
酸化LDL/β2GPI複合体に対する抗体及びその用途
【課題】本発明は、特異的な粥状動脈硬化(粥腫ともいう)の存在部位の特定、治療効果モニタリングなどの非侵襲的な動脈硬化の診断法を提供することを課題とする。
【解決手段】 本発明者らは、動脈硬化の粥腫の構成物を鑑みて、酸化LDL/β2GPI複合体抗体が、in vivoで動脈硬化の粥腫に特異的に結合するか、かつ、体外からその病変を特異的に検出可能か検討した。
すなわち、生体中での、動脈硬化巣特に動脈硬化粥腫の存在位置及びその大きさのイメージングに適応可能な抗体を特定し、その特異性を解析した。その結果、動脈硬化切片の免疫染色ができる酸化LDL/β2GPI複合体抗体の中でも、特定のエピトープに特異性を有する酸化LDL/β2GPI複合体抗体の標識体が、粥状動脈硬化の存在部位の特異的なイメージングに有効であることが明らかとなった。このことは、粥腫という病変に特異的なイメージングを可能とする。
【解決手段】 本発明者らは、動脈硬化の粥腫の構成物を鑑みて、酸化LDL/β2GPI複合体抗体が、in vivoで動脈硬化の粥腫に特異的に結合するか、かつ、体外からその病変を特異的に検出可能か検討した。
すなわち、生体中での、動脈硬化巣特に動脈硬化粥腫の存在位置及びその大きさのイメージングに適応可能な抗体を特定し、その特異性を解析した。その結果、動脈硬化切片の免疫染色ができる酸化LDL/β2GPI複合体抗体の中でも、特定のエピトープに特異性を有する酸化LDL/β2GPI複合体抗体の標識体が、粥状動脈硬化の存在部位の特異的なイメージングに有効であることが明らかとなった。このことは、粥腫という病変に特異的なイメージングを可能とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、酸化LDL/β2GPI複合体に対する抗体、及び該抗体を利用した粥状動脈硬化の存在部位の特定、治療効果モニタリングなどの非侵襲的な動脈硬化の診断法に関する。
【背景技術】
【0002】
動脈硬化の状態を観察する現在実用化されている診断方法としては、以下の4種類の方法が例示される。
「足関節上腕血圧比」:横になった状態で両腕、両足の血圧を測定すると、正常では足首の方がやや高い値になる。ところが血管につまりがある場合、そこから下流の血圧が低下し、足関節の血圧/上腕の血圧の比(ABI)が低くなる。ABIの低下は、下肢動脈だけでなく、全身に動脈硬化があることを推定する。
「脈波速度検査」:動脈の硬さを調べる方法で動脈硬化の進行を推定する方法。健康人は血管に弾力性があるため、振動が血管壁で吸収され、脈波の速度は遅い。動脈硬化が進むと、脈波は速くなるので、速度を指標として動脈硬化の進行を推定することができる。
「頸動脈超音波検査」:皮膚表面近くを走る頸動脈は内部の様子を超音波で観察しやすいので、この頸動脈を調べ、全身の動脈硬化の進行状況を推定する方法。
「MR血管造影(MRA)」:「CT血管造影(CTA)」血管疾患の画像診断法として従来血管造影が主流であったが、より侵襲が少なく血管造影にほぼ匹敵する画像情報が用いられるようになった。CTAの利点は、(1)空間分解能が高い、(2)検査が簡便、(3)石灰化病変の検出に優れる点が挙げられる。
上記「足関節上腕血圧比」、「脈波速度検査」では、粥状化位置特定と個別箇所ごとの進行状況の診断はできず、間接的に数値として評価せざるを得ない。
また、「頸動脈超音波検査」は、脈波速度検査などと違い、実際に血管の内部を画像として直接的に診ることができる点が優れている。しかし、超音波画像の濃淡や形から血管壁の状況を読みとる必要があるため、検査を行う医師や検査技師に技量が求められ、頸動脈以外の部位の、粥状化位置特定と個別箇所ごとの進行状況の診断はできない。
【0003】
また、動脈硬化進行状況をモニタリングする方法としては、血中の酸化LDL/β2GPI複合体を測定することによる、ELISA系が存在する(特許3370334号、特許3898680号、WO2003/022866、WO2004/023141)。しかしながら、従来の酸化LDL/β2GPI複合体測定ELISAでは、動脈硬化巣の大きさを推定することは出来るが、動脈硬化巣の存在部位は特定できない。
さらに、MRIや放射ラベルを用いたイメージングでも画像の濃淡や形から血管壁の状況を読みとる必要があるため、検査を行う医師や検査技師に技量が求められる(米国特許6716410号、米国特許6375925号)。
なお、以下に本発明に関連する先行技術文献を示す。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特許3370334号
【特許文献2】特許3898680号
【特許文献3】WO2003/022866
【特許文献4】WO2004/023141
【特許文献5】特表2001−506983
【特許文献6】特許第4044972
【特許文献7】米国特許6716410号
【特許文献8】米国特許6375925号
【非特許文献】
【0005】
【非特許文献1】Journal of Biological Chemistry 269, 15274-15279,1994
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、特異的な粥状動脈硬化(粥腫ともいう)の存在部位の特定、治療効果モニタリングなどの非侵襲的な動脈硬化の診断法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、動脈硬化の粥腫の構成物を鑑みて、酸化LDL/β2GPI複合体抗体が、in vivoで動脈硬化の粥腫に特異的に結合するか、かつ、体外からその病変を特異的に検出可能か検討した。
すなわち、生体中での、動脈硬化巣特に動脈硬化粥腫の存在位置及びその大きさのイメージングに適応可能な抗体を特定し、その特異性を解析した。その結果、動脈硬化切片の免疫染色ができる酸化LDL/β2GPI複合体抗体の中でも、特定のエピトープに特異性を有する酸化LDL/β2GPI複合体抗体の標識体が、粥状動脈硬化の存在部位の特異的なイメージングに有効であることが明らかとなった。このことは、粥腫という病変に特異的なイメージングを可能とする。
即ち本発明は、以下〔1〕〜〔12〕を提供するものである。
〔1〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する、下記(a)〜(j)のいずれかに記載の抗体;
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体、
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
〔2〕 〔1〕に記載の抗体が結合するエピトープと同じエピトープに結合する抗体。
〔3〕 ヒト化抗体またはキメラ抗体である、〔1〕または〔2〕に記載の抗体。
〔4〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング剤。
〔5〕 〔1〕〜〔3〕のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング剤。
〔6〕 動脈硬化において、粥腫の位置及び/または大きさをイメージングする、〔4〕または〔5〕のイメージング剤。
〔7〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング用キット。
〔8〕 〔1〕〜〔3〕のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング用キット。
〔9〕 以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法:
(a)〔1〕〜〔3〕のいずれかに記載の抗体が投与された動脈硬化疾患モデル非ヒト動物に、候補化合物を投与する工程、
(b)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣の大きさまたは存在部位を比較する工程、及び
(d)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
〔10〕 〔1〕〜〔3〕に記載の抗体を含む、動脈硬化の存在部位のイメージング方法。
〔11〕 動脈硬化の存在部位のイメージング剤の製造における、〔1〕〜〔3〕に記載の抗体の使用。
〔12〕 動脈硬化の存在部位のイメージング方法に使用するための、〔1〕〜〔3〕に記載の抗体。
【図面の簡単な説明】
【0008】
【図1】BALB/cマウスに抗原として酸化LDL/β2GPI複合体を免疫して得られたモノクローナル抗体の、固相抗原に対する反応性を示す図である。ヨコ軸は抗体濃度、縦軸には吸光度を示す。
【図2】液中の抗原との競合性をアッセイ(競合阻害試験)するための模式図である。
【図3】抗原による競合阻害試験を示すグラフである。ヨコ軸は液中の抗原濃度、縦軸には阻害抗原が存在しない場合の吸光度を100%としたときの阻害された%を示す。3H3、および4C12は、酸化LDLに結合したβ2GPIを認識する抗体で、遊離のβ2GPIを認識していない。一方、2H6、3D4、2A12は、遊離のβ2GPIに反応する抗体である。
【図4】動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色写真である。A)DAPI:核の染色、B)Mac3:マクロファージに特異的抗体、C)3H3抗体、D)control。C57BL6系統のマウスを普通飼料で飼育したものに対し、3H3抗体。Mac3を用いる免疫蛍光染色では、泡沫化マクロファージが集族してできた粥腫が染まっている。3H3によっても同様な部位が染まっている。
【図5】動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色写真である。酸化LDL/β2GPI複合体抗体を用いた他の抗体の蛍光免疫染色の結果を示す。粥腫の染色陽性例は、3H3および抗体Aのみである。
【図6】IVIS 200による特異抗体を用いた蛍光イメージング(反射蛍光観察)写真である。in vivo:高脂肪食6ヵ月以上負荷したApoE-/-マウスに、イメージング剤を、脂肪食負荷ApoE-/-マウスの尾静脈より投与し、2〜24時間後に、IVIS 200にて麻酔吸入下でin vivo蛍光を観察・撮影した。その際ApoE-/-マウスでは黒い体毛が蛍光を吸収するため体毛を剃って観察した。 ex vivo:マウスを安楽死させ、開胸して心臓と大動脈を露出させ、右心耳に小さく切り込みを入れた後、左心室に針を挿入して冷PBS 10 mlで心灌流し、心臓および大動脈を摘出して、IVIS 200により反射蛍光画像を得た。
【図7】IVIS200によるイメージング写真である(Excitation:640 nm, Emission:720 nm.)。実験1:高脂肪食負荷apoE-/-マウスに生理食塩水(PBS、対照)、Cy5.5標識抗体A、Cy5.5標識3H3抗体を尾静脈より投与。24時間後に胸部皮膚を剥がし、生きた状態で全身を撮影。更に胸部大動脈が繋がった状態で心臓を摘出して撮影。実験2:PBS、Cy5.5標識2A12抗体、Cy5.5標識3H3抗体投与マウスから摘出した心臓と胸部大動脈。3H3投与で大動脈起始部が強く染まる。抗体Aでも幾分染まるが3H3の場合ほど蛍光強度は強くない。2A12では全く染まらない。
【図8】特異抗体を用いた動脈硬化の3次元画像イメージング写真である。(A) IVIS 200による特異抗体を用いた蛍光イメージング(反射)、(B) IVIS200による透過光 3D 画像(左図)と統合前のCT-3D像(中央)と統合後の3D画像(右図)、(C) IVISによる蛍光シグナルと3次元CTの統合画像である。
【図9】統合前のIVIS 200による蛍光3次元画像(上段:A)及びIVIS 200による蛍光シグナルと3次元CTの統合画像(下段:B)の写真である。
【図10】IVIS200で測定した際の大動脈起始部周辺のCy5.5に依存する蛍光量を示す図である。大動脈起始部の一定面積あたりの蛍光強度を測定した。PBSを投与した対照マウスの蛍光を1.Oとした。3H3投与で、対照の3倍程度の蛍光が確認されたその他の抗体投与では蛍光強度に大きな変化は見られなかった。
【図11】3H3抗体アミノ酸配列図である。各CDRを下線で示した。
【図12】マウスH鎖からヒト化 H鎖への変換に関する図である。(A)マウスH鎖と移植先ヒト H鎖のFR 部分の相同性を比較したものである。すなわちマウス3H3抗体の重鎖(H鎖)とヒト抗体AY882577のアミノ酸を比較した図である。H鎖のFR 部分については、87アミノ酸中80アミノ酸が一致しており、91.9%の相同性があった。(B)マウスH鎖からヒト化 H鎖への変換過程を示す。マウス3H3抗体の重鎖をKabat則によりナンバリングし、CDR1〜CDR 3、FR1〜FR 4を同定した。Kabat則の詳細については、http://www.bioinf.org.uk/abs/numbering.htmlを参照のこと。マウス3H3抗体FR1〜FR4と相同性のあるヒトFR1〜FR4を有するAY882577を同定し、マウス3H3 CDR1〜CDR 3を移植した3H3RHAを設計した。図中の3H3 VHはマウス3H3抗体の重鎖であり、3H3CDRは3H3重鎖のCDR1〜CDR 3であり、AY882577 FRはAY882577のCDRを除いた重鎖フレームワークである。
【図13】マウスL鎖からヒト化 L鎖への変換に関する図である。(A)マウスL鎖と移植先ヒト L鎖FR 部分の相同性を比較したものである。すなわちマウス3H3抗体の軽鎖(L鎖)とヒト抗体AY942004のアミノ酸を比較した図である。L鎖のFR 部分については、80アミノ酸中65アミノ酸が一致しており、81.3%の相同性があった。(B)マウスL鎖からヒト化 L鎖への変換過程を示す。マウス3H3抗体の軽鎖をKabat則によりナンバリングし、CDR1〜CDR 3、FR1〜FR 4を同定した。Kabat則の詳細については、http://www.bioinf.org.uk/abs/numbering.htmlを参照のこと。マウス3H3抗体FR1〜FR4と相同性のあるヒトFR1〜FR4を有するAY942004を同定し、マウス3H3 CDR1〜CDR 3を移植した3H3RKAを設計した。図中の3H3 VKはマウス3H3抗体の軽鎖であり、3H3CDRは3H3 L鎖のCDR1〜CDR 3であり、AY942004FRはAY942004のCDRを除いた軽鎖フレームワークである。
【図14】3H3RHAの塩基配列及びアミノ酸配列を示す図である。シグナル配列を含めた、3H3RHAの塩基配列及びアミノ酸配列を示す。シグナル配列は下線で示した。
【図15】3H3RKAの塩基配列及びアミノ酸配列を示す図である。シグナル配列を含めた、3H3RKAの塩基配列及びアミノ酸配列を示す。シグナル配列は下線で示した。
【図16】抗原に対するELISA活性(ヒト化IgG1とキメラIgG1の比較)を示すグラフである。横軸に抗体濃度、縦軸にELISA で測定された吸光度(650nm)を示す。ヒト化IgG1とキメラIgG1とマウス3H3は結合性に差はなかった。コントロールとしては、293細胞の培養上清を用いた。
【図17】変異型IgG4抗体の作製を示す図である。定常領域のアミノ酸配列中、IgG1,2と異なるKabat則241番目のセリンがIgG4の不安定化につながるという報告があるため、この位置をプロリンに変換した定常領域を作製した。
【図18】変異型IgG4抗体の結合活性比較図である。横軸に抗体濃度、縦軸にELISA で測定された吸光度(650nm)を示す。ヒト化IgG4とキメラIgG1とヒト化IgG1の結合性に差はなかった。コントロールとしては、293細胞の培養上清を用いた。
【発明を実施するための形態】
【0009】
本発明は、酸化変性LDL(酸化LDL)とβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を提供する。動脈硬化巣で酸化LDLと血漿糖タンパク質であるβ2GPIとの複合体が形成される。本願発明に包含される抗体は、この複合体に結合することを特徴とする。
【0010】
本願発明に包含される抗体としては、具体的には以下の抗体が挙げられるが、これらに限定されるものではない。
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体。
【0011】
また、本発明には、「(a)〜(e)に記載の抗体において1若しくは複数のアミノ酸が置換、欠失、付加および/または挿入された抗体であって、(a)〜(e)に記載の抗体と同等の活性を有し、CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖、並びに、CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖の対を有する抗体」も包含されるが、これに限定されない。
ポリペプチドに変異を導入する方法は、あるポリペプチドと機能的に同等なポリペプチドを調製するための、当業者によく知られた方法の一つである。例えば、当業者であれば、部位特異的変異誘発法(Hashimoto-Gotoh, T. et al. (1995) Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Methods Enzymol. 100, 468-500、Kramer, W. et al. (1984) Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Proc Natl Acad Sci USA. 82, 488-492、Kunkel (1988) Methods Enzymol. 85, 2763-2766)などを用いて、本発明の抗体に適宜変異を導入することにより、該抗体と機能的に同等な抗体を調製することができる。また、アミノ酸の変異は自然界においても生じうる。このように、本発明の抗体のアミノ酸配列において1もしくは複数のアミノ酸が変異したアミノ酸配列を有し、該抗体と機能的に同等な抗体もまた本発明の抗体に含まれる。
このような変異体における、変異するアミノ酸数は、通常、50 20アミノ酸以内であり、好ましくは30 15 アミノ酸以内であり、さらに好ましくは10アミノ酸以内(例えば、5アミノ酸以内)である。尚、変異はFR(フレームワーク)内であることが好ましい。
変異するアミノ酸残基においては、アミノ酸側鎖の性質が保存されている別のアミノ酸に変異されることが望ましい。例えばアミノ酸側鎖の性質に基づいて、次のような分類が確立している。
疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、
親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、
脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、
水酸基含有側鎖を有するアミノ酸(S、T、Y)、
硫黄原子含有側鎖を有するアミノ酸(C、M)、
カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、
塩基含有側鎖を有するアミノ酸(R、K、H)、
芳香族含有側鎖を有するアミノ酸(H、F、Y、W)
(括弧内はいずれもアミノ酸の一文字標記を表す)
あるアミノ酸配列に対する1又は複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するポリペプチドがその生物学的活性を維持することはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA (1984) 81, 5662-5666 、Zoller, M. J. and Smith, M., Nucleic Acids Research (1982) 10, 6487-6500 、Wang, A. et al., Science 224, 1431-1433 、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA (1982) 79, 6409-6413 )。すなわち、一般に、あるポリペプチドを構成するアミノ酸配列中、各群に分類されたアミノ酸は、相互に置換したときに、当該ポリペプチドの活性が維持される可能性が高いとされている。本発明において、上記アミノ酸群の群内のアミノ酸間の置換を保存的置換という。
【0012】
また本発明は、酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する本願発明の抗体が結合するエピトープと同じエピトープに結合する抗体も提供する。この抗体は、酸化LDLと複合体を形成している酸化LDL/β2GPI分子上の特定のエピトープを認識する。
この限りではないが、例えばある抗体が他の抗体と同じエピトープを認識するか否かは、両者のエピトープに対する競合によって確認することができる。抗体間の競合は、競合結合アッセイによって評価することができ、その手段としてELISA、蛍光エネルギー転移測定法(FRET)や蛍光微量測定技術(FMAT(登録商標))などが挙げられる。抗原に結合した該抗体の量は、同じエピトープへの結合に対して競合する候補競合抗体(被検抗体)の結合能に間接的に相関している。すなわち、同じエピトープに対する被検抗体の量や親和性が大きくなるほど、該抗体の抗原への結合量は低下し、抗原への被検抗体の結合量は増加する。具体的には、抗原に対し、適当な標識をした該抗体と評価すべき抗体を同時に添加し、標識を利用して結合している該抗体を検出する。抗原に結合した該抗体量は、該抗体を予め標識しておくことで、容易に測定できる。この標識は特には制限されないが、手法に応じた標識方法を選択する。標識方法は、具体的には蛍光標識、放射標識、酵素標識などが挙げられる。
ここでいう「同じエピトープを認識する抗体」とは、標識該抗体に対して、非標識の該抗体の結合により結合量を50%低下させる濃度(IC50)に対して、被検抗体が非標識該抗体のIC50の通常、100倍、好ましくは80倍、さらに好ましくは50倍、さらに好ましくは30倍、より好ましくは10倍高い濃度で少なくとも50%、標識該抗体の結合量を低下させることができる抗体である。
【0013】
本発明の抗体には、ポリクローナル抗体およびモノクローナル抗体の両方が含まれる。モノクローナル抗体およびポリクローナル抗体の調製および精製方法は、当分野で知られており、例えば Harlow and Lane, Antibodies: A Laboratory Manual (New York: Cold Spring Harbor Laboratory Press, 1988)に記載されている。また、本発明の抗体は、アイソタイプ(IgG、IgM、IgA、IgE、IgDなど)を問わない。
【0014】
本発明の抗体には、ヒト化(humanized)抗体またはキメラ(chimeric)抗体などの組換え抗体も含まれる。「ヒト化抗体」とは、ヒトの抗体に構造を類似させた抗体のことをいう。このようなヒト化抗体またはキメラ抗体には、ヒト型キメラ抗体(例えば抗体の一部がヒト化された抗体、CH2領域がヒト化された抗体、Fc領域がヒト化された抗体、定常領域がヒト化された抗体)、及び定常領域及び可変領域に存在するCDR(相補性決定領域)以外の部分がヒト化されたヒト型CDR移植(CDR-grafted)抗体(P.T.Johons et al., Nature 321,522(1986))、完全ヒト化抗体などが含まれる。ヒト型CDR移植抗体の抗原結合活性を高めるため、マウス抗体と相同性の高いヒト抗体FR(フレームワーク)を選択する方法、相同性の高いヒト化抗体を作製する方法、ヒト抗体にマウスCDRを移植した後さらにFRのアミノ酸を置換する方法の改良技術もすでに開発され(米国特許第5585089号、米国特許第5693761号、米国特許第5693762号、米国特許第6180370号、欧州特許第451216号、欧州特許第682040号、特許第2828340号などを参照)、本発明のヒト型CDR移植抗体の作製に利用することもできる。ヒト化の際に用いられるヒト抗体は、IgG、IgM、IgA、IgE、IgDなど如何なるアイソタイプのヒト抗体でもよい。
本願発明に包含される「ヒト化抗体」としては、具体的には以下の抗体が挙げられるが、これらに限定されるものではない。
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
【0015】
また、本発明には、「(f)〜(j)に記載の抗体において1若しくは複数のアミノ酸が置換、欠失、付加および/または挿入された抗体であって、(f)〜(j)に記載の抗体と同等の活性を有し、CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖、並びに、CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖の対を有する抗体」も包含されるが、これに限定されない。
【0016】
ヒト型キメラ抗体は例えば、上記のH鎖可変領域の構造及び/又はL鎖可変領域の構造を有する抗体の定常領域をヒト抗体の定常領域に置換することにより作製することができる。ヒト抗体の定常領域としては公知のものを採用することができる。以下に、ヒト型キメラ抗体の作製方法の一例を示す。
【0017】
まず、特定の対象抗原に対するマウス抗体を産生するハイブリドーマよりmRNAを抽出し、常法に従ってcDNAを合成する。合成したcDNAをベクターに組み込みcDNAライブラリーを構築する。このcDNAライブラリーから、H鎖遺伝子フラグメント及びL鎖遺伝子フラグメントをプローブとして用いることにより、H鎖遺伝子及びL鎖遺伝子を含有するベクターを選択する。選択されたベクターの挿入配列のシークエンシングを行うことにより、H鎖可変領域及びL鎖可変領域の遺伝子の配列が決定される。このようにして得られた配列データを基にH鎖可変領域をコードするDNAを化学合成、生化学的切断/再結合等により作製する。得られたH鎖可変領域をコードするDNAを、ヒトH鎖定常領域をコードするDNAとライゲーションして発現用ベクターに組込むことによりH鎖発現ベクターを作製する。発現ベクターとしては例えばSV40 virus basedベクター、EB virus basedベクター、BPV(パピローマウイルス)basedベクターなどを用いることができるが、これらに限定されるものではない。一方、同様の方法によりL鎖発現ベクターを作製する。これらH鎖発現ベクター及びL鎖発現ベクターにより宿主細胞を共形質転換する。宿主細胞としてはCHO細胞(チャイニーズハムスター卵巣)(A.Wright & S.L.Morrison, J.Immunol.160, 3393-3402 (1998))、SP2/0細胞(マウスミエローマ)(K.Motmans et al., Eur.J.Cancer Prev.5,512-519 (1996),R.P.Junghans et al., Cancer Res.50,1495-1502 (1990))などが好適に用いられる。また、形質転換にはリポフェクチン法(R.W.Malone et al.,Proc.Natl.Acad.Sci.USA 86,6077 (1989), P.L.Felgner et al., Proc.Natl.Acad.Sci.USA 84,7413 (1987))、エレクトロポレーション法、リン酸カルシウム法(F.L.Graham & A.J.van der Eb,Virology 52,456-467(1973))、DEAE-Dextran法等が好適に用いられる。
【0018】
形質転換体を培養した後、形質転換体の細胞内又は培養液よりヒト型キメラ抗体を分離する。抗体の分離、精製には、遠心分離、硫安分画、塩析、限外濾過、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーなどの方法を適宜組み合わせて利用することができる。
【0019】
一方、ヒト型CDR移植抗体は例えば以下の方法により作製することができる。まず、上記キメラ抗体の製造方法の欄で述べた方法により、特定の抗原に対する抗体のH鎖可変領域及びL鎖可変領域のアミノ酸配列及びそれをコードする塩基配列を決定する。併せて各CDR領域のアミノ酸配列及び塩基配列を決定する。
【0020】
次に、CDR領域を挟んで存在するFR(フレームワーク領域)を選択する。FRの選択には、およそ三つの方法が採用できる。1つめの方法は、NEWM、REIなど既に三次元構造の明らかとなったヒト抗体フレームを用いる方法である(Riechmann L. et al., Nature 332, 323-3Z7 (1988); Tempst, PR. et al., Protein Engineering 7, 1501-1507 (1994); Ellis JH. et al., J. Immunol 155, 925-937 (1995))。2つめの方法は、目的のマウス抗体可変領域と最も高いホモロジーを持つヒト抗体可変領域をデータベースより選択し、そのFRを用いる方法である(Queen C. et al., Proc Natl Acad SciUSA 86, 10029-10033 (1989); Rozak MJ. et al., J Biol Chem 271, 22611-22618 (1996); Shearman CW. et al., J.Immunol 147, 4366-4373 (1991))。3つめの方法は、ヒト抗体のFRで最も共通に用いられるアミノ酸を選択する方法である(Sato K. et al., Mol Immunol 31, 371-381 (1994); Kobinger F. et al., Protein Engineering 6, 971-980 (1993); Kettleborough CA. et al., Protein Engineering 4, 773-783 (1991))。本発明ではこれらいずれの方法を用いることもできる。
尚、選択されたヒトFRのアミノ酸配列を改変したアミノ酸配列であっても、最終的に得られるヒト型CDR移植抗体が対象抗原に対する特異的結合性を有する限り、FRのアミノ酸配列として利用することができる。特に、選択されたヒトFRのアミノ酸の一部をCDRの由来となった抗体のFRのアミノ酸に変更した場合、抗体の特性が維持される可能性が高い。改変されるアミノ酸の数は好ましくはFR全体の30%以下であり、更に好ましくはFR全体の20%以下であり、更に好ましくはFR全体の10%以下である。
【0021】
次に、これらいずれかの方法により選択したFRと上記CDRとを組み合わせることによりH鎖可変領域及びL鎖可変領域をコードするDNAを設計する。この設計を基にH鎖可変領域をコードするDNAとL鎖可変領域をコードするDNAを化学合成、生化学的切断/再結合等によりそれぞれ作製する。そしてH鎖可変領域をコードするDNAを、ヒト免疫グロブリンH鎖定常領域をコードするDNAとともに発現ベクターに組み込みH鎖発現ベクターを構築する。同様に、L鎖可変領域をコードするDNAを、ヒト免疫グロブリンL鎖定常領域をコードするDNAとともに発現ベクターに組み込みL鎖発現ベクターを構築する。発現ベクターとしては例えばSV40 virus basedベクター、EB virus basedベクター、BPV(パピローマウイルス)basedベクターなどを用いることができるが、これらに限定されるものではない。
【0022】
以上の方法で作製されたH鎖発現ベクター及びL鎖発現ベクターにより宿主細胞を共形質転換する。宿主細胞としてはCHO細胞(チャイニーズハムスター卵巣)(A.Wright& S.L.Morrison, J.Immunol.160, 3393-3402 (1998))、SP2/0細胞(マウスミエローマ)(K.Motmans et al., Eur.J.Cancer Prev.5,512-519 (1996),R.P.Junghans et al.,Cancer Res.50,1495-1502 (1990))などが好適に用いられる。また、形質転換にはリポフェクチン法(R.W.Malone et al.,Proc.Natl.Acad.Sci.USA 86,6077 (1989), P.L.Felgner et al.,Proc.Natl.Acad.Sci.USA 84,7413 (1987))、エレクトロポレーション法、リン酸カルシウム法(F.L.Graham & A.J.van der Eb,Virology 52,456-467(1973))、DEAE-Dextran法等が好適に用いられる。
【0023】
形質転換体を培養した後、形質転換体の細胞内又は培養液よりヒト型CDR移植抗体を分離する。抗体の分離、精製は、遠心分離、硫安分画、塩析、限外濾過、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーなどの方法を適宜組み合わせて行うことができる。
【0024】
また、ヒト抗体の取得方法も知られている。例えば、ヒトリンパ球をin vitroで所望の抗原または所望の抗原を発現する細胞で感作し、感作リンパ球をヒトミエローマ細胞、例えばU266と融合させ、抗原への結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878参照)。また、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物を所望の抗原で免疫することで所望のヒト抗体を取得することができる(国際特許出願公開番号WO 93/12227, WO 92/03918,WO 94/02602, WO 94/25585,WO 96/34096, WO 96/33735参照)。
【0025】
更なる実施態様では、抗体又は抗体断片は、McCafferty等(Nature, 348:552-554 (1990))に記載された技術を使用して産生される抗体ファージライブラリーから分離することができる。Clackson等(Nature, 352:624-628 (1991))及び Marks等(J. Mol. Biol., 222:581-597 (1991))は、ファージライブラリーを使用したマウス及びヒト抗体の分離を記述している。続く刊行物は、鎖シャフリングによる高親和性(nM範囲)のヒト抗体の生成(Marks等, Bio/Technology, 10:779-783[1992])、並びに非常に大きなファージライブラリーを構築するための方策としてコンビナトリアル感染とインビボ組換え(Waterhouse等, Nuc. Acids. Res., 21:2265-2266[1993])を記述している。従って、これらの技術はモノクローナル抗体の分離に対する伝統的なモノクローナル抗体ハイブリドーマ法に対する実行可能な別法である。
【0026】
この点において、バクテリオファージ(ファージ)ディスプレイは、大きなオリゴペプチドライブラリーを検索して、ポリペプチド標的に特異的に結合する能力のあるこれらライブラリーのメンバーを同定することを可能にするよく知られた技術の一つである。ファージディスプレイは、様々なポリペプチドがバクテリオファージ粒子の表面上のコートタンパク質に融合タンパク質として提示されることによる技術である(Scott,J.K.及びSmith G. P. (1990) Science 249:386)。ファージディスプレイの有用性は、選択的にランダム化されたタンパク質変異体(又はランダムクローンcDNA)の大きなライブラリーを標的分子に高い親和性で結合するこれらの配列について素早く効果的に分類することができる点にある。ファージでのペプチド(Cwirla,S.E.等 (1990) Proc. Natl. Acad. Sci. USA, 87:6378)又はタンパク質(Lowman,H.B.ら (1991) Biochemistry, 30:10832; Clackson,T.ら (1991) Nature, 352: 624; Marks,J.D.等 (1991), J. Mol. Biol., 222:581; Kang,A.S.等 (1991) Proc. Natl. Acad. Sci. USA, 88:8363)ライブラリーのディスプレイは、特異的に結合する特性を有するものについて無数のポリペプチド又はオリゴペプチドをスクリーニングするために使用されている(Smith, G.P. (1991) Current Opin. Biotechnol., 2:668)。ランダム突然変異体のファージライブラリーの分類は、多数の変異体を構築して増殖させる方法、標的レセプターを用いた親和性精製の方法、及び結合増強の結果を評価する手段を必要とする(米国特許第5223409号、同第5403484号、同第5571689号、及び同第5663143号を参照)。
【0027】
ほとんどのファージディスプレイ法は繊維状ファージを使用していたが、λファージディスプレイシステム(WO95/34683;米国特許第5627024号)、T4ファージディスプレイシステム(RenJ.らGene 215:439 (1998); Zhuら Cancer Researc, 58(15):3209-3214 (1998); Jiang等 Infection & Immunity, 65(11): 4770-4777 (1997); Ren等, Gene, 195(2):303-311 (1997); Ren, Protein Sci. 5:1833 (1996); Efimov等 Virus Genes 10:173(1995)及びT7ファージディスプレイシステム(Smith及びScott Methods in Enzymology,217, 228-257(1993); 米国特許第5766905号)も知られている。
【0028】
現在、基礎的なファージディスプレイ法は多くの改良及び変形が開発されている。これらの改良により、選択された標的分子との結合性など、ペプチドライブラリーやタンパク質ライブラリーから特性、能力などに基づいてスクリーニングする方法が改善された。ファージディスプレイ法のための組み換え反応手段については、WO98/14277に記載がある。ファージディスプレイライブラリーは、二分子相互作用(WO98/20169;WO98/20159)及び拘束性へリックスペプチドの特性(WO98/20036)を分析及び制御するために使用されている。WO97/35196には、リガンドが標的分子に結合しうる第一の溶液、及び親和性リガンドが標的分子に結合しない第二の溶液とファージディスプレイライブラリーを接触させて結合リガンドを選択的に単離する、親和性リガンドの単離方法が記載されている。WO97/46251は、親和性精製抗体でランダムファージディスプレイライブラリーをバイオパニングし、次いで結合ファージを単離し、続いてマイクロプレートのウェルでマイクロパニングして高親和性結合ファージを単離する方法を記載する。黄色ブドウ球菌(Staphlylococcus aureus)タンパク質Aの親和性タグとしての使用も報告されている(Li等, (1998) Mol Biotech., 9:187)。WO97/47314は、ファージディスプレイライブラリーでもよいコンビナトリアルライブラリーを用いて酵素特異性を識別するための基質サブトラクションライブラリーの使用を記載している。ファージディスプレイに用いる洗浄剤における使用に適した酵素を選択する方法はWO97/09446に記載される。特異的に結合するタンパク質を選択する更なる方法は、米国特許第5498538号、同第5432018号、及びWO98/15833に記載されている。ペプチドライブラリーの作製及びこれらのライブラリーのスクリーニングの方法は、米国特許第5723286号、同第5432018号、同第5580717号、同第5427908号、同第5498530号、同第5770434号、同第5734018号、同第5698426号、同第5763192号、及び同第5723323号に記載される。
【0029】
さらに、ヒト抗体ライブラリーを用いて、パニングによりヒト抗体を取得する技術も知られている。例えば、ヒト抗体の可変領域を一本鎖抗体(scFv)としてファージディスプレイ法によりファージの表面に発現させ、抗原に結合するファージを選択することができる。選択されたファージの遺伝子を解析すれば、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。抗原に結合するscFvのDNA配列が明らかになれば、当該配列を有する適当な発現ベクターを作製し、適当な宿主に導入して発現させることによりヒト抗体を取得することができる。これらの方法は既に周知であり、WO 92/01047、WO 92/20791、WO 93/06213、WO 93/11236、WO 93/19172、WO 95/01438、WO 95/15388を参考に実施することができる。
【0030】
別法として、ファージディスプレイ技術(McCafferty等, Nature 348:552-553[1990])を使用して、非免疫化ドナーの免疫グロブリン可変(V)ドメイン遺伝子レパートリーから、インビトロでヒト抗体及び抗体断片を産出させることができる。この技術によれば、抗体Vドメイン遺伝子を、フレーム単位で、繊維状バクテリオファージ、例えばM13又はfdのコートタンパク質遺伝子のどちらかでクローニングし、ファージ粒子の表面で機能的抗体断片として提示させる。繊維状粒子がファージゲノムの一本鎖DNAコピーを含むので、抗体の機能特性に基づいた選択に基づいても、結果としてこれらの特性を示す抗体をコードする遺伝子の選択が成される。よって、このファージはB細胞のいくつかの特性を模倣している。ファージディスプレイは多様な形式で行うことができる;例えばJohnson, Kevin S. 及びChiswell, David J., Current Opinion in Structural Biology 3:564-571(1993)を参照。V-遺伝子セグメントのいくつかの供給源を、ファージディスプレイのために使用できる。Clackson等, Nature, 352:624-628(1991)は、免疫化したマウス脾臓由来のV遺伝子の小さいランダムなコンビナトリアルライブラリーから、多様で多くの抗-オキサゾロン抗体を単離した。非免疫化ヒトドナーのV遺伝子のレパートリーが構成可能であり、多様で多くの抗原(自己抗原を含む)に対する抗体は、Marks等, J. Mol. Biol. 222:581-597(1991)、又はGriffith等, EMBO J. 12:725-734(1993)に記載の技術にそのまま従うことで単離することができる。また、米国特許第5565332号及び同5573905号を参照のこと。
【0031】
本発明の抗体には、Fab、Fab'、F(ab')2、Fv、scFv、dsFv、Diabody及びsc(Fv)2等の、抗体の機能的断片も含まれる。また、これら機能的断片の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本発明の抗体に含まれる。
Fabは、IgGをシステイン存在下パパイン消化することにより得られる、L鎖とH鎖可変領域、並びにCH1ドメイン及びヒンジ部の一部からなるH鎖フラグメントとから構成される分子量約5万の断片である。本発明では、上記抗体をパパイン消化することにより得ることができる。また、上記抗体のH鎖の一部及びL鎖をコードするDNAを適当なベクターに組み込み、当該ベクターを用いて形質転換した形質転換体よりFabを調製することもできる。
Fab'は、後述のF(ab')2のH鎖間のジスルフィド結合を切断することにより得られる分子量が約5万の断片である。本発明では、上記抗体をペプシン消化し、還元剤を用いてジスルフィド結合を切断することにより得られる。また、Fab同様に、Fab'をコードするDNAを用いて遺伝子工学的に調製することもできる。
F(ab')2は、IgGをペプシン消化することにより得られる、L鎖とH鎖可変領域、並びにCH1ドメイン及びヒンジ部の一部からなるH鎖フラグメントとから構成される断片(Fab')がジスルフィド結合で結合した分子量約10万の断片である。本発明では、上記抗体をペプシン消化することにより得られる。また、Fab同様に、F(ab')2をコードするDNAを用いて遺伝子工学的に調製することもできる。
Fvは、抗体を酵素、例えばパパイン、ペプシンで処理し抗体断片を生成させるか、または、これら抗体断片をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現できる(例えば、Co, M.S. et al., J. Immunol.(1994)152, 2968-2976、Better, M. & Horwitz, A. H. Methods in Enzymology(1989)178, 476-496、Plueckthun, A. ; Skerra, A. Methods in Enzymology(1989)178, 476-496、Lamoyi, E., Methods in Enzymology(1989)121, 652-663、Rousseaux, J. et al., Methods in Enzymology(1989)121, 663-669、Bird, R. E. et al., TIBTECH(1991)9, 132-137参照)。
scFvは、H鎖可変領域とL鎖可変領域とからなるFvを、片方の鎖のC末端と他方のN末端とを適当なペプチドリンカーで連結し一本鎖化した抗体断片である。ペプチドリンカーとしては例えば柔軟性の高い(GGGGS)3などを用いることができる。例えば、上記抗体のH鎖可変領域及びL鎖可変領域をコードするDNAとペプチドリンカーをコードするDNAを用いてscFv抗体をコードするDNAを構築し、これを適当なベクターに組み込み、当該ベクターを用いて形質転換した形質転換体よりscFvを調製することができる。
dsFvは、H鎖可変領域及びL鎖可変領域の適切な位置にCys残基を導入し、H鎖可変領域とL鎖可変領域とをジスルフィド結合により安定化させたFv断片である。各鎖におけるCys残基の導入位置は分子モデリングにより予測される立体構造に基づき決定することができる。本発明では例えば上記抗体のH鎖可変領域及びL鎖可変領域のアミノ酸配列から立体構造を予測し、かかる予測に基づき変異を導入したH鎖可変領域及びL鎖可変領域をそれぞれコードするDNAを構築し、これを適当なベクターに組み込み、そして当該ベクターを用いて形質転換した形質転換体よりdsFvを調製することができる。
尚、適当なリンカーを用いてscFv抗体、dsFv抗体などを連結させたり、ストレプトアビジンを融合させたりして抗体断片を多量体化することもできる。本発明の抗体(抗体断片を含む)に低分子化合物、タンパク質、標識物質などを融合又は結合させることにより、融合抗体又は標識化抗体を構成することができる。標識物質としては125I等の放射性物質、などを用いることができる。
【0032】
Diabodyは、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(Holliger P et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。Diabodyは、2本のポリペプチド鎖から構成されるダイマーであり、通常、ポリペプチド鎖は各々、同じ鎖中でVL及びVHが、互いに結合できない位に短い、例えば、5残基程度のリンカーにより結合されている。同一ポリペプチド鎖上にコードされるVLとVHとは、その間のリンカーが短いため単鎖可変領域フラグメントを形成することが出来ず二量体を形成するため、Diabodyは2つの抗原結合部位を有することとなる。Diabodyは、抗体を酵素、例えば、パパイン、ペプシンなどで処理し、抗体断片を生成させるか、又はこれら抗体断片をコードするDNAを構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させればよい(例えば、Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976; Better, M. and Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515; Lamoyi, E., Methods Enzymol. (1986) 121, 652-663 ; Rousseaux, J. et al., Methods Enzymol. (1986) 121, 663-669; Bird, R. E. and Walker, B. W., Trends Biotechnol. (1991) 9, 132-137参照)。
sc(Fv)2は、2つのVH及び2つのVLをリンカー等で結合して一本鎖にした低分子化抗体である(Hudson et al、J Immunol. Methods 1999;231:177-189)。sc(Fv)2は、例えば、scFvをリンカーで結ぶことによって作製できる。
【0033】
本発明の抗体には、本発明の抗体と他のペプチド又はタンパク質とが融合した融合タンパク質も含まれる。融合タンパク質を作製する方法は、本発明の抗体をコードするポリヌクレオチドと他のペプチド又はポリペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明の抗体との融合に付される他のペプチド又はポリペプチドとしては、例えば、FLAG(Hopp, T. P. et al., BioTechnology (1988) 6, 1204-1210 )、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein C の断片等の公知のペプチドを使用することができる。また、本発明の抗体との融合に付される他のポリペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。
【0034】
本発明の抗体には、標識物質が結合した抗体も含まれる。
標識物質としては、酵素発光(ルシフェラーゼ)による発光、発光低分子を利用するものと、蛍光タンパクや蛍光低分子)を用いるもの、放射性核種等が挙げられるがこれらには限定されない。放射性核種としては51Cr、59Fe、57Co、67Ga、75Se、81mKr、99mTc、111In、125I、131I、133Xe、201Tlなどのγ線放出核種や、11C、13N、15O、18F、35mCl、76Br、 45Ti、48V、60Cu、61Cu、62Cu、66Ga、89Zr、94mTc、124I、123Iなどの陽電子放出核種などが挙げられるがこれらには限定されない。なお当業者には自明であるが"m"とは核異性体を示す。
蛍光標識や発光標識としては酵素発光(ルシフェラーゼ)による発光を利用するものと、蛍光(GFP、DsRed、クサビラオレンジ等の蛍光タンパク質やFITCやCy5.5、Alexa Fluor 750などの蛍光性低分子)を用いるものとのが使用できる。
酵素発光(ルシフェラーゼ)による発光の場合は基質投与が別途必要である。
特に、動物本来の自家蛍光による影響を軽減したものが好ましく、さらには皮膚透過性の高いシグナルを発する標識が好ましい。
【0035】
また本発明は、本発明の抗体をコードするDNA、該DNAが挿入されたベクター、及び該ベクターが導入された形質転換細胞を提供する。ベクターの例としては、M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Scriptなどが挙げられる。また、cDNAのサブクローニング、切り出しを目的とした場合、上記ベクターの他に、例えば、pGEM-T、pDIRECT、pT7などが挙げられる。本発明の抗体をコードするDNA、該DNAが挿入されたベクター、及び該ベクターが導入された形質転換細胞は公知の技術を用いて作成される。
【0036】
本発明の酸化LDL/β2GPI複合体に結合する抗体をコードするDNAとしては、以下のDNAが挙げられる。
(a)配列番号:5に記載の塩基配列を有する、重鎖をコードするDNA
(b)配列番号:10に記載の塩基配列を有する、軽鎖をコードするDNA
(c)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖をコードするDNA
(d)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖をコードするDNA
(e)配列番号:45に記載の塩基配列を有する、重鎖をコードするDNA
(f)配列番号:53に記載の塩基配列を有する、軽鎖をコードするDNA
【0037】
発現ベクターとしては、例えば、大腸菌での発現を目的とした場合は、ベクターが大腸菌で増幅されるような上記特徴を持つほかに、宿主をJM109、DH5α、HB101、XL1-Blueなどの大腸菌とした場合においては、大腸菌で効率よく発現できるようなプロモーター、例えば、lacZプロモーター(Wardら, Nature (1989) 341, 544-546;FASEB J. (1992) 6, 2422-2427)、araBプロモーター(Betterら, Science (1988) 240, 1041-1043 )、またはT7プロモーターなどを持っていることが不可欠である。このようなベクターとしては、上記ベクターの他にpGEX-5X-1(ファルマシア社製)、「QIAexpress system」(キアゲン社製)、pEGFP、またはpET(この場合、宿主はT7 RNAポリメラーゼを発現しているBL21が好ましい)などが挙げられる。
【0038】
また、ベクターには、ポリペプチド分泌のためのシグナル配列が含まれていてもよい。蛋白質分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(Lei, S. P. et al J. Bacteriol. (1987) 169, 4379)を使用すればよい。宿主細胞へのベクターの導入は、例えば塩化カルシウム法、エレクトロポレーション法を用いて行うことができる。
【0039】
大腸菌以外にも、例えば、哺乳動物由来の発現ベクター(例えば、pcDNA3 (インビトロゲン社製)や、pEGF-BOS (Nucleic Acids. Res.1990, 18(17),p5322)、pEF 、pCDM8)、昆虫細胞由来の発現ベクター(例えば「Bac-to-BAC baculovairus expression system」(ギブコBRL社製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来の発現ベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来の発現ベクター(例えば、pZIPneo)、酵母由来の発現ベクター(例えば、「Pichia Expression Kit」(インビトロゲン社製)、pNV11、SP-Q01)、枯草菌由来の発現ベクター(例えば、pPL608、pKTH50)が挙げられる。
【0040】
CHO細胞、COS細胞、NIH3T3細胞等の動物細胞での発現を目的とした場合には、細胞内で発現させるために必要なプロモーター、例えばSV40プロモーター(Mulliganら, Nature (1979) 277, 108)、MMTV-LTRプロモーター、EF1αプロモーター(Mizushimaら, Nucleic Acids Res. (1990) 18, 5322)、CMVプロモーターなどを持っていることが不可欠であり、細胞への形質転換を選抜するための遺伝子(例えば、薬剤(ネオマイシン、G418など)により判別できるような薬剤耐性遺伝子)を有すればさらに好ましい。このような特性を有するベクターとしては、例えば、pMAM、pDR2、pBK-RSV、pBK-CMV、pOPRSV、pOP13などが挙げられる。
【0041】
さらに、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする場合には、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pCHOIなど)を導入し、メトトレキセート(MTX)により増幅させる方法が挙げられ、また、遺伝子の一過性の発現を目的とする場合には、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。
【0042】
ベクターが導入される宿主細胞としては特に制限はなく、例えば、大腸菌や種々の動物細胞などを用いることが可能である。宿主細胞は、例えば、本発明の抗体の製造や発現のための産生系として使用することができる。ポリペプチド製造のための産生系は、in vitroおよびin vivoの産生系がある。in vitroの産生系としては、真核細胞を使用する産生系や原核細胞を使用する産生系が挙げられる。
【0043】
真核細胞を使用する場合、例えば、動物細胞、植物細胞、真菌細胞を宿主に用いることができる。動物細胞としては、哺乳類細胞、例えば、CHO(J. Exp. Med. (1995) 108, 945)、COS、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero、両生類細胞、例えばアフリカツメガエル卵母細胞(Valle, et al., Nature (1981) 291, 358-340)、あるいは昆虫細胞、例えば、 Sf9、 Sf21、 Tn5が知られている。本発明においては、CHO-DG44、CHO-DXB11、COS7細胞、BHK細胞が好適に用いられる。動物細胞において、大量発現を目的とする場合には特にCHO細胞が好ましい。宿主細胞へのベクターの導入は、例えば、リン酸カルシウム法、DEAEデキストラン法、カチオニックリボソームDOTAP(ベーリンガーマンハイム社製)を用いた方法、エレクトロポーレーション法、リポフェクションなどの方法で行うことが可能である。
【0044】
植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞が蛋白質生産系として知られており、これをカルス培養すればよい。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属、例えば、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・ポンベ(Saccharomyces pombe)、糸状菌、例えば、アスペルギルス(Aspergillus)属、例えば、アスペルギルス・ニガー(Aspergillus niger)が知られている。
【0045】
原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、大腸菌(E. coli)、例えば、JM109、DH5α、HB101等が挙げられ、その他、枯草菌が知られている。本発明のDNAにより形質転換された細胞をin vitroで培養し、当業者が通常行う方法によって精製することによって、本発明の抗体を得ることが可能である。
【0046】
また本発明は、本発明の抗体をコードする核酸を含むベクターを有する宿主生物を提供する。本発明の宿主生物は、組換え型抗体の産生に有用である。本発明における宿主生物としては、ヤギなどが挙げられる。例えば、本発明のトランスジェニックヤギの作成は次のようにして行うことが可能である。即ち、抗体遺伝子が乳汁中に固有に産生されるタンパク質(ヤギβカゼインなど)をコードする遺伝子の内部にインフレームで挿入された融合遺伝子を構築する。抗体遺伝子が挿入された融合遺伝子を含むDNA断片をヤギの胚へ注入すれば、該注入胚が雌のヤギへ導入される。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、本発明の抗体を取得することができる。トランスジェニックヤギから産生される本発明の抗体を含む乳汁量を増加させるために、ホルモンをトランスジェニックヤギに適宜使用することも可能である(Ebert, K.M. et al., Bio/Technology(1994)12, 699-702)。
【0047】
本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を含む、動脈硬化の存在部位のイメージング剤を提供する。また、本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を哺乳動物に投与する工程を含む、動脈硬化の存在部位のイメージング方法を提供する。本発明のイメージング剤は、動脈硬化の存在部位をイメージングするために哺乳動物に投与される。哺乳動物としては、ヒト、非ヒト哺乳動物(例えばマウス、ラット、ハムスター、ウサギ、ブタ、サルなど)が挙げられる。本発明のイメージング剤は、動脈硬化の診断において有用である。本発明のイメージング剤はin vivo用、in vitro用のどちらとしても利用できる。
より好ましい態様としては、本発明のイメージング剤はin vivo用イメージング剤である。in vivo用イメージング剤とは、酸化LDL/β2GPI複合体に結合する抗体を含むイメージング剤を注射などの方法により動物個体の血管内に投与し、動脈硬化の存在部位を個体生存化で観察するものである。そのため、in vivo用イメージング剤には生体の健康上に悪影響を及ぼす組成物は極力除かれる。また、in vivo用イメージング剤は皮下染色部位の観察(体内の酸化LDL/β2GPI複合体に結合する抗体存在部位の検出)は体外から検出が行われるため、酸化LDL/β2GPI複合体に結合する抗体に対し、生体に吸収されにくい波長である近赤外の蛍光標識か、放射線核種による標識が用いられる。蛍光標識として、Alexa Fluor 750標識は、779 nmの最大放出蛍光を持ち、実施例に示すとおり高感度、好適に用いられる。また、ヒトに対してin vivo用イメージング剤を適用する場合は、放射線核種が好ましい。さらに、ex vivoイメージングに用いる場合もある。ex vivoイメージングとは、マウスモデル動物などの非ヒト哺乳動物へ生前にイメージング剤を投与しておき、堵殺後、酸化LDL/β2GPI複合体に結合する抗体存在部位を観察する方法である。
動脈硬化には大きく分け、粥腫と石灰化病変の症状が存在するが、本発明のイメージング剤によって、特に、動脈硬化粥腫部位が染色される。
粥腫は、マクロファージが、コレステロールを多量に含有する酸化LDLをレセプターを介して特異的に取り込み、泡沫化することが知られており、それが血管内膜に蓄積、プラーク(粥腫)を形成する、動脈硬化の一病態である。血管内膜は、血管内皮細胞層によって覆われているが、in vivo用イメージング剤は血管内投与により、血管内膜下(血管内皮細胞下)のプラーク部位をイメージングできる。
【0048】
本発明のイメージング剤は、酸化LDL/β2GPI複合体に結合する抗体、特に好ましくは3H3抗体に、直接、あるいは間接的に追尾できる、イメージング用標識、プローブを結合させたものである。
前記プローブを生体に生体内投与(例えば静脈内投与)した後、PETやSPECT、CCDカメラなどの画像計測装置を用いて蓄積量や分布を測定することができる。
【0049】
また、近年、透過性線源により、物体を走査し、そのデータをコンピュータを用いて処理することで、物体の内部画像を構成する技術コンピュータ断層撮影(Computed Tomography:以下、「CT」との記載もComputed Tomographyを意味する)の病状診断、臨床への応用がされている。
そのようなCT技術は、ポジトロン断層法 (PET)や単一光子放射断層撮影 (SPECT)、や核磁気共鳴画像法 (MRI) などの、断面像を得て、物体の(輪切りなどの)2次元断面画像を得る技術であるが、これらの検査技術は単に断面画像として用いられるだけでなく、コンピュータ画像処理技術向上によってその2次元画像を統合し3次元グラフィックスとして表示されることも多く、3次元的な病変の位置の特定、診断、手術方針決定等に有力な技術となっている。
例えば、単純CTは、造影剤を使用せずにX線照射等の撮影を行うものであり、組織の浮腫、骨の形態異常、形態など造影剤を用いなくても充分に観察でき、一方、造影CTとは、X線吸収率の高い造影剤等を血管内に注射してから撮影を行うものをいい、血管や血流が豊富な組織の形状を観察することができる。さらに、いわゆる次世代CT、例えば、線源がらせん状に動くヘリカルCT、検出器自体を対軸方向に、分割した、多列検出器CT (MDCT)(マルチスライスCT(MSCT)とも呼ぶ)なども、開発され、それらはいずれも特に制限なく、単独又は併用で発明のイメージング剤の検出に適用可能である。
イメージング用標識プローブ(本発明のイメージング剤)がX線吸収率の高い放射線核種である場合は、検出器としてのCTの単独使用も可能である。
【0050】
標識物質としては酵素発光(ルシフェラーゼ)による発光、発光低分子を利用するものと、蛍光タンパクや蛍光低分子)を用いるもの、放射性核種等が挙げられるがこれらには限定されない。放射性核種としては51Cr、59Fe、57Co、67Ga、75Se、81mKr、 99mTc、111In、125I、131I、133Xe、201Tlなどのγ線放出核種や、11C、13N、15O、18F、35mCl、76Br、 45Ti、48V、60Cu、61Cu、62Cu、66Ga、89Zr、94mTc、124I、123Iなどの陽電子放出核種などが挙げられるがこれらには限定されない。なお当業者には自明であるが"m"とは核異性体を示す。特に、インジウム−111、テクネチウム−99mまたはヨウ素−131は、平面走査またはシングルフォトン断層撮影(SPECT)のために特に好ましく使用できる。陽電子放射標識の例えばフッ素−19は、陽電子放射断層法において特に好ましく使用できる。常磁性イオンの例えばガドリニウム(III)またはマンガン(II)は特に好ましく磁気共鳴映像法(MRI)において使用できる。
【0051】
蛍光標識や酵素(ルシフェラーゼ)による発光系を利用するものと、蛍光(GFP、DsRed、クサビラオレンジ等の蛍光タンパク質やFITCやCy5.5、Alexa Fluor 750などの蛍光性低分子)を用いるものとのが使用できる。
酵素発光(ルシフェラーゼ)による発光の場合は基質投与が別途必要である。
特に、動物本来の自家蛍光による影響を軽減したものが好ましく、さらには皮膚透過性の高いシグナルを発する標識が好ましい。
【0052】
イメージング検出器としては磁気共鳴映像法(MRI)PETやSPECTが用いられるが、特に、蛍光標識プローブの観察において、低侵しゅう性の観点で好ましく測定機器は、CCDカメラである。
それゆえ、CCDカメラで補足可能な波長、例えばおよそ350〜900 nmの光を、発する標識が好ましい。さらにCCDカメラによる測定動物の体表面における測定値から生体内の光源の強さを測定できる機械が好ましい。蛍光標識の場合、反射蛍光画像、透過蛍光画像のいずれでも良いが、両方の画像を捉えることが好ましい。また、蛍光画像の多方面(反射、透過を問わず)重ね合わせ、それに線源情報を統合して処理し、立体画像(3次元)として捉えることが、3次元的な位置、分布を正確に構成でき、好ましい。こうして得た3次元画像は、さらにCT画像とさらに統合することもできる。
【0053】
イメージング用標識プローブがX線吸収率の高い放射線核種を結合したものである場合、前述したように イメージング検出器としての、CTの単独でも十分使用可能(例えばPETやSPECT)で動脈硬化病巣の位置、蓄積量や分布を測定することもできる。
【0054】
一方、前記イメージング用標識プローブを生体に生体内投与(例えば静脈内投与)した後、標識された前記プローブのCT観察、又はCCDとの併用観察も可能である。併用観察の例として、蛍光標識したプローブのCCD像を、単純CT(及び/又は造影CTの像と重ね合わせる)を用いることができる。即ち 単純CTにより骨、肺などの臓器像の抽出(及び/又は造影CTの血管、組織像抽出)を行って得られたCT画像と、心臓など主要な動脈疾患部分のCCDカメラによる蛍光プローブ画像を統合し、その動脈硬化病巣の位置、蓄積量や分布を3次元的な組織、血管との相対的な位置関係、動脈硬化病巣の3次元(局在)イメージをより正確に把握することができる。
【0055】
本発明のイメージング剤は、抗体に加えて医薬的に許容し得る担体を導入し、公知の方法で製剤化することが可能である。例えば、水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、又は懸濁液剤の注射剤の形で非経口的に使用できる。例えば、薬理学上許容される担体もしくは媒体、具体的には、滅菌水や生理食塩水、植物油、乳化剤、懸濁剤、界面活性剤、安定剤、香味剤、賦形剤、ベヒクル、防腐剤、結合剤などと適宜組み合わせて、一般に認められた製薬実施に要求される単位用量形態で混和することによって製剤化することが考えられる。これら製剤における有効成分量は指示された範囲の適当な容量が得られるようにするものである。
【0056】
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施に従って処方することができる。
注射用の水溶液としては、例えば生理食塩水、ブドウ糖やその他の補助薬を含む等張液、例えばD-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウムが挙げられ、適当な溶解補助剤、例えばアルコール、具体的にはエタノール、ポリアルコール、例えばプロピレングリコール、ポリエチレングリコール、非イオン性界面活性剤、例えばポリソルベート80(TM)、HCO-50と併用してもよい。
【0057】
油性液としてはゴマ油、大豆油があげられ、溶解補助剤として安息香酸ベンジル、ベンジルアルコールと併用してもよい。また、緩衝剤、例えばリン酸塩緩衝液、酢酸ナトリウム緩衝液、無痛化剤、例えば、塩酸プロカイン、安定剤、例えばベンジルアルコール、フェノール、酸化防止剤と配合してもよい。調製された注射液は通常、適当なアンプルに充填させる。
【0058】
投与は好ましくは非経口投与であり、具体的には、注射剤型、経鼻投与剤型、経肺投与剤型、経皮投与型などが挙げられる。注射剤型の例としては、例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などにより全身または局部的に投与することができる。
【0059】
また、患者の年齢、症状により適宜投与方法を選択することができる。本発明のイメージング剤の投与量としては、例えば、一回につき体重1kgあたり0.0001mgから1000mgの範囲で選ぶことが可能である。あるいは、例えば、対象あたり0.001から100000mg/bodyの範囲で投与量を選ぶことができるが、これらの数値に必ずしも制限されるものではない。
投与量、投与方法は、対象の体重や年齢、症状、mg抗体あたりの蛍光標識の強度・検出機械の検出感度などにより変動するが、当業者であれば適宜選択することが可能である。
【0060】
また本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を含む、動脈硬化の存在部位のイメージング用キットを提供する。本発明のキットは、対象に投与されることにより、動脈硬化の存在部位をイメージングすることを特徴とする。上記キットには、本発明の抗体に加えて、例えば、イメージング剤投与用の注射(点滴)器具、非吸着吸着を抑える補助剤(例えばアルブミン等)等が含まれるが、これらに制限されるものではない。
また、上記キットには、イメージングに用いるための指示書、適当な容器、コントロール試薬等、通常のキットに含まれるものを含んでいてもよい。
【0061】
本発明は、以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法を提供する。
(a)本発明の酸化LDL/β2GPI複合体に結合する抗体、及び、候補化合物を動脈硬化疾患モデル非ヒト動物に投与する工程、例えば、本発明の酸化LDL/β2GPI複合体に結合する抗体が投与された動脈硬化疾患モデル非ヒト動物に候補化合物を投与する工程、
(b)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣(例えば、動脈硬化巣の大きさまたは存在部位)を比較する工程、及び
(d)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
各工程は、上述した技術または公知の技術を用いて実施される。
【0062】
本発明のスクリーニング方法において利用することができる候補化合物には、精製蛋白質(抗体を含む)、遺伝子ライブラリーの発現産物、合成ペプチドのライブラリー、DNAまたはRNAライブラリー(アプタマー、siRNAなど機能性核酸を含む)、細胞抽出液、細胞培養上清、及び合成低分子化合物のライブラリー等が挙げられるが、これらに制限されない。
【0063】
本発明のスクリーニング方法において利用することができる疾患モデル非ヒト動物としては、マウス、ハムスター、ラット、ウサギ、ブタ、サル等が挙げられるが、これらに制限されない。
例えば、動脈硬化疾患モデルマウスとしては遺伝子を過剰発現させるトランスジェニックマウスと、ある遺伝子を欠損させるジーンターゲッティングによるノックアウトマウス、例えば、動脈硬化モデルとしては悪玉コレステロールとして知られているLDLを形作っているタンパク質(アポリポタンパク質E)のアポE欠失(apoE-/-)、LDL受容体欠失(LDLR-/-)、ヒト型アポB導入、ドミナント変異アポE導入モデルがある。さらには、2型糖尿病モデルマウス(KKAy)、あるいは、マウスのなかでもっとも動脈硬化になりやすいC57BL6系統で、この系統では高コレステロール食のみで動脈硬化巣がみられる場合もあり、高コレステロール食等、その餌により作製された、動脈硬化モデルマウスも用いることができる。
ウサギは高コレステロール食により約2ヶ月半で動脈硬化巣がみられる場合もあり、さらにLDL受容体欠損の動脈硬化疾患モデルウサギとしては、WHHLウサギなどが知られている。
ブタでもアポBのLDL受容体結合部分のアミノ酸配列異常により、動脈硬化をおこしやすいモデルがしられており、当業者であれば、血栓症・動脈硬化モデル動物作製法 編著 鈴木 宏治(株式会社 金芳堂)など参照して、動脈硬化モデル動物を作製、本発明に利用できる。
【0064】
本発明のスクリーニング方法によって選択される動脈硬化巣を減少または消失させる化合物は、動脈硬化治療剤の候補化合物となる。即ち、本発明は、本発明のスクリーニングによって選択された物質を有効成分として含有する動脈硬化治療剤を提供する。また本発明は、本発明のスクリーニング方法によって選択された化合物の動脈硬化治療剤製造における使用に関する。本発明のスクリーニング法により単離される物質を、治療剤として用いる場合には、公知の製剤学的製造法により製剤化して用いることができる。例えば、薬理学上許容される担体または媒体(生理食塩水、植物油、懸濁剤、界面活性剤、安定剤など)とともに患者に投与する。投与は、物質の性質に応じて、経皮的、鼻腔内的、経気管支的、筋内的、静脈内、または経口的に行われる。投与量は、患者の年齢、体重、症状、投与方法等により変動するが、当業者であれば適宜適当な投与量を選択することができる。
【0065】
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<3H3抗体>
配列番号1:重鎖可変領域のアミノ酸配列
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号5:重鎖可変領域の塩基配列
配列番号6:軽鎖可変領域のアミノ酸配列
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号10:軽鎖可変領域の塩基配列
配列番号55:重鎖FR1のアミノ酸配列
配列番号56:重鎖FR2のアミノ酸配列
配列番号57:重鎖FR3のアミノ酸配列
配列番号58:重鎖FR4のアミノ酸配列
配列番号59:軽鎖FR1のアミノ酸配列
配列番号60:軽鎖FR2のアミノ酸配列
配列番号61:軽鎖FR3のアミノ酸配列
配列番号62:軽鎖FR4のアミノ酸配列
<ヒト化3H3抗体>
配列番号44:重鎖可変領域のアミノ酸配列
配列番号46:重鎖のアミノ酸配列(シグナル配列を含む重鎖)
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号39:重鎖FR1のアミノ酸配列
配列番号40:重鎖FR2のアミノ酸配列
配列番号41:重鎖FR3のアミノ酸配列
配列番号42:重鎖FR4のアミノ酸配列
配列番号45:重鎖の塩基配列(シグナル配列を含む重鎖)
配列番号52:軽鎖可変領域のアミノ酸配列
配列番号54:軽鎖のアミノ酸配列(シグナル配列を含む軽鎖)
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号47:軽鎖FR1のアミノ酸配列
配列番号48:軽鎖FR2のアミノ酸配列
配列番号49:軽鎖FR3のアミノ酸配列
配列番号50:軽鎖FR4のアミノ酸配列
配列番号53:軽鎖の塩基配列(シグナル配列を含む軽鎖)
なお、本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0066】
以下、本発明を実施例によって詳細に説明するが、本発明の範囲はこれらの実施例に記載された態様に限定されるものではない。
[実施例1]
酸化LDL/β2GPI複合体の調製
600μgのヒトLDL(Organon Teknika Corp,Durham,NC)を、5μM CuSO4 を含むPBS(2mL)中で37℃、12時間酸化処理した。1 mMのEDTAを添加することによって酸化を停止させた。
上記、酸化LDL 0.2 mg/mLをヒトβ2GPI(Affinity Biologicalsより購入)とともに終濃度 0.2mg/mL、37℃条件下で、16時間インキュベートし、酸化LDL/β2GPI複合体を得た。
【0067】
[実施例2]
抗原免疫
ヒト酸化LDL/β2GPI複合体精製タンパク質を同量のコンプリートアジュバント(SIGMA;F5881)と混合してエマルジョンにし、BALB/cマウス(メス)のfoot-padに5〜50μg/headで3から7日おきに数回免疫した。最終免疫の3〜5日後にマウスから鼠径リンパ節を摘出し、マウス骨髄腫細胞P3U1(P3-X63Ag8U1)との細胞融合を行った。
【0068】
[実施例3]
細胞融合、及びモノクローナル抗体産生細胞の選択と取得
細胞融合は次に示す一般的な方法を基本として行った。全ての培地中のFetal Bovine Serum(FBS)は、56℃で30min保温する処理により、非働化したものを使用した。P3U1は、RPMI1640-10%FBS(Penicillin, Streptomycin 含)で培養して準備した。
摘出したマウスの鼠径リンパ節細胞とP3U1を10:1〜2:1の割合で混合し、遠心した。沈殿した細胞に50%ポリエチレングリコール4000(Merck社、ガスクロマトグラフィー用 PEG4000、カタログNo. 9727)を融合促進剤として徐々に加えながら穏やかに混合し、細胞融合を行った。さらにRPMI1640を徐々に加えながら穏やかに混合後、遠心した。沈殿した融合細胞を、15% FCSを含むHAT培地(RPMI1640, HAT-supplement(Invitrogen;11067-030), Penicillin, Streptomycin)で適宜希釈し、200 μL/wellで96穴のマイクロプレートに播種した。
融合細胞をCO2インキュベータ(5% CO2、37 ℃)中で培養し、コロニーが十分に形成されたところで、培養上清をサンプリングしてスクリーニングを行った。
スクリーニングは、免疫に使用したヒト酸化LDL/β2GPI複合体抗原を感作した96穴プレートに対するELISA(実施例4に記載)で陽性となったものを選別した。15% FCSを含むHT培地(RPMI1640, HT-supplement(Invitrogen;21060-017), Penicillin, Streptomycin)でこれらを拡大培養後、限界希釈(limiting dilution)法により単クローン化した。このようにして、抗ヒト酸化LDL/β2GPI複合体抗体を免疫原としてクローン3H3を含むハイブリドーマクローン他、7種類を得た。
【0069】
[実施例4]
ヒト酸化LDL/β2GPI複合体及びβ2GPIに対する反応性(ELISA)
抗ヒト酸化LDL/β2GPI複合体抗体を検出するためのELISAは、以下の方法によって行った。すなわち、酸化LDL/β2GPIをマイクロプレート(Nunc社 Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20mM HEPES (pH 7.4))を用いて、各横軸に記載の抗体濃度に希釈したサンプル50μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/ PBS で洗浄した後、基質TMB (MOSS社;TMBZ) を50 μL添加し、室温にて 3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した(図1A)。
β2GPIとの反応性を検出するためのELISAは、以下の方法によって行った。すなわち、β2GPIをマイクロプレート(Nunc社Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4 ℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20 mM HEPES (pH 7.4))を用いて、各横軸に記載の抗体濃度に希釈したサンプル50 μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBS で洗浄した後、基質TMB (MOSS社; TMBZ) を50 μL添加し、室温にて3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した(図1B)。
さらに、β2GPIをマイクロプレート(nunc社 Maxisorp)に〜50 μg/mLまで濃度を変え (50 μL/ウエル)入れて、4℃で一晩インキュベートすることによって吸着させ、同様に抗体反応性を試験した(図示さず)。
その結果、固相化酸化LDL/β2GPI複合体に対しては、2H6 > 3H3, 2A12, 3D4 > 4C12, 1H4の反応性を示した。また、固相化β2GPIに対しては、2H6, 3D4 > 2A12, 4F10を示し、3H3, 4C12については反応性を示さなかった(図1A,B)。
しかし、マイクロタイタープレートへの感作濃度をあげると3H3なども反応性を示した(図示さず)。
抗体の反応性を確認する方法として次に、遊離の抗原で阻害をかける試験を行い、さらに、各抗体の特異性を見た。
【0070】
[実施例5]
溶液中の遊離のβ2GPI、酸化LDL/β2GPI複合体に対する競合反応性(ELISA)
固相化ヒト酸化LDL/β2GPI複合体及びβ2GPIに対する反応性(ELISA)試験において、各抗体と反応させる際に、酸化LDL/β2GPI複合体またはβ2GPIを共存させて反応することにより、固相抗原に対する阻害反応を行った(図2に測定系模式図を示す)。
すなわち、β2GPIをマイクロプレート(Nunc社; Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4 ℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20 mM HEPES (pH 7.4))を用いて、適当な濃度に希釈した抗体サンプル25 μL及び、競合させる抗原である酸化LDL/β2GPI複合体またはβ2GPIを各横軸に記載の抗原濃度にAssay bufferを用いて希釈したサンプル25 μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL; Code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBS で洗浄した後、基質TMB (MOSS社; TMBZ) を50 μL添加し、室温にて 3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した。
その結果、3H3, 4C12, 2A12ではELISA中に混在させた遊離の抗原が酸化LDL/β2GPI複合体の場合、固相の酸化LDL/β2GPIへの結合阻害が顕著であり、β2GPIでは阻害がかからなかった。2H6では、フリーの抗原が酸化LDL/β2GPI複合体でも阻害が起こる一方でβ2GPIの混在でも若干阻害が見られた。3D4では、遊離の抗原が酸化LDL/β2GPI複合体よりも、β2GPIによる阻害の方が高い阻害が見られた(図3)。
これらの結果より、各抗体の反応性は表1にまとめられる(実施例7に示す)。3H3は4C12と近い反応性を示すが同一の反応性ではなく、異なる特異性を有していた。
【0071】
[実施例6]
抗体の動脈硬化巣免疫組織染色
apoE-/-マウスおよび LDLR-/-マウス(Jackson Labより入手し、岡山大学内動物実験施設にて系統維持)を8週齢までは、普通飼料で飼育し(オリエンタル酵母NMF)、それ以降に高脂肪食(コレステロール1%、コール酸1%、無塩バター15%を普通飼料に配合)を4-6ヶ月負荷すると、胸部あるいは腹部大動脈に動脈硬化巣(プラーク)が現れ、肥厚、粥腫が出現する。そこで、8ヶ月齢のこれらマウスを屠殺し、特に今回は、胸部大動脈、大動脈の起始部、および大動脈弁の凍結切片を作製し、標本とし観察した。
蛍光免疫染色抗体法のためには、凍結切片を調製後、パラホルムアルデヒド固定し実験に供した。
モノクローナル抗体のCy5.5標識
0.1 M炭酸緩衝液(pH9.3)に対し、4 ℃で一晩透析した各種モノクローナル抗体(1mg/ml)をFluorolink Cy5.5 monofunctional dye (1 tube)に入れ、室温で30分間反応させ標識した。反応後、Sephadex G-25カラムにてCy5.5標識抗体を得た。
凍結切片を用いた免疫蛍光染色
1%パラホルムアルデヒドで5分間固定した切片に各種モノクローナル抗体を4℃で一晩反応させた。洗浄後、FITC標識抗マウスIgGあるいはIgM抗体(2次抗体)を室温で1時間反応させた。DAPlおよびRhodamine Phalloidin染色は、この2次抗体の反応時に添加した。その後、蛍光顕微鏡にて観察、撮影した。
免疫組織染色
結果、C57BL6系統のマウスを普通飼料で飼育したものに対し、3H3抗体、Mac3を用いる免疫蛍光染色では、泡沫化マクロファージが集族してできた粥腫が染まった。3H3によっても同様な部位が染まっている(図4)。
動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色を酸化LDL/β2GPI複合体を認識する他の抗体で行ったものと比較した。粥腫の染色陽性例は、粥腫の染色陽性例は、3H3および抗体Aのみである(図5)。
Cy5.5、Alexaなどを標識した粥腫に特異的な種々のモノクローナル抗体で動脈硬化巣の特異的な免疫染色が可能となった。
【0072】
[実施例7]
イメージング
in vivoイメージング:
Xenogen社のIVIS(登録商標) Imaging System、IVIS200により、イメージングを行った(Excitation:640 nm, Emission: 720 nm.)。
実験1:実施例6と同様に作製した高脂肪食負荷apoE-/-マウスに尾静脈より、Cy5.5標識モノクローナル抗体(0.25 mg/ml)を0.15 ml/head投与した。生理食塩水(PBS、対照)、Cy5.5標識抗体A、Cy5.5標識3H3抗体の3種類を投与した。投与して24時間後に胸部皮膚を剥がし、生きた状態で全身を撮影した(図7)。
実験2:更に胸部大動脈が繋がった状態で心臓を摘出して撮影した(図7)。3H3投与で大動脈起始部が強く染まる。抗体Aでも幾分染まるが3H3の場合ほど蛍光強度は強くない。2A12では全く染まらない。抗体Aでは十分に粥種が観察できず、その他取得された抗体クローンでも同様であり、粥種のイメージングの使用には適さなかった。
大動脈起始部の一定面積あたりの蛍光強度を測定した。PBSを投与した対照マウスの蛍光を1.0とした。3H3投与で、対照の3倍程度の蛍光が確認された。その他の抗体投与ではPBSを投与した対照マウスと蛍光強度に大きな変化は見られなかった(図10)。尚、この蛍光部位を解剖後確認したところ、臨床所見上の粥腫と一致していた。
以上、今回の抗体群の特異性試験を表1にまとめた。
【0073】
【表1】

固相酸化LDL/β2GPI複合体或いは固相β2GPI ELISA液中に競合阻害抗原として、β2GPIとの阻害性を確認したところ、固相酸化LDL/β2GPI複合体の場合:3D4>2H6>4C12>3H3、固相β2GPIの場合:2H6>3D4(4C12、3H3は固相β2GPIに弱くしか反応せず)。3H3は液中フリーの酸化LDL/β2GPI複合体(非変性)への特異性が高い。
【0074】
[実施例8]
酸化LDL/β2GPI複合体を認識するマウスモノクローナル抗体の可変領域遺伝子の解析
解析したモノクローナル抗体は、4種類のClone (3H3, 4C12, 2H6, 3D4)である。
これら4種類のCloneの抗体サブグラスは、3H3と4C12がIgG2b、2H6と3D4はIgG1である。
(L鎖可変領域遺伝子の解析)
4種(3H3, 4C12, 2H6, 3D4)のモノクローナル抗体を産生するハイブリドーマ細胞をRPMI1640+10%FCS培地でそれぞれ培養した。そのハイブリドーマ細胞からQuickPrep micro mRNA purification kit (Amersham Biosciences, code 27-9255-01)を用いてmRNAを得た。そのmRNAをFirst-Strand cDNA Synthesis kit (Amersham Biosciences, code 27-9261-01)を用いてcDNAとした。このcDNAを鋳型としてPCR法で遺伝子を増幅させるのであるが、以下に示す11パターンのPrimerの組み合わせによりPCR反応を行った。ここで、MKV1〜MKV11のPrimer配列は、多くのモノクローナル抗体のシグナル配列を解析することで、ほぼ全てのモノクローナル抗体のL鎖シグナルは配列をこの11種類のPrimer配列で網羅するように設定されている。この11種類のMKV primerとマウスL鎖定常領域配列に相当するMKC primer間における11パターンのPCR反応から少なくとも1種類のPCR反応で目的のL鎖可変領域が増幅されてくる。
【0075】
PCR反応条件は、以下のとおり。
マウスハイブリドーマからのcDNA 4 μL
2.5 mM dNTPs 4 μL
MKV1〜MKV11 primer(20μM)の11種類中の1つ 2.5 μL
MKC primer(20μM) 2.5 μL
DMSO 2.5 μL
×10 pfu polymerase Buffer 5 μL
pfu polymerase 1 μL
滅菌水 28.5 μL
Total 50 μL
94℃ 2 min
94℃ 1 min 55℃ 2min 72℃ 2min (30サイクル)
72℃ 4min
4℃ 無制限時間
【0076】
PrimerのDNA配列は以下を参照。
MKV1 primer:ATGAAGTTGCCTGTTAGGCTGTTGGTGCTG(配列番号:11)
MKV2 primer:ATGGAGWCAGACACACTCCTGYTATGGGTG(配列番号:12)
MKV3 primer:ATGAGTGTGCTCACTCAGGTCCTGGSGTTG(配列番号:13)
MKV4 primer:ATGAGGRCCCCTGCTCAGWTTYTTGGMWTCTTG(配列番号:14)
MKV5 primer:ATGGATTTWCAGGTGCAGATTWTCAGCTTC(配列番号:15)
MKV6 primer:ATGAGGTKCYYTGYTSAGYTYCTGRGG(配列番号:16)
MKV7 primer:ATGGGCWTCAAGATGGAGTCACAKWYYCWGG(配列番号:17)
MKV8 primer:ATGTGGGGAYCTKTTTYCMMTTTTTCAATTG(配列番号:18)
MKV9 primer:ATGGTRTCCWCASCTCAGTTCCTTG(配列番号:19)
MKV10 primer:ATGTATATATGTTTGTTGTCTATTTCT(配列番号:20)
MKV11 primer:ATGGAAGCCCCAGCTCAGCTTCTCTTCC(配列番号:21)
MKC primer:ACTGGATGGTGGGAAGATGG(配列番号:22)
(M=A or C, R=A orG, W=A orT, S=C or G, Y=C or T, K=G orT)
【0077】
PCR反応によりL鎖可変領域が増幅してきたPCR primerの組み合わせは、以下になる。
3H3:MKV7 - MKC
4C12:MKV7 - MKC
2H6:MKV5 - MKC
3D4:MKV4 - MKC
PCR増幅したL鎖可変領域遺伝子はpCR2.1ベクター(Invitogen)に挿入された。
L鎖可変領域遺伝子が挿入されたPCR2.1ベクターは、DNAシークエンサー(Applied Biosystems製:3130 Genatic Analyzer)によりDNA塩基配列が解読された。
【0078】
(H鎖可変領域遺伝子の解析)
4種(3H3, 4C12, 2H6, 3D4)のモノクローナル抗体を産生するハイブリドーマ細胞をRPMI1640+10%FCS培地でそれぞれ培養した。そのハイブリドーマ細胞からQuickPrep micro mRNA purification kit (Amersham Biosciences, code 27-9255-01)を用いてmRNAを得た。そのmRNAをFirst-Strand cDNA Synthesis kit (Amersham Biosciences, code 27-9261-01)を用いてcDNAとした。このcDNAを鋳型としてPCR法でH鎖可変領域遺伝子を増幅させるのであるが、以下に示す12パターンのPrimerの組み合わせによりPCR反応を行った。ここで、MHV1〜MHV12のPrimer配列は、多くのモノクローナル抗体のシグナル配列を解析することで、ほぼ全てのモノクローナル抗体のH鎖シグナルは配列をこの12種類のPrimer配列で網羅するように設定されている。この12種類のMHV primerとマウスH鎖定常領域配列に相当するMHCG2bあるいはMHCG1 primer間における12パターンのPCR反応から少なくとも1種類のPCR反応で目的のH鎖可変領域が増幅されてくる。ここで、MHCG2b Primerは、マウスIgG2bのH鎖定常領域に相当する配列を持ち、MHCG1 Primerは、マウスIgG1のH鎖定常領域に相当する配列である。それゆえ、IgG2b サブクラスである3H3 cloneと4C12 cloneのPCR増幅には、MHCG2b Primerが用いられ、IgG1サブクラスである2H6 cloneと3D4 cloneのPCR増幅には、MHCG1 Primerが用いられた。
【0079】
PCR反応条件は、以下のとおり。
マウスハイブリドーマからのcDNA 4 μL
2.5 mM dNTPs 4 μL
MHV1〜MHV12 primer(20μM)の12種類中の1つ 2.5 μL
MHCG2b あるいはMHCG1 primer(20μM) 2.5 μL
DMSO 2.5 μL
×10 pfu polymerase Buffer 5 μL
pfu polymerase 1 μL
滅菌水 28.5 μL
Total 50 μL
94℃ 2 min
94℃ 1 min 55℃ 2min 72℃ 2min (30サイクル)
72℃ 4min
4℃ 無制限時間
【0080】
PrimerのDNA配列は以下を参照。
MHV1 primer:ATGAAATGCAGCTGGGGCATSTTCTTC(配列番号:23)
MHV2 primer:ATGGGATGGAGCTRTATCATSYTCTT(配列番号:24)
MHV3 primer:ATGAAGWTGTGGTTAAACTGGGTTTTT(配列番号:25)
MHV4 primer:ATGRACTTTGGGYTCAGCTTGRTTT(配列番号:26)
MHV5 primer:ATGGACTCCAGGCTCAATTTAGTTTTCCTT(配列番号:27)
MHV6 primer:ATGGCTGTCYTRGSGCTRCTCTTCTGC(配列番号:28)
MHV7 primer:ATGGRATGGAGCKGGRTCTTTMTCTT(配列番号:29)
MHV8 primer:ATGAGAGTGCTGATTCTTTTGTG(配列番号:30)
MHV9 primer:ATGGMTTGGGTGTGGAMCTTGCTATTCCTG(配列番号:31)
MHV10 primer:ATGGGCAGACTTACATTCTCATTCCTG(配列番号:32)
MHV11 primer:ATGGATTTTGGGCTGATTTTTTTTATTG(配列番号:33)
MHV12 primer:ATGATGGTGTTAAGTCTTCTGTACCTG(配列番号:34)
MHCG2b primer:CAGTGGATAGACTGATGGGGG(配列番号:35)
MHCG1 primer:CAGTGGATAGACAGATGGGGG(配列番号:36)
(M=A or C, R=A orG, W=A orT, S=C or G, Y=C or T, K=G orT)
【0081】
PCR反応によりH鎖可変領域が増幅してきたPCR primerの組み合わせは、以下になる。
3H3:MHV4 - MHCG2b
4C12:MKV4 - MHCG2b
2H6:MHV4 - MHCG1
3D4:MHV1 - MHCG1
【0082】
PCR増幅したH鎖可変領域遺伝子はpCR2.1ベクター(Invitogen)に挿入された。
H鎖可変領域遺伝子が挿入されたPCR2.1ベクターは、DNAシークエンサー(Applied Biosystems製:3130 Genatic Analyzer)によりDNA塩基配列が解読された。
その結果、本発明に用いることのできる3H3のアミノ酸配列、CDRが明らかになった(図11)。
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<3H3抗体>
配列番号1:重鎖可変領域のアミノ酸配列
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号5:重鎖可変領域の塩基配列
配列番号6:軽鎖可変領域のアミノ酸配列
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号10:軽鎖可変領域の塩基配列
配列番号55:重鎖FR1のアミノ酸配列
配列番号56:重鎖FR2のアミノ酸配列
配列番号57:重鎖FR3のアミノ酸配列
配列番号58:重鎖FR4のアミノ酸配列
配列番号59:軽鎖FR1のアミノ酸配列
配列番号60:軽鎖FR2のアミノ酸配列
配列番号61:軽鎖FR3のアミノ酸配列
配列番号62:軽鎖FR4のアミノ酸配列
【0083】
[実施例9]
IVIS 200および3次元CTを用いた画像解析に関する検討
Computed Tomography(CT)画像を統合することによる、動脈硬化病巣の3次元(局在)イメージ得るための実験を実施した。
In vivo蛍光イメージング:
IVIS 200 Imaging System(Xenogen社)を用いて、蛍光イメージングを行った(Cy5.5の場合Excitation:640 nm、Emission: 720 nm、Alexa Fluor 750の場合Excitation:745 nm、Emission: 800 nm)。Cy5.5標識3H3抗体(IgG)0.25 mg/ml、0.15 ml/1匹、またはAlexa Fluor750標識3H3抗体1.0〜1.5 mg/ml、0.15 ml/1匹を高脂肪食負荷ApoE-/-マウスの尾静脈より投与し、2〜24時間後に、IVIS 200にて麻酔吸入下でin vivo蛍光を観察・撮影した。ApoE-/-マウスでは黒い体毛が蛍光を吸収するため体毛を剃って観察した。はじめに反射光による蛍光観察を行い、その後、透過光による蛍光観察を行って、マウスの3次元(3D)画像を作成し、光源情報と融合させた(図9 A :統合前のIVISによる3次元画像)。 図の赤い点の部分が3H3に標識した蛍光シグナルであり、濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化していることを示す。
Ex vivoイメージング:
3D-CT解析の終了後マウスを安楽死させ、PBS 10 mlで心灌流し、心臓および大動脈を摘出し、IVIS 200により反射蛍光画像を得た。
CT イメージング:
eXplore Locus CT System (GE Healthcare)によりCTイメージングを行った。IVIS 200によりイメージングを行ったのと同じマウス個体を用い、麻酔吸入下にX線照射を行い、CT画像を得た。
蛍光とCTの画像統合:
IVIS 200により検出した蛍光イメージング画像と、CTを用いて撮影した画像を、汎用3D 可視化ソフト(Amira、Mercury Computer Systems)を用いて統合させた(図9B:統合後の3次元CT画像)。
なお、一連の手順を模式的に示した(図8)。
(A) IVIS 200による特異抗体を用いた蛍光イメージング(反射)。
(B)統合前のIVIS 200による特異抗体を用いた蛍光イメージング(透過光;左)と CT像(中) 統合図(右):蛍光イメージング(透過光;左)中の赤い点の部分について濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化集中した部分(3H3結合 反応性が高い部位)であることを示す。
(C) IVISによる蛍光シグナルと3次元CTの統合画像:コンピューターの仮想空間上に3次元像を動画として構成(3次元グラフィックスのアニメーション)として、多方面からその標識された位置を確認した写真である。
可視光は生体内で吸収されてしまうためin vivo イメージングを行うには、生体に吸収されにくい波長である近赤外の蛍光標識が適している。今回は、Cy5.5またはAlexa Fluor 750により標識した抗体をマウスの尾静脈より投与して、IVIS200により蛍光を測定し、反射および透過による蛍光測定条件の検討を行った。Cy5.5標識抗体によるin vivoイメージングの反射蛍光画像では動脈硬化発症ApoE-/-マウスの大動脈弁および胸部大動脈の強いシグナルが観察された。さらに、ex vivoイメージングおよびex vivo蛍光顕微鏡観察により、静脈投与した蛍光標識抗体の動脈硬化巣への局在が確認できた。しかしながら、Cy5.5標識抗体を用いる場合、透過蛍光のシグナルが弱く、立体画像(3次元)での蛍光位置特定が容易ではなかった。一方、Alexa Fluor 750標識抗体では、静脈内投与後2時間後で強い特異的なシグナルが反射蛍光画像、透過蛍光画像の両方で得られ、立体画像を構築してみると胸の内部に強い蛍光シグナルが認められた(図8A、B中の左図)。続いて同じマウスについてCT撮影を行い、CT画像より骨と肺の臓器抽出を行って得られた画像(図8B中と、IVIS200により構築した蛍光イメージング画像をAmiraにより統合させた、統合3D画像(図8B右)を得たところ、心臓とその近傍の位置に蛍光シグナルが観察された。図の赤い点の部分について濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化していることを示す。CT(中央)と3D-CT統合の画像を示した(図8B右)。コンピューターの仮想空間上に3次元像を動画として構成(3次元グラフィックスのアニメーション)として、多方面からその標識された位置を確認した(図8C)。
【0084】
上記実験の結果、蛍光標識3H3抗体によるin vivoイメージングの反射蛍光画像では動脈硬化発症ApoE-/-マウスの大動脈弁および胸部大動脈の強いシグナルが観察された。さらに、ex vivoイメージングおよびex vivo蛍光顕微鏡観察により、静脈投与した蛍光標識抗体の動脈硬化巣への局在が確認できた。
【0085】
[実施例10]
キメラ抗体発現ベクターの作製
マウスH鎖の重鎖可変領域DNA(配列番号:5)を、HindIII, XhoIで切断して精製し、Lonza社のpEE6.4ベクター(ヒトIgG1定常領域をマルチクローニングサイトで組み込んである)に挿入し、キメラH鎖ベクターを作製した。同様に、マウスL鎖の軽鎖可変領域DNA(配列番号:10)をHindIII, BsiWI で切断して精製し、Lonza社のpEE14.4ベクター(ヒトKappa定常領域をマルチクローニングサイトに組み込んである)に挿入し、キメラL鎖ベクターを作製した。なお、重鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MELGLRWVFLVAILEGVQC(配列番号:37)のシグナル配列を利用し、さらに軽鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MDMRVPAQLLGLLLLWLPGARC(配列番号:38)のシグナル配列を利用した。
【0086】
[実施例11]
ヒト型化抗体可変領域の設計
CDR-grafting法に従い、ヒトフレームワークを選択し、マウスモノクローナル抗体3H3のCDRに置き換えた。
まず、H鎖可変領域フレームワーク、L鎖可変領域フレームワークそれぞれについてホモロジー検索を行った。
その結果、3H3抗体H鎖可変領域のフレームワーク(FR)領域(FR1領域(配列番号:55)、FR2領域(配列番号:56)、FR3領域(配列番号:57)、FR4領域(配列番号:58))は、ヒト抗体(Accession number AY882577(配列番号:43)のFR領域(FR1領域(配列番号:39)、FR2領域(配列番号:40)、FR3領域(配列番号:41)、FR4領域(配列番号:42))と高い相同性を持つことが分かった(図12(A)参照)。具体的には、H鎖について、マウス3H3抗体のFRと、CDR移植先ヒト抗体のFR 部分の相同性は 80/87=91.9%であった。
次に、これらのヒト抗体(Accession number AY882577)のFR領域に、3H3の重鎖CDR1〜CDR3(配列番号:2〜配列番号:4)が適切に移植されるように、アミノ酸配列を設計し、ヒト化抗体重鎖可変領域アミノ酸配列(配列番号:44)を設計した(図12(B)参照)。
重鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MELGLRWVFLVAILEGVQCのシグナル配列を利用した。
3H3抗体H鎖可変領域と同様に、3H3抗体L鎖可変領域のフレームワーク(FR)領域(FR1領域(配列番号:59)、FR2領域(配列番号:60)、FR3領域(配列番号:61)、FR4領域(配列番号:62))も、ヒト抗体(Accession number AY942004(配列番号:51))のFR領域(FR1領域(配列番号:47)、FR2領域(配列番号:48)、FR3領域(配列番号:49)、FR4領域(配列番号:50))と高い相同性を持つことが分かった(図13(A)参照)。具体的には、L鎖について、マウス3H3抗体のFRとCDR移植先ヒト抗体のFR 部分の相同性は 65/80=81.3%であった。
次に、ヒト抗体(Accession number AY942004)のFR領域に、3H3の軽鎖CDR1〜CDR3(配列番号:7〜配列番号:9)が適切に移植されるように、アミノ酸配列を設計し、ヒト化抗体軽鎖可変領域アミノ酸配列(配列番号:52)を設計した(図13(B)参照)。
軽鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MDMRVPAQLLGLLLLWLPGARCのシグナル配列を利用した。
ヒト化抗体の重鎖及び軽鎖可変領域をコードするDNAは、50base程度の合成オリゴDNAを約20base程度ハイブリダイズするように設計し、これらの合成オリゴDNAをPCR法によりアッセンブリさせることにより作製した。重鎖可変領域をコードするDNAを、5'端の合成オリゴDNAの末端に挿入した制限酵素HindIII部位および3'端の合成オリゴDNAの 末端に挿入したXhoI部位で切断し、上記Lonza社のヒトIgG1定常領域がクローニングされたpEE6.4ベクターへクローニングした。また、軽鎖可変領域をコードするDNAを、5'端の合成オリゴDNAの末端に挿入した制限酵素HindIII部位および3'端の合成オリゴDNAの 末端に挿入したBsiWI部位で切断し、上記ヒトκ鎖定常領域がクローニングされたpEE14.4ベクターへクローニングした。
上記の手法で作製したヒト化抗体3H3抗体の重鎖を3H3RHAと命名した(図14、シグナル配列-重鎖をコードする配列、塩基配列は配列番号:45、アミノ酸配列は配列番号:46)。同様に、ヒト化抗体3H3抗体の軽鎖を3H3RKAと命名した(図15、シグナル配列-軽鎖をコードする配列、塩基配列は配列番号:53、アミノ酸配列は配列番号:54)。
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<ヒト化3H3抗体>
配列番号44:重鎖可変領域のアミノ酸配列
配列番号46:重鎖のアミノ酸配列(シグナル配列を含む重鎖)
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号39:重鎖FR1のアミノ酸配列
配列番号40:重鎖FR2のアミノ酸配列
配列番号41:重鎖FR3のアミノ酸配列
配列番号42:重鎖FR4のアミノ酸配列
配列番号45:重鎖の塩基配列(シグナル配列を含む重鎖)
配列番号52:軽鎖可変領域のアミノ酸配列
配列番号54:軽鎖のアミノ酸配列(シグナル配列を含む軽鎖)
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号47:軽鎖FR1のアミノ酸配列
配列番号48:軽鎖FR2のアミノ酸配列
配列番号49:軽鎖FR3のアミノ酸配列
配列番号50:軽鎖FR4のアミノ酸配列
配列番号53:軽鎖の塩基配列(シグナル配列を含む軽鎖)
【0087】
[実施例12]
トランスフェクション、発現
キメラ抗体のH鎖ベクター及びL鎖ベクター、ヒト化3H3抗体のH鎖ベクター及びL鎖ベクターについて、構築したH鎖ベクターとL鎖ベクターをそれぞれNotI、SalIで消化し、断片同士をライゲーションし、一つのベクターを構築した。その後、構築したDNAをPvuI切断により直鎖化し、エレクトロポレーションによりCHOK1SV細胞に導入した。エレクトロポレーション後、死細胞を含む細胞溶液をグルタミンフリーの10%FBSを含むグルタミンフリーDMEMに加え、50μlずつ96穴プレートに撒いた。24時間培養後、セレクション試薬であるMSXを最終濃度25μMになるように、グルタミンフリーDMEM/10%FBS培地に溶解し、サプリメントとして、GS supplement(50倍溶液)を同培地に溶解し、先述した96穴プレートに150μlずつ添加した。その後、ELISAにて最も発現量の多いクローンを選択し、安定細胞株を樹立した。
培養上清中のIgG濃度は、培養上清を回収し、Goat anti-human IgG antibody, Fcγ fragment-specific (Stratech Scientific)、と Human IgG1/Kappa antibody (Binding site)でのサンドイッチELISAを行い、検出用として、Goat anti-human kappa light chain peroxidase conjugate (Sigma)を用い、市販の精製ヒトIgG(Cappel社)と の比較により定量した。
【0088】
[実施例13]
抗原に対するELISA活性(ヒト化IgG1とキメラIgG1の比較;結合活性比較)
一部の条件を除き[実施例4]に記載の方法と同じ方法でキメラ抗体-IgG1、ヒト化3H3(3H3RHA、3H3RKA)-IgG1の結合活性の濃度依存性を確認した。本方法は、吸収波長を650nmに変更して検出し、IgG濃度が測定された培養上清を各横軸に記載の抗体濃度に希釈したサンプルとした点において[実施例4]に記載の方法と異なる。その結果、ヒト化IgG1はマウス可変領域を有するキメラIgG1とELISA結合活性の抗体濃度依存性が同等であった。なお、コントロールとしては293T細胞の培養上清を用いた(図16)。
【0089】
[実施例14]
Antibody-dependent Cellular Cytotoxicity (ADCC)活性の低いIgG4型への変換
イメージング用途としては、IgG1型より、ADCC活性、CDC活性等の生理活性の低いIgG2又は4型が望ましいと考えられるため、ヒト化3H3(3H3RHA、3H3RKA)-IgG1の定常領域をIgG4型へ変換した抗体を作製した。IgG4は不安定でヘテロな複合体を形成し、定常領域におけるKabat則によるナンバリングで241番目のアミノ酸であるセリンがその安定性に関わることが報告されており、このアミノ酸をIgG1型、IgG2型のプロリンへ変換することで安定性が改善されることが報告されている。そこで、241番目のアミノ酸をプロリンに変換した変異型IgG4をIgG1に換えて組み込んだpEE6.4ベクターを構築し、上記と同様の方法でヒト化3H3(3H3RHA、3H3RLA)- IgG4を作成した(図17)。抗体発現、抗体濃度を測定後、結合活性比較を行った。
抗原に対するELISA活性(ヒト化変異型IgG4とキメラIgG1、ヒト化IgG1の比較;結合活性比較)
実施例13と同様の方法でヒト化3H3(3H3RHA、3H3RLA)- IgG4抗体の結合活性をキメラ-IgG1、ヒト化3H3(3H3RHA、3H3RLA)-IgG1 と比較したところ、ELISA結合活性は同等であった(図18)。
【0090】
[実施例15]
免疫染色/イメージング
上記実施例において作成された、キメラ-IgG1、ヒト化3H3(3H3RHA、3H3RLA)- IgG1抗体、ヒト化3H3(3H3RHA、3H3RLA)- IgG4抗体は[実施例6]の免疫染色、[実施例7]のイメージングと同様の試験を行うことにより、マウス3H3抗体と同等のイメージング活性を示した活性を有することが確認できる。
【0091】
本実験により、近赤外蛍光物質(Cy5.5、Alexa750)標識抗体を用いることで、マウスにおける動脈硬化のイメージングが可能となったばかりではなく、3次元CTによる画像との統合も可能であることが判明した。またこの種の抗体がヒトの動脈硬化巣を検出できることも明らかになっており、本実験結果は臨床用の画像診断技術に繋がる。さらに、当該マウスイメージング技術は、創薬のためのスクリーニング系として既に実用可能なものである。
【産業上の利用可能性】
【0092】
従来の動脈硬化検査法では、動脈硬化の発症部位(存在部位)が特定できない。これに対し、本発明は、動脈硬化巣(特に粥腫、粥状動脈硬化)の存在部位やその大きさを視覚的に捉えることの出来る非侵襲的診断法を提供するものである。
動脈硬化好発モデルマウス(例えば、アポE欠失(ApoE-/-)マウス:高い血漿コレステロール濃度を維持し、自然発生アテローム性動脈硬化様病変を発症する)と、当該イメージング用の抗体を用いることで、粥状動脈硬化の治療薬のスクリーニング系が構築できる。
さらには、当該抗体をヒト化抗体に変換して、臨床診断用イメージングシステムを構築することが可能である。従って、動脈粥腫からのプラーク遊離などが動脈塞栓を起因とした脳塞栓症、心筋梗塞の原因となることが知られており、慢性的かつ無症候性に潜行進展するヒトの粥状動脈硬化のモニタリング法は、生活習慣病の予防、治療方針の一助になるといえる。
【技術分野】
【0001】
本発明は、酸化LDL/β2GPI複合体に対する抗体、及び該抗体を利用した粥状動脈硬化の存在部位の特定、治療効果モニタリングなどの非侵襲的な動脈硬化の診断法に関する。
【背景技術】
【0002】
動脈硬化の状態を観察する現在実用化されている診断方法としては、以下の4種類の方法が例示される。
「足関節上腕血圧比」:横になった状態で両腕、両足の血圧を測定すると、正常では足首の方がやや高い値になる。ところが血管につまりがある場合、そこから下流の血圧が低下し、足関節の血圧/上腕の血圧の比(ABI)が低くなる。ABIの低下は、下肢動脈だけでなく、全身に動脈硬化があることを推定する。
「脈波速度検査」:動脈の硬さを調べる方法で動脈硬化の進行を推定する方法。健康人は血管に弾力性があるため、振動が血管壁で吸収され、脈波の速度は遅い。動脈硬化が進むと、脈波は速くなるので、速度を指標として動脈硬化の進行を推定することができる。
「頸動脈超音波検査」:皮膚表面近くを走る頸動脈は内部の様子を超音波で観察しやすいので、この頸動脈を調べ、全身の動脈硬化の進行状況を推定する方法。
「MR血管造影(MRA)」:「CT血管造影(CTA)」血管疾患の画像診断法として従来血管造影が主流であったが、より侵襲が少なく血管造影にほぼ匹敵する画像情報が用いられるようになった。CTAの利点は、(1)空間分解能が高い、(2)検査が簡便、(3)石灰化病変の検出に優れる点が挙げられる。
上記「足関節上腕血圧比」、「脈波速度検査」では、粥状化位置特定と個別箇所ごとの進行状況の診断はできず、間接的に数値として評価せざるを得ない。
また、「頸動脈超音波検査」は、脈波速度検査などと違い、実際に血管の内部を画像として直接的に診ることができる点が優れている。しかし、超音波画像の濃淡や形から血管壁の状況を読みとる必要があるため、検査を行う医師や検査技師に技量が求められ、頸動脈以外の部位の、粥状化位置特定と個別箇所ごとの進行状況の診断はできない。
【0003】
また、動脈硬化進行状況をモニタリングする方法としては、血中の酸化LDL/β2GPI複合体を測定することによる、ELISA系が存在する(特許3370334号、特許3898680号、WO2003/022866、WO2004/023141)。しかしながら、従来の酸化LDL/β2GPI複合体測定ELISAでは、動脈硬化巣の大きさを推定することは出来るが、動脈硬化巣の存在部位は特定できない。
さらに、MRIや放射ラベルを用いたイメージングでも画像の濃淡や形から血管壁の状況を読みとる必要があるため、検査を行う医師や検査技師に技量が求められる(米国特許6716410号、米国特許6375925号)。
なお、以下に本発明に関連する先行技術文献を示す。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特許3370334号
【特許文献2】特許3898680号
【特許文献3】WO2003/022866
【特許文献4】WO2004/023141
【特許文献5】特表2001−506983
【特許文献6】特許第4044972
【特許文献7】米国特許6716410号
【特許文献8】米国特許6375925号
【非特許文献】
【0005】
【非特許文献1】Journal of Biological Chemistry 269, 15274-15279,1994
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、特異的な粥状動脈硬化(粥腫ともいう)の存在部位の特定、治療効果モニタリングなどの非侵襲的な動脈硬化の診断法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、動脈硬化の粥腫の構成物を鑑みて、酸化LDL/β2GPI複合体抗体が、in vivoで動脈硬化の粥腫に特異的に結合するか、かつ、体外からその病変を特異的に検出可能か検討した。
すなわち、生体中での、動脈硬化巣特に動脈硬化粥腫の存在位置及びその大きさのイメージングに適応可能な抗体を特定し、その特異性を解析した。その結果、動脈硬化切片の免疫染色ができる酸化LDL/β2GPI複合体抗体の中でも、特定のエピトープに特異性を有する酸化LDL/β2GPI複合体抗体の標識体が、粥状動脈硬化の存在部位の特異的なイメージングに有効であることが明らかとなった。このことは、粥腫という病変に特異的なイメージングを可能とする。
即ち本発明は、以下〔1〕〜〔12〕を提供するものである。
〔1〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する、下記(a)〜(j)のいずれかに記載の抗体;
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体、
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
〔2〕 〔1〕に記載の抗体が結合するエピトープと同じエピトープに結合する抗体。
〔3〕 ヒト化抗体またはキメラ抗体である、〔1〕または〔2〕に記載の抗体。
〔4〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング剤。
〔5〕 〔1〕〜〔3〕のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング剤。
〔6〕 動脈硬化において、粥腫の位置及び/または大きさをイメージングする、〔4〕または〔5〕のイメージング剤。
〔7〕 酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング用キット。
〔8〕 〔1〕〜〔3〕のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング用キット。
〔9〕 以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法:
(a)〔1〕〜〔3〕のいずれかに記載の抗体が投与された動脈硬化疾患モデル非ヒト動物に、候補化合物を投与する工程、
(b)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣の大きさまたは存在部位を比較する工程、及び
(d)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
〔10〕 〔1〕〜〔3〕に記載の抗体を含む、動脈硬化の存在部位のイメージング方法。
〔11〕 動脈硬化の存在部位のイメージング剤の製造における、〔1〕〜〔3〕に記載の抗体の使用。
〔12〕 動脈硬化の存在部位のイメージング方法に使用するための、〔1〕〜〔3〕に記載の抗体。
【図面の簡単な説明】
【0008】
【図1】BALB/cマウスに抗原として酸化LDL/β2GPI複合体を免疫して得られたモノクローナル抗体の、固相抗原に対する反応性を示す図である。ヨコ軸は抗体濃度、縦軸には吸光度を示す。
【図2】液中の抗原との競合性をアッセイ(競合阻害試験)するための模式図である。
【図3】抗原による競合阻害試験を示すグラフである。ヨコ軸は液中の抗原濃度、縦軸には阻害抗原が存在しない場合の吸光度を100%としたときの阻害された%を示す。3H3、および4C12は、酸化LDLに結合したβ2GPIを認識する抗体で、遊離のβ2GPIを認識していない。一方、2H6、3D4、2A12は、遊離のβ2GPIに反応する抗体である。
【図4】動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色写真である。A)DAPI:核の染色、B)Mac3:マクロファージに特異的抗体、C)3H3抗体、D)control。C57BL6系統のマウスを普通飼料で飼育したものに対し、3H3抗体。Mac3を用いる免疫蛍光染色では、泡沫化マクロファージが集族してできた粥腫が染まっている。3H3によっても同様な部位が染まっている。
【図5】動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色写真である。酸化LDL/β2GPI複合体抗体を用いた他の抗体の蛍光免疫染色の結果を示す。粥腫の染色陽性例は、3H3および抗体Aのみである。
【図6】IVIS 200による特異抗体を用いた蛍光イメージング(反射蛍光観察)写真である。in vivo:高脂肪食6ヵ月以上負荷したApoE-/-マウスに、イメージング剤を、脂肪食負荷ApoE-/-マウスの尾静脈より投与し、2〜24時間後に、IVIS 200にて麻酔吸入下でin vivo蛍光を観察・撮影した。その際ApoE-/-マウスでは黒い体毛が蛍光を吸収するため体毛を剃って観察した。 ex vivo:マウスを安楽死させ、開胸して心臓と大動脈を露出させ、右心耳に小さく切り込みを入れた後、左心室に針を挿入して冷PBS 10 mlで心灌流し、心臓および大動脈を摘出して、IVIS 200により反射蛍光画像を得た。
【図7】IVIS200によるイメージング写真である(Excitation:640 nm, Emission:720 nm.)。実験1:高脂肪食負荷apoE-/-マウスに生理食塩水(PBS、対照)、Cy5.5標識抗体A、Cy5.5標識3H3抗体を尾静脈より投与。24時間後に胸部皮膚を剥がし、生きた状態で全身を撮影。更に胸部大動脈が繋がった状態で心臓を摘出して撮影。実験2:PBS、Cy5.5標識2A12抗体、Cy5.5標識3H3抗体投与マウスから摘出した心臓と胸部大動脈。3H3投与で大動脈起始部が強く染まる。抗体Aでも幾分染まるが3H3の場合ほど蛍光強度は強くない。2A12では全く染まらない。
【図8】特異抗体を用いた動脈硬化の3次元画像イメージング写真である。(A) IVIS 200による特異抗体を用いた蛍光イメージング(反射)、(B) IVIS200による透過光 3D 画像(左図)と統合前のCT-3D像(中央)と統合後の3D画像(右図)、(C) IVISによる蛍光シグナルと3次元CTの統合画像である。
【図9】統合前のIVIS 200による蛍光3次元画像(上段:A)及びIVIS 200による蛍光シグナルと3次元CTの統合画像(下段:B)の写真である。
【図10】IVIS200で測定した際の大動脈起始部周辺のCy5.5に依存する蛍光量を示す図である。大動脈起始部の一定面積あたりの蛍光強度を測定した。PBSを投与した対照マウスの蛍光を1.Oとした。3H3投与で、対照の3倍程度の蛍光が確認されたその他の抗体投与では蛍光強度に大きな変化は見られなかった。
【図11】3H3抗体アミノ酸配列図である。各CDRを下線で示した。
【図12】マウスH鎖からヒト化 H鎖への変換に関する図である。(A)マウスH鎖と移植先ヒト H鎖のFR 部分の相同性を比較したものである。すなわちマウス3H3抗体の重鎖(H鎖)とヒト抗体AY882577のアミノ酸を比較した図である。H鎖のFR 部分については、87アミノ酸中80アミノ酸が一致しており、91.9%の相同性があった。(B)マウスH鎖からヒト化 H鎖への変換過程を示す。マウス3H3抗体の重鎖をKabat則によりナンバリングし、CDR1〜CDR 3、FR1〜FR 4を同定した。Kabat則の詳細については、http://www.bioinf.org.uk/abs/numbering.htmlを参照のこと。マウス3H3抗体FR1〜FR4と相同性のあるヒトFR1〜FR4を有するAY882577を同定し、マウス3H3 CDR1〜CDR 3を移植した3H3RHAを設計した。図中の3H3 VHはマウス3H3抗体の重鎖であり、3H3CDRは3H3重鎖のCDR1〜CDR 3であり、AY882577 FRはAY882577のCDRを除いた重鎖フレームワークである。
【図13】マウスL鎖からヒト化 L鎖への変換に関する図である。(A)マウスL鎖と移植先ヒト L鎖FR 部分の相同性を比較したものである。すなわちマウス3H3抗体の軽鎖(L鎖)とヒト抗体AY942004のアミノ酸を比較した図である。L鎖のFR 部分については、80アミノ酸中65アミノ酸が一致しており、81.3%の相同性があった。(B)マウスL鎖からヒト化 L鎖への変換過程を示す。マウス3H3抗体の軽鎖をKabat則によりナンバリングし、CDR1〜CDR 3、FR1〜FR 4を同定した。Kabat則の詳細については、http://www.bioinf.org.uk/abs/numbering.htmlを参照のこと。マウス3H3抗体FR1〜FR4と相同性のあるヒトFR1〜FR4を有するAY942004を同定し、マウス3H3 CDR1〜CDR 3を移植した3H3RKAを設計した。図中の3H3 VKはマウス3H3抗体の軽鎖であり、3H3CDRは3H3 L鎖のCDR1〜CDR 3であり、AY942004FRはAY942004のCDRを除いた軽鎖フレームワークである。
【図14】3H3RHAの塩基配列及びアミノ酸配列を示す図である。シグナル配列を含めた、3H3RHAの塩基配列及びアミノ酸配列を示す。シグナル配列は下線で示した。
【図15】3H3RKAの塩基配列及びアミノ酸配列を示す図である。シグナル配列を含めた、3H3RKAの塩基配列及びアミノ酸配列を示す。シグナル配列は下線で示した。
【図16】抗原に対するELISA活性(ヒト化IgG1とキメラIgG1の比較)を示すグラフである。横軸に抗体濃度、縦軸にELISA で測定された吸光度(650nm)を示す。ヒト化IgG1とキメラIgG1とマウス3H3は結合性に差はなかった。コントロールとしては、293細胞の培養上清を用いた。
【図17】変異型IgG4抗体の作製を示す図である。定常領域のアミノ酸配列中、IgG1,2と異なるKabat則241番目のセリンがIgG4の不安定化につながるという報告があるため、この位置をプロリンに変換した定常領域を作製した。
【図18】変異型IgG4抗体の結合活性比較図である。横軸に抗体濃度、縦軸にELISA で測定された吸光度(650nm)を示す。ヒト化IgG4とキメラIgG1とヒト化IgG1の結合性に差はなかった。コントロールとしては、293細胞の培養上清を用いた。
【発明を実施するための形態】
【0009】
本発明は、酸化変性LDL(酸化LDL)とβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を提供する。動脈硬化巣で酸化LDLと血漿糖タンパク質であるβ2GPIとの複合体が形成される。本願発明に包含される抗体は、この複合体に結合することを特徴とする。
【0010】
本願発明に包含される抗体としては、具体的には以下の抗体が挙げられるが、これらに限定されるものではない。
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体。
【0011】
また、本発明には、「(a)〜(e)に記載の抗体において1若しくは複数のアミノ酸が置換、欠失、付加および/または挿入された抗体であって、(a)〜(e)に記載の抗体と同等の活性を有し、CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖、並びに、CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖の対を有する抗体」も包含されるが、これに限定されない。
ポリペプチドに変異を導入する方法は、あるポリペプチドと機能的に同等なポリペプチドを調製するための、当業者によく知られた方法の一つである。例えば、当業者であれば、部位特異的変異誘発法(Hashimoto-Gotoh, T. et al. (1995) Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Methods Enzymol. 100, 468-500、Kramer, W. et al. (1984) Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Proc Natl Acad Sci USA. 82, 488-492、Kunkel (1988) Methods Enzymol. 85, 2763-2766)などを用いて、本発明の抗体に適宜変異を導入することにより、該抗体と機能的に同等な抗体を調製することができる。また、アミノ酸の変異は自然界においても生じうる。このように、本発明の抗体のアミノ酸配列において1もしくは複数のアミノ酸が変異したアミノ酸配列を有し、該抗体と機能的に同等な抗体もまた本発明の抗体に含まれる。
このような変異体における、変異するアミノ酸数は、通常、50 20アミノ酸以内であり、好ましくは30 15 アミノ酸以内であり、さらに好ましくは10アミノ酸以内(例えば、5アミノ酸以内)である。尚、変異はFR(フレームワーク)内であることが好ましい。
変異するアミノ酸残基においては、アミノ酸側鎖の性質が保存されている別のアミノ酸に変異されることが望ましい。例えばアミノ酸側鎖の性質に基づいて、次のような分類が確立している。
疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、
親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、
脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、
水酸基含有側鎖を有するアミノ酸(S、T、Y)、
硫黄原子含有側鎖を有するアミノ酸(C、M)、
カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、
塩基含有側鎖を有するアミノ酸(R、K、H)、
芳香族含有側鎖を有するアミノ酸(H、F、Y、W)
(括弧内はいずれもアミノ酸の一文字標記を表す)
あるアミノ酸配列に対する1又は複数個のアミノ酸残基の欠失、付加及び/又は他のアミノ酸による置換により修飾されたアミノ酸配列を有するポリペプチドがその生物学的活性を維持することはすでに知られている(Mark, D. F. et al., Proc. Natl. Acad. Sci. USA (1984) 81, 5662-5666 、Zoller, M. J. and Smith, M., Nucleic Acids Research (1982) 10, 6487-6500 、Wang, A. et al., Science 224, 1431-1433 、Dalbadie-McFarland, G. et al., Proc. Natl. Acad. Sci. USA (1982) 79, 6409-6413 )。すなわち、一般に、あるポリペプチドを構成するアミノ酸配列中、各群に分類されたアミノ酸は、相互に置換したときに、当該ポリペプチドの活性が維持される可能性が高いとされている。本発明において、上記アミノ酸群の群内のアミノ酸間の置換を保存的置換という。
【0012】
また本発明は、酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する本願発明の抗体が結合するエピトープと同じエピトープに結合する抗体も提供する。この抗体は、酸化LDLと複合体を形成している酸化LDL/β2GPI分子上の特定のエピトープを認識する。
この限りではないが、例えばある抗体が他の抗体と同じエピトープを認識するか否かは、両者のエピトープに対する競合によって確認することができる。抗体間の競合は、競合結合アッセイによって評価することができ、その手段としてELISA、蛍光エネルギー転移測定法(FRET)や蛍光微量測定技術(FMAT(登録商標))などが挙げられる。抗原に結合した該抗体の量は、同じエピトープへの結合に対して競合する候補競合抗体(被検抗体)の結合能に間接的に相関している。すなわち、同じエピトープに対する被検抗体の量や親和性が大きくなるほど、該抗体の抗原への結合量は低下し、抗原への被検抗体の結合量は増加する。具体的には、抗原に対し、適当な標識をした該抗体と評価すべき抗体を同時に添加し、標識を利用して結合している該抗体を検出する。抗原に結合した該抗体量は、該抗体を予め標識しておくことで、容易に測定できる。この標識は特には制限されないが、手法に応じた標識方法を選択する。標識方法は、具体的には蛍光標識、放射標識、酵素標識などが挙げられる。
ここでいう「同じエピトープを認識する抗体」とは、標識該抗体に対して、非標識の該抗体の結合により結合量を50%低下させる濃度(IC50)に対して、被検抗体が非標識該抗体のIC50の通常、100倍、好ましくは80倍、さらに好ましくは50倍、さらに好ましくは30倍、より好ましくは10倍高い濃度で少なくとも50%、標識該抗体の結合量を低下させることができる抗体である。
【0013】
本発明の抗体には、ポリクローナル抗体およびモノクローナル抗体の両方が含まれる。モノクローナル抗体およびポリクローナル抗体の調製および精製方法は、当分野で知られており、例えば Harlow and Lane, Antibodies: A Laboratory Manual (New York: Cold Spring Harbor Laboratory Press, 1988)に記載されている。また、本発明の抗体は、アイソタイプ(IgG、IgM、IgA、IgE、IgDなど)を問わない。
【0014】
本発明の抗体には、ヒト化(humanized)抗体またはキメラ(chimeric)抗体などの組換え抗体も含まれる。「ヒト化抗体」とは、ヒトの抗体に構造を類似させた抗体のことをいう。このようなヒト化抗体またはキメラ抗体には、ヒト型キメラ抗体(例えば抗体の一部がヒト化された抗体、CH2領域がヒト化された抗体、Fc領域がヒト化された抗体、定常領域がヒト化された抗体)、及び定常領域及び可変領域に存在するCDR(相補性決定領域)以外の部分がヒト化されたヒト型CDR移植(CDR-grafted)抗体(P.T.Johons et al., Nature 321,522(1986))、完全ヒト化抗体などが含まれる。ヒト型CDR移植抗体の抗原結合活性を高めるため、マウス抗体と相同性の高いヒト抗体FR(フレームワーク)を選択する方法、相同性の高いヒト化抗体を作製する方法、ヒト抗体にマウスCDRを移植した後さらにFRのアミノ酸を置換する方法の改良技術もすでに開発され(米国特許第5585089号、米国特許第5693761号、米国特許第5693762号、米国特許第6180370号、欧州特許第451216号、欧州特許第682040号、特許第2828340号などを参照)、本発明のヒト型CDR移植抗体の作製に利用することもできる。ヒト化の際に用いられるヒト抗体は、IgG、IgM、IgA、IgE、IgDなど如何なるアイソタイプのヒト抗体でもよい。
本願発明に包含される「ヒト化抗体」としては、具体的には以下の抗体が挙げられるが、これらに限定されるものではない。
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
【0015】
また、本発明には、「(f)〜(j)に記載の抗体において1若しくは複数のアミノ酸が置換、欠失、付加および/または挿入された抗体であって、(f)〜(j)に記載の抗体と同等の活性を有し、CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖、並びに、CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖の対を有する抗体」も包含されるが、これに限定されない。
【0016】
ヒト型キメラ抗体は例えば、上記のH鎖可変領域の構造及び/又はL鎖可変領域の構造を有する抗体の定常領域をヒト抗体の定常領域に置換することにより作製することができる。ヒト抗体の定常領域としては公知のものを採用することができる。以下に、ヒト型キメラ抗体の作製方法の一例を示す。
【0017】
まず、特定の対象抗原に対するマウス抗体を産生するハイブリドーマよりmRNAを抽出し、常法に従ってcDNAを合成する。合成したcDNAをベクターに組み込みcDNAライブラリーを構築する。このcDNAライブラリーから、H鎖遺伝子フラグメント及びL鎖遺伝子フラグメントをプローブとして用いることにより、H鎖遺伝子及びL鎖遺伝子を含有するベクターを選択する。選択されたベクターの挿入配列のシークエンシングを行うことにより、H鎖可変領域及びL鎖可変領域の遺伝子の配列が決定される。このようにして得られた配列データを基にH鎖可変領域をコードするDNAを化学合成、生化学的切断/再結合等により作製する。得られたH鎖可変領域をコードするDNAを、ヒトH鎖定常領域をコードするDNAとライゲーションして発現用ベクターに組込むことによりH鎖発現ベクターを作製する。発現ベクターとしては例えばSV40 virus basedベクター、EB virus basedベクター、BPV(パピローマウイルス)basedベクターなどを用いることができるが、これらに限定されるものではない。一方、同様の方法によりL鎖発現ベクターを作製する。これらH鎖発現ベクター及びL鎖発現ベクターにより宿主細胞を共形質転換する。宿主細胞としてはCHO細胞(チャイニーズハムスター卵巣)(A.Wright & S.L.Morrison, J.Immunol.160, 3393-3402 (1998))、SP2/0細胞(マウスミエローマ)(K.Motmans et al., Eur.J.Cancer Prev.5,512-519 (1996),R.P.Junghans et al., Cancer Res.50,1495-1502 (1990))などが好適に用いられる。また、形質転換にはリポフェクチン法(R.W.Malone et al.,Proc.Natl.Acad.Sci.USA 86,6077 (1989), P.L.Felgner et al., Proc.Natl.Acad.Sci.USA 84,7413 (1987))、エレクトロポレーション法、リン酸カルシウム法(F.L.Graham & A.J.van der Eb,Virology 52,456-467(1973))、DEAE-Dextran法等が好適に用いられる。
【0018】
形質転換体を培養した後、形質転換体の細胞内又は培養液よりヒト型キメラ抗体を分離する。抗体の分離、精製には、遠心分離、硫安分画、塩析、限外濾過、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーなどの方法を適宜組み合わせて利用することができる。
【0019】
一方、ヒト型CDR移植抗体は例えば以下の方法により作製することができる。まず、上記キメラ抗体の製造方法の欄で述べた方法により、特定の抗原に対する抗体のH鎖可変領域及びL鎖可変領域のアミノ酸配列及びそれをコードする塩基配列を決定する。併せて各CDR領域のアミノ酸配列及び塩基配列を決定する。
【0020】
次に、CDR領域を挟んで存在するFR(フレームワーク領域)を選択する。FRの選択には、およそ三つの方法が採用できる。1つめの方法は、NEWM、REIなど既に三次元構造の明らかとなったヒト抗体フレームを用いる方法である(Riechmann L. et al., Nature 332, 323-3Z7 (1988); Tempst, PR. et al., Protein Engineering 7, 1501-1507 (1994); Ellis JH. et al., J. Immunol 155, 925-937 (1995))。2つめの方法は、目的のマウス抗体可変領域と最も高いホモロジーを持つヒト抗体可変領域をデータベースより選択し、そのFRを用いる方法である(Queen C. et al., Proc Natl Acad SciUSA 86, 10029-10033 (1989); Rozak MJ. et al., J Biol Chem 271, 22611-22618 (1996); Shearman CW. et al., J.Immunol 147, 4366-4373 (1991))。3つめの方法は、ヒト抗体のFRで最も共通に用いられるアミノ酸を選択する方法である(Sato K. et al., Mol Immunol 31, 371-381 (1994); Kobinger F. et al., Protein Engineering 6, 971-980 (1993); Kettleborough CA. et al., Protein Engineering 4, 773-783 (1991))。本発明ではこれらいずれの方法を用いることもできる。
尚、選択されたヒトFRのアミノ酸配列を改変したアミノ酸配列であっても、最終的に得られるヒト型CDR移植抗体が対象抗原に対する特異的結合性を有する限り、FRのアミノ酸配列として利用することができる。特に、選択されたヒトFRのアミノ酸の一部をCDRの由来となった抗体のFRのアミノ酸に変更した場合、抗体の特性が維持される可能性が高い。改変されるアミノ酸の数は好ましくはFR全体の30%以下であり、更に好ましくはFR全体の20%以下であり、更に好ましくはFR全体の10%以下である。
【0021】
次に、これらいずれかの方法により選択したFRと上記CDRとを組み合わせることによりH鎖可変領域及びL鎖可変領域をコードするDNAを設計する。この設計を基にH鎖可変領域をコードするDNAとL鎖可変領域をコードするDNAを化学合成、生化学的切断/再結合等によりそれぞれ作製する。そしてH鎖可変領域をコードするDNAを、ヒト免疫グロブリンH鎖定常領域をコードするDNAとともに発現ベクターに組み込みH鎖発現ベクターを構築する。同様に、L鎖可変領域をコードするDNAを、ヒト免疫グロブリンL鎖定常領域をコードするDNAとともに発現ベクターに組み込みL鎖発現ベクターを構築する。発現ベクターとしては例えばSV40 virus basedベクター、EB virus basedベクター、BPV(パピローマウイルス)basedベクターなどを用いることができるが、これらに限定されるものではない。
【0022】
以上の方法で作製されたH鎖発現ベクター及びL鎖発現ベクターにより宿主細胞を共形質転換する。宿主細胞としてはCHO細胞(チャイニーズハムスター卵巣)(A.Wright& S.L.Morrison, J.Immunol.160, 3393-3402 (1998))、SP2/0細胞(マウスミエローマ)(K.Motmans et al., Eur.J.Cancer Prev.5,512-519 (1996),R.P.Junghans et al.,Cancer Res.50,1495-1502 (1990))などが好適に用いられる。また、形質転換にはリポフェクチン法(R.W.Malone et al.,Proc.Natl.Acad.Sci.USA 86,6077 (1989), P.L.Felgner et al.,Proc.Natl.Acad.Sci.USA 84,7413 (1987))、エレクトロポレーション法、リン酸カルシウム法(F.L.Graham & A.J.van der Eb,Virology 52,456-467(1973))、DEAE-Dextran法等が好適に用いられる。
【0023】
形質転換体を培養した後、形質転換体の細胞内又は培養液よりヒト型CDR移植抗体を分離する。抗体の分離、精製は、遠心分離、硫安分画、塩析、限外濾過、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーなどの方法を適宜組み合わせて行うことができる。
【0024】
また、ヒト抗体の取得方法も知られている。例えば、ヒトリンパ球をin vitroで所望の抗原または所望の抗原を発現する細胞で感作し、感作リンパ球をヒトミエローマ細胞、例えばU266と融合させ、抗原への結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878参照)。また、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物を所望の抗原で免疫することで所望のヒト抗体を取得することができる(国際特許出願公開番号WO 93/12227, WO 92/03918,WO 94/02602, WO 94/25585,WO 96/34096, WO 96/33735参照)。
【0025】
更なる実施態様では、抗体又は抗体断片は、McCafferty等(Nature, 348:552-554 (1990))に記載された技術を使用して産生される抗体ファージライブラリーから分離することができる。Clackson等(Nature, 352:624-628 (1991))及び Marks等(J. Mol. Biol., 222:581-597 (1991))は、ファージライブラリーを使用したマウス及びヒト抗体の分離を記述している。続く刊行物は、鎖シャフリングによる高親和性(nM範囲)のヒト抗体の生成(Marks等, Bio/Technology, 10:779-783[1992])、並びに非常に大きなファージライブラリーを構築するための方策としてコンビナトリアル感染とインビボ組換え(Waterhouse等, Nuc. Acids. Res., 21:2265-2266[1993])を記述している。従って、これらの技術はモノクローナル抗体の分離に対する伝統的なモノクローナル抗体ハイブリドーマ法に対する実行可能な別法である。
【0026】
この点において、バクテリオファージ(ファージ)ディスプレイは、大きなオリゴペプチドライブラリーを検索して、ポリペプチド標的に特異的に結合する能力のあるこれらライブラリーのメンバーを同定することを可能にするよく知られた技術の一つである。ファージディスプレイは、様々なポリペプチドがバクテリオファージ粒子の表面上のコートタンパク質に融合タンパク質として提示されることによる技術である(Scott,J.K.及びSmith G. P. (1990) Science 249:386)。ファージディスプレイの有用性は、選択的にランダム化されたタンパク質変異体(又はランダムクローンcDNA)の大きなライブラリーを標的分子に高い親和性で結合するこれらの配列について素早く効果的に分類することができる点にある。ファージでのペプチド(Cwirla,S.E.等 (1990) Proc. Natl. Acad. Sci. USA, 87:6378)又はタンパク質(Lowman,H.B.ら (1991) Biochemistry, 30:10832; Clackson,T.ら (1991) Nature, 352: 624; Marks,J.D.等 (1991), J. Mol. Biol., 222:581; Kang,A.S.等 (1991) Proc. Natl. Acad. Sci. USA, 88:8363)ライブラリーのディスプレイは、特異的に結合する特性を有するものについて無数のポリペプチド又はオリゴペプチドをスクリーニングするために使用されている(Smith, G.P. (1991) Current Opin. Biotechnol., 2:668)。ランダム突然変異体のファージライブラリーの分類は、多数の変異体を構築して増殖させる方法、標的レセプターを用いた親和性精製の方法、及び結合増強の結果を評価する手段を必要とする(米国特許第5223409号、同第5403484号、同第5571689号、及び同第5663143号を参照)。
【0027】
ほとんどのファージディスプレイ法は繊維状ファージを使用していたが、λファージディスプレイシステム(WO95/34683;米国特許第5627024号)、T4ファージディスプレイシステム(RenJ.らGene 215:439 (1998); Zhuら Cancer Researc, 58(15):3209-3214 (1998); Jiang等 Infection & Immunity, 65(11): 4770-4777 (1997); Ren等, Gene, 195(2):303-311 (1997); Ren, Protein Sci. 5:1833 (1996); Efimov等 Virus Genes 10:173(1995)及びT7ファージディスプレイシステム(Smith及びScott Methods in Enzymology,217, 228-257(1993); 米国特許第5766905号)も知られている。
【0028】
現在、基礎的なファージディスプレイ法は多くの改良及び変形が開発されている。これらの改良により、選択された標的分子との結合性など、ペプチドライブラリーやタンパク質ライブラリーから特性、能力などに基づいてスクリーニングする方法が改善された。ファージディスプレイ法のための組み換え反応手段については、WO98/14277に記載がある。ファージディスプレイライブラリーは、二分子相互作用(WO98/20169;WO98/20159)及び拘束性へリックスペプチドの特性(WO98/20036)を分析及び制御するために使用されている。WO97/35196には、リガンドが標的分子に結合しうる第一の溶液、及び親和性リガンドが標的分子に結合しない第二の溶液とファージディスプレイライブラリーを接触させて結合リガンドを選択的に単離する、親和性リガンドの単離方法が記載されている。WO97/46251は、親和性精製抗体でランダムファージディスプレイライブラリーをバイオパニングし、次いで結合ファージを単離し、続いてマイクロプレートのウェルでマイクロパニングして高親和性結合ファージを単離する方法を記載する。黄色ブドウ球菌(Staphlylococcus aureus)タンパク質Aの親和性タグとしての使用も報告されている(Li等, (1998) Mol Biotech., 9:187)。WO97/47314は、ファージディスプレイライブラリーでもよいコンビナトリアルライブラリーを用いて酵素特異性を識別するための基質サブトラクションライブラリーの使用を記載している。ファージディスプレイに用いる洗浄剤における使用に適した酵素を選択する方法はWO97/09446に記載される。特異的に結合するタンパク質を選択する更なる方法は、米国特許第5498538号、同第5432018号、及びWO98/15833に記載されている。ペプチドライブラリーの作製及びこれらのライブラリーのスクリーニングの方法は、米国特許第5723286号、同第5432018号、同第5580717号、同第5427908号、同第5498530号、同第5770434号、同第5734018号、同第5698426号、同第5763192号、及び同第5723323号に記載される。
【0029】
さらに、ヒト抗体ライブラリーを用いて、パニングによりヒト抗体を取得する技術も知られている。例えば、ヒト抗体の可変領域を一本鎖抗体(scFv)としてファージディスプレイ法によりファージの表面に発現させ、抗原に結合するファージを選択することができる。選択されたファージの遺伝子を解析すれば、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。抗原に結合するscFvのDNA配列が明らかになれば、当該配列を有する適当な発現ベクターを作製し、適当な宿主に導入して発現させることによりヒト抗体を取得することができる。これらの方法は既に周知であり、WO 92/01047、WO 92/20791、WO 93/06213、WO 93/11236、WO 93/19172、WO 95/01438、WO 95/15388を参考に実施することができる。
【0030】
別法として、ファージディスプレイ技術(McCafferty等, Nature 348:552-553[1990])を使用して、非免疫化ドナーの免疫グロブリン可変(V)ドメイン遺伝子レパートリーから、インビトロでヒト抗体及び抗体断片を産出させることができる。この技術によれば、抗体Vドメイン遺伝子を、フレーム単位で、繊維状バクテリオファージ、例えばM13又はfdのコートタンパク質遺伝子のどちらかでクローニングし、ファージ粒子の表面で機能的抗体断片として提示させる。繊維状粒子がファージゲノムの一本鎖DNAコピーを含むので、抗体の機能特性に基づいた選択に基づいても、結果としてこれらの特性を示す抗体をコードする遺伝子の選択が成される。よって、このファージはB細胞のいくつかの特性を模倣している。ファージディスプレイは多様な形式で行うことができる;例えばJohnson, Kevin S. 及びChiswell, David J., Current Opinion in Structural Biology 3:564-571(1993)を参照。V-遺伝子セグメントのいくつかの供給源を、ファージディスプレイのために使用できる。Clackson等, Nature, 352:624-628(1991)は、免疫化したマウス脾臓由来のV遺伝子の小さいランダムなコンビナトリアルライブラリーから、多様で多くの抗-オキサゾロン抗体を単離した。非免疫化ヒトドナーのV遺伝子のレパートリーが構成可能であり、多様で多くの抗原(自己抗原を含む)に対する抗体は、Marks等, J. Mol. Biol. 222:581-597(1991)、又はGriffith等, EMBO J. 12:725-734(1993)に記載の技術にそのまま従うことで単離することができる。また、米国特許第5565332号及び同5573905号を参照のこと。
【0031】
本発明の抗体には、Fab、Fab'、F(ab')2、Fv、scFv、dsFv、Diabody及びsc(Fv)2等の、抗体の機能的断片も含まれる。また、これら機能的断片の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本発明の抗体に含まれる。
Fabは、IgGをシステイン存在下パパイン消化することにより得られる、L鎖とH鎖可変領域、並びにCH1ドメイン及びヒンジ部の一部からなるH鎖フラグメントとから構成される分子量約5万の断片である。本発明では、上記抗体をパパイン消化することにより得ることができる。また、上記抗体のH鎖の一部及びL鎖をコードするDNAを適当なベクターに組み込み、当該ベクターを用いて形質転換した形質転換体よりFabを調製することもできる。
Fab'は、後述のF(ab')2のH鎖間のジスルフィド結合を切断することにより得られる分子量が約5万の断片である。本発明では、上記抗体をペプシン消化し、還元剤を用いてジスルフィド結合を切断することにより得られる。また、Fab同様に、Fab'をコードするDNAを用いて遺伝子工学的に調製することもできる。
F(ab')2は、IgGをペプシン消化することにより得られる、L鎖とH鎖可変領域、並びにCH1ドメイン及びヒンジ部の一部からなるH鎖フラグメントとから構成される断片(Fab')がジスルフィド結合で結合した分子量約10万の断片である。本発明では、上記抗体をペプシン消化することにより得られる。また、Fab同様に、F(ab')2をコードするDNAを用いて遺伝子工学的に調製することもできる。
Fvは、抗体を酵素、例えばパパイン、ペプシンで処理し抗体断片を生成させるか、または、これら抗体断片をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現できる(例えば、Co, M.S. et al., J. Immunol.(1994)152, 2968-2976、Better, M. & Horwitz, A. H. Methods in Enzymology(1989)178, 476-496、Plueckthun, A. ; Skerra, A. Methods in Enzymology(1989)178, 476-496、Lamoyi, E., Methods in Enzymology(1989)121, 652-663、Rousseaux, J. et al., Methods in Enzymology(1989)121, 663-669、Bird, R. E. et al., TIBTECH(1991)9, 132-137参照)。
scFvは、H鎖可変領域とL鎖可変領域とからなるFvを、片方の鎖のC末端と他方のN末端とを適当なペプチドリンカーで連結し一本鎖化した抗体断片である。ペプチドリンカーとしては例えば柔軟性の高い(GGGGS)3などを用いることができる。例えば、上記抗体のH鎖可変領域及びL鎖可変領域をコードするDNAとペプチドリンカーをコードするDNAを用いてscFv抗体をコードするDNAを構築し、これを適当なベクターに組み込み、当該ベクターを用いて形質転換した形質転換体よりscFvを調製することができる。
dsFvは、H鎖可変領域及びL鎖可変領域の適切な位置にCys残基を導入し、H鎖可変領域とL鎖可変領域とをジスルフィド結合により安定化させたFv断片である。各鎖におけるCys残基の導入位置は分子モデリングにより予測される立体構造に基づき決定することができる。本発明では例えば上記抗体のH鎖可変領域及びL鎖可変領域のアミノ酸配列から立体構造を予測し、かかる予測に基づき変異を導入したH鎖可変領域及びL鎖可変領域をそれぞれコードするDNAを構築し、これを適当なベクターに組み込み、そして当該ベクターを用いて形質転換した形質転換体よりdsFvを調製することができる。
尚、適当なリンカーを用いてscFv抗体、dsFv抗体などを連結させたり、ストレプトアビジンを融合させたりして抗体断片を多量体化することもできる。本発明の抗体(抗体断片を含む)に低分子化合物、タンパク質、標識物質などを融合又は結合させることにより、融合抗体又は標識化抗体を構成することができる。標識物質としては125I等の放射性物質、などを用いることができる。
【0032】
Diabodyは、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(Holliger P et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。Diabodyは、2本のポリペプチド鎖から構成されるダイマーであり、通常、ポリペプチド鎖は各々、同じ鎖中でVL及びVHが、互いに結合できない位に短い、例えば、5残基程度のリンカーにより結合されている。同一ポリペプチド鎖上にコードされるVLとVHとは、その間のリンカーが短いため単鎖可変領域フラグメントを形成することが出来ず二量体を形成するため、Diabodyは2つの抗原結合部位を有することとなる。Diabodyは、抗体を酵素、例えば、パパイン、ペプシンなどで処理し、抗体断片を生成させるか、又はこれら抗体断片をコードするDNAを構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させればよい(例えば、Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976; Better, M. and Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515; Lamoyi, E., Methods Enzymol. (1986) 121, 652-663 ; Rousseaux, J. et al., Methods Enzymol. (1986) 121, 663-669; Bird, R. E. and Walker, B. W., Trends Biotechnol. (1991) 9, 132-137参照)。
sc(Fv)2は、2つのVH及び2つのVLをリンカー等で結合して一本鎖にした低分子化抗体である(Hudson et al、J Immunol. Methods 1999;231:177-189)。sc(Fv)2は、例えば、scFvをリンカーで結ぶことによって作製できる。
【0033】
本発明の抗体には、本発明の抗体と他のペプチド又はタンパク質とが融合した融合タンパク質も含まれる。融合タンパク質を作製する方法は、本発明の抗体をコードするポリヌクレオチドと他のペプチド又はポリペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明の抗体との融合に付される他のペプチド又はポリペプチドとしては、例えば、FLAG(Hopp, T. P. et al., BioTechnology (1988) 6, 1204-1210 )、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein C の断片等の公知のペプチドを使用することができる。また、本発明の抗体との融合に付される他のポリペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。
【0034】
本発明の抗体には、標識物質が結合した抗体も含まれる。
標識物質としては、酵素発光(ルシフェラーゼ)による発光、発光低分子を利用するものと、蛍光タンパクや蛍光低分子)を用いるもの、放射性核種等が挙げられるがこれらには限定されない。放射性核種としては51Cr、59Fe、57Co、67Ga、75Se、81mKr、99mTc、111In、125I、131I、133Xe、201Tlなどのγ線放出核種や、11C、13N、15O、18F、35mCl、76Br、 45Ti、48V、60Cu、61Cu、62Cu、66Ga、89Zr、94mTc、124I、123Iなどの陽電子放出核種などが挙げられるがこれらには限定されない。なお当業者には自明であるが"m"とは核異性体を示す。
蛍光標識や発光標識としては酵素発光(ルシフェラーゼ)による発光を利用するものと、蛍光(GFP、DsRed、クサビラオレンジ等の蛍光タンパク質やFITCやCy5.5、Alexa Fluor 750などの蛍光性低分子)を用いるものとのが使用できる。
酵素発光(ルシフェラーゼ)による発光の場合は基質投与が別途必要である。
特に、動物本来の自家蛍光による影響を軽減したものが好ましく、さらには皮膚透過性の高いシグナルを発する標識が好ましい。
【0035】
また本発明は、本発明の抗体をコードするDNA、該DNAが挿入されたベクター、及び該ベクターが導入された形質転換細胞を提供する。ベクターの例としては、M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Scriptなどが挙げられる。また、cDNAのサブクローニング、切り出しを目的とした場合、上記ベクターの他に、例えば、pGEM-T、pDIRECT、pT7などが挙げられる。本発明の抗体をコードするDNA、該DNAが挿入されたベクター、及び該ベクターが導入された形質転換細胞は公知の技術を用いて作成される。
【0036】
本発明の酸化LDL/β2GPI複合体に結合する抗体をコードするDNAとしては、以下のDNAが挙げられる。
(a)配列番号:5に記載の塩基配列を有する、重鎖をコードするDNA
(b)配列番号:10に記載の塩基配列を有する、軽鎖をコードするDNA
(c)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖をコードするDNA
(d)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖をコードするDNA
(e)配列番号:45に記載の塩基配列を有する、重鎖をコードするDNA
(f)配列番号:53に記載の塩基配列を有する、軽鎖をコードするDNA
【0037】
発現ベクターとしては、例えば、大腸菌での発現を目的とした場合は、ベクターが大腸菌で増幅されるような上記特徴を持つほかに、宿主をJM109、DH5α、HB101、XL1-Blueなどの大腸菌とした場合においては、大腸菌で効率よく発現できるようなプロモーター、例えば、lacZプロモーター(Wardら, Nature (1989) 341, 544-546;FASEB J. (1992) 6, 2422-2427)、araBプロモーター(Betterら, Science (1988) 240, 1041-1043 )、またはT7プロモーターなどを持っていることが不可欠である。このようなベクターとしては、上記ベクターの他にpGEX-5X-1(ファルマシア社製)、「QIAexpress system」(キアゲン社製)、pEGFP、またはpET(この場合、宿主はT7 RNAポリメラーゼを発現しているBL21が好ましい)などが挙げられる。
【0038】
また、ベクターには、ポリペプチド分泌のためのシグナル配列が含まれていてもよい。蛋白質分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(Lei, S. P. et al J. Bacteriol. (1987) 169, 4379)を使用すればよい。宿主細胞へのベクターの導入は、例えば塩化カルシウム法、エレクトロポレーション法を用いて行うことができる。
【0039】
大腸菌以外にも、例えば、哺乳動物由来の発現ベクター(例えば、pcDNA3 (インビトロゲン社製)や、pEGF-BOS (Nucleic Acids. Res.1990, 18(17),p5322)、pEF 、pCDM8)、昆虫細胞由来の発現ベクター(例えば「Bac-to-BAC baculovairus expression system」(ギブコBRL社製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来の発現ベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来の発現ベクター(例えば、pZIPneo)、酵母由来の発現ベクター(例えば、「Pichia Expression Kit」(インビトロゲン社製)、pNV11、SP-Q01)、枯草菌由来の発現ベクター(例えば、pPL608、pKTH50)が挙げられる。
【0040】
CHO細胞、COS細胞、NIH3T3細胞等の動物細胞での発現を目的とした場合には、細胞内で発現させるために必要なプロモーター、例えばSV40プロモーター(Mulliganら, Nature (1979) 277, 108)、MMTV-LTRプロモーター、EF1αプロモーター(Mizushimaら, Nucleic Acids Res. (1990) 18, 5322)、CMVプロモーターなどを持っていることが不可欠であり、細胞への形質転換を選抜するための遺伝子(例えば、薬剤(ネオマイシン、G418など)により判別できるような薬剤耐性遺伝子)を有すればさらに好ましい。このような特性を有するベクターとしては、例えば、pMAM、pDR2、pBK-RSV、pBK-CMV、pOPRSV、pOP13などが挙げられる。
【0041】
さらに、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする場合には、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pCHOIなど)を導入し、メトトレキセート(MTX)により増幅させる方法が挙げられ、また、遺伝子の一過性の発現を目的とする場合には、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。
【0042】
ベクターが導入される宿主細胞としては特に制限はなく、例えば、大腸菌や種々の動物細胞などを用いることが可能である。宿主細胞は、例えば、本発明の抗体の製造や発現のための産生系として使用することができる。ポリペプチド製造のための産生系は、in vitroおよびin vivoの産生系がある。in vitroの産生系としては、真核細胞を使用する産生系や原核細胞を使用する産生系が挙げられる。
【0043】
真核細胞を使用する場合、例えば、動物細胞、植物細胞、真菌細胞を宿主に用いることができる。動物細胞としては、哺乳類細胞、例えば、CHO(J. Exp. Med. (1995) 108, 945)、COS、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero、両生類細胞、例えばアフリカツメガエル卵母細胞(Valle, et al., Nature (1981) 291, 358-340)、あるいは昆虫細胞、例えば、 Sf9、 Sf21、 Tn5が知られている。本発明においては、CHO-DG44、CHO-DXB11、COS7細胞、BHK細胞が好適に用いられる。動物細胞において、大量発現を目的とする場合には特にCHO細胞が好ましい。宿主細胞へのベクターの導入は、例えば、リン酸カルシウム法、DEAEデキストラン法、カチオニックリボソームDOTAP(ベーリンガーマンハイム社製)を用いた方法、エレクトロポーレーション法、リポフェクションなどの方法で行うことが可能である。
【0044】
植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞が蛋白質生産系として知られており、これをカルス培養すればよい。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属、例えば、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、サッカロミセス・ポンベ(Saccharomyces pombe)、糸状菌、例えば、アスペルギルス(Aspergillus)属、例えば、アスペルギルス・ニガー(Aspergillus niger)が知られている。
【0045】
原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、大腸菌(E. coli)、例えば、JM109、DH5α、HB101等が挙げられ、その他、枯草菌が知られている。本発明のDNAにより形質転換された細胞をin vitroで培養し、当業者が通常行う方法によって精製することによって、本発明の抗体を得ることが可能である。
【0046】
また本発明は、本発明の抗体をコードする核酸を含むベクターを有する宿主生物を提供する。本発明の宿主生物は、組換え型抗体の産生に有用である。本発明における宿主生物としては、ヤギなどが挙げられる。例えば、本発明のトランスジェニックヤギの作成は次のようにして行うことが可能である。即ち、抗体遺伝子が乳汁中に固有に産生されるタンパク質(ヤギβカゼインなど)をコードする遺伝子の内部にインフレームで挿入された融合遺伝子を構築する。抗体遺伝子が挿入された融合遺伝子を含むDNA断片をヤギの胚へ注入すれば、該注入胚が雌のヤギへ導入される。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、本発明の抗体を取得することができる。トランスジェニックヤギから産生される本発明の抗体を含む乳汁量を増加させるために、ホルモンをトランスジェニックヤギに適宜使用することも可能である(Ebert, K.M. et al., Bio/Technology(1994)12, 699-702)。
【0047】
本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を含む、動脈硬化の存在部位のイメージング剤を提供する。また、本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を哺乳動物に投与する工程を含む、動脈硬化の存在部位のイメージング方法を提供する。本発明のイメージング剤は、動脈硬化の存在部位をイメージングするために哺乳動物に投与される。哺乳動物としては、ヒト、非ヒト哺乳動物(例えばマウス、ラット、ハムスター、ウサギ、ブタ、サルなど)が挙げられる。本発明のイメージング剤は、動脈硬化の診断において有用である。本発明のイメージング剤はin vivo用、in vitro用のどちらとしても利用できる。
より好ましい態様としては、本発明のイメージング剤はin vivo用イメージング剤である。in vivo用イメージング剤とは、酸化LDL/β2GPI複合体に結合する抗体を含むイメージング剤を注射などの方法により動物個体の血管内に投与し、動脈硬化の存在部位を個体生存化で観察するものである。そのため、in vivo用イメージング剤には生体の健康上に悪影響を及ぼす組成物は極力除かれる。また、in vivo用イメージング剤は皮下染色部位の観察(体内の酸化LDL/β2GPI複合体に結合する抗体存在部位の検出)は体外から検出が行われるため、酸化LDL/β2GPI複合体に結合する抗体に対し、生体に吸収されにくい波長である近赤外の蛍光標識か、放射線核種による標識が用いられる。蛍光標識として、Alexa Fluor 750標識は、779 nmの最大放出蛍光を持ち、実施例に示すとおり高感度、好適に用いられる。また、ヒトに対してin vivo用イメージング剤を適用する場合は、放射線核種が好ましい。さらに、ex vivoイメージングに用いる場合もある。ex vivoイメージングとは、マウスモデル動物などの非ヒト哺乳動物へ生前にイメージング剤を投与しておき、堵殺後、酸化LDL/β2GPI複合体に結合する抗体存在部位を観察する方法である。
動脈硬化には大きく分け、粥腫と石灰化病変の症状が存在するが、本発明のイメージング剤によって、特に、動脈硬化粥腫部位が染色される。
粥腫は、マクロファージが、コレステロールを多量に含有する酸化LDLをレセプターを介して特異的に取り込み、泡沫化することが知られており、それが血管内膜に蓄積、プラーク(粥腫)を形成する、動脈硬化の一病態である。血管内膜は、血管内皮細胞層によって覆われているが、in vivo用イメージング剤は血管内投与により、血管内膜下(血管内皮細胞下)のプラーク部位をイメージングできる。
【0048】
本発明のイメージング剤は、酸化LDL/β2GPI複合体に結合する抗体、特に好ましくは3H3抗体に、直接、あるいは間接的に追尾できる、イメージング用標識、プローブを結合させたものである。
前記プローブを生体に生体内投与(例えば静脈内投与)した後、PETやSPECT、CCDカメラなどの画像計測装置を用いて蓄積量や分布を測定することができる。
【0049】
また、近年、透過性線源により、物体を走査し、そのデータをコンピュータを用いて処理することで、物体の内部画像を構成する技術コンピュータ断層撮影(Computed Tomography:以下、「CT」との記載もComputed Tomographyを意味する)の病状診断、臨床への応用がされている。
そのようなCT技術は、ポジトロン断層法 (PET)や単一光子放射断層撮影 (SPECT)、や核磁気共鳴画像法 (MRI) などの、断面像を得て、物体の(輪切りなどの)2次元断面画像を得る技術であるが、これらの検査技術は単に断面画像として用いられるだけでなく、コンピュータ画像処理技術向上によってその2次元画像を統合し3次元グラフィックスとして表示されることも多く、3次元的な病変の位置の特定、診断、手術方針決定等に有力な技術となっている。
例えば、単純CTは、造影剤を使用せずにX線照射等の撮影を行うものであり、組織の浮腫、骨の形態異常、形態など造影剤を用いなくても充分に観察でき、一方、造影CTとは、X線吸収率の高い造影剤等を血管内に注射してから撮影を行うものをいい、血管や血流が豊富な組織の形状を観察することができる。さらに、いわゆる次世代CT、例えば、線源がらせん状に動くヘリカルCT、検出器自体を対軸方向に、分割した、多列検出器CT (MDCT)(マルチスライスCT(MSCT)とも呼ぶ)なども、開発され、それらはいずれも特に制限なく、単独又は併用で発明のイメージング剤の検出に適用可能である。
イメージング用標識プローブ(本発明のイメージング剤)がX線吸収率の高い放射線核種である場合は、検出器としてのCTの単独使用も可能である。
【0050】
標識物質としては酵素発光(ルシフェラーゼ)による発光、発光低分子を利用するものと、蛍光タンパクや蛍光低分子)を用いるもの、放射性核種等が挙げられるがこれらには限定されない。放射性核種としては51Cr、59Fe、57Co、67Ga、75Se、81mKr、 99mTc、111In、125I、131I、133Xe、201Tlなどのγ線放出核種や、11C、13N、15O、18F、35mCl、76Br、 45Ti、48V、60Cu、61Cu、62Cu、66Ga、89Zr、94mTc、124I、123Iなどの陽電子放出核種などが挙げられるがこれらには限定されない。なお当業者には自明であるが"m"とは核異性体を示す。特に、インジウム−111、テクネチウム−99mまたはヨウ素−131は、平面走査またはシングルフォトン断層撮影(SPECT)のために特に好ましく使用できる。陽電子放射標識の例えばフッ素−19は、陽電子放射断層法において特に好ましく使用できる。常磁性イオンの例えばガドリニウム(III)またはマンガン(II)は特に好ましく磁気共鳴映像法(MRI)において使用できる。
【0051】
蛍光標識や酵素(ルシフェラーゼ)による発光系を利用するものと、蛍光(GFP、DsRed、クサビラオレンジ等の蛍光タンパク質やFITCやCy5.5、Alexa Fluor 750などの蛍光性低分子)を用いるものとのが使用できる。
酵素発光(ルシフェラーゼ)による発光の場合は基質投与が別途必要である。
特に、動物本来の自家蛍光による影響を軽減したものが好ましく、さらには皮膚透過性の高いシグナルを発する標識が好ましい。
【0052】
イメージング検出器としては磁気共鳴映像法(MRI)PETやSPECTが用いられるが、特に、蛍光標識プローブの観察において、低侵しゅう性の観点で好ましく測定機器は、CCDカメラである。
それゆえ、CCDカメラで補足可能な波長、例えばおよそ350〜900 nmの光を、発する標識が好ましい。さらにCCDカメラによる測定動物の体表面における測定値から生体内の光源の強さを測定できる機械が好ましい。蛍光標識の場合、反射蛍光画像、透過蛍光画像のいずれでも良いが、両方の画像を捉えることが好ましい。また、蛍光画像の多方面(反射、透過を問わず)重ね合わせ、それに線源情報を統合して処理し、立体画像(3次元)として捉えることが、3次元的な位置、分布を正確に構成でき、好ましい。こうして得た3次元画像は、さらにCT画像とさらに統合することもできる。
【0053】
イメージング用標識プローブがX線吸収率の高い放射線核種を結合したものである場合、前述したように イメージング検出器としての、CTの単独でも十分使用可能(例えばPETやSPECT)で動脈硬化病巣の位置、蓄積量や分布を測定することもできる。
【0054】
一方、前記イメージング用標識プローブを生体に生体内投与(例えば静脈内投与)した後、標識された前記プローブのCT観察、又はCCDとの併用観察も可能である。併用観察の例として、蛍光標識したプローブのCCD像を、単純CT(及び/又は造影CTの像と重ね合わせる)を用いることができる。即ち 単純CTにより骨、肺などの臓器像の抽出(及び/又は造影CTの血管、組織像抽出)を行って得られたCT画像と、心臓など主要な動脈疾患部分のCCDカメラによる蛍光プローブ画像を統合し、その動脈硬化病巣の位置、蓄積量や分布を3次元的な組織、血管との相対的な位置関係、動脈硬化病巣の3次元(局在)イメージをより正確に把握することができる。
【0055】
本発明のイメージング剤は、抗体に加えて医薬的に許容し得る担体を導入し、公知の方法で製剤化することが可能である。例えば、水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、又は懸濁液剤の注射剤の形で非経口的に使用できる。例えば、薬理学上許容される担体もしくは媒体、具体的には、滅菌水や生理食塩水、植物油、乳化剤、懸濁剤、界面活性剤、安定剤、香味剤、賦形剤、ベヒクル、防腐剤、結合剤などと適宜組み合わせて、一般に認められた製薬実施に要求される単位用量形態で混和することによって製剤化することが考えられる。これら製剤における有効成分量は指示された範囲の適当な容量が得られるようにするものである。
【0056】
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施に従って処方することができる。
注射用の水溶液としては、例えば生理食塩水、ブドウ糖やその他の補助薬を含む等張液、例えばD-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウムが挙げられ、適当な溶解補助剤、例えばアルコール、具体的にはエタノール、ポリアルコール、例えばプロピレングリコール、ポリエチレングリコール、非イオン性界面活性剤、例えばポリソルベート80(TM)、HCO-50と併用してもよい。
【0057】
油性液としてはゴマ油、大豆油があげられ、溶解補助剤として安息香酸ベンジル、ベンジルアルコールと併用してもよい。また、緩衝剤、例えばリン酸塩緩衝液、酢酸ナトリウム緩衝液、無痛化剤、例えば、塩酸プロカイン、安定剤、例えばベンジルアルコール、フェノール、酸化防止剤と配合してもよい。調製された注射液は通常、適当なアンプルに充填させる。
【0058】
投与は好ましくは非経口投与であり、具体的には、注射剤型、経鼻投与剤型、経肺投与剤型、経皮投与型などが挙げられる。注射剤型の例としては、例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などにより全身または局部的に投与することができる。
【0059】
また、患者の年齢、症状により適宜投与方法を選択することができる。本発明のイメージング剤の投与量としては、例えば、一回につき体重1kgあたり0.0001mgから1000mgの範囲で選ぶことが可能である。あるいは、例えば、対象あたり0.001から100000mg/bodyの範囲で投与量を選ぶことができるが、これらの数値に必ずしも制限されるものではない。
投与量、投与方法は、対象の体重や年齢、症状、mg抗体あたりの蛍光標識の強度・検出機械の検出感度などにより変動するが、当業者であれば適宜選択することが可能である。
【0060】
また本発明は、本発明の酸化LDL/β2GPI複合体に結合する抗体を含む、動脈硬化の存在部位のイメージング用キットを提供する。本発明のキットは、対象に投与されることにより、動脈硬化の存在部位をイメージングすることを特徴とする。上記キットには、本発明の抗体に加えて、例えば、イメージング剤投与用の注射(点滴)器具、非吸着吸着を抑える補助剤(例えばアルブミン等)等が含まれるが、これらに制限されるものではない。
また、上記キットには、イメージングに用いるための指示書、適当な容器、コントロール試薬等、通常のキットに含まれるものを含んでいてもよい。
【0061】
本発明は、以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法を提供する。
(a)本発明の酸化LDL/β2GPI複合体に結合する抗体、及び、候補化合物を動脈硬化疾患モデル非ヒト動物に投与する工程、例えば、本発明の酸化LDL/β2GPI複合体に結合する抗体が投与された動脈硬化疾患モデル非ヒト動物に候補化合物を投与する工程、
(b)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣(例えば、動脈硬化巣の大きさまたは存在部位)を比較する工程、及び
(d)該抗体、及び、該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該抗体を投与されているが該候補化合物は投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
各工程は、上述した技術または公知の技術を用いて実施される。
【0062】
本発明のスクリーニング方法において利用することができる候補化合物には、精製蛋白質(抗体を含む)、遺伝子ライブラリーの発現産物、合成ペプチドのライブラリー、DNAまたはRNAライブラリー(アプタマー、siRNAなど機能性核酸を含む)、細胞抽出液、細胞培養上清、及び合成低分子化合物のライブラリー等が挙げられるが、これらに制限されない。
【0063】
本発明のスクリーニング方法において利用することができる疾患モデル非ヒト動物としては、マウス、ハムスター、ラット、ウサギ、ブタ、サル等が挙げられるが、これらに制限されない。
例えば、動脈硬化疾患モデルマウスとしては遺伝子を過剰発現させるトランスジェニックマウスと、ある遺伝子を欠損させるジーンターゲッティングによるノックアウトマウス、例えば、動脈硬化モデルとしては悪玉コレステロールとして知られているLDLを形作っているタンパク質(アポリポタンパク質E)のアポE欠失(apoE-/-)、LDL受容体欠失(LDLR-/-)、ヒト型アポB導入、ドミナント変異アポE導入モデルがある。さらには、2型糖尿病モデルマウス(KKAy)、あるいは、マウスのなかでもっとも動脈硬化になりやすいC57BL6系統で、この系統では高コレステロール食のみで動脈硬化巣がみられる場合もあり、高コレステロール食等、その餌により作製された、動脈硬化モデルマウスも用いることができる。
ウサギは高コレステロール食により約2ヶ月半で動脈硬化巣がみられる場合もあり、さらにLDL受容体欠損の動脈硬化疾患モデルウサギとしては、WHHLウサギなどが知られている。
ブタでもアポBのLDL受容体結合部分のアミノ酸配列異常により、動脈硬化をおこしやすいモデルがしられており、当業者であれば、血栓症・動脈硬化モデル動物作製法 編著 鈴木 宏治(株式会社 金芳堂)など参照して、動脈硬化モデル動物を作製、本発明に利用できる。
【0064】
本発明のスクリーニング方法によって選択される動脈硬化巣を減少または消失させる化合物は、動脈硬化治療剤の候補化合物となる。即ち、本発明は、本発明のスクリーニングによって選択された物質を有効成分として含有する動脈硬化治療剤を提供する。また本発明は、本発明のスクリーニング方法によって選択された化合物の動脈硬化治療剤製造における使用に関する。本発明のスクリーニング法により単離される物質を、治療剤として用いる場合には、公知の製剤学的製造法により製剤化して用いることができる。例えば、薬理学上許容される担体または媒体(生理食塩水、植物油、懸濁剤、界面活性剤、安定剤など)とともに患者に投与する。投与は、物質の性質に応じて、経皮的、鼻腔内的、経気管支的、筋内的、静脈内、または経口的に行われる。投与量は、患者の年齢、体重、症状、投与方法等により変動するが、当業者であれば適宜適当な投与量を選択することができる。
【0065】
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<3H3抗体>
配列番号1:重鎖可変領域のアミノ酸配列
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号5:重鎖可変領域の塩基配列
配列番号6:軽鎖可変領域のアミノ酸配列
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号10:軽鎖可変領域の塩基配列
配列番号55:重鎖FR1のアミノ酸配列
配列番号56:重鎖FR2のアミノ酸配列
配列番号57:重鎖FR3のアミノ酸配列
配列番号58:重鎖FR4のアミノ酸配列
配列番号59:軽鎖FR1のアミノ酸配列
配列番号60:軽鎖FR2のアミノ酸配列
配列番号61:軽鎖FR3のアミノ酸配列
配列番号62:軽鎖FR4のアミノ酸配列
<ヒト化3H3抗体>
配列番号44:重鎖可変領域のアミノ酸配列
配列番号46:重鎖のアミノ酸配列(シグナル配列を含む重鎖)
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号39:重鎖FR1のアミノ酸配列
配列番号40:重鎖FR2のアミノ酸配列
配列番号41:重鎖FR3のアミノ酸配列
配列番号42:重鎖FR4のアミノ酸配列
配列番号45:重鎖の塩基配列(シグナル配列を含む重鎖)
配列番号52:軽鎖可変領域のアミノ酸配列
配列番号54:軽鎖のアミノ酸配列(シグナル配列を含む軽鎖)
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号47:軽鎖FR1のアミノ酸配列
配列番号48:軽鎖FR2のアミノ酸配列
配列番号49:軽鎖FR3のアミノ酸配列
配列番号50:軽鎖FR4のアミノ酸配列
配列番号53:軽鎖の塩基配列(シグナル配列を含む軽鎖)
なお、本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0066】
以下、本発明を実施例によって詳細に説明するが、本発明の範囲はこれらの実施例に記載された態様に限定されるものではない。
[実施例1]
酸化LDL/β2GPI複合体の調製
600μgのヒトLDL(Organon Teknika Corp,Durham,NC)を、5μM CuSO4 を含むPBS(2mL)中で37℃、12時間酸化処理した。1 mMのEDTAを添加することによって酸化を停止させた。
上記、酸化LDL 0.2 mg/mLをヒトβ2GPI(Affinity Biologicalsより購入)とともに終濃度 0.2mg/mL、37℃条件下で、16時間インキュベートし、酸化LDL/β2GPI複合体を得た。
【0067】
[実施例2]
抗原免疫
ヒト酸化LDL/β2GPI複合体精製タンパク質を同量のコンプリートアジュバント(SIGMA;F5881)と混合してエマルジョンにし、BALB/cマウス(メス)のfoot-padに5〜50μg/headで3から7日おきに数回免疫した。最終免疫の3〜5日後にマウスから鼠径リンパ節を摘出し、マウス骨髄腫細胞P3U1(P3-X63Ag8U1)との細胞融合を行った。
【0068】
[実施例3]
細胞融合、及びモノクローナル抗体産生細胞の選択と取得
細胞融合は次に示す一般的な方法を基本として行った。全ての培地中のFetal Bovine Serum(FBS)は、56℃で30min保温する処理により、非働化したものを使用した。P3U1は、RPMI1640-10%FBS(Penicillin, Streptomycin 含)で培養して準備した。
摘出したマウスの鼠径リンパ節細胞とP3U1を10:1〜2:1の割合で混合し、遠心した。沈殿した細胞に50%ポリエチレングリコール4000(Merck社、ガスクロマトグラフィー用 PEG4000、カタログNo. 9727)を融合促進剤として徐々に加えながら穏やかに混合し、細胞融合を行った。さらにRPMI1640を徐々に加えながら穏やかに混合後、遠心した。沈殿した融合細胞を、15% FCSを含むHAT培地(RPMI1640, HAT-supplement(Invitrogen;11067-030), Penicillin, Streptomycin)で適宜希釈し、200 μL/wellで96穴のマイクロプレートに播種した。
融合細胞をCO2インキュベータ(5% CO2、37 ℃)中で培養し、コロニーが十分に形成されたところで、培養上清をサンプリングしてスクリーニングを行った。
スクリーニングは、免疫に使用したヒト酸化LDL/β2GPI複合体抗原を感作した96穴プレートに対するELISA(実施例4に記載)で陽性となったものを選別した。15% FCSを含むHT培地(RPMI1640, HT-supplement(Invitrogen;21060-017), Penicillin, Streptomycin)でこれらを拡大培養後、限界希釈(limiting dilution)法により単クローン化した。このようにして、抗ヒト酸化LDL/β2GPI複合体抗体を免疫原としてクローン3H3を含むハイブリドーマクローン他、7種類を得た。
【0069】
[実施例4]
ヒト酸化LDL/β2GPI複合体及びβ2GPIに対する反応性(ELISA)
抗ヒト酸化LDL/β2GPI複合体抗体を検出するためのELISAは、以下の方法によって行った。すなわち、酸化LDL/β2GPIをマイクロプレート(Nunc社 Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20mM HEPES (pH 7.4))を用いて、各横軸に記載の抗体濃度に希釈したサンプル50μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/ PBS で洗浄した後、基質TMB (MOSS社;TMBZ) を50 μL添加し、室温にて 3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した(図1A)。
β2GPIとの反応性を検出するためのELISAは、以下の方法によって行った。すなわち、β2GPIをマイクロプレート(Nunc社Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4 ℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20 mM HEPES (pH 7.4))を用いて、各横軸に記載の抗体濃度に希釈したサンプル50 μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBS で洗浄した後、基質TMB (MOSS社; TMBZ) を50 μL添加し、室温にて3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した(図1B)。
さらに、β2GPIをマイクロプレート(nunc社 Maxisorp)に〜50 μg/mLまで濃度を変え (50 μL/ウエル)入れて、4℃で一晩インキュベートすることによって吸着させ、同様に抗体反応性を試験した(図示さず)。
その結果、固相化酸化LDL/β2GPI複合体に対しては、2H6 > 3H3, 2A12, 3D4 > 4C12, 1H4の反応性を示した。また、固相化β2GPIに対しては、2H6, 3D4 > 2A12, 4F10を示し、3H3, 4C12については反応性を示さなかった(図1A,B)。
しかし、マイクロタイタープレートへの感作濃度をあげると3H3なども反応性を示した(図示さず)。
抗体の反応性を確認する方法として次に、遊離の抗原で阻害をかける試験を行い、さらに、各抗体の特異性を見た。
【0070】
[実施例5]
溶液中の遊離のβ2GPI、酸化LDL/β2GPI複合体に対する競合反応性(ELISA)
固相化ヒト酸化LDL/β2GPI複合体及びβ2GPIに対する反応性(ELISA)試験において、各抗体と反応させる際に、酸化LDL/β2GPI複合体またはβ2GPIを共存させて反応することにより、固相抗原に対する阻害反応を行った(図2に測定系模式図を示す)。
すなわち、β2GPIをマイクロプレート(Nunc社; Maxisorp)に1 μg/mL (50 μL/ウエル)入れて、4 ℃で一晩インキュベートすることによって吸着させ、このプレートを1% BSAを用いてブロッキングした。Assay buffer(1% BSA, 0.15 M NaCl/20 mM HEPES (pH 7.4))を用いて、適当な濃度に希釈した抗体サンプル25 μL及び、競合させる抗原である酸化LDL/β2GPI複合体またはβ2GPIを各横軸に記載の抗原濃度にAssay bufferを用いて希釈したサンプル25 μLを入れて、ウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBSで洗浄した後、次いでHRP標識した抗マウスIgG (MBL; Code 330) 2000倍希釈 50 μLをプレートに添加しウエル中で30分間インキュベートした。液を捨て、0.1% Tween20/PBS で洗浄した後、基質TMB (MOSS社; TMBZ) を50 μL添加し、室温にて 3分間インキュベートし、0.18 M 硫酸 50 μL を添加して反応停止後、吸光度 450 nmで検出した。
その結果、3H3, 4C12, 2A12ではELISA中に混在させた遊離の抗原が酸化LDL/β2GPI複合体の場合、固相の酸化LDL/β2GPIへの結合阻害が顕著であり、β2GPIでは阻害がかからなかった。2H6では、フリーの抗原が酸化LDL/β2GPI複合体でも阻害が起こる一方でβ2GPIの混在でも若干阻害が見られた。3D4では、遊離の抗原が酸化LDL/β2GPI複合体よりも、β2GPIによる阻害の方が高い阻害が見られた(図3)。
これらの結果より、各抗体の反応性は表1にまとめられる(実施例7に示す)。3H3は4C12と近い反応性を示すが同一の反応性ではなく、異なる特異性を有していた。
【0071】
[実施例6]
抗体の動脈硬化巣免疫組織染色
apoE-/-マウスおよび LDLR-/-マウス(Jackson Labより入手し、岡山大学内動物実験施設にて系統維持)を8週齢までは、普通飼料で飼育し(オリエンタル酵母NMF)、それ以降に高脂肪食(コレステロール1%、コール酸1%、無塩バター15%を普通飼料に配合)を4-6ヶ月負荷すると、胸部あるいは腹部大動脈に動脈硬化巣(プラーク)が現れ、肥厚、粥腫が出現する。そこで、8ヶ月齢のこれらマウスを屠殺し、特に今回は、胸部大動脈、大動脈の起始部、および大動脈弁の凍結切片を作製し、標本とし観察した。
蛍光免疫染色抗体法のためには、凍結切片を調製後、パラホルムアルデヒド固定し実験に供した。
モノクローナル抗体のCy5.5標識
0.1 M炭酸緩衝液(pH9.3)に対し、4 ℃で一晩透析した各種モノクローナル抗体(1mg/ml)をFluorolink Cy5.5 monofunctional dye (1 tube)に入れ、室温で30分間反応させ標識した。反応後、Sephadex G-25カラムにてCy5.5標識抗体を得た。
凍結切片を用いた免疫蛍光染色
1%パラホルムアルデヒドで5分間固定した切片に各種モノクローナル抗体を4℃で一晩反応させた。洗浄後、FITC標識抗マウスIgGあるいはIgM抗体(2次抗体)を室温で1時間反応させた。DAPlおよびRhodamine Phalloidin染色は、この2次抗体の反応時に添加した。その後、蛍光顕微鏡にて観察、撮影した。
免疫組織染色
結果、C57BL6系統のマウスを普通飼料で飼育したものに対し、3H3抗体、Mac3を用いる免疫蛍光染色では、泡沫化マクロファージが集族してできた粥腫が染まった。3H3によっても同様な部位が染まっている(図4)。
動脈硬化好発モデルマウス(高脂肪食を負荷したapoE-/-)の大動脈弁の免疫蛍光染色を酸化LDL/β2GPI複合体を認識する他の抗体で行ったものと比較した。粥腫の染色陽性例は、粥腫の染色陽性例は、3H3および抗体Aのみである(図5)。
Cy5.5、Alexaなどを標識した粥腫に特異的な種々のモノクローナル抗体で動脈硬化巣の特異的な免疫染色が可能となった。
【0072】
[実施例7]
イメージング
in vivoイメージング:
Xenogen社のIVIS(登録商標) Imaging System、IVIS200により、イメージングを行った(Excitation:640 nm, Emission: 720 nm.)。
実験1:実施例6と同様に作製した高脂肪食負荷apoE-/-マウスに尾静脈より、Cy5.5標識モノクローナル抗体(0.25 mg/ml)を0.15 ml/head投与した。生理食塩水(PBS、対照)、Cy5.5標識抗体A、Cy5.5標識3H3抗体の3種類を投与した。投与して24時間後に胸部皮膚を剥がし、生きた状態で全身を撮影した(図7)。
実験2:更に胸部大動脈が繋がった状態で心臓を摘出して撮影した(図7)。3H3投与で大動脈起始部が強く染まる。抗体Aでも幾分染まるが3H3の場合ほど蛍光強度は強くない。2A12では全く染まらない。抗体Aでは十分に粥種が観察できず、その他取得された抗体クローンでも同様であり、粥種のイメージングの使用には適さなかった。
大動脈起始部の一定面積あたりの蛍光強度を測定した。PBSを投与した対照マウスの蛍光を1.0とした。3H3投与で、対照の3倍程度の蛍光が確認された。その他の抗体投与ではPBSを投与した対照マウスと蛍光強度に大きな変化は見られなかった(図10)。尚、この蛍光部位を解剖後確認したところ、臨床所見上の粥腫と一致していた。
以上、今回の抗体群の特異性試験を表1にまとめた。
【0073】
【表1】
固相酸化LDL/β2GPI複合体或いは固相β2GPI ELISA液中に競合阻害抗原として、β2GPIとの阻害性を確認したところ、固相酸化LDL/β2GPI複合体の場合:3D4>2H6>4C12>3H3、固相β2GPIの場合:2H6>3D4(4C12、3H3は固相β2GPIに弱くしか反応せず)。3H3は液中フリーの酸化LDL/β2GPI複合体(非変性)への特異性が高い。
【0074】
[実施例8]
酸化LDL/β2GPI複合体を認識するマウスモノクローナル抗体の可変領域遺伝子の解析
解析したモノクローナル抗体は、4種類のClone (3H3, 4C12, 2H6, 3D4)である。
これら4種類のCloneの抗体サブグラスは、3H3と4C12がIgG2b、2H6と3D4はIgG1である。
(L鎖可変領域遺伝子の解析)
4種(3H3, 4C12, 2H6, 3D4)のモノクローナル抗体を産生するハイブリドーマ細胞をRPMI1640+10%FCS培地でそれぞれ培養した。そのハイブリドーマ細胞からQuickPrep micro mRNA purification kit (Amersham Biosciences, code 27-9255-01)を用いてmRNAを得た。そのmRNAをFirst-Strand cDNA Synthesis kit (Amersham Biosciences, code 27-9261-01)を用いてcDNAとした。このcDNAを鋳型としてPCR法で遺伝子を増幅させるのであるが、以下に示す11パターンのPrimerの組み合わせによりPCR反応を行った。ここで、MKV1〜MKV11のPrimer配列は、多くのモノクローナル抗体のシグナル配列を解析することで、ほぼ全てのモノクローナル抗体のL鎖シグナルは配列をこの11種類のPrimer配列で網羅するように設定されている。この11種類のMKV primerとマウスL鎖定常領域配列に相当するMKC primer間における11パターンのPCR反応から少なくとも1種類のPCR反応で目的のL鎖可変領域が増幅されてくる。
【0075】
PCR反応条件は、以下のとおり。
マウスハイブリドーマからのcDNA 4 μL
2.5 mM dNTPs 4 μL
MKV1〜MKV11 primer(20μM)の11種類中の1つ 2.5 μL
MKC primer(20μM) 2.5 μL
DMSO 2.5 μL
×10 pfu polymerase Buffer 5 μL
pfu polymerase 1 μL
滅菌水 28.5 μL
Total 50 μL
94℃ 2 min
94℃ 1 min 55℃ 2min 72℃ 2min (30サイクル)
72℃ 4min
4℃ 無制限時間
【0076】
PrimerのDNA配列は以下を参照。
MKV1 primer:ATGAAGTTGCCTGTTAGGCTGTTGGTGCTG(配列番号:11)
MKV2 primer:ATGGAGWCAGACACACTCCTGYTATGGGTG(配列番号:12)
MKV3 primer:ATGAGTGTGCTCACTCAGGTCCTGGSGTTG(配列番号:13)
MKV4 primer:ATGAGGRCCCCTGCTCAGWTTYTTGGMWTCTTG(配列番号:14)
MKV5 primer:ATGGATTTWCAGGTGCAGATTWTCAGCTTC(配列番号:15)
MKV6 primer:ATGAGGTKCYYTGYTSAGYTYCTGRGG(配列番号:16)
MKV7 primer:ATGGGCWTCAAGATGGAGTCACAKWYYCWGG(配列番号:17)
MKV8 primer:ATGTGGGGAYCTKTTTYCMMTTTTTCAATTG(配列番号:18)
MKV9 primer:ATGGTRTCCWCASCTCAGTTCCTTG(配列番号:19)
MKV10 primer:ATGTATATATGTTTGTTGTCTATTTCT(配列番号:20)
MKV11 primer:ATGGAAGCCCCAGCTCAGCTTCTCTTCC(配列番号:21)
MKC primer:ACTGGATGGTGGGAAGATGG(配列番号:22)
(M=A or C, R=A orG, W=A orT, S=C or G, Y=C or T, K=G orT)
【0077】
PCR反応によりL鎖可変領域が増幅してきたPCR primerの組み合わせは、以下になる。
3H3:MKV7 - MKC
4C12:MKV7 - MKC
2H6:MKV5 - MKC
3D4:MKV4 - MKC
PCR増幅したL鎖可変領域遺伝子はpCR2.1ベクター(Invitogen)に挿入された。
L鎖可変領域遺伝子が挿入されたPCR2.1ベクターは、DNAシークエンサー(Applied Biosystems製:3130 Genatic Analyzer)によりDNA塩基配列が解読された。
【0078】
(H鎖可変領域遺伝子の解析)
4種(3H3, 4C12, 2H6, 3D4)のモノクローナル抗体を産生するハイブリドーマ細胞をRPMI1640+10%FCS培地でそれぞれ培養した。そのハイブリドーマ細胞からQuickPrep micro mRNA purification kit (Amersham Biosciences, code 27-9255-01)を用いてmRNAを得た。そのmRNAをFirst-Strand cDNA Synthesis kit (Amersham Biosciences, code 27-9261-01)を用いてcDNAとした。このcDNAを鋳型としてPCR法でH鎖可変領域遺伝子を増幅させるのであるが、以下に示す12パターンのPrimerの組み合わせによりPCR反応を行った。ここで、MHV1〜MHV12のPrimer配列は、多くのモノクローナル抗体のシグナル配列を解析することで、ほぼ全てのモノクローナル抗体のH鎖シグナルは配列をこの12種類のPrimer配列で網羅するように設定されている。この12種類のMHV primerとマウスH鎖定常領域配列に相当するMHCG2bあるいはMHCG1 primer間における12パターンのPCR反応から少なくとも1種類のPCR反応で目的のH鎖可変領域が増幅されてくる。ここで、MHCG2b Primerは、マウスIgG2bのH鎖定常領域に相当する配列を持ち、MHCG1 Primerは、マウスIgG1のH鎖定常領域に相当する配列である。それゆえ、IgG2b サブクラスである3H3 cloneと4C12 cloneのPCR増幅には、MHCG2b Primerが用いられ、IgG1サブクラスである2H6 cloneと3D4 cloneのPCR増幅には、MHCG1 Primerが用いられた。
【0079】
PCR反応条件は、以下のとおり。
マウスハイブリドーマからのcDNA 4 μL
2.5 mM dNTPs 4 μL
MHV1〜MHV12 primer(20μM)の12種類中の1つ 2.5 μL
MHCG2b あるいはMHCG1 primer(20μM) 2.5 μL
DMSO 2.5 μL
×10 pfu polymerase Buffer 5 μL
pfu polymerase 1 μL
滅菌水 28.5 μL
Total 50 μL
94℃ 2 min
94℃ 1 min 55℃ 2min 72℃ 2min (30サイクル)
72℃ 4min
4℃ 無制限時間
【0080】
PrimerのDNA配列は以下を参照。
MHV1 primer:ATGAAATGCAGCTGGGGCATSTTCTTC(配列番号:23)
MHV2 primer:ATGGGATGGAGCTRTATCATSYTCTT(配列番号:24)
MHV3 primer:ATGAAGWTGTGGTTAAACTGGGTTTTT(配列番号:25)
MHV4 primer:ATGRACTTTGGGYTCAGCTTGRTTT(配列番号:26)
MHV5 primer:ATGGACTCCAGGCTCAATTTAGTTTTCCTT(配列番号:27)
MHV6 primer:ATGGCTGTCYTRGSGCTRCTCTTCTGC(配列番号:28)
MHV7 primer:ATGGRATGGAGCKGGRTCTTTMTCTT(配列番号:29)
MHV8 primer:ATGAGAGTGCTGATTCTTTTGTG(配列番号:30)
MHV9 primer:ATGGMTTGGGTGTGGAMCTTGCTATTCCTG(配列番号:31)
MHV10 primer:ATGGGCAGACTTACATTCTCATTCCTG(配列番号:32)
MHV11 primer:ATGGATTTTGGGCTGATTTTTTTTATTG(配列番号:33)
MHV12 primer:ATGATGGTGTTAAGTCTTCTGTACCTG(配列番号:34)
MHCG2b primer:CAGTGGATAGACTGATGGGGG(配列番号:35)
MHCG1 primer:CAGTGGATAGACAGATGGGGG(配列番号:36)
(M=A or C, R=A orG, W=A orT, S=C or G, Y=C or T, K=G orT)
【0081】
PCR反応によりH鎖可変領域が増幅してきたPCR primerの組み合わせは、以下になる。
3H3:MHV4 - MHCG2b
4C12:MKV4 - MHCG2b
2H6:MHV4 - MHCG1
3D4:MHV1 - MHCG1
【0082】
PCR増幅したH鎖可変領域遺伝子はpCR2.1ベクター(Invitogen)に挿入された。
H鎖可変領域遺伝子が挿入されたPCR2.1ベクターは、DNAシークエンサー(Applied Biosystems製:3130 Genatic Analyzer)によりDNA塩基配列が解読された。
その結果、本発明に用いることのできる3H3のアミノ酸配列、CDRが明らかになった(図11)。
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<3H3抗体>
配列番号1:重鎖可変領域のアミノ酸配列
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号5:重鎖可変領域の塩基配列
配列番号6:軽鎖可変領域のアミノ酸配列
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号10:軽鎖可変領域の塩基配列
配列番号55:重鎖FR1のアミノ酸配列
配列番号56:重鎖FR2のアミノ酸配列
配列番号57:重鎖FR3のアミノ酸配列
配列番号58:重鎖FR4のアミノ酸配列
配列番号59:軽鎖FR1のアミノ酸配列
配列番号60:軽鎖FR2のアミノ酸配列
配列番号61:軽鎖FR3のアミノ酸配列
配列番号62:軽鎖FR4のアミノ酸配列
【0083】
[実施例9]
IVIS 200および3次元CTを用いた画像解析に関する検討
Computed Tomography(CT)画像を統合することによる、動脈硬化病巣の3次元(局在)イメージ得るための実験を実施した。
In vivo蛍光イメージング:
IVIS 200 Imaging System(Xenogen社)を用いて、蛍光イメージングを行った(Cy5.5の場合Excitation:640 nm、Emission: 720 nm、Alexa Fluor 750の場合Excitation:745 nm、Emission: 800 nm)。Cy5.5標識3H3抗体(IgG)0.25 mg/ml、0.15 ml/1匹、またはAlexa Fluor750標識3H3抗体1.0〜1.5 mg/ml、0.15 ml/1匹を高脂肪食負荷ApoE-/-マウスの尾静脈より投与し、2〜24時間後に、IVIS 200にて麻酔吸入下でin vivo蛍光を観察・撮影した。ApoE-/-マウスでは黒い体毛が蛍光を吸収するため体毛を剃って観察した。はじめに反射光による蛍光観察を行い、その後、透過光による蛍光観察を行って、マウスの3次元(3D)画像を作成し、光源情報と融合させた(図9 A :統合前のIVISによる3次元画像)。 図の赤い点の部分が3H3に標識した蛍光シグナルであり、濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化していることを示す。
Ex vivoイメージング:
3D-CT解析の終了後マウスを安楽死させ、PBS 10 mlで心灌流し、心臓および大動脈を摘出し、IVIS 200により反射蛍光画像を得た。
CT イメージング:
eXplore Locus CT System (GE Healthcare)によりCTイメージングを行った。IVIS 200によりイメージングを行ったのと同じマウス個体を用い、麻酔吸入下にX線照射を行い、CT画像を得た。
蛍光とCTの画像統合:
IVIS 200により検出した蛍光イメージング画像と、CTを用いて撮影した画像を、汎用3D 可視化ソフト(Amira、Mercury Computer Systems)を用いて統合させた(図9B:統合後の3次元CT画像)。
なお、一連の手順を模式的に示した(図8)。
(A) IVIS 200による特異抗体を用いた蛍光イメージング(反射)。
(B)統合前のIVIS 200による特異抗体を用いた蛍光イメージング(透過光;左)と CT像(中) 統合図(右):蛍光イメージング(透過光;左)中の赤い点の部分について濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化集中した部分(3H3結合 反応性が高い部位)であることを示す。
(C) IVISによる蛍光シグナルと3次元CTの統合画像:コンピューターの仮想空間上に3次元像を動画として構成(3次元グラフィックスのアニメーション)として、多方面からその標識された位置を確認した写真である。
可視光は生体内で吸収されてしまうためin vivo イメージングを行うには、生体に吸収されにくい波長である近赤外の蛍光標識が適している。今回は、Cy5.5またはAlexa Fluor 750により標識した抗体をマウスの尾静脈より投与して、IVIS200により蛍光を測定し、反射および透過による蛍光測定条件の検討を行った。Cy5.5標識抗体によるin vivoイメージングの反射蛍光画像では動脈硬化発症ApoE-/-マウスの大動脈弁および胸部大動脈の強いシグナルが観察された。さらに、ex vivoイメージングおよびex vivo蛍光顕微鏡観察により、静脈投与した蛍光標識抗体の動脈硬化巣への局在が確認できた。しかしながら、Cy5.5標識抗体を用いる場合、透過蛍光のシグナルが弱く、立体画像(3次元)での蛍光位置特定が容易ではなかった。一方、Alexa Fluor 750標識抗体では、静脈内投与後2時間後で強い特異的なシグナルが反射蛍光画像、透過蛍光画像の両方で得られ、立体画像を構築してみると胸の内部に強い蛍光シグナルが認められた(図8A、B中の左図)。続いて同じマウスについてCT撮影を行い、CT画像より骨と肺の臓器抽出を行って得られた画像(図8B中と、IVIS200により構築した蛍光イメージング画像をAmiraにより統合させた、統合3D画像(図8B右)を得たところ、心臓とその近傍の位置に蛍光シグナルが観察された。図の赤い点の部分について濃度が濃いほど、蛍光強度が強い、即ち、イメージング剤が局在化していることを示す。CT(中央)と3D-CT統合の画像を示した(図8B右)。コンピューターの仮想空間上に3次元像を動画として構成(3次元グラフィックスのアニメーション)として、多方面からその標識された位置を確認した(図8C)。
【0084】
上記実験の結果、蛍光標識3H3抗体によるin vivoイメージングの反射蛍光画像では動脈硬化発症ApoE-/-マウスの大動脈弁および胸部大動脈の強いシグナルが観察された。さらに、ex vivoイメージングおよびex vivo蛍光顕微鏡観察により、静脈投与した蛍光標識抗体の動脈硬化巣への局在が確認できた。
【0085】
[実施例10]
キメラ抗体発現ベクターの作製
マウスH鎖の重鎖可変領域DNA(配列番号:5)を、HindIII, XhoIで切断して精製し、Lonza社のpEE6.4ベクター(ヒトIgG1定常領域をマルチクローニングサイトで組み込んである)に挿入し、キメラH鎖ベクターを作製した。同様に、マウスL鎖の軽鎖可変領域DNA(配列番号:10)をHindIII, BsiWI で切断して精製し、Lonza社のpEE14.4ベクター(ヒトKappa定常領域をマルチクローニングサイトに組み込んである)に挿入し、キメラL鎖ベクターを作製した。なお、重鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MELGLRWVFLVAILEGVQC(配列番号:37)のシグナル配列を利用し、さらに軽鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MDMRVPAQLLGLLLLWLPGARC(配列番号:38)のシグナル配列を利用した。
【0086】
[実施例11]
ヒト型化抗体可変領域の設計
CDR-grafting法に従い、ヒトフレームワークを選択し、マウスモノクローナル抗体3H3のCDRに置き換えた。
まず、H鎖可変領域フレームワーク、L鎖可変領域フレームワークそれぞれについてホモロジー検索を行った。
その結果、3H3抗体H鎖可変領域のフレームワーク(FR)領域(FR1領域(配列番号:55)、FR2領域(配列番号:56)、FR3領域(配列番号:57)、FR4領域(配列番号:58))は、ヒト抗体(Accession number AY882577(配列番号:43)のFR領域(FR1領域(配列番号:39)、FR2領域(配列番号:40)、FR3領域(配列番号:41)、FR4領域(配列番号:42))と高い相同性を持つことが分かった(図12(A)参照)。具体的には、H鎖について、マウス3H3抗体のFRと、CDR移植先ヒト抗体のFR 部分の相同性は 80/87=91.9%であった。
次に、これらのヒト抗体(Accession number AY882577)のFR領域に、3H3の重鎖CDR1〜CDR3(配列番号:2〜配列番号:4)が適切に移植されるように、アミノ酸配列を設計し、ヒト化抗体重鎖可変領域アミノ酸配列(配列番号:44)を設計した(図12(B)参照)。
重鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MELGLRWVFLVAILEGVQCのシグナル配列を利用した。
3H3抗体H鎖可変領域と同様に、3H3抗体L鎖可変領域のフレームワーク(FR)領域(FR1領域(配列番号:59)、FR2領域(配列番号:60)、FR3領域(配列番号:61)、FR4領域(配列番号:62))も、ヒト抗体(Accession number AY942004(配列番号:51))のFR領域(FR1領域(配列番号:47)、FR2領域(配列番号:48)、FR3領域(配列番号:49)、FR4領域(配列番号:50))と高い相同性を持つことが分かった(図13(A)参照)。具体的には、L鎖について、マウス3H3抗体のFRとCDR移植先ヒト抗体のFR 部分の相同性は 65/80=81.3%であった。
次に、ヒト抗体(Accession number AY942004)のFR領域に、3H3の軽鎖CDR1〜CDR3(配列番号:7〜配列番号:9)が適切に移植されるように、アミノ酸配列を設計し、ヒト化抗体軽鎖可変領域アミノ酸配列(配列番号:52)を設計した(図13(B)参照)。
軽鎖のシグナル配列についてはヒトgermlineより選択したシグナル配列MDMRVPAQLLGLLLLWLPGARCのシグナル配列を利用した。
ヒト化抗体の重鎖及び軽鎖可変領域をコードするDNAは、50base程度の合成オリゴDNAを約20base程度ハイブリダイズするように設計し、これらの合成オリゴDNAをPCR法によりアッセンブリさせることにより作製した。重鎖可変領域をコードするDNAを、5'端の合成オリゴDNAの末端に挿入した制限酵素HindIII部位および3'端の合成オリゴDNAの 末端に挿入したXhoI部位で切断し、上記Lonza社のヒトIgG1定常領域がクローニングされたpEE6.4ベクターへクローニングした。また、軽鎖可変領域をコードするDNAを、5'端の合成オリゴDNAの末端に挿入した制限酵素HindIII部位および3'端の合成オリゴDNAの 末端に挿入したBsiWI部位で切断し、上記ヒトκ鎖定常領域がクローニングされたpEE14.4ベクターへクローニングした。
上記の手法で作製したヒト化抗体3H3抗体の重鎖を3H3RHAと命名した(図14、シグナル配列-重鎖をコードする配列、塩基配列は配列番号:45、アミノ酸配列は配列番号:46)。同様に、ヒト化抗体3H3抗体の軽鎖を3H3RKAと命名した(図15、シグナル配列-軽鎖をコードする配列、塩基配列は配列番号:53、アミノ酸配列は配列番号:54)。
本明細書で開示した抗体の塩基配列およびアミノ酸配列は、以下の配列番号に従って配列表に記載されている。
<ヒト化3H3抗体>
配列番号44:重鎖可変領域のアミノ酸配列
配列番号46:重鎖のアミノ酸配列(シグナル配列を含む重鎖)
配列番号2:重鎖CDR1のアミノ酸配列
配列番号3:重鎖CDR2のアミノ酸配列
配列番号4:重鎖CDR3のアミノ酸配列
配列番号39:重鎖FR1のアミノ酸配列
配列番号40:重鎖FR2のアミノ酸配列
配列番号41:重鎖FR3のアミノ酸配列
配列番号42:重鎖FR4のアミノ酸配列
配列番号45:重鎖の塩基配列(シグナル配列を含む重鎖)
配列番号52:軽鎖可変領域のアミノ酸配列
配列番号54:軽鎖のアミノ酸配列(シグナル配列を含む軽鎖)
配列番号7:軽鎖CDR1のアミノ酸配列
配列番号8:軽鎖CDR2のアミノ酸配列
配列番号9:軽鎖CDR3のアミノ酸配列
配列番号47:軽鎖FR1のアミノ酸配列
配列番号48:軽鎖FR2のアミノ酸配列
配列番号49:軽鎖FR3のアミノ酸配列
配列番号50:軽鎖FR4のアミノ酸配列
配列番号53:軽鎖の塩基配列(シグナル配列を含む軽鎖)
【0087】
[実施例12]
トランスフェクション、発現
キメラ抗体のH鎖ベクター及びL鎖ベクター、ヒト化3H3抗体のH鎖ベクター及びL鎖ベクターについて、構築したH鎖ベクターとL鎖ベクターをそれぞれNotI、SalIで消化し、断片同士をライゲーションし、一つのベクターを構築した。その後、構築したDNAをPvuI切断により直鎖化し、エレクトロポレーションによりCHOK1SV細胞に導入した。エレクトロポレーション後、死細胞を含む細胞溶液をグルタミンフリーの10%FBSを含むグルタミンフリーDMEMに加え、50μlずつ96穴プレートに撒いた。24時間培養後、セレクション試薬であるMSXを最終濃度25μMになるように、グルタミンフリーDMEM/10%FBS培地に溶解し、サプリメントとして、GS supplement(50倍溶液)を同培地に溶解し、先述した96穴プレートに150μlずつ添加した。その後、ELISAにて最も発現量の多いクローンを選択し、安定細胞株を樹立した。
培養上清中のIgG濃度は、培養上清を回収し、Goat anti-human IgG antibody, Fcγ fragment-specific (Stratech Scientific)、と Human IgG1/Kappa antibody (Binding site)でのサンドイッチELISAを行い、検出用として、Goat anti-human kappa light chain peroxidase conjugate (Sigma)を用い、市販の精製ヒトIgG(Cappel社)と の比較により定量した。
【0088】
[実施例13]
抗原に対するELISA活性(ヒト化IgG1とキメラIgG1の比較;結合活性比較)
一部の条件を除き[実施例4]に記載の方法と同じ方法でキメラ抗体-IgG1、ヒト化3H3(3H3RHA、3H3RKA)-IgG1の結合活性の濃度依存性を確認した。本方法は、吸収波長を650nmに変更して検出し、IgG濃度が測定された培養上清を各横軸に記載の抗体濃度に希釈したサンプルとした点において[実施例4]に記載の方法と異なる。その結果、ヒト化IgG1はマウス可変領域を有するキメラIgG1とELISA結合活性の抗体濃度依存性が同等であった。なお、コントロールとしては293T細胞の培養上清を用いた(図16)。
【0089】
[実施例14]
Antibody-dependent Cellular Cytotoxicity (ADCC)活性の低いIgG4型への変換
イメージング用途としては、IgG1型より、ADCC活性、CDC活性等の生理活性の低いIgG2又は4型が望ましいと考えられるため、ヒト化3H3(3H3RHA、3H3RKA)-IgG1の定常領域をIgG4型へ変換した抗体を作製した。IgG4は不安定でヘテロな複合体を形成し、定常領域におけるKabat則によるナンバリングで241番目のアミノ酸であるセリンがその安定性に関わることが報告されており、このアミノ酸をIgG1型、IgG2型のプロリンへ変換することで安定性が改善されることが報告されている。そこで、241番目のアミノ酸をプロリンに変換した変異型IgG4をIgG1に換えて組み込んだpEE6.4ベクターを構築し、上記と同様の方法でヒト化3H3(3H3RHA、3H3RLA)- IgG4を作成した(図17)。抗体発現、抗体濃度を測定後、結合活性比較を行った。
抗原に対するELISA活性(ヒト化変異型IgG4とキメラIgG1、ヒト化IgG1の比較;結合活性比較)
実施例13と同様の方法でヒト化3H3(3H3RHA、3H3RLA)- IgG4抗体の結合活性をキメラ-IgG1、ヒト化3H3(3H3RHA、3H3RLA)-IgG1 と比較したところ、ELISA結合活性は同等であった(図18)。
【0090】
[実施例15]
免疫染色/イメージング
上記実施例において作成された、キメラ-IgG1、ヒト化3H3(3H3RHA、3H3RLA)- IgG1抗体、ヒト化3H3(3H3RHA、3H3RLA)- IgG4抗体は[実施例6]の免疫染色、[実施例7]のイメージングと同様の試験を行うことにより、マウス3H3抗体と同等のイメージング活性を示した活性を有することが確認できる。
【0091】
本実験により、近赤外蛍光物質(Cy5.5、Alexa750)標識抗体を用いることで、マウスにおける動脈硬化のイメージングが可能となったばかりではなく、3次元CTによる画像との統合も可能であることが判明した。またこの種の抗体がヒトの動脈硬化巣を検出できることも明らかになっており、本実験結果は臨床用の画像診断技術に繋がる。さらに、当該マウスイメージング技術は、創薬のためのスクリーニング系として既に実用可能なものである。
【産業上の利用可能性】
【0092】
従来の動脈硬化検査法では、動脈硬化の発症部位(存在部位)が特定できない。これに対し、本発明は、動脈硬化巣(特に粥腫、粥状動脈硬化)の存在部位やその大きさを視覚的に捉えることの出来る非侵襲的診断法を提供するものである。
動脈硬化好発モデルマウス(例えば、アポE欠失(ApoE-/-)マウス:高い血漿コレステロール濃度を維持し、自然発生アテローム性動脈硬化様病変を発症する)と、当該イメージング用の抗体を用いることで、粥状動脈硬化の治療薬のスクリーニング系が構築できる。
さらには、当該抗体をヒト化抗体に変換して、臨床診断用イメージングシステムを構築することが可能である。従って、動脈粥腫からのプラーク遊離などが動脈塞栓を起因とした脳塞栓症、心筋梗塞の原因となることが知られており、慢性的かつ無症候性に潜行進展するヒトの粥状動脈硬化のモニタリング法は、生活習慣病の予防、治療方針の一助になるといえる。
【特許請求の範囲】
【請求項1】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する、下記(a)〜(j)のいずれかに記載の抗体;
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体、
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
【請求項2】
請求項1に記載の抗体が結合するエピトープと同じエピトープに結合する抗体。
【請求項3】
ヒト化抗体またはキメラ抗体である、請求項1又は2に記載の抗体。
【請求項4】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング剤。
【請求項5】
請求項1〜3のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング剤。
【請求項6】
動脈硬化において、粥腫の位置及び/または大きさをイメージングする、請求項4または5のイメージング剤。
【請求項7】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング用キット。
【請求項8】
請求項1〜3のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング用キット。
【請求項9】
以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法:
(a)請求項1〜3のいずれかに記載の抗体が投与された動脈硬化疾患モデル非ヒト動物に、候補化合物を投与する工程、
(b)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣の大きさまたは存在部位を比較する工程、及び
(d)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
【請求項1】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する、下記(a)〜(j)のいずれかに記載の抗体;
(a)CDR1として配列番号:2に記載のアミノ酸配列、CDR2として配列番号:3に記載アミノ酸配列、及びCDR3として配列番号:4に記載のアミノ酸配列を有する重鎖を含む抗体、
(b)重鎖可変領域として配列番号:1に記載のアミノ酸配列を有する重鎖を含む抗体、
(c)CDR1として配列番号:7に記載のアミノ酸配列、CDR2として配列番号:8に記載アミノ酸配列、及びCDR3として配列番号:9に記載のアミノ酸配列を有する軽鎖を含む抗体、
(d)軽鎖可変領域として配列番号:6に記載のアミノ酸配列を有する軽鎖を含む抗体、
(e)上記(a)または(b)に記載の重鎖、および、上記(c)または(d)に記載の軽鎖の対を有する抗体、
(f)重鎖可変領域として配列番号:44に記載のアミノ酸配列を有する重鎖を含む抗体、
(g)配列番号:46に記載のアミノ酸配列を有する重鎖を含む抗体、
(h)軽鎖可変領域として配列番号:52に記載のアミノ酸配列を有する軽鎖を含む抗体、
(i)配列番号:54に記載のアミノ酸配列を有する軽鎖を含む抗体、
(j)上記(f)または(g)に記載の重鎖、および、上記(h)または(i)に記載の軽鎖の対を有する抗体。
【請求項2】
請求項1に記載の抗体が結合するエピトープと同じエピトープに結合する抗体。
【請求項3】
ヒト化抗体またはキメラ抗体である、請求項1又は2に記載の抗体。
【請求項4】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング剤。
【請求項5】
請求項1〜3のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング剤。
【請求項6】
動脈硬化において、粥腫の位置及び/または大きさをイメージングする、請求項4または5のイメージング剤。
【請求項7】
酸化LDLとβ2-グリコプロテインIの複合体(酸化LDL/β2GPI複合体)に結合する抗体を含む、動脈硬化の存在部位のイメージング用キット。
【請求項8】
請求項1〜3のいずれかに記載の抗体を含む、動脈硬化の存在部位のイメージング用キット。
【請求項9】
以下に記載の工程を含む、動脈硬化治療剤の候補化合物のスクリーニング方法:
(a)請求項1〜3のいずれかに記載の抗体が投与された動脈硬化疾患モデル非ヒト動物に、候補化合物を投与する工程、
(b)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣をイメージングする工程、
(c)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物と、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物において、動脈硬化巣の大きさまたは存在部位を比較する工程、及び
(d)該候補化合物を投与された動脈硬化疾患モデル非ヒト動物において、該候補化合物を投与されていない動脈硬化疾患モデル非ヒト動物と比較して、動脈硬化巣が減少または消失する候補化合物を選択する工程。
【図1】

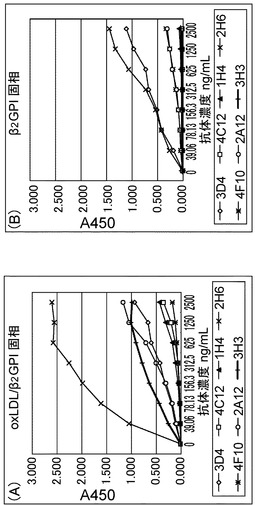
【図2】
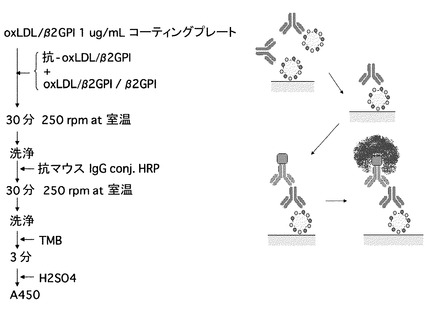
【図3】
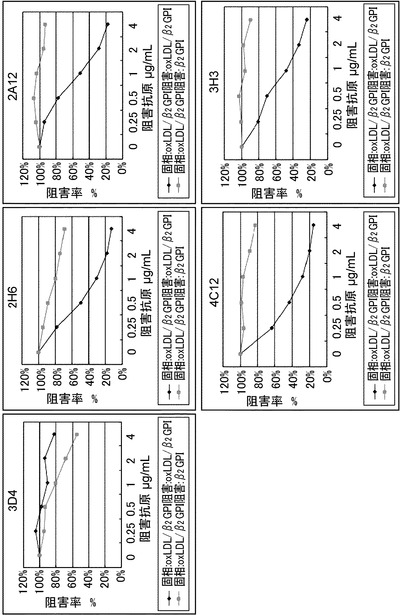
【図4】
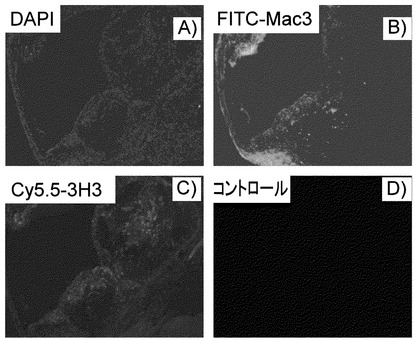
【図5】

【図6】

【図7】
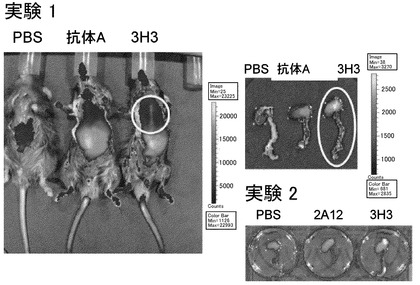
【図8】

【図9】

【図10】
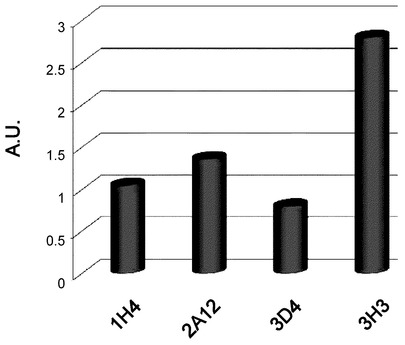
【図11】
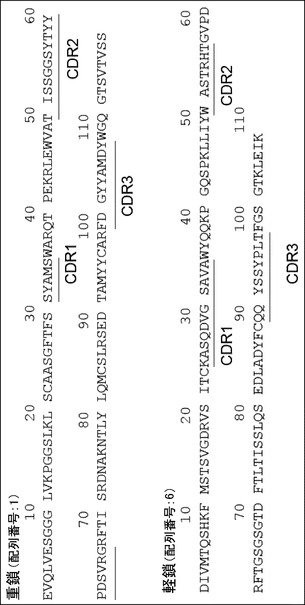
【図12】
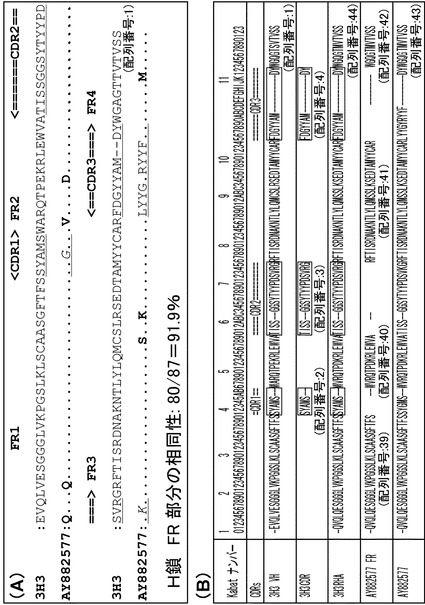
【図13】

【図14】

【図15】

【図16】

【図17】
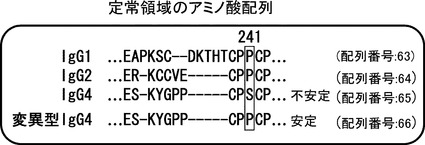
【図18】
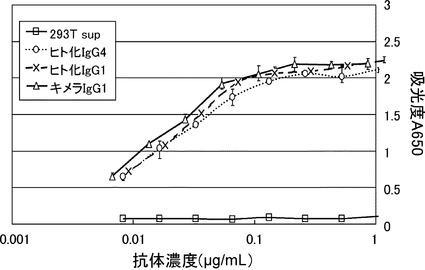
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2011−42581(P2011−42581A)
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2009−146783(P2009−146783)
【出願日】平成21年6月19日(2009.6.19)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年3月11日、財団法人岡山医学振興会(メディカルテクノおかやま)発行の「平成19年度文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」研究成果報告会」に発表。 平成20年3月11日、メディカルテクノおかやま主催の「平成19年度文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」研究成果報告会」において、文書をもって発表。 平成20年3月13日、http://www.optic.or.jp/medical/itaku/dengen/080311.pdfを通じて発表。 平成20年3月17日、財団法人岡山医学振興会発行の「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」に発表。 平成21年4月20日、株式会社岡山日日新聞新社発行の「平成21年4月20日付岡山日日新聞の第3ページの『岡大発 医学・医療の最前線29 石灰化小球に特殊な脂質』」に発表。 平成21年5月25日、株式会社岡山日日新聞新社発行の「平成21年5月25日付岡山日日新聞の第3ページの『岡大発 医学・医療の最前線30 動脈硬化にも標的医療を適用』」に発表。 平成21年4月24日、http://www.okayama−u.net/medic/icont/materials/results/0904vol29.pdfを通じて発表。 平成21年5月27日、http://www.okayama−u.net/medic/icont/materials/results/0905vol30.pdfを通じて発表。 平成21年3月25日、http://www.optic.or.jp/medical/itaku/dengen/matsuura.pdfを通じて発表。 平成21年3月10日、メディカルテクノおかやま、岡山県、岡山大学ナノバイオ標的医療イノベーションセンター主催の「『ナノバイオ標的医療の融合的創出拠点の形成』事業『ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究』事業合同成果報告会」において、文書をもって発表。 平成21年3月10日、メディカルテクノおかやま発行の「平成20年度特別電源所在県科学技術振興事業『ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究』研究成果報告会 要旨集」に発表。
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度及び平成20年度、文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究事業」に係る委託研究、産業技術力強化法第19条の適用を受けるもの
【出願人】(504147243)国立大学法人 岡山大学 (444)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成21年6月19日(2009.6.19)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年3月11日、財団法人岡山医学振興会(メディカルテクノおかやま)発行の「平成19年度文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」研究成果報告会」に発表。 平成20年3月11日、メディカルテクノおかやま主催の「平成19年度文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」研究成果報告会」において、文書をもって発表。 平成20年3月13日、http://www.optic.or.jp/medical/itaku/dengen/080311.pdfを通じて発表。 平成20年3月17日、財団法人岡山医学振興会発行の「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究」に発表。 平成21年4月20日、株式会社岡山日日新聞新社発行の「平成21年4月20日付岡山日日新聞の第3ページの『岡大発 医学・医療の最前線29 石灰化小球に特殊な脂質』」に発表。 平成21年5月25日、株式会社岡山日日新聞新社発行の「平成21年5月25日付岡山日日新聞の第3ページの『岡大発 医学・医療の最前線30 動脈硬化にも標的医療を適用』」に発表。 平成21年4月24日、http://www.okayama−u.net/medic/icont/materials/results/0904vol29.pdfを通じて発表。 平成21年5月27日、http://www.okayama−u.net/medic/icont/materials/results/0905vol30.pdfを通じて発表。 平成21年3月25日、http://www.optic.or.jp/medical/itaku/dengen/matsuura.pdfを通じて発表。 平成21年3月10日、メディカルテクノおかやま、岡山県、岡山大学ナノバイオ標的医療イノベーションセンター主催の「『ナノバイオ標的医療の融合的創出拠点の形成』事業『ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究』事業合同成果報告会」において、文書をもって発表。 平成21年3月10日、メディカルテクノおかやま発行の「平成20年度特別電源所在県科学技術振興事業『ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究』研究成果報告会 要旨集」に発表。
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度及び平成20年度、文部科学省特別電源所在県科学技術振興事業「ナノバイオ標的医療等の新たな医療の創造とその基盤技術研究事業」に係る委託研究、産業技術力強化法第19条の適用を受けるもの
【出願人】(504147243)国立大学法人 岡山大学 (444)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
[ Back to top ]