
抗悪性腫瘍活性を有するPFKFB3阻害物質ファミリー
6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)の阻害方法及び、阻害化合物を提供する。また、細胞増殖の阻害方法、癌の治療方法及びPFKFB3阻害能を測定するための化合物のスクリーニング方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2007年6月18日に出願された米国仮出願60/936,030に基づく優先権を主張し、これは本明細書に引用される。
本明細書に開示した発明は米国防総省により与えられた助成金番号PCF OGMB04-1166のもとで支援された米国政府によって為された。従って、米国政府は、本明細書に開示した発明の一部の権利を有する。
本願発明は、解糖フラックス(流量)を減少させる方法及び化合物に関する。より詳細には、本願発明は、6−ホスホフルクトー2−キナーゼ(PFK−2)の一誘導酵素である6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(以下「PFKFB3」という。)を阻害する、アザカルコン及びこれらの類似化合物に関係する。解糖フラックスを減少させ、細胞増殖を阻害し及び癌を治療する化合物を使用する方法を記述する。更に、PFKFB3の阻害能をもつ化合物のスクリーニング方法を記述する。
【背景技術】
【0002】
解糖経路は真核細胞における糖代謝の基本的嫌気的経路である。解糖は2重の働きを有し、即ち糖を分解してエネルギー(ATP)を産生し及び合成反応に使われる単純化合物を提供する。グルコースからピルビン酸塩への変換速度は、これら2種の主要な細胞必要物を満たすように調節されている。より詳細には、解糖系過程における不可逆反応を触媒するヘキソキナーゼ、ホスホフルクトキナーゼ及びピルビン酸キナーゼのような酵素は、解糖における制御点として調節されている。
【0003】
1930年にワールブルグは、腫瘍は亢進した嫌気的解糖を行い、また相対的に酸素濃度が高い場合でも、解糖速度の減少を示さないことを指摘した。この調節的制御の喪失(即ち、パスツール効果)は、ワールブルグ効果と呼ばれ始めた。以来、その後の研究により、癌に特有な以下の性質が、常に確認された:a)グルコースの急速な消費;b)活?な解糖活性(非特許文献1参照);c)早い細胞増殖(非特許文献2参照);d)乳酸産生及び蓄積(非特許文献3参照);及びe)腫瘍周囲に外接して、低グルコース濃度を伴う細胞外の低pH。特許文献1を参照のこと。
【0004】
癌細胞の代謝が、エネルギー及び高分子前駆体産生に関して、解糖フラックスが亢進した状態にシフトしているので、解糖の阻害を標的とする可能性を有する抗悪性腫瘍戦略として浮かび上がった。解糖経路の阻害効果による抗腫瘍特性を有するものとして、過去数十年にわたり、様々な小分子が同定されてきた。特に2種の分子、3−ブロモピルビン酸塩(3−BrPA)及び2−デオキシグルコース(2−DG)の両者は、ヘキソキナーゼを標的とし、ミトコンドリア呼吸欠失した、又は低酸素下のトランスフォーム細胞に対して細胞毒性を示した。非特許文献4、非特許文献5及び非特許文献6を参照のこと。抗悪性腫瘍剤Imatinib(商品名Gleevec(R)、Novartis Pharmaceuticals Incorporation,East Hanover, New Jersey, USA)もまたヘキソキナーゼを抑制することが示された。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願公開20060035981号
【非特許文献】
【0006】
【非特許文献1】Maublantほか、Bull Cancer, 85,935-950(1998)
【非特許文献2】Chesneyほか、Proc Natl Acad Sci USA、96,3047-3052(1999)
【非特許文献3】Baggetto、Biochimie,74,959-974(1992)
【非特許文献4】Xuほか、Cancer Res.,65,613-621(2005)
【非特許文献5】Luiほか、Biochemistry,40,5542-5547(2001)
【非特許文献6】Maherほか、Cancer Chemother.Pharmacol.,53,116-122(2004)
【発明の概要】
【発明が解決しようとする課題】
【0007】
さらなる抗癌治療薬の要請があり、特に、癌と関連する亢進した解糖フラックスに関係するメカニズムを通して悪性細胞を標的とする抗ガン治療薬が要請されている。また、解糖に関わる酵素の阻害能を測定して化合物をスクリーニングするさらなる方法が要請されている。
【課題を解決するための手段】
【0008】
本明細書における開示の対象である発明は、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)を阻害化合物と接触させることから成るPFKFB3を阻害する方法を提供する。
【0009】
幾つかの実施態様において、この方法は、PFKFB3を化学式(I):
【化1】

(式中、XはC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1は、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2はアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群より独立に選択される。)
で表わされる化合物接触させることから成る。
【0010】
幾つかの実施態様において、Ar1及びAr2の少なくとも1つは、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、ピリジン基、置換ピリジン基、キノリン基、置換キノリン基、イソキノリン基及び置換イソキノリン基からなる群から選択される。
幾つかの実施態様において、L1は存在しない。幾つかの実施態様において、L2は存在して、C2アルキレン基である。
【0011】
幾つかの実施態様において、この化学式(I)で表わされる化合物は、化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、それぞれ独立にN又はCR12であり、式中、各R12は水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5はO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13は、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つはNである。)
で表わされる構造を有する化合物又はこれらの医薬的に許可された塩である。
【0012】
幾つかの実施態様において、X1はOであり、かつXはCである。
幾つかの実施態様において、Ar2は:
【化5】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基、からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、化学式(II)で表わされる化合物は化学式(III)で表わされる構造:
【化6】
を有する。
【0013】
幾つかの実施態様において、X1はOである。幾つかの実施態様において、R10及びR11はそれぞれ水素原子である。
幾つかの実施態様において、化学式(III)で表わされる化合物は、以下の化学構造:
【化7】
からなる群から選択される。
【0014】
幾つかの実施態様において、Ar2は:
【化8】
であり、
X、X2及びX3のそれぞれは、Cであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は化学式(IV):
【化9】
で表わされる構造を有する。
【0015】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11は、それぞれ水素原子である。幾つかの実施態様において、化学式(IV)で表わされる化合物は、
【化10】
である。
【0016】
幾つかの実施態様において、Ar2は、
【化11】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は化学式(V)で表わされる構造:
【化12】
を有する。
【0017】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11は、それぞれ水素原子である。幾つかの実施態様において、化学式(V)で表わされる化合物は、
【化13】
である。
【0018】
幾つかの実施態様において、本明細書において開示した発明は、細胞を有効量のPFKFB3阻害剤と接触させることから成る、細胞内の解糖フラックスを減少させる方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、細胞を有効量の化学式(I)で表わされる化合物と接触させることから成る、細胞内の解糖フラックスを減少させる方法である。
【0019】
幾つかの実施態様において、本明細書で開示した発明は、細胞に有効量のPFKFB3阻害剤を接触させることから成る、細胞の増殖能を減少させる方法を提供する。幾つかの実施態様において、本明細書で開示した発明は、細胞に有効量の化学式(II)で表わされる化合物を接触させることから成る、細胞の増殖能を減少させる方法を提供する。幾つかの実施態様において、該化合物は、以下の構造:
【化3】
を有する置換基を含み、式中、X4は、NR14,式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;X6は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、アルキル基、アラルキル基及びアリール基からなる群より独立に選択され;及びX6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。幾つかの実施態様において、X5は、Oであり、X6は水素原子、アルキル基、アラルキル基及びアリール基から選択される。
【0020】
幾つかの実施態様において、該化合物は、下記化学式(IIIa)、(IIIb)、(IIIc)及び(IIId):
【化14】
【化15】
【化16】
及び
【化17】
の中の一つである。
【0021】
幾つかの実施態様において、該細胞は、哺乳動物細胞である。幾つかの実施態様において、該細胞はヒト細胞である。幾つかの実施態様において、該細胞は、癌細胞である。
幾つかの実施態様において、本明細書に開示した発明は、有効量のPFKFB3阻害剤を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、有効量の化学式(II)で表わされる化合物を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法を提供する。幾つかの実施態様において、該化合物は化学式(IIIa)、(IIIb)、(IIIc)及び(IIId)の中の一つで表わされる化合物である。幾つかの実施態様において、該患者は、哺乳動物である。幾つかの実施態様において、該患者はヒトである。幾つかの実施態様において、該癌は、乳癌、肺癌、子宮頸癌、皮膚癌及び白血病からなる群から選択される。
【0022】
幾つかの実施態様において、該方法は更に、患者に1又は2以上の付加的な治療用化合物を投与することを含む。幾つかの実施態様において、1又は2以上の付加的治療用化合物は、抗癌治療薬である。幾つかの実施態様において、1又は2以上の付加的治療用化合物は、シスプラチン及びタキソールからなる群から選択される。
【0023】
幾つかの実施態様において、本明細書に開示した発明は、PFKFB3を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、腫瘍増殖を阻害する能力を有する化合物をスクリーニングする方法を提供する。幾つかの実施態様において、この化合物をスクリーニングすることが、PFKFB3のモデルに対して多くの化合物をスクリーニングして、それぞれの化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定することから成る。
【0024】
従って、PFKFB3を阻害し、それにより解糖フラックスを調節する化合物を提供することが、本明細書に開示した発明の目的である。
上記のように記述した本明細書に開示した発明の目的及び本明細書に開示した発明により全体的に又は部分的に到達された他の目的は、以下に最善に記載して添付した図表を参照して、明細書が進むに従い明らかとなろう。
【図面の簡単な説明】
【0025】
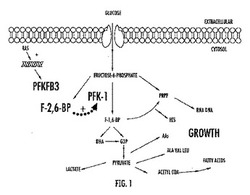
【図1】図1はこの経路により、フルクトース−2,6−ビスリン酸塩によるPFK−1の活性化を経由して、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)が解糖フラックスを亢進させることを示す、経路の概略図である。
【図2】図2Aは、2次元構造で描写した6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)のホモロジーモデルの概略図である。この概略図はまた、結合部位における3−(3−ピリジニル)−1−(4−ピリジニル)−2−プロペン−1−オン(3PO)を示す。図2Bは、2.5Å以内の残基を示すPFKFB3結合ポケットの側面図の図解である。図2Cは、棒状の表示で示す前面残基を伴う結合ポケット残基のConnelly表面(溶媒接触表面)を示す、PFKFB3結合ポケットの端面図の図解である。
【図3】図3Aはフルクトース−6−リン酸塩濃度(60,80,160,240,320,又は400μM)の関数として、PFKFB3酵素活性のLineweaver−Burke2重逆数プロットを示すグラフである。キナーゼ検定は、DMSO(●)、60μM 3PO(O),100μM 3PO(▼)及び150μM 3PO(▽)存在下で行った。3POは、化合物1、3−(3−ピリジニル)−1−(4−ピリジニル)―2−プロペン−1−オンを表す。図3Bは、PFKFB3の比活性(S.A),フルクトース−6−リン酸塩のKm及び3POのKiを含む酵素反応速度パラメーターの表である。
【図4】図4Aは、1,3,10又は33μM 3PO又は溶媒(V)のみで処理後48時間のNHBE細胞(グラフの左側)及びトランスフォームNHBE細胞(ht/Lt/ras,グラフの右側)の細胞増殖を示す棒グラフである。非処理対照細胞分布(C)もまた示す。各棒は、代表的実験の3回の測定の平均細胞数(×103)を示す。誤差バーは、±1標準偏差(STD)を示し;*はp−値<0.01を示す。図4Bは、様々な固形及び血液細胞株における化合物1(即ち、3PO)のIC50値(μM)の表である。各値は、溶媒のみによる対照細胞増殖の50%に必要な1の濃度である。
【図5】図5Aは増加する濃度(0.3,1,3,10及び33μM)の3POを36時間処理した Jurkat細胞の細胞増殖の棒グラフである。溶媒処理細胞(V)及び非処理対照 Jurkat細胞(C)の細胞増殖もまた示す。各棒は、代表的実験の3回の測定の平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示し;*はp−値<0.01を示す。図5Bは、溶媒(●)又は10μM 3PO(○)存在下の Jurkat細胞の時間依存的増殖の測定を示すグラフである。各データポイントは、3回の独立した実験において測定した平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示す。図5Cは、溶媒(●)又は10μM 3PO(○)処理した、Jurkat細胞の経時的なG2/M期細胞集団パーセンテージ(%)を示すグラフである。各データポイントは、3回の独立した実験において測定した平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示す。図5Dは、溶媒(実践)又は10μM 3PO(点線)で36時間処理したJurkat細胞のフローサイトメトリーにより測定した細胞周期ヒストグラムの合成画像である。図5Eは、10μM 3PO存在下又は不存在下での、時間の関数で表す、乳酸塩分泌(▼)、2−デオキシグルコース(2−DG)取り込み(●)及びFru−2,6−BP産生(○)を示すグラフである。誤差バーは±1標準偏差(STD)を示す。図5Fは、10μM 3PO存在下又は不存在下での、時間の関数で表す細胞全体のNAD+(●)、NADH(○)及びATP(▼)濃度を示すグラフである。誤差バーは±1標準偏差(STD)を示す。図5Gは、溶媒(左側スペクトル)又は3PO(右側スペクトル)存在下、アラニン(Ala)及び細胞内乳酸塩(Lac)への13C取り込みを示す、2次元(2D)核磁気共鳴(NMR)スペクトルである。 3回の独立した実験からの代表的スペクトルを示す。 点線ボックスの縁は、代表的代謝物の13Cピークに対応し、これらは中心の内因性12Cピークを囲む。
【図6】図6Aは、その後ヒトテロメラーゼ(ht)、ラージT抗原(LT)及び腫瘍遺伝子H−rasv12で不死化し、またトランスフォームした野生型(+/+)又はハプロ不全(+/−)PFKFB3マウスからの繊維芽細胞で測定した、F2,5BPの細胞内濃度を表す棒グラフである。挿画は、繊維芽細胞におけるPFKFB3発現のウェスターンブロット解析を示す。図6Bは、その後ヒトテロメラーゼ(ht)、ラージT抗原(LT)及び腫瘍遺伝子H−rasv12で不死化し、またトランスフォームした野生型(+/+)又はハプロ不全(+/−)PFKFB3マウスからの繊維芽細胞に対する3POの増殖抑制効果を示す棒グラフである。トランスフォームした野生型細胞(+/+)に対する細胞数(対照細胞の細胞数に対する%として表わす)を実線バーで表し、またハプロ不全細胞(+/−)に対する細胞数をストライプバーで表す。グラフの底に示すように、細胞を溶媒又は増加する3PO量(5,10,20,30,40又は60μM)とインキュベートして、生存細胞数を48時間後に計数する。バーは、代表的実験からの3回の値の平均値±STDを示す。*p−値<0.01は、溶媒対照及び3PO処理試料の間の統計的差異を表す。図6Cは、Tet−onシステム(+PFKFB3)及び空ベクター(Vector)を含む対照細胞を用いて、ドキシサイクリン処理によりPFKFB3を過剰発現するよう加工したJurkat細胞を使って測定したF2,5BPの細胞濃度の棒グラフである。細胞を1μg/mlのドキシサイクリンで処理し、PFKFB3蛋白質発現を誘導した。挿図は細胞内のPFKFB3発現のウェスターンブロット解析である。図6Dは、Tet−onシステムを用いて、ドキシサイクリン処理でPFKFB3を過剰発現するよう加工したJurkat細胞(+PFKFB3、ストライプバー)での3POの増殖抑制効果を示すう棒グラフである。3PO処理24時間前に、PFKFB3蛋白質レベルを誘導するために、1μg/mLのドキシサイクリンをPFKFB3発現のためのベクターを含むJurkat細胞とインキュベートする。空ベクターを含む対照細胞(―PFKFB3,実線バー)を同様にドキシサイクリン処理し、バックグランドPFKFB3発現として用いた。その後、両種の細胞を異なる濃度の3PO(1,3,10,又は33μM)又は溶媒(V)とインキュベートし、生存細胞数を48時間後に解析した。バーは、代表的実験からの3回の値の平均値±STDを示す。
【図7】図7Aは、C57/Blk6マウスに異種移植したLewis Lung カルシノーマ腫瘍の増殖に対する3PO処理の効果を示すグラフである。平滑末端のノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=11)又は3PO処理群(○、n=14)に分けた。実験マウスの体重を測定し、毎日、表示した時点において、50μL DMSO、又は0.07mg/g 3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は毎日の対照又は3PO投与を示す。最初の注射(2日目、p−値<0.0003)後に、DMSO及び3PO群の間に統計的に有意の差異が得られた。 図7Bは、Balb/c胸腺欠損マウスに異種移植した、株化したMDA−MB231乳腺癌腫瘍の増殖に対する3PO処理の効果を示すグラフである。ノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=14)又は3PO処理群(○、n=13)に分けた。実験マウスの体重を測定し、研究期間(14日)の間、継続する3日間の投与の後3日間は無投与という周期的投与計画に従い、50μL DMSO、又は0.07mg/g3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は対照又は3PO投与を示す。2日目以後、DMSO対照群及び3PO実験群の間に統計的な差異が得られた(p−値<0.0001)。 図7Cは、Balb/c胸腺欠損マウスに異種移植した株化したHL−60急性前骨髄性白血病細胞の増殖への3PO処理効果を示すグラフである。ノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=11)又は3PO処理群(○、n=12)に分けた。実験マウスの体重を測定し、研究期間(14日)の間、継続する2日間のDMSO又は3POの投与、その後7日間の休息期間という周期的投与計画に従い、50μL DMSO、又は0.07mg/g 3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は対照又は3PO投与を示す。最初の注射後、p−値<0.0001をもって、統計的な差異が得られた。
【図8】図8は、化合物2、3−(4−クロロフェニル)−1−(3−ピリジニル)−2−プロペン−1−オンのPFKFB3阻害活性を示す棒グラフである。150μM 2で処理した遺伝子組み換えPFKFB3の反応速度は、同容量の溶媒(EtOH)処理のPFKFB3で観測される反応速度の39.8%であった。
【図9】図9は、溶媒(DMSO)処理PFKFB3酵素の活性と比較した、化合物3 (即ち、3−(1−ナフチル)−1−(4−ピリジニル)−2−プロペン−1−オン),4 (即ち1−(3−ピリジニル)−3−(2−キノリニル)−2−プロペン−1−オン),5 (即ち、N−{4−[3−オキソ−3−(4−ピリジニル)−1−プロペン−1−イル]フェニル}アセトアミド),6 (即ち、3−(2−クロロフェニル)−1−(2−ピリジニル)−2−プロペン−1−オン)及び7 (即ち、3−(2−クロロフェニル)−1−(3−ピリジニル)−2−プロペン−1−オン)のPFKFB3阻害活性を示す棒グラフである。
【図10】図10は、異なる濃度(1,3,10又は33μM)の化合物2、又は溶媒(EtOH)処理した、HL−60細胞(灰色バー)及びK562細胞(白色バー)の細胞増殖を示す棒グラフである。バーは、代表的実験からの3回の値の平均値±STDを示す。
【図11】図11は0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物3をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図12】図12は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物4をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図13】図13は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物5をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図14】図14は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物6をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図15】図15は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物7をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図16】図16は、化合物2〜7(0.3〜33μMにおける)存在下でのMDA−MB−231乳腺癌細胞の増殖と生存率を示すグラフである。
【図17】図17は、化合物2〜7(0.3〜33μMにおける)存在下でのA5491肺腺癌細胞の増殖と生存率を示すグラフである。
【図18】図18は、化合物2〜7(0.3〜33μMにおける)存在下でのK562慢性骨髄性白血病細胞の増殖と生存率を示すグラフである。
【図19】図19は、化合物2〜7(0.3〜33μMにおける)存在下でのHL−60前骨髄性白血病細胞の増殖と生存率を示すグラフである。
【図20】図20は、化合物2〜7(0.3〜33μMにおける)存在下でのCRL−11174メラノーマ細胞の増殖と生存率を示すグラフである。
【図21】図21は、化合物2〜7(0.3〜33μMにおける)存在下でのHeLa子宮頸癌細胞の増殖と生存率を示すグラフである。
【図22】図22は、Lewis肺癌細胞異種移植片を有するマウスに取り込まれた18F−2−デオキシグルコースのマイクロポジトロン発光断層撮影(トモグラフィー)画像を示す。マウスには、150μキュリーの18F−フルオロ−2−デオキシグルコースの腹腔注射(i.p.)30分前に、50μL DMSO(溶媒)又は0.07mg/g 3PO(+3PO)を含むDMSOのi.p.を行った。矢印は、右側腹部における心臓及び腫瘍異種移植片の位置を示す。
【図23】図23は、生後4ヶ月のトランスジェニックMMTV−Neu乳腫瘍マウスにおける腫瘍に対する3PO処理の効果を示すグラフである。該マウスは、無作為に、等しい腫瘍負荷を有する2群に分けた。第1群(+3PO)には、3日間投与(矢印で示す)及び3日間非投与の周期で、3PO(DMSO中、0.07mg/g;i.p.)を投与した。第2群(溶媒)には、DMSOのみ注射した。
【発明を実施するための最良の形態】
【0026】
本明細書において開示した発明を、代表的な実施態様を示す、実施例を参照しつつより詳細に以下に記載する。しかしながら、本明細書に開示した発明は、異なった形に具体化可能であり、本明細書に記載したように実施態様を制限すると解釈してはならない。むしろ、これらの実施態様は、開示が十分であり、完全であるように提供されており、当業者には、実施態様の範囲が十分に伝達されるであろう。
もし別に定義しなければ、本明細書で用いる技術及び科学用語は、本明細書に開示した発明が属する当業者の1人に一般的に理解されるのと同じ意味を有する。全ての出版物、特許出願、特許、本明細書に述べた参考文献は、その全体が参考文献に取り込まれている。
仕様及び特許請求の範囲を通して、与えられた化学式又は化学名は、光学及び立体異性体、並びに異性体及び混合物が属するラセミ混合物を包含する。
【0027】
この明細書では下記の略号を用いる:
2−DG=2−デオキシグルコース
3−BrPA=3−ブロモピルビン酸塩
3PO=3−(3−ピリジニル)−1−(4−ピリジニル)−2−プロペン−1−オン
AA=アミノ酸
ATP=アデノシン3リン酸
Ci=キュリー
DMSO=ジメチルスルフォキシド
DNA=デオキシリボ核酸
DMEM=ダルベッコの改変イーグル培地
EDTA=エチレンジアミンテトラ酢酸
F2,6BP=フルクトース−2,6−ビスリン酸塩
F6P=フルクトース−6−リン酸塩
HIF−1=低酸素症誘導因子1
ht=ヒトテロメラーゼ
LDH=乳酸デヒドロゲナーゼ
LT=ラージT抗原
μM=マイクロモル
mg=ミリグラム
mL=ミリリットル
min=分
ng=ナノグラム
NHBE=正常ヒト気管支上皮
Ni=ニッケル
nm=ナノメートル
NTA=ニトリロ三酢酸
PBS=リン酸緩衝生理食塩水
PEP=ホスホエノールピルビン酸塩
PFK−1=6−ホスホフルクト−1−キナーゼ
PFK−2=6−ホスホフルクト−2−キナーゼ
PFKFB3=6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3
PK=ピルビン酸キナーゼ
PMSF=フェニルメチルスルフォニルフッ化物
PRPP=5−ホスホ−D−リボシル−1−ピロリン酸塩
RNA=リボ核酸
SLN=Sybylライン表記法
STD=標準偏差
【0028】
1.定義
本明細書で用いる用語"癌"は、無制御な細胞分裂及び細胞の転移力、又は他の位置での新増殖を確立する力により引き起こされる病気を表わす。用語"悪性な"、"悪性"、"新生物"、"腫瘍"及び様々なこれらの変形語は、癌細胞又は癌細胞群を表す。
癌の特定の種類としては、皮膚癌、結合組織癌、脂肪腫、乳癌、肺癌、胃癌、膵臓癌、卵巣癌、子宮頸癌、子宮癌、肛門性器癌、腎臓癌、膀胱癌、大腸癌、前立腺癌、中枢神経(CNS)癌、網膜癌、血液及びリンパ球癌があるが、これらに制限されない。
【0029】
用語"競争的阻害剤"は、酵素に結合することにより、酵素の正常な基質の結合を阻害する阻害剤を表す。
本明細書で用いるように、用語"アルキル基"は、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert−ブチル基、ペンチル基、ヘキシル基、オクチル基、エテニル基、プロペニル基、ブテニル基、ペンテニル基、ヘキセニル基、オクテニル基、ブタジエニル基、プロピニル基、ブチニル基、ペンチニル基、ヘキシニル基、ヘプチニル基及びアレニル基を含む、直線(即ち、"直鎖")の、分枝の、又は環状の、飽和の、又は少なくとも部分的に及びある場合は完全に不飽和な(即ち、アルケニル基及びアルキニル基)炭化水素鎖を含むC1−20を表わす。"分枝状の"は、メチル基、エチル基又はプロピル基のような、低級アルキル基が直線型アルキル鎖に付加したアルキル基を表す。"低級アルキル基"は1から約8個の炭素原子(即ちC1−8アルキル基)、例えば、1,2,3,4,5,6,7又は8炭素原子、を有するアルキル基を表す。"高級アルキル基"は、約10から約20炭素原子、例えば、10,11,12,13,14,15,16,17,18,19,又は20炭素原子を有するアルキル基を表す。ある実施態様において、"アルキル基"は、特に、C1−8直鎖型アルキル基を表す。他の実施態様において、"アルキル基"は、特に、C1−8分枝型アルキル基を表す。
【0030】
アルキル基は、任意に、同一又は異なる、1又は2以上のアルキル基置換体により置換することができる("置換アルキル基")。用語"アルキル基置換体"は、アルキル基、置換アルキル基、ハロゲン原子、アリールアミノ基、アシル基、ヒドロキシル基、アリールオキシル基、アルコキシル基、アルキルチオ基、アリールチオ基、アラルキルオキシル基、アラルキルチオ基、カルボキシル基、アルコキシカルボニル基、オキソ基及びシクロアルキル基を含むが、これらに制限されない。アルキル鎖には、任意に、1又は2以上の酸素、硫黄、又は置換又は非置換窒素原子を挿入できるが、ここで窒素置換基は、水素原子、低級アルキル基(本明細書では、"アルキルアミノアルキル基"と表わす)、又はアリール基である。
従って、本明細書に用いるように、用語"置換アルキル基"には、例えば、アルキル基、置換アルキル基、ハロゲン原子、アリール基、置換アリール基、アルコキシル基、ヒドロキシル基、ニトロ基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、硫酸基及びメルカプト基が含まれるように、アルキル基の1又は2以上の原子又は官能基が他の原子又は官能基に置き変えられたアルキル基が含まれる。
【0031】
本明細書で用いる、用語"アリール基"は、共有結合で連結した、又はメチレン基又はエチレン基の部分又はこれらに制限されない普通の置換基に連結した、単一の芳香族環、又は互いに縮合した多環芳香族であることが可能な芳香族置換体を表す。普通の連結基としてはまた、ベンゾフェノンにおけるようなカルボニル基、又はジフェニルエーテルにおけるような酸素原子、又はジフェニルアミンにおけるような窒素原子であることができる。用語"アリール基"は、特に複素環式芳香族化合物も包含する。該芳香族環は、特に、フェニル基、ナフチル基、ビフェニル基、ジフェニルエーテル基、ジフェニルアミン基及びベンゾフェノン基を含むことができる。特定の実施態様において、用語"アリール基"は、約5から約10個の炭素原子、即ち、5,6,7,8,9,又は10炭素原子、を含み、また5及び6員炭化水素及び複素環式環芳香族基を含む、環状芳香族基を意味する。
【0032】
該アリール基は、任意に、同一又は異なる、1又は2以上のアリール基置換体により置換することができて、ここで"アリール基置換体"は、アルキル基、置換アルキル基、アリール基、置換アリール基、アラルキル基、ヒドロキシル基、アルコキシル基、アリールオキシル基、アラルキルオキシル基、カルボキシル基、アシル基、ハロゲン、ニトロ基、アルコキシカルボニル基、アリールオキシカルボニル基、アラルコキシカルボニル基、アシルオキシル基、アシルアミノ基、アロイルアミノ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、アリールチオ基、アルキルチオ基、アルキレン基及び−NR'R"を含み、ここでR'及びR"は、それぞれ独立に、水素、アルキル基、置換アルキル基、アリール基、置換アリール基及びアラルキル基であることができる。
【0033】
従って、本明細書に用いるように、用語"置換アリール基"は、例えば、アルキル基、置換アルキル基、ハロゲン、アリール基、置換アリール基、アルコキシル基、ヒドロキシル基、ニトロ基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、硫酸基及びメルカプト基が含まれるように、アリール基の1又は2以上の原子又は官能基が他の原子又は官能基に置き変えられたアリール基が含まれる。
アリール基の特別な例としては、シクロペンタジエニル基、フェニル基、フラン基、チオフェン基、ピロール基、ピラン基、ピリジン基、イミダゾール基、ベンズイミダゾール基、イソチアゾール基、イソクサゾール基、ピラゾール基、ピラジン基、トリアジン基、ピリミジン基、キノリン基、イソキノリン基、インドール基、カルバゾール基等々が含まれるが、これらに制限されない。
【0034】
本明細書で用いるように、用語"アザ基"は、少なくとも1個の窒素原子を含む複素環式環状構造を表す。アザ基の例としては、ピロリジン基、ピペリジン基、キヌクリジン基、ピリジン基、ピロール基、インドール基、プリン基、ピリダジン基、ピリミジン基及びピラジン基を含むが、これらに制限されない。
用語"アザアリール基"は、アリール環又は環類の1又は2以上の原子が、窒素原子である複素環式アリール基を表す。アザアリール基の例としては単環又は二環のモノ−又はジアザアリール基(即ち、2個の窒素原子を含むアリール基)を含み、このアザアリール基は、非置換型、又は置換型であり、置換型の場合、例えばメチル基のような低級アルキル基;例えばメトキシ基のような低級アルコキシ基及び/又は例えば、塩素、又は臭素原子の様なハロゲン原子からなる群から選択された群の一つにより置換される。従って、該用語"アザアリール基"は、ピリジン基、ピリダジン基、ピリミジン基、ピラジン基、キノリン基、キナリジン基、キノクサリン基及びこれらの置換型の類似体を含む置換基を表わすが、これらに制限されない。幾つかの実施態様において、アザアリール基は、例えば、2−、3−、又は4−ピリジル基のようなピリジル基;例えば、4−キノリニル基又は1−イソキノリニル基のようなキノリニル基又はイソキノリニル基;イミダゾイル基;例えば、2−又は4−ピリミジニル基のようなピリミジニル基;例えば、3−ピリダジニル基のようなピリダジニル基;又は例えば、2−ピラジニル基のようなピラジニル基である。
【0035】
本明細書において用いられるように以下:
【化18】
のような化学式で一般的に表される構造は環状構造と呼ばれ、例えば、3炭素、4炭素、5炭素、6炭素等々、脂肪族及び/又は芳香族環状化合物であるが、これらに制限されず、またこれらはR置換基を含み、Rは存在しても不存在でもよい、存在する場合は、1又は2以上のR基は、環状構造の1又は2以上の可能な炭素原子上で置換されることができる。R基の存在又は不存在及びR基の数は、整数nの値により決定される。もし1以上の場合、各R基は、他のR基上より、環状構造の可能な炭素上で置換される。例えば
【化19】
式中、nは0から2の間の整数であり、上記の構造は、以下の構造群:
【化20】
を含む化合物群を含むが、これらに制限されない。
【0036】
化学構造において、結合を示す点線は、該結合が存在する、又は不存在であることを示す。例えば、下記:
【化21】
の置換基は、1重結合又は2重結合からなる置換基を表すことができる。
命名された原子又は置換基が"不存在"と定義される時、命名された原子又は置換基は、直接の結合により置換される。連結基又はスペーサー基が不存在であると定義される場合、連結基又はスペーサー基は直接結合により置換される。
【0037】
幾つかの実施態様において、本明細書において開示した発明の化合物は、連結基を含む。本明細書で用いるように、用語"連結基"は、2又は3個の他の分子部分と結合して、安定な構造を形成するアルキレン基、又はこれに制限されない置換基、を含む2価の分子部分を含む。幾つかの実施態様において、O又はSの様な単一原子は、連結基として働く。幾つかの実施態様において、連結基は、例えば、メチレンジオキシル(-O-CH2-O-)又はエチレンジオキシル(-O-CH2CH2-O-)のようなオキシ基又はチオ基を含むことができる。幾つかの実施態様において、該連結基は、フェニレン基、フラニル基、チエニル基、又はピロリール基のような、2価のアリール基であることができる。
【0038】
用語"3価連結基"は、1又は2以上の化学置換基上で、3個の置換基又は3個の部位(例えば、原子)を連結する連結基を表す。例えば、3価の連結基は以下の化学式:
【化22】
であってもよく、式中、各Rは、独立に、1又は2以上のO、S,窒素、又は置換窒素原子、又は不飽和結合を含むことができる、C0〜C6アルキル基である。
【0039】
"アルキレン基"は、1から約20個の炭素原子、例えば、1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,又は20炭素原子を有する直鎖型又は分枝型2価脂肪族炭化水素を表す。アルキレン基は、直鎖型、分枝型、又は環状である。アルキレン基は、また任意に、不飽和であり及び/又は、1又は2以上の、ヒドロキシル基、ハロゲン、ニトロ基、アルキル基、アリール基、アラルキル基、カルボキシル基、等々を含む、"アルキル基置換体"と置換されることができる。アルキレン基上に、1又は2以上の酸素、硫黄、又は置換又は非置換窒素原子(本明細書ではまた"アルキルアミノアルキル基"と表わす)を任意に挿入することができるが、ここで窒素置換基は、前述のようにアルキル基である。アルキレン基の例としては、メチレン基(−CH2−);エチレン基(−CH2−CH2−);プロピレン基(−(CH2)3−);シクロへキシレン基(−C6H10−);−CH=CH−CH=CH−;−CH=CH−CH2−;−(CH2)q−N(R)−(CH2)r―を含むが、式中、各q及びrは、独立に0から約20までの整数であり、例えば、0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19、又は20であり、またRは、水素又は低級アルキル基;メチレンジオキシル基(−O−CH2−O−);及びエチレンジオキシル基(−O−(CH2)2−O−)である。アルキレン基は、約2から約3個の炭素原子を有することができて、また更に6〜20個の炭素を有することができる。
【0040】
本明細書で用いるように、用語"アシル基"は、有機カルボン酸基を表し、ここでカルボキシル基の−OHは他の置換体(即ち、RCO−で表せるように、ここでRは、アルキル基、アラルキル基、又は、置換アルキル基、アラルキル基及びアリール基を含む、本明細書で定義されたアリール基である)で置換される。従って、用語"アシル基"は、特に、アセチルフラン基及びフェナシル基の様な、アリールアシル基を含む。アシル基の特別な例として、アセチル基及びベンゾイル基が含まれる。
【0041】
"環状"及び"シクロアルキル基"は、約3から約10個の炭素原子、例えば、3,4,5,6,7,8,9又は10炭素原子の、非芳香属性単環式又は多環式環状システムを表す。該シクロアルキル基は、任意に、部分的に不飽和であることができる。シクロアルキル基は、任意に、本明細書で定義したように、オキソ基及び/又はアルキレン基の様な、アルキル基置換体により置換されることができる。環状アルキル鎖に沿って、任意に、1又は2以上の酸素、硫黄、又は置換型の又は非置換型の窒素原子を挿入することができて、式中、窒素置換基は、水素、アルキル基、置換アルキル基、アリール基、又は置換アリール基であり、従って、複素環式置換基を提供する。代表的な単環式シクロアルキル環は、シクロペンチル基、シクロヘキシル基及びシクロヘプチル基を含む。多環式シクロアルキル環は、アダマンチル、オクタヒドロナフチル基、デカリン、樟脳、カンファン及びノラダマンチルを含む。
【0042】
"アルコキシル基"は、アルキル−O−基を表し、ここでアルキル基は、既述の通りである。本明細書で用いる、用語"アルコキシル基"は、例えば、メトキシル基、エトキシル基、プロポキシル基、イソプロポキシル基、ブトキシル基、t−ブトキシル基及びペントキシル基と表すことができる。用語"オキシアルキル基"は、"アルコキシル基"と互換的に用いることができる。
"アリールオキシル基"は、アリール−O−基を表し、ここでアリール基は、置換アリール基を含み、既述の通りである。本明細書で用いる用語"アリールオキシル基"は、フェニルオキシル基又はヘキシルオキシル基及びアルキル基、置換アルキル基、ハロゲン原子、又はアルコキシル置換フェニルオキシル基又はヘキシルオキシル基と表すことができる。
"アラルキル基"は、アリール−アルキル基を表し、ここでアリール基及びアルキル基は既述通りであり、また置換アリール基及び置換アルキル基を含む。アラルキル基の例として、ベンジル基、フェニルエチル基及びナフチルメチル基が含まれる。
"アラルキルオキシル基"は、アラルキル−O−基を表し、ここでアラルキル基は既述通りである。アラルキルオキシル基の例として、ベンジルオキシル基がある。
"ジアルキルアミノ基"は−NRR'基を表し、ここでR及びR'のそれぞれは、既述のように、独立に、アルキル基及び/又は置換アルキル基である。アルキルアミノ基の例としては、エチルメチルアミノ基、ジメチルアミノ基及びジエチルアミノ基が含まれる。
【0043】
"アルコキシカルボニル基"は、アルキル−O−CO−基を表す。アルコキシカルボニル基の例として、メトキシカルボニル基、エトキシカルボニル基、ブチルオキシカルボニル基及びt−ブチルオキシカルボニル基が含まれる。
"アリールオキシカルボニル基"は、アリール−O−CO−基を表す。アリールオキシカルボニル基の例として、フェノキシ−及びナフトキシ−カルボニルが含まれる。
"アラルコキシカルボニル基"は、アラルキル−O−CO−基を表す。アラルコキシカルボニル基の例としては、ベンジルオキシカルボニル基がある。
"カルバモイル基"は、H2N−CO−基である。
"アルキルカルバモイル基"はR'RN−CO−基を表し、ここでR及びR'の中の一方は、水素であり、R及びR'の中の他方は、既述のように、アルキル基及び/又は置換アルキル基である。
"ジアルキルカルバモイル基"は、R'RN−CO基を表し、ここでR及びR'のそれぞれは、独立に、既述のようなアルキル基及び/又は置換アルキル基である。
"アシルオキシル基"は、アシル−O−基を表し、ここでアシル基は既述の通りである。
"アシルアミノ基"は、アシル−NR−基を表し、ここでアシル基は、既述の通りであり、Rは、水素原子又はアルキル基である。従って、該"アシルアミノ基"は、構造−NR−C(=O)−R'を有することができて、R'は、アルキル基、アリール基、アラルキル基等々である。
【0044】
用語"アミノ基"は、−NH2基を表す。
用語"カルボニル基"は、−(C=O)−基を表す。
用語"カルボキシル基"は、−COOH基を表す。
本明細書で用いる様に、用語"ハロ"、"ハロゲン化物"又は"ハロゲン"は、フッ素、塩素、臭素及び沃素基を表す。
用語"ヒドロキシル基"は、−OH基を表す。
用語"ヒドロキシアルキル基"は、−OH基で置換されたアルキル基を表す。
用語"メルカプト基"は、−SH基を表す。
用語"オキソ基"は、本明細書で既述の化合物を表し、ここで炭素原子は酸素原子に置換される。
用語 "アザ基"は、炭素原子が窒素原子に置換された化合物を表す。
用語"ニトロ基"は、−NO2基を表す。
用語"チオ基"は、本明細書で既述の化合物を表し、ここで炭素又は酸素原子がイオウ原子に置換される。
用語"硫酸基"は、−SO4基を表す。
用語"カルコン"又は"カルコーン"は、1,3−ジフェニル−1−プロペン−3−オン:
【化23】
及びこの類似体(例えば、ジアリールプロペノン類、ジアリールプロペノン及びジアリールプロパンのチア及びアザ類似体)を表す。用語"アザカルコン"は、1又は2以上の窒素原子を含むカルコンを表す。特に該アザカルコンは、1又は2以上のアザアリール基を含むことができる。
【0045】
用語"独立に選択される"を用いた場合、関係する置換体(例えば、R1又はR2基、又はX及びY基のようなR基)は同一であるか、又は異なる。例えば、R1及びR2の両者は、アルキル基により置換される、又はR1は水素であることができて及びR2は置換アルキル基等々であることができる。
名付けられた"R"、"X" 、"A" 、"Ar"、"Y" 、又は"L" 基は、本明細書において特に他の名前で特定されなければ、一般的に当業者にその名前を有する置換基に対応すると認識される構造を有する。描画の目的のために、既述の代表的"R"、"X" 、"Y"、 "L"、"Ar"及び"A" 基 は以下のように定義される。これらの定義は、補充と描画のために意図され、本明細書開示を閲覧した当業者に明らかな定義を排除しない。
【0046】
II.一般的考察
癌組織において解糖フラックスが増加の方向へシフトする正確なメカニズムを理解することは、ある時は、重要の研究の焦点であった。解糖の増加が起こるには、様々な因子:a)基質利用可能性、b)解糖フラックスのために必要な代謝酵素の発現及びc)経路の特定の作用点で働くアロステリック活性化因子及び阻害剤のレベル、が含まれる。例えば、GLUT1の様な、グルコース輸送体の過剰発現は、亢進した解糖速度を維持するために必要な過剰なグルコースの供給を提供すると考えられており、他方ヘキソキナーゼ及びホスホフルクト−1−キナーゼ(PFK1)の様な鍵となる律速酵素は、癌細胞において亢進していることが明らかとなっている。Vora ほか, Cancer Res., 45(7), 2993-3001 (1985); Macheda ほか, J. Cell Physiol., 202(3), 654-662 (2005); 及び Mathupala ほか, Oncogene, 25(34), 4777-4786 (2006)を参照されたい。最後に、解糖酵素のアロステリック調節酵素は、特にPFK1が関わる最初の不可逆的な、委ねられた律速段階において、グルコースフラックスに大きな役割を果たす。Van Schaftingen, ほか., Proc. Natl. Acad. Sci. USA, 78(6), 3483-3486 (1981)を参照されたい。
【0047】
PFK1活性は、原発腫瘍組織ばかりではなく、腫瘍細胞において増加すると観測されてきた。さらに、腫瘍遺伝子ras又はsrcをRat−1繊維芽細胞又はニワトリ胎児繊維芽細胞に導入すると、それぞれ、高いPFK1活性をもたらす。Hennipman ほか., Tumour Biol., 8(5), 251-263 (1987); Kole,ほか, Arch. Biochem. Biophys. 286(2), 586-590 (1991); 及びSanchez-Martinez 及びAragon, FEBS Lett., 409(1), 86-90 (1997)を参照されたい。しかしながら、形質転換細胞内の高いレベルのPFK−1活性は、蛋白質発現の増加によるのではなく、フルクトース−2,6−ビスリン酸塩(F2,6BP)の増加によることが分かった。Kole, ほか., Arch. Biochem. Biophys. 286(2), 586-590 (1991)を参照されたい。1980年代の初め、F2,6BPに、PFK1のアロステリック活性化因子の可能性が見出され、基質である、フルクトース−6−リン酸塩に対して低親和性から高親和性へ、PFK1のコンホメーションの平衡をシフトすることにより、ATPの阻害効果を取り除くことができる。Van Schaftingen, ほか., Biochem. J., 192(3), 887-895 (1980); Van Schaftingen, ほか., Biochem. J., 192(3), 897-901 (1980); 及び Van Schaftingen, ほか, Proc. Natl. Acad. Sci., USA, 78(6), 3483-3486 (1981)を参照されたい。従って、F2,6BPレベルの亢進調節により、高エネルギー産生にも拘わらず(即ち、生理的ATPレベル)、癌組織が高い解糖的産生を維持することを可能にする。図1は、癌遺伝子rasが、如何に、癌細胞においてPFKFB3の発現及び活性を増加させ、従ってF2,6BP産生を亢進させ、律速PFK−1の活性化及び、究極的に、高い解糖フラックスをもたらすと信じられ、またRNA,DNA、アミノ酸(AAs)及び脂肪酸を含む高分子の産生を増加させるかを表す。
【0048】
細胞内のF2,6BPレベルは、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ(PFK−2/FBPases)と名付けられた二機能性の酵素ファミリーにより維持され、また4種の遺伝子PFKFB1〜4によりコードされている。PFKFB1,2及び4は、それぞれ、肝臓/筋肉、腎臓/心臓及び精巣内に発現されると報告されており、ホスファターゼ活性に等しいキナーゼ活性を示す。PFKFB3遺伝子によりコードされているPFK−2の誘導型イソ型(iPFK2、胎盤PFK2,ユビキタス(遍在)PFK2及びPGR1と報告されている)は、分裂、炎症及び低酸素刺激により亢進されることが示されてきた、またホスファターゼ領域内の突然変異により、キナーゼ:ホスファターゼ比が740:1であることを示す。
【0049】
PFKFB3は、大腸、前立腺、肺、乳房、膵臓及び卵巣を含む多くの種類の癌組織において、過剰発現していることが観測されてきた。Atsumi, ほか., Cancer Res., 62, 5881-5887 (2002)を参照されたい。不死化したマウス繊維芽細胞に腫瘍遺伝子rasを導入すると、PFKFB3発現が高まり、PFKFB3は癌化形質転換において必要不可欠の成分であるかもしれないことを示唆する。Chesney及び共同研究者等は、PFKFB3に対するアンチセンスオリゴヌクレオチドを用いて、無胸腺マウスに異種移植したK562白血病細胞の抑制を示し、一方また、ヌクレオチド生合成の前駆体である5−ホスホリボシル−1−ピロリン酸塩(PRPP)の減少を観測した。ヒトテロメラーゼ、ラージT抗原及び癌遺伝子H−rasv12により、不死化し、続いてトランスフォームした異形接合のPFKFB3(+/−)マウス繊維芽細胞において、F2,6BP産生は減少し、グルコースの乳酸への解糖フラックスは抑制された。より重要なことは、rasトランスフォーム繊維芽細胞においてPFKFB3レベルが低いために、軟寒天中の基質非依存性増殖の喪失がもたらされ、また無胸腺マウスにおける腫瘍のin vivo増殖が顕著に減少する。Telang ほか., Oncogene, 25, 7225-7234 (2006)を参照されたい。これとは別に、siRNAによるA549肺癌細胞におけるPFKFB3の抑制は、F2,6BPの減少をもたらし、軟寒天中のコロニー形成の喪失をもたらす。
小分子阻害剤による選択的なPFKFB3の標的化が、新しい抗ガン治療の戦略であると言う所見を、本明細書にはじめて開示した。
【0050】
III.PFKFB3阻害剤
本明細書に開示した発明は、PFKFB3の阻害方法を提供し、該方法はPFKFB3と阻害化合物との接触を含む。幾つかの実施態様において、該方法はPFKFB3と小分子阻害剤との接触を含む。
【0051】
幾つかの実施態様において、本明細書に開示した発明は、PFKFB3と、PFKFB3を阻害する又は、そうでなければPFKFB3活性を変える化合物とを接触させる方法を提供し、式中、該化合物は以下の化学式(I):
【化1】
で表わされる化合物であり、
式中、Xは、C又はCHであり;
X1はO、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、夫々独立に、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から選択され、またR2及びR7は、夫々独立に、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から選択され;
L1は、存在又は不存在であってもよく、存在の場合、O、S、NR8、アルキレン基、置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在の場合、O、S、NR9、アルキレン基及び置換アルキレン基及び3価連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また3価連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及び、Ar1及びAr2からなる群の一つと結合する第3の原子を含む。
Ar1及びAr2は,独立にアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群から選択される。
【0052】
幾つかの実施態様において、化学式(I)で表わされる化合物は、アザカルコンである。
幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の両者は、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、ピリジン、置換ピリジン、キノリン、置換キノリン、イソキノリン及び置換イソキノリンからなる群から選択される。
【0053】
幾つかの実施態様において、Ar1及びAr2の少なくとも一つはアシルアミノ基により置換される。幾つかの実施態様において、化学式(I)で表わされる化合物は、アシルアミノ置換アザカルコンである。
幾つかの実施態様において、Ar1は、アザアリール基である。幾つかの実施態様において、Ar1は、2−、3−又は4−ピリジン又は置換2−、3−、又は4−ピリジンである。
【0054】
幾つかの実施態様において、L1は不存在である。幾つかの実施態様において、L2は、存在して、C2アルキレン基である。
幾つかの実施態様において、化学式(II)で表わされる化合物は、化学式(II):
【化2】
で表わされる構造を有し、式中、
X、X2及びX3は、それぞれC又はCHである。
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6のそれぞれは、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群より独立に選択され、また各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、夫々独立に、N又はCR12であり、式中、各R12は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び、次の構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4は、NR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;又は
式中、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は以下の構造:
【化4】
から選択され、式中、
各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4は、NR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び、
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアルキル基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;又は式中、R10及び1個のR13はともにアルキレン基であり;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つは、Nであり;
又はこれらの医薬的に許容された塩である。
【0055】
幾つかの実施態様において、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の2個は、Nである。
幾つかの実施態様において、X1は、Oであり、XはCである。
幾つかの実施態様において、Ar2は、次の構造:
【化5】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は、化学式(III)で表わされる構造:
【化6】
を有する。
【0056】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11のそれぞれは、水素原子である。
幾つかの実施態様において、Y1,Y2,Y3,Y4及びY5の中の少なくとも1つは、CR13であり、式中、R13は、構造:
【化3】
を有する置換基である。
幾つかの実施態様において、X5は、Oであり、X6は、水素原子、アルキル基、アラルキル基及びアリール基より選択され、またAr2は、アシルアミノ置換アリール基又はヘテロアリール基である。
【0057】
幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIa):
【化14】
で表わされる化合物である。
幾つかの実施態様において、化学式(IIIa)で表わされる化合物のA3はNである。幾つかの実施態様において、A3はNであり、A1,A2,A4及びA5のそれぞれは、CR12であり、またY1,Y2,Y4及びY5は、CR13である。
【0058】
幾つかの実施態様において、化学式(III)で表わされる化合物は、少なくとも1個のハロ置換体を含む。幾つかの実施態様において、該ハロ置換体はClである。幾つかの実施態様において、該ハロ置換体は、アルケンに付加する炭素に対してオルソ位置である。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIb):
【化15】
で表わされる化合物である。幾つかの実施態様において、化学式(IIIb)で表わされる化合物のA4及びA5の中の1つは、Nである。幾つかの実施態様において、A4及びA5の中の一つは、Nであり、A1,A2及びA3のそれぞれは、CR12であり、またY2,Y3,Y4及びY5のそれぞれは、CR13である。
【0059】
幾つかの実施態様において、ハロ置換体は、アルケンに付加する炭素に対してパラ位置である。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIc):
【化16】
で表わされる化合物である。幾つかの実施態様において、化学式(IIIc)で表わされる化合物のA2は、Nである。幾つかの実施態様において、A2はNであり、A1,A3,A4及びA5のそれぞれは、CR12であり、またY1,Y2,Y4及びY5のそれぞれは、CR13である。
【0060】
幾つかの実施態様において、化学式(III)で表わされる化合物は、2個のアザアリール基を含む。幾つかの実施態様において、該化合物は、1個の4−ピリジル基及び1個の3−ピリジル基を含む。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIId):
【化17】
で表わされる化合物である。幾つかの実施態様において、A1,A2,A4及びA5のそれぞれは、CR12であり、またY1,Y3,Y4及びY5のそれぞれは、CR13である。
【0061】
幾つかの実施態様において、化学式(III)で表わされる化合物は、以下の化学式:
【化7】
からなる群から選択される。
【0062】
幾つかの実施態様において、化学式(II)で表わされる化合物は、Ar2が:
【化8】
である化合物であり、式中、
X、X2及びX3のそれぞれは、Cであり;
X1はO、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群より独立に選択され;及び化学式(II)で表わされる化合物は、化学式(IV):
【化9】
で表わされる構造を有する。
【0063】
幾つかの実施態様において、化学式(IV)で表わされる化合物は、2個のアザアリール基を含む。幾つかの実施態様において、Y6及びA3のそれぞれは、Nである。幾つかの実施態様において、A1,A2,A4及びA5のそれぞれは、CR12であり、またY7,Y8,Y9,Y10、Y11及びY12のそれぞれは、CR13である。幾つかの実施態様において化学式(IV)で表わされる化合物は:
【化10】
である。
【0064】
幾つかの実施態様において、化学式(II)で表わされる化合物は、Ar2が:
【化11】
であり、
X、X2及びX3のそれぞれがCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び該化合物は、化学式(V):
【化12】
で表わされる構造を有する。
【0065】
幾つかの実施態様において、A3はNである。幾つかの実施態様において、化学式(V)で表わされる化合物は:
【化13】
である。
【0066】
幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、in vitroにおいて、PFKFB3を阻害するために用いることができて、又はそうでなければ、PFKFB3に干渉又は相互作用する為に用いることができる。幾つかの実施態様において、該化合物は、PFKFB3の競合的阻害剤である。幾つかの実施態様において、該化合物は、細胞又はPFKFB3を含む細胞抽出液と接触させるために用いることができる。幾つかの実施態様において、該化合物は、組織、組織抽出物、又は血液試料のような他の生物由来の試料と接触するために用いることができる。幾つかの実施態様において、哺乳動物又はトリのような生存被験者にPFKFB3が存在する場合、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、in vivoでPFKFB3に接触するために用いることができる。幾つかの実施態様において、哺乳動物はヒトである。PFKFB3活性を阻害することにより、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、又はこれらの医薬学的許容物を用いて、細胞中の解糖フラックスを減少させることができて、幾つかの場合特に腫瘍細胞の場合、解糖フラックスの減少により、細胞内乳酸塩及びフルクトース−2,6−ビスリン酸塩レベルを減少させる。
【0067】
IV.医薬処方物
化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、これらの医薬学的に許容された塩類、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物に対応する前駆薬剤及びこれらの医薬学的に許容された塩類の全ては、本明細書において、"活性化合物"と呼ぶ。前述の活性化合物を含む医薬処方物もまた、本明細書に提供されている。本明細書において記載されるように、これらの医薬処方物は、医薬学的に許容された担体の中に、活性化合物を含む。以下により詳細に考察する様に、医薬処方物を、経口、静脈内、又は噴霧剤として投与することができる。また、本明細書に開示した発明は、凍結乾燥し、再構成して投与のための医薬学的に許容された処方物(ヒトに対して医薬学的に許容された処方物も含め)を作成できる、活性化合物を提供する。
【0068】
活性化合物の使用は、本明細書に記載された実施態様の範囲内であるが、全ての活性化合物の治療上有効な投与量は、化合物によって、また患者によって異なり、また患者の健康状態及び投与経路にも依存するであろう。一般的な計画としては、約0.1から約50mg/kgの投与量は、治療効果を有するであろう、但しここで全ての重量は、塩類を用いた場合も含めて、活性化合物の重量に基づいて計算されてある。高濃度における毒性を考慮して、静脈内投与量を、約10mg/kgまでのように、低濃度に制限することができる、但しここで全ての重量は、塩類を用いた場合も含めて、活性化合物の重量に基づいて計算されてある。経口投与の場合、約10mg/kgから約50mg/kgの投与量を採用することができる。一般的に、筋肉内注射に対して、約0.5mg/kgから約5mg/kgの投与量を採用することができる。静脈内又は経口投与に対して、好ましい化合物の投与量は、1μmol/kgから50μmol/kgであり、より好ましくは、22μmol/kg及び33μmol/kgである。治療期間は、通常、2から3週間の期間に対して1日1回であるか、又は健康状態が本質的に改善されるまでである。低頻度で低濃度の投与量を、感染の再発防止、又は発生率の低下のために、予防的に用いることができる。
【0069】
本明細書に開示した方法に従い、本明細書に記載した医薬学的に活性化合物は固体又は液体として、経口的に投与できて、又は溶液、懸濁液又は乳状液として、筋肉内又は静脈内に投与することができる。あるいは、該化合物又は塩を、リポソーム懸濁物として、吸入、静脈内、又は筋肉内に投与することができる。吸入により投与する場合、活性化合物又は塩は、粒子サイズが約0.5から約5μm及び好ましくは、約1から約2μmである、複数の固体粒子又はドロップの形をとる必要がある。
【0070】
静脈内又は筋肉内注射に適した医薬処方物は、本明細書に提供されたさらなる実施態様である。該医薬処方物は、本明細書に記載した、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、本明細書に記載した前駆薬剤、又は医薬学的に許容されたこれらの塩を、全ての医薬学的に許容された担体の中に含む。もし溶液が望まれるならば、水溶性化合物又は塩に関しては、水が選択された担体である。水溶性の化合物又は塩に関して、グリセロール、プロピレングリコール、ポリエチレングリコール又はこれらの混合物のような有機溶媒が適切であることができる。後者の例において、有機溶媒は、多量の水を含むことができる。上記二例における溶液は、当業者には既知の適切な手段で、一般的には、0.22μmフィルターを通して、無菌化できる。無菌化に続いて、該溶液を、脱パイロジェン化したガラスバイアルのような、適切な容器に分配することができる。この分配を、好ましくは、無菌法で行う。次ぎに殺菌した栓をバイアル上に置くことができて、必要に応じて、バイアル内容物を凍結乾燥できる。
【0071】
化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物又はこれらの塩又は前駆薬剤に加えて、医薬処方物は、pH調整添加剤のような他の添加剤を含むことができる。特に、有用なpH調整試薬としては、塩酸のような酸、乳酸ナトリウム、酢酸ナトリウム、リン酸ナトリウム、クエン酸ナトリウム、ホウ酸ナトリウム、又はグルコン酸ナトリウムのような、塩又は緩衝剤が含まれる。更に、該処方物は、抗菌保存剤を含むことができる。有用な抗菌保存剤としては、メチルパラベン、プロピルパラベン及びベンジルアルコールが含まれる。抗菌保存剤は、一般的に、処方物が多回数投与量使用を表すバイアルに入れられる場合に、使われる。本明細書に記載される医薬処方物は、当業者に既知の技術を用いて凍結乾燥される。
【0072】
本明細書に記載する発明の他の実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、又はこれらの塩を含む注射可能な、安定な、滅菌した処方物を提供する。該化合物又は塩は、密封した凍結乾燥物の形で提供される。該化合物又は塩を、適切な医薬学的に許容された担体と共に再構成可能であり、患者への注射に適した液体処方物を形成することができる凍結乾燥物として提供する。一般的に一回投与の形式は、約10mgから約10gの化合物塩を含む。化合物又は塩が、実質的に、非水溶性の場合、生理学的に許容された、十分量の乳化剤が十分量使われ、該化合物又は塩を水溶性担体に乳化することができる。このような有用な乳化剤としては、ホスファチジルコリンがある。
【0073】
他の医薬処方物を、本明細書に開示した、水溶性塩基乳化剤のような、非水溶性化合物又はこれらの塩から作成することができる。このような例の場合、該処方物は、十分量の医薬学的に許容された乳化剤を含み、必要量の化合物又はこれらの塩を乳化する。特に有用な乳化試薬は、ホスファチジルコリン及びレシチンを含む。
【0074】
本明細書に提供された更なる実施態様には、本明細書に開示した活性化合物のリポソーム処方物が含まれる。リポソーム懸濁物を作成する技術は、既知の技法である。化合物が、水溶性塩である場合、従来のリポソーム技術を用いて、脂質小胞にこれを取り込むことができる。このような例において、活性化合物が水溶性であるために、該活性化合物は、実質的に親水性中心、又はリポソームのコアに取り込まれる。用いられる脂質層は、従来型のどの様な組成も可能であり、コレステロールを含む、又は含めないことができる。注目する活性化合物が非水溶性の場合、再び、従来型のリポソーム形成技術を用いて、塩は、リポソームの構造を作る疎水性脂質二重膜内に、実質的に取り込まれる。いずれにしても標準的超音波及びホモジナイズ技術により、作成されたリポソームのサイズを、小さくすることができる。
【0075】
本明細書に開示された活性化合物を含むリポソーム処方物は、凍結乾燥により、凍結乾燥物を作ることができて、これは、水のような医薬学的に許容された担体を用いて、再構成して、リポソーム懸濁物を再生することができる。
【0076】
また吸入による煙霧剤として投与に適した医薬処方物として提供される。これらの処方物は、本明細書に記載した望ましい化合物、又はこれらの塩、又は化合物又は塩の複数の固体粒子の溶液又は懸濁物を含む。望みの処方物を、小さな容器に入れ、霧状にすることができる。噴霧化は、圧縮空気又は超音波エネルギーによって行われ、化合物又は塩を含む複数の液体ドロップ又は固体粒子を形成する。液体ドロップ又は固体粒子のサイズは、約0.5から約10μmの範囲、より好ましくは、約0.5から約5μmの範囲にあるべきである。固体粒子は、微粉化のような既知のいかなる方法によっても、固体化合物又はこれらの塩を処理して得ることができる。最も好ましくは、固体粒子又はドロップのサイズは、約1から約2μmであろう。この点において、この目的を達するために、商品化された噴霧器を用いることができる。その開示物は本明細書参照文献にその全体が取り込まれているが、合衆国特許番号5628984で公にされた方法で吸入可能な粒子の煙霧懸濁液を通して、該化合物を投与することができる。
【0077】
煙霧剤としての投与に適した医薬処方物が液体の場合、該処方物は、水を含む担体中の水溶性活性化合物を含むであろう。界面活性剤は、存在できて、処方物の表面張力を十分に低下させて、噴霧化した場合、望ましいサイズ範囲の小滴をもたらす。
【0078】
既述の様に、水溶性及び非水溶性活性化合物が提供される。本明細書で用いるように、用語"水溶性"は約50mg/mL又はそれ以上の量が水に溶ける全ての化合物を定義するつもりである。また、本明細書で用いるように、用語"非水溶性"は約20mg/mL以下の水への溶解度を有する全ての組成物を定義するつもりである。幾つかの実施態様において、水溶性化合物又は塩が好ましいが、他方、他の実施態様において、非水溶性化合物又は塩が好ましい。
【0079】
V.PFKFB3阻害剤で細胞増殖阻害し、癌を治療する方法
本明細書に開示した発明は、細胞増殖を阻害するための方法及び組成物を提供する。より詳細には、本明細書に開示した発明は、PFKFB3活性を阻害し、解糖フラックスを中断させる方法を提供する。解糖活性を中断することにより、細胞増殖は停止し、また、幾つかの場合、アポトーシスが引き起こされる。従って、本明細書に開示した発明は、望ましくない解糖活性及び/又は望ましくない細胞増殖が関与する、癌を含む病気を治療する方法を提供する。
【0080】
幾つかの実施態様において、細胞増殖の阻害又は癌の治療のための方法は、本明細書に記載した活性化合物を必要とする患者に対する、活性化合物の投与を含む。既述のように、これらの活性化合物は、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、これらの前駆薬剤及び医薬学的に許容されたこれら化合物と前駆薬剤の塩を含む。幾つかの実施態様において、該活性化合物は、既述のように、医薬処方物の中に存在する。
【0081】
本明細書に開示した化合物は、皮膚癌、結合組織癌、脂肪腫、乳癌、肺癌、胃癌、膵臓癌、卵巣癌、子宮頸癌、子宮癌、肛門性器癌、腎臓癌、膀胱癌、大腸癌、前立腺癌、中枢神経癌(CNS),網膜癌、血液及びリンパ液癌を含む、多種の腫瘍及び癌に対する治療を提供することができる。
【0082】
癌の治療に関係して、本明細書で定義される"有効量"、は、癌細胞又は腫瘍の増殖を減らす、低減する、阻害する、又は他の方法で排除する量である。幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、患者の身体の特に病期に冒された、部位又は領域に局部的に輸送することができる。このような治療がより適切と考えられた幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物は、全身的に投与できる。例えば、該化合物を経口又は静脈内に投与できる。
【0083】
さらに、癌の治療に対する治療上の恩恵は、PFKFB3阻害化合物又は、本明細書で開示した他の化合物と、1又は2以上の他の抗癌剤又は治療と組み合わせた治療により実現することができる。その様な組合せの選択は、病気の種類、年齢及び患者の一般的健康状態、病気進行の攻撃性及び組合せに含められた試薬に対する患者の耐久力を含む、しかしこれらに限定されない、様々な因子に依存するであろう。例えば、PFKFB3阻害剤は、腫瘍サイズを減少させる上で有効な他の試薬及び治療計画(例えば、放射線、手術、化学療法、ホルモン治療及び/又は遺伝子治療)と組み合わせることができる。さらに、幾つかの実施態様において、PFKFB3阻害剤化合物を、病気の副次的影響又は治療試薬の一つの副作用を治療する、1又は2以上の薬剤と組み合わせることが望ましくできる、例えば、患者に、鎮痛剤、又は患者自身の免疫応答を高めるに有効な薬剤(例えば、コロニー刺激因子)を提供することである。
【0084】
幾つかの実施態様において、本明細書に開示した方法及び化合物は、我々が本明細書では"好気性治療"として、全体的に又は個別的に表す、好気性の急速に増殖する細胞に焦点を当てた化学療法及び/又は放射線療法と結びつけて用いることができる。幾つかの実施態様において、本明細書で開示したPFKFB3阻害剤の使用は、通常最も抵抗性であり、また従って、好気的治療を用いてでは根絶することが最も難しい、固形腫瘍の内部コアに見られる嫌気的な遅速増殖腫瘍細胞を選択的に殺すことにより、癌治療の有効性を付け加える。
【0085】
従って、本明細書で記載された発明の1又は2以上のPFKFB3阻害化合物と組み合わせて、"抗癌剤"又は"化学療法剤"としてもまた記載される様々な化学化合物を用いることができる。このような化合物としては、アルキル化剤、DNAインターカレーター、蛋白質合成阻害剤、DNA又はRNA合成阻害剤、DNA塩基類似体、トポイソメラーゼ阻害剤、抗脈管形成薬剤及びテロメラーゼ阻害剤又はテロメアのDNA結合化合物が含まれるが、これらに制限されない。例えば、適切なアルキル化剤としては、ブスルファン、インプロスルファン及びピポスルファンのようなスルホン酸アルキル;ベンゾジゼパ、カルボコン、メツレデパ及びウレデパのようなアジリジン;アルトレタミン、トリエチレンメラミン、トリエチレンホスホラミド、トリエチレンティオホスホラミド及びトリメチルオルメラミンのようなエチレニミン及びメチルメラミン;クロランブシル、クロルナファジン、サイクロホスファミド、エストラムスティン、イホスファミド、メクロレタミン、メクロレタミンオキシド塩酸塩、メルファラン、ノブエンビチン(Novembichine)、フェネステリン、プレドニムスチン、トロホスファミド及びウラシルマスタードのような、ナイトロジェンマスタード;カルムスチン、クロロゾトシン、フォテムスチン、ロムスチン、ニムスチン及びラニムスチンのようなニトロソウレアが含まれる。
【0086】
癌の治療に用いられる抗生物質としては、ダクチノマイシン、ダウノルビシン、ドクソルビシン、イダルビシン、硫酸ブレオマイシン、マイトマイシン、プリカマイシン及びストレプトゾシンが含まれる。化学療法に使われる抗代謝薬剤としては、メルカプトプリン、チオグアニン、クラドリビン、リン酸フルダラビン、フルオロウラシル(5−FU),フロクスウリジン、シタラビン、ペントスタチン、メトトレクセート及びアザチオプリン、アシクロビル、アデニンβ−1−D−アラビノシド、アメトプテリン、アミノプテリン、2−アミノプリン、アフィジコリン、8−アザグアニン、アザセリン、6−アザウラシル、2'−アジド−2'−デオキシヌクレオシド、5−ブロモデオキシシチジン、シトシンβ−1−D−アラビノシド、ジアゾオキシノルロイシン、ジデオキシヌクレオシド、5−フルオロデオキシシチジン、5−フルオロデオキシウリジン及びヒドロキシウレアが含まれる。
【0087】
化学療法のための蛋白質合成阻害剤としては、アブリン、オーリントリカルボン酸、クロランフェニコール、コリシンE3,シクロヘキシミド、ジフテリア毒素、エデインA,エメチン、エリスロマイシン、エチオニン、フッ化物、5−フルオロトリプトファン、フシジン酸、ジホスホン酸グアニリルメチレン及びジホスホン酸グアニリルイミド、カナマイシン、カスガマイシン、キッロマイシン及びO−メチルスレオニンがある。さらなる蛋白質合成阻害剤としては、モデシン、ネオマイシン、ノルバリン、パクタマイシン、パラモマイシン、ピューロマイシン、リシン、シガ毒素、ショウドマイシン、スパルソマイシン、スペクチノマイシン、ストレプトマイシン、テトラサイクリン、チオストレプトン及びトリメトプリムがある。硫酸ジメチル;マイトマイシンC;ナイトロジェンマスタード及びサルファーマスタードのようなアルキル化剤;アクリジン色素、アクチノマイシン、アドリアマイシン、アントラセン、ベンゾピレン、エシジウムブロマイド、2ヨウ化プロピジウムのようなインターカレート試薬;ジスタマイシン及びネトロプシンのような薬剤;等のDNA合成阻害剤もまた、化学療法組成物中で、本明細書に開示した発明の化合物と組み合わせることができる。クメルマイシン、ナリジクス酸、ノボビオシン及びオキソリン酸のようなトポイソメラーゼ阻害剤;コルセミド、コルヒチン、ビンブラスチン及びビンクリスチンを含む細胞分裂阻害剤;及びアクチノマイシンD,α−アマニチン及び他のカビのアマトキシン、コルディセピン(3'−デオキシアデノシン)、ジクロロリボフラノシルベンズイミダゾール、リファンピシン、ストレプトバリシン及びストレプトリジギンを含むRNA合成阻害剤もまた、本明細書に開示した発明のPFKFB3阻害剤化合物と組み合わせて、適切な癌治療に提供することができる。
【0088】
従って、本明細書に開示した発明のPFKFB3阻害剤と組合せて治療に用いることができる現在の化学療法薬剤としては、アドリアマイシン、5−フルオロウラシル(5FU),エトポシド、カンプトテシン、アクチノマイシン−D,マイトマイシン、シスプラチン、過酸化水素、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、イホスファミド、メルファラン、クロランブシル、ビサルファン、ニトロソウレア、ダクチノマイシン、ダウノルビシン、ドクソルビシン、ブレオマイシン、プリコマイシン、タモキシフェン、タキソール、トランスプラチナ、ビンブラスチン及びメトトレクセート等々がある。
【0089】
PFKFB3阻害化合物及び、他の化学療法薬剤のような、他の治療薬剤を含む組合せ治療は、細胞を、PFKFB3阻害剤及び他の薬剤と同時に接触させることにより達成することができる。このような組合せ治療は、該細胞を、両薬剤を含む単一組成物又は医薬処方物を接触させる、又は該細胞を、一方の組成物がPFKFB3阻害剤を含み、また他方が他の薬剤を含む、2種の別個の組成物又は処方物と同時に接触させることで達成することができる。
【0090】
あるいは、PFKFB3阻害化合物を用いる治療は、分から週単位の範囲の間隔で、他の薬剤治療の前、また後に適用することができる。他の薬剤及びPFKFB3阻害剤治療が、細胞に対して別々に行われる実施態様において、該薬剤及びPFKFB3阻害剤治療が、細胞上に有利な組合せ効果を示すことができる様に一般的に守られることは、両治療の間に長期の時間間隔が経ってしまいそれぞれの治療の間で失効しないことである。この様な場合、人は細胞を約12〜24時間以内に及び任意に、約6〜12時間以内に、両治療法に接触させることになる。幾つかの状況において、治療の期間をかなり延長することが望まれるが、それぞれの投与間隔を、数日間(2,3,4,5,6又は7日間)から数週間(1,2,3,4,5,6,7又は8週間)に延長される。また、幾つかの状況下で、PFKFB3阻害剤又は他薬剤の1回以上の投与が望まれる。
【0091】
他の実施態様において、本明細書に開示した発明のPFKFB3阻害剤化合物、又はPFKFB3阻害剤と組み合わせて用いる他の抗癌化合物は、単一クローン抗体(例えば、齧歯動物又はヒト単一クローン抗体)のような標的薬剤と結合した細胞毒性試薬と組み合わされ、又はこの細胞性毒試薬と共有結合する。後者の組合せにより、より大きな特異性を持って癌細胞に細胞毒性薬剤を導入することができることが分かるであろう。従って、細胞毒性薬剤の活性型(例えば、フリーな形)は、抗体により標的とされた細胞の中にのみ存在することになろう。
【0092】
他の癌治療法を、PFKFB3阻害剤化合物の投与と組み合わせて用いることができる。例えば、本明細書に開示した発明のPFKFB3阻害剤化合物は、さらに外科的に取り除いた部分、又は全ての癌の増殖に対する試みを含む治療コースの一部として用いることができる。例えば、本明細書に開示した発明のPFKFB3阻害剤を、患者の外科的治療の後に投与して、全ての残された癌、又は転移した細胞を治療することができる。本明細書で開示した発明のPFKFB3阻害薬剤による治療は、手術に先んじて行い、腫瘍のサイズを縮退させ、切除する組織量を減らす試みができて、これにより手術をより非浸襲的及び非衝撃的にする。
【0093】
本明細書に開示した発明のPFKFB3阻害薬剤による癌の治療は、放射線治療作用物を伴う1又は2以上の治療コースをさらに含み、DNA損傷をもたらすことができる。放射線治療作用物には、例えば、ガンマ線照射、X−線、UV−照射、マイクロ波、電子発光、放射性同位元素、等々がある。治療は、上記の形の放射線を局所的腫瘍部位に照射することで達成することができる。
【0094】
組合せ治療は、また、腫瘍細胞の表面に見出される腫瘍抗原に向けられた免疫治療を含めることができる。本明細書に開示したPFKFB3阻害剤で癌を治療することは、さらに、遺伝子治療に基づく治療と組み合わせることができて、p53,p16,p21、Rb,APC,DCC,NF-1,NF-2,BRCA2,FHIT,WT-1,MEN-I,MEN-II,BRCA1,VHL,FCC,MCC,ras,myc,neu,raf,erb,src,fms,jun,trk,ret,gsp,hst,bcl及びablのような、腫瘍遺伝子及び/又は細胞周期調節遺伝子、これらはしばしば癌組織中の正常細胞対応物の突然変異バージョンであるが、を標的とすることができる。
【0095】
本明細書に開示した発明のPFKFB3阻害剤化合物は、試験を行い、癌細胞の増殖阻害能、癌細胞のアポトーシス誘導能、腫瘍負荷の減少能及び転移阻害能を測定することができる。例えば、MTT試験により、細胞増殖を測定することができる。MTT試験により測定した増殖試験は、広く既知である。MTT試験において、癌細胞を様々な濃度の抗ガン化合物とインキュベートし、細胞生存能をテトラゾリウム塩、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロム化物(MTT)の着色したホルマザン塩の形成を測定して決める。細胞死及び、又は細胞増殖を測定する他の既知の試験もまた用いることができる。
【0096】
例えば、OVCAR−5腫瘍細胞をヌードマウスに移植したマウス異種移植モデルを用いてin vivo試験を行うことができて、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物で処理したこのマウスは、平均して、最初の投与後ある時期の間増殖するが、処理を続けると、腫瘍塊が縮小することが期待される。それに対して、対照(例えばDMSO)処理を行ったマウスは、腫瘍塊が増殖し続けると期待される。本明細書に開示した化合物の抗癌効果を測定する更なる方法を、本明細書の以下の実施例において記載する。
【0097】
VI.スクリーニング方法
本明細書に開示した発明は、化合物の持つPFKFB3への妨害(例えば、阻害又は競合的な阻害)能についてスクリーニングする方法を提供する。より詳細には、該方法は、(1)PFKFB3のF6P結合ポケットを含むPFKFB3の3Dモデルの提供;(2)化合物(例えば、小分子)の3Dモデル;及び(3)PFKFB3モデルのF6P結合ポケットの中の1又は2以上の溶媒接触表面との相互作用能を有する化合物のスクリーニングを含む。幾つかの実施態様において、該方法は、計算機(例えば、コンピューター内での)を用いて行うことができる。幾つかの実施態様において、該方法は、(a)複数の化合物(例えば、化合物のライブラリー又はデータベース)の3Dモデルの提供及び(b)各化合物の個別のスクリーニング及びその後、(c)複数の化合物中のある一個の化合物のF6P結合ポケットとの相互作用能と、他の化合物の相互作用能との比較;を含むことができる。
【0098】
従って、一態様において、化合物をスクリーニングする方法は、一般的に、PFKFB3のF6P結合ポケットの計算機モデルに対する選択した化合物(化学的実体)が持つ結合能を、計算機によって評価することを含む。例えば、この方法は、(a)選択した化合物とPFKFB3のF6P結合ポケットの間の嵌め込み操作を行うための計算機によるアプローチを用いるステップ;(b)嵌め込み操作の結果を解析して、化合物と結合ポケットの間の結合を定量化するステップを含む。幾つかの実施態様において、目視検査、又は計算機を用いたドッキング(結合)実験を用いて、ある分子、又は分子のライブラリーを、PFKFB3のF6P結合ポケットとの結合親和性について試験することができる。
【0099】
幾つかの実施態様において、PFKFB3の計算機モデルは、PFKFB3アイソザイムの既知のX−線構造から導かれることができる。例えば、PFKFB3のあるモデルは、PFKFB3の残基配列をヒト肝PFKFB1及びラット精巣PFKFB4アイソザイムの同型配列に揃えて並べることにより導くことができる。PFKFB3自体の結晶構造もまた記載されている。Kimほか,J.Biol.Chem.,281(5)2939-2944(2006)を参照されたい。
【0100】
用語"結合ポケット"は、その形状の結果として、他の化合物(例えば、F6P又は阻害剤)と都合良く結合する分子又は、分子複合体のある領域を表す。用語"ポケット"は、裂け目、チャンネル、又は部位を含む、がこれらに制限されない。結合ポケットの形状は、大体は化合物との結合前にあらかじめ形成され、化合物との結合と同時に形成され、又は分子の異なる結合ポケットへ他の化学物質が結合し、このことが逆に結合ポケットの形状の変化を誘導する、ことにより形成されるかも知れない。
【0101】
用語"ドッキング(結合)"は、ディスタンスジオメトリー又はエネルギーに基づいて、結合ポケット、領域、分子、又は分子複合体又はこれらの部分における、化合物の配向;回転;平行移動を表す。ドッキングは、ディスタンスジオメトリー法によって行うことができて、この方法は結合ポケット、領域、分子又は分子複合体又はこれらの部分の球状プローブの中心の集合にマッチ(適合)する化合物の原子集合を見出す。Mengほか,J.Comp.Chem.,4,505-524(1992)を参照されたい。球状プローブ中心は、結合ポケット、領域、分子又は分子複合体又はこれらの部分において、原子(水素原子を除く)からの所与の距離に追加の半径を加えることで作り出される。リアルタイム相互作用エネルギー計算、エネルギー最小化、又は剛体最小化(Gschwend ほか,J.Mol Recognition,9:175-186(1996)参照)を行って、化合物を配向させて、ドッキングを容易にすることができる。例えば、相互作用をするドッキング実験を計画して、最小抵抗の経路に従うように設計することができる。相互作用的ドッキング実験において、使用者がエネルギー増加の方向へ動くならば、該システムは、その運動に抵抗する。しかしながら、もし該使用者が、エネルギー減少の方へ動くなら、該システムは、応答性が増加して動くことを好む。(Cohen,ほか,J.Med.Chem.,33,889-894(1990))ドッキングはまた、分子親和性ポテンシャルを用いて、MonteCarlo探索技術と早いエネルギー評価を組み合わせることにより為し遂げることができる。Goodsell及びOlson,Proteins:Structure,Function and Genetics 8,195-202 (1990)を参照されたい。ドッキング機能を行うソフトウェアプログラムとしては、MATCHMOL(Cory,ほか.J.Mol Graphics,2,39(1984));MOLFIT(Redington,Comput Chem.16,217(1992)参照)及びDOCK(Mengほか、上記参照)が含まれる、がこれらに制限されない。
【0102】
用語"3次元(D)構造の創出"又は"3次元表示の創出"は、構造座標のリストを3次元の構造モデル又はグラフによる表出に変換することを表す。これは、商業的に、又は公共で入手可能なソフトウェアにより行われる。従って、分子又は分子複合体の3次元構造のモデルは、構造座標が与えられ、また正しいソフトウェアをインストールした計算機により、計算機のスクリーン上に構築することができる。該3次元構造を表示すること、又は計算機モデリング又は当て嵌め操作を行うために用いることができる。さらに、表示されたモデルは無くとも、構造座標それ自体を用いて、計算機に基づくモデリング及び当て嵌め操作を行うことができる。
【0103】
幾つかの実施態様において化合物が持つ、結合ポケット又は溶媒接触表面との相互作用能を定量することができる。用語"接触スコア"は、化合物及び結合ポケットの間での形状相補性の指標を表し、これは、化合物の原子の全て又は部分及び結合ポケット内に結合したリガンドの原子の全て又は部分の間の最小自乗スーパーインポジション(重ね合わせ;superimposition)から得られる、RMSD値と相関する。接触スコア又はRMSD値により、ドッキング過程を容易にすることができる。例えば、もし化合物が高いRMSDを有する位置に動くならば、システムはその動きに抵抗するであろう。化合物の配置の集合は、接触スコアにより、ランク付けすることができる。より低RMSD値はより高い接触スコアを与えるであろう。Mengほか.J.Comp.Chem.,4,505−524(1992)を参照されたい。
【0104】
用語"二乗平均偏差"又は"RMSD"は、平均値からの変位の二乗の算術平均の平方根を意味する。これは、傾向又は対象からの変位又は偏差を表現する一方法である。用語"構造座標"は、結晶型のタンパク質又はタンパク質複合体の原子(散乱中心)による、X−線の単色光線の回折から得られたパターンに関係する数式から導かれたCartesian(デカルト)座標を表す。回折データを用いて、結晶の繰り返し単位の電子密度地図を計算する。次ぎに、電子密度地図を用いて、分子又は分子複合体の個々の原子の位置を確定する。
【0105】
本明細書の発明を閲覧した当業者には理解できることであるが、計算機スクリーニング法のために、様々な小分子(即ち、1000原子質量単位(amu)又はそれ以下の分子量を有する化合物、750amu又はそれ以下、又は500amu又はそれ以下)ライブラリーが、入手できる。用いることができる化学構造データベースは、ACD(Molecular Designs Ltd,現在 Elsevier MDL, San Ramon, California, USA),NCI(National Cancer Institute, Fredrick, Maryland, USA), CCDC(Cambridge Crystallographic Data Center, Cambridge,英国),CAST(Chemical Abstract Service, Columbus, Ohio, USA), Maybridge(Maybridge Chemical Company Ltd, Tintagel,英国),Aldrich(Aldrich Chemical Company, Milwaukee, Wisconsin)及び Directory of Natural Products(天然物要覧、Chapman & Hall, London,英国)を含むが、これらに制限されない。
【0106】
CONCORD(Tripos Associates,St.Louis,Missouri, USA)又はDB-Converter(Molecular Simulations Ltd.,San Leandro,California, USA)のような計算機プログラムを用いて、2次元に表示されたデータセットを3Dに表示されたデータセットに変換するために用いることができる。望みのファーマコフォア(薬理原子団)を有する分子を同定するための3Dデタベースを探索するために適したプログラムとしては、MACCS-3D及び ISIS/3D(Molecular Design Ltd.,現在 Elsevier MDL, San Ramon California, USA) ChemDBS-3D(Chemical Design Ltd., Oxford,英国)及びSybyl/3 DB Unity(Tripos Associates, St.Louis, Missouri, USA)がある。本明細書で用いるように、"ファーマコフォア"又は"薬作用発生パターン"は、結合に対して重要であると信じられている、化合物の特徴の幾何学的配置である。
【0107】
化合物はまた、細胞をベースとしたスクリーニング法を用いて、PFKFB3の阻害能についてスクリーニングすることができる。従って、幾つかの実施態様において細胞増殖停止能及び/又はPFKFB3発現細胞における乳酸塩産生能について、化合物をスクリーニングすることができる。幾つかの実施態様において、細胞をベースとしたスクリーニング法を用いて、前述のin silicoスクリーニング法で同定した化合物のPFKFB3阻害能をさらに証明するために化合物をスクリーニングすることができる。
【実施例】
【0108】
一般的方法
対照と3PO処理の間の増殖阻害、乳酸塩産生及びin vivo研究についての統計的有意さは、Graph Pad Prism Version3.0(Graph Pad Software,San Diego,California, USA)を用いた2試料t−検定により定められた。p値<0.01は、統計的に有意であると考える。
【0109】
実施例1
PFKFB3分子モデリング及び化合物スクリーニング
計算機によるモデリングは、Silicon Graphics Array(1TB以上の記憶装置を備えた71×R12000及びR140000プロセッサー),32P R14000 500MHz Origin 2000サーバー及び2P R12000 300MHz OCTANEグラフィックワークステーション(SGI,Sunnyvale,California, USA)を用いて行った。PFKFB3ホモロジーモデルは、構造上の原型としてラット精巣PFKFB4(PDBコード1B1F)アイソザイムのX線構造を用いた。ClustalWを用いてアラインメントを作り出した。Chennaほか,Nucleic Acids Res.,31,3497-3500(2003)を参照されたい。4種のホモロジーモデルをModeller(Sali及びBlundell, J.Mol.Biol.,234,779-815(1993)参照)を用いて作成し、またPFKFB3結合部位(Chesney ほか,Proc.Natl.Acad Sci,96,3047-3052(1999)及びBertrandほか.,Eur.J.Biochem.,254,490-496(1998)参照)を最も良く再現した構造を更なる使用のために選択した。PFKFB3のためのリガンド結合及びタンパク質活性に重要な残基(Chesney ほか,Proc.Natl.Acad.Sci,96,3047-3052(1999)及びBertrandほか,Eur.J.Biochem.,254,490-496(1998)参照)は、コンセンサス(共通)構造における等価な残基数と相関した。該モデルをInsightII (Accelrys, San Diego California, USA)に読み込み、また3種の重要な残基Arg66,Tyr161及びThr94を仮想スクリーニング運転の重心標的として選択した。PFKFB3ホモロジーモデルの描画を図2A,2B及び2Cに示す。
【0110】
ホモロジーモデル(配列番号1)に対するアミノ酸配列は、Homo sapiens由来の6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビホスファターゼ3(GENEBANK(R)登録番号NP_004557)のアミノ酸配列34〜466のアミノ酸に対応する。配列番号1は、GENBANK(R)登録番号、NM_004566(配列番号2)のヌクレオチド429〜1727によりコードされている。
【0111】
配列番号1は:
SPTVIVMVGLPARGKTYISKKLTRYLNWIGVPTKVFNVGEYRREAVKQYSSYNFFRPDNEEAMKVRKQCALAALRDVKSYLAKEGGQIAVFDATNTTRERRHMILHFAKENDFKAFFIESVCDDPTVVASNIMEVKISSPDYKDCNSAEAMDDFMKRISCYEASYQPLDPDKCDRDLSLIKVIDVGRRFLVNRVQDHIQSRIVYYLMNIHVQPRTIYLCRHGENEHNLQGRIGGDSGLSSRGKKFASALSKFVEEQNLKDLRVWTSQLKSTIQTAEALRLPYEQWKALNEIDAGVCEELTYEEIRDTYPEEYALREQDKYYYRYPTGESYQDLVQRLEPVIMELERQENVLVICHQAVLRCLLAYFLDKSAEEMPYLKCPLHTVLKLTPVAYGCRVESIYLNVESVCTHRERSEDAKKGPNPLMRRNSVTPLAである。
【0112】
Ludi(Accelrys,San Diego,California, USA)仮想スクリーニングプログラムを用いて、ChemNavigator iResearch library(ChemNavigator,San Diego,California, USA)を処理した。これらのスクリーニング運転の完了後、Ludiのスコアリングシステムを用いて500以上のスコアの分子を、タンパク質の活性位置内で仮想的吟味を行い解析した。活性ポケット内に正しく結合したリガンドは、スクリーニングのために用いた標的及びライブラリーに従って目録に入れた。200個のスコアが最高の分子を、Scifinder Scholarを用いて同定し、またトップ45個の分子を実験検定可能あるものと選択した。
【0113】
PFKFB3 F6P結合ポケット中に結合ポテンシャルを有すると決定された化合物の幾つかの構造を以下の表1に示す。本明細書に開示した発明の化合物は、Sigma-Aldrich(St.Louis,Missouri, USA)及びChembridge Corporation(San Diego,California, USA)の様な、しかしこれらに制限されない、適切な市販供給源から得ることができる。
【0114】
あるいは、カルコン、アザカルコン及びこれらの類似体もまた既知の方法で合成することができる。例えば、カルコン及びアザカルコンは、適切なアリールアルデヒド及びアリールケトンを縮合させ、その後脱水させ(H2Oの除去)、エノンを作成する。該縮合は、酸又は塩基を触媒とすることができる。幾つかの実施態様において、ベンズアルデヒド、置換ベンズアルデヒド又はこれらのアザ類似体を、アセトフェノン、置換アセトフェノン又はこれらのアザ類似体と、塩基(例えば、NaOH水溶液)存在下に、適切な温度で、ある時間の間(例えば、分、1から数時間、1日又は2日以上)混ぜ合わせることができる。該溶液を疎水性溶媒を用いて抽出し、濃縮してカルコン及びアザカルコンを提供することができる。幾つかの実施態様において、該抽出物をさらに、クロマトグラフィー又は再結晶のようないかなる方法を用いても、精製することができる。
【0115】
【表1】
【0116】
実施例2
In vitro組み変え酵素検定
PFKFB3クローニング、発現及び精製: ヒトPFKFB3 cDNAを以下のプライマー:
PFKFB3F: 5'-CTTCATATGCCGTTGGAACTGACGCA-3' (配列番号3)
PFKFB3R: 5'-CTTCTCGAGGTGTTTCCTGGAGGAGTCAGC-3' (配列番号4)
を用いて、既存の哺乳動物発現プラスミドから増幅した。
該PCA産物は、XhoI及びNdeI制限酵素(Promega, Madison, Wisconsin, USA)で消化し、またpET−30b(+)ベクター(Novagen, San Diego, California, USA)の対応する位置にクローン化した。該pET−30b(+)−PFKFB3C−termHis plasmidをBL21(DE3)E. coli コンピテント細胞(Novagen, San Diego, California, USA)に形質導入した。
【0117】
PFKFB3の発現及び精製のために、BL21−PFKFB3形質転換細胞の1L培養を16時間、37℃で振盪した。16時間後に、この培養液に、さらに1Lの2mMイソプロピルβ−D−1−チオガラクトピラノシド(IPTG;最終濃度=1mM)を含むLB培地を加えた、そして4時間30℃で振盪した。細菌を遠心で集菌し、タンパク質精製をQiagen(Venlo,オランダ)に記載されたプロトコールに従い、自然条件下で行った。手短に言うと、細菌ペレットを1回Dulbeccoのリン酸緩衝液(PBS;Invitrogen,Carlsbad,California, USA)で洗浄し、ペレットのグラム当たり2mLの溶解緩衝液に懸濁し、この溶解緩衝液は、5mM β−メルカプトエタノール、1mg/mL リゾチーム、1mM フェニルメチルスルフォニルフッ化物(PMSF)を含みさらにプロテアーゼ阻害カクテル(Sigma-Aldrich,St.Louis,Missouri, USA)1:100稀釈液を添加したものである。次ぎに該細菌混合物を、10秒間、8回超音波に懸け、遠心により上清を得た。溶解物を1時間、3mLの50% Ni−NTAとインキュベートし、該混合物をPoly−Prepクロマトグラフィーカラム(BioRad,Hercules,California, USA)に載せた。ビーズを1mM PMSF及びプロテアーゼ阻害カクテルの1:100稀釈液を含む15mLの6xHis洗浄緩衝液で洗浄し、PFKFB3を、200mMイミダゾールを含む1.5mLの溶出緩衝液で溶出した。さらなる精製のために、溶出分画を16時間20mMTris−HCl、200mM NaCl(pH 7.4)緩衝液に対して透析して、その後Sephadex S200カラム(Amersham,Piscataway,New Jersey, USA)によりゲル濾過した。
【0118】
PFKFB3酵素検定: 既述のように、PFKFB3活性を、ピルビン酸キナーゼ及び乳酸デヒドロゲナーゼを用いた酵素共役のキネティック検定により測定した。Bucher及びPfleiderer, Methods in Enzymology, 1, 435-440 (1955)を参照されたい。該検定は、化学量論的に1mol ATP:1mol 消費NADHに従い、340nmでの吸光度の減少から分光学的にNADH消費を測定する。該検定を、37℃で、最終200μLの反応溶液を用いて、96ウェルプレートを用いて行ったが、該反応溶液には、1xPFKFB3 反応緩衝液(100mM Tris−HCl、100μM EDTA, 5mM KH2PO4;pH=7.4)、420μM ホスホエノールピルビン酸(PEP),5mM ATP, 400μM NADH,613ユニットのピルビン酸キナーゼ(PK)、1092ユニットの乳酸デヒドロゲナーゼ(LDH),様々な濃度のフルクトース−6−リン酸(40〜400μM)及び15μgの精製PFKFB3が含まれた。酵素検定のための対照反応として、F6P無しの活性PFKFB3を用いた。化合物1阻害に対し、60μM,100μM又は150μM阻害剤存在下で、反応を行った。化合物1阻害に対する対照反応は、PFKFB3の添加無しで、様々な濃度の1を用いた。検定は、BioTek POWERWAVE(R)プレートリーダー(BioTek Instruments,Inc.,Winooski,Vermont, USA)を用いて、15分以上のキネティックモードで、測定して、PFKFB3の反応速度を測定した。SigmaPlot(R)9.0(SYSTAT Software,Inc., San Jose, California, USA)に対する酵素キネティック測定規準を用いて、PFKFB3及び1阻害に対するキネティックパラメーター(Vmax,Km及びKi)を計算した。Vmaxは、PFKFB3の比活性を、nmolF6P×min―1×mg−1で表し、Km及びKiは、それぞれ、F6P又は1に対して、μMで表される。表示したデータは、2回の独立した実験の、3通りの実験の平均値±STDである。
【0119】
図3Aに示すように、Lineweaver-Burke2重逆数プロットにより、1はPFKFB3活性を競合的及び非競合的な、阻害機構によりPFKFB3活性を阻害していることが明らかとなる。1とのインキュベーションにより、低濃度のF6Pで、PFKFB3酵素活性の顕著な用量依存的減少がもたらされる。しかしながら、この阻害は、F6P量の増加により克服されるので、PFKFB3結合位置において、1とF6Pの間で競合が在ることが示唆される。これらの研究より、酵素反応速度パラメーターを、PFKFB3活性及び化合物1阻害に対して計算した。組み換えPFKFB3の比活性は、277±9nmol F6P×min―1×mg−1であることが分かり、またF6Pに対するKmは97μMと決定された。図3Bを参照されたい。1に対するKiは、25±9μMと決定され、この値は、F6Pに対するKmと比較して、約4分の1に減少した。これらの研究により、1は主にF6Pとの競合によるPFKFB3の阻害剤であり、競合的阻害剤に対する酵素基質結合位置の分子標的は、抗癌治療同定するための重要な方法であることが、確認できた。
【0120】
実施例3
形質転換した腫瘍細胞における化合物1の毒性
哺乳動物発現に対するFLAG−PFKFB3構築体の作成: PFKFB3全コード配列及びN−末端にFLAG−エピトープを含むFLAG−PFKFB3をレトロウィルスTet応答性ベクターpRevTRE(Clontech,Mountain View,California, USA)内のBamH1/HindIII制限部位にサブクローン化した。組み換えレトロウィルスは、Lipofectamine(Invitrogen, Carlsbad, California, USA)を仲立ちとするPT67パッケージ細胞株にpRevTRE−FLAG−PFKFB3構築体を形質導入することより作り出した。誘導可能FLAG−PFKFB3を安定的に組み込み、また発現するJurkat細胞株を作るために、細胞をFLAG−PFKFB3を含む組み換えレトロウィルスで感染させ、400μg/mL ヒグロマイシン(Clontech, Mountain View, California, USA)存在下で安定なクローンを選択した。
【0121】
細胞培養: K562,HL−60,MDA−MB231及びメラノーマ(CRL−11174)ヒト癌細胞株をATCC(American Tissue Type Culture Collection,Manassas,Virginia, USA)から購入した。HeLa,A549,Lewis Lung Carcinoma,MDA−MB231及びメラノーマ細胞を10% 仔牛胎児血清(Hyclone,Logan,Utah, USA)及び50μg/mL 硫酸ゲンタマイシン(Invitrogen, Carlsbad, California, USA)を補強したDulbeccoの改変Eagle培地(DMEM)(Hyclone, Logan, Utah, USA)中で増殖させた。HL−60、K562及びJurkat細胞は、10% 仔牛胎児血清及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI−1640培地(Hyclone,Logan,Utah, USA)で増殖させた。初代正常ヒト気管上皮(NHBE)細胞及びヒトテロメラーゼ(ht)及びラージT抗原(LT)で不死化し、また突然変異したrasでトランスフォームした細胞(ht/LT/ras細胞)は、52μg/mL 仔牛脳下垂体抽出物、0.5μg/mL ハイドロコーチゾン、0.5ng/mL ヒト上皮成長因子、0.5μg/mL エピネフィリン、10μg/mL トランスフェリン、5μg/mL インシュリン、0.1ng/mL レチノイン酸、6.5ng/mL 三ヨウ化チロニン、50μg/mL ゲンタマイシン及び50ng/mL アンフォテリシンB(SingleQuots, Cambrex, Walkersville, Maryland, USA)を補強したBronchial Epithelial Cell Basal Growth Medium(Cambrex, Walkersville, Maryland, USA)中で培養した。全ての細胞株は、37℃で、5% CO2下で維持した。
【0122】
細胞周期解析及びフローサイトメトリー: Jurkat細胞を、10% FBS及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI培地中で1×105細胞/mLの密度で蒔種した。細胞周期をVybrant DyeCycle Orange染色(Molecular Probes-Invitrogen, Eugene, Oregon, USA)を用い製造者の指針に従って行った。
【0123】
In Vitro増殖抑制: 全ての細胞株を1×105細胞/wellの密度で、適切な培地中に蒔種した。懸濁細胞に対して、様々な濃度の1を直ちに培地に加えた、それに対して、接着細胞に対しては、化合物1処理は、翌日に開始した。用量依存性研究に於いて、36時間にわたって様々な濃度の化合物1を加えた。時間依存性研究に於いて、10μMの化合物1を、0,4,8,16,24,又は36時間目に加えた。PFKFB3過剰発現研究のために、FLAG―PFKFB3発現ベクター又は対照プラスミドを含むJurkat細胞を、化合物1とのインキュベーションの24時間前にドキシサイクリン(1?g/mL;Clontech,Mountain View,California, USA)投与により誘導した。
細胞を処理48時間後に集め、細胞数及び生存率をトリパンブルー排除法により測定した。溶媒処理細胞増殖の50%増殖に必要な化合物1濃度として、IC50値を計算した。表示したデータは、3回の独立した実験からの3通りの測定の平均値±STDである。
【0124】
乳酸塩測定: 1×105Jurkat細胞を様々な濃度の1とインキュベートした。望みの時点で、培地試料を集め、製造者(Trinity, St.Louis, Missouri, USA)の指針に従い、乳酸オキシダーゼをベースとした540nmで測定の比色検定により乳酸塩レベルを測定し、タンパク質濃度に対して規格化した。
【0125】
フルクトース−2,6−ビスリン酸塩検定: Jurkat細胞を1×105細胞/mLの密度で蒔種して、直ちに10μM 化合物1と0,4,8,16,24又は36時間インキュベートした。F2,6BP検定は、既述のように行った。Van Shaftingenほか,Eur.J.Biochem,129,191-195(1982)を参照されたい。
【0126】
2−デオキシ−グルコース取り込み: Jurkat細胞を1×105細胞/mLの密度で、10%FBS及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI培地に蒔種した。細胞を直ちに、溶媒(DMSO)又は10μM 化合物1で、36時間までの間、処理し、その後、グルコースを欠いたRPMI培地に30分間置いた。14C−2−DG(0.25?Ci/mL;Perkin Elmer, Waltham, Massachusetts, USA)を60分間加えた。その後、細胞を3回氷冷したグルコースを欠いたRPMIで洗浄した。細胞溶解液を500μLの0.1%SDS中に集め、溶解液400μLについてシンチレーション(cpm)を計数した。計数値をタンパク質濃度について規格化した。2回の独立した実験について、2回の測定値の平均値±STDで、データを表した。
【0127】
全細胞ATP: Jurkat細胞を1×105細胞/mLの密度で蒔種し、様々な時点について10μM 化合物1とインキュベートした。ATPレベルを、Molecular Probes-Invitrogen(Eugene, Oregon, USA)から得たATP測定キットを用いて、製造者の指針に従って測定した。
【0128】
NAD+及びNADHレベル: Jurkat細胞を1×105細胞/mLの密度で蒔種し、直ちに10μM化合物1と、0,4,8,16,24又は36時間インキュベートした。NAD+及びNADHレベルを、BioAssay Systems(Hayward,California, USA)から得たEnzyChrom NAD+/NADH検定キットを用いて、製造者の指針に従い測定した。
【0129】
NMR: Jurkat細胞を、13C−グルコース存在下で、36時間、溶媒(DMSO)又は10μM 化合物1で処理した。同数の細胞をペレットにし、2回冷却したPBSで洗浄して、残った培地を除き、最終的にペレットにし、液体窒素中で瞬時に凍結した。冷却したペレットから氷冷した10%TCAで2回抽出し、その後凍結乾燥した。乾燥した抽出物を、0.35mL D2Oに再溶解し5mm Shigemiチューブに入れた。NMRスペクトルは、Varian Inova NMR スペクトロメーター(Varian,Inc., Palo Alto, California, USA)上で、20℃で、90°励起パルスを用いて、14.1Tで、記録した。2次元TOCSY及びHSQC実験を行った。代謝物を、1H及び13C化学シフト及びTOCSYコネクティヴィティパターンを基に同定した。代謝物は、TOCSY実験で積分により定量した。
【0130】
タンパク質抽出及びウェスターンブロット解析: タンパク質抽出及びウェスターンブロット解析は、以前の記載のように行った。Telangほか, Oncogene, 25, 7225-7234 (2006)を参照されたい。ブロットは、抗−PFKFB3抗体(Abjent, Inc., San Diego, California, USA)及び抗−β−アクチン抗体(Sigma, St.Louis, Missouri, USA)を用いて、先ずPFKFB3に対して探索し、その後プローブを外し、β−アクチンに対して再探索した。
【0131】
結果: 解糖フラックスの増加がトランスフォームした細胞増殖に必要と仮定すると、特別の理論に囚われることなく、PFKFB3の阻害は、選択的に腫瘍細胞増殖を標的とする。トランスフォームしたNHBE細胞(ht/LT/ras)は、遺伝的に一致する初代細胞と比較して、1に対してより大きな感受性があった。図4Aを参照のこと。初代NHBE細胞は、1とのインキュベーションに対して、試験した最高濃度(33μM)で、顕著な感受性を示さなかった。しかしながら、トランスフォームした細胞を1μM以下の濃度で1と処理すると、細胞増殖停止を示し、また約10μMで完全に細胞毒性を示した。NHBE−ht/LT/ras細胞に対する1のIC50は1.5μMと測定された。従って、化合物1は、他の解糖阻害剤、2−DG及び3−BrPA、(それぞれ、低mM濃度から、100μMで有効であった)に比べて10倍以上の阻害能を示した。
【0132】
固形腫瘍及び血液由来疾患由来の細胞株を含め、トランスフォームした細胞に対する1の効果を、さらに様々な腫瘍細胞に対する1のIC50を計算することにより吟味した。白血病細胞株は、接着細胞株に比べて、1の処理に対して増加した感受性(平均して約7倍)を示した。図4Bを参照されたい。K562、Jurkat及びHL−60白血病細胞に対するIC50値は、それぞれ、3.2,1.4及び4.5μMと決定された。接着細胞において、IC50の差異は大きかった。2種の肺癌モデル、ヒトA549及びマウスLLC,では、IC50値は、それぞれ、24及び19μMであり、他方HeLa子宮頸癌細胞のIC50値は,24μMであった。MDA−MB231乳腺癌細胞は化合物1との処理に対して、他の固形癌由来細胞株と比較して特に感受性が高く、計算したIC50値は、4.7μMであった。最後に、メラノーマ細胞株のIC50値は15μMであった。従って、1は、腫瘍細胞増殖を選択的に標的とする抗癌化合物であろう。
【0133】
1とのインキュベーションは、JurkatT細胞白血病細胞の増殖の用量依存的低下をもたらした。図5Aを参照されたい。0.3μMほどの3POが、細胞増殖を低下させて、また10μM 3POは、36時間にわたって、完全に細胞増殖を阻害した。図5A及び5Bを参照されたい。細胞増殖の抑制は、ヨウ化プロピジウム染色で測定したように、G2/M期の細胞周期停止の結果であるようである。図5C及び5Dを参照されたい。またFru−2,6−BP産生、2−デオキシグルコース(2−DG)取り込み、乳酸塩分泌に対する10μM 1の効果も調べた。図5Eを参照されたい。2−DG取り込み及びFru−2,6−BPは、4時間の処理の間に顕著に減少した。これらの代謝上の変化の後に、乳酸塩分泌(図5E;8時間を参照)、NADH(図5F;16時間参照)、NAD+(図5F;24時間参照)及びATP(図5F;24時間参照)の減少が続く。1との処理の間、Jurkat細胞を完全に標識した13C−グルコースでパルスし、またNMR分光で13C原子の運命を調べることにより、解糖の乳酸塩への直接のフラックスが抑制された。図5Gを参照されたい。
【0134】
1が細胞内のFru−2,6−BP,2−DG取り込み及び乳酸塩分泌をG2/M期停止前に減少させると言う観察により、特定の理論に囚われることなく、1がエネルギー的及び同化的代謝の中断により細胞増殖を阻害すると言う仮説が、相当自然に支持される。HL−60前骨髄球性白血病細胞において以前観察された様に、1によるG2/M期停止は、細胞周期のG2/M期間のATPの高い要求性と比べると、補助的でありうる。Sweet及びSingh,Cancer Res.,55,5164-5167(1995)を参照されたい。13C−グルコースのNMR分光による追跡から、解糖のアラニンではなく、乳酸塩へのフラックスが阻害されたことが明らかとなった。アラニン及び乳酸塩は同じピルビン酸プールを分かち合うが、乳酸デヒドロゲナーゼは、グリセルアルデヒド−3−リン酸デヒドロゲナーゼを経る解糖フラックスの産物である、NADHの即応の補給を必要とするので、グルコースに由来する乳酸塩産生は、特に解糖阻害により影響を受ける。
【0135】
新しい抗ガン治療の最も困難な課題の一つは、in situの標的検証である。この問題に取り組むために、異なるPFKFB3発現レベルを有する2種の異なる細胞株を研究した。もし、PFKFB3が真に1の標的であるならば、該酵素の発現が低下した細胞は、1の処理に対してより感受性があるか、又は逆である。従って、PFKFB3半数不足型(+/−)繊維芽細胞をヒトテロメラーゼ(ht)及びラージT抗原(LT)で不死化させ、突然変異型rasの発現によりトランスフォームさせた(PFKFB3 +/−ht/LT/ras)これらの繊維芽細胞は、約50%のPFKFB3発現及び低下したF2,6BP産生を有することが示された。該+/−ht/LT/rasトランスフォーム細胞及びこれらの野生型の遺伝子にマッチした対応細胞(+/+ht/LT/ras)を様々な濃度の1とインキュベートした。
【0136】
図6Aに示すように、PFKFB3+/− LT/rasトランスフォーム繊維芽細胞は、野生型対応細胞に比べて、低下したPFKFB3タンパク質及び低下した細胞内F2,6BPを発現する。野生型細胞における、F2,6BP濃度は、60.3±3.7 pmol/mgであり、他方PFKFB3+/−LT/rasトランスフォーム繊維芽細胞では、F2,6BP濃度は、10.3±1.5 pmol/mgであった。図6Bに示すように、PFKFB3異形接合繊維芽細胞は野生型トランスフォーム細胞に比べて、1の処理に対してより感受性が高かった。+/−繊維芽細胞に対する計算したIC50値(26μM)は、+/+繊維芽細胞に対するIC50値(49μM)の約半分であった。
【0137】
逆に、PFKFB3の過剰発現は、細胞と1のインキュベーションによる細胞増殖の低下を抑制するはずである。Jurkat細胞において、ドキシサイクリン応答性PFKFB3 Tet−ONシステムの影響下で、PFKFB3レベルを操作した。ドキシサイクリン処理した、PFKFB3発現ベクターを持たない対照 Jurkat細胞に比較して、ドキシサイクリン処理によりPFKFB3発現が増加する。PFKFB3タンパク質の異所性の発現は、F2,6BP濃度を増加させる。図6Cに示すように、+PFKFB3細胞におけるF2,6BP濃度は、38.1±3.4pmol/mgであり、他方、対照細胞において、F2,6BPの濃度は、9.2±0.95pmol/mgであった。両種の細胞を様々な濃度の1とインキュベートした、そして、予想されたように、過剰発現したPFKFB3は、Jurkat細胞への阻害剤の効果を抑制した(図6D)。PFKFB3過剰発現細胞に対するIC50値は、19.3μMと決定されたが、PFKFB3発現レベルの変わらぬJurkat細胞のIC50は8.9μMと計算された。
総合すると、これらの研究は、PFKFB3発現を調節することにより、1の細胞阻害効果を変えることができることを示し、PFKFB3が1の有効なin situ標的であることを示唆する。
【0138】
実施例4
化合物1を用いたIn Vivo研究
対数増殖期のMDA−MB231及びHL−60細胞を適切な培地に集め、2回洗浄して、20×107細胞/mLの密度でPBSに再懸濁した。次ぎに細胞を1:1の割合でMatrigelマトリクス(BD Biosciences, Bedford, Massachusetts, USA)と混ぜ、0.1mLの細胞懸濁液(1×107細胞)を雌のBalb/cヌードマウス(20g)の皮下に注射した。対数増殖期のLewis肺癌細胞を集め、2回洗浄し、PBSに懸濁した(1×107細胞/mL)。C57Blk/6雌マウス(20g)の皮下に0.1mLの容積で1×106細胞を注射した。体重及び腫瘍増殖を研究の間中、毎日観察した。重量(mg)=[幅(mm2)×長さ(mm)]/2の式を用いて、腫瘍重量をノギスで測定した。Taetleほか,Cancer Treat.Rep.,71,297−304(1987)を参照されたい。樹立した腫瘍を有するマウス(130mg〜190mg)を無作為に溶媒対照又は化合物1処理群に分けた。指示した時点で、溶媒対照グループには、50μLジメチルスルホキシド(DMSO)を腹腔注射(i.p.)し、他方、処理群には、0.07mg/g 化合物1を含む50μL DMSOをi.p.注射した。
【0139】
結果:1の毒性研究により、副作用の発現の無かった最高の容認された投与量は、0.07mg/g を含むDMSOの腹腔(IP)注射であった。試験した第1の腫瘍モデルは、C57/Blk6マウスに異種移植増殖するマウスLewis肺癌細胞からなる。この研究のために、マウスにDMSO又は計算した投与量の1を研究の期間(14日間)、毎日一度、IP注射した。図7Aに示したように、1の投与は、顕著にDMSO対照群と比較して、Lewis肺癌細胞異種移植体の増殖を73%抑えた。別個に、MDA−MB231ヒト乳腺癌をBalb/cヌードマウスに樹立した。このモデルにおいて、処理の低下が、Lewis肺癌モデルで見られたと同じ効果をもたらすかどうか決めるために、投与量の回数を減らした。DMSO(対照)及び化合物1処理群の両者のマウスに対して、3日間毎日の注射後、3日間の無処理という周期的な投薬計画を行った。DMSO対照と比較して、MDA−MB231細胞の異種移植体増殖の全阻害は最大66%観察された(図7B).3番目のin vivoモデルは、Balb/cヌードマウスに異種移植したHL−60白血病細胞である。処理計画はさらに減らして、2日間の連続注射後、7日間連続の無処理とした。図7Cに示すように、化合物1処理により、制限された投与にもかかわらず、DMSO対照と比較して、HL−60腫瘍増殖が74%以上阻害された。さらに、2回目の投与計画の効率は9日目の投与後の増殖低下が、増殖曲線上で見られる。Lewis 肺癌及びHL−60異種移植モデルに対する化合物1の効果は、腫瘍増殖の後退が見られないことから、細胞増殖停止と思われる。しかしながら、MDA−MB231モデルでは、化合物1阻害が、初期の細胞増殖停止効果から初期の腫瘍後退を導き、その後他の2モデルで見られるような細胞増殖停止をカバーするようである。まとめると、上記のデータから、化合物1がin vivo腫瘍増殖を減少させる能力を有する抗癌剤の可能性を有するものと支持される。
【0140】
実施例5
化合物2〜7によるPFKFB3の阻害
化合物2〜7がin vitroでPFKFB3活性の阻害能を有することは、上記実施例2に記載した酵素検定法を用いて測定した。150μM化合物2の処理により、溶媒(EtOH)処理と比較してPFKFB3の反応速度を39.8%減少させた。図8を参照されたい。溶媒(DMSO)処理と比較した、化合物3〜7の阻害効果は、図9に示されている。化合物4が最もPFKFB3を低下させ、80%以上であった。
【0141】
実施例6
トランスフォーム腫瘍細胞における化合物2〜7の毒性
様々なトランスフォーム細胞株に対する化合物2〜7の毒性は、上記実施例3に記載したように検定した。化合物2は、2種の白血病細胞HL−60及びK562細胞において、用量依存的細胞毒性を示した。図10を参照されたい。Jurkat白血病細胞増殖への様々な濃度の化合物3〜7の効果を図11〜15に示す。
図16〜21は、様々なトランスフォーム細胞において、化合物濃度0.3,1,3,10,又は33μMで処理した化合物2〜7の効果を示す。化合物2〜7の6種全ては、検定した固形腫瘍及び血液細胞タイプのそれぞれの増殖を阻害した。
【0142】
実施例7
Micro−Petにより観察したグルコース取り込みに対する化合物1の効果
C57Bl/6雌マウス(20g)に、1×106Lewis肺癌細胞を皮下に注射した。異種移植体の重量を測定して150及び180mgの間になった時、該マウスを2群に分けた。1群には、0.07mg/gの化合物1をi.p.注射した。他の群を対照群として用い、50μL DMSOをi.p.注射した。注射4時間後腫瘍を取り除き、等容量の0.1M及び0.05M NaOH中でホモジナイズした。F2,6BPアッセイを既述のように行った。Van Shaftingenほか.,Eur.J.Biochem,129,191-195 (1982)を参照されたい。化合物1処理により、腫瘍異種移植体中のF2,6BP産生は溶媒処理の対照群と比較して、顕著に減少した。化合物1処理のマウス異種移植体中のF2,6BP濃度は8.5±1.7pmol/mgであり、他方、対照マウス異種移植体中のF2,6BP濃度は、13.1±1.9pmol/mgであった。
【0143】
化合物1の効果をさらに調べるために、化合物1−処理マウスにおけるグルコースの取り込みをミクローポジトロン発光断層撮影法(micro―Pet)を用いて研究した。Lewis肺癌異種移植体を担うマウスに50μLのDMSO又は0.07mg/g 化合物1のDMSO溶液をi.p.注射した。最初の注射30分後に、各マウスに2−(18F)−フルオロ−2−デオキシグルコース(18F−2−DG;150μCurie、100μL H2O中)をi.p.注射した。さらに15分後に、マウスを酸素中2% イソフルランで麻酔し、R-4 Rodent Scanner Micro-Pet(CTI Concorde Microsystems, Inc.; Knoxville, Tennessee, USA)に移した。各群(即ち化合物1処置又は対照)3匹のマウスを研究した。図22は、代表的な化合物1−処理マウス及び代表的対照群マウスから取ったmicro―Pet画像である。
【0144】
図22に示すように、化合物1処理により、異種移植体内の18F−2−DG取り込みは、対照群マウスと比較して著しく減少した。化合物1処理マウス及び対照群マウスの間で、心臓の18F−2−DG取り込みにも差異が観測された。超音波心臓診断法を、Dawnほか,Proc.Natl.Acad.Aci.,U.S.A.,102,3766-3771(2005)に記載されたように行った。マウスの心臓機能の超音波心臓診断により、駆出分画に急性の変化は見られなかった(対照(溶媒):69%±4%;化合物1:71%±6%)。従って、心臓グルコース取り込みの差異は、化合物1の心臓毒性によるものではないようである。まとめると、1はin vivoでPFK2活性を標的とし、解糖表現型を低下させる。
【0145】
実施例8
トランスジェニックマウスの腫瘍量への化合物1の効果
トランスジェニックMMTV−Neu乳腫瘍マウスは、乳腺癌を生後3〜4ヶ月以内に発症し、マウス乳癌ウィルスプロモーター/エンハンサーの調節の下の癌遺伝子Neuのトランスジェニック発現による。これらのマウスは、これらの腫瘍増殖の性質の陰険さ及び組織培養条件に起因する人為性がない結果として、異種移植体モデルと比べて、ヒト癌により密接に類似する。
トランスジェニックMMTV−Neu乳癌マウスへの化合物1の効果を測るために、トランスジェニックマウスにおける相対的腫瘍率及び大きさを4ヶ月後に測定し、同等な腫瘍負荷になる様に無作為に2群に分けた。1群のマウスは、化合物1を3日間毎日の投与(0.07mg/g、DMSO中;腹腔投与;矢印を参照;腫瘍量が体重の10%を越えるまで)、その後3日間無処理の繰り返しサイクル投与を行った。対照マウスには、溶媒対照(DMSOのみ)で注射した。腫瘍量は、次の確立した式:重量(mg)=(幅、mm2×長さ,mm)/2に従い測定した。統計的な有意性を得るために、(α値が0.05及び指数が0.90と仮定して)各群に30匹のマウスを必要とした。長期にわたる腫瘍量への化合物1処理の効果を図23に示す。
【0146】
本明細書に開示した対照事項の様々な詳細を、本明細書に開示した発明の範囲から離れることなく、変更できる。さらに、既に述べた記載は、説明の目的のためにのみ有り、制限付けの目的のためではない。
【技術分野】
【0001】
本出願は、2007年6月18日に出願された米国仮出願60/936,030に基づく優先権を主張し、これは本明細書に引用される。
本明細書に開示した発明は米国防総省により与えられた助成金番号PCF OGMB04-1166のもとで支援された米国政府によって為された。従って、米国政府は、本明細書に開示した発明の一部の権利を有する。
本願発明は、解糖フラックス(流量)を減少させる方法及び化合物に関する。より詳細には、本願発明は、6−ホスホフルクトー2−キナーゼ(PFK−2)の一誘導酵素である6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(以下「PFKFB3」という。)を阻害する、アザカルコン及びこれらの類似化合物に関係する。解糖フラックスを減少させ、細胞増殖を阻害し及び癌を治療する化合物を使用する方法を記述する。更に、PFKFB3の阻害能をもつ化合物のスクリーニング方法を記述する。
【背景技術】
【0002】
解糖経路は真核細胞における糖代謝の基本的嫌気的経路である。解糖は2重の働きを有し、即ち糖を分解してエネルギー(ATP)を産生し及び合成反応に使われる単純化合物を提供する。グルコースからピルビン酸塩への変換速度は、これら2種の主要な細胞必要物を満たすように調節されている。より詳細には、解糖系過程における不可逆反応を触媒するヘキソキナーゼ、ホスホフルクトキナーゼ及びピルビン酸キナーゼのような酵素は、解糖における制御点として調節されている。
【0003】
1930年にワールブルグは、腫瘍は亢進した嫌気的解糖を行い、また相対的に酸素濃度が高い場合でも、解糖速度の減少を示さないことを指摘した。この調節的制御の喪失(即ち、パスツール効果)は、ワールブルグ効果と呼ばれ始めた。以来、その後の研究により、癌に特有な以下の性質が、常に確認された:a)グルコースの急速な消費;b)活?な解糖活性(非特許文献1参照);c)早い細胞増殖(非特許文献2参照);d)乳酸産生及び蓄積(非特許文献3参照);及びe)腫瘍周囲に外接して、低グルコース濃度を伴う細胞外の低pH。特許文献1を参照のこと。
【0004】
癌細胞の代謝が、エネルギー及び高分子前駆体産生に関して、解糖フラックスが亢進した状態にシフトしているので、解糖の阻害を標的とする可能性を有する抗悪性腫瘍戦略として浮かび上がった。解糖経路の阻害効果による抗腫瘍特性を有するものとして、過去数十年にわたり、様々な小分子が同定されてきた。特に2種の分子、3−ブロモピルビン酸塩(3−BrPA)及び2−デオキシグルコース(2−DG)の両者は、ヘキソキナーゼを標的とし、ミトコンドリア呼吸欠失した、又は低酸素下のトランスフォーム細胞に対して細胞毒性を示した。非特許文献4、非特許文献5及び非特許文献6を参照のこと。抗悪性腫瘍剤Imatinib(商品名Gleevec(R)、Novartis Pharmaceuticals Incorporation,East Hanover, New Jersey, USA)もまたヘキソキナーゼを抑制することが示された。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願公開20060035981号
【非特許文献】
【0006】
【非特許文献1】Maublantほか、Bull Cancer, 85,935-950(1998)
【非特許文献2】Chesneyほか、Proc Natl Acad Sci USA、96,3047-3052(1999)
【非特許文献3】Baggetto、Biochimie,74,959-974(1992)
【非特許文献4】Xuほか、Cancer Res.,65,613-621(2005)
【非特許文献5】Luiほか、Biochemistry,40,5542-5547(2001)
【非特許文献6】Maherほか、Cancer Chemother.Pharmacol.,53,116-122(2004)
【発明の概要】
【発明が解決しようとする課題】
【0007】
さらなる抗癌治療薬の要請があり、特に、癌と関連する亢進した解糖フラックスに関係するメカニズムを通して悪性細胞を標的とする抗ガン治療薬が要請されている。また、解糖に関わる酵素の阻害能を測定して化合物をスクリーニングするさらなる方法が要請されている。
【課題を解決するための手段】
【0008】
本明細書における開示の対象である発明は、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)を阻害化合物と接触させることから成るPFKFB3を阻害する方法を提供する。
【0009】
幾つかの実施態様において、この方法は、PFKFB3を化学式(I):
【化1】
(式中、XはC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1は、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2はアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群より独立に選択される。)
で表わされる化合物接触させることから成る。
【0010】
幾つかの実施態様において、Ar1及びAr2の少なくとも1つは、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、ピリジン基、置換ピリジン基、キノリン基、置換キノリン基、イソキノリン基及び置換イソキノリン基からなる群から選択される。
幾つかの実施態様において、L1は存在しない。幾つかの実施態様において、L2は存在して、C2アルキレン基である。
【0011】
幾つかの実施態様において、この化学式(I)で表わされる化合物は、化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、それぞれ独立にN又はCR12であり、式中、各R12は水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5はO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13は、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つはNである。)
で表わされる構造を有する化合物又はこれらの医薬的に許可された塩である。
【0012】
幾つかの実施態様において、X1はOであり、かつXはCである。
幾つかの実施態様において、Ar2は:
【化5】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基、からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、化学式(II)で表わされる化合物は化学式(III)で表わされる構造:
【化6】
を有する。
【0013】
幾つかの実施態様において、X1はOである。幾つかの実施態様において、R10及びR11はそれぞれ水素原子である。
幾つかの実施態様において、化学式(III)で表わされる化合物は、以下の化学構造:
【化7】
からなる群から選択される。
【0014】
幾つかの実施態様において、Ar2は:
【化8】
であり、
X、X2及びX3のそれぞれは、Cであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は化学式(IV):
【化9】
で表わされる構造を有する。
【0015】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11は、それぞれ水素原子である。幾つかの実施態様において、化学式(IV)で表わされる化合物は、
【化10】
である。
【0016】
幾つかの実施態様において、Ar2は、
【化11】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は化学式(V)で表わされる構造:
【化12】
を有する。
【0017】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11は、それぞれ水素原子である。幾つかの実施態様において、化学式(V)で表わされる化合物は、
【化13】
である。
【0018】
幾つかの実施態様において、本明細書において開示した発明は、細胞を有効量のPFKFB3阻害剤と接触させることから成る、細胞内の解糖フラックスを減少させる方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、細胞を有効量の化学式(I)で表わされる化合物と接触させることから成る、細胞内の解糖フラックスを減少させる方法である。
【0019】
幾つかの実施態様において、本明細書で開示した発明は、細胞に有効量のPFKFB3阻害剤を接触させることから成る、細胞の増殖能を減少させる方法を提供する。幾つかの実施態様において、本明細書で開示した発明は、細胞に有効量の化学式(II)で表わされる化合物を接触させることから成る、細胞の増殖能を減少させる方法を提供する。幾つかの実施態様において、該化合物は、以下の構造:
【化3】
を有する置換基を含み、式中、X4は、NR14,式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;X6は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、アルキル基、アラルキル基及びアリール基からなる群より独立に選択され;及びX6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。幾つかの実施態様において、X5は、Oであり、X6は水素原子、アルキル基、アラルキル基及びアリール基から選択される。
【0020】
幾つかの実施態様において、該化合物は、下記化学式(IIIa)、(IIIb)、(IIIc)及び(IIId):
【化14】
【化15】
【化16】
及び
【化17】
の中の一つである。
【0021】
幾つかの実施態様において、該細胞は、哺乳動物細胞である。幾つかの実施態様において、該細胞はヒト細胞である。幾つかの実施態様において、該細胞は、癌細胞である。
幾つかの実施態様において、本明細書に開示した発明は、有効量のPFKFB3阻害剤を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、有効量の化学式(II)で表わされる化合物を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法を提供する。幾つかの実施態様において、該化合物は化学式(IIIa)、(IIIb)、(IIIc)及び(IIId)の中の一つで表わされる化合物である。幾つかの実施態様において、該患者は、哺乳動物である。幾つかの実施態様において、該患者はヒトである。幾つかの実施態様において、該癌は、乳癌、肺癌、子宮頸癌、皮膚癌及び白血病からなる群から選択される。
【0022】
幾つかの実施態様において、該方法は更に、患者に1又は2以上の付加的な治療用化合物を投与することを含む。幾つかの実施態様において、1又は2以上の付加的治療用化合物は、抗癌治療薬である。幾つかの実施態様において、1又は2以上の付加的治療用化合物は、シスプラチン及びタキソールからなる群から選択される。
【0023】
幾つかの実施態様において、本明細書に開示した発明は、PFKFB3を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法を提供する。幾つかの実施態様において、本明細書に開示した発明は、腫瘍増殖を阻害する能力を有する化合物をスクリーニングする方法を提供する。幾つかの実施態様において、この化合物をスクリーニングすることが、PFKFB3のモデルに対して多くの化合物をスクリーニングして、それぞれの化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定することから成る。
【0024】
従って、PFKFB3を阻害し、それにより解糖フラックスを調節する化合物を提供することが、本明細書に開示した発明の目的である。
上記のように記述した本明細書に開示した発明の目的及び本明細書に開示した発明により全体的に又は部分的に到達された他の目的は、以下に最善に記載して添付した図表を参照して、明細書が進むに従い明らかとなろう。
【図面の簡単な説明】
【0025】
【図1】図1はこの経路により、フルクトース−2,6−ビスリン酸塩によるPFK−1の活性化を経由して、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)が解糖フラックスを亢進させることを示す、経路の概略図である。
【図2】図2Aは、2次元構造で描写した6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)のホモロジーモデルの概略図である。この概略図はまた、結合部位における3−(3−ピリジニル)−1−(4−ピリジニル)−2−プロペン−1−オン(3PO)を示す。図2Bは、2.5Å以内の残基を示すPFKFB3結合ポケットの側面図の図解である。図2Cは、棒状の表示で示す前面残基を伴う結合ポケット残基のConnelly表面(溶媒接触表面)を示す、PFKFB3結合ポケットの端面図の図解である。
【図3】図3Aはフルクトース−6−リン酸塩濃度(60,80,160,240,320,又は400μM)の関数として、PFKFB3酵素活性のLineweaver−Burke2重逆数プロットを示すグラフである。キナーゼ検定は、DMSO(●)、60μM 3PO(O),100μM 3PO(▼)及び150μM 3PO(▽)存在下で行った。3POは、化合物1、3−(3−ピリジニル)−1−(4−ピリジニル)―2−プロペン−1−オンを表す。図3Bは、PFKFB3の比活性(S.A),フルクトース−6−リン酸塩のKm及び3POのKiを含む酵素反応速度パラメーターの表である。
【図4】図4Aは、1,3,10又は33μM 3PO又は溶媒(V)のみで処理後48時間のNHBE細胞(グラフの左側)及びトランスフォームNHBE細胞(ht/Lt/ras,グラフの右側)の細胞増殖を示す棒グラフである。非処理対照細胞分布(C)もまた示す。各棒は、代表的実験の3回の測定の平均細胞数(×103)を示す。誤差バーは、±1標準偏差(STD)を示し;*はp−値<0.01を示す。図4Bは、様々な固形及び血液細胞株における化合物1(即ち、3PO)のIC50値(μM)の表である。各値は、溶媒のみによる対照細胞増殖の50%に必要な1の濃度である。
【図5】図5Aは増加する濃度(0.3,1,3,10及び33μM)の3POを36時間処理した Jurkat細胞の細胞増殖の棒グラフである。溶媒処理細胞(V)及び非処理対照 Jurkat細胞(C)の細胞増殖もまた示す。各棒は、代表的実験の3回の測定の平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示し;*はp−値<0.01を示す。図5Bは、溶媒(●)又は10μM 3PO(○)存在下の Jurkat細胞の時間依存的増殖の測定を示すグラフである。各データポイントは、3回の独立した実験において測定した平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示す。図5Cは、溶媒(●)又は10μM 3PO(○)処理した、Jurkat細胞の経時的なG2/M期細胞集団パーセンテージ(%)を示すグラフである。各データポイントは、3回の独立した実験において測定した平均細胞数(×104)を示す。誤差バーは、±1標準偏差(STD)を示す。図5Dは、溶媒(実践)又は10μM 3PO(点線)で36時間処理したJurkat細胞のフローサイトメトリーにより測定した細胞周期ヒストグラムの合成画像である。図5Eは、10μM 3PO存在下又は不存在下での、時間の関数で表す、乳酸塩分泌(▼)、2−デオキシグルコース(2−DG)取り込み(●)及びFru−2,6−BP産生(○)を示すグラフである。誤差バーは±1標準偏差(STD)を示す。図5Fは、10μM 3PO存在下又は不存在下での、時間の関数で表す細胞全体のNAD+(●)、NADH(○)及びATP(▼)濃度を示すグラフである。誤差バーは±1標準偏差(STD)を示す。図5Gは、溶媒(左側スペクトル)又は3PO(右側スペクトル)存在下、アラニン(Ala)及び細胞内乳酸塩(Lac)への13C取り込みを示す、2次元(2D)核磁気共鳴(NMR)スペクトルである。 3回の独立した実験からの代表的スペクトルを示す。 点線ボックスの縁は、代表的代謝物の13Cピークに対応し、これらは中心の内因性12Cピークを囲む。
【図6】図6Aは、その後ヒトテロメラーゼ(ht)、ラージT抗原(LT)及び腫瘍遺伝子H−rasv12で不死化し、またトランスフォームした野生型(+/+)又はハプロ不全(+/−)PFKFB3マウスからの繊維芽細胞で測定した、F2,5BPの細胞内濃度を表す棒グラフである。挿画は、繊維芽細胞におけるPFKFB3発現のウェスターンブロット解析を示す。図6Bは、その後ヒトテロメラーゼ(ht)、ラージT抗原(LT)及び腫瘍遺伝子H−rasv12で不死化し、またトランスフォームした野生型(+/+)又はハプロ不全(+/−)PFKFB3マウスからの繊維芽細胞に対する3POの増殖抑制効果を示す棒グラフである。トランスフォームした野生型細胞(+/+)に対する細胞数(対照細胞の細胞数に対する%として表わす)を実線バーで表し、またハプロ不全細胞(+/−)に対する細胞数をストライプバーで表す。グラフの底に示すように、細胞を溶媒又は増加する3PO量(5,10,20,30,40又は60μM)とインキュベートして、生存細胞数を48時間後に計数する。バーは、代表的実験からの3回の値の平均値±STDを示す。*p−値<0.01は、溶媒対照及び3PO処理試料の間の統計的差異を表す。図6Cは、Tet−onシステム(+PFKFB3)及び空ベクター(Vector)を含む対照細胞を用いて、ドキシサイクリン処理によりPFKFB3を過剰発現するよう加工したJurkat細胞を使って測定したF2,5BPの細胞濃度の棒グラフである。細胞を1μg/mlのドキシサイクリンで処理し、PFKFB3蛋白質発現を誘導した。挿図は細胞内のPFKFB3発現のウェスターンブロット解析である。図6Dは、Tet−onシステムを用いて、ドキシサイクリン処理でPFKFB3を過剰発現するよう加工したJurkat細胞(+PFKFB3、ストライプバー)での3POの増殖抑制効果を示すう棒グラフである。3PO処理24時間前に、PFKFB3蛋白質レベルを誘導するために、1μg/mLのドキシサイクリンをPFKFB3発現のためのベクターを含むJurkat細胞とインキュベートする。空ベクターを含む対照細胞(―PFKFB3,実線バー)を同様にドキシサイクリン処理し、バックグランドPFKFB3発現として用いた。その後、両種の細胞を異なる濃度の3PO(1,3,10,又は33μM)又は溶媒(V)とインキュベートし、生存細胞数を48時間後に解析した。バーは、代表的実験からの3回の値の平均値±STDを示す。
【図7】図7Aは、C57/Blk6マウスに異種移植したLewis Lung カルシノーマ腫瘍の増殖に対する3PO処理の効果を示すグラフである。平滑末端のノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=11)又は3PO処理群(○、n=14)に分けた。実験マウスの体重を測定し、毎日、表示した時点において、50μL DMSO、又は0.07mg/g 3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は毎日の対照又は3PO投与を示す。最初の注射(2日目、p−値<0.0003)後に、DMSO及び3PO群の間に統計的に有意の差異が得られた。 図7Bは、Balb/c胸腺欠損マウスに異種移植した、株化したMDA−MB231乳腺癌腫瘍の増殖に対する3PO処理の効果を示すグラフである。ノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=14)又は3PO処理群(○、n=13)に分けた。実験マウスの体重を測定し、研究期間(14日)の間、継続する3日間の投与の後3日間は無投与という周期的投与計画に従い、50μL DMSO、又は0.07mg/g3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は対照又は3PO投与を示す。2日目以後、DMSO対照群及び3PO実験群の間に統計的な差異が得られた(p−値<0.0001)。 図7Cは、Balb/c胸腺欠損マウスに異種移植した株化したHL−60急性前骨髄性白血病細胞の増殖への3PO処理効果を示すグラフである。ノギスを用いて毎日腫瘍を測定し、樹立した腫瘍(130〜190mg)を盲検的に無作為にDMSO対照群(●、n=11)又は3PO処理群(○、n=12)に分けた。実験マウスの体重を測定し、研究期間(14日)の間、継続する2日間のDMSO又は3POの投与、その後7日間の休息期間という周期的投与計画に従い、50μL DMSO、又は0.07mg/g 3POを含む50μL DMSOを腹腔内注射した。矢印(↓)は対照又は3PO投与を示す。最初の注射後、p−値<0.0001をもって、統計的な差異が得られた。
【図8】図8は、化合物2、3−(4−クロロフェニル)−1−(3−ピリジニル)−2−プロペン−1−オンのPFKFB3阻害活性を示す棒グラフである。150μM 2で処理した遺伝子組み換えPFKFB3の反応速度は、同容量の溶媒(EtOH)処理のPFKFB3で観測される反応速度の39.8%であった。
【図9】図9は、溶媒(DMSO)処理PFKFB3酵素の活性と比較した、化合物3 (即ち、3−(1−ナフチル)−1−(4−ピリジニル)−2−プロペン−1−オン),4 (即ち1−(3−ピリジニル)−3−(2−キノリニル)−2−プロペン−1−オン),5 (即ち、N−{4−[3−オキソ−3−(4−ピリジニル)−1−プロペン−1−イル]フェニル}アセトアミド),6 (即ち、3−(2−クロロフェニル)−1−(2−ピリジニル)−2−プロペン−1−オン)及び7 (即ち、3−(2−クロロフェニル)−1−(3−ピリジニル)−2−プロペン−1−オン)のPFKFB3阻害活性を示す棒グラフである。
【図10】図10は、異なる濃度(1,3,10又は33μM)の化合物2、又は溶媒(EtOH)処理した、HL−60細胞(灰色バー)及びK562細胞(白色バー)の細胞増殖を示す棒グラフである。バーは、代表的実験からの3回の値の平均値±STDを示す。
【図11】図11は0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物3をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図12】図12は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物4をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図13】図13は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物5をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図14】図14は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物6をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図15】図15は、0.1(中程度灰色バー)、0.3(無色バー)、1.0(斜めストライプバー)、3.0(ダイアモンド塗りバー)、10.0μM(水平ストライプバー)又は30.0μM(点状バー)の化合物7をグラフの最上部に示すように、48時間又は72時間処理したJurkat細胞の細胞増殖を示す棒グラフである。48時間、又は72時間DMSO処理したJurkat細胞の細胞増殖(暗色バー)もまた表示する。
【図16】図16は、化合物2〜7(0.3〜33μMにおける)存在下でのMDA−MB−231乳腺癌細胞の増殖と生存率を示すグラフである。
【図17】図17は、化合物2〜7(0.3〜33μMにおける)存在下でのA5491肺腺癌細胞の増殖と生存率を示すグラフである。
【図18】図18は、化合物2〜7(0.3〜33μMにおける)存在下でのK562慢性骨髄性白血病細胞の増殖と生存率を示すグラフである。
【図19】図19は、化合物2〜7(0.3〜33μMにおける)存在下でのHL−60前骨髄性白血病細胞の増殖と生存率を示すグラフである。
【図20】図20は、化合物2〜7(0.3〜33μMにおける)存在下でのCRL−11174メラノーマ細胞の増殖と生存率を示すグラフである。
【図21】図21は、化合物2〜7(0.3〜33μMにおける)存在下でのHeLa子宮頸癌細胞の増殖と生存率を示すグラフである。
【図22】図22は、Lewis肺癌細胞異種移植片を有するマウスに取り込まれた18F−2−デオキシグルコースのマイクロポジトロン発光断層撮影(トモグラフィー)画像を示す。マウスには、150μキュリーの18F−フルオロ−2−デオキシグルコースの腹腔注射(i.p.)30分前に、50μL DMSO(溶媒)又は0.07mg/g 3PO(+3PO)を含むDMSOのi.p.を行った。矢印は、右側腹部における心臓及び腫瘍異種移植片の位置を示す。
【図23】図23は、生後4ヶ月のトランスジェニックMMTV−Neu乳腫瘍マウスにおける腫瘍に対する3PO処理の効果を示すグラフである。該マウスは、無作為に、等しい腫瘍負荷を有する2群に分けた。第1群(+3PO)には、3日間投与(矢印で示す)及び3日間非投与の周期で、3PO(DMSO中、0.07mg/g;i.p.)を投与した。第2群(溶媒)には、DMSOのみ注射した。
【発明を実施するための最良の形態】
【0026】
本明細書において開示した発明を、代表的な実施態様を示す、実施例を参照しつつより詳細に以下に記載する。しかしながら、本明細書に開示した発明は、異なった形に具体化可能であり、本明細書に記載したように実施態様を制限すると解釈してはならない。むしろ、これらの実施態様は、開示が十分であり、完全であるように提供されており、当業者には、実施態様の範囲が十分に伝達されるであろう。
もし別に定義しなければ、本明細書で用いる技術及び科学用語は、本明細書に開示した発明が属する当業者の1人に一般的に理解されるのと同じ意味を有する。全ての出版物、特許出願、特許、本明細書に述べた参考文献は、その全体が参考文献に取り込まれている。
仕様及び特許請求の範囲を通して、与えられた化学式又は化学名は、光学及び立体異性体、並びに異性体及び混合物が属するラセミ混合物を包含する。
【0027】
この明細書では下記の略号を用いる:
2−DG=2−デオキシグルコース
3−BrPA=3−ブロモピルビン酸塩
3PO=3−(3−ピリジニル)−1−(4−ピリジニル)−2−プロペン−1−オン
AA=アミノ酸
ATP=アデノシン3リン酸
Ci=キュリー
DMSO=ジメチルスルフォキシド
DNA=デオキシリボ核酸
DMEM=ダルベッコの改変イーグル培地
EDTA=エチレンジアミンテトラ酢酸
F2,6BP=フルクトース−2,6−ビスリン酸塩
F6P=フルクトース−6−リン酸塩
HIF−1=低酸素症誘導因子1
ht=ヒトテロメラーゼ
LDH=乳酸デヒドロゲナーゼ
LT=ラージT抗原
μM=マイクロモル
mg=ミリグラム
mL=ミリリットル
min=分
ng=ナノグラム
NHBE=正常ヒト気管支上皮
Ni=ニッケル
nm=ナノメートル
NTA=ニトリロ三酢酸
PBS=リン酸緩衝生理食塩水
PEP=ホスホエノールピルビン酸塩
PFK−1=6−ホスホフルクト−1−キナーゼ
PFK−2=6−ホスホフルクト−2−キナーゼ
PFKFB3=6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3
PK=ピルビン酸キナーゼ
PMSF=フェニルメチルスルフォニルフッ化物
PRPP=5−ホスホ−D−リボシル−1−ピロリン酸塩
RNA=リボ核酸
SLN=Sybylライン表記法
STD=標準偏差
【0028】
1.定義
本明細書で用いる用語"癌"は、無制御な細胞分裂及び細胞の転移力、又は他の位置での新増殖を確立する力により引き起こされる病気を表わす。用語"悪性な"、"悪性"、"新生物"、"腫瘍"及び様々なこれらの変形語は、癌細胞又は癌細胞群を表す。
癌の特定の種類としては、皮膚癌、結合組織癌、脂肪腫、乳癌、肺癌、胃癌、膵臓癌、卵巣癌、子宮頸癌、子宮癌、肛門性器癌、腎臓癌、膀胱癌、大腸癌、前立腺癌、中枢神経(CNS)癌、網膜癌、血液及びリンパ球癌があるが、これらに制限されない。
【0029】
用語"競争的阻害剤"は、酵素に結合することにより、酵素の正常な基質の結合を阻害する阻害剤を表す。
本明細書で用いるように、用語"アルキル基"は、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert−ブチル基、ペンチル基、ヘキシル基、オクチル基、エテニル基、プロペニル基、ブテニル基、ペンテニル基、ヘキセニル基、オクテニル基、ブタジエニル基、プロピニル基、ブチニル基、ペンチニル基、ヘキシニル基、ヘプチニル基及びアレニル基を含む、直線(即ち、"直鎖")の、分枝の、又は環状の、飽和の、又は少なくとも部分的に及びある場合は完全に不飽和な(即ち、アルケニル基及びアルキニル基)炭化水素鎖を含むC1−20を表わす。"分枝状の"は、メチル基、エチル基又はプロピル基のような、低級アルキル基が直線型アルキル鎖に付加したアルキル基を表す。"低級アルキル基"は1から約8個の炭素原子(即ちC1−8アルキル基)、例えば、1,2,3,4,5,6,7又は8炭素原子、を有するアルキル基を表す。"高級アルキル基"は、約10から約20炭素原子、例えば、10,11,12,13,14,15,16,17,18,19,又は20炭素原子を有するアルキル基を表す。ある実施態様において、"アルキル基"は、特に、C1−8直鎖型アルキル基を表す。他の実施態様において、"アルキル基"は、特に、C1−8分枝型アルキル基を表す。
【0030】
アルキル基は、任意に、同一又は異なる、1又は2以上のアルキル基置換体により置換することができる("置換アルキル基")。用語"アルキル基置換体"は、アルキル基、置換アルキル基、ハロゲン原子、アリールアミノ基、アシル基、ヒドロキシル基、アリールオキシル基、アルコキシル基、アルキルチオ基、アリールチオ基、アラルキルオキシル基、アラルキルチオ基、カルボキシル基、アルコキシカルボニル基、オキソ基及びシクロアルキル基を含むが、これらに制限されない。アルキル鎖には、任意に、1又は2以上の酸素、硫黄、又は置換又は非置換窒素原子を挿入できるが、ここで窒素置換基は、水素原子、低級アルキル基(本明細書では、"アルキルアミノアルキル基"と表わす)、又はアリール基である。
従って、本明細書に用いるように、用語"置換アルキル基"には、例えば、アルキル基、置換アルキル基、ハロゲン原子、アリール基、置換アリール基、アルコキシル基、ヒドロキシル基、ニトロ基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、硫酸基及びメルカプト基が含まれるように、アルキル基の1又は2以上の原子又は官能基が他の原子又は官能基に置き変えられたアルキル基が含まれる。
【0031】
本明細書で用いる、用語"アリール基"は、共有結合で連結した、又はメチレン基又はエチレン基の部分又はこれらに制限されない普通の置換基に連結した、単一の芳香族環、又は互いに縮合した多環芳香族であることが可能な芳香族置換体を表す。普通の連結基としてはまた、ベンゾフェノンにおけるようなカルボニル基、又はジフェニルエーテルにおけるような酸素原子、又はジフェニルアミンにおけるような窒素原子であることができる。用語"アリール基"は、特に複素環式芳香族化合物も包含する。該芳香族環は、特に、フェニル基、ナフチル基、ビフェニル基、ジフェニルエーテル基、ジフェニルアミン基及びベンゾフェノン基を含むことができる。特定の実施態様において、用語"アリール基"は、約5から約10個の炭素原子、即ち、5,6,7,8,9,又は10炭素原子、を含み、また5及び6員炭化水素及び複素環式環芳香族基を含む、環状芳香族基を意味する。
【0032】
該アリール基は、任意に、同一又は異なる、1又は2以上のアリール基置換体により置換することができて、ここで"アリール基置換体"は、アルキル基、置換アルキル基、アリール基、置換アリール基、アラルキル基、ヒドロキシル基、アルコキシル基、アリールオキシル基、アラルキルオキシル基、カルボキシル基、アシル基、ハロゲン、ニトロ基、アルコキシカルボニル基、アリールオキシカルボニル基、アラルコキシカルボニル基、アシルオキシル基、アシルアミノ基、アロイルアミノ基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、アリールチオ基、アルキルチオ基、アルキレン基及び−NR'R"を含み、ここでR'及びR"は、それぞれ独立に、水素、アルキル基、置換アルキル基、アリール基、置換アリール基及びアラルキル基であることができる。
【0033】
従って、本明細書に用いるように、用語"置換アリール基"は、例えば、アルキル基、置換アルキル基、ハロゲン、アリール基、置換アリール基、アルコキシル基、ヒドロキシル基、ニトロ基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、硫酸基及びメルカプト基が含まれるように、アリール基の1又は2以上の原子又は官能基が他の原子又は官能基に置き変えられたアリール基が含まれる。
アリール基の特別な例としては、シクロペンタジエニル基、フェニル基、フラン基、チオフェン基、ピロール基、ピラン基、ピリジン基、イミダゾール基、ベンズイミダゾール基、イソチアゾール基、イソクサゾール基、ピラゾール基、ピラジン基、トリアジン基、ピリミジン基、キノリン基、イソキノリン基、インドール基、カルバゾール基等々が含まれるが、これらに制限されない。
【0034】
本明細書で用いるように、用語"アザ基"は、少なくとも1個の窒素原子を含む複素環式環状構造を表す。アザ基の例としては、ピロリジン基、ピペリジン基、キヌクリジン基、ピリジン基、ピロール基、インドール基、プリン基、ピリダジン基、ピリミジン基及びピラジン基を含むが、これらに制限されない。
用語"アザアリール基"は、アリール環又は環類の1又は2以上の原子が、窒素原子である複素環式アリール基を表す。アザアリール基の例としては単環又は二環のモノ−又はジアザアリール基(即ち、2個の窒素原子を含むアリール基)を含み、このアザアリール基は、非置換型、又は置換型であり、置換型の場合、例えばメチル基のような低級アルキル基;例えばメトキシ基のような低級アルコキシ基及び/又は例えば、塩素、又は臭素原子の様なハロゲン原子からなる群から選択された群の一つにより置換される。従って、該用語"アザアリール基"は、ピリジン基、ピリダジン基、ピリミジン基、ピラジン基、キノリン基、キナリジン基、キノクサリン基及びこれらの置換型の類似体を含む置換基を表わすが、これらに制限されない。幾つかの実施態様において、アザアリール基は、例えば、2−、3−、又は4−ピリジル基のようなピリジル基;例えば、4−キノリニル基又は1−イソキノリニル基のようなキノリニル基又はイソキノリニル基;イミダゾイル基;例えば、2−又は4−ピリミジニル基のようなピリミジニル基;例えば、3−ピリダジニル基のようなピリダジニル基;又は例えば、2−ピラジニル基のようなピラジニル基である。
【0035】
本明細書において用いられるように以下:
【化18】
のような化学式で一般的に表される構造は環状構造と呼ばれ、例えば、3炭素、4炭素、5炭素、6炭素等々、脂肪族及び/又は芳香族環状化合物であるが、これらに制限されず、またこれらはR置換基を含み、Rは存在しても不存在でもよい、存在する場合は、1又は2以上のR基は、環状構造の1又は2以上の可能な炭素原子上で置換されることができる。R基の存在又は不存在及びR基の数は、整数nの値により決定される。もし1以上の場合、各R基は、他のR基上より、環状構造の可能な炭素上で置換される。例えば
【化19】
式中、nは0から2の間の整数であり、上記の構造は、以下の構造群:
【化20】
を含む化合物群を含むが、これらに制限されない。
【0036】
化学構造において、結合を示す点線は、該結合が存在する、又は不存在であることを示す。例えば、下記:
【化21】
の置換基は、1重結合又は2重結合からなる置換基を表すことができる。
命名された原子又は置換基が"不存在"と定義される時、命名された原子又は置換基は、直接の結合により置換される。連結基又はスペーサー基が不存在であると定義される場合、連結基又はスペーサー基は直接結合により置換される。
【0037】
幾つかの実施態様において、本明細書において開示した発明の化合物は、連結基を含む。本明細書で用いるように、用語"連結基"は、2又は3個の他の分子部分と結合して、安定な構造を形成するアルキレン基、又はこれに制限されない置換基、を含む2価の分子部分を含む。幾つかの実施態様において、O又はSの様な単一原子は、連結基として働く。幾つかの実施態様において、連結基は、例えば、メチレンジオキシル(-O-CH2-O-)又はエチレンジオキシル(-O-CH2CH2-O-)のようなオキシ基又はチオ基を含むことができる。幾つかの実施態様において、該連結基は、フェニレン基、フラニル基、チエニル基、又はピロリール基のような、2価のアリール基であることができる。
【0038】
用語"3価連結基"は、1又は2以上の化学置換基上で、3個の置換基又は3個の部位(例えば、原子)を連結する連結基を表す。例えば、3価の連結基は以下の化学式:
【化22】
であってもよく、式中、各Rは、独立に、1又は2以上のO、S,窒素、又は置換窒素原子、又は不飽和結合を含むことができる、C0〜C6アルキル基である。
【0039】
"アルキレン基"は、1から約20個の炭素原子、例えば、1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,又は20炭素原子を有する直鎖型又は分枝型2価脂肪族炭化水素を表す。アルキレン基は、直鎖型、分枝型、又は環状である。アルキレン基は、また任意に、不飽和であり及び/又は、1又は2以上の、ヒドロキシル基、ハロゲン、ニトロ基、アルキル基、アリール基、アラルキル基、カルボキシル基、等々を含む、"アルキル基置換体"と置換されることができる。アルキレン基上に、1又は2以上の酸素、硫黄、又は置換又は非置換窒素原子(本明細書ではまた"アルキルアミノアルキル基"と表わす)を任意に挿入することができるが、ここで窒素置換基は、前述のようにアルキル基である。アルキレン基の例としては、メチレン基(−CH2−);エチレン基(−CH2−CH2−);プロピレン基(−(CH2)3−);シクロへキシレン基(−C6H10−);−CH=CH−CH=CH−;−CH=CH−CH2−;−(CH2)q−N(R)−(CH2)r―を含むが、式中、各q及びrは、独立に0から約20までの整数であり、例えば、0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19、又は20であり、またRは、水素又は低級アルキル基;メチレンジオキシル基(−O−CH2−O−);及びエチレンジオキシル基(−O−(CH2)2−O−)である。アルキレン基は、約2から約3個の炭素原子を有することができて、また更に6〜20個の炭素を有することができる。
【0040】
本明細書で用いるように、用語"アシル基"は、有機カルボン酸基を表し、ここでカルボキシル基の−OHは他の置換体(即ち、RCO−で表せるように、ここでRは、アルキル基、アラルキル基、又は、置換アルキル基、アラルキル基及びアリール基を含む、本明細書で定義されたアリール基である)で置換される。従って、用語"アシル基"は、特に、アセチルフラン基及びフェナシル基の様な、アリールアシル基を含む。アシル基の特別な例として、アセチル基及びベンゾイル基が含まれる。
【0041】
"環状"及び"シクロアルキル基"は、約3から約10個の炭素原子、例えば、3,4,5,6,7,8,9又は10炭素原子の、非芳香属性単環式又は多環式環状システムを表す。該シクロアルキル基は、任意に、部分的に不飽和であることができる。シクロアルキル基は、任意に、本明細書で定義したように、オキソ基及び/又はアルキレン基の様な、アルキル基置換体により置換されることができる。環状アルキル鎖に沿って、任意に、1又は2以上の酸素、硫黄、又は置換型の又は非置換型の窒素原子を挿入することができて、式中、窒素置換基は、水素、アルキル基、置換アルキル基、アリール基、又は置換アリール基であり、従って、複素環式置換基を提供する。代表的な単環式シクロアルキル環は、シクロペンチル基、シクロヘキシル基及びシクロヘプチル基を含む。多環式シクロアルキル環は、アダマンチル、オクタヒドロナフチル基、デカリン、樟脳、カンファン及びノラダマンチルを含む。
【0042】
"アルコキシル基"は、アルキル−O−基を表し、ここでアルキル基は、既述の通りである。本明細書で用いる、用語"アルコキシル基"は、例えば、メトキシル基、エトキシル基、プロポキシル基、イソプロポキシル基、ブトキシル基、t−ブトキシル基及びペントキシル基と表すことができる。用語"オキシアルキル基"は、"アルコキシル基"と互換的に用いることができる。
"アリールオキシル基"は、アリール−O−基を表し、ここでアリール基は、置換アリール基を含み、既述の通りである。本明細書で用いる用語"アリールオキシル基"は、フェニルオキシル基又はヘキシルオキシル基及びアルキル基、置換アルキル基、ハロゲン原子、又はアルコキシル置換フェニルオキシル基又はヘキシルオキシル基と表すことができる。
"アラルキル基"は、アリール−アルキル基を表し、ここでアリール基及びアルキル基は既述通りであり、また置換アリール基及び置換アルキル基を含む。アラルキル基の例として、ベンジル基、フェニルエチル基及びナフチルメチル基が含まれる。
"アラルキルオキシル基"は、アラルキル−O−基を表し、ここでアラルキル基は既述通りである。アラルキルオキシル基の例として、ベンジルオキシル基がある。
"ジアルキルアミノ基"は−NRR'基を表し、ここでR及びR'のそれぞれは、既述のように、独立に、アルキル基及び/又は置換アルキル基である。アルキルアミノ基の例としては、エチルメチルアミノ基、ジメチルアミノ基及びジエチルアミノ基が含まれる。
【0043】
"アルコキシカルボニル基"は、アルキル−O−CO−基を表す。アルコキシカルボニル基の例として、メトキシカルボニル基、エトキシカルボニル基、ブチルオキシカルボニル基及びt−ブチルオキシカルボニル基が含まれる。
"アリールオキシカルボニル基"は、アリール−O−CO−基を表す。アリールオキシカルボニル基の例として、フェノキシ−及びナフトキシ−カルボニルが含まれる。
"アラルコキシカルボニル基"は、アラルキル−O−CO−基を表す。アラルコキシカルボニル基の例としては、ベンジルオキシカルボニル基がある。
"カルバモイル基"は、H2N−CO−基である。
"アルキルカルバモイル基"はR'RN−CO−基を表し、ここでR及びR'の中の一方は、水素であり、R及びR'の中の他方は、既述のように、アルキル基及び/又は置換アルキル基である。
"ジアルキルカルバモイル基"は、R'RN−CO基を表し、ここでR及びR'のそれぞれは、独立に、既述のようなアルキル基及び/又は置換アルキル基である。
"アシルオキシル基"は、アシル−O−基を表し、ここでアシル基は既述の通りである。
"アシルアミノ基"は、アシル−NR−基を表し、ここでアシル基は、既述の通りであり、Rは、水素原子又はアルキル基である。従って、該"アシルアミノ基"は、構造−NR−C(=O)−R'を有することができて、R'は、アルキル基、アリール基、アラルキル基等々である。
【0044】
用語"アミノ基"は、−NH2基を表す。
用語"カルボニル基"は、−(C=O)−基を表す。
用語"カルボキシル基"は、−COOH基を表す。
本明細書で用いる様に、用語"ハロ"、"ハロゲン化物"又は"ハロゲン"は、フッ素、塩素、臭素及び沃素基を表す。
用語"ヒドロキシル基"は、−OH基を表す。
用語"ヒドロキシアルキル基"は、−OH基で置換されたアルキル基を表す。
用語"メルカプト基"は、−SH基を表す。
用語"オキソ基"は、本明細書で既述の化合物を表し、ここで炭素原子は酸素原子に置換される。
用語 "アザ基"は、炭素原子が窒素原子に置換された化合物を表す。
用語"ニトロ基"は、−NO2基を表す。
用語"チオ基"は、本明細書で既述の化合物を表し、ここで炭素又は酸素原子がイオウ原子に置換される。
用語"硫酸基"は、−SO4基を表す。
用語"カルコン"又は"カルコーン"は、1,3−ジフェニル−1−プロペン−3−オン:
【化23】
及びこの類似体(例えば、ジアリールプロペノン類、ジアリールプロペノン及びジアリールプロパンのチア及びアザ類似体)を表す。用語"アザカルコン"は、1又は2以上の窒素原子を含むカルコンを表す。特に該アザカルコンは、1又は2以上のアザアリール基を含むことができる。
【0045】
用語"独立に選択される"を用いた場合、関係する置換体(例えば、R1又はR2基、又はX及びY基のようなR基)は同一であるか、又は異なる。例えば、R1及びR2の両者は、アルキル基により置換される、又はR1は水素であることができて及びR2は置換アルキル基等々であることができる。
名付けられた"R"、"X" 、"A" 、"Ar"、"Y" 、又は"L" 基は、本明細書において特に他の名前で特定されなければ、一般的に当業者にその名前を有する置換基に対応すると認識される構造を有する。描画の目的のために、既述の代表的"R"、"X" 、"Y"、 "L"、"Ar"及び"A" 基 は以下のように定義される。これらの定義は、補充と描画のために意図され、本明細書開示を閲覧した当業者に明らかな定義を排除しない。
【0046】
II.一般的考察
癌組織において解糖フラックスが増加の方向へシフトする正確なメカニズムを理解することは、ある時は、重要の研究の焦点であった。解糖の増加が起こるには、様々な因子:a)基質利用可能性、b)解糖フラックスのために必要な代謝酵素の発現及びc)経路の特定の作用点で働くアロステリック活性化因子及び阻害剤のレベル、が含まれる。例えば、GLUT1の様な、グルコース輸送体の過剰発現は、亢進した解糖速度を維持するために必要な過剰なグルコースの供給を提供すると考えられており、他方ヘキソキナーゼ及びホスホフルクト−1−キナーゼ(PFK1)の様な鍵となる律速酵素は、癌細胞において亢進していることが明らかとなっている。Vora ほか, Cancer Res., 45(7), 2993-3001 (1985); Macheda ほか, J. Cell Physiol., 202(3), 654-662 (2005); 及び Mathupala ほか, Oncogene, 25(34), 4777-4786 (2006)を参照されたい。最後に、解糖酵素のアロステリック調節酵素は、特にPFK1が関わる最初の不可逆的な、委ねられた律速段階において、グルコースフラックスに大きな役割を果たす。Van Schaftingen, ほか., Proc. Natl. Acad. Sci. USA, 78(6), 3483-3486 (1981)を参照されたい。
【0047】
PFK1活性は、原発腫瘍組織ばかりではなく、腫瘍細胞において増加すると観測されてきた。さらに、腫瘍遺伝子ras又はsrcをRat−1繊維芽細胞又はニワトリ胎児繊維芽細胞に導入すると、それぞれ、高いPFK1活性をもたらす。Hennipman ほか., Tumour Biol., 8(5), 251-263 (1987); Kole,ほか, Arch. Biochem. Biophys. 286(2), 586-590 (1991); 及びSanchez-Martinez 及びAragon, FEBS Lett., 409(1), 86-90 (1997)を参照されたい。しかしながら、形質転換細胞内の高いレベルのPFK−1活性は、蛋白質発現の増加によるのではなく、フルクトース−2,6−ビスリン酸塩(F2,6BP)の増加によることが分かった。Kole, ほか., Arch. Biochem. Biophys. 286(2), 586-590 (1991)を参照されたい。1980年代の初め、F2,6BPに、PFK1のアロステリック活性化因子の可能性が見出され、基質である、フルクトース−6−リン酸塩に対して低親和性から高親和性へ、PFK1のコンホメーションの平衡をシフトすることにより、ATPの阻害効果を取り除くことができる。Van Schaftingen, ほか., Biochem. J., 192(3), 887-895 (1980); Van Schaftingen, ほか., Biochem. J., 192(3), 897-901 (1980); 及び Van Schaftingen, ほか, Proc. Natl. Acad. Sci., USA, 78(6), 3483-3486 (1981)を参照されたい。従って、F2,6BPレベルの亢進調節により、高エネルギー産生にも拘わらず(即ち、生理的ATPレベル)、癌組織が高い解糖的産生を維持することを可能にする。図1は、癌遺伝子rasが、如何に、癌細胞においてPFKFB3の発現及び活性を増加させ、従ってF2,6BP産生を亢進させ、律速PFK−1の活性化及び、究極的に、高い解糖フラックスをもたらすと信じられ、またRNA,DNA、アミノ酸(AAs)及び脂肪酸を含む高分子の産生を増加させるかを表す。
【0048】
細胞内のF2,6BPレベルは、6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ(PFK−2/FBPases)と名付けられた二機能性の酵素ファミリーにより維持され、また4種の遺伝子PFKFB1〜4によりコードされている。PFKFB1,2及び4は、それぞれ、肝臓/筋肉、腎臓/心臓及び精巣内に発現されると報告されており、ホスファターゼ活性に等しいキナーゼ活性を示す。PFKFB3遺伝子によりコードされているPFK−2の誘導型イソ型(iPFK2、胎盤PFK2,ユビキタス(遍在)PFK2及びPGR1と報告されている)は、分裂、炎症及び低酸素刺激により亢進されることが示されてきた、またホスファターゼ領域内の突然変異により、キナーゼ:ホスファターゼ比が740:1であることを示す。
【0049】
PFKFB3は、大腸、前立腺、肺、乳房、膵臓及び卵巣を含む多くの種類の癌組織において、過剰発現していることが観測されてきた。Atsumi, ほか., Cancer Res., 62, 5881-5887 (2002)を参照されたい。不死化したマウス繊維芽細胞に腫瘍遺伝子rasを導入すると、PFKFB3発現が高まり、PFKFB3は癌化形質転換において必要不可欠の成分であるかもしれないことを示唆する。Chesney及び共同研究者等は、PFKFB3に対するアンチセンスオリゴヌクレオチドを用いて、無胸腺マウスに異種移植したK562白血病細胞の抑制を示し、一方また、ヌクレオチド生合成の前駆体である5−ホスホリボシル−1−ピロリン酸塩(PRPP)の減少を観測した。ヒトテロメラーゼ、ラージT抗原及び癌遺伝子H−rasv12により、不死化し、続いてトランスフォームした異形接合のPFKFB3(+/−)マウス繊維芽細胞において、F2,6BP産生は減少し、グルコースの乳酸への解糖フラックスは抑制された。より重要なことは、rasトランスフォーム繊維芽細胞においてPFKFB3レベルが低いために、軟寒天中の基質非依存性増殖の喪失がもたらされ、また無胸腺マウスにおける腫瘍のin vivo増殖が顕著に減少する。Telang ほか., Oncogene, 25, 7225-7234 (2006)を参照されたい。これとは別に、siRNAによるA549肺癌細胞におけるPFKFB3の抑制は、F2,6BPの減少をもたらし、軟寒天中のコロニー形成の喪失をもたらす。
小分子阻害剤による選択的なPFKFB3の標的化が、新しい抗ガン治療の戦略であると言う所見を、本明細書にはじめて開示した。
【0050】
III.PFKFB3阻害剤
本明細書に開示した発明は、PFKFB3の阻害方法を提供し、該方法はPFKFB3と阻害化合物との接触を含む。幾つかの実施態様において、該方法はPFKFB3と小分子阻害剤との接触を含む。
【0051】
幾つかの実施態様において、本明細書に開示した発明は、PFKFB3と、PFKFB3を阻害する又は、そうでなければPFKFB3活性を変える化合物とを接触させる方法を提供し、式中、該化合物は以下の化学式(I):
【化1】
で表わされる化合物であり、
式中、Xは、C又はCHであり;
X1はO、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、夫々独立に、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から選択され、またR2及びR7は、夫々独立に、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から選択され;
L1は、存在又は不存在であってもよく、存在の場合、O、S、NR8、アルキレン基、置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在の場合、O、S、NR9、アルキレン基及び置換アルキレン基及び3価連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また3価連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及び、Ar1及びAr2からなる群の一つと結合する第3の原子を含む。
Ar1及びAr2は,独立にアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群から選択される。
【0052】
幾つかの実施態様において、化学式(I)で表わされる化合物は、アザカルコンである。
幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の両者は、アザアリール基である。幾つかの実施態様において、Ar1及びAr2の少なくとも一つは、ピリジン、置換ピリジン、キノリン、置換キノリン、イソキノリン及び置換イソキノリンからなる群から選択される。
【0053】
幾つかの実施態様において、Ar1及びAr2の少なくとも一つはアシルアミノ基により置換される。幾つかの実施態様において、化学式(I)で表わされる化合物は、アシルアミノ置換アザカルコンである。
幾つかの実施態様において、Ar1は、アザアリール基である。幾つかの実施態様において、Ar1は、2−、3−又は4−ピリジン又は置換2−、3−、又は4−ピリジンである。
【0054】
幾つかの実施態様において、L1は不存在である。幾つかの実施態様において、L2は、存在して、C2アルキレン基である。
幾つかの実施態様において、化学式(II)で表わされる化合物は、化学式(II):
【化2】
で表わされる構造を有し、式中、
X、X2及びX3は、それぞれC又はCHである。
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6のそれぞれは、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群より独立に選択され、また各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、夫々独立に、N又はCR12であり、式中、各R12は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び、次の構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4は、NR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;又は
式中、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は以下の構造:
【化4】
から選択され、式中、
各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4は、NR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び、
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアルキル基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;又は式中、R10及び1個のR13はともにアルキレン基であり;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つは、Nであり;
又はこれらの医薬的に許容された塩である。
【0055】
幾つかの実施態様において、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の2個は、Nである。
幾つかの実施態様において、X1は、Oであり、XはCである。
幾つかの実施態様において、Ar2は、次の構造:
【化5】
であり;
X、X2及びX3は、それぞれCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物は、化学式(III)で表わされる構造:
【化6】
を有する。
【0056】
幾つかの実施態様において、X1は、Oである。幾つかの実施態様において、R10及びR11のそれぞれは、水素原子である。
幾つかの実施態様において、Y1,Y2,Y3,Y4及びY5の中の少なくとも1つは、CR13であり、式中、R13は、構造:
【化3】
を有する置換基である。
幾つかの実施態様において、X5は、Oであり、X6は、水素原子、アルキル基、アラルキル基及びアリール基より選択され、またAr2は、アシルアミノ置換アリール基又はヘテロアリール基である。
【0057】
幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIa):
【化14】
で表わされる化合物である。
幾つかの実施態様において、化学式(IIIa)で表わされる化合物のA3はNである。幾つかの実施態様において、A3はNであり、A1,A2,A4及びA5のそれぞれは、CR12であり、またY1,Y2,Y4及びY5は、CR13である。
【0058】
幾つかの実施態様において、化学式(III)で表わされる化合物は、少なくとも1個のハロ置換体を含む。幾つかの実施態様において、該ハロ置換体はClである。幾つかの実施態様において、該ハロ置換体は、アルケンに付加する炭素に対してオルソ位置である。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIb):
【化15】
で表わされる化合物である。幾つかの実施態様において、化学式(IIIb)で表わされる化合物のA4及びA5の中の1つは、Nである。幾つかの実施態様において、A4及びA5の中の一つは、Nであり、A1,A2及びA3のそれぞれは、CR12であり、またY2,Y3,Y4及びY5のそれぞれは、CR13である。
【0059】
幾つかの実施態様において、ハロ置換体は、アルケンに付加する炭素に対してパラ位置である。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIIc):
【化16】
で表わされる化合物である。幾つかの実施態様において、化学式(IIIc)で表わされる化合物のA2は、Nである。幾つかの実施態様において、A2はNであり、A1,A3,A4及びA5のそれぞれは、CR12であり、またY1,Y2,Y4及びY5のそれぞれは、CR13である。
【0060】
幾つかの実施態様において、化学式(III)で表わされる化合物は、2個のアザアリール基を含む。幾つかの実施態様において、該化合物は、1個の4−ピリジル基及び1個の3−ピリジル基を含む。幾つかの実施態様において、化学式(III)で表わされる化合物は、化学式(IIId):
【化17】
で表わされる化合物である。幾つかの実施態様において、A1,A2,A4及びA5のそれぞれは、CR12であり、またY1,Y3,Y4及びY5のそれぞれは、CR13である。
【0061】
幾つかの実施態様において、化学式(III)で表わされる化合物は、以下の化学式:
【化7】
からなる群から選択される。
【0062】
幾つかの実施態様において、化学式(II)で表わされる化合物は、Ar2が:
【化8】
である化合物であり、式中、
X、X2及びX3のそれぞれは、Cであり;
X1はO、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群より独立に選択され;及び化学式(II)で表わされる化合物は、化学式(IV):
【化9】
で表わされる構造を有する。
【0063】
幾つかの実施態様において、化学式(IV)で表わされる化合物は、2個のアザアリール基を含む。幾つかの実施態様において、Y6及びA3のそれぞれは、Nである。幾つかの実施態様において、A1,A2,A4及びA5のそれぞれは、CR12であり、またY7,Y8,Y9,Y10、Y11及びY12のそれぞれは、CR13である。幾つかの実施態様において化学式(IV)で表わされる化合物は:
【化10】
である。
【0064】
幾つかの実施態様において、化学式(II)で表わされる化合物は、Ar2が:
【化11】
であり、
X、X2及びX3のそれぞれがCであり;
X1は、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1は、水素原子及びアルキル基からなる群から選択され、また各R2は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び該化合物は、化学式(V):
【化12】
で表わされる構造を有する。
【0065】
幾つかの実施態様において、A3はNである。幾つかの実施態様において、化学式(V)で表わされる化合物は:
【化13】
である。
【0066】
幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、in vitroにおいて、PFKFB3を阻害するために用いることができて、又はそうでなければ、PFKFB3に干渉又は相互作用する為に用いることができる。幾つかの実施態様において、該化合物は、PFKFB3の競合的阻害剤である。幾つかの実施態様において、該化合物は、細胞又はPFKFB3を含む細胞抽出液と接触させるために用いることができる。幾つかの実施態様において、該化合物は、組織、組織抽出物、又は血液試料のような他の生物由来の試料と接触するために用いることができる。幾つかの実施態様において、哺乳動物又はトリのような生存被験者にPFKFB3が存在する場合、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、in vivoでPFKFB3に接触するために用いることができる。幾つかの実施態様において、哺乳動物はヒトである。PFKFB3活性を阻害することにより、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、又はこれらの医薬学的許容物を用いて、細胞中の解糖フラックスを減少させることができて、幾つかの場合特に腫瘍細胞の場合、解糖フラックスの減少により、細胞内乳酸塩及びフルクトース−2,6−ビスリン酸塩レベルを減少させる。
【0067】
IV.医薬処方物
化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、これらの医薬学的に許容された塩類、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物に対応する前駆薬剤及びこれらの医薬学的に許容された塩類の全ては、本明細書において、"活性化合物"と呼ぶ。前述の活性化合物を含む医薬処方物もまた、本明細書に提供されている。本明細書において記載されるように、これらの医薬処方物は、医薬学的に許容された担体の中に、活性化合物を含む。以下により詳細に考察する様に、医薬処方物を、経口、静脈内、又は噴霧剤として投与することができる。また、本明細書に開示した発明は、凍結乾燥し、再構成して投与のための医薬学的に許容された処方物(ヒトに対して医薬学的に許容された処方物も含め)を作成できる、活性化合物を提供する。
【0068】
活性化合物の使用は、本明細書に記載された実施態様の範囲内であるが、全ての活性化合物の治療上有効な投与量は、化合物によって、また患者によって異なり、また患者の健康状態及び投与経路にも依存するであろう。一般的な計画としては、約0.1から約50mg/kgの投与量は、治療効果を有するであろう、但しここで全ての重量は、塩類を用いた場合も含めて、活性化合物の重量に基づいて計算されてある。高濃度における毒性を考慮して、静脈内投与量を、約10mg/kgまでのように、低濃度に制限することができる、但しここで全ての重量は、塩類を用いた場合も含めて、活性化合物の重量に基づいて計算されてある。経口投与の場合、約10mg/kgから約50mg/kgの投与量を採用することができる。一般的に、筋肉内注射に対して、約0.5mg/kgから約5mg/kgの投与量を採用することができる。静脈内又は経口投与に対して、好ましい化合物の投与量は、1μmol/kgから50μmol/kgであり、より好ましくは、22μmol/kg及び33μmol/kgである。治療期間は、通常、2から3週間の期間に対して1日1回であるか、又は健康状態が本質的に改善されるまでである。低頻度で低濃度の投与量を、感染の再発防止、又は発生率の低下のために、予防的に用いることができる。
【0069】
本明細書に開示した方法に従い、本明細書に記載した医薬学的に活性化合物は固体又は液体として、経口的に投与できて、又は溶液、懸濁液又は乳状液として、筋肉内又は静脈内に投与することができる。あるいは、該化合物又は塩を、リポソーム懸濁物として、吸入、静脈内、又は筋肉内に投与することができる。吸入により投与する場合、活性化合物又は塩は、粒子サイズが約0.5から約5μm及び好ましくは、約1から約2μmである、複数の固体粒子又はドロップの形をとる必要がある。
【0070】
静脈内又は筋肉内注射に適した医薬処方物は、本明細書に提供されたさらなる実施態様である。該医薬処方物は、本明細書に記載した、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、本明細書に記載した前駆薬剤、又は医薬学的に許容されたこれらの塩を、全ての医薬学的に許容された担体の中に含む。もし溶液が望まれるならば、水溶性化合物又は塩に関しては、水が選択された担体である。水溶性の化合物又は塩に関して、グリセロール、プロピレングリコール、ポリエチレングリコール又はこれらの混合物のような有機溶媒が適切であることができる。後者の例において、有機溶媒は、多量の水を含むことができる。上記二例における溶液は、当業者には既知の適切な手段で、一般的には、0.22μmフィルターを通して、無菌化できる。無菌化に続いて、該溶液を、脱パイロジェン化したガラスバイアルのような、適切な容器に分配することができる。この分配を、好ましくは、無菌法で行う。次ぎに殺菌した栓をバイアル上に置くことができて、必要に応じて、バイアル内容物を凍結乾燥できる。
【0071】
化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物又はこれらの塩又は前駆薬剤に加えて、医薬処方物は、pH調整添加剤のような他の添加剤を含むことができる。特に、有用なpH調整試薬としては、塩酸のような酸、乳酸ナトリウム、酢酸ナトリウム、リン酸ナトリウム、クエン酸ナトリウム、ホウ酸ナトリウム、又はグルコン酸ナトリウムのような、塩又は緩衝剤が含まれる。更に、該処方物は、抗菌保存剤を含むことができる。有用な抗菌保存剤としては、メチルパラベン、プロピルパラベン及びベンジルアルコールが含まれる。抗菌保存剤は、一般的に、処方物が多回数投与量使用を表すバイアルに入れられる場合に、使われる。本明細書に記載される医薬処方物は、当業者に既知の技術を用いて凍結乾燥される。
【0072】
本明細書に記載する発明の他の実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、又はこれらの塩を含む注射可能な、安定な、滅菌した処方物を提供する。該化合物又は塩は、密封した凍結乾燥物の形で提供される。該化合物又は塩を、適切な医薬学的に許容された担体と共に再構成可能であり、患者への注射に適した液体処方物を形成することができる凍結乾燥物として提供する。一般的に一回投与の形式は、約10mgから約10gの化合物塩を含む。化合物又は塩が、実質的に、非水溶性の場合、生理学的に許容された、十分量の乳化剤が十分量使われ、該化合物又は塩を水溶性担体に乳化することができる。このような有用な乳化剤としては、ホスファチジルコリンがある。
【0073】
他の医薬処方物を、本明細書に開示した、水溶性塩基乳化剤のような、非水溶性化合物又はこれらの塩から作成することができる。このような例の場合、該処方物は、十分量の医薬学的に許容された乳化剤を含み、必要量の化合物又はこれらの塩を乳化する。特に有用な乳化試薬は、ホスファチジルコリン及びレシチンを含む。
【0074】
本明細書に提供された更なる実施態様には、本明細書に開示した活性化合物のリポソーム処方物が含まれる。リポソーム懸濁物を作成する技術は、既知の技法である。化合物が、水溶性塩である場合、従来のリポソーム技術を用いて、脂質小胞にこれを取り込むことができる。このような例において、活性化合物が水溶性であるために、該活性化合物は、実質的に親水性中心、又はリポソームのコアに取り込まれる。用いられる脂質層は、従来型のどの様な組成も可能であり、コレステロールを含む、又は含めないことができる。注目する活性化合物が非水溶性の場合、再び、従来型のリポソーム形成技術を用いて、塩は、リポソームの構造を作る疎水性脂質二重膜内に、実質的に取り込まれる。いずれにしても標準的超音波及びホモジナイズ技術により、作成されたリポソームのサイズを、小さくすることができる。
【0075】
本明細書に開示された活性化合物を含むリポソーム処方物は、凍結乾燥により、凍結乾燥物を作ることができて、これは、水のような医薬学的に許容された担体を用いて、再構成して、リポソーム懸濁物を再生することができる。
【0076】
また吸入による煙霧剤として投与に適した医薬処方物として提供される。これらの処方物は、本明細書に記載した望ましい化合物、又はこれらの塩、又は化合物又は塩の複数の固体粒子の溶液又は懸濁物を含む。望みの処方物を、小さな容器に入れ、霧状にすることができる。噴霧化は、圧縮空気又は超音波エネルギーによって行われ、化合物又は塩を含む複数の液体ドロップ又は固体粒子を形成する。液体ドロップ又は固体粒子のサイズは、約0.5から約10μmの範囲、より好ましくは、約0.5から約5μmの範囲にあるべきである。固体粒子は、微粉化のような既知のいかなる方法によっても、固体化合物又はこれらの塩を処理して得ることができる。最も好ましくは、固体粒子又はドロップのサイズは、約1から約2μmであろう。この点において、この目的を達するために、商品化された噴霧器を用いることができる。その開示物は本明細書参照文献にその全体が取り込まれているが、合衆国特許番号5628984で公にされた方法で吸入可能な粒子の煙霧懸濁液を通して、該化合物を投与することができる。
【0077】
煙霧剤としての投与に適した医薬処方物が液体の場合、該処方物は、水を含む担体中の水溶性活性化合物を含むであろう。界面活性剤は、存在できて、処方物の表面張力を十分に低下させて、噴霧化した場合、望ましいサイズ範囲の小滴をもたらす。
【0078】
既述の様に、水溶性及び非水溶性活性化合物が提供される。本明細書で用いるように、用語"水溶性"は約50mg/mL又はそれ以上の量が水に溶ける全ての化合物を定義するつもりである。また、本明細書で用いるように、用語"非水溶性"は約20mg/mL以下の水への溶解度を有する全ての組成物を定義するつもりである。幾つかの実施態様において、水溶性化合物又は塩が好ましいが、他方、他の実施態様において、非水溶性化合物又は塩が好ましい。
【0079】
V.PFKFB3阻害剤で細胞増殖阻害し、癌を治療する方法
本明細書に開示した発明は、細胞増殖を阻害するための方法及び組成物を提供する。より詳細には、本明細書に開示した発明は、PFKFB3活性を阻害し、解糖フラックスを中断させる方法を提供する。解糖活性を中断することにより、細胞増殖は停止し、また、幾つかの場合、アポトーシスが引き起こされる。従って、本明細書に開示した発明は、望ましくない解糖活性及び/又は望ましくない細胞増殖が関与する、癌を含む病気を治療する方法を提供する。
【0080】
幾つかの実施態様において、細胞増殖の阻害又は癌の治療のための方法は、本明細書に記載した活性化合物を必要とする患者に対する、活性化合物の投与を含む。既述のように、これらの活性化合物は、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物、これらの前駆薬剤及び医薬学的に許容されたこれら化合物と前駆薬剤の塩を含む。幾つかの実施態様において、該活性化合物は、既述のように、医薬処方物の中に存在する。
【0081】
本明細書に開示した化合物は、皮膚癌、結合組織癌、脂肪腫、乳癌、肺癌、胃癌、膵臓癌、卵巣癌、子宮頸癌、子宮癌、肛門性器癌、腎臓癌、膀胱癌、大腸癌、前立腺癌、中枢神経癌(CNS),網膜癌、血液及びリンパ液癌を含む、多種の腫瘍及び癌に対する治療を提供することができる。
【0082】
癌の治療に関係して、本明細書で定義される"有効量"、は、癌細胞又は腫瘍の増殖を減らす、低減する、阻害する、又は他の方法で排除する量である。幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物を、患者の身体の特に病期に冒された、部位又は領域に局部的に輸送することができる。このような治療がより適切と考えられた幾つかの実施態様において、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物は、全身的に投与できる。例えば、該化合物を経口又は静脈内に投与できる。
【0083】
さらに、癌の治療に対する治療上の恩恵は、PFKFB3阻害化合物又は、本明細書で開示した他の化合物と、1又は2以上の他の抗癌剤又は治療と組み合わせた治療により実現することができる。その様な組合せの選択は、病気の種類、年齢及び患者の一般的健康状態、病気進行の攻撃性及び組合せに含められた試薬に対する患者の耐久力を含む、しかしこれらに限定されない、様々な因子に依存するであろう。例えば、PFKFB3阻害剤は、腫瘍サイズを減少させる上で有効な他の試薬及び治療計画(例えば、放射線、手術、化学療法、ホルモン治療及び/又は遺伝子治療)と組み合わせることができる。さらに、幾つかの実施態様において、PFKFB3阻害剤化合物を、病気の副次的影響又は治療試薬の一つの副作用を治療する、1又は2以上の薬剤と組み合わせることが望ましくできる、例えば、患者に、鎮痛剤、又は患者自身の免疫応答を高めるに有効な薬剤(例えば、コロニー刺激因子)を提供することである。
【0084】
幾つかの実施態様において、本明細書に開示した方法及び化合物は、我々が本明細書では"好気性治療"として、全体的に又は個別的に表す、好気性の急速に増殖する細胞に焦点を当てた化学療法及び/又は放射線療法と結びつけて用いることができる。幾つかの実施態様において、本明細書で開示したPFKFB3阻害剤の使用は、通常最も抵抗性であり、また従って、好気的治療を用いてでは根絶することが最も難しい、固形腫瘍の内部コアに見られる嫌気的な遅速増殖腫瘍細胞を選択的に殺すことにより、癌治療の有効性を付け加える。
【0085】
従って、本明細書で記載された発明の1又は2以上のPFKFB3阻害化合物と組み合わせて、"抗癌剤"又は"化学療法剤"としてもまた記載される様々な化学化合物を用いることができる。このような化合物としては、アルキル化剤、DNAインターカレーター、蛋白質合成阻害剤、DNA又はRNA合成阻害剤、DNA塩基類似体、トポイソメラーゼ阻害剤、抗脈管形成薬剤及びテロメラーゼ阻害剤又はテロメアのDNA結合化合物が含まれるが、これらに制限されない。例えば、適切なアルキル化剤としては、ブスルファン、インプロスルファン及びピポスルファンのようなスルホン酸アルキル;ベンゾジゼパ、カルボコン、メツレデパ及びウレデパのようなアジリジン;アルトレタミン、トリエチレンメラミン、トリエチレンホスホラミド、トリエチレンティオホスホラミド及びトリメチルオルメラミンのようなエチレニミン及びメチルメラミン;クロランブシル、クロルナファジン、サイクロホスファミド、エストラムスティン、イホスファミド、メクロレタミン、メクロレタミンオキシド塩酸塩、メルファラン、ノブエンビチン(Novembichine)、フェネステリン、プレドニムスチン、トロホスファミド及びウラシルマスタードのような、ナイトロジェンマスタード;カルムスチン、クロロゾトシン、フォテムスチン、ロムスチン、ニムスチン及びラニムスチンのようなニトロソウレアが含まれる。
【0086】
癌の治療に用いられる抗生物質としては、ダクチノマイシン、ダウノルビシン、ドクソルビシン、イダルビシン、硫酸ブレオマイシン、マイトマイシン、プリカマイシン及びストレプトゾシンが含まれる。化学療法に使われる抗代謝薬剤としては、メルカプトプリン、チオグアニン、クラドリビン、リン酸フルダラビン、フルオロウラシル(5−FU),フロクスウリジン、シタラビン、ペントスタチン、メトトレクセート及びアザチオプリン、アシクロビル、アデニンβ−1−D−アラビノシド、アメトプテリン、アミノプテリン、2−アミノプリン、アフィジコリン、8−アザグアニン、アザセリン、6−アザウラシル、2'−アジド−2'−デオキシヌクレオシド、5−ブロモデオキシシチジン、シトシンβ−1−D−アラビノシド、ジアゾオキシノルロイシン、ジデオキシヌクレオシド、5−フルオロデオキシシチジン、5−フルオロデオキシウリジン及びヒドロキシウレアが含まれる。
【0087】
化学療法のための蛋白質合成阻害剤としては、アブリン、オーリントリカルボン酸、クロランフェニコール、コリシンE3,シクロヘキシミド、ジフテリア毒素、エデインA,エメチン、エリスロマイシン、エチオニン、フッ化物、5−フルオロトリプトファン、フシジン酸、ジホスホン酸グアニリルメチレン及びジホスホン酸グアニリルイミド、カナマイシン、カスガマイシン、キッロマイシン及びO−メチルスレオニンがある。さらなる蛋白質合成阻害剤としては、モデシン、ネオマイシン、ノルバリン、パクタマイシン、パラモマイシン、ピューロマイシン、リシン、シガ毒素、ショウドマイシン、スパルソマイシン、スペクチノマイシン、ストレプトマイシン、テトラサイクリン、チオストレプトン及びトリメトプリムがある。硫酸ジメチル;マイトマイシンC;ナイトロジェンマスタード及びサルファーマスタードのようなアルキル化剤;アクリジン色素、アクチノマイシン、アドリアマイシン、アントラセン、ベンゾピレン、エシジウムブロマイド、2ヨウ化プロピジウムのようなインターカレート試薬;ジスタマイシン及びネトロプシンのような薬剤;等のDNA合成阻害剤もまた、化学療法組成物中で、本明細書に開示した発明の化合物と組み合わせることができる。クメルマイシン、ナリジクス酸、ノボビオシン及びオキソリン酸のようなトポイソメラーゼ阻害剤;コルセミド、コルヒチン、ビンブラスチン及びビンクリスチンを含む細胞分裂阻害剤;及びアクチノマイシンD,α−アマニチン及び他のカビのアマトキシン、コルディセピン(3'−デオキシアデノシン)、ジクロロリボフラノシルベンズイミダゾール、リファンピシン、ストレプトバリシン及びストレプトリジギンを含むRNA合成阻害剤もまた、本明細書に開示した発明のPFKFB3阻害剤化合物と組み合わせて、適切な癌治療に提供することができる。
【0088】
従って、本明細書に開示した発明のPFKFB3阻害剤と組合せて治療に用いることができる現在の化学療法薬剤としては、アドリアマイシン、5−フルオロウラシル(5FU),エトポシド、カンプトテシン、アクチノマイシン−D,マイトマイシン、シスプラチン、過酸化水素、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、イホスファミド、メルファラン、クロランブシル、ビサルファン、ニトロソウレア、ダクチノマイシン、ダウノルビシン、ドクソルビシン、ブレオマイシン、プリコマイシン、タモキシフェン、タキソール、トランスプラチナ、ビンブラスチン及びメトトレクセート等々がある。
【0089】
PFKFB3阻害化合物及び、他の化学療法薬剤のような、他の治療薬剤を含む組合せ治療は、細胞を、PFKFB3阻害剤及び他の薬剤と同時に接触させることにより達成することができる。このような組合せ治療は、該細胞を、両薬剤を含む単一組成物又は医薬処方物を接触させる、又は該細胞を、一方の組成物がPFKFB3阻害剤を含み、また他方が他の薬剤を含む、2種の別個の組成物又は処方物と同時に接触させることで達成することができる。
【0090】
あるいは、PFKFB3阻害化合物を用いる治療は、分から週単位の範囲の間隔で、他の薬剤治療の前、また後に適用することができる。他の薬剤及びPFKFB3阻害剤治療が、細胞に対して別々に行われる実施態様において、該薬剤及びPFKFB3阻害剤治療が、細胞上に有利な組合せ効果を示すことができる様に一般的に守られることは、両治療の間に長期の時間間隔が経ってしまいそれぞれの治療の間で失効しないことである。この様な場合、人は細胞を約12〜24時間以内に及び任意に、約6〜12時間以内に、両治療法に接触させることになる。幾つかの状況において、治療の期間をかなり延長することが望まれるが、それぞれの投与間隔を、数日間(2,3,4,5,6又は7日間)から数週間(1,2,3,4,5,6,7又は8週間)に延長される。また、幾つかの状況下で、PFKFB3阻害剤又は他薬剤の1回以上の投与が望まれる。
【0091】
他の実施態様において、本明細書に開示した発明のPFKFB3阻害剤化合物、又はPFKFB3阻害剤と組み合わせて用いる他の抗癌化合物は、単一クローン抗体(例えば、齧歯動物又はヒト単一クローン抗体)のような標的薬剤と結合した細胞毒性試薬と組み合わされ、又はこの細胞性毒試薬と共有結合する。後者の組合せにより、より大きな特異性を持って癌細胞に細胞毒性薬剤を導入することができることが分かるであろう。従って、細胞毒性薬剤の活性型(例えば、フリーな形)は、抗体により標的とされた細胞の中にのみ存在することになろう。
【0092】
他の癌治療法を、PFKFB3阻害剤化合物の投与と組み合わせて用いることができる。例えば、本明細書に開示した発明のPFKFB3阻害剤化合物は、さらに外科的に取り除いた部分、又は全ての癌の増殖に対する試みを含む治療コースの一部として用いることができる。例えば、本明細書に開示した発明のPFKFB3阻害剤を、患者の外科的治療の後に投与して、全ての残された癌、又は転移した細胞を治療することができる。本明細書で開示した発明のPFKFB3阻害薬剤による治療は、手術に先んじて行い、腫瘍のサイズを縮退させ、切除する組織量を減らす試みができて、これにより手術をより非浸襲的及び非衝撃的にする。
【0093】
本明細書に開示した発明のPFKFB3阻害薬剤による癌の治療は、放射線治療作用物を伴う1又は2以上の治療コースをさらに含み、DNA損傷をもたらすことができる。放射線治療作用物には、例えば、ガンマ線照射、X−線、UV−照射、マイクロ波、電子発光、放射性同位元素、等々がある。治療は、上記の形の放射線を局所的腫瘍部位に照射することで達成することができる。
【0094】
組合せ治療は、また、腫瘍細胞の表面に見出される腫瘍抗原に向けられた免疫治療を含めることができる。本明細書に開示したPFKFB3阻害剤で癌を治療することは、さらに、遺伝子治療に基づく治療と組み合わせることができて、p53,p16,p21、Rb,APC,DCC,NF-1,NF-2,BRCA2,FHIT,WT-1,MEN-I,MEN-II,BRCA1,VHL,FCC,MCC,ras,myc,neu,raf,erb,src,fms,jun,trk,ret,gsp,hst,bcl及びablのような、腫瘍遺伝子及び/又は細胞周期調節遺伝子、これらはしばしば癌組織中の正常細胞対応物の突然変異バージョンであるが、を標的とすることができる。
【0095】
本明細書に開示した発明のPFKFB3阻害剤化合物は、試験を行い、癌細胞の増殖阻害能、癌細胞のアポトーシス誘導能、腫瘍負荷の減少能及び転移阻害能を測定することができる。例えば、MTT試験により、細胞増殖を測定することができる。MTT試験により測定した増殖試験は、広く既知である。MTT試験において、癌細胞を様々な濃度の抗ガン化合物とインキュベートし、細胞生存能をテトラゾリウム塩、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロム化物(MTT)の着色したホルマザン塩の形成を測定して決める。細胞死及び、又は細胞増殖を測定する他の既知の試験もまた用いることができる。
【0096】
例えば、OVCAR−5腫瘍細胞をヌードマウスに移植したマウス異種移植モデルを用いてin vivo試験を行うことができて、化学式(I),(II),(III),(IIIa)、(IIIb)、(IIIc)、(IIId)、(IV)又は(V)で表わされる化合物で処理したこのマウスは、平均して、最初の投与後ある時期の間増殖するが、処理を続けると、腫瘍塊が縮小することが期待される。それに対して、対照(例えばDMSO)処理を行ったマウスは、腫瘍塊が増殖し続けると期待される。本明細書に開示した化合物の抗癌効果を測定する更なる方法を、本明細書の以下の実施例において記載する。
【0097】
VI.スクリーニング方法
本明細書に開示した発明は、化合物の持つPFKFB3への妨害(例えば、阻害又は競合的な阻害)能についてスクリーニングする方法を提供する。より詳細には、該方法は、(1)PFKFB3のF6P結合ポケットを含むPFKFB3の3Dモデルの提供;(2)化合物(例えば、小分子)の3Dモデル;及び(3)PFKFB3モデルのF6P結合ポケットの中の1又は2以上の溶媒接触表面との相互作用能を有する化合物のスクリーニングを含む。幾つかの実施態様において、該方法は、計算機(例えば、コンピューター内での)を用いて行うことができる。幾つかの実施態様において、該方法は、(a)複数の化合物(例えば、化合物のライブラリー又はデータベース)の3Dモデルの提供及び(b)各化合物の個別のスクリーニング及びその後、(c)複数の化合物中のある一個の化合物のF6P結合ポケットとの相互作用能と、他の化合物の相互作用能との比較;を含むことができる。
【0098】
従って、一態様において、化合物をスクリーニングする方法は、一般的に、PFKFB3のF6P結合ポケットの計算機モデルに対する選択した化合物(化学的実体)が持つ結合能を、計算機によって評価することを含む。例えば、この方法は、(a)選択した化合物とPFKFB3のF6P結合ポケットの間の嵌め込み操作を行うための計算機によるアプローチを用いるステップ;(b)嵌め込み操作の結果を解析して、化合物と結合ポケットの間の結合を定量化するステップを含む。幾つかの実施態様において、目視検査、又は計算機を用いたドッキング(結合)実験を用いて、ある分子、又は分子のライブラリーを、PFKFB3のF6P結合ポケットとの結合親和性について試験することができる。
【0099】
幾つかの実施態様において、PFKFB3の計算機モデルは、PFKFB3アイソザイムの既知のX−線構造から導かれることができる。例えば、PFKFB3のあるモデルは、PFKFB3の残基配列をヒト肝PFKFB1及びラット精巣PFKFB4アイソザイムの同型配列に揃えて並べることにより導くことができる。PFKFB3自体の結晶構造もまた記載されている。Kimほか,J.Biol.Chem.,281(5)2939-2944(2006)を参照されたい。
【0100】
用語"結合ポケット"は、その形状の結果として、他の化合物(例えば、F6P又は阻害剤)と都合良く結合する分子又は、分子複合体のある領域を表す。用語"ポケット"は、裂け目、チャンネル、又は部位を含む、がこれらに制限されない。結合ポケットの形状は、大体は化合物との結合前にあらかじめ形成され、化合物との結合と同時に形成され、又は分子の異なる結合ポケットへ他の化学物質が結合し、このことが逆に結合ポケットの形状の変化を誘導する、ことにより形成されるかも知れない。
【0101】
用語"ドッキング(結合)"は、ディスタンスジオメトリー又はエネルギーに基づいて、結合ポケット、領域、分子、又は分子複合体又はこれらの部分における、化合物の配向;回転;平行移動を表す。ドッキングは、ディスタンスジオメトリー法によって行うことができて、この方法は結合ポケット、領域、分子又は分子複合体又はこれらの部分の球状プローブの中心の集合にマッチ(適合)する化合物の原子集合を見出す。Mengほか,J.Comp.Chem.,4,505-524(1992)を参照されたい。球状プローブ中心は、結合ポケット、領域、分子又は分子複合体又はこれらの部分において、原子(水素原子を除く)からの所与の距離に追加の半径を加えることで作り出される。リアルタイム相互作用エネルギー計算、エネルギー最小化、又は剛体最小化(Gschwend ほか,J.Mol Recognition,9:175-186(1996)参照)を行って、化合物を配向させて、ドッキングを容易にすることができる。例えば、相互作用をするドッキング実験を計画して、最小抵抗の経路に従うように設計することができる。相互作用的ドッキング実験において、使用者がエネルギー増加の方向へ動くならば、該システムは、その運動に抵抗する。しかしながら、もし該使用者が、エネルギー減少の方へ動くなら、該システムは、応答性が増加して動くことを好む。(Cohen,ほか,J.Med.Chem.,33,889-894(1990))ドッキングはまた、分子親和性ポテンシャルを用いて、MonteCarlo探索技術と早いエネルギー評価を組み合わせることにより為し遂げることができる。Goodsell及びOlson,Proteins:Structure,Function and Genetics 8,195-202 (1990)を参照されたい。ドッキング機能を行うソフトウェアプログラムとしては、MATCHMOL(Cory,ほか.J.Mol Graphics,2,39(1984));MOLFIT(Redington,Comput Chem.16,217(1992)参照)及びDOCK(Mengほか、上記参照)が含まれる、がこれらに制限されない。
【0102】
用語"3次元(D)構造の創出"又は"3次元表示の創出"は、構造座標のリストを3次元の構造モデル又はグラフによる表出に変換することを表す。これは、商業的に、又は公共で入手可能なソフトウェアにより行われる。従って、分子又は分子複合体の3次元構造のモデルは、構造座標が与えられ、また正しいソフトウェアをインストールした計算機により、計算機のスクリーン上に構築することができる。該3次元構造を表示すること、又は計算機モデリング又は当て嵌め操作を行うために用いることができる。さらに、表示されたモデルは無くとも、構造座標それ自体を用いて、計算機に基づくモデリング及び当て嵌め操作を行うことができる。
【0103】
幾つかの実施態様において化合物が持つ、結合ポケット又は溶媒接触表面との相互作用能を定量することができる。用語"接触スコア"は、化合物及び結合ポケットの間での形状相補性の指標を表し、これは、化合物の原子の全て又は部分及び結合ポケット内に結合したリガンドの原子の全て又は部分の間の最小自乗スーパーインポジション(重ね合わせ;superimposition)から得られる、RMSD値と相関する。接触スコア又はRMSD値により、ドッキング過程を容易にすることができる。例えば、もし化合物が高いRMSDを有する位置に動くならば、システムはその動きに抵抗するであろう。化合物の配置の集合は、接触スコアにより、ランク付けすることができる。より低RMSD値はより高い接触スコアを与えるであろう。Mengほか.J.Comp.Chem.,4,505−524(1992)を参照されたい。
【0104】
用語"二乗平均偏差"又は"RMSD"は、平均値からの変位の二乗の算術平均の平方根を意味する。これは、傾向又は対象からの変位又は偏差を表現する一方法である。用語"構造座標"は、結晶型のタンパク質又はタンパク質複合体の原子(散乱中心)による、X−線の単色光線の回折から得られたパターンに関係する数式から導かれたCartesian(デカルト)座標を表す。回折データを用いて、結晶の繰り返し単位の電子密度地図を計算する。次ぎに、電子密度地図を用いて、分子又は分子複合体の個々の原子の位置を確定する。
【0105】
本明細書の発明を閲覧した当業者には理解できることであるが、計算機スクリーニング法のために、様々な小分子(即ち、1000原子質量単位(amu)又はそれ以下の分子量を有する化合物、750amu又はそれ以下、又は500amu又はそれ以下)ライブラリーが、入手できる。用いることができる化学構造データベースは、ACD(Molecular Designs Ltd,現在 Elsevier MDL, San Ramon, California, USA),NCI(National Cancer Institute, Fredrick, Maryland, USA), CCDC(Cambridge Crystallographic Data Center, Cambridge,英国),CAST(Chemical Abstract Service, Columbus, Ohio, USA), Maybridge(Maybridge Chemical Company Ltd, Tintagel,英国),Aldrich(Aldrich Chemical Company, Milwaukee, Wisconsin)及び Directory of Natural Products(天然物要覧、Chapman & Hall, London,英国)を含むが、これらに制限されない。
【0106】
CONCORD(Tripos Associates,St.Louis,Missouri, USA)又はDB-Converter(Molecular Simulations Ltd.,San Leandro,California, USA)のような計算機プログラムを用いて、2次元に表示されたデータセットを3Dに表示されたデータセットに変換するために用いることができる。望みのファーマコフォア(薬理原子団)を有する分子を同定するための3Dデタベースを探索するために適したプログラムとしては、MACCS-3D及び ISIS/3D(Molecular Design Ltd.,現在 Elsevier MDL, San Ramon California, USA) ChemDBS-3D(Chemical Design Ltd., Oxford,英国)及びSybyl/3 DB Unity(Tripos Associates, St.Louis, Missouri, USA)がある。本明細書で用いるように、"ファーマコフォア"又は"薬作用発生パターン"は、結合に対して重要であると信じられている、化合物の特徴の幾何学的配置である。
【0107】
化合物はまた、細胞をベースとしたスクリーニング法を用いて、PFKFB3の阻害能についてスクリーニングすることができる。従って、幾つかの実施態様において細胞増殖停止能及び/又はPFKFB3発現細胞における乳酸塩産生能について、化合物をスクリーニングすることができる。幾つかの実施態様において、細胞をベースとしたスクリーニング法を用いて、前述のin silicoスクリーニング法で同定した化合物のPFKFB3阻害能をさらに証明するために化合物をスクリーニングすることができる。
【実施例】
【0108】
一般的方法
対照と3PO処理の間の増殖阻害、乳酸塩産生及びin vivo研究についての統計的有意さは、Graph Pad Prism Version3.0(Graph Pad Software,San Diego,California, USA)を用いた2試料t−検定により定められた。p値<0.01は、統計的に有意であると考える。
【0109】
実施例1
PFKFB3分子モデリング及び化合物スクリーニング
計算機によるモデリングは、Silicon Graphics Array(1TB以上の記憶装置を備えた71×R12000及びR140000プロセッサー),32P R14000 500MHz Origin 2000サーバー及び2P R12000 300MHz OCTANEグラフィックワークステーション(SGI,Sunnyvale,California, USA)を用いて行った。PFKFB3ホモロジーモデルは、構造上の原型としてラット精巣PFKFB4(PDBコード1B1F)アイソザイムのX線構造を用いた。ClustalWを用いてアラインメントを作り出した。Chennaほか,Nucleic Acids Res.,31,3497-3500(2003)を参照されたい。4種のホモロジーモデルをModeller(Sali及びBlundell, J.Mol.Biol.,234,779-815(1993)参照)を用いて作成し、またPFKFB3結合部位(Chesney ほか,Proc.Natl.Acad Sci,96,3047-3052(1999)及びBertrandほか.,Eur.J.Biochem.,254,490-496(1998)参照)を最も良く再現した構造を更なる使用のために選択した。PFKFB3のためのリガンド結合及びタンパク質活性に重要な残基(Chesney ほか,Proc.Natl.Acad.Sci,96,3047-3052(1999)及びBertrandほか,Eur.J.Biochem.,254,490-496(1998)参照)は、コンセンサス(共通)構造における等価な残基数と相関した。該モデルをInsightII (Accelrys, San Diego California, USA)に読み込み、また3種の重要な残基Arg66,Tyr161及びThr94を仮想スクリーニング運転の重心標的として選択した。PFKFB3ホモロジーモデルの描画を図2A,2B及び2Cに示す。
【0110】
ホモロジーモデル(配列番号1)に対するアミノ酸配列は、Homo sapiens由来の6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビホスファターゼ3(GENEBANK(R)登録番号NP_004557)のアミノ酸配列34〜466のアミノ酸に対応する。配列番号1は、GENBANK(R)登録番号、NM_004566(配列番号2)のヌクレオチド429〜1727によりコードされている。
【0111】
配列番号1は:
SPTVIVMVGLPARGKTYISKKLTRYLNWIGVPTKVFNVGEYRREAVKQYSSYNFFRPDNEEAMKVRKQCALAALRDVKSYLAKEGGQIAVFDATNTTRERRHMILHFAKENDFKAFFIESVCDDPTVVASNIMEVKISSPDYKDCNSAEAMDDFMKRISCYEASYQPLDPDKCDRDLSLIKVIDVGRRFLVNRVQDHIQSRIVYYLMNIHVQPRTIYLCRHGENEHNLQGRIGGDSGLSSRGKKFASALSKFVEEQNLKDLRVWTSQLKSTIQTAEALRLPYEQWKALNEIDAGVCEELTYEEIRDTYPEEYALREQDKYYYRYPTGESYQDLVQRLEPVIMELERQENVLVICHQAVLRCLLAYFLDKSAEEMPYLKCPLHTVLKLTPVAYGCRVESIYLNVESVCTHRERSEDAKKGPNPLMRRNSVTPLAである。
【0112】
Ludi(Accelrys,San Diego,California, USA)仮想スクリーニングプログラムを用いて、ChemNavigator iResearch library(ChemNavigator,San Diego,California, USA)を処理した。これらのスクリーニング運転の完了後、Ludiのスコアリングシステムを用いて500以上のスコアの分子を、タンパク質の活性位置内で仮想的吟味を行い解析した。活性ポケット内に正しく結合したリガンドは、スクリーニングのために用いた標的及びライブラリーに従って目録に入れた。200個のスコアが最高の分子を、Scifinder Scholarを用いて同定し、またトップ45個の分子を実験検定可能あるものと選択した。
【0113】
PFKFB3 F6P結合ポケット中に結合ポテンシャルを有すると決定された化合物の幾つかの構造を以下の表1に示す。本明細書に開示した発明の化合物は、Sigma-Aldrich(St.Louis,Missouri, USA)及びChembridge Corporation(San Diego,California, USA)の様な、しかしこれらに制限されない、適切な市販供給源から得ることができる。
【0114】
あるいは、カルコン、アザカルコン及びこれらの類似体もまた既知の方法で合成することができる。例えば、カルコン及びアザカルコンは、適切なアリールアルデヒド及びアリールケトンを縮合させ、その後脱水させ(H2Oの除去)、エノンを作成する。該縮合は、酸又は塩基を触媒とすることができる。幾つかの実施態様において、ベンズアルデヒド、置換ベンズアルデヒド又はこれらのアザ類似体を、アセトフェノン、置換アセトフェノン又はこれらのアザ類似体と、塩基(例えば、NaOH水溶液)存在下に、適切な温度で、ある時間の間(例えば、分、1から数時間、1日又は2日以上)混ぜ合わせることができる。該溶液を疎水性溶媒を用いて抽出し、濃縮してカルコン及びアザカルコンを提供することができる。幾つかの実施態様において、該抽出物をさらに、クロマトグラフィー又は再結晶のようないかなる方法を用いても、精製することができる。
【0115】
【表1】
【0116】
実施例2
In vitro組み変え酵素検定
PFKFB3クローニング、発現及び精製: ヒトPFKFB3 cDNAを以下のプライマー:
PFKFB3F: 5'-CTTCATATGCCGTTGGAACTGACGCA-3' (配列番号3)
PFKFB3R: 5'-CTTCTCGAGGTGTTTCCTGGAGGAGTCAGC-3' (配列番号4)
を用いて、既存の哺乳動物発現プラスミドから増幅した。
該PCA産物は、XhoI及びNdeI制限酵素(Promega, Madison, Wisconsin, USA)で消化し、またpET−30b(+)ベクター(Novagen, San Diego, California, USA)の対応する位置にクローン化した。該pET−30b(+)−PFKFB3C−termHis plasmidをBL21(DE3)E. coli コンピテント細胞(Novagen, San Diego, California, USA)に形質導入した。
【0117】
PFKFB3の発現及び精製のために、BL21−PFKFB3形質転換細胞の1L培養を16時間、37℃で振盪した。16時間後に、この培養液に、さらに1Lの2mMイソプロピルβ−D−1−チオガラクトピラノシド(IPTG;最終濃度=1mM)を含むLB培地を加えた、そして4時間30℃で振盪した。細菌を遠心で集菌し、タンパク質精製をQiagen(Venlo,オランダ)に記載されたプロトコールに従い、自然条件下で行った。手短に言うと、細菌ペレットを1回Dulbeccoのリン酸緩衝液(PBS;Invitrogen,Carlsbad,California, USA)で洗浄し、ペレットのグラム当たり2mLの溶解緩衝液に懸濁し、この溶解緩衝液は、5mM β−メルカプトエタノール、1mg/mL リゾチーム、1mM フェニルメチルスルフォニルフッ化物(PMSF)を含みさらにプロテアーゼ阻害カクテル(Sigma-Aldrich,St.Louis,Missouri, USA)1:100稀釈液を添加したものである。次ぎに該細菌混合物を、10秒間、8回超音波に懸け、遠心により上清を得た。溶解物を1時間、3mLの50% Ni−NTAとインキュベートし、該混合物をPoly−Prepクロマトグラフィーカラム(BioRad,Hercules,California, USA)に載せた。ビーズを1mM PMSF及びプロテアーゼ阻害カクテルの1:100稀釈液を含む15mLの6xHis洗浄緩衝液で洗浄し、PFKFB3を、200mMイミダゾールを含む1.5mLの溶出緩衝液で溶出した。さらなる精製のために、溶出分画を16時間20mMTris−HCl、200mM NaCl(pH 7.4)緩衝液に対して透析して、その後Sephadex S200カラム(Amersham,Piscataway,New Jersey, USA)によりゲル濾過した。
【0118】
PFKFB3酵素検定: 既述のように、PFKFB3活性を、ピルビン酸キナーゼ及び乳酸デヒドロゲナーゼを用いた酵素共役のキネティック検定により測定した。Bucher及びPfleiderer, Methods in Enzymology, 1, 435-440 (1955)を参照されたい。該検定は、化学量論的に1mol ATP:1mol 消費NADHに従い、340nmでの吸光度の減少から分光学的にNADH消費を測定する。該検定を、37℃で、最終200μLの反応溶液を用いて、96ウェルプレートを用いて行ったが、該反応溶液には、1xPFKFB3 反応緩衝液(100mM Tris−HCl、100μM EDTA, 5mM KH2PO4;pH=7.4)、420μM ホスホエノールピルビン酸(PEP),5mM ATP, 400μM NADH,613ユニットのピルビン酸キナーゼ(PK)、1092ユニットの乳酸デヒドロゲナーゼ(LDH),様々な濃度のフルクトース−6−リン酸(40〜400μM)及び15μgの精製PFKFB3が含まれた。酵素検定のための対照反応として、F6P無しの活性PFKFB3を用いた。化合物1阻害に対し、60μM,100μM又は150μM阻害剤存在下で、反応を行った。化合物1阻害に対する対照反応は、PFKFB3の添加無しで、様々な濃度の1を用いた。検定は、BioTek POWERWAVE(R)プレートリーダー(BioTek Instruments,Inc.,Winooski,Vermont, USA)を用いて、15分以上のキネティックモードで、測定して、PFKFB3の反応速度を測定した。SigmaPlot(R)9.0(SYSTAT Software,Inc., San Jose, California, USA)に対する酵素キネティック測定規準を用いて、PFKFB3及び1阻害に対するキネティックパラメーター(Vmax,Km及びKi)を計算した。Vmaxは、PFKFB3の比活性を、nmolF6P×min―1×mg−1で表し、Km及びKiは、それぞれ、F6P又は1に対して、μMで表される。表示したデータは、2回の独立した実験の、3通りの実験の平均値±STDである。
【0119】
図3Aに示すように、Lineweaver-Burke2重逆数プロットにより、1はPFKFB3活性を競合的及び非競合的な、阻害機構によりPFKFB3活性を阻害していることが明らかとなる。1とのインキュベーションにより、低濃度のF6Pで、PFKFB3酵素活性の顕著な用量依存的減少がもたらされる。しかしながら、この阻害は、F6P量の増加により克服されるので、PFKFB3結合位置において、1とF6Pの間で競合が在ることが示唆される。これらの研究より、酵素反応速度パラメーターを、PFKFB3活性及び化合物1阻害に対して計算した。組み換えPFKFB3の比活性は、277±9nmol F6P×min―1×mg−1であることが分かり、またF6Pに対するKmは97μMと決定された。図3Bを参照されたい。1に対するKiは、25±9μMと決定され、この値は、F6Pに対するKmと比較して、約4分の1に減少した。これらの研究により、1は主にF6Pとの競合によるPFKFB3の阻害剤であり、競合的阻害剤に対する酵素基質結合位置の分子標的は、抗癌治療同定するための重要な方法であることが、確認できた。
【0120】
実施例3
形質転換した腫瘍細胞における化合物1の毒性
哺乳動物発現に対するFLAG−PFKFB3構築体の作成: PFKFB3全コード配列及びN−末端にFLAG−エピトープを含むFLAG−PFKFB3をレトロウィルスTet応答性ベクターpRevTRE(Clontech,Mountain View,California, USA)内のBamH1/HindIII制限部位にサブクローン化した。組み換えレトロウィルスは、Lipofectamine(Invitrogen, Carlsbad, California, USA)を仲立ちとするPT67パッケージ細胞株にpRevTRE−FLAG−PFKFB3構築体を形質導入することより作り出した。誘導可能FLAG−PFKFB3を安定的に組み込み、また発現するJurkat細胞株を作るために、細胞をFLAG−PFKFB3を含む組み換えレトロウィルスで感染させ、400μg/mL ヒグロマイシン(Clontech, Mountain View, California, USA)存在下で安定なクローンを選択した。
【0121】
細胞培養: K562,HL−60,MDA−MB231及びメラノーマ(CRL−11174)ヒト癌細胞株をATCC(American Tissue Type Culture Collection,Manassas,Virginia, USA)から購入した。HeLa,A549,Lewis Lung Carcinoma,MDA−MB231及びメラノーマ細胞を10% 仔牛胎児血清(Hyclone,Logan,Utah, USA)及び50μg/mL 硫酸ゲンタマイシン(Invitrogen, Carlsbad, California, USA)を補強したDulbeccoの改変Eagle培地(DMEM)(Hyclone, Logan, Utah, USA)中で増殖させた。HL−60、K562及びJurkat細胞は、10% 仔牛胎児血清及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI−1640培地(Hyclone,Logan,Utah, USA)で増殖させた。初代正常ヒト気管上皮(NHBE)細胞及びヒトテロメラーゼ(ht)及びラージT抗原(LT)で不死化し、また突然変異したrasでトランスフォームした細胞(ht/LT/ras細胞)は、52μg/mL 仔牛脳下垂体抽出物、0.5μg/mL ハイドロコーチゾン、0.5ng/mL ヒト上皮成長因子、0.5μg/mL エピネフィリン、10μg/mL トランスフェリン、5μg/mL インシュリン、0.1ng/mL レチノイン酸、6.5ng/mL 三ヨウ化チロニン、50μg/mL ゲンタマイシン及び50ng/mL アンフォテリシンB(SingleQuots, Cambrex, Walkersville, Maryland, USA)を補強したBronchial Epithelial Cell Basal Growth Medium(Cambrex, Walkersville, Maryland, USA)中で培養した。全ての細胞株は、37℃で、5% CO2下で維持した。
【0122】
細胞周期解析及びフローサイトメトリー: Jurkat細胞を、10% FBS及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI培地中で1×105細胞/mLの密度で蒔種した。細胞周期をVybrant DyeCycle Orange染色(Molecular Probes-Invitrogen, Eugene, Oregon, USA)を用い製造者の指針に従って行った。
【0123】
In Vitro増殖抑制: 全ての細胞株を1×105細胞/wellの密度で、適切な培地中に蒔種した。懸濁細胞に対して、様々な濃度の1を直ちに培地に加えた、それに対して、接着細胞に対しては、化合物1処理は、翌日に開始した。用量依存性研究に於いて、36時間にわたって様々な濃度の化合物1を加えた。時間依存性研究に於いて、10μMの化合物1を、0,4,8,16,24,又は36時間目に加えた。PFKFB3過剰発現研究のために、FLAG―PFKFB3発現ベクター又は対照プラスミドを含むJurkat細胞を、化合物1とのインキュベーションの24時間前にドキシサイクリン(1?g/mL;Clontech,Mountain View,California, USA)投与により誘導した。
細胞を処理48時間後に集め、細胞数及び生存率をトリパンブルー排除法により測定した。溶媒処理細胞増殖の50%増殖に必要な化合物1濃度として、IC50値を計算した。表示したデータは、3回の独立した実験からの3通りの測定の平均値±STDである。
【0124】
乳酸塩測定: 1×105Jurkat細胞を様々な濃度の1とインキュベートした。望みの時点で、培地試料を集め、製造者(Trinity, St.Louis, Missouri, USA)の指針に従い、乳酸オキシダーゼをベースとした540nmで測定の比色検定により乳酸塩レベルを測定し、タンパク質濃度に対して規格化した。
【0125】
フルクトース−2,6−ビスリン酸塩検定: Jurkat細胞を1×105細胞/mLの密度で蒔種して、直ちに10μM 化合物1と0,4,8,16,24又は36時間インキュベートした。F2,6BP検定は、既述のように行った。Van Shaftingenほか,Eur.J.Biochem,129,191-195(1982)を参照されたい。
【0126】
2−デオキシ−グルコース取り込み: Jurkat細胞を1×105細胞/mLの密度で、10%FBS及び50μg/mL 硫酸ゲンタマイシンを補強したRPMI培地に蒔種した。細胞を直ちに、溶媒(DMSO)又は10μM 化合物1で、36時間までの間、処理し、その後、グルコースを欠いたRPMI培地に30分間置いた。14C−2−DG(0.25?Ci/mL;Perkin Elmer, Waltham, Massachusetts, USA)を60分間加えた。その後、細胞を3回氷冷したグルコースを欠いたRPMIで洗浄した。細胞溶解液を500μLの0.1%SDS中に集め、溶解液400μLについてシンチレーション(cpm)を計数した。計数値をタンパク質濃度について規格化した。2回の独立した実験について、2回の測定値の平均値±STDで、データを表した。
【0127】
全細胞ATP: Jurkat細胞を1×105細胞/mLの密度で蒔種し、様々な時点について10μM 化合物1とインキュベートした。ATPレベルを、Molecular Probes-Invitrogen(Eugene, Oregon, USA)から得たATP測定キットを用いて、製造者の指針に従って測定した。
【0128】
NAD+及びNADHレベル: Jurkat細胞を1×105細胞/mLの密度で蒔種し、直ちに10μM化合物1と、0,4,8,16,24又は36時間インキュベートした。NAD+及びNADHレベルを、BioAssay Systems(Hayward,California, USA)から得たEnzyChrom NAD+/NADH検定キットを用いて、製造者の指針に従い測定した。
【0129】
NMR: Jurkat細胞を、13C−グルコース存在下で、36時間、溶媒(DMSO)又は10μM 化合物1で処理した。同数の細胞をペレットにし、2回冷却したPBSで洗浄して、残った培地を除き、最終的にペレットにし、液体窒素中で瞬時に凍結した。冷却したペレットから氷冷した10%TCAで2回抽出し、その後凍結乾燥した。乾燥した抽出物を、0.35mL D2Oに再溶解し5mm Shigemiチューブに入れた。NMRスペクトルは、Varian Inova NMR スペクトロメーター(Varian,Inc., Palo Alto, California, USA)上で、20℃で、90°励起パルスを用いて、14.1Tで、記録した。2次元TOCSY及びHSQC実験を行った。代謝物を、1H及び13C化学シフト及びTOCSYコネクティヴィティパターンを基に同定した。代謝物は、TOCSY実験で積分により定量した。
【0130】
タンパク質抽出及びウェスターンブロット解析: タンパク質抽出及びウェスターンブロット解析は、以前の記載のように行った。Telangほか, Oncogene, 25, 7225-7234 (2006)を参照されたい。ブロットは、抗−PFKFB3抗体(Abjent, Inc., San Diego, California, USA)及び抗−β−アクチン抗体(Sigma, St.Louis, Missouri, USA)を用いて、先ずPFKFB3に対して探索し、その後プローブを外し、β−アクチンに対して再探索した。
【0131】
結果: 解糖フラックスの増加がトランスフォームした細胞増殖に必要と仮定すると、特別の理論に囚われることなく、PFKFB3の阻害は、選択的に腫瘍細胞増殖を標的とする。トランスフォームしたNHBE細胞(ht/LT/ras)は、遺伝的に一致する初代細胞と比較して、1に対してより大きな感受性があった。図4Aを参照のこと。初代NHBE細胞は、1とのインキュベーションに対して、試験した最高濃度(33μM)で、顕著な感受性を示さなかった。しかしながら、トランスフォームした細胞を1μM以下の濃度で1と処理すると、細胞増殖停止を示し、また約10μMで完全に細胞毒性を示した。NHBE−ht/LT/ras細胞に対する1のIC50は1.5μMと測定された。従って、化合物1は、他の解糖阻害剤、2−DG及び3−BrPA、(それぞれ、低mM濃度から、100μMで有効であった)に比べて10倍以上の阻害能を示した。
【0132】
固形腫瘍及び血液由来疾患由来の細胞株を含め、トランスフォームした細胞に対する1の効果を、さらに様々な腫瘍細胞に対する1のIC50を計算することにより吟味した。白血病細胞株は、接着細胞株に比べて、1の処理に対して増加した感受性(平均して約7倍)を示した。図4Bを参照されたい。K562、Jurkat及びHL−60白血病細胞に対するIC50値は、それぞれ、3.2,1.4及び4.5μMと決定された。接着細胞において、IC50の差異は大きかった。2種の肺癌モデル、ヒトA549及びマウスLLC,では、IC50値は、それぞれ、24及び19μMであり、他方HeLa子宮頸癌細胞のIC50値は,24μMであった。MDA−MB231乳腺癌細胞は化合物1との処理に対して、他の固形癌由来細胞株と比較して特に感受性が高く、計算したIC50値は、4.7μMであった。最後に、メラノーマ細胞株のIC50値は15μMであった。従って、1は、腫瘍細胞増殖を選択的に標的とする抗癌化合物であろう。
【0133】
1とのインキュベーションは、JurkatT細胞白血病細胞の増殖の用量依存的低下をもたらした。図5Aを参照されたい。0.3μMほどの3POが、細胞増殖を低下させて、また10μM 3POは、36時間にわたって、完全に細胞増殖を阻害した。図5A及び5Bを参照されたい。細胞増殖の抑制は、ヨウ化プロピジウム染色で測定したように、G2/M期の細胞周期停止の結果であるようである。図5C及び5Dを参照されたい。またFru−2,6−BP産生、2−デオキシグルコース(2−DG)取り込み、乳酸塩分泌に対する10μM 1の効果も調べた。図5Eを参照されたい。2−DG取り込み及びFru−2,6−BPは、4時間の処理の間に顕著に減少した。これらの代謝上の変化の後に、乳酸塩分泌(図5E;8時間を参照)、NADH(図5F;16時間参照)、NAD+(図5F;24時間参照)及びATP(図5F;24時間参照)の減少が続く。1との処理の間、Jurkat細胞を完全に標識した13C−グルコースでパルスし、またNMR分光で13C原子の運命を調べることにより、解糖の乳酸塩への直接のフラックスが抑制された。図5Gを参照されたい。
【0134】
1が細胞内のFru−2,6−BP,2−DG取り込み及び乳酸塩分泌をG2/M期停止前に減少させると言う観察により、特定の理論に囚われることなく、1がエネルギー的及び同化的代謝の中断により細胞増殖を阻害すると言う仮説が、相当自然に支持される。HL−60前骨髄球性白血病細胞において以前観察された様に、1によるG2/M期停止は、細胞周期のG2/M期間のATPの高い要求性と比べると、補助的でありうる。Sweet及びSingh,Cancer Res.,55,5164-5167(1995)を参照されたい。13C−グルコースのNMR分光による追跡から、解糖のアラニンではなく、乳酸塩へのフラックスが阻害されたことが明らかとなった。アラニン及び乳酸塩は同じピルビン酸プールを分かち合うが、乳酸デヒドロゲナーゼは、グリセルアルデヒド−3−リン酸デヒドロゲナーゼを経る解糖フラックスの産物である、NADHの即応の補給を必要とするので、グルコースに由来する乳酸塩産生は、特に解糖阻害により影響を受ける。
【0135】
新しい抗ガン治療の最も困難な課題の一つは、in situの標的検証である。この問題に取り組むために、異なるPFKFB3発現レベルを有する2種の異なる細胞株を研究した。もし、PFKFB3が真に1の標的であるならば、該酵素の発現が低下した細胞は、1の処理に対してより感受性があるか、又は逆である。従って、PFKFB3半数不足型(+/−)繊維芽細胞をヒトテロメラーゼ(ht)及びラージT抗原(LT)で不死化させ、突然変異型rasの発現によりトランスフォームさせた(PFKFB3 +/−ht/LT/ras)これらの繊維芽細胞は、約50%のPFKFB3発現及び低下したF2,6BP産生を有することが示された。該+/−ht/LT/rasトランスフォーム細胞及びこれらの野生型の遺伝子にマッチした対応細胞(+/+ht/LT/ras)を様々な濃度の1とインキュベートした。
【0136】
図6Aに示すように、PFKFB3+/− LT/rasトランスフォーム繊維芽細胞は、野生型対応細胞に比べて、低下したPFKFB3タンパク質及び低下した細胞内F2,6BPを発現する。野生型細胞における、F2,6BP濃度は、60.3±3.7 pmol/mgであり、他方PFKFB3+/−LT/rasトランスフォーム繊維芽細胞では、F2,6BP濃度は、10.3±1.5 pmol/mgであった。図6Bに示すように、PFKFB3異形接合繊維芽細胞は野生型トランスフォーム細胞に比べて、1の処理に対してより感受性が高かった。+/−繊維芽細胞に対する計算したIC50値(26μM)は、+/+繊維芽細胞に対するIC50値(49μM)の約半分であった。
【0137】
逆に、PFKFB3の過剰発現は、細胞と1のインキュベーションによる細胞増殖の低下を抑制するはずである。Jurkat細胞において、ドキシサイクリン応答性PFKFB3 Tet−ONシステムの影響下で、PFKFB3レベルを操作した。ドキシサイクリン処理した、PFKFB3発現ベクターを持たない対照 Jurkat細胞に比較して、ドキシサイクリン処理によりPFKFB3発現が増加する。PFKFB3タンパク質の異所性の発現は、F2,6BP濃度を増加させる。図6Cに示すように、+PFKFB3細胞におけるF2,6BP濃度は、38.1±3.4pmol/mgであり、他方、対照細胞において、F2,6BPの濃度は、9.2±0.95pmol/mgであった。両種の細胞を様々な濃度の1とインキュベートした、そして、予想されたように、過剰発現したPFKFB3は、Jurkat細胞への阻害剤の効果を抑制した(図6D)。PFKFB3過剰発現細胞に対するIC50値は、19.3μMと決定されたが、PFKFB3発現レベルの変わらぬJurkat細胞のIC50は8.9μMと計算された。
総合すると、これらの研究は、PFKFB3発現を調節することにより、1の細胞阻害効果を変えることができることを示し、PFKFB3が1の有効なin situ標的であることを示唆する。
【0138】
実施例4
化合物1を用いたIn Vivo研究
対数増殖期のMDA−MB231及びHL−60細胞を適切な培地に集め、2回洗浄して、20×107細胞/mLの密度でPBSに再懸濁した。次ぎに細胞を1:1の割合でMatrigelマトリクス(BD Biosciences, Bedford, Massachusetts, USA)と混ぜ、0.1mLの細胞懸濁液(1×107細胞)を雌のBalb/cヌードマウス(20g)の皮下に注射した。対数増殖期のLewis肺癌細胞を集め、2回洗浄し、PBSに懸濁した(1×107細胞/mL)。C57Blk/6雌マウス(20g)の皮下に0.1mLの容積で1×106細胞を注射した。体重及び腫瘍増殖を研究の間中、毎日観察した。重量(mg)=[幅(mm2)×長さ(mm)]/2の式を用いて、腫瘍重量をノギスで測定した。Taetleほか,Cancer Treat.Rep.,71,297−304(1987)を参照されたい。樹立した腫瘍を有するマウス(130mg〜190mg)を無作為に溶媒対照又は化合物1処理群に分けた。指示した時点で、溶媒対照グループには、50μLジメチルスルホキシド(DMSO)を腹腔注射(i.p.)し、他方、処理群には、0.07mg/g 化合物1を含む50μL DMSOをi.p.注射した。
【0139】
結果:1の毒性研究により、副作用の発現の無かった最高の容認された投与量は、0.07mg/g を含むDMSOの腹腔(IP)注射であった。試験した第1の腫瘍モデルは、C57/Blk6マウスに異種移植増殖するマウスLewis肺癌細胞からなる。この研究のために、マウスにDMSO又は計算した投与量の1を研究の期間(14日間)、毎日一度、IP注射した。図7Aに示したように、1の投与は、顕著にDMSO対照群と比較して、Lewis肺癌細胞異種移植体の増殖を73%抑えた。別個に、MDA−MB231ヒト乳腺癌をBalb/cヌードマウスに樹立した。このモデルにおいて、処理の低下が、Lewis肺癌モデルで見られたと同じ効果をもたらすかどうか決めるために、投与量の回数を減らした。DMSO(対照)及び化合物1処理群の両者のマウスに対して、3日間毎日の注射後、3日間の無処理という周期的な投薬計画を行った。DMSO対照と比較して、MDA−MB231細胞の異種移植体増殖の全阻害は最大66%観察された(図7B).3番目のin vivoモデルは、Balb/cヌードマウスに異種移植したHL−60白血病細胞である。処理計画はさらに減らして、2日間の連続注射後、7日間連続の無処理とした。図7Cに示すように、化合物1処理により、制限された投与にもかかわらず、DMSO対照と比較して、HL−60腫瘍増殖が74%以上阻害された。さらに、2回目の投与計画の効率は9日目の投与後の増殖低下が、増殖曲線上で見られる。Lewis 肺癌及びHL−60異種移植モデルに対する化合物1の効果は、腫瘍増殖の後退が見られないことから、細胞増殖停止と思われる。しかしながら、MDA−MB231モデルでは、化合物1阻害が、初期の細胞増殖停止効果から初期の腫瘍後退を導き、その後他の2モデルで見られるような細胞増殖停止をカバーするようである。まとめると、上記のデータから、化合物1がin vivo腫瘍増殖を減少させる能力を有する抗癌剤の可能性を有するものと支持される。
【0140】
実施例5
化合物2〜7によるPFKFB3の阻害
化合物2〜7がin vitroでPFKFB3活性の阻害能を有することは、上記実施例2に記載した酵素検定法を用いて測定した。150μM化合物2の処理により、溶媒(EtOH)処理と比較してPFKFB3の反応速度を39.8%減少させた。図8を参照されたい。溶媒(DMSO)処理と比較した、化合物3〜7の阻害効果は、図9に示されている。化合物4が最もPFKFB3を低下させ、80%以上であった。
【0141】
実施例6
トランスフォーム腫瘍細胞における化合物2〜7の毒性
様々なトランスフォーム細胞株に対する化合物2〜7の毒性は、上記実施例3に記載したように検定した。化合物2は、2種の白血病細胞HL−60及びK562細胞において、用量依存的細胞毒性を示した。図10を参照されたい。Jurkat白血病細胞増殖への様々な濃度の化合物3〜7の効果を図11〜15に示す。
図16〜21は、様々なトランスフォーム細胞において、化合物濃度0.3,1,3,10,又は33μMで処理した化合物2〜7の効果を示す。化合物2〜7の6種全ては、検定した固形腫瘍及び血液細胞タイプのそれぞれの増殖を阻害した。
【0142】
実施例7
Micro−Petにより観察したグルコース取り込みに対する化合物1の効果
C57Bl/6雌マウス(20g)に、1×106Lewis肺癌細胞を皮下に注射した。異種移植体の重量を測定して150及び180mgの間になった時、該マウスを2群に分けた。1群には、0.07mg/gの化合物1をi.p.注射した。他の群を対照群として用い、50μL DMSOをi.p.注射した。注射4時間後腫瘍を取り除き、等容量の0.1M及び0.05M NaOH中でホモジナイズした。F2,6BPアッセイを既述のように行った。Van Shaftingenほか.,Eur.J.Biochem,129,191-195 (1982)を参照されたい。化合物1処理により、腫瘍異種移植体中のF2,6BP産生は溶媒処理の対照群と比較して、顕著に減少した。化合物1処理のマウス異種移植体中のF2,6BP濃度は8.5±1.7pmol/mgであり、他方、対照マウス異種移植体中のF2,6BP濃度は、13.1±1.9pmol/mgであった。
【0143】
化合物1の効果をさらに調べるために、化合物1−処理マウスにおけるグルコースの取り込みをミクローポジトロン発光断層撮影法(micro―Pet)を用いて研究した。Lewis肺癌異種移植体を担うマウスに50μLのDMSO又は0.07mg/g 化合物1のDMSO溶液をi.p.注射した。最初の注射30分後に、各マウスに2−(18F)−フルオロ−2−デオキシグルコース(18F−2−DG;150μCurie、100μL H2O中)をi.p.注射した。さらに15分後に、マウスを酸素中2% イソフルランで麻酔し、R-4 Rodent Scanner Micro-Pet(CTI Concorde Microsystems, Inc.; Knoxville, Tennessee, USA)に移した。各群(即ち化合物1処置又は対照)3匹のマウスを研究した。図22は、代表的な化合物1−処理マウス及び代表的対照群マウスから取ったmicro―Pet画像である。
【0144】
図22に示すように、化合物1処理により、異種移植体内の18F−2−DG取り込みは、対照群マウスと比較して著しく減少した。化合物1処理マウス及び対照群マウスの間で、心臓の18F−2−DG取り込みにも差異が観測された。超音波心臓診断法を、Dawnほか,Proc.Natl.Acad.Aci.,U.S.A.,102,3766-3771(2005)に記載されたように行った。マウスの心臓機能の超音波心臓診断により、駆出分画に急性の変化は見られなかった(対照(溶媒):69%±4%;化合物1:71%±6%)。従って、心臓グルコース取り込みの差異は、化合物1の心臓毒性によるものではないようである。まとめると、1はin vivoでPFK2活性を標的とし、解糖表現型を低下させる。
【0145】
実施例8
トランスジェニックマウスの腫瘍量への化合物1の効果
トランスジェニックMMTV−Neu乳腫瘍マウスは、乳腺癌を生後3〜4ヶ月以内に発症し、マウス乳癌ウィルスプロモーター/エンハンサーの調節の下の癌遺伝子Neuのトランスジェニック発現による。これらのマウスは、これらの腫瘍増殖の性質の陰険さ及び組織培養条件に起因する人為性がない結果として、異種移植体モデルと比べて、ヒト癌により密接に類似する。
トランスジェニックMMTV−Neu乳癌マウスへの化合物1の効果を測るために、トランスジェニックマウスにおける相対的腫瘍率及び大きさを4ヶ月後に測定し、同等な腫瘍負荷になる様に無作為に2群に分けた。1群のマウスは、化合物1を3日間毎日の投与(0.07mg/g、DMSO中;腹腔投与;矢印を参照;腫瘍量が体重の10%を越えるまで)、その後3日間無処理の繰り返しサイクル投与を行った。対照マウスには、溶媒対照(DMSOのみ)で注射した。腫瘍量は、次の確立した式:重量(mg)=(幅、mm2×長さ,mm)/2に従い測定した。統計的な有意性を得るために、(α値が0.05及び指数が0.90と仮定して)各群に30匹のマウスを必要とした。長期にわたる腫瘍量への化合物1処理の効果を図23に示す。
【0146】
本明細書に開示した対照事項の様々な詳細を、本明細書に開示した発明の範囲から離れることなく、変更できる。さらに、既に述べた記載は、説明の目的のためにのみ有り、制限付けの目的のためではない。
【特許請求の範囲】
【請求項1】
6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)を阻害化合物と接触させることから成るPFKFB3を阻害する方法。
【請求項2】
前記阻害化合物が化学式(I):
【化1】
(式中、XはC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1は、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2はアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群より独立に選択される。)
で表わされる化合物である請求項1に記載の方法。
【請求項3】
Ar1及びAr2の中の少なくとも一つがアザアリール基である請求項2に記載の方法。
【請求項4】
L1が不存在である請求項2に記載の方法。
【請求項5】
L2が存在し、L2がC2アルキレン基である請求項2に記載の方法。
【請求項6】
前記化学式(I)で表わされる化合物が化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、それぞれ独立にN又はCR12であり、式中、各R12は水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5はO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13は、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つはNである。)で表わされる構造を有する化合物又はこれらの医薬的に許可された塩である請求項2に記載の方法。
【請求項7】
X1がOであり、XがCである請求項6に記載の方法。
【請求項8】
Ar2が:
【化5】
で表わされ;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、かつ各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、前記化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項6に記載の方法。
【請求項9】
前記化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項8に記載の方法。
【請求項10】
Ar2が:
【化8】
で表わされ、
X、X2及びX3が、それぞれCであり;
X1がO、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群より独立に選択され;及び化学式(II)で表わされる化合物が、化学式(IV)で表わされる構造:
【化9】
を有する請求項6に記載の方法。
【請求項11】
前記化学式(IV)で表わされる化合物が、
【化10】
である請求項10に記載の方法。
【請求項12】
Ar2が:
【化11】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項6に記載の方法。
【請求項13】
前記化学式(V)で表わされる化合物が:
【化13】
である請求項12に記載の方法。
【請求項14】
細胞を有効量のPFKFB3阻害剤と接触させることから成る、細胞内の解糖フラックスを減少させる方法。
【請求項15】
細胞を有効量の化学式(I):
【化1】
(式中、XがC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1が、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8が水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2が、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9が水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基が、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2が、アリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群から独立に選択される。)
で表わされる化合物と接触させることから成る、細胞内の解糖フラックスを減少させる方法。
【請求項16】
Ar1及びAr2の中の少なくとも一つがアザアリール基である請求項15に記載の方法。
【請求項17】
L1が不存在である請求項15に記載の方法。
【請求項18】
L2が存在し、L2がC2アルキレン基である請求項15に記載の方法。
【請求項19】
化学式(I)で表わされる化合物が化学式(II)で表わされる構造:
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
を有する化合物又はこれらの医薬的に許可された塩である請求項15に記載の方法。
【請求項20】
X1がOであり、XがCである請求項19に記載の方法。
【請求項21】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基、からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項19に記載の方法。
【請求項22】
前記化学式(III)で表わされる化合物が、以下の化合物:
【化7】
からなる群から選択される請求項21に記載の方法。
【請求項23】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され、化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項19に記載の方法。
【請求項24】
化学式(IV)で表わされる化合物が:
【化10】
である請求項23に記載の方法。
【請求項25】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され、
化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項19に記載の方法。
【請求項26】
化学式(V)で表わされる化合物が:
【化13】
である請求項25に記載の方法。
【請求項27】
細胞に有効量のPFKFB3阻害剤を接触させることから成る、細胞の増殖能を減少させる方法。
【請求項28】
細胞に有効量の化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又が、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
で表わされる化合物又はこれらの医薬的に許可された塩を接触させることから成る、細胞の増殖能を減少させる方法。
【請求項29】
前記細胞が哺乳動物細胞である請求項28に記載の方法。
【請求項30】
前記細胞が癌細胞である請求項28に記載の方法。
【請求項31】
Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つが、CR13であり、式中、R13が構造:
【化3】
(式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。)
を有する置換基である請求項28に記載の方法。
【請求項32】
X1がOであり、XがCである請求項28に記載の方法。
【請求項33】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項28に記載の方法。
【請求項34】
Y1,Y2,Y3,Y4及びY5の中の少なくとも一つが、CR13であり、式中、R13が構造:
【化3】
(式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。)
を有する置換基である請求項33に記載の方法。
【請求項35】
X5がOであり、X6が水素原子、アルキル基、アラルキル基及びアリール基から選択される請求項34に記載の方法。
【請求項36】
化学式(III)で表わされる化合物が化学式(IIIa):
【化14】
で表わされる化合物である請求項33に記載の方法。
【請求項37】
A3がNである請求項36に記載の方法。
【請求項38】
化学式(III)で表わされる化合物が化学式(IIIb):
【化15】
で表わされる化合物である請求項33に記載の方法。
【請求項39】
A4及びA5の中の一つがNである請求項38に記載の方法。
【請求項40】
化学式(III)で表わされる化合物が化学式(IIIc):
【化16】
で表わされる化合物である請求項33に記載の方法。
【請求項41】
A2がNである請求項40に記載の方法。
【請求項42】
化学式(III)で表わされる化合物が化学式(IIId):
【化17】
で表わされる化合物である請求項33に記載の方法。
【請求項43】
化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項33に記載の方法。
【請求項44】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項28に記載の方法。
【請求項45】
Y6及びA3が、それぞれNである請求項44に記載の方法。
【請求項46】
化学式(IV)で表わされる化合物が:
【化10】
である請求項45に記載の方法。
【請求項47】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項28に記載の方法。
【請求項48】
A3がNである請求項47に記載の方法。
【請求項49】
化学式(V)で表わされる化合物が:
【化13】
である請求項48に記載の方法。
【請求項50】
1又は2以上の癌細胞において解糖フラックスを直接阻害するために、有効量のPFKFB3阻害剤を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法。
【請求項51】
有効量の化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、
各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
で表わされる化合物又はこれらの医薬的に許可された塩を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法。
【請求項52】
Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つが、CR13であり、式中、R13が、次の化学構造:
【化3】
(式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択される。)
で表わされる請求項51に記載の方法。
【請求項53】
X1がOであり、XがCである請求項51に記載の方法。
【請求項54】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基、からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、
化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項51に記載の方法。
【請求項55】
Y1,Y2,Y3,Y4及びY5の中の少なくとも一つが、CR13であり、式中、R13が、次の化学構造:
【化3】
(式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択される。)
で表わされる請求項54に記載の方法。
【請求項56】
X5がOであり、X6が水素原子、アルキル基、アラルキル基及びアリール基から選択される請求項55に記載の方法。
【請求項57】
化学式(III)で表わされる化合物が化学式(IIIa):
【化14】
で表わされる化合物である請求項54に記載の方法。
【請求項58】
Ar2がNである請求項57に記載の方法。
【請求項59】
化学式(III)で表わされる化合物が化学式(IIIb):
【化15】
で表わされる化合物である請求項54に記載の方法。
【請求項60】
A4及びA5の中の一つがNである請求項59に記載の方法。
【請求項61】
化学式(III)で表わされる化合物が化学式(IIIc):
【化16】
で表わされる化合物である請求項54に記載の方法。
【請求項62】
A2がNである請求項61に記載の方法。
【請求項63】
化学式(III)で表わされる化合物が化学式(IIId):
【化17】
で表わされる化合物である請求項54に記載の方法。
【請求項64】
化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項54に記載の方法。
【請求項65】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項51に記載の方法。
【請求項66】
Y6及びA3がそれぞれNである請求項65に記載の方法。
【請求項67】
化学式(IV)で表わされる化合物が:
【化10】
である請求項66に記載の方法。
【請求項68】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項51に記載の方法。
【請求項69】
A3がNである請求項68に記載の方法。
【請求項70】
化学式(V)で表わされる化合物が:
【化13】
である請求項69に記載の方法。
【請求項71】
前記患者が哺乳動物である請求項51に記載の方法。
【請求項72】
前記癌が乳癌、肺癌、子宮頸癌、皮膚癌及び白血病からなる群から選択される請求項51に記載の方法。
【請求項73】
更に患者に1又は2以上の治療用化合物を投与することを含む請求項51に記載の方法。
【請求項74】
前記1又は2以上の治療用化合物がシスプラチン及びタキソールからなる群から選択される請求項73に記載の方法。
【請求項75】
6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ(PFKFB3)を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法。
【請求項76】
腫瘍増殖を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法。
【請求項77】
化合物をスクリーニングすることが、PFKFB3のモデルに対して多くの化合物をスクリーニングして、それぞれの化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定することから成る請求項76に記載の方法。
【請求項1】
6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ3(PFKFB3)を阻害化合物と接触させることから成るPFKFB3を阻害する方法。
【請求項2】
前記阻害化合物が化学式(I):
【化1】
(式中、XはC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1は、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2は、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9は水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基は、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2はアリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群より独立に選択される。)
で表わされる化合物である請求項1に記載の方法。
【請求項3】
Ar1及びAr2の中の少なくとも一つがアザアリール基である請求項2に記載の方法。
【請求項4】
L1が不存在である請求項2に記載の方法。
【請求項5】
L2が存在し、L2がC2アルキレン基である請求項2に記載の方法。
【請求項6】
前記化学式(I)で表わされる化合物が化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1は、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6は、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7は、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11は、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5は、それぞれ独立にN又はCR12であり、式中、各R12は水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5は、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12は、一緒にアルキレン基を形成し;
Ar2は、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19は、N及びCR13からなる群から独立に選択され、式中、各R13は、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4はNR14であり、式中、R14は、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5はO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15は、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6は、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13は、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つはNである。)で表わされる構造を有する化合物又はこれらの医薬的に許可された塩である請求項2に記載の方法。
【請求項7】
X1がOであり、XがCである請求項6に記載の方法。
【請求項8】
Ar2が:
【化5】
で表わされ;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、かつ各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、前記化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項6に記載の方法。
【請求項9】
前記化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項8に記載の方法。
【請求項10】
Ar2が:
【化8】
で表わされ、
X、X2及びX3が、それぞれCであり;
X1がO、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群より独立に選択され;及び化学式(II)で表わされる化合物が、化学式(IV)で表わされる構造:
【化9】
を有する請求項6に記載の方法。
【請求項11】
前記化学式(IV)で表わされる化合物が、
【化10】
である請求項10に記載の方法。
【請求項12】
Ar2が:
【化11】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項6に記載の方法。
【請求項13】
前記化学式(V)で表わされる化合物が:
【化13】
である請求項12に記載の方法。
【請求項14】
細胞を有効量のPFKFB3阻害剤と接触させることから成る、細胞内の解糖フラックスを減少させる方法。
【請求項15】
細胞を有効量の化学式(I):
【化1】
(式中、XがC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、但し式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群からそれぞれ独立して選択され、R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群からそれぞれ独立して選択され;
L1が、存在又は不存在であってもよく、存在する場合は、O、S、NR8、アルキレン基及び置換アルキレン基からなる群から選択され、式中、R8が水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され;
L2が、存在又は不存在であってもよく、存在する場合は、O、S、NR9、アルキレン基、置換アルキレン基及び三価の連結基からなる群から選択され、式中、R9が水素原子、アルキル基、アリール基及びアラルキル基からなる群から選択され、また三価の連結基が、Ar2に結合する第1の原子、Xに結合する第2の原子及びAr1及びAr2からなる群の一つと結合する第3の原子からなり;及び
Ar1及びAr2が、アリール基、置換アリール基、ヘテロアリール基及び置換ヘテロアリール基からなる群から独立に選択される。)
で表わされる化合物と接触させることから成る、細胞内の解糖フラックスを減少させる方法。
【請求項16】
Ar1及びAr2の中の少なくとも一つがアザアリール基である請求項15に記載の方法。
【請求項17】
L1が不存在である請求項15に記載の方法。
【請求項18】
L2が存在し、L2がC2アルキレン基である請求項15に記載の方法。
【請求項19】
化学式(I)で表わされる化合物が化学式(II)で表わされる構造:
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
を有する化合物又はこれらの医薬的に許可された塩である請求項15に記載の方法。
【請求項20】
X1がOであり、XがCである請求項19に記載の方法。
【請求項21】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基、からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項19に記載の方法。
【請求項22】
前記化学式(III)で表わされる化合物が、以下の化合物:
【化7】
からなる群から選択される請求項21に記載の方法。
【請求項23】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され、化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項19に記載の方法。
【請求項24】
化学式(IV)で表わされる化合物が:
【化10】
である請求項23に記載の方法。
【請求項25】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され、
化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項19に記載の方法。
【請求項26】
化学式(V)で表わされる化合物が:
【化13】
である請求項25に記載の方法。
【請求項27】
細胞に有効量のPFKFB3阻害剤を接触させることから成る、細胞の増殖能を減少させる方法。
【請求項28】
細胞に有効量の化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又が、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
で表わされる化合物又はこれらの医薬的に許可された塩を接触させることから成る、細胞の増殖能を減少させる方法。
【請求項29】
前記細胞が哺乳動物細胞である請求項28に記載の方法。
【請求項30】
前記細胞が癌細胞である請求項28に記載の方法。
【請求項31】
Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つが、CR13であり、式中、R13が構造:
【化3】
(式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。)
を有する置換基である請求項28に記載の方法。
【請求項32】
X1がOであり、XがCである請求項28に記載の方法。
【請求項33】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項28に記載の方法。
【請求項34】
Y1,Y2,Y3,Y4及びY5の中の少なくとも一つが、CR13であり、式中、R13が構造:
【化3】
(式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択される。)
を有する置換基である請求項33に記載の方法。
【請求項35】
X5がOであり、X6が水素原子、アルキル基、アラルキル基及びアリール基から選択される請求項34に記載の方法。
【請求項36】
化学式(III)で表わされる化合物が化学式(IIIa):
【化14】
で表わされる化合物である請求項33に記載の方法。
【請求項37】
A3がNである請求項36に記載の方法。
【請求項38】
化学式(III)で表わされる化合物が化学式(IIIb):
【化15】
で表わされる化合物である請求項33に記載の方法。
【請求項39】
A4及びA5の中の一つがNである請求項38に記載の方法。
【請求項40】
化学式(III)で表わされる化合物が化学式(IIIc):
【化16】
で表わされる化合物である請求項33に記載の方法。
【請求項41】
A2がNである請求項40に記載の方法。
【請求項42】
化学式(III)で表わされる化合物が化学式(IIId):
【化17】
で表わされる化合物である請求項33に記載の方法。
【請求項43】
化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項33に記載の方法。
【請求項44】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項28に記載の方法。
【請求項45】
Y6及びA3が、それぞれNである請求項44に記載の方法。
【請求項46】
化学式(IV)で表わされる化合物が:
【化10】
である請求項45に記載の方法。
【請求項47】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項28に記載の方法。
【請求項48】
A3がNである請求項47に記載の方法。
【請求項49】
化学式(V)で表わされる化合物が:
【化13】
である請求項48に記載の方法。
【請求項50】
1又は2以上の癌細胞において解糖フラックスを直接阻害するために、有効量のPFKFB3阻害剤を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法。
【請求項51】
有効量の化学式(II)
【化2】
(式中、X、X2及びX3が、それぞれC又はCHであり;
X1が、O、S、NR1、C(R2)2、OR3、SR4、NR5R6及びC(R7)3からなる群から選択され、式中、R1、R3、R4、R5及びR6が、水素原子、アルキル基、アリール基、アラルキル基及びアシル基からなる群から、それぞれ独立に選択され、各R2及びR7が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;
R10が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
R11が、水素原子、アルキル基、ハロゲン原子、シアノ基、ヒドロキシル基、アリール基及びアラルキル基からなる群から選択され;
A1,A2,A3,A4及びA5が、それぞれ独立にN又はCR12であり、式中、各R12が水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され:
X5が、O、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基から選択され;
又は、R10及び1個のR12が、一緒にアルキレン基を形成し;
Ar2が、次の化学式:
【化4】
からなる群から選択され、
式中、
各Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19が、N及びCR13からなる群から独立に選択され、式中、各R13が、水素原子、アルキル基、ハロゲン原子、ニトロ基、シアノ基、ヒドロキシル基、メルカプト基、アミノ基、アルキルアミノ基、ジアルキルアミノ基、カルボキシル基、アシル基、カルバモイル基、アルキルカルバモイル基、ジアルキルカルバモイル基、硫酸基及び次の化学構造:
【化3】
を有する置換基からなる群から独立に選択され、
式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択され;
又は式中、R10及び1個のR13が、一緒にアルキレン基を形成し;及び
式中、A1,A2,A3,A4,A5,Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14、Y15,Y16,Y17,Y18及びY19の少なくとも一つが、Nである。)
で表わされる化合物又はこれらの医薬的に許可された塩を患者へ投与することから成る、治療を必要とする患者の癌を治療する方法。
【請求項52】
Y1,Y2,Y3,Y4,Y5,Y6,Y7,Y8,Y9,Y10,Y11,Y12,Y13,Y14,Y15,Y16,Y17,Y18及びY19の中の少なくとも一つが、CR13であり、式中、R13が、次の化学構造:
【化3】
(式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択される。)
で表わされる請求項51に記載の方法。
【請求項53】
X1がOであり、XがCである請求項51に記載の方法。
【請求項54】
Ar2が:
【化5】
であり;
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基、からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び、
化学式(II)で表わされる化合物が化学式(III)で表わされる構造:
【化6】
を有する請求項51に記載の方法。
【請求項55】
Y1,Y2,Y3,Y4及びY5の中の少なくとも一つが、CR13であり、式中、R13が、次の化学構造:
【化3】
(式中、X4がNR14であり、式中、R14が、水素原子、アルキル基、ヒドロキシル基、アラルキル基及びアリール基からなる群から選択され;
X5がO、S、C(R15)2及びNR14からなる群から選択され、式中、各R15が、水素原子、ヒドロキシル基、アルコキシ基、アルキル基、アラルキル基及びアリール基からなる群から独立に選択され;及び
X6が、水素原子、アルキル基、アラルキル基、アリール基、ヘテロアリール基、アルキルアミノ基、ジアルキルアミノ基及びアルコキシ基からなる群から選択される。)
で表わされる請求項54に記載の方法。
【請求項56】
X5がOであり、X6が水素原子、アルキル基、アラルキル基及びアリール基から選択される請求項55に記載の方法。
【請求項57】
化学式(III)で表わされる化合物が化学式(IIIa):
【化14】
で表わされる化合物である請求項54に記載の方法。
【請求項58】
Ar2がNである請求項57に記載の方法。
【請求項59】
化学式(III)で表わされる化合物が化学式(IIIb):
【化15】
で表わされる化合物である請求項54に記載の方法。
【請求項60】
A4及びA5の中の一つがNである請求項59に記載の方法。
【請求項61】
化学式(III)で表わされる化合物が化学式(IIIc):
【化16】
で表わされる化合物である請求項54に記載の方法。
【請求項62】
A2がNである請求項61に記載の方法。
【請求項63】
化学式(III)で表わされる化合物が化学式(IIId):
【化17】
で表わされる化合物である請求項54に記載の方法。
【請求項64】
化学式(III)で表わされる化合物が以下の化合物:
【化7】
からなる群から選択される請求項54に記載の方法。
【請求項65】
Ar2が:
【化8】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(IV)で表わされる構造:
【化9】
を有する請求項51に記載の方法。
【請求項66】
Y6及びA3がそれぞれNである請求項65に記載の方法。
【請求項67】
化学式(IV)で表わされる化合物が:
【化10】
である請求項66に記載の方法。
【請求項68】
Ar2が:
【化11】
であり、
X、X2及びX3が、それぞれCであり;
X1が、O、S、NR1及びC(R2)2からなる群から選択され、式中、R1が、水素原子及びアルキル基からなる群から選択され、また各R2が、水素原子、ハロゲン原子、ヒドロキシル基、アルコキシ基及びアルキル基からなる群から独立に選択され;及び
化学式(II)で表わされる化合物が化学式(V)で表わされる構造:
【化12】
を有する請求項51に記載の方法。
【請求項69】
A3がNである請求項68に記載の方法。
【請求項70】
化学式(V)で表わされる化合物が:
【化13】
である請求項69に記載の方法。
【請求項71】
前記患者が哺乳動物である請求項51に記載の方法。
【請求項72】
前記癌が乳癌、肺癌、子宮頸癌、皮膚癌及び白血病からなる群から選択される請求項51に記載の方法。
【請求項73】
更に患者に1又は2以上の治療用化合物を投与することを含む請求項51に記載の方法。
【請求項74】
前記1又は2以上の治療用化合物がシスプラチン及びタキソールからなる群から選択される請求項73に記載の方法。
【請求項75】
6−ホスホフルクト−2−キナーゼ/フルクトース−2,6−ビスホスファターゼ(PFKFB3)を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法。
【請求項76】
腫瘍増殖を阻害する能力を有する化合物をスクリーニングする方法であって、(a)PFKFB3の3Dモデルを提供すること、但し該PFKFB3モデルはフルクトース−6−リン酸塩(F6P)結合ポケットを含む;(b)ある化合物の3Dモデルを提供すること;及び(c)この化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定するために、PFKFB3のモデルに対してこの化合物をスクリーニングすること、から成る方法。
【請求項77】
化合物をスクリーニングすることが、PFKFB3のモデルに対して多くの化合物をスクリーニングして、それぞれの化合物が、該PFKFB3のF6P結合ポケット中の1又は2以上の溶媒接触部位に接触する能力を決定することから成る請求項76に記載の方法。
【図1】


【図2A】

【図2B】

【図2C】

【図3A】

【図3B】

【図4A】

【図4B】

【図5A】

【図5B】

【図5C】

【図5D】

【図5E】

【図5F】

【図5G】

【図6A】

【図6B】

【図6C】

【図6D】

【図7A】

【図7B】

【図7C】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図5G】
【図6A】
【図6B】
【図6C】
【図6D】
【図7A】
【図7B】
【図7C】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2010−531304(P2010−531304A)
【公表日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願番号】特願2010−513237(P2010−513237)
【出願日】平成20年6月18日(2008.6.18)
【国際出願番号】PCT/US2008/007594
【国際公開番号】WO2008/156783
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(509347206)ユニバーシティ オブ ルイビル リサーチ ファウンデーション、インコーポレイテッド (1)
【氏名又は名称原語表記】UNIVERSITY OF LOUISVILLE RESEARCH FOUNDATION,INC.
【Fターム(参考)】
【公表日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願日】平成20年6月18日(2008.6.18)
【国際出願番号】PCT/US2008/007594
【国際公開番号】WO2008/156783
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(509347206)ユニバーシティ オブ ルイビル リサーチ ファウンデーション、インコーポレイテッド (1)
【氏名又は名称原語表記】UNIVERSITY OF LOUISVILLE RESEARCH FOUNDATION,INC.
【Fターム(参考)】
[ Back to top ]