
癌免疫療法におけるP53を発現する改変・ワクシニア・アンカラ
腫瘍抑制タンパク質 p53に対する突然変異はすべてのヒト癌の40-60%において観察されている。これら突然変異にはしばしば高いp53の核内および細胞質濃度が伴う。多くの腫瘍は非常に高いp53レベルを示すため、このタンパク質は癌免疫療法の魅力的な標的である。残念なことに、p53は、免疫系によって自己-タンパク質として寛容されそうである自己抗原である。本発明はp53 (rMVAp53)をコードする核酸を含む組換え・改変・ワクシニア・アンカラ (MVA) の投与によりこの自己寛容が克服されるという発見に基づく。本発明はrMVAp53を含む組成物の投与による突然変異p53を発現する腫瘍細胞に対するp53-特異的CTL応答の生成方法を開示する。rMVAp53の投与は様々な悪性細胞型における腫瘍発生、腫瘍増殖、および死亡率を低下させる。これらの効果は、CTLA-4遮断薬および/またはCpGオリゴデオキシヌクレオチド免疫調節物質の投与によって増強される。
【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
本出願は2002年12月23日出願の米国仮特許出願第06/436268号(Ellenhorn and Diamond)および2003年4月30日出願の第06/466607号(Ellenhorn and Diamond)からの優先権を主張し、その開示全体を引用により本出願に含める。
【0002】
連邦政府援助の研究に関する出願の記載
本発明は一部、NIH、Division of AIDS (RO1-AI43267およびR21-AI44313)および NCI: RO1-CA77544、PO1-CA30206、R29-CA70819、およびCA33572からの助成により政府援助によりなされた。政府は本発明における一定の権利を有しうる。
【0003】
発明の分野
本発明は、ウイルス学、分子生物学、および腫瘍免疫学の分野に関する。具体的には、本発明はp53を発現する悪性腫瘍に対して有効な免疫応答を誘発するための組成物および方法に関する。
【背景技術】
【0004】
背景
p53は細胞周期停止またはアポトーシスに必要な一定の遺伝子の発現を制御する腫瘍抑制タンパク質である。p53をコードする腫瘍抑制遺伝子は、DNA損傷、細胞ストレス、または特定の癌遺伝子の異常発現によって活性化する(Levine 1997)。いったん活性化すると、野生型p53 (wt p53)は一時的に細胞周期を停止させる役割を果たし、DNA修復の時間を与え、損傷したDNAを有する細胞が制御を失って増殖することを防ぐ(Levine 1997)。p53はまた一定のタイプの生理的損傷を有する細胞におけるアポトーシスの誘導にも関与している(Levine 1997)。
【0005】
p53の増殖抑制能を機能的に不活性化させるp53における突然変異はすべてのヒトの癌の40-60%において観察されており、悪性表現型と関係がある(Hainaut 2000)。p53に対する突然変異は腫瘍形成の初期現象として起こり(Millikan 1995; Querzoli 1998; Allred 1993)、該タンパク質が細胞分裂を抑制する能力を失わせる (Finlay 1989; Eliyahu 1989)。細胞におけるp53発現の制御は、p53 mRNA量のレベルまたは53タンパク質量のレベルにて起こりうる。p53の突然変異は、突然変異タンパク質の半減期の延長により、しばしばp53タンパク質の高い核内および細胞質濃度をともなう。多くの腫瘍は非悪性細胞と比べて細胞内p53レベルが非常に高いことによって特徴づけられる。腫瘍のなかには多量の突然変異p53を合成するが、細胞内p53レベルは低い、即ち通常の定常状態レベルを下回るものもあり、これはおそらくタンパク質の細胞内分解の加速の結果と考えられる。p53の過剰発現は以下の独立の前兆である:より悪性の癌 (Turner 2000; Elkhuizen 2000; Zellars 2000)、リンパ節転移(Pratap 1998)、標準的治療に対する応答の欠損 (Berns 1998; Berns 2000)、および死亡率 (Sirvent 2001; Querzoli 2001)。
【0006】
ミスセンス点突然変異は癌におけるp53突然変異でもっとも頻繁なものであり、ほとんどのp53タンパク質は野生型形態(wt p53)のままである。p53突然変異は真の腫瘍特異的抗原を表すこともあるが、かかる突然変異のほとんどは、T細胞によって認識される免疫性エピトープには対応しない部位において起こる(Wiedenfeld 1994)。このため、広く利用できるp53に向けられた免疫療法はいずれもwt p53を標的としなければならない。実験モデルにおいて、p53を標的とすることが可能であった。なぜなら突然変異分子はp53タンパク質の高い核内および細胞質濃度を伴うからである(Finlay 1988)。p53は適応免疫応答にとって魅力的な標的である。なぜなら健康細胞における非突然変異p53の細胞内濃度は非常に低いからである(Zambetti 1993; Reich 1984)。これは非-突然変異体 p53を発現する健康細胞は過剰発現した突然変異体 p53に対する増強した免疫応答をおそらくまぬがれるであろうということを意味する(Offringa 2000)。
【0007】
p53は、細胞性免疫応答によって認識されうるほとんどの腫瘍関連抗原と同様に、自己抗原である(Rosenberg 2001)。p53は発生の過程で広く発現した自己抗原であるという事実と(Schmid 1991)、腫瘍において発現している突然変異p53のほとんどは野生型タンパク質と同じ構造を有するという事実とを組み合わせると、腫瘍によって発現したp53が免疫系によっておそらく自己-タンパク質として寛容されるということを意味する。マウスにおける機能および四量体研究によって細胞障害性Tリンパ球レベル(CTL)で存在することが示されたこの寛容は(Theobald 1997; Erdile 2000)、p53に向けた免疫療法の有効性を制限する。成功するためには、有効な免疫療法は正常細胞および組織に対する自己免疫を誘発することなくこの寛容を克服しなければならない (Theobald 1997; Erdile 2000; Hernandez 2000)。少数の自己反応性T細胞は免疫寛容に関与するプロセスにおいて回避する。
【発明の開示】
【発明が解決しようとする課題】
【0008】
p53を過剰発現する腫瘍は、エピトープ特異的CTL(Vierboom 2000a; Vierboom 2000b; Vierboom 1997; Hilburger 2001)、エピトープで刺激した樹状細胞(DC) (Mayordomo 1996)、または突然変異体p53エピトープとIL-12 (Noguchi 1995)の全身投与によってマウスモデルにおいて排除された。これら戦略はそれぞれ臨床適用の面でかなりの問題を有する。CTL 注入およびエピトープで刺激した樹状細胞の注入は時間がかかり高価である。というのは細胞の単離、培養および再注入は各々の患者にとって別々に行わなければならないからである。逆に、なんらかの効果を得るためには、従来用いられていた無細胞ワクチン接種戦略は、腫瘍内注射または腫瘍攻撃に先立つワクチン接種のいずれかを必要としそのいずれも臨床使用では実践的なアプローチではない。したがって簡便、有効かつ広く適用できる癌の治療における免疫療法戦略が必要とされている。
【課題を解決するための手段】
【0009】
発明の概要
p53 遺伝子産物は大多数の癌で過剰発現しており、したがってそれは癌免疫療法にとって理想的な標的となる。しかしこれら療法の効力は制限されてきた。というのは腫瘍によって発現されるp53は自己-タンパク質として免疫系によって寛容されるようであるという事実に基づく。本発明はこの自己寛容はp53をコードする核酸を含む組換えMVA (rMVAp53)の投与によって克服されうるという発見に基づく。p53の投与は様々な悪性細胞型で攻撃したマウスにおける腫瘍発生、腫瘍増殖、および死亡率を大幅に減少することが示されている。rMVAp53の治療効果はCTLA-4遮断薬またはCpGオリゴデオキシヌクレオチド (CpG ODN) 免疫調節物質の投与により増強されることも示される。この増強は両方の免疫調節物質を投与した場合にもっとも大きくなる。本発明は癌の治療における使用のための組換えMVA 組成物、この組成物を用いる癌の治療方法およびこの組成物の投与のためのキットを提供する。
【0010】
第一の態様において、本発明はp53をコードする核酸を含む組換えMVA を含む組成物を提供する。好ましくは、組換えMVA によってコードされるp53はwt ヒト p53である。本発明によると、組成物はCTLA-4遮断薬および/またはCpG ODNを含んでいてもよい。
【0011】
別の態様において、本発明はp53-発現悪性腫瘍を有する対象の治療方法を提供する。この方法は、p53をコードする核酸を含む組換えMVAの投与に基づく。好ましくは、該方法は免疫調節物質としてCTLA-4遮断薬および/または CpG ODNの投与も要求する。第三の態様において、本発明は、p53-発現悪性腫瘍を治療するためのキットを提供する。このキットはp53をコードする核酸を含む組換えMVAを含み、免疫調節物質としてCTLA-4遮断薬および/または CpG ODNを含んでいてもよい。最後の態様において、本発明は、wt ヒト p53をコードする核酸インサートを含むMVA組換えプラスミドを提供する。
【0012】
図面の簡単な説明
図 1: pLW22-hup53 コンストラクトのPCR分析。
rMVAhup53注入(レーン 1、2) および wtMVA注入(レーン 3、4) BHK 細胞を全DNA抽出および wtMVA (レーン 2、4)またはhup53 (レーン 1、3) 特異的プライマーを用いたPCR 増幅に供した。rMVAhup53 産物は汚染wtMVAを有さないことが示された。
【0013】
図 2: rMVAmup53で感染した細胞によるmup53の発現。
rMVAmup53で感染させた細胞は mup53を高レベルで発現し、MVAが好適なワクチンベクターであることが確認された。細胞可溶化液をSDS-PAGEおよびウェスタンブロッティングに供した。レーンを以下のように指定する: 1) Meth A、非操作 Meth A 肉腫細胞、2) HCMV IE1 エキソン4-rMVA感染BHK細胞、3-4) rMVAmup53感染BHK細胞 (それぞれ0.125μl、0.25μl 細胞可溶化液をローディング)、5) rAdp53、および6) rAdpp65感染HEK293細胞。特に断りのない限りすべてのレーンに20μlのサンプルをローディングした。
【0014】
図 3: インビトロでのrMVAmup53によるp53-特異的CTL応答の生成。
rMVAmup53 による一回の腹腔内 (i.p.) ワクチン接種により、p53を過剰発現する細胞を有効に殺すp53 特異的CTL応答が生じる。(a) rMVAmup53で処理したマウスからの脾細胞を14日目に回収し、インビトロで6日間 rAdp53感染同系 LPS 芽球で再刺激した。CTL活性をrVVp53 (実線)またはrVVpp65 (破線)感染10.1 細胞を用いて標準的 4-h 51Cr 放出アッセイで評価した。(b) rMVAmup53 (実線)またはrMVApp65 (破線) ワクチン接種マウスからの脾細胞をワクチン接種の14日後に回収し、インビトロで6日間 rAdp53感染同系 LPS 芽球で再刺激した。細胞障害性をrVVp53感染10.1 細胞に対して測定した。 (c) rMVAmup53 (実線)またはrMVApp65 (破線) ワクチン接種の14日後に回収した脾細胞をインビトロで6日間 rMVAp53で感染した同系 LPS 芽球を用いて再刺激した。細胞障害性を標準的 4-h 51Cr 放出アッセイによってMeth A 細胞に対して測定した。
【0015】
図 4: rMVAmup53によるワクチン接種の Meth A 腫瘍阻止に対する効果。
Balb/c マウスの皮下に (s.c.) 5 x105 Meth A 細胞を注射した。5日目に、マウスを5 x107 pfu のrMVAmup53 (MVAp53) (n = 16)、5 x107 pfuのrMVApp65 (MVApp65) (n = 16)、またはPBS (n = 12)でワクチン接種した。生存プロットは、各群における生存動物の比率を腫瘍攻撃後の日数の関数として示す。rMVAmup53をワクチン接種したマウスの両方の対照群と比較しての改善はlog順位検定により測定して統計的に有意である(P < 1)。
【0016】
図 5: rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した Meth A 腫瘍に対する効果。
マウスに迅速に死に至る用量の106 Meth A 細胞を皮下注射した。6、9、および12日目に、マウスに抗-CTLA-4 mAb (CTLA4mAb)またはコントロール mAbのいずれかを腹腔内注射した。7日目に、マウスに5 x107 pfuのrMVAp53 (MVAp53)または5 x107 pfuのrMVApp65 (MVAapp65)のいずれかをワクチン接種した。生存プロットは各群における生存動物の比率を示す。rMVAp53 + 抗-CTLA-4 mAb (n=14)をワクチン接種したマウスの、rMVApp65 + CTLA-4 (n=14)、rMVAp53 + コントロール ab (n=14)、またはrMVApp65 + コントロール ab (n=6) を与えられたコントロール動物に対する生存の向上はlog順位検定によって判定して統計的に有意である(P < 0.001)。
【0017】
図 6: rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウスに2 x 106の11A-1 細胞(rMVAmup53 + 抗-CTLA-4 mAbを他のすべての群と比較してp = 0.00044)を皮下注射した。抗-CTLA-4 mAb 9H10 (CTLA4 mAb)またはコントロールハムスターアイソタイプ一致ポリクローナル抗体 (アイソタイプ一致 Ab)を4、7、および10日目にそれぞれ100、50、および50μgの用量で腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65 (MVApp65)、またはPBSを腹腔内にワクチン接種した。それぞれの線は8匹のマウスの平均および標準偏差を表す。
【0018】
図 7: rMVAmup53 + 抗-CTLA-4 mAbのワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した (rMVAmup53 + 抗-CTLA-4 mAb をその他の全ての群と比較してp = 0.0001)。抗-CTLA-4 mAb 9H10 (CTLA4 mAb)またはコントロールハムスターアイソタイプ一致ポリクローナル抗体 (アイソタイプ一致 Ab)を4、7、および10日目にそれぞれ100、50、および50μgの用量で腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65 (MVApp65)、またはPBSのいずれかを腹腔内にワクチン接種した。各線は8匹のマウスの平均および標準偏差を表す。
【0019】
図 8: rMVAmup53 + CpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウスに2 x 106 11A-1 細胞を皮下注射した(rMVAmup53 + CpG ODNをその他のすべての群と比較してp = 0.00002)。15 nmolesの CpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0020】
図 9: rMVAmup53 + CpG ODNによるワクチン接種の確立したMeth A 腫瘍に対する効果。
Balb/c マウスに1 x 106 Meth A 細胞を皮下注射した(MVAmup53 + CpG ODN をすべての他の群と比較してp = 0.0015)。15 nmoles のCpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0021】
図 10: rMVAmup53 + CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウスに1 x 106 MC-38 細胞(rMVAmup53 + CpG ODNをその他のすべての群と比較してp = 0.0004)を注射した。15 nmolesのCpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0022】
図 11: rMVAmup53 + 抗-CTLA-4 mAbおよびCpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウス (n = 8)に2 x 106 11A-1 細胞を皮下注射した。抗-CTLA-4 mAb (CTLA4 mAb)を14、17、および20日目にそれぞれ100、50、50μg用量で腹腔内注射した。15 nmolesのCpG ODN (CpG)を14、19、および24日目に腹腔内注射した。15日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。生存プロットは腫瘍攻撃後の日数の関数としての各群における生存動物の比率を示す。CpG ODN と抗-CTLA-4 mAbとの組み合わせをCpG ODNのみに対して比較してp = 0.02、およびCpG ODNと抗-CTLA-4 mAbの組み合わせを抗-CTLA-4 mAbのみと比較してp = 0.01。
【0023】
図 12: rMVAmup53 + 抗-CTLA-4 mAb および CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウス (n = 8)に1 x 106 MC-38 細胞を皮下注射した。抗-CTLA-4 mAbを4、7、および10日目にそれぞれ100、50、および50μg用量で腹腔内注射した。15 nmolesのCpG ODNを4、9および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53、5 x107 pfuのMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。生存プロットは腫瘍攻撃後の日数の関数としての各群における生存動物の比率を示す。CpG ODNおよび抗-CTLA-4 mAbの組み合わせをCpGのみと比較してp = 0.002、およびCpG ODN および抗-CTLA-4 mAbの組み合わせを抗-CTLA-4 mAbのみと比較してp = 0.001。
【0024】
図 13: Meth A 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求。
Balb/c マウス (a)またはIFN-γKO Balb/c マウス (b)に迅速に死に至る用量の106 Meth A 細胞を皮下注射した。両集団からのマウスの群に枯渇させる用量(depleting dose)の抗-CD4、抗-CD8、抗-NK1.1、またはコントロール mAbを-1、1、3、および10日目、そしてその後毎週、腹腔内注射した。6、9、および12日目にマウスに抗-CTLA-4 mAb (CTLA4mAb)またはコントロール mAbのいずれかを腹腔内注射した。7日目に、マウスに5 x107pfuのrMVAp53 (MVAp53)または5 x107 pfuのrMVApp65 (MVAapp65)のいずれかをワクチン接種した。(a) 平均腫瘍増殖をBalb/c マウスの各群について計算し、エラーバーは標準偏差を示す。各線の最後のデータポイントは最初の死亡を示す。(b)生存IFN-γKO Balb/c マウスの比率をプロットする。
【0025】
図 14: 11A-1 腫瘍に対するCpG ODN 免疫調節物質効果の細胞による要求。
Balb/c マウスに2 x 106 11A-1 細胞を皮下注射した。15 nmoles のCpG ODNを4、9、および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53を腹腔内にワクチン接種した。マウスに枯渇させる用量の抗-CD4 (CD4)、抗-CD8 (CD8)、抗-NK1.1 (NK)、またはコントロール mAbを4、6、8および15日目、そしてその後7日ごとに腹腔内注射した。腫瘍を三次元で週に2回測定した。CD8+枯渇をすべてのその他の群と両側ウイルコクソン検定によって比較してp = 0.004。抗-NK1.1 を抗-CD4およびコントロール mAbと比較してp = 0.007。
【0026】
図 15:11A-1 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求。
マウスに2 x 106 11A-1 細胞を皮下注射した。抗-CTLA-4 mAbを4、7、および10日目にそれぞれ100、50、および50μg/用量で腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内にワクチン接種した。4、6、8、および15日目、そしてその後7日ごとに対応するmAbまたはコントロール mAbを腹腔内注射することによってマウスからCD8+、CD4+、またはNK 細胞を枯渇させた。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。CD8+ 枯渇をその他のすべての群と比較してp = 0.004。CD4+枯渇をNK枯渇およびコントロール群と比較してp = 0.008。
【0027】
図 16: CpG ODN 免疫調節物質効果に対するTLR 9の寄与。
TLR9-/- (抗-CTLA-4 mAbをCpG ODN 群と比較してp = 0.0009) 突然変異体 C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した。マウスを4、7、および10日目にそれぞれ100、50および50μg/用量で抗-CTLA-4 mAb (CTLA4 mAb)で処理するかあるいは15 nmolesのCpG ODNで4、9、および14日目に処理した。5日目に、すべてのマウスに5 x107 pfuのrMVAmup53を腹腔内にワクチン接種した。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。
【0028】
図 17: IL-6 の CpG ODN 免疫調節物質効果に対する寄与。
IL-6-/- (ウィルコクスン両側順位和検定によって抗-CTLA-4 mAbをCpG ODN 群と比較してp = 0.02) 突然変異体 C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した。マウスを 抗-CTLA-4 mAb (CTLA4 mAb) で4、7、および10日目にそれぞれ100、50、および50μg/用量であるいは15 nmolesのCpG ODNで4、9、および14日目に処理した。5日目に、すべてのマウスに5 x107 pfu のrMVAmup53を腹腔内にワクチン接種した。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。
【0029】
図 18: rMVAhup53を感染した細胞によるhup53の発現。
BHK細胞に精製rMVAhup53 (MVA/p53)を注入した。hup53の発現を24および48時間後に測定した。細胞可溶化液をSDS-PAGEおよびウェスタンブロッティングに供した。レーン1: コントロール MVA を注入したBHK細胞;レーン2: rMVAhup53を24時間注入したBHK細胞;レーン3: rMVAhup53を48時間注入したBHK細胞。すべてのレーンに20μlのサンプルをローディングした。
【0030】
図 19: rMVAhup53 + 抗-CTLA-4 mAbおよびCpG ODNのワクチン接種の確立した4T1/hup53 腫瘍に対する効果。
マウスに5 x104 4T1/hup53を皮下注射し、次いで107 pfuのrMVAhup53またはPBS コントロールを6日目に腹腔内にワクチン接種した。16日目に、マウスにrMVAhup53 またはPBSの追加免疫注射を15 nmoleのCpG ODNおよび50μgの抗-CTLA-4 mAbとともに与えた。rMVAhup53ワクチン接種マウスはPBS コントロールと比較して生存において有意な向上を示した(p < 0.05、両側T検定)。
【0031】
詳細な説明
本発明は正常および癌細胞の両方に発現するタンパク質に対する自己寛容が克服され、強い抗-腫瘍免疫応答が、腫瘍内投与を必要とせず、全身毒性または自己免疫を引き起こす、生成させうるという発見に基づく。本発明は新規な無細胞組成物および多様なヒ悪性腫瘍に対する有効な免疫応答を、対象のハプロタイプまたはジェノタイプに関わりなく生成させる方法を提供する。以下に記載する実施例は、野生型マウスまたは野生型ヒト p53 (rMVAmup53またはrMVAhup53)を発現するよう操作した改変・ワクシニア・アンカラ・ベクターでのワクチン接種により、激しいp53-特異的CTL応答が刺激されることを示す。この応答はCTLA-4遮断薬および/または CpG ODNからなる免疫調節物質の共投与により増強されうる。
【0032】
MVAウイルス (GenBank Accession Number U94848)は一次ニワトリ胚線維芽細胞の570回以上の連続継代に由来するワクシニアウイルスのアンカラ株のバリアントである。弱毒化ポックスウイルスとしてのMVAのいくつかの特性により、それはp53を発現する腫瘍に対する治療応答の生成のために理想的となる。MVAの一つの利点は哺乳類細胞においてそのDNAを効率的に複製できることであるが、それは無毒であり伝播しない。この形質は、ニワトリ胚線維芽細胞の継代の過程で生じる少なくとも25のさらなる突然変異および欠失のなかで2つの重要な宿主範囲遺伝子が失われた結果である (Meyer 1991; Antoine 1998)。NYVAC (弱毒化コペンハーゲン株)およびALVAC (宿主範囲制限アビポックス)と異なり、MVAにおける初期および後期転写はともに損なわれておらず、ウイルス生活環にわたって連続して遺伝子を発現することが可能である(Carroll 1997a; Carroll 1997b; Blanchard 1998; Sutter 1992)。 MVAはウエスタンリザーブ (WR) 株よりも免疫原性が強いことが判明しており、既存のポックスウイルス免疫性の条件において利用できる(Ramirez 2000a; Ramirez 2000b)。組換えワクチン送達媒体としてのMVAの好ましい臨床特性はその天然痘ワクチンとしての良性安全性特性としてヨーロッパにおいて1970年代後半に支持されている(Mayr 1999; Mayr 1978)。MVAは老人および若者を含む120,000を超える高リスク個体に重篤な副作用無く投与された (Mayr 1978)。より最近では、MVAは免疫不全の非-ヒト霊長類に不利な結果をもたらすことなく投与されてきた(Stittelaar 2001)。これはその他のベクター、例えば、ヒト宿主に危険をもたらすレトロウイルスおよびアデノウイルスとは全く対照的である。用いられるベクター、アジュバント、または免疫調節物質の免疫毒性は、癌の免疫療法において特に問題となる点である。というのはほとんどの癌患者は化学療法、放射線療法、または癌自体の免疫抑制効果によってひどい免疫不全となっているからである。非-弱毒化ポックスウイルス、例えば、WR 株は免疫不全の個体に安全に投与することができないことが明らかになった後、MVAは1990年代前半にワクチン媒体として最初に開発された(Redfield 1987; Collier 1991)。要約すると、MVAの発現ベクターとしての能力と、霊長類およびヒトにおけるその安全特性とを組み合わせると、それは癌遺伝子のための送達系として非常に魅力的である。
【0033】
rMVAmup53およびrMVAhup53の構築は当業者に周知の組換えDNA技術により達成される(Sambrook et al.、Molecular Cloning、Cold Spring Harbor Laboratory、2001; Ausubel et al.、Current Protocols in Molecular Biology、John Wiley & Sons、1986 and 2000)。野生型p53のコード配列は便宜にp53-特異的 プライマーを用いたRT-PCRによって得ることが出来る。これらプライマーはDNAにハイブリダイズし、DNA合成の開始部位として役立つ。ヌクレオチドプライマーは二本鎖の反対側の離れた部位に結合するよう設計され、それによって増幅される部分としての介在配列が規定される。プライマーとして用いる核酸分子は一般に増幅すべきDNAセグメントに相補的な少なくとも10塩基対の配列を含む。プライマーの選択は当業者に周知である。wt mup53またはwt hup53の増幅用プライマーは好適なMVA組換えプラスミド、例えば、pMCO3、pLW22、pLW51、pUCII LZまたはその他の当該技術分野で周知のMVA送達ベクターへのサブクローニングのための適当な制限部位を含むように設計すればよい。組換えプラスミドは、外来遺伝子インサートの発現に必要な配列、およびMVAゲノムにおける欠失した選択部位への相同組換えに必要な隣接配列を含む。組換えMVAウイルスを作るために、細胞にMVAウイルスを感染させ、外来遺伝子インサートを含む組換えプラスミドをトランスフェクトする。ウイルスとプラスミドの間の相同組換えを起こらせた後、インサート遺伝子を発現する組換えMVAを単離する。
【0034】
rMVAmup53またはrMVAhup53による感染後のp53タンパク質の細胞による発現を分析し、組換えウイルスからのその発現の忠実度と程度を判定した。突然変異p53を過剰発現するMeth A 細胞をポジティブコントロールとして用い、HCMV IE1 エキソン4 rMVA 感染BHK細胞をネガティブコントロールとして用いた。ウェスタンブロット分析によりrMVAmup53またはrMVAhup53で感染した細胞およびMeth A 細胞による豊富なp53 発現が明らかとなった。HCMV IE1 エキソン 4-rMVA 感染BHK細胞によるp53の検出可能な発現は観察されなかった。rMVAp53 感染BHK細胞による高レベルのp53発現は、蛍光顕微鏡観察によっても観察された。その他のウイルスおよび細胞形態と比較したrMVAmup53およびrMVAhup53 により示される高レベルのp53 発現は、ワクチン接種プロトコールにおけるその有用性を示す。
【0035】
動物実験モデルにおいて、MVAに基づくワクチンは腫瘍特異的CTL活性を刺激し(Espenschied 2003; Drexler 1999)、確立した腫瘍の退行をもたらす(Espenschied 2003; Carroll 1997b; Mulryan 2002; Rosales 2000)。MVAにおいて発現する全タンパク質による免疫化には多数の利点がある。ペプチド免疫化と異なり、複数のエピトープの発現が可能であり、ポリクローナル宿主応答が刺激できる。CTL応答の伝播に必須な抗原-特異的同族援助(cognate help)は、MVAにおけるタンパク質の発現を介して達成できる。さらに、全タンパク質の発現の結果、そうでなければ陰性のエピトープに対する応答の刺激が起こりう得る。組換えウイルスによる免疫化は、抗原-特異的細胞の拡張および養子移入を伴う複雑かつ高価なアプローチの必要性、または特定の癌患者のための個別化ワクチンの作成の必要性を避けうる。組換えワクチンアプローチのこの利点により、疾患の初期段階の患者における再発を予防するより広範な臨床使用が促される。
【0036】
インビトロ実験を行って、rMVAmup53によるワクチン接種がp53 寛容に打ち勝つかを調べたところ、p53-特異的CTLが生じた。rMVAmup53による単回腹腔内 (i.p.) ワクチン接種の後マウスから脾細胞を回収し、インビトロでp53 過剰発現細胞で再刺激した。脾細胞は wt p53過剰発現標的を認識し、溶解した。一方、rMVApp65をワクチン接種したマウスからの脾細胞は強いpp65 特異的CTL応答を刺激したが、p53過剰発現標的は認識せず、リンパ球応答の特異性が示された。rMVAmup53 ワクチン接種はまた、突然変異p53を発現するMeth A 細胞のCTL認識も刺激し得る。rMVAmup53をワクチン接種したマウスからの再刺激した脾細胞は突然変異体 p53 過剰発現Meth Aを認識したが、rMVApp65をワクチン接種したコントロールマウスは認識しなかった。
【0037】
rMVAmup53による単回ワクチン接種の結果CTL応答が増強したので、インビボでのMeth A 腫瘍細胞の増殖に対するrMVAmup53 ワクチン接種の効果を調べることには十分な正当性があった。rMVAmup53の投与は突然変異体 p53を過剰発現する免疫原性腫瘍細胞株であるマウス肉腫 Meth Aの増殖を阻害することが示された。致死量の Meth A 腫瘍細胞を接種され、腹腔内注射によりrMVAmup53をワクチン接種したマウスは3日後にコントロール動物と比べてより遅い腫瘍増殖および高い生存率を示した。ほとんどのワクチン接種マウスは完全に腫瘍を発達させることが出来ず、これらマウスは52日後のMeth Aによる再攻撃に対して耐性であった (Espenschied 2003)。
【0038】
上記の結果は悪性腫瘍によって発現するp53の標的化における新規なrMVAmup53 無細胞ワクチンの有効性を示す。さらなる実験を行って、この効果がCTLA-4遮断薬またはCpG ODN 免疫調節物質の添加によって増強されるかを調べた。ワクシニアウイルスコンストラクトによる免疫化の結果、DCによりウイルスタンパク質が取り込まれ、提示される(Norbury 2002)。流入領域リンパ節において、DCは抗原を未処置のCD8+T細胞に提示し、その結果T 細胞活性化が起こり、続いて免疫応答が伝播される(Norbury 2002)。免疫調節物質実験を、応答の開始とその伝播の両方に取り組むことによって、rMVAp53に対する応答を増強させることができるかを調べるために設計した。
【0039】
腫瘍ワクチンの最適化の1つの強力な戦略は、CTLA-4とリガンドとの結合をブロックする分子を用いることによりT 細胞応答性の負の制御を操作することを含み、この現象は「CTLA-4遮断」と称される。CTLA-4はT 細胞上にみられる細胞表面受容体である。CTLA-4の活性化はT 細胞応答の阻害を導く。CTLA-4は末梢T-細胞寛容の制御に重要な役割を果たし、それはT-細胞活性化を受動的および能動的機構の両方を介して干渉することによる (Egen 2002)。CTLA-4遮断薬の腫瘍関連自己抗原を発現する癌ワクチンと組み合わせての使用は、場合によっては、腫瘍拒絶と寛容の打破をもたらすが、自己免疫が同時に誘導される(Espenschied 2003; Hurwitz 2000; van Elsas 1999)。インビトロでは、CTLA-4遮断はT-細胞活性化閾値を低め、CTLA-4の軽減効果を除く。CTLA-4遮断はまた、Treg 細胞活性をインビボで阻害する(Read 2000)。 GM-CSF産生腫瘍細胞ワクチンと組み合わせた場合、CTLA-4遮断の結果、確立された免疫原性の低いメラノーマ、乳癌、および前立腺癌移植片の拒絶がもたらされる(Hurwitz 1998; Hurwitz 2000; van Elsas 1999)。これは腫瘍関連抗原に対する寛容の打破をともなうプロセスを介して起こる。CTLA-4遮断薬はCTLA-4 受容体に特異的に結合し、CTLA-4のそのカウンター-受容体に対する結合を阻害する分子である。CTLA-4遮断薬は以下のいずれであってもよい:モノクローナルまたはポリクローナル抗体、抗体の断片、ペプチド、有機低分子、ペプチド疑似体(peptidomimetic)、核酸、例えば、干渉RNA (iRNA)またはアンチセンス分子、アプタマー、CTLA-4 リガンド、例えば、CTLA-4 リガンドのB7ファミリーのメンバーからのあらゆるドメイン、ここで該リガンドは好ましくは、免疫細胞上に存在するCTLA-4に結合することが出来、CTLA-4機能をブロックすることが出来る組換え可溶性タンパク質として合成されたものであり得る。抗-CTLA-4抗体は、宿主動物をCTLA-4 タンパク質またはCTLA-4を発現する細胞で免疫することによって作成できる。CTLA-4に対するモノクローナル抗体(抗-CTLA-4 mAb)は常套技術によって産生でき、即ち、ハイブリドーマ細胞と抗-CTLA-4 抗体を産生する哺乳類免疫細胞を融合させることによって産生できる。抗-CTLA-4 mAbの作成に用いられる哺乳類細胞には以下の細胞が含まれる:ラット、マウス、ハムスター、ヒツジ、またはヒト細胞。 抗-CTLA-4 mAbはハイブリドーマ細胞上清または腹水から精製すればよい。抗-CTLA-4抗体は、トランスジェニック動物(Bruggemann 1991; Mendez 1997)またはヒト免疫グロブリンファージディスプレーライブラリー (Winter 1994)を用いて作製されるヒト抗体であってよい。抗-CTLA-4 抗体はまたキメラ抗体およびヒト化 (または「新形態」)抗体も含む。CTLA-4に対するキメラ抗体は、組換え方法を用いて、非-ヒト抗体のCTLA-4 結合ドメインとヒト抗体の定常ドメインを含むように作ればよい。CTLA-4に対するヒト化抗体は、組換え方法により、ヒト抗体構造フレームワーク上に非ヒト抗-CTLA-4 抗体のCDR領域のみを含むように作ればよい(Jones 1986; Low 1986)。非-ヒト領域における個々の残基をヒト抗体フレームワークからの残基と置換すればよい。逆に、ヒト抗体フレームワークにおける個々の残基を非-ヒト抗体からの残基と置換してもよい(Foote 1992)。かかる置換を用いて、ヒト化抗体の結合能を上昇させ、あるいは抗体に対する免疫応答を低減することが出来る。CTLA-4に対するヒト化抗体は、トランスジェニックヒト免疫グロブリン定常領域遺伝子を有する動物の産物であってもよい。組換えDNA技術によってそれらを操作して、CH1、CH2、CH3、ヒンジドメインまたは対応のヒト配列を有するその他のドメインを当該技術分野で公知の方法により置換することができる。
【0040】
非メチル化 CpG (シトシン-フォスフェート-グアニン) モチーフを含むオリゴデオキシヌクレオチドは、ワクチン効力を高める強力な免疫賦活剤である(Krieg 2002)。CpG ODNによる免疫活性化はB 細胞およびプラズマ細胞様(plasmacytoid)樹状細胞におけるToll-様受容体-9 (TLR9)への特異的結合から開始する(Krieg 2002)。DCにおけるTLR9連結の結果、リンパ球、マクロファージ、単球、ナチュラルキラー (NK)、およびT-細胞集団が二次的に活性化する。これはTH1 サイトカイン環境を生成するサイトカインの合成を介して起こる(Krieg 2003)。この結果、NK活性が上昇し、抗原提示が上昇し、およびT細胞援助が起こり、これによって体液性および細胞-媒介免疫応答の両方が増強しうる。さらに、TLR9 連結の結果、DCによるIL-6産生が起こり、これは、CD4+CD25+ Treg 細胞の抑制効果に打ち勝つのに役立つ(Pasare 2003)。CpG ODNの単独での投与は、中程度の抗-腫瘍効果を多数のマウス腫瘍モデルにおいて発揮することが示されている (Carpentier 1999; Kawarada 2001; Ballas 2001; Baines 2003; Sharma 2003)。 CpG ODNはマウスにおける多数の実験的腫瘍ワクチンの有効なアジュバントであることが示されている。それは少なくともフロイントアジュバントと同程度に有効であるが、TH1活性がより高く、毒性が少ない(Chu 1997; Weiner 1997)。CpG ODNはペプチド (Davila 2000; Stern 2002)、タンパク質 (Kim 2002)、DC (Heckelsmiller 2002)、イディオタイプ (Baral 2003)、およびGM-CSF分泌性腫瘍細胞ワクチン (Sandler 2003)の効果を増強しうる。CpG ODNが TH1 応答とNK 細胞の刺激を開始させる能力はおそらくこれらワクチンアプローチおよび以下の記載における、免疫調節物質活性の原因であろう。
【0041】
rMVAmup53ワクチン接種と組み合わせてのCTLA-4遮断薬の投与が有益であるか、または自己免疫疾患を引き起こすかを調べるために、CTLA-4に特異的なモノクローナル抗体(抗-CTLA-4 mAb)を用いた。rMVAmup53と抗-CTLA-4 mAbによるワクチン接種は、確立した、触診可能なMeth A 腫瘍の拒絶に効果があることが示された。高用量のMeth Aを注射され、rMVAmup53と抗-CTLA-4 mAb (9H10)をワクチン接種したマウスは触診可能な腫瘍小結節の形成のすぐ後、完全な腫瘍退行および持続的な腫瘍免疫性を示した。インビボ抗体枯渇研究により、この抗腫瘍効果は主にCD8+、そして程度は低いがCD4+依存的であることが確認された。
【0042】
上記結果は腫瘍特異的ではないことを確認するために、rMVAmup53およびCTLA-4遮断薬免疫調節物質によるワクチン接種を11A-1またはMC-38 腫瘍細胞を注入されたマウスに行った。11A-1は迅速に増殖する悪性細胞株であり、免疫原性は低い。MC-38は結腸癌細胞株である。11A-1またはMC-38腫瘍細胞を注入され、4日後にrMVAmup53および抗-CTLA-4 mAbをワクチン接種したマウスはそれらの腫瘍を拒絶した。抗-CTLA-4 mAbの代わりにCpG ODNを用いた場合も同様の結果が見られた。rMVAmup53とCpG ODNで処理したほとんどのマウスは触診可能な腫瘍を発症せず、持続性腫瘍免疫性を発達させ、60日後の再攻撃を拒絶した。
【0043】
抗-CTLA-4 mAbおよびCpG ODN 免疫調節物質のさらなる可能性のある効果を、両方の免疫調節物質をrMVAmup53と組み合わせて、触診可能な腫瘍を有する11A-1注射マウスに投与することによって調べた。腫瘍拒絶および生存の延長がrMVAmup53と組み合わせて両方の免疫調節物質を与えられたマウスのほとんどにおいて観察された。rMVAと組み合わせて一方の免疫調節物質のみを与えられたマウスは一方で、すべて最終的には腫瘍増殖に屈した。両方の免疫調節物質の組み合わせは単独で作用する免疫調節物質よりもより有益であるだけでなく、その組み合わせ効果は、免疫調節物質の効果の単純な和よりも大きかった。MC 38 腫瘍を担持するマウスにおいても同様の結果が見られた。
【0044】
ヒト p53 配列を含む組換えMVAの効力を調べるために、rMVAhup53をヒトp53をトランスフェクトした4T1(H-2d) 細胞を注射したhupki マウスに投与した。4T1(H-2d)はマウス乳癌細胞株である。4T1 細胞の注射の6日後にマウスにrMVAhup53をワクチン接種し、10日後に再びワクチン接種した。二回目のワクチン接種の際に、CpG ODNおよび抗-CTLA-4 mAbもともに投与した。ワクチンと両方の免疫調節物質で処置されたマウスは、統計的に有意な生存の向上を示した。
【0045】
上記結果は様々な悪性腫瘍型におけるp53を標的とする免疫応答の誘発における新規rMVAmup53またはrMVAhup53 無細胞ワクチンの効力を示すと共に、このワクチンに対する抗-CTLA-4 mAbおよびCpG ODNの免疫調節物質としての効力を示す。したがって、本発明はp53を発現するように操作された組換えMVAウイルス(rMVAp53)を含む組成物を提供する。本発明はさらにrMVAp53の投与による多様なp53-発現悪性腫瘍に対する免疫応答を誘発させる免疫療法を提供する。
【0046】
rMVAp53の対象への導入は当業者に知られたいずれの方法で行ってもよく、効力または安全性について、腫瘍小結節の位置に依存しない。したがって、rMVAp53は送達のいくつかの可能な態様を挙げると、血管内、皮下、腹膜、筋肉内、皮内または経皮注射によって投与できる。rMVAp53は医薬上許容される媒体、例えば通常の生理食塩水、植物油、鉱油、PBS等中に有効な用量にて製剤として調製すればよい。治療用調製物は、生理的に許容される液体、ゲルまたは固体担体、希釈剤、アジュバントおよび賦形剤を含んでいてもよい。添加剤としては殺菌薬、等張を維持する添加剤(例えば、NaCl、マンニトール)、化学的安定性を維持する添加剤(例えば、緩衝剤、保存料)およびその他の成分が挙げられる。非経口投与のためには、rMVAp53は溶液、懸濁液、乳濁液または凍結乾燥粉末として、医薬上許容される非経口媒体と共に製剤すればよい。リポソームまたは非水性媒体、例えば、不揮発性油を用いてもよい。製剤は当該技術分野で知られた方法によって滅菌すればよい。
【0047】
rMVAp53製剤は共刺激物質、例えば、サイトカイン、腫瘍抗原、病原体由来抗原、または免疫調節物質によってさらに増強してもよい。共刺激物質はrMVAp53と組み合わせて免疫応答を直接的または間接的に刺激するいずれの薬剤でもよく、APCまたはT-細胞機能を調節するその能力について選択すればよい。例えば、MVAを操作してGM-CSF、IL-12、またはその他の刺激性サイトカインを発現させ、共刺激物質を産生してもよく、rMVAp53と共刺激物質 (ここで:刺激性サイトカインを発現するMVA)の組み合わせを対象に導入してもよい。処置は抗原提示細胞を刺激する以下のようなサイトカインの投与と組み合わせて行ってもよい。例えば、顆粒球-マクロファージコロニー刺激因子 (GM-CSF)、マクロファージコロニー刺激因子 (M-CSF)、顆粒球コロニー刺激因子 (G-CSF)、インターロイキン 3 (IL-3)、インターロイキン 12 (IL-12)、および当該技術分野で周知のその他のもの。その他の共刺激物質としては、サイトカイン-形質導入腫瘍細胞、例えば、GM-CSFを形質導入された腫瘍細胞、および照射および/またはエキソビボまたはインビボで化学療法薬で処置された腫瘍細胞が挙げられる。化学療法薬または放射療法薬は共刺激物質のさらなる例である。したがって、rMVAp53は当業者に知られた様々な共刺激物質と組み合わせて投与すればよい。
【0048】
製剤はT 細胞の抗原刺激に対する応答を上昇させるのに有効な用量で投与する。T細胞応答の測定は治療される症状によって異なる。T細胞活性の有用な尺度は増殖、サイトカイン、例えば、IL-2、IFNγ、TNFαなどの放出; CD25およびCD69などのマーカーのT細胞発現;および当該技術分野で知られたその他のT 細胞活性の尺度。治療用製剤の用量右派、以下に応じて広範に変動しうる:癌のステージ、投与頻度、投与方法または目的、対象からのrMVAp53のクリアランスおよびその他の因子。投与される用量は以下の公知の因子に応じて変動する。例えば、特定の薬剤の薬力学特性、投与態様および経路、レシピエントの年齢、健康状態および体重、症状の性質および程度、併用療法、治療頻度、および所望の効果。用量は週に1回または2週間に1回ほどの低頻度で投与すればよく、小用量に分けて、毎日、週に2回などを投与して、効果的な用量レベルを維持すればよい。
【0049】
一般に、活性成分の一日用量は約 106-1011 IU (感染単位)/kg 体重とすればよい。内部投与に好適な用量形態は一般に約 106〜1012 IU の活性成分/単位を含む。活性成分は組成物の総重量に対して0.5〜95重量%の範囲でよい。場合によっては、過剰のT 細胞増殖によって治療期間を制限することが望ましいこともある。該制限は患者の治療に対する応答、患者におけるT 細胞数などに応じて経験的に決定される。T細胞の数は患者において当該技術分野で知られた方法によりモニターでき、例えばT細胞特異的抗体による染色やフローサイトメトリーが挙げられる。
【0050】
本発明の好適な態様において、rMVAp53は免疫調節物質、具体的には、CTLA-4遮断薬またはCpG ODNを組み合わせて投与する。rMVAp53およびCTLA-4遮断薬である抗-CTLA-4 mAbの併用投与は、処置せずに放置すると直ちに死に至らしめる進行した腫瘍を退行させることにおいて予期に反し強力である。rMVAp53と CpG ODNとの併用投与についても同じことがいえる。両方の免疫調節物質をrMVAp53と組み合わせて投与した場合に強度はさらに強くなる。さらに、抗-CTLA-4 mAb CpG ODN 免疫調節物質は対象に対して非毒性であり、rMVAp53と組み合わせて投与した場合に腫瘍細胞による致死性の攻撃に対する長期持続性の免疫性を生成することができる。rMVAp53のみの場合と同様に、rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNの対象への導入は、当業者に知られた手順で行えばよく、効力または安全性について腫瘍小結節の位置に依存しない。したがって、rMVAp53、抗-CTLA-4 mAbおよびCpG ODNは、可能性のあるいくつかの送達態様を挙げると、血管内、皮下、腹膜、筋肉内、皮内または経皮注射によって投与できる。rMVAp53、抗-CTLA-4 mAbおよびCpG ODNは共に投与しても別々に投与してもよく、いずれの順で逐次に投与してもよく、同じ投与経路で投与しても異なる経路で投与してもよい。rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNは医薬上許容される媒体中で有効用量において製剤として調製してもよく、医薬上許容される媒体としては例えば、通常の生理的食塩水、植物油、鉱油、PBS等が挙げられる。治療用調製物は生理的に許容される液体、ゲルまたは固体担体、希釈剤、アジュバントおよび賦形剤を含んでいてもよい。添加剤としては、殺菌薬、等張性を維持する添加剤、例えば、NaCl、マンニトール;および化学安定性を維持する添加剤、例えば、緩衝剤および保存料およびその他の成分を含んでいてもよい。 rMVAmup53 + 抗-CTLA-4 mAb および/または CpG ODNは混合物または単一の薬剤として投与してもよい。非経口投与のために、抗-CTLA-4 mAbおよびCpG ODNは医薬上許容される非経口媒体と共に、溶液、懸濁液、乳濁液または凍結乾燥粉末として製剤すればよい。リポソームまたは非水性媒体、例えば、不揮発性油を用いてもよい。製剤は当該技術分野で知られた技術によって滅菌すればよい。
【0051】
rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNの組み合わせは共刺激物質、例えば、サイトカイン、腫瘍抗原、または病原体由来抗原によってさらに増強してもよい。共刺激物質はrMVAp53またはrMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNと組み合わせて免疫応答を直接的または間接的に刺激するいずれの薬剤でもよい。例えば、MVAを操作してGM-CSF、IL-12、またはその他の刺激性サイトカインを発現させ、共刺激物質を産生してもよく、rMVAp53と共刺激物質 (ここで:刺激性サイトカインを発現するMVA)またはrMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNと共刺激物質の組み合わせを対象に導入してもよい。処置は抗原提示細胞を刺激する以下のようなサイトカインの投与と組み合わせて行ってもよい。例えば、顆粒球-マクロファージコロニー刺激因子 (GM-CSF)、マクロファージコロニー刺激因子 (M-CSF)、顆粒球コロニー刺激因子 (G-CSF)、インターロイキン 3 (IL-3)、インターロイキン 12 (IL-12)、および当該技術分野で周知のその他のもの。その他の共刺激物質としては、サイトカイン-形質導入腫瘍細胞、例えば、GM-CSFを形質導入された腫瘍細胞、および照射および/またはエキソビボまたはインビボで化学療法薬で処置された腫瘍細胞が挙げられる。化学療法薬または放射療法薬は共刺激物質のさらなる例である。したがって、rMVAp53は単独でまたは抗-CTLA-4 mAb および/または CpG ODNと組み合わせて当業者に知られた様々な共刺激物質と組み合わせて投与すればよい。
【0052】
治療用製剤の用量は、以下に応じて広範に変動しうる:癌のステージ、投与頻度、投与方法または目的、対象からのrMVAp53、抗-CTLA-4 mAbおよびCpG ODNのクリアランスおよびその他の因子。投与される用量は以下の公知の因子に応じて変動する。例えば、特定の薬剤の薬力学特性、投与態様および経路、レシピエントの年齢、健康状態および体重、症状の性質および程度、併用療法、治療頻度、および所望の効果。用量は週に1回または2週間に1回ほどの低頻度で投与すればよく、小用量に分けて、毎日、週に2回などを投与して、効果的な用量レベルを維持すればよい。
【0053】
一般に、活性成分(抗体)の一日用量は約0.1 〜 100 mg /kg 体重とすればよい。内部投与に好適な用量形態は一般に約 0.1 mg 〜 500 mgの活性成分/単位を含む。活性成分は組成物の総重量に対して0.5〜95重量%の範囲でよい。場合によっては、過剰のT 細胞増殖によって治療期間を制限することが望ましいこともある。該制限は患者の治療に対する応答、患者におけるT 細胞数などに応じて経験的に決定される。T細胞の数は患者において当該技術分野で知られた方法によりモニターでき、例えばT細胞特異的抗体による染色やフローサイトメトリーが挙げられる。製剤はT 細胞の抗原刺激に対する応答を上昇させるのに有効な用量で投与する。T 細胞応答の判定は治療すべき症状によって異なる。T 細胞活性の有用な尺度は増殖、サイトカイン、例えば、IL-2、IFNγ、TNFαなどの放出; CD25およびCD69などのマーカーのT 細胞発現;およびその他の当該技術分野に知られたT 細胞活性の尺度である。
【0054】
本発明はさらに当業者にp53-発現悪性腫瘍に対する免疫応答を誘発するための免疫治療計画の準備を可能とするキットを提供する。キットの例は、rMVAp53、CTLA-4遮断薬および/またはCpG ODN、これら化合物は対象におけるp53-発現悪性腫瘍に対する免疫応答を誘発するために用いるという指示書を含む。キットはさらに1以上の医薬上許容される担体を含んでいてもよい。投与する場合、キットの組成物は医薬上許容される調製物において投与される。「投与」、「投与する」および「導入する」の語は、本発明の組成物をp53-発現悪性腫瘍の治療または予防を必要とする個体に対して医薬として提供することをいう。主成分または活性成分として本発明の組成物を含むこの医薬は、広範な治療用量形態で局所、経口、全身、局部、および非経口投与用の常套の媒体中で投与すればよい。したがって、本発明のキットは非経口投与用の組成物を提供し、これは許容される担体、好ましくは水性担体中に溶解または懸濁された組成物の溶液を含む。組成物は医薬上許容される補助剤を適当な生理条件に応じて所望により含んでいてもよく、例えば、pH調整剤および緩衝剤、浸透圧調整剤、湿潤剤など、例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウム、モノラウリン酸ソルビタン、オレイン酸トリエタノールアミンおよびその他の多くのものが挙げられる。非経口投与のための化合物を調製する実際の方法は当業者に知られているか明らかであり、より詳細には、例えば、Remington: The Science and Practice of Pharmacy ("Remington's Pharmaceutical Sciences") Gennaro AR ed. 20th edition、2000: Williams & Wilkins PA、USAに記載されており、これは引用により本出願に含める。
【0055】
かかる調製物は常套的に医薬上許容される濃度の塩、緩衝剤、保存料、許容される担体、補助的免疫増強剤、例えば、アジュバントおよびサイトカインおよび所望によりその他の治療薬を含んでいてもよい。本発明の調製物はすべて有効量で投与される。有効量は医薬調製物の単独、またはさらなる用量と組み合わせて所望の応答を刺激する量である。癌の治療の場合、所望の応答は癌の発症または進行の阻害または癌を退行させることである。これは一時的に疾患の進行を遅くさせることも含むが、しかしより好ましくは、疾患の進行を永久に停止させることを含む。これらの所望の応答は常套方法によってモニターしてもよいし、本明細書に記載する本発明の診断方法にしたがってモニターしてもよい。投与態様に応じて104 IU/kg 〜 1011 IU/kgの免疫原の用量範囲が有効であると考えられる。好ましい範囲は、106 IU〜109 IU/kgであると考えられる。絶対量は様々な因子に依存し、例えば、投与に選択された組み合わせ、投与が単一用量であるか複数用量であるか、そして個々の患者パラメーター、例えば、年齢、身体状態、身長、体重および疾患のステージが挙げられる。これらの因子は当業者に周知であり、常套の実験によって決定しうる。
【0056】
以下の実施例は特許請求の範囲に記載の本発明をさらに説明するために提供するものであり、本発明の範囲を限定するものではない。特定の材料が言及される範囲は単に例示の目的であり、本発明を限定する意図のものではない。特に断りのない限り、一般的クローニング方法、例えば、Sambrook et al.、Molecular Cloning、Cold Spring Harbor Laboratory (2001)、Ausubel et al. (Eds.) Current Protocols in Molecular Biology、John Wiley & Sons (1986、2000)に記載のものを用いる。当業者であれば、本発明を実施することなく、本発明の範囲を逸脱することなく、同等の手段または反応材料を開発することが出来よう。
【0057】
多くの改変が本発明に拘束されつつここに記載した手順においてなすことができることが理解できよう。同様に、遺伝コードの縮重のために、ここに開示のものと同等のコドンを有する核酸配列がここに開示されたものと機能的に同等または同一のタンパク質をコードすることが理解される。かかる改変は本発明の範囲内に含まれると言うことが本発明者らの意図である。
【実施例】
【0058】
実施例
材料および方法
動物
雌性6-8週齡Balb/c、C57BL/6、B6.129S2-IL6tm1Kopf (IL-6-/-、およびIFN-γ ノックアウト (IFN-γKO) マウス(Balb/cバックグラウンド)は、The Jackson Laboratory (Bar Harbor、Maine)から得た。TLR9-/- マウスは、Dr. Shizuo Akira (Osaka University、Osaka、Japan)から譲り受けた。マウスを特定病原体除去環境に維持した。すべての研究は、Research Animal Care Committee of the City of Hope National Medical Centerにより認可を受け、AAALACガイドラインにしたがって行った。
【0059】
細胞株
CV-1 (Kit 1965)、TK- (Berson 1996)、および仔ハムスター腎臓細胞(BHK-21) (Macpherson 1962)をAmerican Type Culture Collection (ATCC) (Manassas、VA)から購入し、非必須アミノ酸、L-グルタミン、および10% FCS を追加したMEM中で培養した。11A-1 (Selvanayagam 1995)は、Dr. R.L. Ullrich (University of Texas Medical Branch、Galveston、TX)から譲り受けた。Hek 293 細胞およびp53null 10.1 細胞は、Dr. K.K. Wong および Dr. Susan Kane (City of Hope National Medical Center、Duarte、CA)から譲り受けた。MC-38 (Tan 1976)は、Dr. S.A. Rosenberg (National Cancer Institute、Bethesda、MD)から譲り受けた。Meth A 肉腫細胞(Meth A) (DeLeo 1977)は、Dr. L. J. Old (Memorial Sloan-Kettering Cancer Center、New York、NY)から譲り受けた。Meth Aを腹水腫瘍として継代した。細胞を収集し、計数し、使用前にPBSで洗浄した。Meth A、11A-1、およびMC-38 腫瘍細胞株の特徴を以下の表に要約する:
【0060】
【表1】

【0061】
抗体
抗-CD4 (GK1.5) (Dialynas 1983)および抗-NK1.1 (PK136) (Koo 1984)をATCCから購入した。抗-CD8 (H35) (Miconnet 2001)および抗-CTLA-4 mAb (9H10) (Krummel 1995)はJames P. Allison (University of California、Berkeley、CA)から譲り受けた。抗体はCELLine Device (BD Biosciences、Bedford、MA)を用いて産生した。IgG 抗体はタンパク質G-セファロース (Amersham、Uppsala、Sweden)に吸収し、次いで0.1M Glycine-HCl、pH 2.7での溶出により精製した。産物を次いでリン酸緩衝生理食塩水 (PBS)で透析し、Centriplus 遠心ろ過装置 (Millipore、Bedford、MA)を用いて濃縮した。コントロールシリアンハムスター IgGをJackson Immuno Research (West Grove、PA)から得た。
【0062】
ウイルスコンストラクト
マウス p53を発現するrMVA (rMVAmup53):
野生型 MVA (wtMVA)をDr. Bernard Moss および Dr. Linda Wyatt (National Institutes of Health Bethesda、MD)から得た。mup53 を含む組換えMVA (rMVA)の作成のためのwtMVA ストックを特定病原体除去ニワトリ胚線維芽細胞(SPF/CEF)上で増殖させる。wtMVA ストックを免疫染色により力価測定し、アリコートに分け、-80℃で保存する。
【0063】
マウス p53 (mup53)はヒトp53と類似であり、80% 配列相同性を有する(Halevy 1991; Sukumar 1995)。全長野生型mup53のmRNA コード配列を配列番号1に示す。マウスとヒト p53とのこの相同性のレベルにより、マウス系は良好なp53に対する寛容に打ち勝つ免疫学的アプローチの評価の前臨床モデルとなる。マウス p53を発現するrMVA をEspenschied 2003に記載のようにwtMVAとマウス p53 インサートを含むpMCO3 挿入ベクターとの相同組換えにより作成した。マウス野生型p53の全長cDNAをマウス脾細胞から得たmRNAのPCRにより増幅した。マウス p53 PCR 産物をMVA 発現ベクター pMCO3のクローニング部位にライゲーションした(これもDr. Moss および Dr. Wyattから得た)。このベクターはMVAゲノムのデリーションIIIに挿入する配列を含み、また、gus(大腸菌β-グルクロニダーゼ)オペロンをスクリーニングの目的で含む(Ourmanov 2000)。組換えMVAの作成は、BHK-21 細胞の単層上で達成した (Espenschied 2003)。簡単に説明すると、BHK-21 細胞にLipofectin (Invitrogen、Carlsbad、CA)を用いて20 μgのプラスミド DNAをトランスフェクトし、wtMVAをmoi 0.01にて感染させた。感染細胞を48時間インキュベートし、次いで収集し、ペレットにし、3サイクルの凍結乾燥および超音波処理に供し、細胞を溶解した。マウス p53を発現するrMVAウイルス (rMVAmup53)をX-GlcA (5-ブロモ-4-クロロ-3-インドリルβ-D-グルクロニド、Sigma-Aldrich、St Louis、MO)の添加によるgus 発現によってスクリーニングした。10ラウンドの精製後、rMVAmup53をBHK-21 単層上に播いた。rMVAmup53 力価はVectastain Elite ABC Kit (ベクター Laboratories、Burlingame、CA)を用いて感染培養の免疫染色によって測定した。
【0064】
ヒトp53を発現するrMVA (rMVAhup53):
ヒト p53を発現するrMVA (rMVAhup53)の2種類のコンストラクトを作った。全長野生型 hup53をコードするmRNA 配列を配列番号2に示す。第一のものはpLW51 挿入プラスミドを用いて作り、第二のものはpLW22 挿入プラスミドを用いて作った。第一のコンストラクトを作るために用いたwtMVAをSPF/CEF上で増殖させた。第二のコンストラクトを作るために用いたwtMVAをBHK-21 (BHK) 細胞上で増殖させた。wtMVA ストックを免疫染色により力価を測定し、アリコートに分け、-80℃で保存した。
【0065】
pLW51を第一の rMVAhup53 コンストラクトを作るための挿入プラスミドとして用いた。pLW51は4つの重要な特徴を有する。第一にそれはデリーションIII のMVA 隣接領域を含み、それによって相同組換えを介してMVAのデリーションIII 領域に挿入される。第二に、それは発色スクリーニングマーカー遺伝子、β-グルクロニダーゼ (gus)をP11と称されるワクシニアプロモーターの制御下に含む。第三に、それはMVA 配列から構成される2つの直接反復を(DR1およびDR2と称される) gus スクリーニングマーカー遺伝子に隣接して含むため、gus遺伝子を組換えMVAから除くことが可能である。最後に、それは2つのワクシニアプロモーター (PSYNおよびP7.5) と2つのマルチクローニング部位(MCS)を含むため、2つの別々の外来遺伝子をPSYNおよびP7.5 プロモーターの制御下に挿入することが可能である。第一の MCSは初期/後期 PSYN プロモーターの下流にあり、第二の MCSは初期/後期 PmH5 プロモーターを用いる。これにより、隣接する直接繰返し配列のセットを介した組換えによりgus マーカー遺伝子を除くことが可能である。最初のrMVA ストックの作成をBHK細胞について以前に記載されている方法を用いてCEFに対して行ったが、医薬品安全性試験実施基準 (GLP) 条件とするために改変した。正しい組換えが起こるかを確認するために約40-50の病巣が第一ラウンドのスクリーニングから引き出され、その後5-10を各次なるラウンドにおいて引き出され得る。各選択ラウンドの後、免疫染色または免疫蛍光のいずれかを各プラグ(plug)に対して行って、プラグがhup53遺伝子を発現するかを確認する。細菌遺伝子マーカーのウイルスからの除去を達成するためにhup53を発現する精製MVAを、24 ウェルプレート中に低希釈度で播く。gus遺伝子を示す着色反応を有さないウェルをさらにhup53 遺伝子産物の存在について分析する。これは細胞からのウイルスの回収を可能とする条件を用いて抗体染色によって達成する。着色反応の非存在下でhup53 免疫染色を示すウェルをさらに増殖させて正確な表現型を確認する。gusマーカーの非存在のスクリーニングの最終ラウンドから引き出されるウイルスプラグの一部をCEFの100 mm 組織培養皿で拡張する。この後DNA抽出およびPCR分析を行う(以下に記載)。
【0066】
pLW22を第二の rMVAhup53 コンストラクトの作成のための挿入プラスミドとして用いた。pLW22は相同組換えを介するMVAへの挿入を可能とするMVA 隣接領域を有する。それはまた、発色スクリーニングマーカー遺伝子、β-ガラクトシダーゼを有する。wt hup53をコードするDNAを得るために、大腸菌におけるHp53B プラスミドをATCCから得た(#57254)。Hup53をpHp53B プラスミドから配列番号3のフォワードプライマーと配列番号4のリバースプライマーを用いて増幅した。増幅した wt hup53 DNAをpLW22 ベクターの制限部位Pme-とAsc-1の間に挿入し、pLW22-hup53を作成した。pLW22-hup53のプラスミド配列を配列番号5に示す。
【0067】
rMVAの作成はBHK細胞の単層上で行った。BHK細胞にLipofectin (Invitrogen、Carlsbad、CA)を用いて20μgのプラスミド DNAをトランスフェクトし、wtMVA、moi 0.01で感染した。感染細胞を48 時間インキュベートし、収集し、ペレットにし、三回の凍結乾燥サイクルおよび超音波処理に供し、細胞を溶解した。hup53を発現するrMVAをBluo-galTM基質(Sigma-Aldrich、St Louis、MO) (Chakrabarti 1985)の存在の添加によりβ-gal 発現についてスクリーニングした。10ラウンドの精製の後、rMVAhup53を BHK 単層上に広げた。rMVA 力価は免疫染色感染培養物によってVectastain Elite ABC kit (Vector Laboratories、Burlingame、CA)を用いて測定した。
【0068】
両方のコンストラクトについて、標準的DNA抽出を行う。50μLの細胞可溶化液のエタノール沈殿により、wtMVAの汚染が無いことを確認するためのPCR反応を行うのに十分なDNAが得られた。一セットのPCR プライマーを遺伝子が挿入された組換え部位の隣接領域の外側に設計する。非修飾wtMVA 配列の存在により500 bpのPCR産物が生じるが、hup53を含む配列の挿入は標準的 PCR条件下では増幅するのが通常困難であるより大きな断片(>6 kb)を有する。第二のセットのPCR プライマーはhup53 インサート内の配列を増幅するように設計する。hup53 インサートの存在により、300 bpのPCR 産物が生じる。PCR サンプルを1% アガロースゲルで泳動し、さらなるスクリーニングが残りのwtMVAを除去するために必要であるかを判定するために分析する。ヒト p53を含む精製MVAの例は完全に均質であることが示された(図1)。
【0069】
pp65を発現するrMVA (rMVApp65):
CMV テグメント・タンパク質、pp65を発現するrMVA (rMVApp65)を、rMVAmup53 (Gibson)の構築に用いたものと類似の技術を用いて構築した。
【0070】
マウス p53またはpp65を発現するrVV:
マウス 野生型p53またはpp65 (rVVp53、rVVpp65)を発現する組換えウエスタンリザーブ株ワクシニアウイルスを公表された技術を用いて構築した(Diamond 1997)。
【0071】
マウス p53を発現するrAd:
野生型マウス p53を発現する組換えアデノウイルス(rAd-mup53)をpAd Easyシステム(He 1998)を用いて構築した。pAd Track-CMVとpAd Easy-1 プラスミドとは両方 、Dr. Bert Vogelstein (Johns Hopkins Oncology Center、Baltimore、MD)から譲り受けた。野生型マウス p53 cDNAをCMV プロモーター (p53-pAd Track-CMV)とともに緑色蛍光タンパク質 (GFP)を含むpAd Track-CMV シャトルベクターのBgl II部位とXba I 部位にクローニングした。p53-pAd Track-CMV をpAd Easy-1 プラスミドと共にBJ5183 細胞に共形質転換し、相同組換えによりp53 組換えアデノウイルスコンストラクトを作成した。組換え体におけるp53遺伝子の存在はDNA 配列決定により確認した。p53 組換えアデノウイルスコンストラクトをPac Iで切断し、HEK-293 細胞にトランスフェクトした。rAd-mup53をトランスフェクションの5日後に収集し、p53タンパク質発現をウェスタンブロットにより確認した。アデノウイルスをHEK-293細胞上に広げ、塩化セシウム勾配によって精製した。精製ウイルスをPBSで透析し、HEK-293 細胞上で力価を測定し、20%グリセロール中で-80℃で保存した。
【0072】
オリゴデオキシヌクレオチド(ODN)
CpG モチーフを有する合成ODN 1826 (配列番号6)および非-CpG ODN 1982 (配列番号7) (Moldoveanu 1998)をヌクレアーゼ耐性ホスホロチオエート骨格を用いてTrilink (San Diego、CA)によって合成した。ODNのNa+塩を10 mM Tris (pH 7.0) 1 mM EDTA中5 mg ml-1に再懸濁し、50 μlのアリコートとして-20℃で保存し、注射の前に0.9% 塩化ナトリウム水溶液中に希釈した。
【0073】
実施例1: rMVAmup53によるマウスp53タンパク質の発現:
rMVAmup53による感染後のマウス p53タンパク質の発現を分析し、組換えウイルスからのその発現の忠実度および程度を調べた。rMVAmup53で感染したBHKまたはHEK 293 細胞から可溶化液を調製し、SDS-PAGEおよびウェスタンブロッティングに供した。標準的ウェスタンブロッティング技術をECL ウェスタンブロットキット(Amersham Pharmacia Biotech、England)を用いて行った。サンプルを精製マウス 抗-p53 モノクローナル抗体、PAb 122 (Gurney 1980)とともにインキュベートし、次いで、ECL ウェスタンブロットキット中のペルオキシダーゼ標識ヤギ抗-マウス二次抗体とインキュベーションした。rMVAmup53で感染したBHK細胞のウェスタンブロット分析は豊富なp53 発現を示した(図 2)。その他のウイルスおよび細胞内形態と比較してrMVAmup53によって示された顕著な発現レベルはそのワクチン接種プロトコールにおける有用性を示す。図 1に示すように、rMVAmup53 レーンにおける容積はその他のレーンにおいてゲルにアプライされた容積の80-160分の1であったが、バンドの強度は数倍強かった。これはrMVAmup53による非常に高レベルのp53 発現を示す。Meth A 細胞をポジティブコントロールとして用い、HCMV IE1 エキソン 4 rMVAで感染したBHK細胞をネガティブコントロールとして用いた。Meth Aは Balb/c由来の、腫瘍原性 3-メチルコラントレン誘導性肉腫であり、突然変異p53を過剰発現する。53 キロダルトンバンドが、p53過剰発現Meth A 肉腫とrMVAmup53 感染BHK細胞の両方に観察された(図 1)。これはHCMV IE1 エキソン 4-rMVA 感染BHK細胞において検出可能なp53発現がないことと対照的である。強いp53 発現はrMVAmup53で感染したBHK細胞において蛍光顕微鏡によっても観察された(データ示さず)。
【0074】
実施例2: rMVAmup53によるp53-特異的CTL応答のインビトロでの生成:
ウイルス性および腫瘍関連抗原を発現するrMVA によるマウスのワクチン接種の結果、抗原特異的CTL応答が増強する。この実施例の1つの目的は、rMVAmup53によるワクチン接種がp53 寛容を打ち破り、p53-特異的CTLを生成させることができるかを調べることであった。マウスに5 x107 pfuのrMVAmup53またはrMVApp65のいずれかを腹腔内にワクチン接種した。2週間後、脾臓を収集して分離し、脾細胞を洗浄し、計数した。脾細胞をインビトロで6日間 rAd-mup53またはrMVAmup53で感染した同系 LPS 芽球で再刺激した。wt p53を過剰発現するNa-51CrO4-標識化標的細胞を96 ウェルプレートにエフェクターとともに三連で、様々なエフェクター:標的比で、200μlの完全培地中に添加した。プレートを4 時間37℃でインキュベートし、上清を収集し分析した。パーセント特異的溶解を次式を用いて計算した: パーセント特異的放出 = (実験的放出 自発的放出) / (全放出 - 自発的放出) X 100。rMVAmup53をワクチン接種した脾細胞は、wt p53を過剰発現する標的細胞を認識し、溶解した(図 3)。一方、rMVApp65をワクチン接種したマウスからの脾細胞は激しいpp65 特異的CTL応答を刺激するが、p53を過剰発現する標的は認識せず(図 3B)、リンパ球応答の特異性を示した。rMVAmup53 ワクチン接種は突然変異p53を担持する細胞株であるMeth AのCTL認識も刺激することが出来る。rMVAmup53をワクチン接種した再刺激脾細胞は突然変異体 p53を過剰発現するMeth A 細胞を認識したが、rMVAmup53をワクチン接種した脾細胞は認識しなかった(図 3c)。
【0075】
実施例3: インビボ rMVAmup53 腫瘍攻撃実験:
rMVAmup53による単回ワクチン接種の結果、CTL応答が増強したので、腫瘍細胞のインビボでの増殖に対するrMVAmup53 ワクチン接種の効果を調べるのには十分な正当性があった。
【0076】
統計的方法
初期に屠殺する必要がある、いくらかの腫瘍の増殖速度の実験のために、増殖曲線はlog順位検定を用いて一定のサイズになるときまでに比較した。1つの群のその他の全ての群との比較は単一のオムニバス検査の後に行った。細胞枯渇実験のために、すべてのマウスを一定の時間追跡し、最終的な腫瘍サイズは、3以上の群がある場合には有意Kruskal-Wallis検定の後にウィルコクスン順位和検定によって比較した。生存実験のためには、log順位検定を用いた。
【0077】
rMVAmup53 対Meth A 細胞
6週齡雌性 Balb/c マウスに、皮下 (s.c.)経路によって左脇腹に5 x105 Meth A 細胞を注射した。Meth A 細胞を皮下注射したマウスには迅速に増殖する線維肉腫が発生し、21 日以内にマウスのほとんどが死に至った(図 3)。3日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内 (i.p.) 注射によってワクチン接種した。ネガティブコントロールマウスには5 x107 rMVApp65またはPBSを注射した。皮下腫瘍のサイズをキャリパーを用いて3次元で週に2回測定した。rMVAmup53 処置動物における腫瘍は、コントロール動物と比較してかなりゆっくりと増殖した。14日目、rMVAmup53 処置群(n=16)についての平均皮下腫瘍体積はrMVApp65 (n=16)およびPBS (n=12) コントロールの両方と比較して劇的に小さかった(22mm3対348 mm3、p<0.001および 22mm3対252mm3、p<0.001、スチューデントt検定)。rMVAmup53 処置動物の生存もいずれのコントロール群と比較しても有意に延長していた(図 4)。16匹のrMVAmup53 免疫化マウスのうち12匹ではまったく腫瘍を発達させることができなかった。この12匹の腫瘍の無いrMVAmup53 処置動物を52日目に5 x105 Meth A 腫瘍細胞で再攻撃した。すべての動物は30日の観察期間にわたって腫瘍が無いままであった(データ示さず)。
【0078】
rMVAmup53 + 抗-CTLA-4 mAb対Meth A 細胞
腫瘍ワクチンを至適化する1つの強力な戦略には、CTLA-4のリガンドへの結合をブロックする抗体を用いるT 細胞応答性の負の制御が含まれる。この現象はCTLA-4遮断と称される。抗-CTLA-4 mAbの腫瘍関連自己抗原を発現する癌ワクチンとの組み合わせての使用は、場合によっては寛容に打ち勝つ腫瘍拒絶と自己免疫の誘導をもたらす。それゆえ、CTLA-4に特異的なmAbをrMVAmup53 ワクチン接種に加えて、それがインビボでMeth Aに対する抗-腫瘍活性を協同および増強するかを調べた。CTLA-4遮断のみの強力な抗腫瘍効果に打ち勝つためにより厳しい腫瘍モデルを設計した。6週齡 Balb/c マウスの左脇腹に5 x105 Meth A 細胞ではなく、106 Meth A 細胞を皮下注射し、処置を触診可能な腫瘍小結節が同定されるまで延長した(6日目)。このより厳しいモデルはCTLA-4遮断の効果に打ち勝ち、抗-CTLA-4 mAb処置にもかかわらずほとんどのマウスにおいて迅速に致死性の腫瘍が生じた(図 5)。7日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内注射した。コントロールは上記の通りとした。抗-CTLA-4 mAb 抗体またはコントロールハムスター Abを6、9および12日目にそれぞれ 100、50および50μgの用量で腹腔内注射した。rMVAmup53 + 抗-CTLA-4 mAbで免疫した14匹のマウスのうち11匹が腫瘍を拒絶し、60日の観察期間の持続中腫瘍無しで生存した(図 5)。一方、rMVApp65およびコントロール抗体で処置したマウは進行性腫瘍によりすぐに死にいたり(図 5)、PBS処置コントロールも同様であった(データ示さず)。11匹の腫瘍の無い、rMVAmup53 + 抗-CTLA-4 mAb 処置マウスは60日目の106 Meth A 腫瘍細胞での再攻撃も拒絶し、30日間の観察期間の持続中、腫瘍の無いままであった (データ示さず)。
【0079】
rMVAmup53 + 抗-CTLA-4 mAb対11A-1 細胞
6週齡 Balb/c マウスの左脇腹に2 x 106 11A-1 細胞を皮下注射したた。11A-1は迅速に増殖する免疫原性の弱い悪性細胞株である。106の放射線照射した11A-1 腫瘍細胞をワクチン接種したマウスは11A-1による続く攻撃を拒絶することができなかった (データ示さず)。抗-CTLA-4 mAbまたはコントロールハムスター抗体を4、7、および10日目に100、50、および50μg/用量でそれぞれ腹腔内注射した。5日目に、マウスに5 x107 pfuの rMVAmup53、5 x107 MVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍を三次元でキャリパーを用いて週に2回測定した。rMVAmup53 + 抗-CTLA-4 mAbをワクチン接種したマウスはそれら腫瘍を拒絶した(図 6)。抗-CTLA-4 mAbのみまたはコントロール MVA ワクチンで処置した動物は迅速に進行性致死性腫瘍を発達させた(p = 0.00044、抗-CTLA-4 mAb 遮断を伴うrMVAmup53をコントロール群と比較)。
【0080】
rMVAmup53 + 抗-CTLA-4 mAb 対 MC-38 細胞
6週齡 C57BL/6 マウス、TLR9-/-、またはIL-6-/- マウスの左脇腹に1 x 106 MC-38 細胞を皮下注射した。抗-CTLA-4 mAbまたはコントロールハムスター抗体を4、7および10日目にそれぞれ100、50、および50μg/用量で腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53、5 x107rMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍をキャリパーを用いて三次元で週に2回測定した。rMVAmup53 + 抗-CTLA-4 mAbをワクチン接種したマウスはそれら腫瘍を拒絶したが、抗-CTLA-4 mAbのみ、またはコントロール MVA ワクチンで処置したマウスは迅速に進行性腫瘍を発達させた(p = 0.0001、抗-CTLA-4 mAb をともなうrMVAmup53をコントロール群と比較) (図 7)。
【0081】
rMVAmup53 + CpG ODN 対 11A-1 細胞
CpG ODN処置は多くの実験腫瘍ワクチンモデルにおいて有効な免疫調節物質であることが示されている(Krieg 2002)。マウスを上記のように11A-1 腫瘍で攻撃した。15 nmoles の CpG ODNまたは非-CpG ODN コントロールを4、9、および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfuの rMVAmup53、5 x107 rMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍をキャリパーを用いて三次元で週に2回測定した。rMVAmup53とCpG ODNはそれぞれ別々にわずかな腫瘍増殖の低減をもたらしたが、すべての動物は次第に致死性の腫瘍を発達させた。CpG ODNとrMVAmup53の組み合わせてのワクチン接種の結果、腫瘍増殖が有意に低減した(p = 0.00002) (図 8)。rMVAmup53 + CpG ODNで処置した8匹の動物のうち6匹は触診可能な腫瘍を発達させず、腫瘍免疫性が持続し、60日目の11A-1による再攻撃を拒絶した(データ示さず)。
【0082】
rMVAmup53 + CpG ODN対Meth A 細胞
11A-1についてのものと類似の腫瘍拒絶のパターンがBalb/c マウスにおける初期に確立したMeth A 腫瘍の処置後にみられた(p = 0.0015) (図 9)。
【0083】
rMVAmup53 + CpG ODN 対 MC-38 細胞
rMVAmup53 ワクチン接種に対するCpG ODNの免疫調節効果が株特異的ではないことを示すために、ワクチン接種戦略を初期に確立したMC38 大腸癌を担持するC57BL/6 マウスにおいて繰り返した。rMVAmup53 + CpG ODNによるワクチン接種の結果、腫瘍増殖が有意に抑制された(p = 0.0004) (図 10)。
【0084】
rMVAmup53 + 抗-CTLA-4 mAb + CpG ODN 対 11A-1 細胞
CpG ODNと抗-CTLA-4 mAbのrMVAmup53 ワクチン接種に対する強力な追加的効果を評価するためにより厳しい腫瘍モデルを設計した。6週齡 Balb/c マウスの左脇腹に2 x 106 11A-1 細胞を皮下注射し、触診可能な腫瘍が存在するまで2週間追跡した。抗-CTLA-4 mAb またはコントロールハムスター抗体を14、17、および20日目に、100、50、および50μg/用量にて腹腔内注射した。15 nmolesのCpG ODNを14、19、および24日目に腹腔内注射した。15日目に、マウスに5 x107 pfu のrMVAmup53、5 x107 MVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。
【0085】
抗-CTLA-4 mAbまたはCpG ODN 免疫調節物質と組み合わせたrMVAmup53 ワクチン接種の結果、生存の延長が起こったが、すべての動物において最終的には、進行性腫瘍が増殖した。抗-CTLA-4 mAbおよびCpG ODN 投与とrMVAmup53 ワクチン接種の組み合わせの結果、ほとんどの処置動物において腫瘍拒絶および生存の延長が起こった(図 11)。抗-CTLA-4 mAbおよびCpG ODNの組み合わせはCpG ODNのみ(p = 0.02)または抗-CTLA-4 mAbのみ(p = 0.01)と比べてよりよい免疫調節活性を提供した。抗-CTLA-4 mAbおよびCpG ODNの組み合わせ投与の効果は、60日目の生存に関して、両方の免疫調節物質の単独での単純な添加と比べてより大きい利益をもたらす。
【0086】
rMVAmup53 + 抗-CTLA-4 mAb + CpG ODN 対 MC-38 細胞
類似のパターンがMC 38 腫瘍を担持するC57BL/6 マウスにおいてみられた(図 12)。MC-38 腫瘍を担持するC57BL/6 マウスをrMVAmup53 +抗-CTLA-4 mAbおよびCpG ODNの組み合わせで11A-1について記載したように処置した。この腫瘍モデルにおいて、抗-CTLA-4 mAbおよびCpG ODNの組み合わせはCpG ODNのみ(p = 0.002)または抗-CTLA-4 mAbのみ(p = 0.001)よりも良好な免疫調節活性を提供した。両方の腫瘍モデルにおけるこの組み合わせ効果は単に相加効果ではない。というのは、CpG ODN と抗-CTLA-4 mAbはともにすでに最大効力の用量で投与されているからである。両方の免疫調節物質を少なくとも2種類の腫瘍においてともに用いた場合にみられた活性の顕著な上昇は、併用療法がヒトにおいて保証されることの研究をさらに示唆する。
【0087】
実施例4: 抗-CTLA-4 mAbおよびCpG ODN 免疫調節効果の細胞による要求:
抗-CTLA-4 mAbおよびCpG ODNの免疫調節効果の細胞による要求を調べるために、Balb/cマウスからワクチン接種の前にCD4+、CD8+、またはNK 細胞を枯渇させた。枯渇は200μg のCD4+、CD8+、またはNK1.1 細胞 特異的 mAb、あるいはコントロール mAbの腹腔内注射により達成した。注射は、-1、1、3、4、6、8および15日目に行い、動物が死ぬまで7日ごとに投与を維持した。この投与計画は処置動物の末梢血のフローサイトメトリーに基づくと、(>95%) Balb/c マウスからCD4+、CD8+、またはNK 1.1 細胞を枯渇させることが示された(データ示さず)。
【0088】
rMVAmup53 ワクチン接種に対するCTLA-4遮断の免疫調節効果の細胞による要求をBalb/c マウスにおけるMeth A 腫瘍モデルを用いて評価した。CD8+ T 細胞またはCD4+および CD8+ T 細胞を同時に枯渇させたマウスでは迅速に致死性の腫瘍が発達した。これら腫瘍はrMVAmup53 および 抗-CTLA-4 mAbによるワクチン接種に抵抗性である。一方、CD4+ T 細胞枯渇の結果、ワクチンへの応答が部分的にのみ抑止された。NK1.1 細胞枯渇はワクチン接種マウスが Meth Aを拒絶する能力にほとんど効果を有さなかった(図 13a)。結果は枯渇に用いるmAbをワクチンおよび抗-CTLA-4 mAb処置後に投与した場合も同じであった (データ示さず)。同様の結果がMeth A 腫瘍モデルではなく11A-1 腫瘍モデルを用いた場合にも得られた。rMVAmup53および抗-CTLA-4 mAbの治療効果は、枯渇させる用量の抗-CD8+ mAbの投与により排除できた (p = 0.004) (図 15)。抗腫瘍効果は枯渇させるための抗-CD4+ mAbの投与により部分的に阻害され(p = 0.008)、NK 枯渇に用いるmAbの投与による影響はなかった。これらの結果は抗-CTLA-4 mAbの免疫調節効果が完全にCD8+ 細胞に依存し、部分的にCD4+ 細胞に依存し、まったくNK 細胞には依存しないことを示す(Espenschied 2003)。
【0089】
rMVAmup53 ワクチン接種に対するCpG ODNの免疫調節効果の細胞による要求を4日間確率 11A-1 腫瘍を有するBalb/c マウスを用いて評価した。抗-CTLA-4 mAbと同様にCpG ODNの MVAmup53 ワクチン接種に対する免疫調節効果は枯渇させるための CD8+ mAbの投与によって完全に抑止できた(p = 0.004) (図 14)。しかし抗-CTLA-4 mAbと異なり、CpG ODNの免疫調節効果はCD4+ 枯渇によっては影響を受けず、一方NK 細胞の枯渇は部分的にワクチン効果を抑止した(p = 0.007、NK枯渇をCD4+およびコントロール抗体枯渇と比較)。CD4+ とNKの細胞による要求の抗-CTLA-4 mAbおよびCpG ODNでの差は驚くべきである。というのは両方の免疫調節物質が等価なレベルの拒絶をもたらすからである。これらの結果は、これら2つの免疫調節物質が異なる免疫学的機構で作用することを示唆する。この情報は、抗-CTLA-4 mAb /CpG ODN 組み合わせ投与のrMVAmup53に対する効果に関するデータとともに、これら2つの免疫調節物質による腫瘍増殖に対する相乗効果を示唆する。
【0090】
IFN-γの寄与
CTLA-4遮断およびrMVAmup53 ワクチン接種の効果に対するIFN-γ 分泌の寄与をIFN-γKO マウスにおいて評価した。非ワクチン接種マウスおよびrMVApp65および抗-CTLA-4 mAbをワクチン接種したマウスの両方は正常Balb/c マウスにみられるのと同様の速度で致死性の腫瘍を発達させた(図 13b)。 rMVAmup53および抗-CTLA-4 mAb をワクチン接種した5匹のIFN-γKO マウスのうち3匹は致死性の腫瘍増殖を発達させ、IFN-γのワクチン/CTLA-4遮断効果への寄与が確認された。
【0091】
TLR 9の寄与
細胞サブセット枯渇研究は、CTLA-4遮断とCpG ODNの免疫調節活性の機構が異なることを示唆する。CpG ODN 活性はTLR9 受容体との相互作用を介するB-細胞とプラズマ細胞様 樹状細胞の刺激に起因する(Chu 1997)。CpG処置は、TH1 サイトカイン環境およびNK 細胞増殖の刺激に偏らせ、これによって腫瘍拒絶の部分的効果が説明される。 CpG ODN と CTLA-4遮断の免疫調節効果に関与する異なる経路をさらに説明するために、MC-38 腫瘍攻撃実験をTLR9-/- マウスにおいて行った。TLR9-/- マウス はCpG ODN 投与に対して免疫学的に応答することができない(Hemmi 2000)。予測されるように、初期確立MC-38 腫瘍を担持するTLR9-/- マウス はCpG ODNおよびrMVAmup53 ワクチン接種に対して免疫学的に応答することができなかった(図 16)。一方、抗-CTLA-4 mAbをrMVAmup53 ワクチン接種に含めた場合、TLR9-/- マウスにおける腫瘍拒絶が起こり(p = 0.0009)、これは wt C57BL/6 マウスにおいてみられるのと同様であった(図 16、図 7)。
【0092】
IL-6の寄与
CpG ODNおよびCTLA-4遮断はともにCD25+ CD4+ 抑制因子または調節性 T 細胞(Treg)を阻害し、この効果が記載した腫瘍モデルにおけるそれらの免疫調節活性の原因であり得る。CTLA-4の遮断はTregに対する直接の阻害効果を有すると考えられ、Treg のほとんどは構成的にCTLA-4を発現する(Read 2000)。一方、CpG ODNはDCによるIL-6の分泌を介してTreg 活性を阻害する(Pasare 2003)。IL-6の CpG ODおよび抗-CTLA-4 mAb 免疫調節効果に対する役割を評価するために、腫瘍攻撃実験をIL-6-/- マウスにおいて行った。初期確立 MC-38 腫瘍を担持するIL-6-/- マウスは腫瘍の拒絶によりCpG ODNとのrMVAmup53 ワクチン接種に免疫学的に応答することができなかった(図 17)。これはCpG ODNがその免疫調節効果を少なくとも部分的にTreg 細胞阻害のIL-6依存的経路を介して媒介していることを示唆する。一方、抗-CTLA-4 mABをrMVAmup53 ワクチン接種に含めた場合wt C57BL/6 マウスにおいてみられたのと同程度にIL-6-/- マウスにおける腫瘍拒絶が起こった(p = 0.02) (図 17、図 7)。
【0093】
実施例5: rMVAhup53によるヒト p53の発現:
BHK細胞に精製rMVAhup53を感染させた。hup53の発現を24および 48 時間目に測定し、ウェスタンブロットおよび免疫組織化学によって分析した。感染したrMVAhup53 細胞は両方の時点で激しいhup53の発現を示した(図 18)。
【0094】
実施例6: インビボ rMVAhu53 腫瘍攻撃実験:
ヒト p53を発現する新規なマウスノックインモデルであるHupki マウスを129/Sv 遺伝的背景においてDr. Monica Hollstein (DKFZ、Heidelberg、Germany)から得た。このマウスを4世代にわたってBalb/c(H-2d) バックグラウンドに対して戻し交雑し、腫瘍およびその他の試薬が容易に入手できるマウスバックグラウンドにおいてノックイン導入遺伝子の利益を享受できるようにした。Balb/c バックグラウンドにおけるhupki マウスを戻し交雑してPCR分析によって確認してホモ接合性とした。これには近親交配効果を最小とする交雑手段を用いた (データ示さず)。4T1(H-2d) マウス乳癌細胞株をヒト p53で安定にトランスフェクトし、hupki マウスの脇腹皮下に5 x104 4T1/hup53を注射した。4T1/hup53を注射したマウスでは進行性腫瘍が増殖し、大部分はこれら腫瘍によって35日目までに死んだ。rMVAhup53の効力を試験するために、マウスに4T1/hup53 注射の6日目に腹腔内注射によって107 pfu rMVAhup53をワクチン接種した。10日後、マウスに、CpG-ODN (15 nmoleのODN 1826)および抗-CTLA-4 mAb (50 μg /マウス)とともにrMVAhup53 追加免疫注射を行った。rMVAhup53 ワクチン接種はPBS コントロールと比較して生存において統計的に有意な向上をもたらした(p < 0.05、両側T検定)(図 19)。
【0095】
上記のように、記載は本発明の様々な態様の単に例示の目的である。したがって上記の特定の改変は本発明の範囲を限定するものではない。当業者には、様々な同等物、変更および改変が本発明の枠を逸脱することなく行えることが明らかであり、かかる同等の態様は本発明に含まれると理解すべきである。本明細書で引用したすべての文献を明細書に完全に開示したかのように引用により本明細書に含める。
【0096】
使用した略語:
GFP、緑色蛍光タンパク質; DC、樹状細胞; IFN-γKO、IFN-γ ノックアウト; MVA、改変ワクシニアウイルスアンカラ; rMVA組換え改変ワクシニアウイルスアンカラ; rAd-mup53、マウス 野生型p53を発現する組換えアデノウイルス; hup53、野生型ヒト p53; mup53、野生型マウス p53; rMVAp53、p53を発現する組換えMVA; rMVAmup53、野生型マウス p53を発現する組換えMVA; rMVAhup53、野生型ヒト p53を発現する組換えMVA; rMVApp65、pp65を発現する組換えMVA; rVVmup53、マウス野生型p53を発現する組換えワクシニアウイルス; rVVpp65、pp65を発現する組換えワクシニアウイルス; wtMVA、野生型 MVA; WR、ウエスタンリザーブ; i.p.、腹腔内; s.c.、皮下; mAb、モノクローナル抗体。
【0097】
参考文献
1.Allred, D.C., O'Connell, P., Fuqua, S.A. 1993. Biomarkers in early breast neoplasia. J Cell Biochem Suppl 17G:125-131.
2.Antoine, G., Scheiflinger, F., Dorner, F., Falkner, F.G. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365-396.
3. Baines, J., Celis, E. 2003. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin Cancer Res 9:2693-2700.
4.Ballas, Z.K., et al. 2001. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J Immunol 167:4878-4886.
5.Baral, R.N., et al. 2003. Immunostimulatory CpG oligonucleotides enhance the immune response of anti-idiotype vaccine that mimics carcinoembryonic antigen. Cancer Immunol Immunother 52:317-327.
6.Berns, E.M., et al. 1998. p53 protein accumulation predicts poor response to tamoxifen therapy of patients with recurrent breast cancer. J Clin Oncol 16:121-127.
7.Berns, E.M., et al. 2000. Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res 60:2155-2162.
8.Berson, J.F., et al. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strain. J Virol 70:6288-6295.
9.Blanchard, T.J., Alcami, A, Andrea, P., Smith, G.L. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol 79(Pt 5):1159-1167.
10.Bruggemann, M., et al. 1991. Human antibody production in transgenic mice: expression from 100 kb of the human IgH locus. Eur J Immunol 5:1323-1326.
11.Carpentier, A.F., Chen, L., Maltonti, F., Delattre, J.Y. 1999. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res 59:5429-5432.
12.Carroll, M.W., Moss, B. 1997a. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198-211.
13.Carroll, M.W., et al. 1997b. Highly attenuated modified vaccinia virus Ankara (MVA) as an effective recombinant vector: a murine tumor model. Vaccine 15:387-394.
14.Chakrabarti, S., Brechling, K., Moss, B. 1985. Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol 5:3403-3409.
15.Chu, R.S., et al. 1997. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med 186:1623-1631.
16.Collier, L.H. 1991. Safety of recombinant vaccinia vaccines. Lancet 337:1035-1036.
17.Davila, E., Celis, E. 2000. Repeated administration of cytosine-phosphorothiolated guanine-containing oligonucleotides together with peptide/protein immunization results in enhanced CTL responses with anti-tumor activity. J Immunol 165:539-547.
18.DeLeo, A.B., et al. 1977. Cell surface antigens of chemically induced sarcomas of the mouse. I. Murine leukemia virus-related antigens and alloantigens on cultured fibroblasts and sarcoma cells: description of a unique antigen on BALB/c Meth A sarcoma. J Exp Med 146:720-734.
19.Dialynas, D.P., et al. 1983. Characterization of the murine antigenic determinant, designated L3T4a, recognized by monoclonal antibody GK1.5: expression of L3T4a by functional T cell clones appears to correlate primarily with class II MHC antigen-reactivity. Immunol Rev 74:29-56.
20.Diamond, D.J., et al. 1997. Development of a candidate HLA A*0201 restricted peptide-based vaccine against human cytomegalovirus infection. Blood 90:1751-1767.
21.Drexler, I., et al. 1999. Modified vaccinia virus Ankara for delivery of human tyrosinase as melanoma-associated antigen: induction of tyrosi. Cancer Res 59:4955-4963.
22.Egen, J.G., Kuhns, M.S., Allison, J.P. 2002. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol 3:611-618.
23.Eliyahu, D., et al. 1989. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci USA 86:8763-8767.
24.Elkhuizen, P.H., et al. 2000. High local recurrence risk after breast-conserving therapy in node-negative premenopausal breast cancer patients is greatly reduced by one course of perioperative chemotherapy: A European Organization for Research and Treatment of Cancer Breast Cancer Cooperative Group Study. J Clin Oncol 18:1075-1083.
25.Erdile, L.F., Smith, D. 2000. CD40 activation enhances the magnitude of cellular immune responses against p53 but not the avidity of the effectors. Cancer Immunol Immunother 49:410-416.
26.Espenschied, J., et al. 2003. CTLA-4 blockade enhances the therapeutic effect of an attenuated poxvirus vaccine targeting p53 in an established murine tumor model. J Immunol 170:3401-3407.
27.Finlay, C.A., et al. 1988. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol 8:531-539.
28.Finlay, C.A., Hinds, P.W., Levine, A.J. 1989. The p53 proto-oncogene can act as a suppressor of transformation. Cell 57:1083-1093.
29.Foote, J., Winter, G. 1992. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol 224(2):487-499.
30.Gibson, L., et al. Human Cytomegalovirus Proteins pp65 and IE1 are Common Targets for CD8+ T cell Responses in Children with Congenital and Postnatal HCMV infection. J Immunol, in press.
31.Gurney, E.G., Harrison, R.O., Fenno, J. 1980. Monoclonal antibodies against simian virus 40 T antigens: evidence for distinct subclasses of large T antigen and for similarities among nonviral T antigens. J Virol 34:752-763.
32.Hainaut, P., Hollstein, M. 2000. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 77:81-137.
33.Halevy, O., Rodel, J., Peled, A., Oren, M. 1991. Frequent p53 mutations in chemically induced murine fibrosarcoma. Oncogene 6:1593-1600.
34.He, T.C., et al. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95:2509-2514.
35.Heckelsmiller, K., et al. 2002. Combined dendritic cell- and CpG oligonucleotide-based immune therapy cures large murine tumors that resist chemotherapy. Eur J Immunol 32:3235-3245.
36.Hemmi, H., et al. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740-745.
37.Hernandez, J., Lee, P.P., Davis, M.M., Sherman, L.A. 2000. The use of HLA A2.1/p53 peptide tetramers to visualize the impact of self tolerance on the TCR repertoire. J Immunol 164:596-602.
38.Hilburger, R.M., Abrams, S.I. 2001. Characterization of CD8+ cytotoxic T lymphocyte/tumor cell interactions reflecting recognition of an endogenously expressed murine wild-type p53 determinant. Cancer Immunol Immunother 49:603-612.
39.Hurwitz, A.A., Yu, T.F., Leach, D.R., Allison, J.P. 1998. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci USA 95:10067-10071.
40.Hurwitz, A.A., et al. 2000. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res 60:2444-2448.
41.Jones, P.T., Dear, P.H., Foote, J., Neuberger, M.S., Winter, G. 1986. Replacing complementarity-determining regions in a human antibody with those from a mouse. Nature 321(6069):522-525.
42.Kawarada, Y., et al. 2001. NK-and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol 167:5247-5253.
43.Kim, T.Y., et al. 2002. Both E7 and CpG-oligodeoxynucleotide are required for protective immunity against challenge with human papillomavirus 16 (E6/E7) immortalized tumor cells: involvement of CD4+ and CD8+ T cells in protection. Cancer Res 62:7234-7240.
44.Kit, S., Dubbs, D.R., DeTorres, R.A., Melnick, J.L. 1965. Enhanced thymidine kinase activity following infection of green monkey kidney cells by simian adenoviruses, simian papovavirus SV40, and an adenovirus-SV40 "hybrid". Virology 27:453-457.
45.Koo, G.C., Peppard, J.R. 1984. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma 3:301-303.
46.Krieg, A.M. 2002. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20:709-760.
47.Krieg, A.M. 2003. CpG motifs: the active ingredient in bacterial extracts? Nat Med 9:831-835.
48.Krummel, M.F., Allison, J.P. 1995. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182:459-465.
49.Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331.
50.Low, N.M., Holliger, P.H., Winter, G. 1986. Mimicking somatic hypermutation: affinity maturation. J Mol Biol 260:359-368.
51.Macpherson, I., Stoker, M. 1962. Polyoma transformation of hamster cell clones an investigation of genetic factors affecting cell competence. Virology 16:147-151.
52.Mayordomo, J.I., et al. 1996. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med 183:1357-1365.
53.Mayr, A., et al. 1978. [The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author's transl)]. Zentralbl Bakteriol [B] 167:375-390.
54.Mayr, A. 1999. [Historical review of smallpox, the eradication of smallpox and the attenuated smallpox MVA vaccine]. Berlin Munch Tierarztl Wochenschr 112:322-328.
55.Mendez, M.J., et al. 1997. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet 2:146-156.
56.Meyer, H., Sutter, G., Mayr, A. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol 72(Pt 5):1031-1038.
57.Miconnet, I., et al. 2001. Cancer vaccine design: a novel bacterial adjuvant for peptide-specific CTL induction. J Immunol 166:4612-4619.
58.Millikan, R., et al. 1995. p53 mutations in benign breast tissue. J Clin Oncol 13:2293-2300.
59.Moldoveanu, Z., Love-Homan, L., Huang, W.Q., Krieg, A.M. 1998. CpG DNA, a novel immune enhancer for systemic and mucosal immunization with influenza virus. Vaccine 16:1216-1224.
60.Mulryan, K., et al. Attenuated recombinant vaccinia virus expressing oncofetal antigen (tumor-associated antigen) 5T4 induces active therapy of established tumors. Mol Cancer Ther 1:1129-1137.
61.Noguchi, Y., Richards, E.C., Chen, Y.T., Old, L.J. 1995. Influence of interleukin 12 on p53 peptide vaccination against established Meth A sarcoma. Proc Natl Acad Sci USA 92:2219-2223.
62.Norbury, C.C., et al. 2002. Visualizing priming of virus-specific CD8+ cells by infected dendritic cells in vivo. Nat Immunol 3:265-271.
63.Offringa, R., et al. p53: a potential target antigen for immunotherapy of cancer. Ann N Y Acad Sci 910:223-233.
64.Ourmanov, I., et al. 2000. Comparative efficacy of recombinant modified vaccinia virus Ankara expressing simian immunodeficiency virus (SIV) Gag-Pol and/or Env in macaques challenged with pathogenic SIV. J Virol 74:2740-2751.
65.Pasare, C., Medzhitov, R. 2003. Toll pathway-dependent blockade of CD4+CD25+ T-cell mediated suppression by dendritic cells. Science 299:1033-1036.
66.Pratap, R., Shousha, S. 1998. Breast carcinoma in women under the age of 50: relationship between p53 immunostaining, tumour grade, and axillary lymph node status. Breast Cancer Res Treat 49:35-39.
67.Querzoli, P., et al. 1998. Modulation of biomarkers in minimal breast carcinoma: a model for human breast carcinoma progression. Cancer 83:89-97.
68.Querzoli, P., et al. 2001. Biophenotypes and survival of BRCA1 and TP53 deleted breast cancer in young women. Breast Cancer Res Treat 66:135-142.
69.Ramirez, J.C., Gherardi, M.M., Rodriguez, D., Esteban, M. 2000a. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. J Virol 74:7651-7655.
70.Ramirez, J.C., Gherardi, M.M., Esteban, M. 2000b. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation. J Virol 74:923-933.
71.Read, S., Malmstrom, V., Powrie, F. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med 192:295-302.
72.Redfield, R.R., et al. 1987. Disseminated vaccinia in a military recruit with human immunodeficiency virus (HIV) disease. N Engl J Med 316:673-676.
73.Reich, N.C., Levine, A.J. 1984. Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature 308:199-201.
74.Rosales, C., et al. A recombinant vaccinia virus containing the papilloma E2 protein promotes tumor regression by stimulating macrophage antibody-dependent cytotoxicity. Cancer Immunol Immunother 49:347-360.
75.Rosenberg, S.A. 2001. Progress in human tumour immunology and immunotherapy. Nature 411:380-384.
76.Sandler, A.D., et al. 2003. CpG oligonucleotides enhance the tumor antigen-specific immune response of a granulocyte macrophage colony-stimulating factor-based vaccine strategy in neuroblastoma. Cancer Res 63:394-399.
77.Schmid, P., Lorenz, A., Hameister, H., Montenarh, M. 1991. Expression of p53 during mouse embryogenesis. Development 113:857-865.
78.Selvanayagam, C.S., Davis, C.M., Cornforth, M.N., Ullrich, R.L. 1995. Latent expression of p53 mutations and radiation-induced mammary cancer. Cancer Res 55:3310-3317.
79.Sharma, S., et al. 2003. Intra-tumoral injection of CpG results in the inhibition of tumor growth in murine Colon-26 and B-16 tumors. Biotechnol Lett 25:149-153.
80.Sirvent, J.J., Fortuna-Mar, A., Olona, M., Orti, A. 2001. Prognostic value of p53 protein expression and clinicopathological factors in infiltrating ductal carcinoma of the breast. Histol Histopathol 16:99-106.
81.Stern, B.V., Boehm, B.O., Tary-Lehmann, M. 2002. Vaccination with tumor peptide in CpG adjuvant protects via IFN-gamma-dependent CD4 cell immunity. J Immunol 168:6099-6105.
82.Stittelaar, K.J., et al. 2001. Safety of a modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 19:3700-3709.
83.Sukumar, S., McKenzie, K., Chen, Y. 1995. Animal models for breast cancer. Mutat Res 333:37-44.
84.Sutter, G., Moss, B. 1992. Nonreplicating vaccinia virus vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA 89:10847-10851.
85.Tan, M.H., Holyoke, E.D., Goldrosen, M.H. 1976. Murine colon adenocarcinomas: methods for selective culture in vitro. J Natl Cancer Inst 56:871-873.
86.Theobald, M., et al. 1997. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med 185:833-841.
87.Turner, B.C., et al. 2000. Mutant p53 protein overexpression in women with ipsilateral breast tumor recurrence following lumpectomy and radiation therapy. Cancer 88:1091-1098.
88.van Elsas, A., Hurwitz, A.A., Allison, J.P. 1999. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190:355-366.
89.Vierboom, M.P., et al. 2000a. High steady-state levels of p53 are not a prerequisite for tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. Cancer Res 60:5508-5513.
90.Vierboom, M.P., et al. 2000b. Cyclophosphamide enhances anti-tumor effect of wild-type p53-specific CTL. Int J Cancer 87:253-260.
91.Vierboom, M.P., et al. 1997. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med 186:695-704.
92.Weiner, G.J., et al. 1997. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci USA 94:10833-10837.
93.Wiedenfeld, E.A., Fernandez-Vina, M., Berzofsky, J.A., Carbone, D.P. 1994. Evidence for selection against human lung cancers bearing p53 missense mutations which occur within the HLA A*0201 peptide consensus motif. Cancer Res 54:1175-1177.
94.Winter, G., Griffiths, A.D., Hawkins, R.E., Hoogenboom, H.R. 1994. Making antibodies by phage display technology. Annu Rev Immunol 12:433-455.
95.Zambetti, G.P., Levine, A.J. 1993. A comparison of the biological activities of wild-type and mutant p53. FASEB J 7:855-865.
96.Zellars, R.C., et al. 2000. Prognostic value of p53 for local failure in mastectomy-treated breast cancer patients. J Clin Oncol 18:1906-1913.
【図面の簡単な説明】
【0098】
【図1】図 1は、 pLW22-hup53 コンストラクトのPCR分析である。
【図2】図 2は、rMVAmup53で感染した細胞によるmup53の発現である。
【図3】図 3は、インビトロでのrMVAmup53によるp53-特異的CTL応答の生成である。
【図4】図 4は、rMVAmup53によるワクチン接種の Meth A 腫瘍阻止に対する効果である。
【図5】図 5は、rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した Meth A 腫瘍に対する効果である。
【図6】図 6は、rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した11A-1 腫瘍に対する効果である。
【図7】図 7は、rMVAmup53 + 抗-CTLA-4 mAbのワクチン接種の確立したMC-38 腫瘍に対する効果である。
【図8】図 8は、rMVAmup53 + CpG ODNによるワクチン接種の確立した11A-1 腫瘍に
【図9】図 9は、rMVAmup53 + CpG ODNによるワクチン接種の確立したMeth A 腫瘍に対する効果である。
【図10】図 10は、rMVAmup53 + CpG ODNによるワクチン接種の確立したMC-38 腫
【図11】図 11は、 rMVAmup53 + 抗-CTLA-4 mAbおよびCpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果である。
【図12】図 12は、rMVAmup53 + 抗-CTLA-4 mAb and CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果である。
【図13】図 13は、Meth A 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求である。
【図14】図 14は、11A-1 腫瘍に対するCpG ODN 免疫調節物質効果の細胞による要求である。
【図15】図 15は、11A-1 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求である。
【図16】図 16は、CpG ODN 免疫調節物質効果に対するTLR 9の寄与である。
【図17】図 17は、IL-6 の CpG ODN 免疫調節物質効果に対する寄与である。
【配列表】
【技術分野】
【0001】
発明の背景
本出願は2002年12月23日出願の米国仮特許出願第06/436268号(Ellenhorn and Diamond)および2003年4月30日出願の第06/466607号(Ellenhorn and Diamond)からの優先権を主張し、その開示全体を引用により本出願に含める。
【0002】
連邦政府援助の研究に関する出願の記載
本発明は一部、NIH、Division of AIDS (RO1-AI43267およびR21-AI44313)および NCI: RO1-CA77544、PO1-CA30206、R29-CA70819、およびCA33572からの助成により政府援助によりなされた。政府は本発明における一定の権利を有しうる。
【0003】
発明の分野
本発明は、ウイルス学、分子生物学、および腫瘍免疫学の分野に関する。具体的には、本発明はp53を発現する悪性腫瘍に対して有効な免疫応答を誘発するための組成物および方法に関する。
【背景技術】
【0004】
背景
p53は細胞周期停止またはアポトーシスに必要な一定の遺伝子の発現を制御する腫瘍抑制タンパク質である。p53をコードする腫瘍抑制遺伝子は、DNA損傷、細胞ストレス、または特定の癌遺伝子の異常発現によって活性化する(Levine 1997)。いったん活性化すると、野生型p53 (wt p53)は一時的に細胞周期を停止させる役割を果たし、DNA修復の時間を与え、損傷したDNAを有する細胞が制御を失って増殖することを防ぐ(Levine 1997)。p53はまた一定のタイプの生理的損傷を有する細胞におけるアポトーシスの誘導にも関与している(Levine 1997)。
【0005】
p53の増殖抑制能を機能的に不活性化させるp53における突然変異はすべてのヒトの癌の40-60%において観察されており、悪性表現型と関係がある(Hainaut 2000)。p53に対する突然変異は腫瘍形成の初期現象として起こり(Millikan 1995; Querzoli 1998; Allred 1993)、該タンパク質が細胞分裂を抑制する能力を失わせる (Finlay 1989; Eliyahu 1989)。細胞におけるp53発現の制御は、p53 mRNA量のレベルまたは53タンパク質量のレベルにて起こりうる。p53の突然変異は、突然変異タンパク質の半減期の延長により、しばしばp53タンパク質の高い核内および細胞質濃度をともなう。多くの腫瘍は非悪性細胞と比べて細胞内p53レベルが非常に高いことによって特徴づけられる。腫瘍のなかには多量の突然変異p53を合成するが、細胞内p53レベルは低い、即ち通常の定常状態レベルを下回るものもあり、これはおそらくタンパク質の細胞内分解の加速の結果と考えられる。p53の過剰発現は以下の独立の前兆である:より悪性の癌 (Turner 2000; Elkhuizen 2000; Zellars 2000)、リンパ節転移(Pratap 1998)、標準的治療に対する応答の欠損 (Berns 1998; Berns 2000)、および死亡率 (Sirvent 2001; Querzoli 2001)。
【0006】
ミスセンス点突然変異は癌におけるp53突然変異でもっとも頻繁なものであり、ほとんどのp53タンパク質は野生型形態(wt p53)のままである。p53突然変異は真の腫瘍特異的抗原を表すこともあるが、かかる突然変異のほとんどは、T細胞によって認識される免疫性エピトープには対応しない部位において起こる(Wiedenfeld 1994)。このため、広く利用できるp53に向けられた免疫療法はいずれもwt p53を標的としなければならない。実験モデルにおいて、p53を標的とすることが可能であった。なぜなら突然変異分子はp53タンパク質の高い核内および細胞質濃度を伴うからである(Finlay 1988)。p53は適応免疫応答にとって魅力的な標的である。なぜなら健康細胞における非突然変異p53の細胞内濃度は非常に低いからである(Zambetti 1993; Reich 1984)。これは非-突然変異体 p53を発現する健康細胞は過剰発現した突然変異体 p53に対する増強した免疫応答をおそらくまぬがれるであろうということを意味する(Offringa 2000)。
【0007】
p53は、細胞性免疫応答によって認識されうるほとんどの腫瘍関連抗原と同様に、自己抗原である(Rosenberg 2001)。p53は発生の過程で広く発現した自己抗原であるという事実と(Schmid 1991)、腫瘍において発現している突然変異p53のほとんどは野生型タンパク質と同じ構造を有するという事実とを組み合わせると、腫瘍によって発現したp53が免疫系によっておそらく自己-タンパク質として寛容されるということを意味する。マウスにおける機能および四量体研究によって細胞障害性Tリンパ球レベル(CTL)で存在することが示されたこの寛容は(Theobald 1997; Erdile 2000)、p53に向けた免疫療法の有効性を制限する。成功するためには、有効な免疫療法は正常細胞および組織に対する自己免疫を誘発することなくこの寛容を克服しなければならない (Theobald 1997; Erdile 2000; Hernandez 2000)。少数の自己反応性T細胞は免疫寛容に関与するプロセスにおいて回避する。
【発明の開示】
【発明が解決しようとする課題】
【0008】
p53を過剰発現する腫瘍は、エピトープ特異的CTL(Vierboom 2000a; Vierboom 2000b; Vierboom 1997; Hilburger 2001)、エピトープで刺激した樹状細胞(DC) (Mayordomo 1996)、または突然変異体p53エピトープとIL-12 (Noguchi 1995)の全身投与によってマウスモデルにおいて排除された。これら戦略はそれぞれ臨床適用の面でかなりの問題を有する。CTL 注入およびエピトープで刺激した樹状細胞の注入は時間がかかり高価である。というのは細胞の単離、培養および再注入は各々の患者にとって別々に行わなければならないからである。逆に、なんらかの効果を得るためには、従来用いられていた無細胞ワクチン接種戦略は、腫瘍内注射または腫瘍攻撃に先立つワクチン接種のいずれかを必要としそのいずれも臨床使用では実践的なアプローチではない。したがって簡便、有効かつ広く適用できる癌の治療における免疫療法戦略が必要とされている。
【課題を解決するための手段】
【0009】
発明の概要
p53 遺伝子産物は大多数の癌で過剰発現しており、したがってそれは癌免疫療法にとって理想的な標的となる。しかしこれら療法の効力は制限されてきた。というのは腫瘍によって発現されるp53は自己-タンパク質として免疫系によって寛容されるようであるという事実に基づく。本発明はこの自己寛容はp53をコードする核酸を含む組換えMVA (rMVAp53)の投与によって克服されうるという発見に基づく。p53の投与は様々な悪性細胞型で攻撃したマウスにおける腫瘍発生、腫瘍増殖、および死亡率を大幅に減少することが示されている。rMVAp53の治療効果はCTLA-4遮断薬またはCpGオリゴデオキシヌクレオチド (CpG ODN) 免疫調節物質の投与により増強されることも示される。この増強は両方の免疫調節物質を投与した場合にもっとも大きくなる。本発明は癌の治療における使用のための組換えMVA 組成物、この組成物を用いる癌の治療方法およびこの組成物の投与のためのキットを提供する。
【0010】
第一の態様において、本発明はp53をコードする核酸を含む組換えMVA を含む組成物を提供する。好ましくは、組換えMVA によってコードされるp53はwt ヒト p53である。本発明によると、組成物はCTLA-4遮断薬および/またはCpG ODNを含んでいてもよい。
【0011】
別の態様において、本発明はp53-発現悪性腫瘍を有する対象の治療方法を提供する。この方法は、p53をコードする核酸を含む組換えMVAの投与に基づく。好ましくは、該方法は免疫調節物質としてCTLA-4遮断薬および/または CpG ODNの投与も要求する。第三の態様において、本発明は、p53-発現悪性腫瘍を治療するためのキットを提供する。このキットはp53をコードする核酸を含む組換えMVAを含み、免疫調節物質としてCTLA-4遮断薬および/または CpG ODNを含んでいてもよい。最後の態様において、本発明は、wt ヒト p53をコードする核酸インサートを含むMVA組換えプラスミドを提供する。
【0012】
図面の簡単な説明
図 1: pLW22-hup53 コンストラクトのPCR分析。
rMVAhup53注入(レーン 1、2) および wtMVA注入(レーン 3、4) BHK 細胞を全DNA抽出および wtMVA (レーン 2、4)またはhup53 (レーン 1、3) 特異的プライマーを用いたPCR 増幅に供した。rMVAhup53 産物は汚染wtMVAを有さないことが示された。
【0013】
図 2: rMVAmup53で感染した細胞によるmup53の発現。
rMVAmup53で感染させた細胞は mup53を高レベルで発現し、MVAが好適なワクチンベクターであることが確認された。細胞可溶化液をSDS-PAGEおよびウェスタンブロッティングに供した。レーンを以下のように指定する: 1) Meth A、非操作 Meth A 肉腫細胞、2) HCMV IE1 エキソン4-rMVA感染BHK細胞、3-4) rMVAmup53感染BHK細胞 (それぞれ0.125μl、0.25μl 細胞可溶化液をローディング)、5) rAdp53、および6) rAdpp65感染HEK293細胞。特に断りのない限りすべてのレーンに20μlのサンプルをローディングした。
【0014】
図 3: インビトロでのrMVAmup53によるp53-特異的CTL応答の生成。
rMVAmup53 による一回の腹腔内 (i.p.) ワクチン接種により、p53を過剰発現する細胞を有効に殺すp53 特異的CTL応答が生じる。(a) rMVAmup53で処理したマウスからの脾細胞を14日目に回収し、インビトロで6日間 rAdp53感染同系 LPS 芽球で再刺激した。CTL活性をrVVp53 (実線)またはrVVpp65 (破線)感染10.1 細胞を用いて標準的 4-h 51Cr 放出アッセイで評価した。(b) rMVAmup53 (実線)またはrMVApp65 (破線) ワクチン接種マウスからの脾細胞をワクチン接種の14日後に回収し、インビトロで6日間 rAdp53感染同系 LPS 芽球で再刺激した。細胞障害性をrVVp53感染10.1 細胞に対して測定した。 (c) rMVAmup53 (実線)またはrMVApp65 (破線) ワクチン接種の14日後に回収した脾細胞をインビトロで6日間 rMVAp53で感染した同系 LPS 芽球を用いて再刺激した。細胞障害性を標準的 4-h 51Cr 放出アッセイによってMeth A 細胞に対して測定した。
【0015】
図 4: rMVAmup53によるワクチン接種の Meth A 腫瘍阻止に対する効果。
Balb/c マウスの皮下に (s.c.) 5 x105 Meth A 細胞を注射した。5日目に、マウスを5 x107 pfu のrMVAmup53 (MVAp53) (n = 16)、5 x107 pfuのrMVApp65 (MVApp65) (n = 16)、またはPBS (n = 12)でワクチン接種した。生存プロットは、各群における生存動物の比率を腫瘍攻撃後の日数の関数として示す。rMVAmup53をワクチン接種したマウスの両方の対照群と比較しての改善はlog順位検定により測定して統計的に有意である(P < 1)。
【0016】
図 5: rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した Meth A 腫瘍に対する効果。
マウスに迅速に死に至る用量の106 Meth A 細胞を皮下注射した。6、9、および12日目に、マウスに抗-CTLA-4 mAb (CTLA4mAb)またはコントロール mAbのいずれかを腹腔内注射した。7日目に、マウスに5 x107 pfuのrMVAp53 (MVAp53)または5 x107 pfuのrMVApp65 (MVAapp65)のいずれかをワクチン接種した。生存プロットは各群における生存動物の比率を示す。rMVAp53 + 抗-CTLA-4 mAb (n=14)をワクチン接種したマウスの、rMVApp65 + CTLA-4 (n=14)、rMVAp53 + コントロール ab (n=14)、またはrMVApp65 + コントロール ab (n=6) を与えられたコントロール動物に対する生存の向上はlog順位検定によって判定して統計的に有意である(P < 0.001)。
【0017】
図 6: rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウスに2 x 106の11A-1 細胞(rMVAmup53 + 抗-CTLA-4 mAbを他のすべての群と比較してp = 0.00044)を皮下注射した。抗-CTLA-4 mAb 9H10 (CTLA4 mAb)またはコントロールハムスターアイソタイプ一致ポリクローナル抗体 (アイソタイプ一致 Ab)を4、7、および10日目にそれぞれ100、50、および50μgの用量で腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65 (MVApp65)、またはPBSを腹腔内にワクチン接種した。それぞれの線は8匹のマウスの平均および標準偏差を表す。
【0018】
図 7: rMVAmup53 + 抗-CTLA-4 mAbのワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した (rMVAmup53 + 抗-CTLA-4 mAb をその他の全ての群と比較してp = 0.0001)。抗-CTLA-4 mAb 9H10 (CTLA4 mAb)またはコントロールハムスターアイソタイプ一致ポリクローナル抗体 (アイソタイプ一致 Ab)を4、7、および10日目にそれぞれ100、50、および50μgの用量で腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65 (MVApp65)、またはPBSのいずれかを腹腔内にワクチン接種した。各線は8匹のマウスの平均および標準偏差を表す。
【0019】
図 8: rMVAmup53 + CpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウスに2 x 106 11A-1 細胞を皮下注射した(rMVAmup53 + CpG ODNをその他のすべての群と比較してp = 0.00002)。15 nmolesの CpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0020】
図 9: rMVAmup53 + CpG ODNによるワクチン接種の確立したMeth A 腫瘍に対する効果。
Balb/c マウスに1 x 106 Meth A 細胞を皮下注射した(MVAmup53 + CpG ODN をすべての他の群と比較してp = 0.0015)。15 nmoles のCpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0021】
図 10: rMVAmup53 + CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウスに1 x 106 MC-38 細胞(rMVAmup53 + CpG ODNをその他のすべての群と比較してp = 0.0004)を注射した。15 nmolesのCpG ODN (CpG)を4、9、および14日目に腹腔内注射した。5日目に、マウスの腹腔内に5 x107 pfuのrMVAmup53 (MVAp53)、5 x107 pfu のrMVApp65 (MVApp65)、またはPBSのいずれかで免疫した。
【0022】
図 11: rMVAmup53 + 抗-CTLA-4 mAbおよびCpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果。
Balb/c マウス (n = 8)に2 x 106 11A-1 細胞を皮下注射した。抗-CTLA-4 mAb (CTLA4 mAb)を14、17、および20日目にそれぞれ100、50、50μg用量で腹腔内注射した。15 nmolesのCpG ODN (CpG)を14、19、および24日目に腹腔内注射した。15日目に、マウスに5 x107 pfu のrMVAmup53 (MVAp53)、5 x107 pfuのrMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。生存プロットは腫瘍攻撃後の日数の関数としての各群における生存動物の比率を示す。CpG ODN と抗-CTLA-4 mAbとの組み合わせをCpG ODNのみに対して比較してp = 0.02、およびCpG ODNと抗-CTLA-4 mAbの組み合わせを抗-CTLA-4 mAbのみと比較してp = 0.01。
【0023】
図 12: rMVAmup53 + 抗-CTLA-4 mAb および CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果。
C57BL/6 マウス (n = 8)に1 x 106 MC-38 細胞を皮下注射した。抗-CTLA-4 mAbを4、7、および10日目にそれぞれ100、50、および50μg用量で腹腔内注射した。15 nmolesのCpG ODNを4、9および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53、5 x107 pfuのMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。生存プロットは腫瘍攻撃後の日数の関数としての各群における生存動物の比率を示す。CpG ODNおよび抗-CTLA-4 mAbの組み合わせをCpGのみと比較してp = 0.002、およびCpG ODN および抗-CTLA-4 mAbの組み合わせを抗-CTLA-4 mAbのみと比較してp = 0.001。
【0024】
図 13: Meth A 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求。
Balb/c マウス (a)またはIFN-γKO Balb/c マウス (b)に迅速に死に至る用量の106 Meth A 細胞を皮下注射した。両集団からのマウスの群に枯渇させる用量(depleting dose)の抗-CD4、抗-CD8、抗-NK1.1、またはコントロール mAbを-1、1、3、および10日目、そしてその後毎週、腹腔内注射した。6、9、および12日目にマウスに抗-CTLA-4 mAb (CTLA4mAb)またはコントロール mAbのいずれかを腹腔内注射した。7日目に、マウスに5 x107pfuのrMVAp53 (MVAp53)または5 x107 pfuのrMVApp65 (MVAapp65)のいずれかをワクチン接種した。(a) 平均腫瘍増殖をBalb/c マウスの各群について計算し、エラーバーは標準偏差を示す。各線の最後のデータポイントは最初の死亡を示す。(b)生存IFN-γKO Balb/c マウスの比率をプロットする。
【0025】
図 14: 11A-1 腫瘍に対するCpG ODN 免疫調節物質効果の細胞による要求。
Balb/c マウスに2 x 106 11A-1 細胞を皮下注射した。15 nmoles のCpG ODNを4、9、および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfu のrMVAmup53を腹腔内にワクチン接種した。マウスに枯渇させる用量の抗-CD4 (CD4)、抗-CD8 (CD8)、抗-NK1.1 (NK)、またはコントロール mAbを4、6、8および15日目、そしてその後7日ごとに腹腔内注射した。腫瘍を三次元で週に2回測定した。CD8+枯渇をすべてのその他の群と両側ウイルコクソン検定によって比較してp = 0.004。抗-NK1.1 を抗-CD4およびコントロール mAbと比較してp = 0.007。
【0026】
図 15:11A-1 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求。
マウスに2 x 106 11A-1 細胞を皮下注射した。抗-CTLA-4 mAbを4、7、および10日目にそれぞれ100、50、および50μg/用量で腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内にワクチン接種した。4、6、8、および15日目、そしてその後7日ごとに対応するmAbまたはコントロール mAbを腹腔内注射することによってマウスからCD8+、CD4+、またはNK 細胞を枯渇させた。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。CD8+ 枯渇をその他のすべての群と比較してp = 0.004。CD4+枯渇をNK枯渇およびコントロール群と比較してp = 0.008。
【0027】
図 16: CpG ODN 免疫調節物質効果に対するTLR 9の寄与。
TLR9-/- (抗-CTLA-4 mAbをCpG ODN 群と比較してp = 0.0009) 突然変異体 C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した。マウスを4、7、および10日目にそれぞれ100、50および50μg/用量で抗-CTLA-4 mAb (CTLA4 mAb)で処理するかあるいは15 nmolesのCpG ODNで4、9、および14日目に処理した。5日目に、すべてのマウスに5 x107 pfuのrMVAmup53を腹腔内にワクチン接種した。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。
【0028】
図 17: IL-6 の CpG ODN 免疫調節物質効果に対する寄与。
IL-6-/- (ウィルコクスン両側順位和検定によって抗-CTLA-4 mAbをCpG ODN 群と比較してp = 0.02) 突然変異体 C57BL/6 マウスに1 x 106 MC-38 細胞を皮下注射した。マウスを 抗-CTLA-4 mAb (CTLA4 mAb) で4、7、および10日目にそれぞれ100、50、および50μg/用量であるいは15 nmolesのCpG ODNで4、9、および14日目に処理した。5日目に、すべてのマウスに5 x107 pfu のrMVAmup53を腹腔内にワクチン接種した。腫瘍をキャリパーを用いて三次元で週に2回測定した。各曲線は8匹のマウスの平均および標準偏差を表す。
【0029】
図 18: rMVAhup53を感染した細胞によるhup53の発現。
BHK細胞に精製rMVAhup53 (MVA/p53)を注入した。hup53の発現を24および48時間後に測定した。細胞可溶化液をSDS-PAGEおよびウェスタンブロッティングに供した。レーン1: コントロール MVA を注入したBHK細胞;レーン2: rMVAhup53を24時間注入したBHK細胞;レーン3: rMVAhup53を48時間注入したBHK細胞。すべてのレーンに20μlのサンプルをローディングした。
【0030】
図 19: rMVAhup53 + 抗-CTLA-4 mAbおよびCpG ODNのワクチン接種の確立した4T1/hup53 腫瘍に対する効果。
マウスに5 x104 4T1/hup53を皮下注射し、次いで107 pfuのrMVAhup53またはPBS コントロールを6日目に腹腔内にワクチン接種した。16日目に、マウスにrMVAhup53 またはPBSの追加免疫注射を15 nmoleのCpG ODNおよび50μgの抗-CTLA-4 mAbとともに与えた。rMVAhup53ワクチン接種マウスはPBS コントロールと比較して生存において有意な向上を示した(p < 0.05、両側T検定)。
【0031】
詳細な説明
本発明は正常および癌細胞の両方に発現するタンパク質に対する自己寛容が克服され、強い抗-腫瘍免疫応答が、腫瘍内投与を必要とせず、全身毒性または自己免疫を引き起こす、生成させうるという発見に基づく。本発明は新規な無細胞組成物および多様なヒ悪性腫瘍に対する有効な免疫応答を、対象のハプロタイプまたはジェノタイプに関わりなく生成させる方法を提供する。以下に記載する実施例は、野生型マウスまたは野生型ヒト p53 (rMVAmup53またはrMVAhup53)を発現するよう操作した改変・ワクシニア・アンカラ・ベクターでのワクチン接種により、激しいp53-特異的CTL応答が刺激されることを示す。この応答はCTLA-4遮断薬および/または CpG ODNからなる免疫調節物質の共投与により増強されうる。
【0032】
MVAウイルス (GenBank Accession Number U94848)は一次ニワトリ胚線維芽細胞の570回以上の連続継代に由来するワクシニアウイルスのアンカラ株のバリアントである。弱毒化ポックスウイルスとしてのMVAのいくつかの特性により、それはp53を発現する腫瘍に対する治療応答の生成のために理想的となる。MVAの一つの利点は哺乳類細胞においてそのDNAを効率的に複製できることであるが、それは無毒であり伝播しない。この形質は、ニワトリ胚線維芽細胞の継代の過程で生じる少なくとも25のさらなる突然変異および欠失のなかで2つの重要な宿主範囲遺伝子が失われた結果である (Meyer 1991; Antoine 1998)。NYVAC (弱毒化コペンハーゲン株)およびALVAC (宿主範囲制限アビポックス)と異なり、MVAにおける初期および後期転写はともに損なわれておらず、ウイルス生活環にわたって連続して遺伝子を発現することが可能である(Carroll 1997a; Carroll 1997b; Blanchard 1998; Sutter 1992)。 MVAはウエスタンリザーブ (WR) 株よりも免疫原性が強いことが判明しており、既存のポックスウイルス免疫性の条件において利用できる(Ramirez 2000a; Ramirez 2000b)。組換えワクチン送達媒体としてのMVAの好ましい臨床特性はその天然痘ワクチンとしての良性安全性特性としてヨーロッパにおいて1970年代後半に支持されている(Mayr 1999; Mayr 1978)。MVAは老人および若者を含む120,000を超える高リスク個体に重篤な副作用無く投与された (Mayr 1978)。より最近では、MVAは免疫不全の非-ヒト霊長類に不利な結果をもたらすことなく投与されてきた(Stittelaar 2001)。これはその他のベクター、例えば、ヒト宿主に危険をもたらすレトロウイルスおよびアデノウイルスとは全く対照的である。用いられるベクター、アジュバント、または免疫調節物質の免疫毒性は、癌の免疫療法において特に問題となる点である。というのはほとんどの癌患者は化学療法、放射線療法、または癌自体の免疫抑制効果によってひどい免疫不全となっているからである。非-弱毒化ポックスウイルス、例えば、WR 株は免疫不全の個体に安全に投与することができないことが明らかになった後、MVAは1990年代前半にワクチン媒体として最初に開発された(Redfield 1987; Collier 1991)。要約すると、MVAの発現ベクターとしての能力と、霊長類およびヒトにおけるその安全特性とを組み合わせると、それは癌遺伝子のための送達系として非常に魅力的である。
【0033】
rMVAmup53およびrMVAhup53の構築は当業者に周知の組換えDNA技術により達成される(Sambrook et al.、Molecular Cloning、Cold Spring Harbor Laboratory、2001; Ausubel et al.、Current Protocols in Molecular Biology、John Wiley & Sons、1986 and 2000)。野生型p53のコード配列は便宜にp53-特異的 プライマーを用いたRT-PCRによって得ることが出来る。これらプライマーはDNAにハイブリダイズし、DNA合成の開始部位として役立つ。ヌクレオチドプライマーは二本鎖の反対側の離れた部位に結合するよう設計され、それによって増幅される部分としての介在配列が規定される。プライマーとして用いる核酸分子は一般に増幅すべきDNAセグメントに相補的な少なくとも10塩基対の配列を含む。プライマーの選択は当業者に周知である。wt mup53またはwt hup53の増幅用プライマーは好適なMVA組換えプラスミド、例えば、pMCO3、pLW22、pLW51、pUCII LZまたはその他の当該技術分野で周知のMVA送達ベクターへのサブクローニングのための適当な制限部位を含むように設計すればよい。組換えプラスミドは、外来遺伝子インサートの発現に必要な配列、およびMVAゲノムにおける欠失した選択部位への相同組換えに必要な隣接配列を含む。組換えMVAウイルスを作るために、細胞にMVAウイルスを感染させ、外来遺伝子インサートを含む組換えプラスミドをトランスフェクトする。ウイルスとプラスミドの間の相同組換えを起こらせた後、インサート遺伝子を発現する組換えMVAを単離する。
【0034】
rMVAmup53またはrMVAhup53による感染後のp53タンパク質の細胞による発現を分析し、組換えウイルスからのその発現の忠実度と程度を判定した。突然変異p53を過剰発現するMeth A 細胞をポジティブコントロールとして用い、HCMV IE1 エキソン4 rMVA 感染BHK細胞をネガティブコントロールとして用いた。ウェスタンブロット分析によりrMVAmup53またはrMVAhup53で感染した細胞およびMeth A 細胞による豊富なp53 発現が明らかとなった。HCMV IE1 エキソン 4-rMVA 感染BHK細胞によるp53の検出可能な発現は観察されなかった。rMVAp53 感染BHK細胞による高レベルのp53発現は、蛍光顕微鏡観察によっても観察された。その他のウイルスおよび細胞形態と比較したrMVAmup53およびrMVAhup53 により示される高レベルのp53 発現は、ワクチン接種プロトコールにおけるその有用性を示す。
【0035】
動物実験モデルにおいて、MVAに基づくワクチンは腫瘍特異的CTL活性を刺激し(Espenschied 2003; Drexler 1999)、確立した腫瘍の退行をもたらす(Espenschied 2003; Carroll 1997b; Mulryan 2002; Rosales 2000)。MVAにおいて発現する全タンパク質による免疫化には多数の利点がある。ペプチド免疫化と異なり、複数のエピトープの発現が可能であり、ポリクローナル宿主応答が刺激できる。CTL応答の伝播に必須な抗原-特異的同族援助(cognate help)は、MVAにおけるタンパク質の発現を介して達成できる。さらに、全タンパク質の発現の結果、そうでなければ陰性のエピトープに対する応答の刺激が起こりう得る。組換えウイルスによる免疫化は、抗原-特異的細胞の拡張および養子移入を伴う複雑かつ高価なアプローチの必要性、または特定の癌患者のための個別化ワクチンの作成の必要性を避けうる。組換えワクチンアプローチのこの利点により、疾患の初期段階の患者における再発を予防するより広範な臨床使用が促される。
【0036】
インビトロ実験を行って、rMVAmup53によるワクチン接種がp53 寛容に打ち勝つかを調べたところ、p53-特異的CTLが生じた。rMVAmup53による単回腹腔内 (i.p.) ワクチン接種の後マウスから脾細胞を回収し、インビトロでp53 過剰発現細胞で再刺激した。脾細胞は wt p53過剰発現標的を認識し、溶解した。一方、rMVApp65をワクチン接種したマウスからの脾細胞は強いpp65 特異的CTL応答を刺激したが、p53過剰発現標的は認識せず、リンパ球応答の特異性が示された。rMVAmup53 ワクチン接種はまた、突然変異p53を発現するMeth A 細胞のCTL認識も刺激し得る。rMVAmup53をワクチン接種したマウスからの再刺激した脾細胞は突然変異体 p53 過剰発現Meth Aを認識したが、rMVApp65をワクチン接種したコントロールマウスは認識しなかった。
【0037】
rMVAmup53による単回ワクチン接種の結果CTL応答が増強したので、インビボでのMeth A 腫瘍細胞の増殖に対するrMVAmup53 ワクチン接種の効果を調べることには十分な正当性があった。rMVAmup53の投与は突然変異体 p53を過剰発現する免疫原性腫瘍細胞株であるマウス肉腫 Meth Aの増殖を阻害することが示された。致死量の Meth A 腫瘍細胞を接種され、腹腔内注射によりrMVAmup53をワクチン接種したマウスは3日後にコントロール動物と比べてより遅い腫瘍増殖および高い生存率を示した。ほとんどのワクチン接種マウスは完全に腫瘍を発達させることが出来ず、これらマウスは52日後のMeth Aによる再攻撃に対して耐性であった (Espenschied 2003)。
【0038】
上記の結果は悪性腫瘍によって発現するp53の標的化における新規なrMVAmup53 無細胞ワクチンの有効性を示す。さらなる実験を行って、この効果がCTLA-4遮断薬またはCpG ODN 免疫調節物質の添加によって増強されるかを調べた。ワクシニアウイルスコンストラクトによる免疫化の結果、DCによりウイルスタンパク質が取り込まれ、提示される(Norbury 2002)。流入領域リンパ節において、DCは抗原を未処置のCD8+T細胞に提示し、その結果T 細胞活性化が起こり、続いて免疫応答が伝播される(Norbury 2002)。免疫調節物質実験を、応答の開始とその伝播の両方に取り組むことによって、rMVAp53に対する応答を増強させることができるかを調べるために設計した。
【0039】
腫瘍ワクチンの最適化の1つの強力な戦略は、CTLA-4とリガンドとの結合をブロックする分子を用いることによりT 細胞応答性の負の制御を操作することを含み、この現象は「CTLA-4遮断」と称される。CTLA-4はT 細胞上にみられる細胞表面受容体である。CTLA-4の活性化はT 細胞応答の阻害を導く。CTLA-4は末梢T-細胞寛容の制御に重要な役割を果たし、それはT-細胞活性化を受動的および能動的機構の両方を介して干渉することによる (Egen 2002)。CTLA-4遮断薬の腫瘍関連自己抗原を発現する癌ワクチンと組み合わせての使用は、場合によっては、腫瘍拒絶と寛容の打破をもたらすが、自己免疫が同時に誘導される(Espenschied 2003; Hurwitz 2000; van Elsas 1999)。インビトロでは、CTLA-4遮断はT-細胞活性化閾値を低め、CTLA-4の軽減効果を除く。CTLA-4遮断はまた、Treg 細胞活性をインビボで阻害する(Read 2000)。 GM-CSF産生腫瘍細胞ワクチンと組み合わせた場合、CTLA-4遮断の結果、確立された免疫原性の低いメラノーマ、乳癌、および前立腺癌移植片の拒絶がもたらされる(Hurwitz 1998; Hurwitz 2000; van Elsas 1999)。これは腫瘍関連抗原に対する寛容の打破をともなうプロセスを介して起こる。CTLA-4遮断薬はCTLA-4 受容体に特異的に結合し、CTLA-4のそのカウンター-受容体に対する結合を阻害する分子である。CTLA-4遮断薬は以下のいずれであってもよい:モノクローナルまたはポリクローナル抗体、抗体の断片、ペプチド、有機低分子、ペプチド疑似体(peptidomimetic)、核酸、例えば、干渉RNA (iRNA)またはアンチセンス分子、アプタマー、CTLA-4 リガンド、例えば、CTLA-4 リガンドのB7ファミリーのメンバーからのあらゆるドメイン、ここで該リガンドは好ましくは、免疫細胞上に存在するCTLA-4に結合することが出来、CTLA-4機能をブロックすることが出来る組換え可溶性タンパク質として合成されたものであり得る。抗-CTLA-4抗体は、宿主動物をCTLA-4 タンパク質またはCTLA-4を発現する細胞で免疫することによって作成できる。CTLA-4に対するモノクローナル抗体(抗-CTLA-4 mAb)は常套技術によって産生でき、即ち、ハイブリドーマ細胞と抗-CTLA-4 抗体を産生する哺乳類免疫細胞を融合させることによって産生できる。抗-CTLA-4 mAbの作成に用いられる哺乳類細胞には以下の細胞が含まれる:ラット、マウス、ハムスター、ヒツジ、またはヒト細胞。 抗-CTLA-4 mAbはハイブリドーマ細胞上清または腹水から精製すればよい。抗-CTLA-4抗体は、トランスジェニック動物(Bruggemann 1991; Mendez 1997)またはヒト免疫グロブリンファージディスプレーライブラリー (Winter 1994)を用いて作製されるヒト抗体であってよい。抗-CTLA-4 抗体はまたキメラ抗体およびヒト化 (または「新形態」)抗体も含む。CTLA-4に対するキメラ抗体は、組換え方法を用いて、非-ヒト抗体のCTLA-4 結合ドメインとヒト抗体の定常ドメインを含むように作ればよい。CTLA-4に対するヒト化抗体は、組換え方法により、ヒト抗体構造フレームワーク上に非ヒト抗-CTLA-4 抗体のCDR領域のみを含むように作ればよい(Jones 1986; Low 1986)。非-ヒト領域における個々の残基をヒト抗体フレームワークからの残基と置換すればよい。逆に、ヒト抗体フレームワークにおける個々の残基を非-ヒト抗体からの残基と置換してもよい(Foote 1992)。かかる置換を用いて、ヒト化抗体の結合能を上昇させ、あるいは抗体に対する免疫応答を低減することが出来る。CTLA-4に対するヒト化抗体は、トランスジェニックヒト免疫グロブリン定常領域遺伝子を有する動物の産物であってもよい。組換えDNA技術によってそれらを操作して、CH1、CH2、CH3、ヒンジドメインまたは対応のヒト配列を有するその他のドメインを当該技術分野で公知の方法により置換することができる。
【0040】
非メチル化 CpG (シトシン-フォスフェート-グアニン) モチーフを含むオリゴデオキシヌクレオチドは、ワクチン効力を高める強力な免疫賦活剤である(Krieg 2002)。CpG ODNによる免疫活性化はB 細胞およびプラズマ細胞様(plasmacytoid)樹状細胞におけるToll-様受容体-9 (TLR9)への特異的結合から開始する(Krieg 2002)。DCにおけるTLR9連結の結果、リンパ球、マクロファージ、単球、ナチュラルキラー (NK)、およびT-細胞集団が二次的に活性化する。これはTH1 サイトカイン環境を生成するサイトカインの合成を介して起こる(Krieg 2003)。この結果、NK活性が上昇し、抗原提示が上昇し、およびT細胞援助が起こり、これによって体液性および細胞-媒介免疫応答の両方が増強しうる。さらに、TLR9 連結の結果、DCによるIL-6産生が起こり、これは、CD4+CD25+ Treg 細胞の抑制効果に打ち勝つのに役立つ(Pasare 2003)。CpG ODNの単独での投与は、中程度の抗-腫瘍効果を多数のマウス腫瘍モデルにおいて発揮することが示されている (Carpentier 1999; Kawarada 2001; Ballas 2001; Baines 2003; Sharma 2003)。 CpG ODNはマウスにおける多数の実験的腫瘍ワクチンの有効なアジュバントであることが示されている。それは少なくともフロイントアジュバントと同程度に有効であるが、TH1活性がより高く、毒性が少ない(Chu 1997; Weiner 1997)。CpG ODNはペプチド (Davila 2000; Stern 2002)、タンパク質 (Kim 2002)、DC (Heckelsmiller 2002)、イディオタイプ (Baral 2003)、およびGM-CSF分泌性腫瘍細胞ワクチン (Sandler 2003)の効果を増強しうる。CpG ODNが TH1 応答とNK 細胞の刺激を開始させる能力はおそらくこれらワクチンアプローチおよび以下の記載における、免疫調節物質活性の原因であろう。
【0041】
rMVAmup53ワクチン接種と組み合わせてのCTLA-4遮断薬の投与が有益であるか、または自己免疫疾患を引き起こすかを調べるために、CTLA-4に特異的なモノクローナル抗体(抗-CTLA-4 mAb)を用いた。rMVAmup53と抗-CTLA-4 mAbによるワクチン接種は、確立した、触診可能なMeth A 腫瘍の拒絶に効果があることが示された。高用量のMeth Aを注射され、rMVAmup53と抗-CTLA-4 mAb (9H10)をワクチン接種したマウスは触診可能な腫瘍小結節の形成のすぐ後、完全な腫瘍退行および持続的な腫瘍免疫性を示した。インビボ抗体枯渇研究により、この抗腫瘍効果は主にCD8+、そして程度は低いがCD4+依存的であることが確認された。
【0042】
上記結果は腫瘍特異的ではないことを確認するために、rMVAmup53およびCTLA-4遮断薬免疫調節物質によるワクチン接種を11A-1またはMC-38 腫瘍細胞を注入されたマウスに行った。11A-1は迅速に増殖する悪性細胞株であり、免疫原性は低い。MC-38は結腸癌細胞株である。11A-1またはMC-38腫瘍細胞を注入され、4日後にrMVAmup53および抗-CTLA-4 mAbをワクチン接種したマウスはそれらの腫瘍を拒絶した。抗-CTLA-4 mAbの代わりにCpG ODNを用いた場合も同様の結果が見られた。rMVAmup53とCpG ODNで処理したほとんどのマウスは触診可能な腫瘍を発症せず、持続性腫瘍免疫性を発達させ、60日後の再攻撃を拒絶した。
【0043】
抗-CTLA-4 mAbおよびCpG ODN 免疫調節物質のさらなる可能性のある効果を、両方の免疫調節物質をrMVAmup53と組み合わせて、触診可能な腫瘍を有する11A-1注射マウスに投与することによって調べた。腫瘍拒絶および生存の延長がrMVAmup53と組み合わせて両方の免疫調節物質を与えられたマウスのほとんどにおいて観察された。rMVAと組み合わせて一方の免疫調節物質のみを与えられたマウスは一方で、すべて最終的には腫瘍増殖に屈した。両方の免疫調節物質の組み合わせは単独で作用する免疫調節物質よりもより有益であるだけでなく、その組み合わせ効果は、免疫調節物質の効果の単純な和よりも大きかった。MC 38 腫瘍を担持するマウスにおいても同様の結果が見られた。
【0044】
ヒト p53 配列を含む組換えMVAの効力を調べるために、rMVAhup53をヒトp53をトランスフェクトした4T1(H-2d) 細胞を注射したhupki マウスに投与した。4T1(H-2d)はマウス乳癌細胞株である。4T1 細胞の注射の6日後にマウスにrMVAhup53をワクチン接種し、10日後に再びワクチン接種した。二回目のワクチン接種の際に、CpG ODNおよび抗-CTLA-4 mAbもともに投与した。ワクチンと両方の免疫調節物質で処置されたマウスは、統計的に有意な生存の向上を示した。
【0045】
上記結果は様々な悪性腫瘍型におけるp53を標的とする免疫応答の誘発における新規rMVAmup53またはrMVAhup53 無細胞ワクチンの効力を示すと共に、このワクチンに対する抗-CTLA-4 mAbおよびCpG ODNの免疫調節物質としての効力を示す。したがって、本発明はp53を発現するように操作された組換えMVAウイルス(rMVAp53)を含む組成物を提供する。本発明はさらにrMVAp53の投与による多様なp53-発現悪性腫瘍に対する免疫応答を誘発させる免疫療法を提供する。
【0046】
rMVAp53の対象への導入は当業者に知られたいずれの方法で行ってもよく、効力または安全性について、腫瘍小結節の位置に依存しない。したがって、rMVAp53は送達のいくつかの可能な態様を挙げると、血管内、皮下、腹膜、筋肉内、皮内または経皮注射によって投与できる。rMVAp53は医薬上許容される媒体、例えば通常の生理食塩水、植物油、鉱油、PBS等中に有効な用量にて製剤として調製すればよい。治療用調製物は、生理的に許容される液体、ゲルまたは固体担体、希釈剤、アジュバントおよび賦形剤を含んでいてもよい。添加剤としては殺菌薬、等張を維持する添加剤(例えば、NaCl、マンニトール)、化学的安定性を維持する添加剤(例えば、緩衝剤、保存料)およびその他の成分が挙げられる。非経口投与のためには、rMVAp53は溶液、懸濁液、乳濁液または凍結乾燥粉末として、医薬上許容される非経口媒体と共に製剤すればよい。リポソームまたは非水性媒体、例えば、不揮発性油を用いてもよい。製剤は当該技術分野で知られた方法によって滅菌すればよい。
【0047】
rMVAp53製剤は共刺激物質、例えば、サイトカイン、腫瘍抗原、病原体由来抗原、または免疫調節物質によってさらに増強してもよい。共刺激物質はrMVAp53と組み合わせて免疫応答を直接的または間接的に刺激するいずれの薬剤でもよく、APCまたはT-細胞機能を調節するその能力について選択すればよい。例えば、MVAを操作してGM-CSF、IL-12、またはその他の刺激性サイトカインを発現させ、共刺激物質を産生してもよく、rMVAp53と共刺激物質 (ここで:刺激性サイトカインを発現するMVA)の組み合わせを対象に導入してもよい。処置は抗原提示細胞を刺激する以下のようなサイトカインの投与と組み合わせて行ってもよい。例えば、顆粒球-マクロファージコロニー刺激因子 (GM-CSF)、マクロファージコロニー刺激因子 (M-CSF)、顆粒球コロニー刺激因子 (G-CSF)、インターロイキン 3 (IL-3)、インターロイキン 12 (IL-12)、および当該技術分野で周知のその他のもの。その他の共刺激物質としては、サイトカイン-形質導入腫瘍細胞、例えば、GM-CSFを形質導入された腫瘍細胞、および照射および/またはエキソビボまたはインビボで化学療法薬で処置された腫瘍細胞が挙げられる。化学療法薬または放射療法薬は共刺激物質のさらなる例である。したがって、rMVAp53は当業者に知られた様々な共刺激物質と組み合わせて投与すればよい。
【0048】
製剤はT 細胞の抗原刺激に対する応答を上昇させるのに有効な用量で投与する。T細胞応答の測定は治療される症状によって異なる。T細胞活性の有用な尺度は増殖、サイトカイン、例えば、IL-2、IFNγ、TNFαなどの放出; CD25およびCD69などのマーカーのT細胞発現;および当該技術分野で知られたその他のT 細胞活性の尺度。治療用製剤の用量右派、以下に応じて広範に変動しうる:癌のステージ、投与頻度、投与方法または目的、対象からのrMVAp53のクリアランスおよびその他の因子。投与される用量は以下の公知の因子に応じて変動する。例えば、特定の薬剤の薬力学特性、投与態様および経路、レシピエントの年齢、健康状態および体重、症状の性質および程度、併用療法、治療頻度、および所望の効果。用量は週に1回または2週間に1回ほどの低頻度で投与すればよく、小用量に分けて、毎日、週に2回などを投与して、効果的な用量レベルを維持すればよい。
【0049】
一般に、活性成分の一日用量は約 106-1011 IU (感染単位)/kg 体重とすればよい。内部投与に好適な用量形態は一般に約 106〜1012 IU の活性成分/単位を含む。活性成分は組成物の総重量に対して0.5〜95重量%の範囲でよい。場合によっては、過剰のT 細胞増殖によって治療期間を制限することが望ましいこともある。該制限は患者の治療に対する応答、患者におけるT 細胞数などに応じて経験的に決定される。T細胞の数は患者において当該技術分野で知られた方法によりモニターでき、例えばT細胞特異的抗体による染色やフローサイトメトリーが挙げられる。
【0050】
本発明の好適な態様において、rMVAp53は免疫調節物質、具体的には、CTLA-4遮断薬またはCpG ODNを組み合わせて投与する。rMVAp53およびCTLA-4遮断薬である抗-CTLA-4 mAbの併用投与は、処置せずに放置すると直ちに死に至らしめる進行した腫瘍を退行させることにおいて予期に反し強力である。rMVAp53と CpG ODNとの併用投与についても同じことがいえる。両方の免疫調節物質をrMVAp53と組み合わせて投与した場合に強度はさらに強くなる。さらに、抗-CTLA-4 mAb CpG ODN 免疫調節物質は対象に対して非毒性であり、rMVAp53と組み合わせて投与した場合に腫瘍細胞による致死性の攻撃に対する長期持続性の免疫性を生成することができる。rMVAp53のみの場合と同様に、rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNの対象への導入は、当業者に知られた手順で行えばよく、効力または安全性について腫瘍小結節の位置に依存しない。したがって、rMVAp53、抗-CTLA-4 mAbおよびCpG ODNは、可能性のあるいくつかの送達態様を挙げると、血管内、皮下、腹膜、筋肉内、皮内または経皮注射によって投与できる。rMVAp53、抗-CTLA-4 mAbおよびCpG ODNは共に投与しても別々に投与してもよく、いずれの順で逐次に投与してもよく、同じ投与経路で投与しても異なる経路で投与してもよい。rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNは医薬上許容される媒体中で有効用量において製剤として調製してもよく、医薬上許容される媒体としては例えば、通常の生理的食塩水、植物油、鉱油、PBS等が挙げられる。治療用調製物は生理的に許容される液体、ゲルまたは固体担体、希釈剤、アジュバントおよび賦形剤を含んでいてもよい。添加剤としては、殺菌薬、等張性を維持する添加剤、例えば、NaCl、マンニトール;および化学安定性を維持する添加剤、例えば、緩衝剤および保存料およびその他の成分を含んでいてもよい。 rMVAmup53 + 抗-CTLA-4 mAb および/または CpG ODNは混合物または単一の薬剤として投与してもよい。非経口投与のために、抗-CTLA-4 mAbおよびCpG ODNは医薬上許容される非経口媒体と共に、溶液、懸濁液、乳濁液または凍結乾燥粉末として製剤すればよい。リポソームまたは非水性媒体、例えば、不揮発性油を用いてもよい。製剤は当該技術分野で知られた技術によって滅菌すればよい。
【0051】
rMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNの組み合わせは共刺激物質、例えば、サイトカイン、腫瘍抗原、または病原体由来抗原によってさらに増強してもよい。共刺激物質はrMVAp53またはrMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNと組み合わせて免疫応答を直接的または間接的に刺激するいずれの薬剤でもよい。例えば、MVAを操作してGM-CSF、IL-12、またはその他の刺激性サイトカインを発現させ、共刺激物質を産生してもよく、rMVAp53と共刺激物質 (ここで:刺激性サイトカインを発現するMVA)またはrMVAp53 + 抗-CTLA-4 mAb および/または CpG ODNと共刺激物質の組み合わせを対象に導入してもよい。処置は抗原提示細胞を刺激する以下のようなサイトカインの投与と組み合わせて行ってもよい。例えば、顆粒球-マクロファージコロニー刺激因子 (GM-CSF)、マクロファージコロニー刺激因子 (M-CSF)、顆粒球コロニー刺激因子 (G-CSF)、インターロイキン 3 (IL-3)、インターロイキン 12 (IL-12)、および当該技術分野で周知のその他のもの。その他の共刺激物質としては、サイトカイン-形質導入腫瘍細胞、例えば、GM-CSFを形質導入された腫瘍細胞、および照射および/またはエキソビボまたはインビボで化学療法薬で処置された腫瘍細胞が挙げられる。化学療法薬または放射療法薬は共刺激物質のさらなる例である。したがって、rMVAp53は単独でまたは抗-CTLA-4 mAb および/または CpG ODNと組み合わせて当業者に知られた様々な共刺激物質と組み合わせて投与すればよい。
【0052】
治療用製剤の用量は、以下に応じて広範に変動しうる:癌のステージ、投与頻度、投与方法または目的、対象からのrMVAp53、抗-CTLA-4 mAbおよびCpG ODNのクリアランスおよびその他の因子。投与される用量は以下の公知の因子に応じて変動する。例えば、特定の薬剤の薬力学特性、投与態様および経路、レシピエントの年齢、健康状態および体重、症状の性質および程度、併用療法、治療頻度、および所望の効果。用量は週に1回または2週間に1回ほどの低頻度で投与すればよく、小用量に分けて、毎日、週に2回などを投与して、効果的な用量レベルを維持すればよい。
【0053】
一般に、活性成分(抗体)の一日用量は約0.1 〜 100 mg /kg 体重とすればよい。内部投与に好適な用量形態は一般に約 0.1 mg 〜 500 mgの活性成分/単位を含む。活性成分は組成物の総重量に対して0.5〜95重量%の範囲でよい。場合によっては、過剰のT 細胞増殖によって治療期間を制限することが望ましいこともある。該制限は患者の治療に対する応答、患者におけるT 細胞数などに応じて経験的に決定される。T細胞の数は患者において当該技術分野で知られた方法によりモニターでき、例えばT細胞特異的抗体による染色やフローサイトメトリーが挙げられる。製剤はT 細胞の抗原刺激に対する応答を上昇させるのに有効な用量で投与する。T 細胞応答の判定は治療すべき症状によって異なる。T 細胞活性の有用な尺度は増殖、サイトカイン、例えば、IL-2、IFNγ、TNFαなどの放出; CD25およびCD69などのマーカーのT 細胞発現;およびその他の当該技術分野に知られたT 細胞活性の尺度である。
【0054】
本発明はさらに当業者にp53-発現悪性腫瘍に対する免疫応答を誘発するための免疫治療計画の準備を可能とするキットを提供する。キットの例は、rMVAp53、CTLA-4遮断薬および/またはCpG ODN、これら化合物は対象におけるp53-発現悪性腫瘍に対する免疫応答を誘発するために用いるという指示書を含む。キットはさらに1以上の医薬上許容される担体を含んでいてもよい。投与する場合、キットの組成物は医薬上許容される調製物において投与される。「投与」、「投与する」および「導入する」の語は、本発明の組成物をp53-発現悪性腫瘍の治療または予防を必要とする個体に対して医薬として提供することをいう。主成分または活性成分として本発明の組成物を含むこの医薬は、広範な治療用量形態で局所、経口、全身、局部、および非経口投与用の常套の媒体中で投与すればよい。したがって、本発明のキットは非経口投与用の組成物を提供し、これは許容される担体、好ましくは水性担体中に溶解または懸濁された組成物の溶液を含む。組成物は医薬上許容される補助剤を適当な生理条件に応じて所望により含んでいてもよく、例えば、pH調整剤および緩衝剤、浸透圧調整剤、湿潤剤など、例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウム、モノラウリン酸ソルビタン、オレイン酸トリエタノールアミンおよびその他の多くのものが挙げられる。非経口投与のための化合物を調製する実際の方法は当業者に知られているか明らかであり、より詳細には、例えば、Remington: The Science and Practice of Pharmacy ("Remington's Pharmaceutical Sciences") Gennaro AR ed. 20th edition、2000: Williams & Wilkins PA、USAに記載されており、これは引用により本出願に含める。
【0055】
かかる調製物は常套的に医薬上許容される濃度の塩、緩衝剤、保存料、許容される担体、補助的免疫増強剤、例えば、アジュバントおよびサイトカインおよび所望によりその他の治療薬を含んでいてもよい。本発明の調製物はすべて有効量で投与される。有効量は医薬調製物の単独、またはさらなる用量と組み合わせて所望の応答を刺激する量である。癌の治療の場合、所望の応答は癌の発症または進行の阻害または癌を退行させることである。これは一時的に疾患の進行を遅くさせることも含むが、しかしより好ましくは、疾患の進行を永久に停止させることを含む。これらの所望の応答は常套方法によってモニターしてもよいし、本明細書に記載する本発明の診断方法にしたがってモニターしてもよい。投与態様に応じて104 IU/kg 〜 1011 IU/kgの免疫原の用量範囲が有効であると考えられる。好ましい範囲は、106 IU〜109 IU/kgであると考えられる。絶対量は様々な因子に依存し、例えば、投与に選択された組み合わせ、投与が単一用量であるか複数用量であるか、そして個々の患者パラメーター、例えば、年齢、身体状態、身長、体重および疾患のステージが挙げられる。これらの因子は当業者に周知であり、常套の実験によって決定しうる。
【0056】
以下の実施例は特許請求の範囲に記載の本発明をさらに説明するために提供するものであり、本発明の範囲を限定するものではない。特定の材料が言及される範囲は単に例示の目的であり、本発明を限定する意図のものではない。特に断りのない限り、一般的クローニング方法、例えば、Sambrook et al.、Molecular Cloning、Cold Spring Harbor Laboratory (2001)、Ausubel et al. (Eds.) Current Protocols in Molecular Biology、John Wiley & Sons (1986、2000)に記載のものを用いる。当業者であれば、本発明を実施することなく、本発明の範囲を逸脱することなく、同等の手段または反応材料を開発することが出来よう。
【0057】
多くの改変が本発明に拘束されつつここに記載した手順においてなすことができることが理解できよう。同様に、遺伝コードの縮重のために、ここに開示のものと同等のコドンを有する核酸配列がここに開示されたものと機能的に同等または同一のタンパク質をコードすることが理解される。かかる改変は本発明の範囲内に含まれると言うことが本発明者らの意図である。
【実施例】
【0058】
実施例
材料および方法
動物
雌性6-8週齡Balb/c、C57BL/6、B6.129S2-IL6tm1Kopf (IL-6-/-、およびIFN-γ ノックアウト (IFN-γKO) マウス(Balb/cバックグラウンド)は、The Jackson Laboratory (Bar Harbor、Maine)から得た。TLR9-/- マウスは、Dr. Shizuo Akira (Osaka University、Osaka、Japan)から譲り受けた。マウスを特定病原体除去環境に維持した。すべての研究は、Research Animal Care Committee of the City of Hope National Medical Centerにより認可を受け、AAALACガイドラインにしたがって行った。
【0059】
細胞株
CV-1 (Kit 1965)、TK- (Berson 1996)、および仔ハムスター腎臓細胞(BHK-21) (Macpherson 1962)をAmerican Type Culture Collection (ATCC) (Manassas、VA)から購入し、非必須アミノ酸、L-グルタミン、および10% FCS を追加したMEM中で培養した。11A-1 (Selvanayagam 1995)は、Dr. R.L. Ullrich (University of Texas Medical Branch、Galveston、TX)から譲り受けた。Hek 293 細胞およびp53null 10.1 細胞は、Dr. K.K. Wong および Dr. Susan Kane (City of Hope National Medical Center、Duarte、CA)から譲り受けた。MC-38 (Tan 1976)は、Dr. S.A. Rosenberg (National Cancer Institute、Bethesda、MD)から譲り受けた。Meth A 肉腫細胞(Meth A) (DeLeo 1977)は、Dr. L. J. Old (Memorial Sloan-Kettering Cancer Center、New York、NY)から譲り受けた。Meth Aを腹水腫瘍として継代した。細胞を収集し、計数し、使用前にPBSで洗浄した。Meth A、11A-1、およびMC-38 腫瘍細胞株の特徴を以下の表に要約する:
【0060】
【表1】
【0061】
抗体
抗-CD4 (GK1.5) (Dialynas 1983)および抗-NK1.1 (PK136) (Koo 1984)をATCCから購入した。抗-CD8 (H35) (Miconnet 2001)および抗-CTLA-4 mAb (9H10) (Krummel 1995)はJames P. Allison (University of California、Berkeley、CA)から譲り受けた。抗体はCELLine Device (BD Biosciences、Bedford、MA)を用いて産生した。IgG 抗体はタンパク質G-セファロース (Amersham、Uppsala、Sweden)に吸収し、次いで0.1M Glycine-HCl、pH 2.7での溶出により精製した。産物を次いでリン酸緩衝生理食塩水 (PBS)で透析し、Centriplus 遠心ろ過装置 (Millipore、Bedford、MA)を用いて濃縮した。コントロールシリアンハムスター IgGをJackson Immuno Research (West Grove、PA)から得た。
【0062】
ウイルスコンストラクト
マウス p53を発現するrMVA (rMVAmup53):
野生型 MVA (wtMVA)をDr. Bernard Moss および Dr. Linda Wyatt (National Institutes of Health Bethesda、MD)から得た。mup53 を含む組換えMVA (rMVA)の作成のためのwtMVA ストックを特定病原体除去ニワトリ胚線維芽細胞(SPF/CEF)上で増殖させる。wtMVA ストックを免疫染色により力価測定し、アリコートに分け、-80℃で保存する。
【0063】
マウス p53 (mup53)はヒトp53と類似であり、80% 配列相同性を有する(Halevy 1991; Sukumar 1995)。全長野生型mup53のmRNA コード配列を配列番号1に示す。マウスとヒト p53とのこの相同性のレベルにより、マウス系は良好なp53に対する寛容に打ち勝つ免疫学的アプローチの評価の前臨床モデルとなる。マウス p53を発現するrMVA をEspenschied 2003に記載のようにwtMVAとマウス p53 インサートを含むpMCO3 挿入ベクターとの相同組換えにより作成した。マウス野生型p53の全長cDNAをマウス脾細胞から得たmRNAのPCRにより増幅した。マウス p53 PCR 産物をMVA 発現ベクター pMCO3のクローニング部位にライゲーションした(これもDr. Moss および Dr. Wyattから得た)。このベクターはMVAゲノムのデリーションIIIに挿入する配列を含み、また、gus(大腸菌β-グルクロニダーゼ)オペロンをスクリーニングの目的で含む(Ourmanov 2000)。組換えMVAの作成は、BHK-21 細胞の単層上で達成した (Espenschied 2003)。簡単に説明すると、BHK-21 細胞にLipofectin (Invitrogen、Carlsbad、CA)を用いて20 μgのプラスミド DNAをトランスフェクトし、wtMVAをmoi 0.01にて感染させた。感染細胞を48時間インキュベートし、次いで収集し、ペレットにし、3サイクルの凍結乾燥および超音波処理に供し、細胞を溶解した。マウス p53を発現するrMVAウイルス (rMVAmup53)をX-GlcA (5-ブロモ-4-クロロ-3-インドリルβ-D-グルクロニド、Sigma-Aldrich、St Louis、MO)の添加によるgus 発現によってスクリーニングした。10ラウンドの精製後、rMVAmup53をBHK-21 単層上に播いた。rMVAmup53 力価はVectastain Elite ABC Kit (ベクター Laboratories、Burlingame、CA)を用いて感染培養の免疫染色によって測定した。
【0064】
ヒトp53を発現するrMVA (rMVAhup53):
ヒト p53を発現するrMVA (rMVAhup53)の2種類のコンストラクトを作った。全長野生型 hup53をコードするmRNA 配列を配列番号2に示す。第一のものはpLW51 挿入プラスミドを用いて作り、第二のものはpLW22 挿入プラスミドを用いて作った。第一のコンストラクトを作るために用いたwtMVAをSPF/CEF上で増殖させた。第二のコンストラクトを作るために用いたwtMVAをBHK-21 (BHK) 細胞上で増殖させた。wtMVA ストックを免疫染色により力価を測定し、アリコートに分け、-80℃で保存した。
【0065】
pLW51を第一の rMVAhup53 コンストラクトを作るための挿入プラスミドとして用いた。pLW51は4つの重要な特徴を有する。第一にそれはデリーションIII のMVA 隣接領域を含み、それによって相同組換えを介してMVAのデリーションIII 領域に挿入される。第二に、それは発色スクリーニングマーカー遺伝子、β-グルクロニダーゼ (gus)をP11と称されるワクシニアプロモーターの制御下に含む。第三に、それはMVA 配列から構成される2つの直接反復を(DR1およびDR2と称される) gus スクリーニングマーカー遺伝子に隣接して含むため、gus遺伝子を組換えMVAから除くことが可能である。最後に、それは2つのワクシニアプロモーター (PSYNおよびP7.5) と2つのマルチクローニング部位(MCS)を含むため、2つの別々の外来遺伝子をPSYNおよびP7.5 プロモーターの制御下に挿入することが可能である。第一の MCSは初期/後期 PSYN プロモーターの下流にあり、第二の MCSは初期/後期 PmH5 プロモーターを用いる。これにより、隣接する直接繰返し配列のセットを介した組換えによりgus マーカー遺伝子を除くことが可能である。最初のrMVA ストックの作成をBHK細胞について以前に記載されている方法を用いてCEFに対して行ったが、医薬品安全性試験実施基準 (GLP) 条件とするために改変した。正しい組換えが起こるかを確認するために約40-50の病巣が第一ラウンドのスクリーニングから引き出され、その後5-10を各次なるラウンドにおいて引き出され得る。各選択ラウンドの後、免疫染色または免疫蛍光のいずれかを各プラグ(plug)に対して行って、プラグがhup53遺伝子を発現するかを確認する。細菌遺伝子マーカーのウイルスからの除去を達成するためにhup53を発現する精製MVAを、24 ウェルプレート中に低希釈度で播く。gus遺伝子を示す着色反応を有さないウェルをさらにhup53 遺伝子産物の存在について分析する。これは細胞からのウイルスの回収を可能とする条件を用いて抗体染色によって達成する。着色反応の非存在下でhup53 免疫染色を示すウェルをさらに増殖させて正確な表現型を確認する。gusマーカーの非存在のスクリーニングの最終ラウンドから引き出されるウイルスプラグの一部をCEFの100 mm 組織培養皿で拡張する。この後DNA抽出およびPCR分析を行う(以下に記載)。
【0066】
pLW22を第二の rMVAhup53 コンストラクトの作成のための挿入プラスミドとして用いた。pLW22は相同組換えを介するMVAへの挿入を可能とするMVA 隣接領域を有する。それはまた、発色スクリーニングマーカー遺伝子、β-ガラクトシダーゼを有する。wt hup53をコードするDNAを得るために、大腸菌におけるHp53B プラスミドをATCCから得た(#57254)。Hup53をpHp53B プラスミドから配列番号3のフォワードプライマーと配列番号4のリバースプライマーを用いて増幅した。増幅した wt hup53 DNAをpLW22 ベクターの制限部位Pme-とAsc-1の間に挿入し、pLW22-hup53を作成した。pLW22-hup53のプラスミド配列を配列番号5に示す。
【0067】
rMVAの作成はBHK細胞の単層上で行った。BHK細胞にLipofectin (Invitrogen、Carlsbad、CA)を用いて20μgのプラスミド DNAをトランスフェクトし、wtMVA、moi 0.01で感染した。感染細胞を48 時間インキュベートし、収集し、ペレットにし、三回の凍結乾燥サイクルおよび超音波処理に供し、細胞を溶解した。hup53を発現するrMVAをBluo-galTM基質(Sigma-Aldrich、St Louis、MO) (Chakrabarti 1985)の存在の添加によりβ-gal 発現についてスクリーニングした。10ラウンドの精製の後、rMVAhup53を BHK 単層上に広げた。rMVA 力価は免疫染色感染培養物によってVectastain Elite ABC kit (Vector Laboratories、Burlingame、CA)を用いて測定した。
【0068】
両方のコンストラクトについて、標準的DNA抽出を行う。50μLの細胞可溶化液のエタノール沈殿により、wtMVAの汚染が無いことを確認するためのPCR反応を行うのに十分なDNAが得られた。一セットのPCR プライマーを遺伝子が挿入された組換え部位の隣接領域の外側に設計する。非修飾wtMVA 配列の存在により500 bpのPCR産物が生じるが、hup53を含む配列の挿入は標準的 PCR条件下では増幅するのが通常困難であるより大きな断片(>6 kb)を有する。第二のセットのPCR プライマーはhup53 インサート内の配列を増幅するように設計する。hup53 インサートの存在により、300 bpのPCR 産物が生じる。PCR サンプルを1% アガロースゲルで泳動し、さらなるスクリーニングが残りのwtMVAを除去するために必要であるかを判定するために分析する。ヒト p53を含む精製MVAの例は完全に均質であることが示された(図1)。
【0069】
pp65を発現するrMVA (rMVApp65):
CMV テグメント・タンパク質、pp65を発現するrMVA (rMVApp65)を、rMVAmup53 (Gibson)の構築に用いたものと類似の技術を用いて構築した。
【0070】
マウス p53またはpp65を発現するrVV:
マウス 野生型p53またはpp65 (rVVp53、rVVpp65)を発現する組換えウエスタンリザーブ株ワクシニアウイルスを公表された技術を用いて構築した(Diamond 1997)。
【0071】
マウス p53を発現するrAd:
野生型マウス p53を発現する組換えアデノウイルス(rAd-mup53)をpAd Easyシステム(He 1998)を用いて構築した。pAd Track-CMVとpAd Easy-1 プラスミドとは両方 、Dr. Bert Vogelstein (Johns Hopkins Oncology Center、Baltimore、MD)から譲り受けた。野生型マウス p53 cDNAをCMV プロモーター (p53-pAd Track-CMV)とともに緑色蛍光タンパク質 (GFP)を含むpAd Track-CMV シャトルベクターのBgl II部位とXba I 部位にクローニングした。p53-pAd Track-CMV をpAd Easy-1 プラスミドと共にBJ5183 細胞に共形質転換し、相同組換えによりp53 組換えアデノウイルスコンストラクトを作成した。組換え体におけるp53遺伝子の存在はDNA 配列決定により確認した。p53 組換えアデノウイルスコンストラクトをPac Iで切断し、HEK-293 細胞にトランスフェクトした。rAd-mup53をトランスフェクションの5日後に収集し、p53タンパク質発現をウェスタンブロットにより確認した。アデノウイルスをHEK-293細胞上に広げ、塩化セシウム勾配によって精製した。精製ウイルスをPBSで透析し、HEK-293 細胞上で力価を測定し、20%グリセロール中で-80℃で保存した。
【0072】
オリゴデオキシヌクレオチド(ODN)
CpG モチーフを有する合成ODN 1826 (配列番号6)および非-CpG ODN 1982 (配列番号7) (Moldoveanu 1998)をヌクレアーゼ耐性ホスホロチオエート骨格を用いてTrilink (San Diego、CA)によって合成した。ODNのNa+塩を10 mM Tris (pH 7.0) 1 mM EDTA中5 mg ml-1に再懸濁し、50 μlのアリコートとして-20℃で保存し、注射の前に0.9% 塩化ナトリウム水溶液中に希釈した。
【0073】
実施例1: rMVAmup53によるマウスp53タンパク質の発現:
rMVAmup53による感染後のマウス p53タンパク質の発現を分析し、組換えウイルスからのその発現の忠実度および程度を調べた。rMVAmup53で感染したBHKまたはHEK 293 細胞から可溶化液を調製し、SDS-PAGEおよびウェスタンブロッティングに供した。標準的ウェスタンブロッティング技術をECL ウェスタンブロットキット(Amersham Pharmacia Biotech、England)を用いて行った。サンプルを精製マウス 抗-p53 モノクローナル抗体、PAb 122 (Gurney 1980)とともにインキュベートし、次いで、ECL ウェスタンブロットキット中のペルオキシダーゼ標識ヤギ抗-マウス二次抗体とインキュベーションした。rMVAmup53で感染したBHK細胞のウェスタンブロット分析は豊富なp53 発現を示した(図 2)。その他のウイルスおよび細胞内形態と比較してrMVAmup53によって示された顕著な発現レベルはそのワクチン接種プロトコールにおける有用性を示す。図 1に示すように、rMVAmup53 レーンにおける容積はその他のレーンにおいてゲルにアプライされた容積の80-160分の1であったが、バンドの強度は数倍強かった。これはrMVAmup53による非常に高レベルのp53 発現を示す。Meth A 細胞をポジティブコントロールとして用い、HCMV IE1 エキソン 4 rMVAで感染したBHK細胞をネガティブコントロールとして用いた。Meth Aは Balb/c由来の、腫瘍原性 3-メチルコラントレン誘導性肉腫であり、突然変異p53を過剰発現する。53 キロダルトンバンドが、p53過剰発現Meth A 肉腫とrMVAmup53 感染BHK細胞の両方に観察された(図 1)。これはHCMV IE1 エキソン 4-rMVA 感染BHK細胞において検出可能なp53発現がないことと対照的である。強いp53 発現はrMVAmup53で感染したBHK細胞において蛍光顕微鏡によっても観察された(データ示さず)。
【0074】
実施例2: rMVAmup53によるp53-特異的CTL応答のインビトロでの生成:
ウイルス性および腫瘍関連抗原を発現するrMVA によるマウスのワクチン接種の結果、抗原特異的CTL応答が増強する。この実施例の1つの目的は、rMVAmup53によるワクチン接種がp53 寛容を打ち破り、p53-特異的CTLを生成させることができるかを調べることであった。マウスに5 x107 pfuのrMVAmup53またはrMVApp65のいずれかを腹腔内にワクチン接種した。2週間後、脾臓を収集して分離し、脾細胞を洗浄し、計数した。脾細胞をインビトロで6日間 rAd-mup53またはrMVAmup53で感染した同系 LPS 芽球で再刺激した。wt p53を過剰発現するNa-51CrO4-標識化標的細胞を96 ウェルプレートにエフェクターとともに三連で、様々なエフェクター:標的比で、200μlの完全培地中に添加した。プレートを4 時間37℃でインキュベートし、上清を収集し分析した。パーセント特異的溶解を次式を用いて計算した: パーセント特異的放出 = (実験的放出 自発的放出) / (全放出 - 自発的放出) X 100。rMVAmup53をワクチン接種した脾細胞は、wt p53を過剰発現する標的細胞を認識し、溶解した(図 3)。一方、rMVApp65をワクチン接種したマウスからの脾細胞は激しいpp65 特異的CTL応答を刺激するが、p53を過剰発現する標的は認識せず(図 3B)、リンパ球応答の特異性を示した。rMVAmup53 ワクチン接種は突然変異p53を担持する細胞株であるMeth AのCTL認識も刺激することが出来る。rMVAmup53をワクチン接種した再刺激脾細胞は突然変異体 p53を過剰発現するMeth A 細胞を認識したが、rMVAmup53をワクチン接種した脾細胞は認識しなかった(図 3c)。
【0075】
実施例3: インビボ rMVAmup53 腫瘍攻撃実験:
rMVAmup53による単回ワクチン接種の結果、CTL応答が増強したので、腫瘍細胞のインビボでの増殖に対するrMVAmup53 ワクチン接種の効果を調べるのには十分な正当性があった。
【0076】
統計的方法
初期に屠殺する必要がある、いくらかの腫瘍の増殖速度の実験のために、増殖曲線はlog順位検定を用いて一定のサイズになるときまでに比較した。1つの群のその他の全ての群との比較は単一のオムニバス検査の後に行った。細胞枯渇実験のために、すべてのマウスを一定の時間追跡し、最終的な腫瘍サイズは、3以上の群がある場合には有意Kruskal-Wallis検定の後にウィルコクスン順位和検定によって比較した。生存実験のためには、log順位検定を用いた。
【0077】
rMVAmup53 対Meth A 細胞
6週齡雌性 Balb/c マウスに、皮下 (s.c.)経路によって左脇腹に5 x105 Meth A 細胞を注射した。Meth A 細胞を皮下注射したマウスには迅速に増殖する線維肉腫が発生し、21 日以内にマウスのほとんどが死に至った(図 3)。3日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内 (i.p.) 注射によってワクチン接種した。ネガティブコントロールマウスには5 x107 rMVApp65またはPBSを注射した。皮下腫瘍のサイズをキャリパーを用いて3次元で週に2回測定した。rMVAmup53 処置動物における腫瘍は、コントロール動物と比較してかなりゆっくりと増殖した。14日目、rMVAmup53 処置群(n=16)についての平均皮下腫瘍体積はrMVApp65 (n=16)およびPBS (n=12) コントロールの両方と比較して劇的に小さかった(22mm3対348 mm3、p<0.001および 22mm3対252mm3、p<0.001、スチューデントt検定)。rMVAmup53 処置動物の生存もいずれのコントロール群と比較しても有意に延長していた(図 4)。16匹のrMVAmup53 免疫化マウスのうち12匹ではまったく腫瘍を発達させることができなかった。この12匹の腫瘍の無いrMVAmup53 処置動物を52日目に5 x105 Meth A 腫瘍細胞で再攻撃した。すべての動物は30日の観察期間にわたって腫瘍が無いままであった(データ示さず)。
【0078】
rMVAmup53 + 抗-CTLA-4 mAb対Meth A 細胞
腫瘍ワクチンを至適化する1つの強力な戦略には、CTLA-4のリガンドへの結合をブロックする抗体を用いるT 細胞応答性の負の制御が含まれる。この現象はCTLA-4遮断と称される。抗-CTLA-4 mAbの腫瘍関連自己抗原を発現する癌ワクチンとの組み合わせての使用は、場合によっては寛容に打ち勝つ腫瘍拒絶と自己免疫の誘導をもたらす。それゆえ、CTLA-4に特異的なmAbをrMVAmup53 ワクチン接種に加えて、それがインビボでMeth Aに対する抗-腫瘍活性を協同および増強するかを調べた。CTLA-4遮断のみの強力な抗腫瘍効果に打ち勝つためにより厳しい腫瘍モデルを設計した。6週齡 Balb/c マウスの左脇腹に5 x105 Meth A 細胞ではなく、106 Meth A 細胞を皮下注射し、処置を触診可能な腫瘍小結節が同定されるまで延長した(6日目)。このより厳しいモデルはCTLA-4遮断の効果に打ち勝ち、抗-CTLA-4 mAb処置にもかかわらずほとんどのマウスにおいて迅速に致死性の腫瘍が生じた(図 5)。7日目に、マウスに5 x107 pfuのrMVAmup53を腹腔内注射した。コントロールは上記の通りとした。抗-CTLA-4 mAb 抗体またはコントロールハムスター Abを6、9および12日目にそれぞれ 100、50および50μgの用量で腹腔内注射した。rMVAmup53 + 抗-CTLA-4 mAbで免疫した14匹のマウスのうち11匹が腫瘍を拒絶し、60日の観察期間の持続中腫瘍無しで生存した(図 5)。一方、rMVApp65およびコントロール抗体で処置したマウは進行性腫瘍によりすぐに死にいたり(図 5)、PBS処置コントロールも同様であった(データ示さず)。11匹の腫瘍の無い、rMVAmup53 + 抗-CTLA-4 mAb 処置マウスは60日目の106 Meth A 腫瘍細胞での再攻撃も拒絶し、30日間の観察期間の持続中、腫瘍の無いままであった (データ示さず)。
【0079】
rMVAmup53 + 抗-CTLA-4 mAb対11A-1 細胞
6週齡 Balb/c マウスの左脇腹に2 x 106 11A-1 細胞を皮下注射したた。11A-1は迅速に増殖する免疫原性の弱い悪性細胞株である。106の放射線照射した11A-1 腫瘍細胞をワクチン接種したマウスは11A-1による続く攻撃を拒絶することができなかった (データ示さず)。抗-CTLA-4 mAbまたはコントロールハムスター抗体を4、7、および10日目に100、50、および50μg/用量でそれぞれ腹腔内注射した。5日目に、マウスに5 x107 pfuの rMVAmup53、5 x107 MVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍を三次元でキャリパーを用いて週に2回測定した。rMVAmup53 + 抗-CTLA-4 mAbをワクチン接種したマウスはそれら腫瘍を拒絶した(図 6)。抗-CTLA-4 mAbのみまたはコントロール MVA ワクチンで処置した動物は迅速に進行性致死性腫瘍を発達させた(p = 0.00044、抗-CTLA-4 mAb 遮断を伴うrMVAmup53をコントロール群と比較)。
【0080】
rMVAmup53 + 抗-CTLA-4 mAb 対 MC-38 細胞
6週齡 C57BL/6 マウス、TLR9-/-、またはIL-6-/- マウスの左脇腹に1 x 106 MC-38 細胞を皮下注射した。抗-CTLA-4 mAbまたはコントロールハムスター抗体を4、7および10日目にそれぞれ100、50、および50μg/用量で腹腔内注射した。5日目に、マウスに5 x107 pfuのrMVAmup53、5 x107rMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍をキャリパーを用いて三次元で週に2回測定した。rMVAmup53 + 抗-CTLA-4 mAbをワクチン接種したマウスはそれら腫瘍を拒絶したが、抗-CTLA-4 mAbのみ、またはコントロール MVA ワクチンで処置したマウスは迅速に進行性腫瘍を発達させた(p = 0.0001、抗-CTLA-4 mAb をともなうrMVAmup53をコントロール群と比較) (図 7)。
【0081】
rMVAmup53 + CpG ODN 対 11A-1 細胞
CpG ODN処置は多くの実験腫瘍ワクチンモデルにおいて有効な免疫調節物質であることが示されている(Krieg 2002)。マウスを上記のように11A-1 腫瘍で攻撃した。15 nmoles の CpG ODNまたは非-CpG ODN コントロールを4、9、および14日目に腹腔内注射した。5日目に、マウスに5 x107 pfuの rMVAmup53、5 x107 rMVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。皮下腫瘍をキャリパーを用いて三次元で週に2回測定した。rMVAmup53とCpG ODNはそれぞれ別々にわずかな腫瘍増殖の低減をもたらしたが、すべての動物は次第に致死性の腫瘍を発達させた。CpG ODNとrMVAmup53の組み合わせてのワクチン接種の結果、腫瘍増殖が有意に低減した(p = 0.00002) (図 8)。rMVAmup53 + CpG ODNで処置した8匹の動物のうち6匹は触診可能な腫瘍を発達させず、腫瘍免疫性が持続し、60日目の11A-1による再攻撃を拒絶した(データ示さず)。
【0082】
rMVAmup53 + CpG ODN対Meth A 細胞
11A-1についてのものと類似の腫瘍拒絶のパターンがBalb/c マウスにおける初期に確立したMeth A 腫瘍の処置後にみられた(p = 0.0015) (図 9)。
【0083】
rMVAmup53 + CpG ODN 対 MC-38 細胞
rMVAmup53 ワクチン接種に対するCpG ODNの免疫調節効果が株特異的ではないことを示すために、ワクチン接種戦略を初期に確立したMC38 大腸癌を担持するC57BL/6 マウスにおいて繰り返した。rMVAmup53 + CpG ODNによるワクチン接種の結果、腫瘍増殖が有意に抑制された(p = 0.0004) (図 10)。
【0084】
rMVAmup53 + 抗-CTLA-4 mAb + CpG ODN 対 11A-1 細胞
CpG ODNと抗-CTLA-4 mAbのrMVAmup53 ワクチン接種に対する強力な追加的効果を評価するためにより厳しい腫瘍モデルを設計した。6週齡 Balb/c マウスの左脇腹に2 x 106 11A-1 細胞を皮下注射し、触診可能な腫瘍が存在するまで2週間追跡した。抗-CTLA-4 mAb またはコントロールハムスター抗体を14、17、および20日目に、100、50、および50μg/用量にて腹腔内注射した。15 nmolesのCpG ODNを14、19、および24日目に腹腔内注射した。15日目に、マウスに5 x107 pfu のrMVAmup53、5 x107 MVApp65、またはPBSのいずれかを腹腔内にワクチン接種した。
【0085】
抗-CTLA-4 mAbまたはCpG ODN 免疫調節物質と組み合わせたrMVAmup53 ワクチン接種の結果、生存の延長が起こったが、すべての動物において最終的には、進行性腫瘍が増殖した。抗-CTLA-4 mAbおよびCpG ODN 投与とrMVAmup53 ワクチン接種の組み合わせの結果、ほとんどの処置動物において腫瘍拒絶および生存の延長が起こった(図 11)。抗-CTLA-4 mAbおよびCpG ODNの組み合わせはCpG ODNのみ(p = 0.02)または抗-CTLA-4 mAbのみ(p = 0.01)と比べてよりよい免疫調節活性を提供した。抗-CTLA-4 mAbおよびCpG ODNの組み合わせ投与の効果は、60日目の生存に関して、両方の免疫調節物質の単独での単純な添加と比べてより大きい利益をもたらす。
【0086】
rMVAmup53 + 抗-CTLA-4 mAb + CpG ODN 対 MC-38 細胞
類似のパターンがMC 38 腫瘍を担持するC57BL/6 マウスにおいてみられた(図 12)。MC-38 腫瘍を担持するC57BL/6 マウスをrMVAmup53 +抗-CTLA-4 mAbおよびCpG ODNの組み合わせで11A-1について記載したように処置した。この腫瘍モデルにおいて、抗-CTLA-4 mAbおよびCpG ODNの組み合わせはCpG ODNのみ(p = 0.002)または抗-CTLA-4 mAbのみ(p = 0.001)よりも良好な免疫調節活性を提供した。両方の腫瘍モデルにおけるこの組み合わせ効果は単に相加効果ではない。というのは、CpG ODN と抗-CTLA-4 mAbはともにすでに最大効力の用量で投与されているからである。両方の免疫調節物質を少なくとも2種類の腫瘍においてともに用いた場合にみられた活性の顕著な上昇は、併用療法がヒトにおいて保証されることの研究をさらに示唆する。
【0087】
実施例4: 抗-CTLA-4 mAbおよびCpG ODN 免疫調節効果の細胞による要求:
抗-CTLA-4 mAbおよびCpG ODNの免疫調節効果の細胞による要求を調べるために、Balb/cマウスからワクチン接種の前にCD4+、CD8+、またはNK 細胞を枯渇させた。枯渇は200μg のCD4+、CD8+、またはNK1.1 細胞 特異的 mAb、あるいはコントロール mAbの腹腔内注射により達成した。注射は、-1、1、3、4、6、8および15日目に行い、動物が死ぬまで7日ごとに投与を維持した。この投与計画は処置動物の末梢血のフローサイトメトリーに基づくと、(>95%) Balb/c マウスからCD4+、CD8+、またはNK 1.1 細胞を枯渇させることが示された(データ示さず)。
【0088】
rMVAmup53 ワクチン接種に対するCTLA-4遮断の免疫調節効果の細胞による要求をBalb/c マウスにおけるMeth A 腫瘍モデルを用いて評価した。CD8+ T 細胞またはCD4+および CD8+ T 細胞を同時に枯渇させたマウスでは迅速に致死性の腫瘍が発達した。これら腫瘍はrMVAmup53 および 抗-CTLA-4 mAbによるワクチン接種に抵抗性である。一方、CD4+ T 細胞枯渇の結果、ワクチンへの応答が部分的にのみ抑止された。NK1.1 細胞枯渇はワクチン接種マウスが Meth Aを拒絶する能力にほとんど効果を有さなかった(図 13a)。結果は枯渇に用いるmAbをワクチンおよび抗-CTLA-4 mAb処置後に投与した場合も同じであった (データ示さず)。同様の結果がMeth A 腫瘍モデルではなく11A-1 腫瘍モデルを用いた場合にも得られた。rMVAmup53および抗-CTLA-4 mAbの治療効果は、枯渇させる用量の抗-CD8+ mAbの投与により排除できた (p = 0.004) (図 15)。抗腫瘍効果は枯渇させるための抗-CD4+ mAbの投与により部分的に阻害され(p = 0.008)、NK 枯渇に用いるmAbの投与による影響はなかった。これらの結果は抗-CTLA-4 mAbの免疫調節効果が完全にCD8+ 細胞に依存し、部分的にCD4+ 細胞に依存し、まったくNK 細胞には依存しないことを示す(Espenschied 2003)。
【0089】
rMVAmup53 ワクチン接種に対するCpG ODNの免疫調節効果の細胞による要求を4日間確率 11A-1 腫瘍を有するBalb/c マウスを用いて評価した。抗-CTLA-4 mAbと同様にCpG ODNの MVAmup53 ワクチン接種に対する免疫調節効果は枯渇させるための CD8+ mAbの投与によって完全に抑止できた(p = 0.004) (図 14)。しかし抗-CTLA-4 mAbと異なり、CpG ODNの免疫調節効果はCD4+ 枯渇によっては影響を受けず、一方NK 細胞の枯渇は部分的にワクチン効果を抑止した(p = 0.007、NK枯渇をCD4+およびコントロール抗体枯渇と比較)。CD4+ とNKの細胞による要求の抗-CTLA-4 mAbおよびCpG ODNでの差は驚くべきである。というのは両方の免疫調節物質が等価なレベルの拒絶をもたらすからである。これらの結果は、これら2つの免疫調節物質が異なる免疫学的機構で作用することを示唆する。この情報は、抗-CTLA-4 mAb /CpG ODN 組み合わせ投与のrMVAmup53に対する効果に関するデータとともに、これら2つの免疫調節物質による腫瘍増殖に対する相乗効果を示唆する。
【0090】
IFN-γの寄与
CTLA-4遮断およびrMVAmup53 ワクチン接種の効果に対するIFN-γ 分泌の寄与をIFN-γKO マウスにおいて評価した。非ワクチン接種マウスおよびrMVApp65および抗-CTLA-4 mAbをワクチン接種したマウスの両方は正常Balb/c マウスにみられるのと同様の速度で致死性の腫瘍を発達させた(図 13b)。 rMVAmup53および抗-CTLA-4 mAb をワクチン接種した5匹のIFN-γKO マウスのうち3匹は致死性の腫瘍増殖を発達させ、IFN-γのワクチン/CTLA-4遮断効果への寄与が確認された。
【0091】
TLR 9の寄与
細胞サブセット枯渇研究は、CTLA-4遮断とCpG ODNの免疫調節活性の機構が異なることを示唆する。CpG ODN 活性はTLR9 受容体との相互作用を介するB-細胞とプラズマ細胞様 樹状細胞の刺激に起因する(Chu 1997)。CpG処置は、TH1 サイトカイン環境およびNK 細胞増殖の刺激に偏らせ、これによって腫瘍拒絶の部分的効果が説明される。 CpG ODN と CTLA-4遮断の免疫調節効果に関与する異なる経路をさらに説明するために、MC-38 腫瘍攻撃実験をTLR9-/- マウスにおいて行った。TLR9-/- マウス はCpG ODN 投与に対して免疫学的に応答することができない(Hemmi 2000)。予測されるように、初期確立MC-38 腫瘍を担持するTLR9-/- マウス はCpG ODNおよびrMVAmup53 ワクチン接種に対して免疫学的に応答することができなかった(図 16)。一方、抗-CTLA-4 mAbをrMVAmup53 ワクチン接種に含めた場合、TLR9-/- マウスにおける腫瘍拒絶が起こり(p = 0.0009)、これは wt C57BL/6 マウスにおいてみられるのと同様であった(図 16、図 7)。
【0092】
IL-6の寄与
CpG ODNおよびCTLA-4遮断はともにCD25+ CD4+ 抑制因子または調節性 T 細胞(Treg)を阻害し、この効果が記載した腫瘍モデルにおけるそれらの免疫調節活性の原因であり得る。CTLA-4の遮断はTregに対する直接の阻害効果を有すると考えられ、Treg のほとんどは構成的にCTLA-4を発現する(Read 2000)。一方、CpG ODNはDCによるIL-6の分泌を介してTreg 活性を阻害する(Pasare 2003)。IL-6の CpG ODおよび抗-CTLA-4 mAb 免疫調節効果に対する役割を評価するために、腫瘍攻撃実験をIL-6-/- マウスにおいて行った。初期確立 MC-38 腫瘍を担持するIL-6-/- マウスは腫瘍の拒絶によりCpG ODNとのrMVAmup53 ワクチン接種に免疫学的に応答することができなかった(図 17)。これはCpG ODNがその免疫調節効果を少なくとも部分的にTreg 細胞阻害のIL-6依存的経路を介して媒介していることを示唆する。一方、抗-CTLA-4 mABをrMVAmup53 ワクチン接種に含めた場合wt C57BL/6 マウスにおいてみられたのと同程度にIL-6-/- マウスにおける腫瘍拒絶が起こった(p = 0.02) (図 17、図 7)。
【0093】
実施例5: rMVAhup53によるヒト p53の発現:
BHK細胞に精製rMVAhup53を感染させた。hup53の発現を24および 48 時間目に測定し、ウェスタンブロットおよび免疫組織化学によって分析した。感染したrMVAhup53 細胞は両方の時点で激しいhup53の発現を示した(図 18)。
【0094】
実施例6: インビボ rMVAhu53 腫瘍攻撃実験:
ヒト p53を発現する新規なマウスノックインモデルであるHupki マウスを129/Sv 遺伝的背景においてDr. Monica Hollstein (DKFZ、Heidelberg、Germany)から得た。このマウスを4世代にわたってBalb/c(H-2d) バックグラウンドに対して戻し交雑し、腫瘍およびその他の試薬が容易に入手できるマウスバックグラウンドにおいてノックイン導入遺伝子の利益を享受できるようにした。Balb/c バックグラウンドにおけるhupki マウスを戻し交雑してPCR分析によって確認してホモ接合性とした。これには近親交配効果を最小とする交雑手段を用いた (データ示さず)。4T1(H-2d) マウス乳癌細胞株をヒト p53で安定にトランスフェクトし、hupki マウスの脇腹皮下に5 x104 4T1/hup53を注射した。4T1/hup53を注射したマウスでは進行性腫瘍が増殖し、大部分はこれら腫瘍によって35日目までに死んだ。rMVAhup53の効力を試験するために、マウスに4T1/hup53 注射の6日目に腹腔内注射によって107 pfu rMVAhup53をワクチン接種した。10日後、マウスに、CpG-ODN (15 nmoleのODN 1826)および抗-CTLA-4 mAb (50 μg /マウス)とともにrMVAhup53 追加免疫注射を行った。rMVAhup53 ワクチン接種はPBS コントロールと比較して生存において統計的に有意な向上をもたらした(p < 0.05、両側T検定)(図 19)。
【0095】
上記のように、記載は本発明の様々な態様の単に例示の目的である。したがって上記の特定の改変は本発明の範囲を限定するものではない。当業者には、様々な同等物、変更および改変が本発明の枠を逸脱することなく行えることが明らかであり、かかる同等の態様は本発明に含まれると理解すべきである。本明細書で引用したすべての文献を明細書に完全に開示したかのように引用により本明細書に含める。
【0096】
使用した略語:
GFP、緑色蛍光タンパク質; DC、樹状細胞; IFN-γKO、IFN-γ ノックアウト; MVA、改変ワクシニアウイルスアンカラ; rMVA組換え改変ワクシニアウイルスアンカラ; rAd-mup53、マウス 野生型p53を発現する組換えアデノウイルス; hup53、野生型ヒト p53; mup53、野生型マウス p53; rMVAp53、p53を発現する組換えMVA; rMVAmup53、野生型マウス p53を発現する組換えMVA; rMVAhup53、野生型ヒト p53を発現する組換えMVA; rMVApp65、pp65を発現する組換えMVA; rVVmup53、マウス野生型p53を発現する組換えワクシニアウイルス; rVVpp65、pp65を発現する組換えワクシニアウイルス; wtMVA、野生型 MVA; WR、ウエスタンリザーブ; i.p.、腹腔内; s.c.、皮下; mAb、モノクローナル抗体。
【0097】
参考文献
1.Allred, D.C., O'Connell, P., Fuqua, S.A. 1993. Biomarkers in early breast neoplasia. J Cell Biochem Suppl 17G:125-131.
2.Antoine, G., Scheiflinger, F., Dorner, F., Falkner, F.G. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365-396.
3. Baines, J., Celis, E. 2003. Immune-mediated tumor regression induced by CpG-containing oligodeoxynucleotides. Clin Cancer Res 9:2693-2700.
4.Ballas, Z.K., et al. 2001. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J Immunol 167:4878-4886.
5.Baral, R.N., et al. 2003. Immunostimulatory CpG oligonucleotides enhance the immune response of anti-idiotype vaccine that mimics carcinoembryonic antigen. Cancer Immunol Immunother 52:317-327.
6.Berns, E.M., et al. 1998. p53 protein accumulation predicts poor response to tamoxifen therapy of patients with recurrent breast cancer. J Clin Oncol 16:121-127.
7.Berns, E.M., et al. 2000. Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res 60:2155-2162.
8.Berson, J.F., et al. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strain. J Virol 70:6288-6295.
9.Blanchard, T.J., Alcami, A, Andrea, P., Smith, G.L. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol 79(Pt 5):1159-1167.
10.Bruggemann, M., et al. 1991. Human antibody production in transgenic mice: expression from 100 kb of the human IgH locus. Eur J Immunol 5:1323-1326.
11.Carpentier, A.F., Chen, L., Maltonti, F., Delattre, J.Y. 1999. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res 59:5429-5432.
12.Carroll, M.W., Moss, B. 1997a. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198-211.
13.Carroll, M.W., et al. 1997b. Highly attenuated modified vaccinia virus Ankara (MVA) as an effective recombinant vector: a murine tumor model. Vaccine 15:387-394.
14.Chakrabarti, S., Brechling, K., Moss, B. 1985. Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol 5:3403-3409.
15.Chu, R.S., et al. 1997. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med 186:1623-1631.
16.Collier, L.H. 1991. Safety of recombinant vaccinia vaccines. Lancet 337:1035-1036.
17.Davila, E., Celis, E. 2000. Repeated administration of cytosine-phosphorothiolated guanine-containing oligonucleotides together with peptide/protein immunization results in enhanced CTL responses with anti-tumor activity. J Immunol 165:539-547.
18.DeLeo, A.B., et al. 1977. Cell surface antigens of chemically induced sarcomas of the mouse. I. Murine leukemia virus-related antigens and alloantigens on cultured fibroblasts and sarcoma cells: description of a unique antigen on BALB/c Meth A sarcoma. J Exp Med 146:720-734.
19.Dialynas, D.P., et al. 1983. Characterization of the murine antigenic determinant, designated L3T4a, recognized by monoclonal antibody GK1.5: expression of L3T4a by functional T cell clones appears to correlate primarily with class II MHC antigen-reactivity. Immunol Rev 74:29-56.
20.Diamond, D.J., et al. 1997. Development of a candidate HLA A*0201 restricted peptide-based vaccine against human cytomegalovirus infection. Blood 90:1751-1767.
21.Drexler, I., et al. 1999. Modified vaccinia virus Ankara for delivery of human tyrosinase as melanoma-associated antigen: induction of tyrosi. Cancer Res 59:4955-4963.
22.Egen, J.G., Kuhns, M.S., Allison, J.P. 2002. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol 3:611-618.
23.Eliyahu, D., et al. 1989. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci USA 86:8763-8767.
24.Elkhuizen, P.H., et al. 2000. High local recurrence risk after breast-conserving therapy in node-negative premenopausal breast cancer patients is greatly reduced by one course of perioperative chemotherapy: A European Organization for Research and Treatment of Cancer Breast Cancer Cooperative Group Study. J Clin Oncol 18:1075-1083.
25.Erdile, L.F., Smith, D. 2000. CD40 activation enhances the magnitude of cellular immune responses against p53 but not the avidity of the effectors. Cancer Immunol Immunother 49:410-416.
26.Espenschied, J., et al. 2003. CTLA-4 blockade enhances the therapeutic effect of an attenuated poxvirus vaccine targeting p53 in an established murine tumor model. J Immunol 170:3401-3407.
27.Finlay, C.A., et al. 1988. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol 8:531-539.
28.Finlay, C.A., Hinds, P.W., Levine, A.J. 1989. The p53 proto-oncogene can act as a suppressor of transformation. Cell 57:1083-1093.
29.Foote, J., Winter, G. 1992. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol 224(2):487-499.
30.Gibson, L., et al. Human Cytomegalovirus Proteins pp65 and IE1 are Common Targets for CD8+ T cell Responses in Children with Congenital and Postnatal HCMV infection. J Immunol, in press.
31.Gurney, E.G., Harrison, R.O., Fenno, J. 1980. Monoclonal antibodies against simian virus 40 T antigens: evidence for distinct subclasses of large T antigen and for similarities among nonviral T antigens. J Virol 34:752-763.
32.Hainaut, P., Hollstein, M. 2000. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 77:81-137.
33.Halevy, O., Rodel, J., Peled, A., Oren, M. 1991. Frequent p53 mutations in chemically induced murine fibrosarcoma. Oncogene 6:1593-1600.
34.He, T.C., et al. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95:2509-2514.
35.Heckelsmiller, K., et al. 2002. Combined dendritic cell- and CpG oligonucleotide-based immune therapy cures large murine tumors that resist chemotherapy. Eur J Immunol 32:3235-3245.
36.Hemmi, H., et al. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740-745.
37.Hernandez, J., Lee, P.P., Davis, M.M., Sherman, L.A. 2000. The use of HLA A2.1/p53 peptide tetramers to visualize the impact of self tolerance on the TCR repertoire. J Immunol 164:596-602.
38.Hilburger, R.M., Abrams, S.I. 2001. Characterization of CD8+ cytotoxic T lymphocyte/tumor cell interactions reflecting recognition of an endogenously expressed murine wild-type p53 determinant. Cancer Immunol Immunother 49:603-612.
39.Hurwitz, A.A., Yu, T.F., Leach, D.R., Allison, J.P. 1998. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci USA 95:10067-10071.
40.Hurwitz, A.A., et al. 2000. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res 60:2444-2448.
41.Jones, P.T., Dear, P.H., Foote, J., Neuberger, M.S., Winter, G. 1986. Replacing complementarity-determining regions in a human antibody with those from a mouse. Nature 321(6069):522-525.
42.Kawarada, Y., et al. 2001. NK-and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol 167:5247-5253.
43.Kim, T.Y., et al. 2002. Both E7 and CpG-oligodeoxynucleotide are required for protective immunity against challenge with human papillomavirus 16 (E6/E7) immortalized tumor cells: involvement of CD4+ and CD8+ T cells in protection. Cancer Res 62:7234-7240.
44.Kit, S., Dubbs, D.R., DeTorres, R.A., Melnick, J.L. 1965. Enhanced thymidine kinase activity following infection of green monkey kidney cells by simian adenoviruses, simian papovavirus SV40, and an adenovirus-SV40 "hybrid". Virology 27:453-457.
45.Koo, G.C., Peppard, J.R. 1984. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma 3:301-303.
46.Krieg, A.M. 2002. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20:709-760.
47.Krieg, A.M. 2003. CpG motifs: the active ingredient in bacterial extracts? Nat Med 9:831-835.
48.Krummel, M.F., Allison, J.P. 1995. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182:459-465.
49.Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331.
50.Low, N.M., Holliger, P.H., Winter, G. 1986. Mimicking somatic hypermutation: affinity maturation. J Mol Biol 260:359-368.
51.Macpherson, I., Stoker, M. 1962. Polyoma transformation of hamster cell clones an investigation of genetic factors affecting cell competence. Virology 16:147-151.
52.Mayordomo, J.I., et al. 1996. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med 183:1357-1365.
53.Mayr, A., et al. 1978. [The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author's transl)]. Zentralbl Bakteriol [B] 167:375-390.
54.Mayr, A. 1999. [Historical review of smallpox, the eradication of smallpox and the attenuated smallpox MVA vaccine]. Berlin Munch Tierarztl Wochenschr 112:322-328.
55.Mendez, M.J., et al. 1997. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet 2:146-156.
56.Meyer, H., Sutter, G., Mayr, A. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol 72(Pt 5):1031-1038.
57.Miconnet, I., et al. 2001. Cancer vaccine design: a novel bacterial adjuvant for peptide-specific CTL induction. J Immunol 166:4612-4619.
58.Millikan, R., et al. 1995. p53 mutations in benign breast tissue. J Clin Oncol 13:2293-2300.
59.Moldoveanu, Z., Love-Homan, L., Huang, W.Q., Krieg, A.M. 1998. CpG DNA, a novel immune enhancer for systemic and mucosal immunization with influenza virus. Vaccine 16:1216-1224.
60.Mulryan, K., et al. Attenuated recombinant vaccinia virus expressing oncofetal antigen (tumor-associated antigen) 5T4 induces active therapy of established tumors. Mol Cancer Ther 1:1129-1137.
61.Noguchi, Y., Richards, E.C., Chen, Y.T., Old, L.J. 1995. Influence of interleukin 12 on p53 peptide vaccination against established Meth A sarcoma. Proc Natl Acad Sci USA 92:2219-2223.
62.Norbury, C.C., et al. 2002. Visualizing priming of virus-specific CD8+ cells by infected dendritic cells in vivo. Nat Immunol 3:265-271.
63.Offringa, R., et al. p53: a potential target antigen for immunotherapy of cancer. Ann N Y Acad Sci 910:223-233.
64.Ourmanov, I., et al. 2000. Comparative efficacy of recombinant modified vaccinia virus Ankara expressing simian immunodeficiency virus (SIV) Gag-Pol and/or Env in macaques challenged with pathogenic SIV. J Virol 74:2740-2751.
65.Pasare, C., Medzhitov, R. 2003. Toll pathway-dependent blockade of CD4+CD25+ T-cell mediated suppression by dendritic cells. Science 299:1033-1036.
66.Pratap, R., Shousha, S. 1998. Breast carcinoma in women under the age of 50: relationship between p53 immunostaining, tumour grade, and axillary lymph node status. Breast Cancer Res Treat 49:35-39.
67.Querzoli, P., et al. 1998. Modulation of biomarkers in minimal breast carcinoma: a model for human breast carcinoma progression. Cancer 83:89-97.
68.Querzoli, P., et al. 2001. Biophenotypes and survival of BRCA1 and TP53 deleted breast cancer in young women. Breast Cancer Res Treat 66:135-142.
69.Ramirez, J.C., Gherardi, M.M., Rodriguez, D., Esteban, M. 2000a. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. J Virol 74:7651-7655.
70.Ramirez, J.C., Gherardi, M.M., Esteban, M. 2000b. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation. J Virol 74:923-933.
71.Read, S., Malmstrom, V., Powrie, F. 2000. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med 192:295-302.
72.Redfield, R.R., et al. 1987. Disseminated vaccinia in a military recruit with human immunodeficiency virus (HIV) disease. N Engl J Med 316:673-676.
73.Reich, N.C., Levine, A.J. 1984. Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature 308:199-201.
74.Rosales, C., et al. A recombinant vaccinia virus containing the papilloma E2 protein promotes tumor regression by stimulating macrophage antibody-dependent cytotoxicity. Cancer Immunol Immunother 49:347-360.
75.Rosenberg, S.A. 2001. Progress in human tumour immunology and immunotherapy. Nature 411:380-384.
76.Sandler, A.D., et al. 2003. CpG oligonucleotides enhance the tumor antigen-specific immune response of a granulocyte macrophage colony-stimulating factor-based vaccine strategy in neuroblastoma. Cancer Res 63:394-399.
77.Schmid, P., Lorenz, A., Hameister, H., Montenarh, M. 1991. Expression of p53 during mouse embryogenesis. Development 113:857-865.
78.Selvanayagam, C.S., Davis, C.M., Cornforth, M.N., Ullrich, R.L. 1995. Latent expression of p53 mutations and radiation-induced mammary cancer. Cancer Res 55:3310-3317.
79.Sharma, S., et al. 2003. Intra-tumoral injection of CpG results in the inhibition of tumor growth in murine Colon-26 and B-16 tumors. Biotechnol Lett 25:149-153.
80.Sirvent, J.J., Fortuna-Mar, A., Olona, M., Orti, A. 2001. Prognostic value of p53 protein expression and clinicopathological factors in infiltrating ductal carcinoma of the breast. Histol Histopathol 16:99-106.
81.Stern, B.V., Boehm, B.O., Tary-Lehmann, M. 2002. Vaccination with tumor peptide in CpG adjuvant protects via IFN-gamma-dependent CD4 cell immunity. J Immunol 168:6099-6105.
82.Stittelaar, K.J., et al. 2001. Safety of a modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 19:3700-3709.
83.Sukumar, S., McKenzie, K., Chen, Y. 1995. Animal models for breast cancer. Mutat Res 333:37-44.
84.Sutter, G., Moss, B. 1992. Nonreplicating vaccinia virus vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA 89:10847-10851.
85.Tan, M.H., Holyoke, E.D., Goldrosen, M.H. 1976. Murine colon adenocarcinomas: methods for selective culture in vitro. J Natl Cancer Inst 56:871-873.
86.Theobald, M., et al. 1997. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med 185:833-841.
87.Turner, B.C., et al. 2000. Mutant p53 protein overexpression in women with ipsilateral breast tumor recurrence following lumpectomy and radiation therapy. Cancer 88:1091-1098.
88.van Elsas, A., Hurwitz, A.A., Allison, J.P. 1999. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190:355-366.
89.Vierboom, M.P., et al. 2000a. High steady-state levels of p53 are not a prerequisite for tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. Cancer Res 60:5508-5513.
90.Vierboom, M.P., et al. 2000b. Cyclophosphamide enhances anti-tumor effect of wild-type p53-specific CTL. Int J Cancer 87:253-260.
91.Vierboom, M.P., et al. 1997. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med 186:695-704.
92.Weiner, G.J., et al. 1997. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci USA 94:10833-10837.
93.Wiedenfeld, E.A., Fernandez-Vina, M., Berzofsky, J.A., Carbone, D.P. 1994. Evidence for selection against human lung cancers bearing p53 missense mutations which occur within the HLA A*0201 peptide consensus motif. Cancer Res 54:1175-1177.
94.Winter, G., Griffiths, A.D., Hawkins, R.E., Hoogenboom, H.R. 1994. Making antibodies by phage display technology. Annu Rev Immunol 12:433-455.
95.Zambetti, G.P., Levine, A.J. 1993. A comparison of the biological activities of wild-type and mutant p53. FASEB J 7:855-865.
96.Zellars, R.C., et al. 2000. Prognostic value of p53 for local failure in mastectomy-treated breast cancer patients. J Clin Oncol 18:1906-1913.
【図面の簡単な説明】
【0098】
【図1】図 1は、 pLW22-hup53 コンストラクトのPCR分析である。
【図2】図 2は、rMVAmup53で感染した細胞によるmup53の発現である。
【図3】図 3は、インビトロでのrMVAmup53によるp53-特異的CTL応答の生成である。
【図4】図 4は、rMVAmup53によるワクチン接種の Meth A 腫瘍阻止に対する効果である。
【図5】図 5は、rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した Meth A 腫瘍に対する効果である。
【図6】図 6は、rMVAmup53 + 抗-CTLA-4 mAbによるワクチン接種の確立した11A-1 腫瘍に対する効果である。
【図7】図 7は、rMVAmup53 + 抗-CTLA-4 mAbのワクチン接種の確立したMC-38 腫瘍に対する効果である。
【図8】図 8は、rMVAmup53 + CpG ODNによるワクチン接種の確立した11A-1 腫瘍に
【図9】図 9は、rMVAmup53 + CpG ODNによるワクチン接種の確立したMeth A 腫瘍に対する効果である。
【図10】図 10は、rMVAmup53 + CpG ODNによるワクチン接種の確立したMC-38 腫
【図11】図 11は、 rMVAmup53 + 抗-CTLA-4 mAbおよびCpG ODNによるワクチン接種の確立した11A-1 腫瘍に対する効果である。
【図12】図 12は、rMVAmup53 + 抗-CTLA-4 mAb and CpG ODNによるワクチン接種の確立したMC-38 腫瘍に対する効果である。
【図13】図 13は、Meth A 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求である。
【図14】図 14は、11A-1 腫瘍に対するCpG ODN 免疫調節物質効果の細胞による要求である。
【図15】図 15は、11A-1 腫瘍に対する抗-CTLA-4 mAb 免疫調節物質効果の細胞による要求である。
【図16】図 16は、CpG ODN 免疫調節物質効果に対するTLR 9の寄与である。
【図17】図 17は、IL-6 の CpG ODN 免疫調節物質効果に対する寄与である。
【配列表】
【特許請求の範囲】
【請求項1】
p53をコードする核酸配列を含む組換えMVAウイルス (rMVA)を含む組成物。
【請求項2】
該p53が野生型マウスp53である請求項1の組成物。
【請求項3】
該核酸配列が配列番号1のヌクレオチド配列を含む請求項2の組成物。
【請求項4】
該p53が野生型ヒト p53である請求項1の組成物。
【請求項5】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項4の組成物。
【請求項6】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドの両方を含む免疫調節物質をさらに含む請求項1の組成物。
【請求項7】
該CTLA-4遮断薬が抗体である請求項6の組成物。
【請求項8】
該抗体がモノクローナル抗体である請求項7の組成物。
【請求項9】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物を対象に導入することを含むp53-発現悪性腫瘍を有する対象の治療方法。
【請求項10】
該対象がヒトである請求項9の方法。
【請求項11】
該組成物の導入が該p53-発現悪性腫瘍に対して有効な免疫応答を誘発する請求項9の方法。
【請求項12】
該p53が野生型ヒト p53である請求項9の方法。
【請求項13】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項12の方法。
【請求項14】
該組成物の導入を1回以上繰り返す請求項9の方法。
【請求項15】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質を該対象に導入することをさらに含む請求項9の方法。
【請求項16】
該CTLA-4遮断薬が抗体である請求項15の方法。
【請求項17】
該抗体がモノクローナル抗体である請求項16の方法。
【請求項18】
該免疫調節物質の導入を、組換えMVAウイルスを含む組成物の導入の前、同時または後に行う請求項15の方法。
【請求項19】
該免疫調節物質の導入を1回以上繰り返す請求項15の方法。
【請求項20】
該導入の方法が皮下、経皮、皮内、腹腔内、筋肉内、腫瘍内および静脈内注射からなる群から選択される請求項9または15の方法。
【請求項21】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物および該組成物の投与の説明書を含む、p53-発現悪性腫瘍を有する対象の治療用キット、この場合、該投与が該p53-発現悪性腫瘍に有効な免疫応答を誘発する。
【請求項22】
該p53がヒト p53である請求項21のキット。
【請求項23】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項22のキット。
【請求項24】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質をさらに含む請求項21のキット。
【請求項25】
該CTLA-4遮断薬が抗体である請求項24のキット。
【請求項26】
該抗体がモノクローナル抗体である請求項25のキット。
【請求項27】
該対象がヒトである請求項21のキット。
【請求項28】
p53をコードする核酸インサートを含むMVA組換えプラスミドを含むベクター。
【請求項29】
該MVA組換えプラスミドがpLW22である請求項28のベクター。
【請求項30】
該核酸インサートがヒト野生型p53をコードする請求項28のベクター。
【請求項31】
該ベクターが配列番号5の配列を含む請求項30のベクター。
【請求項32】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物を投与することを含む、突然変異体 p53を過剰発現する細胞に対するp53 特異的細胞障害性Tリンパ球(CTL)応答を生じさせる方法。
【請求項33】
p53をコードする該核酸配列がマウス野生型p53をコードする請求項32の方法。
【請求項34】
該核酸配列が配列番号1のヌクレオチド配列を含む請求項33の方法。
【請求項35】
p53をコードする該核酸配列がヒト野生型p53をコードする請求項32の方法。
【請求項36】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項35の方法。
【請求項37】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質を投与することをさらに含む請求項32の方法。
【請求項38】
該CTLA-4遮断薬が抗体である請求項37の方法。
【請求項39】
該抗体がモノクローナル抗体である請求項38の方法。
【請求項40】
該免疫調節物質の投与を組換えMVAウイルスを含む組成物の導入の前、同時または後に行う請求項37の方法。
【請求項41】
該免疫調節物質の導入を1回以上繰り返す請求項37の方法。
【請求項1】
p53をコードする核酸配列を含む組換えMVAウイルス (rMVA)を含む組成物。
【請求項2】
該p53が野生型マウスp53である請求項1の組成物。
【請求項3】
該核酸配列が配列番号1のヌクレオチド配列を含む請求項2の組成物。
【請求項4】
該p53が野生型ヒト p53である請求項1の組成物。
【請求項5】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項4の組成物。
【請求項6】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドの両方を含む免疫調節物質をさらに含む請求項1の組成物。
【請求項7】
該CTLA-4遮断薬が抗体である請求項6の組成物。
【請求項8】
該抗体がモノクローナル抗体である請求項7の組成物。
【請求項9】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物を対象に導入することを含むp53-発現悪性腫瘍を有する対象の治療方法。
【請求項10】
該対象がヒトである請求項9の方法。
【請求項11】
該組成物の導入が該p53-発現悪性腫瘍に対して有効な免疫応答を誘発する請求項9の方法。
【請求項12】
該p53が野生型ヒト p53である請求項9の方法。
【請求項13】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項12の方法。
【請求項14】
該組成物の導入を1回以上繰り返す請求項9の方法。
【請求項15】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質を該対象に導入することをさらに含む請求項9の方法。
【請求項16】
該CTLA-4遮断薬が抗体である請求項15の方法。
【請求項17】
該抗体がモノクローナル抗体である請求項16の方法。
【請求項18】
該免疫調節物質の導入を、組換えMVAウイルスを含む組成物の導入の前、同時または後に行う請求項15の方法。
【請求項19】
該免疫調節物質の導入を1回以上繰り返す請求項15の方法。
【請求項20】
該導入の方法が皮下、経皮、皮内、腹腔内、筋肉内、腫瘍内および静脈内注射からなる群から選択される請求項9または15の方法。
【請求項21】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物および該組成物の投与の説明書を含む、p53-発現悪性腫瘍を有する対象の治療用キット、この場合、該投与が該p53-発現悪性腫瘍に有効な免疫応答を誘発する。
【請求項22】
該p53がヒト p53である請求項21のキット。
【請求項23】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項22のキット。
【請求項24】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質をさらに含む請求項21のキット。
【請求項25】
該CTLA-4遮断薬が抗体である請求項24のキット。
【請求項26】
該抗体がモノクローナル抗体である請求項25のキット。
【請求項27】
該対象がヒトである請求項21のキット。
【請求項28】
p53をコードする核酸インサートを含むMVA組換えプラスミドを含むベクター。
【請求項29】
該MVA組換えプラスミドがpLW22である請求項28のベクター。
【請求項30】
該核酸インサートがヒト野生型p53をコードする請求項28のベクター。
【請求項31】
該ベクターが配列番号5の配列を含む請求項30のベクター。
【請求項32】
p53をコードする核酸配列を含む組換えMVAウイルスを含む組成物を投与することを含む、突然変異体 p53を過剰発現する細胞に対するp53 特異的細胞障害性Tリンパ球(CTL)応答を生じさせる方法。
【請求項33】
p53をコードする該核酸配列がマウス野生型p53をコードする請求項32の方法。
【請求項34】
該核酸配列が配列番号1のヌクレオチド配列を含む請求項33の方法。
【請求項35】
p53をコードする該核酸配列がヒト野生型p53をコードする請求項32の方法。
【請求項36】
該核酸配列が配列番号2のヌクレオチド配列を含む請求項35の方法。
【請求項37】
CTLA-4遮断薬、CpGオリゴデオキシヌクレオチド、またはCTLA-4遮断薬とCpGオリゴデオキシヌクレオチドとの両方を含む免疫調節物質を投与することをさらに含む請求項32の方法。
【請求項38】
該CTLA-4遮断薬が抗体である請求項37の方法。
【請求項39】
該抗体がモノクローナル抗体である請求項38の方法。
【請求項40】
該免疫調節物質の投与を組換えMVAウイルスを含む組成物の導入の前、同時または後に行う請求項37の方法。
【請求項41】
該免疫調節物質の導入を1回以上繰り返す請求項37の方法。
【図1】


【図2】
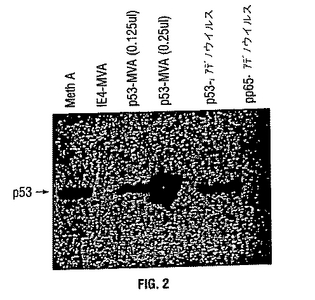
【図3−A】

【図3−B】

【図3−C】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図2】
【図3−A】
【図3−B】
【図3−C】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公表番号】特表2006−512097(P2006−512097A)
【公表日】平成18年4月13日(2006.4.13)
【国際特許分類】
【出願番号】特願2005−510053(P2005−510053)
【出願日】平成15年12月22日(2003.12.22)
【国際出願番号】PCT/US2003/041053
【国際公開番号】WO2004/058801
【国際公開日】平成16年7月15日(2004.7.15)
【出願人】(598004424)シティ・オブ・ホープ (15)
【氏名又は名称原語表記】City of Hope
【住所又は居所原語表記】1500 East,Duarte Road,Duarte,California 91010−0269,United States of America
【Fターム(参考)】
【公表日】平成18年4月13日(2006.4.13)
【国際特許分類】
【出願日】平成15年12月22日(2003.12.22)
【国際出願番号】PCT/US2003/041053
【国際公開番号】WO2004/058801
【国際公開日】平成16年7月15日(2004.7.15)
【出願人】(598004424)シティ・オブ・ホープ (15)
【氏名又は名称原語表記】City of Hope
【住所又は居所原語表記】1500 East,Duarte Road,Duarte,California 91010−0269,United States of America
【Fターム(参考)】
[ Back to top ]