
有機分子固定化強磁性ナノ粒子並びにその製造方法及び分離方法
【課題】室温においてナノサイズでの磁気分離が可能な、有機分子で修飾された強磁性ナノ粒子を提供する。
【解決手段】強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子;強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することにより前記有機分子固定化強磁性ナノ粒子を製造する方法;並びに前記有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【解決手段】強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子;強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することにより前記有機分子固定化強磁性ナノ粒子を製造する方法;並びに前記有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【発明の詳細な説明】
【技術分野】
【0001】
有機分子で修飾された強磁性ナノ粒子並びにその製造方法及び分離方法に関する。
【背景技術】
【0002】
近年ナノ粒子は生物分離や各種測定、ドラッグデリバリーシステム等バイオ分野での幅広い応用が期待されている。ナノ粒子の特徴としては、粒径がナノサイズになることによる体積の減少から用途の拡大、生体適合性の増大が見込め、比表面積の増大から認識能の向上、認識時間の短縮が見込める。そのため、ナノ粒子の開発は大きなテーマとなっており、特にナノ粒子を磁性体とした磁性ナノ粒子はMRIによる測定、磁気誘導によるドラッグデリバリーシステムの構築等、その磁気特性を利用した分野での応用が期待されている(非特許文献1)。
【0003】
しかしながら、現状酸化鉄ナノ粒子に代表されるように、磁性粒子の体積を減少させていくと、室温では熱エネルギーによって磁気モーメントの向きが無秩序となる超常磁性体となり、その磁気特性が失われ、ナノサイズでの磁気分離などが不可能とされてきた。
【0004】
一般的に磁気異方性エネルギーKuと磁性体の体積Vの積が熱エネルギーKTを下回ると、磁性体としての性質を失ってしまうので、室温磁性ナノ粒子の候補としては、磁気異方性エネルギーKuの高いものであることが必要不可欠である。
【0005】
【非特許文献1】応用物理第72巻第7号909頁(2003)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、室温においてナノサイズでの磁気分離が可能な、有機分子で修飾された強磁性ナノ粒子を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明の要旨は以下のとおりである。
(1)強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子。
(2)強磁性FePtナノ粒子がL10規則構造を有する前記(1)に記載の有機分子固定化強磁性ナノ粒子。
(3)強磁性FePtナノ粒子が、Fe錯体及びPt錯体を沸点300℃以上のポリオール系溶媒で処理することにより得られるものである前記(1)又は(2)に記載の有機分子固定化強磁性ナノ粒子。
(4)強磁性FePtナノ粒子に固定化される有機分子が、特異的結合のパートナー及び生理活性物質から選ばれる前記(1)〜(3)のいずれかに記載の有機分子固定化強磁性ナノ粒子。
(5)強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することを特徴とする、前記(1)〜(4)のいずれかに記載の有機分子固定化強磁性ナノ粒子の製造方法。
(6)カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物の少なくとも一方が特異的結合のパートナー、DNA、酵素及び生理活性物質から選ばれる残基を有する前記(5)に記載の製造方法。
(7)前記(5)又は(6)に記載の製造方法により得られた有機分子固定化強磁性ナノ粒子。
(8)前記(1)〜(4)及び(7)のいずれかに記載の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【発明の効果】
【0008】
本発明によれば、室温においてナノサイズでの磁気分離が可能な、有機分子で修飾された強磁性ナノ粒子を提供することができる。
【発明を実施するための最良の形態】
【0009】
本発明に用いる強磁性FePtナノ粒子とは、Fe原子及びPt原子を主成分(通常、全金属原子中Fe原子及びPt原子が合わせて50モル%以上、好ましくは90モル%以上)とする1〜10nm程度の粒径を有する粒子で室温にて磁気ヒステリシスを示すものをいう。
【0010】
L10規則構造を示すFePt合金においては高い結晶磁気異方性を有する(International Center For Diffraction Data, Powder Diffraction File SET43 Inorganic and organic, 43-1539)。この高い結晶磁気異方性は1Tbit/inch2以上の超高密度記録が原理的に可能であるとされ(K. Inomata, T. Sawa, S. Hashimoto, J. Appl. Phys. 1996, 64, 2537)、将来の超高密度記録媒体への応用が期待されている。
【0011】
本発明では、高い結晶磁気異方性エネルギーKuを有していることから、ナノオーダーでの強磁性を示すFePtナノ粒子を用いる。
【0012】
FePt合金は安定相であるL10規則構造と準安定構造であるfcc構造の2種の構造を取りうる。ここでL10規則構造はc軸に磁化容易軸があり、高い結晶磁気異方性を有することから強磁性を示すのに対し、準安定構造であるfcc構造は磁化容易軸が存在せず、常磁性を示す(図1)。
【0013】
以下、L10−FePt合金の高い結晶磁気異方性の起源を説明する。
4d遷移金属Pd、5d遷移金属Ptは白金族原子と呼ばれ、バルクの単体では常磁性を示すが、強磁性体直前の金属であることが知られている。また、この白金族は鉄族との合金化によって大きなスピン偏極を示すことが知られている。
【0014】
図2にFePt合金の状態密度図を示した(A. Sakuma, J. Phys. Soc. Jpn. 1994, 63, 3053)。
FeとPtのmajority spinバンドはフェルミエネルギーEf付近でFe3dバンドとPt4dバンドの混成化に由来する重なりが見られる。この混成化によってPtのスピン磁気モーメントが誘起され、縮退により消滅していたFeの軌道磁気モーメントが復活するといわれている。表1にFe及びPtのスピン軌道結合エネルギーζ、スピン磁気モーメントS、軌道磁気モーメントL、合成磁気モーメントMtotal、磁気モーメントの実測値Mexを示した。Fe及びPtの合金化による、Ptの磁気モーメントの誘起を確認することができる。このPtの磁気モーメントの誘起による大きなスピン軌道結合定数により、FePt合金は大きなスピン軌道相互作用を持つことが分かり、高い磁気異方性の起源となっていることを知ることができる。
【0015】
【表1】

【0016】
FePt合金は一般にFe組成がモル比で45%〜64%のとき、L10規則構造を有する。しかしながら、化学合成などによりFePt合金を作成した場合、準安定相であるfcc不規則相をとることが多い。2000年、S.Sunらによってポリオールプロセスと呼ばれる化学的手法により粒径分布のほとんどないFePtナノ粒子の合成法が報告され(S. Sun, C. B. Murray, D. Weller, L. Folks, A. Moser, Science 2000, 287, 1989; S. Sun, Adv. Mater. 2006, 18, 393)、FePt合金に関する研究が加速し、磁気記録デバイスへの実用化が近いものと期待された。しかしながら、このポリオールプロセスにより合成したFePtナノ粒子もやはり準安定相である不規則相を取っており、高い磁気異方性を有するL10構造への構造転移には600℃程度の熱処理による規則化プロセスが必要であった。この熱処理プロセスはFePtナノ粒子同士の焼結や凝集を招き、これが実用化への大きな障壁となっている。このような問題を解決するために、この熱処理による規則化温度の低減もしくは、熱処理時の焼結を防ぐ試みがなされており、以下に、いくつかの方法を例示する。
【0017】
(1)高沸点エチレングリコール系溶媒等のポリオール系溶媒を用いた液層直接合成(K. Sato, B. Jeyadevan, K. Tohji, J. Magn. Magn. Mater. 2005, 289, 1;R. Minami, Y. Kitamoto, T.Chikata, S. Kato, Electrochimica Acta 2005, 51, 864)
この手法は、Fe及びPt化合物の前駆体(錯体、塩など)を、ポリオール系溶媒を還元剤及び溶媒として還元する手法である。エチレングリコール系溶媒等のポリオール系溶媒は、Fe及びPtの還元に際した反応速度を低下させることで、規則化温度を300℃以下に低下させることが知られている。更に、ポリオール系溶媒の一種であり、300℃以上の沸点を持つテトラエチレングリコール等の高沸点ポリオール系溶媒を溶媒として用いることで液層において還元と規則化を同時に起こし、L10規則相を直接合成する手法である。この手法では、L10構造への完全な規則化には至らず、部分的な規則化に留まるという欠点があるものの、得られるFePt合金において高い磁気異方性が確認されており、有効な手法の一つである。
【0018】
また、この手法を用いれば、600℃におけるアニーリングというプロセスを省くことができ、更に液相中での合成が可能なことから、合成後の化学的な表面修飾が可能であるという利点がある。本発明にとって、これは大きなアドバンテージであり、後述する実施例で採用した合成法がこの手法である。
【0019】
ここで用いるFe及びPt化合物の前駆体(錯体、塩など)としてはFe元素及びPt元素を含む錯体や金属塩であれば制限はなく、例えばアセチルアセトネート(acac)錯体、カルボニル錯体、エトキシド化合物、塩化物などが挙げられる。
【0020】
ここで用いるポリオール系溶媒としては沸点が300℃以上のものであれば制限はないが、好ましくはテトラエチレングリコール、ヘキサエチレングリコール、オクタエチレングリコール、又は高沸点有機溶媒とエチレングリコール系溶媒の混合物等の高沸点エチレングリコール系溶媒が挙げられる。
【0021】
Fe錯体とPt錯体の割合は、好ましくは、モル比で45:55〜64:36である。ポリオール系溶媒の使用量は、好ましくは、金属イオン1mmol当たり100ml程度である。
【0022】
(2)第三元素の添加による規則化温度の低減(J. W. Harrell, D. E. Nikles, S. S. Kang, X. C. Sun, Z. Jia, J. Magn. Soc. Jpn. 2004, 28, 847; O. Kitamura, Y. Shimada, K. Oikawa, H. Daimon, K. Fukamichi, Appl. Phys. Lett. 2002, 80, 2147; T. Maeda, T. Kai, A. Kikitu, T. Nagase, J. Akiyama, Appl. Phys. Lett. 2001, 78, 1104; K. M. Park, K. H. Na, J. G. Na, P. W. Jang, H. J. Kim, S. R. Lee, IEEE Trans. Magn. 2000, 38, 1961)
この手法は、Fe及びPtの他に第三の元素を添加することにより、原子拡散を増大させて規則化を促そうという手法である。Ag,Au,Cuといった元素を添加することでL10規則相への構造転移温度が低下するという報告がなされている。この中でも特に大きな規則化温度の低減を齎すのがCuであり、Ag,Auにおいて規則化温度が400℃まで低下するのに対し、Cuの添加は規則化温度を300℃まで低下させている。Ag,Auにおいては熱処理後の元素の析出が観測されているが、Cuにおいては観測されていない。つまり、Au及びAgについては析出により原子の拡散を促し規則化温度を低下させているのに対し、CuはFePt合金中へ固溶することによって、規則相のポテンシャルエネルギーを下げ、規則化を促しているわけである。
【0023】
本発明においては、この手法を用いることもできる。更に、SiO2ナノリアクター法を用いることもできる。
【0024】
前述したようにFePt合金は安定相であるL10規則相と準安定相であるfcc不規則相の2種の構造を取りうる。したがって、FePt合金を用いる時には、構造転移の証明や、この構造転移の割合を示すオーダーパラメーターを知ることが大変重要である。これらを知るための手法としてはX線による回折パターンの解析が極めて有効である。L10規則相においては、不規則相であるfcc構造の回折線に加え、規則配置に起因する余分の回折線が出現する。この余分の回折線を超格子反射と呼ぶ。超格子反射の出現は、原子配置の規則化により、消滅則が異なってくることに由来するものである。
【0025】
不規則相であるfcc構造の消滅則は、
【数1】
で与えられることからも分かるように、fcc構造においてはz=1、z=0の面からの回折線の位相とz=1/2の面からの回折線の位相は半波長分だけずれるため、回折線は打ち消しあって(001)面からのブラッグ反射は起こらない。一方、規則相であるL10構造の消滅則は、
【0026】
【数2】
で与えられる。この消滅則からも分かるようにz=0、z=1の面とz=1/2の面では散乱因子が異なっており、回折線は完全には打ち消しあわず、結果として超格子反射が観測される。更に、このような(001)面などからの超格子反射の出現に加え、正方晶への変化に伴う(002)、(200)面からの回折線の分裂が生じる。
【0027】
したがって、X線回折パターンを解析し、超格子反射の出現を観測することで、構造転移の証明が可能となる。
【0028】
本発明において強磁性FePtナノ粒子に固定化させる有機分子は、目的に応じて適宜選択すればよく、例えば特異的結合のパートナー、DNA、酵素、生理活性物質が挙げられる。また、前記有機分子に加えて、目的に応じて、例えば親水性、生体適合性、分散性、各種溶媒への親媒性等を付与する目的でポリエチレングリコール鎖、イオン基を有するアルキル鎖等を組み合わせて固定化することもできる。
【0029】
前記特異的結合のパートナーとは、関与する分子の三次元構造に依存する特異的な非共有相互作用により相互作用する分子の対のメンバーをいい、当該特異的結合のパートナーの典型的な対としては、例えば抗原−抗体、ハプテン−抗体、ホルモン−レセプター、核酸鎖−相補的核酸鎖、基質−酵素、基質類似体−酵素、インヒビター−酵素、炭水化物−レクチン、ビオチン−アビジン及びウイルス−細胞レセプターが挙げられる。
【0030】
生理活性物質としては、例えば薬物が挙げられる。
例えば、抗体を固定化した強磁性ナノ粒子は細胞分離、化学物質の検出・回収に用いることができ、DNA鎖を固定化した強磁性ナノ粒子はSNPs検出、遺伝子診断、品種識別に用いることができ、酵素を固定化した強磁性ナノ粒子は酵素反応センシング・回収再利用に用いることができ、ビオチンを固定化した強磁性ナノ粒子は、各種測定方法、又はビオチン−アビジン結合を介した各種物質の固定化に用いることができ、薬物を固定化した強磁性ナノ粒子は、ドラッグデリバリーシステムに用いることができる。
【0031】
強磁性FePtナノ粒子に有機分子を固定化させるためには、目的とする有機分子を含む配位子がFeとPtに対して高いアフィニティを有するカルボキシル基、アミノ基、メルカプト基、水酸基等を末端にもたなければならない。また、バイオ磁性ナノ粒子として、生体内で応用する場合には、親水性、生体適合性の付与、及び目的とする各有機分子間の立体的混雑を避けるスペーサーとして、ポリエチレングリコール鎖(PEG)(好ましくは重合度10〜20)を配位子中に含ませることが好ましい。
【0032】
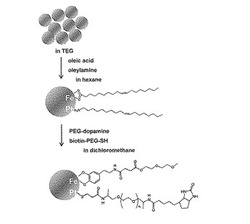
そのため、後述する実施例では、PEGの両末端にビオチン及びメルカプト基を持つビオチン−PEG−SH及び片方の末端にドパミン基を持つドパミン−ポリエチレングリコールモノメチルエーテル(mPEG)という二つの配位子を設計・合成した。ここでビオチン−PEG−SHはPtに配位し、ドパミン−mPEGはFeに配位する。ドパミン−mPEGはビオチン基を持たないものの、親水性の向上を目的としており、粒子表面のFeの酸化をも同時に防いでいる。
【0033】
次いで、合成した配位子を強磁性FePtナノ粒子上へ固定化させるが、前記のポリオール系溶媒を用いた液層直接合成により強磁性FePtナノ粒子を合成する場合には、合成直後の強磁性FePtナノ粒子は大量のポリオール系溶媒中に存在するため、生成量が非常に少ない合成した配位子を直接固定化させることが困難なことから、一度別の有機配位子、例えば、オクタン酸等の炭素数6〜20の脂肪酸及びオレイルアミン等の炭素数6〜20の脂肪族アミンをFePtナノ粒子上に固定化させ、配位子交換反応を用いて目的の合成した配位子、例えばカテコール残基を有する有機化合物(例えば、ドパミン−mPEG)及びメルカプト基を有する有機化合物(例えば、ビオチン−PEG−SH)をFePtナノ粒子上に固定化させることが好ましい(Rui Hong, Nicholas O. Fischer, Todd Emrick, Vincent M. Rotello, chem. Mater, 2005, 17, 4617)。
【0034】
前記脂肪酸の使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)であり、脂肪族アミンの使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)であり、目的の合成した配位子の使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)である。
本発明の有機分子固定化強磁性ナノ粒子は、磁気分離が可能である。したがって、本発明の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離する方法に適用することができる。
【0035】
例えば、特定抗体が固定化された強磁性ナノ粒子を診断薬として用いる場合、検体にもし特定の抗原が存在すれば、抗原は抗体が固定化された強磁性ナノ粒子に吸着される。吸着後、標識(発色団など)された抗体(二次抗体)を加え、磁石により検体から分離し、分離された強磁性ナノ粒子を緩衝溶液などで洗浄し、余分な標識抗体を取り除く、次いで、得られた強磁性ナノ粒子を再度緩衝溶液などで希釈し標識部に分光工学的な手法などを用いて抗原の定量が行える。
【実施例】
【0036】
以下、実施例を挙げて本発明を具体的に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
以下の実施例で使用した測定機器の一覧を表2に示した。
【0037】
【表2】
【0038】
なお、UV−Visは紫外可視分光光度計を、XRDは粉末X線回折装置を、SQUIDは超伝導量子干渉素子磁力計を、TEMは透過型電子顕微鏡をそれぞれ示し、以後これらの略語を用いて表記する。
【0039】
(実施例1)ビオチン化FePtナノ粒子の合成
1.有機配位子の合成
FePtナノ粒子に表面修飾する有機配位子としてビオチン−PEG−SH及びドパミン−mPEGの二つの配位子を既報(Rui Hong, Nicholas O. Fischer, Todd Emrick, Vincent M. Rotello, chem. Mater, 2005, 17, 4617; Bryan Parrish and Todd Emrick, Macromplecules, 2004, 37, 5863; Karl Kaiser, Markus Marek, Thomas Haselgrubler, Hansgeorg Schindler, and Hermann J. Gruber ,Bioconjugate chem. 1997, 8, 54)に従い、設計・合成した。全体のスキームは以下の通りとなる。
【0040】
ドパミン−mPEGの全体合成反応式
【化1】
【0041】
ビオチン−PEG−SHの全体合成反応式
【化2】
【0042】
以下、各合成段階の詳細を述べる。
(1)ポリ(エチレングリコール)−コハク酸エステルの合成
【化3】
【0043】
クロロホルム100mlの中にポリ(エチレングリコール)(2.759g,5mmol)、無水コハク酸(1.2525g,12.5mmol)、4−(ジメチルアミノ)ピリジン(0.0648g,0.5mmol)を溶解させ、12時間加熱還流した。その後溶媒を減圧留去し、水に溶解させた後、ヘキサン:酢酸エチル=1:1の混合比で混ぜた溶液で洗浄、ジクロロメタンで抽出した。ジクロロメタン抽出液を硫酸マグネシウムで脱水乾燥させ、硫酸マグネシウムを濾別した後、ジクロロメタンを減圧留去し一昼夜真空乾燥を行いポリ(エチレングリコール)−コハク酸エステルを得た(3.3718g,5mmol,100%)。
【0044】
(2)ドパミン−ポリ(エチレングリコール)の合成
【化4】
【0045】
N,N−ジメチルホルムアミド10ml中に、ポリ(エチレングリコール)−コハク酸エステル(1.3202g,2mmol)、1−ヒドロキシベンゾトリアゾール(0.2702g,2mmol)、N,N−ジイソプロピルエチルアミン(0.52g,4mmol)を0℃で加え、攪拌した。その後1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(3.902g,20mmol)、ドパミン塩酸塩(0.4025g,2.1mmol)を5分間で加えた。この溶液を窒素パージした後4℃で一夜攪拌し、N,N−ジメチルホルムアミドを減圧留去し、150mlのクロロホルムに溶解させた。1Mの冷やした塩酸、飽和炭酸水素ナトリウム水溶液、水、brineで洗浄し、硫酸マグネシウムで脱水乾燥させ、硫酸マグネシウムを濾別した後、クロロホルムを減圧留去し、真空乾燥させドパミン−ポリ(エチレングリコール)を得た(1.28g,1.6mmol,80%)。
【0046】
(3)ビオチン−NHSの合成
【化5】
【0047】
N,N−ジメチルホルムアミド中にD−ビオチン(0.5292g,2.17mmol)、N−ヒドロキシスクシンイミド(0.3259g,2.83mmol)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(0.53g,2.76mmol)を窒素パージして一昼夜攪拌した。N,N−ジメチルホルムアミドを減圧留去し、濾過、過剰の水とメタノールで洗浄し、ビオチン−NHSの白色結晶を得た(0.4858g,1.423mmol,65.7%)。
【0048】
(4)CH3COOH・NH2−PEG800−NH−Bocの合成
【化6】
【0049】
O,O’−ビス−(2−アミノプロピル)-ポリプロピレングリコール800(50.7g,63.4mmol)を50mlのメタノールに溶解させ、30分かけて50mlのメタノールに溶解させたdi-tert-butyl pyrocarbonate(Boc2)(12.04g,55.2mmol)を滴下し、窒素でバブリングパージ、一昼夜攪拌した。その後、酢酸3mlとトルエン100mlを加え、50ml以下になるまで減圧留去し、またトルエン100mlを加え50ml以下になるまで減圧留去という作業を二回繰り返し、冷凍保存した。次に反応物の半分をカラムクロマトグラフィー(シリカゲル、展開溶媒クロロホルム/メタノール/酢酸=90:10:0.1)で展開し、TLC(展開溶媒クロロホルム/メタノール/酢酸=100:30:2)でRf値0.29〜0.4の部分を取り出して、前記のトルエンを使用した減圧留去の操作を繰り返し、冷凍保存した。そして、それに水300mlを加え、塩化ナトリウムを過剰に投入、攪拌し濾過、未溶解の塩化ナトリウムを除去した。その後、3回クロロホルム 200mlで抽出を行い、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、真空乾燥によって目的となるCH3COOH・NH2−PEG800−NH−Bocを得た。
【0050】
(5)ビオチン−NH−PEG800−NH−Bocの合成
【化7】
【0051】
先に合成したCH3COOH・NH2−PEG800−NH−Boc(1.39g)とビオチン−NHS(0.59g,1.73mmol)を脱水したDMF12mlとトリエチルアミン(Et3N)0.3mlに加え、アルゴンでパージし一昼夜室温で攪拌した。それに水12mlを加えて2時間攪拌し、水を減圧留去、200mMの炭酸ナトリウム水溶液15mlを加え、濾過した。その後、塩化ナトリウムを飽和するまで加え、ジクロロメタン75mlで3回抽出し、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、ジクロロメタンを減圧留去し、真空乾燥することで、目的となるビオチン−NH−PEG800−NH−Bocを得た。
【0052】
(6)ビオチン−NH−PEG800−NH2・HClの合成
【化8】
【0053】
先に合成したビオチン−NH−PEG800−NH−Bocを12mlのギ酸と水0.3mlに溶解させ、窒素パージし、一昼夜攪拌することにより黄色の溶液が橙色へと変化した。その後、トルエン30mlを加えて減圧留去する操作を二回繰り返し、7mlの水に溶解させた。次に、窒素雰囲気下でイオン交換クロマトグラフィーを行い(Sephadex C−25,水で充填後、窒素バブリングした50mM塩化ナトリウム水溶液で展開)、ニンヒドリン反応で反応したアミノ基をもつ部分を取り出し、減圧留去、brineに溶解させ、80mlのクロロホルムで3回抽出し、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、真空乾燥によって目的となるビオチン−NH−PEG800−NH2・HCl(1.37g,1.3mmol)を得た。
【0054】
(7)ビオチン−NH−PEG800−SHの合成
【化9】
【0055】
ビオチン−NH−PEG800−NH2・HCl(930mg,0.78mmol)と3,3’−ジチオ(スクシンイミジルプロピオナート)(800mg,2mmol)、トリエチルアミン500μlをテトラヒドロフラン100mlに窒素雰囲気下で溶解させ、一昼夜攪拌した。その後、溶媒を減圧留去しbufferA(100mM塩化ナトリウム、50mM リン酸二水素ナトリウム、1mMエチレンジアミン四酢酸を水酸化ナトリウムでpH7.5に調整)50mlを加えて濾過した。濾液を2分間窒素バブリングし、1,4−ジチオトレイトール(620mg,4mmol)を加えて更に15分間バブリングした。その後、0.5Mの酢酸を加えてpH4.5とし、溶媒を50mlまで減圧留去した。そして溶液を25ml取り出し、窒素雰囲気下でカラムクロマトグラフィーを行い(Sephadex G−25,展開溶媒bufferB(100mM塩化ナトリウム、20mM酢酸ナトリウムを塩酸でpH4.5に調整)、赤色のアビジン−HABA溶液が黄色に変色したビオチン含有部分を取り出した。その後、クロロホルム150mlで3回抽出を行い、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、目的とするビオチン−NH−PEG800−SHを得た。
【0056】
2.FePtナノ粒子の合成
本実施例ではポリオールプロセスによる液相直接合成法を採用してFePtナノ粒子を合成した。ポリオールを還元剤及び溶媒として使用し、テトラエチレングリコールを用いることでFe及びPtの還元に際した反応速度を低下させ、液層において還元と規則化を同時に起こし、L10規則相を直接合成する手法である。また、FePt合金への前駆体としての有機金属錯体にはアセチルアセトネート(acac)錯体を使用した。実際には以下のようなプロセスで反応が行われていると考えられている(K. Sato, B. Jeyadevan, K. Tohji, J. Magn. Magn. Mater. 2005, 289, 1)(図3)。
【0057】
以下に、実験方法を具体的に記す。
Fe(acac)3(0.106g,0.3mmol)とPt(acac)2(0.118mg,0.3mmol)をテトラエチレングリコール80mlに溶解させ、窒素又はアルゴン置換させた後、マントルヒーターで約300℃に加熱、3.5時間加熱還流した。反応後、室温まで空冷し、少量のオレイルアミン及びオクタン酸を反応溶液に加え、ヘキサンを大量に加えた後、窒素もしくはアルゴンパージし、一昼夜油浴中約100℃で加熱還流した。その後ヘキサン層を分取し、ヘキサンを減圧留去、残留物にエタノール約100mlを加えて一夜冷蔵保存した。冷やしたエタノール溶液は遠心分離にかけ(1500rpm,12分)、上澄みを除去し、またエタノールを加えて同様に遠心分離、上澄みを除去し、底に残った黒い粒子を真空乾燥させ、目的のオレイルアミン及びオクタン酸が固定化されたFePtナノ粒子を得た。
【0058】
3.配位子交換反応によるビオチン化FePtナノ粒子の合成
配位子交換反応によるビオチン化FePtナノ粒子の合成過程を図4に示す。以下のようにして、配位子交換反応によりビオチン化FePtナノ粒子を合成した。
オレイルアミン及びオクタン酸を固定化したFePtナノ粒子(38mg)、ドパミン−mPEG(32mg)、ビオチン−PEG−SH(57mg)を8mlのジクロロメタンに溶解させ、窒素又はアルゴン雰囲気下で2日間加熱攪拌した(30〜40℃)。反応後、一度ジクロロメタンを減圧留去し、ジクロロメタンを少量とジエチルエーテルを加えて遠心分離した(1500rpm,12分)。上澄みは捨て、もう一度少量のジクロロメタンとジエチルエーテルを加えた後、もう一度遠心分離し(1500rpm,12分)、上澄みを捨て、沈殿物に目的とする黒色のビオチン化FePtナノ粒子を得た。
【0059】
(実施例2)FePtナノ粒子の評価
SQUIDによる磁化測定及びXRDによるX線回折パターン測定、TEMによる粒径分布の観察を通してFePtナノ粒子の評価を行った。
【0060】
(1)SQUIDによる磁化測定
FePtナノ粒子の磁化測定に際しては、図5に示すように、セロハンテープでFePtナノ粒子を包み込み、ストローで上下を挟み込んだ測定サンプル作成を行った。このとき、FePtナノ粒子は磁化が非常に大きいので、ストローによる反磁性はほぼ無視できると判断した。
磁化曲線を測定した。すなわち、磁化の磁場依存性を測定した。
更に、ZFC−FCによる磁化測定を行った。
【0061】
このときの磁化測定の結果を図6に示す。
この結果に見られるように、300Kにおいても明瞭な履歴曲線が観測され、残留磁化及び保磁力が存在することから、室温強磁性ナノ粒子の生成が確認される。実際の値は表3に示すとおりである。
【0062】
【表3】
【0063】
図7にZFC−FC磁化曲線の測定結果を示す。
この結果より、400Kにおいても、室温強磁性ナノ粒子の生成が確認される。
【0064】
(2)XRDによるX線回折パターンの測定
合成したFePtナノ粒子がL10規則相を取っているかどうかを調べるには、XRDによるX線回折パターンを調べるのが非常に有効である。このとき、FePtナノ粒子がL10規則相であるならば、消滅則によって本来消えるはずである、fcc構造では確認されない格子のピークが観測されるはずである。つまり、fcc構造では本来観測されない(001),(110),(210),(112)面からの格子反射ピークが観測されれば、そのFePtナノ粒子はL10規則相をとっているということができる。ここで実際に測定したFePtナノ粒子のXRDによる測定結果を図8に示す。
【0065】
この結果より、(001),(110),(210),(112)というfcc構造では本来見られない超格子ピークが観測されたことから、作成したFePtナノ粒子はL10規則相を取っていることが分かる。
また、格子定数を以下の式を使って計算したところ、a軸は3.85Åとなった。
【0066】
【数3】
【0067】
また、このとき、c軸はFeとPtが交互に層状に重なっているので、a軸に比べて少し値が小さくなっており、下の計算式を用いて計算すると、c軸は3.743Åであった。
【0068】
【数4】
【0069】
更に、以下に示すScherrerの式により、平均粒子径を計算したところ、平均粒子径は6.36nmとなり、XRDによる測定からナノ粒子の生成が確認された。
【0070】
【数5】
θ:回折角(°),λ:X線波長(1.54Å),h,k,l:ミュラー指数,
d:面間隔(Å),k:固有係数(約0.9),β:半幅値(rad)
【0071】
(3)TEMによるFePtナノ粒子の観測
ナノ粒子の粒径分布及び分散を確認するため、TEMによる観測を行った。
観測方法は、グリッドに、作成したオレイルアミン及びオクタン酸を固定化したFePtナノ粒子にヘキサンを加えて分散させ、滴下することで試料を作成した。また、配位子交換法によってビオチンを固定化したFePtナノ粒子については、アセトンを加えて分散させ、同じく滴下することで試料を作成した。観測の結果は以下の図9に示すとおりである。
【0072】
これらのTEM画像より、FePtナノ粒子が均一な粒子間距離をもって分散していることが確認され、粒子が凝集状態にないことから、有機配位子が各粒子間のスペーサーとしての役割を果たしているということが確認される。よってFePtナノ粒子上への有機配位子の固定化がここで確認される。
また、各TEM画像より粒径を測定し、分布及び平均粒子径を求めたものを図10に示す。
これより、FePtナノ粒子の合成が確認され、またこれはXRDによるScherrerの式の計算値と大きく違わないことが明らかとなった。
【0073】
(実施例3)IRスペクトルによる配位子交換の評価
IRスペクトルによって、配位子交換反応が進んだかどうかを検証した。
ここで、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子はKBrによる錠剤法で測定を行い、ビオチン化FePtナノ粒子はCaF2基盤にキャスティングして測定、ビオチン配位子はクロロホルムに溶解させ、CaF2による液体セルでの測定を行った。
【0074】
(1)ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルの比較
前記した方法により測定した、ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルを図11に示す。
【0075】
ここで、ビオチン配位子はクロロホルムに溶解させたため、3000及び2400cm−1等にクロロホルムの吸収がでている。そこで、配位子交換が行われているかどうかを確認するのに最適な吸収であるSH伸縮振動を比較するため、2500〜2700cm−1の吸収の拡大図を図12に示す。
【0076】
これより、ビオチン配位子には見られる2585cm−1のSH伸縮振動が、ビオチン化FePtナノ粒子には見られないことから、ビオチン配位子のFePtナノ粒子上への固定化が確認され、またPt−S結合は以下のメカニズムで生成するということが説明されるので、ここからPt−S結合の生成が示唆される。
R−SH + Pt → R−S−Pt + 1/2H2
【0077】
(2)配位子交換反応前後でのFePtナノ粒子のIRスペクトルの比較
前記(1)でFePtナノ粒子の方は、配位子のSH伸縮振動の消滅が確認されたが、それは単に配位子が存在していないだけである、という可能性が存在するため、更に、配位子交換反応前後でのIRスペクトルの違いを比較してビオチン配位子の存在を確認するため、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子とビオチン化FePtナノ粒子のIRスペクトルを測定した(図13)。
【0078】
図13において、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子には見られない、ビオチン配位子由来の1950cm−1の1,2,4−三置換ベンゼン環倍音、1350cm−1のグリコール変角振動が配位子交換反応後のFePtナノ粒子には見られることから、配位子交換反応後のFePtナノ粒子上にビオチン配位子が存在することが確認された。
これにより、配位子交換反応後のFePtナノ粒子にはビオチン配位子が結合していることがIRスペクトルより示唆された。
【0079】
(実施例4)ゼータ電位によるビオチン化FePtナノ粒子の評価
FePtナノ粒子にビオチン配位子を固定化することで、親水性の付与に成功したかを調べるため、ゼータ電位による微粒子表面の電荷状態を調べた。
【0080】
ここで、FePtナノ粒子にビオチン配位子が固定化されていれば、その配位子に存在するPEG鎖によって負電荷を帯びているはずであり、それによりFePtナノ粒子に親水性が付与されているということが示唆される(図14)。
【0081】
結果としては、FePtナノ粒子の表面ゼータ電位は−13.74mVであると測定することができたことからFePtナノ粒子のPEG鎖に基づく親水性の付与に成功したことが伺える。
【0082】
(実施例5)アビジン−HABA法によるビオチン化FePtナノ粒子の評価
1.溶液中でのビオチン化FePtナノ粒子の定量
ゼータ電位の測定により、ビオチン化FePtナノ粒子の親水性付与が確認され、水中に安定に分散することが確認されたので、次にFePtに結合しているビオチンがアビジンと定量的に結合するかどうかの確認をアビジン−HABA法により行った。
以下の方法で水中に分散させたビオチン化FePtナノ粒子上のビオチンの定量を行った。
ここで測定した試料はビオチン化FePtナノ粒子を分散させた溶液及び対照としてのビオチン溶液である。
【0083】
(1)アビジン−HABA法によるビオチン化FePtナノ粒子の定量
0.1Mリン酸二水素ナトリウム及び0.1Mリン酸水素二ナトリウムを作成し、この二つを混合してpH7.0としたリン酸緩衝液を作成した。以降は溶媒としてこれを用いた。
(i)アビジン−HABA溶液の作成
1.8mgのHABA色素(4−ヒドロキシアゾベンゼン−2’−カルボン酸)を50mlのメスフラスコで調整し、これにアビジン10mgを加えて、アビジン−HABA溶液とした。
(ii)ビオチン溶液の作成
D−ビオチン(122mg,0.5mmol)を200mlメスフラスコで調整し、そこから4ml取り出して50mlのメスフラスコで調整したものをビオチン溶液とした。
(iii)ビオチン及びビオチン化FePtナノ粒子の定量
作成したアビジン−HABA溶液2mlをセルに入れ、UV−visスペクトルで測定した後、ビオチン及びビオチン化FePt溶液を10μlずつ加えて、そのスペクトル変化を観測した。
結果を図15に示す。
【0084】
次に500nmにおけるビオチン添加による吸収の減少度を調べるため、500nmにおける吸収の検量線を図16に示す。
これにより、ビオチン化FePtナノ粒子がビオチンと同じように、定量的に500nmの吸収が降下していることが確認されるので、ビオチン化FePtナノ粒子溶液はアビジン−HABA法で定量できることと、アビジンと定量的に結合できることが明らかとなった。
【0085】
この方法は、ビオチンが存在しているかどうかの確認方法として迅速かつ簡便にでき、後述するFePtナノ粒子の溶液中での磁気分離が可能かどうかの判断材料として用いることができる。
【0086】
2.溶液中でのビオチン化FePtナノ粒子の磁気分離
前記1.での実験により、ビオチン化FePtナノ粒子表面のビオチンがアビジンと問題なく定量的に結合し、アビジン−HABA法を用いることでビオチン化FePtナノ粒子上のビオチンの存在を確認することが可能であることが明らかとなったので、ここでは溶液中に分散させたビオチン化FePtナノ粒子の磁気分離が可能であるかどうかを調べた。実験の概要は図17に示す通りである。
【0087】
この方法により、磁気分離したFePtナノ粒子を、前記1.と同じようにアビジン−HABA溶液2mlのセル中に滴下させ、UV−visスペクトルでその吸収の変化を調べた。ここで500nmの吸収が滴定により減少すれば、ビオチンの存在、つまりビオチン化FePtナノ粒子の存在が明らかとなり、ビオチン化FePtナノ粒子の磁気分離が可能なことが明らかとなる。
結果を図18に示す。
【0088】
この結果より、滴定後には500nmのアビジン−HABA複合体の吸収が減少していることが確認される。これにより、磁気回収したものにビオチンが含まれていることが明らかとなるので、ビオチン化FePtナノ粒子が回収されたことが確認される。よって、この結果より溶液中でビオチン化FePtナノ粒子を磁気回収することが可能であることが明らかとなった。
【図面の簡単な説明】
【0089】
【図1】FePt合金における構造転移と磁気特性の関係を示す図である。
【図2】FePt合金の状態密度図である。
【図3】ポリオールプロセスによるFePtナノ粒子の液相直接合成法を示す図である。
【図4】配位子交換反応によるビオチン化FePtナノ粒子の合成過程を示す図である。
【図5】FePtナノ粒子の磁化測定における測定サンプルの状態を示す図である。
【図6】外部磁場変化による磁化測定の結果を示す図である。上は、全体図、下は−10000〜10000Gまでの拡大図である。
【図7】ZFC−FC磁化曲線の測定結果を示す図である。
【図8】FePtナノ粒子のXRDによる測定結果を示す図である。
【図9】異なる倍率によるFePtナノ粒子のTEMによる観測結果を示す図である。上は、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子、下はビオチン化FePtナノ粒子のものである。
【図10】FePtナノ粒子の粒径分布を示す図である。左は、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子、右はビオチン化FePtナノ粒子のものである。
【図11】ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルを示す図である。
【図12】ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルの拡大図である。
【図13】オレイルアミン及びオクタン酸を固定化したFePtナノ粒子とビオチン化FePtナノ粒子のIRスペクトルを示す図である。
【図14】ビオチン化FePtナノ粒子の構造を示す図である。
【図15】ビオチン化FePtナノ粒子及びビオチンの定量結果を示す図である。
【図16】ビオチン化FePtナノ粒子及びビオチンの500nmにおける吸収の検量線を示す図である。
【図17】ビオチン化FePtナノ粒子の溶液からの磁気分離方法の概要を示す図である。
【図18】磁気分離したビオチン化FePtナノ粒子の滴下によるスペクトルの変化を示す図である。
【技術分野】
【0001】
有機分子で修飾された強磁性ナノ粒子並びにその製造方法及び分離方法に関する。
【背景技術】
【0002】
近年ナノ粒子は生物分離や各種測定、ドラッグデリバリーシステム等バイオ分野での幅広い応用が期待されている。ナノ粒子の特徴としては、粒径がナノサイズになることによる体積の減少から用途の拡大、生体適合性の増大が見込め、比表面積の増大から認識能の向上、認識時間の短縮が見込める。そのため、ナノ粒子の開発は大きなテーマとなっており、特にナノ粒子を磁性体とした磁性ナノ粒子はMRIによる測定、磁気誘導によるドラッグデリバリーシステムの構築等、その磁気特性を利用した分野での応用が期待されている(非特許文献1)。
【0003】
しかしながら、現状酸化鉄ナノ粒子に代表されるように、磁性粒子の体積を減少させていくと、室温では熱エネルギーによって磁気モーメントの向きが無秩序となる超常磁性体となり、その磁気特性が失われ、ナノサイズでの磁気分離などが不可能とされてきた。
【0004】
一般的に磁気異方性エネルギーKuと磁性体の体積Vの積が熱エネルギーKTを下回ると、磁性体としての性質を失ってしまうので、室温磁性ナノ粒子の候補としては、磁気異方性エネルギーKuの高いものであることが必要不可欠である。
【0005】
【非特許文献1】応用物理第72巻第7号909頁(2003)
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、室温においてナノサイズでの磁気分離が可能な、有機分子で修飾された強磁性ナノ粒子を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明の要旨は以下のとおりである。
(1)強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子。
(2)強磁性FePtナノ粒子がL10規則構造を有する前記(1)に記載の有機分子固定化強磁性ナノ粒子。
(3)強磁性FePtナノ粒子が、Fe錯体及びPt錯体を沸点300℃以上のポリオール系溶媒で処理することにより得られるものである前記(1)又は(2)に記載の有機分子固定化強磁性ナノ粒子。
(4)強磁性FePtナノ粒子に固定化される有機分子が、特異的結合のパートナー及び生理活性物質から選ばれる前記(1)〜(3)のいずれかに記載の有機分子固定化強磁性ナノ粒子。
(5)強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することを特徴とする、前記(1)〜(4)のいずれかに記載の有機分子固定化強磁性ナノ粒子の製造方法。
(6)カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物の少なくとも一方が特異的結合のパートナー、DNA、酵素及び生理活性物質から選ばれる残基を有する前記(5)に記載の製造方法。
(7)前記(5)又は(6)に記載の製造方法により得られた有機分子固定化強磁性ナノ粒子。
(8)前記(1)〜(4)及び(7)のいずれかに記載の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【発明の効果】
【0008】
本発明によれば、室温においてナノサイズでの磁気分離が可能な、有機分子で修飾された強磁性ナノ粒子を提供することができる。
【発明を実施するための最良の形態】
【0009】
本発明に用いる強磁性FePtナノ粒子とは、Fe原子及びPt原子を主成分(通常、全金属原子中Fe原子及びPt原子が合わせて50モル%以上、好ましくは90モル%以上)とする1〜10nm程度の粒径を有する粒子で室温にて磁気ヒステリシスを示すものをいう。
【0010】
L10規則構造を示すFePt合金においては高い結晶磁気異方性を有する(International Center For Diffraction Data, Powder Diffraction File SET43 Inorganic and organic, 43-1539)。この高い結晶磁気異方性は1Tbit/inch2以上の超高密度記録が原理的に可能であるとされ(K. Inomata, T. Sawa, S. Hashimoto, J. Appl. Phys. 1996, 64, 2537)、将来の超高密度記録媒体への応用が期待されている。
【0011】
本発明では、高い結晶磁気異方性エネルギーKuを有していることから、ナノオーダーでの強磁性を示すFePtナノ粒子を用いる。
【0012】
FePt合金は安定相であるL10規則構造と準安定構造であるfcc構造の2種の構造を取りうる。ここでL10規則構造はc軸に磁化容易軸があり、高い結晶磁気異方性を有することから強磁性を示すのに対し、準安定構造であるfcc構造は磁化容易軸が存在せず、常磁性を示す(図1)。
【0013】
以下、L10−FePt合金の高い結晶磁気異方性の起源を説明する。
4d遷移金属Pd、5d遷移金属Ptは白金族原子と呼ばれ、バルクの単体では常磁性を示すが、強磁性体直前の金属であることが知られている。また、この白金族は鉄族との合金化によって大きなスピン偏極を示すことが知られている。
【0014】
図2にFePt合金の状態密度図を示した(A. Sakuma, J. Phys. Soc. Jpn. 1994, 63, 3053)。
FeとPtのmajority spinバンドはフェルミエネルギーEf付近でFe3dバンドとPt4dバンドの混成化に由来する重なりが見られる。この混成化によってPtのスピン磁気モーメントが誘起され、縮退により消滅していたFeの軌道磁気モーメントが復活するといわれている。表1にFe及びPtのスピン軌道結合エネルギーζ、スピン磁気モーメントS、軌道磁気モーメントL、合成磁気モーメントMtotal、磁気モーメントの実測値Mexを示した。Fe及びPtの合金化による、Ptの磁気モーメントの誘起を確認することができる。このPtの磁気モーメントの誘起による大きなスピン軌道結合定数により、FePt合金は大きなスピン軌道相互作用を持つことが分かり、高い磁気異方性の起源となっていることを知ることができる。
【0015】
【表1】
【0016】
FePt合金は一般にFe組成がモル比で45%〜64%のとき、L10規則構造を有する。しかしながら、化学合成などによりFePt合金を作成した場合、準安定相であるfcc不規則相をとることが多い。2000年、S.Sunらによってポリオールプロセスと呼ばれる化学的手法により粒径分布のほとんどないFePtナノ粒子の合成法が報告され(S. Sun, C. B. Murray, D. Weller, L. Folks, A. Moser, Science 2000, 287, 1989; S. Sun, Adv. Mater. 2006, 18, 393)、FePt合金に関する研究が加速し、磁気記録デバイスへの実用化が近いものと期待された。しかしながら、このポリオールプロセスにより合成したFePtナノ粒子もやはり準安定相である不規則相を取っており、高い磁気異方性を有するL10構造への構造転移には600℃程度の熱処理による規則化プロセスが必要であった。この熱処理プロセスはFePtナノ粒子同士の焼結や凝集を招き、これが実用化への大きな障壁となっている。このような問題を解決するために、この熱処理による規則化温度の低減もしくは、熱処理時の焼結を防ぐ試みがなされており、以下に、いくつかの方法を例示する。
【0017】
(1)高沸点エチレングリコール系溶媒等のポリオール系溶媒を用いた液層直接合成(K. Sato, B. Jeyadevan, K. Tohji, J. Magn. Magn. Mater. 2005, 289, 1;R. Minami, Y. Kitamoto, T.Chikata, S. Kato, Electrochimica Acta 2005, 51, 864)
この手法は、Fe及びPt化合物の前駆体(錯体、塩など)を、ポリオール系溶媒を還元剤及び溶媒として還元する手法である。エチレングリコール系溶媒等のポリオール系溶媒は、Fe及びPtの還元に際した反応速度を低下させることで、規則化温度を300℃以下に低下させることが知られている。更に、ポリオール系溶媒の一種であり、300℃以上の沸点を持つテトラエチレングリコール等の高沸点ポリオール系溶媒を溶媒として用いることで液層において還元と規則化を同時に起こし、L10規則相を直接合成する手法である。この手法では、L10構造への完全な規則化には至らず、部分的な規則化に留まるという欠点があるものの、得られるFePt合金において高い磁気異方性が確認されており、有効な手法の一つである。
【0018】
また、この手法を用いれば、600℃におけるアニーリングというプロセスを省くことができ、更に液相中での合成が可能なことから、合成後の化学的な表面修飾が可能であるという利点がある。本発明にとって、これは大きなアドバンテージであり、後述する実施例で採用した合成法がこの手法である。
【0019】
ここで用いるFe及びPt化合物の前駆体(錯体、塩など)としてはFe元素及びPt元素を含む錯体や金属塩であれば制限はなく、例えばアセチルアセトネート(acac)錯体、カルボニル錯体、エトキシド化合物、塩化物などが挙げられる。
【0020】
ここで用いるポリオール系溶媒としては沸点が300℃以上のものであれば制限はないが、好ましくはテトラエチレングリコール、ヘキサエチレングリコール、オクタエチレングリコール、又は高沸点有機溶媒とエチレングリコール系溶媒の混合物等の高沸点エチレングリコール系溶媒が挙げられる。
【0021】
Fe錯体とPt錯体の割合は、好ましくは、モル比で45:55〜64:36である。ポリオール系溶媒の使用量は、好ましくは、金属イオン1mmol当たり100ml程度である。
【0022】
(2)第三元素の添加による規則化温度の低減(J. W. Harrell, D. E. Nikles, S. S. Kang, X. C. Sun, Z. Jia, J. Magn. Soc. Jpn. 2004, 28, 847; O. Kitamura, Y. Shimada, K. Oikawa, H. Daimon, K. Fukamichi, Appl. Phys. Lett. 2002, 80, 2147; T. Maeda, T. Kai, A. Kikitu, T. Nagase, J. Akiyama, Appl. Phys. Lett. 2001, 78, 1104; K. M. Park, K. H. Na, J. G. Na, P. W. Jang, H. J. Kim, S. R. Lee, IEEE Trans. Magn. 2000, 38, 1961)
この手法は、Fe及びPtの他に第三の元素を添加することにより、原子拡散を増大させて規則化を促そうという手法である。Ag,Au,Cuといった元素を添加することでL10規則相への構造転移温度が低下するという報告がなされている。この中でも特に大きな規則化温度の低減を齎すのがCuであり、Ag,Auにおいて規則化温度が400℃まで低下するのに対し、Cuの添加は規則化温度を300℃まで低下させている。Ag,Auにおいては熱処理後の元素の析出が観測されているが、Cuにおいては観測されていない。つまり、Au及びAgについては析出により原子の拡散を促し規則化温度を低下させているのに対し、CuはFePt合金中へ固溶することによって、規則相のポテンシャルエネルギーを下げ、規則化を促しているわけである。
【0023】
本発明においては、この手法を用いることもできる。更に、SiO2ナノリアクター法を用いることもできる。
【0024】
前述したようにFePt合金は安定相であるL10規則相と準安定相であるfcc不規則相の2種の構造を取りうる。したがって、FePt合金を用いる時には、構造転移の証明や、この構造転移の割合を示すオーダーパラメーターを知ることが大変重要である。これらを知るための手法としてはX線による回折パターンの解析が極めて有効である。L10規則相においては、不規則相であるfcc構造の回折線に加え、規則配置に起因する余分の回折線が出現する。この余分の回折線を超格子反射と呼ぶ。超格子反射の出現は、原子配置の規則化により、消滅則が異なってくることに由来するものである。
【0025】
不規則相であるfcc構造の消滅則は、
【数1】
で与えられることからも分かるように、fcc構造においてはz=1、z=0の面からの回折線の位相とz=1/2の面からの回折線の位相は半波長分だけずれるため、回折線は打ち消しあって(001)面からのブラッグ反射は起こらない。一方、規則相であるL10構造の消滅則は、
【0026】
【数2】
で与えられる。この消滅則からも分かるようにz=0、z=1の面とz=1/2の面では散乱因子が異なっており、回折線は完全には打ち消しあわず、結果として超格子反射が観測される。更に、このような(001)面などからの超格子反射の出現に加え、正方晶への変化に伴う(002)、(200)面からの回折線の分裂が生じる。
【0027】
したがって、X線回折パターンを解析し、超格子反射の出現を観測することで、構造転移の証明が可能となる。
【0028】
本発明において強磁性FePtナノ粒子に固定化させる有機分子は、目的に応じて適宜選択すればよく、例えば特異的結合のパートナー、DNA、酵素、生理活性物質が挙げられる。また、前記有機分子に加えて、目的に応じて、例えば親水性、生体適合性、分散性、各種溶媒への親媒性等を付与する目的でポリエチレングリコール鎖、イオン基を有するアルキル鎖等を組み合わせて固定化することもできる。
【0029】
前記特異的結合のパートナーとは、関与する分子の三次元構造に依存する特異的な非共有相互作用により相互作用する分子の対のメンバーをいい、当該特異的結合のパートナーの典型的な対としては、例えば抗原−抗体、ハプテン−抗体、ホルモン−レセプター、核酸鎖−相補的核酸鎖、基質−酵素、基質類似体−酵素、インヒビター−酵素、炭水化物−レクチン、ビオチン−アビジン及びウイルス−細胞レセプターが挙げられる。
【0030】
生理活性物質としては、例えば薬物が挙げられる。
例えば、抗体を固定化した強磁性ナノ粒子は細胞分離、化学物質の検出・回収に用いることができ、DNA鎖を固定化した強磁性ナノ粒子はSNPs検出、遺伝子診断、品種識別に用いることができ、酵素を固定化した強磁性ナノ粒子は酵素反応センシング・回収再利用に用いることができ、ビオチンを固定化した強磁性ナノ粒子は、各種測定方法、又はビオチン−アビジン結合を介した各種物質の固定化に用いることができ、薬物を固定化した強磁性ナノ粒子は、ドラッグデリバリーシステムに用いることができる。
【0031】
強磁性FePtナノ粒子に有機分子を固定化させるためには、目的とする有機分子を含む配位子がFeとPtに対して高いアフィニティを有するカルボキシル基、アミノ基、メルカプト基、水酸基等を末端にもたなければならない。また、バイオ磁性ナノ粒子として、生体内で応用する場合には、親水性、生体適合性の付与、及び目的とする各有機分子間の立体的混雑を避けるスペーサーとして、ポリエチレングリコール鎖(PEG)(好ましくは重合度10〜20)を配位子中に含ませることが好ましい。
【0032】
そのため、後述する実施例では、PEGの両末端にビオチン及びメルカプト基を持つビオチン−PEG−SH及び片方の末端にドパミン基を持つドパミン−ポリエチレングリコールモノメチルエーテル(mPEG)という二つの配位子を設計・合成した。ここでビオチン−PEG−SHはPtに配位し、ドパミン−mPEGはFeに配位する。ドパミン−mPEGはビオチン基を持たないものの、親水性の向上を目的としており、粒子表面のFeの酸化をも同時に防いでいる。
【0033】
次いで、合成した配位子を強磁性FePtナノ粒子上へ固定化させるが、前記のポリオール系溶媒を用いた液層直接合成により強磁性FePtナノ粒子を合成する場合には、合成直後の強磁性FePtナノ粒子は大量のポリオール系溶媒中に存在するため、生成量が非常に少ない合成した配位子を直接固定化させることが困難なことから、一度別の有機配位子、例えば、オクタン酸等の炭素数6〜20の脂肪酸及びオレイルアミン等の炭素数6〜20の脂肪族アミンをFePtナノ粒子上に固定化させ、配位子交換反応を用いて目的の合成した配位子、例えばカテコール残基を有する有機化合物(例えば、ドパミン−mPEG)及びメルカプト基を有する有機化合物(例えば、ビオチン−PEG−SH)をFePtナノ粒子上に固定化させることが好ましい(Rui Hong, Nicholas O. Fischer, Todd Emrick, Vincent M. Rotello, chem. Mater, 2005, 17, 4617)。
【0034】
前記脂肪酸の使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)であり、脂肪族アミンの使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)であり、目的の合成した配位子の使用量は、好ましくは、目的とする粒子の表面原子数の10倍以上(モル比)である。
本発明の有機分子固定化強磁性ナノ粒子は、磁気分離が可能である。したがって、本発明の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離する方法に適用することができる。
【0035】
例えば、特定抗体が固定化された強磁性ナノ粒子を診断薬として用いる場合、検体にもし特定の抗原が存在すれば、抗原は抗体が固定化された強磁性ナノ粒子に吸着される。吸着後、標識(発色団など)された抗体(二次抗体)を加え、磁石により検体から分離し、分離された強磁性ナノ粒子を緩衝溶液などで洗浄し、余分な標識抗体を取り除く、次いで、得られた強磁性ナノ粒子を再度緩衝溶液などで希釈し標識部に分光工学的な手法などを用いて抗原の定量が行える。
【実施例】
【0036】
以下、実施例を挙げて本発明を具体的に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
以下の実施例で使用した測定機器の一覧を表2に示した。
【0037】
【表2】
【0038】
なお、UV−Visは紫外可視分光光度計を、XRDは粉末X線回折装置を、SQUIDは超伝導量子干渉素子磁力計を、TEMは透過型電子顕微鏡をそれぞれ示し、以後これらの略語を用いて表記する。
【0039】
(実施例1)ビオチン化FePtナノ粒子の合成
1.有機配位子の合成
FePtナノ粒子に表面修飾する有機配位子としてビオチン−PEG−SH及びドパミン−mPEGの二つの配位子を既報(Rui Hong, Nicholas O. Fischer, Todd Emrick, Vincent M. Rotello, chem. Mater, 2005, 17, 4617; Bryan Parrish and Todd Emrick, Macromplecules, 2004, 37, 5863; Karl Kaiser, Markus Marek, Thomas Haselgrubler, Hansgeorg Schindler, and Hermann J. Gruber ,Bioconjugate chem. 1997, 8, 54)に従い、設計・合成した。全体のスキームは以下の通りとなる。
【0040】
ドパミン−mPEGの全体合成反応式
【化1】
【0041】
ビオチン−PEG−SHの全体合成反応式
【化2】
【0042】
以下、各合成段階の詳細を述べる。
(1)ポリ(エチレングリコール)−コハク酸エステルの合成
【化3】
【0043】
クロロホルム100mlの中にポリ(エチレングリコール)(2.759g,5mmol)、無水コハク酸(1.2525g,12.5mmol)、4−(ジメチルアミノ)ピリジン(0.0648g,0.5mmol)を溶解させ、12時間加熱還流した。その後溶媒を減圧留去し、水に溶解させた後、ヘキサン:酢酸エチル=1:1の混合比で混ぜた溶液で洗浄、ジクロロメタンで抽出した。ジクロロメタン抽出液を硫酸マグネシウムで脱水乾燥させ、硫酸マグネシウムを濾別した後、ジクロロメタンを減圧留去し一昼夜真空乾燥を行いポリ(エチレングリコール)−コハク酸エステルを得た(3.3718g,5mmol,100%)。
【0044】
(2)ドパミン−ポリ(エチレングリコール)の合成
【化4】
【0045】
N,N−ジメチルホルムアミド10ml中に、ポリ(エチレングリコール)−コハク酸エステル(1.3202g,2mmol)、1−ヒドロキシベンゾトリアゾール(0.2702g,2mmol)、N,N−ジイソプロピルエチルアミン(0.52g,4mmol)を0℃で加え、攪拌した。その後1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(3.902g,20mmol)、ドパミン塩酸塩(0.4025g,2.1mmol)を5分間で加えた。この溶液を窒素パージした後4℃で一夜攪拌し、N,N−ジメチルホルムアミドを減圧留去し、150mlのクロロホルムに溶解させた。1Mの冷やした塩酸、飽和炭酸水素ナトリウム水溶液、水、brineで洗浄し、硫酸マグネシウムで脱水乾燥させ、硫酸マグネシウムを濾別した後、クロロホルムを減圧留去し、真空乾燥させドパミン−ポリ(エチレングリコール)を得た(1.28g,1.6mmol,80%)。
【0046】
(3)ビオチン−NHSの合成
【化5】
【0047】
N,N−ジメチルホルムアミド中にD−ビオチン(0.5292g,2.17mmol)、N−ヒドロキシスクシンイミド(0.3259g,2.83mmol)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(0.53g,2.76mmol)を窒素パージして一昼夜攪拌した。N,N−ジメチルホルムアミドを減圧留去し、濾過、過剰の水とメタノールで洗浄し、ビオチン−NHSの白色結晶を得た(0.4858g,1.423mmol,65.7%)。
【0048】
(4)CH3COOH・NH2−PEG800−NH−Bocの合成
【化6】
【0049】
O,O’−ビス−(2−アミノプロピル)-ポリプロピレングリコール800(50.7g,63.4mmol)を50mlのメタノールに溶解させ、30分かけて50mlのメタノールに溶解させたdi-tert-butyl pyrocarbonate(Boc2)(12.04g,55.2mmol)を滴下し、窒素でバブリングパージ、一昼夜攪拌した。その後、酢酸3mlとトルエン100mlを加え、50ml以下になるまで減圧留去し、またトルエン100mlを加え50ml以下になるまで減圧留去という作業を二回繰り返し、冷凍保存した。次に反応物の半分をカラムクロマトグラフィー(シリカゲル、展開溶媒クロロホルム/メタノール/酢酸=90:10:0.1)で展開し、TLC(展開溶媒クロロホルム/メタノール/酢酸=100:30:2)でRf値0.29〜0.4の部分を取り出して、前記のトルエンを使用した減圧留去の操作を繰り返し、冷凍保存した。そして、それに水300mlを加え、塩化ナトリウムを過剰に投入、攪拌し濾過、未溶解の塩化ナトリウムを除去した。その後、3回クロロホルム 200mlで抽出を行い、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、真空乾燥によって目的となるCH3COOH・NH2−PEG800−NH−Bocを得た。
【0050】
(5)ビオチン−NH−PEG800−NH−Bocの合成
【化7】
【0051】
先に合成したCH3COOH・NH2−PEG800−NH−Boc(1.39g)とビオチン−NHS(0.59g,1.73mmol)を脱水したDMF12mlとトリエチルアミン(Et3N)0.3mlに加え、アルゴンでパージし一昼夜室温で攪拌した。それに水12mlを加えて2時間攪拌し、水を減圧留去、200mMの炭酸ナトリウム水溶液15mlを加え、濾過した。その後、塩化ナトリウムを飽和するまで加え、ジクロロメタン75mlで3回抽出し、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、ジクロロメタンを減圧留去し、真空乾燥することで、目的となるビオチン−NH−PEG800−NH−Bocを得た。
【0052】
(6)ビオチン−NH−PEG800−NH2・HClの合成
【化8】
【0053】
先に合成したビオチン−NH−PEG800−NH−Bocを12mlのギ酸と水0.3mlに溶解させ、窒素パージし、一昼夜攪拌することにより黄色の溶液が橙色へと変化した。その後、トルエン30mlを加えて減圧留去する操作を二回繰り返し、7mlの水に溶解させた。次に、窒素雰囲気下でイオン交換クロマトグラフィーを行い(Sephadex C−25,水で充填後、窒素バブリングした50mM塩化ナトリウム水溶液で展開)、ニンヒドリン反応で反応したアミノ基をもつ部分を取り出し、減圧留去、brineに溶解させ、80mlのクロロホルムで3回抽出し、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、真空乾燥によって目的となるビオチン−NH−PEG800−NH2・HCl(1.37g,1.3mmol)を得た。
【0054】
(7)ビオチン−NH−PEG800−SHの合成
【化9】
【0055】
ビオチン−NH−PEG800−NH2・HCl(930mg,0.78mmol)と3,3’−ジチオ(スクシンイミジルプロピオナート)(800mg,2mmol)、トリエチルアミン500μlをテトラヒドロフラン100mlに窒素雰囲気下で溶解させ、一昼夜攪拌した。その後、溶媒を減圧留去しbufferA(100mM塩化ナトリウム、50mM リン酸二水素ナトリウム、1mMエチレンジアミン四酢酸を水酸化ナトリウムでpH7.5に調整)50mlを加えて濾過した。濾液を2分間窒素バブリングし、1,4−ジチオトレイトール(620mg,4mmol)を加えて更に15分間バブリングした。その後、0.5Mの酢酸を加えてpH4.5とし、溶媒を50mlまで減圧留去した。そして溶液を25ml取り出し、窒素雰囲気下でカラムクロマトグラフィーを行い(Sephadex G−25,展開溶媒bufferB(100mM塩化ナトリウム、20mM酢酸ナトリウムを塩酸でpH4.5に調整)、赤色のアビジン−HABA溶液が黄色に変色したビオチン含有部分を取り出した。その後、クロロホルム150mlで3回抽出を行い、硫酸ナトリウムで一昼夜脱水乾燥した。硫酸ナトリウムを濾別した後、クロロホルムを減圧留去し、目的とするビオチン−NH−PEG800−SHを得た。
【0056】
2.FePtナノ粒子の合成
本実施例ではポリオールプロセスによる液相直接合成法を採用してFePtナノ粒子を合成した。ポリオールを還元剤及び溶媒として使用し、テトラエチレングリコールを用いることでFe及びPtの還元に際した反応速度を低下させ、液層において還元と規則化を同時に起こし、L10規則相を直接合成する手法である。また、FePt合金への前駆体としての有機金属錯体にはアセチルアセトネート(acac)錯体を使用した。実際には以下のようなプロセスで反応が行われていると考えられている(K. Sato, B. Jeyadevan, K. Tohji, J. Magn. Magn. Mater. 2005, 289, 1)(図3)。
【0057】
以下に、実験方法を具体的に記す。
Fe(acac)3(0.106g,0.3mmol)とPt(acac)2(0.118mg,0.3mmol)をテトラエチレングリコール80mlに溶解させ、窒素又はアルゴン置換させた後、マントルヒーターで約300℃に加熱、3.5時間加熱還流した。反応後、室温まで空冷し、少量のオレイルアミン及びオクタン酸を反応溶液に加え、ヘキサンを大量に加えた後、窒素もしくはアルゴンパージし、一昼夜油浴中約100℃で加熱還流した。その後ヘキサン層を分取し、ヘキサンを減圧留去、残留物にエタノール約100mlを加えて一夜冷蔵保存した。冷やしたエタノール溶液は遠心分離にかけ(1500rpm,12分)、上澄みを除去し、またエタノールを加えて同様に遠心分離、上澄みを除去し、底に残った黒い粒子を真空乾燥させ、目的のオレイルアミン及びオクタン酸が固定化されたFePtナノ粒子を得た。
【0058】
3.配位子交換反応によるビオチン化FePtナノ粒子の合成
配位子交換反応によるビオチン化FePtナノ粒子の合成過程を図4に示す。以下のようにして、配位子交換反応によりビオチン化FePtナノ粒子を合成した。
オレイルアミン及びオクタン酸を固定化したFePtナノ粒子(38mg)、ドパミン−mPEG(32mg)、ビオチン−PEG−SH(57mg)を8mlのジクロロメタンに溶解させ、窒素又はアルゴン雰囲気下で2日間加熱攪拌した(30〜40℃)。反応後、一度ジクロロメタンを減圧留去し、ジクロロメタンを少量とジエチルエーテルを加えて遠心分離した(1500rpm,12分)。上澄みは捨て、もう一度少量のジクロロメタンとジエチルエーテルを加えた後、もう一度遠心分離し(1500rpm,12分)、上澄みを捨て、沈殿物に目的とする黒色のビオチン化FePtナノ粒子を得た。
【0059】
(実施例2)FePtナノ粒子の評価
SQUIDによる磁化測定及びXRDによるX線回折パターン測定、TEMによる粒径分布の観察を通してFePtナノ粒子の評価を行った。
【0060】
(1)SQUIDによる磁化測定
FePtナノ粒子の磁化測定に際しては、図5に示すように、セロハンテープでFePtナノ粒子を包み込み、ストローで上下を挟み込んだ測定サンプル作成を行った。このとき、FePtナノ粒子は磁化が非常に大きいので、ストローによる反磁性はほぼ無視できると判断した。
磁化曲線を測定した。すなわち、磁化の磁場依存性を測定した。
更に、ZFC−FCによる磁化測定を行った。
【0061】
このときの磁化測定の結果を図6に示す。
この結果に見られるように、300Kにおいても明瞭な履歴曲線が観測され、残留磁化及び保磁力が存在することから、室温強磁性ナノ粒子の生成が確認される。実際の値は表3に示すとおりである。
【0062】
【表3】
【0063】
図7にZFC−FC磁化曲線の測定結果を示す。
この結果より、400Kにおいても、室温強磁性ナノ粒子の生成が確認される。
【0064】
(2)XRDによるX線回折パターンの測定
合成したFePtナノ粒子がL10規則相を取っているかどうかを調べるには、XRDによるX線回折パターンを調べるのが非常に有効である。このとき、FePtナノ粒子がL10規則相であるならば、消滅則によって本来消えるはずである、fcc構造では確認されない格子のピークが観測されるはずである。つまり、fcc構造では本来観測されない(001),(110),(210),(112)面からの格子反射ピークが観測されれば、そのFePtナノ粒子はL10規則相をとっているということができる。ここで実際に測定したFePtナノ粒子のXRDによる測定結果を図8に示す。
【0065】
この結果より、(001),(110),(210),(112)というfcc構造では本来見られない超格子ピークが観測されたことから、作成したFePtナノ粒子はL10規則相を取っていることが分かる。
また、格子定数を以下の式を使って計算したところ、a軸は3.85Åとなった。
【0066】
【数3】
【0067】
また、このとき、c軸はFeとPtが交互に層状に重なっているので、a軸に比べて少し値が小さくなっており、下の計算式を用いて計算すると、c軸は3.743Åであった。
【0068】
【数4】
【0069】
更に、以下に示すScherrerの式により、平均粒子径を計算したところ、平均粒子径は6.36nmとなり、XRDによる測定からナノ粒子の生成が確認された。
【0070】
【数5】
θ:回折角(°),λ:X線波長(1.54Å),h,k,l:ミュラー指数,
d:面間隔(Å),k:固有係数(約0.9),β:半幅値(rad)
【0071】
(3)TEMによるFePtナノ粒子の観測
ナノ粒子の粒径分布及び分散を確認するため、TEMによる観測を行った。
観測方法は、グリッドに、作成したオレイルアミン及びオクタン酸を固定化したFePtナノ粒子にヘキサンを加えて分散させ、滴下することで試料を作成した。また、配位子交換法によってビオチンを固定化したFePtナノ粒子については、アセトンを加えて分散させ、同じく滴下することで試料を作成した。観測の結果は以下の図9に示すとおりである。
【0072】
これらのTEM画像より、FePtナノ粒子が均一な粒子間距離をもって分散していることが確認され、粒子が凝集状態にないことから、有機配位子が各粒子間のスペーサーとしての役割を果たしているということが確認される。よってFePtナノ粒子上への有機配位子の固定化がここで確認される。
また、各TEM画像より粒径を測定し、分布及び平均粒子径を求めたものを図10に示す。
これより、FePtナノ粒子の合成が確認され、またこれはXRDによるScherrerの式の計算値と大きく違わないことが明らかとなった。
【0073】
(実施例3)IRスペクトルによる配位子交換の評価
IRスペクトルによって、配位子交換反応が進んだかどうかを検証した。
ここで、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子はKBrによる錠剤法で測定を行い、ビオチン化FePtナノ粒子はCaF2基盤にキャスティングして測定、ビオチン配位子はクロロホルムに溶解させ、CaF2による液体セルでの測定を行った。
【0074】
(1)ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルの比較
前記した方法により測定した、ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルを図11に示す。
【0075】
ここで、ビオチン配位子はクロロホルムに溶解させたため、3000及び2400cm−1等にクロロホルムの吸収がでている。そこで、配位子交換が行われているかどうかを確認するのに最適な吸収であるSH伸縮振動を比較するため、2500〜2700cm−1の吸収の拡大図を図12に示す。
【0076】
これより、ビオチン配位子には見られる2585cm−1のSH伸縮振動が、ビオチン化FePtナノ粒子には見られないことから、ビオチン配位子のFePtナノ粒子上への固定化が確認され、またPt−S結合は以下のメカニズムで生成するということが説明されるので、ここからPt−S結合の生成が示唆される。
R−SH + Pt → R−S−Pt + 1/2H2
【0077】
(2)配位子交換反応前後でのFePtナノ粒子のIRスペクトルの比較
前記(1)でFePtナノ粒子の方は、配位子のSH伸縮振動の消滅が確認されたが、それは単に配位子が存在していないだけである、という可能性が存在するため、更に、配位子交換反応前後でのIRスペクトルの違いを比較してビオチン配位子の存在を確認するため、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子とビオチン化FePtナノ粒子のIRスペクトルを測定した(図13)。
【0078】
図13において、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子には見られない、ビオチン配位子由来の1950cm−1の1,2,4−三置換ベンゼン環倍音、1350cm−1のグリコール変角振動が配位子交換反応後のFePtナノ粒子には見られることから、配位子交換反応後のFePtナノ粒子上にビオチン配位子が存在することが確認された。
これにより、配位子交換反応後のFePtナノ粒子にはビオチン配位子が結合していることがIRスペクトルより示唆された。
【0079】
(実施例4)ゼータ電位によるビオチン化FePtナノ粒子の評価
FePtナノ粒子にビオチン配位子を固定化することで、親水性の付与に成功したかを調べるため、ゼータ電位による微粒子表面の電荷状態を調べた。
【0080】
ここで、FePtナノ粒子にビオチン配位子が固定化されていれば、その配位子に存在するPEG鎖によって負電荷を帯びているはずであり、それによりFePtナノ粒子に親水性が付与されているということが示唆される(図14)。
【0081】
結果としては、FePtナノ粒子の表面ゼータ電位は−13.74mVであると測定することができたことからFePtナノ粒子のPEG鎖に基づく親水性の付与に成功したことが伺える。
【0082】
(実施例5)アビジン−HABA法によるビオチン化FePtナノ粒子の評価
1.溶液中でのビオチン化FePtナノ粒子の定量
ゼータ電位の測定により、ビオチン化FePtナノ粒子の親水性付与が確認され、水中に安定に分散することが確認されたので、次にFePtに結合しているビオチンがアビジンと定量的に結合するかどうかの確認をアビジン−HABA法により行った。
以下の方法で水中に分散させたビオチン化FePtナノ粒子上のビオチンの定量を行った。
ここで測定した試料はビオチン化FePtナノ粒子を分散させた溶液及び対照としてのビオチン溶液である。
【0083】
(1)アビジン−HABA法によるビオチン化FePtナノ粒子の定量
0.1Mリン酸二水素ナトリウム及び0.1Mリン酸水素二ナトリウムを作成し、この二つを混合してpH7.0としたリン酸緩衝液を作成した。以降は溶媒としてこれを用いた。
(i)アビジン−HABA溶液の作成
1.8mgのHABA色素(4−ヒドロキシアゾベンゼン−2’−カルボン酸)を50mlのメスフラスコで調整し、これにアビジン10mgを加えて、アビジン−HABA溶液とした。
(ii)ビオチン溶液の作成
D−ビオチン(122mg,0.5mmol)を200mlメスフラスコで調整し、そこから4ml取り出して50mlのメスフラスコで調整したものをビオチン溶液とした。
(iii)ビオチン及びビオチン化FePtナノ粒子の定量
作成したアビジン−HABA溶液2mlをセルに入れ、UV−visスペクトルで測定した後、ビオチン及びビオチン化FePt溶液を10μlずつ加えて、そのスペクトル変化を観測した。
結果を図15に示す。
【0084】
次に500nmにおけるビオチン添加による吸収の減少度を調べるため、500nmにおける吸収の検量線を図16に示す。
これにより、ビオチン化FePtナノ粒子がビオチンと同じように、定量的に500nmの吸収が降下していることが確認されるので、ビオチン化FePtナノ粒子溶液はアビジン−HABA法で定量できることと、アビジンと定量的に結合できることが明らかとなった。
【0085】
この方法は、ビオチンが存在しているかどうかの確認方法として迅速かつ簡便にでき、後述するFePtナノ粒子の溶液中での磁気分離が可能かどうかの判断材料として用いることができる。
【0086】
2.溶液中でのビオチン化FePtナノ粒子の磁気分離
前記1.での実験により、ビオチン化FePtナノ粒子表面のビオチンがアビジンと問題なく定量的に結合し、アビジン−HABA法を用いることでビオチン化FePtナノ粒子上のビオチンの存在を確認することが可能であることが明らかとなったので、ここでは溶液中に分散させたビオチン化FePtナノ粒子の磁気分離が可能であるかどうかを調べた。実験の概要は図17に示す通りである。
【0087】
この方法により、磁気分離したFePtナノ粒子を、前記1.と同じようにアビジン−HABA溶液2mlのセル中に滴下させ、UV−visスペクトルでその吸収の変化を調べた。ここで500nmの吸収が滴定により減少すれば、ビオチンの存在、つまりビオチン化FePtナノ粒子の存在が明らかとなり、ビオチン化FePtナノ粒子の磁気分離が可能なことが明らかとなる。
結果を図18に示す。
【0088】
この結果より、滴定後には500nmのアビジン−HABA複合体の吸収が減少していることが確認される。これにより、磁気回収したものにビオチンが含まれていることが明らかとなるので、ビオチン化FePtナノ粒子が回収されたことが確認される。よって、この結果より溶液中でビオチン化FePtナノ粒子を磁気回収することが可能であることが明らかとなった。
【図面の簡単な説明】
【0089】
【図1】FePt合金における構造転移と磁気特性の関係を示す図である。
【図2】FePt合金の状態密度図である。
【図3】ポリオールプロセスによるFePtナノ粒子の液相直接合成法を示す図である。
【図4】配位子交換反応によるビオチン化FePtナノ粒子の合成過程を示す図である。
【図5】FePtナノ粒子の磁化測定における測定サンプルの状態を示す図である。
【図6】外部磁場変化による磁化測定の結果を示す図である。上は、全体図、下は−10000〜10000Gまでの拡大図である。
【図7】ZFC−FC磁化曲線の測定結果を示す図である。
【図8】FePtナノ粒子のXRDによる測定結果を示す図である。
【図9】異なる倍率によるFePtナノ粒子のTEMによる観測結果を示す図である。上は、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子、下はビオチン化FePtナノ粒子のものである。
【図10】FePtナノ粒子の粒径分布を示す図である。左は、オレイルアミン及びオクタン酸を固定化したFePtナノ粒子、右はビオチン化FePtナノ粒子のものである。
【図11】ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルを示す図である。
【図12】ビオチン配位子及びビオチン化FePtナノ粒子のIRスペクトルの拡大図である。
【図13】オレイルアミン及びオクタン酸を固定化したFePtナノ粒子とビオチン化FePtナノ粒子のIRスペクトルを示す図である。
【図14】ビオチン化FePtナノ粒子の構造を示す図である。
【図15】ビオチン化FePtナノ粒子及びビオチンの定量結果を示す図である。
【図16】ビオチン化FePtナノ粒子及びビオチンの500nmにおける吸収の検量線を示す図である。
【図17】ビオチン化FePtナノ粒子の溶液からの磁気分離方法の概要を示す図である。
【図18】磁気分離したビオチン化FePtナノ粒子の滴下によるスペクトルの変化を示す図である。
【特許請求の範囲】
【請求項1】
強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子。
【請求項2】
強磁性FePtナノ粒子がL10規則構造を有する請求項1記載の有機分子固定化強磁性ナノ粒子。
【請求項3】
強磁性FePtナノ粒子が、Fe錯体及びPt錯体を沸点300℃以上のポリオール系溶媒で処理することにより得られるものである請求項1又は2記載の有機分子固定化強磁性ナノ粒子。
【請求項4】
強磁性FePtナノ粒子に固定化される有機分子が、特異的結合のパートナー及び生理活性物質から選ばれる請求項1〜3のいずれか1項に記載の有機分子固定化強磁性ナノ粒子。
【請求項5】
強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することを特徴とする、請求項1〜4のいずれか1項に記載の有機分子固定化強磁性ナノ粒子の製造方法。
【請求項6】
カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物の少なくとも一方が特異的結合のパートナー、DNA、酵素及び生理活性物質から選ばれる残基を有する請求項5記載の製造方法。
【請求項7】
請求項5又は6記載の製造方法により得られた有機分子固定化強磁性ナノ粒子。
【請求項8】
請求項1〜4及び7のいずれか1項に記載の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【請求項1】
強磁性FePtナノ粒子に有機分子を固定化してなる有機分子固定化強磁性ナノ粒子。
【請求項2】
強磁性FePtナノ粒子がL10規則構造を有する請求項1記載の有機分子固定化強磁性ナノ粒子。
【請求項3】
強磁性FePtナノ粒子が、Fe錯体及びPt錯体を沸点300℃以上のポリオール系溶媒で処理することにより得られるものである請求項1又は2記載の有機分子固定化強磁性ナノ粒子。
【請求項4】
強磁性FePtナノ粒子に固定化される有機分子が、特異的結合のパートナー及び生理活性物質から選ばれる請求項1〜3のいずれか1項に記載の有機分子固定化強磁性ナノ粒子。
【請求項5】
強磁性FePtナノ粒子を脂肪酸及び脂肪族アミンで処理して、強磁性FePtナノ粒子に脂肪酸及び脂肪族アミンを固定化させた後、カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物で処理することを特徴とする、請求項1〜4のいずれか1項に記載の有機分子固定化強磁性ナノ粒子の製造方法。
【請求項6】
カテコール残基を有する有機化合物及びメルカプト基を有する有機化合物の少なくとも一方が特異的結合のパートナー、DNA、酵素及び生理活性物質から選ばれる残基を有する請求項5記載の製造方法。
【請求項7】
請求項5又は6記載の製造方法により得られた有機分子固定化強磁性ナノ粒子。
【請求項8】
請求項1〜4及び7のいずれか1項に記載の有機分子固定化強磁性ナノ粒子、及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を含む溶液中から磁気により前記有機分子固定化強磁性ナノ粒子及び/又は該有機分子固定化強磁性ナノ粒子に他の物質が結合した粒子を分離することを特徴とする有機分子固定化強磁性ナノ粒子の分離方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2008−260724(P2008−260724A)
【公開日】平成20年10月30日(2008.10.30)
【国際特許分類】
【出願番号】特願2007−105415(P2007−105415)
【出願日】平成19年4月13日(2007.4.13)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 研究集会名:慶應義塾大学理工学部化学科 平成18年度卒業論文発表会 開催日:2007年2月2日、3日 開催場所:慶應義塾大学(矢上キャンパス) 卒業論文要旨集発行日:2007年1月31日
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
【公開日】平成20年10月30日(2008.10.30)
【国際特許分類】
【出願日】平成19年4月13日(2007.4.13)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 研究集会名:慶應義塾大学理工学部化学科 平成18年度卒業論文発表会 開催日:2007年2月2日、3日 開催場所:慶應義塾大学(矢上キャンパス) 卒業論文要旨集発行日:2007年1月31日
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
[ Back to top ]