.9のメトキシル置換度及び約0.19から約0.24のヒドロキシプロポキシルモル置換度を有する、請求項29の方法。
【請求項31】
ヒドロキシプロピルメチルセルロースが、約1.4のメトキシル置換度及び約0.21のヒドロキシプロポキシルモル置換度を有する、請求項30の方法。
【請求項32】
ヒドロキシプロピルメチルセルロースが約11,000から約22,000mPasの粘度を有する、請求項29の方法。
【請求項33】
ヒドロキシプロピルメチルセルロースが約13,000から約18,000mPasの粘度を有する、請求項32の方法。
【請求項34】
医薬剤形がコーティングをさらに含む、請求項23の方法。
【請求項35】
前記コーティングが、医薬剤形の重量を約1.5から約8.0%増加させるために塗布される色彩塗料である、請求項34の医薬組成物。
【請求項36】
前記コーティングが、医薬剤形の重量を約1.75から約5.0%増加させるために適用される色彩塗料である、請求項35の医薬組成物。
【請求項37】
直接圧縮ナイアシン錠剤を調製する方法であり
(a)ナイアシン約70%から約92%w/w、放出遅延剤約7%から約25%w/w、結合剤約0.1%から約4.3%w/w、及び潤滑剤約0.5%から約1.5%w/wの混合物を混合する段階、
(b)段階(a)の混合物を錠剤へと圧縮する段階
を含む、方法。
【請求項38】
前記ナイアシン錠剤が1000mgナイアシン投与製剤である、請求項37の方法。
【請求項39】
錠剤を被覆することをさらに含む、請求項37の方法。
【請求項40】
錠剤の重量を約1.5から約8.0%増加させるために色彩塗料で錠剤を被覆することを含む、請求項37の方法。
【請求項41】
前記色彩塗料が約1.75から5.0%重量増加となる、請求項40の方法。
【請求項42】
前記放出遅延剤が、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC又はヒプロメロース)、メチルセルロース(MC)、ヒドロキシエチルセルロース(HEC)、ポリビニルピロリドン(PVP)、トリメチルアンモニオエチルメタクリレートとのメタクリレートコポリマー(EUDRAGIT RS(R)、EUDRAGIT RL(R))及びキサンタンガム又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項43】
前記結合剤が、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、エチルセルロース、ポリメタクリレート及びワックス又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項44】
前記潤滑剤が、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及び硬化植物油又はそれらの混合物からなる群から選択される、請求項40の方法。
【請求項45】
前記錠剤が、ナイアシン約76%から約88%w/w、放出遅延剤約11.0%から約20%w/w、結合剤約0.2%から約3.25%w/w、及び潤滑剤約0.75%から約1.25%w/wを含む、請求項40の方法。
【請求項46】
前記錠剤が、ナイアシン約78%から約82%w/w、放出遅延剤約14%から約18%w/w、結合剤約2.5%から約3.0%w/w、及び潤滑剤約0.95%から約1.05%w/wと、を含む、請求項45の方法。
【請求項47】
前記放出遅延剤がヒドロキシプロピルメチルセルロースであり、前記結合剤がポリビニルピロリドンであり、前記潤滑剤がステアリン酸であり、並びにヒドロキシプロピルメチルセルロースが約1.2から約2.0のメトキシル置換度及び約0.1から約0.3のヒドロキシプロポキシルモル置換度を有する、請求項37の方法。
【請求項48】
(a)ナイアシン約65%から約85%w/w、
(b)放出遅延剤約20%から約32%w/w、
(c)結合剤約2%から約3%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項49】
(a)ナイアシン約68%から約75%w/w、
(b)放出遅延剤約24%から約29%w/w、
(c)結合剤約2.25%から約2.75%w/w、及び
(d)潤滑剤約0.95%から約1.05%w/w、
を含む、請求項48に記載の直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項50】
コーティングをさらに含み、該コーティングが約1.5から約8.0%重量増加となる、請求項48に記載の直接圧縮500mgナイアシン徐放錠剤製剤。
【請求項51】
(a)ナイアシン約74%から約80%w/w、
(b)放出遅延剤約16%から約22%w/w、
(c)結合剤約2.5%から約2.75%w/w、及び
(d)潤滑剤約0.75%から約1.25%w/w、
を含む、直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項52】
(a)ナイアシン約76%から約79%w/w、
(b)放出遅延剤約18%から約21%w/w、
(c)結合剤約2.5%から約2.7%w/w、及び
(d)潤滑剤約0.95%から約1.05%w/w、
を含む、請求項51に記載の直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項53】
コーティングをさらに含み、該コーティングが約1.5から約8.0%の重量増加となる、請求項52に記載の直接圧縮750mgナイアシン徐放錠剤製剤。
【請求項54】
抗高脂血剤をさらに含む、請求項2の医薬組成物。
【請求項55】
抗高脂血剤がHMG−CoA還元酵素阻害剤である、請求項54の医薬組成物。
【請求項56】
紅潮抑制剤をさらに含む、請求項55の医薬組成物。
【請求項57】
紅潮抑制剤をさらに含む、請求項2の医薬組成物。
【請求項58】
紅潮抑制剤が非ステロイド抗炎症薬(NSAID)である、請求項57の医薬組成物。
【請求項59】
紅潮抑制剤がアスピリン(ASA)である、請求項58の医薬組成物。
【請求項60】
紅潮抑制剤がプロスタグランジンD2受容体アンタゴニストである、請求項57の医薬組成物。
【請求項61】
プロスタグランジンD2受容体アンタゴニストがMK−0524である、請求項60の医薬組成物。
【請求項62】
ナイアシンが粒状ナイアシンである、請求項23、37、48又は51の何れか一項の方法。
【請求項63】
粒状ナイアシン粒子サイズが、ふるい分画100−425μmに対してNLT85%(w/w)であり、及びダスト<100μmに対してNMT10%(w/w)である、請求項62の方法。
【請求項64】
1000mg徐放ナイアシン医薬組成物であり、これを必要とする患者に2個の1000mg錠剤を単回投与として投与する場合、次の生物学的利用率パラメータ
(a)2601.8ng/mLのNUA Cmax;
(b)60.5%の尿中ナイアシン総回収;
(c)4958.9ng/mLのナイアシンCmax;及び
(d)12414.5ng/mLのナイアシンAUC
の少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物。
【請求項65】
自然対数変換比が90%から115%内である、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項66】
自然対数変換比が95%から110%内である、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項67】
紅潮抑制剤及び抗高脂血剤からなる群から選択される少なくとも1つのさらなる治療剤をさらに含む、請求項64の1000mg徐放ナイアシン医薬組成物。
【請求項68】
請求項77の1000mg徐放ナイアシン医薬組成物であり、前記組成物が前記患者に1日1回摂取される場合、このような治療を中止する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項69】
1000mg徐放ナイアシン医薬組成物であり、4個の500mg NIASPAN(R)錠剤の単回投与を前記1000mg徐放ナイアシン組成物2個の単回投与と比較する生物学的同等性試験において対象に投与される場合、適切な生物学的利用率パラメータの自然対数変換比に対する90%CIが80%から125%の間隔内である、医薬組成物。
【請求項70】
前記生物学的利用率パラメータが、NUA Cmax(ng/mL)及び総回収又はナイアシンCmax(ng/mL)及びナイアシンAUCである、請求項69に記載の1000mg徐放ナイアシン医薬組成物。
【請求項71】
1000mg徐放ナイアシン医薬組成物であり、これを必要とする患者に2個の1000mg錠剤の単回投与として投与される場合、次の生物学的利用率パラメータ
(a)約2111.0ng/mLから約3253ng/mL NUA Cmax;
(b)約49.24%から約70.23%の尿中ナイアシンの総回収;
(c)約3096ng/mLから約6750ng/mLのナイアシンCmax;及び
(d)約6723ng/mLから約18643ng/mLのナイアシンAUC
の少なくとも1つについて、自然対数変換比に対する90%CIが80%から125%間隔内であるインビボ血漿プロファイルを与える、1000mg徐放ナイアシン医薬組成物。
【請求項72】
請求項71の1000mg徐放ナイアシン医薬組成物であり、前記組成物が前記患者に1日1回摂取される場合、治療を中止する必要がある、治療を制限する(i)肝毒性及び(ii)尿酸レベルもしくはグルコースレベル又は両方の上昇を引き起こすことなく、血清脂質を低下させるのに有効である、医薬組成物。
【請求項73】
ナイアシン放出が遅延される、請求項3の医薬組成物。
【請求項74】
即放型紅潮抑制剤をさらに含む、請求項73の医薬組成物。
【請求項75】
紅潮抑制剤がプロスタグランジンD2受容体である、請求項74の医薬組成物。
【請求項76】
プロスタグランジンD2受容体がMK−0524である、請求項75の医薬組成物。
【請求項77】
紅潮抑制剤が非ステロイド抗炎症薬(NSAID)である、請求項74の医薬組成物。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】
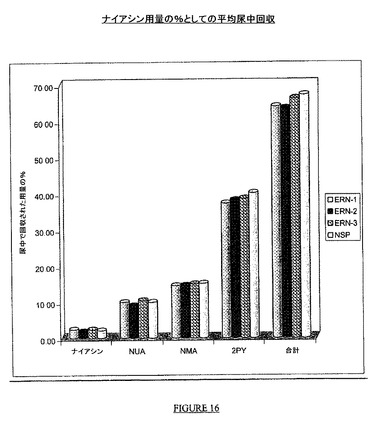
【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2009−527477(P2009−527477A)
【公表日】平成21年7月30日(2009.7.30)
【国際特許分類】
【出願番号】特願2008−555379(P2008−555379)
【出願日】平成19年2月15日(2007.2.15)
【国際出願番号】PCT/US2007/004105
【国際公開番号】WO2007/120385
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(508247903)アボツト・レスピラトリー・エル・エル・シー (1)
【Fターム(参考)】
【公表日】平成21年7月30日(2009.7.30)
【国際特許分類】
【出願日】平成19年2月15日(2007.2.15)
【国際出願番号】PCT/US2007/004105
【国際公開番号】WO2007/120385
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(508247903)アボツト・レスピラトリー・エル・エル・シー (1)
【Fターム(参考)】
[ Back to top ]