
NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の製造方法、それにより得られるタンパク質およびその使用
【課題】NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の製造方法、それにより得られるタンパク質およびその使用の提供
【解決手段】SNI、その部分ペプチドまたはそれらの発現ベクターを含む、医薬(例、連鎖球菌感染症、自己免疫疾患、多発性骨髄腫等の疾患の治療薬)および試薬;連鎖球菌由来NADaseおよびSNIの共発現ベクター、それを含む形質転換体、当該形質転換体を利用する連鎖球菌由来NADaseの製造方法、当該製造方法により得られるタンパク質;SNIおよびNADaseを含む複合体:NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:完全長SLOまたは大腸菌に毒性を示し得るその部分ペプチドの発現ベクター、それを含む大腸菌、当該大腸菌を用いる当該SLOまたは領域の製造方法:SLOに特異的な抗体など。
【解決手段】SNI、その部分ペプチドまたはそれらの発現ベクターを含む、医薬(例、連鎖球菌感染症、自己免疫疾患、多発性骨髄腫等の疾患の治療薬)および試薬;連鎖球菌由来NADaseおよびSNIの共発現ベクター、それを含む形質転換体、当該形質転換体を利用する連鎖球菌由来NADaseの製造方法、当該製造方法により得られるタンパク質;SNIおよびNADaseを含む複合体:NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:完全長SLOまたは大腸菌に毒性を示し得るその部分ペプチドの発現ベクター、それを含む大腸菌、当該大腸菌を用いる当該SLOまたは領域の製造方法:SLOに特異的な抗体など。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現する各タンパク質の製造方法、当該製造方法により得られるタンパク質およびその抗体、該タンパク質をコードするポリヌクレオチドを含むベクターおよびその形質転換体、ならびにそれらの使用などを提供する。
【背景技術】
【0002】
溶血性連鎖球菌(hemolytic streptococci)は、咽頭炎、皮膚感染症、猩紅熱、糸球体腎炎、リウマチ熱、敗血症などの原因となる病原細菌であり、赤血球を含む血液寒天平板培地上で溶血斑を形成し、細胞壁に局在する多糖抗原の免疫化学的特異性に基づいてA〜G群に分けられている。溶血性連鎖球菌感染症は、小児感染症として派生頻度の高い疾患である。また、1980年代以降、日本や欧米などの先進各国で、溶血性連鎖球菌による極めて急激な筋肉組織の壊死、多臓器に及ぶ障害、ショック症状などを伴う死亡率の高い劇症型感染症が発生しており、そのためヒト喰いバクテリアとしてたびたび報道などされている。
【0003】
溶血性連鎖球菌は、20種類以上にも及ぶさまざまな生理活性物質を菌体外に産生しているが、中でも溶血毒素であるストレプトリシンO(SLO)は古くから重要な病原性因子であると考えられ詳細な研究が進められてきた。最近、SLOにより宿主の細胞膜上に形成された孔からNADaseなどの病原性因子が宿主細胞内に移行することが確認された。この発見は、腸管出血性大腸菌などのグラム陰性病原菌で高度に保存されているタイプIII分泌機構がグラム陽性菌である溶血性連鎖球菌においても存在することを示すものとして大きな反響を呼び注目されている。
【0004】
本発明者らは、これまでに、SLO遺伝子がその上流に位置するNADase遺伝子(nga)からのreadthroughによりポリシストロニックに転写されていることを明らかにしている(非特許文献1参照)。このnga−sloオペロンは、A群およびC群溶血性連鎖球菌間で高度に保存されており、溶血性連鎖球菌感染症において重要な役割を果たしていると考えられるが、ngaとslo遺伝子との間には、遺伝子orf1が存在することがわかっていた。しかしながら、この遺伝子orf1の機能は全く不明である。
【0005】
NADaseは、その活性として、(a)β−ニコチンアミド−アデニン−ジヌクレオチド(NAD)を加水分解し、アデノシンジホスホリボース(ADPR)およびニコチンアミドを生成する活性(NADグリコヒドロラーゼ活性)、(b)NADからサイクリックADPリボース(cADPR)を合成する活性(ADPリボシルシクラーゼ活性)、および(c)cADPRを加水分解し、ADPRを生成する活性(cADPRヒドロラーゼ活性)を有する。NADaseは、SLOにより宿主細胞膜上に形成された孔から宿主細胞内に侵入し、これらの活性によりその毒性を発揮し、連鎖球菌感染症の病態を担い得ると考えられるので、その解析は連鎖球菌感染症を理解する上で非常に重要である。
【0006】
ところで、NADaseの製造方法としては、これまでに幾つかの報告がある。
例えば、非特許文献2および3には、溶血性連鎖球菌の培養上清からNADaseをその活性を保持した状態で精製したことが記載されている。しかしながら、大腸菌等の異種宿主細胞でのNADaseの発現は未だ成功していない。これは、NADaseが異種宿主細胞に対して極めて強い毒性を示すためである。
【0007】
また、SLO遺伝子は、異なる溶血性連鎖球菌群で高度に保存され、特にA群(GAS)とC群(GCS)では両者のSLOに本質的な相違はみられないことが知られている。両群のSLOは、N末端シグナルペプチドを含む31アミノ酸残基(3.3kDa)が外れて分泌されるとともに、自身が分泌するプロテアーゼにより次第に低分子化する。SLOは抗原性が強く中和抗体がつくられやすく、感染に際して血清中の抗ストレプトリジンO抗体(ASLO)が上昇するため、本菌による感染の診断に抗体価の測定が広く用いられている(抗SLO試験)。SLOは、抗SLO試験において試験抗原として使用されているだけでなく、巨大なリング状の孔を細胞膜上に形成し、生細胞内にタンパク質レベルの大きさの分子を送り込むことができるので、細胞膜の透過性を高めるツールとして近年多用されている。また、SLOはASLO精製用の抗原としても利用可能である。従って、SLOを大量かつ安価に製造する方法の開発が求められている。
【0008】
また、ASLOは臨床検査試薬等の試薬として有用であるが、通常市販されているASLOは溶血性連鎖球菌の培養上清から部分精製したSLOを抗原として用いて作製されている。ところが、培養上清から部分精製された調製物中にはSLOに加え、NADaseも混在しているため、このような調製物を用いて作製された抗体は、SLOのみならずNADaseとも交差反応することが知られている(非特許文献1、4)。従って、交差反応性が低い抗体の作製という観点からも、高純度のSLOを製造する方法の開発が求められている。
【0009】
SLOの製造方法としては、これまでに幾つかの報告がある。
例えば、特許文献1には、溶血性連鎖球菌の培養上清から完全長SLOを製造(精製)する方法が記載されている。しかしながら、溶血性連鎖球菌のSLO産生量は低く、また、自身が産生するプロテアーゼによる分解のために、溶血性連鎖球菌の培養上清からの完全長SLOの精製収率が低いという問題がある。
【0010】
また、宿主として大腸菌を用いるSLOの製造方法としては、以下の報告がある。
非特許文献5には、SLO遺伝子を高コピー数プラスミドにサブクローニングしたところ、宿主である大腸菌は溶菌しやすく、また増殖も遅く、さらにはSLO遺伝子自体が不安定であり脱落が生じたこと、ならびに、SLO遺伝子を低コピー数プラスミドに移したところ、SLO遺伝子は安定化したがSLOの産生量はわずかであったことが記載されている。
非特許文献6には、特定のN末端欠失型SLO(シグナルペプチド配列を含むN末端側の75アミノ酸残基を除いた496アミノ酸残基に相当するSLO)をマルトース結合タンパク質(MBP,42kDa)との融合タンパク質とすることで、SLOの大腸菌での大量発現に成功したことが記載されている。これは、完全長ではないが、大腸菌で初めてSLOの大量発現に成功した例である。
非特許文献7には、大腸菌における特定のN末端欠失型SLO(シグナルペプチド配列を含むN末端側の103アミノ酸残基を除いた468アミノ酸残基に相当する領域をコードするSLO)の大量発現に成功したことが記載されている。
しかしながら、大腸菌での完全長SLOの大量発現は未だ成功していない。これは、SLOのN末端近傍の32−75アミノ酸残基付近の領域が大腸菌に対して何らかの毒性を示すと考えられるためである(非特許文献5−7)。
【0011】
さらに、宿主として枯草菌(Bacillus subtilis)を用いる完全長SLOの大量発現について幾つかの報告がある(例えば、非特許文献8、特許文献2および3)。しかしながら、枯草菌はプロテアーゼの産生能力が高く、複数のプロテアーゼを産生するにもかかわらず、完全なプロテアーゼ欠損株の作製は未だ成功していないことから、SLOの分解が問題となっている。さらには、枯草菌は多様なタンパク質を分泌するため、SLOの精製過程が複雑になり、高純度のSLOを入手し難いという問題がある。
【0012】
【特許文献1】特開平5−331198号公報
【特許文献2】特開平5−184372号公報
【特許文献3】特開平6−237775号公報
【非特許文献1】Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005)
【非特許文献2】Karasawa et al., FEMS Microbiology Letters 130: 201-204 (1995)
【非特許文献3】Gerlach et al., FEMS Microbiology Letters 136: 71-78 (1996)
【非特許文献4】Madden et al., Cell 104: 143-152 (2001)
【非特許文献5】Kehoe et al., Infect Immun 43: 804-810 (1984)
【非特許文献6】Weller et al., Eur J Biochem 236: 34-39 (1996)
【非特許文献7】Yamamoto et al., Biosci Biotechnol Biochem 65: 2682-2689 (2001)
【非特許文献8】Yamada et al., Biosci Biotechnol Biochem 59: 363-366 (1995)
【発明の開示】
【発明が解決しようとする課題】
【0013】
上記の通り連鎖球菌由来NADaseおよびSLOの製造方法が幾つか報告されている。組換えタンパク質を高純度かつ安価に製造する方法としては大腸菌等の異種宿主細胞を利用する方法が知られている。ところが、完全長のNADaseおよびSLOを高純度で大量かつ安価に製造する方法は、以下の理由により未だ開発されていない。
【0014】
連鎖球菌由来NADaseは、大腸菌等の異種宿主細胞に対して強い毒性を示すため、完全長NADaseまたはその毒性を示す領域を含むNADase断片を、異種宿主細胞を用いて大量に製造する方法は開発されていない。NADaseの解析は連鎖球菌感染症の理解やその応用に非常に重要であると考えられるため、NADaseを大量に製造する方法の開発が求められている。
【0015】
連鎖球菌由来SLOは、そのN末端近傍の32−75アミノ酸残基付近の領域が大腸菌に対して何らかの毒性を示すと考えられ、完全長SLOまたは上記領域を含むSLO断片を、大腸菌を用いて大量かつ安価に製造方法は開発されていない。
SLOのようなコレステロール依存性の細胞障害毒素(cholesterol-dependent cytolysin)は、分子構造と機能が類似した大きなグループを形成しており、化膿レンサ球菌である Streptococcus属だけでなく、Bacillus、Clostridium、Listeria属などの細菌も産生すること、およびそれらのアミノ酸配列は高い相同性を示すことが知られている。しかしながら、SLOはこれらグループの中で最も分子量が大きく、また、そのN末端領域は他のグループには存在せず、溶血性連鎖球菌に特徴的である。このようなことから、完全長SLOおよびそのN末端断片は溶血性連鎖球菌感染症のための臨床検査薬(抗体作製用の抗原としても)だけでなく、病原機構を分子レベルで明らかにするための研究ツールとしても有用であると考えられる。従って、完全長SLOまたは上記領域を含むSLO断片の製造方法が開発できれば、純度の高い抗原が大量に得られ、また、交差反応性を実質的に有しないASLOの作製も可能になると考えられることから、それらを高純度で大量かつ安価に製造する方法の開発が求められている。
【課題を解決するための手段】
【0016】
本発明者らは、鋭意検討した結果、異種宿主細胞においてNADase遺伝子(nga)を、その下流に存在する未知遺伝子orf1とともに発現させることにより、完全長NADaseの大量発現に成功した。この結果に基づき、orf1遺伝子産物をさらに解析したところ、宿主細胞内でNADaseと複合体を形成することによりNADaseのインヒビターとして作用し得ることを見出した。本発明者らは、このorf1遺伝子産物を、連鎖球菌NADaseインヒビター(streptococcal NADase inhibitor:SNI)と標記することを提案する。NADaseと共発現し、複合体化するSNIは、その保有菌をNAD枯渇から保護し得ると考えられる。この知見に基づけば、連鎖球菌内のSNI・NADase複合体を解離させる物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。また、SNIは、NADaseと非常に強固かつ特異的に結合することから、現在広く用いられているHisタグ等の精製用タグと同様の効果が期待できると考えられる。さらに、SNIは熱に非常に安定性であった。また、SNIは、細菌由来NADaseのみではなく、哺乳動物由来NADaseも阻害した。従って、哺乳動物由来NADaseが関与し得る疾患の予防・治療にもSNIが応用可能と考えられる。以上のように、本発明者らの知見によれば、SNIを利用して種々の応用が可能である。
【0017】
また、本発明者らは、大腸菌での遺伝子発現を厳格に制御可能である誘導プロモーターを用いて、基底レベルの遺伝子発現を効果的に抑制し安定な培養を可能とするとともに、該プロモーターに対する誘導剤を併用することにより、完全長SLOの大量発現に成功した。本発明者はまた、SLOに適切なタグを付加することにより、高純度の完全長SLOを簡便かつ安価に製造することに成功した。本発明者らはさらに、上記のように調製された高純度の完全長SLOを用いることにより、NADaseに対する交差反応性を示さない抗SLO抗体を作製することに成功した。
【0018】
以上に基づき、本発明者らは、本発明を完成するに至った。即ち、本発明は、下記の発明などを提供する:
〔1〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、医薬。
〔2〕連鎖球菌感染症の治療薬である、上記〔1〕の医薬。
〔3〕自己免疫疾患、炎症性疾患または多発性骨髄腫の治療薬である、上記〔1〕の医薬。
〔4〕NADaseが細菌または動物由来NADaseである、上記〔1〕の医薬。
〔5〕細菌または動物由来NADaseが、連鎖球菌由来NADase、あるいはCD38またはCD157である、上記〔4〕の医薬。
〔6〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、NADase阻害剤。
〔7〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、白血球の走化性の阻害剤。
〔8〕白血球が好中球である、上記〔7〕の剤。
〔9〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、試薬。
〔10〕連鎖球菌由来NADaseの組換えタンパク質であって、天然型NADaseに対して1以上のアミノ酸の欠失、置換、付加および挿入からなる群より選ばれる1以上のアミノ酸の修飾が施された、組換えタンパク質。
〔11〕該1以上の修飾が精製用タグの付加を少なくとも含む、上記〔10〕の組換えタンパク質。
〔12〕上記〔10〕の組換えタンパク質を含む、医薬または試薬。
〔13〕上記〔10〕の組換えタンパク質をコードするポリヌクレオチド。
〔14〕第1および第2の発現ユニットの共発現ベクターであって、
第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み、
第1の発現ユニットが、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドであり、
第2の発現ユニットが、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドである、共発現ベクター。
〔15〕NADaseが細菌または動物由来NADaseである、上記〔14〕の共発現ベクター。
〔16〕NADaseが連鎖球菌由来NADaseである、上記〔14〕の共発現ベクター。
〔17〕NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを発現する、形質転換体。
〔18〕形質転換体が大腸菌である、上記〔17〕の形質転換体。
〔19〕上記〔17〕の形質転換体を培養し、培養上清からNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドを回収することを含む、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造方法。
〔20〕NADaseとの結合能および/またはNADase阻害活性を有するSNIの部分ペプチド。
〔21〕上記〔20〕の部分ペプチドをコードするポリヌクレオチド。
〔22〕上記〔21〕のポリヌクレオチドを含む、発現ベクター。
〔23〕SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む、複合体。
〔24〕NADaseが連鎖球菌由来NADaseである、上記〔23〕の複合体。
〔25〕SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法。
〔26〕以下(a)〜(d)からなる群より選ばれる構成要素を含む、SNIまたはNADaseとの結合能を有するその部分ペプチドとNADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成剤:
(a)SNI;
(b)NADaseとの結合能を有するSNIの部分ペプチド;
(c)NADase;
(d)SNIとの結合能を有するNADaseの部分ペプチド。
〔27〕以下(a)および/または(b)を含む、SNIまたはNADaseに親和性を有する融合タンパク質の調製剤:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター;
(b)NADaseまたはSNIとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター。
〔28〕以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたは該連鎖球菌由来NADaseとの結合能を有するその部分ペプチドと、該連鎖球菌由来NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成を抑制し得るか否かを評価する工程;
(b)該複合体の形成を抑制し得る被験物質を選択する工程。
〔29〕以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、該連鎖球菌由来NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
〔30〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターであって、
該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、ベクター。
〔31〕溶血性連鎖球菌がA群またはC群連鎖球菌である、上記〔30〕のベクター。
〔32〕該SLOが精製用タグを有する、上記〔30〕のベクター。
〔33〕該誘導プロモーターがアラビノースオペロン制御プロモーターである、上記〔30〕のベクター。
〔34〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターで形質転換された大腸菌であって、
該発現ベクターは、該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、大腸菌。
〔35〕大腸菌が、プロテアーゼの欠損株、および/または誘導プロモーターに対する誘導剤の代謝能の欠損株である、上記〔34〕の大腸菌。
〔36〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの製造方法であって、誘導プロモーターに対する誘導剤の存在下において上記〔34〕の大腸菌を培養し、培養上清から該SLOまたはその部分ペプチドを回収することを含む、方法。
〔37〕溶血性連鎖球菌由来SLOに対する特異的な反応性を示し、かつ溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体。
【発明の効果】
【0019】
本発明のポリペプチドは、種々の応用が可能である。詳細には、本発明のSNIおよびその部分ペプチドならびにそれらの発現ベクターは、例えば、連鎖球菌等の細菌の感染症、および感染症以外の種々の疾患の治療、ならびにNADaseの阻害および白血球の走化性の阻害に有用である。本発明の非天然型NADaseおよびその部分ペプチドならびにそれらの発現ベクターは、例えば、連鎖球菌NADaseの研究および当該NADaseを用いるその毒性を抑制し得る医薬の開発、ならびに抗菌または抗細胞剤として有用である。
本発明の発現ベクターは、例えば、医薬または試薬、あるいは目的ポリペプチドの大量発現に有用である。詳細には、本発明のSNIおよびNADase発現ベクターは、上記の通り医薬および/または試薬として使用できるのみならず、宿主細胞でのSNIおよびNADaseの大量発現、ならびに自身が精製用タグとして機能し得る融合タンパク質の作製などにも有用である。本発明のSLO発現ベクターは、完全長SLOまたはその毒性領域を含む部分ペプチドの大量発現などに有用である。従って、このような発現ベクターを含む形質転換体および当該形質転換体を用いる目的ポリペプチドの製造方法もまた有用である。
本発明の抗体は、例えば、SNI、NADaseまたはSLOの検出・定量、あるいはそれらの機能阻害による医薬または試薬などとして有用である。詳細には、本発明のSLO抗体は、SLOに対する特異性が高く交差反応しないため、試薬として特に有用である。
本発明の複合体およびその製造方法は、例えば、NADaseの製造、NADaseまたはSNIあるいはそれらの部分ペプチドを有するタンパク質の精製、ならびに本発明のスクリーニング方法に有用である。
本発明のスクリーニング方法は、例えば、連鎖球菌感染症の治療薬の開発に有用である。
【発明を実施するための最良の形態】
【0020】
1.ポリペプチドおよびその部分ペプチド
本発明は、連鎖球菌NADaseインヒビター(streptococcal NADase inhibitor:SNI)またはその部分ペプチドを提供する。本発明者らは、SNIの発現に初めて成功し、また、その機能の実証にも併せて成功した。NADaseに対する細菌性インヒビターの存在は、Bacillus subtilis、Proteus vulgaris、Proteus rettgeriおよびMycobacterium butyricumでは知られていたが、連鎖球菌では初めてである。しかも、熱安定性に関して、従来のNADaseインヒビターは、60℃で15分間加熱することにより完全に不活化される(Acta Path. Microbiol. Scandinav. 69: 277-286 (1967))のに対し、SNIはそのような条件でも活性を保持し得る。本発明のSNIは、NADase阻害活性を有する連鎖球菌由来ポリペプチドである限り特に限定されないが、例えば、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチドであり得る。
【0021】
本発明はまた、連鎖球菌由来の非天然型NADaseまたはその部分ペプチドを提供する。これまでに、溶血性連鎖球菌の培養上清からNADaseをその活性を保持した状態で精製したことが報告されている(例えば、FEMS Microbiology Letters 130: 201-204 (1995); FEMS Microbiology Letters 136: 71-78 (1996)参照)。しかしながら、連鎖球菌由来NADaseは、大腸菌等の宿主細胞に対して強い毒性を示すため、宿主細胞でのNADaseの発現、即ち、組換えタンパク質の調製には未だ成功していない。今回、本発明者らは、連鎖球菌由来NADaseの組換えタンパク質の調製に初めて成功した。本発明者らの研究成果により、宿主細胞を用いた連鎖球菌由来天然型NADaseの組換えタンパク質の調製のみならず、非天然型NADase(変異型NADaseまたは遺伝子改変NADase)の組換えタンパク質の調製が初めて可能となる。また、このように組換え技術を利用することで、NADaseの部分ペプチドの調製も容易になる。本発明の非天然型NADaseは、既報の天然型NADase以外のNADaseであって、かつNADase活性を有するポリペプチドである限り特に限定されないが、例えば、配列番号4で表されるアミノ酸配列、または配列番号4で表されるアミノ酸配列において分泌シグナル配列をコードするアミノ酸配列が除去されたアミノ酸配列(以下、必要に応じて、「配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列」と省略する)と実質的に同一のアミノ酸配列(但し、配列番号4で表されるアミノ酸配列、またはその分泌シグナル除去アミノ酸配列と同一のアミノ酸配列は除く)からなるポリペプチドであり得る。
【0022】
本明細書中で使用される場合、「NADase活性」とは、NADaseが有する任意の活性をいい、特に断らない限り、(a)ニコチンアミド−アデニン−ジヌクレオチド(NAD)を加水分解し、アデノシンジホスホリボース(ADPR)およびニコチンアミドを生成する活性(NADグリコヒドロラーゼ活性)、(b)NADからサイクリックADPリボース(cADPR)を合成する活性(ADPリボシルシクラーゼ活性)、および(c)cADPRを加水分解し、ADPRを生成する活性(cADPRヒドロラーゼ活性)のいずれかの活性を意味する(図1参照)。これは、連鎖球菌由来NADaseは上記(a)〜(c)の活性を有すると考えられるためである。
【0023】
本明細書中で使用される場合、「NADase阻害活性」とは、NADaseが有する任意の活性を抑制する活性をいい、特に断らない限り、(d)NADグリコヒドロラーゼ活性の阻害活性、(e)ADPリボシルシクラーゼ活性の阻害活性、および(f)cADPRヒドロラーゼ活性の阻害活性のいずれかの阻害活性を意味する。これは、SNIは、連鎖球菌由来NADaseと非常に安定かつ強固な複合体を形成することによりNADaseの活性を阻害することから、上記(d)〜(f)の阻害活性を有すると考えられるためである。
【0024】
NADグリコヒドロラーゼ活性を有するNADaseは、細菌から哺乳動物までの多くの生物に存在する。多くの真核生物NADaseは、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性をさらに併有する。cADPRは、シグナル伝達の調節に関与しており、実際、真核生物細胞の細胞膜表面NADase(例、CD38、CD157)がNADからcADPRを合成すると、細胞内貯蔵Ca2+濃度が細胞質に遊離され、このカルシウムイオンの上昇がシグナルとなって種々の生物学的応答が生じる。殆どの原核生物由来NADaseは、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性を有しない。しかし、本明細書に開示のある連鎖球菌NADaseは、原核生物由来NADseであるにもかかわらず、これらの活性を併有することが知られている(FEMS Microbiol. Lett. 130:201-204 (1995)参照)。また、この連鎖球菌NADaseは、分泌タンパク質である点で、細胞質、細胞膜等に局在する通常のNADaseとは異なる。なお、連鎖球菌由来NADaseの詳細については、例えば、FEMS Microbiol. Lett. 191: 235-241 (2000) を参照のこと。
【0025】
一実施形態では、配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と実質的に同一のアミノ酸配列は、配列番号2で表されるアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と所定のアミノ酸配列同一性を有するアミノ酸配列であり得る。アミノ酸配列同一性の程度は、例えば約70%、好ましくは約80%、より好ましくは約90%、さらにより好ましくは約95%、最も好ましくは約97%、約98%または約99%以上であり得る。アミノ酸配列同一性は自体公知の方法により決定できる。例えば、アミノ酸配列同一性(%)は、当該分野で慣用のプログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。また、別の局面では、同一性(%)は、当該分野で公知の任意のアルゴリズム、例えば、Needlemanら(1970) (J. Mol. Biol. 48: 444-453)、Myers及びMiller (CABIOS, 1988, 4: 11-17)のアルゴリズム等を使用して決定することができる。Needlemanらのアルゴリズムは、GCGソフトウェアパッケージ(www.gcg.comで入手可能)のGAPプログラムに組み込まれており、同一性(%)は、例えば、BLOSUM 62 matrix又はPAM250 matrix、並びにgap weight: 16、14、12、10、8、6若しくは4、及びlength weight: 1、2、3、4、5若しくは6のいずれかを使用することによって決定することができる。また、Myers及びMillerのアルゴリズムは、GCG配列アラインメントソフトウェアパッケージの一部であるALIGNプログラムに組み込まれている。アミノ酸配列を比較するためにALIGNプログラムを利用する場合、例えば、PAM120 weight residue table、gap length penalty 12、gap penalty 4を用いることができる。アミノ酸配列同一性は、上記の任意の方法で決定されたものであれば構わないが、好ましくは計算に際して、上記の方法のなかで最も低い値を示す方法が採用され得る。
【0026】
別の実施形態では、配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と実質的に同一のアミノ酸配列は、配列番号2で表されるアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列において1以上のアミノ酸が置換、付加、欠失および/または挿入されたアミノ酸配列であり得る。置換、付加、欠失および/または挿入されるアミノ酸の数は、1以上であれば特に限定されないが、例えば1〜約100個、好ましくは1〜約50個、より好ましくは1〜約30個、さらにより好ましくは1〜約20個、最も好ましくは1〜約10個、1〜約5個あるいは1または2個であり得る。
【0027】
本発明のSNIは、NADase阻害活性、NADase結合活性を有し得る。本発明のSNIが配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列からなるポリペプチドである場合、活性の程度は、配列番号2で表されるアミノ酸配列と同一のアミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0028】
本発明のSNIは、また、熱安定性が高いという特徴を有する。従来のNADaseインヒビターは、60℃で15分間加熱することにより完全に不活化されていた。しかし、本発明のSNIはこのような条件でも活性を保持し得る。より詳細には、本発明のSNIは、15分間の60℃での熱処理後には80%以上、100℃での熱処理後には70%以上の活性を保持し得る。
【0029】
本発明の非天然型NADaseは、NADase活性(NADグリコヒドロラーゼ活性、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性)、SNI結合活性を有し得る。本発明の非天然型NADaseが配列番号4で表されるアミノ酸配列と実質的に同一のアミノ酸配列からなるポリペプチドである場合、活性の程度は、配列番号4で表されるアミノ酸配列と同一のアミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0030】
本発明の非天然型NADaseは、酵素学的特性として既報の連鎖球菌由来の天然型NADaseと同様の特性を有し得る。
【0031】
本発明の部分ペプチドは、本発明のSNIポリペプチドの部分ペプチド、または本発明のNADaseポリペプチドの部分ペプチドであり得る。本発明の部分ペプチドの長さは、所定の活性を有する限り特に限定されないが、例えば、上記ポリペプチドをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。詳細には、本発明の部分ペプチドとしては、例えば、NADaseとの結合能を有するSNIの部分ペプチド、SNIとの結合能を有するNADase(天然型、変異型を含む)の部分ペプチドが挙げられる。
【0032】
また、精製用タグなどを付加したポリペプチドまたはその部分ペプチドを形質転換体に産生させ、当該タグに親和性を有する物質(例えば、Ni2+レジン、タグに特異的な抗体)を用いることで、本発明のポリペプチドまたはその部分ペプチドをより簡便に単離、精製することができる。従って、本発明のポリペプチドまたはその部分ペプチドは、必要に応じて、このような精製用タグを有し得る。精製用タグについては後述する。
【0033】
本発明のポリペプチドおよびその部分ペプチドは、自体公知の方法により作製できる。その詳細については後述する。
【0034】
2.ポリヌクレオチドおよびその部分ヌクレオチド
本発明は、本発明のSNIをコードするポリヌクレオチドあるいはその部分ヌクレオチドを提供する。本発明のSNIポリヌクレオチドは、配列番号1で表されるヌクレオチド配列と同一または実質的に同一のヌクレオチド配列であり得る。本発明のSNIポリヌクレオチドあるいはその部分ヌクレオチドは、例えば、本発明のSNIまたはその部分ペプチドの作製に有用であり得る。
【0035】
本発明はまた、天然型NADaseをコードするポリヌクレオチド、および本発明の非天然型NADaseをコードするポリヌクレオチド(以下、必要に応じて、本発明のNADaseポリヌクレオチドと省略する)、ならびにそれらの部分ヌクレオチドを提供する。これまで、連鎖球菌由来NADaseは、大腸菌等の異種宿主細胞に対して強い毒性を示すため、異種宿主細胞でのNADaseの発現、即ち、組換えタンパク質の調製には未だ成功していなかった。今回、本発明者らは、連鎖球菌由来NADaseの組換えタンパク質の調製に初めて成功した。本発明者らの研究成果により、異種宿主細胞を用いた連鎖球菌由来天然型NADaseおよび非天然型NADaseの組換えタンパク質の調製に使用できるという、本発明のポリヌクレオチドの意義ある利用が初めて可能になった。本発明のNADaseポリヌクレオチドは、例えば、配列番号3で表されるヌクレオチド配列、または配列番号3で表されるヌクレオチド配列において分泌シグナル配列をコードするヌクレオチド配列が除去されたヌクレオチド配列(以下、必要に応じて、「配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列」と省略する)と同一または実質的に同一のヌクレオチド配列からなるポリヌクレオチドであり得る。本発明のNADaseポリヌクレオチドまたはその部分ヌクレオチドは、例えば、本発明のNADaseまたはその部分ペプチドの作製に有用であり得る。
【0036】
一実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と所定の配列同一性を有するヌクレオチド配列であり得る。ヌクレオチド配列同一性の程度は、例えば約70%、好ましくは約80%、より好ましくは約90%、さらにより好ましくは約95%、最も好ましくは約97%、約98%または約99%以上であり得る。ヌクレオチド配列同一性は自体公知の方法により決定できる。例えば、ヌクレオチド配列同一性(%)は、上述したアミノ酸配列同一性(%)と同様の方法により決定できる。
【0037】
別の実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列において1以上のヌクレオチドが置換、付加、欠失および/または挿入されたヌクレオチド配列であり得る。置換、付加、欠失および/または挿入されるヌクレオチドの数は、1以上であれば特に限定されないが、例えば1〜約300個、好ましくは1〜約150個、より好ましくは1〜約100個、さらにより好ましくは1〜約50個、最も好ましくは1〜約30個、1〜約20個、1〜約10個または1〜約5個であり得る。
【0038】
さらに別の実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列の相補配列に対してハイストリンジェント条件下でハイブリダイズし得るポリヌクレオチドであり得る(但し、配列番号1で表されるヌクレオチド配列、、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列を除く)。ハイストリンジェント条件下でのハイブリダイゼーションの条件は、既報の条件を参考に設定することができる(Current Protocols in Molecular Biology, John Wiley & Sons, 6.3.1-6.3.6, 1999)。例えば、ハイストリンジェント条件下でのハイブリダイゼーションの条件としては、6×SSC(sodium chloride/sodium citrate)/45℃でのハイブリダイゼーション、次いで0.2×SSC/0.1%SDS/50〜65℃での1回または2回以上の洗浄が挙げられる。
【0039】
本発明のNADaseポリヌクレオチドは、本発明の非天然型NADase、または天然型NADaseと同様の活性を有するポリペプチドをコードし得る。本発明のNADaseポリヌクレオチドが配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列からなるポリヌクレオチドである場合、当該ポリヌクレオチドによりコードされるポリペプチドの活性の程度は、配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0040】
本発明の部分ヌクレオチドは、本発明のSNI部分ペプチド、または本発明のNADase部分ペプチドをコードするヌクレオチドであり得る。詳細には、本発明の部分ヌクレオチドは、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と同一または実質的に同一のヌクレオチド配列からなるポリヌクレオチドの部分ヌクレオチドであり得る。本発明の部分ヌクレオチドとしては、例えば、NADaseとの結合能を有するSNIの部分ペプチドをコードするポリヌクレオチド、SNIとの結合能を有するNADase(天然型、変異型を含む)の部分ペプチドをコードするポリヌクレオチドが挙げられる。
【0041】
本発明のポリヌクレオチドは、自体公知の方法により作製できる。例えば、配列番号1で表されるヌクレオチド配列と同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と同一のヌクレオチド配列からなるポリヌクレオチドは、連鎖球菌から総RNAを抽出し、mRNAからcDNAを調製した後、適切なプライマーを用いてPCRを行うことによりクローニングできる。また、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列からなるポリヌクレオチドは、上記の通りクローニングしたポリヌクレオチドに変異を導入することにより作製できる。変異導入法としては、例えば、合成オリゴヌクレオチド指定突然変異導入法、gapped duplex法、ランダムに点突然変異を導入する方法(例えば、亜硝酸若しくは亜硫酸での処理)、カセット変異法、リンカースキャニング法、ミスマッチプライマー法などの方法が挙げられる。
【0042】
3.発現ベクター
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の調製に有用な発現ベクターを提供する。なお、本発明の発現ベクターは、下記を参照することで、自体公知の分子生物学的手法により作製できる。例えば、分子生物学的手法については、Molecular Cloning, 2nd edition, J. Sambrook et al., Cold Spring Harbor Lab. Press (1989)を参照のこと。
【0043】
3.1.SNIまたはその部分ペプチドの発現ベクター
本発明は、SNIまたはその部分ペプチドの発現ベクター(以下、必要に応じてSNI発現ベクターと省略する)を提供する。
【0044】
一実施形態では、SNI発現ベクターは、宿主細胞でのSNIまたはその部分ペプチド自体の発現を目的とする発現ベクターであり得る。SNI発現ベクターとしては、原核細胞および/または真核細胞の各種の宿主細胞中で本発明のSNIポリペプチドまたはその部分ペプチドをコードする遺伝子を発現し、これらを産生できるものであれば特に制限されないが、例えば、プラスミドベクター、ウイルスベクターが挙げられる。発現ベクターを医薬として用いる場合、ヒト等の哺乳動物への投与に好適なベクターとしては、アデノウイルス、レトロウイルス、アデノ随伴ウイルス、ヘルペスウイルス、ワクシニアウイルス、ポックスウイルス、ポリオウイルス、シンドビスウイルス、センダイウイルス、エプスタイン・バー・ウイルス等のウイルスベクターが挙げられる。
【0045】
SNI産生用宿主細胞として細菌(例えば、大腸菌)等の原核生物細胞を用いる場合、発現ベクターは、例えば、プロモーター−オペレーター領域、開始コドン、本発明のSNIポリペプチドまたはその部分ペプチドをコードするポリヌクレオチド、終止コドン、ターミネーター領域、複製起点等のエレメントを含み得る。細菌中で本発明のポリペプチドを発現させるためのプロモーター−オペレーター領域は、プロモーター、オペレーター及びShine-Dalgarno (SD) 配列を含むものである。これらのエレメントについては、自体公知のものを用いることができる。
【0046】
また、SNI発現ベクターを医薬または研究用試薬として使用する場合、酵母、動物細胞または昆虫細胞等の真核生物細胞を宿主細胞として利用可能な発現ベクターが用いられ得る。この場合、使用されるプロモーターは、投与対象である哺乳動物等の真核生物で機能し得るものであれば特に制限されない。例えば、医薬として使用する場合、SV40由来初期プロモーター、サイトメガロウイルスLTR、ラウス肉腫ウイルスLTR、MoMuLV由来LTR、アデノウイルス由来初期プロモーター等のウイルスプロモーター、並びにβ−アクチン遺伝子プロモーター、PGK遺伝子プロモーター、トランスフェリン遺伝子プロモーター等の哺乳動物の構成蛋白質遺伝子プロモーターなどが挙げられる。
【0047】
SNI発現ベクターは、さらに、選択マーカー遺伝子(テトラサイクリン、アンピシリン、カナマイシン、ハイグロマイシン、ホスフィノスリシン等の薬剤に対する抵抗性を付与する遺伝子、栄養要求性変異を相補する遺伝子等)をさらに含んでいてもよい。
【0048】
別の実施形態では、SNI発現ベクターは、融合タンパク質発現用ベクターであり得る。この場合、SNIまたはその部分ペプチドは、精製用タグとして用いられ得る。従って、SNIの部分ペプチドは、NADaseとの結合能を有するSNIの部分ペプチドであり得る。本発明のSNIは、3M NaClという高濃度の塩の存在下であってもNADaseと結合し得ることから、NADaseまたはSNIとの結合能を有するその部分ペプチドを結合対として用いることにより、SNIまたはその部分ペプチドが付加された融合タンパク質の精製が可能となる。このようなSNI発現ベクターは、融合されるべき目的タンパク質のクローニングを容易とするため、SNIまたはその部分ペプチドをコードするポリヌクレオチドの上流または下流にマルチクローニングサイト(MCS)を含み得る。
【0049】
なお、SNI発現ベクターは、後述するNADase発現ベクター、あるいは後述するSLO発現ベクターと同様の形態であってもよい。即ち、後に詳述される、節「3.2.NADaseまたはその部分ペプチドの発現ベクター」における「NADase」、および節「3.3.SLOまたはその部分ペプチドの発現ベクター」における「SLO」は、「SNI」と読み替え可能であり、そのような発現ベクターもまた、「SNIまたはその部分ペプチドの発現ベクター」であり得る。
【0050】
3.2.NADaseまたはその部分ペプチドの発現ベクター
本発明は、NADaseまたはその部分ペプチドの発現ベクター(以下、必要に応じてNADase発現ベクターと省略する)を提供する。
【0051】
一実施形態では、NADase発現ベクターは、宿主細胞でのNADaseまたはその部分ペプチド自体の発現を可能とする共発現ベクターであり得る。NADaseは全般的に宿主細胞に毒性を示すため宿主細胞でのNADaseの発現は困難であり、特に連鎖球菌由来NADaseは大腸菌等の宿主細胞に対する強い毒性を示すため連鎖球菌由来NADaseの組換えタンパク質の調製には未だ成功していない。しかし、本発明者らは、宿主細胞でのNADaseの製造を可能にするためには、NADaseを、SNIまたは宿主細胞に対する毒性作用に対する中和作用を有するその部分ペプチドと共発現させればよいことを見出した。
【0052】
従って、NADase発現ベクターは、第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み得る。第1および第2の発現ユニットは、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドであり得る。プロモーターの機能的連結とは、プロモーターが、その制御下にある遺伝子の発現を可能とするように、該遺伝子をコードするポリヌクレオチドに結合していることを意味する。
【0053】
NADase発現ベクターが導入され得る宿主細胞としては、後述する任意の宿主細胞である限り特に限定されないが、例えば、大腸菌、バチルス属菌(例、枯草菌)等の原核生物細胞が好ましい。宿主細胞に対する毒性作用としては、例えば、NADase活性が挙げられる。宿主細胞に対する毒性作用に対する中和作用としては、例えば、NADase阻害活性が挙げられる。
【0054】
より詳細には、NADase発現ベクターは、特にNADaseが連鎖球菌由来NADaseである場合、ポリシストロニックmRNA発現ベクターであり得る。ポリシストロニックmRNA発現ベクターは、第1および第2の発現ユニットのポリシストロニックmRNAの発現を可能とする、第1および第2の発現ユニットをコードするポリヌクレオチド、ならびに当該ポリシストロニックmRNAの発現を可能とするように該ポリヌクレオチドに機能的に連結されたプロモーターを含み得る。ポリシストロニックmRNA発現ベクターにおいて、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドをコードするポリヌレオチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドをコードするポリヌレオチドは、いずれが上流または下流に位置してもよいが、天然状態(即ち、nga−sloオペロン)を反映させるという観点からは、前者が上流に位置するのが好ましい。
【0055】
NADase発現ベクターはまた、非ポリシストロニックmRNA発現ベクターであり得る。非ポリシストロニックmRNA発現ベクターは、第1の発現ユニットをコードするポリヌクレオチドおよび該ポリヌクレオチドに機能的に連結された第1のプロモーター、ならびに第2の発現ユニットをコードするポリヌクレオチドおよび該ポリヌクレオチドに機能的に連結された第2のプロモーターを含み得る。
【0056】
NADase発現ベクターは、例えば、上述したポリシストロニックmRNA発現ベクターまたは非ポリシストロニックmRNA発現ベクターであり得るが、好ましくは、ポリシストロニックmRNA発現ベクターであり得る。
【0057】
NADase発現ベクターは、上述の特徴以外は、SNI発現ベクターの特徴と同様であり得る。即ち、本発明の共発現ベクターは、プラスミドベクター、ウイルスベクターであり得、また、SNIまたはその部分ペプチドの発現ベクターと同様のプロモーター等のエレメント、選択マーカー遺伝子を含み得る。
【0058】
別の実施形態では、NADase発現ベクターは、融合タンパク質発現用ベクターであり得る。この場合、NADaseまたはその部分ペプチドは、精製用タグとして用いられ得る。従って、NADaseの部分ペプチドは、SNIとの結合能を有するNADaseの部分ペプチドであり得る。NADaseは、3M NaClという高濃度の塩の存在下であってもSNIと結合し得ることから、SNIまたはNADaseとの結合能を有するその部分ペプチドを結合対として用いることにより、NADaseまたはその部分ペプチドが付加された融合タンパク質の精製が可能となる。このような発現ベクターは、融合されるべき目的タンパク質のクローニングを容易とするため、NADaseまたはその部分ペプチドをコードするポリヌクレオチドの上流または下流にマルチクローニングサイト(MCS)を含み得る。
【0059】
なお、NADase発現ベクターは、上述したSNI発現ベクター、あるいは後述するSLO発現ベクターと同様の形態であってもよい。即ち、先に詳述した節「3.1.SNIまたはその部分ペプチドの発現ベクター」における「SNI」、および後に詳述する節「3.3.SLOまたはその部分ペプチドの発現ベクター」における「SLO」は、「NADase」と読み替え可能であり、そのような発現ベクターもまた、「NADaseまたはその部分ペプチドの発現ベクター」であり得る。
【0060】
3.3.SLOまたはその部分ペプチドの発現ベクター
本発明は、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクター(以下、必要に応じてSLO発現ベクターと省略する)を提供する。SLO発現ベクターは、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含む。
【0061】
ストレプトリシンO(SLO)は、5つの属〔連鎖球菌属(streptococcus)、バチルス属(Bacillus)、クロストリジウス属(Clostridium)、リステリア属(Listeria)、アルカノバクテリウム属(Arcanobacterium)〕の種に見出されるサイトリシンの高度に相同な単一ファミリーに属する(例えば、Pharmacol Ther 11: 661-717 (1980); Int J Med Microbiol 290: 351-356 (2000); FEMS Microbiol Lett 182: 197-205 (2000); Arch Microbiol 165: 73-79 (1996); Infect Immun 47, 52-60 (1985); Toxicon 39: 1681-1689 (2001)参照)。本発明で対象となり得るSLOは、上記由来のSLOである限り特に限定されないが、SLO遺伝子は異なる溶血性連鎖球菌群間で高度に保存されていることから、溶血性連鎖球菌由来SLOが好ましい。溶血性連鎖球菌としては、A群〜C群、G群の連鎖球菌が挙げられるが、化膿性咽頭炎、創傷感染および連鎖球菌トキシックショック様症候群、猩紅熱、急性糸球体腎炎などの発症因子としての報告(例えば、Clin Microbiol Rev 13: 470-511 (2000); FEMS Immunol Med Microbiol 34: 159-167 (2002); J Clin Microbiol 42: 186-192 (2004)参照)があるA群連鎖球菌(GAS)、C群連鎖球菌(GCS)、G群連鎖球菌(GGS)が好ましい。なお、GASおよびGCSでは、いずれのSLOも、N末端シグナルペプチドを含む31個のアミノ酸残基が除去されて分泌されるとともに、自身が分泌するプロテアーゼにより次第に低分子化することが報告されている(例えば、Infect Immun 61: 2727-2731 (1993); Infect Immun 63: 2776-2779 (1995)参照)。このような理由により、GASおよびGCSの完全長SLOは精製が特に困難であり得るが、本発明者らの開発した方法によればこれらの完全長SLOの精製が可能である。従って、上記の連鎖球菌に由来するSLOのなかでも、GASおよびGCS由来のSLOがより好ましい。ところで、GASおよびGCSには幾つかの亜種が知られている。本発明で対象となり得るSLOが由来するGASの亜種としては、例えば、D58X株(ATCC12383)が挙げられる。本発明で対象となり得るSLOが由来するGCSの亜種としては、例えば、H46A株(ATCC12449)が挙げられる。
【0062】
完全長SLOとは、N末端の分泌シグナルペプチド除去後のSLOを意味する。
【0063】
大腸菌に毒性を示し得る領域を含む完全長SLOの部分ペプチドとは、大腸菌に対する毒性を有し得るペプチド部分を含む任意の断片を意味する。例えば、GAS、GCS等の溶血性連鎖球菌に由来するSLOは、そのN末端近傍の領域、特にその約32−約75位のアミノ酸残基からなるポリペプチドの領域中に、このような領域を有すると考えられる(例えば、Biosci Biotechnol Biochem 65: 2682-2689 (2001) 参照)。従って、大腸菌に毒性を示し得る領域を含む部分ペプチドは、このようなポリペプチド中に含まれる大腸菌に毒性を示し得るペプチド部分を含むものであり得る。
【0064】
SLO発現ベクターから発現され得る完全長SLOまたはその部分ペプチドは、好ましくは精製用タグを有する。従って、SLO発現ベクターは、精製用タグ融合SLOの発現を可能とする、精製用タグをコードするポリヌクレオチドとSLOをコードするポリヌクレオチドとのin frame連結物を含み得る。精製用タグは、本発明の発現ベクターが導入された形質転換体の培養上清からの、SLOの精製を容易とするものである限り特に限定されず、例えば、Hisタグ、マルトース結合タンパク(MBP)、グルタチオン−S−トランスフェラーゼ(GST)、カルモジュリン結合ペプチド(CBP)、Strep−tag II、FLAG、heayy chain of protein C(HPC)が挙げられるが、付加されるアミノ酸配列が短く、多くの場合除去する必要がない、および特異的な抗体や精製担体も複数の市販品が入手可能であり、安価である等の利点より、Hisタグが好ましい。
【0065】
誘導プロモーターとは、誘導剤の存在下でその制御下にある遺伝子の発現を増強するプロモーターを意味する。SLO発現ベクターでは、誘導プロモーターは、SLO遺伝子をコードするポリヌクレオチドに機能的に連結され得る。
【0066】
SLO発現ベクターに含まれる誘導プロモーターはまた、大腸菌における基底レベルの遺伝子発現を抑制し得る。基底レベルの遺伝子発現とは、非誘導条件下での遺伝子発現を意味する。SLOは大腸菌に毒性を示し得る領域を含み得るため、その基底レベルのSLO遺伝子の発現量が多いと大腸菌が傷害を受け易くなることから、このような領域を含むSLOの大量発現が難しいという問題があった。しかし、本発明者らは、基底レベルの遺伝子発現を厳格に抑制できる誘導プロモーターを用いることにより、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドを大腸菌で大量発現させることに成功した。大腸菌における基底レベルの遺伝子発現を抑制し得るこのような誘導プロモーターとしては、例えば、アラビノースオペロン制御プロモーター(例、L−アラビノースを誘導剤とするaraBAD:例えば、pBADシリーズのベクターを利用可能)、キシロースオペロン制御プロモーター(例、キシロースを誘導剤とするxylAプロモーター:例えば、pWH1520を利用可能)、サリチル酸塩で制御可能なプロモーター(例、サリチル酸塩を誘導剤とするPmプロモーター:例えば、pCASシリーズのベクターを利用可能)、低温処理で制御可能なプロモーター(例、低温処理により誘導され得るcspAプロモーター:例えば、pColdシリーズのベクターを利用可能)が挙げられるが、使い易さ、発現レベル等の観点より、アラビノースオペロン制御プロモーターが好ましい。
【0067】
SLO発現ベクターはまた、転写開始および転写終結のための部位、および転写領域において翻訳に必要とされ得るリボソーム結合部位、複製起点ならびに選択マーカー遺伝子などを含み得る。選択マーカー遺伝子としては、宿主として大腸菌を用いた場合に、形質転換された大腸菌を選別できるものである限り特に限定されないが、例えば、アンピシリン、テトラサイクリン、カナマイシン、スペクチノマイシン、エリスロマイシン、クロラムフェニコール、カルベニシリン等の抗生物質に対する耐性遺伝子が挙げられる。
【0068】
4.形質転換体
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の調製に有用な形質転換体、またはこのようなタンパク質の解析を可能とする形質転換体を提供する。
【0069】
本発明の形質転換体は、上述した本発明の発現ベクターまたはその他の発現ベクターを宿主細胞に導入することにより作製できる。形質転換体の作製に用いられる宿主細胞としては、前記の発現ベクターに適合し、形質転換され得るものであれば特に限定されず、本発明の技術分野において通常使用される天然細胞あるいは人工的に樹立された細胞株など種々の細胞が用いられ得るが、それぞれの形質転換体に応じた好ましい宿主細胞については以下に述べる。それぞれの形質転換体は、自体公知の方法により培養できる(例えば、後述の培養法を参照)。
【0070】
4.1.SNIまたはその部分ペプチドを発現し得る形質転換体
本発明は、SNIまたはその部分ペプチドを発現し得る形質転換体(以下、必要に応じてSNI形質転換体と省略する)を提供する。
【0071】
SNI形質転換体は、本発明のSNIまたはその部分ペプチドの発現ベクターで宿主細胞を形質転換することにより作製できる。
【0072】
SNI形質転換体の宿主細胞としては、例えば、大腸菌(例、「4.3.SLOまたはその部分ペプチドを発現し得る形質転換体」で詳述される大腸菌)、バチルス属菌(例、枯草菌)、放線菌等の原核生物細胞、ならびに酵母、昆虫細胞、鳥類細胞、哺乳動物細胞(例、ヒト、サル等の霊長類、マウス、ラット等のげっ歯類に由来する細胞)等の真核生物細胞が挙げられる。なお、SNIまたはその部分ペプチドを大量に製造することを目的とする場合には、原核生物細胞を宿主細胞として用いることが好ましい。
【0073】
4.2.NADaseまたはその部分ペプチドを発現し得る形質転換体
本発明は、NADaseまたはその部分ペプチドを発現し得る形質転換体(以下、必要に応じてNADase形質転換体と省略する)を提供する。NADaseは全般的に宿主細胞に毒性を示すため宿主細胞でのNADaseの発現は困難であり、特に連鎖球菌由来NADaseは大腸菌等の宿主細胞に対する強い毒性を示すため連鎖球菌由来NADaseの組換えタンパク質の調製には未だ成功していない。しかし、本発明者らは、宿主細胞でのNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造を可能にするためには、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを共発現する形質転換体を作製すればよいことを見出した。
【0074】
NADase形質転換体は、NADase発現ベクター(例、上述したポリシストロニックmRNA発現ベクターおよび非ポリシストロニックmRNA発現ベクター)で宿主細胞を形質転換することにより作製できる。NADase形質転換体はまた、NADaseのみを発現し、SNIを発現しないベクター(即ち、NADase単独発現ベクター)、およびSNI発現ベクターの双方で宿主細胞を形質転換することにより作製できる。
【0075】
NADase形質転換体の宿主細胞としては、例えば、上述したSNI形質転換体と同様のものが挙げられる。なお、NADaseまたはその部分ペプチドを大量に製造することを目的とする場合には、原核生物細胞を宿主細胞として用いることが好ましい。
【0076】
4.3.SLOまたはその部分ペプチドを発現し得る形質転換体
本発明は、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドを大量に発現し得る形質転換体(以下、必要に応じてSLO形質転換体と省略する)を提供する。
【0077】
SLO形質転換体は、本発明のSLOまたはその部分ペプチドの発現ベクターで宿主細胞を形質転換することにより作製できる。
【0078】
SLO形質転換体の宿主細胞としては、その目的に鑑みれば、大腸菌が好ましい。
【0079】
宿主として用いられ得る大腸菌は、完全長SLOを大量かつ高純度で製造する(換言すれば、SLOの切断を抑える)という本発明の一つの目的を考慮すれば、プロテアーゼの欠損株であることが好ましい。大腸菌において欠損されるべきこのようなプロテアーゼとしては、例えば、lon、ompTが挙げられる。本発明において宿主として用いられ得るプロテアーゼ欠損株としては、例えば、BL21株が挙げられる。
【0080】
また、宿主として用いられ得る大腸菌は、誘導プロモーターの能力を十分に発揮させ、大腸菌に毒性を示し得る領域を含むSLOを大量に製造するという本発明の別の目的を考慮すれば、誘導プロモーターに対する誘導剤の代謝能を欠損していることが好ましい。欠損されるべき代謝能は、本発明で用いられる誘導プロモーターの種類に応じて異なるが、例えば、上述した誘導プロモーターの誘導剤の代謝能であり得る。
【0081】
5.形質転換体を用いるポリペプチドまたはその部分ペプチドの製造方法
本発明は、本発明の形質転換体を用いるポリペプチドまたはその部分ペプチドの製造方法を提供する。例えば、本発明の製造方法は、本発明の形質転換体を培養し、目的のポリペプチドまたはその部分ペプチドを産生させ、次いで回収することを含む。本発明の製造方法はまた、本発明の形質転換体が誘導プロモーターを有する発現ベクターを含む場合、誘導プロモーターに対する誘導剤を培地に添加し、培養することを含み得る。
【0082】
形質転換体の培養は、自体公知の方法により液体培地等の栄養培地中で行われ得る。培地は、形質転換体の生育に必要な炭素源、窒素源、無機物などを含有することが好ましい。ここで、炭素源としては、例えば、グルコース、デキストリン、可溶性澱粉、ショ糖などが挙げられ、窒素源としては、例えば、アンモニウム塩類、硝酸塩類、コーンスチープ・リカー、ペプトン、カゼイン、肉エキス、大豆粕、バレイショ抽出液などの無機または有機物質が挙げられ、無機物としては、例えば、塩化カルシウム、リン酸二水素ナトリウム、塩化マグネシウムなどがそれぞれ挙げられる。また、培地には、酵母エキス、ビタミン類などを添加してもよい。培養条件、例えば温度、培地のpHおよび培養時間は、本発明のポリペプチドが大量に生産されるように適宜選択される。培養温度は、例えば30〜37℃である。
【0083】
本発明の形質転換体が誘導プロモーターを有する発現ベクターを含む場合、本発明の形質転換体は、誘導プロモーターに対する誘導剤の存在下で培養され得る。誘導剤の存在下における形質転換体の培養は、非誘導条件下において大量発現に適切な数に増殖した形質転換体を用いて、誘導剤の種類に応じた適切な条件下において行われ得る。例えば、形質転換体の宿主細胞として大腸菌を用いる完全長SLOまたはその部分ペプチドの製造方法では、誘導プロモーターがアラビノースオペロン制御プロモーターである場合には、培地に添加されるL−アラビノースの濃度が0.002%(W/V)以上であるときに、完全長SLOまたはその部分ペプチドの十分な発現が確認されている。従って、培地に添加されるL−アラビノースの濃度は完全長SLOまたはその部分ペプチドの効率的な発現を可能とする限り特に限定されないものの、好ましくは約0.002%(W/V)以上、より好ましくは約0.02%(W/V)以上であり得る。なお、L−アラビノースの上限濃度は特に限定されないが、例えば0.2%(W/V)以下であり得る。また、本発明の大腸菌が誘導条件下におかれる時間としては、完全長SLOまたはその部分ペプチドの効率的な発現を可能とする限り特に限定されないが、例えば、約1〜約24時間が挙げられる。なお、誘導条件下における培養は、誘導剤を所定の条件下で用いること以外は、非誘導条件下における培養と同様に行われ得る。
【0084】
形質転換体の培養により産生されたポリペプチドまたはその部分ペプチドは、回収、好ましくは単離、精製され得る。例えば、SNIまたはその部分ペプチドは、培養された形質転換体を集め、溶解処理(例、リゾチーム、ソニケーション等による処理)した後、溶解液中から回収され得る。また、NADase、SLOまたはそれらの部分ペプチドは、その培養上清から回収され得る。
【0085】
単離、精製方法としては、例えば塩析、溶媒沈澱法等の溶解度を利用する方法、透析、限外濾過、ゲル濾過、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動など分子量の差を利用する方法、イオン交換クロマトグラフィーやヒドロキシルアパタイトクロマトグラフィーなどの荷電を利用する方法、アフィニティークロマトグラフィーなどの特異的親和性を利用する方法、逆相高速液体クロマトグラフィーなどの疎水性の差を利用する方法、等電点電気泳動などの等電点の差を利用する方法などが挙げられる。
【0086】
本発明の製造方法によれば、SNI、NADase、SLOおよびそれらの部分ペプチドを高純度で大量かつ安価に製造することが可能である。例えば、溶血性連鎖球菌に由来する完全長SLOまたはその部分ペプチドの製造方法では、用いる培地や誘導条件によっても異なるが、LB培地1リットルあたり例えば約1mg以上、好ましくは約1mg〜約30mg、より好ましくは約5mg〜約30mg、さらにより好ましくは約5mg〜約20mg、最も好ましくは約5mg〜約10mgの収量を可能とする。また、本発明の製造方法により得られるSLOは、異種の宿主で産生された組換えタンパク質であるにもかかわらず、溶血性連鎖球菌の培養上清から得られる天然タンパク質と同様の比活性を有し得る。
【0087】
6.抗体
【0088】
本発明は、SNI、NADase、SLOおよびそれらの部分ペプチドに対する抗体を提供する。
【0089】
本発明の抗体は、ポリクローナル抗体、モノクローナル抗体のいずれであってもよく、周知の免疫学的手法により作製することができる。この抗体は、完全な抗体分子だけでなく、本発明のポリペプチドに対する抗原結合部位(CDR)を有する限りいかなるフラグメントであってもよく、例えば、Fab、F(ab')2、ScFv、minibody等が挙げられる。
【0090】
例えば、ポリクローナル抗体は、上記抗原(必要に応じて、ウシ血清アルブミン、KLH(Keyhole Limpet Hemocyanin)等のキャリア蛋白質に架橋した複合体とすることもできる)を、市販のアジュバント(例えば、完全または不完全フロイントアジュバント)とともに、動物の皮下あるいは腹腔内に2〜3週間おきに2〜4回程度投与し(部分採血した血清の抗体価を公知の抗原抗体反応により測定し、その上昇を確認しておく)、最終免疫から約3〜10日後に全血を採取して抗血清を精製することにより取得できる。抗原を投与する動物としては、ラット、マウス、ウサギ、ヤギ、モルモット、ハムスターなどの哺乳動物が挙げられる。
【0091】
また、モノクローナル抗体は、細胞融合法により作成することができる。例えば、マウスに上記抗原を市販のアジュバントと共に2〜4回皮下あるいは腹腔内に投与し、最終投与の3日後に脾臓あるいはリンパ節を採取し、白血球を採取する。この白血球と骨髄腫細胞(例えば、NS-1, P3X63Ag8など)を細胞融合して該因子に対するモノクローナル抗体を産生するハイブリドーマを得る。細胞融合はPEG法でも電圧パルス法であってもよい。所望のモノクローナル抗体を産生するハイブリドーマは、周知のEIAまたはRIA法等を用いて抗原と特異的に結合する抗体を、培養上清中から検出することにより選択できる。モノクローナル抗体を産生するハイブリドーマの培養は、インビトロ、またはマウスもしくはラット、好ましくはマウス腹水中等のインビボで行うことができ、抗体はそれぞれハイブリドーマの培養上清および動物の腹水から取得することができる。
【0092】
さらに、本発明の抗体は、キメラ抗体(chimeric antibody)、ヒト化抗体(humanized antibody)、ヒト型抗体(human antibody)であってもよい。キメラ抗体は、例えば「実験医学(臨時増刊号), Vol.6, No.10, 1988」、特公平3-73280号公報等を、ヒト化抗体は、例えば特表平4-506458号公報、特開昭62-296890号公報等を、ヒト抗体は、例えば「Nature Genetics, Vol.15, p.146-156, 1997」、「Nature Genetics, Vol.7, p.13-21, 1994」、特表平4-504365号公報、国際出願公開WO94/25585号公報、「日経サイエンス、6月号、第40〜第50頁、1995年」、「Nature, Vol.368, p.856-859, 1994」、特表平6-500233号公報等を参考にそれぞれ作製することができる。例えば、本発明の抗体を医薬として用いる場合、ヒト化抗体およびヒト型抗体が好ましい。
【0093】
本発明の抗体(特に、後述のSLO抗体)は、臨床検査試薬、研究用試薬等の試薬および医薬などとして有用である。本発明の抗体が試薬として用いられる場合、例えば、酵素免疫測定法(EIA)、放射免疫測定法(RIA)、蛍光免疫測定法(FIA)、免疫クロマト法、ルミネッセンス免疫測定法、スピン免疫測定法、ウエスタンブロット法による抗原(特にSLO)の検出および定量に用いられ得る。EIA法としては、例えば、直接競合ELISA、間接競合ELISA、サンドイッチELISAが挙げられる。一方、本発明の抗体が医薬として用いられる場合、例えば、溶血性連鎖球菌感染症の治療薬として用いられ得る。
【0094】
本発明の抗体が医薬として使用される場合、本発明の医薬は、医薬上許容される担体が配合されたものであり得る。医薬上許容される担体としては、後述するものが挙げられる。本発明の抗体は、好ましくは、後述の投与量において後述の方法に従い非経口投与され得る。
【0095】
6.1.SNIまたはその部分ペプチドに対する抗体
本発明は、SNIまたはその部分ペプチドに対する抗体(以下、必要に応じてSNI抗体と省略する)を提供する。
【0096】
SNI抗体の作製に用いられる抗原としては、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチド、あるいはそれらの部分ペプチドを用いることができる。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列、あるいは上述のドメインをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。
【0097】
また、中和抗体を得ることを目的とする場合には、抗原として、NADase阻害活性を有する部分ペプチドを用いることが好ましく、後述する本発明の複合体の解離剤を得ることを目的とする場合には、抗原として、NADaseとの結合能を有するその部分ペプチドを用いることが好ましい。
【0098】
6.2.NADaseまたはその部分ペプチドに対する抗体
本発明は、NADaseまたはその部分ペプチドに対する抗体(以下、必要に応じてNADase抗体と省略する)を提供する。本発明者らの研究成果により、宿主細胞を用いた連鎖球菌由来天然型NADaseの組換えタンパク質の調製のみならず、宿主細胞を用いた非天然型NADase(変異型NADaseまたは遺伝子改変NADase)の組換えタンパク質の調製が初めて可能となる。また、このように組換え技術を利用することで、NADaseの部分ペプチドの調製も容易になる。従って、従来よりも、NADase抗体の作製が容易になり得る。
【0099】
NADase抗体の作製に用いられる抗原としては、配列番号4で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチド、あるいはそれらの部分ペプチドを用いることができる。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、配列番号4で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列、あるいは上述のドメインをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。
【0100】
また、中和抗体を得ることを目的とする場合には、抗原として、NADase活性を有する部分ペプチドを用いることが好ましく、後述する本発明の複合体の解離剤を得ることを目的とする場合には、抗原として、SNIとの結合能を有するその部分ペプチドを用いることが好ましい。
【0101】
6.3.SLOまたはその部分ペプチドに対する抗体
本発明は、SLOまたはその部分ペプチドに対する抗体(以下、必要に応じてSLO抗体と省略する)を提供する。
【0102】
SLO抗体の作製に用いられる抗原としては、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチド(例えば、約32〜約75位のアミノ酸残基を含むポリペプチドの部分ペプチド)が用いられ得る。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、上記SLOの任意の部分から選ばれる少なくとも8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個以上の連続したアミノ酸からなるポリペプチドであり得る。
【0103】
SLO抗体は、例えば、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドに対する特異的な反応性を示し、かつNADase、特に溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体であり得る。本発明の抗体はまた、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの活性、特に溶血活性の中和抗体であり得る。
【0104】
7.SNIに関連するその他の発明
本発明は、SNIポリヌクレオチドまたはその部分ヌクレオチドに相補的な核酸(アンチセンス核酸)、RNAi誘導核酸(siRNA)、リボザイム、増幅用プライマー対などを提供する。これらは、例えば、本発明のSNIの解析、変異型連鎖球菌の作製などに有用であり得る。
【0105】
8.医薬または試薬
本発明はまた、SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む医薬または試薬を提供する。本発明者らは、未知遺伝子SNIの機能の一端を解明することに初めて成功した。より詳細には、本発明者らは、SNIがNADaseを阻害し得ることを見出した。従って、本発明者らは、SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを、医薬または試薬として用いることを初めて可能とした。
【0106】
本発明の医薬は、例えば、連鎖球菌等の細菌の感染症の治療薬として有用である。分泌タンパク質である連鎖球菌NADaseは、血液等の生体液中に分泌され、SLOにより宿主細胞膜上に形成された孔から宿主細胞内に侵入し、NADase活性によりその毒性を発揮することで、連鎖球菌感染症の病態を担い得ると考えられる。従って、SNIまたはNADase阻害活性を有するその部分ペプチドが生体内に存在すれば、連鎖球菌NADaseとSNIまたはその部分ペプチドとの安定複合体が形成され、以って連鎖球菌NADaseのNADase活性を低減し得ることにより、連鎖球菌感染症の病態を軽減し得ると考えられる。本発明の医薬が適用され得る感染症としては、NADaseにより毒性作用を発揮する細菌により引き起こされ得る感染症である限り特に限定されないが、例えば、A群、C群およびG群溶血性連鎖球菌により引き起こされる感染症が挙げられる。
【0107】
本発明の医薬はまた、非感染症の治療に有用である。本発明者らは、SNIが細菌由来NADaseのみならず、哺乳動物由来NADaseも阻害し得ることを見出した。従って、SNIまたはNADase阻害活性を有するその部分ペプチドは、哺乳動物由来NADase(例、CD38、CD157)の機能亢進によりもたらされ得る疾患、あるいは哺乳動物由来NADaseの機能阻害により対症療法的に治療され得る疾患(例えば、哺乳動物由来NADaseの機能阻害により症状が改善、緩和または軽減され得る疾患)に対して有効であると考えられる。このような疾患としては、例えば、自己免疫疾患(例、潰瘍性大腸炎)、炎症性疾患(例、成人呼吸促迫症候群(ARDS:adult respiratory distress syndrome)、関節炎)、アレルギー疾患、多発性骨髄腫、虚血、喘息、糖尿病、移植拒絶反応、神経変性疾患が挙げられるがこれらに限定されない(例えば、WO2002/032288;Nature Medicine 7(11): 1209-16(2001);The Journal of Immunology 172: 1896-1906 (2004) 参照)。
【0108】
本発明の医薬または試薬はさらに、NADase阻害剤として有用である。NADase阻害剤とは、上述したNADase阻害活性を有するものをいう。NADaseは細菌から哺乳動物まで広く認められること、ならびにSNIは細菌および哺乳動物NADaseを阻害したことから、SNIは、あらゆる種に由来するNADseを阻害し得ると考えられる。SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得るNADseとしては、原核生物から真核生物にまで至る任意のNADaseが挙げられるが、例えば、細菌(例、連鎖球菌)、哺乳動物(例、ヒト、サル、チンパンジー、ウサギ、ウシ)、昆虫、鳥類、魚類、軟体動物(例、アメフラシ)、植物が挙げられ、細菌およびヒト等の哺乳動物が好ましい。詳細には、SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得る連鎖球菌由来NADaseとしては、A群、C群およびG群連鎖球菌由来NADaseが挙げられる。SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得る哺乳動物由来NADaseとしては、CD38、CD157、アナウサギ属ADPリボシルシクラーゼ、ウシNAD+グリコヒドロラーゼが挙げられるが、CD38、CD157が好ましい。NADaseはまた、その局在に応じて、分泌型、細胞膜結合型、細胞質局在型などに分類できる。SNIまたはNADase阻害活性を有するその部分ペプチドは、これらの任意の型のNADaseを阻害し得るが、好ましくは分泌型または細胞膜結合型のNADaseを阻害し得る。また、CD38の酵素活性(およびcADPRの生成)は、細胞内Ca2+貯蔵からのCa2+の放出、細胞増殖、アポトーシスなどに関与し得るので(例えば、WO2002/032288参照)、本発明の医薬および試薬はこれらの作用を阻害し得る。本発明のNADase阻害剤はまた、細胞系(例、本発明の形質転換体の使用)または無細胞系(例、本発明の共発現ベクターの使用、あるいはNADase発現ベクターおよびSNI発現ベクターの組合せの使用、あるいはNADase合成系におけるSNIの添加)におけるNADaseの調製剤として用いることができる。
【0109】
本発明の医薬または試薬はまた、白血球の走化性の阻害剤として有用である。白血球の走化性とは、白血球が炎症部位に動員されることをいう。CD38、CD157は、白血球の走化性に重要な役割を果たし得ることが知られている(例えば、Blood vol.104(13): 4269-4278 (2004)、Nat Med. vol.7(11): 1209-16 (2001))。SNIまたはNADase阻害活性を有するその部分ペプチドにより走化性が阻害され得る白血球としては、例えば、好中球、好酸球、単球、リンパ球、マクロファージ、樹状細胞が挙げられるが(例えば、WO2002/032288参照)、好中球が好ましい。
【0110】
本発明の医薬は、医薬上許容される担体が配合されたものであり得る。医薬上許容される担体としては、例えば、ショ糖、デンプン、マンニット、ソルビット、乳糖、グルコース、セルロース、タルク、リン酸カルシウム、炭酸カルシウム等の賦形剤、セルロース、メチルセルロース、ヒドロキシプロピルセルロース、ポリプロピルピロリドン、ゼラチン、アラビアゴム、ポリエチレングリコール、ショ糖、デンプン等の結合剤、デンプン、カルボキシメチルセルロース、ヒドロキシプロピルスターチ、ナトリウム−グリコール−スターチ、炭酸水素ナトリウム、リン酸カルシウム、クエン酸カルシウム等の崩壊剤、ステアリン酸マグネシウム、エアロジル、タルク、ラウリル硫酸ナトリウム等の滑剤、クエン酸、メントール、グリシルリシン・アンモニウム塩、グリシン、オレンジ粉等の芳香剤、安息香酸ナトリウム、亜硫酸水素ナトリウム、メチルパラベン、プロピルパラベン等の保存剤、クエン酸、クエン酸ナトリウム、酢酸等の安定剤、メチルセルロース、ポリビニルピロリドン、ステアリン酸アルミニウム等の懸濁剤、界面活性剤等の分散剤、水、生理食塩水、オレンジジュース等の希釈剤、カカオ脂、ポリエチレングリコール、白灯油等のベースワックスなどが挙げられるが、それらに限定されるものではない。
【0111】
経口投与に好適な製剤は、水、生理食塩水、オレンジジュースのような希釈液に有効量のリガンドを溶解させた液剤、有効量のリガンドを固体や顆粒として含んでいるカプセル剤、サッシェ剤または錠剤、適当な分散媒中に有効量の有効成分を懸濁させた懸濁液剤、有効量の有効成分を溶解させた溶液を適当な分散媒中に分散させ乳化させた乳剤等である。
【0112】
非経口的な投与(例えば、静脈内注射、皮下注射、筋肉注射、局所注入、腹腔内投与など)に好適な製剤としては、水性および非水性の等張な無菌の注射液剤があり、これには抗酸化剤、緩衝液、制菌剤、等張化剤等が含まれていてもよい。また、水性および非水性の無菌の懸濁液剤が挙げられ、これには懸濁剤、可溶化剤、増粘剤、安定化剤、防腐剤等が含まれていてもよい。当該製剤は、アンプルやバイアルのように単位投与量あるいは複数回投与量ずつ容器に封入することができる。また、有効成分および医薬上許容される担体を凍結乾燥し、使用直前に適当な無菌のビヒクルに溶解または懸濁すればよい状態で保存することもできる。
【0113】
本発明の製剤の投与量は、有効成分の種類・活性、病気の重篤度、投与対象となる動物種、投与対象の薬物受容性、体重、年齢等によって異なるが、通常、成人1日あたり有効成分量として約0.001〜約2.0g/kgであり得る。
【0114】
9.複合体およびその製造方法
本発明はまた、SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADase(例、連鎖球菌由来NADase)またはSNIとの結合能を有するその部分ペプチドとを含む複合体を提供する。本発明者らは、SNIがNADaseと結合して複合体を形成し、NADase活性を抑制することを見出した。
【0115】
本発明の複合体において、SNIまたはNADaseとの結合能を有するその部分ペプチド、ならびにNADaseまたはSNIとの結合能を有するその部分ペプチドは、上述の通りであり得るが、それぞれ標識されていてもよい。標識としては、それらの複合体の形成を妨げない限り特に限定されないが、上述した精製用タグによる標識、放射性同位体(例えば、35S等の放射性同位体)による標識が挙げられる。複合体における構成成分の未標識・標識パターンは以下(a)〜(d)の通りである:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチド(未標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(未標識)との組合せ;
(b)SNIまたはNADaseとの結合能を有するその部分ペプチド(標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(未標識)との組合せ;
(c)SNIまたはNADaseとの結合能を有するその部分ペプチド(未標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(標識)との組合せ;
(d)SNIまたはNADaseとの結合能を有するその部分ペプチド(標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(標識)との組合せ。
また、双方が標識される場合、標識の種類は同じであっても異なっていてもよいが、好ましくは異なり得る。従って、この場合、異なる精製用タグによる標識、異なる放射性同位体による標識、精製用タグと放射性同位体による標識が利用され得る。
【0116】
精製用タグで標識されたタンパク質またはペプチドは、自体公知の方法により作製できる。例えば、精製用タグをコードするポリヌクレオチドに目的タンパク質またはペプチドをコードするポリヌクレオチドを適切に連結してDNA構築物を作製し、このDNA構築物を宿主細胞で発現させることにより、精製用タグで標識されたタンパク質またはペプチドを作製できる。一方、放射性同位体で標識されたタンパク質またはペプチドは、その発現細胞を放射性同位体含有培地で培養することにより作製できる。
【0117】
本発明の複合体は、非常に強固かつ特異的に結合する。例えば、本発明の複合体は3M NaClという高濃度の塩の存在下であっても解離し得ない。従って、本発明の複合体は非常に安定であり得る。
【0118】
本発明はまた、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法を提供する。このような製造方法は、例えば、SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む。
【0119】
より詳細には、本発明の複合体を作製するための接触は、(a)単離されたSNIまたはその部分ペプチドと単離されたNADaseまたはその部分ペプチドとの接触、(b)SNIまたはその部分ペプチドの発現細胞に対する、NADaseまたはその部分ペプチドの発現ベクターの導入、(c)NADaseまたはその部分ペプチドの発現細胞に対する、SNIまたはその部分ペプチドの発現ベクターの導入、(d)宿主細胞に対する、SNIまたはその部分ペプチドの発現ベクターおよびNADaseまたはその部分ペプチドの発現ベクターの導入、あるいは(e)宿主細胞に対する、SNIまたはその部分ペプチドおよびNADaseまたはその部分ペプチドの同時発現ベクター(即ち、上述の本発明のベクター)の導入などにより達成できる。
【0120】
本発明の複合体およびその製造方法は、例えば、連鎖球菌感染症の治療薬の開発、あるいは本発明の複合体におけるSNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとの解離剤の開発に有用である。従って、本発明はまた、このような治療薬および解離剤の開発を可能とする、複合体の形成を調節し得る物質のスクリーニング方法を提供する。
【0121】
10.スクリーニング方法
本発明は、連鎖球菌感染症の治療薬または本発明の複合体の解離剤の開発を可能とするスクリーニング方法、ならびに当該スクリーニング方法による得られる物質、および当該物質を含む剤を提供する。
【0122】
一実施形態では、本発明のスクリーニング方法は、以下の工程(a)および(b)を含み得る:
(a)被験物質が本発明の複合体の形成を抑制し得るか否かを評価する工程;
(b)本発明の複合体の形成を抑制し得る被験物質を選択する工程。
以下、本スクリーニング方法を、必要に応じてスクリーニング方法Iと省略する。
【0123】
本発明のスクリーニング方法Iの工程(a)で用いられる被験物質は、任意の公知化合物および新規化合物であり得、例えば、有機低分子化合物、コンビナトリアルケミストリー技術を用いて作製された化合物ライブラリー、核酸(例、ヌクレオシド、オリゴヌクレオチド、ポリヌクレオチド)、糖質(例、単糖、二糖、オリゴ糖、多糖)、脂質(例、飽和または不飽和の直鎖、分岐鎖および/または環を含む脂肪酸)、アミノ酸、蛋白質(例、オリゴペプチド、ポリペプチド)、固相合成やファージディスプレイ法により作製されたランダムペプチドライブラリー、あるいは微生物、動植物、海洋生物等由来の天然成分等が挙げられる。
【0124】
工程(a)は被験物質による本発明の複合体の形成の抑制を評価可能である限り如何なる様式でも行われ得るが、例えば、再構成系(即ち、非細胞系)、細胞系(例、ツーハイブリッドシステム、酵母、細菌等を用いる細胞表層工学的手法)を用いて行われ得る。この場合、工程(a)は、例えば、以下の工程(a1)、(a2)を含み得る:
(a1)被験物質、SNIまたはNADaseとの結合能を有するその部分ペプチド、およびNADaseまたはSNIとの結合能を有するその部分ペプチドを接触させる工程;
(a2)上記(a1)により形成された本発明の複合体量を測定し、該複合体量を被験物質の不在下で形成された本発明の複合体量と比較する工程。
【0125】
上記工程(a1)では、接触は、本発明の複合体の形成を可能とする自体公知の方法により行なわれ得る。また、被験物質、SNIまたはその部分ペプチド、およびNADaseまたはその部分ペプチドの接触は同時に行われても、順次行われてもよい。例えば、先ず、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを接触させて本発明の複合体を形成させた後、被験物質を本発明の複合体と接触させてもよい。このような態様もまた、本工程で好ましく行われ得る。
【0126】
上記工程(a2)では、先ず、被験物質の存在下で形成された本発明の複合体量が測定される。複合体量の測定は、自体公知の方法により行うことができ、例えば、免疫学的手法(例、免疫沈降法、ELISA)、表面プラズモン共鳴を利用する相互作用解析法(例、BiacoreTMの使用)、ツーハイブリッドシステム、酵母、細菌等を用いる細胞表層工学的手法が挙げられる。
【0127】
次いで、被験物質の存在下で形成された複合体量が、被験物質の不在下で形成される複合体量と比較される。複合体量の比較は、好ましくは、有意差の有無に基づいて行なわれる。被験物質の不在下で形成される複合体量は、被験物質の存在下で形成された複合体量の測定に対し、事前に測定した複合体量であっても、同時に測定した複合体量であってもよいが、実験の精度、再現性の観点から同時に測定した複合体量であることが好ましい。
【0128】
本発明のスクリーニング方法Iの工程(b)では、本発明の複合体量を減少させる(複合体の形成を抑制する)被験物質が選択され得る。このように選択された被験物質は、例えば、NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療のため、あるいは本発明の複合体を解離させるために有用である。
【0129】
本発明のスクリーニング方法Iが連鎖球菌感染症(例えば、上述した連鎖球菌感染症)の治療薬の開発を特に意図して行われる場合、対象となり得る連鎖球菌は、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌であり得る。連鎖球菌は、NADaseをSNIと共発現することにより、自身をNAD枯渇から保護していると考えられる。この知見に基づけば、連鎖球菌内のSNI・NADase複合体を解離させる物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。NADase遺伝子およびSNI遺伝子を併有する連鎖球菌としては、例えば、A群、C群およびG群連鎖球菌が挙げられる。
【0130】
また、本発明のスクリーニング方法Iが連鎖球菌感染症の治療薬の開発を意図して行われる場合、より生理条件を反映させるという観点から、NADaseとの結合能を有するSNIの部分ペプチドは、さらにNADase阻害活性を保持することが好ましい。同様の観点から、SNIとの結合能を有するNADaseの部分ペプチドは、さらにNADase活性を保持することが好ましい。より好ましくは、本発明のスクリーニング方法Iは、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌由来のNADaseおよびSNIを用いて行われ得る。
【0131】
別の実施形態では、本発明のスクリーニング方法はまた、以下の工程(a)および(b)を含み得る:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
以下、本スクリーニング方法を、必要に応じてスクリーニング方法IIと省略する。
【0132】
本発明のスクリーニング方法IIの工程(a)で用いられる被験物質は、上述のスクリーニング方法Iで用いられるものと同様であり得る。
【0133】
工程(a)は、上述したNADase活性について、被験物質による抑制の解除を評価可能である限り如何なる様式でも行われ得るが、例えば、再構成系(即ち、非細胞系)、細胞系(例、酵母、細菌等を用いる細胞表層工学的手法)を用いて行われ得る。この場合、工程(a)は、例えば、以下の工程(a1)、(a2)を含み得る:
(a1)被験物質、基質、SNIまたはNADase阻害活性を有するその部分ペプチド、およびNADaseまたはNADase活性を有するその部分ペプチドを接触させる工程;
(a2)被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下でNADaseまたはNADase活性を有するその部分ペプチドのNADase活性を測定し、該活性を被験物質の不在下およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下での該NADase活性と比較する工程。
【0134】
上記工程(a1)では、接触は、NADase活性の検出を可能とする自体公知の方法により行なわれ得る。また、被験物質、基質、SNIまたはその部分ペプチド、およびNADaseまたはその部分ペプチドの接触は活性の測定に適切である限り同時に行われても、順次行われてもよい。基質としては、例えば、NADが用いられ得る。
【0135】
上記工程(a2)では、先ず、被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下でのNADase活性が測定される。NADase活性の測定は、自体公知の方法により行うことができる。例えば、NADase活性は、基質の減少量、または検出可能な生成物の増加量に基づき評価できる。基質から変換され得る検出可能な生成物としては、例えば、ADPR、ニコチンアミド、cADPRが挙げられる。NADase活性の測定については、例えば、FEMS Microbiol Lett 136, 71-78 (1996); J. Exp. Med. 104: 577-587 (1956); Appl. Microbiol. 14: 391-393 (1966) を参照のこと。
【0136】
次いで、被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下で測定されたNADase活性が、被験物質の不在下およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下で測定されたNADase活性と比較される。NADase活性の比較は、好ましくは、有意差の有無に基づいて行なわれる。被験物質の不在下で測定されるNADase活性は、被験物質の存在下で測定されたNADase活性の測定に対し、事前に測定したNADase活性であっても、同時に測定したNADase活性であってもよいが、実験の精度、再現性の観点から同時に測定したNADase活性であることが好ましい。
【0137】
本発明のスクリーニング方法IIの工程(b)では、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得る被験物質が選択され得る。このように選択された被験物質は、例えば、NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療のため、あるいは本発明の複合体の解離のために有用である。
【0138】
本発明のスクリーニング方法IIが連鎖球菌感染症(例えば、上述した連鎖球菌感染症)の治療薬の開発を特に意図して行われる場合、対象となり得る連鎖球菌は、NADase遺伝子およびSNI遺伝子を併有する上述の連鎖球菌であり得る。連鎖球菌は、NADaseをSNIと共発現することにより、自身をNAD枯渇から保護していると考えられる。この知見に基づけば、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得る物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。好ましくは、本発明のスクリーニング方法IIは、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌由来のNADaseおよびSNIを用いて行われ得る。
【0139】
本明細書中で挙げられた特許および特許出願明細書を含む全ての刊行物に記載された内容は、本明細書での引用により、その全てが明示されたと同程度に本明細書に組み込まれるものである。
【0140】
以下に実施例を挙げ、本発明を更に詳しく説明するが、本発明は下記実施例等に何ら制約されるものではない。なお、「材料および方法」から実施例9まではSNIおよびNADase関連発明に関する実施例であり、実施例10〜11はSLO関連発明の実施例である。
【実施例】
【0141】
材料および方法
(1)材料
ムタノリシン(Mutanolysin)およびリゾチームは、Sigma Chemical Co. (St. Louis, MO. USA) から購入した。アンピシリンナトリウムおよびL(+)-アラビノースは、Wako Pure Chemical Industries, Ltd. (Osaka, Japan) から入手した。DNA修飾および制限酵素は、Takara Shuzo Co., Ltd. (Kyoto, Japan)、Toyobo Co., Ltd. (Osaka, Japan)、またはNippon gene (Toyama, Japan) から得た。タンパク質分子標品は、Bio-Rad (California, USA) およびAmersham Biosciences Corp. (NJ, USA) から購入した。酵母抽出物およびトリプトンは、Difco Laboratories (Detroit, Mich. USA) から購入した。西洋ワサビペルオキシダーゼ・コンジュゲート・Ni-NTAは、Qiagen (Hilden, Germany) から入手した。他の全ての化学物質は、試薬グレードまたは分子生物学グレードであった。
【0142】
(2)菌株および培養条件
Streptococcus dysgalactiae subsp. equisimilis H46A (Christensen, L.R. (1945) J Gen Physiol 28, 363-383) は、1% 酵母抽出物 (yeast extract) を補充したTodd Hewitt broth (Becton Dickinson, Cockeysville, Md. USA) (THY培地) 中で37℃にて増殖させた。Escherichia coli TOP10 (Invitrogen, Frederick, USA) およびE.coli BL21 (Novagen, Madison, Wisconsin) を、組換えプラスミドのクローニング用宿主として用いた。これらの株は、Luria-Bertani Broth (LB) 中で37℃にて培養した。Escherichia coli TOP10は、L-アラビノースの輸送能を有するが、その代謝能は有しない。エレクトロポレーションを、プラスミドDNAのE.coli TOP10およびBL21への導入のために用いた (J Biochem (Tokyo) 122: 237-242 (1997))。形質転換体を、100μg/mlのアンピシリンを含有するLBプレート上で選択した。
【0143】
(3)ポリメラーゼ連鎖反応(PCR)
Expand High Fidelity PLUS PCR Systemは、Roche Diagnostics (Penzberg, Germany) からの製品である。酵素およびバッファーを製造業者の推奨に従って用いた。PCR混合液の総量は約50μlであった。通常通り、初期変性 (94℃で90秒)、鎖の変性 (94℃で30秒)、アニーリング (55℃で30秒)、および伸長 (72℃で1kbあたり60秒) の条件下で40サイクルの増幅が行われた。
【0144】
(4)DNA操作
E.coli TOP10のプラスミドDNAを、QIA prep Spin Miniprep Kit (Qiagen, Hilden, Germany) を用いて抽出した。PCR用のstreptococciゲノムDNAを既報 (Biochim Biophys Acta 1681: 134-149 (2005)) に従い調製した。
【0145】
(5)プラスミドおよび合成オリゴヌクレオチドプライマー
プラスミドpBAD/Hisシリーズは、Invitrogen (Groningen, The Netherlands) から購入した。nga-orf1-sloオペロンのnga-orf1領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより、S. dysgalactiae subsp. equisimilis H46Aの染色体DNAから増幅した。
5’-CGGGATCCGTTAGTGGCAAAGAAGGTAAAAAAAGCG-3’(下線部はBamHI部位:配列番号7)
5’-GGGGTACCCTAAAATGTTTCTATTGTTCTTTCGAC-3’(下線部はKpnI部位:配列番号8)
次いで、このPCR産物をBamHIおよびKpnIで消化し、BglIIおよびKpnIで前処理したpBAD/HisBに連結し、プラスミドpBAD/HisIを作製した(図2A)。
同様に、nga-orf1-sloオペロンのnga領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより増幅した。
5’-CGGGATCCGTTAGTGGCAAAGAAGGTAAAAAAAGCG-3’(下線部はBamHI部位:配列番号9)
5’-GGGGTACCTTACTTCCTATCTTGCATTTTCTTAATTTTG-3’(下線部はKpnI部位:配列番号10)
次いで、このPCR産物をpBAD/HisBに連結し、プラスミドpBAD/HisIIを作製した(図2A)。
プラスミドpQE30は、Qiagen (Hilden, Germany) から購入した。nga-orf1-sloオペロンのorf1領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより、S. dysgalactiae subsp. equisimilis H46Aの染色体DNAから増幅した。
5’-CGGGATCCTATAAGGTGCCAAAGGGTTTAGAAC-3’(下線部はBamHI部位:配列番号11)
5’-GGGGTACCCTAAAATGTTTCTATTGTTCTTTCGAC-3’(下線部はKpnI部位:配列番号12)
次いで、このPCR産物をBamHIおよびKpnIで消化し、BamHIおよびKpnIで前処理したpQE30に連結し、6個の連続するヒスチジンタグに融合したOrf1を作製した。
【0146】
(6)DNAシークエンシングおよび配列データ解析
ヌクレオチド配列は、適切なプライマーおよびABI PARISMTM 3100 genetic analyzer (PE Applied Biosystems, California, USA) を用いてジデオキシ法により決定した。ヌクレオチド配列データを、GENETYXコンピュータソフトウェア (Software development Co. Ltd., Tokyo, Japan) を用いて解析した。染色体DNA配列データベースを、BLASTN検索アルゴリズム(http://www.ncbi.nlm.hih.gov/BLAST/) を用いて検索した。
(7)ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)
酵素のSDS-PAGEを、Laemmliの方法 (Nature 227, 680-685 (1970)) により還元条件下で行った。タンパク質バンドを、Coomassie Brilliant Blue (CBB) R-250で染色した。E.coliの全細胞溶解物 (whole cell lysate) を調製するため、0.5 mlの培養物を遠心分離 (16,000×g, 3分, 4℃) し、得られた細菌ペレットを0.1 mlの1×サンプルバッファー (組成:1% SDS, 1% 2-メルカプトエタノール, 20% グリセロール, 50 mM Tris-HCl, pH 6.8) で溶解し、この溶解物を100℃で3分間インキュベートした。残存した細菌粒子および非溶解細胞を、遠心分離 (16,000×g, 5分, 室温) により除去した。同様に、5 mlのS. dysgalactiae subsp. equisimilis H46A培養物を遠心分離 (4,000×g, 15分, 4℃) し、得られた細菌ペレットを、溶解バッファー (組成: 100 U/mlムタノリシン, 10 mg/mlリゾチーム, 50 mMグルコース, 10 mM EDTA,25 mM Tris-HCl, pH 8.0) 中で37℃にて30分間インキュベートした。インキュベーション後、細胞を遠心分離 (4,000×g, 5分, 4℃) により回収し、ペレットを0.1 mlの超純粋中に懸濁させた。次いで、0.1 mlの2×サンプルバッファーを加え、懸濁液を100℃で3分間インキュベートした。残存した細菌粒子および非溶解細胞を遠心分離 (16,000×g, 5分, 室温) により除去した。
【0147】
(8)NH2末端アミノ酸配列
E.Coli BL21中で過剰発現させたインタクトOrf1 (成熟型, Hisタグなし) およびS. dysgalactiae subsp. equisimilis H46Aから高度に精製されたインタクトOrf1を12.5%および15% SDS-PAGEに供し、PVDF膜に転写した。PVDF膜をCBB R-250により染色し、19kDaのタンパク質バンドを膜から切り出した。2つのバンドのタンパク質シークエンシングを、PE Applied Biosystems model 491 Procise protein sequencing system (PE Applied Biosystems, California, USA) を用いた自動化エドマン分解により行った。
【0148】
(9)NADaseおよびOrf1の過剰発現
プラスミドpBAD/HisB (ベクターのみ)、pBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1) またはpBAD/HisII (Hisタグ融合NADase単独) を担持するE.coli TOP10の一晩培養物を、新鮮培地中に1:20で希釈し、OD660が0.5に達するまで増殖させた。次いで、培養物を0.002%または0.02% L-アラビノースで4時間誘導した。細胞を遠心分離 (5,000×g, 10分, 4℃) により採取し、リン酸緩衝化生理食塩水で洗浄し、BugBusterTMタンパク質抽出試薬 (Novagen, Madison, Wisconsin) 中に室温にて再懸濁した。BugBuster試薬を製造業者の推奨に従い用いた。タンパク質抽出後、不溶性細胞片を遠心分離 (12,000×g, 30分間) により除去し、上清をタンパク質源として用いた。同様に、組換えプラスミドpQE30 (Hisタグ融合Orf1) を担持するE.coli BL21の一晩培養物を、新鮮培地中に1:20で希釈し、OD660が0.5に達するまで増殖させた。次いで、培養物を1 mM IPTGで3時間誘導し、Hisタグ融合Orf1をBugBuster試薬を用いて抽出した。
【0149】
(10)TSKgel BioAssist Ni2+キレートカラム
0.01% アジ化ナトリウムを含有する50 mMリン酸ナトリウムバッファー (pH 7.4) (バッファーA) で予め洗浄したTSKgel BioAssistキレートカラム (Tosoh Co., Tokyo, Japan) に、4 mlの0.1 M Ni2SO4溶液をロードし、次いで500 mMイミダゾールを含有する10 mlのバッファーAで洗浄し、PEEK型PPCMポンプを備えたTosoh HPLCシステム (Tosoh Co., Tokyo, Japan) を用いて、流速1 ml/分により、20 mMイミダゾールを含有するバッファーAで室温にて平衡化した。
【0150】
(11)Hisタグ融合NADase/Orf1複合体の精製
組換えプラスミドpBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、抽出液3.5 mlを、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムに繰り返しロードした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01%アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。溶出は、流速1.0 ml/分で20℃で行った。タンパク質溶出プロフィールは、UV8000 (Tosoh Co., Tokyo, Japan) を用いて280 nmでの吸光度をモニターすることにより得た。Hisタグ融合NADase/Orf1複合体は、30分間にわたる、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。複合体画分をプールし、バッファーAに対して透析した。
【0151】
(12)インタクトOrf1 (成熟型, Hisタグなし) およびHisタグ融合NADaseの精製
組換えプラスミドpBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、透析した抽出液を、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムにアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。Orf1を、3 Mチオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する3 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) での溶出により回収した。精製したOrf1を等量の水で希釈し、バッファーAに対して透析した。次いで、カラムに吸着したHisタグ融合NADaseを、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製酵素をプールし、バッファーAに対して透析した。
【0152】
(13)Hisタグ融合Orf1の精製
E.coli細胞のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、透析した抽出液を、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムにアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。Hisタグ融合Orf1を、30分間にわたる、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製したHisタグ融合Orf1をプールし、バッファーAに対して透析した。
【0153】
(14)S. dysgalactiae subsp. equisimilis H46A培養物からのインタクトNADase (成熟型, Hisタグなし) の精製
株H46Aの培養上清に、硫酸アンモニウムを80%飽和となるように加え、得られた沈殿を遠心分離(10,000×g, 20分, 4℃)により回収した。ペレットを0.01%アジ化ナトリウムを含有する0.1 Mリン酸ナトリウムバッファー (pH 7.4) 中に溶解し、同バッファーに対して4℃で一晩透析した。遠心分離 (10,000×g, 20分) により得られた、透析した清澄な上清を、ToyoScreen 5ml column (Tosoh Co., Tokyo, Japan) に充填したヒドロキシアパタイトBio-gel HT gel (Bio-Rad Laboratories, CA) に、流速1.0 ml/分で室温にてアプライした。NADaseを、30分間にわたる、0.01% アジ化ナトリウムを含有する0.1 Mから0.5 Mのリン酸ナトリウムバッファー (pH 7.4) の線形勾配により回収した。活性画分をプールし、バッファーAに対して透析した。透析し、部分精製したNADaseをTSKgel BioAssist Ni2+キレート-Hisタグ融合Orf1アフィニティーカラムにアプライした。このカラムには、1 mgの精製Hisタグ融合Orf1が予めロードされ、20 mMイミダゾールを含有するバッファーAで平衡化されていた。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。インタクトNADaseを、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する3 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を流速0.5 ml/分でロードすることにより回収した。各画分 (0.5 ml) を0.5 mlの水を含有するチューブ中に回収し、高濃度のNaSCNによるタンパク質の変性を防いだ。NADase活性画分をプールし、バッファーAに対して可能な限り透析した。
【0154】
(15)インタクトNADase (成熟型, Hisタグなし)、Orf1およびNADase/Orf1複合体のゲル透過クロマトグラフィー
TSKgel BioAssist G3SWXLカラム (Tosoh Co., Tokyo, Japan) を用いて、インタクトNADase (成熟型, Hisタグなし) Orf1およびNADase/Orf1複合体のゲル透過クロマトグラフィーを行った。カラムを、0.15 M リン酸ナトリウムバッファー (pH 6.6) により流速0.5 ml/分で室温にて展開した。タンパク質溶出プロフィールを、280 nm の吸光度をモニターすることにより得た。
【0155】
(16)S. dysgalactiae subsp. equisimilis H46Aの細胞内Orf1の解析
溶解溶液 (組成:100 U/ml ムタノリシン, 10 mg/ml リゾチーム, 50mM グルコース, 10 mM EDTA, 25mM Tris-HCl, pH 8.0) 中での消化およびソニケーションにより得た細胞抽出液を、抗Orf1抗体コンジュゲートHiTrap NHS活性化HPカラム (Amersham Biosciences Corp., NJ, USA) にアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) をアプライし、次いでOrf1を、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収した。Orf1画分をプールし、水に対して透析した。透析物をSpeedvac Concentrator (Sarvant Instrument Inc., NY, USA) を用いて濃縮し、SDS-PAGEに供した。ゲル中のタンパク質をPVDF膜に転写し、CBB R-250で染色した。タンパク質バンドを切り出し、PE Applied Biosystems model 491 Procise protein sequencing systems (PE Applied Biosystems, California, USA) により解析した。
【0156】
(17)抗体
Hisタグ融合Orf1をウサギ抗Orf1抗体の惹起に用いた。簡潔には、完全フロインドアジュバントと混合した約2 mgのHisタグ融合Orf1をウサギに皮下注射した。1 mgのタンパク質および不完全フロインドアジュバントからなるブースター注射を2週間の間隔で3回与えた。イムノグロブリンG画分を、プロテインA-セファロース (Amersham Biosciences Corp., NJ, USA) を用いたアフィニティークロマトグラフィーにより精製した。
ウサギポリクローナル抗SLO IgG (抗NADase抗体含有) は、American Research Product Inc. (Belmont, MA. USA) から購入した。
【0157】
(18)イムノブロット解析(ウエスタンブロット解析)
SDS-PAGE後、タンパク質を、電気ブロッティングによりSequi-Blot PVDF膜 (Bio-Rad, California, USA) 上に転写した。膜を、0.05% Tween 20を含有するリン酸緩衝化生理食塩水 (PBST) において5% スキムミルクおよび1% ウシ血清アルブミン (BSA) で1時間ブロッキングした。膜をPBSTで洗浄し、次いで一次抗体 (PBST中で1:3,000に希釈) を加え、ブロットを1時間インキュベートした。次に、ヤギaffini-pure IgG ペルオキシダーゼ・コンジュゲート・抗ウサギIgG (Wako Pure Chemical Industries, Ltd) をPBST中で1:3,000希釈したものを加え、1時間インキュベートし、抗体抗原複合体を、製造業者により記載(J Immunol Methods 95, 71-77 (1986)) されるようにPOD免疫染色セット (Wako Pure Chemical Industries, Ltd) により可視化した。Ni-NTA HRPコンジュゲート (Qiagen, Hilden, Germany) (西洋ワサビペルオキシダーゼとカップリングしたニッケル-ニトリロトリ酢酸 (Ni-NTA)) を、Hisタグを有する組換えタンパク質の直接的な検出のために使用し、製造業者の推奨に従い使用した。
【0158】
(19)Orf1・細胞外NADase間の相互作用のファーウエスタン解析
S. dysgalactiae subsp. equisimilis H46Aの培養上清および培地単独を、SDS-PAGE (10% アクリルアミド) 上で泳動し、PVDF膜に転写し、精製Hisタグ融合Orf1 (PBST中10μg/ml) の存在下または不在下で室温にて1時間インキュベートした。精製Hisタグ融合Orf1のNADaseへの結合は、「イムノブロット解析」に記載されるような抗Orf1抗体を用いるウエスタンブロット解析によりアッセイした。簡潔には、膜をPBSTで洗浄し、次いでウサギ抗Orf1抗血清を加え、ブロットを1時間インキュベートした。次いで、ヤギaffii-pureIgGペルオキシダーゼ・コンジュゲート・抗ウサギIgGを加え、1時間インキュベートし、抗体抗原複合体をPOD免疫染色により可視化した。培養上清中のNADaseおよび培地単独(コントロール)を、抗NADase抗体を含有する抗SLOポリクローナル抗体によるウエスタンブロット解析によりアッセイした。
【0159】
(20)NADaseアッセイ
NADaseアッセイを、Gerlach et al. (FEMS Microbiol Lett 136, 71-78 (1996)) の方法により行った。1単位の酵素は、37℃では0.010μM NADを7.5分で切断する。
・全細胞抽出物の調製:
対数増殖期の中期培養物 (10 ml) の細胞を、溶解溶液 (組成:100 U/ml ムタノリシン, 5 mg/ml リゾチーム, 0.01% アジ化ナトリウム, 150 mM リン酸ナトリウム, pH 6.6) 中で、37℃にて30分間インキュベートした。インキュベーション後、細胞ソニケートし、溶解物を低温で遠心分離し、上清をNADaseアッセイに用いた。
・温度安定性:
インタクトOrf1 (成熟型, Hisタグなし) の温度安定性を、0.15 M リン酸ナトリウムバッファー (pH 7.4) において38μg/ml (2.0μM) のタンパク質濃度で決定した。Orf1溶液をEppendorfチューブ中に密閉し、20-100℃で15分間インキュベートした。インキュベーションを、溶液を氷水中で冷却することにより停止し、0.5μlの各溶液を、50 mM リン酸カリウムバッファー (pH 7.5) 中に70μg/ml (1μM) のBSAを含有する10μlの5μg/ml (0.1μM) インタクトNADase (成熟型, Hisタグなし) 溶液に加えた。次いで、混合液を室温で10分間インキュベートし (Orf1とNADaseの最終モル比は1:2)、残存したヒドロラーゼ活性をNADで測定した。コントロールとして、インキュベートしたOrf1溶液の代わりに、BSAのみを含有する50mMリン酸カリウムバッファー (pH 7.5) を用いた。
【0160】
実施例1:E.coliでの連鎖球菌NADaseの発現
連鎖球菌NADグリコヒドロラーゼ (NADase) - ストレプトリシンO (SLO) オペロンの調節に関する以前の研究では、ベクターとしてpHY300PLKを使用することにより、E.coliでの遺伝子クラスターのサブクローニングが試みられた。活性SLOの過剰発現はE.coliで達成されたが (Biosci Bisotechnol Biochem 65, 2682-2689 (2001))、NADase遺伝子インサートを有するコロニーは全く得られなかった (Biochim Biophys Acta 1681: 134-149 (2005))。この酵素の潜在的宿主毒性を考慮して、E.coliでの遺伝子発現を厳格に制御するaraBADプロモーター (pBAD) を有する発現ベクターpBADに、NADase遺伝子ngaをサブクローニングした(図2A)。しかし、NADase遺伝子インサートのみを有するE.coli TOP10 (pBAD/HisII) では、L-アラビノース処理による誘導の際に、この酵素タンパク質の産生は生じなかった(図2B)。この組換え株の増殖は誘導剤の不在下でさえも遅く、細胞の一部は凝集した(データ示さず)。一方、ngaからorf1の3’端にわたるより長いインサートを有するpBAD/HisIを担持するE.coli組換え株は、有意な量のNADaseを産生した(図2B)。データベース検索をすると、A群連鎖球菌 (GAS) 株ではOrf1が同様に保存されていたが、他の細菌株では欠失していた(データ示さず)。全長のNADase-SLOオペロンをpBAD制御下に配置し、かつE.coliで発現させた場合、NADaseおよびOrf1に加え、活性SLOが産生された(データ示さず)。
これらの結果は、細胞内連鎖球菌NADaseがE.coliのみではなく、連鎖球菌自体にもおそらく毒性を示すこと、ならびに共発現されたOrf1がその保有菌をNAD枯渇から保護し得ることを示唆する。この可能性を検証するため、NADase-SLOオペロンの未知遺伝子産物Orf1の遺伝的および生化学的性質をその後の実験で調べた。
【0161】
実施例2:Orf1の発現およびそのN末端領域のシークエンシング
E.coliにおいてPBAD制御下で共発現させた場合、SDS-PAGEプロフィールにより判定すると、Orf1の量はNADaseのそれよりも多かった(図3A)。Orf1バンドをゲルから切り出し、N末端アミノ酸シークエンシングに直接供した。得られた配列MYKVPKGLEHYQKM(配列番号15)は、NADase-SLOオペロン中のorf1遺伝子のヌクレオチドアライメントより推定したものと同一であった(図3B)。この遺伝子は483塩基対からなり、推定161アミノ酸をコードしていた。ヌクレオチド配列から、このOrf1タンパク質の分子量は18,800であると推定された。
S. dysgalactiae subsp. equisimilis H46A中のorf1の発現を実証するため、組換えE.coli細胞で産生されたHisタグ融合Orf1を用いてウサギを免疫することにより、抗Orf1抗体を調製した(図4A)。この抗体を用いたウエスタンブロット解析は、Orf1タンパク質が産生され、H46A球菌内に専ら局在したことを示した(図4B)。Orf1の分子量は、E.coli中でNADaseと共発現したタンパク質の分子量と一致した(図4B)。SLOおよびNADaseとは異なり、Orf1は連鎖球菌培養上清中に検出されなかった(図4B)。
【0162】
実施例3:S. dysgalactiae subsp. equisimilis H46A由来のOrf1の精製およびアミノ末端シークエンシング
Orf1タンパク質をS. dysgalactiae subsp. equisimilis H46Aから抽出し、抗Orf1抗体固定化カラムを用いたアフィニティークロマトグラフィーにより精製した(図5)。この溶出画分をSDS-PAGEに供し、PVDF膜上にブロットした。CBB R-250染色により、細胞内Orf1複合体形態におそらく由来する5つのバンドを検出した(図5)。アミノ末端シークエンシングは、19kDaのバンドがOrf1であることを示した:その配列MYKVPKGLEHYQKM(配列番号15)ならびに分子サイズは、ヌクレオチド配列から推定したもの、および組換えE.coli細胞中で発現したOrf1タンパク質のものと同じであった。計算値50 kDaの2個のバンドは、NADaseの前駆体かもしれなかったが、それらのアミノ酸シークエンシングは、おそらくアミノ末端ブロッキングのため、成功しなかった。翻訳後、約2 kDaの領域がNADaseのC末端からタンパク質分解により除去されることが最近見出されている (Biochim Biophys Acta 1681: 134-149 (2005))。約50 kDaのバンドがNADaseである場合、C末端の除去が球菌細胞で生じているに違いない。計算値34 kDaの濃いバンドは、N末端の187個のアミノ酸が欠失したNADaseタンパク質 (ΔN187) であった。この欠失が、異常な転写または球菌抽出物の調製の際のタンパク質分解によって生じるかは不明である。しかしながら、この短縮型分子が抗Orf1抗体固定化カラムに吸着したという事実は、NADaseのN末端領域がOrf1との相互作用に必須ではないことを示す。16 kDaのバンドは連鎖球菌細胞の溶解に使用された卵白リゾチームであった。非生理学的ではあるが、この溶解酵素はインビトロでOrf1、NADaseまたはOrf1-NADase複合体のいずれかと相互作用するようであった。SDS-PAGEプロフィール(図5)から推測されるように、NADase前駆体に対するOrf1の比率は、E.coliでの比率に対し、連鎖球菌ではそれほど高くなかった(図3A)。
NADase-SLOオペロンのメンバーOrf1は、組換えE.coliおよび連鎖球菌株H46Aにおいて同じ開始コドンから翻訳された。E.coliでの連鎖球菌NADaseの産生がOrf1の共発現を必要とするという事実は、潜在的な毒性を示すNADase活性の抑制におけるこのタンパク質の自己保護的な役割を示唆する。従って、NADaseおよびOrf1のインビトロでの物理的および機能的相互作用を解析するために、以下の実験を行った。
【0163】
実施例4:Orf1・NADase間の相互作用の解析
NADaseとの相互作用の解析のため、Hisタグ融合タンパク質としてE.coliで産生されたOrf1を、ニッケル−キレートカラムクロマトグラフィーにより精製した(図4A)。S. dysgalactiae subsp. equisimilis H46Aの培養前後のTHY培地をSDS-PAGEに供し、PVDF膜上にブロットし、2部に分割した。膜の一方を抗SLOポリクローナルIgGを用いるウエスタンブロッティングに直接供し、他の膜は精製Orf1で処理し、次いで抗Orf1抗血清を用いたウエスタンブロッティングに供した(図6)。図4Bに示される通り、連鎖球菌H46A培養物からの消費培地はOrf1およびその交差反応性物質を含有しなかった。NADaseの検出のため、この酵素タンパク質と交差反応する市販の抗SLOポリクローナルIgGを用いた (Cell 104: 143-152 (2001); Biochim Biophys Acta 1681: 134-149 (2005))。
ファーウエスタンブロッティングにより、Orf1は、Orf1処理したPBDF膜で、NADaseバンドに対応するまさにその位置に検出された。この結果はNADase・Orf1間の物理的相互作用を示しているので、NADase活性に対する後者のタンパク質の効果を続いて調べた。
【0164】
実施例5:Orf1によるNADaseの阻害
Orf1は細胞内タンパク質であり、その細胞含量はそれ程高くない(図5)。このタンパク質を過剰産生させるため、NADaseとの共発現を、pBAD/HisIを担持するE.coliで試みた。図4Bおよび図5に示されるように、E.coli由来のOrf1の電気泳動プロフィールおよびN末端配列は、H46A由来のものと同じであった。同時に、E.coliでOrf1と共発現したHisタグ融合NADaseを、ニッケル-キレートカラムクロマトグラフィーにより精製した。共発現したOrf1分子は、予期された通り、NADaseとの複合体として存在し、相互の分離は、3M NaCl処理でさえも達成できなかった(図7A)。3M NaSCNを用いてこの複合体を解離させ(図7B)、ゲル透過クロマトグラフィーによりOrf1をさらに精製した。組換えE.coli抽出物中のNADaseは、Orf1との酵素学的に不活性な複合体として存在した。N末端シークエンシングおよび分子サイズの決定は、このNADaseタンパク質が前駆体形態であることを示す。Orf1から解離した場合、NADase前駆体は分泌成熟形態の酵素の比活性とほぼ同じ比活性を示した(データ示さず)。NADase前駆体は少量であり、かつ分泌成熟酵素もまたOrf1と不活性複合体を形成したので、細胞外形態を以下の実験で用いた。従って、NADaseは、Hisタグ融合タンパク質としてE.coliで過剰産生されたOrf1が固定されたカラムを用いたアフィニティーカラムクロマトグラフィーにより、S. dysgalactiae subsp. equisimilis H46Aの培養上清から精製した(図7C)。精製したNADaseをOrf1と混合し、室温で10分間インキュベートした場合、この酵素活性は当価なモル比においてほぼ完全にブロックされた(図7D)。この阻害反応は非常に迅速であり、また、0℃でさえも生じた(データ示さず)。
連鎖球菌内のNADase前駆体がOrf1に結合した不活性複合体として存在する可能性を検証するため、H46A溶解物を等容量の6M NaSCNと混合した。解離処理後、Orf1を抗Orf1抗血清で中和した。表1に示されるように、活性NADaseは連鎖球菌溶解物中に検出できなかったが、NaSCN、次いで抗Orf1抗血清で処理したサンプル1 g (湿重量) あたり、約104ユニットの活性が認められた。活性化抽出物を水で希釈した場合、NADase活性は再度マスクされた(データ示さず)。このことは、NADaseがOrf1と再会合したことを示唆する。これらの結果は、初期のNADaseが、組換えE.coliにおける場合と同様に、連鎖球菌内でOrf1と不活性複合体を形成することを示す。
【0165】
【表1】

【0166】
a 活性は、3つの実験からの平均±S.D.で示した。
b 全細胞抽出物は、材料および方法における「NADaseアッセイ」に記載の通り調製し、10μlの抽出物を、10μlの0.1 M Na2HPO4(pH 7.4) および80μlの超純粋と混合した。
c 10μlの全細胞抽出物を、10μlの6 M NaSCN, 0.1M Na2HPO4 (pH 7.4) と室温にて混合し、5秒間のボルテックス後、80μlの超純粋を即座に加えた。
d 10μlの全細胞抽出物を、10μlの0.1 M Na2HPO4 (pH 7.4)、1μlの抗Orf1抗血清、および79μlの超純粋と混合した。
e 1μlの抗Orf1抗血清を、10μlの0.1 M Na2HPO4 (pH 7.4) および89μlの超純粋と混合した。
f 10μlの全細胞抽出物を、10μlの6 M NaSCN, 0.1M Na2HPO4 (pH 7.4) と室温にて十分に混合し、5秒間のボルテックス後、1μlの抗Orf1抗血清および79μlの超純粋を即座に加えた。
【0167】
実施例6:NADase・Orf1複合体の分子サイズの決定
NADase・Orf1複合体の分子サイズを決定するため、TSKgel BioAssist G3SWXLカラム (Tosho Co., Tokyo, Japan) を用いて、ゲル透過クロマトグラフィーを行った。図8Aに示されるように、NADase、Orf1およびNADase・Orf1複合体の各々を、明瞭な単一ピークとしてそれぞれ検出した。各保持時間から見積もった分子量は、24 kDa (Orf1)、54 kDa (NADase)および69 kDa (NADase・Orf1複合体) であった。これらの値(図8B)は幾らか高いが、各ヌクレオチド配列から推定した値に近似していた。これらの結果は、NADaseおよびOrf1がモノマータンパク質として存在し、混合した場合に、ヘテロダイマーとしての複合体を形成することを示す。
【0168】
実施例7:温度安定性およびプロテアーゼ感受性
これまでに記載した細菌NADaesは、細胞内酵素であり、それらの阻害タンパク質は熱に不安定である(Methods Enzymol 66; 137-144 (1980); Science 123: 50-53 (1956))。興味深いことに、Orf1は熱に安定であり、15分間の熱処理後に残存する相対阻害活性は、60℃では計算値で82%、100℃では計算値で72%であった(図9)。精製したOrf1はトリプシンに感受性であり、リシルエンドペプチダーゼにより不活化された(データ示さず)。
【0169】
実施例1〜7に関する考察
NADase-ストレプトリシンOオペロンは、AおよびC群の連鎖球菌で高度に保存されている (Cell 104: 143-152 (2001); Biochim Biophys Acta 1681: 134-149 (2005); Infect Immun 70: 2730-2733 (2002))。Ngaおよびsloに加えて、このオペロンは、未知遺伝子orf1を含んでいた。上記データは、この遺伝子が連鎖球菌NADaseインヒビター (streptococcal NADase inhibitor:SNI) として機能する18.8 kDaタンパク質をコードすることを示す。従って、この遺伝子をsniと標記することは適切であると考えられる。E.coliでの単一ngaのサブクローニングの失敗 (Biochim Biophys Acta 1681: 134-149 (2005)) とは対照的に、この遺伝子のorf1 (sni) との共発現は、nga-orf1インサートを有する組換えプラスミドを担持する異種宿主において安定的に生じた。菌体内での活性NADaseの産生は、酸化還元に必須のNADの分解による毒性効果を引き起こし得る。この潜在的に有害な酵素に対する細菌性インヒビターの存在は、Bacillus subtilis、Proteus vulgaris、Proteus rettgeriおよびMycobacterium butyricumで知られている。これらの細菌のNADaseは、熱不安定性のインヒビターと複合体化した細胞内熱安定タンパク質である (Methods Enzymol 66: 137-144 (1980); Science 123: 50-53 (1956))。これらのインヒビターの遺伝的および機能的調査はこれまでに行なわれていない。一方、溶血性連鎖球菌は、培養培地中に熱不安定性NADaseを分泌する (J Exp Med 104: 577-587 (1956); J Exp Med 106: 15-26 (1957); FEMS Microbiol Lett 136: 71-78 (1996))。細胞外NADaseを産生する連鎖球菌細胞は、この酵素に対するインヒビタータンパク質を含む (Acta Path. Microbiol. Scandinav. 69: 277-286 (1967))。細胞内局在、分子量およびトリプシン感受性は、SNIのそれらに準じる。しかし、熱安定性に関しては、HolmおよびKaijser (Acta Path. Microbiol. Scandinav. 69: 277-286 (1967)) により報告されるインヒビターは、SNIとは異なる。それらの調製は、60℃で15分間加熱することにより完全に不活化されるのに対し、SNIはむしろ熱安定性であった(図9)。
【0170】
SNIをコードする遺伝子 (sni) は、nga (spn) プロモーターから、ポリシストロニックmRNAとして、sloと共転写される (Biochim Biophys Acta 1681: 134-149 (2005))。nga-sloオペロンの翻訳の際、NADase前駆体の自己毒性は、モル比1:1での、SNIとの複合体の形成により迅速にブロックされるようである(図10)。ウエスタンブロット解析より実証されるように、SNIの局在は完全に細胞内である(図4B)。この結果と一致して、シグナルペプチド領域は、SOSUI (Bioinformatics 14: 378-379 (1998); Bioinformatics 18: 608-616 (2002)) およびSignalP (Int J Neural Dsyst 8: 581-599 (1997)) 膜貫通推定アルゴリズムを用いるインシリコ解析では、SNIに見出されなかった。球菌体内のNADase前駆体およびSLOの量はわずかであった(データ示さず)。このことは、合成されたタンパク質が培地中へと迅速に分泌されることを示唆する。しかし、NADase前駆体は安定な細胞内複合体を形成するので、この複合体は、球菌エンベロープを介する排出中に解離するはずである。この複合体の解離が幾つかの膜成分との相互作用により、SNIのタンパク質分解排除により、または他の機構により引き起こされるか否かは、解明すべき課題である。
【0171】
実施例8:Orf1によるCD38、CD157の阻害
ヒト由来CD38およびCD157の遺伝子を組み込んだpQE30-His-タグ-CD38およびpQE30-His-タグ-CD157で形質転換した大腸菌を1リットルのアンピシリンを含むLB液体培地で培養し、1mM IPTGを加えて3時間37℃で培養した菌体を遠心分離で集めた。菌体に20 mlのBugBuster (菌体内タンパク質抽出液) を加えて、His-タグ-CD38およびHis-タグ-CD157をそれぞれ抽出した。この原液をTSKgel BioAssist Ni-キレートクロマトグラフィーに供して、His-タグ-CD38およびHis-タグ-CD157をそれぞれ部分精製した。そのNADase活性を有する画分を酵素試料 (20units/ml、注意:CD38, 157とも菌に対する毒性が強くわずかしか発現していない。そのためキレートカラムを用いた上記の精製でも純粋なものは得られていないので比活性 (units/mg) として表すことができず、units/mlで代用している) として用いた。酵素溶液20units/mlに等量のAssay Mixture (2μM SNIを含む) を加えて反応を開始した。反応は20℃で5分間行った。すなわち酵素活性は10units/mlに薄まり、SNIは1μMになっている。この条件でNADase活性はほぼ100%阻害された(図11および12参照)。
以上より、Orf1は、ヒト由来CD38およびCD157を阻害することが示された。
【0172】
実施例9:無細胞系によるNADaseタンパク質合成
本発明者らは、溶血性連鎖球菌由来NADaseが強い細胞毒性を示すことから、無細胞系によるin vitroタンパク質合成を試みた。無細胞系としては、RTSスケラーブル・セルフリータンパク質発現システム(ロシュ製)を、その使用説明書に従って用いた。その結果、無細胞系でも、NADaseタンパク質を合成できなかった。
宿主細胞内における、毒素としてのNADaseの機能に関しては、未だに殆どのことが明らかにされていない。しかしながら、NADは生体内に最も大量に存在する補酵素であり、エネルギー産生に非常に重要であることから、NADaseによる細胞内NAD濃度の減少がタンパク質の生合成を停止させると考えられる。実際、DNA損傷によりポリADP-リボース合成酵素が過剰に活性化されると、その基質であるNADの細胞内濃度が低下することにより細胞死を引き起こすことはよく知られている事実である。
無細胞系はNADを含む。NADは翻訳系に直接必要ではないが、NAD濃度が低下するとエネルギー産生系に影響を及ぼし、結果としてATP等の濃度も減少するので、タンパク質合成が低下する。従って、上述したように宿主細胞を用いた場合は勿論のこと、無細胞系を用いてもやはりNADaseタンパク質の合成は困難であると考えられ、また、本実施例の結果はこのような考えを支持する。
以上より、本発明者らが開発した共発現ベクター、それが導入された形質転換体、および該形質転換体を用いるNADaseタンパク質の製造方法は極めて有用と考えられる。また、これらを利用することで、連鎖球菌由来NADaseの非天然型タンパク質の製造も初めて可能になると考えられる。
【0173】
実施例10:大腸菌における完全長SLOの大量発現
本発明者らによるSLO遺伝子のプロモーター解析 (Biochim Biophys Acta 1681: 134-149 (2005)) からの知見より、SLO遺伝子は自身のプロモーターを持たず、基本的に上流に位置しているNADase遺伝子 (nga) のプロモーターからのreadthroughにより転写が行われるオペロンの一部であることが判明している。そこで、本発明者らは、SLO遺伝子が自身のプロモーターを持っていないこと、およびSLOの細胞毒性を考慮して、遺伝子の発現を大腸菌で厳密に制御できるaraBAD プロモーター (PBAD) を利用した発現ベクターにより、Hisタグを融合させた完全長SLOの大量発現を試みた。Hisタグ法で付加されるアミノ酸配列は短く、多くの場合除去する必要がない。また、特異抗体や精製担体も複数の市販品が入手可能でありコストが安いなどの利点がある。さらに、尿素や塩酸グアニジンのようなタンパク質の変性剤の存在下でも精製が可能で、カラムに結合させたままの状態でリフォールディングできるなどの大きな利点がある (Protein Expr Purif 41: 98-105 (2005))。
【0174】
先ず、本発明者らは、医薬品としてすでに使われているストレプトキナーゼやストレプトドルナーゼの工業生産株としてよく知られているStreptococcus dysgalactiae subsp. equisimilis H46A (J Gen Physiol 28: 363-383 (1945)) の染色体DNAからPCR法でSLO遺伝子を増幅し、大腸菌での遺伝子発現を極めて厳密に実行するためHisタグタンパク質発現ベクターpBAD/HisB (Invitrogen) にクローニングした(図13)。pBAD/His BのBamHIおよびKpnI部位に、シグナルペプチド配列を含むN末端側の31アミノ酸残基 (3.3 kDa) を除いた540アミノ酸残基 (60.4 kDa) に相当する完全長 SLO遺伝子の領域をin frameで挿入した。この組み換えプラスミドを大腸菌TOP10株 (L-アラビノース代謝能欠損株) に導入し、遺伝子発現の誘導物質であるL-アラビノースによるHisタグ融合完全長SLOの発現実験を行った。その結果、培地中のL-アラビノースの濃度が0.002% (w/v) からSLOの十分な発現が確認された(図14)。イムノブロット解析の結果、大腸菌で発現したSLOの分子量は約70 kDaと推定され、ベクター側のHisタグを含む4.1 kDaの領域を合わせて予想される64.5 kDaよりも少し大きかった。SLOの電気泳動解析から求めた分子量が予想値よりも増大することは、Gerlachらも報告している (Infect Immun 61: 2727-2731 (1993))。
次に、本発明者らは、大腸菌をTOP10株から、lonおよびompTの両プロテアーゼ欠損株であるBL21株に変更してSLOを大量発現させた。精製には市販のTSKgel BioAssist Ni2+-chelate column (Tosoh Co., Tokyo, Japan) を用い、このアフィニティーカラムクロマトグラフィーにより一段階で高純度のSLOを精製した(図15)。Hisタグ融合完全長SLOの発現量は、タンパク質量として、LB培地1リットルあたり約6 mg(活性タンパク質量では約3 mg)の収量であり、その比活性は748,000 HU/mgタンパク質でGerlach らがStreptococcus dysgalactiae subsp. equisimilis H46Aの培養上清から精製したSLOの750,000 HU/mgとほぼ一致した (Infect Immun 61: 2727-2731 (1993))。なお、本実験で得られたタンパク質量は上記の通りであったが、培養条件の適切な変更(例、培地の至適化による菌密度の向上、および誘導物質による誘導時間の延長)により少なくとも約10-30 mgのタンパク質量を得ることが可能と考えられる。
【0175】
実施例11:大腸菌における完全長SLOの大量発現
現在市販されている抗ストレプトリジンO (ASLO) 抗体は溶血性連鎖球菌の培養上清から部分精製したSLOを抗原として用いているため、同じく培養上清中に分泌されるNADグリコヒドロラーゼ (NADase) と交差反応することが知られている (Biochim Biophys Acta 1681: 134-149 (2005); Cell 104: 143-152 (2001))。そこで、本発明者らは、精製したSLOをウサギに免役することによりASLOを作製し、GAS Sa株および GCS H46A株の培養上清をイムノブロット解析することによりASLOの抗原特異性を評価した(図16)。
その結果、本発明者らが作製したASLOにより、GCS H46A株の培養上清中のSLOが明確なバンドとして高感度で検出された。メジャーバンドの下に見える2本の弱いバンドは、低分子化したSLO分子と思われる。一方、GAS Sa株ではSLOを検出できなかった。これは、GASのSLO産生量は一般に少なく、また自身が産生するプロテアーゼにより速やかに低分子化するためと考えられる。実際、本発明者らは、GAS Sa株におけるSLOの検出や精製は困難であることを報告している (Kimoto et al., Biosci Biotechnol Biochem 67: 2203-2209 (2003); Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005))。なお、このASLOは、大腸菌から精製したHisタグ融合完全長SLO、および溶血性連鎖球菌の産生したSLOの溶血活性を、いずれも中和した (データ示さず)。
次にASLOのNADaseとの交差反応を調べるために、GAS Sa 株および GCS H46A株の培養上清を用いて、抗NADase抗血清によるイムノブロット解析を行った。
その結果、GAS Sa株および GCS H46A株の両培養上清中にNADaseを確認した。両者間でNADaseの分子量がやや異なるのは、GCS H46A株が産生するNADaseは翻訳後にC末端領域が分解されるためである (Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005))。いずれにしても、SLOとNADaseは予想された分子量でバンドが検出され、両者間の大きさは明らかに異なっていた。また、免疫していないコントロール血清では、いずれのバンドも検出されなかった。
以上より、今回作製したASLOは、これまでの市販品とは異なり、SLOを特異的かつ高感度に検出し得ることが示された。
【図面の簡単な説明】
【0176】
【図1】NADaseの活性の様式を示す図である。
【図2A】S. dysgalactiae subsp. equisimilis H46AのNADaseストレプトリシンOオペロンの模式図である。プロモーターおよびターミネーターをそれぞれ、「P」および「T」で示している。「orf1(sni)」で表示された矢印は、データベース中の如何なる遺伝子にも類似していなかった。NADase発現プラスミドpBAD/HisIおよびIIはそれぞれ、NADaseからorf1までの染色体領域およびNADase領域のみを含む。
【図2B】E.coli TOP10全細胞溶解物における連鎖球菌NADaseのウエスタンブロット解析を示す図である。E.coli TOP10細胞を、6個の連続するヒスチジン残基 (Hisタグ) を含むように改変された、示された領域をコードする発現プラスミドで形質転換し、L-アラビノースを用いて誘導し、次いでNADaseの合成についてアッセイした。pBAD/His B (ベクターのみ) を担持するE.coli TOP10の溶解物をコントロールとして用いた。NADaseの相対発現レベルを決定するため、全細胞溶解物をウエスタンブロット解析に供した。
【図3A】H46AのOrf1タンパク質の検出を示す図である。pBAD/His Iを担持する組換えE. coli TOP10の全細胞溶解物中のタンパク質をSDS-PAGEにより解析した。NH2末端アミノ酸酸列を括弧で示す。
【図3B】H46Aのorf1のヌクレオチド配列および推定アミノ酸配列を示す図である。下線を付したアミノ酸は、Orf1のNH2末端アミノ酸シークエンシングにより決定した。
【図4A】Hisタグ融合Orf1の精製を示す図である。組換えプラスミドpQE30 (Hisタグ融合Orf1) を担持するE.coli BL21細胞からの抽出物を、TSKgel BioAssist Ni2+キレートカラム (Tosoh Co., Tokyo, Japan) にアプライした。Hisタグ融合Orf1を、イミダゾールの20-250 mM の線形勾配により回収し、各溶出画分をSDS-PAGEにより調べた。
【図4B】組換えE.coli細胞およびS. dysgalactiae subsp. equisimilis H46A培養上清中のorf1産物のウエスタンブロット解析を示す図である。Orf1の発現を確認するため、全細胞溶解物および培養上清を、上記の通り精製したOrf1を用いた免疫により得られるウサギ抗Orf1抗血清を用いて、ウエスタンブロット解析に供した。
【図5】H46A由来のOrf1の精製およびアミノ末端シークエンシングを示す図である。H46A由来の全細胞抽出物を、抗Orf1抗体・コンジュゲート・HiTrap NHS活性化HPカラム (Amercham Biosciences Corp., NJ, USA) にアプライし、Orf1を、3 M チオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する4 ml の0.1 M リン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収した。Orf画分をプールし、水に対して透析し、次いで濃縮し、SDS-PAGEに供した。ゲル中のタンパク質をPVDF膜に転写し、CBB R-250で染色した。タンパク質バンドを切り出し、PE Applied Biosystems model 491 Procise protein sequencer (Foster City, CA, USA) で解析した。
【図6】H46AのOrf1・細胞外NADase間の相互作用のファーウエスタン解析。H46Aの培養上清および培地単独を、SDS-PAGE上で泳動し、PVDF膜に転写し、精製Hisタグ融合Orf1の存在下または非存在下でインキュベートした。培養上清中のNADaseを、抗NADase抗体を含有する抗SLOポリクローナルIgGによるウエスタンブロット解析によりアッセイした(左パネル)。精製Hisタグ融合Orf1のNADaseへの結合を、抗Orf1抗血清を用いてファーウエスタン解析によりアッセイした(右パネル)。
【図7A】TSKgel BioAssist Ni2+キレートカラムを用いたHisタグ融合NADaseおよびインタクトOrf1 (成熟型, Hisタグなし) の共精製を示す図である。組換えプラスミドpBAD/His I (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10由来の細胞抽出物を、TSKgel BioAssist Ni2+キレートカラムにアプライし、Hisタグ融合NADase/ORF1複合体を、イミダゾールの20-250 mM線形勾配により回収した。精製したNADase/Orf1複合体画分をプールし、透析し、SDS-PAGEに供した。
【図7B】TSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによるインタクトOrf1の精製を示す図である。組換えプラスミドpBAD/His I (Hisタグ融合NADaseおよびインタクトOrf1をコード)を担持するE.coli TOP10由来の細胞抽出物を、TSKgel BioAssist Ni2+キレートカラムにアプライした。Orf1を、3 M チオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する3 ml の0.1 M リン酸ナトリウムバッファー (pH 7.4) での溶出により回収した。次いで、カラムに吸着したHisタグ融合NADaseを、50 mM リン酸ナトリウムバッファー (pH 7.4) におけるイミダゾールの20-250 mM 線形勾配により回収した。精製酵素をプールし、0.01% アジ化ナトリウムを含有する50 mM リン酸ナトリウムバッファー (pH 7.4) に対して透析し、SDS-PAGEに供した。
【図7C】Hisタグ融合Orf1が固定されたカラムを用いたアフィニティークロマトグラフィーによるS. dysgalactiae subsp. equisimilis H46Aの培養上清からのインタクトNADase (成熟型, Hisタグなし) の精製を示す図である。硫酸アンモニウム沈殿および透析後、上清からのタンパク質を、TSKgel BioAssist Ni2+キレート-Hisタグ融合Orf1アフィニティーカラムにアプライした。NADaseを、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する0.1 M リン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収し、透析した。
【図7D】Orf1によるNADaseの阻害を示す図である。精製したインタクトNADaseを、インタクトOrf1と混合し、室温で10分間インキュベートし、残存するNADase活性を測定した。
【図8A】NADase、Orf1およびNADase-Orf1複合体のTSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによる解析を示す図である。ウシ血清アルブミン (BSA) ダイマー (134 kDa)、モノマー (67 kDa)、オブアルブミン (OV, 43 kDa)、炭酸脱水酵素 (CA, 29 kDa) およびリゾチーム (14.4 kDa) を、分子量マーカーとして使用した。VoおよびVcは空容量およびカラム容量をそれぞれ示す。Hisタグ融合Orf1固定カラムおよび抗Orf1抗体固定カラムを用いたアフィニティークロマトグラフィーにより精製されたインタクトNADaseおよびOrf1 (成熟型, Hisタグなし) を、0.15 M リン酸ナトリウムバッファー (pH 6.6) において、TSKgel BioAssist G3SWXLゲル透過クロマトグラフィーに供した。
【図8B】NADase、Orf1およびNADase-Orf1複合体のTSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによる解析を示す図である。ウシ血清アルブミン (BSA) ダイマー (134 kDa)、モノマー (67 kDa)、オブアルブミン (OV, 43 kDa)、炭酸脱水酵素 (CA, 29 kDa) およびリゾチーム (14.4 kDa) を、分子量マーカーとして使用した。VoおよびVcは空容量およびカラム容量をそれぞれ示す。精製したNADaseおよびOrf1を混合し、室温で10分間インキュベートし、TSKgel BioAssist G3SWXLカラムにアプライし、0.15 M リン酸ナトリウムバッファー (pH 6.6) で展開した。
【図9】インタクトOrf1 (Hisタグなし) の温度安定性を示す図である。Orf1溶液を20-100℃で15分間インキュベートし、氷水に浸した。各溶液を精製したHisタグを有しない成熟型NADaseに加えた。次いで、混合液を10分間室温でインキュベートし(Orf1とNADaseの最終モル比は1:2)、残存するヒドロラーゼ活性をNADで測定した。コントロールとして、BSAを含有する50 mMリン酸ナトリウムバッファー (pH 7.4) を、Orf1溶液の代わりに使用した。
【図10】nga-ni-sloオペロンの産物間の相互作用のモデル図である。SNI をコードする遺伝子(sni) は、ポリシストロニックmRNAとして、ngaプロモーターからnga (NADase) およびsloと共に転写される。翻訳の際、NADaseの自己毒性は、モル比1:1での、SNIとの安定な細胞内複合体の形成により迅速にブロックされ得る。NADase-SNI複合体は、連鎖球菌のエンベロープを介した排出の間に、おそらく幾つかの膜成分との相互作用により、SNIのタンパク質分解除去により、または他の機構により解離する。
【図11】pQE30-ヒト-His-タグ-CD38/1l LB/Amp/1 mM IPTG (3h誘導) 細胞を遠心分離により回収し、25 ml BugBusterにより抽出した。粗抽出液 (30 ml, 15 mgタンパク質/ml) を、室温にて流速1 ml/分でTSKゲルBioAssist Ni-キレートクロマトグラフィーにアプライした。スターティングバッファーは、50 mMリン酸ナトリウム(pH7.4)、20 mMイミダゾールおよび0.01%アジ化ナトリウムであった。20分間にわたるスターティングバッファーから50 mMリン酸ナトリウム(pH7.4)、250 mMイミダゾールおよび0.01%アジ化ナトリウムの線形勾配を行った。NADase活性 (units/ml) をSNI (アッセイ混合液中0.1μM) の存在下(◇)および非存在下(◆)で測定した。
【図12】pQE30-ヒト-His-タグ-CD157/1l LB/Amp/1 mM IPTG (3h誘導) 細胞を遠心分離により回収し、25 ml BugBusterにより抽出した。粗抽出液 (30 ml, 11 mgタンパク質/ml) を、室温にて流速1 ml/分でTSKゲルBioAssist Ni-キレートクロマトグラフィーにアプライした。スターティングバッファーは、50 mMリン酸ナトリウム(pH7.4)、20 mMイミダゾールおよび0.01%アジ化ナトリウムであった。20分間にわたるスターティングバッファーから50 mMリン酸ナトリウム(pH7.4)、250 mMイミダゾールおよび0.01%アジ化ナトリウムの線形勾配を行った。NADase活性 (units/ml) をSNI (アッセイ混合液中0.1μM) の存在下(◇)および非存在下(◆)で測定した。
【図13】ストレプトリシンO遺伝子 (SLO) およびSLO発現用ベクターの構築を示す模式図である。BamHIおよびKpnI部位を含むセンスプライマー (5’-cgGGATCCgactcgaacaaacaaaacactgcc-3’:配列番号13) およびアンチセンスプライマー (5’-cggGGTACCcttttatgttatgctttgcttg-3’:配列番号14) を、NH2末端シグナルペプチドを欠くHisタグ融合SLO配列の発現ベクターpBAD/HisB (Invitrogen, Frederick, USA) へのクローニングを可能とするように設計した。
【図14】E.coli TOP10全細胞抽出物における連鎖球菌SLOのイムノブロット解析を示す図である。プレ染色タンパク質分子標品の位置および計算分子量はパネルの左に示す。サイズはキロダルトン (kDa) で示す。矢印はSLOの位置を示す。E.coli. TOP10細胞を、6個の連続するヒスチジン残基 (Hisタグ) を含むように改変したSLO領域をコードする発現プラスミドで形質転換し、次いでSLOの合成についてアッセイした。L-アラビノースの存在下では、pBADからの発現がオンになるが、L-アラビノースの不在下では、pBADからの転写レベルは非常に小さくなる。培養物を0.00002% - 0.02%のL-アラビノースで37℃にて4時間誘導した。pBAD/HisB (ベクターのみ) を担持するE. coli TOP10の溶解物はコントロール(0.02% L-アラビノースで誘導)とした。SLOの相対発現レベルを決定するため、Ni-NTA HRPコンジュゲート (Qiagen, Hilden, Germany) を用いて、全細胞溶解物をウエスタンブロット解析に供した。このコンジュゲート〔西洋ワサビペルオキシダーゼにカップリングさせたニッケル−ニトリロトリ酢酸 (Ni-NTA)〕は、Hisタグを有する組換えタンパク質の直接的な検出のために用いた。レーン1:0.00002% L-アラビノース;レーン2:0.0002% L-アラビノース;レーン3:0.002% L-アラビノース;レーン4:0.02% L-アラビノース;レーン5:コントロール (ベクターのみ, 0.02% L-アラビノースで誘導)
【図15】Hisタグ融合SLOの精製を示す図である。pBAD/His B-sloを含む E. Coli BL21の培養物を、0.2% L-アラビノースで37℃にて4時間誘導した。細胞抽出物を、20 mMイミダゾール(バッファーA)を含有する50 mMリン酸ナトリウムバッファー (pH 7.4) で平衡化したTSKgel BioAssist Ni2+-キレートカラム (Tosoh Co., Tokyo, Japan) にアプライした。非特異的吸着物をカラムから洗浄して除くために、3 M NaClを含む0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。このHisタグ融合SLOを、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製したHisタグ融合SLOをプールし、バッファーAに対して透析した。透析物をSDS-PAGEに供し、クマシーブリリアントブルー (CBB) R-250で染色した。タンパク質分子標品の位置および各計算分子量をパネルの左に示す。サイズはキロダルトン (kDa) で示す。矢印はSLOの位置を表す。
【図16】GAS株SaおよびGCS株H46A由来の培養上清のイムノブロット解析を示す図である。精製したHisタグ融合SLOを用いて、ウサギ抗SLO抗体を惹起した。約2 mgのHisタグ融合SLO (完全フロインドアジュバントと混合) をウサギに皮下注射した。1 mgのタンパク質と不完全フロインドアジュバントからなるブースター注射を2週間の間隔で3回投与した。プロテインA-セファロース (Amersham Biosciences Corp., NJ, USA) のアフィニティークロマトグラフィーにより、イムノグロブリンG画分を精製した。精製H46A NADase (Gerlach et al., 1996) での免疫により作製したウサギ抗血清は、Friedrich-Schiller大学 (ドイツ) のD. Gerlach博士からご提供頂いた。連鎖球菌の培養上清をSDS-PAGE (10% 総アクリルアミド) に供し、電気ブロッティングによりタンパク質をSequi-Blot PVDF膜 (Bio-Rad, CA, USA) 上に転写した。この膜を、0.05% Tween 20を含有するリン酸緩衝化生理食塩水 (PBST) において、5% スキムミルクおよび1% ウシ血清アルブミン (BSA) で1時間ブロッキングした。膜をPBSTで洗浄し、次いで各一次抗体を添加し (PBSTで1:3,000に希釈) 、ブロットを1時間インキュベートした。次いで、抗ウサギIgGにコンジュゲートしたヤギaffini pure IgGペルオキシダーゼ (Wako Pure Chemical Industries, Ltd., Osaka, Japan) を、PBSTで1:3,000に希釈して添加し、1時間インキュベートし、抗体・抗原複合体を、製造業者により記載される通りにPOD免疫染色セット (Wako Pure Chemical Industries, Ltd., Osaka, Japan) を用いて可視化した。プレ染色タンパク質分子標品の位置および各計算分子量はパネルの左に示す。サイズはキロダルトン (kDa) で示す。
【技術分野】
【0001】
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現する各タンパク質の製造方法、当該製造方法により得られるタンパク質およびその抗体、該タンパク質をコードするポリヌクレオチドを含むベクターおよびその形質転換体、ならびにそれらの使用などを提供する。
【背景技術】
【0002】
溶血性連鎖球菌(hemolytic streptococci)は、咽頭炎、皮膚感染症、猩紅熱、糸球体腎炎、リウマチ熱、敗血症などの原因となる病原細菌であり、赤血球を含む血液寒天平板培地上で溶血斑を形成し、細胞壁に局在する多糖抗原の免疫化学的特異性に基づいてA〜G群に分けられている。溶血性連鎖球菌感染症は、小児感染症として派生頻度の高い疾患である。また、1980年代以降、日本や欧米などの先進各国で、溶血性連鎖球菌による極めて急激な筋肉組織の壊死、多臓器に及ぶ障害、ショック症状などを伴う死亡率の高い劇症型感染症が発生しており、そのためヒト喰いバクテリアとしてたびたび報道などされている。
【0003】
溶血性連鎖球菌は、20種類以上にも及ぶさまざまな生理活性物質を菌体外に産生しているが、中でも溶血毒素であるストレプトリシンO(SLO)は古くから重要な病原性因子であると考えられ詳細な研究が進められてきた。最近、SLOにより宿主の細胞膜上に形成された孔からNADaseなどの病原性因子が宿主細胞内に移行することが確認された。この発見は、腸管出血性大腸菌などのグラム陰性病原菌で高度に保存されているタイプIII分泌機構がグラム陽性菌である溶血性連鎖球菌においても存在することを示すものとして大きな反響を呼び注目されている。
【0004】
本発明者らは、これまでに、SLO遺伝子がその上流に位置するNADase遺伝子(nga)からのreadthroughによりポリシストロニックに転写されていることを明らかにしている(非特許文献1参照)。このnga−sloオペロンは、A群およびC群溶血性連鎖球菌間で高度に保存されており、溶血性連鎖球菌感染症において重要な役割を果たしていると考えられるが、ngaとslo遺伝子との間には、遺伝子orf1が存在することがわかっていた。しかしながら、この遺伝子orf1の機能は全く不明である。
【0005】
NADaseは、その活性として、(a)β−ニコチンアミド−アデニン−ジヌクレオチド(NAD)を加水分解し、アデノシンジホスホリボース(ADPR)およびニコチンアミドを生成する活性(NADグリコヒドロラーゼ活性)、(b)NADからサイクリックADPリボース(cADPR)を合成する活性(ADPリボシルシクラーゼ活性)、および(c)cADPRを加水分解し、ADPRを生成する活性(cADPRヒドロラーゼ活性)を有する。NADaseは、SLOにより宿主細胞膜上に形成された孔から宿主細胞内に侵入し、これらの活性によりその毒性を発揮し、連鎖球菌感染症の病態を担い得ると考えられるので、その解析は連鎖球菌感染症を理解する上で非常に重要である。
【0006】
ところで、NADaseの製造方法としては、これまでに幾つかの報告がある。
例えば、非特許文献2および3には、溶血性連鎖球菌の培養上清からNADaseをその活性を保持した状態で精製したことが記載されている。しかしながら、大腸菌等の異種宿主細胞でのNADaseの発現は未だ成功していない。これは、NADaseが異種宿主細胞に対して極めて強い毒性を示すためである。
【0007】
また、SLO遺伝子は、異なる溶血性連鎖球菌群で高度に保存され、特にA群(GAS)とC群(GCS)では両者のSLOに本質的な相違はみられないことが知られている。両群のSLOは、N末端シグナルペプチドを含む31アミノ酸残基(3.3kDa)が外れて分泌されるとともに、自身が分泌するプロテアーゼにより次第に低分子化する。SLOは抗原性が強く中和抗体がつくられやすく、感染に際して血清中の抗ストレプトリジンO抗体(ASLO)が上昇するため、本菌による感染の診断に抗体価の測定が広く用いられている(抗SLO試験)。SLOは、抗SLO試験において試験抗原として使用されているだけでなく、巨大なリング状の孔を細胞膜上に形成し、生細胞内にタンパク質レベルの大きさの分子を送り込むことができるので、細胞膜の透過性を高めるツールとして近年多用されている。また、SLOはASLO精製用の抗原としても利用可能である。従って、SLOを大量かつ安価に製造する方法の開発が求められている。
【0008】
また、ASLOは臨床検査試薬等の試薬として有用であるが、通常市販されているASLOは溶血性連鎖球菌の培養上清から部分精製したSLOを抗原として用いて作製されている。ところが、培養上清から部分精製された調製物中にはSLOに加え、NADaseも混在しているため、このような調製物を用いて作製された抗体は、SLOのみならずNADaseとも交差反応することが知られている(非特許文献1、4)。従って、交差反応性が低い抗体の作製という観点からも、高純度のSLOを製造する方法の開発が求められている。
【0009】
SLOの製造方法としては、これまでに幾つかの報告がある。
例えば、特許文献1には、溶血性連鎖球菌の培養上清から完全長SLOを製造(精製)する方法が記載されている。しかしながら、溶血性連鎖球菌のSLO産生量は低く、また、自身が産生するプロテアーゼによる分解のために、溶血性連鎖球菌の培養上清からの完全長SLOの精製収率が低いという問題がある。
【0010】
また、宿主として大腸菌を用いるSLOの製造方法としては、以下の報告がある。
非特許文献5には、SLO遺伝子を高コピー数プラスミドにサブクローニングしたところ、宿主である大腸菌は溶菌しやすく、また増殖も遅く、さらにはSLO遺伝子自体が不安定であり脱落が生じたこと、ならびに、SLO遺伝子を低コピー数プラスミドに移したところ、SLO遺伝子は安定化したがSLOの産生量はわずかであったことが記載されている。
非特許文献6には、特定のN末端欠失型SLO(シグナルペプチド配列を含むN末端側の75アミノ酸残基を除いた496アミノ酸残基に相当するSLO)をマルトース結合タンパク質(MBP,42kDa)との融合タンパク質とすることで、SLOの大腸菌での大量発現に成功したことが記載されている。これは、完全長ではないが、大腸菌で初めてSLOの大量発現に成功した例である。
非特許文献7には、大腸菌における特定のN末端欠失型SLO(シグナルペプチド配列を含むN末端側の103アミノ酸残基を除いた468アミノ酸残基に相当する領域をコードするSLO)の大量発現に成功したことが記載されている。
しかしながら、大腸菌での完全長SLOの大量発現は未だ成功していない。これは、SLOのN末端近傍の32−75アミノ酸残基付近の領域が大腸菌に対して何らかの毒性を示すと考えられるためである(非特許文献5−7)。
【0011】
さらに、宿主として枯草菌(Bacillus subtilis)を用いる完全長SLOの大量発現について幾つかの報告がある(例えば、非特許文献8、特許文献2および3)。しかしながら、枯草菌はプロテアーゼの産生能力が高く、複数のプロテアーゼを産生するにもかかわらず、完全なプロテアーゼ欠損株の作製は未だ成功していないことから、SLOの分解が問題となっている。さらには、枯草菌は多様なタンパク質を分泌するため、SLOの精製過程が複雑になり、高純度のSLOを入手し難いという問題がある。
【0012】
【特許文献1】特開平5−331198号公報
【特許文献2】特開平5−184372号公報
【特許文献3】特開平6−237775号公報
【非特許文献1】Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005)
【非特許文献2】Karasawa et al., FEMS Microbiology Letters 130: 201-204 (1995)
【非特許文献3】Gerlach et al., FEMS Microbiology Letters 136: 71-78 (1996)
【非特許文献4】Madden et al., Cell 104: 143-152 (2001)
【非特許文献5】Kehoe et al., Infect Immun 43: 804-810 (1984)
【非特許文献6】Weller et al., Eur J Biochem 236: 34-39 (1996)
【非特許文献7】Yamamoto et al., Biosci Biotechnol Biochem 65: 2682-2689 (2001)
【非特許文献8】Yamada et al., Biosci Biotechnol Biochem 59: 363-366 (1995)
【発明の開示】
【発明が解決しようとする課題】
【0013】
上記の通り連鎖球菌由来NADaseおよびSLOの製造方法が幾つか報告されている。組換えタンパク質を高純度かつ安価に製造する方法としては大腸菌等の異種宿主細胞を利用する方法が知られている。ところが、完全長のNADaseおよびSLOを高純度で大量かつ安価に製造する方法は、以下の理由により未だ開発されていない。
【0014】
連鎖球菌由来NADaseは、大腸菌等の異種宿主細胞に対して強い毒性を示すため、完全長NADaseまたはその毒性を示す領域を含むNADase断片を、異種宿主細胞を用いて大量に製造する方法は開発されていない。NADaseの解析は連鎖球菌感染症の理解やその応用に非常に重要であると考えられるため、NADaseを大量に製造する方法の開発が求められている。
【0015】
連鎖球菌由来SLOは、そのN末端近傍の32−75アミノ酸残基付近の領域が大腸菌に対して何らかの毒性を示すと考えられ、完全長SLOまたは上記領域を含むSLO断片を、大腸菌を用いて大量かつ安価に製造方法は開発されていない。
SLOのようなコレステロール依存性の細胞障害毒素(cholesterol-dependent cytolysin)は、分子構造と機能が類似した大きなグループを形成しており、化膿レンサ球菌である Streptococcus属だけでなく、Bacillus、Clostridium、Listeria属などの細菌も産生すること、およびそれらのアミノ酸配列は高い相同性を示すことが知られている。しかしながら、SLOはこれらグループの中で最も分子量が大きく、また、そのN末端領域は他のグループには存在せず、溶血性連鎖球菌に特徴的である。このようなことから、完全長SLOおよびそのN末端断片は溶血性連鎖球菌感染症のための臨床検査薬(抗体作製用の抗原としても)だけでなく、病原機構を分子レベルで明らかにするための研究ツールとしても有用であると考えられる。従って、完全長SLOまたは上記領域を含むSLO断片の製造方法が開発できれば、純度の高い抗原が大量に得られ、また、交差反応性を実質的に有しないASLOの作製も可能になると考えられることから、それらを高純度で大量かつ安価に製造する方法の開発が求められている。
【課題を解決するための手段】
【0016】
本発明者らは、鋭意検討した結果、異種宿主細胞においてNADase遺伝子(nga)を、その下流に存在する未知遺伝子orf1とともに発現させることにより、完全長NADaseの大量発現に成功した。この結果に基づき、orf1遺伝子産物をさらに解析したところ、宿主細胞内でNADaseと複合体を形成することによりNADaseのインヒビターとして作用し得ることを見出した。本発明者らは、このorf1遺伝子産物を、連鎖球菌NADaseインヒビター(streptococcal NADase inhibitor:SNI)と標記することを提案する。NADaseと共発現し、複合体化するSNIは、その保有菌をNAD枯渇から保護し得ると考えられる。この知見に基づけば、連鎖球菌内のSNI・NADase複合体を解離させる物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。また、SNIは、NADaseと非常に強固かつ特異的に結合することから、現在広く用いられているHisタグ等の精製用タグと同様の効果が期待できると考えられる。さらに、SNIは熱に非常に安定性であった。また、SNIは、細菌由来NADaseのみではなく、哺乳動物由来NADaseも阻害した。従って、哺乳動物由来NADaseが関与し得る疾患の予防・治療にもSNIが応用可能と考えられる。以上のように、本発明者らの知見によれば、SNIを利用して種々の応用が可能である。
【0017】
また、本発明者らは、大腸菌での遺伝子発現を厳格に制御可能である誘導プロモーターを用いて、基底レベルの遺伝子発現を効果的に抑制し安定な培養を可能とするとともに、該プロモーターに対する誘導剤を併用することにより、完全長SLOの大量発現に成功した。本発明者はまた、SLOに適切なタグを付加することにより、高純度の完全長SLOを簡便かつ安価に製造することに成功した。本発明者らはさらに、上記のように調製された高純度の完全長SLOを用いることにより、NADaseに対する交差反応性を示さない抗SLO抗体を作製することに成功した。
【0018】
以上に基づき、本発明者らは、本発明を完成するに至った。即ち、本発明は、下記の発明などを提供する:
〔1〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、医薬。
〔2〕連鎖球菌感染症の治療薬である、上記〔1〕の医薬。
〔3〕自己免疫疾患、炎症性疾患または多発性骨髄腫の治療薬である、上記〔1〕の医薬。
〔4〕NADaseが細菌または動物由来NADaseである、上記〔1〕の医薬。
〔5〕細菌または動物由来NADaseが、連鎖球菌由来NADase、あるいはCD38またはCD157である、上記〔4〕の医薬。
〔6〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、NADase阻害剤。
〔7〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、白血球の走化性の阻害剤。
〔8〕白血球が好中球である、上記〔7〕の剤。
〔9〕SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、試薬。
〔10〕連鎖球菌由来NADaseの組換えタンパク質であって、天然型NADaseに対して1以上のアミノ酸の欠失、置換、付加および挿入からなる群より選ばれる1以上のアミノ酸の修飾が施された、組換えタンパク質。
〔11〕該1以上の修飾が精製用タグの付加を少なくとも含む、上記〔10〕の組換えタンパク質。
〔12〕上記〔10〕の組換えタンパク質を含む、医薬または試薬。
〔13〕上記〔10〕の組換えタンパク質をコードするポリヌクレオチド。
〔14〕第1および第2の発現ユニットの共発現ベクターであって、
第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み、
第1の発現ユニットが、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドであり、
第2の発現ユニットが、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドである、共発現ベクター。
〔15〕NADaseが細菌または動物由来NADaseである、上記〔14〕の共発現ベクター。
〔16〕NADaseが連鎖球菌由来NADaseである、上記〔14〕の共発現ベクター。
〔17〕NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを発現する、形質転換体。
〔18〕形質転換体が大腸菌である、上記〔17〕の形質転換体。
〔19〕上記〔17〕の形質転換体を培養し、培養上清からNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドを回収することを含む、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造方法。
〔20〕NADaseとの結合能および/またはNADase阻害活性を有するSNIの部分ペプチド。
〔21〕上記〔20〕の部分ペプチドをコードするポリヌクレオチド。
〔22〕上記〔21〕のポリヌクレオチドを含む、発現ベクター。
〔23〕SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む、複合体。
〔24〕NADaseが連鎖球菌由来NADaseである、上記〔23〕の複合体。
〔25〕SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法。
〔26〕以下(a)〜(d)からなる群より選ばれる構成要素を含む、SNIまたはNADaseとの結合能を有するその部分ペプチドとNADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成剤:
(a)SNI;
(b)NADaseとの結合能を有するSNIの部分ペプチド;
(c)NADase;
(d)SNIとの結合能を有するNADaseの部分ペプチド。
〔27〕以下(a)および/または(b)を含む、SNIまたはNADaseに親和性を有する融合タンパク質の調製剤:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター;
(b)NADaseまたはSNIとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター。
〔28〕以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたは該連鎖球菌由来NADaseとの結合能を有するその部分ペプチドと、該連鎖球菌由来NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成を抑制し得るか否かを評価する工程;
(b)該複合体の形成を抑制し得る被験物質を選択する工程。
〔29〕以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、該連鎖球菌由来NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
〔30〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターであって、
該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、ベクター。
〔31〕溶血性連鎖球菌がA群またはC群連鎖球菌である、上記〔30〕のベクター。
〔32〕該SLOが精製用タグを有する、上記〔30〕のベクター。
〔33〕該誘導プロモーターがアラビノースオペロン制御プロモーターである、上記〔30〕のベクター。
〔34〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターで形質転換された大腸菌であって、
該発現ベクターは、該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、大腸菌。
〔35〕大腸菌が、プロテアーゼの欠損株、および/または誘導プロモーターに対する誘導剤の代謝能の欠損株である、上記〔34〕の大腸菌。
〔36〕溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの製造方法であって、誘導プロモーターに対する誘導剤の存在下において上記〔34〕の大腸菌を培養し、培養上清から該SLOまたはその部分ペプチドを回収することを含む、方法。
〔37〕溶血性連鎖球菌由来SLOに対する特異的な反応性を示し、かつ溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体。
【発明の効果】
【0019】
本発明のポリペプチドは、種々の応用が可能である。詳細には、本発明のSNIおよびその部分ペプチドならびにそれらの発現ベクターは、例えば、連鎖球菌等の細菌の感染症、および感染症以外の種々の疾患の治療、ならびにNADaseの阻害および白血球の走化性の阻害に有用である。本発明の非天然型NADaseおよびその部分ペプチドならびにそれらの発現ベクターは、例えば、連鎖球菌NADaseの研究および当該NADaseを用いるその毒性を抑制し得る医薬の開発、ならびに抗菌または抗細胞剤として有用である。
本発明の発現ベクターは、例えば、医薬または試薬、あるいは目的ポリペプチドの大量発現に有用である。詳細には、本発明のSNIおよびNADase発現ベクターは、上記の通り医薬および/または試薬として使用できるのみならず、宿主細胞でのSNIおよびNADaseの大量発現、ならびに自身が精製用タグとして機能し得る融合タンパク質の作製などにも有用である。本発明のSLO発現ベクターは、完全長SLOまたはその毒性領域を含む部分ペプチドの大量発現などに有用である。従って、このような発現ベクターを含む形質転換体および当該形質転換体を用いる目的ポリペプチドの製造方法もまた有用である。
本発明の抗体は、例えば、SNI、NADaseまたはSLOの検出・定量、あるいはそれらの機能阻害による医薬または試薬などとして有用である。詳細には、本発明のSLO抗体は、SLOに対する特異性が高く交差反応しないため、試薬として特に有用である。
本発明の複合体およびその製造方法は、例えば、NADaseの製造、NADaseまたはSNIあるいはそれらの部分ペプチドを有するタンパク質の精製、ならびに本発明のスクリーニング方法に有用である。
本発明のスクリーニング方法は、例えば、連鎖球菌感染症の治療薬の開発に有用である。
【発明を実施するための最良の形態】
【0020】
1.ポリペプチドおよびその部分ペプチド
本発明は、連鎖球菌NADaseインヒビター(streptococcal NADase inhibitor:SNI)またはその部分ペプチドを提供する。本発明者らは、SNIの発現に初めて成功し、また、その機能の実証にも併せて成功した。NADaseに対する細菌性インヒビターの存在は、Bacillus subtilis、Proteus vulgaris、Proteus rettgeriおよびMycobacterium butyricumでは知られていたが、連鎖球菌では初めてである。しかも、熱安定性に関して、従来のNADaseインヒビターは、60℃で15分間加熱することにより完全に不活化される(Acta Path. Microbiol. Scandinav. 69: 277-286 (1967))のに対し、SNIはそのような条件でも活性を保持し得る。本発明のSNIは、NADase阻害活性を有する連鎖球菌由来ポリペプチドである限り特に限定されないが、例えば、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチドであり得る。
【0021】
本発明はまた、連鎖球菌由来の非天然型NADaseまたはその部分ペプチドを提供する。これまでに、溶血性連鎖球菌の培養上清からNADaseをその活性を保持した状態で精製したことが報告されている(例えば、FEMS Microbiology Letters 130: 201-204 (1995); FEMS Microbiology Letters 136: 71-78 (1996)参照)。しかしながら、連鎖球菌由来NADaseは、大腸菌等の宿主細胞に対して強い毒性を示すため、宿主細胞でのNADaseの発現、即ち、組換えタンパク質の調製には未だ成功していない。今回、本発明者らは、連鎖球菌由来NADaseの組換えタンパク質の調製に初めて成功した。本発明者らの研究成果により、宿主細胞を用いた連鎖球菌由来天然型NADaseの組換えタンパク質の調製のみならず、非天然型NADase(変異型NADaseまたは遺伝子改変NADase)の組換えタンパク質の調製が初めて可能となる。また、このように組換え技術を利用することで、NADaseの部分ペプチドの調製も容易になる。本発明の非天然型NADaseは、既報の天然型NADase以外のNADaseであって、かつNADase活性を有するポリペプチドである限り特に限定されないが、例えば、配列番号4で表されるアミノ酸配列、または配列番号4で表されるアミノ酸配列において分泌シグナル配列をコードするアミノ酸配列が除去されたアミノ酸配列(以下、必要に応じて、「配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列」と省略する)と実質的に同一のアミノ酸配列(但し、配列番号4で表されるアミノ酸配列、またはその分泌シグナル除去アミノ酸配列と同一のアミノ酸配列は除く)からなるポリペプチドであり得る。
【0022】
本明細書中で使用される場合、「NADase活性」とは、NADaseが有する任意の活性をいい、特に断らない限り、(a)ニコチンアミド−アデニン−ジヌクレオチド(NAD)を加水分解し、アデノシンジホスホリボース(ADPR)およびニコチンアミドを生成する活性(NADグリコヒドロラーゼ活性)、(b)NADからサイクリックADPリボース(cADPR)を合成する活性(ADPリボシルシクラーゼ活性)、および(c)cADPRを加水分解し、ADPRを生成する活性(cADPRヒドロラーゼ活性)のいずれかの活性を意味する(図1参照)。これは、連鎖球菌由来NADaseは上記(a)〜(c)の活性を有すると考えられるためである。
【0023】
本明細書中で使用される場合、「NADase阻害活性」とは、NADaseが有する任意の活性を抑制する活性をいい、特に断らない限り、(d)NADグリコヒドロラーゼ活性の阻害活性、(e)ADPリボシルシクラーゼ活性の阻害活性、および(f)cADPRヒドロラーゼ活性の阻害活性のいずれかの阻害活性を意味する。これは、SNIは、連鎖球菌由来NADaseと非常に安定かつ強固な複合体を形成することによりNADaseの活性を阻害することから、上記(d)〜(f)の阻害活性を有すると考えられるためである。
【0024】
NADグリコヒドロラーゼ活性を有するNADaseは、細菌から哺乳動物までの多くの生物に存在する。多くの真核生物NADaseは、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性をさらに併有する。cADPRは、シグナル伝達の調節に関与しており、実際、真核生物細胞の細胞膜表面NADase(例、CD38、CD157)がNADからcADPRを合成すると、細胞内貯蔵Ca2+濃度が細胞質に遊離され、このカルシウムイオンの上昇がシグナルとなって種々の生物学的応答が生じる。殆どの原核生物由来NADaseは、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性を有しない。しかし、本明細書に開示のある連鎖球菌NADaseは、原核生物由来NADseであるにもかかわらず、これらの活性を併有することが知られている(FEMS Microbiol. Lett. 130:201-204 (1995)参照)。また、この連鎖球菌NADaseは、分泌タンパク質である点で、細胞質、細胞膜等に局在する通常のNADaseとは異なる。なお、連鎖球菌由来NADaseの詳細については、例えば、FEMS Microbiol. Lett. 191: 235-241 (2000) を参照のこと。
【0025】
一実施形態では、配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と実質的に同一のアミノ酸配列は、配列番号2で表されるアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と所定のアミノ酸配列同一性を有するアミノ酸配列であり得る。アミノ酸配列同一性の程度は、例えば約70%、好ましくは約80%、より好ましくは約90%、さらにより好ましくは約95%、最も好ましくは約97%、約98%または約99%以上であり得る。アミノ酸配列同一性は自体公知の方法により決定できる。例えば、アミノ酸配列同一性(%)は、当該分野で慣用のプログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。また、別の局面では、同一性(%)は、当該分野で公知の任意のアルゴリズム、例えば、Needlemanら(1970) (J. Mol. Biol. 48: 444-453)、Myers及びMiller (CABIOS, 1988, 4: 11-17)のアルゴリズム等を使用して決定することができる。Needlemanらのアルゴリズムは、GCGソフトウェアパッケージ(www.gcg.comで入手可能)のGAPプログラムに組み込まれており、同一性(%)は、例えば、BLOSUM 62 matrix又はPAM250 matrix、並びにgap weight: 16、14、12、10、8、6若しくは4、及びlength weight: 1、2、3、4、5若しくは6のいずれかを使用することによって決定することができる。また、Myers及びMillerのアルゴリズムは、GCG配列アラインメントソフトウェアパッケージの一部であるALIGNプログラムに組み込まれている。アミノ酸配列を比較するためにALIGNプログラムを利用する場合、例えば、PAM120 weight residue table、gap length penalty 12、gap penalty 4を用いることができる。アミノ酸配列同一性は、上記の任意の方法で決定されたものであれば構わないが、好ましくは計算に際して、上記の方法のなかで最も低い値を示す方法が採用され得る。
【0026】
別の実施形態では、配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列と実質的に同一のアミノ酸配列は、配列番号2で表されるアミノ酸配列、あるいは配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列において1以上のアミノ酸が置換、付加、欠失および/または挿入されたアミノ酸配列であり得る。置換、付加、欠失および/または挿入されるアミノ酸の数は、1以上であれば特に限定されないが、例えば1〜約100個、好ましくは1〜約50個、より好ましくは1〜約30個、さらにより好ましくは1〜約20個、最も好ましくは1〜約10個、1〜約5個あるいは1または2個であり得る。
【0027】
本発明のSNIは、NADase阻害活性、NADase結合活性を有し得る。本発明のSNIが配列番号2で表されるアミノ酸配列と実質的に同一のアミノ酸配列からなるポリペプチドである場合、活性の程度は、配列番号2で表されるアミノ酸配列と同一のアミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0028】
本発明のSNIは、また、熱安定性が高いという特徴を有する。従来のNADaseインヒビターは、60℃で15分間加熱することにより完全に不活化されていた。しかし、本発明のSNIはこのような条件でも活性を保持し得る。より詳細には、本発明のSNIは、15分間の60℃での熱処理後には80%以上、100℃での熱処理後には70%以上の活性を保持し得る。
【0029】
本発明の非天然型NADaseは、NADase活性(NADグリコヒドロラーゼ活性、ADPリボシルシクラーゼ活性、およびcADPRヒドロラーゼ活性)、SNI結合活性を有し得る。本発明の非天然型NADaseが配列番号4で表されるアミノ酸配列と実質的に同一のアミノ酸配列からなるポリペプチドである場合、活性の程度は、配列番号4で表されるアミノ酸配列と同一のアミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0030】
本発明の非天然型NADaseは、酵素学的特性として既報の連鎖球菌由来の天然型NADaseと同様の特性を有し得る。
【0031】
本発明の部分ペプチドは、本発明のSNIポリペプチドの部分ペプチド、または本発明のNADaseポリペプチドの部分ペプチドであり得る。本発明の部分ペプチドの長さは、所定の活性を有する限り特に限定されないが、例えば、上記ポリペプチドをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。詳細には、本発明の部分ペプチドとしては、例えば、NADaseとの結合能を有するSNIの部分ペプチド、SNIとの結合能を有するNADase(天然型、変異型を含む)の部分ペプチドが挙げられる。
【0032】
また、精製用タグなどを付加したポリペプチドまたはその部分ペプチドを形質転換体に産生させ、当該タグに親和性を有する物質(例えば、Ni2+レジン、タグに特異的な抗体)を用いることで、本発明のポリペプチドまたはその部分ペプチドをより簡便に単離、精製することができる。従って、本発明のポリペプチドまたはその部分ペプチドは、必要に応じて、このような精製用タグを有し得る。精製用タグについては後述する。
【0033】
本発明のポリペプチドおよびその部分ペプチドは、自体公知の方法により作製できる。その詳細については後述する。
【0034】
2.ポリヌクレオチドおよびその部分ヌクレオチド
本発明は、本発明のSNIをコードするポリヌクレオチドあるいはその部分ヌクレオチドを提供する。本発明のSNIポリヌクレオチドは、配列番号1で表されるヌクレオチド配列と同一または実質的に同一のヌクレオチド配列であり得る。本発明のSNIポリヌクレオチドあるいはその部分ヌクレオチドは、例えば、本発明のSNIまたはその部分ペプチドの作製に有用であり得る。
【0035】
本発明はまた、天然型NADaseをコードするポリヌクレオチド、および本発明の非天然型NADaseをコードするポリヌクレオチド(以下、必要に応じて、本発明のNADaseポリヌクレオチドと省略する)、ならびにそれらの部分ヌクレオチドを提供する。これまで、連鎖球菌由来NADaseは、大腸菌等の異種宿主細胞に対して強い毒性を示すため、異種宿主細胞でのNADaseの発現、即ち、組換えタンパク質の調製には未だ成功していなかった。今回、本発明者らは、連鎖球菌由来NADaseの組換えタンパク質の調製に初めて成功した。本発明者らの研究成果により、異種宿主細胞を用いた連鎖球菌由来天然型NADaseおよび非天然型NADaseの組換えタンパク質の調製に使用できるという、本発明のポリヌクレオチドの意義ある利用が初めて可能になった。本発明のNADaseポリヌクレオチドは、例えば、配列番号3で表されるヌクレオチド配列、または配列番号3で表されるヌクレオチド配列において分泌シグナル配列をコードするヌクレオチド配列が除去されたヌクレオチド配列(以下、必要に応じて、「配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列」と省略する)と同一または実質的に同一のヌクレオチド配列からなるポリヌクレオチドであり得る。本発明のNADaseポリヌクレオチドまたはその部分ヌクレオチドは、例えば、本発明のNADaseまたはその部分ペプチドの作製に有用であり得る。
【0036】
一実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と所定の配列同一性を有するヌクレオチド配列であり得る。ヌクレオチド配列同一性の程度は、例えば約70%、好ましくは約80%、より好ましくは約90%、さらにより好ましくは約95%、最も好ましくは約97%、約98%または約99%以上であり得る。ヌクレオチド配列同一性は自体公知の方法により決定できる。例えば、ヌクレオチド配列同一性(%)は、上述したアミノ酸配列同一性(%)と同様の方法により決定できる。
【0037】
別の実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列において1以上のヌクレオチドが置換、付加、欠失および/または挿入されたヌクレオチド配列であり得る。置換、付加、欠失および/または挿入されるヌクレオチドの数は、1以上であれば特に限定されないが、例えば1〜約300個、好ましくは1〜約150個、より好ましくは1〜約100個、さらにより好ましくは1〜約50個、最も好ましくは1〜約30個、1〜約20個、1〜約10個または1〜約5個であり得る。
【0038】
さらに別の実施形態では、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列は、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列の相補配列に対してハイストリンジェント条件下でハイブリダイズし得るポリヌクレオチドであり得る(但し、配列番号1で表されるヌクレオチド配列、、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列を除く)。ハイストリンジェント条件下でのハイブリダイゼーションの条件は、既報の条件を参考に設定することができる(Current Protocols in Molecular Biology, John Wiley & Sons, 6.3.1-6.3.6, 1999)。例えば、ハイストリンジェント条件下でのハイブリダイゼーションの条件としては、6×SSC(sodium chloride/sodium citrate)/45℃でのハイブリダイゼーション、次いで0.2×SSC/0.1%SDS/50〜65℃での1回または2回以上の洗浄が挙げられる。
【0039】
本発明のNADaseポリヌクレオチドは、本発明の非天然型NADase、または天然型NADaseと同様の活性を有するポリペプチドをコードし得る。本発明のNADaseポリヌクレオチドが配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列からなるポリヌクレオチドである場合、当該ポリヌクレオチドによりコードされるポリペプチドの活性の程度は、配列番号4で表されるアミノ酸配列またはその分泌シグナル除去アミノ酸配列からなるポリペプチドと定量的に同等であり得るが、許容し得る範囲(例えば約0.1〜約5倍、好ましくは約0.5〜約2倍)で異なっていてもよい。
【0040】
本発明の部分ヌクレオチドは、本発明のSNI部分ペプチド、または本発明のNADase部分ペプチドをコードするヌクレオチドであり得る。詳細には、本発明の部分ヌクレオチドは、配列番号1で表されるヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と同一または実質的に同一のヌクレオチド配列からなるポリヌクレオチドの部分ヌクレオチドであり得る。本発明の部分ヌクレオチドとしては、例えば、NADaseとの結合能を有するSNIの部分ペプチドをコードするポリヌクレオチド、SNIとの結合能を有するNADase(天然型、変異型を含む)の部分ペプチドをコードするポリヌクレオチドが挙げられる。
【0041】
本発明のポリヌクレオチドは、自体公知の方法により作製できる。例えば、配列番号1で表されるヌクレオチド配列と同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と同一のヌクレオチド配列からなるポリヌクレオチドは、連鎖球菌から総RNAを抽出し、mRNAからcDNAを調製した後、適切なプライマーを用いてPCRを行うことによりクローニングできる。また、配列番号1で表されるヌクレオチド配列と実質的に同一のヌクレオチド配列、あるいは配列番号3で表されるヌクレオチド配列またはその分泌シグナル除去ヌクレオチド配列と実質的に同一のヌクレオチド配列からなるポリヌクレオチドは、上記の通りクローニングしたポリヌクレオチドに変異を導入することにより作製できる。変異導入法としては、例えば、合成オリゴヌクレオチド指定突然変異導入法、gapped duplex法、ランダムに点突然変異を導入する方法(例えば、亜硝酸若しくは亜硫酸での処理)、カセット変異法、リンカースキャニング法、ミスマッチプライマー法などの方法が挙げられる。
【0042】
3.発現ベクター
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の調製に有用な発現ベクターを提供する。なお、本発明の発現ベクターは、下記を参照することで、自体公知の分子生物学的手法により作製できる。例えば、分子生物学的手法については、Molecular Cloning, 2nd edition, J. Sambrook et al., Cold Spring Harbor Lab. Press (1989)を参照のこと。
【0043】
3.1.SNIまたはその部分ペプチドの発現ベクター
本発明は、SNIまたはその部分ペプチドの発現ベクター(以下、必要に応じてSNI発現ベクターと省略する)を提供する。
【0044】
一実施形態では、SNI発現ベクターは、宿主細胞でのSNIまたはその部分ペプチド自体の発現を目的とする発現ベクターであり得る。SNI発現ベクターとしては、原核細胞および/または真核細胞の各種の宿主細胞中で本発明のSNIポリペプチドまたはその部分ペプチドをコードする遺伝子を発現し、これらを産生できるものであれば特に制限されないが、例えば、プラスミドベクター、ウイルスベクターが挙げられる。発現ベクターを医薬として用いる場合、ヒト等の哺乳動物への投与に好適なベクターとしては、アデノウイルス、レトロウイルス、アデノ随伴ウイルス、ヘルペスウイルス、ワクシニアウイルス、ポックスウイルス、ポリオウイルス、シンドビスウイルス、センダイウイルス、エプスタイン・バー・ウイルス等のウイルスベクターが挙げられる。
【0045】
SNI産生用宿主細胞として細菌(例えば、大腸菌)等の原核生物細胞を用いる場合、発現ベクターは、例えば、プロモーター−オペレーター領域、開始コドン、本発明のSNIポリペプチドまたはその部分ペプチドをコードするポリヌクレオチド、終止コドン、ターミネーター領域、複製起点等のエレメントを含み得る。細菌中で本発明のポリペプチドを発現させるためのプロモーター−オペレーター領域は、プロモーター、オペレーター及びShine-Dalgarno (SD) 配列を含むものである。これらのエレメントについては、自体公知のものを用いることができる。
【0046】
また、SNI発現ベクターを医薬または研究用試薬として使用する場合、酵母、動物細胞または昆虫細胞等の真核生物細胞を宿主細胞として利用可能な発現ベクターが用いられ得る。この場合、使用されるプロモーターは、投与対象である哺乳動物等の真核生物で機能し得るものであれば特に制限されない。例えば、医薬として使用する場合、SV40由来初期プロモーター、サイトメガロウイルスLTR、ラウス肉腫ウイルスLTR、MoMuLV由来LTR、アデノウイルス由来初期プロモーター等のウイルスプロモーター、並びにβ−アクチン遺伝子プロモーター、PGK遺伝子プロモーター、トランスフェリン遺伝子プロモーター等の哺乳動物の構成蛋白質遺伝子プロモーターなどが挙げられる。
【0047】
SNI発現ベクターは、さらに、選択マーカー遺伝子(テトラサイクリン、アンピシリン、カナマイシン、ハイグロマイシン、ホスフィノスリシン等の薬剤に対する抵抗性を付与する遺伝子、栄養要求性変異を相補する遺伝子等)をさらに含んでいてもよい。
【0048】
別の実施形態では、SNI発現ベクターは、融合タンパク質発現用ベクターであり得る。この場合、SNIまたはその部分ペプチドは、精製用タグとして用いられ得る。従って、SNIの部分ペプチドは、NADaseとの結合能を有するSNIの部分ペプチドであり得る。本発明のSNIは、3M NaClという高濃度の塩の存在下であってもNADaseと結合し得ることから、NADaseまたはSNIとの結合能を有するその部分ペプチドを結合対として用いることにより、SNIまたはその部分ペプチドが付加された融合タンパク質の精製が可能となる。このようなSNI発現ベクターは、融合されるべき目的タンパク質のクローニングを容易とするため、SNIまたはその部分ペプチドをコードするポリヌクレオチドの上流または下流にマルチクローニングサイト(MCS)を含み得る。
【0049】
なお、SNI発現ベクターは、後述するNADase発現ベクター、あるいは後述するSLO発現ベクターと同様の形態であってもよい。即ち、後に詳述される、節「3.2.NADaseまたはその部分ペプチドの発現ベクター」における「NADase」、および節「3.3.SLOまたはその部分ペプチドの発現ベクター」における「SLO」は、「SNI」と読み替え可能であり、そのような発現ベクターもまた、「SNIまたはその部分ペプチドの発現ベクター」であり得る。
【0050】
3.2.NADaseまたはその部分ペプチドの発現ベクター
本発明は、NADaseまたはその部分ペプチドの発現ベクター(以下、必要に応じてNADase発現ベクターと省略する)を提供する。
【0051】
一実施形態では、NADase発現ベクターは、宿主細胞でのNADaseまたはその部分ペプチド自体の発現を可能とする共発現ベクターであり得る。NADaseは全般的に宿主細胞に毒性を示すため宿主細胞でのNADaseの発現は困難であり、特に連鎖球菌由来NADaseは大腸菌等の宿主細胞に対する強い毒性を示すため連鎖球菌由来NADaseの組換えタンパク質の調製には未だ成功していない。しかし、本発明者らは、宿主細胞でのNADaseの製造を可能にするためには、NADaseを、SNIまたは宿主細胞に対する毒性作用に対する中和作用を有するその部分ペプチドと共発現させればよいことを見出した。
【0052】
従って、NADase発現ベクターは、第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み得る。第1および第2の発現ユニットは、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドであり得る。プロモーターの機能的連結とは、プロモーターが、その制御下にある遺伝子の発現を可能とするように、該遺伝子をコードするポリヌクレオチドに結合していることを意味する。
【0053】
NADase発現ベクターが導入され得る宿主細胞としては、後述する任意の宿主細胞である限り特に限定されないが、例えば、大腸菌、バチルス属菌(例、枯草菌)等の原核生物細胞が好ましい。宿主細胞に対する毒性作用としては、例えば、NADase活性が挙げられる。宿主細胞に対する毒性作用に対する中和作用としては、例えば、NADase阻害活性が挙げられる。
【0054】
より詳細には、NADase発現ベクターは、特にNADaseが連鎖球菌由来NADaseである場合、ポリシストロニックmRNA発現ベクターであり得る。ポリシストロニックmRNA発現ベクターは、第1および第2の発現ユニットのポリシストロニックmRNAの発現を可能とする、第1および第2の発現ユニットをコードするポリヌクレオチド、ならびに当該ポリシストロニックmRNAの発現を可能とするように該ポリヌクレオチドに機能的に連結されたプロモーターを含み得る。ポリシストロニックmRNA発現ベクターにおいて、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドをコードするポリヌレオチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドをコードするポリヌレオチドは、いずれが上流または下流に位置してもよいが、天然状態(即ち、nga−sloオペロン)を反映させるという観点からは、前者が上流に位置するのが好ましい。
【0055】
NADase発現ベクターはまた、非ポリシストロニックmRNA発現ベクターであり得る。非ポリシストロニックmRNA発現ベクターは、第1の発現ユニットをコードするポリヌクレオチドおよび該ポリヌクレオチドに機能的に連結された第1のプロモーター、ならびに第2の発現ユニットをコードするポリヌクレオチドおよび該ポリヌクレオチドに機能的に連結された第2のプロモーターを含み得る。
【0056】
NADase発現ベクターは、例えば、上述したポリシストロニックmRNA発現ベクターまたは非ポリシストロニックmRNA発現ベクターであり得るが、好ましくは、ポリシストロニックmRNA発現ベクターであり得る。
【0057】
NADase発現ベクターは、上述の特徴以外は、SNI発現ベクターの特徴と同様であり得る。即ち、本発明の共発現ベクターは、プラスミドベクター、ウイルスベクターであり得、また、SNIまたはその部分ペプチドの発現ベクターと同様のプロモーター等のエレメント、選択マーカー遺伝子を含み得る。
【0058】
別の実施形態では、NADase発現ベクターは、融合タンパク質発現用ベクターであり得る。この場合、NADaseまたはその部分ペプチドは、精製用タグとして用いられ得る。従って、NADaseの部分ペプチドは、SNIとの結合能を有するNADaseの部分ペプチドであり得る。NADaseは、3M NaClという高濃度の塩の存在下であってもSNIと結合し得ることから、SNIまたはNADaseとの結合能を有するその部分ペプチドを結合対として用いることにより、NADaseまたはその部分ペプチドが付加された融合タンパク質の精製が可能となる。このような発現ベクターは、融合されるべき目的タンパク質のクローニングを容易とするため、NADaseまたはその部分ペプチドをコードするポリヌクレオチドの上流または下流にマルチクローニングサイト(MCS)を含み得る。
【0059】
なお、NADase発現ベクターは、上述したSNI発現ベクター、あるいは後述するSLO発現ベクターと同様の形態であってもよい。即ち、先に詳述した節「3.1.SNIまたはその部分ペプチドの発現ベクター」における「SNI」、および後に詳述する節「3.3.SLOまたはその部分ペプチドの発現ベクター」における「SLO」は、「NADase」と読み替え可能であり、そのような発現ベクターもまた、「NADaseまたはその部分ペプチドの発現ベクター」であり得る。
【0060】
3.3.SLOまたはその部分ペプチドの発現ベクター
本発明は、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクター(以下、必要に応じてSLO発現ベクターと省略する)を提供する。SLO発現ベクターは、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含む。
【0061】
ストレプトリシンO(SLO)は、5つの属〔連鎖球菌属(streptococcus)、バチルス属(Bacillus)、クロストリジウス属(Clostridium)、リステリア属(Listeria)、アルカノバクテリウム属(Arcanobacterium)〕の種に見出されるサイトリシンの高度に相同な単一ファミリーに属する(例えば、Pharmacol Ther 11: 661-717 (1980); Int J Med Microbiol 290: 351-356 (2000); FEMS Microbiol Lett 182: 197-205 (2000); Arch Microbiol 165: 73-79 (1996); Infect Immun 47, 52-60 (1985); Toxicon 39: 1681-1689 (2001)参照)。本発明で対象となり得るSLOは、上記由来のSLOである限り特に限定されないが、SLO遺伝子は異なる溶血性連鎖球菌群間で高度に保存されていることから、溶血性連鎖球菌由来SLOが好ましい。溶血性連鎖球菌としては、A群〜C群、G群の連鎖球菌が挙げられるが、化膿性咽頭炎、創傷感染および連鎖球菌トキシックショック様症候群、猩紅熱、急性糸球体腎炎などの発症因子としての報告(例えば、Clin Microbiol Rev 13: 470-511 (2000); FEMS Immunol Med Microbiol 34: 159-167 (2002); J Clin Microbiol 42: 186-192 (2004)参照)があるA群連鎖球菌(GAS)、C群連鎖球菌(GCS)、G群連鎖球菌(GGS)が好ましい。なお、GASおよびGCSでは、いずれのSLOも、N末端シグナルペプチドを含む31個のアミノ酸残基が除去されて分泌されるとともに、自身が分泌するプロテアーゼにより次第に低分子化することが報告されている(例えば、Infect Immun 61: 2727-2731 (1993); Infect Immun 63: 2776-2779 (1995)参照)。このような理由により、GASおよびGCSの完全長SLOは精製が特に困難であり得るが、本発明者らの開発した方法によればこれらの完全長SLOの精製が可能である。従って、上記の連鎖球菌に由来するSLOのなかでも、GASおよびGCS由来のSLOがより好ましい。ところで、GASおよびGCSには幾つかの亜種が知られている。本発明で対象となり得るSLOが由来するGASの亜種としては、例えば、D58X株(ATCC12383)が挙げられる。本発明で対象となり得るSLOが由来するGCSの亜種としては、例えば、H46A株(ATCC12449)が挙げられる。
【0062】
完全長SLOとは、N末端の分泌シグナルペプチド除去後のSLOを意味する。
【0063】
大腸菌に毒性を示し得る領域を含む完全長SLOの部分ペプチドとは、大腸菌に対する毒性を有し得るペプチド部分を含む任意の断片を意味する。例えば、GAS、GCS等の溶血性連鎖球菌に由来するSLOは、そのN末端近傍の領域、特にその約32−約75位のアミノ酸残基からなるポリペプチドの領域中に、このような領域を有すると考えられる(例えば、Biosci Biotechnol Biochem 65: 2682-2689 (2001) 参照)。従って、大腸菌に毒性を示し得る領域を含む部分ペプチドは、このようなポリペプチド中に含まれる大腸菌に毒性を示し得るペプチド部分を含むものであり得る。
【0064】
SLO発現ベクターから発現され得る完全長SLOまたはその部分ペプチドは、好ましくは精製用タグを有する。従って、SLO発現ベクターは、精製用タグ融合SLOの発現を可能とする、精製用タグをコードするポリヌクレオチドとSLOをコードするポリヌクレオチドとのin frame連結物を含み得る。精製用タグは、本発明の発現ベクターが導入された形質転換体の培養上清からの、SLOの精製を容易とするものである限り特に限定されず、例えば、Hisタグ、マルトース結合タンパク(MBP)、グルタチオン−S−トランスフェラーゼ(GST)、カルモジュリン結合ペプチド(CBP)、Strep−tag II、FLAG、heayy chain of protein C(HPC)が挙げられるが、付加されるアミノ酸配列が短く、多くの場合除去する必要がない、および特異的な抗体や精製担体も複数の市販品が入手可能であり、安価である等の利点より、Hisタグが好ましい。
【0065】
誘導プロモーターとは、誘導剤の存在下でその制御下にある遺伝子の発現を増強するプロモーターを意味する。SLO発現ベクターでは、誘導プロモーターは、SLO遺伝子をコードするポリヌクレオチドに機能的に連結され得る。
【0066】
SLO発現ベクターに含まれる誘導プロモーターはまた、大腸菌における基底レベルの遺伝子発現を抑制し得る。基底レベルの遺伝子発現とは、非誘導条件下での遺伝子発現を意味する。SLOは大腸菌に毒性を示し得る領域を含み得るため、その基底レベルのSLO遺伝子の発現量が多いと大腸菌が傷害を受け易くなることから、このような領域を含むSLOの大量発現が難しいという問題があった。しかし、本発明者らは、基底レベルの遺伝子発現を厳格に抑制できる誘導プロモーターを用いることにより、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドを大腸菌で大量発現させることに成功した。大腸菌における基底レベルの遺伝子発現を抑制し得るこのような誘導プロモーターとしては、例えば、アラビノースオペロン制御プロモーター(例、L−アラビノースを誘導剤とするaraBAD:例えば、pBADシリーズのベクターを利用可能)、キシロースオペロン制御プロモーター(例、キシロースを誘導剤とするxylAプロモーター:例えば、pWH1520を利用可能)、サリチル酸塩で制御可能なプロモーター(例、サリチル酸塩を誘導剤とするPmプロモーター:例えば、pCASシリーズのベクターを利用可能)、低温処理で制御可能なプロモーター(例、低温処理により誘導され得るcspAプロモーター:例えば、pColdシリーズのベクターを利用可能)が挙げられるが、使い易さ、発現レベル等の観点より、アラビノースオペロン制御プロモーターが好ましい。
【0067】
SLO発現ベクターはまた、転写開始および転写終結のための部位、および転写領域において翻訳に必要とされ得るリボソーム結合部位、複製起点ならびに選択マーカー遺伝子などを含み得る。選択マーカー遺伝子としては、宿主として大腸菌を用いた場合に、形質転換された大腸菌を選別できるものである限り特に限定されないが、例えば、アンピシリン、テトラサイクリン、カナマイシン、スペクチノマイシン、エリスロマイシン、クロラムフェニコール、カルベニシリン等の抗生物質に対する耐性遺伝子が挙げられる。
【0068】
4.形質転換体
本発明は、NADase、SNIおよびSLO遺伝子を含むオペロンから発現するタンパク質の調製に有用な形質転換体、またはこのようなタンパク質の解析を可能とする形質転換体を提供する。
【0069】
本発明の形質転換体は、上述した本発明の発現ベクターまたはその他の発現ベクターを宿主細胞に導入することにより作製できる。形質転換体の作製に用いられる宿主細胞としては、前記の発現ベクターに適合し、形質転換され得るものであれば特に限定されず、本発明の技術分野において通常使用される天然細胞あるいは人工的に樹立された細胞株など種々の細胞が用いられ得るが、それぞれの形質転換体に応じた好ましい宿主細胞については以下に述べる。それぞれの形質転換体は、自体公知の方法により培養できる(例えば、後述の培養法を参照)。
【0070】
4.1.SNIまたはその部分ペプチドを発現し得る形質転換体
本発明は、SNIまたはその部分ペプチドを発現し得る形質転換体(以下、必要に応じてSNI形質転換体と省略する)を提供する。
【0071】
SNI形質転換体は、本発明のSNIまたはその部分ペプチドの発現ベクターで宿主細胞を形質転換することにより作製できる。
【0072】
SNI形質転換体の宿主細胞としては、例えば、大腸菌(例、「4.3.SLOまたはその部分ペプチドを発現し得る形質転換体」で詳述される大腸菌)、バチルス属菌(例、枯草菌)、放線菌等の原核生物細胞、ならびに酵母、昆虫細胞、鳥類細胞、哺乳動物細胞(例、ヒト、サル等の霊長類、マウス、ラット等のげっ歯類に由来する細胞)等の真核生物細胞が挙げられる。なお、SNIまたはその部分ペプチドを大量に製造することを目的とする場合には、原核生物細胞を宿主細胞として用いることが好ましい。
【0073】
4.2.NADaseまたはその部分ペプチドを発現し得る形質転換体
本発明は、NADaseまたはその部分ペプチドを発現し得る形質転換体(以下、必要に応じてNADase形質転換体と省略する)を提供する。NADaseは全般的に宿主細胞に毒性を示すため宿主細胞でのNADaseの発現は困難であり、特に連鎖球菌由来NADaseは大腸菌等の宿主細胞に対する強い毒性を示すため連鎖球菌由来NADaseの組換えタンパク質の調製には未だ成功していない。しかし、本発明者らは、宿主細胞でのNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造を可能にするためには、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを共発現する形質転換体を作製すればよいことを見出した。
【0074】
NADase形質転換体は、NADase発現ベクター(例、上述したポリシストロニックmRNA発現ベクターおよび非ポリシストロニックmRNA発現ベクター)で宿主細胞を形質転換することにより作製できる。NADase形質転換体はまた、NADaseのみを発現し、SNIを発現しないベクター(即ち、NADase単独発現ベクター)、およびSNI発現ベクターの双方で宿主細胞を形質転換することにより作製できる。
【0075】
NADase形質転換体の宿主細胞としては、例えば、上述したSNI形質転換体と同様のものが挙げられる。なお、NADaseまたはその部分ペプチドを大量に製造することを目的とする場合には、原核生物細胞を宿主細胞として用いることが好ましい。
【0076】
4.3.SLOまたはその部分ペプチドを発現し得る形質転換体
本発明は、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドを大量に発現し得る形質転換体(以下、必要に応じてSLO形質転換体と省略する)を提供する。
【0077】
SLO形質転換体は、本発明のSLOまたはその部分ペプチドの発現ベクターで宿主細胞を形質転換することにより作製できる。
【0078】
SLO形質転換体の宿主細胞としては、その目的に鑑みれば、大腸菌が好ましい。
【0079】
宿主として用いられ得る大腸菌は、完全長SLOを大量かつ高純度で製造する(換言すれば、SLOの切断を抑える)という本発明の一つの目的を考慮すれば、プロテアーゼの欠損株であることが好ましい。大腸菌において欠損されるべきこのようなプロテアーゼとしては、例えば、lon、ompTが挙げられる。本発明において宿主として用いられ得るプロテアーゼ欠損株としては、例えば、BL21株が挙げられる。
【0080】
また、宿主として用いられ得る大腸菌は、誘導プロモーターの能力を十分に発揮させ、大腸菌に毒性を示し得る領域を含むSLOを大量に製造するという本発明の別の目的を考慮すれば、誘導プロモーターに対する誘導剤の代謝能を欠損していることが好ましい。欠損されるべき代謝能は、本発明で用いられる誘導プロモーターの種類に応じて異なるが、例えば、上述した誘導プロモーターの誘導剤の代謝能であり得る。
【0081】
5.形質転換体を用いるポリペプチドまたはその部分ペプチドの製造方法
本発明は、本発明の形質転換体を用いるポリペプチドまたはその部分ペプチドの製造方法を提供する。例えば、本発明の製造方法は、本発明の形質転換体を培養し、目的のポリペプチドまたはその部分ペプチドを産生させ、次いで回収することを含む。本発明の製造方法はまた、本発明の形質転換体が誘導プロモーターを有する発現ベクターを含む場合、誘導プロモーターに対する誘導剤を培地に添加し、培養することを含み得る。
【0082】
形質転換体の培養は、自体公知の方法により液体培地等の栄養培地中で行われ得る。培地は、形質転換体の生育に必要な炭素源、窒素源、無機物などを含有することが好ましい。ここで、炭素源としては、例えば、グルコース、デキストリン、可溶性澱粉、ショ糖などが挙げられ、窒素源としては、例えば、アンモニウム塩類、硝酸塩類、コーンスチープ・リカー、ペプトン、カゼイン、肉エキス、大豆粕、バレイショ抽出液などの無機または有機物質が挙げられ、無機物としては、例えば、塩化カルシウム、リン酸二水素ナトリウム、塩化マグネシウムなどがそれぞれ挙げられる。また、培地には、酵母エキス、ビタミン類などを添加してもよい。培養条件、例えば温度、培地のpHおよび培養時間は、本発明のポリペプチドが大量に生産されるように適宜選択される。培養温度は、例えば30〜37℃である。
【0083】
本発明の形質転換体が誘導プロモーターを有する発現ベクターを含む場合、本発明の形質転換体は、誘導プロモーターに対する誘導剤の存在下で培養され得る。誘導剤の存在下における形質転換体の培養は、非誘導条件下において大量発現に適切な数に増殖した形質転換体を用いて、誘導剤の種類に応じた適切な条件下において行われ得る。例えば、形質転換体の宿主細胞として大腸菌を用いる完全長SLOまたはその部分ペプチドの製造方法では、誘導プロモーターがアラビノースオペロン制御プロモーターである場合には、培地に添加されるL−アラビノースの濃度が0.002%(W/V)以上であるときに、完全長SLOまたはその部分ペプチドの十分な発現が確認されている。従って、培地に添加されるL−アラビノースの濃度は完全長SLOまたはその部分ペプチドの効率的な発現を可能とする限り特に限定されないものの、好ましくは約0.002%(W/V)以上、より好ましくは約0.02%(W/V)以上であり得る。なお、L−アラビノースの上限濃度は特に限定されないが、例えば0.2%(W/V)以下であり得る。また、本発明の大腸菌が誘導条件下におかれる時間としては、完全長SLOまたはその部分ペプチドの効率的な発現を可能とする限り特に限定されないが、例えば、約1〜約24時間が挙げられる。なお、誘導条件下における培養は、誘導剤を所定の条件下で用いること以外は、非誘導条件下における培養と同様に行われ得る。
【0084】
形質転換体の培養により産生されたポリペプチドまたはその部分ペプチドは、回収、好ましくは単離、精製され得る。例えば、SNIまたはその部分ペプチドは、培養された形質転換体を集め、溶解処理(例、リゾチーム、ソニケーション等による処理)した後、溶解液中から回収され得る。また、NADase、SLOまたはそれらの部分ペプチドは、その培養上清から回収され得る。
【0085】
単離、精製方法としては、例えば塩析、溶媒沈澱法等の溶解度を利用する方法、透析、限外濾過、ゲル濾過、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動など分子量の差を利用する方法、イオン交換クロマトグラフィーやヒドロキシルアパタイトクロマトグラフィーなどの荷電を利用する方法、アフィニティークロマトグラフィーなどの特異的親和性を利用する方法、逆相高速液体クロマトグラフィーなどの疎水性の差を利用する方法、等電点電気泳動などの等電点の差を利用する方法などが挙げられる。
【0086】
本発明の製造方法によれば、SNI、NADase、SLOおよびそれらの部分ペプチドを高純度で大量かつ安価に製造することが可能である。例えば、溶血性連鎖球菌に由来する完全長SLOまたはその部分ペプチドの製造方法では、用いる培地や誘導条件によっても異なるが、LB培地1リットルあたり例えば約1mg以上、好ましくは約1mg〜約30mg、より好ましくは約5mg〜約30mg、さらにより好ましくは約5mg〜約20mg、最も好ましくは約5mg〜約10mgの収量を可能とする。また、本発明の製造方法により得られるSLOは、異種の宿主で産生された組換えタンパク質であるにもかかわらず、溶血性連鎖球菌の培養上清から得られる天然タンパク質と同様の比活性を有し得る。
【0087】
6.抗体
【0088】
本発明は、SNI、NADase、SLOおよびそれらの部分ペプチドに対する抗体を提供する。
【0089】
本発明の抗体は、ポリクローナル抗体、モノクローナル抗体のいずれであってもよく、周知の免疫学的手法により作製することができる。この抗体は、完全な抗体分子だけでなく、本発明のポリペプチドに対する抗原結合部位(CDR)を有する限りいかなるフラグメントであってもよく、例えば、Fab、F(ab')2、ScFv、minibody等が挙げられる。
【0090】
例えば、ポリクローナル抗体は、上記抗原(必要に応じて、ウシ血清アルブミン、KLH(Keyhole Limpet Hemocyanin)等のキャリア蛋白質に架橋した複合体とすることもできる)を、市販のアジュバント(例えば、完全または不完全フロイントアジュバント)とともに、動物の皮下あるいは腹腔内に2〜3週間おきに2〜4回程度投与し(部分採血した血清の抗体価を公知の抗原抗体反応により測定し、その上昇を確認しておく)、最終免疫から約3〜10日後に全血を採取して抗血清を精製することにより取得できる。抗原を投与する動物としては、ラット、マウス、ウサギ、ヤギ、モルモット、ハムスターなどの哺乳動物が挙げられる。
【0091】
また、モノクローナル抗体は、細胞融合法により作成することができる。例えば、マウスに上記抗原を市販のアジュバントと共に2〜4回皮下あるいは腹腔内に投与し、最終投与の3日後に脾臓あるいはリンパ節を採取し、白血球を採取する。この白血球と骨髄腫細胞(例えば、NS-1, P3X63Ag8など)を細胞融合して該因子に対するモノクローナル抗体を産生するハイブリドーマを得る。細胞融合はPEG法でも電圧パルス法であってもよい。所望のモノクローナル抗体を産生するハイブリドーマは、周知のEIAまたはRIA法等を用いて抗原と特異的に結合する抗体を、培養上清中から検出することにより選択できる。モノクローナル抗体を産生するハイブリドーマの培養は、インビトロ、またはマウスもしくはラット、好ましくはマウス腹水中等のインビボで行うことができ、抗体はそれぞれハイブリドーマの培養上清および動物の腹水から取得することができる。
【0092】
さらに、本発明の抗体は、キメラ抗体(chimeric antibody)、ヒト化抗体(humanized antibody)、ヒト型抗体(human antibody)であってもよい。キメラ抗体は、例えば「実験医学(臨時増刊号), Vol.6, No.10, 1988」、特公平3-73280号公報等を、ヒト化抗体は、例えば特表平4-506458号公報、特開昭62-296890号公報等を、ヒト抗体は、例えば「Nature Genetics, Vol.15, p.146-156, 1997」、「Nature Genetics, Vol.7, p.13-21, 1994」、特表平4-504365号公報、国際出願公開WO94/25585号公報、「日経サイエンス、6月号、第40〜第50頁、1995年」、「Nature, Vol.368, p.856-859, 1994」、特表平6-500233号公報等を参考にそれぞれ作製することができる。例えば、本発明の抗体を医薬として用いる場合、ヒト化抗体およびヒト型抗体が好ましい。
【0093】
本発明の抗体(特に、後述のSLO抗体)は、臨床検査試薬、研究用試薬等の試薬および医薬などとして有用である。本発明の抗体が試薬として用いられる場合、例えば、酵素免疫測定法(EIA)、放射免疫測定法(RIA)、蛍光免疫測定法(FIA)、免疫クロマト法、ルミネッセンス免疫測定法、スピン免疫測定法、ウエスタンブロット法による抗原(特にSLO)の検出および定量に用いられ得る。EIA法としては、例えば、直接競合ELISA、間接競合ELISA、サンドイッチELISAが挙げられる。一方、本発明の抗体が医薬として用いられる場合、例えば、溶血性連鎖球菌感染症の治療薬として用いられ得る。
【0094】
本発明の抗体が医薬として使用される場合、本発明の医薬は、医薬上許容される担体が配合されたものであり得る。医薬上許容される担体としては、後述するものが挙げられる。本発明の抗体は、好ましくは、後述の投与量において後述の方法に従い非経口投与され得る。
【0095】
6.1.SNIまたはその部分ペプチドに対する抗体
本発明は、SNIまたはその部分ペプチドに対する抗体(以下、必要に応じてSNI抗体と省略する)を提供する。
【0096】
SNI抗体の作製に用いられる抗原としては、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチド、あるいはそれらの部分ペプチドを用いることができる。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、配列番号2で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列、あるいは上述のドメインをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。
【0097】
また、中和抗体を得ることを目的とする場合には、抗原として、NADase阻害活性を有する部分ペプチドを用いることが好ましく、後述する本発明の複合体の解離剤を得ることを目的とする場合には、抗原として、NADaseとの結合能を有するその部分ペプチドを用いることが好ましい。
【0098】
6.2.NADaseまたはその部分ペプチドに対する抗体
本発明は、NADaseまたはその部分ペプチドに対する抗体(以下、必要に応じてNADase抗体と省略する)を提供する。本発明者らの研究成果により、宿主細胞を用いた連鎖球菌由来天然型NADaseの組換えタンパク質の調製のみならず、宿主細胞を用いた非天然型NADase(変異型NADaseまたは遺伝子改変NADase)の組換えタンパク質の調製が初めて可能となる。また、このように組換え技術を利用することで、NADaseの部分ペプチドの調製も容易になる。従って、従来よりも、NADase抗体の作製が容易になり得る。
【0099】
NADase抗体の作製に用いられる抗原としては、配列番号4で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列からなるポリペプチド、あるいはそれらの部分ペプチドを用いることができる。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、配列番号4で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列、あるいは上述のドメインをコードするアミノ酸配列から選ばれる少なくとも約8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個、最も好ましくは20個以上の連続したアミノ酸からなるペプチドであり得る。
【0100】
また、中和抗体を得ることを目的とする場合には、抗原として、NADase活性を有する部分ペプチドを用いることが好ましく、後述する本発明の複合体の解離剤を得ることを目的とする場合には、抗原として、SNIとの結合能を有するその部分ペプチドを用いることが好ましい。
【0101】
6.3.SLOまたはその部分ペプチドに対する抗体
本発明は、SLOまたはその部分ペプチドに対する抗体(以下、必要に応じてSLO抗体と省略する)を提供する。
【0102】
SLO抗体の作製に用いられる抗原としては、溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチド(例えば、約32〜約75位のアミノ酸残基を含むポリペプチドの部分ペプチド)が用いられ得る。部分ペプチドとしては、免疫原性を有する限り特に限定されないが、例えば、上記SLOの任意の部分から選ばれる少なくとも8個、好ましくは少なくとも10個、より好ましくは少なくとも12個、さらにより好ましくは少なくとも15個以上の連続したアミノ酸からなるポリペプチドであり得る。
【0103】
SLO抗体は、例えば、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドに対する特異的な反応性を示し、かつNADase、特に溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体であり得る。本発明の抗体はまた、完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの活性、特に溶血活性の中和抗体であり得る。
【0104】
7.SNIに関連するその他の発明
本発明は、SNIポリヌクレオチドまたはその部分ヌクレオチドに相補的な核酸(アンチセンス核酸)、RNAi誘導核酸(siRNA)、リボザイム、増幅用プライマー対などを提供する。これらは、例えば、本発明のSNIの解析、変異型連鎖球菌の作製などに有用であり得る。
【0105】
8.医薬または試薬
本発明はまた、SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む医薬または試薬を提供する。本発明者らは、未知遺伝子SNIの機能の一端を解明することに初めて成功した。より詳細には、本発明者らは、SNIがNADaseを阻害し得ることを見出した。従って、本発明者らは、SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを、医薬または試薬として用いることを初めて可能とした。
【0106】
本発明の医薬は、例えば、連鎖球菌等の細菌の感染症の治療薬として有用である。分泌タンパク質である連鎖球菌NADaseは、血液等の生体液中に分泌され、SLOにより宿主細胞膜上に形成された孔から宿主細胞内に侵入し、NADase活性によりその毒性を発揮することで、連鎖球菌感染症の病態を担い得ると考えられる。従って、SNIまたはNADase阻害活性を有するその部分ペプチドが生体内に存在すれば、連鎖球菌NADaseとSNIまたはその部分ペプチドとの安定複合体が形成され、以って連鎖球菌NADaseのNADase活性を低減し得ることにより、連鎖球菌感染症の病態を軽減し得ると考えられる。本発明の医薬が適用され得る感染症としては、NADaseにより毒性作用を発揮する細菌により引き起こされ得る感染症である限り特に限定されないが、例えば、A群、C群およびG群溶血性連鎖球菌により引き起こされる感染症が挙げられる。
【0107】
本発明の医薬はまた、非感染症の治療に有用である。本発明者らは、SNIが細菌由来NADaseのみならず、哺乳動物由来NADaseも阻害し得ることを見出した。従って、SNIまたはNADase阻害活性を有するその部分ペプチドは、哺乳動物由来NADase(例、CD38、CD157)の機能亢進によりもたらされ得る疾患、あるいは哺乳動物由来NADaseの機能阻害により対症療法的に治療され得る疾患(例えば、哺乳動物由来NADaseの機能阻害により症状が改善、緩和または軽減され得る疾患)に対して有効であると考えられる。このような疾患としては、例えば、自己免疫疾患(例、潰瘍性大腸炎)、炎症性疾患(例、成人呼吸促迫症候群(ARDS:adult respiratory distress syndrome)、関節炎)、アレルギー疾患、多発性骨髄腫、虚血、喘息、糖尿病、移植拒絶反応、神経変性疾患が挙げられるがこれらに限定されない(例えば、WO2002/032288;Nature Medicine 7(11): 1209-16(2001);The Journal of Immunology 172: 1896-1906 (2004) 参照)。
【0108】
本発明の医薬または試薬はさらに、NADase阻害剤として有用である。NADase阻害剤とは、上述したNADase阻害活性を有するものをいう。NADaseは細菌から哺乳動物まで広く認められること、ならびにSNIは細菌および哺乳動物NADaseを阻害したことから、SNIは、あらゆる種に由来するNADseを阻害し得ると考えられる。SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得るNADseとしては、原核生物から真核生物にまで至る任意のNADaseが挙げられるが、例えば、細菌(例、連鎖球菌)、哺乳動物(例、ヒト、サル、チンパンジー、ウサギ、ウシ)、昆虫、鳥類、魚類、軟体動物(例、アメフラシ)、植物が挙げられ、細菌およびヒト等の哺乳動物が好ましい。詳細には、SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得る連鎖球菌由来NADaseとしては、A群、C群およびG群連鎖球菌由来NADaseが挙げられる。SNIまたはNADase阻害活性を有するその部分ペプチドにより阻害され得る哺乳動物由来NADaseとしては、CD38、CD157、アナウサギ属ADPリボシルシクラーゼ、ウシNAD+グリコヒドロラーゼが挙げられるが、CD38、CD157が好ましい。NADaseはまた、その局在に応じて、分泌型、細胞膜結合型、細胞質局在型などに分類できる。SNIまたはNADase阻害活性を有するその部分ペプチドは、これらの任意の型のNADaseを阻害し得るが、好ましくは分泌型または細胞膜結合型のNADaseを阻害し得る。また、CD38の酵素活性(およびcADPRの生成)は、細胞内Ca2+貯蔵からのCa2+の放出、細胞増殖、アポトーシスなどに関与し得るので(例えば、WO2002/032288参照)、本発明の医薬および試薬はこれらの作用を阻害し得る。本発明のNADase阻害剤はまた、細胞系(例、本発明の形質転換体の使用)または無細胞系(例、本発明の共発現ベクターの使用、あるいはNADase発現ベクターおよびSNI発現ベクターの組合せの使用、あるいはNADase合成系におけるSNIの添加)におけるNADaseの調製剤として用いることができる。
【0109】
本発明の医薬または試薬はまた、白血球の走化性の阻害剤として有用である。白血球の走化性とは、白血球が炎症部位に動員されることをいう。CD38、CD157は、白血球の走化性に重要な役割を果たし得ることが知られている(例えば、Blood vol.104(13): 4269-4278 (2004)、Nat Med. vol.7(11): 1209-16 (2001))。SNIまたはNADase阻害活性を有するその部分ペプチドにより走化性が阻害され得る白血球としては、例えば、好中球、好酸球、単球、リンパ球、マクロファージ、樹状細胞が挙げられるが(例えば、WO2002/032288参照)、好中球が好ましい。
【0110】
本発明の医薬は、医薬上許容される担体が配合されたものであり得る。医薬上許容される担体としては、例えば、ショ糖、デンプン、マンニット、ソルビット、乳糖、グルコース、セルロース、タルク、リン酸カルシウム、炭酸カルシウム等の賦形剤、セルロース、メチルセルロース、ヒドロキシプロピルセルロース、ポリプロピルピロリドン、ゼラチン、アラビアゴム、ポリエチレングリコール、ショ糖、デンプン等の結合剤、デンプン、カルボキシメチルセルロース、ヒドロキシプロピルスターチ、ナトリウム−グリコール−スターチ、炭酸水素ナトリウム、リン酸カルシウム、クエン酸カルシウム等の崩壊剤、ステアリン酸マグネシウム、エアロジル、タルク、ラウリル硫酸ナトリウム等の滑剤、クエン酸、メントール、グリシルリシン・アンモニウム塩、グリシン、オレンジ粉等の芳香剤、安息香酸ナトリウム、亜硫酸水素ナトリウム、メチルパラベン、プロピルパラベン等の保存剤、クエン酸、クエン酸ナトリウム、酢酸等の安定剤、メチルセルロース、ポリビニルピロリドン、ステアリン酸アルミニウム等の懸濁剤、界面活性剤等の分散剤、水、生理食塩水、オレンジジュース等の希釈剤、カカオ脂、ポリエチレングリコール、白灯油等のベースワックスなどが挙げられるが、それらに限定されるものではない。
【0111】
経口投与に好適な製剤は、水、生理食塩水、オレンジジュースのような希釈液に有効量のリガンドを溶解させた液剤、有効量のリガンドを固体や顆粒として含んでいるカプセル剤、サッシェ剤または錠剤、適当な分散媒中に有効量の有効成分を懸濁させた懸濁液剤、有効量の有効成分を溶解させた溶液を適当な分散媒中に分散させ乳化させた乳剤等である。
【0112】
非経口的な投与(例えば、静脈内注射、皮下注射、筋肉注射、局所注入、腹腔内投与など)に好適な製剤としては、水性および非水性の等張な無菌の注射液剤があり、これには抗酸化剤、緩衝液、制菌剤、等張化剤等が含まれていてもよい。また、水性および非水性の無菌の懸濁液剤が挙げられ、これには懸濁剤、可溶化剤、増粘剤、安定化剤、防腐剤等が含まれていてもよい。当該製剤は、アンプルやバイアルのように単位投与量あるいは複数回投与量ずつ容器に封入することができる。また、有効成分および医薬上許容される担体を凍結乾燥し、使用直前に適当な無菌のビヒクルに溶解または懸濁すればよい状態で保存することもできる。
【0113】
本発明の製剤の投与量は、有効成分の種類・活性、病気の重篤度、投与対象となる動物種、投与対象の薬物受容性、体重、年齢等によって異なるが、通常、成人1日あたり有効成分量として約0.001〜約2.0g/kgであり得る。
【0114】
9.複合体およびその製造方法
本発明はまた、SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADase(例、連鎖球菌由来NADase)またはSNIとの結合能を有するその部分ペプチドとを含む複合体を提供する。本発明者らは、SNIがNADaseと結合して複合体を形成し、NADase活性を抑制することを見出した。
【0115】
本発明の複合体において、SNIまたはNADaseとの結合能を有するその部分ペプチド、ならびにNADaseまたはSNIとの結合能を有するその部分ペプチドは、上述の通りであり得るが、それぞれ標識されていてもよい。標識としては、それらの複合体の形成を妨げない限り特に限定されないが、上述した精製用タグによる標識、放射性同位体(例えば、35S等の放射性同位体)による標識が挙げられる。複合体における構成成分の未標識・標識パターンは以下(a)〜(d)の通りである:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチド(未標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(未標識)との組合せ;
(b)SNIまたはNADaseとの結合能を有するその部分ペプチド(標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(未標識)との組合せ;
(c)SNIまたはNADaseとの結合能を有するその部分ペプチド(未標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(標識)との組合せ;
(d)SNIまたはNADaseとの結合能を有するその部分ペプチド(標識)と、NADaseまたはSNIとの結合能を有するその部分ペプチド(標識)との組合せ。
また、双方が標識される場合、標識の種類は同じであっても異なっていてもよいが、好ましくは異なり得る。従って、この場合、異なる精製用タグによる標識、異なる放射性同位体による標識、精製用タグと放射性同位体による標識が利用され得る。
【0116】
精製用タグで標識されたタンパク質またはペプチドは、自体公知の方法により作製できる。例えば、精製用タグをコードするポリヌクレオチドに目的タンパク質またはペプチドをコードするポリヌクレオチドを適切に連結してDNA構築物を作製し、このDNA構築物を宿主細胞で発現させることにより、精製用タグで標識されたタンパク質またはペプチドを作製できる。一方、放射性同位体で標識されたタンパク質またはペプチドは、その発現細胞を放射性同位体含有培地で培養することにより作製できる。
【0117】
本発明の複合体は、非常に強固かつ特異的に結合する。例えば、本発明の複合体は3M NaClという高濃度の塩の存在下であっても解離し得ない。従って、本発明の複合体は非常に安定であり得る。
【0118】
本発明はまた、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法を提供する。このような製造方法は、例えば、SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む。
【0119】
より詳細には、本発明の複合体を作製するための接触は、(a)単離されたSNIまたはその部分ペプチドと単離されたNADaseまたはその部分ペプチドとの接触、(b)SNIまたはその部分ペプチドの発現細胞に対する、NADaseまたはその部分ペプチドの発現ベクターの導入、(c)NADaseまたはその部分ペプチドの発現細胞に対する、SNIまたはその部分ペプチドの発現ベクターの導入、(d)宿主細胞に対する、SNIまたはその部分ペプチドの発現ベクターおよびNADaseまたはその部分ペプチドの発現ベクターの導入、あるいは(e)宿主細胞に対する、SNIまたはその部分ペプチドおよびNADaseまたはその部分ペプチドの同時発現ベクター(即ち、上述の本発明のベクター)の導入などにより達成できる。
【0120】
本発明の複合体およびその製造方法は、例えば、連鎖球菌感染症の治療薬の開発、あるいは本発明の複合体におけるSNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとの解離剤の開発に有用である。従って、本発明はまた、このような治療薬および解離剤の開発を可能とする、複合体の形成を調節し得る物質のスクリーニング方法を提供する。
【0121】
10.スクリーニング方法
本発明は、連鎖球菌感染症の治療薬または本発明の複合体の解離剤の開発を可能とするスクリーニング方法、ならびに当該スクリーニング方法による得られる物質、および当該物質を含む剤を提供する。
【0122】
一実施形態では、本発明のスクリーニング方法は、以下の工程(a)および(b)を含み得る:
(a)被験物質が本発明の複合体の形成を抑制し得るか否かを評価する工程;
(b)本発明の複合体の形成を抑制し得る被験物質を選択する工程。
以下、本スクリーニング方法を、必要に応じてスクリーニング方法Iと省略する。
【0123】
本発明のスクリーニング方法Iの工程(a)で用いられる被験物質は、任意の公知化合物および新規化合物であり得、例えば、有機低分子化合物、コンビナトリアルケミストリー技術を用いて作製された化合物ライブラリー、核酸(例、ヌクレオシド、オリゴヌクレオチド、ポリヌクレオチド)、糖質(例、単糖、二糖、オリゴ糖、多糖)、脂質(例、飽和または不飽和の直鎖、分岐鎖および/または環を含む脂肪酸)、アミノ酸、蛋白質(例、オリゴペプチド、ポリペプチド)、固相合成やファージディスプレイ法により作製されたランダムペプチドライブラリー、あるいは微生物、動植物、海洋生物等由来の天然成分等が挙げられる。
【0124】
工程(a)は被験物質による本発明の複合体の形成の抑制を評価可能である限り如何なる様式でも行われ得るが、例えば、再構成系(即ち、非細胞系)、細胞系(例、ツーハイブリッドシステム、酵母、細菌等を用いる細胞表層工学的手法)を用いて行われ得る。この場合、工程(a)は、例えば、以下の工程(a1)、(a2)を含み得る:
(a1)被験物質、SNIまたはNADaseとの結合能を有するその部分ペプチド、およびNADaseまたはSNIとの結合能を有するその部分ペプチドを接触させる工程;
(a2)上記(a1)により形成された本発明の複合体量を測定し、該複合体量を被験物質の不在下で形成された本発明の複合体量と比較する工程。
【0125】
上記工程(a1)では、接触は、本発明の複合体の形成を可能とする自体公知の方法により行なわれ得る。また、被験物質、SNIまたはその部分ペプチド、およびNADaseまたはその部分ペプチドの接触は同時に行われても、順次行われてもよい。例えば、先ず、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを接触させて本発明の複合体を形成させた後、被験物質を本発明の複合体と接触させてもよい。このような態様もまた、本工程で好ましく行われ得る。
【0126】
上記工程(a2)では、先ず、被験物質の存在下で形成された本発明の複合体量が測定される。複合体量の測定は、自体公知の方法により行うことができ、例えば、免疫学的手法(例、免疫沈降法、ELISA)、表面プラズモン共鳴を利用する相互作用解析法(例、BiacoreTMの使用)、ツーハイブリッドシステム、酵母、細菌等を用いる細胞表層工学的手法が挙げられる。
【0127】
次いで、被験物質の存在下で形成された複合体量が、被験物質の不在下で形成される複合体量と比較される。複合体量の比較は、好ましくは、有意差の有無に基づいて行なわれる。被験物質の不在下で形成される複合体量は、被験物質の存在下で形成された複合体量の測定に対し、事前に測定した複合体量であっても、同時に測定した複合体量であってもよいが、実験の精度、再現性の観点から同時に測定した複合体量であることが好ましい。
【0128】
本発明のスクリーニング方法Iの工程(b)では、本発明の複合体量を減少させる(複合体の形成を抑制する)被験物質が選択され得る。このように選択された被験物質は、例えば、NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療のため、あるいは本発明の複合体を解離させるために有用である。
【0129】
本発明のスクリーニング方法Iが連鎖球菌感染症(例えば、上述した連鎖球菌感染症)の治療薬の開発を特に意図して行われる場合、対象となり得る連鎖球菌は、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌であり得る。連鎖球菌は、NADaseをSNIと共発現することにより、自身をNAD枯渇から保護していると考えられる。この知見に基づけば、連鎖球菌内のSNI・NADase複合体を解離させる物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。NADase遺伝子およびSNI遺伝子を併有する連鎖球菌としては、例えば、A群、C群およびG群連鎖球菌が挙げられる。
【0130】
また、本発明のスクリーニング方法Iが連鎖球菌感染症の治療薬の開発を意図して行われる場合、より生理条件を反映させるという観点から、NADaseとの結合能を有するSNIの部分ペプチドは、さらにNADase阻害活性を保持することが好ましい。同様の観点から、SNIとの結合能を有するNADaseの部分ペプチドは、さらにNADase活性を保持することが好ましい。より好ましくは、本発明のスクリーニング方法Iは、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌由来のNADaseおよびSNIを用いて行われ得る。
【0131】
別の実施形態では、本発明のスクリーニング方法はまた、以下の工程(a)および(b)を含み得る:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
以下、本スクリーニング方法を、必要に応じてスクリーニング方法IIと省略する。
【0132】
本発明のスクリーニング方法IIの工程(a)で用いられる被験物質は、上述のスクリーニング方法Iで用いられるものと同様であり得る。
【0133】
工程(a)は、上述したNADase活性について、被験物質による抑制の解除を評価可能である限り如何なる様式でも行われ得るが、例えば、再構成系(即ち、非細胞系)、細胞系(例、酵母、細菌等を用いる細胞表層工学的手法)を用いて行われ得る。この場合、工程(a)は、例えば、以下の工程(a1)、(a2)を含み得る:
(a1)被験物質、基質、SNIまたはNADase阻害活性を有するその部分ペプチド、およびNADaseまたはNADase活性を有するその部分ペプチドを接触させる工程;
(a2)被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下でNADaseまたはNADase活性を有するその部分ペプチドのNADase活性を測定し、該活性を被験物質の不在下およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下での該NADase活性と比較する工程。
【0134】
上記工程(a1)では、接触は、NADase活性の検出を可能とする自体公知の方法により行なわれ得る。また、被験物質、基質、SNIまたはその部分ペプチド、およびNADaseまたはその部分ペプチドの接触は活性の測定に適切である限り同時に行われても、順次行われてもよい。基質としては、例えば、NADが用いられ得る。
【0135】
上記工程(a2)では、先ず、被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下でのNADase活性が測定される。NADase活性の測定は、自体公知の方法により行うことができる。例えば、NADase活性は、基質の減少量、または検出可能な生成物の増加量に基づき評価できる。基質から変換され得る検出可能な生成物としては、例えば、ADPR、ニコチンアミド、cADPRが挙げられる。NADase活性の測定については、例えば、FEMS Microbiol Lett 136, 71-78 (1996); J. Exp. Med. 104: 577-587 (1956); Appl. Microbiol. 14: 391-393 (1966) を参照のこと。
【0136】
次いで、被験物質およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下で測定されたNADase活性が、被験物質の不在下およびSNIまたはNADase阻害活性を有するその部分ペプチドの存在下で測定されたNADase活性と比較される。NADase活性の比較は、好ましくは、有意差の有無に基づいて行なわれる。被験物質の不在下で測定されるNADase活性は、被験物質の存在下で測定されたNADase活性の測定に対し、事前に測定したNADase活性であっても、同時に測定したNADase活性であってもよいが、実験の精度、再現性の観点から同時に測定したNADase活性であることが好ましい。
【0137】
本発明のスクリーニング方法IIの工程(b)では、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得る被験物質が選択され得る。このように選択された被験物質は、例えば、NADaseおよびSNI遺伝子を併有する連鎖球菌による感染症の治療のため、あるいは本発明の複合体の解離のために有用である。
【0138】
本発明のスクリーニング方法IIが連鎖球菌感染症(例えば、上述した連鎖球菌感染症)の治療薬の開発を特に意図して行われる場合、対象となり得る連鎖球菌は、NADase遺伝子およびSNI遺伝子を併有する上述の連鎖球菌であり得る。連鎖球菌は、NADaseをSNIと共発現することにより、自身をNAD枯渇から保護していると考えられる。この知見に基づけば、SNIまたはNADase阻害活性を有するその部分ペプチドによる、NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得る物質をスクリーニングすることで、連鎖球菌感染症の治療薬が開発可能になると考えられる。好ましくは、本発明のスクリーニング方法IIは、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌由来のNADaseおよびSNIを用いて行われ得る。
【0139】
本明細書中で挙げられた特許および特許出願明細書を含む全ての刊行物に記載された内容は、本明細書での引用により、その全てが明示されたと同程度に本明細書に組み込まれるものである。
【0140】
以下に実施例を挙げ、本発明を更に詳しく説明するが、本発明は下記実施例等に何ら制約されるものではない。なお、「材料および方法」から実施例9まではSNIおよびNADase関連発明に関する実施例であり、実施例10〜11はSLO関連発明の実施例である。
【実施例】
【0141】
材料および方法
(1)材料
ムタノリシン(Mutanolysin)およびリゾチームは、Sigma Chemical Co. (St. Louis, MO. USA) から購入した。アンピシリンナトリウムおよびL(+)-アラビノースは、Wako Pure Chemical Industries, Ltd. (Osaka, Japan) から入手した。DNA修飾および制限酵素は、Takara Shuzo Co., Ltd. (Kyoto, Japan)、Toyobo Co., Ltd. (Osaka, Japan)、またはNippon gene (Toyama, Japan) から得た。タンパク質分子標品は、Bio-Rad (California, USA) およびAmersham Biosciences Corp. (NJ, USA) から購入した。酵母抽出物およびトリプトンは、Difco Laboratories (Detroit, Mich. USA) から購入した。西洋ワサビペルオキシダーゼ・コンジュゲート・Ni-NTAは、Qiagen (Hilden, Germany) から入手した。他の全ての化学物質は、試薬グレードまたは分子生物学グレードであった。
【0142】
(2)菌株および培養条件
Streptococcus dysgalactiae subsp. equisimilis H46A (Christensen, L.R. (1945) J Gen Physiol 28, 363-383) は、1% 酵母抽出物 (yeast extract) を補充したTodd Hewitt broth (Becton Dickinson, Cockeysville, Md. USA) (THY培地) 中で37℃にて増殖させた。Escherichia coli TOP10 (Invitrogen, Frederick, USA) およびE.coli BL21 (Novagen, Madison, Wisconsin) を、組換えプラスミドのクローニング用宿主として用いた。これらの株は、Luria-Bertani Broth (LB) 中で37℃にて培養した。Escherichia coli TOP10は、L-アラビノースの輸送能を有するが、その代謝能は有しない。エレクトロポレーションを、プラスミドDNAのE.coli TOP10およびBL21への導入のために用いた (J Biochem (Tokyo) 122: 237-242 (1997))。形質転換体を、100μg/mlのアンピシリンを含有するLBプレート上で選択した。
【0143】
(3)ポリメラーゼ連鎖反応(PCR)
Expand High Fidelity PLUS PCR Systemは、Roche Diagnostics (Penzberg, Germany) からの製品である。酵素およびバッファーを製造業者の推奨に従って用いた。PCR混合液の総量は約50μlであった。通常通り、初期変性 (94℃で90秒)、鎖の変性 (94℃で30秒)、アニーリング (55℃で30秒)、および伸長 (72℃で1kbあたり60秒) の条件下で40サイクルの増幅が行われた。
【0144】
(4)DNA操作
E.coli TOP10のプラスミドDNAを、QIA prep Spin Miniprep Kit (Qiagen, Hilden, Germany) を用いて抽出した。PCR用のstreptococciゲノムDNAを既報 (Biochim Biophys Acta 1681: 134-149 (2005)) に従い調製した。
【0145】
(5)プラスミドおよび合成オリゴヌクレオチドプライマー
プラスミドpBAD/Hisシリーズは、Invitrogen (Groningen, The Netherlands) から購入した。nga-orf1-sloオペロンのnga-orf1領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより、S. dysgalactiae subsp. equisimilis H46Aの染色体DNAから増幅した。
5’-CGGGATCCGTTAGTGGCAAAGAAGGTAAAAAAAGCG-3’(下線部はBamHI部位:配列番号7)
5’-GGGGTACCCTAAAATGTTTCTATTGTTCTTTCGAC-3’(下線部はKpnI部位:配列番号8)
次いで、このPCR産物をBamHIおよびKpnIで消化し、BglIIおよびKpnIで前処理したpBAD/HisBに連結し、プラスミドpBAD/HisIを作製した(図2A)。
同様に、nga-orf1-sloオペロンのnga領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより増幅した。
5’-CGGGATCCGTTAGTGGCAAAGAAGGTAAAAAAAGCG-3’(下線部はBamHI部位:配列番号9)
5’-GGGGTACCTTACTTCCTATCTTGCATTTTCTTAATTTTG-3’(下線部はKpnI部位:配列番号10)
次いで、このPCR産物をpBAD/HisBに連結し、プラスミドpBAD/HisIIを作製した(図2A)。
プラスミドpQE30は、Qiagen (Hilden, Germany) から購入した。nga-orf1-sloオペロンのorf1領域を、以下のオリゴヌクレオチドプライマーを用いてPCRにより、S. dysgalactiae subsp. equisimilis H46Aの染色体DNAから増幅した。
5’-CGGGATCCTATAAGGTGCCAAAGGGTTTAGAAC-3’(下線部はBamHI部位:配列番号11)
5’-GGGGTACCCTAAAATGTTTCTATTGTTCTTTCGAC-3’(下線部はKpnI部位:配列番号12)
次いで、このPCR産物をBamHIおよびKpnIで消化し、BamHIおよびKpnIで前処理したpQE30に連結し、6個の連続するヒスチジンタグに融合したOrf1を作製した。
【0146】
(6)DNAシークエンシングおよび配列データ解析
ヌクレオチド配列は、適切なプライマーおよびABI PARISMTM 3100 genetic analyzer (PE Applied Biosystems, California, USA) を用いてジデオキシ法により決定した。ヌクレオチド配列データを、GENETYXコンピュータソフトウェア (Software development Co. Ltd., Tokyo, Japan) を用いて解析した。染色体DNA配列データベースを、BLASTN検索アルゴリズム(http://www.ncbi.nlm.hih.gov/BLAST/) を用いて検索した。
(7)ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)
酵素のSDS-PAGEを、Laemmliの方法 (Nature 227, 680-685 (1970)) により還元条件下で行った。タンパク質バンドを、Coomassie Brilliant Blue (CBB) R-250で染色した。E.coliの全細胞溶解物 (whole cell lysate) を調製するため、0.5 mlの培養物を遠心分離 (16,000×g, 3分, 4℃) し、得られた細菌ペレットを0.1 mlの1×サンプルバッファー (組成:1% SDS, 1% 2-メルカプトエタノール, 20% グリセロール, 50 mM Tris-HCl, pH 6.8) で溶解し、この溶解物を100℃で3分間インキュベートした。残存した細菌粒子および非溶解細胞を、遠心分離 (16,000×g, 5分, 室温) により除去した。同様に、5 mlのS. dysgalactiae subsp. equisimilis H46A培養物を遠心分離 (4,000×g, 15分, 4℃) し、得られた細菌ペレットを、溶解バッファー (組成: 100 U/mlムタノリシン, 10 mg/mlリゾチーム, 50 mMグルコース, 10 mM EDTA,25 mM Tris-HCl, pH 8.0) 中で37℃にて30分間インキュベートした。インキュベーション後、細胞を遠心分離 (4,000×g, 5分, 4℃) により回収し、ペレットを0.1 mlの超純粋中に懸濁させた。次いで、0.1 mlの2×サンプルバッファーを加え、懸濁液を100℃で3分間インキュベートした。残存した細菌粒子および非溶解細胞を遠心分離 (16,000×g, 5分, 室温) により除去した。
【0147】
(8)NH2末端アミノ酸配列
E.Coli BL21中で過剰発現させたインタクトOrf1 (成熟型, Hisタグなし) およびS. dysgalactiae subsp. equisimilis H46Aから高度に精製されたインタクトOrf1を12.5%および15% SDS-PAGEに供し、PVDF膜に転写した。PVDF膜をCBB R-250により染色し、19kDaのタンパク質バンドを膜から切り出した。2つのバンドのタンパク質シークエンシングを、PE Applied Biosystems model 491 Procise protein sequencing system (PE Applied Biosystems, California, USA) を用いた自動化エドマン分解により行った。
【0148】
(9)NADaseおよびOrf1の過剰発現
プラスミドpBAD/HisB (ベクターのみ)、pBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1) またはpBAD/HisII (Hisタグ融合NADase単独) を担持するE.coli TOP10の一晩培養物を、新鮮培地中に1:20で希釈し、OD660が0.5に達するまで増殖させた。次いで、培養物を0.002%または0.02% L-アラビノースで4時間誘導した。細胞を遠心分離 (5,000×g, 10分, 4℃) により採取し、リン酸緩衝化生理食塩水で洗浄し、BugBusterTMタンパク質抽出試薬 (Novagen, Madison, Wisconsin) 中に室温にて再懸濁した。BugBuster試薬を製造業者の推奨に従い用いた。タンパク質抽出後、不溶性細胞片を遠心分離 (12,000×g, 30分間) により除去し、上清をタンパク質源として用いた。同様に、組換えプラスミドpQE30 (Hisタグ融合Orf1) を担持するE.coli BL21の一晩培養物を、新鮮培地中に1:20で希釈し、OD660が0.5に達するまで増殖させた。次いで、培養物を1 mM IPTGで3時間誘導し、Hisタグ融合Orf1をBugBuster試薬を用いて抽出した。
【0149】
(10)TSKgel BioAssist Ni2+キレートカラム
0.01% アジ化ナトリウムを含有する50 mMリン酸ナトリウムバッファー (pH 7.4) (バッファーA) で予め洗浄したTSKgel BioAssistキレートカラム (Tosoh Co., Tokyo, Japan) に、4 mlの0.1 M Ni2SO4溶液をロードし、次いで500 mMイミダゾールを含有する10 mlのバッファーAで洗浄し、PEEK型PPCMポンプを備えたTosoh HPLCシステム (Tosoh Co., Tokyo, Japan) を用いて、流速1 ml/分により、20 mMイミダゾールを含有するバッファーAで室温にて平衡化した。
【0150】
(11)Hisタグ融合NADase/Orf1複合体の精製
組換えプラスミドpBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、抽出液3.5 mlを、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムに繰り返しロードした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01%アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。溶出は、流速1.0 ml/分で20℃で行った。タンパク質溶出プロフィールは、UV8000 (Tosoh Co., Tokyo, Japan) を用いて280 nmでの吸光度をモニターすることにより得た。Hisタグ融合NADase/Orf1複合体は、30分間にわたる、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。複合体画分をプールし、バッファーAに対して透析した。
【0151】
(12)インタクトOrf1 (成熟型, Hisタグなし) およびHisタグ融合NADaseの精製
組換えプラスミドpBAD/HisI (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、透析した抽出液を、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムにアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。Orf1を、3 Mチオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する3 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) での溶出により回収した。精製したOrf1を等量の水で希釈し、バッファーAに対して透析した。次いで、カラムに吸着したHisタグ融合NADaseを、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製酵素をプールし、バッファーAに対して透析した。
【0152】
(13)Hisタグ融合Orf1の精製
E.coli細胞のBugBuster抽出物を、2LのバッファーAに対し4℃にて一晩透析した。遠心分離 (10,000×g, 20分) による変性物の除去後、透析した抽出液を、20 mMイミダゾールを含有するバッファーAで平衡化したTSKgel BioAssist Ni2+キレートカラムにアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。Hisタグ融合Orf1を、30分間にわたる、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製したHisタグ融合Orf1をプールし、バッファーAに対して透析した。
【0153】
(14)S. dysgalactiae subsp. equisimilis H46A培養物からのインタクトNADase (成熟型, Hisタグなし) の精製
株H46Aの培養上清に、硫酸アンモニウムを80%飽和となるように加え、得られた沈殿を遠心分離(10,000×g, 20分, 4℃)により回収した。ペレットを0.01%アジ化ナトリウムを含有する0.1 Mリン酸ナトリウムバッファー (pH 7.4) 中に溶解し、同バッファーに対して4℃で一晩透析した。遠心分離 (10,000×g, 20分) により得られた、透析した清澄な上清を、ToyoScreen 5ml column (Tosoh Co., Tokyo, Japan) に充填したヒドロキシアパタイトBio-gel HT gel (Bio-Rad Laboratories, CA) に、流速1.0 ml/分で室温にてアプライした。NADaseを、30分間にわたる、0.01% アジ化ナトリウムを含有する0.1 Mから0.5 Mのリン酸ナトリウムバッファー (pH 7.4) の線形勾配により回収した。活性画分をプールし、バッファーAに対して透析した。透析し、部分精製したNADaseをTSKgel BioAssist Ni2+キレート-Hisタグ融合Orf1アフィニティーカラムにアプライした。このカラムには、1 mgの精製Hisタグ融合Orf1が予めロードされ、20 mMイミダゾールを含有するバッファーAで平衡化されていた。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。インタクトNADaseを、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する3 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) を流速0.5 ml/分でロードすることにより回収した。各画分 (0.5 ml) を0.5 mlの水を含有するチューブ中に回収し、高濃度のNaSCNによるタンパク質の変性を防いだ。NADase活性画分をプールし、バッファーAに対して可能な限り透析した。
【0154】
(15)インタクトNADase (成熟型, Hisタグなし)、Orf1およびNADase/Orf1複合体のゲル透過クロマトグラフィー
TSKgel BioAssist G3SWXLカラム (Tosoh Co., Tokyo, Japan) を用いて、インタクトNADase (成熟型, Hisタグなし) Orf1およびNADase/Orf1複合体のゲル透過クロマトグラフィーを行った。カラムを、0.15 M リン酸ナトリウムバッファー (pH 6.6) により流速0.5 ml/分で室温にて展開した。タンパク質溶出プロフィールを、280 nm の吸光度をモニターすることにより得た。
【0155】
(16)S. dysgalactiae subsp. equisimilis H46Aの細胞内Orf1の解析
溶解溶液 (組成:100 U/ml ムタノリシン, 10 mg/ml リゾチーム, 50mM グルコース, 10 mM EDTA, 25mM Tris-HCl, pH 8.0) 中での消化およびソニケーションにより得た細胞抽出液を、抗Orf1抗体コンジュゲートHiTrap NHS活性化HPカラム (Amersham Biosciences Corp., NJ, USA) にアプライした。カラムの非特異的吸着物を洗浄するために、3 M NaClおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) をアプライし、次いでOrf1を、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する4 mlの0.1 Mリン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収した。Orf1画分をプールし、水に対して透析した。透析物をSpeedvac Concentrator (Sarvant Instrument Inc., NY, USA) を用いて濃縮し、SDS-PAGEに供した。ゲル中のタンパク質をPVDF膜に転写し、CBB R-250で染色した。タンパク質バンドを切り出し、PE Applied Biosystems model 491 Procise protein sequencing systems (PE Applied Biosystems, California, USA) により解析した。
【0156】
(17)抗体
Hisタグ融合Orf1をウサギ抗Orf1抗体の惹起に用いた。簡潔には、完全フロインドアジュバントと混合した約2 mgのHisタグ融合Orf1をウサギに皮下注射した。1 mgのタンパク質および不完全フロインドアジュバントからなるブースター注射を2週間の間隔で3回与えた。イムノグロブリンG画分を、プロテインA-セファロース (Amersham Biosciences Corp., NJ, USA) を用いたアフィニティークロマトグラフィーにより精製した。
ウサギポリクローナル抗SLO IgG (抗NADase抗体含有) は、American Research Product Inc. (Belmont, MA. USA) から購入した。
【0157】
(18)イムノブロット解析(ウエスタンブロット解析)
SDS-PAGE後、タンパク質を、電気ブロッティングによりSequi-Blot PVDF膜 (Bio-Rad, California, USA) 上に転写した。膜を、0.05% Tween 20を含有するリン酸緩衝化生理食塩水 (PBST) において5% スキムミルクおよび1% ウシ血清アルブミン (BSA) で1時間ブロッキングした。膜をPBSTで洗浄し、次いで一次抗体 (PBST中で1:3,000に希釈) を加え、ブロットを1時間インキュベートした。次に、ヤギaffini-pure IgG ペルオキシダーゼ・コンジュゲート・抗ウサギIgG (Wako Pure Chemical Industries, Ltd) をPBST中で1:3,000希釈したものを加え、1時間インキュベートし、抗体抗原複合体を、製造業者により記載(J Immunol Methods 95, 71-77 (1986)) されるようにPOD免疫染色セット (Wako Pure Chemical Industries, Ltd) により可視化した。Ni-NTA HRPコンジュゲート (Qiagen, Hilden, Germany) (西洋ワサビペルオキシダーゼとカップリングしたニッケル-ニトリロトリ酢酸 (Ni-NTA)) を、Hisタグを有する組換えタンパク質の直接的な検出のために使用し、製造業者の推奨に従い使用した。
【0158】
(19)Orf1・細胞外NADase間の相互作用のファーウエスタン解析
S. dysgalactiae subsp. equisimilis H46Aの培養上清および培地単独を、SDS-PAGE (10% アクリルアミド) 上で泳動し、PVDF膜に転写し、精製Hisタグ融合Orf1 (PBST中10μg/ml) の存在下または不在下で室温にて1時間インキュベートした。精製Hisタグ融合Orf1のNADaseへの結合は、「イムノブロット解析」に記載されるような抗Orf1抗体を用いるウエスタンブロット解析によりアッセイした。簡潔には、膜をPBSTで洗浄し、次いでウサギ抗Orf1抗血清を加え、ブロットを1時間インキュベートした。次いで、ヤギaffii-pureIgGペルオキシダーゼ・コンジュゲート・抗ウサギIgGを加え、1時間インキュベートし、抗体抗原複合体をPOD免疫染色により可視化した。培養上清中のNADaseおよび培地単独(コントロール)を、抗NADase抗体を含有する抗SLOポリクローナル抗体によるウエスタンブロット解析によりアッセイした。
【0159】
(20)NADaseアッセイ
NADaseアッセイを、Gerlach et al. (FEMS Microbiol Lett 136, 71-78 (1996)) の方法により行った。1単位の酵素は、37℃では0.010μM NADを7.5分で切断する。
・全細胞抽出物の調製:
対数増殖期の中期培養物 (10 ml) の細胞を、溶解溶液 (組成:100 U/ml ムタノリシン, 5 mg/ml リゾチーム, 0.01% アジ化ナトリウム, 150 mM リン酸ナトリウム, pH 6.6) 中で、37℃にて30分間インキュベートした。インキュベーション後、細胞ソニケートし、溶解物を低温で遠心分離し、上清をNADaseアッセイに用いた。
・温度安定性:
インタクトOrf1 (成熟型, Hisタグなし) の温度安定性を、0.15 M リン酸ナトリウムバッファー (pH 7.4) において38μg/ml (2.0μM) のタンパク質濃度で決定した。Orf1溶液をEppendorfチューブ中に密閉し、20-100℃で15分間インキュベートした。インキュベーションを、溶液を氷水中で冷却することにより停止し、0.5μlの各溶液を、50 mM リン酸カリウムバッファー (pH 7.5) 中に70μg/ml (1μM) のBSAを含有する10μlの5μg/ml (0.1μM) インタクトNADase (成熟型, Hisタグなし) 溶液に加えた。次いで、混合液を室温で10分間インキュベートし (Orf1とNADaseの最終モル比は1:2)、残存したヒドロラーゼ活性をNADで測定した。コントロールとして、インキュベートしたOrf1溶液の代わりに、BSAのみを含有する50mMリン酸カリウムバッファー (pH 7.5) を用いた。
【0160】
実施例1:E.coliでの連鎖球菌NADaseの発現
連鎖球菌NADグリコヒドロラーゼ (NADase) - ストレプトリシンO (SLO) オペロンの調節に関する以前の研究では、ベクターとしてpHY300PLKを使用することにより、E.coliでの遺伝子クラスターのサブクローニングが試みられた。活性SLOの過剰発現はE.coliで達成されたが (Biosci Bisotechnol Biochem 65, 2682-2689 (2001))、NADase遺伝子インサートを有するコロニーは全く得られなかった (Biochim Biophys Acta 1681: 134-149 (2005))。この酵素の潜在的宿主毒性を考慮して、E.coliでの遺伝子発現を厳格に制御するaraBADプロモーター (pBAD) を有する発現ベクターpBADに、NADase遺伝子ngaをサブクローニングした(図2A)。しかし、NADase遺伝子インサートのみを有するE.coli TOP10 (pBAD/HisII) では、L-アラビノース処理による誘導の際に、この酵素タンパク質の産生は生じなかった(図2B)。この組換え株の増殖は誘導剤の不在下でさえも遅く、細胞の一部は凝集した(データ示さず)。一方、ngaからorf1の3’端にわたるより長いインサートを有するpBAD/HisIを担持するE.coli組換え株は、有意な量のNADaseを産生した(図2B)。データベース検索をすると、A群連鎖球菌 (GAS) 株ではOrf1が同様に保存されていたが、他の細菌株では欠失していた(データ示さず)。全長のNADase-SLOオペロンをpBAD制御下に配置し、かつE.coliで発現させた場合、NADaseおよびOrf1に加え、活性SLOが産生された(データ示さず)。
これらの結果は、細胞内連鎖球菌NADaseがE.coliのみではなく、連鎖球菌自体にもおそらく毒性を示すこと、ならびに共発現されたOrf1がその保有菌をNAD枯渇から保護し得ることを示唆する。この可能性を検証するため、NADase-SLOオペロンの未知遺伝子産物Orf1の遺伝的および生化学的性質をその後の実験で調べた。
【0161】
実施例2:Orf1の発現およびそのN末端領域のシークエンシング
E.coliにおいてPBAD制御下で共発現させた場合、SDS-PAGEプロフィールにより判定すると、Orf1の量はNADaseのそれよりも多かった(図3A)。Orf1バンドをゲルから切り出し、N末端アミノ酸シークエンシングに直接供した。得られた配列MYKVPKGLEHYQKM(配列番号15)は、NADase-SLOオペロン中のorf1遺伝子のヌクレオチドアライメントより推定したものと同一であった(図3B)。この遺伝子は483塩基対からなり、推定161アミノ酸をコードしていた。ヌクレオチド配列から、このOrf1タンパク質の分子量は18,800であると推定された。
S. dysgalactiae subsp. equisimilis H46A中のorf1の発現を実証するため、組換えE.coli細胞で産生されたHisタグ融合Orf1を用いてウサギを免疫することにより、抗Orf1抗体を調製した(図4A)。この抗体を用いたウエスタンブロット解析は、Orf1タンパク質が産生され、H46A球菌内に専ら局在したことを示した(図4B)。Orf1の分子量は、E.coli中でNADaseと共発現したタンパク質の分子量と一致した(図4B)。SLOおよびNADaseとは異なり、Orf1は連鎖球菌培養上清中に検出されなかった(図4B)。
【0162】
実施例3:S. dysgalactiae subsp. equisimilis H46A由来のOrf1の精製およびアミノ末端シークエンシング
Orf1タンパク質をS. dysgalactiae subsp. equisimilis H46Aから抽出し、抗Orf1抗体固定化カラムを用いたアフィニティークロマトグラフィーにより精製した(図5)。この溶出画分をSDS-PAGEに供し、PVDF膜上にブロットした。CBB R-250染色により、細胞内Orf1複合体形態におそらく由来する5つのバンドを検出した(図5)。アミノ末端シークエンシングは、19kDaのバンドがOrf1であることを示した:その配列MYKVPKGLEHYQKM(配列番号15)ならびに分子サイズは、ヌクレオチド配列から推定したもの、および組換えE.coli細胞中で発現したOrf1タンパク質のものと同じであった。計算値50 kDaの2個のバンドは、NADaseの前駆体かもしれなかったが、それらのアミノ酸シークエンシングは、おそらくアミノ末端ブロッキングのため、成功しなかった。翻訳後、約2 kDaの領域がNADaseのC末端からタンパク質分解により除去されることが最近見出されている (Biochim Biophys Acta 1681: 134-149 (2005))。約50 kDaのバンドがNADaseである場合、C末端の除去が球菌細胞で生じているに違いない。計算値34 kDaの濃いバンドは、N末端の187個のアミノ酸が欠失したNADaseタンパク質 (ΔN187) であった。この欠失が、異常な転写または球菌抽出物の調製の際のタンパク質分解によって生じるかは不明である。しかしながら、この短縮型分子が抗Orf1抗体固定化カラムに吸着したという事実は、NADaseのN末端領域がOrf1との相互作用に必須ではないことを示す。16 kDaのバンドは連鎖球菌細胞の溶解に使用された卵白リゾチームであった。非生理学的ではあるが、この溶解酵素はインビトロでOrf1、NADaseまたはOrf1-NADase複合体のいずれかと相互作用するようであった。SDS-PAGEプロフィール(図5)から推測されるように、NADase前駆体に対するOrf1の比率は、E.coliでの比率に対し、連鎖球菌ではそれほど高くなかった(図3A)。
NADase-SLOオペロンのメンバーOrf1は、組換えE.coliおよび連鎖球菌株H46Aにおいて同じ開始コドンから翻訳された。E.coliでの連鎖球菌NADaseの産生がOrf1の共発現を必要とするという事実は、潜在的な毒性を示すNADase活性の抑制におけるこのタンパク質の自己保護的な役割を示唆する。従って、NADaseおよびOrf1のインビトロでの物理的および機能的相互作用を解析するために、以下の実験を行った。
【0163】
実施例4:Orf1・NADase間の相互作用の解析
NADaseとの相互作用の解析のため、Hisタグ融合タンパク質としてE.coliで産生されたOrf1を、ニッケル−キレートカラムクロマトグラフィーにより精製した(図4A)。S. dysgalactiae subsp. equisimilis H46Aの培養前後のTHY培地をSDS-PAGEに供し、PVDF膜上にブロットし、2部に分割した。膜の一方を抗SLOポリクローナルIgGを用いるウエスタンブロッティングに直接供し、他の膜は精製Orf1で処理し、次いで抗Orf1抗血清を用いたウエスタンブロッティングに供した(図6)。図4Bに示される通り、連鎖球菌H46A培養物からの消費培地はOrf1およびその交差反応性物質を含有しなかった。NADaseの検出のため、この酵素タンパク質と交差反応する市販の抗SLOポリクローナルIgGを用いた (Cell 104: 143-152 (2001); Biochim Biophys Acta 1681: 134-149 (2005))。
ファーウエスタンブロッティングにより、Orf1は、Orf1処理したPBDF膜で、NADaseバンドに対応するまさにその位置に検出された。この結果はNADase・Orf1間の物理的相互作用を示しているので、NADase活性に対する後者のタンパク質の効果を続いて調べた。
【0164】
実施例5:Orf1によるNADaseの阻害
Orf1は細胞内タンパク質であり、その細胞含量はそれ程高くない(図5)。このタンパク質を過剰産生させるため、NADaseとの共発現を、pBAD/HisIを担持するE.coliで試みた。図4Bおよび図5に示されるように、E.coli由来のOrf1の電気泳動プロフィールおよびN末端配列は、H46A由来のものと同じであった。同時に、E.coliでOrf1と共発現したHisタグ融合NADaseを、ニッケル-キレートカラムクロマトグラフィーにより精製した。共発現したOrf1分子は、予期された通り、NADaseとの複合体として存在し、相互の分離は、3M NaCl処理でさえも達成できなかった(図7A)。3M NaSCNを用いてこの複合体を解離させ(図7B)、ゲル透過クロマトグラフィーによりOrf1をさらに精製した。組換えE.coli抽出物中のNADaseは、Orf1との酵素学的に不活性な複合体として存在した。N末端シークエンシングおよび分子サイズの決定は、このNADaseタンパク質が前駆体形態であることを示す。Orf1から解離した場合、NADase前駆体は分泌成熟形態の酵素の比活性とほぼ同じ比活性を示した(データ示さず)。NADase前駆体は少量であり、かつ分泌成熟酵素もまたOrf1と不活性複合体を形成したので、細胞外形態を以下の実験で用いた。従って、NADaseは、Hisタグ融合タンパク質としてE.coliで過剰産生されたOrf1が固定されたカラムを用いたアフィニティーカラムクロマトグラフィーにより、S. dysgalactiae subsp. equisimilis H46Aの培養上清から精製した(図7C)。精製したNADaseをOrf1と混合し、室温で10分間インキュベートした場合、この酵素活性は当価なモル比においてほぼ完全にブロックされた(図7D)。この阻害反応は非常に迅速であり、また、0℃でさえも生じた(データ示さず)。
連鎖球菌内のNADase前駆体がOrf1に結合した不活性複合体として存在する可能性を検証するため、H46A溶解物を等容量の6M NaSCNと混合した。解離処理後、Orf1を抗Orf1抗血清で中和した。表1に示されるように、活性NADaseは連鎖球菌溶解物中に検出できなかったが、NaSCN、次いで抗Orf1抗血清で処理したサンプル1 g (湿重量) あたり、約104ユニットの活性が認められた。活性化抽出物を水で希釈した場合、NADase活性は再度マスクされた(データ示さず)。このことは、NADaseがOrf1と再会合したことを示唆する。これらの結果は、初期のNADaseが、組換えE.coliにおける場合と同様に、連鎖球菌内でOrf1と不活性複合体を形成することを示す。
【0165】
【表1】
【0166】
a 活性は、3つの実験からの平均±S.D.で示した。
b 全細胞抽出物は、材料および方法における「NADaseアッセイ」に記載の通り調製し、10μlの抽出物を、10μlの0.1 M Na2HPO4(pH 7.4) および80μlの超純粋と混合した。
c 10μlの全細胞抽出物を、10μlの6 M NaSCN, 0.1M Na2HPO4 (pH 7.4) と室温にて混合し、5秒間のボルテックス後、80μlの超純粋を即座に加えた。
d 10μlの全細胞抽出物を、10μlの0.1 M Na2HPO4 (pH 7.4)、1μlの抗Orf1抗血清、および79μlの超純粋と混合した。
e 1μlの抗Orf1抗血清を、10μlの0.1 M Na2HPO4 (pH 7.4) および89μlの超純粋と混合した。
f 10μlの全細胞抽出物を、10μlの6 M NaSCN, 0.1M Na2HPO4 (pH 7.4) と室温にて十分に混合し、5秒間のボルテックス後、1μlの抗Orf1抗血清および79μlの超純粋を即座に加えた。
【0167】
実施例6:NADase・Orf1複合体の分子サイズの決定
NADase・Orf1複合体の分子サイズを決定するため、TSKgel BioAssist G3SWXLカラム (Tosho Co., Tokyo, Japan) を用いて、ゲル透過クロマトグラフィーを行った。図8Aに示されるように、NADase、Orf1およびNADase・Orf1複合体の各々を、明瞭な単一ピークとしてそれぞれ検出した。各保持時間から見積もった分子量は、24 kDa (Orf1)、54 kDa (NADase)および69 kDa (NADase・Orf1複合体) であった。これらの値(図8B)は幾らか高いが、各ヌクレオチド配列から推定した値に近似していた。これらの結果は、NADaseおよびOrf1がモノマータンパク質として存在し、混合した場合に、ヘテロダイマーとしての複合体を形成することを示す。
【0168】
実施例7:温度安定性およびプロテアーゼ感受性
これまでに記載した細菌NADaesは、細胞内酵素であり、それらの阻害タンパク質は熱に不安定である(Methods Enzymol 66; 137-144 (1980); Science 123: 50-53 (1956))。興味深いことに、Orf1は熱に安定であり、15分間の熱処理後に残存する相対阻害活性は、60℃では計算値で82%、100℃では計算値で72%であった(図9)。精製したOrf1はトリプシンに感受性であり、リシルエンドペプチダーゼにより不活化された(データ示さず)。
【0169】
実施例1〜7に関する考察
NADase-ストレプトリシンOオペロンは、AおよびC群の連鎖球菌で高度に保存されている (Cell 104: 143-152 (2001); Biochim Biophys Acta 1681: 134-149 (2005); Infect Immun 70: 2730-2733 (2002))。Ngaおよびsloに加えて、このオペロンは、未知遺伝子orf1を含んでいた。上記データは、この遺伝子が連鎖球菌NADaseインヒビター (streptococcal NADase inhibitor:SNI) として機能する18.8 kDaタンパク質をコードすることを示す。従って、この遺伝子をsniと標記することは適切であると考えられる。E.coliでの単一ngaのサブクローニングの失敗 (Biochim Biophys Acta 1681: 134-149 (2005)) とは対照的に、この遺伝子のorf1 (sni) との共発現は、nga-orf1インサートを有する組換えプラスミドを担持する異種宿主において安定的に生じた。菌体内での活性NADaseの産生は、酸化還元に必須のNADの分解による毒性効果を引き起こし得る。この潜在的に有害な酵素に対する細菌性インヒビターの存在は、Bacillus subtilis、Proteus vulgaris、Proteus rettgeriおよびMycobacterium butyricumで知られている。これらの細菌のNADaseは、熱不安定性のインヒビターと複合体化した細胞内熱安定タンパク質である (Methods Enzymol 66: 137-144 (1980); Science 123: 50-53 (1956))。これらのインヒビターの遺伝的および機能的調査はこれまでに行なわれていない。一方、溶血性連鎖球菌は、培養培地中に熱不安定性NADaseを分泌する (J Exp Med 104: 577-587 (1956); J Exp Med 106: 15-26 (1957); FEMS Microbiol Lett 136: 71-78 (1996))。細胞外NADaseを産生する連鎖球菌細胞は、この酵素に対するインヒビタータンパク質を含む (Acta Path. Microbiol. Scandinav. 69: 277-286 (1967))。細胞内局在、分子量およびトリプシン感受性は、SNIのそれらに準じる。しかし、熱安定性に関しては、HolmおよびKaijser (Acta Path. Microbiol. Scandinav. 69: 277-286 (1967)) により報告されるインヒビターは、SNIとは異なる。それらの調製は、60℃で15分間加熱することにより完全に不活化されるのに対し、SNIはむしろ熱安定性であった(図9)。
【0170】
SNIをコードする遺伝子 (sni) は、nga (spn) プロモーターから、ポリシストロニックmRNAとして、sloと共転写される (Biochim Biophys Acta 1681: 134-149 (2005))。nga-sloオペロンの翻訳の際、NADase前駆体の自己毒性は、モル比1:1での、SNIとの複合体の形成により迅速にブロックされるようである(図10)。ウエスタンブロット解析より実証されるように、SNIの局在は完全に細胞内である(図4B)。この結果と一致して、シグナルペプチド領域は、SOSUI (Bioinformatics 14: 378-379 (1998); Bioinformatics 18: 608-616 (2002)) およびSignalP (Int J Neural Dsyst 8: 581-599 (1997)) 膜貫通推定アルゴリズムを用いるインシリコ解析では、SNIに見出されなかった。球菌体内のNADase前駆体およびSLOの量はわずかであった(データ示さず)。このことは、合成されたタンパク質が培地中へと迅速に分泌されることを示唆する。しかし、NADase前駆体は安定な細胞内複合体を形成するので、この複合体は、球菌エンベロープを介する排出中に解離するはずである。この複合体の解離が幾つかの膜成分との相互作用により、SNIのタンパク質分解排除により、または他の機構により引き起こされるか否かは、解明すべき課題である。
【0171】
実施例8:Orf1によるCD38、CD157の阻害
ヒト由来CD38およびCD157の遺伝子を組み込んだpQE30-His-タグ-CD38およびpQE30-His-タグ-CD157で形質転換した大腸菌を1リットルのアンピシリンを含むLB液体培地で培養し、1mM IPTGを加えて3時間37℃で培養した菌体を遠心分離で集めた。菌体に20 mlのBugBuster (菌体内タンパク質抽出液) を加えて、His-タグ-CD38およびHis-タグ-CD157をそれぞれ抽出した。この原液をTSKgel BioAssist Ni-キレートクロマトグラフィーに供して、His-タグ-CD38およびHis-タグ-CD157をそれぞれ部分精製した。そのNADase活性を有する画分を酵素試料 (20units/ml、注意:CD38, 157とも菌に対する毒性が強くわずかしか発現していない。そのためキレートカラムを用いた上記の精製でも純粋なものは得られていないので比活性 (units/mg) として表すことができず、units/mlで代用している) として用いた。酵素溶液20units/mlに等量のAssay Mixture (2μM SNIを含む) を加えて反応を開始した。反応は20℃で5分間行った。すなわち酵素活性は10units/mlに薄まり、SNIは1μMになっている。この条件でNADase活性はほぼ100%阻害された(図11および12参照)。
以上より、Orf1は、ヒト由来CD38およびCD157を阻害することが示された。
【0172】
実施例9:無細胞系によるNADaseタンパク質合成
本発明者らは、溶血性連鎖球菌由来NADaseが強い細胞毒性を示すことから、無細胞系によるin vitroタンパク質合成を試みた。無細胞系としては、RTSスケラーブル・セルフリータンパク質発現システム(ロシュ製)を、その使用説明書に従って用いた。その結果、無細胞系でも、NADaseタンパク質を合成できなかった。
宿主細胞内における、毒素としてのNADaseの機能に関しては、未だに殆どのことが明らかにされていない。しかしながら、NADは生体内に最も大量に存在する補酵素であり、エネルギー産生に非常に重要であることから、NADaseによる細胞内NAD濃度の減少がタンパク質の生合成を停止させると考えられる。実際、DNA損傷によりポリADP-リボース合成酵素が過剰に活性化されると、その基質であるNADの細胞内濃度が低下することにより細胞死を引き起こすことはよく知られている事実である。
無細胞系はNADを含む。NADは翻訳系に直接必要ではないが、NAD濃度が低下するとエネルギー産生系に影響を及ぼし、結果としてATP等の濃度も減少するので、タンパク質合成が低下する。従って、上述したように宿主細胞を用いた場合は勿論のこと、無細胞系を用いてもやはりNADaseタンパク質の合成は困難であると考えられ、また、本実施例の結果はこのような考えを支持する。
以上より、本発明者らが開発した共発現ベクター、それが導入された形質転換体、および該形質転換体を用いるNADaseタンパク質の製造方法は極めて有用と考えられる。また、これらを利用することで、連鎖球菌由来NADaseの非天然型タンパク質の製造も初めて可能になると考えられる。
【0173】
実施例10:大腸菌における完全長SLOの大量発現
本発明者らによるSLO遺伝子のプロモーター解析 (Biochim Biophys Acta 1681: 134-149 (2005)) からの知見より、SLO遺伝子は自身のプロモーターを持たず、基本的に上流に位置しているNADase遺伝子 (nga) のプロモーターからのreadthroughにより転写が行われるオペロンの一部であることが判明している。そこで、本発明者らは、SLO遺伝子が自身のプロモーターを持っていないこと、およびSLOの細胞毒性を考慮して、遺伝子の発現を大腸菌で厳密に制御できるaraBAD プロモーター (PBAD) を利用した発現ベクターにより、Hisタグを融合させた完全長SLOの大量発現を試みた。Hisタグ法で付加されるアミノ酸配列は短く、多くの場合除去する必要がない。また、特異抗体や精製担体も複数の市販品が入手可能でありコストが安いなどの利点がある。さらに、尿素や塩酸グアニジンのようなタンパク質の変性剤の存在下でも精製が可能で、カラムに結合させたままの状態でリフォールディングできるなどの大きな利点がある (Protein Expr Purif 41: 98-105 (2005))。
【0174】
先ず、本発明者らは、医薬品としてすでに使われているストレプトキナーゼやストレプトドルナーゼの工業生産株としてよく知られているStreptococcus dysgalactiae subsp. equisimilis H46A (J Gen Physiol 28: 363-383 (1945)) の染色体DNAからPCR法でSLO遺伝子を増幅し、大腸菌での遺伝子発現を極めて厳密に実行するためHisタグタンパク質発現ベクターpBAD/HisB (Invitrogen) にクローニングした(図13)。pBAD/His BのBamHIおよびKpnI部位に、シグナルペプチド配列を含むN末端側の31アミノ酸残基 (3.3 kDa) を除いた540アミノ酸残基 (60.4 kDa) に相当する完全長 SLO遺伝子の領域をin frameで挿入した。この組み換えプラスミドを大腸菌TOP10株 (L-アラビノース代謝能欠損株) に導入し、遺伝子発現の誘導物質であるL-アラビノースによるHisタグ融合完全長SLOの発現実験を行った。その結果、培地中のL-アラビノースの濃度が0.002% (w/v) からSLOの十分な発現が確認された(図14)。イムノブロット解析の結果、大腸菌で発現したSLOの分子量は約70 kDaと推定され、ベクター側のHisタグを含む4.1 kDaの領域を合わせて予想される64.5 kDaよりも少し大きかった。SLOの電気泳動解析から求めた分子量が予想値よりも増大することは、Gerlachらも報告している (Infect Immun 61: 2727-2731 (1993))。
次に、本発明者らは、大腸菌をTOP10株から、lonおよびompTの両プロテアーゼ欠損株であるBL21株に変更してSLOを大量発現させた。精製には市販のTSKgel BioAssist Ni2+-chelate column (Tosoh Co., Tokyo, Japan) を用い、このアフィニティーカラムクロマトグラフィーにより一段階で高純度のSLOを精製した(図15)。Hisタグ融合完全長SLOの発現量は、タンパク質量として、LB培地1リットルあたり約6 mg(活性タンパク質量では約3 mg)の収量であり、その比活性は748,000 HU/mgタンパク質でGerlach らがStreptococcus dysgalactiae subsp. equisimilis H46Aの培養上清から精製したSLOの750,000 HU/mgとほぼ一致した (Infect Immun 61: 2727-2731 (1993))。なお、本実験で得られたタンパク質量は上記の通りであったが、培養条件の適切な変更(例、培地の至適化による菌密度の向上、および誘導物質による誘導時間の延長)により少なくとも約10-30 mgのタンパク質量を得ることが可能と考えられる。
【0175】
実施例11:大腸菌における完全長SLOの大量発現
現在市販されている抗ストレプトリジンO (ASLO) 抗体は溶血性連鎖球菌の培養上清から部分精製したSLOを抗原として用いているため、同じく培養上清中に分泌されるNADグリコヒドロラーゼ (NADase) と交差反応することが知られている (Biochim Biophys Acta 1681: 134-149 (2005); Cell 104: 143-152 (2001))。そこで、本発明者らは、精製したSLOをウサギに免役することによりASLOを作製し、GAS Sa株および GCS H46A株の培養上清をイムノブロット解析することによりASLOの抗原特異性を評価した(図16)。
その結果、本発明者らが作製したASLOにより、GCS H46A株の培養上清中のSLOが明確なバンドとして高感度で検出された。メジャーバンドの下に見える2本の弱いバンドは、低分子化したSLO分子と思われる。一方、GAS Sa株ではSLOを検出できなかった。これは、GASのSLO産生量は一般に少なく、また自身が産生するプロテアーゼにより速やかに低分子化するためと考えられる。実際、本発明者らは、GAS Sa株におけるSLOの検出や精製は困難であることを報告している (Kimoto et al., Biosci Biotechnol Biochem 67: 2203-2209 (2003); Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005))。なお、このASLOは、大腸菌から精製したHisタグ融合完全長SLO、および溶血性連鎖球菌の産生したSLOの溶血活性を、いずれも中和した (データ示さず)。
次にASLOのNADaseとの交差反応を調べるために、GAS Sa 株および GCS H46A株の培養上清を用いて、抗NADase抗血清によるイムノブロット解析を行った。
その結果、GAS Sa株および GCS H46A株の両培養上清中にNADaseを確認した。両者間でNADaseの分子量がやや異なるのは、GCS H46A株が産生するNADaseは翻訳後にC末端領域が分解されるためである (Kimoto et al., Biochim Biophys Acta 1681: 134-149 (2005))。いずれにしても、SLOとNADaseは予想された分子量でバンドが検出され、両者間の大きさは明らかに異なっていた。また、免疫していないコントロール血清では、いずれのバンドも検出されなかった。
以上より、今回作製したASLOは、これまでの市販品とは異なり、SLOを特異的かつ高感度に検出し得ることが示された。
【図面の簡単な説明】
【0176】
【図1】NADaseの活性の様式を示す図である。
【図2A】S. dysgalactiae subsp. equisimilis H46AのNADaseストレプトリシンOオペロンの模式図である。プロモーターおよびターミネーターをそれぞれ、「P」および「T」で示している。「orf1(sni)」で表示された矢印は、データベース中の如何なる遺伝子にも類似していなかった。NADase発現プラスミドpBAD/HisIおよびIIはそれぞれ、NADaseからorf1までの染色体領域およびNADase領域のみを含む。
【図2B】E.coli TOP10全細胞溶解物における連鎖球菌NADaseのウエスタンブロット解析を示す図である。E.coli TOP10細胞を、6個の連続するヒスチジン残基 (Hisタグ) を含むように改変された、示された領域をコードする発現プラスミドで形質転換し、L-アラビノースを用いて誘導し、次いでNADaseの合成についてアッセイした。pBAD/His B (ベクターのみ) を担持するE.coli TOP10の溶解物をコントロールとして用いた。NADaseの相対発現レベルを決定するため、全細胞溶解物をウエスタンブロット解析に供した。
【図3A】H46AのOrf1タンパク質の検出を示す図である。pBAD/His Iを担持する組換えE. coli TOP10の全細胞溶解物中のタンパク質をSDS-PAGEにより解析した。NH2末端アミノ酸酸列を括弧で示す。
【図3B】H46Aのorf1のヌクレオチド配列および推定アミノ酸配列を示す図である。下線を付したアミノ酸は、Orf1のNH2末端アミノ酸シークエンシングにより決定した。
【図4A】Hisタグ融合Orf1の精製を示す図である。組換えプラスミドpQE30 (Hisタグ融合Orf1) を担持するE.coli BL21細胞からの抽出物を、TSKgel BioAssist Ni2+キレートカラム (Tosoh Co., Tokyo, Japan) にアプライした。Hisタグ融合Orf1を、イミダゾールの20-250 mM の線形勾配により回収し、各溶出画分をSDS-PAGEにより調べた。
【図4B】組換えE.coli細胞およびS. dysgalactiae subsp. equisimilis H46A培養上清中のorf1産物のウエスタンブロット解析を示す図である。Orf1の発現を確認するため、全細胞溶解物および培養上清を、上記の通り精製したOrf1を用いた免疫により得られるウサギ抗Orf1抗血清を用いて、ウエスタンブロット解析に供した。
【図5】H46A由来のOrf1の精製およびアミノ末端シークエンシングを示す図である。H46A由来の全細胞抽出物を、抗Orf1抗体・コンジュゲート・HiTrap NHS活性化HPカラム (Amercham Biosciences Corp., NJ, USA) にアプライし、Orf1を、3 M チオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する4 ml の0.1 M リン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収した。Orf画分をプールし、水に対して透析し、次いで濃縮し、SDS-PAGEに供した。ゲル中のタンパク質をPVDF膜に転写し、CBB R-250で染色した。タンパク質バンドを切り出し、PE Applied Biosystems model 491 Procise protein sequencer (Foster City, CA, USA) で解析した。
【図6】H46AのOrf1・細胞外NADase間の相互作用のファーウエスタン解析。H46Aの培養上清および培地単独を、SDS-PAGE上で泳動し、PVDF膜に転写し、精製Hisタグ融合Orf1の存在下または非存在下でインキュベートした。培養上清中のNADaseを、抗NADase抗体を含有する抗SLOポリクローナルIgGによるウエスタンブロット解析によりアッセイした(左パネル)。精製Hisタグ融合Orf1のNADaseへの結合を、抗Orf1抗血清を用いてファーウエスタン解析によりアッセイした(右パネル)。
【図7A】TSKgel BioAssist Ni2+キレートカラムを用いたHisタグ融合NADaseおよびインタクトOrf1 (成熟型, Hisタグなし) の共精製を示す図である。組換えプラスミドpBAD/His I (Hisタグ融合NADaseおよびインタクトOrf1をコード) を担持するE.coli TOP10由来の細胞抽出物を、TSKgel BioAssist Ni2+キレートカラムにアプライし、Hisタグ融合NADase/ORF1複合体を、イミダゾールの20-250 mM線形勾配により回収した。精製したNADase/Orf1複合体画分をプールし、透析し、SDS-PAGEに供した。
【図7B】TSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによるインタクトOrf1の精製を示す図である。組換えプラスミドpBAD/His I (Hisタグ融合NADaseおよびインタクトOrf1をコード)を担持するE.coli TOP10由来の細胞抽出物を、TSKgel BioAssist Ni2+キレートカラムにアプライした。Orf1を、3 M チオシアン酸ナトリウム (NaSCN) および0.01% アジ化ナトリウムを含有する3 ml の0.1 M リン酸ナトリウムバッファー (pH 7.4) での溶出により回収した。次いで、カラムに吸着したHisタグ融合NADaseを、50 mM リン酸ナトリウムバッファー (pH 7.4) におけるイミダゾールの20-250 mM 線形勾配により回収した。精製酵素をプールし、0.01% アジ化ナトリウムを含有する50 mM リン酸ナトリウムバッファー (pH 7.4) に対して透析し、SDS-PAGEに供した。
【図7C】Hisタグ融合Orf1が固定されたカラムを用いたアフィニティークロマトグラフィーによるS. dysgalactiae subsp. equisimilis H46Aの培養上清からのインタクトNADase (成熟型, Hisタグなし) の精製を示す図である。硫酸アンモニウム沈殿および透析後、上清からのタンパク質を、TSKgel BioAssist Ni2+キレート-Hisタグ融合Orf1アフィニティーカラムにアプライした。NADaseを、3 M NaSCNおよび0.01% アジ化ナトリウムを含有する0.1 M リン酸ナトリウムバッファー (pH 7.8) をロードすることにより回収し、透析した。
【図7D】Orf1によるNADaseの阻害を示す図である。精製したインタクトNADaseを、インタクトOrf1と混合し、室温で10分間インキュベートし、残存するNADase活性を測定した。
【図8A】NADase、Orf1およびNADase-Orf1複合体のTSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによる解析を示す図である。ウシ血清アルブミン (BSA) ダイマー (134 kDa)、モノマー (67 kDa)、オブアルブミン (OV, 43 kDa)、炭酸脱水酵素 (CA, 29 kDa) およびリゾチーム (14.4 kDa) を、分子量マーカーとして使用した。VoおよびVcは空容量およびカラム容量をそれぞれ示す。Hisタグ融合Orf1固定カラムおよび抗Orf1抗体固定カラムを用いたアフィニティークロマトグラフィーにより精製されたインタクトNADaseおよびOrf1 (成熟型, Hisタグなし) を、0.15 M リン酸ナトリウムバッファー (pH 6.6) において、TSKgel BioAssist G3SWXLゲル透過クロマトグラフィーに供した。
【図8B】NADase、Orf1およびNADase-Orf1複合体のTSKgel BioAssist G3SWXLゲル透過クロマトグラフィーによる解析を示す図である。ウシ血清アルブミン (BSA) ダイマー (134 kDa)、モノマー (67 kDa)、オブアルブミン (OV, 43 kDa)、炭酸脱水酵素 (CA, 29 kDa) およびリゾチーム (14.4 kDa) を、分子量マーカーとして使用した。VoおよびVcは空容量およびカラム容量をそれぞれ示す。精製したNADaseおよびOrf1を混合し、室温で10分間インキュベートし、TSKgel BioAssist G3SWXLカラムにアプライし、0.15 M リン酸ナトリウムバッファー (pH 6.6) で展開した。
【図9】インタクトOrf1 (Hisタグなし) の温度安定性を示す図である。Orf1溶液を20-100℃で15分間インキュベートし、氷水に浸した。各溶液を精製したHisタグを有しない成熟型NADaseに加えた。次いで、混合液を10分間室温でインキュベートし(Orf1とNADaseの最終モル比は1:2)、残存するヒドロラーゼ活性をNADで測定した。コントロールとして、BSAを含有する50 mMリン酸ナトリウムバッファー (pH 7.4) を、Orf1溶液の代わりに使用した。
【図10】nga-ni-sloオペロンの産物間の相互作用のモデル図である。SNI をコードする遺伝子(sni) は、ポリシストロニックmRNAとして、ngaプロモーターからnga (NADase) およびsloと共に転写される。翻訳の際、NADaseの自己毒性は、モル比1:1での、SNIとの安定な細胞内複合体の形成により迅速にブロックされ得る。NADase-SNI複合体は、連鎖球菌のエンベロープを介した排出の間に、おそらく幾つかの膜成分との相互作用により、SNIのタンパク質分解除去により、または他の機構により解離する。
【図11】pQE30-ヒト-His-タグ-CD38/1l LB/Amp/1 mM IPTG (3h誘導) 細胞を遠心分離により回収し、25 ml BugBusterにより抽出した。粗抽出液 (30 ml, 15 mgタンパク質/ml) を、室温にて流速1 ml/分でTSKゲルBioAssist Ni-キレートクロマトグラフィーにアプライした。スターティングバッファーは、50 mMリン酸ナトリウム(pH7.4)、20 mMイミダゾールおよび0.01%アジ化ナトリウムであった。20分間にわたるスターティングバッファーから50 mMリン酸ナトリウム(pH7.4)、250 mMイミダゾールおよび0.01%アジ化ナトリウムの線形勾配を行った。NADase活性 (units/ml) をSNI (アッセイ混合液中0.1μM) の存在下(◇)および非存在下(◆)で測定した。
【図12】pQE30-ヒト-His-タグ-CD157/1l LB/Amp/1 mM IPTG (3h誘導) 細胞を遠心分離により回収し、25 ml BugBusterにより抽出した。粗抽出液 (30 ml, 11 mgタンパク質/ml) を、室温にて流速1 ml/分でTSKゲルBioAssist Ni-キレートクロマトグラフィーにアプライした。スターティングバッファーは、50 mMリン酸ナトリウム(pH7.4)、20 mMイミダゾールおよび0.01%アジ化ナトリウムであった。20分間にわたるスターティングバッファーから50 mMリン酸ナトリウム(pH7.4)、250 mMイミダゾールおよび0.01%アジ化ナトリウムの線形勾配を行った。NADase活性 (units/ml) をSNI (アッセイ混合液中0.1μM) の存在下(◇)および非存在下(◆)で測定した。
【図13】ストレプトリシンO遺伝子 (SLO) およびSLO発現用ベクターの構築を示す模式図である。BamHIおよびKpnI部位を含むセンスプライマー (5’-cgGGATCCgactcgaacaaacaaaacactgcc-3’:配列番号13) およびアンチセンスプライマー (5’-cggGGTACCcttttatgttatgctttgcttg-3’:配列番号14) を、NH2末端シグナルペプチドを欠くHisタグ融合SLO配列の発現ベクターpBAD/HisB (Invitrogen, Frederick, USA) へのクローニングを可能とするように設計した。
【図14】E.coli TOP10全細胞抽出物における連鎖球菌SLOのイムノブロット解析を示す図である。プレ染色タンパク質分子標品の位置および計算分子量はパネルの左に示す。サイズはキロダルトン (kDa) で示す。矢印はSLOの位置を示す。E.coli. TOP10細胞を、6個の連続するヒスチジン残基 (Hisタグ) を含むように改変したSLO領域をコードする発現プラスミドで形質転換し、次いでSLOの合成についてアッセイした。L-アラビノースの存在下では、pBADからの発現がオンになるが、L-アラビノースの不在下では、pBADからの転写レベルは非常に小さくなる。培養物を0.00002% - 0.02%のL-アラビノースで37℃にて4時間誘導した。pBAD/HisB (ベクターのみ) を担持するE. coli TOP10の溶解物はコントロール(0.02% L-アラビノースで誘導)とした。SLOの相対発現レベルを決定するため、Ni-NTA HRPコンジュゲート (Qiagen, Hilden, Germany) を用いて、全細胞溶解物をウエスタンブロット解析に供した。このコンジュゲート〔西洋ワサビペルオキシダーゼにカップリングさせたニッケル−ニトリロトリ酢酸 (Ni-NTA)〕は、Hisタグを有する組換えタンパク質の直接的な検出のために用いた。レーン1:0.00002% L-アラビノース;レーン2:0.0002% L-アラビノース;レーン3:0.002% L-アラビノース;レーン4:0.02% L-アラビノース;レーン5:コントロール (ベクターのみ, 0.02% L-アラビノースで誘導)
【図15】Hisタグ融合SLOの精製を示す図である。pBAD/His B-sloを含む E. Coli BL21の培養物を、0.2% L-アラビノースで37℃にて4時間誘導した。細胞抽出物を、20 mMイミダゾール(バッファーA)を含有する50 mMリン酸ナトリウムバッファー (pH 7.4) で平衡化したTSKgel BioAssist Ni2+-キレートカラム (Tosoh Co., Tokyo, Japan) にアプライした。非特異的吸着物をカラムから洗浄して除くために、3 M NaClを含む0.1 Mリン酸ナトリウムバッファー (pH 7.8) を通過させた。このHisタグ融合SLOを、バッファーAにおける20-250 mMのイミダゾールの線形勾配により回収した。精製したHisタグ融合SLOをプールし、バッファーAに対して透析した。透析物をSDS-PAGEに供し、クマシーブリリアントブルー (CBB) R-250で染色した。タンパク質分子標品の位置および各計算分子量をパネルの左に示す。サイズはキロダルトン (kDa) で示す。矢印はSLOの位置を表す。
【図16】GAS株SaおよびGCS株H46A由来の培養上清のイムノブロット解析を示す図である。精製したHisタグ融合SLOを用いて、ウサギ抗SLO抗体を惹起した。約2 mgのHisタグ融合SLO (完全フロインドアジュバントと混合) をウサギに皮下注射した。1 mgのタンパク質と不完全フロインドアジュバントからなるブースター注射を2週間の間隔で3回投与した。プロテインA-セファロース (Amersham Biosciences Corp., NJ, USA) のアフィニティークロマトグラフィーにより、イムノグロブリンG画分を精製した。精製H46A NADase (Gerlach et al., 1996) での免疫により作製したウサギ抗血清は、Friedrich-Schiller大学 (ドイツ) のD. Gerlach博士からご提供頂いた。連鎖球菌の培養上清をSDS-PAGE (10% 総アクリルアミド) に供し、電気ブロッティングによりタンパク質をSequi-Blot PVDF膜 (Bio-Rad, CA, USA) 上に転写した。この膜を、0.05% Tween 20を含有するリン酸緩衝化生理食塩水 (PBST) において、5% スキムミルクおよび1% ウシ血清アルブミン (BSA) で1時間ブロッキングした。膜をPBSTで洗浄し、次いで各一次抗体を添加し (PBSTで1:3,000に希釈) 、ブロットを1時間インキュベートした。次いで、抗ウサギIgGにコンジュゲートしたヤギaffini pure IgGペルオキシダーゼ (Wako Pure Chemical Industries, Ltd., Osaka, Japan) を、PBSTで1:3,000に希釈して添加し、1時間インキュベートし、抗体・抗原複合体を、製造業者により記載される通りにPOD免疫染色セット (Wako Pure Chemical Industries, Ltd., Osaka, Japan) を用いて可視化した。プレ染色タンパク質分子標品の位置および各計算分子量はパネルの左に示す。サイズはキロダルトン (kDa) で示す。
【特許請求の範囲】
【請求項1】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、医薬。
【請求項2】
連鎖球菌感染症の治療薬である、請求項1記載の医薬。
【請求項3】
自己免疫疾患、炎症性疾患または多発性骨髄腫の治療薬である、請求項1記載の医薬。
【請求項4】
NADaseが細菌または動物由来NADaseである、請求項1記載の医薬。
【請求項5】
細菌または動物由来NADaseが、連鎖球菌由来NADase、あるいはCD38またはCD157である、請求項4記載の医薬。
【請求項6】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、NADase阻害剤。
【請求項7】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、白血球の走化性の阻害剤。
【請求項8】
白血球が好中球である、請求項7記載の剤。
【請求項9】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、試薬。
【請求項10】
連鎖球菌由来NADaseの組換えタンパク質であって、天然型NADaseに対して1以上のアミノ酸の欠失、置換、付加および挿入からなる群より選ばれる1以上のアミノ酸の修飾が施された、組換えタンパク質。
【請求項11】
該1以上の修飾が精製用タグの付加を少なくとも含む、請求項10記載の組換えタンパク質。
【請求項12】
請求項10記載の組換えタンパク質を含む、医薬または試薬。
【請求項13】
請求項10記載の組換えタンパク質をコードするポリヌクレオチド。
【請求項14】
第1および第2の発現ユニットの共発現ベクターであって、
第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み、
第1の発現ユニットが、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドであり、
第2の発現ユニットが、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドである、共発現ベクター。
【請求項15】
NADaseが細菌または動物由来NADaseである、請求項14記載の共発現ベクター。
【請求項16】
NADaseが連鎖球菌由来NADaseである、請求項14記載の共発現ベクター。
【請求項17】
NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを発現する、形質転換体。
【請求項18】
形質転換体が大腸菌である、請求項17記載の形質転換体。
【請求項19】
請求項17記載の形質転換体を培養し、培養上清からNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドを回収することを含む、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造方法。
【請求項20】
NADaseとの結合能および/またはNADase阻害活性を有するSNIの部分ペプチド。
【請求項21】
請求項20記載の部分ペプチドをコードするポリヌクレオチド。
【請求項22】
請求項21記載のポリヌクレオチドを含む、発現ベクター。
【請求項23】
SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む、複合体。
【請求項24】
NADaseが連鎖球菌由来NADaseである、請求項23記載の複合体。
【請求項25】
SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法。
【請求項26】
以下(a)〜(d)からなる群より選ばれる構成要素を含む、SNIまたはNADaseとの結合能を有するその部分ペプチドとNADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成剤:
(a)SNI;
(b)NADaseとの結合能を有するSNIの部分ペプチド;
(c)NADase;
(d)SNIとの結合能を有するNADaseの部分ペプチド。
【請求項27】
以下(a)および/または(b)を含む、SNIまたはNADaseに親和性を有する融合タンパク質の調製剤:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター;
(b)NADaseまたはSNIとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター。
【請求項28】
以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたは該連鎖球菌由来NADaseとの結合能を有するその部分ペプチドと、該連鎖球菌由来NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成を抑制し得るか否かを評価する工程;
(b)該複合体の形成を抑制し得る被験物質を選択する工程。
【請求項29】
以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、該連鎖球菌由来NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
【請求項30】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターであって、
該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、ベクター。
【請求項31】
溶血性連鎖球菌がA群またはC群連鎖球菌である、請求項30記載のベクター。
【請求項32】
該SLOが精製用タグを有する、請求項30記載のベクター。
【請求項33】
該誘導プロモーターがアラビノースオペロン制御プロモーターである、請求項30記載のベクター。
【請求項34】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターで形質転換された大腸菌であって、
該発現ベクターは、該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、大腸菌。
【請求項35】
大腸菌が、プロテアーゼの欠損株、および/または誘導プロモーターに対する誘導剤の代謝能の欠損株である、請求項34記載の大腸菌。
【請求項36】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの製造方法であって、誘導プロモーターに対する誘導剤の存在下において請求項34記載の大腸菌を培養し、培養上清から該SLOまたはその部分ペプチドを回収することを含む、方法。
【請求項37】
溶血性連鎖球菌由来SLOに対する特異的な反応性を示し、かつ溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体。
【請求項1】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、医薬。
【請求項2】
連鎖球菌感染症の治療薬である、請求項1記載の医薬。
【請求項3】
自己免疫疾患、炎症性疾患または多発性骨髄腫の治療薬である、請求項1記載の医薬。
【請求項4】
NADaseが細菌または動物由来NADaseである、請求項1記載の医薬。
【請求項5】
細菌または動物由来NADaseが、連鎖球菌由来NADase、あるいはCD38またはCD157である、請求項4記載の医薬。
【請求項6】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、NADase阻害剤。
【請求項7】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、白血球の走化性の阻害剤。
【請求項8】
白血球が好中球である、請求項7記載の剤。
【請求項9】
SNI、NADase阻害活性を有するその部分ペプチドまたはそれらの発現ベクターを含む、試薬。
【請求項10】
連鎖球菌由来NADaseの組換えタンパク質であって、天然型NADaseに対して1以上のアミノ酸の欠失、置換、付加および挿入からなる群より選ばれる1以上のアミノ酸の修飾が施された、組換えタンパク質。
【請求項11】
該1以上の修飾が精製用タグの付加を少なくとも含む、請求項10記載の組換えタンパク質。
【請求項12】
請求項10記載の組換えタンパク質を含む、医薬または試薬。
【請求項13】
請求項10記載の組換えタンパク質をコードするポリヌクレオチド。
【請求項14】
第1および第2の発現ユニットの共発現ベクターであって、
第1および第2の発現ユニットをコードするポリヌクレオチド、および該発現ユニットをコードするポリヌクレオチドに機能的に連結されたプロモーターを含み、
第1の発現ユニットが、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドであり、
第2の発現ユニットが、SNIまたは該毒性作用に対する中和作用を有するその部分ペプチドである、共発現ベクター。
【請求項15】
NADaseが細菌または動物由来NADaseである、請求項14記載の共発現ベクター。
【請求項16】
NADaseが連鎖球菌由来NADaseである、請求項14記載の共発現ベクター。
【請求項17】
NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチド、およびSNIまたは該毒性作用に対する中和作用を有するその部分ペプチドを発現する、形質転換体。
【請求項18】
形質転換体が大腸菌である、請求項17記載の形質転換体。
【請求項19】
請求項17記載の形質転換体を培養し、培養上清からNADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドを回収することを含む、NADaseまたは宿主細胞に対する毒性作用を有するその部分ペプチドの製造方法。
【請求項20】
NADaseとの結合能および/またはNADase阻害活性を有するSNIの部分ペプチド。
【請求項21】
請求項20記載の部分ペプチドをコードするポリヌクレオチド。
【請求項22】
請求項21記載のポリヌクレオチドを含む、発現ベクター。
【請求項23】
SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む、複合体。
【請求項24】
NADaseが連鎖球菌由来NADaseである、請求項23記載の複合体。
【請求項25】
SNIまたはNADaseとの結合能を有するその部分ペプチドと、NADaseまたはSNIとの結合能を有するその部分ペプチドとを接触させることを含む、SNIまたはその部分ペプチドとNADaseまたはその部分ペプチドとを含む複合体の製造方法。
【請求項26】
以下(a)〜(d)からなる群より選ばれる構成要素を含む、SNIまたはNADaseとの結合能を有するその部分ペプチドとNADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成剤:
(a)SNI;
(b)NADaseとの結合能を有するSNIの部分ペプチド;
(c)NADase;
(d)SNIとの結合能を有するNADaseの部分ペプチド。
【請求項27】
以下(a)および/または(b)を含む、SNIまたはNADaseに親和性を有する融合タンパク質の調製剤:
(a)SNIまたはNADaseとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター;
(b)NADaseまたはSNIとの結合能を有するその部分ペプチドをコードするポリヌクレオチドを含む発現ベクター。
【請求項28】
以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたは該連鎖球菌由来NADaseとの結合能を有するその部分ペプチドと、該連鎖球菌由来NADaseまたはSNIとの結合能を有するその部分ペプチドとを含む複合体の形成を抑制し得るか否かを評価する工程;
(b)該複合体の形成を抑制し得る被験物質を選択する工程。
【請求項29】
以下の工程(a)、(b)を含む、NADase遺伝子およびSNI遺伝子を併有する連鎖球菌による感染症の治療薬のスクリーニング方法:
(a)被験物質が、SNIまたはNADase阻害活性を有するその部分ペプチドによる、該連鎖球菌由来NADaseまたはNADase活性を有するその部分ペプチドのNADase活性の抑制を解除し得るか否かを評価する工程;
(b)該抑制を解除し得る被験物質を選択する工程。
【請求項30】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターであって、
該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、ベクター。
【請求項31】
溶血性連鎖球菌がA群またはC群連鎖球菌である、請求項30記載のベクター。
【請求項32】
該SLOが精製用タグを有する、請求項30記載のベクター。
【請求項33】
該誘導プロモーターがアラビノースオペロン制御プロモーターである、請求項30記載のベクター。
【請求項34】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの発現ベクターで形質転換された大腸菌であって、
該発現ベクターは、該SLOまたはその部分ペプチドをコードするポリヌクレオチド、および該ポリヌクレオチドに機能的に連結された誘導プロモーターを含み、
該誘導プロモーターが、大腸菌における基底レベルの遺伝子発現を抑制し得る誘導プロモーターである、大腸菌。
【請求項35】
大腸菌が、プロテアーゼの欠損株、および/または誘導プロモーターに対する誘導剤の代謝能の欠損株である、請求項34記載の大腸菌。
【請求項36】
溶血性連鎖球菌に由来する完全長SLOまたは大腸菌に毒性を示し得る領域を含むその部分ペプチドの製造方法であって、誘導プロモーターに対する誘導剤の存在下において請求項34記載の大腸菌を培養し、培養上清から該SLOまたはその部分ペプチドを回収することを含む、方法。
【請求項37】
溶血性連鎖球菌由来SLOに対する特異的な反応性を示し、かつ溶血性連鎖球菌由来NADaseに対する交差反応性を示さない抗体。
【図1】

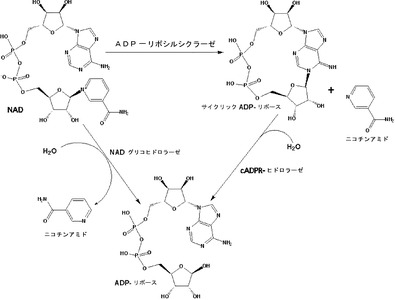
【図2A】

【図3B】

【図7A】

【図7B】

【図7C】

【図7D】

【図8A】

【図8B】

【図11】

【図12】

【図13】

【図2B】
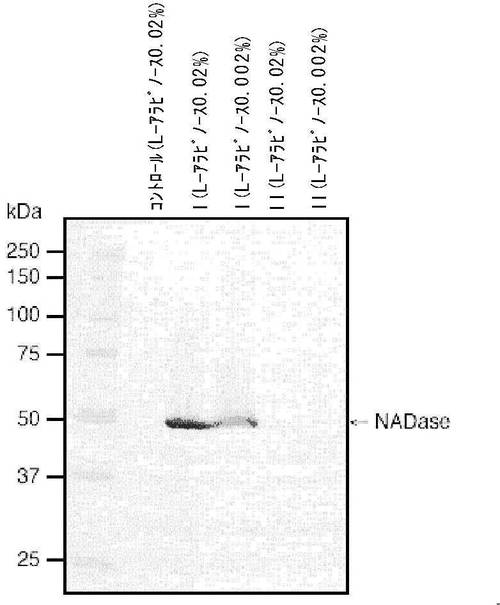
【図3A】

【図4A】

【図4B】

【図5】

【図6】

【図9】
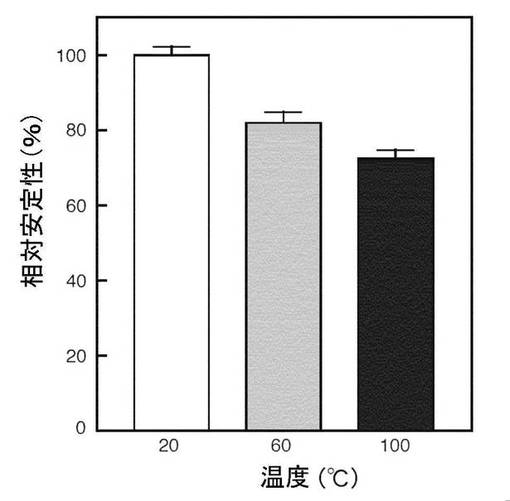
【図10】

【図14】

【図15】

【図16】

【図2A】
【図3B】
【図7A】
【図7B】
【図7C】
【図7D】
【図8A】
【図8B】
【図11】
【図12】
【図13】
【図2B】
【図3A】
【図4A】
【図4B】
【図5】
【図6】
【図9】
【図10】
【図14】
【図15】
【図16】
【公開番号】特開2007−61066(P2007−61066A)
【公開日】平成19年3月15日(2007.3.15)
【国際特許分類】
【出願番号】特願2005−254512(P2005−254512)
【出願日】平成17年9月2日(2005.9.2)
【出願人】(504145320)国立大学法人福井大学 (287)
【Fターム(参考)】
【公開日】平成19年3月15日(2007.3.15)
【国際特許分類】
【出願日】平成17年9月2日(2005.9.2)
【出願人】(504145320)国立大学法人福井大学 (287)
【Fターム(参考)】
[ Back to top ]