
ストレス誘導性リガンド依存性EGFR活性化の阻害

【課題】本発明は、レセプターチロシンキナーゼのリガンド、特に細胞外リガンドを阻害することによるストレス誘導性レセプターチロシンキナーゼ活性の阻害のための新規薬剤または方法を提供する。
【解決手段】浸透ストレスまたは酸化ストレスに応答するEGFR活性化がメタロプロテアーゼに媒介されるproHB-EGFの切断に関わるという新たな知見により、前記新規薬剤または方法が発明された。
【解決手段】浸透ストレスまたは酸化ストレスに応答するEGFR活性化がメタロプロテアーゼに媒介されるproHB-EGFの切断に関わるという新たな知見により、前記新規薬剤または方法が発明された。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、レセプターチロシンキナーゼのリガンド、特に細胞外リガンドを阻害することによるストレス誘導性レセプターチロシンキナーゼ活性の阻害に関する。
【背景技術】
【0002】
高浸透圧および酸化剤、電離線または紫外線のような環境ストレスに哺乳類の細胞が暴露されると、様々なシグナルトランスダクションのカスケードが活性化される。活性酸素種(ROS)がセカンドメッセンジャーとして関わっているとされているが、その無制御の生産とオキシダントへの暴露による酸化ストレスが細胞損傷と癌のような生理学的疾患に関わっている(Finkel, 1998(非特許文献1); Kamata and Hirata, 1999(非特許文献2))。さらに、哺乳類の細胞は増大するモル浸透圧濃度を含む細胞外環境の変化に適応しなくてはならず、このことが結果として細胞収縮と低分子の増加合成を生じ、細胞内と細胞外の状態を等しくする(de Nadal et al., 2002(非特許文献3))。
【0003】
浸透ストレスまたは酸化ストレスは、種々のレセプターチロシンキナーゼを活性化し、最も際だったものは上皮成長因子レセプター(EGFR)である(King et al., 1989(非特許文献4); Knebel et al., 1996(非特許文献5); Rao, 1996(非特許文献6); Rosette and Karin, 1996(非特許文献7))。EGFRは、細胞増殖、細胞分化、細胞移動またはアンチアポトーシスシグナルを含む重要な生物学的応答の過多を制御するので、頻繁に様々なヒトの疾患に影響を与えてきた(Prenzel et al., 2001(非特許文献8)。リガンド依存性およびリガンド非依存性レセプター活性化メカニズムは、EGFRに関して記載されてきている。リガンドに媒介されたレセプター活性化は、EGF、HB-EGF、アンフィレグリンまたはTGF−αのようなEGF様リガンドがレセプターのエクトドメインに結合し、2つのリガンドレセプターの二量化がヘテロダイマーを生じ、引き続き固有のキナーゼ活性化とオートリン酸化を活性化することにより生じる(Schlessinger, 2002(非特許文献9)。リガンド非依存性レセプター活性化は、その触媒ポケット内のクリティカルなシステイン残基の酸化に関わるホスファターゼの不活性化により生じることが提唱されている(Knebel et al., 1996(非特許文献5))。よって、レセプターリン酸化の平衡は、非リン酸化状態からリン酸化状態にシフトする。リガンド非依存性レセプター活性化のその他のメカニズムは、非特異的クラスター化とEGFRの内在化に関わると示唆されてきた(Rosette and Karin, 1996(非特許文献7))。さらに、c-Srcのような細胞質のノンレセプターチロシンキナーゼがEGFRをリン酸化することが示された(Biscardi et al., 1999(非特許文献10) Tice et al., 1999(非特許文献11))。
【0004】
EGFRのチロシンリン酸化は、Gタンパク質共役レセプター(GPCR)刺激によっても誘導することができ、このプロセスはEGFRトランスアクチべーションと名付けられた(Daub et al., 1996(非特許文献12))。このメカニズムは当初、専らリガンド非依存性プロセスによるものとされていた。しかし、多くの細胞系ではEGFRトランスアクチべーションはメタロプロテアーゼに媒介される膜貫通型EGF−様リガンドのシェディングにより生じ、活性になるためにプロセシングしなくてはならない(Prenzel et al., 1999(非特許文献13))。メタロプロテアーゼのADAMファミリーの極めて最近のメンバーは、GPCR−誘導性proHB-EGFとpro-ARプロセシングに必要なシェダーゼとして同定された(Gschwind et al., 2003(非特許文献14); Yan et al., 2002(非特許文献15))。とりわけ、TNF-α変換酵素(TACE)とも称されるADAM17、ならびにADAM10とADAM12も関わることが示された(Asakura et al., 2002(非特許文献16); Gschwind et al., 2003(非特許文献14); Yan et al., 2002(非特許文献15))。リガンド依存性EGFRトランスアクチベーションが関わる異所のシグナリングプロセスは、頭頚部の扁平上皮癌、心臓および胃腸肥大および嚢胞性線維症のような種々のヒトの疾患に関わってきている(Asakura et al., 2002(非特許文献16); Gschwind et al., 2002(非特許文献17); Keates et al., 2001(非特許文献18); Lemjabbar and Basbaum, 2002(非特許文献19)。EGFRトランスアクチベーションとは別に、ADAM9は、TPA刺激に応答してproHB-EGFをプロセシングすることが示された(Izumi et al., 1998)のに対して、ADAM17よるproTGF-α、proHB-EGFおよびプロアンフィレグリンの切断が報告されている(Merlos-Suarez et al., 2001(非特許文献20); Peschon et al., 1998(非特許文献21); Sunnarborg et al., 2002(非特許文献22))、TGF-αの場合の腫瘍形成を示唆している(Borrell-Pages et al., 2003(非特許文献23))。
【0005】
RTKリン酸化の他に、環境ストレスはマイトジェン活性化タンパク質キナーゼ(MAPKs)の活性化を導き、これは転写因子の活性化により細胞外刺激を核へカップリングさせる細胞内シグナルトランスダクションカスケードの主な部分である。従って、MAPK経路は、増殖、移動、分化およびストレス応答のような重要なプロセスを制御する(Chen et al., 2001b(非特許文献24); Johnson and Lapadat, 2002(非特許文献25))。しかし、転写因子を活性化する以外に、MAPKsである細胞外シグナル制御キナーゼ−1/2(ERK1/2)とp38は、ADAM17の細胞内ドメインのリン酸化により膜貫通型タンパク質(Fan and Derynck, 1999(非特許文献26))のプロセシングの制御に関わっていた(Diaz-Rodriguez et al., 2002(非特許文献27); Fan et al., 2003(非特許文献28))。細胞の運命がこれらのシグナリング経路間の連絡により決定されるので、徹底的な研究は、浸透ストレスと酸化ストレスによるMAPK活性化のメカニズムに注目した(Kyriakis and Avruch, 2001(非特許文献29))。以前の報告は、ストレス剤ならびに足場タンパク質機能の変異によるMAPK活性化におけるホスファターゼの不活性化またはダウンレギュレーションが、シグナリング複合体の会合または解離を導くと提唱していた(Benhar et al., 2002(非特許文献30); de Nadal et al., 2002(非特許文献2))。さらに、小さなGタンパク質は浸透ストレスによりMAPKの活性化に働いていることが証明された(de Nadal et al., 2002(非特許文献2))。多くの証拠は特にROS、ストレス活性化キナーゼp38とJNKおよびストレスシグナリングがアポトーシスへの癌細胞の感受性ならびに増殖応答と関わると示唆しているので、ストレス刺激によるMAPKの活性化は、ヒトの癌の発生と進行に過酷な結果をもたらす。近年の研究は、抗ガン剤がストレスシグナリングカスケードを活性化すると示しているので、EGFRを介するストレスシグナリングは、癌治療に影響を及ぼすことができる(Benhar et al., 2002(非特許文献30)で調査)。
【0006】
酸化ストレスと浸透ストレスに応答するRTKとMAPK活性化のメカニズムが集中的に研究されてきたが、しかしこれまでにこれらの制御経路はヒト癌細胞では一般にリガンド非依存性プロセスであると記載されていた。ADAMメタロプロテアーゼによるEGFR活性化の制御の見解における近年の進歩は、EGF様リガンドプロセシングの潜在的な関連性についてストレス誘導性EGFRと、MAPK刺激のメカニズムを研究するきっかけとなった。
【0007】
Fischer et al.(Poster PS01-0916, Eur. J. Biochem. (非特許文献31))は、p38とADAMファミリーのメタロプロテアーゼがヒト癌細胞の下流のシグナリングにおいてストレス誘導性リガンド依存性EGFR活性化を制御することを記載している。しかし、特異的なメタロプロテアーゼは同定されていない。
【0008】
Takenobu et al. (J. Biol. Chem. 278, (2003), 1725-1762(非特許文献32))は、HB-EGF様成長因子のストレスおよび炎症サイトカイン誘導性エクトドメイン・シェディングがp38 MAPK により媒介されていることを開示している。メタロプロテアーゼADAM9がVeroH細胞ではストレス誘導性pro HB-EGFのシェディングに不必要であることが分かった。
【0009】
Herrlich et al. (FASEB J. 16 (2002), A56(非特許文献33))およびTschumperlin et al.(FASEB J.16 (2002), A1150(非特許文献34))は、HER2/HER3またはEGFR活性化の刺激が、それぞれ浸透ストレスまたは機械的ストレスによってヘレグリンシェディングに媒介されることを開示している。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Finkel, 1998
【非特許文献2】Kamata and Hirata, 1999
【非特許文献3】de Nadal et al., 2002
【非特許文献4】King et al., 1989
【非特許文献5】Knebel et al., 1996
【非特許文献6】Rao, 1996
【非特許文献7】Rosette and Karin, 1996
【非特許文献8】Prenzel et al., 2001
【非特許文献9】Schlessinger, 2002
【非特許文献10】Biscardi et al., 1999
【非特許文献11】Tice et al., 1999
【非特許文献12】Daub et al., 1996
【非特許文献13】Prenzel et al., 1999
【非特許文献14】Gschwind et al., 2003
【非特許文献15】Yan et al., 2002
【非特許文献16】Asakura et al., 2002
【非特許文献17】Gschwind et al., 2002
【非特許文献18】Keates et al., 2001
【非特許文献19】Lemjabbar and Basbaum, 2002
【非特許文献20】Merlos-Suarez et al., 2001
【非特許文献21】Peschon et al., 1998
【非特許文献22】Sunnarborg et al., 2002
【非特許文献23】Borrell-Pages et al., 2003
【非特許文献24】Chen et al., 2001b
【非特許文献25】Johnson and Lapadat, 2002
【非特許文献26】Fan and Derynck, 1999
【非特許文献27】Diaz-Rodriguez et al., 2002
【非特許文献28】Fan et al., 2003
【非特許文献29】Kyriakis and Avruch, 2001
【非特許文献30】Benhar et al., 2002
【非特許文献31】Fischer et al.(Poster PS01-0916, Eur. J. Biochem.
【非特許文献32】Takenobu et al. J. Biol. Chem. 278, (2003), 1725-1762
【非特許文献33】Herrlich et al. FASEB J. 16 (2002), A56
【非特許文献34】Tschumperlin et al. FASEB J.16 (2002), A1150
【発明の概要】
【発明が解決しようとする課題】
【0011】
以上の背景技術の下、本発明は、レセプターチロシンキナーゼのリガンド、特に細胞外リガンドを阻害することによるストレス誘導性レセプターチロシンキナーゼ活性の阻害のための新規薬剤または方法を提供する。
【課題を解決するための手段】
【0012】
本発明では、浸透ストレスまたは酸化ストレスに応答するEGFR活性化がメタロプロテアーゼに媒介されるproHB-EGFの切断に関わることを証明する。重要なメタロプロテアーゼは、proHB-EGF をプロセシングするADAMファミリー、特にADAM9、ADAM10およびADAM17のメンバーを含む。さらに、ストレス誘導性リガンド依存性EGFR活性化は、MAPKs であるERK1/2およびJNKと結びつくことができる。我々は、ストレス活性化EGFRリン酸化がp38活性に依存しており、p38がヒト癌細胞のストレス応答においてADAMメタロプロテアーゼの上流アクチベーターとして関わっている証拠を提供する。
【0013】
さらに化学療法剤、例えば、p38活性化を誘導するドキソルビシンでの腫瘍細胞の治療と、細胞死を著しく促進するHB-EGF作用の遮断との組合せは、ドキソルビシンだけの治療と比較した場合に、著しく細胞死を促進した。この結果は、化学療法に誘導される細胞死を逃れる腫瘍細胞のシグナルメカニズムの役割を示唆している。
【0014】
従って、細胞、この好ましくは哺乳類細胞、より好ましくはヒトの細胞、例えば、腫瘍細胞、特に過剰増殖性細胞またはアポトーシス耐性細胞で、レセプターチロシンキナーゼまたはRTK−媒介シグナリング経路のストレス誘導性活性化の調節を可能にする方法を提供し、前記方法は該レセプターチロシンキナーゼのリガンドの活性の阻害を含む。
【0015】
本発明の第一の点は、少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療するための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0016】
本発明のもう1つの点は、過剰増殖性疾患に対する治療の効果を上げるためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0017】
本発明のもう1つの点は、照射療法および/または薬剤治療に対する過剰増殖性疾患の感受性を上げるための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0018】
本発明のもう1つの点は、レセプターチロシンキナーゼのストレス誘導性活性化により生じるか、またはこれと関連する疾患を予防または治療するための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0019】
また本発明のもう1つの点は、活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキットである。
【発明の効果】
【0020】
我々の知る限りでは、これがヒト癌細胞でEGFRのリガンド依存性活性化と、それに続くMAPKp38により制御される浸透ストレスと酸化ストレス剤による下流のシグナリングを証明する初めての報告である。ますます増える証拠は、心臓血管疾患、神経変性疾患または癌のような過剰増殖性疾患のように多種の様々なヒトの疾患で過剰のROS生産により引き起こされる酸化ストレスに特に関わる。ゆえに、ストレス剤により引き起こされる細胞損傷に関わる病態生理学的疾患と各治療アプローチにとって特別に重要である。
【0021】
本明細書で記載したデータは、哺乳類細胞、特にヒト癌腫でのストレス刺激のシグナリングメカニズムにおける以前の結果に及ぶ。我々の発見は、ヒト癌細胞でのストレス応答の重要なメディエーターとしてADAMファミリープロテアーゼとHB-EGFの重要性を実証する。さらに、我々のデータはMAPKの種々のグループ間でのクロスコミュニケーションがADAMプロテアーゼとEGFRをシグナリング媒介物として使用していることを示唆する。これに関連して、ERK1/2とJNK活性の間のバランスは、細胞の運命にとって特に重要である。他のEGFR下流シグナリング事象ならびにストレス剤に応答する癌細胞の強化増殖または移動のような可能性のある病理学的応答に焦点をおいて将来の研究が成されるべきである。
【図面の簡単な説明】
【0022】
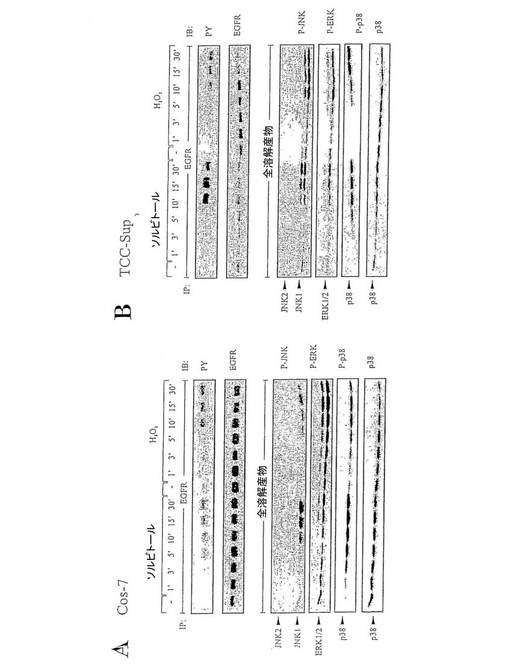
【図1】図1は、種々の細胞系におけるストレス誘導性EGFRおよびMAPKリン酸化の経時変化を表す図である。(A)浸透ストレスと酸化ストレスに応答するEGFRおよびMAPKリン酸化。(B)ヒト膀胱癌細胞系におけるストレス誘導性EGFRリン酸化。
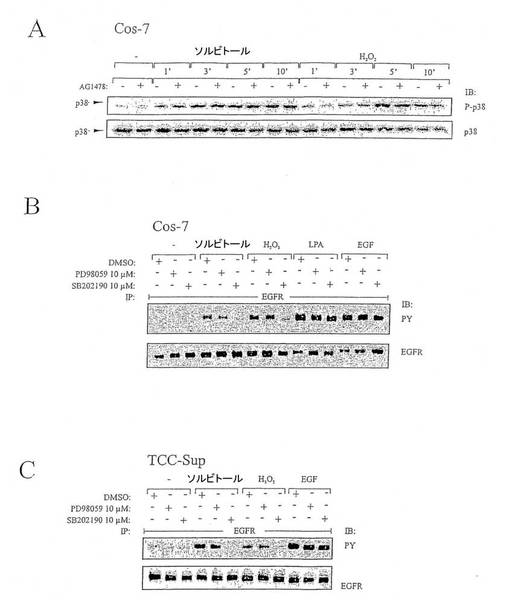
【図2】図2は、ストレス誘導性EGFRリン酸化を媒介するp38を表す図である。(A)EGFRキナーゼ機能に依存するp38リン酸化の経時変化。(B)Cos-7細胞ではストレス誘導性EGFR活性化はp38に依存し、ERK活性には依存しない。(C)TCC-Sup癌細胞ではストレス誘導性EGFR活性化はp38活性に依存する。
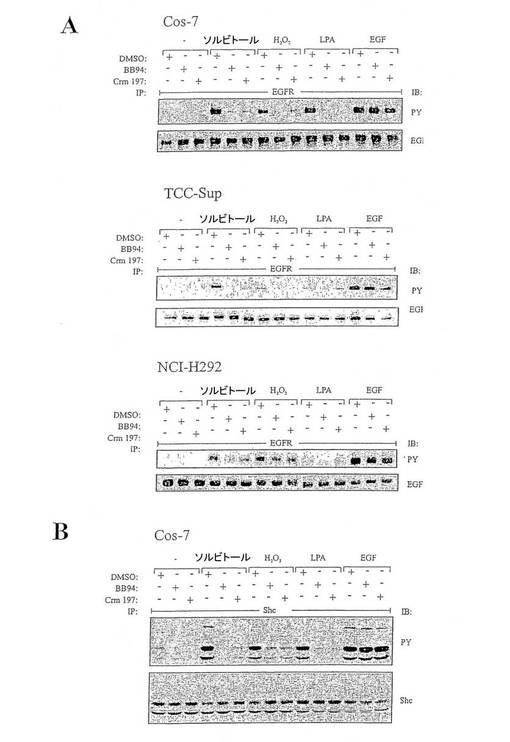
【図3】図3は、リガンド依存性である浸透ストレスと酸化ストレスに応答するEGFR活性化を表す図である。(A)EGFRリン酸化におけるメタロプロテアーゼとHB-EGF阻害の効果。(B)ストレス剤に応答するShcリン酸化の分析。
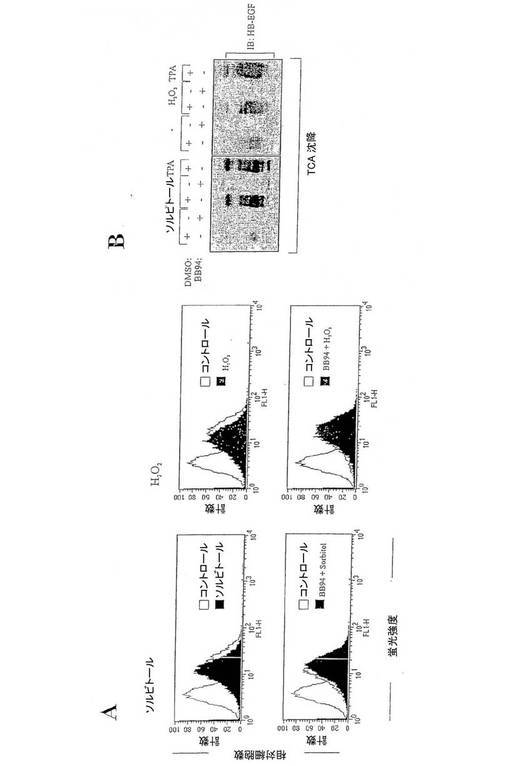
【図4】図4は、ストレス剤に応答するproHB-EGF放出の分析を表す図である。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。(B)条件培地の免疫ブロット分析。
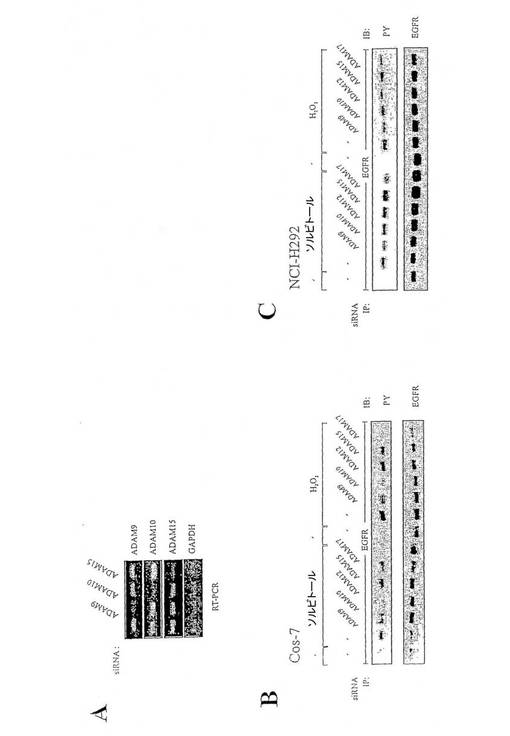
【図5】図5は、ストレス誘導性EGFRリン酸化における種々のADAMタンパク質に対するsiRNAsの効果を表す図である。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。(B)Cos-7細胞。(C)NCl-H292細胞。
【図6】図6は、ストレス剤に応答するMAPK活性化およびEGFR、メタロプロテアーゼおよびHB-EGF機能の遮断を表す図である。
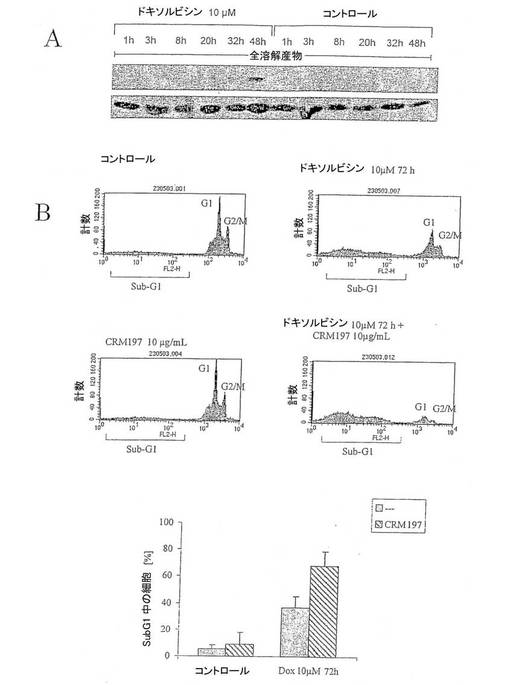
【図7】図7は、TCC-Sup癌細胞のドキソルビシン誘導性細胞死を表す図である。(A)ドキソルビシン処理に応答するp38活性化。(B)ドキソルビシンに応答して細胞死を促進するHB-EGF機能の遮断。
【発明を実施するための形態】
【0023】
レセプターチロシンキナーゼリガンドは、好ましくは細胞外レセプターチロシンキナーゼドメインに結合する分子である。適切なレセプターチロシンキナーゼリガンドの例は、HB−EGF、EGF、アンフィレグリン、ベータセルリン、エピレグリン、TGF−α、ニューレグリンまたはヘレグリンである。さらに好ましくは、レセプターチロシンキナーゼリガンドはHB-EGFである。
【0024】
レセプターチロシンキナーゼリガンドの活性の阻害は、メタロプロテアーゼおよび/またはレセプターチロシンキナーゼの活性化によるリガンドの前駆体、特にリガンドの膜結合前駆体の切断の阻害に関するのが好ましい(例えば、リガンドによるEGFR)。本発明のメタロプロテアーゼ阻害剤は、好ましくはリガンド前駆体の切断およびその放出を阻害できる。適切なメタロプロテアーゼの例は、ADAM9、10および17、特に細胞ストレス応答の重要な媒介物であるADAM 17である。二者択一的に、本発明の阻害剤はレセプターチロシンキナーゼリガンド、特にEGFRチロシンリン酸化、下流のマイトジェンシグナリング事象の生物活性を阻害することもできる。例えば、ERK1/2および/またはJNKのようなマイトジェン活性化タンパク質キナーゼ(MAPKs)の活性化、細胞増殖および/または移動を阻害することができる。
【0025】
レセプターチロシンキナーゼリガンドの阻害剤は、疾患、特に過剰増殖性疾患の予防または治療に使用することもできる。好ましくは、疾患はレセプターチロシンキナーゼのストレス誘導性活性化により引き起こされるか、またはこれに関連する。このストレスは、好ましくは酸化ストレスおよび/または浸透ストレスである。より好ましくは、このストレスはp38に媒介されるストレスである。このようなタイプの疾患の存在は、例えば、mRNAレベル(cDNA 配列分析、SAGE、ノーザンブロットなど)で、および/またはタンパク質レベル(ウエスタンブロット分析、免疫蛍光顕微鏡検査法、in situハイブリダイゼーション技法など)でp38発現を測定することにより決定できる。このようなタイプの疾患の存在は、p38をコード化するゲノムおよび/またはmRNA分子中の活性突然変異の発生を試験することにより決定することもできる。さらに血清および/または疾患に影響された組織内で、p38アゴニストの高いレベルを決定することもできる。このタイプの疾患がレセプターチロシンキナーゼ発現強化と関連する必要がないことを示すべきである。
【0026】
例えば、この疾患は癌、例えば、乳癌、胃癌、前立腺癌、膀胱癌、卵巣癌、肺癌、肝臓癌、腎臓癌または膵臓癌、グリオーム、黒色腫、白血病などのような過剰増殖性疾患または過剰増殖性皮膚病、例えば、乾癬または炎症性疾患のような他の疾患であることができる。
【0027】
レセプターチロシンキナーゼリガンドの活性(好ましくは直接に、またはメタロプロテアーゼ阻害を介して)は、核酸レベルで、例えば、遺伝子レベルまたは転写レベルで阻害することができる。
【0028】
遺伝子レベルでの阻害は、遺伝子破壊による部分的または完全な遺伝子不活性化を含んでいてもよい。他方で、阻害は転写レベルで例えばアンチセンス分子、例えば、DNA分子、RNA分子または核酸類似物、リボザイム、例えば、RNA干渉(RNAi)が可能なRNA分子または核酸類似物または低分子RNA、例えば、メタロプロテアーゼおよび/またはリガンドmRNAと反対と向いたRNA分子または核酸類似物の利用によりで起こってもよい。
【0029】
さらに、活性はタンパク質レベルで例えばメタロプロテアーゼおよび/またはリガンド活性の特異的阻害を結果として生じる化合物の利用により阻害されてもよい。タンパク質レベルの阻害は、例えば、ADAM9、10または17のようなメタロプロテアーゼまたはHB-EGFまたはPreHB-EGFのようなリガンドもしくはリガンド前駆体と反対を向いた抗体または抗体断片の利用を含んでいてもよい。抗体はポリクロナール抗体またはモノクロナール抗体、組換え抗体、例えば、少なくとも1つの抗原結合部位を有する一本鎖抗体またはこのような抗体の断片、例えば、Fab、Fab’またはF(ab’)2断片のようなタンパク質分解性抗体断片、またはscFv断片のような組換え抗体断片であってよい。治療目的、特にヒトの治療のために、キメラ抗体、ヒト化抗体またはヒト抗体の利用がとりわけ有利である。
【0030】
さらに、メタロプロテアーゼおよび/またはリガンドのタンパク様または低分子量の阻害剤を使用してもよい。このような阻害剤の例は、CRM197、バチマスタット、マリマスタット、ヘパリンまたはリガンドに対する遮断抗体である。さらなる抗体は以下に記載されているようなスクリーニング法により同定してもよい。
【0031】
治療法に関して、付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有する製剤学的組成物の形で薬剤を投与することができる。
【0032】
本発明で使用するために適切な製剤学的組成物には、活性成分が意図する目的を達成するために効果的な量で含有されている組成物が含まれる。治療学的に有効な投与量とは、患者において症状の回復または生存の延長を結果として生じる化合物の量を意味する。このような化合物の毒性および治療的有効度は、細胞培養または実験動物において標準的な製剤学的方法、例えば、LD50(母集団の50%致死量)とED50(母集団の50%治療的有効量)を測定することにより決定することができる。本発明の方法で使用する化合物に関して、治療的有効量は細胞培養アッセイから初めに概算することができる。例えば、薬用量を動物モデルにおいて調製して、細胞培養で決定したようなIC50を含めた濃度幅の循環を達成することができる(すなわち、成長因子レセプター活性の最大の半分の阻害を達成する試験化合物の濃度)。このような情報を使用して、ヒトで有効な薬用量を正確に決定することができる。毒性と治療効果の間の薬用量比は、治療係数であり、かつLD50とED50の比として表現することができる。高い治療係数を示す化合物が好ましい。正確な調製物、投与経路および投与量は患者の状態を考慮して各々の内科医により選択することができる(例えば、Fingl et al., 1975, "The Pharmacological Basis of Therapeutics", 第1章、1頁参照)。投与量と間隔をそれぞれ調節して、レセプター調節効果、または最小有効濃度(MEC)を維持するために十分である活性部分の血しょうレベルを提供してもよい。MECは、各化合物に関して変化するが、in vitroでのデータから概算することもできる。例えば、この濃度は、本明細書中で記載されたアッセイで使用するレセプターの50〜90%阻害を達成するために必要である。化合物は、時間の10〜90%、好ましくは30〜90%、最も好ましくは50〜90%MECを上回って血しょうレベルを保つレジュメを使用して投与すべきである。MECを達成するために必要な投与量は、各々の特徴と投与経路に応じる。局所投与または選択的摂取の場合には、薬剤の効果的な局所濃度は血しょう濃度に関係ない。
【0033】
投与される組成物の正確な量は、自明ながら治療される被験者、被験者の重量、病気の度合い、投与方法および処方する内科医の判断による。抗体または治療的に活性な核酸分子、および他の化合物については、例えば1日あたり0.001〜100mg/kg、特に0.01〜10mg/kgの日用量が適切である。
【0034】
適切な投与経路には、例えば、経口、直腸、粘膜または腸管投与;筋肉内、皮下、髄内注射、ならびに髄腔内、脳室内、静脈内、腹腔内、鼻腔内または眼球内注射を含む非経口輸送が含まれる。
【0035】
二者択一的に、全身的な方法よりも局所で化合物を投与してもよい。例えば、多くの場合デポー剤または持続放出性調製物の形で注射することにより充実性腫瘍に化合物を直接投与することができる。
【0036】
さらに、薬剤をターゲットをしぼったドラックデリバリーシステムの形で、例えば、腫瘍に特異的な抗体で被覆したリポソームの形で投与することもできる。リポソームは、ターゲットをしぼり、腫瘍により選択的に摂取される。
【0037】
上記のように、本発明は特に治療抵抗性の過剰増殖性疾患、好ましくは治療抵抗性タイプの癌、例えば、照射および/または薬剤耐性タイプの癌の治療または予防に適切である。
【0038】
照射および/または細胞増殖抑制剤および/または細胞毒剤を用いる癌の治療は、ストレスキナーゼp38を活性化することが示された。意外にも、HB-EGFのようなレセプターチロシンキナーゼリガンドの阻害は、照射および/または化学療法の治療活性、特にそのアポトーシス誘導活性を強く促進した。従って、ダイレクトなレセプターチロシンキナーゼリガンド阻害剤、またはリガンド前駆体シェディングを防ぐ阻害剤と、更なる治療法および/または薬剤との共同適用は、結果として前記の方法および/または薬剤の適用に対して疾患の感受性を実質的に増大させ、ひいては前記の更なる治療法および/または薬剤の有効性を増強する。上記のようなリガンド阻害剤の投与は、少なくとも部分的に照射療法および/または細胞増殖抑制剤および/または細胞毒性薬剤に耐性である疾患、特に少なくとも部分的にアポトーシス誘導法と薬剤に対して耐性である疾患の治療または予防に特に適切である。
【0039】
本発明の好ましい実施態様では、レセプターチロシンキナーゼリガンド阻害剤を、照射療法、特にγ線照射療法と共同適用する。さらに好ましい実施態様では、レセプターチロシンキナーゼリガンド阻害剤を更なる抗ガン剤、特にアポトーシス誘導剤と共同適用する。適切な抗ガン剤の好ましい例は、ドキソルビシン、タキサン、シス/トランス−プラチンまたはこれらの誘導体、5−フルオロウラシル、マイトマイシンD、パクリタキセル、エトポシド、シクロホスホアミド、ドセタキセルまたは他のアポトーシス誘導剤または抗体のようなタンパク質である。
【0040】
リガンド阻害剤および更なる方法および/または薬剤の共同適用は、同時におよび/または連続的に行うことができる。リガンンド阻害剤の適用は、他の治療の適用に対して治療すべき疾患の感受性の増大を導く。特に、照射および/または化学療法に対する腫瘍耐性が減少する。
【0041】
従って、本発明のもう1つの点は、活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキットである。
【0042】
好ましくは、該組成物またはキットは付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有してよい。
【0043】
さらに、ストレス誘導性レセプターチロシンキナーゼ活性、例えば、p38−誘導活性のモジュレーターの同定を可能にする方法が提供する。前記方法は、試験化合物がレセプターチロシンキナーゼのリガンドの活性を阻害できるかを決定することを含む。この方法は、ストレス誘導性シグナルトランスダクションを調節できる新規化合物または化合物のクラスを同定するためのスクリーニング法、例えば、ハイスループットスクリーニング法として適切である。さらに、この方法は、化合物の製剤学的有効性および/または副作用を特徴付けるための確証法として適切である。この方法は、単離タンパク質、細胞抽出物、組換え細胞または人間以外のトランスジェニック動物の使用を含んでいてもよい。組換え細胞または人間以外のトランスジェニック動物は、相応の野生型細胞または動物と比べて変化したメタロプロテアーゼおよび/またはリガンド発現を示すのが好ましい。
【0044】
さらに、本発明を以下の図と実施例により説明する。
【0045】
図の説明文
図1:種々の細胞系におけるストレス誘導性EGFRおよびMAPKリン酸化の経時変化。(A)浸透ストレスと酸化ストレスに応答するEGFRおよびMAPKリン酸化。Cos−7細胞は、ソルビトール(0.3M)と過酸化水素(200μM)で記載された時間の間処理した。抗−EGFR抗体タンパク質を用いる細胞抽出物の免疫沈降(IP)に続いて、抗−ホスホチロシン抗体で免疫ブロットし(IB)、抗−EGFR抗体でリプローブした。リン酸化したMAPKsは、全体の溶解産物を抗−ホスホERK抗体、抗−ホスホ−JNKおよび抗−ホスホ−p38抗体で免疫ブロットすることにより検出された。同じフィルターを抗−p38抗体でリプローブした。(B)ヒト膀胱癌細胞系におけるストレス誘導性EGFRリン酸化。TCC−Sup細胞は(A)で示したように処理した。
【0046】
図2:ストレス誘導性EGFRリン酸化を媒介するp38。(A)EGFRキナーゼ機能に依存するp38リン酸化の経時変化。Cos−7細胞をAG1478(250nM)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールまたは200μM過酸化水素で記載された時間の間刺激した。細胞抽出物を抗−ホスホ−p38抗体で免疫ブロットし、同じフィルターをポリクロナール抗−p38抗体でリプローブした。(B)Cos-7細胞ではストレス誘導性EGFR活性化はp38に依存し、ERK活性には依存しない。Cos-7細胞をPD98059(50μM)、SB202190(10μM)または当量の空ベヒクル(DMSO)で30分間前処理し、0.3Mソルビトールと200μM過酸化水素で10分間刺激し、陽性コントロールとしてGPCRアゴニストのLPA(10μM)またはEGF(2ng/mL)で3分間刺激した。細胞抽出物をEGFRチロシンリン酸化内容物に関してアッセイした。(C)TCC-Sup癌細胞ではストレス誘導性EGFR活性化はp38活性に依存する。TCC-Sup細胞を(A)で記載したように前処理し、0.3Mソルビトールと200μM過酸化水素で10分間刺激し、陽性コントロールとしてEGF(2ng/mL)で3分間刺激した。細胞抽出物を溶解後、EGFRチロシンリン酸化内容物に関してアッセイした。
【0047】
図3:リガンド依存性である浸透ストレスと酸化ストレスに応答するEGFR活性化。(A)EGFRリン酸化におけるメタロプロテアーゼとHB-EGF阻害の効果。Cos-7、NCl-H292およびTCC-Sup細胞を24時間血清飢餓状態にし、BB94(10μM)、ジフテリア毒素変異体Crm197(10μg/mL)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトール、200μM過酸化水素、10μM LPAまたは2ng/mL EGFで10分間刺激した。抗−EGFR抗体タンパク質を用いる細胞抽出物の免疫沈降に続いて、抗−ホスホチロシン抗体で免疫ブロットし、抗−EGFR抗体でリプローブした。(B)ストレス剤に応答するShcリン酸化の分析。Cos-7細胞は(A)で記載したように処理した。ポリクロナール抗−Shc抗体タンパク質を用いる細胞抽出物からShcを免疫沈降した後に、抗−ホスホチロシン抗体で免疫ブロットし、かつ抗−Shc抗体でリプローブした。
【0048】
図4:ストレス剤に応答するproHB-EGF放出の分析。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。Cos-7細胞をBB94(10μM)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールと200μM過酸化水素で30分間刺激した。細胞を回収し、表面のproHB-EGFを染色し、フローサイトメトリーにより分析した。(B)条件培地の免疫ブロット分析。Cos-7細胞をproHB-EGF cDNAで一時的にトランスフェクションした。24時間血清飢餓状態にした後に、細胞をソルビトール(0.3M)または過酸化水素(200μM)で20分間刺激し、上澄培地内のタンパク質をトリクロロ酸(TCA)を用いて沈殿させた。沈殿したタンパク質をSchaegger-Jagowのプロトコールに従ってトリシン−ドデシル硫酸ナトリウムゲル電気泳動にかけ、続いて抗HB-EGF抗体を用いて免疫ブロット分析した。TPA刺激は陽性コントロールとして含めた。
【0049】
図5:ストレス誘導性EGFRリン酸化における種々のADAMタンパク質に対するsiRNAsの効果。(A)RNA干渉(RNAi)によるADAMメタロプロテアーゼ発現の遮断。NCl-H292細胞をADAM9、ADAM10またはADAM15に対するsiRNAでトランスフェクションし、2日間培養し、示したようにRT-PCRにより遺伝子発現を分析した。(B)Cos-7細胞をADAM9、−10、−12、−15および−17に対するsiRNAsでトランスフェクションし、24時間血清飢餓状態にさせ、0.3Mソルビトールまたは200μM過酸化水素で10分間刺激し、EGFRチロシンリン酸化内容物をアッセイした。(C)NCl-H292細胞を(B)で記載したように処理した。
【0050】
図6:ストレス剤に応答するMAPK活性化およびEGFR、メタロプロテアーゼおよびHB-EGF機能の遮断。(A)pcDNA3-HA-ERK2、TCC-SupおよびNCl-H292細胞で一時的にトランスフェクションしたCos-7細胞をAG1478(250nM)、BB94(10μM)、Crm197(10μg/mL)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールまたは200μM過酸化水素で30分間刺激した。全体の溶解産物が溶解した後に、抗−ホスホ−ERK抗体で免疫ブロットし、続いて同じ膜をポリクロナール抗−ERK抗体でリプローブした。FUJI LAS1000イメージングシステムを使用して3つの独立の試験からERKリン酸化を定量分析した(平均±標準偏差)。(B)Cos-7とNCl-H292細胞を(A)で記載したように処理した。溶解後、抗JNK抗体を用いてJNKを免疫沈降させ、基質としてGST-c-JUN融合タンパク質を用いてJNK活性をアッセイした。リン酸化したGST-c-JUNをオートラジオグラフィーにより視覚化し、かつポリクロナールJNK抗体を用いて、JNKを平行に免疫ブロットした。FUJI LAS1000イメージングシステムを使用して3つの独立の試験からGST-c-JUNリン酸化を定量分析した(平均±標準偏差)。TCC-Sup細胞を(A)で記載したように処理した。細胞溶解後、抗−ホスホ−JNK抗体で細胞抽出物を免疫ブロッティングすることによりJNKリン酸化をアッセイし、同じフィルターを抗−JNK抗体でリプローブした。(C)p38リン酸化はEGFR、メタロプロテアーゼおよびHB-EGF機能とは独立である。Cos-7細胞を(A)で記載したように処理した。抗−ホスホ−p38抗体で細胞抽出物を免疫ブロッティングすることによりp38リン酸化をアッセイし、かつ同じフィルターを抗−p38抗体でリプローブした。
【0051】
図7:TCC-Sup癌細胞のドキソルビシン誘導性細胞死。(A)ドキソルビシン処理に応答するp38活性化。TCC-Sup細胞を播種し、記載した時間の間ドキソルビシンで処理した。細胞溶解後、抗−ホスホ−p38抗体で細胞抽出物を免疫ブロッティングしてp38活性化を評価し、かつ同じフィルターを抗−p38抗体でリプローブした。(B)ドキソルビシンに応答して細胞死を促進するHB-EGF機能の遮断。細胞を10μMドキソルビシンと10μg/mL Crm197で記載されているように24時間ごとに72時間処理した。アッセイ緩衝液中の細胞を回収した後に、 Plで核を染め、フローサイトメトリー分析により分析した。
【実施例】
【0052】
実施例
1.材料と方法
1.1細胞培養、プラスミドおよびトランスフェクション
提供者の指示書に従い、全ての細胞系(American Type Culture Collection, Manassas, VA)を決まった手順で成長させた。メーカーのプロトコールに従い、リポフェクタミン(Invitrogen)を用いてCos-7細胞のトランスフェクションを行った。簡単には、一時的なトランスフェクションのために6cm皿中の細胞を、1皿あたり20μLリポフェクタミンと全体のプラスミドDNA2μg含有の無血清培地2mL中で4時間インキュベートした。次に、トランスフェクション混合物を当量の20%胎児ウシ血清で補足し、20時間後に細胞を洗浄し、刺激の前に無血清培地中でさらに24時間培養した。阻害剤AG1478(Alexis Biochemicals)、バチマスタット(BB94, British Biotech, Oxford, UK)、Crm197(Quadratech Ltd., UK)、SB202190(Calbiochem)およびPD98059(Alexis Biochemicals)を各刺激の前に血清飢餓状態の細胞に加えた。
【0053】
プロ−およびメタロプロテアーゼドメインが欠けたADAM10、12、15および17から成っているドミナントネガティブプロテアーゼは、予め記載してある(Gschwind et al., 2003)。プラスミドpcDNA3-HA-ERK2(Daub et al., 1997)とpcDNA3-proHB-EGF-VSV(Prenzel et al., 1999)をこの試験で使用した。
【0054】
1.2タンパク質分析
細胞を溶解し、かつ上記のようにタンパク質を免疫沈降させた(Daub et al., 1997)。溶解前に、80%コンフルエンスまで成長させた細胞を図中の説明文に示されているように阻害剤とアゴニストで処理し、次に50μM HEPES、pH7.5、150mM NaCl、1%トリトンX-100、1mM EDTA、10%グリセロール、10mM ピロリン酸ナトリウム、2mM オルトバナジウム酸ナトリウム、10mM フッ化ナトリウム、1mM フェニルメチルスルホニルフルオリド、10μg/mLアプロチニン含有の緩衝液中、氷上で10分間溶解した。溶解産物を13000rpmにて4℃で10分間の遠心分離により予めきれいにした。予めきれいにした分解遺産物を各抗体およびプロテインA−セファローズ20μLを用いて4℃で4時間免疫沈降した。沈殿物をHNTG緩衝液0.5mLで3回洗浄し、2×SDS試料緩衝液中に懸濁させ、3分間沸騰させ、ゲル電気泳動にかけた。SDS−ポリアクリルアミドゲル電気泳動に続いて、タンパク質をニトロセルロース膜に移した。従来の方法に従って、ウェスタンブロットを行った。ヒトEGFR(108.1)とSHCに対する抗体は前に特徴付けてある(Prenzel et al., 1999)。4G10モノクロナール抗体を用いてホスホチロシンを検出した(UBl, Lake Placid, NY)。ポリクロナール抗−ホスホ−p44/p42(Thr202/Tyr204)MAPK抗体、抗−ホスホ−JNK(Thr183/Tyr185)および抗−ホスホ−p38(Thr180/Tyr182)抗体はNew England Biolabs(Beverly, MA)から購入した。ポリクロナール抗−ERK2、抗−JNK1および抗−p38抗体はSanta Cruz Biotechnology(Santa Cruz, CA)から購入した。
【0055】
1.3 JNK活性のアッセイ
上記のようにJNK活性をアッセイした(Sudo and Karin, 2000)。培養した細胞を、20mM トリス(pH 7.6)、0.5%Nonidet P-40、250mM NaCl、3mM EDTA、1mM ジチオトレイトール、0.5mM フェニルメチルスルホニルフルオリド、20mM β−グリセロホスフェート、1mM オルトバナジウム酸ナトリウムおよび1μ/mLロイペプチン含有の溶解緩衝液中で溶解した。ポリクロナール抗−JNK抗体を用いて6ウェル皿から得られた溶解産物よりJNKを免疫沈降した。免疫沈降物を溶解緩衝液で2回洗浄し、かつキナーゼアッセイ緩衝液(25mM HEPES(pH 7.5)、20mM β−グリセロホスフェート、20mM PNPP、20mM MgCl2、2mMジチオトレイトールおよび0.1mM オルトバナジウム酸ナトリウム)で2回洗浄した。1μgGST-c-Jun(aa1-79)、20μMの冷たいATPおよび[y-32P]ATP5μCiを補足したキナーゼ緩衝液30μL中30℃で30分間キナーゼ反応を行った。Laemmli緩衝液30μLを添加することにより反応を中止し、12.5%ゲル上でゲル電気泳動にかけた。標識したGST-c-Junをホスホイメージャー(Fuji)で定量した。
【0056】
1.4 フローサイトメトリー分析
FACS分析を上記のように行った(Prenzel et al., 1999)。簡単には、細胞を播種し、20時間成長させ、24時間血清飢餓状態にした。上記のように阻害剤で細胞を処理し、かつ刺激した。回収した後、proHB-EGFに対するエクトドメインに特異的な抗体で細胞を45分間染色した。PBSで洗浄後、細胞をFITC共役二次抗体で15分間インキュベートし、PBSで再び洗浄した。細胞をBecton Dickinson FACS caliburフローサイトメーターで分析した。
【0057】
1.5 HB-EGFのTCA沈殿
pcDNA3-proHB-EGF-VSVで一時的にトランスフェクションしたCos-7を24時間血清飢餓状態にした。刺激の前に細胞を洗浄し、BB94(10μM)でプレインキュベートし、かつ記載したように刺激した。刺激後、上澄液を回収し、デソキシコリン酸ナトリウム(100μg/mL)を添加し、氷上での10分間のインキュベーションに続いて、溶液にトリクロロ酢酸(TCA)を加えて10%TCAの最終濃度にした。氷上で30分間インキュベーションした後、試料を遠心分離し、上澄液を捨て、かつ沈殿物をSchaegger-Jargow試料緩衝液中で再懸濁させた。Tris-HCl(pH8.8)を用いてTCAを中和し、トリシンSDSゲル電気泳動プロトコールを用いて試料を分離した(Schaegger and von Jargow, 1987)。
【0058】
1.6 RNA干渉とRT-PCR分析
内因性遺伝子をターゲティングするための21-ヌクレオチドsiRNA二本鎖(Dharmacon Research, Lafayette, CO, USA)のトランスフェクションを、NCl-H292細胞用のオリゴフェクタミン(Invitrogen)と6ウェルプレート当たり4.2μgのsiRNA二本鎖を用いて前記のように行った(Elbashir et al., 2001)。メーカーのプロトコールに従ってリポフェクタミン2000(Invitrogen)を用いてCos-7細胞をトランスフェクションした。簡単には、6cm皿あたり8.4μgのsiRNA二本鎖を無血清培地1mL中のリポフェクタミン2000 10μLと一緒に20分間インキュベートした。トランスフェクション混合物を血清含有の細胞培養培地に加え、6時間後に細胞を洗浄し、かつ血清含有の培地中で一晩インキュベートした。NCl-H292とCos-7細胞を血清餓死にし、トランスフェクション後2日目にアッセイした。有利な遺伝子の種々の領域をターゲティングするsiRNA二本鎖の混合物を使用することによりターゲット遺伝子のサイレンシングにおいて最も高い効率が得られた。使用したsiRNAの配列は以下のものである。
【0059】
【表1】

【0060】
ADAM12とADAM17に対するsiRNA二本鎖は予め記載してある(Gschwind et al., 2003)。
【0061】
ターゲット遺伝子の特異的サイレンシングをRT-PCR分析により確認した。RNeasy Mini Kit(Qiagen, Hilden, Germany)を用いて単離されたRNAをAMV逆転写酵素(Roche, Mannheim, Germany)を用いて逆転写した。PuReTaq Ready-To-Go PCR Beads(Amersham Biosciences, Piscataway, NJ)をPCR増幅に使用した。以下がプライマー(Sigma Ark, Steinheim, Germany)である:
【0062】
【表2】
【0063】
PCR製品を2.5%アガロースゲル上で電気泳動にかけ、DNAを臭化エチジウム染色により視覚化した。
【0064】
1.7 アポトーシスアッセイ
TCC-膀胱癌細胞を播種し、20時間成長させ、かつ記載したように10μM ドキソルビシンとCrm197で72時間処理した。細胞をヨウ化プロピジウム(PI)含有のアッセイ緩衝液中で回収し、4℃で3時間インキュベートした。核PI染色をBecton Dickinson FACS caliburフローサイトメターで分析した。
【0065】
2. 結果
2.1 Cos-7とヒト癌細胞系におけるEGFRとMAPK活性化の特異な動力学
浸透ストレスと酸化ストレスは、幅広い種々の細胞系においてEGFRとMAPKsのリン酸化を導いた(Carpenter, 1999; de Nadal et al., 2002; King et al., 1989)。潜在的なメカニズムを調べるために、免疫ブロット分析によりCos-7とTCC-Sup腎臓癌細胞系において経時変化実験を行った。図1に示されるように(上のパネル)、ストレス剤であるソルビトール(0.3M)と過酸化水素(200μM)に応答するEGFRのチロシンリン酸化は両方の細胞系で5〜10分以内に生じたのに対して、MAPKsであるERK1/2およびJNKはそれぞれ10分後と15分後にリン酸化された(図1A/B、下のパネル)。ERK1/2の弱い活性化は既に1分後に検出されたのに対して、リン酸化は10分後に著しく増大した。これとは対照的に、p38のリン酸化はストレス剤に即座に応答した。経時変化実験の結果は、EGFRがストレス刺激に応答してERK1/2とJNK活性化に関わっていることを示唆している。これとは対照的に、p38の活性化がEGFR刺激に先行した。興味深いことに、レセプターの活性化は、リガンド非依存性の活性化メカニズムを可能にするだけではなく、リガンド依存性の活性化メカニズムを可能にする時間の尺度で検出可能であった。
【0066】
2.2 p38は、浸透ストレスと酸化ストレスによるEGFR活性化を制御する
p38活性化がEGFRチロシンリン酸化に先行したという発見は(図1)、p38がCos-7とヒト癌細胞系においてEGFR刺激の上流レギュレーターとして働いているのかどうかという疑問を生じた。
【0067】
選択的EGFR―キナーゼ阻害剤AG1478とのCos-7細胞のプレインキュベーションは、ストレス剤に応答するp38リン酸化に影響を与えないので(図2A)、p38活性化がEGFR活性とは独立に起きたことを証明している。p38がEGFRの上流に位置しているのかどうかという疑問を処理するために、p38に特異的な阻害剤であるSB202190を使用してストレス誘導性EGFR活性化におけるp38活性の遮断効果を調べた。図2Bに示されているように、SB202190とのCos-7細胞のプレインキュベーションは、ストレス誘導性EGFR活性化を完全に排除し、リゾホスファチド酸(LPA)とEGF誘導性レセプターのリン酸化に影響を与えなかった。これとは異なり、MEK1/2阻害剤であるPD98059は、EGFRリン酸化に影響を与えなかった。似た結果は、腎臓癌細胞系TCC-Supで得られた(図2C)。まとめると、これらの結果はp38がCos-7とTCC-Sup腎臓癌細胞においてストレス誘導性EGFR活性化の上流メディエーターであることを証明している。
【0068】
2.3 メタロプロテアーゼ活性とHB-EGF機能に依存するストレス誘導性EGFRリン酸化
近年の研究はEGFRシグナリング、特に以前はリガンド非依存性であると考えられていたGPCRによるEGFRシグナルトランスアクチベーションのようなシグナルトランスダクション事象におけるEGF様リガンドのタンパク分解の重要性をはっきりと示している(Prenzel et al., 1999)。これらの発見に基づき、ストレス刺激によるEGFR活性化がリガンド依存性メカニズムに関わるかどうかという疑問に取り組んだ。従って、EGF様リガンドプロセシングとそれに続くEGFRトランスアクチベーションを阻害することが示されたメタロプロテアーゼ阻害剤であるバチマスタット(BB94)と一緒に細胞をプレインキュベートした(Prenzel et al., 1999)。EGFRチロシンリン酸化は、免疫ブロット分析を用いるストレス剤で刺激した後にモニターした。図3Aに示されるように、Cos-7およびTCC-Sup腎臓癌細胞においてBB94は殆ど完全にソルビトールまたは過酸化水素−誘導性EGFRリン酸化を遮断した(上と真ん中のパネル)。肺癌細胞系NCl-H292では、BB94はEGFR活性化を50%まで阻害したので、このことは二者択一的な平行の活性化メカニズムを示唆している(図3A、下のパネル)。
【0069】
次に、ストレス活性化EGFRチロシンリン酸化においてHB-EGF機能を特に遮断するジフテリア毒素変異体Crm197の効果を調べた。実に、Crm197はBB94と同じ程度にEGFRリン酸化を阻害したので、これらの3つの細胞系ではHB-EGFがストレス誘導性EGFR活性化に決定的に関わっていることを示唆している。陽性コントロールとしてのLPAによるレセプター活性化は、これらの阻害剤により完全に妨げられたのに対して、EGFを用いた直接の刺激は影響されなかった。
【0070】
さらに、このリガンド依存性がEGFRの下流のシグナリングパートナーに従うことができるかどうかは特別に興味のあることであった。Shcアダプタータンパク質は、レセプターがRas/Raf/ERK-MAPKカスケードの活性化とリンクするEGFRの顕著なシグナリングアダプターである。実際に、図3B(下のパネル)で示されているデータは、ストレス刺激に応答するShcタンパク質のリン酸化内容物がEGFR自体の内容物と似ていることを証明している。従って、ストレス刺激によるShcのリン酸化はリガンド依存性EGFRリン酸化メカニズムに極めて依存している。
【0071】
2.4 Cos-7細胞において浸透ストレスと酸化ストレスに応答して誘導されるproHB-EGFのエクトドメインシェディング
このリガンド依存性EGFR刺激メカニズムにおけるHB-EGFの役割を更に実証するために、リガンド自体のレベルで高浸透圧と過酸化水素処理に応答するproHB-EGFのシェディングを調べた。従って、ソルビトールまたは過酸化水素で刺激する前と後にCos-7細胞の細胞表面に存在するproHB-EGFの量をフローサイトメトメトリー分析により分析した。図4Aに示されているように、両方の刺激は細胞表面上でHB-EGF前駆体の著しい減少を導いた。さらに、図3Aに示されている結果と一致して、メタロプロテアーゼ阻害剤BB94を用いる予備処理は、proHB-EGFプロセシングを排除した。HB-EGF前駆体の減少の他に、proHB-EGFを異所性発現してCos-7細胞の細胞表面から細胞培養培地に放出された可溶性の成熟型HB-EGFの量を測定した。図4Bに示されるように、高浸透圧と過酸化水素を用いる両方の処理は、免疫ブロット分析により測定されたように調整培地中でHB-EGFの増大を誘導した。さらにまた、HB-EGF放出がメタロプロテアーゼ阻害剤BB94でのプレインキュベーションにより遮断され、メタロプロテアーゼ活性の関わりを確証している。
【0072】
2.5 浸透ストレスと酸化ストレスによりEGFR活性化を媒介するADAMファミリーのメタロプロテアーゼ
メタロプロテアーゼ依存性メカニズムがストレス誘導性EGFRとShc活性化に著しく寄与するという発見は、どのメタロプロテアーゼが関係しているのかという疑問を生じた。RNA干渉法を使用して、すでにEGF様リガンドの切断に関わっていたADAMプロテアーゼであるADAM9、10、12、15および17の内因発現を阻害した。図5Aは、ADAM9、10および15に対するsiRNAによるターゲット遺伝子発現の効果的かつ特異的ノックダウンをRT-PCRで証明している。ADAM12と17に対するsiRNAsは既に記載してある(Gschwind et al., 2003)。各プロテアーゼに対するsiRNAsを用いる一時的なトランスフェクション、およびそれに引き続くソルビトールまたは過酸化水素で刺激した後のEGFRの免疫ブロット分析は、Cos-7細胞では両方のストレス刺激後にADAM10とADAM17がEGFR活性化に関わっていることを明らかにした(図5B)。しかし、ヒトの癌細胞におけるこれらのプロセス制御に興味があったので、ヒト肺ガン細胞系NCl-H292におけるADAMファミリーメンバーの関係をさらに調べた。すわなち、図5Cで証明されているように、Cos-7細胞で得られた結果と類似して、ADAM17もストレス誘導性EGFR活性化に関わっている。Cos-7細胞とは対照的に、ADAM9および過酸化水素処理の場合でも、ADAM12は、NCl-H292細胞のストレス応答に関わっている。全体として、細胞タイプにより、メタロプロテアーゼの種々のADAMファミリーのメンバー、特にADAM17は、ストレス剤に応答するHB-EGF依存性EGFR活性化において中心的な役割を担っている。
【0073】
2.6 HB-EGF依存性EGFR活性化に媒介される高浸透圧ストレスと酸化ストレスに応答するMAPKsERK1/2とJNKの活性化
MAPKs であるERK1/2とJNKが高浸透圧と活性酸素種により活性化されるので(de Nadal et al., 2002; Kyriakis and Avruch, 2001)、リガンド依存性EGFRリン酸化がストレス刺激によるMAPK活性化の誘導に寄与するかどうかを調べた。ゆえに、選択的EGFR tyrphostin AG1478を使用して、EGFRキナーゼ活性におけるストレス誘導性MAPK活性化の全体的な依存性を調べた。さらに、この効果をBB94とCrm197によるリガンド依存性EGFR活性化プロセスの阻害と比較した。図6A(上のパネル)に示されているように、ERK1/2のソルビトールと過酸化水素誘導性活性化は、AG1478により遮断された。さらに、BB94とCrm197はERK1/2リン酸化の遮断において殆ど有効であった。これらのデータは、浸透ストレスと酸化ストレスに応答するCos-7細胞におけるERK1/2活性化がリガンド依存性メカニズムに大いに帰因しているEGFR活性化に殆ど完全に依存していることを示唆している。同じ実験構成を使用して、このコンセプトをヒト癌細胞系におけるストレスシグナリングにまで延ばすことができるかどうかという問題に的をしぼった。しかし、TCC-SupおよびNCl-H292細胞における高浸透圧および酸化ストレス誘導性ERK1/2活性化がAG1478、BB94およびCrm197により実質的に遮断されることが分かった(図6A真ん中と下のパネル)。興味深いことに、NCl-H292細胞において酸化ストレスがリガンド依存性メカニズムにより部分的にだけEGFRリン酸化を誘導するにもかかわらず、この細胞タイプではERK活性化はproHB-EGFプロセシングに大いに依存する。
【0074】
ERK1/2とは別に、JNKの活性化を導くシグナリングメカニズムに興味があった。図6B(上のパネル)には、AG1478、BB94およびCrm197を用いる処理に依存してJNK活性が浸透ストレスと酸化ストレスにより誘導されていることが示されている。Cos-7細胞では、ソルビトール誘導性JNK活性がEGFRに大いに依存しているのに対して、酸化ストレスに応答してJNK活性はHB-EGFによるEGFR活性化に50%依存している。似た結果は、図6B(真ん中のパネル)に示されているように腎臓癌細胞系TCC-Supで得られている。これとは対照的に、肺ガン細胞系NCl-H292では過酸化水素に刺激されたJNK活性化がEGFRに非依存的であるように見えたのに対して、高浸透圧に応答するJNK活性は部分的にリガンド依存性EGFR活性化経路に依存していた。p38がストレス誘導性EGFR活性化の上流メディエーターとして働く(図2)という発見と一致して、p38活性は、EGFRキナーゼ活性にもメタロプロテアーゼまたはHB-EGF機能にも依存していない(図6C)。
【0075】
化学療法を用いる腫瘍細胞の治療は、ストレスキナーゼp38とJNKを活性化して細胞死を導くことを示した。しかし、p38活性化がHB-EGF依存性EGFR活性化とErk1/2活性化を導くので、TCC-Sup腎臓癌細胞のドキソルビシン誘導性アポトーシスにおいてHB-EGF機能を遮断する効果を調べた。図7Aは、ドキソルビシン処理がp38活性化を誘導したことを証明している。図7B/Cには興味深いことに、HB-EGF阻害剤CRM197でのTCC-Sup細胞処理は、ドキソルビシンだけと比較した場合にドキソルビシンに応答してアポトーシスを著しく促進したのに対して、CRM197だけで処理した場合にはマイナーな効果だけを発揮したことが示されている。
【0076】
全体として、これらのデータはヒト癌細胞において、ストレス剤によるリガンド依存性EGFRチロシンリン酸化は、浸透ストレスと酸化ストレスに応答するMAPKsERK1/2とJNKの活性化において重要な役割を担っていることを証明している。さらに、これらの結果は、ここで示されているシグナリングメカニズムが腫瘍細胞により使用され化学療法誘導性細胞死を回避する経路に帰因するとしている。
【0077】
3. 考察
徹底的な調査は、どのように哺乳類細胞がストレス剤に対処しているのかという問題に焦点をおいた。本研究は、ヒト癌細胞において浸透ストレスと酸化ストレスに応答するEGFRとそれに続くMAPK活性化を導く成長因子依存性メカニズムにおいて新たな洞察を提供した。
【0078】
ここでは、浸透ストレスと酸化ストレスの両方によって誘導されるEGFRリン酸化が、Cos-7細胞とヒト癌細胞系においてメタロプロテアーゼ活性とHB-EGFの放出を必要とすることを証明した。この結果は、EGFRレベル(図3)とEGF様リガンドHB-EGFレベル(図4)の両方で得られた。以前の報告は、高浸透圧と酸化ストレスがレセプター凝集と二量化によりEGFR活性化を導くことを証明している(Rosette and Karin, 1996)。二者択一的に、ホスファターゼの不活性化は、それぞれ細胞内キナーゼまたはレセプターの刺激を導き(Blanchetot et al., 2002; Knebel et al., 1996)、これによりEGFRリン酸化を誘導することを提唱した。これに関連して、NCl-H292細胞においてストレス誘導性EGFRリン酸化が部分的にリガンド依存性メカニズムに依存し、かつ部分的にリガンド非依存性メカニズムに依存しているという発見は(図3)、両方のメカニズムがレセプター活性化と協力していると思われるので、以前の研究と一致している。Cos-7とTCC-Sup腎臓癌細胞ではリガンド依存性メカニズムが浸透ストレスと酸化ストレスによるレセプターリン酸化によるレセプターリン酸化の主因であるので、これらのメカニズムのそれぞれの寄与は細胞の状況に依存しているようである(図3)。ストレス刺激がラット胃上皮細胞でHB-EGFとアンフィレグリンの発現を誘導することを証明する以前の研究により我々の結果は更に支持された(Miyazaki et al., 2001)。さらに、Frank et al.(Frank et al., 2003)による近年の報告は、血管平滑筋細胞での過酸化水素誘導性EGFRリン酸化におけるHB-EGFに関する。高浸透圧に応答するproTGF-αのシェディングは、内因性EGFRが欠けたCHO細胞で報告されていた(Fan and Derynck, 1999; Montero et al., 2002)。Chen et al.(Chen et al., 2001a)による報告は、Cos-7細胞における過酸化水素誘導性EGFR活性化がc-Srcによるリガンド非依存性メカニズムによるとしている。これとは対照的に、同じ細胞系では、Src阻害剤であるPP1とPP2を用いる酸化ストレスによってEGFR活性化の特異的阻害が達成されなかった(未発表のデータ)。
【0079】
ますます増える証拠は、亜鉛依存性プロテアーゼのADAMファミリーがEGF様リガンドプロセシングの重要なメディエーターであることを示唆している。これらの報告と一致して、我々の結果はADAMプロテアーゼがストレス刺激により誘導されるproHB-EGFのシェディングに関わっていることを証明している。興味深いことに、EGFRトランスアクティべーションに関連して、1つ1つの個別のADAMプロテアーゼが活性化されるのに対して(Gschwind et al., 2003: Yan et al., 2002)、我々は細胞系に応じて2つ以上のADAMプロテアーゼが活性になることを見つけた(図5)。重要なことは、一般にADAM17がストレス刺激されたシェディング事象に関わっているようだが、ADAM9、ADAM10およびADAM12も関わることができることである(図5)。これらの全ての酵素は以前にEGF様リガンドシェディングに関わっていた(Asakura et al., 2002; Izumi et al., 1998; Lemjabbar and Basbaum, 2002; Yan et al., 2002; Gschwind et al., 2003)。種々のADAMプロテアーゼが活性化されても、proHB-EGFだけが切断された発見は、アンフィレグリンとTGF-αもNCl-H292細胞内で発現されたので意外であった(データの発表なし)。もう一方で、VeroH細胞ではTPA刺激に応答してADAM9がproHB-EGFを切断するのに対して(Izumi et al., 1998)、同じ細胞系ではLPA刺激後のproHB-EGFプロセシングがADAM9とは独立であることを証明する以前の報告により(Umata et al., 2001)、我々の結果は裏付けされ、proHB-EGFシェダーゼが細胞の状況と刺激の両方により限定されることを示唆している。さらに、ADAM9ノックアウトマウスは、ある明確な表現型が欠損しているが、ADAM9−/−フィブロブラストはproHB-EGFプロセシングにおいて何の欠陥も生じない(Weskamp et al., 2002)。このことは、in vivoでのproHB-EGF切断酵素間の機能的な余剰を示している。
【0080】
どのようにADAMファミリーのメタロプロテアーゼが活性化され、ついにはEGFRリン酸化と下流のシグナリング応答を導くのであろうか。以前の報告は、MAPK ERK1/2により成長因子とTPAに応答して、膜貫通型タンパク質のメタロプロテアーゼに媒介されるエクトドメインの切断を制御しているのに対して、エクトドメインシェディングの基礎レベルはp38活性に帰因していたことを証明した(Fan and Derynck, 1999; Gechtman et al., 1999; Rizoli et al., 1999)。さらに、形質転換されていないヒトケラチノサイトでのソルビトール誘導性EGFR活性化において、p38はEGFRの上流メディエーターとして関連しているとされた(Chang et al., 2002)。我々の結果とは対照的に、著者は培地移動実験(medium transfer experiments)に基づいて、リガンド依存性メカニズムを排除している。放出されたEGF様リガンドがヘパリンスルフェートグリカンに結合して細胞外マトリックス中に保留していたので、リガンド依存性EGFR活性化の関わりを除くことができない。これらの報告と我々のシステムにおいて、p38がEGFRリン酸化状態とは独立である発見(図2と図6C)は、p38がストレス剤によるリガンド依存性EGFR活性化と下流のシグナリングを制御する上流のシグナリング要素であるのかという疑問を投げかけた。実に、特異的p38阻害剤とのプレインキュベーションがストレス誘導性EGFR活性化を排除したのに対して、ERK1/2活性化の遮断はEGFRリン酸化に影響しないままであることを発見した(図2B)。これに反して、ストレス剤に応答するp38活性化自体は、EGFR選択阻害剤であるAG1478を用いて査定したようにEGFRとは独立であった(図2A)。さらに、経時変化実験は、p38活性化がEGFRリン酸化に先行し、このことはp38がEGFRの上流に位置するために必要な前提条件であることを明らかにした。一方で、ERK1/2とJNK活性化はもっと後で生じる(図1)。全体として、これらのデータは、p38がリガンド依存性EGFR活性化の上流インデューサーであり、かつそれに続く下流のシグナリングであることを示唆している。
【0081】
ストレス応答でのp38の重要な役割は、発生の全体に渡ってよく保護されている。サッカロミセス属cerevisaeでは、高浸透圧への応答が集中的に研究されてきた。中心的役割は、HOGキナーゼ、ヒトp38の酵母ホモログが担っている。HOGは浸透圧を変化させるためのセンサー分子により活性化されるが、ヒトにはない。これらのタンパク質によるHOGの活性化は酵母で順応応答を導き高浸透圧を対処する(de Nadal et al., 2002)。
【0082】
酸化ストレスと浸透ストレスに応答するERK1/2とJNKの活性化は、細胞ストレス応答において重要な段階を示す(Kyriakis and Avruch, 2001で調査)。MAPKシグナリングの潜在的なミディエーターとして種々のレセプターチロシンキナーゼのリン酸化がストレス剤により刺激されている一方で、様々な報告はストレス刺激によるMAPK活性化にEGFRが際だった役割をを有することを明らかにしている(Carpenter, 1999で調査)。癌細胞が高レベルのROSを頻繁に生産するので、近年の研究は抗ガン治療でMAPKによるストレスシグナリングに厳格な因果関係の証拠を提供している(Burdon, 1995; Szatrowski and Nathan, 1991)。さらに、抗ガン剤または照射療法はストレスシグナリングカスケードの活性化を導き(Benhar et al., 2002)、これが、これらの薬剤により引き起こされるROS生産に帰因してきた。さらに、EGFR依存性MAPKシグナリングは、Bcl-2ファミリーのようなアポトーシスレギュレーターの発現レベルに影響を与えると報告されている(Jost et al., 2001)。これらの理由から、治療の影響力ゆえにストレスシグナリングメカニズムとそれらの複雑な相互作用を解明することに注目が集まっている。
【0083】
ここでは、ストレス誘導性リガンド依存性EGFR活性化がERK1/2の場合には決定的に、かつJNKの場合に高い度合いでEGFR活性化に依存している下流のシグナリング事象を導くことを示された(図6)。以前の報告は、ストレス誘導性MAPK活性化で種々のリガンド非依存性活性化メカニズムを示唆していた。これらの経路はホスフェートならびに他のレドックスの影響を受けやすいタンパク質の不活性化とダウンレギュレーションに関わり、足場タンパク質と小さなGタンパク質に影響を与える。EGF様リガンド依存性メカニズムと非依存性メカニズムの両方がMAPK活性化で協力することを証明したので、我々のデータはこれらの結果に及ぶ(図6)。これらの経路の各々の寄与は、特定の細胞状況に依存しているようである。リガンド依存性EGFR活性化は、種々のMAPKシグナリング経路間の連絡を媒介しているようである。さらに、腎臓癌細胞中のドキソルビシン誘導性アポトーシスをHB-EGF機能の遮断により強く促進できるのに対して(図7)、ドキソルビシン処理がp38活性化を誘導することを証明した。化学療法剤がストレスシグナリングカスケードを活性化することが以前に示されたので(Benhar et al., 2002)、このシグナリングメカニズムは、薬剤誘導性細胞死を避ける腫瘍細胞の分子の真相を提供する。
【産業上の利用可能性】
【0084】
我々の知る限りでは、これがヒト癌細胞でEGFRのリガンド依存性活性化と、それに続くMAPKp38により制御される浸透ストレスと酸化ストレス剤による下流のシグナリングを証明する初めての報告である。ますます増える証拠は、心臓血管疾患、神経変性疾患または癌のような過剰増殖性疾患のように多種の様々なヒトの疾患で過剰のROS生産により引き起こされる酸化ストレスに特に関わる。ゆえに、ストレス剤により引き起こされる細胞損傷に関わる病態生理学的疾患と各治療アプローチにとって特別に重要である。
【0085】
本明細書で記載したデータは、哺乳類細胞、特にヒト癌腫でのストレス刺激のシグナリングメカニズムにおける以前の結果に及ぶ。我々の発見は、ヒト癌細胞でのストレス応答の重要なメディエーターとしてADAMファミリープロテアーゼとHB-EGFの重要性を実証する。さらに、我々のデータはMAPKの種々のグループ間でのクロスコミュニケーションがADAMプロテアーゼとEGFRをシグナリング媒介物として使用していることを示唆する。これに関連して、ERK1/2とJNK活性の間のバランスは、細胞の運命にとって特に重要である。他のEGFR下流シグナリング事象ならびにストレス剤に応答する癌細胞の強化増殖または移動のような可能性のある病理学的応答に焦点をおいて将来の研究が成されるべきである。
【0086】
【表3】
【0087】
【表4】
【0088】
【表5】
【0089】
【表6】
【0090】
【表7】
【0091】
【表8】
【0092】
【表9】
【技術分野】
【0001】
本発明は、レセプターチロシンキナーゼのリガンド、特に細胞外リガンドを阻害することによるストレス誘導性レセプターチロシンキナーゼ活性の阻害に関する。
【背景技術】
【0002】
高浸透圧および酸化剤、電離線または紫外線のような環境ストレスに哺乳類の細胞が暴露されると、様々なシグナルトランスダクションのカスケードが活性化される。活性酸素種(ROS)がセカンドメッセンジャーとして関わっているとされているが、その無制御の生産とオキシダントへの暴露による酸化ストレスが細胞損傷と癌のような生理学的疾患に関わっている(Finkel, 1998(非特許文献1); Kamata and Hirata, 1999(非特許文献2))。さらに、哺乳類の細胞は増大するモル浸透圧濃度を含む細胞外環境の変化に適応しなくてはならず、このことが結果として細胞収縮と低分子の増加合成を生じ、細胞内と細胞外の状態を等しくする(de Nadal et al., 2002(非特許文献3))。
【0003】
浸透ストレスまたは酸化ストレスは、種々のレセプターチロシンキナーゼを活性化し、最も際だったものは上皮成長因子レセプター(EGFR)である(King et al., 1989(非特許文献4); Knebel et al., 1996(非特許文献5); Rao, 1996(非特許文献6); Rosette and Karin, 1996(非特許文献7))。EGFRは、細胞増殖、細胞分化、細胞移動またはアンチアポトーシスシグナルを含む重要な生物学的応答の過多を制御するので、頻繁に様々なヒトの疾患に影響を与えてきた(Prenzel et al., 2001(非特許文献8)。リガンド依存性およびリガンド非依存性レセプター活性化メカニズムは、EGFRに関して記載されてきている。リガンドに媒介されたレセプター活性化は、EGF、HB-EGF、アンフィレグリンまたはTGF−αのようなEGF様リガンドがレセプターのエクトドメインに結合し、2つのリガンドレセプターの二量化がヘテロダイマーを生じ、引き続き固有のキナーゼ活性化とオートリン酸化を活性化することにより生じる(Schlessinger, 2002(非特許文献9)。リガンド非依存性レセプター活性化は、その触媒ポケット内のクリティカルなシステイン残基の酸化に関わるホスファターゼの不活性化により生じることが提唱されている(Knebel et al., 1996(非特許文献5))。よって、レセプターリン酸化の平衡は、非リン酸化状態からリン酸化状態にシフトする。リガンド非依存性レセプター活性化のその他のメカニズムは、非特異的クラスター化とEGFRの内在化に関わると示唆されてきた(Rosette and Karin, 1996(非特許文献7))。さらに、c-Srcのような細胞質のノンレセプターチロシンキナーゼがEGFRをリン酸化することが示された(Biscardi et al., 1999(非特許文献10) Tice et al., 1999(非特許文献11))。
【0004】
EGFRのチロシンリン酸化は、Gタンパク質共役レセプター(GPCR)刺激によっても誘導することができ、このプロセスはEGFRトランスアクチべーションと名付けられた(Daub et al., 1996(非特許文献12))。このメカニズムは当初、専らリガンド非依存性プロセスによるものとされていた。しかし、多くの細胞系ではEGFRトランスアクチべーションはメタロプロテアーゼに媒介される膜貫通型EGF−様リガンドのシェディングにより生じ、活性になるためにプロセシングしなくてはならない(Prenzel et al., 1999(非特許文献13))。メタロプロテアーゼのADAMファミリーの極めて最近のメンバーは、GPCR−誘導性proHB-EGFとpro-ARプロセシングに必要なシェダーゼとして同定された(Gschwind et al., 2003(非特許文献14); Yan et al., 2002(非特許文献15))。とりわけ、TNF-α変換酵素(TACE)とも称されるADAM17、ならびにADAM10とADAM12も関わることが示された(Asakura et al., 2002(非特許文献16); Gschwind et al., 2003(非特許文献14); Yan et al., 2002(非特許文献15))。リガンド依存性EGFRトランスアクチベーションが関わる異所のシグナリングプロセスは、頭頚部の扁平上皮癌、心臓および胃腸肥大および嚢胞性線維症のような種々のヒトの疾患に関わってきている(Asakura et al., 2002(非特許文献16); Gschwind et al., 2002(非特許文献17); Keates et al., 2001(非特許文献18); Lemjabbar and Basbaum, 2002(非特許文献19)。EGFRトランスアクチベーションとは別に、ADAM9は、TPA刺激に応答してproHB-EGFをプロセシングすることが示された(Izumi et al., 1998)のに対して、ADAM17よるproTGF-α、proHB-EGFおよびプロアンフィレグリンの切断が報告されている(Merlos-Suarez et al., 2001(非特許文献20); Peschon et al., 1998(非特許文献21); Sunnarborg et al., 2002(非特許文献22))、TGF-αの場合の腫瘍形成を示唆している(Borrell-Pages et al., 2003(非特許文献23))。
【0005】
RTKリン酸化の他に、環境ストレスはマイトジェン活性化タンパク質キナーゼ(MAPKs)の活性化を導き、これは転写因子の活性化により細胞外刺激を核へカップリングさせる細胞内シグナルトランスダクションカスケードの主な部分である。従って、MAPK経路は、増殖、移動、分化およびストレス応答のような重要なプロセスを制御する(Chen et al., 2001b(非特許文献24); Johnson and Lapadat, 2002(非特許文献25))。しかし、転写因子を活性化する以外に、MAPKsである細胞外シグナル制御キナーゼ−1/2(ERK1/2)とp38は、ADAM17の細胞内ドメインのリン酸化により膜貫通型タンパク質(Fan and Derynck, 1999(非特許文献26))のプロセシングの制御に関わっていた(Diaz-Rodriguez et al., 2002(非特許文献27); Fan et al., 2003(非特許文献28))。細胞の運命がこれらのシグナリング経路間の連絡により決定されるので、徹底的な研究は、浸透ストレスと酸化ストレスによるMAPK活性化のメカニズムに注目した(Kyriakis and Avruch, 2001(非特許文献29))。以前の報告は、ストレス剤ならびに足場タンパク質機能の変異によるMAPK活性化におけるホスファターゼの不活性化またはダウンレギュレーションが、シグナリング複合体の会合または解離を導くと提唱していた(Benhar et al., 2002(非特許文献30); de Nadal et al., 2002(非特許文献2))。さらに、小さなGタンパク質は浸透ストレスによりMAPKの活性化に働いていることが証明された(de Nadal et al., 2002(非特許文献2))。多くの証拠は特にROS、ストレス活性化キナーゼp38とJNKおよびストレスシグナリングがアポトーシスへの癌細胞の感受性ならびに増殖応答と関わると示唆しているので、ストレス刺激によるMAPKの活性化は、ヒトの癌の発生と進行に過酷な結果をもたらす。近年の研究は、抗ガン剤がストレスシグナリングカスケードを活性化すると示しているので、EGFRを介するストレスシグナリングは、癌治療に影響を及ぼすことができる(Benhar et al., 2002(非特許文献30)で調査)。
【0006】
酸化ストレスと浸透ストレスに応答するRTKとMAPK活性化のメカニズムが集中的に研究されてきたが、しかしこれまでにこれらの制御経路はヒト癌細胞では一般にリガンド非依存性プロセスであると記載されていた。ADAMメタロプロテアーゼによるEGFR活性化の制御の見解における近年の進歩は、EGF様リガンドプロセシングの潜在的な関連性についてストレス誘導性EGFRと、MAPK刺激のメカニズムを研究するきっかけとなった。
【0007】
Fischer et al.(Poster PS01-0916, Eur. J. Biochem. (非特許文献31))は、p38とADAMファミリーのメタロプロテアーゼがヒト癌細胞の下流のシグナリングにおいてストレス誘導性リガンド依存性EGFR活性化を制御することを記載している。しかし、特異的なメタロプロテアーゼは同定されていない。
【0008】
Takenobu et al. (J. Biol. Chem. 278, (2003), 1725-1762(非特許文献32))は、HB-EGF様成長因子のストレスおよび炎症サイトカイン誘導性エクトドメイン・シェディングがp38 MAPK により媒介されていることを開示している。メタロプロテアーゼADAM9がVeroH細胞ではストレス誘導性pro HB-EGFのシェディングに不必要であることが分かった。
【0009】
Herrlich et al. (FASEB J. 16 (2002), A56(非特許文献33))およびTschumperlin et al.(FASEB J.16 (2002), A1150(非特許文献34))は、HER2/HER3またはEGFR活性化の刺激が、それぞれ浸透ストレスまたは機械的ストレスによってヘレグリンシェディングに媒介されることを開示している。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Finkel, 1998
【非特許文献2】Kamata and Hirata, 1999
【非特許文献3】de Nadal et al., 2002
【非特許文献4】King et al., 1989
【非特許文献5】Knebel et al., 1996
【非特許文献6】Rao, 1996
【非特許文献7】Rosette and Karin, 1996
【非特許文献8】Prenzel et al., 2001
【非特許文献9】Schlessinger, 2002
【非特許文献10】Biscardi et al., 1999
【非特許文献11】Tice et al., 1999
【非特許文献12】Daub et al., 1996
【非特許文献13】Prenzel et al., 1999
【非特許文献14】Gschwind et al., 2003
【非特許文献15】Yan et al., 2002
【非特許文献16】Asakura et al., 2002
【非特許文献17】Gschwind et al., 2002
【非特許文献18】Keates et al., 2001
【非特許文献19】Lemjabbar and Basbaum, 2002
【非特許文献20】Merlos-Suarez et al., 2001
【非特許文献21】Peschon et al., 1998
【非特許文献22】Sunnarborg et al., 2002
【非特許文献23】Borrell-Pages et al., 2003
【非特許文献24】Chen et al., 2001b
【非特許文献25】Johnson and Lapadat, 2002
【非特許文献26】Fan and Derynck, 1999
【非特許文献27】Diaz-Rodriguez et al., 2002
【非特許文献28】Fan et al., 2003
【非特許文献29】Kyriakis and Avruch, 2001
【非特許文献30】Benhar et al., 2002
【非特許文献31】Fischer et al.(Poster PS01-0916, Eur. J. Biochem.
【非特許文献32】Takenobu et al. J. Biol. Chem. 278, (2003), 1725-1762
【非特許文献33】Herrlich et al. FASEB J. 16 (2002), A56
【非特許文献34】Tschumperlin et al. FASEB J.16 (2002), A1150
【発明の概要】
【発明が解決しようとする課題】
【0011】
以上の背景技術の下、本発明は、レセプターチロシンキナーゼのリガンド、特に細胞外リガンドを阻害することによるストレス誘導性レセプターチロシンキナーゼ活性の阻害のための新規薬剤または方法を提供する。
【課題を解決するための手段】
【0012】
本発明では、浸透ストレスまたは酸化ストレスに応答するEGFR活性化がメタロプロテアーゼに媒介されるproHB-EGFの切断に関わることを証明する。重要なメタロプロテアーゼは、proHB-EGF をプロセシングするADAMファミリー、特にADAM9、ADAM10およびADAM17のメンバーを含む。さらに、ストレス誘導性リガンド依存性EGFR活性化は、MAPKs であるERK1/2およびJNKと結びつくことができる。我々は、ストレス活性化EGFRリン酸化がp38活性に依存しており、p38がヒト癌細胞のストレス応答においてADAMメタロプロテアーゼの上流アクチベーターとして関わっている証拠を提供する。
【0013】
さらに化学療法剤、例えば、p38活性化を誘導するドキソルビシンでの腫瘍細胞の治療と、細胞死を著しく促進するHB-EGF作用の遮断との組合せは、ドキソルビシンだけの治療と比較した場合に、著しく細胞死を促進した。この結果は、化学療法に誘導される細胞死を逃れる腫瘍細胞のシグナルメカニズムの役割を示唆している。
【0014】
従って、細胞、この好ましくは哺乳類細胞、より好ましくはヒトの細胞、例えば、腫瘍細胞、特に過剰増殖性細胞またはアポトーシス耐性細胞で、レセプターチロシンキナーゼまたはRTK−媒介シグナリング経路のストレス誘導性活性化の調節を可能にする方法を提供し、前記方法は該レセプターチロシンキナーゼのリガンドの活性の阻害を含む。
【0015】
本発明の第一の点は、少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療するための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0016】
本発明のもう1つの点は、過剰増殖性疾患に対する治療の効果を上げるためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0017】
本発明のもう1つの点は、照射療法および/または薬剤治療に対する過剰増殖性疾患の感受性を上げるための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0018】
本発明のもう1つの点は、レセプターチロシンキナーゼのストレス誘導性活性化により生じるか、またはこれと関連する疾患を予防または治療するための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用に関する。
【0019】
また本発明のもう1つの点は、活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキットである。
【発明の効果】
【0020】
我々の知る限りでは、これがヒト癌細胞でEGFRのリガンド依存性活性化と、それに続くMAPKp38により制御される浸透ストレスと酸化ストレス剤による下流のシグナリングを証明する初めての報告である。ますます増える証拠は、心臓血管疾患、神経変性疾患または癌のような過剰増殖性疾患のように多種の様々なヒトの疾患で過剰のROS生産により引き起こされる酸化ストレスに特に関わる。ゆえに、ストレス剤により引き起こされる細胞損傷に関わる病態生理学的疾患と各治療アプローチにとって特別に重要である。
【0021】
本明細書で記載したデータは、哺乳類細胞、特にヒト癌腫でのストレス刺激のシグナリングメカニズムにおける以前の結果に及ぶ。我々の発見は、ヒト癌細胞でのストレス応答の重要なメディエーターとしてADAMファミリープロテアーゼとHB-EGFの重要性を実証する。さらに、我々のデータはMAPKの種々のグループ間でのクロスコミュニケーションがADAMプロテアーゼとEGFRをシグナリング媒介物として使用していることを示唆する。これに関連して、ERK1/2とJNK活性の間のバランスは、細胞の運命にとって特に重要である。他のEGFR下流シグナリング事象ならびにストレス剤に応答する癌細胞の強化増殖または移動のような可能性のある病理学的応答に焦点をおいて将来の研究が成されるべきである。
【図面の簡単な説明】
【0022】
【図1】図1は、種々の細胞系におけるストレス誘導性EGFRおよびMAPKリン酸化の経時変化を表す図である。(A)浸透ストレスと酸化ストレスに応答するEGFRおよびMAPKリン酸化。(B)ヒト膀胱癌細胞系におけるストレス誘導性EGFRリン酸化。
【図2】図2は、ストレス誘導性EGFRリン酸化を媒介するp38を表す図である。(A)EGFRキナーゼ機能に依存するp38リン酸化の経時変化。(B)Cos-7細胞ではストレス誘導性EGFR活性化はp38に依存し、ERK活性には依存しない。(C)TCC-Sup癌細胞ではストレス誘導性EGFR活性化はp38活性に依存する。
【図3】図3は、リガンド依存性である浸透ストレスと酸化ストレスに応答するEGFR活性化を表す図である。(A)EGFRリン酸化におけるメタロプロテアーゼとHB-EGF阻害の効果。(B)ストレス剤に応答するShcリン酸化の分析。
【図4】図4は、ストレス剤に応答するproHB-EGF放出の分析を表す図である。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。(B)条件培地の免疫ブロット分析。
【図5】図5は、ストレス誘導性EGFRリン酸化における種々のADAMタンパク質に対するsiRNAsの効果を表す図である。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。(B)Cos-7細胞。(C)NCl-H292細胞。
【図6】図6は、ストレス剤に応答するMAPK活性化およびEGFR、メタロプロテアーゼおよびHB-EGF機能の遮断を表す図である。
【図7】図7は、TCC-Sup癌細胞のドキソルビシン誘導性細胞死を表す図である。(A)ドキソルビシン処理に応答するp38活性化。(B)ドキソルビシンに応答して細胞死を促進するHB-EGF機能の遮断。
【発明を実施するための形態】
【0023】
レセプターチロシンキナーゼリガンドは、好ましくは細胞外レセプターチロシンキナーゼドメインに結合する分子である。適切なレセプターチロシンキナーゼリガンドの例は、HB−EGF、EGF、アンフィレグリン、ベータセルリン、エピレグリン、TGF−α、ニューレグリンまたはヘレグリンである。さらに好ましくは、レセプターチロシンキナーゼリガンドはHB-EGFである。
【0024】
レセプターチロシンキナーゼリガンドの活性の阻害は、メタロプロテアーゼおよび/またはレセプターチロシンキナーゼの活性化によるリガンドの前駆体、特にリガンドの膜結合前駆体の切断の阻害に関するのが好ましい(例えば、リガンドによるEGFR)。本発明のメタロプロテアーゼ阻害剤は、好ましくはリガンド前駆体の切断およびその放出を阻害できる。適切なメタロプロテアーゼの例は、ADAM9、10および17、特に細胞ストレス応答の重要な媒介物であるADAM 17である。二者択一的に、本発明の阻害剤はレセプターチロシンキナーゼリガンド、特にEGFRチロシンリン酸化、下流のマイトジェンシグナリング事象の生物活性を阻害することもできる。例えば、ERK1/2および/またはJNKのようなマイトジェン活性化タンパク質キナーゼ(MAPKs)の活性化、細胞増殖および/または移動を阻害することができる。
【0025】
レセプターチロシンキナーゼリガンドの阻害剤は、疾患、特に過剰増殖性疾患の予防または治療に使用することもできる。好ましくは、疾患はレセプターチロシンキナーゼのストレス誘導性活性化により引き起こされるか、またはこれに関連する。このストレスは、好ましくは酸化ストレスおよび/または浸透ストレスである。より好ましくは、このストレスはp38に媒介されるストレスである。このようなタイプの疾患の存在は、例えば、mRNAレベル(cDNA 配列分析、SAGE、ノーザンブロットなど)で、および/またはタンパク質レベル(ウエスタンブロット分析、免疫蛍光顕微鏡検査法、in situハイブリダイゼーション技法など)でp38発現を測定することにより決定できる。このようなタイプの疾患の存在は、p38をコード化するゲノムおよび/またはmRNA分子中の活性突然変異の発生を試験することにより決定することもできる。さらに血清および/または疾患に影響された組織内で、p38アゴニストの高いレベルを決定することもできる。このタイプの疾患がレセプターチロシンキナーゼ発現強化と関連する必要がないことを示すべきである。
【0026】
例えば、この疾患は癌、例えば、乳癌、胃癌、前立腺癌、膀胱癌、卵巣癌、肺癌、肝臓癌、腎臓癌または膵臓癌、グリオーム、黒色腫、白血病などのような過剰増殖性疾患または過剰増殖性皮膚病、例えば、乾癬または炎症性疾患のような他の疾患であることができる。
【0027】
レセプターチロシンキナーゼリガンドの活性(好ましくは直接に、またはメタロプロテアーゼ阻害を介して)は、核酸レベルで、例えば、遺伝子レベルまたは転写レベルで阻害することができる。
【0028】
遺伝子レベルでの阻害は、遺伝子破壊による部分的または完全な遺伝子不活性化を含んでいてもよい。他方で、阻害は転写レベルで例えばアンチセンス分子、例えば、DNA分子、RNA分子または核酸類似物、リボザイム、例えば、RNA干渉(RNAi)が可能なRNA分子または核酸類似物または低分子RNA、例えば、メタロプロテアーゼおよび/またはリガンドmRNAと反対と向いたRNA分子または核酸類似物の利用によりで起こってもよい。
【0029】
さらに、活性はタンパク質レベルで例えばメタロプロテアーゼおよび/またはリガンド活性の特異的阻害を結果として生じる化合物の利用により阻害されてもよい。タンパク質レベルの阻害は、例えば、ADAM9、10または17のようなメタロプロテアーゼまたはHB-EGFまたはPreHB-EGFのようなリガンドもしくはリガンド前駆体と反対を向いた抗体または抗体断片の利用を含んでいてもよい。抗体はポリクロナール抗体またはモノクロナール抗体、組換え抗体、例えば、少なくとも1つの抗原結合部位を有する一本鎖抗体またはこのような抗体の断片、例えば、Fab、Fab’またはF(ab’)2断片のようなタンパク質分解性抗体断片、またはscFv断片のような組換え抗体断片であってよい。治療目的、特にヒトの治療のために、キメラ抗体、ヒト化抗体またはヒト抗体の利用がとりわけ有利である。
【0030】
さらに、メタロプロテアーゼおよび/またはリガンドのタンパク様または低分子量の阻害剤を使用してもよい。このような阻害剤の例は、CRM197、バチマスタット、マリマスタット、ヘパリンまたはリガンドに対する遮断抗体である。さらなる抗体は以下に記載されているようなスクリーニング法により同定してもよい。
【0031】
治療法に関して、付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有する製剤学的組成物の形で薬剤を投与することができる。
【0032】
本発明で使用するために適切な製剤学的組成物には、活性成分が意図する目的を達成するために効果的な量で含有されている組成物が含まれる。治療学的に有効な投与量とは、患者において症状の回復または生存の延長を結果として生じる化合物の量を意味する。このような化合物の毒性および治療的有効度は、細胞培養または実験動物において標準的な製剤学的方法、例えば、LD50(母集団の50%致死量)とED50(母集団の50%治療的有効量)を測定することにより決定することができる。本発明の方法で使用する化合物に関して、治療的有効量は細胞培養アッセイから初めに概算することができる。例えば、薬用量を動物モデルにおいて調製して、細胞培養で決定したようなIC50を含めた濃度幅の循環を達成することができる(すなわち、成長因子レセプター活性の最大の半分の阻害を達成する試験化合物の濃度)。このような情報を使用して、ヒトで有効な薬用量を正確に決定することができる。毒性と治療効果の間の薬用量比は、治療係数であり、かつLD50とED50の比として表現することができる。高い治療係数を示す化合物が好ましい。正確な調製物、投与経路および投与量は患者の状態を考慮して各々の内科医により選択することができる(例えば、Fingl et al., 1975, "The Pharmacological Basis of Therapeutics", 第1章、1頁参照)。投与量と間隔をそれぞれ調節して、レセプター調節効果、または最小有効濃度(MEC)を維持するために十分である活性部分の血しょうレベルを提供してもよい。MECは、各化合物に関して変化するが、in vitroでのデータから概算することもできる。例えば、この濃度は、本明細書中で記載されたアッセイで使用するレセプターの50〜90%阻害を達成するために必要である。化合物は、時間の10〜90%、好ましくは30〜90%、最も好ましくは50〜90%MECを上回って血しょうレベルを保つレジュメを使用して投与すべきである。MECを達成するために必要な投与量は、各々の特徴と投与経路に応じる。局所投与または選択的摂取の場合には、薬剤の効果的な局所濃度は血しょう濃度に関係ない。
【0033】
投与される組成物の正確な量は、自明ながら治療される被験者、被験者の重量、病気の度合い、投与方法および処方する内科医の判断による。抗体または治療的に活性な核酸分子、および他の化合物については、例えば1日あたり0.001〜100mg/kg、特に0.01〜10mg/kgの日用量が適切である。
【0034】
適切な投与経路には、例えば、経口、直腸、粘膜または腸管投与;筋肉内、皮下、髄内注射、ならびに髄腔内、脳室内、静脈内、腹腔内、鼻腔内または眼球内注射を含む非経口輸送が含まれる。
【0035】
二者択一的に、全身的な方法よりも局所で化合物を投与してもよい。例えば、多くの場合デポー剤または持続放出性調製物の形で注射することにより充実性腫瘍に化合物を直接投与することができる。
【0036】
さらに、薬剤をターゲットをしぼったドラックデリバリーシステムの形で、例えば、腫瘍に特異的な抗体で被覆したリポソームの形で投与することもできる。リポソームは、ターゲットをしぼり、腫瘍により選択的に摂取される。
【0037】
上記のように、本発明は特に治療抵抗性の過剰増殖性疾患、好ましくは治療抵抗性タイプの癌、例えば、照射および/または薬剤耐性タイプの癌の治療または予防に適切である。
【0038】
照射および/または細胞増殖抑制剤および/または細胞毒剤を用いる癌の治療は、ストレスキナーゼp38を活性化することが示された。意外にも、HB-EGFのようなレセプターチロシンキナーゼリガンドの阻害は、照射および/または化学療法の治療活性、特にそのアポトーシス誘導活性を強く促進した。従って、ダイレクトなレセプターチロシンキナーゼリガンド阻害剤、またはリガンド前駆体シェディングを防ぐ阻害剤と、更なる治療法および/または薬剤との共同適用は、結果として前記の方法および/または薬剤の適用に対して疾患の感受性を実質的に増大させ、ひいては前記の更なる治療法および/または薬剤の有効性を増強する。上記のようなリガンド阻害剤の投与は、少なくとも部分的に照射療法および/または細胞増殖抑制剤および/または細胞毒性薬剤に耐性である疾患、特に少なくとも部分的にアポトーシス誘導法と薬剤に対して耐性である疾患の治療または予防に特に適切である。
【0039】
本発明の好ましい実施態様では、レセプターチロシンキナーゼリガンド阻害剤を、照射療法、特にγ線照射療法と共同適用する。さらに好ましい実施態様では、レセプターチロシンキナーゼリガンド阻害剤を更なる抗ガン剤、特にアポトーシス誘導剤と共同適用する。適切な抗ガン剤の好ましい例は、ドキソルビシン、タキサン、シス/トランス−プラチンまたはこれらの誘導体、5−フルオロウラシル、マイトマイシンD、パクリタキセル、エトポシド、シクロホスホアミド、ドセタキセルまたは他のアポトーシス誘導剤または抗体のようなタンパク質である。
【0040】
リガンド阻害剤および更なる方法および/または薬剤の共同適用は、同時におよび/または連続的に行うことができる。リガンンド阻害剤の適用は、他の治療の適用に対して治療すべき疾患の感受性の増大を導く。特に、照射および/または化学療法に対する腫瘍耐性が減少する。
【0041】
従って、本発明のもう1つの点は、活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキットである。
【0042】
好ましくは、該組成物またはキットは付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有してよい。
【0043】
さらに、ストレス誘導性レセプターチロシンキナーゼ活性、例えば、p38−誘導活性のモジュレーターの同定を可能にする方法が提供する。前記方法は、試験化合物がレセプターチロシンキナーゼのリガンドの活性を阻害できるかを決定することを含む。この方法は、ストレス誘導性シグナルトランスダクションを調節できる新規化合物または化合物のクラスを同定するためのスクリーニング法、例えば、ハイスループットスクリーニング法として適切である。さらに、この方法は、化合物の製剤学的有効性および/または副作用を特徴付けるための確証法として適切である。この方法は、単離タンパク質、細胞抽出物、組換え細胞または人間以外のトランスジェニック動物の使用を含んでいてもよい。組換え細胞または人間以外のトランスジェニック動物は、相応の野生型細胞または動物と比べて変化したメタロプロテアーゼおよび/またはリガンド発現を示すのが好ましい。
【0044】
さらに、本発明を以下の図と実施例により説明する。
【0045】
図の説明文
図1:種々の細胞系におけるストレス誘導性EGFRおよびMAPKリン酸化の経時変化。(A)浸透ストレスと酸化ストレスに応答するEGFRおよびMAPKリン酸化。Cos−7細胞は、ソルビトール(0.3M)と過酸化水素(200μM)で記載された時間の間処理した。抗−EGFR抗体タンパク質を用いる細胞抽出物の免疫沈降(IP)に続いて、抗−ホスホチロシン抗体で免疫ブロットし(IB)、抗−EGFR抗体でリプローブした。リン酸化したMAPKsは、全体の溶解産物を抗−ホスホERK抗体、抗−ホスホ−JNKおよび抗−ホスホ−p38抗体で免疫ブロットすることにより検出された。同じフィルターを抗−p38抗体でリプローブした。(B)ヒト膀胱癌細胞系におけるストレス誘導性EGFRリン酸化。TCC−Sup細胞は(A)で示したように処理した。
【0046】
図2:ストレス誘導性EGFRリン酸化を媒介するp38。(A)EGFRキナーゼ機能に依存するp38リン酸化の経時変化。Cos−7細胞をAG1478(250nM)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールまたは200μM過酸化水素で記載された時間の間刺激した。細胞抽出物を抗−ホスホ−p38抗体で免疫ブロットし、同じフィルターをポリクロナール抗−p38抗体でリプローブした。(B)Cos-7細胞ではストレス誘導性EGFR活性化はp38に依存し、ERK活性には依存しない。Cos-7細胞をPD98059(50μM)、SB202190(10μM)または当量の空ベヒクル(DMSO)で30分間前処理し、0.3Mソルビトールと200μM過酸化水素で10分間刺激し、陽性コントロールとしてGPCRアゴニストのLPA(10μM)またはEGF(2ng/mL)で3分間刺激した。細胞抽出物をEGFRチロシンリン酸化内容物に関してアッセイした。(C)TCC-Sup癌細胞ではストレス誘導性EGFR活性化はp38活性に依存する。TCC-Sup細胞を(A)で記載したように前処理し、0.3Mソルビトールと200μM過酸化水素で10分間刺激し、陽性コントロールとしてEGF(2ng/mL)で3分間刺激した。細胞抽出物を溶解後、EGFRチロシンリン酸化内容物に関してアッセイした。
【0047】
図3:リガンド依存性である浸透ストレスと酸化ストレスに応答するEGFR活性化。(A)EGFRリン酸化におけるメタロプロテアーゼとHB-EGF阻害の効果。Cos-7、NCl-H292およびTCC-Sup細胞を24時間血清飢餓状態にし、BB94(10μM)、ジフテリア毒素変異体Crm197(10μg/mL)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトール、200μM過酸化水素、10μM LPAまたは2ng/mL EGFで10分間刺激した。抗−EGFR抗体タンパク質を用いる細胞抽出物の免疫沈降に続いて、抗−ホスホチロシン抗体で免疫ブロットし、抗−EGFR抗体でリプローブした。(B)ストレス剤に応答するShcリン酸化の分析。Cos-7細胞は(A)で記載したように処理した。ポリクロナール抗−Shc抗体タンパク質を用いる細胞抽出物からShcを免疫沈降した後に、抗−ホスホチロシン抗体で免疫ブロットし、かつ抗−Shc抗体でリプローブした。
【0048】
図4:ストレス剤に応答するproHB-EGF放出の分析。(A)proHB-EGFプロセッシングのフローサイトメトリー分析。Cos-7細胞をBB94(10μM)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールと200μM過酸化水素で30分間刺激した。細胞を回収し、表面のproHB-EGFを染色し、フローサイトメトリーにより分析した。(B)条件培地の免疫ブロット分析。Cos-7細胞をproHB-EGF cDNAで一時的にトランスフェクションした。24時間血清飢餓状態にした後に、細胞をソルビトール(0.3M)または過酸化水素(200μM)で20分間刺激し、上澄培地内のタンパク質をトリクロロ酸(TCA)を用いて沈殿させた。沈殿したタンパク質をSchaegger-Jagowのプロトコールに従ってトリシン−ドデシル硫酸ナトリウムゲル電気泳動にかけ、続いて抗HB-EGF抗体を用いて免疫ブロット分析した。TPA刺激は陽性コントロールとして含めた。
【0049】
図5:ストレス誘導性EGFRリン酸化における種々のADAMタンパク質に対するsiRNAsの効果。(A)RNA干渉(RNAi)によるADAMメタロプロテアーゼ発現の遮断。NCl-H292細胞をADAM9、ADAM10またはADAM15に対するsiRNAでトランスフェクションし、2日間培養し、示したようにRT-PCRにより遺伝子発現を分析した。(B)Cos-7細胞をADAM9、−10、−12、−15および−17に対するsiRNAsでトランスフェクションし、24時間血清飢餓状態にさせ、0.3Mソルビトールまたは200μM過酸化水素で10分間刺激し、EGFRチロシンリン酸化内容物をアッセイした。(C)NCl-H292細胞を(B)で記載したように処理した。
【0050】
図6:ストレス剤に応答するMAPK活性化およびEGFR、メタロプロテアーゼおよびHB-EGF機能の遮断。(A)pcDNA3-HA-ERK2、TCC-SupおよびNCl-H292細胞で一時的にトランスフェクションしたCos-7細胞をAG1478(250nM)、BB94(10μM)、Crm197(10μg/mL)または当量の空ベヒクル(DMSO)で20分間前処理し、0.3Mソルビトールまたは200μM過酸化水素で30分間刺激した。全体の溶解産物が溶解した後に、抗−ホスホ−ERK抗体で免疫ブロットし、続いて同じ膜をポリクロナール抗−ERK抗体でリプローブした。FUJI LAS1000イメージングシステムを使用して3つの独立の試験からERKリン酸化を定量分析した(平均±標準偏差)。(B)Cos-7とNCl-H292細胞を(A)で記載したように処理した。溶解後、抗JNK抗体を用いてJNKを免疫沈降させ、基質としてGST-c-JUN融合タンパク質を用いてJNK活性をアッセイした。リン酸化したGST-c-JUNをオートラジオグラフィーにより視覚化し、かつポリクロナールJNK抗体を用いて、JNKを平行に免疫ブロットした。FUJI LAS1000イメージングシステムを使用して3つの独立の試験からGST-c-JUNリン酸化を定量分析した(平均±標準偏差)。TCC-Sup細胞を(A)で記載したように処理した。細胞溶解後、抗−ホスホ−JNK抗体で細胞抽出物を免疫ブロッティングすることによりJNKリン酸化をアッセイし、同じフィルターを抗−JNK抗体でリプローブした。(C)p38リン酸化はEGFR、メタロプロテアーゼおよびHB-EGF機能とは独立である。Cos-7細胞を(A)で記載したように処理した。抗−ホスホ−p38抗体で細胞抽出物を免疫ブロッティングすることによりp38リン酸化をアッセイし、かつ同じフィルターを抗−p38抗体でリプローブした。
【0051】
図7:TCC-Sup癌細胞のドキソルビシン誘導性細胞死。(A)ドキソルビシン処理に応答するp38活性化。TCC-Sup細胞を播種し、記載した時間の間ドキソルビシンで処理した。細胞溶解後、抗−ホスホ−p38抗体で細胞抽出物を免疫ブロッティングしてp38活性化を評価し、かつ同じフィルターを抗−p38抗体でリプローブした。(B)ドキソルビシンに応答して細胞死を促進するHB-EGF機能の遮断。細胞を10μMドキソルビシンと10μg/mL Crm197で記載されているように24時間ごとに72時間処理した。アッセイ緩衝液中の細胞を回収した後に、 Plで核を染め、フローサイトメトリー分析により分析した。
【実施例】
【0052】
実施例
1.材料と方法
1.1細胞培養、プラスミドおよびトランスフェクション
提供者の指示書に従い、全ての細胞系(American Type Culture Collection, Manassas, VA)を決まった手順で成長させた。メーカーのプロトコールに従い、リポフェクタミン(Invitrogen)を用いてCos-7細胞のトランスフェクションを行った。簡単には、一時的なトランスフェクションのために6cm皿中の細胞を、1皿あたり20μLリポフェクタミンと全体のプラスミドDNA2μg含有の無血清培地2mL中で4時間インキュベートした。次に、トランスフェクション混合物を当量の20%胎児ウシ血清で補足し、20時間後に細胞を洗浄し、刺激の前に無血清培地中でさらに24時間培養した。阻害剤AG1478(Alexis Biochemicals)、バチマスタット(BB94, British Biotech, Oxford, UK)、Crm197(Quadratech Ltd., UK)、SB202190(Calbiochem)およびPD98059(Alexis Biochemicals)を各刺激の前に血清飢餓状態の細胞に加えた。
【0053】
プロ−およびメタロプロテアーゼドメインが欠けたADAM10、12、15および17から成っているドミナントネガティブプロテアーゼは、予め記載してある(Gschwind et al., 2003)。プラスミドpcDNA3-HA-ERK2(Daub et al., 1997)とpcDNA3-proHB-EGF-VSV(Prenzel et al., 1999)をこの試験で使用した。
【0054】
1.2タンパク質分析
細胞を溶解し、かつ上記のようにタンパク質を免疫沈降させた(Daub et al., 1997)。溶解前に、80%コンフルエンスまで成長させた細胞を図中の説明文に示されているように阻害剤とアゴニストで処理し、次に50μM HEPES、pH7.5、150mM NaCl、1%トリトンX-100、1mM EDTA、10%グリセロール、10mM ピロリン酸ナトリウム、2mM オルトバナジウム酸ナトリウム、10mM フッ化ナトリウム、1mM フェニルメチルスルホニルフルオリド、10μg/mLアプロチニン含有の緩衝液中、氷上で10分間溶解した。溶解産物を13000rpmにて4℃で10分間の遠心分離により予めきれいにした。予めきれいにした分解遺産物を各抗体およびプロテインA−セファローズ20μLを用いて4℃で4時間免疫沈降した。沈殿物をHNTG緩衝液0.5mLで3回洗浄し、2×SDS試料緩衝液中に懸濁させ、3分間沸騰させ、ゲル電気泳動にかけた。SDS−ポリアクリルアミドゲル電気泳動に続いて、タンパク質をニトロセルロース膜に移した。従来の方法に従って、ウェスタンブロットを行った。ヒトEGFR(108.1)とSHCに対する抗体は前に特徴付けてある(Prenzel et al., 1999)。4G10モノクロナール抗体を用いてホスホチロシンを検出した(UBl, Lake Placid, NY)。ポリクロナール抗−ホスホ−p44/p42(Thr202/Tyr204)MAPK抗体、抗−ホスホ−JNK(Thr183/Tyr185)および抗−ホスホ−p38(Thr180/Tyr182)抗体はNew England Biolabs(Beverly, MA)から購入した。ポリクロナール抗−ERK2、抗−JNK1および抗−p38抗体はSanta Cruz Biotechnology(Santa Cruz, CA)から購入した。
【0055】
1.3 JNK活性のアッセイ
上記のようにJNK活性をアッセイした(Sudo and Karin, 2000)。培養した細胞を、20mM トリス(pH 7.6)、0.5%Nonidet P-40、250mM NaCl、3mM EDTA、1mM ジチオトレイトール、0.5mM フェニルメチルスルホニルフルオリド、20mM β−グリセロホスフェート、1mM オルトバナジウム酸ナトリウムおよび1μ/mLロイペプチン含有の溶解緩衝液中で溶解した。ポリクロナール抗−JNK抗体を用いて6ウェル皿から得られた溶解産物よりJNKを免疫沈降した。免疫沈降物を溶解緩衝液で2回洗浄し、かつキナーゼアッセイ緩衝液(25mM HEPES(pH 7.5)、20mM β−グリセロホスフェート、20mM PNPP、20mM MgCl2、2mMジチオトレイトールおよび0.1mM オルトバナジウム酸ナトリウム)で2回洗浄した。1μgGST-c-Jun(aa1-79)、20μMの冷たいATPおよび[y-32P]ATP5μCiを補足したキナーゼ緩衝液30μL中30℃で30分間キナーゼ反応を行った。Laemmli緩衝液30μLを添加することにより反応を中止し、12.5%ゲル上でゲル電気泳動にかけた。標識したGST-c-Junをホスホイメージャー(Fuji)で定量した。
【0056】
1.4 フローサイトメトリー分析
FACS分析を上記のように行った(Prenzel et al., 1999)。簡単には、細胞を播種し、20時間成長させ、24時間血清飢餓状態にした。上記のように阻害剤で細胞を処理し、かつ刺激した。回収した後、proHB-EGFに対するエクトドメインに特異的な抗体で細胞を45分間染色した。PBSで洗浄後、細胞をFITC共役二次抗体で15分間インキュベートし、PBSで再び洗浄した。細胞をBecton Dickinson FACS caliburフローサイトメーターで分析した。
【0057】
1.5 HB-EGFのTCA沈殿
pcDNA3-proHB-EGF-VSVで一時的にトランスフェクションしたCos-7を24時間血清飢餓状態にした。刺激の前に細胞を洗浄し、BB94(10μM)でプレインキュベートし、かつ記載したように刺激した。刺激後、上澄液を回収し、デソキシコリン酸ナトリウム(100μg/mL)を添加し、氷上での10分間のインキュベーションに続いて、溶液にトリクロロ酢酸(TCA)を加えて10%TCAの最終濃度にした。氷上で30分間インキュベーションした後、試料を遠心分離し、上澄液を捨て、かつ沈殿物をSchaegger-Jargow試料緩衝液中で再懸濁させた。Tris-HCl(pH8.8)を用いてTCAを中和し、トリシンSDSゲル電気泳動プロトコールを用いて試料を分離した(Schaegger and von Jargow, 1987)。
【0058】
1.6 RNA干渉とRT-PCR分析
内因性遺伝子をターゲティングするための21-ヌクレオチドsiRNA二本鎖(Dharmacon Research, Lafayette, CO, USA)のトランスフェクションを、NCl-H292細胞用のオリゴフェクタミン(Invitrogen)と6ウェルプレート当たり4.2μgのsiRNA二本鎖を用いて前記のように行った(Elbashir et al., 2001)。メーカーのプロトコールに従ってリポフェクタミン2000(Invitrogen)を用いてCos-7細胞をトランスフェクションした。簡単には、6cm皿あたり8.4μgのsiRNA二本鎖を無血清培地1mL中のリポフェクタミン2000 10μLと一緒に20分間インキュベートした。トランスフェクション混合物を血清含有の細胞培養培地に加え、6時間後に細胞を洗浄し、かつ血清含有の培地中で一晩インキュベートした。NCl-H292とCos-7細胞を血清餓死にし、トランスフェクション後2日目にアッセイした。有利な遺伝子の種々の領域をターゲティングするsiRNA二本鎖の混合物を使用することによりターゲット遺伝子のサイレンシングにおいて最も高い効率が得られた。使用したsiRNAの配列は以下のものである。
【0059】
【表1】
【0060】
ADAM12とADAM17に対するsiRNA二本鎖は予め記載してある(Gschwind et al., 2003)。
【0061】
ターゲット遺伝子の特異的サイレンシングをRT-PCR分析により確認した。RNeasy Mini Kit(Qiagen, Hilden, Germany)を用いて単離されたRNAをAMV逆転写酵素(Roche, Mannheim, Germany)を用いて逆転写した。PuReTaq Ready-To-Go PCR Beads(Amersham Biosciences, Piscataway, NJ)をPCR増幅に使用した。以下がプライマー(Sigma Ark, Steinheim, Germany)である:
【0062】
【表2】
【0063】
PCR製品を2.5%アガロースゲル上で電気泳動にかけ、DNAを臭化エチジウム染色により視覚化した。
【0064】
1.7 アポトーシスアッセイ
TCC-膀胱癌細胞を播種し、20時間成長させ、かつ記載したように10μM ドキソルビシンとCrm197で72時間処理した。細胞をヨウ化プロピジウム(PI)含有のアッセイ緩衝液中で回収し、4℃で3時間インキュベートした。核PI染色をBecton Dickinson FACS caliburフローサイトメターで分析した。
【0065】
2. 結果
2.1 Cos-7とヒト癌細胞系におけるEGFRとMAPK活性化の特異な動力学
浸透ストレスと酸化ストレスは、幅広い種々の細胞系においてEGFRとMAPKsのリン酸化を導いた(Carpenter, 1999; de Nadal et al., 2002; King et al., 1989)。潜在的なメカニズムを調べるために、免疫ブロット分析によりCos-7とTCC-Sup腎臓癌細胞系において経時変化実験を行った。図1に示されるように(上のパネル)、ストレス剤であるソルビトール(0.3M)と過酸化水素(200μM)に応答するEGFRのチロシンリン酸化は両方の細胞系で5〜10分以内に生じたのに対して、MAPKsであるERK1/2およびJNKはそれぞれ10分後と15分後にリン酸化された(図1A/B、下のパネル)。ERK1/2の弱い活性化は既に1分後に検出されたのに対して、リン酸化は10分後に著しく増大した。これとは対照的に、p38のリン酸化はストレス剤に即座に応答した。経時変化実験の結果は、EGFRがストレス刺激に応答してERK1/2とJNK活性化に関わっていることを示唆している。これとは対照的に、p38の活性化がEGFR刺激に先行した。興味深いことに、レセプターの活性化は、リガンド非依存性の活性化メカニズムを可能にするだけではなく、リガンド依存性の活性化メカニズムを可能にする時間の尺度で検出可能であった。
【0066】
2.2 p38は、浸透ストレスと酸化ストレスによるEGFR活性化を制御する
p38活性化がEGFRチロシンリン酸化に先行したという発見は(図1)、p38がCos-7とヒト癌細胞系においてEGFR刺激の上流レギュレーターとして働いているのかどうかという疑問を生じた。
【0067】
選択的EGFR―キナーゼ阻害剤AG1478とのCos-7細胞のプレインキュベーションは、ストレス剤に応答するp38リン酸化に影響を与えないので(図2A)、p38活性化がEGFR活性とは独立に起きたことを証明している。p38がEGFRの上流に位置しているのかどうかという疑問を処理するために、p38に特異的な阻害剤であるSB202190を使用してストレス誘導性EGFR活性化におけるp38活性の遮断効果を調べた。図2Bに示されているように、SB202190とのCos-7細胞のプレインキュベーションは、ストレス誘導性EGFR活性化を完全に排除し、リゾホスファチド酸(LPA)とEGF誘導性レセプターのリン酸化に影響を与えなかった。これとは異なり、MEK1/2阻害剤であるPD98059は、EGFRリン酸化に影響を与えなかった。似た結果は、腎臓癌細胞系TCC-Supで得られた(図2C)。まとめると、これらの結果はp38がCos-7とTCC-Sup腎臓癌細胞においてストレス誘導性EGFR活性化の上流メディエーターであることを証明している。
【0068】
2.3 メタロプロテアーゼ活性とHB-EGF機能に依存するストレス誘導性EGFRリン酸化
近年の研究はEGFRシグナリング、特に以前はリガンド非依存性であると考えられていたGPCRによるEGFRシグナルトランスアクチベーションのようなシグナルトランスダクション事象におけるEGF様リガンドのタンパク分解の重要性をはっきりと示している(Prenzel et al., 1999)。これらの発見に基づき、ストレス刺激によるEGFR活性化がリガンド依存性メカニズムに関わるかどうかという疑問に取り組んだ。従って、EGF様リガンドプロセシングとそれに続くEGFRトランスアクチベーションを阻害することが示されたメタロプロテアーゼ阻害剤であるバチマスタット(BB94)と一緒に細胞をプレインキュベートした(Prenzel et al., 1999)。EGFRチロシンリン酸化は、免疫ブロット分析を用いるストレス剤で刺激した後にモニターした。図3Aに示されるように、Cos-7およびTCC-Sup腎臓癌細胞においてBB94は殆ど完全にソルビトールまたは過酸化水素−誘導性EGFRリン酸化を遮断した(上と真ん中のパネル)。肺癌細胞系NCl-H292では、BB94はEGFR活性化を50%まで阻害したので、このことは二者択一的な平行の活性化メカニズムを示唆している(図3A、下のパネル)。
【0069】
次に、ストレス活性化EGFRチロシンリン酸化においてHB-EGF機能を特に遮断するジフテリア毒素変異体Crm197の効果を調べた。実に、Crm197はBB94と同じ程度にEGFRリン酸化を阻害したので、これらの3つの細胞系ではHB-EGFがストレス誘導性EGFR活性化に決定的に関わっていることを示唆している。陽性コントロールとしてのLPAによるレセプター活性化は、これらの阻害剤により完全に妨げられたのに対して、EGFを用いた直接の刺激は影響されなかった。
【0070】
さらに、このリガンド依存性がEGFRの下流のシグナリングパートナーに従うことができるかどうかは特別に興味のあることであった。Shcアダプタータンパク質は、レセプターがRas/Raf/ERK-MAPKカスケードの活性化とリンクするEGFRの顕著なシグナリングアダプターである。実際に、図3B(下のパネル)で示されているデータは、ストレス刺激に応答するShcタンパク質のリン酸化内容物がEGFR自体の内容物と似ていることを証明している。従って、ストレス刺激によるShcのリン酸化はリガンド依存性EGFRリン酸化メカニズムに極めて依存している。
【0071】
2.4 Cos-7細胞において浸透ストレスと酸化ストレスに応答して誘導されるproHB-EGFのエクトドメインシェディング
このリガンド依存性EGFR刺激メカニズムにおけるHB-EGFの役割を更に実証するために、リガンド自体のレベルで高浸透圧と過酸化水素処理に応答するproHB-EGFのシェディングを調べた。従って、ソルビトールまたは過酸化水素で刺激する前と後にCos-7細胞の細胞表面に存在するproHB-EGFの量をフローサイトメトメトリー分析により分析した。図4Aに示されているように、両方の刺激は細胞表面上でHB-EGF前駆体の著しい減少を導いた。さらに、図3Aに示されている結果と一致して、メタロプロテアーゼ阻害剤BB94を用いる予備処理は、proHB-EGFプロセシングを排除した。HB-EGF前駆体の減少の他に、proHB-EGFを異所性発現してCos-7細胞の細胞表面から細胞培養培地に放出された可溶性の成熟型HB-EGFの量を測定した。図4Bに示されるように、高浸透圧と過酸化水素を用いる両方の処理は、免疫ブロット分析により測定されたように調整培地中でHB-EGFの増大を誘導した。さらにまた、HB-EGF放出がメタロプロテアーゼ阻害剤BB94でのプレインキュベーションにより遮断され、メタロプロテアーゼ活性の関わりを確証している。
【0072】
2.5 浸透ストレスと酸化ストレスによりEGFR活性化を媒介するADAMファミリーのメタロプロテアーゼ
メタロプロテアーゼ依存性メカニズムがストレス誘導性EGFRとShc活性化に著しく寄与するという発見は、どのメタロプロテアーゼが関係しているのかという疑問を生じた。RNA干渉法を使用して、すでにEGF様リガンドの切断に関わっていたADAMプロテアーゼであるADAM9、10、12、15および17の内因発現を阻害した。図5Aは、ADAM9、10および15に対するsiRNAによるターゲット遺伝子発現の効果的かつ特異的ノックダウンをRT-PCRで証明している。ADAM12と17に対するsiRNAsは既に記載してある(Gschwind et al., 2003)。各プロテアーゼに対するsiRNAsを用いる一時的なトランスフェクション、およびそれに引き続くソルビトールまたは過酸化水素で刺激した後のEGFRの免疫ブロット分析は、Cos-7細胞では両方のストレス刺激後にADAM10とADAM17がEGFR活性化に関わっていることを明らかにした(図5B)。しかし、ヒトの癌細胞におけるこれらのプロセス制御に興味があったので、ヒト肺ガン細胞系NCl-H292におけるADAMファミリーメンバーの関係をさらに調べた。すわなち、図5Cで証明されているように、Cos-7細胞で得られた結果と類似して、ADAM17もストレス誘導性EGFR活性化に関わっている。Cos-7細胞とは対照的に、ADAM9および過酸化水素処理の場合でも、ADAM12は、NCl-H292細胞のストレス応答に関わっている。全体として、細胞タイプにより、メタロプロテアーゼの種々のADAMファミリーのメンバー、特にADAM17は、ストレス剤に応答するHB-EGF依存性EGFR活性化において中心的な役割を担っている。
【0073】
2.6 HB-EGF依存性EGFR活性化に媒介される高浸透圧ストレスと酸化ストレスに応答するMAPKsERK1/2とJNKの活性化
MAPKs であるERK1/2とJNKが高浸透圧と活性酸素種により活性化されるので(de Nadal et al., 2002; Kyriakis and Avruch, 2001)、リガンド依存性EGFRリン酸化がストレス刺激によるMAPK活性化の誘導に寄与するかどうかを調べた。ゆえに、選択的EGFR tyrphostin AG1478を使用して、EGFRキナーゼ活性におけるストレス誘導性MAPK活性化の全体的な依存性を調べた。さらに、この効果をBB94とCrm197によるリガンド依存性EGFR活性化プロセスの阻害と比較した。図6A(上のパネル)に示されているように、ERK1/2のソルビトールと過酸化水素誘導性活性化は、AG1478により遮断された。さらに、BB94とCrm197はERK1/2リン酸化の遮断において殆ど有効であった。これらのデータは、浸透ストレスと酸化ストレスに応答するCos-7細胞におけるERK1/2活性化がリガンド依存性メカニズムに大いに帰因しているEGFR活性化に殆ど完全に依存していることを示唆している。同じ実験構成を使用して、このコンセプトをヒト癌細胞系におけるストレスシグナリングにまで延ばすことができるかどうかという問題に的をしぼった。しかし、TCC-SupおよびNCl-H292細胞における高浸透圧および酸化ストレス誘導性ERK1/2活性化がAG1478、BB94およびCrm197により実質的に遮断されることが分かった(図6A真ん中と下のパネル)。興味深いことに、NCl-H292細胞において酸化ストレスがリガンド依存性メカニズムにより部分的にだけEGFRリン酸化を誘導するにもかかわらず、この細胞タイプではERK活性化はproHB-EGFプロセシングに大いに依存する。
【0074】
ERK1/2とは別に、JNKの活性化を導くシグナリングメカニズムに興味があった。図6B(上のパネル)には、AG1478、BB94およびCrm197を用いる処理に依存してJNK活性が浸透ストレスと酸化ストレスにより誘導されていることが示されている。Cos-7細胞では、ソルビトール誘導性JNK活性がEGFRに大いに依存しているのに対して、酸化ストレスに応答してJNK活性はHB-EGFによるEGFR活性化に50%依存している。似た結果は、図6B(真ん中のパネル)に示されているように腎臓癌細胞系TCC-Supで得られている。これとは対照的に、肺ガン細胞系NCl-H292では過酸化水素に刺激されたJNK活性化がEGFRに非依存的であるように見えたのに対して、高浸透圧に応答するJNK活性は部分的にリガンド依存性EGFR活性化経路に依存していた。p38がストレス誘導性EGFR活性化の上流メディエーターとして働く(図2)という発見と一致して、p38活性は、EGFRキナーゼ活性にもメタロプロテアーゼまたはHB-EGF機能にも依存していない(図6C)。
【0075】
化学療法を用いる腫瘍細胞の治療は、ストレスキナーゼp38とJNKを活性化して細胞死を導くことを示した。しかし、p38活性化がHB-EGF依存性EGFR活性化とErk1/2活性化を導くので、TCC-Sup腎臓癌細胞のドキソルビシン誘導性アポトーシスにおいてHB-EGF機能を遮断する効果を調べた。図7Aは、ドキソルビシン処理がp38活性化を誘導したことを証明している。図7B/Cには興味深いことに、HB-EGF阻害剤CRM197でのTCC-Sup細胞処理は、ドキソルビシンだけと比較した場合にドキソルビシンに応答してアポトーシスを著しく促進したのに対して、CRM197だけで処理した場合にはマイナーな効果だけを発揮したことが示されている。
【0076】
全体として、これらのデータはヒト癌細胞において、ストレス剤によるリガンド依存性EGFRチロシンリン酸化は、浸透ストレスと酸化ストレスに応答するMAPKsERK1/2とJNKの活性化において重要な役割を担っていることを証明している。さらに、これらの結果は、ここで示されているシグナリングメカニズムが腫瘍細胞により使用され化学療法誘導性細胞死を回避する経路に帰因するとしている。
【0077】
3. 考察
徹底的な調査は、どのように哺乳類細胞がストレス剤に対処しているのかという問題に焦点をおいた。本研究は、ヒト癌細胞において浸透ストレスと酸化ストレスに応答するEGFRとそれに続くMAPK活性化を導く成長因子依存性メカニズムにおいて新たな洞察を提供した。
【0078】
ここでは、浸透ストレスと酸化ストレスの両方によって誘導されるEGFRリン酸化が、Cos-7細胞とヒト癌細胞系においてメタロプロテアーゼ活性とHB-EGFの放出を必要とすることを証明した。この結果は、EGFRレベル(図3)とEGF様リガンドHB-EGFレベル(図4)の両方で得られた。以前の報告は、高浸透圧と酸化ストレスがレセプター凝集と二量化によりEGFR活性化を導くことを証明している(Rosette and Karin, 1996)。二者択一的に、ホスファターゼの不活性化は、それぞれ細胞内キナーゼまたはレセプターの刺激を導き(Blanchetot et al., 2002; Knebel et al., 1996)、これによりEGFRリン酸化を誘導することを提唱した。これに関連して、NCl-H292細胞においてストレス誘導性EGFRリン酸化が部分的にリガンド依存性メカニズムに依存し、かつ部分的にリガンド非依存性メカニズムに依存しているという発見は(図3)、両方のメカニズムがレセプター活性化と協力していると思われるので、以前の研究と一致している。Cos-7とTCC-Sup腎臓癌細胞ではリガンド依存性メカニズムが浸透ストレスと酸化ストレスによるレセプターリン酸化によるレセプターリン酸化の主因であるので、これらのメカニズムのそれぞれの寄与は細胞の状況に依存しているようである(図3)。ストレス刺激がラット胃上皮細胞でHB-EGFとアンフィレグリンの発現を誘導することを証明する以前の研究により我々の結果は更に支持された(Miyazaki et al., 2001)。さらに、Frank et al.(Frank et al., 2003)による近年の報告は、血管平滑筋細胞での過酸化水素誘導性EGFRリン酸化におけるHB-EGFに関する。高浸透圧に応答するproTGF-αのシェディングは、内因性EGFRが欠けたCHO細胞で報告されていた(Fan and Derynck, 1999; Montero et al., 2002)。Chen et al.(Chen et al., 2001a)による報告は、Cos-7細胞における過酸化水素誘導性EGFR活性化がc-Srcによるリガンド非依存性メカニズムによるとしている。これとは対照的に、同じ細胞系では、Src阻害剤であるPP1とPP2を用いる酸化ストレスによってEGFR活性化の特異的阻害が達成されなかった(未発表のデータ)。
【0079】
ますます増える証拠は、亜鉛依存性プロテアーゼのADAMファミリーがEGF様リガンドプロセシングの重要なメディエーターであることを示唆している。これらの報告と一致して、我々の結果はADAMプロテアーゼがストレス刺激により誘導されるproHB-EGFのシェディングに関わっていることを証明している。興味深いことに、EGFRトランスアクティべーションに関連して、1つ1つの個別のADAMプロテアーゼが活性化されるのに対して(Gschwind et al., 2003: Yan et al., 2002)、我々は細胞系に応じて2つ以上のADAMプロテアーゼが活性になることを見つけた(図5)。重要なことは、一般にADAM17がストレス刺激されたシェディング事象に関わっているようだが、ADAM9、ADAM10およびADAM12も関わることができることである(図5)。これらの全ての酵素は以前にEGF様リガンドシェディングに関わっていた(Asakura et al., 2002; Izumi et al., 1998; Lemjabbar and Basbaum, 2002; Yan et al., 2002; Gschwind et al., 2003)。種々のADAMプロテアーゼが活性化されても、proHB-EGFだけが切断された発見は、アンフィレグリンとTGF-αもNCl-H292細胞内で発現されたので意外であった(データの発表なし)。もう一方で、VeroH細胞ではTPA刺激に応答してADAM9がproHB-EGFを切断するのに対して(Izumi et al., 1998)、同じ細胞系ではLPA刺激後のproHB-EGFプロセシングがADAM9とは独立であることを証明する以前の報告により(Umata et al., 2001)、我々の結果は裏付けされ、proHB-EGFシェダーゼが細胞の状況と刺激の両方により限定されることを示唆している。さらに、ADAM9ノックアウトマウスは、ある明確な表現型が欠損しているが、ADAM9−/−フィブロブラストはproHB-EGFプロセシングにおいて何の欠陥も生じない(Weskamp et al., 2002)。このことは、in vivoでのproHB-EGF切断酵素間の機能的な余剰を示している。
【0080】
どのようにADAMファミリーのメタロプロテアーゼが活性化され、ついにはEGFRリン酸化と下流のシグナリング応答を導くのであろうか。以前の報告は、MAPK ERK1/2により成長因子とTPAに応答して、膜貫通型タンパク質のメタロプロテアーゼに媒介されるエクトドメインの切断を制御しているのに対して、エクトドメインシェディングの基礎レベルはp38活性に帰因していたことを証明した(Fan and Derynck, 1999; Gechtman et al., 1999; Rizoli et al., 1999)。さらに、形質転換されていないヒトケラチノサイトでのソルビトール誘導性EGFR活性化において、p38はEGFRの上流メディエーターとして関連しているとされた(Chang et al., 2002)。我々の結果とは対照的に、著者は培地移動実験(medium transfer experiments)に基づいて、リガンド依存性メカニズムを排除している。放出されたEGF様リガンドがヘパリンスルフェートグリカンに結合して細胞外マトリックス中に保留していたので、リガンド依存性EGFR活性化の関わりを除くことができない。これらの報告と我々のシステムにおいて、p38がEGFRリン酸化状態とは独立である発見(図2と図6C)は、p38がストレス剤によるリガンド依存性EGFR活性化と下流のシグナリングを制御する上流のシグナリング要素であるのかという疑問を投げかけた。実に、特異的p38阻害剤とのプレインキュベーションがストレス誘導性EGFR活性化を排除したのに対して、ERK1/2活性化の遮断はEGFRリン酸化に影響しないままであることを発見した(図2B)。これに反して、ストレス剤に応答するp38活性化自体は、EGFR選択阻害剤であるAG1478を用いて査定したようにEGFRとは独立であった(図2A)。さらに、経時変化実験は、p38活性化がEGFRリン酸化に先行し、このことはp38がEGFRの上流に位置するために必要な前提条件であることを明らかにした。一方で、ERK1/2とJNK活性化はもっと後で生じる(図1)。全体として、これらのデータは、p38がリガンド依存性EGFR活性化の上流インデューサーであり、かつそれに続く下流のシグナリングであることを示唆している。
【0081】
ストレス応答でのp38の重要な役割は、発生の全体に渡ってよく保護されている。サッカロミセス属cerevisaeでは、高浸透圧への応答が集中的に研究されてきた。中心的役割は、HOGキナーゼ、ヒトp38の酵母ホモログが担っている。HOGは浸透圧を変化させるためのセンサー分子により活性化されるが、ヒトにはない。これらのタンパク質によるHOGの活性化は酵母で順応応答を導き高浸透圧を対処する(de Nadal et al., 2002)。
【0082】
酸化ストレスと浸透ストレスに応答するERK1/2とJNKの活性化は、細胞ストレス応答において重要な段階を示す(Kyriakis and Avruch, 2001で調査)。MAPKシグナリングの潜在的なミディエーターとして種々のレセプターチロシンキナーゼのリン酸化がストレス剤により刺激されている一方で、様々な報告はストレス刺激によるMAPK活性化にEGFRが際だった役割をを有することを明らかにしている(Carpenter, 1999で調査)。癌細胞が高レベルのROSを頻繁に生産するので、近年の研究は抗ガン治療でMAPKによるストレスシグナリングに厳格な因果関係の証拠を提供している(Burdon, 1995; Szatrowski and Nathan, 1991)。さらに、抗ガン剤または照射療法はストレスシグナリングカスケードの活性化を導き(Benhar et al., 2002)、これが、これらの薬剤により引き起こされるROS生産に帰因してきた。さらに、EGFR依存性MAPKシグナリングは、Bcl-2ファミリーのようなアポトーシスレギュレーターの発現レベルに影響を与えると報告されている(Jost et al., 2001)。これらの理由から、治療の影響力ゆえにストレスシグナリングメカニズムとそれらの複雑な相互作用を解明することに注目が集まっている。
【0083】
ここでは、ストレス誘導性リガンド依存性EGFR活性化がERK1/2の場合には決定的に、かつJNKの場合に高い度合いでEGFR活性化に依存している下流のシグナリング事象を導くことを示された(図6)。以前の報告は、ストレス誘導性MAPK活性化で種々のリガンド非依存性活性化メカニズムを示唆していた。これらの経路はホスフェートならびに他のレドックスの影響を受けやすいタンパク質の不活性化とダウンレギュレーションに関わり、足場タンパク質と小さなGタンパク質に影響を与える。EGF様リガンド依存性メカニズムと非依存性メカニズムの両方がMAPK活性化で協力することを証明したので、我々のデータはこれらの結果に及ぶ(図6)。これらの経路の各々の寄与は、特定の細胞状況に依存しているようである。リガンド依存性EGFR活性化は、種々のMAPKシグナリング経路間の連絡を媒介しているようである。さらに、腎臓癌細胞中のドキソルビシン誘導性アポトーシスをHB-EGF機能の遮断により強く促進できるのに対して(図7)、ドキソルビシン処理がp38活性化を誘導することを証明した。化学療法剤がストレスシグナリングカスケードを活性化することが以前に示されたので(Benhar et al., 2002)、このシグナリングメカニズムは、薬剤誘導性細胞死を避ける腫瘍細胞の分子の真相を提供する。
【産業上の利用可能性】
【0084】
我々の知る限りでは、これがヒト癌細胞でEGFRのリガンド依存性活性化と、それに続くMAPKp38により制御される浸透ストレスと酸化ストレス剤による下流のシグナリングを証明する初めての報告である。ますます増える証拠は、心臓血管疾患、神経変性疾患または癌のような過剰増殖性疾患のように多種の様々なヒトの疾患で過剰のROS生産により引き起こされる酸化ストレスに特に関わる。ゆえに、ストレス剤により引き起こされる細胞損傷に関わる病態生理学的疾患と各治療アプローチにとって特別に重要である。
【0085】
本明細書で記載したデータは、哺乳類細胞、特にヒト癌腫でのストレス刺激のシグナリングメカニズムにおける以前の結果に及ぶ。我々の発見は、ヒト癌細胞でのストレス応答の重要なメディエーターとしてADAMファミリープロテアーゼとHB-EGFの重要性を実証する。さらに、我々のデータはMAPKの種々のグループ間でのクロスコミュニケーションがADAMプロテアーゼとEGFRをシグナリング媒介物として使用していることを示唆する。これに関連して、ERK1/2とJNK活性の間のバランスは、細胞の運命にとって特に重要である。他のEGFR下流シグナリング事象ならびにストレス剤に応答する癌細胞の強化増殖または移動のような可能性のある病理学的応答に焦点をおいて将来の研究が成されるべきである。
【0086】
【表3】
【0087】
【表4】
【0088】
【表5】
【0089】
【表6】
【0090】
【表7】
【0091】
【表8】
【0092】
【表9】
【特許請求の範囲】
【請求項1】
少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療するための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項2】
疾患は癌である、請求項1に記載の使用。
【請求項3】
疾患は少なくとも部分的に照射および/または薬剤耐性癌である、請求項1または2に記載の使用。
【請求項4】
疾患は少なくとも部分的にアポトーシス誘導治療に対して耐性である、請求項1から3までのいずれか1項に記載の使用。
【請求項5】
疾患は、細胞増殖抑制剤および/または細胞毒性剤、特にアポトーシス誘導剤の投与に対して少なくとも部分的に耐性である、請求項1から4までのいずれか1項に記載の使用。
【請求項6】
レセプターチロシンキナーゼリガンドの阻害剤は、更なる治療法および/または薬剤と共同適用される、請求項1から5までのいずれか1項に記載の使用。
【請求項7】
薬剤は照射療法と共同適用される、請求項6に記載の使用。
【請求項8】
薬剤は、更なる抗ガン剤、特に化学療法剤または抗腫瘍抗体と共同適用される、請求項6または7に記載の使用。
【請求項9】
更なる抗癌剤は、ドキソルビシン、タキサン、シス/トランス−プラチンまたはこれらの誘導体、5−フルオロウラシル、マイトマイシンD、パクリタキセル、エトポシド、シクロホスホアミド、ドセタキセルまたは他のアポトーシス誘導剤またはタンパク質、特に抗体から選択される、請求項8に記載の使用。
【請求項10】
過剰増殖性疾患に対する治療の効率を上げるための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項11】
照射療法および/または薬物治療に対する過剰増殖性疾患の感受性を上げるための薬剤を製造するための、レセプターチロシンキナーゼの阻害剤の使用。
【請求項12】
レセプターチロシンキナーゼのストレス誘導性活性化により引き起こされるか、または関連する過剰増殖性疾患を予防または治療するための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項13】
ストレスは酸化ストレスおよび/または浸透ストレスである、請求項12に記載の使用。
【請求項14】
ストレスはp38媒介ストレスである、請求項12または13に記載の使用。
【請求項15】
疾患は癌である、請求項12から14までのいずれか1項に記載の使用。
【請求項16】
レセプターチロシンキナーゼは、EGFRおよび他のEGFRファミリーのメンバーから選択される、請求項1から15までのいずれか1項に記載の使用。
【請求項17】
レセプターはEGFRである、請求項1から6までのいずれか1項に記載の方法。
【請求項18】
レセプターチロシンキナーゼリガンドは、該レセプターチロシンキナーゼの細胞外ドメインに結合するリガンドである、請求項1から17までのいずれか1項に記載の使用。
【請求項19】
レセプターチロシンキナーゼリガンドは、HB−EGF、EGF、アンフィレグリン、ベータセルリン、エピレグリン、TGF-α、ニューレグリンまたはヘレグリンから選択される、請求項1から18までのいずれか1項に記載の使用。
【請求項20】
レセプターチロシンキナーゼリガンドは、HB−EGFである、請求項19に記載の使用。
【請求項21】
阻害剤は、レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤である、請求項1から20までのいずれか1項に記載の使用。
【請求項22】
阻害剤は、レセプターチロシンキナーゼリガンドの直接的な阻害剤である、請求項1から12までのいずれか1項に記載の使用。
【請求項23】
阻害剤は核酸レベルで働く、請求項1から22までのいずれか1項に記載の使用。
【請求項24】
阻害剤は、特異的な転写阻害剤、特にアンチセンス分子、リボザイムまたはRNAi分子から選択される、請求項23に記載の使用。
【請求項25】
阻害剤は遺伝子不活性化因子である、請求項24に記載の使用。
【請求項26】
阻害剤は、タンパク質レベルで働く、請求項1から22までのいずれか1項に記載の使用。
【請求項27】
阻害剤は、特異的タンパク質阻害剤であり、特に抗体または抗体断片および/またはタンパク質分解性阻害剤または低分子量阻害剤から選択される、請求項26に記載の使用。
【請求項28】
活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキット。
【請求項29】
付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有する請求項28に記載の組成物またはキット。
【請求項30】
レセプターチロシンキナーゼリガンドの阻害剤を、これを必要とする被験者に投与することを含む、少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療する方法。
【請求項1】
少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療するための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項2】
疾患は癌である、請求項1に記載の使用。
【請求項3】
疾患は少なくとも部分的に照射および/または薬剤耐性癌である、請求項1または2に記載の使用。
【請求項4】
疾患は少なくとも部分的にアポトーシス誘導治療に対して耐性である、請求項1から3までのいずれか1項に記載の使用。
【請求項5】
疾患は、細胞増殖抑制剤および/または細胞毒性剤、特にアポトーシス誘導剤の投与に対して少なくとも部分的に耐性である、請求項1から4までのいずれか1項に記載の使用。
【請求項6】
レセプターチロシンキナーゼリガンドの阻害剤は、更なる治療法および/または薬剤と共同適用される、請求項1から5までのいずれか1項に記載の使用。
【請求項7】
薬剤は照射療法と共同適用される、請求項6に記載の使用。
【請求項8】
薬剤は、更なる抗ガン剤、特に化学療法剤または抗腫瘍抗体と共同適用される、請求項6または7に記載の使用。
【請求項9】
更なる抗癌剤は、ドキソルビシン、タキサン、シス/トランス−プラチンまたはこれらの誘導体、5−フルオロウラシル、マイトマイシンD、パクリタキセル、エトポシド、シクロホスホアミド、ドセタキセルまたは他のアポトーシス誘導剤またはタンパク質、特に抗体から選択される、請求項8に記載の使用。
【請求項10】
過剰増殖性疾患に対する治療の効率を上げるための薬剤を製造するための、レセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項11】
照射療法および/または薬物治療に対する過剰増殖性疾患の感受性を上げるための薬剤を製造するための、レセプターチロシンキナーゼの阻害剤の使用。
【請求項12】
レセプターチロシンキナーゼのストレス誘導性活性化により引き起こされるか、または関連する過剰増殖性疾患を予防または治療するための薬剤を製造するためのレセプターチロシンキナーゼリガンドの阻害剤の使用。
【請求項13】
ストレスは酸化ストレスおよび/または浸透ストレスである、請求項12に記載の使用。
【請求項14】
ストレスはp38媒介ストレスである、請求項12または13に記載の使用。
【請求項15】
疾患は癌である、請求項12から14までのいずれか1項に記載の使用。
【請求項16】
レセプターチロシンキナーゼは、EGFRおよび他のEGFRファミリーのメンバーから選択される、請求項1から15までのいずれか1項に記載の使用。
【請求項17】
レセプターはEGFRである、請求項1から6までのいずれか1項に記載の方法。
【請求項18】
レセプターチロシンキナーゼリガンドは、該レセプターチロシンキナーゼの細胞外ドメインに結合するリガンドである、請求項1から17までのいずれか1項に記載の使用。
【請求項19】
レセプターチロシンキナーゼリガンドは、HB−EGF、EGF、アンフィレグリン、ベータセルリン、エピレグリン、TGF-α、ニューレグリンまたはヘレグリンから選択される、請求項1から18までのいずれか1項に記載の使用。
【請求項20】
レセプターチロシンキナーゼリガンドは、HB−EGFである、請求項19に記載の使用。
【請求項21】
阻害剤は、レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤である、請求項1から20までのいずれか1項に記載の使用。
【請求項22】
阻害剤は、レセプターチロシンキナーゼリガンドの直接的な阻害剤である、請求項1から12までのいずれか1項に記載の使用。
【請求項23】
阻害剤は核酸レベルで働く、請求項1から22までのいずれか1項に記載の使用。
【請求項24】
阻害剤は、特異的な転写阻害剤、特にアンチセンス分子、リボザイムまたはRNAi分子から選択される、請求項23に記載の使用。
【請求項25】
阻害剤は遺伝子不活性化因子である、請求項24に記載の使用。
【請求項26】
阻害剤は、タンパク質レベルで働く、請求項1から22までのいずれか1項に記載の使用。
【請求項27】
阻害剤は、特異的タンパク質阻害剤であり、特に抗体または抗体断片および/またはタンパク質分解性阻害剤または低分子量阻害剤から選択される、請求項26に記載の使用。
【請求項28】
活性成分として
a)レセプターチロシンキナーゼリガンドを切断できるメタロプロテアーゼの阻害剤であるか、またはメタロプロテアーゼの上流の調節段階の阻害剤であるレセプターチロシンキナーゼリガンドの阻害剤および
b)過剰増殖性疾患を治療するための更なる薬剤
を有する製剤学的組成物またはキット。
【請求項29】
付加的に製剤学的に認容性のキャリア、希釈剤および/または助剤を有する請求項28に記載の組成物またはキット。
【請求項30】
レセプターチロシンキナーゼリガンドの阻害剤を、これを必要とする被験者に投与することを含む、少なくとも部分的に治療抵抗性の過剰増殖性疾患を予防または治療する方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】

【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−84580(P2011−84580A)
【公開日】平成23年4月28日(2011.4.28)
【国際特許分類】
【出願番号】特願2011−15188(P2011−15188)
【出願日】平成23年1月27日(2011.1.27)
【分割の表示】特願2006−518114(P2006−518114)の分割
【原出願日】平成16年7月5日(2004.7.5)
【出願人】(390040420)マックス−プランク−ゲゼルシャフト・ツア・フェルデルング・デア・ヴィッセンシャフテン・エー・ファオ (54)
【氏名又は名称原語表記】Max−Planck−Gesellschaft zur Foerderung der Wissenschaften e.V.
【Fターム(参考)】
【公開日】平成23年4月28日(2011.4.28)
【国際特許分類】
【出願日】平成23年1月27日(2011.1.27)
【分割の表示】特願2006−518114(P2006−518114)の分割
【原出願日】平成16年7月5日(2004.7.5)
【出願人】(390040420)マックス−プランク−ゲゼルシャフト・ツア・フェルデルング・デア・ヴィッセンシャフテン・エー・ファオ (54)
【氏名又は名称原語表記】Max−Planck−Gesellschaft zur Foerderung der Wissenschaften e.V.
【Fターム(参考)】
[ Back to top ]