
N−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩を、3つの多形(A型、B型、およびC型)の形で本明細書に提供する。A型、B型、およびC型は、粉末X線回折パターンのそれらのピーク、それらの赤外吸収スペクトルのそれらの吸収ピーク、それらのラマンスペクトルのそれらのピーク、およびそれらの融点により特定される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、Reichweinらの2006年3月13日に提出された、“N−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形”という表題の、米国仮特許出願第60/781,861号に対する優先権を主張する。上の関連する出願の開示を本明細書において参照として援用する。
【0002】
(発明の技術分野)
本明細書において、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形、およびそれらの製造法を提供する。
【背景技術】
【0003】
(背景)
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウム塩は、ペプチドのエンドセリンファミリーの活性を調節し、そのためエンドセリンを介した障害の治療に有用である。これらの障害の性質のため、医薬製品としての本化合物の使用は、長期間の保存を必要とすると考えられる。したがって、保存期間中の熱および湿気に対する本化合物(医薬用化学製品の原体)の安定性は、非常に重要である。そのため本化合物のより安定な形が所望される。
【発明の開示】
【課題を解決するための手段】
【0004】
(概要)
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形であるA型およびB型;メチルt−ブチルエーテル溶媒和物であるC型;ならびにアモルファス型は、適当な溶媒および条件からの結晶化により産業スケールで選択的に生産することができることを発見した。さらにB型、ならびにA型およびB型の混合物は、適切な条件下でより安定なA型に相互変換することができる。
【0005】
シタキセンタンナトリウムのアモルファス型はかなり吸湿性であるのに対して、結晶型はそうではない(アモルファスは、95%RHでその総重量の22%増量する;結晶は、95%RHでその重量の1.5%未満しか増量しない)。相互変換の研究により、A型の多形がより熱力学的に安定な形であることを見出した。理論に拘束されるものでは決してないが、アモルファス状態は、時間と共に多形の混合物に変換されると考えられる。
【0006】
特に以下の化学構造:
【0007】
【化1】
【0008】
を有する、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形であるA型およびB型は、選択的に生成することができ、それらの粉末X線回折(XRPD)パターン、赤外線吸収スペクトル、ラマンスペクトル、および融点におけるそれらの特徴的なピークに基づいて識別することができる。
【0009】
XRPDパターンの測定のための方法および条件
測定の方法
XRPD分析は、以下の条件によりサンプルについてShimadzu XRD-6000 粉末X線回折計にて行った。
【0010】
測定の条件
【0011】
【化2】
【0012】
赤外線吸収の測定のための方法および条件
熱重量赤外線(TG/IR)吸収スペクトルは、Nicolet モデル560フーリエ変換赤外線(FT−IR)分光光度計とインターフェースしたTA Instrument TGA 2050にて得た。
【0013】
ラマン吸収の測定のための方法および条件
ラマンスペクトルは、Nicolet Magna 860 FT−IR分光光度計にインターフェースしたラマンベンチ(bench)にて得た。
【0014】
多形A(A型)
角度2θにおいて表わされるA型のXRPDパターンの主要なピークは、およそ6.72、15.96、22.38、23.38および26.22にある。
【0015】
図1−8は、A型のXRPDパターンを示す。
A型のラマンスペクトルの主要なピーク(cm−1)は、およそ1697.4、1602.1、1489.8および1402.2cm−1にある。
【0016】
図9は、A型のラマンスペクトルを示す。
特徴付けのデータに基づくと、A型は、およそ200℃で分解する結晶性、非吸湿性の固体であると思われる。
【0017】
多形B(B型)
角度2θにおいて表わされるB型のXRPDパターンの主要なピークは、およそ6.6、15.52、18.38、18.94および22.72にある。
【0018】
図1は、B型のXRPDパターンを示す。
B型のラマンスペクトルの主要なピーク(cm−1)は、およそ1696.9、1594.7、1490.2および1397.8cm−1にある。
【0019】
図22は、B型のラマンスペクトルを示す。
特徴付けのデータに基づくと、B型は、203℃付近で分解する非吸湿性の結晶材料であると思われる。
【0020】
多形C(C型)
角度2θにおいて表わされるC型のXRPDパターンの主要なピークは、およそ5.14、23.48、および26.78にある。
【0021】
図1は、C型のXRPDパターンを示す。
図23は、C型の赤外線吸収スペクトルを示す。
特徴付けのデータに基づくと、C型は、化合物のメチルt−ブチルエーテル溶媒和物であると思われる。
【0022】
(詳細な説明)
A.定義
他に定義していなければ、本明細書で使用するすべての技術的用語および科学的用語は、本主題の物質の属する当業者により共通して理解されているのと同じ意味を有する。本明細書に述べたすべての特許および公開を参照として援用する。
【0023】
本明細書において使用する場合、“シタキセンタンナトリウム”はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウムをいう。シタキセンタンナトリウムの他の化学名として、4−クロロ−3−メチル−5−(2−(2−メチル−(6−メチルベンゾ[d][1,3]ジオキソル−5−イル)アセチル)−3−チエニルスルホンアミド)イソキサゾール、ナトリウム、およびN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[3,4−(メチレンジオキシ)−6−メチルフェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウムを含む。シタキセンタンナトリウム塩の化学構造は、本明細書において他所に記載している。
【0024】
本明細書において使用する場合、エンドセリン(ET)ペプチドは、エンドセリン−1、エンドセリン−2、またはエンドセリン−3のアミノ酸配列を実質的に有するペプチド、および強力な内在性血管収縮ペプチドとして作用するペプチドを含む。
【0025】
本明細書において使用する場合、エンドセリンを介した状態は、異常なエンドセリン活性によって引き起こされる状態、またはエンドセリン活性を阻害する化合物が治療としての用途を持つ状態である。そのような疾患として、高血圧症、心血管疾患、喘息、炎症性疾患、眼科的疾患、月経障害、産科的状態、胃腸の疾患、腎不全、肺高血圧症、間質性肺疾患、拡張期心不全、エンドトキシンショック、アナフィラキシーショック、または出血性ショックを含むがこれに限定されない。エンドトキシンを介した状態はまた、エンドトキシンレベルを上昇させる薬剤、例えばエリスロポエチンおよび免疫抑制薬による治療に起因する状態を含む。
【0026】
本明細書において使用する場合、特定の疾患を治療するための化合物の有効量は、疾患に関連する症状を寛解させる、または何らかの様式で低減するために十分である量である。そのような量は、それが有効となる、単回投与として投与してもよいし、または投与計画に従って投与してもよい。その量は当該疾患を治癒してもよい。もう1つの態様においてその量は、当該疾患の1つまたはそれより多くの症状を寛解させる目的で投与する。その他の態様において、症状の所望の寛解を達成するために繰り返し投与が必要とされる。
【0027】
本明細書において使用する場合、エンドセリンアゴニストは、エンドセリンペプチドに関連するまたはエンドセリンペプチドの保有する生物学的活性を、増強するまたは示す化合物である。
【0028】
本明細書において使用する場合、エンドセリンアンタゴニストは、エンドセリンに刺激される血管収縮(vasoconstriction)および収縮(contraction)、ならびにその他のエンドセリンを介した生理学的応答を阻害する薬剤または抗体というような化合物である。アンタゴニストは、エンドセリンとエンドセリン特異的受容体との相互作用を妨害することにより、または血管収縮のようなエンドセリンイソペプチドへの生理学的応答もしくはエンドセリンイソペプチドの生理活性を妨害することにより、作用してよい。したがって本明細書において使用する場合、エンドセリンアンタゴニストは、当業者に公知のアッセイにより評価した時に、エンドセリンに刺激される血管収縮もしくはその他の応答を妨害する、またはエンドセリンとエンドセリン特異的受容体、例えばETA受容体との相互作用を妨害する。
【0029】
潜在的アゴニストおよびアンタゴニストの有効性は、当業者に公知の方法を使用して評価することができる。例えばエンドセリンアゴニストの活性は、単離したラットの胸部大動脈または門脈の環状部分の血管収縮を刺激するその能力により、同定することができる(Borges et al. (1989) "Tissue selectivity of endothelin" Eur. J. Pharmacol. 165: 223-230)。
【0030】
本明細書において使用する場合、ETA選択的であるスルホンアミドは、ETA受容体に関してETB受容体と比べて少なくとも約1/10低いIC50を示すスルホンアミドをいう。
【0031】
本明細書において使用する場合、ETB選択的であるスルホンアミドは、ETB受容体に関してETA受容体と比べて少なくとも約1/10低いIC50を示すスルホンアミドをいう。
【0032】
本明細書において使用する場合、治療は、状態、障害もしくは疾患の症状が寛解される、またはそうでなければ有利に変化するあらゆる様式を意味する。治療はまた、本明細書における組成物のあらゆる医薬的使用、例えば避妊薬としての使用も包括的に含む。
【0033】
本明細書において使用する場合、特定の医薬組成物の投与による特定の障害の症状の寛解は、永久的または一時的、持続的または一過性的あるにせよ、当該組成物の投与に起因または関連し得るあらゆる緩和をいう。
【0034】
本明細書において使用する場合、実質的に純粋であることは、そのような純度を評価するために当業者によって使用される標準的な分析法、例えば薄相クロマトグラフィー(TLC)、ゲル電気泳動、および高速液体クロマトグラフィー(HPLC)により決定されるような、容易に検出され得る不純物を含んでいないと思われる十分な均一性、またはさらに精製しても、その物質の物理的特性および化学的特性、例えば酵素活性および生物学的活性を検出可能なほど変化させることがないというように十分に純粋であること、を意味する。実質的に化学的に純粋な化合物を生成するための化合物の精製方法は、当業者に公知である。しかし実質的に化学的に純粋な化合物は、立体異性体の混合物であってよい。そのような場合は、さらなる精製が化合物の特異的活性を増大させることになるかもしれない。
【0035】
本明細書において使用する場合、生物学的活性は、化合物、組成物またはその他の混合物のin vivo投与の結果得られる、化合物のin vivo活性または生理学的応答をいう。したがって生物学的活性は、そのような化合物、組成物および混合物の治療効果および医薬的活性を包括的に含む。
【0036】
本明細書において使用する場合、製剤の増加した安定性は、当業者に公知のアッセイ、例えば高速液体クロマトグラフィー、ガスクロマトグラフィーなどによって決定した場合に、製剤の調製後の所定の時間での、製剤中に存在する活性成分のパーセントが、製剤の調製後の同じ時間での別の製剤中に存在する活性成分のパーセントより、有意に高いことを意味する。この場合、前者の製剤は後者の製剤に比して増加した安定性を有すると言われる。
【0037】
B.分析方法
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の結晶化サンプルは、それらの多形(A型またはB型)、水和物および溶媒和物(C型)を決定するため、それらのXRPD、赤外線吸収スペクトル、ラマンスペクトル、融点、示差走査熱量測定(DSC)、熱重量測定(TG)、高温顕微鏡、および自動吸湿/放湿により分析した。
【0038】
1.XRPD
XRPD分析は、Cu Kα線を使用してShimadzu XRD-6000粉末X線回折計にて行った。この装置にファインフォーカスのX線管球を装備した。管球の出力およびアンペア数は、各々40kVおよび40mAに設定した。発散スリットおよび散乱スリットは1°に設定し、受光スリットは0.15mmに設定した。回折光は、Nalシンチレーションカウンターにより検出した。2.5°(2θ)から40°(2θ)について3°/分(0.4秒/0.02°ステップ)でθ−2θ継続スキャンを使用した。シリコンスタンダードを毎日分析し、装置の調整をチェックした。各サンプルは、クォーツのサンプルホルダーに置くことにより、分析用に調製した。優先配向の効果を低減するため、3つのサンプルをスピンさせながら(25rpm)分析した。スキャンは0.5°/分に調整して、スピン速度について補正した。
【0039】
2.TG/IR
TG/IR吸収は、Nicolet モデル560フーリエ変換赤外線(FT−IR)分光光度計とインターフェースしたTA Instrument TGA 2050にて得た。この装置に、黒体輻射光源(globar source)、Ge/KBrビームスプリッタ、重水素化トリグリセリン硫酸塩(DTGS)検出器を装備した。IR分光光度計は、スタンダードとして使用日毎にポリスチレンを用いて、一方TGは1週間毎にニッケルおよびアルメルを使用して波長を較正した。およそ5mgのサンプルをプラチナのパンに量りとり、ヘリウムパージして、20℃から150℃まで、20℃/分の比率で記録した。IRスペクトルは、4cm−1の解像度で8回の加算したスキャンを表す各スペクトルの連続したものとして得た。揮発性物質は、HR Nicolet TGA 気相スペクトルライブラリを検索して同定した。
【0040】
3.ラマンスペクトル
ラマンスペクトルは、Nicolet Magna 860 FT-IR分光光度計にインターフェースしたラマンベンチにて得た。この装置は、1064nmの励起波長、およびおよそ0.5WのNd:YAGレーザー出力を利用した。スペクトルは、4cm−1の解像度で得た32または64回加算したスキャンを表す。サンプルは、材料をガラス管にいれ、このガラス管を分光光度計にセットすることによる分析用に調製した。分光光度計は、使用時にイオウおよびシクロヘキサンを用いて、(波長を)較正した。
【0041】
4.示差走査熱量計(DSC)
示差走査熱量計のデータは、TA Instruments Differential Scanning Calorimeter 2920にて得た。使用した較正用スタンダードは、インジウムであった。およそ2から5mgのサンプルをDSCパンに置き、重さを正確に測定し記録した。パンは密封して封入し、圧を放出させるためにピンホールを使用した。サンプルは、窒素下で10℃/分の比率で、最終温度300℃まで加熱した。アモルファス材料のガラス転位温度(Tg)の研究用には、サンプルは窒素下、10℃/分の比率で125℃まで加熱した。サンプルをこの温度で15分間保持した後、放置して冷却し、25℃での平衡状態とした。サンプルを再び10℃/分の比率で125℃まで加熱し、この温度で15分間保持した後、冷却し、25℃で15分間平衡状態とした。次にこのサンプルを10℃/分で最終温度200℃まで加熱した。
【0042】
5.熱重量(TG)分析
サンプルの熱重量(TG)分析は、TA Instruments Thermogravimetric Analyzer 2050 または 2950にて行った。使用した較正用スタンダードは、ニッケルおよびアルメル(Alumel、商標)であった。およそ2から5mgのサンプルをパンに置き、正確に重量測定し、TG炉に入れた。その後サンプルを窒素下、10℃/分の比率で最終温度300℃まで加熱した。
【0043】
6.高温顕微鏡
高温顕微鏡測定は、Leica顕微鏡にて、Kofler高温ステージにのせて行った。高温ステージの温度は、Testo 6000-903熱電対およびTesto 720デジタル読み取り器を使用して測定した。温度はUSPのスタンダードを用いて較正した。
【0044】
7.吸湿/放湿
吸湿/放湿データは、VT SGA-100平衡含湿システム(moisture balance system)にて集めた。吸湿等温線として、10%RH増加における5から95%相対湿度(RH)の吸湿範囲、および95から5%RHの放湿範囲を分析用に使用した。サンプルは分析前に乾燥させなかった。分析に関して使用した平衡の基準は、重量の判定基準が一致しない場合、3時間の最大平衡時間内で5分間に0.0100%未満の重量変化とした。データは、サンプルの最初の湿度含有量については補正しなかった。
【0045】
8.多形のスクリーニング
多形のスクリーニングは、できるだけ多くの固体の形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩を作成する試みにおいて行った。この技術は、様々な条件下での固体の作成、およびその後のXRPDによる特徴づけを伴った。3つの区別される形、ならびにアモルファスの形を表す3つの区別されるXRPDパターンが、このスクリーニングにおいて見出された。結晶のパターンはA型、B型、およびC型と命名した。A型は、高温溶液の緩やかな冷却、スラリー化、または貧溶媒を用いての沈殿より得た。B型は、高温溶液の緩やかな冷却、および貧溶媒による結晶化より得た。C型はメチルt−ブチルエーテルからの貧溶媒による結晶化より得ており、したがってN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のメチルt−ブチルエーテル溶媒和物であると思われる。アモルファスの材料は、溶液を緩やかにおよび速く蒸発させることにより生成した。
【0046】
9.結晶化の方法
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の秤量したサンプル(通常30mg)を、検査溶媒(試薬グレードまたはHPLCグレード)を一定分量ずつ加えて処理し、20から200μLの溶液を生成した。これらの溶液を超音波処理し、すべての固体が溶解した(目視)時点で、溶液を濾過し、周囲条件下でふたをしないバイアル中に放置(速い蒸発)するか、またはピンホールのあるアルミホイルで覆った(緩やかな蒸発)。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。この化合物の固体サンプルはまた、上の濾過した室温溶液を−78℃に急速に冷却する(急激な冷却)ことによっても生成した。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0047】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の秤量したサンプルはまた、高温で検査溶媒を一定分量ずつ加えて処理した。これらのサンプルおよび溶媒は、45℃または80℃のどちらかに保温したホットプレート上で加熱し、得られた溶液を、同じホットプレート上に保持したバイアル中に素早く濾過した。熱源のスイッチを切り、ホットプレートおよびバイアルを放置して周囲温度に冷却(緩やかな冷却)し、一晩そのまま静置した。析出した固体の有無を調べた;固体が存在しない場合、またはXRPD分析には固体の量が少なすぎると判断した場合には、バイアルを一晩冷蔵庫に置いた。再び析出した固体の有無を調べ、固体が認められない場合にはバイアルを冷凍庫に一晩おいた。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0048】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の溶解度は、溶液を得るために使用した溶媒総量に基づいて実験から見積もった。実際の溶解度は、一定分量ずつという多過ぎる溶媒を使用していること、または遅い溶解速度のために、算出された値より大きいと考えられる。実験中に溶解が起こらなかった場合は、溶解度は“より小さい”と表す。一定分量の溶媒をすべて加えてしまう前に固体が溶解した場合は、溶解度は“より大きい”と列記する。
【0049】
貧溶媒の実験は、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体サンプルを検査溶媒中に溶解させ、得られた溶液を貧溶媒中に濾過することにより行った。固体が形成された場合、“急激な結晶化”と呼ぶ;そして固体が、溶液を冷却した後、またはカバーしてそのまま静置した後に形成された場合、“沈殿”と呼ぶ。直ちに固体が形成されなかった場合は、サンプルは、固体が見られるまで周囲条件下に放置した。形成されたいかなる固体も濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0050】
スラリーの実験は、過剰な固体を含有する、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を作製することにより行った。これらのスラリーは、周囲温度で3日間撹拌した。析出した固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0051】
蒸気拡散の実験は、バイアル中にN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を入れ、次にそれを、貧溶媒を含有するより大きなバイアルに入れることにより行った。次により大きなバイアルを密封し、周囲温度に維持した。固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0052】
液体拡散の実験は、バイアル中にN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を入れ、混和しない貧溶媒を加えることにより行った。沈殿した固体の有無を調べた。固体が形成された場合は、溶媒をデカントし、固体を集めた。固体が形成されなかった場合は、バイアルに蓋をして、周囲温度でそのまま静置した。形成されたあらゆる固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0053】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体サンプルはまた、この化合物の融液の急速な冷却(−78℃)により作成した。
【0054】
C.多形A、B、Cおよびアモルファス材料
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形スクリーニングで得た固体の形を表1から3にまとめる。A型、B型、およびC型と命名した、3つの区別される形を表す3つの区別されるXRPDパターンが見出された。A型は、緩やかな冷却、スラリー化、または貧溶媒による結晶化より得た。B型は、高温溶液の緩やかな冷却、および貧溶媒による結晶化より得た。C型はメチルt−ブチルエーテルからの貧溶媒による結晶化より得た。アモルファスの材料は、溶液を緩やかにおよび速く蒸発させることにより生成した。
【0055】
【表1−1】
【0056】
【表1−2】
【0057】
表2は貧溶媒による再結晶化の結果を示す。
【0058】
【表2−1】
【0059】
【表2−2】
【0060】
【表2−3】
【0061】
表3は蒸気拡散実験に関する結果を示す。
【0062】
【表3−1】
【0063】
【表3−2】
【0064】
a. A型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のA型は、XRPD、DSC、TG、高温顕微鏡、および吸湿/放湿を用いて特徴づけを行い、データを図1から18、表4(高温顕微鏡の研究)、表5(吸湿/放湿データ)、表6(XRPDピーク)、表7(ラマンスペクトルのピーク)および表8(IRスペクトルのピーク)にまとめる。発熱分解は200℃付近で見られ、高温顕微鏡により確認した。TG曲線は175℃で最小の重量変化を示した。吸湿/放湿データでは、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のサンプルが、5%RHで最小量の重量を喪失することを示し、少量の最初の揮発物質のみが低RH条件下で除去されたことを指摘する。サンプルは、95%RHでその重量の1.5%未満が増量するが、この値は0.5水和物(hemihydrate)の形成に関する増量の理論値(1.87%)より低い。重量のほとんどは放湿曲線の75%RHまでに失われ、材料は35%RH以下で、非溶媒和状態の平衡状態に戻る。実験完了後のサンプルのXRPDパターンは、材料がA型であることを示す。A型の材料は、吸湿/放湿データに基づくと、75%RHまでは非吸湿性であると思われる。
【0065】
【表4】
【0066】
【表5】
【0067】
【表6】
【0068】
【表7−1】
【0069】
【表7−2】
【0070】
【表8−1】
【0071】
【表8−2】
【0072】
特徴付けのデータに基づくと、A型は、200℃以上で分解する、非溶媒和物、非吸湿性の結晶材料であると思われる。
b. B型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のB型は、通常貧溶媒による結晶化より得、XRPD、DSC、TG、高温顕微鏡、および吸湿/放湿を用いて特徴づけを行い、データを図1および19−22、表4(高温顕微鏡の研究)、表5(吸湿/放湿データ)、表6(XRPDピーク)、表7(ラマンスペクトルのピーク)および表8(IRスペクトルのピーク)にまとめる。
【0073】
B型に関する熱データは、図19および20に示す。DSCは、高温顕微鏡データより、分解に起因する205℃での緩やかな発熱を示す。TG曲線は175℃での最小量の重量喪失を示す。B型は吸湿/放湿実験の間に、最小量の重量の増減を示す。実験完了後にサンプルを集めたXRPDパターンは、材料がB型であったことを示す。B型のサンプルを、そのナトリウム含有量(4.85%)について分析したところ、この値はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の理論値(4.82%)と一致し、この塩がインタクトであることを示した。
【0074】
特徴付けのデータに基づくと、B型は、203℃付近で分解する非溶媒和の結晶材料であると思われる。
c. C型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のC型は、溶媒としてメタノールまたはエタノール、そして貧溶媒としてメチル−t−ブチルを使用して、貧溶媒による結晶化より、単独でまたはB型との混合物として得た。同じ条件下での繰り返しての結晶化は、ほとんどの場合B型またはB型とC型の混合物を得た。C型は、XRPD、TG/IRおよびTGを用いて特徴づけを行い、データを図1および23−24、ならびに表4に示す。
【0075】
C型および少量のB型を含有するサンプルのTGデータは、175℃で22.4%の重量喪失を示し、これはN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩の8水和物(理論値23.2%)またはメチル−t−ブチルエーテレート(21.7%、2薬剤分子当たり溶媒3分子)としての理論値に近似する。類似サンプルの元素分析から、4.19%のナトリウム含有量を得たが、これは、上に記載した8水和物(3.7%)またはメチル−t−ブチルエーテレート(3.8%)として算出した値よりわずかに高い。C型のサンプルをTG/IRを用いて分析し、メチル−t−ブチルエーテレートを含有することを見出し、C型がN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のメチル−t−ブチルエーテル溶媒和物であることを確認した。実験後に集めた材料をXRPDにより分析し、C型を維持していることを発見した。
【0076】
2.結晶化の研究
A型およびB型の、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形を調製するための結晶化の研究および詳細な製法を、以下に記載する。これらの研究は、これらの多形を適当な条件下で選択的に生成することができることを実証している。さらにB型、およびA型とB型との混合物は、A型に相互変換することができ、A型がより安定な種であることを示唆する。
【0077】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体結晶の形、すなわちA型およびB型のXRPDパターンを、各々図1および4に示す。これらのXRPDパターンを使用して、以下に記載する結晶化および工程の研究より得た固体の形を同定した。
【0078】
3.近似の溶解度
溶解度は、溶液を得るために使用した総溶媒量に基づいて、実験より見積もった。実際の溶解度は、一定分量ずつという多過ぎる溶媒を使用したこと、または遅い溶解速度のため、算出された値より大きいと考えられる。実験中に溶解が起こらなかった場合は、溶解度は“より小さい”と表す。一定分量の溶媒をすべて加えてしまう前に固体が溶解していた場合は、溶解度は“より大きい”と列記する。
【0079】
周囲温度での多様な溶媒中の、A型のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の近似の溶解度を、表9にまとめる。A型はN,N−ジメチルホルムアミド(228mg/mL)中で最も溶解度が高く、続いてメタノール(160mg/mL)、アセトン(96mg/mL)、テトラヒドロフラン(86mg/mL)、エタノール(60mg/mL)、水(48mg/mL)、およびメチルエチルケトン(34mg/mL)であった。A型は、クロロホルム、ジクロロメタン、およびメチルt−ブチルエーテル(<3mg/mL)には難溶性であった。
【0080】
【表9】
【0081】
4.相互変換の研究
A型およびB型の相互変換は、酢酸エチルおよび95%イソプロパノール:水を使用して行った。A型は、95%イソプロパノール:水中で、熱力学的により安定であると思われる。酢酸エチル中の相互変換ではA型およびB型の混合物を得たが、これはおそらく材料の低溶解度のためである。これらの結果はB型が、熱力学的により不安定な形の形成の方が通常選ばれる貧溶媒による結晶化によって形成された、という事実によって裏付けられる。
【0082】
D.多形の調製のための製法
酢酸エチル中での相互変換の研究に基づくと、A型はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の最も安定な形であると思われる。A型は、緩やかな冷却、スラリー化、または貧溶媒による結晶化より得た。B型は、熱力学的により不安定な形の方の形成が通常選ばれる、貧溶媒による結晶化より得た。C型は、メチルt−ブチルエーテルから貧溶媒による結晶化より得、そしてアモルファス材料は、溶液を緩やかにおよび速く蒸発させることにより得た。
【0083】
ある種の態様において、本明細書において提供したシタキセンタンナトリウムの結晶化の工程は、多形AおよびBの混合物を生成する。ある種の態様において、この混合物は多形AおよびBを約60:40の比率で含有する。他の態様において多形A対Bの比率は、約65:35、70:30,80:20、85:15、90:10、92:8、93:7、94:6、95:5、98:2、96:4、97:3、もしくは99:1である、またはそれより大きい、または等しい。1つの態様において、本明細書に提供する工程は、約100%の多形Aを生成する。1つの態様において、本明細書に提供する工程は、約100%の多形Bを生成する。
【0084】
E.組成物の製剤および投与
当該多形の製剤を本明細書において提供する。製剤は、本明細書に提供する多形の投与のためにデザインされた組成物である。組成物は、経口および非経口投与に適する。そのような組成物は、溶液、懸濁液、錠剤、分散性錠剤、ピル、カプセル、粉末、徐放性製剤、およびその他のあらゆる適切な製剤を含む。1つの態様において、組成物はピルまたは錠剤の形をとることになる。錠剤、カプセル、およびその他のそのような製剤の製造方法は、当業者に公知である(例えばAnsel, H.C. (1885) Forms, Introduction to Pharmaceutical Dosage Forms, 第4版, pp. 126-163を参照のこと)。
【0085】
本明細書に提供する製剤において、有効濃度の多形または多形の混合物を、適切な医薬用担体またはビヒクルとともに混合する。製剤中の多形の濃度は、投与して、エンドセリンを介した疾患の症状を寛解させる量を送達するための有効な量である。ある種の態様において、組成物は単回投与の投与用に製剤化する。組成物を製剤化するため、化合物のある重量画分を、治療する状態が軽減または寛解されるような有効濃度で、選択されたビヒクル中に溶解、懸濁、分散、またはそうでなければ混合する。本明細書に提供する化合物の投与に適する医薬的担体またはビヒクルは、投与の特定の様式に適することが当業者に知られているあらゆるそのような担体を含む。
【0086】
加えて当該化合物は、組成物中のただ1つの医薬的に活性な成分として製剤化してもよいし、または他の活性な成分と組み合わせてもよい。組織を標的とするリポソームを含むリポソーム懸濁液もまた、医薬的に重要可能な担体として適すると考えられる。これらは、当業者に公知の方法に従って調製してよい。例えばリポソーム製剤は、米国特許第4,522,811号に記載されているように調製してよい。
【0087】
多形または多形の混合物としての活性化合物は、治療する患者における望ましくない副作用なしに、治療上有用な効果を発するために十分な量で、医薬的に重要可能な担体中に含まれる。治療有効濃度は、公知のin vitroおよびin vivoの系において化合物を検査することにより経験的に決定し(例えばIshikawaらに対する米国特許第5,114,918号;BANYU PHARMACEUTICAL CO., LTD に対するEP A1 0 436 189 (1991年10月7日); Borges, et al. (1989) Eur. J. Pharm. 165: 223-230; Filep et al. (1991) Biochem. Biophys. Res. Commun. 177: 171-176を参照のこと)、その後それからヒトのための投与量を外挿してよい。
【0088】
薬剤組成物中の活性化合物の多形または多形混合物の濃度は、活性化合物の吸収、不活性化および排泄の速度、化合物の物理化学的特徴、投与計画、ならびに投与する量、同様に当業者に公知のその他の因子に依存することになる。例えば送達される量は、高血圧症の症状を治療するために十分な量である。エンドセリンを介した障害を治療するための有功量は、細菌感染を治療するために投与されるスルホンアミド化合物の量より多いと予想される。
【0089】
1つの態様において治療上有効な投与量は、約0.1ng/mlから約50−100μg/mlの活性成分の血清濃度を生成しなければならない。医薬的投与ユニット剤形は、1投与ユニット剤形当たり、約20mgから約300mgおよび約25mgから約200mg、または約25mgから約100mgの必須活性成分または必須活性成分の組み合わせを提供するように調製する。
【0090】
活性成分は一度に投与してもよいし、または何回かのより少ない用量に分割して時間の間隔をおいて投与してもよい。正確な投与量および治療期間は、治療する疾患の関数であり、公知の試験プロトコルを使用して、またはin vitroもしくはin vivoの検査データからの外挿により決定してよい。濃度および投与量の数値はまた、緩和すべき状態の重症度に伴って変化させてよいことも注意しなければならない。あらゆる特定の被験者に対して、具体的な投与計画が、個人の必要性、および組成物を投与する人または組成物の投与を監督する人の専門的判断に従って、長期にわたり調整されなければならないこと、そして本明細書に記述する濃度範囲は、具体例に過ぎず、主張した組成物の範囲または実践を限定する意図はないことは、さらに理解されなければならない。
【0091】
医薬的に受容可能な誘導体には、酸、塩、エステル、水和物、溶媒和物、およびプロドラッグの形を含む。誘導体は、対応する中性の化合物より安定な形であるよう選択される。
【0092】
したがって本明細書に提供する多形もしくは多形の混合物、または医薬的に受容可能なその誘導体の有効濃度または有効量を、全身、局所的または局部的な投与のための適切な医薬的担体またはビヒクルと共に混合して、医薬組成物を形成する。
【0093】
組成物は、治療する障害に依存して、経口、非経口、経直腸、ならびに局所的および局部的を含む、適切な経路により投与することを意図する。例えば眼科的障害、例えば緑内障の治療には、眼球内、また硝子体内への注射用の製剤を意図する。1つの態様において、カプセルおよび錠剤は経口投与用に使用する。本明細書に記載したように調製した凍結乾燥粉末の再構成は、非経口投与用に使用してよい。液体、半液体、または固体の剤形の化合物は、各投与経路に適する様式で製剤化する。投与の様式は、非経口および経口の投与様式を含む。
【0094】
非経口、皮内、皮下、または局所塗布に使用する溶液または懸濁液は、以下の成分:滅菌希釈剤、例えば注射用水、生理食塩水溶液、固定油、ポリエチレングリコール、グリセリン、プロピレングリコール、またはその他の合成溶媒;抗生剤、例えばベンジルアルコールおよびメチルパラベン;抗酸化剤、例えばアスコルビン酸および亜硫酸水素ナトリウム;キレート剤、例えばエチレンジアミン四酢酸(EDTA);バッファー、例えば酢酸塩、クエン酸塩、およびリン酸塩;浸透圧の調整剤、例えば塩化ナトリウムまたはブドウ糖、のいずれかを含むことができる。非経口調製物は、アンプル、使い捨ての注射器、またはガラス、プラスチックもしくはその他の適切な材料で作られた単回用量もしくは多数回用量のバイアル中に封入することができる。
【0095】
化合物が十分な溶解度を示さない場合、化合物を可溶化するための方法を使用してよい。そのような方法は当業者に公知であり、共溶媒、例えばジメチルスルホキシド(DMSO)の使用、界面活性剤 例えばツイーンの使用、または重炭酸ナトリウム水溶液中での溶解を含むがこれに限定されない。化合物の誘導体、例えば化合物のプロドラッグもまた、有効な医薬組成物の製剤化において使用してよい。
【0096】
スルホンアミド化合物(1つまたは複数)のナトリウム塩を混合または添加することで得られる混合物は、溶液、懸濁液、エマルジョン等であってよい。得られる混合物の形は、意図した投与様式、および選択した担体またはビヒクル中の化合物の溶解度を含む、いくつかの因子に依存する。有効濃度は、治療する疾患、障害または状態の症状を寛解させるための十分な量であり、経験的に決定してよい。
【0097】
製剤は、ヒトおよび動物への投与用に、適切な量の化合物、特に医薬的に重要可能なその塩、例えばナトリウム塩のを含有する、ユニット投与剤形で、例えば錠剤、カプセル、ピル、粉末、顆粒、滅菌非経口溶液または懸濁液、および経口溶液または懸濁液、および油−水エマルジョンの形で提供する。医薬的、治療的に活性な化合物およびその誘導体は、ある種の態様において、ユニット投与剤形または複数回投与剤形にて製剤化し、投与する。本明細書において使用する場合ユニット用量剤形は、ヒトおよび動物の被験者に適する、そして当該技術で公知であるような個々に包装された、物理的に個別のユニットをいう。各ユニット用量は、必要な医薬的担体、ビヒクル、または希釈剤と共同して、所望の治療効果を得るために十分な、治療的に活性な化合物のあらかじめ決定された量を含有する。ユニット用量の剤形の例として、アンプルおよび注射器、個々に包装された錠剤またはカプセルを含む。ユニット−用量剤形は、その分画またはその複数個で投与してもよい。複数回−用量の剤形は、分離されたユニット−用量剤形で投与される、1つの容器内に包装された複数個の個別ユニット−投与剤形である。複数回−用量の剤形の例は、バイアル、錠剤もしくはカプセルのビン、またはパイントもしくはガロンのビンを含む。この場合、複数回用量の剤形は、包装内で分離されていない複数のユニット−用量である。
【0098】
組成物は活性成分と共に:希釈剤、例えばラクトース、ショ糖、リン酸二カルシウム、またはカルボキシメチルセルロース;滑剤、例えばステアリン酸マグネシウム、ステアリン酸カルシウムおよびタルク;ならびに結合剤、例えばスターチ、天然のガム 例えばアカシアゼラチンガム、グルコース、糖蜜、ポリビニルピロリジン、セルロースおよびその誘導体、ポビドン、クロスポビドン、および当業者に公知のその他のそのような結合剤、を含有することができる。液体の医薬的に投与可能な組成物は、例えば上に定義したような活性化合物および所望による医薬的なアジュバントを、担体中、例えば水、生理食塩水、ブドウ糖水溶液、グリセロール、グリコール、エタノール、等の中で、溶かす、分散させる、またはそうでなければ混合することにより調製して、それにより溶液または懸濁液を形成することができる。所望であれば、投与する医薬組成物はまた、少量の非毒性の補助的物質、例えば湿潤剤、乳化剤、または可溶化剤、pH緩衝化剤等、例えばアセテート、クエン酸ナトリウム、シクロデキストリン誘導体、モノラウリン酸ソルビタン、酢酸ナトリウムトリエタノールアミン(triethanolamine sodium acetate)、オレイン酸トリエタノールアミン、およびその他のそのような物質を含有してよい。そのような投与剤形を調製する実際の方法は、当業者に公知である、または明らかになるだろう;例えばRemington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa. 第15版, 1975を参照のこと。投与する組成物または製剤は、いずれにしても、治療する被験者の症状を緩和するための十分な量の、一定量の活性化合物を含有することになる。
【0099】
非毒性担体で重量調製した0.005%から100%の範囲の活性成分を含有する投与剤形または組成物を調製してよい。経口投与用に、医薬的に受容可能な非毒性組成物は、一般に利用される賦形剤、例えば医薬グレードのマンニトール、ラクトース、スターチ、ステアリン酸マグネシウム、滑石、セルロース誘導体、クロスカルメロースナトリウム、グルコース、ショ糖、炭酸マグネシウム、サッカリンナトリウム、のいずれかを組み込むことにより形成する。そのような組成物として、溶液、懸濁液、錠剤、カプセル、粉末、ならびに徐放性製剤、例えば非限定的に、植込錠およびマイクロカプセル化送達システム、および生分解性、生体適合性のポリマー 例えばコラーゲンエチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、ポリオルトエステル、ポリ乳酸、およびその他の物質を含む。これらの製剤の調製法は、当業者に公知である。ある態様において意図した組成物は、0.001%−100%の活性成分を、もう1つの態様において0.1−85%、もう1つの態様において75−95%の活性成分を含有してよい。
【0100】
組成物は、体からの急速な排出に対して当該化合物を保護する担体、例えば時間で放出される製剤またはコーティングを用いて調製してよい。
製剤は、所望の組み合わせの特性を得るため、他の活性化合物を含んでよい。多形はまた、治療または予防の目的のため、本明細書で上述した1つまたはそれより多くの疾患または医学的状態を治療する上で、価値があることが一般に知られている別の薬学的物質、例えばベータ−アドレナリン遮断薬(例えばアテノロール)、カルシウムチャネル遮断薬(例えばニフェジピン)、アンジオテンシン変換酵素(ACE)阻害薬(例えばリシノプリル)、利尿薬(例えばフロセミドまたはヒドロクロロチアジド)、エンドセリン変換酵素(ECE)阻害薬(例えばホスホラミドン)、中性エンドペプチダーゼ(NEP)阻害薬、HMGCoA還元酵素阻害薬、一酸化窒素供与体、抗酸化薬、血管拡張薬、ドーパミンアゴニスト、神経保護薬、ステロイド、ベータ−アゴニスト、抗凝血薬、または血栓溶解薬、と合わせて有利となるように投与してよい。そのような併用療法は、本明細書に提供する治療の組成物および方法のさらなる側面を構成することは、理解されなければならない。
【0101】
本明細書に提供するラクトースを含まない組成物は、当該技術において周知されている賦形剤、および例えばU.S. Pharmacopeia (USP) 25-NF20 (2002)に列記されている賦形剤を含有することができる。一般にラクトースを含まない組成物は、医薬的に適合性のあるそして医薬的に受容可能な量の、活性成分、結合剤/フィラー、および滑剤を含有する。特定のラクトースを含まない投与剤形は、活性成分、微結晶セルロース、アルファ化スターチ、およびステアリン酸マグネシウムを含有する。
【0102】
水は一部の化合物の分解を促進する可能性があるため、活性成分を包含する無水の医薬組成物および投与剤形をさらに提供する。例えば水の添加(例えば5%)は、時間にわたる有効期間または安定性といった特徴を決定する目的で、長期的保存をシュミレートする手段として医薬技術において広く受け入れられている。例えばJens T. Carstensen, Drug Stability: Principles & Practice, 第2版, Marcel Dekker, NY, NY, 1995, pp. 379-80を参照のこと。事実、水および熱は一部の化合物の分解を加速する。したがって湿度および/または湿気には普通、製剤の製造、取り扱い、包装、保存、出荷、および使用時に遭遇するため、製剤における水の影響は非常に重要となり得る。
【0103】
本明細書に提供する無水の医薬組成物および投与剤形は、無水または低湿度を含有する成分、および低湿度または低い湿気の条件を使用して調製することができる。
無水医薬組成物は、その無水という性質が維持されるように調製し保存しなければならない。したがって無水組成物は一般に、適切な製剤キットに含めることができるような、水への暴露を防ぐことが知られている材料を使用して包装される。適切な包装の例として、密封されたホイル、プラスチック、ユニット用量の容器(例えばバイアル)、ブリスター包装、およびストリップ包装を含むがこれに限定されない。
【0104】
1.経口投与用製剤
経口医薬投与剤形は、固体、ゲル、または液体のどれかである。固体の投与剤形は、錠剤、カプセル、顆粒、および原末である。経口錠剤のタイプには、腸溶コーティング、糖衣、またはフィルムコーティングであってよい、圧縮型の咀嚼可能なロゼンジおよび錠剤を含む。カプセルは、ハードまたはソフトのゼラチンカプセルであってよく、一方顆粒および粉末は、当業者に公知の他の成分と組み合わせて、非発泡性または発泡性の剤形で提供してよい。そのような投与剤形は、予め決定された量の活性成分を含有し、当業者に周知の薬学の方法により調製してよい。一般的には、Remington’s Pharmaceutical Sciences, 第20版, Mack Publishing, Easton PA (2000)を参照のこと。
【0105】
ある種の態様において、製剤は固体の剤形、例えばカプセルまたは錠剤である。錠剤、ピル、カプセル、トローチ等は、以下の成分、または類似の性質の複合体(conjugate);結合剤;フィラー、希釈剤;崩壊剤;滑剤;潤沢剤;甘味剤;および香味剤のいずれかを含有することができる。本明細書に提供する経口投与剤形に使用することのできる賦形剤の例は、結合剤、フィラー、崩壊剤、および滑剤を含むが、これに限定されない。医薬組成物および投与剤形における使用に適する結合剤は、コーンスターチ、ポテトスターチ、またはその他のスターチ、ゼラチン、天然および合成のガム 例えばアカシア、アルギン酸ナトリウム、アルギン酸、その他のアルギン酸塩、粉末トラガカント、グアーガム、セルロースおよびその誘導体(例えばエチルセルロース、酢酸セルロース、カルボキシメチルセルロールカルシウム、カルボキシメチルセルロースナトリウム)、ポリビニルピロリドン、メチルセルロース、アルファ化スターチ、ヒドロキシプロピルメチルセルロース、(例えばNo2208、No2906、No2910)、微結晶セルロース、およびそれらの混合物を含むが、これに限定されない。
【0106】
微結晶セルロースの適切な形は、AVICEL-PH-101、AVICEL-PH-103、AVICEL RC-581、AVICEL-PH-105(FMC Corporation、American Viscose Division、Avicel Sales、Marcus Hook、PAより入手可能)として販売されている材料、およびそれらの混合物を含むがこれに限定されない。具体的な結合剤は、微結晶セルロース、およびAVICEL RC-581として販売されているカルボキシメチルセルロースナトリウムの混合物である。適切な無水または低湿度の賦形剤または添加剤は、AVICEL-PH-103 および Starch 1500 LMを含む。
【0107】
本明細書に開示する医薬組成物および投与剤形における使用に適するフィラーの例は、タルク、炭酸カルシウム(例えば顆粒または粉末)、微結晶セルロース、粉末セルロース、デキストレート、カオリン、マンニトール、ケイ酸、ソルビトール、スターチ、アルファ化スターチ、およびそれらの混合物、を含むがこれに限定されない。本明細書における医薬組成物中の結合剤またはフィラーは、典型的には医薬組成物または投与剤形の約50から約99重量パーセントで存在する。
【0108】
崩壊剤は、水性環境に暴露された時に崩壊する錠剤を提供するため、本明細書に提供する組成物中に使用する。あまりにも多量の崩壊剤を含有する錠剤は、保存中に崩壊してしまうかもしれないが、一方であまりにも少量しか含有しないものは、所望の速度でまたは所望の条件下で崩壊しないかもしれない。したがって活性成分の放出を有害に変化させる程多過ぎも少な過ぎもしない十分な量の崩壊剤を使用して、本明細書に提供する固形の経口投与剤形を形成しなければならない。使用する崩壊剤の量は、製剤のタイプに基づいて変動するが、当業者に容易に識別できよう。典型的な医薬組成物は、約0.5から約15重量パーセントの崩壊剤、または約1から約5重量パーセントの崩壊剤を含有する。
【0109】
本明細書に提供する医薬組成物および投与剤形中に使用することのできる崩壊剤は、寒天(agar-agar)、アルギン酸、炭酸カルシウム、微結晶セルロース、クロスカルメロースナトリウム、クロスポビドン、ポラクリリンカリウム、グルコール酸スターチナトリウム、ポテトまたはタピオカのスターチ、その他のスターチ、アルファ化スターチ、その他のスターチ、粘土(clay)、その他のアルギン、その他のセルロース、ガム、およびそれらの混合物を含むがこれに限定されない。
【0110】
本明細書に提供する医薬組成物および投与剤形中に使用することのできる滑剤は、ステアリン酸カルシウム、ステアリン酸マグネシウム、鉱油、軽油、グリセリン、ソルビトール、マンニトール、ポリエチレングリコール、その他のグリコール、ステアリン酸、ラウリル硫酸ナトリウム、タルク、水素化植物油(例えばピーナツ油、綿実油、ヒマワリ油、ゴマ油、オリーブ油、コーン油、および大豆油)、ステアリン酸亜鉛、オレイン酸エチル、ラウリン酸エチル、寒天(agar)、およびそれらの混合物を含むがこれに限定されない。付加的な滑剤として、例えばsyloidシリカゲル(W.R. Grace Co. of Baltimore, MD により製造されたAEROSIL(登録商標)200)、合成シリカの凝集エアロゾル(coagulated aerosol)(Degussa Co. of Plano, TXより市販されている)、CAB-O-SIL (Cabot Co. of Boston, MA より販売されている発熱性シリコン二酸化物の製品)、およびそれらの混合物を含む。仮に使用する場合には、滑剤は典型的には、それらが組み込まれる医薬組成物または投与剤形の約1重量パーセントより少ない量で使用する。
【0111】
経口投与が所望される場合、多形および多形の混合物は、腸溶性コーティング錠、糖衣錠、フィルムコーティグ錠、または多層圧縮錠として形成される組成物中に提供することができる。腸溶コーティング錠は、胃の酸性環境から活性成分を保護する。糖衣錠は、医薬的に受容可能な物質の別々の層が塗布されている圧縮錠である。フィルムコーティング錠は、ポリマーまたはその他の適切なコーティングで覆われた圧縮錠である。多層圧縮錠は、先に述べた医薬的に受容可能な物質を利用して、1回より多くの圧縮サイクルにより作製された圧縮錠である。着色剤もまた、上の投与剤形に使用してよい。香味剤および甘味剤は、圧縮錠、糖衣錠、多層圧縮錠、および咀嚼可能な錠剤において使用される。香味剤および甘味剤は、咀嚼可能な錠剤およびロゼンジの形成においてとくに有用である。組成物はまた、制酸薬またはその他のそのような成分と組み合わせて製剤化してもよい。
【0112】
投与ユニット剤形がカプセルである場合、上のタイプの材料に加えて液体の担体、例えば脂肪油を含有することができる。ゼラチンカプセルにおいては、例えばポリエチレンカーボネート、植物油、またはトリグリセリド中に、シテキセンタンナトリウムを含有する溶液または懸濁液を、カプセル内に封入する。そのような溶液、ならびにその調製および封入は、米国特許第4,328,245号;4,409,239号;および4,410,545号に開示されている。
【0113】
活性成分はまた、所望の作用を損なうことのない他の活性材料と共に、または所望の作用を補う材料、例えば制酸薬、H2遮断薬、および利尿薬と共に混合することができる。活性成分の重量の約98%までの高濃度を含んでよい。
【0114】
液体経口投与剤形は、水性の溶液、エマルジョン、懸濁液、非発泡性の顆粒から再構成される溶液および/または懸濁液、ならびに発泡性の顆粒から再構成される発泡性の調製物を含む。水性溶液として、例えばエリキシルおよびシロップを含む。エリキシルは透明な甘みを加えた含水アルコール調製物である。エリキシル中に使用する医薬的に受容可能な担体は、溶媒を含む。シロップは、糖分、例えばショ糖の濃縮水溶液であり、保存剤を含有してよい。
【0115】
エマルジョンは、一方の液体がもう一方の液体全体に小さな球状の形で分散している、2相系である。エマルジョン中に使用する医薬的に受容可能な担体は、非水性の液体、乳化剤および保存剤である。懸濁液は、医薬的に受容可能な懸濁剤および保存剤を使用する。
液体の経口投与剤形に再構成される非発泡性顆粒中に使用する医薬的に受容可能な物質は、希釈剤、甘味剤、および湿潤剤を含む。液体の経口投与剤形に再構成される発泡性顆粒中に使用する医薬的に受容可能な物質は、有機酸および二酸化炭素の原料を含む。着色剤および香味剤は、上の投与剤形のすべてにおいて使用される。
【0116】
溶媒は、グリセリン、ソルビトール、エチルアルコール、およびシロップを含む。保存剤の例は、グリセリン、メチルパラベンおよびプロピルパラベン、安息香酸、安息香酸ナトリウムおよびアルコールを含む。エマルジョンに利用する非水性液体の例は、鉱物油および綿実油を含む。乳化剤の例は、ゼラチン、アカシア、トラガカント、ベントナイト、および界面活性剤 例えばポリオキシエチレンソルビタンモノオレエートを含む。懸濁剤は、カルボキシメチルセルロースナトリウム、ペクチン、トラガカント、ヴィーガム(Veegum)およびアカシアを含む。
【0117】
希釈剤は、ラクトースおよびショ糖を含む。甘味剤は、ショ糖、シロップ、グリセリンおよび合成甘味剤 例えばサッカリンを含む。湿潤剤は、モノステアリン酸プロピレングリコール、モノオレイン酸ソルビタン、モノラウリン酸ジエチレングリコール、ポリオキシエチレンラウリルエーテルを含む。有機酸は、クエン酸および酒石酸を含む。二酸化炭素の原料は、炭酸水素ナトリウムおよび炭酸ナトリウムを含む。着色剤は、認可され保証された水溶性FDおよびC色素のいずれか、およびそれらの混合物を含む。香味剤は、植物 例えば果物から抽出された天然の香味剤、および心地よい味覚を生じる化合物の合成ブレンドを含む。
【0118】
ミセル型の活性成分を含有する医薬組成物は、米国特許第6,350,458号に記載されているように調製することができる。そのような医薬組成物は、経口、経鼻、および頬側塗布において特に有用である。
【0119】
ある種の態様において、製剤は、本明細書に提供する多形または多形の混合物、1,2−ジメトキシメタン、ジグリム、トリグリム、テトラグリム、ポリエチレングリコール−350−ジメチルエーテル、ポリエチレングリコール−550−ジメチルエーテル、ポリエチレングリコール−750−ジメチルエーテル(ここで350、550および750はポリエチレングリコールのおよその平均分子量をいう)を含むがこれに限定されないジアルキル化モノ−またはポリ−アルキレングリコール、そして1つまたはそれより多くの抗酸化剤、例えばブチル化ヒドロキシトルエン(BHT)、ブチル化ヒドロキシアニソール(BHA)、没食子酸プロピル、ビタミンE、ヒドロキノン、ヒドロキシクマリン、エタノールアミン、レシチン、セファリン、アスコルビン酸、リンゴ酸、ソルビトール、リン酸、チオジプロピオン酸およびそのエステル、ならびにジチオカルバメート、を含有するものを含むがこれに限定されない。
【0120】
他の製剤は、医薬的に受容可能なアセタールを含む水性アルコール性溶液、を含むがこれに限定されない。これらの製剤中に使用するアルコールは、プロピレングリコールおよびエタノールを含むがこれに限定されない、1つまたはそれより多くのヒドロキシル基を有する、あらゆる医薬的に受容可能な水混和性の溶媒である。アセタールは、低級アルキルアルデヒドのジ(低級アルキル)アセタール、例えばアセトアルデヒドジエチルアセタールを含むがこれに限定されない。
【0121】
ある種の態様において、多形および多形の混合物は、約50mg、約75mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mgの活性成分、を含有する経口錠剤として製剤化する。カプセルは、不活性成分、例えばポリエチレングリコール400、ポリソルベート20、ポビドン、およびブチル化ヒドロキシアミソールを含有することができる。カプセルの殻は、ゼラチン、ソルビトールと特定のゼラチンのブレンドおよび二酸化チタンを含有することができる。
【0122】
具体例としての経口錠剤の製剤
ある種の態様において、本明細書に提供する方法は、本明細書に提供する多形または多形の混合物を含有する経口錠剤の投与に関与する。1つの態様において、経口錠剤はさらに、バッファーを含有する。1つの態様において、経口錠剤はさらに抗酸化剤を含有する。1つの態様において、経口錠剤はさらに湿度をバリアーするコーティングを含有することができる。
【0123】
一部の態様において錠剤は、抗酸化剤、例えばアスコルビン酸ナトリウム、グリシン、メタ重亜硫酸ナトリウム、パルミチン酸アスコルビル、エデト酸二ナトリウム(EDTA)、またはそれらの組み合わせ;結合剤、例えばヒドロキシプロピルメチルセルロース;希釈剤、例えばラクトース一水和物fast flo(粒内)およびラクトース一水和物fast flo(粒外)を含むラクトース一水和物、ならびに微結晶セルロース、ならびにバッファー、例えばリン酸バッファー、を含むがこれに限定されない賦形剤を含有する。錠剤はさらに、滑剤、崩壊剤および充填剤より選択される1つまたはそれより多くの賦形剤を含有することができる。
【0124】
ある種の態様において、経口錠剤中のシタキセンタンナトリウムの量は、組成物の総重量の約5%から約40%である。ある種の態様において、シタキセンタンナトリウムの量は、組成物の総重量の約7%から約35%、10%から約30%、12%から約32%、15%から約30%、17%から約27%、15%から約25%である。ある種の態様において、シタキセンタンナトリウムの量は、組成物の総重量の約5%、7%、9%、10%、12%、15%、17%、20%、22%、25%、27%、30%、35%、または40%である。ある種の態様において、シタキセンタンナトリウムの量は約20%である。
【0125】
ある種の態様において経口錠剤は、約10mg、20mg、25mg、30mg、40mg、50mg、60mg、70mg、80mg、90mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mg、280mg、300mg、または350mgのシタキセンタンナトリウムを含有する。
【0126】
ある種の態様において錠剤は、2つの抗酸化剤の組み合わせ、例えばパルミチン酸アスコルビルおよびEDTA、二ナトリウムを含有する。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、錠剤の総重量の約0.05%から約3%の範囲にある。他の態様において、パルミチン酸アスコルビルの量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1%である。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、錠剤の総重量の約0.2%である。
【0127】
ある種の態様において経口錠剤中のパルミチン酸アスコルビルの量は、約0.1mgから約5mg、約0.5mgから約4mg、約0.7mgから約3mg、または約1mgから約2mgである。ある種の態様において経口錠剤中のパルミチン酸アスコルビルの量は、約0.1mg、0.5mg、0.7mg、1mg、1.3mg、1.5mg、1.7mg、2mg、2.5mg、または約3mgである。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は約1mgである。
【0128】
ある種の態様において製剤中のEDTA、二ナトリウムの量は、錠剤の総重量の約0.05%から約3%の範囲にある。他の態様においてEDTA、二ナトリウムの量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において製剤中のEDTA、二ナトリウムの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1%である。ある種の態様において錠剤中のEDTA、二ナトリウムの量は、錠剤の総重量の約0.2%である。
【0129】
ある種の態様において経口錠剤中のEDTA、二ナトリウムの量は、約0.1mgから約5mg、約0.5mgから約4mg、約0.7mgから約3mg、または約1mgから約2mgである。ある種の態様において経口錠剤中のEDTA、二ナトリウムの量は、約0.1mg、0.5mg、0.7mg、1mg、1.3mg、1.5mg、1.7mg、2mg、2.5mg、または約3mgである。ある種の態様において、経口錠剤中のEDTA、二ナトリウムの量は約1mgである。
【0130】
ある種の態様において、錠剤は希釈剤の組み合わせ、例えば微結晶セルロース(AVICEL PH 102)、ラクトース一水和物fast flo(粒内)およびラクトース一水和物fast flo(粒外)を含有する。ある種の態様において、経口錠剤中のラクトース一水和物fast flo(粒内)の量は、組成物の総重量の約5%から約30%である。ある種の態様において、ラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約7%から約25%、約10%から約20%、約13%から約20%である。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約5%、7%、10%、13%、14%、15%、15.5%、16%、16.1%、16.2%、16.3%、16.4%、16.5%、16.6%、16.7%、16.8%、16.9%、17%、17.5%、18%、18.5%、19%、20%、25%または30%である。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約16.9%である。
【0131】
ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約40mgから約100mg、約45mgから約95mg、約50mgから約90mgである。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約40mg、45mg、50mg、55mg、60mg、65mg、70mg、75mg、80mg、81mg、82mg、83mg、83.5mg、84mg、84.1mg、84.2mg、84.3mg、84.4mg、84.5mg、84.6mg、84.7mg、85mg、85.5mg、90mg、90.5mg、または100mgである。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約84.3mgである。
【0132】
ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約7%から約25%、約10%から約20%、約13%から約20%である。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約5%、7%、10%、13%、14%、15%、15.5%、16%、16.1%、16.2%、16.3%、16.4%、16.5%、16.6%、16.7%、16.8%、16.9%、17%、17.5%、18%、18.5%、19%、20%、25%または30%である。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約16.4%である。ある種の態様において、経口錠剤中のラクトース一水和物fast flo(粒外)の量は、約40mgから約100mg、約45mgから約95mg、約50mgから約90mgである。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、約40mg、45mg、50mg、55mg、60mg、65mg、70mg、75mg、80mg、81mg、81.3mg、81.5mg、81.8mg、82mg、82.3mg、82.5mg、82.7mg、83mg、83.5mg、84mg、85mg、85.5mg、90mg、90.5mg、または100mgである。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、約82mgである。
【0133】
ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、組成物の総重量の約10%から約50%である。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約15%から約45%、約20%から約43%、約25%から約40%である。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約15%、17%、20%、23%、25%、27%、30%、32%、34%、35%、37%、40%、42%、45%、または50%である。ある種の態様において微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約35%である。
【0134】
ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、約130mgから300mgである。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、約140mgから約275mg、または約150mgから約250mgである。ある種の態様において微結晶セルロース(AVICEL PH 102)の量は、約150mg、160mg、165mg、170mg、175mg、180mg、185mg、190mg、または200mgである。ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、約175mgである。
【0135】
ある種の態様において、結合剤はヒドロキシプロピルメチルセルロース(E−5P)である。ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、組成物の総重量の約0.5%から約20%である。ある種の態様において、ヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約1%から約15%、約2%から約10%、約3%から約8%である。ある種の態様においてヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約1%、2%、3%、4%、5%、6%、7%、8%、9%、または10%である。ある種の態様においてヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約5%である。
【0136】
ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約5mgから約50mg、約10mgから約40mg、約15mgから約30mgである。ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約10mg、15mg、20mg、22mg、25mg、27mg、30mg、35mg、または約40mgである。ある種の態様において錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約25mgである。
【0137】
本明細書に提供するシタキセンタンナトリウムの製剤は、中性のpHで安定である。ある種の態様において、バッファー剤の混合物、例えば一塩基性リン酸ナトリウム一水和物、および二塩基性リン酸ナトリウム無水物を使用して、錠剤中の薬剤の安定性を改善することができる。ある種の態様において一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.05重量%から約3重量%の範囲にある。他の態様において一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において、製剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1.%である。ある種の態様において、製剤中の一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.1%である。
【0138】
ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.1mgから約3mg、約0.2mgから約2.5mg、約0.5mgから約2mg、または約0.6mgから約1mgである。ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.1mg、0.2mg、0.3mg、0.4mg、0.5mg、0.6mg、0.7mg、0.8mg、0.9mg、または1mgである。ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.6mgである。
【0139】
ある種の態様において二塩基性リン酸ナトリウム無水物の量は、錠剤の総重量の約0.05重量%から約3重量%の範囲にある。他の態様において二塩基性リン酸ナトリウムの量は、錠剤の総重量の約0.07%から約1.5%、約0.1%から約1%、約0.15%から約0.5%の範囲にある。ある種の態様において、製剤中の二塩基性リン酸ナトリウムの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1.%である。ある種の態様において製剤中の二塩基性リン酸ナトリウムの量は、錠剤の総重量の約0.2%である。
【0140】
ある種の態様において、経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約0.1mgから約3.5mg、約0.5mgから約2.5mg、または約0.7mgから約2mgである。ある種の態様において、経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約0.1mg、0.3mg、0.5mg、0.7mg、0.9mg、1mg、1.1mg、1.3mg、1.5mg、1.7mg、または2mgである。経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約1.1mgである。
【0141】
ある種の態様において錠剤は、崩壊剤、例えばナトリウムスターチグリコレート(粒内)およびナトリウムスターチグリコレート(粒外)を含有する。ある種の態様において錠剤中のナトリウムスターチグリコレート(粒内)の量は、組成物の総重量の約0.1%から約10%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約0.5%から約8%、約1%から約5%、約2%から約4%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約0.5%、1%、1.5%、1.7%、2%、2.3%、2.5%、2.7%、3%、3.5%、4%、または5%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約2.5%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約30mgから約5mg、約20mgから約10mg、約15mgから約10mgである。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約5mg、7mg、10mg、11mg、11.5mg、12mg、12.5mg、13mg、15mg、または20mgである。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約12.5mgである。
【0142】
ある種の態様において錠剤中のナトリウムスターチグリコレート(粒外)の量は、組成物の総重量の約0.1%から約10%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約0.5%から約8%、約1%から約5%、約2%から約4%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約0.5%、1%、1.5%、1.7%、2%、2.3%、2.5%、2.7%、3%、3.5%、4%、または5%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約2.5%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約30mgから約5mg、約20mgから約10mg、約15mgから約10mgである。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約5mg、7mg、10mg、11mg、11.5mg、12mg、12.5mg、13mg、15mg、または20mgである。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約12.5mgである。
【0143】
ある種の態様において、錠剤は滑剤、例えばステアリン酸マグネシウムを含有する。ある種の態様において錠剤中のステアリン酸マグネシウムの量は、組成物の総重量の約0.1%から約8%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約0.5%から約6%、約0.7%から約5%、約1%から約4%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約0.5%、0.7%、1%、1.2%、1.5%、1.7%、2%、2.5%、または3%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約2.5%である。ある種の態様において錠剤中のステアリン酸マグネシウムの量は、約15mgから約1mgである。ある種の態様においてステアリン酸マグネシウムの量は、約10mgから約3mg、または約7mgから約5mgである。ある種の態様においてステアリン酸マグネシウムの量は、約3mg、4mg、4.5mg、5mg、6mg、7mg、8mg、9mgまたは10mgである。ある種の態様においてステアリン酸マグネシウムの量は、約5mgである。
【0144】
1つの態様において、本明細書に提供する錠剤製剤は、湿度をバリアーするコーティングを含有する。適切なコーティング材料は当該技術において公知であり、セルロース由来 例えばセルロースフタレート(セピフィルム(Sepifilm)、ファーマコート(Pharmacoat))、またはポリビニル由来のセピフィルムECLタイプ、またはショ糖由来 例えばセピスパース(Sepisperse) DR、AS、AP OR K (着色)タイプの糖衣用の糖、例えばセピスパース Dry 3202 Yellow、Blue Opadry、Eudragit EPO およびOpadry AMBのどれかのコーティング剤を含むがこれに限定されない。コーティングは、シタキセンタンナトリウムの酸化を阻止するための湿度のバリアーとして役立つ。ある種の態様においてコーティング材料は、セピフィルムLP014/セピスパース Dry 3202 Yellow (セピフィルム/セピスパース) (3/2wt/wt)を、約1から約7%、または約4%の錠剤の重量増量である。ある種の態様においてコーティング材料は、セピフィルムLP 014/セピスパース Dry 3202 Yellow (セピフィルム/セピスパース)である。ある種の態様においてセピフィルム/セピスパースの比率は、1:2、1:1、または3:2wt/wtである。ある種の態様においてセピフィルム/セピスパースコーティングは、約1%、2%、3%、4%、5%、6%、または7%の錠剤の重量増量である。ある種の態様においてセピフィルム/セピスパースコーティングは、約1.6%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、約0.5%、0.8%、1%、1.3%、1.6%、2%、2.4%、2.5%、3%、または4%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、約2.4%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、1錠当たり約1mg、3mg、5mg、6mg、7mg、8mg、9mg、10mg、13mg、15mg、または20mgである。ある種の態様においてセピスパース Dry 3202(黄色)は、1錠あたり約8mgである。ある種の態様においてセピフィルムLP 014は、約0.5%、1%、1.5%、2%、2.2%、2.4%、2.6%、3%、3.5%、または4%の錠剤の重量増量である。ある種の態様においてセピフィルムLP 014は、約2.4%の錠剤の重量増量である。ある種の態様においてセピフィルムLP 014は、1錠当たり約5mg、7mg、9mg、10mg、11mg、12mg、13mg、15mg、17mg、または20mgである。ある種の態様においてセピフィルムLP 014コーティングは、1錠当たり約12mgである。
【0145】
ある種の態様において錠剤は、シタキセンタンナトリウム、微結晶セルロース、ラクトース一水和物fast flo(粒内)、ラクトース一水和物fast flo(粒外)、ヒドロキシプロピルメチルセルロースE−5P、パルミチン酸アスコルビル、EDTA二ナトリウム、一塩基性リン酸ナトリウム一水和物、二塩基性リン酸ナトリウム無水物、ナトリウムスターチグリコレート(粒内)、ナトリウムスターチグリコレート(粒外)、ステアリン酸マグネシウム、およびセピフィルムLP014/セピスパース Dry 3202 Yellowのコーティングを含有する。
【0146】
ある種の態様において錠剤は、約20%のシタキセンタンナトリウム、約35%の微結晶セルロース、約16.9%のラクトース一水和物fast flo(粒内)、約16.4%のラクトース一水和物fast flo(粒外)、約5.0%のヒドロキシプロピルメチルセルロースE−5P、約0.2%のパルミチン酸アスコルビル、約0.2%のエデト酸二ナトリウム(EDTA)、約0.1%の一塩基性リン酸ナトリウム一水和物、約0.2%の二塩基性リン酸ナトリウム無水物、約2.5%のナトリウムスターチグリコレート(粒外)、約2.5%のナトリウムスターチグリコレート(粒内)、および約1%のステアリン酸マグネシウムを含有する。錠剤はさらに、セピフィルムLP014のコーティングを約2.4%の重量増量にて、およびセピスパース Dry 3202 Yellowのコーティングを約1.6%の重量増量にて含有する。
【0147】
ある種の態様において、本明細書に提供する経口錠剤は、約100mgのシタキセンタンナトリウム、約1.0mgのパルミチン酸アスコルビル、約1.0mgのエデト酸二ナトリウム(EDTA)、約25mgのヒドロキシプロピルメチルセルロースE−5P、約84.3mgのラクトース一水和物fast flo(粒外)、約82mgのラクトース一水和物fast flo(粒内)、約175mgの微結晶セルロース、約0.6mgの一塩基性リン酸ナトリウム一水和物、約1.1mgの二塩基性リン酸ナトリウム無水物、約12.5mgのナトリウムスターチグリコレート(粒内)、約12.5mgのナトリウムスターチグリコレート(粒外)、約5mgのステアリン酸マグネシウム、non-bovine、および約192.5mgの純水を含有する、500mgの錠剤である。錠剤はさらにセピフィルムLP014のコーティングを約12mgで、セピスパース Dry 3202 Yellowのコーティングを約8mgで含有する。
【0148】
b.徐放性投与剤形
本明細書に提供する多形は、コントロールされた放出手段により、または当業者に周知されている送達デバイスにより投与することができる。例として、米国特許第3,845,770号;3,916,899号;3,536,809号;3,598,123号;および4,008,719号、5,674,533号、5,059,595号、5,591,767号、5,120,548号、5,073,543号、5,639,476号、5,354,556号、5,639,480号、5,733,566号、5,739,108号、5,891,474号、5,922,356号、5,972,891号、5,980,945号、5,993,855号、6,045,830号、6,087,324号、6,113,943号、6,197,350号、6,248,363号、6,264,970号、6,267,981号、6,376,461号、6,419,961号、6,589,548号、6,613,358号、6,699,500号、および6,740,634号に記載されているものを含むが、これに限定されない。またこれらの各文献を本明細書において参照として援用する。そのような投与剤形を使用して、例えばヒドロキシプロピルメチルセルロース、他のポリマーマトリクス、ゲル、透過可能な膜、浸透圧システム、多層コーティング、微粒子、リポソーム、マイクロスフェア、またはそれらを組み合わせを使用することで、1つまたはそれより多くの活性成分の緩やかなまたはコントロールされた放出を提供することができ、これらの比率を変化させて所望の放出プロフィールを提供することができる。本明細書に記載するものを含む、当業者に公知の適切なコントロールされた放出の製剤は、本明細書に提供する活性成分と共に使用するために容易に選択することができる。
【0149】
すべてのコントロールされた放出の医薬製品は、それらのコントロールされていない対応製品によって達成されるものを上回る薬剤療法の改善という、共通の目標を有する。理想的には、医学的治療における所望によりデザインされたコントロールされた放出の調製物の使用は、最小量の薬剤物質を利用して、最小時間で状態を治癒またはコントロールすることを特徴とする。コントロールされた放出の製剤の利点として、延長される薬剤の活性、低減される投与頻度、および増加される患者のコンプライアンスを含む。加えてコントロールされた放出の製剤を使用して、作用またはその他の特徴、例えば薬剤の血液レベルの開始時間に影響を及ぼすことができ、したがって副作用(例えば有害な影響)の発生に影響を及ぼすことができる。
【0150】
最もコントロールされた放出の製剤は、即時に所望の治療効果を生む一定量の薬剤(活性成分)を最初に放出し、そして延長された時間にわたりこのレベルの治療効果または予防効果を維持するために、別の量の薬剤を徐々にそして継続的に放出するようにデザインされる。この一定レベルの薬剤を体内に維持する目的のため、薬剤は、代謝され体内から排泄される薬剤の量と置き換わる速度で、投与剤形から放出されなければならない。活性成分のコントロールされた放出は、pH、温度、酵素、水、またはその他の生理学的条件もしくは化合物を含むがこれに限定されない様々な条件によって、刺激され得る。
【0151】
ある種の態様において当該多形または多形の混合物は、静脈内注入、植え込み型浸透圧ポンプ、経皮パッチ、リポソーム、またはその他の投与様式を使用して投与してよい。1つの態様において、ポンプを使用してよい(Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al., Surgery 88:507 (1980); Saudek et al., N. Engl. J. Med. 321:574 (1989)を参照のこと)。もう1つの態様において、ポリマー材料を使用することができる。なおもう1つの態様において、コントロールされた放出システムは、治療標的の近位に置く、すなわち、したがって全身用量の一分画のみしか必要としないことが可能である(例えばGoodson, Medical Applications of Controlled Release, vol. 2, pp. 115-138 (1984)を参照のこと)。一部の態様において、コントロールされた放出のデバイスは、被験者の体内に不適当な免疫活性化部位または腫瘍部位の近位に導入する。その他のコントロールされた放出システムは、Langerによる総説(Science 249:1527-1533 (1990)において議論されている。活性成分は、固体の内側のマトリクス中に、例えばポリメチルメタクリレート、ポリブチルメタクリレート、可塑化または非可塑化ポリ塩化ビニル、可塑化ナイロン、可塑化ポリエチレンテレフタレート、天然ゴム、ポリイソプレン、ポリイソブチレン、ポリブタジエン、ポリエチレン、エチレン−ビニルアセテートコポリマー、シリコンゴム、ポリジメチルシロキサン、炭酸シリコンコポリマー、親水性ポリマー 例えばアクリル酸およびメタクリル酸のエステルのヒドロゲル、コラーゲン、架橋したポリビニルアルコールおよび架橋し部分的に加水分解したポリビニル酢酸の中に分散させることができ、これを、外側のポリマーの膜、例えば体液中で不溶性であるポリエチレン、ポリプロピレン、エチレン/プロピレンコポリマー、エチレン/アクリル酸エチルコポリマー、エチレン/酢酸ビニルコポリマー、シリコンゴム、ポリジメチルシロキサン、ネオプレンゴム、塩素化ポリエチレン、ポリ塩化ビニル、塩化ビニルと酢酸ビニルとのコポリマー、塩化ビニリデン、エチレンおよびプロピレン、イオノマーのポリエチレンテレフタレート、ブチルゴム、エピクロロヒドリンゴム、エチレン/ビニルアルコールコポリマー、エチレン/酢酸ビニル/ビニルアルコールターポリマー、およびエチレン/ビニルオキシエタノールコポリマーで覆う。その後活性成分は、放出速度コントロールステップにおいて、外側のポリマーの膜を通って拡散する。そのような非経口組成物中に含有される活性成分のパーセンテージは、その具体的な性質、ならびに被験者の必要性にかなり依存する。
【0152】
c.非経口投与
皮下、筋肉内、または静脈内のどちらかへの注射を一般に特徴とする非経口投与もまた、本明細書において意図する。注射剤は、液体の溶液もしくは懸濁液、注射前に液体中での溶液もしくは懸濁液に適する固体として、またはエマルジョンとしてのどちらかで、従来の剤形において調製することができる。適切な賦形剤は、例えば水、生理食塩水、ブドウ糖、グリセロール、またはエタノールである。加えて所望であれば、投与する医薬組成物はまた、微量の非毒性の補助的物質、例えば湿潤剤または乳化剤、pH緩衝化剤、安定剤、溶解度増強剤、ならびに他のそのような物質、例えば酢酸ナトリウム、モノラウリル酸ソルビタン、オレイン酸トリエタノールアミン、およびシクロデキストリンを含有してよい。
【0153】
当該組成物の非経口投与は、静脈内、皮下、および筋肉内への投与を含む。非経口投与のための調製物は、注射用に用意された滅菌溶液、滅菌乾燥の可溶性製品、例えば皮下注射用錠剤を含む、使用直前に溶媒と組み合わるように用意された凍結乾燥粉末、注射用に用意された滅菌懸濁液、使用直前にビヒクルと組み合わせるように用意された滅菌乾燥の非可溶性製品、および滅菌エマルジョンを含む。溶液は水溶性でも非水溶性でもよい。
【0154】
静脈内に投与する場合、適切な担体は、生理学的食塩水またはリン酸緩衝化食塩水(RBS)、ならびに増粘剤および可溶化剤、例えばグルコース、ポリエチレングリコール、およびポリプロピレングリコール、およびそれらの混合物を含有する溶液を含む。
【0155】
非経口調製物中に使用する医薬的に受容可能な担体は、水性ビヒクル、非水性ビヒクル、抗菌剤、等張剤、バッファー、抗酸化剤、局所麻酔薬、懸濁剤および分散剤、乳化剤、金属イオン封鎖剤またはキレート剤、およびその他の医薬的に受容可能な物質を含む。
【0156】
水性ビヒクルの例は、塩化ナトリウム注射液(Sodium Chloride Injection)、リンゲル注射液(Ringers Injection)、等張性ブドウ糖注射液(Isotonic Dextrose Injection)、滅菌水注射液(Sterile Water Injection)、ブドウ糖および乳酸加リンガー注射液(Dextrose and Lactated Ringers Injection)を含む。非水溶性非経口用ビヒクルは、植物由来の固定油、すなわち綿実油、コーン油、ゴマ油、およびピーナツ油を含む。フェノールまたはクレゾール、水銀剤、ベンジルアルコール、クロロブタノール、メチルおよびプロピルp−ヒドロキシ安息香酸エステル、チメロサール、ベンザルコニウムクロリド、ならびにベンズエトニウムクロリドを含む多数回−用量の容器中に包装される非経口調製剤に、静菌的または静真菌的濃度の抗生物質が、加えられなければならない。等張剤は、塩化ナトリウムおよびブドウ糖を含む。バッファーはリン酸塩およびクエン酸塩を含む。抗酸化剤は、重硫酸ナトリウムを含む。局所鎮静剤は、プロカイン塩酸塩を含む。懸濁剤および分散剤は、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルメチルセルロース、およびポリビニルピロリドンを含む。乳化剤は、ポリソルベート80(TWEEN(登録商標)80)を含む。金属イオン封鎖剤または金属イオンのキレート剤は、EDTAを含む。医薬的担体はまた、水混和性ビヒクルとしてエチルアルコール、ポリエチレングリコール、およびプロピレングリコール、ならびにpH調節剤として水酸化ナトリウム、塩酸、クエン酸、または乳酸を含む。
【0157】
シタキセンタンナトリウムの濃度は、注射が、所望の薬学的効果を得られる有効量を提供するように調整する。正確な用量は、当該技術において公知であるように、患者または動物の年齢、体重および状態に依存する。
【0158】
ユニット用量の非経口調製物は、アンプル、バイアル、または針付き注射器内に充填する。非経口投与のすべての調製物は、当該技術において知られており実践されているように、滅菌しなければならない。
【0159】
実例として、活性成分を含有する滅菌水溶液の静脈内注入または動脈内注入は、有効な投与様式である。もう1つの態様は、所望の薬学的効果を得るために必要時に注射する、活性材料を含有する滅菌の水性または油性の溶液または懸濁液である。
【0160】
注射液は、局所または全身への投与用にデザインする。典型的には治療上有効な投与量は、治療する組織(1つまたは複数)に、少なくとも約0.1%w/wから約90%w/wもしくはそれ以上までの、または1%w/wより多くの濃度のシタキセンタンを含有するように製剤化する。活性成分は、一度に投与してもよいし、または何回かのより少量の用量に分割して、時間の間隔をおいて投与してもよい。正確な投与量および治療期間は、治療する組織の関数であり、公知の試験プロトコルを使用して、またはin vivo もしくはin vitroの検査データからの外挿により経験的に決定してよいことは、理解されよう。濃度および投与量の数値はまた、治療する個体の年齢によって変更してよいことには注意しなければならない。あらゆる特定の被験者に対して、具体的な投与計画が、個々の必要性、および投与する人もしくは製剤の投与を監督する人の専門的判断に従って、時間にわたり調整されなければならないこと、そして本明細書に述べた濃度範囲は、具体例に過ぎず、主張した製剤の範囲または実践を限定する意図はないことは、さらに理解されなければならない。
【0161】
当該多形または多形の混合物は、微粒子化した形または他の適切な形で懸濁してもよいし、または誘導体化して、より可溶性の活性製品を生成するもしくはプロドラッグを生成してもよい。得られる混合物の形は、意図した投与様式、および選択した担体またはビヒクルにおけるシタキセンタンナトリウムの溶解度を含む、いくつかの因子に依存する。有効濃度は、状態の症状を緩和するために十分な濃度であり、経験的に決定してよい。
【0162】
d.凍結乾燥粉末
凍結乾燥粉末もまた本明細書において提供するが、これは、溶液、エマルジョンおよびその他の混合物として投与用に再構成することができる。これらはまた、固体またはゲルとして再構成および製剤化してよい。
【0163】
滅菌の凍結乾燥粉末は、活性成分または医薬的に受容可能なその塩を、適切な溶媒中に溶かすことによって調製する。溶媒は、溶解度を改善する賦形剤、または粉末のその他の医薬成分、または粉末から調製されて再構成される溶液を含有してよい。使用してよい賦形剤は、ブドウ糖、ソルビトール、果糖、コーンシロップ、キシリトール、グリセリン、グルコース、ショ糖、またはその他の適切な物質を含んでよいが、これに限定されない。溶媒はまた、バッファー、例えばクエン酸塩、リン酸ナトリウムもしくはリン酸カリウム、または当業者に公知の、典型的にはほぼ中性のpHのその他のそのようなバッファーを含有してよい。その後の溶液の滅菌濾過に続いて、当業者に公知の標準的な条件下での凍結乾燥により、所望の製剤を提供する。一般に得られた溶液は、凍結乾燥用のバイアル内に分注することになる。各バイアルは、シタキセンタンナトリウムの単回投与量(10mg−350mgもしくは100−300mg)、または複数回投与量を含有することになる。凍結乾燥粉末は、適当な条件下、例えば約4℃から室温までで保存することができる。
【0164】
この凍結乾燥粉末の注射用水を用いての再構成は、非経口投与で使用するための製剤を提供する。再構成には、1mL滅菌水または他の適切な担体当たり、約1−50mg、5−35mg、または約9−30mgの凍結乾燥粉末を加える。正確な量は、選択される複合体(conjugate)に依存する。そのような量は経験的に決定することができる。
【0165】
具体例としての凍結乾燥製剤
ある種の態様において、シタキセンタンナトリウムの安定な凍結乾燥粉末を本明細書において提供する。凍結乾燥粉末は、抗酸化剤、バッファー、および充填剤を含有する。本明細書に提供する凍結乾燥粉末において、存在するシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約25%から約60%の範囲にある。ある種の態様においてシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約30%から約50%、または約35%から約45%である。ある種の態様においてシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約30%、33%、35%、37%、40%、41%、43%、45%、47%、50%、53%、55%、または60%である。1つの態様において凍結乾燥粉末中のシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約41%である。
【0166】
ある種の態様において凍結乾燥粉末は、抗酸化剤、例えば亜硫酸ナトリウム、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、モノチオグリセロール、アスコルビン酸、またはそれらの組み合わせを含有する。1つの態様において、抗酸化剤はモノチオグリセロールである。1つの態様において抗酸化剤は、アスコルビン酸、亜硫酸ナトリウム、および亜硫酸水素ナトリウムを組み合わせたものである。ある種の態様において、本明細書に提供する凍結乾燥製剤は、公知のシタキセンタンナトリウムの凍結乾燥製剤(WO 98/49162を参照のこと)と比較して、再構成時に改善された安定性を有する。
【0167】
ある種の態様において、抗酸化剤はモノチオグリセロールである。ある種の態様においてモノチオグリセロールは、凍結乾燥粉末の総重量の約10%から約30%の範囲である量で存在する。ある種の態様においてモノチオグリセロールは、凍結乾燥粉末の総重量の約12%から約25%、または約15%から約20%の範囲である量で存在する。ある種の態様において、凍結乾燥粉末中のモノチオグリセロールの量は、凍結乾燥粉末の総重量の約10%、12%、14%、15%、15.5%、16%、16.2%、16.4%、16.8%、17%、17.5%、19%、22%、25%、または30%である。ある種の態様においてモノチオグリセロールの量は、凍結乾燥粉末の総重量の約16.4%である。
【0168】
ある種の態様において亜硫酸ナトリウムは、凍結乾燥粉末の総重量の約1%から約6%の量で存在する。他の態様において亜硫酸ナトリウムは、約1.5%から約5%、または約2%から約4%の量で存在する。ある種の態様において亜硫酸ナトリウムの量は、凍結乾燥粉末の総重量の約1%、1.5%、2%、2.5%、3%、3.3%、3.5%、3.8%、4%、4.5%、または5%である。1つの態様において亜硫酸ナトリウムの量は、凍結乾燥粉末の総重量の約3.3%である。
【0169】
ある種の態様においてアスコルビン酸は、凍結乾燥粉末の総重量の約1%から約6%の量で存在する。他の態様においてアスコルビン酸は、約1.5%から約5%、または約2%から約4%の量で存在する。ある種の態様においてアスコルビン酸の量は、凍結乾燥粉末の総重量の約1%、1.5%、2%、2.5%、3%、3.3%、3.5%、3.8%、4%、4.5%、または5%である。1つの態様においてアスコルビン酸の量は、凍結乾燥粉末の総重量の約3.3%である。
【0170】
ある種の態様において亜硫酸水素ナトリウムは、凍結乾燥粉末の総重量の約5%から約15%、または約8%から約12%の量で存在する。ある種の態様において亜硫酸水素ナトリウムの量は、凍結乾燥粉末の総重量の約5%、6%、7%、8%、9%、10%、10.3%、10.5%、10.8%、11%、11.5%、12%、または15%の量で存在する。1つの態様において亜硫酸水素ナトリウムの量は、凍結乾燥粉末の総重量の約10.8%である。
【0171】
1つの態様において抗酸化剤は、アスコルビン酸、亜硫酸ナトリウム、および亜硫酸水素ナトリウムの組み合わせである。1つの態様において凍結乾燥粉末中のアスコルビン酸の量は、凍結乾燥粉末の総重量の約3.3%、亜硫酸ナトリウムの量は約3.3%、および亜硫酸水素ナトリウムの量は約10.8%である。
【0172】
1つの態様において凍結乾燥粉末はまた、以下の賦形剤:バッファー、例えばリン酸ナトリウムもしくはリン酸カリウム、またはクエン酸塩;および充填剤、例えばグルコース、ブドウ糖、マルトース、ショ糖、ラクトース、ソルビトール、マンニトール、グリシン、ポリビニルピロリドン、デキストラン、の1つまたはそれより多くを含有する。1つの態様において充填剤は、ブドウ糖、D−マンニトール、またはソルビトールより選択する。
【0173】
ある種の態様において、本明細書に提供する凍結乾燥粉末は、リン酸バッファーを含有する。ある種の態様においてリン酸バッファーは、約10mM、約15mM、約20mM、約25mM、または約30mMの濃度で存在する。ある種の態様において、リン酸バッファーは20mMの濃度で存在する。ある種の態様において、リン酸バッファーは約20mMの濃度で存在し、そして構成された製剤は約7のpHを有する。
【0174】
ある種の態様において、本明細書に提供する凍結乾燥粉末は、クエン酸バッファーを含有する。1つの態様においてクエン酸バッファーは、クエン酸ナトリウム二水和物である。ある種の態様においてクエン酸ナトリウム二水和物の量は、凍結乾燥粉末の総重量の約5%から約15%、約6%から約12%、または約7%から約10%である。ある種の態様において、凍結乾燥粉末中のクエン酸ナトリウム二水和物の量は、凍結乾燥粉末の総重量の約5%、6%、7%、7.5%、8%、8.3%、8.5%、8.8%、9%、9.5%、10%、12%、または約15%である。ある種の態様において構成される製剤は、約5から10、または約6のpHを有する。
【0175】
ある種の態様において本明細書に提供する凍結乾燥粉末は、ブドウ糖を、凍結乾燥粉末の総重量の約30%から約60%の範囲である量で含有する。ある種の態様においてブドウ糖の量は、凍結乾燥粉末の総重量の約30%、35%、40%、45%、50%、または60%である。ある種の態様においてブドウ糖の量は、凍結乾燥粉末の総重量の約40%である。ある種の態様において本明細書に提供する凍結乾燥粉末は、マンニトールを、凍結乾燥粉末の総重量の約20%から約50%の範囲である量で含有する。ある種の態様においてマンニトールの量は、凍結乾燥粉末の総重量の約20%、25%、30%、32%、32.5%、32.8%、33%、34%、37%、40%、45%、または50%、である。ある種の態様においてマンニトールの量は、凍結乾燥粉末の総重量の約32.8%である。
【0176】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約41%のシタキセンタンナトリウム、約3.3%のアスコルビン酸、約3.3%の亜硫酸ナトリウム、および約10.8%の亜硫酸水素ナトリウム、約8.8%のクエン酸ナトリウム二水和物、および約32.8%のマンニトールを含有する。ある種の態様において、凍結乾燥粉末は以下の組成を有する:
【0177】
【表10】
【0178】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約40から約30%のシタキセンタンナトリウム、約4から約6%のアスコルビン酸、約6から約8%のクエン酸ナトリウム二水和物、約50から約60%のD−マンニトール、および約1から約2%のクエン酸一水和物を含有する。ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約33%のシタキセンタンナトリウム、約5.3%のアスコルビン酸、約7.6%のクエン酸ナトリウム二水和物、約53%のD−マンニトール、および約0.13%のクエン酸一水和物を含有する。1つの態様において、凍結乾燥粉末は以下の組成を有する:
【0179】
【表11】
【0180】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約40から約30%のシタキセンタンナトリウム、約4から約6%のアスコルビン酸、約3から約4%の二塩基性リン酸ナトリウム七水和物、約50から約60%のD−マンニトール、および約1.5から約2.5%の一塩基性リン酸ナトリウム一水和物を含有する。ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約34%のシタキセンタンナトリウム、約5.5%のアスコルビン酸、約3.7%の二塩基性リン酸ナトリウム七水和物、約55%のD−マンニトール、および約1.9%の一塩基性リン酸ナトリウム一水和物を含有する。1つの態様において、凍結乾燥粉末は以下の組成を有する:
【0181】
【表12】
【0182】
本明細書に提供するシタキセンタンナトリウムの凍結乾燥製剤は、本明細書に記載する方法を含むがこれに限定されない、シタキセンタンナトリウムを送達するための標準的な治療法を使用して、それを必要とする患者に投与することができる。1つの態様において凍結乾燥シタキセンタンナトリウムは、本明細書に提供する凍結乾燥シタキセンタンナトリウムの治療有効量を、医薬的に受容可能な溶媒中に溶かして医薬的に受容可能な溶液を生成し、その溶液を(例えば静脈内注射により)患者に投与することにより、投与する。
【0183】
本明細書に提供する凍結乾燥シタキセンタンナトリウム製剤は、あらゆる医薬的に受容可能な希釈剤を使用して、患者への非経口投与用に構成することができる。そのような希釈剤として、注射用滅菌水(Sterile Water for Injection)、USP、注射用滅菌静菌水(Sterile Bacteriostatic Water for Injection)、生理食塩水、USP(保存剤としてベンジルアルコールまたはパラベン)を含むがこれに限定されない。注射用の適切な溶液が調製されるように、あらゆる量の希釈剤を使用して、凍結乾燥シタキセンタンナトリウム製剤を構成してよい。したがって希釈剤の量は、凍結乾燥シタキセンタンナトリウムを溶解させるための十分量でなければならない。典型的には10−50mL、または10から20mLの希釈剤を使用して、凍結乾燥シタキセンタンナトリウム製剤を構成し、約1−50mg/mL、約5−40mg/mL、約10−30mg/mL、または10−25mg/mLの最終濃度を得る。ある種の態様において、再構成される溶液中のシタキセンタンナトリウムの最終濃度は、約25mg/mL、または約12.5mg/mLである。正確な量は、治療する適応症に依存する。そのような量は経験的に決定することができる。一部の態様において、再構成される溶液のpHは約5から約10まで、または約6から約8までである。一部の態様において、再構成される溶液のpHは約5、6、7、8、9、または10である。
【0184】
凍結乾燥シタキセンタンナトリウムの構成される溶液は、構成後直ちに患者に投与することができる。あるいは構成された溶液を保存して、約1−72時間、約1−48時間、または約1−24時間以内に使用することができる。一部の態様において、溶液は調製の1時間以内に使用する。
【0185】
e.局所投与
局所用混合物は、局在的投与および全身投与用として記載したように調製する。得られる混合物は、溶液、懸濁液、エマルジョン、等であってよく、クリーム、ゲル、軟膏、エマルジョン、溶液、エリキシル、ローション、懸濁液、チンキ、ペースト、フォーム、エアゾール、灌水、スプレー、座薬、絆創膏、皮膚パッチ、または局所投与に適するあらゆるその他の製剤であってよい。
【0186】
シタキセンタンナトリウムは局在的または局所的投与用に、例えば皮膚および粘膜への局所塗布用に、ゲル、クリームおよびローションの剤形で製剤化してよい。局所投与は、経皮送達として、およびまた粘膜投与として、または吸入療法として意図する。
【0187】
f.他の投与経路に関する組成物
その他の投与経路、例えば局所塗布、経皮パッチ、および直腸投与もまた本明細書において意図する。例えば直腸投与用の医薬投与剤形は、全身への効果のための直腸用座薬、カプセル、および錠剤である。本明細書において使用する直腸用座薬は、体温で融けるかまたは軟化して、1つまたはそれより多くの医薬的にまたは治療的に活性な成分を放出する、直腸内への挿入のための固形のボディーを意味する。直腸用座薬中に利用する医薬的に受容可能な物質は、ベースまたはビヒクル、および融点を上昇させる剤である。ベースの例は、カカオバター(cocoa butter)(カカオ脂(theobroma oil))、グリセリン−ゼラチン、カーボワックス(ポリオキシエチレングリコール)、ならびに脂肪酸のモノ−、ジ−、およびトリ−グリセリドの適当な混合物を含む。様々なベースの組み合わせを使用してよい。座薬の融点を上昇させる剤は、鯨ろうおよびワックスを含む。直腸用座薬は、圧縮法または成形のどちらかにより調製してよい。直腸用座薬の典型的な重量は、約2から3グラムである。
【0188】
直腸投与用錠剤およびカプセルは、経口投与用製剤と同じ医薬的に受容可能な物質を使用して、そして同じ方法により製造する。
g.製造品
当該多形または多形の混合物は、シタキセンタンナトリウムを拡張期心不全を治療するために使用することを指示する、包装材料およびラベルを含有する製造品として包装してよい。本明細書に提供する製造品は、包装材料を含有する。医薬品の包装に使用するための包装材料は、当業者に周知されている。例えば米国特許第5,323,907号、5,052,558号および5,033,352号を参照のこと。医薬用包装材料の例として、ブリスターパック、瓶、管、吸入器、ポンプ、袋、バイアル、容器、注射器、瓶、ならびに選択した製剤および意図した投与様式および治療様式に適するあらゆる包装材料を含むが、これに限定されない。本明細書に提供するシタキセンタンの広く多数の製剤を、本明細書において意図する。
【0189】
投与量
ヒトの治療において医師は、予防処置または治癒処置に従って、そして治療する被験者に特定の年齢、体重、疾患の段階、およびその他の因子に従って、最も適当である投与計画を決定することになる。ある種の態様において、シタキセンタンナトリウムの用量のレートは、成人に関して1日当たり約1から約300mg、1日当たり約5から約250mg、1日当たり約5から約250mg、または1日当たり約10から50mgである。1日当たり約50から約300mgの用量のレートもまた本明細書において意図する。ある種の態様において用量は、成人1日当たり約5mg、10mg、15mg、20mg、25mg、30mg、35mg、40mg、45mg、50mg、60mg、70mg、80mg、100mg、125mg、150mg、175mg、または200mgである。
【0190】
拡張期心不全、またはその1つもしくはそれより多くの症状の予防または治療において有効となる、本明細書に提供する製剤中のシタキセンタンナトリウムの量は、疾患または状態の性質および重症度、および活性成分を投与する経路に伴って変わることになる。頻度および投与量もまた、投与する具体的な治療(例えば治療薬または予防薬)、障害、疾患、または状態の重症度、投与経路、ならびに被験者の年齢、体重、応答および過去の病歴に依存する、各被験者に特定な因子に従って変わることになる。
【0191】
製剤の具体例としての用量は、被験者またはサンプルの体重1キログラム当たりの活性化合物のミリグラムまたはマイクログラムの量(例えば1キログラム当たり約1マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約10マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約100マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約100マイクログラムから1キログラム当たり約2ミリグラム)を含む。ある種の態様において、投与するシタキセンタンナトリウムの量は、それを必要とする被験者に対して約0.01から約3mg/kgである。ある種の態様において、投与するシタキセンタンナトリウムの量は、約0.01、0.05、0.1、0.2、0.4、0.8、1.5、2、3mg/被験者の体重1kgである。ある種の態様においてシタキセンタンナトリウムの投与は、静脈内注射による。
【0192】
当業者には明らかとなるように、一部の場合には本明細書に開示する範囲外で活性成分の投与量を使用することが必要となるかもしれない。さらに臨床医または担当医は、被験者の応答を考慮しながら、どのようにそしていつ治療を中断、調整、または終了するかを知ることになることも言及しておく。
【0193】
拡張期心不全の症状を予防、管理、治療、または緩和するためには十分であるが、本明細書に提供する組成物に関連する有害作用の原因となる程ではない、または有害作用を低減するには十分である量もまた、上に記載した投与量および用量の頻度の計画により包括的に含まれる。さらに被験者に、本明細書に提供する組成物を多数回投与量を投与する場合、投与量のすべてが同一である必要はない。例えば被験者に投与する投与量は、組成物の予防的効果または治療的効果を改善するために増量してもよいし、または特定の被験者が経験している1つまたはそれより多くの副作用を低減するために減量してもよい。
【0194】
もう1つの態様において本明細書に提供する製剤の投与量は、被験者の拡張期心不全の症状を予防、治療、管理、または緩和するために、約1mgから300mg、50mgから250mg、または75mgから200mgのユニット用量で投与する。
【0195】
ある種の態様において、本明細書に提供する同じ製剤の投与を繰り返してもよく、投与は少なくとも1日、2日、3日、5日、10日、15日、30日、45日、2ヶ月、75日、3ヶ月、または6ヶ月まで間隔をあけてよい。
【0196】
F.活性の評価
本明細書に提供する方法におけるシタキセンタンナトリウムの効能を検査するための、標準的な生理学的、薬理学的、および生化学的な方法を利用することができ、そしてそれらは当業者に公知である。例えば米国特許第5,114,918号、EP 0 436 189 A1、Borges, et al. (1989) Eur. J. Pharm. 165: 223-230; Filip et al. (1991) Biochem. Biophys. Res. Commun. 177: 171-176を参照のこと。例えばDHFの治療におけるシタキセンタンナトリウムの効能の評価は、治療期間中に周期的間隔で行うトレッドミル運動検査;心エコー(ECHO)による心室の構造および機能(すなわち左心室の質量)における効果の決定;ドップラーECHOおよび組織ドップラー画像(TDI)による経僧帽弁流入速度(E)対 拡張早期僧帽弁輪部速度(E’)の比率の決定;Minnesota Living with Heart Failure questionnaire (MLHF)によって測定された生活の質(QOL)における変化の決定;および心機能分類評価(NYHA)、を含むがこれに限定されないルーチン検査により行うことができる。例えば、Zile らのHeart failure with a normal ejection fraction: is measurement of diastolic heart failure necessary to make the diagnosis of diastolic heart failure?(正常な駆出率を伴う心不全:拡張期心不全の測定は、拡張期心不全の診断を行うために必要か?)、Circulation 2001;104:779-782 、およびMiguel らのRecommendations for quantification of Doppler echocardiography(ドップラー心エコーの定量化に関する推奨):a report from the Doppler quantification task force of the nomenclature and standards committee of the American society of echocardiography(アメリカ心エコー学会の命名法および基準委員会のドップラー定量化特別委員会からの報告), J. Am. Soc. Echocardiogr. 2002; 15: 167-84を参照のこと。
【0197】
G.併用療法
本明細書に提供する方法において、当該多形および多形の混合物は、例えば単独で、1つもしくはそれより多くの他のエンドセリンアンタゴニストと組み合わせて、または拡張期心不全の治療に有用な別の化合物または治療と組み合わせて使用してよい。例えば当該製剤は、エンドセリン受容体の活性を調節することが知られているその他の化合物、例えば米国特許第6,432,994号;6,683,103号;6,686,382号;6,248,767号;6,852,745号;5,783,705号;5,962,490号;5,594,021号;5,571,821号;5,591,761号;5,514,691号に記載されている化合物と組み合わせて投与することができる。いくつかのその他のエンドセリンアンタゴニストは、上に記載した文献に記載されている。
【0198】
一部の態様において、当該方法は、拡張期心不全の治療において使用する他の化合物と組み合わせてのシタキセンタンナトリウムの投与を伴う。そのような薬剤として、ループ系利尿薬、例えばBumex(登録商標)(ブメタニド)、Lasix(登録商標)(フロセミド)、Demadex(登録商標)(トルセミド);チアジド系利尿薬、例えばHygroton(登録商標)(クロルタリドン)、Hydrodiuril(登録商標)、Esidrix(登録商標)(HCTZ、ヒドロクロロチアジド)、Amiloride、Aldactone(登録商標)(スピロノラクトン);長期作用型硝酸薬、例えばIsordil(登録商標)、Sorbitrate(登録商標)(二硝酸イソソルビド)、Imdur(登録商標)(一硝酸イソソルビド);β−遮断薬、例えばフマル酸ビソプロロール、プロプラノロール、アテノロール、ラベタロール、ソタロール、カルベジロール;カルシウムチャネル遮断薬、例えばNorvasc(登録商標)(アムロジピン)、Cardizem(登録商標)(ジルチアゼム)、Isoptin(登録商標)(ベラパミル)、Procardia(登録商標)(ニフェジピン);腎動脈狭窄症(RAS)阻害薬およびアンジオテンシン変換酵素(ACE)阻害薬、例えばカプトプリル、フォシノプリル、ベナゼプリル、エナラプリル、リシノプリル、モエキシプリル、ペリンドプリル、キナプリル、ラミプリル、スピラプリル、トランドラプリル;アンジオテンシン受容体遮断薬(ARB類)、例えばロサルタン、バルサルタン、イルベサルタン、テルメサルタン(telmesartan)、およびアルドステロンアンタゴニスト、を含むがこれに限定されない。
【0199】
一部の態様において当該方法は、間質性肺疾患の治療に使用される他の化合物、例えば
副腎皮質ステロイド、例えばプレドニソンまたはメチルプレドニソン(活性な進行中の肺胞および間質の炎症および傷害を抑制するために、そして間質性肺疾患の患者を治療する上で使用される)と組み合わせての、シタキセンタンナトリウムの投与を伴う。
【0200】
さらに本明細書に提供する多形は、当該技術において公知のエンドセリンアンタゴニストと組み合わせて利用することができる。これらアンタゴニストは、環式ペンタペプチド、すなわちシクロ(D−Glu−L−Ala−allo−D−lle−L−Leu−D−Trp)であるBE−18257Bと命名されたStreptomyces misakiensisの発酵生成物;BE−18257Bに関連する環式ペンタペプチド、例えばシクロ(D−Asp−Pro−D−Val−Leu−D−Trp)(BQ−123)(Ishikawa らに対する米国特許第5,114,918号を参照のこと;またBANYU PHARMACEUTICAL CO., LTD に対するEP A1 0 436 189(1991年10月7日)を参照のこと)を含むがこれに限定されない;そしてその他のペプチドおよび非ペプチドETAアンタゴニストは、例えば米国特許第6,432,994号;6,683,103号;6,686,382号;6,248,767号;6,852,745号;5,783,705号;5,962,490号;5,594,021号;5,571,821号;5,591,761号;5,514,691号;5,352,800号;5,334,598号;5,352,659号;5,248,807号;5,240,910号;5,198,548号;5,187,195号;5,082,838号;6,953,780号;6,946,481号;6,852,745号;6,835,741号;6,673,824号;6,670,367号;および6,670,362号において同定されている。これらは、他の環式ペンタペプチド、アクリルトリペプチド、ヘキサペプチド類似体、ある種のアントラキノン誘導体、インダンカルボン酸、ある種のN−ピリミニル(pyriminyl)ベンゼンスルホンアミド、ある種のベンゼンスルホンアミド、およびある種のナフタレンスルホンアミドを含む(Nakajima et al. (1991) J. Antibiot. 44:1348-1356;Miyata et al. (1992) J. Antibiot. 45:74-8;Ishikawa et al. (1992) J .Med. Chem. 35:2139-2142;Ishikawa et al.に対する米国特許第5,114,918号;EP A1 0 569 193;EP A1 0 558 258;BANYU PHARMACEUTICAL CO., LTDに対するEP A1 0 436 189(1991年10月7日);カナダ特許出願2,067,288号;カナダ特許出願2,071,193号;米国特許第5,208,243号;米国特許第5,270,313号;米国特許第5,612,359号、米国特許第5,514,696号、米国特許第5,378,715号; Cody et al. (1993) Med. Chem. Res. 3:154-162;Miyata et al. (1992) J. Antibiot 45:1041-1046;Miyata et al. (1992) J. Antibiot 45:1029-1040、Fujimoto et al. (1992) FEBS Lett. 305:41-44;Oshashi et al. (1002) J. Antibiot 45:1684-1685;EP A1 0 496 452;Clozel et al. (1993) Nature 365:759-761;国際特許出願WO93/08799;Nishikibe et al. (1993) Life Sci. 52:717-724;およびBenigni et al. (1993) Kidney Int. 44:440-444)。エンドセリンペプチドアンタゴニストである多数のスルホンアミドもまた、米国特許第5,464,853号;5,594,021号;5,591,761号;5,571,821号;5,514,691号;5,464,853号;国際PCT出願第96/31492号;および国際PCT出願第WO07/27979号に記載されている。
【0201】
以下の文献に記載されているさらなるエンドセリンアンタゴニスト(それらの全内容において本明細書にて参照として援用する)は、本明細書に提供する多形と組み合わせて使用することを意図する薬剤の具体例である:米国特許第5,420,123号;米国特許第5,965,732号;米国特許第6,080,774号;米国特許第5,780,473号;米国特許第5,543,521号;WO 96/06095;WO 95/08550;WO 95/26716;WO 96/11914;WO 95/26360;EP 601386;EP 633259;米国特許第5,292,740号;EP 510526;EP 526708;WO 93/25580;WO 93/23404;WO 96/04905;WO 94/21259;GB 2276383;WO 95/03044;EP 617001;WO 95/03295;GB 2275926;WO 95/08989;GB 2266890;EP 496452;WO 94/21590;WO 94/21259;GB 2277446;WO 95/13262;WO 96/12706;WO 94/24084;WO 94/25013;米国特許第5,571,821号;WO 95/04534;WO 95/04530;WO 94/02474;WO 94/14434;WO 96/07653;WO 93/08799;WO 95/05376;WO 95/12611;DE 4341663;WO 95/15963;WO 95/15944;EP 658548;EP 555537;WO 95/05374;WO 95/05372;米国特許第5,389,620号;EP 628569;JP 6256261;WO 94/03483;EP 552417;WO 93/21219;EP 436189;WO 96/11927;JP 6122625;JP 7330622;WO 96/23773;WO 96/33170;WO 96/15109;WO 96/33190;米国特許第5,541,186号;WO 96/19459;WO 96/19455;EP 713875;WO 95/26360;WO 96/20177;JP 7133254;WO 96/08486;WO 96/09818;WO 96/08487;WO 96/04905;EP 733626;WO 96/22978;WO 96/08483;JP 8059635;JP 7316188;WO 95/33748;WO 96/30358;米国特許第5,559,105号;WO 95/35107;JP 7258098;米国特許第5,482,960号;EP 682016;GB 2295616;WO 95/26957;WO 95/33752;EP 743307;およびWO 96/31492;例えば列挙した文献に記載されている以下の化合物:BQ−123(Ihara, M.ら、“ETA受容体に選択的なきわめて強力な新規エンドセリンアンタゴニストの生物学的プロフィール”, Life Sciences, Vol. 50(4), pp. 247-255 (1992));PD 156707(Reynolds, E.ら、 “経口活性のETA受容体アンタゴニストであるPD 156707の薬理学的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 273(3), pp. 1410-1417 (1995));L−754,142(Williams, D. L.ら、“極めて強力な経口活性の非ペプチジルエンドセリンアンタゴニストであるL−754,142の薬理学”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 275(3), pp. 1518-1526 (1995));SB 209670(Ohlstein, E. H.ら、“合理的にデザインされた強力な非ペプチドエンドセリン受容体アンタゴニストであるSB 209670”, Proc. Natl. Acad. Sci. USA, Vol. 91, pp. 8052-8056 (1994));SB 217242(Ohlstein, E. H.ら、 “非ペプチドエンドセリン受容体アンタゴニストVI:強力なそして高い生体利用可能性のあるエンドセリン受容体アンタゴニストである、SB 217242の薬理学的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 276(2), pp. 609-615 (1996));A−127722(Opgenorth, T. J.ら、“A−127722の薬理学的特徴付け:経口活性の極めて強力なE.sub.TA−選択的受容体アンタゴニスト”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 276(2), pp.473-481 (1996));TAK−044(Masuda, Y.ら、“ヒトエンドセリンA受容体およびエンドセリンB受容体における、新規エンドセリン受容体アンタゴニストであるTAK−044{シクロ [D−α−アスパルチル−3−[(4−フェニルピペラジン−1−イル)カルボニル]−L−アラニルーL−α−アスパルチル−D−2−(2−チエニル)グリシル−L−ロイシル−D−トリプロフィル]二ナトリウム塩}の受容体への結合およびアンタゴニストの特性”、The Journal of Pharmacology and Experimental Therapeutics, Vol. 279(2), pp. 675-685 (1996));ボセンタン(Ro 47-0203, Clozel, M.ら、“新規の強力な経口活性非ペプチドエンドセリン受容体アンタゴニストであるボセンタンの医薬的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 270(1), pp. 228-235 (1994))。
【0202】
本明細書に提供する多形はまた、他のクラスの化合物と組み合わせて投与することができる。本明細書における組み合わせのための化合物の具体例としてのクラスには、エンドセリン変換酵素(ECE)阻害薬、例えばホスホルアミドン;トロンボキサン受容体アンタゴニスト、例えばイフェトロバン;カリウムチャネルオープナー;トロンビン阻害薬(例えばヒルジン等);増殖因子阻害薬、例えばPDGF活性の調節薬、;血小板活性化因子(PAF)アンタゴニスト;抗血小板薬、例えばGPIIb/IIIa遮断薬(例えばアブドキシマブ(abdximab)、エプチフィバチド、およびチロフィバン)、P2Y(AC)アンタゴニスト(例えばクロピドグレル、チクロピジン、およびCS−747)、およびアスピリン;抗凝固薬、例えばワルファリン、低分子量ヘパリン 例えばエノキサパリン、第VIIa因子阻害薬、および第Xa因子阻害薬、レニン阻害薬;アンジオテンシン変換酵素(ACE)阻害薬 例えばカプトプリル、ゾフェノプリル、フォシノプリル、セラナプリル、アラセプリル、エナラプリル、デラプリル、ペントプリル、キナプリル、ラミプリル、リシノプリル、およびそのような化合物の塩;中性エンドペプチダーゼ(NEP)阻害薬;バソペプシダーゼ(vasopepsidase)阻害薬(二重のNEP-ACE阻害薬)例えばオマパトリラトおよびゲモパトリラト;HMG CoA還元酵素阻害薬 例えばプラバスタチン、ロバスタチン、アトルバスタチン、シンバスタチン、NK−104(別名イタバスタチン、またはニスバスタチン(nisvastatin もしくは nisbastatin))およびZD−4522(ロスバスタチン、またはアタバスタチン(atavastatin)もしくはビサスタチン(atavastatin)としても知られている);スクアレン合成酵素阻害薬;フィブラート系薬剤;胆汁酸捕捉薬、例えばクエストラン;ナイアシン;抗アテローム硬化性薬剤、例えばACAT阻害薬;MTP阻害薬;カルシウムチャネル遮断薬、例えばベシル酸アムロジピン;カリウムチャネル活性化薬;アルファ−アドレナリン遮断薬(alpha-adrenergic agents)、ベータ−アドレナリン遮断薬(beta-adrenergic agents)、例えばカルベジロールおよびメトプロロール;抗不整脈薬;利尿薬、例えばクロロチアジド、ヒドロクロロロチアジド、フルメチアジド、ヒドロフルメチアジド、ベンドロフルメチアジド、メチルクロロチアジド、トリクロロメチアジド、ポリチアジド、またはベンゾチアジド、同様にエタクリン酸、トリクリナフェン、クロルタリドン、フロセニルデ、ムソリミン、ブメタニド、トリアムテレン、アミロリド、ならびにスピロノラクトンおよびそのような化合物の塩;血栓溶解薬、例えば組織プラスミノーゲン活性化因子(tPA)、リコンビナントtPA、ストレプトキナーゼ、ウロキナーゼ、プロウロキナーゼ、およびアニソイル化(anisoylated) プラスミノーゲンストレプトキナーゼ活性化因子複合体(APSAC);抗糖尿病薬、例えばビグアナイド系(例えばメトホルミン)、グルコシダーゼ阻害薬(例えばアカルボース)、インスリン、メグリチニド(例えばレパグリニド)、スルホニル尿素(例えばグリメピリド、グリブリド、およびグリピジド)、チアゾリジネジオン(例えばトログリタゾン、ロシグリタゾン、およびピオグリタゾン)ならびにPPAR−ガンマアゴニスト;鉱質コルチコイド受容体アンタゴニスト、例えばスピロノラクトンおよびエプレレノン;成長ホルモン分泌促進薬;aP2阻害薬;非ステロイド系抗炎症薬(NSAIDS)、例えばアスピリンおよびイブプロフェン;ホスホジエステラーゼ阻害薬、例えばPDE III阻害薬(例えばシロスタゾール)およびPDE V阻害薬(例えばシルデナフィル、タダラフィル、バルデナフィル);タンパク質チロシンキナーゼ阻害薬;抗炎症薬;抗増殖薬、例えばメトトレキサート、FK506(タクロリムス、プログラフ)、ミコフェノレート、およびモフェチル;化学療法薬;免疫抑制薬;抗癌薬および細胞傷害性薬剤(例えばアルキル化薬、例えば窒素マスタード、スルホン酸アルキル、ニトロソ尿素、エチレンイミン、およびトリアゼン);代謝拮抗薬、例えば葉酸アンタゴニスト、プリン類似体、およびピリジン類似体;抗生物質、例えばアントラサイクリン系、ブレオマイシン、マイトマイシン、ダクチノマイシン、およびプリカマイシン;酵素、例えばL−アスパラギナーゼ;ファルネシル−タンパク質トランスフェラーゼ阻害薬;ホルモン薬、例えば糖質コルチコイド(例えばコルチゾン)、エストロゲン/抗エストロゲン薬、アンドロゲン/抗アンドロゲン薬、プロゲスチン、および黄体形成ホルモン放出ホルモンアンタゴニスト、酢酸オクトレオチド;微小管重合阻害薬、例えばエクテイナシジン(ecteinascidins)またはそれらの類似体および誘導体:微小管安定化薬、例えばパシタキセル(pacitaxel)(Taxol(登録商標))、ドセタキセル(Taxotere(登録商標))、およびエポチロンA−Fまたはそれらの類似体もしくは誘導体;植物由来産生物、例えばビンカアルカロイド系、エピポドフィロトキシン系、タキサン系;およびトポイソメラーゼ阻害薬:タンパク質のプレニル転移酵素阻害薬:および種々の薬剤、例えばヒドロキシ尿素、プロカルバジン、ミトタン、ヘキサメチルメラミン、白金配位錯体 例えばシスプラチン、サトラプラチン、およびカルボプラチン);シクロスポリン;ステロイド 例えばプレドニゾンまたはデキサメタゾン;金化合物;細胞傷害性薬剤 例えばアザチプリンおよびシクロホスファミド;TNF−アルファ阻害薬 例えばテニダップ;抗TNF抗体または可溶性TNF受容体 例えばエタネルセプト(Enbrel)、ラパマイシン(シロリムスまたはRapamune)、レフルニミド(leflunimide)(Arava);そしてシクロオキシゲナーゼ−2(COX−2)阻害薬 例えばセレコキシブ(Celebrex)およびロフェコキシブ(Vioxx)を含む。
【0203】
上のその他の治療薬は、例えばPhysicians' Desk Reference(PDR)に指示されている量で、またはそうでなければ当業者により決定されたように使用してよい。
H. N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形の使用法
多形A、BおよびCのN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩は、エンドセリンを介した疾患の治療において有用である。これらの治療には、多形A、B型またはC型の有効量を被験者に投与することを包括的に含み、その場合その有効量は、疾患の1つまたはそれより多くの症状を寛解させるのに十分な量である。
【0204】
多形A、BおよびCは、高血圧症、心血管疾患、心筋梗塞を含む心疾患、肺高血圧症、新生児肺高血圧症、エリスロポエチンを介した高血圧症、喘息、気管支収縮を含む呼吸器疾患および炎症性疾患、緑内障および不適切な網膜の灌流を含む眼科的疾患、胃腸疾患、腎不全、エンドトキシンショック、月経障害、産科的状態、創傷、蹄葉炎、勃起不全、月経閉止、骨粗しょう症および代謝性骨疾患、中年女性の卵巣機能低下に関連するのぼせ、異常な血塊パターン、泌尿生殖器の不快感、および心血管疾患の増加する発生率、および中年女性における卵巣機能低下に伴う他の障害、妊娠中毒症、妊娠時のコントロールおよび出産の管理、一酸化窒素減弱障害(nitric oxide attenuated disorders)、アナフィラキシーショック、または出血性ショック、間質性肺疾患、拡張期心不全、ならびに免疫抑制を介した腎血管収縮、の治療に有効である。1つの態様において、疾患は肺高血圧症である。
【0205】
多形A、BおよびCはまた、エンドセリンA(ETA)受容体またはエンドセリンB(ETB)受容体へのエンドセリンペプチドの結合を阻害するために有用である。この阻害は、受容体を、多形A、BもしくはC、または医薬的に受容可能なその誘導体のいずれかと接触させることを包括的に含み、その場合この接触は、受容体とエンドセリンペプチドとの接触前、接触時時、または接触後において達成される。
【0206】
多形A、BおよびCはまた、エンドセリン受容体を介した活性を変化させるために有用である。この変化には、エンドセリン受容体を、多形A、BまたはCと接触させることを包括的に含む。
【0207】
以下の実施例は説明の目的のみとして含んでいるに過ぎず、主張した主題事項の範囲を限定する意図はない。N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩(シタキセンタンナトリウム)は、国際(PCT)特許出願公開第WO 98/49162号に記載されている方法により調製してよい。
【実施例】
【0208】
実施例1
MeOHを用いての生成法:
10gのシタキセンタンナトリウムを、40mLのiPrOAc、30mLのEtOHおよび30mLのMeOH中に懸濁し、透明な溶液が得られるまで75℃で加熱した。この溶液を放置して室温まで冷却したが、溶液は1時間透明なままであった。50mLのMTBEを加え、時間を経て黄色固体が形成された。固体を濾過して集め、MTBEで洗浄し、真空下で乾燥させ、6.4gのシタキセンタンナトリウムを主に多形Aとして得た。
【0209】
実施例2
MeOHを用いない生成法:
10gのシタキセンタンナトリウムを100mLのiPrOAcおよび80mLのEtOH中に懸濁させ、透明な溶液が得られるまで90℃(還流)に加熱した(この溶媒の量を、還流で透明な溶液を得るために必要とした)。溶液を放置して室温まで冷却したが、溶液は1時間透明なままであった。140mLのMTBEを加え、時間を経て非常に少量の淡黄色固体が形成された(より少量のMTBEを加えると、固体の形成には至らなかった)。100mLのMTBEを加え、時間を経てより多くの淡黄色固体が形成された。これらの固体を溶液のデカントにより得(これらの固体はうまく濾過できなかった)、MTBEで洗浄し、真空下で乾燥させ、5.4gのシタキセンタンナトリウムを主に多形Bとして得た。
【0210】
実施例3
湿ったiPrOAcからの再結晶化:
10gのシタキセンタンナトリウムを100mLのiPrOAcおよび5mLの水中に懸濁させ、透明な溶液が得られるまで100℃(還流)に加熱した。溶液を放置して淡黄色固体が形成されるまで、室温まで冷却した。これらの固体を濾過して集め、iPrOHで洗浄し、真空下で乾燥させ、6.4gのシタキセンタンナトリウムのロットを主に多形Aとして得た。
【0211】
実施例4
シタキセンタンナトリウム多形Bの再結晶化:
実施例5からの1.0gのシタキセンタンナトリウムを、1.6mLのiPrOAc、1.6mLのMeOHおよび1.6mLのEtOH中に懸濁し、予め65℃に加熱した油浴に入れた。完全な溶解は5分で得られた。溶液を放置して室温まで冷却したが、溶液は透明なままだった。数日間そのまま静置すると淡黄色固体が形成され、これを濾過して集め、MTBEで洗浄し、真空下で乾燥させ、シタキセンタンナトリウムを〜94%多形Aとして得た。
【0212】
実施例5
シタキセンタンナトリウム多形Aの再結晶化:
688gのシタキセンタンナトリウムを、30分間、60℃で、7.8LのEtOH中で加熱した後、10℃に冷却した。MTBE(35L)を加え、混合液を10℃で濾過した。固体を真空中で乾燥させ、およそ86:14の多形A:多形Bを得た。
【0213】
実施例6
387gのシタキセンタンナトリウムを、3.0Lのイソプロパノール中で2時間スラリーとした後、2日間5℃で冷却した。固体を濾過し、真空中で乾燥させて、344gのシタキセンタンナトリウム得た。この材料を1.72Lのイソプロパノール中で室温で30分間スラリーとした後、5℃に45分間冷却した。生成物を濾過し、真空中で乾燥させて、主に多形Bを得た。
【0214】
実施例7
シタキセンタンナトリウム(23.4kg)を、酢酸イソプロピル(32.8kg)、エタノール(30kg)、およびメタノール(30kg)中に懸濁し、65℃に加熱した。固体が溶解した後、溶液を0.45ミクロンフィルターを通して熱濾過した。濾液を撹拌し、45℃に冷却した。シタキセンタンナトリウムの種結晶を加え、内容物の撹拌を45℃の温度で3時間続けた。MTBE(164.3kg)を45±5℃で、1分当たり1kgを超える速度で、ゆっくり加えた。内容物を、4.5時間にわたり0℃までゆっくり冷却した。0℃でさらに4.5時間、撹拌を続けた。得られた結晶を濾過し、湿ったケークをMTBE(93.6kg)で洗浄した。湿ったケークを、脱液化(de-liquored)するまで窒素下に保持した。この湿ったケークを、残存するMTBEのレベルが500ppmより少なくなるまで、Filter/Dryer中で40℃で緩やかに撹拌しながら乾燥させた。得られた材料は、主に多形A(95:5±3 多形A:多形B)であった。
【0215】
実施例8
比較例
シタキセンタンナトリウムの形成:
50mLのDCM中の10gのシタキセンタンナトリウムの十分に撹拌した懸濁液に、50mLの2NのHClを加え、続いて透明な溶液が得られるまでMeOHを加えた。二層分離し、有機層をMgSO4上で乾燥、濃縮し、真空中で乾燥を完了させて、シタキセンタン(〜9g)を乾燥黄色泡状物質として得た。
【0216】
A.最初の結晶化
シタキセンタンを100mLのEtOAc中に再度溶かし、3×50mLの飽和NaHCO3、食塩水で洗浄し、MgSO4上で乾燥させ、真空中で濃縮、乾燥させた。この材料をDCM中に再度懸濁したところ、濁った溶液を形成し、淡黄色固体が形成された後に5分間撹拌した。150mLのEt2Oを加えた。固体を濾過して集め、1対2のDCM対Et2Oで洗浄し、真空下で乾燥させてシタキセンタンナトリウムを主にアモルファスの材料として得た。
【0217】
B.2回目の結晶化
シタキセンタンナトリウムを200mLの水に再度溶解させ、濃HClでpH〜2に酸性化したところ、非常に淡い黄色の固体が形成された。この固体を濾過して得た。この材料を、100mLのEtOAcに再度溶かし、50mLの食塩水、2×50mLの飽和NaHCO3、食塩水で洗浄し、MgSO4上で乾燥させ、真空中で乾燥、濃縮した。この材料をDCM中に再度懸濁したところ、濁った溶液を形成し、淡黄色固体が形成された後に5分間撹拌した。150mLのEt2Oを加えた。固体を濾過して集め、1対2のDCM対Et2Oで洗浄し、真空下で乾燥させて、6.1gのシタキセンタンナトリウムを主にアモルファスの材料として得た。
【0218】
C.3回目の結晶化
1.0シタキセンタンナトリウムを、10mLのEtOHに懸濁し、透明溶液が得られるまで加熱還流した(この溶媒の量を、透明溶液を得るために必要とした)。この溶液を放置して室温まで冷却したが、1時間透明のままであった。この時点で15mLのMTBEを加えたところ、溶液は混濁した(さらに10mLのMTBEを加えても、30分以内に固体の形成には至らなかった)。この溶液を加熱還流したが、固体のcrash outは防げなかった。固体を濾過して集め、MTBEで洗浄し、真空下で乾燥させ、0.64gのシタキセンタンナトリウムを、アモルファスおよび結晶材料の混合物として得た。
【0219】
修飾は当業者にとって明白であろう故、主張した主題事項は添付の請求項の範囲によってのみ限定されることを意図する。
【図面の簡単な説明】
【0220】
【図1】図1は、多形A、B、Cおよびアモルファス型のXRPDパターンである。
【図2】図2は、多形A、サンプルロットIのXRPDパターンである。
【図3】図3は、多形A、サンプルロットIIのXRPDパターンである。
【図4】図4は、多形A、サンプルロットIIIのXRPDパターンである。
【図5】図5は、多形A、サンプルロットIVのXRPDパターンである。
【図6】図6は、多形A、サンプルロットVのXRPDパターンである。
【図7】図7は、多形A、サンプルロットVIのXRPDパターンである。
【図8】図8は、多形A、サンプルロットVIIのXRPDパターンである。
【図9】図9は、多形Aのラマン吸収スペクトルである。
【図10】図10は、多形A、サンプルロットIのDSCである。
【図11】図11は、多形A、サンプルロットIVのDSCである。
【図12】図12は、多形A、サンプルロットIIIのDSCである。
【図13】図13は、多形A、サンプルロットIのTGである。
【図14】図14は、多形A、サンプルロットIVのTGである。
【図15】図15は、多形A、サンプルロットIIIのTGである。
【図16】図16は、多形A、サンプルロットIの吸湿/放湿である。
【図17】図17は、多形A、サンプルロットIVの吸湿/放湿である。
【図18】図18は、多形A、サンプルロットIIIの吸湿/放湿である。
【図19】図19は、多形BのDSCである。
【図20】図20は、多形BのTGである。
【図21】図21は、多形Bの吸湿/放湿である。
【図22】図22は、多形Bのラマン吸収スペクトルである。
【図23】図23は、多形CのTG/IR吸収スペクトルである。
【図24】図24は、多形CのTGである。
【図25】図25は、多形AのTG/IR吸収スペクトルである。
【図26】図26は、多形BのTG/IR吸収スペクトルである。
【図27】図27は、多形B、サンプルロットIのXRPDパターンである。
【図28】図28は、多形B、サンプルロットIIのXRPDパターンである。
【図29】図29は、多形B、サンプルロットIIIのXRPDパターンである。
【技術分野】
【0001】
本発明は、Reichweinらの2006年3月13日に提出された、“N−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形”という表題の、米国仮特許出願第60/781,861号に対する優先権を主張する。上の関連する出願の開示を本明細書において参照として援用する。
【0002】
(発明の技術分野)
本明細書において、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形、およびそれらの製造法を提供する。
【背景技術】
【0003】
(背景)
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウム塩は、ペプチドのエンドセリンファミリーの活性を調節し、そのためエンドセリンを介した障害の治療に有用である。これらの障害の性質のため、医薬製品としての本化合物の使用は、長期間の保存を必要とすると考えられる。したがって、保存期間中の熱および湿気に対する本化合物(医薬用化学製品の原体)の安定性は、非常に重要である。そのため本化合物のより安定な形が所望される。
【発明の開示】
【課題を解決するための手段】
【0004】
(概要)
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形であるA型およびB型;メチルt−ブチルエーテル溶媒和物であるC型;ならびにアモルファス型は、適当な溶媒および条件からの結晶化により産業スケールで選択的に生産することができることを発見した。さらにB型、ならびにA型およびB型の混合物は、適切な条件下でより安定なA型に相互変換することができる。
【0005】
シタキセンタンナトリウムのアモルファス型はかなり吸湿性であるのに対して、結晶型はそうではない(アモルファスは、95%RHでその総重量の22%増量する;結晶は、95%RHでその重量の1.5%未満しか増量しない)。相互変換の研究により、A型の多形がより熱力学的に安定な形であることを見出した。理論に拘束されるものでは決してないが、アモルファス状態は、時間と共に多形の混合物に変換されると考えられる。
【0006】
特に以下の化学構造:
【0007】
【化1】
【0008】
を有する、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形であるA型およびB型は、選択的に生成することができ、それらの粉末X線回折(XRPD)パターン、赤外線吸収スペクトル、ラマンスペクトル、および融点におけるそれらの特徴的なピークに基づいて識別することができる。
【0009】
XRPDパターンの測定のための方法および条件
測定の方法
XRPD分析は、以下の条件によりサンプルについてShimadzu XRD-6000 粉末X線回折計にて行った。
【0010】
測定の条件
【0011】
【化2】
【0012】
赤外線吸収の測定のための方法および条件
熱重量赤外線(TG/IR)吸収スペクトルは、Nicolet モデル560フーリエ変換赤外線(FT−IR)分光光度計とインターフェースしたTA Instrument TGA 2050にて得た。
【0013】
ラマン吸収の測定のための方法および条件
ラマンスペクトルは、Nicolet Magna 860 FT−IR分光光度計にインターフェースしたラマンベンチ(bench)にて得た。
【0014】
多形A(A型)
角度2θにおいて表わされるA型のXRPDパターンの主要なピークは、およそ6.72、15.96、22.38、23.38および26.22にある。
【0015】
図1−8は、A型のXRPDパターンを示す。
A型のラマンスペクトルの主要なピーク(cm−1)は、およそ1697.4、1602.1、1489.8および1402.2cm−1にある。
【0016】
図9は、A型のラマンスペクトルを示す。
特徴付けのデータに基づくと、A型は、およそ200℃で分解する結晶性、非吸湿性の固体であると思われる。
【0017】
多形B(B型)
角度2θにおいて表わされるB型のXRPDパターンの主要なピークは、およそ6.6、15.52、18.38、18.94および22.72にある。
【0018】
図1は、B型のXRPDパターンを示す。
B型のラマンスペクトルの主要なピーク(cm−1)は、およそ1696.9、1594.7、1490.2および1397.8cm−1にある。
【0019】
図22は、B型のラマンスペクトルを示す。
特徴付けのデータに基づくと、B型は、203℃付近で分解する非吸湿性の結晶材料であると思われる。
【0020】
多形C(C型)
角度2θにおいて表わされるC型のXRPDパターンの主要なピークは、およそ5.14、23.48、および26.78にある。
【0021】
図1は、C型のXRPDパターンを示す。
図23は、C型の赤外線吸収スペクトルを示す。
特徴付けのデータに基づくと、C型は、化合物のメチルt−ブチルエーテル溶媒和物であると思われる。
【0022】
(詳細な説明)
A.定義
他に定義していなければ、本明細書で使用するすべての技術的用語および科学的用語は、本主題の物質の属する当業者により共通して理解されているのと同じ意味を有する。本明細書に述べたすべての特許および公開を参照として援用する。
【0023】
本明細書において使用する場合、“シタキセンタンナトリウム”はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウムをいう。シタキセンタンナトリウムの他の化学名として、4−クロロ−3−メチル−5−(2−(2−メチル−(6−メチルベンゾ[d][1,3]ジオキソル−5−イル)アセチル)−3−チエニルスルホンアミド)イソキサゾール、ナトリウム、およびN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[3,4−(メチレンジオキシ)−6−メチルフェニルアセチル]−チオフェン−3−スルホンアミド、ナトリウムを含む。シタキセンタンナトリウム塩の化学構造は、本明細書において他所に記載している。
【0024】
本明細書において使用する場合、エンドセリン(ET)ペプチドは、エンドセリン−1、エンドセリン−2、またはエンドセリン−3のアミノ酸配列を実質的に有するペプチド、および強力な内在性血管収縮ペプチドとして作用するペプチドを含む。
【0025】
本明細書において使用する場合、エンドセリンを介した状態は、異常なエンドセリン活性によって引き起こされる状態、またはエンドセリン活性を阻害する化合物が治療としての用途を持つ状態である。そのような疾患として、高血圧症、心血管疾患、喘息、炎症性疾患、眼科的疾患、月経障害、産科的状態、胃腸の疾患、腎不全、肺高血圧症、間質性肺疾患、拡張期心不全、エンドトキシンショック、アナフィラキシーショック、または出血性ショックを含むがこれに限定されない。エンドトキシンを介した状態はまた、エンドトキシンレベルを上昇させる薬剤、例えばエリスロポエチンおよび免疫抑制薬による治療に起因する状態を含む。
【0026】
本明細書において使用する場合、特定の疾患を治療するための化合物の有効量は、疾患に関連する症状を寛解させる、または何らかの様式で低減するために十分である量である。そのような量は、それが有効となる、単回投与として投与してもよいし、または投与計画に従って投与してもよい。その量は当該疾患を治癒してもよい。もう1つの態様においてその量は、当該疾患の1つまたはそれより多くの症状を寛解させる目的で投与する。その他の態様において、症状の所望の寛解を達成するために繰り返し投与が必要とされる。
【0027】
本明細書において使用する場合、エンドセリンアゴニストは、エンドセリンペプチドに関連するまたはエンドセリンペプチドの保有する生物学的活性を、増強するまたは示す化合物である。
【0028】
本明細書において使用する場合、エンドセリンアンタゴニストは、エンドセリンに刺激される血管収縮(vasoconstriction)および収縮(contraction)、ならびにその他のエンドセリンを介した生理学的応答を阻害する薬剤または抗体というような化合物である。アンタゴニストは、エンドセリンとエンドセリン特異的受容体との相互作用を妨害することにより、または血管収縮のようなエンドセリンイソペプチドへの生理学的応答もしくはエンドセリンイソペプチドの生理活性を妨害することにより、作用してよい。したがって本明細書において使用する場合、エンドセリンアンタゴニストは、当業者に公知のアッセイにより評価した時に、エンドセリンに刺激される血管収縮もしくはその他の応答を妨害する、またはエンドセリンとエンドセリン特異的受容体、例えばETA受容体との相互作用を妨害する。
【0029】
潜在的アゴニストおよびアンタゴニストの有効性は、当業者に公知の方法を使用して評価することができる。例えばエンドセリンアゴニストの活性は、単離したラットの胸部大動脈または門脈の環状部分の血管収縮を刺激するその能力により、同定することができる(Borges et al. (1989) "Tissue selectivity of endothelin" Eur. J. Pharmacol. 165: 223-230)。
【0030】
本明細書において使用する場合、ETA選択的であるスルホンアミドは、ETA受容体に関してETB受容体と比べて少なくとも約1/10低いIC50を示すスルホンアミドをいう。
【0031】
本明細書において使用する場合、ETB選択的であるスルホンアミドは、ETB受容体に関してETA受容体と比べて少なくとも約1/10低いIC50を示すスルホンアミドをいう。
【0032】
本明細書において使用する場合、治療は、状態、障害もしくは疾患の症状が寛解される、またはそうでなければ有利に変化するあらゆる様式を意味する。治療はまた、本明細書における組成物のあらゆる医薬的使用、例えば避妊薬としての使用も包括的に含む。
【0033】
本明細書において使用する場合、特定の医薬組成物の投与による特定の障害の症状の寛解は、永久的または一時的、持続的または一過性的あるにせよ、当該組成物の投与に起因または関連し得るあらゆる緩和をいう。
【0034】
本明細書において使用する場合、実質的に純粋であることは、そのような純度を評価するために当業者によって使用される標準的な分析法、例えば薄相クロマトグラフィー(TLC)、ゲル電気泳動、および高速液体クロマトグラフィー(HPLC)により決定されるような、容易に検出され得る不純物を含んでいないと思われる十分な均一性、またはさらに精製しても、その物質の物理的特性および化学的特性、例えば酵素活性および生物学的活性を検出可能なほど変化させることがないというように十分に純粋であること、を意味する。実質的に化学的に純粋な化合物を生成するための化合物の精製方法は、当業者に公知である。しかし実質的に化学的に純粋な化合物は、立体異性体の混合物であってよい。そのような場合は、さらなる精製が化合物の特異的活性を増大させることになるかもしれない。
【0035】
本明細書において使用する場合、生物学的活性は、化合物、組成物またはその他の混合物のin vivo投与の結果得られる、化合物のin vivo活性または生理学的応答をいう。したがって生物学的活性は、そのような化合物、組成物および混合物の治療効果および医薬的活性を包括的に含む。
【0036】
本明細書において使用する場合、製剤の増加した安定性は、当業者に公知のアッセイ、例えば高速液体クロマトグラフィー、ガスクロマトグラフィーなどによって決定した場合に、製剤の調製後の所定の時間での、製剤中に存在する活性成分のパーセントが、製剤の調製後の同じ時間での別の製剤中に存在する活性成分のパーセントより、有意に高いことを意味する。この場合、前者の製剤は後者の製剤に比して増加した安定性を有すると言われる。
【0037】
B.分析方法
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の結晶化サンプルは、それらの多形(A型またはB型)、水和物および溶媒和物(C型)を決定するため、それらのXRPD、赤外線吸収スペクトル、ラマンスペクトル、融点、示差走査熱量測定(DSC)、熱重量測定(TG)、高温顕微鏡、および自動吸湿/放湿により分析した。
【0038】
1.XRPD
XRPD分析は、Cu Kα線を使用してShimadzu XRD-6000粉末X線回折計にて行った。この装置にファインフォーカスのX線管球を装備した。管球の出力およびアンペア数は、各々40kVおよび40mAに設定した。発散スリットおよび散乱スリットは1°に設定し、受光スリットは0.15mmに設定した。回折光は、Nalシンチレーションカウンターにより検出した。2.5°(2θ)から40°(2θ)について3°/分(0.4秒/0.02°ステップ)でθ−2θ継続スキャンを使用した。シリコンスタンダードを毎日分析し、装置の調整をチェックした。各サンプルは、クォーツのサンプルホルダーに置くことにより、分析用に調製した。優先配向の効果を低減するため、3つのサンプルをスピンさせながら(25rpm)分析した。スキャンは0.5°/分に調整して、スピン速度について補正した。
【0039】
2.TG/IR
TG/IR吸収は、Nicolet モデル560フーリエ変換赤外線(FT−IR)分光光度計とインターフェースしたTA Instrument TGA 2050にて得た。この装置に、黒体輻射光源(globar source)、Ge/KBrビームスプリッタ、重水素化トリグリセリン硫酸塩(DTGS)検出器を装備した。IR分光光度計は、スタンダードとして使用日毎にポリスチレンを用いて、一方TGは1週間毎にニッケルおよびアルメルを使用して波長を較正した。およそ5mgのサンプルをプラチナのパンに量りとり、ヘリウムパージして、20℃から150℃まで、20℃/分の比率で記録した。IRスペクトルは、4cm−1の解像度で8回の加算したスキャンを表す各スペクトルの連続したものとして得た。揮発性物質は、HR Nicolet TGA 気相スペクトルライブラリを検索して同定した。
【0040】
3.ラマンスペクトル
ラマンスペクトルは、Nicolet Magna 860 FT-IR分光光度計にインターフェースしたラマンベンチにて得た。この装置は、1064nmの励起波長、およびおよそ0.5WのNd:YAGレーザー出力を利用した。スペクトルは、4cm−1の解像度で得た32または64回加算したスキャンを表す。サンプルは、材料をガラス管にいれ、このガラス管を分光光度計にセットすることによる分析用に調製した。分光光度計は、使用時にイオウおよびシクロヘキサンを用いて、(波長を)較正した。
【0041】
4.示差走査熱量計(DSC)
示差走査熱量計のデータは、TA Instruments Differential Scanning Calorimeter 2920にて得た。使用した較正用スタンダードは、インジウムであった。およそ2から5mgのサンプルをDSCパンに置き、重さを正確に測定し記録した。パンは密封して封入し、圧を放出させるためにピンホールを使用した。サンプルは、窒素下で10℃/分の比率で、最終温度300℃まで加熱した。アモルファス材料のガラス転位温度(Tg)の研究用には、サンプルは窒素下、10℃/分の比率で125℃まで加熱した。サンプルをこの温度で15分間保持した後、放置して冷却し、25℃での平衡状態とした。サンプルを再び10℃/分の比率で125℃まで加熱し、この温度で15分間保持した後、冷却し、25℃で15分間平衡状態とした。次にこのサンプルを10℃/分で最終温度200℃まで加熱した。
【0042】
5.熱重量(TG)分析
サンプルの熱重量(TG)分析は、TA Instruments Thermogravimetric Analyzer 2050 または 2950にて行った。使用した較正用スタンダードは、ニッケルおよびアルメル(Alumel、商標)であった。およそ2から5mgのサンプルをパンに置き、正確に重量測定し、TG炉に入れた。その後サンプルを窒素下、10℃/分の比率で最終温度300℃まで加熱した。
【0043】
6.高温顕微鏡
高温顕微鏡測定は、Leica顕微鏡にて、Kofler高温ステージにのせて行った。高温ステージの温度は、Testo 6000-903熱電対およびTesto 720デジタル読み取り器を使用して測定した。温度はUSPのスタンダードを用いて較正した。
【0044】
7.吸湿/放湿
吸湿/放湿データは、VT SGA-100平衡含湿システム(moisture balance system)にて集めた。吸湿等温線として、10%RH増加における5から95%相対湿度(RH)の吸湿範囲、および95から5%RHの放湿範囲を分析用に使用した。サンプルは分析前に乾燥させなかった。分析に関して使用した平衡の基準は、重量の判定基準が一致しない場合、3時間の最大平衡時間内で5分間に0.0100%未満の重量変化とした。データは、サンプルの最初の湿度含有量については補正しなかった。
【0045】
8.多形のスクリーニング
多形のスクリーニングは、できるだけ多くの固体の形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩を作成する試みにおいて行った。この技術は、様々な条件下での固体の作成、およびその後のXRPDによる特徴づけを伴った。3つの区別される形、ならびにアモルファスの形を表す3つの区別されるXRPDパターンが、このスクリーニングにおいて見出された。結晶のパターンはA型、B型、およびC型と命名した。A型は、高温溶液の緩やかな冷却、スラリー化、または貧溶媒を用いての沈殿より得た。B型は、高温溶液の緩やかな冷却、および貧溶媒による結晶化より得た。C型はメチルt−ブチルエーテルからの貧溶媒による結晶化より得ており、したがってN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のメチルt−ブチルエーテル溶媒和物であると思われる。アモルファスの材料は、溶液を緩やかにおよび速く蒸発させることにより生成した。
【0046】
9.結晶化の方法
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の秤量したサンプル(通常30mg)を、検査溶媒(試薬グレードまたはHPLCグレード)を一定分量ずつ加えて処理し、20から200μLの溶液を生成した。これらの溶液を超音波処理し、すべての固体が溶解した(目視)時点で、溶液を濾過し、周囲条件下でふたをしないバイアル中に放置(速い蒸発)するか、またはピンホールのあるアルミホイルで覆った(緩やかな蒸発)。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。この化合物の固体サンプルはまた、上の濾過した室温溶液を−78℃に急速に冷却する(急激な冷却)ことによっても生成した。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0047】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の秤量したサンプルはまた、高温で検査溶媒を一定分量ずつ加えて処理した。これらのサンプルおよび溶媒は、45℃または80℃のどちらかに保温したホットプレート上で加熱し、得られた溶液を、同じホットプレート上に保持したバイアル中に素早く濾過した。熱源のスイッチを切り、ホットプレートおよびバイアルを放置して周囲温度に冷却(緩やかな冷却)し、一晩そのまま静置した。析出した固体の有無を調べた;固体が存在しない場合、またはXRPD分析には固体の量が少なすぎると判断した場合には、バイアルを一晩冷蔵庫に置いた。再び析出した固体の有無を調べ、固体が認められない場合にはバイアルを冷凍庫に一晩おいた。固体は濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0048】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の溶解度は、溶液を得るために使用した溶媒総量に基づいて実験から見積もった。実際の溶解度は、一定分量ずつという多過ぎる溶媒を使用していること、または遅い溶解速度のために、算出された値より大きいと考えられる。実験中に溶解が起こらなかった場合は、溶解度は“より小さい”と表す。一定分量の溶媒をすべて加えてしまう前に固体が溶解した場合は、溶解度は“より大きい”と列記する。
【0049】
貧溶媒の実験は、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体サンプルを検査溶媒中に溶解させ、得られた溶液を貧溶媒中に濾過することにより行った。固体が形成された場合、“急激な結晶化”と呼ぶ;そして固体が、溶液を冷却した後、またはカバーしてそのまま静置した後に形成された場合、“沈殿”と呼ぶ。直ちに固体が形成されなかった場合は、サンプルは、固体が見られるまで周囲条件下に放置した。形成されたいかなる固体も濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0050】
スラリーの実験は、過剰な固体を含有する、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を作製することにより行った。これらのスラリーは、周囲温度で3日間撹拌した。析出した固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0051】
蒸気拡散の実験は、バイアル中にN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を入れ、次にそれを、貧溶媒を含有するより大きなバイアルに入れることにより行った。次により大きなバイアルを密封し、周囲温度に維持した。固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0052】
液体拡散の実験は、バイアル中にN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の飽和溶液を入れ、混和しない貧溶媒を加えることにより行った。沈殿した固体の有無を調べた。固体が形成された場合は、溶媒をデカントし、固体を集めた。固体が形成されなかった場合は、バイアルに蓋をして、周囲温度でそのまま静置した。形成されたあらゆる固体を濾過して取り出し、空気乾燥し、XRPDにより分析した。
【0053】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体サンプルはまた、この化合物の融液の急速な冷却(−78℃)により作成した。
【0054】
C.多形A、B、Cおよびアモルファス材料
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形スクリーニングで得た固体の形を表1から3にまとめる。A型、B型、およびC型と命名した、3つの区別される形を表す3つの区別されるXRPDパターンが見出された。A型は、緩やかな冷却、スラリー化、または貧溶媒による結晶化より得た。B型は、高温溶液の緩やかな冷却、および貧溶媒による結晶化より得た。C型はメチルt−ブチルエーテルからの貧溶媒による結晶化より得た。アモルファスの材料は、溶液を緩やかにおよび速く蒸発させることにより生成した。
【0055】
【表1−1】
【0056】
【表1−2】
【0057】
表2は貧溶媒による再結晶化の結果を示す。
【0058】
【表2−1】
【0059】
【表2−2】
【0060】
【表2−3】
【0061】
表3は蒸気拡散実験に関する結果を示す。
【0062】
【表3−1】
【0063】
【表3−2】
【0064】
a. A型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のA型は、XRPD、DSC、TG、高温顕微鏡、および吸湿/放湿を用いて特徴づけを行い、データを図1から18、表4(高温顕微鏡の研究)、表5(吸湿/放湿データ)、表6(XRPDピーク)、表7(ラマンスペクトルのピーク)および表8(IRスペクトルのピーク)にまとめる。発熱分解は200℃付近で見られ、高温顕微鏡により確認した。TG曲線は175℃で最小の重量変化を示した。吸湿/放湿データでは、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のサンプルが、5%RHで最小量の重量を喪失することを示し、少量の最初の揮発物質のみが低RH条件下で除去されたことを指摘する。サンプルは、95%RHでその重量の1.5%未満が増量するが、この値は0.5水和物(hemihydrate)の形成に関する増量の理論値(1.87%)より低い。重量のほとんどは放湿曲線の75%RHまでに失われ、材料は35%RH以下で、非溶媒和状態の平衡状態に戻る。実験完了後のサンプルのXRPDパターンは、材料がA型であることを示す。A型の材料は、吸湿/放湿データに基づくと、75%RHまでは非吸湿性であると思われる。
【0065】
【表4】
【0066】
【表5】
【0067】
【表6】
【0068】
【表7−1】
【0069】
【表7−2】
【0070】
【表8−1】
【0071】
【表8−2】
【0072】
特徴付けのデータに基づくと、A型は、200℃以上で分解する、非溶媒和物、非吸湿性の結晶材料であると思われる。
b. B型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のB型は、通常貧溶媒による結晶化より得、XRPD、DSC、TG、高温顕微鏡、および吸湿/放湿を用いて特徴づけを行い、データを図1および19−22、表4(高温顕微鏡の研究)、表5(吸湿/放湿データ)、表6(XRPDピーク)、表7(ラマンスペクトルのピーク)および表8(IRスペクトルのピーク)にまとめる。
【0073】
B型に関する熱データは、図19および20に示す。DSCは、高温顕微鏡データより、分解に起因する205℃での緩やかな発熱を示す。TG曲線は175℃での最小量の重量喪失を示す。B型は吸湿/放湿実験の間に、最小量の重量の増減を示す。実験完了後にサンプルを集めたXRPDパターンは、材料がB型であったことを示す。B型のサンプルを、そのナトリウム含有量(4.85%)について分析したところ、この値はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の理論値(4.82%)と一致し、この塩がインタクトであることを示した。
【0074】
特徴付けのデータに基づくと、B型は、203℃付近で分解する非溶媒和の結晶材料であると思われる。
c. C型
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のC型は、溶媒としてメタノールまたはエタノール、そして貧溶媒としてメチル−t−ブチルを使用して、貧溶媒による結晶化より、単独でまたはB型との混合物として得た。同じ条件下での繰り返しての結晶化は、ほとんどの場合B型またはB型とC型の混合物を得た。C型は、XRPD、TG/IRおよびTGを用いて特徴づけを行い、データを図1および23−24、ならびに表4に示す。
【0075】
C型および少量のB型を含有するサンプルのTGデータは、175℃で22.4%の重量喪失を示し、これはN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩の8水和物(理論値23.2%)またはメチル−t−ブチルエーテレート(21.7%、2薬剤分子当たり溶媒3分子)としての理論値に近似する。類似サンプルの元素分析から、4.19%のナトリウム含有量を得たが、これは、上に記載した8水和物(3.7%)またはメチル−t−ブチルエーテレート(3.8%)として算出した値よりわずかに高い。C型のサンプルをTG/IRを用いて分析し、メチル−t−ブチルエーテレートを含有することを見出し、C型がN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩のメチル−t−ブチルエーテル溶媒和物であることを確認した。実験後に集めた材料をXRPDにより分析し、C型を維持していることを発見した。
【0076】
2.結晶化の研究
A型およびB型の、N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形を調製するための結晶化の研究および詳細な製法を、以下に記載する。これらの研究は、これらの多形を適当な条件下で選択的に生成することができることを実証している。さらにB型、およびA型とB型との混合物は、A型に相互変換することができ、A型がより安定な種であることを示唆する。
【0077】
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の固体結晶の形、すなわちA型およびB型のXRPDパターンを、各々図1および4に示す。これらのXRPDパターンを使用して、以下に記載する結晶化および工程の研究より得た固体の形を同定した。
【0078】
3.近似の溶解度
溶解度は、溶液を得るために使用した総溶媒量に基づいて、実験より見積もった。実際の溶解度は、一定分量ずつという多過ぎる溶媒を使用したこと、または遅い溶解速度のため、算出された値より大きいと考えられる。実験中に溶解が起こらなかった場合は、溶解度は“より小さい”と表す。一定分量の溶媒をすべて加えてしまう前に固体が溶解していた場合は、溶解度は“より大きい”と列記する。
【0079】
周囲温度での多様な溶媒中の、A型のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の近似の溶解度を、表9にまとめる。A型はN,N−ジメチルホルムアミド(228mg/mL)中で最も溶解度が高く、続いてメタノール(160mg/mL)、アセトン(96mg/mL)、テトラヒドロフラン(86mg/mL)、エタノール(60mg/mL)、水(48mg/mL)、およびメチルエチルケトン(34mg/mL)であった。A型は、クロロホルム、ジクロロメタン、およびメチルt−ブチルエーテル(<3mg/mL)には難溶性であった。
【0080】
【表9】
【0081】
4.相互変換の研究
A型およびB型の相互変換は、酢酸エチルおよび95%イソプロパノール:水を使用して行った。A型は、95%イソプロパノール:水中で、熱力学的により安定であると思われる。酢酸エチル中の相互変換ではA型およびB型の混合物を得たが、これはおそらく材料の低溶解度のためである。これらの結果はB型が、熱力学的により不安定な形の形成の方が通常選ばれる貧溶媒による結晶化によって形成された、という事実によって裏付けられる。
【0082】
D.多形の調製のための製法
酢酸エチル中での相互変換の研究に基づくと、A型はN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の最も安定な形であると思われる。A型は、緩やかな冷却、スラリー化、または貧溶媒による結晶化より得た。B型は、熱力学的により不安定な形の方の形成が通常選ばれる、貧溶媒による結晶化より得た。C型は、メチルt−ブチルエーテルから貧溶媒による結晶化より得、そしてアモルファス材料は、溶液を緩やかにおよび速く蒸発させることにより得た。
【0083】
ある種の態様において、本明細書において提供したシタキセンタンナトリウムの結晶化の工程は、多形AおよびBの混合物を生成する。ある種の態様において、この混合物は多形AおよびBを約60:40の比率で含有する。他の態様において多形A対Bの比率は、約65:35、70:30,80:20、85:15、90:10、92:8、93:7、94:6、95:5、98:2、96:4、97:3、もしくは99:1である、またはそれより大きい、または等しい。1つの態様において、本明細書に提供する工程は、約100%の多形Aを生成する。1つの態様において、本明細書に提供する工程は、約100%の多形Bを生成する。
【0084】
E.組成物の製剤および投与
当該多形の製剤を本明細書において提供する。製剤は、本明細書に提供する多形の投与のためにデザインされた組成物である。組成物は、経口および非経口投与に適する。そのような組成物は、溶液、懸濁液、錠剤、分散性錠剤、ピル、カプセル、粉末、徐放性製剤、およびその他のあらゆる適切な製剤を含む。1つの態様において、組成物はピルまたは錠剤の形をとることになる。錠剤、カプセル、およびその他のそのような製剤の製造方法は、当業者に公知である(例えばAnsel, H.C. (1885) Forms, Introduction to Pharmaceutical Dosage Forms, 第4版, pp. 126-163を参照のこと)。
【0085】
本明細書に提供する製剤において、有効濃度の多形または多形の混合物を、適切な医薬用担体またはビヒクルとともに混合する。製剤中の多形の濃度は、投与して、エンドセリンを介した疾患の症状を寛解させる量を送達するための有効な量である。ある種の態様において、組成物は単回投与の投与用に製剤化する。組成物を製剤化するため、化合物のある重量画分を、治療する状態が軽減または寛解されるような有効濃度で、選択されたビヒクル中に溶解、懸濁、分散、またはそうでなければ混合する。本明細書に提供する化合物の投与に適する医薬的担体またはビヒクルは、投与の特定の様式に適することが当業者に知られているあらゆるそのような担体を含む。
【0086】
加えて当該化合物は、組成物中のただ1つの医薬的に活性な成分として製剤化してもよいし、または他の活性な成分と組み合わせてもよい。組織を標的とするリポソームを含むリポソーム懸濁液もまた、医薬的に重要可能な担体として適すると考えられる。これらは、当業者に公知の方法に従って調製してよい。例えばリポソーム製剤は、米国特許第4,522,811号に記載されているように調製してよい。
【0087】
多形または多形の混合物としての活性化合物は、治療する患者における望ましくない副作用なしに、治療上有用な効果を発するために十分な量で、医薬的に重要可能な担体中に含まれる。治療有効濃度は、公知のin vitroおよびin vivoの系において化合物を検査することにより経験的に決定し(例えばIshikawaらに対する米国特許第5,114,918号;BANYU PHARMACEUTICAL CO., LTD に対するEP A1 0 436 189 (1991年10月7日); Borges, et al. (1989) Eur. J. Pharm. 165: 223-230; Filep et al. (1991) Biochem. Biophys. Res. Commun. 177: 171-176を参照のこと)、その後それからヒトのための投与量を外挿してよい。
【0088】
薬剤組成物中の活性化合物の多形または多形混合物の濃度は、活性化合物の吸収、不活性化および排泄の速度、化合物の物理化学的特徴、投与計画、ならびに投与する量、同様に当業者に公知のその他の因子に依存することになる。例えば送達される量は、高血圧症の症状を治療するために十分な量である。エンドセリンを介した障害を治療するための有功量は、細菌感染を治療するために投与されるスルホンアミド化合物の量より多いと予想される。
【0089】
1つの態様において治療上有効な投与量は、約0.1ng/mlから約50−100μg/mlの活性成分の血清濃度を生成しなければならない。医薬的投与ユニット剤形は、1投与ユニット剤形当たり、約20mgから約300mgおよび約25mgから約200mg、または約25mgから約100mgの必須活性成分または必須活性成分の組み合わせを提供するように調製する。
【0090】
活性成分は一度に投与してもよいし、または何回かのより少ない用量に分割して時間の間隔をおいて投与してもよい。正確な投与量および治療期間は、治療する疾患の関数であり、公知の試験プロトコルを使用して、またはin vitroもしくはin vivoの検査データからの外挿により決定してよい。濃度および投与量の数値はまた、緩和すべき状態の重症度に伴って変化させてよいことも注意しなければならない。あらゆる特定の被験者に対して、具体的な投与計画が、個人の必要性、および組成物を投与する人または組成物の投与を監督する人の専門的判断に従って、長期にわたり調整されなければならないこと、そして本明細書に記述する濃度範囲は、具体例に過ぎず、主張した組成物の範囲または実践を限定する意図はないことは、さらに理解されなければならない。
【0091】
医薬的に受容可能な誘導体には、酸、塩、エステル、水和物、溶媒和物、およびプロドラッグの形を含む。誘導体は、対応する中性の化合物より安定な形であるよう選択される。
【0092】
したがって本明細書に提供する多形もしくは多形の混合物、または医薬的に受容可能なその誘導体の有効濃度または有効量を、全身、局所的または局部的な投与のための適切な医薬的担体またはビヒクルと共に混合して、医薬組成物を形成する。
【0093】
組成物は、治療する障害に依存して、経口、非経口、経直腸、ならびに局所的および局部的を含む、適切な経路により投与することを意図する。例えば眼科的障害、例えば緑内障の治療には、眼球内、また硝子体内への注射用の製剤を意図する。1つの態様において、カプセルおよび錠剤は経口投与用に使用する。本明細書に記載したように調製した凍結乾燥粉末の再構成は、非経口投与用に使用してよい。液体、半液体、または固体の剤形の化合物は、各投与経路に適する様式で製剤化する。投与の様式は、非経口および経口の投与様式を含む。
【0094】
非経口、皮内、皮下、または局所塗布に使用する溶液または懸濁液は、以下の成分:滅菌希釈剤、例えば注射用水、生理食塩水溶液、固定油、ポリエチレングリコール、グリセリン、プロピレングリコール、またはその他の合成溶媒;抗生剤、例えばベンジルアルコールおよびメチルパラベン;抗酸化剤、例えばアスコルビン酸および亜硫酸水素ナトリウム;キレート剤、例えばエチレンジアミン四酢酸(EDTA);バッファー、例えば酢酸塩、クエン酸塩、およびリン酸塩;浸透圧の調整剤、例えば塩化ナトリウムまたはブドウ糖、のいずれかを含むことができる。非経口調製物は、アンプル、使い捨ての注射器、またはガラス、プラスチックもしくはその他の適切な材料で作られた単回用量もしくは多数回用量のバイアル中に封入することができる。
【0095】
化合物が十分な溶解度を示さない場合、化合物を可溶化するための方法を使用してよい。そのような方法は当業者に公知であり、共溶媒、例えばジメチルスルホキシド(DMSO)の使用、界面活性剤 例えばツイーンの使用、または重炭酸ナトリウム水溶液中での溶解を含むがこれに限定されない。化合物の誘導体、例えば化合物のプロドラッグもまた、有効な医薬組成物の製剤化において使用してよい。
【0096】
スルホンアミド化合物(1つまたは複数)のナトリウム塩を混合または添加することで得られる混合物は、溶液、懸濁液、エマルジョン等であってよい。得られる混合物の形は、意図した投与様式、および選択した担体またはビヒクル中の化合物の溶解度を含む、いくつかの因子に依存する。有効濃度は、治療する疾患、障害または状態の症状を寛解させるための十分な量であり、経験的に決定してよい。
【0097】
製剤は、ヒトおよび動物への投与用に、適切な量の化合物、特に医薬的に重要可能なその塩、例えばナトリウム塩のを含有する、ユニット投与剤形で、例えば錠剤、カプセル、ピル、粉末、顆粒、滅菌非経口溶液または懸濁液、および経口溶液または懸濁液、および油−水エマルジョンの形で提供する。医薬的、治療的に活性な化合物およびその誘導体は、ある種の態様において、ユニット投与剤形または複数回投与剤形にて製剤化し、投与する。本明細書において使用する場合ユニット用量剤形は、ヒトおよび動物の被験者に適する、そして当該技術で公知であるような個々に包装された、物理的に個別のユニットをいう。各ユニット用量は、必要な医薬的担体、ビヒクル、または希釈剤と共同して、所望の治療効果を得るために十分な、治療的に活性な化合物のあらかじめ決定された量を含有する。ユニット用量の剤形の例として、アンプルおよび注射器、個々に包装された錠剤またはカプセルを含む。ユニット−用量剤形は、その分画またはその複数個で投与してもよい。複数回−用量の剤形は、分離されたユニット−用量剤形で投与される、1つの容器内に包装された複数個の個別ユニット−投与剤形である。複数回−用量の剤形の例は、バイアル、錠剤もしくはカプセルのビン、またはパイントもしくはガロンのビンを含む。この場合、複数回用量の剤形は、包装内で分離されていない複数のユニット−用量である。
【0098】
組成物は活性成分と共に:希釈剤、例えばラクトース、ショ糖、リン酸二カルシウム、またはカルボキシメチルセルロース;滑剤、例えばステアリン酸マグネシウム、ステアリン酸カルシウムおよびタルク;ならびに結合剤、例えばスターチ、天然のガム 例えばアカシアゼラチンガム、グルコース、糖蜜、ポリビニルピロリジン、セルロースおよびその誘導体、ポビドン、クロスポビドン、および当業者に公知のその他のそのような結合剤、を含有することができる。液体の医薬的に投与可能な組成物は、例えば上に定義したような活性化合物および所望による医薬的なアジュバントを、担体中、例えば水、生理食塩水、ブドウ糖水溶液、グリセロール、グリコール、エタノール、等の中で、溶かす、分散させる、またはそうでなければ混合することにより調製して、それにより溶液または懸濁液を形成することができる。所望であれば、投与する医薬組成物はまた、少量の非毒性の補助的物質、例えば湿潤剤、乳化剤、または可溶化剤、pH緩衝化剤等、例えばアセテート、クエン酸ナトリウム、シクロデキストリン誘導体、モノラウリン酸ソルビタン、酢酸ナトリウムトリエタノールアミン(triethanolamine sodium acetate)、オレイン酸トリエタノールアミン、およびその他のそのような物質を含有してよい。そのような投与剤形を調製する実際の方法は、当業者に公知である、または明らかになるだろう;例えばRemington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa. 第15版, 1975を参照のこと。投与する組成物または製剤は、いずれにしても、治療する被験者の症状を緩和するための十分な量の、一定量の活性化合物を含有することになる。
【0099】
非毒性担体で重量調製した0.005%から100%の範囲の活性成分を含有する投与剤形または組成物を調製してよい。経口投与用に、医薬的に受容可能な非毒性組成物は、一般に利用される賦形剤、例えば医薬グレードのマンニトール、ラクトース、スターチ、ステアリン酸マグネシウム、滑石、セルロース誘導体、クロスカルメロースナトリウム、グルコース、ショ糖、炭酸マグネシウム、サッカリンナトリウム、のいずれかを組み込むことにより形成する。そのような組成物として、溶液、懸濁液、錠剤、カプセル、粉末、ならびに徐放性製剤、例えば非限定的に、植込錠およびマイクロカプセル化送達システム、および生分解性、生体適合性のポリマー 例えばコラーゲンエチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、ポリオルトエステル、ポリ乳酸、およびその他の物質を含む。これらの製剤の調製法は、当業者に公知である。ある態様において意図した組成物は、0.001%−100%の活性成分を、もう1つの態様において0.1−85%、もう1つの態様において75−95%の活性成分を含有してよい。
【0100】
組成物は、体からの急速な排出に対して当該化合物を保護する担体、例えば時間で放出される製剤またはコーティングを用いて調製してよい。
製剤は、所望の組み合わせの特性を得るため、他の活性化合物を含んでよい。多形はまた、治療または予防の目的のため、本明細書で上述した1つまたはそれより多くの疾患または医学的状態を治療する上で、価値があることが一般に知られている別の薬学的物質、例えばベータ−アドレナリン遮断薬(例えばアテノロール)、カルシウムチャネル遮断薬(例えばニフェジピン)、アンジオテンシン変換酵素(ACE)阻害薬(例えばリシノプリル)、利尿薬(例えばフロセミドまたはヒドロクロロチアジド)、エンドセリン変換酵素(ECE)阻害薬(例えばホスホラミドン)、中性エンドペプチダーゼ(NEP)阻害薬、HMGCoA還元酵素阻害薬、一酸化窒素供与体、抗酸化薬、血管拡張薬、ドーパミンアゴニスト、神経保護薬、ステロイド、ベータ−アゴニスト、抗凝血薬、または血栓溶解薬、と合わせて有利となるように投与してよい。そのような併用療法は、本明細書に提供する治療の組成物および方法のさらなる側面を構成することは、理解されなければならない。
【0101】
本明細書に提供するラクトースを含まない組成物は、当該技術において周知されている賦形剤、および例えばU.S. Pharmacopeia (USP) 25-NF20 (2002)に列記されている賦形剤を含有することができる。一般にラクトースを含まない組成物は、医薬的に適合性のあるそして医薬的に受容可能な量の、活性成分、結合剤/フィラー、および滑剤を含有する。特定のラクトースを含まない投与剤形は、活性成分、微結晶セルロース、アルファ化スターチ、およびステアリン酸マグネシウムを含有する。
【0102】
水は一部の化合物の分解を促進する可能性があるため、活性成分を包含する無水の医薬組成物および投与剤形をさらに提供する。例えば水の添加(例えば5%)は、時間にわたる有効期間または安定性といった特徴を決定する目的で、長期的保存をシュミレートする手段として医薬技術において広く受け入れられている。例えばJens T. Carstensen, Drug Stability: Principles & Practice, 第2版, Marcel Dekker, NY, NY, 1995, pp. 379-80を参照のこと。事実、水および熱は一部の化合物の分解を加速する。したがって湿度および/または湿気には普通、製剤の製造、取り扱い、包装、保存、出荷、および使用時に遭遇するため、製剤における水の影響は非常に重要となり得る。
【0103】
本明細書に提供する無水の医薬組成物および投与剤形は、無水または低湿度を含有する成分、および低湿度または低い湿気の条件を使用して調製することができる。
無水医薬組成物は、その無水という性質が維持されるように調製し保存しなければならない。したがって無水組成物は一般に、適切な製剤キットに含めることができるような、水への暴露を防ぐことが知られている材料を使用して包装される。適切な包装の例として、密封されたホイル、プラスチック、ユニット用量の容器(例えばバイアル)、ブリスター包装、およびストリップ包装を含むがこれに限定されない。
【0104】
1.経口投与用製剤
経口医薬投与剤形は、固体、ゲル、または液体のどれかである。固体の投与剤形は、錠剤、カプセル、顆粒、および原末である。経口錠剤のタイプには、腸溶コーティング、糖衣、またはフィルムコーティングであってよい、圧縮型の咀嚼可能なロゼンジおよび錠剤を含む。カプセルは、ハードまたはソフトのゼラチンカプセルであってよく、一方顆粒および粉末は、当業者に公知の他の成分と組み合わせて、非発泡性または発泡性の剤形で提供してよい。そのような投与剤形は、予め決定された量の活性成分を含有し、当業者に周知の薬学の方法により調製してよい。一般的には、Remington’s Pharmaceutical Sciences, 第20版, Mack Publishing, Easton PA (2000)を参照のこと。
【0105】
ある種の態様において、製剤は固体の剤形、例えばカプセルまたは錠剤である。錠剤、ピル、カプセル、トローチ等は、以下の成分、または類似の性質の複合体(conjugate);結合剤;フィラー、希釈剤;崩壊剤;滑剤;潤沢剤;甘味剤;および香味剤のいずれかを含有することができる。本明細書に提供する経口投与剤形に使用することのできる賦形剤の例は、結合剤、フィラー、崩壊剤、および滑剤を含むが、これに限定されない。医薬組成物および投与剤形における使用に適する結合剤は、コーンスターチ、ポテトスターチ、またはその他のスターチ、ゼラチン、天然および合成のガム 例えばアカシア、アルギン酸ナトリウム、アルギン酸、その他のアルギン酸塩、粉末トラガカント、グアーガム、セルロースおよびその誘導体(例えばエチルセルロース、酢酸セルロース、カルボキシメチルセルロールカルシウム、カルボキシメチルセルロースナトリウム)、ポリビニルピロリドン、メチルセルロース、アルファ化スターチ、ヒドロキシプロピルメチルセルロース、(例えばNo2208、No2906、No2910)、微結晶セルロース、およびそれらの混合物を含むが、これに限定されない。
【0106】
微結晶セルロースの適切な形は、AVICEL-PH-101、AVICEL-PH-103、AVICEL RC-581、AVICEL-PH-105(FMC Corporation、American Viscose Division、Avicel Sales、Marcus Hook、PAより入手可能)として販売されている材料、およびそれらの混合物を含むがこれに限定されない。具体的な結合剤は、微結晶セルロース、およびAVICEL RC-581として販売されているカルボキシメチルセルロースナトリウムの混合物である。適切な無水または低湿度の賦形剤または添加剤は、AVICEL-PH-103 および Starch 1500 LMを含む。
【0107】
本明細書に開示する医薬組成物および投与剤形における使用に適するフィラーの例は、タルク、炭酸カルシウム(例えば顆粒または粉末)、微結晶セルロース、粉末セルロース、デキストレート、カオリン、マンニトール、ケイ酸、ソルビトール、スターチ、アルファ化スターチ、およびそれらの混合物、を含むがこれに限定されない。本明細書における医薬組成物中の結合剤またはフィラーは、典型的には医薬組成物または投与剤形の約50から約99重量パーセントで存在する。
【0108】
崩壊剤は、水性環境に暴露された時に崩壊する錠剤を提供するため、本明細書に提供する組成物中に使用する。あまりにも多量の崩壊剤を含有する錠剤は、保存中に崩壊してしまうかもしれないが、一方であまりにも少量しか含有しないものは、所望の速度でまたは所望の条件下で崩壊しないかもしれない。したがって活性成分の放出を有害に変化させる程多過ぎも少な過ぎもしない十分な量の崩壊剤を使用して、本明細書に提供する固形の経口投与剤形を形成しなければならない。使用する崩壊剤の量は、製剤のタイプに基づいて変動するが、当業者に容易に識別できよう。典型的な医薬組成物は、約0.5から約15重量パーセントの崩壊剤、または約1から約5重量パーセントの崩壊剤を含有する。
【0109】
本明細書に提供する医薬組成物および投与剤形中に使用することのできる崩壊剤は、寒天(agar-agar)、アルギン酸、炭酸カルシウム、微結晶セルロース、クロスカルメロースナトリウム、クロスポビドン、ポラクリリンカリウム、グルコール酸スターチナトリウム、ポテトまたはタピオカのスターチ、その他のスターチ、アルファ化スターチ、その他のスターチ、粘土(clay)、その他のアルギン、その他のセルロース、ガム、およびそれらの混合物を含むがこれに限定されない。
【0110】
本明細書に提供する医薬組成物および投与剤形中に使用することのできる滑剤は、ステアリン酸カルシウム、ステアリン酸マグネシウム、鉱油、軽油、グリセリン、ソルビトール、マンニトール、ポリエチレングリコール、その他のグリコール、ステアリン酸、ラウリル硫酸ナトリウム、タルク、水素化植物油(例えばピーナツ油、綿実油、ヒマワリ油、ゴマ油、オリーブ油、コーン油、および大豆油)、ステアリン酸亜鉛、オレイン酸エチル、ラウリン酸エチル、寒天(agar)、およびそれらの混合物を含むがこれに限定されない。付加的な滑剤として、例えばsyloidシリカゲル(W.R. Grace Co. of Baltimore, MD により製造されたAEROSIL(登録商標)200)、合成シリカの凝集エアロゾル(coagulated aerosol)(Degussa Co. of Plano, TXより市販されている)、CAB-O-SIL (Cabot Co. of Boston, MA より販売されている発熱性シリコン二酸化物の製品)、およびそれらの混合物を含む。仮に使用する場合には、滑剤は典型的には、それらが組み込まれる医薬組成物または投与剤形の約1重量パーセントより少ない量で使用する。
【0111】
経口投与が所望される場合、多形および多形の混合物は、腸溶性コーティング錠、糖衣錠、フィルムコーティグ錠、または多層圧縮錠として形成される組成物中に提供することができる。腸溶コーティング錠は、胃の酸性環境から活性成分を保護する。糖衣錠は、医薬的に受容可能な物質の別々の層が塗布されている圧縮錠である。フィルムコーティング錠は、ポリマーまたはその他の適切なコーティングで覆われた圧縮錠である。多層圧縮錠は、先に述べた医薬的に受容可能な物質を利用して、1回より多くの圧縮サイクルにより作製された圧縮錠である。着色剤もまた、上の投与剤形に使用してよい。香味剤および甘味剤は、圧縮錠、糖衣錠、多層圧縮錠、および咀嚼可能な錠剤において使用される。香味剤および甘味剤は、咀嚼可能な錠剤およびロゼンジの形成においてとくに有用である。組成物はまた、制酸薬またはその他のそのような成分と組み合わせて製剤化してもよい。
【0112】
投与ユニット剤形がカプセルである場合、上のタイプの材料に加えて液体の担体、例えば脂肪油を含有することができる。ゼラチンカプセルにおいては、例えばポリエチレンカーボネート、植物油、またはトリグリセリド中に、シテキセンタンナトリウムを含有する溶液または懸濁液を、カプセル内に封入する。そのような溶液、ならびにその調製および封入は、米国特許第4,328,245号;4,409,239号;および4,410,545号に開示されている。
【0113】
活性成分はまた、所望の作用を損なうことのない他の活性材料と共に、または所望の作用を補う材料、例えば制酸薬、H2遮断薬、および利尿薬と共に混合することができる。活性成分の重量の約98%までの高濃度を含んでよい。
【0114】
液体経口投与剤形は、水性の溶液、エマルジョン、懸濁液、非発泡性の顆粒から再構成される溶液および/または懸濁液、ならびに発泡性の顆粒から再構成される発泡性の調製物を含む。水性溶液として、例えばエリキシルおよびシロップを含む。エリキシルは透明な甘みを加えた含水アルコール調製物である。エリキシル中に使用する医薬的に受容可能な担体は、溶媒を含む。シロップは、糖分、例えばショ糖の濃縮水溶液であり、保存剤を含有してよい。
【0115】
エマルジョンは、一方の液体がもう一方の液体全体に小さな球状の形で分散している、2相系である。エマルジョン中に使用する医薬的に受容可能な担体は、非水性の液体、乳化剤および保存剤である。懸濁液は、医薬的に受容可能な懸濁剤および保存剤を使用する。
液体の経口投与剤形に再構成される非発泡性顆粒中に使用する医薬的に受容可能な物質は、希釈剤、甘味剤、および湿潤剤を含む。液体の経口投与剤形に再構成される発泡性顆粒中に使用する医薬的に受容可能な物質は、有機酸および二酸化炭素の原料を含む。着色剤および香味剤は、上の投与剤形のすべてにおいて使用される。
【0116】
溶媒は、グリセリン、ソルビトール、エチルアルコール、およびシロップを含む。保存剤の例は、グリセリン、メチルパラベンおよびプロピルパラベン、安息香酸、安息香酸ナトリウムおよびアルコールを含む。エマルジョンに利用する非水性液体の例は、鉱物油および綿実油を含む。乳化剤の例は、ゼラチン、アカシア、トラガカント、ベントナイト、および界面活性剤 例えばポリオキシエチレンソルビタンモノオレエートを含む。懸濁剤は、カルボキシメチルセルロースナトリウム、ペクチン、トラガカント、ヴィーガム(Veegum)およびアカシアを含む。
【0117】
希釈剤は、ラクトースおよびショ糖を含む。甘味剤は、ショ糖、シロップ、グリセリンおよび合成甘味剤 例えばサッカリンを含む。湿潤剤は、モノステアリン酸プロピレングリコール、モノオレイン酸ソルビタン、モノラウリン酸ジエチレングリコール、ポリオキシエチレンラウリルエーテルを含む。有機酸は、クエン酸および酒石酸を含む。二酸化炭素の原料は、炭酸水素ナトリウムおよび炭酸ナトリウムを含む。着色剤は、認可され保証された水溶性FDおよびC色素のいずれか、およびそれらの混合物を含む。香味剤は、植物 例えば果物から抽出された天然の香味剤、および心地よい味覚を生じる化合物の合成ブレンドを含む。
【0118】
ミセル型の活性成分を含有する医薬組成物は、米国特許第6,350,458号に記載されているように調製することができる。そのような医薬組成物は、経口、経鼻、および頬側塗布において特に有用である。
【0119】
ある種の態様において、製剤は、本明細書に提供する多形または多形の混合物、1,2−ジメトキシメタン、ジグリム、トリグリム、テトラグリム、ポリエチレングリコール−350−ジメチルエーテル、ポリエチレングリコール−550−ジメチルエーテル、ポリエチレングリコール−750−ジメチルエーテル(ここで350、550および750はポリエチレングリコールのおよその平均分子量をいう)を含むがこれに限定されないジアルキル化モノ−またはポリ−アルキレングリコール、そして1つまたはそれより多くの抗酸化剤、例えばブチル化ヒドロキシトルエン(BHT)、ブチル化ヒドロキシアニソール(BHA)、没食子酸プロピル、ビタミンE、ヒドロキノン、ヒドロキシクマリン、エタノールアミン、レシチン、セファリン、アスコルビン酸、リンゴ酸、ソルビトール、リン酸、チオジプロピオン酸およびそのエステル、ならびにジチオカルバメート、を含有するものを含むがこれに限定されない。
【0120】
他の製剤は、医薬的に受容可能なアセタールを含む水性アルコール性溶液、を含むがこれに限定されない。これらの製剤中に使用するアルコールは、プロピレングリコールおよびエタノールを含むがこれに限定されない、1つまたはそれより多くのヒドロキシル基を有する、あらゆる医薬的に受容可能な水混和性の溶媒である。アセタールは、低級アルキルアルデヒドのジ(低級アルキル)アセタール、例えばアセトアルデヒドジエチルアセタールを含むがこれに限定されない。
【0121】
ある種の態様において、多形および多形の混合物は、約50mg、約75mg、約100mg、約150mg、約200mg、約250mg、約300mg、約350mgの活性成分、を含有する経口錠剤として製剤化する。カプセルは、不活性成分、例えばポリエチレングリコール400、ポリソルベート20、ポビドン、およびブチル化ヒドロキシアミソールを含有することができる。カプセルの殻は、ゼラチン、ソルビトールと特定のゼラチンのブレンドおよび二酸化チタンを含有することができる。
【0122】
具体例としての経口錠剤の製剤
ある種の態様において、本明細書に提供する方法は、本明細書に提供する多形または多形の混合物を含有する経口錠剤の投与に関与する。1つの態様において、経口錠剤はさらに、バッファーを含有する。1つの態様において、経口錠剤はさらに抗酸化剤を含有する。1つの態様において、経口錠剤はさらに湿度をバリアーするコーティングを含有することができる。
【0123】
一部の態様において錠剤は、抗酸化剤、例えばアスコルビン酸ナトリウム、グリシン、メタ重亜硫酸ナトリウム、パルミチン酸アスコルビル、エデト酸二ナトリウム(EDTA)、またはそれらの組み合わせ;結合剤、例えばヒドロキシプロピルメチルセルロース;希釈剤、例えばラクトース一水和物fast flo(粒内)およびラクトース一水和物fast flo(粒外)を含むラクトース一水和物、ならびに微結晶セルロース、ならびにバッファー、例えばリン酸バッファー、を含むがこれに限定されない賦形剤を含有する。錠剤はさらに、滑剤、崩壊剤および充填剤より選択される1つまたはそれより多くの賦形剤を含有することができる。
【0124】
ある種の態様において、経口錠剤中のシタキセンタンナトリウムの量は、組成物の総重量の約5%から約40%である。ある種の態様において、シタキセンタンナトリウムの量は、組成物の総重量の約7%から約35%、10%から約30%、12%から約32%、15%から約30%、17%から約27%、15%から約25%である。ある種の態様において、シタキセンタンナトリウムの量は、組成物の総重量の約5%、7%、9%、10%、12%、15%、17%、20%、22%、25%、27%、30%、35%、または40%である。ある種の態様において、シタキセンタンナトリウムの量は約20%である。
【0125】
ある種の態様において経口錠剤は、約10mg、20mg、25mg、30mg、40mg、50mg、60mg、70mg、80mg、90mg、100mg、125mg、150mg、175mg、200mg、225mg、250mg、275mg、280mg、300mg、または350mgのシタキセンタンナトリウムを含有する。
【0126】
ある種の態様において錠剤は、2つの抗酸化剤の組み合わせ、例えばパルミチン酸アスコルビルおよびEDTA、二ナトリウムを含有する。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、錠剤の総重量の約0.05%から約3%の範囲にある。他の態様において、パルミチン酸アスコルビルの量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1%である。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は、錠剤の総重量の約0.2%である。
【0127】
ある種の態様において経口錠剤中のパルミチン酸アスコルビルの量は、約0.1mgから約5mg、約0.5mgから約4mg、約0.7mgから約3mg、または約1mgから約2mgである。ある種の態様において経口錠剤中のパルミチン酸アスコルビルの量は、約0.1mg、0.5mg、0.7mg、1mg、1.3mg、1.5mg、1.7mg、2mg、2.5mg、または約3mgである。ある種の態様において、製剤中のパルミチン酸アスコルビルの量は約1mgである。
【0128】
ある種の態様において製剤中のEDTA、二ナトリウムの量は、錠剤の総重量の約0.05%から約3%の範囲にある。他の態様においてEDTA、二ナトリウムの量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において製剤中のEDTA、二ナトリウムの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1%である。ある種の態様において錠剤中のEDTA、二ナトリウムの量は、錠剤の総重量の約0.2%である。
【0129】
ある種の態様において経口錠剤中のEDTA、二ナトリウムの量は、約0.1mgから約5mg、約0.5mgから約4mg、約0.7mgから約3mg、または約1mgから約2mgである。ある種の態様において経口錠剤中のEDTA、二ナトリウムの量は、約0.1mg、0.5mg、0.7mg、1mg、1.3mg、1.5mg、1.7mg、2mg、2.5mg、または約3mgである。ある種の態様において、経口錠剤中のEDTA、二ナトリウムの量は約1mgである。
【0130】
ある種の態様において、錠剤は希釈剤の組み合わせ、例えば微結晶セルロース(AVICEL PH 102)、ラクトース一水和物fast flo(粒内)およびラクトース一水和物fast flo(粒外)を含有する。ある種の態様において、経口錠剤中のラクトース一水和物fast flo(粒内)の量は、組成物の総重量の約5%から約30%である。ある種の態様において、ラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約7%から約25%、約10%から約20%、約13%から約20%である。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約5%、7%、10%、13%、14%、15%、15.5%、16%、16.1%、16.2%、16.3%、16.4%、16.5%、16.6%、16.7%、16.8%、16.9%、17%、17.5%、18%、18.5%、19%、20%、25%または30%である。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、錠剤の総重量の約16.9%である。
【0131】
ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約40mgから約100mg、約45mgから約95mg、約50mgから約90mgである。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約40mg、45mg、50mg、55mg、60mg、65mg、70mg、75mg、80mg、81mg、82mg、83mg、83.5mg、84mg、84.1mg、84.2mg、84.3mg、84.4mg、84.5mg、84.6mg、84.7mg、85mg、85.5mg、90mg、90.5mg、または100mgである。ある種の態様においてラクトース一水和物fast flo(粒内)の量は、約84.3mgである。
【0132】
ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約7%から約25%、約10%から約20%、約13%から約20%である。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約5%、7%、10%、13%、14%、15%、15.5%、16%、16.1%、16.2%、16.3%、16.4%、16.5%、16.6%、16.7%、16.8%、16.9%、17%、17.5%、18%、18.5%、19%、20%、25%または30%である。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、錠剤の総重量の約16.4%である。ある種の態様において、経口錠剤中のラクトース一水和物fast flo(粒外)の量は、約40mgから約100mg、約45mgから約95mg、約50mgから約90mgである。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、約40mg、45mg、50mg、55mg、60mg、65mg、70mg、75mg、80mg、81mg、81.3mg、81.5mg、81.8mg、82mg、82.3mg、82.5mg、82.7mg、83mg、83.5mg、84mg、85mg、85.5mg、90mg、90.5mg、または100mgである。ある種の態様においてラクトース一水和物fast flo(粒外)の量は、約82mgである。
【0133】
ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、組成物の総重量の約10%から約50%である。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約15%から約45%、約20%から約43%、約25%から約40%である。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約15%、17%、20%、23%、25%、27%、30%、32%、34%、35%、37%、40%、42%、45%、または50%である。ある種の態様において微結晶セルロース(AVICEL PH 102)の量は、錠剤の総重量の約35%である。
【0134】
ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、約130mgから300mgである。ある種の態様において、微結晶セルロース(AVICEL PH 102)の量は、約140mgから約275mg、または約150mgから約250mgである。ある種の態様において微結晶セルロース(AVICEL PH 102)の量は、約150mg、160mg、165mg、170mg、175mg、180mg、185mg、190mg、または200mgである。ある種の態様において、経口錠剤中の微結晶セルロース(AVICEL PH 102)の量は、約175mgである。
【0135】
ある種の態様において、結合剤はヒドロキシプロピルメチルセルロース(E−5P)である。ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、組成物の総重量の約0.5%から約20%である。ある種の態様において、ヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約1%から約15%、約2%から約10%、約3%から約8%である。ある種の態様においてヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約1%、2%、3%、4%、5%、6%、7%、8%、9%、または10%である。ある種の態様においてヒドロキシプロピルメチルセルロース(E−5P)の量は、錠剤の総重量の約5%である。
【0136】
ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約5mgから約50mg、約10mgから約40mg、約15mgから約30mgである。ある種の態様において、錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約10mg、15mg、20mg、22mg、25mg、27mg、30mg、35mg、または約40mgである。ある種の態様において錠剤中のヒドロキシプロピルメチルセルロース(E−5P)の量は、約25mgである。
【0137】
本明細書に提供するシタキセンタンナトリウムの製剤は、中性のpHで安定である。ある種の態様において、バッファー剤の混合物、例えば一塩基性リン酸ナトリウム一水和物、および二塩基性リン酸ナトリウム無水物を使用して、錠剤中の薬剤の安定性を改善することができる。ある種の態様において一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.05重量%から約3重量%の範囲にある。他の態様において一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.07%から約1.5%、0.1%から約1%、0.15%から約0.5%の範囲にある。ある種の態様において、製剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1.%である。ある種の態様において、製剤中の一塩基性リン酸ナトリウム一水和物の量は、錠剤の総重量の約0.1%である。
【0138】
ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.1mgから約3mg、約0.2mgから約2.5mg、約0.5mgから約2mg、または約0.6mgから約1mgである。ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.1mg、0.2mg、0.3mg、0.4mg、0.5mg、0.6mg、0.7mg、0.8mg、0.9mg、または1mgである。ある種の態様において、経口錠剤中の一塩基性リン酸ナトリウム一水和物の量は、約0.6mgである。
【0139】
ある種の態様において二塩基性リン酸ナトリウム無水物の量は、錠剤の総重量の約0.05重量%から約3重量%の範囲にある。他の態様において二塩基性リン酸ナトリウムの量は、錠剤の総重量の約0.07%から約1.5%、約0.1%から約1%、約0.15%から約0.5%の範囲にある。ある種の態様において、製剤中の二塩基性リン酸ナトリウムの量は、約0.05%、0.07%、0.09%、0.1%、0.12%、0.15%、0.17%、0.18%、0.2%、0.23%、0.25%、0.27%、0.3%、0.35%、0.4%、0.45%、0.5%、0.7%、または1.%である。ある種の態様において製剤中の二塩基性リン酸ナトリウムの量は、錠剤の総重量の約0.2%である。
【0140】
ある種の態様において、経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約0.1mgから約3.5mg、約0.5mgから約2.5mg、または約0.7mgから約2mgである。ある種の態様において、経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約0.1mg、0.3mg、0.5mg、0.7mg、0.9mg、1mg、1.1mg、1.3mg、1.5mg、1.7mg、または2mgである。経口錠剤中の二塩基性リン酸ナトリウム無水物の量は、約1.1mgである。
【0141】
ある種の態様において錠剤は、崩壊剤、例えばナトリウムスターチグリコレート(粒内)およびナトリウムスターチグリコレート(粒外)を含有する。ある種の態様において錠剤中のナトリウムスターチグリコレート(粒内)の量は、組成物の総重量の約0.1%から約10%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約0.5%から約8%、約1%から約5%、約2%から約4%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約0.5%、1%、1.5%、1.7%、2%、2.3%、2.5%、2.7%、3%、3.5%、4%、または5%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、錠剤の総重量の約2.5%である。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約30mgから約5mg、約20mgから約10mg、約15mgから約10mgである。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約5mg、7mg、10mg、11mg、11.5mg、12mg、12.5mg、13mg、15mg、または20mgである。ある種の態様においてナトリウムスターチグリコレート(粒内)の量は、約12.5mgである。
【0142】
ある種の態様において錠剤中のナトリウムスターチグリコレート(粒外)の量は、組成物の総重量の約0.1%から約10%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約0.5%から約8%、約1%から約5%、約2%から約4%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約0.5%、1%、1.5%、1.7%、2%、2.3%、2.5%、2.7%、3%、3.5%、4%、または5%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、錠剤の総重量の約2.5%である。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約30mgから約5mg、約20mgから約10mg、約15mgから約10mgである。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約5mg、7mg、10mg、11mg、11.5mg、12mg、12.5mg、13mg、15mg、または20mgである。ある種の態様においてナトリウムスターチグリコレート(粒外)の量は、約12.5mgである。
【0143】
ある種の態様において、錠剤は滑剤、例えばステアリン酸マグネシウムを含有する。ある種の態様において錠剤中のステアリン酸マグネシウムの量は、組成物の総重量の約0.1%から約8%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約0.5%から約6%、約0.7%から約5%、約1%から約4%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約0.5%、0.7%、1%、1.2%、1.5%、1.7%、2%、2.5%、または3%である。ある種の態様においてステアリン酸マグネシウムの量は、錠剤の総重量の約2.5%である。ある種の態様において錠剤中のステアリン酸マグネシウムの量は、約15mgから約1mgである。ある種の態様においてステアリン酸マグネシウムの量は、約10mgから約3mg、または約7mgから約5mgである。ある種の態様においてステアリン酸マグネシウムの量は、約3mg、4mg、4.5mg、5mg、6mg、7mg、8mg、9mgまたは10mgである。ある種の態様においてステアリン酸マグネシウムの量は、約5mgである。
【0144】
1つの態様において、本明細書に提供する錠剤製剤は、湿度をバリアーするコーティングを含有する。適切なコーティング材料は当該技術において公知であり、セルロース由来 例えばセルロースフタレート(セピフィルム(Sepifilm)、ファーマコート(Pharmacoat))、またはポリビニル由来のセピフィルムECLタイプ、またはショ糖由来 例えばセピスパース(Sepisperse) DR、AS、AP OR K (着色)タイプの糖衣用の糖、例えばセピスパース Dry 3202 Yellow、Blue Opadry、Eudragit EPO およびOpadry AMBのどれかのコーティング剤を含むがこれに限定されない。コーティングは、シタキセンタンナトリウムの酸化を阻止するための湿度のバリアーとして役立つ。ある種の態様においてコーティング材料は、セピフィルムLP014/セピスパース Dry 3202 Yellow (セピフィルム/セピスパース) (3/2wt/wt)を、約1から約7%、または約4%の錠剤の重量増量である。ある種の態様においてコーティング材料は、セピフィルムLP 014/セピスパース Dry 3202 Yellow (セピフィルム/セピスパース)である。ある種の態様においてセピフィルム/セピスパースの比率は、1:2、1:1、または3:2wt/wtである。ある種の態様においてセピフィルム/セピスパースコーティングは、約1%、2%、3%、4%、5%、6%、または7%の錠剤の重量増量である。ある種の態様においてセピフィルム/セピスパースコーティングは、約1.6%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、約0.5%、0.8%、1%、1.3%、1.6%、2%、2.4%、2.5%、3%、または4%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、約2.4%の錠剤の重量増量である。ある種の態様においてセピスパース Dry 3202(黄色)は、1錠当たり約1mg、3mg、5mg、6mg、7mg、8mg、9mg、10mg、13mg、15mg、または20mgである。ある種の態様においてセピスパース Dry 3202(黄色)は、1錠あたり約8mgである。ある種の態様においてセピフィルムLP 014は、約0.5%、1%、1.5%、2%、2.2%、2.4%、2.6%、3%、3.5%、または4%の錠剤の重量増量である。ある種の態様においてセピフィルムLP 014は、約2.4%の錠剤の重量増量である。ある種の態様においてセピフィルムLP 014は、1錠当たり約5mg、7mg、9mg、10mg、11mg、12mg、13mg、15mg、17mg、または20mgである。ある種の態様においてセピフィルムLP 014コーティングは、1錠当たり約12mgである。
【0145】
ある種の態様において錠剤は、シタキセンタンナトリウム、微結晶セルロース、ラクトース一水和物fast flo(粒内)、ラクトース一水和物fast flo(粒外)、ヒドロキシプロピルメチルセルロースE−5P、パルミチン酸アスコルビル、EDTA二ナトリウム、一塩基性リン酸ナトリウム一水和物、二塩基性リン酸ナトリウム無水物、ナトリウムスターチグリコレート(粒内)、ナトリウムスターチグリコレート(粒外)、ステアリン酸マグネシウム、およびセピフィルムLP014/セピスパース Dry 3202 Yellowのコーティングを含有する。
【0146】
ある種の態様において錠剤は、約20%のシタキセンタンナトリウム、約35%の微結晶セルロース、約16.9%のラクトース一水和物fast flo(粒内)、約16.4%のラクトース一水和物fast flo(粒外)、約5.0%のヒドロキシプロピルメチルセルロースE−5P、約0.2%のパルミチン酸アスコルビル、約0.2%のエデト酸二ナトリウム(EDTA)、約0.1%の一塩基性リン酸ナトリウム一水和物、約0.2%の二塩基性リン酸ナトリウム無水物、約2.5%のナトリウムスターチグリコレート(粒外)、約2.5%のナトリウムスターチグリコレート(粒内)、および約1%のステアリン酸マグネシウムを含有する。錠剤はさらに、セピフィルムLP014のコーティングを約2.4%の重量増量にて、およびセピスパース Dry 3202 Yellowのコーティングを約1.6%の重量増量にて含有する。
【0147】
ある種の態様において、本明細書に提供する経口錠剤は、約100mgのシタキセンタンナトリウム、約1.0mgのパルミチン酸アスコルビル、約1.0mgのエデト酸二ナトリウム(EDTA)、約25mgのヒドロキシプロピルメチルセルロースE−5P、約84.3mgのラクトース一水和物fast flo(粒外)、約82mgのラクトース一水和物fast flo(粒内)、約175mgの微結晶セルロース、約0.6mgの一塩基性リン酸ナトリウム一水和物、約1.1mgの二塩基性リン酸ナトリウム無水物、約12.5mgのナトリウムスターチグリコレート(粒内)、約12.5mgのナトリウムスターチグリコレート(粒外)、約5mgのステアリン酸マグネシウム、non-bovine、および約192.5mgの純水を含有する、500mgの錠剤である。錠剤はさらにセピフィルムLP014のコーティングを約12mgで、セピスパース Dry 3202 Yellowのコーティングを約8mgで含有する。
【0148】
b.徐放性投与剤形
本明細書に提供する多形は、コントロールされた放出手段により、または当業者に周知されている送達デバイスにより投与することができる。例として、米国特許第3,845,770号;3,916,899号;3,536,809号;3,598,123号;および4,008,719号、5,674,533号、5,059,595号、5,591,767号、5,120,548号、5,073,543号、5,639,476号、5,354,556号、5,639,480号、5,733,566号、5,739,108号、5,891,474号、5,922,356号、5,972,891号、5,980,945号、5,993,855号、6,045,830号、6,087,324号、6,113,943号、6,197,350号、6,248,363号、6,264,970号、6,267,981号、6,376,461号、6,419,961号、6,589,548号、6,613,358号、6,699,500号、および6,740,634号に記載されているものを含むが、これに限定されない。またこれらの各文献を本明細書において参照として援用する。そのような投与剤形を使用して、例えばヒドロキシプロピルメチルセルロース、他のポリマーマトリクス、ゲル、透過可能な膜、浸透圧システム、多層コーティング、微粒子、リポソーム、マイクロスフェア、またはそれらを組み合わせを使用することで、1つまたはそれより多くの活性成分の緩やかなまたはコントロールされた放出を提供することができ、これらの比率を変化させて所望の放出プロフィールを提供することができる。本明細書に記載するものを含む、当業者に公知の適切なコントロールされた放出の製剤は、本明細書に提供する活性成分と共に使用するために容易に選択することができる。
【0149】
すべてのコントロールされた放出の医薬製品は、それらのコントロールされていない対応製品によって達成されるものを上回る薬剤療法の改善という、共通の目標を有する。理想的には、医学的治療における所望によりデザインされたコントロールされた放出の調製物の使用は、最小量の薬剤物質を利用して、最小時間で状態を治癒またはコントロールすることを特徴とする。コントロールされた放出の製剤の利点として、延長される薬剤の活性、低減される投与頻度、および増加される患者のコンプライアンスを含む。加えてコントロールされた放出の製剤を使用して、作用またはその他の特徴、例えば薬剤の血液レベルの開始時間に影響を及ぼすことができ、したがって副作用(例えば有害な影響)の発生に影響を及ぼすことができる。
【0150】
最もコントロールされた放出の製剤は、即時に所望の治療効果を生む一定量の薬剤(活性成分)を最初に放出し、そして延長された時間にわたりこのレベルの治療効果または予防効果を維持するために、別の量の薬剤を徐々にそして継続的に放出するようにデザインされる。この一定レベルの薬剤を体内に維持する目的のため、薬剤は、代謝され体内から排泄される薬剤の量と置き換わる速度で、投与剤形から放出されなければならない。活性成分のコントロールされた放出は、pH、温度、酵素、水、またはその他の生理学的条件もしくは化合物を含むがこれに限定されない様々な条件によって、刺激され得る。
【0151】
ある種の態様において当該多形または多形の混合物は、静脈内注入、植え込み型浸透圧ポンプ、経皮パッチ、リポソーム、またはその他の投与様式を使用して投与してよい。1つの態様において、ポンプを使用してよい(Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al., Surgery 88:507 (1980); Saudek et al., N. Engl. J. Med. 321:574 (1989)を参照のこと)。もう1つの態様において、ポリマー材料を使用することができる。なおもう1つの態様において、コントロールされた放出システムは、治療標的の近位に置く、すなわち、したがって全身用量の一分画のみしか必要としないことが可能である(例えばGoodson, Medical Applications of Controlled Release, vol. 2, pp. 115-138 (1984)を参照のこと)。一部の態様において、コントロールされた放出のデバイスは、被験者の体内に不適当な免疫活性化部位または腫瘍部位の近位に導入する。その他のコントロールされた放出システムは、Langerによる総説(Science 249:1527-1533 (1990)において議論されている。活性成分は、固体の内側のマトリクス中に、例えばポリメチルメタクリレート、ポリブチルメタクリレート、可塑化または非可塑化ポリ塩化ビニル、可塑化ナイロン、可塑化ポリエチレンテレフタレート、天然ゴム、ポリイソプレン、ポリイソブチレン、ポリブタジエン、ポリエチレン、エチレン−ビニルアセテートコポリマー、シリコンゴム、ポリジメチルシロキサン、炭酸シリコンコポリマー、親水性ポリマー 例えばアクリル酸およびメタクリル酸のエステルのヒドロゲル、コラーゲン、架橋したポリビニルアルコールおよび架橋し部分的に加水分解したポリビニル酢酸の中に分散させることができ、これを、外側のポリマーの膜、例えば体液中で不溶性であるポリエチレン、ポリプロピレン、エチレン/プロピレンコポリマー、エチレン/アクリル酸エチルコポリマー、エチレン/酢酸ビニルコポリマー、シリコンゴム、ポリジメチルシロキサン、ネオプレンゴム、塩素化ポリエチレン、ポリ塩化ビニル、塩化ビニルと酢酸ビニルとのコポリマー、塩化ビニリデン、エチレンおよびプロピレン、イオノマーのポリエチレンテレフタレート、ブチルゴム、エピクロロヒドリンゴム、エチレン/ビニルアルコールコポリマー、エチレン/酢酸ビニル/ビニルアルコールターポリマー、およびエチレン/ビニルオキシエタノールコポリマーで覆う。その後活性成分は、放出速度コントロールステップにおいて、外側のポリマーの膜を通って拡散する。そのような非経口組成物中に含有される活性成分のパーセンテージは、その具体的な性質、ならびに被験者の必要性にかなり依存する。
【0152】
c.非経口投与
皮下、筋肉内、または静脈内のどちらかへの注射を一般に特徴とする非経口投与もまた、本明細書において意図する。注射剤は、液体の溶液もしくは懸濁液、注射前に液体中での溶液もしくは懸濁液に適する固体として、またはエマルジョンとしてのどちらかで、従来の剤形において調製することができる。適切な賦形剤は、例えば水、生理食塩水、ブドウ糖、グリセロール、またはエタノールである。加えて所望であれば、投与する医薬組成物はまた、微量の非毒性の補助的物質、例えば湿潤剤または乳化剤、pH緩衝化剤、安定剤、溶解度増強剤、ならびに他のそのような物質、例えば酢酸ナトリウム、モノラウリル酸ソルビタン、オレイン酸トリエタノールアミン、およびシクロデキストリンを含有してよい。
【0153】
当該組成物の非経口投与は、静脈内、皮下、および筋肉内への投与を含む。非経口投与のための調製物は、注射用に用意された滅菌溶液、滅菌乾燥の可溶性製品、例えば皮下注射用錠剤を含む、使用直前に溶媒と組み合わるように用意された凍結乾燥粉末、注射用に用意された滅菌懸濁液、使用直前にビヒクルと組み合わせるように用意された滅菌乾燥の非可溶性製品、および滅菌エマルジョンを含む。溶液は水溶性でも非水溶性でもよい。
【0154】
静脈内に投与する場合、適切な担体は、生理学的食塩水またはリン酸緩衝化食塩水(RBS)、ならびに増粘剤および可溶化剤、例えばグルコース、ポリエチレングリコール、およびポリプロピレングリコール、およびそれらの混合物を含有する溶液を含む。
【0155】
非経口調製物中に使用する医薬的に受容可能な担体は、水性ビヒクル、非水性ビヒクル、抗菌剤、等張剤、バッファー、抗酸化剤、局所麻酔薬、懸濁剤および分散剤、乳化剤、金属イオン封鎖剤またはキレート剤、およびその他の医薬的に受容可能な物質を含む。
【0156】
水性ビヒクルの例は、塩化ナトリウム注射液(Sodium Chloride Injection)、リンゲル注射液(Ringers Injection)、等張性ブドウ糖注射液(Isotonic Dextrose Injection)、滅菌水注射液(Sterile Water Injection)、ブドウ糖および乳酸加リンガー注射液(Dextrose and Lactated Ringers Injection)を含む。非水溶性非経口用ビヒクルは、植物由来の固定油、すなわち綿実油、コーン油、ゴマ油、およびピーナツ油を含む。フェノールまたはクレゾール、水銀剤、ベンジルアルコール、クロロブタノール、メチルおよびプロピルp−ヒドロキシ安息香酸エステル、チメロサール、ベンザルコニウムクロリド、ならびにベンズエトニウムクロリドを含む多数回−用量の容器中に包装される非経口調製剤に、静菌的または静真菌的濃度の抗生物質が、加えられなければならない。等張剤は、塩化ナトリウムおよびブドウ糖を含む。バッファーはリン酸塩およびクエン酸塩を含む。抗酸化剤は、重硫酸ナトリウムを含む。局所鎮静剤は、プロカイン塩酸塩を含む。懸濁剤および分散剤は、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルメチルセルロース、およびポリビニルピロリドンを含む。乳化剤は、ポリソルベート80(TWEEN(登録商標)80)を含む。金属イオン封鎖剤または金属イオンのキレート剤は、EDTAを含む。医薬的担体はまた、水混和性ビヒクルとしてエチルアルコール、ポリエチレングリコール、およびプロピレングリコール、ならびにpH調節剤として水酸化ナトリウム、塩酸、クエン酸、または乳酸を含む。
【0157】
シタキセンタンナトリウムの濃度は、注射が、所望の薬学的効果を得られる有効量を提供するように調整する。正確な用量は、当該技術において公知であるように、患者または動物の年齢、体重および状態に依存する。
【0158】
ユニット用量の非経口調製物は、アンプル、バイアル、または針付き注射器内に充填する。非経口投与のすべての調製物は、当該技術において知られており実践されているように、滅菌しなければならない。
【0159】
実例として、活性成分を含有する滅菌水溶液の静脈内注入または動脈内注入は、有効な投与様式である。もう1つの態様は、所望の薬学的効果を得るために必要時に注射する、活性材料を含有する滅菌の水性または油性の溶液または懸濁液である。
【0160】
注射液は、局所または全身への投与用にデザインする。典型的には治療上有効な投与量は、治療する組織(1つまたは複数)に、少なくとも約0.1%w/wから約90%w/wもしくはそれ以上までの、または1%w/wより多くの濃度のシタキセンタンを含有するように製剤化する。活性成分は、一度に投与してもよいし、または何回かのより少量の用量に分割して、時間の間隔をおいて投与してもよい。正確な投与量および治療期間は、治療する組織の関数であり、公知の試験プロトコルを使用して、またはin vivo もしくはin vitroの検査データからの外挿により経験的に決定してよいことは、理解されよう。濃度および投与量の数値はまた、治療する個体の年齢によって変更してよいことには注意しなければならない。あらゆる特定の被験者に対して、具体的な投与計画が、個々の必要性、および投与する人もしくは製剤の投与を監督する人の専門的判断に従って、時間にわたり調整されなければならないこと、そして本明細書に述べた濃度範囲は、具体例に過ぎず、主張した製剤の範囲または実践を限定する意図はないことは、さらに理解されなければならない。
【0161】
当該多形または多形の混合物は、微粒子化した形または他の適切な形で懸濁してもよいし、または誘導体化して、より可溶性の活性製品を生成するもしくはプロドラッグを生成してもよい。得られる混合物の形は、意図した投与様式、および選択した担体またはビヒクルにおけるシタキセンタンナトリウムの溶解度を含む、いくつかの因子に依存する。有効濃度は、状態の症状を緩和するために十分な濃度であり、経験的に決定してよい。
【0162】
d.凍結乾燥粉末
凍結乾燥粉末もまた本明細書において提供するが、これは、溶液、エマルジョンおよびその他の混合物として投与用に再構成することができる。これらはまた、固体またはゲルとして再構成および製剤化してよい。
【0163】
滅菌の凍結乾燥粉末は、活性成分または医薬的に受容可能なその塩を、適切な溶媒中に溶かすことによって調製する。溶媒は、溶解度を改善する賦形剤、または粉末のその他の医薬成分、または粉末から調製されて再構成される溶液を含有してよい。使用してよい賦形剤は、ブドウ糖、ソルビトール、果糖、コーンシロップ、キシリトール、グリセリン、グルコース、ショ糖、またはその他の適切な物質を含んでよいが、これに限定されない。溶媒はまた、バッファー、例えばクエン酸塩、リン酸ナトリウムもしくはリン酸カリウム、または当業者に公知の、典型的にはほぼ中性のpHのその他のそのようなバッファーを含有してよい。その後の溶液の滅菌濾過に続いて、当業者に公知の標準的な条件下での凍結乾燥により、所望の製剤を提供する。一般に得られた溶液は、凍結乾燥用のバイアル内に分注することになる。各バイアルは、シタキセンタンナトリウムの単回投与量(10mg−350mgもしくは100−300mg)、または複数回投与量を含有することになる。凍結乾燥粉末は、適当な条件下、例えば約4℃から室温までで保存することができる。
【0164】
この凍結乾燥粉末の注射用水を用いての再構成は、非経口投与で使用するための製剤を提供する。再構成には、1mL滅菌水または他の適切な担体当たり、約1−50mg、5−35mg、または約9−30mgの凍結乾燥粉末を加える。正確な量は、選択される複合体(conjugate)に依存する。そのような量は経験的に決定することができる。
【0165】
具体例としての凍結乾燥製剤
ある種の態様において、シタキセンタンナトリウムの安定な凍結乾燥粉末を本明細書において提供する。凍結乾燥粉末は、抗酸化剤、バッファー、および充填剤を含有する。本明細書に提供する凍結乾燥粉末において、存在するシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約25%から約60%の範囲にある。ある種の態様においてシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約30%から約50%、または約35%から約45%である。ある種の態様においてシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約30%、33%、35%、37%、40%、41%、43%、45%、47%、50%、53%、55%、または60%である。1つの態様において凍結乾燥粉末中のシタキセンタンナトリウムの量は、凍結乾燥粉末の総重量の約41%である。
【0166】
ある種の態様において凍結乾燥粉末は、抗酸化剤、例えば亜硫酸ナトリウム、亜硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、モノチオグリセロール、アスコルビン酸、またはそれらの組み合わせを含有する。1つの態様において、抗酸化剤はモノチオグリセロールである。1つの態様において抗酸化剤は、アスコルビン酸、亜硫酸ナトリウム、および亜硫酸水素ナトリウムを組み合わせたものである。ある種の態様において、本明細書に提供する凍結乾燥製剤は、公知のシタキセンタンナトリウムの凍結乾燥製剤(WO 98/49162を参照のこと)と比較して、再構成時に改善された安定性を有する。
【0167】
ある種の態様において、抗酸化剤はモノチオグリセロールである。ある種の態様においてモノチオグリセロールは、凍結乾燥粉末の総重量の約10%から約30%の範囲である量で存在する。ある種の態様においてモノチオグリセロールは、凍結乾燥粉末の総重量の約12%から約25%、または約15%から約20%の範囲である量で存在する。ある種の態様において、凍結乾燥粉末中のモノチオグリセロールの量は、凍結乾燥粉末の総重量の約10%、12%、14%、15%、15.5%、16%、16.2%、16.4%、16.8%、17%、17.5%、19%、22%、25%、または30%である。ある種の態様においてモノチオグリセロールの量は、凍結乾燥粉末の総重量の約16.4%である。
【0168】
ある種の態様において亜硫酸ナトリウムは、凍結乾燥粉末の総重量の約1%から約6%の量で存在する。他の態様において亜硫酸ナトリウムは、約1.5%から約5%、または約2%から約4%の量で存在する。ある種の態様において亜硫酸ナトリウムの量は、凍結乾燥粉末の総重量の約1%、1.5%、2%、2.5%、3%、3.3%、3.5%、3.8%、4%、4.5%、または5%である。1つの態様において亜硫酸ナトリウムの量は、凍結乾燥粉末の総重量の約3.3%である。
【0169】
ある種の態様においてアスコルビン酸は、凍結乾燥粉末の総重量の約1%から約6%の量で存在する。他の態様においてアスコルビン酸は、約1.5%から約5%、または約2%から約4%の量で存在する。ある種の態様においてアスコルビン酸の量は、凍結乾燥粉末の総重量の約1%、1.5%、2%、2.5%、3%、3.3%、3.5%、3.8%、4%、4.5%、または5%である。1つの態様においてアスコルビン酸の量は、凍結乾燥粉末の総重量の約3.3%である。
【0170】
ある種の態様において亜硫酸水素ナトリウムは、凍結乾燥粉末の総重量の約5%から約15%、または約8%から約12%の量で存在する。ある種の態様において亜硫酸水素ナトリウムの量は、凍結乾燥粉末の総重量の約5%、6%、7%、8%、9%、10%、10.3%、10.5%、10.8%、11%、11.5%、12%、または15%の量で存在する。1つの態様において亜硫酸水素ナトリウムの量は、凍結乾燥粉末の総重量の約10.8%である。
【0171】
1つの態様において抗酸化剤は、アスコルビン酸、亜硫酸ナトリウム、および亜硫酸水素ナトリウムの組み合わせである。1つの態様において凍結乾燥粉末中のアスコルビン酸の量は、凍結乾燥粉末の総重量の約3.3%、亜硫酸ナトリウムの量は約3.3%、および亜硫酸水素ナトリウムの量は約10.8%である。
【0172】
1つの態様において凍結乾燥粉末はまた、以下の賦形剤:バッファー、例えばリン酸ナトリウムもしくはリン酸カリウム、またはクエン酸塩;および充填剤、例えばグルコース、ブドウ糖、マルトース、ショ糖、ラクトース、ソルビトール、マンニトール、グリシン、ポリビニルピロリドン、デキストラン、の1つまたはそれより多くを含有する。1つの態様において充填剤は、ブドウ糖、D−マンニトール、またはソルビトールより選択する。
【0173】
ある種の態様において、本明細書に提供する凍結乾燥粉末は、リン酸バッファーを含有する。ある種の態様においてリン酸バッファーは、約10mM、約15mM、約20mM、約25mM、または約30mMの濃度で存在する。ある種の態様において、リン酸バッファーは20mMの濃度で存在する。ある種の態様において、リン酸バッファーは約20mMの濃度で存在し、そして構成された製剤は約7のpHを有する。
【0174】
ある種の態様において、本明細書に提供する凍結乾燥粉末は、クエン酸バッファーを含有する。1つの態様においてクエン酸バッファーは、クエン酸ナトリウム二水和物である。ある種の態様においてクエン酸ナトリウム二水和物の量は、凍結乾燥粉末の総重量の約5%から約15%、約6%から約12%、または約7%から約10%である。ある種の態様において、凍結乾燥粉末中のクエン酸ナトリウム二水和物の量は、凍結乾燥粉末の総重量の約5%、6%、7%、7.5%、8%、8.3%、8.5%、8.8%、9%、9.5%、10%、12%、または約15%である。ある種の態様において構成される製剤は、約5から10、または約6のpHを有する。
【0175】
ある種の態様において本明細書に提供する凍結乾燥粉末は、ブドウ糖を、凍結乾燥粉末の総重量の約30%から約60%の範囲である量で含有する。ある種の態様においてブドウ糖の量は、凍結乾燥粉末の総重量の約30%、35%、40%、45%、50%、または60%である。ある種の態様においてブドウ糖の量は、凍結乾燥粉末の総重量の約40%である。ある種の態様において本明細書に提供する凍結乾燥粉末は、マンニトールを、凍結乾燥粉末の総重量の約20%から約50%の範囲である量で含有する。ある種の態様においてマンニトールの量は、凍結乾燥粉末の総重量の約20%、25%、30%、32%、32.5%、32.8%、33%、34%、37%、40%、45%、または50%、である。ある種の態様においてマンニトールの量は、凍結乾燥粉末の総重量の約32.8%である。
【0176】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約41%のシタキセンタンナトリウム、約3.3%のアスコルビン酸、約3.3%の亜硫酸ナトリウム、および約10.8%の亜硫酸水素ナトリウム、約8.8%のクエン酸ナトリウム二水和物、および約32.8%のマンニトールを含有する。ある種の態様において、凍結乾燥粉末は以下の組成を有する:
【0177】
【表10】
【0178】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約40から約30%のシタキセンタンナトリウム、約4から約6%のアスコルビン酸、約6から約8%のクエン酸ナトリウム二水和物、約50から約60%のD−マンニトール、および約1から約2%のクエン酸一水和物を含有する。ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約33%のシタキセンタンナトリウム、約5.3%のアスコルビン酸、約7.6%のクエン酸ナトリウム二水和物、約53%のD−マンニトール、および約0.13%のクエン酸一水和物を含有する。1つの態様において、凍結乾燥粉末は以下の組成を有する:
【0179】
【表11】
【0180】
ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約40から約30%のシタキセンタンナトリウム、約4から約6%のアスコルビン酸、約3から約4%の二塩基性リン酸ナトリウム七水和物、約50から約60%のD−マンニトール、および約1.5から約2.5%の一塩基性リン酸ナトリウム一水和物を含有する。ある種の態様において本明細書に提供する凍結乾燥粉末は、凍結乾燥粉末の総重量の約34%のシタキセンタンナトリウム、約5.5%のアスコルビン酸、約3.7%の二塩基性リン酸ナトリウム七水和物、約55%のD−マンニトール、および約1.9%の一塩基性リン酸ナトリウム一水和物を含有する。1つの態様において、凍結乾燥粉末は以下の組成を有する:
【0181】
【表12】
【0182】
本明細書に提供するシタキセンタンナトリウムの凍結乾燥製剤は、本明細書に記載する方法を含むがこれに限定されない、シタキセンタンナトリウムを送達するための標準的な治療法を使用して、それを必要とする患者に投与することができる。1つの態様において凍結乾燥シタキセンタンナトリウムは、本明細書に提供する凍結乾燥シタキセンタンナトリウムの治療有効量を、医薬的に受容可能な溶媒中に溶かして医薬的に受容可能な溶液を生成し、その溶液を(例えば静脈内注射により)患者に投与することにより、投与する。
【0183】
本明細書に提供する凍結乾燥シタキセンタンナトリウム製剤は、あらゆる医薬的に受容可能な希釈剤を使用して、患者への非経口投与用に構成することができる。そのような希釈剤として、注射用滅菌水(Sterile Water for Injection)、USP、注射用滅菌静菌水(Sterile Bacteriostatic Water for Injection)、生理食塩水、USP(保存剤としてベンジルアルコールまたはパラベン)を含むがこれに限定されない。注射用の適切な溶液が調製されるように、あらゆる量の希釈剤を使用して、凍結乾燥シタキセンタンナトリウム製剤を構成してよい。したがって希釈剤の量は、凍結乾燥シタキセンタンナトリウムを溶解させるための十分量でなければならない。典型的には10−50mL、または10から20mLの希釈剤を使用して、凍結乾燥シタキセンタンナトリウム製剤を構成し、約1−50mg/mL、約5−40mg/mL、約10−30mg/mL、または10−25mg/mLの最終濃度を得る。ある種の態様において、再構成される溶液中のシタキセンタンナトリウムの最終濃度は、約25mg/mL、または約12.5mg/mLである。正確な量は、治療する適応症に依存する。そのような量は経験的に決定することができる。一部の態様において、再構成される溶液のpHは約5から約10まで、または約6から約8までである。一部の態様において、再構成される溶液のpHは約5、6、7、8、9、または10である。
【0184】
凍結乾燥シタキセンタンナトリウムの構成される溶液は、構成後直ちに患者に投与することができる。あるいは構成された溶液を保存して、約1−72時間、約1−48時間、または約1−24時間以内に使用することができる。一部の態様において、溶液は調製の1時間以内に使用する。
【0185】
e.局所投与
局所用混合物は、局在的投与および全身投与用として記載したように調製する。得られる混合物は、溶液、懸濁液、エマルジョン、等であってよく、クリーム、ゲル、軟膏、エマルジョン、溶液、エリキシル、ローション、懸濁液、チンキ、ペースト、フォーム、エアゾール、灌水、スプレー、座薬、絆創膏、皮膚パッチ、または局所投与に適するあらゆるその他の製剤であってよい。
【0186】
シタキセンタンナトリウムは局在的または局所的投与用に、例えば皮膚および粘膜への局所塗布用に、ゲル、クリームおよびローションの剤形で製剤化してよい。局所投与は、経皮送達として、およびまた粘膜投与として、または吸入療法として意図する。
【0187】
f.他の投与経路に関する組成物
その他の投与経路、例えば局所塗布、経皮パッチ、および直腸投与もまた本明細書において意図する。例えば直腸投与用の医薬投与剤形は、全身への効果のための直腸用座薬、カプセル、および錠剤である。本明細書において使用する直腸用座薬は、体温で融けるかまたは軟化して、1つまたはそれより多くの医薬的にまたは治療的に活性な成分を放出する、直腸内への挿入のための固形のボディーを意味する。直腸用座薬中に利用する医薬的に受容可能な物質は、ベースまたはビヒクル、および融点を上昇させる剤である。ベースの例は、カカオバター(cocoa butter)(カカオ脂(theobroma oil))、グリセリン−ゼラチン、カーボワックス(ポリオキシエチレングリコール)、ならびに脂肪酸のモノ−、ジ−、およびトリ−グリセリドの適当な混合物を含む。様々なベースの組み合わせを使用してよい。座薬の融点を上昇させる剤は、鯨ろうおよびワックスを含む。直腸用座薬は、圧縮法または成形のどちらかにより調製してよい。直腸用座薬の典型的な重量は、約2から3グラムである。
【0188】
直腸投与用錠剤およびカプセルは、経口投与用製剤と同じ医薬的に受容可能な物質を使用して、そして同じ方法により製造する。
g.製造品
当該多形または多形の混合物は、シタキセンタンナトリウムを拡張期心不全を治療するために使用することを指示する、包装材料およびラベルを含有する製造品として包装してよい。本明細書に提供する製造品は、包装材料を含有する。医薬品の包装に使用するための包装材料は、当業者に周知されている。例えば米国特許第5,323,907号、5,052,558号および5,033,352号を参照のこと。医薬用包装材料の例として、ブリスターパック、瓶、管、吸入器、ポンプ、袋、バイアル、容器、注射器、瓶、ならびに選択した製剤および意図した投与様式および治療様式に適するあらゆる包装材料を含むが、これに限定されない。本明細書に提供するシタキセンタンの広く多数の製剤を、本明細書において意図する。
【0189】
投与量
ヒトの治療において医師は、予防処置または治癒処置に従って、そして治療する被験者に特定の年齢、体重、疾患の段階、およびその他の因子に従って、最も適当である投与計画を決定することになる。ある種の態様において、シタキセンタンナトリウムの用量のレートは、成人に関して1日当たり約1から約300mg、1日当たり約5から約250mg、1日当たり約5から約250mg、または1日当たり約10から50mgである。1日当たり約50から約300mgの用量のレートもまた本明細書において意図する。ある種の態様において用量は、成人1日当たり約5mg、10mg、15mg、20mg、25mg、30mg、35mg、40mg、45mg、50mg、60mg、70mg、80mg、100mg、125mg、150mg、175mg、または200mgである。
【0190】
拡張期心不全、またはその1つもしくはそれより多くの症状の予防または治療において有効となる、本明細書に提供する製剤中のシタキセンタンナトリウムの量は、疾患または状態の性質および重症度、および活性成分を投与する経路に伴って変わることになる。頻度および投与量もまた、投与する具体的な治療(例えば治療薬または予防薬)、障害、疾患、または状態の重症度、投与経路、ならびに被験者の年齢、体重、応答および過去の病歴に依存する、各被験者に特定な因子に従って変わることになる。
【0191】
製剤の具体例としての用量は、被験者またはサンプルの体重1キログラム当たりの活性化合物のミリグラムまたはマイクログラムの量(例えば1キログラム当たり約1マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約10マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約100マイクログラムから1キログラム当たり約3ミリグラム、1キログラム当たり約100マイクログラムから1キログラム当たり約2ミリグラム)を含む。ある種の態様において、投与するシタキセンタンナトリウムの量は、それを必要とする被験者に対して約0.01から約3mg/kgである。ある種の態様において、投与するシタキセンタンナトリウムの量は、約0.01、0.05、0.1、0.2、0.4、0.8、1.5、2、3mg/被験者の体重1kgである。ある種の態様においてシタキセンタンナトリウムの投与は、静脈内注射による。
【0192】
当業者には明らかとなるように、一部の場合には本明細書に開示する範囲外で活性成分の投与量を使用することが必要となるかもしれない。さらに臨床医または担当医は、被験者の応答を考慮しながら、どのようにそしていつ治療を中断、調整、または終了するかを知ることになることも言及しておく。
【0193】
拡張期心不全の症状を予防、管理、治療、または緩和するためには十分であるが、本明細書に提供する組成物に関連する有害作用の原因となる程ではない、または有害作用を低減するには十分である量もまた、上に記載した投与量および用量の頻度の計画により包括的に含まれる。さらに被験者に、本明細書に提供する組成物を多数回投与量を投与する場合、投与量のすべてが同一である必要はない。例えば被験者に投与する投与量は、組成物の予防的効果または治療的効果を改善するために増量してもよいし、または特定の被験者が経験している1つまたはそれより多くの副作用を低減するために減量してもよい。
【0194】
もう1つの態様において本明細書に提供する製剤の投与量は、被験者の拡張期心不全の症状を予防、治療、管理、または緩和するために、約1mgから300mg、50mgから250mg、または75mgから200mgのユニット用量で投与する。
【0195】
ある種の態様において、本明細書に提供する同じ製剤の投与を繰り返してもよく、投与は少なくとも1日、2日、3日、5日、10日、15日、30日、45日、2ヶ月、75日、3ヶ月、または6ヶ月まで間隔をあけてよい。
【0196】
F.活性の評価
本明細書に提供する方法におけるシタキセンタンナトリウムの効能を検査するための、標準的な生理学的、薬理学的、および生化学的な方法を利用することができ、そしてそれらは当業者に公知である。例えば米国特許第5,114,918号、EP 0 436 189 A1、Borges, et al. (1989) Eur. J. Pharm. 165: 223-230; Filip et al. (1991) Biochem. Biophys. Res. Commun. 177: 171-176を参照のこと。例えばDHFの治療におけるシタキセンタンナトリウムの効能の評価は、治療期間中に周期的間隔で行うトレッドミル運動検査;心エコー(ECHO)による心室の構造および機能(すなわち左心室の質量)における効果の決定;ドップラーECHOおよび組織ドップラー画像(TDI)による経僧帽弁流入速度(E)対 拡張早期僧帽弁輪部速度(E’)の比率の決定;Minnesota Living with Heart Failure questionnaire (MLHF)によって測定された生活の質(QOL)における変化の決定;および心機能分類評価(NYHA)、を含むがこれに限定されないルーチン検査により行うことができる。例えば、Zile らのHeart failure with a normal ejection fraction: is measurement of diastolic heart failure necessary to make the diagnosis of diastolic heart failure?(正常な駆出率を伴う心不全:拡張期心不全の測定は、拡張期心不全の診断を行うために必要か?)、Circulation 2001;104:779-782 、およびMiguel らのRecommendations for quantification of Doppler echocardiography(ドップラー心エコーの定量化に関する推奨):a report from the Doppler quantification task force of the nomenclature and standards committee of the American society of echocardiography(アメリカ心エコー学会の命名法および基準委員会のドップラー定量化特別委員会からの報告), J. Am. Soc. Echocardiogr. 2002; 15: 167-84を参照のこと。
【0197】
G.併用療法
本明細書に提供する方法において、当該多形および多形の混合物は、例えば単独で、1つもしくはそれより多くの他のエンドセリンアンタゴニストと組み合わせて、または拡張期心不全の治療に有用な別の化合物または治療と組み合わせて使用してよい。例えば当該製剤は、エンドセリン受容体の活性を調節することが知られているその他の化合物、例えば米国特許第6,432,994号;6,683,103号;6,686,382号;6,248,767号;6,852,745号;5,783,705号;5,962,490号;5,594,021号;5,571,821号;5,591,761号;5,514,691号に記載されている化合物と組み合わせて投与することができる。いくつかのその他のエンドセリンアンタゴニストは、上に記載した文献に記載されている。
【0198】
一部の態様において、当該方法は、拡張期心不全の治療において使用する他の化合物と組み合わせてのシタキセンタンナトリウムの投与を伴う。そのような薬剤として、ループ系利尿薬、例えばBumex(登録商標)(ブメタニド)、Lasix(登録商標)(フロセミド)、Demadex(登録商標)(トルセミド);チアジド系利尿薬、例えばHygroton(登録商標)(クロルタリドン)、Hydrodiuril(登録商標)、Esidrix(登録商標)(HCTZ、ヒドロクロロチアジド)、Amiloride、Aldactone(登録商標)(スピロノラクトン);長期作用型硝酸薬、例えばIsordil(登録商標)、Sorbitrate(登録商標)(二硝酸イソソルビド)、Imdur(登録商標)(一硝酸イソソルビド);β−遮断薬、例えばフマル酸ビソプロロール、プロプラノロール、アテノロール、ラベタロール、ソタロール、カルベジロール;カルシウムチャネル遮断薬、例えばNorvasc(登録商標)(アムロジピン)、Cardizem(登録商標)(ジルチアゼム)、Isoptin(登録商標)(ベラパミル)、Procardia(登録商標)(ニフェジピン);腎動脈狭窄症(RAS)阻害薬およびアンジオテンシン変換酵素(ACE)阻害薬、例えばカプトプリル、フォシノプリル、ベナゼプリル、エナラプリル、リシノプリル、モエキシプリル、ペリンドプリル、キナプリル、ラミプリル、スピラプリル、トランドラプリル;アンジオテンシン受容体遮断薬(ARB類)、例えばロサルタン、バルサルタン、イルベサルタン、テルメサルタン(telmesartan)、およびアルドステロンアンタゴニスト、を含むがこれに限定されない。
【0199】
一部の態様において当該方法は、間質性肺疾患の治療に使用される他の化合物、例えば
副腎皮質ステロイド、例えばプレドニソンまたはメチルプレドニソン(活性な進行中の肺胞および間質の炎症および傷害を抑制するために、そして間質性肺疾患の患者を治療する上で使用される)と組み合わせての、シタキセンタンナトリウムの投与を伴う。
【0200】
さらに本明細書に提供する多形は、当該技術において公知のエンドセリンアンタゴニストと組み合わせて利用することができる。これらアンタゴニストは、環式ペンタペプチド、すなわちシクロ(D−Glu−L−Ala−allo−D−lle−L−Leu−D−Trp)であるBE−18257Bと命名されたStreptomyces misakiensisの発酵生成物;BE−18257Bに関連する環式ペンタペプチド、例えばシクロ(D−Asp−Pro−D−Val−Leu−D−Trp)(BQ−123)(Ishikawa らに対する米国特許第5,114,918号を参照のこと;またBANYU PHARMACEUTICAL CO., LTD に対するEP A1 0 436 189(1991年10月7日)を参照のこと)を含むがこれに限定されない;そしてその他のペプチドおよび非ペプチドETAアンタゴニストは、例えば米国特許第6,432,994号;6,683,103号;6,686,382号;6,248,767号;6,852,745号;5,783,705号;5,962,490号;5,594,021号;5,571,821号;5,591,761号;5,514,691号;5,352,800号;5,334,598号;5,352,659号;5,248,807号;5,240,910号;5,198,548号;5,187,195号;5,082,838号;6,953,780号;6,946,481号;6,852,745号;6,835,741号;6,673,824号;6,670,367号;および6,670,362号において同定されている。これらは、他の環式ペンタペプチド、アクリルトリペプチド、ヘキサペプチド類似体、ある種のアントラキノン誘導体、インダンカルボン酸、ある種のN−ピリミニル(pyriminyl)ベンゼンスルホンアミド、ある種のベンゼンスルホンアミド、およびある種のナフタレンスルホンアミドを含む(Nakajima et al. (1991) J. Antibiot. 44:1348-1356;Miyata et al. (1992) J. Antibiot. 45:74-8;Ishikawa et al. (1992) J .Med. Chem. 35:2139-2142;Ishikawa et al.に対する米国特許第5,114,918号;EP A1 0 569 193;EP A1 0 558 258;BANYU PHARMACEUTICAL CO., LTDに対するEP A1 0 436 189(1991年10月7日);カナダ特許出願2,067,288号;カナダ特許出願2,071,193号;米国特許第5,208,243号;米国特許第5,270,313号;米国特許第5,612,359号、米国特許第5,514,696号、米国特許第5,378,715号; Cody et al. (1993) Med. Chem. Res. 3:154-162;Miyata et al. (1992) J. Antibiot 45:1041-1046;Miyata et al. (1992) J. Antibiot 45:1029-1040、Fujimoto et al. (1992) FEBS Lett. 305:41-44;Oshashi et al. (1002) J. Antibiot 45:1684-1685;EP A1 0 496 452;Clozel et al. (1993) Nature 365:759-761;国際特許出願WO93/08799;Nishikibe et al. (1993) Life Sci. 52:717-724;およびBenigni et al. (1993) Kidney Int. 44:440-444)。エンドセリンペプチドアンタゴニストである多数のスルホンアミドもまた、米国特許第5,464,853号;5,594,021号;5,591,761号;5,571,821号;5,514,691号;5,464,853号;国際PCT出願第96/31492号;および国際PCT出願第WO07/27979号に記載されている。
【0201】
以下の文献に記載されているさらなるエンドセリンアンタゴニスト(それらの全内容において本明細書にて参照として援用する)は、本明細書に提供する多形と組み合わせて使用することを意図する薬剤の具体例である:米国特許第5,420,123号;米国特許第5,965,732号;米国特許第6,080,774号;米国特許第5,780,473号;米国特許第5,543,521号;WO 96/06095;WO 95/08550;WO 95/26716;WO 96/11914;WO 95/26360;EP 601386;EP 633259;米国特許第5,292,740号;EP 510526;EP 526708;WO 93/25580;WO 93/23404;WO 96/04905;WO 94/21259;GB 2276383;WO 95/03044;EP 617001;WO 95/03295;GB 2275926;WO 95/08989;GB 2266890;EP 496452;WO 94/21590;WO 94/21259;GB 2277446;WO 95/13262;WO 96/12706;WO 94/24084;WO 94/25013;米国特許第5,571,821号;WO 95/04534;WO 95/04530;WO 94/02474;WO 94/14434;WO 96/07653;WO 93/08799;WO 95/05376;WO 95/12611;DE 4341663;WO 95/15963;WO 95/15944;EP 658548;EP 555537;WO 95/05374;WO 95/05372;米国特許第5,389,620号;EP 628569;JP 6256261;WO 94/03483;EP 552417;WO 93/21219;EP 436189;WO 96/11927;JP 6122625;JP 7330622;WO 96/23773;WO 96/33170;WO 96/15109;WO 96/33190;米国特許第5,541,186号;WO 96/19459;WO 96/19455;EP 713875;WO 95/26360;WO 96/20177;JP 7133254;WO 96/08486;WO 96/09818;WO 96/08487;WO 96/04905;EP 733626;WO 96/22978;WO 96/08483;JP 8059635;JP 7316188;WO 95/33748;WO 96/30358;米国特許第5,559,105号;WO 95/35107;JP 7258098;米国特許第5,482,960号;EP 682016;GB 2295616;WO 95/26957;WO 95/33752;EP 743307;およびWO 96/31492;例えば列挙した文献に記載されている以下の化合物:BQ−123(Ihara, M.ら、“ETA受容体に選択的なきわめて強力な新規エンドセリンアンタゴニストの生物学的プロフィール”, Life Sciences, Vol. 50(4), pp. 247-255 (1992));PD 156707(Reynolds, E.ら、 “経口活性のETA受容体アンタゴニストであるPD 156707の薬理学的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 273(3), pp. 1410-1417 (1995));L−754,142(Williams, D. L.ら、“極めて強力な経口活性の非ペプチジルエンドセリンアンタゴニストであるL−754,142の薬理学”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 275(3), pp. 1518-1526 (1995));SB 209670(Ohlstein, E. H.ら、“合理的にデザインされた強力な非ペプチドエンドセリン受容体アンタゴニストであるSB 209670”, Proc. Natl. Acad. Sci. USA, Vol. 91, pp. 8052-8056 (1994));SB 217242(Ohlstein, E. H.ら、 “非ペプチドエンドセリン受容体アンタゴニストVI:強力なそして高い生体利用可能性のあるエンドセリン受容体アンタゴニストである、SB 217242の薬理学的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 276(2), pp. 609-615 (1996));A−127722(Opgenorth, T. J.ら、“A−127722の薬理学的特徴付け:経口活性の極めて強力なE.sub.TA−選択的受容体アンタゴニスト”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 276(2), pp.473-481 (1996));TAK−044(Masuda, Y.ら、“ヒトエンドセリンA受容体およびエンドセリンB受容体における、新規エンドセリン受容体アンタゴニストであるTAK−044{シクロ [D−α−アスパルチル−3−[(4−フェニルピペラジン−1−イル)カルボニル]−L−アラニルーL−α−アスパルチル−D−2−(2−チエニル)グリシル−L−ロイシル−D−トリプロフィル]二ナトリウム塩}の受容体への結合およびアンタゴニストの特性”、The Journal of Pharmacology and Experimental Therapeutics, Vol. 279(2), pp. 675-685 (1996));ボセンタン(Ro 47-0203, Clozel, M.ら、“新規の強力な経口活性非ペプチドエンドセリン受容体アンタゴニストであるボセンタンの医薬的特徴付け”, The Journal of Pharmacology and Experimental Therapeutics, Vol. 270(1), pp. 228-235 (1994))。
【0202】
本明細書に提供する多形はまた、他のクラスの化合物と組み合わせて投与することができる。本明細書における組み合わせのための化合物の具体例としてのクラスには、エンドセリン変換酵素(ECE)阻害薬、例えばホスホルアミドン;トロンボキサン受容体アンタゴニスト、例えばイフェトロバン;カリウムチャネルオープナー;トロンビン阻害薬(例えばヒルジン等);増殖因子阻害薬、例えばPDGF活性の調節薬、;血小板活性化因子(PAF)アンタゴニスト;抗血小板薬、例えばGPIIb/IIIa遮断薬(例えばアブドキシマブ(abdximab)、エプチフィバチド、およびチロフィバン)、P2Y(AC)アンタゴニスト(例えばクロピドグレル、チクロピジン、およびCS−747)、およびアスピリン;抗凝固薬、例えばワルファリン、低分子量ヘパリン 例えばエノキサパリン、第VIIa因子阻害薬、および第Xa因子阻害薬、レニン阻害薬;アンジオテンシン変換酵素(ACE)阻害薬 例えばカプトプリル、ゾフェノプリル、フォシノプリル、セラナプリル、アラセプリル、エナラプリル、デラプリル、ペントプリル、キナプリル、ラミプリル、リシノプリル、およびそのような化合物の塩;中性エンドペプチダーゼ(NEP)阻害薬;バソペプシダーゼ(vasopepsidase)阻害薬(二重のNEP-ACE阻害薬)例えばオマパトリラトおよびゲモパトリラト;HMG CoA還元酵素阻害薬 例えばプラバスタチン、ロバスタチン、アトルバスタチン、シンバスタチン、NK−104(別名イタバスタチン、またはニスバスタチン(nisvastatin もしくは nisbastatin))およびZD−4522(ロスバスタチン、またはアタバスタチン(atavastatin)もしくはビサスタチン(atavastatin)としても知られている);スクアレン合成酵素阻害薬;フィブラート系薬剤;胆汁酸捕捉薬、例えばクエストラン;ナイアシン;抗アテローム硬化性薬剤、例えばACAT阻害薬;MTP阻害薬;カルシウムチャネル遮断薬、例えばベシル酸アムロジピン;カリウムチャネル活性化薬;アルファ−アドレナリン遮断薬(alpha-adrenergic agents)、ベータ−アドレナリン遮断薬(beta-adrenergic agents)、例えばカルベジロールおよびメトプロロール;抗不整脈薬;利尿薬、例えばクロロチアジド、ヒドロクロロロチアジド、フルメチアジド、ヒドロフルメチアジド、ベンドロフルメチアジド、メチルクロロチアジド、トリクロロメチアジド、ポリチアジド、またはベンゾチアジド、同様にエタクリン酸、トリクリナフェン、クロルタリドン、フロセニルデ、ムソリミン、ブメタニド、トリアムテレン、アミロリド、ならびにスピロノラクトンおよびそのような化合物の塩;血栓溶解薬、例えば組織プラスミノーゲン活性化因子(tPA)、リコンビナントtPA、ストレプトキナーゼ、ウロキナーゼ、プロウロキナーゼ、およびアニソイル化(anisoylated) プラスミノーゲンストレプトキナーゼ活性化因子複合体(APSAC);抗糖尿病薬、例えばビグアナイド系(例えばメトホルミン)、グルコシダーゼ阻害薬(例えばアカルボース)、インスリン、メグリチニド(例えばレパグリニド)、スルホニル尿素(例えばグリメピリド、グリブリド、およびグリピジド)、チアゾリジネジオン(例えばトログリタゾン、ロシグリタゾン、およびピオグリタゾン)ならびにPPAR−ガンマアゴニスト;鉱質コルチコイド受容体アンタゴニスト、例えばスピロノラクトンおよびエプレレノン;成長ホルモン分泌促進薬;aP2阻害薬;非ステロイド系抗炎症薬(NSAIDS)、例えばアスピリンおよびイブプロフェン;ホスホジエステラーゼ阻害薬、例えばPDE III阻害薬(例えばシロスタゾール)およびPDE V阻害薬(例えばシルデナフィル、タダラフィル、バルデナフィル);タンパク質チロシンキナーゼ阻害薬;抗炎症薬;抗増殖薬、例えばメトトレキサート、FK506(タクロリムス、プログラフ)、ミコフェノレート、およびモフェチル;化学療法薬;免疫抑制薬;抗癌薬および細胞傷害性薬剤(例えばアルキル化薬、例えば窒素マスタード、スルホン酸アルキル、ニトロソ尿素、エチレンイミン、およびトリアゼン);代謝拮抗薬、例えば葉酸アンタゴニスト、プリン類似体、およびピリジン類似体;抗生物質、例えばアントラサイクリン系、ブレオマイシン、マイトマイシン、ダクチノマイシン、およびプリカマイシン;酵素、例えばL−アスパラギナーゼ;ファルネシル−タンパク質トランスフェラーゼ阻害薬;ホルモン薬、例えば糖質コルチコイド(例えばコルチゾン)、エストロゲン/抗エストロゲン薬、アンドロゲン/抗アンドロゲン薬、プロゲスチン、および黄体形成ホルモン放出ホルモンアンタゴニスト、酢酸オクトレオチド;微小管重合阻害薬、例えばエクテイナシジン(ecteinascidins)またはそれらの類似体および誘導体:微小管安定化薬、例えばパシタキセル(pacitaxel)(Taxol(登録商標))、ドセタキセル(Taxotere(登録商標))、およびエポチロンA−Fまたはそれらの類似体もしくは誘導体;植物由来産生物、例えばビンカアルカロイド系、エピポドフィロトキシン系、タキサン系;およびトポイソメラーゼ阻害薬:タンパク質のプレニル転移酵素阻害薬:および種々の薬剤、例えばヒドロキシ尿素、プロカルバジン、ミトタン、ヘキサメチルメラミン、白金配位錯体 例えばシスプラチン、サトラプラチン、およびカルボプラチン);シクロスポリン;ステロイド 例えばプレドニゾンまたはデキサメタゾン;金化合物;細胞傷害性薬剤 例えばアザチプリンおよびシクロホスファミド;TNF−アルファ阻害薬 例えばテニダップ;抗TNF抗体または可溶性TNF受容体 例えばエタネルセプト(Enbrel)、ラパマイシン(シロリムスまたはRapamune)、レフルニミド(leflunimide)(Arava);そしてシクロオキシゲナーゼ−2(COX−2)阻害薬 例えばセレコキシブ(Celebrex)およびロフェコキシブ(Vioxx)を含む。
【0203】
上のその他の治療薬は、例えばPhysicians' Desk Reference(PDR)に指示されている量で、またはそうでなければ当業者により決定されたように使用してよい。
H. N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩の多形の使用法
多形A、BおよびCのN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニル−アセチル]チオフェン−3−スルホンアミド、ナトリウム塩は、エンドセリンを介した疾患の治療において有用である。これらの治療には、多形A、B型またはC型の有効量を被験者に投与することを包括的に含み、その場合その有効量は、疾患の1つまたはそれより多くの症状を寛解させるのに十分な量である。
【0204】
多形A、BおよびCは、高血圧症、心血管疾患、心筋梗塞を含む心疾患、肺高血圧症、新生児肺高血圧症、エリスロポエチンを介した高血圧症、喘息、気管支収縮を含む呼吸器疾患および炎症性疾患、緑内障および不適切な網膜の灌流を含む眼科的疾患、胃腸疾患、腎不全、エンドトキシンショック、月経障害、産科的状態、創傷、蹄葉炎、勃起不全、月経閉止、骨粗しょう症および代謝性骨疾患、中年女性の卵巣機能低下に関連するのぼせ、異常な血塊パターン、泌尿生殖器の不快感、および心血管疾患の増加する発生率、および中年女性における卵巣機能低下に伴う他の障害、妊娠中毒症、妊娠時のコントロールおよび出産の管理、一酸化窒素減弱障害(nitric oxide attenuated disorders)、アナフィラキシーショック、または出血性ショック、間質性肺疾患、拡張期心不全、ならびに免疫抑制を介した腎血管収縮、の治療に有効である。1つの態様において、疾患は肺高血圧症である。
【0205】
多形A、BおよびCはまた、エンドセリンA(ETA)受容体またはエンドセリンB(ETB)受容体へのエンドセリンペプチドの結合を阻害するために有用である。この阻害は、受容体を、多形A、BもしくはC、または医薬的に受容可能なその誘導体のいずれかと接触させることを包括的に含み、その場合この接触は、受容体とエンドセリンペプチドとの接触前、接触時時、または接触後において達成される。
【0206】
多形A、BおよびCはまた、エンドセリン受容体を介した活性を変化させるために有用である。この変化には、エンドセリン受容体を、多形A、BまたはCと接触させることを包括的に含む。
【0207】
以下の実施例は説明の目的のみとして含んでいるに過ぎず、主張した主題事項の範囲を限定する意図はない。N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩(シタキセンタンナトリウム)は、国際(PCT)特許出願公開第WO 98/49162号に記載されている方法により調製してよい。
【実施例】
【0208】
実施例1
MeOHを用いての生成法:
10gのシタキセンタンナトリウムを、40mLのiPrOAc、30mLのEtOHおよび30mLのMeOH中に懸濁し、透明な溶液が得られるまで75℃で加熱した。この溶液を放置して室温まで冷却したが、溶液は1時間透明なままであった。50mLのMTBEを加え、時間を経て黄色固体が形成された。固体を濾過して集め、MTBEで洗浄し、真空下で乾燥させ、6.4gのシタキセンタンナトリウムを主に多形Aとして得た。
【0209】
実施例2
MeOHを用いない生成法:
10gのシタキセンタンナトリウムを100mLのiPrOAcおよび80mLのEtOH中に懸濁させ、透明な溶液が得られるまで90℃(還流)に加熱した(この溶媒の量を、還流で透明な溶液を得るために必要とした)。溶液を放置して室温まで冷却したが、溶液は1時間透明なままであった。140mLのMTBEを加え、時間を経て非常に少量の淡黄色固体が形成された(より少量のMTBEを加えると、固体の形成には至らなかった)。100mLのMTBEを加え、時間を経てより多くの淡黄色固体が形成された。これらの固体を溶液のデカントにより得(これらの固体はうまく濾過できなかった)、MTBEで洗浄し、真空下で乾燥させ、5.4gのシタキセンタンナトリウムを主に多形Bとして得た。
【0210】
実施例3
湿ったiPrOAcからの再結晶化:
10gのシタキセンタンナトリウムを100mLのiPrOAcおよび5mLの水中に懸濁させ、透明な溶液が得られるまで100℃(還流)に加熱した。溶液を放置して淡黄色固体が形成されるまで、室温まで冷却した。これらの固体を濾過して集め、iPrOHで洗浄し、真空下で乾燥させ、6.4gのシタキセンタンナトリウムのロットを主に多形Aとして得た。
【0211】
実施例4
シタキセンタンナトリウム多形Bの再結晶化:
実施例5からの1.0gのシタキセンタンナトリウムを、1.6mLのiPrOAc、1.6mLのMeOHおよび1.6mLのEtOH中に懸濁し、予め65℃に加熱した油浴に入れた。完全な溶解は5分で得られた。溶液を放置して室温まで冷却したが、溶液は透明なままだった。数日間そのまま静置すると淡黄色固体が形成され、これを濾過して集め、MTBEで洗浄し、真空下で乾燥させ、シタキセンタンナトリウムを〜94%多形Aとして得た。
【0212】
実施例5
シタキセンタンナトリウム多形Aの再結晶化:
688gのシタキセンタンナトリウムを、30分間、60℃で、7.8LのEtOH中で加熱した後、10℃に冷却した。MTBE(35L)を加え、混合液を10℃で濾過した。固体を真空中で乾燥させ、およそ86:14の多形A:多形Bを得た。
【0213】
実施例6
387gのシタキセンタンナトリウムを、3.0Lのイソプロパノール中で2時間スラリーとした後、2日間5℃で冷却した。固体を濾過し、真空中で乾燥させて、344gのシタキセンタンナトリウム得た。この材料を1.72Lのイソプロパノール中で室温で30分間スラリーとした後、5℃に45分間冷却した。生成物を濾過し、真空中で乾燥させて、主に多形Bを得た。
【0214】
実施例7
シタキセンタンナトリウム(23.4kg)を、酢酸イソプロピル(32.8kg)、エタノール(30kg)、およびメタノール(30kg)中に懸濁し、65℃に加熱した。固体が溶解した後、溶液を0.45ミクロンフィルターを通して熱濾過した。濾液を撹拌し、45℃に冷却した。シタキセンタンナトリウムの種結晶を加え、内容物の撹拌を45℃の温度で3時間続けた。MTBE(164.3kg)を45±5℃で、1分当たり1kgを超える速度で、ゆっくり加えた。内容物を、4.5時間にわたり0℃までゆっくり冷却した。0℃でさらに4.5時間、撹拌を続けた。得られた結晶を濾過し、湿ったケークをMTBE(93.6kg)で洗浄した。湿ったケークを、脱液化(de-liquored)するまで窒素下に保持した。この湿ったケークを、残存するMTBEのレベルが500ppmより少なくなるまで、Filter/Dryer中で40℃で緩やかに撹拌しながら乾燥させた。得られた材料は、主に多形A(95:5±3 多形A:多形B)であった。
【0215】
実施例8
比較例
シタキセンタンナトリウムの形成:
50mLのDCM中の10gのシタキセンタンナトリウムの十分に撹拌した懸濁液に、50mLの2NのHClを加え、続いて透明な溶液が得られるまでMeOHを加えた。二層分離し、有機層をMgSO4上で乾燥、濃縮し、真空中で乾燥を完了させて、シタキセンタン(〜9g)を乾燥黄色泡状物質として得た。
【0216】
A.最初の結晶化
シタキセンタンを100mLのEtOAc中に再度溶かし、3×50mLの飽和NaHCO3、食塩水で洗浄し、MgSO4上で乾燥させ、真空中で濃縮、乾燥させた。この材料をDCM中に再度懸濁したところ、濁った溶液を形成し、淡黄色固体が形成された後に5分間撹拌した。150mLのEt2Oを加えた。固体を濾過して集め、1対2のDCM対Et2Oで洗浄し、真空下で乾燥させてシタキセンタンナトリウムを主にアモルファスの材料として得た。
【0217】
B.2回目の結晶化
シタキセンタンナトリウムを200mLの水に再度溶解させ、濃HClでpH〜2に酸性化したところ、非常に淡い黄色の固体が形成された。この固体を濾過して得た。この材料を、100mLのEtOAcに再度溶かし、50mLの食塩水、2×50mLの飽和NaHCO3、食塩水で洗浄し、MgSO4上で乾燥させ、真空中で乾燥、濃縮した。この材料をDCM中に再度懸濁したところ、濁った溶液を形成し、淡黄色固体が形成された後に5分間撹拌した。150mLのEt2Oを加えた。固体を濾過して集め、1対2のDCM対Et2Oで洗浄し、真空下で乾燥させて、6.1gのシタキセンタンナトリウムを主にアモルファスの材料として得た。
【0218】
C.3回目の結晶化
1.0シタキセンタンナトリウムを、10mLのEtOHに懸濁し、透明溶液が得られるまで加熱還流した(この溶媒の量を、透明溶液を得るために必要とした)。この溶液を放置して室温まで冷却したが、1時間透明のままであった。この時点で15mLのMTBEを加えたところ、溶液は混濁した(さらに10mLのMTBEを加えても、30分以内に固体の形成には至らなかった)。この溶液を加熱還流したが、固体のcrash outは防げなかった。固体を濾過して集め、MTBEで洗浄し、真空下で乾燥させ、0.64gのシタキセンタンナトリウムを、アモルファスおよび結晶材料の混合物として得た。
【0219】
修飾は当業者にとって明白であろう故、主張した主題事項は添付の請求項の範囲によってのみ限定されることを意図する。
【図面の簡単な説明】
【0220】
【図1】図1は、多形A、B、Cおよびアモルファス型のXRPDパターンである。
【図2】図2は、多形A、サンプルロットIのXRPDパターンである。
【図3】図3は、多形A、サンプルロットIIのXRPDパターンである。
【図4】図4は、多形A、サンプルロットIIIのXRPDパターンである。
【図5】図5は、多形A、サンプルロットIVのXRPDパターンである。
【図6】図6は、多形A、サンプルロットVのXRPDパターンである。
【図7】図7は、多形A、サンプルロットVIのXRPDパターンである。
【図8】図8は、多形A、サンプルロットVIIのXRPDパターンである。
【図9】図9は、多形Aのラマン吸収スペクトルである。
【図10】図10は、多形A、サンプルロットIのDSCである。
【図11】図11は、多形A、サンプルロットIVのDSCである。
【図12】図12は、多形A、サンプルロットIIIのDSCである。
【図13】図13は、多形A、サンプルロットIのTGである。
【図14】図14は、多形A、サンプルロットIVのTGである。
【図15】図15は、多形A、サンプルロットIIIのTGである。
【図16】図16は、多形A、サンプルロットIの吸湿/放湿である。
【図17】図17は、多形A、サンプルロットIVの吸湿/放湿である。
【図18】図18は、多形A、サンプルロットIIIの吸湿/放湿である。
【図19】図19は、多形BのDSCである。
【図20】図20は、多形BのTGである。
【図21】図21は、多形Bの吸湿/放湿である。
【図22】図22は、多形Bのラマン吸収スペクトルである。
【図23】図23は、多形CのTG/IR吸収スペクトルである。
【図24】図24は、多形CのTGである。
【図25】図25は、多形AのTG/IR吸収スペクトルである。
【図26】図26は、多形BのTG/IR吸収スペクトルである。
【図27】図27は、多形B、サンプルロットIのXRPDパターンである。
【図28】図28は、多形B、サンプルロットIIのXRPDパターンである。
【図29】図29は、多形B、サンプルロットIIIのXRPDパターンである。
【特許請求の範囲】
【請求項1】
多形Aの形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の化合物。
【請求項2】
多形Aの量が約80%より多い、請求項1に記載の化合物。
【請求項3】
多形Aの量が約85%より多い、請求項1または2に記載の化合物。
【請求項4】
多形Aの量が約90%より多い、請求項1−3のいずれかに記載の化合物。
【請求項5】
多形Aの量が約95%より多い、請求項1−4のいずれかに記載の化合物。
【請求項6】
多形Aの量が約98%より多い、請求項1−5のいずれかに記載の化合物。
【請求項7】
多形Aの量が約99%より多い、請求項1−6のいずれかに記載の化合物。
【請求項8】
多形Aの量が約100%である、請求項1−7のいずれかに記載の化合物。
【請求項9】
多形AおよびBの混合物の形の化合物N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩であって、ここで多形A:Bの比率が約80:20より大きいかまたは等しい前記化合物。
【請求項10】
A:Bの比率が約86:14より大きいかまたは等しい、請求項9のいずれかに記載の化合物。
【請求項11】
A:Bの比率が約90:10より大きいかまたは等しい、請求項9または10のいずれかに記載の化合物。
【請求項12】
A:Bの比率が約91:9より大きいかまたは等しい、請求項9−11のいずれかに記載の化合物。
【請求項13】
A:Bの比率が約92:8より大きいかまたは等しい、請求項9−12のいずれかに記載の化合物。
【請求項14】
A:Bの比率が約93:7より大きいかまたは等しい、請求項9−13のいずれかに記載の化合物。
【請求項15】
A:Bの比率が約94:6より大きいかまたは等しい、請求項9−14のいずれかに記載の化合物。
【請求項16】
A:Bの比率が約95:5より大きいかまたは等しい、請求項9−15のいずれかに記載の化合物。
【請求項17】
A:Bの比率が約96:4より大きいかまたは等しい、請求項9−16のいずれかに記載の化合物。
【請求項18】
A:Bの比率が約97:3より大きいかまたは等しい、請求項9−17のいずれかに記載の化合物。
【請求項19】
A:Bの比率が約98:2より大きいかまたは等しい、請求項9−18のいずれかに記載の化合物。
【請求項20】
A:Bの比率が約99:1より大きいかまたは等しい、請求項9−19のいずれかに記載の化合物。
【請求項21】
多形Aが、XRPDパターンにおける2θのおよそ22.38および23.38度のピークを特徴とする、請求項1−20のいずれかに記載の化合物。
【請求項22】
多形Aが、XRPDパターンにおける2θのおよそ6.72、15.96、22.38、23.38および26.22度のピークを特徴とする、請求項1−21のいずれかに記載の化合物。
【請求項23】
多形Aが、ラマンスペクトルにおけるおよそ1602.1cm−1のピークを特徴とする、請求項1-22のいずれかに記載の化合物。
【請求項24】
多形Aが、ラマンスペクトルにおけるおよそ1697.4、1602.1、1489.8および1402.2cm−1のピークを特徴とする、請求項1-23のいずれかに記載の化合物。
【請求項25】
請求項1−24のいずれかに定義したような多形Aを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウムを温めた溶媒中に溶解させて、飽和溶液をとする;そして
飽和溶液を冷却して固体の沈殿物を得る;
ことを包含する、前記製法。
【請求項26】
溶媒が、アセトニトリル、クロロホルム、ジクロロメタン、エタノール、酢酸エチル、ヘキサン、イソプロパノール、酢酸イソプロピル、メチルt−ブチルエーテル、メチルエチルケトン、トルエン、またはテトラヒドロフランである、請求項25に記載の製法。
【請求項27】
溶媒がエタノールであり、そして飽和溶液を緩やかに周囲温度まで冷却する、請求項25または26に記載の製法。
【請求項28】
飽和溶液がスラリーである、請求項25−27のいずれかに記載の製法。
【請求項29】
溶媒がエタノールであり、そして固体の沈殿物が沈殿した後1時間またはそれより多くの時間内にそれを濾過する、請求項25−28のいずれかに記載の製法。
【請求項30】
請求項1−24のいずれかに定義したような多形Aを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩を溶媒中に溶解させて、飽和溶液とする;そして
貧溶媒を加える;
ことを包含する、前記製法。
【請求項31】
溶媒がテトラヒドロフランであり、そして貧溶媒がヘキサンである、請求項30に記載の製法。
【請求項32】
溶媒がメタノールであり、そして貧溶媒がトルエンである、請求項31に記載の製法。
【請求項33】
溶媒が酢酸イソプロピル、エタノール、およびメタノールを包含する、請求項31に記載の製法。
【請求項34】
溶媒を約65℃まで加熱する工程をさらに包含する、請求項33に記載の製法。
【請求項35】
貧溶媒が、メチルt−ブチルエーテルである、請求項33または34に記載の製法。
【請求項36】
メチルt−ブチルエーテルを約45±5℃の温度で加える、請求項35に記載の製法。
【請求項37】
約0℃まで冷却する工程をさらに包含する、請求項36に記載の製法。
【請求項38】
冷却工程を約3.5から4.5時間の間にわたり行う、請求項37に記載の製法。
【請求項39】
多形Bの形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の化合物であって、ここで化合物中の多形Bの量が約70%より多い、前記化合物。
【請求項40】
多形Bの量が約80%より多い、請求項39に記載の化合物。
【請求項41】
多形Bの量が約85%より多い、請求項39または40に記載の化合物。
【請求項42】
多形Bの量が約90%より多い、請求項39−41のいずれかに記載の化合物。
【請求項43】
多形Bの量が約95%より多い、請求項39−42のいずれかに記載の化合物。
【請求項44】
多形Bの量が約98%より多い、請求項39−43のいずれかに記載の化合物。
【請求項45】
多形Bの量が約99%より多い、請求項39−44のいずれかに記載の化合物。
【請求項46】
多形Bの量が約100%である、請求項39−45のいずれかに記載の化合物。
【請求項47】
多形Bが、XRPDパターンにおける2θのおよそ22.72度のピークを特徴とする、請求項39−46のいずれかに記載の化合物。
【請求項48】
多形Bが、XRPDパターンにおける2θのおよそ6.6、15.52、18.38、18.94および22.72度のピークを特徴とする、請求項39−47のいずれかに記載の化合物。
【請求項49】
多形Bが、ラマンスペクトルにおけるおよそ1594.7cm−1のピークを特徴とする、請求項39−48のいずれかに記載の化合物。
【請求項50】
多形Bが、ラマンスペクトルにおけるおよそ1696.9、1594.7、1490.2および1397.8cm−1のピークを特徴とする、請求項39−49のいずれかに記載の化合物。
【請求項51】
請求項39−50のいずれかに定義したような多形Bを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウムを溶媒中に溶解させて、飽和溶液とする;そして
貧溶媒を加えて固体の沈殿物を得る;
ことを包含する、前記製法。
【請求項52】
溶媒が酢酸エチルであり、そして貧溶媒がヘキサン、メチルt−ブチルエーテル、またはトルエンである、請求項51に記載の製法。
【請求項53】
溶媒がアセトンであり、そして貧溶媒がジクロロメタン、メチルt−ブチルエーテル、またはトルエンである、請求項51に記載の製法。
【請求項54】
溶媒がテトラヒドロフランであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項55】
溶媒が酢酸イソプロピルであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項56】
溶媒がエタノールであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項57】
溶媒がメタノールであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項58】
貧溶媒を約20℃の温度で加える、請求項51−57のいずれかに記載の製法。
【請求項59】
冷却の工程をさらに包含する、請求項58に記載の製法。
【請求項60】
冷却を、0℃まで約3時間の間にわたり行う、請求項59に記載の製法。
【請求項61】
固体の沈殿物が沈殿した後1時間またはそれより多くの時間内にそれを濾過する、請求項60に記載の製法。
【請求項62】
請求項1−24および39−50のいずれかに記載の多形の有効量を被験者に投与することを包含する、エンドセリンを介した疾患の治療のための方法。
【請求項63】
疾患が、高血圧症、心血管疾患、心疾患、肺高血圧症、新生児肺高血圧症、エリスロポエチンを介した高血圧症、呼吸器疾患、炎症性疾患、眼科的疾患、胃腸疾患、腎不全、エンドトキシンショック、月経障害、産科的状態、創傷、蹄葉炎、勃起不全、月経閉止、骨粗しょう症、代謝性骨疾患、更年期障害、中年女性の卵巣機能低下に関連する障害、妊娠中毒症、妊娠時の出産の管理、一酸化窒素減弱性障害、アナフィラキシーショック、間質性肺疾患、拡張期心不全、出血性ショック、および免疫抑制を介した腎血管収縮、から成る群より選択される、請求項62に記載の方法。
【請求項64】
疾患が、肺高血圧症、間質性肺疾患、および拡張期心不全から成る群より選択される、請求項62または63に記載の方法。
【請求項65】
疾患が肺高血圧症である、請求項62−64に記載の方法。
【請求項66】
エンドセリンA(ETA)受容体またはエンドセリンB(ETB)受容体へのエンドセリンペプチドの結合を阻害するための方法であって、該受容体を、請求項1−24および39−50のいずれかに記載の多形と接触させることを包含し、ここで:
当該接触は、受容体とエンドセリンペプチドとの接触前、接触時、または接触後において達成される、前記方法。
【請求項67】
エンドセリン受容体を、請求項1−24および39−50のいずれかに記載の多形と接触させることを包含する、エンドセリン受容体を介した活性を変化させるための方法。
【請求項68】
医薬的に受容可能な担体中に、請求項1−24および39−50のいずれかに記載の多形の有効量を包含する、医薬組成物。
【請求項69】
請求項1−24および39−50のいずれかに記載の多形、および医薬的に受容可能な担体を包含する、医薬組成物。
【請求項70】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の70%よりほぼ多い量で存在する、請求項69に記載の医薬組成物。
【請求項71】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の80%よりほぼ多い量で存在する、請求項69または70に記載の医薬組成物。
【請求項72】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の85%よりほぼ多い量で存在する、請求項69−71のいずれかに記載の医薬組成物。
【請求項73】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の90%よりほぼ多い量で存在する、請求項69−72のいずれかに記載の医薬組成物。
【請求項74】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の95%よりほぼ多い量で存在する、請求項69−73のいずれかに記載の医薬組成物。
【請求項75】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99%よりほぼ多い量で存在する、請求項69−74のいずれかに記載の医薬組成物。
【請求項76】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99.5%よりほぼ多い量で存在する、請求項69−75のいずれかに記載の医薬組成物。
【請求項77】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99.9%より多くの量で存在する、請求項69−76のいずれかに記載の医薬組成物。
【請求項78】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の約100%である量で存在する、請求項69−77のいずれかに記載の医薬組成物。
【請求項79】
単回投与または複数回投与の投与用に製剤化される、請求項68−78のいずれかに記載の組成物。
【請求項80】
経口錠剤として製剤化される、請求項68−79のいずれかに記載の医薬組成物。
【請求項81】
抗酸化剤、結合剤、希釈剤、バッファー、および耐湿コーティングをさらに包含する、請求項80に記載の経口錠剤。
【請求項82】
請求項1−24および39−50のいずれかに記載の多形を包含する凍結乾燥粉末。
【請求項83】
抗酸化剤、バッファー、および充填剤をさらに包含する、請求項81に記載の凍結乾燥粉末。
【請求項84】
製造品であって、包装材料および該包装材料内に含有される請求項1−24および39−50のいずれかに記載の多形を包含し、ここで多形は、エンドセリンの効果に拮抗する、エンドセリンを介した障害の症状を緩和する、またはエンドセリンペプチドのET受容体への結合を阻害するために有効であり、そして該包装材料は、該多形が、エンドセリンの効果に拮抗する、エンドセリンのエンドセリン受容体への結合を阻害する、またはエンドセリンを介した障害を治療するために使用されることを指摘するラベルを含む、前記製造品。
【請求項1】
多形Aの形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の化合物。
【請求項2】
多形Aの量が約80%より多い、請求項1に記載の化合物。
【請求項3】
多形Aの量が約85%より多い、請求項1または2に記載の化合物。
【請求項4】
多形Aの量が約90%より多い、請求項1−3のいずれかに記載の化合物。
【請求項5】
多形Aの量が約95%より多い、請求項1−4のいずれかに記載の化合物。
【請求項6】
多形Aの量が約98%より多い、請求項1−5のいずれかに記載の化合物。
【請求項7】
多形Aの量が約99%より多い、請求項1−6のいずれかに記載の化合物。
【請求項8】
多形Aの量が約100%である、請求項1−7のいずれかに記載の化合物。
【請求項9】
多形AおよびBの混合物の形の化合物N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩であって、ここで多形A:Bの比率が約80:20より大きいかまたは等しい前記化合物。
【請求項10】
A:Bの比率が約86:14より大きいかまたは等しい、請求項9のいずれかに記載の化合物。
【請求項11】
A:Bの比率が約90:10より大きいかまたは等しい、請求項9または10のいずれかに記載の化合物。
【請求項12】
A:Bの比率が約91:9より大きいかまたは等しい、請求項9−11のいずれかに記載の化合物。
【請求項13】
A:Bの比率が約92:8より大きいかまたは等しい、請求項9−12のいずれかに記載の化合物。
【請求項14】
A:Bの比率が約93:7より大きいかまたは等しい、請求項9−13のいずれかに記載の化合物。
【請求項15】
A:Bの比率が約94:6より大きいかまたは等しい、請求項9−14のいずれかに記載の化合物。
【請求項16】
A:Bの比率が約95:5より大きいかまたは等しい、請求項9−15のいずれかに記載の化合物。
【請求項17】
A:Bの比率が約96:4より大きいかまたは等しい、請求項9−16のいずれかに記載の化合物。
【請求項18】
A:Bの比率が約97:3より大きいかまたは等しい、請求項9−17のいずれかに記載の化合物。
【請求項19】
A:Bの比率が約98:2より大きいかまたは等しい、請求項9−18のいずれかに記載の化合物。
【請求項20】
A:Bの比率が約99:1より大きいかまたは等しい、請求項9−19のいずれかに記載の化合物。
【請求項21】
多形Aが、XRPDパターンにおける2θのおよそ22.38および23.38度のピークを特徴とする、請求項1−20のいずれかに記載の化合物。
【請求項22】
多形Aが、XRPDパターンにおける2θのおよそ6.72、15.96、22.38、23.38および26.22度のピークを特徴とする、請求項1−21のいずれかに記載の化合物。
【請求項23】
多形Aが、ラマンスペクトルにおけるおよそ1602.1cm−1のピークを特徴とする、請求項1-22のいずれかに記載の化合物。
【請求項24】
多形Aが、ラマンスペクトルにおけるおよそ1697.4、1602.1、1489.8および1402.2cm−1のピークを特徴とする、請求項1-23のいずれかに記載の化合物。
【請求項25】
請求項1−24のいずれかに定義したような多形Aを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウムを温めた溶媒中に溶解させて、飽和溶液をとする;そして
飽和溶液を冷却して固体の沈殿物を得る;
ことを包含する、前記製法。
【請求項26】
溶媒が、アセトニトリル、クロロホルム、ジクロロメタン、エタノール、酢酸エチル、ヘキサン、イソプロパノール、酢酸イソプロピル、メチルt−ブチルエーテル、メチルエチルケトン、トルエン、またはテトラヒドロフランである、請求項25に記載の製法。
【請求項27】
溶媒がエタノールであり、そして飽和溶液を緩やかに周囲温度まで冷却する、請求項25または26に記載の製法。
【請求項28】
飽和溶液がスラリーである、請求項25−27のいずれかに記載の製法。
【請求項29】
溶媒がエタノールであり、そして固体の沈殿物が沈殿した後1時間またはそれより多くの時間内にそれを濾過する、請求項25−28のいずれかに記載の製法。
【請求項30】
請求項1−24のいずれかに定義したような多形Aを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩を溶媒中に溶解させて、飽和溶液とする;そして
貧溶媒を加える;
ことを包含する、前記製法。
【請求項31】
溶媒がテトラヒドロフランであり、そして貧溶媒がヘキサンである、請求項30に記載の製法。
【請求項32】
溶媒がメタノールであり、そして貧溶媒がトルエンである、請求項31に記載の製法。
【請求項33】
溶媒が酢酸イソプロピル、エタノール、およびメタノールを包含する、請求項31に記載の製法。
【請求項34】
溶媒を約65℃まで加熱する工程をさらに包含する、請求項33に記載の製法。
【請求項35】
貧溶媒が、メチルt−ブチルエーテルである、請求項33または34に記載の製法。
【請求項36】
メチルt−ブチルエーテルを約45±5℃の温度で加える、請求項35に記載の製法。
【請求項37】
約0℃まで冷却する工程をさらに包含する、請求項36に記載の製法。
【請求項38】
冷却工程を約3.5から4.5時間の間にわたり行う、請求項37に記載の製法。
【請求項39】
多形Bの形のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウム塩の化合物であって、ここで化合物中の多形Bの量が約70%より多い、前記化合物。
【請求項40】
多形Bの量が約80%より多い、請求項39に記載の化合物。
【請求項41】
多形Bの量が約85%より多い、請求項39または40に記載の化合物。
【請求項42】
多形Bの量が約90%より多い、請求項39−41のいずれかに記載の化合物。
【請求項43】
多形Bの量が約95%より多い、請求項39−42のいずれかに記載の化合物。
【請求項44】
多形Bの量が約98%より多い、請求項39−43のいずれかに記載の化合物。
【請求項45】
多形Bの量が約99%より多い、請求項39−44のいずれかに記載の化合物。
【請求項46】
多形Bの量が約100%である、請求項39−45のいずれかに記載の化合物。
【請求項47】
多形Bが、XRPDパターンにおける2θのおよそ22.72度のピークを特徴とする、請求項39−46のいずれかに記載の化合物。
【請求項48】
多形Bが、XRPDパターンにおける2θのおよそ6.6、15.52、18.38、18.94および22.72度のピークを特徴とする、請求項39−47のいずれかに記載の化合物。
【請求項49】
多形Bが、ラマンスペクトルにおけるおよそ1594.7cm−1のピークを特徴とする、請求項39−48のいずれかに記載の化合物。
【請求項50】
多形Bが、ラマンスペクトルにおけるおよそ1696.9、1594.7、1490.2および1397.8cm−1のピークを特徴とする、請求項39−49のいずれかに記載の化合物。
【請求項51】
請求項39−50のいずれかに定義したような多形Bを生成するための製法であって、以下の工程:
N−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミド、ナトリウムを溶媒中に溶解させて、飽和溶液とする;そして
貧溶媒を加えて固体の沈殿物を得る;
ことを包含する、前記製法。
【請求項52】
溶媒が酢酸エチルであり、そして貧溶媒がヘキサン、メチルt−ブチルエーテル、またはトルエンである、請求項51に記載の製法。
【請求項53】
溶媒がアセトンであり、そして貧溶媒がジクロロメタン、メチルt−ブチルエーテル、またはトルエンである、請求項51に記載の製法。
【請求項54】
溶媒がテトラヒドロフランであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項55】
溶媒が酢酸イソプロピルであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項56】
溶媒がエタノールであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項57】
溶媒がメタノールであり、そして貧溶媒がメチルt−ブチルエーテルである、請求項51に記載の製法。
【請求項58】
貧溶媒を約20℃の温度で加える、請求項51−57のいずれかに記載の製法。
【請求項59】
冷却の工程をさらに包含する、請求項58に記載の製法。
【請求項60】
冷却を、0℃まで約3時間の間にわたり行う、請求項59に記載の製法。
【請求項61】
固体の沈殿物が沈殿した後1時間またはそれより多くの時間内にそれを濾過する、請求項60に記載の製法。
【請求項62】
請求項1−24および39−50のいずれかに記載の多形の有効量を被験者に投与することを包含する、エンドセリンを介した疾患の治療のための方法。
【請求項63】
疾患が、高血圧症、心血管疾患、心疾患、肺高血圧症、新生児肺高血圧症、エリスロポエチンを介した高血圧症、呼吸器疾患、炎症性疾患、眼科的疾患、胃腸疾患、腎不全、エンドトキシンショック、月経障害、産科的状態、創傷、蹄葉炎、勃起不全、月経閉止、骨粗しょう症、代謝性骨疾患、更年期障害、中年女性の卵巣機能低下に関連する障害、妊娠中毒症、妊娠時の出産の管理、一酸化窒素減弱性障害、アナフィラキシーショック、間質性肺疾患、拡張期心不全、出血性ショック、および免疫抑制を介した腎血管収縮、から成る群より選択される、請求項62に記載の方法。
【請求項64】
疾患が、肺高血圧症、間質性肺疾患、および拡張期心不全から成る群より選択される、請求項62または63に記載の方法。
【請求項65】
疾患が肺高血圧症である、請求項62−64に記載の方法。
【請求項66】
エンドセリンA(ETA)受容体またはエンドセリンB(ETB)受容体へのエンドセリンペプチドの結合を阻害するための方法であって、該受容体を、請求項1−24および39−50のいずれかに記載の多形と接触させることを包含し、ここで:
当該接触は、受容体とエンドセリンペプチドとの接触前、接触時、または接触後において達成される、前記方法。
【請求項67】
エンドセリン受容体を、請求項1−24および39−50のいずれかに記載の多形と接触させることを包含する、エンドセリン受容体を介した活性を変化させるための方法。
【請求項68】
医薬的に受容可能な担体中に、請求項1−24および39−50のいずれかに記載の多形の有効量を包含する、医薬組成物。
【請求項69】
請求項1−24および39−50のいずれかに記載の多形、および医薬的に受容可能な担体を包含する、医薬組成物。
【請求項70】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の70%よりほぼ多い量で存在する、請求項69に記載の医薬組成物。
【請求項71】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の80%よりほぼ多い量で存在する、請求項69または70に記載の医薬組成物。
【請求項72】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の85%よりほぼ多い量で存在する、請求項69−71のいずれかに記載の医薬組成物。
【請求項73】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の90%よりほぼ多い量で存在する、請求項69−72のいずれかに記載の医薬組成物。
【請求項74】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の95%よりほぼ多い量で存在する、請求項69−73のいずれかに記載の医薬組成物。
【請求項75】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99%よりほぼ多い量で存在する、請求項69−74のいずれかに記載の医薬組成物。
【請求項76】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99.5%よりほぼ多い量で存在する、請求項69−75のいずれかに記載の医薬組成物。
【請求項77】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)−2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の99.9%より多くの量で存在する、請求項69−76のいずれかに記載の医薬組成物。
【請求項78】
多形Aが、組成物中のN−(4−クロロ−3−メチル−5−イソキサゾリル)2−[2−メチル−4,5−(メチレンジオキシ)フェニルアセチル]チオフェン−3−スルホンアミドナトリウムの総重量の約100%である量で存在する、請求項69−77のいずれかに記載の医薬組成物。
【請求項79】
単回投与または複数回投与の投与用に製剤化される、請求項68−78のいずれかに記載の組成物。
【請求項80】
経口錠剤として製剤化される、請求項68−79のいずれかに記載の医薬組成物。
【請求項81】
抗酸化剤、結合剤、希釈剤、バッファー、および耐湿コーティングをさらに包含する、請求項80に記載の経口錠剤。
【請求項82】
請求項1−24および39−50のいずれかに記載の多形を包含する凍結乾燥粉末。
【請求項83】
抗酸化剤、バッファー、および充填剤をさらに包含する、請求項81に記載の凍結乾燥粉末。
【請求項84】
製造品であって、包装材料および該包装材料内に含有される請求項1−24および39−50のいずれかに記載の多形を包含し、ここで多形は、エンドセリンの効果に拮抗する、エンドセリンを介した障害の症状を緩和する、またはエンドセリンペプチドのET受容体への結合を阻害するために有効であり、そして該包装材料は、該多形が、エンドセリンの効果に拮抗する、エンドセリンのエンドセリン受容体への結合を阻害する、またはエンドセリンを介した障害を治療するために使用されることを指摘するラベルを含む、前記製造品。
【図1】


【図2】

【図3】
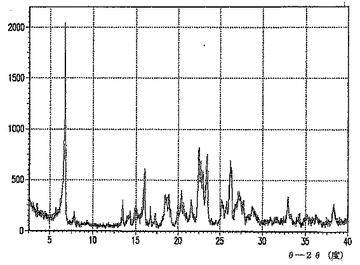
【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図24】

【図25】

【図26】

【図27】

【図28】
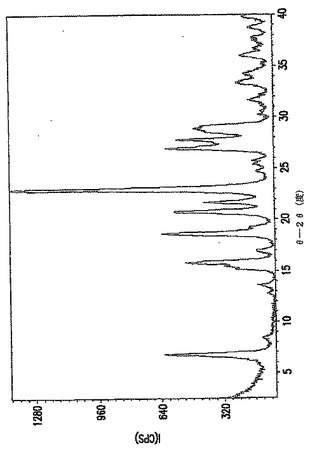
【図29】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【公表番号】特表2009−530279(P2009−530279A)
【公表日】平成21年8月27日(2009.8.27)
【国際特許分類】
【出願番号】特願2009−500424(P2009−500424)
【出願日】平成19年3月12日(2007.3.12)
【国際出願番号】PCT/US2007/006277
【国際公開番号】WO2007/106467
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(508339666)エンサイシブ・ファーマシューティカルズ・インコーポレイテッド (9)
【Fターム(参考)】
【公表日】平成21年8月27日(2009.8.27)
【国際特許分類】
【出願日】平成19年3月12日(2007.3.12)
【国際出願番号】PCT/US2007/006277
【国際公開番号】WO2007/106467
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(508339666)エンサイシブ・ファーマシューティカルズ・インコーポレイテッド (9)
【Fターム(参考)】
[ Back to top ]