
物質を血液−脳関門を渡って輸送するための人工低密度リポタンパク質キャリア
【課題】治療剤を血液−脳関門(BBB)を渡ってターゲットにデリバーするための高効率の人工低密度リポタンパク質(LDL)キャリアシステムの提供。
【解決手段】3つの脂質エレメント:ホスファチジルコリン、脂肪酸−アシル−コレステロールエステル、および少なくとも一のアポリポタンパク質を含む人工LDL粒子の提供。更に、脳疾患を予防および治療するために薬剤をBBBへ向けBBBを通過させるための人工LDL粒子を含む組成物、方法およびキット。
【解決手段】3つの脂質エレメント:ホスファチジルコリン、脂肪酸−アシル−コレステロールエステル、および少なくとも一のアポリポタンパク質を含む人工LDL粒子の提供。更に、脳疾患を予防および治療するために薬剤をBBBへ向けBBBを通過させるための人工LDL粒子を含む組成物、方法およびキット。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2002年12月3日に出願された米国仮出願シリアル番号60/430,476の優先権を主張する。
【0002】
本発明は、物質を効率的にターゲットにし血液−脳関門(BBB)を渡ってデリバーする人工低密度リポタンパク質(LDL)粒子の使用に関する。更に本発明は、種々の脳疾患を予防および治療するための人工LDL粒子の組成物、方法、およびキットを提供する。
【背景技術】
【0003】
血液−脳関門(BBB)は、循環する毒素に対して脳を効果的に保護するが、アルツハイマー病、パーキンソン病、および脳腫瘍などの脳疾患を薬理学的に治療する際に大きな難題を引き起こす。多くの荷電分子、およびサイズ700ダルトンを超える多くの分子は、その関門を通過することができず、より小さい分子は、肝臓でコンジュゲートされ得る。これらの要因が、アルツハイマー病、パーキンソン病、細菌およびウイルス感染、および癌などの脳および中枢神経系(CNS)の疾患を薬理学的に治療する際に大きな難題を引き起こす。
【0004】
脳およびCNSの疾患および障害を治療するための多くの治療剤は、充分に親水性であり、BBBを渡る直接的な輸送を阻まれる。更にこれら薬剤および作用物質は、血液および末梢組織での分解を受けやすく、治療上効果的な血清濃度を達成するのに必要な用量を増大させる。本発明は、人工低密度リポタンパク質粒子(LDL)に治療剤をカプセル化することにより、BBBへ治療剤をデリバーする方法を提供する。本発明のLDLは、トランスサイトーシスによりBBBを渡る治療剤の輸送を促進する。親水性であるためにBBBを通過することができない多くの薬剤および治療剤は、親水性であるためにLDL粒子に組み込むことができず、そのため本発明は、LDL粒子への組み込み、BBBを渡る輸送、およびその後の治療剤の細胞への放出を促進するLDL構成成分と治療剤とのコンジュゲートを製造するための方法を提供する。
【0005】
BBBを渡って薬剤をデリバーするための従来の方法は、3つの一般的なカテゴリーを含む:(1)治療剤がキャリア内にカプセル化されているリポソームベースの方法;(2)合成ポリマーを用いて粒子を作成し、精密に規定されたサイズ特性を達成する合成ポリマーベースの方法;および(3)治療剤がインスリンなどのキャリアに共有結合している、キャリアの薬剤への直接コンジュゲーション。
【0006】
A.リポソーム
リポソームは、リン脂質を水溶液中で音波処理したときに自然に形成される小粒子であり、水性環境を取り囲む中空球として構成されるシンメトリー脂質二重層から成る。そのリン脂質は形質膜に吸収され、これによりリポソームの内容物が自動的にサイトゾルへ放出されるため、リポソームは、水溶性薬剤を細胞膜を通過させて輸送する手段として魅力を有する。この技術の成功したバリエーションには、陽イオン性脂質の使用が挙げられ、これは、膜にナノ孔を協同的に作り出すことができる。陽イオン性脂質は、細胞培養において広く使用され、DNA分子などの水溶性物質を実験のために培養細胞に導入する。
【0007】
リポソームは、その大きな運搬能力のために、BBBを渡って薬剤を輸送するのに魅力的である。しかし、リポソームは、一般に大きすぎてBBBを効率よく通過することができず、本質的に不安定であり、その構成脂質は、血漿において脂質結合タンパク質による吸収により徐々に失われる。たとえば、幾つかの研究において、大きなサイズのリポソームを使用すると、ミクロ塞栓症を引き起こし、これが脳への取込みの誤った印象を与えた。幾つかの研究において、リポソームは、安定化剤として、BBBを破壊可能な洗浄剤、ポリソルベート80と共に注入した。これらの研究においてポリソルベート80によるBBBの破壊は、観察されるBBBを渡る輸送すべてに貢献し得る。
【0008】
その結果、リポソームは、BBBを渡って薬剤を輸送するための輸送手段(vehicle)として変化に富んだ経歴がある。リポソームを特定の細胞ターゲットに向かわせるために幾つかの試みが為されている。BBBの内在性レセプターをターゲットとするペプチド様mAbsを使用して、レセプター介在性の取込みを達成する目的で、種々のBBBレセプターにPEG化(pegylated)イムノリポソームを向ける。しかし、このアプローチは、費用のかかるモノクローナル抗体の作成、試験および政府の認可が必要である。mAbsは、典型的にマウスで作成し、分解を受けやすいため、ペプチド様mAbの採用は、重大な規制の障害に直面するだけでなく、患者環境において活用することが難しいことが分かる。
【0009】
イムノリポソームは、たとえば、リポソーム表面にモノクローナル抗体(mAbs)を共有結合させることを含むプロセスで構築されている。これらイムノリポソームは、mAbコンジュゲートリポソームを強力に破壊するシステムである細網内皮系(RES)による取込みを誘発する血漿タンパク質で直に被覆されているため、イムノリポソームは、PEG化(pegylation)として知られているプロセスにおいて、ポリエチレングリコール(PEG)で処理される。残念なことに、PEG分子はmAbを妨害し、立体的妨害によりそれらを非特異的にする。Huwyler et al. (1996) Proc. Nat'l. Acad. Sci USA 93: 14164-14169は、PEGテイルの先端にマレイミド部分を備え、これによりチオール化(thiolated)mAbとコンジュゲート可能なイムノリポソームを作成することにより、この問題を回避した。内部にダウノマイシンを備えて調製されるこれらPEG化OX26イムノリポソームは、遊離の治療剤または単純な非PEG化リポソームより血漿中で安定であることが示された。しかし、共焦点顕微鏡により、リポソームはラットの脳毛細血管にエンドサイトースされるが、脳細胞に到達せず内皮細胞に結合したままであることが示された。よって、PEG化されマレイミド処理されたリポソームは、薬剤デリバリー輸送手段としてどちらかといえば効果的でないようである。
【0010】
1997年にDehouck et al.は、ApoEを結合するLDLレセプターが、BBBを渡るLDLのトランスサイトーシスに関与することを発見した。一連の3つの文献において、Versluis et al.は、マウスにおいてダウノルビシンを癌細胞にデリバーするためにApoEリッチなリポソームを使用することを記載している。腫瘍細胞がその膜において高レベルのLDLレセプターを発現するという発見に基づいて、LDLレセプターターゲッティングタンパク質としてApoEを選択した。またVersluis et al. (1998) は、天然のLDLを使用することを提案したが、この試みは試されておらず、その後の論文は、ApoEリッチなリポソームにのみ焦点をあてている。Versluis et al. (1999) は、ダウノルビシンの組織分布を調査したが、脳の取込みに関するデータはなく、このことは、この方法がBBBを渡ってダウノルビシンを輸送するための手段として考えられていなかったことを示す。
【0011】
加えて、Versluis et al.により用いられたコンジュゲーション化学は、本発明で使用されるものとは異なる。薬剤をリポソーム膜に固定するために、著者らは、3α-O-(オレオイル)-4β-コラン酸 (リトコール酸のエステル) をテトラペプチドAla-Leu-Ala-Leuに連結し、次にこれを親水性抗腫瘍剤ダウノルビシンに共有結合した。このように、コンジュゲートされ遊離していないダウノルビシンにより腫瘍は治療された。リトコール酸は、活性化可能な酸の基を予め含有するステロイドであるが、この酸の基は、3-OH位ではなくステロイド側鎖に位置し、これにより、望ましくない特徴の反応生成物が得られる。遊離のダウノルビシンは、高酸性リソソームに見られるプロテアーゼによって切断された後にのみつくることができ、これは、コンジュゲート薬剤または作用物質を、プロテアーゼ、酸性およびその他の加水分解酵素による分解に晒すものである。その後、治療剤は、リソソーム内空間に放出され、ここで更なる分解を受け細胞から放出される。
【0012】
対照的に、本発明のコンジュゲートは、好ましくは、エステル結合を介した治療剤の結合を提供し、これは、細胞質ゾルで容易に切断され、その結果、Versluis et al.の方法に必要とされる過酷なリソソーム状態を避けることができる。よって、本発明の方法によりコンジュゲートされる治療剤は、そのターゲットへの道のりを生き残りやすく、ターゲットにおいて効率的な手法で放出される可能性が高い。また、本発明の方法によりコンジュゲートされる治療剤は、リポソームよりもBBBを渡って輸送されやすい。
【0013】
また、Versluis et al.の方法は、テトラペプチドを合成するために多数の固相ペプチド化学の工程が必要であり、それをFMOCとコンジュゲートし、そのコンジュゲートをリトコール酸と反応させ最後に薬剤と反応させるために幾つかの追加の工程が必要である。本発明は、ずっと少ない工程数を使用し、その各々によりほぼ定量的な収率が得られる。このように本発明は、高効率と低コストを提供する。
【0014】
デオキシルビシンの他のリポソーム製剤が、現在、見込みのある癌治療剤として臨床で使用されているが、LDLを使用した製品は売り出されていない。
【0015】
Demeule et al.は、タンパク質メラノトランスフェリン(p97)がBBBを渡ってトランスサイトーシスにより輸送されることを見出し、LDLレセプターが関与していると断定し、これにより、このタンパク質が薬剤デリバリーシステムとして採用されることが示唆された。
【0016】
B.合成ポリマー
ポリソルベート80で被覆したポリ(ブチルシアノアクリレート)またはポリアクリルアミドなどの合成ポリマーも試験してみた。これらポリマーは、その粒子が充分親水性で水に可溶であり、更に長時間にわたってその構造形態を維持することができ、これにより、天然の解毒プロセスを受ける肝臓および腎臓への取込みから治療剤は保護されるため、魅力的である。いずれのケースにおいても、取込みは、細胞膜を渡る受動拡散により起こるか、あるいはクラスリン被覆小胞による防御的取込みとして起こると一般に想像される。その後、前者のケースにおいて治療剤は、以前ほどターゲットにさほど近くない内皮細胞にトラップされるが、後者のケースにおいて治療剤は、プロテアーゼや胃の内容物に類似した他の消化酵素を含有する細胞における高酸性コンパートメントであるリソソームに輸送される。よって後者のケースでは、治療剤は、より過酷な状況の間ずっと安定なまま留まらなければならない。いずれのケースも、薬剤は細胞を渡って運搬されず、所望の結果である脳実質組織への排出は起こらない。よって、これら二つの方法のいずれもが、さほど臨床上使用されていないことは驚くことではない。
【0017】
多くの研究者らは、上述のアプローチの種々の改変を試みたところ、ある程度の成果で、BBBを渡るキャリアの取込みは改良された。たとえば、Kreuter et al. (2002) J. Drug Target 10(4): 317-25は、BBBに位置するアポリポタンパク質レセプターに結合する種々のアポリポタンパク質を含有する合成粒子を設計した。彼らは、ポリ(ブチルシアノアクリレート)ナノ粒子に結合し、ポリソルベート80で被覆された薬剤の輸送を実証した。取込みには、ポリソルベート80、ApoEまたはApoBでのコーティングが必要であった。アポリポタンパク質All、Cll、またはJコーティングは、機能しなかった。しかし、これらナノ粒子は天然に存在しないため、望ましくない副作用があるかもしれない。アクリレートポリマーは、自己免疫応答を開始させることで特に有名であり;化学的に類縁のポリマー、ポリ(アクリルアミド)がしばしばアジュバントとして使用される。
【0018】
Alyaudin et al. (2001) J. Drug Target 9(3): 209-21は、ポリソルベート80でオーバーコートしたポリ(ブチルシアノアクリレート)ナノ粒子を使用して、BBBを渡って[3H]-ダラルジン(dalargin)を輸送し、そのプロセスがエンドサイトーシスとその後のトランスサイトーシスの一つであると推量した。このポリマーは、更に免疫学的な混乱をきたすかもしれない。
【0019】
C.治療剤コンジュゲート
BBBを渡って輸送可能な物質、たとえばインスリンと薬理学的作用物質の直接的コンジュゲーションも試みられている。インスリンおよびインスリン様増殖因子は、特殊な促進拡散システムにより血液脳関門を通過することが知られている(Reinhardt et al. (1994) Endocrinology 135(5): 1753-1761)。インスリンは、内皮インスリンレセプターにより媒介されるトランスサイトーシスにより吸収される(Pardridge et al. (1986) Ann. Intern. Med. 105(1): 82-95)。また、グルコースやトリプトファンなどの大きなアミノ酸のための特定のトランスポーターが存在する。しかし、インスリントランスポーターはその特異性が高いため、インスリンに共有結合した薬理学的作用物質を脳へ通過させることができないことが分かっている。同様の結果が、グルコースやアミノ酸コンジュゲートでも得られており、その取込みは、他の低分子量物質と同じ一般的原理に従うことが観察されており、700 Da以下の非電荷分子のみが脳への有意なアクセスを達成する。コンジュゲート形態の生体分子の化学合成を考え出す煩わしさ、予期しない毒性効果を引き起こす
リスク、および完全に新規な化合物についてFDA認可を得なければならないかもしれない必要性が、このアプローチに対する熱意を失わせる。
【0020】
陽イオン化アルブミンなどのタンパク質である輸送ベクター、またはトランスフェリンレセプターに対するOX26モノクローナル抗体は、それぞれ、吸収力介在性トランスサイトーシスおよびレセプター介在性トランスサイトーシスを受けBBBを通過する。これらは、少量の薬剤を輸送するために使用される。このプロセスは、費用が高く、モノクローナル抗体や陽イオン化アルブミンを作成する難しさがあり、他のタイプの分子に適用することができない。また、陽イオン化タンパク質は、その免疫原性や腎臓で沈殿する免疫複合体の形成のために毒性があることが示されている。
【0021】
Wu et al. (2002) J. Drug Target 10(3): 239-45は、ストレプトアビジン(SA)とラットトランスフェリンレセプターに対するマウスOX26モノクローナル抗体と、ベクターに結合したビオチン化bFGF(bio-bFGF)のコンジュゲートから成り、bio-bFGF/OX26-SAと称される薬剤デリバリーベクターを用いて、ヒト塩基性繊維芽細胞増殖因子(bFGF)、タンパク質神経保護剤がBBBを渡って輸送されることを示した。彼らは、[125I]標識bio-bFGFの末梢器官への強力な取込みを示したが、脳1グラムにつき注入量の0.01%しか吸収されなかった。また、この手段は、薬剤の共有結合修飾を必要とするため、限られたクラスの薬剤についてのみ有効である。また、キャリアが構成成分としてマウスモノクローナル抗体を含有するため、これが、患者に免疫応答を引き起こすであろう。
【0022】
また、Kang et al. (2000) J. Drug Target 8(6): 425-34は、BBBを渡ってペプチドを輸送するためにアビジン−ビオチン連結キメラペプチドを使用したが、組織1グラムにつき注入量の0.12%しか吸収されなかった。Kang and Pardridge (Pharm. Res. 11: 1257-1264) は、陽イオン化ヒト血清アルブミンとニュートラルライトアビジンとをコンジュゲートし、それを放射性標識ビオチンに結合させた。ビオチン/cHSA/NLA複合体は、血液中で24時間まで安定であったが、そのコンジュゲートは脳で選択的に分解され、遊離のビオチンを放出した。上述のとおり、陽イオン化タンパク質は、その免疫原性のために毒性があることが示されている。
【0023】
また、陽イオン化モノクローナル抗体(mAbs)が使用されている。Pardridge (J. Neur
ochem. 70: 1781-2) は、天然のヒト化4D5 MAbは、そのタンパク質を陽イオン化した後のみ、吸収力介在性トランスサイトーシスによりBBBを通過することを共焦点顕微鏡により示した。しかし、このプロセスは、モノクローナル抗体を作成し化学的に修飾する費用が高く、他のタイプの分子に適用することができない。
【0024】
Witt et al. (2000) J. Pharmacol. Exp. Ther. 295(3): 972-8 は、デルタ−オピオイドレセプター−選択的ペプチドD-ペニシラミン(DPDPE)、Met-エンケファリン類似体を、BBBを渡って輸送するためにインスリンを使用した。しかし、インスリンは、治療ストラテジーとしてのその使用を制限する多くの危険を提示する。また、他の研究者らは、インスリンレセプターが極めて選択的であることを見出した。よって、キメラペプチドを作成する難しさに加えて、このストラテジーは、狭いクラスの薬学的作用物質に限定される。
【0025】
他の研究者らは、たとえばグリコペプチドを用いて薬剤をグルコースにコンジュゲートすることを試みた。しかし、BBB Glut1トランスポーターを介したグリコペプチドの有意な輸送はこれまで実証されていない。Glut1グルコーストランスポーター、コリントランスポーター、またはLAT1大アミノ酸トランスポーターなどのキャリア介在性トランスポーターの高い輸送速度を使用する試みは、キャリアトランスポーターが非常に選択的であるためコンジュゲート物質を受け入れることができないという問題で失敗した。また、これら試みは、多剤耐性遺伝子のメンバーであるp-糖タンパク質が、関門を通過させたい薬剤などの多くの低分子を活発に脳から除去するために迅速に機能するという問題がある。
【0026】
LDLレセプターに加えて、BBBは、タイプIIスカベンジャーレセプター(SR)を含有し、これは高いアフィニティーでLDLを結合する。このスカベンジャーレセプターは、アセチル化LDLなどの修飾型LDLととりわけ親和性がある。SRへの結合により、エンドサイトーシスのみが起こり、所望のトランスサイトーシスは起こらない。Rigotti et al. (1995) J. Biol. Chem. 270: 16221-4は、アセチル化LDLはBBBを渡って輸送されないが、陽イオン化ウシIgGが更に効果的であることを見出した Bickel et al. (1993) Adv. Drug. Del. Rev. 10: 205-245。アセチル化LDLを用いてトランスサイトーシスを実証できなかったことにより、LDLを用いて更なる実験を試みる気力を多くの研究者から奪った。
【0027】
Protter et al. (WO 87/02061) は、薬学的作用物質または作用物質含有キャリアに共有結合したApoEおよびApoBなどのアポリポタンパク質に由来するペプチドを使用した薬剤デリバリーシステムを記載している。しかし、分子コンジュゲートの使用は、少数の薬剤クラスにのみ限定され、上述したのと同じ問題を多く有する。
【0028】
Muller et al. (US特許第6,288,040号) は、ApoE分子が共有結合した合成ポリ(ブチルシアノアクリレート)粒子の使用を記載している。これら粒子の表面は、界面活性剤により、または親水性ポリマーの共有結合により更に修飾される。上述のとおり、これら粒子は天然に存在しないため、種々の望ましくない副作用があるかもしれない。
【0029】
Samain et al. (WO 92/21330) は、固体の親水性コアに共有結合し、腫瘍またはマクロファージへ物質をデリバーするためのApoBを有する、脂質を含有する合成粒子キャリアの使用を記載している。しかし彼らは、BBBを渡って薬剤をデリバーするためにかかるベクターが有用であることをまったく開示していない。
【発明の開示】
【0030】
本発明は、物質をターゲットにし血液脳関門を渡って脳へデリバーするために、低密度リポタンパク質(LDL)粒子を使用することに関する。本発明の更なる目的は、構造的に安定であり、免疫原性がなく、かつ分解、不活性化、加水分解、コンジュゲーション、および非ターゲット組織への取込みから広範囲の薬剤を保護するLDLキャリアを合成することである。本発明は、治療剤のキャリアとして使用されるLDL粒子を提供するとともに、従来の方法と比較して改善された、かかる薬剤および作用物質をBBBを渡ってデリバーする方法を提供する。リポソームとは異なり、たとえば、LDL粒子は固形粒子であるためリポソームより構造的安定性が高い。本発明の更なる目的は、広範囲の脳疾患を治療および予防するためのLDLキャリアを含む組成物、方法、およびキットを提供することである。
【0031】
本発明は、親水性治療剤をコレステロールとコンジュゲートし、コンジュゲート治療剤を本発明の人工LDL粒子に組込み易くするための方法を提供する。好ましい態様において、本発明は、コレステロールがコンジュゲートしたアドリアマイシンおよびテトラサイクリンを提供する。当該方法、得られたコレステロールコンジュゲート、およびかかるコンジュゲートの組成物は、トランスサイトーシス(これは、コレステロールや必須脂肪酸を脳に取込む手段として脳の毛細血管内皮細胞で機能するレセプター介在性プロセスである)により脳関門を渡って治療剤を輸送する目的でLDL粒子を提供する際に有用であり、これが、脳およびCNSの種々の疾患および障害の治療を促進し改良するであろう。あるいは、本発明のコレステロールコンジュゲートは、このコンジュゲートを本発明のLDL粒子に組み込むことなく、対応の治療剤をBBBを渡ってデリバーするために有効である。
【0032】
好ましい態様において、本発明のコレステロールコンジュゲートは、エステル結合を介して連結され、これが、ユビキタスな内在性エステラーゼの作用により当該コンジュゲートから治療剤の放出を可能にする。本発明のLDL粒子に本発明のコレステロールコンジュゲートを封入することにより、コレステロールコンジュゲートは、これらエステラーゼによる加水分解から保護される。
【0033】
本発明は、卵黄ホスファチジルコリン(EYPC)、コレステロールオレアート、およびアポリポタンパク質E3(ApoE3)を含む人工LDL粒子を更に提供する。構成成分の脂質は、3つの層を含有する固形粒子を形成する:コレステロール、コレステロールエステル、および活性物質から成る固体の脂質コア;ホスファチジルコリンの脂肪酸鎖の混合物から成る中間の界面層;およびリン脂質の頭部の基およびApoE3から成る表面層。
【0034】
本発明のLDL粒子は、毛細血管内皮細胞への活性物質のターゲッティングを有意に増大させ、トランスサイトーシスによる血液脳接合部を渡る輸送を促進する。また、治療剤を取り囲むタンパク質とリン脂質の構成成分は、分解や非ターゲット細胞への取込みから治療剤を保護するために機能する。
【0035】
また本発明は、人工LDL粒子により促進される作用物質の移動に基づいて、疾患、病気およびコンディションを治療する方法に関する。たとえば、本発明は、本発明の人工LDL粒子を用いて特定の作用物質を脳組織へ向けることを含む、種々の脳疾患を治療するための薬学的組成物および方法を提供する。
【0036】
本発明は、外側のリン脂質単一層と固体の脂質コアとを含む人工LDL粒子であって、外側のリン脂質単一層が少なくとも一のアポリポタンパク質を含み、固体の脂質コアが少なくとも一の治療剤を含有する人工LDL粒子を提供する。一つの態様において、少なくとも一のアポリポタンパク質は、ApoA、ApoB、ApoC、ApoD、またはApoE、または前記アポリポタンパク質の一つのアイソフォーム、またはリポタンパク質および/またはアイソフォームの組合せから成る群より選択される。好ましい態様において、少なくとも一のアポリポタンパク質はApoEである。更に好ましい態様において、少なくとも一のアポリポタンパク質は、ApoE2、ApoE3およびApoE4から成る群より選択される。また本発明は、脳組織へのLDL複合体のターゲットデリバリーを増大させる追加のターゲッティング分子または作用物質を更に含む人工LDL粒子に関する。最も好ましい態様において、少なくとも一のアポリポタンパク質はApoE3であり、ApoE3は単独であるか、または一以上のオキシステロールおよび/またはApoBおよびApoE4から成る群より選択される追加のアポリポタンパク質と組み合わせられる。
【0037】
本発明は、血液−脳関門を渡って治療剤を輸送するための人工LDL粒子を提供する。好ましい態様において、少なくとも一の治療剤は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される。別の態様において、少なくとも一の治療剤は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される作用物質とコレステロールとの間で形成されたコンジュゲートである。好ましい態様において、少なくとも一の治療剤は、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される。
【0038】
人工LDL粒子の外側のリン脂質単一層は、ホルファチジン酸、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルグリセロールなどを含むがこれらに限定されない任意のリン脂質またはリン脂質の組合せを含み得る。好ましい態様において、外側のリン脂質単一層は、ホスファチジルコリンおよび少なくとも一のアポリポタンパク質を含む。
【0039】
本発明の人工LDL粒子は、好ましくは直径約15〜50 nmの粒子である。更に好ましい態様において、人工LDL粒子は、直径約20〜30 nmである。本発明の人工LDL粒子は、好ましくは密度約1.00〜1.07 g/mLである。更に好ましい態様において、人工LDL粒子は、密度約1.02〜1.06 g/mLである。更に本発明の人工LDL粒子は、少なくとも2時間の血清安定性を有する。
【0040】
本発明は、トランスサイトーシスによりBBBを渡って輸送される人工LDL粒子を提供する。好ましい態様において、本発明の粒子は、脳に対する取込み特異性が肝臓に比べて少なくとも3倍高い。
【0041】
本発明の人工LDL粒子の固体の脂質コアは、トリアシルグリセロール、コレステロール、コレステロールエステル、脂肪酸アシルエステルなどを含むがこれらに限定されない一以上の脂質を含み得る。好ましい態様において、固体の脂質コアは、ミリステート、パルミテート、ステアレート、アラキデート、リグノセレートなどを含むがこれらに限定されない飽和脂肪酸、またはパルミトレアート、オレアート、バクセネート、リノレアート、リノレネート、アラキドネートなどを含むがこれらに限定されない不飽和脂肪酸とコレステロールがエステル化したコレステロールエステルを含む。更に好ましい態様において、固体の脂質コアは、コレステロールエステルであるコレステロールオレアートを含む。好ましい態様において、本発明の人工LDL粒子の固体の脂質コアは、アドリアマイシンまたはテトラサイクリンとコレステロールとの間で形成されたコンジュゲートである少なくとも一の治療剤を含む。好ましい態様において、コンジュゲートのコレステロールと治療剤は、エステル結合により連結される。
【0042】
また本発明は、本発明の人工LDL粒子および薬学的に許容可能なキャリアを含む、治療剤を血液−脳関門を渡ってデリバーするための組成物を提供する。
【0043】
また本発明は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される治療剤に連結されたコレステロールを含むコンジュゲートを提供する。好ましい態様において、治療剤は、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される。更に好ましい態様において、本発明のコンジュゲートの治療剤は、アドリアマイシンまたはテトラサイクリンである。最も好ましい態様において、本発明のコンジュゲートのコレステロールと治療剤は、エステル結合により連結される。本発明のコンジュゲートの各々は、本明細書に記載されるとおり、薬学的に許容可能なキャリアと組合せて、本発明の薬剤デリバリーの方法の何れかにおいて使用することができる。
【0044】
また本発明は、1)コンジュゲートまたは非コンジュゲート治療剤を含有するリン脂質を緩衝液中に懸濁する工程;2)当該溶液を音波処理し、外側のリン脂質単一層と固体の脂質コアを形成する工程;および3)少なくとも一のアポリポタンパク質を含む溶液を添加し、当該アポリポタンパク質が前記外側のリン脂質単一層に取り込まれる工程を含む、本発明の人工LDL粒子を製造する方法を提供する。好ましい態様において、本発明の方法により製造される人工LDL粒子は、直径10〜50 nmである。
【0045】
また本発明は、効果的な量の本発明の組成物の何れかを、それを必要とする哺乳類に投与することを含む、血液−脳関門を渡って物質をデリバーするための方法を提供する。
【0046】
また本発明は、本発明の組成物の何れかを含有する容器と使用説明書を含む、血液−脳関門を渡って物質をデリバーするためのキットを提供する。
【0047】
また本発明は、アルツハイマー病、パーキンソン病、細菌およびウイルス感染、および癌などの脳および中枢神経系(CNS)の疾患を治療するための医薬の製造における、本発明のコンジュゲート、人工LDL粒子、および組成物の使用を提供する。
【発明の詳細な説明】
【0048】
定義
本明細書で使用される「人工LDL粒子」の用語は、球状のリン脂質単一層と固体の脂質コアを含む構造体を意味する。
【0049】
本明細書で使用される「リポソーム」の用語は、球状の脂質二重層と水性コアを含む構造体を意味する。
【0050】
本明細書で使用される「取込み特異性」の用語は、脳および肝臓における脂質粒子(外側のリン脂質単一層にアポタンパク質が含まれないことを除いて人工LDL粒子と同じ粒子)の取込みに対する人工LDL粒子の取込みの比をいう。人工LDL粒子と脂質粒子の取込みは、本明細書に記載されるとおり、Sprague-Dawleyラットに注入してから2時間後、脳および肝臓の両方で測定する。取込み特異性は、脳における脂質粒子の取込みに対する人工LDL粒子の取込みの比を、肝臓における脂質粒子の取込みに対する人工LDL粒子の取込みの比で割ることにより計算する。
【0051】
本明細書で使用される「血清安定性」の用語は、注入された人工LDL粒子の少なくとも75%が血漿中に残存する時間の長さを意味する。
【0052】
本明細書で使用される「アポリポタンパク質」および「アポタンパク質」の用語は、リポタンパク質のリン脂質単一層と結びついたタンパク質を意味し、ApoA;ApoB;ApoC;ApoD;ApoE;および各々のアイソフォームのすべてを含むがこれらに限定されない。
【0053】
本明細書で使用される「ApoE」の用語は、ApoEの一以上のアイソフォームを意味し、ApoE2、ApoE3およびApoE4を含むがこれらに限定されない。
【0054】
本明細書で使用される「ApoB」の用語は、ApoBの一以上のアイソフォームを意味し、ApoB48およびApoB-100を含むがこれらに限定されない。
【0055】
本明細書で使用される「外側のリン脂質単一層」の用語は、リン脂質のホスフェートの頭部の基が外側に向けられ、脂肪酸アシル鎖が固体の脂質コアに向かって内側に向けられた少なくとも一のリン脂質を含む単一層を意味する。リン脂質は、ホスファチジン酸、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルグリセロールなどを含むがこれらに限定されない。
【0056】
本明細書で使用される「固体のコア」の用語は、球状のリン脂質単一層により取り囲まれた人工LDL粒子の該当部分を意味する。固体のコアは、トリアシルグリセロール、コレステロール、コレステロールエステル、脂肪酸アシルエステルなどを含むがこれらに限定されない一以上の脂質を含む。本明細書で使用される「コレステロールエステル」の用語は、ミリステート、パルミテート、ステアレート、アラキデート、リグノセレートなどを含むがこれらに限定されない飽和脂肪酸、またはパルミトレアート、オレアート、バクセネート、リノレアート、リノレネート、アラキドネートなどを含むがこれらに限定されない不飽和脂肪酸とエステル化したコレステロールをいう。
【0057】
本明細書で使用される「治療剤」の用語は、治療上有用なアミノ酸、ペプチド、タンパク質、ポリヌクレオチド、オリゴヌクレオチド、遺伝子などを含むがこれらに限定されない核酸、炭水化物、および脂質を意味する。本発明の治療剤は、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤などを含む。本発明の治療剤は、当該治療剤がターゲット組織にデリバーされたときに活性化され得る、薬剤、プロドラッグ、および前駆体を含む。
【0058】
本明細書で使用される「薬学的に許容可能なキャリア」の用語は、有効成分と組合せることができ、その組合せに従って有効成分を対象に投与するために使用することができる化学組成物または化合物を意味する。本明細書で使用される「生理学的に許容可能な」エステルまたは塩の用語は、薬学的組成物の他の任意の成分と適合可能な有効成分のエステルまたは塩の形態であって、組成物を投与する対象に対して有害でないものを意味する。また本明細書で使用される「薬学的に許容可能なキャリア」は、一以上の以下のものを含むがこれらに限定されない:賦形剤;界面活性剤;分散剤;不活性な希釈剤;顆粒化および粉状化剤;結合剤;潤滑剤;甘味剤;香味剤;着色剤;保存剤;ゼラチンなどの生理的に分解可能な組成物;水性賦形剤および溶媒;油性賦形剤および溶媒;懸濁剤;分散または湿潤剤;乳化剤、粘滑剤;緩衝剤;塩;増粘剤;充填剤;乳化剤;抗酸化剤;安定化剤;および薬学的に許容可能なポリマーまたは疎水性材料。本発明の薬学的組成物に含有され得るその他の「追加の成分」は、当該技術分野で公知であり、たとえばGenaro ed., 1985, Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa.に記載され、これは参照により本明細書の開示内容の一部とする。
【0059】
本明細書で使用される「効果的な量」は、治療応答を引き起こすのに充分な量である。
【0060】
LDL粒子およびアポタンパク質の特性
本発明は、人工LDL粒子が、トランスサイトーシスとして公知の活性レセプター介在性プロセスにより、物質を効率的にターゲットにし血液−脳関門を渡ってデリバーするという発見に関する。トランスサイトーシスは、コレステロールおよび必須脂肪酸を脳へ輸送する手段として、脳の毛細血管内皮細胞に天然に存在する。従って、本発明の人工LDLキャリアシステムは、BBBの破壊またはその他の望ましくない副作用を最小限にして、薬剤を脳へ効率的に向けデリバーする手段を提供する。
【0061】
天然のLDL粒子は、平均約22 nmの直径を有する。内部コアは、約150-200のトリグリセリド分子および1500-2000のコレステリルエステル分子から成る。粒子の表面は、約450のリン脂質分子、185分子のスフィンゴミエリン、および1分子のアポタンパク質、典型的にはApoB-100の単一層を含有する(Hevonoja et al.(2000) Biochim Biophys Acta 1488(3): 189-210)。天然のLDL分子は、約600分子の非エステルコレステロールおよび少量のリゾホスファチジルコリン、ホスファチジルエタノールアミン、ジアシルグリセロール、セラミド、およびホスファチジルイノシトール、並びに微量のその他の生物学的脂質を含んでいてもよい(Hevonoja et al.(2000) Biochim Biophys Acta 1488(3): 189-210)。ApoEなどのその他のアポタンパク質は、LDL、VLVL、およびHDLにみられるが、種々のレセプター結合特性を有する(Bradly et al.(1984) J. Biol. Chem. 259(23): 14728-35)。
【0062】
従って、LDL分子の表面は、アポタンパク質で不均一に被覆されているが、種々の物理的特性を備えた種々の領域から成る。アポタンパク質分子は、粒子の構造の完全性を維持することに貢献するとともに、肝臓、腎臓、および血液−脳関門でリポタンパク質レセプターに結合することに貢献する。アポタンパク質は、脂質構成成分の状態に依存する構造的変化を受ける(Mims et al.(1990) Biochemistry 29(28): 6639-47)。
【0063】
アポタンパク質E(ApoE)は、体全体においてコレステロール輸送および血漿リポタンパク質代謝に関与するタンパク質である。末梢細胞においてApoEは、その輸送を指示することによりコレステロールの細胞濃度に影響を及ぼす。ニューロンにおいてコレステロールレベルの変化は、アルツハイマー病で変化するのと同じ部位で微小管結合タンパク質tauのリン酸化状態に影響を及ぼす。ApoEは、3つの主なアイソフォーム:ApoE4、ApoE3、およびApoE2を有し、これらは単一のアミノ酸置換により異なる。ApoE3は、正常のアイソフォームであるが、ApoE4およびApoE2は機能不全である。ApoE2は、タイプIII高リポ蛋白血症と関連する。ApoE4は、アテローム性動脈硬化症およびアルツハイマー病のリスクの増大、認知機能の衰え、および神経突起の伸出の低下と関連する。年齢を除けば、ApoE4は、散発性アルツハイマー病の最も重要な危険因子である。ApoE4は、カルシウムに依存した毒性効果があるかもしれないが(Veinbergs et al.(2002) J. Neurosci. Res. 67(3): 379-87)、その主要な効果は、ApoE3によるベータ−アミロイドの除去を低下させるようである(Holzman et al.(2001) J. Mol. Neurosci. 17(2): 147-55)。このことは、血液−脳関門で起こることが見出されており(Shibata et al. (2000) J. Clin. Invest. 106(12): 1489-99)、そのため重要な治療用途とすることができる。
【0064】
人工LDL粒子の調製
好ましい態様において、本発明の人工LDL粒子は、卵黄ホスファチジルコリン(EYPC)、コレステロールオレアート、およびApoE3の混合物を含む。構成成分の脂質は、3つの層から成る固形粒子を形成する(Hevonoja et al.(2000) Biochim. Biophys. Acta 1488: 189-210):コレステロール、コレステロールエステル、およびコレステロールにコンジュゲートされていてもコンジュゲートされていなくてもよい薬理学的活性物質を含有する固体の脂質コア;ホスファチジルコリンの脂肪酸鎖の混合物を含有する中間の界面層;およびリン脂質の頭部の基およびApoE3を含有する表面層。
【0065】
固体のコアおよびApoE3の存在により、本発明のLDL粒子はリポソーム(これは、水性コアを取り囲む球状脂質二重層から成り不安定である)と区別される。加えて、LDL粒子は、天然の非免疫原性構成成分からつくられており、これにより、人工ナノ粒子、分子的または化学的コンジュゲート、またはコロイド懸濁液と本発明のLDL粒子は区別される。ApoE3は、活性細胞−介在性トランスサイトーシスプロセスにより、毛細血管内皮細胞上の特定のレセプターに結合する。ひとたび脳内に治療剤が入ると、コレステロールおよびリン脂質が脳によって徐々に吸収され利用されるのに伴って、治療剤がLDL粒子から自然に放出される。
【0066】
上述の脂質構成成分が好ましいが、本発明は、他の脂質、たとえば、化学的に修飾された脂質などの異なる脂質組成のLDL粒子、または天然に存在する他の親油性分子の混合物も同様に機能し得ることを想定する。当業者であれば、LDLキャリアシステムを特定の治療剤または治療用途に適合させるために改変可能であることを認識するでしょう。
【0067】
好ましくは、LDL粒子は、人工LDLおよびクローンApoE3を用いて調製される。これにより、治療剤は、LDLの脂質中心に効率よく安定して組み込まれ易くなり、ヒトドナーから精製される翻訳後修飾されたApoE3タンパク質、変異型ApoE3タンパク質、または純粋でないApoE3タンパク質による抗原性の問題が回避される。また、HIVまたは他のウイルスなどの血液媒介疾患によるApoE3または脂質の不慮のコンタミネーションの可能性が回避される。かかるコンタミネーションは、ヒト由来の原料を使用した場合に常に生じる重大な欠点である。
【0068】
好ましい態様において、本発明は、人工LDL粒子を調製する改変ミクロエマルジョン法であって、コンジュゲートまたは非コンジュゲート薬剤を含有する脂質をリン酸緩衝生理食塩水(PBS)中に懸濁する工程と、少なくとも約25ワットをデリバーすることができるソニケーター(22kHzでプローブの18μm振幅)を用いて、54℃で窒素雰囲気下において1時間、当該溶液を音波処理する工程とを含む方法に関する。このパワーレベルは、BBBを渡る輸送を促進するのに適したサイズ、好ましくは直径50 nm未満、より好ましくは直径30 nm未満のLDL粒子を作成するために重要である。LDL粒子を含有するサンプルは、水ジャケット装備の音波処理チャンバーを使用することにより、一定の温度、好ましくは約53〜56℃の間に維持される。音波処理の後、脂質溶液をApoEとインキュベートし、臭化カリウム(KBr)ステップ勾配で285,000 gで超遠心分離することにより、産生されたリポタンパク質粒子を分離する。その後、PBSに対する透析によりKBrを除去する。その粒子を、後の使用のために好ましくは2週間まで4℃で保存することができる。
【0069】
当業者であれば、当該方法の種々の面(aspect)を置き換え可能であることを認識するでしょう。例えば、他の適切な密度勾配、たとえば塩化セシウムまたはスクロースが代わりに使用されてもよい。別の態様において、本発明のLDL粒子は、遠心分離の代わりにサイズ排除クロマトグラフィー、電気泳動、またはその他の手段により単離することができる。
【0070】
本明細書に記載される調製方法は、BBBを通過するのに適し、共に組込まれた(co-incorporated)不安定な分子の活性と構造安定性を維持するのに適したサイズの薬剤含有LDL粒子を製造する。脳へのデリバリーのために、LDL粒子は、一般に50 nm未満のサイズ、好ましくは20〜30 nmの範囲の直径にすべきである。
【0071】
別の好ましい態様において、本発明のLDL粒子は、EYPC、コレステロールオレアート、およびApoE3の混合物を、リポソームのグラムあたり0.02〜0.2グラムの比で含み、効率的な治療剤の組込みおよびBBBを渡るトランスサイトーシスを提供する。更に好ましい態様において、その範囲は、リポソームのグラムあたり0.08〜0.12グラムである。更に好ましくは、EYPC対コレステロールオレアート対ApoE3のモル比(molar ratio of EYPC to cholesterol oleate to ApoE3)は、重量ベースで23:2.2である。
【0072】
本発明の更なる態様において、LDLベースのキャリアシステムは、脳への作用物質の輸送およびデリバリーを更に促進するために、表面層に共に組込まれた追加のターゲッティング分子を含有してもよい。例として、コレステロールの酸化誘導体(オキシステロール)、たとえばコレステロールヒドロペルオキシド、コレステロールエポキシド、およびヒドロキシコレステロール誘導体を使用して、取込みを改善してもよい。また、LDL粒子は、ApoBまたはApoE4などの他のアポタンパク質を組み込んでもよい。
【0073】
また本発明は、種々の脳疾患または障害を治療するための治療剤など一以上の広範囲の物質の組込みに関する。LDLキャリアシステムの利点の一つは、化学的に不安定なもの、活性が高いもの、または水溶液中で容易に加水分解されるものなど広範囲の薬剤クラスを安全かつ自然にデリバーすることができる能力である。
【0074】
考えられる作用物質のリストは、以下のものを含むがこれらに限定されない:脳傷害および神経変性疾患を治療するための向神経性因子、たとえばNGF、またはアミロイド前駆タンパク質から産生される向神経性フラグメント;遺伝的欠陥により失われた酵素に取って代わるための酵素、たとえばフェニルアラニンヒドロキシラーゼ;パーキンソン病で失われたドーパミンを再生するための酵素、たとえばチロシンヒドロキシラーゼおよびDOPAデカルボキシラーゼ;失った生合成機能を回復するための酵素アクチベーターまたはインヒビター;感染症を治療するための抗生物質、たとえばテトラサイクリン;痛み、または行動、認知、および振る舞いを含むコンディションを治療するための神経伝達物質および神経調節物質;耐量を全身に投与したときに充分な量が脳に到達しない作用物質を用いて脳腫瘍または他のコンディションを治療するための化学療法剤および抗AIDS剤、たとえばエトポシド、リバビリン、または抗ヒスタミン薬、たとえばロラタジン、フェキソフェナジン、またはセルチリジン(Zyrtec(登録商標));診断用薬、たとえばガドリニウム誘導体、イオヘキソール、またはイオキサグレートなどの造影剤、または全身投与で脳への浸透が低いために現在使用されない作用物質;治療用または診断用タンパク質、たとえば人工および天然の抗体;および遺伝子またはタンパク質、またはDNA、RNA、またはアミノ酸を含むその一部をコードする治療用配列であって、BBBを渡って非侵襲的に導入することができるもの。
【0075】
LDLキャリアに存在する治療剤の量は、分子のタイプに依存して大きく変化する。LDLキャリアにおける最適な取込みのために、治療剤の量は、0.1モル/モルコレステロール未満とすべきである。より高いレベルは、LDL粒子を不安定にすることが予測される。本発明は、複数の薬剤または追加の作用物質が、同粒子中に存在してもよいことを更に想定する。
【0076】
一つの態様において、活性物質が充分に疎水性で、コレステロールとのミクロエマルジョンとして残るときには、活性物質の非共有結合修飾が、本発明のLDL粒子における取込みのために必要である。このことは、多くの中性および適度に荷電した分子にあてはまる。
【0077】
別の態様において、たとえば治療剤がDNAまたはペプチドのように高い電荷をもつか、あるいは拡散を妨害する場合、治療剤は、コレステロールに共有結合することができる。好ましい態様において、共有後結合はエステル結合でなされ、これにより、脳組織でみられるユビキタスなエステラーゼにより治療剤は放出され得る(Yordanov et al.,(2002) J. Med. Chem. 45(11): 2283-8;Yang et al.(1995) Pharm. Res. 12(3): 329-36)。あるいは、当業者であれば、同様に機能するその他の結合モード、なかでも例示的には、ホスファチジルコリン、その他の親油性化合物、またはApoE自体への共有結合を選択することができるであろう。
【0078】
上述のとおり、本発明のLDL粒子は、PEG化リポソーム、ナノ粒子、および同様の人工物質などの他のキャリアと比較したときに幾つかの利点を有する。本発明のLDL粒子は、血液血漿の標準の構成成分からつくられ、その天然のレセプターに結合し、外来性必須脂肪酸に対する脳の生来の要求の一環として標準の経路により細胞に輸送される。従って、その毒性は、ポリマーベースのキャリアや細胞膜の完全な状態を破壊する作用物質より有意に低い。薬剤を脳へデリバーするために用いられる現在の方法は、マンニトールによるBBBの浸透圧性破壊である。しかし、BBBの破壊は重大な欠点を有し、とりわけ所望の治療剤と共にトキシンやウイルスまでも脳に侵入させるという事実を有し、これは重大な付随効果を引き起こし得る。
【0079】
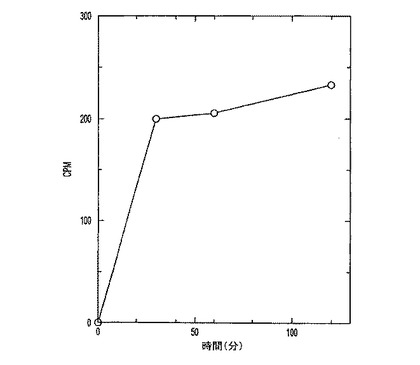
また本発明の人工LDLキャリアシステムは、少なくとも2時間血漿中に高レベルで残存するという利点を提供する(図4)。これは、脳への物質の充分な取込みやデリバリーにとって充分効果的な血漿濃度(しばしば「血漿濃度曲線下面積」すなわちAUCと称される)を維持する際に重要である。
【0080】
例3に示されるとおり、脳組織を特異的にターゲットとすることにより、薬剤が肝臓に取り込まれP450などの肝臓酵素による不活性化を含む解毒経路により不活性化されにくいため、本発明のLDL粒子は、薬剤の治療効果を有意に増大させる。
【0081】
人工LDL粒子を含む組成物
本発明のLDL粒子は、治療する脳疾患のタイプによって種々の方法で製剤化され得る。従って、本方法は、ヒトまたは動物薬に使用するために製剤化された少なくとも一の薬剤含有LDL粒子複合体を含む薬学的組成物をその範囲内に含む。かかる組成物は、薬学的に許容可能なキャリアまたは賦形剤、必要に応じて補助的薬剤と共に使用に供されてもよい。また慣用的なキャリアも本発明とともに使用することができる。許容可能なキャリアには、グルコース、生理食塩水、およびリン酸緩衝生理食塩水が含まれるがこれらに限定されない。
【0082】
処理により遊離の治療剤またはその他の組込まれなかった分子を除去した後、LDL粒子懸濁液を、患者に投与するために薬学的に許容可能なキャリア中に所望の濃度にする。LDL粒子は大きすぎて非経口的に効率よく吸収されないため、組成物は、静脈内使用が意図されているが、筋内または動脈内に投与されてもよいし、あるいは適切な改変を加えて非経口的または経口的に投与されてもよいことが考えられる。
【0083】
このように、本発明は、LDL粒子中に含有される活性物質の溶液および薬学的に許容可能なキャリア、好ましくは水性キャリアを含む、BBBをターゲットとして投与するための組成物を提供する。得られた組成物は、滅菌してキットで使用するためにパッケージ化してもよいし、あるいは無菌条件下で濾過して凍結乾燥してもよい。凍結乾燥された調製物を含有する静脈内投与のためのキットは、投与前に混合するための無菌水溶液を含んでいてもよい。
【0084】
本組成物は、おおよその生理学的条件に必要とされる薬学的に許容可能な補助的物質、たとえば、pH調整および緩衝剤、張度調整剤など、たとえば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなどを含有していてもよい。
【0085】
これら製剤中のLDL粒子の濃度は、大きく変化させることができ、すなわち約0.5%未満、通常約1%または少なくとも約1%から、5〜10重量%まで変化させることができる。静脈内に投与可能なLDL粒子製剤を調製するための更なる方法は、当業者に公知である。かかる方法は、たとえば、Goodman & Gilman, The Pharmacological Basis of Therapeutics by Joel G. Hardman (Editor), Lee E. Limbird McGraw-Hill Professional; ISBN: 0071354697; 10th edition (August 13, 2001) に詳細に記載されている。
【0086】
人工LDLキャリアを使用する治療方法
更なる態様において、人工LDL粒子を、治療を必要としている哺乳類宿主に投与して、作用物質をBBBを渡って脳へ効果的にデリバーすることができる。治療用としては、効果的な量の薬剤含有LDL粒子を、LDL粒子を毛細血管内皮細胞に取込むことが可能な任意の様式により対象に投与することができる。
【0087】
臨床上の適用において、LDL粒子デリバリーシステムは、アルツハイマー病、パーキンソン病、および脳腫瘍などに用いられる薬剤の治療効果を有意に高める。たとえば、神経成長因子などの向神経性因子をLDL粒子に組込むことができ、当該ペプチドの脳への取込みを可能にする。これにより、多くの神経変性疾患において有益である新規神経突起の成長が引き起こされる。本明細書に記載されるとおり、当業者であれば、本発明のLDLキャリアシステムを用いた広範囲のその他の臨床上の適用が存在することを認識するでしょう。
【0088】
治療剤のコレステロール−コンジュゲート
コレステロールは、相対的に化学的に不活性な分子である。その結果、コレステロールをアミン含有治療剤と反応させる前に、コレステロールを活性化しなければならない。たとえば、コレステロールは、環状無水物、たとえばカルボキシル基を含有するフタル酸エステルまたはコハク酸エステルを産生する無水フタル酸または無水コハク酸と反応させることにより活性化することができる。カルボキシル基は、その後ペンタフルオロフェノールとの反応により活性化され、更にジイソプロピルカルボジイミドと反応して、アミン含有治療剤とアミド結合をつくる。得られた生成物は、治療剤がエステル結合を介してコレステロールにコンジュゲートしたコレステロール−治療剤コンジュゲートである。コレステロール−治療剤コンジュゲートは、充分親油性であるため本発明の人工LDL粒子に組込み可能である。また、好ましい結合がエステル結合であるため、治療剤は、ユビキタスな内在性エステラーゼの作用によりコレステロール部分から放出され得る。このように、コンジュゲートからの治療剤の放出は、過酷な状況やリソソームにみられる非特異的ペプチダーゼに依存しない。コレステロールと治療剤との間の好ましい結合はエステル結合であるが、本発明は、エーテル、アミドおよびペプチド結合を含むがこれらに限定されない他の結合を想定する。
【0089】
あるいは、アミンまたはヒドロキシル含有治療剤は、チオコレステロール、治療剤および二官能性架橋試薬(たとえばPMPIであるがこれに限定されない)の反応により、チオコレステロールにコンジュゲートすることができる。得られるコンジュゲートは、チオエーテル結合により連結されたコレステロールと治療剤のコンジュゲートである。治療剤がアミノ基を含有する場合、多くの商業的に入手可能な二官能性架橋剤の一つ(スクシンイミジル 4-[N-マレイミドメチル]シクロヘキサン-1-カルボキシレートを含むがこれに限定されない)を用いて、チオコレステロールにコンジュゲートすることができる。
【0090】
コレステロール−治療剤のコンジュゲートが一旦形成されると、本明細書に記載されるとおり、非コンジュゲートコレステロールオレアート、リン脂質、リポタンパク質と混合し、これにより本発明の人工LDL粒子を作成することができる。
【0091】
上述の各引用文献は、その全体を参照により本明細書の開示内容の一部とする。
【0092】
実施例
以下の実施例は、本発明を更に説明するためのものであり、本発明の範囲を限定するものと解釈すべきでない。
【0093】
例1:全長アポタンパク質E(ApoE)の精製
Avanti Polar Lipids, Inc.から入手したDMPC (1,2-ジミリストイル-sn-グリセロ-3-ホスホコリン) を、ガラス管内でベンゼン中に100 mg/mlの濃度で懸濁し、ベンチトップソニケーターを用いて音波処理する。DMPC懸濁液をシェル凍結し、一晩凍結乾燥した後、30 ml 標準バッファー (10 mM Tris-HCl. pH 7.6. 0.15 M NaCI, 1 mM EDTA. 0.0005% NaN3) 中に再懸濁し、10-20 mg/ml DMPCを作成し、プラスチック製50 ml円錐管に移す。円錐管を水浴に置き、マイクロチップを備えたソニケーターを用いて、4回の10分間隔で、2-3 分の冷却を散在させて音波処理する。音波処理は、溶液がほぼ透明になるまで繰り返す。その後、音波処理されたDMPCを2000 rpmで20分間遠心分離し、音波処理の間に分離した(slough off)かもしれないチタンを除去する。
【0094】
クローンApoEを発現する細菌細胞を遠心分離により集め、2分の冷却間隔により隔てられた4ピリオドの強力音波処理を30秒間用いて、大きなチップを備えたBranson Sonifierで氷上において音波処理する。音波処理された懸濁液を4℃で20分間39.000 x gで遠心分離し、細胞破片を除去し、上清を元の培地1リットルあたり100 mgでDMPCと混合する。その混合物を水浴において24℃(DMPCの遷移温度)で一晩インキュベートする。
【0095】
固体KBrの添加によりDMPC-ApoE混合物の密度を1.21 g/mlに上昇させ、15℃において60 Ti Beckman固定アングルローターで55,000 rpmで一晩、ステップ勾配(1.21、1.10、および1.006 g/ml)で遠心分離する。遊離のDMPCを含有する管の頂上近くの白色バンドを捨て、1.009 mg/mlの密度で浮遊するその下のApoE-DMPC複合体を取り出す。ApoEの存在をゲル電気泳動により確認する。その後、適切なフラクションを0.1M NH4HC03および0.0005% NaN3に対して4℃において3回のバッファー交換で透析する。
【0096】
その後、タンパク質をトロンビンで分解処理し、ベクターにより残されたポリヒスチジンタグを除去する。活性化トロンビンとタンパク質を、1:500のトロンビン:ApoE、w/wの比で混合し、37℃で少なくとも1時間インキュベートし、その一部をゲル電気泳動で分析して、タンパク質が完全に切断されたことを保証する。2つの切断部位が存在するため、不完全な切断により、より大きな分子量の第二のバンドが得られる。完全な切断が証明されたらすぐβ−メルカプトエタノールを1%最終濃度まで添加し、反応を停止させる。その後ApoEを、50 ml酸洗浄管においてシェル凍結し、一晩凍結乾燥する。
【0097】
その後ApoEを、30 ml冷却クロロホルム/メタノール(2:1 v/v)で洗浄し、J6 Beckman
遠心機において4℃で10分間1500 rpmで遠心分離することにより集める。沈殿を少量の冷却メタノールに再懸濁し、ボルテックスにかけ、その後、管を冷却メタノールで満たし、遠心分離する。この工程により、残存するクロロホルムが除去される。メタノールを注ぎ出し、ボルテックスにかけられる少量を管に残し、沈殿からペーストを作成する。6M グアニジン-HCl、0.1M Tris HCl、pH 7.4、0.01% EDTA、1% β-メルカプトエタノール、および0005% アジ化ナトリウムを含有する溶液5 mlを添加し、4M グアニジン-HCl、0.1M Tris-HCl、pH 7.4、0.1% β-メルカプトエタノール、および0.0005% NaN3で平衡化したSephacryl S-300カラムにロードする。0.1% β-メルカプトエタノールおよび0.0005% アジ化ナトリウムを含有する4M グアニジンバッファーを用いてタンパク質を溶出する。フラクションを0.1M NH4HC03および0.0005% NaN3に対して4回のバッファー交換で透析する。精製されたApoEを、YM10 Amicon Centriprepコンセントレーター(Millipore)を用いて、最終濃度1-2 mg/mlに濃縮し、-20℃に保存する。
【0098】
例2:LDL-リポソームのApoE強化(enrichment)
1.脂質ケーキの調製: 卵黄ホスファチジルコリン(25 mg)およびコレステリルオレアート(2 mg)を、メタノール/クロロホルム(2:3)に溶解する(比25:1)。溶媒を4℃で窒素下において蒸発させる。
【0099】
2.リポソームの調製: 脂質ケーキを、予め窒素ガス下でバブリングした0.1 M塩化カリウムを含有する12 mlの10 mM Tris-HClバッファー pH 8.0中で水和させた。その混合物を、54℃で窒素の流れの下において18-umのアウトプットで1時間音波処理し、遠心分離して、音波処理の間に生成したチタン粒子を除去する。
【0100】
3.人工LDLの調製: ステップ2で調製されたリポソームを、ApoEタンパク質とともに1/10(w/w)タンパク質/脂質の比で37℃において30分間インキュベートする。その後リポソームを、1.064、1.019、および1.006 g/cm3の密度の3層KClステップ勾配を用いて、4℃で18時間285,000 × gで密度勾配超遠心分離をすることにより精製し濃縮する。KBrをリポソーム溶液に添加し、その密度を1.21に上昇させ、遠心管の底に適用する。Optiseal管(Beckman)が超遠心分離ステップに適している。遠心後、リポソームは管の上端から約1/4の位置に細い乳白色層として観察される。この層を取り出し、1 mM EDTAを含有するPBS(リン酸緩衝化生理食塩水)に対して4℃で一晩透析する。LDL懸濁液は少なくとも7時間安定であり、アルゴンまたは窒素の下で-20℃で保存することができる。
【0101】
例3:脳における人工LDL粒子の取込み
約1 mgの脂質を含有する14C-LDLのPBS懸濁液を、Sprague-Dawleyラット(150 g)の尾部静脈に注射した。種々の間隔で、EGTAを含有するシリンジを用いた心臓穿刺により血液サンプルを得た。血漿、RBC、および組織を、水中でホモジナイズし、14Cをシンチレーションカウンターで測定した。
【0102】
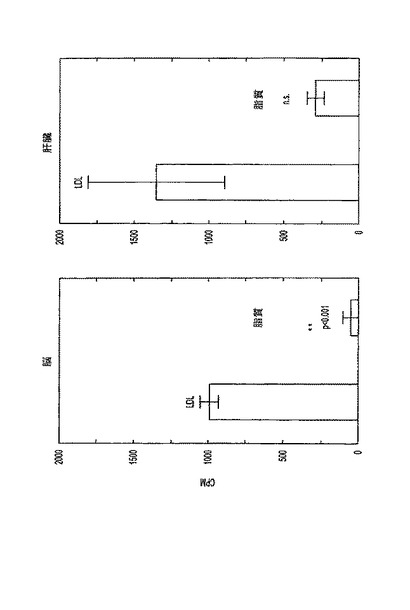
図2は、脳(左)および肝臓(右)におけるLDLおよび脂質の取込みの結果を示す。オスSprague-Dawleyラットに、14C-コレステロールを含有するLDLまたは脂質粒子を静脈内に注射し、脳および肝臓における放射能を2時間後に測定した。脳および肝臓は、ApoEの存在を除けば同一の組成を有する脂質粒子と比較して、LDLからそれぞれ19.8倍および4.7倍のラベルを取込んだ。このことは、この取込みがLDLレセプター介在性トランスサイトーシスにより引き起こされることを示す(p(>T)=0.00055)。
【0103】
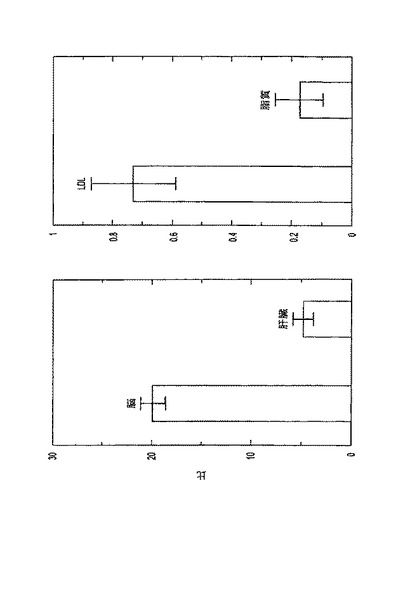
図3(左)は、脳および肝臓における脂質粒子取込みに対するLDL取込みの比を示す。脳は、肝臓と比べて、脂質粒子に対するLDLの取込みパーセンテージが高く(p(>T)=0.0003)、肝臓と比べてターゲット器官として4倍高い特異性を示す。図3(右)は、LDLおよび脂質粒子に関する、肝臓の取込みに対する脳の取込みの比を示す。器官特異性の別の指標である脳:肝臓の比は、LDLに関しては脂質粒子より高かった(p(>T)=0.0003)。すなわち、脂質粒子は、脳よりも肝臓に5.88倍多く取込まれたが、LDL粒子は脳よりも肝臓に1.36倍多くだけ取込まれた(p(>T)=0.034)。
【0104】
図4は、14C標識LDL粒子の血漿レベルの時間的経過を示す。血中14Cは、LDLの注射後少なくとも2時間は一定のままであり、これは、標識が他の器官により循環系から取り出されないことを示す。
【0105】
例4:第一級アミンの14Cコレステロールとのコンジュゲーション
コレステロール−フタレートエステルの調製: コレステロール(1 mg)を、窒素を用いて蒸発させ、凍結乾燥させてエタノールを除去する。その固体を最少体積のTHFに溶解し、ガラス反応バイアルに移す。固体の無水フタル酸(1-2 mg, >4当量)を添加し、その後50 Et3Nを添加する。その混合物を100℃で5分間加熱し、〜20μlピリジンを添加して、懸濁液を透明にする。その混合物を100℃で30分間、溶液が赤くなるまで加熱し、TLC(Silica/UV254プレート)でEtOH/トルエン(2:1)を用いて精製する。最も濃いUVバンド(RF=0.74)をプレートからスクラップし、生成物をTHFで溶出する(収率:98=100%)。
【0106】
ペンタフルオロフェノールを用いたカルボキシル基の活性化: THF中のペンタフルオロフェノール(PPP)10 mgを、コレステロールフタレートに添加し、その後5μlのDIIC(ジイソプロピルカルボジイミド)を添加する。その溶液を反応バイアル中において室温(RT)で1時間反応させ、活性化されたコレステロール−フタレート−PFPをTLC(CHCl3/CH3OH 30:5)により精製し、THFで溶出する(収率:100%)。
【0107】
活性化されたアミドの生成: THF中の活性化されたコレステロール−フタレート−PFPを10μlまで蒸発させ、DMF中に溶解した2当量の第一級アミンを添加する。DMFは、副反応を引き起こし得るが、過剰なアミンが存在する場合これは問題ではない。メタノールまたはエタノールなどのアルコールの添加は、その収率を大きく低下させる。3マイクロリットルのDIICを添加し、溶液をRTで一晩反応させ、TLC(CHCl3/CH3OH 30:5)により精製する(収率 98%)。重要なことに、Silica Gel G-25 UV254プレート(Alltech 809021)だけが精製のために使用された。Silica gel 60プレートでは、精製されていないあいまいで不明瞭なバンドが生じる。
【0108】
例5:アドリアマイシン−コレステロール(I)の合成
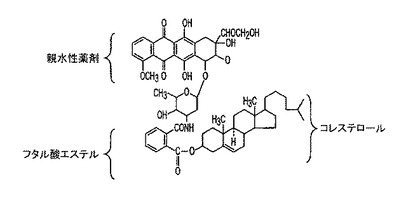
THF中の活性化されたコレステロール−フタレート−PFPを10μlまで蒸発させる。DMF中に溶解した2当量のアドリアマイシンを添加する。DMFによる副反応は、過剰のアミンを用いることにより低減することができる。メタノールまたはエタノールなどのアルコールの添加は、その収率を大きく低下させる。3マイクロリットルのDIICを添加し、溶液をRTで一晩反応させ、TLC(CHCl3/CH3OH 30:5)により精製する(収率 95%)。このバンドは、アドリアマイシン−DIICコンジュゲートと生成物を含有する。コレステロール−アドリアマイシンコンジュゲートは、100% CH3OH中でC18−逆相HPLCにより単離することができる(流速=0.5 ml/分、検出=A471)。生成物は、コレステロールとアドリアマイシンのおよそ中間の4.7分で溶出する。全収率:95%。アドリアマイシン−コレステロールコンジュゲート(I)の構造を図5に示す。
【0109】
例6:ヒドロキシ含有化合物の14Cコレステロールとのコンジュゲーション
N-[p-マレイミドフェニル]イソシアネート (PMPI) (5 mg) を、200μl DMSO中で1当量のヒドロキシ含有化合物と混合し、室温で30分間反応させる。チオコレステロール (6.4 mg) を添加する。その混合物を室温で120分間インキュベートし、生成物を、0.1 M EDTA pH 8.0で下塗りしたSilica Gel G UV 254プレートを用いて、EtOH/H2O 1:1を溶媒として用いた薄層クロマトグラフィーにより単離する。
【0110】
例7:テトラサクリン−コレステロール(II)の合成
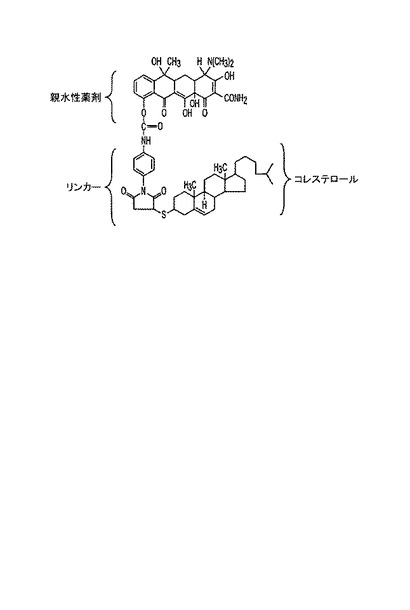
テトラサイクリンを凍結乾燥させて溶媒を除去し、200μl DMSO中の2.2 mgテトラサイクリンを5 mg PMPIと混合し、室温で30分間反応させる。チオコレステロール(6.4 mg)を添加する。その混合物を室温で120分間インキュベートし、生成物を、0.1 M EDTA pH 8.0で下塗りしたSilica Gel G UV 254プレートを用いて、EtOH/H2O 1:1を溶媒として用いた薄層クロマトグラフィーにより単離する。テトラサクリン−コレステロールコンジュゲート(II)の構造を図6に示す。
【0111】
参照文献
下記参照文献のそれぞれは、その全体を参照により本明細書の開示内容の一部とする。
【参照文献】
【0112】

【図面の簡単な説明】
【0113】
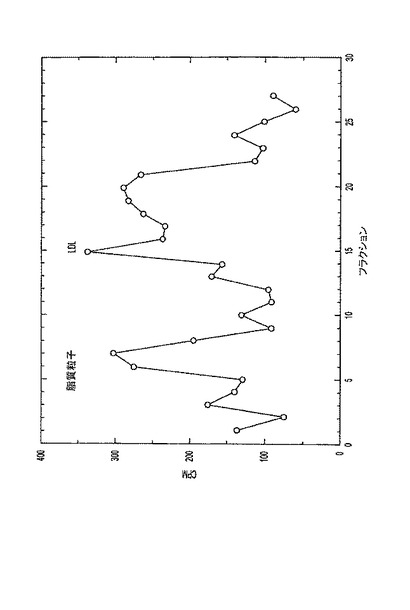
【図1】放射性標識脂質およびLDLの臭化カリウム密度勾配プロファイル。
【図2】脳(左)および肝臓(右)におけるLDLおよび脂質の取込み。
【図3】(左)脳および肝臓における脂質粒子の取込みに対するLDLの取込みの比。(右)LDLおよび脂質粒子の脳への取込みの比。
【図4】14C標識LDL粒子の血漿レベルの時間的経過。
【図5】フタレートエステルを介してコレステロールに連結された親水性分子の化学構造。示される具体的な親水性分子はアドリアマイシンである。コンジュゲーションすることなくアドリアマイシン単独では、親水性であるためにLDL粒子に組み込むことはできない。
【図6】チオエーテルエステル介してコレステロールに連結された親水性分子の化学構造。示される具体的な親水性分子はテトラサイクリンである。コンジュゲーションすることなくテトラサイクリン単独では、親水性であるためにLDL粒子に組み込むことはできない。
【技術分野】
【0001】
本出願は、2002年12月3日に出願された米国仮出願シリアル番号60/430,476の優先権を主張する。
【0002】
本発明は、物質を効率的にターゲットにし血液−脳関門(BBB)を渡ってデリバーする人工低密度リポタンパク質(LDL)粒子の使用に関する。更に本発明は、種々の脳疾患を予防および治療するための人工LDL粒子の組成物、方法、およびキットを提供する。
【背景技術】
【0003】
血液−脳関門(BBB)は、循環する毒素に対して脳を効果的に保護するが、アルツハイマー病、パーキンソン病、および脳腫瘍などの脳疾患を薬理学的に治療する際に大きな難題を引き起こす。多くの荷電分子、およびサイズ700ダルトンを超える多くの分子は、その関門を通過することができず、より小さい分子は、肝臓でコンジュゲートされ得る。これらの要因が、アルツハイマー病、パーキンソン病、細菌およびウイルス感染、および癌などの脳および中枢神経系(CNS)の疾患を薬理学的に治療する際に大きな難題を引き起こす。
【0004】
脳およびCNSの疾患および障害を治療するための多くの治療剤は、充分に親水性であり、BBBを渡る直接的な輸送を阻まれる。更にこれら薬剤および作用物質は、血液および末梢組織での分解を受けやすく、治療上効果的な血清濃度を達成するのに必要な用量を増大させる。本発明は、人工低密度リポタンパク質粒子(LDL)に治療剤をカプセル化することにより、BBBへ治療剤をデリバーする方法を提供する。本発明のLDLは、トランスサイトーシスによりBBBを渡る治療剤の輸送を促進する。親水性であるためにBBBを通過することができない多くの薬剤および治療剤は、親水性であるためにLDL粒子に組み込むことができず、そのため本発明は、LDL粒子への組み込み、BBBを渡る輸送、およびその後の治療剤の細胞への放出を促進するLDL構成成分と治療剤とのコンジュゲートを製造するための方法を提供する。
【0005】
BBBを渡って薬剤をデリバーするための従来の方法は、3つの一般的なカテゴリーを含む:(1)治療剤がキャリア内にカプセル化されているリポソームベースの方法;(2)合成ポリマーを用いて粒子を作成し、精密に規定されたサイズ特性を達成する合成ポリマーベースの方法;および(3)治療剤がインスリンなどのキャリアに共有結合している、キャリアの薬剤への直接コンジュゲーション。
【0006】
A.リポソーム
リポソームは、リン脂質を水溶液中で音波処理したときに自然に形成される小粒子であり、水性環境を取り囲む中空球として構成されるシンメトリー脂質二重層から成る。そのリン脂質は形質膜に吸収され、これによりリポソームの内容物が自動的にサイトゾルへ放出されるため、リポソームは、水溶性薬剤を細胞膜を通過させて輸送する手段として魅力を有する。この技術の成功したバリエーションには、陽イオン性脂質の使用が挙げられ、これは、膜にナノ孔を協同的に作り出すことができる。陽イオン性脂質は、細胞培養において広く使用され、DNA分子などの水溶性物質を実験のために培養細胞に導入する。
【0007】
リポソームは、その大きな運搬能力のために、BBBを渡って薬剤を輸送するのに魅力的である。しかし、リポソームは、一般に大きすぎてBBBを効率よく通過することができず、本質的に不安定であり、その構成脂質は、血漿において脂質結合タンパク質による吸収により徐々に失われる。たとえば、幾つかの研究において、大きなサイズのリポソームを使用すると、ミクロ塞栓症を引き起こし、これが脳への取込みの誤った印象を与えた。幾つかの研究において、リポソームは、安定化剤として、BBBを破壊可能な洗浄剤、ポリソルベート80と共に注入した。これらの研究においてポリソルベート80によるBBBの破壊は、観察されるBBBを渡る輸送すべてに貢献し得る。
【0008】
その結果、リポソームは、BBBを渡って薬剤を輸送するための輸送手段(vehicle)として変化に富んだ経歴がある。リポソームを特定の細胞ターゲットに向かわせるために幾つかの試みが為されている。BBBの内在性レセプターをターゲットとするペプチド様mAbsを使用して、レセプター介在性の取込みを達成する目的で、種々のBBBレセプターにPEG化(pegylated)イムノリポソームを向ける。しかし、このアプローチは、費用のかかるモノクローナル抗体の作成、試験および政府の認可が必要である。mAbsは、典型的にマウスで作成し、分解を受けやすいため、ペプチド様mAbの採用は、重大な規制の障害に直面するだけでなく、患者環境において活用することが難しいことが分かる。
【0009】
イムノリポソームは、たとえば、リポソーム表面にモノクローナル抗体(mAbs)を共有結合させることを含むプロセスで構築されている。これらイムノリポソームは、mAbコンジュゲートリポソームを強力に破壊するシステムである細網内皮系(RES)による取込みを誘発する血漿タンパク質で直に被覆されているため、イムノリポソームは、PEG化(pegylation)として知られているプロセスにおいて、ポリエチレングリコール(PEG)で処理される。残念なことに、PEG分子はmAbを妨害し、立体的妨害によりそれらを非特異的にする。Huwyler et al. (1996) Proc. Nat'l. Acad. Sci USA 93: 14164-14169は、PEGテイルの先端にマレイミド部分を備え、これによりチオール化(thiolated)mAbとコンジュゲート可能なイムノリポソームを作成することにより、この問題を回避した。内部にダウノマイシンを備えて調製されるこれらPEG化OX26イムノリポソームは、遊離の治療剤または単純な非PEG化リポソームより血漿中で安定であることが示された。しかし、共焦点顕微鏡により、リポソームはラットの脳毛細血管にエンドサイトースされるが、脳細胞に到達せず内皮細胞に結合したままであることが示された。よって、PEG化されマレイミド処理されたリポソームは、薬剤デリバリー輸送手段としてどちらかといえば効果的でないようである。
【0010】
1997年にDehouck et al.は、ApoEを結合するLDLレセプターが、BBBを渡るLDLのトランスサイトーシスに関与することを発見した。一連の3つの文献において、Versluis et al.は、マウスにおいてダウノルビシンを癌細胞にデリバーするためにApoEリッチなリポソームを使用することを記載している。腫瘍細胞がその膜において高レベルのLDLレセプターを発現するという発見に基づいて、LDLレセプターターゲッティングタンパク質としてApoEを選択した。またVersluis et al. (1998) は、天然のLDLを使用することを提案したが、この試みは試されておらず、その後の論文は、ApoEリッチなリポソームにのみ焦点をあてている。Versluis et al. (1999) は、ダウノルビシンの組織分布を調査したが、脳の取込みに関するデータはなく、このことは、この方法がBBBを渡ってダウノルビシンを輸送するための手段として考えられていなかったことを示す。
【0011】
加えて、Versluis et al.により用いられたコンジュゲーション化学は、本発明で使用されるものとは異なる。薬剤をリポソーム膜に固定するために、著者らは、3α-O-(オレオイル)-4β-コラン酸 (リトコール酸のエステル) をテトラペプチドAla-Leu-Ala-Leuに連結し、次にこれを親水性抗腫瘍剤ダウノルビシンに共有結合した。このように、コンジュゲートされ遊離していないダウノルビシンにより腫瘍は治療された。リトコール酸は、活性化可能な酸の基を予め含有するステロイドであるが、この酸の基は、3-OH位ではなくステロイド側鎖に位置し、これにより、望ましくない特徴の反応生成物が得られる。遊離のダウノルビシンは、高酸性リソソームに見られるプロテアーゼによって切断された後にのみつくることができ、これは、コンジュゲート薬剤または作用物質を、プロテアーゼ、酸性およびその他の加水分解酵素による分解に晒すものである。その後、治療剤は、リソソーム内空間に放出され、ここで更なる分解を受け細胞から放出される。
【0012】
対照的に、本発明のコンジュゲートは、好ましくは、エステル結合を介した治療剤の結合を提供し、これは、細胞質ゾルで容易に切断され、その結果、Versluis et al.の方法に必要とされる過酷なリソソーム状態を避けることができる。よって、本発明の方法によりコンジュゲートされる治療剤は、そのターゲットへの道のりを生き残りやすく、ターゲットにおいて効率的な手法で放出される可能性が高い。また、本発明の方法によりコンジュゲートされる治療剤は、リポソームよりもBBBを渡って輸送されやすい。
【0013】
また、Versluis et al.の方法は、テトラペプチドを合成するために多数の固相ペプチド化学の工程が必要であり、それをFMOCとコンジュゲートし、そのコンジュゲートをリトコール酸と反応させ最後に薬剤と反応させるために幾つかの追加の工程が必要である。本発明は、ずっと少ない工程数を使用し、その各々によりほぼ定量的な収率が得られる。このように本発明は、高効率と低コストを提供する。
【0014】
デオキシルビシンの他のリポソーム製剤が、現在、見込みのある癌治療剤として臨床で使用されているが、LDLを使用した製品は売り出されていない。
【0015】
Demeule et al.は、タンパク質メラノトランスフェリン(p97)がBBBを渡ってトランスサイトーシスにより輸送されることを見出し、LDLレセプターが関与していると断定し、これにより、このタンパク質が薬剤デリバリーシステムとして採用されることが示唆された。
【0016】
B.合成ポリマー
ポリソルベート80で被覆したポリ(ブチルシアノアクリレート)またはポリアクリルアミドなどの合成ポリマーも試験してみた。これらポリマーは、その粒子が充分親水性で水に可溶であり、更に長時間にわたってその構造形態を維持することができ、これにより、天然の解毒プロセスを受ける肝臓および腎臓への取込みから治療剤は保護されるため、魅力的である。いずれのケースにおいても、取込みは、細胞膜を渡る受動拡散により起こるか、あるいはクラスリン被覆小胞による防御的取込みとして起こると一般に想像される。その後、前者のケースにおいて治療剤は、以前ほどターゲットにさほど近くない内皮細胞にトラップされるが、後者のケースにおいて治療剤は、プロテアーゼや胃の内容物に類似した他の消化酵素を含有する細胞における高酸性コンパートメントであるリソソームに輸送される。よって後者のケースでは、治療剤は、より過酷な状況の間ずっと安定なまま留まらなければならない。いずれのケースも、薬剤は細胞を渡って運搬されず、所望の結果である脳実質組織への排出は起こらない。よって、これら二つの方法のいずれもが、さほど臨床上使用されていないことは驚くことではない。
【0017】
多くの研究者らは、上述のアプローチの種々の改変を試みたところ、ある程度の成果で、BBBを渡るキャリアの取込みは改良された。たとえば、Kreuter et al. (2002) J. Drug Target 10(4): 317-25は、BBBに位置するアポリポタンパク質レセプターに結合する種々のアポリポタンパク質を含有する合成粒子を設計した。彼らは、ポリ(ブチルシアノアクリレート)ナノ粒子に結合し、ポリソルベート80で被覆された薬剤の輸送を実証した。取込みには、ポリソルベート80、ApoEまたはApoBでのコーティングが必要であった。アポリポタンパク質All、Cll、またはJコーティングは、機能しなかった。しかし、これらナノ粒子は天然に存在しないため、望ましくない副作用があるかもしれない。アクリレートポリマーは、自己免疫応答を開始させることで特に有名であり;化学的に類縁のポリマー、ポリ(アクリルアミド)がしばしばアジュバントとして使用される。
【0018】
Alyaudin et al. (2001) J. Drug Target 9(3): 209-21は、ポリソルベート80でオーバーコートしたポリ(ブチルシアノアクリレート)ナノ粒子を使用して、BBBを渡って[3H]-ダラルジン(dalargin)を輸送し、そのプロセスがエンドサイトーシスとその後のトランスサイトーシスの一つであると推量した。このポリマーは、更に免疫学的な混乱をきたすかもしれない。
【0019】
C.治療剤コンジュゲート
BBBを渡って輸送可能な物質、たとえばインスリンと薬理学的作用物質の直接的コンジュゲーションも試みられている。インスリンおよびインスリン様増殖因子は、特殊な促進拡散システムにより血液脳関門を通過することが知られている(Reinhardt et al. (1994) Endocrinology 135(5): 1753-1761)。インスリンは、内皮インスリンレセプターにより媒介されるトランスサイトーシスにより吸収される(Pardridge et al. (1986) Ann. Intern. Med. 105(1): 82-95)。また、グルコースやトリプトファンなどの大きなアミノ酸のための特定のトランスポーターが存在する。しかし、インスリントランスポーターはその特異性が高いため、インスリンに共有結合した薬理学的作用物質を脳へ通過させることができないことが分かっている。同様の結果が、グルコースやアミノ酸コンジュゲートでも得られており、その取込みは、他の低分子量物質と同じ一般的原理に従うことが観察されており、700 Da以下の非電荷分子のみが脳への有意なアクセスを達成する。コンジュゲート形態の生体分子の化学合成を考え出す煩わしさ、予期しない毒性効果を引き起こす
リスク、および完全に新規な化合物についてFDA認可を得なければならないかもしれない必要性が、このアプローチに対する熱意を失わせる。
【0020】
陽イオン化アルブミンなどのタンパク質である輸送ベクター、またはトランスフェリンレセプターに対するOX26モノクローナル抗体は、それぞれ、吸収力介在性トランスサイトーシスおよびレセプター介在性トランスサイトーシスを受けBBBを通過する。これらは、少量の薬剤を輸送するために使用される。このプロセスは、費用が高く、モノクローナル抗体や陽イオン化アルブミンを作成する難しさがあり、他のタイプの分子に適用することができない。また、陽イオン化タンパク質は、その免疫原性や腎臓で沈殿する免疫複合体の形成のために毒性があることが示されている。
【0021】
Wu et al. (2002) J. Drug Target 10(3): 239-45は、ストレプトアビジン(SA)とラットトランスフェリンレセプターに対するマウスOX26モノクローナル抗体と、ベクターに結合したビオチン化bFGF(bio-bFGF)のコンジュゲートから成り、bio-bFGF/OX26-SAと称される薬剤デリバリーベクターを用いて、ヒト塩基性繊維芽細胞増殖因子(bFGF)、タンパク質神経保護剤がBBBを渡って輸送されることを示した。彼らは、[125I]標識bio-bFGFの末梢器官への強力な取込みを示したが、脳1グラムにつき注入量の0.01%しか吸収されなかった。また、この手段は、薬剤の共有結合修飾を必要とするため、限られたクラスの薬剤についてのみ有効である。また、キャリアが構成成分としてマウスモノクローナル抗体を含有するため、これが、患者に免疫応答を引き起こすであろう。
【0022】
また、Kang et al. (2000) J. Drug Target 8(6): 425-34は、BBBを渡ってペプチドを輸送するためにアビジン−ビオチン連結キメラペプチドを使用したが、組織1グラムにつき注入量の0.12%しか吸収されなかった。Kang and Pardridge (Pharm. Res. 11: 1257-1264) は、陽イオン化ヒト血清アルブミンとニュートラルライトアビジンとをコンジュゲートし、それを放射性標識ビオチンに結合させた。ビオチン/cHSA/NLA複合体は、血液中で24時間まで安定であったが、そのコンジュゲートは脳で選択的に分解され、遊離のビオチンを放出した。上述のとおり、陽イオン化タンパク質は、その免疫原性のために毒性があることが示されている。
【0023】
また、陽イオン化モノクローナル抗体(mAbs)が使用されている。Pardridge (J. Neur
ochem. 70: 1781-2) は、天然のヒト化4D5 MAbは、そのタンパク質を陽イオン化した後のみ、吸収力介在性トランスサイトーシスによりBBBを通過することを共焦点顕微鏡により示した。しかし、このプロセスは、モノクローナル抗体を作成し化学的に修飾する費用が高く、他のタイプの分子に適用することができない。
【0024】
Witt et al. (2000) J. Pharmacol. Exp. Ther. 295(3): 972-8 は、デルタ−オピオイドレセプター−選択的ペプチドD-ペニシラミン(DPDPE)、Met-エンケファリン類似体を、BBBを渡って輸送するためにインスリンを使用した。しかし、インスリンは、治療ストラテジーとしてのその使用を制限する多くの危険を提示する。また、他の研究者らは、インスリンレセプターが極めて選択的であることを見出した。よって、キメラペプチドを作成する難しさに加えて、このストラテジーは、狭いクラスの薬学的作用物質に限定される。
【0025】
他の研究者らは、たとえばグリコペプチドを用いて薬剤をグルコースにコンジュゲートすることを試みた。しかし、BBB Glut1トランスポーターを介したグリコペプチドの有意な輸送はこれまで実証されていない。Glut1グルコーストランスポーター、コリントランスポーター、またはLAT1大アミノ酸トランスポーターなどのキャリア介在性トランスポーターの高い輸送速度を使用する試みは、キャリアトランスポーターが非常に選択的であるためコンジュゲート物質を受け入れることができないという問題で失敗した。また、これら試みは、多剤耐性遺伝子のメンバーであるp-糖タンパク質が、関門を通過させたい薬剤などの多くの低分子を活発に脳から除去するために迅速に機能するという問題がある。
【0026】
LDLレセプターに加えて、BBBは、タイプIIスカベンジャーレセプター(SR)を含有し、これは高いアフィニティーでLDLを結合する。このスカベンジャーレセプターは、アセチル化LDLなどの修飾型LDLととりわけ親和性がある。SRへの結合により、エンドサイトーシスのみが起こり、所望のトランスサイトーシスは起こらない。Rigotti et al. (1995) J. Biol. Chem. 270: 16221-4は、アセチル化LDLはBBBを渡って輸送されないが、陽イオン化ウシIgGが更に効果的であることを見出した Bickel et al. (1993) Adv. Drug. Del. Rev. 10: 205-245。アセチル化LDLを用いてトランスサイトーシスを実証できなかったことにより、LDLを用いて更なる実験を試みる気力を多くの研究者から奪った。
【0027】
Protter et al. (WO 87/02061) は、薬学的作用物質または作用物質含有キャリアに共有結合したApoEおよびApoBなどのアポリポタンパク質に由来するペプチドを使用した薬剤デリバリーシステムを記載している。しかし、分子コンジュゲートの使用は、少数の薬剤クラスにのみ限定され、上述したのと同じ問題を多く有する。
【0028】
Muller et al. (US特許第6,288,040号) は、ApoE分子が共有結合した合成ポリ(ブチルシアノアクリレート)粒子の使用を記載している。これら粒子の表面は、界面活性剤により、または親水性ポリマーの共有結合により更に修飾される。上述のとおり、これら粒子は天然に存在しないため、種々の望ましくない副作用があるかもしれない。
【0029】
Samain et al. (WO 92/21330) は、固体の親水性コアに共有結合し、腫瘍またはマクロファージへ物質をデリバーするためのApoBを有する、脂質を含有する合成粒子キャリアの使用を記載している。しかし彼らは、BBBを渡って薬剤をデリバーするためにかかるベクターが有用であることをまったく開示していない。
【発明の開示】
【0030】
本発明は、物質をターゲットにし血液脳関門を渡って脳へデリバーするために、低密度リポタンパク質(LDL)粒子を使用することに関する。本発明の更なる目的は、構造的に安定であり、免疫原性がなく、かつ分解、不活性化、加水分解、コンジュゲーション、および非ターゲット組織への取込みから広範囲の薬剤を保護するLDLキャリアを合成することである。本発明は、治療剤のキャリアとして使用されるLDL粒子を提供するとともに、従来の方法と比較して改善された、かかる薬剤および作用物質をBBBを渡ってデリバーする方法を提供する。リポソームとは異なり、たとえば、LDL粒子は固形粒子であるためリポソームより構造的安定性が高い。本発明の更なる目的は、広範囲の脳疾患を治療および予防するためのLDLキャリアを含む組成物、方法、およびキットを提供することである。
【0031】
本発明は、親水性治療剤をコレステロールとコンジュゲートし、コンジュゲート治療剤を本発明の人工LDL粒子に組込み易くするための方法を提供する。好ましい態様において、本発明は、コレステロールがコンジュゲートしたアドリアマイシンおよびテトラサイクリンを提供する。当該方法、得られたコレステロールコンジュゲート、およびかかるコンジュゲートの組成物は、トランスサイトーシス(これは、コレステロールや必須脂肪酸を脳に取込む手段として脳の毛細血管内皮細胞で機能するレセプター介在性プロセスである)により脳関門を渡って治療剤を輸送する目的でLDL粒子を提供する際に有用であり、これが、脳およびCNSの種々の疾患および障害の治療を促進し改良するであろう。あるいは、本発明のコレステロールコンジュゲートは、このコンジュゲートを本発明のLDL粒子に組み込むことなく、対応の治療剤をBBBを渡ってデリバーするために有効である。
【0032】
好ましい態様において、本発明のコレステロールコンジュゲートは、エステル結合を介して連結され、これが、ユビキタスな内在性エステラーゼの作用により当該コンジュゲートから治療剤の放出を可能にする。本発明のLDL粒子に本発明のコレステロールコンジュゲートを封入することにより、コレステロールコンジュゲートは、これらエステラーゼによる加水分解から保護される。
【0033】
本発明は、卵黄ホスファチジルコリン(EYPC)、コレステロールオレアート、およびアポリポタンパク質E3(ApoE3)を含む人工LDL粒子を更に提供する。構成成分の脂質は、3つの層を含有する固形粒子を形成する:コレステロール、コレステロールエステル、および活性物質から成る固体の脂質コア;ホスファチジルコリンの脂肪酸鎖の混合物から成る中間の界面層;およびリン脂質の頭部の基およびApoE3から成る表面層。
【0034】
本発明のLDL粒子は、毛細血管内皮細胞への活性物質のターゲッティングを有意に増大させ、トランスサイトーシスによる血液脳接合部を渡る輸送を促進する。また、治療剤を取り囲むタンパク質とリン脂質の構成成分は、分解や非ターゲット細胞への取込みから治療剤を保護するために機能する。
【0035】
また本発明は、人工LDL粒子により促進される作用物質の移動に基づいて、疾患、病気およびコンディションを治療する方法に関する。たとえば、本発明は、本発明の人工LDL粒子を用いて特定の作用物質を脳組織へ向けることを含む、種々の脳疾患を治療するための薬学的組成物および方法を提供する。
【0036】
本発明は、外側のリン脂質単一層と固体の脂質コアとを含む人工LDL粒子であって、外側のリン脂質単一層が少なくとも一のアポリポタンパク質を含み、固体の脂質コアが少なくとも一の治療剤を含有する人工LDL粒子を提供する。一つの態様において、少なくとも一のアポリポタンパク質は、ApoA、ApoB、ApoC、ApoD、またはApoE、または前記アポリポタンパク質の一つのアイソフォーム、またはリポタンパク質および/またはアイソフォームの組合せから成る群より選択される。好ましい態様において、少なくとも一のアポリポタンパク質はApoEである。更に好ましい態様において、少なくとも一のアポリポタンパク質は、ApoE2、ApoE3およびApoE4から成る群より選択される。また本発明は、脳組織へのLDL複合体のターゲットデリバリーを増大させる追加のターゲッティング分子または作用物質を更に含む人工LDL粒子に関する。最も好ましい態様において、少なくとも一のアポリポタンパク質はApoE3であり、ApoE3は単独であるか、または一以上のオキシステロールおよび/またはApoBおよびApoE4から成る群より選択される追加のアポリポタンパク質と組み合わせられる。
【0037】
本発明は、血液−脳関門を渡って治療剤を輸送するための人工LDL粒子を提供する。好ましい態様において、少なくとも一の治療剤は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される。別の態様において、少なくとも一の治療剤は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される作用物質とコレステロールとの間で形成されたコンジュゲートである。好ましい態様において、少なくとも一の治療剤は、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される。
【0038】
人工LDL粒子の外側のリン脂質単一層は、ホルファチジン酸、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルグリセロールなどを含むがこれらに限定されない任意のリン脂質またはリン脂質の組合せを含み得る。好ましい態様において、外側のリン脂質単一層は、ホスファチジルコリンおよび少なくとも一のアポリポタンパク質を含む。
【0039】
本発明の人工LDL粒子は、好ましくは直径約15〜50 nmの粒子である。更に好ましい態様において、人工LDL粒子は、直径約20〜30 nmである。本発明の人工LDL粒子は、好ましくは密度約1.00〜1.07 g/mLである。更に好ましい態様において、人工LDL粒子は、密度約1.02〜1.06 g/mLである。更に本発明の人工LDL粒子は、少なくとも2時間の血清安定性を有する。
【0040】
本発明は、トランスサイトーシスによりBBBを渡って輸送される人工LDL粒子を提供する。好ましい態様において、本発明の粒子は、脳に対する取込み特異性が肝臓に比べて少なくとも3倍高い。
【0041】
本発明の人工LDL粒子の固体の脂質コアは、トリアシルグリセロール、コレステロール、コレステロールエステル、脂肪酸アシルエステルなどを含むがこれらに限定されない一以上の脂質を含み得る。好ましい態様において、固体の脂質コアは、ミリステート、パルミテート、ステアレート、アラキデート、リグノセレートなどを含むがこれらに限定されない飽和脂肪酸、またはパルミトレアート、オレアート、バクセネート、リノレアート、リノレネート、アラキドネートなどを含むがこれらに限定されない不飽和脂肪酸とコレステロールがエステル化したコレステロールエステルを含む。更に好ましい態様において、固体の脂質コアは、コレステロールエステルであるコレステロールオレアートを含む。好ましい態様において、本発明の人工LDL粒子の固体の脂質コアは、アドリアマイシンまたはテトラサイクリンとコレステロールとの間で形成されたコンジュゲートである少なくとも一の治療剤を含む。好ましい態様において、コンジュゲートのコレステロールと治療剤は、エステル結合により連結される。
【0042】
また本発明は、本発明の人工LDL粒子および薬学的に許容可能なキャリアを含む、治療剤を血液−脳関門を渡ってデリバーするための組成物を提供する。
【0043】
また本発明は、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される治療剤に連結されたコレステロールを含むコンジュゲートを提供する。好ましい態様において、治療剤は、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される。更に好ましい態様において、本発明のコンジュゲートの治療剤は、アドリアマイシンまたはテトラサイクリンである。最も好ましい態様において、本発明のコンジュゲートのコレステロールと治療剤は、エステル結合により連結される。本発明のコンジュゲートの各々は、本明細書に記載されるとおり、薬学的に許容可能なキャリアと組合せて、本発明の薬剤デリバリーの方法の何れかにおいて使用することができる。
【0044】
また本発明は、1)コンジュゲートまたは非コンジュゲート治療剤を含有するリン脂質を緩衝液中に懸濁する工程;2)当該溶液を音波処理し、外側のリン脂質単一層と固体の脂質コアを形成する工程;および3)少なくとも一のアポリポタンパク質を含む溶液を添加し、当該アポリポタンパク質が前記外側のリン脂質単一層に取り込まれる工程を含む、本発明の人工LDL粒子を製造する方法を提供する。好ましい態様において、本発明の方法により製造される人工LDL粒子は、直径10〜50 nmである。
【0045】
また本発明は、効果的な量の本発明の組成物の何れかを、それを必要とする哺乳類に投与することを含む、血液−脳関門を渡って物質をデリバーするための方法を提供する。
【0046】
また本発明は、本発明の組成物の何れかを含有する容器と使用説明書を含む、血液−脳関門を渡って物質をデリバーするためのキットを提供する。
【0047】
また本発明は、アルツハイマー病、パーキンソン病、細菌およびウイルス感染、および癌などの脳および中枢神経系(CNS)の疾患を治療するための医薬の製造における、本発明のコンジュゲート、人工LDL粒子、および組成物の使用を提供する。
【発明の詳細な説明】
【0048】
定義
本明細書で使用される「人工LDL粒子」の用語は、球状のリン脂質単一層と固体の脂質コアを含む構造体を意味する。
【0049】
本明細書で使用される「リポソーム」の用語は、球状の脂質二重層と水性コアを含む構造体を意味する。
【0050】
本明細書で使用される「取込み特異性」の用語は、脳および肝臓における脂質粒子(外側のリン脂質単一層にアポタンパク質が含まれないことを除いて人工LDL粒子と同じ粒子)の取込みに対する人工LDL粒子の取込みの比をいう。人工LDL粒子と脂質粒子の取込みは、本明細書に記載されるとおり、Sprague-Dawleyラットに注入してから2時間後、脳および肝臓の両方で測定する。取込み特異性は、脳における脂質粒子の取込みに対する人工LDL粒子の取込みの比を、肝臓における脂質粒子の取込みに対する人工LDL粒子の取込みの比で割ることにより計算する。
【0051】
本明細書で使用される「血清安定性」の用語は、注入された人工LDL粒子の少なくとも75%が血漿中に残存する時間の長さを意味する。
【0052】
本明細書で使用される「アポリポタンパク質」および「アポタンパク質」の用語は、リポタンパク質のリン脂質単一層と結びついたタンパク質を意味し、ApoA;ApoB;ApoC;ApoD;ApoE;および各々のアイソフォームのすべてを含むがこれらに限定されない。
【0053】
本明細書で使用される「ApoE」の用語は、ApoEの一以上のアイソフォームを意味し、ApoE2、ApoE3およびApoE4を含むがこれらに限定されない。
【0054】
本明細書で使用される「ApoB」の用語は、ApoBの一以上のアイソフォームを意味し、ApoB48およびApoB-100を含むがこれらに限定されない。
【0055】
本明細書で使用される「外側のリン脂質単一層」の用語は、リン脂質のホスフェートの頭部の基が外側に向けられ、脂肪酸アシル鎖が固体の脂質コアに向かって内側に向けられた少なくとも一のリン脂質を含む単一層を意味する。リン脂質は、ホスファチジン酸、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルグリセロールなどを含むがこれらに限定されない。
【0056】
本明細書で使用される「固体のコア」の用語は、球状のリン脂質単一層により取り囲まれた人工LDL粒子の該当部分を意味する。固体のコアは、トリアシルグリセロール、コレステロール、コレステロールエステル、脂肪酸アシルエステルなどを含むがこれらに限定されない一以上の脂質を含む。本明細書で使用される「コレステロールエステル」の用語は、ミリステート、パルミテート、ステアレート、アラキデート、リグノセレートなどを含むがこれらに限定されない飽和脂肪酸、またはパルミトレアート、オレアート、バクセネート、リノレアート、リノレネート、アラキドネートなどを含むがこれらに限定されない不飽和脂肪酸とエステル化したコレステロールをいう。
【0057】
本明細書で使用される「治療剤」の用語は、治療上有用なアミノ酸、ペプチド、タンパク質、ポリヌクレオチド、オリゴヌクレオチド、遺伝子などを含むがこれらに限定されない核酸、炭水化物、および脂質を意味する。本発明の治療剤は、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤などを含む。本発明の治療剤は、当該治療剤がターゲット組織にデリバーされたときに活性化され得る、薬剤、プロドラッグ、および前駆体を含む。
【0058】
本明細書で使用される「薬学的に許容可能なキャリア」の用語は、有効成分と組合せることができ、その組合せに従って有効成分を対象に投与するために使用することができる化学組成物または化合物を意味する。本明細書で使用される「生理学的に許容可能な」エステルまたは塩の用語は、薬学的組成物の他の任意の成分と適合可能な有効成分のエステルまたは塩の形態であって、組成物を投与する対象に対して有害でないものを意味する。また本明細書で使用される「薬学的に許容可能なキャリア」は、一以上の以下のものを含むがこれらに限定されない:賦形剤;界面活性剤;分散剤;不活性な希釈剤;顆粒化および粉状化剤;結合剤;潤滑剤;甘味剤;香味剤;着色剤;保存剤;ゼラチンなどの生理的に分解可能な組成物;水性賦形剤および溶媒;油性賦形剤および溶媒;懸濁剤;分散または湿潤剤;乳化剤、粘滑剤;緩衝剤;塩;増粘剤;充填剤;乳化剤;抗酸化剤;安定化剤;および薬学的に許容可能なポリマーまたは疎水性材料。本発明の薬学的組成物に含有され得るその他の「追加の成分」は、当該技術分野で公知であり、たとえばGenaro ed., 1985, Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa.に記載され、これは参照により本明細書の開示内容の一部とする。
【0059】
本明細書で使用される「効果的な量」は、治療応答を引き起こすのに充分な量である。
【0060】
LDL粒子およびアポタンパク質の特性
本発明は、人工LDL粒子が、トランスサイトーシスとして公知の活性レセプター介在性プロセスにより、物質を効率的にターゲットにし血液−脳関門を渡ってデリバーするという発見に関する。トランスサイトーシスは、コレステロールおよび必須脂肪酸を脳へ輸送する手段として、脳の毛細血管内皮細胞に天然に存在する。従って、本発明の人工LDLキャリアシステムは、BBBの破壊またはその他の望ましくない副作用を最小限にして、薬剤を脳へ効率的に向けデリバーする手段を提供する。
【0061】
天然のLDL粒子は、平均約22 nmの直径を有する。内部コアは、約150-200のトリグリセリド分子および1500-2000のコレステリルエステル分子から成る。粒子の表面は、約450のリン脂質分子、185分子のスフィンゴミエリン、および1分子のアポタンパク質、典型的にはApoB-100の単一層を含有する(Hevonoja et al.(2000) Biochim Biophys Acta 1488(3): 189-210)。天然のLDL分子は、約600分子の非エステルコレステロールおよび少量のリゾホスファチジルコリン、ホスファチジルエタノールアミン、ジアシルグリセロール、セラミド、およびホスファチジルイノシトール、並びに微量のその他の生物学的脂質を含んでいてもよい(Hevonoja et al.(2000) Biochim Biophys Acta 1488(3): 189-210)。ApoEなどのその他のアポタンパク質は、LDL、VLVL、およびHDLにみられるが、種々のレセプター結合特性を有する(Bradly et al.(1984) J. Biol. Chem. 259(23): 14728-35)。
【0062】
従って、LDL分子の表面は、アポタンパク質で不均一に被覆されているが、種々の物理的特性を備えた種々の領域から成る。アポタンパク質分子は、粒子の構造の完全性を維持することに貢献するとともに、肝臓、腎臓、および血液−脳関門でリポタンパク質レセプターに結合することに貢献する。アポタンパク質は、脂質構成成分の状態に依存する構造的変化を受ける(Mims et al.(1990) Biochemistry 29(28): 6639-47)。
【0063】
アポタンパク質E(ApoE)は、体全体においてコレステロール輸送および血漿リポタンパク質代謝に関与するタンパク質である。末梢細胞においてApoEは、その輸送を指示することによりコレステロールの細胞濃度に影響を及ぼす。ニューロンにおいてコレステロールレベルの変化は、アルツハイマー病で変化するのと同じ部位で微小管結合タンパク質tauのリン酸化状態に影響を及ぼす。ApoEは、3つの主なアイソフォーム:ApoE4、ApoE3、およびApoE2を有し、これらは単一のアミノ酸置換により異なる。ApoE3は、正常のアイソフォームであるが、ApoE4およびApoE2は機能不全である。ApoE2は、タイプIII高リポ蛋白血症と関連する。ApoE4は、アテローム性動脈硬化症およびアルツハイマー病のリスクの増大、認知機能の衰え、および神経突起の伸出の低下と関連する。年齢を除けば、ApoE4は、散発性アルツハイマー病の最も重要な危険因子である。ApoE4は、カルシウムに依存した毒性効果があるかもしれないが(Veinbergs et al.(2002) J. Neurosci. Res. 67(3): 379-87)、その主要な効果は、ApoE3によるベータ−アミロイドの除去を低下させるようである(Holzman et al.(2001) J. Mol. Neurosci. 17(2): 147-55)。このことは、血液−脳関門で起こることが見出されており(Shibata et al. (2000) J. Clin. Invest. 106(12): 1489-99)、そのため重要な治療用途とすることができる。
【0064】
人工LDL粒子の調製
好ましい態様において、本発明の人工LDL粒子は、卵黄ホスファチジルコリン(EYPC)、コレステロールオレアート、およびApoE3の混合物を含む。構成成分の脂質は、3つの層から成る固形粒子を形成する(Hevonoja et al.(2000) Biochim. Biophys. Acta 1488: 189-210):コレステロール、コレステロールエステル、およびコレステロールにコンジュゲートされていてもコンジュゲートされていなくてもよい薬理学的活性物質を含有する固体の脂質コア;ホスファチジルコリンの脂肪酸鎖の混合物を含有する中間の界面層;およびリン脂質の頭部の基およびApoE3を含有する表面層。
【0065】
固体のコアおよびApoE3の存在により、本発明のLDL粒子はリポソーム(これは、水性コアを取り囲む球状脂質二重層から成り不安定である)と区別される。加えて、LDL粒子は、天然の非免疫原性構成成分からつくられており、これにより、人工ナノ粒子、分子的または化学的コンジュゲート、またはコロイド懸濁液と本発明のLDL粒子は区別される。ApoE3は、活性細胞−介在性トランスサイトーシスプロセスにより、毛細血管内皮細胞上の特定のレセプターに結合する。ひとたび脳内に治療剤が入ると、コレステロールおよびリン脂質が脳によって徐々に吸収され利用されるのに伴って、治療剤がLDL粒子から自然に放出される。
【0066】
上述の脂質構成成分が好ましいが、本発明は、他の脂質、たとえば、化学的に修飾された脂質などの異なる脂質組成のLDL粒子、または天然に存在する他の親油性分子の混合物も同様に機能し得ることを想定する。当業者であれば、LDLキャリアシステムを特定の治療剤または治療用途に適合させるために改変可能であることを認識するでしょう。
【0067】
好ましくは、LDL粒子は、人工LDLおよびクローンApoE3を用いて調製される。これにより、治療剤は、LDLの脂質中心に効率よく安定して組み込まれ易くなり、ヒトドナーから精製される翻訳後修飾されたApoE3タンパク質、変異型ApoE3タンパク質、または純粋でないApoE3タンパク質による抗原性の問題が回避される。また、HIVまたは他のウイルスなどの血液媒介疾患によるApoE3または脂質の不慮のコンタミネーションの可能性が回避される。かかるコンタミネーションは、ヒト由来の原料を使用した場合に常に生じる重大な欠点である。
【0068】
好ましい態様において、本発明は、人工LDL粒子を調製する改変ミクロエマルジョン法であって、コンジュゲートまたは非コンジュゲート薬剤を含有する脂質をリン酸緩衝生理食塩水(PBS)中に懸濁する工程と、少なくとも約25ワットをデリバーすることができるソニケーター(22kHzでプローブの18μm振幅)を用いて、54℃で窒素雰囲気下において1時間、当該溶液を音波処理する工程とを含む方法に関する。このパワーレベルは、BBBを渡る輸送を促進するのに適したサイズ、好ましくは直径50 nm未満、より好ましくは直径30 nm未満のLDL粒子を作成するために重要である。LDL粒子を含有するサンプルは、水ジャケット装備の音波処理チャンバーを使用することにより、一定の温度、好ましくは約53〜56℃の間に維持される。音波処理の後、脂質溶液をApoEとインキュベートし、臭化カリウム(KBr)ステップ勾配で285,000 gで超遠心分離することにより、産生されたリポタンパク質粒子を分離する。その後、PBSに対する透析によりKBrを除去する。その粒子を、後の使用のために好ましくは2週間まで4℃で保存することができる。
【0069】
当業者であれば、当該方法の種々の面(aspect)を置き換え可能であることを認識するでしょう。例えば、他の適切な密度勾配、たとえば塩化セシウムまたはスクロースが代わりに使用されてもよい。別の態様において、本発明のLDL粒子は、遠心分離の代わりにサイズ排除クロマトグラフィー、電気泳動、またはその他の手段により単離することができる。
【0070】
本明細書に記載される調製方法は、BBBを通過するのに適し、共に組込まれた(co-incorporated)不安定な分子の活性と構造安定性を維持するのに適したサイズの薬剤含有LDL粒子を製造する。脳へのデリバリーのために、LDL粒子は、一般に50 nm未満のサイズ、好ましくは20〜30 nmの範囲の直径にすべきである。
【0071】
別の好ましい態様において、本発明のLDL粒子は、EYPC、コレステロールオレアート、およびApoE3の混合物を、リポソームのグラムあたり0.02〜0.2グラムの比で含み、効率的な治療剤の組込みおよびBBBを渡るトランスサイトーシスを提供する。更に好ましい態様において、その範囲は、リポソームのグラムあたり0.08〜0.12グラムである。更に好ましくは、EYPC対コレステロールオレアート対ApoE3のモル比(molar ratio of EYPC to cholesterol oleate to ApoE3)は、重量ベースで23:2.2である。
【0072】
本発明の更なる態様において、LDLベースのキャリアシステムは、脳への作用物質の輸送およびデリバリーを更に促進するために、表面層に共に組込まれた追加のターゲッティング分子を含有してもよい。例として、コレステロールの酸化誘導体(オキシステロール)、たとえばコレステロールヒドロペルオキシド、コレステロールエポキシド、およびヒドロキシコレステロール誘導体を使用して、取込みを改善してもよい。また、LDL粒子は、ApoBまたはApoE4などの他のアポタンパク質を組み込んでもよい。
【0073】
また本発明は、種々の脳疾患または障害を治療するための治療剤など一以上の広範囲の物質の組込みに関する。LDLキャリアシステムの利点の一つは、化学的に不安定なもの、活性が高いもの、または水溶液中で容易に加水分解されるものなど広範囲の薬剤クラスを安全かつ自然にデリバーすることができる能力である。
【0074】
考えられる作用物質のリストは、以下のものを含むがこれらに限定されない:脳傷害および神経変性疾患を治療するための向神経性因子、たとえばNGF、またはアミロイド前駆タンパク質から産生される向神経性フラグメント;遺伝的欠陥により失われた酵素に取って代わるための酵素、たとえばフェニルアラニンヒドロキシラーゼ;パーキンソン病で失われたドーパミンを再生するための酵素、たとえばチロシンヒドロキシラーゼおよびDOPAデカルボキシラーゼ;失った生合成機能を回復するための酵素アクチベーターまたはインヒビター;感染症を治療するための抗生物質、たとえばテトラサイクリン;痛み、または行動、認知、および振る舞いを含むコンディションを治療するための神経伝達物質および神経調節物質;耐量を全身に投与したときに充分な量が脳に到達しない作用物質を用いて脳腫瘍または他のコンディションを治療するための化学療法剤および抗AIDS剤、たとえばエトポシド、リバビリン、または抗ヒスタミン薬、たとえばロラタジン、フェキソフェナジン、またはセルチリジン(Zyrtec(登録商標));診断用薬、たとえばガドリニウム誘導体、イオヘキソール、またはイオキサグレートなどの造影剤、または全身投与で脳への浸透が低いために現在使用されない作用物質;治療用または診断用タンパク質、たとえば人工および天然の抗体;および遺伝子またはタンパク質、またはDNA、RNA、またはアミノ酸を含むその一部をコードする治療用配列であって、BBBを渡って非侵襲的に導入することができるもの。
【0075】
LDLキャリアに存在する治療剤の量は、分子のタイプに依存して大きく変化する。LDLキャリアにおける最適な取込みのために、治療剤の量は、0.1モル/モルコレステロール未満とすべきである。より高いレベルは、LDL粒子を不安定にすることが予測される。本発明は、複数の薬剤または追加の作用物質が、同粒子中に存在してもよいことを更に想定する。
【0076】
一つの態様において、活性物質が充分に疎水性で、コレステロールとのミクロエマルジョンとして残るときには、活性物質の非共有結合修飾が、本発明のLDL粒子における取込みのために必要である。このことは、多くの中性および適度に荷電した分子にあてはまる。
【0077】
別の態様において、たとえば治療剤がDNAまたはペプチドのように高い電荷をもつか、あるいは拡散を妨害する場合、治療剤は、コレステロールに共有結合することができる。好ましい態様において、共有後結合はエステル結合でなされ、これにより、脳組織でみられるユビキタスなエステラーゼにより治療剤は放出され得る(Yordanov et al.,(2002) J. Med. Chem. 45(11): 2283-8;Yang et al.(1995) Pharm. Res. 12(3): 329-36)。あるいは、当業者であれば、同様に機能するその他の結合モード、なかでも例示的には、ホスファチジルコリン、その他の親油性化合物、またはApoE自体への共有結合を選択することができるであろう。
【0078】
上述のとおり、本発明のLDL粒子は、PEG化リポソーム、ナノ粒子、および同様の人工物質などの他のキャリアと比較したときに幾つかの利点を有する。本発明のLDL粒子は、血液血漿の標準の構成成分からつくられ、その天然のレセプターに結合し、外来性必須脂肪酸に対する脳の生来の要求の一環として標準の経路により細胞に輸送される。従って、その毒性は、ポリマーベースのキャリアや細胞膜の完全な状態を破壊する作用物質より有意に低い。薬剤を脳へデリバーするために用いられる現在の方法は、マンニトールによるBBBの浸透圧性破壊である。しかし、BBBの破壊は重大な欠点を有し、とりわけ所望の治療剤と共にトキシンやウイルスまでも脳に侵入させるという事実を有し、これは重大な付随効果を引き起こし得る。
【0079】
また本発明の人工LDLキャリアシステムは、少なくとも2時間血漿中に高レベルで残存するという利点を提供する(図4)。これは、脳への物質の充分な取込みやデリバリーにとって充分効果的な血漿濃度(しばしば「血漿濃度曲線下面積」すなわちAUCと称される)を維持する際に重要である。
【0080】
例3に示されるとおり、脳組織を特異的にターゲットとすることにより、薬剤が肝臓に取り込まれP450などの肝臓酵素による不活性化を含む解毒経路により不活性化されにくいため、本発明のLDL粒子は、薬剤の治療効果を有意に増大させる。
【0081】
人工LDL粒子を含む組成物
本発明のLDL粒子は、治療する脳疾患のタイプによって種々の方法で製剤化され得る。従って、本方法は、ヒトまたは動物薬に使用するために製剤化された少なくとも一の薬剤含有LDL粒子複合体を含む薬学的組成物をその範囲内に含む。かかる組成物は、薬学的に許容可能なキャリアまたは賦形剤、必要に応じて補助的薬剤と共に使用に供されてもよい。また慣用的なキャリアも本発明とともに使用することができる。許容可能なキャリアには、グルコース、生理食塩水、およびリン酸緩衝生理食塩水が含まれるがこれらに限定されない。
【0082】
処理により遊離の治療剤またはその他の組込まれなかった分子を除去した後、LDL粒子懸濁液を、患者に投与するために薬学的に許容可能なキャリア中に所望の濃度にする。LDL粒子は大きすぎて非経口的に効率よく吸収されないため、組成物は、静脈内使用が意図されているが、筋内または動脈内に投与されてもよいし、あるいは適切な改変を加えて非経口的または経口的に投与されてもよいことが考えられる。
【0083】
このように、本発明は、LDL粒子中に含有される活性物質の溶液および薬学的に許容可能なキャリア、好ましくは水性キャリアを含む、BBBをターゲットとして投与するための組成物を提供する。得られた組成物は、滅菌してキットで使用するためにパッケージ化してもよいし、あるいは無菌条件下で濾過して凍結乾燥してもよい。凍結乾燥された調製物を含有する静脈内投与のためのキットは、投与前に混合するための無菌水溶液を含んでいてもよい。
【0084】
本組成物は、おおよその生理学的条件に必要とされる薬学的に許容可能な補助的物質、たとえば、pH調整および緩衝剤、張度調整剤など、たとえば、酢酸ナトリウム、乳酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムなどを含有していてもよい。
【0085】
これら製剤中のLDL粒子の濃度は、大きく変化させることができ、すなわち約0.5%未満、通常約1%または少なくとも約1%から、5〜10重量%まで変化させることができる。静脈内に投与可能なLDL粒子製剤を調製するための更なる方法は、当業者に公知である。かかる方法は、たとえば、Goodman & Gilman, The Pharmacological Basis of Therapeutics by Joel G. Hardman (Editor), Lee E. Limbird McGraw-Hill Professional; ISBN: 0071354697; 10th edition (August 13, 2001) に詳細に記載されている。
【0086】
人工LDLキャリアを使用する治療方法
更なる態様において、人工LDL粒子を、治療を必要としている哺乳類宿主に投与して、作用物質をBBBを渡って脳へ効果的にデリバーすることができる。治療用としては、効果的な量の薬剤含有LDL粒子を、LDL粒子を毛細血管内皮細胞に取込むことが可能な任意の様式により対象に投与することができる。
【0087】
臨床上の適用において、LDL粒子デリバリーシステムは、アルツハイマー病、パーキンソン病、および脳腫瘍などに用いられる薬剤の治療効果を有意に高める。たとえば、神経成長因子などの向神経性因子をLDL粒子に組込むことができ、当該ペプチドの脳への取込みを可能にする。これにより、多くの神経変性疾患において有益である新規神経突起の成長が引き起こされる。本明細書に記載されるとおり、当業者であれば、本発明のLDLキャリアシステムを用いた広範囲のその他の臨床上の適用が存在することを認識するでしょう。
【0088】
治療剤のコレステロール−コンジュゲート
コレステロールは、相対的に化学的に不活性な分子である。その結果、コレステロールをアミン含有治療剤と反応させる前に、コレステロールを活性化しなければならない。たとえば、コレステロールは、環状無水物、たとえばカルボキシル基を含有するフタル酸エステルまたはコハク酸エステルを産生する無水フタル酸または無水コハク酸と反応させることにより活性化することができる。カルボキシル基は、その後ペンタフルオロフェノールとの反応により活性化され、更にジイソプロピルカルボジイミドと反応して、アミン含有治療剤とアミド結合をつくる。得られた生成物は、治療剤がエステル結合を介してコレステロールにコンジュゲートしたコレステロール−治療剤コンジュゲートである。コレステロール−治療剤コンジュゲートは、充分親油性であるため本発明の人工LDL粒子に組込み可能である。また、好ましい結合がエステル結合であるため、治療剤は、ユビキタスな内在性エステラーゼの作用によりコレステロール部分から放出され得る。このように、コンジュゲートからの治療剤の放出は、過酷な状況やリソソームにみられる非特異的ペプチダーゼに依存しない。コレステロールと治療剤との間の好ましい結合はエステル結合であるが、本発明は、エーテル、アミドおよびペプチド結合を含むがこれらに限定されない他の結合を想定する。
【0089】
あるいは、アミンまたはヒドロキシル含有治療剤は、チオコレステロール、治療剤および二官能性架橋試薬(たとえばPMPIであるがこれに限定されない)の反応により、チオコレステロールにコンジュゲートすることができる。得られるコンジュゲートは、チオエーテル結合により連結されたコレステロールと治療剤のコンジュゲートである。治療剤がアミノ基を含有する場合、多くの商業的に入手可能な二官能性架橋剤の一つ(スクシンイミジル 4-[N-マレイミドメチル]シクロヘキサン-1-カルボキシレートを含むがこれに限定されない)を用いて、チオコレステロールにコンジュゲートすることができる。
【0090】
コレステロール−治療剤のコンジュゲートが一旦形成されると、本明細書に記載されるとおり、非コンジュゲートコレステロールオレアート、リン脂質、リポタンパク質と混合し、これにより本発明の人工LDL粒子を作成することができる。
【0091】
上述の各引用文献は、その全体を参照により本明細書の開示内容の一部とする。
【0092】
実施例
以下の実施例は、本発明を更に説明するためのものであり、本発明の範囲を限定するものと解釈すべきでない。
【0093】
例1:全長アポタンパク質E(ApoE)の精製
Avanti Polar Lipids, Inc.から入手したDMPC (1,2-ジミリストイル-sn-グリセロ-3-ホスホコリン) を、ガラス管内でベンゼン中に100 mg/mlの濃度で懸濁し、ベンチトップソニケーターを用いて音波処理する。DMPC懸濁液をシェル凍結し、一晩凍結乾燥した後、30 ml 標準バッファー (10 mM Tris-HCl. pH 7.6. 0.15 M NaCI, 1 mM EDTA. 0.0005% NaN3) 中に再懸濁し、10-20 mg/ml DMPCを作成し、プラスチック製50 ml円錐管に移す。円錐管を水浴に置き、マイクロチップを備えたソニケーターを用いて、4回の10分間隔で、2-3 分の冷却を散在させて音波処理する。音波処理は、溶液がほぼ透明になるまで繰り返す。その後、音波処理されたDMPCを2000 rpmで20分間遠心分離し、音波処理の間に分離した(slough off)かもしれないチタンを除去する。
【0094】
クローンApoEを発現する細菌細胞を遠心分離により集め、2分の冷却間隔により隔てられた4ピリオドの強力音波処理を30秒間用いて、大きなチップを備えたBranson Sonifierで氷上において音波処理する。音波処理された懸濁液を4℃で20分間39.000 x gで遠心分離し、細胞破片を除去し、上清を元の培地1リットルあたり100 mgでDMPCと混合する。その混合物を水浴において24℃(DMPCの遷移温度)で一晩インキュベートする。
【0095】
固体KBrの添加によりDMPC-ApoE混合物の密度を1.21 g/mlに上昇させ、15℃において60 Ti Beckman固定アングルローターで55,000 rpmで一晩、ステップ勾配(1.21、1.10、および1.006 g/ml)で遠心分離する。遊離のDMPCを含有する管の頂上近くの白色バンドを捨て、1.009 mg/mlの密度で浮遊するその下のApoE-DMPC複合体を取り出す。ApoEの存在をゲル電気泳動により確認する。その後、適切なフラクションを0.1M NH4HC03および0.0005% NaN3に対して4℃において3回のバッファー交換で透析する。
【0096】
その後、タンパク質をトロンビンで分解処理し、ベクターにより残されたポリヒスチジンタグを除去する。活性化トロンビンとタンパク質を、1:500のトロンビン:ApoE、w/wの比で混合し、37℃で少なくとも1時間インキュベートし、その一部をゲル電気泳動で分析して、タンパク質が完全に切断されたことを保証する。2つの切断部位が存在するため、不完全な切断により、より大きな分子量の第二のバンドが得られる。完全な切断が証明されたらすぐβ−メルカプトエタノールを1%最終濃度まで添加し、反応を停止させる。その後ApoEを、50 ml酸洗浄管においてシェル凍結し、一晩凍結乾燥する。
【0097】
その後ApoEを、30 ml冷却クロロホルム/メタノール(2:1 v/v)で洗浄し、J6 Beckman
遠心機において4℃で10分間1500 rpmで遠心分離することにより集める。沈殿を少量の冷却メタノールに再懸濁し、ボルテックスにかけ、その後、管を冷却メタノールで満たし、遠心分離する。この工程により、残存するクロロホルムが除去される。メタノールを注ぎ出し、ボルテックスにかけられる少量を管に残し、沈殿からペーストを作成する。6M グアニジン-HCl、0.1M Tris HCl、pH 7.4、0.01% EDTA、1% β-メルカプトエタノール、および0005% アジ化ナトリウムを含有する溶液5 mlを添加し、4M グアニジン-HCl、0.1M Tris-HCl、pH 7.4、0.1% β-メルカプトエタノール、および0.0005% NaN3で平衡化したSephacryl S-300カラムにロードする。0.1% β-メルカプトエタノールおよび0.0005% アジ化ナトリウムを含有する4M グアニジンバッファーを用いてタンパク質を溶出する。フラクションを0.1M NH4HC03および0.0005% NaN3に対して4回のバッファー交換で透析する。精製されたApoEを、YM10 Amicon Centriprepコンセントレーター(Millipore)を用いて、最終濃度1-2 mg/mlに濃縮し、-20℃に保存する。
【0098】
例2:LDL-リポソームのApoE強化(enrichment)
1.脂質ケーキの調製: 卵黄ホスファチジルコリン(25 mg)およびコレステリルオレアート(2 mg)を、メタノール/クロロホルム(2:3)に溶解する(比25:1)。溶媒を4℃で窒素下において蒸発させる。
【0099】
2.リポソームの調製: 脂質ケーキを、予め窒素ガス下でバブリングした0.1 M塩化カリウムを含有する12 mlの10 mM Tris-HClバッファー pH 8.0中で水和させた。その混合物を、54℃で窒素の流れの下において18-umのアウトプットで1時間音波処理し、遠心分離して、音波処理の間に生成したチタン粒子を除去する。
【0100】
3.人工LDLの調製: ステップ2で調製されたリポソームを、ApoEタンパク質とともに1/10(w/w)タンパク質/脂質の比で37℃において30分間インキュベートする。その後リポソームを、1.064、1.019、および1.006 g/cm3の密度の3層KClステップ勾配を用いて、4℃で18時間285,000 × gで密度勾配超遠心分離をすることにより精製し濃縮する。KBrをリポソーム溶液に添加し、その密度を1.21に上昇させ、遠心管の底に適用する。Optiseal管(Beckman)が超遠心分離ステップに適している。遠心後、リポソームは管の上端から約1/4の位置に細い乳白色層として観察される。この層を取り出し、1 mM EDTAを含有するPBS(リン酸緩衝化生理食塩水)に対して4℃で一晩透析する。LDL懸濁液は少なくとも7時間安定であり、アルゴンまたは窒素の下で-20℃で保存することができる。
【0101】
例3:脳における人工LDL粒子の取込み
約1 mgの脂質を含有する14C-LDLのPBS懸濁液を、Sprague-Dawleyラット(150 g)の尾部静脈に注射した。種々の間隔で、EGTAを含有するシリンジを用いた心臓穿刺により血液サンプルを得た。血漿、RBC、および組織を、水中でホモジナイズし、14Cをシンチレーションカウンターで測定した。
【0102】
図2は、脳(左)および肝臓(右)におけるLDLおよび脂質の取込みの結果を示す。オスSprague-Dawleyラットに、14C-コレステロールを含有するLDLまたは脂質粒子を静脈内に注射し、脳および肝臓における放射能を2時間後に測定した。脳および肝臓は、ApoEの存在を除けば同一の組成を有する脂質粒子と比較して、LDLからそれぞれ19.8倍および4.7倍のラベルを取込んだ。このことは、この取込みがLDLレセプター介在性トランスサイトーシスにより引き起こされることを示す(p(>T)=0.00055)。
【0103】
図3(左)は、脳および肝臓における脂質粒子取込みに対するLDL取込みの比を示す。脳は、肝臓と比べて、脂質粒子に対するLDLの取込みパーセンテージが高く(p(>T)=0.0003)、肝臓と比べてターゲット器官として4倍高い特異性を示す。図3(右)は、LDLおよび脂質粒子に関する、肝臓の取込みに対する脳の取込みの比を示す。器官特異性の別の指標である脳:肝臓の比は、LDLに関しては脂質粒子より高かった(p(>T)=0.0003)。すなわち、脂質粒子は、脳よりも肝臓に5.88倍多く取込まれたが、LDL粒子は脳よりも肝臓に1.36倍多くだけ取込まれた(p(>T)=0.034)。
【0104】
図4は、14C標識LDL粒子の血漿レベルの時間的経過を示す。血中14Cは、LDLの注射後少なくとも2時間は一定のままであり、これは、標識が他の器官により循環系から取り出されないことを示す。
【0105】
例4:第一級アミンの14Cコレステロールとのコンジュゲーション
コレステロール−フタレートエステルの調製: コレステロール(1 mg)を、窒素を用いて蒸発させ、凍結乾燥させてエタノールを除去する。その固体を最少体積のTHFに溶解し、ガラス反応バイアルに移す。固体の無水フタル酸(1-2 mg, >4当量)を添加し、その後50 Et3Nを添加する。その混合物を100℃で5分間加熱し、〜20μlピリジンを添加して、懸濁液を透明にする。その混合物を100℃で30分間、溶液が赤くなるまで加熱し、TLC(Silica/UV254プレート)でEtOH/トルエン(2:1)を用いて精製する。最も濃いUVバンド(RF=0.74)をプレートからスクラップし、生成物をTHFで溶出する(収率:98=100%)。
【0106】
ペンタフルオロフェノールを用いたカルボキシル基の活性化: THF中のペンタフルオロフェノール(PPP)10 mgを、コレステロールフタレートに添加し、その後5μlのDIIC(ジイソプロピルカルボジイミド)を添加する。その溶液を反応バイアル中において室温(RT)で1時間反応させ、活性化されたコレステロール−フタレート−PFPをTLC(CHCl3/CH3OH 30:5)により精製し、THFで溶出する(収率:100%)。
【0107】
活性化されたアミドの生成: THF中の活性化されたコレステロール−フタレート−PFPを10μlまで蒸発させ、DMF中に溶解した2当量の第一級アミンを添加する。DMFは、副反応を引き起こし得るが、過剰なアミンが存在する場合これは問題ではない。メタノールまたはエタノールなどのアルコールの添加は、その収率を大きく低下させる。3マイクロリットルのDIICを添加し、溶液をRTで一晩反応させ、TLC(CHCl3/CH3OH 30:5)により精製する(収率 98%)。重要なことに、Silica Gel G-25 UV254プレート(Alltech 809021)だけが精製のために使用された。Silica gel 60プレートでは、精製されていないあいまいで不明瞭なバンドが生じる。
【0108】
例5:アドリアマイシン−コレステロール(I)の合成
THF中の活性化されたコレステロール−フタレート−PFPを10μlまで蒸発させる。DMF中に溶解した2当量のアドリアマイシンを添加する。DMFによる副反応は、過剰のアミンを用いることにより低減することができる。メタノールまたはエタノールなどのアルコールの添加は、その収率を大きく低下させる。3マイクロリットルのDIICを添加し、溶液をRTで一晩反応させ、TLC(CHCl3/CH3OH 30:5)により精製する(収率 95%)。このバンドは、アドリアマイシン−DIICコンジュゲートと生成物を含有する。コレステロール−アドリアマイシンコンジュゲートは、100% CH3OH中でC18−逆相HPLCにより単離することができる(流速=0.5 ml/分、検出=A471)。生成物は、コレステロールとアドリアマイシンのおよそ中間の4.7分で溶出する。全収率:95%。アドリアマイシン−コレステロールコンジュゲート(I)の構造を図5に示す。
【0109】
例6:ヒドロキシ含有化合物の14Cコレステロールとのコンジュゲーション
N-[p-マレイミドフェニル]イソシアネート (PMPI) (5 mg) を、200μl DMSO中で1当量のヒドロキシ含有化合物と混合し、室温で30分間反応させる。チオコレステロール (6.4 mg) を添加する。その混合物を室温で120分間インキュベートし、生成物を、0.1 M EDTA pH 8.0で下塗りしたSilica Gel G UV 254プレートを用いて、EtOH/H2O 1:1を溶媒として用いた薄層クロマトグラフィーにより単離する。
【0110】
例7:テトラサクリン−コレステロール(II)の合成
テトラサイクリンを凍結乾燥させて溶媒を除去し、200μl DMSO中の2.2 mgテトラサイクリンを5 mg PMPIと混合し、室温で30分間反応させる。チオコレステロール(6.4 mg)を添加する。その混合物を室温で120分間インキュベートし、生成物を、0.1 M EDTA pH 8.0で下塗りしたSilica Gel G UV 254プレートを用いて、EtOH/H2O 1:1を溶媒として用いた薄層クロマトグラフィーにより単離する。テトラサクリン−コレステロールコンジュゲート(II)の構造を図6に示す。
【0111】
参照文献
下記参照文献のそれぞれは、その全体を参照により本明細書の開示内容の一部とする。
【参照文献】
【0112】
【図面の簡単な説明】
【0113】
【図1】放射性標識脂質およびLDLの臭化カリウム密度勾配プロファイル。
【図2】脳(左)および肝臓(右)におけるLDLおよび脂質の取込み。
【図3】(左)脳および肝臓における脂質粒子の取込みに対するLDLの取込みの比。(右)LDLおよび脂質粒子の脳への取込みの比。
【図4】14C標識LDL粒子の血漿レベルの時間的経過。
【図5】フタレートエステルを介してコレステロールに連結された親水性分子の化学構造。示される具体的な親水性分子はアドリアマイシンである。コンジュゲーションすることなくアドリアマイシン単独では、親水性であるためにLDL粒子に組み込むことはできない。
【図6】チオエーテルエステル介してコレステロールに連結された親水性分子の化学構造。示される具体的な親水性分子はテトラサイクリンである。コンジュゲーションすることなくテトラサイクリン単独では、親水性であるためにLDL粒子に組み込むことはできない。
【特許請求の範囲】
【請求項1】
外側のリン脂質単一層と固体の脂質コアとを含む人工LDL粒子であって、前記外側のリン脂質単一層が少なくとも一のアポリポタンパク質を含み、前記固体の脂質コアが少なくとも一の治療剤を含有する人工LDL粒子。
【請求項2】
前記少なくとも一のアポリポタンパク質がApoEである、請求項1に記載の人工LDL粒子。
【請求項3】
前記少なくとも一のアポリポタンパク質がApoE3である、請求項2に記載の人工LDL粒子。
【請求項4】
前記外側のリン脂質単一層が、一以上のオキシステロールおよび/またはApoBおよびApoE4から成る群より選択される追加のアポリポタンパク質を更に含む、請求項3に記載の人工LDL粒子。
【請求項5】
前記少なくとも一の治療剤が、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される、請求項1に記載の人工LDL粒子。
【請求項6】
前記少なくとも一の治療剤が、アミノ酸、ペプチド、タンパク質、核酸、炭水化物および脂質から成る群より選択される作用物質とコレステロールとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項7】
前記治療剤が、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項5に記載の人工LDL粒子。
【請求項8】
前記治療剤が、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項6に記載の人工LDL粒子。
【請求項9】
前記外側のリン脂質単一層が、ホスファチジルコリンおよび少なくとも一のアポリポタンパク質を含む、請求項1に記載の人工LDL粒子。
【請求項10】
前記少なくとも一のアポリポタンパク質がApoEである、請求項9に記載の人工LDL粒子。
【請求項11】
前記粒子が直径約15〜50 nmである、請求項1に記載の人工LDL粒子。
【請求項12】
前記粒子が直径約20〜30 nmである、請求項1に記載の人工LDL粒子。
【請求項13】
前記粒子が密度約1.00〜1.07 g/mLである、請求項1に記載の人工LDL粒子。
【請求項14】
前記粒子が密度約1.02〜1.06 g/mLである、請求項1に記載の人工LDL粒子。
【請求項15】
前記粒子が少なくとも2時間の血清安定性を有する、請求項1に記載の人工LDL粒子。
【請求項16】
前記粒子が、トランスサイトーシスにより血液−脳関門(BBB)を渡って輸送される、請求項1に記載の人工LDL粒子。
【請求項17】
前記粒子の脳に対する取込み特異性が、肝臓に比べて少なくとも3倍高い、請求項1に記載の人工LDL粒子。
【請求項18】
前記少なくとも一の治療剤が、コレステロールとアドリアマイシンとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項19】
前記少なくとも一の治療剤が、コレステロールとテトラサイクリンとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項20】
前記コンジュゲートのコレステロールとアドリアマイシンが、エステル結合により連結される、請求項18に記載の人工LDL粒子。
【請求項21】
前記コンジュゲートのコレステロールとテトラサイクリンが、エステル結合により連結される、請求項19に記載の人工LDL粒子。
【請求項22】
外側のホスファチジルコリン単一層、脂肪酸アシル−コレステロールエステルを含む固体の脂質コア、および前記外側単一層におけるApoEを含む、作用物質を血液−脳関門を渡ってデリバーするための人工LDL粒子。
【請求項23】
前記固体の脂質コアが、コレステロールを更に含む、請求項22に記載の人工LDL粒子。
【請求項24】
前記外側単一層におけるApoEが、ApoE3である、請求項22に記載の人工LDL粒子。
【請求項25】
請求項1に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項26】
請求項4に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項27】
請求項5に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項28】
アミノ酸、ペプチド、タンパク質、核酸、炭水化物および脂質から成る群より選択される治療剤に連結されたコレステロールを含むコンジュゲート。
【請求項29】
前記治療剤が、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項28に記載のコンジュゲート。
【請求項30】
前記治療剤がアドリアマイシンである、請求項29に記載のコンジュゲート。
【請求項31】
前記アドリアマイシンとコレステロールがエステル結合により連結される、請求項30に記載のコンジュゲート。
【請求項32】
前記治療剤がテトラサイクリンである、請求項29に記載のコンジュゲート。
【請求項33】
前記テトラサイクリンとコレステロールがエステル結合により連結される、請求項32に記載のコンジュゲート。
【請求項34】
1)コンジュゲートまたは非コンジュゲート治療剤を含有するリン脂質を緩衝液中に懸濁する工程;
2)前記溶液を音波処理し、外側のリン脂質単一層と固体の脂質コアを形成する工程;
および
3)少なくとも一のアポリポタンパク質を含む溶液を添加し、前記アポリポタンパク質が前記外側のリン脂質単一層に取り込まれる工程
を含む、請求項1に記載の人工LDL粒子を製造する方法。
【請求項35】
製造される前記人工LDL粒子が、直径10〜50 nmである、請求項34に記載の方法。
【請求項36】
効果的な量の請求項25に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項37】
効果的な量の請求項26に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項38】
効果的な量の請求項27に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項39】
請求項25に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【請求項40】
請求項26に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【請求項41】
請求項27に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【請求項1】
外側のリン脂質単一層と固体の脂質コアとを含む人工LDL粒子であって、前記外側のリン脂質単一層が少なくとも一のアポリポタンパク質を含み、前記固体の脂質コアが少なくとも一の治療剤を含有する人工LDL粒子。
【請求項2】
前記少なくとも一のアポリポタンパク質がApoEである、請求項1に記載の人工LDL粒子。
【請求項3】
前記少なくとも一のアポリポタンパク質がApoE3である、請求項2に記載の人工LDL粒子。
【請求項4】
前記外側のリン脂質単一層が、一以上のオキシステロールおよび/またはApoBおよびApoE4から成る群より選択される追加のアポリポタンパク質を更に含む、請求項3に記載の人工LDL粒子。
【請求項5】
前記少なくとも一の治療剤が、アミノ酸、ペプチド、タンパク質、炭水化物および脂質から成る群より選択される、請求項1に記載の人工LDL粒子。
【請求項6】
前記少なくとも一の治療剤が、アミノ酸、ペプチド、タンパク質、核酸、炭水化物および脂質から成る群より選択される作用物質とコレステロールとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項7】
前記治療剤が、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項5に記載の人工LDL粒子。
【請求項8】
前記治療剤が、向神経性因子、成長因子、酵素、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項6に記載の人工LDL粒子。
【請求項9】
前記外側のリン脂質単一層が、ホスファチジルコリンおよび少なくとも一のアポリポタンパク質を含む、請求項1に記載の人工LDL粒子。
【請求項10】
前記少なくとも一のアポリポタンパク質がApoEである、請求項9に記載の人工LDL粒子。
【請求項11】
前記粒子が直径約15〜50 nmである、請求項1に記載の人工LDL粒子。
【請求項12】
前記粒子が直径約20〜30 nmである、請求項1に記載の人工LDL粒子。
【請求項13】
前記粒子が密度約1.00〜1.07 g/mLである、請求項1に記載の人工LDL粒子。
【請求項14】
前記粒子が密度約1.02〜1.06 g/mLである、請求項1に記載の人工LDL粒子。
【請求項15】
前記粒子が少なくとも2時間の血清安定性を有する、請求項1に記載の人工LDL粒子。
【請求項16】
前記粒子が、トランスサイトーシスにより血液−脳関門(BBB)を渡って輸送される、請求項1に記載の人工LDL粒子。
【請求項17】
前記粒子の脳に対する取込み特異性が、肝臓に比べて少なくとも3倍高い、請求項1に記載の人工LDL粒子。
【請求項18】
前記少なくとも一の治療剤が、コレステロールとアドリアマイシンとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項19】
前記少なくとも一の治療剤が、コレステロールとテトラサイクリンとの間で形成されたコンジュゲートである、請求項1に記載の人工LDL粒子。
【請求項20】
前記コンジュゲートのコレステロールとアドリアマイシンが、エステル結合により連結される、請求項18に記載の人工LDL粒子。
【請求項21】
前記コンジュゲートのコレステロールとテトラサイクリンが、エステル結合により連結される、請求項19に記載の人工LDL粒子。
【請求項22】
外側のホスファチジルコリン単一層、脂肪酸アシル−コレステロールエステルを含む固体の脂質コア、および前記外側単一層におけるApoEを含む、作用物質を血液−脳関門を渡ってデリバーするための人工LDL粒子。
【請求項23】
前記固体の脂質コアが、コレステロールを更に含む、請求項22に記載の人工LDL粒子。
【請求項24】
前記外側単一層におけるApoEが、ApoE3である、請求項22に記載の人工LDL粒子。
【請求項25】
請求項1に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項26】
請求項4に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項27】
請求項5に記載の人工LDL粒子と薬学的に許容可能なキャリアを含む、作用物質を血液−脳関門を渡ってデリバーするための組成物。
【請求項28】
アミノ酸、ペプチド、タンパク質、核酸、炭水化物および脂質から成る群より選択される治療剤に連結されたコレステロールを含むコンジュゲート。
【請求項29】
前記治療剤が、向神経性因子、成長因子、酵素、抗体、神経伝達物質、神経調節物質、抗生物質、抗ウイルス剤、抗真菌剤および化学療法剤から成る群より選択される、請求項28に記載のコンジュゲート。
【請求項30】
前記治療剤がアドリアマイシンである、請求項29に記載のコンジュゲート。
【請求項31】
前記アドリアマイシンとコレステロールがエステル結合により連結される、請求項30に記載のコンジュゲート。
【請求項32】
前記治療剤がテトラサイクリンである、請求項29に記載のコンジュゲート。
【請求項33】
前記テトラサイクリンとコレステロールがエステル結合により連結される、請求項32に記載のコンジュゲート。
【請求項34】
1)コンジュゲートまたは非コンジュゲート治療剤を含有するリン脂質を緩衝液中に懸濁する工程;
2)前記溶液を音波処理し、外側のリン脂質単一層と固体の脂質コアを形成する工程;
および
3)少なくとも一のアポリポタンパク質を含む溶液を添加し、前記アポリポタンパク質が前記外側のリン脂質単一層に取り込まれる工程
を含む、請求項1に記載の人工LDL粒子を製造する方法。
【請求項35】
製造される前記人工LDL粒子が、直径10〜50 nmである、請求項34に記載の方法。
【請求項36】
効果的な量の請求項25に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項37】
効果的な量の請求項26に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項38】
効果的な量の請求項27に記載の組成物を、それを必要とする哺乳類に投与することを含む、物質を血液−脳関門を渡ってデリバーするための方法。
【請求項39】
請求項25に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【請求項40】
請求項26に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【請求項41】
請求項27に記載の組成物を含有する容器と使用説明書を含む、物質を血液−脳関門を渡ってデリバーするためのキット。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−131799(P2012−131799A)
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−21053(P2012−21053)
【出願日】平成24年2月2日(2012.2.2)
【分割の表示】特願2004−556682(P2004−556682)の分割
【原出願日】平成15年12月2日(2003.12.2)
【出願人】(503310224)ブランシェット・ロックフェラー・ニューロサイエンスィズ・インスティテュート (25)
【Fターム(参考)】
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願番号】特願2012−21053(P2012−21053)
【出願日】平成24年2月2日(2012.2.2)
【分割の表示】特願2004−556682(P2004−556682)の分割
【原出願日】平成15年12月2日(2003.12.2)
【出願人】(503310224)ブランシェット・ロックフェラー・ニューロサイエンスィズ・インスティテュート (25)
【Fターム(参考)】
[ Back to top ]