
希土類発光プローブ
【課題】アミノ酸もしくはペプチドの切断が蛍光により検出可能な化合物ないし蛍光プローブを提供する。
【解決手段】一般式(I)
R−CONH−Ar−R1−(アミン系配位子−希土類錯体) (I)
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
【解決手段】一般式(I)
R−CONH−Ar−R1−(アミン系配位子−希土類錯体) (I)
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、発光プローブとして有用な新規化合物および発光プローブ、加水分解酵素活性測定法、さらにその手法を用いた加水分解酵素阻害剤のスクリーニング方法に関する。
【背景技術】
【0002】
現在、加水分解酵素のアッセイには、クマリン(MCA)色素を用いたペプチド基質が用いられることが多い。MCA 基質を用いたアッセイは、定常状態蛍光を測定するアッセイであることから試料の自家蛍光やその他の蛍光物質などにより影響を受ける。そこで、近年では希土類金属イオン由来の長寿命蛍光を利用したアッセイ法が注目されている。希土類蛍光を利用したアッセイの中で最も有名なのは、PerkinElmer 社のDELFIA システムであろう。このシステムは96 ウェルまたは384 ウェルのプレートを用いて時間分解蛍光測定を行う手法で、高い検出感度を有することから汎用されている。加水分解酵素の活性を検出する測定法に関しては、希土類蛍光と蛍光共鳴エネルギー移動を用いたTR-FRET (Time-resolved Fluorescence Resonance Energy Transfer)システム(代表的なものとして、Invitrogen 社のLanthaScreen)が挙げられる。しかしながら、TR-FRET 法は基質ペプチドの両側にFRET のドナーである希土類錯体とアクセプターの色素を結合させる必要があり、合成が煩雑となる。また、ある一定の濃度を超えると、分子間エネルギー移動が起こる可能性も指摘されている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、アミノ酸もしくはペプチドの切断が蛍光により検出可能な化合物ないし蛍光プローブを提供することを目的とする。
【0004】
また、本発明はペプチドの切断が容易に検出できる蛍光プローブを用いた加水分解酵素活性の時間分解蛍光測定法、ならびにその手法を応用した阻害剤のスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、上記目的を達成すべく鋭意研究を重ねた結果、特定の基(アンテナ分子)を介してペプチドを結合させた希土類錯体が、ペプチドの切断により蛍光を発光することを見出した。
【0006】
本発明は、以下の化合物ないし蛍光プローブ、加水分解酵素活性測定法、および加水分解酵素阻害剤のスクリーニング方法を提供することを目的とする。
項1.
一般式(I)
【0007】
【化1】

【0008】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
項2.
前記アミン系配位子が、4個のアミノ基と3個のカルボキシル基、あるいは3個のアミノ基と4個のカルボキシル基を有する7配位の配位子である、項1に記載の化合物。
項3.
前記希土類錯体のリガンドである「R−CONH−Ar−R1−(アミン系配位子)」が、下記式(IIA)、(IIB)のいずれかで表される、項1又は2に記載の化合物:
【0009】
【化2】
【0010】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
項4.
希土類が、Tb,Sm,Eu、DyおよびNdからなる群から選ばれる、項1〜3のいずれかに記載の錯体。
項5.
Arが−C6H4−であり、R1が−CH2−である、項1〜4のいずれかに記載の錯体。
項6.
「アミン系配位子−希土類錯体」が以下の構造を有する、項1〜5のいずれかに記載の化合物:
【0011】
【化3】
【0012】
(式中、Lnはランタノイドを示す。)
項7.
下記式
【0013】
【化4】
【0014】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。R−COは、アミノ酸もしくはペプチド基を示す。Lnは、ランタノイドを示す。)
で表される項1〜6のいずれかに記載の化合物。
項8.
R−COが、加水分解酵素の認識配列を含む、項1〜7のいずれかに記載の化合物。
項9.
項1〜8のいずれかに記載の化合物に、加水分解酵素阻害の候補物質の存在下で加水分解酵素を作用させ、前記候補物質の加水分解酵素の阻害作用を、前記化合物のペプチドの切断による発光を用いてスクリーニングすることを特徴とする、加水分解酵素阻害剤のスクリーニング方法。
項10.
項1〜8のいずれかに記載の化合物を含む、希土類発光プローブ
項11.
下記式(IIC)で表される化合物:
【0015】
【化5】
【0016】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。)
【発明の効果】
【0017】
本発明の化合物は、ペプチドの切断により蛍光が著しく増強されるため、ペプチドの切断を蛍光により容易に検出可能である。
【0018】
本発明の化合物は、シンプルな構造を持ち、加水分解酵素によってペプチド配列が切断されることで切断前と比較して蛍光強度が大きく増大する。この蛍光は長寿命であり、時間分解蛍光測定を行うことによって、高いシグナル/ノイズ比で加水分解酵素活性の検出を行うことができる。
【0019】
アミノ酸/ペプチドを特定の加水分解酵素の認識配列とすることで、さまざまな種類の加水分解酵素の活性測定に適用可能であり、またこの測定法を用いてそれらの加水分解酵素の阻害剤を容易にスクリーニングすることができる。
【発明を実施するための最良の形態】
【0020】
本発明は、一般式(I)
R−CONH−Ar−R1−(アミン系配位子−希土類錯体) (I)
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは 置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物を提供する。
【0021】
R1は、2価の連結器であれば特に限定されず、CH2,CH2CH2、CH2CH2CH2、CH2CH(CH3)、CH(CH3)CH2、CH2CH2CH2CH2などの直鎖又は分枝を有するアルキレン基、CH2NH、CH2NHCO、CH2CO、CH2CONH、CH2CH2NH、CH2CH2NHCO、CH2CH2CO、CH2CH2CONH、CH2CH2NHCOC≡C、CH2CH2NHCOCX=CH(XはH,CH3、Cl,F、CNなど)などの1〜8個、好ましくは1〜6個の鎖状の炭素原子又はヘテロ原子(O,N,S)を介してArとアミン系配位子を連結する基を表す。ここで、「鎖状の炭素原子又はヘテロ原子(O,N,S)を介する」とは、側鎖の基は含まず、例えばCH2CH(CH3)は2個の炭素原子を介した結合であり、CH2CH2NHCOは、C,C,N,Cの4個の原子を介して結合しているとみなす。R1が長すぎると、希土類元素とArが離れすぎ、エネルギー移動が起こりにくくなり、蛍光が弱くなる可能性がある。
【0022】
R1は、アミン系配位子の窒素原子と結合するのが望ましいが、配位子のアミノ基をつなぐアルキレン基(好ましくはエチレン基)の炭素原子に結合させてもよい。
【0023】
Arは、置換されていてもよい(アリーレン基、ヘテロアリーレン基、アラルキレン基またはヘテロアラルキレン基)を示す。
【0024】
アリーレン基としては、アリール基(フェニル、ナフチル、アントラセニル、フェナントリル、トルイル、キシリル)の芳香環に結合した1個の水素原子を除いた基を表し、具体的にはフェニレン、ナフチレン、アントラセニレン、フェナントリレン、トルイレン、キシリレンなどが挙げられる。
【0025】
フェニレンは、オルト、メタ、パラのいずれでもよいが、パラフェニレンが好ましい。同様にナフチレン、アントラセニレン、フェナントリレン、トルイレン、キシリレンについても、芳香環における2個の結合の位置関係は任意であり、例えばナフチレンについては、1,2−、1,3−、1,4−、1,5−、1,6−、1,7−、1,8−、2,3−、2,4−、2,5−、2,6−、2,7−などの任意の結合位置の組み合わせが可能であり、Arで表される他の基も同様である。
【0026】
ヘテロアリーレン基としては、チエニレン、ピロリレン、フラニレン、イミダゾリレン、ピラゾリレン、チアゾリレン、イソチアゾリレン、オキサゾリレン、イソオキサゾリレン、インドリレン、イソインドリレン、ベンゾチエニレン、ベンゾフラニレン、ベンゾイミダゾリレン、ベンゾピラゾリレン、ベンゾチアゾリレン、ベンゾイソチアゾリレン、ベンゾオキサゾリレン、ベンゾイソオキサゾリレン、ピリジレン、ピラゾリジニレン、ピリミジニレン、ピリダジニレン、キノリレン、イソキノリレン、フタラジニレン、ナフチリジニレン、キノキサリニレン、キナゾリニレンなどが挙げられる。
【0027】
アラルキニレンとしては、(アリーレン)−(アルキレン)が挙げられ、アリーレンは前記に定義されるとおりであり、アルキレンは、メチレン、エチレン、プロピレン、イソプロピレン、ブチレンなどのC1〜C4の直鎖又は分枝を有するアルキレンが挙げられる。
【0028】
ヘテロアラルキニレンとしては、(ヘテロアリーレン)−(アルキレン)が挙げられ、ヘテロアリーレンは前記に定義されるとおりであり、アルキレンは、メチレン、エチレン、プロピレン、イソプロピレン、ブチレンなどのC1〜C4の直鎖又は分枝を有するアルキレンが挙げられる。
【0029】
アリーレン基、ヘテロアリーレン基、アラルキレン基またはヘテロアラルキレン基の置換基としては、水酸基、チオール、シアノ,ニトロ、C1〜C4の直鎖又は分枝を有するアルキル、C1〜C4の直鎖又は分枝を有するアルコキシ、アミノ、C1〜C4の直鎖又は分枝を有するアルキルでモノ置換もしくはジ置換されたアミノ、アセチル、アセチルアミノ、COOH、C1〜C4の直鎖又は分枝を有するアルコキシカルボニル、ハロゲン原子などが挙げられ、これらの置換基で1〜3置換、特に、1又は2置換され得る。
【0030】
アミン系配位子は、希土類に対し、配位可能なアミノ基を3個以上(好ましくは3個又は4個、特に4個)、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上(好ましくは2〜6個、さらに好ましくは3〜5個、特に3個又は4個)含み、かつ、5配位から9配位(好ましくは6配位〜9配位、より好ましくは7配位〜9配位、特に7配位又は8配位)のいずれかの配位数を有する配位子である。
【0031】
アミン系配位子は、隣接するアミノ基がCH2CH2で連結された、3個又は4個のアミノ基を有する環状又は鎖状(開環状)の構造を有し、アミノ基には、好ましくはCH2COOH、CH2(PO3H2)、CH2CH2COOH、CH2CH2(PO3H2)、のように、アルキレン基(メチレン基又はエチレン基)を介してカルボン酸又はホスホン酸のいずれかの酸性基が連結したものが好ましく例示される。カルボン酸又はホスホン酸のいずれかの酸性基は、1〜6個配位することができるが、希土類が3価のカチオンとして配位するので、2〜5個、好ましくは2〜4個、特に3個又は4個配位する配位子が好ましい。なお、アミノ基の1つには、R−CONH−Ar−R1が好ましく結合されるので、本発明の好ましい配位子は、アミノ基が3個又は4個有する環状又は鎖状の構造を有し、カルボキシル基又はホスホン酸基(好ましくはカルボキシル基)は、環状テトラアミンで3個(合計7配位)、鎖状テトラアミンで5個(合計9配位)、鎖状トリアミンで4個(合計7配位)の構造を有する。
【0032】
本発明の特に好ましいアミン系配位子を以下に示す。
【0033】
【化6】
【0034】
「R−CONH−Ar−R1−(アミン系配位子)」で表されるリガンドとしては、
【0035】
【化7】
【0036】
(式中、Ar,R1,Rは、前記に定義されるとおりである。)
が挙げられる。
【0037】
なお、R1の部分にカルボニル基(CO)、アミド基(CONHもしくはNHCO)、カルボキシル基(COOH)がある場合には、これらの基が希土類に配位する可能性があり、これらの基が配位した化合物も、本発明の化合物に包含される。
【0038】
希土類としては、Sc,Y,La,Ce,Pr,Nd,Pm,Sm,Eu,Gd,Tb,Dy,Ho,Er,Tm,Yb,Luが挙げられ、好ましくはTb,Sm,Eu,Dy,Nd、より好ましくはTb,Sm,Eu,Dy,さらに好ましくはTb,Sm,Eu,最も好ましくはTbが挙げられる。希土類は、通常3価のイオンとして、アミン系配位子に配位する。
【0039】
R−COとしては、加水分解酵素で全体が除去され得るアミノ酸もしくはペプチド基を表し、具体的には以下の表の組み合わせが挙げられる。
【0040】
【表1−1】
【0041】
【表1−2】
【0042】
本発明の好ましい化合物は、以下のものである。
【0043】
【化8】
【0044】
本発明では、Arの部分に置換基を導入するか、Arと結合するR1の部分に共役系(二重結合、三重結合など)を導入することで、蛍光波長を長波長シフトさせることができる。
【0045】
本発明の化合物は、以下に記載される合成スキーム、あるいは、公知文献、ペプチド合成やアミン系配位子の合成に関する書籍、文献などを参考にして、当業者であれば容易に製造することができる。
【0046】
本発明の具体的な化合物の一般的な合成法を以下に例示する。
一般的な合成法
1.配位子の合成
(1)DO3Aの合成
1の合成
Cyclen (2.42 g, 14.1 mmol, 1.0 eq.)、NaHCO3 (5.91 g, 70.0 mmol, 5.0 eq.) を80 mlのacetonitrileに加え、0℃で30分撹拌した。反応液にtert-butyl bromoacetate (9.05 g, 46.4 mmol, 3.3 eq.)を60分間かけて滴下し、室温で24時間撹拌した後、濾過を行った。濾液を減圧蒸留し、真空乾燥を行い、tolueneで再結晶を行い、化合物1・HBr(3.88 g)を得た。収率 54 %。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.88-2.93 (m, 12H), 3.11 (s, 4H), 3.29 (s, 2H), 3.38 (s, 4H); MS (ESI+) m/z: 515.37 ([M+H]+).
【0047】
【化9】
【0048】
(2)2-aminoethylDO3Aの合成
2の合成
2−ブロモエチルアンモニウムブロミド 2 g(10 mmol)を1 N NaOH 水溶液20 mL に溶かし、Z-クロリド1.56 ml(10 mmol)をゆっくりと滴下した。その後40分間激しく攪拌した。その後、CH2Cl2を40 ml加え攪拌し、1 N NaOH水溶液を取り除いた。有機相を1 N HClとH2Oで2回洗い、Na2SO4で脱水した。Na2SO4をろ過した後、溶媒を減圧蒸留で留去し、化合物14を1.86 g得た。収率 72%。
1H-NMR (400 MHz, CDCl3) δ3.43 (d, J=5.2 Hz, 2H), 3.53 (d, J=5.2 Hz, 2H), 5.09 (s, 1H), 7.31-7.35 (m, 5H)
MS (ES+) m/e calcd for [M+H]+258.01, found 257.94。
【0049】
3の合成
化合物2 1 g (3.89 mmol)をトルエン45 mlを加え、アルゴン置換した後にcyclen 1.68 g (9.73 mmol)をゆっくりと滴下し、12時間加熱還流した。放冷後にH2Oで3回洗った後、CH2Cl2で3回抽出した。Na2SO4で脱水、ろ過した後、溶媒を減圧蒸留で留去した。722 mg、収率 53%。
1H-NMR (400 MHz, CD3OD) δ2.36-2.79 (m, 20H), 5.09 (s, 2H), 7.32-7.36 (m, 5H)
MS (FAB+) m/e calcd for [M+H]+ 350.25, found 350.25。
【0050】
4の合成
化合物3 700 mg (2.00 mmol)、Na2CO32.12 g (20.0 mmol)、tert-butylbromoacetate 1.47 ml(10.0 mmol)にCH3CN 35 ml を加え、アルゴン置換した後に、24時間加熱還流した。放冷後にHexaneで3回洗った後、Na2SO4で脱水した。ろ過後、溶媒を減圧蒸留で留去し、カラムクロマトグラフィーにより精製を行った。609 mg、収率 44 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.34-3.68 (m, 28H), 5.09 (s, 2H), 7.32-7.37 (m, 5H)
MS (ESI+) m/e calcd for [M+H]+ 692.45, Found 692.39。
【0051】
5の合成
化合物4 550 mg(0.796 mmol)を80 mlのMeOHにとかし、Pd/C 4 mgを加えた後、水素置換後を行った。12時間激しく攪拌した後、Pd/Cをろ過し溶媒を減圧蒸留で留去した。368 mg、収率 83 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.35-3.50 (m, 28H)
MS (ESI+) m/e calcd for [M+H]+ 558.42, Found 558.43.
【0052】
【化10】
【0053】
2.アンテナ配位子縮合体の合成
(1)DO3Aと縮合させる場合
化合物1 (1当量)、NaHCO3 (4当量)、2-Bromo-4-nitroacetophenone (1当量) を適量のacetonitrileに加え、アルゴン置換したのち24時間加熱還流した。放冷してから濾過を行い、濾液を減圧蒸留した。その後、シリカゲルクロマトグラフィーにより精製を行い、アンテナ配位子縮合体を得た。
【0054】
【化11】
【0055】
(2)2-aminoethylDO3Aと縮合させる場合
ニトロ基を持つアンテナ前駆体化合物O2N-Ar-R1-OH (1当量)、HBTU (1.2当量)、 HOBt・H2O (1.2当量)、化合物5 (1.2当量) を適量のDMFに溶かし、ジイソプロピルエチルアミン (1.2当量) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗った。その後カラムクロマトグラフィーで精製を行い、アンテナ配位子縮合体を得た。
【0056】
【化12】
【0057】
3.ニトロ基の還元
上記2.で合成したニトロ化合物をエタノール又はメタノールに溶解し、触媒量のPd/Cの存在下、水素雰囲気下、室温で激しく撹拌した。原料の消失後、反応液をろ過し、ろ液を留去しアミノ体を得た。
【0058】
4.アミノ酸(ペプチド)の修飾・脱保護
保護アミノ酸 (1当量)、HBTU (1.5当量)、 HOBt・H2O (1.5当量)、上記3で合成したアミノ化合物 (1当量) を適量のDMFに溶かし、DIEA (2等量) を加え室温で3時間攪拌した。溶媒を減圧留去した。ペプチドの場合は、脱保護、アミノ酸の付加を繰り返した後、乾燥後TFA 10 mlに溶かし室温で12時間攪拌した。精製はHPLCで行い、アミノ酸又はペプチド修飾化合物を得た。
【0059】
5.希土類イオンの配位
錯形成はスペクトル測定前に、リガンドと当量の希土類金属硝酸塩を100 mM HEPES緩衝液 (pH 7.4 )中で混合し、2~16時間かけて配位させた。
【0060】
【化13】
【実施例】
【0061】
以下、本発明を実施例によってさらに具体的に説明するが、本発明はこれら実施例により何ら限定されるものではない。
実施例1
1.Aminobenzyl-DO3A(ABD)誘導体の合成
スキーム1に示す方法で、Aminobenzyl-DO3A (ABD)錯体とそれをアセチル化したAcetylaminobenzyl-DO3A-Tb (Ac-ABD-Tb) を合成した。各化合物及びその中間体の同定は、基本的に1H-NMR、13C-NMR、及び各種MSによって行った。
【0062】
スキーム1:ABD-Ln及びAc-ABD-Lnの合成
【0063】
【化14】
【0064】
6の合成
1・HBr (3.60 g, 7.00 mmol 1.0 eq.)および4-nitrobenzyl bromide (1.51 g, 7.00 mmol, 1.0 eq.)をアルゴン雰囲気下70 mLのアセトニトリルに溶解させた。NaHCO3 (2.94 g, 35.0 mmol, 5.0 eq) を溶液に加え、100℃で8時間加熱還流した後ろ過し、ろ液を留去した。残渣をシリカゲルカラムクロマトグラフィーで精製し、6を得た (4.25 g, y. 94%). 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.19-3.50 (m, 24H), 7.72 (d, J = 8.0 Hz, 2H), 8.18 (d, J = 8.0 Hz, 2H); MS (ESI+) m/z : 650.37 ([M+H]+)。
【0065】
7の合成
6 (1.40 g, 2.16 mmol) を10 mLのメタノールに溶解し10% Pd/C (40 mg)を加え、水素雰囲気下で12時間激しく撹拌した。反応液をろ過した後、減圧留去により化合物7をえた。(1.26 g, 95%).
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.21-3.67 (m, 26H), 6.62 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H); MS (ESI+) m/z: 620.42 ([M+H]+)。
【0066】
ABDの合成
7 (962 mg, 1.55 mmol) をトリフルオロ酢酸(15 mL) に溶解し、室温で9時間撹拌を行った。溶媒を減圧留去し、ジエチルエーテルで結晶化させてABD 645 mgを得た。収率92%。
1H NMR (400 MHz, D2O) δ 2.79-3.78 (m, 24H), 7.01 (d, J= 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 43.70, 49.92, 50.30, 52.42, 54.14, 56.55, 59.25, 116.67, 119.58, 133.69, 162.60, 162.95; HRMS (ESI+) m/z: 452.2462 (Calcd for [M+H]+: 452.2509)。
【0067】
8の合成
6(594 mg, 0.914 mmol) を無水酢酸/酢酸 (v/v= 2/1, 6 mL)に溶解し、10% Pd/C (26 mg) を加え、室温水素雰囲気下で40時間撹拌を行った。反応液をろ過し、溶媒を留去した後、残渣をシリカゲルカラムクロマトグラフィーで精製し、8 268 mgを得た。収率44%。
1H NMR (270 MHz, CDCl3) δ 1.46 (s, 3H), 1.51 (s, 30H), 2.28-3.47(m, 24H), 7.45 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 7.8 Hz, 2H); MS (ESI+) m/z: 662.44 ([M+H]+)。
【0068】
Ac-ABDの合成
8(962 mg, 1.55 mmol) をTFA (15 mL)に溶解し、室温で9時間撹拌した。溶媒を減圧留去した後、ジエチルエーテルで結晶化させ、化合物Ac-ABD 645 mgを得た。収率92%。
1H NMR (270 MHz, D2O) δ 2.03 (s, 3H), 2.89-3.80 (m, 24H), 7.33 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H); 13C-NMR (64.5 MHz, CD3OD) δ 23.95, 30.71, 50.58, 51.47, 52.18, 55.68, 57.09, 58.50, 121.74, 132.14, 139.96, 171.15, 171.45, 209.85; HRMS (ESI+) m/z: 494.2570 (Calcd for [M+H]+: 494.2615)。
【0069】
2.ロイシンアミノペプチダーゼ(LAP)を標的としたABD-Tbプローブ
標的とするプロテアーゼとして、まず最初にLAP (leucine aminopeptidase)を選択した。LAPは、APM (aminopeptidase M) やAPN (aminopeptidase N) 、CD13などとも呼ばれ、ペプチドやタンパク質のN末端からロイシン(Leu)を始めとする疎水性アミノ酸を1残基切断する酵素である。ヒトにおいても様々な臓器に存在することが知られている。臨床的には、肝・胆道系疾患において血清中の活性値が上昇することが知られており、実際に臨床検査においてもその活性は測定されている。また最近では、癌の転移や自己免疫疾患との関係も指摘されている。現在までに報告されている時間分解蛍光測定可能な長寿命発光プローブは消光型のみであり、発光増大型プローブは報告されていない。
【0070】
具体的なプローブのデザインをスキーム2に示した。プローブの基本骨格としては、前節で述べたABD-Tb錯体を用いることにした。LAPの基質配列であるL-Leuをアンテナ部分にアミド結合でつなげた錯体Leu-ABD-TbからLAPによってLeuが切り出されると、アンテナ部分がアニリンの錯体ABD-Tbと変化する。前節においてアンテナ部分がアシルアミノベンジル基の時に光らず、アミノベンジル基の時に良く光ることが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想される。合成はスキーム3に従い2ステップで行った。
【0071】
スキーム2:LAPプローブのデザイン
【0072】
【化15】
【0073】
スキーム3:Leu-ABD-Tbの合成
【0074】
【化16】
【0075】
3.カルパインを標的としたABD-Tbプローブ
次に標的とするプロテアーゼとして、カルパインを選択した。カルパインはシステインプロテアーゼであり、ペプチドやタンパク質のN末端からLY(Leu-Tyr)配列などを切断する酵素である。ヒトにおいても細胞プロセスに関わっていることが知られている。いくつかの疾患に対して関与していることが知られており、アルツハイマー病や、パーキンソン病に関与している。現在までに時間分解蛍光測定可能な長寿命発光プローブは報告されていない。
【0076】
具体的なプローブのデザインをスキーム4に示した。プローブの基本骨格としては、前節で述べたABD-Tb錯体を用いることにした。カルパインの基質配列であるSuc-LYをアンテナ部分にアミド結合でつなげた錯体Suc-LY-ABD-TbからカルパインによってSuc-LYが切り出されると、アンテナ部分がアニリンの錯体ABD-Tbと変化する。前節においてアンテナ部分がアシルアミノベンジル基の時に光らず、アミノベンジル基の時に良く光ることが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想される。合成はスキーム5に従い2ステップで行った。
【0077】
スキーム4:カルパインプローブのデザイン
【0078】
【化17】
【0079】
スキーム5:Suc-LY-ABD-Tbの合成
【0080】
【化18】
【0081】
10の合成
化合物7 (718 mg, 1.16 mmol)をDMF (25 mL)に加えた後、Z-Tyr(tBu)-OH・DCHA (706 mg, 1.28 mmol), HOBt・6H2O (355 mg, 2.32 mmol), HBTU・PF6 (879 mg, 2.32 mmol)とDIEAを加え、室温で終夜攪拌した。溶液を飽和NaHCO3水溶液で希釈し、酢酸エチルで抽出した。有機層を飽和NaHCO3水溶液、水、飽和食塩水で洗い、MgSO4 で脱水した後、溶媒を減圧留去した。得られた租生成物をシリカゲルカラムクロマトグラフィーで精製し、化合物10を得た。892 mg。収率79%。
1H NMR (400 MHz, CDCl3) δ 1.31 (s, 9H), 1.48 (s, 27H,), 2.53-3.69 (m, 28H), 4.47 (m, 1H), 6.60 (d, J= 8.0 Hz, 2H), 6.90 (d, J= 8.0 Hz, 2H), 7.13 (d, J= 8.0 Hz, 2H), 7.20 (d, J= 8.0 Hz, 1H), 7.31-7.35 (m, 5H), 7.43 (d, J= 8.0 Hz, 2H), 7.86 (s, 1H)
MS (ESI+): 973.63 ([M+H]+)。
【0082】
11の合成
化合物10 (832 mg, 0.855 mmol)をメタノール(50 mL)に加えた。10% Pd/C (27 mg)を加え、水素置換した後、室温で12時間攪拌した。溶液をろ過した後、溶媒を減圧留去し化合物11を得た。695 mg。収率97%。
1H NMR (400 MHz, CDCl3) δ 1.33 (s, 9H), 1.46 (s, 27H), 2.21-3.73 (m, 31H), 6.94 (d, J= 8.4 Hz, 2H), 7.17 (d, J= 8.4 Hz, 2H), 7.40 (d, J= 8.4 Hz, 2H), 7.60 (d, J= 8.0 Hz, 2H), 9.47 (s, 1H)
MS (ESI+): 839.56 ([M+H]+)。
【0083】
12の合成
化合物11 (647 mg, 0.772 mmol)をDMF (15 mL)に加えた後、Z-Leu-OH (246 mg, 0.946 mmol, 1.2 eq), HOBt・6H2O (236 mg, 1.54 mmol, 2.0 eq) HBTU・PF6(584 mg, 1.54 mmol, 2 eq) とDIEAを加え、室温で終夜攪拌した。溶液を飽和NaHCO3水溶液で希釈し、酢酸エチルで抽出した。有機層を飽和NaHCO3水溶液、水、飽和食塩水で洗い、MgSO4 で脱水した後、溶媒を減圧留去した。得られた租生成物をシリカゲルカラムクロマトグラフィーで精製し、化合物12 を得た。388 mg。収率46%
1H NMR (400 MHz, CDCl3) δ 0.87-0.90 (m, 9H), 1.30 (s, 9H), 1.46 (s, 27H), 2.21-3.49 (m, 28H), 4.06 (m, 1H); 4.68 (m, 1H); 6.53 (d, J= 8.0 Hz, 1H); 6.90 (d, J= 8.4 Hz, 2H); 6.92 (d, J= 8.4 Hz, 2H), 7.08 (d, J= 8.4 Hz, 2H), 7.11 (d, J= 8.4 Hz, 2H), 7.34 (m, 5H), 7.59 (d, J= 8.0 Hz, 1H), 8.86 (s, 1H).
MS (ESI+): 1086.68 ([M+H]+)。
【0084】
13の合成
化合物12 (355 mg, 0.327 mmol)をメタノール(40 mL)に加えた。10% Pd/C (13 mg)を加え、水素置換した後、室温で28時間攪拌した。溶液をろ過した後、溶媒を減圧留去し化合物13を得た。295 mg。収率95%
1H NMR (400 MHz, CDCl3) δ 0.86-0.99 (m, 9H), 1.31 (s, 9H), 1.46 (s, 27H), 2.09-3.49 (m, 28H), 4.06 (m, 1H), 4.68 (m, 1H), 6.92 (d, J= 8.4 Hz, 2H), 7.15 (d, J= 8.4 Hz, 2H), 7.36 (d, J= 8.4 Hz, 2H), 7.48 (d, J= 8.4 Hz, 2H), 7.98 (d, J= 8.0 Hz, 1H), 8.61 (s, 1H).
MS (ESI+): 952.63 ([M+H]+)。
【0085】
14の合成
化合物13 (275 mg, 0.289 mmol)をDMF (10 mL)に加えた後、succinic anhydride (31.8 mg, 0.318 mmol)とDIEAを加えた後室温で7時間攪拌した。溶媒を減圧留去した後、シリカゲルカラムクロマトグラフィーで精製し、化合物14を得た。176 mg収率58%
1H NMR (400 MHz, CDCl3) δ 0.83-0.96 (m, 9H), 1.30 (s, 9H), 1.47 (s, 27H), 1.62-3.90 (m, 28H), 4.13 (m, 1H), 4.75 (m, 1H), 6.86 (d, J= 8.6 Hz, 2H), 6.92 (d, J= 8.6 Hz, 2H), 7.50 (d, J= 8.4 Hz, 2H), 7.62 (d, 1H), 7.86(d, J= 8.4 Hz, 2H), 7.92 (d, J= 8.4 Hz, 2H), 8.55 (s, 1H), 9.17 (s, 1H)
MS (ESI+): 1052.67 ([M+H]+)。
【0086】
Suc-LY-ABDの合成
化合物14 (25.0 mg, 0.0238 mmol)をTFA (2 mL)に加え、室温で24時間攪拌した。溶媒を減圧留去した後、逆相HPLCで精製を行い化合物Suc-LY-ABDを得た。14.0 mg収率71 %。
1H NMR (400 MHz, CD3OD) δ 0.85 (d, J= 6.6 Hz, 3H), 0.90 (d, J= 6.6 Hz, 3H), 1.39 (m, 2H), 1.43 (m, 1H), 1.94 (s, 3H), 2.92-3.53 (m, 30H), 4.17 (m, 1H), 4.58 (m, 1H), 6.69 (d, J= 8.8 Hz, 2H), 7.09 (d, J= 8.8 Hz, 2H), 7.47 (d, J= 8.6 Hz, 2H), 7.70 (d, J= 8.6 Hz, 2H).
13C NMR (100 MHz, CD3OD) δ 21.88, 22.51, 23.37, 25.82, 32.17, 32.22, 37.58, 41.22, 50.35, 51.79, 52.94, 54.35, 57.14, 57.55, 58.59, 116.16, 116.44, 121.70, 129.28, 131.19, 132.37, 139.79, 157.12, 171.85, 172.77, 174.81, 176.42, 177.52, 178.41.
HRMS (ESI-): 826.4012 (Calcd for [M-H]-: 826.3987)。
【0087】
Suc-LY-ABD-Tbの合成
Suc-LY-ABDをメタノールに溶解した後、Tb(NO3)3・6H2OとNa2CO3を加え、室温で1時間攪拌した。溶媒を減圧留去した後、ゲルろ過を行い、Suc-LY-Abd-Tbを得た。
HRMS (FAB+) m/z: 984.3154 (Calcd for [M+H]+: 984.3162)。
【0088】
4.p-aminobenzamide-DO3A(pABAD)誘導体の合成
スキーム6:pABAD-Lnの合成
【0089】
【化19】
【0090】
15の合成
p-ニトロ安息香酸 100 mg (0.599 mmol)、HBTU 272 mg (0.719 mmol)、 HOBt・H2O 97.1 mg (0.719mmol)、化合物5 400 mg (0.719 mmol) を25mLのDMFに溶かし、ジイソプロピルエチルアミン125 μL (0.719 mmol) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗浄した。その後カラムクロマトグラフィーで精製を行い15を得た。194 mg、収率46%。
1H-NMR(400 MHz, CDCl3) δ 1.44 (s, 27H), 2.34-3.83 (m, 28H), 8.27 (d, J = 8.0 Hz, 2H), 8.43 (d, J = 8.0 Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 707.43, Found: 707.46。
【0091】
16の合成
化合物15 160 mg (0.227 mmol)を50 mLのMeOHにとかし、Pd/C 8mgを加えた後、室温水素雰囲気下で6時間激しく撹拌を行った。Pd/Cをろ過後、ろ液を減圧留去し、16を得た。110 mg、収率 72 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.27-3.79 (m, 28H), 7.33 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 677.45, Found: 677.45。
【0092】
pABADの合成
化合物16 40 mg (0.0592 mmol)を3 mLのTFAにとかし、12時間攪拌した。その後、溶媒を減圧留去し、残渣を逆層HPLCによって精製しpABADを得た。9.9 mg、収率 33 %。
1H-NMR(400 MHz, D2O) δ2.67-3.89 (m, 28H), 7.35 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4Hz, 2H).
13C-NMR (100MHz, CD3OD) δ 36.04, 52.67, 54.03, 56.20, 114.82, 121.92, 130.22, 153.22, 162.49, 170.80.
MS (FAB+) m/z calcd for [M+H]+: 509.2645, Found: 509.2720。
【0093】
17の合成
化合物16 50 mg (0.0740 mmol)を10 mLのTHFにとかし、acetic anhydride 14 mL (0.148 mmol)とpyridine (0.185 mmol)を加え、アルゴン雰囲気下、室温で12時間攪拌した。その後、溶媒を減圧蒸留で留去し、シリカゲルカラムクロマトグラフィーにより精製を行った。34.5 mg、収率 65 %。
1H-NMR (400 MHz, CDCl3) δ 1.43 (s, 27H), 2.02 (s, 3H),2.99-3.95 (m, 28H), 7.35 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 719.46, Found: 719.37。
【0094】
Ac-pABADの合成
化合物17 より化合物pABADと同様の方法で化合物Ac-pABADを合成した。10 mg、収率 44 %。
1H-NMR(400 MHz, D2O) δ2.02 (s, 3H),2.92-3.87 (m, 28H), 7.33 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H).
13C-NMR (100MHz, CD3OD) δ 17.38, 41.04, 50.32, 51.37, 57.60, 121.22, 128.83, 142.63, 169.67, 174.57.
MS (FAB+) m/e calcd for [M+H]+: 551.2751, Found: 551.2825。
【0095】
5. LAPを標的としたpABAD-Tbプローブ
pABD-Tbで示したのと同様に、pABAD-Tbを基本骨格として有するプロテアーゼ活性検出発光プローブ開発を行った。標的とするプロテアーゼとして、pABD-Tbで用いたのと同様にLAP (leucine aminopeptidase)を選択した。
【0096】
具体的なプローブのデザインをスキーム7に示した。LAPの基質配列であるL-Leucineをアンテナ部分にアミド結合でつなげた錯体Leu-pABAD-Tb3+からLAPによってLeuが切り出されると、アンテナ部分がアニリンの錯体pABAD-Tb3+と変化する。アンテナ部分がアシルアミノベンジル基の時では蛍光は弱く、アミノベンジル基の時に強く蛍光を発することが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想された。合成はスキーム8に従って行った。
【0097】
スキーム7
【0098】
【化20】
【0099】
スキーム8
【0100】
【化21】
【0101】
Leu-pABADの合成
pABAD 60 mg (0.089mmol)、HBTU 272 mg (0.133 mmol)、 HOBt・H2O 97.1 mg (0.133 mmol)、Boc-Leu・H2O 43.5 mg (0.133 mmol) を25mLのDMFに溶かし、DIEA 25 μL (0.133 mmol) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗浄し、溶媒を減圧蒸留で留去し18を得た。18は精製を行わずにTFAに溶かし、12時間攪拌した。TFAを減圧留去した後、HPLCで精製を行いLeu-pABADを得た。4.6 mg、収率8.3%。
1H-NMR(400 MHz, D2O) δ0.84 (s, 6H), 1.68-1.72 (m, 3H), 2.92-3.87 (m, 29H), 7.44 (d, J = 8.4Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H).
13C-NMR (100MHz, CD3OD) δ21.93, 23.29, 36.52, 41.68, 50.70, 51.16, 53.81, 54.71, 120.51, 120.51, 124.54, 129.56, 142.55, 162.70, 169.31, 172.67.
MS (ESI+) m/z calcd for [M+H]+: 620.3486, Found: 620.2712。
【0102】
実施例2:測定
測定は、以下の条件下に行った。
【0103】
測定機器
紫外可視分光光度計UV-1650PC (Shimadzu)
蛍光光度計 F4500 (Hitachi)
時間分解蛍光測定 低温りん光測定 発光寿命測定 SPEX (Horiba)。
【0104】
紫外吸収スペクトル
サンプル10 mM の100 mM HEPES buffer 溶液を測定。測定は1 cm石英セルを25 ℃で測定した。
【0105】
蛍光測定
サンプル10 mM の100 mM HEPES buffer 溶液を測定。測定は1 cm石英セルを25 ℃で測定した。励起光スリット5 nm、発光スリット 5 nm。フォトマル電圧 700 Vで測定した。
【0106】
量子収率測定
量子収率は相対法を用いて算出した。標準物質には1N H2SO4 に溶かした硫酸キニーネを用い、その量子収率をfst = 0.546と参照とした。測定はF4500 (Hitachi) にて行った。
【0107】
発光寿命測定
すべてのサンプルは100 mM HEPES buffer (pH = 7.4) 中で測定した。 データはsingle exponentialで以下の式でフィッティングを行い、t の値を発光寿命とした。
【0108】
【数1】
【0109】
測定はSPEX (Horiba)にて行った。
【0110】
・ABD誘導体の分光学的特性
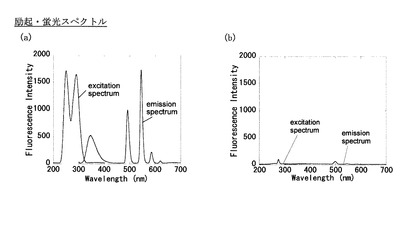
各化合物のTbおよびEu錯体において、中性水系緩衝液中で吸光スペクトルを測定したところ図1のようになった。次に励起および蛍光スペクトルを測定したところ, Ac-ABD-TbよりABD-Tbの法が25倍以上も発効強度が高いことがわかった(図2)。この結果はプロテアーゼによる脱アシル化によって発光強度が増加する可能性を強く示唆するものである。
【0111】
発光寿命測定
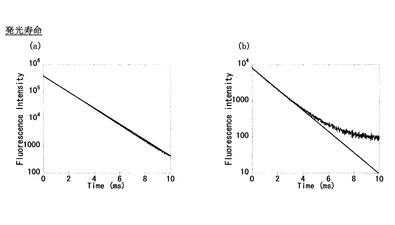
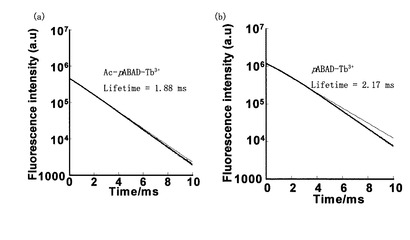
Ac-ABD-Tb錯体、ABD-Tb錯体において中性水系緩衝液中で発光寿命測定を行ったところ図3のようになった。グラフを、
【0112】
【数2】
【0113】
という式でフィッティングして発光寿命(τ)を求めると、ABD-Tbはτ =1.47 ms、Ac-ABD-Tbはτ =1.49 msと求まり、二つの錯体の値は大差ないものとなった。
【0114】
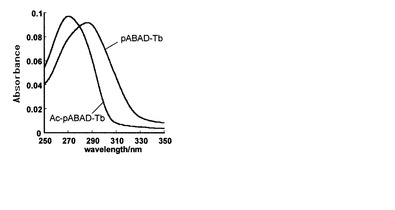
・pABAD誘導体の分光学的特性
各化合物のTb錯体において、中性水系緩衝液中で吸光スペクトルを測定したところ図4のようになった。pABAD-Tb3+ではアンテナのアミノ基をアセチル化したAc-pABAD-Tb3+より長波長側にシフトした。また、2種の化合物はモル吸光係数がTD-Tb3+、SD-Tb3+よりも高いことも確認され、Tb3+由来の蛍光強度が高くなる可能性があることも示唆された。
【0115】
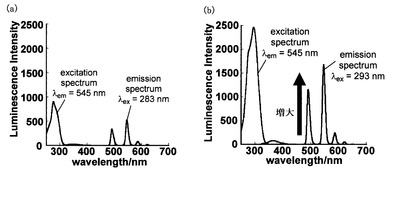
次に蛍光・励起スペクトルを測定したところ, Ac-pABAD-Tb3+よりpABAD-Tb3+の方が3倍以上も蛍光強度が高いことがわかった(図5)。この結果はプロテアーゼの反応による脱アシル化によって蛍光強度が増加する可能性を強く示唆するものである。
【0116】
・ pABAD誘導体の蛍光寿命測定
Ac-pABAD-Tb3+錯体、pABAD-Tb3+錯体において中性水系緩衝液中で発光寿命測定を行ったところ図6のようになった。両者において希土類金属に特徴的な長寿命な蛍光が得られ、蛍光強度の大きかったpABAD-Tb3+の方が発光強度の小さいAc-pABAD-Tb3+より蛍光寿命が長いことがわかった。
【0117】
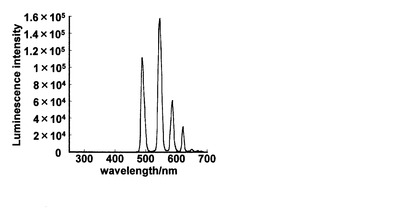
・ pABAD-Tb3+の時間分解測定
5 μMのpABAD-Tb3+ (100 mM HEPES Buffer, pH 7.4中)を実際に時間分解蛍光測定によって計測し、選択的に希土類イオンのみが観測できるかを調べた。その結果を図7に示す。示したスペクトルはdelaytime 30 μsのスペクトルである。これより、pABAD-Tb3+はTb3+由来の発光のみが選択的に観測できていることがわかる。このことから蛍光ノイズの大きい生体試料などでこの方法を用いることで高いS/N比での観察が可能になることが明らかにされた。
【0118】
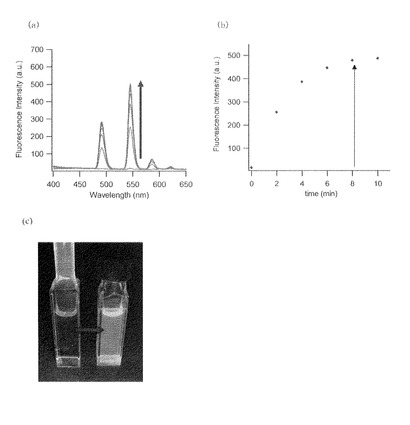
・酵素反応(I)(Leu-ABD-TbによるLAP活性の検出)
Leu-ABD-Tbの酵素との反応性について検討した。中性HEPES緩衝液中、5 μM のLeu-ABD-TbにLAP (from hog kidney) 0.1 Uを添加したところ、図8(a)のような蛍光スペクトルの変化が観測された。反応前後の蛍光強度変化は非常に大きく、肉眼でも捉えられる程であった(図8(c))。波長545 nmの発光強度を時間に対してプロットすると図8(b)のように約10分で飽和に達した。
【0119】
・酵素反応(II)(Suc-LY-ABD-Tbによるカルパイン活性の検出)
中性HEPES緩衝液中、10 μM のSuc-LY-ABD-Tb3+にカルパイン 90 μg/mLを添加したところ、図9 (a)のように、時間経過による蛍光強度の増加が観測された。次に、Delaytime 60 μsec、Gatetime 2.0 msecの条件で酵素反応を時間分解測定で追跡した場合でも、時間経過による蛍光強度の増加が観測され(図9(b))、また、図9(a)で示した通常の蛍光スペクトルと比較によりアンテナ分子の蛍光スペクトルが消えていることから、Suc-LY-ABD-Tb3+を用いることで、LAPの活性測定を時間分解測定によって行えることが明らかになった。
【0120】
・酵素反応(III)(Leu-pABAD-TbによるLAP活性の検出)
次に酵素との反応性について検討した。中性HEPES緩衝液中、5 μM のLeu-pABAD-TbにLAP (from hog kidney) 0.04 Uを添加したところ、図10 (a)のように、時間経過による蛍光強度の増加が観測された。次に、遅延時間30 μsec、ゲート時間0.45 msecの条件で酵素反応を時間分解測定で追跡した場合でも、時間経過による蛍光強度の増加が観測され(図10(b))、また、図10(c)で示した通常の蛍光スペクトルと比較によりアンテナ分子の蛍光スペクトルが消えていることから、Leu-pABAD-Tb3+を用いることで、LAPの活性測定を時間分解測定によって行えることが明らかになった。
【図面の簡単な説明】
【0121】
【図1】Absorption spectra of in 0.1 M HEPES buffer (pH 7.4)中、25 ℃で、100 μM の(a)ABD-Tb及びAc-ABD-Tb (b)ABD-Eu及びAc-ABD-Euの吸収スペクトル
【図2】0.1 M HEPES buffer (pH 7.4)中、25℃、10μMの(a)ABD-Tb (b)Ac-ABD-Tbの、250nmで励起し、545nmでモニターされた励起スペクトル及び発光スペクトル。
【図3】0.1 M HEPES buffer (pH 7.4)中、25 ℃、10 μM (a)ABD-Tb (b)Ac-ABD-Tbの励起状態の寿命。250nmで励起、545nmでモニター。
【図4】pABAD-Tb3+(赤)およびAc-pABAD-Tb3+(青)の吸収スペクトル。
【図5】(a) Ac-pABAD-Tb3+、(b) pABAD-Tb3+の励起(赤)および蛍光(青)スペクトル
【図6】(a) pABAD-Tb3+, (b) Ac-pABAD-Tb3+の蛍光寿命測定
【図7】pABAD-Tb3+の時間分解蛍光スペクトル。
【図8】(a) Reaction of Leu-ABD-TbとLAPの反応。LAP (0.1 U)の添加0, 2, 4, 6, 8, 10分後の5 μM Leu-ABD-Tbの発光スペクトル。反応は、100 mM HEPES buffer (pH 7.4)、37 oCで行った。励起波長は250nm。(b)545 nm (ex. 250 nm)の蛍光強度をプロットした。(c) 酵素反応の前(左)及び60分後(右)のLeu-ABD-Tb (5 μM) の蛍光強度の比較。サンプルは、UVランプ(254 nm)で照射した。
【図9】カルパインとSuc-LY-ABD-Tbの反応。カルパイン90 μg/mLの添加後0, 10, 20, 30, 40, 50, 60, 70, 80, 90分での10 μM のSuc-LY-ABD-Tbの発光スペクトル。反応は、2.5 mM 2-メルカプトエタノール, 5 mM CaCl2, 1 mM EDTA, 1mM EGTAを含む100 mM HEPES buffer (pH 7.4)中、30℃で行った。(a)は、250 nmで励起された発光スペクトル。(b)は、250 nmで励起された時間分解発光スペクトル (遅延時間: 60 μs. ゲート時間: 2.0 ms.)。
【図10】(a)Leu-pABAD-TbとLAPの反応。LAP (0.04 U)の添加後0, 1, 2, 3, 4, 6, 8, 10, 12, 14分での5 μM Leu-pABAD-Tb3+の発光スペクトル。100 mM HEPES buffer (pH 7.4)中、37 oCで反応を行った。励起波長は293nmであった。(b) Leu-pABAD-TbとLAPの反応。LAP (0.005 U) の添加後0, 10, 20, 30, 40分での5 μM Leu-ABD-Tb3+の発光スペクトル。時間分解測定は、遅延時間30 μsおよびゲート時間0.1 msで行った。(c) Leu- pABAD-Tb3+とLAPの反応。LAP (0.005 U)の添加後12時間での5 μM Leu- pABAD-Tb3+ の発光スペクトル。
【技術分野】
【0001】
本発明は、発光プローブとして有用な新規化合物および発光プローブ、加水分解酵素活性測定法、さらにその手法を用いた加水分解酵素阻害剤のスクリーニング方法に関する。
【背景技術】
【0002】
現在、加水分解酵素のアッセイには、クマリン(MCA)色素を用いたペプチド基質が用いられることが多い。MCA 基質を用いたアッセイは、定常状態蛍光を測定するアッセイであることから試料の自家蛍光やその他の蛍光物質などにより影響を受ける。そこで、近年では希土類金属イオン由来の長寿命蛍光を利用したアッセイ法が注目されている。希土類蛍光を利用したアッセイの中で最も有名なのは、PerkinElmer 社のDELFIA システムであろう。このシステムは96 ウェルまたは384 ウェルのプレートを用いて時間分解蛍光測定を行う手法で、高い検出感度を有することから汎用されている。加水分解酵素の活性を検出する測定法に関しては、希土類蛍光と蛍光共鳴エネルギー移動を用いたTR-FRET (Time-resolved Fluorescence Resonance Energy Transfer)システム(代表的なものとして、Invitrogen 社のLanthaScreen)が挙げられる。しかしながら、TR-FRET 法は基質ペプチドの両側にFRET のドナーである希土類錯体とアクセプターの色素を結合させる必要があり、合成が煩雑となる。また、ある一定の濃度を超えると、分子間エネルギー移動が起こる可能性も指摘されている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、アミノ酸もしくはペプチドの切断が蛍光により検出可能な化合物ないし蛍光プローブを提供することを目的とする。
【0004】
また、本発明はペプチドの切断が容易に検出できる蛍光プローブを用いた加水分解酵素活性の時間分解蛍光測定法、ならびにその手法を応用した阻害剤のスクリーニング方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、上記目的を達成すべく鋭意研究を重ねた結果、特定の基(アンテナ分子)を介してペプチドを結合させた希土類錯体が、ペプチドの切断により蛍光を発光することを見出した。
【0006】
本発明は、以下の化合物ないし蛍光プローブ、加水分解酵素活性測定法、および加水分解酵素阻害剤のスクリーニング方法を提供することを目的とする。
項1.
一般式(I)
【0007】
【化1】
【0008】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
項2.
前記アミン系配位子が、4個のアミノ基と3個のカルボキシル基、あるいは3個のアミノ基と4個のカルボキシル基を有する7配位の配位子である、項1に記載の化合物。
項3.
前記希土類錯体のリガンドである「R−CONH−Ar−R1−(アミン系配位子)」が、下記式(IIA)、(IIB)のいずれかで表される、項1又は2に記載の化合物:
【0009】
【化2】
【0010】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
項4.
希土類が、Tb,Sm,Eu、DyおよびNdからなる群から選ばれる、項1〜3のいずれかに記載の錯体。
項5.
Arが−C6H4−であり、R1が−CH2−である、項1〜4のいずれかに記載の錯体。
項6.
「アミン系配位子−希土類錯体」が以下の構造を有する、項1〜5のいずれかに記載の化合物:
【0011】
【化3】
【0012】
(式中、Lnはランタノイドを示す。)
項7.
下記式
【0013】
【化4】
【0014】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。R−COは、アミノ酸もしくはペプチド基を示す。Lnは、ランタノイドを示す。)
で表される項1〜6のいずれかに記載の化合物。
項8.
R−COが、加水分解酵素の認識配列を含む、項1〜7のいずれかに記載の化合物。
項9.
項1〜8のいずれかに記載の化合物に、加水分解酵素阻害の候補物質の存在下で加水分解酵素を作用させ、前記候補物質の加水分解酵素の阻害作用を、前記化合物のペプチドの切断による発光を用いてスクリーニングすることを特徴とする、加水分解酵素阻害剤のスクリーニング方法。
項10.
項1〜8のいずれかに記載の化合物を含む、希土類発光プローブ
項11.
下記式(IIC)で表される化合物:
【0015】
【化5】
【0016】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。)
【発明の効果】
【0017】
本発明の化合物は、ペプチドの切断により蛍光が著しく増強されるため、ペプチドの切断を蛍光により容易に検出可能である。
【0018】
本発明の化合物は、シンプルな構造を持ち、加水分解酵素によってペプチド配列が切断されることで切断前と比較して蛍光強度が大きく増大する。この蛍光は長寿命であり、時間分解蛍光測定を行うことによって、高いシグナル/ノイズ比で加水分解酵素活性の検出を行うことができる。
【0019】
アミノ酸/ペプチドを特定の加水分解酵素の認識配列とすることで、さまざまな種類の加水分解酵素の活性測定に適用可能であり、またこの測定法を用いてそれらの加水分解酵素の阻害剤を容易にスクリーニングすることができる。
【発明を実施するための最良の形態】
【0020】
本発明は、一般式(I)
R−CONH−Ar−R1−(アミン系配位子−希土類錯体) (I)
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは 置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物を提供する。
【0021】
R1は、2価の連結器であれば特に限定されず、CH2,CH2CH2、CH2CH2CH2、CH2CH(CH3)、CH(CH3)CH2、CH2CH2CH2CH2などの直鎖又は分枝を有するアルキレン基、CH2NH、CH2NHCO、CH2CO、CH2CONH、CH2CH2NH、CH2CH2NHCO、CH2CH2CO、CH2CH2CONH、CH2CH2NHCOC≡C、CH2CH2NHCOCX=CH(XはH,CH3、Cl,F、CNなど)などの1〜8個、好ましくは1〜6個の鎖状の炭素原子又はヘテロ原子(O,N,S)を介してArとアミン系配位子を連結する基を表す。ここで、「鎖状の炭素原子又はヘテロ原子(O,N,S)を介する」とは、側鎖の基は含まず、例えばCH2CH(CH3)は2個の炭素原子を介した結合であり、CH2CH2NHCOは、C,C,N,Cの4個の原子を介して結合しているとみなす。R1が長すぎると、希土類元素とArが離れすぎ、エネルギー移動が起こりにくくなり、蛍光が弱くなる可能性がある。
【0022】
R1は、アミン系配位子の窒素原子と結合するのが望ましいが、配位子のアミノ基をつなぐアルキレン基(好ましくはエチレン基)の炭素原子に結合させてもよい。
【0023】
Arは、置換されていてもよい(アリーレン基、ヘテロアリーレン基、アラルキレン基またはヘテロアラルキレン基)を示す。
【0024】
アリーレン基としては、アリール基(フェニル、ナフチル、アントラセニル、フェナントリル、トルイル、キシリル)の芳香環に結合した1個の水素原子を除いた基を表し、具体的にはフェニレン、ナフチレン、アントラセニレン、フェナントリレン、トルイレン、キシリレンなどが挙げられる。
【0025】
フェニレンは、オルト、メタ、パラのいずれでもよいが、パラフェニレンが好ましい。同様にナフチレン、アントラセニレン、フェナントリレン、トルイレン、キシリレンについても、芳香環における2個の結合の位置関係は任意であり、例えばナフチレンについては、1,2−、1,3−、1,4−、1,5−、1,6−、1,7−、1,8−、2,3−、2,4−、2,5−、2,6−、2,7−などの任意の結合位置の組み合わせが可能であり、Arで表される他の基も同様である。
【0026】
ヘテロアリーレン基としては、チエニレン、ピロリレン、フラニレン、イミダゾリレン、ピラゾリレン、チアゾリレン、イソチアゾリレン、オキサゾリレン、イソオキサゾリレン、インドリレン、イソインドリレン、ベンゾチエニレン、ベンゾフラニレン、ベンゾイミダゾリレン、ベンゾピラゾリレン、ベンゾチアゾリレン、ベンゾイソチアゾリレン、ベンゾオキサゾリレン、ベンゾイソオキサゾリレン、ピリジレン、ピラゾリジニレン、ピリミジニレン、ピリダジニレン、キノリレン、イソキノリレン、フタラジニレン、ナフチリジニレン、キノキサリニレン、キナゾリニレンなどが挙げられる。
【0027】
アラルキニレンとしては、(アリーレン)−(アルキレン)が挙げられ、アリーレンは前記に定義されるとおりであり、アルキレンは、メチレン、エチレン、プロピレン、イソプロピレン、ブチレンなどのC1〜C4の直鎖又は分枝を有するアルキレンが挙げられる。
【0028】
ヘテロアラルキニレンとしては、(ヘテロアリーレン)−(アルキレン)が挙げられ、ヘテロアリーレンは前記に定義されるとおりであり、アルキレンは、メチレン、エチレン、プロピレン、イソプロピレン、ブチレンなどのC1〜C4の直鎖又は分枝を有するアルキレンが挙げられる。
【0029】
アリーレン基、ヘテロアリーレン基、アラルキレン基またはヘテロアラルキレン基の置換基としては、水酸基、チオール、シアノ,ニトロ、C1〜C4の直鎖又は分枝を有するアルキル、C1〜C4の直鎖又は分枝を有するアルコキシ、アミノ、C1〜C4の直鎖又は分枝を有するアルキルでモノ置換もしくはジ置換されたアミノ、アセチル、アセチルアミノ、COOH、C1〜C4の直鎖又は分枝を有するアルコキシカルボニル、ハロゲン原子などが挙げられ、これらの置換基で1〜3置換、特に、1又は2置換され得る。
【0030】
アミン系配位子は、希土類に対し、配位可能なアミノ基を3個以上(好ましくは3個又は4個、特に4個)、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上(好ましくは2〜6個、さらに好ましくは3〜5個、特に3個又は4個)含み、かつ、5配位から9配位(好ましくは6配位〜9配位、より好ましくは7配位〜9配位、特に7配位又は8配位)のいずれかの配位数を有する配位子である。
【0031】
アミン系配位子は、隣接するアミノ基がCH2CH2で連結された、3個又は4個のアミノ基を有する環状又は鎖状(開環状)の構造を有し、アミノ基には、好ましくはCH2COOH、CH2(PO3H2)、CH2CH2COOH、CH2CH2(PO3H2)、のように、アルキレン基(メチレン基又はエチレン基)を介してカルボン酸又はホスホン酸のいずれかの酸性基が連結したものが好ましく例示される。カルボン酸又はホスホン酸のいずれかの酸性基は、1〜6個配位することができるが、希土類が3価のカチオンとして配位するので、2〜5個、好ましくは2〜4個、特に3個又は4個配位する配位子が好ましい。なお、アミノ基の1つには、R−CONH−Ar−R1が好ましく結合されるので、本発明の好ましい配位子は、アミノ基が3個又は4個有する環状又は鎖状の構造を有し、カルボキシル基又はホスホン酸基(好ましくはカルボキシル基)は、環状テトラアミンで3個(合計7配位)、鎖状テトラアミンで5個(合計9配位)、鎖状トリアミンで4個(合計7配位)の構造を有する。
【0032】
本発明の特に好ましいアミン系配位子を以下に示す。
【0033】
【化6】
【0034】
「R−CONH−Ar−R1−(アミン系配位子)」で表されるリガンドとしては、
【0035】
【化7】
【0036】
(式中、Ar,R1,Rは、前記に定義されるとおりである。)
が挙げられる。
【0037】
なお、R1の部分にカルボニル基(CO)、アミド基(CONHもしくはNHCO)、カルボキシル基(COOH)がある場合には、これらの基が希土類に配位する可能性があり、これらの基が配位した化合物も、本発明の化合物に包含される。
【0038】
希土類としては、Sc,Y,La,Ce,Pr,Nd,Pm,Sm,Eu,Gd,Tb,Dy,Ho,Er,Tm,Yb,Luが挙げられ、好ましくはTb,Sm,Eu,Dy,Nd、より好ましくはTb,Sm,Eu,Dy,さらに好ましくはTb,Sm,Eu,最も好ましくはTbが挙げられる。希土類は、通常3価のイオンとして、アミン系配位子に配位する。
【0039】
R−COとしては、加水分解酵素で全体が除去され得るアミノ酸もしくはペプチド基を表し、具体的には以下の表の組み合わせが挙げられる。
【0040】
【表1−1】
【0041】
【表1−2】
【0042】
本発明の好ましい化合物は、以下のものである。
【0043】
【化8】
【0044】
本発明では、Arの部分に置換基を導入するか、Arと結合するR1の部分に共役系(二重結合、三重結合など)を導入することで、蛍光波長を長波長シフトさせることができる。
【0045】
本発明の化合物は、以下に記載される合成スキーム、あるいは、公知文献、ペプチド合成やアミン系配位子の合成に関する書籍、文献などを参考にして、当業者であれば容易に製造することができる。
【0046】
本発明の具体的な化合物の一般的な合成法を以下に例示する。
一般的な合成法
1.配位子の合成
(1)DO3Aの合成
1の合成
Cyclen (2.42 g, 14.1 mmol, 1.0 eq.)、NaHCO3 (5.91 g, 70.0 mmol, 5.0 eq.) を80 mlのacetonitrileに加え、0℃で30分撹拌した。反応液にtert-butyl bromoacetate (9.05 g, 46.4 mmol, 3.3 eq.)を60分間かけて滴下し、室温で24時間撹拌した後、濾過を行った。濾液を減圧蒸留し、真空乾燥を行い、tolueneで再結晶を行い、化合物1・HBr(3.88 g)を得た。収率 54 %。
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.88-2.93 (m, 12H), 3.11 (s, 4H), 3.29 (s, 2H), 3.38 (s, 4H); MS (ESI+) m/z: 515.37 ([M+H]+).
【0047】
【化9】
【0048】
(2)2-aminoethylDO3Aの合成
2の合成
2−ブロモエチルアンモニウムブロミド 2 g(10 mmol)を1 N NaOH 水溶液20 mL に溶かし、Z-クロリド1.56 ml(10 mmol)をゆっくりと滴下した。その後40分間激しく攪拌した。その後、CH2Cl2を40 ml加え攪拌し、1 N NaOH水溶液を取り除いた。有機相を1 N HClとH2Oで2回洗い、Na2SO4で脱水した。Na2SO4をろ過した後、溶媒を減圧蒸留で留去し、化合物14を1.86 g得た。収率 72%。
1H-NMR (400 MHz, CDCl3) δ3.43 (d, J=5.2 Hz, 2H), 3.53 (d, J=5.2 Hz, 2H), 5.09 (s, 1H), 7.31-7.35 (m, 5H)
MS (ES+) m/e calcd for [M+H]+258.01, found 257.94。
【0049】
3の合成
化合物2 1 g (3.89 mmol)をトルエン45 mlを加え、アルゴン置換した後にcyclen 1.68 g (9.73 mmol)をゆっくりと滴下し、12時間加熱還流した。放冷後にH2Oで3回洗った後、CH2Cl2で3回抽出した。Na2SO4で脱水、ろ過した後、溶媒を減圧蒸留で留去した。722 mg、収率 53%。
1H-NMR (400 MHz, CD3OD) δ2.36-2.79 (m, 20H), 5.09 (s, 2H), 7.32-7.36 (m, 5H)
MS (FAB+) m/e calcd for [M+H]+ 350.25, found 350.25。
【0050】
4の合成
化合物3 700 mg (2.00 mmol)、Na2CO32.12 g (20.0 mmol)、tert-butylbromoacetate 1.47 ml(10.0 mmol)にCH3CN 35 ml を加え、アルゴン置換した後に、24時間加熱還流した。放冷後にHexaneで3回洗った後、Na2SO4で脱水した。ろ過後、溶媒を減圧蒸留で留去し、カラムクロマトグラフィーにより精製を行った。609 mg、収率 44 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.34-3.68 (m, 28H), 5.09 (s, 2H), 7.32-7.37 (m, 5H)
MS (ESI+) m/e calcd for [M+H]+ 692.45, Found 692.39。
【0051】
5の合成
化合物4 550 mg(0.796 mmol)を80 mlのMeOHにとかし、Pd/C 4 mgを加えた後、水素置換後を行った。12時間激しく攪拌した後、Pd/Cをろ過し溶媒を減圧蒸留で留去した。368 mg、収率 83 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.35-3.50 (m, 28H)
MS (ESI+) m/e calcd for [M+H]+ 558.42, Found 558.43.
【0052】
【化10】
【0053】
2.アンテナ配位子縮合体の合成
(1)DO3Aと縮合させる場合
化合物1 (1当量)、NaHCO3 (4当量)、2-Bromo-4-nitroacetophenone (1当量) を適量のacetonitrileに加え、アルゴン置換したのち24時間加熱還流した。放冷してから濾過を行い、濾液を減圧蒸留した。その後、シリカゲルクロマトグラフィーにより精製を行い、アンテナ配位子縮合体を得た。
【0054】
【化11】
【0055】
(2)2-aminoethylDO3Aと縮合させる場合
ニトロ基を持つアンテナ前駆体化合物O2N-Ar-R1-OH (1当量)、HBTU (1.2当量)、 HOBt・H2O (1.2当量)、化合物5 (1.2当量) を適量のDMFに溶かし、ジイソプロピルエチルアミン (1.2当量) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗った。その後カラムクロマトグラフィーで精製を行い、アンテナ配位子縮合体を得た。
【0056】
【化12】
【0057】
3.ニトロ基の還元
上記2.で合成したニトロ化合物をエタノール又はメタノールに溶解し、触媒量のPd/Cの存在下、水素雰囲気下、室温で激しく撹拌した。原料の消失後、反応液をろ過し、ろ液を留去しアミノ体を得た。
【0058】
4.アミノ酸(ペプチド)の修飾・脱保護
保護アミノ酸 (1当量)、HBTU (1.5当量)、 HOBt・H2O (1.5当量)、上記3で合成したアミノ化合物 (1当量) を適量のDMFに溶かし、DIEA (2等量) を加え室温で3時間攪拌した。溶媒を減圧留去した。ペプチドの場合は、脱保護、アミノ酸の付加を繰り返した後、乾燥後TFA 10 mlに溶かし室温で12時間攪拌した。精製はHPLCで行い、アミノ酸又はペプチド修飾化合物を得た。
【0059】
5.希土類イオンの配位
錯形成はスペクトル測定前に、リガンドと当量の希土類金属硝酸塩を100 mM HEPES緩衝液 (pH 7.4 )中で混合し、2~16時間かけて配位させた。
【0060】
【化13】
【実施例】
【0061】
以下、本発明を実施例によってさらに具体的に説明するが、本発明はこれら実施例により何ら限定されるものではない。
実施例1
1.Aminobenzyl-DO3A(ABD)誘導体の合成
スキーム1に示す方法で、Aminobenzyl-DO3A (ABD)錯体とそれをアセチル化したAcetylaminobenzyl-DO3A-Tb (Ac-ABD-Tb) を合成した。各化合物及びその中間体の同定は、基本的に1H-NMR、13C-NMR、及び各種MSによって行った。
【0062】
スキーム1:ABD-Ln及びAc-ABD-Lnの合成
【0063】
【化14】
【0064】
6の合成
1・HBr (3.60 g, 7.00 mmol 1.0 eq.)および4-nitrobenzyl bromide (1.51 g, 7.00 mmol, 1.0 eq.)をアルゴン雰囲気下70 mLのアセトニトリルに溶解させた。NaHCO3 (2.94 g, 35.0 mmol, 5.0 eq) を溶液に加え、100℃で8時間加熱還流した後ろ過し、ろ液を留去した。残渣をシリカゲルカラムクロマトグラフィーで精製し、6を得た (4.25 g, y. 94%). 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.19-3.50 (m, 24H), 7.72 (d, J = 8.0 Hz, 2H), 8.18 (d, J = 8.0 Hz, 2H); MS (ESI+) m/z : 650.37 ([M+H]+)。
【0065】
7の合成
6 (1.40 g, 2.16 mmol) を10 mLのメタノールに溶解し10% Pd/C (40 mg)を加え、水素雰囲気下で12時間激しく撹拌した。反応液をろ過した後、減圧留去により化合物7をえた。(1.26 g, 95%).
1H NMR (400 MHz, CDCl3) δ 1.46 (s, 27H), 2.21-3.67 (m, 26H), 6.62 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H); MS (ESI+) m/z: 620.42 ([M+H]+)。
【0066】
ABDの合成
7 (962 mg, 1.55 mmol) をトリフルオロ酢酸(15 mL) に溶解し、室温で9時間撹拌を行った。溶媒を減圧留去し、ジエチルエーテルで結晶化させてABD 645 mgを得た。収率92%。
1H NMR (400 MHz, D2O) δ 2.79-3.78 (m, 24H), 7.01 (d, J= 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 43.70, 49.92, 50.30, 52.42, 54.14, 56.55, 59.25, 116.67, 119.58, 133.69, 162.60, 162.95; HRMS (ESI+) m/z: 452.2462 (Calcd for [M+H]+: 452.2509)。
【0067】
8の合成
6(594 mg, 0.914 mmol) を無水酢酸/酢酸 (v/v= 2/1, 6 mL)に溶解し、10% Pd/C (26 mg) を加え、室温水素雰囲気下で40時間撹拌を行った。反応液をろ過し、溶媒を留去した後、残渣をシリカゲルカラムクロマトグラフィーで精製し、8 268 mgを得た。収率44%。
1H NMR (270 MHz, CDCl3) δ 1.46 (s, 3H), 1.51 (s, 30H), 2.28-3.47(m, 24H), 7.45 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 7.8 Hz, 2H); MS (ESI+) m/z: 662.44 ([M+H]+)。
【0068】
Ac-ABDの合成
8(962 mg, 1.55 mmol) をTFA (15 mL)に溶解し、室温で9時間撹拌した。溶媒を減圧留去した後、ジエチルエーテルで結晶化させ、化合物Ac-ABD 645 mgを得た。収率92%。
1H NMR (270 MHz, D2O) δ 2.03 (s, 3H), 2.89-3.80 (m, 24H), 7.33 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H); 13C-NMR (64.5 MHz, CD3OD) δ 23.95, 30.71, 50.58, 51.47, 52.18, 55.68, 57.09, 58.50, 121.74, 132.14, 139.96, 171.15, 171.45, 209.85; HRMS (ESI+) m/z: 494.2570 (Calcd for [M+H]+: 494.2615)。
【0069】
2.ロイシンアミノペプチダーゼ(LAP)を標的としたABD-Tbプローブ
標的とするプロテアーゼとして、まず最初にLAP (leucine aminopeptidase)を選択した。LAPは、APM (aminopeptidase M) やAPN (aminopeptidase N) 、CD13などとも呼ばれ、ペプチドやタンパク質のN末端からロイシン(Leu)を始めとする疎水性アミノ酸を1残基切断する酵素である。ヒトにおいても様々な臓器に存在することが知られている。臨床的には、肝・胆道系疾患において血清中の活性値が上昇することが知られており、実際に臨床検査においてもその活性は測定されている。また最近では、癌の転移や自己免疫疾患との関係も指摘されている。現在までに報告されている時間分解蛍光測定可能な長寿命発光プローブは消光型のみであり、発光増大型プローブは報告されていない。
【0070】
具体的なプローブのデザインをスキーム2に示した。プローブの基本骨格としては、前節で述べたABD-Tb錯体を用いることにした。LAPの基質配列であるL-Leuをアンテナ部分にアミド結合でつなげた錯体Leu-ABD-TbからLAPによってLeuが切り出されると、アンテナ部分がアニリンの錯体ABD-Tbと変化する。前節においてアンテナ部分がアシルアミノベンジル基の時に光らず、アミノベンジル基の時に良く光ることが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想される。合成はスキーム3に従い2ステップで行った。
【0071】
スキーム2:LAPプローブのデザイン
【0072】
【化15】
【0073】
スキーム3:Leu-ABD-Tbの合成
【0074】
【化16】
【0075】
3.カルパインを標的としたABD-Tbプローブ
次に標的とするプロテアーゼとして、カルパインを選択した。カルパインはシステインプロテアーゼであり、ペプチドやタンパク質のN末端からLY(Leu-Tyr)配列などを切断する酵素である。ヒトにおいても細胞プロセスに関わっていることが知られている。いくつかの疾患に対して関与していることが知られており、アルツハイマー病や、パーキンソン病に関与している。現在までに時間分解蛍光測定可能な長寿命発光プローブは報告されていない。
【0076】
具体的なプローブのデザインをスキーム4に示した。プローブの基本骨格としては、前節で述べたABD-Tb錯体を用いることにした。カルパインの基質配列であるSuc-LYをアンテナ部分にアミド結合でつなげた錯体Suc-LY-ABD-TbからカルパインによってSuc-LYが切り出されると、アンテナ部分がアニリンの錯体ABD-Tbと変化する。前節においてアンテナ部分がアシルアミノベンジル基の時に光らず、アミノベンジル基の時に良く光ることが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想される。合成はスキーム5に従い2ステップで行った。
【0077】
スキーム4:カルパインプローブのデザイン
【0078】
【化17】
【0079】
スキーム5:Suc-LY-ABD-Tbの合成
【0080】
【化18】
【0081】
10の合成
化合物7 (718 mg, 1.16 mmol)をDMF (25 mL)に加えた後、Z-Tyr(tBu)-OH・DCHA (706 mg, 1.28 mmol), HOBt・6H2O (355 mg, 2.32 mmol), HBTU・PF6 (879 mg, 2.32 mmol)とDIEAを加え、室温で終夜攪拌した。溶液を飽和NaHCO3水溶液で希釈し、酢酸エチルで抽出した。有機層を飽和NaHCO3水溶液、水、飽和食塩水で洗い、MgSO4 で脱水した後、溶媒を減圧留去した。得られた租生成物をシリカゲルカラムクロマトグラフィーで精製し、化合物10を得た。892 mg。収率79%。
1H NMR (400 MHz, CDCl3) δ 1.31 (s, 9H), 1.48 (s, 27H,), 2.53-3.69 (m, 28H), 4.47 (m, 1H), 6.60 (d, J= 8.0 Hz, 2H), 6.90 (d, J= 8.0 Hz, 2H), 7.13 (d, J= 8.0 Hz, 2H), 7.20 (d, J= 8.0 Hz, 1H), 7.31-7.35 (m, 5H), 7.43 (d, J= 8.0 Hz, 2H), 7.86 (s, 1H)
MS (ESI+): 973.63 ([M+H]+)。
【0082】
11の合成
化合物10 (832 mg, 0.855 mmol)をメタノール(50 mL)に加えた。10% Pd/C (27 mg)を加え、水素置換した後、室温で12時間攪拌した。溶液をろ過した後、溶媒を減圧留去し化合物11を得た。695 mg。収率97%。
1H NMR (400 MHz, CDCl3) δ 1.33 (s, 9H), 1.46 (s, 27H), 2.21-3.73 (m, 31H), 6.94 (d, J= 8.4 Hz, 2H), 7.17 (d, J= 8.4 Hz, 2H), 7.40 (d, J= 8.4 Hz, 2H), 7.60 (d, J= 8.0 Hz, 2H), 9.47 (s, 1H)
MS (ESI+): 839.56 ([M+H]+)。
【0083】
12の合成
化合物11 (647 mg, 0.772 mmol)をDMF (15 mL)に加えた後、Z-Leu-OH (246 mg, 0.946 mmol, 1.2 eq), HOBt・6H2O (236 mg, 1.54 mmol, 2.0 eq) HBTU・PF6(584 mg, 1.54 mmol, 2 eq) とDIEAを加え、室温で終夜攪拌した。溶液を飽和NaHCO3水溶液で希釈し、酢酸エチルで抽出した。有機層を飽和NaHCO3水溶液、水、飽和食塩水で洗い、MgSO4 で脱水した後、溶媒を減圧留去した。得られた租生成物をシリカゲルカラムクロマトグラフィーで精製し、化合物12 を得た。388 mg。収率46%
1H NMR (400 MHz, CDCl3) δ 0.87-0.90 (m, 9H), 1.30 (s, 9H), 1.46 (s, 27H), 2.21-3.49 (m, 28H), 4.06 (m, 1H); 4.68 (m, 1H); 6.53 (d, J= 8.0 Hz, 1H); 6.90 (d, J= 8.4 Hz, 2H); 6.92 (d, J= 8.4 Hz, 2H), 7.08 (d, J= 8.4 Hz, 2H), 7.11 (d, J= 8.4 Hz, 2H), 7.34 (m, 5H), 7.59 (d, J= 8.0 Hz, 1H), 8.86 (s, 1H).
MS (ESI+): 1086.68 ([M+H]+)。
【0084】
13の合成
化合物12 (355 mg, 0.327 mmol)をメタノール(40 mL)に加えた。10% Pd/C (13 mg)を加え、水素置換した後、室温で28時間攪拌した。溶液をろ過した後、溶媒を減圧留去し化合物13を得た。295 mg。収率95%
1H NMR (400 MHz, CDCl3) δ 0.86-0.99 (m, 9H), 1.31 (s, 9H), 1.46 (s, 27H), 2.09-3.49 (m, 28H), 4.06 (m, 1H), 4.68 (m, 1H), 6.92 (d, J= 8.4 Hz, 2H), 7.15 (d, J= 8.4 Hz, 2H), 7.36 (d, J= 8.4 Hz, 2H), 7.48 (d, J= 8.4 Hz, 2H), 7.98 (d, J= 8.0 Hz, 1H), 8.61 (s, 1H).
MS (ESI+): 952.63 ([M+H]+)。
【0085】
14の合成
化合物13 (275 mg, 0.289 mmol)をDMF (10 mL)に加えた後、succinic anhydride (31.8 mg, 0.318 mmol)とDIEAを加えた後室温で7時間攪拌した。溶媒を減圧留去した後、シリカゲルカラムクロマトグラフィーで精製し、化合物14を得た。176 mg収率58%
1H NMR (400 MHz, CDCl3) δ 0.83-0.96 (m, 9H), 1.30 (s, 9H), 1.47 (s, 27H), 1.62-3.90 (m, 28H), 4.13 (m, 1H), 4.75 (m, 1H), 6.86 (d, J= 8.6 Hz, 2H), 6.92 (d, J= 8.6 Hz, 2H), 7.50 (d, J= 8.4 Hz, 2H), 7.62 (d, 1H), 7.86(d, J= 8.4 Hz, 2H), 7.92 (d, J= 8.4 Hz, 2H), 8.55 (s, 1H), 9.17 (s, 1H)
MS (ESI+): 1052.67 ([M+H]+)。
【0086】
Suc-LY-ABDの合成
化合物14 (25.0 mg, 0.0238 mmol)をTFA (2 mL)に加え、室温で24時間攪拌した。溶媒を減圧留去した後、逆相HPLCで精製を行い化合物Suc-LY-ABDを得た。14.0 mg収率71 %。
1H NMR (400 MHz, CD3OD) δ 0.85 (d, J= 6.6 Hz, 3H), 0.90 (d, J= 6.6 Hz, 3H), 1.39 (m, 2H), 1.43 (m, 1H), 1.94 (s, 3H), 2.92-3.53 (m, 30H), 4.17 (m, 1H), 4.58 (m, 1H), 6.69 (d, J= 8.8 Hz, 2H), 7.09 (d, J= 8.8 Hz, 2H), 7.47 (d, J= 8.6 Hz, 2H), 7.70 (d, J= 8.6 Hz, 2H).
13C NMR (100 MHz, CD3OD) δ 21.88, 22.51, 23.37, 25.82, 32.17, 32.22, 37.58, 41.22, 50.35, 51.79, 52.94, 54.35, 57.14, 57.55, 58.59, 116.16, 116.44, 121.70, 129.28, 131.19, 132.37, 139.79, 157.12, 171.85, 172.77, 174.81, 176.42, 177.52, 178.41.
HRMS (ESI-): 826.4012 (Calcd for [M-H]-: 826.3987)。
【0087】
Suc-LY-ABD-Tbの合成
Suc-LY-ABDをメタノールに溶解した後、Tb(NO3)3・6H2OとNa2CO3を加え、室温で1時間攪拌した。溶媒を減圧留去した後、ゲルろ過を行い、Suc-LY-Abd-Tbを得た。
HRMS (FAB+) m/z: 984.3154 (Calcd for [M+H]+: 984.3162)。
【0088】
4.p-aminobenzamide-DO3A(pABAD)誘導体の合成
スキーム6:pABAD-Lnの合成
【0089】
【化19】
【0090】
15の合成
p-ニトロ安息香酸 100 mg (0.599 mmol)、HBTU 272 mg (0.719 mmol)、 HOBt・H2O 97.1 mg (0.719mmol)、化合物5 400 mg (0.719 mmol) を25mLのDMFに溶かし、ジイソプロピルエチルアミン125 μL (0.719 mmol) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗浄した。その後カラムクロマトグラフィーで精製を行い15を得た。194 mg、収率46%。
1H-NMR(400 MHz, CDCl3) δ 1.44 (s, 27H), 2.34-3.83 (m, 28H), 8.27 (d, J = 8.0 Hz, 2H), 8.43 (d, J = 8.0 Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 707.43, Found: 707.46。
【0091】
16の合成
化合物15 160 mg (0.227 mmol)を50 mLのMeOHにとかし、Pd/C 8mgを加えた後、室温水素雰囲気下で6時間激しく撹拌を行った。Pd/Cをろ過後、ろ液を減圧留去し、16を得た。110 mg、収率 72 %。
1H-NMR(400 MHz, CDCl3) δ 1.43 (s, 27H), 2.27-3.79 (m, 28H), 7.33 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 677.45, Found: 677.45。
【0092】
pABADの合成
化合物16 40 mg (0.0592 mmol)を3 mLのTFAにとかし、12時間攪拌した。その後、溶媒を減圧留去し、残渣を逆層HPLCによって精製しpABADを得た。9.9 mg、収率 33 %。
1H-NMR(400 MHz, D2O) δ2.67-3.89 (m, 28H), 7.35 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4Hz, 2H).
13C-NMR (100MHz, CD3OD) δ 36.04, 52.67, 54.03, 56.20, 114.82, 121.92, 130.22, 153.22, 162.49, 170.80.
MS (FAB+) m/z calcd for [M+H]+: 509.2645, Found: 509.2720。
【0093】
17の合成
化合物16 50 mg (0.0740 mmol)を10 mLのTHFにとかし、acetic anhydride 14 mL (0.148 mmol)とpyridine (0.185 mmol)を加え、アルゴン雰囲気下、室温で12時間攪拌した。その後、溶媒を減圧蒸留で留去し、シリカゲルカラムクロマトグラフィーにより精製を行った。34.5 mg、収率 65 %。
1H-NMR (400 MHz, CDCl3) δ 1.43 (s, 27H), 2.02 (s, 3H),2.99-3.95 (m, 28H), 7.35 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H).
MS (ESI+) m/z calcd for [M+H]+: 719.46, Found: 719.37。
【0094】
Ac-pABADの合成
化合物17 より化合物pABADと同様の方法で化合物Ac-pABADを合成した。10 mg、収率 44 %。
1H-NMR(400 MHz, D2O) δ2.02 (s, 3H),2.92-3.87 (m, 28H), 7.33 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H).
13C-NMR (100MHz, CD3OD) δ 17.38, 41.04, 50.32, 51.37, 57.60, 121.22, 128.83, 142.63, 169.67, 174.57.
MS (FAB+) m/e calcd for [M+H]+: 551.2751, Found: 551.2825。
【0095】
5. LAPを標的としたpABAD-Tbプローブ
pABD-Tbで示したのと同様に、pABAD-Tbを基本骨格として有するプロテアーゼ活性検出発光プローブ開発を行った。標的とするプロテアーゼとして、pABD-Tbで用いたのと同様にLAP (leucine aminopeptidase)を選択した。
【0096】
具体的なプローブのデザインをスキーム7に示した。LAPの基質配列であるL-Leucineをアンテナ部分にアミド結合でつなげた錯体Leu-pABAD-Tb3+からLAPによってLeuが切り出されると、アンテナ部分がアニリンの錯体pABAD-Tb3+と変化する。アンテナ部分がアシルアミノベンジル基の時では蛍光は弱く、アミノベンジル基の時に強く蛍光を発することが確かめられている。このことから、このプローブは反応前後で大きな発光強度の変化を示すことが予想された。合成はスキーム8に従って行った。
【0097】
スキーム7
【0098】
【化20】
【0099】
スキーム8
【0100】
【化21】
【0101】
Leu-pABADの合成
pABAD 60 mg (0.089mmol)、HBTU 272 mg (0.133 mmol)、 HOBt・H2O 97.1 mg (0.133 mmol)、Boc-Leu・H2O 43.5 mg (0.133 mmol) を25mLのDMFに溶かし、DIEA 25 μL (0.133 mmol) を加え室温で12時間攪拌した。溶媒を減圧蒸留で留去し、酢酸エチルを加え、NaHCO3水溶液、H2Oで3回洗浄し、溶媒を減圧蒸留で留去し18を得た。18は精製を行わずにTFAに溶かし、12時間攪拌した。TFAを減圧留去した後、HPLCで精製を行いLeu-pABADを得た。4.6 mg、収率8.3%。
1H-NMR(400 MHz, D2O) δ0.84 (s, 6H), 1.68-1.72 (m, 3H), 2.92-3.87 (m, 29H), 7.44 (d, J = 8.4Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H).
13C-NMR (100MHz, CD3OD) δ21.93, 23.29, 36.52, 41.68, 50.70, 51.16, 53.81, 54.71, 120.51, 120.51, 124.54, 129.56, 142.55, 162.70, 169.31, 172.67.
MS (ESI+) m/z calcd for [M+H]+: 620.3486, Found: 620.2712。
【0102】
実施例2:測定
測定は、以下の条件下に行った。
【0103】
測定機器
紫外可視分光光度計UV-1650PC (Shimadzu)
蛍光光度計 F4500 (Hitachi)
時間分解蛍光測定 低温りん光測定 発光寿命測定 SPEX (Horiba)。
【0104】
紫外吸収スペクトル
サンプル10 mM の100 mM HEPES buffer 溶液を測定。測定は1 cm石英セルを25 ℃で測定した。
【0105】
蛍光測定
サンプル10 mM の100 mM HEPES buffer 溶液を測定。測定は1 cm石英セルを25 ℃で測定した。励起光スリット5 nm、発光スリット 5 nm。フォトマル電圧 700 Vで測定した。
【0106】
量子収率測定
量子収率は相対法を用いて算出した。標準物質には1N H2SO4 に溶かした硫酸キニーネを用い、その量子収率をfst = 0.546と参照とした。測定はF4500 (Hitachi) にて行った。
【0107】
発光寿命測定
すべてのサンプルは100 mM HEPES buffer (pH = 7.4) 中で測定した。 データはsingle exponentialで以下の式でフィッティングを行い、t の値を発光寿命とした。
【0108】
【数1】
【0109】
測定はSPEX (Horiba)にて行った。
【0110】
・ABD誘導体の分光学的特性
各化合物のTbおよびEu錯体において、中性水系緩衝液中で吸光スペクトルを測定したところ図1のようになった。次に励起および蛍光スペクトルを測定したところ, Ac-ABD-TbよりABD-Tbの法が25倍以上も発効強度が高いことがわかった(図2)。この結果はプロテアーゼによる脱アシル化によって発光強度が増加する可能性を強く示唆するものである。
【0111】
発光寿命測定
Ac-ABD-Tb錯体、ABD-Tb錯体において中性水系緩衝液中で発光寿命測定を行ったところ図3のようになった。グラフを、
【0112】
【数2】
【0113】
という式でフィッティングして発光寿命(τ)を求めると、ABD-Tbはτ =1.47 ms、Ac-ABD-Tbはτ =1.49 msと求まり、二つの錯体の値は大差ないものとなった。
【0114】
・pABAD誘導体の分光学的特性
各化合物のTb錯体において、中性水系緩衝液中で吸光スペクトルを測定したところ図4のようになった。pABAD-Tb3+ではアンテナのアミノ基をアセチル化したAc-pABAD-Tb3+より長波長側にシフトした。また、2種の化合物はモル吸光係数がTD-Tb3+、SD-Tb3+よりも高いことも確認され、Tb3+由来の蛍光強度が高くなる可能性があることも示唆された。
【0115】
次に蛍光・励起スペクトルを測定したところ, Ac-pABAD-Tb3+よりpABAD-Tb3+の方が3倍以上も蛍光強度が高いことがわかった(図5)。この結果はプロテアーゼの反応による脱アシル化によって蛍光強度が増加する可能性を強く示唆するものである。
【0116】
・ pABAD誘導体の蛍光寿命測定
Ac-pABAD-Tb3+錯体、pABAD-Tb3+錯体において中性水系緩衝液中で発光寿命測定を行ったところ図6のようになった。両者において希土類金属に特徴的な長寿命な蛍光が得られ、蛍光強度の大きかったpABAD-Tb3+の方が発光強度の小さいAc-pABAD-Tb3+より蛍光寿命が長いことがわかった。
【0117】
・ pABAD-Tb3+の時間分解測定
5 μMのpABAD-Tb3+ (100 mM HEPES Buffer, pH 7.4中)を実際に時間分解蛍光測定によって計測し、選択的に希土類イオンのみが観測できるかを調べた。その結果を図7に示す。示したスペクトルはdelaytime 30 μsのスペクトルである。これより、pABAD-Tb3+はTb3+由来の発光のみが選択的に観測できていることがわかる。このことから蛍光ノイズの大きい生体試料などでこの方法を用いることで高いS/N比での観察が可能になることが明らかにされた。
【0118】
・酵素反応(I)(Leu-ABD-TbによるLAP活性の検出)
Leu-ABD-Tbの酵素との反応性について検討した。中性HEPES緩衝液中、5 μM のLeu-ABD-TbにLAP (from hog kidney) 0.1 Uを添加したところ、図8(a)のような蛍光スペクトルの変化が観測された。反応前後の蛍光強度変化は非常に大きく、肉眼でも捉えられる程であった(図8(c))。波長545 nmの発光強度を時間に対してプロットすると図8(b)のように約10分で飽和に達した。
【0119】
・酵素反応(II)(Suc-LY-ABD-Tbによるカルパイン活性の検出)
中性HEPES緩衝液中、10 μM のSuc-LY-ABD-Tb3+にカルパイン 90 μg/mLを添加したところ、図9 (a)のように、時間経過による蛍光強度の増加が観測された。次に、Delaytime 60 μsec、Gatetime 2.0 msecの条件で酵素反応を時間分解測定で追跡した場合でも、時間経過による蛍光強度の増加が観測され(図9(b))、また、図9(a)で示した通常の蛍光スペクトルと比較によりアンテナ分子の蛍光スペクトルが消えていることから、Suc-LY-ABD-Tb3+を用いることで、LAPの活性測定を時間分解測定によって行えることが明らかになった。
【0120】
・酵素反応(III)(Leu-pABAD-TbによるLAP活性の検出)
次に酵素との反応性について検討した。中性HEPES緩衝液中、5 μM のLeu-pABAD-TbにLAP (from hog kidney) 0.04 Uを添加したところ、図10 (a)のように、時間経過による蛍光強度の増加が観測された。次に、遅延時間30 μsec、ゲート時間0.45 msecの条件で酵素反応を時間分解測定で追跡した場合でも、時間経過による蛍光強度の増加が観測され(図10(b))、また、図10(c)で示した通常の蛍光スペクトルと比較によりアンテナ分子の蛍光スペクトルが消えていることから、Leu-pABAD-Tb3+を用いることで、LAPの活性測定を時間分解測定によって行えることが明らかになった。
【図面の簡単な説明】
【0121】
【図1】Absorption spectra of in 0.1 M HEPES buffer (pH 7.4)中、25 ℃で、100 μM の(a)ABD-Tb及びAc-ABD-Tb (b)ABD-Eu及びAc-ABD-Euの吸収スペクトル
【図2】0.1 M HEPES buffer (pH 7.4)中、25℃、10μMの(a)ABD-Tb (b)Ac-ABD-Tbの、250nmで励起し、545nmでモニターされた励起スペクトル及び発光スペクトル。
【図3】0.1 M HEPES buffer (pH 7.4)中、25 ℃、10 μM (a)ABD-Tb (b)Ac-ABD-Tbの励起状態の寿命。250nmで励起、545nmでモニター。
【図4】pABAD-Tb3+(赤)およびAc-pABAD-Tb3+(青)の吸収スペクトル。
【図5】(a) Ac-pABAD-Tb3+、(b) pABAD-Tb3+の励起(赤)および蛍光(青)スペクトル
【図6】(a) pABAD-Tb3+, (b) Ac-pABAD-Tb3+の蛍光寿命測定
【図7】pABAD-Tb3+の時間分解蛍光スペクトル。
【図8】(a) Reaction of Leu-ABD-TbとLAPの反応。LAP (0.1 U)の添加0, 2, 4, 6, 8, 10分後の5 μM Leu-ABD-Tbの発光スペクトル。反応は、100 mM HEPES buffer (pH 7.4)、37 oCで行った。励起波長は250nm。(b)545 nm (ex. 250 nm)の蛍光強度をプロットした。(c) 酵素反応の前(左)及び60分後(右)のLeu-ABD-Tb (5 μM) の蛍光強度の比較。サンプルは、UVランプ(254 nm)で照射した。
【図9】カルパインとSuc-LY-ABD-Tbの反応。カルパイン90 μg/mLの添加後0, 10, 20, 30, 40, 50, 60, 70, 80, 90分での10 μM のSuc-LY-ABD-Tbの発光スペクトル。反応は、2.5 mM 2-メルカプトエタノール, 5 mM CaCl2, 1 mM EDTA, 1mM EGTAを含む100 mM HEPES buffer (pH 7.4)中、30℃で行った。(a)は、250 nmで励起された発光スペクトル。(b)は、250 nmで励起された時間分解発光スペクトル (遅延時間: 60 μs. ゲート時間: 2.0 ms.)。
【図10】(a)Leu-pABAD-TbとLAPの反応。LAP (0.04 U)の添加後0, 1, 2, 3, 4, 6, 8, 10, 12, 14分での5 μM Leu-pABAD-Tb3+の発光スペクトル。100 mM HEPES buffer (pH 7.4)中、37 oCで反応を行った。励起波長は293nmであった。(b) Leu-pABAD-TbとLAPの反応。LAP (0.005 U) の添加後0, 10, 20, 30, 40分での5 μM Leu-ABD-Tb3+の発光スペクトル。時間分解測定は、遅延時間30 μsおよびゲート時間0.1 msで行った。(c) Leu- pABAD-Tb3+とLAPの反応。LAP (0.005 U)の添加後12時間での5 μM Leu- pABAD-Tb3+ の発光スペクトル。
【特許請求の範囲】
【請求項1】
一般式(I)
【化1】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
【請求項2】
前記アミン系配位子が、4個のアミノ基と3個のカルボキシル基、あるいは3個のアミノ基と4個のカルボキシル基を有する7配位の配位子である、請求項1に記載の化合物。
【請求項3】
前記希土類錯体のリガンドである「R−CONH−Ar−R1−(アミン系配位子)」が、下記式(IIA)、(IIB)のいずれかで表される、請求項1又は2に記載の化合物:
【化2】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
【請求項4】
希土類が、Tb,Sm,Eu、DyおよびNdからなる群から選ばれる、請求項1〜3のいずれかに記載の錯体。
【請求項5】
Arが−C6H4−であり、R1が−CH2−である、請求項1〜4のいずれかに記載の錯体。
【請求項6】
「アミン系配位子−希土類錯体」が以下の構造を有する、請求項1〜5のいずれかに記載の化合物:
【化3】
(式中、Lnはランタノイドを示す。)
【請求項7】
下記式
【化4】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。R−COは、アミノ酸もしくはペプチド基を示す。Lnは、ランタノイドを示す。)
で表される請求項1〜6のいずれかに記載の化合物。
【請求項8】
R−COが、加水分解酵素の認識配列を含む、請求項1〜7のいずれかに記載の化合物。
【請求項9】
請求項1〜8のいずれかに記載の化合物に、加水分解酵素阻害の候補物質の存在下で加水分解酵素を作用させ、前記候補物質の加水分解酵素の阻害作用を、前記化合物のペプチドの切断による発光を用いてスクリーニングすることを特徴とする、加水分解酵素阻害剤のスクリーニング方法。
【請求項10】
請求項1〜8のいずれかに記載の化合物を含む、希土類発光プローブ。
【請求項11】
下記式(IIC)で表される化合物:
【化5】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。)
【請求項1】
一般式(I)
【化1】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
で表される化合物。
【請求項2】
前記アミン系配位子が、4個のアミノ基と3個のカルボキシル基、あるいは3個のアミノ基と4個のカルボキシル基を有する7配位の配位子である、請求項1に記載の化合物。
【請求項3】
前記希土類錯体のリガンドである「R−CONH−Ar−R1−(アミン系配位子)」が、下記式(IIA)、(IIB)のいずれかで表される、請求項1又は2に記載の化合物:
【化2】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。「アミン系配位子」は、希土類に対し、配位可能なアミノ基を3個以上、カルボキシル基及びホスホン酸基からなる群から選ばれる配位可能な酸性基を1個以上含み、かつ、5配位から9配位のいずれかの配位数を有する配位子である。R−COは、アミノ酸もしくはペプチド基を示す。)
【請求項4】
希土類が、Tb,Sm,Eu、DyおよびNdからなる群から選ばれる、請求項1〜3のいずれかに記載の錯体。
【請求項5】
Arが−C6H4−であり、R1が−CH2−である、請求項1〜4のいずれかに記載の錯体。
【請求項6】
「アミン系配位子−希土類錯体」が以下の構造を有する、請求項1〜5のいずれかに記載の化合物:
【化3】
(式中、Lnはランタノイドを示す。)
【請求項7】
下記式
【化4】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。R−COは、アミノ酸もしくはペプチド基を示す。Lnは、ランタノイドを示す。)
で表される請求項1〜6のいずれかに記載の化合物。
【請求項8】
R−COが、加水分解酵素の認識配列を含む、請求項1〜7のいずれかに記載の化合物。
【請求項9】
請求項1〜8のいずれかに記載の化合物に、加水分解酵素阻害の候補物質の存在下で加水分解酵素を作用させ、前記候補物質の加水分解酵素の阻害作用を、前記化合物のペプチドの切断による発光を用いてスクリーニングすることを特徴とする、加水分解酵素阻害剤のスクリーニング方法。
【請求項10】
請求項1〜8のいずれかに記載の化合物を含む、希土類発光プローブ。
【請求項11】
下記式(IIC)で表される化合物:
【化5】
(式中、R1はR−CONH−Arとアミン系配位子を連結する2価の連結基を示す。Arは置換されていてもよいアリーレン基、置換されていてもよいヘテロアリーレン基、置換されていてもよいアラルキレン基または置換されていてもよいヘテロアラルキレン基を示す。)
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】

【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2009−215238(P2009−215238A)
【公開日】平成21年9月24日(2009.9.24)
【国際特許分類】
【出願番号】特願2008−61320(P2008−61320)
【出願日】平成20年3月11日(2008.3.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度、文部科学省、科学技術総合研究による委託研究「科学技術連携施策群の効果的・効率的な推進生体内分子を可視化するナノセンサ分子開発」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504176911)国立大学法人大阪大学 (1,536)
【Fターム(参考)】
【公開日】平成21年9月24日(2009.9.24)
【国際特許分類】
【出願日】平成20年3月11日(2008.3.11)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度、文部科学省、科学技術総合研究による委託研究「科学技術連携施策群の効果的・効率的な推進生体内分子を可視化するナノセンサ分子開発」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504176911)国立大学法人大阪大学 (1,536)
【Fターム(参考)】
[ Back to top ]