
癌を治療するためのキラヤサポニンを含む脂質含有粒子の使用
本発明は、癌治療用の医薬を製造するための、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えばリポソーム、イスコムおよび/またはイスコムマトリックス、およびPosintros(ポシントロス)の使用に関する。サポニンは好ましくはQuillaja Saponaria Molina由来である。さらに、該粒子は、補完メカニズムを用いる癌治療のための1または数種の化合物の送達系でもある。さらに、本発明は、少なくとも2の部分を含む部分のキットであって、1つの部分が、癌細胞の殺滅作用を有する疎水性の少なくとも1のサポニン分画を含み、その他の部分が免疫反応を刺激および調節する比較的親水性の少なくとも1のサポニン分画を含むキットを開示する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、癌治療用の医薬を製造するための少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、リポソーム、イスコム(iscom)および/またはイスコムマトリックス、およびポシントロス(posintros)の使用に関する。該粒子は、補完メカニズム(complementary mechanism)を用いる癌治療用の1または数種の化合物のデリバリーシステム(送達系)でもある。
【0002】
本発明は、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を癌治療が必要な個体に投与する癌の治療方法にも関する。
【0003】
さらに、本発明は、1つの部分が癌細胞に対する殺滅効果を有する疎水性の、少なくとも1のサポニン分画を含む脂質含有粒子を含み、その他の部分が、免疫応答、例えば抗体産生および細胞介在性免疫を刺激および調節する比較的親水性の、少なくとも1のサポニン分画を有する脂質含有粒子を含む、少なくとも2の部分を含む部分のキットに関する。
【0004】
本発明は、粒子製剤中の選択したキラヤ(Quillaja)成分が腫瘍細胞を殺し、その増殖を阻害する(以後、KGIという)という発見に関する。粒子製剤は、生体的利用可能性が高いので好ましい。粒子製剤は、標的化分子を用いて製剤化することができ、また、遊離形の溶解作用により生じる副作用なしにヒトや動物によく受け入れられるように製剤化することができる。
【背景技術】
【0005】
脂質含有粒子、例えば、リポソームやイスコムは、抗原の担体やアジュバントとして記載されている。
【0006】
キラヤサポニンの免疫刺激特性は、以前から(Ramon 1926)知られており、キラヤサポニンは、1950年代から市販ワクチンに遊離形で、時にAl(OH)3と組み合わせて用いられてきた(Dalsgaard 1978)、Ma et al. (Ma、Bulger et al. 1994)、(Espinet 1951)。従来の遊離形に比べてキラヤサポニンの実質的により効率的な用途が、Morein et al.により記載されている(Morein、Sundquist et al. 1984)-イスコム技術 (EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1)、および数年後にイスコム-マトリックス技術 (Lovgren and Morein 1988)、(EP 0 436 620 B1))。イスコム技術を用いてワクチン抗原は、キラヤサポニン、コレステロール、およびリン脂質からなる40nmの複合体に組み込まれる。
【0007】
キラヤサポニンは抗癌活性を示すことが知られている。しかし、粗または分画形のサポニンは、疎水性溶解作用があり、投与部位で部分的捕捉を生じるため一般的副作用がある。したがって、遊離形のサポニンは、癌治療には現実的でない。したがって、サポニンは、有用な癌治療薬には発展しなかった。
【0008】
さらに、イスコム技術は、イスコム複合体に統合された癌抗原を含む癌ワクチンに発展した。しかし、これらワクチン中の抗原はある抗体および免疫介在反応を生じるが、イスコム複合体は、分解し、個体が将来癌細胞に侵されたときには存在しないだろう。
【0009】
今回、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、イスコムおよびイスコムマトリックスを癌治療用の医薬を製造するのに用いることができることが解った。
【0010】
癌細胞は、正常細胞より本発明の脂質およびサポニン含有粒子に30〜40倍感受性がある。脂質およびサポニン含有粒子は癌細胞を殺すアポトーシスを誘導する。
【0011】
殺滅作用は、優れたアポトーシス誘導物質(inducer)による。高濃度ではより早いアポトーシスが誘導される。本発明の脂質含有粒子で処置後、細胞は細胞周期にとどまらず、細胞は第二周期を超えない。すなわち、癌細胞の死は不可逆的である。生産期(production phase)はIL-8産生に次いでアポトーシスが生じるという事実により示される。
【0012】
癌細胞の長期培養後、癌細胞は、処置後、より顕著には断続的低生理学的用量に曝露後でも複製には戻らない。
【0013】
癌細胞の死は、「材料と方法」に記載のトリパンブルー染色、アラマーブルー法による酵素代謝阻害を含む種々の方法、プロピジウムアイオダイド染色により可視化される壊死変化、およびアネキシンV染色によるアポトーシスにより分析した。
【発明の概要】
【発明が解決しようとする課題】
【0014】
(発明の要約)
本発明は、癌治療用の医薬を製造するための、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、リポソーム、イスコムおよび/またはイスコムマトリックス、およびポシントロスの使用に関する。
【0015】
本発明は、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を、癌治療を必要とする個体に投与する癌の治療方法にも関する。
【0016】
さらに、本発明は、1つの部分が癌細胞の殺滅作用を有する疎水性の少なくとも1のサポニン分画を含む脂質含有粒子を含み、他の部分が免疫応答、例えば、抗体産生および細胞介在性免疫を刺激および調節する親水性の少なくとも1のサポニン分画を有する脂質含有粒子を含む、少なくとも2の部分を含む部分のキットに関する。
【課題を解決するための手段】
【0017】
本発明は、選択したサポニン、例えば、キラヤ成分、特に脂質含有製剤が腫瘍細胞に対する殺滅および成長阻害作用を有するという発見に関する(以後、KGIおよびBBEという)。サポニンまたはサポニン分画を、腫瘍細胞の殺滅または成長阻害能で選択する。微粒子製剤は、高いバイオアベイラビリティにより選択され、該粒子は、粗サポニンの遊離形またはサポニンの遊離形に比べて、副作用がなく、個体、例えばヒトまたは動物によく受け入れられるように製剤化することができる。
【0018】
他のサポニンまたはサポニン分画(Quillaja Saponaria Molina(バラ科キラヤ)から得られる分画QHAを含む)は、そのようなKGI作用を示すかまたは示さないことがあるが、KGIに対する強力な中和、阻害、および平衡化作用を示すので、選択することができる。これら分画は、KGI粒子の部分として微粒子形でかまたは別個のBBE粒子で微粒子または非微粒子GHCとの相乗作用である種の癌細胞を殺すことができる。該阻害および平衡化(balancing)作用を以後BBEという。KGIおよびBBE粒子は、KGI粒子で処理し、殺された細胞から放出され、交差提示により抗原を提示することができるか、またはBBEが抗原提示細胞(APC)を直接刺激して抗腫瘍作用をもたらすことができるという事実により、腫瘍抗原に対する免疫保護反応を刺激および調節する。
【0019】
本発明をさらに以下の図面により説明する。
【図面の簡単な説明】
【0020】
【図1.1】キラヤサポニンのトリテルペノイド構造。
【図1.2】キラヤサポニンの逆相プロフィール。分画CはKGI 1の主要活性成分であり、分画AはBBEの主要活性成分である。
【図1.3】KGI 1の電子顕微鏡像。
【図2.1】QHCとQHAの構造的違い。細胞膜に対するQHCの高い溶解作用は、マークしたポイント3の右側の脂肪アシル鎖に関連する。QHAは脂肪アシル鎖を欠き、それをより親水性にすることにより溶解しにくくする。QHCおよびQHAは、ともに非分画化キラヤサポニンの天然成分である(図1.2のHPLCによる分離パターン参照)。
【図3.1】KGI 1は、低濃度で癌細胞U937を殺す(アラマーブルー(AlamarBlue)法により測定)。
【図3.2】正常ヒト樹状細胞(DC)を殺すには高用量のKGI 1が必要である。
【図4.1】BBEは、U937腫瘍細胞に無毒性である。
【図4.2】BBEは、正常ヒト樹状細胞(DC)に無毒性である。
【0021】
【図5.1】BBEとKGI 1の比10〜1において、BBEは、KGI 1による殺滅作用を阻害する。この試験はKGI 1の固定濃度(すなわち77μg/ml)およびX軸に示すBBEの増加する濃度で行った。
【図6.1】KGI 2は、1および同じ粒子中に2つのサポニン成分(種々の比(すなわち、9.5:0.5、7.5:2.5、および7.0:3.0)のQHAおよびQHC)を有する。KGI 2のU937細胞に対する癌殺滅能は、QHCの割合の増加に伴って増加する。
【図6.2】1および同じ粒子中に比7:3の2つのサポニン成分(QHAおよびQHC)を有するKGI 2は、U937癌細胞を殺すのにKGI 1より高い濃度の活性物質QHCを必要とする(実施例3中の図3.1参照)。
【図6.3】1および同じ粒子中に比7:3の2つのサポニン成分(QHAおよびQHC)を有するKGI 2は、U937癌細胞を殺すより正常ヒトDCを殺すのにKGI 1より高い濃度の活性物質QHCを必要とする(実施例3中の図3.1参照)。
【図6.4】種々のKGIおよびBBE製剤は、単球由来未成熟DCを活性化して成熟させ、分化および活性化のためにリンパ球と連絡をとり、エフェクター細胞にする活性化DCの分子であるDCマーカーCD86を発現する。
【0022】
【図7.1】KGI 1粒子はU937癌細胞の複製を阻害する。該細胞をマイクロタイタープレートに播き、次いで9日間の実験培養期間中2μg/ml (M2) KGI 1に曝露し、生細胞数をトリパンブルー染色後に顕微鏡で毎日カウントした。
【図7.2】KGI 1粒子は、KGI 1への曝露を中断した後でもU937癌細胞の複製を阻害する。該細胞を培養し、図7.1に記載のごとく2μg/ml (M2)のKGI 1に9日間曝露した。KGI 1を3日間インキュベーションした後に除去した。矢印で示す時点で培養液を交換した。コントロール細胞をKGI 1なしで培養した。
【図7.3】図7.1に記載のごとく培養したU937癌細胞を、高用量の25μg/mlの遊離形のKGI 1(すなわちQuill AのQHC分画)(F)、または粒子として25μg/mlのKGI 1(M)に曝露し、図に示すごとくサンプリングした。細胞をトリパンブルーで染色した(「材料と方法」参照)。細胞の生存性を生コントロール細胞のパーセントで表す。この高用量で、遊離形のKGI 1、すなわちQuill AのQHC分画は、急速、すなわち3時間以内に細胞を殺したが、KGI 1粒子は癌細胞を高率に殺すのにより長い時間、すなわち24時間を必要とした。
【図7.4】U937癌細胞を、低生理学的用量の2μg/mlの遊離形のKGI 1(すなわちQuill AのQHC分画)(F)、または粒子として2μg/mlのKGI 1(M)に曝露し、図に示すごとくサンプリングした。細胞をトリパンブルーで染色した(「材料と方法」参照)。生存性を生存コントロール細胞のパーセントで表す。この低用量で、遊離形のKGI 1、すなわちQuill AのQHC分画は、培養60時間内に細胞を殺さなかったが、KGI 1粒子は24時間後に癌細胞を殺し始めた。
【図7.5】非常に低用量のKGI 1粒子はU937癌細胞の増殖を阻害する。該細胞を、図に示すように、0.5μg/ml (M0.5)の低用量または2μg/ml (M2)のKGI 1に12日間曝露した。細胞数をトリパンブルー法で染色後にカウントした(「材料と方法」参照)。0.5μg/mlの低用量のKGIは該細胞数を無処置細胞に比べて減少させたが、2μg/ml用量のKGI 1粒子は、培養12日間内に全ての癌細胞を殺した。
【図7.6】KGI 1はU937癌細胞のアポトーシスを誘導した。該細胞を培養液中、2μg/ml (M2)または25μg/ml (M25)の濃度のKGI 1に120時間曝露した。アネキシンV陽性細胞数をFACSで測定した(「材料と方法」参照)。2μg/mlの濃度で、24時間曝露後にピークレベルを有するアポトーシス細胞ポピュレーションの増加が生じた。より高濃度、すなわち25μg/ml KGI 1では、さらに12および24時間曝露後にピークレベルを有するアポトーシス細胞の割合が増加した。
【図7.7】KGI 1は、壊死U937癌細胞数の増加をもたらさない。該細胞を、図に示すように培養液中2μg/ml (M2)〜50μg/ml (M50)の濃度のKGI 1に120時間曝露した。該細胞を培養し、図7.1に記載のごとくサンプリングし、プロピジウムアイオダイドで染色し、次いで壊死細胞数をFACSで測定した(「材料と方法」参照)。種々の用量のKGI 1で処理した細胞とKGI 1に曝露しなかったコントロール細胞の壊死組織の割合に差はなかった。
【図7.8】KGI 1は、アネキシンV (アポトーシス)とプロピジウムアイオダイド (壊死)の両方で染色されるU937癌細胞を徐々にもたらした。該細胞を図7.1に記載のごとく増殖させ、培養液中2μg/ml (M2)〜50μg/ml (M50)の濃度のKGI 1に120時間曝露した。該細胞をサンプリングし、図に示すようにプロピジウムアイオダイドおよびアネキシンVで染色した。罹患細胞の割合をFACSで測定した。濃度の増加は、壊死およびアポトーシス作用について染色された細胞の割合の増加をもたらした。
【0023】
【図8.1】KGI 1は、癌細胞U937の増殖を阻害し、該細胞は12日間の培養期間中増殖に復帰しない。細胞を12日間までの培養中に0.5μg/ml (M0.5)および2μg/ml (M2)のKGI 1に曝露し、図に示すように試料を回収した。細胞増殖の減少への変換点は1〜3日間細胞をKGI 1に曝露した後にみられる。トリパンブルー染色後に生細胞をカウントした。
【図8.2】KGI 1は癌細胞U937の増殖を阻害し、細胞はKGI 1除去後も増殖に戻らない。最初に細胞を22時間飢餓させ、細胞を細胞周期に同期化させた(明細書参照)。次に、細胞を培養中2μg/mlのKGI 1で12日間まで曝露し、試料を回収し、次いで培地を3日毎に交換した。第3日にKGI 1を細胞から除去した。トリパンブルー染色後に生細胞をカウントした。
【0024】
【図9.1】細胞周期の説明図。チミジンキナーゼ(TK)活性はS期、すなわちDNA複製期に先立つ。細胞増殖に対するKGI 1の阻害作用は少なくとも低用量では細胞増殖周期の後期に生じるようである。
【図9.2】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)または25μg/ml (M25)のKGI 1で5日間処理した後毎日の細胞溶解で測定した。細胞培養液を無処置細胞の活性の低下を説明するためこの実験期間中交換しなかった。処理細胞のTK活性の低下を5日間の培養中無処置コントロールと比較した。TK活性の低下は、高用量の25μg/mlのKGI 1では24時間曝露後、低用量の2μg/mlのKGI 1では2日間後に顕著になる。
【図9.3】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)または25μg/ml (M25)のKGI 1で5日間処置後の細胞溶解物で測定した。TK活性は非処理細胞のそれのパーセンテージで表す(図9.2も参照)。
【図9.4】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)、10μg/ml (M10)、25μg/ml (M25)、50μg/ml (M50)の濃度の微粒子KGI 1で処置後120時間毎日の細胞溶解物で測定した。微粒子KGI 1で処理後のTK活性の減少を、Fで示した同じ濃度で試験した遊離、すなわち無微粒子KGI 1のそれと比較した。低生理学的用量では、細胞を48時間曝露後に減少が顕著になるが(M2)、遊離KGI 1では減少は顕著でない(F2)。高用量の25μg/mlまたはより高濃度のKGI 1で処理後のTK活性の減少は、24時間曝露後に顕著である。高用量の遊離KGI、すなわち、25μg/mlおよび50μg/ml細胞培養液で処理した細胞は、検出可能なTK活性を示さなかった(図9.5も参照のこと)。
【図9.5】TK活性を、106/mlのU937癌細胞を2μg/ml (M2)、10μg/ml (M10)、25μg/ml (M25)、50μg/ml (M50)の濃度の微粒子KGI 1で5日間処理した後に毎日細胞培養液で測定した。TK活性は、微粒子KGI 1で処理した細胞の培地中には検出されなかった。25μg/mlおよび50μg/mlの濃度で試験した遊離、すなわち非微粒子KGI 1処置細胞(Fで示す)は、培養液にTKを放出した(図9.4も参照)。細胞ではなく細胞培養液中にのみみられるTK活性は、漏出および細胞膜損傷を示す。
【図9.6】U937癌細胞のチミジンキナーゼ(TK)活性を、22時間、細胞を飢餓させて細胞周期を同期化させた後に測定した(本文参照)。次に、該細胞を2μg/mlのKGI 1に0、2、8、18、および24時間曝露した(本文参照)。無処置コントロールを0、8、および24時間にサンプリングした(本文参照)。KGI 1 (2μg/ml、すなわちM2)は、18および24時間処理した細胞試料を用いて記録したU937癌細胞のチミジンキナーゼ活性を低下させる。結果は、最初の8時間は低用量ではTK活性の阻害は起きなかった(18時間後は除く)ことを示す。
【図9.7】KGI 1 (2μg/ml)は、18時間の曝露後に検出された癌細胞U937の増殖を阻害する。最初に、細胞を22時間飢餓させて細胞周期を同期化させた(本文参照)。図9.6に示すように、18時間処理後の細胞増殖の低下はTK活性の減少と一致する。トリパンブルー染色後に生細胞をカウントした。
【図9.8】細胞代謝阻害(アラマーブルー)および細胞殺滅(トリパンブルー)を、細胞を22時間飢餓させて(本文参照)細胞周期を同期化させた後に測定した。次に、細胞を2μg/mlまたは0.5μg/mlのKGI 1に24時間曝露した。無処置コントロールを24時間でサンプリングした。KGI 1(2μg/ml、すなわちM2)および遊離KGI(2μg/ml、すなわちF2)は24時間処理後の細胞生存率を減少させた。濃度0.5μg/mlのKGI 1または遊離KGI 1は24時間処理後の細胞生存率を減少させた。代謝阻害は、KGI 1で処理後のほうが遊離形で処理後より顕著であった。
【0025】
【図10.1】KGI 1は、LC50濃度3μg/mlで癌細胞U937に781pg/mlのIL-8の産生を誘導した。
【図10.2】KGI 2は、LC50濃度19 μg/mlで癌細胞U937に880pg/mlのIL-8の産生を誘導した。
【図10.3】KGI 3は、14 μg/mlで癌細胞U937に917pg /mlのIL-8の産生を誘導した。
【発明を実施するための形態】
【0026】
本発明は、癌治療用の医薬を製造するための少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子の使用に関する。すなわち、本発明は、癌治療用の少なくとも1の脂質および少なくとも1のサポニンを含む医薬に関する。
【0027】
本発明によれば、該医薬は脂質粒子自体、および殺癌作用をもたらすサポニンである。遊離サポニン自体でも癌細胞を殺すことができるが、正常細胞に負の影響があることがわかった。脂質と共にかまたは脂質粒子と統合することにより、正常細胞に毒性がある遊離サポニンの濃度より30倍低い濃度で癌細胞に対する作用が得られる。
【0028】
脂質含有粒子は、リポソーム、イスコムおよび/またはイスコムマトリックス、およびポシントロスから選ぶことができる。
リポソーム
【0029】
リポソームは、一般的に、典型的には1またはそれ以上の同心層、例えば単層、二層、または多層の形の、脂溶性部分を含む両親媒性化合物の球状または回転楕円状クラスターまたは凝集物である。リポソームは、本明細書では脂質粒子ともいうことがある。リポソームは、例えばイオン性脂質および/または非イオン性脂質から製剤化することができる。非イオン性脂質から製剤化したリポソームは、ニオソーム(niosome)ということがある。少なくとも部分がカチオン性脂質またはアニオン性脂質から製剤化されたリポソームは、コクレエート(cochleate)ということがある。リポソームは、例えばLipfordおよびWagner(Lipford、Wagner et al. 1994)およびGregoriadis、G.(Gregoriadis、McCormack et al. 1999)、O´Hagan、DT(2001)に記載のごとく製造することができる。
【0030】
本発明に関連するリポソーム組成物の製造に用いるのに適合した一般的リポソーム製造技術は例えば米国特許4,728,578、4,728,575、4,737,323、4,533,254、4,162,282、4,310,505、および4,921,706;英国特許出願GB 2193095A;国際出願No. PCT/US85/01161およびPCT/US89/05040;Mayer et al.(Mayer、Hope et al. 1986);(Hope et al. 1985);Mayhew et al.(Mayhew、Conroy et al. 1987);Mayhew et al.(Mayhew、Lazo et al. 1984);Cheng et al、(Cheng、Seltzer et al. 1987);およびリポソーム技術、Gregoriadis、G.(Gregoriadis、G.編、1984)に記載されている(これらの内容は本明細書の一部を構成する)。したがって、リポソーム組成物は、当業者に知られている、例えば溶媒透析、フレンチプレス、押し出し(凍結-解凍ありまたはなし)、逆相蒸発、単純な凍結-解凍、ソニケーション、キレート透析、ホモゲナイゼーション、溶媒注入、ミクロエマルジョン化、自然形成、溶媒蒸発、溶媒透析、フレンチプレッシャー細胞技術、制御界面活性剤透析その他(それぞれ、種々の方法での該組成物の製造を含む)を含む当業者に明らかな種々の常套的リポソーム製造技術のいずれかを用いて製造することができる。例えば、Madden et al.、(Madden、Harrigan et al. 1990)参照(この内容は本明細書の一部を構成する)。
【0031】
適切な凍結-解凍技術は、例えばWO出願no. PCT/US89/05040(1989年11月8日出願)に記載されている(この内容は本明細書の一部を構成する)。凍結-解凍技術を含む方法は、リポソームの製造に関して好ましい。リポソームの製造は、溶液、例えば水性生理食塩水溶液、水性リン酸緩衝溶液、または無菌水中で行うことができる。リポソームは、振盪または渦撹拌(vortexing)を含む種々の方法で製造することもでき、これは例えば、以下の機械式振盪器を用いて達成することができる:Wig-L-Bug(登録商標)(Crescent Dental、Lyons、III.)、Mixomat(Degussa AG Frankfurt、Germany)、Capmix(Espe Fabrik Pharmazeutischer Praeparate GMBH & Co.、Seefeld、Oberay Germany)、Silamat Plus(Vivadent、Lechtenstein)、またはVibros(Quayle Dental、Sussex、England)。常套的ミクロエマルジョン化装置、例えばMicrofluidizer(登録商標)(Microfluidics、Woburn、Mass)を用いることもできる。
イスコムおよびイスコムマトリックス
【0032】
イスコムは、少なくとも1のサポニン、例えば、少なくとも1のグリコシド、少なくとも1の脂質および少なくとも1種類の抗原物質を含む。該脂質は少なくともステロール、例えば、コレステロール、所望によりホスファチジルコリンである。この複合体は、1またはそれ以上の他の免疫調節(アジュバント活性)物質を含んでいてもよく、EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1に記載のごとく製造することができる。
【0033】
本発明の組成物中のイスコムマトリックス複合体は、少なくとも1のグリコシドおよび少なくとも1の脂質を含む。脂質は少なくともステロール、例えば、コレステロール、および所望によりホスファチジルコリンである。マトリックスは、同時に投与した抗原物質に対する免疫増強作用を有する。イスコム複合体は、1またはそれ以上の他の免疫調節(アジュバント活性)物質(必ずしもサポニンではない)を含むこともでき、EP 0 436 620 B1に記載のごとく製造することができ、本特許に記載のごとく製造することができる。
【0034】
1またはそれ以上のイスコム粒子、1またはそれ以上のイスコムマトリックス粒子、またはその6ナノメーター環のあらゆるサブフラグメントを用いることができる。そのようなイスコムマトリックス、粒子またはサブフラグメントのあらゆる混合物を用いることができる。
Posintros(ポシントロス)
【0035】
Posintrosは、i)少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロール(ここで、少なくとも1の第2ステロールは、静電気的相互作用および疎水性相互作用から選ばれる相互作用により異種抗原、好ましくは核酸と接触することができ、少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロールは、少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニンと複合体を形成することができる)、およびii)少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニン(ここで、少なくとも1の第2サポニンは、静電気的相互作用および疎水性相互作用から選ばれる相互作用により遺伝的決定因子と接触することができ、少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニンは少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロールと複合体を形成することができる)、および所望によりiii)静電気的相互作用および疎水性相互作用から選ばれる相互作用により遺伝的決定因子と接触させるための少なくとも1の接触基(contacting group)を含む複合体である。ただし、第2ステロールが複合体およびさらに所望によりi)少なくとも1の脂溶性部分中に存在しない時は少なくとも1の接触基が存在する。
【0036】
Posintrosは、免疫刺激複合体(イスコム)として知られているものと同様のケージ様マトリックスの形の微粒子構造を採用することができる。イスコム構造に加えて、ステロールとサポニンの間の相互作用は、構成要素、例えば、格子、ハニカム、棒、および非晶質(amorphic)粒子を含む種々の異なる構造的構成要素を生じることが報告されている(これらすべての構造的構成要素は本発明に含まれる)。
Posintrosは、WO特許出願No.WO 2002/080981およびWO 2004/030696に記載されている。
脂質
【0037】
用いる脂質は、特に本願出願人の特許EP 0 109 942 B1(特に3頁)および特許EP 0 436 620 B1(7頁7〜24行)に記載のものである。特に、ステロール、例えばコレステロールおよびリン脂質、例えばホスファチジルエタノールアミンおよびホスファチジルコリンを用いる。ガングリオシドGM1である、コレラ毒素レセプターを含む、細胞結合成分、例えば糖脂質と結合する脂質含有レセプター、およびフコース化血液群抗原を用いることができる。次に、細胞結合成分は、粘液標的化分子として機能し、それを含む複合体と単純に混合することにより脂質含有物質と結合することができる。そのようなレセプターを含むイスコム複合体およびレセプターはWO 97/30728に記載されている。
サポニン
【0038】
サポニンはあらゆるサポニンでありうる。本発明のある局面によれば、サポニンは植物から得られるグリコシドである。植物グリコシドは、1またはそれ以上の糖部分を有するサポゲニンおよびプロサポゲニンから選ぶことができる。グリコシドはQuillaja Saponaria Molina由来の粗サポニン分画またはその亜分画でありうる。
【0039】
キラヤサポニンおよびその種々の分画は、50年代からアジュバントとしておよび種々のアジュバント製剤に用いられており、ほとんどの疎水性分画のうち例えばQS21は動物ワクチンおよび種々のヒト臨床試験に用いられてきた(Kersten、Spiekstra et al. 1991);(Kensil、Patel et al. 1991)。イスコムまたはイスコムマトリックスは、種々のキラヤ分画、または分画またはより粗なキラヤサポニンの種々の混合物を用いて形成されている。すべての場合、イスコムまたはイスコムマトリックス製剤は、遊離形より局所反応が少ない。最近の開発品では、すぐれた免疫増強能を有する、アジュバントとして用いたいかなる他のキラヤサポニン製剤よりはるかに耐容性のある製剤が設計された。(Morein、Sundquist et al. 1984)。この耐容性のよいキラヤサポニン製剤の成分を、本発明では癌細胞殺滅(KGI)および該効果の平衡化(BBE)のために用いる。
【0040】
サポニンは、1またはそれ以上の糖鎖と結合したアグリコンからなる分子複合体である。サポニンは、構造の部分として有機酸、例えば、酢酸、マロン酸でアシル化することができる(Hostettmann K.およびMarston A. 1995;Rouhi A.M. 1995;Leung A Y.およびFoster S. 1996)。これら複合体の分子量は、600から2000kd以上に及ぶ。疎水性アグリカンおよび親水性糖部分は両親媒性特性をもたらす。特に、トリテルペングリコシドが興味深い。そのアグリコンにより特徴付けられる他のサポニンは、ステロイドグリコシドおよびステロイドアルカロイドグリコシドである。
【0041】
粗キラヤサポニンは、1887年に初めてKobert、R.により単離された(Arch. Exp. Pathol. Pharmakol. 23: 233-272、1887)。後にDalsgaardはラヤサポニンを精製した(Dalsgaard 1974)。Higuchi、R.(Higuchi、R. 1988)は、2つの異なる位置に2つの糖部分が結合したアグルコン(トリテルペノイドキラヤ酸)を認識するキラヤサポニンの完全な構造を報告した。有用なグリコシドはEP 0 109 924 B1に記載されている。サポニンおよびトリテルペンサポニンが好ましい。それらは、Quillaja Saponaria Molina由来の生抽出物(Dalsgaard 1974)、またはそのあらゆる亜分画の形であり得る。それらはKensil et al.に対するPCT/US/88101842(Kensil、Patel et al. 1991)、(Kersten、Spiekstra et al. 1991)「Aspects of Iscoms. Analytical、Pharmaceutical and Adjuvant Properties」;論文、University of Utrecht、EP 0 362 279 B2、およびEP 0 555 276 B1に記載されている。
【0042】
用語「Quillaja Saponaria Molina由来の1サポニン分画」は、本明細書および特許請求の範囲を通してQuillaja Saponariaの半精製または限定サポニン分画または実質的に純粋な分画の一般的説明として用いる。該分画は実質的に1分画を含むイスコムまたはイスコムマトリックスの混合物を用いる時に得られる良好な結果に悪影響を与えるいかなる他の分画も含まないことが重要である。所望によりサポニン製剤は、他の化合物、例えば他のサポニン、または他のアジュバント物質を小量、例えば40重量%以下、例えば30重量%以下、25重量%以下、20重量%以下、15重量%以下、10重量%以下、7重量%以下、5重量%以下、2重量%以下、1重量%以下、0,5重量%以下、0,1重量%以下含みうる。
【0043】
本発明のサポニン分画は、WO 96/11711に記載のA、B、およびC分画、EP 0 436 620に記載のB3、B4、およびB4b分画、EP 0 3632 279 B2に記載の分画 QA1-22、Q-VAC(Nor-Feed、AS Denmark)、Quillaja Saponaria Molina Spikoside(Isconova AB、Ultunaallen 2B、756 51 Uppsala、Sweden)でありうる。EP 0 3632 279 B2の分画 QA-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-16-17-18-19-20-21、および22、特にQA-7、17-18、および21も用いることができる。それらはEP 0 3632 279 B2の特に6頁、8〜9頁の実施例1に記載されている。WO 96/11711に記載の分画A、B、およびCは、粗水性Quillaja Saponaria Molina抽出物のクロマトグラフィ分離を用い、次いで水中70%アセトニトリルで溶出して脂溶性分画を回収して得られた脂溶性分画から製造する。次に、この脂溶性分画を酸性水中25%〜60%アセトニトリルの勾配を用いて溶出するセミプレパラティブHPLCにより分離する。「分画A」または「QH-A」として本明細書で言及している分画は、約39%アセトニトリルで溶出した分画であるかまたはそれに相当する。「分画B」または「QH-B」として本明細書で言及している分画は、約47%アセトニトリルで溶出した分画であるかまたはそれに相当する。「分画C」または「QH-C」として本明細書で言及している分画は、約49%アセトニトリルで溶出した分画であるかまたはそれに相当する。
【0044】
Quillaja Saponaria Molina由来のサポニンは、2つの異なるカテゴリーに分けることができる。すなわち、
(I) より疎水性の分画は、4位に脂肪酸アシル鎖を有する。これらサポニン分画は細胞膜に小さな約12nmの穴を作ることにより強い溶解作用を示す。そのようなサポニン分画は、遊離形で不可逆的に細胞を殺すが、本発明に記載の、および中等度の濃度(Ronnberg、Fekadu et al. 1997)の微粒子形の統合抗原を有する免疫刺激複合体(ISCOM)、または統合抗原を持たない同様の粒子、すなわちイスコムマトリックスでは必ずしもそうではない。
(II) より親水性のキラヤサポニンは、細胞溶解作用を示す前に10倍またはそれ以上の高濃度で与えることができる。微粒子形ではこれらのサポニン分画は、細胞毒性作用またはin vivo毒性作用が実質的にない。
【0045】
より疎水性のおよびより親水性の微粒子形は、イスコムマトリックスといわれ、6nmの環形成サブフラグメントにより形成された40nmの球である(Ronnberg、Fekadu et al. 1995)、(Lovgren and Morein 1991)。
【0046】
脂質粒子、例えば脂肪酸を含む疎水性サポニンを含むイスコムおよびイスコムマトリックスは、本発明ではKGI 粒子(腫瘍細胞を殺し、増殖を阻害する)という。そのようなサポニンは、例えばQuillaja Saponaria Molina由来のサポニンのトリテルペノイドアグリコンの4位に脂肪アシルを含む分画、例えば、Quil Aの分画CおよびB、または分画AとBの間の領域由来の分画、およびEP 0 3632 279 B2に記載の分画15〜21であってよく、特に分画16、17、18が適している。
【0047】
脂質粒子、例えば、親水性サポニンを含むサポニンを含む、例えば脂肪酸を含むイスコムおよびイスコムマトリックスは、BBE粒子(平衡化作用および癌細胞殺滅作用を阻害(ブロック)する)と呼ばれる。Quil Aの分画4〜15、特にEP 0 3632 279 B2に記載の7〜14および分画A(QHA)が本発明では好ましい。
【0048】
脂質粒子は、少なくとも1の疎水性サポニンを含みうる。脂質粒子は少なくとも1の親水性サポニンも含みうる。少なくとも1の親水性サポニンおよび少なくとも1の疎水性サポニンは、1および同じ、または異なる脂質含有粒子中にあってよい。
【0049】
Quillaja Saponaria Molina由来のQHA分画は、細胞殺滅作用を有さず、KGI製剤(formulation)に対する強力な中和、阻害、またはより重要なのは平衡化(例えば細胞の殺滅と分化の調節の間の平衡)作用を有するので、それを選択した。阻害および平衡化作用を以後BBEと略す。KGIおよびBBE粒子は抗原に対する免疫保護反応を刺激および調節する。したがって、これら粒子は、KGI粒子により殺された細胞から放出された腫瘍抗原に対する免疫反応を刺激し、それにより交差提示は抗原を提示することができると予想される。あるいはまた、BBEは、抗腫瘍作用に対する抗原提示細胞(APC)の刺激および獲得抗腫瘍免疫反応の誘導を直接増強することができる。
【0050】
すなわち、本発明においてその作用で名付けられたKGIおよびBBE粒子は異なる特性を有し、KGIは、比較的低濃度、すなわち初代ヒトまたはネズミ細胞に対するものより30〜40倍低い濃度で不可逆的に細胞増殖を阻害し、癌細胞を殺すことができる。さらに、KGIはBBEのように組み込んだ抗原または細胞からその環境に放出されるか同時に投与された抗原に対する免疫増強作用を有する。BBE粒子は、腫瘍抗原が統合されたKGI粒子と同時に投与するか、または例えばKGIにより破壊された癌細胞から腫瘍抗原を自然に得るか、または抗原物質と同時に投与することができる(EP 0 436 620 B1参照)。
【0051】
KGIおよびBBEはいずれも少なくとも1のサポニン、例えば、グリコシドおよび少なくとも1の脂質を含む。それらがイスコムおよびイスコムマトリックスである場合は、WO/1990/003184に記載のごとく脂質コレステロールも含む。
【0052】
癌細胞に対する殺滅作用を有する疎水性サポニンを含む脂質含有粒子は、さらに親水性サポニンも含みうる。
【0053】
脂質含有粒子は、1つおよび同じ脂質含有粒子中に少なくとも2の異なるサポニン分画を含みうる。
【0054】
脂質含有粒子は、少なくとも2の異なるサポニン分画の1つが1つの脂質含有粒子中の結合複合体であり、少なくとも2の異なるサポニン分画の他のものが別の物理的に異なる脂質含有粒子中の結合複合体である、少なくとも2の異なるサポニン分画も含むことができる。
【0055】
異なるサポニンはそれぞれ親水性および疎水性サポニンでありうる。該粒子は、少なくとも分画Cまたは少なくとも分画Bまたは少なくともQuil Aの分画CとBの間のあらゆる分画、およびQuil Aの少なくとも1の他の分画を含むことができる。すなわち、1つの粒子が、分画Cのみ;分画CおよびQuil Aの少なくとも1の他の分画;分画CおよびQuil Aの1またはそれ以上の分画;分画CおよびQuil Aの分画A;粗Quil Aを含みうる。該粒子は、分画Bのみ;分画BおよびQuil Aの少なくとも1の他の分画;分画BおよびQuil Aの1またはそれ以上の分画;分画BおよびQuil Aの分画Aも含みうる。上記の分画混合物は、異なる脂質粒子または1つまたは同じ脂質粒子中に存在しうる。実施例1のKGI 1、KGI 2、およびKGI 3粒子がそのような脂質粒子の例である。
【0056】
本発明のある局面では、KGI粒子は、サポニンまたはその半精製形の混合物を含むQuil Aの粗または生抽出物、例えば、Quillaja Powder Extract(キラヤ粉末エキス)(Berghausen、USA)、Quillaja Ultra Powder(キラヤ微粉末)QP UF 300、Quillaja Ultra Powder QP UF 1000、またはVax-Sap(3つすべてNatural Responses、Chileから)を含みうる。本発明では精製サポニン分画CおよびB単独またはAとの組み合わせをKGI粒子に用い、AをBBE粒子に用いる。BおよびC分画はWO 96/11711に記載されており、B3、B4、およびB4b分画はEP 0 436 620に記載されている。分画QA1-22はEP 0 3632 279 B2に記載、Q-VAC(Nor-Feed、AS Denmark)、Quillaja Saponaria Molina Spikoside(Isconova AB、Uppsala Science Park、751 83、Uppsala、Sweden)。そのようなKGI粒子は、実施例ではKGI 3を示す。
【0057】
KGI粒子の有用なサポニンの例は、上記Quil AのQHC分画およびQuil AのQHCおよびQHA分画の異なる組み合わせである。実施例において、KGI 1粒子は分画QHCのみを含む。KGI 2粒子は、30%のQHCと70%のQHAを含む。上記Quil A(キラヤサポニン)分画の他の組み合わせすべても用いることができる。
【0058】
腫瘍細胞は急速に増殖する未分化細胞である。したがって、腫瘍細胞はある種の細胞毒性物質に感受性がある。本発明の概念は、悪性癌(良性癌を除外しない)のような急速に増殖する細胞に対する毒性および/または調節効果を有するQuillaja Saponaria Molinaの分画または分画の組み合わせに基づく微粒子形の物質(作業名:KGI 1、KGI 2、およびKGI 3、ならびにBBE)を用いることである。毒性または調節作用は細胞レベルで測定することができる。細胞毒性−細胞調節物質、すなわちサポニンは、1またはそれ以上のデリバリー粒子中に組み込まれる。別の粒子は、毒性または細胞調節作用に加えて、毒性を阻害するのに用いることもできる(作業名BBE)。すなわち、腫瘍細胞の平衡化殺滅系を作製することができる。デリバリーシステムにおいて、生存を刺激し、細胞の分化を活性化する免疫モジュレーター(調節物質)を組み込むことができる。さらなる刺激には、癌を除去する主要な免疫防御細胞種である細胞障害性T細胞の誘導が含まれうる。リンパ系の死にかけの細胞もいわゆる交差提示により生DCの刺激に寄与しうる。単球由来の単芽球様細胞はリンパ肉腫腫瘍細胞を表し、正常細胞は単球起源の樹状細胞である。
【0059】
Quillaja Saponaria Molinaから調製したKGIおよびBBE複合体の組み合わせにより、種々の補完的特性、例えばKGI粒子より低い細胞毒性、補完的細胞活性化および分化、および顕著な免疫調節作用を有する製剤を製造することができる。KGIおよびBBE粒子の作用は、癌細胞に対する細胞毒性を生じるKGIに対するBBEによる阻害作用により強調されるようにレセプター介在性である。すなわち、本発明でモデルとして用いたU937細胞に対するKGIの癌細胞殺滅作用は、BBE粒子において確認されない。しかし、BBEは、ある種の他の癌細胞に対する殺滅作用を有する。共通レセプターは、癌細胞の活性化と分化をもたらし、これは、正常細胞ではサイトカイン産生と例えばコミュニケーション分子、例えばCD86の発現をもたらすアジュバント活性と適合性または部分的に適合性である。CD86は、樹状細胞とリンパ球ポピュレーションをコミュニケーションさせ、抗原特異反応をもたらす。これの欠如は、例えば、新生児または高齢者の免疫反応を妨げる。本発明者らは、分画QHA(BBEおよびKGI 2中の成分)およびQHC(KGI 1およびKGI 2中の成分)が、先天性および後天性の未熟または不完全に分化した免疫系を有する新生児の免疫反応を活性化および分化させることを発見した(Hu et al. 2004、Morein & Hu 2007)。アポトーシスを介して癌細胞の殺滅をもたらすレセプターはKGI 粒子上に存在するが、BBE 粒子上にはみられない。しかしながら、BBEが他の癌細胞にアポトーシスを引き起こすことを排除できない。癌細胞を殺すレセプターがそれ自体でか第二のレセプターと共同して副作用を生じうることは排除できない。特に、レセプター活性またはレセプター活性の組み合わせが副作用を生じさせる種差があると考える必要がある。そのような問題に対処することができる系が望まれ、KGIとBBEの組み合わせがその可能性をもたらす。
【0060】
すなわち、細胞のKGIによる殺滅は、この作用がBBEにより阻害することができるのでレセプター介在性である。KGIおよびBBEに対する共通(阻害)レセプターは、サポニン分画GHCのみを含むKGI 1によるU937細胞のレセプター介在性殺滅のそれと異なると考えられる。そうでなければ、BBEもこれらの実験に用いたU937癌細胞殺滅物質であるはずである。BBE中の活性物質はQHAである。それがKGI2と呼ばれるQHCと同じ粒子中に存在するときは、「QHC中の殺滅レセプター」の希釈、または構造修飾により細胞の殺滅作用を抑え、細胞の殺滅に活性なレセプターとリガンドの親和性の低下をもたらす。これに対し、異なる粒子では共通タンパク質による阻害がみられる。
【0061】
本発明のサポニン製剤の使用は、耐容性が増加し、バイオアベイラビリティ、免疫原性が増加した生成物をもたらす。該製剤は、炎症、過敏症、およびアレルギー反応の調節の増大を含む免疫原性を調整する方法に用いることができる。この調整は種依存性であり、毒性、耐容性、および免疫原性に影響を及ぼしうる。
【0062】
少なくとも1の親水性サポニン、例えば、Quil A由来の分画A(例えばBBE粒子)を含む脂質含有粒子の混合物を少なくとも1の疎水性サポニン、例えばQuil Aの分画C(例えばKGI 1、KGI 2、およびKGI 3)を含む脂質含有粒子と一緒に用いると、相乗的抗癌作用が得られることがわかった。
【0063】
少なくとも1の親水性サポニンは、Quil Aの分画4、5、6、7、8、9、10、11、12、13、14、および15、特にEP 0 3632 279 B2に記載の分画7、8、9、10、11、12、13、および14、およびQuil Aの分画Aまたは粗Quil Aの1またはそれ以上でありうる。
【0064】
少なくとも1の疎水性サポニンは、例えばQuillaja Saponaria Molina由来のサポニンのトリテルペノイドアグリコンの4位に脂肪アシルを含む1またはそれ以上のサポニン、例えば、Quil Aの分画CおよびB、または分画AおよびBの間の領域由来の分画ならびにEP 0 3632 279 B2に記載の分画15、16、17、18、19、10および21であってよく、特に分画17および18がここでは適切である。
【0065】
そのような共生(symbiotic)作用のための脂質含有粒子は、イスコムおよびイスコムマトリックス粒子、リポソームおよびPosintrosから選ぶことができる。
【0066】
あらゆる比の親水性および疎水性サポニン、例えば、Quillaja Saponaria Molinaサポニンのサブフラグメントを用いることができる。また、Quillaja Saponaria Molinaの異なる親水性および疎水性サポニンサブフラグメントのあらゆる組み合わせを用いることができる。すなわち、1、2、またはそれ以上の親水性および疎水性サポニン、例えば、サブフラグメントQuillaja Saponaria Molinaサポニンは、それぞれ物理的に1つおよび同じのまたは物理的に分離した脂質含有粒子に統合することができる。
【0067】
それぞれ親水性サポニン、例えばQuillaja Saponaria Molinaの分画Aおよび疎水性サポニン、例えばQuillaja Saponaria Molinaの分画Cのそれらの含有量に基づき異なる脂質含有粒子、例えば、イスコム、イスコムマトリックス複合体、リポソームまたはPosintrosの重量%のあらゆる組み合わせを用いることができる。該混合物は、0.1〜99.9重量%、5〜95重量%、10〜90重量%、15〜85重量%、20〜80重量%、25〜75重量%、30〜70重量%、35〜65重量%、40〜60重量%、45〜55重量%、40〜60重量%、50〜50重量%、55〜45重量%、60〜40重量%、65〜35重量%、70〜30重量%、75〜25重量%、80〜20重量%、85〜15重量%、90〜10重量%、95〜05重量%の脂質含有粒子、例えば親水性サポニン、例えばQuillaja Saponaria Molinaの分画Aを含むイスコム複合体、および残りをそれぞれ100%にする親水性および疎水性サポニン、例えばイスコム複合体中のQuillaja Saponaria Molinaの分画AおよびCの合計の含有量を計算して脂質含有粒子、例えば疎水性サポニン、例えばQuillaja Saponaria Molinaの分画Cを含むイスコム複合体を含むことができる。これは、親水性および疎水性サポニンの両方を含む脂質含有粒子、または疎水性または親水性サポニンのみを含む脂質含有粒子の混合物に適用される。
【0068】
すなわち、脂質含有粒子は、75%〜99.5重量%の親水性サポニン、例えばQuil Aの分画A、および0.5%〜25重量%の疎水性サポニン、例えばQuil Aの分画C;80%〜95%の親水性サポニンおよび5〜20%の疎水性サポニン;85%〜90%の親水性サポニンおよび10〜15%の疎水性サポニン、例えば、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、96%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99,5重量%の親水性サポニン、例えば分画A、および0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%、25重量%の疎水性サポニン、例えば分画Cを含むことができる。
【0069】
上記のすべての間隔を、あらゆる種類のヒトまたは動物種に投与するための製剤中のQuillaja Saponaria Molinaのあらゆる分画のあらゆる組み合わせに用いることができる。本発明の製剤を投与することができる動物種の例には、コンパニオンアニマル、例えばネコ、イヌ、ウマ、鳥、例えばオウム、産業的に重要な種、例えばウシ(例えばウシ種)、ブタ、ヒツジ、ヤギがある。好ましくは、50重量%以上の分画Cを他のあらゆる分画と組み合わせて、特に分画Aと組み合わせて用いる。すなわち、50.5〜99.5重量%のCおよび0,5〜49,5重量%のAを用いることができる。
【0070】
本明細書に記載のごとく製造するとき、Quillaja Saponaria Molinaの分画A、B、およびCは、それぞれ、定義可能な特性を有する化学的に密接に関連した分子の群またはファミリーを表す。それらが得られるクロマトグラフィー条件は、溶出プロフィールと生物活性についてバッチ間の再現性の一貫性が高いものである。
脂質含有粒子中の抗原
【0071】
脂質含有粒子(例えば、リポソーム、Posintros、イスコム、イスコムマトリックス、BBE、および/またはKGI)は、該粒子に統合されるか、該粒子と結合するか、または脂質含有粒子と混合された癌抗原を含むことができる。これら癌抗原は、抗癌免疫を誘発するのに用いることができる。
【0072】
腫瘍抗原は、治療下の腫瘍のものであるか、または脂質含有粒子、例えばKGIは、腫瘍細胞を殺した後に腫瘍抗原を放出させ、バイスタンダー抗原提示細胞(APC)に対する交差提示により抗腫瘍免疫反応の誘発をもたらすかまたは増強することができよう。また、BBEは、選択した腫瘍抗原を含み、統合もしくは同時投与されるか、または例えばKGI粒子が殺す腫瘍細胞により放出される自然の腫瘍抗原に対する免疫反応を誘発することができる。抗原を含む脂質含有粒子、例えばイスコムと、抗原を含まない脂質含有粒子、例えばイスコムマトリックスの両方を本発明に用いることができる。イスコムのような抗原も含む脂質含有粒子は主として樹立癌細胞に対する活性を意図する。抗原を含むイスコムマトリックスのような脂質含有粒子は、ある態様において少なくとも1の癌抗原を含みうる。別の態様によれば、それらは癌抗原を含まない。
【0073】
イスコムに組み込まれる免疫原は、イスコムマトリックスと関連させることができ、限定されるものではないが、細菌、ウイルス、マイコプラズマ、または他の微生物に対する液性および/または細胞性免疫反応を含む免疫反応を、個体、例えば(限定されるものではないが)ヒトまたは他の動物に誘導することができるあらゆる化学的存在でありうる。特異的免疫原は、タンパク質もしくはペプチド、炭化水素、多糖、リポ多糖、またはリポペプチドであるか、またはそれらのあらゆる組み合わせでありうる。
【0074】
特に、特異的免疫原は、天然タンパク質またはタンパク質断片、または合成タンパク質もしくはタンパク質断片またはペプチドを含むことができ;糖タンパク質、糖ペプチド、リポタンパク質、リポペプチド、核タンパク質、核ペプチドを含むことができ;ペプチド−ペプチドコンジュゲートを含むことができ;組換え核酸発現産物を含むことができる。
【0075】
そのような免疫原の例は、EP 0 109 942 B1に記載されており、限定されるものではないが、ウイルス性または細菌性肝炎、インフルエンザ、ジフテリア、破傷風、百日咳、はしか、おたふく風邪、風疹、ポリオ、肺炎球菌、ヘルペス、RS(呼吸器合胞体)ウイルス、ヘモフィルスインフルエンザ、クラミジア、水痘帯状疱疹ウイルス、狂犬病、またはヒト免疫不全ウイルスに対する免疫反応を誘発することができるものを含む。
【0076】
該抗原は、イスコムに組み込むか、イスコムもしくはイスコムマトリックスと結合させるか、またはイスコムおよび/またはイスコムマトリックスと混合することができる。そのようなイスコムまたはイスコムマトリックスのあらゆる混合物を用いることができる。1またはそれ以上の抗原を用いることができ、トランスポートおよびパッセンジャー抗原をEP 9600647-3(PCT/SE97/00289)に記載のごとく用いることができる。
アジュバント
【0077】
脂質含有粒子を他の成分のデリバリーシステムとして用いることができる。脂質含有粒子中に送達するかまたは脂質含有粒子と混合することができるそのような成分の1タイプがアジュバントである。すなわち、サポニン以外のさらなるアジュバントを脂質含有粒子に統合し、該粒子と結合させ、またはそれと混合することができる。アジュバント効果は癌治療で検討され、治療薬として開発中である(例えば、ホルボールエステル、ビタミンA2、およびビタミンD3)。
【0078】
本発明の該粒子は、サポニン以外の免疫刺激および増強成分、例えばリポ多糖(LPS)、脂質A、CTB、CTAまたはCTA1-DDを含むことができる。BBEおよびKGIは、他の癌細胞殺滅剤または細胞毒性物質、例えばコレラ毒素(CT)もしくはその分画、易熱性大腸菌毒素(LT)またはそのサブフラクションを含むこともできる。
【0079】
さらに、上記の全タイプのサポニンをそのようなさらなるアジュバントとして用いることができる。
【0080】
溶液またはサスペンジョンは、少なくとも1の以下のアジュバントを含むこともある:無菌希釈剤、例えば、注射用水、生理食塩水、固定油、ポリエチレングリコール、グリセロール、プロピレングリコール、またはまたは他の合成溶媒、抗菌剤、例えばベンジルアルコール、メチルパラベン、抗酸化剤、例えば、アスコルビン酸もしくは重亜硫酸ナトリウム、キレート剤、例えば、エチレンジアミン四酢酸、緩衝剤、例えば酢酸塩、クエン酸塩、またはリン酸塩、および等張性調節剤、例えば塩化ナトリウムもしくはデキストロース。非経口用製剤は、ガラス製またはプラスチック製のアンプル、ディスポーザブルシリンジ、または複数の用量容器に入れられるであろう。
【0081】
イスコムおよびイスコムマトリックス中に組み込むことができる他のアジュバントの例には、望ましい免疫調節作用を有する天然または合成のあらゆるアジュバント、例えばムラミルジペプチド(MDP)誘導体、例えば脂肪酸、置換MDP、MDPのトレオニルアナログ;DDA、ポリアニオン、例えば硫酸デキストラン、リポ多糖、例えば、サポニン(Quil A以外)がある。ワクチンアジュバントの未来の可能性(WarrenおよびChedid 1988);「無毒性モノホスホリル脂質Aの特性」(Johnson、Tomai et al. 1987);「合成イムノモジュレーターの開発状態」(BerendtおよびIves 1985);「免疫増強コンジュゲート」(Stewart-Tull 1985)、(Morein et al. 2007)。
がある。
抗癌剤
【0082】
脂質含有粒子は、抗癌剤のデリバリーシステムとして用いることもできる。抗癌剤は、脂質含有粒子中に送達されるかまたは脂質含有粒子と混合することができる。KGIおよびBBEは、特に他のメカニズムにより殺す他の抗癌剤のデリバリーシステムにも用いることができる。KGIおよびBBEは、アポトーシスをもたらす活性化−分化によるサイレントキリングに寄与する。他の治療剤は、他の癌細胞殺滅作用を有する。該組み合わせは、癌細胞を治療に耐性にする癌細胞の逆転を避けるのに確実に寄与するであろう。
【0083】
さらなる抗癌剤は、好ましくは白金配位化合物、タキサン化合物、カンプトテシン化合物、抗腫瘍ビンカアルカロイド、抗腫瘍ヌクレオシド誘導体、ナイトロジェンマスタードまたはニトロソウレアアルキル化剤、抗腫瘍アントラサイクリン誘導体、トラストズマブおよび抗腫瘍ポドフィロトキシン誘導体から選ばれる。
【0084】
本明細書で用いている用語「白金配位化合物(platinum coordination compound)」は、イオン形の白金を提供するあらゆる腫瘍細胞の増殖を阻害する白金配位化合物を表す。好ましい白金配位化合物には、シスプラチン、カルボプラチン、クロロ(ジエチレントリアミン)-塩化白金(II);ジクロロ(エチレンジアミン)-白金(II);ジアミン(1,1-シクロブタンジカルボキシラト)-白金(II)(カルボプラチン);スピロプラチン;イプロプラチン;ジアミン(2-エチルマロナト)-白金(II);(1,2-ジアミノシクロヘキサン) マロナト白金(II);(4-カルボキシフタロ)(1,2-ジアミノシクロヘキサン)白金(II);(1,2-ジアミノシクロヘキサン)-(イソシトラト)白金(II);(1,2-ジアミノシクロヘキサン)-cis-(ピルバト)白金(II);および(1,2-ジアミノシクロヘキサン)-オキサラト-白金(II);オルマプラチン、およびテトラプラチンが含まれる。
【0085】
シスプラチンは、例えばBristol Myers Squibb Corporationから商標名Platinolで、水、無菌生理食塩水、または他の適切なビークルで再構成する粉末として市販されている。他の白金配位化合物およびその医薬組成物は、市販され、および/または常套的技術により製造することができる。
【0086】
タキサン化合物は、Bristol Myers Squibbから商標名Taxolで販売されているものであってよく、ドセタキセルは、Rhone-Poulenc Rorerから商標名Taxotereで市販されている。両化合物および他のタキサン化合物は、例えばEP 253738、EP 253739およびWO 92/09589に記載しているような常套的方法、またはそれと類似の方法で製造することができる。
【0087】
カンプトテシン化合物には、イリノテカンおよびトポテカンが含まれる。イリノテカンは、例えばRhone-Poulenc Rorerから商標名Camptoで市販されており、例えば欧州特許明細書No.137145に記載のごとくまたはそれと類似の方法により製造することができる。トポテカンは例えばSmithKline Beechamから商標名Hycamtinで市販されており、例えば欧州特許明細書No. 321122に記載のごとくまたはそれと類似の方法により製造することができる。他のカンプトテシン化合物は常套的方法で、例えばイリノテカンおよびトポテカンについて上記したのと同じ方法により製造することができる。
【0088】
抗腫瘍ビンカアルカロイドには、上記ビンブラスチン、ビンクリスチンおよびビノレルビンが含まれる。ビンブラスチンは例えば注射用硫酸塩としてEli Lilly and Coから商標名Velbanで市販されており、例えばドイツ特許明細書No. 2124023に記載のごとくまたはそれと類似の方法により製造することができる。ビンクリスチンは例えば注射用硫酸塩としてEli Lilly and Coから商標名Oncovinで市販されており、例えば上記ドイツ特許明細書No. 2124023に記載のごとくまたはそれと類似の方法により製造することができる。ビノレルビンは、例えば注射用酒石酸塩としてGlaxo Wellcomeから商標名Navelbineで市販されており、例えば米国特許明細書No. 4307100に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ビンカアルカロイドは常套的方法で、例えばビノブラスチン、ビンクリスチンおよびビノレルビンについて記載したのと類似の方法により製造することができる。
【0089】
抗腫瘍ヌクレオシド誘導体には、上記5-フルオロウラシル、ゲムシタビンおよびカペシタビンが含まれる。5-フルオロウラシルは、広く市販されており、例えば米国特許No. 2802005に記載のごとく製造することができる。ゲムシタビンは例えばEli Lillyから商標名Gemzarで市販されており、例えば欧州特許出願No. 122707に記載のごとくまたはそれと類似の方法により製造することができる。
【0090】
カペシタビンは、例えばHoffman-La Rocheから商標名Xelodaで市販されており、例えば欧州特許明細書No. 698611に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ヌクレオシド誘導体は常套的方法で、例えばカペシタビンおよびゲムシタビンについて上記したのと類似の方法により製造することができる。
【0091】
ナイトロジェンマスタード化合物には、シクロホスファミドおよびクロラムブシルが含まれる。シクロホスファミドは例えばBristol-Myers Squibbから商標名Cytoxanで市販されており、例えば英国特許明細書No. 1235022に記載のごとくまたはそれと類似の方法により製造することができる。クロラムブシルは例えばGlaxo Welcomeから商標名Leukeranで市販されており、例えば米国特許明細書No. 3046301に記載のごとく、またはそれと類似の方法により製造することができる。本発明に用いる好ましいニトロソウレア化合物には、上記カルムスチンおよびロムスチンが含まれる。カルムスチンは例えばBristol-Myers Squibbから商標名BiCNUで市販されており、例えば欧州特許明細書No. 902015に記載のごとく,またはそれと類似の方法により製造することができる。ロムスチンは例えばBristol-Myers Squibbから商標名CeeNUで市販されており、例えば米国特許明細書No. 4377687に記載のごとくまたはそれと類似の方法により製造することができる。
【0092】
抗腫瘍アントラサイクリン誘導体には、上記ダウノルビシン、ドキソルビシンおよびイダルビシンが含まれる。ダウノルビシンは、例えば塩酸塩としてBedford Laboratoriesから商標名Cerubidineで市販されており、例えば米国特許明細書No. 4020270に記載のごとくまたはそれと類似の方法により製造することができる。
【0093】
ドキソルビシンは例えば塩酸塩としてAstraから市販されており、例えば米国特許明細書No. 3803124に記載のごとくまたはそれと類似の方法により製造することができる。イダルビシンは例えば塩酸塩としてPharmacia & Upjohnから商標名Idamycinで市販されており、例えば米国特許明細書No. 4046878に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍アントラサイクリン誘導体は、常套的方法で、例えばダウノルビシン、ドキソルビシンおよびイダルビシンについて上記したのと類似の方法により製造することができる。
【0094】
トラストズマブはGenentechから商標名Herceptinで市販されており、米国特許明細書No. 5821337またはPCT特許明細書WO 94/04679およびWO 92/22653に記載のごとく得ることができる。
【0095】
抗腫瘍ポドフィロトキシン誘導体には、エトポシドおよびテニポシドが含まれる。エトポシドは例えばBristol-Myers Squibbから商標名VePesidで市販されており、例えば欧州特許明細書No. 111058に記載のごとくまたはそれと類似の方法により製造することができる。テニポシドは例えばBristol-Myers Squibbから商標名Vumonで市販されており、例えばPCT特許明細書No. WO 93/02094に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ポドフィロトキシン誘導体は常套的方法で、例えばエトポシドおよびテニポシドについて上記したのと類似の方法により製造することができる。
【0096】
上記のような粗な形のサポニンまたはその分画を遊離形で、すなわち脂質含有粒子に統合しないで抗癌剤として用いることもできる。これら抗癌化合物は、脂質含有粒子、例えば、リポソーム、イスコムおよび/またはイスコムマトリックス、およびPosintrosと混合し、結合させ、または統合することができる。
【0097】
統合するときはそれらが疎水性であれば適切である。そうでなければ、EP 242380に記載のごとく脂質含有粒子に疎水性基を結合させることができる。
【0098】
非疎水性化合物、特にタンパク質またはペプチドは、それらと疎水性基を結合させることで疎水性にすることができる。
【0099】
非疎水性化合物と結合させることができる疎水性基は、好ましくは炭素数1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30の直鎖、分岐鎖、飽和、または不飽和の脂肪族鎖、または疎水性アミノ酸またはペプチド、または他の疎水性構造、例えばステロイドである。疎水性構造の長さは、タンパク質の大きさと性質に適合させる。一例として、10-15アミノ酸のペプチド(口蹄疫ウイルス)は、アミノまたはカルボキシ末端に2個のチロシンを適切に伴うことが挙げられる。分子量70,000ダルトンのタンパク質は、約20個の疎水性アミノ酸を必要とする。検査は実験的に行われる。すなわち、ある者は、特にTrp、Ile、Phe、Pro、Tyr、Leu、Valから選ばれる(特にTyr)1〜20アミノ酸、好ましくは1、2、3、4、5アミノ酸を有するペプチド;コレステロール誘導体、例えば、コリン酸、ウルソデソキシコリン酸を用いる。
【0100】
これら疎水性基は、非疎水性タンパク質または化合物と結合することができる基、例えば、カルボキシル-、アミノ-、ジスルフィド-、ヒドロキシル-、スロヒドリル-およびカルボニル基、例えば、アルデヒド基と結合する必要がある。
【0101】
結合することができる疎水性基は、好ましくは、メタン、エタン、プロパン、ブタン、ヘキサン、ヘプタン、オクタン、およびCys、Asp、Glu、Lysを含むペプチド、好ましくはオクタナル(octanal)およびTyr.Tyr.Tyr-Cys、-Asp、または-Gluのカルボキシル、アルデヒド、アミノ、ヒドロキシル、およびジスルフィド誘導体から選ばれる。結合することができる基を有する疎水性基は、溶解する物質に応じて、例えば上記可溶化剤および界面活性剤、または塩酸、酢酸、67容量%の酢酸、苛性液(caustic liquor)、アンモニアの助けをかりて水に溶解する必要がある。次に、pHを物質を沈殿させずに中性へと調整するが、疎水性基を結合させるタンパク質を変性させるpH値にならないように確認する。脂質は可溶性を増大させ得る。
【0102】
疎水性分子は、分子比10:1〜0.1:1、好ましくは1:1で非疎水性化合物に加えることができる。
【0103】
結合分子としてカルボキシル基を有する疎水性基は、水溶性カルボジイミドまたは合成無水物を介してタンパク質と結合することができる。第1の場合は、カルボキシル基をpH5でカルボジイミドで活性化し、高ホスフェート含有量の緩衝液(pH8)に溶解したタンパク質と混合する。後者の場合は、カルボキシ化合物を、チオキサンまたはアセトニトリル中のトリエチルアミンの存在下でイソブチルクロロホルメートと反応させ、得られた無水物をpH8〜9でタンパク質に加える。ヒドラジンを有するカルボキシル基をヒドラジドに変換し、これと該タンパク質中のペリオデート酸化糖単位中のアルデヒドおよびケトンが一緒になってヒドラゾン結合を得ることもできる。
【0104】
亜硝酸を有するアミノ基を低温でジアゾニウム塩に変換し、Tyr、HisおよびLysとアゾ結合を得ることができる。
【0105】
スクシン酸無水物を有する水酸基をヘミスクシネート誘導体に変換し、これをカルボキシル基として結合させることができる。
【0106】
アルデヒド基をタンパク質中のアミノ基と反応させてシッフ塩基とすることができる。
【0107】
種々のカップリング基および方法は、Journal of Immunological Methods(BlairおよびGhose 1983)、(Conradie, Govender et al. 1983)、Methods in Enzymology(Ghose, Blair et al. 1983)、およびAnalytical Biochemistry(DavisおよびPreston 1981)に記載されている(これらの内容は本明細書の一部を構成する)。
【0108】
そのように製造した疎水性基を受け取ったタンパク質、ペプチド、または化合物は、a)に記載のごとくグリコシドと複合結合する(complex-bonded)が、ここでは細胞断片を除去するための精製工程を省略することができる。
【0109】
開示した疎水性基を有する親水性タンパク質は、タンパク質を静かに変性させることにより、すなわち、約2.5の低pH、3M尿素、または70℃以上の高温で疎水性基を利用しやすくすることにより疎水性にすることができる。そのようなタンパク質は、免疫グロブリン、例えば、IgG、IgM、IgA、IgD、およびIgEでありうる。免疫グロブリンを抗イデオタイプ抗体として用いることができる。タンパク質は、(b)に記載のごとくタンパク質として精製され、次いで(a)に記載のごとくグリコシドと複合結合するが、細胞断片を除去するための精製工程は省略される。
脂質含有粒子のための標的化分子
【0110】
脂質含有粒子は、さらに癌標的化分子、例えば、癌細胞由来の表面抗原、ウイルス表面抗原およびインフルエンザ抗原を含みうる。
【0111】
本願は、脂質含有粒子、例えばKGIおよびBBE粒子が生理的低用量で多くの種々の癌細胞を殺し、またはその増殖を阻害することを示す。イスコムおよびイスコムマトリックス製剤にアジュバントとして用いるこれらのタイプの粒子も良好なバイオアベイラビリティおよびリンパ系、特に樹状細胞に対する標的化能も示す(参考としてMorein et al. 2007参照)。標的化作用をさらに増加させるために、in vivo標的化分子を種々の方法により組み込むことができる。微生物膜由来の表面分子をMorein et al.(1984)およびEP 242380に最初に記載のごとく疎水性相互作用により組み込むことができる。例えばrDNA技術により製造するかまたは合成的に製造した他の分子をWO 2002/080981およびWO 2004/030696に記載のごとく組み込むことができる。
【0112】
そのような標的化分子には、ウイルス、例えば呼吸器、例えば肺癌の標的形に親和性を有するインフルエンザおよびRSウイルス由来のエンベロープタンパク質、またはLycke N.(2004)およびMowat et al.(2001)に記載のごとくKGIまたはBBE製剤に組み込まれたコレラ毒素のAサブユニットのA1部分であるCTA1DDが含まれる。CTA1DDは、それぞれ補完的作用に寄与する3つの主要成分に合理的に設計される。CTA1は、A2およびBサブユニットから分離することにより無毒性に変換されるコレラ毒素の酵素的に活性なサブユニットである。Staphylococcus aureus由来のプロテインA由来のDDと融合すると、CTA1はB細胞を標的化する。すなわち、CTA1はB細胞リンパ肉腫に特に適している。CTA1は、免疫刺激増強のためにB細胞を標的化するためにすでにイスコムに組み込まれている。イスコムではその標的化作用に加え、それはイスコムまたはKGIまたはBBE粒子の活性化および分化作用を補う活性化および分化作用も有する。純粋な標的化作用は、おそらく他の治療用医薬と補い合うBBEまたはKGI粒子に対する標的化部分としてCTA1の代替物であるStaphylococcus aureus由来のプロテインAのDDサブユニット分子から得られる。より一般的には、モノおよびポリクローナル抗体は、EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1に記載のごとく脂質含有粒子、例えばKGIおよびBBE粒子に組み込むことができる。
他の添加剤
【0113】
本発明の組成物は、さらに医薬的に許容される担体、希釈剤、賦形剤、または添加剤を含みうる。
【0114】
適切な医薬的に許容される担体、および/または希釈剤には、あらゆるおよび全ての常套的溶媒、分散媒質、充填剤、固体担体、水性溶液、コーティング、抗菌および抗真菌剤、等張化および吸着遅延剤などが含まれる。医薬的活性物質へのそのような媒質および物質の使用は、当業者でよく知られており、例えばRemington's Pharmaceutical Sciences, 第18版, Mack Publishing Company, Pennsylvania, USA.に記載されている。如何なる常套的媒質または物質も活性物質と不適合性である場合を除き、本発明の医薬組成物へのそれらの使用が予期される。補助活性成分を該組成物に組み込むこともできる。
医薬品形態
【0115】
脂質含有粒子はヒトおよび動物にあらゆる経路で投与することができる。非経口経路を用いることができる。本明細書で用いている用語非経口的には、皮下注射、静脈内、筋肉内、無針注射のための注入技術の皮内注射−ジェット注射および経口エアロゾル投与が含まれる。
【0116】
それぞれ本質的に少なくとも1つのタイプのサポニンを含む本発明の脂質含有粒子は、混合物として組成物で、または同じまたは異なる回数で同じ投与部位または異なる投与部位に異なる組成物で別々に投与することができる。Quillaja Saponaria Molinaの異なる分画を異なるイスコム複合体およびマトリックス複合体および異なる組成物で用いることができる。
【0117】
一般的には、本発明の脂質含有粒子は、医薬的有効量で投与される。実際に投与される粒子の量は、典型的には治療する病状、選んだ投与経路、実際に投与される化合物、個々の患者の年齢、体重および反応、患者の症状の重症度などを含む関連環境に照らして医師が決定するであろう。
【0118】
ヒトに用いる用量は、含まれる他の化合物に応じて異なりうる。治療の持続時間を考慮して、用量は<50μg〜1mg/日またはそれ以上の範囲でありうる。20% KGIおよび80% BBEの混合物を含む最高に耐容性のある製剤では18gマウスに50μgの用量で投与しても副作用を生じず、必要であれば非常に高用量を用いることができる。
部分のキット
【0119】
したがって、本発明は、少なくとも2つの部分を含む部分のキットであって、1部分が、少なくとも1の、癌細胞に対する殺滅作用を有する疎水性のサポニン分画、を含む脂質含有粒子を含み、他の部分が、少なくとも1の、免疫反応、例えば抗体産生および細胞介在性免疫を刺激および調節する親水性のサポニン分画、を含むキットにも関する。
【0120】
少なくとも1の疎水性のサポニン分画を含む脂質含有粒子を含む部分が、さらに少なくとも1の親水性のサポニン分画を有する粒子を含むこともある。
【0121】
本発明の組成物およびキットは、Quillaja Saponaria Molina由来の分画以外の少なくとも1の他のアジュバントを含むこともある。これらアジュバントは、イスコムおよび/またはイスコムマトリックス複合体と混合するか、または該複合体と統合するか、またはそれ自体として遊離形で与えることができる。
治療方法
【0122】
本明細書および特許請求の範囲を通して、文脈上他の意味に解すべき場合を除き、用語「含む(包含する)(「comprise」、またはその変化、例えば「comprises」または「comprising」)」は、記載した成分もしくは成分群を含むが、如何なる他の成分もしくは成分群を排除しないことを意味すると理解される。
【0123】
本明細書に記載のすべての刊行物は本明細書の一部を構成する。本発明は以下の非限定的実施例により説明する。
【実施例1】
【0124】
KGI 1(QHC)、KGI 2(703-マトリックス・イスコム)、KGI 3(QHA〜QHCを含むすべてのQuill A分画)、およびBBE(QHA)の製剤
QHA分画がQHCに存在するアシル鎖を欠くことを示すキラヤサポニンの構造を図1.1に示す。KGI 1粒子は、図1.2に示す右領域のQHCに基づく。このサポニン分画は、図1.2に示す逆クロマトグラフィの右領域のBBE中の塩基性サポニンであるQHAよりも疎水性および溶解性である。
【0125】
KGIは、定義したキラヤサポニン分画または種々のキラヤサポニン分画の混合物から製剤化することができる。すなわち、KGI 1はQHC分画から製造され、KGI 2はQHA(7部)およびQHC(3部)の混合物から製造され、KGI 3は、非分離キラヤ分画の混合物である。また、QHBは、KGI癌殺滅粒子になることができる。BBEはQHAから製造される。それらの割合は、望む殺滅特性と低い細胞毒性特性をもたらし、分化特性を強調するよう変更させることができる。
結 果
【0126】
KGIおよびBBE粒子の形成は上記材料と方法に記載されており、イスコムの製剤化に関する研究(Morein, Sundquist et al. 1984)および後者はイスコムマトリックスに関する研究(Morein et al. 2007)に基づく。直径30〜40nmの典型的ケージ様イスコム構造を電子顕微鏡(EM)により可視化した(図1.3)。超遠心後、約20Sの沈降係数を有するケージ様イスコム構造を含む分画がEMにより観察される(「材料と方法」参照)。
結 論
【0127】
種々の薬理作用を有するが同じ形態を持つ粒子を形成し、容易にEMにより予測し、以下の実施例に記載のごとく用いる勾配遠心により定義する。これら粒子は、免疫反応/癌殺滅特性を修飾するために、そして癌療法の分野で種々の分子用の送達用粒子として用いることができる。
【実施例2】
【0128】
赤血球(RBC)および有核細胞に対する溶解作用
サポニンは、細胞溶解作用を有することがよく知られているので、癌治療の候補薬として試験されている(Wang, Z.P. 2005)。HPLCによる分画化後のキラヤサポニンパターンを図1.1に示す。本発明において、製剤KGI 1、2、および3粒子に用いるQHCは、脂肪アシル鎖により高溶解性である(図2.1)。反対に、製剤BBE、KGI 2、および3粒子に用いるQHAは、脂肪アシル鎖を欠くので、実質的に非溶解性である。RBCを用いて、分光光度法により容易に測定されるヘモグロビンのサスペンジョン液中への漏出をもたらす損傷を引き起こすその細胞膜に対する物質の溶解作用を測定する(「材料と方法」参照)。該方法は、好感度で再現性がある。有核細胞の細胞膜に対する物質の溶解作用は種々の方法で試験する必要がある。本発明者らはトリパンブルー染色を用いた。遊離KGIは、サポニンの特性を有し、細胞膜中のコレステロールと相互作用することにより溶解し、6nmの6角微細孔を生じる。色素は細胞膜のこの微細孔を通して損傷細胞に入る。この微細孔は、瞬時に細胞溶解を生じ、有核細胞を殺すであろう。すなわち、溶解濃度の遊離サポニンは、数分以内で速やかな細胞死をもたらし、例えば10分間で、数時間または数日を必要とする他の細胞殺滅メカニズム、例えばアポトーシスの溶解作用を描出する。微粒子KGIにおいて、サポニンはコレステロールと強く結合し、KGI中のサポニンが細胞膜のコレステロールと相互作用するのを抑制し、微細孔の形成および溶解作用を抑制する。
【0129】
有核癌細胞U937およびヒト血液由来の正常好中球を遊離形および微粒子形のKGIおよびBBEに曝露した。
結 果
【0130】
溶血分析の結果を表2.1に示す。BBE粒子の原料である遊離BBEは、Roennberg et.al.(Ronnberg, Fekadu et al. 1995)により報告されたGHAの用量と同様の50μg/mlまでの用量では溶解作用を生じなかった。BBE粒子は、100μg/mlまでの濃度でRBCを溶解しなかった。
【0131】
遊離KGI 1の原料であり、微粒子KGI 1中に存在するQHCは、5μg/mlの濃度でRBCを溶解した。サポニン分画(QHA、BおよびC)を含む遊離KGI3は、20μg/mlの濃度でRBCを溶解した。KGI 3粒子は、100μg/mlの濃度でRBCの溶解を生じなかった(表2.1参照)。
【0132】
表2.1 種々の遊離形のキラヤサポニンおよび特定の形、すなわち、KGIおよびBBE粒子の細胞毒性および溶血活性
【表1】
【0133】
表2.2において、KGI製剤に10分間曝露し、次いでトリパンブルーで染色した細胞の溶解作用を示す。癌細胞の処理に用いた最高濃度は50μg/mlであり、この時間中に有核細胞は溶解または殺滅されなかった。反対に、遊離形は好中球細胞は17μg/mlで、U937癌細胞は27μg/mlで有核細胞を溶解した。有核細胞は、微粒子KGI 1に曝露して10分以内では殺されず、1時間後でも殺されなかった(示さず)。
【0134】
表2.2 トリパンブルー染色で検出したU937癌細胞および好中球に対する遊離KGI 1および微粒子 KGI 1の溶解作用。細胞をKGI製剤と10分間インキュベーションした。
【表2】
考察および結論
【0135】
癌の処置に用いた化合物は正常細胞より癌細胞を選択的に殺し、癌細胞の殺滅はin vivoで標的部位に対して高濃度になる問題を考慮して低用量で有効であることが必須である。該溶解作用は、遊離形のサポニンに特徴的であり、癌殺滅に対する好ましい細胞毒性作用ではない。さらに、該遊離形は、脂溶性であり、投与部位の細胞膜と相互作用し、局所的細胞破壊をもたらし、サポニンの一部はその部位に捕捉される。KGIおよびBBE粒子は、「生理学的」用量ではRBCまたは有核細胞の細胞膜を溶解しない。実際に、試験した最高用量、すなわち50μg/mlでは溶解または即時的毒性作用は生じなかったが、非微粒子(遊離)形のキラヤサポニンは、かなり低濃度で溶解および細胞毒性作用を生じた。
【0136】
遊離形のKGIは、比較的低用量で10分間以内に有核細胞を殺し、RBCを溶解する。実施例7に示すように、KGI 1は、2μg/mlの低い生理学的用量、またはそれ以下、溶解または膜損傷濃度以下の遊離サポニン形でも癌細胞を殺す。さらに高用量の50μg/mlのKGI 1でも、癌細胞の殺滅に数時間を必要とし、溶解膜損傷作用より癌細胞殺滅に関与する別のメカニズムを強調した。細胞溶解作用は癌細胞と正常好中球細胞で同様であったことは注目される。すなわち、微粒子形は、正常細胞と癌細胞の速やかな溶解性の無差別的殺滅を回避する大きな利点がある。さらに、溶解作用は、注射部位に注射したかなりの量の化合物が捕捉されることによる局所的副作用を生じ、バイオアベイラビリティが低くなる。すなわち、それがキラヤサポニン微粒子を癌治療用にする革新的特徴である。したがって、該微粒子形も薬理学的デリバリーシステムである。
【実施例3】
【0137】
KGI 1は選択的に癌細胞を殺す
本実施例は、KGI 1粒子が正常細胞より腫瘍細胞を選択的に殺す(アラマーブルー法で測定した)ことを証明する。悪性単芽球細胞系(U937)を、正常細胞、すなわち単球由来樹状細胞(DC)と比較するための腫瘍細胞に選んだ(「材料と方法」参照)。KGI 1中の活性サポニン成分は、Quillaja Saponaria Molinaに由来する市販Quill Aから単離したQHC(遊離KGI 1)である。図3.1(Quill AのHPLCクロマトグラム)に示すように、遊離KGI、すなわち分画QHCは、分画QHA、すなわち遊離BBEと比べて疎水性が高い。QHCは、GHAのアシル鎖を欠くことによりGHAと異なり、これはQHCのより高い疎水性とその溶解作用を説明する(図3.2)。
【0138】
腫瘍細胞:Academic hospital Uppsalaから得た単芽球細胞系(THP-1、U937およびU-937-1)およびミエローマ細胞系(LP-1およびJurkat)をKGI 1に曝露した。
結 果
【0139】
材料と方法に記載のごとく製造したKGI 1は、上記および表3.1に記載の腫瘍を殺す。記載した5つの腫瘍細胞系は、0.8〜10μg/mlの範囲のLC50濃度のKGI 1により殺された(表3.1および図3.1)が、正常DCのLC50は24.3である(図3.2)。すなわち、正常細胞を殺すには30倍高濃度のKGI 1が必要である。
【0140】
表3.1 サポニン製剤による腫瘍細胞または正常細胞の増殖の阻害
【表3】
考察と結論
【0141】
本発明者らは、微粒子形のキラヤサポニン分画、KGI 1が選択的に腫瘍細胞を殺すことを確認した。この場合、単芽球細胞U937は、正常細胞を殺すのに必要な濃度より30倍低い濃度で殺された。高殺滅作用は、キラヤサポニンの逆相ダイアグラムの疎水性分画に局在し得る。これに対し、より親水性の分画QHA(図1.1および1.2)は、関連用量で癌細胞や正常細胞を殺さない。
【0142】
すなわち、KGI 1は、癌治療の候補薬であり、さらに以下の実施例で立証される。
【実施例4】
【0143】
BBEは有核細胞に無毒性である
逆相ダイアグラム由来の比較的親水性の分画QHA(遊離BBE)(図1.1および1.2)をBBE粒子に組み込み、癌細胞または正常細胞に対する殺滅作用を試験し、無毒性であることがわかった(図4.1および4.2)。
結 果
【0144】
BBE粒子は、関連用量、すなわち試験した1920μg/mlまでの濃度で癌(図4.1)または正常(図4.2)細胞を殺さなかった。
考察と結論
【0145】
本実施例は、BBE粒子が実質的に細胞毒性がない、すなわち正常および腫瘍細胞によりよく許容されることを示す。BBE粒子は、1920μg/mlまでのあらゆる濃度で腫瘍または正常細胞を殺さない(図4.1および4.2)。1つの違いは、BBE中のサポニンは脂肪族鎖を欠くことであり、これは遊離形の活性物質QHAの低溶解作用を説明する(実施例2、表2.1および2.2参照)。BBE粒子は、その調節作用により、例えばサイトカイン産生を刺激することによりKGI粒子の活性を抑えるかまたは調節するのに有用であろう。
【実施例5】
【0146】
BBEはKGIの細胞殺滅作用を阻害する
本実施例は、BBEがKGI 1の殺滅作用を阻害することを示す。最も顕著な違いは、KGI 1中のサポニンは、アシル鎖を有するが、BBE中のサポニンは該鎖を欠く(より詳細には実施例1、2、および4参照)。
結 果
【0147】
77μg/mlの一定濃度のKGI 1を、増加する濃度のBBEとインキュベーションし、U937癌細胞に適用した。KGI 1の細胞毒性作用は比10:1(KGI 1:BBE)で阻害された。阻害作用は、図5.1の曲線が示すようにレセプターにより仲介されるようである。
【0148】
阻害作用は、U937細胞に関するそのLC50がわずか0.8μg/mlであることを考えると、用いた77μg/mlの一定の高KGI 1濃度に照らして効果的である。
考察と結論
【0149】
本発明者らは、腫瘍細胞を選択的に殺すKGI 1粒子の形の物質を確認した。本発明者は、腫瘍細胞および正常細胞の両方でKGI 1による殺滅作用を阻害するBBE粒子として製剤化された別の物質を確認した(示さず)。すなわち、毒性が生じるとそれを効果的に抑えることができる腫瘍殺滅系が作出される。本実施例中の活性物質QHCおよびQHAは、異なる粒子、すなわちKGI 1およびBBE中に存在することに注目すべきである。癌細胞死を促す阻害レセプターは正常および癌細胞の活性化と分化を促すレセプターと異なるようである。
【実施例6】
【0150】
KGIおよびBBEは、1または2の異なる粒子中の活性物質の提示作用を正常および癌細胞で分析するデリバリーシステムである
実施例5は、別個のBBE粒子中の活性物質QHAは、別の活性サポニン、すなわちGHCを含むKGI 1の細胞毒性作用を阻害することを示す。この阻害効果は、1またはそれ以上のレセプターを介する阻害によるようである。
【0151】
KGI 2は、2つの活性サポニン物質QHC(KGI 1の活性物質)とQHA(BBEの活性サポニン)を有する。本実施例は、1および同じ粒子中のこれら2成分は該成分それぞれの作用を一体化し、抑えることを示す。本実施例は、別個の粒子で同時に投与した同じ成分は曝露した細胞の反応を抑え、調節することも示す。KGI 2のような粒子は、種々の割合の出発物質を含むKGI 2(703)について記載したように製造した比7:3、7.5:2.5、または9.5:0.5の異なる割合のQHA/QHCを有した。細胞の生存はアラマーブルーで測定した(「材料と方法」参照)。
【0152】
図6.2に示す実験において、KGI 2(703)を、U937癌細胞と48時間、およびヒト未成熟DCと同様にインキュベーションした(図6.3)。
【0153】
正常哺乳類細胞は動物またはヒトの生命を維持するための種々の役割を持つ。あるDCポピュレーションは単球に由来する。本実施例では、ヒト末梢血細胞をIL-4およびGMC-SFで処理し、3H biomedical(Uppsala, Sweden)から得られる単球由来未成熟DCポピュレーションを得た。本実施例で用いた詳細な情報については「材料と方法」参照。本実施例は、種々のKGIおよびBBE製剤がヒト未成熟DCを活性化して成熟させ、リンパ球と連絡をとってエフェクター細胞に分化および活性化させる活性化DCの分子であるDCマーカーのCD86を発現させることを示す。
【0154】
異なるKGIおよびBBE製剤は、KGI 1のみを含むKGI 1;1および同じ粒子中の70%のQHA(BBEの原料)および30%のQHC(KGI 1の原料)から製造されたKGI 2;Quill Aの形のキラヤサポニン分画の混合物を含むKGI 3;BBEのみがあり、BBE + KGI 1製剤は80%のBBE粒子および20% KGI 1粒子(すなわち別個の粒子中の各化合物)からなる。最初の研究は、KGI 1, KGI 2およびKGI 3については処理濃度を1μg/ml細胞培養液に、BBEおよびBBE + KGI 1については処理濃度を10μg/mlで用いるべきであることを示した。詳細は「材料と方法」参照。
結 果
【0155】
7:3〜7.5:2.5の範囲の種々の割合のQHA:QHC分画を含むKGI 2のような製剤を20〜100μg/mlで2日間インキュベーションするとU937癌細胞を殺した。活性QHC分画をQHA分画で0.5:9.5に希釈すると48時間以内のインキュベーション時間で細胞毒性活性が完全に消失した(図6.1)。これらの結果は実施例5で得られた結果と比較すべきである。
【0156】
第2実験は、QHAおよびQHC サポニン分画を1および同じ粒子にQHA:QHC 7:3の比で混合したKGI 2は、18.7μg/mlのLC50でU937腫瘍細胞を殺し(図6.2)、これはKGI 1のLC50より23倍高い。正常ヒトDCはU937癌細胞より細胞死に44倍高いKGI 2濃度を必要とした(すなわち、LC 50は826μg/ml)(図6.3)。
【0157】
表3.1に、アラマーブルー法で測定した種々の癌細胞に対する種々のKGI 粒子の癌細胞殺滅効果(正常ヒト好中球細胞についてはトリパンブルーで測定)をまとめる。一般的には、いくつかの異なる癌細胞はKGI製剤に感受性であり、U937細胞よりも遙かに感受性である。本願ではU937細胞を基礎研究のモデルに用いた。
【0158】
活性化および分化活性を図6.4に示す。1μg/mlの低生理学的用量のKGI 2は、最も高い割合のCD86陽性細胞(67.3%)を示し、1および同じ粒子、すなわちKGI 2粒子におけるKGI 1およびBBE成分の相乗効果を示した。また、KGIおよびBBE 粒子は担体送達粒子である、すなわちKGI 2は2成分を送達することを示す。BBEは、LPSと同じ割合のCD86陽性細胞を誘導したが、製剤BBE + KGI 1は65.8%を誘導した。後者の2つの製剤はKGI 2より10倍高用量、すなわち10μg/mlを必要とした。
考察と結論
【0159】
KGI 1、KGI 2、およびKGI 3のような製剤は、2および可能性としてそれ以上の異なる成分を異なる割合で組み合わせるデリバリーシステムである。本実施例において、2の異なる特性を組み合わせて完全に異なる癌殺滅作用を得た。簡単には、実施例4に記載の異なる構成要素(粒子)に閉じこめられた2の異なる化合物、すなわちKGI 1およびBBEの混合は、阻害により活性を抑える。1および同じKGI粒子(KGI 2)中の2つの物質の組み合わせは、1および同じ粒子中の異なる成分で阻害を達成することはできないので、希釈法、すなわちKGI 1リガンド密度の減少により毒性を低下させた。示した作用を癌細胞と正常細胞の両方について記録した。癌細胞に対するKGI 2の毒性はKGI 1粒子のそれに比べて何倍も低下したが、癌細胞(U937)に対しての正常ヒト細胞(DC)に対する毒性作用のゆとりは、30倍(KGI 1)〜40倍(KGI 2)に増加した。希釈効果は、細胞の殺滅を誘導するリガンド-レセプター相互作用の数が低下するので、より低い結合力で生じる。細胞毒性作用は、阻害作用を仲介するものと異なるリガンド-レセプター配置(constellation)により発揮される可能性が高い(most probably)。KGI 2粒子または他の同様の粒子において、LC50をKGI 1の0.8μg/mlからKGI 2の18.7μg/mlに増加させることによりBBEの調節能とKGI 1の殺滅効果を組み合わせて、癌細胞と正常細胞の毒性の間に大きなゆとりを保つか、または増す(例えば40倍の差)ことができる。それはin vivoで臨床的関連があるようである。KGI 1の癌細胞殺滅作用は、BBEの阻害作用により直線的に増加するが、両粒子により発揮される分化能は、今まで行われた実験からわかるように実質的に変化しない。これらの知見の柔軟性は、癌細胞の毒性および活性化-分化をアポトーシスのプログラムされた死を伴う正規化細胞挙動に合わせることを可能にする。両製剤、すなわち、同じ粒子中の2つの物質または別の粒子の2つの物質は、臨床的価値を持つようであることは注目に値する。例えば、KGI 1は、他の特性、例えばプログラムされた細胞死−アポトーシスで終わる癌細胞の分化、活性化を妨げるかもしれない低すぎる濃度で癌細胞を殺し、これはバイスタンダー効果、すなわち、直接曝露されていない近傍の細胞に対する効果に関与するかもしれない(実施例7)。例えば、免疫系において殺されたDCは近傍のDCを交差刺激する成分を放出することが十分立証される。
【0160】
アポトーシス(実施例7参照)は、個体にとっては壊死による細胞死より劇的ではなく、処置された個体に病気をもたらす細胞毒性作用を回避する。したがって、癌細胞のアポトーシスは、癌療法において魅力的である。これに関連して、殺された癌細胞は、KGIおよびBBEを含む粒子中に保持されるもののような能動免疫を増強する成分の影響下で免疫防御を開始することができる免疫原性成分を放出することを予期しうる。すなわち、KGI粒子は、細胞死を調整し、種々の抗癌活性を有する他の物質を統合する可能性を開くデリバリーシステムである。そのような物質、例えば細胞骨格の損傷を介して細胞を殺すタキソール、またはKGIまたはBBE粒子と異なるメカニズムで分化を促すビタミンAまたはDは、他の原理で作用することが好ましい。KGI粒子は、本実施例で用いる成分に限定されない送達能を示す。結論として、種々のKGI、または遊離または微粒子形のBBEのような他の化合物とのKGIの併用は、正常細胞より癌細胞を選択的に殺す(表3.1)。
【0161】
KGI 1製剤は、癌細胞U937を活性化−刺激してIL8およびアポトーシスをもたらすが、壊死により特徴づけられる細胞死はもたらさない(実施例7参照)。KGI 2および3のような他のKGI製剤は、正常および癌細胞を殺すのに遙かに高濃度が必要であり、正常細胞に対してまだ安全性に30〜40倍のゆとりがある(実施例5および6参照)。本実施例では、高い割合で正常な未成熟DCが刺激されて成熟DCとなり、CD86を発現することを示す。最も興味深いことに、活性化合物QHAまたはBBEを、1および同じ粒子中のKGI 1のサポニン成分であるQHCと組み合わせることにより相乗効果が記録された、すなわち、低用量1μg/mlのKGI 2は最も高い割合のCD86陽性細胞が誘導されたことがわかる。それ自体10μg/mlを必要とするBBEは、KGI 2製剤中、0.2μg/mlの活性KGI 1化合物と組み合わせて濃度0.8μg/ml培養液の活性化合物を用いた。これら活性成分はいずれも成分それぞれそれ自体で用いるときに必要とするよりかなり低かった。すなわち、BBE化合物の濃度は10倍以上の低く、活性KGI化合物の濃度は、効果の増加をもたらすKGI 2粒子において5倍減少した。多くのキラヤサポニン分画を含むKGI 3は、本実施例ではCD86発現を誘導せず、むしろそれを阻害した。CD86の発現に関してKGI 3の阻害作用があるか否かを確認するにはさらなる実験が必要である。
【0162】
結論として、ヒトDCの成熟により測定された予期しない相乗作用が、KGI 2粒子中の2成分が細胞と相互作用するとみられ、KGIおよびBBE粒子の、レセプターを介して細胞を標的化する担体系として価値がある可能性を強調する。KGIおよび/またはBBE 粒子による癌殺滅作用は、活性化および分化を介して作用することが提唱されるので、正常細胞で分析することも可能なはずである。癌細胞は非常に異なり、標的化特性を変化させることができる癌細胞殺滅系は癌治療の新しい可能性をもたらすことに注目すべきである。
【実施例7】
【0163】
種々の検出系を適用することによるKGI 1の癌殺滅作用の分析
微粒子形のKGI中のキラヤサポニンは、正常細胞より30〜40倍低い濃度で癌細胞を殺す(実施例3)。本実施例では、癌細胞死を、「材料と方法」に詳述したトリパンブルー染色、アラマーブルー法による酵素代謝阻害、プロピジウムアイオダイド染色により可視化した壊死変化、およびアネキシンV染色によるアポトーシスを含む種々の方法で分析した。処置した細胞のトリパンブルー染色は細胞数を確認するのに有益であり、生存性を9日間まで測定した(図7.1、7.2)。低用量のKGI 1(2μg/ml)では、より長い期間後に癌細胞死を生じたが、高用量(25μg/ml)では短期間内に死をもたらし(図7.3)、より速やかなアポトーシスを引き起こした(図7.6)。本実施例では、トリパンブルー法により測定した細胞死は、細胞が細胞周期から抜け出した後のプログラムされた細胞死、すなわちアポトーシスと相関した。細胞周期は、細胞複製の基礎であり、癌細胞では、連続的複製のために細胞周期に留まることが重要である。該周期から抜け出すことは、細胞が産生、この場合はサイトカイン産生(実施例10)に向かって活性化を開始し、最終的に本実施例で示されたプログラムされた細胞死に進みうることを意味する。本発明者らはチミジンキナーゼ(TK)試験を用いて、処置細胞が複製期、すなわち細胞周期にあるか、または該周期から抜け出し(実施例9)、産生能力および最終的にアポトーシスをもたらす経路に入ったか否かを示した。
【0164】
KGI 1の癌細胞U937を殺す能力を以下の実験においてトリパンブルー染色法(「材料と方法」参照)により分析する。
【0165】
U937細胞を2μg/ml KGI 1を含むマイクロタイタープレートに播いた。培養のあるセットにおいて、該細胞をKGI 1に9日間連続して曝露した(図7.1)。細胞数を3日間毎に同じ濃度のKGI 1を含む新鮮培地で1x106/mlに調整した。各機会に累積細胞数を計算した。培養の第2セットにおいて、3日間後にKGI 1を含まない培地に交換してKGI処置を終了した(図7.2)。細胞数を培地交換時の各機会に1x106/mlに調整した。累積細胞数を計算した。
【0166】
U937癌細胞を高用量の25μg/mlの遊離形または粒子形のKGI 1に曝露し、図7.3に示すようにサンプリングした。細胞をトリパンブルーで染色し、生存性を顕微鏡で数えた。細胞の生存性は細胞コントロールの生細胞のパーセントで表す。
【0167】
細胞毒性を比較するため、U937癌細胞を、図7.4に示すように低生理学的用量の2μg/mlの遊離または粒子形のKGI 1に曝露した。細胞をトリパンブルーで染色した。生存性を細胞コントロールの生細胞のパーセントで表す。
【0168】
U937癌細胞を、図7.5に示すように低用量の0.5μg/mlまたは2μg/mlのKGI 1に12日間曝露した。トリパンブルー染色法で染色後に細胞数を数えた。
【0169】
微粒子KGI 1のアポトーシスを誘導する能力をU937癌細胞で分析した。該細胞を、濃度2μg/mlまたは25μg/mlのKGI 1に120時間曝露した(図7.6)。アネキシンV陽性細胞(図7.6)および壊死、すなわち、プロピジウムアイオダイド(PI)染色細胞(図7.7)の数をFACSで同時に測定した(「材料と方法」参照)。U937癌細胞を、濃度2μg/ml(M2)〜50μg/ml(M50)のKGI 1に120時間曝露した。
【0170】
図7.8において、U937細胞を、濃度2μg/ml〜50μg/mlのKGI 1に120時間曝露した。細胞をサンプリングし、図に示すようにプロピジウムアイオダイドおよびアネキシンVで染色した。影響のあった細胞の割合をFACSで計算した。詳細は「材料と方法」に記載している。
結 果
【0171】
KGI 1で持続的に処理した後のU937癌細胞の生存性を図7.1および7.2に示す。低生理学的用量の2μg/mlのKGI 1による癌細胞の断続処理は、連続処理と同様に癌細胞の増殖の阻害および殺滅に有効であった。細胞数は、9日間のインキュベーション後に実質的に生細胞がなくなるまで減少した。非処理細胞数は、3日間の培養中に1x106/mlから3x106/mlに増加した。処理細胞数は3日間培養後に0.5x106/ml以下に減少し、9日間培養中にさらに減少した(図7.1および7.2)。
【0172】
高用量の25μg/mlの遊離形のKGI 1、すなわちQuill AのQHC分画は、該細胞を速やか、すなわち3時間以内に殺したが、その濃度のKGI 1粒子は高率に癌細胞を殺すにはより長い時間、すなわち24時間必要であった。
【0173】
低生理学的用量の2μg/mlの遊離形のKGI 1、すなわちQuill AのQHC分画は、60時間以内に細胞を殺さなかったが、その濃度のKGI 1粒子は24時間培養後に癌細胞を殺し始め、60時間の実験培養期間中継続し、細胞生存性は20%に低下した。この濃度で遊離非粒子形は60時間の培養期間中癌細胞数を減少させることができなかった。この濃度は細胞溶解性ではない。
【0174】
非常に低用量の0.5μg/ml KGI 1粒子は、無処置細胞に比べて細胞数を減少させたが、生理学的用量の2μg/mlのKGI 1粒子は、培養12日間以内に全ての癌細胞を殺した(図7.5)。
【0175】
プログラムされた(アポトーシス)細胞死の誘導を図7.6に示す。2μg/mlの濃度のKGIは、U937細胞のアポトーシスポピュレーションの増加をもたらし、24時間曝露後にピークレベルに達した。より高用量、すなわち25μg/mlのKGIは、アポトーシス細胞の割合をさらに増加させ、12および24時間曝露後にピークレベルに達した。
【0176】
壊死細胞の誘導(図7.7)。アポトーシスの誘導に対するKGI 1粒子の作用とは対照的に、壊死細胞死に対する効果はなかった。すなわち、種々の用量の微粒子KGI 1で処理した細胞とKGI 1に曝露していないコントロール細胞の壊死細胞の割合には差はなかった。
【0177】
図7.8は、KGI 1処理U937癌細胞が2μg/ml〜50μg/mlの濃度で120時間の実験期間中に次第にプロピジウムアイオダイド(PI)およびアネキシンVの両方に染まることを示す。濃度の増加および時間の経過と共に壊死およびアポトーシス作用の両方について染色された細胞ポピュレーションの増加を生じた。コントロールは、アポトーシスおよび壊死について共に染色された細胞の割合がわずかなことを示した。最初に、KGI 1処理細胞は、主としてアネキシンVのみに染まったが、時間と共に細胞は壊死し、二重に染色された。
考察と結論
【0178】
U937細胞の低用量(2μg/ml)のKGI 1処理は、3〜6日間後に細胞数を急激に減少させる。12日間後、実質的に残った生癌細胞はない。高用量のKGI(25μg/ml)は、36時間以内に全ての癌細胞を殺す。実施例3では、U937癌細胞は正常細胞より30〜40倍感受性が高いことを示した。癌細胞の殺滅が誘導されると(図7.2および8.1参照)、培養からKGI 1を除去しても細胞死の進行は停止しないので、回復しない(実施例4も参照)。正常細胞は、有害な症状を生じないプログラムされた死(アポトーシス)の運命を有する。本実施例では、顕著なアポトーシスがKGI 1により誘導されたことを示す。アポトーシス細胞の割合は曝露の24時間までに増加した。低用量の2μg/mlのKGI 1は、24時間インキュベーション後に最も顕著なアポトーシスを引き起こした。
【0179】
時間とともに、アネキシンV陽性細胞の割合は減少し、アネキシンVおよびPIの両方に染まる細胞の割合は増加した。KGI 1処理は、無処理培養よりかなり大きな二重陽性細胞ポピュレーションをもたらした。二重染色細胞ポピュレーションは壊死またはアポトーシス細胞に由来する。最初は、処理および非処理細胞ポピュレーションの両方で同じくPI陽性細胞の割合が低かった。したがって、KGI処理後の二重陽性細胞の割合が高いことは初期のアポトーシスポピュレーションに由来するようである。
【0180】
KGI 1に曝露後のTK活性の中断(実施例9)は、U937癌細胞がサイトカインIL-8の産生を開始する作用と一致する。IL-8産生を、実施例10に示すKGI 1により引き起こされる産生期に癌細胞を活性化するための指標として用いた。刺激作用は、KGI 1が癌細胞を殺す濃度に近い。革新的で興味深いシナリオは、刺激作用は細胞周期をはずれたKGI 1処理細胞を産生期へと、最終的プログラムされたアポトーシス死への正常産生細胞の不可避の運命へと向かわせるというものである。KGI製剤は決して細胞増殖を刺激することができないことに注意すべきである。癌細胞を殺す工程は、抗癌剤(cytostatica)または放射線照射による癌細胞の殺滅より大きな利点があり、無症状であるか、少なくとも副作用が最小限である。
【実施例8】
【0181】
KGI 1は細胞が無制御な複製に戻ることを許さない癌細胞死をもたらす
癌治療に用いる薬剤は、初期によい効果があることがあるが、連続治療後に癌細胞殺滅作用が取り消されることがあり、細胞は無制御な複製を始める。すなわち、処置した癌細胞は無制御な複製に戻らないことが必須である。実施例7は、KGI 1がアポトーシスのメカニズムを含めて癌細胞を殺すことを示す。本実施例は、KGI 1で処置したU937癌細胞の持続的培養後、より重要なことは低生理学的用量での断続処置後に癌細胞は複製に戻らないことを示す。
【0182】
U937細胞を、0.5μg/mlまたは2μg/mlのKGI 1粒子で12日間培養し、「材料と方法」に記載のごとくトリパンブルー染色により、該細胞の生存率を同じ条件下で培養した非処置細胞と比較した。
【0183】
以下の実験において(図8.2)、濃度2μg/mlのKGI 1による同期化U937細胞の処置は、72時間培養後にKGI 1含有培地をKGI 1不含新鮮培地に交換することにより中断した。3日毎に、培地を交換して細胞増殖を促進した。12日間培養後、生細胞数をトリパンブルー染色後に数えた。
結 果
【0184】
濃度2μg/mlのKGI 1に連続的に曝露したU937癌細胞の数は最初増加し、次いで1〜3日間の時点から徐々に減少した。12日間の実験期間の終わりに、生細胞10%以下が記録された(図8.1)。0.5μg/mlのKGI 1で処置後、細胞数の初期増加がみられ、次いで出発点後第3日からその半分に減少し、その後細胞数は無処置コントロール細胞より有意に低かった。コントロール細胞は、細胞数が出発時から3倍に達した。すなわち、初期複製期間後にKGI 1で細胞を処置後、生細胞数は低生理学的用量のKGI 1を用いた後に減少した。
【0185】
図8.2は、KGI 1が増殖を阻害し、癌細胞U937を殺し、該細胞が12日間の培養期間中、第3日にKGI 1を除去した後でも増殖に戻らないことを示す。
考察と結論
【0186】
実施例3は、KGI 1により発揮される癌殺滅作用を示し、考察する。本実施例から、低および生理学的用量の2μg/mlのKGI 1で処置した細胞は、処置を中断した後でも細胞増殖に戻らないと結論することができる。
【0187】
0.5μg/mlのKGI 1による処置は、無処置コントロール細胞に比べて細胞数を有意に減少させた。初期複製期間後、生細胞数は減少した。低濃度(0.5μg/ml)のKGI 1で処置した培養中の生細胞数は複製しない生存細胞を示すようである。
【0188】
結論として、低生理学的用量のKGI 1に曝露したU937癌細胞は、無制御な複製に戻らないが、細胞数は徐々に減少する。
【実施例9】
【0189】
KGI 1で処置した癌細胞は細胞周期を抜け出す
実施例7は、KGI 1がアポトーシスのプロセスを含み癌細胞を殺すことを示し、実施例8は、癌細胞の殺滅が不可逆的であることを示す。実施例10は、KGI 1が、他の薬剤では必要とするホルボール-12-ミレステート-13-アセテート(PMA)のような細胞活性化または分化剤を加えることなくU937細胞が産生期に入るのを誘導することを示す(Baldridge, Cluff et al. 2002)。細胞周期は細胞の複製に影響する(図9.1)。該周期の初期であるS期に娘細胞のDNAが構築される。DNA構築ブロックの1つにヌクレオチドチミジンがある。チミジンは、そのリン酸化に酵素チミジンキナーゼ(TK)を必要とする。すなわち、この酵素は、S期に先立って利用可能でなければならない。ここでは、TK活性を用いて、KGI 1がU937細胞の細胞周期に影響を及ぼすか、KGI 1処置細胞が該周期に留まるかまたは該周期から抜け出すかを検討した。本実施例では、細胞周期中のKGI 1処置細胞の持続性を、経時的にTK活性を検出し、TK活性と癌細胞代謝の阻害(アラマーブルー試験により記録)、殺滅(トリパンブルー排除染色により測定)、および(考察では)アポトーシスを関連づけることにより分析した。非処置細胞の細胞代謝、複製、およびTK活性を用いて該周期に残っている癌細胞を記録した。
【0190】
1x106/mlのU937細胞を本実施例で行う実験のためにマイクロタイタープレートに播いた。細胞周期中のU937細胞を同期化させる試みは、「材料と方法」に記載のごとく細胞培養液中のウシ胎児血清濃度を10%から0.5%に減らして細胞を22時間飢餓させることにより行った。TK活性を測定し、2μg/mlまたは25μg/mlの濃度の微粒子KGI 1で処理した細胞と無処置細胞の間で比較した。図9.2および9.3に記載のとごく5日間までの間隔で測定し、効果を結果に記載する。図9.4において、細胞溶解物で測定したTK活性の低下を、微粒子 KGI 1処理U937細胞の処置後に同じ濃度の遊離、すなわち非微粒子 KGI 1処理のものと比較した。106/mlの細胞を2μg/ml(M2)、10μg/ml(M10)、25μg/ml(M25)、50μg/ml(M50)で処置した後120時間毎日細胞溶解物を用いて試料を試験した。図9.5において、TK活性を、図9.4に示すように遊離および微粒子KGI 1で処置後の細胞培養液を用いて測定した。図9.6では、TK活性を、2μg/mlのKGI 1に0、2、8、18および24時間曝露した同期化U937細胞において分析した。無処置コントロールを0、8および24時間にサンプリングした。
【0191】
図9.7. 同期化U937細胞を2μg/mlのKGI 1に2、8、18および24時間曝露した。コントロールを8および24時間にサンプリングした。生細胞をトリパンブルー染色後に数えた。
【0192】
図9.8. 細胞代謝阻害(アラマーブルー)および細胞の殺滅(トリパンブルー)を、0.5μg/mlまたは2μg/mlの微粒子KGI 1または遊離KGIで24時間処置後の同期化U937細胞で測定した。
結 果
【0193】
処置細胞のTK活性は5日間の培養中無処置コントロールに比べて低下した。高用量の25μg/mlのKGI 1で処置後のTK活性の低下は、24時間に、低用量の2μg/mlのKGI 1では48時間後に顕著であった(図9.2)。処置細胞のTK活性も非処置細胞のパーセントで記録する。図9.3. 3日間後、25μg/mlのKGI 1で処置した細胞からTK活性は検出されなかった。低用量の2μg/mlのKGI 1による細胞の処置は、5日間後にTK活性を10%に減少させた。
【0194】
図9.4は、低生理学的用量の2μg/mlのKGI 1は、癌細胞を48時間曝露した後にTK活性の顕著な減少をもたらすが、72時間曝露後に最も顕著であることを示す。TK活性の減少は、遊離KGI 1処置後は顕著ではない。高用量の25または50μg/mlのKGI 1で処置後のTK活性の減少は、初期、すなわち24時間曝露後に顕著である。高用量、すなわち25および50μg/mlのKGI 1で処置した細胞は、6日間の培養中全ての時点で検出可能なTK活性がすべて消失したが、培養液中にTK活性が検出され(図9.5)、細胞膜の損傷を示唆した。これらの濃度の遊離KGI 1は溶解作用を有することに注意すべきである(実施例2、表2.1)。対照的に、微粒子KGI 1は検出可能な量のTKを細胞培養液に放出させなかった。
【0195】
図9.6は、低生理学的用量の2μg/mlのKGI 1が18および24時間処置した(すなわち、第2細胞周期の複製期に入る前の)細胞試料で記録される同期化U937細胞のTK活性を減少させることを示す。TK活性の阻害は低濃度では最初の8時間中生じなかった。
【0196】
図9.7は、生理学的低用量の2μg/mlのKGI 1は増殖を阻害し、培養中の細胞の生存性で測定した18時間曝露後に検出される癌細胞数を減少させることを示し、これは、図9.6に示すようにTK活性の低下と一致する。
【0197】
2μg/mlの濃度の微粒子および遊離KGI 1は、本実験で測定した、U937細胞を24時間後の細胞生存性と代謝を減少させた(図9.8)。0.5μg/mlの濃度の微粒子または遊離KGI 1は、24時間の処置期間後の細胞生存性を減少させた。0.5μg/mlによる細胞代謝の阻害は、遊離KGI 1処置後よりKGI 1処置後に顕著であった。一般に、微粒子KGI 1は、細胞代謝と生存性の両方で測定した癌細胞増殖のより有効な阻害剤であった。生細胞数と細胞代謝活性のよい相関もみられた。
考察と結論
【0198】
低用量の2μg/mlのKGI 1で処置したU937癌細胞は、非同期化細胞では24時間後に測定したときTK活性が減少せず、48時間後では減少したが、これはKGI 1阻害効果が第1周期の終わりであることを反映した(図9.2〜9.5)。同調化細胞ではTK活性の阻害はより早いようである(図9.6)。TK活性は、細胞増殖周期初期である。高濃度の25μg/mlのKGI 1は、TK活性をより早く減少させる。おそらく、KGI 1処置は、該細胞を細胞周期から抜け出させる。一般に、TK活性は、生理学的用量のKGI 1で5日間処置後に消失した。測定されたTK活性は、新たに産生されたチミジンキナーゼと、以前の周期からしばらくの間持続すると予想される残存するチミジンキナーゼの合計を含むことに注意すべきである。高用量の25μg/mlのKGI 1は、24時間のインキュベーション後に記録されるTK活性を50%に減少させ、高用量が細胞増殖周期のより初期に細胞増殖を妨げることを示唆する。TK活性の減少は代謝阻害(アラマーブルー分析)を反映し、それと一致し、トリパンブルー染色により測定した生細胞数を減少させる(図9.7および9.8)。同期化細胞培養の18時間KGI 1処置後の癌細胞数の減少は、第1細胞周期におけるKGI 1による阻害効果を示唆する。
【0199】
アポトーシスによる癌細胞の殺滅(実施例7)は、24時間に向かって引き起こされ、アポトーシスによる殺滅は、TK活性に対する高および低用量のKGI 1の初期作用と一致する。KGI 1誘導癌細胞殺滅の初期において、壊死細胞数は、非処置細胞数を超えなかった(実施例7参照)。PI(壊死)およびアネキシンV(アポトーシス)の両方で染色した累積細胞数は、KGI 1により引き起こされるアポトーシス細胞に大部分由来する。
【0200】
しかしながら、より高用量のKGI 1の初期作用が細胞周期のS期に先立つTK産生に直接影響を有することは排除できない。
【0201】
遊離KGI 1の癌殺滅作用は、試験したより高い濃度、すなわち25μg/mlおよび50μg/mlで特に顕著であるが、これらの濃度ではTKが培養液に放出され(図9.4)、細胞膜の損傷が示唆される。より低用量では遊離形は微粒子KGI 1よりTK活性を減少させる能力は低かった。
【0202】
高用量の25μg/mlおよび50μg/mlによるKGI 1の初期癌殺滅作用(すなわち24時間前)は、おそらくTK産生後の細胞に影響を及ぼす低濃度の2μg/mlおよび10μg/mlより細胞周期の初期に発揮される異なるメカニズムを指摘する。より高い濃度の25μg/mlおよび50μg/mlの微粒子KGI 1は、遊離形のKGI 1、すなわちQuill AのQHC分画のようには溶解性でない(実施例2)。
【0203】
結論として、微粒子KGI 1は、高用量でも一次(primary)壊死を生じること無く癌細胞を殺す。TK活性は、アポトーシス死とほぼ同時に妨げられ、アラマーブルーで測定した細胞代謝の阻害およびトリパンブルー染色で測定した細胞の殺滅と一致する。KGI 1は試験した用量で溶解作用を示さない。持続培養でも逆転はみられない。KGI 1による癌細胞殺滅作用は、細胞周期から抜け出す概念に従い、活性化および産生期、最終的にアポトーシスをもたらす。これに対して、遊離形のKGI 1、すなわちQuill AのQHC分画は、アポトーシスに加えて、壊死、高用量では溶解作用により殺す。遊離KGI 1は、低生理学的用量で用いると癌細胞の殺滅に有効性が低い。高有効濃度の遊離KGI 1は副作用を引き起こす。
【実施例10】
【0204】
KGI製剤はU937細胞にIL-8サイトカイン産生を誘導する
リポ多糖(LPS)化合物は、KGI 1の用量を超える用量を用いてU937細胞のサイトカイン産生(IL-8)を刺激することが示された。別の実験では、Baldridgeら(Baldridge, Cluff et al. 2002)は、アミノアシルグルコサミニド 4-ホスフェート(APG)がサイトカイン産生で測定したU937細胞に対するそのような刺激作用を有することをクレームした。しかしながら、その能力を達成するには、培養をホルボール-12-ミレステート-13-アセテート(PMA)で前処置して分化および活性化を促進する必要があった。本実施例は、KGI 1、2、および3が、サイトカイン産生期に入るためにさらなる活性化−分化用化合物を必要とせず、必要なKGI用量は低いことを示す。さらに、実施例7および9は、KGI 1が、U937癌細胞を、KGI 1で処置しない限り閉じこめられる細胞複製周期から抜け出させることを示す。実施例7および9は、KGI 1がU937細胞をアポトーシスにすることも示す。アポトーシスは、正常細胞がその仕事、例えばサイトカインの産生が尽きた時の最終段階である。本実施例では、U937細胞にサイトカインIL-8の産生を誘導する3つのKGI製剤、すなわちKGI 1、2、および3の能力を試験する。
【0205】
KGI 1、2、および3は、「材料と方法」に記載したように製剤化し、図10.1、10.2、および10.3に示す濃度でマイクロタイタープレート中の106/mlのU937細胞に加えた。37℃で2日間インキュベーションした後、上清中のIL-8を測定した。LC50でのIL-8産生もこれらの図に示すように計算した。
結 果
【0206】
IL-8の産生は、KGI 1についてはLC50で781 pg/ml(Fig.10.1)、KGI 2については881 pg/ml(図10.2)、およびKGI 3については916 pg/mlであった(図10.3)。異なるKGI製剤はこれらの図に示すように程度の異なる細胞毒性も生じる。
考察と結論
【0207】
U937細胞は、サイトカインを自発的に産生しない。Baldridgeら(Baldridge, Cluff et al. 2002)は、高用量のある種のAPGがU937細胞のサイトカイン産生を刺激しうる(例えば高用量の(MPA)の前処置を必要とする)ことをクレームした。本実施例は、サイトカイン産生期に入るためにKGI製剤がMPAまたは同様の処置を必要としないことを示す。さらに、実施例9は、KGI 1がU937細胞をKGI 1で処理しない限り癌細胞が捕らわれる細胞複製周期から出させることを示す。実施例4は、KGI 1がU937細胞をアポトーシスにすることを示す。アポトーシスは、正常細胞がその仕事、例えばサイトカインの産生が尽きた時の最終段階である。本実施例では、U937細胞にサイトカインIL-8の産生を誘導する3つのKGI製剤、すなわちKGI 1、2、および3の能力を試験する。結論として、種々のKGI製剤はアポトーシスに先立ってサイトカイン8の産生を誘導する。また、BBEもIL-8産生を誘導する(示さず)。
【実施例11】
【0208】
マウスにおけるKGI 1およびBBEの急性毒性
癌治療を意図する薬剤は、その大部分が、癌治療に可能性がある化合物が臨床試験に進行しないかまたは市販されるようにならない最も一般的な原因であるバイオアベイラビリティの低さと共に副作用を生じる。キラヤサポニンは動物に遊離形で50年間以上用いられてきた。腫脹、発赤、および圧痛(壊死も)の形の局所反応が副作用として現れ、用量を制限することはよく知られている。その種の副作用は、QHCをイスコムおよびイスコムマトリックス製剤に組み込んだ後は同様の用量を用いても記録されなかった。10μgの遊離QHCは、マウスに筋肉内注射すると局所反応を誘導するが、イスコムまたはKGI製剤中に組み込まれたものは誘導しない。遊離サポニンはコレステロールと相互作用して、イスコムおよびKGI製剤中に組み込まれたサポニンと結合するコレステロールの阻害作用により避けられる細胞膜の損傷を生じることも良く認識されている。本実施例では、KGI 1およびBBEの全身毒性をBALB/cマウスで試験した。急性毒性は、「材料と方法」に記載のごとくBALB/cマウスにKGI 1およびBBEを皮下投与後に試験した。マウスを4日間記録した。試験結果を表11.1にまとめ、不活発度のスコアを表11.2に示す。
【0209】
本実施例では、KGI 1およびBBEの一般毒性を、組み込まれたサポニンの遊離形、すなわちKGI 1に組み込まれたキラヤサポニンの分画CおよびBBEに組み込まれた分画Aと比較検討する。用量10、30、および50μgのKGI 1、50および100μgのBBEをBALB/cマウスに皮下注射し、4日間観察した。
【0210】
表11.1 遊離および微粒子KGIの毒性の比較
【表4】
【0211】
表11.2 不活発度のスコア
【表5】
結 果
【0212】
効果を表11.1にまとめる。10μg用量のKGI 1では、マウスで如何なる検出可能な副作用も生じなかった。30μg用量は、3段階スケールで肝臓肥大0.13、脾臓肥大0.75、脾臓の黒さ(darkness)0.65と、ほとんど反応を生じなかった。また、50μg用量のKGI 1は、脾臓の肥大(2.0)および脾臓の黒さ(1.63)を除きわずかな変化のみ生じた。KGI 1は、アジュバント作用(すなわち免疫増強)を有し、脾臓の反応は正常反応由来であることに注意すべきである。これに対して、50μg用量の遊離形分画Cは、死亡率38%および高い不活発度スコア(1.8)、脾臓肥大(2.1)、脾臓の黒さ(2.8)、および下痢を含む重度の副作用を生じた。BBE粒子は、実質的に無毒性であることが知られており、唯一注目される反応はそのアジュバント作用によるものと思われる脾臓肥大である。
考察と結論
【0213】
計算したヒト用量(100μgのKGI 1)に近い50μgのKGI 1の全身反応は、60kgのヒトを考慮した18g BALB/cマウスにおいて副作用は低度であり、死亡はみられなかった。これに対し、この用量(50μg)の、KGI 1中のサポニンである遊離形のキラヤ分画Cは死亡と下痢をもたらした。100μg用量のKGI 1も死亡を生じ、KGI 1粒子のバイオアベイラビリティは分画Cより優れていることを考慮すべきである。BBE粒子は、実質的に副作用がないことが解った。唯一の顕著な作用は、その強い免疫調節作用を反映した脾臓肥大のスコアであった。先の研究は、QS 21およびQHCのような遊離形のキラヤサポニン分画は局所反応を生じ、これは、微粒子形により、KGIおよびBBE粒子および全ての形のイスコム粒子中のサポニンと結合したコレステロールの阻害効果により回避されることを示した。すなわち、記載した粒子中のサポニンは細胞膜中のコレステロールと相互作用することができない。
【実施例12】
【0214】
Killcan(キルキャン)は、免疫刺激を生じさせる立証されたイスコムデリバリーシステムに基づく癌治療のための新規用途および新規概念を確立する。伝統的癌殺滅系は激しく、場合により細胞正常細胞にも影響を及ぼす抗癌剤、細胞正常細胞にも影響を及ぼす放射線療法、および限界がある外科手術では重度の副作用が生じる。癌療法の現代的概念は、実施例7および9で考察したような複製の生理学を妨げることである。無制御な複製は、ほとんどの癌形にとって癌細胞の生存および病原性の主な原動力である。Killcan概念では、市販製品として、標的化細胞ポピュレーションおよび標的化細胞内コンパートメント、例えばエンド−およびリソゾームおよびサイトゾルによる化合物のデリバリーシステムとして証明された良く確立されたイスコム系を用いる。イスコム系を用いることにより、Killcan概念は、調節、活性化、分化により作用する正常な細胞の発達のための推進力であり、明確な無症候性の最後であるプログラムされた細胞死(アポトーシス)をもたらす。すなわち、本発明の癌療法概念では、優れたバイオアベイラビリティ、細胞および細胞内送達の標的化、低毒性、および他の生体機能特性について十分試験された300の刊行物に記載されたイスコム技術の得られた経験を用いる。資料は、Moreinらの最初の刊行物(Morein, 1984)で始まり、最近の包括的総説(Morein, 2007)まである。
【0215】
種々のBBEおよびKGI製剤の今回の分析は、Clinical Pharmacology, Uppsala UniversityのRolf Lasson教授のグループが彼らの技術(Dhar, 1996)を用いて行った。種々の組み合わせによる殺滅、増殖阻害、および相乗効果は、実際にミエローマ、リンパ肉腫、および固形癌に由来する癌細胞に対して予期しない包括的効果であった。さらに、治療できないかまたは治療しにくい腫瘍由来の回避突然変異および癌細胞は1またはそれ以上のKGIまたはBBE製剤に感受性であり、これは実際に予期できないことであった。
ヒト腫瘍細胞系パネル
【0216】
薬剤の活性パターンを評価するため、4つの感受性親細胞系、異なる耐性メカニズムを示す5つの薬剤耐性亜系、一次耐性を有する1細胞系のヒト細胞系パネル(Dhar, 1998)を用いた。含まれた細胞系は、ミエローマ細胞系RPMI 8226/Sおよびその亜系8226/Dox40および8226/LR-5(W.S. Dalton, Dept of Medicine, Arizona Cancer Center, University of Arizona, Tucson, AZから分与)、リンパ肉腫細胞系 U-937 GTBおよびU-937-Vcr(K. Nilsson, Dept of Pathology, University of Uppsala, Swedenから分与)、SCLC細胞系NCI-H69およびその亜系H69AR、乳癌MCF-7および頸癌Hela細胞系(American Type Culture Collection;ATCC, Rockville, MD)、腎臓腺癌細胞系ACHN(ATCC)および白血病細胞系CCRF-CEMおよびその亜系CEM/VM-1(W.T. Beck, Dept of Pharmacology, College of Medicine, University of Tennessee, Memphis, TNより分与)であった。
【0217】
8226/Dox40はドキソルビシン耐性について選択し、P-糖タンパク質170(Pgp)を過剰発現する古典的MDR表現型を示す。8226/LR-5は、GSHレベルの増加に関連することが提唱されているメルファラン耐性について選択した。U-937-Vcrは、チューブリンに関連することが提唱されているビンクリスチン耐性について選択した。ドキソルビシン耐性について選択したH69ARは、MRPにより仲介されることが提唱されているMDR表現型を発現する。テニポシド耐性について選択したCEM/VM-1は、トポイソメラーゼII(topoII)に関連することが提唱されている非定型的MDRを発現する。一次耐性ACHN細胞系に対する耐性の正確なメカニズムは知られておらず、多因子的であるかもしれない。
【0218】
該細胞系は、5% CO2を含む加湿大気中、37℃で3.2項に記載の完全培養液中で増殖させた。8226/Dox40は1月に1度0.24μg/mlのドキソルビシンで処理し、8226/LR-5は1.53μg/mlのメルファランで培地交換毎に処理した。U-937-Vcrは、10ng/mlのビンクリスチン存在下で連続培養し、H69ARは、薬剤不含培地および0.46μg/mlのドキソルビシンを含む培地で交互に培養した。CEM/VM-1細胞系は、6〜8月間如何なる耐性の損失もなしに薬剤不含培地中で培養された。該細胞系の耐性パターンはコントロール実験で日常的に確認した。
【0219】
表12.1. 本試験に用いたヒト腫瘍細胞系
【表6】
試薬および薬剤
【0220】
10%不活化FCS、2mMグルタミン、50μg/mlのストレプトマイシン、および60μg/mlのペニシリンを添加した炭酸緩衝培養液RPMI-1640(HyClone, Cramlington, UK)からなる完全培地を全体で用いた。FDA(Sigma, St Louis, MO)をDMSOに溶解し、遮光してストック溶液として凍結した(-20℃)。
【0221】
被検化合物は、DMSO中の10 mMストック溶液としてDueCom ABから得た。ストック溶液をリン酸緩衝生理食塩水(PBS;Sigma Aldrich)で10倍に希釈した。BIOMEK-2000ロボットシステムを用いて薬剤をさらに希釈し(10倍連続希釈により)、384ウェルのマイクロタイタープレート(NUNC)にプレートした。
蛍光分析的微量培養細胞毒性アッセイ(FMCA)
【0222】
腫瘍細胞を5,000細胞/ウェルの細胞密度で、薬剤を入れた384ウェルのマイクロタイタープレートに播いた。蛍光分析的微量培養細胞毒性アッセイ(FMCA)は、完全な原形質膜を持つ細胞によるFDAのフルオレセインへの加水分解により生じる蛍光の測定に基づき、これは先に詳述されている[14]。プレートを5% CO2を含む加湿大気中、37℃で72時間インキュベーションした。インキュベーション期間の終わりに吸引して除去した。PBSで1回洗浄後、生理学的緩衝液に溶解した50μl/ウェルのFDA(10μg/ml)を加えた。プレートを45分間インキュベーションし、各ウェルから生じた傾向を384ウェルスキャンニング蛍光測定器で測定した。蛍光はウェル中の完全な細胞の数に比例する。
【0223】
うまくいった分析の品質基準には、コントロールウェルの蛍光信号の5回以上の平均ブランク値、コントロールウェル中の平均変動係数(CV)30%以下が含まれた。
FMCA成績の定量
【0224】
細胞の生存は、ブランクウェルの値を引いた、実験ウェルの蛍光のコントロールウェルの蛍光に対するパーセントで定義した生存指数(SI)で示す。
結 果
【0225】
種々の組み合わせのKGIおよびBBE製剤は、3種類の試験した癌、すなわち、リンパ肉腫、ミエローマ、および固形癌に由来する癌細胞を殺した。試験した異なる製剤は、表12.2に示すように癌細胞の殺滅または成長阻害の異なる局面に及ぶ。
・KGI 1は、リンパ肉腫、ミエローマ、および小肺癌細胞H69ARの回避突然変異体を含む11種の被検細胞中7種に殺滅または成長阻害作用を有した。
・BBEは、上記モデルで使用したU937細胞および試験した正常細胞に実質的に無毒性である。予期しないことに、BBEは、2細胞、すなわちBBE耐性U937/GTB由来の回避であるU937/vcrに対して殺滅的/成長阻害的であった。より注目に値するのは、ACHN細胞は、KGI 1に耐性であったが、BBE、ならびにBBEの活性成分であるGHAも含むKGI 2に感受性であった。
・KGI 3は、11種の試験細胞中6種に殺滅または成長阻害作用を有した。白血病細胞がKGI 3製剤に最も感受性であった。KGI 2と共に、KGI 3は、乳癌HELA細胞に対する効果があった唯一の被検製剤であり、他の製剤に耐性であるときにも強い効果がみられた。
・KGI 2は、11種の試験した細胞中5種に殺滅または成長阻害作用を有した。KGI 3と共に、KGI 2は、乳癌HELA細胞に効果(強い効果)があった唯一の製剤であった。
・BBE/KGI 1は、11種の試験細胞中5種に殺滅または成長阻害作用を有した。KGI 1に耐性であった一次耐性ACHN細胞および耐性ミエローマ細胞8226/dox40に対する強い効果は顕著であり、さらに、これら細胞はKGI 2にも感受性であったが、他の被検製剤には感受性でなかった。
考察と結論
【0226】
細胞調節、活性化、分化およびアポトーシス特性に基づく種々の製剤は、驚くほど広範囲の癌殺滅または成長阻害特性を示す。該成分はサポニンであるがサポニンの溶解作用を持たないという事実にも関わらず、該成分は、全ての製剤が試験した異なる癌細胞に対する作用に関してはっきりと異なるプロフィールを持つことで生じる明らかな補完的効果を有する。癌細胞に対する殺滅または成長阻害について試験した製剤による全プロフィールは、11種の試験癌細胞のうち10種に及んだ。全く非感受性の唯一の細胞は、小肺癌細胞系H69であった。回避突然変異体は程度が限られるがKGI 1およびKGI 3に感受性であった。
【0227】
結論として、正常細胞により十分許容されることが証明された十分確認された系は、広範囲の種類の癌に及ぶ強力な癌殺滅または増殖阻害特性を有することが予期せずわかった。癌を殺す粒子のよく確認された送達特性の観点から、該系はサポニン物質ならびに好ましくは他のメカニズムにより作用する他の抗癌剤との組み合わせ療法に十分適している。この予測された効果の裏付けは、癌細胞が最初に感受性であった他の化合物により引き起こされた回避突然変異体に対する種々の製剤の能力である。
【0228】
表12.2:IC50(μg/ml)で表した、リンパ性、骨髄性、および固形腫瘍(表12.1)由来の種々の癌細胞の殺滅/成長阻害。1および同じ粒子中の単独化合物、すなわちBBEおよびKGI 1、1粒子中の種々のキラヤサポニン分画の組み合わせ(KGI 3)、1および同じ粒子中の2成分(QHAおよびQHC、すなわちKGI 2)、および別の粒子中のKGI 1と混合したBBE粒子(比4:1 BBE/KGI 1)の種々の効果
【表7】
【0229】
参考文献
Baldridge, J. R., C. W. Cluff, et al.(2002). "Immunostimulatory activity of aminoalkyl glucosaminide 4-phosphates(AGPs): induction of protective innate immune responses by RC-524 and RC-529." J Endotoxin Res 8(6): 453-8.
Berendt, M. J. and J. L. Ives(1985). "Developmental status of synthetic immunomodulators." Year Immunol: 193-201.
Blair, A. H. and T. I. Ghose(1983). "Linkage of cytotoxic agents to immunoglobulins." J Immunol Methods 59(2): 129-43.
Cheng, K. T., S. E. Seltzer, et al.(1987). "The production and evaluation of contrast-carrying liposomes made with an automatic high-pressure system." Invest Radiol 22(1): 47-55.
Conradie, J. D., M. Govender, et al.(1983). "ELISA solid phase: partial denaturation of coating antibody yields a more efficient solid phase." J Immunol Methods 59(3): 289-99.
Dalsgaard(1978). "A study of the isolation and characterization of the saponin Quil A. Evaluation of its adjuvant activity, with a special reference to the application in the vaccination of cattle against foot-and-mouth disease." Acta Vet Scand Suppl(69): 7-40.
Dalsgaard, K.(1974). "Saponin adjuvants. 3. Isolation of a substance from Quillaja saponaria Molina with adjuvant activity in food-and-mouth disease vaccines." Arch Gesamte Virusforsch 44(3): 243-54.
Davis, M. T. and J. F. Preston(1981). "A simple modified carbodiimide method for conjugation of small-molecular-weight compounds to immunoglobulin G with minimal protein crosslinking." Anal Biochem 116(2): 402-7.
Dhar, S., P. Nygren, et al.(1996). "Anti-cancer drug characterisation using a human cell line panel representing defined types of drug resistance." Br J Cancer 74(6): 888-96.
Dhar, S., P. Nygren, et al.(1998). "Relationship between cytotoxic drug response patterns and activity of drug efflux transporters mediating multidrug resistance." Eur J Pharmacol 346(2-3): 315-22.
Espinet, R. G.(1951). "Nuevo tipo de vacuna antiaftosa a complejo glucovirico." Gac. Vet. 74: 1-13.
Ghose, T. I., A. H. Blair, et al.(1983). "Preparation of antibody-linked cytotoxic agents." Methods Enzymol 93: 280-333.
Gregoriadis, G., ed.(1984). Liposome Technology Vol. l, 29-31,51-67 and 79-108 (CRC Press Inc., Boca Raton, Fla.)
Gregoriadis, G., B. McCormack, et al.(1999). "Vaccine entrapment in liposomes." Methods 19(1): 156-62.
Higuchi, R. et al.(1988). An Acylated Triterpenoid Saponin From Quillaja saponaria, Phytochemistry 27(4):1165-1168.
Hope et al.(1985). Biochimica et Biophysica Acta, Vol. 812, pp. 55-65.
Hostettmann, K.; Marston, A.(1995). Saponins. Cambridge, U.K.: Cambridge Univ. Press.
Johnson, A. G., M. Tomai, et al.(1987). "Characterization of a nontoxic monophosphoryl lipid A." Rev Infect Dis 9 Suppl 5: S512-6.
Kensil, C. R., U. Patel, et al.(1991). "Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex." J Immunol 146(2): 431-7.
Kersten, G. F., A. Spiekstra, et al.(1991). "On the structure of immune-stimulating saponin-lipid complexes(iscoms)." Biochim Biophys Acta 1062(2): 165-71.
Leung A Y., and Foster S.(1996). Encyclopedia of Common Natural Ingredients Used in Food, Drugs and Cosmetics, 2nd ed., John Wiley and sons(Wiley Interscience), New York.
Lipford, G. B., H. Wagner, et al.(1994). "Vaccination with immunodominant peptides encapsulated in Quil A-containing liposomes induces peptide-specific primary CD8+ cytotoxic T cells." Vaccine 12(1): 73-80.
Lovgren, K. and B. Morein(1988). "The requirement of lipids for the formation of immunostimulating complexes(iscoms)." Biotechnol Appl Biochem 10(2): 161-72.
Lovgren, K. and B. Morein(1991). "The ISCOM: an antigen delivery system with built-in adjuvant." Mol Immunol 28(3): 285-6.
Lycke, N.(2004). "From toxin to adjuvant: the rational design of a vaccine adjuvant vector, CTA1-DD/ISCOM." Cell Microbiol 6(1): 23-32.
Ma, J., P. A. Bulger, et al.(1994). "Impact of the saponin adjuvant QS-21 and aluminium hydroxide on the immunogenicity of recombinant OspA and OspB of Borrelia burgdorferi." Vaccine 12(10): 925-32.
Madden, T. D., P. R. Harrigan, et al.(1990). "The accumulation of drugs within large unilamellar vesicles exhibiting a proton gradient: a survey." Chem Phys Lipids 53(1): 37-46.
Martin, R. G. and B. N. Ames(1961). "A method for determining the sedimentation behavior of enzymes: application to protein mixtures." J Biol Chem 236: 1372-9.
Mayer, L. D., M. J. Hope, et al.(1986). "Vesicles of variable sizes produced by a rapid extrusion procedure." Biochim Biophys Acta 858(1): 161-8.
Mayhew, E., S. Conroy, et al.(1987). "High-pressure continuous-flow system for drug entrapment in liposomes." Methods Enzymol 149: 64-77.
Mayhew, E., R. Lazo, et al.(1984). "Characterization of liposomes prepared using a microemulsifier." Biochim Biophys Acta 775(2): 169-74.
Morein, B., B. Sundquist, et al.(1984). "Iscom, a novel structure for antigenic presentation of membrane proteins from enveloped viruses." Nature 308(5958): 457-60.
Morein, B., Hu, K. et al.(2007). "New Iscoms Meet Unsettled Vaccine Demands". Vaccine Adjuvants and Delivery Systems, Chapter 9: 191-222
Mowat, A. M., A. M. Donachie, et al.(2001). "CTA1-DD-immune stimulating complexes: a novel, rationally designed combined mucosal vaccine adjuvant effective with nanogram doses of antigen." J Immunol 167(6): 3398-405.
O´Hagan, DT. "Recent developments in vaccine delivery systems". Curr Drug Targets Infect Disord. 2001 Nov;1(3):273-86.
Ramon, G.(1926). Proc d s pour accroitre la production des antitoxines, Ann Inst Pasteur, 40, 1-10.
Rouhi A.M.(1995). Chem. Eng. News 73(37): 28-35.
Ronnberg, B., M. Fekadu, et al.(1997). "Effects of carbohydrate modification of Quillaja saponaria Molina QH-B fraction on adjuvant activity, cholesterol-binding capacity and toxicity." Vaccine 15(17-18): 1820-6.
Ronnberg, B., M. Fekadu, et al.(1995). "Adjuvant activity of non-toxic Quillaja saponaria Molina components for use in ISCOM matrix." Vaccine 13(14): 1375-82.
Stewart-Tull, D. E.(1985). "Immunopotentiating conjugates." Vaccine 3(1): 40-4.
Wang, Z.P.(2005). United States Patent 20050175623
Warren, H. S. and L. A. Chedid(1988). "Future prospects for vaccine adjuvants." Crit Rev Immunol 8(2): 83-101.
【技術分野】
【0001】
本発明は、癌治療用の医薬を製造するための少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、リポソーム、イスコム(iscom)および/またはイスコムマトリックス、およびポシントロス(posintros)の使用に関する。該粒子は、補完メカニズム(complementary mechanism)を用いる癌治療用の1または数種の化合物のデリバリーシステム(送達系)でもある。
【0002】
本発明は、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を癌治療が必要な個体に投与する癌の治療方法にも関する。
【0003】
さらに、本発明は、1つの部分が癌細胞に対する殺滅効果を有する疎水性の、少なくとも1のサポニン分画を含む脂質含有粒子を含み、その他の部分が、免疫応答、例えば抗体産生および細胞介在性免疫を刺激および調節する比較的親水性の、少なくとも1のサポニン分画を有する脂質含有粒子を含む、少なくとも2の部分を含む部分のキットに関する。
【0004】
本発明は、粒子製剤中の選択したキラヤ(Quillaja)成分が腫瘍細胞を殺し、その増殖を阻害する(以後、KGIという)という発見に関する。粒子製剤は、生体的利用可能性が高いので好ましい。粒子製剤は、標的化分子を用いて製剤化することができ、また、遊離形の溶解作用により生じる副作用なしにヒトや動物によく受け入れられるように製剤化することができる。
【背景技術】
【0005】
脂質含有粒子、例えば、リポソームやイスコムは、抗原の担体やアジュバントとして記載されている。
【0006】
キラヤサポニンの免疫刺激特性は、以前から(Ramon 1926)知られており、キラヤサポニンは、1950年代から市販ワクチンに遊離形で、時にAl(OH)3と組み合わせて用いられてきた(Dalsgaard 1978)、Ma et al. (Ma、Bulger et al. 1994)、(Espinet 1951)。従来の遊離形に比べてキラヤサポニンの実質的により効率的な用途が、Morein et al.により記載されている(Morein、Sundquist et al. 1984)-イスコム技術 (EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1)、および数年後にイスコム-マトリックス技術 (Lovgren and Morein 1988)、(EP 0 436 620 B1))。イスコム技術を用いてワクチン抗原は、キラヤサポニン、コレステロール、およびリン脂質からなる40nmの複合体に組み込まれる。
【0007】
キラヤサポニンは抗癌活性を示すことが知られている。しかし、粗または分画形のサポニンは、疎水性溶解作用があり、投与部位で部分的捕捉を生じるため一般的副作用がある。したがって、遊離形のサポニンは、癌治療には現実的でない。したがって、サポニンは、有用な癌治療薬には発展しなかった。
【0008】
さらに、イスコム技術は、イスコム複合体に統合された癌抗原を含む癌ワクチンに発展した。しかし、これらワクチン中の抗原はある抗体および免疫介在反応を生じるが、イスコム複合体は、分解し、個体が将来癌細胞に侵されたときには存在しないだろう。
【0009】
今回、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、イスコムおよびイスコムマトリックスを癌治療用の医薬を製造するのに用いることができることが解った。
【0010】
癌細胞は、正常細胞より本発明の脂質およびサポニン含有粒子に30〜40倍感受性がある。脂質およびサポニン含有粒子は癌細胞を殺すアポトーシスを誘導する。
【0011】
殺滅作用は、優れたアポトーシス誘導物質(inducer)による。高濃度ではより早いアポトーシスが誘導される。本発明の脂質含有粒子で処置後、細胞は細胞周期にとどまらず、細胞は第二周期を超えない。すなわち、癌細胞の死は不可逆的である。生産期(production phase)はIL-8産生に次いでアポトーシスが生じるという事実により示される。
【0012】
癌細胞の長期培養後、癌細胞は、処置後、より顕著には断続的低生理学的用量に曝露後でも複製には戻らない。
【0013】
癌細胞の死は、「材料と方法」に記載のトリパンブルー染色、アラマーブルー法による酵素代謝阻害を含む種々の方法、プロピジウムアイオダイド染色により可視化される壊死変化、およびアネキシンV染色によるアポトーシスにより分析した。
【発明の概要】
【発明が解決しようとする課題】
【0014】
(発明の要約)
本発明は、癌治療用の医薬を製造するための、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子、例えば、リポソーム、イスコムおよび/またはイスコムマトリックス、およびポシントロスの使用に関する。
【0015】
本発明は、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を、癌治療を必要とする個体に投与する癌の治療方法にも関する。
【0016】
さらに、本発明は、1つの部分が癌細胞の殺滅作用を有する疎水性の少なくとも1のサポニン分画を含む脂質含有粒子を含み、他の部分が免疫応答、例えば、抗体産生および細胞介在性免疫を刺激および調節する親水性の少なくとも1のサポニン分画を有する脂質含有粒子を含む、少なくとも2の部分を含む部分のキットに関する。
【課題を解決するための手段】
【0017】
本発明は、選択したサポニン、例えば、キラヤ成分、特に脂質含有製剤が腫瘍細胞に対する殺滅および成長阻害作用を有するという発見に関する(以後、KGIおよびBBEという)。サポニンまたはサポニン分画を、腫瘍細胞の殺滅または成長阻害能で選択する。微粒子製剤は、高いバイオアベイラビリティにより選択され、該粒子は、粗サポニンの遊離形またはサポニンの遊離形に比べて、副作用がなく、個体、例えばヒトまたは動物によく受け入れられるように製剤化することができる。
【0018】
他のサポニンまたはサポニン分画(Quillaja Saponaria Molina(バラ科キラヤ)から得られる分画QHAを含む)は、そのようなKGI作用を示すかまたは示さないことがあるが、KGIに対する強力な中和、阻害、および平衡化作用を示すので、選択することができる。これら分画は、KGI粒子の部分として微粒子形でかまたは別個のBBE粒子で微粒子または非微粒子GHCとの相乗作用である種の癌細胞を殺すことができる。該阻害および平衡化(balancing)作用を以後BBEという。KGIおよびBBE粒子は、KGI粒子で処理し、殺された細胞から放出され、交差提示により抗原を提示することができるか、またはBBEが抗原提示細胞(APC)を直接刺激して抗腫瘍作用をもたらすことができるという事実により、腫瘍抗原に対する免疫保護反応を刺激および調節する。
【0019】
本発明をさらに以下の図面により説明する。
【図面の簡単な説明】
【0020】
【図1.1】キラヤサポニンのトリテルペノイド構造。
【図1.2】キラヤサポニンの逆相プロフィール。分画CはKGI 1の主要活性成分であり、分画AはBBEの主要活性成分である。
【図1.3】KGI 1の電子顕微鏡像。
【図2.1】QHCとQHAの構造的違い。細胞膜に対するQHCの高い溶解作用は、マークしたポイント3の右側の脂肪アシル鎖に関連する。QHAは脂肪アシル鎖を欠き、それをより親水性にすることにより溶解しにくくする。QHCおよびQHAは、ともに非分画化キラヤサポニンの天然成分である(図1.2のHPLCによる分離パターン参照)。
【図3.1】KGI 1は、低濃度で癌細胞U937を殺す(アラマーブルー(AlamarBlue)法により測定)。
【図3.2】正常ヒト樹状細胞(DC)を殺すには高用量のKGI 1が必要である。
【図4.1】BBEは、U937腫瘍細胞に無毒性である。
【図4.2】BBEは、正常ヒト樹状細胞(DC)に無毒性である。
【0021】
【図5.1】BBEとKGI 1の比10〜1において、BBEは、KGI 1による殺滅作用を阻害する。この試験はKGI 1の固定濃度(すなわち77μg/ml)およびX軸に示すBBEの増加する濃度で行った。
【図6.1】KGI 2は、1および同じ粒子中に2つのサポニン成分(種々の比(すなわち、9.5:0.5、7.5:2.5、および7.0:3.0)のQHAおよびQHC)を有する。KGI 2のU937細胞に対する癌殺滅能は、QHCの割合の増加に伴って増加する。
【図6.2】1および同じ粒子中に比7:3の2つのサポニン成分(QHAおよびQHC)を有するKGI 2は、U937癌細胞を殺すのにKGI 1より高い濃度の活性物質QHCを必要とする(実施例3中の図3.1参照)。
【図6.3】1および同じ粒子中に比7:3の2つのサポニン成分(QHAおよびQHC)を有するKGI 2は、U937癌細胞を殺すより正常ヒトDCを殺すのにKGI 1より高い濃度の活性物質QHCを必要とする(実施例3中の図3.1参照)。
【図6.4】種々のKGIおよびBBE製剤は、単球由来未成熟DCを活性化して成熟させ、分化および活性化のためにリンパ球と連絡をとり、エフェクター細胞にする活性化DCの分子であるDCマーカーCD86を発現する。
【0022】
【図7.1】KGI 1粒子はU937癌細胞の複製を阻害する。該細胞をマイクロタイタープレートに播き、次いで9日間の実験培養期間中2μg/ml (M2) KGI 1に曝露し、生細胞数をトリパンブルー染色後に顕微鏡で毎日カウントした。
【図7.2】KGI 1粒子は、KGI 1への曝露を中断した後でもU937癌細胞の複製を阻害する。該細胞を培養し、図7.1に記載のごとく2μg/ml (M2)のKGI 1に9日間曝露した。KGI 1を3日間インキュベーションした後に除去した。矢印で示す時点で培養液を交換した。コントロール細胞をKGI 1なしで培養した。
【図7.3】図7.1に記載のごとく培養したU937癌細胞を、高用量の25μg/mlの遊離形のKGI 1(すなわちQuill AのQHC分画)(F)、または粒子として25μg/mlのKGI 1(M)に曝露し、図に示すごとくサンプリングした。細胞をトリパンブルーで染色した(「材料と方法」参照)。細胞の生存性を生コントロール細胞のパーセントで表す。この高用量で、遊離形のKGI 1、すなわちQuill AのQHC分画は、急速、すなわち3時間以内に細胞を殺したが、KGI 1粒子は癌細胞を高率に殺すのにより長い時間、すなわち24時間を必要とした。
【図7.4】U937癌細胞を、低生理学的用量の2μg/mlの遊離形のKGI 1(すなわちQuill AのQHC分画)(F)、または粒子として2μg/mlのKGI 1(M)に曝露し、図に示すごとくサンプリングした。細胞をトリパンブルーで染色した(「材料と方法」参照)。生存性を生存コントロール細胞のパーセントで表す。この低用量で、遊離形のKGI 1、すなわちQuill AのQHC分画は、培養60時間内に細胞を殺さなかったが、KGI 1粒子は24時間後に癌細胞を殺し始めた。
【図7.5】非常に低用量のKGI 1粒子はU937癌細胞の増殖を阻害する。該細胞を、図に示すように、0.5μg/ml (M0.5)の低用量または2μg/ml (M2)のKGI 1に12日間曝露した。細胞数をトリパンブルー法で染色後にカウントした(「材料と方法」参照)。0.5μg/mlの低用量のKGIは該細胞数を無処置細胞に比べて減少させたが、2μg/ml用量のKGI 1粒子は、培養12日間内に全ての癌細胞を殺した。
【図7.6】KGI 1はU937癌細胞のアポトーシスを誘導した。該細胞を培養液中、2μg/ml (M2)または25μg/ml (M25)の濃度のKGI 1に120時間曝露した。アネキシンV陽性細胞数をFACSで測定した(「材料と方法」参照)。2μg/mlの濃度で、24時間曝露後にピークレベルを有するアポトーシス細胞ポピュレーションの増加が生じた。より高濃度、すなわち25μg/ml KGI 1では、さらに12および24時間曝露後にピークレベルを有するアポトーシス細胞の割合が増加した。
【図7.7】KGI 1は、壊死U937癌細胞数の増加をもたらさない。該細胞を、図に示すように培養液中2μg/ml (M2)〜50μg/ml (M50)の濃度のKGI 1に120時間曝露した。該細胞を培養し、図7.1に記載のごとくサンプリングし、プロピジウムアイオダイドで染色し、次いで壊死細胞数をFACSで測定した(「材料と方法」参照)。種々の用量のKGI 1で処理した細胞とKGI 1に曝露しなかったコントロール細胞の壊死組織の割合に差はなかった。
【図7.8】KGI 1は、アネキシンV (アポトーシス)とプロピジウムアイオダイド (壊死)の両方で染色されるU937癌細胞を徐々にもたらした。該細胞を図7.1に記載のごとく増殖させ、培養液中2μg/ml (M2)〜50μg/ml (M50)の濃度のKGI 1に120時間曝露した。該細胞をサンプリングし、図に示すようにプロピジウムアイオダイドおよびアネキシンVで染色した。罹患細胞の割合をFACSで測定した。濃度の増加は、壊死およびアポトーシス作用について染色された細胞の割合の増加をもたらした。
【0023】
【図8.1】KGI 1は、癌細胞U937の増殖を阻害し、該細胞は12日間の培養期間中増殖に復帰しない。細胞を12日間までの培養中に0.5μg/ml (M0.5)および2μg/ml (M2)のKGI 1に曝露し、図に示すように試料を回収した。細胞増殖の減少への変換点は1〜3日間細胞をKGI 1に曝露した後にみられる。トリパンブルー染色後に生細胞をカウントした。
【図8.2】KGI 1は癌細胞U937の増殖を阻害し、細胞はKGI 1除去後も増殖に戻らない。最初に細胞を22時間飢餓させ、細胞を細胞周期に同期化させた(明細書参照)。次に、細胞を培養中2μg/mlのKGI 1で12日間まで曝露し、試料を回収し、次いで培地を3日毎に交換した。第3日にKGI 1を細胞から除去した。トリパンブルー染色後に生細胞をカウントした。
【0024】
【図9.1】細胞周期の説明図。チミジンキナーゼ(TK)活性はS期、すなわちDNA複製期に先立つ。細胞増殖に対するKGI 1の阻害作用は少なくとも低用量では細胞増殖周期の後期に生じるようである。
【図9.2】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)または25μg/ml (M25)のKGI 1で5日間処理した後毎日の細胞溶解で測定した。細胞培養液を無処置細胞の活性の低下を説明するためこの実験期間中交換しなかった。処理細胞のTK活性の低下を5日間の培養中無処置コントロールと比較した。TK活性の低下は、高用量の25μg/mlのKGI 1では24時間曝露後、低用量の2μg/mlのKGI 1では2日間後に顕著になる。
【図9.3】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)または25μg/ml (M25)のKGI 1で5日間処置後の細胞溶解物で測定した。TK活性は非処理細胞のそれのパーセンテージで表す(図9.2も参照)。
【図9.4】TK活性は、106/mlのU937癌細胞を2μg/ml (M2)、10μg/ml (M10)、25μg/ml (M25)、50μg/ml (M50)の濃度の微粒子KGI 1で処置後120時間毎日の細胞溶解物で測定した。微粒子KGI 1で処理後のTK活性の減少を、Fで示した同じ濃度で試験した遊離、すなわち無微粒子KGI 1のそれと比較した。低生理学的用量では、細胞を48時間曝露後に減少が顕著になるが(M2)、遊離KGI 1では減少は顕著でない(F2)。高用量の25μg/mlまたはより高濃度のKGI 1で処理後のTK活性の減少は、24時間曝露後に顕著である。高用量の遊離KGI、すなわち、25μg/mlおよび50μg/ml細胞培養液で処理した細胞は、検出可能なTK活性を示さなかった(図9.5も参照のこと)。
【図9.5】TK活性を、106/mlのU937癌細胞を2μg/ml (M2)、10μg/ml (M10)、25μg/ml (M25)、50μg/ml (M50)の濃度の微粒子KGI 1で5日間処理した後に毎日細胞培養液で測定した。TK活性は、微粒子KGI 1で処理した細胞の培地中には検出されなかった。25μg/mlおよび50μg/mlの濃度で試験した遊離、すなわち非微粒子KGI 1処置細胞(Fで示す)は、培養液にTKを放出した(図9.4も参照)。細胞ではなく細胞培養液中にのみみられるTK活性は、漏出および細胞膜損傷を示す。
【図9.6】U937癌細胞のチミジンキナーゼ(TK)活性を、22時間、細胞を飢餓させて細胞周期を同期化させた後に測定した(本文参照)。次に、該細胞を2μg/mlのKGI 1に0、2、8、18、および24時間曝露した(本文参照)。無処置コントロールを0、8、および24時間にサンプリングした(本文参照)。KGI 1 (2μg/ml、すなわちM2)は、18および24時間処理した細胞試料を用いて記録したU937癌細胞のチミジンキナーゼ活性を低下させる。結果は、最初の8時間は低用量ではTK活性の阻害は起きなかった(18時間後は除く)ことを示す。
【図9.7】KGI 1 (2μg/ml)は、18時間の曝露後に検出された癌細胞U937の増殖を阻害する。最初に、細胞を22時間飢餓させて細胞周期を同期化させた(本文参照)。図9.6に示すように、18時間処理後の細胞増殖の低下はTK活性の減少と一致する。トリパンブルー染色後に生細胞をカウントした。
【図9.8】細胞代謝阻害(アラマーブルー)および細胞殺滅(トリパンブルー)を、細胞を22時間飢餓させて(本文参照)細胞周期を同期化させた後に測定した。次に、細胞を2μg/mlまたは0.5μg/mlのKGI 1に24時間曝露した。無処置コントロールを24時間でサンプリングした。KGI 1(2μg/ml、すなわちM2)および遊離KGI(2μg/ml、すなわちF2)は24時間処理後の細胞生存率を減少させた。濃度0.5μg/mlのKGI 1または遊離KGI 1は24時間処理後の細胞生存率を減少させた。代謝阻害は、KGI 1で処理後のほうが遊離形で処理後より顕著であった。
【0025】
【図10.1】KGI 1は、LC50濃度3μg/mlで癌細胞U937に781pg/mlのIL-8の産生を誘導した。
【図10.2】KGI 2は、LC50濃度19 μg/mlで癌細胞U937に880pg/mlのIL-8の産生を誘導した。
【図10.3】KGI 3は、14 μg/mlで癌細胞U937に917pg /mlのIL-8の産生を誘導した。
【発明を実施するための形態】
【0026】
本発明は、癌治療用の医薬を製造するための少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子の使用に関する。すなわち、本発明は、癌治療用の少なくとも1の脂質および少なくとも1のサポニンを含む医薬に関する。
【0027】
本発明によれば、該医薬は脂質粒子自体、および殺癌作用をもたらすサポニンである。遊離サポニン自体でも癌細胞を殺すことができるが、正常細胞に負の影響があることがわかった。脂質と共にかまたは脂質粒子と統合することにより、正常細胞に毒性がある遊離サポニンの濃度より30倍低い濃度で癌細胞に対する作用が得られる。
【0028】
脂質含有粒子は、リポソーム、イスコムおよび/またはイスコムマトリックス、およびポシントロスから選ぶことができる。
リポソーム
【0029】
リポソームは、一般的に、典型的には1またはそれ以上の同心層、例えば単層、二層、または多層の形の、脂溶性部分を含む両親媒性化合物の球状または回転楕円状クラスターまたは凝集物である。リポソームは、本明細書では脂質粒子ともいうことがある。リポソームは、例えばイオン性脂質および/または非イオン性脂質から製剤化することができる。非イオン性脂質から製剤化したリポソームは、ニオソーム(niosome)ということがある。少なくとも部分がカチオン性脂質またはアニオン性脂質から製剤化されたリポソームは、コクレエート(cochleate)ということがある。リポソームは、例えばLipfordおよびWagner(Lipford、Wagner et al. 1994)およびGregoriadis、G.(Gregoriadis、McCormack et al. 1999)、O´Hagan、DT(2001)に記載のごとく製造することができる。
【0030】
本発明に関連するリポソーム組成物の製造に用いるのに適合した一般的リポソーム製造技術は例えば米国特許4,728,578、4,728,575、4,737,323、4,533,254、4,162,282、4,310,505、および4,921,706;英国特許出願GB 2193095A;国際出願No. PCT/US85/01161およびPCT/US89/05040;Mayer et al.(Mayer、Hope et al. 1986);(Hope et al. 1985);Mayhew et al.(Mayhew、Conroy et al. 1987);Mayhew et al.(Mayhew、Lazo et al. 1984);Cheng et al、(Cheng、Seltzer et al. 1987);およびリポソーム技術、Gregoriadis、G.(Gregoriadis、G.編、1984)に記載されている(これらの内容は本明細書の一部を構成する)。したがって、リポソーム組成物は、当業者に知られている、例えば溶媒透析、フレンチプレス、押し出し(凍結-解凍ありまたはなし)、逆相蒸発、単純な凍結-解凍、ソニケーション、キレート透析、ホモゲナイゼーション、溶媒注入、ミクロエマルジョン化、自然形成、溶媒蒸発、溶媒透析、フレンチプレッシャー細胞技術、制御界面活性剤透析その他(それぞれ、種々の方法での該組成物の製造を含む)を含む当業者に明らかな種々の常套的リポソーム製造技術のいずれかを用いて製造することができる。例えば、Madden et al.、(Madden、Harrigan et al. 1990)参照(この内容は本明細書の一部を構成する)。
【0031】
適切な凍結-解凍技術は、例えばWO出願no. PCT/US89/05040(1989年11月8日出願)に記載されている(この内容は本明細書の一部を構成する)。凍結-解凍技術を含む方法は、リポソームの製造に関して好ましい。リポソームの製造は、溶液、例えば水性生理食塩水溶液、水性リン酸緩衝溶液、または無菌水中で行うことができる。リポソームは、振盪または渦撹拌(vortexing)を含む種々の方法で製造することもでき、これは例えば、以下の機械式振盪器を用いて達成することができる:Wig-L-Bug(登録商標)(Crescent Dental、Lyons、III.)、Mixomat(Degussa AG Frankfurt、Germany)、Capmix(Espe Fabrik Pharmazeutischer Praeparate GMBH & Co.、Seefeld、Oberay Germany)、Silamat Plus(Vivadent、Lechtenstein)、またはVibros(Quayle Dental、Sussex、England)。常套的ミクロエマルジョン化装置、例えばMicrofluidizer(登録商標)(Microfluidics、Woburn、Mass)を用いることもできる。
イスコムおよびイスコムマトリックス
【0032】
イスコムは、少なくとも1のサポニン、例えば、少なくとも1のグリコシド、少なくとも1の脂質および少なくとも1種類の抗原物質を含む。該脂質は少なくともステロール、例えば、コレステロール、所望によりホスファチジルコリンである。この複合体は、1またはそれ以上の他の免疫調節(アジュバント活性)物質を含んでいてもよく、EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1に記載のごとく製造することができる。
【0033】
本発明の組成物中のイスコムマトリックス複合体は、少なくとも1のグリコシドおよび少なくとも1の脂質を含む。脂質は少なくともステロール、例えば、コレステロール、および所望によりホスファチジルコリンである。マトリックスは、同時に投与した抗原物質に対する免疫増強作用を有する。イスコム複合体は、1またはそれ以上の他の免疫調節(アジュバント活性)物質(必ずしもサポニンではない)を含むこともでき、EP 0 436 620 B1に記載のごとく製造することができ、本特許に記載のごとく製造することができる。
【0034】
1またはそれ以上のイスコム粒子、1またはそれ以上のイスコムマトリックス粒子、またはその6ナノメーター環のあらゆるサブフラグメントを用いることができる。そのようなイスコムマトリックス、粒子またはサブフラグメントのあらゆる混合物を用いることができる。
Posintros(ポシントロス)
【0035】
Posintrosは、i)少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロール(ここで、少なくとも1の第2ステロールは、静電気的相互作用および疎水性相互作用から選ばれる相互作用により異種抗原、好ましくは核酸と接触することができ、少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロールは、少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニンと複合体を形成することができる)、およびii)少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニン(ここで、少なくとも1の第2サポニンは、静電気的相互作用および疎水性相互作用から選ばれる相互作用により遺伝的決定因子と接触することができ、少なくとも1の第1サポニンおよび/または少なくとも1の第2サポニンは少なくとも1の第1ステロールおよび/または少なくとも1の第2ステロールと複合体を形成することができる)、および所望によりiii)静電気的相互作用および疎水性相互作用から選ばれる相互作用により遺伝的決定因子と接触させるための少なくとも1の接触基(contacting group)を含む複合体である。ただし、第2ステロールが複合体およびさらに所望によりi)少なくとも1の脂溶性部分中に存在しない時は少なくとも1の接触基が存在する。
【0036】
Posintrosは、免疫刺激複合体(イスコム)として知られているものと同様のケージ様マトリックスの形の微粒子構造を採用することができる。イスコム構造に加えて、ステロールとサポニンの間の相互作用は、構成要素、例えば、格子、ハニカム、棒、および非晶質(amorphic)粒子を含む種々の異なる構造的構成要素を生じることが報告されている(これらすべての構造的構成要素は本発明に含まれる)。
Posintrosは、WO特許出願No.WO 2002/080981およびWO 2004/030696に記載されている。
脂質
【0037】
用いる脂質は、特に本願出願人の特許EP 0 109 942 B1(特に3頁)および特許EP 0 436 620 B1(7頁7〜24行)に記載のものである。特に、ステロール、例えばコレステロールおよびリン脂質、例えばホスファチジルエタノールアミンおよびホスファチジルコリンを用いる。ガングリオシドGM1である、コレラ毒素レセプターを含む、細胞結合成分、例えば糖脂質と結合する脂質含有レセプター、およびフコース化血液群抗原を用いることができる。次に、細胞結合成分は、粘液標的化分子として機能し、それを含む複合体と単純に混合することにより脂質含有物質と結合することができる。そのようなレセプターを含むイスコム複合体およびレセプターはWO 97/30728に記載されている。
サポニン
【0038】
サポニンはあらゆるサポニンでありうる。本発明のある局面によれば、サポニンは植物から得られるグリコシドである。植物グリコシドは、1またはそれ以上の糖部分を有するサポゲニンおよびプロサポゲニンから選ぶことができる。グリコシドはQuillaja Saponaria Molina由来の粗サポニン分画またはその亜分画でありうる。
【0039】
キラヤサポニンおよびその種々の分画は、50年代からアジュバントとしておよび種々のアジュバント製剤に用いられており、ほとんどの疎水性分画のうち例えばQS21は動物ワクチンおよび種々のヒト臨床試験に用いられてきた(Kersten、Spiekstra et al. 1991);(Kensil、Patel et al. 1991)。イスコムまたはイスコムマトリックスは、種々のキラヤ分画、または分画またはより粗なキラヤサポニンの種々の混合物を用いて形成されている。すべての場合、イスコムまたはイスコムマトリックス製剤は、遊離形より局所反応が少ない。最近の開発品では、すぐれた免疫増強能を有する、アジュバントとして用いたいかなる他のキラヤサポニン製剤よりはるかに耐容性のある製剤が設計された。(Morein、Sundquist et al. 1984)。この耐容性のよいキラヤサポニン製剤の成分を、本発明では癌細胞殺滅(KGI)および該効果の平衡化(BBE)のために用いる。
【0040】
サポニンは、1またはそれ以上の糖鎖と結合したアグリコンからなる分子複合体である。サポニンは、構造の部分として有機酸、例えば、酢酸、マロン酸でアシル化することができる(Hostettmann K.およびMarston A. 1995;Rouhi A.M. 1995;Leung A Y.およびFoster S. 1996)。これら複合体の分子量は、600から2000kd以上に及ぶ。疎水性アグリカンおよび親水性糖部分は両親媒性特性をもたらす。特に、トリテルペングリコシドが興味深い。そのアグリコンにより特徴付けられる他のサポニンは、ステロイドグリコシドおよびステロイドアルカロイドグリコシドである。
【0041】
粗キラヤサポニンは、1887年に初めてKobert、R.により単離された(Arch. Exp. Pathol. Pharmakol. 23: 233-272、1887)。後にDalsgaardはラヤサポニンを精製した(Dalsgaard 1974)。Higuchi、R.(Higuchi、R. 1988)は、2つの異なる位置に2つの糖部分が結合したアグルコン(トリテルペノイドキラヤ酸)を認識するキラヤサポニンの完全な構造を報告した。有用なグリコシドはEP 0 109 924 B1に記載されている。サポニンおよびトリテルペンサポニンが好ましい。それらは、Quillaja Saponaria Molina由来の生抽出物(Dalsgaard 1974)、またはそのあらゆる亜分画の形であり得る。それらはKensil et al.に対するPCT/US/88101842(Kensil、Patel et al. 1991)、(Kersten、Spiekstra et al. 1991)「Aspects of Iscoms. Analytical、Pharmaceutical and Adjuvant Properties」;論文、University of Utrecht、EP 0 362 279 B2、およびEP 0 555 276 B1に記載されている。
【0042】
用語「Quillaja Saponaria Molina由来の1サポニン分画」は、本明細書および特許請求の範囲を通してQuillaja Saponariaの半精製または限定サポニン分画または実質的に純粋な分画の一般的説明として用いる。該分画は実質的に1分画を含むイスコムまたはイスコムマトリックスの混合物を用いる時に得られる良好な結果に悪影響を与えるいかなる他の分画も含まないことが重要である。所望によりサポニン製剤は、他の化合物、例えば他のサポニン、または他のアジュバント物質を小量、例えば40重量%以下、例えば30重量%以下、25重量%以下、20重量%以下、15重量%以下、10重量%以下、7重量%以下、5重量%以下、2重量%以下、1重量%以下、0,5重量%以下、0,1重量%以下含みうる。
【0043】
本発明のサポニン分画は、WO 96/11711に記載のA、B、およびC分画、EP 0 436 620に記載のB3、B4、およびB4b分画、EP 0 3632 279 B2に記載の分画 QA1-22、Q-VAC(Nor-Feed、AS Denmark)、Quillaja Saponaria Molina Spikoside(Isconova AB、Ultunaallen 2B、756 51 Uppsala、Sweden)でありうる。EP 0 3632 279 B2の分画 QA-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-16-17-18-19-20-21、および22、特にQA-7、17-18、および21も用いることができる。それらはEP 0 3632 279 B2の特に6頁、8〜9頁の実施例1に記載されている。WO 96/11711に記載の分画A、B、およびCは、粗水性Quillaja Saponaria Molina抽出物のクロマトグラフィ分離を用い、次いで水中70%アセトニトリルで溶出して脂溶性分画を回収して得られた脂溶性分画から製造する。次に、この脂溶性分画を酸性水中25%〜60%アセトニトリルの勾配を用いて溶出するセミプレパラティブHPLCにより分離する。「分画A」または「QH-A」として本明細書で言及している分画は、約39%アセトニトリルで溶出した分画であるかまたはそれに相当する。「分画B」または「QH-B」として本明細書で言及している分画は、約47%アセトニトリルで溶出した分画であるかまたはそれに相当する。「分画C」または「QH-C」として本明細書で言及している分画は、約49%アセトニトリルで溶出した分画であるかまたはそれに相当する。
【0044】
Quillaja Saponaria Molina由来のサポニンは、2つの異なるカテゴリーに分けることができる。すなわち、
(I) より疎水性の分画は、4位に脂肪酸アシル鎖を有する。これらサポニン分画は細胞膜に小さな約12nmの穴を作ることにより強い溶解作用を示す。そのようなサポニン分画は、遊離形で不可逆的に細胞を殺すが、本発明に記載の、および中等度の濃度(Ronnberg、Fekadu et al. 1997)の微粒子形の統合抗原を有する免疫刺激複合体(ISCOM)、または統合抗原を持たない同様の粒子、すなわちイスコムマトリックスでは必ずしもそうではない。
(II) より親水性のキラヤサポニンは、細胞溶解作用を示す前に10倍またはそれ以上の高濃度で与えることができる。微粒子形ではこれらのサポニン分画は、細胞毒性作用またはin vivo毒性作用が実質的にない。
【0045】
より疎水性のおよびより親水性の微粒子形は、イスコムマトリックスといわれ、6nmの環形成サブフラグメントにより形成された40nmの球である(Ronnberg、Fekadu et al. 1995)、(Lovgren and Morein 1991)。
【0046】
脂質粒子、例えば脂肪酸を含む疎水性サポニンを含むイスコムおよびイスコムマトリックスは、本発明ではKGI 粒子(腫瘍細胞を殺し、増殖を阻害する)という。そのようなサポニンは、例えばQuillaja Saponaria Molina由来のサポニンのトリテルペノイドアグリコンの4位に脂肪アシルを含む分画、例えば、Quil Aの分画CおよびB、または分画AとBの間の領域由来の分画、およびEP 0 3632 279 B2に記載の分画15〜21であってよく、特に分画16、17、18が適している。
【0047】
脂質粒子、例えば、親水性サポニンを含むサポニンを含む、例えば脂肪酸を含むイスコムおよびイスコムマトリックスは、BBE粒子(平衡化作用および癌細胞殺滅作用を阻害(ブロック)する)と呼ばれる。Quil Aの分画4〜15、特にEP 0 3632 279 B2に記載の7〜14および分画A(QHA)が本発明では好ましい。
【0048】
脂質粒子は、少なくとも1の疎水性サポニンを含みうる。脂質粒子は少なくとも1の親水性サポニンも含みうる。少なくとも1の親水性サポニンおよび少なくとも1の疎水性サポニンは、1および同じ、または異なる脂質含有粒子中にあってよい。
【0049】
Quillaja Saponaria Molina由来のQHA分画は、細胞殺滅作用を有さず、KGI製剤(formulation)に対する強力な中和、阻害、またはより重要なのは平衡化(例えば細胞の殺滅と分化の調節の間の平衡)作用を有するので、それを選択した。阻害および平衡化作用を以後BBEと略す。KGIおよびBBE粒子は抗原に対する免疫保護反応を刺激および調節する。したがって、これら粒子は、KGI粒子により殺された細胞から放出された腫瘍抗原に対する免疫反応を刺激し、それにより交差提示は抗原を提示することができると予想される。あるいはまた、BBEは、抗腫瘍作用に対する抗原提示細胞(APC)の刺激および獲得抗腫瘍免疫反応の誘導を直接増強することができる。
【0050】
すなわち、本発明においてその作用で名付けられたKGIおよびBBE粒子は異なる特性を有し、KGIは、比較的低濃度、すなわち初代ヒトまたはネズミ細胞に対するものより30〜40倍低い濃度で不可逆的に細胞増殖を阻害し、癌細胞を殺すことができる。さらに、KGIはBBEのように組み込んだ抗原または細胞からその環境に放出されるか同時に投与された抗原に対する免疫増強作用を有する。BBE粒子は、腫瘍抗原が統合されたKGI粒子と同時に投与するか、または例えばKGIにより破壊された癌細胞から腫瘍抗原を自然に得るか、または抗原物質と同時に投与することができる(EP 0 436 620 B1参照)。
【0051】
KGIおよびBBEはいずれも少なくとも1のサポニン、例えば、グリコシドおよび少なくとも1の脂質を含む。それらがイスコムおよびイスコムマトリックスである場合は、WO/1990/003184に記載のごとく脂質コレステロールも含む。
【0052】
癌細胞に対する殺滅作用を有する疎水性サポニンを含む脂質含有粒子は、さらに親水性サポニンも含みうる。
【0053】
脂質含有粒子は、1つおよび同じ脂質含有粒子中に少なくとも2の異なるサポニン分画を含みうる。
【0054】
脂質含有粒子は、少なくとも2の異なるサポニン分画の1つが1つの脂質含有粒子中の結合複合体であり、少なくとも2の異なるサポニン分画の他のものが別の物理的に異なる脂質含有粒子中の結合複合体である、少なくとも2の異なるサポニン分画も含むことができる。
【0055】
異なるサポニンはそれぞれ親水性および疎水性サポニンでありうる。該粒子は、少なくとも分画Cまたは少なくとも分画Bまたは少なくともQuil Aの分画CとBの間のあらゆる分画、およびQuil Aの少なくとも1の他の分画を含むことができる。すなわち、1つの粒子が、分画Cのみ;分画CおよびQuil Aの少なくとも1の他の分画;分画CおよびQuil Aの1またはそれ以上の分画;分画CおよびQuil Aの分画A;粗Quil Aを含みうる。該粒子は、分画Bのみ;分画BおよびQuil Aの少なくとも1の他の分画;分画BおよびQuil Aの1またはそれ以上の分画;分画BおよびQuil Aの分画Aも含みうる。上記の分画混合物は、異なる脂質粒子または1つまたは同じ脂質粒子中に存在しうる。実施例1のKGI 1、KGI 2、およびKGI 3粒子がそのような脂質粒子の例である。
【0056】
本発明のある局面では、KGI粒子は、サポニンまたはその半精製形の混合物を含むQuil Aの粗または生抽出物、例えば、Quillaja Powder Extract(キラヤ粉末エキス)(Berghausen、USA)、Quillaja Ultra Powder(キラヤ微粉末)QP UF 300、Quillaja Ultra Powder QP UF 1000、またはVax-Sap(3つすべてNatural Responses、Chileから)を含みうる。本発明では精製サポニン分画CおよびB単独またはAとの組み合わせをKGI粒子に用い、AをBBE粒子に用いる。BおよびC分画はWO 96/11711に記載されており、B3、B4、およびB4b分画はEP 0 436 620に記載されている。分画QA1-22はEP 0 3632 279 B2に記載、Q-VAC(Nor-Feed、AS Denmark)、Quillaja Saponaria Molina Spikoside(Isconova AB、Uppsala Science Park、751 83、Uppsala、Sweden)。そのようなKGI粒子は、実施例ではKGI 3を示す。
【0057】
KGI粒子の有用なサポニンの例は、上記Quil AのQHC分画およびQuil AのQHCおよびQHA分画の異なる組み合わせである。実施例において、KGI 1粒子は分画QHCのみを含む。KGI 2粒子は、30%のQHCと70%のQHAを含む。上記Quil A(キラヤサポニン)分画の他の組み合わせすべても用いることができる。
【0058】
腫瘍細胞は急速に増殖する未分化細胞である。したがって、腫瘍細胞はある種の細胞毒性物質に感受性がある。本発明の概念は、悪性癌(良性癌を除外しない)のような急速に増殖する細胞に対する毒性および/または調節効果を有するQuillaja Saponaria Molinaの分画または分画の組み合わせに基づく微粒子形の物質(作業名:KGI 1、KGI 2、およびKGI 3、ならびにBBE)を用いることである。毒性または調節作用は細胞レベルで測定することができる。細胞毒性−細胞調節物質、すなわちサポニンは、1またはそれ以上のデリバリー粒子中に組み込まれる。別の粒子は、毒性または細胞調節作用に加えて、毒性を阻害するのに用いることもできる(作業名BBE)。すなわち、腫瘍細胞の平衡化殺滅系を作製することができる。デリバリーシステムにおいて、生存を刺激し、細胞の分化を活性化する免疫モジュレーター(調節物質)を組み込むことができる。さらなる刺激には、癌を除去する主要な免疫防御細胞種である細胞障害性T細胞の誘導が含まれうる。リンパ系の死にかけの細胞もいわゆる交差提示により生DCの刺激に寄与しうる。単球由来の単芽球様細胞はリンパ肉腫腫瘍細胞を表し、正常細胞は単球起源の樹状細胞である。
【0059】
Quillaja Saponaria Molinaから調製したKGIおよびBBE複合体の組み合わせにより、種々の補完的特性、例えばKGI粒子より低い細胞毒性、補完的細胞活性化および分化、および顕著な免疫調節作用を有する製剤を製造することができる。KGIおよびBBE粒子の作用は、癌細胞に対する細胞毒性を生じるKGIに対するBBEによる阻害作用により強調されるようにレセプター介在性である。すなわち、本発明でモデルとして用いたU937細胞に対するKGIの癌細胞殺滅作用は、BBE粒子において確認されない。しかし、BBEは、ある種の他の癌細胞に対する殺滅作用を有する。共通レセプターは、癌細胞の活性化と分化をもたらし、これは、正常細胞ではサイトカイン産生と例えばコミュニケーション分子、例えばCD86の発現をもたらすアジュバント活性と適合性または部分的に適合性である。CD86は、樹状細胞とリンパ球ポピュレーションをコミュニケーションさせ、抗原特異反応をもたらす。これの欠如は、例えば、新生児または高齢者の免疫反応を妨げる。本発明者らは、分画QHA(BBEおよびKGI 2中の成分)およびQHC(KGI 1およびKGI 2中の成分)が、先天性および後天性の未熟または不完全に分化した免疫系を有する新生児の免疫反応を活性化および分化させることを発見した(Hu et al. 2004、Morein & Hu 2007)。アポトーシスを介して癌細胞の殺滅をもたらすレセプターはKGI 粒子上に存在するが、BBE 粒子上にはみられない。しかしながら、BBEが他の癌細胞にアポトーシスを引き起こすことを排除できない。癌細胞を殺すレセプターがそれ自体でか第二のレセプターと共同して副作用を生じうることは排除できない。特に、レセプター活性またはレセプター活性の組み合わせが副作用を生じさせる種差があると考える必要がある。そのような問題に対処することができる系が望まれ、KGIとBBEの組み合わせがその可能性をもたらす。
【0060】
すなわち、細胞のKGIによる殺滅は、この作用がBBEにより阻害することができるのでレセプター介在性である。KGIおよびBBEに対する共通(阻害)レセプターは、サポニン分画GHCのみを含むKGI 1によるU937細胞のレセプター介在性殺滅のそれと異なると考えられる。そうでなければ、BBEもこれらの実験に用いたU937癌細胞殺滅物質であるはずである。BBE中の活性物質はQHAである。それがKGI2と呼ばれるQHCと同じ粒子中に存在するときは、「QHC中の殺滅レセプター」の希釈、または構造修飾により細胞の殺滅作用を抑え、細胞の殺滅に活性なレセプターとリガンドの親和性の低下をもたらす。これに対し、異なる粒子では共通タンパク質による阻害がみられる。
【0061】
本発明のサポニン製剤の使用は、耐容性が増加し、バイオアベイラビリティ、免疫原性が増加した生成物をもたらす。該製剤は、炎症、過敏症、およびアレルギー反応の調節の増大を含む免疫原性を調整する方法に用いることができる。この調整は種依存性であり、毒性、耐容性、および免疫原性に影響を及ぼしうる。
【0062】
少なくとも1の親水性サポニン、例えば、Quil A由来の分画A(例えばBBE粒子)を含む脂質含有粒子の混合物を少なくとも1の疎水性サポニン、例えばQuil Aの分画C(例えばKGI 1、KGI 2、およびKGI 3)を含む脂質含有粒子と一緒に用いると、相乗的抗癌作用が得られることがわかった。
【0063】
少なくとも1の親水性サポニンは、Quil Aの分画4、5、6、7、8、9、10、11、12、13、14、および15、特にEP 0 3632 279 B2に記載の分画7、8、9、10、11、12、13、および14、およびQuil Aの分画Aまたは粗Quil Aの1またはそれ以上でありうる。
【0064】
少なくとも1の疎水性サポニンは、例えばQuillaja Saponaria Molina由来のサポニンのトリテルペノイドアグリコンの4位に脂肪アシルを含む1またはそれ以上のサポニン、例えば、Quil Aの分画CおよびB、または分画AおよびBの間の領域由来の分画ならびにEP 0 3632 279 B2に記載の分画15、16、17、18、19、10および21であってよく、特に分画17および18がここでは適切である。
【0065】
そのような共生(symbiotic)作用のための脂質含有粒子は、イスコムおよびイスコムマトリックス粒子、リポソームおよびPosintrosから選ぶことができる。
【0066】
あらゆる比の親水性および疎水性サポニン、例えば、Quillaja Saponaria Molinaサポニンのサブフラグメントを用いることができる。また、Quillaja Saponaria Molinaの異なる親水性および疎水性サポニンサブフラグメントのあらゆる組み合わせを用いることができる。すなわち、1、2、またはそれ以上の親水性および疎水性サポニン、例えば、サブフラグメントQuillaja Saponaria Molinaサポニンは、それぞれ物理的に1つおよび同じのまたは物理的に分離した脂質含有粒子に統合することができる。
【0067】
それぞれ親水性サポニン、例えばQuillaja Saponaria Molinaの分画Aおよび疎水性サポニン、例えばQuillaja Saponaria Molinaの分画Cのそれらの含有量に基づき異なる脂質含有粒子、例えば、イスコム、イスコムマトリックス複合体、リポソームまたはPosintrosの重量%のあらゆる組み合わせを用いることができる。該混合物は、0.1〜99.9重量%、5〜95重量%、10〜90重量%、15〜85重量%、20〜80重量%、25〜75重量%、30〜70重量%、35〜65重量%、40〜60重量%、45〜55重量%、40〜60重量%、50〜50重量%、55〜45重量%、60〜40重量%、65〜35重量%、70〜30重量%、75〜25重量%、80〜20重量%、85〜15重量%、90〜10重量%、95〜05重量%の脂質含有粒子、例えば親水性サポニン、例えばQuillaja Saponaria Molinaの分画Aを含むイスコム複合体、および残りをそれぞれ100%にする親水性および疎水性サポニン、例えばイスコム複合体中のQuillaja Saponaria Molinaの分画AおよびCの合計の含有量を計算して脂質含有粒子、例えば疎水性サポニン、例えばQuillaja Saponaria Molinaの分画Cを含むイスコム複合体を含むことができる。これは、親水性および疎水性サポニンの両方を含む脂質含有粒子、または疎水性または親水性サポニンのみを含む脂質含有粒子の混合物に適用される。
【0068】
すなわち、脂質含有粒子は、75%〜99.5重量%の親水性サポニン、例えばQuil Aの分画A、および0.5%〜25重量%の疎水性サポニン、例えばQuil Aの分画C;80%〜95%の親水性サポニンおよび5〜20%の疎水性サポニン;85%〜90%の親水性サポニンおよび10〜15%の疎水性サポニン、例えば、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、96%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99,5重量%の親水性サポニン、例えば分画A、および0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%、25重量%の疎水性サポニン、例えば分画Cを含むことができる。
【0069】
上記のすべての間隔を、あらゆる種類のヒトまたは動物種に投与するための製剤中のQuillaja Saponaria Molinaのあらゆる分画のあらゆる組み合わせに用いることができる。本発明の製剤を投与することができる動物種の例には、コンパニオンアニマル、例えばネコ、イヌ、ウマ、鳥、例えばオウム、産業的に重要な種、例えばウシ(例えばウシ種)、ブタ、ヒツジ、ヤギがある。好ましくは、50重量%以上の分画Cを他のあらゆる分画と組み合わせて、特に分画Aと組み合わせて用いる。すなわち、50.5〜99.5重量%のCおよび0,5〜49,5重量%のAを用いることができる。
【0070】
本明細書に記載のごとく製造するとき、Quillaja Saponaria Molinaの分画A、B、およびCは、それぞれ、定義可能な特性を有する化学的に密接に関連した分子の群またはファミリーを表す。それらが得られるクロマトグラフィー条件は、溶出プロフィールと生物活性についてバッチ間の再現性の一貫性が高いものである。
脂質含有粒子中の抗原
【0071】
脂質含有粒子(例えば、リポソーム、Posintros、イスコム、イスコムマトリックス、BBE、および/またはKGI)は、該粒子に統合されるか、該粒子と結合するか、または脂質含有粒子と混合された癌抗原を含むことができる。これら癌抗原は、抗癌免疫を誘発するのに用いることができる。
【0072】
腫瘍抗原は、治療下の腫瘍のものであるか、または脂質含有粒子、例えばKGIは、腫瘍細胞を殺した後に腫瘍抗原を放出させ、バイスタンダー抗原提示細胞(APC)に対する交差提示により抗腫瘍免疫反応の誘発をもたらすかまたは増強することができよう。また、BBEは、選択した腫瘍抗原を含み、統合もしくは同時投与されるか、または例えばKGI粒子が殺す腫瘍細胞により放出される自然の腫瘍抗原に対する免疫反応を誘発することができる。抗原を含む脂質含有粒子、例えばイスコムと、抗原を含まない脂質含有粒子、例えばイスコムマトリックスの両方を本発明に用いることができる。イスコムのような抗原も含む脂質含有粒子は主として樹立癌細胞に対する活性を意図する。抗原を含むイスコムマトリックスのような脂質含有粒子は、ある態様において少なくとも1の癌抗原を含みうる。別の態様によれば、それらは癌抗原を含まない。
【0073】
イスコムに組み込まれる免疫原は、イスコムマトリックスと関連させることができ、限定されるものではないが、細菌、ウイルス、マイコプラズマ、または他の微生物に対する液性および/または細胞性免疫反応を含む免疫反応を、個体、例えば(限定されるものではないが)ヒトまたは他の動物に誘導することができるあらゆる化学的存在でありうる。特異的免疫原は、タンパク質もしくはペプチド、炭化水素、多糖、リポ多糖、またはリポペプチドであるか、またはそれらのあらゆる組み合わせでありうる。
【0074】
特に、特異的免疫原は、天然タンパク質またはタンパク質断片、または合成タンパク質もしくはタンパク質断片またはペプチドを含むことができ;糖タンパク質、糖ペプチド、リポタンパク質、リポペプチド、核タンパク質、核ペプチドを含むことができ;ペプチド−ペプチドコンジュゲートを含むことができ;組換え核酸発現産物を含むことができる。
【0075】
そのような免疫原の例は、EP 0 109 942 B1に記載されており、限定されるものではないが、ウイルス性または細菌性肝炎、インフルエンザ、ジフテリア、破傷風、百日咳、はしか、おたふく風邪、風疹、ポリオ、肺炎球菌、ヘルペス、RS(呼吸器合胞体)ウイルス、ヘモフィルスインフルエンザ、クラミジア、水痘帯状疱疹ウイルス、狂犬病、またはヒト免疫不全ウイルスに対する免疫反応を誘発することができるものを含む。
【0076】
該抗原は、イスコムに組み込むか、イスコムもしくはイスコムマトリックスと結合させるか、またはイスコムおよび/またはイスコムマトリックスと混合することができる。そのようなイスコムまたはイスコムマトリックスのあらゆる混合物を用いることができる。1またはそれ以上の抗原を用いることができ、トランスポートおよびパッセンジャー抗原をEP 9600647-3(PCT/SE97/00289)に記載のごとく用いることができる。
アジュバント
【0077】
脂質含有粒子を他の成分のデリバリーシステムとして用いることができる。脂質含有粒子中に送達するかまたは脂質含有粒子と混合することができるそのような成分の1タイプがアジュバントである。すなわち、サポニン以外のさらなるアジュバントを脂質含有粒子に統合し、該粒子と結合させ、またはそれと混合することができる。アジュバント効果は癌治療で検討され、治療薬として開発中である(例えば、ホルボールエステル、ビタミンA2、およびビタミンD3)。
【0078】
本発明の該粒子は、サポニン以外の免疫刺激および増強成分、例えばリポ多糖(LPS)、脂質A、CTB、CTAまたはCTA1-DDを含むことができる。BBEおよびKGIは、他の癌細胞殺滅剤または細胞毒性物質、例えばコレラ毒素(CT)もしくはその分画、易熱性大腸菌毒素(LT)またはそのサブフラクションを含むこともできる。
【0079】
さらに、上記の全タイプのサポニンをそのようなさらなるアジュバントとして用いることができる。
【0080】
溶液またはサスペンジョンは、少なくとも1の以下のアジュバントを含むこともある:無菌希釈剤、例えば、注射用水、生理食塩水、固定油、ポリエチレングリコール、グリセロール、プロピレングリコール、またはまたは他の合成溶媒、抗菌剤、例えばベンジルアルコール、メチルパラベン、抗酸化剤、例えば、アスコルビン酸もしくは重亜硫酸ナトリウム、キレート剤、例えば、エチレンジアミン四酢酸、緩衝剤、例えば酢酸塩、クエン酸塩、またはリン酸塩、および等張性調節剤、例えば塩化ナトリウムもしくはデキストロース。非経口用製剤は、ガラス製またはプラスチック製のアンプル、ディスポーザブルシリンジ、または複数の用量容器に入れられるであろう。
【0081】
イスコムおよびイスコムマトリックス中に組み込むことができる他のアジュバントの例には、望ましい免疫調節作用を有する天然または合成のあらゆるアジュバント、例えばムラミルジペプチド(MDP)誘導体、例えば脂肪酸、置換MDP、MDPのトレオニルアナログ;DDA、ポリアニオン、例えば硫酸デキストラン、リポ多糖、例えば、サポニン(Quil A以外)がある。ワクチンアジュバントの未来の可能性(WarrenおよびChedid 1988);「無毒性モノホスホリル脂質Aの特性」(Johnson、Tomai et al. 1987);「合成イムノモジュレーターの開発状態」(BerendtおよびIves 1985);「免疫増強コンジュゲート」(Stewart-Tull 1985)、(Morein et al. 2007)。
がある。
抗癌剤
【0082】
脂質含有粒子は、抗癌剤のデリバリーシステムとして用いることもできる。抗癌剤は、脂質含有粒子中に送達されるかまたは脂質含有粒子と混合することができる。KGIおよびBBEは、特に他のメカニズムにより殺す他の抗癌剤のデリバリーシステムにも用いることができる。KGIおよびBBEは、アポトーシスをもたらす活性化−分化によるサイレントキリングに寄与する。他の治療剤は、他の癌細胞殺滅作用を有する。該組み合わせは、癌細胞を治療に耐性にする癌細胞の逆転を避けるのに確実に寄与するであろう。
【0083】
さらなる抗癌剤は、好ましくは白金配位化合物、タキサン化合物、カンプトテシン化合物、抗腫瘍ビンカアルカロイド、抗腫瘍ヌクレオシド誘導体、ナイトロジェンマスタードまたはニトロソウレアアルキル化剤、抗腫瘍アントラサイクリン誘導体、トラストズマブおよび抗腫瘍ポドフィロトキシン誘導体から選ばれる。
【0084】
本明細書で用いている用語「白金配位化合物(platinum coordination compound)」は、イオン形の白金を提供するあらゆる腫瘍細胞の増殖を阻害する白金配位化合物を表す。好ましい白金配位化合物には、シスプラチン、カルボプラチン、クロロ(ジエチレントリアミン)-塩化白金(II);ジクロロ(エチレンジアミン)-白金(II);ジアミン(1,1-シクロブタンジカルボキシラト)-白金(II)(カルボプラチン);スピロプラチン;イプロプラチン;ジアミン(2-エチルマロナト)-白金(II);(1,2-ジアミノシクロヘキサン) マロナト白金(II);(4-カルボキシフタロ)(1,2-ジアミノシクロヘキサン)白金(II);(1,2-ジアミノシクロヘキサン)-(イソシトラト)白金(II);(1,2-ジアミノシクロヘキサン)-cis-(ピルバト)白金(II);および(1,2-ジアミノシクロヘキサン)-オキサラト-白金(II);オルマプラチン、およびテトラプラチンが含まれる。
【0085】
シスプラチンは、例えばBristol Myers Squibb Corporationから商標名Platinolで、水、無菌生理食塩水、または他の適切なビークルで再構成する粉末として市販されている。他の白金配位化合物およびその医薬組成物は、市販され、および/または常套的技術により製造することができる。
【0086】
タキサン化合物は、Bristol Myers Squibbから商標名Taxolで販売されているものであってよく、ドセタキセルは、Rhone-Poulenc Rorerから商標名Taxotereで市販されている。両化合物および他のタキサン化合物は、例えばEP 253738、EP 253739およびWO 92/09589に記載しているような常套的方法、またはそれと類似の方法で製造することができる。
【0087】
カンプトテシン化合物には、イリノテカンおよびトポテカンが含まれる。イリノテカンは、例えばRhone-Poulenc Rorerから商標名Camptoで市販されており、例えば欧州特許明細書No.137145に記載のごとくまたはそれと類似の方法により製造することができる。トポテカンは例えばSmithKline Beechamから商標名Hycamtinで市販されており、例えば欧州特許明細書No. 321122に記載のごとくまたはそれと類似の方法により製造することができる。他のカンプトテシン化合物は常套的方法で、例えばイリノテカンおよびトポテカンについて上記したのと同じ方法により製造することができる。
【0088】
抗腫瘍ビンカアルカロイドには、上記ビンブラスチン、ビンクリスチンおよびビノレルビンが含まれる。ビンブラスチンは例えば注射用硫酸塩としてEli Lilly and Coから商標名Velbanで市販されており、例えばドイツ特許明細書No. 2124023に記載のごとくまたはそれと類似の方法により製造することができる。ビンクリスチンは例えば注射用硫酸塩としてEli Lilly and Coから商標名Oncovinで市販されており、例えば上記ドイツ特許明細書No. 2124023に記載のごとくまたはそれと類似の方法により製造することができる。ビノレルビンは、例えば注射用酒石酸塩としてGlaxo Wellcomeから商標名Navelbineで市販されており、例えば米国特許明細書No. 4307100に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ビンカアルカロイドは常套的方法で、例えばビノブラスチン、ビンクリスチンおよびビノレルビンについて記載したのと類似の方法により製造することができる。
【0089】
抗腫瘍ヌクレオシド誘導体には、上記5-フルオロウラシル、ゲムシタビンおよびカペシタビンが含まれる。5-フルオロウラシルは、広く市販されており、例えば米国特許No. 2802005に記載のごとく製造することができる。ゲムシタビンは例えばEli Lillyから商標名Gemzarで市販されており、例えば欧州特許出願No. 122707に記載のごとくまたはそれと類似の方法により製造することができる。
【0090】
カペシタビンは、例えばHoffman-La Rocheから商標名Xelodaで市販されており、例えば欧州特許明細書No. 698611に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ヌクレオシド誘導体は常套的方法で、例えばカペシタビンおよびゲムシタビンについて上記したのと類似の方法により製造することができる。
【0091】
ナイトロジェンマスタード化合物には、シクロホスファミドおよびクロラムブシルが含まれる。シクロホスファミドは例えばBristol-Myers Squibbから商標名Cytoxanで市販されており、例えば英国特許明細書No. 1235022に記載のごとくまたはそれと類似の方法により製造することができる。クロラムブシルは例えばGlaxo Welcomeから商標名Leukeranで市販されており、例えば米国特許明細書No. 3046301に記載のごとく、またはそれと類似の方法により製造することができる。本発明に用いる好ましいニトロソウレア化合物には、上記カルムスチンおよびロムスチンが含まれる。カルムスチンは例えばBristol-Myers Squibbから商標名BiCNUで市販されており、例えば欧州特許明細書No. 902015に記載のごとく,またはそれと類似の方法により製造することができる。ロムスチンは例えばBristol-Myers Squibbから商標名CeeNUで市販されており、例えば米国特許明細書No. 4377687に記載のごとくまたはそれと類似の方法により製造することができる。
【0092】
抗腫瘍アントラサイクリン誘導体には、上記ダウノルビシン、ドキソルビシンおよびイダルビシンが含まれる。ダウノルビシンは、例えば塩酸塩としてBedford Laboratoriesから商標名Cerubidineで市販されており、例えば米国特許明細書No. 4020270に記載のごとくまたはそれと類似の方法により製造することができる。
【0093】
ドキソルビシンは例えば塩酸塩としてAstraから市販されており、例えば米国特許明細書No. 3803124に記載のごとくまたはそれと類似の方法により製造することができる。イダルビシンは例えば塩酸塩としてPharmacia & Upjohnから商標名Idamycinで市販されており、例えば米国特許明細書No. 4046878に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍アントラサイクリン誘導体は、常套的方法で、例えばダウノルビシン、ドキソルビシンおよびイダルビシンについて上記したのと類似の方法により製造することができる。
【0094】
トラストズマブはGenentechから商標名Herceptinで市販されており、米国特許明細書No. 5821337またはPCT特許明細書WO 94/04679およびWO 92/22653に記載のごとく得ることができる。
【0095】
抗腫瘍ポドフィロトキシン誘導体には、エトポシドおよびテニポシドが含まれる。エトポシドは例えばBristol-Myers Squibbから商標名VePesidで市販されており、例えば欧州特許明細書No. 111058に記載のごとくまたはそれと類似の方法により製造することができる。テニポシドは例えばBristol-Myers Squibbから商標名Vumonで市販されており、例えばPCT特許明細書No. WO 93/02094に記載のごとくまたはそれと類似の方法により製造することができる。他の抗腫瘍ポドフィロトキシン誘導体は常套的方法で、例えばエトポシドおよびテニポシドについて上記したのと類似の方法により製造することができる。
【0096】
上記のような粗な形のサポニンまたはその分画を遊離形で、すなわち脂質含有粒子に統合しないで抗癌剤として用いることもできる。これら抗癌化合物は、脂質含有粒子、例えば、リポソーム、イスコムおよび/またはイスコムマトリックス、およびPosintrosと混合し、結合させ、または統合することができる。
【0097】
統合するときはそれらが疎水性であれば適切である。そうでなければ、EP 242380に記載のごとく脂質含有粒子に疎水性基を結合させることができる。
【0098】
非疎水性化合物、特にタンパク質またはペプチドは、それらと疎水性基を結合させることで疎水性にすることができる。
【0099】
非疎水性化合物と結合させることができる疎水性基は、好ましくは炭素数1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29または30の直鎖、分岐鎖、飽和、または不飽和の脂肪族鎖、または疎水性アミノ酸またはペプチド、または他の疎水性構造、例えばステロイドである。疎水性構造の長さは、タンパク質の大きさと性質に適合させる。一例として、10-15アミノ酸のペプチド(口蹄疫ウイルス)は、アミノまたはカルボキシ末端に2個のチロシンを適切に伴うことが挙げられる。分子量70,000ダルトンのタンパク質は、約20個の疎水性アミノ酸を必要とする。検査は実験的に行われる。すなわち、ある者は、特にTrp、Ile、Phe、Pro、Tyr、Leu、Valから選ばれる(特にTyr)1〜20アミノ酸、好ましくは1、2、3、4、5アミノ酸を有するペプチド;コレステロール誘導体、例えば、コリン酸、ウルソデソキシコリン酸を用いる。
【0100】
これら疎水性基は、非疎水性タンパク質または化合物と結合することができる基、例えば、カルボキシル-、アミノ-、ジスルフィド-、ヒドロキシル-、スロヒドリル-およびカルボニル基、例えば、アルデヒド基と結合する必要がある。
【0101】
結合することができる疎水性基は、好ましくは、メタン、エタン、プロパン、ブタン、ヘキサン、ヘプタン、オクタン、およびCys、Asp、Glu、Lysを含むペプチド、好ましくはオクタナル(octanal)およびTyr.Tyr.Tyr-Cys、-Asp、または-Gluのカルボキシル、アルデヒド、アミノ、ヒドロキシル、およびジスルフィド誘導体から選ばれる。結合することができる基を有する疎水性基は、溶解する物質に応じて、例えば上記可溶化剤および界面活性剤、または塩酸、酢酸、67容量%の酢酸、苛性液(caustic liquor)、アンモニアの助けをかりて水に溶解する必要がある。次に、pHを物質を沈殿させずに中性へと調整するが、疎水性基を結合させるタンパク質を変性させるpH値にならないように確認する。脂質は可溶性を増大させ得る。
【0102】
疎水性分子は、分子比10:1〜0.1:1、好ましくは1:1で非疎水性化合物に加えることができる。
【0103】
結合分子としてカルボキシル基を有する疎水性基は、水溶性カルボジイミドまたは合成無水物を介してタンパク質と結合することができる。第1の場合は、カルボキシル基をpH5でカルボジイミドで活性化し、高ホスフェート含有量の緩衝液(pH8)に溶解したタンパク質と混合する。後者の場合は、カルボキシ化合物を、チオキサンまたはアセトニトリル中のトリエチルアミンの存在下でイソブチルクロロホルメートと反応させ、得られた無水物をpH8〜9でタンパク質に加える。ヒドラジンを有するカルボキシル基をヒドラジドに変換し、これと該タンパク質中のペリオデート酸化糖単位中のアルデヒドおよびケトンが一緒になってヒドラゾン結合を得ることもできる。
【0104】
亜硝酸を有するアミノ基を低温でジアゾニウム塩に変換し、Tyr、HisおよびLysとアゾ結合を得ることができる。
【0105】
スクシン酸無水物を有する水酸基をヘミスクシネート誘導体に変換し、これをカルボキシル基として結合させることができる。
【0106】
アルデヒド基をタンパク質中のアミノ基と反応させてシッフ塩基とすることができる。
【0107】
種々のカップリング基および方法は、Journal of Immunological Methods(BlairおよびGhose 1983)、(Conradie, Govender et al. 1983)、Methods in Enzymology(Ghose, Blair et al. 1983)、およびAnalytical Biochemistry(DavisおよびPreston 1981)に記載されている(これらの内容は本明細書の一部を構成する)。
【0108】
そのように製造した疎水性基を受け取ったタンパク質、ペプチド、または化合物は、a)に記載のごとくグリコシドと複合結合する(complex-bonded)が、ここでは細胞断片を除去するための精製工程を省略することができる。
【0109】
開示した疎水性基を有する親水性タンパク質は、タンパク質を静かに変性させることにより、すなわち、約2.5の低pH、3M尿素、または70℃以上の高温で疎水性基を利用しやすくすることにより疎水性にすることができる。そのようなタンパク質は、免疫グロブリン、例えば、IgG、IgM、IgA、IgD、およびIgEでありうる。免疫グロブリンを抗イデオタイプ抗体として用いることができる。タンパク質は、(b)に記載のごとくタンパク質として精製され、次いで(a)に記載のごとくグリコシドと複合結合するが、細胞断片を除去するための精製工程は省略される。
脂質含有粒子のための標的化分子
【0110】
脂質含有粒子は、さらに癌標的化分子、例えば、癌細胞由来の表面抗原、ウイルス表面抗原およびインフルエンザ抗原を含みうる。
【0111】
本願は、脂質含有粒子、例えばKGIおよびBBE粒子が生理的低用量で多くの種々の癌細胞を殺し、またはその増殖を阻害することを示す。イスコムおよびイスコムマトリックス製剤にアジュバントとして用いるこれらのタイプの粒子も良好なバイオアベイラビリティおよびリンパ系、特に樹状細胞に対する標的化能も示す(参考としてMorein et al. 2007参照)。標的化作用をさらに増加させるために、in vivo標的化分子を種々の方法により組み込むことができる。微生物膜由来の表面分子をMorein et al.(1984)およびEP 242380に最初に記載のごとく疎水性相互作用により組み込むことができる。例えばrDNA技術により製造するかまたは合成的に製造した他の分子をWO 2002/080981およびWO 2004/030696に記載のごとく組み込むことができる。
【0112】
そのような標的化分子には、ウイルス、例えば呼吸器、例えば肺癌の標的形に親和性を有するインフルエンザおよびRSウイルス由来のエンベロープタンパク質、またはLycke N.(2004)およびMowat et al.(2001)に記載のごとくKGIまたはBBE製剤に組み込まれたコレラ毒素のAサブユニットのA1部分であるCTA1DDが含まれる。CTA1DDは、それぞれ補完的作用に寄与する3つの主要成分に合理的に設計される。CTA1は、A2およびBサブユニットから分離することにより無毒性に変換されるコレラ毒素の酵素的に活性なサブユニットである。Staphylococcus aureus由来のプロテインA由来のDDと融合すると、CTA1はB細胞を標的化する。すなわち、CTA1はB細胞リンパ肉腫に特に適している。CTA1は、免疫刺激増強のためにB細胞を標的化するためにすでにイスコムに組み込まれている。イスコムではその標的化作用に加え、それはイスコムまたはKGIまたはBBE粒子の活性化および分化作用を補う活性化および分化作用も有する。純粋な標的化作用は、おそらく他の治療用医薬と補い合うBBEまたはKGI粒子に対する標的化部分としてCTA1の代替物であるStaphylococcus aureus由来のプロテインAのDDサブユニット分子から得られる。より一般的には、モノおよびポリクローナル抗体は、EP 0 109 942 B1、EP 0 242 380 B1、およびEP 0 180 564 B1に記載のごとく脂質含有粒子、例えばKGIおよびBBE粒子に組み込むことができる。
他の添加剤
【0113】
本発明の組成物は、さらに医薬的に許容される担体、希釈剤、賦形剤、または添加剤を含みうる。
【0114】
適切な医薬的に許容される担体、および/または希釈剤には、あらゆるおよび全ての常套的溶媒、分散媒質、充填剤、固体担体、水性溶液、コーティング、抗菌および抗真菌剤、等張化および吸着遅延剤などが含まれる。医薬的活性物質へのそのような媒質および物質の使用は、当業者でよく知られており、例えばRemington's Pharmaceutical Sciences, 第18版, Mack Publishing Company, Pennsylvania, USA.に記載されている。如何なる常套的媒質または物質も活性物質と不適合性である場合を除き、本発明の医薬組成物へのそれらの使用が予期される。補助活性成分を該組成物に組み込むこともできる。
医薬品形態
【0115】
脂質含有粒子はヒトおよび動物にあらゆる経路で投与することができる。非経口経路を用いることができる。本明細書で用いている用語非経口的には、皮下注射、静脈内、筋肉内、無針注射のための注入技術の皮内注射−ジェット注射および経口エアロゾル投与が含まれる。
【0116】
それぞれ本質的に少なくとも1つのタイプのサポニンを含む本発明の脂質含有粒子は、混合物として組成物で、または同じまたは異なる回数で同じ投与部位または異なる投与部位に異なる組成物で別々に投与することができる。Quillaja Saponaria Molinaの異なる分画を異なるイスコム複合体およびマトリックス複合体および異なる組成物で用いることができる。
【0117】
一般的には、本発明の脂質含有粒子は、医薬的有効量で投与される。実際に投与される粒子の量は、典型的には治療する病状、選んだ投与経路、実際に投与される化合物、個々の患者の年齢、体重および反応、患者の症状の重症度などを含む関連環境に照らして医師が決定するであろう。
【0118】
ヒトに用いる用量は、含まれる他の化合物に応じて異なりうる。治療の持続時間を考慮して、用量は<50μg〜1mg/日またはそれ以上の範囲でありうる。20% KGIおよび80% BBEの混合物を含む最高に耐容性のある製剤では18gマウスに50μgの用量で投与しても副作用を生じず、必要であれば非常に高用量を用いることができる。
部分のキット
【0119】
したがって、本発明は、少なくとも2つの部分を含む部分のキットであって、1部分が、少なくとも1の、癌細胞に対する殺滅作用を有する疎水性のサポニン分画、を含む脂質含有粒子を含み、他の部分が、少なくとも1の、免疫反応、例えば抗体産生および細胞介在性免疫を刺激および調節する親水性のサポニン分画、を含むキットにも関する。
【0120】
少なくとも1の疎水性のサポニン分画を含む脂質含有粒子を含む部分が、さらに少なくとも1の親水性のサポニン分画を有する粒子を含むこともある。
【0121】
本発明の組成物およびキットは、Quillaja Saponaria Molina由来の分画以外の少なくとも1の他のアジュバントを含むこともある。これらアジュバントは、イスコムおよび/またはイスコムマトリックス複合体と混合するか、または該複合体と統合するか、またはそれ自体として遊離形で与えることができる。
治療方法
【0122】
本明細書および特許請求の範囲を通して、文脈上他の意味に解すべき場合を除き、用語「含む(包含する)(「comprise」、またはその変化、例えば「comprises」または「comprising」)」は、記載した成分もしくは成分群を含むが、如何なる他の成分もしくは成分群を排除しないことを意味すると理解される。
【0123】
本明細書に記載のすべての刊行物は本明細書の一部を構成する。本発明は以下の非限定的実施例により説明する。
【実施例1】
【0124】
KGI 1(QHC)、KGI 2(703-マトリックス・イスコム)、KGI 3(QHA〜QHCを含むすべてのQuill A分画)、およびBBE(QHA)の製剤
QHA分画がQHCに存在するアシル鎖を欠くことを示すキラヤサポニンの構造を図1.1に示す。KGI 1粒子は、図1.2に示す右領域のQHCに基づく。このサポニン分画は、図1.2に示す逆クロマトグラフィの右領域のBBE中の塩基性サポニンであるQHAよりも疎水性および溶解性である。
【0125】
KGIは、定義したキラヤサポニン分画または種々のキラヤサポニン分画の混合物から製剤化することができる。すなわち、KGI 1はQHC分画から製造され、KGI 2はQHA(7部)およびQHC(3部)の混合物から製造され、KGI 3は、非分離キラヤ分画の混合物である。また、QHBは、KGI癌殺滅粒子になることができる。BBEはQHAから製造される。それらの割合は、望む殺滅特性と低い細胞毒性特性をもたらし、分化特性を強調するよう変更させることができる。
結 果
【0126】
KGIおよびBBE粒子の形成は上記材料と方法に記載されており、イスコムの製剤化に関する研究(Morein, Sundquist et al. 1984)および後者はイスコムマトリックスに関する研究(Morein et al. 2007)に基づく。直径30〜40nmの典型的ケージ様イスコム構造を電子顕微鏡(EM)により可視化した(図1.3)。超遠心後、約20Sの沈降係数を有するケージ様イスコム構造を含む分画がEMにより観察される(「材料と方法」参照)。
結 論
【0127】
種々の薬理作用を有するが同じ形態を持つ粒子を形成し、容易にEMにより予測し、以下の実施例に記載のごとく用いる勾配遠心により定義する。これら粒子は、免疫反応/癌殺滅特性を修飾するために、そして癌療法の分野で種々の分子用の送達用粒子として用いることができる。
【実施例2】
【0128】
赤血球(RBC)および有核細胞に対する溶解作用
サポニンは、細胞溶解作用を有することがよく知られているので、癌治療の候補薬として試験されている(Wang, Z.P. 2005)。HPLCによる分画化後のキラヤサポニンパターンを図1.1に示す。本発明において、製剤KGI 1、2、および3粒子に用いるQHCは、脂肪アシル鎖により高溶解性である(図2.1)。反対に、製剤BBE、KGI 2、および3粒子に用いるQHAは、脂肪アシル鎖を欠くので、実質的に非溶解性である。RBCを用いて、分光光度法により容易に測定されるヘモグロビンのサスペンジョン液中への漏出をもたらす損傷を引き起こすその細胞膜に対する物質の溶解作用を測定する(「材料と方法」参照)。該方法は、好感度で再現性がある。有核細胞の細胞膜に対する物質の溶解作用は種々の方法で試験する必要がある。本発明者らはトリパンブルー染色を用いた。遊離KGIは、サポニンの特性を有し、細胞膜中のコレステロールと相互作用することにより溶解し、6nmの6角微細孔を生じる。色素は細胞膜のこの微細孔を通して損傷細胞に入る。この微細孔は、瞬時に細胞溶解を生じ、有核細胞を殺すであろう。すなわち、溶解濃度の遊離サポニンは、数分以内で速やかな細胞死をもたらし、例えば10分間で、数時間または数日を必要とする他の細胞殺滅メカニズム、例えばアポトーシスの溶解作用を描出する。微粒子KGIにおいて、サポニンはコレステロールと強く結合し、KGI中のサポニンが細胞膜のコレステロールと相互作用するのを抑制し、微細孔の形成および溶解作用を抑制する。
【0129】
有核癌細胞U937およびヒト血液由来の正常好中球を遊離形および微粒子形のKGIおよびBBEに曝露した。
結 果
【0130】
溶血分析の結果を表2.1に示す。BBE粒子の原料である遊離BBEは、Roennberg et.al.(Ronnberg, Fekadu et al. 1995)により報告されたGHAの用量と同様の50μg/mlまでの用量では溶解作用を生じなかった。BBE粒子は、100μg/mlまでの濃度でRBCを溶解しなかった。
【0131】
遊離KGI 1の原料であり、微粒子KGI 1中に存在するQHCは、5μg/mlの濃度でRBCを溶解した。サポニン分画(QHA、BおよびC)を含む遊離KGI3は、20μg/mlの濃度でRBCを溶解した。KGI 3粒子は、100μg/mlの濃度でRBCの溶解を生じなかった(表2.1参照)。
【0132】
表2.1 種々の遊離形のキラヤサポニンおよび特定の形、すなわち、KGIおよびBBE粒子の細胞毒性および溶血活性
【表1】
【0133】
表2.2において、KGI製剤に10分間曝露し、次いでトリパンブルーで染色した細胞の溶解作用を示す。癌細胞の処理に用いた最高濃度は50μg/mlであり、この時間中に有核細胞は溶解または殺滅されなかった。反対に、遊離形は好中球細胞は17μg/mlで、U937癌細胞は27μg/mlで有核細胞を溶解した。有核細胞は、微粒子KGI 1に曝露して10分以内では殺されず、1時間後でも殺されなかった(示さず)。
【0134】
表2.2 トリパンブルー染色で検出したU937癌細胞および好中球に対する遊離KGI 1および微粒子 KGI 1の溶解作用。細胞をKGI製剤と10分間インキュベーションした。
【表2】
考察および結論
【0135】
癌の処置に用いた化合物は正常細胞より癌細胞を選択的に殺し、癌細胞の殺滅はin vivoで標的部位に対して高濃度になる問題を考慮して低用量で有効であることが必須である。該溶解作用は、遊離形のサポニンに特徴的であり、癌殺滅に対する好ましい細胞毒性作用ではない。さらに、該遊離形は、脂溶性であり、投与部位の細胞膜と相互作用し、局所的細胞破壊をもたらし、サポニンの一部はその部位に捕捉される。KGIおよびBBE粒子は、「生理学的」用量ではRBCまたは有核細胞の細胞膜を溶解しない。実際に、試験した最高用量、すなわち50μg/mlでは溶解または即時的毒性作用は生じなかったが、非微粒子(遊離)形のキラヤサポニンは、かなり低濃度で溶解および細胞毒性作用を生じた。
【0136】
遊離形のKGIは、比較的低用量で10分間以内に有核細胞を殺し、RBCを溶解する。実施例7に示すように、KGI 1は、2μg/mlの低い生理学的用量、またはそれ以下、溶解または膜損傷濃度以下の遊離サポニン形でも癌細胞を殺す。さらに高用量の50μg/mlのKGI 1でも、癌細胞の殺滅に数時間を必要とし、溶解膜損傷作用より癌細胞殺滅に関与する別のメカニズムを強調した。細胞溶解作用は癌細胞と正常好中球細胞で同様であったことは注目される。すなわち、微粒子形は、正常細胞と癌細胞の速やかな溶解性の無差別的殺滅を回避する大きな利点がある。さらに、溶解作用は、注射部位に注射したかなりの量の化合物が捕捉されることによる局所的副作用を生じ、バイオアベイラビリティが低くなる。すなわち、それがキラヤサポニン微粒子を癌治療用にする革新的特徴である。したがって、該微粒子形も薬理学的デリバリーシステムである。
【実施例3】
【0137】
KGI 1は選択的に癌細胞を殺す
本実施例は、KGI 1粒子が正常細胞より腫瘍細胞を選択的に殺す(アラマーブルー法で測定した)ことを証明する。悪性単芽球細胞系(U937)を、正常細胞、すなわち単球由来樹状細胞(DC)と比較するための腫瘍細胞に選んだ(「材料と方法」参照)。KGI 1中の活性サポニン成分は、Quillaja Saponaria Molinaに由来する市販Quill Aから単離したQHC(遊離KGI 1)である。図3.1(Quill AのHPLCクロマトグラム)に示すように、遊離KGI、すなわち分画QHCは、分画QHA、すなわち遊離BBEと比べて疎水性が高い。QHCは、GHAのアシル鎖を欠くことによりGHAと異なり、これはQHCのより高い疎水性とその溶解作用を説明する(図3.2)。
【0138】
腫瘍細胞:Academic hospital Uppsalaから得た単芽球細胞系(THP-1、U937およびU-937-1)およびミエローマ細胞系(LP-1およびJurkat)をKGI 1に曝露した。
結 果
【0139】
材料と方法に記載のごとく製造したKGI 1は、上記および表3.1に記載の腫瘍を殺す。記載した5つの腫瘍細胞系は、0.8〜10μg/mlの範囲のLC50濃度のKGI 1により殺された(表3.1および図3.1)が、正常DCのLC50は24.3である(図3.2)。すなわち、正常細胞を殺すには30倍高濃度のKGI 1が必要である。
【0140】
表3.1 サポニン製剤による腫瘍細胞または正常細胞の増殖の阻害
【表3】
考察と結論
【0141】
本発明者らは、微粒子形のキラヤサポニン分画、KGI 1が選択的に腫瘍細胞を殺すことを確認した。この場合、単芽球細胞U937は、正常細胞を殺すのに必要な濃度より30倍低い濃度で殺された。高殺滅作用は、キラヤサポニンの逆相ダイアグラムの疎水性分画に局在し得る。これに対し、より親水性の分画QHA(図1.1および1.2)は、関連用量で癌細胞や正常細胞を殺さない。
【0142】
すなわち、KGI 1は、癌治療の候補薬であり、さらに以下の実施例で立証される。
【実施例4】
【0143】
BBEは有核細胞に無毒性である
逆相ダイアグラム由来の比較的親水性の分画QHA(遊離BBE)(図1.1および1.2)をBBE粒子に組み込み、癌細胞または正常細胞に対する殺滅作用を試験し、無毒性であることがわかった(図4.1および4.2)。
結 果
【0144】
BBE粒子は、関連用量、すなわち試験した1920μg/mlまでの濃度で癌(図4.1)または正常(図4.2)細胞を殺さなかった。
考察と結論
【0145】
本実施例は、BBE粒子が実質的に細胞毒性がない、すなわち正常および腫瘍細胞によりよく許容されることを示す。BBE粒子は、1920μg/mlまでのあらゆる濃度で腫瘍または正常細胞を殺さない(図4.1および4.2)。1つの違いは、BBE中のサポニンは脂肪族鎖を欠くことであり、これは遊離形の活性物質QHAの低溶解作用を説明する(実施例2、表2.1および2.2参照)。BBE粒子は、その調節作用により、例えばサイトカイン産生を刺激することによりKGI粒子の活性を抑えるかまたは調節するのに有用であろう。
【実施例5】
【0146】
BBEはKGIの細胞殺滅作用を阻害する
本実施例は、BBEがKGI 1の殺滅作用を阻害することを示す。最も顕著な違いは、KGI 1中のサポニンは、アシル鎖を有するが、BBE中のサポニンは該鎖を欠く(より詳細には実施例1、2、および4参照)。
結 果
【0147】
77μg/mlの一定濃度のKGI 1を、増加する濃度のBBEとインキュベーションし、U937癌細胞に適用した。KGI 1の細胞毒性作用は比10:1(KGI 1:BBE)で阻害された。阻害作用は、図5.1の曲線が示すようにレセプターにより仲介されるようである。
【0148】
阻害作用は、U937細胞に関するそのLC50がわずか0.8μg/mlであることを考えると、用いた77μg/mlの一定の高KGI 1濃度に照らして効果的である。
考察と結論
【0149】
本発明者らは、腫瘍細胞を選択的に殺すKGI 1粒子の形の物質を確認した。本発明者は、腫瘍細胞および正常細胞の両方でKGI 1による殺滅作用を阻害するBBE粒子として製剤化された別の物質を確認した(示さず)。すなわち、毒性が生じるとそれを効果的に抑えることができる腫瘍殺滅系が作出される。本実施例中の活性物質QHCおよびQHAは、異なる粒子、すなわちKGI 1およびBBE中に存在することに注目すべきである。癌細胞死を促す阻害レセプターは正常および癌細胞の活性化と分化を促すレセプターと異なるようである。
【実施例6】
【0150】
KGIおよびBBEは、1または2の異なる粒子中の活性物質の提示作用を正常および癌細胞で分析するデリバリーシステムである
実施例5は、別個のBBE粒子中の活性物質QHAは、別の活性サポニン、すなわちGHCを含むKGI 1の細胞毒性作用を阻害することを示す。この阻害効果は、1またはそれ以上のレセプターを介する阻害によるようである。
【0151】
KGI 2は、2つの活性サポニン物質QHC(KGI 1の活性物質)とQHA(BBEの活性サポニン)を有する。本実施例は、1および同じ粒子中のこれら2成分は該成分それぞれの作用を一体化し、抑えることを示す。本実施例は、別個の粒子で同時に投与した同じ成分は曝露した細胞の反応を抑え、調節することも示す。KGI 2のような粒子は、種々の割合の出発物質を含むKGI 2(703)について記載したように製造した比7:3、7.5:2.5、または9.5:0.5の異なる割合のQHA/QHCを有した。細胞の生存はアラマーブルーで測定した(「材料と方法」参照)。
【0152】
図6.2に示す実験において、KGI 2(703)を、U937癌細胞と48時間、およびヒト未成熟DCと同様にインキュベーションした(図6.3)。
【0153】
正常哺乳類細胞は動物またはヒトの生命を維持するための種々の役割を持つ。あるDCポピュレーションは単球に由来する。本実施例では、ヒト末梢血細胞をIL-4およびGMC-SFで処理し、3H biomedical(Uppsala, Sweden)から得られる単球由来未成熟DCポピュレーションを得た。本実施例で用いた詳細な情報については「材料と方法」参照。本実施例は、種々のKGIおよびBBE製剤がヒト未成熟DCを活性化して成熟させ、リンパ球と連絡をとってエフェクター細胞に分化および活性化させる活性化DCの分子であるDCマーカーのCD86を発現させることを示す。
【0154】
異なるKGIおよびBBE製剤は、KGI 1のみを含むKGI 1;1および同じ粒子中の70%のQHA(BBEの原料)および30%のQHC(KGI 1の原料)から製造されたKGI 2;Quill Aの形のキラヤサポニン分画の混合物を含むKGI 3;BBEのみがあり、BBE + KGI 1製剤は80%のBBE粒子および20% KGI 1粒子(すなわち別個の粒子中の各化合物)からなる。最初の研究は、KGI 1, KGI 2およびKGI 3については処理濃度を1μg/ml細胞培養液に、BBEおよびBBE + KGI 1については処理濃度を10μg/mlで用いるべきであることを示した。詳細は「材料と方法」参照。
結 果
【0155】
7:3〜7.5:2.5の範囲の種々の割合のQHA:QHC分画を含むKGI 2のような製剤を20〜100μg/mlで2日間インキュベーションするとU937癌細胞を殺した。活性QHC分画をQHA分画で0.5:9.5に希釈すると48時間以内のインキュベーション時間で細胞毒性活性が完全に消失した(図6.1)。これらの結果は実施例5で得られた結果と比較すべきである。
【0156】
第2実験は、QHAおよびQHC サポニン分画を1および同じ粒子にQHA:QHC 7:3の比で混合したKGI 2は、18.7μg/mlのLC50でU937腫瘍細胞を殺し(図6.2)、これはKGI 1のLC50より23倍高い。正常ヒトDCはU937癌細胞より細胞死に44倍高いKGI 2濃度を必要とした(すなわち、LC 50は826μg/ml)(図6.3)。
【0157】
表3.1に、アラマーブルー法で測定した種々の癌細胞に対する種々のKGI 粒子の癌細胞殺滅効果(正常ヒト好中球細胞についてはトリパンブルーで測定)をまとめる。一般的には、いくつかの異なる癌細胞はKGI製剤に感受性であり、U937細胞よりも遙かに感受性である。本願ではU937細胞を基礎研究のモデルに用いた。
【0158】
活性化および分化活性を図6.4に示す。1μg/mlの低生理学的用量のKGI 2は、最も高い割合のCD86陽性細胞(67.3%)を示し、1および同じ粒子、すなわちKGI 2粒子におけるKGI 1およびBBE成分の相乗効果を示した。また、KGIおよびBBE 粒子は担体送達粒子である、すなわちKGI 2は2成分を送達することを示す。BBEは、LPSと同じ割合のCD86陽性細胞を誘導したが、製剤BBE + KGI 1は65.8%を誘導した。後者の2つの製剤はKGI 2より10倍高用量、すなわち10μg/mlを必要とした。
考察と結論
【0159】
KGI 1、KGI 2、およびKGI 3のような製剤は、2および可能性としてそれ以上の異なる成分を異なる割合で組み合わせるデリバリーシステムである。本実施例において、2の異なる特性を組み合わせて完全に異なる癌殺滅作用を得た。簡単には、実施例4に記載の異なる構成要素(粒子)に閉じこめられた2の異なる化合物、すなわちKGI 1およびBBEの混合は、阻害により活性を抑える。1および同じKGI粒子(KGI 2)中の2つの物質の組み合わせは、1および同じ粒子中の異なる成分で阻害を達成することはできないので、希釈法、すなわちKGI 1リガンド密度の減少により毒性を低下させた。示した作用を癌細胞と正常細胞の両方について記録した。癌細胞に対するKGI 2の毒性はKGI 1粒子のそれに比べて何倍も低下したが、癌細胞(U937)に対しての正常ヒト細胞(DC)に対する毒性作用のゆとりは、30倍(KGI 1)〜40倍(KGI 2)に増加した。希釈効果は、細胞の殺滅を誘導するリガンド-レセプター相互作用の数が低下するので、より低い結合力で生じる。細胞毒性作用は、阻害作用を仲介するものと異なるリガンド-レセプター配置(constellation)により発揮される可能性が高い(most probably)。KGI 2粒子または他の同様の粒子において、LC50をKGI 1の0.8μg/mlからKGI 2の18.7μg/mlに増加させることによりBBEの調節能とKGI 1の殺滅効果を組み合わせて、癌細胞と正常細胞の毒性の間に大きなゆとりを保つか、または増す(例えば40倍の差)ことができる。それはin vivoで臨床的関連があるようである。KGI 1の癌細胞殺滅作用は、BBEの阻害作用により直線的に増加するが、両粒子により発揮される分化能は、今まで行われた実験からわかるように実質的に変化しない。これらの知見の柔軟性は、癌細胞の毒性および活性化-分化をアポトーシスのプログラムされた死を伴う正規化細胞挙動に合わせることを可能にする。両製剤、すなわち、同じ粒子中の2つの物質または別の粒子の2つの物質は、臨床的価値を持つようであることは注目に値する。例えば、KGI 1は、他の特性、例えばプログラムされた細胞死−アポトーシスで終わる癌細胞の分化、活性化を妨げるかもしれない低すぎる濃度で癌細胞を殺し、これはバイスタンダー効果、すなわち、直接曝露されていない近傍の細胞に対する効果に関与するかもしれない(実施例7)。例えば、免疫系において殺されたDCは近傍のDCを交差刺激する成分を放出することが十分立証される。
【0160】
アポトーシス(実施例7参照)は、個体にとっては壊死による細胞死より劇的ではなく、処置された個体に病気をもたらす細胞毒性作用を回避する。したがって、癌細胞のアポトーシスは、癌療法において魅力的である。これに関連して、殺された癌細胞は、KGIおよびBBEを含む粒子中に保持されるもののような能動免疫を増強する成分の影響下で免疫防御を開始することができる免疫原性成分を放出することを予期しうる。すなわち、KGI粒子は、細胞死を調整し、種々の抗癌活性を有する他の物質を統合する可能性を開くデリバリーシステムである。そのような物質、例えば細胞骨格の損傷を介して細胞を殺すタキソール、またはKGIまたはBBE粒子と異なるメカニズムで分化を促すビタミンAまたはDは、他の原理で作用することが好ましい。KGI粒子は、本実施例で用いる成分に限定されない送達能を示す。結論として、種々のKGI、または遊離または微粒子形のBBEのような他の化合物とのKGIの併用は、正常細胞より癌細胞を選択的に殺す(表3.1)。
【0161】
KGI 1製剤は、癌細胞U937を活性化−刺激してIL8およびアポトーシスをもたらすが、壊死により特徴づけられる細胞死はもたらさない(実施例7参照)。KGI 2および3のような他のKGI製剤は、正常および癌細胞を殺すのに遙かに高濃度が必要であり、正常細胞に対してまだ安全性に30〜40倍のゆとりがある(実施例5および6参照)。本実施例では、高い割合で正常な未成熟DCが刺激されて成熟DCとなり、CD86を発現することを示す。最も興味深いことに、活性化合物QHAまたはBBEを、1および同じ粒子中のKGI 1のサポニン成分であるQHCと組み合わせることにより相乗効果が記録された、すなわち、低用量1μg/mlのKGI 2は最も高い割合のCD86陽性細胞が誘導されたことがわかる。それ自体10μg/mlを必要とするBBEは、KGI 2製剤中、0.2μg/mlの活性KGI 1化合物と組み合わせて濃度0.8μg/ml培養液の活性化合物を用いた。これら活性成分はいずれも成分それぞれそれ自体で用いるときに必要とするよりかなり低かった。すなわち、BBE化合物の濃度は10倍以上の低く、活性KGI化合物の濃度は、効果の増加をもたらすKGI 2粒子において5倍減少した。多くのキラヤサポニン分画を含むKGI 3は、本実施例ではCD86発現を誘導せず、むしろそれを阻害した。CD86の発現に関してKGI 3の阻害作用があるか否かを確認するにはさらなる実験が必要である。
【0162】
結論として、ヒトDCの成熟により測定された予期しない相乗作用が、KGI 2粒子中の2成分が細胞と相互作用するとみられ、KGIおよびBBE粒子の、レセプターを介して細胞を標的化する担体系として価値がある可能性を強調する。KGIおよび/またはBBE 粒子による癌殺滅作用は、活性化および分化を介して作用することが提唱されるので、正常細胞で分析することも可能なはずである。癌細胞は非常に異なり、標的化特性を変化させることができる癌細胞殺滅系は癌治療の新しい可能性をもたらすことに注目すべきである。
【実施例7】
【0163】
種々の検出系を適用することによるKGI 1の癌殺滅作用の分析
微粒子形のKGI中のキラヤサポニンは、正常細胞より30〜40倍低い濃度で癌細胞を殺す(実施例3)。本実施例では、癌細胞死を、「材料と方法」に詳述したトリパンブルー染色、アラマーブルー法による酵素代謝阻害、プロピジウムアイオダイド染色により可視化した壊死変化、およびアネキシンV染色によるアポトーシスを含む種々の方法で分析した。処置した細胞のトリパンブルー染色は細胞数を確認するのに有益であり、生存性を9日間まで測定した(図7.1、7.2)。低用量のKGI 1(2μg/ml)では、より長い期間後に癌細胞死を生じたが、高用量(25μg/ml)では短期間内に死をもたらし(図7.3)、より速やかなアポトーシスを引き起こした(図7.6)。本実施例では、トリパンブルー法により測定した細胞死は、細胞が細胞周期から抜け出した後のプログラムされた細胞死、すなわちアポトーシスと相関した。細胞周期は、細胞複製の基礎であり、癌細胞では、連続的複製のために細胞周期に留まることが重要である。該周期から抜け出すことは、細胞が産生、この場合はサイトカイン産生(実施例10)に向かって活性化を開始し、最終的に本実施例で示されたプログラムされた細胞死に進みうることを意味する。本発明者らはチミジンキナーゼ(TK)試験を用いて、処置細胞が複製期、すなわち細胞周期にあるか、または該周期から抜け出し(実施例9)、産生能力および最終的にアポトーシスをもたらす経路に入ったか否かを示した。
【0164】
KGI 1の癌細胞U937を殺す能力を以下の実験においてトリパンブルー染色法(「材料と方法」参照)により分析する。
【0165】
U937細胞を2μg/ml KGI 1を含むマイクロタイタープレートに播いた。培養のあるセットにおいて、該細胞をKGI 1に9日間連続して曝露した(図7.1)。細胞数を3日間毎に同じ濃度のKGI 1を含む新鮮培地で1x106/mlに調整した。各機会に累積細胞数を計算した。培養の第2セットにおいて、3日間後にKGI 1を含まない培地に交換してKGI処置を終了した(図7.2)。細胞数を培地交換時の各機会に1x106/mlに調整した。累積細胞数を計算した。
【0166】
U937癌細胞を高用量の25μg/mlの遊離形または粒子形のKGI 1に曝露し、図7.3に示すようにサンプリングした。細胞をトリパンブルーで染色し、生存性を顕微鏡で数えた。細胞の生存性は細胞コントロールの生細胞のパーセントで表す。
【0167】
細胞毒性を比較するため、U937癌細胞を、図7.4に示すように低生理学的用量の2μg/mlの遊離または粒子形のKGI 1に曝露した。細胞をトリパンブルーで染色した。生存性を細胞コントロールの生細胞のパーセントで表す。
【0168】
U937癌細胞を、図7.5に示すように低用量の0.5μg/mlまたは2μg/mlのKGI 1に12日間曝露した。トリパンブルー染色法で染色後に細胞数を数えた。
【0169】
微粒子KGI 1のアポトーシスを誘導する能力をU937癌細胞で分析した。該細胞を、濃度2μg/mlまたは25μg/mlのKGI 1に120時間曝露した(図7.6)。アネキシンV陽性細胞(図7.6)および壊死、すなわち、プロピジウムアイオダイド(PI)染色細胞(図7.7)の数をFACSで同時に測定した(「材料と方法」参照)。U937癌細胞を、濃度2μg/ml(M2)〜50μg/ml(M50)のKGI 1に120時間曝露した。
【0170】
図7.8において、U937細胞を、濃度2μg/ml〜50μg/mlのKGI 1に120時間曝露した。細胞をサンプリングし、図に示すようにプロピジウムアイオダイドおよびアネキシンVで染色した。影響のあった細胞の割合をFACSで計算した。詳細は「材料と方法」に記載している。
結 果
【0171】
KGI 1で持続的に処理した後のU937癌細胞の生存性を図7.1および7.2に示す。低生理学的用量の2μg/mlのKGI 1による癌細胞の断続処理は、連続処理と同様に癌細胞の増殖の阻害および殺滅に有効であった。細胞数は、9日間のインキュベーション後に実質的に生細胞がなくなるまで減少した。非処理細胞数は、3日間の培養中に1x106/mlから3x106/mlに増加した。処理細胞数は3日間培養後に0.5x106/ml以下に減少し、9日間培養中にさらに減少した(図7.1および7.2)。
【0172】
高用量の25μg/mlの遊離形のKGI 1、すなわちQuill AのQHC分画は、該細胞を速やか、すなわち3時間以内に殺したが、その濃度のKGI 1粒子は高率に癌細胞を殺すにはより長い時間、すなわち24時間必要であった。
【0173】
低生理学的用量の2μg/mlの遊離形のKGI 1、すなわちQuill AのQHC分画は、60時間以内に細胞を殺さなかったが、その濃度のKGI 1粒子は24時間培養後に癌細胞を殺し始め、60時間の実験培養期間中継続し、細胞生存性は20%に低下した。この濃度で遊離非粒子形は60時間の培養期間中癌細胞数を減少させることができなかった。この濃度は細胞溶解性ではない。
【0174】
非常に低用量の0.5μg/ml KGI 1粒子は、無処置細胞に比べて細胞数を減少させたが、生理学的用量の2μg/mlのKGI 1粒子は、培養12日間以内に全ての癌細胞を殺した(図7.5)。
【0175】
プログラムされた(アポトーシス)細胞死の誘導を図7.6に示す。2μg/mlの濃度のKGIは、U937細胞のアポトーシスポピュレーションの増加をもたらし、24時間曝露後にピークレベルに達した。より高用量、すなわち25μg/mlのKGIは、アポトーシス細胞の割合をさらに増加させ、12および24時間曝露後にピークレベルに達した。
【0176】
壊死細胞の誘導(図7.7)。アポトーシスの誘導に対するKGI 1粒子の作用とは対照的に、壊死細胞死に対する効果はなかった。すなわち、種々の用量の微粒子KGI 1で処理した細胞とKGI 1に曝露していないコントロール細胞の壊死細胞の割合には差はなかった。
【0177】
図7.8は、KGI 1処理U937癌細胞が2μg/ml〜50μg/mlの濃度で120時間の実験期間中に次第にプロピジウムアイオダイド(PI)およびアネキシンVの両方に染まることを示す。濃度の増加および時間の経過と共に壊死およびアポトーシス作用の両方について染色された細胞ポピュレーションの増加を生じた。コントロールは、アポトーシスおよび壊死について共に染色された細胞の割合がわずかなことを示した。最初に、KGI 1処理細胞は、主としてアネキシンVのみに染まったが、時間と共に細胞は壊死し、二重に染色された。
考察と結論
【0178】
U937細胞の低用量(2μg/ml)のKGI 1処理は、3〜6日間後に細胞数を急激に減少させる。12日間後、実質的に残った生癌細胞はない。高用量のKGI(25μg/ml)は、36時間以内に全ての癌細胞を殺す。実施例3では、U937癌細胞は正常細胞より30〜40倍感受性が高いことを示した。癌細胞の殺滅が誘導されると(図7.2および8.1参照)、培養からKGI 1を除去しても細胞死の進行は停止しないので、回復しない(実施例4も参照)。正常細胞は、有害な症状を生じないプログラムされた死(アポトーシス)の運命を有する。本実施例では、顕著なアポトーシスがKGI 1により誘導されたことを示す。アポトーシス細胞の割合は曝露の24時間までに増加した。低用量の2μg/mlのKGI 1は、24時間インキュベーション後に最も顕著なアポトーシスを引き起こした。
【0179】
時間とともに、アネキシンV陽性細胞の割合は減少し、アネキシンVおよびPIの両方に染まる細胞の割合は増加した。KGI 1処理は、無処理培養よりかなり大きな二重陽性細胞ポピュレーションをもたらした。二重染色細胞ポピュレーションは壊死またはアポトーシス細胞に由来する。最初は、処理および非処理細胞ポピュレーションの両方で同じくPI陽性細胞の割合が低かった。したがって、KGI処理後の二重陽性細胞の割合が高いことは初期のアポトーシスポピュレーションに由来するようである。
【0180】
KGI 1に曝露後のTK活性の中断(実施例9)は、U937癌細胞がサイトカインIL-8の産生を開始する作用と一致する。IL-8産生を、実施例10に示すKGI 1により引き起こされる産生期に癌細胞を活性化するための指標として用いた。刺激作用は、KGI 1が癌細胞を殺す濃度に近い。革新的で興味深いシナリオは、刺激作用は細胞周期をはずれたKGI 1処理細胞を産生期へと、最終的プログラムされたアポトーシス死への正常産生細胞の不可避の運命へと向かわせるというものである。KGI製剤は決して細胞増殖を刺激することができないことに注意すべきである。癌細胞を殺す工程は、抗癌剤(cytostatica)または放射線照射による癌細胞の殺滅より大きな利点があり、無症状であるか、少なくとも副作用が最小限である。
【実施例8】
【0181】
KGI 1は細胞が無制御な複製に戻ることを許さない癌細胞死をもたらす
癌治療に用いる薬剤は、初期によい効果があることがあるが、連続治療後に癌細胞殺滅作用が取り消されることがあり、細胞は無制御な複製を始める。すなわち、処置した癌細胞は無制御な複製に戻らないことが必須である。実施例7は、KGI 1がアポトーシスのメカニズムを含めて癌細胞を殺すことを示す。本実施例は、KGI 1で処置したU937癌細胞の持続的培養後、より重要なことは低生理学的用量での断続処置後に癌細胞は複製に戻らないことを示す。
【0182】
U937細胞を、0.5μg/mlまたは2μg/mlのKGI 1粒子で12日間培養し、「材料と方法」に記載のごとくトリパンブルー染色により、該細胞の生存率を同じ条件下で培養した非処置細胞と比較した。
【0183】
以下の実験において(図8.2)、濃度2μg/mlのKGI 1による同期化U937細胞の処置は、72時間培養後にKGI 1含有培地をKGI 1不含新鮮培地に交換することにより中断した。3日毎に、培地を交換して細胞増殖を促進した。12日間培養後、生細胞数をトリパンブルー染色後に数えた。
結 果
【0184】
濃度2μg/mlのKGI 1に連続的に曝露したU937癌細胞の数は最初増加し、次いで1〜3日間の時点から徐々に減少した。12日間の実験期間の終わりに、生細胞10%以下が記録された(図8.1)。0.5μg/mlのKGI 1で処置後、細胞数の初期増加がみられ、次いで出発点後第3日からその半分に減少し、その後細胞数は無処置コントロール細胞より有意に低かった。コントロール細胞は、細胞数が出発時から3倍に達した。すなわち、初期複製期間後にKGI 1で細胞を処置後、生細胞数は低生理学的用量のKGI 1を用いた後に減少した。
【0185】
図8.2は、KGI 1が増殖を阻害し、癌細胞U937を殺し、該細胞が12日間の培養期間中、第3日にKGI 1を除去した後でも増殖に戻らないことを示す。
考察と結論
【0186】
実施例3は、KGI 1により発揮される癌殺滅作用を示し、考察する。本実施例から、低および生理学的用量の2μg/mlのKGI 1で処置した細胞は、処置を中断した後でも細胞増殖に戻らないと結論することができる。
【0187】
0.5μg/mlのKGI 1による処置は、無処置コントロール細胞に比べて細胞数を有意に減少させた。初期複製期間後、生細胞数は減少した。低濃度(0.5μg/ml)のKGI 1で処置した培養中の生細胞数は複製しない生存細胞を示すようである。
【0188】
結論として、低生理学的用量のKGI 1に曝露したU937癌細胞は、無制御な複製に戻らないが、細胞数は徐々に減少する。
【実施例9】
【0189】
KGI 1で処置した癌細胞は細胞周期を抜け出す
実施例7は、KGI 1がアポトーシスのプロセスを含み癌細胞を殺すことを示し、実施例8は、癌細胞の殺滅が不可逆的であることを示す。実施例10は、KGI 1が、他の薬剤では必要とするホルボール-12-ミレステート-13-アセテート(PMA)のような細胞活性化または分化剤を加えることなくU937細胞が産生期に入るのを誘導することを示す(Baldridge, Cluff et al. 2002)。細胞周期は細胞の複製に影響する(図9.1)。該周期の初期であるS期に娘細胞のDNAが構築される。DNA構築ブロックの1つにヌクレオチドチミジンがある。チミジンは、そのリン酸化に酵素チミジンキナーゼ(TK)を必要とする。すなわち、この酵素は、S期に先立って利用可能でなければならない。ここでは、TK活性を用いて、KGI 1がU937細胞の細胞周期に影響を及ぼすか、KGI 1処置細胞が該周期に留まるかまたは該周期から抜け出すかを検討した。本実施例では、細胞周期中のKGI 1処置細胞の持続性を、経時的にTK活性を検出し、TK活性と癌細胞代謝の阻害(アラマーブルー試験により記録)、殺滅(トリパンブルー排除染色により測定)、および(考察では)アポトーシスを関連づけることにより分析した。非処置細胞の細胞代謝、複製、およびTK活性を用いて該周期に残っている癌細胞を記録した。
【0190】
1x106/mlのU937細胞を本実施例で行う実験のためにマイクロタイタープレートに播いた。細胞周期中のU937細胞を同期化させる試みは、「材料と方法」に記載のごとく細胞培養液中のウシ胎児血清濃度を10%から0.5%に減らして細胞を22時間飢餓させることにより行った。TK活性を測定し、2μg/mlまたは25μg/mlの濃度の微粒子KGI 1で処理した細胞と無処置細胞の間で比較した。図9.2および9.3に記載のとごく5日間までの間隔で測定し、効果を結果に記載する。図9.4において、細胞溶解物で測定したTK活性の低下を、微粒子 KGI 1処理U937細胞の処置後に同じ濃度の遊離、すなわち非微粒子 KGI 1処理のものと比較した。106/mlの細胞を2μg/ml(M2)、10μg/ml(M10)、25μg/ml(M25)、50μg/ml(M50)で処置した後120時間毎日細胞溶解物を用いて試料を試験した。図9.5において、TK活性を、図9.4に示すように遊離および微粒子KGI 1で処置後の細胞培養液を用いて測定した。図9.6では、TK活性を、2μg/mlのKGI 1に0、2、8、18および24時間曝露した同期化U937細胞において分析した。無処置コントロールを0、8および24時間にサンプリングした。
【0191】
図9.7. 同期化U937細胞を2μg/mlのKGI 1に2、8、18および24時間曝露した。コントロールを8および24時間にサンプリングした。生細胞をトリパンブルー染色後に数えた。
【0192】
図9.8. 細胞代謝阻害(アラマーブルー)および細胞の殺滅(トリパンブルー)を、0.5μg/mlまたは2μg/mlの微粒子KGI 1または遊離KGIで24時間処置後の同期化U937細胞で測定した。
結 果
【0193】
処置細胞のTK活性は5日間の培養中無処置コントロールに比べて低下した。高用量の25μg/mlのKGI 1で処置後のTK活性の低下は、24時間に、低用量の2μg/mlのKGI 1では48時間後に顕著であった(図9.2)。処置細胞のTK活性も非処置細胞のパーセントで記録する。図9.3. 3日間後、25μg/mlのKGI 1で処置した細胞からTK活性は検出されなかった。低用量の2μg/mlのKGI 1による細胞の処置は、5日間後にTK活性を10%に減少させた。
【0194】
図9.4は、低生理学的用量の2μg/mlのKGI 1は、癌細胞を48時間曝露した後にTK活性の顕著な減少をもたらすが、72時間曝露後に最も顕著であることを示す。TK活性の減少は、遊離KGI 1処置後は顕著ではない。高用量の25または50μg/mlのKGI 1で処置後のTK活性の減少は、初期、すなわち24時間曝露後に顕著である。高用量、すなわち25および50μg/mlのKGI 1で処置した細胞は、6日間の培養中全ての時点で検出可能なTK活性がすべて消失したが、培養液中にTK活性が検出され(図9.5)、細胞膜の損傷を示唆した。これらの濃度の遊離KGI 1は溶解作用を有することに注意すべきである(実施例2、表2.1)。対照的に、微粒子KGI 1は検出可能な量のTKを細胞培養液に放出させなかった。
【0195】
図9.6は、低生理学的用量の2μg/mlのKGI 1が18および24時間処置した(すなわち、第2細胞周期の複製期に入る前の)細胞試料で記録される同期化U937細胞のTK活性を減少させることを示す。TK活性の阻害は低濃度では最初の8時間中生じなかった。
【0196】
図9.7は、生理学的低用量の2μg/mlのKGI 1は増殖を阻害し、培養中の細胞の生存性で測定した18時間曝露後に検出される癌細胞数を減少させることを示し、これは、図9.6に示すようにTK活性の低下と一致する。
【0197】
2μg/mlの濃度の微粒子および遊離KGI 1は、本実験で測定した、U937細胞を24時間後の細胞生存性と代謝を減少させた(図9.8)。0.5μg/mlの濃度の微粒子または遊離KGI 1は、24時間の処置期間後の細胞生存性を減少させた。0.5μg/mlによる細胞代謝の阻害は、遊離KGI 1処置後よりKGI 1処置後に顕著であった。一般に、微粒子KGI 1は、細胞代謝と生存性の両方で測定した癌細胞増殖のより有効な阻害剤であった。生細胞数と細胞代謝活性のよい相関もみられた。
考察と結論
【0198】
低用量の2μg/mlのKGI 1で処置したU937癌細胞は、非同期化細胞では24時間後に測定したときTK活性が減少せず、48時間後では減少したが、これはKGI 1阻害効果が第1周期の終わりであることを反映した(図9.2〜9.5)。同調化細胞ではTK活性の阻害はより早いようである(図9.6)。TK活性は、細胞増殖周期初期である。高濃度の25μg/mlのKGI 1は、TK活性をより早く減少させる。おそらく、KGI 1処置は、該細胞を細胞周期から抜け出させる。一般に、TK活性は、生理学的用量のKGI 1で5日間処置後に消失した。測定されたTK活性は、新たに産生されたチミジンキナーゼと、以前の周期からしばらくの間持続すると予想される残存するチミジンキナーゼの合計を含むことに注意すべきである。高用量の25μg/mlのKGI 1は、24時間のインキュベーション後に記録されるTK活性を50%に減少させ、高用量が細胞増殖周期のより初期に細胞増殖を妨げることを示唆する。TK活性の減少は代謝阻害(アラマーブルー分析)を反映し、それと一致し、トリパンブルー染色により測定した生細胞数を減少させる(図9.7および9.8)。同期化細胞培養の18時間KGI 1処置後の癌細胞数の減少は、第1細胞周期におけるKGI 1による阻害効果を示唆する。
【0199】
アポトーシスによる癌細胞の殺滅(実施例7)は、24時間に向かって引き起こされ、アポトーシスによる殺滅は、TK活性に対する高および低用量のKGI 1の初期作用と一致する。KGI 1誘導癌細胞殺滅の初期において、壊死細胞数は、非処置細胞数を超えなかった(実施例7参照)。PI(壊死)およびアネキシンV(アポトーシス)の両方で染色した累積細胞数は、KGI 1により引き起こされるアポトーシス細胞に大部分由来する。
【0200】
しかしながら、より高用量のKGI 1の初期作用が細胞周期のS期に先立つTK産生に直接影響を有することは排除できない。
【0201】
遊離KGI 1の癌殺滅作用は、試験したより高い濃度、すなわち25μg/mlおよび50μg/mlで特に顕著であるが、これらの濃度ではTKが培養液に放出され(図9.4)、細胞膜の損傷が示唆される。より低用量では遊離形は微粒子KGI 1よりTK活性を減少させる能力は低かった。
【0202】
高用量の25μg/mlおよび50μg/mlによるKGI 1の初期癌殺滅作用(すなわち24時間前)は、おそらくTK産生後の細胞に影響を及ぼす低濃度の2μg/mlおよび10μg/mlより細胞周期の初期に発揮される異なるメカニズムを指摘する。より高い濃度の25μg/mlおよび50μg/mlの微粒子KGI 1は、遊離形のKGI 1、すなわちQuill AのQHC分画のようには溶解性でない(実施例2)。
【0203】
結論として、微粒子KGI 1は、高用量でも一次(primary)壊死を生じること無く癌細胞を殺す。TK活性は、アポトーシス死とほぼ同時に妨げられ、アラマーブルーで測定した細胞代謝の阻害およびトリパンブルー染色で測定した細胞の殺滅と一致する。KGI 1は試験した用量で溶解作用を示さない。持続培養でも逆転はみられない。KGI 1による癌細胞殺滅作用は、細胞周期から抜け出す概念に従い、活性化および産生期、最終的にアポトーシスをもたらす。これに対して、遊離形のKGI 1、すなわちQuill AのQHC分画は、アポトーシスに加えて、壊死、高用量では溶解作用により殺す。遊離KGI 1は、低生理学的用量で用いると癌細胞の殺滅に有効性が低い。高有効濃度の遊離KGI 1は副作用を引き起こす。
【実施例10】
【0204】
KGI製剤はU937細胞にIL-8サイトカイン産生を誘導する
リポ多糖(LPS)化合物は、KGI 1の用量を超える用量を用いてU937細胞のサイトカイン産生(IL-8)を刺激することが示された。別の実験では、Baldridgeら(Baldridge, Cluff et al. 2002)は、アミノアシルグルコサミニド 4-ホスフェート(APG)がサイトカイン産生で測定したU937細胞に対するそのような刺激作用を有することをクレームした。しかしながら、その能力を達成するには、培養をホルボール-12-ミレステート-13-アセテート(PMA)で前処置して分化および活性化を促進する必要があった。本実施例は、KGI 1、2、および3が、サイトカイン産生期に入るためにさらなる活性化−分化用化合物を必要とせず、必要なKGI用量は低いことを示す。さらに、実施例7および9は、KGI 1が、U937癌細胞を、KGI 1で処置しない限り閉じこめられる細胞複製周期から抜け出させることを示す。実施例7および9は、KGI 1がU937細胞をアポトーシスにすることも示す。アポトーシスは、正常細胞がその仕事、例えばサイトカインの産生が尽きた時の最終段階である。本実施例では、U937細胞にサイトカインIL-8の産生を誘導する3つのKGI製剤、すなわちKGI 1、2、および3の能力を試験する。
【0205】
KGI 1、2、および3は、「材料と方法」に記載したように製剤化し、図10.1、10.2、および10.3に示す濃度でマイクロタイタープレート中の106/mlのU937細胞に加えた。37℃で2日間インキュベーションした後、上清中のIL-8を測定した。LC50でのIL-8産生もこれらの図に示すように計算した。
結 果
【0206】
IL-8の産生は、KGI 1についてはLC50で781 pg/ml(Fig.10.1)、KGI 2については881 pg/ml(図10.2)、およびKGI 3については916 pg/mlであった(図10.3)。異なるKGI製剤はこれらの図に示すように程度の異なる細胞毒性も生じる。
考察と結論
【0207】
U937細胞は、サイトカインを自発的に産生しない。Baldridgeら(Baldridge, Cluff et al. 2002)は、高用量のある種のAPGがU937細胞のサイトカイン産生を刺激しうる(例えば高用量の(MPA)の前処置を必要とする)ことをクレームした。本実施例は、サイトカイン産生期に入るためにKGI製剤がMPAまたは同様の処置を必要としないことを示す。さらに、実施例9は、KGI 1がU937細胞をKGI 1で処理しない限り癌細胞が捕らわれる細胞複製周期から出させることを示す。実施例4は、KGI 1がU937細胞をアポトーシスにすることを示す。アポトーシスは、正常細胞がその仕事、例えばサイトカインの産生が尽きた時の最終段階である。本実施例では、U937細胞にサイトカインIL-8の産生を誘導する3つのKGI製剤、すなわちKGI 1、2、および3の能力を試験する。結論として、種々のKGI製剤はアポトーシスに先立ってサイトカイン8の産生を誘導する。また、BBEもIL-8産生を誘導する(示さず)。
【実施例11】
【0208】
マウスにおけるKGI 1およびBBEの急性毒性
癌治療を意図する薬剤は、その大部分が、癌治療に可能性がある化合物が臨床試験に進行しないかまたは市販されるようにならない最も一般的な原因であるバイオアベイラビリティの低さと共に副作用を生じる。キラヤサポニンは動物に遊離形で50年間以上用いられてきた。腫脹、発赤、および圧痛(壊死も)の形の局所反応が副作用として現れ、用量を制限することはよく知られている。その種の副作用は、QHCをイスコムおよびイスコムマトリックス製剤に組み込んだ後は同様の用量を用いても記録されなかった。10μgの遊離QHCは、マウスに筋肉内注射すると局所反応を誘導するが、イスコムまたはKGI製剤中に組み込まれたものは誘導しない。遊離サポニンはコレステロールと相互作用して、イスコムおよびKGI製剤中に組み込まれたサポニンと結合するコレステロールの阻害作用により避けられる細胞膜の損傷を生じることも良く認識されている。本実施例では、KGI 1およびBBEの全身毒性をBALB/cマウスで試験した。急性毒性は、「材料と方法」に記載のごとくBALB/cマウスにKGI 1およびBBEを皮下投与後に試験した。マウスを4日間記録した。試験結果を表11.1にまとめ、不活発度のスコアを表11.2に示す。
【0209】
本実施例では、KGI 1およびBBEの一般毒性を、組み込まれたサポニンの遊離形、すなわちKGI 1に組み込まれたキラヤサポニンの分画CおよびBBEに組み込まれた分画Aと比較検討する。用量10、30、および50μgのKGI 1、50および100μgのBBEをBALB/cマウスに皮下注射し、4日間観察した。
【0210】
表11.1 遊離および微粒子KGIの毒性の比較
【表4】
【0211】
表11.2 不活発度のスコア
【表5】
結 果
【0212】
効果を表11.1にまとめる。10μg用量のKGI 1では、マウスで如何なる検出可能な副作用も生じなかった。30μg用量は、3段階スケールで肝臓肥大0.13、脾臓肥大0.75、脾臓の黒さ(darkness)0.65と、ほとんど反応を生じなかった。また、50μg用量のKGI 1は、脾臓の肥大(2.0)および脾臓の黒さ(1.63)を除きわずかな変化のみ生じた。KGI 1は、アジュバント作用(すなわち免疫増強)を有し、脾臓の反応は正常反応由来であることに注意すべきである。これに対して、50μg用量の遊離形分画Cは、死亡率38%および高い不活発度スコア(1.8)、脾臓肥大(2.1)、脾臓の黒さ(2.8)、および下痢を含む重度の副作用を生じた。BBE粒子は、実質的に無毒性であることが知られており、唯一注目される反応はそのアジュバント作用によるものと思われる脾臓肥大である。
考察と結論
【0213】
計算したヒト用量(100μgのKGI 1)に近い50μgのKGI 1の全身反応は、60kgのヒトを考慮した18g BALB/cマウスにおいて副作用は低度であり、死亡はみられなかった。これに対し、この用量(50μg)の、KGI 1中のサポニンである遊離形のキラヤ分画Cは死亡と下痢をもたらした。100μg用量のKGI 1も死亡を生じ、KGI 1粒子のバイオアベイラビリティは分画Cより優れていることを考慮すべきである。BBE粒子は、実質的に副作用がないことが解った。唯一の顕著な作用は、その強い免疫調節作用を反映した脾臓肥大のスコアであった。先の研究は、QS 21およびQHCのような遊離形のキラヤサポニン分画は局所反応を生じ、これは、微粒子形により、KGIおよびBBE粒子および全ての形のイスコム粒子中のサポニンと結合したコレステロールの阻害効果により回避されることを示した。すなわち、記載した粒子中のサポニンは細胞膜中のコレステロールと相互作用することができない。
【実施例12】
【0214】
Killcan(キルキャン)は、免疫刺激を生じさせる立証されたイスコムデリバリーシステムに基づく癌治療のための新規用途および新規概念を確立する。伝統的癌殺滅系は激しく、場合により細胞正常細胞にも影響を及ぼす抗癌剤、細胞正常細胞にも影響を及ぼす放射線療法、および限界がある外科手術では重度の副作用が生じる。癌療法の現代的概念は、実施例7および9で考察したような複製の生理学を妨げることである。無制御な複製は、ほとんどの癌形にとって癌細胞の生存および病原性の主な原動力である。Killcan概念では、市販製品として、標的化細胞ポピュレーションおよび標的化細胞内コンパートメント、例えばエンド−およびリソゾームおよびサイトゾルによる化合物のデリバリーシステムとして証明された良く確立されたイスコム系を用いる。イスコム系を用いることにより、Killcan概念は、調節、活性化、分化により作用する正常な細胞の発達のための推進力であり、明確な無症候性の最後であるプログラムされた細胞死(アポトーシス)をもたらす。すなわち、本発明の癌療法概念では、優れたバイオアベイラビリティ、細胞および細胞内送達の標的化、低毒性、および他の生体機能特性について十分試験された300の刊行物に記載されたイスコム技術の得られた経験を用いる。資料は、Moreinらの最初の刊行物(Morein, 1984)で始まり、最近の包括的総説(Morein, 2007)まである。
【0215】
種々のBBEおよびKGI製剤の今回の分析は、Clinical Pharmacology, Uppsala UniversityのRolf Lasson教授のグループが彼らの技術(Dhar, 1996)を用いて行った。種々の組み合わせによる殺滅、増殖阻害、および相乗効果は、実際にミエローマ、リンパ肉腫、および固形癌に由来する癌細胞に対して予期しない包括的効果であった。さらに、治療できないかまたは治療しにくい腫瘍由来の回避突然変異および癌細胞は1またはそれ以上のKGIまたはBBE製剤に感受性であり、これは実際に予期できないことであった。
ヒト腫瘍細胞系パネル
【0216】
薬剤の活性パターンを評価するため、4つの感受性親細胞系、異なる耐性メカニズムを示す5つの薬剤耐性亜系、一次耐性を有する1細胞系のヒト細胞系パネル(Dhar, 1998)を用いた。含まれた細胞系は、ミエローマ細胞系RPMI 8226/Sおよびその亜系8226/Dox40および8226/LR-5(W.S. Dalton, Dept of Medicine, Arizona Cancer Center, University of Arizona, Tucson, AZから分与)、リンパ肉腫細胞系 U-937 GTBおよびU-937-Vcr(K. Nilsson, Dept of Pathology, University of Uppsala, Swedenから分与)、SCLC細胞系NCI-H69およびその亜系H69AR、乳癌MCF-7および頸癌Hela細胞系(American Type Culture Collection;ATCC, Rockville, MD)、腎臓腺癌細胞系ACHN(ATCC)および白血病細胞系CCRF-CEMおよびその亜系CEM/VM-1(W.T. Beck, Dept of Pharmacology, College of Medicine, University of Tennessee, Memphis, TNより分与)であった。
【0217】
8226/Dox40はドキソルビシン耐性について選択し、P-糖タンパク質170(Pgp)を過剰発現する古典的MDR表現型を示す。8226/LR-5は、GSHレベルの増加に関連することが提唱されているメルファラン耐性について選択した。U-937-Vcrは、チューブリンに関連することが提唱されているビンクリスチン耐性について選択した。ドキソルビシン耐性について選択したH69ARは、MRPにより仲介されることが提唱されているMDR表現型を発現する。テニポシド耐性について選択したCEM/VM-1は、トポイソメラーゼII(topoII)に関連することが提唱されている非定型的MDRを発現する。一次耐性ACHN細胞系に対する耐性の正確なメカニズムは知られておらず、多因子的であるかもしれない。
【0218】
該細胞系は、5% CO2を含む加湿大気中、37℃で3.2項に記載の完全培養液中で増殖させた。8226/Dox40は1月に1度0.24μg/mlのドキソルビシンで処理し、8226/LR-5は1.53μg/mlのメルファランで培地交換毎に処理した。U-937-Vcrは、10ng/mlのビンクリスチン存在下で連続培養し、H69ARは、薬剤不含培地および0.46μg/mlのドキソルビシンを含む培地で交互に培養した。CEM/VM-1細胞系は、6〜8月間如何なる耐性の損失もなしに薬剤不含培地中で培養された。該細胞系の耐性パターンはコントロール実験で日常的に確認した。
【0219】
表12.1. 本試験に用いたヒト腫瘍細胞系
【表6】
試薬および薬剤
【0220】
10%不活化FCS、2mMグルタミン、50μg/mlのストレプトマイシン、および60μg/mlのペニシリンを添加した炭酸緩衝培養液RPMI-1640(HyClone, Cramlington, UK)からなる完全培地を全体で用いた。FDA(Sigma, St Louis, MO)をDMSOに溶解し、遮光してストック溶液として凍結した(-20℃)。
【0221】
被検化合物は、DMSO中の10 mMストック溶液としてDueCom ABから得た。ストック溶液をリン酸緩衝生理食塩水(PBS;Sigma Aldrich)で10倍に希釈した。BIOMEK-2000ロボットシステムを用いて薬剤をさらに希釈し(10倍連続希釈により)、384ウェルのマイクロタイタープレート(NUNC)にプレートした。
蛍光分析的微量培養細胞毒性アッセイ(FMCA)
【0222】
腫瘍細胞を5,000細胞/ウェルの細胞密度で、薬剤を入れた384ウェルのマイクロタイタープレートに播いた。蛍光分析的微量培養細胞毒性アッセイ(FMCA)は、完全な原形質膜を持つ細胞によるFDAのフルオレセインへの加水分解により生じる蛍光の測定に基づき、これは先に詳述されている[14]。プレートを5% CO2を含む加湿大気中、37℃で72時間インキュベーションした。インキュベーション期間の終わりに吸引して除去した。PBSで1回洗浄後、生理学的緩衝液に溶解した50μl/ウェルのFDA(10μg/ml)を加えた。プレートを45分間インキュベーションし、各ウェルから生じた傾向を384ウェルスキャンニング蛍光測定器で測定した。蛍光はウェル中の完全な細胞の数に比例する。
【0223】
うまくいった分析の品質基準には、コントロールウェルの蛍光信号の5回以上の平均ブランク値、コントロールウェル中の平均変動係数(CV)30%以下が含まれた。
FMCA成績の定量
【0224】
細胞の生存は、ブランクウェルの値を引いた、実験ウェルの蛍光のコントロールウェルの蛍光に対するパーセントで定義した生存指数(SI)で示す。
結 果
【0225】
種々の組み合わせのKGIおよびBBE製剤は、3種類の試験した癌、すなわち、リンパ肉腫、ミエローマ、および固形癌に由来する癌細胞を殺した。試験した異なる製剤は、表12.2に示すように癌細胞の殺滅または成長阻害の異なる局面に及ぶ。
・KGI 1は、リンパ肉腫、ミエローマ、および小肺癌細胞H69ARの回避突然変異体を含む11種の被検細胞中7種に殺滅または成長阻害作用を有した。
・BBEは、上記モデルで使用したU937細胞および試験した正常細胞に実質的に無毒性である。予期しないことに、BBEは、2細胞、すなわちBBE耐性U937/GTB由来の回避であるU937/vcrに対して殺滅的/成長阻害的であった。より注目に値するのは、ACHN細胞は、KGI 1に耐性であったが、BBE、ならびにBBEの活性成分であるGHAも含むKGI 2に感受性であった。
・KGI 3は、11種の試験細胞中6種に殺滅または成長阻害作用を有した。白血病細胞がKGI 3製剤に最も感受性であった。KGI 2と共に、KGI 3は、乳癌HELA細胞に対する効果があった唯一の被検製剤であり、他の製剤に耐性であるときにも強い効果がみられた。
・KGI 2は、11種の試験した細胞中5種に殺滅または成長阻害作用を有した。KGI 3と共に、KGI 2は、乳癌HELA細胞に効果(強い効果)があった唯一の製剤であった。
・BBE/KGI 1は、11種の試験細胞中5種に殺滅または成長阻害作用を有した。KGI 1に耐性であった一次耐性ACHN細胞および耐性ミエローマ細胞8226/dox40に対する強い効果は顕著であり、さらに、これら細胞はKGI 2にも感受性であったが、他の被検製剤には感受性でなかった。
考察と結論
【0226】
細胞調節、活性化、分化およびアポトーシス特性に基づく種々の製剤は、驚くほど広範囲の癌殺滅または成長阻害特性を示す。該成分はサポニンであるがサポニンの溶解作用を持たないという事実にも関わらず、該成分は、全ての製剤が試験した異なる癌細胞に対する作用に関してはっきりと異なるプロフィールを持つことで生じる明らかな補完的効果を有する。癌細胞に対する殺滅または成長阻害について試験した製剤による全プロフィールは、11種の試験癌細胞のうち10種に及んだ。全く非感受性の唯一の細胞は、小肺癌細胞系H69であった。回避突然変異体は程度が限られるがKGI 1およびKGI 3に感受性であった。
【0227】
結論として、正常細胞により十分許容されることが証明された十分確認された系は、広範囲の種類の癌に及ぶ強力な癌殺滅または増殖阻害特性を有することが予期せずわかった。癌を殺す粒子のよく確認された送達特性の観点から、該系はサポニン物質ならびに好ましくは他のメカニズムにより作用する他の抗癌剤との組み合わせ療法に十分適している。この予測された効果の裏付けは、癌細胞が最初に感受性であった他の化合物により引き起こされた回避突然変異体に対する種々の製剤の能力である。
【0228】
表12.2:IC50(μg/ml)で表した、リンパ性、骨髄性、および固形腫瘍(表12.1)由来の種々の癌細胞の殺滅/成長阻害。1および同じ粒子中の単独化合物、すなわちBBEおよびKGI 1、1粒子中の種々のキラヤサポニン分画の組み合わせ(KGI 3)、1および同じ粒子中の2成分(QHAおよびQHC、すなわちKGI 2)、および別の粒子中のKGI 1と混合したBBE粒子(比4:1 BBE/KGI 1)の種々の効果
【表7】
【0229】
参考文献
Baldridge, J. R., C. W. Cluff, et al.(2002). "Immunostimulatory activity of aminoalkyl glucosaminide 4-phosphates(AGPs): induction of protective innate immune responses by RC-524 and RC-529." J Endotoxin Res 8(6): 453-8.
Berendt, M. J. and J. L. Ives(1985). "Developmental status of synthetic immunomodulators." Year Immunol: 193-201.
Blair, A. H. and T. I. Ghose(1983). "Linkage of cytotoxic agents to immunoglobulins." J Immunol Methods 59(2): 129-43.
Cheng, K. T., S. E. Seltzer, et al.(1987). "The production and evaluation of contrast-carrying liposomes made with an automatic high-pressure system." Invest Radiol 22(1): 47-55.
Conradie, J. D., M. Govender, et al.(1983). "ELISA solid phase: partial denaturation of coating antibody yields a more efficient solid phase." J Immunol Methods 59(3): 289-99.
Dalsgaard(1978). "A study of the isolation and characterization of the saponin Quil A. Evaluation of its adjuvant activity, with a special reference to the application in the vaccination of cattle against foot-and-mouth disease." Acta Vet Scand Suppl(69): 7-40.
Dalsgaard, K.(1974). "Saponin adjuvants. 3. Isolation of a substance from Quillaja saponaria Molina with adjuvant activity in food-and-mouth disease vaccines." Arch Gesamte Virusforsch 44(3): 243-54.
Davis, M. T. and J. F. Preston(1981). "A simple modified carbodiimide method for conjugation of small-molecular-weight compounds to immunoglobulin G with minimal protein crosslinking." Anal Biochem 116(2): 402-7.
Dhar, S., P. Nygren, et al.(1996). "Anti-cancer drug characterisation using a human cell line panel representing defined types of drug resistance." Br J Cancer 74(6): 888-96.
Dhar, S., P. Nygren, et al.(1998). "Relationship between cytotoxic drug response patterns and activity of drug efflux transporters mediating multidrug resistance." Eur J Pharmacol 346(2-3): 315-22.
Espinet, R. G.(1951). "Nuevo tipo de vacuna antiaftosa a complejo glucovirico." Gac. Vet. 74: 1-13.
Ghose, T. I., A. H. Blair, et al.(1983). "Preparation of antibody-linked cytotoxic agents." Methods Enzymol 93: 280-333.
Gregoriadis, G., ed.(1984). Liposome Technology Vol. l, 29-31,51-67 and 79-108 (CRC Press Inc., Boca Raton, Fla.)
Gregoriadis, G., B. McCormack, et al.(1999). "Vaccine entrapment in liposomes." Methods 19(1): 156-62.
Higuchi, R. et al.(1988). An Acylated Triterpenoid Saponin From Quillaja saponaria, Phytochemistry 27(4):1165-1168.
Hope et al.(1985). Biochimica et Biophysica Acta, Vol. 812, pp. 55-65.
Hostettmann, K.; Marston, A.(1995). Saponins. Cambridge, U.K.: Cambridge Univ. Press.
Johnson, A. G., M. Tomai, et al.(1987). "Characterization of a nontoxic monophosphoryl lipid A." Rev Infect Dis 9 Suppl 5: S512-6.
Kensil, C. R., U. Patel, et al.(1991). "Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex." J Immunol 146(2): 431-7.
Kersten, G. F., A. Spiekstra, et al.(1991). "On the structure of immune-stimulating saponin-lipid complexes(iscoms)." Biochim Biophys Acta 1062(2): 165-71.
Leung A Y., and Foster S.(1996). Encyclopedia of Common Natural Ingredients Used in Food, Drugs and Cosmetics, 2nd ed., John Wiley and sons(Wiley Interscience), New York.
Lipford, G. B., H. Wagner, et al.(1994). "Vaccination with immunodominant peptides encapsulated in Quil A-containing liposomes induces peptide-specific primary CD8+ cytotoxic T cells." Vaccine 12(1): 73-80.
Lovgren, K. and B. Morein(1988). "The requirement of lipids for the formation of immunostimulating complexes(iscoms)." Biotechnol Appl Biochem 10(2): 161-72.
Lovgren, K. and B. Morein(1991). "The ISCOM: an antigen delivery system with built-in adjuvant." Mol Immunol 28(3): 285-6.
Lycke, N.(2004). "From toxin to adjuvant: the rational design of a vaccine adjuvant vector, CTA1-DD/ISCOM." Cell Microbiol 6(1): 23-32.
Ma, J., P. A. Bulger, et al.(1994). "Impact of the saponin adjuvant QS-21 and aluminium hydroxide on the immunogenicity of recombinant OspA and OspB of Borrelia burgdorferi." Vaccine 12(10): 925-32.
Madden, T. D., P. R. Harrigan, et al.(1990). "The accumulation of drugs within large unilamellar vesicles exhibiting a proton gradient: a survey." Chem Phys Lipids 53(1): 37-46.
Martin, R. G. and B. N. Ames(1961). "A method for determining the sedimentation behavior of enzymes: application to protein mixtures." J Biol Chem 236: 1372-9.
Mayer, L. D., M. J. Hope, et al.(1986). "Vesicles of variable sizes produced by a rapid extrusion procedure." Biochim Biophys Acta 858(1): 161-8.
Mayhew, E., S. Conroy, et al.(1987). "High-pressure continuous-flow system for drug entrapment in liposomes." Methods Enzymol 149: 64-77.
Mayhew, E., R. Lazo, et al.(1984). "Characterization of liposomes prepared using a microemulsifier." Biochim Biophys Acta 775(2): 169-74.
Morein, B., B. Sundquist, et al.(1984). "Iscom, a novel structure for antigenic presentation of membrane proteins from enveloped viruses." Nature 308(5958): 457-60.
Morein, B., Hu, K. et al.(2007). "New Iscoms Meet Unsettled Vaccine Demands". Vaccine Adjuvants and Delivery Systems, Chapter 9: 191-222
Mowat, A. M., A. M. Donachie, et al.(2001). "CTA1-DD-immune stimulating complexes: a novel, rationally designed combined mucosal vaccine adjuvant effective with nanogram doses of antigen." J Immunol 167(6): 3398-405.
O´Hagan, DT. "Recent developments in vaccine delivery systems". Curr Drug Targets Infect Disord. 2001 Nov;1(3):273-86.
Ramon, G.(1926). Proc d s pour accroitre la production des antitoxines, Ann Inst Pasteur, 40, 1-10.
Rouhi A.M.(1995). Chem. Eng. News 73(37): 28-35.
Ronnberg, B., M. Fekadu, et al.(1997). "Effects of carbohydrate modification of Quillaja saponaria Molina QH-B fraction on adjuvant activity, cholesterol-binding capacity and toxicity." Vaccine 15(17-18): 1820-6.
Ronnberg, B., M. Fekadu, et al.(1995). "Adjuvant activity of non-toxic Quillaja saponaria Molina components for use in ISCOM matrix." Vaccine 13(14): 1375-82.
Stewart-Tull, D. E.(1985). "Immunopotentiating conjugates." Vaccine 3(1): 40-4.
Wang, Z.P.(2005). United States Patent 20050175623
Warren, H. S. and L. A. Chedid(1988). "Future prospects for vaccine adjuvants." Crit Rev Immunol 8(2): 83-101.
【特許請求の範囲】
【請求項1】
癌治療用の医薬を製造するための、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子の使用。
【請求項2】
サポニンが植物から得られるグリコシドである請求項1〜4のいずれかに記載の使用。
【請求項3】
植物グリコシドが1またはそれ以上の糖部分を有するサポゲニンおよびプロサポゲニンから選ばれる請求項2記載の使用。
【請求項4】
該グリコシドがQuillaja Saponaria Molina由来の粗サポニン分画またはその亜分画である請求項3記載の使用。
【請求項5】
サポニンが、Quillaja Saponaria Molinaの分画A、分画B、分画C、分画QA 1〜22、Spicoside(スピコシド)、およびQ VACから選ばれる請求項4記載の使用。
【請求項6】
サポニン分画が、親水性の、例えば、脂肪酸を含まない分画、例えば分画A、Quil 4-15であり、抗体産生および細胞介在性免疫のような免疫反応を刺激および調節するものである請求項2〜5のいずれかに記載の使用。
【請求項7】
サポニン分画が、疎水性の、例えばサポニンの4位に脂肪酸を含む分画、例えばQuil Aの分画CおよびB、または分画AとBの間の領域由来の分画、Quillaja Saponaria Molinaの分画QA15〜21であり、癌細胞の殺滅作用を有するものである請求項2〜5のいずれかに記載の使用。
【請求項8】
脂質含有粒子がさらに親水性サポニンを含む請求項7記載の使用。
【請求項9】
脂質含有粒子が1つおよび同じ脂質含有粒子中に少なくとも2の異なるサポニン分画を含む請求項1〜8のいずれかに記載の使用。
【請求項10】
脂質含有粒子が少なくとも2の異なるサポニン分画を含み、該少なくとも2の異なるサポニン分画の1つが1つの脂質含有粒子中に複合結合しており、該少なくとも2の異なるサポニン分画の他の1つ(他のもの)が別の(他の)物理的に異なる脂質含有粒子中に複合結合している、請求項1〜9のいずれかに記載の使用。
【請求項11】
該異なるサポニンが親水性および疎水性サポニンである請求項12または13記載の使用。
【請求項12】
少なくとも1の親水性サポニン、例えばQuil A由来の分画Aを含む脂質含有粒子の混合物を、相乗的抗癌作用をうるために少なくとも1の疎水性サポニン、例えばQuil A由来の分画Cを含む脂質含有粒子と一緒に用いる請求項11記載の使用。
【請求項13】
該脂質含有粒子が該粒子に統合され、該粒子と結合し、または該脂質含有粒子と混合された癌抗原を含む請求項1〜12のいずれかに記載の使用。
【請求項14】
さらに癌治療用化合物を脂質含有粒子と一緒に用いる請求項1〜13のいずれかに記載の使用。
【請求項15】
該癌治療用化合物が、白金配位化合物、タキサン化合物、カンプトテシン化合物、抗腫瘍ビンカアルカロイド、抗腫瘍ヌクレオシド誘導体、ナイトロジェンマスタードまたはニトロソウレアアルキル化剤、抗腫瘍アントラサイクリン誘導体、トラストズマブおよび抗腫瘍ポドフィロトキシン誘導体、Quila A、ならびにそれらのサブフラグメントから選ばれる請求項14記載の使用。
【請求項16】
さらにアジュバントが該粒子中に統合され、該粒子と結合し、または該脂質含有粒子と混合されている請求項1〜15のいずれかに記載の使用。
【請求項17】
該脂質含有粒子が、癌標的化分子、例えば、癌細胞由来の表面抗原、ウイルス表面抗原、およびインフルエンザ抗原を含む請求項1〜16のいずれかに記載の使用。
【請求項18】
該粒子がリポソーム、イスコムおよび/またはイスコムマトリックス、およびPosintros(ポシントロス)から選ばれる請求項1〜17のいずれかに記載の使用。
【請求項19】
少なくとも1のサポニン、少なくとも1の脂質、および少なくとも1種類の抗原物質を含むイスコムを用いる請求項18記載の使用。
【請求項20】
少なくとも1のサポニンおよび少なくとも1の脂質を含むイスコムマトリックスを用いる請求項18記載の使用。
【請求項21】
少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を癌治療を必要とする個体に投与する癌の治療方法。
【請求項22】
少なくとも2の部分を含む部分のキットであって、1つの部分が、癌細胞の殺滅作用を有する疎水性の少なくとも1のサポニン分画を含む脂質含有粒子を含み、その他の部分が免疫反応、例えば抗体産生および細胞介在性免疫を刺激および調節する親水性の少なくとも1のサポニン分画を有する脂質含有粒子を含むキット。
【請求項1】
癌治療用の医薬を製造するための、少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子の使用。
【請求項2】
サポニンが植物から得られるグリコシドである請求項1〜4のいずれかに記載の使用。
【請求項3】
植物グリコシドが1またはそれ以上の糖部分を有するサポゲニンおよびプロサポゲニンから選ばれる請求項2記載の使用。
【請求項4】
該グリコシドがQuillaja Saponaria Molina由来の粗サポニン分画またはその亜分画である請求項3記載の使用。
【請求項5】
サポニンが、Quillaja Saponaria Molinaの分画A、分画B、分画C、分画QA 1〜22、Spicoside(スピコシド)、およびQ VACから選ばれる請求項4記載の使用。
【請求項6】
サポニン分画が、親水性の、例えば、脂肪酸を含まない分画、例えば分画A、Quil 4-15であり、抗体産生および細胞介在性免疫のような免疫反応を刺激および調節するものである請求項2〜5のいずれかに記載の使用。
【請求項7】
サポニン分画が、疎水性の、例えばサポニンの4位に脂肪酸を含む分画、例えばQuil Aの分画CおよびB、または分画AとBの間の領域由来の分画、Quillaja Saponaria Molinaの分画QA15〜21であり、癌細胞の殺滅作用を有するものである請求項2〜5のいずれかに記載の使用。
【請求項8】
脂質含有粒子がさらに親水性サポニンを含む請求項7記載の使用。
【請求項9】
脂質含有粒子が1つおよび同じ脂質含有粒子中に少なくとも2の異なるサポニン分画を含む請求項1〜8のいずれかに記載の使用。
【請求項10】
脂質含有粒子が少なくとも2の異なるサポニン分画を含み、該少なくとも2の異なるサポニン分画の1つが1つの脂質含有粒子中に複合結合しており、該少なくとも2の異なるサポニン分画の他の1つ(他のもの)が別の(他の)物理的に異なる脂質含有粒子中に複合結合している、請求項1〜9のいずれかに記載の使用。
【請求項11】
該異なるサポニンが親水性および疎水性サポニンである請求項12または13記載の使用。
【請求項12】
少なくとも1の親水性サポニン、例えばQuil A由来の分画Aを含む脂質含有粒子の混合物を、相乗的抗癌作用をうるために少なくとも1の疎水性サポニン、例えばQuil A由来の分画Cを含む脂質含有粒子と一緒に用いる請求項11記載の使用。
【請求項13】
該脂質含有粒子が該粒子に統合され、該粒子と結合し、または該脂質含有粒子と混合された癌抗原を含む請求項1〜12のいずれかに記載の使用。
【請求項14】
さらに癌治療用化合物を脂質含有粒子と一緒に用いる請求項1〜13のいずれかに記載の使用。
【請求項15】
該癌治療用化合物が、白金配位化合物、タキサン化合物、カンプトテシン化合物、抗腫瘍ビンカアルカロイド、抗腫瘍ヌクレオシド誘導体、ナイトロジェンマスタードまたはニトロソウレアアルキル化剤、抗腫瘍アントラサイクリン誘導体、トラストズマブおよび抗腫瘍ポドフィロトキシン誘導体、Quila A、ならびにそれらのサブフラグメントから選ばれる請求項14記載の使用。
【請求項16】
さらにアジュバントが該粒子中に統合され、該粒子と結合し、または該脂質含有粒子と混合されている請求項1〜15のいずれかに記載の使用。
【請求項17】
該脂質含有粒子が、癌標的化分子、例えば、癌細胞由来の表面抗原、ウイルス表面抗原、およびインフルエンザ抗原を含む請求項1〜16のいずれかに記載の使用。
【請求項18】
該粒子がリポソーム、イスコムおよび/またはイスコムマトリックス、およびPosintros(ポシントロス)から選ばれる請求項1〜17のいずれかに記載の使用。
【請求項19】
少なくとも1のサポニン、少なくとも1の脂質、および少なくとも1種類の抗原物質を含むイスコムを用いる請求項18記載の使用。
【請求項20】
少なくとも1のサポニンおよび少なくとも1の脂質を含むイスコムマトリックスを用いる請求項18記載の使用。
【請求項21】
少なくとも1の脂質および少なくとも1のサポニンを含む脂質含有粒子を癌治療を必要とする個体に投与する癌の治療方法。
【請求項22】
少なくとも2の部分を含む部分のキットであって、1つの部分が、癌細胞の殺滅作用を有する疎水性の少なくとも1のサポニン分画を含む脂質含有粒子を含み、その他の部分が免疫反応、例えば抗体産生および細胞介在性免疫を刺激および調節する親水性の少なくとも1のサポニン分画を有する脂質含有粒子を含むキット。
【図1.1】


【図1.2】
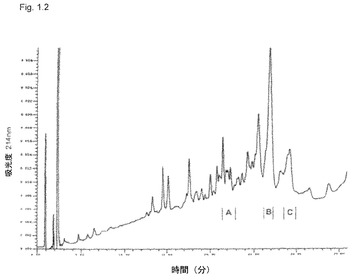
【図1.3】

【図2.1】
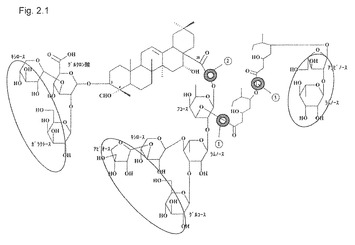
【図3.1】
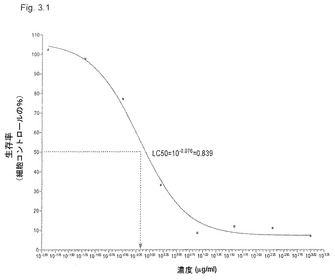
【図3.2】
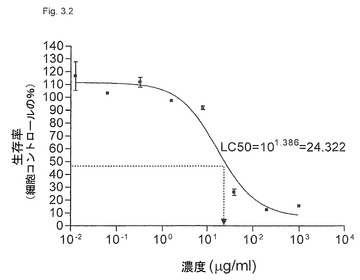
【図4.1】

【図4.2】

【図5.1】

【図6.1】

【図6.2】
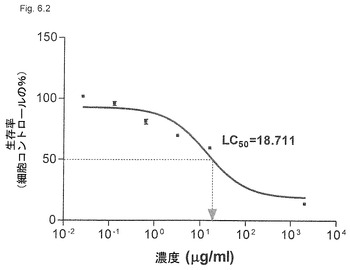
【図6.3】

【図6.4】

【図7.1】
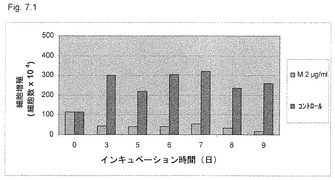
【図7.2】
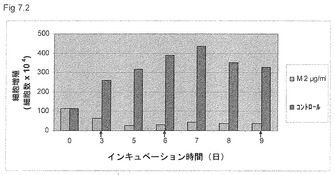
【図7.3】
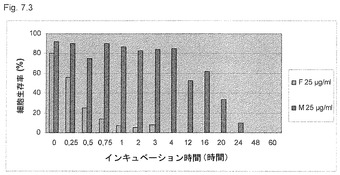
【図7.4】
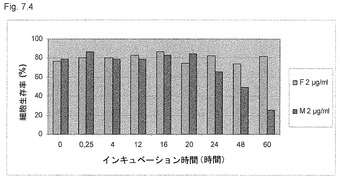
【図7.5】
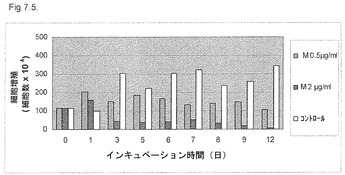
【図7.6】

【図7.7】

【図7.8】
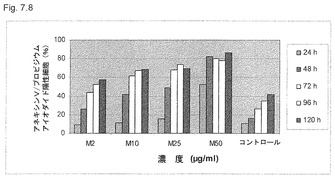
【図8.1】
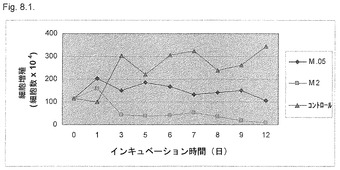
【図8.2】
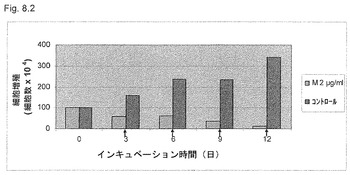
【図9.1】

【図9.2】
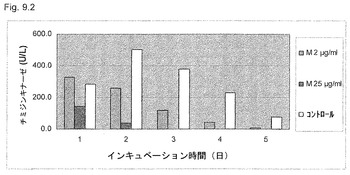
【図9.3】

【図9.4】
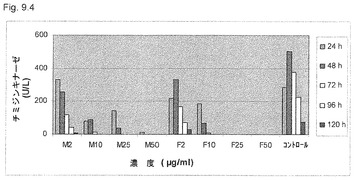
【図9.5】
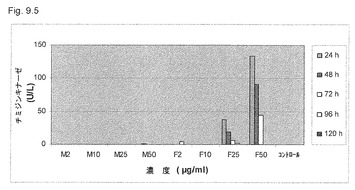
【図9.6】
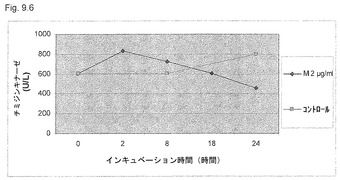
【図9.7】
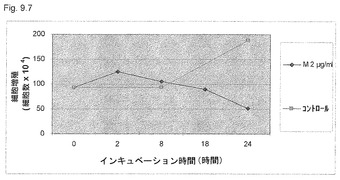
【図9.8】
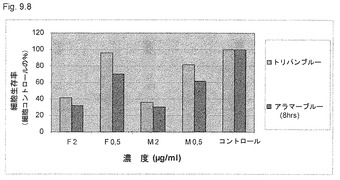
【図10.1】
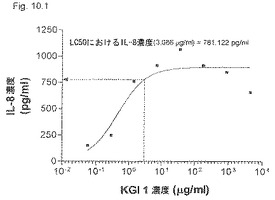
【図10.2】
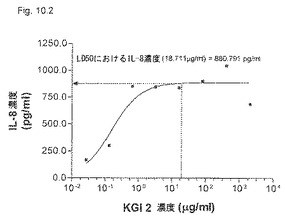
【図10.3】
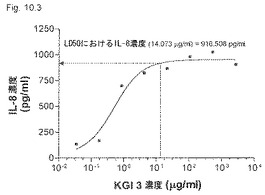
【図1.2】
【図1.3】
【図2.1】
【図3.1】
【図3.2】
【図4.1】
【図4.2】
【図5.1】
【図6.1】
【図6.2】
【図6.3】
【図6.4】
【図7.1】
【図7.2】
【図7.3】
【図7.4】
【図7.5】
【図7.6】
【図7.7】
【図7.8】
【図8.1】
【図8.2】
【図9.1】
【図9.2】
【図9.3】
【図9.4】
【図9.5】
【図9.6】
【図9.7】
【図9.8】
【図10.1】
【図10.2】
【図10.3】
【公表番号】特表2010−510308(P2010−510308A)
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願番号】特願2009−538372(P2009−538372)
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/SE2007/050878
【国際公開番号】WO2008/063129
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(509141899)
【氏名又は名称原語表記】DUECOM
【Fターム(参考)】
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/SE2007/050878
【国際公開番号】WO2008/063129
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(509141899)
【氏名又は名称原語表記】DUECOM
【Fターム(参考)】
[ Back to top ]