
重硫酸アタザナビルおよび新規形態の製造方法
HIVプロテアーゼ阻害剤である重硫酸アタザナビルを製造するための方法を提供し、その方法においては、アタザナビル遊離塩基の溶液を遊離塩基の約15重量%以下と反応する量で濃硫酸と反応し、A型結晶の重硫酸アタザナビルの種晶を反応混合物に加え、重硫酸塩の結晶が形成するように重硫酸アタザナビルのA型結晶形成を行うために、追加の濃硫酸を三次方程式に従った増加速度で多段階にて加える。また、Cパターン物質である重硫酸アタザナビルを調製するための方法を提供する。エタノールから重硫酸塩の高度に結晶性のトリエタノレート溶媒和物であるE3型である重硫酸アタザナビルの新規形態もまた提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2004年5月4日に出願した米国仮出願第60/568,043号、および2004年9月7日に出願した第60/607,533号からの優先権を利用し、その開示は参照によって本明細書に引用される。本発明は、HIVプロテアーゼ阻害剤である重硫酸アタザナビルおよびその新規形態の製造方法に関する。
【背景技術】
【0002】
Fasslerらの米国特許第5,849,911号は、以下の構造:
【化1】

[式中、
R1は、低級アルコキシカルボニルであり、
R2は、二級または三級低級アルキルまたは低級アルキルチオール−低級アルキルであり、
R3は、1またはそれ以上の低級アルコキシ基、またはC4−C8シクロアルキルによって非置換または置換されるフェニルであり、
R4は、フェニルまたはシクロヘキシルであり、それぞれが環の炭素原子を経由して結合し、5〜8個の環の原子を有し、窒素、酸素、硫黄、スルフィニル(−SO−)およびスルホニル(−SO2−)から選択される1〜4個のヘテロ原子を含み、低級アルキルまたはフェニル−低級アルキルによって非置換または置換される不飽和ヘテロシクリルによってその4位で置換され、
R5は、R2とは独立して、R2に記載される意味の一つを有し、および
R6は、R1とは独立して、低級アルコキシカルボニル、またはその塩であるが、但し、少なくとも一つの塩を形成する基が存在する]および種々のその医薬的に許容される酸付加塩を有するアザペプチドHIVプロテアーゼ阻害剤(アタザナビルを含む)のシリーズを開示する。
【0003】
R1およびR6、ならびにR2およびR5が、それぞれ二つの同一基であり、構造(a)
【化2】
のジアミノ化合物が構造(b)
【化3】
[式中、R1’およびR2’は、それぞれR1およびR6、ならびにR2およびR5で定義される]
の酸またはその活性な酸誘導体と縮合される化合物の製造を含む、アザペプチドを製造するためのいくつかの方法が提供される。
【0004】
上記の方法を用いるアタザナビルの生成において、構造式:
【化4】
を有するジアミノ化合物(a)は、イソプロピルアルコールの存在下で、エポキシド体:
【化5】
とヒドラジノカーバメート体:
【化6】
とカップリングして、保護ジアミン体:
【化7】
を形成し、テトラヒドロフランのような溶媒の存在下で、塩酸と処理し、ジアミン体(a):
【化8】
を形成することによって製造される。該ジアミン体を単離し、O−(1,2−ジヒドロ−2−オキソ−1−ピリジル)−N,N,N1,N1−テトラメチルウロニウムテトラフルオロボレート(TPTU)のようなカップリング剤を用いて、酸(b)
【化9】
または、その活性エステル体と反応させる、次のカップリング工程に用いる。
【0005】
該ジアミン遊離塩基は不安定であり、それ故、アタザナビル遊離塩基の製造において、使用するためには望ましくないことがわかっている。
【0006】
Singhらの米国特許第6,087,383号は、該構造:
【化10】
(また、重硫酸アタザナビルまたは硫酸アタザナビルとも呼ばれる)を有するアタザナビルとして知られるアザペプチドHIVプロテアーゼ阻害剤の重硫酸塩を開示する。
【0007】
Singhらの実施例3は、水和化した吸湿性で結晶性形状であるII型結晶の形態、および無水/脱溶媒和結晶形であることが明らかであるI型結晶の形態の重硫酸アタザナビルの製造を記載する。
【0008】
(発明の簡単な説明)
本発明によれば、Cパターン物質およびE3型を含む、重硫酸アタザナビルの新規形態が提供される。Cパターン物質が好ましい。
【0009】
さらに本発明によれば、A型結晶(バルク薬)(Singhらの米国特許第6,087,383号の実施例3でI型結晶と呼ばれる)の形態の、重硫酸アタザナビルを製造するための方法を提供する。本発明の方法によって製造されるA型結晶は、所望の実質的に整った粒子径分布および実質的に整った平均粒子径を有し、医薬製剤を調製するための種々の賦形剤と製剤化される部分的な結晶物質であるCパターン物質への変換に用いられる。
【0010】
重硫酸アタザナビル塩のA型結晶を調製するための本発明の方法は、硫酸が三次方程式(以下に記載)に従う増加速度で加える改良立方結晶化技術を用いて、アタザナビル遊離塩基の有機溶媒溶液(該重硫酸アタザナビル塩が実質的に不溶である)をアタザナビル遊離塩基の約15%重量以下、好ましくは約12重量%以下で反応するための量の濃硫酸の最初の一部と反応させ、重硫酸アタザナビルのA型結晶の種晶を反応混合物に加え、重硫酸アタザナビルの結晶が形成するようにA型結晶の形成を行うために三次方程式による増加速度で多段階にて、追加の濃硫酸を加える工程を含む。
【0011】
さらに本発明によれば、重硫酸アタザナビルから生じ、重硫酸アタザナビルを含むアタザナビルの形態を製造するための方法が提供され、それは、Cパターン物質と称される。Cパターンは、水中でA型結晶を懸濁し、乾燥することで生じ得る。または、Cパターン物質は、約95%RH(水蒸気)より高い相対湿度で少なくとも24時間、A型結晶を付すことによって形成されうる。Cパターン物質はまた、重硫酸アタザナビル、または重硫酸アタザナビルと賦形剤の組合せを湿式造粒し、湿式造粒物を乾燥することによっても形成されうる。
【0012】
好ましい態様において、A型結晶は、例えばラクトースのような1またはそれ以上の充填剤、クロスポビドンのような1またはそれ以上の崩壊剤のような製剤用賦形剤と混合し、湿式造粒して賦形剤との混合においてCパターン物質を直接形成する。
【0013】
さらに本発明によれば、重硫酸アタザナビルの新規形態、すなわち重硫酸アタザナビルのトリエタノレート溶媒和物の高度な結晶形である、E3形が提供される。
【0014】
E3型は、エタノール中でアタザナビル遊離塩基をスラリーにし、該スラリーを濃硫酸で処理し、生じた溶液を加熱し、エタノールで湿ったE3結晶で種晶添加し、該混合物をヘプタン(またはトルエンまたはヘキサンのような他の溶媒)で処理し、濾過し、乾燥することにより調製される。
【0015】
さらにまた本発明によれば、重硫酸アタザナビルのA型結晶を調製するための方法が提供され、その方法は、構造:
【化11】
のトリアミン塩(好ましくはHCl(3モル)塩)を調製し、該トリアミン塩を単離することなく、塩基および有機溶媒の存在下、活性エステル体、好ましくは構造式:
【化12】
の活性エステル体と反応させ、アタザナビル遊離塩基を形成し、単離することなく、本明細書で述べる改良立方結晶化技術によって重硫酸アタザナビルへと変換される工程を含む。
【0016】
さらに本発明によれば、A型結晶またはCパターン物質である重硫酸アタザナビル、およびその医薬的に許容される担体を含む新規な重硫酸アタザナビルの組成物を提供する。該医薬的に許容される担体は、充填剤、結合剤、崩壊剤、滑沢剤、および他の通常の賦形剤を含んでもよい。
【0017】
本発明による重硫酸アタザナビルの種々の形態は、種々の技術、当該技術分野における通常の技術を有する者に周知である操作を用いて測定されうる。該形態は、固定された分析温度で形態の単結晶の単位格子測定に基づく、単結晶X線回折を用いて測定され、区別される。単位格子の詳細な説明は、Stout & Jensen, X-Ray Structure Determination: A Practical Guide, Macmillan Co., New York (1968), Chapter 3 内で提供され、それは参照によって本明細書に引用される。または、結晶格子内の空間的関係における独特の原子配列は、観察された原子分率座標によって測定されうる。結晶構造を測定する他の手段は、実験的または観察された回折プロフィールが純粋な粉末物質を表す模擬プロフィールと比較する粉末X線回折分析によって行われ、両方のランは同一の分析温度で、一連の2θ値として特徴付けられる対象の形態のために測定する。
【0018】
固体状態核磁気共鳴(SSNMR)、示差走査熱量測定(DSC)および熱重量分析(TGA)のような形態を測定する他の手段が使用されてもよい。これらのパラメーターはまた、対象の形態を測定するために組み合わせて使用されてもよい。
【0019】
A型結晶は、下記:
[格子サイズ:
a=9.86(5)Å
b=29.245(6)Å
c=8.327(2)Å
α=93.56(2)°
β=114.77(3)°
γ=80.49(3)°
空間群1
分子/非対称単位2]
と実質的に同等である単位格子パラメーターによって特徴付けられ、結晶形態が約+22℃で存在する。
【0020】
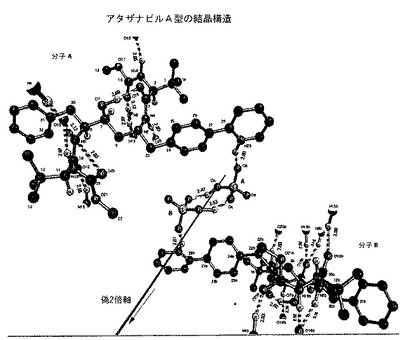
A型は、実質的に表3に記載される原子分率座標および実質的に図2で示される結晶構造によって特徴付けられうる。
【0021】
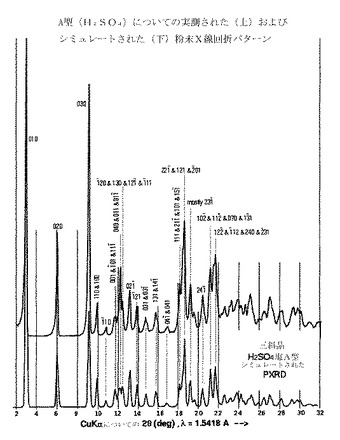
A型は、実質的に図1で示されるシミュレートされたおよび実測された粉末X線回折パターンによって特徴付けられうる。
【0022】
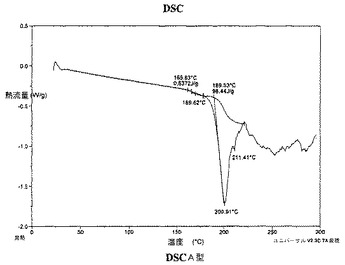
A型は、実質的に図3で示される約165.6℃でのピーク発現と共に吸熱を有する示差走査熱量測定(DSC)サーモグラムによって特徴付けられうる。
【0023】
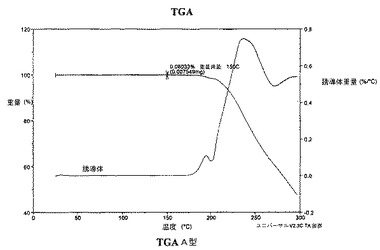
A型は、実質的に図4で示される約100℃〜150℃までにごく僅かな減量を有する熱重量分析(TGA)曲線によって特徴付けられうる。
【0024】
A型は、実質的に表4で示される固体状態NMR(SSNMR)の化学シフトおよび実質的に図5で示されるスペクトルによって特徴付けられうる。
【0025】
A型は、実質的に表5で示される原子分率座標によって特徴付けられうる。
【0026】
A型塩は、25℃で25〜75%RHの範囲において、約0.1%の重量増加と共に吸湿等温線によって特徴付けられうる。
【0027】
本発明の一態様において、Cパターンは、図5で示される観察された粉末X線回折パターンによって実質的に特徴付けられうる。
【0028】
本発明の異なった態様において、Cパターンは、約76.7〜約96.6℃および約156.8〜約165.9℃の範囲内で典型的に吸熱を有する、図7で示される示差走査熱量サーモグラムによって実質的に特徴付けられうる。
【0029】
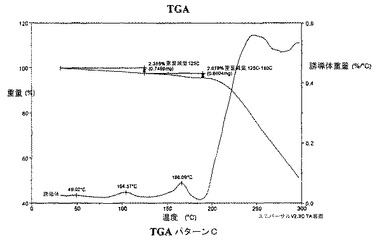
本発明の異なった態様において、Cパターンは、実質的に図8で示される約125℃で約2.4%の重量減量、および約190℃までで約4.4%の減量を有する熱重量分析曲線によって特徴付けられうる。
【0030】
本発明によれば、E3型は、下記:
[a=10.749Å
b=13.450(4)Å
c=9.250(2)Å
α=98.33(2)°
β=95.92(3)°
γ=102.82(3)°
空間群P1
分子/非対称単位1]
と実質的に同等である表5に示される結晶学的データによって特徴付けられ、結晶形が約−23℃で存在する。
【0031】
本発明の異なった態様において、E3型は、実質的に表6で記載される原子分率座標によって特徴付けられうる。
【0032】
本発明の異なった態様において、E3型は、実質的に図9で示されるシミュレートされたおよび実測された粉末X線回折パターンによって特徴付けられうる。
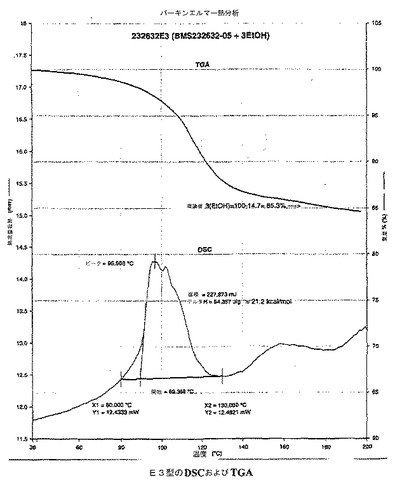
【0033】
本発明の異なった態様において、E3型は、実質的に図11で示される約89.4〜約96.6℃の範囲内で典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられうる。
【0034】
本発明の異なった態様において、E3型は、実質的に表8で示される約150℃で約14.7%の重量減量を有する熱重量分析曲線によって特徴付けられうる。
【0035】
本発明の異なった態様において、E3型は、実質的に図10で示される結晶構造によって特徴付けられうる。
【0036】
(発明の詳細な説明)
本発明は、少なくとも一部分で重硫酸アタザナビルの形態、すなわち新規材料として、特に医薬的に許容される形態でE3型およびCパターンを提供する。本明細書で用いられる用語「医薬的に許容される」とは、合理的な利点/リスク比に相応して過剰毒性、刺激作用、アレルギー反応、または他の問題のある合併症がなく、正しい医療判断の範囲内で、ヒトおよび動物組織と接するのに適した、それらの化合物、物質、組成物および/または製剤を意味する。ある好ましい態様において、その遊離塩基Iおよびその塩の結晶形は、実質的に純粋な形態である。本明細書で用いられる用語「実質的に純粋」とは、例えば約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、および約100%を含む、約90%以上の純度を有する化合物を意味する。
【0037】
本明細書で用いられる「多形」とは、同一の化学的組成物を有する結晶形ではあるが、結晶を形成する分子、原子および/またはイオンが異なった空間的配置をとることを意味する。
【0038】
本明細書で用いられる「溶媒和物」とは、結晶構造に組み込まれる溶媒の分子がさらに含まれる、分子、原子、および/またはイオンの結晶形を意味する。溶媒和物における溶媒分子は、規則的配列および/または不規則な配列で存在されうる。該溶媒和物は、化学量論量または非化学量論量のいずれかの溶媒分子を含んでもよい。例えば、非化学量論量の溶媒分子を有する溶媒和物は、溶媒和物から溶媒の部分的な損失から生じうる。
【0039】
結晶形のサンプルは、優位な量の単一の結晶形および場合により微量の1またはそれ以上の他の結晶形の存在を示す、実質的に純粋な相均質性で提供されうる。サンプル内の1より多い結晶形の存在は、粉末X線回折(PXRD)または固体状態核磁気共鳴スペクトル(SSNMR)のような技術によって決定されうる。例えば、シミュレートされたPXRDパターンで実験的に測定されたPXRDパターンの比較において、余分なピークの存在は、サンプル内に1より多い結晶形の存在を示しうる。シミュレートされたPXRDは、単結晶X線データから計算されうる。Smith, D.K.の書籍:A FORTRAN Program for Calculating X-Ray Powder Diffraction Patterns, Lawrence Radiation Laboratory, Livermore, California, UCRL-7196 (April 1963)も参照されたい。好ましくは、該結晶形はシミュレートされたPXRDパターンに存在しない余分なピークから生じる実験的に測定したPXRDパターンにおいて、全ピーク面積の10%以下、好ましくは5%以下、およびより好ましくは2%以下で示される実質的に純粋な相均質性を有する。最も好ましくは、シミュレートされたPXRDパターンに存在しない余分なピークから生じる実験的に測定されたPXRDパターンにおいて、全ピーク面積の1%以下の実質的に純粋な相均質性を有する結晶形である。
【0040】
結晶形の調製のための手順は、当該技術分野で公知である。該結晶形は、例えば適当な溶媒からの結晶化または再結晶化、昇華、融解からの成長、他の相からの固体状態への変化、超臨界液体による結晶化、およびジェットスプレーを含む多様な方法によって調製されうる。溶媒混合物から結晶形の結晶化または再結晶化のための技術には、例えば溶媒のエバポレート、溶媒混合物の温度減少、分子および/または塩の過飽和の溶媒混合物への種晶添加、溶媒混合物の凍結乾燥、および貧溶媒(対溶媒)の溶媒混合物への添加が含まれる。
【0041】
多形、製造方法、および薬物結晶の特徴を含む薬物の結晶は、Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, West Lafayette, Indiana (1999) で論じられている。
【0042】
溶媒を用いる結晶化技術に関して、溶媒の選択は、化合物の溶解性、結晶化技術、および溶媒の蒸気圧のような1またはそれ以上の要素に典型的に依存する。溶媒の組合せも使用してもよく、例えば化合物を第一の溶媒に溶解し、溶液とし、続いて貧溶媒の添加により溶液中の化合物の溶解性を減少させ、結晶を形成させてもよい。貧溶媒は、該化合物が低溶解性を有する溶媒である。結晶を調製するための適当な溶媒には、極性および非極性溶媒が含まれる。
【0043】
結晶を調製する一方法において、重硫酸アタザナビルは、適当な溶媒中で懸濁および/または攪拌してスラリーが得られ、該スラリーは溶解を促進するために加熱してもよい。本明細書で用いられる用語「スラリー」とは、重硫酸アタザナビルまたはその塩の飽和溶液を意味し、所定温度で重硫酸アタザナビルまたはその塩と溶媒との不均一混合物を与える追加量の重硫酸アタザナビルまたはその塩も含み得る。この点における適当な溶媒には、例えば、極性非プロトン性溶媒、および極性プロトン性溶媒、およびここで開示される2またはそれ以上のこれらの混合物が含まれる。
【0044】
種晶は、結晶化を促進するためにいずれの結晶化混合物に加えられてもよい。熟練した技術者に自明であるように、種晶添加は、特定の結晶形の成長を制御する手段として、または結晶生成物の粒子径分布を制御する手段として使用される。従って、必要な種晶量の計算は、例えば、J.W. Mullin および J. Nyvlt の文献:Programmed cooling of batch crystallizers, Chemical Engineering Science (1971) 26:369-377.に記載されているような入手可能な種晶の大きさおよび平均粒子生成物の所望の大きさに依存する。一般に、小サイズの種晶は、バッチにおいて結晶の成長を有効に制御することが必要である。小サイズの種晶は、より大きい結晶をふるいにかけるか、破砕するか、または微粉化することによって、あるいは溶液の微結晶化によって生成されうる。結晶の破砕または微粉化することが所望の結晶形からの結晶度におけるいずれの変化(すなわち、アモルファスまたは他の多形への変化)も生じないことを配慮されるべきである。
【0045】
冷却した混合物を減圧下で濾過し、単離した固体を冷却した再結晶溶媒のような適当な溶媒で洗浄し、窒素パージ下で乾燥し、所望の結晶形を得てもよい。単離した固体は、SSNMR、DSC、PXRDなどのような適当な分光学的または分析技術によって分析し、生成物の好ましい結晶形の形成を確認してもよい。生じた結晶形は、典型的に約70重量%以上の単離収率の量で生じるが、好ましくは結晶化法を初めから用いて重硫酸アタザナビルの重量に基づいて90重量%以上で生じる。該生成物は、必要なら生成物を塊にならないように破砕するか、または網目のふるいにかけてもよい。
【0046】
結晶形は、重硫酸アタザナビルを調製するためのプロセスの最終工程の反応系から直接調製されうる。これは、例えばプロセスの最終工程において、重硫酸アタザナビルが結晶化され得る溶媒または溶媒混合物を用いることによって達成されうる。または結晶形は、蒸留または溶媒添加技術によって得られうる。この目的のための適当な溶媒には、アルコールのようなプロトン性極性溶媒、およびケトンのような非プロトン性極性溶媒を含む、本明細書に記載されたいずれの溶媒も含まれる。
【0047】
一般的な指図を経由して、該反応混合物は、いずれの不要な不純物、無機塩なども除去するために濾過し、続いて反応溶媒または結晶化溶媒で洗浄してもよい。生じた溶液は、過剰量の溶媒または気体成分を除去するために濃縮してもよい。蒸留を用いるなら、回収された留分の最終量は、例えば容器の大きさ、攪拌能力などを含むプロセス要因に依存して変更可能である。一般的な指図によって、該反応溶液は、最初の量である約{フラクション(1/10)}まで蒸留した後、溶媒置換を実施した。標準的なプロセス技術に従って、反応の程度および生成物の重量%を決定するために、該反応液をサンプリングしアッセイしてもよい。必要なら追加の反応溶媒を加えるか、または反応濃度を最適化するために除去してもよい。好ましくは、最終濃度が典型的にスラリーを生じる点において、約50重量%に調整される。
【0048】
反応混合物を蒸留することなく反応容器に直接溶媒を加えることが好ましい。この目的のための好ましい溶媒は、溶媒交換に関連して、上記の通り結晶格子内で最終的に共存してもよいものである。最終濃度は、所望の純度、回収率などに依存して変更可能であるけれど、溶液中の遊離塩基Iの最終濃度は、約4%〜約7%が好ましい。該反応混合物は、攪拌し、続いて溶媒添加し、同時に温めてもよい。例として該反応混合物は、約70℃まで温めながら約1時間攪拌してもよい。該反応液は、好ましくは熱時濾過し、反応溶媒、加えた溶媒、またはその組合せのどれかで洗浄する。種晶は、結晶化を開始するために、いずれの結晶化溶液に加えてもよい。
【0049】
本明細書に記載の種々の形態は、当該技術分野における通常の知識を有する者に公知の種々の分析技術の使用を通じてお互いから区別されうる。かかる技術には、これに限らないが、固体状態核磁気共鳴(SSNMR)スペクトル、X線粉末回折(PXRD)、示差走査熱量測定(DSC)、および/または熱重量分析(TGA)が含まれる。
【0050】
X線回折パターンが、使用する測定条件に依存する測定誤差を含んで得られうることは当該技術分野で通常の技術を有する者に理解されている。特に、X線回折パターンにおける強度は、用いた測定条件および結晶形または多形に依存して変動することが一般的に知られる。相対強度もまた、実験条件に依存して変動し得ることはさらに理解されるべきであり、従って正確な強度のオーダーは考慮されるべきではない。従って、通常のX線回折パターンのための回折角の測定誤差は、典型的に約0.2%またはそれ以下であり、好ましくは約0.1%(下文に記載)であり、測定誤差のかかる程度は、前述の回折角に関連するとして考慮されるべきである。その結果、本発明の結晶形は、本明細書に開示される添付図に記載されるX線回折パターンと完全に同一であるX線回折パターンを提供する結晶形に限定されないことが理解されるべきである。添付図に開示されたパターンと実質的に同一であるX線回折パターンを提供するいずれの結晶形も本発明の範囲に含まれる。X線回折パターンの実質的な同一性を確認する能力は、当該技術分野で通常の技術を有する者の範囲内である。
【0051】
A型およびE3型に関して、本明細書で用いられる用語「形態、形」とは、均一な結晶構造を意味する。
【0052】
Cパターン物質に関して、本明細書で用いられる用語「パターン」とは、特徴的なX線回折パターンを意味する。
【0053】
本明細書で用いられる用語「重硫酸アタザナビル」とは、重硫酸アタザナビルならびに硫酸アタザナビルを意味する。
【0054】
重硫酸アタザナビル塩のA型結晶を調製するための本発明の方法の実施において、改良した立方結晶化技術が用いられ、その中でアタザナビル遊離塩基は、重硫酸アタザナビル塩が実質的に不溶であり、アセトン、アセトンおよびN−メチルピロリドン混合液、エタノール、エタノールおよびアセトン混合液などを含む有機溶媒で溶解され、アタザナビル遊離塩基の約6.5〜約9.7重量%、好ましくは約6.9〜約8.1重量%の範囲内のアタザナビル遊離塩基の濃度を有する溶液が得られる。
【0055】
アタザナビル遊離塩基の溶液は、約35〜約55℃、好ましくは約40〜約50℃の温度範囲で加熱し、相当量の濃硫酸(約95〜約100%H2SO4を含む)と反応し、アタザナビル遊離塩基の全重量の約15%以下、好ましくは約5〜約12%以下、より好ましくは約8〜約10%と反応する。従って、アタザナビル遊離塩基の出発溶液は、用いる硫酸の全量の約15%以下、好ましくは約5〜約12%と最初に反応される。反応の間、該反応混合物は、約35〜約55℃、好ましくは約40〜約50℃の範囲内で維持される。
【0056】
該反応を約12〜約60分間、好ましくは約15〜約30分間継続させる。
【0057】
該反応混合物は、反応混合物を約35〜約55℃、好ましくは約40〜約50℃の温度範囲を維持しながら、反応混合物に残存するアタザナビル遊離塩基の重量に基づいて、約0.1〜約80重量%、好ましくは約3〜約8重量%の範囲内の量で相当量の種晶を用いる重硫酸アタザナビルのA型結晶で種晶添加される。
【0058】
該反応は、結晶化を開始するまで継続させる。その後硫酸を下記のような三次方程式に従う増加速度で多段階にて加え、重硫酸アタザナビルを形成し、乾燥してA型結晶が生じる。
【0059】
形成される重硫酸アタザナビル塩の結晶粒子径および多形は、硫酸の添加速度に依存し、結晶化速度を決定する。改良「立方」結晶化技術(三次方程式に従う増加速度で加えた酸)は、定常添加速度での結晶化よりも、より狭い粒子径範囲およびより僅かな細かさを伴う相対的により大きく、より良く定義された重硫酸アタザナビルの結晶を与えることが見出されている。ゆっくりとした最初の酸の流速は、二次的な核形成にわたる結晶成長に有利にはたらくことが示されている。従って、粒子径と共に表面積が増加するにつれて、種晶床は、二次的な核形成の誘発を含まない酸の流速を増加することを受け入れることができる。ゆっくりとした最初の添加速度は、結晶がより大きく成長するための時間を許容し、平均サイズを増加させる。該立方結晶化は、あまり圧縮性のない濾過ケークを与え、該ケークは、有効なケークの脱液および洗浄、ならびに定常添加速度で結晶化する生成物よりも固くない塊を有し、より容易に乾燥する生成物を与えることを促進する。
【0060】
用いた立方結晶化方法は、Mullinの書籍:Crystallization, 3rd Ed., 1993, Butterworth-Heineman, Pubs.に由来する温度制御した結晶化であり、下記の単純化した式で定義される:
【数1】
【0061】
重硫酸アタザナビルの結晶化は、硫酸の添加速度によって制御されるので、温度変数は、式(1)の酸の量で置き換えられる。この式において最小量を表す変数は、除去される。
【数2】
式(2)は、「三次方程式」と称する。
【0062】
この式を用いて結晶化速度を制御することによって、核形成はシステムが一定の低レベルの過飽和を維持するので許容限界内で制御される。
【0063】
A型結晶は、図1および2のそれぞれで示される粉末X線回折パターンおよび結晶構造で同定される。
【0064】
重硫酸アタザナビルのA型結晶またはCパターン物質、および上記のように調製されるE3型は、最終の重硫酸アタザナビルであって、患者に投与するための製剤として使用され得る。
【0065】
本発明の方法に従って、Cパターン物質は、A型結晶を水に曝露し、続いて乾燥することによって調製されうる。
【0066】
本発明に従う別の方法において、Cパターン物質は、A型結晶を約95%RH以上、好ましくは約95〜約100%RH(水蒸気)の高い相対湿度に、少なくとも24時間、好ましくは約24〜約48時間曝露することによって形成されうる。
【0067】
本発明の別の態様において、Cパターン物質は、重硫酸アタザナビルのA型を湿式造粒することによって、重硫酸アタザナビルの造粒物を生じ、次いで該造粒物を乾燥することによって調製される。
【0068】
湿式造粒法の実施において、重硫酸アタザナビルは、水中で造粒し、約40〜約80℃、好ましくは約50〜約60℃の温度範囲で乾燥する。乾燥工程は、少なくとも約2時間、約20時間まで、好ましくは約8〜約10時間実施される。
【0069】
Cパターン物質はまた、例えば1またはそれ以上の充填剤、好ましくはラクトース、1またはそれ以上の崩壊剤、好ましくはクロスポビドンのような通常の医薬賦形剤の存在下で重硫酸アタザナビルのA型を湿式造粒し、上記の通り乾燥し、賦形剤との混合物のCパターン物質を形成することで形成してもよい。
【0070】
それはCパターン物質、A型またはE3型、好ましくはCパターン物質であり、下文に記載されるウイルスによって引き起こされる疾病の治療において、投与するために製剤化される。
【0071】
Cパターン物質は図3に示される、その粉末X線回折パターンによって特徴付けられる。
【0072】
E3型は、アタザナビル遊離塩基をエタノール中でスラリーにし、該スラリーを酸:遊離塩基が約1:1〜約1.1:1の範囲のモル比を用いて濃硫酸と処理し、約30〜約40℃で生じた溶液を加熱し、該溶液をエタノールで湿らせた硫酸アタザナビルのE3結晶で種晶添加し、該混合物をヘプタン(またはヘキサンまたはトルエンのような他の溶媒)で処理し、濾過し、乾燥して重硫酸アタザナビルのE3型(トリエタノール溶媒和物)を得ることにより調製される。
【0073】
種晶添加工程は、E3結晶を形成する相当量の種晶、例えば、重硫酸アタザナビルE−3種晶:遊離塩基が約0.02:1〜約0.04:1の範囲内のモル比を用いる。
【0074】
E3型は、図7に示される粉末X線回折パターンおよび図6に示される結晶構造によって同定される。
【0075】
本発明に従って、その遊離塩基の形態におけるアタザナビルは、構造:
【化13】
[式中、PGは、t−ブチルオキシカルボニル(Boc)またはトリフルオロアセチル、好ましくはBocのような保護基を表す]
の保護されたトリアミン塩溶液を、塩化メチレン、テトラヒドロフラン、またはメタノールのような有機溶媒、好ましくは塩化メチレンの存在下、約25〜約50℃、好ましくは約30〜約40℃の温度範囲で、酸、好ましくは塩酸(Bocが用いられる場合)、または塩基(トリフルオロアセチルが用いられる)と処理し、トリアミン酸塩、好ましくは構造:
【化14】
の塩酸塩を形成することで、および
トリアミン酸塩を単離することなく、該トリアミン酸塩を構造:
【化15】
の酸の活性エステル体、好ましくは構造:
【化16】
の活性エステル体とK2HPO4、ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸ナトリウム、または炭酸カリウムのような塩基、好ましくはK2HPO4の存在下、塩化メチレン、酢酸エチルおよび酢酸ブチルの混合液、アセトニトリルまたは酢酸エチルのような有機溶媒、好ましくは塩化メチレンの存在下で、約25〜約50℃の温度範囲内好ましくは約30〜約40℃にて反応し、アタザナビル遊離塩基を形成することにより調製される。
【0076】
該保護されたトリアミンの出発物質をN−(tert−ブチルオキシカルボニル)−2(S)−アミノ−1−フェニル−3(R)−3,4−エポキシブタンのようなエポキシド体:
【化17】
[式中、PGは、好ましくはBoc基である]
をヒドラジンカーバメート体:
【化18】
[PGは、好ましくはBoc基である]
と、イソプロピルアルコールまたはエタノールまたはブタノールのような他のアルコールの存在下で反応することにより調製される。
【0077】
重硫酸アタザナビルは、レトロウイルスプロテアーゼ、とりわけHIV−1またはHIV−II・gagプロテアーゼのようなレトロウイルスアスパラギン酸プロテアーゼ阻害に応答する疾病、例えばAIDSまたはその予備段階のようなレトロウイルスの疾病の治療または予防のために、温血動物、とりわけヒトに投与するのに有用である。
【0078】
重硫酸アタザナビル、とりわけCパターン物質、A型またはE3型、好ましくはCパターン物質またはA型は、ウイルス、とりわけレトロウイルスによって引き起こされる疾病、とりわけAIDSまたはその予備段階の治療方法に使用され、治療上の有効量のCパターン物質、A型またはE3型の重硫酸アタザナビルが上述の疾病の一つ、とりわけAIDSまたはその予備段階でかかる治療が必要であるとりわけ温血動物、例えばヒトに該疾病の治療に有効な投与量で投与される。温血動物、例えばおよそ70kg体重のヒトに投与される好ましい投与量は、1日あたりヒトに約3mg〜約1.5g、好ましくは約10mg〜約1.25g、例えば約50mg〜約600mgであり、好ましくは、例えば同量で1〜4回の単回投与に分けられる。通常、子供は大人の投与量の半分を受ける。好ましくは経口投与である。
【0079】
Cパターン物質、A型またはE3型の重硫酸アタザナビルは、上記の医薬用途のために使用される。経口投与用のCパターン物質またはA型もしくはE3型を含む適当な組成物には、錠剤、粉剤、カプセル剤、およびエリキシルが含まれる。約10〜600mgの活性成分は、許容される医薬技術によって必要とされる単位投与量において、生理的に許容されるベヒクル、担体、賦形剤、結合剤、保存料、安定化剤、香料などと配合される。
【0080】
経口投与用の医薬組成物は、必要なら生じた混合物を造粒し、該混合物を処理し、所望または必要なら適当な賦形剤を、錠剤、ドラジェ(dragee)コア、カプセル剤または経口使用のための粉剤に添加後、活性成分を固体担体と組み合わせることにより得ることができる。活性成分が測定される量で拡散または放出させる可塑性担体に導入することも可能である。
【0081】
充填剤または増量剤は、組成物の約0〜約95重量%、好ましくは約10〜約85重量%の範囲内の量で本発明の医薬組成物中に存在する。本明細書で使用するために適当な充填剤または増量剤の具体例には、これに限らないが、微結晶セルロースまたは木質セルロースのようなセルロース誘導体、ラクトース、ショ糖、デンプン、プレゼラチン化したデンプン、デキストロース、マンニトール、フルクトース、キシリトール、ソルビトール、トウモロコシデンプン、変性トウモロコシデンプン、炭酸カルシウム、リン酸カルシウム、リン酸二カルシウム、硫酸カルシウムのような無機塩、デキストリン/デキストレート、マルトデキストリン、圧縮性糖類、および他の公知の充填剤または増量剤、および/または2またはそれ以上の混合物が含まれ、好ましくはラクトースがよい。
【0082】
結合剤は、組成物の約0〜約20重量%、好ましくは約1〜約10重量%の範囲内の量で本発明の医薬組成物中に適宜存在する。本明細書の使用に適した結合剤の具体例には、これに限らないが、ヒドロキシプロピルセルロース、トウモロコシデンプン、プレゼラチン化したデンプン、変性トウモロコシデンプン、ポリビニルピロリドン(PVP)(約5000〜約80000の範囲の分子量、好ましくは約40000である)、ヒドロキシプロピルメチルセルロース(HPMC)、ラクトース、アカシアゴム、エチルセルロース、酢酸セルロース、並びにカルナウバロウ、パラフィン、鯨ロウ、ポリエチレンまたは微結晶性ワックスのようなロウ性結合剤、ならびに他の通常の結合剤および/または2またはそれ以上のそれらの混合物が含まれ、好ましくはヒドロキシプロピルセルロースがよい。
【0083】
崩壊剤は、組成物の約0〜約20重量%、好ましくは約0.25〜約15重量%の範囲内の量で、本発明の医薬組成物中に適宜存在する。本明細書の使用に適した崩壊剤の具体例には、これに限らないが、クロスカルメロースナトリウム、クロスポビドン、ジャガイモデンプン、プレゼラチン化したデンプン、トウモロコシデンプン、グリコール酸デンプンナトリウム、微結晶性セルロース、または他の公知の崩壊剤が含まれ、好ましくはクロスカルメロースナトリウムがよい。
【0084】
滑沢剤は、組成物の約0.1〜約4重量%、好ましくは約0.2〜約2重量%の範囲内の量で、本発明の医薬組成物に適宜存在する。本明細書の使用に適した打錠滑沢剤の具体例には、これに限らないが、ステアリン酸マグネシウム、ステアリン酸亜鉛、ステアリン酸カルシウム、タルク、カルナウバロウ、ステアリン酸、パルミチン酸、ステアリルフマル酸ナトリウム、または水素化された植物油および脂肪、または他の公知の打錠滑沢剤、および/または2またはそれ以上のそれらの混合物が含まれ、好ましくはステアリン酸マグネシウムがよい。
【0085】
カプセル剤は、グリセロールまたはソルビトールのようなゼラチンおよび可塑剤で製造された硬ゼラチンカプセルおよびソフトシールカプセルである。該硬ゼラチンカプセルは、例えばラクトースのような充填剤、デンプン、クロスポビドンのような結合剤および/またはタルクまたはステアリン酸マグネシウムのような流動促進剤、および必要なら安定化剤と一緒に顆粒形態の活性成分を含んでいてもよい。ソフトゼラチンカプセルの活性成分は、好ましくは脂肪油、パラフィンオイルまたは液体ポリエチレングリコールのような適当な油状賦形剤に溶解または懸濁され、安定化剤および/または抗菌剤を加えることが同様に可能である。
【0086】
以下の実施例は、本発明の好適な態様を示す。
【0087】
実施例1
1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4−(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・重硫酸塩(A型)(重硫酸アタザナビルA型)
【化19】
【0088】
(1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス[tert−ブチルオキシカルボニル)アミノ]−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・3HCl(トリアミン・3HCl塩))
メカニカルスターラー、窒素吸気口および温度プローブを取り付けた1000mLの3頸丸底フラスコに、保護トリアミン体、1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス[tert−ブチルオキシカルボニル)アミノ]−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン:
【化20】
(100g,0.178mol)、およびCH2Cl2(500mL;5mL/gの保護トリアミン体)(Z. Xuらの文献:Process Research and Development for an Efficient Synthesis of the HIV Protease Inhibitor BMS-232,632, Organic Process Research and Development, 6, 323-328 (2002)に記載されたとおり製造した)を加え、生じたスラリーを約5〜約22℃の温度を維持しながら攪拌した。
【0089】
濃塩酸(68mL,0.82mol、4.6当量)を反応混合物の温度が5〜30℃を維持するような速度で該反応混合物に加えた。該反応混合物を30〜40℃に加熱し、反応がHPLCアッセイによって完了したと判断するまで攪拌した。
【0090】
水(70〜210mL、0.7〜2.1mL/gの保護したトリアミン体の投入量)を反応混合物に加え、該反応混合物を15分間攪拌し、層を分離させた。上層の生成物(トリアミン・3HCl塩)リッチな水性油状物を滴下漏斗に移した。
【0091】
B.
【化21】
(N−メトキシカルボニル−L−tert−ロイシン:
【化22】
の活性エステル体)
メカニカルスターラー、滴下ロート、窒素吸気口、および温度プローブを取り付けた3000mLの3頸丸底フラスコに、N−メトキシカルボニル−L−tert−ロイシン(77.2g,0.408mol,2.30当量)、1−ヒドロキシベンゾトリアゾール(HOBT)(60.8g,0.450mol,2.53当量)、およびN−エチル N’−ジメチルアミノプロピルカルボジイミド(EDAC)(82.0g,0.430mol,2.42当量)、続いて、CH2Cl2(880mL;8.8mL/gの保護したトリアミン体の投入量)を加え、該活性エステル体の形成がHPLCで完了したと判断するまで、該混合物を周囲温度(18〜25℃)で攪拌した。
【0092】
C.1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン(アタザナビル遊離塩基)
無水第二リン酸カリウム(K2HPO4;226g,1.30mol,保護トリアミン体に対して7.30当量)を1130mLの水(11.3mL/gの保護アミン体;5mL/gのK2HPO4)に溶解した。
【0093】
該K2HPO4溶液をパートBで調製した活性エステル体溶液に加えた。攪拌した活性エステル体/K2HPO4水溶液混合物に、攪拌および5〜20℃の容器温度を維持しながら1.5〜2.0時間以上かけてパートAの塩酸塩水溶液をゆっくり加えた。
【0094】
パートAの塩酸塩溶液の添加が完了した後、該反応混合物(カップリング反応)を30〜40℃に加熱し、カップリング反応がHPLCアッセイによって完了したと判断するまで攪拌した。
【0095】
該カップリング混合物を15〜20℃まで冷却し、下層の生成物リッチな有機層を下層から分離し、水層を廃棄した。
【0096】
該生成物リッチな有機層を1MのNaH2PO4(880mL;pH=1.5;8.8mL/gの保護アミン体投入量;保護アミン体に対して5モル当量)で洗浄し、該層を分離させ、不要な水層を除去した。
【0097】
生成物リッチな有機層のHPLCアッセイで、それぞれ活性エステル体が0.3I.I.以下を示すまで、洗浄した生成物リッチな有機層を0.5NのNaOH(800mL;8mL/gの保護アミン投入量)と共に攪拌した。該層を分離させて、不要な水層を除去した。
【0098】
該生成物リッチな有機層を5%NaH2PO4(450mL,4.5mL/gの保護トリアミン体投入量;pH=4.3)で洗浄し、該層を分離させて、不要な水層を除去した。
【0099】
該生成物リッチな有機層を10w/v%のNaCl(475mL,4.75mL/gの保護トリアミン体投入量)で洗浄し、不要な水層を除去した。
【0100】
標記の遊離塩基溶液の濃度は、製造過程中の計算収率が95〜100mol%である、120〜150mg/mLであった。
【0101】
D.CH2Cl2からアセトン/N−メチルピロリドンへの溶媒交換
メカニカルスターラー、温度プローブ、および蒸留冷却管を取り付けた3000mLの3頸丸底フラスコ中のパートCの遊離塩基リッチな溶液に、N−メチルピロリドン(148mL;製造過程の定量アッセイに基づく1.25mL/gのパートCの遊離塩基)を加えた。該溶液を70℃またはそれ以下のジャケット温度を用いて、約360mL(2.5〜3.5mL/gのパートCの遊離塩基)まで濃縮し;500mLのアセトン(4〜5mL/gのパートCの遊離塩基)を濃縮液に加え、該混合液を約400mLまたはそれ以下の量まで蒸留した。
【0102】
CH2Cl2レベルを示す製造過程中のアッセイが目的の終点に達するまで、アセトン添加および蒸留を繰り返した。結晶化量において、生成物リッチな有機溶液におけるCH2Cl2量が0.77v/v%であった。アセトンを全量が16mL/gの遊離塩基に達するように濃縮した遊離塩基溶液に加えた。浴温は、遊離塩基の結晶化を防ぐために40〜50℃に維持した。温度を40〜50℃に維持しながら、該溶液を10ミクロンまたはそれより細かいフィルターでポリッシュフィルターにて濾過した。該ポリッシュフィルターをアセトン(125mL,1.0mL/gの遊離塩基)でリンスし、該リンス液を遊離塩基リッチなアセトン/N−メチルピロリドン溶液に加え、次工程に使用した。
【0103】
E.1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・重硫酸塩
40〜50℃の温度を維持しながら、表面添加によって濃硫酸(19g,1.10当量)の総添加量の約10%(2g)をパートDの遊離塩基のアセトン/N−メチルピロリドン溶液に加えた。
【0104】
該反応混合物を重硫酸塩の5.0wt%(計算した遊離塩基溶液に対して)を種晶添加した。重硫酸塩が、この時間中不透明度が増加する混合物から明らかなように、結晶化を開始した時間において、種晶を加えた混合物を40〜50℃で少なくとも30分間攪拌した。
【0105】
残る硫酸(17.8g)を40〜50℃の温度を保ちながら、三次方程式によって定義される下記のプロトコルに従って、五段階で約5時間かけて添加した。
【0106】
各添加段階の速度は、上記の三次方程式に従って決定し、以下の表に示される。
表1
【0107】
H2SO4の添加が完了した後、スラリーを攪拌しながら、少なくとも1時間、20〜25℃まで冷却した。該スラリーを少なくとも1時間、20〜25℃で攪拌した。該重硫酸塩を濾過し、母液は、完全に移行する必要があるため回収した。濾過ケークをアセトン(5〜10mL/gの遊離塩基;1200mLのアセトン)で洗浄した。該重硫酸塩をLOD<1%までNMT55℃にて減圧下で乾燥し、結晶物質を得る。
【0108】
該結晶生成物をPXRD、DSCおよびTGAパターンおよびSSNMRスペクトルによって分析し、標記の重硫酸塩(図1〜5を参照されたい)のA型結晶(無溶媒和物)であることがわかった。
【表1】
【表2】
【表3】
【表4】
異方性の精製された原子は、(4/3)*[a2*B(1,1)+b2*B(2,2)+c2*B(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)]として定義される、等価等方性原子変位パラメーターの形態で与えられる。
【0109】
A型は、図3に示される約165.6℃〜約200.9℃の範囲内で、典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられる。
【0110】
A型はまた、約100〜150℃まででごく僅かな減量を有する熱重量分析曲線によっても特徴付けられる。
【0111】
H2SO4が上記の三次方程式に従う増加速度で加えられる立方結晶化によって生じる結晶は、一定の添加速度による結晶化を用いて得られた結晶より相対的に大きく、明確で、狭い粒子径範囲を有し、ほとんど細かくなかった。
【0112】
立方結晶化技術を用いて得られた濾過ケークは、一定の添加速度による結晶化を用いて得られたものより圧縮性が小さく、有効なケークの脱液および洗浄に役立ち、均一生成物を生じた。
【表5】
【0113】
実施例2
重硫酸アタザナビル−Cパターン物質
方法A:
重硫酸アタザナビルのA型結晶(実施例1に記載されるように調製した)(25.33g)を200mLの水に懸濁し、該混合物を機械的に攪拌し、厚いゲルを生じ乾燥した。
【0114】
乾燥した混合物をスパチュラですりつぶし、Cパターン物質を生じた。Cパターン物質の粉末X線回折パターンは、図6に示す。
【0115】
方法B:
重硫酸アタザナビルのA型結晶は、適当な混合造粒機において、充分な量の水(約40%w/w)を用いて湿式造粒した。湿塊をオーブンで乾燥した。該生成物を適当なふるいを用いて、等しい大きさにした。生じた生成物のX線回折パターンは、図6に示されるCパターン物質と一致する。
【0116】
Cパターンは、約76.7〜約96.6℃および約156.8〜約165.9℃の範囲内で典型的な吸熱を有する、図7に示される示差走査熱量サーモグラムによって特徴付けられる。
【0117】
Cパターンはまた、図8に示される約125℃で約2.4%および約190℃で約4.4%の重量減量を有する熱重量分析曲線によっても特徴付けられる。
【0118】
実施例3
重硫酸アタザナビル−E3型(トリエタノール溶媒和物)
メカニカルスターラー、温度プローブ、および圧力を等しくする液体滴下ロートを取り付けた100mLの3頸丸底フラスコにおいて、アタザナビル遊離塩基(実施例1、パートCに記載されるように調製)(3.0g,4.26mmol)を無水の200プルーフ(proof)のエタノール(20.25mL,6.75mL/gの遊離塩基)中でスラリーにした。
【0119】
濃H2SO4(0.25mL,0.46g,4.69mmol,1.1当量)を20〜25℃で維持したアタザナビル遊離塩基のスラリーに加えた。生じた溶液(0.2〜1.0%水分量のKF)をポリッシュフィルター(Whatman ♯1ペーパー)で濾過し、フィルターを2.25mLの無水エタノールでリンスし、該リンス液を濾過液に加えた。該溶液を37℃に加熱し、E3型結晶から(E3型結晶を周囲温度に曝すことによって)生じる10mgのアモルファスの重硫酸アタザナビルで種晶添加し、該混合物を15分間攪拌した。ヘプタン(380mL,8.25mL/gの遊離塩基)を1時間以上かけて加えた。生じた結晶化混合物を15〜25℃で8時間攪拌した。結晶化した重硫酸アタザナビルをブフナー漏斗で濾過した。該生成物ケークを184mL(4mL/gの遊離塩基)のエタノール:ヘプタン(1:1)で洗浄した。該生成物ケークを、46mL(1mL/gの遊離塩基)のヘプタンで洗浄した。生じた生成物をLOD=0.97%となるまで、40〜50℃で減圧下にて乾燥した。生成物の収量は、HPLC HI=100.0(図9および10を参照されたい)である重硫酸アタザナビルE3型(トリエタノール溶媒和物)の47.7g(0.0594mol,74.3mol%)であった。
【表6】
【表7】
【表8】
異方性の精密な原子は、(4/3)*[a2*B(1,1)+b2*B(2,2)+c2*B(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)]として定義される、等価等方性原子変位パラメーターの形態で与えられる。
【0120】
E3型は、図11に示される、約89.4〜約96.6の範囲内で典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられる。
【0121】
E3型はまた、図11に示される約150℃で約14.7%の重量減量を有する熱重量分析曲線によっても特徴付けられる。
【0122】
実施例4
下記の組成物を有するC形の重硫酸アタザナビルカプセル製剤は、下記の通り調製された。
【表9】
【0123】
重硫酸アタザナビルの保存顆粒は、下記のように調製され、Cパターン物質が形成された。
【0124】
A型の重硫酸アタザナビル、含水ラクトース、およびクロスポビドンの一部(クロスポビドンの全量の3%)をプラネタリーミキサーで混合した。生じた混合物をA型からCパターン物質に変換するために精製水で湿式造粒した。湿式造粒を棚型乾燥機で乾燥し、ハンマーミルを用いて等しい大きさにした。残りのクロスポビドンを粉砕した造粒に加え、該混合物をPK V−混合機で混合した。ステアリン酸マグネシウムを加え、該混合物を実質的に均一な保存顆粒を形成するまで混合した。
【0125】
適当な重量の保存顆粒をカプセルに充填し、重硫酸アタザナビルを含む、50mg、100mgおよび200mgカプセルを生成した。
【0126】
実施例5
下記の組成物を有する経口用途製剤のための重硫酸アタザナビルのA型物質の粉末は、下記のように調製される。
【0127】
重硫酸アタザナビルA型は、適当な混合機でアスパルテーム、オレンジバニラフレーバーおよびショ糖と混合する。該混合物はハンマーミルを用いて粉砕し、続いて、二次混合操作を行って均一な混合物を得る。生成物は、高密度のポリエチレン瓶に充填する。
【図面の簡単な説明】
【0128】
【図1】図1は、A型の計算した(シミュレートした)(22℃)および実測した(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図2】図2は、A型の結晶構造を示し;
【図3】図3は、A型の示差走査熱量測定(DSC)サーモグラムを示し;
【図4】図4は、A型の熱重量分析曲線(TGA)を示し;
【図5】図5は、A型の固体状態C−13 NMRを示し;
【図6】図6は、Cパターンの実測された(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図7】図7は、Cパターンの示差走査熱量サーモグラムを示し;
【図8】図8は、Cパターンの熱重量分析曲線を示し;
【図9】図9は、E3型の計算された(シミュレートした)(22℃)および実測された(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図10】図10は、E3型の結晶構造を示し;および
【図11】図11は、E3型の示差走査熱量測定(DSC)サーモグラム、およびE3型の熱重量分析曲線を示す。
【技術分野】
【0001】
本出願は、2004年5月4日に出願した米国仮出願第60/568,043号、および2004年9月7日に出願した第60/607,533号からの優先権を利用し、その開示は参照によって本明細書に引用される。本発明は、HIVプロテアーゼ阻害剤である重硫酸アタザナビルおよびその新規形態の製造方法に関する。
【背景技術】
【0002】
Fasslerらの米国特許第5,849,911号は、以下の構造:
【化1】
[式中、
R1は、低級アルコキシカルボニルであり、
R2は、二級または三級低級アルキルまたは低級アルキルチオール−低級アルキルであり、
R3は、1またはそれ以上の低級アルコキシ基、またはC4−C8シクロアルキルによって非置換または置換されるフェニルであり、
R4は、フェニルまたはシクロヘキシルであり、それぞれが環の炭素原子を経由して結合し、5〜8個の環の原子を有し、窒素、酸素、硫黄、スルフィニル(−SO−)およびスルホニル(−SO2−)から選択される1〜4個のヘテロ原子を含み、低級アルキルまたはフェニル−低級アルキルによって非置換または置換される不飽和ヘテロシクリルによってその4位で置換され、
R5は、R2とは独立して、R2に記載される意味の一つを有し、および
R6は、R1とは独立して、低級アルコキシカルボニル、またはその塩であるが、但し、少なくとも一つの塩を形成する基が存在する]および種々のその医薬的に許容される酸付加塩を有するアザペプチドHIVプロテアーゼ阻害剤(アタザナビルを含む)のシリーズを開示する。
【0003】
R1およびR6、ならびにR2およびR5が、それぞれ二つの同一基であり、構造(a)
【化2】
のジアミノ化合物が構造(b)
【化3】
[式中、R1’およびR2’は、それぞれR1およびR6、ならびにR2およびR5で定義される]
の酸またはその活性な酸誘導体と縮合される化合物の製造を含む、アザペプチドを製造するためのいくつかの方法が提供される。
【0004】
上記の方法を用いるアタザナビルの生成において、構造式:
【化4】
を有するジアミノ化合物(a)は、イソプロピルアルコールの存在下で、エポキシド体:
【化5】
とヒドラジノカーバメート体:
【化6】
とカップリングして、保護ジアミン体:
【化7】
を形成し、テトラヒドロフランのような溶媒の存在下で、塩酸と処理し、ジアミン体(a):
【化8】
を形成することによって製造される。該ジアミン体を単離し、O−(1,2−ジヒドロ−2−オキソ−1−ピリジル)−N,N,N1,N1−テトラメチルウロニウムテトラフルオロボレート(TPTU)のようなカップリング剤を用いて、酸(b)
【化9】
または、その活性エステル体と反応させる、次のカップリング工程に用いる。
【0005】
該ジアミン遊離塩基は不安定であり、それ故、アタザナビル遊離塩基の製造において、使用するためには望ましくないことがわかっている。
【0006】
Singhらの米国特許第6,087,383号は、該構造:
【化10】
(また、重硫酸アタザナビルまたは硫酸アタザナビルとも呼ばれる)を有するアタザナビルとして知られるアザペプチドHIVプロテアーゼ阻害剤の重硫酸塩を開示する。
【0007】
Singhらの実施例3は、水和化した吸湿性で結晶性形状であるII型結晶の形態、および無水/脱溶媒和結晶形であることが明らかであるI型結晶の形態の重硫酸アタザナビルの製造を記載する。
【0008】
(発明の簡単な説明)
本発明によれば、Cパターン物質およびE3型を含む、重硫酸アタザナビルの新規形態が提供される。Cパターン物質が好ましい。
【0009】
さらに本発明によれば、A型結晶(バルク薬)(Singhらの米国特許第6,087,383号の実施例3でI型結晶と呼ばれる)の形態の、重硫酸アタザナビルを製造するための方法を提供する。本発明の方法によって製造されるA型結晶は、所望の実質的に整った粒子径分布および実質的に整った平均粒子径を有し、医薬製剤を調製するための種々の賦形剤と製剤化される部分的な結晶物質であるCパターン物質への変換に用いられる。
【0010】
重硫酸アタザナビル塩のA型結晶を調製するための本発明の方法は、硫酸が三次方程式(以下に記載)に従う増加速度で加える改良立方結晶化技術を用いて、アタザナビル遊離塩基の有機溶媒溶液(該重硫酸アタザナビル塩が実質的に不溶である)をアタザナビル遊離塩基の約15%重量以下、好ましくは約12重量%以下で反応するための量の濃硫酸の最初の一部と反応させ、重硫酸アタザナビルのA型結晶の種晶を反応混合物に加え、重硫酸アタザナビルの結晶が形成するようにA型結晶の形成を行うために三次方程式による増加速度で多段階にて、追加の濃硫酸を加える工程を含む。
【0011】
さらに本発明によれば、重硫酸アタザナビルから生じ、重硫酸アタザナビルを含むアタザナビルの形態を製造するための方法が提供され、それは、Cパターン物質と称される。Cパターンは、水中でA型結晶を懸濁し、乾燥することで生じ得る。または、Cパターン物質は、約95%RH(水蒸気)より高い相対湿度で少なくとも24時間、A型結晶を付すことによって形成されうる。Cパターン物質はまた、重硫酸アタザナビル、または重硫酸アタザナビルと賦形剤の組合せを湿式造粒し、湿式造粒物を乾燥することによっても形成されうる。
【0012】
好ましい態様において、A型結晶は、例えばラクトースのような1またはそれ以上の充填剤、クロスポビドンのような1またはそれ以上の崩壊剤のような製剤用賦形剤と混合し、湿式造粒して賦形剤との混合においてCパターン物質を直接形成する。
【0013】
さらに本発明によれば、重硫酸アタザナビルの新規形態、すなわち重硫酸アタザナビルのトリエタノレート溶媒和物の高度な結晶形である、E3形が提供される。
【0014】
E3型は、エタノール中でアタザナビル遊離塩基をスラリーにし、該スラリーを濃硫酸で処理し、生じた溶液を加熱し、エタノールで湿ったE3結晶で種晶添加し、該混合物をヘプタン(またはトルエンまたはヘキサンのような他の溶媒)で処理し、濾過し、乾燥することにより調製される。
【0015】
さらにまた本発明によれば、重硫酸アタザナビルのA型結晶を調製するための方法が提供され、その方法は、構造:
【化11】
のトリアミン塩(好ましくはHCl(3モル)塩)を調製し、該トリアミン塩を単離することなく、塩基および有機溶媒の存在下、活性エステル体、好ましくは構造式:
【化12】
の活性エステル体と反応させ、アタザナビル遊離塩基を形成し、単離することなく、本明細書で述べる改良立方結晶化技術によって重硫酸アタザナビルへと変換される工程を含む。
【0016】
さらに本発明によれば、A型結晶またはCパターン物質である重硫酸アタザナビル、およびその医薬的に許容される担体を含む新規な重硫酸アタザナビルの組成物を提供する。該医薬的に許容される担体は、充填剤、結合剤、崩壊剤、滑沢剤、および他の通常の賦形剤を含んでもよい。
【0017】
本発明による重硫酸アタザナビルの種々の形態は、種々の技術、当該技術分野における通常の技術を有する者に周知である操作を用いて測定されうる。該形態は、固定された分析温度で形態の単結晶の単位格子測定に基づく、単結晶X線回折を用いて測定され、区別される。単位格子の詳細な説明は、Stout & Jensen, X-Ray Structure Determination: A Practical Guide, Macmillan Co., New York (1968), Chapter 3 内で提供され、それは参照によって本明細書に引用される。または、結晶格子内の空間的関係における独特の原子配列は、観察された原子分率座標によって測定されうる。結晶構造を測定する他の手段は、実験的または観察された回折プロフィールが純粋な粉末物質を表す模擬プロフィールと比較する粉末X線回折分析によって行われ、両方のランは同一の分析温度で、一連の2θ値として特徴付けられる対象の形態のために測定する。
【0018】
固体状態核磁気共鳴(SSNMR)、示差走査熱量測定(DSC)および熱重量分析(TGA)のような形態を測定する他の手段が使用されてもよい。これらのパラメーターはまた、対象の形態を測定するために組み合わせて使用されてもよい。
【0019】
A型結晶は、下記:
[格子サイズ:
a=9.86(5)Å
b=29.245(6)Å
c=8.327(2)Å
α=93.56(2)°
β=114.77(3)°
γ=80.49(3)°
空間群1
分子/非対称単位2]
と実質的に同等である単位格子パラメーターによって特徴付けられ、結晶形態が約+22℃で存在する。
【0020】
A型は、実質的に表3に記載される原子分率座標および実質的に図2で示される結晶構造によって特徴付けられうる。
【0021】
A型は、実質的に図1で示されるシミュレートされたおよび実測された粉末X線回折パターンによって特徴付けられうる。
【0022】
A型は、実質的に図3で示される約165.6℃でのピーク発現と共に吸熱を有する示差走査熱量測定(DSC)サーモグラムによって特徴付けられうる。
【0023】
A型は、実質的に図4で示される約100℃〜150℃までにごく僅かな減量を有する熱重量分析(TGA)曲線によって特徴付けられうる。
【0024】
A型は、実質的に表4で示される固体状態NMR(SSNMR)の化学シフトおよび実質的に図5で示されるスペクトルによって特徴付けられうる。
【0025】
A型は、実質的に表5で示される原子分率座標によって特徴付けられうる。
【0026】
A型塩は、25℃で25〜75%RHの範囲において、約0.1%の重量増加と共に吸湿等温線によって特徴付けられうる。
【0027】
本発明の一態様において、Cパターンは、図5で示される観察された粉末X線回折パターンによって実質的に特徴付けられうる。
【0028】
本発明の異なった態様において、Cパターンは、約76.7〜約96.6℃および約156.8〜約165.9℃の範囲内で典型的に吸熱を有する、図7で示される示差走査熱量サーモグラムによって実質的に特徴付けられうる。
【0029】
本発明の異なった態様において、Cパターンは、実質的に図8で示される約125℃で約2.4%の重量減量、および約190℃までで約4.4%の減量を有する熱重量分析曲線によって特徴付けられうる。
【0030】
本発明によれば、E3型は、下記:
[a=10.749Å
b=13.450(4)Å
c=9.250(2)Å
α=98.33(2)°
β=95.92(3)°
γ=102.82(3)°
空間群P1
分子/非対称単位1]
と実質的に同等である表5に示される結晶学的データによって特徴付けられ、結晶形が約−23℃で存在する。
【0031】
本発明の異なった態様において、E3型は、実質的に表6で記載される原子分率座標によって特徴付けられうる。
【0032】
本発明の異なった態様において、E3型は、実質的に図9で示されるシミュレートされたおよび実測された粉末X線回折パターンによって特徴付けられうる。
【0033】
本発明の異なった態様において、E3型は、実質的に図11で示される約89.4〜約96.6℃の範囲内で典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられうる。
【0034】
本発明の異なった態様において、E3型は、実質的に表8で示される約150℃で約14.7%の重量減量を有する熱重量分析曲線によって特徴付けられうる。
【0035】
本発明の異なった態様において、E3型は、実質的に図10で示される結晶構造によって特徴付けられうる。
【0036】
(発明の詳細な説明)
本発明は、少なくとも一部分で重硫酸アタザナビルの形態、すなわち新規材料として、特に医薬的に許容される形態でE3型およびCパターンを提供する。本明細書で用いられる用語「医薬的に許容される」とは、合理的な利点/リスク比に相応して過剰毒性、刺激作用、アレルギー反応、または他の問題のある合併症がなく、正しい医療判断の範囲内で、ヒトおよび動物組織と接するのに適した、それらの化合物、物質、組成物および/または製剤を意味する。ある好ましい態様において、その遊離塩基Iおよびその塩の結晶形は、実質的に純粋な形態である。本明細書で用いられる用語「実質的に純粋」とは、例えば約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、および約100%を含む、約90%以上の純度を有する化合物を意味する。
【0037】
本明細書で用いられる「多形」とは、同一の化学的組成物を有する結晶形ではあるが、結晶を形成する分子、原子および/またはイオンが異なった空間的配置をとることを意味する。
【0038】
本明細書で用いられる「溶媒和物」とは、結晶構造に組み込まれる溶媒の分子がさらに含まれる、分子、原子、および/またはイオンの結晶形を意味する。溶媒和物における溶媒分子は、規則的配列および/または不規則な配列で存在されうる。該溶媒和物は、化学量論量または非化学量論量のいずれかの溶媒分子を含んでもよい。例えば、非化学量論量の溶媒分子を有する溶媒和物は、溶媒和物から溶媒の部分的な損失から生じうる。
【0039】
結晶形のサンプルは、優位な量の単一の結晶形および場合により微量の1またはそれ以上の他の結晶形の存在を示す、実質的に純粋な相均質性で提供されうる。サンプル内の1より多い結晶形の存在は、粉末X線回折(PXRD)または固体状態核磁気共鳴スペクトル(SSNMR)のような技術によって決定されうる。例えば、シミュレートされたPXRDパターンで実験的に測定されたPXRDパターンの比較において、余分なピークの存在は、サンプル内に1より多い結晶形の存在を示しうる。シミュレートされたPXRDは、単結晶X線データから計算されうる。Smith, D.K.の書籍:A FORTRAN Program for Calculating X-Ray Powder Diffraction Patterns, Lawrence Radiation Laboratory, Livermore, California, UCRL-7196 (April 1963)も参照されたい。好ましくは、該結晶形はシミュレートされたPXRDパターンに存在しない余分なピークから生じる実験的に測定したPXRDパターンにおいて、全ピーク面積の10%以下、好ましくは5%以下、およびより好ましくは2%以下で示される実質的に純粋な相均質性を有する。最も好ましくは、シミュレートされたPXRDパターンに存在しない余分なピークから生じる実験的に測定されたPXRDパターンにおいて、全ピーク面積の1%以下の実質的に純粋な相均質性を有する結晶形である。
【0040】
結晶形の調製のための手順は、当該技術分野で公知である。該結晶形は、例えば適当な溶媒からの結晶化または再結晶化、昇華、融解からの成長、他の相からの固体状態への変化、超臨界液体による結晶化、およびジェットスプレーを含む多様な方法によって調製されうる。溶媒混合物から結晶形の結晶化または再結晶化のための技術には、例えば溶媒のエバポレート、溶媒混合物の温度減少、分子および/または塩の過飽和の溶媒混合物への種晶添加、溶媒混合物の凍結乾燥、および貧溶媒(対溶媒)の溶媒混合物への添加が含まれる。
【0041】
多形、製造方法、および薬物結晶の特徴を含む薬物の結晶は、Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, West Lafayette, Indiana (1999) で論じられている。
【0042】
溶媒を用いる結晶化技術に関して、溶媒の選択は、化合物の溶解性、結晶化技術、および溶媒の蒸気圧のような1またはそれ以上の要素に典型的に依存する。溶媒の組合せも使用してもよく、例えば化合物を第一の溶媒に溶解し、溶液とし、続いて貧溶媒の添加により溶液中の化合物の溶解性を減少させ、結晶を形成させてもよい。貧溶媒は、該化合物が低溶解性を有する溶媒である。結晶を調製するための適当な溶媒には、極性および非極性溶媒が含まれる。
【0043】
結晶を調製する一方法において、重硫酸アタザナビルは、適当な溶媒中で懸濁および/または攪拌してスラリーが得られ、該スラリーは溶解を促進するために加熱してもよい。本明細書で用いられる用語「スラリー」とは、重硫酸アタザナビルまたはその塩の飽和溶液を意味し、所定温度で重硫酸アタザナビルまたはその塩と溶媒との不均一混合物を与える追加量の重硫酸アタザナビルまたはその塩も含み得る。この点における適当な溶媒には、例えば、極性非プロトン性溶媒、および極性プロトン性溶媒、およびここで開示される2またはそれ以上のこれらの混合物が含まれる。
【0044】
種晶は、結晶化を促進するためにいずれの結晶化混合物に加えられてもよい。熟練した技術者に自明であるように、種晶添加は、特定の結晶形の成長を制御する手段として、または結晶生成物の粒子径分布を制御する手段として使用される。従って、必要な種晶量の計算は、例えば、J.W. Mullin および J. Nyvlt の文献:Programmed cooling of batch crystallizers, Chemical Engineering Science (1971) 26:369-377.に記載されているような入手可能な種晶の大きさおよび平均粒子生成物の所望の大きさに依存する。一般に、小サイズの種晶は、バッチにおいて結晶の成長を有効に制御することが必要である。小サイズの種晶は、より大きい結晶をふるいにかけるか、破砕するか、または微粉化することによって、あるいは溶液の微結晶化によって生成されうる。結晶の破砕または微粉化することが所望の結晶形からの結晶度におけるいずれの変化(すなわち、アモルファスまたは他の多形への変化)も生じないことを配慮されるべきである。
【0045】
冷却した混合物を減圧下で濾過し、単離した固体を冷却した再結晶溶媒のような適当な溶媒で洗浄し、窒素パージ下で乾燥し、所望の結晶形を得てもよい。単離した固体は、SSNMR、DSC、PXRDなどのような適当な分光学的または分析技術によって分析し、生成物の好ましい結晶形の形成を確認してもよい。生じた結晶形は、典型的に約70重量%以上の単離収率の量で生じるが、好ましくは結晶化法を初めから用いて重硫酸アタザナビルの重量に基づいて90重量%以上で生じる。該生成物は、必要なら生成物を塊にならないように破砕するか、または網目のふるいにかけてもよい。
【0046】
結晶形は、重硫酸アタザナビルを調製するためのプロセスの最終工程の反応系から直接調製されうる。これは、例えばプロセスの最終工程において、重硫酸アタザナビルが結晶化され得る溶媒または溶媒混合物を用いることによって達成されうる。または結晶形は、蒸留または溶媒添加技術によって得られうる。この目的のための適当な溶媒には、アルコールのようなプロトン性極性溶媒、およびケトンのような非プロトン性極性溶媒を含む、本明細書に記載されたいずれの溶媒も含まれる。
【0047】
一般的な指図を経由して、該反応混合物は、いずれの不要な不純物、無機塩なども除去するために濾過し、続いて反応溶媒または結晶化溶媒で洗浄してもよい。生じた溶液は、過剰量の溶媒または気体成分を除去するために濃縮してもよい。蒸留を用いるなら、回収された留分の最終量は、例えば容器の大きさ、攪拌能力などを含むプロセス要因に依存して変更可能である。一般的な指図によって、該反応溶液は、最初の量である約{フラクション(1/10)}まで蒸留した後、溶媒置換を実施した。標準的なプロセス技術に従って、反応の程度および生成物の重量%を決定するために、該反応液をサンプリングしアッセイしてもよい。必要なら追加の反応溶媒を加えるか、または反応濃度を最適化するために除去してもよい。好ましくは、最終濃度が典型的にスラリーを生じる点において、約50重量%に調整される。
【0048】
反応混合物を蒸留することなく反応容器に直接溶媒を加えることが好ましい。この目的のための好ましい溶媒は、溶媒交換に関連して、上記の通り結晶格子内で最終的に共存してもよいものである。最終濃度は、所望の純度、回収率などに依存して変更可能であるけれど、溶液中の遊離塩基Iの最終濃度は、約4%〜約7%が好ましい。該反応混合物は、攪拌し、続いて溶媒添加し、同時に温めてもよい。例として該反応混合物は、約70℃まで温めながら約1時間攪拌してもよい。該反応液は、好ましくは熱時濾過し、反応溶媒、加えた溶媒、またはその組合せのどれかで洗浄する。種晶は、結晶化を開始するために、いずれの結晶化溶液に加えてもよい。
【0049】
本明細書に記載の種々の形態は、当該技術分野における通常の知識を有する者に公知の種々の分析技術の使用を通じてお互いから区別されうる。かかる技術には、これに限らないが、固体状態核磁気共鳴(SSNMR)スペクトル、X線粉末回折(PXRD)、示差走査熱量測定(DSC)、および/または熱重量分析(TGA)が含まれる。
【0050】
X線回折パターンが、使用する測定条件に依存する測定誤差を含んで得られうることは当該技術分野で通常の技術を有する者に理解されている。特に、X線回折パターンにおける強度は、用いた測定条件および結晶形または多形に依存して変動することが一般的に知られる。相対強度もまた、実験条件に依存して変動し得ることはさらに理解されるべきであり、従って正確な強度のオーダーは考慮されるべきではない。従って、通常のX線回折パターンのための回折角の測定誤差は、典型的に約0.2%またはそれ以下であり、好ましくは約0.1%(下文に記載)であり、測定誤差のかかる程度は、前述の回折角に関連するとして考慮されるべきである。その結果、本発明の結晶形は、本明細書に開示される添付図に記載されるX線回折パターンと完全に同一であるX線回折パターンを提供する結晶形に限定されないことが理解されるべきである。添付図に開示されたパターンと実質的に同一であるX線回折パターンを提供するいずれの結晶形も本発明の範囲に含まれる。X線回折パターンの実質的な同一性を確認する能力は、当該技術分野で通常の技術を有する者の範囲内である。
【0051】
A型およびE3型に関して、本明細書で用いられる用語「形態、形」とは、均一な結晶構造を意味する。
【0052】
Cパターン物質に関して、本明細書で用いられる用語「パターン」とは、特徴的なX線回折パターンを意味する。
【0053】
本明細書で用いられる用語「重硫酸アタザナビル」とは、重硫酸アタザナビルならびに硫酸アタザナビルを意味する。
【0054】
重硫酸アタザナビル塩のA型結晶を調製するための本発明の方法の実施において、改良した立方結晶化技術が用いられ、その中でアタザナビル遊離塩基は、重硫酸アタザナビル塩が実質的に不溶であり、アセトン、アセトンおよびN−メチルピロリドン混合液、エタノール、エタノールおよびアセトン混合液などを含む有機溶媒で溶解され、アタザナビル遊離塩基の約6.5〜約9.7重量%、好ましくは約6.9〜約8.1重量%の範囲内のアタザナビル遊離塩基の濃度を有する溶液が得られる。
【0055】
アタザナビル遊離塩基の溶液は、約35〜約55℃、好ましくは約40〜約50℃の温度範囲で加熱し、相当量の濃硫酸(約95〜約100%H2SO4を含む)と反応し、アタザナビル遊離塩基の全重量の約15%以下、好ましくは約5〜約12%以下、より好ましくは約8〜約10%と反応する。従って、アタザナビル遊離塩基の出発溶液は、用いる硫酸の全量の約15%以下、好ましくは約5〜約12%と最初に反応される。反応の間、該反応混合物は、約35〜約55℃、好ましくは約40〜約50℃の範囲内で維持される。
【0056】
該反応を約12〜約60分間、好ましくは約15〜約30分間継続させる。
【0057】
該反応混合物は、反応混合物を約35〜約55℃、好ましくは約40〜約50℃の温度範囲を維持しながら、反応混合物に残存するアタザナビル遊離塩基の重量に基づいて、約0.1〜約80重量%、好ましくは約3〜約8重量%の範囲内の量で相当量の種晶を用いる重硫酸アタザナビルのA型結晶で種晶添加される。
【0058】
該反応は、結晶化を開始するまで継続させる。その後硫酸を下記のような三次方程式に従う増加速度で多段階にて加え、重硫酸アタザナビルを形成し、乾燥してA型結晶が生じる。
【0059】
形成される重硫酸アタザナビル塩の結晶粒子径および多形は、硫酸の添加速度に依存し、結晶化速度を決定する。改良「立方」結晶化技術(三次方程式に従う増加速度で加えた酸)は、定常添加速度での結晶化よりも、より狭い粒子径範囲およびより僅かな細かさを伴う相対的により大きく、より良く定義された重硫酸アタザナビルの結晶を与えることが見出されている。ゆっくりとした最初の酸の流速は、二次的な核形成にわたる結晶成長に有利にはたらくことが示されている。従って、粒子径と共に表面積が増加するにつれて、種晶床は、二次的な核形成の誘発を含まない酸の流速を増加することを受け入れることができる。ゆっくりとした最初の添加速度は、結晶がより大きく成長するための時間を許容し、平均サイズを増加させる。該立方結晶化は、あまり圧縮性のない濾過ケークを与え、該ケークは、有効なケークの脱液および洗浄、ならびに定常添加速度で結晶化する生成物よりも固くない塊を有し、より容易に乾燥する生成物を与えることを促進する。
【0060】
用いた立方結晶化方法は、Mullinの書籍:Crystallization, 3rd Ed., 1993, Butterworth-Heineman, Pubs.に由来する温度制御した結晶化であり、下記の単純化した式で定義される:
【数1】
【0061】
重硫酸アタザナビルの結晶化は、硫酸の添加速度によって制御されるので、温度変数は、式(1)の酸の量で置き換えられる。この式において最小量を表す変数は、除去される。
【数2】
式(2)は、「三次方程式」と称する。
【0062】
この式を用いて結晶化速度を制御することによって、核形成はシステムが一定の低レベルの過飽和を維持するので許容限界内で制御される。
【0063】
A型結晶は、図1および2のそれぞれで示される粉末X線回折パターンおよび結晶構造で同定される。
【0064】
重硫酸アタザナビルのA型結晶またはCパターン物質、および上記のように調製されるE3型は、最終の重硫酸アタザナビルであって、患者に投与するための製剤として使用され得る。
【0065】
本発明の方法に従って、Cパターン物質は、A型結晶を水に曝露し、続いて乾燥することによって調製されうる。
【0066】
本発明に従う別の方法において、Cパターン物質は、A型結晶を約95%RH以上、好ましくは約95〜約100%RH(水蒸気)の高い相対湿度に、少なくとも24時間、好ましくは約24〜約48時間曝露することによって形成されうる。
【0067】
本発明の別の態様において、Cパターン物質は、重硫酸アタザナビルのA型を湿式造粒することによって、重硫酸アタザナビルの造粒物を生じ、次いで該造粒物を乾燥することによって調製される。
【0068】
湿式造粒法の実施において、重硫酸アタザナビルは、水中で造粒し、約40〜約80℃、好ましくは約50〜約60℃の温度範囲で乾燥する。乾燥工程は、少なくとも約2時間、約20時間まで、好ましくは約8〜約10時間実施される。
【0069】
Cパターン物質はまた、例えば1またはそれ以上の充填剤、好ましくはラクトース、1またはそれ以上の崩壊剤、好ましくはクロスポビドンのような通常の医薬賦形剤の存在下で重硫酸アタザナビルのA型を湿式造粒し、上記の通り乾燥し、賦形剤との混合物のCパターン物質を形成することで形成してもよい。
【0070】
それはCパターン物質、A型またはE3型、好ましくはCパターン物質であり、下文に記載されるウイルスによって引き起こされる疾病の治療において、投与するために製剤化される。
【0071】
Cパターン物質は図3に示される、その粉末X線回折パターンによって特徴付けられる。
【0072】
E3型は、アタザナビル遊離塩基をエタノール中でスラリーにし、該スラリーを酸:遊離塩基が約1:1〜約1.1:1の範囲のモル比を用いて濃硫酸と処理し、約30〜約40℃で生じた溶液を加熱し、該溶液をエタノールで湿らせた硫酸アタザナビルのE3結晶で種晶添加し、該混合物をヘプタン(またはヘキサンまたはトルエンのような他の溶媒)で処理し、濾過し、乾燥して重硫酸アタザナビルのE3型(トリエタノール溶媒和物)を得ることにより調製される。
【0073】
種晶添加工程は、E3結晶を形成する相当量の種晶、例えば、重硫酸アタザナビルE−3種晶:遊離塩基が約0.02:1〜約0.04:1の範囲内のモル比を用いる。
【0074】
E3型は、図7に示される粉末X線回折パターンおよび図6に示される結晶構造によって同定される。
【0075】
本発明に従って、その遊離塩基の形態におけるアタザナビルは、構造:
【化13】
[式中、PGは、t−ブチルオキシカルボニル(Boc)またはトリフルオロアセチル、好ましくはBocのような保護基を表す]
の保護されたトリアミン塩溶液を、塩化メチレン、テトラヒドロフラン、またはメタノールのような有機溶媒、好ましくは塩化メチレンの存在下、約25〜約50℃、好ましくは約30〜約40℃の温度範囲で、酸、好ましくは塩酸(Bocが用いられる場合)、または塩基(トリフルオロアセチルが用いられる)と処理し、トリアミン酸塩、好ましくは構造:
【化14】
の塩酸塩を形成することで、および
トリアミン酸塩を単離することなく、該トリアミン酸塩を構造:
【化15】
の酸の活性エステル体、好ましくは構造:
【化16】
の活性エステル体とK2HPO4、ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸ナトリウム、または炭酸カリウムのような塩基、好ましくはK2HPO4の存在下、塩化メチレン、酢酸エチルおよび酢酸ブチルの混合液、アセトニトリルまたは酢酸エチルのような有機溶媒、好ましくは塩化メチレンの存在下で、約25〜約50℃の温度範囲内好ましくは約30〜約40℃にて反応し、アタザナビル遊離塩基を形成することにより調製される。
【0076】
該保護されたトリアミンの出発物質をN−(tert−ブチルオキシカルボニル)−2(S)−アミノ−1−フェニル−3(R)−3,4−エポキシブタンのようなエポキシド体:
【化17】
[式中、PGは、好ましくはBoc基である]
をヒドラジンカーバメート体:
【化18】
[PGは、好ましくはBoc基である]
と、イソプロピルアルコールまたはエタノールまたはブタノールのような他のアルコールの存在下で反応することにより調製される。
【0077】
重硫酸アタザナビルは、レトロウイルスプロテアーゼ、とりわけHIV−1またはHIV−II・gagプロテアーゼのようなレトロウイルスアスパラギン酸プロテアーゼ阻害に応答する疾病、例えばAIDSまたはその予備段階のようなレトロウイルスの疾病の治療または予防のために、温血動物、とりわけヒトに投与するのに有用である。
【0078】
重硫酸アタザナビル、とりわけCパターン物質、A型またはE3型、好ましくはCパターン物質またはA型は、ウイルス、とりわけレトロウイルスによって引き起こされる疾病、とりわけAIDSまたはその予備段階の治療方法に使用され、治療上の有効量のCパターン物質、A型またはE3型の重硫酸アタザナビルが上述の疾病の一つ、とりわけAIDSまたはその予備段階でかかる治療が必要であるとりわけ温血動物、例えばヒトに該疾病の治療に有効な投与量で投与される。温血動物、例えばおよそ70kg体重のヒトに投与される好ましい投与量は、1日あたりヒトに約3mg〜約1.5g、好ましくは約10mg〜約1.25g、例えば約50mg〜約600mgであり、好ましくは、例えば同量で1〜4回の単回投与に分けられる。通常、子供は大人の投与量の半分を受ける。好ましくは経口投与である。
【0079】
Cパターン物質、A型またはE3型の重硫酸アタザナビルは、上記の医薬用途のために使用される。経口投与用のCパターン物質またはA型もしくはE3型を含む適当な組成物には、錠剤、粉剤、カプセル剤、およびエリキシルが含まれる。約10〜600mgの活性成分は、許容される医薬技術によって必要とされる単位投与量において、生理的に許容されるベヒクル、担体、賦形剤、結合剤、保存料、安定化剤、香料などと配合される。
【0080】
経口投与用の医薬組成物は、必要なら生じた混合物を造粒し、該混合物を処理し、所望または必要なら適当な賦形剤を、錠剤、ドラジェ(dragee)コア、カプセル剤または経口使用のための粉剤に添加後、活性成分を固体担体と組み合わせることにより得ることができる。活性成分が測定される量で拡散または放出させる可塑性担体に導入することも可能である。
【0081】
充填剤または増量剤は、組成物の約0〜約95重量%、好ましくは約10〜約85重量%の範囲内の量で本発明の医薬組成物中に存在する。本明細書で使用するために適当な充填剤または増量剤の具体例には、これに限らないが、微結晶セルロースまたは木質セルロースのようなセルロース誘導体、ラクトース、ショ糖、デンプン、プレゼラチン化したデンプン、デキストロース、マンニトール、フルクトース、キシリトール、ソルビトール、トウモロコシデンプン、変性トウモロコシデンプン、炭酸カルシウム、リン酸カルシウム、リン酸二カルシウム、硫酸カルシウムのような無機塩、デキストリン/デキストレート、マルトデキストリン、圧縮性糖類、および他の公知の充填剤または増量剤、および/または2またはそれ以上の混合物が含まれ、好ましくはラクトースがよい。
【0082】
結合剤は、組成物の約0〜約20重量%、好ましくは約1〜約10重量%の範囲内の量で本発明の医薬組成物中に適宜存在する。本明細書の使用に適した結合剤の具体例には、これに限らないが、ヒドロキシプロピルセルロース、トウモロコシデンプン、プレゼラチン化したデンプン、変性トウモロコシデンプン、ポリビニルピロリドン(PVP)(約5000〜約80000の範囲の分子量、好ましくは約40000である)、ヒドロキシプロピルメチルセルロース(HPMC)、ラクトース、アカシアゴム、エチルセルロース、酢酸セルロース、並びにカルナウバロウ、パラフィン、鯨ロウ、ポリエチレンまたは微結晶性ワックスのようなロウ性結合剤、ならびに他の通常の結合剤および/または2またはそれ以上のそれらの混合物が含まれ、好ましくはヒドロキシプロピルセルロースがよい。
【0083】
崩壊剤は、組成物の約0〜約20重量%、好ましくは約0.25〜約15重量%の範囲内の量で、本発明の医薬組成物中に適宜存在する。本明細書の使用に適した崩壊剤の具体例には、これに限らないが、クロスカルメロースナトリウム、クロスポビドン、ジャガイモデンプン、プレゼラチン化したデンプン、トウモロコシデンプン、グリコール酸デンプンナトリウム、微結晶性セルロース、または他の公知の崩壊剤が含まれ、好ましくはクロスカルメロースナトリウムがよい。
【0084】
滑沢剤は、組成物の約0.1〜約4重量%、好ましくは約0.2〜約2重量%の範囲内の量で、本発明の医薬組成物に適宜存在する。本明細書の使用に適した打錠滑沢剤の具体例には、これに限らないが、ステアリン酸マグネシウム、ステアリン酸亜鉛、ステアリン酸カルシウム、タルク、カルナウバロウ、ステアリン酸、パルミチン酸、ステアリルフマル酸ナトリウム、または水素化された植物油および脂肪、または他の公知の打錠滑沢剤、および/または2またはそれ以上のそれらの混合物が含まれ、好ましくはステアリン酸マグネシウムがよい。
【0085】
カプセル剤は、グリセロールまたはソルビトールのようなゼラチンおよび可塑剤で製造された硬ゼラチンカプセルおよびソフトシールカプセルである。該硬ゼラチンカプセルは、例えばラクトースのような充填剤、デンプン、クロスポビドンのような結合剤および/またはタルクまたはステアリン酸マグネシウムのような流動促進剤、および必要なら安定化剤と一緒に顆粒形態の活性成分を含んでいてもよい。ソフトゼラチンカプセルの活性成分は、好ましくは脂肪油、パラフィンオイルまたは液体ポリエチレングリコールのような適当な油状賦形剤に溶解または懸濁され、安定化剤および/または抗菌剤を加えることが同様に可能である。
【0086】
以下の実施例は、本発明の好適な態様を示す。
【0087】
実施例1
1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4−(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・重硫酸塩(A型)(重硫酸アタザナビルA型)
【化19】
【0088】
(1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス[tert−ブチルオキシカルボニル)アミノ]−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・3HCl(トリアミン・3HCl塩))
メカニカルスターラー、窒素吸気口および温度プローブを取り付けた1000mLの3頸丸底フラスコに、保護トリアミン体、1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス[tert−ブチルオキシカルボニル)アミノ]−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン:
【化20】
(100g,0.178mol)、およびCH2Cl2(500mL;5mL/gの保護トリアミン体)(Z. Xuらの文献:Process Research and Development for an Efficient Synthesis of the HIV Protease Inhibitor BMS-232,632, Organic Process Research and Development, 6, 323-328 (2002)に記載されたとおり製造した)を加え、生じたスラリーを約5〜約22℃の温度を維持しながら攪拌した。
【0089】
濃塩酸(68mL,0.82mol、4.6当量)を反応混合物の温度が5〜30℃を維持するような速度で該反応混合物に加えた。該反応混合物を30〜40℃に加熱し、反応がHPLCアッセイによって完了したと判断するまで攪拌した。
【0090】
水(70〜210mL、0.7〜2.1mL/gの保護したトリアミン体の投入量)を反応混合物に加え、該反応混合物を15分間攪拌し、層を分離させた。上層の生成物(トリアミン・3HCl塩)リッチな水性油状物を滴下漏斗に移した。
【0091】
B.
【化21】
(N−メトキシカルボニル−L−tert−ロイシン:
【化22】
の活性エステル体)
メカニカルスターラー、滴下ロート、窒素吸気口、および温度プローブを取り付けた3000mLの3頸丸底フラスコに、N−メトキシカルボニル−L−tert−ロイシン(77.2g,0.408mol,2.30当量)、1−ヒドロキシベンゾトリアゾール(HOBT)(60.8g,0.450mol,2.53当量)、およびN−エチル N’−ジメチルアミノプロピルカルボジイミド(EDAC)(82.0g,0.430mol,2.42当量)、続いて、CH2Cl2(880mL;8.8mL/gの保護したトリアミン体の投入量)を加え、該活性エステル体の形成がHPLCで完了したと判断するまで、該混合物を周囲温度(18〜25℃)で攪拌した。
【0092】
C.1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン(アタザナビル遊離塩基)
無水第二リン酸カリウム(K2HPO4;226g,1.30mol,保護トリアミン体に対して7.30当量)を1130mLの水(11.3mL/gの保護アミン体;5mL/gのK2HPO4)に溶解した。
【0093】
該K2HPO4溶液をパートBで調製した活性エステル体溶液に加えた。攪拌した活性エステル体/K2HPO4水溶液混合物に、攪拌および5〜20℃の容器温度を維持しながら1.5〜2.0時間以上かけてパートAの塩酸塩水溶液をゆっくり加えた。
【0094】
パートAの塩酸塩溶液の添加が完了した後、該反応混合物(カップリング反応)を30〜40℃に加熱し、カップリング反応がHPLCアッセイによって完了したと判断するまで攪拌した。
【0095】
該カップリング混合物を15〜20℃まで冷却し、下層の生成物リッチな有機層を下層から分離し、水層を廃棄した。
【0096】
該生成物リッチな有機層を1MのNaH2PO4(880mL;pH=1.5;8.8mL/gの保護アミン体投入量;保護アミン体に対して5モル当量)で洗浄し、該層を分離させ、不要な水層を除去した。
【0097】
生成物リッチな有機層のHPLCアッセイで、それぞれ活性エステル体が0.3I.I.以下を示すまで、洗浄した生成物リッチな有機層を0.5NのNaOH(800mL;8mL/gの保護アミン投入量)と共に攪拌した。該層を分離させて、不要な水層を除去した。
【0098】
該生成物リッチな有機層を5%NaH2PO4(450mL,4.5mL/gの保護トリアミン体投入量;pH=4.3)で洗浄し、該層を分離させて、不要な水層を除去した。
【0099】
該生成物リッチな有機層を10w/v%のNaCl(475mL,4.75mL/gの保護トリアミン体投入量)で洗浄し、不要な水層を除去した。
【0100】
標記の遊離塩基溶液の濃度は、製造過程中の計算収率が95〜100mol%である、120〜150mg/mLであった。
【0101】
D.CH2Cl2からアセトン/N−メチルピロリドンへの溶媒交換
メカニカルスターラー、温度プローブ、および蒸留冷却管を取り付けた3000mLの3頸丸底フラスコ中のパートCの遊離塩基リッチな溶液に、N−メチルピロリドン(148mL;製造過程の定量アッセイに基づく1.25mL/gのパートCの遊離塩基)を加えた。該溶液を70℃またはそれ以下のジャケット温度を用いて、約360mL(2.5〜3.5mL/gのパートCの遊離塩基)まで濃縮し;500mLのアセトン(4〜5mL/gのパートCの遊離塩基)を濃縮液に加え、該混合液を約400mLまたはそれ以下の量まで蒸留した。
【0102】
CH2Cl2レベルを示す製造過程中のアッセイが目的の終点に達するまで、アセトン添加および蒸留を繰り返した。結晶化量において、生成物リッチな有機溶液におけるCH2Cl2量が0.77v/v%であった。アセトンを全量が16mL/gの遊離塩基に達するように濃縮した遊離塩基溶液に加えた。浴温は、遊離塩基の結晶化を防ぐために40〜50℃に維持した。温度を40〜50℃に維持しながら、該溶液を10ミクロンまたはそれより細かいフィルターでポリッシュフィルターにて濾過した。該ポリッシュフィルターをアセトン(125mL,1.0mL/gの遊離塩基)でリンスし、該リンス液を遊離塩基リッチなアセトン/N−メチルピロリドン溶液に加え、次工程に使用した。
【0103】
E.1−[4−(ピリジン−2−イル)フェニル]−5(S)−2,5−ビス{[N−(メトキシカルボニル)−L−tert−ロイシニル]アミノ}−4(S)−ヒドロキシ−6−フェニル−2−アザヘキサン・重硫酸塩
40〜50℃の温度を維持しながら、表面添加によって濃硫酸(19g,1.10当量)の総添加量の約10%(2g)をパートDの遊離塩基のアセトン/N−メチルピロリドン溶液に加えた。
【0104】
該反応混合物を重硫酸塩の5.0wt%(計算した遊離塩基溶液に対して)を種晶添加した。重硫酸塩が、この時間中不透明度が増加する混合物から明らかなように、結晶化を開始した時間において、種晶を加えた混合物を40〜50℃で少なくとも30分間攪拌した。
【0105】
残る硫酸(17.8g)を40〜50℃の温度を保ちながら、三次方程式によって定義される下記のプロトコルに従って、五段階で約5時間かけて添加した。
【0106】
各添加段階の速度は、上記の三次方程式に従って決定し、以下の表に示される。
表1
【0107】
H2SO4の添加が完了した後、スラリーを攪拌しながら、少なくとも1時間、20〜25℃まで冷却した。該スラリーを少なくとも1時間、20〜25℃で攪拌した。該重硫酸塩を濾過し、母液は、完全に移行する必要があるため回収した。濾過ケークをアセトン(5〜10mL/gの遊離塩基;1200mLのアセトン)で洗浄した。該重硫酸塩をLOD<1%までNMT55℃にて減圧下で乾燥し、結晶物質を得る。
【0108】
該結晶生成物をPXRD、DSCおよびTGAパターンおよびSSNMRスペクトルによって分析し、標記の重硫酸塩(図1〜5を参照されたい)のA型結晶(無溶媒和物)であることがわかった。
【表1】
【表2】
【表3】
【表4】
異方性の精製された原子は、(4/3)*[a2*B(1,1)+b2*B(2,2)+c2*B(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)]として定義される、等価等方性原子変位パラメーターの形態で与えられる。
【0109】
A型は、図3に示される約165.6℃〜約200.9℃の範囲内で、典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられる。
【0110】
A型はまた、約100〜150℃まででごく僅かな減量を有する熱重量分析曲線によっても特徴付けられる。
【0111】
H2SO4が上記の三次方程式に従う増加速度で加えられる立方結晶化によって生じる結晶は、一定の添加速度による結晶化を用いて得られた結晶より相対的に大きく、明確で、狭い粒子径範囲を有し、ほとんど細かくなかった。
【0112】
立方結晶化技術を用いて得られた濾過ケークは、一定の添加速度による結晶化を用いて得られたものより圧縮性が小さく、有効なケークの脱液および洗浄に役立ち、均一生成物を生じた。
【表5】
【0113】
実施例2
重硫酸アタザナビル−Cパターン物質
方法A:
重硫酸アタザナビルのA型結晶(実施例1に記載されるように調製した)(25.33g)を200mLの水に懸濁し、該混合物を機械的に攪拌し、厚いゲルを生じ乾燥した。
【0114】
乾燥した混合物をスパチュラですりつぶし、Cパターン物質を生じた。Cパターン物質の粉末X線回折パターンは、図6に示す。
【0115】
方法B:
重硫酸アタザナビルのA型結晶は、適当な混合造粒機において、充分な量の水(約40%w/w)を用いて湿式造粒した。湿塊をオーブンで乾燥した。該生成物を適当なふるいを用いて、等しい大きさにした。生じた生成物のX線回折パターンは、図6に示されるCパターン物質と一致する。
【0116】
Cパターンは、約76.7〜約96.6℃および約156.8〜約165.9℃の範囲内で典型的な吸熱を有する、図7に示される示差走査熱量サーモグラムによって特徴付けられる。
【0117】
Cパターンはまた、図8に示される約125℃で約2.4%および約190℃で約4.4%の重量減量を有する熱重量分析曲線によっても特徴付けられる。
【0118】
実施例3
重硫酸アタザナビル−E3型(トリエタノール溶媒和物)
メカニカルスターラー、温度プローブ、および圧力を等しくする液体滴下ロートを取り付けた100mLの3頸丸底フラスコにおいて、アタザナビル遊離塩基(実施例1、パートCに記載されるように調製)(3.0g,4.26mmol)を無水の200プルーフ(proof)のエタノール(20.25mL,6.75mL/gの遊離塩基)中でスラリーにした。
【0119】
濃H2SO4(0.25mL,0.46g,4.69mmol,1.1当量)を20〜25℃で維持したアタザナビル遊離塩基のスラリーに加えた。生じた溶液(0.2〜1.0%水分量のKF)をポリッシュフィルター(Whatman ♯1ペーパー)で濾過し、フィルターを2.25mLの無水エタノールでリンスし、該リンス液を濾過液に加えた。該溶液を37℃に加熱し、E3型結晶から(E3型結晶を周囲温度に曝すことによって)生じる10mgのアモルファスの重硫酸アタザナビルで種晶添加し、該混合物を15分間攪拌した。ヘプタン(380mL,8.25mL/gの遊離塩基)を1時間以上かけて加えた。生じた結晶化混合物を15〜25℃で8時間攪拌した。結晶化した重硫酸アタザナビルをブフナー漏斗で濾過した。該生成物ケークを184mL(4mL/gの遊離塩基)のエタノール:ヘプタン(1:1)で洗浄した。該生成物ケークを、46mL(1mL/gの遊離塩基)のヘプタンで洗浄した。生じた生成物をLOD=0.97%となるまで、40〜50℃で減圧下にて乾燥した。生成物の収量は、HPLC HI=100.0(図9および10を参照されたい)である重硫酸アタザナビルE3型(トリエタノール溶媒和物)の47.7g(0.0594mol,74.3mol%)であった。
【表6】
【表7】
【表8】
異方性の精密な原子は、(4/3)*[a2*B(1,1)+b2*B(2,2)+c2*B(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)]として定義される、等価等方性原子変位パラメーターの形態で与えられる。
【0120】
E3型は、図11に示される、約89.4〜約96.6の範囲内で典型的に吸熱を有する示差走査熱量サーモグラムによって特徴付けられる。
【0121】
E3型はまた、図11に示される約150℃で約14.7%の重量減量を有する熱重量分析曲線によっても特徴付けられる。
【0122】
実施例4
下記の組成物を有するC形の重硫酸アタザナビルカプセル製剤は、下記の通り調製された。
【表9】
【0123】
重硫酸アタザナビルの保存顆粒は、下記のように調製され、Cパターン物質が形成された。
【0124】
A型の重硫酸アタザナビル、含水ラクトース、およびクロスポビドンの一部(クロスポビドンの全量の3%)をプラネタリーミキサーで混合した。生じた混合物をA型からCパターン物質に変換するために精製水で湿式造粒した。湿式造粒を棚型乾燥機で乾燥し、ハンマーミルを用いて等しい大きさにした。残りのクロスポビドンを粉砕した造粒に加え、該混合物をPK V−混合機で混合した。ステアリン酸マグネシウムを加え、該混合物を実質的に均一な保存顆粒を形成するまで混合した。
【0125】
適当な重量の保存顆粒をカプセルに充填し、重硫酸アタザナビルを含む、50mg、100mgおよび200mgカプセルを生成した。
【0126】
実施例5
下記の組成物を有する経口用途製剤のための重硫酸アタザナビルのA型物質の粉末は、下記のように調製される。
【0127】
重硫酸アタザナビルA型は、適当な混合機でアスパルテーム、オレンジバニラフレーバーおよびショ糖と混合する。該混合物はハンマーミルを用いて粉砕し、続いて、二次混合操作を行って均一な混合物を得る。生成物は、高密度のポリエチレン瓶に充填する。
【図面の簡単な説明】
【0128】
【図1】図1は、A型の計算した(シミュレートした)(22℃)および実測した(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図2】図2は、A型の結晶構造を示し;
【図3】図3は、A型の示差走査熱量測定(DSC)サーモグラムを示し;
【図4】図4は、A型の熱重量分析曲線(TGA)を示し;
【図5】図5は、A型の固体状態C−13 NMRを示し;
【図6】図6は、Cパターンの実測された(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図7】図7は、Cパターンの示差走査熱量サーモグラムを示し;
【図8】図8は、Cパターンの熱重量分析曲線を示し;
【図9】図9は、E3型の計算された(シミュレートした)(22℃)および実測された(室温での実験)粉末X線回折パターン(CuKαλ=1.5418Å)を示し;
【図10】図10は、E3型の結晶構造を示し;および
【図11】図11は、E3型の示差走査熱量測定(DSC)サーモグラム、およびE3型の熱重量分析曲線を示す。
【特許請求の範囲】
【請求項1】
A型結晶形の重硫酸アタザナビルの製造方法であって、アタザナビルの重硫酸塩が実質的に不溶である有機溶媒中で、アタザナビル遊離塩基溶液をアタザナビル遊離塩基の約15重量%以下が反応する量の濃硫酸の一部と最初反応させ、重硫酸アタザナビルのA型結晶の種晶を反応混合物に加え、重硫酸アタザナビルの結晶が形成するように重硫酸アタザナビルの結晶形成を行うために多段階で追加の濃硫酸を加え、得られた重硫酸アタザナビルを乾燥して、A型結晶を形成することを特徴とする方法。
【請求項2】
該アタザナビル遊離塩基の溶液が、用いられる硫酸の全重量の約5〜約15%と最初に反応する、請求項1に記載の方法。
【請求項3】
該アタザナビル遊離塩基の溶液が、用いられる硫酸の全重量の約8〜約12%と最初に反応する、請求項1に記載の方法。
【請求項4】
該アタザナビル遊離塩基を、約35〜約55℃の温度範囲で硫酸の一部と最初に反応させる、請求項1に記載の方法。
【請求項5】
アタザナビル遊離塩基を、約35〜約55℃の温度範囲に加熱した後、硫酸と反応させる、請求項1に記載の方法。
【請求項6】
アタザナビル遊離塩基および硫酸の反応混合物にアタザナビル遊離塩基の重量に基づいて、A型結晶の約0.1〜約80重量%で種晶添加される、請求項1に記載の方法。
【請求項7】
種晶添加された反応混合物を、約35〜約55℃の温度範囲で加熱する、請求項1に記載の方法。
【請求項8】
アタザナビル遊離塩基のための有機溶媒がアセトン、アセトンおよびN−メチルピロリドンの混合物、エタノール、またはエタノールおよびアセトンの混合物である、請求項1に記載の方法。
【請求項9】
重硫酸アタザナビルのCパターン物質。
【請求項10】
図6に示されるパターンに実質的に一致する粉末X線回折パターンによってCパターン物質の重硫酸アタザナビルの特徴である、請求項9に記載の化合物。
【請求項11】
図7に示されるサーモグラムに実質的に一致する示差走査熱量サーモグラムによって特徴付けられる、請求項9に記載の化合物。
【請求項12】
図8に示される曲線は実質的に一致する熱重量分析曲線によって特徴付けられる、請求項9に記載の化合物。
【請求項13】
水中で重硫酸アタザナビルのA型結晶を懸濁すること、または相対湿度が少なくとも約95%RHで少なくとも24時間、重硫酸アタザナビルのA型結晶を付すこと、または重硫酸アタザナビルのA型結晶を湿式造粒し、次いで乾燥することにより調製される、請求項9に記載の化合物。
【請求項14】
請求項24の方法によって調製されるCパターン物質。
【請求項15】
重硫酸アタザナビルのA型結晶を、1またはそれ以上の製剤用賦形剤および水と混合し、続いて乾燥することによって調製される医薬組成物の形態である、請求項9に記載の化合物。
【請求項16】
重硫酸アタザナビルのE3型結晶。
【請求項17】
重硫酸アタザナビルのトリエタノレート溶媒和物として調製される、請求項16に記載の化合物。
【請求項18】
図9に示されるパターンに実質的に一致する粉末X線回折パターンによって特徴付けられる、請求項16に記載の化合物。
【請求項19】
実質的に図10に示される結晶構造を有する、請求項16に記載の化合物。
【請求項20】
実質的に表6に記載される原子分率座標によって特徴付けられる、請求項16に記載の化合物。
【請求項21】
下記:
[格子サイズ:
a=10.749(5)Å
b=13.450(4)Å
c=9.250(2)Å
α=98.33(2)°
β=95.92(3)°
γ=102.82(3)°
空間群P1
分子/非対称単位1]
と実質的に同等である結晶学的データによって特徴付けられ、該結晶形が約−23℃で存在する、請求項16に記載の化合物。
【請求項22】
図11で示されるサーモグラムに実質的に一致する示差走査熱量サーモグラムによって特徴付けられる、請求項16に記載の化合物。
【請求項23】
図11で示される曲線に実質的に一致する熱重量分析曲線によって特徴付けられる、請求項16に記載の化合物。
【請求項24】
(a)水中で、重硫酸アタザナビルのA型結晶を懸濁し、該懸濁液を乾燥して、Cパターン物質を形成するか;または
(b)95%RHより高い相対湿度で少なくとも24時間、重硫酸アタザナビルのA型結晶を付し、Cパターン物質を形成するか;または
(c)重硫酸アタザナビルを湿式造粒し、該湿造粒物を乾燥し、Cパターン物質を形成するか;または
(d)A型結晶を1またはそれ以上の製剤用賦形剤と混合し、生じた混合物を湿式造粒し、賦形剤との混合物であるCパターン物質を直接的に形成すること
を含む、請求項9に記載された重硫酸アタザナビルのCパターン物質の製造方法。
【請求項25】
A型結晶形の重硫酸アタザナビル:
【化1】
を製造する方法であって、構造式:
【化2】
のトリアミン塩を調製し、トリアミン塩を単離することなく、該トリアミン塩を有機溶媒の存在下で、構造式:
【化3】
の酸の活性エステル体および塩基と反応し、構造式:
【化4】
のアタザナビル遊離塩基の溶液を形成し、遊離塩基を対応する重硫酸塩に変換すること
を含む方法。
【請求項26】
トリアミン塩が、塩酸塩:
【化5】
である、請求項25に記載の方法。
【請求項27】
該酸の活性エステル体が、構造式:
【化6】
を有する、請求項25に記載の方法。
【請求項28】
該塩基がアルカリ金属ヒドロキシド、アルカリ土類金属ヒドロキシド、アルカリ金属カーボネート、アルカリ土類金属カーボネート、アルカリ金属ホスフェート、アルカリ土類金属ホスフェートまたは有機塩基である、請求項25に記載の方法。
【請求項29】
該塩基がNaOH、KOH、Mg(OH)2、K2HPO4、MgCO3、Na2CO3、K2CO3、トリエチルアミン、ジイソプロピルエチルアミンまたはN−メチルモルホリンであり、該有機溶媒が塩化メチレン、酢酸エチル、ジクロロエタン、テトラヒドロフラン、アセトニトリルまたはN,N−ジメチルホルムアミドである、請求項27に記載の方法。
【請求項30】
該トリアミン塩および該活性エステル体を、約30〜約40℃の温度範囲で反応させる、請求項25に記載の方法。
【請求項31】
該トリアミン塩および該活性エステル体を、塩基としてK2HPO4の存在下で、溶媒として塩化メチレン中反応させる、請求項30に記載の方法。
【請求項32】
該遊離塩基が遊離塩基の塩化メチレン溶液をN−メチルピロリドンおよびアセトンで処理し、上記の混合物を加熱し、塩化メチレンを除去して、上記混合物を硫酸で処理し、遊離塩基の重硫酸塩を形成することによって対応する重硫酸塩に変換される、請求項25に記載の方法。
【請求項33】
遊離塩基、アセトンおよびN−メチルピロリドンの混合物に重硫酸アタザナビルの結晶の種晶を添加する工程を含む、請求項32に記載の方法。
【請求項34】
該硫酸が、下式:
【数1】
[式中、
Vtime=経過時間内に加えられた硫酸の量
Vtotal=90%添加を表す酸の全量
time=結晶化における経過時間
timetotal=結晶化の全時間または酸を添加するための全時間]
に従う増加速度で加えられる、請求項25に記載の方法。
【請求項35】
重硫酸アタザナビル:
【化7】
を製造する方法であって、構造式:
【化8】
のトリアミン塩酸塩を調製し、
該トリメチル塩酸塩を塩化メチレン中、構造式:
【化9】
の活性エステル体およびK2HPO4と反応し、構造式:
【化10】
の遊離塩基の塩化メチレン溶液を形成し、
立方結晶化技術によって該遊離塩基を対応する重硫酸塩に変換することを含む方法。
【請求項36】
請求項9に記載される重硫酸アタザナビルのCパターン物質およびその医薬的に許容される担体を含む医薬製剤。
【請求項37】
重硫酸アタザナビルのCパターン物質、1またはそれ以上の充填剤、1またはそれ以上の崩壊剤、任意の1またはそれ以上の結合剤、および任意の1またはそれ以上の流動促進剤または滑沢剤を含む、請求項36に記載の製剤。
【請求項38】
重硫酸アタザナビルのCパターン物質、ラクトース、クロスポビドン、およびステアリン酸マグネシウムを含む、請求項36に記載の製剤。
【請求項39】
請求項16に記載される重硫酸アタザナビルのE3型または重硫酸アタザナビルのA型、およびその医薬的に許容される担体を含む医薬製剤。
【請求項40】
治療上の有効量の請求項9に記載される重硫酸アタザナビルのCパターン物質またはA型もしくはE3型を、治療の必要なヒトの患者に投与することからなるレトロウイルスによって引き起こされる疾病の治療方法。
【請求項1】
A型結晶形の重硫酸アタザナビルの製造方法であって、アタザナビルの重硫酸塩が実質的に不溶である有機溶媒中で、アタザナビル遊離塩基溶液をアタザナビル遊離塩基の約15重量%以下が反応する量の濃硫酸の一部と最初反応させ、重硫酸アタザナビルのA型結晶の種晶を反応混合物に加え、重硫酸アタザナビルの結晶が形成するように重硫酸アタザナビルの結晶形成を行うために多段階で追加の濃硫酸を加え、得られた重硫酸アタザナビルを乾燥して、A型結晶を形成することを特徴とする方法。
【請求項2】
該アタザナビル遊離塩基の溶液が、用いられる硫酸の全重量の約5〜約15%と最初に反応する、請求項1に記載の方法。
【請求項3】
該アタザナビル遊離塩基の溶液が、用いられる硫酸の全重量の約8〜約12%と最初に反応する、請求項1に記載の方法。
【請求項4】
該アタザナビル遊離塩基を、約35〜約55℃の温度範囲で硫酸の一部と最初に反応させる、請求項1に記載の方法。
【請求項5】
アタザナビル遊離塩基を、約35〜約55℃の温度範囲に加熱した後、硫酸と反応させる、請求項1に記載の方法。
【請求項6】
アタザナビル遊離塩基および硫酸の反応混合物にアタザナビル遊離塩基の重量に基づいて、A型結晶の約0.1〜約80重量%で種晶添加される、請求項1に記載の方法。
【請求項7】
種晶添加された反応混合物を、約35〜約55℃の温度範囲で加熱する、請求項1に記載の方法。
【請求項8】
アタザナビル遊離塩基のための有機溶媒がアセトン、アセトンおよびN−メチルピロリドンの混合物、エタノール、またはエタノールおよびアセトンの混合物である、請求項1に記載の方法。
【請求項9】
重硫酸アタザナビルのCパターン物質。
【請求項10】
図6に示されるパターンに実質的に一致する粉末X線回折パターンによってCパターン物質の重硫酸アタザナビルの特徴である、請求項9に記載の化合物。
【請求項11】
図7に示されるサーモグラムに実質的に一致する示差走査熱量サーモグラムによって特徴付けられる、請求項9に記載の化合物。
【請求項12】
図8に示される曲線は実質的に一致する熱重量分析曲線によって特徴付けられる、請求項9に記載の化合物。
【請求項13】
水中で重硫酸アタザナビルのA型結晶を懸濁すること、または相対湿度が少なくとも約95%RHで少なくとも24時間、重硫酸アタザナビルのA型結晶を付すこと、または重硫酸アタザナビルのA型結晶を湿式造粒し、次いで乾燥することにより調製される、請求項9に記載の化合物。
【請求項14】
請求項24の方法によって調製されるCパターン物質。
【請求項15】
重硫酸アタザナビルのA型結晶を、1またはそれ以上の製剤用賦形剤および水と混合し、続いて乾燥することによって調製される医薬組成物の形態である、請求項9に記載の化合物。
【請求項16】
重硫酸アタザナビルのE3型結晶。
【請求項17】
重硫酸アタザナビルのトリエタノレート溶媒和物として調製される、請求項16に記載の化合物。
【請求項18】
図9に示されるパターンに実質的に一致する粉末X線回折パターンによって特徴付けられる、請求項16に記載の化合物。
【請求項19】
実質的に図10に示される結晶構造を有する、請求項16に記載の化合物。
【請求項20】
実質的に表6に記載される原子分率座標によって特徴付けられる、請求項16に記載の化合物。
【請求項21】
下記:
[格子サイズ:
a=10.749(5)Å
b=13.450(4)Å
c=9.250(2)Å
α=98.33(2)°
β=95.92(3)°
γ=102.82(3)°
空間群P1
分子/非対称単位1]
と実質的に同等である結晶学的データによって特徴付けられ、該結晶形が約−23℃で存在する、請求項16に記載の化合物。
【請求項22】
図11で示されるサーモグラムに実質的に一致する示差走査熱量サーモグラムによって特徴付けられる、請求項16に記載の化合物。
【請求項23】
図11で示される曲線に実質的に一致する熱重量分析曲線によって特徴付けられる、請求項16に記載の化合物。
【請求項24】
(a)水中で、重硫酸アタザナビルのA型結晶を懸濁し、該懸濁液を乾燥して、Cパターン物質を形成するか;または
(b)95%RHより高い相対湿度で少なくとも24時間、重硫酸アタザナビルのA型結晶を付し、Cパターン物質を形成するか;または
(c)重硫酸アタザナビルを湿式造粒し、該湿造粒物を乾燥し、Cパターン物質を形成するか;または
(d)A型結晶を1またはそれ以上の製剤用賦形剤と混合し、生じた混合物を湿式造粒し、賦形剤との混合物であるCパターン物質を直接的に形成すること
を含む、請求項9に記載された重硫酸アタザナビルのCパターン物質の製造方法。
【請求項25】
A型結晶形の重硫酸アタザナビル:
【化1】
を製造する方法であって、構造式:
【化2】
のトリアミン塩を調製し、トリアミン塩を単離することなく、該トリアミン塩を有機溶媒の存在下で、構造式:
【化3】
の酸の活性エステル体および塩基と反応し、構造式:
【化4】
のアタザナビル遊離塩基の溶液を形成し、遊離塩基を対応する重硫酸塩に変換すること
を含む方法。
【請求項26】
トリアミン塩が、塩酸塩:
【化5】
である、請求項25に記載の方法。
【請求項27】
該酸の活性エステル体が、構造式:
【化6】
を有する、請求項25に記載の方法。
【請求項28】
該塩基がアルカリ金属ヒドロキシド、アルカリ土類金属ヒドロキシド、アルカリ金属カーボネート、アルカリ土類金属カーボネート、アルカリ金属ホスフェート、アルカリ土類金属ホスフェートまたは有機塩基である、請求項25に記載の方法。
【請求項29】
該塩基がNaOH、KOH、Mg(OH)2、K2HPO4、MgCO3、Na2CO3、K2CO3、トリエチルアミン、ジイソプロピルエチルアミンまたはN−メチルモルホリンであり、該有機溶媒が塩化メチレン、酢酸エチル、ジクロロエタン、テトラヒドロフラン、アセトニトリルまたはN,N−ジメチルホルムアミドである、請求項27に記載の方法。
【請求項30】
該トリアミン塩および該活性エステル体を、約30〜約40℃の温度範囲で反応させる、請求項25に記載の方法。
【請求項31】
該トリアミン塩および該活性エステル体を、塩基としてK2HPO4の存在下で、溶媒として塩化メチレン中反応させる、請求項30に記載の方法。
【請求項32】
該遊離塩基が遊離塩基の塩化メチレン溶液をN−メチルピロリドンおよびアセトンで処理し、上記の混合物を加熱し、塩化メチレンを除去して、上記混合物を硫酸で処理し、遊離塩基の重硫酸塩を形成することによって対応する重硫酸塩に変換される、請求項25に記載の方法。
【請求項33】
遊離塩基、アセトンおよびN−メチルピロリドンの混合物に重硫酸アタザナビルの結晶の種晶を添加する工程を含む、請求項32に記載の方法。
【請求項34】
該硫酸が、下式:
【数1】
[式中、
Vtime=経過時間内に加えられた硫酸の量
Vtotal=90%添加を表す酸の全量
time=結晶化における経過時間
timetotal=結晶化の全時間または酸を添加するための全時間]
に従う増加速度で加えられる、請求項25に記載の方法。
【請求項35】
重硫酸アタザナビル:
【化7】
を製造する方法であって、構造式:
【化8】
のトリアミン塩酸塩を調製し、
該トリメチル塩酸塩を塩化メチレン中、構造式:
【化9】
の活性エステル体およびK2HPO4と反応し、構造式:
【化10】
の遊離塩基の塩化メチレン溶液を形成し、
立方結晶化技術によって該遊離塩基を対応する重硫酸塩に変換することを含む方法。
【請求項36】
請求項9に記載される重硫酸アタザナビルのCパターン物質およびその医薬的に許容される担体を含む医薬製剤。
【請求項37】
重硫酸アタザナビルのCパターン物質、1またはそれ以上の充填剤、1またはそれ以上の崩壊剤、任意の1またはそれ以上の結合剤、および任意の1またはそれ以上の流動促進剤または滑沢剤を含む、請求項36に記載の製剤。
【請求項38】
重硫酸アタザナビルのCパターン物質、ラクトース、クロスポビドン、およびステアリン酸マグネシウムを含む、請求項36に記載の製剤。
【請求項39】
請求項16に記載される重硫酸アタザナビルのE3型または重硫酸アタザナビルのA型、およびその医薬的に許容される担体を含む医薬製剤。
【請求項40】
治療上の有効量の請求項9に記載される重硫酸アタザナビルのCパターン物質またはA型もしくはE3型を、治療の必要なヒトの患者に投与することからなるレトロウイルスによって引き起こされる疾病の治療方法。
【図1】

【図2】
【図3】
【図4】
【図5】
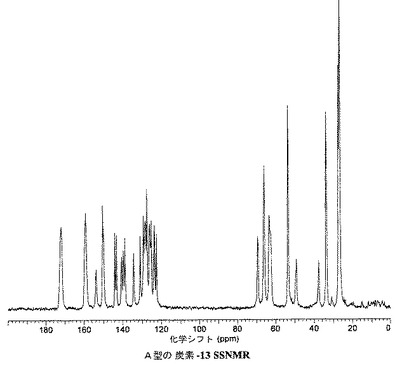
【図6】
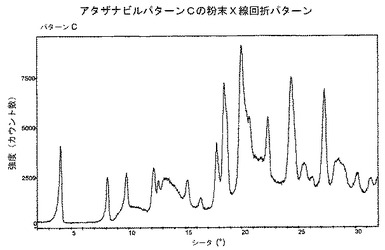
【図7】

【図8】
【図9】
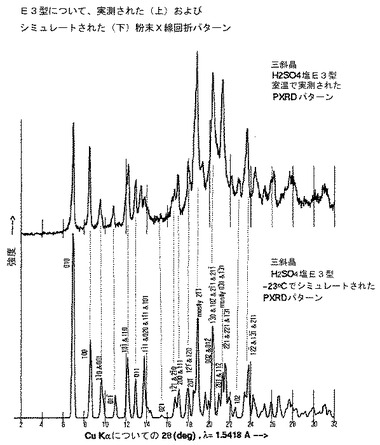
【図10】

【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2007−536245(P2007−536245A)
【公表日】平成19年12月13日(2007.12.13)
【国際特許分類】
【出願番号】特願2007−511502(P2007−511502)
【出願日】平成17年5月3日(2005.5.3)
【国際出願番号】PCT/US2005/015333
【国際公開番号】WO2005/108349
【国際公開日】平成17年11月17日(2005.11.17)
【出願人】(391015708)ブリストル−マイヤーズ スクイブ カンパニー (494)
【氏名又は名称原語表記】BRISTOL−MYERS SQUIBB COMPANY
【Fターム(参考)】
【公表日】平成19年12月13日(2007.12.13)
【国際特許分類】
【出願日】平成17年5月3日(2005.5.3)
【国際出願番号】PCT/US2005/015333
【国際公開番号】WO2005/108349
【国際公開日】平成17年11月17日(2005.11.17)
【出願人】(391015708)ブリストル−マイヤーズ スクイブ カンパニー (494)
【氏名又は名称原語表記】BRISTOL−MYERS SQUIBB COMPANY
【Fターム(参考)】
[ Back to top ]