
癌の処置方法およびその関連方法
【課題】癌を処置するための方法を提供すること。
【解決手段】4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンを用いた癌処置の方法が提供される。特に、その方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、または骨髄増殖病を含む固形腫瘍や白血病の処置に有効である。さらに、4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンの量を測定し、その結果、代謝プロフィールを決定する方法が提供される。
【解決手段】4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンを用いた癌処置の方法が提供される。特に、その方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、または骨髄増殖病を含む固形腫瘍や白血病の処置に有効である。さらに、4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンの量を測定し、その結果、代謝プロフィールを決定する方法が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2003年6月16日に提出された米国仮出願番号60/478,916;2003年4月3日に提出された米国仮出願番号60/460,369;2003年4月3日に提出された米国仮出願番号60/460,327;2003年4月3日に提出された米国仮出願番号60/460,493;2003年4月3日に提出された米国仮出願番号60/460,328;2002年11月13日に提出された米国仮出願番号60/426,204;2002年11月13日に提出された米国仮出願番号60/426,226;2002年11月13日に提出された米国仮出願番号60/426,282;2002年11月13日に提出された米国仮出願番号60/426,107および2003年11月7日に提出された「Methods of Treating Cancer and Related Methods」と題された米国仮出願に対する優先権を主張し、それらの各々が、十分に本明細書の前に記載されているかのように、その全体が、全ての目的のために参考文献として本明細書に緩用される。
(発明の分野)
本発明は、受容体チロシンキナ−ゼインヒビターを用いた癌の処置方法に関する。本発明はまた、被験体へのインヒビターの投与後の、そのインヒビターおよびその代謝物の量および濃度の測定方法に関する。
【背景技術】
【0002】
(発明の背景)
毛細血管は、人体のほぼ全ての組織に達し、組織に酸素および栄養物を供給し、老廃物の除去を行う。ヒトの成体では、典型的な条件下で、毛細血管の内部を覆う内皮細胞は分裂せず、それゆえ毛細血管は、通常数が増えないし、大きくもならない。しかし、ある通常の条件下で、組織が損傷されたような場合、または月経周期のある部分の間に、毛細血管は迅速に増殖し始める。既存の血管からの、新しい毛細血管の形成過程は、新脈管形成または新生血管形成として公知である。Folkman,J.Scientific American 275,150−154(1996)を参照のこと。創傷治癒中の新脈管形成は、成人生活における、病態生理的な新生血管形成の一例である。創傷治癒の間に、追加の毛細血管は酸素および栄養物の供給を提供し、肉芽組織を促進し、老廃物の除去を補助する。治癒過程の終結後、毛細血管は通常は消退する。Lymboussaki,A.”Vascular Endothelial Growth Factors and their Receptors in Embryos, Adults,and in Tumors”Academic Dissertation,University of Helsinki,Molecular/Cancer Biology Laboratory and Department of Pathology,Haartman Institute,(1999)。
【0003】
新血管形成はまた、癌細胞の増殖に重要な役割を果たす。癌細胞の巣窟は、一度ある一定の大きさ、大まかには直径1〜2ミリメートルの直径に達すれば、癌細胞に十分量の酸素および栄養物を供給するには拡散が十分でないので、腫瘍がより大きく成長するのに、癌細胞は血液供給を発達させなければならないことが公知である。
【0004】
レセプターチロシンキナーゼ(RTK)は、発生段階にある細胞の増殖および分化、そして成体の組織のリモデリングおよび再生を調節する膜貫通ポリペプチドである。Mustonen,T.ら、J.Cell Biology 129,895−898(1995);van der Geer,P.ら、Ann Rev.Cell Biol.10,251−337(1994)。増殖因子またはサイトカインとして公知のポリペプチドのリガンドは、RTKを活性化することが公知である。シグナル伝達をするRTKは、リガンドの結合およびレセプターを二量体化するレセプターの外部ドメインのコンフォメーション変化を含む。Lymboussaki,A.”Vascular Endothelial Growth Factors and their Receptors in Embryos, Adults, and in Tumors”Academic Dissertation,University of Helsinki,Molecular/Cancer Biology Laboratory and Department of Pathology,Haartman Institute,(1999);Ullrich,A.ら,Cell 61,203−212(1990)。リガンドのRTKへの結合は、レセプターの特定のチロシン残基でのリン酸転移反応を生じ、、細胞質に存在する基質のリン酸化の触媒ドメインの次の活性化に帰着する。
【0005】
RTKの二つのサブファミリーは、血管内皮細胞に特有である。これらは、血管内皮増殖因子(VEGF)サブファミリーおよびTieレセプターファミリーを含む。クラスIII型RTKは、VEGFR−1、VEGFR−2およびVEGFR−3を含む。Shibuya,Mら、Oncogene 5,519−525(1990);Terman,Bら、Oncogene 6,1677−1683(1991);Aprelikova,Oら、Cancer Res.52,746−748(1992)。
【0006】
VEGFサブファミリーのメンバーは、血管透過性および内皮細胞の増殖を誘導し得るとして記載され、さらに新脈管形成および脈管形成の主要な誘導物質として同定されている。Ferrara,N.ら、Endocrinol.Rev.18,4−25(1997)。VEGFは、VEGFR−1およびVEGFR−2を誘導するRTKに特異的に結合することが公知である。DeVries,C.ら、Science 255,989−991(1992);Quinn,Tら、Proc.Natl.Acad.Sci.90,7533−7537(1993)。VEGFは内皮細胞の移動および増殖を刺激し、インビトロおよびインビボの両方で新脈管形成を誘導する。Connolly,D.ら、J.Biol.Chem.264,20017−20024(1989);Connolly,D.ら、J.Clin.Invest.84,1470−1478(1989);Ferrara,N.ら、Endocrino.Rew.18,4−25(1997);Leung,D.ら、Science 246,1306−1309(1989);Plouet,J.ら、EMBO J8、3801−3806(1989)。
【0007】
新脈管形成は、癌の増殖に決定的であるということならびにVEGFおよびVEGF−RTKにより制御されることが公知であるので、VEGF−RTKのアンタゴニストであり、それゆえ新脈管形成を阻害し、または遅らせ、望ましくは腫瘍の増殖を妨害し、または停止させる治療剤を開発するのにかなりの努力が保証された。
【0008】
増殖因子受容体キナーゼ(PDGFRK)由来の血小板は、RTKの別のタイプである。PDGFの発現は、グリア芽細胞腫から前立腺癌までの、多数の様々な固形腫瘍に示されている。これらの様々な腫瘍タイプでは、PDGFシグナル伝達の生物学的な役割は、癌細胞の増殖のオートクリン刺激から、隣のストローマおよび新脈管形成を含む、より微妙なパラクリン相互作用まで変化し得る。従って、PDGFRキナーゼの活性の小分子による抑制は、腫瘍の増殖および新脈管形成を妨害し得る。
【0009】
4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンは、VEGFーRTK、PDGF−RTKおよび線維芽細胞増殖因子受容体(FGF−RTK)のような他の受容体チロシンキナーゼの小分子インヒビターである。この化合物は、一つの特許およびいくつかの特許出願に記述されており、その開示全体は、参考文献としておよび全ての目的で本明細書中に援用される:米国特許第6,605,617号、U.S.S.N.10/644,055、米国仮出願番号60/405,729、60/428,210および60/484,048。この潜在的抗癌薬剤の代謝プロフィールの決定のための方法が必要であるように、本化合物を投与する特定の方法が必要である。
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の要約)
本発明は、白血病および固形腫瘍を含む癌の処置方法を提供する。特に、被験体で癌の増殖を抑制する十分な血液レベルの4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンを得るための方法が提供される。この化合物は、受容体チロシンキナーゼのインヒビターである。バイオマーカーとしての化合物およびそのような化合物を用いて被験体中でのインヒビターの分布および代謝をモニターするための方法がさらに提供される。さらに、本発明は、薬学的組成物ならびにインヒビターを含む医薬およびそれらの使用方法を提供する。
【0011】
本発明は、例えば、以下を提供する。
(項目1)
癌処置のための単位投薬形態の医薬の調製における、式
【化1】

の化合物、その薬学的に受容可能な塩、その互変異性体、または該互変異性体の薬学的に受容可能な塩の使用であって、ここで、該医薬の各々の単位用量は、以下:
(a)該化合物が被験体に投与された場合、該被験体の血漿中での、約20ng/mL〜4000ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約40ng/mL〜8000ng/mLの該化合物のCmax値、
(b)被験体への投与の24時間後の、該被験体の血漿中での、約10ng/mL〜2,000ng/mLの該化合物もしくは被験体への投与の24時間後の該被験体の血液中での、約20ng/mL〜4,000ng/mLの該化合物、または
(c)該化合物が被験体に投与された場合、該被験体の血漿中での約500ng*h/mL〜60,000ng*h/mLの該化合物もしくは該被験体の血液中での約750ng*h/mLから120,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目2)
項目1に記載の使用であって、ここで、各々の単位用量は、以下:
(a)前記被験体の血漿中での、約50ng/mL〜500ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約100ng/mL〜1000ng/mLの該化合物のCmax値、
(b)投与の24時間後の、該被験体の血漿中での、約20ng/mL〜1,000ng/mLの該化合物もしくは投与の24時間後の、該被験体の血液中での、約40ng/mL〜2,000ng/mLの該化合物、または
(c)該被験体の血漿中での約1,000ng*h/mL〜30,000ng*h/mLの該化合物もしくは該被験体の血液中での約1,500ng*h/mL〜60,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目3)
項目1に記載の使用であって、ここで、各々の単位用量は、以下:
(a)前記被験体の血漿中での、約50ng/mL〜250ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約100ng/mL〜500ng/mLの該化合物のCmax値、
(b)該被験体への投与の24時間後の、該被験体の血漿中での、約40ng/mL〜500ng/mLの該化合物もしくは該被験体への投与の24時間後の、該被験体の血液中での、約80ng/mL〜1,000ng/mLの該化合物、または
(c)該被験体の血漿中での約2,000ng*h/mL〜15,000ng*h/mLの該化合物もしくは該被験体の血液中での約3,000ng*h/mL〜30,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目4)
項目1の使用であって、ここで、各々の単位用量は、以下:
(a)該被験体の血漿中での、約75ng/mL〜150ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約150ng/mL〜300ng/mLの該化合物のCmax値、または
(b)投与の24時間後の、該被験体の血漿中での、約40ng/mL〜250ng/mLの該化合物もしくは投与の24時間後の、該被験体の血液中での、約80ng/mL〜500ng/mLの該化合物、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目5)
項目1に記載の使用であって、ここで、各々の単位用量が、前記被験体の血漿中で約100ng/mL〜2000ng/mLの化合物のCmax値または該被験体の血液中で約200ng/mL〜4000ng/mLの化合物のCmax値を提供するのに十分である、使用。
(項目6)
項目1に記載の使用であって、ここで、各々の単位用量が前記被験者の血漿中での100ng/mL〜1000ng/mLの前記化合物のCmax値または該被験体の血液中での200ng/mL〜2000ng/mLの該化合物のCmax値を提供するのに十分である、使用。
(項目7)
項目1〜6のいずれか1項に記載の使用であって、ここで、化合物の乳酸塩が前記医薬を調製するために使用される、使用。
(項目8)
項目1〜7のいずれか1項に記載の使用であって、ここで、前記医薬が経口投与に適切である、使用。
(項目9)
項目8に記載の使用であって、ここで、前記医薬の前記単位投与形態が、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁剤または水溶液である、使用。
(項目10)
項目1〜7のいずれか1項に記載の使用であって、ここで、前記医薬が、短いボーラス、遅い注入または長期の注入としての注射による投与に適している、使用。
(項目11)
項目1〜10のいずれか1項に記載の使用であって、ここで、前記医薬の各々の単位用量が、前記被験体の体重に基づいて、体重1kg当たり0.25mg〜30mgの前記化合物、互変異性体、および/または塩を含む、使用。
(項目12)
項目1〜10のいずれか1項に記載の使用であって、ここで、前記医薬の各々の単位用量が、25mg〜1500mgの範囲の量の前記化合物、互変異性体および/または塩を含む、使用。
(項目13)
項目1〜12のいずれか1項に記載の使用であって、ここで、前記医薬が、7個、14個、21個または28個の毎日の量の上記単位用量を含むキットに配置され、該キットが、7日間、14日間、21日間または28日間の処置周期での使用に適する、各々の前記化合物の前記毎日量の投与、続く該化合物の投与なしの7日または14日を含む、使用。
(項目14)
式
【化2】
を有する化合物、その薬学的に受容可能な塩、その互変異性体または該互変異性体の薬学的に受容可能な塩であって、該化合物が、癌処置の方法に使用するためのものであり、該癌処置の方法が、上記化合物のある量を癌患者に投与することを含み、その量が、
(a)該化合物が被験体に投与された場合、該被験体の血漿中で約20ng/mL〜4000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約40ng/mL〜8000ng/mLの該化合物のCmax値、
(b)被験体への投与の24時間後の該被験体の血漿中で約10ng/mL〜2,000ng/mLの該化合物もしくは該被験体への投与の24時間後の該被験体の血液中で約20ng/mL〜4,000ng/mLの該化合物、または
(c)該化合物が被験体に投与された場合、該被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物もしくは該被験体の血液中で約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目15)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約50ng/mL〜500ng/mLの該化合物のCmax値もしくは該被験体の血液中で約100ng/mL〜1000ng/mLの該化合物のCmax値、
(b)投与の24時間後の該被験体の血漿中で約20ng/mL〜1,000ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約40ng/mL〜2,000ng/mLの該化合物、または
(c)該被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの該化合物もしくは該被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの該化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目16)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約50ng/mL〜250ng/mLの該化合物のCmax値もしくは該被験体の血液中で約100ng/mL〜500ng/mLの該化合物のCmax値、
(b)投与の24時間後の該被験体の血漿中で約40ng/mL〜500ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約80ng/mL〜1,000ng/mLの該化合物、または
(c)該被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの該化合物もしくは該被験体の血液中で約3,000ng*h/mL〜30,000ng*h/mLの該化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目17)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約75ng/mL〜150ng/mLの該化合物のCmax値もしくは該被験体の血液中で約150ng/mL〜300ng/mLの該化合物のCmax値、または
(b)投与の24時間後の該被験体の血漿中で約40ng/mL〜250ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約80ng/mL〜500ng/mLの該化合物、
の少なくとも一つを提供するのに十分である、化合物。
(項目18)
項目14に記載の化合物であって、ここで、各々の単位用量が、前記被験体の血漿中で100ng/mL〜2000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約200ng/mL〜4000ng/mLの該化合物のCmax値を提供するのに十分である、化合物。
(項目19)
項目14に記載の化合物であって、ここで、各々の単位用量が、前記被験体の血漿中で100ng/mL〜1000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約200ng/mL〜2000ng/mLの該化合物のCmax値を提供するのに十分である、化合物。
(項目20)
被験体中の、以下の式
【化3】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の代謝プロフィールを決定するための、代謝物の使用であって、該代謝物が、以下の式
【化4】
のN−オキシド化合物または以下の式
【化5】
N−デスメチル化合物の少なくとも一つを含む、使用。
(項目21)
被験体中の、以下の式:
【化6】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の代謝プロフィールを決定する方法であって、該方法が、該被験体から採られた尿、血液または組織の一つ以上のサンプル中の、該化合物の代謝物の少なくとも一つの量を測定することを含む、方法。
(項目22)
項目21に記載の方法であって、ここで、前記少なくとも一つの代謝物が、以下の式:
【化7】
を有するN−オキシド化合物であるか、または
以下の式:
【化8】
を有するN−デスメチル化合物である、方法。
(項目23)
項目22に記載の方法であって、ここで、該方法が、前記N−オキシド化合物および前記N−デスメチル化合物の両方の前記量を測定することを含む、方法。
(項目24)
項目21〜23のいずれかに記載の方法であって、ここで、前記代謝物が、紫外線分光器法または液体クロマトグラフィー−質量分光法により測定される、方法。
(項目25)
被験体中の、以下の式:
【化9】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または該互変異性体の薬学的に受容可能な塩の前記量を測定する方法であって、該方法は、該化合物が該被験体に投与された後、該被験体から採られる尿、血液、または組織のサンプル中の該化合物の量を測定することを含む。
(項目26)
項目25に記載の方法であって、該方法が、前記サンプル中の前記化合物の代謝物の量を測定することをさらに含む、方法。
(項目27)
項目26に記載の方法であって、ここで、前記代謝物が以下の式:
【化10】
を有するN−オキシド化合物である、方法。
(項目28)
項目26に記載の方法であって、ここで、前記代謝物が、以下の式:
【化11】
を有するN−デスメチル化合物である、方法。
(項目29)
項目25に記載の方法であって、前記被験体に前記化合物が投与された後、該被験体から異なる時点に二つ以上のサンプルを抜き出すことをさらに含む、方法。
(項目30)
癌処置のための単位投与形態の、医薬の前記調製における、以下の式:
【化12】
の化合物、その薬学的に受容可能な塩、その互変異性体、該互変異性体の薬学的に受容可能な塩の使用であって、ここで、該医薬の各々の単位用量は、25mg〜1000mgの量の該化合物を含む、使用。
【0012】
従って、本発明に従って、癌を処置するための方法が提供され、この方法は、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmax値もしくは被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax値を提供するのに十分な量の式Iの化合物:
【0013】
【化13】
その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩を、癌を有する被験体に対して投与する工程を含有する。ある実施形態では、投与される化合物の量は、被験体の血漿中で約35ng/mL〜2,000ng/mLの化合物のCmax値もしくは被験体の血液中で約70ng/mL〜4,000ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約50ng/mL〜500ng/mLの化合物のCmax値もしくは被験体の血液中で約100ng/mL〜1,000ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約50ng/mL〜250ng/mLの化合物のCmax値もしくは被験体の血液中で約100ng/mL〜500ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値もしくは被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約100ng/mL〜2,000ng/mLの化合物のCmax値もしくは被験体の血液中で約200ng/mL〜4,000ng/mLの化合物のCmax値を与えるのに十分な量である。式Iの化合物の乳酸塩は、ある実施形態で被験体に投与され、そしていくつかのそのような実施形態において被験体はヒトである。化合物、互変異性体、またはその塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液、水溶液、または本明細書に記載の他の形態として処方され得る。いくつかのこのような実施形態では、乳酸塩は水溶液であり、ヒト被験体に経口投与される。他の実施形態では、化合物は注射により投与され得る。
【0014】
さらなる局面では、本発明は、癌を処置する方法を提供し、この方法は、投与の24時間後に被験体の血漿中に約10ng/mL〜2,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約20ng/mL〜4,000ng/mLの化合物を提供するのに十分な量の式Iの化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩を、癌を有する被験体へ投与する工程を含有する。いくつかの実施形態では、投与される化合物量は、投与の24時間後に被験体の血漿中に約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約40ng/mL〜2,000ng/mLの化合物、投与の24時間後に被験体の血漿中に約40ng/mL〜500ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約80ng/mL〜1,000ng/mLの化合物、または投与の24時間後に被験体の血漿中に約40ng/mL〜250ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約80ng/mL〜500ng/mLの化合物を提供するのに十分な量である。いくつかの実施形態では、被験体はヒトである。一般に、癌処置の本方法では、式Iの化合物の乳酸塩は被験体に投与される。いくつかのそのような実施形態では、乳酸塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液または水溶液であり、ヒト被験体に経口投与される。
【0015】
従って、癌の本処置方法ある実施形態では、式Iの化合物は、フルクトースを含む薬学的組成物または医薬として投与される。そのような実施形態では、薬学的組成物はさらにテトラロームマンダリン風味のような矯味矯臭剤を含む。他の実施形態では、薬学的化合物は、さらに水を含む。従って、癌処置の本方法は、式Iの固形化合物を水と混和し、水性混合物を形成するために、式Iの固形化合物と水とを混和することをさらに含み得る。その発明は、式Iの化合物の用途をさらに提供する。本発明は、癌処置に使用するための医薬の調合をする際の、式Iの化合物の使用をさらに提供する。
【0016】
本明細書に記載の癌の処置方法に関する他の実施形態では、化合物は、顆粒剤、散剤、懸濁剤、錠剤、丸剤、カプセル剤、ゲルキャップ、キャプレット、エマルジョン、、シロップ剤、エリキシル剤、懸濁液、スプレー、エアロゾル、座薬または水剤から選択された薬学的組成物として投与される。好ましくは、薬学的組成物は錠剤、癌剤、カプセル剤、ゲルキャップまたはキャプレットから選択される。
【0017】
本明細書に記載の癌処置の本方法に関する、さらに別の実施形態では、化合物は、短いボーラス、遅い注入、または長期の注入として注射により投与される。注射は、日に一回、二回、三回または四回投与され得る。
【0018】
癌処置の本方法のある実施形態では、被験体に投与される式Iの化合物の量は、被験体の体重により、0.25mg/kg〜0.30mg/kgの範囲に及ぶ。他の実施形態では、被験体に投与される化合物の量は、一日当たり約25mg〜1,500mgであり、好ましくは一日当たり約200mg〜500mgの範囲である。
【0019】
癌の本処置方法は、処置されるべき癌が固形腫瘍または白血病である人々を含む、広い様々な種類の癌に対して有効である。特に、本方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖病、非小細胞肺癌、小細胞肺癌、慢性リンパ性白血病、肉腫、黒色腫、リンパ腫、甲状腺腫、神経内分泌の癌、腎細胞癌、胃癌、消化管間質潰瘍、神経膠腫、脳腫瘍または膀胱癌のような癌を処置するのに使われ得る。
【0020】
いくつかの実施形態では、本明細書に記載の癌の処置方法は、処置周期の一部としての式Iの化合物の投与をさらに含む。従って、処置周期は、その量の式Iの化合物の7日間、14日間、21日間または28日間にわたる毎日の投与、続く化合物の投与のない7日間または14日間を含み得る。いくつかの実施形態では、処置周期は、その量の化合物の7日間にわたる毎日の投与、続く化合物の投与のない7日間を含む。処置の過程を提供するため、処置周期は、一回以上繰り返され得る。さらに、化合物は、処置周期の投与段階の間、日に一回、二回、三回または四回投与され得る。他の実施形態では、その方法は、処置の過程の間に、一日または一日おきに一回、二回、三回または四回のその化合物の量の投与をさらに含む。
【0021】
さらに、癌処置の方法が提供され、その方法は、被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物または被験体の血液中で約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値を提供するのに十分な量の式
【0022】
【化14】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩を、癌を有する被験体へ投与する工程を含有する。他のそのような実施形態では、投与される化合物の量は、被験体の血漿中で、約1,000ng*h/mL〜30,000ng*h/mLの化合物または被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値を提供するのに十分である。他のそのような実施形態では、AUC値は、被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの化合物または被験体の血液中での3,000ng*h/mL〜30,000ng*h/mLの化合物である。
【0023】
本発明は、癌の処置方法をさらに提供し、その方法は、式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体で薬学的に受容可能な塩を、癌を有する被験体に投与することを含む。ここで、第1の処置周期での投与される化合物の量は、一日当たり25mgであり、そして投与される化合物の量は、一日当たり1500mgの化合物が被験体に投与されるまでか、または被験体に用量限界の毒性が観察されるまで、各処置周期につれて増える。概してそのような方法では、投与される化合物の量は、第1の周期の後、処置周期毎に投与される化合物の量が倍加する。いくつかの実施形態では、処置周期は、7日間にわたる毎日の等量の化合物の投与、続く化合物が投与されない7日間を含む。
【0024】
他の局面では、本発明は、癌の処置方法を提供し、その方法は、式I
【0025】
【化15】
の化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分量を、癌を有する被験体へ投与すること、および以下の式
【0026】
【化16】
から選択される式IIならびに式IIIの一つあるいは両方の、被験体への曝露を含む。ここで、式IIおよび式IIIの一つあるいは両方が、被験体による式Iの化合物の代謝により産生されて、被験体の血漿中で約20ng/mL〜約4000ng/mLの範囲に及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の合計Cmax値または被験体の血液中で約40ng/mL〜約8000ng/mLの範囲に及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の合計Cmax値を提供する。
【0027】
さらに別の局面では、本発明は、癌処置の方法を提供し、その方法は、以下
【0028】
【化17】
から選択される式を有する一つ以上の化合物、その活性代謝物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分量に、癌を有する被験体を曝すことを含み、それらの化合物は、被験体の血漿中での、約20ng/mL〜4000ng/mLの一つ以上の化合物の合計Cmax値または被験体の血液中での約40ng/mL〜8000ng/mLの一つ以上の化合物の合計Cmax値を提供するのに十分な量である。いくつかの実施形態では、一つ以上の化合物の量は、被験体の血漿中での、約35ng/mL〜2600ng/mLの一つの化合物のCmax値または被験体の血液中での、35ng/mL〜6000ng/mLの一つの化合物のCmax値を提供する。他の実施形態では、一つ以上の化合物の量は、被験体の血漿中での、約35ng/mL〜1200ng/mLの化合物の一つのCmax値または被験体の血液中での、約50ng/mL〜2400ng/mLの化合物の一つのCmax値を提供する。
【0029】
本発明の他の局面では、式Iの化合物、その薬学的に受容可能な塩、その互変異性体またはその互変異性体の薬学的に受容可能な塩の代謝プロフィールを被験体において決定する方法が提供される。その方法は、被験体から採られた尿、血液、または組織の一つ以上のサンプルの化合物の少なくとも一つの代謝物の量を測定することを含む。いくつかのそのような実施形態では、少なくとも一つの代謝物は、以下の式II
【0030】
【化18】
を有するN−オキシド化合物である。
【0031】
他のそのような実施形態では、少なくとも一つの代謝物は、式III
【0032】
【化19】
を有するN−デスメチル化合物である。
【0033】
いくつかのそのような実施形態では、少なくとも一つの代謝物は、式IIのN−オキシド化合物である第2の代謝物をさらに含む。代謝物の量は、紫外線(UV)分光学および/または液体クロマトグラフィー−質量分光学(LC−MS)を含む技術を用いて測定され得る。
【0034】
本発明の他の局面では、被験体中の式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の量を決定する方法が提供される。この方法は、その化合物が被験体に投与された後に、被験体から採られた尿、血液または組織の一つのサンプル中の化合物の量を測定することを含む。この方法は、サンプル中の化合物の代謝物の量を測定することをさらに含み得る。測定され得る代謝物としては、式IIのN−オキシド化合物および/または式IIIを有するN−デスメチル化合物が挙げられ得るが、これらに限定されない。いくつかの実施形態では、その方法は、式Iの化合物が被験体へ投与された後の異なる時点で被験体からの二つ以上のサンプルを回収することをさらに含む。
【0035】
本発明の他の目的、特徴および利点は、以下の詳細な説明から明らかになる。しかし、発明の精神および範囲内の様々な変更および改変は詳細な説明から当業者に明らかになるので、詳細な説明および具体的実施例は、本発明のある実施形態を示していると同時に、単なる例示として与えられる、ということは理解されるべきである。
【図面の簡単な説明】
【0036】
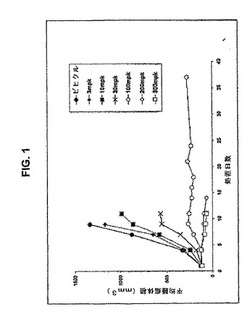
【図1】図1は、式Iの化合物によるKM12L4aの腫瘍抑制を示している。
【図2】図2は、Cmax値およびAUC値対KM12L4a腫瘍保有マウスでのKM12L4a腫瘍の増殖の抑制パーセントを示している。
【発明を実施するための形態】
【0037】
(詳細な説明)
本発明は、式Iの化合物を用いた癌の処置方法、被験体から採られる生物学的サンプル中の式Iの化合物および/またはその代謝物の量を測定する方法、ならびに式Iの化合物を含む薬学的組成物および医薬、そしてそれらの使用方法に関する。
【0038】
本明細書中で定義される以下の専門用語および語句は、本明細書を通して使用される。
【0039】
本明細書で用いられる「AUC」は、時間の経過に対する血漿中の化合物の濃度のグラフ中での曲線下の面積のことを指す。
【0040】
本明細書で用いられているように、「Cmax」は、化合物が投与された被験体の血漿中、組織中、血液中の化合物の最高濃度を指す。概してCmaxは、被験体への化合物の投与の数時間以内に生じる。
【0041】
毒性を制限する用量は、Common Terminology Criteria of Adverse Events Version 3.0(CTCAE)に従って規定される。従って、薬物処置周期内に次の事象のいずれかが観察される場合、毒性制御用量は、化合物の被験者への投与と共に生じる:5日間以上の連続した日数の段階4の好中球減少症(すなわち、絶対好中球数(ANC)≦500個細胞/mm3)または発熱好中球減少症(すなわち、発熱≧38.5℃かつANC≦1000個細胞/mm3);段階4の血小板減少症(すなわち、≦25,000個細胞/mm3または血小板輸血を必要とする出血エピソード);段階4の疲労、またはECOGパフォーマンスステータスにおける二点減少;十分/最大の医学的介入にも関わらない、段階3以上の悪心、下痢、嘔吐および/または筋痛;段階3以上の非血液学的毒性(疲労を除く);化合物1を用いた処置に関連した、毒性からの遅れた回復のための、2週間より長い再処置の遅延;段階2以上の、臨床的に重要な心毒性(例えば、休止駆出率の40%≦50%の低下または短縮画分の15%≦24%の低下;心トロポニンT≧0.05ng/mL)。
【0042】
薬学的に受容可能な塩としては、無機塩基、有機塩基、無機酸、有機酸または塩基性もしくは酸性アミノ酸が挙げられる。無機塩基の塩として、本発明は、例えば、ナトリウムまたはカリウムのようなアルカリ金属、カルシウムおよびマグネシウムようなアルカリ土類金属、アルミニウム、ならびにアンモニアを含む。有機塩基の塩として、本発明は、例えば、トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、エタノールアミン、ジエタノールアミン、およびトリエタノールアミンを含む。無機酸の塩として、本発明は、例えば塩酸、臭化水素酸、硝酸、硫酸、およびリン酸を含む。有機酸の塩として、本発明は、例えば、乳酸、ギ酸、酢酸、トリフルオロ酢酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホニル酸、ベンゼンスルホン酸およびp−トルエンスルホン酸を含む。塩基性アミノ酸の塩として、本発明は例えば、アルギニン、リジンおよびオルニチンを含む。酸性アミノ酸は、例えば、アスパラギン酸およびグルタミン酸を含む。
【0043】
本発明に従うその有機化合物が、互変異性の現象を示し得るということは理解されるべきである。本明細書内の化学構造は、一時点で起こり得る互変異性体形態の一つのみを表し得るので、本発明は描かれた構造の任意の互変異性体形態を含有するということは理解されるべきである。例えば、式Iの化合物は、一つの互変異性体である互変異性体Iaと共に以下:
【0044】
【化20】
に示されている。
【0045】
式Iの化合物の他の互変異性体である互変異性体Ibおよび互変異性体Icが以下:
【0046】
【化21】
に示されている。
【0047】
当業者が容易に理解するように、幅広い様々な官能基および他の構造は、互変異性を示し得、そして式Iを有する化合物の全ての互変異性体は、本発明の範囲内である。
【0048】
「被験体」という用語は、本明細書で用いられている場合、本発明の方法の有益な効果を経験し得る任意の動物を指す。従って、式Iの化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩は、本発明により提供される癌の処置方法に従って、その化合物の有益な効果を経験し得る任意の動物に投与され得る。好ましくは、その動物は哺乳動物、特にヒトであるが、本発明はそのように制限されるように意図されていない。他の適切な動物の例としては、ラット、マウス、サル、イヌ、ネコ、ウシ、ウマ、ブタ、ヒツジなどが挙げられるが、これらに制限されていない。
【0049】
本発明の文脈において、「処置」は、障害もしくは疾患と関連する症状の軽減、それらの症状のさらなる進行もしくは悪化の停止、または疾患もしくは障害の防止もしくは予防を意味する。例えば、癌の文脈において、好結果の処置は、腫瘍の増殖速度の減少、腫瘍の増殖の停止、腫瘍の大きさの減少、癌の部分的寛解もしくは完全寛解、または生存率もしくは臨床的利点の増加から測定されるように、症状の軽減または疾患の進行の停止を含み得る。
【0050】
一つの局面では、本発明は癌処置の方法を提供し、その方法は癌を有する被験体に、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmax値または被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax値を提供する、式Iの化合物:
【0051】
【化22】
その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分な量を投与することを含む。いくつかの実施形態では、式Iを有する化合物の投与量は、被験体の血漿中で約35ng/mL〜2000ng/mLの化合物のCmax値または被験体の血液中で約70ng/mL〜4000ng/mLの化合物のCmax値、被験体の血漿中で約50ng/mL〜500ng/mLの化合物のCmax値または被験体の血液中で約100ng/mL〜1000ng/mLの化合物のCmax値、被験体の血漿中で約50ng/mL〜250ng/mLの化合物のCmax値または被験体の血液中で約100ng/mL〜500ng/mLの化合物のCmax値、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値または被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値、被験体の血漿中で約100ng/mL〜2000ng/mLの化合物のCmax値または被験体の血液中で約200ng/mL〜4000ng/mLの化合物のCmax値、被験体の血漿中で約100ng/mL〜1000ng/mLの化合物のCmax値または被験体の血液中で約200ng/mL〜2000ng/mLの化合物のCmax値を提供するのに十分である。好ましくは、投与される化合物の量は、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値または被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値を提供するのに十分である。従って、式Iの化合物、互変異性体およびその塩の十分な量により提供されるCmax値は所定の範囲内に入るということが理解されるべきである。
【0052】
さらなる局面では、本発明は、癌の処置方法を提供し、その方法は、式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の十分量を、癌を有する被験体に対して投与することを含み、この量は、投与の24時間後に被験体の血漿中で約10ng/mL〜2,000ng/mLの化合物を、もしくは投与の24時間後に被験体の血液中で約20ng/mL〜4,000ng/mLの化合物を提供する。いくつかの実施形態では、投与される化合物の量は、投与の24時間後に被験体の血漿中に約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約40ng/mL〜2,000ng/mLの化合物、投与の24時間後に被験体の血漿中に約40ng/mL〜500ng/mLの化合物もしくは投与の24時間後に被験体の血液中に80ng/mL〜1,000ng/mLの化合物、または投与の24時間後に被験体の血漿中に40ng/mL〜250ng/mLの化合物もしくは投与の24時間後に被験体の血液中に80ng/mL〜500ng/mLの化合物を提供するのに十分な量である。
【0053】
概して、本明細書に記載の癌の処置方法では、式Iの化合物またはその互変異性体は、薬学的に受容可能な塩として投与される。乳酸塩、リンゴ酸塩、メシラート、酢酸塩、酒石酸塩、リン酸塩、硫酸塩、硝酸塩、塩酸塩、クエン酸塩またはマレイン酸塩のような塩は、様々なモル比で、エナンチオマー形態またはラセミ体形態で適切である。好ましくは、式Iの化合物の乳酸塩は、ヒト被験体のような被験体に投与される。その乳酸塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液、または水溶液として患者に都合良く投与され経口投与される。他の実施形態では、化合物または塩は、以下に記述されるように、注射により投与され得る。
【0054】
従って、癌処置の本方法のいくつかの実施形態では、式Iの化合物は、フルクトースを含む薬学的組成物または医薬として投与される。そのような組成物はまた、テトラロームマンダリン風味などのような矯味矯臭剤および/または水のような希釈剤を含み得る。従って、癌処置の本方法は、式Iの固形化合物を水と混和し、そして被験体にその化合物を投与する前に水性混合物を形成することをさらに含み得る。本発明は、癌処置に用いるための医薬の調製における式Iの化合物1の使用をさらに提供する。
【0055】
癌処置の本方法のいくつかの実施形態において、被験体に投与される式Iの化合物の量は、被験体の体重1kg当たり0.25mg〜30mgに及ぶ。他の実施形態では、被験体に投与される化合物の量は、一日当たり約25mg/被験体〜1500mg/被験体、一日当たり約100mg/被験体〜1000mg/被験体、または一日当たり200mg/被験体〜500mg/被験体の範囲である。
【0056】
癌処置の本方法は、処置されるべき癌が固形腫瘍または白血病であるものを含む、幅広い様々な癌に対して有効である。特に、本方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖病、非小細胞肺癌、小細胞肺癌、慢性リンパ性白血病、肉腫、黒色腫、リンパ腫、甲状腺腫、神経内分泌の癌、腎細胞癌、胃癌、消化管間質腫瘍、神経膠腫、脳腫瘍または膀胱癌のような癌の処置に使用され得る。理論に縛られることは望まないが、癌処置の本方法は、固形腫瘍に有効であると考えられている。なぜなら、式Iの化合物は、新脈管形成の阻害剤として作用するからである。より正確にいえば、式Iの化合物およびその活性代謝物は、腫瘍の新脈管形成および白血病に関与している特定のレセプターチロシンキナーゼを選択的に阻害すると考えられている。
【0057】
いくつかの実施形態では、癌処置の本方法は、式Iの化合物の処置周期の一部としての投与をさらに含む。処置周期は、化合物が被験体に規則正しい基準で与えられる投与段階および化合物が投与されない休日を含む。例えば、処置周期は、7日間、14日間、21日間または28日間にわたる、毎日の式Iの化合物のその量の投与、続く化合物が投与されない7日もしくは14日を含み得る。いくつかの実施形態では、処置周期は、7日間にわたる毎日のその量の化合物の投与、続く化合物が投与なしの7日間を含む。処置周期は、一回以上繰り返され得、処置過程を提供し得る。さらに、その化合物は、処置周期の投与段階の間、1日に一回、二回、三回または四回投与され得る。他の実施形態では、その方法は処置の過程の間に、1日にまたは隔日に一回、二回、三回または四回のその量の化合物の投与を含み得る。処置の過程は、被験体が、本方法によって癌処置を受けている時間に言及している。従って、処置の過程は、一回以上の処置周期に拡張し得るか、または被験体が毎日もしくは断続的に式Iの化合物の用量を受け取っている時間に言及し得る。
【0058】
さらに癌の処置方法が提供され、その方法は、式Iの化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の、十分量を癌を有する被験体に投与することを含み、それらの量は、被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物または被験体の血液中に約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値を提供する。そのような他の実施形態では、投与される化合物の量は、被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの化合物または被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値を提供するのに十分な量である。そのような他の実施形態では、AUC値は、被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの化合物または被験体の血液中で約3,000ng*h/mL〜30,000ng*h/mLの化合物である。
【0059】
本発明の別の局面では、癌処置のための単位投薬形態での医薬の調製における、式Iの化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の使用が、提供される。ここで、各々の医薬の単位用量は、以下:
(a)化合物が被験体に投与された場合の、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmaxもしくは被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax、
(b)被験体への投与の24時間後の被験体の血漿中での約10ng/mL〜2,000ng/mLの化合物もしくは被験体への投与の24時間後の被験体の血液中での約20ng/mL〜4,000ng/mLの化合物、または
(c)化合物が被験体に投与された場合、被験体の血漿中での約500ng*h/mL〜60,000ng*h/mLの化合物もしくは被験体の血液中での約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である。
【0060】
癌処置のための医薬の調製における、式Iの化合物の使用のいくつかの実施形態では、各々の単位用量は以下:
(a)被験体の血漿中での約50ng/mL〜500ng/mLの化合物のCmax値もしくは被験体の血液中で100ng/mL〜1000ng/mLの化合物のCmax値
(b)投与の24時間後での被験体の血漿中の約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後での被験体の血液中の約40ng/mL〜2,000ng/mLの化合物、または
(c)被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの化合物のAUC値もしくは被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である。
【0061】
他の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜250ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜500ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ40〜500ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜1,000ng/mLの化合物、または
(c)被験体の血漿中の化合物のおよそ2,000〜15,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ3,000〜30,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0062】
さらに他の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ75〜150ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ150〜300ng/mLのCmax、もしくは
(b)投与24時間後の被験体の血漿中のおよそ40〜250ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜500ng/mLの化合物
のうちの少なくとも一つを提供するのに十分である。
【0063】
別の実施形態において、各単位用量は、被験体の血漿中の化合物のおよそ100〜2000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜4000ng/mLのCmax;または被験体の血漿中の化合物の100〜1000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜2000ng/mLのCmaxを提供するのに十分である。
【0064】
代表的に、本明細書中に記載される式Iの化合物の使用において、医薬の調製のために化合物の乳酸塩が使用される。このような医薬は、経口投与に適している。医薬の単位投薬形態としては、丸剤、カプセル剤、錠剤、ゲルキャップ剤、キャプレット剤、懸濁液もしくは水溶液が挙げられるが、これらに限定されない。さらに、医薬は、短時間のボーラス、ゆっくりした輸液、もしくは長時間の輸液のような注入による投与に適している。
【0065】
式I、式IIおよび式IIIの化合物には、本発明の方法のいずれかを記載する説明書が付随し得る。したがって、いくつかの実施形態においては、本発明は、癌の処置法もしくは式Iの化合物の代謝様式の分析法における使用説明書との組み合わせで、式I、式IIおよび/もしくは式IIIの少なくとも一つの化合物を提供する。
【0066】
本発明はさらに、癌を有する被験体に、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩を投与する工程を包含する、癌を処置するための方法を提供し、この処置法においては、第一の処置サイクルで投与される化合物の量は一日25mgであり、かつ一日化合物1500mgが被験体に投与されるまでもしくは被験体に用量制限毒性が観察されるまでのどちらかまで、投与される化合物の量が、その後に続く各処置サイクルにより増加される。代表的に、このような方法において、投与される化合物の量は、第一の処置サイクルの後に続く各処置サイクルにより倍増される。いくつかの実施形態において、処置サイクルは、一日につき同じ量の化合物を7日間投与し、その後7日間化合物を投与しない工程を包含する。
【0067】
同様に、本明細書において記載される医薬の調製における、式Iの化合物の使用のいくつかの実施形態において、医薬の各単位用量は、被験体の体重に基づいて0.25〜30mg/kgの化合物、互変異性体、および/または塩を含む。さらに、医薬の各単位用量は、25〜1500mgに及ぶ量の化合物、互変異性体、および/または塩を含み得る。医薬は、7日、14日,21日もしくは28日分の量の前記単位用量を含有するキットに配合され得、処置サイクルにおける使用に適したこのキットは、各7日,14日,21日もしくは28日間、一日分量の化合物を投与し、その後7日もしくは14日間化合物を投与しない工程を包含する。
【0068】
別の局面において、本発明は癌を処置する方法を提供し、この方法は、癌を有する被験体に十分量の式I
【0069】
【化23】
を有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能なその互変異性体の塩を投与する工程、および被験体を以下:
【0070】
【化24】
から選択される式IIおよび式IIIの化合物の一方もしくは両方に曝露する工程であって、それによって、式IIおよび式IIIの化合物の一方もしくは両方が、被験体による式Iの化合物の代謝により生成される工程を包含し、被験体の血漿中における、およそ20〜4000ng/mLに及ぶ式I、式IIおよび式IIIの化合物の一つ以上の複合Cmaxもしくは被験体の血液中における、およそ40〜8000ng/mLに及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の複合Cmaxを提供する。
【0071】
さらに別の局面において、本発明は、癌を有する被験体を、十分量の一つ以上の以下:
【0072】
【化25】
から選択される式を有する化合物、活性なその代謝物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異異性体の塩であって、被験体の血漿中の一つ以上の化合物のおよそ20〜4000ng/mLの複合Cmaxもしくは被験体の血液中の一つ以上の化合物のおよそ40〜8000ng/mLの複合Cmaxを提供するのに十分である化合物に曝露する工程を包含する、癌を処置するための方法を提供する。いくつかの実施形態において、一つ以上の化合物は、被験体の血漿中のおよそ35〜2600ng/mLの一つの化合物のCmaxもしくは被験体の血液中のおよそ35〜6000ng/mLの一つの化合物のCmaxを提供する。他の実施形態において、一つ以上の化合物の量は、被験体の血漿中のおよそ35〜1200ng/mLの一つの化合物のCmaxもしくは被験体の血液中のおよそ50〜2400ng/mLの一つの化合物のCmaxを提供する。いくつかの実施形態において、式:
【0073】
【化26】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。別の実施形態において、式:
【0074】
【化27】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。さらに別の実施形態において、式:
【0075】
【化28】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。
【0076】
特定の疾患に対する式Iの化合物の安全性および/もしくは効力の決定において、重要なことは、この化合物の投与後、被験体におけるこの化合物の薬物動態および薬理作用をモニタリング可能であることである。そこで、本発明の一つの局面にしたがって、被験体における式Iの化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の代謝様式を決定するための提供される方法が存在する。この方法は、被験体から得られる尿、血液、もしくは組織の一つ以上のサンプルにおいて、この化合物の少なくとも一つの代謝物の量を測定する工程を包含する。この方法により測定され得る代謝物としては、式II:
【0077】
【化29】
を有するN−オキシド化合物であって、4−アミノ−5−フルオロ−3−[6−(4−メチル−4−オキシドピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オンとしてもまた公知の化合物、および式III:
【0078】
【化30】
を有するN−デスメチル化合物であって、4−アミノ−5−フルオロ−3−[6−(ピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オンとしてもまた公知の化合物が挙げられる。式Iの化合物の代謝様式を決定するためのいくつかのこのような方法において、これらの方法は、式IIの代謝物および式IIIの代謝物の量を測定する工程を包含する。代謝物の量は、紫外(UV)分光学もしくは液体クロマトグラフィー−質量分析(LC−MS)を含む当業者に周知の技術を用いて測定され得る。
【0079】
本発明の別の局面において、被験体における式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の量を決定する方法が提供される。この方法は、化合物が被験体に投与された後に被験体から得られる尿、血液、もしくは組織のサンプルにおける化合物の量を測定する工程を包含する。この方法は、サンプル中の化合物の代謝物の量を測定する工程をさらに包含し得る。測定され得る代謝物としては、式IIのN−オキシド化合物および/もしくは式IIIのN−デスメチル化合物が挙げられるが、これらに限定されない。いくつかの実施形態において、この方法は、式Iの化合物が被験体に投与された後の異なる時点に、被験体から二つ以上のサンプルを回収する工程をさらに包含する。
【0080】
別の局面において、癌を処置する方法における使用のための式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が提供され、この方法は、以下
(a)化合物が被験体に投与される場合における、被験体の血漿中の化合物のおよそ20〜4000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ40〜8000ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ10〜2,000ng/mLの化合物もしくは被験体への投与24時間後の被験体の血液中のおよそ20〜4,000ng/mLの化合物、または
(c)化合物が被験体に投与される場合における、被験体の血漿中の化合物のおよそ500〜60,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ750〜120,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分な量において、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩を被験体に投与する工程を包含する。
【0081】
癌を処置する方法における使用のためのこの化合物のいくつかの実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜500ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜1000ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ20〜1,000ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ40〜2,000ng/mLの化合物、または
(c)被験体の血漿中のこの化合物のおよそ1,000〜30,000ng×h/mLのAUCもしくは被験体の血液中のこの化合物のおよそ1,500〜60,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0082】
癌を処置する方法における使用のためのこの化合物の別の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜250ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜500ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ40〜500ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜1,000ng/mLの化合物、または
(c)被験体の血漿中の化合物のおよそ2,000〜15,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ3,000〜30,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0083】
癌を処置する方法における使用のためのこの化合物のさらに別の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ75〜150ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ150〜300ng/mLのCmax、または
(b)投与24時間後の被験体の血漿中のおよそ40〜250ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜500ng/mLの化合物
のうちの少なくとも一つを提供するのに十分である。
なお別の実施形態において、各単位用量は、被験体の血漿中の化合物のおよそ100〜2000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜4000ng/mLのCmaxを提供するのに十分である;または各単位用量は、被験体の血漿中の化合物のおよそ100〜1000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜2000ng/mLのCmaxを提供するのに十分である。
【0084】
被験体における、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の代謝様式の決定のための代謝物の使用がさらに提供され、この代謝物は、式:
【0085】
【化31】
のN−オキシド化合物もしくは式:
【0086】
【化32】
のN−デスメチル化合物のうちの少なくとも一つを含む。
【0087】
本発明はまた、一つ以上の本発明の化合物、もしくは薬学的に受容可能なその塩もしくはその互変異性体を、薬学的に受容可能なキャリアー、賦形剤、結合剤、希釈剤などと混合することにより調製され得る種々の疾患を処置もしくは改善するための組成物を提供する。このような疾患の例としては、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖性疾患、非小細胞肺癌、小細胞肺癌、慢性リンパ球白血病、肉腫、黒色腫、リンパ腫、甲状腺癌、神経内分泌癌、腎細胞癌、胃癌、消化管間質癌、神経膠腫、脳癌、または膀胱癌を含む癌が挙げられるが、これらに限定されない。処置上有効な用量は、結果としてこの疾患の症状の改善をもたらすのに十分な本発明の一つ以上の化合物の量に、さらに言及する。本発明の薬学的組成物は、とりわけ、通常の顆粒化、混合、溶解、封入、凍結乾燥、乳化もしくは微粒子化プロセスのような当該分野で周知の方法により製造され得る。この組成物は、例えば、顆粒剤、粉剤、錠剤、カプセル剤、シロップ剤、坐剤、注射剤、乳化剤、エリキシル剤、懸濁剤もしくは液剤のような形状を取り得る。本発明の組成物は、例えば、経口投与、鼻腔内投与、経粘膜投与、直腸投与、または皮下投与および鞘内、静脈内、筋肉内、腹腔内、鼻腔内、眼内もしくは心室内注射の様な種々の投与経路に処方され得る。本発明の化合物もしくは化合物はまた、全身型よりむしろ、徐放性処方物としての注射のような局所型において投与され得る。以下の投薬形態は、例として与えられ、かつ本発明を限定するものとして解釈されるべきではない。
【0088】
経口、頬側および舌下投与において、粉剤、懸濁剤、顆粒剤、錠剤、丸剤、カプセル剤、ゲルキャップ剤、およびキャプレット剤が固体の投薬形態として受容可能である。これらは、例えば、一つ以上の本発明の化合物、もしくは薬学的に受容可能な塩もしくはその互変異性体を、添加剤またはデンプンもしくは他の添加剤のような賦形剤のうちの少なくとも一つと混合することにより調製され得る。適切な添加剤もしくは賦形剤は、フルクトース、スクロース、ラクトース、セルロース糖、マンニトール、マルチトール、デキストラン、ソルビトール、デンプン、寒天、アルギン酸塩、キチン、キトサン、ペクチン、トラガカントゴム、アラビンゴム、ゼラチン、コラーゲン、カゼイン、アルブミン、合成もしくは半合成ポリマーまたはグリセリド、メチルセルロース、ヒドロキシプロピルメチル‐セルロース、および/またはポリビニルピロリドンである。必要に応じて、経口投薬形態は、不活性な賦形剤、またはステアリン酸マグネシウムのような滑沢剤、またはパラベンもしくはソルビン酸のような防腐剤、またはアスコルビン酸、トコフェロールもしくはシステインのような酸化防止剤、崩壊剤、結合剤、濃化剤、緩衝剤、甘味剤、風味剤または芳香剤のような、投与において補助となる他の成分を含み得る。さらに、識別のために染料もしくは色素が添加され得る。錠剤および丸剤は、当該分野において公知の適切な塗膜物質によりさらに処理され得る。
【0089】
経口投与のための液体投薬形態は、薬学的に受容可能な乳化剤、シロップ剤、エリキシル剤、懸濁剤、スラリー剤および液剤の形態を取り得、これらは水のような不活性な賦形剤を含み得る。薬学的処方物は、油、水、アルコール、およびこれらの組み合わせなどであるがこれらに限定されない滅菌の液体を使用して、液体懸濁剤もしくは液剤として調製され得る。薬学的に適切な界面活性剤、懸濁化剤、乳化剤は、経口投与もしくは非経口投与のために添加され得る。
【0090】
上記のように、懸濁剤は、油を含み得る。このような油としては、ピーナッツ油、ゴマ油、綿実油、トウモロコシ油およびオリーブ油が挙げられるが、これらに限定されない。懸濁調製液はまた、オレイン酸エチル、ミリスチン酸イソプロピル、脂肪酸グリセリドおよびアセチル化脂肪酸グリセリドのような脂肪酸のエステルを含み得る。懸濁剤処方物は、エタノール、イソプロピルアルコール、ヘキサデシルアルコール、グリセロールおよびプロピレングリコールなどである、これらに限定されないアルコールを含み得る。ポリ(エチレングリコール)のような、しかしこれに限定されないエーテル、鉱油およびペトロラタムのような石油炭化水素;および水はまた、懸濁剤処方物において使用され得る。
【0091】
鼻腔内投与(例えば、化合物を脳に送達するため)、もしくは吸入投与(例えば、肺を介して化合物を送達するため)の場合、薬学的処方物は、液剤、噴霧剤、ドライパウダー、もしくはあらゆる適切な溶媒および安定化剤、殺菌物剤、酸化防止剤、pH改変剤、界面活性剤、バイオアベイラビリティ改変剤およびこれらの組み合わせのような、しかしこれらに限定されない任意の別の化合物を含むエアロゾルであり得る。鼻腔内処方物および鼻腔内投与方法の例は、WO01/41782、WO00/33813,WO91/97947、米国特許第6,180,603号および米国特許第5,624,898号に見出され得る。エアロゾル処方の推進剤としては、圧縮された空気、窒素、二酸化炭素、もしくは炭化水素ベースの低沸点溶媒が挙げられ得る。本発明の化合物は、好都合には、エアロゾル噴霧提供の形態で噴霧器などから輸送される。
【0092】
注射投薬形態は、一般に、適切な分散剤もしくは加湿剤および懸濁剤を用いて調製され得る水性懸濁液もしくは油性懸濁液を含み得る。注射可能形態は、溶液相または溶媒もしくは希釈剤により調製される懸濁形態を取り得る。受容可能な溶媒もしくはビヒクルとしては、滅菌水、リンガー溶液、もしくは等張生理食塩水溶液が挙げられる。代替的に、溶媒もしくは懸濁剤として滅菌油が採択され得る。好ましくは、この油もしくは脂肪酸としては、揮発性でない、天然もしくは合成の油、脂肪酸、モノ−グリセリド、ジ−グリセリドもしくはトリ−グリセリドが挙げられる。
【0093】
注射の場合、薬学的処方物は、上記のような適切な溶液による再形成のために適切な粉末であり得る。これらの例としては、凍結乾燥、回転乾燥もしくは噴霧乾燥された粉末、不定形粉末、顆粒、沈殿物、もしくは微粒子があげられるが、これらに限定されない。注射の場合、処方物は、安定化剤、pH改変剤、界面活性剤、バイオアベイラビリティ改変剤およびこれらの組み合わせを任意に含み得る。これらの化合物は、ボーラス注射もしくは連続的な輸液のような注射による、非経口投与のために処方され得る。注射のための単位用量形態は、アンプルもしくはマルチ用量容器を取り得る。
【0094】
このように、本明細書中に記載される癌を処置するための本方法において、化合物は、短時間のボーラス、ゆっくりした輸液、もしくは長時間の輸液として注射によって投与され得る。注射は、一日に一回、二回、三回、もしくは四回投与され得る。短時間のボーラスは、通常、およそ1〜30分間にわたって投与される;ゆっくりとした輸液は、通常、30分〜6時間にわたって投与される;そして長時間の輸液は、通常、6時間〜およそ7日までにわたって投与される。
【0095】
直腸投与の場合、薬学的処方物は、腸、S状結腸および/もしくは直腸における化合物の放出のため、坐剤、軟膏、浣腸、錠剤もしくはクリームの形態を取り得る。直腸坐剤は本発明の化合物、薬学的に受容可能な塩もしくはこの化合物の互変異性体のうちの一つ以上を、受容可能な賦形剤、例えば、ココアバターもしくはポリエチレングリコールと共に混合することにより調製され、これらは、通常の保管温度においては固相で存在し、かつ直腸内のような体内における薬物の放出に適切な温度においては液相で存在する。油はまた、軟ゼラチン型および坐剤の処方の調製において採択され得る。水、生理食塩水、ブドウ糖水溶液および関連する糖溶液、ならびにグリセロールは、ペクチン、カルボマー、メチルセルロース、ヒドロキシプロピルセルロースもしくはカルボキシメチルセルロースのような懸濁剤、ならびに緩衝剤および防腐剤をも含み得る懸濁処方物の調製において採択され得る。
【0096】
それらの上記の代表的な投薬形態に加えて、薬学的に受容可能な賦形剤およびキャリアーは、通常、当業者に公知であり、したがって本発明に含まれる。このような賦形剤およびキャリアーは、例えば、本明細書中に参考として援用される「Remingtons Pharmaceutical Sciences」Mark Pub.Co.,New
Jersey(1991)に記載される。
【0097】
本発明の処方物は、以下に記載する、短時間の作用、迅速な放出、長時間の作用、および持続的な放出のために設計され得る。このように、薬学的処方物はまた、制御された放出もしくはゆっくりとした放出のために処方され得る。
【0098】
本組成物はまた、例えば、ミセルもしくはリポソーム、または何らかの別の被包性の形態を含み得、あるいは持続的な蓄積および/もしくは送達効果を提供するために、長期間の放出形態において投与され得る。したがって、薬学的処方物は、ペレット剤もしくはシリンダーに圧縮され得、そして筋肉内もしくは皮下に、蓄積注射もしくはステントのような移植片として移植され得る。このような移植物は、シリコーンおよび生分解性ポリマーのような公知の不活性な物質を採択し得る。
【0099】
処置上有効な用量は、結果として症状の改善をもたらす化合物の量をいう。特定の投薬量は、疾患の状態、被験体の年齢、体重、通常の健康状態、性別、食事、用量間隔、投与経路、排出率、および薬物の組み合わせに基づいて調整され得る。有効量を含む上記のいずれの投薬形態も、慣用的な実験の境界内に十分含まれ、そしてそれゆえ、本発明の範囲内に十分含まれる。処置上有効な用量は、投薬経路および投薬形態に依存して変化し得る。本発明の好ましい化合物は、高い処置指数を示す処方物である。処置指数は、LD50とED50との比として表され得る、毒性効果と処置効果との間の用量比である。LD50は、母集団の50%に致命的な量であり、そしてED50は、母集団の50%に処置上有効な量である。LD50およびED50は、動物細胞培養物もしくは実験動物における標準的な薬学的手順により決定される。
【0100】
本発明はまた、ヒトもしくは非ヒト動物において抗癌活性を促進する方法を提供する。この方法は、有効量の本発明の化合物、もしくは組成物を、前記哺乳動物もしくは非ヒト動物へ投与する工程を包含する。本発明の化合物の有効量は、RTKを阻害する量を含み、これは、例えば、本明細書中に記載のアッセイ、もしくは当業者に公知のシグナル伝達を検出する他のあらゆるアッセイにより、例えば、コントロールモデルと比較したcAMP上昇レベルの測定による、G−タンパク質共役受容体の活性化を介する生物化学的経路において、検出可能である。有効量はまた、RTKの阻害により処置可能なRTK疾患の症状を緩和する量を含み得る。
【0101】
提供されるこれらの方法により処置され得るRTK疾患、もしくはRTKが媒介する疾患は、RTKが関与するか、またはRTKの阻害およびRTKが、疾患もしくは疾患の状態において欠損している生物化学的経路を増強する任意の生物学的疾患もしくは疾患を含む。このような疾患の例は、前立腺癌、大腸癌、乳癌、多発性骨髄腫、膵臓、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、もしくは骨髄増殖性疾患のような癌である。
【0102】
化合物Iの合成は、米国特許第6,605,617号において開示されている。化合物2および3の同一性の確認のために、化合物Iの代謝物、化合物IIおよびIIIは、実施例6において示されるように、独立して合成された。
【0103】
本発明は、このように一般的に記載される、以下の実施例の参照によってより容易に理解され、以下の実施例は実例として提供され、そして本発明を限定するものとして意図されない。
【実施例】
【0104】
以下の略語および用語が、実施例において使用される:
ATP: アデノシン三リン酸
AUC: 曲線下面積
BSA: ウシ胎児アルブミン
DMSO: ジメチルスルホキシド
EDTA: エチレンジアミン四酢酸
ERK: 細胞外制御キナーゼ
Hepes: N−(2−ヒドロキシエチル)ピペラジン−N’−(2−エタンスルホン酸)
HPLC: 高速液体クロマトグラフィー
HMVEC: ヒト微小管内皮細胞
kg: キログラム
LC: 液体クロマトグラフィー
MAPK: マイトジェン活性化プロテインキナーゼ
MS: 質量分析
MeOH: メタノール
mg: ミリグラム
mL: ミリリットル
MTS: [3−(4,5−ジメチル−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム]、分子内塩
nM: ナノモーラー
PBS: リン酸緩衝化生理食塩水
NMR: 核磁気共鳴分析
RT−PCR: 逆転写酵素ポリメラーゼ連鎖反応
SCF: 幹細胞因子
TFA: トリフルオロ酢酸
T1/2: 半減期‐生物学的系から化合物の50%が排出されるのに必要な時間
μg: マイクログラム
μL: マイクロリットル
μM: マイクロモーラー
UV: 紫外線分光法
(実施例1)
多数の癌細胞株および初代非悪性細胞株に対して、4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オン(化合物1)の抗増殖活性を試験した。方法は、以下のように行った:細胞を96−ウェルプレートにプレートした;接着細胞株のための3〜5時間のゲル化時間の後、化合物の希釈溶液(delusioin)を添加し、3日後、MTS溶液(Promega)の添加により生細胞を決定した。490nmで吸光度を測定し、非直線回帰を用いてEC50値を算出した。HMVECアッセイのために、3日間、5ng/mLの組換え型VEGFの存在下、細胞と共に化合物をインキュベートした。SCF/c−KITアッセイのために、3日間、それぞれ40ng/mLおよび100ng/mLの組換え型SCF存在下、TF−1細胞およびH526細胞をインキュベートした。MTS溶液の添加および490nmでの吸光度の測定により、増殖をアッセイした。非直線回帰により、EC50を算出した。結果を表1に示す。
【0105】
癌細胞株および内皮細胞の部分集合において、KM12L4a細胞株を除いて、EC50が50nM以下で増殖が阻害され、これは化合物1により標的とされるRTKへのそれらの依存と矛盾しない(MV4;11:構成的に活性なFLT3の発現;HMVEC:VEGFR2に媒介される増殖;TF−1:c−KITに媒介される増殖)。この細胞株が、いくつかの標的とされるRTK(例えば、RT−PCRにより決定されるVEGF1/2およびPDGFR)を発現する場合においても、実験は、これらの個々のRTKの阻害が、化合物1に観察される強力な抗増殖効果を完全には説明しないことを示した。この発見は、複数のRTKの阻害もしくは今は未同定な効果のどちらかが、この細胞株において化合物1により媒介される抗増殖効果に関与し得ることを示唆する。
【0106】
1μMと10μMとの間のEC50を有する化合物1と共にインキュベートされる場合、二つの初代細胞株、HMEC(ヒト正常乳房上皮細胞)およびPrEC(正常ヒト前立腺上皮細胞)を含む大多数の細胞株は、抗増殖応答を示した。インビトロにおける結果と一致して、インビボにおいて、化合物1がマウスにおけるKM12L4a異種移植片とMV4;11異種移植片との両方の生育を効果的に阻害した。
【0107】
【表1】
(実施例2)
2週間の毒性研究からのプールされたラットの血漿において、化合物1の二つの代謝物が同定され、そして部分的に特徴付けられた。一日一回、30mg/kgもしくは80mg/kg、経口投与の投与群から、UVおよびLC/MSにより、1日目および14日目の投与された動物の血漿が分析された。二つの同定された代謝物は、式IIのピペラジンN−オキシド化合物(化合物2)および式IIIのN−脱メチル化合物(化合物3)であった(これらの化合物の合成および特徴付けについては、実施例6を参照)。(UV吸光度に基づき、かつ以前の分析からの同じサンプルにおいて定量された既知のレベルの化合物1に比較して)見積もられた代謝物のレベルを、表2に示す。投与後プールされた血漿の全てのサンプルにおいて、N−デスメチル代謝物が化合物1よりもかなり低い濃度であることが見出された。80mg/kgの投与群における14日目の24時間および30mg/kgの投与群における1日目の1〜2時間を除いて、N−オキシド代謝物が化合物1よりも低い濃度で存在することが観察された(表2)。代謝様式は、投与量および投与の継続によって変化しない。通常、代謝物のレベルは化合物1のレベルの投与量の増加に伴って上昇する。
【0108】
両方の投与群で、1日目と14日目を比較して、投与の継続は、代謝物のみの血漿レベルにおいて(表2)もしくは化合物1のレベルと比較して、結果として上昇を生じないことが明らかである。化合物1のレベルは、投与の継続とともに減少し、これは代謝物レベルの減少によっても反映される。このことは、化合物1の曝露における時間依存的な減少は、上昇する代謝物に反映されないことを示唆する。14日目の24時間のサンプルは、1日目の24時間のサンプルよりも低いレベルの化合物1および代謝物を含み、このことは、30mg/kgもしくは80mg/kgの一日一回の投与規則によっては代謝物および化合物1は蓄積されないことを示す。80mg/kg投与群の全てのアッセイされた時点において、そして30mg/kg投与群の1日目、24時間を除く全ての時点において、N−オキシド代謝物はN−デスメチル代謝物よりも高い濃度で存在している。N−デスメチル代謝物レベルは、化合物1のレベルよりもゆっくりと低下することが明らかであり、このことは、より長いT1/2を示唆し、そしてこの代謝物の血漿レベルが、N−オキシドが対照的にそうあり得るようにその形成速度ではなく、その排出率によって決定され得ることを示す。
【0109】
【表2】
(実施例3)
レセプターチロシンキナーゼのためのインビトロキナーゼアッセイ
多くのタンパク質チロシンキナーゼのキナーゼ活性を、ATPおよびリン酸化のためのチロシンアミノ酸残基を含む適切なペプチドもしくはタンパク質を提供すること、そしてチロシン残基へのリン酸エステル分子の転移を分析することにより測定した。FLT−1(VEGFR1)、VEGFR2、VEGFR3、Tie−2、PDGFRα、PDGFRβ、およびFGFR1レセプターの細胞質ドメインに対応する組換え型タンパク質を、バキュロウィルス発現系(InVitrogen)を用いてSf9昆虫細胞において発現させ、そしてGlu抗体相互作用(Glu−エピトープ標識化組換え体について)を介してもしくは金属イオンクロマトグラフィー(His6(配列番号1)標識化組換え体について)によって精製し得る。各アッセイのために、試験化合物を順にDMSOに希釈し、そして次にATPを加えた適切なキナーゼ反応緩衝液と混合した。キナーゼタンパク質および適切なビオチン化ペプチド基質を添加し、最終容量を50〜100μLとし、反応液を1〜3時間、室温でインキュベートし、そして次に45mMのEDTA、50mMのHepes pH7.5を含有する25〜50μLを添加することにより反応を停止した。停止された反応混合液(75μL)を、ストレプトアビジン塗膜マイクロタイタープレート(Boehringer Mannheim)に移し、1時間インキュベートした。リン酸化されたペプチド生成物を、ユーロピウム標識された抗リン酸化チロシン抗体PT66を用いて、DELFIA時間分解蛍光システム(WallacまたはPE Biosciences)により測定し、抗体の希釈のためにDELFIAアッセイ緩衝液に1mMのMgCl2を補充するという改変を施した。Wallac 1232 DELFIA蛍光計およびPE Victor II多重信号読み取り機において時間分解蛍光を読み取った。XL Fitデータ分析ソフトウェアを用い、非直線回帰を採用して50%阻害のための各化合物の濃度(IC50)を算出した。
【0110】
FLT−1、VEGFR2、VEGFR3、FGFR3、Tie−2、およびFGFR1キナーゼを、50mMのHepes pH7.0、2mMのMgCl2、10mMのMnCl2、1mMのNaF、1mMのDTT、1mg/mLのBSA、2μMのATP、および0.20〜0.50μMの対応するビオチン化ペプチド基質中において分析した。FLT−1、VEGFR2、VEGFR3、Tie−2、およびFGFR1キナーゼを、それぞれ0.1μg/mL、0.05μg/mL、もしくは0.1μg/mL添加した。PDGFRキナーゼアッセイのために、ATP濃度およびペプチド基質濃度を1.4μMのATP、および0.25μMのビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2)ペプチド基質に変換すること以外は上記と同じ緩衝液条件で、120μg/mLの酵素を使用した。FLT−1、VEGFR2、VEGFR3、FGFRに関しては、上記の化合物の各々が10μMより低いIC50値を示した。
【0111】
組換え型かつ活性なチロシンキナーゼFyn、およびLckは商業上入手可能であり、そしてこれらをUpstate Biotechnologyから購入した。各アッセイのために、試験化合物を順にDMSOに希釈し、そして次に10nMの33P γ−標識化ATPを加えた適切なキナーゼ反応緩衝液と混合した。キナーゼタンパク質および適切なビオチン化ペプチド基質を添加し、最終容量を150μLとした。反応液を、3〜4時間、室温でインキュベートし、そして次に100mMのEDTAおよび50μMの非標識化ATPを含有する100μLの反応停止緩衝液を含む、ストレプトアビジン塗膜ホワイトマイクロタイタープレート(Thermo Labsystems)に移すことにより反応を停止させた。1時間のインキュベート後、ストレプトアビジンプレートをPBSで洗浄し、そして各ウェルにつき200μLのMicroscint 20シンチレーション流体を添加した。このプレートを密閉し、そしてTopCountを用いて計数した。XL Fitデータ分析ソフトウェアを用い、非直線回帰を採用して50%阻害のための各化合物の濃度(IC50)を算出した。
【0112】
Fyn、Lckおよびc−ABLのためのキナーゼ反応緩衝液は、50mM Tris−HCl pH7.5、15mM MgCl2、30mM MnCl2、 2mM DTT、2mM EDTA、25mM β―グリセロールリン酸塩、0.01% BSA/PBS、0.5μMの適当なペプチドの基質(ビオチン標識されたSrcのペプチドの基質:FynおよびLckについてはビオチン―GGGGKVEKIGEGTYGVVYK−NH2(配列番号3))、1μM 非標識ATPおよび1nMキナーゼを含んだ。
【0113】
c−KitおよびFLT−3のキナーゼ活性を、ATP、およびリン酸化のためのチロシンアミノ酸残基を含むペプチドまたはタンパク質を提供、およびチロシン残基に対してリン酸基の転移のためにアッセイすることにより、計測される。c−KitおよびFLT−3受容体の細胞質ドメインに対応した組み換えタンパク質は、購入した(Proquinase)。テストのために、模範的な化合物、例えば、4−アミノ−5−フルオロ−3−[5−(4−メチルピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)―オンは、DMSOで希釈し、それから下記に説明されたキナーゼ反応緩衝液およびATPと混合した。キナーゼタンパク質、c−KitまたはFLT−3およびビオチン化されたペプチドの基質(ビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2)は、100μLの最終容量を提供するよう添加した。これらの反応を、室温で2時間インキュベートし、次いで50μLの45mM EDTAおよび50mM HEPES、pH7.5を添加することにより停止した。停止された反応混合液(75μL)を、ストレプトアビジン被覆のマイクロタイタープレート(Boehringer Mannheim)に移行され、1時間インキュベートした。リン酸化されたペプチド産物を、DELPHIA経時分離時間分解蛍光システム(WallacまたはPE Biosciences)を使用し、ユーロピウム標識された抗ホスホチロシン抗体PT66を使用して、抗体希釈のためにDELFIAアッセイ緩衝液に1mM MgCl2を追加したという変更を伴って計測した。時間分解蛍光値を、Wallac 1232 DELFIA 蛍光計測器またはPE Victor II多重シグナル解読器で決定した。50%の阻害(IC50)に対するそれぞれの化合物の濃度IC50を、XL Fitデータ解析ソフトウェアを用いる非線形回帰を使用し計算した。
【0114】
FLT−3およびc−Kitキナーゼを、50mM Hepes pH7.5、1mM
NaF、2mM MgCl2、10mM MnCl2、および1mg/mL BSA、8μM ATPおよび1μMの対応するビオチン化されたペプチド基質(ビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2))においてアッセイした。FLT−3およびc−Kitキナーゼの濃度を、2nMでアッセイした。
IC50を、化合物1の代謝物において計測し、比較のために化合物1のIC50を一緒に表3に示す。
【0115】
【表3】
(実施例4)
この単物質の研究は、KM12L4aのヒト結腸の腫瘍モデルにおいて、化合物1の毎日の経口投薬を評価した。
【0116】
生後7、8週間の(Charles River)雌のNu/Nuマウスに、右側腹部において、2×106のKM12L4a 細胞を皮下に移植した。処置は、腫瘍の容量の平均が125mm3であるときより7日後に開始した。これを、一日目の研究と名付けた。化合物1を、10mM H3PO4で溶液として処方し、そして経口栄養補給によって投与した。
【0117】
7つの処置群を、研究に含めた、(n=10/群):ビヒクル(水)p.o.、q.d.;および化合物1に含めた投薬の6群:3mg/kg、10mg/kg、30mg/kg、100mg/kg、200mg/kg、300mg/kgのp.o.、q.d.。
【0118】
血漿サンプルを、様々な日数におけるそれぞれの投薬群において、腫瘍保有マウスにおいて化合物1の薬物動態を特徴付けるために衛星動物(satellite animal)から引き出した(N=2/時点/投薬群)。化合物1の組織濃度および腫瘍濃度を、22日目の8時間および24時間の投薬後に100mg/kgおよび200mg/kgの投薬群において、動物から収集されたサンプルにおいて決定した(N=2/時点/投薬群)。
【0119】
血漿化合物1の濃度を、1ng/mLから8000ng/mLの較正の範囲および1ng/mLの定量限界(LLOQ)の検証されていないLC/MS/MSアッセイによって決定した(Charles River Laboratories、Worcester、MA)。組織および腫瘍の化合物1の濃度もまた、20ng/gから43740ng/gの較正の範囲および20ng/gのLLOQの検証されていないLC/MS/MSアッセイを使用して決定した。
【0120】
複雑な薬物動態パラメーター(CmaxおよびAUC値)を、それぞれのサンプリング日におけるそれぞれの投薬群について、血漿化合物の濃度―時間のデータの平均から、標準的な非コンパートメント分析を使用して得た(WinNonlin Professional、version4)。報告したAUC値を、3つの濃度―時間のデータ点を使用して決定した。投薬前の濃度値を、投薬の直前に対して直接に観察された濃度値のとして報告した。
【0121】
腫瘍の成長において有意な用量依存性の阻害を、4日〜7日の処置による全ての投薬で観察した(表4参照)。計算したED50は、17mg/kgであった。初期サイズの50%より大きい腫瘍の退化が、200mg/kgおよび300mg/kgにおいて、化合物1を投薬された大部分のマウスにおいて観察された。しかし、これらの投薬は、全体の研究期間において、耐性を示されなかった。12日〜16日の間まで、300mg/kgで処置したマウスは、体重の20%〜30%を失い、安楽死させた。200mg/kgで処置されたマウスにおいて、10匹の内1匹は、22%体重減少したので14日目に安楽死させ、そして残存したマウスは、25%より大きく体重減少したので21日〜24日目に安楽死させた。マウスを、100mg/kgで37日間投薬し、最初の体重の98%に維持した;この投薬において、腫瘍は安定なままであった(図1)。ビヒクル群を、9日で屠殺し、そして腫瘍成長の阻害(TGI)を計算した(表4)。
【0122】
【表4】
投薬2日目(2日目)において、化合物1の血漿濃度は、全ての投薬群において用量に対し比例的に増加した(表5)。少なくとも2週間の間での多重の投薬に続いて、血漿濃度は2日目の投薬に匹敵し得た。これはマウスおいて日に一度の投薬における蓄積が無いことを示唆する(表5)。同様に、3日目、8日目および15日目に収集された化合物1の投薬前血漿濃度は、それぞれの投薬群と類似しており、これは、2日目以降に定常状態に達したことを示唆する。以上のことから、これらのデータは、化合物1が、腫瘍保有マウスにおいて、投薬および時間に非依存性の薬物動態に従うことを示唆する。
【0123】
35%〜60%の腫瘍成長阻害が、10mg/kgおよび30mg/kgの用量においてそれぞれ観察された。対応する化合物1の血漿曝露は、CmaxおよびAUC値によって評価された場合に、それぞれ163ng/mL〜742ng/mLおよび1420ng×hr/mL〜5540ng×hr/mLの範囲であった。(図2)。対応する血漿投薬前濃度値は、2ng/mL〜135ng/mLの範囲であった。
【0124】
【表5】
それぞれ2つのサンプリング時点(投薬後8時間および24時間)で100mg/kgおよび200mg/kgの投薬群において、22日目における化合物1の組織中の濃度は、化合物1の血漿中の濃度より高かった(表6)。8時間または24時間の投薬後における脳または心臓中の化合物1の濃度は、これらの二つの投薬群において、血漿の濃度よりも13倍から34倍高かった;一方で肝臓、肺および腎臓の濃度は、これらの二つの投薬群において投薬後で8時間または24時間の血漿濃度より40倍から126倍高かった。一般的に、8時間における組織濃度の血漿濃度に対する割合は、24時間でのそれに匹敵した。さらに、24時間における組織濃度は、8時間におけるそれと比較して一貫して低かった。総合すれば、これらの結果は、化合物1の組織濃度が、血漿におけるそれと平行して減少するようであったことを示唆する。それゆえに、化合物1は、血漿に関連した組織(脳を含む)に広範に分布されるようであるが、多重の経口投薬に続いて、組織においては蓄積されないようである。
【0125】
【表6】
それぞれの2つのサンプリング時点(投薬後8時間および24時間)において100mg/kgおよび200mg/kgの投薬群において、22日目における化合物1の腫瘍中の濃度は、血漿中の濃度よりも37倍から354倍高かった。しかし、24時間における腫瘍の濃度は、これらの2つの投薬群においての投薬後8時間の濃度よりも17%〜65%だけ低くなっていた。これは、他の正常な組織(脳、心臓、肝臓、肺、および腎臓のような)からの排出速度と比較して、腫瘍からのいくらかより低い排出速度を示唆する。それゆえに、化合物1は、血漿と比較して腫瘍に広範に分布しているようである。しかし、血漿または正常の組織と比較して腫瘍において優先的な保持を示し得る。
【0126】
要約すると、化合物1の効力および耐性は、処置の4日〜7日後の有意な阻害を伴って、その用量に関係した。腫瘍抑制は、300mg/kgおよび200mg/kgで観察された;これらの用量では、およそ14日間および21日間においては、それぞれ耐性を示した。体重減少は、毒性に関連する臨床徴候であった。100mg/kgの投薬は、有害な臨床徴候なしに、コントロールと比較して80%の腫瘍成長阻害を伴い、37日間の間、耐性があった。30mg/kgは成長を60%阻害した。化合物1は、腫瘍保有マウスにおいて、用量依存的かつ時間依存的な薬物動態を実証した。35%〜60%の腫瘍成長阻害に関連した血漿中の化合物1のCmax値、AUC値およびCmin値は、それぞれ163ng/mL〜742ng/mL、1420ng×hr/mL〜5540ng×hr/mLおよび2ng/mL〜135ng/mLの範囲であった。化合物1は、組織に対し広範に分布したが、複数の経口投薬後では組織に蓄積しないようであった。経口の投薬後に、他の組織と比較して、腫瘍において、化合物1を優先的に保持する傾向があった。
(実施例5)
今回の一つの物質の研究では、PC3ヒト前立腺腫瘍モデルにおいて化合物1の断続的な経口投薬を評価した。
【0127】
9〜10週齢の雄のSCIDマウスに、5,000,000個のPC3ヒト前立腺細胞を皮下の腹部に移植した。腫瘍が150mm3に達したとき、処置を開始した。これを研究一日目と名付けた。化合物1を水性の溶液として処方し、そして経口栄養補給によって投与した。
【0128】
5つの処置群を研究に含めた、(n=10/群):ビヒクル(水)p.o.、q.d.;および100mg/kg;q.d.、q.2.d.、q.3.d.、q.4.d.の化合物1投薬の4群。
【0129】
表7に示すように、有意でありかつ類似した腫瘍阻害の結果が、全ての処置群で観察された。研究を、11日目に毎日の投薬群において停止した。残りの群において研究は、研究25日目で終了し、そして平均の腫瘍容量を計測し、ビヒクルと比較した。毒性の臨床的指標として、体重損失の百分率を、それぞれの群において計測した。
【0130】
【表7】
(実施例6)
化合物1の同定された代謝物の構造を確認するために、この代謝物を独立して合成した。
【0131】
化合物1のN−オキシド代謝物である化合物2を、下記のスキームに示すように合成した。化合物1をエタノール、脱メチルアセトアミドおよび過酸化水素の混合物中で加熱した。反応終了時において、化合物2を遠心によって単離し、そしてエタノールで洗浄した。必要とする場合、生成物を、カラムクロマトグラフィーによってさらに精製し得る。
【0132】
【化33】
化合物1のN−デスメチル代謝物である化合物3を、下記のスキームに示すように合成した。5―クロロ―2―ニトロアミンを、ピペラジンを使用して処理し、4を得た。4を、引き続いて、ブチルカルボニル(Boc)基で保護し、5を得た。ニトロ基の還元、引き続く3―エトキシ―3―イミノプロピオン酸エチルエステルとの縮合により、6を得た。塩基として、カリウムヘキサメチルジシルアジドを使用した6―フルオロアントラニロニトリルと6により、7を得た。粗製の7を水性のHClで処理し、精製後に黄色/茶色の固体として望ましい代謝物を得た。
【0133】
【化34】
(実施例7)
(化合物1の乳酸塩の調製)
【0134】
【化35】
ジャック付の3000mLの4つ口フラスコに、冷却器、温度計、N2ガス挿入口および機械的攪拌器を備え付けた。この反応容器を、少なくとも15分間N2でパージし、それから4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H−ベンゾイミダゾール―2―イル]―1H―キノリン―2―オン(484g、1.23mol)を充填した。D,L―乳酸(243.3g、モノマーの1.72mol、下記の段落参照)、水(339mL)ならびにエタノール(1211mL)の溶液を調製し、次いで反応フラスコに充填した。攪拌を、中間の速度で開始し、そしてその反応物を内部温度68℃〜72℃で加熱した。この反応物の内部温度を15分間〜45分間の間68℃〜72℃で維持し、次いで加熱を中止した。生じた混合物を、10〜20ミクロンのフリットを通して濾過し、12リットルのフラスコ内に濾液を収集した。この12リットルのフラスコに内部温度計、還流冷却器、添加漏斗、気体の挿入および排出口、ならびにオーバーヘッド攪拌器を備え付けた。次いで、攪拌物を中間の速度で攪拌し、加熱還流した(約78℃の内部温度)。穏やかな還流を維持しながら、エタノール(3,596mL)を約20分間にわたってフラスコに充填した。次いで、この反応フラスコを15分間〜25分間以内で約64℃〜70℃の範囲の内部温度に冷却し、そしてこの温度を約30分間の間維持した。反応器を結晶について検査した。結晶が存在しない場合、4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H―ベンゾイミダゾール―2―イル]―1H―キノリン―2―オンの乳酸塩の結晶(484mg、0.1モル%)をフラスコに添加し、そしてその反応物を30分間64℃〜70℃で攪拌し、その後、結晶についてそのフラスコを検査した。結晶が存在するとすぐに、攪拌を低速度に減少させ、そしてその反応物を、さらに90分間64℃〜70℃で攪拌した。次いで、この反応物を、約2時間の間にわたって約0℃で冷却し、そして、生じた混合物を25ミクロン〜50ミクロンのフリットのフィルターを通して濾過した。反応器をエタノール(484mL)で洗浄し、そして内部温度が約0℃になるまで攪拌した。冷エタノールを使用して、フィルターケーキを洗浄し、この手順を、さらに2回繰り返した。収集した固体を、真空オーブン内で真空下50℃で一定の質量まで乾燥して、結晶性の黄色い4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H―ベンゾイミダゾール―2―イル]―1H―キノリン―2―オンの乳酸塩510.7g(85.7%)を収集した。ラバーダムまたは不活性化状態を、主として濾過プロセスの間に使用した。乾燥した固体が、さほど湿潤でない一方、湿ったフィルターケーキは、水を吸収し、粘度を増す傾向にある。予防策を、大気に対する濾過ケーキの長期の曝露を避けるために実行した。
【0135】
市販の乳酸は、一般的に約8%w/w〜12%w/wの水を含み、モノマー乳酸に加えて、ダイマーおよびトリマーを含む。乳酸ダイマー対乳酸モノマーのモル比は、一般的に約1.0:4.7である。商業的な等級の乳酸は、モノ乳酸塩が、反応混合物から優先的に沈殿するように、上記段落に説明されたプロセスにおいて使用され得る。乳酸モノマーを以下の手順に従って、精製する。
(実施例8)
この研究では、FGFを補充したマトリゲルモデル(Matrigel model)において化合物1の抗血行性の可能性を評価した。
【0136】
11〜12週齢の雌のBDF1マウス(Charles River、Wilmington、MA)に、2μgのFGF―2を追加した0.5mLのマトリゲル(BD Biosciences、Bedford、MA)を皮下に移植した。血管形成(新血管新生または脈管形成)に追加されたFGF―2を、動物からそれらの除去後にマトリゲルプラグにおいてヘモグロビンレベルを計測することで定量した。
【0137】
テスト物品の経口投与を、マトリゲル移植の一日前に開始し、毎日1回で8回の投薬を連続して行った。化合物1を10mM H3PO4中の溶液として処方した。12の処理群が含まれた:ビヒクル(10mM H3PO4)のp.o.×8日間、q.d.×8日間(2つのコントロール群);補充していないマトリゲル(標準ヘモグロビンレベル)またはFGFを補充したマトリゲル(ポジティブコントロール)を移植したマウス;化合物1を3mg/kg、10mg/kg、30mg/kg、100mg/kg、200mg/kg、300mg/kgでp.o.×8日間、q.d.×8日間投与。1群あたり4匹を使用する200mg/kg、300mg/kgで投薬するマウスを除き、1群あたり8匹のマウスを使用した。
【0138】
ビヒクルで処理されたマウスと比較した、化合物で処理されたマウスにおけるヘモグロビンレベルのパーセンテージの阻害は、この化合物の抗脈管形成能を示す。結果を、単位マトリゲルプラグあたりの総ヘモグロビン量(mg/dL)で示す。ED50を、脈管形成を効果的に約50%阻害する用量として定義する。ヘモグロビン濃度を、Drabkin試薬(Sigma Diagnostics、ST.Louis MO)を使用する吸光度分光法を使用して、マウスから取り出し瞬間冷却した均質化マトリゲルプラグにおいて決定した。
【0139】
化合物1の血漿の曝露を評価するため、血液を、8つの持続的な投薬(8日目)後、2時間および24時間後に採取した。200mg/kgおよび300mg/kgの投薬群においては、血液を2時間の時点でのみ採取した。1の血漿濃度を、1ng/mL〜8000ng/mLの較正の範囲および1ng/mLの定量限界(LLOQ)の検証されていないLC/MS/MSアッセイによって決定した(Charles River Laboratories、Worcester、MA)。
【0140】
8日目において、マトリゲルプラグにおけるヘモグロビンレベルおよび化合物1の血漿濃度を計測した。研究の間に動物を観察し、そして体重を計測した。
【0141】
ビヒクル処置動物からのプラグと比較して評価したそれぞれの用量において、化合物1は、マトリゲルプラグにおけるヘモグロビン濃度の有意な阻害を生じた(表8)。計算したED50は、2.6mg/kgであった。3mg/kgおよび10mg/kgの用量は、54%および57%の阻害をそれぞれ結果として生じ、一方で、30mg/kg、100mg/kg、200mg/kgおよび300mg/kgの用量は、FGFを補充されていないマトリゲルのレベルまでヘモグロビンを減少させ、FGFを補充されたコントロールに対して、70%から92%の阻害を結果として生じた。8日目における投薬後2時間での化合物1の血漿濃度は、44ng/mLかつ3mg/kgから3920ng/mLかつ300mg/kgまでの濃度範囲に対して用量に比例した増加を示した(表9)。全ての用量では、良好な耐性を示し、体重の損失は、観察されなかった。
【0142】
【表8】
【0143】
【表9】
1の血漿濃度(投薬後2時間)は用量に比例して増加した。マトリゲルプラグのヘモグロビン量において、用量および血漿濃度依存的な減少が、観察された。44ng/mgの血漿濃度(投薬後2時間、8日目)は、このモデルにおいて抗脈管形成活性に関連するようである。
【0144】
要約すると、化合物1のヘモグロビン阻害は、8日間の処置後有意な阻害を伴って用量依存的であった。統計的に、有意なヘモグロビン阻害が、化合物1の全ての用量において観察された。全ての用量は、体重の損失または観察された有害な臨床徴候のない良好な耐性を示した。このモデルにおいて44ng/mLの化合物1の血漿濃度(投薬後2時間)は、抗脈管形成活性と関連していた。
(実施例9)
5mg/kgのBIDの複数の経口投薬の研究に由来するサルの血漿における化合物1の代謝物のプロフィールを、投薬1日目および14日目のサンプルにおいて決定した。ある代謝物は、LC/UVおよびLD/MS/MSによって脱メチル化から生じた(化合物3)と同定され、特徴付けられた。親化合物1(P)は、クロマトグラフィーの保存時間が18.3分においてm/z=393.3で(M+H+)イオンを産生した。脱メチル化された代謝物(P−CH3)は、m/z=379.3(M+H+)およびクロマトグラフィーの保存時間が18.1分で同定された。その代謝物と化合物1の間の14ダルトンの質量の違いは、脱メチル化された化合物1と一致する。代謝物の質量およびクロマトグラフィーの保存は、独立に合成された化合物3と同じであった。化合物1のピペラジンN−オキシド(N−オキシドは化合物2)に対応する代謝物は、この投薬レベルにおいて血漿で検出されなかった。356nmにおける吸光度クロマトグラムで17.7分および18.5分でUV信号を発生する成分は、化合物1に対するUVスペクトルの比較に基づいて、そしてブランク血漿(投薬1日目の時刻0)におけるそれらの存在に起因してマトリックス成分であって代謝物ではないと決定された。
【0145】
脱メチル化された代謝産物の推定レベルを表1に示す。代謝物(化合物1の相当量における)の推定レベルは、この分析で得られた化合物1のUV吸着ピークの高さに対する、代謝物のUV吸光度ピークの高さの比に基づき、そして以前の定量分析研究において同様のサンプルで決定された既知のレベルの化合物1に対して吸光度比を考慮に入れることよって外挿される。親化合物は、全ての蓄積された時点で代謝物よりも、より大きな吸光度を示すことが判明した。化合物1のレベルは、基本的に検出できないN−脱メチル代謝物に平行して、14日目のサンプルにおいて十分に低くなることが見出された。共役したフェーズII(PhaseII)タイプの代謝物(グルクロニドまたはサルフェイト)を含む他の代謝物は、用量投薬の1日目または14日目においてこれらの血漿サンプルに内に検出されなかった。
【0146】
【表10】
(実施例10)
化合物1での処理後にマウスから収集された血漿および腫瘍を用いる研究を、潜在的な薬力学の最終点を評価するために行った。化合物1の処理後にKM12L4a腫瘍における標的調節の分析は、VEGFR1、VEGFR2、PDGFRβおよびFGFR1のリン酸化が、時間および用量依存的様式で阻害されることを示した。例えば、HMVEC細胞は、約0.1μMのIC50でVEGFによって媒介されるVEGFR2リン酸化の阻害を示した。さらに、化合物1を用いる内皮細胞の処理は、VEGFを介すことでMAPKおよびAktのリン酸化を阻害した。
【0147】
さらに、ERK(MAPK)の活性化の時間および用量依存的阻害、レセプターチロシンキナーゼの下流の標的が、KM12L4A細胞において0.1μMから0.5μMの範囲のIC50で観察された(KM12L4A細胞はそれらの表面においてPDGFRβおよびVEGFR1/2を発現する)。KM12L4A細胞を、無血清DMEM中で化合物1とともに3時間インキュベートした。収集後、溶解物を、SDS−Pageによって分離し、ホスホ−ERK1/2およびERK1/2抗体でプローブした。検出において、ECL試薬(Amersham)を使用した。レセプターのリン酸化およびERKの活性化に対する化合物1の阻害効果は、処置後24時間維持された。MV4〜MV11細胞におけるERK1/2のリン酸化は、用量依存的様式で0.01μM〜0.1μMのIC50において1によって阻害された。
【0148】
有意な活性が、HCT116ヒト結腸腫瘍モデルにおいてインビボで観察された。HCT116腫瘍において、化合物1は、用量および時間依存的様式におけるERK(MAPK)のリン酸化を阻害し、そして、腫瘍の組織学解析において有意な変化が観察された。
【0149】
前臨床モデルにおいて、これらのPK/PD評価は、化合物1が、標的レセプターおよび下流のシグナル分子ERK(MAPK)の両方の用量および時間依存的阻害を示したを示す。これらの研究は、潜在的バイオマーカーの同定を補助し、その結果、臨床的試行における化合物1の生物学的活性を観測することを補助する。
(実施例11)
雄および雌のスプレイグドーリー(SD)ラットに対する、14Cで標識した化合物1(キノリノン環の第4位)の一回の経口(PO)投薬(5mg/kg)後の、組織における放射能の分布を、全身オートラジオグラフィー(WBA)によって定量した。WBAのための血液および屠体を、投薬後24時間を通して特定の時点で収集した。屠体をヘキサン/ドライアイスバスで凍結させ、水分を排出させ、吸い取り紙で乾燥させて、そしてドライアイス上に配置したか、もしくは少なくとも2時間、約−70℃で保管した。凍結した屠体を、冷却したカルボキシメチルセルロースに埋め込み、埋め込まれたオートラジオグラフィー標準と一緒に固形に凍結させ、そして分析まで−20℃で保管した。40μmの厚さの適当な断片を矢状平面における、目的の5レベルで粘着性のテープの上に収集した。全ての主要な組織、器官および生物学的流体が標本にされた。蛍光検出画面に、その断片を表示し、走査し、そして14C−1の組織濃度内挿をするために標準曲線を形成した。血漿を、液体シンチレーションカウント(LSC)によって放射能の濃度について分析した。例示的な結果を表11に示す。
【0150】
14C−1の経口投与後に、14C−1由来の放射能は、投薬後1時間で全ての組織にわたって広範囲に分布し、そして投薬後4時間でほとんどの組織においてCmaxに達した。雄および雌の組織における放射能の全体的な分布は類似した。14C−1由来の放射能は、血清よりも組織からよりゆっくりと除去された。雄および雌において、24時間を通して胃腸路を除いた14C−1の最も高い組織中の濃度を、ハーダー腺、副腎腺、腎髄質、眼窪内の涙腺および眼窪外の涙腺で検出した。14C−1由来の放射能は、経口投薬投与後に、血液脳の関門を横切った。
【表11A】
【表11B】
【0151】
本発明は、本明細書中に記載される実施形態に限定されず、添付の特許請求の範囲の範囲内に入るような全ての形式を包括する、と理解される。
【技術分野】
【0001】
本出願は、2003年6月16日に提出された米国仮出願番号60/478,916;2003年4月3日に提出された米国仮出願番号60/460,369;2003年4月3日に提出された米国仮出願番号60/460,327;2003年4月3日に提出された米国仮出願番号60/460,493;2003年4月3日に提出された米国仮出願番号60/460,328;2002年11月13日に提出された米国仮出願番号60/426,204;2002年11月13日に提出された米国仮出願番号60/426,226;2002年11月13日に提出された米国仮出願番号60/426,282;2002年11月13日に提出された米国仮出願番号60/426,107および2003年11月7日に提出された「Methods of Treating Cancer and Related Methods」と題された米国仮出願に対する優先権を主張し、それらの各々が、十分に本明細書の前に記載されているかのように、その全体が、全ての目的のために参考文献として本明細書に緩用される。
(発明の分野)
本発明は、受容体チロシンキナ−ゼインヒビターを用いた癌の処置方法に関する。本発明はまた、被験体へのインヒビターの投与後の、そのインヒビターおよびその代謝物の量および濃度の測定方法に関する。
【背景技術】
【0002】
(発明の背景)
毛細血管は、人体のほぼ全ての組織に達し、組織に酸素および栄養物を供給し、老廃物の除去を行う。ヒトの成体では、典型的な条件下で、毛細血管の内部を覆う内皮細胞は分裂せず、それゆえ毛細血管は、通常数が増えないし、大きくもならない。しかし、ある通常の条件下で、組織が損傷されたような場合、または月経周期のある部分の間に、毛細血管は迅速に増殖し始める。既存の血管からの、新しい毛細血管の形成過程は、新脈管形成または新生血管形成として公知である。Folkman,J.Scientific American 275,150−154(1996)を参照のこと。創傷治癒中の新脈管形成は、成人生活における、病態生理的な新生血管形成の一例である。創傷治癒の間に、追加の毛細血管は酸素および栄養物の供給を提供し、肉芽組織を促進し、老廃物の除去を補助する。治癒過程の終結後、毛細血管は通常は消退する。Lymboussaki,A.”Vascular Endothelial Growth Factors and their Receptors in Embryos, Adults,and in Tumors”Academic Dissertation,University of Helsinki,Molecular/Cancer Biology Laboratory and Department of Pathology,Haartman Institute,(1999)。
【0003】
新血管形成はまた、癌細胞の増殖に重要な役割を果たす。癌細胞の巣窟は、一度ある一定の大きさ、大まかには直径1〜2ミリメートルの直径に達すれば、癌細胞に十分量の酸素および栄養物を供給するには拡散が十分でないので、腫瘍がより大きく成長するのに、癌細胞は血液供給を発達させなければならないことが公知である。
【0004】
レセプターチロシンキナーゼ(RTK)は、発生段階にある細胞の増殖および分化、そして成体の組織のリモデリングおよび再生を調節する膜貫通ポリペプチドである。Mustonen,T.ら、J.Cell Biology 129,895−898(1995);van der Geer,P.ら、Ann Rev.Cell Biol.10,251−337(1994)。増殖因子またはサイトカインとして公知のポリペプチドのリガンドは、RTKを活性化することが公知である。シグナル伝達をするRTKは、リガンドの結合およびレセプターを二量体化するレセプターの外部ドメインのコンフォメーション変化を含む。Lymboussaki,A.”Vascular Endothelial Growth Factors and their Receptors in Embryos, Adults, and in Tumors”Academic Dissertation,University of Helsinki,Molecular/Cancer Biology Laboratory and Department of Pathology,Haartman Institute,(1999);Ullrich,A.ら,Cell 61,203−212(1990)。リガンドのRTKへの結合は、レセプターの特定のチロシン残基でのリン酸転移反応を生じ、、細胞質に存在する基質のリン酸化の触媒ドメインの次の活性化に帰着する。
【0005】
RTKの二つのサブファミリーは、血管内皮細胞に特有である。これらは、血管内皮増殖因子(VEGF)サブファミリーおよびTieレセプターファミリーを含む。クラスIII型RTKは、VEGFR−1、VEGFR−2およびVEGFR−3を含む。Shibuya,Mら、Oncogene 5,519−525(1990);Terman,Bら、Oncogene 6,1677−1683(1991);Aprelikova,Oら、Cancer Res.52,746−748(1992)。
【0006】
VEGFサブファミリーのメンバーは、血管透過性および内皮細胞の増殖を誘導し得るとして記載され、さらに新脈管形成および脈管形成の主要な誘導物質として同定されている。Ferrara,N.ら、Endocrinol.Rev.18,4−25(1997)。VEGFは、VEGFR−1およびVEGFR−2を誘導するRTKに特異的に結合することが公知である。DeVries,C.ら、Science 255,989−991(1992);Quinn,Tら、Proc.Natl.Acad.Sci.90,7533−7537(1993)。VEGFは内皮細胞の移動および増殖を刺激し、インビトロおよびインビボの両方で新脈管形成を誘導する。Connolly,D.ら、J.Biol.Chem.264,20017−20024(1989);Connolly,D.ら、J.Clin.Invest.84,1470−1478(1989);Ferrara,N.ら、Endocrino.Rew.18,4−25(1997);Leung,D.ら、Science 246,1306−1309(1989);Plouet,J.ら、EMBO J8、3801−3806(1989)。
【0007】
新脈管形成は、癌の増殖に決定的であるということならびにVEGFおよびVEGF−RTKにより制御されることが公知であるので、VEGF−RTKのアンタゴニストであり、それゆえ新脈管形成を阻害し、または遅らせ、望ましくは腫瘍の増殖を妨害し、または停止させる治療剤を開発するのにかなりの努力が保証された。
【0008】
増殖因子受容体キナーゼ(PDGFRK)由来の血小板は、RTKの別のタイプである。PDGFの発現は、グリア芽細胞腫から前立腺癌までの、多数の様々な固形腫瘍に示されている。これらの様々な腫瘍タイプでは、PDGFシグナル伝達の生物学的な役割は、癌細胞の増殖のオートクリン刺激から、隣のストローマおよび新脈管形成を含む、より微妙なパラクリン相互作用まで変化し得る。従って、PDGFRキナーゼの活性の小分子による抑制は、腫瘍の増殖および新脈管形成を妨害し得る。
【0009】
4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンは、VEGFーRTK、PDGF−RTKおよび線維芽細胞増殖因子受容体(FGF−RTK)のような他の受容体チロシンキナーゼの小分子インヒビターである。この化合物は、一つの特許およびいくつかの特許出願に記述されており、その開示全体は、参考文献としておよび全ての目的で本明細書中に援用される:米国特許第6,605,617号、U.S.S.N.10/644,055、米国仮出願番号60/405,729、60/428,210および60/484,048。この潜在的抗癌薬剤の代謝プロフィールの決定のための方法が必要であるように、本化合物を投与する特定の方法が必要である。
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の要約)
本発明は、白血病および固形腫瘍を含む癌の処置方法を提供する。特に、被験体で癌の増殖を抑制する十分な血液レベルの4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンズイミダゾール−2−イル]キノリン−2(1H)−オンを得るための方法が提供される。この化合物は、受容体チロシンキナーゼのインヒビターである。バイオマーカーとしての化合物およびそのような化合物を用いて被験体中でのインヒビターの分布および代謝をモニターするための方法がさらに提供される。さらに、本発明は、薬学的組成物ならびにインヒビターを含む医薬およびそれらの使用方法を提供する。
【0011】
本発明は、例えば、以下を提供する。
(項目1)
癌処置のための単位投薬形態の医薬の調製における、式
【化1】
の化合物、その薬学的に受容可能な塩、その互変異性体、または該互変異性体の薬学的に受容可能な塩の使用であって、ここで、該医薬の各々の単位用量は、以下:
(a)該化合物が被験体に投与された場合、該被験体の血漿中での、約20ng/mL〜4000ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約40ng/mL〜8000ng/mLの該化合物のCmax値、
(b)被験体への投与の24時間後の、該被験体の血漿中での、約10ng/mL〜2,000ng/mLの該化合物もしくは被験体への投与の24時間後の該被験体の血液中での、約20ng/mL〜4,000ng/mLの該化合物、または
(c)該化合物が被験体に投与された場合、該被験体の血漿中での約500ng*h/mL〜60,000ng*h/mLの該化合物もしくは該被験体の血液中での約750ng*h/mLから120,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目2)
項目1に記載の使用であって、ここで、各々の単位用量は、以下:
(a)前記被験体の血漿中での、約50ng/mL〜500ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約100ng/mL〜1000ng/mLの該化合物のCmax値、
(b)投与の24時間後の、該被験体の血漿中での、約20ng/mL〜1,000ng/mLの該化合物もしくは投与の24時間後の、該被験体の血液中での、約40ng/mL〜2,000ng/mLの該化合物、または
(c)該被験体の血漿中での約1,000ng*h/mL〜30,000ng*h/mLの該化合物もしくは該被験体の血液中での約1,500ng*h/mL〜60,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目3)
項目1に記載の使用であって、ここで、各々の単位用量は、以下:
(a)前記被験体の血漿中での、約50ng/mL〜250ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約100ng/mL〜500ng/mLの該化合物のCmax値、
(b)該被験体への投与の24時間後の、該被験体の血漿中での、約40ng/mL〜500ng/mLの該化合物もしくは該被験体への投与の24時間後の、該被験体の血液中での、約80ng/mL〜1,000ng/mLの該化合物、または
(c)該被験体の血漿中での約2,000ng*h/mL〜15,000ng*h/mLの該化合物もしくは該被験体の血液中での約3,000ng*h/mL〜30,000ng*h/mLの該化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目4)
項目1の使用であって、ここで、各々の単位用量は、以下:
(a)該被験体の血漿中での、約75ng/mL〜150ng/mLの該化合物のCmax値もしくは該被験体の血液中での、約150ng/mL〜300ng/mLの該化合物のCmax値、または
(b)投与の24時間後の、該被験体の血漿中での、約40ng/mL〜250ng/mLの該化合物もしくは投与の24時間後の、該被験体の血液中での、約80ng/mL〜500ng/mLの該化合物、
のうちの少なくとも一つを提供するのに十分である、使用。
(項目5)
項目1に記載の使用であって、ここで、各々の単位用量が、前記被験体の血漿中で約100ng/mL〜2000ng/mLの化合物のCmax値または該被験体の血液中で約200ng/mL〜4000ng/mLの化合物のCmax値を提供するのに十分である、使用。
(項目6)
項目1に記載の使用であって、ここで、各々の単位用量が前記被験者の血漿中での100ng/mL〜1000ng/mLの前記化合物のCmax値または該被験体の血液中での200ng/mL〜2000ng/mLの該化合物のCmax値を提供するのに十分である、使用。
(項目7)
項目1〜6のいずれか1項に記載の使用であって、ここで、化合物の乳酸塩が前記医薬を調製するために使用される、使用。
(項目8)
項目1〜7のいずれか1項に記載の使用であって、ここで、前記医薬が経口投与に適切である、使用。
(項目9)
項目8に記載の使用であって、ここで、前記医薬の前記単位投与形態が、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁剤または水溶液である、使用。
(項目10)
項目1〜7のいずれか1項に記載の使用であって、ここで、前記医薬が、短いボーラス、遅い注入または長期の注入としての注射による投与に適している、使用。
(項目11)
項目1〜10のいずれか1項に記載の使用であって、ここで、前記医薬の各々の単位用量が、前記被験体の体重に基づいて、体重1kg当たり0.25mg〜30mgの前記化合物、互変異性体、および/または塩を含む、使用。
(項目12)
項目1〜10のいずれか1項に記載の使用であって、ここで、前記医薬の各々の単位用量が、25mg〜1500mgの範囲の量の前記化合物、互変異性体および/または塩を含む、使用。
(項目13)
項目1〜12のいずれか1項に記載の使用であって、ここで、前記医薬が、7個、14個、21個または28個の毎日の量の上記単位用量を含むキットに配置され、該キットが、7日間、14日間、21日間または28日間の処置周期での使用に適する、各々の前記化合物の前記毎日量の投与、続く該化合物の投与なしの7日または14日を含む、使用。
(項目14)
式
【化2】
を有する化合物、その薬学的に受容可能な塩、その互変異性体または該互変異性体の薬学的に受容可能な塩であって、該化合物が、癌処置の方法に使用するためのものであり、該癌処置の方法が、上記化合物のある量を癌患者に投与することを含み、その量が、
(a)該化合物が被験体に投与された場合、該被験体の血漿中で約20ng/mL〜4000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約40ng/mL〜8000ng/mLの該化合物のCmax値、
(b)被験体への投与の24時間後の該被験体の血漿中で約10ng/mL〜2,000ng/mLの該化合物もしくは該被験体への投与の24時間後の該被験体の血液中で約20ng/mL〜4,000ng/mLの該化合物、または
(c)該化合物が被験体に投与された場合、該被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物もしくは該被験体の血液中で約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目15)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約50ng/mL〜500ng/mLの該化合物のCmax値もしくは該被験体の血液中で約100ng/mL〜1000ng/mLの該化合物のCmax値、
(b)投与の24時間後の該被験体の血漿中で約20ng/mL〜1,000ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約40ng/mL〜2,000ng/mLの該化合物、または
(c)該被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの該化合物もしくは該被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの該化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目16)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約50ng/mL〜250ng/mLの該化合物のCmax値もしくは該被験体の血液中で約100ng/mL〜500ng/mLの該化合物のCmax値、
(b)投与の24時間後の該被験体の血漿中で約40ng/mL〜500ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約80ng/mL〜1,000ng/mLの該化合物、または
(c)該被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの該化合物もしくは該被験体の血液中で約3,000ng*h/mL〜30,000ng*h/mLの該化合物のAUC値、
の少なくとも一つを提供するのに十分である、化合物。
(項目17)
項目14に記載の化合物であって、ここで、各々の単位用量が、以下:
(a)前記被験体の血漿中で約75ng/mL〜150ng/mLの該化合物のCmax値もしくは該被験体の血液中で約150ng/mL〜300ng/mLの該化合物のCmax値、または
(b)投与の24時間後の該被験体の血漿中で約40ng/mL〜250ng/mLの該化合物もしくは投与の24時間後の該被験体の血液中で約80ng/mL〜500ng/mLの該化合物、
の少なくとも一つを提供するのに十分である、化合物。
(項目18)
項目14に記載の化合物であって、ここで、各々の単位用量が、前記被験体の血漿中で100ng/mL〜2000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約200ng/mL〜4000ng/mLの該化合物のCmax値を提供するのに十分である、化合物。
(項目19)
項目14に記載の化合物であって、ここで、各々の単位用量が、前記被験体の血漿中で100ng/mL〜1000ng/mLの該化合物のCmax値もしくは該被験体の血液中で約200ng/mL〜2000ng/mLの該化合物のCmax値を提供するのに十分である、化合物。
(項目20)
被験体中の、以下の式
【化3】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の代謝プロフィールを決定するための、代謝物の使用であって、該代謝物が、以下の式
【化4】
のN−オキシド化合物または以下の式
【化5】
N−デスメチル化合物の少なくとも一つを含む、使用。
(項目21)
被験体中の、以下の式:
【化6】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の代謝プロフィールを決定する方法であって、該方法が、該被験体から採られた尿、血液または組織の一つ以上のサンプル中の、該化合物の代謝物の少なくとも一つの量を測定することを含む、方法。
(項目22)
項目21に記載の方法であって、ここで、前記少なくとも一つの代謝物が、以下の式:
【化7】
を有するN−オキシド化合物であるか、または
以下の式:
【化8】
を有するN−デスメチル化合物である、方法。
(項目23)
項目22に記載の方法であって、ここで、該方法が、前記N−オキシド化合物および前記N−デスメチル化合物の両方の前記量を測定することを含む、方法。
(項目24)
項目21〜23のいずれかに記載の方法であって、ここで、前記代謝物が、紫外線分光器法または液体クロマトグラフィー−質量分光法により測定される、方法。
(項目25)
被験体中の、以下の式:
【化9】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、または該互変異性体の薬学的に受容可能な塩の前記量を測定する方法であって、該方法は、該化合物が該被験体に投与された後、該被験体から採られる尿、血液、または組織のサンプル中の該化合物の量を測定することを含む。
(項目26)
項目25に記載の方法であって、該方法が、前記サンプル中の前記化合物の代謝物の量を測定することをさらに含む、方法。
(項目27)
項目26に記載の方法であって、ここで、前記代謝物が以下の式:
【化10】
を有するN−オキシド化合物である、方法。
(項目28)
項目26に記載の方法であって、ここで、前記代謝物が、以下の式:
【化11】
を有するN−デスメチル化合物である、方法。
(項目29)
項目25に記載の方法であって、前記被験体に前記化合物が投与された後、該被験体から異なる時点に二つ以上のサンプルを抜き出すことをさらに含む、方法。
(項目30)
癌処置のための単位投与形態の、医薬の前記調製における、以下の式:
【化12】
の化合物、その薬学的に受容可能な塩、その互変異性体、該互変異性体の薬学的に受容可能な塩の使用であって、ここで、該医薬の各々の単位用量は、25mg〜1000mgの量の該化合物を含む、使用。
【0012】
従って、本発明に従って、癌を処置するための方法が提供され、この方法は、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmax値もしくは被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax値を提供するのに十分な量の式Iの化合物:
【0013】
【化13】
その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩を、癌を有する被験体に対して投与する工程を含有する。ある実施形態では、投与される化合物の量は、被験体の血漿中で約35ng/mL〜2,000ng/mLの化合物のCmax値もしくは被験体の血液中で約70ng/mL〜4,000ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約50ng/mL〜500ng/mLの化合物のCmax値もしくは被験体の血液中で約100ng/mL〜1,000ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約50ng/mL〜250ng/mLの化合物のCmax値もしくは被験体の血液中で約100ng/mL〜500ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値もしくは被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値を提供するか、被験体の血漿中で約100ng/mL〜2,000ng/mLの化合物のCmax値もしくは被験体の血液中で約200ng/mL〜4,000ng/mLの化合物のCmax値を与えるのに十分な量である。式Iの化合物の乳酸塩は、ある実施形態で被験体に投与され、そしていくつかのそのような実施形態において被験体はヒトである。化合物、互変異性体、またはその塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液、水溶液、または本明細書に記載の他の形態として処方され得る。いくつかのこのような実施形態では、乳酸塩は水溶液であり、ヒト被験体に経口投与される。他の実施形態では、化合物は注射により投与され得る。
【0014】
さらなる局面では、本発明は、癌を処置する方法を提供し、この方法は、投与の24時間後に被験体の血漿中に約10ng/mL〜2,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約20ng/mL〜4,000ng/mLの化合物を提供するのに十分な量の式Iの化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩を、癌を有する被験体へ投与する工程を含有する。いくつかの実施形態では、投与される化合物量は、投与の24時間後に被験体の血漿中に約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約40ng/mL〜2,000ng/mLの化合物、投与の24時間後に被験体の血漿中に約40ng/mL〜500ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約80ng/mL〜1,000ng/mLの化合物、または投与の24時間後に被験体の血漿中に約40ng/mL〜250ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約80ng/mL〜500ng/mLの化合物を提供するのに十分な量である。いくつかの実施形態では、被験体はヒトである。一般に、癌処置の本方法では、式Iの化合物の乳酸塩は被験体に投与される。いくつかのそのような実施形態では、乳酸塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液または水溶液であり、ヒト被験体に経口投与される。
【0015】
従って、癌の本処置方法ある実施形態では、式Iの化合物は、フルクトースを含む薬学的組成物または医薬として投与される。そのような実施形態では、薬学的組成物はさらにテトラロームマンダリン風味のような矯味矯臭剤を含む。他の実施形態では、薬学的化合物は、さらに水を含む。従って、癌処置の本方法は、式Iの固形化合物を水と混和し、水性混合物を形成するために、式Iの固形化合物と水とを混和することをさらに含み得る。その発明は、式Iの化合物の用途をさらに提供する。本発明は、癌処置に使用するための医薬の調合をする際の、式Iの化合物の使用をさらに提供する。
【0016】
本明細書に記載の癌の処置方法に関する他の実施形態では、化合物は、顆粒剤、散剤、懸濁剤、錠剤、丸剤、カプセル剤、ゲルキャップ、キャプレット、エマルジョン、、シロップ剤、エリキシル剤、懸濁液、スプレー、エアロゾル、座薬または水剤から選択された薬学的組成物として投与される。好ましくは、薬学的組成物は錠剤、癌剤、カプセル剤、ゲルキャップまたはキャプレットから選択される。
【0017】
本明細書に記載の癌処置の本方法に関する、さらに別の実施形態では、化合物は、短いボーラス、遅い注入、または長期の注入として注射により投与される。注射は、日に一回、二回、三回または四回投与され得る。
【0018】
癌処置の本方法のある実施形態では、被験体に投与される式Iの化合物の量は、被験体の体重により、0.25mg/kg〜0.30mg/kgの範囲に及ぶ。他の実施形態では、被験体に投与される化合物の量は、一日当たり約25mg〜1,500mgであり、好ましくは一日当たり約200mg〜500mgの範囲である。
【0019】
癌の本処置方法は、処置されるべき癌が固形腫瘍または白血病である人々を含む、広い様々な種類の癌に対して有効である。特に、本方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖病、非小細胞肺癌、小細胞肺癌、慢性リンパ性白血病、肉腫、黒色腫、リンパ腫、甲状腺腫、神経内分泌の癌、腎細胞癌、胃癌、消化管間質潰瘍、神経膠腫、脳腫瘍または膀胱癌のような癌を処置するのに使われ得る。
【0020】
いくつかの実施形態では、本明細書に記載の癌の処置方法は、処置周期の一部としての式Iの化合物の投与をさらに含む。従って、処置周期は、その量の式Iの化合物の7日間、14日間、21日間または28日間にわたる毎日の投与、続く化合物の投与のない7日間または14日間を含み得る。いくつかの実施形態では、処置周期は、その量の化合物の7日間にわたる毎日の投与、続く化合物の投与のない7日間を含む。処置の過程を提供するため、処置周期は、一回以上繰り返され得る。さらに、化合物は、処置周期の投与段階の間、日に一回、二回、三回または四回投与され得る。他の実施形態では、その方法は、処置の過程の間に、一日または一日おきに一回、二回、三回または四回のその化合物の量の投与をさらに含む。
【0021】
さらに、癌処置の方法が提供され、その方法は、被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物または被験体の血液中で約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値を提供するのに十分な量の式
【0022】
【化14】
を有する化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩を、癌を有する被験体へ投与する工程を含有する。他のそのような実施形態では、投与される化合物の量は、被験体の血漿中で、約1,000ng*h/mL〜30,000ng*h/mLの化合物または被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値を提供するのに十分である。他のそのような実施形態では、AUC値は、被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの化合物または被験体の血液中での3,000ng*h/mL〜30,000ng*h/mLの化合物である。
【0023】
本発明は、癌の処置方法をさらに提供し、その方法は、式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体で薬学的に受容可能な塩を、癌を有する被験体に投与することを含む。ここで、第1の処置周期での投与される化合物の量は、一日当たり25mgであり、そして投与される化合物の量は、一日当たり1500mgの化合物が被験体に投与されるまでか、または被験体に用量限界の毒性が観察されるまで、各処置周期につれて増える。概してそのような方法では、投与される化合物の量は、第1の周期の後、処置周期毎に投与される化合物の量が倍加する。いくつかの実施形態では、処置周期は、7日間にわたる毎日の等量の化合物の投与、続く化合物が投与されない7日間を含む。
【0024】
他の局面では、本発明は、癌の処置方法を提供し、その方法は、式I
【0025】
【化15】
の化合物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分量を、癌を有する被験体へ投与すること、および以下の式
【0026】
【化16】
から選択される式IIならびに式IIIの一つあるいは両方の、被験体への曝露を含む。ここで、式IIおよび式IIIの一つあるいは両方が、被験体による式Iの化合物の代謝により産生されて、被験体の血漿中で約20ng/mL〜約4000ng/mLの範囲に及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の合計Cmax値または被験体の血液中で約40ng/mL〜約8000ng/mLの範囲に及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の合計Cmax値を提供する。
【0027】
さらに別の局面では、本発明は、癌処置の方法を提供し、その方法は、以下
【0028】
【化17】
から選択される式を有する一つ以上の化合物、その活性代謝物、その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分量に、癌を有する被験体を曝すことを含み、それらの化合物は、被験体の血漿中での、約20ng/mL〜4000ng/mLの一つ以上の化合物の合計Cmax値または被験体の血液中での約40ng/mL〜8000ng/mLの一つ以上の化合物の合計Cmax値を提供するのに十分な量である。いくつかの実施形態では、一つ以上の化合物の量は、被験体の血漿中での、約35ng/mL〜2600ng/mLの一つの化合物のCmax値または被験体の血液中での、35ng/mL〜6000ng/mLの一つの化合物のCmax値を提供する。他の実施形態では、一つ以上の化合物の量は、被験体の血漿中での、約35ng/mL〜1200ng/mLの化合物の一つのCmax値または被験体の血液中での、約50ng/mL〜2400ng/mLの化合物の一つのCmax値を提供する。
【0029】
本発明の他の局面では、式Iの化合物、その薬学的に受容可能な塩、その互変異性体またはその互変異性体の薬学的に受容可能な塩の代謝プロフィールを被験体において決定する方法が提供される。その方法は、被験体から採られた尿、血液、または組織の一つ以上のサンプルの化合物の少なくとも一つの代謝物の量を測定することを含む。いくつかのそのような実施形態では、少なくとも一つの代謝物は、以下の式II
【0030】
【化18】
を有するN−オキシド化合物である。
【0031】
他のそのような実施形態では、少なくとも一つの代謝物は、式III
【0032】
【化19】
を有するN−デスメチル化合物である。
【0033】
いくつかのそのような実施形態では、少なくとも一つの代謝物は、式IIのN−オキシド化合物である第2の代謝物をさらに含む。代謝物の量は、紫外線(UV)分光学および/または液体クロマトグラフィー−質量分光学(LC−MS)を含む技術を用いて測定され得る。
【0034】
本発明の他の局面では、被験体中の式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の量を決定する方法が提供される。この方法は、その化合物が被験体に投与された後に、被験体から採られた尿、血液または組織の一つのサンプル中の化合物の量を測定することを含む。この方法は、サンプル中の化合物の代謝物の量を測定することをさらに含み得る。測定され得る代謝物としては、式IIのN−オキシド化合物および/または式IIIを有するN−デスメチル化合物が挙げられ得るが、これらに限定されない。いくつかの実施形態では、その方法は、式Iの化合物が被験体へ投与された後の異なる時点で被験体からの二つ以上のサンプルを回収することをさらに含む。
【0035】
本発明の他の目的、特徴および利点は、以下の詳細な説明から明らかになる。しかし、発明の精神および範囲内の様々な変更および改変は詳細な説明から当業者に明らかになるので、詳細な説明および具体的実施例は、本発明のある実施形態を示していると同時に、単なる例示として与えられる、ということは理解されるべきである。
【図面の簡単な説明】
【0036】
【図1】図1は、式Iの化合物によるKM12L4aの腫瘍抑制を示している。
【図2】図2は、Cmax値およびAUC値対KM12L4a腫瘍保有マウスでのKM12L4a腫瘍の増殖の抑制パーセントを示している。
【発明を実施するための形態】
【0037】
(詳細な説明)
本発明は、式Iの化合物を用いた癌の処置方法、被験体から採られる生物学的サンプル中の式Iの化合物および/またはその代謝物の量を測定する方法、ならびに式Iの化合物を含む薬学的組成物および医薬、そしてそれらの使用方法に関する。
【0038】
本明細書中で定義される以下の専門用語および語句は、本明細書を通して使用される。
【0039】
本明細書で用いられる「AUC」は、時間の経過に対する血漿中の化合物の濃度のグラフ中での曲線下の面積のことを指す。
【0040】
本明細書で用いられているように、「Cmax」は、化合物が投与された被験体の血漿中、組織中、血液中の化合物の最高濃度を指す。概してCmaxは、被験体への化合物の投与の数時間以内に生じる。
【0041】
毒性を制限する用量は、Common Terminology Criteria of Adverse Events Version 3.0(CTCAE)に従って規定される。従って、薬物処置周期内に次の事象のいずれかが観察される場合、毒性制御用量は、化合物の被験者への投与と共に生じる:5日間以上の連続した日数の段階4の好中球減少症(すなわち、絶対好中球数(ANC)≦500個細胞/mm3)または発熱好中球減少症(すなわち、発熱≧38.5℃かつANC≦1000個細胞/mm3);段階4の血小板減少症(すなわち、≦25,000個細胞/mm3または血小板輸血を必要とする出血エピソード);段階4の疲労、またはECOGパフォーマンスステータスにおける二点減少;十分/最大の医学的介入にも関わらない、段階3以上の悪心、下痢、嘔吐および/または筋痛;段階3以上の非血液学的毒性(疲労を除く);化合物1を用いた処置に関連した、毒性からの遅れた回復のための、2週間より長い再処置の遅延;段階2以上の、臨床的に重要な心毒性(例えば、休止駆出率の40%≦50%の低下または短縮画分の15%≦24%の低下;心トロポニンT≧0.05ng/mL)。
【0042】
薬学的に受容可能な塩としては、無機塩基、有機塩基、無機酸、有機酸または塩基性もしくは酸性アミノ酸が挙げられる。無機塩基の塩として、本発明は、例えば、ナトリウムまたはカリウムのようなアルカリ金属、カルシウムおよびマグネシウムようなアルカリ土類金属、アルミニウム、ならびにアンモニアを含む。有機塩基の塩として、本発明は、例えば、トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、エタノールアミン、ジエタノールアミン、およびトリエタノールアミンを含む。無機酸の塩として、本発明は、例えば塩酸、臭化水素酸、硝酸、硫酸、およびリン酸を含む。有機酸の塩として、本発明は、例えば、乳酸、ギ酸、酢酸、トリフルオロ酢酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホニル酸、ベンゼンスルホン酸およびp−トルエンスルホン酸を含む。塩基性アミノ酸の塩として、本発明は例えば、アルギニン、リジンおよびオルニチンを含む。酸性アミノ酸は、例えば、アスパラギン酸およびグルタミン酸を含む。
【0043】
本発明に従うその有機化合物が、互変異性の現象を示し得るということは理解されるべきである。本明細書内の化学構造は、一時点で起こり得る互変異性体形態の一つのみを表し得るので、本発明は描かれた構造の任意の互変異性体形態を含有するということは理解されるべきである。例えば、式Iの化合物は、一つの互変異性体である互変異性体Iaと共に以下:
【0044】
【化20】
に示されている。
【0045】
式Iの化合物の他の互変異性体である互変異性体Ibおよび互変異性体Icが以下:
【0046】
【化21】
に示されている。
【0047】
当業者が容易に理解するように、幅広い様々な官能基および他の構造は、互変異性を示し得、そして式Iを有する化合物の全ての互変異性体は、本発明の範囲内である。
【0048】
「被験体」という用語は、本明細書で用いられている場合、本発明の方法の有益な効果を経験し得る任意の動物を指す。従って、式Iの化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩は、本発明により提供される癌の処置方法に従って、その化合物の有益な効果を経験し得る任意の動物に投与され得る。好ましくは、その動物は哺乳動物、特にヒトであるが、本発明はそのように制限されるように意図されていない。他の適切な動物の例としては、ラット、マウス、サル、イヌ、ネコ、ウシ、ウマ、ブタ、ヒツジなどが挙げられるが、これらに制限されていない。
【0049】
本発明の文脈において、「処置」は、障害もしくは疾患と関連する症状の軽減、それらの症状のさらなる進行もしくは悪化の停止、または疾患もしくは障害の防止もしくは予防を意味する。例えば、癌の文脈において、好結果の処置は、腫瘍の増殖速度の減少、腫瘍の増殖の停止、腫瘍の大きさの減少、癌の部分的寛解もしくは完全寛解、または生存率もしくは臨床的利点の増加から測定されるように、症状の軽減または疾患の進行の停止を含み得る。
【0050】
一つの局面では、本発明は癌処置の方法を提供し、その方法は癌を有する被験体に、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmax値または被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax値を提供する、式Iの化合物:
【0051】
【化22】
その薬学的に受容可能な塩、その互変異性体、またはその互変異性体の薬学的に受容可能な塩の十分な量を投与することを含む。いくつかの実施形態では、式Iを有する化合物の投与量は、被験体の血漿中で約35ng/mL〜2000ng/mLの化合物のCmax値または被験体の血液中で約70ng/mL〜4000ng/mLの化合物のCmax値、被験体の血漿中で約50ng/mL〜500ng/mLの化合物のCmax値または被験体の血液中で約100ng/mL〜1000ng/mLの化合物のCmax値、被験体の血漿中で約50ng/mL〜250ng/mLの化合物のCmax値または被験体の血液中で約100ng/mL〜500ng/mLの化合物のCmax値、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値または被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値、被験体の血漿中で約100ng/mL〜2000ng/mLの化合物のCmax値または被験体の血液中で約200ng/mL〜4000ng/mLの化合物のCmax値、被験体の血漿中で約100ng/mL〜1000ng/mLの化合物のCmax値または被験体の血液中で約200ng/mL〜2000ng/mLの化合物のCmax値を提供するのに十分である。好ましくは、投与される化合物の量は、被験体の血漿中で約75ng/mL〜150ng/mLの化合物のCmax値または被験体の血液中で約150ng/mL〜300ng/mLの化合物のCmax値を提供するのに十分である。従って、式Iの化合物、互変異性体およびその塩の十分な量により提供されるCmax値は所定の範囲内に入るということが理解されるべきである。
【0052】
さらなる局面では、本発明は、癌の処置方法を提供し、その方法は、式Iを有する化合物、その薬学的に受容可能な塩、その互変異性体、または互変異性体の薬学的に受容可能な塩の十分量を、癌を有する被験体に対して投与することを含み、この量は、投与の24時間後に被験体の血漿中で約10ng/mL〜2,000ng/mLの化合物を、もしくは投与の24時間後に被験体の血液中で約20ng/mL〜4,000ng/mLの化合物を提供する。いくつかの実施形態では、投与される化合物の量は、投与の24時間後に被験体の血漿中に約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後に被験体の血液中に約40ng/mL〜2,000ng/mLの化合物、投与の24時間後に被験体の血漿中に約40ng/mL〜500ng/mLの化合物もしくは投与の24時間後に被験体の血液中に80ng/mL〜1,000ng/mLの化合物、または投与の24時間後に被験体の血漿中に40ng/mL〜250ng/mLの化合物もしくは投与の24時間後に被験体の血液中に80ng/mL〜500ng/mLの化合物を提供するのに十分な量である。
【0053】
概して、本明細書に記載の癌の処置方法では、式Iの化合物またはその互変異性体は、薬学的に受容可能な塩として投与される。乳酸塩、リンゴ酸塩、メシラート、酢酸塩、酒石酸塩、リン酸塩、硫酸塩、硝酸塩、塩酸塩、クエン酸塩またはマレイン酸塩のような塩は、様々なモル比で、エナンチオマー形態またはラセミ体形態で適切である。好ましくは、式Iの化合物の乳酸塩は、ヒト被験体のような被験体に投与される。その乳酸塩は、丸剤、カプセル剤、錠剤、ゲルキャップ、キャプレット、懸濁液、または水溶液として患者に都合良く投与され経口投与される。他の実施形態では、化合物または塩は、以下に記述されるように、注射により投与され得る。
【0054】
従って、癌処置の本方法のいくつかの実施形態では、式Iの化合物は、フルクトースを含む薬学的組成物または医薬として投与される。そのような組成物はまた、テトラロームマンダリン風味などのような矯味矯臭剤および/または水のような希釈剤を含み得る。従って、癌処置の本方法は、式Iの固形化合物を水と混和し、そして被験体にその化合物を投与する前に水性混合物を形成することをさらに含み得る。本発明は、癌処置に用いるための医薬の調製における式Iの化合物1の使用をさらに提供する。
【0055】
癌処置の本方法のいくつかの実施形態において、被験体に投与される式Iの化合物の量は、被験体の体重1kg当たり0.25mg〜30mgに及ぶ。他の実施形態では、被験体に投与される化合物の量は、一日当たり約25mg/被験体〜1500mg/被験体、一日当たり約100mg/被験体〜1000mg/被験体、または一日当たり200mg/被験体〜500mg/被験体の範囲である。
【0056】
癌処置の本方法は、処置されるべき癌が固形腫瘍または白血病であるものを含む、幅広い様々な癌に対して有効である。特に、本方法は、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖病、非小細胞肺癌、小細胞肺癌、慢性リンパ性白血病、肉腫、黒色腫、リンパ腫、甲状腺腫、神経内分泌の癌、腎細胞癌、胃癌、消化管間質腫瘍、神経膠腫、脳腫瘍または膀胱癌のような癌の処置に使用され得る。理論に縛られることは望まないが、癌処置の本方法は、固形腫瘍に有効であると考えられている。なぜなら、式Iの化合物は、新脈管形成の阻害剤として作用するからである。より正確にいえば、式Iの化合物およびその活性代謝物は、腫瘍の新脈管形成および白血病に関与している特定のレセプターチロシンキナーゼを選択的に阻害すると考えられている。
【0057】
いくつかの実施形態では、癌処置の本方法は、式Iの化合物の処置周期の一部としての投与をさらに含む。処置周期は、化合物が被験体に規則正しい基準で与えられる投与段階および化合物が投与されない休日を含む。例えば、処置周期は、7日間、14日間、21日間または28日間にわたる、毎日の式Iの化合物のその量の投与、続く化合物が投与されない7日もしくは14日を含み得る。いくつかの実施形態では、処置周期は、7日間にわたる毎日のその量の化合物の投与、続く化合物が投与なしの7日間を含む。処置周期は、一回以上繰り返され得、処置過程を提供し得る。さらに、その化合物は、処置周期の投与段階の間、1日に一回、二回、三回または四回投与され得る。他の実施形態では、その方法は処置の過程の間に、1日にまたは隔日に一回、二回、三回または四回のその量の化合物の投与を含み得る。処置の過程は、被験体が、本方法によって癌処置を受けている時間に言及している。従って、処置の過程は、一回以上の処置周期に拡張し得るか、または被験体が毎日もしくは断続的に式Iの化合物の用量を受け取っている時間に言及し得る。
【0058】
さらに癌の処置方法が提供され、その方法は、式Iの化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の、十分量を癌を有する被験体に投与することを含み、それらの量は、被験体の血漿中で約500ng*h/mL〜60,000ng*h/mLの化合物または被験体の血液中に約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値を提供する。そのような他の実施形態では、投与される化合物の量は、被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの化合物または被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値を提供するのに十分な量である。そのような他の実施形態では、AUC値は、被験体の血漿中で約2,000ng*h/mL〜15,000ng*h/mLの化合物または被験体の血液中で約3,000ng*h/mL〜30,000ng*h/mLの化合物である。
【0059】
本発明の別の局面では、癌処置のための単位投薬形態での医薬の調製における、式Iの化合物、その薬学的に受容可能な塩、その互変異性体または互変異性体の薬学的に受容可能な塩の使用が、提供される。ここで、各々の医薬の単位用量は、以下:
(a)化合物が被験体に投与された場合の、被験体の血漿中で約20ng/mL〜4000ng/mLの化合物のCmaxもしくは被験体の血液中で約40ng/mL〜8000ng/mLの化合物のCmax、
(b)被験体への投与の24時間後の被験体の血漿中での約10ng/mL〜2,000ng/mLの化合物もしくは被験体への投与の24時間後の被験体の血液中での約20ng/mL〜4,000ng/mLの化合物、または
(c)化合物が被験体に投与された場合、被験体の血漿中での約500ng*h/mL〜60,000ng*h/mLの化合物もしくは被験体の血液中での約750ng*h/mL〜120,000ng*h/mLの化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である。
【0060】
癌処置のための医薬の調製における、式Iの化合物の使用のいくつかの実施形態では、各々の単位用量は以下:
(a)被験体の血漿中での約50ng/mL〜500ng/mLの化合物のCmax値もしくは被験体の血液中で100ng/mL〜1000ng/mLの化合物のCmax値
(b)投与の24時間後での被験体の血漿中の約20ng/mL〜1,000ng/mLの化合物もしくは投与の24時間後での被験体の血液中の約40ng/mL〜2,000ng/mLの化合物、または
(c)被験体の血漿中で約1,000ng*h/mL〜30,000ng*h/mLの化合物のAUC値もしくは被験体の血液中で約1,500ng*h/mL〜60,000ng*h/mLの化合物のAUC値、
のうちの少なくとも一つを提供するのに十分である。
【0061】
他の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜250ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜500ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ40〜500ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜1,000ng/mLの化合物、または
(c)被験体の血漿中の化合物のおよそ2,000〜15,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ3,000〜30,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0062】
さらに他の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ75〜150ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ150〜300ng/mLのCmax、もしくは
(b)投与24時間後の被験体の血漿中のおよそ40〜250ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜500ng/mLの化合物
のうちの少なくとも一つを提供するのに十分である。
【0063】
別の実施形態において、各単位用量は、被験体の血漿中の化合物のおよそ100〜2000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜4000ng/mLのCmax;または被験体の血漿中の化合物の100〜1000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜2000ng/mLのCmaxを提供するのに十分である。
【0064】
代表的に、本明細書中に記載される式Iの化合物の使用において、医薬の調製のために化合物の乳酸塩が使用される。このような医薬は、経口投与に適している。医薬の単位投薬形態としては、丸剤、カプセル剤、錠剤、ゲルキャップ剤、キャプレット剤、懸濁液もしくは水溶液が挙げられるが、これらに限定されない。さらに、医薬は、短時間のボーラス、ゆっくりした輸液、もしくは長時間の輸液のような注入による投与に適している。
【0065】
式I、式IIおよび式IIIの化合物には、本発明の方法のいずれかを記載する説明書が付随し得る。したがって、いくつかの実施形態においては、本発明は、癌の処置法もしくは式Iの化合物の代謝様式の分析法における使用説明書との組み合わせで、式I、式IIおよび/もしくは式IIIの少なくとも一つの化合物を提供する。
【0066】
本発明はさらに、癌を有する被験体に、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩を投与する工程を包含する、癌を処置するための方法を提供し、この処置法においては、第一の処置サイクルで投与される化合物の量は一日25mgであり、かつ一日化合物1500mgが被験体に投与されるまでもしくは被験体に用量制限毒性が観察されるまでのどちらかまで、投与される化合物の量が、その後に続く各処置サイクルにより増加される。代表的に、このような方法において、投与される化合物の量は、第一の処置サイクルの後に続く各処置サイクルにより倍増される。いくつかの実施形態において、処置サイクルは、一日につき同じ量の化合物を7日間投与し、その後7日間化合物を投与しない工程を包含する。
【0067】
同様に、本明細書において記載される医薬の調製における、式Iの化合物の使用のいくつかの実施形態において、医薬の各単位用量は、被験体の体重に基づいて0.25〜30mg/kgの化合物、互変異性体、および/または塩を含む。さらに、医薬の各単位用量は、25〜1500mgに及ぶ量の化合物、互変異性体、および/または塩を含み得る。医薬は、7日、14日,21日もしくは28日分の量の前記単位用量を含有するキットに配合され得、処置サイクルにおける使用に適したこのキットは、各7日,14日,21日もしくは28日間、一日分量の化合物を投与し、その後7日もしくは14日間化合物を投与しない工程を包含する。
【0068】
別の局面において、本発明は癌を処置する方法を提供し、この方法は、癌を有する被験体に十分量の式I
【0069】
【化23】
を有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能なその互変異性体の塩を投与する工程、および被験体を以下:
【0070】
【化24】
から選択される式IIおよび式IIIの化合物の一方もしくは両方に曝露する工程であって、それによって、式IIおよび式IIIの化合物の一方もしくは両方が、被験体による式Iの化合物の代謝により生成される工程を包含し、被験体の血漿中における、およそ20〜4000ng/mLに及ぶ式I、式IIおよび式IIIの化合物の一つ以上の複合Cmaxもしくは被験体の血液中における、およそ40〜8000ng/mLに及ぶ、式I、式IIおよび式IIIの化合物の一つ以上の複合Cmaxを提供する。
【0071】
さらに別の局面において、本発明は、癌を有する被験体を、十分量の一つ以上の以下:
【0072】
【化25】
から選択される式を有する化合物、活性なその代謝物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異異性体の塩であって、被験体の血漿中の一つ以上の化合物のおよそ20〜4000ng/mLの複合Cmaxもしくは被験体の血液中の一つ以上の化合物のおよそ40〜8000ng/mLの複合Cmaxを提供するのに十分である化合物に曝露する工程を包含する、癌を処置するための方法を提供する。いくつかの実施形態において、一つ以上の化合物は、被験体の血漿中のおよそ35〜2600ng/mLの一つの化合物のCmaxもしくは被験体の血液中のおよそ35〜6000ng/mLの一つの化合物のCmaxを提供する。他の実施形態において、一つ以上の化合物の量は、被験体の血漿中のおよそ35〜1200ng/mLの一つの化合物のCmaxもしくは被験体の血液中のおよそ50〜2400ng/mLの一つの化合物のCmaxを提供する。いくつかの実施形態において、式:
【0073】
【化26】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。別の実施形態において、式:
【0074】
【化27】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。さらに別の実施形態において、式:
【0075】
【化28】
の化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が、被験体に投与される。
【0076】
特定の疾患に対する式Iの化合物の安全性および/もしくは効力の決定において、重要なことは、この化合物の投与後、被験体におけるこの化合物の薬物動態および薬理作用をモニタリング可能であることである。そこで、本発明の一つの局面にしたがって、被験体における式Iの化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の代謝様式を決定するための提供される方法が存在する。この方法は、被験体から得られる尿、血液、もしくは組織の一つ以上のサンプルにおいて、この化合物の少なくとも一つの代謝物の量を測定する工程を包含する。この方法により測定され得る代謝物としては、式II:
【0077】
【化29】
を有するN−オキシド化合物であって、4−アミノ−5−フルオロ−3−[6−(4−メチル−4−オキシドピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オンとしてもまた公知の化合物、および式III:
【0078】
【化30】
を有するN−デスメチル化合物であって、4−アミノ−5−フルオロ−3−[6−(ピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オンとしてもまた公知の化合物が挙げられる。式Iの化合物の代謝様式を決定するためのいくつかのこのような方法において、これらの方法は、式IIの代謝物および式IIIの代謝物の量を測定する工程を包含する。代謝物の量は、紫外(UV)分光学もしくは液体クロマトグラフィー−質量分析(LC−MS)を含む当業者に周知の技術を用いて測定され得る。
【0079】
本発明の別の局面において、被験体における式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の量を決定する方法が提供される。この方法は、化合物が被験体に投与された後に被験体から得られる尿、血液、もしくは組織のサンプルにおける化合物の量を測定する工程を包含する。この方法は、サンプル中の化合物の代謝物の量を測定する工程をさらに包含し得る。測定され得る代謝物としては、式IIのN−オキシド化合物および/もしくは式IIIのN−デスメチル化合物が挙げられるが、これらに限定されない。いくつかの実施形態において、この方法は、式Iの化合物が被験体に投与された後の異なる時点に、被験体から二つ以上のサンプルを回収する工程をさらに包含する。
【0080】
別の局面において、癌を処置する方法における使用のための式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩が提供され、この方法は、以下
(a)化合物が被験体に投与される場合における、被験体の血漿中の化合物のおよそ20〜4000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ40〜8000ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ10〜2,000ng/mLの化合物もしくは被験体への投与24時間後の被験体の血液中のおよそ20〜4,000ng/mLの化合物、または
(c)化合物が被験体に投与される場合における、被験体の血漿中の化合物のおよそ500〜60,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ750〜120,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分な量において、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩を被験体に投与する工程を包含する。
【0081】
癌を処置する方法における使用のためのこの化合物のいくつかの実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜500ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜1000ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ20〜1,000ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ40〜2,000ng/mLの化合物、または
(c)被験体の血漿中のこの化合物のおよそ1,000〜30,000ng×h/mLのAUCもしくは被験体の血液中のこの化合物のおよそ1,500〜60,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0082】
癌を処置する方法における使用のためのこの化合物の別の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ50〜250ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ100〜500ng/mLのCmax、
(b)投与24時間後の被験体の血漿中のおよそ40〜500ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜1,000ng/mLの化合物、または
(c)被験体の血漿中の化合物のおよそ2,000〜15,000ng×h/mLのAUCもしくは被験体の血液中の化合物のおよそ3,000〜30,000ng×h/mLのAUC
のうちの少なくとも一つを提供するのに十分である。
【0083】
癌を処置する方法における使用のためのこの化合物のさらに別の実施形態において、各単位用量は、以下
(a)被験体の血漿中の化合物のおよそ75〜150ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ150〜300ng/mLのCmax、または
(b)投与24時間後の被験体の血漿中のおよそ40〜250ng/mLの化合物もしくは投与24時間後の被験体の血液中のおよそ80〜500ng/mLの化合物
のうちの少なくとも一つを提供するのに十分である。
なお別の実施形態において、各単位用量は、被験体の血漿中の化合物のおよそ100〜2000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜4000ng/mLのCmaxを提供するのに十分である;または各単位用量は、被験体の血漿中の化合物のおよそ100〜1000ng/mLのCmaxもしくは被験体の血液中の化合物のおよそ200〜2000ng/mLのCmaxを提供するのに十分である。
【0084】
被験体における、式Iを有する化合物、薬学的に受容可能なその塩、その互変異性体、もしくは薬学的に受容可能な互変異性体の塩の代謝様式の決定のための代謝物の使用がさらに提供され、この代謝物は、式:
【0085】
【化31】
のN−オキシド化合物もしくは式:
【0086】
【化32】
のN−デスメチル化合物のうちの少なくとも一つを含む。
【0087】
本発明はまた、一つ以上の本発明の化合物、もしくは薬学的に受容可能なその塩もしくはその互変異性体を、薬学的に受容可能なキャリアー、賦形剤、結合剤、希釈剤などと混合することにより調製され得る種々の疾患を処置もしくは改善するための組成物を提供する。このような疾患の例としては、前立腺癌、結腸直腸癌、乳癌、多発性骨髄腫、膵臓癌、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、骨髄増殖性疾患、非小細胞肺癌、小細胞肺癌、慢性リンパ球白血病、肉腫、黒色腫、リンパ腫、甲状腺癌、神経内分泌癌、腎細胞癌、胃癌、消化管間質癌、神経膠腫、脳癌、または膀胱癌を含む癌が挙げられるが、これらに限定されない。処置上有効な用量は、結果としてこの疾患の症状の改善をもたらすのに十分な本発明の一つ以上の化合物の量に、さらに言及する。本発明の薬学的組成物は、とりわけ、通常の顆粒化、混合、溶解、封入、凍結乾燥、乳化もしくは微粒子化プロセスのような当該分野で周知の方法により製造され得る。この組成物は、例えば、顆粒剤、粉剤、錠剤、カプセル剤、シロップ剤、坐剤、注射剤、乳化剤、エリキシル剤、懸濁剤もしくは液剤のような形状を取り得る。本発明の組成物は、例えば、経口投与、鼻腔内投与、経粘膜投与、直腸投与、または皮下投与および鞘内、静脈内、筋肉内、腹腔内、鼻腔内、眼内もしくは心室内注射の様な種々の投与経路に処方され得る。本発明の化合物もしくは化合物はまた、全身型よりむしろ、徐放性処方物としての注射のような局所型において投与され得る。以下の投薬形態は、例として与えられ、かつ本発明を限定するものとして解釈されるべきではない。
【0088】
経口、頬側および舌下投与において、粉剤、懸濁剤、顆粒剤、錠剤、丸剤、カプセル剤、ゲルキャップ剤、およびキャプレット剤が固体の投薬形態として受容可能である。これらは、例えば、一つ以上の本発明の化合物、もしくは薬学的に受容可能な塩もしくはその互変異性体を、添加剤またはデンプンもしくは他の添加剤のような賦形剤のうちの少なくとも一つと混合することにより調製され得る。適切な添加剤もしくは賦形剤は、フルクトース、スクロース、ラクトース、セルロース糖、マンニトール、マルチトール、デキストラン、ソルビトール、デンプン、寒天、アルギン酸塩、キチン、キトサン、ペクチン、トラガカントゴム、アラビンゴム、ゼラチン、コラーゲン、カゼイン、アルブミン、合成もしくは半合成ポリマーまたはグリセリド、メチルセルロース、ヒドロキシプロピルメチル‐セルロース、および/またはポリビニルピロリドンである。必要に応じて、経口投薬形態は、不活性な賦形剤、またはステアリン酸マグネシウムのような滑沢剤、またはパラベンもしくはソルビン酸のような防腐剤、またはアスコルビン酸、トコフェロールもしくはシステインのような酸化防止剤、崩壊剤、結合剤、濃化剤、緩衝剤、甘味剤、風味剤または芳香剤のような、投与において補助となる他の成分を含み得る。さらに、識別のために染料もしくは色素が添加され得る。錠剤および丸剤は、当該分野において公知の適切な塗膜物質によりさらに処理され得る。
【0089】
経口投与のための液体投薬形態は、薬学的に受容可能な乳化剤、シロップ剤、エリキシル剤、懸濁剤、スラリー剤および液剤の形態を取り得、これらは水のような不活性な賦形剤を含み得る。薬学的処方物は、油、水、アルコール、およびこれらの組み合わせなどであるがこれらに限定されない滅菌の液体を使用して、液体懸濁剤もしくは液剤として調製され得る。薬学的に適切な界面活性剤、懸濁化剤、乳化剤は、経口投与もしくは非経口投与のために添加され得る。
【0090】
上記のように、懸濁剤は、油を含み得る。このような油としては、ピーナッツ油、ゴマ油、綿実油、トウモロコシ油およびオリーブ油が挙げられるが、これらに限定されない。懸濁調製液はまた、オレイン酸エチル、ミリスチン酸イソプロピル、脂肪酸グリセリドおよびアセチル化脂肪酸グリセリドのような脂肪酸のエステルを含み得る。懸濁剤処方物は、エタノール、イソプロピルアルコール、ヘキサデシルアルコール、グリセロールおよびプロピレングリコールなどである、これらに限定されないアルコールを含み得る。ポリ(エチレングリコール)のような、しかしこれに限定されないエーテル、鉱油およびペトロラタムのような石油炭化水素;および水はまた、懸濁剤処方物において使用され得る。
【0091】
鼻腔内投与(例えば、化合物を脳に送達するため)、もしくは吸入投与(例えば、肺を介して化合物を送達するため)の場合、薬学的処方物は、液剤、噴霧剤、ドライパウダー、もしくはあらゆる適切な溶媒および安定化剤、殺菌物剤、酸化防止剤、pH改変剤、界面活性剤、バイオアベイラビリティ改変剤およびこれらの組み合わせのような、しかしこれらに限定されない任意の別の化合物を含むエアロゾルであり得る。鼻腔内処方物および鼻腔内投与方法の例は、WO01/41782、WO00/33813,WO91/97947、米国特許第6,180,603号および米国特許第5,624,898号に見出され得る。エアロゾル処方の推進剤としては、圧縮された空気、窒素、二酸化炭素、もしくは炭化水素ベースの低沸点溶媒が挙げられ得る。本発明の化合物は、好都合には、エアロゾル噴霧提供の形態で噴霧器などから輸送される。
【0092】
注射投薬形態は、一般に、適切な分散剤もしくは加湿剤および懸濁剤を用いて調製され得る水性懸濁液もしくは油性懸濁液を含み得る。注射可能形態は、溶液相または溶媒もしくは希釈剤により調製される懸濁形態を取り得る。受容可能な溶媒もしくはビヒクルとしては、滅菌水、リンガー溶液、もしくは等張生理食塩水溶液が挙げられる。代替的に、溶媒もしくは懸濁剤として滅菌油が採択され得る。好ましくは、この油もしくは脂肪酸としては、揮発性でない、天然もしくは合成の油、脂肪酸、モノ−グリセリド、ジ−グリセリドもしくはトリ−グリセリドが挙げられる。
【0093】
注射の場合、薬学的処方物は、上記のような適切な溶液による再形成のために適切な粉末であり得る。これらの例としては、凍結乾燥、回転乾燥もしくは噴霧乾燥された粉末、不定形粉末、顆粒、沈殿物、もしくは微粒子があげられるが、これらに限定されない。注射の場合、処方物は、安定化剤、pH改変剤、界面活性剤、バイオアベイラビリティ改変剤およびこれらの組み合わせを任意に含み得る。これらの化合物は、ボーラス注射もしくは連続的な輸液のような注射による、非経口投与のために処方され得る。注射のための単位用量形態は、アンプルもしくはマルチ用量容器を取り得る。
【0094】
このように、本明細書中に記載される癌を処置するための本方法において、化合物は、短時間のボーラス、ゆっくりした輸液、もしくは長時間の輸液として注射によって投与され得る。注射は、一日に一回、二回、三回、もしくは四回投与され得る。短時間のボーラスは、通常、およそ1〜30分間にわたって投与される;ゆっくりとした輸液は、通常、30分〜6時間にわたって投与される;そして長時間の輸液は、通常、6時間〜およそ7日までにわたって投与される。
【0095】
直腸投与の場合、薬学的処方物は、腸、S状結腸および/もしくは直腸における化合物の放出のため、坐剤、軟膏、浣腸、錠剤もしくはクリームの形態を取り得る。直腸坐剤は本発明の化合物、薬学的に受容可能な塩もしくはこの化合物の互変異性体のうちの一つ以上を、受容可能な賦形剤、例えば、ココアバターもしくはポリエチレングリコールと共に混合することにより調製され、これらは、通常の保管温度においては固相で存在し、かつ直腸内のような体内における薬物の放出に適切な温度においては液相で存在する。油はまた、軟ゼラチン型および坐剤の処方の調製において採択され得る。水、生理食塩水、ブドウ糖水溶液および関連する糖溶液、ならびにグリセロールは、ペクチン、カルボマー、メチルセルロース、ヒドロキシプロピルセルロースもしくはカルボキシメチルセルロースのような懸濁剤、ならびに緩衝剤および防腐剤をも含み得る懸濁処方物の調製において採択され得る。
【0096】
それらの上記の代表的な投薬形態に加えて、薬学的に受容可能な賦形剤およびキャリアーは、通常、当業者に公知であり、したがって本発明に含まれる。このような賦形剤およびキャリアーは、例えば、本明細書中に参考として援用される「Remingtons Pharmaceutical Sciences」Mark Pub.Co.,New
Jersey(1991)に記載される。
【0097】
本発明の処方物は、以下に記載する、短時間の作用、迅速な放出、長時間の作用、および持続的な放出のために設計され得る。このように、薬学的処方物はまた、制御された放出もしくはゆっくりとした放出のために処方され得る。
【0098】
本組成物はまた、例えば、ミセルもしくはリポソーム、または何らかの別の被包性の形態を含み得、あるいは持続的な蓄積および/もしくは送達効果を提供するために、長期間の放出形態において投与され得る。したがって、薬学的処方物は、ペレット剤もしくはシリンダーに圧縮され得、そして筋肉内もしくは皮下に、蓄積注射もしくはステントのような移植片として移植され得る。このような移植物は、シリコーンおよび生分解性ポリマーのような公知の不活性な物質を採択し得る。
【0099】
処置上有効な用量は、結果として症状の改善をもたらす化合物の量をいう。特定の投薬量は、疾患の状態、被験体の年齢、体重、通常の健康状態、性別、食事、用量間隔、投与経路、排出率、および薬物の組み合わせに基づいて調整され得る。有効量を含む上記のいずれの投薬形態も、慣用的な実験の境界内に十分含まれ、そしてそれゆえ、本発明の範囲内に十分含まれる。処置上有効な用量は、投薬経路および投薬形態に依存して変化し得る。本発明の好ましい化合物は、高い処置指数を示す処方物である。処置指数は、LD50とED50との比として表され得る、毒性効果と処置効果との間の用量比である。LD50は、母集団の50%に致命的な量であり、そしてED50は、母集団の50%に処置上有効な量である。LD50およびED50は、動物細胞培養物もしくは実験動物における標準的な薬学的手順により決定される。
【0100】
本発明はまた、ヒトもしくは非ヒト動物において抗癌活性を促進する方法を提供する。この方法は、有効量の本発明の化合物、もしくは組成物を、前記哺乳動物もしくは非ヒト動物へ投与する工程を包含する。本発明の化合物の有効量は、RTKを阻害する量を含み、これは、例えば、本明細書中に記載のアッセイ、もしくは当業者に公知のシグナル伝達を検出する他のあらゆるアッセイにより、例えば、コントロールモデルと比較したcAMP上昇レベルの測定による、G−タンパク質共役受容体の活性化を介する生物化学的経路において、検出可能である。有効量はまた、RTKの阻害により処置可能なRTK疾患の症状を緩和する量を含み得る。
【0101】
提供されるこれらの方法により処置され得るRTK疾患、もしくはRTKが媒介する疾患は、RTKが関与するか、またはRTKの阻害およびRTKが、疾患もしくは疾患の状態において欠損している生物化学的経路を増強する任意の生物学的疾患もしくは疾患を含む。このような疾患の例は、前立腺癌、大腸癌、乳癌、多発性骨髄腫、膵臓、小細胞癌、急性骨髄性白血病、慢性骨髄性白血病、もしくは骨髄増殖性疾患のような癌である。
【0102】
化合物Iの合成は、米国特許第6,605,617号において開示されている。化合物2および3の同一性の確認のために、化合物Iの代謝物、化合物IIおよびIIIは、実施例6において示されるように、独立して合成された。
【0103】
本発明は、このように一般的に記載される、以下の実施例の参照によってより容易に理解され、以下の実施例は実例として提供され、そして本発明を限定するものとして意図されない。
【実施例】
【0104】
以下の略語および用語が、実施例において使用される:
ATP: アデノシン三リン酸
AUC: 曲線下面積
BSA: ウシ胎児アルブミン
DMSO: ジメチルスルホキシド
EDTA: エチレンジアミン四酢酸
ERK: 細胞外制御キナーゼ
Hepes: N−(2−ヒドロキシエチル)ピペラジン−N’−(2−エタンスルホン酸)
HPLC: 高速液体クロマトグラフィー
HMVEC: ヒト微小管内皮細胞
kg: キログラム
LC: 液体クロマトグラフィー
MAPK: マイトジェン活性化プロテインキナーゼ
MS: 質量分析
MeOH: メタノール
mg: ミリグラム
mL: ミリリットル
MTS: [3−(4,5−ジメチル−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム]、分子内塩
nM: ナノモーラー
PBS: リン酸緩衝化生理食塩水
NMR: 核磁気共鳴分析
RT−PCR: 逆転写酵素ポリメラーゼ連鎖反応
SCF: 幹細胞因子
TFA: トリフルオロ酢酸
T1/2: 半減期‐生物学的系から化合物の50%が排出されるのに必要な時間
μg: マイクログラム
μL: マイクロリットル
μM: マイクロモーラー
UV: 紫外線分光法
(実施例1)
多数の癌細胞株および初代非悪性細胞株に対して、4−アミノ−5−フルオロ−3−[6−(4−メチルピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)−オン(化合物1)の抗増殖活性を試験した。方法は、以下のように行った:細胞を96−ウェルプレートにプレートした;接着細胞株のための3〜5時間のゲル化時間の後、化合物の希釈溶液(delusioin)を添加し、3日後、MTS溶液(Promega)の添加により生細胞を決定した。490nmで吸光度を測定し、非直線回帰を用いてEC50値を算出した。HMVECアッセイのために、3日間、5ng/mLの組換え型VEGFの存在下、細胞と共に化合物をインキュベートした。SCF/c−KITアッセイのために、3日間、それぞれ40ng/mLおよび100ng/mLの組換え型SCF存在下、TF−1細胞およびH526細胞をインキュベートした。MTS溶液の添加および490nmでの吸光度の測定により、増殖をアッセイした。非直線回帰により、EC50を算出した。結果を表1に示す。
【0105】
癌細胞株および内皮細胞の部分集合において、KM12L4a細胞株を除いて、EC50が50nM以下で増殖が阻害され、これは化合物1により標的とされるRTKへのそれらの依存と矛盾しない(MV4;11:構成的に活性なFLT3の発現;HMVEC:VEGFR2に媒介される増殖;TF−1:c−KITに媒介される増殖)。この細胞株が、いくつかの標的とされるRTK(例えば、RT−PCRにより決定されるVEGF1/2およびPDGFR)を発現する場合においても、実験は、これらの個々のRTKの阻害が、化合物1に観察される強力な抗増殖効果を完全には説明しないことを示した。この発見は、複数のRTKの阻害もしくは今は未同定な効果のどちらかが、この細胞株において化合物1により媒介される抗増殖効果に関与し得ることを示唆する。
【0106】
1μMと10μMとの間のEC50を有する化合物1と共にインキュベートされる場合、二つの初代細胞株、HMEC(ヒト正常乳房上皮細胞)およびPrEC(正常ヒト前立腺上皮細胞)を含む大多数の細胞株は、抗増殖応答を示した。インビトロにおける結果と一致して、インビボにおいて、化合物1がマウスにおけるKM12L4a異種移植片とMV4;11異種移植片との両方の生育を効果的に阻害した。
【0107】
【表1】
(実施例2)
2週間の毒性研究からのプールされたラットの血漿において、化合物1の二つの代謝物が同定され、そして部分的に特徴付けられた。一日一回、30mg/kgもしくは80mg/kg、経口投与の投与群から、UVおよびLC/MSにより、1日目および14日目の投与された動物の血漿が分析された。二つの同定された代謝物は、式IIのピペラジンN−オキシド化合物(化合物2)および式IIIのN−脱メチル化合物(化合物3)であった(これらの化合物の合成および特徴付けについては、実施例6を参照)。(UV吸光度に基づき、かつ以前の分析からの同じサンプルにおいて定量された既知のレベルの化合物1に比較して)見積もられた代謝物のレベルを、表2に示す。投与後プールされた血漿の全てのサンプルにおいて、N−デスメチル代謝物が化合物1よりもかなり低い濃度であることが見出された。80mg/kgの投与群における14日目の24時間および30mg/kgの投与群における1日目の1〜2時間を除いて、N−オキシド代謝物が化合物1よりも低い濃度で存在することが観察された(表2)。代謝様式は、投与量および投与の継続によって変化しない。通常、代謝物のレベルは化合物1のレベルの投与量の増加に伴って上昇する。
【0108】
両方の投与群で、1日目と14日目を比較して、投与の継続は、代謝物のみの血漿レベルにおいて(表2)もしくは化合物1のレベルと比較して、結果として上昇を生じないことが明らかである。化合物1のレベルは、投与の継続とともに減少し、これは代謝物レベルの減少によっても反映される。このことは、化合物1の曝露における時間依存的な減少は、上昇する代謝物に反映されないことを示唆する。14日目の24時間のサンプルは、1日目の24時間のサンプルよりも低いレベルの化合物1および代謝物を含み、このことは、30mg/kgもしくは80mg/kgの一日一回の投与規則によっては代謝物および化合物1は蓄積されないことを示す。80mg/kg投与群の全てのアッセイされた時点において、そして30mg/kg投与群の1日目、24時間を除く全ての時点において、N−オキシド代謝物はN−デスメチル代謝物よりも高い濃度で存在している。N−デスメチル代謝物レベルは、化合物1のレベルよりもゆっくりと低下することが明らかであり、このことは、より長いT1/2を示唆し、そしてこの代謝物の血漿レベルが、N−オキシドが対照的にそうあり得るようにその形成速度ではなく、その排出率によって決定され得ることを示す。
【0109】
【表2】
(実施例3)
レセプターチロシンキナーゼのためのインビトロキナーゼアッセイ
多くのタンパク質チロシンキナーゼのキナーゼ活性を、ATPおよびリン酸化のためのチロシンアミノ酸残基を含む適切なペプチドもしくはタンパク質を提供すること、そしてチロシン残基へのリン酸エステル分子の転移を分析することにより測定した。FLT−1(VEGFR1)、VEGFR2、VEGFR3、Tie−2、PDGFRα、PDGFRβ、およびFGFR1レセプターの細胞質ドメインに対応する組換え型タンパク質を、バキュロウィルス発現系(InVitrogen)を用いてSf9昆虫細胞において発現させ、そしてGlu抗体相互作用(Glu−エピトープ標識化組換え体について)を介してもしくは金属イオンクロマトグラフィー(His6(配列番号1)標識化組換え体について)によって精製し得る。各アッセイのために、試験化合物を順にDMSOに希釈し、そして次にATPを加えた適切なキナーゼ反応緩衝液と混合した。キナーゼタンパク質および適切なビオチン化ペプチド基質を添加し、最終容量を50〜100μLとし、反応液を1〜3時間、室温でインキュベートし、そして次に45mMのEDTA、50mMのHepes pH7.5を含有する25〜50μLを添加することにより反応を停止した。停止された反応混合液(75μL)を、ストレプトアビジン塗膜マイクロタイタープレート(Boehringer Mannheim)に移し、1時間インキュベートした。リン酸化されたペプチド生成物を、ユーロピウム標識された抗リン酸化チロシン抗体PT66を用いて、DELFIA時間分解蛍光システム(WallacまたはPE Biosciences)により測定し、抗体の希釈のためにDELFIAアッセイ緩衝液に1mMのMgCl2を補充するという改変を施した。Wallac 1232 DELFIA蛍光計およびPE Victor II多重信号読み取り機において時間分解蛍光を読み取った。XL Fitデータ分析ソフトウェアを用い、非直線回帰を採用して50%阻害のための各化合物の濃度(IC50)を算出した。
【0110】
FLT−1、VEGFR2、VEGFR3、FGFR3、Tie−2、およびFGFR1キナーゼを、50mMのHepes pH7.0、2mMのMgCl2、10mMのMnCl2、1mMのNaF、1mMのDTT、1mg/mLのBSA、2μMのATP、および0.20〜0.50μMの対応するビオチン化ペプチド基質中において分析した。FLT−1、VEGFR2、VEGFR3、Tie−2、およびFGFR1キナーゼを、それぞれ0.1μg/mL、0.05μg/mL、もしくは0.1μg/mL添加した。PDGFRキナーゼアッセイのために、ATP濃度およびペプチド基質濃度を1.4μMのATP、および0.25μMのビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2)ペプチド基質に変換すること以外は上記と同じ緩衝液条件で、120μg/mLの酵素を使用した。FLT−1、VEGFR2、VEGFR3、FGFRに関しては、上記の化合物の各々が10μMより低いIC50値を示した。
【0111】
組換え型かつ活性なチロシンキナーゼFyn、およびLckは商業上入手可能であり、そしてこれらをUpstate Biotechnologyから購入した。各アッセイのために、試験化合物を順にDMSOに希釈し、そして次に10nMの33P γ−標識化ATPを加えた適切なキナーゼ反応緩衝液と混合した。キナーゼタンパク質および適切なビオチン化ペプチド基質を添加し、最終容量を150μLとした。反応液を、3〜4時間、室温でインキュベートし、そして次に100mMのEDTAおよび50μMの非標識化ATPを含有する100μLの反応停止緩衝液を含む、ストレプトアビジン塗膜ホワイトマイクロタイタープレート(Thermo Labsystems)に移すことにより反応を停止させた。1時間のインキュベート後、ストレプトアビジンプレートをPBSで洗浄し、そして各ウェルにつき200μLのMicroscint 20シンチレーション流体を添加した。このプレートを密閉し、そしてTopCountを用いて計数した。XL Fitデータ分析ソフトウェアを用い、非直線回帰を採用して50%阻害のための各化合物の濃度(IC50)を算出した。
【0112】
Fyn、Lckおよびc−ABLのためのキナーゼ反応緩衝液は、50mM Tris−HCl pH7.5、15mM MgCl2、30mM MnCl2、 2mM DTT、2mM EDTA、25mM β―グリセロールリン酸塩、0.01% BSA/PBS、0.5μMの適当なペプチドの基質(ビオチン標識されたSrcのペプチドの基質:FynおよびLckについてはビオチン―GGGGKVEKIGEGTYGVVYK−NH2(配列番号3))、1μM 非標識ATPおよび1nMキナーゼを含んだ。
【0113】
c−KitおよびFLT−3のキナーゼ活性を、ATP、およびリン酸化のためのチロシンアミノ酸残基を含むペプチドまたはタンパク質を提供、およびチロシン残基に対してリン酸基の転移のためにアッセイすることにより、計測される。c−KitおよびFLT−3受容体の細胞質ドメインに対応した組み換えタンパク質は、購入した(Proquinase)。テストのために、模範的な化合物、例えば、4−アミノ−5−フルオロ−3−[5−(4−メチルピペラジン−1−イル)−1H−ベンゾイミダゾール−2−イル]キノリン−2(1H)―オンは、DMSOで希釈し、それから下記に説明されたキナーゼ反応緩衝液およびATPと混合した。キナーゼタンパク質、c−KitまたはFLT−3およびビオチン化されたペプチドの基質(ビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2)は、100μLの最終容量を提供するよう添加した。これらの反応を、室温で2時間インキュベートし、次いで50μLの45mM EDTAおよび50mM HEPES、pH7.5を添加することにより停止した。停止された反応混合液(75μL)を、ストレプトアビジン被覆のマイクロタイタープレート(Boehringer Mannheim)に移行され、1時間インキュベートした。リン酸化されたペプチド産物を、DELPHIA経時分離時間分解蛍光システム(WallacまたはPE Biosciences)を使用し、ユーロピウム標識された抗ホスホチロシン抗体PT66を使用して、抗体希釈のためにDELFIAアッセイ緩衝液に1mM MgCl2を追加したという変更を伴って計測した。時間分解蛍光値を、Wallac 1232 DELFIA 蛍光計測器またはPE Victor II多重シグナル解読器で決定した。50%の阻害(IC50)に対するそれぞれの化合物の濃度IC50を、XL Fitデータ解析ソフトウェアを用いる非線形回帰を使用し計算した。
【0114】
FLT−3およびc−Kitキナーゼを、50mM Hepes pH7.5、1mM
NaF、2mM MgCl2、10mM MnCl2、および1mg/mL BSA、8μM ATPおよび1μMの対応するビオチン化されたペプチド基質(ビオチン−GGLFDDPSYVNVQNL−NH2(配列番号2))においてアッセイした。FLT−3およびc−Kitキナーゼの濃度を、2nMでアッセイした。
IC50を、化合物1の代謝物において計測し、比較のために化合物1のIC50を一緒に表3に示す。
【0115】
【表3】
(実施例4)
この単物質の研究は、KM12L4aのヒト結腸の腫瘍モデルにおいて、化合物1の毎日の経口投薬を評価した。
【0116】
生後7、8週間の(Charles River)雌のNu/Nuマウスに、右側腹部において、2×106のKM12L4a 細胞を皮下に移植した。処置は、腫瘍の容量の平均が125mm3であるときより7日後に開始した。これを、一日目の研究と名付けた。化合物1を、10mM H3PO4で溶液として処方し、そして経口栄養補給によって投与した。
【0117】
7つの処置群を、研究に含めた、(n=10/群):ビヒクル(水)p.o.、q.d.;および化合物1に含めた投薬の6群:3mg/kg、10mg/kg、30mg/kg、100mg/kg、200mg/kg、300mg/kgのp.o.、q.d.。
【0118】
血漿サンプルを、様々な日数におけるそれぞれの投薬群において、腫瘍保有マウスにおいて化合物1の薬物動態を特徴付けるために衛星動物(satellite animal)から引き出した(N=2/時点/投薬群)。化合物1の組織濃度および腫瘍濃度を、22日目の8時間および24時間の投薬後に100mg/kgおよび200mg/kgの投薬群において、動物から収集されたサンプルにおいて決定した(N=2/時点/投薬群)。
【0119】
血漿化合物1の濃度を、1ng/mLから8000ng/mLの較正の範囲および1ng/mLの定量限界(LLOQ)の検証されていないLC/MS/MSアッセイによって決定した(Charles River Laboratories、Worcester、MA)。組織および腫瘍の化合物1の濃度もまた、20ng/gから43740ng/gの較正の範囲および20ng/gのLLOQの検証されていないLC/MS/MSアッセイを使用して決定した。
【0120】
複雑な薬物動態パラメーター(CmaxおよびAUC値)を、それぞれのサンプリング日におけるそれぞれの投薬群について、血漿化合物の濃度―時間のデータの平均から、標準的な非コンパートメント分析を使用して得た(WinNonlin Professional、version4)。報告したAUC値を、3つの濃度―時間のデータ点を使用して決定した。投薬前の濃度値を、投薬の直前に対して直接に観察された濃度値のとして報告した。
【0121】
腫瘍の成長において有意な用量依存性の阻害を、4日〜7日の処置による全ての投薬で観察した(表4参照)。計算したED50は、17mg/kgであった。初期サイズの50%より大きい腫瘍の退化が、200mg/kgおよび300mg/kgにおいて、化合物1を投薬された大部分のマウスにおいて観察された。しかし、これらの投薬は、全体の研究期間において、耐性を示されなかった。12日〜16日の間まで、300mg/kgで処置したマウスは、体重の20%〜30%を失い、安楽死させた。200mg/kgで処置されたマウスにおいて、10匹の内1匹は、22%体重減少したので14日目に安楽死させ、そして残存したマウスは、25%より大きく体重減少したので21日〜24日目に安楽死させた。マウスを、100mg/kgで37日間投薬し、最初の体重の98%に維持した;この投薬において、腫瘍は安定なままであった(図1)。ビヒクル群を、9日で屠殺し、そして腫瘍成長の阻害(TGI)を計算した(表4)。
【0122】
【表4】
投薬2日目(2日目)において、化合物1の血漿濃度は、全ての投薬群において用量に対し比例的に増加した(表5)。少なくとも2週間の間での多重の投薬に続いて、血漿濃度は2日目の投薬に匹敵し得た。これはマウスおいて日に一度の投薬における蓄積が無いことを示唆する(表5)。同様に、3日目、8日目および15日目に収集された化合物1の投薬前血漿濃度は、それぞれの投薬群と類似しており、これは、2日目以降に定常状態に達したことを示唆する。以上のことから、これらのデータは、化合物1が、腫瘍保有マウスにおいて、投薬および時間に非依存性の薬物動態に従うことを示唆する。
【0123】
35%〜60%の腫瘍成長阻害が、10mg/kgおよび30mg/kgの用量においてそれぞれ観察された。対応する化合物1の血漿曝露は、CmaxおよびAUC値によって評価された場合に、それぞれ163ng/mL〜742ng/mLおよび1420ng×hr/mL〜5540ng×hr/mLの範囲であった。(図2)。対応する血漿投薬前濃度値は、2ng/mL〜135ng/mLの範囲であった。
【0124】
【表5】
それぞれ2つのサンプリング時点(投薬後8時間および24時間)で100mg/kgおよび200mg/kgの投薬群において、22日目における化合物1の組織中の濃度は、化合物1の血漿中の濃度より高かった(表6)。8時間または24時間の投薬後における脳または心臓中の化合物1の濃度は、これらの二つの投薬群において、血漿の濃度よりも13倍から34倍高かった;一方で肝臓、肺および腎臓の濃度は、これらの二つの投薬群において投薬後で8時間または24時間の血漿濃度より40倍から126倍高かった。一般的に、8時間における組織濃度の血漿濃度に対する割合は、24時間でのそれに匹敵した。さらに、24時間における組織濃度は、8時間におけるそれと比較して一貫して低かった。総合すれば、これらの結果は、化合物1の組織濃度が、血漿におけるそれと平行して減少するようであったことを示唆する。それゆえに、化合物1は、血漿に関連した組織(脳を含む)に広範に分布されるようであるが、多重の経口投薬に続いて、組織においては蓄積されないようである。
【0125】
【表6】
それぞれの2つのサンプリング時点(投薬後8時間および24時間)において100mg/kgおよび200mg/kgの投薬群において、22日目における化合物1の腫瘍中の濃度は、血漿中の濃度よりも37倍から354倍高かった。しかし、24時間における腫瘍の濃度は、これらの2つの投薬群においての投薬後8時間の濃度よりも17%〜65%だけ低くなっていた。これは、他の正常な組織(脳、心臓、肝臓、肺、および腎臓のような)からの排出速度と比較して、腫瘍からのいくらかより低い排出速度を示唆する。それゆえに、化合物1は、血漿と比較して腫瘍に広範に分布しているようである。しかし、血漿または正常の組織と比較して腫瘍において優先的な保持を示し得る。
【0126】
要約すると、化合物1の効力および耐性は、処置の4日〜7日後の有意な阻害を伴って、その用量に関係した。腫瘍抑制は、300mg/kgおよび200mg/kgで観察された;これらの用量では、およそ14日間および21日間においては、それぞれ耐性を示した。体重減少は、毒性に関連する臨床徴候であった。100mg/kgの投薬は、有害な臨床徴候なしに、コントロールと比較して80%の腫瘍成長阻害を伴い、37日間の間、耐性があった。30mg/kgは成長を60%阻害した。化合物1は、腫瘍保有マウスにおいて、用量依存的かつ時間依存的な薬物動態を実証した。35%〜60%の腫瘍成長阻害に関連した血漿中の化合物1のCmax値、AUC値およびCmin値は、それぞれ163ng/mL〜742ng/mL、1420ng×hr/mL〜5540ng×hr/mLおよび2ng/mL〜135ng/mLの範囲であった。化合物1は、組織に対し広範に分布したが、複数の経口投薬後では組織に蓄積しないようであった。経口の投薬後に、他の組織と比較して、腫瘍において、化合物1を優先的に保持する傾向があった。
(実施例5)
今回の一つの物質の研究では、PC3ヒト前立腺腫瘍モデルにおいて化合物1の断続的な経口投薬を評価した。
【0127】
9〜10週齢の雄のSCIDマウスに、5,000,000個のPC3ヒト前立腺細胞を皮下の腹部に移植した。腫瘍が150mm3に達したとき、処置を開始した。これを研究一日目と名付けた。化合物1を水性の溶液として処方し、そして経口栄養補給によって投与した。
【0128】
5つの処置群を研究に含めた、(n=10/群):ビヒクル(水)p.o.、q.d.;および100mg/kg;q.d.、q.2.d.、q.3.d.、q.4.d.の化合物1投薬の4群。
【0129】
表7に示すように、有意でありかつ類似した腫瘍阻害の結果が、全ての処置群で観察された。研究を、11日目に毎日の投薬群において停止した。残りの群において研究は、研究25日目で終了し、そして平均の腫瘍容量を計測し、ビヒクルと比較した。毒性の臨床的指標として、体重損失の百分率を、それぞれの群において計測した。
【0130】
【表7】
(実施例6)
化合物1の同定された代謝物の構造を確認するために、この代謝物を独立して合成した。
【0131】
化合物1のN−オキシド代謝物である化合物2を、下記のスキームに示すように合成した。化合物1をエタノール、脱メチルアセトアミドおよび過酸化水素の混合物中で加熱した。反応終了時において、化合物2を遠心によって単離し、そしてエタノールで洗浄した。必要とする場合、生成物を、カラムクロマトグラフィーによってさらに精製し得る。
【0132】
【化33】
化合物1のN−デスメチル代謝物である化合物3を、下記のスキームに示すように合成した。5―クロロ―2―ニトロアミンを、ピペラジンを使用して処理し、4を得た。4を、引き続いて、ブチルカルボニル(Boc)基で保護し、5を得た。ニトロ基の還元、引き続く3―エトキシ―3―イミノプロピオン酸エチルエステルとの縮合により、6を得た。塩基として、カリウムヘキサメチルジシルアジドを使用した6―フルオロアントラニロニトリルと6により、7を得た。粗製の7を水性のHClで処理し、精製後に黄色/茶色の固体として望ましい代謝物を得た。
【0133】
【化34】
(実施例7)
(化合物1の乳酸塩の調製)
【0134】
【化35】
ジャック付の3000mLの4つ口フラスコに、冷却器、温度計、N2ガス挿入口および機械的攪拌器を備え付けた。この反応容器を、少なくとも15分間N2でパージし、それから4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H−ベンゾイミダゾール―2―イル]―1H―キノリン―2―オン(484g、1.23mol)を充填した。D,L―乳酸(243.3g、モノマーの1.72mol、下記の段落参照)、水(339mL)ならびにエタノール(1211mL)の溶液を調製し、次いで反応フラスコに充填した。攪拌を、中間の速度で開始し、そしてその反応物を内部温度68℃〜72℃で加熱した。この反応物の内部温度を15分間〜45分間の間68℃〜72℃で維持し、次いで加熱を中止した。生じた混合物を、10〜20ミクロンのフリットを通して濾過し、12リットルのフラスコ内に濾液を収集した。この12リットルのフラスコに内部温度計、還流冷却器、添加漏斗、気体の挿入および排出口、ならびにオーバーヘッド攪拌器を備え付けた。次いで、攪拌物を中間の速度で攪拌し、加熱還流した(約78℃の内部温度)。穏やかな還流を維持しながら、エタノール(3,596mL)を約20分間にわたってフラスコに充填した。次いで、この反応フラスコを15分間〜25分間以内で約64℃〜70℃の範囲の内部温度に冷却し、そしてこの温度を約30分間の間維持した。反応器を結晶について検査した。結晶が存在しない場合、4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H―ベンゾイミダゾール―2―イル]―1H―キノリン―2―オンの乳酸塩の結晶(484mg、0.1モル%)をフラスコに添加し、そしてその反応物を30分間64℃〜70℃で攪拌し、その後、結晶についてそのフラスコを検査した。結晶が存在するとすぐに、攪拌を低速度に減少させ、そしてその反応物を、さらに90分間64℃〜70℃で攪拌した。次いで、この反応物を、約2時間の間にわたって約0℃で冷却し、そして、生じた混合物を25ミクロン〜50ミクロンのフリットのフィルターを通して濾過した。反応器をエタノール(484mL)で洗浄し、そして内部温度が約0℃になるまで攪拌した。冷エタノールを使用して、フィルターケーキを洗浄し、この手順を、さらに2回繰り返した。収集した固体を、真空オーブン内で真空下50℃で一定の質量まで乾燥して、結晶性の黄色い4―アミノ―5―フルオロ―3―[6―(4―メチル―ピペラジン―1―イル)―1H―ベンゾイミダゾール―2―イル]―1H―キノリン―2―オンの乳酸塩510.7g(85.7%)を収集した。ラバーダムまたは不活性化状態を、主として濾過プロセスの間に使用した。乾燥した固体が、さほど湿潤でない一方、湿ったフィルターケーキは、水を吸収し、粘度を増す傾向にある。予防策を、大気に対する濾過ケーキの長期の曝露を避けるために実行した。
【0135】
市販の乳酸は、一般的に約8%w/w〜12%w/wの水を含み、モノマー乳酸に加えて、ダイマーおよびトリマーを含む。乳酸ダイマー対乳酸モノマーのモル比は、一般的に約1.0:4.7である。商業的な等級の乳酸は、モノ乳酸塩が、反応混合物から優先的に沈殿するように、上記段落に説明されたプロセスにおいて使用され得る。乳酸モノマーを以下の手順に従って、精製する。
(実施例8)
この研究では、FGFを補充したマトリゲルモデル(Matrigel model)において化合物1の抗血行性の可能性を評価した。
【0136】
11〜12週齢の雌のBDF1マウス(Charles River、Wilmington、MA)に、2μgのFGF―2を追加した0.5mLのマトリゲル(BD Biosciences、Bedford、MA)を皮下に移植した。血管形成(新血管新生または脈管形成)に追加されたFGF―2を、動物からそれらの除去後にマトリゲルプラグにおいてヘモグロビンレベルを計測することで定量した。
【0137】
テスト物品の経口投与を、マトリゲル移植の一日前に開始し、毎日1回で8回の投薬を連続して行った。化合物1を10mM H3PO4中の溶液として処方した。12の処理群が含まれた:ビヒクル(10mM H3PO4)のp.o.×8日間、q.d.×8日間(2つのコントロール群);補充していないマトリゲル(標準ヘモグロビンレベル)またはFGFを補充したマトリゲル(ポジティブコントロール)を移植したマウス;化合物1を3mg/kg、10mg/kg、30mg/kg、100mg/kg、200mg/kg、300mg/kgでp.o.×8日間、q.d.×8日間投与。1群あたり4匹を使用する200mg/kg、300mg/kgで投薬するマウスを除き、1群あたり8匹のマウスを使用した。
【0138】
ビヒクルで処理されたマウスと比較した、化合物で処理されたマウスにおけるヘモグロビンレベルのパーセンテージの阻害は、この化合物の抗脈管形成能を示す。結果を、単位マトリゲルプラグあたりの総ヘモグロビン量(mg/dL)で示す。ED50を、脈管形成を効果的に約50%阻害する用量として定義する。ヘモグロビン濃度を、Drabkin試薬(Sigma Diagnostics、ST.Louis MO)を使用する吸光度分光法を使用して、マウスから取り出し瞬間冷却した均質化マトリゲルプラグにおいて決定した。
【0139】
化合物1の血漿の曝露を評価するため、血液を、8つの持続的な投薬(8日目)後、2時間および24時間後に採取した。200mg/kgおよび300mg/kgの投薬群においては、血液を2時間の時点でのみ採取した。1の血漿濃度を、1ng/mL〜8000ng/mLの較正の範囲および1ng/mLの定量限界(LLOQ)の検証されていないLC/MS/MSアッセイによって決定した(Charles River Laboratories、Worcester、MA)。
【0140】
8日目において、マトリゲルプラグにおけるヘモグロビンレベルおよび化合物1の血漿濃度を計測した。研究の間に動物を観察し、そして体重を計測した。
【0141】
ビヒクル処置動物からのプラグと比較して評価したそれぞれの用量において、化合物1は、マトリゲルプラグにおけるヘモグロビン濃度の有意な阻害を生じた(表8)。計算したED50は、2.6mg/kgであった。3mg/kgおよび10mg/kgの用量は、54%および57%の阻害をそれぞれ結果として生じ、一方で、30mg/kg、100mg/kg、200mg/kgおよび300mg/kgの用量は、FGFを補充されていないマトリゲルのレベルまでヘモグロビンを減少させ、FGFを補充されたコントロールに対して、70%から92%の阻害を結果として生じた。8日目における投薬後2時間での化合物1の血漿濃度は、44ng/mLかつ3mg/kgから3920ng/mLかつ300mg/kgまでの濃度範囲に対して用量に比例した増加を示した(表9)。全ての用量では、良好な耐性を示し、体重の損失は、観察されなかった。
【0142】
【表8】
【0143】
【表9】
1の血漿濃度(投薬後2時間)は用量に比例して増加した。マトリゲルプラグのヘモグロビン量において、用量および血漿濃度依存的な減少が、観察された。44ng/mgの血漿濃度(投薬後2時間、8日目)は、このモデルにおいて抗脈管形成活性に関連するようである。
【0144】
要約すると、化合物1のヘモグロビン阻害は、8日間の処置後有意な阻害を伴って用量依存的であった。統計的に、有意なヘモグロビン阻害が、化合物1の全ての用量において観察された。全ての用量は、体重の損失または観察された有害な臨床徴候のない良好な耐性を示した。このモデルにおいて44ng/mLの化合物1の血漿濃度(投薬後2時間)は、抗脈管形成活性と関連していた。
(実施例9)
5mg/kgのBIDの複数の経口投薬の研究に由来するサルの血漿における化合物1の代謝物のプロフィールを、投薬1日目および14日目のサンプルにおいて決定した。ある代謝物は、LC/UVおよびLD/MS/MSによって脱メチル化から生じた(化合物3)と同定され、特徴付けられた。親化合物1(P)は、クロマトグラフィーの保存時間が18.3分においてm/z=393.3で(M+H+)イオンを産生した。脱メチル化された代謝物(P−CH3)は、m/z=379.3(M+H+)およびクロマトグラフィーの保存時間が18.1分で同定された。その代謝物と化合物1の間の14ダルトンの質量の違いは、脱メチル化された化合物1と一致する。代謝物の質量およびクロマトグラフィーの保存は、独立に合成された化合物3と同じであった。化合物1のピペラジンN−オキシド(N−オキシドは化合物2)に対応する代謝物は、この投薬レベルにおいて血漿で検出されなかった。356nmにおける吸光度クロマトグラムで17.7分および18.5分でUV信号を発生する成分は、化合物1に対するUVスペクトルの比較に基づいて、そしてブランク血漿(投薬1日目の時刻0)におけるそれらの存在に起因してマトリックス成分であって代謝物ではないと決定された。
【0145】
脱メチル化された代謝産物の推定レベルを表1に示す。代謝物(化合物1の相当量における)の推定レベルは、この分析で得られた化合物1のUV吸着ピークの高さに対する、代謝物のUV吸光度ピークの高さの比に基づき、そして以前の定量分析研究において同様のサンプルで決定された既知のレベルの化合物1に対して吸光度比を考慮に入れることよって外挿される。親化合物は、全ての蓄積された時点で代謝物よりも、より大きな吸光度を示すことが判明した。化合物1のレベルは、基本的に検出できないN−脱メチル代謝物に平行して、14日目のサンプルにおいて十分に低くなることが見出された。共役したフェーズII(PhaseII)タイプの代謝物(グルクロニドまたはサルフェイト)を含む他の代謝物は、用量投薬の1日目または14日目においてこれらの血漿サンプルに内に検出されなかった。
【0146】
【表10】
(実施例10)
化合物1での処理後にマウスから収集された血漿および腫瘍を用いる研究を、潜在的な薬力学の最終点を評価するために行った。化合物1の処理後にKM12L4a腫瘍における標的調節の分析は、VEGFR1、VEGFR2、PDGFRβおよびFGFR1のリン酸化が、時間および用量依存的様式で阻害されることを示した。例えば、HMVEC細胞は、約0.1μMのIC50でVEGFによって媒介されるVEGFR2リン酸化の阻害を示した。さらに、化合物1を用いる内皮細胞の処理は、VEGFを介すことでMAPKおよびAktのリン酸化を阻害した。
【0147】
さらに、ERK(MAPK)の活性化の時間および用量依存的阻害、レセプターチロシンキナーゼの下流の標的が、KM12L4A細胞において0.1μMから0.5μMの範囲のIC50で観察された(KM12L4A細胞はそれらの表面においてPDGFRβおよびVEGFR1/2を発現する)。KM12L4A細胞を、無血清DMEM中で化合物1とともに3時間インキュベートした。収集後、溶解物を、SDS−Pageによって分離し、ホスホ−ERK1/2およびERK1/2抗体でプローブした。検出において、ECL試薬(Amersham)を使用した。レセプターのリン酸化およびERKの活性化に対する化合物1の阻害効果は、処置後24時間維持された。MV4〜MV11細胞におけるERK1/2のリン酸化は、用量依存的様式で0.01μM〜0.1μMのIC50において1によって阻害された。
【0148】
有意な活性が、HCT116ヒト結腸腫瘍モデルにおいてインビボで観察された。HCT116腫瘍において、化合物1は、用量および時間依存的様式におけるERK(MAPK)のリン酸化を阻害し、そして、腫瘍の組織学解析において有意な変化が観察された。
【0149】
前臨床モデルにおいて、これらのPK/PD評価は、化合物1が、標的レセプターおよび下流のシグナル分子ERK(MAPK)の両方の用量および時間依存的阻害を示したを示す。これらの研究は、潜在的バイオマーカーの同定を補助し、その結果、臨床的試行における化合物1の生物学的活性を観測することを補助する。
(実施例11)
雄および雌のスプレイグドーリー(SD)ラットに対する、14Cで標識した化合物1(キノリノン環の第4位)の一回の経口(PO)投薬(5mg/kg)後の、組織における放射能の分布を、全身オートラジオグラフィー(WBA)によって定量した。WBAのための血液および屠体を、投薬後24時間を通して特定の時点で収集した。屠体をヘキサン/ドライアイスバスで凍結させ、水分を排出させ、吸い取り紙で乾燥させて、そしてドライアイス上に配置したか、もしくは少なくとも2時間、約−70℃で保管した。凍結した屠体を、冷却したカルボキシメチルセルロースに埋め込み、埋め込まれたオートラジオグラフィー標準と一緒に固形に凍結させ、そして分析まで−20℃で保管した。40μmの厚さの適当な断片を矢状平面における、目的の5レベルで粘着性のテープの上に収集した。全ての主要な組織、器官および生物学的流体が標本にされた。蛍光検出画面に、その断片を表示し、走査し、そして14C−1の組織濃度内挿をするために標準曲線を形成した。血漿を、液体シンチレーションカウント(LSC)によって放射能の濃度について分析した。例示的な結果を表11に示す。
【0150】
14C−1の経口投与後に、14C−1由来の放射能は、投薬後1時間で全ての組織にわたって広範囲に分布し、そして投薬後4時間でほとんどの組織においてCmaxに達した。雄および雌の組織における放射能の全体的な分布は類似した。14C−1由来の放射能は、血清よりも組織からよりゆっくりと除去された。雄および雌において、24時間を通して胃腸路を除いた14C−1の最も高い組織中の濃度を、ハーダー腺、副腎腺、腎髄質、眼窪内の涙腺および眼窪外の涙腺で検出した。14C−1由来の放射能は、経口投薬投与後に、血液脳の関門を横切った。
【表11A】
【表11B】
【0151】
本発明は、本明細書中に記載される実施形態に限定されず、添付の特許請求の範囲の範囲内に入るような全ての形式を包括する、と理解される。
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】


【図2】

【図2】
【公開番号】特開2011−46716(P2011−46716A)
【公開日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願番号】特願2010−208650(P2010−208650)
【出願日】平成22年9月16日(2010.9.16)
【分割の表示】特願2005−507133(P2005−507133)の分割
【原出願日】平成15年11月12日(2003.11.12)
【出願人】(591076811)ノバルティス バクシンズ アンド ダイアグノスティックス,インコーポレーテッド (265)
【Fターム(参考)】
【公開日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願日】平成22年9月16日(2010.9.16)
【分割の表示】特願2005−507133(P2005−507133)の分割
【原出願日】平成15年11月12日(2003.11.12)
【出願人】(591076811)ノバルティス バクシンズ アンド ダイアグノスティックス,インコーポレーテッド (265)
【Fターム(参考)】
[ Back to top ]